WO2017114456A1 - 吗啉衍生物的盐及其晶型、其制备方法及药物组合物、用途 - Google Patents
吗啉衍生物的盐及其晶型、其制备方法及药物组合物、用途 Download PDFInfo
- Publication number
- WO2017114456A1 WO2017114456A1 PCT/CN2016/112966 CN2016112966W WO2017114456A1 WO 2017114456 A1 WO2017114456 A1 WO 2017114456A1 CN 2016112966 W CN2016112966 W CN 2016112966W WO 2017114456 A1 WO2017114456 A1 WO 2017114456A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- morpholine derivative
- morpholine
- tartrate
- free base
- acetone
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/33—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems
- C07C309/34—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems formed by two rings
- C07C309/35—Naphthalene sulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/41—Preparation of salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/43—Separation; Purification; Stabilisation; Use of additives by change of the physical state, e.g. crystallisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C53/00—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen
- C07C53/08—Acetic acid
- C07C53/10—Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/235—Saturated compounds containing more than one carboxyl group
- C07C59/245—Saturated compounds containing more than one carboxyl group containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/235—Saturated compounds containing more than one carboxyl group
- C07C59/245—Saturated compounds containing more than one carboxyl group containing hydroxy or O-metal groups
- C07C59/255—Tartaric acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- the invention belongs to the technical field of medicinal chemistry, in particular to a salt of a morpholine derivative and a crystal form thereof, a preparation method thereof, and a pharmaceutical composition and use thereof.
- CN103562191 discloses a free base form of the morpholine derivative, a process for its preparation, and a renin inhibitory activity.
- the free base is a semi-solid or amorphous powder, is poorly water-soluble, and is more susceptible to oxidation and is not suitable for long-term storage.
- the present invention provides novel pharmaceutically acceptable salts of morpholine derivatives, including malate, tartrate, hydrochloride, acetate and naphthalene diphosphate, of which tartrate has A crystal form (tetrahydrate), B crystal form (anhydrous) and dihydrate three crystalline salt forms, malate, hydrochloride, acetate each have a crystalline salt type, naphthalene Phosphate is amorphous.
- the present invention has one or more improved properties compared to the free base of known morpholine derivatives.
- the present invention further provides a process for the preparation of a salt and a crystalline form of the morpholine derivative, a pharmaceutical composition and use thereof.
- One of the contents of the present invention is to provide a morpholine derivative malate and a process for the preparation thereof.
- the morpholine derivative malate is a compound formed by a morpholine derivative and L-malic acid in a 1:1 molar ratio, and has the following structural formula:
- the morpholine derivative malate has a crystalline form, and its X-ray powder diffraction pattern is 7.767° ⁇ 0.2°, 13.897° ⁇ 0.2°, 14.775° ⁇ 0.2°, 17.098° ⁇ 0.2°, 18.999° ⁇ at 2 ⁇ . Characteristic peaks are present at 0.2°, 20.153 ⁇ 0.2°, 20.960° ⁇ 0.2°, 21.423° ⁇ 0.2°, 26.348° ⁇ 0.2°, 27.892° ⁇ 0.2°.
- the crystalline form of the morpholine derivative malate has an X-ray powder diffraction pattern of 2.598° ⁇ 0.2°, 7.357° ⁇ 0.2°, 7.767° ⁇ 0.2°, 10.395° ⁇ 0.2°, 11.108 at 2 ⁇ .
- the morpholine derivative malate has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative malate has a polarizing light microscope (PLM) pattern as shown in FIG. 2.
- PLM polarizing light microscope
- the morpholine derivative malate has a thermogravimetric analysis (TGA) pattern as shown in FIG. 3, and the TGA pattern shows that the morpholine derivative malate is decomposed at about 185.8 ° C, and the sample has no weight loss before decomposition. .
- TGA thermogravimetric analysis
- the morpholine derivative malate has a differential thermal analysis (DSC) spectrum as shown in Fig. 4, and its DSC spectrum shows an endothermic peak (95 J/g) at around 121 °C.
- DSC differential thermal analysis
- the morpholine derivative malate has a dynamic moisture adsorption analysis (DVS) spectrum as shown in FIG. 5, and the DVS spectrum thereof shows that the morpholine derivative malate is in a range of 20% to 80% humidity.
- the internal weight change is approximately 1.2%.
- the method for preparing the morpholine derivative malate comprises the steps of dissolving a morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran (preferably acetone) to obtain a morpholine-containing compound.
- the molar ratio of the morpholine derivative free base to malic acid is from 1:1.1 to 1:3.3, preferably 1:1.1.
- the morpholine derivative malate has one or more improved properties, for example, a better crystalline state, it is difficult to absorb moisture at 20%-80% RH, and the water solubility is greatly improved. ( ⁇ 100 mg/mL) and good stability under conditions of light and oxidation.
- a second aspect of the present invention provides a morpholine derivative tartrate salt and a crystal form thereof, and a process for the preparation thereof.
- the morpholine derivative tartrate is a compound formed by a morphine ratio of a morpholine derivative and L-tartaric acid, and has the following structural formula:
- the method for preparing the morpholine derivative tartrate comprises the steps of dissolving L-tartaric acid by dissolving the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran (preferably acetone) Water, an aqueous solution of L-tartaric acid was added dropwise to a solution of the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran, and stirred at room temperature overnight to precipitate a white solid.
- the molar ratio of the morpholine derivative to L-tartaric acid is 1:1.03-1:2.2, preferably 1:1.03.
- the morpholine derivative tartrate is a morpholine derivative tartrate crystal form B, and its X-ray powder diffraction pattern is 3.339° ⁇ 0.2°, 6.562° ⁇ 0.2°, 11.331° ⁇ 0.2°, 16.396° ⁇ 0.2 at 2 ⁇ . Characteristic peaks at °, 22.041 ° ⁇ 0.2 °.
- the morpholine derivative tartrate crystal form B has an X-ray powder diffraction pattern at 2.39° ⁇ 0.2°, 5.078° ⁇ 0.2°, 6.562° ⁇ 0.2°, 6.864° ⁇ 0.2°, 8.250° at 2 ⁇ .
- the morpholine derivative tartrate crystal form B has an XRPD pattern as shown in FIG.
- the morpholine derivative tartrate crystal form B has a PLM pattern as shown in FIG.
- the morpholine derivative tartrate crystal form B has a TGA pattern as shown in FIG. 8, and the TGA pattern shows that the morpholine derivative tartrate crystal form B decomposes at around 186.0 ° C, and has a 2.5% slow weight loss before decomposition. (The weight loss starts from around 150 °C).
- the morpholine derivative tartrate crystal form B has a DSC spectrum as shown in FIG. 9 and a DSC spectrum It is shown that the morpholine derivative tartrate crystal form B has an endothermic peak (38 J/g) at around 161.5 °C.
- the morpholine derivative tartrate crystal form B has a DVS spectrum as shown in FIG. 10, and the DVS spectrum shows that the weight change is about 7% in a humidity range of 20% to 80%, which is relatively easy to absorb moisture. It may also become a hydrate.
