WO2017130623A1 - Pd担持Zr系複合酸化物 - Google Patents
Pd担持Zr系複合酸化物 Download PDFInfo
- Publication number
- WO2017130623A1 WO2017130623A1 PCT/JP2016/088772 JP2016088772W WO2017130623A1 WO 2017130623 A1 WO2017130623 A1 WO 2017130623A1 JP 2016088772 W JP2016088772 W JP 2016088772W WO 2017130623 A1 WO2017130623 A1 WO 2017130623A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composite oxide
- supported
- mass
- based composite
- precursor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/92—Chemical or biological purification of waste gases of engine exhaust gases
- B01D53/94—Chemical or biological purification of waste gases of engine exhaust gases by catalytic processes
- B01D53/9445—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC]
- B01D53/945—Simultaneously removing carbon monoxide, hydrocarbons or nitrogen oxides making use of three-way catalysts [TWC] or four-way-catalysts [FWC] characterised by a specific catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/066—Zirconium or hafnium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/10—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of rare earths
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
- B01J35/391—Physical properties of the active metal ingredient
- B01J35/393—Metal or metal oxide crystallite size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/70—Catalysts, in general, characterised by their form or physical properties characterised by their crystalline properties, e.g. semi-crystalline
- B01J35/77—Compounds characterised by their crystallite size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
- B01J37/0018—Addition of a binding agent or of material, later completely removed among others as result of heat treatment, leaching or washing,(e.g. forming of pores; protective layer, desintegrating by heat)
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/024—Multiple impregnation or coating
- B01J37/0244—Coatings comprising several layers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
- B01J37/031—Precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/04—Mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F01—MACHINES OR ENGINES IN GENERAL; ENGINE PLANTS IN GENERAL; STEAM ENGINES
- F01N—GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR MACHINES OR ENGINES IN GENERAL; GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR INTERNAL-COMBUSTION ENGINES
- F01N3/00—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust
- F01N3/08—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous
- F01N3/10—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous by thermal or catalytic conversion of noxious components of exhaust
- F01N3/101—Three-way catalysts
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F01—MACHINES OR ENGINES IN GENERAL; ENGINE PLANTS IN GENERAL; STEAM ENGINES
- F01N—GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR MACHINES OR ENGINES IN GENERAL; GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR INTERNAL-COMBUSTION ENGINES
- F01N3/00—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust
- F01N3/08—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous
- F01N3/10—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous by thermal or catalytic conversion of noxious components of exhaust
- F01N3/24—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous by thermal or catalytic conversion of noxious components of exhaust characterised by constructional aspects of converting apparatus
- F01N3/28—Construction of catalytic reactors
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/10—Noble metals or compounds thereof
- B01D2255/102—Platinum group metals
- B01D2255/1023—Palladium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/206—Rare earth metals
- B01D2255/2061—Yttrium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/206—Rare earth metals
- B01D2255/2063—Lanthanum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/206—Rare earth metals
- B01D2255/2065—Cerium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/207—Transition metals
- B01D2255/20715—Zirconium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/40—Mixed oxides
- B01D2255/407—Zr-Ce mixed oxides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/90—Physical characteristics of catalysts
- B01D2255/908—O2-storage component incorporated in the catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2258/00—Sources of waste gases
- B01D2258/01—Engine exhaust gases
- B01D2258/014—Stoichiometric gasoline engines
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
- B01J2235/15—X-ray diffraction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F01—MACHINES OR ENGINES IN GENERAL; ENGINE PLANTS IN GENERAL; STEAM ENGINES
- F01N—GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR MACHINES OR ENGINES IN GENERAL; GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR INTERNAL-COMBUSTION ENGINES
- F01N2370/00—Selection of materials for exhaust purification
- F01N2370/02—Selection of materials for exhaust purification used in catalytic reactors
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F01—MACHINES OR ENGINES IN GENERAL; ENGINE PLANTS IN GENERAL; STEAM ENGINES
- F01N—GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR MACHINES OR ENGINES IN GENERAL; GAS-FLOW SILENCERS OR EXHAUST APPARATUS FOR INTERNAL-COMBUSTION ENGINES
- F01N3/00—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust
- F01N3/08—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous
- F01N3/10—Exhaust or silencing apparatus having means for purifying, rendering innocuous, or otherwise treating exhaust for rendering innocuous by thermal or catalytic conversion of noxious components of exhaust
Definitions
- the present invention relates to a Pd-supported Zr-based composite oxide.
- it is related with the said complex oxide which can be used conveniently as a component of the exhaust gas purification catalyst of a motor vehicle.
- a three-way catalyst in which a noble metal is supported on an inorganic oxide is known.
- Three-way catalysts are widely used as catalysts that can efficiently remove hydrocarbons (HC), nitrogen oxides (NO x ), and carbon monoxide (CO) simultaneously.
- Patent Documents 1 to 3 research on palladium (Pd) having low heat resistance is active (Patent Documents 1 to 3), and certain results are being achieved.
- the present invention has been made in view of the above circumstances.
- the purpose is to provide a catalyst material for exhaust gas purification that suppresses the deterioration of Pd due to continued use under automobile driving conditions and is excellent in exhaust gas purification performance particularly in a low temperature region.
- the present invention provides the following materials.
- Pd is supported on a composite oxide support containing Zr
- Pd obtained by XAFS (X-ray absorption fine structure) analysis is characterized in that the peak position of the maximum peak within the range of the bond distance of Pd of 2.500 to 3.500 ⁇ is 3.050 to 3.110 ⁇ Supported Zr-based composite oxide.
- a catalyst material for exhaust gas purification that suppresses the deterioration of Pd even if the use is continued under severe conditions such as when the vehicle is running, and is excellent in exhaust gas purification performance particularly in a low temperature region.
- FIG. 1 is a view showing the XAFS spectrum of the Pd-supported Zr-based composite oxide obtained in Example 1 in comparison with the spectrum of palladium oxide.
- FIG. 2 is a diagram showing the XAFS spectrum of the Pd-supported Zr-based composite oxide obtained in Comparative Example 1 in comparison with the spectrum of palladium oxide.
