WO2017196697A1 - Drug delivery system for the delivery of antiviral agents - Google Patents
Drug delivery system for the delivery of antiviral agents Download PDFInfo
- Publication number
- WO2017196697A1 WO2017196697A1 PCT/US2017/031493 US2017031493W WO2017196697A1 WO 2017196697 A1 WO2017196697 A1 WO 2017196697A1 US 2017031493 W US2017031493 W US 2017031493W WO 2017196697 A1 WO2017196697 A1 WO 2017196697A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- drug delivery
- delivery system
- poly
- ethynyl
- fluoro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/50—Materials characterised by their function or physical properties, e.g. injectable or lubricating compositions, shape-memory materials, surface modified materials
- A61L27/54—Biologically active materials, e.g. therapeutic substances
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/173—Purine radicals with 2-deoxyribosyl as the saccharide radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/14—Macromolecular materials
- A61L27/16—Macromolecular materials obtained by reactions only involving carbon-to-carbon unsaturated bonds
Definitions
- HAART regimens have proven to be highly effective treatments, significantly decreasing HIV viral load in HIV-infected patients, thereby slowing the evolution of the illness and reducing HIV-related morbidity and mortality. Yet, the treatment success of HAART is directly related to adherence to the regimen by the patient. Unless appropriate levels of the antiretroviral drug combinations are maintained in the blood, viral mutations will develop, leading to therapy resistance and cross-resistances to molecules of the same therapeutic class, thus placing the long- term efficacy of treatments at risk. Various clinical studies have shown a decline in treatment effectiveness with relatively small lapses in adherence.
- HAART regimens continue to be far from optimal.
- Various characteristics of HAART make adherence particularly difficult.
- Therapeutic regimens are complex, requiring multiple drugs to be taken daily, often at different times of the day, and many with strict requirements on food intake.
- Many HAART medications also have unpleasant side effects, including nausea, diarrhea, headache, and peripheral neuropathy. Social and psychological factors can also negatively impact adherence. Patients report that
- New HIV treatment interventions aim to improve adherence by reducing the complexity of treatments, the frequency of the dosages, and/or the side effects of the
- LAI Long-acting injectable
- This invention relates to novel implant drug delivery systems for long-acting delivery of antiviral drugs. These compositions are useful for the treatment or prevention of human immunodeficiency virus (HIV) infection.
- HIV human immunodeficiency virus
- Figure 1 X-ray image of a barium sulfate containing implant in a rat (image taken after a 6 month duration in vivo).
- This invention relates to novel implant drug delivery systems for long-acting delivery of antiviral drugs.
- the novel implant drug delivery systems comprise a polymer and an antiviral agent. These implant drug delivery systems are useful for the treatment or prevention of human immunodeficiency virus (HIV) infection.
- the invention further relates to methods of treating and preventing HIV infection with the novel implant drug delivery systems described herein.
- novel implant delivery systems of the invention comprise a biocompatible nonerodible polymer to generate monolithic matrices with dispersed or dissolved drug.
- the chemical properties of the polymer matrices are tuned to achieve a range of drug release characteristics, offering the opportunity to extend duration of dosing.
- the novel implant delivery systems are compatible with molecules having a broad spectrum of physicochemical properties, including those of high aqueous solubility or amorphous phases which are unsuitable to formulation as solid drug suspensions.
- this invention relates to novel implant drug delivery systems comprising a biocompatible nonerodible polymer and 4'-ethynyl-2-fluoro-2'-deoxyadenosine wherein said implant drug delivery system is implanted subdermally and 4'-ethynyl-2-fluoro-2'- deoxyadenosine is continually released in vivo at a rate resulting in a plasma concentration between 0.01 ng/mL and 3000.0 ng/mL.
- implant delivery systems are desired and useful for prophylaxis and/or treatment of HIV infection from both compliance and convenience standpoints.
- biocompatible nonerodible polymer refers to polymeric materials that are sufficiently resistant to degradation (both chemical and physical) in the presence of biological systems. Biocompatible nonerodible polymers are sufficiently resistant to chemical and/or physical destruction by the environment of use such that the polymer remains essentially intact throughout the release period.
- the polymer is generally hydrophobic so that it retains its integrity for a suitable period of time when placed in an aqueous
- Nonerodible polymers remain intact in vivo for extended periods of time, typically months or years.
- Drug molecules encapsulated in the polymer are released over time via diffusion through channels and pores in a sustained manner.
- the release rate can be altered by modifying the percent drug loading, porosity of the polymer, structure of the implantable device, or hydrophobicity of the polymer, or by adding a coating to the exterior of the implantable device.
- Biocompatible nonerodible polymers of the instant invention include, but are not limited to, ethylene vinyl acetate copolymer (EVA), poly(urethane), silicone, hydrogels such as crosslinked poly(vinyl alcohol) and poly(hydroxy ethylmethacrylate), acyl substituted cellulose acetates and alkyl derivatives thereof, partially and completely hydrolyzed alkylene-vinyl acetate copolymers, unplasticized polyvinyl chloride, crosslinked homo- and copolymers of polyvinyl acetate, crosslinked polyesters of acrylic acid and/or methacrylic acid, polyvinyl alkyl ethers, polyvinyl fluoride, polycarbonate, polyamide, polysulphones, styrene acrylonitrile copolymers, crosslinked poly(ethylene oxide),
- EVA ethylene vinyl acetate copolymer
- silicone hydrogels such as crosslinked poly(vinyl alcohol) and poly(hydroxy ethylmethacrylate),
- the biocompatible nonerodible polymer is ethylene vinyl acetate copolymer (EVA).
