WO2017204615A2 - Cutinasas recombinantes de aspergillus nidulans para biodegradación de poliésteres - Google Patents
Cutinasas recombinantes de aspergillus nidulans para biodegradación de poliésteres Download PDFInfo
- Publication number
- WO2017204615A2 WO2017204615A2 PCT/MX2017/000056 MX2017000056W WO2017204615A2 WO 2017204615 A2 WO2017204615 A2 WO 2017204615A2 MX 2017000056 W MX2017000056 W MX 2017000056W WO 2017204615 A2 WO2017204615 A2 WO 2017204615A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- recombinant
- degrading
- polyesters
- enzyme
- ancut2
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Definitions
- the present invention relates to recombinant enzymes of Aspergillus nidulans, the methods for their production and their use in a method for the degradation of polyesters through biocatalytic treatment.
- the present invention relates to novel 10-polyester degrading cutinases such as PET and the DNA sequences encoding them, as well as the biocatalytic method for degrading plastics using recombinant enzymes.
- the enzymes had the following molecular weights of 37, 29 and 24 kDa.
- the 37 kDa activity corresponded to the PrtA protease purified by Pe ⁇ a-Montes, the 29 kDa, a new cutinase (ANCUT2) and the 24 kDa, to a post-translational processing product of ANCUT2.
- the ANCUT2 enzyme was purified and an amino acid analysis was carried out, in addition to the crude extract of the native enzyme solvent stability tests and cutin hydrolysis assays were performed.
- Berm ⁇ dez carried out the purification and biochemical characterization of the native cutinase ANCUT2 of A. nidulans PW1 (Berm ⁇ dez, 2013).
- CEH ANCUT1 (AN05309) and ANCUT2 (AN07541)
- ANCUT2 is described as a protein with cutinolithic activity (Castro-Ochoa, 2012; Esqueda-Dominguez; 2012). While the two remaining enzymes remain as hypothetical cutinases and there is still no evidence that clearly defines them.
- Cutinases (EC 3.1.1.74) are enzymes that are classified within the group of carboxylic acid ester hydrolases since they carry out their hydrolysis (Carvahlo, 1999).
- the main substrate of cutinases is cutin, the cuticular polymer of plants, which is a polyester composed of fatty acids (Purdy and Kolattukudy, 1975), and they are also capable of hydrolyzing short chain and long chain esters.
- Cutinase is mainly produced by phytopathogenic fungi as a mechanism of invasion of plants, the most studied system to date being that produced by the pathogen Fusarium solani.
- the work team of the present invention has performed the cloning of esterases produced by A. nidulans using the yeast Pichia pastoris as a heterologous organism for its expression (Pe ⁇ a-Montes et al. 2009). Induction is done when the body grows in media supplemented with methanol.
- the heterologous enzyme in P. pastoris is obtained by secretion in the medium because the enzyme was labeled with the signal sequence called the pre pro-coupling factor of Saccharomyces cerevisiae.
- the vector also has a sequence coding for a histidine tail that can be added to the enzyme in the terminal carboxyl to facilitate purification by means of an affinity column with Nickel or Cobalt. Table 1 shows the characteristics of the vector:
- the heterologous organism can carry out post-translational changes, it was necessary to perform a characterization of the biochemical properties of recombinant ANCUT1 and ANCUT2 cutinases to determine if they change with respect to the biochemical characteristics of native cutinases. Additionally it is important to mention that the new, ANCUT1 and ANCUT2 recombinant enzymes have biochemical properties different from the native ones because their amino acid sequences have been modified. It is even known that changing an amino acid in the sequence of an enzyme can generate changes in biochemical and biocatalytic properties. This is what we observe in the present invention for both enzymes recombinant with respect to the native ones, where several changes were generated as will be mentioned later.
- the present invention describes the biochemical properties of the new recombinant enzymes. It was determined that the optimal pH of the recombinant enzyme ANCUT2 ranges from 7-10, the optimum temperature was 40 ° C, it is a thermoalkaline enzyme since it resists temperatures of 60 ° C and pH 10.0 for up to 1 to 3 hours of incubation respectively, the catalytic capacity was up to 26 times larger than the catalytic capacity of the native enzyme (Morales-Garc ⁇ a, 2015).
- cutinases have potential use in industrial products and processes.
- the use of cutinases is present in the daily industry in the hydrolysis of milk fats, maintaining detergents, oleochemical industry, synthesis of triglycerides, polymers and surfactants, synthesis of ingredients for personal care products, synthesis of pharmaceutical and agrochemical products (Carvalho , 1998). While some of these processes are applied in the industry, others remain under evaluation at the research level.
- cutinases have demonstrated their application potential for surface modification and degradation of aliphatic and aromatic polyesters, especially ethylene polyterephthalate (PET), which is a synthetic aromatic polyester composed of terephthalic acid ( ⁇ ) and ethylene glycol (Dimaronga et ai , 2015).
- PET ethylene polyterephthalate
- ⁇ terephthalic acid
- ethylene glycol ethylene glycol
- ANCUT1 and ANCUT2 recombinant cutinases
- PCL poly (1, 4-butulensuccinate)
- PES polycaprolactone
- PES poly (ethylene succinate)
- PLLA poly (L-lactic)
- PET polyethylene terephthalate
- one of the main objectives of the present invention is to provide new modified cutinases that exhibit increased polyester degradation activity with respect to native enzymes.
- Another particular objective of the present invention is to provide the DNA sequences encoding the modified cutinases with said increased polyester degradation activity.
- Another additional objective of the present invention is to provide expression vectors comprising the DNA sequences encoding the modified cutinases with said increased polyester degradation activity
- a further objective of the present invention is to provide recombinant microorganisms that comprise said expression vectors and that are capable of producing modified cutinases with said increased polyester degradation activity.
- Another particular objective of the present invention is to provide a method for obtaining modified cutinases with said increased polyester degradation activity, using said recombinant microorganisms.
- the main objective of the present invention is to provide a method for the degradation of polyesters contained in urban solid waste such as PET, using modified cutinases and recombinant microorganisms with an increased polyester degradation activity.
- Figure 1 shows a 1% agarose gel stained with ethidium bromide with RNA (ribonucleic acid) obtained from mycelium of A. nidulans.
- Lane A RNA obtained from mycelium of CEHM optimized medium and lane B: RNA obtained from mycelium of medium with cutin.
- Figure 2 shows a 1% agarose gel stained with ethidium bromide in which amplicons of the ancutl and ancut2 cutinase genes are observed.
- Lane ⁇ ANCÜT1 cutinase amplicon (650 bp) obtained by RT-PCR with A. nidulans mycelium cDNA grown in medium with cutin.
- Lane B ANCUT2 cutinase amplicon (750 bp) obtained by RT-PCR with A. nidulans mycelium cDNA grown in CEHM optimized medium.
- MPM Rail lkB DNA molecular weight marker.
- Figure 3 shows the DNA (deoxyribonucleic acid) of the P. pastoris clones with the ANCUT1 or ANCUT2 gene and X-33 (wild strain) in a 1% agarose gel stained with ethidium bromide.
- Lane 1 Molecular weight marker (Gen ruler lkB, Thermo Scientific).
- Lanes 2 and 3 DNA of the clone with ANCÜTl.
- Lane 4 DNA of the clone with ANCUT2.
- Lane 5 X-33 DNA.
- Figure 4A shows the amplification of the ancutl gene with specific primers for this gene by taking the DNA of the clone with ANCÜTl of P. pastoris as tempered on a 1% agarose gel stained with ethidium bromide.
- Lane 1 Molecular weight marker (Gen ruler lkb, Thermo Scientific).
- Lanes 2 and 3 Amplicon of the ancutl gene.
- Figure 4B shows the amplification of the ancutl gene with universal AOX primers in a 1% agarose gel stained with ethidium bromide taking the DNA of P. pastoris as tempered.
- Lane 1 Molecular weight marker (Gen ruler lkb, Thermo Scientific).
- Lanes 2 to 4 Amplicon obtained with DNA from P.
- Lane 5 Amplicon obtained with DNA from P. pastoris clones with the pPICZaB vector without cutinase used as a negative control.
- Figure 5 shows the amplification of the ANCUT2 gene with specific and universal AOX primers in a 1% agarose gel stained with ethidium bromide.
- MPM rail Molecular weight marker (Gen ruler lkb, Thermo Scientific).