- the morpholine derivative tartrate crystal form B is added to a mixed solvent of acetone and water, stirred at room temperature for 2 days, and then filtered to obtain a morpholine derivative tartrate dihydrate; the volume ratio of acetone to water is preferably 30:1.
- the crystalline form of the morpholine derivative tartrate dihydrate has an X-ray powder diffraction pattern of 9.851° ⁇ 0.2°, 13.434° ⁇ 0.2°, 14.410° ⁇ 0.2°, 14.774° ⁇ 0.2° at 2 ⁇ .
- the morpholine derivative tartrate dihydrate has an XRPD pattern as shown in FIG.
- the morpholine derivative tartrate dihydrate has a PLM pattern as shown in FIG.
- the morpholine derivative tartrate dihydrate has a TGA pattern as shown in FIG. 13, and the TGA pattern shows that the morpholine derivative tartrate dihydrate is decomposed at about 189.6 ° C, and has a gradient of 6.95% before decomposition. weightlessness.
- the morpholine derivative tartrate dihydrate has a DSC spectrum as shown in FIG. 14, and the DSC spectrum shows that the morpholine derivative tartrate dihydrate has an endothermic peak at about 29.5 ° C (112 J / g), there is an exothermic peak (27 J/g) around 99 °C, and an endothermic peak (19 J/g) around 154 °C.
- the morpholine derivative tartrate dihydrate has a DVS spectrum as shown in FIG. 15, and the DVS spectrum shows a weight change of about 11.06% in a humidity range of 0% to 80%, which is relatively easy to absorb moisture.
- the invention also discloses a morpholine derivative tartrate tetrahydrate, which is a crystalline form A, and its X-ray powder diffraction pattern is 9.082° ⁇ 0.2°, 14.426° ⁇ 0.2°, 14.802° ⁇ 0.2°, 16.275° ⁇ at 2 ⁇ . There are characteristic peaks at 0.2°, 20.085° ⁇ 0.2°, 20.872° ⁇ 0.2°, 21.978° ⁇ 0.2°, 23.236° ⁇ 0.2°.
- the morpholine derivative tartrate crystal form A has an X-ray powder diffraction pattern at 9.82° ⁇ 0.2°, 11.964° ⁇ 0.2°, 13.558° ⁇ 0.2°, 14.426° ⁇ 0.2°, 14.802° at 2 ⁇ .
- the morpholine derivative tartrate crystal form A has an XRPD pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a PLM pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a TGA pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a DSC pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a DVS pattern as shown in FIG.
- the invention also discloses a preparation method of the morpholine derivative tartrate crystal form A, which comprises the steps of dissolving the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran (preferably Acetone), obtaining a solution of the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran, dissolving L-tartaric acid in water to obtain an aqueous solution of tartaric acid, and adding the aqueous solution of the L-tartaric acid to the solution
- the morpholine derivative free base is stirred in a solution of acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran at room temperature for not less than 48 h, and a white solid is precipitated and filtered.
- the morpholine derivative tartrate has one or more improved properties, for example, both crystal forms A and B have a better crystalline state, but crystal form A is very high at 20%-80% RH. It is difficult to absorb moisture, and Form B is easy to absorb moisture. At the same time, it improves water solubility (50-300mg/mL), and under oxidizing conditions, Form B has better stability than Form A and is stable under light conditions. Sex is equal.
- a third aspect of the present invention provides a morpholine derivative hydrochloride and a process for the preparation thereof.
- the morpholine derivative hydrochloride is a compound formed of a morpholine derivative and hydrochloric acid in a molar ratio of 1:2, and has the following structural formula:
- the morpholine derivative hydrochloride has a crystalline form, and its X-ray powder diffraction pattern is 3.981° ⁇ 0.2°, 7.784° ⁇ 0.2°, 8.667° ⁇ 0.2°, 13.634° ⁇ 0.2°, 18.238° ⁇ at 2 ⁇ . Characteristic peaks are present at 0.2°, 19.620° ⁇ 0.2°, 24.624° ⁇ 0.2°, 24.987° ⁇ 0.2°, 28.072° ⁇ 0.2°, 31.815° ⁇ 0.2°.
- the crystalline form of the morpholine derivative hydrochloride has an X-ray powder diffraction pattern at 2.81 ° ⁇ 0.2 °, 7.784 ° ⁇ 0.2 °, 8.667 ° ⁇ 0.2 °, 10.914 ° ⁇ 0.2 °, 11.557.
- the morpholine derivative hydrochloride has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative hydrochloride has a polarizing light microscope (PLM) pattern as shown in FIG.
- PLM polarizing light microscope
- the morpholine derivative hydrochloride has a thermogravimetric analysis (TGA) spectrum as shown in FIG. 23, and the TGA spectrum thereof shows that the morpholine derivative hydrochloride continuously loses weight during the temperature rise, and the decomposition temperature is 207. °C, there are two stages of weight loss before decomposition, a total of about 9.1%.
- TGA thermogravimetric analysis
- the morpholine derivative hydrochloride has a differential thermal analysis (DSC) spectrum as shown in Fig. 24, and its DSC spectrum shows a broad endothermic peak between 25 and 115 ° C (57.68 J). /g), having an endothermic peak at 133 ° C (57.65 J/g).
- DSC differential thermal analysis
- the morpholine derivative hydrochloride has a dynamic moisture adsorption analysis (DVS) spectrum as shown in FIG. 25, and the DVS spectrum thereof shows that the morpholine derivative hydrochloride is absorbed at 0%-60% RH. It is 15% water, which is deliquescent in this humidity range.
- DVS dynamic moisture adsorption analysis
- the method for preparing the morpholine derivative hydrochloride comprises the steps of dissolving the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran (preferably acetone) to obtain morpholine.
- the free base was stirred in a solution of acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran at room temperature overnight to precipitate a white solid.
- the molar ratio of the morpholine derivative free base to hydrochloric acid is from 1:1.03 to 1:3.5, preferably 1:3.4.
- the morpholine derivative hydrochloride has one or more improved properties, for example, having a better crystalline state, greatly improving water solubility (>500 mg/mL), but very easy It absorbs moisture and has good thermal stability under light conditions.
- a fourth aspect of the present invention provides a morpholine derivative acetate and a process for the preparation thereof.
- the morpholine derivative acetate is a compound formed of a morpholine derivative and acetic acid in a molar ratio of 1:1, and has the following structural formula:
- the morpholine derivative acetate has a crystalline form, and its X-ray powder diffraction pattern is 7.784° ⁇ 0.2°, 11.429° ⁇ 0.2°, 14.455° ⁇ 0.2°, 16.874° ⁇ 0.2°, 19.899° ⁇ at 2 ⁇ . There are characteristic peaks at 0.2°, 21.146° ⁇ 0.2°, and 24.887° ⁇ 0.2°.
- the crystalline form of the morpholine derivative acetate has an X-ray powder diffraction pattern of 6.012° ⁇ 0.2°, 7.457° ⁇ 0.2°, 7.784° ⁇ 0.2°, 10.391° ⁇ 0.2°, 10.768 in 2 ⁇ .
