WO2017164172A1 - クルクミンホウ素錯体及びこれを含有する医薬 - Google Patents
クルクミンホウ素錯体及びこれを含有する医薬 Download PDFInfo
- Publication number
- WO2017164172A1 WO2017164172A1 PCT/JP2017/011232 JP2017011232W WO2017164172A1 WO 2017164172 A1 WO2017164172 A1 WO 2017164172A1 JP 2017011232 W JP2017011232 W JP 2017011232W WO 2017164172 A1 WO2017164172 A1 WO 2017164172A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- salt
- atom
- boron complex
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images














Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to a medicament for preventing or treating diseases involving various pathogenic amyloids.
- proteins fold to form a specific native structure and take on vital functions.
- misfolding may cause aggregation (amyloidation) into fibers rich in ⁇ -sheet structures.
- Aggregates oligomers, protofibrils, fibers
- amyloid diseases include Alzheimer's disease amyloid ⁇ , tau protein, Parkinson's disease ⁇ -synuclein, diabetic amylin, systemic amyloid-cis transthyretin, and Huntington's disease huntingtin.
- amyloid ⁇ (abbreviated as A ⁇ ) which is the causative amyloid of Alzheimer's disease
- a ⁇ amyloid ⁇
- a ⁇ degrading enzyme promoters an inhibitor of an enzyme that produces A ⁇ from a precursor protein, A ⁇ degrading enzyme promoters, immunotherapy, A ⁇ aggregation inhibitors and the like are known.
- a ⁇ a small amount of Met oxygenated product of A ⁇ peptide (oxygenated product of sulfur atom of Met residue of A ⁇ peptide oxygenated (O)) remains in the living body, and the Met oxygenated product.
- O oxygenated product of Met residue of A ⁇ peptide oxygenated
- the object of the present invention is a compound useful as an amyloid oxygenation catalyst applicable to in vivo, selective to amyloid, applicable to not only A ⁇ peptide but also other amyloid, and amyloid using the same
- the purpose is to provide drugs for preventing and treating related diseases.
- X 3 is a bromine atom, an iodine atom, or a selenium atom.
- the curcumin boron complex represented by the following general formula (1) has strong oxygenation activity against A ⁇ peptide and other amyloids by irradiation with light on the long wavelength side, and particularly strong oxygenation activity against highly toxic A ⁇ peptide aggregates. Because it has no oxygenation activity for peptides other than amyloid and is highly stable against water and light irradiation, it should be useful as an in vivo catalyst for producing amyloid oxygenates without aggregation. The present invention has been completed.
- the present invention provides the following [1] to [11].
- X 1 and X 2 are the same or different and each represents a halogenoalkyl group or a halogen atom
- X 3 represents a bromine atom, an iodine atom or a selenium atom
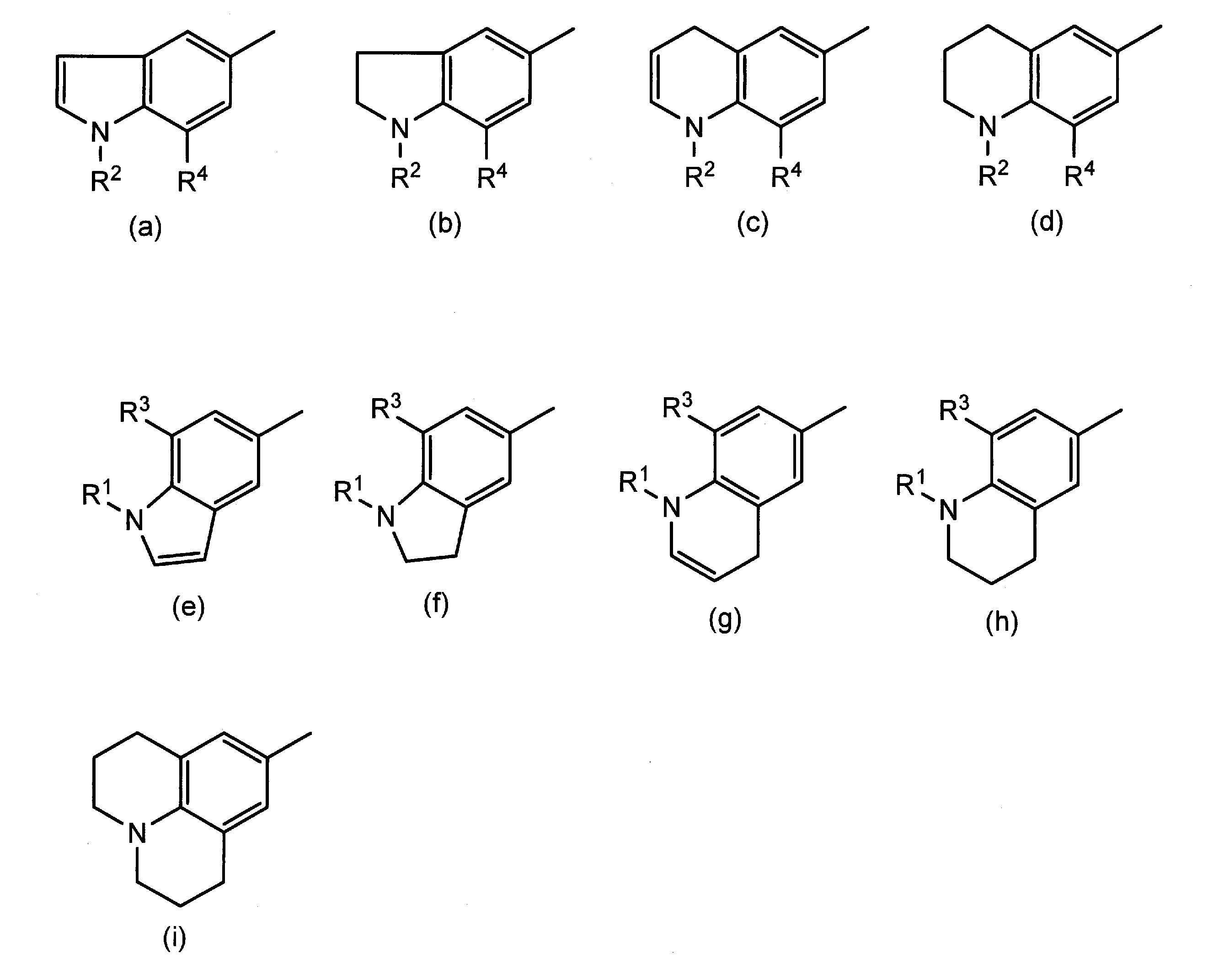
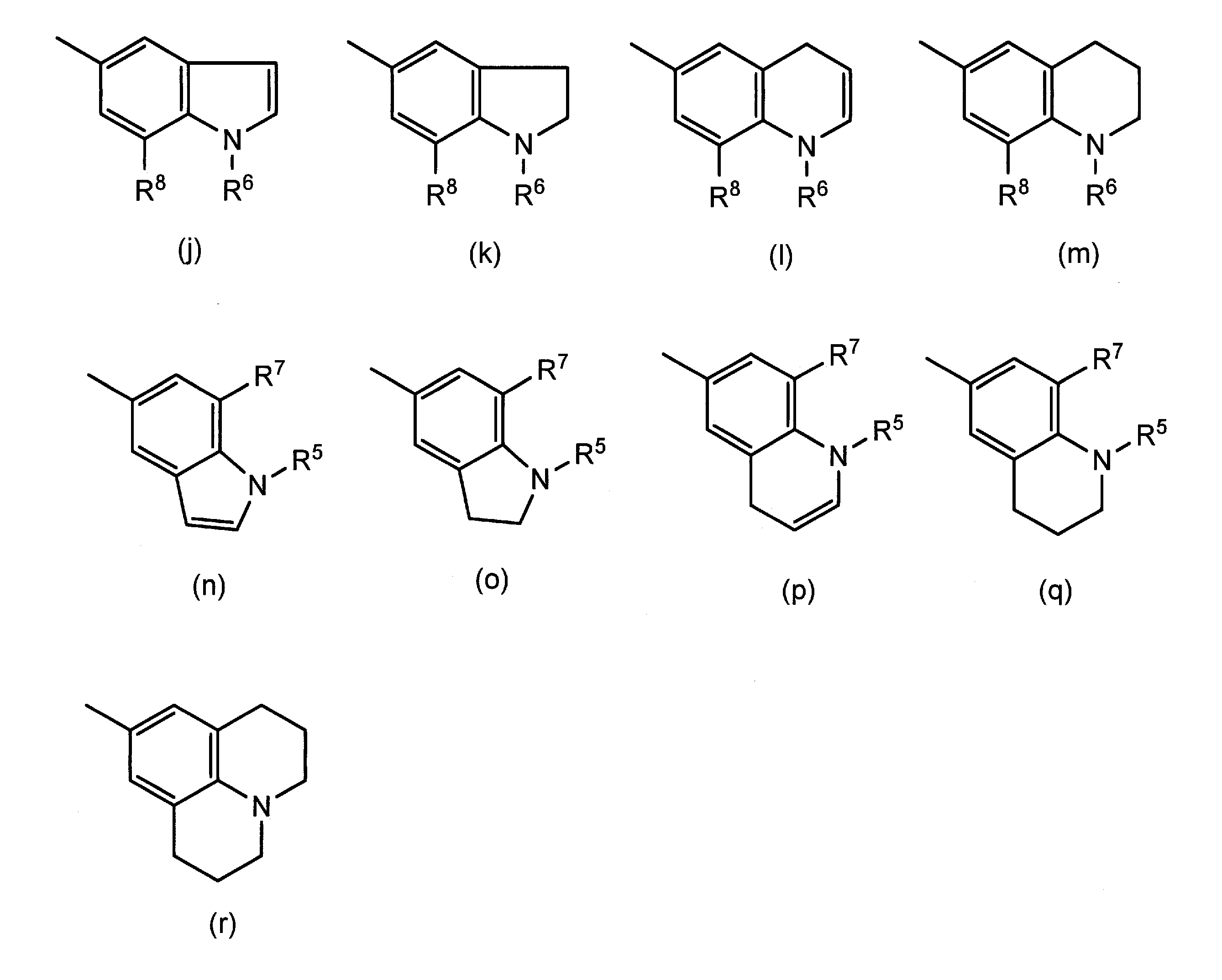
- R 1 and R 2 are the same or different and each represents a hydrogen atom or an optionally substituted alkyl group
- R 3 and R 4 are the same or different and each represents a hydrogen atom, a halogen atom, an alkoxy group or an alkyl group which may have a substituent, or R 1 and R 3 , or R 2 and R 4 together
- R 5 and R 6 are the same or different and each represents a hydrogen atom or an optionally substituted alkyl group
- R 7 and R 8 are the same or different and each represents a hydrogen atom, a halogen atom, an alkoxy group or an alkyl group which may have a substituent, or R 5 and R
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 and the alkyl group which may have a substituent represented by R 8 is a carboxy group, a sulfonic acid group or a hydroxy group.
- the alkylene or alkenylene group formed by combining R 1 and R 3 , R 2 and R 4 , R 5 and R 7 , or R 6 and R 8 together has 2 or 3 carbon atoms
- a medicament comprising the curcumin boron complex or a salt thereof according to any one of [1] to [5] as an active ingredient.