- Lanes 1 and 3 Amplicon obtained by taking DNA from P. pastoris clones with ANCUT2 with specific primers.
- Lane 2 Amplicon obtained by taking as DNA temperate from P. pastoris clones with ANCUT2 with universal primers (AOX).
- Figure 6A shows a sequencing electropherogram in the 5 'to 3' direction of the amplicon obtained with the universal primers from the DNA of the P. pastoris clone with the ancutl gene.
- Figure 6B shows a sequencing electropherogram in the 3 'to 5' direction of the amplicon obtained with the universal primers from the DNA of the P. pastoris clone with the ancutl gene.
- Figure 6C shows a sequencing electropherogram in the 5 'to 3' direction of the amplicon obtained with the universal primers from the DNA of the P. pastoris clone with the ancut2 gene.
- Figure 6D shows a sequencing electropherogram in the 3 'to 5' direction of the amplicon obtained with the universal primers from the DNA of the P. pastoris clone with the ancut2 gene.
- Figure 7 shows the DNA and amino acid sequence of the heterologous protein ANCUT1 inserted into the genome of P. pastoris.
- the signal sequence oc factor is identified
- in dark gray the sequence of an epitope of myc and the polyhistidine stem is identified.
- the catalytic triad within the underlined consensus pentapeptide is indicated with a circle.
- the sequence of nitrogenous bases added to the original sequence of the gene is enclosed with a dotted line and the corresponding amino acid is indicated with light gray.
- the letters of the amino acids identified in the peptides elucidated by the mass spectrometry technique are found in light gray.
- Figure 8 shows the DNA and amino acid sequence of the heterologous protein ANCUT2 inserted into the genome of P. pastoris. Amino acids are found in "bold”. Light gray shaded peptides represent the ⁇ factor. The dark gray shaded peptides correspond to the amino acid sequence of the histidine stem added to the terminal carboxyl for purification. Peptides elucidated by mass spectrometry are observed in deep gray. The light gray letter amino acids are those encoded by the nucleotides added for the ligation of the ⁇ factor to the amino terminal of cutinase 2 of A. nidulans. The amino acids of the catalytic triad are shown in white.
- Figure 9 shows the qualitative activity of ANCÜT1 cutinase during induction.
- Rows A correspond to the crude extracts of the clone with the ANCUT1 gene and rows B correspond to the crude extracts of the wild strain of P. pastoris X-33.
- Lanes 1 to 6 show the different induction times: 0, 24, 48, 72, 96 and 120 h; respectively.
- the color brown (dark gray) indicates the presence of activity and the greater intensity refers to the greater activity.
- Figure 10 shows the qualitative activity of ANCUT2 cutinase during induction.
- the activity in the crude extracts of the P. pastoris clone with ANCUT2 is observed at different induction times: 0, 24, 48, 72, 96 and 120 hours.
- White represents the crude extract of the wild strain of P. pastoris X-33 at zero induction time. Be Observe a positive result with a brown (dark gray) coloration, where greater intensity refers to greater activity.
- Figure 11 shows a graph where the specific activity (U / mg protein) and the amount of protein (mg) in the crude extract of the P. pastoris clone are correlated with ANCUT1, with respect to time. This in order to determine the optimal induction time of ANCUT1.
- Figure 12 shows a graph of the specific activity (U / mg protein) of ANCUT2 in the crude extract of the P. pastoris clone with ANCUT2 at different induction times. This in order to determine the optimal induction time of the ANCUT2.
- Figure 13 shows an extracellular protein profile of crude extracts (EC) of ANCUT1 at different induction times in SDS-PAGE gel stained with Coomassie blue.
- MPM rail Molecular weight marker (Low range, BioRad).
- Lanes 1 to 7 Extracellular protein profiles at different induction times of 0, 24, 48, 72, 96, 120 and 240 hours, respectively. In lane 7, the numbers next to each band observed indicate the molecular weights of the expressed proteins.
- Figure 14 shows an extracellular protein profile of crude ANCUT2 extracts at different induction times in SDS-PAGE gel stained with Coomassie blue.
- MPM lane Molecular weight marker (LowRange BioRad) where the ⁇ band has a molecular weight of 21kDa, B has a molecular weight of 31kDa and C has a molecular weight of 45kDa.
- the following lanes correspond to the extracellular protein profiles precipitated with TCA at the different indicated induction times of 0, 24, 48, 72, 96, 120, 168 and 192 hours.
- the presence of three proteins with molecular weights of 37, 23 and 21 kDa whose production increases with respect to induction time is indicated.
- Figure 15 shows a zymogram ( w in situ activity ") of the cutinase expressed in the crude extract of the P. pastoris clone with ANCÜT1 at different induction times in SDS-PAGE gel revealed with ⁇ naphthyl acetate and Fast Red.
- Lanes 1 to 6 Extracellular cutinase with esterase activity in crude extracts at different induction times of 0, 24, 48, 72, 96, 120 hours, respectively
- Lane 7 Molecular weight marker (kaleidoscopic, BioRad) The only band observed in lanes 2 to 5 corresponds to active cutinase with a molecular weight of 22.7 kDa which corresponds to that observed in figure 13. At 120 hours (lane 6) more bands are observed corresponding to P.
- Figure 16 shows a zymogram ("in situ" activity) of the cutinase expressed in the crude extract of the P. pastoris clone with ANCUT2 at different induction times in SDS-PAGE gel revealed with ⁇ naphthyl acetate and Fast Red.
- Lane MPM Molecular Weight Marker (LowRange BioRad).
- the following lanes correspond to the active cutinase band in the raw extracts at the different indicated induction times of 0, 24, 48, 96, 120, 168, 192 and 240 hours.
- the only band observed at 24 hours corresponds to the active cutinase ANCUT2 with a molecular weight of 37 kDa which corresponds to that observed in Figure 14. From 48 hours and up to 240 hours another band is observed at 23 kDa which corresponds to another form of ANCUT2 hydrolysis product of 37 kDa.
- Figure 17A shows a zymogram of the activity of ANCUT1 cutinase through the SDS-PAGE gel purification process revealed with ot naphthyl acetate and Fast Red.
- Lane 1 Raw extract of P. pastoris X-33 after 24 hours induction
- Lane 2 Crude extract of the P. pastoris clone with ANCUT1 after 24 hours of induction.
- Lane 3 Ultrafiltrate of the crude extract of the P. pastoris clone with ANCUT1 after 24 hours of induction.
- Lane 4 pure ANCUT1. The only band observed in each lane in dark color corresponds to the active cutinase.
- Figure 17B shows a protein profile on an SDS-PAGE gel with silver staining of the ANCUT1 purification process.
- Lane 5 Molecular weight marker (kaleidoscopic, BioRad). Lane 6: pure ANCUT1. Lane 7: Filtration of the crude extract of the P. pastoris clone with ANCUT1 after 24 hours of induction. Lane 8: Crude extract of the P. pastoris clone with ANCUT1 after 24 hours of induction. Lane 9: Raw extract of P. pastoris X-33 after 24 hours of induction. In lanes 6 to 8, the numbers next to each band observed indicate the molecular weights of the expressed proteins.
- Figure 17C shows a Western blot for the identification of pure ANCUT1 by the presence of the polyhistidine stalk in the PVDF membrane recombinant enzyme.
- Lane 1 Molecular weight marker (kaleidoscopic, BioRad).
- Lanes 3, 4 and 6 pure ANCUT1.
- Lanes 2 and 5 They were left empty.
- Figure 18A shows a Western blot for the identification of pure ANCUT2 by the presence of the polyhistidine stem in the recombinant enzyme with specific anti-his-tag antibodies in PVDF membrane.
- MPM rail molecular weight marker (Kaleidosdcopic from BioRad).
- Lanes 1 and 2 pure ANCUT2.
- Figure 18B shows the pure recombinant ANCUT2 enzyme in SDS-PAGE gel, which was revealed with silver staining.
- Rail MPM molecular weight marker (KaRidosdcopic from BioRad). Lanes 1 and 2: pure ANCUT2.
- Figure 19 shows the results of the preliminary evaluation in polyester degradation plate by ANCUT1 (left) and ANCUT2 (right) after 168 hours.
- Block 1 PLLA hydrolysis.
- Block 2 PBS hydrolysis.
- Block 3 PES hydrolysis.
- Block 4 PCL hydrolysis. The degradation is marked with arrows and a hydrolysis halo is observed around the colonies of P. pastoris.