- the morpholine derivative acetate has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative acetate has a polarizing light microscope (PLM) pattern as shown in FIG.
- PLM polarizing light microscope
- the morpholine derivative acetate has a thermogravimetric analysis (TGA) pattern as shown in FIG. 28, and the TGA pattern shows that the morpholine derivative acetate has a step ladder at 50 ° C and 75 ° C respectively. Weight loss was 4.0% and 9.5%, respectively.
- TGA thermogravimetric analysis
- the morpholine derivative acetate has a differential thermal analysis (DSC) spectrum as shown in Fig. 29, and its DSC spectrum shows an endothermic peak (61 J/g) at 95 °C.
- DSC differential thermal analysis
- the morpholine derivative acetate has a dynamic moisture adsorption analysis (DVS) spectrum as shown in FIG. 30, and the DVS spectrum thereof shows that the morpholine derivative hydrochloride is dried at an initial humidity of 0%. At the stage, there is about 5% weight loss, and then 20%-60% of the humidity absorbs 6.4% of the water, and absorbs 40% of the water at 90% humidity, indicating deliquescence.
- DVS dynamic moisture adsorption analysis
- the method for preparing the morpholine derivative acetate comprises the steps of dissolving the morpholine derivative free base in acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran (preferably acetone) to obtain morpholine.
- Derivative free base Acetone, chloroform, acetonitrile, ethyl acetate, methanol or tetrahydrofuran solution
- the acetic acid is dissolved in acetone to obtain an acetone solution of acetic acid
- the acetone solution of the acetic acid is added dropwise to the acetone containing the free base of the morpholine derivative.
- the molar ratio of the morpholine derivative free base to acetic acid is from 1:1.1 to 1:3.1, preferably 1:1.4.
- the morpholine derivative acetate has one or more improved properties, for example, having a better crystalline state and greatly improving the water solubility (150 to 300 mg/mL), but very It is easy to absorb moisture and has good thermal stability under light conditions.
- a fifth aspect of the present invention provides a morpholine derivative naphthalene disulfonate and a process for the preparation thereof.
- the morpholine derivative naphthalene disulfonate is a compound formed by a morpholine derivative and naphthalene disulfonic acid in a 1:1 molar ratio, and has the following structural formula:
- the morpholine derivative naphthalene disulfonate is amorphous.
- the morpholine derivative naphthalene disulfonate has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the preparation method of the morpholine derivative naphthalene disulfonate comprises the steps of dissolving a morpholine derivative free base in ethyl acetate, dissolving naphthalene disulfonic acid in ethanol, and reacting naphthalene disulfonic acid in ethanol.
- the solution was added dropwise to an ethyl acetate solution containing the free base of the morpholine derivative, and stirred at room temperature to obtain a white flocculent precipitate, which was filtered.
- the molar ratio of the morpholine derivative free base to naphthalene disulfonic acid is from 1:1.1 to 1:3, preferably 1:1.4.
- the morpholine derivative naphthalene disulfonate has one or more improved properties, for example, having a better crystalline state and greatly improving water solubility (>500 mg/mL), but It is very easy to absorb moisture and has good thermal stability under light conditions.
- the "stirring" described in any of the above-described preparation methods of the present invention can be accomplished by conventional techniques such as magnetic stirring and mechanical stirring.
- the stirring speed is 50 to 1800 rpm, preferably 300 to 900 rpm.
- room temperature means 15-25 °C.
- overnight means 24 hours or more.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically and/or prophylactically effective amount of one or more morpholine derivative salts of the present invention or a crystalline form thereof, or prepared by the method of the present invention
- the morpholine derivative salt or a crystal form thereof is selected from the group consisting of a malate salt of a morpholine derivative, a tartrate salt, a tartrate salt form A, a tartrate salt form B, a hydrochloride salt, an acetate salt, and a naphthalene disulfonate.
- the acid salt, in addition, the pharmaceutical composition may further comprise other pharmaceutically acceptable salt forms, crystal forms or amorphous forms of the morpholine derivative.
- Excipients in the pharmaceutical composition include sugars, cellulose and derivatives thereof, starch or modified starch, solid inorganic substances such as calcium phosphate, dicalcium phosphate, hydroxyapatite, calcium sulfate, calcium carbonate, semi-solid Such as lipid or paraffin, binders such as microcrystalline cellulose, ethyl cellulose, hydroxymethyl cellulose, hydroxypropyl methyl cellulose, hydroxyethyl cellulose, glidants such as colloidal silica, Light anhydrous silicic acid, crystalline cellulose, talc or magnesium stearate, disintegrants such as sodium starch glycolate, crospovidone, croscarmellose, sodium carboxymethylcellulose, dried corn Starch, lubricants such as stearic acid, magnesium stearate, sodium stearyl fumarate, polyethylene glycol.
- solid inorganic substances such as calcium phosphate, dicalcium phosphate, hydroxyapatite, calcium sulfate
- the pharmaceutical composition may be in a solid or liquid form, such as a solid oral dosage form, including tablets, granules, powders, pills, and capsules; liquid oral dosage forms including solutions, syrups, suspensions, dispersions, and emulsions; Injectable preparations, including solutions, dispersions, and lyophilizates.
- the formulation may be adapted for rapid release, delayed release or modified release of the active ingredient. It may be a conventional, dispersible, chewable, orally dissolved or rapidly melted formulation. Routes of administration include oral, intravenous subcutaneous injection, injection into tissue, transdermal administration, rectal administration, intranasal administration, and the like.
- the pharmaceutical composition can be prepared using methods well known to those skilled in the art.
- the morpholine derivative salt of the present invention or a crystalline form thereof is mixed with one or more pharmaceutically acceptable carriers, optionally with other crystalline forms of pharmaceutically acceptable morpholine derivatives, Other amorphous or salt forms are mixed, optionally in admixture with one or more other active ingredients.
- the solid preparation can be prepared by a process such as direct mixing, granulation, or the like.
- the present invention provides the use of the aforementioned morpholine derivative salt of the present invention or a crystal form thereof as a renin inhibitor, and the use thereof for the preparation of a therapeutic and/or preventive hypertension, cardiac insufficiency, diabetic nephropathy, etc. Use in medicines for diseases.