- the medicament according to [6] which is a preventive or therapeutic agent for a disease associated with pathogenic amyloid.
- a pharmaceutical composition comprising the curcumin boron complex or a salt thereof according to any one of [1] to [5] and a pharmaceutically acceptable carrier.
- curcumin boron complex or a salt thereof for the manufacture of a preventive or therapeutic agent for a disease involving pathogenic amyloid.
- a method for preventing or treating a disease associated with pathogenic amyloid which comprises administering an effective amount of the curcumin boron complex or a salt thereof according to any one of [1] to [5].
- the curcumin boron complex (1) of the present invention has high catalytic activity for oxygenating pathogenic amyloid such as A ⁇ peptide by irradiation with light on the long wavelength side, and suppresses amyloid aggregation by oxygenating amyloid in vivo. In addition, it has high oxygenation activity against aggregated amyloid, no oxygenation activity against peptides other than amyloid, and high stability under water and light irradiation conditions. It is useful as a prophylactic and therapeutic drug for diseases involving As used herein, oxygenation refers to a reaction that combines oxygen atoms, among other oxidations.
- a ⁇ peptide oxygenation activity is shown.
- the A ⁇ peptide oxygenation activity of the compound of the present invention is shown.
- the cytotoxicity of the compound of the present invention under light irradiation conditions is shown.
- action with respect to the cell by A ⁇ 1-42 selective oxygenation of this invention compound is shown.
- the oxygenation activity with respect to the benzoyl methionine of this invention compound is shown.
- the stability of the compound of the present invention under light irradiation conditions is shown.
- the A ⁇ peptide oxygenation activity of the compound of the present invention is shown.
- the oxygenation activity of A (beta) peptide and a non-target molecule by this invention compound is shown.
- the oxygenation activity of the A ⁇ peptide and the non-target molecule by the target compound is shown.
- X 1 and X 2 are the same or different and each represents a halogenoalkyl group or a halogen atom.
- a linear or branched halogeno C 1 -C 6 alkyl group is preferable, and a linear or branched halogeno C 1 -C 4 alkyl group is more preferable.
- Specific examples of the halogenoalkyl group include perfluoro C 1 -C 6 alkyl groups such as a trifluoromethyl group and a pentafluoroethyl group, and a perfluoro C 1 -C 4 alkyl group is more preferable.
- the halogen atom include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- X 1 and X 2 may both be a halogen atom or a halogenoalkyl group, but X 1 is a halogenoalkyl group and X 2 is a halogen atom from the viewpoint of selectivity of oxygenation activity with respect to amyloid.
- X 1 is a perfluoro C 1 -C 6 alkyl group
- X 2 is more preferably a fluorine atom
- X 1 is a trifluoromethyl group or a pentafluoroethyl group
- X 2 is fluorine. More preferably it is an atom.
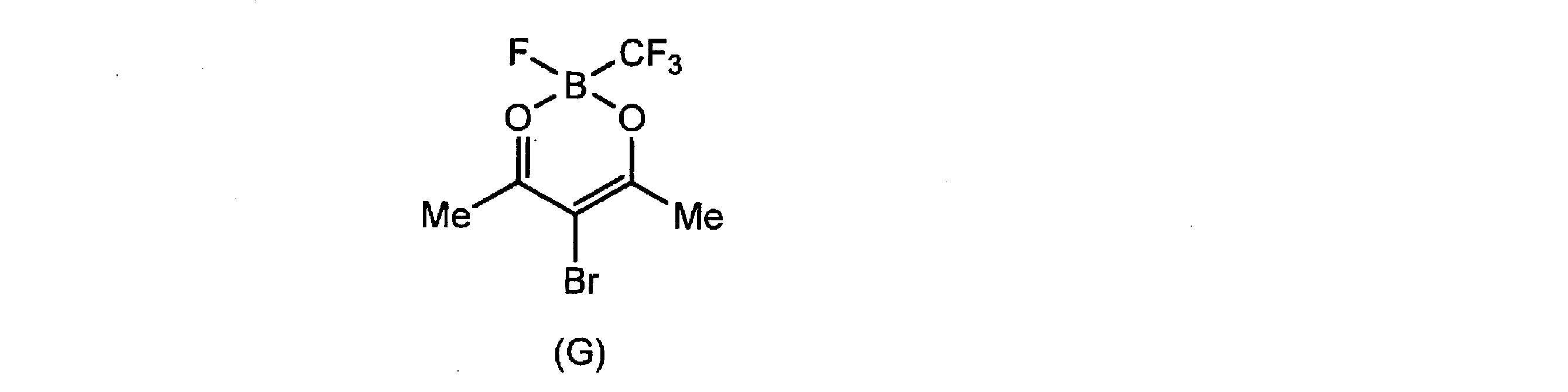
- X 3 represents a bromine atom, an iodine atom or a selenium atom. When X 3 is these heavy atoms, strong oxygenation activity for amyloid is obtained.
- R 1 and R 2 are the same or different and each represents a hydrogen atom or an alkyl group which may have a substituent.
- R 3 and R 4 are the same or different and each represents a hydrogen atom, a halogen atom, an alkoxy group or an alkyl group which may have a substituent, or R 1 and R 3 , or R 2 and R 4 together
- the alkylene group or alkenylene group which may have a substituent may be formed.
- R 5 and R 6 are the same or different and each represents a hydrogen atom or an optionally substituted alkyl group.
- R 7 and R 8 are the same or different and each represents a hydrogen atom, a halogen atom, an alkoxy group or an alkyl group which may have a substituent, or R 5 and R 7 , or R 6 and R 8 together.
- the alkylene group or alkenylene group which may have a substituent may be formed.
- alkyl group for R 1 to R 8 a linear or branched C 1 -C 6 alkyl group is preferable, and a linear or branched C 1 -C 4 alkyl group is more preferable.
- the alkyl group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group and the like.
- 1 to 3 groups selected from a carboxy group, a sulfonic acid group, a hydroxy group, an amino group, —CO—, —CONH—, and a triazole group are preferable.
- a carboxy group, a sulfonic acid group, a hydroxy group, and an amino group are preferable in terms of enhancing water solubility.
- Examples of preferred substituents include — (CH 2 ) 1 — (Y) 0 — (CH 2 ) p —Z.
- Y represents —CO—, —CONH— or a triazole ring.
- O represents a number of 0 or 1.
- Examples of the triazole ring include a 1,2,3-triazole-1,4-diyl group and a 1,2,4-triazole-1,3-diyl group.
- l and p are the same or different and each represents an integer of 1 to 6.
- l is preferably an integer of 1 to 6, more preferably an integer of 1 to 4.
- p is preferably an integer of 1 to 6, and more preferably an integer of 1 to 4.
- Z represents a hydroxy group, an amino group, a carboxyl group (—COOH) or a sulfonic acid group (—SO 3 H).
- the alkoxy group represented by R 3 , R 4 , R 7 and R 8 is preferably a linear or branched C 1 -C 6 alkoxy group, more preferably a C 1 -C 4 alkoxy group. Specific examples include a methoxy group, an ethoxy group, and a propyloxy group.
- the halogen atom include a chlorine atom, a bromine atom, an iodine atom, and a fluorine atom.
- the alkylene group formed by combining R 1 and R 3 , R 2 and R 4 , R 5 and R 7 , or R 6 and R 8 together is preferably a C 2 to C 4 alkylene group, an ethylene group, trimethylene Group and tetramethylene group.
- alkenylene groups include vinylene groups and propenylene groups.
- the ring structure formed by these groups together includes the following.
- R 1 to R 8 represent groups other than those forming an alkylene group or an alkenylene group.
- M and n represent an integer of 1 to 3, preferably 1 or 2, and more preferably 1.
- the present invention includes optical isomers, and includes both optical isomers and racemates.
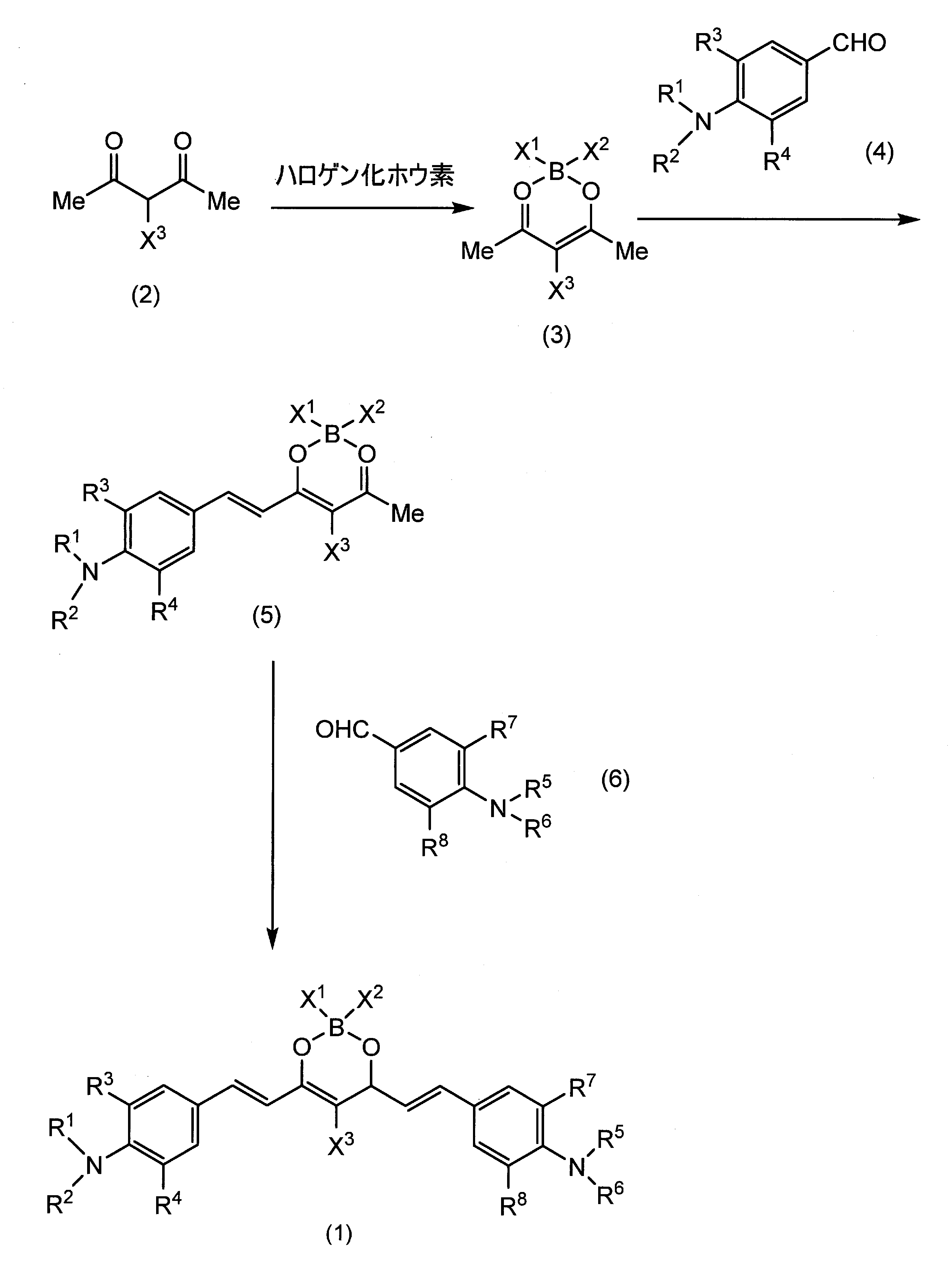
- the compound (1) of the present invention can be produced, for example, according to the following reaction formula.