- FIG. 3 is a diagram showing the Pd crystallite diameter of the Pd-supported Zr-based composite oxide obtained in each example as a relative value with respect to the Pd crystallite diameter of a comparative example having the same composition.
- FIG. 4 is a graph showing a comparison of cold HC emissions for the two-layer catalysts obtained in Example 10 and Comparative Example 10, respectively.
- FIG. 5 is a graph comparing the cold HC emission behavior of the two-layer catalysts obtained in Example 10 and Comparative Example 10, respectively.
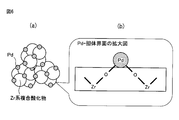
- FIG. 6 is a schematic diagram for explaining the structure of the Pd-supported Zr-based composite oxide.
- Pd-supported Zr-based composite oxide of the present invention Pd is supported on a composite oxide support containing Zr, and a Pd bond distance of 2.500 to 3 is obtained by XAFS (X-ray absorption fine structure) analysis.
- the peak position of the maximum peak within the range of .500 cm is 3.050 cm to 3.110 cm.
- the composite oxide support contains zirconium (Zr), preferably contains cerium (Ce) together with Zr, and may further contain a metal element other than these.
- the metal element other than Zr and Ce is preferably a rare earth element (excluding Ce; hereinafter, the symbol [Ln] may be used when generically referring to rare earth elements excluding Ce).
- rare earth elements include yttrium (Y), lanthanum (La), praseodymium (Pr), neodymium (Nd), samarium (Sm), europium (Eu), gadolinium (Gd), erbium (Er), ytterbium (Yb). ), Lutetium (Lu) and the like. Of these, one or more selected from Y, La, Pr, Nd, and Eu are preferable.
- the complex oxide support in the present invention Preferably composed of a complex oxide containing Zr and Ce; More preferably, it is composed of a composite oxide containing Zr and Ce and a rare earth element excluding Ce; More preferably, it is made of a complex oxide containing Zr and Ce and one or more rare earth elements selected from Y, La, Pr, Nd, and Eu.
- Preferred ratios of the respective metal elements contained in the composite oxide support in the present invention are as follows as oxide conversion values when the total mass of the composite oxide support is 100% by mass.
- Zr 1% by mass or more and 90% by mass or less as ZrO 2 converted value
- Ce 90% by mass or less as CeO 2 converted value
- Rare earth element excluding Ce 1% by mass or more and 20% by mass or less as Ln 2 O 3 converted value
- the total of the Zr content in terms of ZrO 2 and the Ce content in terms of CeO 2 is 80% by mass or more when the total mass of the composite oxide support is 100% by mass.
- the content of Zr in terms of ZrO 2 is more preferably 2 to 85% by mass, further preferably 10 to 75% by mass, particularly preferably 20 to 60% by mass.
- the Ce content in terms of CeO 2 is more preferably 5 to 80% by mass, still more preferably 15 to 70% by mass, particularly preferably 25 to 60% by mass.
- the content of rare earth elements excluding Ce described above in terms of Ln 2 O 3 is more preferably 3 to 18% by mass, and further preferably 5 to 15% by mass. Particularly preferred is 8 to 12% by mass.
- the Pd content (supported amount) in the Pd-supported Zr-based composite oxide of the present invention is 0.75 mass% or more and 3 or more as a Pd metal conversion value when the mass of the composite oxide support is 100 mass%. It is preferable that it is 0.0 mass% or less.
- the Pd particle-support interface of the Pd-supported Zr-based composite oxide of the present invention is viewed microscopically, the Pd atoms on the surface of the Pd metal particles and the Zr atoms on the support surface are chemically bonded via oxygen atoms O. It is verified from the XAFS analysis that a Pd—O—Zr bond is formed. An enlarged schematic view of this state is shown in FIG. Although FIG. 6B is drawn in a form having two Pd—O—Zr bonds per Pd atom, the present invention is not limited to this embodiment. The present invention also includes other forms such as one or three Pd—O—Zr bonds per Pd atom. In the present invention, it is sufficient that the second neighboring element of Pd is mainly Zr.
- the analysis is performed by paying attention to the interatomic distance centered on the Pd atom among the wide variety of information obtained from the XAFS analysis.
- the bond distance between the Pd atom and the oxygen atom which is the first adjacent element of Pd is about 1.5 mm, regardless of the type of the composite oxide. A value is obtained.
- the bond distance between Pd and the second neighboring element takes a value peculiar to the type of M in Pd—OM (where M is the second neighboring element). Therefore, the type of M in Pd—OM can be identified by examining the bond distance of the second adjacent element of Pd and comparing it with the literature value.
- the composite oxide forms a Pd—O—Zr bond.
- the peak of the second adjacent element of Pd in the XAFS analysis appears in the range of approximately 2.500 to 3.500. Therefore, paying attention to the maximum peak in this range, the comparison is made with the above bond distance.
- the peak position of the maximum peak within the range of the Pd bond distance of 2.500 mm to 3.500 mm obtained by the XAFS analysis is 3.050 mm to 3.110 mm.
- the composite oxide may be considered to form a Pd—O—Zr bond.
- the Pd-supported Zr-based composite oxide of the present invention may be manufactured by any method as long as it has the above composition and structure.
- the Pd-supported Zr-based composite oxide of the present invention is, for example, A first step (Zr-containing sol forming step) in which an aqueous solution containing a zirconium oxide precursor (ZrO precursor) is heated to form a Zr-containing sol; A second step (Pd precursor addition step) to obtain a Pd-added Zr-containing sol by adding a palladium precursor (Pd precursor) to the Zr-containing sol obtained in the first step; A third step (precipitate generating step) in which the Pd-added Zr-containing sol obtained in the second step is contacted with a basic compound to obtain a precipitate; A fourth step (firing step) for firing the precipitate obtained in the third step; Can be manufactured by the method including the above in the order described above.
- the first step is a step of forming a Zr-containing sol by heat-treating an aqueous solution containing a ZrO precursor. This heat treatment is considered to convert the ZrO precursor into a sol in which Zr (OH) 4 is highly dispersed.
- Zr (OH) 4 is highly dispersed.
- a Pd—O—Zr bond is efficiently formed by adding a Pd precursor to such a highly dispersed Zr compound.