- Figure 20 shows a graph of the results of the percentage of PBS weight loss after 24, 48 and 72 hours of hydrolysis with the ANCUT1 and ANCUT2 cutinases. For both cutinases, greater weight loss is observed as time increases. The highest hydrolysis is observed with ANCUT1 at 72 hours with a weight loss of 49.86%.
- Figure 21 shows a graph of the results of the percentage of weight loss of PES after 24, 48 and 72 hours of hydrolysis with the ANCUT1 and ANCUT2 cutinases. For both cutinases, greater weight loss is observed as time increases. The highest hydrolysis is observed with ANCUT1 at 72 hours with a weight loss of 87.39%.
- Figure 22 shows a graph of the results of the percentage of PCL weight loss after 24, 48 and 72 hours of hydrolysis with ANCUT1 and ANCUT2 cutinases. For both cutinases, greater weight loss is observed as time increases. The highest hydrolysis is observed with ANCUT1 at 72 hours with a weight loss of 71.99%.
- Figure 23 shows a graph of the results of the percentage of PLLA weight loss after 24, 48 and 72 hours of hydrolysis with the ANCUT1 and ANCUT2 cutinases. For both cutinases, greater weight loss is observed as time increases. The highest hydrolysis is observed with ANCUT1 and ANCUT2 at 72 hours.
- Figure 24 shows a graph of the results of free fatty acid titration after PBS degradation for 24, 48 and 72 hours.
- a graph of the volume of NaOH spent for the coloring beam of the pH indicator (phenolphthalein) against enzymatic hydrolysis reaction time is observed.
- ⁇ After 24 hours of reaction it is observed that the hydrolysis carried out by ANCUT1 required a larger volume of NaOH to titrate the reaction medium (8.87 ml) than that required by the hydrolysis with ANCUT2 (1.47 ml).
- Figure 25 shows a graph of the titration of free fatty acids after PES degradation.
- Figure 26 shows a graph of the titration of free fatty acids after degradation of PCL.
- Figure 27 shows a graph of the titration of free fatty acids after degradation of PLLA.
- Figure 28 shows a graph of the weight loss of the PBS polymer after hydrolysis with ANCUT1 at 40 ° C, 50 ° C and 60 * C and ANCUT2 at 30 ° C, 40 ° C and 50 ° C and different pH values. 7.0, 8.0 and 9.0.
- Figure 29 shows a graph of the weight loss of the PES polymer after hydrolysis with ANCUT1 at 40 ° C, 50 ° C and 60 ° C and ANCUT2 at 30 ° C, 40 ° C and 50 ° C and different pH values. 7.0, 8.0 and 9.0.
- Figure 30 shows a graph of the weight loss (%) of PCL polymer after hydrolysis with ANCÜT1 at 40 ° C, 50 ° C and 60 ° C and with ANCUT2 at 30 ° C, 40 ° C and 50 ° C . Both also evaluated at a different pH of 7.0, 8.0 and 9.0.
- Figure 31 shows a graph of the weight loss of the PLLA polymer after hydrolysis with ANCUT1 at 40 ° C, 50 ° C and 60 ° C and ANCUT2 at 30 ° C, 40 ° C and 50 ° C and different pH of 7.0, 8.0 and 9.0.
- Figure 32 shows a graph of the weight loss of the PBS polymer after its hydrolysis reaction with hexane and toluene (30%) with the enzymes ANCUT1 and ANCUT2.
- Figure 33 shows a graph of the weight loss of the PES polymer after its hydrolysis reaction with hexane and toluene (30%) with the enzymes ANCUT1 and ANCUT2.
- Figure 34 shows a graph of the PCL polymer weight loss after its hydrolysis reaction with hexane and toluene (30%) with the enzymes ANCUT1 and ANCUT2.
- Figure 35 shows the micrographs obtained by scanning electron microscopy (SEM) of the PBS and PCL polyesters after 72 hours of degradation.
- ⁇ control (wild strain X-33).
- B ANCÜT1.
- C ANCUT2.
- Figure 36 shows the micrographs obtained by scanning electron microscopy (SEM) of the PES, PLLA and PET polyesters after 72 hours of degradation.
- the microorganism used to obtain genes was Aspergillus nidulans PW1 (biAl; argB2; methGl; veAl), an arginine auxotrophic strain acquired from the Fungal Genetics Stock Center (Kansas City, KS, USA).
- the spores of A. nidulans were harvested from plates with minimal medium (Kafer, 1977). They were collected in 0.1% NaCl solution and stored at 4 ° C in silica gel (Kawasaki et al. 1995).
- A. nidulans was grown in CEHM optimized medium and in medium with cutin as the only carbon source as already described previously inoculating 1 x 10 6 spores / ml (Pe ⁇ a-Montes et al. 2008; Esqueda-Dominguez 2012).
- E. coli was grown at 37 ° C in Luria-Bertani (LB) medium (1% yeast extract-0.5% Bacto-triptone-1% NaCl) and low-salt LB (with 0.5% NaCl) (Bertani, 1951 ).
- LB Luria-Bertani
- P. pastoris was grown in YEPD medium containing 1% (w / v) yeast extract, 2% ( ⁇ / v) peptone and 2% (w / v) dextrose.
- 1 M sorbitol and 100 ⁇ g / ml zeocin (YEPS medium) were added to the plates with YEPD.
- Clones that secreted the enzyme extracellularly were selected in YEPDT medium with 0.5% (w / v) dextrose, 1% (v / v) tributyrine (substrate for esterases) and 100 ⁇ g I zeocin (YEPT medium).
- the induction of the enzyme in Petri dishes was maintained by adding 200 ⁇ l of methanol every 24 hours.
- BMGY For induction, cells grown in BMGY were centrifuged (3000 x g, 10 minutes) and resuspended in BMMY medium (BMGY with 0.5% (v / v) methanol instead of glycerol).
- the mycelium of A. nidulans grown under the induction conditions was collected by filtration for each enzyme (minimum medium optimized for ANCUT2 (CEHM) and medium with cutin induction for ANCUT1).
- the mycelium was washed with the buffer A (10 mM EDTA, 0.1 M NaCl, 40 mM Tris-HCl pH 8.0) and sprayed in a mortar in the presence of liquid nitrogen until a fine powder was obtained.
- Total RNA was isolated with the "RNeasy Plant Mini Kit” kit (Qiagen, Valencia, CA) and the mRNA was purified with the "PolyATrack mRNA isolation system” kit (Promega, Madison, WI) according to the supplier's instructions.
- CDNA was synthesized with the w cDNA cycle "kit (Invitrogen) and the cDNA obtained was used as tempering for subsequent PCR reactions.
- PCR for amplification gives ancutl and an.ca.t2.
- the genes were amplified using the cDNA obtained as described above.
- the ancut2 gene was amplified from the cDNA obtained mycelium from the CEHM optimized medium (Pe ⁇ a-Montes et al; 2008) and the ancutl gene from the cDNA obtained from the mycelium of the cutin-induced medium ⁇ Esqueda-Dom ⁇ nguez, 2012).
- the following specific primers were used (sequences in the 5'a 3 'direction):
- DH5ot electrocompetent cells were made and transformed by electroporation with either the pPICZarancutl or pPICZaancut2 vector.
- Recombinant clones were grown at 37 ° C in 50 ml of LB medium with zeocin for plasmid extraction with the "Plasmid midi" kit (Qiagen).
- the presence of each gene in both constructs was verified by amplification of each gene with both specific and universal primers (AOX) contained in the vector, as well as by sequencing the amplicons obtained.
- Electrocompetent cells of P. pastoria X-33 were prepared and transformed by electroporation with either the vector p? ICZccancutl or with pPICZotancut2. All incubations were performed from 28 ° C to 30 ° C. Recombinant clones were selected in YEPS medium with zeocin and halo (positive) producing clones were subsequently selected in YEPDT medium, both media described above. The positive transformants were grown in BMGY liquid medium until reaching an OD 6 or ⁇ 2 to 6, centrifuged (4,000 xg, 10 min) and the cell button was transferred to BMMY medium to induce protein expression. Methanol (0.5% v / v) was added daily for induction. The induced extracts were centrifuged (4,000 xg, 10 min) and the supernatant was collected.
- the transforming cells were taken with ANCUT1 or ANCÜT2 and a 50 ml sterile Falcon tube was inoculated with 5 ml of the YEPD medium and incubated between 16 hours at 18 hours from 28 ° C to 30 ° C at 200 to 250 rpm. It was centrifuged for 5 min and the button was resuspended in 1 ml of YEPD-Glycerol 1% medium. 100 ⁇ l aliquots were taken and stored in 200 ⁇ l tubes at -70 ° C.