- halogenoacetylacetone (2) is reacted with a halogenated boric acid compound to obtain compound (3), and benzaldehyde compound (4) is condensed with this to obtain compound (5), and then compound (5) and compound (6 ) Is further condensed to give compound (1).
- Examples of the halogenated boric acid to be reacted with the halogenoacetylacetone (2) include BF 3 .Et 2 O, CF 3 BF 3 K, CF 3 CF 2 BF 3 K, and the like.
- the reaction between the halogenoacetylacetone (2) and the halogenated boric acid can be carried out at 0 ° C. to room temperature in a polar solvent such as acetonitrile in the presence of an acid such as trimethylsilyl trifluoromethanesulfonate.
- the condensation reaction between the compound (3) and the benzaldehyde compound (4) is an aldol condensation reaction.
- the reaction can be carried out in the presence of a boric acid ester such as trimethoxyborane, triethoxyborane and tributoxyborane, and a base such as amine.
- the reaction can be carried out in an inert solvent such as toluene at a temperature of room temperature to about 80 ° C.
- the present compound (1) is obtained by condensing the compound (6) with the obtained compound (5).
- the condensation reaction of compound (5) and compound (6) is also an aldol condensation reaction.
- the reaction can be carried out in the same manner as the condensation reaction of the compound (3) and the benzaldehyde compound (4).
- R 5 or R 6 represents — (CH 2 ) 1 — (Y) 2 O— (CH 2 ) p —Z
- the compound (6) can be produced, for example, according to the following reaction formula.
- Hal represents a halogen atom
- Z 1 represents an alkoxycarbonyl group or an alkylsulfonyl group
- Z, Y, l, o, and p are the same as described above.
- Compound (6) is obtained by reacting tetrahydroquinoline (7) with compound (8), reacting the resulting compound (9) with a formylating agent such as dimethylformamide and hydrolyzing it.
- a formylating agent such as dimethylformamide
- the reaction between tetrahydroquinoline (7) and compound (8) may be carried out at 80 to 100 ° C. in acetonitrile in the presence of a base such as potassium iodide or potassium carbonate.
- the formylation reaction of compound (9) may be performed by reacting dimethylformamide or the like at room temperature in the presence of an acid catalyst such as phosphorus oxychloride.
- the hydrolysis of the Z 1 portion may be performed by reacting a base such as sodium hydroxide or potassium hydroxide.
- Y in the above is a heterocyclic ring such as triazole, it can also be produced according to the following reaction formula.
- the condensation reaction between the compound (5) and the compound (10) is an aldol condensation reaction, and can be performed in the same manner as the reaction between the compound (3) and the aldehyde compound (4).
- the reaction of the compound (11) and the compound (12) is a 1,3-dipolar addition reaction of alkyne and azide, and in the presence of a copper catalyst such as copper sulfate in a polar solvent such as dimethylformamide and water at room temperature. Proceed easily.
- the resulting compound (1) of the present invention can be isolated and purified from the reaction mixture by usual means such as washing, crystallization, recrystallization, chromatography and the like.
- the maximum absorption wavelength of the compound (1) of the present invention is shifted to the longer wavelength side compared to Thioflavin T.
- native A ⁇ decreases with time, and oxygenated A ⁇ with 1 to 4 oxygen atoms added thereto. Increased.
- the oxygenation efficiency was significantly higher than that of thioflavin T.
- the oxygenation reaction by the compound (1) of the present invention is extremely weak against non-amyloid peptides such as angiotensin IV and methionine enkephalin and is selective for A ⁇ .
- the compound (1) of the present invention had higher oxygenation activity against the highly toxic aggregated A ⁇ peptide than the oxygenation activity against the monomeric A ⁇ peptide. Moreover, this invention compound (1) is excellent also in the stability with respect to water and light irradiation conditions. Accordingly, the compound (1) of the present invention acts as a catalyst for selectively oxygenating pathogenic amyloids such as A ⁇ peptide, amylin, transthyretin, ⁇ -synuclein, tau protein, huntingtin and the like. These pathogenic amyloids do not form ⁇ -sheet structure laminates when oxygenated, and therefore do not cause pathogenicity.
- pathogenic amyloids such as A ⁇ peptide, amylin, transthyretin, ⁇ -synuclein, tau protein, huntingtin and the like.
- the compound (1) of the present invention is useful as a prophylactic / therapeutic agent for diseases involving pathogenic amyloid such as Alzheimer's disease, Parkinson's disease, diabetes, Huntington's disease, and systemic amyloidosis in animals including humans.
- the compound (1) of the present invention catalyzes the oxygenation reaction of pathogenic amyloid. This oxygenation reaction proceeds when the compound (1) of the present invention is excited by light and oxygenates amyloid. Accordingly, when the compound (1) of the present invention is used as a medicine, it is preferable to irradiate the patient with light after administering the compound (1) of the present invention. Moreover, since the wavelength of the light for making this invention compound (1) into an excited state is 660 nm or more and a long wavelength, it has the characteristic that it is easy to permeate
- the pharmaceutical composition containing the compound (1) of the present invention can be prepared by selecting an appropriate preparation according to the administration method and preparing various preparations using a pharmaceutically acceptable carrier.
- a pharmaceutically acceptable carrier for example, tablets, powders, granules, capsules, liquids, syrups, elixirs, oily or aqueous suspensions and the like are used as oral preparations. It can be illustrated as
- stabilizers As injections, stabilizers, preservatives, and solubilizing aids may be used in the preparation. After storing a solution that may contain these adjuvants in a container, it may be prepared as a solid preparation by lyophilization or the like. It is good also as a formulation. Further, a single dose may be stored in one container, and multiple doses may be stored in one container.
- liquid preparations suspensions, emulsions, ointments, gels, creams, lotions, sprays, patches and the like can be exemplified as external preparations.
- the solid preparation contains a pharmaceutically acceptable additive together with the compound (1) of the present invention.
- a pharmaceutically acceptable additive for example, fillers, extenders, binders, disintegrants, dissolution promoters, wetting agents, lubricantskinds and the like can be selected and mixed as necessary to prepare a formulation.
- liquid preparations include solutions, suspensions, emulsions and the like, but additives may include suspending agents, emulsifiers and the like.
- the dosage is preferably 1 mg to 1 g, preferably 1 mg to 300 mg per day for an adult.
- Trimethylsilyl trifluoromethanesulfonate (TMS-OTf, 1) was added to an acetonitrile suspension of potassium trifluoro (pentafluoroethyl) borate (CF 3 CF 2 BF 3 K, 0.86 g, 3.8 mmol) cooled to 0 ° C. with ice. 0.03 mL, 5.7 mmol) was added dropwise and the mixture was stirred at 0 ° C. for 30 minutes. To this reaction solution, 3-bromopentane-2,4-dione (0.34 g, 1.9 mmol) was added, and the mixture was warmed to room temperature and stirred overnight.
- TMS-OTf Trimethylsilyl trifluoromethanesulfonate
- Compound G was synthesized in the same manner as Compound C using 3-bromopentane-2,4-dione and potassium trifluoro (trifluoromethyl) borate (CF 3 BF 3 K). Pale brown oily substance (0.85 g, 62%).
- Compound H was synthesized in the same manner as Compound C using 3-iodopentane-2,4-dione and potassium trifluoro (trifluoromethyl) borate (CF 3 BF 3 K). Pale brown oily substance (0.34 g, 51%).
- the intermediate compound was prepared in the same manner as in Synthesis Example 3 using potassium trifluoro (heptafluoropropyl) borate (276.0 mg, 1.0 mmol) instead of potassium trifluoro (pentafluoroethyl) borate. Obtained as a colored oil (79.0 mg, 21%). Using this intermediate compound (37.7 mg, 0.1 mmol) instead of compound C, compound R was obtained as a dark purple solid (20.7 mg, 37%) in the same manner as in Synthesis Example 4.
- Test example 1 In the present invention, in vitro oxygenation experiments were conducted using A ⁇ 1-42 as a substrate to confirm that the compounds actually have A ⁇ oxygenation activity.
- a test compound (each 1 ⁇ M) was added to a phosphate buffer solution (pH 7.4 / cell culture medium (containing 0.1% horse serum) (1: 3)) containing A ⁇ 1-42 (10 ⁇ M), and LED irradiation was performed.
- the reaction was followed with a mass spectrometer (MALDI-TOF MS) after incubation at 37 ° C. (wavelength: 660 nm, 730 nm), and native A ⁇ 1-42 and Na + adducts were mainly observed before light irradiation.
- MALDI-TOF MS mass spectrometer
- Test example 2 The culture medium of rat adrenal medulla-derived pheochromocytoma PC12 cells (purchased from RIKEN) seeded in a poly-D-lysine-coated 96-well plate was replaced with 75 ⁇ L of Dulbecco's modified Eagle medium containing 0.1% horse serum, After incubation at 37 ° C. for 1 day in a 5% CO 2 atmosphere, 25 ⁇ L of phosphate buffered saline (pH 7.4) containing the compound was added thereto. Thereafter, this mixed solution was incubated at 37 ° C. for 5 minutes under LED irradiation (wavelength: 660 nm, 10 mW) (light not irradiated in dark).
- LED irradiation wavelength: 660 nm, 10 mW
- the cell culture plate containing the reaction solution was incubated at 37 ° C. for 48 hours in a 5% CO 2 atmosphere.
- a live cell count reagent SF containing WST-8 (10 ⁇ L: purchased from Nacalai) was added, incubated at 37 ° C. for 3 hours in a 5% CO 2 atmosphere, and then the absorbance at 450 nm (reference wavelength: 655 nm) was determined. Cell viability was measured.
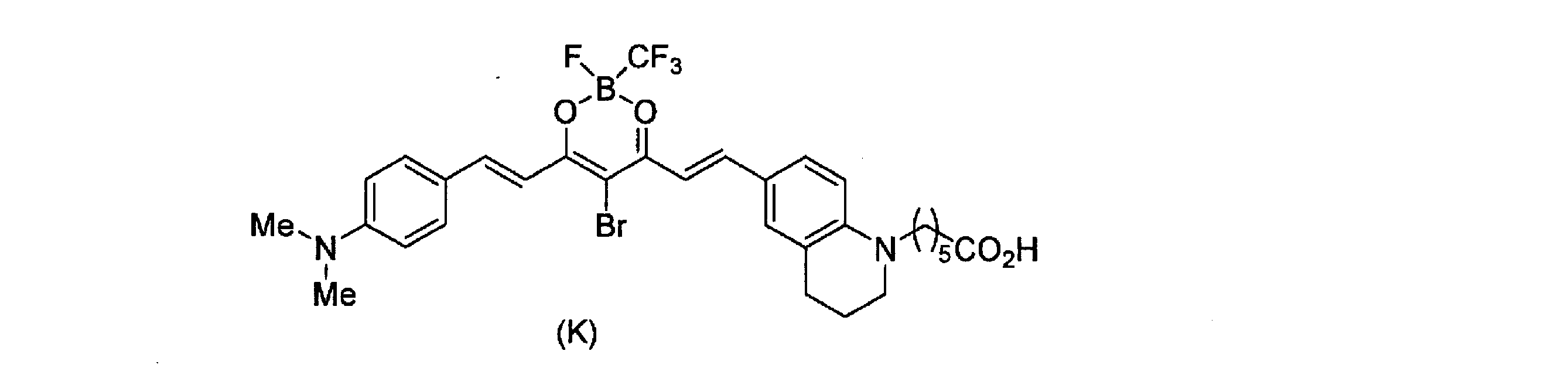
- the compound E had the lowest toxicity under the light irradiation conditions, and the compound J, the compound K, and the compound L increased in this order.