- ZrO precursor used here examples include zirconium nitrate, zirconium oxynitrate, zirconium sulfate, and zirconium acetate. Of these, zirconium oxynitrate is preferable from the viewpoint that a Pd—O—Zr bond can be formed with high efficiency.
- zirconium hydroxide when zirconium hydroxide is used as the ZrO precursor, the formation efficiency of Pd—O—Zr bonds in the obtained Pd-supported Zr-based composite oxide is low.
- the presence of a Zr (OH) 4 sol obtained by heat-treating the preferred aqueous solution of ZrO precursor is considered to be important for the formation of Pd—O—Zr bonds.
- the holding temperature in the heat treatment in the first step is preferably 90 ° C. or higher, more preferably 95 ° C. or higher.
- the upper limit of the heat treatment holding temperature is preferably 120 ° C. or less, and more preferably 100 ° C. or less.
- it is not prohibited to perform a heat treatment at a higher temperature, for example, about 150 ° C.
- the holding time in the heat treatment is desirably 5 hours to 48 hours, more preferably 7 hours to 24 hours. This holding time means the time for maintaining the above heat treatment holding temperature.
- the aqueous solution to be heat-treated may contain a rare earth oxide precursor (LnO precursor) in addition to the ZrO precursor.
- LnO precursor include rare earth nitrates, sulfates, acetates, and the like. Of these, rare earth nitrates are preferred from the viewpoint of easy formation of highly dispersed sols.
- the usage ratio of each precursor should be appropriately selected by those skilled in the art according to the desired composition in the Pd-supported Zr-based composite oxide.
- the first step in the present invention includes May be done in one step;
- the precursor to be added may be divided into a plurality of groups, and the addition and heat treatment may be sequentially performed for each group, and may be performed in multiple stages; or
- the precursor to be added may be divided into a plurality of groups, and heat treatment may be performed for each group, followed by mixing.
- the most preferred embodiment is a method in which the first step is performed in the following two steps.
- First step of the first step a step of preparing an aqueous solution containing a ZrO precursor and an LnO precursor, heat-treating the solution, and a second step of the first step: obtained in the first step A step of adding a precursor of Ce oxide to the heat-treated aqueous solution and heat-treating it.
- the first step when the first step is performed in two steps, the one that has passed through the second step is handled as a “Zr-containing sol” that is used in the second step. Even if the product that has passed through the first stage in the first step becomes a sol, if the second stage is scheduled to be performed, the sol is not a “Zr-containing sol” in the present invention. .
- the heat treatment conditions at each stage can be the preferred modes described above.
- the first-stage heat treatment condition and the second-stage heat treatment condition may be the same or different.
- the second step is a step of adding a palladium precursor (Pd precursor) to the Zr-containing sol obtained in the first step.
- Examples of the Pd precursor used here include palladium nitrate and palladium chloride. Of these, palladium nitrate is preferable from the viewpoint that a Pd—O—Zr bond can be formed with high efficiency.
- the amount of the Pd precursor to be used should be appropriately selected by those skilled in the art depending on the desired composition of the Pd-supported Zr-based composite oxide.
- a rare earth oxide precursor (LnO precursor) excluding ceria may be added.
- LnO precursor for example, a rare earth nitrate is preferable, and lanthanum nitrate is particularly preferable.
- the third step is a step of obtaining a precipitate by bringing the Pd-added Zr-containing sol obtained in the second step into contact with a basic compound.
- a basic compound used here ammonia or an organic base is preferable, for example.
- the organic base include pyridine, triethylamine, diazabicycloundecene, tetraalkylammonium hydroxide and the like.
- tetraalkylammonium hydroxide is preferable, and tetramethylammonium hydroxide is particularly preferable.
- the amount of the basic compound may be excessively large.
- a method of adding or gradually adding the aqueous solution obtained in the second step to an aqueous solution of the basic compound adjusted to a pH of about 13.0 or higher and 14.0 or lower. is preferred.
- the Pd-added Zr-containing sol obtained in the second step is brought into contact with a pore-forming agent together with a basic compound, so that an appropriate pore structure is obtained in the obtained Pd-supported Zr-based composite oxide. It is preferable at the point which forms.
- This pore-forming agent is a compound having a template effect that is taken into the precipitate obtained in the third step, and burns out and dissipates in the subsequent firing step to form pores.
- a pore forming agent for example, a long chain fatty acid is preferably used. Specific examples include capric acid, lauric acid, myristic acid, palmitic acid and the like.
- the pore forming agent may coexist in an aqueous solution of a basic compound.
- the amount used thereof can be 16.4 parts by mass or more and 65.4 parts by mass or less, preferably 24.5 parts by mass or more and 49.0 parts by mass with respect to 100 parts by mass of the Pd-supported Zr-based composite oxide. It is preferable that the amount is not more than part because the pore structure to be formed is suitable.
- the Pd-supported Zr-based composite oxide of the present invention can be obtained by firing the precipitate obtained in the third step.
- the firing temperature can be, for example, 250 ° C. or more and 1,200 ° C. or less, and preferably 300 ° C. or more and 500 ° C. or less.
- the firing time can be, for example, 2 hours or more and 24 hours or less.
- the ambient atmosphere at the time of firing is preferably an oxidizing atmosphere, and can be performed, for example, in the air.
- the Pd-supported Zr-based composite oxide of the present invention is obtained.
- the Pd-supported Zr-based composite oxide of the present invention is one in which deterioration of Pd is suppressed even when continued to be used under severe conditions, and is particularly excellent in HC purification performance in a low temperature region. Therefore, this Pd-supported Zr-based composite oxide can be suitably used, for example, as a component of an automobile exhaust gas purification catalyst.
- an automobile exhaust gas purification catalyst includes the Pd-supported Zr-based composite oxide of the present invention.
- the composite oxide is coated on a suitable substrate and used as a catalyst having a single layer or a multilayer structure. At least one of them is a coating layer of the Pd-supported Zr-based composite oxide of the present invention.
- the layer of the Pd-supported Zr-based composite oxide of the present invention may be in any layer of the multilayer catalyst. It is preferable that the lowermost layer is in direct contact with the material.
- the base material those generally used as a base material for automobile exhaust gas purification catalysts can be used.
