WO2017138068A1 - 1,2-ベンゼンジメタノール化合物の製造方法 - Google Patents
1,2-ベンゼンジメタノール化合物の製造方法 Download PDFInfo
- Publication number
- WO2017138068A1 WO2017138068A1 PCT/JP2016/053650 JP2016053650W WO2017138068A1 WO 2017138068 A1 WO2017138068 A1 WO 2017138068A1 JP 2016053650 W JP2016053650 W JP 2016053650W WO 2017138068 A1 WO2017138068 A1 WO 2017138068A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound represented
- compound
- acetyl
- methylsulfonyloxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c1c(*)c(*)c(CO)c(CO)c1* Chemical compound *c1c(*)c(*)c(CO)c(CO)c1* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/26—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids
- C07C303/30—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfonic acids by reactions not involving the formation of esterified sulfo groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/24—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of esters of sulfuric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/63—Esters of sulfonic acids
- C07C309/64—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms
- C07C309/65—Esters of sulfonic acids having sulfur atoms of esterified sulfo groups bound to acyclic carbon atoms of a saturated carbon skeleton
- C07C309/66—Methanesulfonates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to a method for producing a 1,2-benzenedimethanol compound.
- Non-Patent Documents 1 to 3 a method of treating 1,2-bishalomethylbenzene with a basic aqueous solution has been reported.
- Non-Patent Documents 1 to 3 when trying to industrially produce the compound using the former method, it is necessary to treat the by-product aluminum compound or to carry out the production under water-free conditions, and the compound using the latter method.
- An object of the present invention is to provide a method for efficiently preparing a 1,2-benzenedimethanol compound that is a synthetic intermediate of a piperidine derivative useful as a bactericidal crop protection agent.
- the present invention includes a method for producing the following compounds.
- the halogenating reagent for the halogenation reaction is a chlorinating reagent such as chlorine, sulfuryl chloride or N-chlorosuccinimide, or a brominating reagent such as bromine or N-bromosuccinimide, (5) or (6 ) Manufacturing method.
- a compound represented by the formula [2] is obtained by reacting a compound represented by the formula [3] with a metal acetate, and the compound represented by the formula [2] is isolated.
- the method for producing according to any one of (1) to (3), comprising a step of obtaining a compound represented by the formula [1] by hydrolysis under basic conditions.
- reaction condition is provided by a step of reacting in water or a mixed solvent of water and an organic solvent in the presence of a base or a metal sulfate.
- reaction conditions are provided by a step of reacting in water or a mixed solvent of water and an organic solvent.
- X 1 is —OS (O) 2 R 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me, (1) The production method according to any one of (11).
- X 1 represents —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c— Pr, -OS (O) 2 n-Bu, -OS (O) 2 n-C 8 H 17 , -OS (O) 2 CH 2 CH 2 CF 3
- X 2 and X 3 are hydrogen atoms
- X 4 is a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me, The production method according to (12).
- X 1 is —OS (O) 2 Me; X 4 is a hydrogen atom or a fluorine atom, (13) The manufacturing method as described.
- X 1 is —OS (O) 2
- R 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me
- L 1 and L 2 are each independently a chlorine atom or a bromine atom, (3) The production method according to any one of (11).
- X 1 represents —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c— Pr, -OS (O) 2 n-Bu, -OS (O) 2 n-C 8 H 17 , -OS (O) 2 CH 2 CH 2 CF 3
- X 2 and X 3 is a hydrogen atom
- X 4 is a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me
- L 1 and L 2 are bromine atoms
- X 1 is —OS (O) 2 Me; X 4 is a hydrogen atom or a fluorine atom, (16) The production method according to the above.
- X 1 , X 3 and X 4 are hydrogen atoms, X 2 is —OS (O) 2 R 1 , R 1 is C 1 -C 4 alkyl, (1) The production method according to any one of (11).
- X 1 , X 3 and X 4 are hydrogen atoms
- X 2 is —OS (O) 2 R 1
- R 1 is C 1 ⁇ C 4 alkyl
- L 1 and L 2 are each independently a chlorine atom or a bromine atom
- X 2 is —OS (O) 2 Me; L 1 and L 2 are bromine atoms, (20) The manufacturing method as described.
- X 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, nitro, halogen atom or -OS (O) 2 R 1 , Any one of X 1 , X 2 , X 3 and X 4 is —OS (O) 2 R 1 ; R 1 is C 1 -C 8 alkyl, C 1 -C 8 haloalkyl or C 3 -C 6 cycloalkyl ⁇ Or a salt thereof.
- X 1 represents —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c— Pr, -OS (O) 2 n-Bu, -OS (O) 2 n-C 8 H 17 , -OS (O) 2 CH 2 CH 2 CF 3
- X 2 and X 3 are hydrogen atoms
- X 4 is hydrogen atom, nitro, a halogen atom, difluoromethoxy or -OS (O) 2 Me, The compound according to (22) or a salt thereof.
- X 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, nitro, halogen atom or -OS (O) is 2 R 1, Any one of X 1 , X 2 , X 3 and X 4 is —OS (O) 2 R 1 ; R 1 is C 1 -C 8 alkyl, C 1 -C 8 haloalkyl or C 3 -C 6 cycloalkyl; L 1 and L 2 are each independently a halogen atom ⁇ Or a salt thereof.
- X 1 represents —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c— Pr, -OS (O) 2 n-Bu, -OS (O) 2 n-C 8 H 17 , -OS (O) 2 CH 2 CH 2 CF 3
- X 2 and X 3 are hydrogen atoms
- X 4 is a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me
- L 1 and L 2 are bromine atoms
- a compound or a salt thereof is a compound or a salt thereof.
- a group selected from R 2 and R 3 are each independently C 1 -C 4 alkyl, C 1 -C 4 haloalkyl or a halogen atom
- R 4 , R 5 , R 6 , R 7 and R 8 are each independently a hydrogen atom, halogen atom, cyano, hydroxy, amino, nitro, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 ⁇ C 6 alkynyl, C 1 ⁇ C 6 haloalkyl, C 2 ⁇ C 6 haloalkenyl, C 2 ⁇ C 6 haloalkynyl, C 1 ⁇ C 6 alkoxy, C 1 ⁇ C 6 haloalkoxy, C 3 ⁇ C 6 cycloalkyl C 3 -C 6 halocycloalkyl, C 4 -C 10 cycloalkylalkyl, C 4 -C 10 alkyl cycloalkyl, C 5 -C 10 alkyl
- R 4 , R 6 and R 7 are hydrogen atoms
- R 5 and R 8 are each independently a hydrogen atom, a halogen atom, C 1 -C 6 alkyl or C 1 -C 6 haloalkyl
- R 2 is trifluoromethyl, difluoromethyl or a chlorine atom
- R 3 is a methyl, trifluoromethyl, difluoromethyl or chlorine atom
- R 5 and R 8 are each independently a hydrogen atom, a chlorine atom, trifluoromethyl or methyl
- A is A-1.
- (26) The production method according to any one of (28).
- (30) A is [5-methyl-3- (trifluoromethyl) -1H-pyrazol-1-yl] acetyl or [3,5-bis (difluoromethyl) -1H-pyrazol-1-yl] acetyl , (26)
- X 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, C 1 -C 4 haloalkoxy, nitro, halogen atom or -OS (O) is 2 R 1, Any one of X 1 , X 2 , X 3 and X 4 is —OS (O) 2 R 1 ; R 1 is C 1 -C 8 alkyl, C 1 -C 8 haloalkyl or C 3 -C 6 cycloalkyl, (26) The manufacturing method as described.
- X 1 is —OS (O) 2 R 1 , X 2 , X 3 and X 4 are each independently a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me, (26) or the production method according to (31).
- X 1 represents —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c— Pr, -OS (O) 2 n-Bu, -OS (O) 2 n-C 8 H 17 , -OS (O) 2 CH 2 CH 2 CF 3
- X 2 and X 3 are hydrogen atoms
- X 4 is a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me, (26)
- the manufacturing method according to any one of (31) and (32).
- X 1 is —OS (O) 2 Me
- X 4 is a hydrogen atom or a fluorine atom, (26) and the production method according to any one of (31) to (33).
- X 1 , X 3 and X 4 are hydrogen atoms;
- X 2 is —OS (O) 2 R 1 , R 1 is C 1 -C 4 alkyl, (26) The manufacturing method as described.
- X 2 is -OS (O) 2 Me; (26) or the production method according to (35).
- the compound represented by the formula [5] is 4- [4- (6-Methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1- [2- [5-methyl-3 -(Trifluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine, 4- [4- (6-Ethylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1- [2- [5-methyl-3 -(Trifluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine, 4- [4- (6-Fluoro-9-methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1
- Formula [1] gives a general definition of 1,2-benzenedimethanol compounds that can be prepared according to the present invention. Preferred definitions of groups relating to the formulas shown above and below are described below. Such a definition applies to the final product of formula [1] and likewise applies to all intermediates.
- X 1, X 2, X 3 and X 4 each independently preferably a hydrogen atom, C 1 ⁇ C 4 alkyl, C 1 ⁇ C 4 alkoxy, C 1 ⁇ C 4 haloalkoxy, nitro, halogen atom or —OS (O) 2 R 1 , preferably any one of X 1 , X 2 , X 3 and X 4 is —OS (O) 2 R 1 , more preferably X 1 Is —OS (O) 2 R 1 , more preferably X 2 , X 3 and X 4 are each independently a hydrogen atom, nitro, halogen atom, difluoromethoxy or —OS (O) 2 Me; Particularly preferably, X 1 is —OS (O) 2 Me, —OS (O) 2 Et, —OS (O) 2 n-Pr, —OS (O) 2 i-Pr, —OS (O) 2 c -Pr, -OS (O) 2 n
- R 1 is preferably C 1 -C 8 alkyl, C 1 -C 8 haloalkyl or C 3 -C 6 cycloalkyl, more preferably methyl.
- L 1 and L 2 are each independently preferably a halogen atom, more preferably a chlorine atom or a bromine atom, and particularly preferably a bromine atom.
- A is preferably A-1.
- R 2 and R 3 are each independently preferably a C 1 -C 6 alkyl, C 1 -C 6 haloalkyl or halogen atom, more preferably a methyl, trifluoromethyl, difluoromethyl or chlorine atom.
- R 4 , R 6 and R 7 are preferably hydrogen atoms.
- R 5 and R 8 are each independently preferably a hydrogen atom, a halogen atom, C 1 -C 6 alkyl or C 1 -C 6 haloalkyl, more preferably a hydrogen atom, a chlorine atom, trifluoromethyl or methyl. is there.
- One preferred embodiment is a compound represented by the formula [1] ⁇ wherein at least one of X 1 , X 2 , X 3 and X 4 represents —OS (O) 2 Me ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 Me and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 Et, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 n-Pr, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 i-Pr, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 n-Bu, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 n—C 8 H 17 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- X 1 represents —OS (O) 2 n—C 8 H 17 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 represents —OS (O) 2 CH 2 CH 2 CF 3 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- X 1 represents —OS (O) 2 CH 2 CH 2 CF 3 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents nitro ⁇ . It is.
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a fluorine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a fluorine atom ⁇ .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a chlorine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a chlorine atom ⁇ .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a bromine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a bromine atom ⁇ .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents an iodine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents an iodine atom ⁇ .
- Another preferred embodiment is represented by the formula [1] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents difluoromethoxy ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents difluoromethoxy ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 and X 4 represent —OS (O) 2 Me, and X 2 and X 3 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [1] ⁇ wherein X 1 and X 3 , X 4 represents a hydrogen atom, and X 2 represents —OS (O) 2 Me ⁇ .
- One preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [5-methyl-3- (trifluoromethyl) -1H-pyrazol-1-yl] acetyl ⁇ . is there.
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [3,5-bis (trifluoromethyl) -1H-pyrazol-1-yl] acetyl ⁇ . .
- Another preferred embodiment is a compound represented by the formula [5] (wherein A represents 2- [3,5-bis (difluoromethyl) -1H-pyrazol-1-yl] acetyl).
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (3,5-dimethyl-1H-pyrazol-1-yl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (2,5-dimethylphenyl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (2,5-difluorophenyl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (2,5-dichlorophenyl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (2,5-dibromophenyl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [2,5-bis (trifluoromethyl) phenyl] acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- (5-bromo-2-methylphenyl) acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [2-methyl-5- (trifluoromethyl) phenyl] acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [2-fluoro-5- (trifluoromethyl) phenyl] acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [2-chloro-5- (trifluoromethyl) phenyl] acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein A represents 2- [2-bromo-5- (trifluoromethyl) phenyl] acetyl ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein at least one of X 1 , X 2 , X 3 and X 4 represents —OS (O) 2 Me ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 Me and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 Et, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 n-Pr, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 i-Pr, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 c-Pr, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 n-Bu, and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ . .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 n—C 8 H 17 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- X 1 represents —OS (O) 2 n—C 8 H 17 and X 2 , X 3 and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 CH 2 CH 2 CF 3 , and X 2 , X 3, and X 4 represent a hydrogen atom ⁇ .
- X 1 represents —OS (O) 2 CH 2 CH 2 CF 3
- X 2 , X 3, and X 4 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents nitro ⁇ . It is.
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a fluorine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a fluorine atom ⁇ .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a chlorine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a chlorine atom ⁇ .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents a bromine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents a bromine atom ⁇ .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 is —OS (O) 2 Me, X 2 and X 3 are hydrogen atoms, and X 4 represents an iodine atom ⁇ .
- X 1 is —OS (O) 2 Me
- X 2 and X 3 are hydrogen atoms
- X 4 represents an iodine atom ⁇ .
- Another preferred embodiment is represented by the formula [5] ⁇ wherein X 1 represents —OS (O) 2 Me, X 2 and X 3 represent a hydrogen atom, and X 4 represents difluoromethoxy ⁇ .
- X 1 represents —OS (O) 2 Me
- X 2 and X 3 represent a hydrogen atom
- X 4 represents difluoromethoxy ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 and X 4 are —OS (O) 2 Me and X 2 and X 3 represent a hydrogen atom ⁇ .
- Another preferred embodiment is a compound represented by the formula [5] ⁇ wherein X 1 and X 3 , X 4 represents a hydrogen atom, and X 2 represents —OS (O) 2 Me ⁇ .
- Halogen atom includes fluorine atom, chlorine atom, bromine atom or iodine atom.
- the notation by an element symbol such as C 1 to C 6 and a subscript number indicates that the number of elements of the group described subsequently is in a range indicated by the subscript number.
- the carbon number is 1 to 6
- the C 2 to C 6 notation indicates that the carbon number is 2 to 6.
- C 2 -C 6 alkylcarbonyl indicates that the alkylcarbonyl as a whole has 2 to 6 carbon atoms and includes a propylcarbonyl group and the like.
- alkyl means linear or branched alkyl having 1 to 8 carbon atoms, preferably 1 to 6 carbon atoms, such as methyl, ethyl, n-propyl, isobutyl, n- Butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl, 1-ethylpropyl, 1,1-dimethylpropyl, 1,2-dimethylpropyl, neopentyl, n -Hexyl, 1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 1-ethylbutyl, 2-ethylbutyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3- Dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-d
- alkyls as part of complex substituents such as haloalkyl, alkylthio, alkylcarbonyl, and so on, unless otherwise defined.
- alkyl-terminated composite substituent such as alkylcycloalkyl
- that portion of the cycloalkyl may be independently mono- or polysubstituted by the same or different alkyls.
- Complex substituents terminated with other groups such as alkenyl, alkoxy, hydroxy, halogen, etc. are similarly understood.
- cycloalkyl means a cycloalkyl having a branched chain having 3 to 8 carbon atoms, preferably 3 to 6 carbon atoms, such as cyclopropyl, 1-methylcyclopropyl, 2-methylcyclohexane. Examples include propyl, cyclobutyl, cyclopentyl, cyclohexyl, 4,4-dimethylcyclohexyl and the like. This definition also applies to cycloalkyl as part of a composite substituent such as halocycloalkyl, unless otherwise defined.
- halo in “halo ...” (eg, “haloalkyl”) includes fluorine, chlorine, bromine and iodine.
- Halo substitution represented by the prefix “halo” includes mono- or polysubstitution and preferably includes mono-, di- and tri-substitution.
- haloalkyl means a straight or branched alkyl having 1 to 6 carbon atoms, and a group in which some or all of the hydrogen atoms are substituted with halogen atoms.
- fluoromethyl, chloromethyl, bromomethyl, iodomethyl, difluoromethyl, dichloromethyl, dibromomethyl, diiodomethyl, trifluoromethyl, trichloromethyl, tribromomethyl, triiodomethyl, 1-chloroethyl, 1-bromoethyl, 2-trifluoro Mention may be made of groups such as ethyl, 3-chloropropyl, 3-bromopropyl, 4-chlorobutyl, 4-bromobutyl, 4-trifluorobutyl, 5-chloropentyl, 6-chlorohexyl and the like. This definition also applies to haloalkyls as part of complex substituents such as haloalkyls as part
- alkenyl means straight or branched alkenyl having 2 to 6 carbon atoms, such as vinyl, 1-propenyl, 2-propenyl, isopropenyl, 3-butenyl, 1,3- Mention may be made of groups such as butadienyl, 4-pentenyl, 5-hexenyl and the like. This definition also applies to alkenyl as part of a complex substituent such as haloalkenyl, unless otherwise defined.
- alkynyl means straight or branched alkynyl having 2 to 6 carbon atoms, such as ethynyl, 1-propynyl, 2-propynyl, 3-butynyl, 1-methyl-3-propynyl. , 4-pentynyl, 5-hexynyl and the like. This definition also applies to alkynyls as part of complex substituents such as haloalkynyl, unless otherwise defined.
- Alkoxy means a straight or branched alkoxy having 1 to 6 carbon atoms unless otherwise specified, and includes, for example, methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, Mention may be made of groups such as pentyloxy, hexyloxy and the like. This definition also applies to alkoxy as part of a composite substituent, such as haloalkoxy, alkoxycarbonyl, etc., unless otherwise defined.
- Haloalkoxy means linear or branched alkoxy having 1 to 6 carbon atoms substituted by 1 or more, preferably 1 to 10 halogen atoms, unless otherwise specified, and includes, for example, fluoromethoxy, Chloromethoxy, bromomethoxy, iodomethoxy, difluoromethoxy, dichloromethoxy, dibromomethoxy, diiodomethoxy, trifluoromethoxy, trichloromethoxy, tribromomethoxy, triiodomethoxy, 1-chloroethoxy, 1-bromoethoxy, 2-tri Mention may be made of groups such as fluoroethoxy, 3-chloropropoxy, 3-bromopropoxy, 4-chlorobutoxy, 4-bromobutoxy, 4-trifluorobutoxy, 5-chloropentoxy, 6-chlorohexyloxy and the like. This definition also applies to haloalkoxy as part of complex substituents such as haloalkoxycarbony
- alkylthio means an (alkyl) -S-group having 1 to 6 carbon atoms in which the alkyl moiety has the above meaning, for example, a group such as methylthio, ethylthio, n-propylthio, isopropylthio, etc. Can be mentioned. This definition also applies to alkylthio as part of a complex substituent such as haloalkylthio, unless otherwise defined.
- alkylsulfinyl means an (alkyl) -SO— group having 1 to 6 carbon atoms in which the alkyl moiety has the above meaning, for example, methylsulfinyl, ethylsulfinyl, n-propylsulfinyl, isopropylsulfinyl And the like.
- This definition also applies to alkylsulfinyls as part of complex substituents such as haloalkylsulfinyl, unless otherwise defined.
- alkylsulfonyl means an (alkyl) -SO 2 — group having 1 to 6 carbon atoms in which the alkyl moiety has the above meaning, such as methylsulfonyl, ethylsulfonyl, n-propylsulfonyl, isopropyl Examples include groups such as sulfonyl. This definition also applies to alkylsulfonyls as part of complex substituents such as haloalkylsulfonyl, unless otherwise defined.
- Hydroxyalkyl means a linear or branched alkyl group having 1 to 6 carbon atoms substituted by 1 to 5 hydroxy groups, such as hydroxymethyl, hydroxyethyl, hydroxypropyl, hydroxyisopropyl, etc. Can be mentioned.
- alkylcarbonyl means an (alkyl) -C ( ⁇ O) — group in which the alkyl moiety has the above-mentioned meaning, and examples thereof include groups such as formyl, acetyl, propionyl, butyryl, pivaloyl and the like. it can.
- This definition also applies to alkylcarbonyls as part of complex substituents such as haloalkylcarbonyl, unless otherwise defined.
- alkylcarbonyloxy means an (alkyl) -C ( ⁇ O) O— group in which the alkyl moiety has the above meaning, for example, a group such as methylcarbonyloxy, ethylcarbonyloxy, propylcarbonyloxy, etc. Can be mentioned. This definition also applies to alkylcarbonyloxy as part of a complex substituent such as haloalkylcarbonyloxy, unless otherwise defined.
- the acid used in the reaction according to the present invention means a Bronsted acid that releases protons in the reaction system, and includes inorganic acids such as hydrochloric acid, hydrobromic acid, and sulfuric acid, acetic acid, trifluoro
- inorganic acids such as hydrochloric acid, hydrobromic acid, and sulfuric acid