- the biocompatible nonerodible polymer is selected from the group consisting of ethylene vinyl acetate copolymer (9% vinyl acetate), ethylene vinyl acetate copolymer (15% vinyl acetate), ethylene vinyl acetate copolymer (28% vinyl acetate), and ethylene vinyl acetate copolymer (33% vinyl acetate).
- the biocompatible nonerodible polymer is ethylene vinyl acetate copolymer (9% vinyl acetate).
- the biocompatible nonerodible polymer is ethylene vinyl acetate copolymer (15% vinyl acetate).
- the biocompatible nonerodible polymer is poly(urethane).
- the term "diffusional barrier” refers to a coating that is permeable to the drug and is placed over at least a portion of the device to further regulate the rate of release.
- a coating of biocompatible nonerodible polymeric material e.g., EVA
- a coating of a biocompatible nonerodible polymeric material with a lower drug loading than the remainder of the implant delivery system may be used.
- the diffusional barrier may be formed, for example, by coextrusion with the device.
- Suitable diffusional barriers of the instant invention include, but are not limited to, ethylene vinyl acetate copolymer (EVA), poly(urethane), silicone, hydrogels such as crosslinked poly(vinyl alcohol) and poly(hydroxy ethylmethacrylate), acyl substituted cellulose acetates and alkyl derivatives thereof, partially and completely hydrolyzed alkylene-vinyl acetate copolymers, unplasticized polyvinyl chloride, crosslinked homo- and copolymers of polyvinyl acetate, crosslinked polyesters of acrylic acid and/or methacrylic acid, polyvinyl alkyl ethers, polyvinyl fluoride, polycarbonate, polyamide, polysulphones, styrene acrylonitrile copolymers, crosslinked poly(ethylene oxide), poly(alkylenes), poly(vinyl imidazole), poly(esters), poly(ethylene terephthalate), polyphosphazenes, and chlorosulphonated poly
- the diffusional barrier is poly(urethane). In a class of the invention, the diffusional barrier is ethylene vinyl acetate copolymer (EVA). In another class of the invention, the diffusional barrier is poly(urethane).
- the diffusion barrier contains an antiviral drug.
- the diffusion barrier comprises 4'-ethynyl-2-fluoro-2'- deoxyadenosine.
- the term "dispersed or dissolved in the biocompatible nonerodible polymer” refers to the drug and polymer being mixed and then hot-melt extruded.
- the term “continually released” refers to the drug being released from the biocompatible nonerodible polymer at continuous rates for extended periods of time.
- the implant drug delivery systems of the instant invention generally exhibit linear release kinetics for the drug in vivo, sometimes after an initial burst.
- the novel implant delivery systems of the instant invention can further comprise a radiopaque component.
- the radiopaque component will cause the implant to be X- ray visible.
- the radiopaque component can be any such element known in the art, such as barium sulphate, titanium dioxide, bismuth oxide, tantalum, tungsten or platinum. In a specific embodiment, the radiopaque component is barium sulphate.
- the radiopaque material is about 1% to 30% by weight. In another embodiment, the radiopaque material is about 1% to 20% by weight. In another embodiment, the radiopaque material is about 4% to 25% by weight. In further embodiment, the radiopaque material is about 6% to 20% by weight. In another embodiment, the radiopaque material is about 4% to 15% by weight. In another embodiment, the radiopaque material is about 8% to 15% by weight.
- the radiopaque material does not affect the release of 4'-ethynyl-2-fIuoro-2'- deoxyadenosine from the implant.
- novel implant delivery systems of the invention comprise antiviral agents.
- Suitable antiviral agents include anti-HIV agents.
- the antiviral agent is administered as a monotherapy.
- two or more antiviral agents are administered in combination.
- an "anti-HIV agent” is any agent which is directly or indirectly effective in the inhibition of HIV reverse transcriptase or another enzyme required for HIV replication or infection, or the prophylaxis of HIV infection, and/or the treatment, prophylaxis or delay in the onset or progression of AIDS. It is understood that an anti-HIV agent is effective in treating, preventing, or delaying the onset or progression of HIV infection or AIDS and/or diseases or conditions arising therefrom or associated therewith.
- Suitable anti-viral agents for use in implant drug delivery systems described herein include, for example, those listed in Table A as follows:
- EI entry inhibitor
- FI fusion inhibitor
- Inl integrase inhibitor
- PI protease
- nRTI nucleoside reverse transcriptase inhibitor
- nnRTI non-nucleoside
- drugs listed in the table can be used in a salt form; e.g., abacavir sulfate, delavirdine mesylate, indinavir sulfate, atazanavir sulfate, nelfinavir mesylate, saquinavir mesylate.
- antiviral agents in the implant drug delivery systems described herein are employed in their conventional dosage ranges and regimens as reported in the art, including, for example, the dosages described in editions of the Physicians' Desk
- the antiviral agents in the implant drug delivery systems described herein are employed in lower than their conventional dosage ranges. In other embodiments, the antiviral agents in the implant drug delivery systems described herein are employed in higher than their conventional dosage ranges.
- the antiviral agent can be an entry inhibitor; fusion inhibitor; integrase inhibitor; protease inhibitor; nucleoside reverse transcriptase inhibitor; or non-nucleoside reverse transcriptase inhibitor.
- the antiviral agent is a nucleoside reverse transcriptaseinhibitor.
- the antiviral agent is a nucleoside reverse transciptase inhibitor (NRTI).
