WO2017179536A1 - 硬化性組成物及び接着剤 - Google Patents
硬化性組成物及び接着剤 Download PDFInfo
- Publication number
- WO2017179536A1 WO2017179536A1 PCT/JP2017/014681 JP2017014681W WO2017179536A1 WO 2017179536 A1 WO2017179536 A1 WO 2017179536A1 JP 2017014681 W JP2017014681 W JP 2017014681W WO 2017179536 A1 WO2017179536 A1 WO 2017179536A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mass
- parts
- curable composition
- component
- polymer fine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J163/00—Adhesives based on epoxy resins; Adhesives based on derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J163/00—Adhesives based on epoxy resins; Adhesives based on derivatives of epoxy resins
- C09J163/10—Epoxy resins modified by unsaturated compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/04—Interconnection of layers
- B32B7/12—Interconnection of layers using interposed adhesives or interposed materials with bonding properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/06—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J11/00—Features of adhesives not provided for in group C09J9/00, e.g. additives
- C09J11/08—Macromolecular additives
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J133/00—Adhesives based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Adhesives based on derivatives of such polymers
- C09J133/04—Homopolymers or copolymers of esters
- C09J133/06—Homopolymers or copolymers of esters of esters containing only carbon, hydrogen and oxygen, the oxygen atom being present only as part of the carboxyl radical
- C09J133/10—Homopolymers or copolymers of methacrylic acid esters
- C09J133/12—Homopolymers or copolymers of methyl methacrylate
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J133/00—Adhesives based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Adhesives based on derivatives of such polymers
- C09J133/04—Homopolymers or copolymers of esters
- C09J133/14—Homopolymers or copolymers of esters of esters containing halogen, nitrogen, sulfur or oxygen atoms in addition to the carboxy oxygen
Definitions
- the present invention relates to a curable composition mainly composed of an epoxy resin, which becomes a rubber-like cured product having excellent mechanical strength after curing while having low viscosity before curing, and particularly an adhesive using the curable composition. It is about.
- the cured epoxy resin is excellent in many respects such as dimensional stability, mechanical strength, electrical insulation characteristics, heat resistance, water resistance, and chemical resistance.
- a cured product of an epoxy resin is highly rigid but has a low elongation property and may exhibit a very brittle property, and such a property often causes a problem in applications where high elongation is required.
- Patent Document 4 and Patent Document 5 the toughness and impact resistance of a cured product obtained are improved by dispersing polymer fine particles in a curable resin composition containing a curable resin such as an epoxy resin as a main component.
- a curable resin such as an epoxy resin as a main component.
- Technology is disclosed.
- Patent Literature 4 and Patent Literature 5 are not developed as structural rubber elastic adhesives.
- This invention is made
- a curable composition containing an epoxy resin, polymer fine particles, and an epoxy curing agent (C) As the epoxy resin, a polymer fine particle (B) having a core-shell structure in which the obtained cured product uses a flexible epoxy resin (A) exhibiting rubber elasticity as a main component and the glass transition temperature (Tg) of the core layer is greater than 0 ° C. )
- Tg glass transition temperature
- the present invention relates to 100 parts by mass of a flexible epoxy resin (A), 1 to 150 parts by mass of polymer fine particles (B) having a core-shell structure having at least two layers of a core layer and a shell layer, and epoxy curing A curable composition containing 1 to 200 parts by mass of an agent (C),
- the glass transition temperature (Tg) of the core layer of the component (B) calculated from the following formula (1) is greater than 0 ° C.
- the present invention relates to a curable composition characterized in that the cured product obtained by curing the curable composition has a type A durometer hardness of 5 to 95 at 23 ° C. as defined in JIS K6253-3.
- M i is the weight fraction of each monomer i component selected from butadiene and non-crosslinkable monomers constituting the core layer of component (B), and T g i is a homopolymer of each monomer i.
- T g i is a homopolymer of each monomer i.
- the glass transition temperature (K) is preferably 15 to 150 ° C.
- the epoxy equivalent of the component (A) is preferably 200 to 4000 g / eq. 4)
- the core layer of the component (B) is preferably a (meth) acrylate polymer.
- the core layer of the component (B) is a polymer obtained by polymerizing a monomer mixture comprising 80 to 99% by mass of the non-crosslinkable monomer and 20 to 1% by mass of the crosslinkable monomer. It is preferably a coalescence.
- the shell layer of the component (B) is preferably a (meth) acrylate polymer.
- the shell layer of component (B) preferably has an epoxy group.
- the content of the epoxy group in the shell layer of the component (B) is preferably 0.05 to 3.5 mmol / g.
- (B) component has a shell layer formed by graft-polymerizing the monomer component which has an epoxy group to a core layer.
- the component (B) is preferably dispersed in the form of primary particles in the curable composition.
- it is a cured product obtained by curing any of the curable compositions.
- it is an adhesive agent using any one of the curable compositions described above.
- 13) Preferably, it is an adhesive for vehicles which uses any one of the said curable compositions.
- two substrates made of different materials are laminated adhesive substrates bonded with any one of the curable compositions.
- the laminated adhesive substrate is preferably an exterior panel. 16) Further, at least one of the substrates is preferably at least one selected from a steel plate, an aluminum alloy plate, a titanium alloy plate, a magnesium alloy plate, and a plastic substrate.
- the curable composition of the present invention is excellent in tensile strength while the resulting cured product has rubber elasticity and exhibits high elongation characteristics. Moreover, the curable composition of this invention is the low viscosity and is excellent in the handleability in the preferable aspect.
- the curable composition of the present invention comprises 100 parts by mass of a flexible epoxy resin (A), 1 to 150 parts by mass of polymer fine particles (B) having a core-shell structure in which the glass transition point of the core layer is greater than 0 ° C., epoxy 1 to 200 parts by mass of a curing agent (C). It is essential that the cured product obtained by curing the curable composition exhibits rubber elasticity.
- the type A durometer hardness defined in JIS K6253-3 of the cured product is essential to be 5 to 95 at 23 ° C., preferably 20 to 90, preferably 40 to 87. Is more preferable, and 50 to 85 is particularly preferable. When the type A durometer hardness of the cured product is less than 5, the tensile strength of the cured product may be insufficient, and when it exceeds 95, rubber elasticity may be insufficient and elongation characteristics may be poor.
- the polymer fine particles (B) having a core-shell structure with a glass transition temperature of the core layer greater than 0 ° C. are added to the flexible epoxy resin (A), the mechanism of excellent tensile strength is demonstrated while exhibiting high elongation characteristics.
- the epoxy resin does not have flexibility, even if the polymer fine particles (B) are blended, the cured product becomes hard without showing rubber elasticity, and the polymer fine particles (B) become cracking points at the time of deformation. Become. That is, it is estimated that the tensile strength is rather lowered by the polymer fine particles (B).
- the cured product obtained using the flexible epoxy resin (A) exhibits rubber elasticity and has high followability to deformation, it is estimated that the polymer fine particles (B) are unlikely to become crack generation points. Then, the glass transition temperature of the core layer of the polymer fine particles (B) is made higher than 0 ° C., and the fine particles (B) are dispersed to suppress the deformation of the cured product, and the elastic modulus is maintained while maintaining the high elongation of the cured product. As a result, it is estimated that the tensile strength is increased. On the other hand, when polymer fine particles having a glass transition point of the core layer of less than 0 ° C.
- the fine particles have low elasticity at room temperature, so that they easily deform in accordance with the deformation of the flexible epoxy resin (A) that is a matrix resin. It seems that the tensile strength is not improved.
- grains such as an inorganic filler, the same quality effect as the polymer fine particle (B) whose glass transition temperature of a core layer is larger than 0 degreeC is acquired, the grade is inferior to this polymer fine particle (B). The reason is that the inorganic particles have a small interaction with the matrix resin (A) component, whereas the polymer fine particles (B) having the core-shell structure have a large interaction with the matrix resin. .
- a flexible epoxy resin (A) As a main component of the curable composition of the present invention, a flexible epoxy resin (A) is used.
- a flexible epoxy resin (A) component By using a flexible epoxy resin (A) component, the hardened
- the flexible epoxy resin (A) is cured under the following conditions regardless of the main chain skeleton of the component, the type of epoxy group, the number of epoxy groups per molecule, the size of the molecular weight, the presence or absence of branching, etc.
- the cured product obtained has a type A durometer hardness value of 1 to 95.
- the flexible epoxy resin various flexible epoxy resins can be used.
- a fatty acid-modified epoxy obtained by addition reaction of an aliphatic polybasic acid such as a dimer acid-modified epoxy resin to an epoxy resin.
- Resins such as epoxidized polybutadiene in which epoxy groups are introduced by oxidation of vinyl groups of 1,2-polybutadiene, thiol-modified epoxy resins modified with epoxy resins with dithioether, polyol-modified epoxy resins, ⁇ -caprolactone Examples thereof include modified epoxy resins, rubber-modified epoxy resins, urethane-modified epoxy resins, and epoxy group-containing (meth) acrylic copolymers. These epoxy resins may be used alone or in combination of two or more.
- fatty acid-modified epoxy resins are preferable because of their low viscosity and excellent handleability.
- Fatty acid-modified epoxy resins are cured products.
- the dimer acid-modified epoxy resin is particularly preferable because it has a low viscosity value when polymer fine particles, which will be described later, are dispersed, and is excellent in handleability.
- polyol-modified epoxy resin examples include glycidyl ethers of bisphenol A ethylene oxide adducts such as bisphenol A bis (polyethylene glycol glycidyl ether) ether and glycidyl ethers of bisphenol A propylene oxide adducts such as bisphenol A bis (polypropylene glycol glycidyl ether) ether.
- ⁇ -Caprolactone-modified epoxy resin examples include ⁇ -caprolactone-modified bisphenol A type epoxy resin, ⁇ -caprolactone-modified 3 ′, 4′-epoxycyclohexylmethyl 3,4-epoxycyclohexanecarboxylate and the like. And functional epoxy resins.
- the rubber-modified epoxy resin is a reaction product obtained by reacting rubber and an epoxy group-containing compound.
- the rubber include acrylonitrile butadiene rubber (NBR), styrene butadiene rubber (SBR), and hydrogenated nitrile rubber (HNBR). ), Ethylene propylene rubber (EPDM), acrylic rubber (ACM), butyl rubber (IIR), butadiene rubber, and polyoxyalkylenes such as polypropylene oxide, polyethylene oxide, and polytetramethylene oxide, and the like.
- the rubber polymer is preferably one having a reactive group such as an amino group, a hydroxy group, or a carboxyl group at the terminal.
- the rubber-modified epoxy resin used in the present invention is a product obtained by reacting these rubber-based polymer and epoxy resin at an appropriate blending ratio by a known method.
- acrylonitrile-butadiene rubber-modified epoxy resin and polyoxyalkylene-modified epoxy resin are preferable from the viewpoint of elongation characteristics and tensile strength of the resulting curable composition, and acrylonitrile-butadiene rubber-modified epoxy resin is more preferable.
- the acrylonitrile-butadiene rubber-modified epoxy resin can be obtained, for example, by reacting a carboxyl group-terminated NBR (CTBN) with a bisphenol A type epoxy resin.
- CBN carboxyl group-terminated NBR
- the content of the acrylonitrile monomer component in the acrylonitrile-butadiene rubber is preferably 5 to 40% by mass, more preferably 10 to 35% by mass, from the viewpoint of elongation characteristics and tensile strength of the resulting curable composition. 15 to 30% by mass is more preferable, and 20 to 30% by mass is particularly preferable.
- addition reaction products of amino group-terminated polyoxyalkylenes and epoxy resins are also included in rubber-modified epoxy resins.
- adducts addition reaction products of amino group-terminated polyoxyalkylenes and epoxy resins
- the adduct can be easily manufactured by a known method.
- the epoxy resin used in the production of the adduct include specific examples of the component (A) exemplified in the present invention, and bisphenol A type epoxy resins and bisphenol F type epoxy resins are preferable, and bisphenol A A type epoxy resin is more preferable.
- Examples of commercially available amino group-terminated polyoxyalkylenes used in the production of the adduct include Huntsman's Jeffamine D-230, Jeffamine D-400, Jeffamine D-2000, Jeffamine D-4000, Jeffamine T-5000 and the like.
- the average number of epoxide reactive end groups per molecule in the rubber is preferably 1.5 to 2.5, more preferably 1.8 to 2.2.
- the number average molecular weight of the rubber is preferably 1000 to 8000, more preferably 2000 to 6000, and particularly preferably 3000 to 5000 in terms of polystyrene-converted molecular weight measured by GPC.
- a rubber modified epoxy resin there is no restriction
- the rubber-modified epoxy resin is produced by heating to a temperature of 100 to 250 ° C.
- the epoxy group-containing compound used in producing the rubber-modified epoxy resin is not particularly limited, but bisphenol A type epoxy resin and bisphenol F type epoxy resin are preferable, and bisphenol A type epoxy resin is more preferable.
- the unreacted epoxy group-containing compound remaining after the reaction is included in the rubber-modified epoxy resin of the present invention. , Not included.
- the epoxy resin can be modified by pre-reaction with a bisphenol component.
- the bisphenol component used for the modification is preferably 3 to 35 parts by mass, and more preferably 5 to 25 parts by mass with respect to 100 parts by mass of the rubber component in the rubber-modified epoxy resin.
- a cured product obtained by curing a curable composition containing a modified rubber-modified epoxy resin further exhibits high elongation characteristics and is excellent in tensile strength.
- the glass transition temperature (Tg) of the rubber-modified epoxy resin is not particularly limited, but is preferably ⁇ 25 ° C. or lower, more preferably ⁇ 35 ° C. or lower, still more preferably ⁇ 40 ° C. or lower, and particularly preferably ⁇ 50 ° C. or lower.
- the urethane-modified epoxy resin is a reaction product obtained by reacting a compound containing an epoxy group with a group having reactivity with an isocyanate group and a urethane prepolymer containing an isocyanate group.
- a urethane-modified epoxy resin can be obtained by reacting a hydroxy group-containing epoxy compound with a urethane prepolymer.
- An epoxy group-containing (meth) acrylic copolymer is a (meth) acrylic polymer having an epoxy group in the molecule, and is obtained by copolymerization of an epoxy group-containing monomer and another (meth) acrylic monomer.
- Polymers are preferred. Specific examples thereof include a copolymer of an epoxy group-containing monomer such as glycidyl (meth) acrylate and butyl acrylate or 2-ethylhexyl acrylate.
- the fatty acid-modified epoxy resin is obtained by addition reaction of an aliphatic polybasic acid or the like with an epoxy resin.
- the aliphatic polybasic acids include an unsaturated polyvalent carboxylic acid having no aromatic ring such as maleic acid, maleic anhydride, fumaric acid, itaconic acid, itaconic anhydride, citraconic acid, or the like, or an anhydride thereof, or Tetrahydrophthalic acid, tetrahydrophthalic anhydride, hexahydrophthalic acid, hexahydrophthalic anhydride, cyclohexanedicarboxylic acid, succinic acid, malonic acid, glutaric acid, adipic acid, azelaic acid, sebacic acid, 1,12-docanic acid, Examples thereof include saturated polyvalent carboxylic acids having no aromatic ring such as dimer acid, and anhydrides thereof. Dimer acid is particularly preferable from the viewpoint of elong, maleic anhydride, fumaric
- the epoxy equivalent of the flexible epoxy resin (A) is preferably 200 to 4000 g / eq, more preferably 250 to 2000 g / eq, still more preferably 300 to 1500 g / eq, and particularly preferably 350 to 1200 g / eq. If the epoxy equivalent is less than 200 g / eq, rubber elasticity may be insufficient. If the epoxy equivalent exceeds 4000 g / eq, the viscosity becomes high and the workability of the curable composition may be poor. From the viewpoint of lowering the viscosity of the curable composition, the epoxy equivalent of the flexible epoxy resin (A) may be, for example, 700 g / eq or less, particularly 500 g / eq or less.
- the number average molecular weight of the flexible epoxy resin (A) is preferably from 400 to 8000, more preferably from 500 to 4000, still more preferably from 600 to 3000, and particularly preferably from 700 to 2400 in terms of polystyrene equivalent molecular weight measured by GPC. . If the number average molecular weight is less than 400, rubber elasticity may be insufficient, and if the number average molecular weight exceeds 8000, the viscosity becomes high and the workability of the curable composition may be poor.
- the flexible epoxy resin (A) has at least 1.1 epoxy groups in one molecule. Those having 1.2 or more are preferable from the viewpoint of high reactivity during curing and the cured product easily forms a three-dimensional network, and 1.4 or more and 3 or less epoxy groups in one molecule. More preferably, those having 1.6 or more and 2.7 or less, particularly preferably 1.8 or more and 2.4 or less are more preferable from the viewpoint of elongation and tensile strength of the obtained cured product.
- the flexible epoxy resin (A) is used as the main component of the curable composition of the present invention, but an epoxy resin other than the flexible epoxy resin (hard epoxy resin) to the extent that the effect of the present invention is not lowered. Can also be used in small amounts.
- hard epoxy resin examples include bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol AD type epoxy resin, bisphenol S type epoxy resin, glycidyl ester type epoxy resin, glycidyl amine type epoxy resin, and novolak type epoxy resin.
- Hydrogenated bisphenol A (or F) type epoxy resin fluorinated epoxy resin, flame retardant epoxy resin such as tetrabromobisphenol A glycidyl ether, p-oxybenzoic acid glycidyl ether ester type epoxy resin, m-aminophenol type Epoxy resin, diaminodiphenylmethane epoxy resin, various alicyclic epoxy resins, N, N-diglycidylaniline, N, N-diglycidyl-o-toluidine, triglycidyl isocyanurate, Divinylbenzene dioxide, resorcinol diglycidyl ether, and the like are exemplified, but the invention is not limited to, epoxy resins may be used which are commonly used.
- the resulting cured product has a higher modulus of elasticity and may not exhibit rubber elasticity due to low elongation. 10 parts by weight or less is preferable with respect to 100 parts by weight, 5 parts by weight or less is more preferable, 1 part by weight or less is further preferable, 0.1 part by weight or less is particularly preferable, and substantially no content is most preferable. .
- the curable composition of the present invention uses 1 to 150 parts by mass of polymer fine particles (B) having a core-shell structure with respect to 100 parts by mass of the flexible epoxy resin (A) component.
- the polymer fine particle (B) component is 1-150 in relation to 100 parts by mass of the flexible epoxy resin (A) component.
- Parts by mass preferably 2 to 120 parts by mass, more preferably 3 to 100 parts by mass, and still more preferably 4 to 70 parts by mass. Further, it may be 30 to 70 parts by mass, or 40 to 70 parts by mass.
- the particle diameter of the polymer fine particle is not particularly limited, but considering industrial productivity, the volume average particle diameter (Mv) is preferably 10 to 2000 nm, more preferably 30 to 600 nm, further preferably 50 to 400 nm, and more preferably 100 to 200 nm. Is particularly preferred.
- the volume average particle diameter (Mv) of the polymer fine particles can be measured using Microtrac UPA150 (manufactured by Nikkiso Co., Ltd.).
- the polymer fine particle (B) component has a half width of 0.5 to 1 times the number average particle diameter in the number distribution of the particle diameters obtained.
- the composition is preferred because of its low viscosity and easy handling. From the viewpoint of easily realizing the specific particle size distribution described above, it is preferable that there are two or more maximum values in the particle size distribution of the polymer fine particle (B) component, from the viewpoint of labor and cost during production. More preferably, 2 to 3 local maximum values are present, and 2 local maximum values are even more preferable.
- the polymer fine particle (B) component is preferably dispersed in a primary particle state in the curable composition.
- “polymer fine particles are dispersed in the state of primary particles in the curable composition” means that the polymer fine particles are substantially independent (without contact).