- acetic acid trifluoro
- organic acids such as acetic acid, p-toluenesulfonic acid, trifluoromethanesulfonic acid.
- the Lewis acid used in the reaction according to the present invention means a compound that functions as an electron pair acceptor in the reaction system other than hydrogen ions, such as zinc chloride, aluminum chloride, tin chloride, boron trichloride, boron trifluoride. And trimethylsilyl trifluoromethanesulfonate.
- the base used in the reaction according to the present invention means a compound that receives a proton in the reaction system or a compound that functions as an electron pair donor in the reaction system, and includes triethylamine, pyridine, 4- Organic amines such as dimethylaminopyridine, N, N-dimethylaniline, 1,8-diazabicyclo [5,4,0] -7-undecene; metal carbonates such as sodium carbonate, potassium carbonate, magnesium carbonate, calcium carbonate; Metal hydrogen carbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate; metal carboxylates represented by metal acetates such as sodium acetate, potassium acetate, calcium acetate and magnesium acetate; sodium methoxide, sodium ethoxide and sodium tertiary Butoxide, potassium methoxide, potassium potassium Metal alkoxides such as shary butoxide; metal hydroxides such as sodium hydroxide, potassium hydroxide, calcium hydroxide and magnesium hydroxide; metal hydrides such as
- Examples of the ionic liquid used in the reaction according to the present invention include 1-butyl-3-methylimidazolium tetrafluoroborate, 1-butyl-2,3-dimethylimidazolium tetrafluoroborate, and 1-ethyl-3-methyl.
- Imidazolium tetrafluoroborate 1-hexyl-3-methylimidazolium tetrafluoroborate, 1-ethyl-3-methylimidazolium methanesulfonate, 1-butyl-3-methylimidazolium trifluoromethanesulfonate, 1- Imidazolium salts such as butyl-2,3-dimethylimidazolium trifluoromethanesulfonate, 1-ethyl-3-methylimidazolium trifluoromethanesulfonate, 1-hexyl-3-methylimidazolium trifluoromethanesulfonate; Trimonium fluoride, tetrabutylammonium chloride, tetrabutylammonium bromide, and ammonium salts such as tetrabutylammonium iodide can be exemplified.
- Me represents a methyl group
- Et represents an ethyl group
- n-Pr represents an n-propyl group
- i-Pr represents an isopropyl group
- c-Pr represents a cyclopropyl group
- n-Bu represents an n-butyl group.
- the compound of the formula [3] can be produced by reacting the compound of the formula [4] in a solvent in the presence of a halogenating reagent.
- halogenating reagents such as chlorine, sulfuryl chloride, and N-chlorosuccinimide
- bromination reagents such as bromine and N-bromosuccinimide.
- the amount of the halogenating reagent used in this step may be appropriately selected from the range of 2.0 to 10 mol, preferably 2.0 to 3.0 mol, per 1 mol of the compound of the formula [4]. .
- the solvent that can be used in this step is not particularly limited as long as it does not inhibit the progress of this reaction.
- nitriles such as acetonitrile
- ethers such as diethyl ether, diisopropyl ether, tetrahydrofuran, dioxane, monoglyme, diglyme
- dichloromethane dichloroethane
- Halogenated hydrocarbons such as chloroform, carbon tetrachloride and tetrachloroethane
- aromatic hydrocarbons such as benzene, chlorobenzene, nitrobenzene and toluene
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- Imidazolinones such as 1,3-dimethyl-2-imidazolinone
- sulfur compounds such as dimethyl sulfoxide can be used, and a mixed solvent thereof can also be used.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L with respect to 1 mol of the compound of the formula [4], and preferably 0.1 to 10 L.
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of formula [3], which is the target product of the reaction is collected from the reaction system by a conventional method after completion of the reaction, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [3] obtained by this step can be used for the reaction in Step 2 without performing isolation and purification operations after completion of the reaction.
- the compound of the formula [2] can be produced by reacting the compound represented by the formula [3] in a solvent in the presence of metal acetates.
- metal acetates examples include lithium acetate, sodium acetate, and potassium acetate.
- the amount of the metal acetate used in this step may be appropriately selected from the range of 2.0 to 10 mol, preferably 2.0 to 3.0 mol, relative to 1 mol of the compound of the formula [3]. is there.
- Examples of the solvent that can be used in this step include the same solvents described in Production Method 1 and Step 1.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [3].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [2] obtained by this step is collected from the reaction system by a conventional method after completion of the reaction, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [2] obtained by this step can be used for the reaction in Step 3 without performing isolation and purification operations after completion of the reaction.
- the compound of the formula [1] can be produced by reacting the compound represented by the formula [2] in water and a solvent in the presence of an acid, an acid chloride or a base.
- Examples of the acid that can be used in this step include inorganic acids such as hydrochloric acid, hydrobromic acid, and sulfuric acid; organic acids such as acetic acid, trifluoroacetic acid, p-toluenesulfonic acid, and trifluoromethanesulfonic acid. .
- the amount of acid used may be appropriately selected from the range of 0.1 to 10 mol, preferably 0.1 to 5.0 mol, per 1 mol of the compound of the formula [2].
- the amount of the acid chloride used may be appropriately selected from the range of 0.1 to 10 mol, preferably 0.1 to 5.0 mol, per 1 mol of the compound of the formula [2].
- Examples of the base that can be used in this step include organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- Metal carbonates such as sodium carbonate, potassium carbonate, magnesium carbonate and calcium carbonate
- Metal hydrogen carbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate
- Metal acetates such as sodium acetate, potassium acetate, calcium acetate and magnesium acetate Representative metal carboxylates; metal alkoxides such as sodium methoxide, sodium ethoxide, sodium tertiary butoxide, potassium methoxide, potassium tertiary butoxide; sodium hydroxide, potassium hydroxide, calcium hydroxide, magnesium hydroxide
- Metal hydroxides such as beam; lithium hydride, sodium hydride, metal hydrides such as calcium hydride and the like, preferably sodium carbonate, potassium carbonate, magnesium carbonate, metal carbonates such as calcium carbonate.
- the amount of the base used may be appropriately selected from the range of 0.2 to 10 mol, preferably 0.2 to 5.0 mol, per 1 mol of the compound of the formula [2].
- Examples of the solvent that can be used in this step include water and the same method described in Production Method 1 and Step 1.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [2].
- the amount of water used may be appropriately selected from the range of 0.01 to 100 L with respect to 1 mol of the compound of the formula [2], preferably 0.1 to 10 L.
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [1] which is the target product of the reaction, is collected from the reaction system by a conventional method after completion of the reaction, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [1] is obtained by reacting the compound represented by the formula [3] in a solvent in the presence of metal acetates (step 1), and then adding alcohol, water and a base to the reaction solution of step 1. It can manufacture by making it react (process 2).
- the amount of the metal acetate used in this step may be appropriately selected from the range of 2.0 to 10 mol, preferably 2.0 to 3.0 mol, relative to 1 mol of the compound of the formula [3]. is there.
- Examples of the solvent that can be used in this step include the same solvents described in Production Method 1 and Step 1.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [3].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [1] can be produced by adding alcohol, water and a base to the reaction solution obtained by the production method 2 and the step 1 and reacting them.
- Examples of the alcohol that can be used in this step include methanol and ethanol.
- the amount of alcohol used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [3].
- the amount of water used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [3].
- Examples of the base that can be used in this step include the same ones described in Production Method 1 and Step 3.
- the amount of the base used may be appropriately selected from the range of 0.2 to 10 mol, preferably 0.2 to 5.0 mol, per 1 mol of the compound of the formula [3].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [1] which is the target product of the reaction, is collected from the reaction system by a conventional method after completion of the reaction, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [1] is obtained by reacting the compound of the formula [3] in water or a mixed solvent of water and an organic solvent in the presence or absence of a base, an ionic liquid and a metal sulfate, preferably a base or It can be produced by reacting in water or in a mixed solvent of water and an organic solvent in the presence of a metal sulfate, more preferably in water or in a mixed solvent of water and an organic solvent.
- Examples of the base that can be used in this step include organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- Metal carbonates such as sodium carbonate, potassium carbonate, magnesium carbonate and calcium carbonate
- Metal hydrogen carbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate
- Metal acetates such as sodium acetate, potassium acetate, calcium acetate and magnesium acetate Representative metal carboxylates; metal alkoxides such as sodium methoxide, sodium ethoxide, sodium tertiary butoxide, potassium methoxide, potassium tertiary butoxide; sodium hydroxide, potassium hydroxide, calcium hydroxide, magnesium hydroxide
- Metal hydroxides such as sodium hydride; metal hydrides such as lithium hydride, sodium hydride, calcium hydride and the like, preferably metal carbonates such as sodium carbonate, potassium carbonate, magnesium carbonate, calcium carbonate; Metal hydrogen carbonates such as sodium and potassium hydrogen carbonate.
- the amount of the base used in this reaction may be appropriately selected from the range of 0 to 100 mol, preferably 0 to 10 mol, per 1 mol of the compound of the formula [3].
- Examples of the ionic liquid that can be used in this step include 1-butyl-3-methylimidazolium tetrafluoroborate, 1-butyl-2,3-dimethylimidazolium tetrafluoroborate, and 1-ethyl-3- Methyl imidazolium tetrafluoroborate, 1-hexyl-3-methylimidazolium tetrafluoroborate, 1-ethyl-3-methylimidazolium methanesulfonate, 1-butyl-3-methylimidazolium trifluoromethanesulfonate, 1 Imidazolium salts such as butyl-2,3-dimethylimidazolium trifluoromethanesulfonate, 1-ethyl-3-methylimidazolium trifluoromethanesulfonate, 1-hexyl-3-methylimidazolium trifluoromethanesulfonate; Tetrabutylammonium fluoride,
- the amount of the ionic liquid used in this reaction may be appropriately selected from the range of 0 to 100 mol, preferably 0 to 10 mol, relative to 1 mol of the compound of the formula [3].
- metal sulfates examples include copper (II) sulfate and iron (III) sulfate.
- the amount of the metal sulfate used in this reaction may be appropriately selected from the range of 0 to 100 mol, preferably 0 to 1.0 mol, per 1 mol of the compound of the formula [3].
- Examples of the solvent that can be used in this step include water and the same method described in Production Method 1 and Step 1.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [3].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [1] which is the target product of the reaction, is collected from the reaction system by a conventional method after completion of the reaction, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the formula [5] By reacting the compound represented by the formula [1] and the compound represented by the formula [6] in a solvent in the presence of an acid or a Lewis acid, preferably in the presence of an acid, the formula [5]
- the compound of the present invention represented can be produced.
- the amount of the compound of the formula [1] used in this step may be appropriately selected from the range of 1.0 to 10 mol per 1 mol of the compound of the formula [6], preferably 1.0 to 3. 0 mole.
- Examples of the acid that can be used in this step include inorganic acids such as hydrochloric acid, hydrobromic acid, and sulfuric acid, and organic acids such as acetic acid, trifluoroacetic acid, p-toluenesulfonic acid, and trifluoromethanesulfonic acid. .
- Lewis acids examples include zinc chloride, aluminum chloride, tin chloride, boron trichloride, boron trifluoride, and trimethylsilyl trifluoromethanesulfonate.
- the amount of acid or Lewis acid used may be appropriately selected from the range of 0.01 to 5 mol, preferably 0.1 to 1.0 mol, per 1 mol of the compound of the formula [6].
- Examples of the solvent that can be used in this step include the same solvents described in Production Method 1 and Step 1.
- the amount of the solvent used may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [6].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 150 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [5] which is the target product of the reaction, is collected from the reaction system by a conventional method, and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [6] can be produced by reacting the compound of the formula [7] and the compound of the formula [8] in a solvent in the presence or absence of a base and in the presence of a dehydration condensing agent. it can.
- the amount of the compound of the formula [8] used in this step may be appropriately selected from the range of 0.5 to 10 mol per 1 mol of the compound of the formula [7], preferably 1.0 to 1. 2 moles.
- Examples of the dehydrating condensing agent that can be used in this step include dicyclohexylcarbodiimide (DCC), N- (3-dimethylaminopropyl) -N′-ethylcarbodiimide (EDC or WSC), N, N-carbonyldiimidazole, 2 -Chloro-1,3-dimethylimidazolium chloride, 2-chloro-1-pyridinium iodide, and the like.
- DCC dicyclohexylcarbodiimide
- EDC or WSC N- (3-dimethylaminopropyl) -N′-ethylcarbodiimide
- N N-carbonyldiimidazole
- 2 -Chloro-1,3-dimethylimidazolium chloride 2-chloro-1-pyridinium iodide, and the like.
- the amount of the dehydrating condensing agent used in this reaction may be appropriately selected from the range of 1.0 to 10 mol, preferably 1.0 to 3.0 mol, per 1 mol of the compound of the formula [7]. .
- Examples of the base that can be used in this step include organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- organic amines such as triethylamine, pyridine, 4-dimethylaminopyridine, N, N-dimethylaniline, and 1,8-diazabicyclo [5,4,0] -7-undecene.
- Metal carbonates such as sodium carbonate, potassium carbonate, magnesium carbonate and calcium carbonate
- Metal hydrogen carbonates such as sodium hydrogen carbonate and potassium hydrogen carbonate
- Metal acetates such as sodium acetate, potassium acetate, calcium acetate and magnesium acetate Representative metal carboxylates; metal alkoxides such as sodium methoxide, sodium ethoxide, sodium tertiary butoxide, potassium methoxide, potassium tertiary butoxide; sodium hydroxide, potassium hydroxide, calcium hydroxide, magnesium hydroxide
- Metal hydroxides such as beam; lithium hydride, sodium hydride, and metal hydrides such as calcium hydride.
- the amount of the base used in this reaction may be appropriately selected from the range of 0 to 100 mol, preferably 0 to 10 mol, relative to 1 mol of the compound of the formula [7].
- Examples of the solvent that can be used in this step include the same solvents described in Production Method 1 and Step 1.
- the amount of the solvent used in this reaction may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, per 1 mol of the compound of the formula [7].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [6] which is the target product of the reaction, is collected from the reaction system by a conventional method and can be purified by operations such as column chromatography and recrystallization as necessary.
- the compound of the formula [6] can also be produced by reacting the compound of the formula [7] with the compound of the formula [9] in a solvent in the presence of a base.
- the amount of the compound of the formula [9] used in this step may be appropriately selected from the range of 0.5 to 10 mol per 1 mol of the compound of the formula [7], preferably 1.0 to 1. 2 moles.
- Examples of the base that can be used in this step include those described in Intermediate production method 1 and step 1.
- the amount of the base used in this reaction may be appropriately selected from the range of 0 to 100 mol, preferably 0 to 10 mol, relative to 1 mol of the compound of the formula [7].
- Examples of the solvent that can be used in this step include the same solvents described in Production Method 1 and Step 1.
- the amount of the solvent used in this step may be appropriately selected from the range of 0.01 to 100 L, preferably 0.1 to 10 L, relative to 1 mol of the compound of the formula [7].
- the reaction temperature may be selected from the range of the boiling point of the inert solvent used from ⁇ 20 ° C., preferably 0 ° C. to 100 ° C.
- the reaction time varies depending on the reaction temperature, reaction substrate, reaction amount, etc., but is usually 10 minutes to 48 hours.
- the compound of the formula [6] which is the target product of the reaction, is collected from the reaction system by a conventional method and can be purified by operations such as column chromatography and recrystallization as necessary.
- Step 2 Preparation of 1-methylsulfonyloxy-2,3-bis (bromomethyl) benzene
- the product (16.39 g) obtained in Step 1 of Example 1 above was dissolved in carbon tetrachloride (300 mL), and N -Bromosuccinimide (32.05 g) and 2,2'-azobisisobutyronitrile (671 mg) were added, and the mixture was heated to reflux for 4 hours.
- the reaction mixture was cooled to room temperature, filtered through celite, and the solvent was evaporated under reduced pressure to give the title compound (29.3 g, yield 100%).
- Step 3 Preparation of 1-methylsulfonyloxy-2,3-bis (acetyloxymethyl) benzene
- the product (506 mg) obtained in Step 2 of Example 1 above was converted to N, N-dimethylformamide (2.8 mL).
- sodium acetate (241 mg) was added and stirred overnight at room temperature.
- Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (380 mg, yield 85%).
- the title compound can also be prepared by the following Step 3A and Step 3B.
- Step 3A Preparation of 1-methylsulfonyloxy-2,3-bis (chloromethyl) benzene
- the product (1 g) obtained in Step 1 of Example 1 above was dissolved in chlorobenzene (25 mL) and 2,2 ′ -Azobisisobutyronitrile (0.05 g) was added.
- the temperature of the reaction solution was raised to 90 ° C., and chlorine gas (3.15 equivalents relative to the starting material) was bubbled through the reaction solution. After cooling the reaction solution to room temperature, the solvent was distilled off under reduced pressure to obtain a crude product containing the title compound. This crude product was used in the next step without purification.
- Step 3B Alternative Preparation of 1-Methylsulfonyloxy-2,3-bis (acetyloxymethyl) benzene
- the crude product (630 mg) obtained in Step 3A of Example 1 above was dissolved in toluene (23 mL) Sodium acetate (480 mg) and tetrabutylammonium bromide (75 mg) were added, and the mixture was stirred at 80 ° C. for 7 hours.
- the reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- Step 4 Preparation of 3-methylsulfonyloxy-1,2-benzenedimethanol
- the product (17 g) obtained in Step 3 of Example 1 above was dissolved in methanol (160 mL) and tetrahydrofuran (160 mL) and ice-cooled. Under water (40 mL) and potassium carbonate (15.23 g) were added and stirred for 3 hours. The solvent was distilled off under reduced pressure, water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was distilled off under reduced pressure to obtain the title compound (12.18 g, yield 98%).
- the title compound can also be prepared by the following step 4A.
- Step 4A Alternative Preparation of 3-Methylsulfonyloxy-1,2-benzenedimethanol
- water 5.6 mL
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (325 mg, yield 100%).
- Step 2 Preparation of 1-methylsulfonyloxy-2,3-bis (bromomethyl) -4-nitrobenzene
- the product (2.92 g) obtained in Step 1 of Example 2 above was converted to 1,2-dichloroethane (140 mL).
- N-bromosuccinimide (8.43 g) and 2,2′-azobisisobutyronitrile (195 mg) were added, and the mixture was heated to reflux for 8 hours.
- the reaction mixture was cooled to room temperature and allowed to stand overnight.
- the precipitated solid was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (4.8 g, yield 100%).
- Step 3 Preparation of 1-methylsulfonyloxy-2,3-bis (acetyloxymethyl) -4-nitrobenzene
- the product (4.8 g) obtained in Step 2 of Example 2 above was converted to N, N-dimethylformamide. (50 mL), sodium acetate (1.75 g) was added, and the mixture was stirred overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- Step 4 Preparation of 3-methylsulfonyloxy-6-nitro-1,2-benzenedimethanol
- methanol 80 mL
- tetrahydrofuran 80 mL
- Water 20 mL
- potassium carbonate 1.9 g
- 1N Hydrochloric acid was added to the reaction mixture, the solvent was evaporated under reduced pressure, water was added, and the mixture was extracted with ethyl acetate.