- NRTI nucleoside reverse transciptase inhibitor
- the NRTI is 4'-ethynyl-2-fluoro-2'- deoxyadenosine.
- the antiviral agent is present in the biocompatible nonerodible polymer at about 0.10% - 80% by weight of drug loading. In other embodiments, the antiviral agent is present in the biocompatible nonerodible polymer at about 20%-60% by weight, at about 40%-60% by weight, at about 40%- 50% by weight or at about 40%-45% by weight of drug loading. In a class of the embodiment of the implant drug delivery system described herein, 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 0.10%-80% by weight of drug loading.
- 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 20%-60% by weight of drug loading.
- 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 30%-65% by weight of drug loading.
- 4'- ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 40%-60% by weight of drug loading.
- 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 40%-50% by weight of drug loading.
- 4'- ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at about 40%-45% by weight of drug loading.
- 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at 40% by weight of drug loading.
- 4'-ethynyl-2-fIuoro-2'- deoxyadenosine is present in the biocompatible nonerodible polymer at 45% by weight of drug loading.
- 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at 50% by weight of drug loading. In another example of the embodiment of the implant drug delivery system described herein, 4'-ethynyl-2-fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at 60% by weight of drug loading. In another example of the embodiment of the implant drug delivery system described herein, 4'-ethynyl-2- fluoro-2'-deoxyadenosine is present in the biocompatible nonerodible polymer at 80% by weight of drug loading.
- the implant drug delivery systems of the instant invention may be produced using an extrusion process, wherein ground biocompatible, nonerodible polymer is blended with the antiviral agent, melted and extruded into rod-shaped structures. Rods are cut into individual implantable devices of the desired length, packaged and sterilized prior to use.
- Other methods for encapsulating therapeutic compounds in implantable polymeric, nonerodible matrices are known to those of skill in the art. Such methods include solvent casting (see US Patent Nos. 4,883,666, 5, 114,719 and 5,601835).
- solvent casting see US Patent Nos. 4,883,666, 5, 114,719 and 5,601835.
- One of skill in the art would be able to readily determine an appropriate method of preparing such an implant drug delivery system, depending on the shape, size, drug loading, and release kinetics desired for a particular type of patient or clinical application.
- the size and shape of the implant drug delivery systems may be modified to achieve a desired overall dosage.
- the implant drug delivery systems of the instant invention are often about 0.5 cm to about 10 cm in length. In an embodiment of the invention, the implant drug delivery systems are about 1.5 cm to about 5 cm in length. In a class of the embodiment, the implant drug delivery systems are about 2 cm to about 5 cm in length. In a subclass of the embodiment, the implant drug delivery systems are about 2 cm to about 4 cm in length.
- the implant drug delivery systems of the instant invention are often about 0.5 mm to about 7 mm in diameter. In an embodiment of the invention, the implant drug delivery systems are about 1.5 mm to about 5 mm in diameter. In a class of the embodiment, the implant drug delivery systems are about 2 mm to about 5 mm in diameter. In a subclass of the embodiment, the implant drug delivery systems are about 2 mm to about 4 mm in diameter.
- the implant drug delivery systems described herein are capable of releasing 4'- ethynyl-2-fluoro-2'-deoxyadenosine over a period of 21 days, 28 days, 31 days, 4 weeks, 6 weeks, 8 weeks, 12 weeks, one month, two months, three months, four months, five months, six months, seven months, eight months, nine months, ten months, eleven months, twelve months, eighteen months, twenty-four months or thirty-six months at an average rate of between 0.02-8.0 ng per day.
- the 4'-ethynyl-2-fluoro-2'-deoxyadenosine is released at therapeutic concentrations for a duration from between three months and thirty-six months.
- the 4'-ethynyl-2-fluoro-2'-deoxyadenosine is released at therapeutic concentrations for a duration from between six months and twelve months. In an embodiment of the invention, the 4'-ethynyl-2-fluoro-2'-deoxyadenosine is released at prophylactic concentrations for a duration from between three months and thirty-six months. In a class of the embodiment, the 4'-ethynyl-2-fluoro-2'-deoxyadenosine is released at prophylactic concentrations for a duration from between six months and twelve months.
- One or more implants can be used to achieve the desired therapeutic dose. In an embodiment of the invention, one or more implants can be used to achieve the therapeutic dose for durations of up to 1 year. In another embodiment of the invention, one or more implants can be used to achieve the therapeutic dose for durations of up to 2 years.
- the implant drug delivery systems described herein are capable of releasing 4'- ethynyl-2-fluoro-2'-deoxyadenosine resulting in a plasma concentration of between 0.02-300 ng/mL per day.
- the implant drug delivery systems described herein are capable of releasing 4'-ethynyl-2-fluoro-2'-deoxyadenosine resulting in a plasma concentration of between 0.02-30.0 ng/mL per day.
- the implant drug delivery systems described herein are capable of releasing 4'-ethynyl-2-fluoro-2'- deoxyadenosine resulting in a plasma concentration of between 0.02-15.0 ng/mL per day.
- the implant drug delivery systems described herein are capable of releasing 4'-ethynyl-2-fluoro-2'-deoxyadenosine resulting in a plasma concentration of between 0.02-8.0 ng/mL per day.
- the implant drug delivery systems described herein are capable of releasing 4'-ethynyl-2-fluoro-2'-deoxyadenosine resulting in a plasma concentration of between 0.1-1.0 ng/mL per day.
- Implants were prepared using an extrusion process.
- the micronized polymer, and 4'-ethynyl-2-fluoro-2'-deoxyadenosine were blended at various ratios: 30, 35, 40, 45 and 50wt% drug in EVA.