- Transforming cells were seeded with ANCUT1 or ANCUT2 of P. pastoris in a 250 ml flask with 25 ml of sterile BMGY medium, which was incubated between 16 and 18 hours at 28 ° C at 30 ° C at 200-250-300 rpm (OD from 2 to 6). The contents of the flask were placed in a 50 ml falcon tube and centrifuged at 10,000 rpm for 5 min.
- the cell button was resuspended in 100 ml of BMMY medium (BMGY with 0.5% methanol instead of glycerol) and an aliquot of the medium (2 to 4 ml) was taken as a sample of the zero induction time. It was incubated at 28 ° C at 30 ° C at 200-300 rpm. 0.5 ml of absolute methanol was added every 24 hours and samples were taken at each induction time (2 to 4 ml every 24 hours until reaching 120 h) to determine the optimal production time of each enzyme. At the end of the induction it was centrifuged at 14000 rpm for 15 min at 4 ° C.
- the enzyme supernatant was subsequently used for the purification of the cutinase and the cell button of each of the tubes was stored at -70 ° C for later use in DNA extraction and verification of the presence of the cutinase gene in the P. pastoris genome by PCR amplification and sequencing.
- PBS poly (1, 4-butulen succinate)
- PCL polycaprolactone
- PES poly (ethylene succinate)
- PLS poly (L-lactic)
- PET polyethylene terephthalate
- Emulsion of polyimaroa Emulsion of polyimaroa.
- Emulsions were carried out separately from PBS, PCL, PES and PLLA (Sigma-Aldrich, Missouri, USA). To dissolve the polyester, 200 mg of polymer was added in 5 ml of chloroform. A 0.1% solution of SDS in deionized water was prepared to which the polymer dissolved in chloroform was added. It was emulsified by homogenizing the solution, until a stable phase is obtained. The emulsion was evaporated at 40 ° C to remove the chloroform (Hiraishi, 2008).
- the BMGY 2X medium and 1.5% agar were prepared, a 1: 1 dilution was made by adding the polymer emulsion. Once the plaque was obtained, the P. pastoris clones expressing the ANCUT1, ANCÜT2 enzymes were seeded there and the P. pastoris X-33 strain was also used as a negative control. They were incubated at 29 ° C, adding 100 ⁇ l of 20X Methanol every 24 hours in the lid of the Petri dish. The final concentration of the polymer in the boxes is 0.4%.
- Circular films of approximately 3 mm in diameter and 1 mm thick were prepared, weighing approximately 10 mg.
- the PBS and PCL were melted at approximately 80 ° C and compressed in a mold to obtain small films (Sulaiman, 2012).
- the enzymatic hydrolysis reaction was carried out by placing in a vial:
- the films were washed with ethanol and dried in an oven at 37 ° C overnight. They were weighed in analytical balance and the weight was recorded. The weight loss is determined by subtracting the final weight from the initial weight. Acide., Titrable.
- the degradation reaction supernatant was titrated with 0.01N NaOH and used as a phenolphthalein indicator. My NaOH expense was recorded. Intermediation of the conditions of the reaction: Temperature and pH.
- the vials were incubated at the temperatures described above and at 225 rpm for 24 hours. Since the hydrolysis reaction showed that there is a significant loss of weight. In the case of the ANCUT2 enzyme, 100 ⁇ l of the extract was added every 24 hours.
- the buffers used were 0.1M phosphate buffer (pH 7.2), 0.1M Tris-HCl buffer (pH 8.0) and 0.1M Tris-HCl buffer (pH 9.0).
- Degradation was determined by weight loss after the hydrolysis reaction.
- Hexane and 30% toluene were used as solvents to carry out the hydrolysis reaction at 48 hours, taking samples every 24 hours.
- the reaction was carried out for 96 hours at 37 ° C and 225 rpm, taking samples every 24 hours. After the reaction, the Films were washed with ethanol and dried in an oven at 37 ° C overnight.
- Degradation was determined by quantifying the weight loss after the hydrolysis reaction.
- the assay was extended to 3 weeks, adding ANCUT1 enzyme every 72 hours and ANCUT2 enzyme every 24 hours, taking samples every 72 hours. Degradation was determined by weight loss and scanning electron microscopy (SEM).
- the morphology of the films before and after the hydrolysis reaction was observed by scanning electron microscopy (SEM) (JSM-5900LV of JEOL) at high vacuum at an acceleration voltage of 20 kV.
- SEM scanning electron microscopy
- the movies were coated with a thin layer of gold (10 ⁇ ) before being observed.
- Figure 1 shows the RNA obtained from each of the A. nidulans mycelia grown under induction conditions for each enzyme, that is, in a minimum medium optimized for ANCUT2 (CEHM) (lane A) and medium with cutin induction for ANCUT1 (lane B).
- Figure 2 shows the PCR product (amplicon) of each gene of the ancutl (lane A) and ancut2 (lane B) cutinases obtained by RT-PCR. In the case of the ANCUT1 cutinase an amplicon of 650 bp was obtained and for the ANCUT2 cutinase of 750 bp, as theoretically expected.
- EXAMPLE 2 Verification of the presence of the ANCUT1 6 ANCUT2 gene in the gancma da P. pastoris and gives the expression of the entinases. DNA extraction.
- a sample of a culture of P. pastoris induced cells was centrifuged for the production of the ANCUT1 and ANCUT2 recombinant enzymes, as indicated in the section on obtaining inoculums and induction of recombinant enzymes mentioned above, at 14,000 rpm during 10 minutes at 4 ° C.
- the supernatant was removed and 500 ⁇ l of lysis buffer (2M Tris buffer, pH 8.0, 250 mM NaCl, 25 mM SDS and 0.5% EDTA) and 200 ⁇ l of the equivalent in glass beads were added to help lysate the yeast. It was stirred in a vortex at maximum speed for 3 minutes. It was incubated in a water bath at 65 ° C for 30 minutes.
- FIG. 3 shows the DNA of the P. pastoris X-33 clone with the ANCÜT1 gene (lanes 2 and 3) or ANCUT2 (lane 4) and of the wild strain X-33 (lane 5). DNA concentration resulting in 3,782.3 ng / ⁇ l with a 260/280 ratio of 1.89, which indicates an acceptable purity.
- DNA concentration resulting in 3,782.3 ng / ⁇ l with a 260/280 ratio of 1.89, which indicates an acceptable purity.
- Figure 4A shows a band close to 650 bp corresponding to the theoretical molecular weight of the ancutl gene. PCR was also performed with universal primers for the promoter and terminator region of the aox gene.
- Figure 4B in lanes 2 to 4, the presence of a band close to the theoretical molecular weight of 1200 bp corresponding to the DNA fragment inserted from the construction in plasmid pPICZa (from the promoter region) is observed aox to the terminator region aox) that codes for: 1) the promoter region of the aox gene, 2) the signal sequence of the ccha factor of Sacharomyces Cerevisiae, 3) the ANCÜT1 gene of the cutinase, 4) an epitope myc, 5) histidine stem and 6) the terminator region of the aox gene.
- the additional band to that expected above 1500 bp may correspond to the amplification of the aox2 gene since the primers can hybridize with the promoter and terminator region of the copy of the aox gene (aox2) in the Pichia pastoris DNA ( Cerenghino and Cregg, 2000).
- the other band above 6000 bp may correspond to DNA degradation or nonspecific amplification products. However, a purification was performed to obtain only the band of interest (1200 bp).
- Figure 5 shows the amplification of the ANCUT2 gene with specific and universal AOX primers. It is observed that the PCR amplicon with specific primers has an approximate weight of 700 bp (lanes 1 and 3) that coincides with the expected. On the other hand, the amplicon obtained with the AOX universal primers shows a band that has a approximate weight of 1290 bp (lane 2) corresponding to the theoretical value.
- the supplier of the cloning vector in Pichia pastoris indicates that the AOX region has a size of 592 bp and the weight of the gene inserted in the vector (700 bp) is further considered giving a theoretical total size of 1292 bp.
- the purification of the amplicons was performed with the GenElute Minus EtBr spin columns kit according to the provider's protocol (Sigma-Aldrich). In the end the pellet was resuspended in 50 ⁇ l of TE buffer. Dilutions of the pure amplicon obtained from 50 ng / ⁇ l were prepared and sent for a first sequencing to Macrogen, Korea.