- primary dispersion means that the polymer fine particles are substantially independent (without contact).
- a part of the curable composition is dissolved in a solvent such as methyl ethyl ketone, and the particle size is measured by a particle size measuring device using laser light scattering. It can be confirmed by measuring.
- stable dispersion of polymer fine particles means that the polymer fine particles are not aggregated, separated, or precipitated in the continuous layer, and are constantly under normal conditions over a long period of time. Mean that the distribution of the polymer fine particles in the continuous layer does not substantially change, and the viscosity is lowered by heating these compositions in a range that is not dangerous. It is preferable that “stable dispersion” can be maintained even if stirring is performed.
- the polymer fine particle (B) component may be used alone or in combination of two or more.
- the structure of the polymer fine particle is required to have a core-shell structure having at least two layers of a core layer and a shell layer, and is composed of an intermediate layer that covers the core layer and a shell layer that further covers this intermediate layer. It is also possible to have a structure of three or more layers. Hereinafter, each layer will be specifically described.
- M i is the weight fraction of each monomer i component selected from butadiene and non-crosslinkable monomers constituting the core layer of the polymer fine particle (B) component
- T g i is the Represents the glass transition temperature (K) of the homopolymer.
- the glass transition temperature of the homopolymer of the non-crosslinkable monomer is, for example, J. It can be confirmed by literature and catalogs such as “Polymer Handbook Fourth Edition” written by Brandrup.
- the upper limit of the glass transition temperature of the core layer of the component (B) calculated from the mathematical formula (1) is not particularly limited, it is preferably 300 ° C. or lower (particularly 200 ° C. or lower) from the viewpoint of availability.
- the glass transition temperature of the core layer is more preferably 15 to 200 ° C. (particularly 15 to 150 ° C.), further preferably 30 to 150 ° C., and particularly preferably 50 to 110 ° C. (particularly 25 to 100 ° C. or 40 to 70 ° C.). .
- the viscosity of the curable composition tends to decrease. Further, the higher the glass transition temperature of the core layer, the higher the maximum tensile stress of the cured product.
- the core layer is preferably a (meth) acrylate polymer polymerized using a (meth) acrylate monomer as a main component because the viscosity of the curable composition is low.
- the (meth) acrylate-based polymer the (meth) acrylate-based monomer is, for example, 50% by mass or more, preferably 70% by mass or more, in all the monomers of the core layer.
- non-crosslinkable monomer examples include, for example, methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, octyl (meth) acrylate, dodecyl (meth) ) Acrylates, stearyl (meth) acrylates, isobornyl (meth) acrylates, dicyclopentanyl (meth) acrylates, 1-adamantyl (meth) acrylates, behenyl (meth) acrylates, and other alkyl (meth) acrylates; ) Acrylic ring-containing (meth) acrylates such as acrylate and benzyl (meth) acrylate; hydroxyalkyl (meth) acrylates such as 2-hydroxyethyl (meth) acrylate and 4-hydroxybutyl (meth) acrylate Glycidyl (meth)
- alkyl methacrylates having 1 to 4 carbon atoms, vinyl arenes, and vinyl cyanides are preferable because of their high availability and high Tg of the polymer.
- Methyl methacrylate, ethyl methacrylate, isopropyl methacrylate, isobutyl methacrylate, n-butyl methacrylate, t-butyl methacrylate, styrene, ⁇ -methyl styrene, and acrylonitrile are particularly preferred because of their high availability and high polymer Tg.
- the core layer has a crosslinked structure introduced therein. Moreover, when a crosslinked structure is introduced, the viscosity of the curable composition of the present invention is decreased, and a cured product obtained by curing tends to increase the strength.
- a method for introducing a crosslinked structure a generally used method can be employed. Examples thereof include a method of copolymerizing a non-crosslinkable monomer and a crosslinkable monomer such as a polyfunctional monomer or a mercapto group-containing compound.
- the core layer may be a polymer having only a non-crosslinkable monomer and not having a crosslinked structure, but the non-crosslinkable monomer is 80 to 99.9% by mass and the crosslinkable monomer is 20 to 0.1.
- a polymer having a cross-linked structure obtained by polymerizing a monomer mixture consisting of mass% is preferable, and a single unit consisting of 80 to 99 mass% of a non-crosslinkable monomer and 20 to 1 mass% of a cross-linkable monomer.
- a polymer having a crosslinked structure obtained by polymerizing a monomer mixture is more preferable.
- a monomer mixture comprising 90 to 98% by mass of the non-crosslinkable monomer and 10 to 2% by mass of the crosslinkable monomer is more preferable, and 94 to 97% by mass of the non-crosslinkable monomer and the crosslinkable property. Most preferred is a monomer mixture comprising 6 to 3% by weight of monomers.
- the gel content is preferably 60% by mass or more, more preferably 80% by mass or more, further preferably 90% by mass or more, and 95% by mass. % Or more is particularly preferable.
- the gel content referred to in the present specification means that 0.5 g of crumb obtained by coagulation and drying is immersed in 100 g of toluene and left to stand at 23 ° C. for 24 hours, and then insoluble and soluble components are separated. The ratio of insoluble matter to the total amount of insoluble matter and soluble matter is meant.
- polyfunctional monomer examples include allyl alkyl (meth) acrylates such as allyl (meth) acrylate and allylalkyl (meth) acrylate; allyloxyalkyl (meth) acrylates; (poly) ethylene glycol di (meth) acrylate, Polyfunctional (meth) having two or more (meth) acrylic groups such as butanediol di (meth) acrylate, ethylene glycol di (meth) acrylate, triethylene glycol di (meth) acrylate, tetraethylene glycol di (meth) acrylate Acrylates; Conjugated diene monomers such as 1,3-butadiene, isoprene, 2-chloro-1,3-butadiene, 2-methyl-1,3-butadiene; diallyl phthalate, triallyl cyanurate, triallyl isocyanurate The Nirubenzen, and the like.
- allyl methacrylate particularly preferred are allyl methacrylate, triallyl isocyanurate, butanediol di (meth) acrylate, and divinylbenzene.
- a polyfunctional monomer may be used independently or may be used in combination of 2 or more type.
- the volume average particle diameter of the core layer is preferably 0.03 to 2 ⁇ m, more preferably 0.05 to 1 ⁇ m. In many cases, it is difficult to stably obtain a volume average particle diameter of less than 0.03 ⁇ m, and when it exceeds 2 ⁇ m, the strength of a cured product obtained by curing the curable composition may be reduced.
- the volume average particle diameter can be measured using Microtrac UPA150 (manufactured by Nikkiso Co., Ltd.).
- the core layer preferably does not contain a conjugated diene monomer (particularly 1,3-butadiene).
- the core layer is preferably 40 to 97% by mass, more preferably 60 to 95% by mass, still more preferably 70 to 93% by mass, and particularly preferably 80 to 90% by mass, based on 100% by mass of the entire polymer fine particles. If the core layer is less than 40% by mass, the mechanical strength of the cured product may decrease. When the core layer is larger than 97% by mass, the polymer fine particles are likely to aggregate, and the curable composition has a high viscosity and may be difficult to handle. Increasing the shell layer / core layer ratio (mass ratio) may contribute to lowering the viscosity of the curable composition. The mass ratio may be, for example, 0.20 or more, or 0.25 or more. In the present invention, the core layer often has a single layer structure, but may have a multilayer structure. When the core layer has a multilayer structure, the polymer composition of each layer may be different.
- an intermediate layer may be formed if necessary.
- the following surface cross-linked layer may be formed as the intermediate layer.
- the surface cross-linked layer is obtained by polymerizing a surface cross-linked layer component composed of 30 to 100% by mass of a polyfunctional monomer having two or more radical double bonds in the same molecule and 0 to 70% by mass of other vinyl monomers. It consists of an intermediate layer polymer and has the effect of reducing the viscosity of the curable composition of the present invention and the effect of improving the dispersibility of the polymer fine particles (B) in the flexible epoxy resin (A) component. It also has the effect of increasing the crosslinking density of the core layer and increasing the grafting efficiency of the shell layer.
- Specific examples of the polyfunctional monomer include the same monomers as the above-mentioned polyfunctional monomer, but preferably allyl methacrylate and triallyl isocyanurate.
- the outermost shell layer of the polymer fine particle is obtained by polymerizing the monomer for shell formation, and improves the compatibility between the polymer fine particle (B) component and the flexible epoxy resin (A) component. Or a shell polymer that plays a role of allowing the polymer fine particles (B) to be dispersed in the form of primary particles in the cured product.
- Such a shell polymer is preferably grafted on the core layer and / or the intermediate layer.
- the monomer component used for forming the polymer is graft-polymerized to the core polymer forming the core layer so that the shell polymer and the core polymer are substantially chemically bonded. That is, preferably, the shell polymer is formed by graft polymerization of the shell-forming monomer in the presence of the core polymer, and in this way, the core polymer is graft-polymerized. Covers part or whole. This polymerization operation can be carried out by adding a monomer which is a constituent component of the shell polymer to the core polymer latex prepared and present in an aqueous polymer latex state and polymerizing it.
- the monomer for forming the shell layer from the viewpoint of compatibility and dispersibility of the polymer fine particle (B) component in the curable composition, for example, aromatic vinyl monomer, vinyl cyan monomer, (meth) acrylate monomer are preferable, (Meth) acrylate monomers are more preferred.
- the shell layer is preferably a (meth) acrylate polymer polymerized using a (meth) acrylate monomer as a main component because the viscosity of the curable composition is low. These monomers for forming the shell layer may be used alone or in appropriate combination.
- the (meth) acrylate-based monomer is, for example, 50% by mass or more, and preferably 70% by mass or more in the total monomer of the shell layer.
- the monomer having an epoxy group is preferably used for forming a shell layer, and more preferably used only for the shell layer.
- the shell layer forming monomer when a polyfunctional monomer having two or more radical polymerizable double bonds is used as the shell layer forming monomer, swelling of the polymer fine particles in the curable composition is prevented, and the viscosity of the curable composition is also reduced. Is preferable because it tends to be low and handleability is improved.
- the polyfunctional monomer is preferably contained in 1 to 20% by mass, more preferably 5 to 15% by mass, in 100% by mass of the shell-forming monomer.
- aromatic vinyl monomer examples include vinylbenzenes such as styrene, ⁇ -methylstyrene, p-methylstyrene, and divinylbenzene.
- vinylcyan monomer examples include acrylonitrile and methacrylonitrile.
- the (meth) acrylate monomer examples include (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, and butyl (meth) acrylate; hydroxyethyl (meth) acrylate, hydroxybutyl (meth) ) (Meth) acrylic acid hydroxyalkyl esters such as acrylate.
- the monomer having an epoxy group examples include glycidyl group-containing vinyl monomers such as glycidyl (meth) acrylate, 4-hydroxybutyl (meth) acrylate glycidyl ether, and allyl glycidyl ether.
- Specific examples of the multifunctional monomer having two or more radically polymerizable double bonds include the same monomers as the above-mentioned multifunctional monomer, preferably allyl methacrylate and triallyl isocyanurate.
- the shell layer may be formed including other monomer components in addition to the monomer components.
- the content of the epoxy group in the shell layer of the polymer fine particle (B) component (content per 1 g of the shell layer forming component) is preferably 0.05 to 3.5 mmol / g, preferably 0.1 to 2.0 mmol / g. Is more preferable, 0.2 to 1.0 mmol / g is further preferable, and 0.3 to 0.7 mmol / g is particularly preferable. If the content of the epoxy group in the shell layer is less than 0.05 mmol / g, the mechanical strength of the cured product obtained by curing the curable composition may be lowered. When the content of the epoxy group in the shell layer exceeds 3.5 mmol / g, the viscosity of the composition after storage tends to increase.
- the graft ratio of the shell layer is preferably 70% or more (more preferably 80% or more, and further 90% or more). When the graft ratio is less than 70%, the viscosity of the liquid resin composition may increase.
- the method for calculating the graft ratio is as follows.
- an aqueous latex containing polymer fine particles is coagulated and dehydrated, and finally dried to obtain polymer fine particle powder.
- the MEK soluble component is separated from the MEK insoluble component, and the methanol insoluble component is further separated from the MEK soluble component.
- a graft ratio is calculated by calculating
- ⁇ Method for producing polymer fine particles (Manufacturing method of core layer)
- Formation of the core layer constituting the polymer fine particles used in the present invention can be produced, for example, by emulsion polymerization, suspension polymerization, microsuspension polymerization or the like, and for example, the method described in WO2005 / 028546 pamphlet can be used.
- the polymerization of the core layer forming monomer is preferably carried out by an emulsion polymerization method.
- the intermediate layer can be formed by polymerizing the monomer for forming the intermediate layer by a known radical polymerization.
- the polymerization of the intermediate layer forming monomer is preferably carried out by an emulsion polymerization method.
- the shell layer can be formed by polymerizing a shell layer forming monomer by known radical polymerization.
- the polymerization of the monomer for forming the shell layer is preferably performed by an emulsion polymerization method, for example, WO2005 / 028546 pamphlet.
- alkyl or aryl sulfonic acid represented by dioctylsulfosuccinic acid and dodecylbenzenesulfonic acid
- alkyl or arylether sulfonic acid alkyl or aryl represented by dodecylsulfuric acid, and the like.
- emulsifier dispersant
- an emulsifier (dispersant) is so preferable that the water solubility is high. If the water solubility is high, the emulsifier (dispersant) can be easily removed by washing with water, and adverse effects on the finally obtained cured product can be easily prevented.
- a known initiator that is, 2,2′-azobisisobutyronitrile, hydrogen peroxide, potassium persulfate, ammonium persulfate, or the like can be used as the thermal decomposition type initiator.
- organic peroxides such as t-butylperoxyisopropyl carbonate, paramentane hydroperoxide, cumene hydroperoxide, dicumyl peroxide, t-butyl hydroperoxide, di-t-butyl peroxide, t-hexyl peroxide, etc.
- Oxides such as inorganic peroxides such as hydrogen peroxide, potassium persulfate, and ammonium persulfate; reducing agents such as sodium formaldehyde sulfoxylate and glucose as necessary; and iron sulfate (II as necessary) ),
- a chelating agent such as disodium ethylenediaminetetraacetate if necessary, and a redox type initiator using a phosphorus-containing compound such as sodium pyrophosphate if necessary.
- the polymerization can be performed at a low temperature at which the peroxide is not substantially thermally decomposed, and the polymerization temperature can be set in a wide range, which is preferable.
- organic peroxides such as cumene hydroperoxide, dicumyl peroxide, and t-butyl hydroperoxide are preferably used as the redox initiator.
- the amount of the initiator used, or the redox type initiator is used, the amount of the reducing agent / transition metal salt / chelating agent used may be within a known range.
- a known chain transfer agent can be used within a known range.
- a surfactant can be used, but this is also within a known range.
- the conditions such as polymerization temperature, pressure and deoxygenation during the polymerization can be within the known ranges.
- the polymerization of the intermediate layer forming monomer may be performed in one stage or in two or more stages.
- the method of adding continuously, the core for forming the core layer in the reactor in which the monomer for forming the intermediate layer is previously charged For example, a method of performing polymerization after adding a polymer emulsion may be employed.
- an epoxy curing agent (C) can be used as necessary.
- the curable composition of the present invention is used as a one-component composition (such as a one-component curable composition)
- the adhesive is rapidly cured when heated to a temperature of 80 ° C. or higher, preferably 140 ° C. or higher.
- the epoxy curing agent (C) component is preferably selected.
- epoxy curing agent (C) component a component that is active by heating (sometimes referred to as a latent curing agent) can be used.
- a latent epoxy curing agent an N-containing curing agent such as a specific amine curing agent (including an imine curing agent) can be used, for example, boron trichloride / amine complex, boron trifluoride / amine.
- dicyandiamide isophthalic acid dihydrazide, adipic acid dihydrazide, or 4,4'-diaminodiphenylsulfone, and dicyandiamide is particularly preferable.
- the latent epoxy curing agent is preferable because the curable composition of the present invention can be made into one component.
- other amine-based curing agents including imine-based curing agents
- mercaptan-based curing agents room temperature-curable curing agents.
- an epoxy curing agent (C) component that exhibits activity at a relatively low temperature of about room temperature.
- Examples of the epoxy curing agent (C) component exhibiting activity at a relatively low temperature include chain aliphatic polyamines such as diethylenetriamine, triethylenetetramine, tetraethylenepentamine, dipropylenediamine, diethylaminopropylamine, and hexamethylenediamine.
- N-aminoethylpiverazine bis (4-amino-3-methylcyclohexyl) methane, mensendiamine, isophoronediamine, 4,4'-diaminodicyclohexylmethane, 3,9-bis (3-aminopropyl)- Cycloaliphatic polyamines such as 2,4,8,10-tetraoxaspiro [5.5] undecane (spiroacetal diamine), norbornanediamine, tricyclodecanediamine, 1,3-bisaminomethylcyclohexane; metaxylenediamine Such as fat Aliphatic aromatic amines; polyamine epoxy resin adducts, which are a reaction product of an epoxy resin and excess polyamine; ketimines, which are dehydration reaction products of polyamine and ketones such as methyl ethyl ketone and isobutyl methyl ketone; Polyamidoamines formed by condensation of dimers (dimer acids) and polyamine
- An amine-terminated polyether containing a polyether main chain and having an average of 1 to 4 (preferably 1.5 to 3) amino groups and / or imino groups per molecule is also used as component (C). It can.
- Commercially available amine-terminated polyethers include Huntsman's Jeffamine D-230, Jeffamine D-400, Jeffamine D-2000, Jeffamine D-4000, Jeffamine T-5000, and the like.
- an amine-terminated rubber containing a conjugated diene polymer main chain and having an average of 1 to 4 (more preferably 1.5 to 3) amino groups and / or imino groups per molecule is also (C ) Can be used as a component.
- the main chain of the rubber is preferably a polybutadiene homopolymer or copolymer, more preferably a polybutadiene / acrylonitrile copolymer, and an acrylonitrile monomer content of 5 to 40% by mass (more preferably 10 to 35% by mass, still more preferably 15 to Polybutadiene / acrylonitrile copolymers that are 30% by weight) are particularly preferred.
- Examples of commercially available amine-terminated rubbers include Hypro 1300X16 ATBN manufactured by CVC.
- amine curing agents that exhibit activity at a relatively low temperature such as room temperature
- polyamide amines, amine-terminated polyethers, and amine-terminated rubbers are more preferable.
- Polyamide amines, amine-terminated polyethers, and amine-terminated rubbers are more preferable. It is particularly preferable to use them in combination.
- curing agent (C) component acid anhydrides, phenols, etc. can be used as an epoxy hardening
- acid anhydrides include polysebacic acid polyanhydride, polyazeline acid polyanhydride, succinic anhydride, citraconic acid anhydride, itaconic acid anhydride, alkenyl-substituted succinic acid anhydride, dodecenyl succinic acid anhydride, maleic acid anhydride, Tricarballylic anhydride, nadic anhydride, methyl nadic anhydride, linoleic acid adduct with maleic anhydride, alkylated terminal alkylene tetrahydrophthalic anhydride, methyl tetrahydrophthalic anhydride, tetrahydrophthalic anhydride, hexahydro Phthalic anhydride, pyromellitic dianhydride, trimellitic anhydride, phthalic anhydride, tetrachlorophthalic anhydride, tetrabromophthalic anhydride, dichloromaleic anhydride,
- the epoxy curing agent (C) component may be used alone or in combination of two or more.
- the epoxy curing agent (C) component is used in an amount sufficient to cure the composition. Typically, sufficient curing agent is provided to consume at least 80% of the epoxide groups present in the composition. A large excess over that required for consumption of epoxide groups is usually not necessary.