- Test example 3 Phosphate buffered saline (pH 7.4) containing A ⁇ (40 ⁇ M) was incubated at 37 ° C. for 3 hours. Compound E was then added (4 or 12 ⁇ M). The culture medium of rat adrenal medulla-derived pheochromocytoma PC12 cells (purchased from RIKEN) seeded in a poly-D-lysine-coated 96-well plate was replaced with 75 ⁇ L of Dulbecco's modified Eagle medium containing 0.1% horse serum, After incubation at 37 ° C.
- a live cell count reagent SF containing WST-8 (10 ⁇ L: purchased from Nacalai) was added, incubated at 37 ° C. for 3 hours in a 5% CO 2 atmosphere, and then the absorbance at 450 nm (reference wavelength: 655 nm) was determined. Cell viability was measured. As a result, as shown in FIG. 4, it was decided whether or not the toxicity could be reduced by oxygenating A ⁇ 1-42 in the presence of cells. Rat adrenal medulla-derived neural model cell PC12 was used as the cell, and Compound E was used as the catalyst. The cell viability was obtained by measuring the absorbance at 450 nm by WST-8 with a plate reader.
- Test example 4 Compound K (2 ⁇ M) was added to glycerol / water (0: 100, 10:90, 30:70, 50:50) in which benzoylmethionine (2000 ⁇ M) was dissolved, and after LED irradiation (wavelength 730 nm) at room temperature, LC / The reaction was followed by MS (ESI-Q).
- the benzoylmethionine oxygenate ratio (%) increased as the concentration of highly viscous glycerol increased. By being suppressed, it was suggested that oxygenation activity was expressed through the production of singlet oxygen.
- Test Example 5 The neutral phosphate buffer containing the catalyst was incubated under non-light irradiation or light irradiation conditions (660 nm), and then ethanol containing benzyl alcohol (as an internal standard) was added and analyzed by HPLC. Under the non-irradiation conditions, both catalysts were stable after 1 day. Further, in the light irradiation condition, as shown in FIG. 6, the stability is lowest when the boron center is BF 2 (compound L), and in the order of BFCF 3 (compound K) ⁇ BFCF 2 CF 3 (compound J). Stability increased. In addition, the compounds having julolidine at the donor site (Compound E, Compound I) showed increased stability compared to the corresponding dimethylanilines (Compound K, Compound J).
- Test Example 6 Oxygenation reaction with mouse brain extract Brains isolated from 8-month-old App knock-in (App NL-GF / NL-GF ) mice (Alzheimer model mice), cerebral cortex, hippocampus, and remaining brain Separated into tissues. The cerebral cortex and hippocampus were combined and homogenized in phosphate buffered saline, and the suspension was stored at -80 ° C. Compound E or riboflavin was added to the lysate suspension to a final concentration of 100 ⁇ M or 50 ⁇ M, respectively, and irradiated with light (at a wavelength of 780 nm) or stored in the dark at room temperature.
- Test Example 7 Subcutaneous Oxygenation
- a ⁇ 1-42 (20 ⁇ M, preaggregated for 3 hours
- Compound E or Compound 3 9- (6-Bromo-3-methyl-1,3-benzotriazol-3-ium-2-yl) -julolidine
- a microtube containing a phosphate buffer containing] was embedded in the skin of the back of a wild mouse, and irradiated with light at a wavelength of 780 nm from the outside of the mouse for 30 minutes using an LED lamp (FIG. 10).
- Test Example 8 Conversion of Furfuryl Alcohol or N-Benzoyl-Met under Conditions of Changing Glycerol Concentration
- Furfuryl alcohol or N-benzoyl- in a glycerol-methanol mixed solvent (glycerol: 0, 10, 30, or 50%) Met (2 mM each) and compound E (2 ⁇ M each) were added, and the mixture was irradiated with light (wavelength 780 nm) at room temperature, and furfuryl alcohol or N-benzoyl-Met at 10 minutes, 20 minutes, and 30 minutes after the start.
- the concentration of was quantified with a UV absorbance meter using LC / MS. The results are shown in FIGS.
- Test Example 9 A ⁇ Selectivity of Oxygenation A ⁇ peptide before aggregation (prepared by incubating at 37 ° C. for 1 hour), angiotensin-IV (AT4), Met-enkephalin (ME), or somatostatin (Sst) (each 20 ⁇ M)
- the phosphate buffer (pH 7.4) contained was irradiated with light at 500 nm in the presence of 780 nm (7 mW) or riboflavin (5 ⁇ M) in the presence of Compound E (5 ⁇ M) at 37 ° C. for 30 minutes.
- Compound E 25 mol%) oxygenated 44% of the pre-aggregated A ⁇ peptide (FIG. 14).
- Test Example 10 Comparison of oxygenation activity and toxicity of each photocatalyst for A ⁇ 1-42 (1)
- Oxygenation A ⁇ 1-42 isopeptide concentration of 250 ⁇ M in 0.1% trifluoroacetic acid aqueous solution
- angiotensin IV 200 ⁇ M aqueous solution
- Met-enkephalin 200 ⁇ M aqueous solution
- somatostatin 200 ⁇ M aqueous solution
- phosphate buffer or phosphate buffered saline pH 7.4
- the A ⁇ 1-42 solution was incubated at 37 ° C. for 1-3 hours before being subjected to the oxygenation reaction.
- Dulbecco's modified Eagle's medium (DMEM, Life) containing 0.1% horse serum buffered with 25 mM 4- (2-hydroxyethyl) -1-piperazineethanesulfonic acid (HEPES) in a phosphate buffer containing 40 ⁇ M A ⁇ Technology).
