WO2017151530A1 - Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids - Google Patents
Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids Download PDFInfo
- Publication number
- WO2017151530A1 WO2017151530A1 PCT/US2017/019801 US2017019801W WO2017151530A1 WO 2017151530 A1 WO2017151530 A1 WO 2017151530A1 US 2017019801 W US2017019801 W US 2017019801W WO 2017151530 A1 WO2017151530 A1 WO 2017151530A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- mixture
- alkyl
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c(c(Cl)c(c(C#N)n1)O)c1Cl Chemical compound *c(c(Cl)c(c(C#N)n1)O)c1Cl 0.000 description 7
- OJYYIZCFSNMPOJ-UHFFFAOYSA-N COc1cc(Cl)nc(C#N)c1O Chemical compound COc1cc(Cl)nc(C#N)c1O OJYYIZCFSNMPOJ-UHFFFAOYSA-N 0.000 description 1
- QJBWOIGJIHNUOY-UHFFFAOYSA-N N#Cc1nc(Cl)cc(Cl)c1O Chemical compound N#Cc1nc(Cl)cc(Cl)c1O QJBWOIGJIHNUOY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/803—Processes of preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
Definitions
- the present disclosure concerns a process for the preparation of 4-alkoxy-3- hydroxypicolinic acids. More particularly, the present disclosure concerns a process for the preparation of 4-alkoxy-3-hydroxypicolinic acids from furfural.
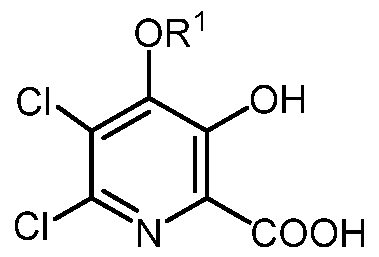
- the present disclosure concerns processes for the preparation of 4-alkoxy-3- hydroxypicolinic acids of Formula A
- R is a C1-C3 alkyl
- the compound of Formula A may be prepared in a multi-step process which comprises the following steps: a) creating a first mixture by combining together the compound of Formula B and a chlorinating agent to form a first mixture; b) isolating a compound of Formula C from the first mixture;
- R is H or CI, and R 1 is a C1-C3 alkyl; f) creating a third mixture containing the compound of Formula E, water, and one of a mineral acid and a strong base; g) heating the third mixture; h) isolating a compound of Formula F from the third mixture
- R is H or CI and R 1 is a C1-C3 alkyl; i) creating a fourth mixture containing the compound of Formula F and a reducing agent; and j) isolating the compound of Formula A from the fourth mixture.
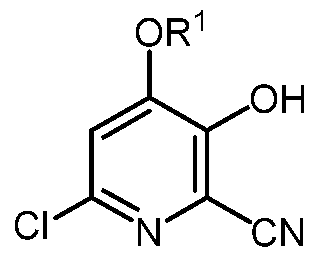
- the present disclosure also concerns a process for the preparation of the trichloro-3 hydroxypicolinonitrile of Formula C
- R 1 is a C1-C3 alkyl
- R 1 is a C1-C3 alkyl
- R 1 is a C1-C3 alkyl
- R 1 is a C1-C3 alkyl
- isolated means to partially or completely remove the desired product from the other components of a finished chemical process mixture using standard methods such as, but not limited to, filtration, extraction, distillation, crystallization, centrifugation, trituration, liquid- liquid phase separation or other methods known to those of ordinary skill in the art.
- the isolated product may have a purity that ranges from ⁇ 50% to > 50%, and may be purified to a higher purity level using standard purification methods.
- the isolated product may also be used in a subsequent process step with or without purification.
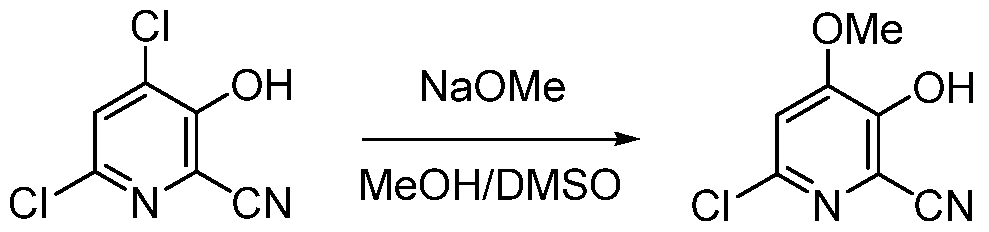
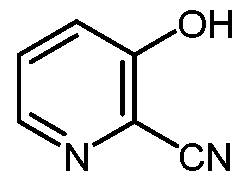
- 3-hydroxypicolinonitrile (B) 4-alkoxy-3-hydroxypicolinic acids of Formula A are prepared from 3-hydroxypicolinonitrile (B) in a series of chemical steps involving chlorination, substitution of a chloro group by an alkoxide group, nitrile hydrolysis, and halogen reduction. Some of the individual steps may be performed in different sequences of order.