- the microorganism with which the experimental work was carried out (P. pastoris) was observed by microscopy, which presented the typical characteristics regarding the shape and size of the yeasts.
- EXAMPLE 4 Kinetics of the production of the «Racoabinantaa Mimas.
- the activity was monitored at all induction times. It was quantified by hydrolysis of p-nitrophenyl acetate. The protein quantification test of the crude extract was performed at each induction time and the specific activity was determined.
- Figure 11 shows the results obtained for ANCUT1, a maximum peak of activity was detected at 24 hours of induction, therefore this time was established as the optimal induction. It is possible to appreciate how the concentration of protein increases throughout the induction time. In this case, a specific activity of 750 LJ / mg was observed for ANCÜT1 at 24 hours.
- EXAMPLE 5 Determination of the protein profile and «laogramee.
- the protein profile in SDS-PAGE gels was determined to observe the presence of the enzymes of interest throughout the induction in the crude extract, loading 10 mg of protein for each induction time ( Figures 13 and 14).
- the ultrafiltrate extract was purified with the Clontech column ** His Heel Gravity column.
- the following shock absorbers were used:
- Wash buffer 50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, pH 7.4, 1L
- Pure recombinant ANCUT1 had a specific activity of 14,823.86 ü / mg, a value that is above those reported in the literature for recombinant pure cutinases such as CUTAB1 of Alternar ⁇ a brassicicola with a value of 4,394 ü / mg or cutinase of F. Solani with a specific activity of 3,990 U / mg both expressed in P. pastoris (Koschorreck et al., Kwon et al., 2009).
- the yield and purification factor of the pure enzyme is 17.3% and 2.4 respectively. The yield can be improved if the enzyme purification process is optimized by means of the cobalt affinity column.
- the inmonublot assay (“Western blot") was performed to verify whether the two proteins found after purification had a polyhistidine stem ( Figure 17C).
- An SDS-PAGE electrophoresis gel was made with 1.5 ⁇ g of protein per lane and subsequently transferred to a PVDF membrane. It was obtained that the 2 bands with molecular weights of 22.7 and 24.8 kDa, which were found in the purification SDS-PAGE gels were detected in the "Western blot" with antibodies against the histidine stem (anti-histag), The result confirms that the recombinant ANCUT1 enzyme has 2 histidine-stemmed forms.
- Peptide identification was performed by the mass spectrometry technique (in the IBT and BioUSAII of the UNAM) of the ANCUT1, in which 9 peptides were identified with a 76.06% coverage of the 22.7 kDa band. All peptides belonged to the amino acid sequence of cutinase ANCUT1 (AN05309). Using ExPASy bioinformatic tools it was determined that the heterologous protein has a theoretical molecular weight of 23.34 kDa and a pl of 5.96. The recombinant protein sequence Verified is seen in Figure 7, as well as the identified peptides.
- EXAMPLE 7 Characterization of those in «imaa pures (AKCUT1 and ANCUT2).
- enzymatic activity was measured in triplicate spectrophotometrically in microplate for 10 min every minute at 30 ° C, 40 ° C, 50 ° C, 60 ° C and 70 ° C at pH 8.0 always running a target of autohydrolysis in each condition. 10 ⁇ l of enzyme in 170 ⁇ l of buffer and 20 ⁇ l of 1 mM p-NPA substrate was added. The reaction was monitored every 2 min for 10 min. Datarmination gives thermal aatability.
- the pure enzyme without substrate was incubated at temperatures of 30 ° C, 40 ° C, 50 ° C, 60 ° C and 70 ° C for time intervals of 15, 30 and 60 min. Subsequently, the solution was allowed to reach room temperature and its activity was monitored against p-NPA at the optimum working temperature.
- the stability of the enzymatic activity against pH was determined using the same buffers as in the previous point.
- the enzyme suspended in each buffer was incubated for 1 and 3 hours. Subsequently, its activity against p-NPA in microplate was determined as in the previous tests at the optimum working pH. Bapacificldad fruit to «uatratoa.
- the specific activity of the enzyme against p-NP short, medium and long chain esters was determined covering a chain length of 2 to 18 carbons (C2-C18). The tests were carried out at pH 8.0 and at 30 ° C to favor dissolution of the substrates without excessive hydrolysis.
- the p-NP esters tested were p-NPA acetate (p-NPA), p-NPButyrate (p-NPB), p-NPMiristate ⁇ p-NPM), p- NPLaurate (p-NPL), p-NP Palmitate (p-NPP ) and p-NPeStearato (p-NPS).
- the activity of the recombinant cutinase was determined in the presence of several metal ions (Berm ⁇ dez, 2013).
- the ions used (Ca 2 * , Mg 2 * 'K *, Na * and Cu 2 *) were added to the enzyme resuspended in 50 mM Tris-HCl buffer pH 8.0 at a concentration of 1 and 10 mM, and the solution was incubated for 24 hours at 4 ° C. After this time, the activity was measured with p-NPA as a substrate in microplate assays as in the other determinations.
- the residual activity was calculated using as 100% the activity of the enzyme in pH 9.0 buffer without added ions with the EDTA chelator at a concentration of 1 mM to eliminate the noise that the possible ions in solution could present.
- Poaiblaa effect of "naturalizers before inactivation" on the recombinant enzyme The effect of denaturing compounds reported for cutlnases (Chen et al., 2010) on the enzymatic activity of recombinant cutinase of A. nidulans was evaluated.
- the compounds used (SDS, PMSF and Tween 80) were added in concentrations of 1 and 10 mM to the enzyme resuspended in Tris-HCl buffer pH 8.0 and incubated for 24 hours at 4 ° C. Subsequently, the activity was quantified with p-NPA and the residual activity was reported considering as 100% the activity obtained without the addition of any surfactant.
- the effect of different solvents on the enzymatic activity of the recombinant cutinase against p-NPA was evaluated.
- the solvents used, acetone, ethanol, methanol, hexane, isopropanol and DMSO were added in 30% concentrations to the cutinase diluted in 50 mM Tris-HCl pH 8.0 buffer and incubated in three different ways: for 24 hours at 4 ° C , 24 hours at 40 ° C and 3 hours at 40 ° C (40 ° C was the optimum assay temperature of the recombinant enzyme) before measuring residual activity spectrophotometrically.
- the activity presented by the enzyme incubated in the same buffer without the addition of any solvent was considered 100% activity. Determination of kinetic parameters.
- Kinetic constants Km, kcat and km / kcat were determined for pure recombinant cutinase.
- the specific activity was determined with p-NPA at different substrate concentrations using the spectrophotometric assay described above.
- the initial velocity was calculated using the linear region of the reaction curve (5 min).
- the apparent kinetic constants were calculated from a double inverse graph (1 / v vs. l / [pNPA]) and are the result of the average of three independent measurements.
- the concentrated crude extracts (1: 5) of ANCUT1 and ANCUT2 were used for the hydrolysis reaction of the PBS, PES, PCL and PLLA films, being determined by the weight loss determination, which the ANCUT1 enzyme degrades at 24 hours almost 24% of PBS and after 72 hours of reaction degrades almost 50% of this same polymer (figure 20).
- the ANCUT1 enzyme degrades at 24 hours almost 24% of PBS and after 72 hours of reaction degrades almost 50% of this same polymer (figure 20).
- PES 62.43% degrades after 24 hours of reaction and 87.39% after 72 hours (Figure 21).
- PCL With PCL, the degradation taken at 24 hours showed 43.89% degradation and 71.99% at 72 hours (Figure 22).
- PLLA at 24 hours managed to degrade 5% and at 72 hours 14.94% (figure 23).
- the hydrolysis was evaluated by titration of fatty acids.
- Figures 24 to 27 show that the results are consistent with those obtained by the weight loss method, that is, the ANCUT1 cutinase is the one that most efficiently hydrolyzes the PBS, PES, PCL and PLLA polymers. An increase is observed with respect to the time of milliliters of NaOH.
- the titration of fatty acids is done indirectly, since initially it is not known that how many mi of "fat or oil" we have but, nevertheless, we know that in the hydrolysis reaction of cutinases with esterase activity and polyesters , a polymer fragmentation is performed, releasing fatty acid chains, whose presence was tried to be determined by the titration of fatty acids, observing a relationship in that, the more the weight loss of the polyesters is, the more the expense of milliliters of NaOH, so it is implied that if hydrolysis is carried out, consequently if a degradation with the cutinases is carried out.