- the amount of the epoxy curing agent (C) component used is preferably 1 to 200 parts by weight, more preferably 2 to 170 parts by weight, with respect to 100 parts by weight of the flexible epoxy resin (A) component. Is more preferable, and 5 to 120 parts by mass is particularly preferable. If it is less than 1 mass part, the sclerosis
- the curing accelerator is a catalyst for accelerating the reaction between the epoxy group and the epoxide reactive group on the other components of the curing agent and the adhesive.
- the curing accelerator include p-chlorophenyl-N, N-dimethylurea (trade name: Monuron), 3-phenyl-1,1-dimethylurea (trade name: Phenuron), 3,4-dichlorophenyl-N, N-dimethylurea (trade name: Diuron), N- (3-chloro-4-methylphenyl) -N ′, N′-dimethylurea (trade name: Chlortoluron), 1,1-dimethylphenylurea (trade name: Ureas such as Dyhard); benzyldimethylamine, 2,4,6-tris (dimethylaminomethyl) phenol, 2- (dimethylaminomethyl) phenol, 2,4 embedded in a poly (
- the catalyst may be encapsulated or may be a potential one that becomes active only when the temperature is raised.
- tertiary amines and imidazoles can improve curing speed, physical properties of cured products, heat resistance, and the like when used in combination with an amine curing agent of the epoxy curing agent (C) component.
- a hardening accelerator may be used independently and may be used together 2 or more types.
- the amount of the curing accelerator used is preferably 0.1 to 10 parts by weight, more preferably 0.2 to 5 parts by weight, and still more preferably 0.5 to 3 parts by weight with respect to 100 parts by weight of component (A). 0.8 to 2 parts by mass is particularly preferable. If it is less than 0.1 mass part, the sclerosis
- silicic acid and / or silicate can be added as an inorganic filler.
- specific examples include dry silica, wet silica, aluminum silicate, magnesium silicate, calcium silicate, wollastonite, talc, and the like.
- the dry silica, also called fumed silica is a surface-untreated hydrophilic fumed silica and a hydrophobic fumed silica produced by chemically treating the silanol group portion of the hydrophilic fumed silica with silane or siloxane. In view of dispersibility in the component (A), hydrophobic fumed silica is preferable.
- inorganic fillers include reinforcing fillers such as dolomite and carbon black; colloidal calcium carbonate, heavy calcium carbonate, magnesium carbonate, titanium oxide, ferric oxide, aluminum fine powder, zinc oxide, activated zinc white, etc. Is mentioned.
- the inorganic filler is preferably surface-treated with a surface treatment agent. The dispersibility of the inorganic filler in the composition is improved by the surface treatment, and as a result, the mechanical properties of the obtained cured product are improved.
- the amount of the inorganic filler used is preferably 1 to 100 parts by weight, more preferably 2 to 70 parts by weight, still more preferably 5 to 40 parts by weight with respect to 100 parts by weight of the flexible epoxy resin (A) component. 7 to 20 parts by mass is particularly preferable.
- An inorganic filler may be used independently and may be used together 2 or more types.
- Calcium oxide can be added to the curable composition of the present invention.
- Calcium oxide removes moisture by reaction with moisture in the curable composition and solves various physical property problems caused by the presence of moisture. For example, it functions as an anti-bubble agent due to moisture removal and suppresses a decrease in adhesive strength.
- Calcium oxide can be surface treated with a surface treating agent. The surface treatment improves the dispersibility of the calcium oxide in the composition. As a result, the mechanical properties of the obtained cured product are improved as compared with the case where calcium oxide not subjected to surface treatment is used.
- the surface treatment agent is not particularly limited, but is preferably a fatty acid.
- the amount of calcium oxide used is preferably 0.1 to 10 parts by mass, more preferably 0.2 to 5 parts by mass, and 0.5 to 3 parts by mass with respect to 100 parts by mass of the flexible epoxy resin (A) component. Part is more preferable, and 1 to 2 parts by mass is particularly preferable. If the amount is less than 0.1 parts by mass, the moisture removing effect may not be sufficient. If the amount is more than 10 parts by mass, the tensile strength of the resulting cured product may be low. Calcium oxide may be used alone or in combination of two or more.
- a monoepoxide can be used as necessary.
- the monoepoxide can function as a reactive diluent.
- Specific examples of monoepoxides include aliphatic glycidyl ethers such as butyl glycidyl ether, or aromatic glycidyl ethers such as phenyl glycidyl ether and cresyl glycidyl ether, such as 2-ethylhexyl glycidyl ether.
- Ethers consisting of alkyl groups and glycidyl groups, for example ethers consisting of phenyl groups having 6 to 12 carbon atoms and glycidyl groups which can be substituted by alkyl groups having 2 to 8 carbon atoms such as p-tertbutylphenylglycidyl ether, such as dodecyl Ethers composed of alkyl groups having 12 to 14 carbon atoms such as glycidyl ether and glycidyl groups; for example, aliphatic glycidyl esters such as glycidyl (meth) acrylate and glycidyl maleate; Glycol ester, neodecanoic acid glycidyl ester, glycidyl esters of aliphatic carboxylic acids having 8 to 12 carbon atoms, such as lauric acid glycidyl ester; and p-t-butylbenzoic acid glycidyl
- the amount used is preferably 0.1 to 20 parts by weight, more preferably 0.5 to 10 parts by weight, based on 100 parts by weight of the flexible epoxy resin (A) component. ⁇ 5 parts by mass is particularly preferred. If the amount is less than 0.1 parts by mass, the effect of reducing the viscosity may not be sufficient. If the amount is more than 20 parts by mass, physical properties such as adhesiveness may be deteriorated.
- a silane coupling agent can be added as necessary. Adhesiveness can be improved by adding a silane coupling agent. Specific examples include ⁇ -isocyanatopropyltrimethoxysilane, ⁇ -isocyanatopropyltriethoxysilane, ⁇ -isocyanatopropylmethyldiethoxysilane, ⁇ -isocyanatopropylmethyldimethoxysilane, (isocyanatemethyl) trimethoxysilane, and (isocyanatemethyl).
- Isocyanate group-containing silanes such as dimethoxymethylsilane, (isocyanatemethyl) triethoxysilane, (isocyanatemethyl) diethoxymethylsilane; ⁇ -aminopropyltrimethoxysilane, ⁇ -aminopropyltriethoxysilane, ⁇ -aminopropyltriiso Propoxysilane, ⁇ -aminopropylmethyldimethoxysilane, ⁇ -aminopropylmethyldiethoxysilane, ⁇ - (2-aminoethyl) aminopropyltrime Xysilane, ⁇ - (2-aminoethyl) aminopropylmethyldimethoxysilane, ⁇ - (2-aminoethyl) aminopropyltriethoxysilane, ⁇ - (2-aminoethyl) aminopropylmethyldiethoxysilane, ⁇ - (2- Aminoethyl)
- silanes such as N- (1,3-dimethylbutylidene) -3- (triethoxysilyl) -1-propanamine; ⁇ -mercaptopropyltrimethoxysilane, ⁇ -mercaptopropyltri Mercapto group-containing silanes such as ethoxysilane, ⁇ -mercaptopropylmethyldimethoxysilane, ⁇ -mercaptopropylmethyldiethoxysilane, mercaptomethyltrimethoxysilane, mercaptomethyltriethoxysilane; ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -glycidoxypropyltriethoxysilane, ⁇ -glycidoxypropylmethyldimethoxysilane, ⁇ - (3,4-epoxycyclohexyl) ethyltrimethoxysilane, ⁇ - (3,4-epoxy
- Epoxy group-containing silanes such as ⁇ -carboxyethyltriethoxysilane, ⁇ -carboxyethylphenylbis (2-methoxyethoxy) silane, N- ⁇ - (carboxymethyl) aminoethyl- ⁇ -aminopropyltrimethoxysilane
- Silanes Vinyl type unsaturated such as vinyltrimethoxysilane, vinyltriethoxysilane, ⁇ -methacryloyloxypropylmethyldimethoxysilane, ⁇ -acryloyloxypropyltriethoxysilane, methacryloyloxymethyltrimethoxysilane It can be mentioned tris (3-trimethoxysilylpropyl) isocyanurate silanes such as isocyanurate; containing silanes; .gamma.
- halogen-containing silanes such as chloropropyl trimethoxy silane.
- the reaction product of aminosilane and epoxysilane, the reaction product of aminosilane and isocyanate silane, partial condensates of various silane coupling agents, and the like can also be used as the silane coupling agent.
- the amount used is preferably 0.1 to 15 parts by weight, more preferably 0.5 to 10 parts by weight, based on 100 parts by weight of the flexible epoxy resin (A) component. 1 to 5 parts by mass is particularly preferred. If the blending amount is below this range, the adhesion may not be sufficient. On the other hand, if the blending amount exceeds this range, the strength of the cured product may decrease.
- compounding component can be used as needed.
- Other compounding components include azo-type chemical foaming agents and expansion agents such as thermally expandable microballoons, fiber pulps such as aramid pulp, colorants such as pigments and dyes, extenders, ultraviolet absorbers, antioxidants, Radical curable resins, photopolymerization initiators, stabilizers (anti-gelling agents), plasticizers, leveling agents, antifoaming agents, antistatic agents, flame retardants, lubricants, viscosity reducers, low shrinkage agents, organic fillers , Thermoplastic resin, desiccant, dispersant and the like.
- azo-type chemical foaming agents and expansion agents such as thermally expandable microballoons, fiber pulps such as aramid pulp, colorants such as pigments and dyes, extenders, ultraviolet absorbers, antioxidants, Radical curable resins, photopolymerization initiators, stabilizers (anti-gelling agents), plasticizers, leveling agents, antifoaming agents,
- the curable composition of the present invention contains polymer fine particles (B) in a curable composition having a flexible epoxy resin (A) component as a resin main component (50% by mass or more in 100% by mass of resin).
- a curable composition having a flexible epoxy resin (A) component as a resin main component 50% by mass or more in 100% by mass of resin.
- it is a composition in which the polymer fine particles (B) are dispersed in the form of primary particles.
- polymer fine particles (B) obtained in an aqueous latex state are flexible.
- a method of removing unnecessary components such as water after contact with the conductive epoxy resin (A) component, and after mixing the polymer fine particles (B) with the flexible epoxy resin (A) component after extraction into an organic solvent
- the method of removing an organic solvent etc. is mentioned, It is preferable to utilize the method as described in WO2005 / 028546 pamphlet.
- an aqueous latex containing polymer fine particles (B) (specifically, a reaction mixture after producing polymer fine particles by emulsion polymerization) has a solubility in water at 20 ° C. of 5% by mass.
- the first step of aggregating the polymer particles, and separating and collecting the agglomerated polymer fine particles (B) from the liquid phase A second step of obtaining an organic solvent solution of polymer fine particles (B) by mixing with an organic solvent, and further mixing the organic solvent solution with a flexible epoxy resin (A) component, followed by distilling off the organic solvent. It is preferable to prepare including three steps.
- the flexible epoxy resin (A) component is preferably in a liquid state at 23 ° C. because the third step becomes easy. “Liquid at 23 ° C.” means that the softening point is 23 ° C. or lower, and indicates fluidity at 23 ° C.
- the flexible epoxy resin (A) component and the epoxy curing agent, which are obtained through the above steps, are further dispersed in the composition in which the polymer fine particles (B) are dispersed in the state of primary particles in the flexible epoxy resin (A) component.
- the curability of the present invention in which the polymer fine particles (B) are dispersed in the form of primary particles by further mixing at least one selected from the component (C) and each component of the other blending components as necessary. A composition is obtained.
- the powdered polymer fine particles (B) obtained by coagulation by a method such as salting out and the like are dried using a disperser having high mechanical shearing force such as three paint rolls, a roll mill, and a kneader.
- a disperser having high mechanical shearing force such as three paint rolls, a roll mill, and a kneader.
- the flexible epoxy resin (A) component and the polymer fine particle (B) component efficiently disperse the polymer fine particle (B) component by applying a mechanical shearing force at a high temperature.
- the temperature at the time of dispersion is preferably 50 to 200 ° C, more preferably 70 to 170 ° C, still more preferably 80 to 150 ° C, and particularly preferably 90 to 120 ° C.
- the curable composition of the present invention can be used as a one-component curable composition that is prepared by preliminarily blending all blending components and then stored in a hermetically sealed state and cured by heating or light irradiation after coating.
- a flexible epoxy resin (A) component as a main component
- a liquid A containing a polymer fine particle (B) component, an epoxy curing agent (C) component and a curing accelerator and further if necessary.
- Prepared as a two-component or multi-component curable composition comprising a separately prepared B liquid containing the polymer fine particle (B) component, and the A liquid and the B liquid are mixed before use, It can also be used.
- the polymer fine particle (B) component and the epoxy curing agent (C) component only have to be contained in at least one of the liquid A and the liquid B, for example, only the liquid A, only the liquid B, It may be contained in both the liquid and the B liquid.
- the present invention includes a cured product obtained by curing the curable composition.
- a curable composition in which polymer fine particles are dispersed in the form of primary particles a cured product in which polymer fine particles are uniformly dispersed can be easily obtained by curing the composition.
- the polymer fine particles hardly swell and the viscosity of the curable composition is low, a cured product can be obtained with good workability.
- the curable composition of the present invention can be applied by any method. It can be applied at a low temperature of about room temperature, and can be applied by heating as necessary.
- the curable composition of the present invention is extruded onto a substrate in the form of a bead, monofilament or swirl using a coating robot, or a mechanical coating method such as a caulking gun or other manual coating means. You can also.
- the composition can also be applied to the substrate using a jet spray method or a streaming method. Bonding is performed by applying the curable composition of the present invention to one or both substrates, bringing the curable composition into contact between the substrates to be bonded, and curing the substrates.
- the viscosity of the curable composition is not particularly limited, and is preferably about 150 to 600 Pa ⁇ s at 45 ° C. in the extrusion bead method, and preferably about 100 Pa ⁇ s at 45 ° C. in the swirl coating method. In a high volume coating method using a high-speed fluidizer, a pressure of about 20 to 400 Pa ⁇ s at 45 ° C. is preferable.
- ⁇ Laminated adhesive substrate> wood, metal, plastic, glass, etc. can be bonded, preferably, such as cold rolled steel plate or hot dip galvanized steel plate Steel plates, aluminum alloy plates such as aluminum alloys and coated aluminum alloys, titanium alloy plates, magnesium alloy plates, general purpose plastics, resin plates such as engineering plastics, composites such as carbon fiber reinforced plastic (CFRP) and glass fiber reinforced plastic (GFRP) Various plastic substrates such as substrates can be bonded. These substrates may be formed from different materials.
- the curable composition of the present invention is suitable for joining different kinds of substrates having different linear expansion coefficients in order to be excellent in elongation physical properties.
- the substrates are bonded together using the curable composition of the present invention, it is preferable to join the automobile parts, more preferably the joining of the automobile frames or the joining of the automobile frame and other automobile parts. More preferably, the exterior panel is manufactured by the following.
- the curable composition of the present invention can also be used for joining aerospace components, particularly exterior metal components (exterior panels).
- the curing temperature of the curable composition of the present invention is not particularly limited, but when used as a one-component curable composition, it is preferably 50 ° C. to 250 ° C., more preferably 80 ° C. to 220 ° C., and 100 It is more preferably from 200 ° C. to 200 ° C., particularly preferably from 130 ° C. to 180 ° C.
- it is not particularly limited, but is preferably 0 ° C to 150 ° C, more preferably 10 ° C to 100 ° C, still more preferably 15 ° C to 80 ° C, and more preferably 20 ° C to 60 ° C. ° C is particularly preferred.
- the curable composition of the present invention is used as an automobile adhesive, after the adhesive is applied to an automobile member, the coating is then applied, and the adhesive is cured at the same time as the coating is baked and cured. Is preferable from the viewpoint of process shortening and simplification.
- the curable composition of the present invention is cured in two stages, it is preferable from the viewpoint of process shortening and simplification.
- the curable composition is heated to 80 ° C. to 130 ° C. for a short time (for example, about 0.5 to 5 minutes) to partially cure the bonded substrates to the extent that they can be temporarily fixed (temporarily bonded).
- a short time for example, about 0.5 to 5 minutes
- the composition of the present invention includes structural adhesives for vehicles and aircraft, adhesives such as structural adhesives for wind power generation, paints, materials for lamination with glass fibers, printed wiring board materials, solder resists, interlayer insulation Films, build-up materials, adhesives for FPC, electrical insulation materials such as encapsulants for electronic components such as semiconductors and LEDs, die bond materials, underfills, semiconductor mounting materials such as ACF, ACP, NCF, NCP, liquid crystal panels, OLEDs It is preferably used for a sealing device for display devices and lighting devices such as lighting and OLED displays. In particular, it is useful as a structural adhesive for vehicles.
- adhesives such as structural adhesives for wind power generation, paints, materials for lamination with glass fibers, printed wiring board materials, solder resists, interlayer insulation Films, build-up materials, adhesives for FPC, electrical insulation materials such as encapsulants for electronic components such as semiconductors and LEDs, die bond materials, underfills, semiconductor mounting materials such as ACF, ACP, NCF, NCP,
- the glass transition temperature of the core layer of the polymer fine particle (B) component in the curable composition is the Kelvin temperature (K) from Equation (1). Calculated and converted to Celsius temperature (° C.).
- K Kelvin temperature
- the Tg of the homopolymer of each non-crosslinkable monomer used at that time was as follows: methyl methacrylate 378K, butyl acrylate 219K, butyl methacrylate 293K, butadiene 188K, styrene 373K.
- M i is the weight fraction of each monomer i component selected from butadiene and non-crosslinkable monomers constituting the core layer of the polymer fine particle (B) component
- T g i is the Represents the glass transition temperature (K) of the homopolymer.
- a core layer monomer methyl methacrylate (MMA) 87 parts by mass, allyl methacrylate (ALMA) 0.4 parts by mass
- CHP cumene hydroperoxide 0.13 parts by mass
- 20 parts by mass of a 5% by mass aqueous solution of SDS was continuously added over 3 hours. Stirring was continued for 1 hour from the end of the monomer mixture addition to complete the polymerization, and a latex containing acrylic polymer fine particles was obtained.
- a mixture of a graft monomer (MMA 3 parts by mass, butyl acrylate (BA) 10 parts by mass) and CHP 0.07 parts by mass was continuously added thereto over 120 minutes.
- 0.07 part by mass of CHP was added, and stirring was further continued for 2 hours to complete the polymerization, thereby obtaining a latex (L-1) containing core-shell polymer fine particles.
- the polymerization conversion rate of the monomer component was 99% or more.
- the volume average particle diameter of the core-shell polymer fine particles contained in the obtained latex was 0.36 ⁇ m.
- Production Example 1-2 Preparation of Core Shell Polymer Latex (L-2)
- BA 10 parts by mass> as a graft monomer
- ⁇ MMA 2 parts by mass BA 10 parts by mass
- GMA glycidyl methacrylate
- a core-shell polymer latex (L-2) was obtained in the same manner as in Production Example 1-1 except that 1 part by mass> was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.36 ⁇ m.
- Production Example 1-3 Preparation of Core Shell Polymer Latex (L-3)
- ⁇ MMA 87 parts by mass ALMA 0.4 parts by mass> as a monomer for the core layer
- ⁇ MMA 87 parts by mass ALMA 4 parts by mass
- ⁇ MMA 2 parts by mass BA 10 parts by mass, GMA 1 part by mass> was used instead of ⁇ MMA 3 parts by mass, BA 10 parts by mass> as the graft monomer.
- Latex (L-3) was obtained.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.35 ⁇ m.