- the preblend was melt extruded with a twin screw extruder at temperatures ranging from 100-140°C, screw speed at 30 rpm, and then pelletized.
- the pellets were then extruded with a single screw extruder with temperatures ranging from 110-140°C, and screw speed at 20-25 rpm to form a 2 ⁇ 0.05mm diameter filament, and then cut to a length of 40 ⁇ 2mm.
- the in vitro release rate of 4'-ethynyl-2-fluoro-2'-deoxyadenosine was determined by incubating the implants segments, approximately 1 cm in length, in a glass vial containing phosphate buffered saline (PBS) at 37°C, and 50 rpm shaking in an Innova 42 incubator.
- PBS phosphate buffered saline
- the volume of PBS was sufficient to maintain sink conditions. Sink conditions are defined as the drug concentration maintained at or below 1/3 of the maximum solubility (drug concentration ⁇ 0.45 mg/mL in PBS at 37°C).
- Samples were removed (0.5 mL) at selected time points, and centrifuged at 20,800xg for 8 min. The supernatant was removed (0.4 mL), diluted 4- fold, and vortexed.
- Implantable devices were prepared using an extrusion process.
- the first step involved mixing the dry, micronized powders of the active compound and the cryomilled EVA using a Turbula T2F mixer.
- Drug and polymer blends were prepared at 50, 60 and 80 wt % drug load.
- the 4'-ethynyl-2-fluoro-2'-deoxyadenosine and polymer blends were hot-melt extruded using a twin screw extruder through a 3 mm diameter die, and pulled to a diameter of approximately 1.9-2.3 mm.
- the screws contained predominately conveying elements with a single 90° mixing section.
- the 1st zone where the drug-polymer blends were introduced was water-cooled and maintained at room temperature. The temperature for zones 2-4 was 100°C .
- Extruded fibers with diameters between 1.9-2.3 mm were cut to a length of approximately 40 mm.
- the in vitro release rate of 4'-ethynyl-2-fluoro-2' -deoxyadenosine was determined by incubating the implants segments, approximately 1 cm in length, in a glass vial containing phosphate buffered saline (PBS) at 37°C, and 50 rpm shaking in an Innova 42 incubator.
- PBS phosphate buffered saline
- the volume of PBS was sufficient to maintain sink conditions. Sink conditions are defined as the drug concentration maintained at or below 1/3 of the maximum solubility (drug concentration ⁇ 0.45 mg/mL in PBS at 37°C).
- Samples were removed (0.5 mL) at selected time points, and centrifuged at 20,800xg for 8 min. The supernatant was removed (0.4 mL), diluted 4- fold, and vortexed.
- Implantable devices were prepared using an extrusion process.
- the first step involved mixing the dry, micronized powders of the active compound and the cryomilled EVA using a Turbula T2F mixer.
- Drug and polymer blends were prepared at 40, 50, 60 and 80 wt % drug load.
- the 4'-ethynyl-2-fluoro-2'-deoxyadenosine and polymer blends were hot-melt extruded using a twin screw extruder through a 3 mm diameter die, and pulled to a diameter of approximately 1.9-2.3 mm.
- the screws contained predominately conveying elements with a single 90° mixing section.
- the 1st zone where the drug-polymer blends were introduced was water-cooled and maintained at room temperature.
- zones 2-4 The temperature for zones 2-4 was 100°C. Extruded fibers with diameters between 1.9-2.3 mm were cut to an appropriate length to achieve the desired amount of drug per implant for in vivo studies. All animal studies were conducted following protocols in accordance with the Institutional Animal Care and Use Committee
- Implants were prepared using an extrusion process.
- the micronized polymer, 4'- ethynyl-2-fluoro-2'-deoxyadenosine, and BaS0 4 were blended at various ratios: 40 and 45wt% drug in EVA, and 35 and 40wt% drug with 10wt% BaS0 4 in EVA.
- the preblend was melt extruded with a twin screw extruder at temperatures ranging from 100-140°C, screw speed at 30 rpm, and then pelletized.
- the pellets were then extruded with a single screw extruder with temperatures ranging from 1 10-140°C, and screw speed at 20-25 rpm to form a 2 ⁇ 0.05mm diameter filament, and then cut to a length of 40 ⁇ 2mm.
- the in vitro release rate of 4'-ethynyl-2-fluoro-2'-deoxyadenosine was determined by incubating the implants segments, approximately 1 cm in length, in a glass vial containing phosphate buffered saline (PBS) at 37°C, and 50 rpm shaking in an Innova 42 incubator.
- PBS phosphate buffered saline
- the volume of PBS was sufficient to maintain sink conditions. Sink conditions are defined as the drug concentration maintained at or below 1/3 of the maximum solubility (drug concentration ⁇ 0.45 mg/mL in PBS at 37°C).
- Samples were removed (0.5 mL) at selected time points, and centrifuged at 20,800xg for 8 min. The supernatant was removed (0.4 mL), diluted 4- fold, and vortexed.
- Implants were prepared using an extrusion process.
- the micronized polymer, 4'- ethynyl-2-fluoro-2'-deoxyadenosine, and BaS0 4 were blended at various ratios: 40 and 45wt% drug in EVA, and 35 and 40wt% drug with 10wt% BaS0 4 in EVA.
- the preblend was melt extruded with a twin screw extruder at temperatures ranging from 100-140°C, screw speed at 30 rpm, and then pelletized.