- 3-Hydroxypicolinonitrile of Formula B is readily prepared from furfural as disclosed in U.S. Application Serial Number 14/794430.
- reaction solvents for the chlorination reaction can be selected from the group including water, acetonitrile, sulfolane, DMF, DMSO and mixtures thereof.
- Typical alkali metal alkoxides useful in this reaction include sodium or potassium, methoxide, ethoxide, 1- propoxide or 2-propoxide.
- the reaction may be carried out in a protic solvent or reaction medium such as methanol (for methoxide), ethanol (for ethoxide), 1-propanol (for 1- propoxide) or 2-propanol (for 2-propoxide), or mixtures of methanol, ethanol, 1-propanol or 2-propanol with a polar, aprotic co-solvent such as DMSO, DMF, sulfolane or NMP.
- a protic solvent or reaction medium such as methanol (for methoxide), ethanol (for ethoxide), 1-propanol (for 1- propoxide) or 2-propanol (for 2-propoxide), or mixtures of methanol, ethanol, 1-propanol or 2-propanol with a polar, aprotic co-solvent such
- the reaction may also be conducted with an alkali metal alkoxide in one or more of the polar, aprotic solvents in the absence of an alcohol co- solvent.
- the temperature at which the reaction is conducted is between about 20 °C and about 150 °C, preferably between about 40 °C and about 100 °C.
- the substitution reaction generally requires from about 1 to about
- the volume percent (vol%) ratio of the protic solvent to the polar aprotic solvent in the total solvent mixture is 80-100 vol% protic solvent to 0-20 vol% polar aprotic solvent, 60-80 vol% protic solvent to 20-40 vol% polar aprotic solvent, 40-60 vol% protic solvent to 40-60 vol% polar aprotic solvent, 20-40 vol% protic solvent to 60-80 vol% polar aprotic solvent, or 0-20 vol% protic solvent to 80-100 vol% polar aprotic solvent.
- Preferable volume percent (vol%) ratios of the protic solvent to the polar aprotic solvent are from about 0.01-10 vol% protic solvent to about 90-99.99 vol% polar aprotic solvent.
- the starting picolinonitriles are typically suspended in a strong, aqueous mineral acid reaction medium and heated for a period of time at elevated temperature with good mixing.
- Strong mineral acids useful in the hydrolysis reaction include sulfuric acid, phosphoric acid, hydrochloric acid and hydrobromic acid.
- Preferred, strong mineral acid reaction mediums include aqueous sulfuric acid mixtures such as about 25%, about 30%, about 35%, about 40%, about 45%, about 50%, about 55%, about 60%, about 65%, about 70%, about 75% or about 80% sulfuric acid in water on a weight basis. Most preferably, from about 25% to about 70% sulfuric acid in water may be used.
- the temperature at which the hydrolysis reaction may be conducted is usually between about 75 °C and about 150 °C and preferably between about 80 °C and about 120 °C.
- the hydrolysis reaction generally requires from about 8 to about 48 hours, preferably from about 8 to about 36 hours, to reach completion. After the reaction is complete, the desired product
- the hydrolysis reaction of the nitrile group of the 4-alkoxy-3- hydroxypicolinonitriles of Formulas D and F to produce the 4-alkoxy-3-hydroxypicolinic acids of Formulas E and A, respectively (Steps b in Scheme ⁇ )
- the starting picolinonitriles are suspended in an aqueous reaction medium containing a strong base, such as an hydroxide of an alkali or alkaline earth metal, and heated for a period of time at elevated temperature with good mixing.
- Strong bases for use in the hydrolysis of the picolinonitriles include sodium hydroxide and potassium hydroxide.
- the concentration of the strong base used in the hydrolysis of the picolinonitriles may range from about 10 to about 40 weight percent (wt %), from about 15 to about 40 wt %, from about 20 to about 40 wt %, from about 30 to about 40 wt %, or from about 15 to about 25 wt%.
- the molar equivalent ratio of strong base to the nitrile starting material for the hydrolysis reaction may range from about 3:1 to about 10:1, preferably from about 4:1 to about 7: 1.
- the temperature at which the strong base hydrolysis reaction may be conducted is usually between about 75 °C and about 150 °C and preferably between about 80 °C and about 120 °C.