- Figure 1 is an XRPD pattern of a morpholine derivative malate
- Figure 2 is a PLM spectrum of a morpholine derivative malate
- Figure 3 is a TGA map of the morpholine derivative malate
- Figure 4 is a DSC spectrum of a morpholine derivative malate
- Figure 5 is a DVS spectrum of the morpholine derivative malate
- Figure 6 is an XRPD pattern of the morpholine derivative tartrate crystal form B
- Figure 7 is a PLM spectrum of the morpholine derivative tartrate crystal form B
- Figure 8 is a TGA spectrum of the morpholine derivative tartrate salt form B
- Figure 9 is a DSC spectrum of the morpholine derivative tartrate crystal form B
- Figure 10 is a DVS spectrum of the morpholine derivative tartrate salt form B
- Figure 11 is an XRPD pattern of the morpholine derivative tartrate dihydrate
- Figure 12 is a PLM spectrum of the morpholine derivative tartrate dihydrate
- Figure 13 is a TGA spectrum of the morpholine derivative tartrate dihydrate
- Figure 14 is a DSC spectrum of the morpholine derivative tartrate dihydrate
- Figure 15 is a DVS spectrum of the morpholine derivative tartrate dihydrate
- Figure 16 is an XRPD pattern of the morpholine derivative tartrate crystal form A
- Figure 17 is a PLM spectrum of the morpholine derivative tartrate crystal form A
- Figure 18 is a TGA spectrum of the morpholine derivative tartrate crystal form A
- Figure 19 is a DSC chart of the morpholine derivative tartrate crystal form A
- Figure 20 is a DVS spectrum of the morpholine derivative tartrate crystal form A
- Figure 21 is an XRPD pattern of the morpholine derivative hydrochloride
- Figure 22 is a PLM spectrum of the morpholine derivative hydrochloride
- Figure 23 is a TGA spectrum of the morpholine derivative hydrochloride
- Figure 24 is a DSC chart of the morpholine derivative hydrochloride
- Figure 25 is a DVS spectrum of the morpholine derivative hydrochloride
- Figure 26 is an XRPD pattern of the morpholine derivative acetate
- Figure 27 is a PLM spectrum of the morpholine derivative acetate
- Figure 28 is a TGA spectrum of the morpholine derivative acetate
- Figure 29 is a DSC spectrum of the morpholine derivative acetate
- Figure 30 is a DVS spectrum of the morpholine derivative acetate
- Figure 31 is an XRPD pattern of the morpholine derivative naphthalene disulfonate.
- the instrument used for X-ray powder diffraction was a Bruker D8 Advance diffractometer with K X-rays with a copper target wavelength of 1.54 nm, a ⁇ -2 ⁇ goniometer, a Mo monochromator at 40 kV and 40 mA operating conditions, Lynxeye detector.
- the instrument is used to correct the peak position with the standard sample supplied with the instrument before use.
- the acquisition software is Diffrac Plus XRD Commander and the analysis software is MDI Jade 5.0.
- the sample is tested at room temperature and the sample to be tested is placed on an organic slide.
- the detailed detection conditions are as follows: angle range: 3 to 4 ° 2 ⁇ ; step size: 0.02 ° 2 ⁇ ; speed: 0.2 s / step. Samples were not ground prior to testing unless otherwise stated.
- Polarized light microscopy (PLM) spectra were taken from an XP-500E polarized light microscope (Shanghai Changfang Optical Instrument Co., Ltd.). Take a small amount of powder sample on the glass slide, add a small amount of mineral oil to better disperse the powder sample, cover the cover glass, and then place the sample on the XP-500E polarized light microscope (Shanghai Changfang Optical Instrument Co., Ltd.) On the stage, select the appropriate magnification to observe the shape of the sample and take a picture.
- PLM Polarized light microscopy
- the differential thermal analysis (DSC) data was taken from the TA Instruments Q200MDSC, the instrument control software was Thermal Advantage, and the analysis software was Universal Analysis. Usually take 1 ⁇ 10 mg of the sample in the aluminum crucible with capping (unless otherwise specified), raise the sample from room temperature to 200 at a heating rate of 10 ° C / min under the protection of 50 mL / min dry N 2 °C or 300 °C, while the TA software records the change in heat during the temperature rise of the sample. In the present application, the melting point is reported as the starting temperature.
- Thermogravimetric analysis (TGA) data was taken from the TA Instruments Q500TGA, the instrument control software was Thermal Advantage, and the analysis software was Universal Analysis. Usually, 5 to 15 mg of the sample is placed in a platinum crucible, and the sample is raised from room temperature to 300 ° C under the protection of 50 mL/min dry N 2 at a heating rate of 10 ° C/min using a segmented high-resolution detection method. At the same time, the TA software records the change in weight of the sample during the heating process.
- Dynamic moisture adsorption analysis (DVS) data was taken from the TA Instruments Q5000TGA, the instrument control software was Thermal Advantage, and the analysis software was Universal Analysis. A sample of 1 to 10 mg is usually placed in a platinum crucible. Typically, the TA software records the change in weight of the sample during a change in relative humidity from 0% to 80% to 0%. Depending on the specifics of the sample, different adsorption and desorption steps are also applied to the sample.
- the starting material morpholine derivative free base of the present invention is prepared according to the method disclosed in the document CN103562191 (WO2012124775), and the morpholine derivative free base in the following examples is referred to as free base.
- the morpholine derivative malate has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative malate has a polarizing light microscope (PLM) pattern as shown in FIG.
- the morpholine derivative malate has a thermogravimetric analysis (TGA) pattern as shown in FIG. 3, and its TGA pattern shows that the morpholine derivative malate decomposes at around 185.8 ° C, and the sample has no weight loss before decomposition.
- TGA thermogravimetric analysis
- the morpholine derivative malate has a differential thermal analysis (DSC) pattern as shown in Fig. 4, and its DSC spectrum shows an endothermic peak (95 J/g) at around 121 °C.
- DSC differential thermal analysis
- DVS dynamic moisture adsorption analysis
- the morpholine derivative tartrate crystal form B has an XRPD pattern as shown in FIG.
- the morpholine derivative tartrate crystal form B has a PLM pattern as shown in FIG.
- the morpholine derivative tartrate crystal form B has a TGA pattern as shown in Fig. 8.
- the TGA pattern shows that the morpholine derivative tartrate crystal form B decomposes at around 186.0 ° C, and has a 2.5% slow weight loss before decomposition (weight loss from Start around 150 °C).
- the morpholine derivative tartrate crystal form B has a DSC pattern as shown in Fig. 9, and the DSC pattern shows that the morpholine derivative tartrate crystal form B has an endothermic peak (38 J/g) at around 161.5 °C.
- the morpholine derivative tartrate crystal form B has a DVS spectrum as shown in FIG. 10, and the DVS spectrum shows a weight change of about 7% in a humidity range of 20% to 80%, which is relatively easy to absorb moisture and may also change. Hydrate.
- the morpholine derivative tartrate salt form B obtained in Example 2 was 10 mg, 50 ml of a mixed solvent of acetone and water in a volume ratio of 30:1 was added, and the mixture was stirred at room temperature for 2 days, and then filtered and characterized.
- the morpholine derivative tartrate dihydrate has an XRPD pattern as shown in FIG.
- the morpholine derivative tartrate dihydrate has a PLM pattern as shown in FIG.
- the morpholine derivative tartrate dihydrate has a TGA pattern as shown in FIG. 13, and the TGA pattern shows that the morpholine derivative tartrate dihydrate is decomposed at about 189.6 ° C, and has a gradient weight loss of 6.95% before decomposition.
- the morpholine derivative tartrate dihydrate has a DSC spectrum as shown in FIG. 14, and the DSC spectrum shows that the morpholine derivative tartrate dihydrate has an endothermic peak (112 J/g) at around 29.5 °C. There is an exothermic peak (27 J/g) around 99 °C and an endothermic peak (19 J/g) around 154 °C.