- a monolith honeycomb substrate can be mentioned.
- the other layer can be, for example, a (composite) oxide catalyst on which a noble metal selected from Rh, Pt, and Pd is supported.
- a (composite) oxide catalyst on which a noble metal selected from Rh, Pt, and Pd is supported.
- the oxide as a carrier in this case include alumina, ceria, zirconia, cerium-zirconium composite oxide, and the like.
- Other layers can be formed on the substrate according to a known method.
- the automobile exhaust gas purification catalyst having the above-described structure can efficiently remove HC, NO x , and CO at the same time even under low temperature conditions, and Pd particles are deteriorated by sintering even if they are used continuously under harsh environments. The life is long because the degree is extremely small.
- Zr-containing sol formation step i) First stage 75 g of an aqueous zirconium oxynitrate solution having a ZrO 2 equivalent concentration of 20% by mass; 5 mL of an aqueous yttrium nitrate solution having a Y 2 O 3 equivalent concentration of 300 g / L, 7.5 mL of nitric acid having a HNO 3 equivalent concentration of 67.5% by mass and a specific gravity of 1.4, 1,000 mL of pure water Were mixed to prepare a Zr—Y aqueous solution. The entire amount of this Zr—Y aqueous solution was placed in a container equipped with a cooling tube and a stirrer, heated and maintained at 98 ° C. with stirring for 8 hours, and then cooled to room temperature.
- Precipitate generation step 10.1 g of lauric acid and 760 mL of an aqueous solution of tetramethylammonium hydroxide (TMAH) having a concentration of 128 g / L are mixed and stirred until the liquid becomes transparent, and a TMAH solution of lauric acid was prepared.
- TMAH tetramethylammonium hydroxide
- the Pd-added Zr-containing sol obtained in the above (3) Pd precursor addition step is added at a rate of 50 mL / min with stirring using a metering pump to have a precipitate. A slurry was obtained.
- Example 2 In the “(1) first stage heating and holding step” of Example 1 above, after the addition of the zirconium oxynitrate aqueous solution, 95 mol% of the Ce ions are tetravalent and the concentration in terms of CeO 2 is 200 g / L. Further, 3.0 mL of an aqueous cerium solution was added, and the same operation as in Example 1 was performed except that the amount of the aqueous cerium nitrate solution used in “(2) Second stage heating and holding step” was changed to 57 mL. I got a thing.
- Example 3 The amount of the zirconium oxynitrate aqueous solution used in “(1) the first stage heating and holding step” in Example 1 was 67.5 g, and the “(2) second stage heating and holding step” added the cerium nitrate aqueous solution.
- a Pd-supported Zr-based composite oxide was obtained in the same manner as in Example 1 except that 7.5 g of zirconium oxynitrate aqueous solution having the same concentration as above was added.
- Example 8 Pd-supported Zr-based composite oxides were obtained in the same manner as in Example 1 except that the amount of each solution used in each step was as shown in Table 1.
- the cerium nitrate solution was not added in “(1) Zr-containing sol formation step ii) Second stage”.
- Example 8 i) after heating and holding at 98 ° C. for 8 hours in the first stage, after cooling to room temperature, ii) without stirring the cerium nitrate solution ii) under stirring at 98 ° C. for 20 hours in the second stage Heat holding was performed.