- the organic layer was dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was distilled off under reduced pressure.
- the title compound can also be prepared by the following step 1A.
- Step 1A Alternative Preparation of 1-Amino-2,3-dimethyl-4-methylsulfonyloxybenzene 2,3-Dimethyl-4-nitrophenol (25.3 g) was dissolved in ethyl acetate (300 mL) and triethylamine ( 16.8 g) was added. Thereafter, methanesulfonyl chloride (18.2 g) was added under ice cooling, and the mixture was stirred at room temperature for 30 minutes. Palladium / carbon (8 g, palladium content 10%, approximately 55% water wet product) was added to the reaction solution, and the mixture was stirred at room temperature for 24 hours under a hydrogen atmosphere. The reaction mixture was filtered off through celite, and the solvent was evaporated under reduced pressure to give the title compound (33 g, yield 100%).
- Step 2 Preparation of 2,3-dimethyl-4-methylsulfonyloxybenzenediazonium tetrafluoroborate
- the product obtained in Step 1 of Example 3 above (15.6 g) was added to a 48% tetrafluoroboric acid aqueous solution (84 mL). ), Dissolved in water (120 mL), an aqueous solution in which sodium nitrite (5.1 g) was dissolved in water (12 mL) was added over 10 minutes under ice cooling, and the mixture was stirred for 1 hour and a half under ice cooling. The reaction mixture was filtered off, and the resulting solid was washed with diethyl ether and dried to give the title compound (19.5 g, yield 86%).
- Step 3 Preparation of 1-fluoro-2,3-dimethyl-4-methylsulfonyloxybenzene
- 1-butyl-3-methylimidazolium tetra Fluoroborate (1.8 g) was added and stirred at 80 ° C. for 2 hours.
- the reaction solution was cooled to room temperature and extracted with toluene.
- the organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (0.29 g, yield 83%). It was.
- the title compound can also be prepared by the following step 3A or step 3B.
- Step 3A Alternative preparation of 1-fluoro-2,3-dimethyl-4-methylsulfonyloxybenzene
- the product obtained in Step 1 of Example 3 above (1 g) was added to hydrogen fluoride pyridine ( 8 mL) was added, and then sodium nitrite (355 mg) was added in small portions and stirred for 30 minutes. Then, it stirred at 55 degreeC for 1 hour and a half.
- the reaction solution was cooled to room temperature and extracted with diethyl ether.
- Step 3B Alternative Preparation of 1-Fluoro-2,3-dimethyl-4-methylsulfonyloxybenzene 2,3-Dimethyl-4-fluorophenol (200 mg) was dissolved in ethyl acetate (5 mL) and triethylamine (174 mg) Was added. Thereafter, methanesulfonyl chloride (180 mg) was added at room temperature and stirred for 30 minutes. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (290 mg, yield 93%).
- Step 4 Preparation of 1-fluoro-2,3-bis (bromomethyl) -4-methylsulfonyloxybenzene
- the product (1.48 g) obtained in Step 3 of Example 3 above was dissolved in chlorobenzene (15 mL).
- Water (8 mL) and 2,2′-azobisisobutyronitrile (0.11 g) were added.
- the temperature of the reaction solution was raised to 80 ° C., a solution of bromine (1 mL) dissolved in chlorobenzene (15 mL) was added over 30 minutes, and the mixture was further stirred at 80 ° C. for 1.5 hours.
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- Step 5 Preparation of 1-fluoro-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the product (16.71 g) obtained in Step 4 of Example 3 above was converted to N, N-dimethyl.
- the title compound (11.81 g, yield 80%) was obtained by dissolving in formamide (50 mL), adding sodium acetate (7.65 g), reacting and purifying as in Step 3 of Example 2, and purifying. Obtained.
- the title compound can also be prepared by the following steps 5A and 5B.
- Step 5A Preparation of 1-fluoro-2,3-bis (chloromethyl) -4-methylsulfonyloxybenzene
- the product (1 g) obtained in Step 3 of Example 3 above was dissolved in chlorobenzene (25 mL). 2,2′-Azobisisobutyronitrile (0.08 g) was added. The temperature of the reaction solution was raised to 88 ° C., and chlorine gas (4.1 equivalent to the starting material) was bubbled through the reaction solution. After cooling the reaction solution to room temperature, the solvent was distilled off under reduced pressure to obtain a crude product containing the title compound. This crude product was used in the next step without purification.
- Step 5B Alternative Preparation of 1-Fluoro-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the crude product (2 g) obtained in Step 5A of Example 3 above was toluene (20 mL).
- sodium acetate (1.32 g) and tetrabutylammonium bromide (225 mg) were added and stirred at 80 ° C. for 5 hours.
- the reaction mixture was cooled to room temperature, saturated brine was added, and the mixture was extracted with toluene.
- the organic layer was dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was distilled off under reduced pressure.
- Step 6 Preparation of 3-fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- methanol 20 mL
- water 4 mL
- potassium carbonate 6.53 g
- the solvent was distilled off under reduced pressure, and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (5.38 g, yield 100%).
- the title compound can also be prepared by the following steps 6A, 6B, 6C, 6D or 6E.
- Step 6A Alternative Preparation of 3-Fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- the product obtained in Step 4 of Example 3 above (10 g) was converted to N, N-dimethylformamide (27 mL).
- the mixture was dissolved in sodium acetate (4.58 g) and stirred at 50 ° C. for 4 hours.
- the reaction mixture was cooled to room temperature, methanol (20 mL), water (5 mL), and potassium carbonate (7.73 g) were added, and the mixture was stirred for 2 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (6.43 g, yield 97%).
- Step 6B Alternative Preparation of 3-Fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- water 10 mL
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (0.23 g, yield 92%).
- Step 6C Alternative Preparation of 3-Fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- the product obtained in Step 4 of Example 3 above (10 g) was converted to N, N-dimethylformamide (27 mL).
- water (240 mL) was added and stirred at 100 ° C. for 3 hours.
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (6.66 g, yield 100%).
- Step 6D Alternative Preparation of 3-Fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- the product obtained in Step 4 of Example 3 above (5 g) was converted to N, N-dimethylformamide (11 mL).
- copper (II) sulfate pentahydrate (234 mg) and water (30 mL) were added, and the mixture was heated to reflux at 110 ° C. for 7 hours.
- the reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (3.1 g, yield 93%).
- Step 6E Alternative Preparation of 3-Fluoro-6-methylsulfonyloxy-1,2-benzenedimethanol
- the product obtained in Step 5 of Example 3 above (4.46 g) was dissolved in methanol (20 mL).
- Acetyl chloride (208 mg) was added and stirred at room temperature overnight.
- Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (2.8 g, yield 84%).
- Step 2 Preparation of 1-chloro-2,3-bis (bromomethyl) -4-methylsulfonyloxybenzene
- the product obtained in Step 1 of Example 4 above (1.86 g) was converted to 1,2-dichloroethane (50 mL).
- N-bromosuccinimide (4.23 g) and 2,2′-azobisisobutyronitrile (130 mg) were added, and the mixture was heated to reflux for 3 hours.
- the reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with chloroform.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (3.11 g, yield 100%).
- Step 3 Preparation of 1-chloro-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the product (3.11 g) obtained in Step 2 of Example 4 above was converted to N, N-dimethyl. It was dissolved in formamide (40 mL), sodium acetate (1.56 g) was added, and the mixture was stirred overnight at room temperature. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give a crude product containing the title compound. This crude product was used in the next step without purification.
- Step 4 Preparation of 3-chloro-6-methylsulfonyloxy-1,2-benzenedimethanol
- methanol 20 mL
- tetrahydrofuran 20 mL
- water 5 mL
- potassium carbonate 2.2 g
- Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- the title compound can also be prepared by the following step 4A.
- Step 4A Alternative Preparation of 3-Chloro-6-methylsulfonyloxy-1,2-benzenedimethanol
- water 42 mL
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (1.11 g, yield 100%).
- Step 2 Preparation of 1-bromo-2,3-bis (bromomethyl) -4-methylsulfonyloxybenzene
- the crude product (1.87 g) obtained in Step 1 of Example 5 above was converted into 1,2-dichloroethane ( 13-mL), N-bromosuccinimide (2.5 g) and 2,2′-azobisisobutyronitrile (110 mg) were added, and the mixture was heated to reflux for 3 hours.
- the reaction mixture was cooled to room temperature, washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- Step 3 Preparation of 1-bromo-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the product (15.28 g) obtained in Step 2 of Example 5 above was converted to N, N-dimethyl. It was dissolved in formamide (100 mL), sodium acetate (6.31 g) was added, and the mixture was stirred at 90 ° C. for 3 hr. The reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with ethyl acetate.
- Step 4 Preparation of 3-bromo-6-methylsulfonyloxy-1,2-benzenedimethanol
- the crude product obtained in Step 3 of Example 5 above was dissolved in methanol (75 mL) and tetrahydrofuran (25 mL). Under ice cooling, water (25 mL) and potassium carbonate (9.04 g) were added and stirred for 3 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- the title compound can also be prepared by the following step 4A.
- Step 4A Alternative Preparation of 3-Bromo-6-methylsulfonyloxy-1,2-benzenedimethanol
- water 45 mL
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (1.35 g, yield 97%).
- Step 1 Preparation of 3-iodo-6-methylsulfonyloxy-1,2-benzenedimethanol (Compound 1-19)
- Step 1 Preparation of 1-iodo-2,3-dimethyl-4-methylsulfonyloxybenzene
- the product (3.44 g) obtained in Step 1 of Example 3 above was dissolved in dimethyl sulfoxide (100 mL), potassium iodide (13.3 g) and sodium nitrite (4.41 g) were added, and ice was added. Under cooling, 48% hydrobromic acid (20 mL) was added little by little and stirred for 3 hours.
- Step 2 Preparation of 1-iodo-2,3-bis (bromomethyl) -4-methylsulfonyloxybenzene
- the product obtained in Step 1 of Example 6 above (2.97 g) was converted to 1,2-dichloroethane (50 mL).
- N-bromosuccinimide (3.89 g) and 2,2′-azobisisobutyronitrile (150 mg) were added, and the mixture was heated to reflux for 3 hours.
- the reaction mixture was cooled to room temperature, water was added, and the mixture was extracted with chloroform.
- Step 3 Preparation of 1-iodo-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the crude product obtained in Step 2 of Example 6 above was converted to N, N-dimethylformamide (50 mL).
- sodium acetate (1.79 g)
- the mixture was stirred overnight at room temperature.
- Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give a crude product containing the title compound. This crude product was used in the next step without purification.
- Step 4 Preparation of 3-iodo-6-methylsulfonyloxy-1,2-benzenedimethanol
- methanol 20 mL
- tetrahydrofuran 20 mL
- water 5 mL
- potassium carbonate 2.52 g
- Step 2 Preparation of 1-difluoromethoxy-2,3-dimethyl-4-methylsulfonyloxybenzene
- the product obtained in Step 1 of Example 7 (200 mg) was dissolved in tetrahydrofuran (20 mL) and triethylamine (178 mg) was dissolved. ) was added. Thereafter, methanesulfonyl chloride (134 mg) was added under ice cooling, and the mixture was stirred at room temperature for 3 hours. The reaction solution was filtered off and the solvent was distilled off under reduced pressure.
- Step 3 Preparation of 1-difluoromethoxy-2,3-bis (bromomethyl) -4-methylsulfonyloxybenzene
- the product (220 mg) obtained in Step 2 of Example 7 above was dissolved in carbon tetrachloride (10 mL).
- N-bromosuccinimide (353 mg) and 2,2′-azobisisobutyronitrile (13.6 mg) were added, and the mixture was heated to reflux for 4 hours. After cooling the reaction solution to room temperature, the solvent was distilled off under reduced pressure.
- Step 4 Preparation of 1-difluoromethoxy-2,3-bis (acetyloxymethyl) -4-methylsulfonyloxybenzene
- the product (340 mg) obtained in Step 3 of Example 7 above was converted to N, N-dimethylformamide. (10 mL), sodium acetate (158 mg) was added, and the mixture was stirred at room temperature overnight. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (306 mg, yield 100%).
- Step 5 Preparation of 3-difluoromethoxy-6-methylsulfonyloxy-1,2-benzenedimethanol
- methanol 8 mL
- tetrahydrofuran 8 mL
- the mixture was dissolved, water (2 mL) and potassium carbonate (222 mg) were added under ice-cooling, and the mixture was stirred for 1 hr.
- Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic matter was filtered off, and the solvent was evaporated under reduced pressure.
- Step 2 Preparation of 1,4-bis (methylsulfonyloxy) -2,3-bis (bromomethyl) benzene
- the product (2 g) obtained in Step 1 of Example 8 above was converted to 1,2-dichloroethane (13 mL).
- N-bromosuccinimide (2.53 g) and 2,2′-azobisisobutyronitrile (112 mg) were added, and the mixture was heated to reflux for 3 hours.
- the reaction mixture was cooled to room temperature, washed with water and saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (2.81 g, yield). 92%).
- Step 3 Preparation of 1,4-bis (methylsulfonyloxy) -2,3-bis (acetyloxymethyl) benzene
- the product (2.68 g) obtained in Step 2 of Example 8 above was converted to N, N—
- the title compound (1.78 g, 60% yield) was obtained by dissolving in dimethylformamide (40 mL), adding sodium acetate (1.07 g), reacting and purifying in the same manner as in Step 3 of Example 2, and purifying. Got.
- Step 4 Preparation of 3,6-bis (methylsulfonyloxy) -1,2-benzenedimethanol
- the product (1.78 g) obtained in Step 3 of Example 8 above was added to methanol (40 mL), tetrahydrofuran (40 mL). ), And water (10 mL) and potassium carbonate (1.1 g) were added under ice-cooling.
- the mixture was reacted and purified in the same manner as in Step 4 of Example 2, and purified to give the title compound (916 mg, yield). 64%).
- the title compound can also be prepared by the following step 4A.
- Step 4A Alternative Preparation of 3,6-Bis (methylsulfonyloxy) -1,2-benzenedimethanol
- water 22 mL
- the reaction solution was cooled to room temperature and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was evaporated under reduced pressure to give the title compound (790 mg, yield 100%).
- Step 2 Preparation of 1-methylsulfonyloxy-3,4-bis (bromomethyl) benzene
- the crude product obtained in Step 1 of Example 9 above was dissolved in carbon tetrachloride (60 mL).
- N-bromosuccinimide (6.4 g) and 2,2′-azobisisobutyronitrile (134 mg) were added, reacted in the same manner as in Step 2 of Example 1, and worked up to give the title compound.
- a crude product containing was obtained. This crude product was used in the next step without purification.
- Step 3 Preparation of 1-methylsulfonyloxy-3,4-bis (acetyloxymethyl) benzene
- the crude product obtained in Step 2 of Example 9 above was dissolved in N, N-dimethylformamide (50 mL).
- Sodium acetate (3.36 g) was added, and the mixture was reacted and purified in the same manner as in Step 3 of Example 2 to obtain the title compound (3.26 g).
- Step 4 Preparation of 4-methylsulfonyloxy-1,2-benzenedimethanol
- the product (3.26 g) obtained in Step 3 of Example 9 above was dissolved in methanol (40 mL) and tetrahydrofuran (40 mL). Under cooling with ice, water (20 mL) and potassium carbonate (2.6 g) were added, and the mixture was reacted and purified in the same manner as in Step 4 of Example 2 to purify the title compound (1.64 g, yield 69%).
- Example 10 4- [4- (6-Methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1-[[5- Preparation of methyl-3- (trifluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine (Compound 2-1) 4- (4-Formyl-2-thiazolyl) -1- [2- [5-methyl -3- (Trifluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine (200 mg) (compound described in WO 2008/013622), the product obtained in Example 1 (121 mg), p -Toluenesulfonic acid monohydrate (20 mg) was dissolved in toluene (20 mL) and heated to reflux for 1 hour using a Dean-Stark apparatus.
- the reaction mixture was cooled to room temperature, diluted with ethyl acetate, and washed with water and saturated brine.
- the organic layer was dried over anhydrous sodium sulfate, the inorganic substance was filtered off, and the solvent was distilled off under reduced pressure.
- the residue was purified using silica gel flash chromatography (ethyl acetate-hexane: eluted with 40% -100%) using a flash automatic purifier (Biotage AB / Isolera TM ) to give the title compound (298 mg, yield 96). %).
- Example 12 4- [4- (6-Methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1-[[3 Preparation of 5-bis (difluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine (Compound 2-15) 4- (4-Formyl-2-thiazolyl) -1- [2- [3,5-bis (Difluoromethyl) -1H-pyrazol-1-yl] acetyl] piperidine (202 mg) (compound described in WO 2010/066353), the product obtained in Example 1 (232 mg), and p-toluenesulfonic acid The title compound (164 mg, yield) was obtained by dissolving monohydrate (5 mg) in toluene (15 mL), reacting and purifying in the same manner as in the preparation of Compound 2-1, and purifying. 53%).
- Example 14 4- [4- (6-Methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1- [2- ( Preparation of 2,5-dimethylphenyl) acetyl] piperidine (Compound 3-1) 4- (4-Formyl-2-thiazolyl) -1- [2- (2,5-dimethylphenyl) acetyl] piperidine (200 mg) and The product (142 mg) obtained in Example 1 and p-toluenesulfonic acid monohydrate (20 mg) were dissolved in toluene (15 mL) and reacted in the same manner as in the preparation of compound 2-1. And the title compound (206 mg, yield 64%) was obtained by purification.
- Example 15 4- [4- (6-Methylsulfonyloxy-1,5-dihydro-3H-2,4-benzodioxepin-3-yl) -2-thiazolyl] -1- [2- ( Preparation of 2,5-dichlorophenyl) acetyl] piperidine (Compound 3-2) Performed with 4- (4-formyl-2-thiazolyl) -1- [2- (2,5-dichlorophenyl) acetyl] piperidine (191 mg) The product obtained in Example 1 (232 mg) and p-toluenesulfonic acid monohydrate (5 mg) were dissolved in toluene (15 mL) and reacted in the same manner as in the preparation of compound 2-1. The title compound (267 mg, yield 72%) was obtained by purification.
- Examples 16 to 18 below show production examples of production starting materials used in the above Examples 1 to 15.
- Step 2 Preparation of 4- (4-formyl-2-thiazolyl) -1- [2- (2,5-dimethylphenyl) acetyl] piperidine
- the product (1 g) obtained in Step 1 of Example 17 above was Dissolve in dichloromethane (50 mL), add 2,5-dimethylphenylacetic acid (919 mg) and 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (1.07 g), and stir at room temperature overnight. did. Water was added to the reaction solution, and the mixture was extracted with chloroform.
- Example 18 Preparation of 4- (4-formyl-2-thiazolyl) -1- [2- (2,5-dichlorophenyl) acetyl] piperidine
- the product obtained in Step 1 of Example 17 above (1. 1 g) was dissolved in dichloromethane (30 mL), 2,5-dichlorophenylacetic acid (1.7 g) and 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (1.18 g) were added, and the reaction was carried out.
- the title compound (1.7 g, yield 79%) was obtained by reacting and purifying in the same manner as in Preparation of Step 2 of Example 17.
- Flowable agent 10 parts of the present compound, 4 parts of polyoxyethylene allyl phenyl ether sulfate, 5 parts of polyoxyethylene alkyl ether, 5 parts of propylene glycol, 0.2 part of a silicone-based antifoaming agent, 0. 8 parts and 50 parts of water were added and mixed, and wet pulverized using a dyno mill to obtain a pulverized suspension.
- Granule wettable powder 10 parts of the present invention compound 20 parts of sodium lignin sulfonate, 10 parts of sodium salt of naphthalene sulfonic acid condensate, 3 parts of sodium alkylbenzene sulfonate, 0.5 part of a silicone-based defoamer, 5 parts of diatomaceous earth, 10 parts of ammonium sulfate, 10 parts of talc, and 31.5 parts of clay part were added, sufficiently mixed and pulverized to obtain a pulverized product. An appropriate amount of water as necessary was added to the pulverized product, granulated with a granulator, and sieved after drying to obtain 10% hydratable fine granules.
- Emulsion agent 10 parts of the present compound, 15 parts of an aromatic hydrocarbon mixture, 2 parts of calcium dodecylbenzenesulfonate, 20 parts of polyoxyethylene castor oil and 4 parts of propylene glycol were dissolved to obtain a mixed solution.
- the mixture was added to 49 parts of water and mixed using a homogenizer to obtain a homogeneous 10% emulsion.
- Microemulsion agent 10 parts of the present compound, 12 parts of fatty acid dimethylamide, 10 parts of cyclohexanone, 15 parts of arylphenol ethoxylate, 10 parts of alcohol ethoxylate and 43 parts of water are added and heated. Stir for several minutes to obtain a stable 10% water-soluble liquid.
- Each cell is filled with 20 ml of contaminated soil in a 31 x 31 mm 2 cell tray, 3 seeded rice persimmons (variety: Koshihikari) are seeded per cell, 5 ml of covering soil is added, and 2.5 ml of the test suspension is irrigated.
- the budding process was performed by incubating for 72 hours in a wet chamber adjusted to 28 ° C. Thereafter, the disease was promoted for 2 days in a low-temperature room at 5 ° C., and then raised in a greenhouse at 25 ° C. for 14 days.
- Disease severity [ ⁇ (number of disease-causing strains by degree ⁇ morbidity index) / (surveyed strain ⁇ 3)] ⁇ 100 [Disease index] 0: healthy strain 1: growth-suppressed strain 3: dead strain
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description