- riboflavin (1 mM in 0.1% trifluoroacetic acid / acetonitrile (1: 1) aqueous solution), compound 3 (1 mM in acetonitrile), CRANAD-2 [(T-4)-[(1E, 6E) -1,7-bis [4- (dimethylamino) phenyl] -1,6-heptadiene-3,5-dionato- ⁇ O 3 , ⁇ O 5 ] difluoroboron] (200 ⁇ M dimethyl sulfoxide solution) or compound E, compound 5-10 (200 ⁇ M dimethyl sulfoxide solution) of I, Compound J, Compound L, Compound K, or Compound S was added.
- the final concentration of the compound was 2 ⁇ M and was used for the evaluation of A ⁇ 1-42 oxygenation. A final concentration of 5 ⁇ M was used for evaluation of peptide selectivity.
- the mixture was irradiated at room temperature with a LED (wavelength 500, 660, or 780 nm) at a distance of about 5-10 cm. A non-control group without light irradiation was also prepared.
- the reaction was subjected to monitor analysis using MALDI-TOF MS.
- compound E ((100 ⁇ M dimethyl sulfoxide solution)) was added to A ⁇ (preaggregated for 3 hours, 40 ⁇ M), phosphate buffered saline containing 25 ⁇ L of this solution (A ⁇ of Final concentration is 10 ⁇ M, final concentration of Compound E is 1 ⁇ M).
- the mixture was irradiated with LED (wavelength 780 nm) from a distance of about 5 cm at 37 ° C. for 5 minutes.
- the cells were incubated for 48 hours in a 5% carbon dioxide atmosphere at 37 ° C.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Emergency Medicine (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Endocrinology (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
一方、Aβに関しては、AβペプチドのMet酸素化体(AβペプチドのMet残基の硫黄原子が酸素化(O)された酸素化体)が生体内に少量残存すること、及び当該Met酸素化体はAβペプチドに比べて凝集性が低いことが報告されている(非特許文献1~3)。かかる観点から、本発明者は、式(a)で表されるAβ結合部位を有するフラビン光触媒を用いてAβペプチドを酸素化するとAβペプチド酸素化体が得られ、当該Aβペプチド酸素化体はAβの凝集を抑制することを報告した(非特許文献4)。
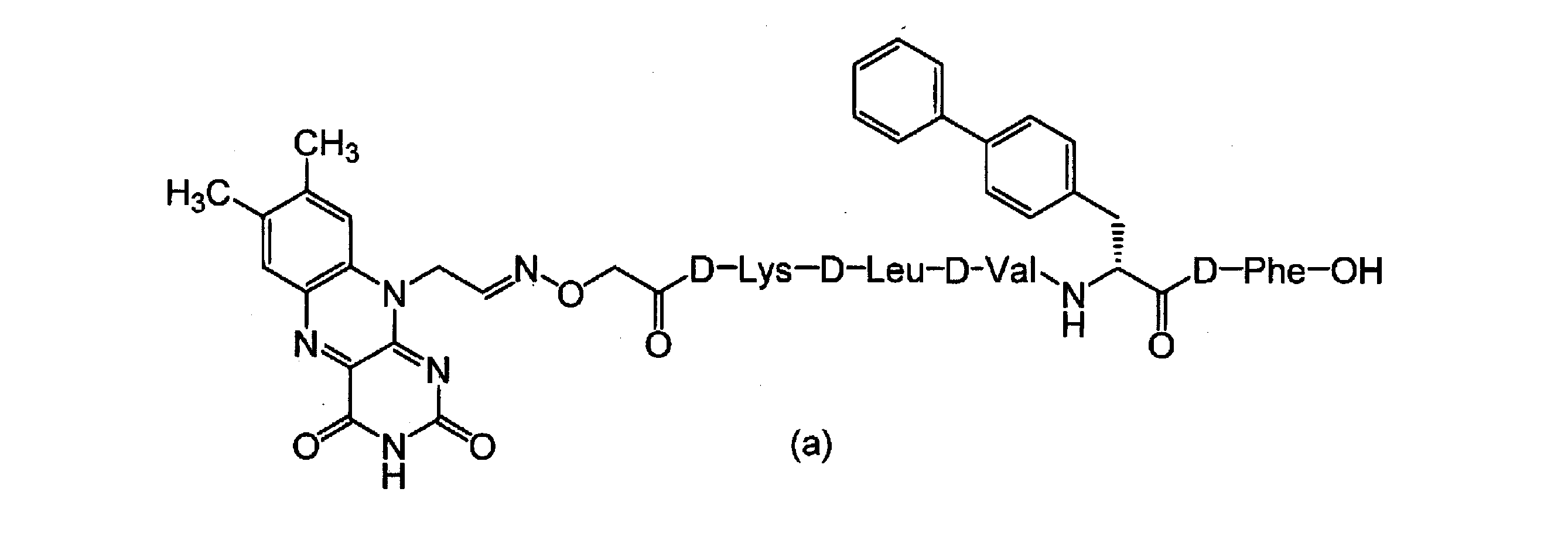
従って、本発明の課題は、生体内に適用可能で、アミロイドに選択的であって、Aβペプチドだけでなく他のアミロイドにも適用可能なアミロイド酸素化触媒として有用な化合物及びこれを用いたアミロイド関連疾患予防治療薬を提供することにある。

X3は、臭素原子、ヨウ素原子又はセレン原子を示し;
R1及びR2は、同一又は異なって、水素原子又は置換基を有していてもよいアルキル基を示し;
R3及びR4は、同一又は異なって、水素原子、ハロゲン原子、アルコキシ基又は置換基を有していてもよいアルキル基を示すか、R1とR3、又はR2とR4が一緒になって、置換基を有していてもよいアルキレン基又はアルケニレン基を形成してもよく;
R5及びR6は、同一又は異なって、水素原子又は置換基を有していてもよいアルキル基を示し;
R7及びR8は、同一又は異なって、水素原子、ハロゲン原子、アルコキシ基又は置換基を有していてもよいアルキル基を示すか、R5とR7、又はR6とR8が一緒になって、置換基を有していてもよいアルキレン基又はアルケニレン基を形成してもよく;
m及びnは、1~3の整数を示す)
で表されるクルクミンホウ素錯体又はその塩。
〔2〕X1がハロゲノアルキル基であり、X2がハロゲン原子である〔1〕記載のクルクミンホウ素錯体又はその塩。
〔3〕m及びnが1である〔1〕又は〔2〕記載のクルクミンホウ素錯体又はその塩。
〔4〕R1、R2、R3、R4、R5、R6、R7及びR8が示す置換基を有していてもよいアルキル基が、カルボキシ基、スルホン酸基、ヒドロキシ基、アミノ基、-CO-、-CONH-及びトリアゾール基から選ばれる1種以上の置換基を有していてもよいアルキル基である〔1〕~〔3〕のいずれかに記載のクルクミンホウ素錯体又はその塩。
〔5〕R1とR3、R2とR4、R5とR7、又はR6とR8が一緒になって形成されるアルキレン基又はアルケニレン基の炭素数が2又は3である〔1〕~〔4〕のいずれかに記載のクルクミンホウ素錯体又はその塩。
〔6〕〔1〕~〔5〕のいずれかに記載のクルクミンホウ素錯体又はその塩を有効成分とする医薬。
〔7〕病原性アミロイドが関連する疾患の予防又は治療薬である〔6〕記載の医薬。
〔8〕〔1〕~〔5〕のいずれかに記載のクルクミンホウ素錯体又はその塩及び薬学的に許容される担体を含有する医薬組成物。
〔9〕病原性アミロイドが関与する疾患の予防又は治療薬製造のための〔1〕~〔5〕のいずれかに記載のクルクミンホウ素錯体又はその塩の使用。
〔10〕病原性アミロイドが関与する疾患を予防又は治療するための、〔1〕~〔5〕のいずれかに記載のクルクミンホウ素錯体又はその塩。
〔11〕〔1〕~〔5〕のいずれかに記載のクルクミンホウ素錯体又はその塩の有効量を投与することを特徴とする病原性アミロイドが関連する疾患を予防又は治療する方法。
l及びpは、同一又は異なって、1~6の整数を示す。lは1~6の整数が好ましく、1~4の整数がより好ましい。pは1~6の整数が好ましく、1~4の整数がより好ましい。
Zは、ヒドロキシ基、アミノ基、カルボキシル基(-COOH)又はスルホン酸基(-SO3H)を示す。
R3、R4、R7及びR8で示されるアルコキシ基としては、直鎖又は分岐鎖のC1-C6アルコキシ基が好ましく、C1-C4アルコキシ基がさらに好ましい。具体的には、メトキシ基、エトキシ基、プロピルオキシ基等が挙げられる。また、ハロゲン原子としては、塩素原子、臭素原子、ヨウ素原子、フッ素原子が挙げられる。


また、Aβに本発明化合物(1)を添加し、生理的条件下で660nm以上の光を照射すると、経時的にネイティブAβが減少するとともに、酸素原子が1~4個付加した酸素化Aβが増加した。その酸素化効率は、チオフラビンTに比べて顕著に高かった。また、本発明化合物(1)による酸素化反応は、アンギオテンシンIVやメチオニンエンケファリン等の非アミロイド性のペプチドに対しては極めて弱く、Aβに選択的である。
さらに、本発明化合物(1)は、単量体のAβペプチドに対する酸素化活性よりも、毒性が強い凝集Aβペプチドに対する酸素化活性が高かった。また、本発明化合物(1)は、水に対する安定性及び光照射条件下での安定性も優れている。
従って、本発明化合物(1)は、Aβペプチド、アミリン、トランスサイレチン、αシヌクレイン、タウ蛋白質、ハンチンチン等の病原性アミロイドを選択的に酸素化する触媒として作用する。これらの病原性アミロイドは、酸素化されるとβ-シート構造の積層体を形成しなくなるため、病原性を生じなくなる。従って、本発明化合物(1)は、ヒトを含む動物のアルツハイマー病、パーキンソン病、糖尿病、ハンチントン病、全身性アミロイド-シス等の病原性アミロイドが関与する疾患の予防治療薬として有用である。
液体製剤としては溶液、懸濁液、乳液剤等を挙げることができるが添加剤として懸濁化剤、乳化剤等を含むこともある。
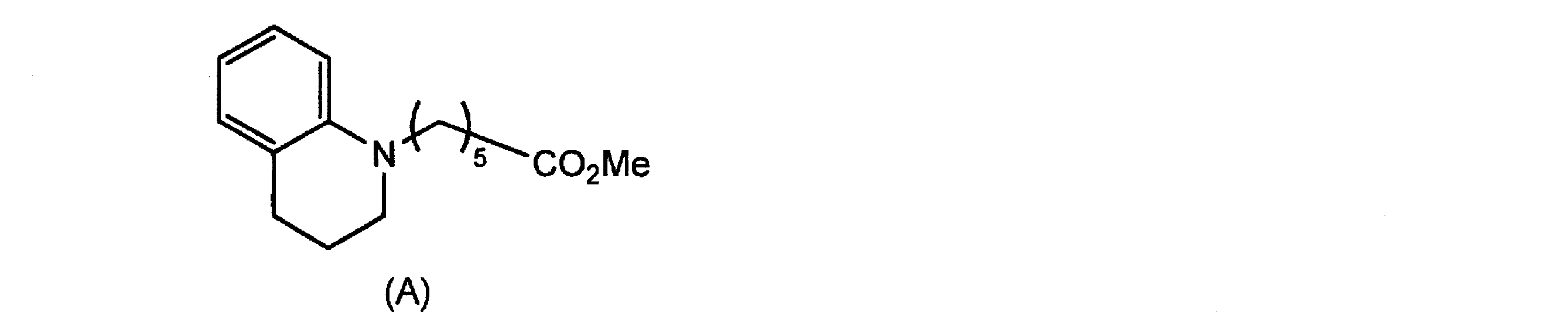
1H NMR (500 MHz, CDCl3) δ 7.02 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 7.5 Hz, 1H), 6.51-6.54 (m, 2H), 3.66 (s, 3H), 3.20-3.26 (m, 4H), 2.73 (t, J = 6.3 Hz, 2H), 2.31 (t, J = 7.5 Hz, 2H), 1.90-1.94 (m, 2H), 1.63-1.69 (m, 2H), 1.56-1.62 (m, 2H), 1.32-1.38 (m, 2H); LRMS (ESI): m/z calcd for [M+H]+: 262; found: 262.
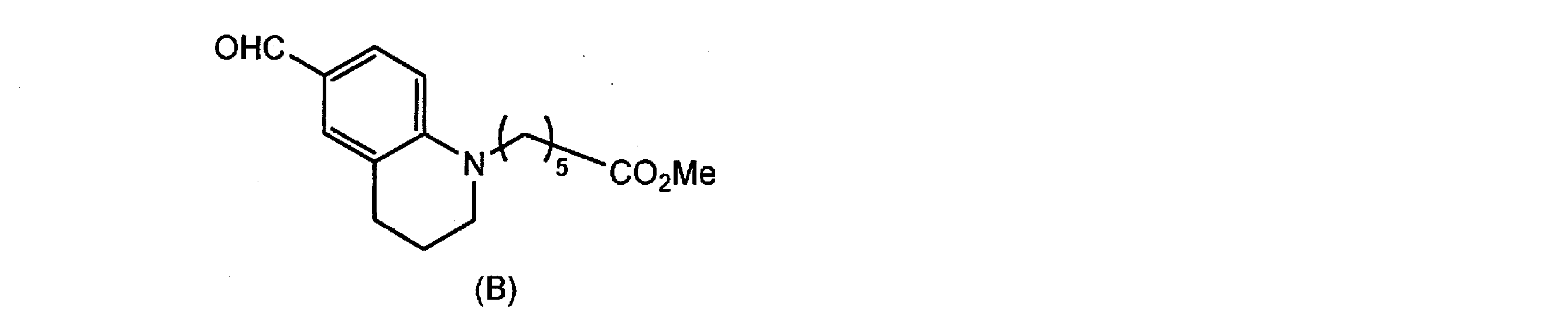
1H NMR (392 MHz, DMSO-d6) δ 12.03 (s, 1H), 9.56 (s, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.35 (s, 1H), 6.64 (d, J = 5.7 Hz, 1H), 3.49 - 3.18 (m, 4H), 2.83 - 2.56 (m,2H), 2.35 - 2.06 (m, 2H), 1.93 - 1.66 (m, 2H), 1.63 - 1.41 (m, 4H), 1.41 - 1.12(m, 2H); LRMS (ESI): m/z calcd for [M+H]+: 276; found: 276.


1H NMR (392 MHz, CDCl3) δ 8.03 (d, J = 14.4 Hz, 1H), 7.19 (s, 2H), 6.91 (d, J =14.4 Hz, 1H), 3.45 - 3.31 (m, 4H), 2.80 - 2.69 (m, 4H), 2.45 (s, 3H), 1.98 (dd, J = 11.2, 5.7 Hz, 4H); LRMS (ESI): m/z calcd for [M+H]+: 510; found: 510.

1H NMR (392 MHz, DMSO-d6) δ 7.66 (dd, J = 14.5, 9.1 Hz, 2H), 7.46 (d, J = 8.9 Hz, 1H), 7.41 (s, 1H), 7.30 (s, 2H), 7.01 (dd, J = 17.4, 14.6 Hz, 2H), 6.69 (d, J= 9.0 Hz, 1H), 3.45 - 3.30 (m, 8H), 2.75 - 2.67 (m, 6H), 2.22 (t, J = 7.3 Hz, 2H), 1.92 - 1.78 (m, 6H), 1.52 (dd, J = 15.1, 7.4 Hz, 4H), 1.32 (dd, J = 15.1, 8.1 Hz, 2H); LRMS (ESI): m/z calcd for [M+H]+: 767; found: 767.



LRMS (ESI): m/z calcd for [M+H]+: 717; found: 717.

LRMS (ESI): m/z calcd for [M+H]+: 715; found: 715.
LRMS (ESI): m/z calcd for [M+H]+: 665; found: 665.

LRMS (ESI): m/z calcd for [M+H]+: 615; found: 615.