- PES is the polymer that shows the greatest weight loss after 72 hours of reaction, followed by PCL with the enzyme ANCUT1, while the others have less than 50% of degradation.
- tables 6 and 7 summarize the optimal degradation conditions of polyesters for the enzymes ANCUT1 and ANCUT2.
- control films that were subjected to reaction with the crude extract of the wild strain X-33 show no apparent degradation, while the degradation and wear of the films that were subjected to the reaction with the enzymes ANCÜT1 and ANCUT2.
- Catalysis B Enzymatic, 63 (3), 121-127.
- thermostable carboxylesterase from an archaeon, Sulfolobus shibatae DSM5389: non-linear kinetic behavior of a hormone-sensitive lipase family enzyrae. Journal of bioscience and bioengineering, 98 ⁇ 6), 445-451.
- Enzymatic surface hydrolysis of PET Effect of structural diversity on kinetic properties of cutinases from Thermobifida. Macromol, 44 (12): 4632-4640.
- Pichia expression kit A manual of methods for expression of recombinant proteins using pPICZ and PICZa in Pichia pastoris. Catalog, (K1740-01).
Landscapes
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Separation, Recovery Or Treatment Of Waste Materials Containing Plastics (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
La presente invención se refiere a nuevos polinucleótidos recombinantes aislados de los genes ancut1 y ancut2, que codifican polipéptidos recombinantes funcionales capaces de degradar plásticos de poliésteres como PET, de enzimas cutinasas de Aspergillus nidulans. También se refiere a los vectores de expresión que comprenden a dichos polinucleótidos, a los microorganismos recombinantes que comprenden a dichos vectores, a los métodos para la producción y purificación de dichos polipéptidos recombinantes funcionales, así como a los métodos para degradar plásticos de poliésteres mediante la aplicación de composiciones que comprenden a dichos microorganismos y polipéptidos recombinantes funcionales.
Description
1
CUTINASAS RECOMBINANTES DE ASPERGILLUS NIDULANS PARA BIODEGRADACIÓN DE POLIÉSTERES
CAMPO DE LA INVENCIÓN
5 La presente invención se refiere a enzimas recombinantes de Aspergillus nidulans, a los métodos para su producción y a su uso en un método para la degradación de poliésteres a través del tratamiento biocatalitico. En particular, la presente invención se refiere a cutinasas novedosas degradadoras de 10 poliésteres como PET y a las secuencias de DNA que las codifican, así como al método biocatalitico para degradar plásticos utilizando las enzimas recombinantes.
ANTECEDENTES DE LA INVENCIÓN
15 El equipo de trabajo de la presente invención, desde hace más de 10 años, inició una línea de investigación sobre hidrolasas de ésteres de ácidos carboxilicos (CEH) debido a la importancia de sus aplicaciones biotecnológicas. Los estudios se enfocaron en el microorganismo filamentoso
20 Aspergillus nidulans PW1.
Kawasaki et al., (1995) reportaron que la actividad lipolítica producida por el hongo, cuando se cultiva en medio conteniendo aceite de olivo como inductor es inhibida por glucosa mediante un mecanismo independiente del gen creA e
identificaron una banda única de 37 kDa con actividad CEH sobre laurato de o-nitrofenilo.
Peña en el 2001 analizó diferentes nutrientes y condiciones de cultivo para optimizar la producción de actividad CEH en A. nidulans. Encontró que la mayor producción de la actividad se obtenía al emplear aceite de olivo (1%) como inductor, almidón (1.5%) como fuente de carbono y extracto de levadura (0.5%) como fuente de nitrógeno. También, purificó y caracterizó la enzima con mayor actividad CEH de 37 kDa reportada por Kawasaki (1995) y producida en este medio (Peña-Montes et al., 2008). Sin embargo, a pesar de que la enzima mostró una alta actividad de carboxiesterasa, la secuencia de aminoácidos deducida de la proteina purificada correspondió a la proteasa codificada por el gen prtA.
Castro-Ochoa en el 2014 realizó pruebas preliminares con la finalidad de identificar la, o las enzimas, con actividad CEH producida por A. nidulans en el medio de cultivo optimizado por Peña (2008) .
Después de realizar una separación electroforética de las proteínas presentes en el filtrado (concentrado) del medio de cultivo, se observaron 3 bandas con dicha actividad catalítica empleando como sustrato acetato de α-naftilo. Las enzimas presentaron los siguientes pesos moleculares de 37, 29 Y 24 kDa. La actividad de 37 kDa correspondía a la
proteasa PrtA purificada por Peña-Montes, la de 29 kDa, una nueva cutinasa (ANCUT2) y la de 24 kDa, a un producto de procesamiento pos-traduccional de ANCUT2. La enzima ANCUT2 fue purificada y se llevó a cabo un análisis de aminoácidos, además con el extracto crudo de la enzima nativa se realizaron pruebas de estabilidad a solventes y ensayos de hidrólisis de cutina.
Posteriormente, Bermúdez llevó a cabo la purificación y caracterización bioquímica de la cutinasa nativa ANCUT2 de A. nidulans PW1 (Bermúdez, 2013).
En experimentos previos a la purificación de esta ANCCJT2 , se habla cultivado al microorganismo en medio mínimo con cutina como única fuente de carbono y se determinó la presencia de actividad de esterasa (Peña, 2001) .
Desde entonces se ha emprendido una importante investigación para la búsqueda de enzimas con actividad de cutinasa que puedan ser útiles en degradación o síntesis de polímeros. Un análisis de las secuencias de hidrolasas de ésteres de ácidos carboxilicos (CEH) que se han reportado en la base de datos del genoma de A. nidulans (Galagan et al., 2005) reveló que existen más de 40 genes que codifican para la producción de esta clase de enzimas (Peña, 2008) . Dentro de estos, se encontraron 4 secuencias de genes que codifican para
cutinasas, su anotación en el genoma, cantidad de aminoácidos (aa) y localización cromosómica es la siguiente:
• AN07541 (257 aa) en el cromosoma IV
AN05309 (213 aa) en el cromosoma V
· AN07180 (221 aa) en el cromosoma IV
AN10346 (223 aa) en el cromosoma VI
De estas cuatro cutinasas, las dos primeras ya se han confirmado como CEH (ANCUT1 (AN05309) y ANCUT2 (AN07541) ) y se describe a ANCUT2 como proteina con actividad cutinolitica (Castro-Ochoa, 2012; Esqueda-Dominguez; 2012) . Mientras que las dos enzimas restantes permanecen como cutinasas hipotéticas y todavía no hay evidencias que las definan claramente.
Castro-Ochoa (2014), identificó una nueva cutinasa (ANCÜT1) , la cual se induce en el medio con cutina como única fuente de carbono. Esqueda-Dominguez (2012) estudió la producción de esta enzima y Vega-Pérez (2013) la purificó y caracterizó. Las cutinasas (E.C. 3.1.1.74) son enzimas que se clasifican dentro del grupo de las hidrolasas de ésteres de ácidos carboxilicos ya que llevan a cabo la hidrólisis de los mismos (Carvahlo, 1999) . El sustrato principal de las cutinasas es la cutina, el polímero cuticular de las plantas, el cual es un poliéster compuesto por ácidos grasos (Purdy and Kolattukudy, 1975) , además también son capaces de hidrolizar
ésteres de cadena corta y de cadena larga. Estas enzimas presentan similitud catalítica con algunas carboxilesterasas como las lipasas y esterasas. La cutinasa es producida principalmente por hongos fitopatógenos como mecanismo de invasión a plantas, siendo el sistema más estudiado a la fecha el producido por el patógeno Fusarium solani.
Debido a la importancia de las potenciales aplicaciones de las cutinasas, estas enzimas ya han sido clonadas y expresadas en hospederos heterólogos (Carvalho, 1998) .
BREVE DESCRIPCIÓN DE LA INVENCIÓN
De conformidad con la presente invención, entre las ventajas que ofrece el obtener una cutinasa recombinante, se encuentran la obtención de una mayor producción de enzima, además de que en la industria química, alimenticia y farmacéutica, los métodos de purificación son más fáciles de realizar.
El equipo de trabajo de la presente invención ha realizado la clonación de esterasas producidas por A. nidulans usando a la levadura Pichia pastoris como organismo heterólogo de expresión de la misma (Peña-Montes et al. 2009) . La inducción se hace cuando el organismo crece en medios suplementados con metanol .