- Production Example 1-4 Preparation of Core Shell Polymer Latex (L-4)
- ⁇ MMA 87 parts by mass ALMA 0.4 parts by mass> as a monomer for the core layer
- ⁇ MMA 72 parts by mass BA 15 parts by mass , 4 parts by weight of ALMA was used
- ⁇ MMA 2 parts by weight BA 10 parts by weight, GMA 1 part by weight> was used instead of ⁇ MMA 3 parts by weight, BA 10 parts by weight> as the graft monomer, in the same manner as in Production Example 1-1.
- a core-shell polymer latex (L-4) was obtained.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.31 ⁇ m.
- Production Example 1-5 Preparation of Core-Shell Polymer Latex (L-5)
- ⁇ MMA 87 parts by mass ALMA 0.4 parts by mass> as a monomer for the core layer
- ⁇ MMA 57 parts by mass BA 30 parts by mass , 4 parts by weight of ALMA was used
- ⁇ MMA 2 parts by weight BA 10 parts by weight, GMA 1 part by weight> was used instead of ⁇ MMA 3 parts by weight, BA 10 parts by weight> as the graft monomer, in the same manner as in Production Example 1-1.
- a core-shell polymer latex (L-5) was obtained.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.35 ⁇ m.
- Production Example 1-6 Preparation of Core Shell Polymer Latex (L-6)
- ALMA 0.4 parts by mass> as a monomer for the core layer
- BA 30 parts by mass A core-shell polymer latex (L-6) was obtained in the same manner as in Production Example 1-1 except that 4 parts by mass of ALMA was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.37 ⁇ m.
- Production Example 1-7 Preparation of Core Shell Polymer Latex (L-7)
- L-7 Preparation of Core Shell Polymer Latex (L-7)
- ALMA 0.4 parts by mass> as a monomer for the core layer
- BA 30 parts by mass 4 parts by mass of ALMA
- ⁇ MMA 1 part by mass BA 10 parts by mass, GMA 2 parts by mass> instead of ⁇ MMA 3 parts by mass, BA 10 parts by mass> as the graft monomer.
- a core-shell polymer latex (L-7) was obtained.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.35 ⁇ m.
- Production Example 1-8 Preparation of Core-Shell Polymer Latex (L-8)
- the initial charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-8) was obtained in the same manner as in Production Example 1-1, except that 6 parts by mass, 13.1 parts by mass of BA, and 1.3 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.09 ⁇ m.
- Production Example 1-9 Preparation of Core-Shell Polymer Latex (L-9)
- the initially charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- ⁇ MMA 66.2 parts by mass, BA 13.8 parts by mass, ALMA 4 parts by mass> are used in place of ⁇ MMA 3 parts by mass, BA 10 parts by mass> instead of ⁇ MMA 3 parts by mass>.
- a core-shell polymer latex (L-9) was obtained in the same manner as in Production Example 1-1, except that 1 part by mass, 15.4 parts by mass of BA, and 1.5 parts by mass of GMA were used.
- the volume average particle size of the core-shell polymer contained in the obtained latex was 0.10 ⁇ m.
- Production Example 1-10 Preparation of Core-Shell Polymer Latex (L-10)
- the initially charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-10) was obtained in the same manner as in Production Example 1-1 except that 5 parts by mass, BA 17.7 parts by mass, and GMA 1.8 parts by mass> were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.09 ⁇ m.
- Production Example 1-11 Preparation of Core-Shell Polymer Latex (L-11)
- the first charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- the core-shell polymer latex (L-11) was obtained in the same manner as in Production Example 1-1, except that the following were used.
- the volume average particle size of the core-shell polymer contained in the obtained latex was 0.10 ⁇ m.
- Production Example 1-12 Preparation of Core-Shell Polymer Latex (L-12)
- the first charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the core layer monomer ⁇ MMA87 ⁇ MMA 52.4 parts by mass, BA 27.6 parts by mass, ALMA 4 parts by mass> instead of ⁇ mass part, ALMA 0.4 parts by mass>, and ⁇ MMA 3 parts by mass instead of ⁇ MMA 3 parts by mass, BA 10 parts by mass> as a graft monomer
- a core-shell polymer latex (L-12) was obtained in the same manner as in Production Example 1-1 except that 1 part by mass, 15.5 parts by mass of BA, and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-13 Preparation of Core Shell Polymer Latex (L-13)
- the first charged SDS was changed to 1.5 parts by mass instead of 0.01 part by mass
- the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-13) was obtained in the same manner as in Production Example 1-1, except that 1 part by mass, 15.5 parts by mass of BA, and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.06 ⁇ m.
- Production Example 1-14 Preparation of Core Shell Polymer Latex (L-14)
- the initially charged SDS was set to 3.0 parts by mass instead of 0.01 part by mass
- the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-14) was obtained in the same manner as in Production Example 1-1 except that 1 part by mass, 15.5 parts by mass of BA, and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.05 ⁇ m.
- Production Example 1-15 Preparation of Core-Shell Polymer Latex (L-15)
- the initial charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- ALMA 4 parts by mass> is used instead of mass parts, ALMA 0.4 parts by mass>, and ⁇ MMA 3 parts by mass, BA 10 parts by mass> as a graft monomer.
- a core-shell polymer latex (L-15) was obtained in the same manner as in Production Example 1-1 except that ⁇ MMA 3 parts by mass, BA 15.5 parts by mass, GMA 1.5 parts by mass> were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-16 Preparation of Core-Shell Polymer Latex (L-16)
- the first charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass
- the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-16) was obtained in the same manner as in Production Example 1-1 except that 1 part by mass, 15.5 parts by mass of BMA, and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-17 Preparation of Core-Shell Polymer Latex (L-17)
- the initially charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- a core-shell polymer latex (L-17) was obtained in the same manner as in Production Example 1-1, except that 5 parts by mass and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.09 ⁇ m.
- Production Example 1-18 Preparation of Core-Shell Polymer Latex (L-18)
- the initially charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the monomer for core layer was ⁇ MMA87.
- the core-shell polymer latex (L-18) was obtained in the same manner as in Production Example 1-1 except that 1 part by mass, 15.5 parts by mass of BA, and 1.5 parts by mass of GMA were used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-19 Preparation of Core-Shell Polymer Latex (L-19)
- the first charged SDS was changed to 0.4 parts by mass instead of 0.01 parts by mass, and the core layer monomer ⁇ MMA87 Instead of ⁇ mass part, ALMA 0.4 part by mass> instead of ⁇ BA 80 part by mass, ALMA 4 part by mass>, instead of ⁇ MMA 3 part by mass, BA 10 part by mass>, ⁇ MMA 3 part by mass, BA 15.5 part by mass>
- a core-shell polymer latex (L-19) was obtained in the same manner as in Production Example 1-1 except that 1.5 parts by mass of GMA was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-20 Preparation of core-shell polymer latex (L-20) In a pressure-resistant polymerization machine, 200 parts by mass of deionized water, 0.03 parts by mass of tripotassium phosphate, 0.002 parts by mass of EDTA, 0.001 part by mass of FE Then, after adding 1.55 parts by mass of SDS and sufficiently purging nitrogen with stirring to remove oxygen, 100 parts by mass of butadiene (BD) was introduced into the system, and the temperature was raised to 45 ° C. Polymerization was started by adding 0.03 parts by mass of paramentane hydroperoxide (PHP) and subsequently 0.10 parts by mass of SFS. 0.025 parts by mass of PHP was added at 3, 5, and 7 hours from the start of polymerization.
- PPP paramentane hydroperoxide
- EDTA 0.0006 parts by mass and FE 0.003 parts by mass were added to polymerization at 4, 6 and 8 hours, respectively.
- the residual monomer was removed by devolatilization under reduced pressure to complete the polymerization, and a polybutadiene rubber latex (R-1) containing polybutadiene rubber as a main component was obtained.
- the volume average particle diameter of the polybutadiene rubber particles contained in the obtained latex was 0.08 ⁇ m.
- aqueous latex L-20 containing a core-shell polymer.
- the polymerization conversion rate of the monomer component was 99% or more.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.09 ⁇ m.
- Production Example 1-21 Preparation of Core-Shell Polymer Latex (L-21) Production Example 1-20 was conducted except that, instead of 100 parts by mass of BD, 75 parts by mass of BD and 25 parts by mass of styrene (ST) were added to the system. In the same manner as in Example 1-20, a styrene-butadiene rubber latex (R-2) was obtained. The volume average particle diameter of the styrene-butadiene rubber particles contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-22 Preparation of Core Shell Polymer Latex (L-22) 21 parts by mass of polybutadiene rubber latex (R-1) obtained in Production Example 1-20 (including 7 parts by mass of polybutadiene rubber) in a pressure-resistant polymerization machine ), 186 parts by mass of deionized water, 0.03 part by mass of tripotassium phosphate, 0.002 part by mass of EDTA, and 0.001 part by mass of FE were added, and after substituting with nitrogen sufficiently to remove oxygen. , 93 parts by mass of BD was added to the system, and the temperature was raised to 45 ° C. Polymerization was started by adding 0.02 parts by weight of PHP and subsequently 0.10 parts by weight of SFS.
- aqueous latex (L-22) containing a core-shell polymer 0.07 part by mass of CHP was added, and stirring was further continued for 2 hours to complete the polymerization, thereby obtaining an aqueous latex (L-22) containing a core-shell polymer.
- the polymerization conversion rate of the monomer component was 99% or more.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- Production Example 1-23 Preparation of Core Shell Polymer Latex (L-23)
- a core-shell polymer latex (L-23) was obtained in the same manner as in Production Example 1-22 except that> was used.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- Production Example 1-24 Preparation of Core Shell Polymer Latex (L-24)
- ⁇ BA 6 parts by mass, GMA 4 parts by mass> was used instead of ⁇ MMA 4 parts by mass, BA 6 parts by mass> as the graft monomer.
- a core-shell polymer latex (L-24) was obtained in the same manner as in Production Example 1-22.
- the volume average particle diameter of the core-shell polymer contained in the obtained latex was 0.21 ⁇ m.
- Production Example 2-1 instead of the core-shell polymer latex (L-1), Production Example 1-2 or A dispersion in which polymer fine particles are dispersed in a flexible epoxy resin in the same manner as in Production Example 2-1 except that the latex (L-2) or latex (L-3) obtained in 1-3 is used. M-2 or M-3) was obtained.
- Production Example 2-14 Preparation of Dispersion (M-14) In Production Example 2-1, instead of the core-shell polymer latex (L-1), the latex (L-12) obtained in Production Example 1-12 was used. In the same manner as in Production Example 2-1, except that 40 g of the flexible epoxy resin (A-1) was mixed instead of 80 g, a dispersion of polymer fine particles dispersed in the flexible epoxy resin (M -14) was obtained.
- the core-shell polymer latex ( In place of L-1), the latexes (L-13) to (L-19) and (L-22) obtained in Preparation Examples 1-13 to 1-19 and 1-22 to 1-24, respectively, (L-24) was used, and polymer fine particles were dispersed in the flexible epoxy resin in the same manner as in Production Example 2-1, except that 60 g of the flexible epoxy resin (A-1) was mixed instead of 80 g.
- Dispersions (M-15 to M-21 and M-24 to M-26) were obtained.
- Production Example 2-1 instead of core-shell polymer latex (L-1), Production Example 1-20 or A dispersion (M) in which polymer fine particles are dispersed in a flexible epoxy resin in the same manner as in Production Example 2-1, except that the latex (L-20) or (L-21) obtained in 1-21 was used. -22 or M-23).
- Table 1 shows the “content of polymer fine particles (B) in the dispersion (M)”. (Weight%) ”,“ core layer composition (parts by weight) of polymer fine particles (B) ”,“ shell layer composition (parts by weight) of polymer fine particles (B) ”,“ particle diameter ( ⁇ m) of polymer fine particles (B) ” ”,“ Tg (° C.) of core layer of polymer fine particle (B) ”,“ Content of epoxy group in shell layer of polymer fine particle (B) (mmol / g) ”,“ 25 ° C. of dispersion (M) ” Or “viscosity at 50 ° C. (Pa ⁇ s)”.
- Made by CVC, Hypro 1300X16 ATBN: Amino group-terminated butadiene-acrylonitrile copolymer), fumed silica (CABOT, CAB-O-SIL TS-720: fumed silica surface-treated with polydimethylsiloxane), heavy calcium carbonate (Shiraishi calcium Manufactured and Whiten SB) were weighed, mixed well and defoamed to obtain a curable composition.
- the cured product obtained by curing the curable composition of the present invention exhibits rubber elasticity, high elongation, and high strength (maximum tensile stress).
- the amount of the flexible epoxy resin (A) component contained in the curable compositions in Tables 2 and 3 and Tables 4, 5, 7 and 8 to be described later is the components added as the epoxy resin and the dispersion of polymer fine particles It is the amount obtained by adding the component contained in the object (M).
- the cured product obtained by curing the curable composition of the present invention exhibits rubber elasticity, high elongation, and high strength (maximum tensile stress).
- Table 6 shows “polymer fine particles (B) in dispersion (M)”. “Content (% by weight)”, “Particle size ( ⁇ m) of polymer fine particles (B)” “Tg (° C.) of core layer of polymer fine particles (B)”, “Epoxy group in shell layer of polymer fine particles (B)” Content (mmol / g) ”and“ viscosity of dispersion (M) at 25 ° C. (Pa ⁇ s) ”.
- the dispersion (M-5) obtained in Production Example 2-5 or the dispersion obtained in Production Example 2-31 which is a mixture of a flexible epoxy resin (A) and polymer fine particles (B) (M-31) and epoxy curing agent (C-3, manufactured by Wako Pure Chemical Industries, Ltd., isophorone diamine (IPDA)), which are components (M-31) and (C), are weighed, mixed thoroughly, and defoamed to obtain a curable composition Obtained.
- Example 44 Comparative Examples 24-28
- a flexible epoxy resin (A-1 component having an epoxy equivalent of 410 g / eq) or a bisphenol A type epoxy resin (A-3, having an epoxy equivalent of 190 g / eq) as component (A)
- the dispersion (M-5) obtained in Production Example 2-5 which is a mixture of a flexible epoxy resin (A) and polymer fine particles (B), and an epoxy curing agent (C— 3; Wako Pure Chemical Industries, Ltd. (isophoronediamine (IPDA)) was weighed, mixed well, and defoamed to obtain a curable composition.
- IPDA isophoronediamine
- Example 44 Comparative Example 24, when the component (A) was used so that the type A durometer hardness was 95 or less at 23 ° C., it exhibited high elasticity and rubber elasticity, and polymer fine particles (B) It can be seen that the strength (maximum tensile stress) of the cured product is remarkably improved by the addition of.