- the strong base hydrolysis reaction generally requires from about 8 to about 48 hours, preferably from about 8 to about 36 hours, to reach completion.
- the desired product may be isolated by acidifying the reaction mixture and employing standard isolation and purification techniques. Removal of the chloro groups from the 5- and 6-positions of the compound of
- Formula E to produce the reduced product of Formula A may be achieved by catalytic reduction using a hydrogen source and a transition metal catalyst.
- suitable hydrogen sources include hydrogen gas or hydrogen transfer reagents such as ammonium, potassium or sodium formate.
- Suitable transition metal catalysts include, but are not limited to, palladium on carbon (Pd/C) and
- Raney nickel Raney nickel (Ra/Ni). These catalysts may be used at levels from about 0.01% to about 10% on a weight basis of the metal to the chloropyridine substrate.
- Exemplary solvents for use in this reaction include methanol, ethanol, isopropanol, ethyl acetate, and acetic acid.
- a soluble base such as, for example, triethylamine is normally used in the catalytic reduction with
- E, R H, CI A hydrogen. From about 2 to about 4 molar equivalents of the soluble base are normally used.
- hydrogen gas used as the hydrogen source, the reduction reaction may be conducted under an atmospheric pressure of hydrogen gas, or at elevated pressures of hydrogen gas such as 10, 20, 40, 60, 80, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000 pounds or more, per square inch (psi) above atmospheric pressure, or incremental hydrogen gas pressures between these values.
- the desired product is recovered by employing standard isolation and purification techniques.
- removal of the chloro groups from the 5- and 6-positions of the compound of Formula D to produce the reduced product of Formula F might be achieved by catalytic reduction using a hydrogen source and a transition metal catalyst.
- the products obtained by any of these processes can be recovered by conventional means, such as evaporation, filtration or extraction, and can be purified by standard procedures, such as by recrystallization or chromatography.
- 3-Hydroxypicolinonitrile 500mg, 4.2 mmol was combined with l,3-dichloro-5,5- dimethylhydantoin (900mg, 4.6mmol) in 2.5 mL of dry acetonitrile and heated at 50°C for 20 h. After cooling, the mixture was stirred with 30 mL of ethyl acetate and 10 mL of 20% sodium bisulfite solution. The organic phase was washed with 10 mL water and 10 mL of sat. NaCl solution, dried (Na2S0 4 ) and evaporated.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description

























Claims













Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17760562.3A EP3422855B1 (en) | 2016-02-29 | 2017-02-28 | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| JP2018545311A JP2019507156A (en) | 2016-02-29 | 2017-02-28 | Process for producing 4-alkoxy-3-hydroxypicolinic acid |
| CA3016013A CA3016013A1 (en) | 2016-02-29 | 2017-02-28 | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
| CN201780025820.8A CN109068653A (en) | 2016-02-29 | 2017-02-28 | The method for preparing 4- alkoxy -3- hydroxy-picolinic acid |
| KR1020187027518A KR20180116371A (en) | 2016-02-29 | 2017-02-28 | Process for producing 4-alkoxy-3-hydroxypicolic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662301107P | 2016-02-29 | 2016-02-29 | |
| US62/301107 | 2016-02-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017151530A1 true WO2017151530A1 (en) | 2017-09-08 |
Family
ID=59679408
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2017/019801 Ceased WO2017151530A1 (en) | 2016-02-29 | 2017-02-28 | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9951018B2 (en) |
| EP (1) | EP3422855B1 (en) |
| JP (1) | JP2019507156A (en) |
| KR (1) | KR20180116371A (en) |
| CN (1) | CN109068653A (en) |
| AR (1) | AR107758A1 (en) |
| CA (1) | CA3016013A1 (en) |
| TW (1) | TW201733985A (en) |
| WO (1) | WO2017151530A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107935921A (en) * | 2017-12-26 | 2018-04-20 | 苏州开元民生科技股份有限公司 | A kind of preparation method of 2,3 dichloropyridine |