X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルである}
で表される化合物の製造方法であって、
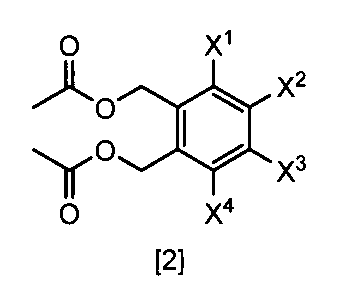
で表される化合物を酸性又は塩基性条件下で加水分解する工程を含む、前記製造方法。

L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物と金属酢酸塩類とを反応させて式[2]で表される化合物を製造する工程をさらに含む、(1)又は(2)に記載の製造方法。

で表される化合物の製造方法であって、

L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物と金属酢酸塩類とを反応させる工程を含む、前記製造方法。

で表される化合物をハロゲン化させて式[3]で表される化合物を製造する工程をさらに含む、(1)~(4)のいずれか1つに記載の製造方法。

L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物の製造方法であって、
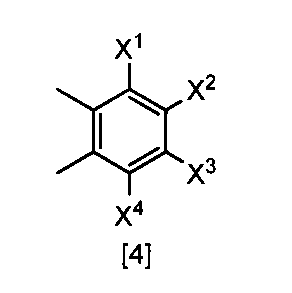
で表される化合物をハロゲン化させる工程を含む、前記製造方法。
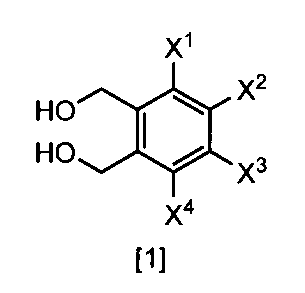
で表される化合物の製造方法であって、
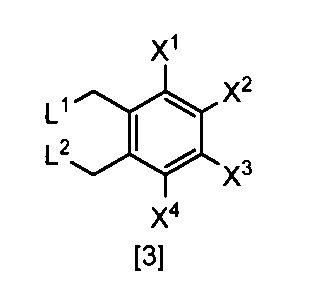
L1及びL2は、(3)に記載した通りである}
で表される化合物を塩基、イオン液体及び金属硫酸塩類の存在下又は非存在下、水中又は水と有機溶媒の混合溶媒中で反応させる工程を含む、前記製造方法。
X2、X3及びX4はそれぞれ独立して水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
(1)~(11)のいずれか1つに記載の製造方法。
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
(12)に記載の製造方法。
X4は水素原子又はフッ素原子である、
(13)に記載の製造方法。
X2、X3及びX4はそれぞれ独立して水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2はそれぞれ独立して塩素原子又は臭素原子である、
(3)~(11)のいずれか1つに記載の製造方法。
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2は臭素原子である、
(15)に記載の製造方法。
X4は水素原子又はフッ素原子である、
(16)に記載の製造方法。
X2は-OS(O)2R1であり、
R1はC1~C4アルキルである、
(1)~(11)のいずれか1つに記載の製造方法。
(18)に記載の製造方法。
X2は-OS(O)2R1であり、
R1はC1~C4アルキルであり、
L1及びL2はそれぞれ独立して塩素原子又は臭素原子である、
(3)~(11)のいずれか1つに記載の製造方法。
L1及びL2は臭素原子である、
(20)に記載の製造方法。

X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルである}
で表される化合物又はその塩。
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
(22)に記載の化合物又はその塩。
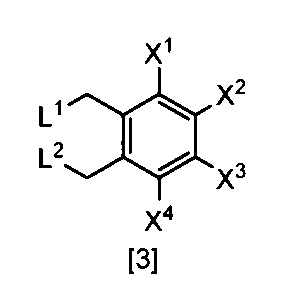
X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルであり、
L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物又はその塩。
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2は臭素原子である、
(24)に記載の化合物又はその塩。
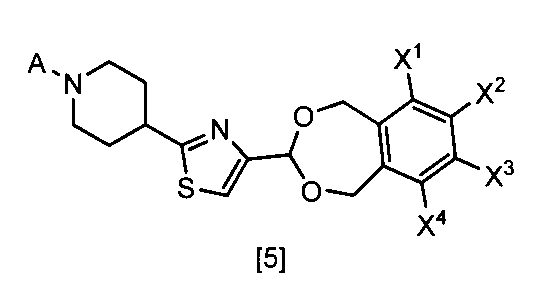

R2及びR3は、それぞれ独立してC1~C4アルキル、C1~C4ハロアルキル又はハロゲン原子であり、
R4、R5、R6、R7及びR8は、それぞれ独立して水素原子、ハロゲン原子、シアノ、ヒドロキシ、アミノ、ニトロ、C1~C6アルキル、C2~C6アルケニル、C2~C6アルキニル、C1~C6ハロアルキル、C2~C6ハロアルケニル、C2~C6ハロアルキニル、C1~C6アルコキシ、C1~C6ハロアルコキシ、C3~C6シクロアルキル、C3~C6ハロシクロアルキル、C4~C10シクロアルキルアルキル、C4~C10アルキルシクロアルキル、C5~C10アルキルシクロアルキルアルキル、C2~C6アルコキシアルキル、C1~C6ヒドロキシアルキル、C1~C6アルキルチオ、C1~C6ハロアルキルチオ、C1~C6アルキルスルフィニル、C1~C6ハロアルキルスルフィニル、C1~C6アルキルスルホニル、C1~C6ハロアルキルスルホニル、C1~C6アルキルアミノ、C2~C8ジアルキルアミノ、C3~C6シクロアルキルアミノ、C2~C6アルキルカルボニル、C2~C6ハロアルキルカルボニル、C2~C6アルコキシカルボニル、C2~C6ハロアルコキシカルボニル、C2~C6アルキルカルボニルオキシ、C2~C6アルキルカルボニルチオ、C2~C6アルキルアミノカルボニル、C2~C8(ジアルキルアミノ)カルボニル、又はC3~C6トリアルキルシリルであり、
X1、X2、X3及びX4は、(1)に定義した通りである}
で表される化合物の製造方法であって、

で表される化合物と、

で表される化合物とを反応させる工程
を含み、式[1]の化合物は前記(1)、(3)、(5)、(8)及び(9)のいずれか1つに記載の工程により製造される、前記製造方法。
R5及びR8は、それぞれ独立して水素原子、ハロゲン原子、C1~C6アルキル又はC1~C6ハロアルキルである、
(26)に記載の製造方法。
R3は、メチル、トリフルオロメチル、ジフルオロメチル又は塩素原子であり、
R5及びR8は、それぞれ独立して水素原子、塩素原子、トリフルオロメチル又はメチルである、
(26)又は(27)に記載の製造方法。
(26)~(28)のいずれか1つに記載の製造方法。
(26)~(29)のいずれか1つに記載の製造方法。
X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルである、
(26)に記載の製造方法。
X2、X3及びX4はそれぞれ独立して水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
(26)又は(31)に記載の製造方法。
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
(26)、(31)及び(32)のいずれか1つに記載の製造方法。
X4は水素原子又はフッ素原子である、
(26)、及び(31)~(33)のいずれか1つに記載の製造方法。
X2は-OS(O)2R1であり、
R1はC1~C4アルキルである、
(26)に記載の製造方法。
(26)又は(35)に記載の製造方法。
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-エチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-シクロプロピルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-プロピルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-オクチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-イソプロピルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-エチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-[6-(1,1,1-トリフルオロプロパン-3-イル)スルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-9-ニトロ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-フルオロ-6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-イソプロピルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-オクチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6,7-ジメチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-クロロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブロモ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-イソプロピルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ヨード-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-[6,9-ビス(メチルスルホニルオキシ)-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(3,5-ジメチル-1H-ピラゾール-1-イル)アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-クロロ-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(3,5-ジクロロ-1H-ピラゾール-1-イル)アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(3,5-ジメチル-1H-ピラゾール-1-イル)アセチル]ピペリジン、
4-[4-(6-クロロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-クロロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジクロロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(3,5-ジメチル-1H-ピラゾール-1-イル)アセチル]ピペリジン、
4-[4-(6,7-ジメチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6,7-ジメチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブロモ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-9-ニトロ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ブロモ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-9-ニトロ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ヨード-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-ヨード-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-フルオロ-6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-フルオロ-6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(7-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-[6-(ジフルオロメトキシ)-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-[6,9-ビス(メチルスルホニルオキシ)-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-[6,9-ビス(メチルスルホニルオキシ)-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-[3,5-ビス(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[5-メチル-3-(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-ブチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-フルオロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-メトキシ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-[2,5-ビス(トリフルオロメチル)フェニル]アセチル]ピペリジン、
4-[4-(6,7-ジメチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6,7-ジメチル-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-ブロモ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-9-ニトロ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-ブロモ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-メチルスルホニルオキシ-9-ニトロ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-クロロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-ヨード-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(6-クロロ-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(7-フルオロ-6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
4-[4-(7-フルオロ-6-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(6-ヨード-9-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、
4-[4-(7-メチルスルホニルオキシ-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル)-2-チアゾリル]-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン、及び
4-[4-[6,9-ビス(メチルスルホニルオキシ)-1,5-ジヒドロ-3H-2,4-ベンゾジオキセピン-3-イル]-2-チアゾリル]-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン、
から選択される(26)~(36)のいずれか1つに記載の製造方法。
Meとはメチル基を示し、
Etとはエチル基を示し、
n-Prとはn-プロピル基を示し、
i-Prとはイソプロピル基を示し、
c-Prとはシクロプロピル基を示し、
n-Buとはn-ブチル基を示す。
式[1]で示される化合物は、下記に例示する反応式からなる方法により製造することができる。