- the curable composition of the present invention is useful as an adhesive, particularly as a structural rubber elastic adhesive, and can be preferably used as an adhesive between various substrates.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Laminated Bodies (AREA)
- Adhesives Or Adhesive Processes (AREA)
- Graft Or Block Polymers (AREA)
- Epoxy Resins (AREA)
Abstract
Description
エポキシ樹脂、ポリマー微粒子、エポキシ硬化剤(C)を含有する硬化性組成物において、
エポキシ樹脂として、得られる硬化物がゴム弾性を示す可とう性エポキシ樹脂(A)を主成分として使用し、コア層のガラス転移温度(Tg)が0℃より大きいコアシェル構造を有するポリマー微粒子(B)と組合せることにより前記課題を解決することを見出し、本発明を完成させた。
下記数式(1)より計算した(B)成分のコア層のガラス転移温度(Tg)が0℃より大きく、かつ、
前記硬化性組成物を硬化して得られる硬化物のJIS K6253-3に規定されるタイプAデュロメータ硬さが23℃で5~95であることを特徴とする硬化性組成物に関する。
1/Tg=Σ(Mi/Tgi) (1)
(式中、Miは(B)成分のコア層を構成するブタジエン及び非架橋性単量体から選ばれる各単量体i成分の重量分率、Tgiは各単量体iのホモポリマーのガラス転移温度(K)を表す。)。
2)さらに、(B)成分のコア層のガラス転移温度(Tg)は、15~150℃であることが好ましい。
3)さらに、(A)成分のエポキシ当量は、200~4000g/eqであることが好ましい。
4)さらに、(B)成分のコア層は、(メタ)アクリレート系重合体であることが好ましい。
5)さらに、(B)成分のコア層は、非架橋性単量体80~99質量%、および、架橋性単量体20~1質量%からなる単量体混合物を重合して得られる重合体であることが好ましい。
6)さらに、(B)成分のシェル層は、(メタ)アクリレート系重合体であることが好ましい。
7)さらに、(B)成分のシェル層は、エポキシ基を有することが好ましい。
8)さらに、(B)成分のシェル層中のエポキシ基の含有量は、0.05~3.5mmol/gであることが好ましい。
9)さらに、(B)成分は、エポキシ基を有するモノマー成分を、コア層にグラフト重合してなるシェル層を有することが好ましい。
10)さらに、(B)成分は、前記の硬化性組成物中で1次粒子の状態で分散していることが好ましい。
11)好ましくは、前記いずれかの硬化性組成物を硬化して得られる硬化物である。
12)好ましくは、前記いずれかの硬化性組成物を用いてなる接着剤である。
13)好ましくは、前記いずれかの硬化性組成物を用いてなる車両用接着剤である。
14)好ましくは、異なる材料からなる2つの基板が、前記いずれかの硬化性組成物で接合された積層接着基板である。
15)さらに前記積層接着基板は、外装パネルであることが好ましい。
16)さらに前記基板の少なくとも一方が、鋼板、アルミニウム合金板、チタニウム合金板、マグネシウム合金板、プラスチック系基板から選ばれる少なくとも1種であることが好ましい。
本発明の硬化性組成物は、可とう性エポキシ樹脂(A)100質量部と、コア層のガラス転移点が0℃より大きいコアシェル構造を有するポリマー微粒子(B)1~150質量部と、エポキシ硬化剤(C)1~200質量部と、を含有する。前記硬化性組成物を硬化して得られる硬化物は、ゴム弾性を示すことが必須である。具体的には、前記硬化物のJIS K6253-3に規定されるタイプAデュロメータ硬さは、23℃で5~95であることが必須であり、20~90であることが好ましく、40~87であることがより好ましく、50~85であることが特に好ましい。前記硬化物のタイプAデュロメータ硬さが、5未満では硬化物の引張強度が不十分となる場合があり、95を超えるとゴム弾性が不十分となり伸び特性が悪い場合がある。
本発明の硬化性組成物の主成分として、可とう性エポキシ樹脂(A)を使用する。
可とう性エポキシ樹脂(A)成分を用いることで、硬化して得られる硬化物が23℃でゴム弾性を示すようになる。可とう性エポキシ樹脂(A)は、成分の主鎖骨格の種類、エポキシ基の種類、1分子あたりのエポキシ基の個数、分子量の大きさ、分岐の有無などに関係なく、以下の条件で硬化して得られる硬化物のタイプAデュロメータ硬さの値が、1~95であるものが好ましい。
[可とう性エポキシ樹脂(A)を硬化して得られる硬化物のタイプAデュロメータ硬さ]エポキシ当量が(α)g/eqである可とう性エポキシ樹脂100質量部に対して、イソフォロンジアミン(1/α×8510)質量部を加えて混合した後、遠心脱泡した混合物を型枠に気泡が入らないように流し込み、23℃で24時間、さらに80℃で40時間養生させて得られる厚さ6mmの硬化物シートを、JIS K-6253に準拠したタイプAデュロメータ硬さ試験機を用いて、23℃50%RHで測定した値。
可とう性エポキシ樹脂としては、各種の可とう性エポキシ樹脂を使用することができ、例えば、ダイマー酸変性エポキシ樹脂等の脂肪族多塩基酸類等をエポキシ樹脂に付加反応させて得られる脂肪酸変性エポキシ樹脂、1,2-ポリブタジエンのビニル基の酸化によりエポキシ基を導入したエポキシ化ポリブタジエンなどのブタジエン系エポキシ樹脂、ジチオエーテル等でエポキシ樹脂を変性したチオール変性エポキシ樹脂、ポリオール変性エポキシ樹脂、ε-カプロラクトン変性エポキシ樹脂、ゴム変性エポキシ樹脂、ウレタン変性エポキシ樹脂、エポキシ基含有(メタ)アクリル系共重合体、等があげられる。これらエポキシ樹脂は単独で用いても良く2種以上併用しても良い。
前記ポリオール変性エポキシ樹脂としては、ビスフェノールAビス(ポリエチレングリコールグリシジルエーテル)エーテル等のビスフェノールAエチレンオキシド付加物のグリシジルエーテル、ビスフェノールAビス(ポリプロピレングリコールグリシジルエーテル)エーテル等のビスフェノールAプロピレンオキシド付加物のグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、ビスフェノール類(ビスフェノールA等)とビニルエーテル類(トリエチレングリコールジビニルエーテル等)とのアセタール化反応物のグリシジルエーテル、脂肪族アルコールまたは脂肪族ポリオールのグリシジルエーテル、などが挙げられる。
前記ε-カプロラクトン変性エポキシ樹脂としては、ε-カプロラクトン変性したビスフェノールA型エポキシ樹脂、ε-カプロラクトン変性した3’,4’-エポキシシクロヘキシルメチル3,4-エポキシシクロヘキサンカルボキシレート等のε-カプロラクトン変性二官能エポキシ樹脂、などが挙げられる。
前記ゴム変性エポキシ樹脂は、ゴムとエポキシ基含有化合物とを反応させて得た反応生成物であり、ゴムとしては,アクリロニトリルブタジエンゴム(NBR),スチレンブタジエンゴム(SBR)、水素添加ニトリルゴム(HNBR)、エチレンプロピレンゴム(EPDM)、アクリルゴム(ACM)、ブチルゴム(IIR)、ブタジエンゴム、ポリプロピレンオキシドやポリエチレンオキシドやポリテトラメチレンオキシド等のポリオキシアルキレン、などのゴム系重合体を挙げることができる。該ゴム系重合体は、アミノ基、ヒドロキシ基、またはカルボキシル基等の反応性基を末端に有するものが好ましい。これらのゴム系重合体とエポキシ樹脂とを公知の方法により適宜の配合比にて反応させた生成物が本発明に使用されるゴム変性エポキシ樹脂である。これらの中でも、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂や、ポリオキシアルキレン変性エポキシ樹脂が、得られる硬化性組成物の伸び特性や引張強度の観点から好ましく、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂がより好ましい。なお、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂は、例えば、カルボキシル基末端NBR(CTBN)とビスフェノールA型エポキシ樹脂との反応により得られる。
ゴム変性エポキシ樹脂のガラス転移温度(Tg)は、特に制限は無いが、-25℃以下が好ましく、-35℃以下がより好ましく、-40℃以下が更に好ましく、-50℃以下が特に好ましい。
ウレタン変性エポキシ樹脂は、イソシアネート基との反応性を有する基とエポキシ基とを含有する化合物と、イソシアネート基を含有するウレタンプレポリマーを反応させて得た反応生成物である。例えば、ヒドロキシ基含有エポキシ化合物とウレタンプレポリマーを反応させることにより、ウレタン変性エポキシ樹脂が得られる。
エポキシ基含有(メタ)アクリル系共重合体は、分子内にエポキシ基を有する(メタ)アクリル系重合体であり、エポキシ基含有モノマーとその他の(メタ)アクリル系モノマーとの共重合により得られる重合体が好ましい。具体的には、グリシジル(メタ)アクリレート等のエポキシ基含有モノマーとブチルアクリレートや2-エチルヘキシルアクリレート等との共重合体、などが挙げられる。
脂肪酸変性エポキシ樹脂は、脂肪族多塩基酸類等をエポキシ樹脂に付加反応させて得られるものである。前記脂肪族多塩基酸類としては、例えば、マレイン酸、無水マレイン酸、フマル酸、イタコン酸、無水イタコン酸、シトラコン酸、等の芳香環を有しない不飽和多価カルボン酸あるいはその無水物、または、テトラヒドロフタル酸、テトラヒドロ無水フタル酸、ヘキサヒドロフタル酸、ヘキサヒドロ無水フタル酸、シクロヘキサンジカルボン酸、コハク酸、マロン酸、グルタル酸、アジピン酸、アゼライン酸、セバシン酸、1,12-ドカン2酸、ダイマー酸、等の芳香環を有しない飽和多価カルボン酸あるいはその無水物などが挙げられる。得られる硬化物の伸び物性の点から、特にダイマー酸が好ましい。
前記の各種の可とう性エポキシ樹脂は、単独でまたは2種以上を組み合わせて使用することができる。
本発明の硬化性組成物の主成分として、可とう性エポキシ樹脂(A)を使用するが、本発明の効果を低下させない程度に、可とう性エポキシ樹脂以外のエポキシ樹脂(硬質のエポキシ樹脂)を少量併用することもできる。
本発明の硬化性組成物は、可とう性エポキシ樹脂(A)成分100質量部に対して、コアシェル構造を有するポリマー微粒子(B)1~150質量部を使用する。ポリマー微粒子(B)成分の添加により、得られる硬化物は機械強度に優れる。
得られる硬化性組成物の取扱いやすさと、得られる硬化物の機械強度改良効果のバランスから、可とう性エポキシ樹脂(A)成分100質量部に対して、ポリマー微粒子(B)成分は1~150質量部であり、2~120質量部が好ましく、3~100質量部がより好ましく、4~70質量部が更に好ましい。また30~70質量部、或いは40~70質量部であってもよい。
上述の特定の粒子径分布を容易に実現する観点から、ポリマー微粒子(B)成分の粒子径の個数分布において、極大値が2個以上存在することが好ましく、製造時の手間やコストの観点から、極大値が2~3個存在することがより好ましく、極大値が2個存在することが更に好ましい。特に、体積平均粒子径が10nm以上150nm未満のポリマー微粒子10~90質量%と、体積平均粒子径が150nm以上2000nm以下のポリマー微粒子90~10質量%を含むことが好ましい。
ポリマー微粒子(B)成分は単独で用いても良く2種以上併用しても良い。
以下、各層について具体的に説明する。
コア層は、本発明の硬化性組成物を硬化して得られる硬化物の機械強度を高める為に、下記数式(1)よりケルビン温度で算出し、セルシウス温度に換算したガラス転移温度(Tg)が0℃より大きいことが必須である。
1/Tg=Σ(Mi/Tgi) (1)
(式中、Miはポリマー微粒子(B)成分のコア層を構成するブタジエン及び非架橋性単量体から選ばれる各単量体i成分の重量分率、Tgiは各単量体iのホモポリマーのガラス転移温度(K)を表す。)
なお、非架橋性単量体のホモポリマーのガラス転移温度は、例えば、J.Brandrup著の「ポリマーハンドブック第4版(POLYMER HANDBOOK Fourth Edition)」等の文献やカタログにより確認することができる。
前記非架橋性単量体の具体例としては、例えば、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、オクチル(メタ)アクリレート、ドデシル(メタ)アクリレート、ステアリル(メタ)アクリレート、イソボルニル(メタ)アクリレート、ジシクロペンタニル(メタ)アクリレート、1-アダマンチル(メタ)アクリレート、ベヘニル(メタ)アクリレートなどのアルキル(メタ)アクリレート類;フェノキシエチル(メタ)アクリレート、ベンジル(メタ)アクリレートなどの芳香環含有(メタ)アクリレート類;2-ヒドロキシエチル(メタ)アクリレート、4-ヒドロキシブチル(メタ)アクリレートなどのヒドロキシアルキル(メタ)アクリレート類;グリシジル(メタ)アクリレート、グリシジルアルキル(メタ)アクリレートなどのグリシジル(メタ)アクリレート類;アルコキシアルキル(メタ)アクリレート類;スチレン、α-メチルスチレン、モノクロロスチレン、ジクロロスチレン、2-ビニルナフタレン、3-メチルスチレン、4-メチルスチレン、2,4-ジメチルスチレン、2,5-ジメチルスチレン、3,5-ジメチルスチレン、2,4,6―トリメチルスチレン、4-メトキシスチレン、4-エトキシスチレン、4-アセトキシスチレン、4-ヒトロキシスチレンなどのビニルアレーン類;ビニルベンゾエート、ビニルシクロヘキサノエート等のビニルエステル類;アクリルアミド、イソプロピルアクリルアミド、N-ビニルピロリドン等のアクリルアミド類;アクリル酸、メタクリル酸などのビニルカルボン酸類;アクリロニトリル、メタクリロニトリルなどのビニルシアン類;塩化ビニル、臭化ビニル、クロロプレンなどのハロゲン化ビニル類;酢酸ビニル;エチレン、プロピレン、ブチレン、イソブチレンなどのアルケン類;などが挙げられる。これらのモノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。これらの中でも、炭素数1~4のアルキルメタクリレート類、ビニルアレーン類、ビニルシアン類、は入手性が高く重合体のTgが高いため好ましい。メチルメタクリレート、エチルメタクリレート、イソプロピルメタクリレート、イソブチルメタクリレート、n-ブチルメタクリレート、t-ブチルメタクリレート、スチレン、α-メチルスチレン、アクリロニトリル、は入手性が高く重合体のTgが高いため特に好ましい。
コア層は、共役ジエン系モノマー(特に1,3-ブタジエン)を含まないのが好ましい。
本発明において、コア層は単層構造であることが多いが、多層構造であってもよい。また、コア層が多層構造の場合は、各層のポリマー組成が各々相違していてもよい。
本発明では、必要により、中間層を形成させてもよい。特に、中間層として、以下の表面架橋層を形成させてもよい。
前記表面架橋層は、同一分子内にラジカル性二重結合を2以上有する多官能性モノマー30~100質量%、及びその他のビニルモノマー0~70質量%からなる表面架橋層成分を重合してなる中間層重合体からなり、本発明の硬化性組成物の粘度を低下させる効果、ポリマー微粒子(B)の可とう性エポキシ樹脂(A)成分への分散性を向上させる効果を有する。また、コア層の架橋密度を上げたりシェル層のグラフト効率を高める効果も有する。
前記多官能性モノマーの具体例としては、上述の多官能性モノマーと同じモノマーが例示されるが、好ましくはアリルメタクリレート、トリアリルイソシアヌレートである。
ポリマー微粒子の最も外側に存在するシェル層は、シェル形成用モノマーを重合したものであるが、ポリマー微粒子(B)成分と可とう性エポキシ樹脂(A)成分との相溶性を向上させ、本発明の硬化性組成物、又はその硬化物中においてポリマー微粒子(B)が一次粒子の状態で分散することを可能にする役割を担うシェルポリマーからなる。
エポキシ基を有するモノマーは、シェル層の形成に使用することが好ましく、シェル層のみに使用することがより好ましい。
多官能性モノマーは、シェル形成用モノマー100質量%中に、1~20質量%含まれていることが好ましく、より好ましくは、5~15質量%である。
前記ビニルシアンモノマーの具体例としては、アクリロニトリル、又はメタクリロニトリル等が挙げられる。
前記ラジカル重合性二重結合を2個以上有する多官能性モノマーの具体例としては、上述の多官能性モノマーと同じモノマーが例示されるが、好ましくはアリルメタクリレート、トリアリルイソシアヌレートである。
シェル層は、上記モノマー成分の他に、他のモノマー成分を含んで形成されてもよい。
ポリマー微粒子(B)成分のシェル層中のエポキシ基の含有量(シェル層形成成分1g当たりの含有量)は、0.05~3.5mmol/gが好ましく、0.1~2.0mmol/gがより好ましく、0.2~1.0mmol/gが更に好ましく、0.3~0.7mmol/gが特に好ましい。シェル層中のエポキシ基の含有量が、0.05mmol/g未満であると、硬化性組成物を硬化して得られる硬化物の機械強度が低下する場合がある。シェル層中のエポキシ基の含有量が、3.5mmol/gを超えると、貯蔵後の組成物の粘度が上昇する傾向がある。
(コア層の製造方法)
本発明で用いるポリマー微粒子を構成するコア層の形成は、例えば、乳化重合、懸濁重合、マイクロサスペンジョン重合などによって製造することができ、例えばWO2005/028546号パンフレットに記載の方法を用いることができる。コア層形成用モノマーの重合は乳化重合法により行うことが好ましい。
中間層は、中間層形成用モノマーを公知のラジカル重合により重合することによって形成することができる。コア層を構成するコアポリマーをエマルジョンとして得た場合には、中間層形成用モノマーの重合は乳化重合法により行うことが好ましい。
シェル層は、シェル層形成用モノマーを、公知のラジカル重合により重合することによって形成することができる。コア層、または、コア層を中間層で被覆して構成されるポリマー粒子前駆体をエマルジョンとして得た場合には、シェル層形成用モノマーの重合は乳化重合法により行うことが好ましく、例えば、WO2005/028546号パンフレットに記載の方法に従って製造することができる。
また、t-ブチルパーオキシイソプロピルカーボネート、パラメンタンハイドロパーオキサイド、クメンハイドロパーオキサイド、ジクミルパーオキサイド、t-ブチルハイドロパーオキサイド、ジ-t-ブチルパーオキサイド、t-ヘキシルパーオキサイドなどの有機過酸化物;過酸化水素、過硫酸カリウム、過硫酸アンモニウムなどの無機過酸化物といった過酸化物と、必要に応じてナトリウムホルムアルデヒドスルホキシレート、グルコースなどの還元剤、および必要に応じて硫酸鉄(II)などの遷移金属塩、さらに必要に応じてエチレンジアミン四酢酸二ナトリウムなどのキレート剤、さらに必要に応じてピロリン酸ナトリウムなどのリン含有化合物などを併用したレドックス型開始剤を使用することもできる。
本発明では、必要に応じてエポキシ硬化剤(C)を使用することができる。
本発明の硬化性組成物を仮に一成分型組成物(一液型硬化性組成物など)として使用する場合、80℃以上、好ましくは140℃以上の温度まで加熱すると接着剤が急速に硬化するようにエポキシ硬化剤(C)成分を選択するのが好ましい。逆に、室温(約22℃)や少なくとも50℃までの温度では硬化するとしても非常にゆっくりとなるよう、エポキシ硬化剤(C)成分および後述の硬化促進剤を選択するのが好ましい。
一方、本発明の硬化性組成物を二成分型又は多成分型組成物として使用する場合、上記以外のアミン系硬化剤(イミン系硬化剤を含む)やメルカプタン系硬化剤(室温硬化性硬化剤と称する場合もある)を、室温程度の比較的低温で活性を示すエポキシ硬化剤(C)成分として選択することができる。
トール油脂肪酸とポリアミンとの縮合により生成するアミドアミン類;ポリメルカプタン類などを挙げることができる。