Además, la enzima heteróloga en P. pastoris es obtenida por secreción en el medio gracias a que la enzima fue marcada con la secuencia señal llamada factor de acoplamiento α pre pro de Saccharomyces cerevisiae. El vector también cuenta con una secuencia que codifica para una cola de histidinas que puede ser añadida a la enzima en el carboxilo terminal para facilitar la purificación por medio de una columna de afinidad con Níquel o Cobalto. En la tabla 1, se muestran las características del vector:
Tabla 1. Características del vector pPICZa (Cregg, 2000).


Debido a que el organismo heterólogo puede llevar a cabo cambios postraduccionales, fue necesario realizar una caracterización de las propiedades bioquímicas de las cutinasas ANCUT1 y ANCUT2 recombinantes para determinar si éstas cambian respecto a las características bioquímicas de las cutinasas nativas. Adicionalmente es importante mencionar, que las nuevas, enzimas recombinantes ANCUT1 y ANCUT2 presentan propiedades bioquímicas distintas a las nativas debido a que sus secuencias de aminoácidos han sido modificadas. Se sabe incluso que el cambio de un aminoácido en la secuencia de una enzima puede generar cambios en las propiedades bioquímicas y biocataliticas . Esto es lo que observamos en la presente invención para ambas enzimas
recombinantes con respecto a las nativas, donde se generaron varios cambios como se mencionará posteriormente.
En la presente invención se describen las propiedades bioquímicas de las nuevas enzimas recombinantes. Se determinó que el pH óptimo de la enzima recombinante ANCUT2 va de un rango de 7-10, la temperatura óptima fue de 40°C, se trata de una enzima termoalcalina ya que resiste temperaturas de 60°C y pH 10.0 hasta por 1 a 3 horas de incubación respectivamente, la capacidad catalítica fue hasta 26 veces más grande que la capacidad catalítica de la enzima nativa (Morales-García, 2015) .
Se determinó que las propiedades catalíticas de la enzima ANCUT1 recombinante mejoraron considerablemente en comparación con la enzima nativa. La recombinante ANCUT1 también presentó una temperatura óptima de 60*C, mostró mayor estabilidad a pH 10.0, el EDTA (quelante) y Ca2+ no disminuyen la actividad de la enzima, el Fe3* aumenta la actividad y la enzima presenta mayor actividad en hexano (Solis-Baez, 2015) . Existen ya reportes de cutinasas recombinantes procedentes de bacterias, levaduras y hongos (Nyyssttlá, et al, 2014; Kwon et al. 2009; Ahmed 2016; Dimarogona, et al 2015) . Sin embargo, la clonación exitosa de las cutinasas descritas en la presente invención, asi como la expresión funcional de las mismas, no se había realizado.
Las reacciones de hidrólisis y sintesis presentadas por las cutinasas tienen uso potencial en productos y procesos industriales. El uso de las cutinasas está presente en la industria diaria en la hidrólisis de grasas de leche, mantener detergentes, industria oleoquimica, sintesis de triglicéridos, polímeros y surfactantes, síntesis de ingredientes para productos de cuidado personal, síntesis de productos farmacéuticos y agroquímicos (Carvalho, 1998) . Si bien, algunos de estos procesos son aplicados en la industria, otros siguen bajo evaluación a nivel de investigación .
Recientemente las cutinasas han demostrado su potencial de aplicación para la modificación de superficie y degradación de poliésteres alifáticos y aromáticos, especialmente politereftalato de etileno (PET) , el cual es un poliéster aromático sintético compuesto de ácido tereftálico (ΤΡΆ) y etilenglicol (Dimaronga et ai, 2015) . Sin embargo, el número de cutinasas que han sido estudiadas con respecto a la modificación de PET sigue siendo limitado, y ésta limitación puede resultar en un retraso de la investigación hacia el uso de las cutinasas (Sulaiman, 2012) .
En la actualidad, el consumo de envases de plásticos tiene gran impacto ambiental mundial, ya que, en el año 2010, la producción de plásticos fue de 265 millones de toneladas, y
sólo en América Latina, se produjo el 5% de estos (PlasticsEurope' s Market Research and Statistics Group (PEMRG) ) .
Mientras que la generación de residuos sólidos en México en el año 2012, referente solamente a plásticos, fue de 4594.90 miles de toneladas, representando el 10.98% del total de residuos urbanos (INEGI, 2013) .
Ya que es un gran problema ambiental, se han buscado alternativas para acelerar la degradación de plásticos, como es la elaboración de polímeros biodegradables, y/o la utilización de enzimas. En este contexto, se han realizado algunas investigaciones, en donde se utilizan los polímeros sintéticos y polímeros biodegradables como sustratos de enzimas, tales como las cutinasas; las cuales ya han sido expresadas en diferentes microorganismos. Este es el caso de las investigaciones de Vertommen (2005), Ronkvist (2009), Herrero (2011) y Sulaiman (2012), entre otros.
En la presente invención se trabajó con las cutinasas recombinantes (ANCUT1 y ANCUT2) , ya que se producen con actividad alta y en mayor cantidad que las enzimas silvestres. Además, se evaluó el potencial de estas enzimas en la degradación de poliésteres, problema ambiental generado por envases de la industria de alimentos, utilizando como modelo los polímeros de poli (1, 4-butulensuccinato) (PBS) ,
policaprolactona (PCL), poli (etilensuccinato) (PES) , poli(L- láctico) (PLLA) y polietilentereftalato (PET) .
De acuerdo a lo descrito anteriormente, existe un número limitado de microorganismos con capacidad para degradar poliésteres y en particular plásticos derivados de estos, y más aún, son escasos los reportes en donde se utilizan enzimas capaces de llevar a cabo dicha degradación. Por lo tanto, existe una necesidad de proporcionar nuevas enzimas y nuevos microorganismos modificados que sean capaces de hidrolizar plásticos más eficientemente, para de esta manera, poder proponer métodos de reciclaje biocatalitico mejorados de estos residuos sólidos urbanos. En consecuencia, uno de los objetivos principales de la presente invención es proporcionar nuevas cutinasas modificadas que presenten una actividad de degradación de poliésteres incrementada con respecto a las enzimas nativas. Otro objetivo particular de la presente invención es proporcionar las secuencias de DNA que codifican para las cutinasas modificadas con dicha actividad de degradación de poliésteres incrementada.
Otro objetivo adicional de la presente invención, es proporcionar vectores de expresión que comprenden las
secuencias de DNA que codifican para las cutinasas modificadas con dicha actividad de degradación de poliésteres incrementada
Un objetivo más de la presente invención es proporcionar microorganismos recombinantes que comprenden dichos vectores de expresión y que son capaces de producir las cutinasas modificadas con dicha actividad de degradación de poliésteres incrementada.
Otro objetivo particular de la presente invención es proporcionar un método para la obtención de las cutinasas modificadas con dicha actividad de degradación de poliésteres incrementada, utilizando dichos microorganismos recombinantes .
Finalmente el objetivo principal de la presente invención es proporcionar un método para la degradación de poliésteres contenidos en residuos sólidos urbanos como PET, utilizando las cutinasas modificadas y los microorganismos recombinantes con una actividad de degradación de poliésteres incrementada. BREVE DESCRIPCIÓN DE LAS FIGURAS
La figura 1 muestra un gel de agarosa al 1% teñido con bromuro de etidio con el RNA (ácido ribonucleico) obtenido de micelio de A. nidulans. Carril A: RNA obtenido de micelio de
medio optimizado CEHM y carril B: RNA obtenido de micelio de medio con cutina.
La figura 2 muestra un gel de agarosa al 1% teñido con bromuro de etidio en el cual se observan los amplicones de los genes de cutinasas ancutl y ancut2. Carril Λ: Amplicón de cutinasa ANCÜT1 (650 bp) obtenido por RT-PCR con cDNA de micelio de A. nidulans crecido en medio con cutina. Carril B: Amplicón de cutinasa ANCUT2 (750 bp) obtenido por RT-PCR con cDNA de micelio de A. nidulans crecido en medio optimizado CEHM. Carril MPM: Marcador de peso molecular lkB DNA.