- ⁇ Comparative Example 1> 30 g of a Zr-based composite oxide (commercially available product) whose composition in terms of oxides is Zr: Ce: La: Y 50: 40: 5: 5 is palladium nitrate having a Pd metal equivalent concentration of 8.2% by mass. After being immersed in 11.31 g of an aqueous solution at 25 ° C. for 1 hour, dried at 100 ° C. for 24 hours, and further fired at 400 ° C. for 5 hours to obtain a Pd-supported Zr-based composite oxide.
- the oxide equivalents of the amount of metal elements in the carrier precursor contained in each solution used in each step and the metal equivalents of the amount of Pd element in the Pd precursor are shown in Table 2, respectively. Indicated.
- the element was identified by calculating the bond distance of the second adjacent element of Pd from the XAFS spectrum after Fourier transform obtained for each sample and comparing it with the above value.
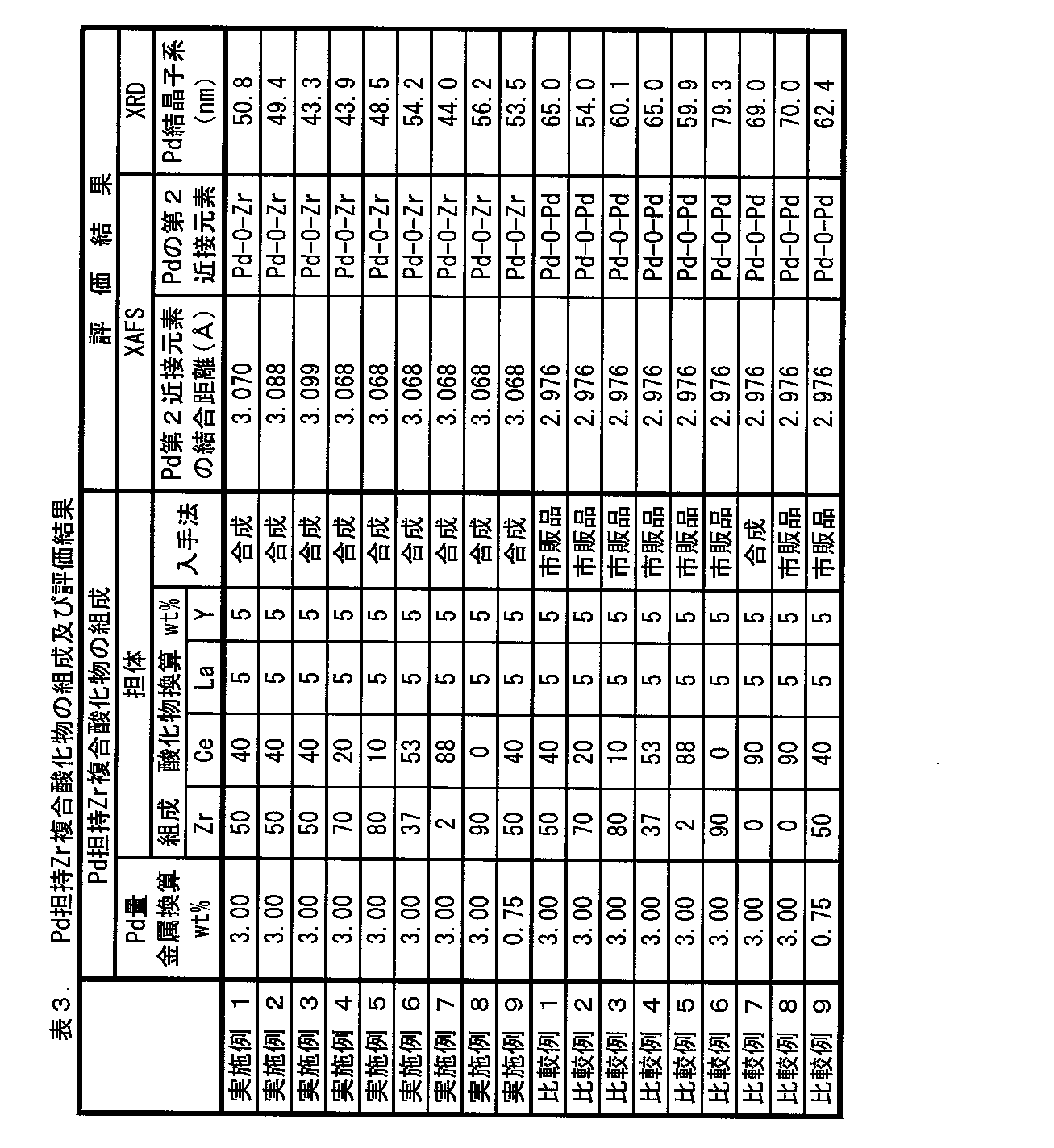
- Table 3 shows the bond distances of the second neighboring elements of Pd obtained for each sample and the types of the second neighboring elements specified therefrom. Moreover, the XAFS spectrum obtained by using each Zr type complex oxide obtained in Example 1 and Comparative Example 1 as a sample is shown in FIGS. 1 and 2 together with the spectrum of commercially available PdO.
- the Pd-supported Zr-based composite oxide of the present invention has a smaller Pd crystallite diameter after durability as compared with the conventional product. It is presumed that the generation of Pd—O—Zr bonds suppressed the deterioration of Pd due to durability.
- FIG. 3 shows the Pd crystallite diameter of the Pd-supported Zr-based composite oxide of each example as a relative value to the Pd crystallite diameter of a comparative example having the same composition. According to the present invention, it was verified that the sintering suppression effect of Pd is expressed. In the case of Comparative Example 7 in which the Zr content is zero, such an effect does not appear. This phenomenon seems to be due to the fact that no Pd—O—Zr bond is formed.
- Example 10 200 g of alumina, 200 g of the Pd-supported Zr-based composite oxide obtained in Example 1, 50 g of barium sulfate, and 2,000 g of water were mixed to prepare a lower layer forming slurry.
- the lower layer forming slurry was coated on a monolith honeycomb substrate having a capacity of 1 L, dried at 250 ° C., and then fired at 500 ° C. for 1 hour.
- a slurry for forming an upper layer was prepared by mixing an aqueous rhodium nitrate solution in an amount equivalent to 0.5 g in terms of Rh metal, 200 g of alumina, 200 g of zirconium oxide, 50 g of barium sulfate, and 2,000 g of water.
- This upper layer forming slurry is coated with the above lower layer forming slurry, further coated on the dried and fired monolith honeycomb substrate, dried at 250 ° C., and then fired at 500 ° C. for 1 hour, whereby the monolith honeycomb substrate is obtained.
- a two-layer catalyst coated on the material was prepared.
- Example 10 is the same as Example 10 except that the same amount of the Pd-supported Zr-based composite oxide obtained in Comparative Example 1 was used instead of the Pd-supported Zr-based composite oxide obtained in Example 1. Thus, a two-layer catalyst coated on the monolith honeycomb substrate was prepared.
- Example 10 has better catalyst performance after endurance than the catalyst of Comparative Example 10 (model of the conventional product). This phenomenon is thought to be due to the suppression of Pd degradation due to the formation of Pd—O—Zr bonds.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Combustion & Propulsion (AREA)