- the pellets were then extruded with a single screw extruder with temperatures ranging from 1 10-140°C, and screw speed at 20-25 rpm to form a 2 ⁇ 0.05mm diameter filament, and then cut to the appropriate length to achieve the desired amount of drug per implant for in vivo studies. All animal studies were conducted following protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) at NIRC and Merck, which adhere to the regulations outlined in the USDA Animal Welfare Act. For each implantation, a Wistar Han rat was anesthetized using isoflurane to effect prior to subcutaneous dose administrations.
- IACUC Institutional Animal Care and Use Committee
- the solid formulation ( ⁇ 2mm in diameter and of varying lengths based on the body weight of the individual animal to achieve the dose appropriate for each group) was placed in the scapular region.
- Figure 1 shows an x-ray image of an implant containing barium sulfate in a rat after a 6 month duration.
- Implants were prepared using an extrusion process.
- the micronized polymer, and 4'-ethynyl-2-fluoro-2'-deoxyadenosine were blended at 45wt% drug in EVA.
- the preblend was melt extruded with a twin screw extruder at temperatures ranging from 100-140°C, screw speed at 30 rpm, and then pelletized.
- the pellets were then extruded with a single screw extruder with temperatures ranging from 1 10-140°C, and screw speed at 20-25 rpm to form a 2 ⁇ 0.05mm diameter filament, and then cut to a length of 40 ⁇ 2mm.
- the implant was placed subcutaneously in the interscapular region.
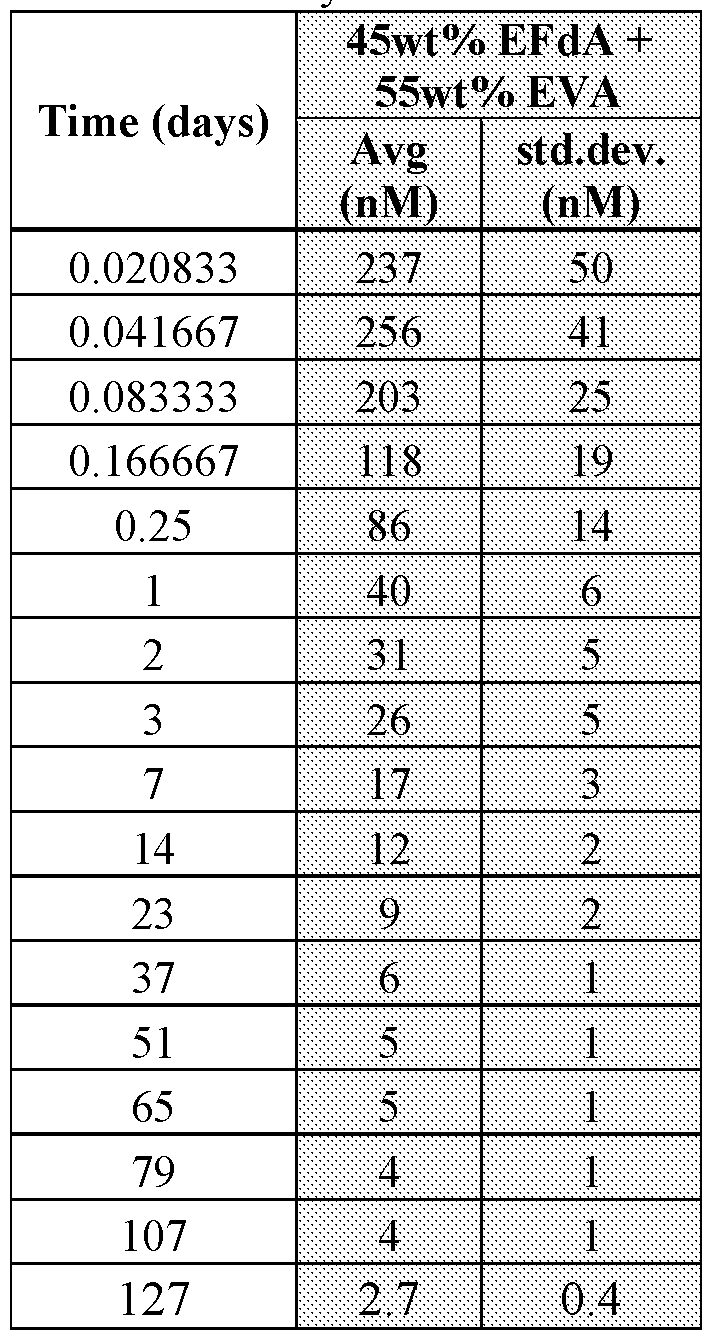
- Implants were prepared by extrusion of a 45 :55 4'-ethynyl-2-fluoro-2'- deoxyadenosineEVA at elevated temperature yielding fibers having diameters between 2.00 ⁇ 0.05 mm that were cut to 40 ⁇ 2mm for in vivo studies. All animal studies were conducted following protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) at NIRC and Merck, which adhere to the regulations outlined in the USDA Animal Welfare Act. For each implantation, a Rhesus monkey was sedated with Ketamine HCl (100 mg/mL) prior to subcutaneous dose administrations. Using an injector device, the implant was placed subcutaneously in the interscapular region. Three animals (all males) were used.