- the morpholine derivative tartrate dihydrate has a DVS spectrum as shown in FIG. 15, and the DVS spectrum shows a weight change of about 11.06% in a humidity range of 0% to 80%, which is relatively easy to absorb moisture.
- the morpholine derivative tartrate crystal form A has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a polarizing light microscope (PLM) pattern as shown in FIG.
- the morpholine derivative tartrate crystal form A has a thermogravimetric analysis (TGA) pattern as shown in Fig. 18, and its TGA pattern shows that the morpholine derivative tartrate tetrahydrate has a weight loss of 10.0-10.4%.
- TGA thermogravimetric analysis
- the morpholine derivative tartrate crystal form A has a differential thermal analysis (DSC) spectrum as shown in FIG. 19, and its DSC spectrum shows an endothermic peak at 62.7 ° C and a melting endothermic peak at 159.3 ° C. Decomposed after 191 °C.
- DSC differential thermal analysis
- the morpholine derivative tartrate crystal form A has a dynamic moisture adsorption analysis (DVS) pattern as shown in FIG. 20, and its DVS spectrum shows that the morpholine derivative tartrate crystal form A is in the range of 20% to 80% humidity.
- the weight change in the range is about 1.5%, which is not easy to absorb moisture.
- the morpholine derivative hydrochloride has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative hydrochloride has a polarizing light microscope (PLM) pattern as shown in FIG.
- PLM polarizing light microscope
- the morpholine derivative hydrochloride has a thermogravimetric analysis (TGA) spectrum as shown in FIG. 23, and the TGA spectrum thereof shows that the morpholine derivative hydrochloride continuously loses weight during the temperature rise, and the decomposition temperature is 207 ° C, and the decomposition There are two sections of weight loss before, totaling about 9.1%.
- TGA thermogravimetric analysis
- the morpholine derivative hydrochloride has a differential thermal analysis (DSC) pattern as shown in Fig. 24, and its DSC spectrum shows a broad endothermic peak (57.68 J/g) between 25 and 115 °C. It has an endothermic peak at 133 ° C (57.65 J / g).
- DSC differential thermal analysis
- DVS dynamic moisture adsorption analysis
- the morpholine derivative acetate has an X-ray powder diffraction (XRPD) pattern as shown in FIG.
- the morpholine derivative acetate has a polarizing light microscope (PLM) pattern as shown in FIG.
- PLM polarizing light microscope
- the morpholine derivative acetate has a thermogravimetric analysis (TGA) spectrum as shown in FIG. 28, and the TGA spectrum thereof shows that the morpholine derivative acetate has a step weight loss at 50 ° C and 75 ° C, respectively. They are 4.0% and 9.5% respectively.
- TGA thermogravimetric analysis
- the morpholine derivative acetate had a differential thermal analysis (DSC) spectrum as shown in Fig. 29, and its DSC spectrum showed an endothermic peak (61 J/g) at 95 °C.
- DSC differential thermal analysis
- the weight loss around % then absorbs 6.4% of the water in the humidity range of 20%-60%, and absorbs 40% of the water in 90% humidity, indicating deliquescence.
- the morpholine derivative naphthalene disulfonate has an X-ray powder diffraction (XRPD) pattern as shown in Fig. 31 and is an amorphous salt.
- XRPD X-ray powder diffraction
- the free base, the salt of the morpholine derivative of Example 1-7 was each taken 5 mg, and pure water was gradually added to each sample at 25 ° C until the sample was completely dissolved, and the sample was calculated according to the actual weight of the sample and the amount of water. Solubility. The results are shown in Table 1. The parallel test showed that the sample did not undergo crystal transformation during the test.
- the salt of the morpholine derivative of the examples was each taken 5 mg, the humidity was increased from 20% to 80%, and the weight change was tested. The results are shown in Table 2.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description