- Health & Medical Sciences (AREA)
- Mechanical Engineering (AREA)
- Toxicology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Environmental & Geological Engineering (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Thermal Sciences (AREA)
- Physics & Mathematics (AREA)
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
- Exhaust Gas After Treatment (AREA)
Abstract
Description
[1] Zrを含む複合酸化物担体上にPdが担持されており、
XAFS(X線吸収微細構造)分析によって得られる、Pdの結合距離2.500Å~3.500Åの範囲内における最大ピークのピーク位置が3.050Å~3.110Åであることを特徴とする、Pd担持Zr系複合酸化物。
[2] 前記複合酸化物担体の全質量を100質量%としたときに、
ZrO2換算のZrの含有量が1質量%以上90質量%以下であり、
CeO2換算のCeの含有量が90質量%以下であり、そして
ZrO2換算のZrの含有量とCeO2換算のCeの含有量との合計が80質量%以上である、[1]に記載のPd担持Zr系複合酸化物。
[3] 前記複合酸化物担体に対するPd金属換算のPd担持量が0.75質量%以上3.0質量%以下である、[1]又は[2]に記載のPd担持Zr系複合酸化物。
[4] [1]~[3]のいずれか一項に記載のPd担持Zr系複合酸化物を含むことを特徴とする、排ガス浄化用触媒。
本発明のPd担持Zr系複合酸化物は、Zrを含む複合酸化物担体上にPdが担持されており、XAFS(X線吸収微細構造)分析によって得られる、Pdの結合距離2.500Å~3.500Åの範囲内における最大ピークのピーク位置が3.050Å~3.110Åであることを特徴とする。
上記複合酸化物担体は、ジルコニウム(Zr)を含み、好ましくはZrとともにセリウム(Ce)を含み、これら以外の金属元素を更に含んでいてもよい。
ZrとCeとを含む複合酸化物から成ることが好ましく;
Zr及びCeと、Ceを除く希土類元素とを含む複合酸化物から成ることがより好ましく;
Zr及びCeと、Y、La、Pr、Nd、及びEuから選択される1種以上の希土類元素と、を含む複合酸化物から成ることが更に好ましい。
Zr:ZrO2換算値として1質量%以上90質量%以下
Ce:CeO2換算値として90質量%以下
Ceを除く希土類元素:Ln2O3換算値として1質量%以上20質量%以下
本発明のPd担持Zr系複合酸化物におけるPdの含有量(担持量)としては、上記複合酸化物担体の質量を100質量%としたときのPd金属換算値として、0.75質量%以上3.0質量%以下であることが好ましい。
本発明におけるPd担持Zr系複合酸化物についてXAFS(X線吸収微細構造)分析を行うと、Pdの結合距離2.500Å~3.500Åの範囲内における最大ピークのピーク位置が、3.050Å~3.110Å、特に3.060Å~3.100Åに見られることとなる。このことは、Pdの第2近接元素が主としてZrであり、Pd-O-Zr結合濃度が高いことを意味するものと考えられる。
(Pdの第2近接元素の結合距離)
Pd-O-Y:2.880Å
Pd-O-Pd:2.976Å
Pd-O-Zr:3.068Å
Pd-O-La:3.220Å
Pd-O-Ce:3.250Å
本発明のPd担持Zr系複合酸化物は、上記のような組成及び構造を有する限り、どのような方法によって製造されたものであってもよい。
ジルコニウム酸化物の前駆体(ZrO前駆体)を含有する水溶液を加熱処理してZr含有ゾルを形成する第1工程(Zr含有ゾル形成工程)と、
前記第1工程で得られたZr含有ゾルにパラジウムの前駆体(Pd前駆体)を添加してPd添加Zr含有ゾルを得る第2工程(Pd前駆体添加工程)と、
前記第2工程で得られたPd添加Zr含有ゾルを、塩基性化合物と接触させて沈殿物を得る第3工程(沈殿物生成工程)と、
前記第3工程で得られた沈殿物を焼成する第4工程(焼成工程)と、
を、上記に記載の順で含む方法により、製造することができる。
第1工程は、ZrO前駆体を含有する水溶液を加熱処理してZr含有ゾルを形成する工程である。この加熱処理により、ZrO前駆体はZr(OH)4が高度に分散したゾルに転化すると考えられる。後述の第2工程において、このような高度の分散状態となったZr化合物にPd前駆体を添加することにより、Pd-O-Zr結合が効率的に形成されるものと考えられる。
1段階で行ってもよいし;
添加する前駆体を複数のグループに分けて、グループごとに添加及び加熱処理を順次に行う態様で、多段階で行ってもよいし;或いは、
添加する前駆体を複数のグループに分けて、グループごとに加熱処理を行った後に、混合する方法によってもよい。
第1工程の第1段階:ZrO前駆体と、LnO前駆体と、を含有する水溶液を調製し、これを加熱処理する段階、並びに
第1工程の第2段階:上記第1段階で得られた加熱処理後の水溶液に、Ce酸化物の前駆体を加え、これを加熱処理する段階。
第2工程は、上記第1工程で得られたZr含有ゾルにパラジウムの前駆体(Pd前駆体)を添加する工程である。
第3工程は、上記第2工程で得られたPd添加Zr含有ゾルを塩基性化合物と接触させて沈殿物を得る工程である。ここで使用される塩基性化合物としては、例えば、アンモニア又は有機塩基が好ましい。有機塩基としては、例えば、ピリジン、トリエチルアミン、ジアザビシクロウンデセン、水酸化テトラアルキルアンモニウム等を挙げることができる。第3工程で使用される塩基性化合物としては、水酸化テトラアルキルアンモニウムが好ましく、特に好ましくは水酸化テトラメチルアンモニウムである。
次いで、第4工程において、上記第3工程で得られた沈殿物を焼成することにより、本発明のPd担持Zr系複合酸化物を得ることができる。
本発明のPd担持Zr系複合酸化物は、過酷な条件下で使用を継続しても、Pdの劣化が抑制されたものであり、特に低温領域におけるHCの浄化性能に優れる。従って、このPd担持Zr系複合酸化物は、例えば、自動車の排ガス浄化用触媒の成分として好適に使用することができる。
<実施例1>
(1)Zr含有ゾル形成工程
i)第1段階
ZrO2換算の濃度が20質量%のオキシ硝酸ジルコニウム水溶液75gと、
Y2O3換算の濃度が300g/Lの硝酸イットリウム水溶液5mLと、
HNO3換算の濃度が67.5質量%であり、比重が1.4である硝酸7.5mLと、
純水1,000mLと、
を混合し、Zr-Y水溶液を調製した。このZr-Y水溶液の全量を、冷却管及び撹拌器を備えた容器に収納し、98℃において8時間撹拌下に加熱保持した後、室温まで冷却した。
上記冷却後のZr-Y水溶液に、Ceイオンの95モル%が4価であり、CeO2換算の濃度が200g/Lの硝酸セリウム水溶液60mLを加えて、Zr-Y-Ce水溶液を調製した。このZr-Y-Ce水溶液の全量を、冷却管及び撹拌器を備えた容器に収納し、98℃において20時間撹拌下に加熱保持してZr含有ゾルとした後、室温まで冷却した。
上記冷却後のZr含有ゾルに、
La2O3換算の濃度が300g/Lの硝酸ランタン水溶液5mLと、
純水1,000mLと、
を添加してスターラー撹拌した後、
Pd金属換算の濃度が8.2質量%の硝酸パラジウム水溶液を11.31g添加して、Pd添加Zr含有ゾルを調製した。
ラウリン酸10.1gと、濃度128g/Lの水酸化テトラメチルアンモニウム(TMAH)水溶液760mLと、を混合し、液が透明になるまで撹拌して、ラウリン酸のTMAH溶液を調製した。
上記スラリー中の沈殿物をヌッチェによりろ取した。得られた沈殿物を、純水1,000mLを用いて洗浄し、大気中、400℃において5時間焼成した後、乳鉢にて粉砕することにより、Pd担持Zr系複合酸化物を得た。