- IACUC Institutional Animal Care and Use Committee
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Dermatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Oral & Maxillofacial Surgery (AREA)
- Transplantation (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Materials For Medical Uses (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description





















Claims
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018558762A JP6934019B2 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for delivering antiviral drugs |
| RU2018141823A RU2755130C2 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for delivery of antiviral drugs |
| KR1020187035360A KR102272235B1 (en) | 2016-05-12 | 2017-05-08 | Drug Delivery Systems for Delivery of Antiviral Agents |
| BR112018072883-7A BR112018072883B1 (en) | 2016-05-12 | 2017-05-08 | 4'-ETHYNYL-2-FLUORO-2'-DEOXYADENOSINE ANTIVIRAL AGENT SUBDERMAL IMPLANT SYSTEM WITH EVA POLYMER AND ITS USE |
| US16/300,121 US11400186B2 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for the delivery of antiviral agents |
| CN202211593835.3A CN115624564A (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for delivering antiviral agents |
| AU2017263253A AU2017263253B2 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for the delivery of antiviral agents |
| MX2018013662A MX386770B (en) | 2016-05-12 | 2017-05-08 | DRUG DELIVERY SYSTEM FOR THE DELIVERY OF ANTIVIRAL AGENTS. |
| CA3023364A CA3023364A1 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for the delivery of antiviral agents |
| EP17796613.2A EP3454868A4 (en) | 2016-05-12 | 2017-05-08 | DRUG DELIVERY SYSTEM FOR DELIVERY OF ANTIVIRAL AGENTS |
| CN201780028478.7A CN109069526A (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for delivering antiviral agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662335319P | 2016-05-12 | 2016-05-12 | |
| US62/335,319 | 2016-05-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017196697A1 true WO2017196697A1 (en) | 2017-11-16 |
Family
ID=60266868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2017/031493 Ceased WO2017196697A1 (en) | 2016-05-12 | 2017-05-08 | Drug delivery system for the delivery of antiviral agents |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US11400186B2 (en) |
| EP (1) | EP3454868A4 (en) |
| JP (1) | JP6934019B2 (en) |
| KR (1) | KR102272235B1 (en) |
| CN (2) | CN109069526A (en) |
| AU (1) | AU2017263253B2 (en) |
| BR (1) | BR112018072883B1 (en) |
| CA (1) | CA3023364A1 (en) |
| MX (1) | MX386770B (en) |
| RU (1) | RU2755130C2 (en) |
| WO (1) | WO2017196697A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020131649A1 (en) | 2018-12-20 | 2020-06-25 | Merck Sharp & Dohme Corp. | Novel crystalline forms of an nrtti compound |
| EP3609508A4 (en) * | 2017-04-10 | 2021-02-10 | Merck Sharp & Dohme Corp. | DRUG DELIVERY SYSTEM FOR THE DELIVERY OF ANTIVIRAL AGENTS |
| WO2021030306A1 (en) * | 2019-08-13 | 2021-02-18 | Merck Sharp & Dohme Corp. | Drug delivery system for the delivery of antiviral agents |
| US20230149296A1 (en) * | 2020-05-05 | 2023-05-18 | Merck Sharp & Dohme Llc | Drug delivery system for the delivery of antiviral agents and contraceptives |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JOP20170038B1 (en) * | 2016-02-12 | 2021-08-17 | Merck Sharp & Dohme | Compounds for use for treatment and prophylaxis of HIV infection |
| JP7402225B2 (en) | 2018-05-11 | 2023-12-20 | ロード アイランド ホスピタル | Compositions and methods for treating joint disorders using nucleoside reverse transcriptase inhibitors |
| SG11202107145SA (en) | 2019-01-25 | 2021-08-30 | Univ Brown | Compositions and methods for treating, preventing or reversing age-associated inflammation and disorders |
| US20250215039A1 (en) * | 2022-03-30 | 2025-07-03 | Primefour Therapeutics, Inc. | Nucleosides for treating cancer |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4883666A (en) | 1987-04-29 | 1989-11-28 | Massachusetts Institute Of Technology | Controlled drug delivery system for treatment of neural disorders |
| US5114719A (en) | 1987-04-29 | 1992-05-19 | Sabel Bernhard A | Extended drug delivery of small, water-soluble molecules |
| US5601835A (en) | 1987-04-29 | 1997-02-11 | Massachusetts Institute Of Technology | Polymeric device for controlled drug delivery to the CNS |
| US20040175426A1 (en) * | 2003-01-24 | 2004-09-09 | Control Delivery Systems, Inc. | Controlled release of highly soluble agents |
| WO2005090349A1 (en) | 2004-03-24 | 2005-09-29 | Yamasa Corporation | 4'-c-substituted 2-haloadenosine derivative |
| WO2009126293A2 (en) | 2008-04-11 | 2009-10-15 | Yale University | Potent chimeric nrti-nnrti bifunctional inhibitors of hiv-1 reverse transcriptase |
| US20100080830A1 (en) * | 2000-04-26 | 2010-04-01 | Psivida Inc. | Systemic delivery of antiviral agents |
| US20110206745A1 (en) * | 2008-06-25 | 2011-08-25 | Endo Pharmaceuticals Solutions Inc. | Octreotide implant having a release agent |
| US20130195950A1 (en) * | 2010-03-16 | 2013-08-01 | Titan Pharmaceuticals, Inc. | Heterogeneous implantable devices for drug delivery |