| 测试样品 | 溶解度(mg/ml) |
| 吗啉衍生物苹果酸盐 | 100 |
| 吗啉衍生物酒石酸盐晶型B | 150-300 |
| 吗啉衍生物酒石酸盐二水合物 | 100 |
| 吗啉衍生物酒石酸盐晶型A | 100 |
| 吗啉衍生物盐酸盐 | >500 |
| 吗啉衍生物醋酸盐 | 150-300 |
| 吗啉衍生物萘二磺酸盐 | >500 |
| 吗啉衍生物游离碱 | 5-10 |
| 测试样品 | 20%~80%吸湿重量变化 | 难易程度 |
| 吗啉衍生物苹果酸盐 | 1.70% | 难 |
| 吗啉衍生物酒石酸盐晶型B | 6.58% | 易 |
| 吗啉衍生物酒石酸盐二水合物 | 9.05% | 易 |
| 吗啉衍生物酒石酸盐晶型A | 1.50% | 难 |
| 吗啉衍生物盐酸盐 | 31.80% | 非常容易 |
| 吗啉衍生物醋酸盐 | 21.30% | 非常容易 |
| 吗啉衍生物萘二磺酸盐 | N/A | 非常容易 |
| 吗啉衍生物游离碱 | N/A | 难 |
| 测试样品 | 分解量(%) | 新杂质种类(种) |
| 吗啉衍生物苹果酸盐 | 8.46% | 2 |
| 吗啉衍生物酒石酸盐晶型B | 3.24% | 4 |
| 吗啉衍生物酒石酸盐晶型A | 17.93% | 6 |
| 吗啉衍生物游离碱 | 38.0% | 6 |
| 吗啉衍生物萘二磺酸盐 | 51.47% | 7 |
| 吗啉衍生物盐酸盐 | 63.53% | 6 |
| 吗啉衍生物醋酸盐 | 95.71% | 5 |
| 吗啉衍生物酒石酸盐二水合物 | N/A | N/A |
| 测试样品 | 分解量(%) |
| 吗啉衍生物苹果酸盐 | 2.05% |
| 吗啉衍生物酒石酸盐晶型B | 3.61% |
| 吗啉衍生物酒石酸盐晶型A | 2.14% |
| 吗啉衍生物盐酸盐 | 5.27% |
| 吗啉衍生物醋酸盐 | 100% |
| 吗啉衍生物萘二磺酸盐 | 12.58% |
| 吗啉衍生物游离碱 | 100% |
Claims (39)
- 一种吗啉衍生物苹果酸盐,其特征在于:所述吗啉衍生物苹果酸盐是吗啉衍生物和L-苹果酸以1:1摩尔比形成的化合物,其结构式如下:

- 如权利要求1所述的吗啉衍生物苹果酸盐,其特征在于:所述吗啉衍生物苹果酸盐为晶型,其X射线粉末衍射图谱在2θ为7.767°±0.2°、13.897°±0.2°、14.775°±0.2°、17.098°±0.2°、18.999°±0.2°、20.153±0.2°、20.960°±0.2°、21.423°±0.2°、26.348°±0.2°、27.892°±0.2°处具有特征峰。
- 如权利要求2所述的吗啉衍生物苹果酸盐,其特征在于:所述的晶型,其X射线粉末衍射图谱还在2θ为5.598°±0.2°、7.357°±0.2°、10.395°±0.2°、11.108°±0.2°、16.037°±0.2°、16.523°±0.2°、19.410°±0.2°、22.645°±0.2°、26.630°±0.2°、26.891°±0.2°、27.380°±0.2°、31.056°±0.2°、33.306°±0.2°、33.775°±0.2°、39.231°±0.2°处具有特征峰。
- 如权利要求2或3所述的吗啉衍生物苹果酸盐,其特征在于:其具有如图1所示的X射线粉末衍射图谱。
- 权利要求1-4中任一项所述吗啉衍生物苹果酸盐的制备方法,其特征在于,包括以下步骤:将吗啉衍生物游离碱溶于丙酮中,将L-苹果酸溶于乙醇中,将L-苹果酸的乙醇溶液滴加到含有吗啉衍生物游离碱的丙酮溶液中,室温下搅拌过夜,析出白色固体,过滤。
- 根据权利要求5所述的制备方法,其特征在于:所述吗啉衍生物游离碱与苹果酸的摩尔比为1:1.1~1:3.3。
- 一种吗啉衍生物酒石酸盐,其特征在于:是吗啉衍生物和L-酒石酸以1:1摩尔比形成的化合物,其结构式如下:
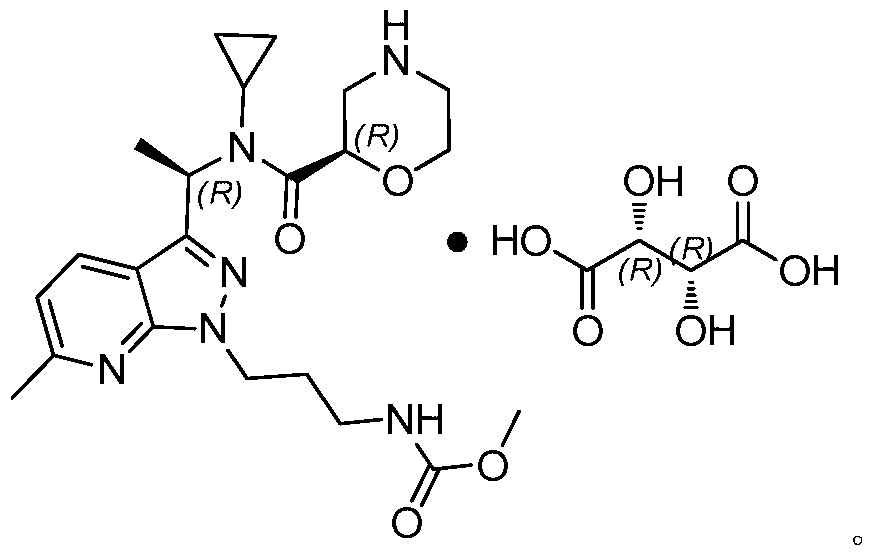
- 如权利要求7所述的吗啉衍生物酒石酸盐,其特征在于:所述吗啉衍生物酒石酸盐为吗啉衍生物酒石酸盐晶型B,其X射线粉末衍射图谱在2θ为3.339°±0.2°、6.562°±0.2°、11.331°±0.2°、16.396°±0.2°、22.041°±0.2°处具有特征峰。
- 如权利要求8所述的吗啉衍生物酒石酸盐,其特征在于:所述的晶型B,其X射线粉末衍射图谱还在2θ为5.078°±0.2°、6.864°±0.2°、8.250°±0.2°、8.444°±0.2°、11.030°±0.2°、12.864°±0.2°、13.907°±0.2°、14.642°±0.2°、19.100°±0.2°、19.359°±0.2°、25.251°±0.2°、26.768°±0.2°、27.894°±0.2°、29.510°±0.2°、38.343°±0.2°处具有特征峰。
- 如权利要求8或9所述的吗啉衍生物酒石酸盐,其特征在于:所述吗啉衍生物酒石酸盐晶型B,具有如图6所示的XRPD图谱。
- 如权利要求7-10中任一项所述的吗啉衍生物酒石酸盐的制备方法,包括以下步骤:将吗啉衍生物游离碱溶于丙酮,将L-酒石酸溶于水,将L-酒石酸的水溶液滴加至吗啉衍生物游离碱的丙酮溶液中,室温搅拌过夜,析出白色固体,过滤。
- 如权利要求11所述的制备方法,其特征在于:所述吗啉衍生物游离碱与L-酒石酸的摩尔比为1:1.03。
- 权利要求7所述的吗啉衍生物酒石酸盐的二水合物,其特征在于:其X射线粉末衍射图谱在2θ为9.851°±0.2°、14.410°±0.2°、14.774°±0.2°、15.052°±0.2°、16.254°±0.2°、20.847°±0.2°、23.225°±0.2°处具有特征峰。
- 如权利要求13所述的吗啉衍生物酒石酸盐的二水合物,其特征在于:其X射线粉 末衍射图谱还在2θ为13.434°±0.2°、15.415°±0.2°、15.701°±0.2°、16.755°±0.2°、17.283°±0.2°、18.079°±0.2°、18.576°±0.2°、20.077°±0.2°、21.960°±0.2°、24.351°±0.2°、27.046°±0.2°、27.865°±0.2°、38.458°±0.2°处具有特征峰。
- 如权利要求13或14所述的吗啉衍生物酒石酸盐的二水合物,其特征在于具有如图11所示的XRPD图谱。
- 如权利要求13、14或15所述的吗啉衍生物酒石酸盐的二水合物的制备方法,其特征在于,包括如下步骤:将吗啉衍生物酒石酸盐晶型B加入丙酮和水的混合溶剂中,室温下搅拌2天后,过滤,得到吗啉衍生物酒石酸盐二水合物。
- 如权利要求16所述的制备方法,其特征在于:所述丙酮和水的体积比为30:1。
- 权利要求7所述的吗啉衍生物酒石酸盐的四水合物,其特征在于:其为吗啉衍生物酒石酸盐晶型A,其X射线粉末衍射图谱在2θ为9.882°±0.2°、14.426°±0.2°、14.802°±0.2°、16.275°±0.2°、20.085°±0.2°、20.872°±0.2°、21.978°±0.2°、23.236°±0.2°处具有特征峰。
- 如权利要求18所述的吗啉衍生物酒石酸盐的四水合物,其特征在于:所述的晶型A,其X射线粉末衍射图谱还在2θ为11.964°±0.2°、13.558°±0.2°、15.076°±0.2°、15.450°±0.2°、16.046°±0.2°、16.754°±0.2°、17.320°±0.2°、18.450°±0.2°、18.790°±0.2°、19.728°±0.2°、20.577°±0.2°、22.426°±0.2°、23.704°±0.2°、24.399°±0.2°、25.346°±0.2°、25.913°±0.2°、26.991°±0.2°、28.199°±0.2°、28.445°±0.2°、29.030°±0.2°、30.209°±0.2°、30.480°±0.2°、32.791°±0.2°、34.796°±0.2°、36.226°±0.2°、38.472°±0.2°处具有特征峰。
- 如权利要求18或19所述的吗啉衍生物酒石酸盐的四水合物,其特征在于具有如图16所示的XRPD图谱。
- 如权利要求18、19或20所述的吗啉衍生物酒石酸盐的四水合物的制备方法,其特征在于,包括如下步骤:将吗啉衍生物游离碱溶于丙酮,将L-酒石酸溶于水,将L-酒石酸的水溶液滴加至吗啉衍生物游离碱的丙酮溶液中,室温搅拌,析出白色固体,过滤。
- 如权利要求21所述的制备方法,其特征在于:所述吗啉衍生物游离碱与L-酒石酸 的摩尔比为1:2.2,所述丙酮与水的体积比为20:1。
- 一种吗啉衍生物盐酸盐,其特征在于:所述吗啉衍生物盐酸盐是吗啉衍生物和盐酸以1:2摩尔比形成的化合物,其结构式如下:

- 如权利要求23所述的吗啉衍生物盐酸盐,其特征在于:所述吗啉衍生物盐酸盐具有晶型形态,其X射线粉末衍射图谱在2θ为3.981°±0.2°、7.784°±0.2°、8.667°±0.2°、13.634°±0.2°、18.238°±0.2°、19.620°±0.2°、24.624°±0.2°、24.987°±0.2°、28.072°±0.2°、31.815°±0.2°处具有特征峰。