また、エポキシ硬化剤(C)成分としては、酸無水物類やフェノール類なども使用できる。酸無水物類やフェノール類などは、アミン系硬化剤と比較して高温を必要とするが、ポットライフが長く、硬化物は電気的特性、化学的特性、機械的特性などの物性バランスが良好である。酸無水物類としては、ポリセバシン酸ポリ無水物、ポリアゼライン酸ポリ無水物、無水コハク酸、シトラコン酸無水物、イタコン酸無水物、アルケニル置換コハク酸無水物、ドデセニルコハク酸無水物、無水マレイン酸、トリカルバリル酸無水物、ナド酸無水物、メチルナド酸無水物、無水マレイン酸によるリノール酸付加物、アルキル化末端アルキレンテトラヒドロフタル酸無水物、メチルテトラヒドロフタル酸無水物、テトラヒドロフタル酸無水物、ヘキサヒドロフタル酸無水物、ピロメリット酸二無水物、トリメリット酸無水物、無水フタル酸、テトラクロロフタル酸無水物、テトラブロモフタル酸無水物、ジクロロマレイン酸無水物、クロロナド酸無水物、およびクロレンド酸無水物、ならびに無水マレイン酸-グラフト化ポリブタジエンなどを挙げることができる。フェノール類としては、フェノールノボラック、ビスフェノールAノボラック、クレゾールノボラックなどを挙げることができる。
エポキシ硬化剤(C)成分は、組成物を硬化させるのに十分な量で使用する。典型的には、組成物中に存在するエポキシド基の少なくとも80%を消費するのに十分な硬化剤を供給する。エポキシド基の消費に必要な量を超える大過剰量は、通常必要ない。エポキシ硬化剤(C)成分の使用量は、可とう性エポキシ樹脂(A)成分100質量部に対して、1~200質量部が好ましく、2~170質量部がより好ましく、3~140質量部が更に好ましく、5~120質量部が特に好ましい。1質量部未満では、本発明の硬化性組成物の硬化性が悪くなる場合がある。200質量部より多いと、本発明の硬化性組成物の貯蔵安定性が悪く、取り扱い難くなる場合がある。
本発明では、必要に応じて硬化促進剤を使用することができる。
硬化促進剤は、エポキシ基と、硬化剤や接着剤の他の成分上のエポキシド反応性基との反応)を促進するための触媒である。
硬化促進剤としては、例えば、p-クロロフェニル-N,N-ジメチル尿素(商品名:Monuron)、3-フェニル-1,1-ジメチル尿素(商品名:Phenuron)、3,4-ジクロロフェニル-N,N-ジメチル尿素(商品名:Diuron)、N-(3-クロロ-4-メチルフェニル)-N’,N’-ジメチル尿素(商品名:Chlortoluron)、1,1-ジメチルフェニルウレア(商品名:Dyhard)などの尿素類;ベンジルジメチルアミン、2,4,6-トリス(ジメチルアミノメチル)フェノール、2-(ジメチルアミノメチル)フェノール、ポリ(p-ビニルフェノール)マトリックスに組み込まれた2,4,6-トリス(ジメチルアミノメチル)フェノール、トリエチレンジアミン、N,N-ジメチルピペリジンなどの三級アミン類;C2-12アルキレンイミダゾール、N-アリールイミダゾール、2-メチルイミダゾール、2-エチル-2-メチルイミダゾール、N-ブチルイミダゾール、1-シアノエチル-2-ウンデシルイミダゾリウム・トリメリテート、エポキシ樹脂とイミダゾールとの付加生成物、などのイミダゾール類;6-カプロラクタム等が挙げられる。触媒は封入されていてもよく、あるいは、温度を上げた場合にのみ活性となる潜在的なものでもよい。
なお、三級アミン類やイミダゾール類は、エポキシ硬化剤(C)成分のアミン系硬化剤と併用することにより、硬化速度、硬化物物性、耐熱性などを向上させることができる。
硬化促進剤は、単独で用いてもよく2種以上併用してもよい。
本発明の硬化性組成物は、無機充填材として、ケイ酸および/またはケイ酸塩を添加することができる。
具体例としては、乾式シリカ、湿式シリカ、ケイ酸アルミニウム、ケイ酸マグネシウム、ケイ酸カルシウム、ウォラストナイト、タルク、などが挙げられる。
前記乾式シリカはヒュームドシリカとも呼ばれ、表面無処理の親水性ヒュームドシリカと、親水性ヒュームドシリカのシラノール基部分にシランやシロキサンで化学的に処理することによって製造した疎水性ヒュームドシリカが挙げられるが、(A)成分への分散性の点から、疎水性ヒュームドシリカが好ましい。
無機充填材は、表面処理剤により表面処理していることが好ましい。表面処理により無機充填材の組成物への分散性が向上し、その結果、得られる硬化物の機械特性が向上する。
無機充填材は単独で用いても良く2種以上併用しても良い。
本発明の硬化性組成物は、酸化カルシウムを添加することができる。
酸化カルシウムは、硬化性組成物中の水分との反応により水分を除去し、水分の存在により引き起こされる種々の物性上の問題を解決する。例えば、水分除去による気泡防止剤として機能し、接着強度の低下を抑制する。
酸化カルシウムは、表面処理剤により表面処理することが可能である。表面処理により酸化カルシウムの組成物への分散性が向上する。その結果、表面処理を施していない酸化カルシウムを使用した場合と比較して、得られる硬化物の機械特性が向上する。前記表面処理剤は、特に制限はないが、脂肪酸が好ましい。
酸化カルシウムは単独で用いても良く2種以上併用しても良い。
本発明では、必要に応じて、モノエポキシドを使用することができる。モノエポキシドは反応性希釈剤として機能しうる。モノエポキシドの具体例としては、例えばブチルグリシジルエーテルなどの脂肪族グリシジルエーテル、あるいは例えばフェニルグリシジルエーテル、クレジルグリシジルエーテルなどの芳香族グリシジルエーテル、例えば2-エチルヘキシルグリシジルエーテルなどの炭素数8~10のアルキル基とグリシジル基とからなるエーテル、例えばp-tertブチルフェニルグリシジルエーテルなどの炭素数2~8のアルキル基で置換され得る炭素数6~12のフェニル基とグリシジル基とからなるエーテル、例えばドデシルグリシジルエーテルなどの炭素数12~14のアルキル基とグリシジル基とからなるエーテル;例えばグリシジル(メタ)アクリレート、グリシジルマレエートなどの脂肪族グリシジルエステル;バーサチック酸グリシジルエステル、ネオデカン酸グリシジルエステル、ラウリン酸グリシジルエステルなどの炭素数8~12の脂肪族カルボン酸のグリシジルエステル;p-t-ブチル安息香酸グリシジルエステルなどが挙げられる。
本発明では、必要に応じて、シランカップリング剤を添加することができる。シランカップリング剤添加により接着性を向上させることができる。具体例としては、γ-イソシアネートプロピルトリメトキシシラン、γ-イソシアネートプロピルトリエトキシシラン、γ-イソシアネートプロピルメチルジエトキシシラン、γ-イソシアネートプロピルメチルジメトキシシラン、(イソシアネートメチル)トリメトキシシラン、(イソシアネートメチル)ジメトキシメチルシラン、(イソシアネートメチル)トリエトキシシラン、(イソシアネートメチル)ジエトキシメチルシラン等のイソシアネート基含有シラン類;γ-アミノプロピルトリメトキシシラン、γ-アミノプロピルトリエトキシシラン、γ-アミノプロピルトリイソプロポキシシラン、γ-アミノプロピルメチルジメトキシシラン、γ-アミノプロピルメチルジエトキシシラン、γ-(2-アミノエチル)アミノプロピルトリメトキシシラン、γ-(2-アミノエチル)アミノプロピルメチルジメトキシシラン、γ-(2-アミノエチル)アミノプロピルトリエトキシシラン、γ-(2-アミノエチル)アミノプロピルメチルジエトキシシラン、γ-(2-アミノエチル)アミノプロピルトリイソプロポキシシラン、γ-(6-アミノヘキシル)アミノプロピルトリメトキシシラン、3-(N-エチルアミノ)-2-メチルプロピルトリメトキシシラン、γ-ウレイドプロピルトリメトキシシラン、γ-ウレイドプロピルトリエトキシシラン、N-フェニル-γ-アミノプロピルトリメトキシシラン、N-ベンジル-γ-アミノプロピルトリメトキシシラン、N-ビニルベンジル-γ-アミノプロピルトリエトキシシラン、N-シクロヘキシルアミノメチルトリエトキシシラン、N-シクロヘキシルアミノメチルジエトキシメチルシラン、N-フェニルアミノメチルトリメトキシシラン、(2-アミノエチル)アミノメチルトリメトキシシラン、N,N‘-ビス[3-(トリメトキシシリル)プロピル]エチレンジアミン等のアミノ基含有シラン類;N-(1,3-ジメチルブチリデン)-3-(トリエトキシシリル)-1-プロパンアミン等のケチミン型シラン類;γ-メルカプトプロピルトリメトキシシラン、γ-メルカプトプロピルトリエトキシシラン、γ-メルカプトプロピルメチルジメトキシシラン、γ-メルカプトプロピルメチルジエトキシシラン、メルカプトメチルトリメトキシシラン、メルカプトメチルトリエトキシシラン等のメルカプト基含有シラン類;γ-グリシドキシプロピルトリメトキシシラン、γ-グリシドキシプロピルトリエトキシシラン、γ-グリシドキシプロピルメチルジメトキシシラン、β-(3,4-エポキシシクロヘキシル)エチルトリメトキシシラン、β-(3,4-エポキシシクロヘキシル)エチルトリエトキシシラン等のエポキシ基含有シラン類;β-カルボキシエチルトリエトキシシラン、β-カルボキシエチルフェニルビス(2-メトキシエトキシ)シラン、N-β-(カルボキシメチル)アミノエチル-γ-アミノプロピルトリメトキシシラン等のカルボキシシラン類;ビニルトリメトキシシラン、ビニルトリエトキシシラン、γ-メタクリロイルオキシプロピルメチルジメトキシシラン、γ-アクリロイルオキシプロピルトリエトキシシラン、メタクリロイルオキシメチルトリメトキシシラン等のビニル型不飽和基含有シラン類;γ-クロロプロピルトリメトキシシラン等のハロゲン含有シラン類;トリス(3-トリメトキシシリルプロピル)イソシアヌレート等のイソシアヌレートシラン類等を挙げることができる。また、上記アミノシランとエポキシシランの反応物、アミノシランとイソシアネートシランの反応物、各種シランカップリング剤の部分縮合体等もシランカップリング剤として用いることができる。
本発明では、必要に応じて、その他の配合成分を使用することができる。その他の配合成分としては、アゾタイプ化学的発泡剤や熱膨張性マイクロバルーンなどの膨張剤、アラミド系パルプなどの繊維パルプ、顔料や染料等の着色剤、体質顔料、紫外線吸収剤、酸化防止剤、ラジカル硬化性樹脂、光重合開始剤、安定化剤(ゲル化防止剤)、可塑剤、レベリング剤、消泡剤、帯電防止剤、難燃剤、滑剤、減粘剤、低収縮剤、有機質充填剤、熱可塑性樹脂、乾燥剤、分散剤等が挙げられる。
本発明の硬化性組成物は、可とう性エポキシ樹脂(A)成分を樹脂主成分(樹脂100質量%中、50質量%以上)とする硬化性組成物中に、ポリマー微粒子(B)を含有する組成物であり、好ましくは、ポリマー微粒子(B)が1次粒子の状態で分散した組成物である。
上記の工程を経て得た、可とう性エポキシ樹脂(A)成分にポリマー微粒子(B)が1次粒子の状態で分散した組成物に、更に可とう性エポキシ樹脂(A)成分、エポキシ硬化剤(C)成分、及び、前記その他配合成分の各成分から選ばれる1種以上を、必要により更に追加混合する事により、ポリマー微粒子(B)が1次粒子の状態で分散した本発明の硬化性組成物が得られる。
本発明の硬化性組成物は、すべての配合成分を予め配合した後密封保存し、塗布後加熱や光照射により硬化する一液型硬化性組成物として使用することができる。また、可とう性エポキシ樹脂(A)成分を主成分とし、さらにポリマー微粒子(B)成分を含有するA液と、エポキシ硬化剤(C)成分や硬化促進剤を含有し、更に必要に応じてポリマー微粒子(B)成分を含有する別途調製したB液からなる、二液型または多液型の硬化性組成物として調製しておき、該A液と該B液を使用前に混合して、使用することもできる。
ポリマー微粒子(B)成分、エポキシ硬化剤(C)成分は、それぞれA液、B液のどちらか少なくとも一方に含まれていればよく、例えば、A液にのみ、B液にのみでもよく、A液とB液の両方に含まれていてもよい。
本発明には、上記硬化性組成物を硬化して得られる硬化物が含まれる。ポリマー微粒子が一次粒子の状態で分散している硬化性組成物の場合には、これを硬化することによって、ポリマー微粒子が均一に分散した硬化物を容易に得ることができる。また、ポリマー微粒子が膨潤し難く、硬化性組成物の粘性が低いことから、硬化物を作業性よく得ることができる。
本発明の硬化性組成物は、任意の方法によって塗布可能である。室温程度の低温で塗布可能であり、必要に応じて加温して塗布することも可能である。
本発明の硬化性組成物は、塗布ロボットを使用してビード状またはモノフィラメント状またはスワール(swirl)状に基板上へ押出したり、コーキングガン等の機械的な塗布方法や他の手動塗布手段を用いたりすることもできる。また、ジェットスプレー法またはストリーミング法を用いて組成物を基板へ塗布することもできる。本発明の硬化性組成物を、一方または両方の基板へ塗布し、接合しようとする基板間に該硬化性組成物が配置されるよう基板同士を接触させ、硬化させることにより接合する。なお、硬化性組成物の粘度は、特に限定は無く、押出しビード法では、45℃で150~600Pa・s程度が好ましく、渦巻き(swirl)塗布法では、45℃で100Pa・s程度が好ましく、高速度流動装置を用いた高体積塗布法では、45℃で20~400Pa・s程度が好ましい。
本発明の組成物を使用して、様々な基板同士を接着させる場合、例えば、木材、金属、プラスチック、ガラス等を接合することができ、好ましくは、冷間圧延鋼板や溶融亜鉛メッキ鋼板などの鋼板、アルミニウム合金や被覆アルミニウム合金などのアルミニウム合金板、チタニウム合金板、マグネシウム合金板、汎用プラスチック、エンジニアリングプラスチック等の樹脂板、炭素繊維強化プラスチック(CFRP)やガラス繊維強化プラスチック(GFRP)等の複合基板等の各種のプラスチック系基板などを接合することができる。これら基板は、異なる材料から形成されていてもよい。本発明の硬化性組成物は、伸び物性に優れる為に、線膨張係数の異なる異種基材間の接合に適している。
本発明の硬化性組成物を使用して基板同士を接着させる場合、自動車部品を接合することが好ましく、自動車フレーム同士の接合または自動車フレームと他の自動車部品との接合がより好ましく、こうした接合技術によって外装パネルを製造するのがさらに好ましい。
また、本発明の硬化性組成物は、航空宇宙用の構成材、特に、外装金属構成材(外装パネル)の接合にも使用できる。
本発明の硬化性組成物の硬化温度は、特に限定はないが、一液型硬化性組成物として使用する場合には、50℃~250℃が好ましく、80℃~220℃がより好ましく、100℃~200℃が更に好ましく、130℃~180℃が特に好ましい。二液型硬化性組成物として使用する場合には、特に限定はないが、0℃~150℃が好ましく、10℃~100℃がより好ましく、15℃~80℃が更に好ましく、20℃~60℃が特に好ましい。
本発明の硬化性組成物を自動車用接着剤として使用する場合、該接着剤を自動車部材へ施工した後、次いでコーティングを塗布し、該コーティングを焼付け・硬化するのと同時に接着剤を硬化させるのが工程短縮・簡便化の観点から好ましい。
本発明の組成物は、車両や航空機向けの構造用接着剤、風力発電用構造接着剤などの接着剤、塗料、ガラス繊維との積層用材料、およびプリント配線基板用材料、ソルダーレジスト、層間絶縁膜、ビルドアップ材料、FPC用接着剤、半導体・LED等電子部品用封止材等の電気絶縁材料、ダイボンド材料、アンダーフィル、ACF、ACP、NCF、NCP等の半導体実装材料、液晶パネル、OLED照明、OLEDディスプレイ等の表示機器・照明機器用封止材の用途に好ましく用いられる。特に、車両用構造接着剤として有用である。
先ず、実施例および比較例によって製造した硬化性組成物の評価方法について、以下説明する。
[1]平均粒子径の測定
水性ラテックスに分散しているポリマー微粒子(B)の体積平均粒子径(Mv)は、マイクロトラックUPA150(日機装株式会社製)を用いて測定した。脱イオン水で希釈したものを測定試料として用いた。測定は、水の屈折率、およびそれぞれのポリマー微粒子の屈折率を入力し、計測時間600秒、Signal Levelが0.6~0.8の範囲内になるように試料濃度を調整して行った。
硬化性組成物中のポリマー微粒子(B)成分のコア層のガラス転移温度は、数式(1)よりケルビン温度(K)で計算し、セルシウス温度(℃)へ換算した。その際に用いた各非架橋性単量体のホモポリマーのTgは次の値を使用した:メチルメタクリレート 378K、ブチルアクリレート 219K、ブチルメタクリレート 293K、ブタジエン 188K、スチレン 373K。
1/Tg=Σ(Mi/Tgi) (1)
(式中、Miはポリマー微粒子(B)成分のコア層を構成するブタジエン及び非架橋性単量体から選ばれる各単量体i成分の重量分率、Tgiは各単量体iのホモポリマーのガラス転移温度(K)を表す。)
硬化性組成物の粘度は、BROOKFIELD社製デジタル粘度計DV-II+Pro型を用いて測定した。スピンドルCPE-41またはCPE-52を使用し、50℃または25℃で、Shear Rate(ずり速度)が10(s-1)における粘度を測定した。
硬化性組成物を、厚み3mmのスペーサーを挟んだ2枚のテフロン(登録商標)コート鋼板の間に注ぎ込み、後述の温度と時間、硬化させ、厚み3mmの硬化板を得た。この硬化板を3号ダンベル型に打ち抜いて、JIS K-6251に従って、23℃にて引っ張り速度200mm/分で引張り試験を行い、100%伸び時の応力(M100)(MPa)、最大引張応力(Tmax)(MPa)、最大引張応力時の伸び(Emax)(%)を測定した。
前記[4]で作成した厚み3mmの硬化板を2枚重ね、JIS K-6253に従って、タイプAの試験機を用いてデュロメータ硬さを、23℃にて測定した。
製造例1-1;コアシェルポリマーラテックス(L-1)の調製
温度計、撹拌機、還流冷却器、窒素流入口、モノマーと乳化剤の添加装置を有するガラス反応器に、脱イオン水181質量部、エチレンジアミン四酢酸二ナトリウム(EDTA)0.006質量部、硫酸第一鉄・7水和塩(FE)0.0015質量部、ナトリウムホルムアルデヒドスルホキシレート(SFS)0.6質量部およびドデシルベンゼンスルホン酸ナトリウム(SDS)0.01質量部を仕込み、窒素気流中で撹拌しながら60℃に昇温した。次にコア層用モノマー(メチルメタクリレート(MMA)87質量部、アリルメタクリレート(ALMA)0.4質量部)、及び、クメンハイドロパーオキサイド(CHP)0.13質量部の混合物を3時間要して滴下した。また、前記のモノマー混合物の添加とともに、SDSの5質量%水溶液20質量部を3時間にわたり連続的に追加した。モノマー混合物添加終了から1時間撹拌を続けて重合を完結し、アクリルポリマー微粒子を含むラテックスを得た。引き続き、そこに、グラフトモノマー(MMA3質量部、ブチルアクリレート(BA)10質量部)、及び、CHP0.07質量部の混合物を120分間かけて連続的に添加した。添加終了後、CHP0.07質量部を添加し、さらに2時間撹拌を続けて重合を完結させ、コアシェルポリマー微粒子を含むラテックス(L-1)を得た。モノマー成分の重合転化率は99%以上であった。得られたラテックスに含まれるコアシェルポリマー微粒子の体積平均粒子径は0.36μmであった。
製造例1-1において、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA2質量部、BA10質量部、グリシジルメタクリレート(GMA)1質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-2)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.36μmであった。
製造例1-1において、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA87質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA2質量部、BA10質量部、GMA1質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-3)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.35μmであった。
製造例1-1において、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA72質量部、BA15質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA2質量部、BA10質量部、GMA1質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-4)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.31μmであった。