| US20150051167A1 (en) * | 2012-03-21 | 2015-02-19 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2015086489A1 (en) | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme B.V. | Drug delivery system for delivery of anti-virals |
| US20150297553A1 (en) * | 2013-03-11 | 2015-10-22 | Del Mar Pharmaceuticals | Compositions and methods to improve the therapeutic benefit of suboptimally administered chemical compounds including substituted hexitols such as dianhydrogalactitol and diacetyldianhydrogalactitol |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8430703D0 (en) * | 1984-12-05 | 1985-01-16 | Lrc Products | Drug release devices |
| US5633000A (en) | 1994-06-23 | 1997-05-27 | Axxia Technologies | Subcutaneous implant |
| US5883144A (en) * | 1994-09-19 | 1999-03-16 | Sentinel Products Corp. | Silane-grafted materials for solid and foam applications |
| WO2003094888A1 (en) * | 2002-05-07 | 2003-11-20 | Control Delivery Systems, Inc. | Processes for forming a drug delivery device |
| TWI336627B (en) * | 2003-05-23 | 2011-02-01 | Organon Nv | Drug delivery system,and use and manufacturing method thereof |
| MXPA05012768A (en) * | 2003-05-30 | 2006-02-22 | Titan Pharmaceuticals Inc | Implantable polymeric device for sustained release of nalmefene. |
| TWI434676B (en) * | 2004-03-19 | 2014-04-21 | Merck Sharp & Dohme | X-ray visible drug delivery device |
| CN100532388C (en) * | 2007-07-16 | 2009-08-26 | 郑州大学 | 2 '-fluoro-4' -substituted-nucleoside analogue, preparation method and application thereof |
| EP2254582B1 (en) * | 2008-01-25 | 2016-01-20 | Chimerix, Inc. | Methods of treating viral infections |
| WO2010072844A1 (en) * | 2008-12-24 | 2010-07-01 | Tibotec Pharmaceuticals | Implantable devices for treating hiv |
| RU2015136769A (en) * | 2013-02-05 | 2017-03-13 | Дзе Попьюлейшн Каунсил, Инк. | INTRAVAGINAL RING FOR DELIVERY OF UNIQUE COMBINATIONS OF ANTI-MICROBIAL COMPOSITIONS |
| WO2016149561A1 (en) | 2015-03-17 | 2016-09-22 | Oak Crest Institute Of Science | Subdermal implants for the sustained delivery of water-soluble drugs |
| US20190388336A1 (en) | 2016-06-20 | 2019-12-26 | Merck Sharp & Dohme Corp. | Drug Delivery System for the Delivery of Antiviral Agents |
| EP3515438B1 (en) | 2016-09-21 | 2022-03-02 | Merck Sharp & Dohme Corp. | Drug delivery system for the delivery of integrase inhibitors |
| WO2018191093A1 (en) | 2017-04-10 | 2018-10-18 | Merck Sharp & Dohme Corp. | Drug delivery system for the delivery of antiviral agents |
-
2017
- 2017-05-08 KR KR1020187035360A patent/KR102272235B1/en not_active Expired - Fee Related
- 2017-05-08 BR BR112018072883-7A patent/BR112018072883B1/en not_active IP Right Cessation
- 2017-05-08 EP EP17796613.2A patent/EP3454868A4/en active Pending
- 2017-05-08 WO PCT/US2017/031493 patent/WO2017196697A1/en not_active Ceased
- 2017-05-08 MX MX2018013662A patent/MX386770B/en unknown
- 2017-05-08 RU RU2018141823A patent/RU2755130C2/en active
- 2017-05-08 US US16/300,121 patent/US11400186B2/en active Active
- 2017-05-08 CA CA3023364A patent/CA3023364A1/en active Pending
- 2017-05-08 CN CN201780028478.7A patent/CN109069526A/en active Pending
- 2017-05-08 CN CN202211593835.3A patent/CN115624564A/en active Pending
- 2017-05-08 AU AU2017263253A patent/AU2017263253B2/en not_active Ceased
- 2017-05-08 JP JP2018558762A patent/JP6934019B2/en not_active Expired - Fee Related
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5114719A (en) | 1987-04-29 | 1992-05-19 | Sabel Bernhard A | Extended drug delivery of small, water-soluble molecules |
| US5601835A (en) | 1987-04-29 | 1997-02-11 | Massachusetts Institute Of Technology | Polymeric device for controlled drug delivery to the CNS |
| US4883666A (en) | 1987-04-29 | 1989-11-28 | Massachusetts Institute Of Technology | Controlled drug delivery system for treatment of neural disorders |
| US20100080830A1 (en) * | 2000-04-26 | 2010-04-01 | Psivida Inc. | Systemic delivery of antiviral agents |
| US20040175426A1 (en) * | 2003-01-24 | 2004-09-09 | Control Delivery Systems, Inc. | Controlled release of highly soluble agents |
| WO2005090349A1 (en) | 2004-03-24 | 2005-09-29 | Yamasa Corporation | 4'-c-substituted 2-haloadenosine derivative |
| US20050215512A1 (en) | 2004-03-24 | 2005-09-29 | Satoru Kohgo | 4' -C-substituted-2-haloadenosine derivative |
| WO2009126293A2 (en) | 2008-04-11 | 2009-10-15 | Yale University | Potent chimeric nrti-nnrti bifunctional inhibitors of hiv-1 reverse transcriptase |
| US20110206745A1 (en) * | 2008-06-25 | 2011-08-25 | Endo Pharmaceuticals Solutions Inc. | Octreotide implant having a release agent |
| US20130195950A1 (en) * | 2010-03-16 | 2013-08-01 | Titan Pharmaceuticals, Inc. | Heterogeneous implantable devices for drug delivery |
| US20150051167A1 (en) * | 2012-03-21 | 2015-02-19 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US20150297553A1 (en) * | 2013-03-11 | 2015-10-22 | Del Mar Pharmaceuticals | Compositions and methods to improve the therapeutic benefit of suboptimally administered chemical compounds including substituted hexitols such as dianhydrogalactitol and diacetyldianhydrogalactitol |