- 如权利要求24所述的吗啉衍生物盐酸盐,其特征在于:所述的晶型,其X射线粉末衍射图谱还在2θ为10.914°±0.2°、11.557°±0.2°、12.211°±0.2°、14.675°±0.2°、15.419°±0.2°、15.817°±0.2°、17.158°±0.2°、19.116°±0.2°、20.618°±0.2°、21.261°±0.2°、21.901°±0.2°、22.428°±0.2°、22.548°±0.2°、23.342°±0.2°、25.902°±0.2°、26.267°±0.2°、26.730°±0.2°、26.946°±0.2°、29.994°±0.2°、31.154°±0.2°、33.220°±0.2°、34.670°±0.2°、35.201°±0.2°处具有特征峰。
- 如权利要求24或25所述的吗啉衍生物盐酸盐,其特征在于所述吗啉衍生物盐酸盐具有如图21所示的XRPD图谱。
- 如权利要求23-26中任一项所述的吗啉衍生物盐酸盐的制备方法,包括以下步骤:将吗啉衍生物游离碱溶于丙酮中,将盐酸溶于丙酮中,然后向含吗啉衍生物游离碱的丙酮溶液中滴加盐酸的丙酮溶液,室温下搅拌过夜,析出白色固体,过滤。
- 如权利要求27所述的制备方法,所述吗啉衍生物游离碱与盐酸的摩尔比为 1:1.03~1:3.5。
- 一种吗啉衍生物醋酸盐,其特在在于:是吗啉衍生物和醋酸以1:1摩尔比形成的化合物,其结构式如下:

- 如权利要求29所述的吗啉衍生物醋酸盐,其特在在于,所述吗啉衍生物醋酸盐具有晶型形态,其X射线粉末衍射图谱在2θ为7.784°±0.2°、11.429°±0.2°、14.455°±0.2°、16.874°±0.2°、19.899°±0.2°、21.146±0.2°、24.887°±0.2°处具有特征峰。
- 如权利要求30所述的吗啉衍生物醋酸盐,其特征在于:所述的晶型,其X射线粉末衍射图谱还在2θ为6.012°±0.2°、7.457°±0.2°、10.391°±0.2°、10.768°±0.2°、13.652°±0.2°、14.089°±0.2°、14.841°±0.2°、15.516°±0.2°、16.301°±0.2°、17.592°±0.2°、18.777°±0.2°、19.375°±0.2°、20.521°±0.2°、21.541°±0.2°、22.346°±0.2°、22.966°±0.2°、23.347°±0.2°、24.585°±0.2°、25.546°±0.2°、26.028°±0.2°、26.328°±0.2°、27.484°±0.2°、27.753°±0.2°、29.206°±0.2°、30.611°±0.2°、30.972°±0.2°、31.233°±0.2°、31.801°±0.2°、33.696°±0.2°、34.699°±0.2°、35.313°±0.2°、36.441°±0.2°、37.961°±0.2°、38.179°±0.2°、39.325°±0.2°处具有特征峰。
- 如权利要求30或31所述的吗啉衍生物醋酸盐,其特在在于,所述吗啉衍生物醋酸盐具有如图26所示的XRPD图谱。
- 权利要求29-32中任一项所述的吗啉衍生物醋酸盐的制备方法,其特征在于,包括如下步骤:将吗啉衍生物游离碱溶于丙酮中,将醋酸溶于丙酮中,然后向含吗啉衍生物游离碱的丙酮溶液中滴加醋酸的丙酮溶液,室温下搅拌过夜,析出白色固体,过滤。
- 如权利要求33所示的制备方法,其特征在于:所述吗啉衍生物游离碱与醋酸的摩尔比为1:1.1~1:3.1。
- 一种吗啉衍生物萘二磺酸盐,其特征在于,是吗啉衍生物和萘二磺酸以1:1摩尔比形成的化合物,其结构式如下:

- 如权利要求35所述吗啉衍生物萘二磺酸盐,其特征在于为无定形盐。
- 如权利要求36所述吗啉衍生物萘二磺酸盐,其特征在于:具有如图31所示的XRPD图谱。
- 如权利要求35-37中任一项所述的吗啉衍生物萘二磺酸盐的制备方法,其特征在于,包括如下步骤:将吗啉衍生物游离碱溶于乙酸乙酯中,将萘二磺酸溶于乙醇中,将萘二磺酸的乙醇溶液滴加到含有吗啉衍生物游离碱的乙酸乙酯溶液中,室温下搅拌,得到白色絮状沉淀,过滤。
- 如权利要求38所述的制备方法,其特征在在于:所述吗啉衍生物游离碱与萘二磺酸的摩尔比为1:1.1~1:3。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES16881245T ES2918674T3 (es) | 2015-12-29 | 2016-12-29 | Sal de derivado de morfolina y forma cristalina de la misma, así como método de preparación, composición farmacéutica y uso de la misma |
| US16/066,490 US10519150B2 (en) | 2015-12-29 | 2016-12-29 | Salts of morpholine derivative, crystal forms thereof, processes for producing the same, pharmaceutical compositions including the same, and use thereof |
| JP2018534034A JP6818031B2 (ja) | 2015-12-29 | 2016-12-29 | モルホリン誘導体の塩、結晶形、その製造方法、これらを含む医薬組成物およびその用途 |
| DK16881245.1T DK3398946T3 (da) | 2015-12-29 | 2016-12-29 | Salt af morpholin-derivat og krystallin form deraf, samt fremstillingsfremgangsmåde, farmaceutisk sammensætning og anvendelse af samme |
| PL16881245.1T PL3398946T3 (pl) | 2015-12-29 | 2016-12-29 | Sól pochodnej morfolinowej i jej postać krystaliczna, jak również sposób jej wytwarzania, kompozycja farmaceutyczna i jej zastosowanie |
| EP16881245.1A EP3398946B1 (en) | 2015-12-29 | 2016-12-29 | Salt of morpholine derivative and crystalline form thereof, as well as preparation method, pharmaceutical composition and use of the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201511016783 | 2015-12-29 | ||
| CN201511016783.3 | 2015-12-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017114456A1 true WO2017114456A1 (zh) | 2017-07-06 |
Family
ID=59224612
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2016/112966 Ceased WO2017114456A1 (zh) | 2015-12-29 | 2016-12-29 | 吗啉衍生物的盐及其晶型、其制备方法及药物组合物、用途 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US10519150B2 (zh) |
| EP (1) | EP3398946B1 (zh) |
| JP (1) | JP6818031B2 (zh) |
| CN (3) | CN113816952A (zh) |
| DK (1) | DK3398946T3 (zh) |
| ES (1) | ES2918674T3 (zh) |
| HU (1) | HUE059444T2 (zh) |
| PL (1) | PL3398946T3 (zh) |
| PT (1) | PT3398946T (zh) |
| TW (1) | TWI705065B (zh) |
| WO (1) | WO2017114456A1 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4209218A4 (en) * | 2020-09-04 | 2024-09-11 | Shanghai Pharmaceuticals Holding Co., Ltd. | Application of nitrogen-containing saturated heterocyclic compound |
| WO2022047730A1 (en) * | 2020-09-04 | 2022-03-10 | Shanghai Pharmaceuticals Holding Co., Ltd. | Methods to treat inflammatory bowel disease |