製造例1-1において、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA57質量部、BA30質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA2質量部、BA10質量部、GMA1質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-5)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.35μmであった。
製造例1-1において、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA57質量部、BA30質量部、ALMA4質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-6)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.37μmであった。
製造例1-1において、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA57質量部、BA30質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA1質量部、BA10質量部、GMA2質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-7)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.35μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA68.7質量部、BA14.3質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA2.6質量部、BA13.1質量部、GMA1.3質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-8)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.09μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA66.2質量部、BA13.8質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3.1質量部、BA15.4質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-9)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.10μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA63.7質量部、BA13.3質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3.5質量部、BA17.7質量部、GMA1.8質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-10)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.09μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA61.2質量部、BA12.8質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA4質量部、BA20質量部、GMA2質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-11)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.10μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA52.4質量部、BA27.6質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-12)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.08μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに1.5質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA52.4質量部、BA27.6質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-13)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.06μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに3.0質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA52.4質量部、BA27.6質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-14)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.05μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA52.4質量部、ブチルメタクリレート(BMA)27.6質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-15)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.08μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA52.4質量部、BMA27.6質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BMA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-16)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.08μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA38質量部、BA42質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-17)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.09μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<MMA17.5質量部、BA62.5質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-18)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.08μmであった。
製造例1-1において、最初に仕込むSDSを0.01質量部の代わりに0.4質量部とし、コア層用モノマーとして<MMA87質量部、ALMA0.4質量部>の代わりに<BA80質量部、ALMA4質量部>を用い、グラフトモノマーとして<MMA3質量部、BA10質量部>の代わりに<MMA3質量部、BA15.5質量部、GMA1.5質量部>を用いたこと以外は製造例1-1と同様にして、コアシェルポリマーのラテックス(L-19)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.08μmであった。
耐圧重合機中に、脱イオン水200質量部、リン酸三カリウム0.03質量部、EDTA0.002質量部、FE0.001質量部、及び、SDS1.55質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、ブタジエン(BD)100質量部を系中に投入し、45℃に昇温した。パラメンタンハイドロパーオキサイド(PHP)0.03質量部、続いてSFS0.10質量部を投入し重合を開始した。重合開始から3、5、7時間目それぞれに、PHP0.025質量部を投入した。また、重合開始4、6、8時間目それぞれに、EDTA0.0006質量部、及びFE0.003質量部を投入した。重合15時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-1)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.08μmであった。
製造例1-20において、BD100質量部の代わりに、BD75質量部およびスチレン(ST)25質量部を系中に投入した以外は製造例1-20と同様にして、スチレン-ブタジエンゴムラテックス(R-2)を得た。得られたラテックスに含まれるスチレン-ブタジエンゴム粒子の体積平均粒子径は0.08μmであった。
耐圧重合機中に、製造例1-20で得たポリブタジエンゴムラテックス(R-1)を21質量部(ポリブタジエンゴム7質量部を含む)、脱イオン水186質量部、リン酸三カリウム0.03質量部、EDTA0.002質量部、及びFE0.001質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、BD93質量部を系中に投入し、45℃に昇温した。PHP0.02質量部、続いてSFS0.10質量部を投入し重合を開始した。重合開始から24時間目まで3時間おきに、それぞれ、PHP0.025質量部、及びEDTA0.0006質量部、及びFE0.003質量部を投入した。重合30時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-3)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.20μmであった。
製造例1-22において、グラフトモノマーとして<MMA4質量部、BA6質量部>の代わりに<MMA3質量部、BA6質量部、GMA1質量部>を用いたこと以外は製造例1-22と同様にして、コアシェルポリマーのラテックス(L-23)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例1-22において、グラフトモノマーとして<MMA4質量部、BA6質量部>の代わりに<BA6質量部、GMA4質量部>を用いたこと以外は製造例1-22と同様にして、コアシェルポリマーのラテックス(L-24)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.21μmであった。
製造例2-1;分散物(M-1)の調製
25℃の1L混合槽にメチルエチルケトン(MEK)120gを導入し、撹拌しながら、前記製造例2-1で得られたコアシェルポリマーラテックス(L-1)を120g(ポリマー微粒子40g相当)投入した。均一に混合後、水180gを80g/分の供給速度で投入した。供給終了後、速やかに撹拌を停止したところ、浮上性の凝集体および有機溶媒を一部含む水相からなるスラリー液を得た。次に、一部の水相を含む凝集体を残し、水相300gを槽下部の払い出し口より排出させた。得られた凝集体にMEK180gを追加して均一に混合し、コアシェルポリマーを均一に分散した分散体を得た。この分散体に、エポキシ樹脂(A)成分である可とう性エポキシ樹脂(A-1;三菱化学社製、JER871:ダイマー酸変性エポキシ樹脂、エポキシ当量は410g/eq)80gを混合した。この混合物から、回転式の蒸発装置で、MEKを除去した。このようにして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-1)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、それぞれ前記製造例1-2又は1-3で得られたラテックス(L-2)又はラテックス(L-3)を用いたこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-2又はM-3)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、それぞれ前記製造例1-3~1-12で得られたラテックス(L-3)~(L-12)を用い、可とう性エポキシ樹脂(A-1)を80gの代わりに60gを混合したこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-4~M-13)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、前記製造例1-12で得られたラテックス(L-12)を用い、可とう性エポキシ樹脂(A-1)を80gの代わりに40gを混合したこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-14)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、それぞれ前記製造例1-13~1-19及び1-22~1-24で得られたラテックス(L-13)~(L-19)及び(L-22)~(L-24)を用い、可とう性エポキシ樹脂(A-1)を80gの代わりに60gを混合したこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-15~M-21及びM-24~M-26)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、それぞれ前記製造例1-20又は1-21で得られたラテックス(L-20)又は(L-21)を用いたこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-22又はM-23)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、それぞれ前記製造例1-8~11で得られたラテックス(L-8)~(L-11)を用い、可とう性エポキシ樹脂(A-1)80gの代わりに、可とう性エポキシ樹脂(A-2;東亞合成製、UG-4010:エポキシ基含有アクリル重合体、エポキシ当量は720g/eq)60gを混合したこと以外は製造例2-1と同様にして、可とう性エポキシ樹脂にポリマー微粒子が分散した分散物(M-27~M-30)を得た。
製造例2-1において、コアシェルポリマーラテックス(L-1)の代わりに、前記製造例1-4で得られたラテックス(L-4)を用い、可とう性エポキシ樹脂(A-1)80gの代わりに、ビスフェノールA型エポキシ樹脂(A-3;三菱化学社製、JER828、エポキシ当量は190g/eq)60gを混合したこと以外は製造例2-1と同様にして、ビスフェノールA型エポキシ樹脂にポリマー微粒子が分散した分散物(M-31)を得た。

表2に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-1;三菱化学社製、JER871:ダイマー酸変性エポキシ樹脂)、可とう性エポキシ樹脂(A)
とポリマー微粒子(B)の混合物である前記製造例2-1~2-26で得られた分散物(M-1~M-26)、(C)成分であるエポキシ硬化剤(C-1;CVC製、Hypro
1300X16 ATBN:アミノ基末端ブタジエン-アクリロニトリル共重合体)、ヒュームドシリカ(CABOT製、CAB-O-SIL TS-720:ポリジメチルシロキサンで表面処理されたヒュームドシリカ)、重質炭酸カルシウム(白石カルシウム製、ホワイトンSB)をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、23℃×48時間+80℃×5時間の硬化条件で得た硬化物の引張物性(100%伸び時の応力:M100、最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表2、表3に示す。
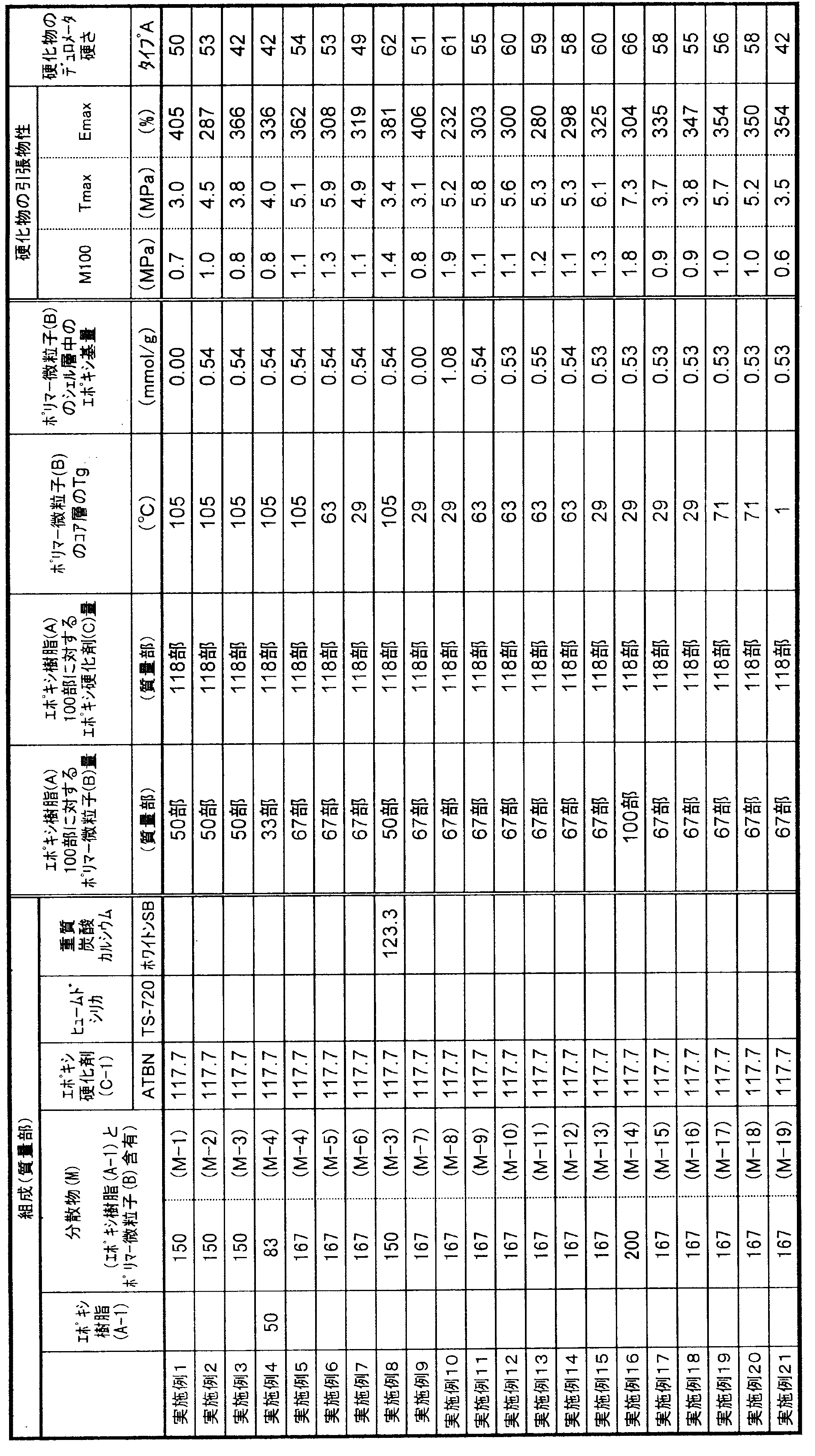
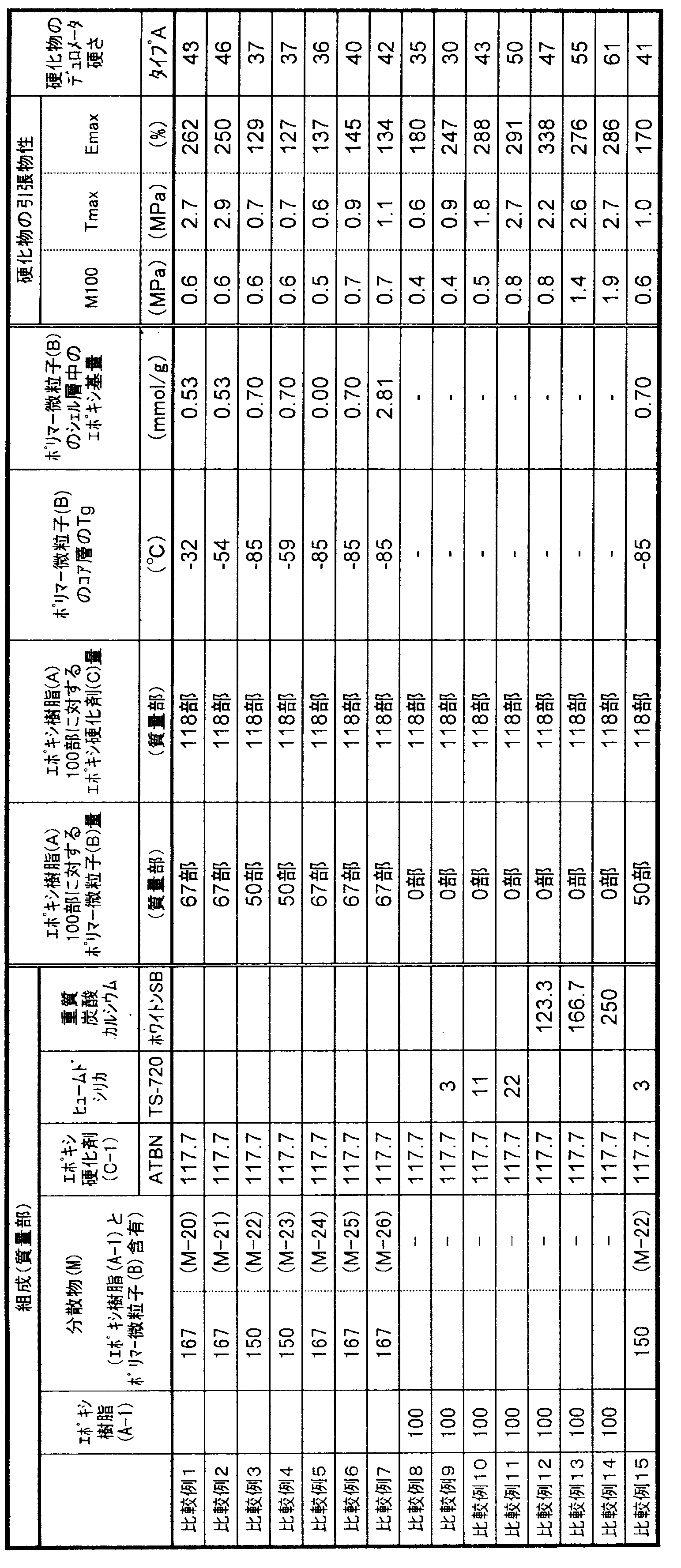
表4に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-1)、可とう性エポキシ樹脂(A)とポリマー微粒子(B)の混合物である前記製造例2-4~2-8,2-13,2-24で得られた分散物(M-4~M-8,M-13,M-24)、(C)成分であるエポキシ硬化剤(C-2;Air Products製、Ancamide 2050:ポリアミドアミン)をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、23℃×48時間+80℃×5時間の硬化条件で得た硬化物の引張物性(最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表4に示す。
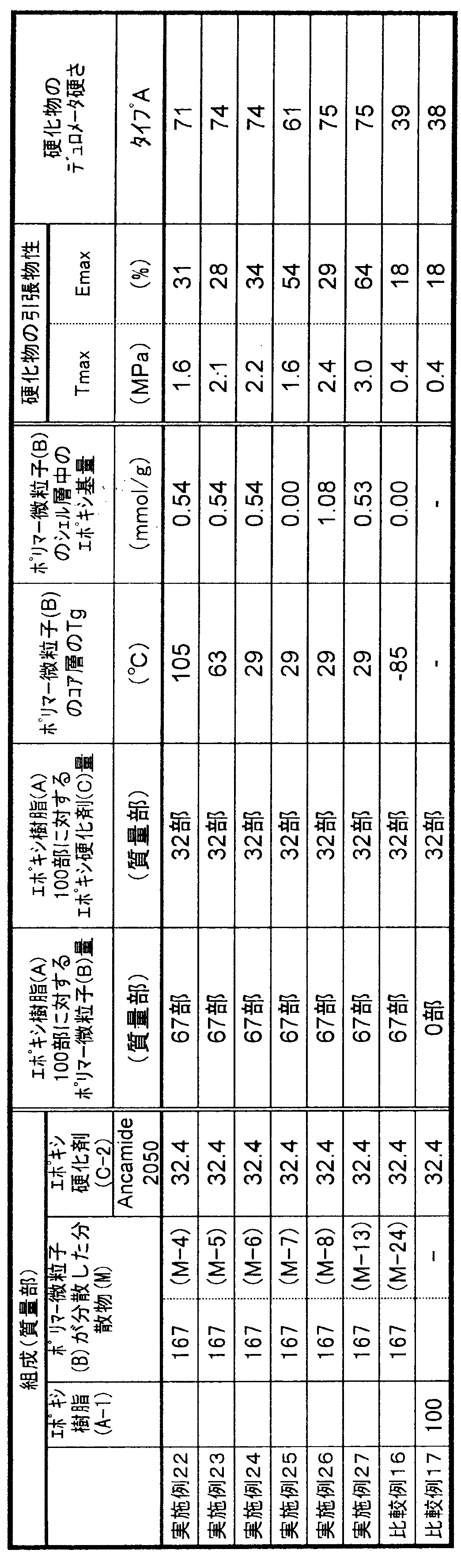
表5に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-1)、可とう性エポキシ樹脂(A)とポリマー微粒子(B)の混合物である前記製造例2-5~2-8,2-13,2-17,2-19,2-21で得られた分散物(M-5~M-8,M-13,M-17,M-19,M-21)、(C)成分であるエポキシ硬化剤(C-3;和光純薬工業製、イソフォロンジアミン(IPDA))をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、23℃×24時間+80℃×40時間の硬化条件で得た硬化物の引張物性(100%伸び時の応力:M100、最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表5に示す。
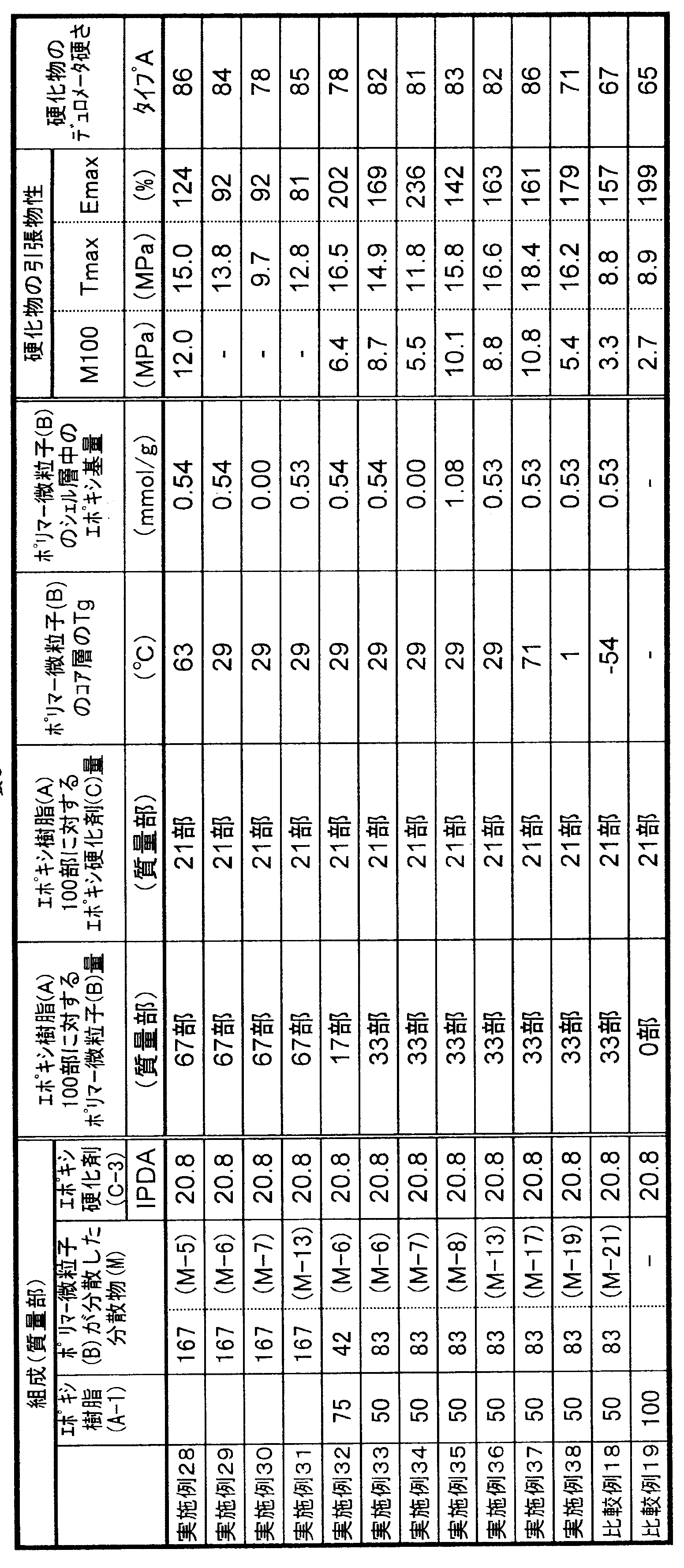

表7に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-2;東亞合成製、UG-4010:エポキシ基含有アクリル重合体)、可とう性エポキシ樹脂(A)とポリマー微粒子(B)の混合物である前記製造例2-27~2-30で得られた分散物(M-27~M-30)、(C)成分であるエポキシ硬化剤(C-1;CVC製、Hypro 1300X16 ATBN:アミノ基末端ブタジエン-アクリロニトリル共重合体)をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、23℃×48時間+80℃×5時間の硬化条件で得た硬化物の引張物性(100%伸び時の応力:M100、最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表7に示す。
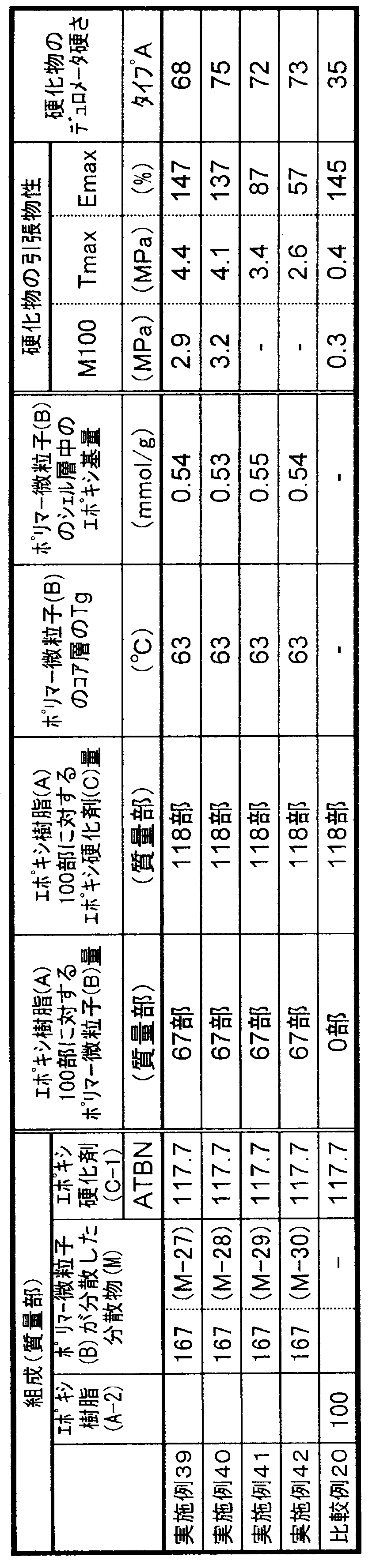
表8に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-1、エポキシ当量は410g/eq)またはビスフェノールA型エポキシ樹脂(A-3、エポキシ当量は190g/eq)、あるいは、可とう性エポキシ樹脂(A)とポリマー微粒子(B)の混合物である前記製造例2-5で得られた分散物(M-5)または前記製造例2-31で得られた分散物(M-31)、(C)成分であるエポキシ硬化剤(C-3;和光純薬工業製、イソフォロンジアミン(IPDA))をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、23℃×24時間+80℃×40時間の硬化条件で得た硬化物の引張物性(100%伸び時の応力:M100、最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表8に示す。
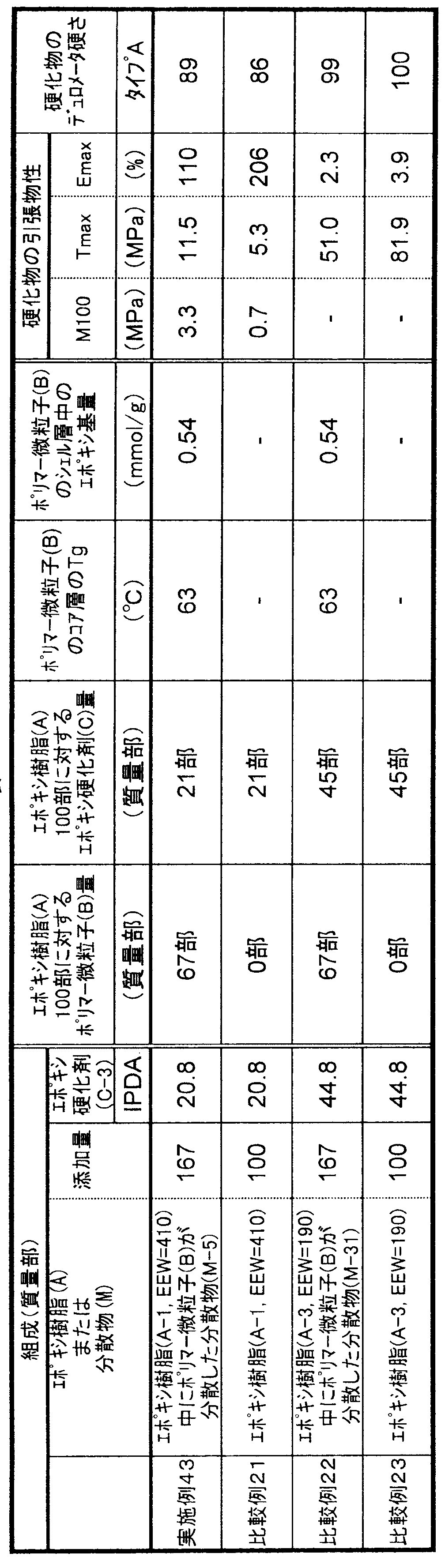
一方、実施例43と比較例21から、可とう性エポキシ樹脂(A-1)を用いた場合、高伸びでゴム弾性を示し、更に、ポリマー微粒子の添加により、硬化物の強度(最大引張応力)が顕著に向上することが判る。
表9に示す処方にしたがって、(A)成分である可とう性エポキシ樹脂(A-1、エポキシ当量は410g/eq)またはビスフェノールA型エポキシ樹脂(A-3、エポキシ当量は190g/eq)、あるいは、可とう性エポキシ樹脂(A)とポリマー微粒子(B)の混合物である前記製造例2-5で得られた分散物(M-5)、(C)成分であるエポキシ硬化剤(C-3;和光純薬工業製、イソフォロンジアミン(IPDA))をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を用いて前記の試験方法に従って、80℃×40時間の硬化条件で得た硬化物の引張物性(最大引張応力:Tmax、最大引張応力時の伸び:Emax)とデュロメータ硬さを測定した。試験結果を表9に示す。