上記実施例1の「(1)1段目の加熱保持工程」において、オキシ硝酸ジルコニウム水溶液の添加後に、Ceイオンの95モル%が4価であり、CeO2換算の濃度が200g/Lの硝酸セリウム水溶液3.0mLを更に加え、「(2)2段目の加熱保持工程」における硝酸セリウム水溶液の使用量を57mLとした他は実施例1と同様に操作して、Pd担持Zr系複合酸化物を得た。
上記実施例1の「(1)1段目の加熱保持工程」におけるオキシ硝酸ジルコニウム水溶液の使用量を67.5gとし、「(2)2段目の加熱保持工程」において、硝酸セリウム水溶液の添加後に上記と同濃度のオキシ硝酸ジルコニウム水溶液7.5gを加えた他は実施例1と同様に操作して、Pd担持Zr系複合酸化物を得た。
各工程における各溶液の使用量を表1のとおりとした他は実施例1と同様に操作して、Pd担持Zr系複合酸化物をそれぞれ得た。
実施例8は、「(1)Zr含有ゾル形成工程 ii)第2段階」において硝酸セリウム溶液を加えなかった。該実施例8では、i)第1段階における98℃、8時間の加熱保持後に、一旦室温まで冷却した後、硝酸セリウム溶液を加えずにii)第2段階における98℃、20時間撹拌下の加熱保持を行った。
各酸化物換算の組成がZr:Ce:La:Y=50:40:5:5であるZr系複合酸化物(市販品)30gを、Pd金属換算の濃度が8.2質量%の硝酸パラジウム水溶液11.31g中に25℃において1時間浸漬した後、100℃において24時間乾燥し、更に400℃において5時間焼成することにより、Pd担持Zr系複合酸化物を得た。
使用したZr系複合酸化物(市販品)の組成、及びPd担持量を、それぞれ、表3のとおりに変更した他は、上記比較例1と同様に操作して、Pd担持Zr系複合酸化物をそれぞれ得た。


上記実施例及び比較例で得られたPd担持Zr系複合酸化物について、X線吸収微細構造(XAFS)分析及びX線回折(XRD)分析を、それぞれ以下の手法により行った。
各Zr系複合酸化物を試料とし、市販の酸化パラジウム(PdO)をリファレンスとして、以下の条件でXAFS分析を行った。
リング名:大型放射光施設Spring-8 BL14-B2
測定装置:XAFS測定装置
測定法:透過法
測定雰囲気:大気中
測定温度:20℃
吸収端:PdK吸収端(25.3keV)近傍
カーブフィッティングの計算プログラム:FEFF
10質量%O2/N2と、5質量%CO/N2と、を交互に切り替えて流速10L/分にて流通させながら、1,000℃において10時間耐久処理を行った後の各Zr系複合酸化物を試料として、XRD測定を行った。この測定には、RIGAKU社製の型式「RINT-2500」を用いた。
[2層触媒の調製]
アルミナ200g、実施例1で得られたPd担持Zr系複合酸化物200g、硫酸バリウム50g、及び水2,000gを混合して、下層形成用スラリーを調製した。この下層形成用スラリーを容量1Lのモノリスハニカム基材にコートし、250℃で乾燥した後、500℃において1時間焼成した。
上記実施例10において、実施例1で得られたPd担持Zr系複合酸化物の代わりに比較例1で得られたPd担持Zr系複合酸化物を同量使用した他は実施例10と同様にして、モノリスハニカム基材にコートされた2層構成の触媒を調製した。
上記実施例10及び比較例10で得られた各触媒を用いて、以下の方法により実車評価を行った。
Claims (4)
- Zrを含む複合酸化物担体上にPdが担持されており、
XAFS(X線吸収微細構造)分析によって得られる、Pdの結合距離2.500Å~3.500Åの範囲内における最大ピークのピーク位置が3.050Å~3.110Åであることを特徴とする、Pd担持Zr系複合酸化物。 - 前記複合酸化物担体の全質量を100質量%としたときに、
ZrO2換算のZrの含有量が1質量%以上90質量%以下であり、
CeO2換算のCeの含有量が90質量%以下であり、そして
ZrO2換算のZrの含有量とCeO2換算のCeの含有量との合計が80質量%以上である、請求項1に記載のPd担持Zr系複合酸化物。 - 前記複合酸化物担体に対するPd金属換算のPd担持量が0.75質量%以上3.0質量%以下である、請求項1又は2に記載のPd担持Zr系複合酸化物。
- 請求項1~3のいずれか一項に記載のPd担持Zr系複合酸化物を含むことを特徴とする、排ガス浄化用触媒。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/061,493 US11084022B2 (en) | 2016-01-28 | 2016-12-26 | Pd-supporting Zr-based composite oxide |
| CN201680080238.7A CN108602049B (zh) | 2016-01-28 | 2016-12-26 | 担载Pd的Zr系复合氧化物 |
| EP16888207.4A EP3409355B1 (en) | 2016-01-28 | 2016-12-26 | Pd-supporting zr-based composite oxide |
| JP2017518566A JP6246424B1 (ja) | 2016-01-28 | 2016-12-26 | Pd担持Zr系複合酸化物 |
| PL16888207T PL3409355T3 (pl) | 2016-01-28 | 2016-12-26 | Niosący Pd tlenek złożony na bazie Zr |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-014461 | 2016-01-28 | ||
| JP2016014461 | 2016-01-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017130623A1 true WO2017130623A1 (ja) | 2017-08-03 |
Family
ID=59398061
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/088772 Ceased WO2017130623A1 (ja) | 2016-01-28 | 2016-12-26 | Pd担持Zr系複合酸化物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11084022B2 (ja) |
| EP (1) | EP3409355B1 (ja) |
| JP (1) | JP6246424B1 (ja) |
| CN (1) | CN108602049B (ja) |
| PL (1) | PL3409355T3 (ja) |
| WO (1) | WO2017130623A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118022724A (zh) * | 2024-01-31 | 2024-05-14 | 浙江工业大学 | 双金属Pd-Zr/CN催化剂及其制备和在加氢抗硫体系中的应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007111004A1 (ja) * | 2006-03-28 | 2007-10-04 | Kabushiki Kaisha Toyota Chuo Kenkyusho | 排ガス浄化用触媒、その再生方法、それを用いた排ガス浄化装置及び排ガス浄化方法 |
| CN100455352C (zh) * | 2007-02-07 | 2009-01-28 | 大连理工大学 | 催化还原氮氧化物的蜂窝状金属丝网载体催化剂及其制法 |
| WO2015083590A1 (ja) * | 2013-12-02 | 2015-06-11 | 田中貴金属工業株式会社 | 排ガス浄化触媒及びその製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1337330A2 (en) * | 2000-09-18 | 2003-08-27 | Valtion Teknillinen Tutkimuskeskus | Catalyst and method for the catalytic reduction of nitrogen oxides |