LRMS (ESI): m/z calcd for [M+H]+: 713; found: 713.

LRMS (ESI): m/z calcd for [M+H]+: 620; found: 620.
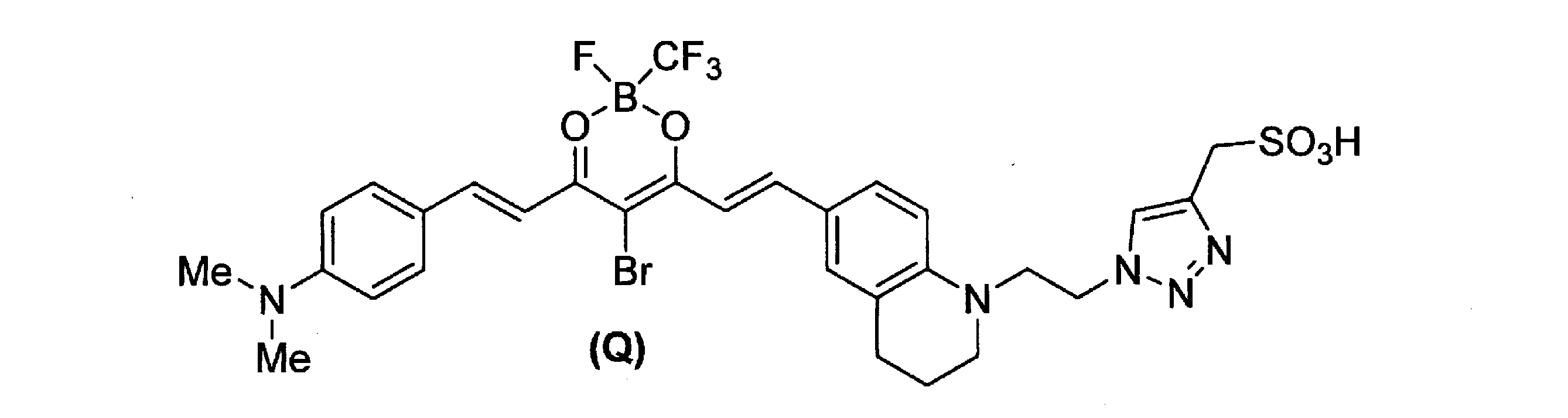
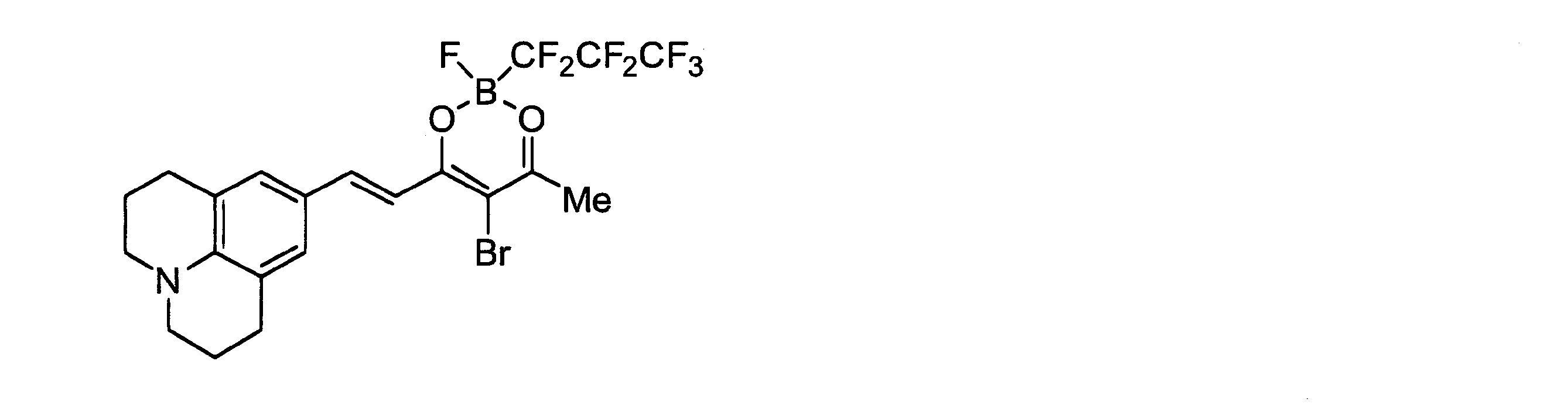
この中間体化合物(37.7mg、0.1mmol)を化合物Cの代わりに用いて合成例4と同様の方法により、化合物Rを濃紫色固体として得た(20.7mg、37%)。
1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 14.4 Hz, 1H), 7.18 (s, 2H), 6.90 (d, J = 14.4 Hz, 1H), 3.58 - 3.33 (m, 4H), 2.88 - 2.72 (m, 4H), 2.44 (s, 3H), 2.10 - 1.90 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 179.74, 174.17, 154.78, 149.42, 132.10, 121.82, 121.64, 108.49, 95.58, 50.55, 27.41, 24.68, 20.90; 19F NMR (369 MHz, CDCl3) δ -81.48(3F), -128.32(2F), -135.96 (2F), -152.51 (1F); 11B NMR (126 MHz, CDCl3) δ 1.07 (s); LRMS (ESI): m/z calcd for [M+H]+ 560.1, found 560.1.

1H NMR (500 MHz, DMSO-d6) δ 7.63 (d, J = 14.7 Hz, 1H), 7.60 (d, J = 14.7 Hz, 1H), 7.39 (d, J = 9.0 Hz, 1H), 7.34 (s, 1H), 7.22 (s, 2H), 6.95 (d, J = 14.7 Hz, 1H), 6.91 (d, J = 14.7 Hz, 1H), 6.62 (d, J = 9.0 Hz, 1H), 3.74 - 3.12 (m, 8H), 2.94 - 2.56 (m, 6H), 2.17 (t, J = 7.3 Hz, 2H), 2.03 - 1.69 (m, 6H), 1.63 - 1.39 (m, 4H), 1.39 - 1.19 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 174.41, 169.03, 168.39, 149.74, 149.05, 148.40, 147.74, 132.38, 130.69, 122.84, 121.79, 121.43, 121.12, 110.94, 110.78, 109.57, 94.61, 50.60, 49.84, 49.35, 33.61, 27.16, 26.75, 25.93, 25.85, 24.34, 20.90, 20.51; 19F NMR (369 MHz, DMSO-d6) δ -81.38 (3F), -128.29 (2F), -135.73 (2F), -154.02 (1F); 11B NMR (126 MHz, DMSO-d6) δ -1.22 (s); LRMS (ESI): m/z calcd for [M+H]+ 817.2, found 817.2.
本発明は化合物が実際にAβ酸素化活性を有することを確認するべくAβ1-42を基質としてin vitro酸素化実験を行った。Aβ1-42(10μM)を含むリン酸緩衝液(pH7.4/細胞培養液(0.1%馬血清を含む)(1:3)に、被検化合物(それぞれ1μM)を加え、LED照射下(波長660nm、730nm)、37℃でインキュベートした後、質量分析装置(MALDI-TOF MS)にて反応を追跡した。光照射前はネイティブAβ1-42およびNa+付加体が主に観測されるが、光照射を行うと経時的に酸素化体の存在を示唆するイオンピークが観測されるようになった(図1、図2)。
その結果、化合物E及び化合物Iは、660nm及び730nmの光照射の両条件においてAβの酸素化が進行した(図1)。また、化合物K及び化合物Mは、730nmの光照射条件においてAβの酸素化が進行した(図2)。
ポリD-リジンコート96穴プレートに播種したラット副腎髄質由来褐色細胞腫PC12細胞(理化学研究所から購入)の培養培地を、0.1%ウマ血清を含むダルベッコ変法イーグル培地75μLに置換し、5%CO2雰囲気下、37℃で1日間インキュベートした後、そこへ化合物を含むリン酸緩衝生理食塩水(pH7.4)25μLを加えた。その後、本混合液を、LED照射下(波長660nm,10mW)(darkでは光非照射)、37℃で5分間インキュベートした。さらに、その反応液を含む細胞培養プレートを5%CO2雰囲気下、37℃で48時間インキュベートした。最後に、WST-8を含む生細胞数測定試薬SF(10μL:ナカライから購入)を加え、5%CO2雰囲気下、37℃で3時間インキュベートした後、450nm(参照波長:655nm)における吸光度から細胞生存率を測定した。
その結果、図3に示すように、光照射条件において、化合物Eの毒性が最も低く、化合物J、化合物K、化合物Lの順に高くなった。この結果から、ホウ素中心がBF2の場合に毒性が最も高く、BFCF3>BFC2F5の順で低くなることが示唆された。また化合物Jと化合物Eの比較から、ジメチルアラニンをジュロリジンに変換することによって、毒性が軽減されることが示唆された。
また、図示していないが、化合物Kと化合物Mの比較から、ヨウ素の方が臭素の場合と比べて、毒性が軽減することが示唆されている。
Aβ(40μM)を含むリン酸緩衝生理食塩水(pH7.4)を37℃にて3時間インキュベートした。次に、化合物Eを加えた(4又は12μM)。ポリD-リジンコート96穴プレートに播種したラット副腎髄質由来褐色細胞腫PC12細胞(理化学研究所から購入)の培養培地を、0.1%ウマ血清を含むダルベッコ変法イーグル培地75μLに置換し、5%CO2雰囲気下、37℃で1日間インキュベートした後、そこへAβと化合物Eを含む上記リン酸緩衝生理食塩水(pH7.4)25μLを加えた。(最終ボリューム:100μL,最終Aβ濃度:10μM,最終化合物Eの濃度:1又は3μM)その後、本混合液を、LED照射下(波長660nm,10mW)(darkでは光非照射)、37℃で5分間インキュベートした。さらに、その反応液を含む細胞培養プレートを5%CO2雰囲気下、37℃で48時間インキュベートした。最後に、WST-8を含む生細胞数測定試薬SF(10μL:ナカライから購入)を加え、5%CO2雰囲気下、37℃で3時間インキュベートした後、450nm(参照波長:655nm)における吸光度から細胞生存率を測定した。
その結果、図4に示すように、細胞存在下Aβ1-42を酸素化することでその毒性を低減することができるか否か検証することとした。細胞にはラット副腎髄質由来の神経モデル細胞PC12を用い、触媒としては化合物Eを用いた。細胞生存率はWST-8による450nmの吸光をプレートリーダーにて測定することで得た。Aβ1-42と化合物Eを添加した場合、光を照射しない条件ではAβ1-42のみを添加した場合と同様に顕著な細胞死が見られた一方で、光照射を行った場合には細胞生存率が有意に向上した。この結果から、触媒がAβ1-42を選択的に酸素化することによって細胞毒性を低減していることが示唆された。
ベンゾイルメチオニン(2000μM)を溶解したグリセロール/水(0:100、10:90、30:70、50:50)に化合物K(2μM)を加え、室温でLED照射(波長730nm)した後、LC/MS(ESI-Q)にて反応を追跡した。
その結果、図5に示すように、グリセロール/水混合溶媒において、粘度の高いグリセロールの濃度増加にしたがって、ベンゾイルメチオニン酸素化体比(%)が増大したことから、化合物Kの分子内回転運動が抑えられることによって、一重項酸素の産生を経て、酸素化活性を発現していることが示唆された。
触媒を含む中性のリン酸緩衝液を、光非照射または光照射条件(660nm)にてインキュベートし、その後ベンジルアルコール(内部標準として)を含むエタノールを加え、HPLCにより分析した。
光非照射条件においては、いずれの触媒も1日後安定であった。
また、光照射条件においては、図6に示すように、ホウ素中心がBF2(化合物L)の場合最も安定性が低く、BFCF3(化合物K)<BFCF2CF3(化合物J)の順で安定性が増大した。また、ドナー部位にジュロリジンを有する化合物(化合物E、化合物I)は、相当するジメチルアニリン体(化合物K、化合物J)と比べて安定性が増大した。
8月齢のApp knock-in(AppNL-G-F/NL-G-F)マウス(アルツハイマー型モデルマウス)から摘出した脳を大脳皮質、海馬、および残りの脳組織に分離した。大脳皮質と海馬は合わせてリン酸緩衝液生理食塩水中でホモジナイズし、懸濁物を-80℃で保管した。化合物Eまたはリボフラビンを溶解物の懸濁液にそれぞれ最終濃度が100μM又は50μMになるように加え、光照射(780nmの波長で)を行うか、または室温で暗所に保管した。任意の時点で、任意の反応混合物の分割量をギ酸で最終濃度が70%になるように希釈し、濃縮し、6Mの尿素水溶液に再溶解した。内部標準(Aβ1-40、ペプチド研究所、日本)を加え、残りの混合物をZipTipにて処理し、ArcticタイプAβ1-38およびAβ1-42をMALDI-TOF MSで分析した。
Aβペプチドの変換はMALDI-TOF MS分析によってピーク強度が低下することにより裏付けられた。反応の30分後、60分後、150分後においてそれぞれArcticタイプAβ1-38では36%/53%/74%、ArcticタイプAβ1-42では43%/60%/83%のピーク強度が低下した(図7)。
暗室において行われた対照実験により、光照射はAβペプチドの低下にとって重要であることが確認された。
Aβ1-38とAβ1-42が化合物Eによってそれぞれ53%、60%の収率で変換されるとき(60分経過時)、非Aβの内因性物質であってその質量スペクトル値がAβの値に近いペプチドA、B、C(分子量がそれぞれ3770、4286、4968のペプチド)は、0分時と比較すると、変換されていなかった(図8)。
対照的に、リボフラビンを用いると、Aβ1-38とAβ1-42だけでなく、非AβのペプチドであるA、B、Cも相当程度消費される(図9)。これらの結果より、Aβの濃度が低く標的外の分子が大量に含まれている脳溶解物であっても、化合物EがAβペプチドを選択的に酸素化することを示す。
生体内におけるアミロイドタンパクの酸素化への化合物Eの有用性を証明するために、マウス皮下(末梢アミロイド疾患の治療モデル)での反応を検証した。Aβ1-42(20μM、3時間事前に凝集したもの)および化合物Eまたは化合物3〔9-(6-ブロモ-3-メチル-1,3-ベンゾトリアゾール-3-イウム-2-イル)-ジュロリジン〕を含むリン酸緩衝液を入れたマイクロチューブを野生マウスの背中の皮内に埋め込み、LEDランプを用いて780nmの波長でマウスの体外より30分間光照射を行った(図10)。対照実験として、同じ成分(Aβ1-42(20μM、3時間事前に凝集したもの)および化合物Eまたは化合物3)を含む別のマイクロチューブに対し、マウス体外にて直接光照射を行った。反応混合物をMALDI-TOF MSで分析後、ZipTip処理を行って解析を行った。酸素化比は、皮膚内部と皮膚外部の酸素化の比率「[マウス皮膚内部での酸素化比/マウス皮膚外部の酸素化比]×100」から計算した。
図11に示すように、化合物Eを用いた酸素化率の比(体外での酸素化率に対して生体内での酸素化率)は、65%であったが、それに対して、化合物3(10μM)では酸素化率の比は12%であった。化合物Eと化合物3による結果の違いは、皮膚を透過した後の光強度の違いである。これらの結果は、NIR光活性化可能な酸素化触媒である化合物Eが、生体内で、毒性のある凝集アミロイドタンパクを酸素化するのに適していることを示す。