式[3]の化合物は、式[4]の化合物をハロゲン化試薬の存在下、溶媒中で反応させることによって製造することができる。
式[2]の化合物は、式[3]で表される化合物を、金属酢酸塩類存在下、溶媒中で反応させることによって製造することができる。
式[1]の化合物は、式[2]で表される化合物を、酸、酸塩化物又は塩基存在下、水及び溶媒中で反応させることによって製造することができる。
式[1]で示される化合物は、下記に例示する反応式からなる方法によっても製造することができる。

本工程で使用できる金属酢酸塩類としては、製造方法1、工程2で説明した同様のものを挙げることができる。
式[1]の化合物は、製造方法2、工程1によって得られた反応液にアルコール、水及び塩基を加え反応させることによって製造することができる。
式[1]で示される化合物は、下記に例示する反応式からなる方法によっても製造することができる。

式[5]で示される本発明化合物は下記に例示する反応式からなる方法により製造することができる。

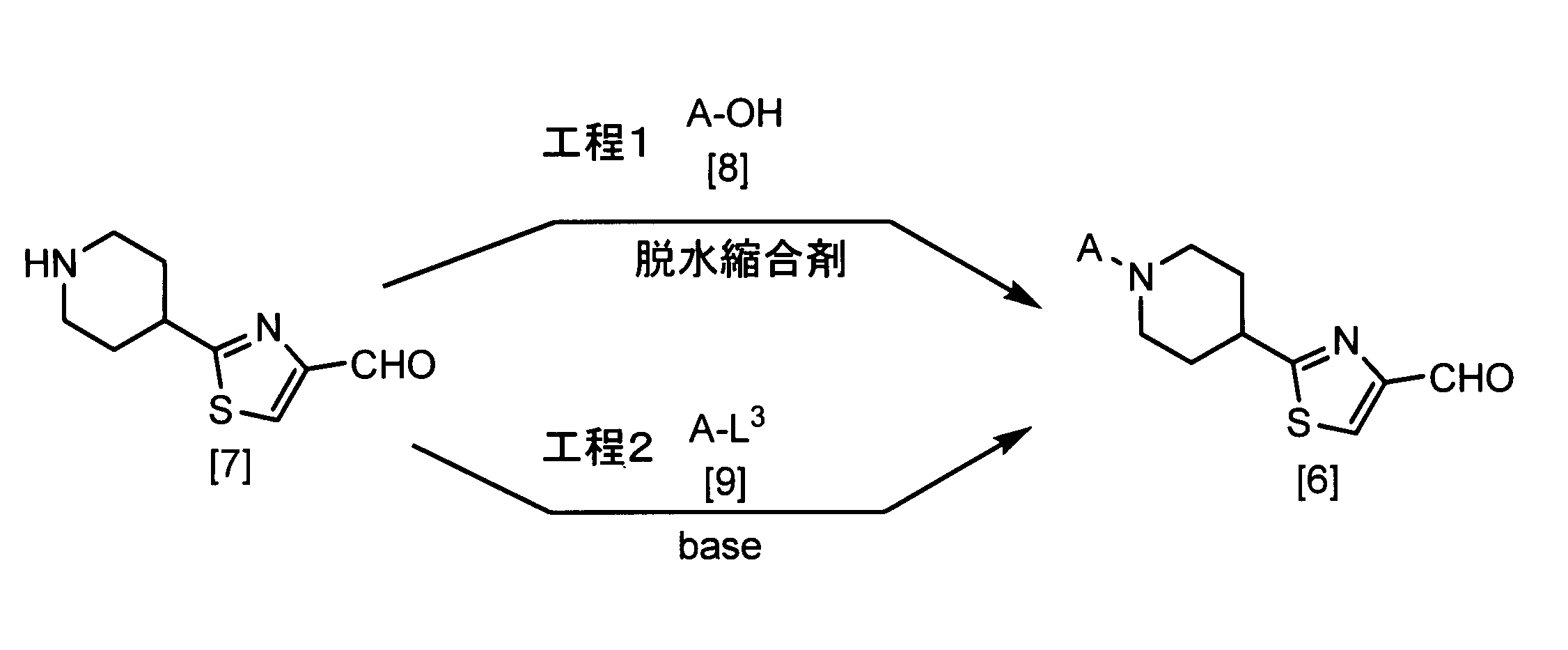
式[6]の化合物は、式[7]の化合物と式[8]の化合物とを、塩基の存在下又は非存在下、脱水縮合剤存在下、溶媒中で反応させることによって製造することができる。
式[6]の化合物は、式[7]の化合物と式[9]の化合物とを、塩基の存在下、溶媒中で反応させることによっても製造することができる。
工程1:1-メチルスルホニルオキシ-2,3-ジメチルベンゼンの調製
2,3-ジメチルフェノール(10g)をテトラヒドロフラン(125mL)に溶解させ、トリエチルアミン(9.1g)を加えた。その後、氷冷下、メタンスルホニルクロリド(10.26g)を加え室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(16.39g、収率100%)を得た。
上記実施例1の工程1において得られた生成物(16.39g)を四塩化炭素(300mL)に溶解させ、N-ブロモスクシンイミド(32.05g)と2,2´-アゾビスイソブチロニトリル(671mg)を加え、4時間加熱還流した。反応液を室温まで冷却後、セライトを用いて濾別し、減圧下溶媒を留去することで、表題化合物(29.3g、収率100%)を得た。
上記実施例1の工程2において得られた生成物(506mg)をN,N-ジメチルホルムアミド(2.8mL)に溶解させ、酢酸ナトリウム(241mg)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(380mg、収率85%)を得た。
上記実施例1の工程1において得られた生成物(1g)をクロロベンゼン(25mL)に溶解させ、2,2´-アゾビスイソブチロニトリル(0.05g)を加えた。反応液を90℃に昇温し、反応液に塩素ガス(出発物質に対して3.15当量)をバブリングした。反応液を室温まで冷却後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例1の工程3Aにおいて得られた粗生成物(630mg)をトルエン(23mL)に溶解させ、酢酸ナトリウム(480mg)とテトラブチルアンモニウムブロミド(75mg)を加え、80℃で7時間撹拌した。反応液を室温まで冷却後、水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:40%で溶出)を用いて精製することで、表題化合物(310mg)を得た。
上記実施例1の工程3において得られた生成物(17g)をメタノール(160mL)、テトラヒドロフラン(160mL)に溶解させ、氷冷下、水(40mL)、炭酸カリウム(15.23g)を加え、3時間撹拌した。減圧下溶媒を留去し、水を加え、酢酸エチルを用いて抽出した。有機層を無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(12.18g、収率98%)を得た。
上記実施例1の工程2において得られた生成物(502mg)に水(5.6mL)を加え、6時間加熱還流した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(325mg、収率100%)を得た。
工程1:1-メチルスルホニルオキシ-2,3-ジメチル-4-ニトロベンゼンの調製
2,3-ジメチル-4-ニトロフェノール(25g)をテトラヒドロフラン(500mL)に溶解させ、トリエチルアミン(16.7g)を加えた。その後、氷冷下、メタンスルホニルクロリド(18.93g)を加え、実施例1の工程1の調製と同様に反応させ、後処理することで、表題化合物(34.98g、収率95%)を得た。
上記実施例2の工程1において得られた生成物(2.92g)を1,2-ジクロロエタン(140mL)に溶解させ、N-ブロモスクシンイミド(8.43g)と2,2´-アゾビスイソブチロニトリル(195mg)を加え、8時間加熱還流した。反応液を室温まで冷却後、一晩静置し、析出した固体を濾別した後、減圧下溶媒を留去することで、表題化合物(4.8g、収率100%)を得た。
上記実施例2の工程2において得られた生成物(4.8g)をN,N-ジメチルホルムアミド(50mL)に溶解させ、酢酸ナトリウム(1.75g)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:40%で溶出)を用いて精製することで、表題化合物(2.8g、収率65%)を得た。
上記実施例2の工程3において得られた生成物(2.8g)をメタノール(80mL)、テトラヒドロフラン(80mL)に溶解させ、氷冷下、水(20mL)、炭酸カリウム(1.9g)を加え、2時間撹拌した。反応液に1規定塩酸を加え、減圧下溶媒を留去し、水を加え、酢酸エチルを用いて抽出した。有機層を無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(1.59g、収率74%)を得た。
工程1:1-アミノ-2,3-ジメチル-4-メチルスルホニルオキシベンゼンの調製
上記実施例2の工程1において得られた生成物(10g)をメタノール(200mL)に溶解させ、パラジウム/炭素(2g、パラジウム含有量10%、約55%水湿潤品)を加え、水素雰囲気下、室温で24時間撹拌した。反応液をセライトを用いて濾別し、減圧下溶媒を留去することで、表題化合物(8.43g、収率96%)を得た。
2,3-ジメチル-4-ニトロフェノール(25.3g)を酢酸エチル(300mL)に溶解させ、トリエチルアミン(16.8g)を加えた。その後、氷冷下、メタンスルホニルクロリド(18.2g)を加え、室温で30分間撹拌した。反応液にパラジウム/炭素(8g、パラジウム含有量10%、約55%水湿潤品)を加え、水素雰囲気下、室温で24時間撹拌した。反応液をセライトを用いて濾別し、減圧下溶媒を留去することで、表題化合物(33g、収率100%)を得た。
上記実施例3の工程1において得られた生成物(15.6g)を48%テトラフルオロホウ酸水溶液(84mL)、水(120mL)に溶解させ、氷冷下、亜硝酸ナトリウム(5.1g)を水(12mL)に溶解させた水溶液を10分間かけて加え、氷冷下、1時間半撹拌した。反応液を濾別し、得られた個体をジエチルエーテルで洗浄した後、乾燥することで、表題化合物(19.5g、収率86%)を得た。
上記実施例3の工程2において得られた生成物(0.5g)に1-ブチル-3-メチルイミダゾリウムテトラフルオロボラート(1.8g)を加え、80℃で2時間撹拌した。反応液を室温まで冷却後、トルエンを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(0.29g、収率83%)を得た。
上記実施例3の工程1において得られた生成物(1g)に、氷冷下、フッ化水素ピリジン(8mL)を加え、その後、亜硝酸ナトリウム(355mg)を少量ずつ加え30分間撹拌した。その後、55℃で1時間半撹拌した。反応液を室温まで冷却後、ジエチルエーテルを用いて抽出した。有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(0.75g、収率74%)を得た。
2,3-ジメチル-4-フルオロフェノール(200mg)を酢酸エチル(5mL)に溶解させ、トリエチルアミン(174mg)を加えた。その後、室温でメタンスルホニルクロリド(180mg)を加え30分間撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(290mg、収率93%)を得た。
上記実施例3の工程3において得られた生成物(1.48g)をクロロベンゼン(15mL)に溶解させ、水(8mL)、2,2´-アゾビスイソブチロニトリル(0.11g)を加えた。反応液を80℃に昇温し、臭素(1mL)をクロロベンゼン(15mL)に溶解させた溶液を30分間かけて加え、さらに80℃で1時間半撹拌した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を水及びチオ硫酸ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(2.45g、収率96%)を得た。
上記実施例3の工程4において得られた生成物(16.71g)をN,N-ジメチルホルムアミド(50mL)に溶解させ、酢酸ナトリウム(7.65g)を加え、実施例2の工程3の調製と同様に反応させ、精製することで、表題化合物(11.81g、収率80%)を得た。
上記実施例3の工程3において得られた生成物(1g)をクロロベンゼン(25mL)に溶解させ、2,2´-アゾビスイソブチロニトリル(0.08g)を加えた。反応液を88℃に昇温し、反応液に塩素ガス(出発物質に対して4.1当量)をバブリングした。反応液を室温まで冷却後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例3の工程5Aにおいて得られた粗生成物(2g)をトルエン(20mL)に溶解させ、酢酸ナトリウム(1.32g)とテトラブチルアンモニウムブロミド(225mg)を加え、80℃で5時間撹拌した。反応液を室温まで冷却後、飽和食塩水を加え、トルエンを用いて抽出した。有機層を無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:35%で溶出)を用いて精製することで、表題化合物(1.47g)を得た。
上記実施例3の工程5において得られた生成物(7.19g)をメタノール(20mL)、に溶解させ、氷冷下、水(4mL)、炭酸カリウム(6.53g)を加え、15分間撹拌した。減圧下溶媒を留去し、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(5.38g、収率100%)を得た。
上記実施例3の工程4において得られた生成物(10g)をN,N-ジメチルホルムアミド(27mL)に溶解させ、酢酸ナトリウム(4.58g)を加え、50℃で4時間撹拌した。反応液を室温まで冷却後メタノール(20mL)、水(5mL)、炭酸カリウム(7.73g)を加え、2時間撹拌した。反応液に水を加えた後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(6.43g、収率97%)を得た。
上記実施例3の工程4において得られた生成物(0.38g)に水(10mL)を加え、6時間加熱還流した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(0.23g、収率92%)を得た。
上記実施例3の工程4において得られた生成物(10g)をN,N-ジメチルホルムアミド(27mL)に溶解させ、水(240mL)を加え、100℃で3時間撹拌した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(6.66g、収率100%)を得た。
上記実施例3の工程4において得られた生成物(5g)をN,N-ジメチルホルムアミド(11mL)に溶解させ、硫酸銅(II)五水和物(234mg)と水(30mL)を加え、110℃で7時間加熱還流した。反応液を室温まで冷却後、水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(3.1g、収率93%)を得た。
上記実施例3の工程5において得られた生成物(4.46g)をメタノール(20mL)に溶解させ、アセチルクロリド(208mg)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(2.8g、収率84%)を得た。
工程1:1-クロロ-2,3-ジメチル-4-メチルスルホニルオキシベンゼンの調製
2,3-ジメチル-4-クロロフェノール(1.81g)をテトラヒドロフラン(20mL)に溶解させ、トリエチルアミン(1.41g)を加えた。その後、氷冷下、メタンスルホニルクロリド(1.45g)を加え、実施例1の工程1の調製と同様に反応させ、後処理することで、表題化合物(1.86g、収率68%)を得た。
上記実施例4の工程1において得られた生成物(1.86g)を1,2-ジクロロエタン(50mL)に溶解させ、N-ブロモスクシンイミド(4.23g)と2,2´-アゾビスイソブチロニトリル(130mg)を加え、3時間加熱還流した。反応液を室温まで冷却後、水を加え、クロロホルムを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(3.11g、収率100%)を得た。
上記実施例4の工程2において得られた生成物(3.11g)をN,N-ジメチルホルムアミド(40mL)に溶解させ、酢酸ナトリウム(1.56g)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例4の工程3において得られた粗生成物をメタノール(20mL)、テトラヒドロフラン(20mL)に溶解させ、氷冷下、水(5mL)、炭酸カリウム(2.2g)を加え、3時間撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:80%で溶出)を用いて精製することで、表題化合物(1.41g)を得た。
上記実施例4の工程2において得られた生成物(1.64g)に水(42mL)を加え、7時間加熱還流した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(1.11g、収率100%)を得た。
工程1:1-ブロモ-2,3-ジメチル-4-メチルスルホニルオキシベンゼンの調製
2,3-ジメチル-4-ブロモフェノール(6.57g)をテトラヒドロフラン(100mL)に溶解させ、トリエチルアミン(3.64g)を加えた。その後、氷冷下、メタンスルホニルクロリド(4.12g)を加え、実施例1の工程1の調製と同様に反応させ、後処理することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例5の工程1において得られた粗生成物(1.87g)を1,2-ジクロロエタン(13mL)に溶解させ、N-ブロモスクシンイミド(2.5g)と2,2´-アゾビスイソブチロニトリル(110mg)を加え、3時間加熱還流した。反応液を室温まで冷却後、水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:30%で溶出)を用いて精製することで、表題化合物(2.66g)を得た。
上記実施例5の工程2において得られた生成物(15.28g)をN,N-ジメチルホルムアミド(100mL)に溶解させ、酢酸ナトリウム(6.31g)を加え、90℃で3時間撹拌した。反応液を室温まで冷却後、水を加え、酢酸エチルを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例5の工程3において得られた粗生成物をメタノール(75mL)、テトラヒドロフラン(25mL)に溶解させ、氷冷下、水(25mL)、炭酸カリウム(9.04g)を加え、3時間撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(7.53g)を得た。
上記実施例5の工程2において得られた生成物(1.95g)に水(45mL)を加え、8時間加熱還流した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(1.35g、収率97%)を得た。
工程1:1-ヨード-2,3-ジメチル-4-メチルスルホニルオキシベンゼンの調製
上記実施例3の工程1において得られた生成物(3.44g)をジメチルスルホキシド(100mL)に溶解させ、ヨウ化カリウム(13.3g)、亜硝酸ナトリウム(4.41g)を加え、氷冷下、48%臭化水素酸(20mL)を少量ずつ加え、3時間撹拌した。反応液にチオ硫酸ナトリウム水溶液を加え、酢酸エチルを用いて抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(2.97g、収率57%)を得た。
上記実施例6の工程1において得られた生成物(2.97g)を1,2-ジクロロエタン(50mL)に溶解させ、N-ブロモスクシンイミド(3.89g)と2,2´-アゾビスイソブチロニトリル(150mg)を加え、3時間加熱還流した。反応液を室温まで冷却後、水を加え、クロロホルムを用いて抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例6の工程2において得られた粗生成物をN,N-ジメチルホルムアミド(50mL)に溶解させ、酢酸ナトリウム(1.79g)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例6の工程3において得られた粗生成物をメタノール(20mL)、テトラヒドロフラン(20mL)に溶解させ、氷冷下、水(5mL)、炭酸カリウム(2.52g)を加え、実施例5の工程4の調製と同様に反応させ、精製することで、表題化合物(652mg)を得た。
工程1:2,3-ジメチル-4-ジフルオロメトキシ-フェノールの調製
2,3-ジメチル-4-[[(1,1-ジメチルエチル)ジメチルシリル]オキシ]フェノール(820mg)(European Journal of Organic Chemistry, (11), 2218-2225; 2010記載の化合物)をアセトニトリル(13mL)に溶解させ、水(13mL)、水酸化カリウム(3.65g)を加え、反応液を-20℃に冷却後、(ブロモジフルオロメチル)ホスホン酸ジエチル(1.74g)を加え、さらに-20℃で1時間撹拌した。その後、室温で2時間半撹拌した後、反応液に水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(200mg、収率33%)を得た。
上記実施例7の工程1において得られた生成物(200mg)をテトラヒドロフラン(20mL)に溶解させ、トリエチルアミン(178mg)を加えた。その後、氷冷下、メタンスルホニルクロリド(134mg)を加え、室温で3時間撹拌した。反応液を濾別し、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(220mg、収率78%)を得た。
上記実施例7の工程2において得られた生成物(220mg)を四塩化炭素(10mL)に溶解させ、N-ブロモスクシンイミド(353mg)と2,2´-アゾビスイソブチロニトリル(13.6mg)を加え、4時間加熱還流した。反応液を室温まで冷却後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:50%で溶出)を用いて精製することで、表題化合物(340mg、収率97%)を得た。
上記実施例7の工程3において得られた生成物(340mg)をN,N-ジメチルホルムアミド(10mL)に溶解させ、酢酸ナトリウム(158mg)を加え、室温で一晩撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(306mg、収率100%)を得た。
上記実施例7の工程4において得られた生成物(306mg)をメタノール(8mL)、テトラヒドロフラン(8mL)に溶解させ、氷冷下、水(2mL)、炭酸カリウム(222mg)を加え、1時間撹拌した。反応液に水を加え、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-メタノール:メタノール15%で溶出)を用いて精製することで、表題化合物(130mg、収率55%)を得た。
工程1:1,4-ビス(メチルスルホニルオキシ)-2,3-ジメチルベンゼンの調製
2,3-ジメチルヒドロキノン(1g)をテトラヒドロフラン(12mL)に溶解させ、トリエチルアミン(1.75g)を加えた。その後、氷冷下、メタンスルホニルクロリド(1.83g)を加え、実施例1の工程1の調製と同様に反応させ、後処理することで、表題化合物(2.13g、収率100%)を得た。
上記実施例8の工程1において得られた生成物(2g)を1,2-ジクロロエタン(13mL)に溶解させ、N-ブロモスクシンイミド(2.53g)と2,2´-アゾビスイソブチロニトリル(112mg)を加え、3時間加熱還流した。反応液を室温まで冷却後、水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(2.81g、収率92%)を得た。
上記実施例8の工程2において得られた生成物(2.68g)をN,N-ジメチルホルムアミド(40mL)に溶解させ、酢酸ナトリウム(1.07g)を加え、実施例2の工程3の調製と同様に反応させ、精製することで、表題化合物(1.78g、収率60%)を得た。
上記実施例8の工程3において得られた生成物(1.78g)をメタノール(40mL)、テトラヒドロフラン(40mL)に溶解させ、氷冷下、水(10mL)、炭酸カリウム(1.1g)を加え、実施例2の工程4の調製と同様に反応させ、精製することで、表題化合物(916mg、収率64%)を得た。
上記実施例8の工程2において得られた生成物(1g)に水(22mL)を加え、7時間加熱還流した。反応液を室温まで冷却後、酢酸エチルを用いて抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(790mg、収率100%)を得た。
工程1:1-メチルスルホニルオキシ-3,4-ジメチルベンゼンの調製
3,4-ジメチルフェノール(2g)をテトラヒドロフラン(25mL)に溶解させ、トリエチルアミン(1.99g)を加えた。その後、氷冷下、メタンスルホニルクロリド(2.06g)を加え、実施例1の工程1の調製と同様に反応させ、後処理することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例9の工程1において得られた粗生成物を四塩化炭素(60mL)に溶解させ、
N-ブロモスクシンイミド(6.4g)と2,2´-アゾビスイソブチロニトリル(134mg)を加え、実施例1の工程2の調製と同様に反応させ、後処理することで、表題化合物を含む粗生成物を得た。この粗生成物を精製せずに次の工程に用いた。
上記実施例9の工程2において得られた粗生成物をN,N-ジメチルホルムアミド(50mL)に溶解させ、酢酸ナトリウム(3.36g)を加え、実施例2の工程3の調製と同様に反応させ、精製することで、表題化合物(3.26g)を得た。
上記実施例9の工程3において得られた生成物(3.26g)をメタノール(40mL)、テトラヒドロフラン(40mL)に溶解させ、氷冷下、水(20mL)、炭酸カリウム(2.6g)を加え、実施例2の工程4の調製と同様に反応させ、精製することで、表題化合物(1.64g、収率69%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン(200mg)(WO2008/013622号公報記載の化合物)と、実施例1において得られた生成物(121mg)と、p-トルエンスルホン酸一水和物(20mg)とを、トルエン(20mL)に溶解し、Dean-Stark装置を用いて1時間加熱還流した。反応液を室温まで冷却後、酢酸エチルで希釈し、水及び飽和食塩水で洗浄した。有機層を無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:40%-100%で溶出)を用いて精製することで、表題化合物(298mg、収率96%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-[5-メチル-3-(トリフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン(220mg)(WO2008/013622号公報記載の化合物)と、実施例3において得られた生成物(150mg)と、p-トルエンスルホン酸一水和物(20mg)とを、トルエン(15mL)に溶解させ、化合物2-1の調製における反応と同様に反応させ、精製することで、表題化合物(297mg、収率84%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン(202mg)(WO2010/066353号公報記載の化合物)と、実施例1において得られた生成物(232mg)と、p-トルエンスルホン酸一水和物(5mg)とを、トルエン(15mL)に溶解させ、化合物2-1の調製における反応と同様に反応させ、精製することで、表題化合物(164mg、収率53%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-[3,5-ビス(ジフルオロメチル)-1H-ピラゾール-1-イル]アセチル]ピペリジン(202mg)(WO2010/066353号公報記載の化合物)と、実施例3において得られた生成物(250mg)と、p-トルエンスルホン酸一水和物(5mg)とを、トルエン(15mL)に溶解させ、化合物2-1の調製における反応と同様に反応させ、精製することで、表題化合物(105mg、収率33%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-(2,5-ジメチルフェニル)アセチル]ピペリジン(200mg)と、実施例1において得られた生成物(142mg)と、p-トルエンスルホン酸一水和物(20mg)とを、トルエン(15mL)に溶解させ、化合物2-1の調製における反応と同様に反応させ、精製することで、表題化合物(206mg、収率64%)を得た。
4-(4-ホルミル-2-チアゾリル)-1-[2-(2,5-ジクロロフェニル)アセチル]ピペリジン(191mg)と、実施例1において得られた生成物(232mg)と、p-トルエンスルホン酸一水和物(5mg)とを、トルエン(15mL)に溶解し、化合物2-1の調製における反応と同様に反応させ、精製することで、表題化合物(267mg、収率72%)を得た。
2,3-ジメチル-4-ニトロソフェノール(500mg)をメタノール(10mL)に懸濁させ、タングステン酸ナトリウム二水和物(54mg)を加え、反応液を60℃に昇温した。ここに30%過酸化水素水溶液(563mg)をゆっくりと加え、60℃で8時間撹拌した。反応液を室温まで冷却し、亜硫酸ナトリウム、水を加え、酢酸エチルを用いて抽出した。有機層を無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去することで、表題化合物(500mg、収率90%)を得た。
工程1:4-(4-ホルミル-2-チアゾリル)ピペリジントリフルオロ酢酸塩の調製
4-(4-ホルミル-2-チアゾリル)ピペリジンカルボン酸1,1-ジメチルエチルエステル(3.9g)(WO2008/013622号公報記載の化合物)をジクロロメタン(65mL)に溶解させ、トリフルオロ酢酸(15.2mL)を加え、室温で一晩撹拌した。ジクロロメタンとトリフルオロ酢酸を減圧下留去することで、表題化合物を得た。
上記実施例17の工程1において得られた生成物(1g)をジクロロメタン(50mL)に溶解させ、2,5-ジメチルフェニル酢酸(919mg)と、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(1.07g)とを加え、室温で一晩撹拌した。反応液に水を加え、クロロホルムを用いて抽出した。有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、無機物を濾別した後、減圧下溶媒を留去した。残渣をフラッシュ自動精製装置(biotage AB社製/IsoleraTM)によるシリカゲルフラッシュクロマトグラフィー(酢酸エチル-ヘキサン:0%-100%で溶出)を用いて精製することで、表題化合物(1.45g、収率83%)を得た。
上記実施例17の工程1において得られた生成物(1.1g)をジクロロメタン(30mL)に溶解させ、2,5-ジクロロフェニル酢酸(1.7g)と、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(1.18g)とを加え、実施例17の工程2の調製と同様に反応させ、精製することで、表題化合物(1.7g、収率79%)を得た。