| JP4204521B2 (ja) | 2004-07-07 | 2009-01-07 | 本田技研工業株式会社 | 排ガス浄化触媒 |
| CN1935361A (zh) * | 2005-09-21 | 2007-03-28 | 卡塔勒公司 | 废气净化催化剂及其制造方法 |
| FR2898887B1 (fr) * | 2006-03-21 | 2008-05-02 | Rhodia Recherches & Tech | Composition a base d'oxyde de zirconium et d'oxyde de cerium a reductibilite elevee et a surface specifique stable procede de preparation et utilisation dans le traitement des gaz d'echappement |
| JP2009078203A (ja) | 2007-09-25 | 2009-04-16 | Mazda Motor Corp | 排ガス浄化用触媒材、同触媒材の製造方法、及び同触媒材を用いた触媒 |
| JP5502971B1 (ja) * | 2012-11-16 | 2014-05-28 | 三井金属鉱業株式会社 | 排気ガス用触媒担体及び排ガス浄化触媒 |
| JP2014223587A (ja) | 2013-05-16 | 2014-12-04 | 新日鉄住金マテリアルズ株式会社 | 排ガス浄化用触媒担体、排ガス浄化用触媒担体の製造方法、および排ガス浄化用触媒、排ガス浄化用触媒の製造方法 |
| US20140369912A1 (en) | 2013-06-13 | 2014-12-18 | Basf Corporation | Integrated Supports for Emission Control Catalysts |
-
2016
- 2016-12-26 EP EP16888207.4A patent/EP3409355B1/en active Active
- 2016-12-26 CN CN201680080238.7A patent/CN108602049B/zh active Active
- 2016-12-26 WO PCT/JP2016/088772 patent/WO2017130623A1/ja not_active Ceased
- 2016-12-26 US US16/061,493 patent/US11084022B2/en active Active
- 2016-12-26 PL PL16888207T patent/PL3409355T3/pl unknown
- 2016-12-26 JP JP2017518566A patent/JP6246424B1/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007111004A1 (ja) * | 2006-03-28 | 2007-10-04 | Kabushiki Kaisha Toyota Chuo Kenkyusho | 排ガス浄化用触媒、その再生方法、それを用いた排ガス浄化装置及び排ガス浄化方法 |
| CN100455352C (zh) * | 2007-02-07 | 2009-01-28 | 大连理工大学 | 催化还原氮氧化物的蜂窝状金属丝网载体催化剂及其制法 |
| WO2015083590A1 (ja) * | 2013-12-02 | 2015-06-11 | 田中貴金属工業株式会社 | 排ガス浄化触媒及びその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3409355A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN108602049A (zh) | 2018-09-28 |
| US20190336945A1 (en) | 2019-11-07 |
| JPWO2017130623A1 (ja) | 2018-02-08 |
| JP6246424B1 (ja) | 2017-12-13 |
| CN108602049B (zh) | 2021-09-03 |
| EP3409355A4 (en) | 2019-07-24 |
| PL3409355T3 (pl) | 2020-08-24 |
| EP3409355A1 (en) | 2018-12-05 |
| US11084022B2 (en) | 2021-08-10 |
| EP3409355B1 (en) | 2020-04-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN104024592B (zh) | 混合相氧化物催化剂 | |
| JP4648089B2 (ja) | 排ガス浄化用触媒 | |
| JP5885004B2 (ja) | 排気ガス浄化触媒、排気ガス浄化モノリス触媒及び排気ガス浄化触媒の製造方法 | |
| JP7078521B2 (ja) | 排ガス浄化用触媒 | |
| CN113042045B (zh) | 排气净化用催化剂 | |
| CN101432069B (zh) | 催化剂载体粒子及其制造方法、以及废气净化催化剂 | |
| CN108883397B (zh) | 排气净化用催化剂及其制造方法以及使用了该排气净化用催化剂的排气净化装置 | |
| JP5583967B2 (ja) | 排ガス浄化用触媒、それを用いた排ガス浄化装置及び排ガス浄化方法 | |
| CN102266772A (zh) | 尾气净化用催化剂 | |
| CN108136374A (zh) | 废气净化用催化剂及其制造方法、以及使用它的废气净化装置 | |
| JP4979900B2 (ja) | 排ガス浄化用触媒 | |
| US11577226B2 (en) | Exhaust gas purification catalyst | |
| JP6246424B1 (ja) | Pd担持Zr系複合酸化物 | |
| JP2001232199A (ja) | 排ガス浄化用触媒 | |
| JP6611623B2 (ja) | 排ガス浄化用触媒 | |
| JP5621109B2 (ja) | 排ガス浄化用触媒 | |
| JP2017180453A (ja) | 排ガス浄化装置及びその製造方法 | |
| WO2022209155A1 (ja) | 排ガス浄化用触媒 | |
| JP2015160207A (ja) | 排ガス浄化用触媒 | |
| JP2016032795A (ja) | 排ガス浄化用触媒 | |
| WO2022209154A1 (ja) | 排ガス浄化用触媒及び排ガス浄化システム | |
| CN121729281A (zh) | 废气净化催化剂 | |
| JP2016140809A (ja) | 排ガス浄化用触媒 | |
| JP2014147877A (ja) | 排ガス浄化用触媒 | |
| JP2016032794A (ja) | 排ガス浄化用触媒 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2017518566 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16888207 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016888207 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016888207 Country of ref document: EP Effective date: 20180828 |