グリセロール-メタノール混合溶媒(グリセロール:0、10、30、または50%)にフルフリルアルコールまたはN-ベンゾイル-Met(それぞれ2mM)および化合物E(2μM)を加え、混合物に室温で光照射(波長780nm)を行い、開始後10分、20分、30分の時点でのフルフリルアルコールまたはN-ベンゾイル-Metの濃度をLC/MSを用いたUV吸光計で定量化した。
結果を図12および13に示す。化合物Eが触媒するフルフリルアルコール(特異的に1O2と反応する)の酸素化反応速度が、溶媒の粘度が上がるにつれて増加した。化合物Eが触媒するN-ベンゾイル-Metの酸素化もまた、溶媒の粘度が高くなるにつれて加速した。これらの観察は、ドナーとアクセプターの間の単結合の自由回転を妨害することで化合物Eの活性化状態の寿命を延ばすと1O2の量子収率が高められることを示唆する。
凝集前のAβペプチド(37℃で1時間インキュベートして調製)、アンギオテンシン-IV(AT4)、Met-エンケファリン(ME)、又はソマトスタチン(Sst)(それぞれ20μM)を含有するリン酸緩衝液(pH7.4)を、化合物E(5μM)存在下で780nm(7mW)またはリボフラビン(5μM)存在下で500nmの光照射を37℃で30分間行った。
化合物E(25mol%)はプレ凝集したAβペプチドを44%酸素化した(図14)。Aβペプチド以外のペプチドでは、同じ反応条件であったが、5%未満の酸素化率であった。クロスβシート認識機能を有しない触媒であるリボフラビンは非選択的であり、酸素化Aβの収率(43%)は、標的外の基質を用いて得られた収率(アンギオテンシン-IV:35%、Met-エンケファリン:50%、ソマトスタチン:60%)とほぼ同一であった。この標的の選択性は、細胞の存在下、脳溶解物において、凝集Aβの選択的酸素化にとって重要である。
(1)酸素化
Aβ1-42イソペプチド(0.1%トリフルオロ酢酸水溶液中に250μMの濃度)、アンギオテンシンIV(200μM水溶液)、Met-エンケファリン(200μM水溶液)、およびソマトスタチン(200μM水溶液)をリン酸緩衝液またはリン酸緩衝生理食塩水(pH7.4)を加えて最終ペプチド濃度が20μMまたは40μM(pH7.4)となるように調整した(必要に応じて、0.1Mの水酸化ナトリウム水溶液を用いた)。Aβ1-42溶液を酸素化反応に付す前に、37℃で1~3時間インキュベートした。40μMのAβを含むリン酸緩衝液を25mMの4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸(HEPES)で緩衝した0.1%ウマ血清を含むダルベッコ変法イーグル培地(DMEM、ライフテクノロジー社)で希釈した。
それぞれの溶液に対し、リボフラビン(0.1%トリフルオロ酢酸/アセトニトリル(1:1)水溶液中に1mM)、化合物3(アセトニトリルに1mM)、CRANAD-2〔(T-4)-[(1E,6E)-1,7-ビス[4-(ジメチルアミノ)フェニル]-1,6-ヘプタジエン-3,5-ジオナト-κO3,κO5]ジフルオロボロン〕(200μMジメチルスルホキシド溶液)又は化合物E、化合物I、化合物J、化合物L、化合物K、もしくは化合物Sの5~10(200μMジメチルスルホキシド溶液)を加えた。化合物の最終濃度を2μMにしたものを、Aβ1-42の酸素化の評価に用いた。最終濃度を5μMにしたものを、ペプチド選択性の評価に用いた。混合物を室温にてLED(波長500、660、又は780nm)で約5~10cmの距離で光照射を行った。光照射を行わない非対照群も用意した。反応はMALDI-TOF MSを用いてモニター分析を行った。酸素化度は酸素化度(%)=(n[O]付加物のMS強度の合計)/(ネイティブのMS強度+n[O]付加物のMS強度の合計)×100の比で求めた。必要に応じ、反応溶液の分配量はZipTip U-C18(メルク・ミリポア)で質量分析前に脱塩処理を行った。結果を以下の表1および2に示す。
ラット褐色細胞腫PC12細胞を5%ウマ血清および10%胎児ウシ血清をポリ-D-リジンコート96穴プレートに1穴あたり10,000細胞の濃度で播種したDMEMに懸濁し、5%二酸化炭素雰囲気下37℃で3日間インキュベートした。溶媒を除去後、細胞を150μLの血清フリーのDMEMで洗浄し、0.1%馬血清を含有するDMEM(75μL)を加えた。その後、96穴プレートを5%二酸化炭素雰囲気下37℃で1日間インキュベートし、酸素化反応に用いた。酸素化反応において、化合物E((100μMジメチルスルホキシド溶液))をAβ(事前に3時間凝集したもの、40μM)、上記の25μLの本溶液を含むリン酸緩衝液生理食塩水に加えた(Aβの最終濃度は10μM、化合物Eの最終濃度は1μM)。混合物をLED(波長780nm)で約5cmの距離から37℃で5分間照射した。細胞を37℃の5%二酸化炭素雰囲気下で48時間インキュベートした。細胞生存率は細胞カウント試薬SF(ナカライテスク、京都、日本)を用いてWST-8(2-(2-メトキシ-4-ニトロフェニル)-3-(4-ニトロフェニル)-5-(2,4-ジスルホフェニル)-2H-テトラゾリウム)で測定した。結果を以下の表1および表2に示す。

b:LC50(暗所)が化合物自身の毒性、LC50(明所)が化合物の光毒性を表す。PC12細胞を暗所または光照射下(波長660nm)光触媒処理し、37℃で5分間、そして5%二酸化炭素雰囲気下37℃で48時間インキュベートし、細胞生存率を分析した(n=5、平均±SEM)。
c:化合物Jは、光照射下(Tukey試験)で化合物Lおよび化合物Kに対してp<0.05であった。

b:PC12細胞を光触媒(1μM)と暗所または光照射(波長780nm)条件で37℃で5分間処置を行った後、細胞生存率を分析した(n=3、平均値±SEM)。
Claims (11)
- 次の一般式(1)

X3は、臭素原子、ヨウ素原子又はセレン原子を示し;
R1及びR2は、同一又は異なって、水素原子又は置換基を有していてもよいアルキル基を示し;
R3及びR4は、同一又は異なって、水素原子、ハロゲン原子、アルコキシ基又は置換基を有していてもよいアルキル基を示すか、R1とR3、又はR2とR4が一緒になって、置換基を有していてもよいアルキレン基又はアルケニレン基を形成してもよく;
R5及びR6は、同一又は異なって、水素原子又は置換基を有していてもよいアルキル基を示し;
R7及びR8は、同一又は異なって、水素原子、ハロゲン原子、アルコキシ基又は置換基を有していてもよいアルキル基を示すか、R5とR7、又はR6とR8が一緒になって、置換基を有していてもよいアルキレン基又はアルケニレン基を形成してもよく;
m及びnは、1~3の整数を示す)
で表されるクルクミンホウ素錯体又はその塩。 - X1がハロゲノアルキル基であり、X2がハロゲン原子である請求項1記載のクルクミンホウ素錯体又はその塩。
- m及びnが1である請求項1又は2記載のクルクミンホウ素錯体又はその塩。
- R1、R2、R3、R4、R5、R6、R7及びR8が示す置換基を有していてもよいアルキル基が、カルボキシ基、スルホン酸基、ヒドロキシ基、アミノ基、-CO-、-CONH-及びトリアゾール基から選ばれる1種以上の置換基を有していてもよいアルキル基である請求項1~3のいずれか1項に記載のクルクミンホウ素錯体又はその塩。
- R1とR3、R2とR4、R5とR7、又はR6とR8が一緒になって形成されるアルキレン基又はアルケニレン基の炭素数が2又は3である請求項1~4のいずれか1項に記載のクルクミンホウ素錯体又はその塩。
- 請求項1~5のいずれか1項に記載のクルクミンホウ素錯体又はその塩を有効成分とする医薬。
- 病原性アミロイドが関連する疾患の予防又は治療薬である請求項6記載の医薬。
- 請求項1~5のいずれか1項に記載のクルクミンホウ素錯体又はその塩及び薬学的に許容される担体を含有する医薬組成物。
- 病原性アミロイドが関与する疾患の予防又は治療薬製造のための請求項1~5のいずれか1項に記載のクルクミンホウ素錯体又はその塩の使用。
- 病原性アミロイドが関与する疾患を予防又は治療するための、請求項1~5のいずれか1項に記載のクルクミンホウ素錯体又はその塩。
- 請求項1~5のいずれか1項に記載のクルクミンホウ素錯体又はその塩の有効量を投与することを特徴とする病原性アミロイドが関連する疾患を予防又は治療する方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780018865.2A CN108884112B (zh) | 2016-03-22 | 2017-03-21 | 姜黄素硼配合物和含有该姜黄素硼配合物的药物 |
| EP17770214.9A EP3434681B1 (en) | 2016-03-22 | 2017-03-21 | Curcumin-boron complex and pharmaceutical containing same |
| JP2018507334A JP6846052B2 (ja) | 2016-03-22 | 2017-03-21 | クルクミンホウ素錯体及びこれを含有する医薬 |
| US16/087,017 US10669287B2 (en) | 2016-03-22 | 2017-03-21 | Curcumin-boron complex and pharmaceutical containing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016056615 | 2016-03-22 | ||
| JP2016-056615 | 2016-03-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017164172A1 true WO2017164172A1 (ja) | 2017-09-28 |
Family
ID=59899637
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/011232 Ceased WO2017164172A1 (ja) | 2016-03-22 | 2017-03-21 | クルクミンホウ素錯体及びこれを含有する医薬 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10669287B2 (ja) |
| EP (1) | EP3434681B1 (ja) |
| JP (1) | JP6846052B2 (ja) |
| CN (1) | CN108884112B (ja) |
| WO (1) | WO2017164172A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2023120277A1 (ja) * | 2021-12-23 | 2023-06-29 | ||
| US12028185B2 (en) | 2020-01-29 | 2024-07-02 | Arris Enterprises Llc | Methods, systems, and devices for steering packets across multiple access technologies |
| JP2024163697A (ja) * | 2023-05-12 | 2024-11-22 | 学校法人近畿大学 | アミロイド線維検出プローブ |
| WO2025206000A1 (ja) * | 2024-03-28 | 2025-10-02 | 国立大学法人 東京大学 | トランスサイレチンアミロイドに対する光酸素化触媒、及びこれを含有する医薬組成物 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114031635B (zh) * | 2021-12-14 | 2024-02-13 | 洛阳师范学院 | 一种二氟硼姜黄素衍生物及其制备方法和应用 |
| CN116969981B (zh) * | 2023-06-25 | 2025-07-25 | 中国药科大学 | 二氢喹啉类化合物及其制备方法、药物组合物和应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016010092A1 (ja) * | 2014-07-16 | 2016-01-21 | 国立研究開発法人科学技術振興機構 | ベンゾチアゾール化合物及びこれを含有する医薬 |