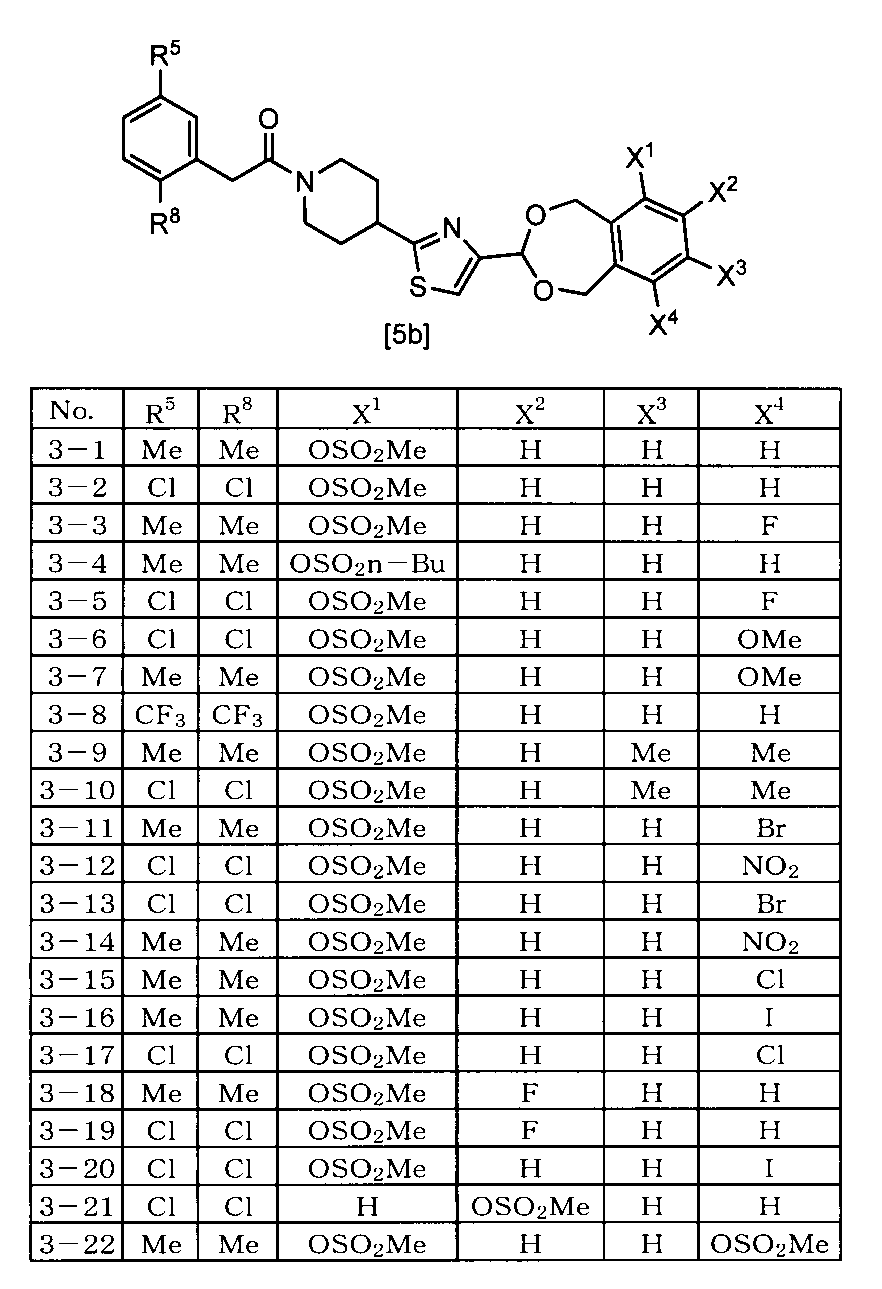
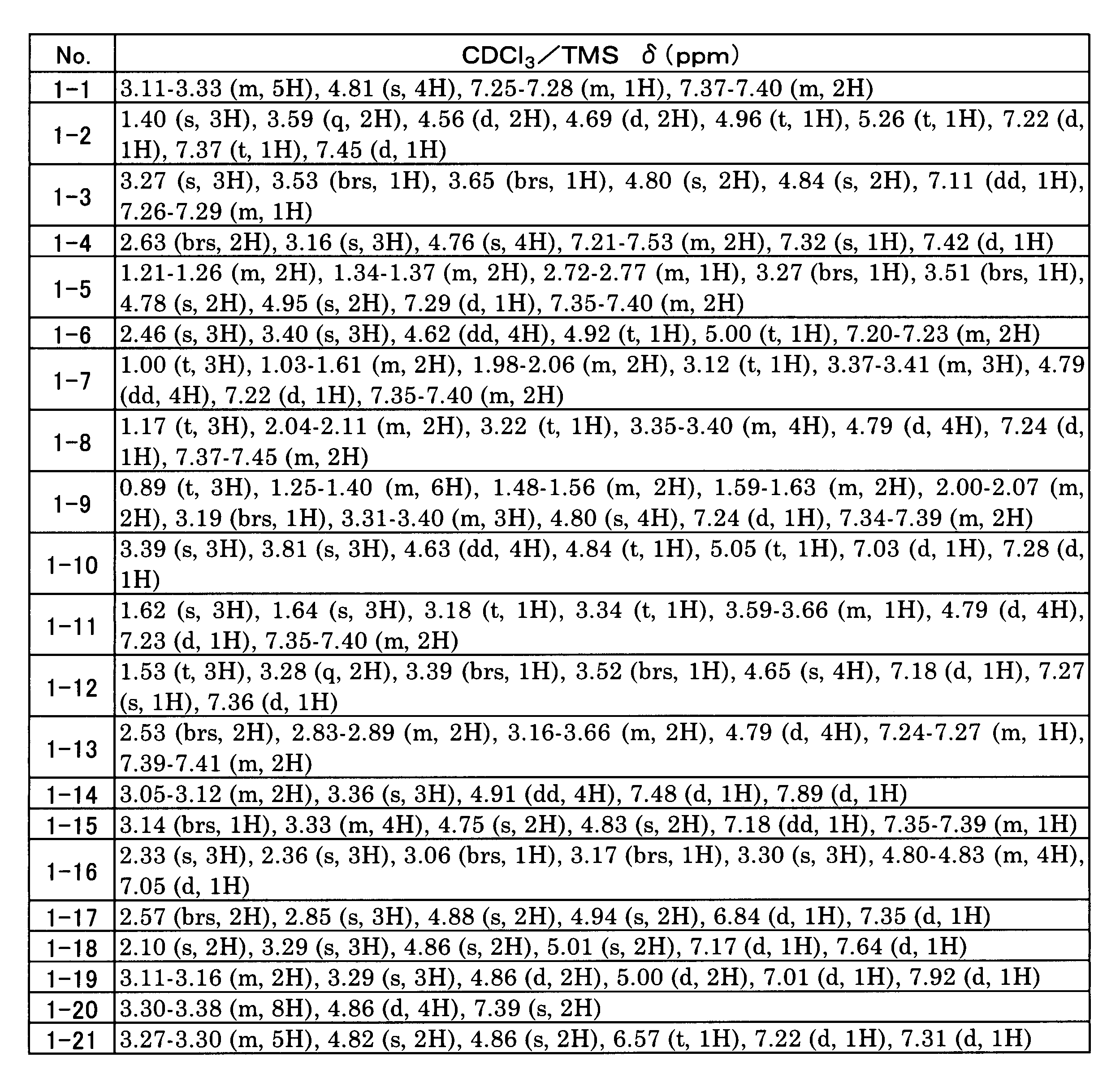