| EP3405034A4 (en) * | 2016-01-22 | 2019-10-23 | Dow Agrosciences LLC | PROCESS FOR THE PREPARATION OF 4-ALCOXY-3-HYDROXYPICOLINIC ACIDS |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UY40356A (en) | 2022-07-18 | 2024-01-31 | Pi Industries Ltd | A PROCESS FOR THE SYNTHESIS OF 4-ALKOXY-3-HYDROXYPICOLINIC ACIDS AND INTERMEDIATES THEREOF. |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3655679A (en) * | 1969-06-25 | 1972-04-11 | Merck & Co Inc | Certain aryl pyridine carboxylic acid derivatives |
| US6521622B1 (en) * | 1999-07-20 | 2003-02-18 | Dow Agrosciences Llc | Fungicidal heterocyclic aromatic amides and their compositions, methods of use and preparation |
| US20140187588A1 (en) * | 2012-12-31 | 2014-07-03 | Dow Agrosciences Llc | Macrocyclic picolinamides as fungicides |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE795518A (en) * | 1972-02-17 | 1973-08-16 | Ciba Geigy | NEW PYRIDINE COMPOUNDS AND THEIR PREPARATION METHODS |
| CN1237051C (en) * | 1999-08-20 | 2006-01-18 | 道农业科学公司 | Fungicidal heterocyclic aromatic amides and their compositions, methods of use and preparation |
| JP2005104838A (en) * | 2003-01-09 | 2005-04-21 | Tanabe Seiyaku Co Ltd | Condensed furan compounds |
| MX369327B (en) * | 2014-07-08 | 2019-11-05 | Dow Agrosciences Llc | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids. |
| ES2726927T3 (en) * | 2014-07-08 | 2019-10-10 | Dow Agrosciences Llc | Process for the preparation of 3-hydroxypicolinic acids |
-
2017
- 2017-02-24 TW TW106106505A patent/TW201733985A/en unknown
- 2017-02-27 US US15/443,446 patent/US9951018B2/en not_active Expired - Fee Related
- 2017-02-28 KR KR1020187027518A patent/KR20180116371A/en not_active Withdrawn
- 2017-02-28 JP JP2018545311A patent/JP2019507156A/en not_active Ceased
- 2017-02-28 WO PCT/US2017/019801 patent/WO2017151530A1/en not_active Ceased
- 2017-02-28 CA CA3016013A patent/CA3016013A1/en not_active Abandoned
- 2017-02-28 CN CN201780025820.8A patent/CN109068653A/en active Pending
- 2017-02-28 EP EP17760562.3A patent/EP3422855B1/en not_active Not-in-force
- 2017-03-01 AR ARP170100499A patent/AR107758A1/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3655679A (en) * | 1969-06-25 | 1972-04-11 | Merck & Co Inc | Certain aryl pyridine carboxylic acid derivatives |
| US6521622B1 (en) * | 1999-07-20 | 2003-02-18 | Dow Agrosciences Llc | Fungicidal heterocyclic aromatic amides and their compositions, methods of use and preparation |
| US20140187588A1 (en) * | 2012-12-31 | 2014-07-03 | Dow Agrosciences Llc | Macrocyclic picolinamides as fungicides |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3405034A4 (en) * | 2016-01-22 | 2019-10-23 | Dow Agrosciences LLC | PROCESS FOR THE PREPARATION OF 4-ALCOXY-3-HYDROXYPICOLINIC ACIDS |
| CN107935921A (en) * | 2017-12-26 | 2018-04-20 | 苏州开元民生科技股份有限公司 | A kind of preparation method of 2,3 dichloropyridine |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170247330A1 (en) | 2017-08-31 |
| TW201733985A (en) | 2017-10-01 |
| EP3422855B1 (en) | 2020-07-08 |
| JP2019507156A (en) | 2019-03-14 |
| US9951018B2 (en) | 2018-04-24 |
| EP3422855A1 (en) | 2019-01-09 |
| CA3016013A1 (en) | 2017-09-08 |
| EP3422855A4 (en) | 2019-08-14 |
| CN109068653A (en) | 2018-12-21 |
| AR107758A1 (en) | 2018-05-30 |
| KR20180116371A (en) | 2018-10-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2954167C (en) | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids | |
| CN114085234B (en) | Process for preparing 1, 3-benzodioxole heterocyclic compounds | |
| EP0300614B1 (en) | Process for the preparation of substituted indolinone derivatives | |
| EP3422855B1 (en) | Process for the preparation of 4-alkoxy-3-hydroxypicolinic acids | |
| US6150528A (en) | Method for producing 5-aminomethyl-2-chloropyridines | |
| EP2943468B1 (en) | A novel process for the preparation of n-(4-nitro-2-sulfamoyl-phenyl)-malonamic acid methyl ester and n-(4-amino-2-sulfamoyl-phenyl)-malonamic acid methyl ester | |
| BR102017003941A2 (en) | METHOD FOR THE PREPARATION OF 4-ALCOXY-3-HYDROXYPICOLINIC ACIDS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 3016013 Country of ref document: CA Ref document number: 2018545311 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20187027518 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017760562 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017760562 Country of ref document: EP Effective date: 20181001 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17760562 Country of ref document: EP Kind code of ref document: A1 |