| WO2016143699A1 (ja) * | 2015-03-06 | 2016-09-15 | 国立研究開発法人科学技術振興機構 | ジピリンホウ素錯体及びこれを含有する医薬 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5679864A (en) | 1995-11-03 | 1997-10-21 | Gene Print Inc. | Process for the synthesis of curcumin-related compounds |
| WO2010017094A2 (en) | 2008-07-31 | 2010-02-11 | The General Hospital Corporation | CURCUMIN DERIVATIVES FOR AMYLOID-β PLAQUE IMAGING |
| US8986657B2 (en) * | 2009-07-31 | 2015-03-24 | The General Hospital Corporation | Methods and system for detecting soluble amyloid-β |
| WO2013167770A1 (es) | 2012-05-08 | 2013-11-14 | Asac Compañía De Biotecnología E Investigacion Sa | Procedimiento para la síntesis de curcumina |
| US9738623B2 (en) * | 2012-08-06 | 2017-08-22 | The General Hospital Corporation | Curcumin analogs |
| US9757477B2 (en) * | 2013-09-06 | 2017-09-12 | The General Hospital Corporation | Imaging brown adipose tissue with curcumin derivatives |
-
2017
- 2017-03-21 WO PCT/JP2017/011232 patent/WO2017164172A1/ja not_active Ceased
- 2017-03-21 JP JP2018507334A patent/JP6846052B2/ja active Active
- 2017-03-21 EP EP17770214.9A patent/EP3434681B1/en active Active
- 2017-03-21 US US16/087,017 patent/US10669287B2/en active Active
- 2017-03-21 CN CN201780018865.2A patent/CN108884112B/zh active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016010092A1 (ja) * | 2014-07-16 | 2016-01-21 | 国立研究開発法人科学技術振興機構 | ベンゾチアゾール化合物及びこれを含有する医薬 |
| WO2016143699A1 (ja) * | 2015-03-06 | 2016-09-15 | 国立研究開発法人科学技術振興機構 | ジピリンホウ素錯体及びこれを含有する医薬 |
Non-Patent Citations (4)
| Title |
|---|
| "29T-pm15 [Development of bio-adaptive photo oxygenation catalyst for pathogenic Temiroid protein]", ABSTRACTS OF 136TH ANNUAL MEETING OF PHARMACEUTICAL SOCIETY OF JAPAN, vol. 136, 19 April 2016 (2016-04-19), pages 29T-pm15, XP009515149, ISSN: 0918-9823 * |
| DAI 32 KAI ABSTRACTS OF SYMPOSIUM ON MEDICINAL CHEMISTRY, 7 November 2014 (2014-11-07), pages 87, XP009515764 * |
| See also references of EP3434681A4 * |
| SOMA YOHEI: "Abstr. 10-17 [Development of highly selective oxygenation catalyst for amyloid] = Amiroido-kō sentaku-tekina sanso-ka shokubai no kaihatsu", PROCEEDINGS OF THE 40TH SYMPOSIUM ON REACTIONS AND SYNTHESIS PROGRESS SYMPOSIUM, vol. 40, 22 October 2014 (2014-10-22), pages 19, XP009512797 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12028185B2 (en) | 2020-01-29 | 2024-07-02 | Arris Enterprises Llc | Methods, systems, and devices for steering packets across multiple access technologies |
| JPWO2023120277A1 (ja) * | 2021-12-23 | 2023-06-29 | ||
| WO2023120277A1 (ja) * | 2021-12-23 | 2023-06-29 | 三井化学株式会社 | 有機エレクトロルミネッセンス素子、薄膜、化合物、遅延蛍光発光体および有機半導体レーザー |
| JP7709649B2 (ja) | 2021-12-23 | 2025-07-17 | 三井化学株式会社 | 有機エレクトロルミネッセンス素子、薄膜、化合物、遅延蛍光発光体および有機半導体レーザー |
| JP2024163697A (ja) * | 2023-05-12 | 2024-11-22 | 学校法人近畿大学 | アミロイド線維検出プローブ |
| JP7713734B2 (ja) | 2023-05-12 | 2025-07-28 | 学校法人近畿大学 | アミロイド線維検出プローブ |
| WO2025206000A1 (ja) * | 2024-03-28 | 2025-10-02 | 国立大学法人 東京大学 | トランスサイレチンアミロイドに対する光酸素化触媒、及びこれを含有する医薬組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3434681B1 (en) | 2023-05-10 |
| US20190100537A1 (en) | 2019-04-04 |
| JP6846052B2 (ja) | 2021-03-24 |
| CN108884112A (zh) | 2018-11-23 |
| EP3434681A1 (en) | 2019-01-30 |
| US10669287B2 (en) | 2020-06-02 |
| JPWO2017164172A1 (ja) | 2019-02-14 |
| CN108884112B (zh) | 2021-03-02 |
| EP3434681A4 (en) | 2019-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6846052B2 (ja) | クルクミンホウ素錯体及びこれを含有する医薬 | |
| Xing et al. | A fluorogenic ONOO–-triggered carbon monoxide donor for mitigating brain ischemic damage | |
| KR102536408B1 (ko) | Hsp90 저해제 및 hsp70 유도제로서의 개질된 에터기를 가진 바이페닐 아마이드 | |
| JP2023106366A (ja) | タウタンパク質分解の化合物 | |
| JP7660063B2 (ja) | サイクリン依存性キナーゼ7のインヒビターおよびそれらの使用 | |
| JP7680999B2 (ja) | サイクリン依存性キナーゼ7のインヒビターおよびそれらの使用 | |
| JP2018502098A (ja) | サイクリン依存性キナーゼ7(cdk7)の阻害剤 | |
| JP6562361B2 (ja) | ベンゾチアゾール化合物及びこれを含有する医薬 | |
| JP6770534B2 (ja) | 治療用化合物 | |
| US10188671B2 (en) | Boron-dipyrrin complex and medicament containing the same | |
| EP3468563B1 (en) | Compounds for delivering glutathione to a target and methods of making and using the same | |
| FR2495155A1 (fr) | Nouveaux 8-aminoalkyl-4-alkylisopsoralenes, leur procede de preparation et composition pharmaceutique les contenant | |
| US20220041538A1 (en) | Cannabigerol quinone acid and salts thereof | |
| JP7652425B2 (ja) | フェノチアジン骨格を有する光酸素化触媒化合物及びこれを含有する医薬 | |
| CN116283927B (zh) | 嘧啶氨基芳基类衍生物及其作为富亮氨酸重复激酶2抑制剂的应用 | |
| US20250313576A1 (en) | Compound for photooxygenation catalyst and medicinal composition containing same | |
| WO2023129116A2 (en) | A drug for the treatment of type 2 diabetes mellitus disease and the nanoparticle thereof | |
| JP5497064B2 (ja) | グルコキナーゼ活性化因子として有用なアリールシクロプロピルアセトアミド誘導体 | |
| US10556872B2 (en) | Fatty acid synthase inhibitors and methods of use | |
| JP7652424B2 (ja) | 光酸素化触媒化合物及びこれを含有する医薬 | |
| WO2023129117A2 (en) | A drug for the treatment of type 2 diabetes mellitus disease and the nanoparticle thereof | |
| EP2927215A1 (en) | Copper chelators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018507334 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017770214 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017770214 Country of ref document: EP Effective date: 20181022 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17770214 Country of ref document: EP Kind code of ref document: A1 |