| CN116425677B (zh) * | 2022-01-11 | 2026-04-14 | 苏州恩华生物医药科技有限公司 | 氨基环丁烷衍生物的盐、其晶型及其制备方法和应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103562191A (zh) * | 2011-03-16 | 2014-02-05 | 上海医药集团股份有限公司 | 含氮的饱和杂环化合物 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1248869A2 (en) * | 2000-01-07 | 2002-10-16 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| WO2014042263A1 (ja) * | 2012-09-14 | 2014-03-20 | 田辺三菱製薬株式会社 | 新規レニン阻害薬 |
| JP5764628B2 (ja) * | 2012-09-14 | 2015-08-19 | 田辺三菱製薬株式会社 | 医薬組成物 |
-
2016
- 2016-12-29 HU HUE16881245A patent/HUE059444T2/hu unknown
- 2016-12-29 TW TW105143910A patent/TWI705065B/zh active
- 2016-12-29 EP EP16881245.1A patent/EP3398946B1/en active Active
- 2016-12-29 CN CN202110801274.0A patent/CN113816952A/zh active Pending
- 2016-12-29 ES ES16881245T patent/ES2918674T3/es active Active
- 2016-12-29 CN CN202110794158.0A patent/CN113816951B/zh active Active
- 2016-12-29 US US16/066,490 patent/US10519150B2/en active Active
- 2016-12-29 WO PCT/CN2016/112966 patent/WO2017114456A1/zh not_active Ceased
- 2016-12-29 JP JP2018534034A patent/JP6818031B2/ja active Active
- 2016-12-29 CN CN201611246909.0A patent/CN106928218B/zh active Active
- 2016-12-29 DK DK16881245.1T patent/DK3398946T3/da active
- 2016-12-29 PL PL16881245.1T patent/PL3398946T3/pl unknown
- 2016-12-29 PT PT168812451T patent/PT3398946T/pt unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103562191A (zh) * | 2011-03-16 | 2014-02-05 | 上海医药集团股份有限公司 | 含氮的饱和杂环化合物 |
Non-Patent Citations (2)
| Title |
|---|
| BERGE, S.M. ET AL.: "Pharmaceutical Salts", JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 66, no. 1, 31 January 1977 (1977-01-31), pages 1 - 19, XP002675560 * |
| See also references of EP3398946A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3398946A4 (en) | 2019-08-28 |
| CN106928218B (zh) | 2021-08-03 |
| ES2918674T3 (es) | 2022-07-19 |
| JP6818031B2 (ja) | 2021-01-20 |
| CN113816951A (zh) | 2021-12-21 |
| EP3398946A1 (en) | 2018-11-07 |
| EP3398946B1 (en) | 2022-05-04 |
| JP2019500385A (ja) | 2019-01-10 |
| PL3398946T3 (pl) | 2022-10-10 |
| US10519150B2 (en) | 2019-12-31 |
| DK3398946T3 (da) | 2022-08-08 |
| TW201722950A (zh) | 2017-07-01 |
| PT3398946T (pt) | 2022-08-09 |
| CN113816952A (zh) | 2021-12-21 |
| TWI705065B (zh) | 2020-09-21 |
| US20190016718A1 (en) | 2019-01-17 |
| CN113816951B (zh) | 2022-09-27 |
| HUE059444T2 (hu) | 2022-11-28 |
| CN106928218A (zh) | 2017-07-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2750676T5 (en) | Crystalline dihydrochloride hydrate salt of omecamtiv mecarbil and process for its preparation | |
| CN105175473A (zh) | 一种奥贝胆酸晶型i及其制备方法、药物组合物和用途 | |
| EP3205653B1 (en) | Crystal form of bisulfate of jak inhibitor and preparation method therefor | |
| JP2003531173A (ja) | ゾルピデムヘミタートレイト | |
| WO2015014315A1 (zh) | 一种抑制剂的晶型及其制备方法和用途 | |
| WO2017114456A1 (zh) | 吗啉衍生物的盐及其晶型、其制备方法及药物组合物、用途 | |
| WO2017160703A1 (en) | Solid state forms of nilotinib salts | |
| CN106279126B (zh) | 阿法替尼酸加成盐及其晶型、其制备方法及药物组合物 | |
| US12384784B2 (en) | Polymorphs of Acalabrutinib, a Bruton's tyrosine kinase inhibitor | |
| AU2012283277B2 (en) | Polymorphs of 6-(piperidin-4-yloxy)-2H-isoquinolin-1-one hydrochloride | |
| CN103819461B (zh) | N-[3-氯-4-(3-氟苄氧基)苯基]-6-[5-[[2-(甲亚磺酰基)乙基]氨基]甲基]-2-呋喃基]-4-喹唑啉胺多晶型物及其制备方法 | |
| EP3517529B1 (en) | Salt of quinazoline derivative, preparation method therefor and application thereof | |
| CN105732596A (zh) | N-[3-氯-4-(3-氟苄氧基)苯基]-6-[5-[[2-(甲亚磺酰基)乙基]氨基]甲基]-2-呋喃基]-4-喹唑啉胺多晶型物及其制备方法 | |
| US20060270685A1 (en) | Anhydrous ziprasidone mesylate and a process for its preparation | |
| HK1218544B (zh) | 奧美卡替莫卡必爾的鹽和製備鹽的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16881245 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018534034 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016881245 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016881245 Country of ref document: EP Effective date: 20180730 |