| WO2015086489A1 (en) | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme B.V. | Drug delivery system for delivery of anti-virals |
Non-Patent Citations (7)
| Title |
|---|
| "Physicians' Desk Reference", 2009 |
| BAERT, L. ET AL.: "Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment", EUROPEAN JOURNAL OF PHARMACEUTICS AND BIOPHARMACEUTICS, vol. 72, no. 3, 2009, pages 502 - 508, XP026218287, DOI: 10.1016/j.ejpb.2009.03.006 |
| MUSIIME, S. ET AL.: "Adherence to Highly Active Antiretroviral Treatment in HIV-Infected Rwandan Women", PLOS ONE, vol. 6, no. 11, 2011, pages 1 - 6 |
| RAJOLI, R. K. R. ET AL.: "Physiologically Based Pharmacokinetic Modelling to Inform Development of Intramuscular Long-Acting Nanoformulations for HIV", CLINICAL PHARMACOKINETICS, vol. 54, no. 6, 2015, pages 639 - 650, XP055394071, DOI: 10.1007/s40262-014-0227-1 |
| See also references of EP3454868A4 |
| SPREEN, W. R. ET AL.: "Long-acting injectable antiretrovirals for HIV treatment and prevention", CURRENT OPINION IN HIV AND AIDS, vol. 8, no. 6, 2013, pages 565 - 571, XP055919554, DOI: 10.1097/COH.0000000000000002 |
| VAN 'T KLOOSTER, G. ET AL.: "Pharmacokinetics and Disposition of Rilpivirine (TMC278) Nanosuspension as a Long-Acting Injectable Antiretroviral Formulation", ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 54, no. 5, 2010, pages 2042 - 2050, XP055190760, DOI: 10.1128/AAC.01529-09 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3609508A4 (en) * | 2017-04-10 | 2021-02-10 | Merck Sharp & Dohme Corp. | DRUG DELIVERY SYSTEM FOR THE DELIVERY OF ANTIVIRAL AGENTS |
| US11419817B2 (en) | 2017-04-10 | 2022-08-23 | Merck Sharp & Dohme Llc | Drug delivery system for the delivery of antiviral agents |
| WO2020131649A1 (en) | 2018-12-20 | 2020-06-25 | Merck Sharp & Dohme Corp. | Novel crystalline forms of an nrtti compound |
| WO2021030306A1 (en) * | 2019-08-13 | 2021-02-18 | Merck Sharp & Dohme Corp. | Drug delivery system for the delivery of antiviral agents |
| US20220362277A1 (en) * | 2019-08-13 | 2022-11-17 | Merck Sharp & Dohme Corp. | Drug delivery system for the delivery of antiviral agents |
| US20230149296A1 (en) * | 2020-05-05 | 2023-05-18 | Merck Sharp & Dohme Llc | Drug delivery system for the delivery of antiviral agents and contraceptives |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112018072883A2 (en) | 2019-03-06 |
| CN109069526A (en) | 2018-12-21 |
| RU2018141823A3 (en) | 2020-09-10 |
| CA3023364A1 (en) | 2017-11-16 |
| US11400186B2 (en) | 2022-08-02 |
| JP6934019B2 (en) | 2021-09-08 |
| MX2018013662A (en) | 2019-03-01 |
| CN115624564A (en) | 2023-01-20 |
| BR112018072883B1 (en) | 2023-01-17 |
| RU2755130C2 (en) | 2021-09-13 |
| RU2018141823A (en) | 2020-06-15 |
| JP2019514992A (en) | 2019-06-06 |
| MX386770B (en) | 2025-03-19 |
| BR112018072883A8 (en) | 2023-01-10 |
| US20190388590A1 (en) | 2019-12-26 |
| KR20190005931A (en) | 2019-01-16 |
| EP3454868A4 (en) | 2019-12-18 |
| EP3454868A1 (en) | 2019-03-20 |
| AU2017263253B2 (en) | 2022-04-14 |
| AU2017263253A1 (en) | 2018-10-25 |
| KR102272235B1 (en) | 2021-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102272235B1 (en) | Drug Delivery Systems for Delivery of Antiviral Agents | |
| EP3471829A1 (en) | Drug delivery system for the delivery of antiviral agents | |
| KR101938662B1 (en) | Pharmaceutical compositions | |
| KR20170056702A (en) | Long acting pharmaceutical compositions | |
| WO2021225869A1 (en) | Drug delivery system for the delivery of antiviral agents and contraceptives | |
| KR20170025011A (en) | Pharmaceutical composition for sustained release of pain-relieving drugs and a device for administration thereof | |
| JP5425890B2 (en) | Method for producing uniform-sized polymer nanoparticles containing poorly soluble drugs | |
| US11419817B2 (en) | Drug delivery system for the delivery of antiviral agents | |
| EP4013407A1 (en) | Drug delivery system for the delivery of antiviral agents | |
| KR101499867B1 (en) | Composition comprising active agent (I) and manufacturing method thereof | |
| EP3515438B1 (en) | Drug delivery system for the delivery of integrase inhibitors | |
| EP4203917A1 (en) | Injectable depot compositions for the delivery of antiviral agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2017263253 Country of ref document: AU Date of ref document: 20170508 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3023364 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2018558762 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018072883 Country of ref document: BR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17796613 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20187035360 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017796613 Country of ref document: EP Effective date: 20181212 |
|
| ENP | Entry into the national phase |
Ref document number: 112018072883 Country of ref document: BR Kind code of ref document: A2 Effective date: 20181107 |