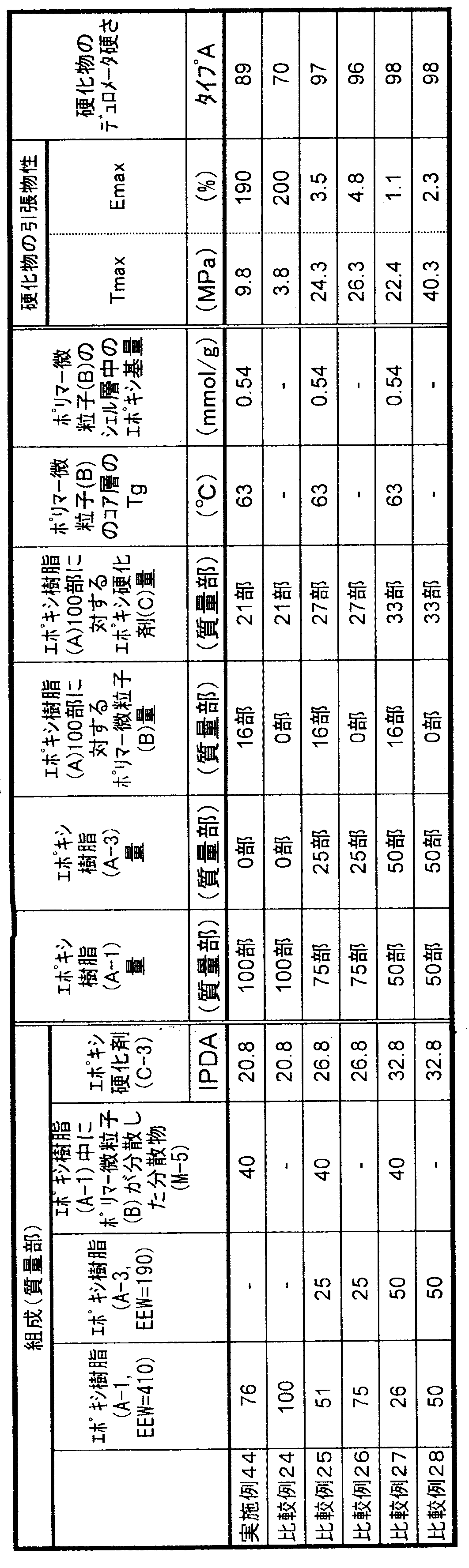
(ビスフェノールA型エポキシ樹脂(A-3、エポキシ当量は190g/eq)55質量部、ゴム変性エポキシ樹脂(EPON Resin 58005、Momentive製、エラストマー濃度:40wt%、ビスフェノールA型エポキシ樹脂濃度:60wt%、エポキシ当量は325~375g/eq)75質量部、(C)成分であるエポキシ硬化剤(C-4;ジシアンジアミド)7質量部、エポキシ硬化助剤(1,1-ジメチル-3-フェニルウレア)1質量部、をそれぞれ計量し、よく混合して脱泡し硬化性組成物を得た。この組成物を、170℃×1時間の硬化条件で硬化物を得た。硬化物をJIS K-7113に従った1号ダンベル型に打ち抜いて、23℃にて引っ張り速度10mm/分で引張り試験を行った結果、最大引張応力時の伸びは2.9%と低伸びであり、デュロメータ硬さは98であった。
Claims (16)
- 可とう性エポキシ樹脂(A)100質量部と、コア層およびシェル層の少なくとも2層を有するコアシェル構造のポリマー微粒子(B)1~150質量部と、エポキシ硬化剤(C)1~200質量部と、を含有する硬化性組成物であって、
下記数式(1)より計算したポリマー微粒子(B)のコア層のガラス転移温度(Tg)が0℃より大きく、かつ、
前記硬化性組成物を硬化して得られる硬化物のJIS K6253-3に規定されるタイプAデュロメータ硬さが23℃で5~95であることを特徴とする硬化性組成物。
1/Tg=Σ(Mi/Tgi) (1)
(式中、Miはポリマー微粒子(B)成分のコア層を構成するブタジエン及び非架橋性単量体から選ばれる各単量体i成分の重量分率、Tgiは各単量体iのホモポリマーのガラス転移温度(K)を表す。) - ポリマー微粒子(B)成分のコア層のガラス転移温度(Tg)が、15~150℃であることを特徴とする請求項1に記載の硬化性組成物。
- 可とう性エポキシ樹脂(A)成分のエポキシ当量が、200~4000g/eqであることを特徴とする請求項1または2に記載の硬化性組成物。
- ポリマー微粒子(B)成分のコア層が、(メタ)アクリレート系重合体であることを特徴とする請求項1から3のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分のコア層が、非架橋性単量体80~99質量%、および、架橋性単量体20~1質量%からなる単量体混合物を重合して得られる重合体であることを特徴とする請求項1から4のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分のシェル層が、(メタ)アクリレート系重合体であることを特徴とする請求項1から5のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分のシェル層が、エポキシ基を有することを特徴とする請求項1から6のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分のシェル層中のエポキシ基の含有量が、0.05~3.5mmol/gであることを特徴とする請求項1から7のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分が、エポキシ基を有するモノマー成分を、コア層にグラフト重合してなるシェル層を有することを特徴とする請求項1から8のいずれかに記載の硬化性組成物。
- ポリマー微粒子(B)成分が、該硬化性組成物中で1次粒子の状態で分散していることを特徴とする請求項1から9のいずれかに記載の硬化性組成物。
- 請求項1から10のいずれかに記載の硬化性組成物を硬化して得られる硬化物。
- 請求項1から10のいずれかに記載の硬化性組成物を用いてなる接着剤。
- 請求項1から10のいずれかに記載の硬化性組成物を用いてなる車両用接着剤。
- 異なる材料からなる2つの基板が、請求項1~10のいずれかに記載の硬化性組成物で接合された積層接着基板。
- 外装パネルである請求項14に記載の積層接着基板。
- 前記基板の少なくとも一方が、鋼板、アルミニウム合金板、チタニウム合金板、マグネシウム合金板、プラスチック系基板から選ばれる少なくとも1種である請求項14または15に記載の積層接着基板。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17782349.9A EP3444301B1 (en) | 2016-04-12 | 2017-04-10 | Curable composition and adhesive |
| JP2018512004A JP6966154B2 (ja) | 2016-04-12 | 2017-04-10 | 硬化性組成物及び接着剤 |
| CN201780022804.3A CN109071920B (zh) | 2016-04-12 | 2017-04-10 | 固化性组合物和粘合剂 |
| US16/157,255 US20190040290A1 (en) | 2016-04-12 | 2018-10-11 | Curable composition and adhesive |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-079836 | 2016-04-12 | ||
| JP2016079836 | 2016-04-12 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/157,255 Continuation US20190040290A1 (en) | 2016-04-12 | 2018-10-11 | Curable composition and adhesive |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017179536A1 true WO2017179536A1 (ja) | 2017-10-19 |
Family
ID=60042503
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/014681 Ceased WO2017179536A1 (ja) | 2016-04-12 | 2017-04-10 | 硬化性組成物及び接着剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20190040290A1 (ja) |
| EP (1) | EP3444301B1 (ja) |
| JP (1) | JP6966154B2 (ja) |
| CN (1) | CN109071920B (ja) |
| WO (1) | WO2017179536A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018053026A (ja) * | 2016-09-27 | 2018-04-05 | 株式会社カネカ | 機械的強度に優れるポリマー微粒子含有硬化性組成物 |
| US20190315979A1 (en) * | 2018-04-16 | 2019-10-17 | Canon Kabushiki Kaisha | Curable resin composition and manufacturing method of three-dimensional object using the same |
| JP2022047219A (ja) * | 2020-09-11 | 2022-03-24 | 国立研究開発法人物質・材料研究機構 | 定着構造体 |
| JPWO2022138808A1 (ja) * | 2020-12-25 | 2022-06-30 | ||
| CN115322657A (zh) * | 2022-08-31 | 2022-11-11 | 常州大学 | 一种含有柔性固化剂的环氧树脂基膨胀防火涂料及其制备方法 |
| WO2023054479A1 (ja) * | 2021-09-30 | 2023-04-06 | 株式会社カネカ | 硬化性樹脂組成物およびその利用 |
| JP2024515381A (ja) * | 2021-08-13 | 2024-04-09 | エルジー・ケム・リミテッド | 硬化性樹脂組成物 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020049516A1 (en) * | 2018-09-06 | 2020-03-12 | Ecole Polytechnique Federale De Lausanne (Epfl) | Composite material |
| CN114921861A (zh) * | 2020-12-22 | 2022-08-19 | 南通新帝克单丝科技股份有限公司 | 一种性能优异的高dpf聚酯工业丝 |
| CN115490966A (zh) * | 2021-12-14 | 2022-12-20 | 江苏锐巴新材料科技有限公司 | 一种高性能连续硫化橡胶材料及其应用 |
| CN119019959B (zh) * | 2024-08-30 | 2025-11-11 | 广东工业大学 | 一种核壳型丙烯酸酯胶粘剂及其制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05214310A (ja) * | 1992-01-31 | 1993-08-24 | Nissan Motor Co Ltd | エポキシ樹脂系接着性組成物 |
| JPH08183836A (ja) * | 1994-12-30 | 1996-07-16 | Nippon Zeon Co Ltd | エポキシ樹脂組成物 |
| JP2010077305A (ja) * | 2008-09-26 | 2010-04-08 | Yokohama Rubber Co Ltd:The | エポキシ樹脂組成物 |
| JP2010090371A (ja) * | 2008-09-11 | 2010-04-22 | Mitsubishi Rayon Co Ltd | エポキシ樹脂組成物、及びこれを硬化したエポキシ硬化物 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5290857A (en) * | 1991-09-04 | 1994-03-01 | Nippon Zeon Co., Ltd. | Epoxy resin adhesive composition |
| EP0754742B1 (en) * | 1995-07-19 | 2001-10-24 | Raytheon Company | Room-temperature stable, one-component, flexible epoxy adhesives |
| US8222324B2 (en) * | 2003-06-09 | 2012-07-17 | Kaneka Corporation | Process for producing modified epoxy resin |
| US8017689B2 (en) * | 2008-08-08 | 2011-09-13 | The Yokohama Rubber Co., Ltd. | Composition of epoxy resin, core-shell particles and curing agent |
| JP4416046B1 (ja) * | 2008-08-08 | 2010-02-17 | 横浜ゴム株式会社 | エポキシ樹脂組成物および構造用接着剤 |
| JP2014141604A (ja) * | 2013-01-25 | 2014-08-07 | Kaneka Corp | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 |
-
2017
- 2017-04-10 JP JP2018512004A patent/JP6966154B2/ja active Active
- 2017-04-10 CN CN201780022804.3A patent/CN109071920B/zh active Active
- 2017-04-10 WO PCT/JP2017/014681 patent/WO2017179536A1/ja not_active Ceased
- 2017-04-10 EP EP17782349.9A patent/EP3444301B1/en active Active
-
2018
- 2018-10-11 US US16/157,255 patent/US20190040290A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05214310A (ja) * | 1992-01-31 | 1993-08-24 | Nissan Motor Co Ltd | エポキシ樹脂系接着性組成物 |
| JPH08183836A (ja) * | 1994-12-30 | 1996-07-16 | Nippon Zeon Co Ltd | エポキシ樹脂組成物 |
| JP2010090371A (ja) * | 2008-09-11 | 2010-04-22 | Mitsubishi Rayon Co Ltd | エポキシ樹脂組成物、及びこれを硬化したエポキシ硬化物 |
| JP2010077305A (ja) * | 2008-09-26 | 2010-04-08 | Yokohama Rubber Co Ltd:The | エポキシ樹脂組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3444301A4 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018053026A (ja) * | 2016-09-27 | 2018-04-05 | 株式会社カネカ | 機械的強度に優れるポリマー微粒子含有硬化性組成物 |
| US20190315979A1 (en) * | 2018-04-16 | 2019-10-17 | Canon Kabushiki Kaisha | Curable resin composition and manufacturing method of three-dimensional object using the same |
| US11613661B2 (en) * | 2018-04-16 | 2023-03-28 | Canon Kabushiki Kaisha | Curable resin composition and manufacturing method of three-dimensional object using the same |
| JP2022047219A (ja) * | 2020-09-11 | 2022-03-24 | 国立研究開発法人物質・材料研究機構 | 定着構造体 |
| JP7686187B2 (ja) | 2020-09-11 | 2025-06-02 | 国立研究開発法人物質・材料研究機構 | 定着構造体 |
| JPWO2022138808A1 (ja) * | 2020-12-25 | 2022-06-30 | ||
| JP2024515381A (ja) * | 2021-08-13 | 2024-04-09 | エルジー・ケム・リミテッド | 硬化性樹脂組成物 |
| JP7688160B2 (ja) | 2021-08-13 | 2025-06-03 | エルジー・ケム・リミテッド | 硬化性樹脂組成物 |
| WO2023054479A1 (ja) * | 2021-09-30 | 2023-04-06 | 株式会社カネカ | 硬化性樹脂組成物およびその利用 |
| CN115322657A (zh) * | 2022-08-31 | 2022-11-11 | 常州大学 | 一种含有柔性固化剂的环氧树脂基膨胀防火涂料及其制备方法 |
| CN115322657B (zh) * | 2022-08-31 | 2024-03-15 | 常州大学 | 一种含有柔性固化剂的环氧树脂基膨胀防火涂料及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3444301A4 (en) | 2019-12-11 |
| EP3444301B1 (en) | 2021-04-07 |
| CN109071920A (zh) | 2018-12-21 |
| US20190040290A1 (en) | 2019-02-07 |
| JPWO2017179536A1 (ja) | 2019-02-28 |
| JP6966154B2 (ja) | 2021-11-10 |
| CN109071920B (zh) | 2021-11-02 |
| EP3444301A1 (en) | 2019-02-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6966154B2 (ja) | 硬化性組成物及び接着剤 | |
| JP6560617B2 (ja) | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6476045B2 (ja) | 接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6924135B2 (ja) | 耐衝撃剥離接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP7403463B2 (ja) | 硬化性エポキシ樹脂組成物、及びそれを用いた積層体 | |
| JP7547031B2 (ja) | エポキシ樹脂組成物及び接着剤 | |
| JP6767758B2 (ja) | 貯蔵安定性および接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6523611B2 (ja) | 異種部材を硬化性樹脂組成物で接合した積層体、および車両用構造パネル | |
| WO2016159223A1 (ja) | チキソトロピー性に優れる硬化性エポキシ樹脂組成物 | |
| JP7853135B2 (ja) | 硬化性樹脂組成物、その硬化物、接着剤および積層体 | |
| JP6722477B2 (ja) | 剥離接着性および耐衝撃剥離接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP7290446B2 (ja) | 硬化性樹脂組成物およびその利用 | |
| WO2019123934A1 (ja) | エポキシ樹脂組成物 | |
| WO2022138807A1 (ja) | 硬化性樹脂組成物及び接着剤 | |
| JP7531354B2 (ja) | エポキシ樹脂組成物及び接着剤 | |
| JP7277241B2 (ja) | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 | |
| WO2023249099A1 (ja) | 硬化性樹脂組成物、硬化物、接着剤および積層体 | |
| WO2024247944A1 (ja) | 一成分型熱硬化性樹脂組成物およびその利用 | |
| CN116490361A (zh) | 单组分型固化性树脂组合物及粘接剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018512004 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017782349 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017782349 Country of ref document: EP Effective date: 20181112 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17782349 Country of ref document: EP Kind code of ref document: A1 |