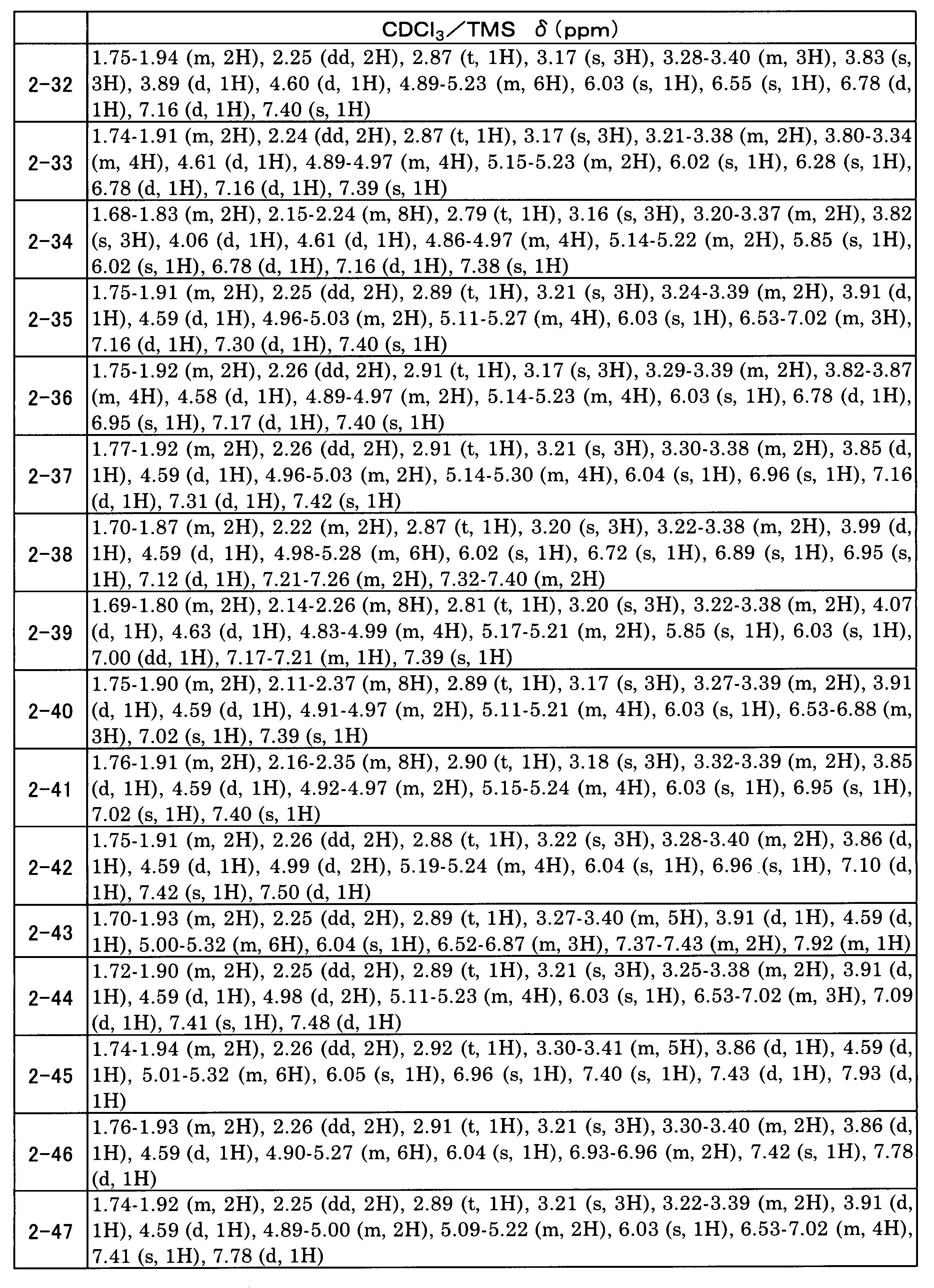



<製剤例1>水和剤
本発明化合物10部を、ラウリル硫酸ナトリウム2部、リグニンスルホン酸ナトリウム4部、ホワイトカーボン20部及びクレー64部を混合し、粉砕して10%水和剤を得た。
本発明化合物10部、ポリオキシエチレンアリルフェニルエーテルサルフェート4部、ポリオキシエチレンアルキルエーテル5部、プロピレングリコール5部、シリコン系消泡剤0.2部、ナトリウムモンモリロナイト0.8部、水50部を加え混合しダイノミルを用いて湿式粉砕して粉砕懸濁液を得た。
本発明化合物10部、ドデシルベンゼンスルホン酸カルシウム2部、ヒマシ油エトキシレート15部を芳香族炭化水素混合物73部と混合し溶解させて均質な10%可乳化油状液体を得た。
本発明化合物10部、リグニンスルホン酸ナトリウム20部、ナフタレンスルホン酸縮合物のナトリウム塩10部、アルキルベンゼンスルホン酸ナトリウム3部、シリコン系泡消剤0.5部、珪藻土5部、硫酸アンモニウム10部、タルク10部、クレー部31.5部添加して十分撹混合し粉砕して粉砕物を得た。粉砕物に必要に応じた適当量の水を加えて造粒機で造粒し、乾燥後に篩分して10%水和性細粒を得た。
本発明化合物10部、芳香族炭化水素混合物15部、ドデシルベンゼンスルホン酸カルシウム2部、ポリオキシエチレンひまし油20部、プロピレングリコール4部加え溶解し混合液を得た。水49部に混合液を添加しホモジナイザーを用いて混合して均質な10%乳濁液体を得た。
本発明化合物10部、ポリカルボン酸型アニオン界面活性剤3部、ジオクチルスルホサクシネートナトリウム0.2部、デキストリン2部、ベントナイトナトリウム15部、炭酸カルシウム69.8部を添加して均一混合した後適当量の水を加えて混練し、バスケット型造粒機で押し出し造粒して乾燥後に篩分して10%細粒を得た。
本発明化合物10部と脂肪酸ジメチルアミド12部、シクロヘキサノン10部アリールフェノールエトキシレート15部を混合し、アルコールエトキシレート10部及び水を43部添加して加温下で数分間撹拌し、安定した10%水溶性液体を得た。
製剤例1に従い作成した10%水和剤を1/5000の濃度に調製したTeen20水溶液で希釈し、式[1]で示される化合物を4ppmの濃度に調製した。また、試験4では式[1]で示される化合物を1000ppmの濃度に調製した。
<試験1 トマト疫病に対する防除効果試験>
5葉期のトマト(品種:レジナ)に試験懸濁液を1苗当たり20ml散布した。散布の1日後に1.0×105個/mlの濃度に調製したフィトフトラ インフェスタンス(Phytophthora infestans)の遊走子懸濁液を噴霧接種し、22℃に調節した湿室に16時間インキュベートした。その後、室内で発病を促し、接種から4日後に葉に生じた病斑面積率を調査し、以下の式を用いて防除価を算出した。
防除価の算出式:防除価値={1-試験薬剤を散布した葉の発病面積率/無処理の発病面積率}×100
2葉期のキュウリ(品種:相模半白)に試験懸濁液を1苗当たり20ml散布した。散布の1日後に1.0×104個/mlの濃度に調製したプセウドペロノスポラ クベンシス(Pseudoperonospora cubensis)の遊走子嚢懸濁液を噴霧接種し、22℃に調節した湿室に16時間インキュベートした。その後、室内で発病を促し、接種から5日後に葉に生じた病斑面積率を調査し、以下の式を用いて防除価を算出した。
防除価の算出式:防除価値={1-試験薬剤を散布した葉の発病面積率/無処理の発病面積率}×100
ブドウ(品種:ネオマスカット)実生に試験懸濁液を1苗当たり20ml散布した。散布の1日後に1.0×104個/mlの濃度に調製したプラズモパラ ビチコーラ(Plasmopara viticola)の遊走子嚢懸濁液を噴霧接種し、22℃に調節した湿室に16時間インキュベートした。その後、室内で発病を促し、接種から5日後に葉に生じた病斑面積率を調査し、以下の式を用いて防除価を算出した。
防除価の算出式:防除価値={1-試験薬剤を散布した葉の発病面積率/無処理の発病面積率}×100
ベントグラス種子培地で培養したピシウム グラミニコラ(Pythium graminicola)の菌叢に蒸留水を加え、ミキサーで撹拌し、土壌1kgに対して菌が5gとなるよう混和し、汚染土を調製した。
発病度=〔Σ(程度別発病株数×発病指数)/(調査した株×3)〕×100
[発病指数]
0:健全株
1:生育抑制株
3:枯死株
防除価の算出式:防除価値={1-試験薬剤を散布した区の発病度/無処理区の発病度}×100
試験1を実施した結果、以下に示す化合物が防除価80以上を示した:
No.2-1~2-55、3-1~3-7、3-10、3-12~3-15、3-17~3-22。
No.2-1~2-55、3-1~3-20、3-22。
No.2-1~2-53、2-55、3-1~3-22。
No.2-1、2-7、2-8、2-14、2-18、2-19、2-23、2-33、2-34、2-38、2-42、2-46、2-48、2-49、2-54、3-1、3-2、3-4、3-8、3-9、3-19、3-20。
Claims (19)
- 式[1]

X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキル、である}
で表される化合物の製造方法であって、
式[2]
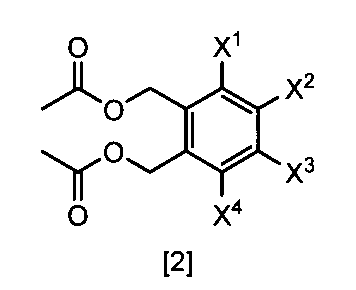
で表される化合物を酸性又は塩基性条件下で加水分解する工程を含む、前記製造方法。 - 前記塩基性条件が、金属炭酸塩類により提供される請求項1に記載の製造方法。
- 式[3]
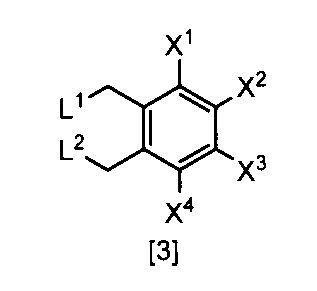
L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物と金属酢酸塩類とを反応させて式[2]で表される化合物を製造する工程をさらに含む、請求項1又は2に記載の製造方法。 - 式[2]

で表される化合物の製造方法であって、
式[3]

L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物と金属酢酸塩類とを反応させる工程を含む、前記製造方法。 - 式[4]
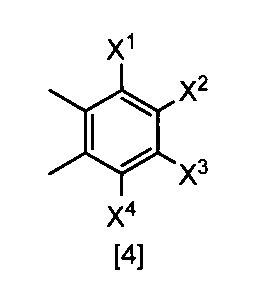
で表される化合物をハロゲン化させて式[3]で表される化合物を製造する工程をさらに含む、請求項1~4のいずれか1項に記載の製造方法。 - 式[3]

L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物の製造方法であって、
式[4]
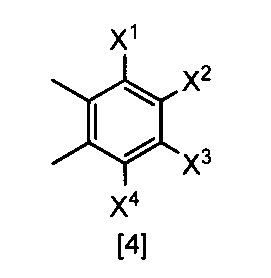
で表される化合物をハロゲン化させる工程を含む、前記製造方法。 - 前記ハロゲン化反応のハロゲン化試薬が、塩素、塩化スルフリル、N-クロロスクシンイミドなどの塩素化試薬、あるいは、臭素、N-ブロモスクシンイミドなどの臭素化試薬である、請求項5又は6に記載の製造方法。
- 式[1]で表される化合物の製造方法であって、式[3]で表される化合物と金属酢酸塩類とを反応させることで式[2]で表される化合物を得て、この式[2]で表される化合物を単離することなく、塩基性条件下、加水分解することにより式[1]で表される化合物を得る工程を含む、請求項1~3のいずれか1項に記載の製造方法。
- 式[1]

で表される化合物の製造方法であって、
式[3]
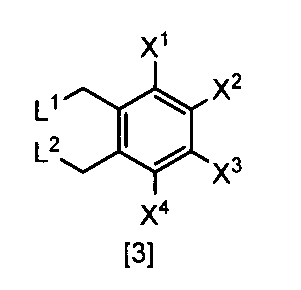
L1及びL2は、請求項3に記載した通りである}
で表される化合物を塩基、イオン液体及び金属硫酸塩類の存在下又は非存在下、水中又は水と有機溶媒の混合溶媒中で反応させる工程を含む、前記製造方法。 - X1は-OS(O)2R1であり、
X2、X3及びX4はそれぞれ独立して水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
請求項1~9のいずれか1項に記載の製造方法。 - X1は-OS(O)2Me、-OS(O)2Et、-OS(O)2n-Pr、-OS(O)2i-Pr、-OS(O)2c-Pr、-OS(O)2n-Bu、-OS(O)2n-C8H17、-OS(O)2CH2CH2CF3であり、
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
請求項10に記載の製造方法。 - X1は-OS(O)2R1であり、
X2、X3及びX4はそれぞれ独立して水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2はそれぞれ独立して塩素原子又は臭素原子である、
請求項3~9のいずれか1項に記載の製造方法。 - X1は-OS(O)2Me、-OS(O)2Et、-OS(O)2n-Pr、-OS(O)2i-Pr、-OS(O)2c-Pr、-OS(O)2n-Bu、-OS(O)2n-C8H17、-OS(O)2CH2CH2CF3であり、
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2は臭素原子である、
請求項12に記載の製造方法。 - X1、X3及びX4は水素原子であり、
X2は-OS(O)2R1であり、
R1はC1~C4アルキルである、
請求項1~9のいずれか1項に記載の製造方法。 - X1、X3及びX4は水素原子であり、
X2は-OS(O)2R1であり、
R1はC1~C4アルキルであり、
L1及びL2はそれぞれ独立して塩素原子又は臭素原子である、
請求項3~9のいずれか1項に記載の製造方法。 - 式[2]
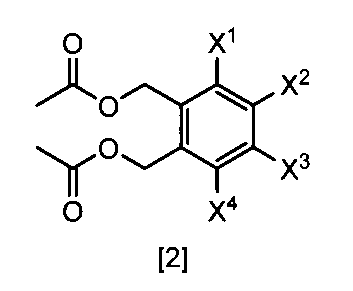
X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルである}
で表される化合物又はその塩。 - X1は-OS(O)2Me、-OS(O)2Et、-OS(O)2n-Pr、-OS(O)2i-Pr、-OS(O)2c-Pr、-OS(O)2n-Bu、-OS(O)2n-C8H17、-OS(O)2CH2CH2CF3であり、
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meである、
請求項16に記載の化合物又はその塩。 - 式[3]

X1、X2、X3及びX4のうちいずれか一つは、-OS(O)2R1であり、
R1はC1~C8アルキル、C1~C8ハロアルキル又はC3~C6シクロアルキルであり、
L1及びL2は、それぞれ独立してハロゲン原子である}
で表される化合物又はその塩。 - X1は-OS(O)2Me、-OS(O)2Et、-OS(O)2n-Pr、-OS(O)2i-Pr、-OS(O)2c-Pr、-OS(O)2n-Bu、-OS(O)2n-C8H17、-OS(O)2CH2CH2CF3であり、
X2及びX3は水素原子であり、
X4は水素原子、ニトロ、ハロゲン原子、ジフルオロメトキシ又は-OS(O)2Meであり、
L1及びL2は臭素原子である、
請求項18に記載の化合物又はその塩。
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL16889770T PL3415496T3 (pl) | 2016-02-08 | 2016-02-08 | Sposób przygotowania związku 1,2-benzenodimetanolu |
| ES16889770T ES2895047T3 (es) | 2016-02-08 | 2016-02-08 | Proceso para la preparación del compuesto 1,2-bencenodimetanol |
| JP2017566254A JP6407457B2 (ja) | 2016-02-08 | 2016-02-08 | 1,2−ベンゼンジメタノール化合物の製造方法 |
| PT16889770T PT3415496T (pt) | 2016-02-08 | 2016-02-08 | Processo para a preparação do composto 1,2-benzenodimetanol |
| CN202210385927.6A CN114702411A (zh) | 2016-02-08 | 2016-02-08 | 1,2-苯二甲醇化合物的制造方法 |
| US16/076,153 US11274076B2 (en) | 2016-02-08 | 2016-02-08 | Process for preparing 1, 2-benzenedimethanol compound |
| EP16889770.0A EP3415496B1 (en) | 2016-02-08 | 2016-02-08 | Process for preparing 1,2-benzenedimethanol compound |
| PCT/JP2016/053650 WO2017138068A1 (ja) | 2016-02-08 | 2016-02-08 | 1,2-ベンゼンジメタノール化合物の製造方法 |
| CN201680081183.1A CN108602765B (zh) | 2016-02-08 | 2016-02-08 | 1,2-苯二甲醇化合物的制造方法 |
| TW106103717A TWI731930B (zh) | 2016-02-08 | 2017-02-03 | 1,2-苯二甲醇化合物之製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2016/053650 WO2017138068A1 (ja) | 2016-02-08 | 2016-02-08 | 1,2-ベンゼンジメタノール化合物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017138068A1 true WO2017138068A1 (ja) | 2017-08-17 |
Family
ID=59562964
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/053650 Ceased WO2017138068A1 (ja) | 2016-02-08 | 2016-02-08 | 1,2-ベンゼンジメタノール化合物の製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11274076B2 (ja) |
| EP (1) | EP3415496B1 (ja) |
| JP (1) | JP6407457B2 (ja) |
| CN (2) | CN108602765B (ja) |
| ES (1) | ES2895047T3 (ja) |
| PL (1) | PL3415496T3 (ja) |
| PT (1) | PT3415496T (ja) |
| TW (1) | TWI731930B (ja) |
| WO (1) | WO2017138068A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10104891B2 (en) | 2014-08-13 | 2018-10-23 | Sds Biotech K.K. | Fused 11-membered compounds and agricultural/horticultural fungicides containing them |
| US11274076B2 (en) | 2016-02-08 | 2022-03-15 | Gowan Company, L.L.C. | Process for preparing 1, 2-benzenedimethanol compound |
| US11903387B2 (en) | 2016-02-08 | 2024-02-20 | Gowan Company, L.L.C. | Fungicidal composition |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108069831B (zh) * | 2018-01-25 | 2020-11-24 | 上海恩氟佳科技有限公司 | 一种合成2,3-二甲基-4-氟苯酚的方法 |
| CN110776441B (zh) * | 2019-11-27 | 2022-03-22 | 西安淳甄新材料有限公司 | 一种4-氟-2,3-二甲基苯基甲磺酸酯的合成方法 |
| CN115448835A (zh) * | 2022-09-08 | 2022-12-09 | 苏州莱克施德药业有限公司 | 一种2-氟-3-(三氟甲基)苯甲酸的合成方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993009113A1 (en) * | 1991-11-09 | 1993-05-13 | Smithkline Beecham Plc | 2-((imidazol-2-yl)methyl)-isoindole derivatives as alpha-2-adrenoceptor antagonists |
| JP2001302658A (ja) * | 2000-04-18 | 2001-10-31 | Showa Denko Kk | 3−イソクロマノン類の製造方法 |
| US20010051620A1 (en) * | 1999-12-29 | 2001-12-13 | American Home Products Corporation | Tricyclic protein kinase inhibitors |
| WO2009131090A1 (ja) * | 2008-04-21 | 2009-10-29 | 旭化成ファーマ株式会社 | アミノ酸化合物 |
| JP2011021013A (ja) * | 2009-06-19 | 2011-02-03 | Ube Industries Ltd | オキシル基を有する芳香族メチルアルコール化合物の製造方法 |
Family Cites Families (71)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4218464A (en) | 1979-03-02 | 1980-08-19 | Smithkline Corporation | 4- and 5-Substituted 2,3-dihydro-1H-isoindoles, pharmaceutical compositions and method of inhibiting phenylethanolamine N-methyltransferase |
| GB8529546D0 (en) | 1985-11-30 | 1986-01-08 | Beecham Group Plc | Compounds |
| EP0313288B1 (en) | 1987-10-19 | 1992-12-30 | Beecham Group Plc | Imidazole derivatives, process for their preparation and their use as alpha 2-adreno-receptor antagonists |
| TWI242011B (en) | 1997-03-31 | 2005-10-21 | Eisai Co Ltd | 1,4-substituted cyclic amine derivatives |
| CA2396579A1 (en) * | 1999-12-29 | 2001-07-05 | Wyeth | Tricyclic protein kinase inhibitors |
| AU2002316734A1 (en) | 2001-07-20 | 2003-03-03 | University Of Rochester | Light-emitting organic oligomer compositions |
| JP3630326B2 (ja) | 2002-08-22 | 2005-03-16 | 三共株式会社 | 水溶性トリアゾール医薬組成物 |
| DE602006010665D1 (en) | 2005-02-07 | 2010-01-07 | Hoffmann La Roche | Heterocyclsche substituierte phenylmethanone als inhibitoren des glycintransporters 1 |
| TW200738701A (en) | 2005-07-26 | 2007-10-16 | Du Pont | Fungicidal carboxamides |
| WO2008013622A2 (en) | 2006-07-27 | 2008-01-31 | E. I. Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| WO2008091594A2 (en) | 2007-01-24 | 2008-07-31 | E. I. Du Pont De Nemours And Company | Fungicidal mixtures |
| JP5337711B2 (ja) | 2007-01-25 | 2013-11-06 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | 殺菌性アミド |
| JP5241834B2 (ja) | 2007-07-19 | 2013-07-17 | メルク・シャープ・アンド・ドーム・コーポレーション | プロテインキナーゼ阻害剤としての複素環アミド化合物 |
| TWI428091B (zh) | 2007-10-23 | 2014-03-01 | Du Pont | 殺真菌劑混合物 |
| US8349870B2 (en) | 2008-01-25 | 2013-01-08 | E I Du Pont De Nemours And Company | Fungicidal hetercyclic compounds |
| EP2238133A2 (en) | 2008-01-25 | 2010-10-13 | E. I. du Pont de Nemours and Company | Fungicidal amides |
| BRPI0910837B1 (pt) | 2008-04-30 | 2017-03-07 | Bayer Cropscience Ag | ésteres e tioésteres tiazol-4-carboxílicos, seus usos, e método e composição para controlar fungos fitopatogênicos prejudiciais |
| EP2334669B1 (de) | 2008-10-01 | 2015-12-23 | Bayer Intellectual Property GmbH | Heterocyclyl substituierte thiazole als pflanzenschutzmittel |
| AU2009322486A1 (en) | 2008-12-02 | 2010-06-10 | E. I. Du Pont De Nemours And Company | Fungicidal heterocyclic compounds |
| CN102292334A (zh) | 2008-12-11 | 2011-12-21 | 拜尔农作物科学股份公司 | 噻唑基肟醚和腙植物保护剂 |
| AU2009333427A1 (en) | 2008-12-17 | 2011-07-28 | Merck Sharp & Dohme Corp. | Imidazolinone derivatives as CGRP receptor antagonists |
| EP2272846A1 (de) | 2009-06-23 | 2011-01-12 | Bayer CropScience AG | Thiazolylpiperidin Derivate als Fungizide |
| EP2464643A1 (en) | 2009-08-12 | 2012-06-20 | Syngenta Participations AG | Microbiocidal heterocycles |
| UA107938C2 (en) | 2009-08-12 | 2015-03-10 | Syngenta Participations Ag | Heterocycles with microbicidal properties |
| WO2011051243A1 (en) | 2009-10-29 | 2011-05-05 | Bayer Cropscience Ag | Active compound combinations |
| KR20120101019A (ko) | 2009-10-30 | 2012-09-12 | 바이엘 크롭사이언스 아게 | 헤테로아릴피페리딘 및 -피페라진 유도체 |
| MX2012007200A (es) | 2009-12-21 | 2012-07-10 | Bayer Cropscience Ag | Bis(difluorometil)pirazoles como fungicidas. |
| US8629286B2 (en) | 2009-12-22 | 2014-01-14 | Syngenta Crop Protection, Llc | Pyrazole derivatives |
| BR112012016733A2 (pt) | 2010-01-07 | 2015-09-01 | Du Pont | "composto, composição fungicida e método" |
| BR112012027762B1 (pt) | 2010-04-28 | 2018-06-05 | Bayer Intellectual Property Gmbh | Derivados de cetoheteroarilpiperidina e - piperazina como fungicidas |
| US8815775B2 (en) | 2010-05-18 | 2014-08-26 | Bayer Cropscience Ag | Bis(difluoromethyl)pyrazoles as fungicides |
| BR112012026708A2 (pt) | 2010-05-20 | 2015-09-22 | Du Pont | composto selecionado, composição fungicida e método para controlar doenças vegetais |
| EP2576539B1 (de) | 2010-05-27 | 2017-12-13 | Bayer CropScience AG | Pyridinylcarbonsäure derivate als fungizide |
| US20120122928A1 (en) | 2010-08-11 | 2012-05-17 | Bayer Cropscience Ag | Heteroarylpiperidine and -Piperazine Derivatives as Fungicides |
| US8759527B2 (en) | 2010-08-25 | 2014-06-24 | Bayer Cropscience Ag | Heteroarylpiperidine and -piperazine derivatives as fungicides |
| EP2423210A1 (de) | 2010-08-25 | 2012-02-29 | Bayer CropScience AG | Heteroarylpiperidin- und -piperazinderivate als Fungizide |
| GB2483736B (en) | 2010-09-16 | 2012-08-29 | Aragon Pharmaceuticals Inc | Estrogen receptor modulators and uses thereof |
| RS58401B1 (sr) | 2010-10-07 | 2019-04-30 | Bayer Cropscience Ag | Sastav fungicida koji sadrži derivat tetrazoliloksima i derivat tiazolilpiperidina |
| EP2632922B1 (de) | 2010-10-27 | 2019-02-27 | Bayer CropScience Aktiengesellschaft | Heteroarylpiperidin und -piperazinderivate als fungizide |
| BR112013012621A2 (pt) | 2010-11-25 | 2016-07-12 | Syngenta Participations Ag | heterociclos microbicidas |
| CN103384470A (zh) | 2010-12-17 | 2013-11-06 | 纳幕尔杜邦公司 | 杀真菌偶氮环酰胺 |
| WO2012104273A1 (de) | 2011-02-01 | 2012-08-09 | Bayer Cropscience Ag | Heteroarylpiperidin und -piperazinderivate als fungizide |
| BR112013020419A2 (pt) | 2011-02-10 | 2016-07-12 | Syngenta Participations Ag | microbicidas derivados de pirazol |
| BR112013020270A2 (pt) | 2011-02-10 | 2016-07-12 | Syngenta Participations Ag | derivados de pirazol microbicidas |
| EP2532233A1 (en) | 2011-06-07 | 2012-12-12 | Bayer CropScience AG | Active compound combinations |
| WO2013000941A1 (en) | 2011-06-30 | 2013-01-03 | Syngenta Participations Ag | Microbiocidal heterocycles |
| MY184916A (en) | 2011-06-30 | 2021-04-30 | Syngenta Participations Ag | Microbiocidal heterocycles |
| US10004232B2 (en) | 2011-09-15 | 2018-06-26 | Bayer Intellectual Property Gmbh | Piperidine pyrazoles as fungicides |
| CN103889982A (zh) | 2011-10-18 | 2014-06-25 | 先正达参股股份有限公司 | 杀微生物的吡唑衍生物 |
| US20140243371A1 (en) | 2011-10-18 | 2014-08-28 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| DK2797899T3 (en) | 2011-12-27 | 2016-04-04 | Bayer Ip Gmbh | Heteroarylpiperidin and -piperazinderivater as fungicides |
| TWI568721B (zh) | 2012-02-01 | 2017-02-01 | 杜邦股份有限公司 | 殺真菌之吡唑混合物 |
| MX360174B (es) | 2012-02-27 | 2018-10-12 | Bayer Ip Gmbh | Combinaciones de compuestos activos que contienen una tiazolilisoxazolina y un fungicida. |
| EP2820011A1 (en) | 2012-03-02 | 2015-01-07 | Syngenta Participations AG | Microbiocidal pyrazole derivatives |
| EP2820012A1 (en) | 2012-03-02 | 2015-01-07 | Syngenta Participations AG | Microbiocidal pyrazole derivatives |
| WO2013127808A1 (en) | 2012-03-02 | 2013-09-06 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| EP2864326A1 (en) | 2012-06-22 | 2015-04-29 | E. I. Du Pont de Nemours and Company | Fungicidal heterocyclic compounds |
| WO2014060176A1 (en) | 2012-10-16 | 2014-04-24 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014075874A1 (en) | 2012-11-13 | 2014-05-22 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014075873A1 (en) | 2012-11-13 | 2014-05-22 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014118142A1 (en) | 2013-02-01 | 2014-08-07 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014118143A1 (en) | 2013-02-04 | 2014-08-07 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014154530A1 (en) | 2013-03-25 | 2014-10-02 | Syngenta Participations Ag | Microbiocidal pyrazole derivatives |
| WO2014179144A1 (en) | 2013-04-29 | 2014-11-06 | E. I. Du Pont De Nemours And Company | Fungicidal heterocyclic compounds |
| WO2014206896A1 (en) | 2013-06-24 | 2014-12-31 | Bayer Cropscience Ag | Piperidinecarboxylic acid derivatives as fungicides |
| WO2015036379A1 (en) | 2013-09-13 | 2015-03-19 | Bayer Cropscience Ag | Fungicidal compositions containing thiazolylisoxazoline fungicide and biological fungicide |
| ES2645448T3 (es) | 2013-10-17 | 2017-12-05 | Bayer Cropscience Aktiengesellschaft | Forma cristalina novedosa de 2-{3-[2-(1-{[3,5-bis(difluorometil)-1H-pirazol-1-il]acetil}piperidin-4-il)-1,3-tiazol-4-il]-4,5-dihidro-1,2-oxazol-5-il}-3-clorofenilmetanosulfonato |
| TWI647215B (zh) | 2013-11-11 | 2019-01-11 | 德商拜耳作物科學股份有限公司 | 自α,α-二鹵胺製備3,5-雙鹵烷基吡唑衍生物之方法 |
| WO2016024350A1 (ja) | 2014-08-13 | 2016-02-18 | 株式会社エス・ディー・エス バイオテック | 縮合11員環化合物及びそれらを含有する農園芸用殺菌剤 |
| KR102605762B1 (ko) | 2016-02-08 | 2023-11-27 | 고완 크롭 프로텍션 리미티드 | 살균성 조성물 |
| CN108602765B (zh) * | 2016-02-08 | 2022-05-03 | 高文有限公司 | 1,2-苯二甲醇化合物的制造方法 |
-
2016
- 2016-02-08 CN CN201680081183.1A patent/CN108602765B/zh not_active Expired - Fee Related
- 2016-02-08 PL PL16889770T patent/PL3415496T3/pl unknown
- 2016-02-08 CN CN202210385927.6A patent/CN114702411A/zh active Pending
- 2016-02-08 JP JP2017566254A patent/JP6407457B2/ja not_active Expired - Fee Related
- 2016-02-08 PT PT16889770T patent/PT3415496T/pt unknown
- 2016-02-08 US US16/076,153 patent/US11274076B2/en not_active Expired - Fee Related
- 2016-02-08 EP EP16889770.0A patent/EP3415496B1/en active Active
- 2016-02-08 ES ES16889770T patent/ES2895047T3/es active Active
- 2016-02-08 WO PCT/JP2016/053650 patent/WO2017138068A1/ja not_active Ceased
-
2017
- 2017-02-03 TW TW106103717A patent/TWI731930B/zh active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993009113A1 (en) * | 1991-11-09 | 1993-05-13 | Smithkline Beecham Plc | 2-((imidazol-2-yl)methyl)-isoindole derivatives as alpha-2-adrenoceptor antagonists |
| US20010051620A1 (en) * | 1999-12-29 | 2001-12-13 | American Home Products Corporation | Tricyclic protein kinase inhibitors |
| JP2001302658A (ja) * | 2000-04-18 | 2001-10-31 | Showa Denko Kk | 3−イソクロマノン類の製造方法 |
| WO2009131090A1 (ja) * | 2008-04-21 | 2009-10-29 | 旭化成ファーマ株式会社 | アミノ酸化合物 |
| JP2011021013A (ja) * | 2009-06-19 | 2011-02-03 | Ube Industries Ltd | オキシル基を有する芳香族メチルアルコール化合物の製造方法 |
Non-Patent Citations (5)
| Title |
|---|
| JOHN E. ANTHONY ET AL.: "Synthesis, optical, thermal, and redox properties of 2,3,9,10- tetrasubstituted-6,13-dialkynyl pentacenes", PROCEEDINGS OF SPIE-THE INTERNATIONAL SOCIETY FOR OPTICAL ENGINEERING, vol. 5940, 31 July 2005 (2005-07-31), pages 594002-1 - 594002-12, XP008067327 * |
| MASANOBU SUZUKI ET AL.: "Synthesis and Absolute Configuration of Pyriculol", AGRICULTURAL AND BIOLOGICAL CHEMISTRY, vol. 51, no. 4, 9 April 1987 (1987-04-09), pages 1121 - 1127, XP055406149 * |
| See also references of EP3415496A4 * |
| THEODORA W. GREENE ET AL.: "Protective Groups in Organic Synthesis. 3rd ed.", 1999, article "PROTECTION FOR THE HYDROXYL GROUP, INCLUDING 1,2- AND 1,3- DIOLS", pages: 198, XP055406157 * |
| VISHWAKARMA SINGH ET AL.: "Oxidative dearomatization and unusual intramolecular Diels-Alder reaction of cyclohexa-2,4-dienone: synthesis and photoreaction of oxa-tricyclo [5.2.2 .01,5] undec-10-ene-8-ones", TETRAHEDRON LETTERS, vol. 56, no. 15, 2015, pages 1982 - 1985, XP029216164 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10104891B2 (en) | 2014-08-13 | 2018-10-23 | Sds Biotech K.K. | Fused 11-membered compounds and agricultural/horticultural fungicides containing them |
| US11274076B2 (en) | 2016-02-08 | 2022-03-15 | Gowan Company, L.L.C. | Process for preparing 1, 2-benzenedimethanol compound |
| US11903387B2 (en) | 2016-02-08 | 2024-02-20 | Gowan Company, L.L.C. | Fungicidal composition |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200399263A1 (en) | 2020-12-24 |
| JP6407457B2 (ja) | 2018-10-17 |
| TWI731930B (zh) | 2021-07-01 |
| US11274076B2 (en) | 2022-03-15 |
| CN114702411A (zh) | 2022-07-05 |
| EP3415496B1 (en) | 2021-07-21 |
| EP3415496A1 (en) | 2018-12-19 |
| JPWO2017138068A1 (ja) | 2018-03-29 |
| TW201800388A (zh) | 2018-01-01 |
| CN108602765A (zh) | 2018-09-28 |
| EP3415496A4 (en) | 2019-08-07 |
| CN108602765B (zh) | 2022-05-03 |
| PL3415496T3 (pl) | 2022-02-07 |
| PT3415496T (pt) | 2021-10-25 |
| ES2895047T3 (es) | 2022-02-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6407457B2 (ja) | 1,2−ベンゼンジメタノール化合物の製造方法 | |
| TWI254708B (en) | 3-phenoxy-4-pyridazinol derivatives and containing thereof for herbicidal compositions | |
| JP4551764B2 (ja) | ピラゾール誘導体及びその製造方法 | |
| JP2009001541A (ja) | 新規ピラゾール化合物を中間体として用いるアントラニルアミド系化合物の製造方法 | |
| JP2004002263A (ja) | 3−フェノキシ−4−ピリダジノール誘導体及びそれを含有する除草剤組成物 | |
| JP2025169362A (ja) | 2-(5-イソオキサゾリル)-フェノールの合成のためのプロセス | |
| JP6371915B2 (ja) | 4−(4−ホルミルチアゾリル)ピペリジン化合物の製造方法 | |
| JP4580175B2 (ja) | 3−フェノキシ−4−ピリダジノール誘導体を含有する除草剤組成物 | |
| JP4549690B2 (ja) | 3−フェノキシ−4−ピリダジノール誘導体を含有する除草性組成物 | |
| JP2004262940A (ja) | 3−フェノキシ−4−ピリダジノール誘導体を含有する除草組成物 | |
| JP4580173B2 (ja) | 3−フェノキシ−4−ピリダジノール化合物を含有する除草組成物 | |
| JP4580174B2 (ja) | 3−フェノキシ−4−ピリダジノール化合物を含有する除草性組成物 | |
| WO2000066569A1 (en) | Benzoxazole compounds, process for the preparation thereof and herbicides | |
| JP2005232114A (ja) | N−シンナモイル−ベンゼンスルホンアミド誘導体及び農園芸用植物病害防除剤 | |
| CA3104048A1 (en) | Pyridone compounds and agricultural and horticultural fungicides containing the same as active ingredients | |
| JP2016079104A (ja) | ジフェニルジスルフィド誘導体およびその製造方法 | |
| WO2011040445A1 (ja) | 3-置換オキシ-4-ピリダジノール誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16889770 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017566254 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016889770 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016889770 Country of ref document: EP Effective date: 20180910 |