La figura 3 muestra el DNA (ácido deoxiribonucléico) de las clonas de P. pastoris con el gen ANCUT1 ó ANCUT2 y de X-33 (cepa silvestre) en un gel de agarosa al 1% teñido con bromuro de etidio. Carril 1: Marcador de peso molecular (Gen ruler lkB, Thermo Scientific) . Carriles 2 y 3: DNA de la clona con ANCÜTl. Carril 4: DNA de la clona con ANCUT2. Carril 5: DNA de X-33.
La figura 4A muestra la amplificación del gen ancutl con cebadores específicos para este gen tomando como templado el DNA de la clona con ANCÜTl de P. pastoris en un gel de agarosa 1% teñido con bromuro de etidio. Carril 1: Marcador de peso molecular (Gen ruler lkb, Thermo Scientific) . Carriles 2 y 3: Amplicón del gen ancutl.
La figura 4B muestra la amplificación del gen ancutl con cebadores universales AOX en un gel de agarosa 1% teñido con bromuro de etidio tomando como templado el DNA de P. pastoris. Carril 1: Marcador de peso molecular (Gen ruler lkb, Thermo Scientific) . Carriles 2 a 4: Amplicón obtenido con DNA de clonas de P. pastoris con ANCUT1. Carril 5 : Amplicón obtenido con DNA de clonas de P. pastoris con el vector pPICZaB sin cutinasa utilizado como control negativo. La figura 5 muestra la amplificación del gen ANCUT2 con cebadores específicos y universales AOX en un gel de agarosa 1% teñido con bromuro de etidio. Carril MPM: Marcador de peso molecular (Gen ruler lkb, Thermo Scientific) . Carriles 1 y 3: Amplicón obtenido tomando como templado DNA de clonas de P. pastoris con ANCUT2 con cebadores específicos. Carril 2: Amplicón obtenido tomando como templado DNA de clonas de P. pastoris con ANCUT2 con cebadores universales (AOX) .
La figura 6A muestra un electroferograma de secuenciación en dirección 5' a 3' del amplicón obtenido con los cebadores universales a partir del DNA de la clona de P. pastoris con el gen ancutl .
La figura 6B muestra un electroferograma de secuenciación en dirección 3' a 5' del amplicón obtenido con los cebadores universales a partir del DNA de la clona de P. pastoris con el gen ancutl.
La figura 6C muestra un electroferograma de secuenciación en dirección 5' a 3' del amplicón obtenido con los cebadores universales a partir del DNA de la clona de P. pastoris con el gen ancut2.
La figura 6D muestra un electroferograma de secuenciación en dirección 3' a 5' del amplicón obtenido con los cebadores universales a partir del DNA de la clona de P. pastoris con el gen ancut2.
La figura 7 muestra la secuencia de DNA y de aminoácidos de la proteina heteróloga ANCUT1 insertada en el genoma de P. pastoris. En color gris claro se identifica la secuencia señal oc factor, en gris obscuro se identifica la secuencia de un Epitope de myc y del tallo de polihistidina. Se señala con un circulo la triada catalitica dentro del pentapéptido consenso subrayado. Se encierra con linea punteada la secuencia de bases nitrogenadas añadida a la secuencia original del gen y se señala con gris claro debajo el aminoácido correspondiente. En gris claro se encuentran las letras de los aminoácidos identificados en los péptidos elucidados por la técnica de espectrometria de masas.
La figura 8 muestra la secuencia de DNA y de aminoácidos de la proteina heteróloga ANCUT2 insertada en el genoma de P. pastoris. Los aminoácidos se encuentran en "negritas". Los péptidos sombreados en gris claro representan el α factor.
Los péptidos sombreados de gris obscuro corresponden con la secuencia de aminoácidos del tallo de histidinas añadidos al carboxilo terminal para su purificación. Se observa en color gris intenso los péptidos elucidados por la técnica de espectrometria de masas. Los aminoácidos con letra en color gris claro son los codificados por los nucleótidos añadidos para la ligación del α factor al amino terminal de la cutinasa 2 de A. nidulans. Los aminoácidos de la triada catalitica se muestran con la letra en color blanco.
La figura 9 muestra la actividad cualitativa de la cutinasa ANCÜT1 durante la inducción. Las filas A corresponden a los extractos crudos de la clona con el gen ANCUT1 y las filas B corresponden a los extractos crudos de la cepa silvestre de P. pastoris X-33. Los carriles 1 a 6 muestran los diferentes tiempos de inducción: 0, 24, 48, 72, 96 y 120 h; respectivamente. El color marrón (gris obscuro) indica la presencia de actividad y la mayor intensidad se refiere a la mayor actividad.
La figura 10 muestra la actividad cualitativa de la cutinasa ANCUT2 durante la inducción. Se observa la actividad en los extractos crudos de la clona de P. pastoris con ANCUT2 a diferentes tiempos de inducción: 0, 24, 48, 72, 96 y 120horas. El blanco representa el extracto crudo de la cepa silvestre de P. pastoris X-33 al tiempo cero de inducción. Se
observa un resultado positivo con una coloración marrón (gris obscuro) , en donde una mayor intensidad refiere una mayor actividad.
La figura 11 muestra un gráfico en donde se correlacionan la actividad especifica (U/mg proteina) y la cantidad de proteina (mg) en el extracto crudo de la clona de P. pastoris con ANCUT1, con respecto al tiempo. Esto con el fin de determinar el tiempo óptimo de inducción de la ANCUT1.
La figura 12 muestra un gráfico de la actividad especifica (U/mg proteina) de la ANCUT2 en el extracto crudo de la clona de P. pastoris con ANCUT2 a diferentes tiempos de inducción. Esto con el fin de determinar el tiempo óptimo de inducción de la ANCUT2.
La figura 13 muestra un perfil de proteínas extracelulares de extractos crudos (EC) de ANCUT1 a diferentes tiempos de inducción en gel SDS-PAGE teñido con Coomassie blue. Carril MPM: Marcador de peso molecular (Low range, BioRad) . Carriles 1 a 7: Perfiles de proteínas extracelulares a los diferentes tiempos de inducción de 0, 24, 48, 72, 96, 120 y 240 horas, respectivamente. En el carril 7, los números al lado de cada banda observada indican los pesos moleculares de las proteínas expresadas.
La figura 14 muestra un perfil de proteínas extracelulares de extractos crudos de ANCUT2 a diferentes tiempos de inducción
en gel SDS-PAGE teñido con Coomassie blue. Carril MPM: Marcador de peso molecular (LowRange BioRad) en donde la banda Ά tiene un peso molecular de 21kDa, B tiene un peso molecular de 31kDa y C tiene un peso molecular de 45kDa. Los siguientes carriles corresponden a los perfiles de proteínas extracelulares precipitadas con TCA a los diferentes tiempos de inducción indicados de 0, 24, 48, 72, 96, 120, 168 y 192 horas. Se señala la presensencia de tres proteínas con pesos moleculares de 37, 23 y 21 kDa cuya producción aumenta con respecto al tiempo de inducción.
La figura 15 muestra un zimograma (actividad win situ") de la cutinasa expresada en el extracto crudo de la clona de P. pastoris con ANCÜT1 a diferentes tiempos de inducción en gel SDS-PAGE revelado con α naftil acetato y Fast Red. Carriles 1 a 6: Cutinasa extracelular con actividad de esterasa en los extractos crudos a diferentes tiempos de inducción de 0, 24, 48, 72, 96, 120horas, respectivamente. Carril 7: Marcador de peso molecular (kaleidoscópico, BioRad) . La única banda observada en los carriles 2 al 5 corresponde a la cutinasa activa con un peso molecular de 22.7 kDa la cual corresponde a la observada en la figura 13. A las 120 horas (carril 6) se observan más bandas que corresponden a esterasas de P. pastoris intracelulares liberadas por lisis de las células.
La figura 16 muestra un zimograma (actividad "in situ") de la cutinasa expresada en el extracto crudo de la clona de P. pastoris con ANCUT2 a diferentes tiempos de inducción en gel SDS-PAGE revelado con α naftil acetato y Fast Red. Carril MPM: Marcador de peso molecular (LowRange BioRad) . Los siguientes carriles corresponden a la banda de cutinasa activa en los extractos crudos a los diferentes tiempos de inducción indicados de 0, 24, 48, 96, 120, 168, 192 y 240 horas. La única banda observada a las 24 horas corresponde a la cutinasa activa ANCUT2 con un peso molecular de 37 kDa la cual corresponde a la observada en la figura 14. A partir de 48 horas y hasta las 240 horas se observa otra banda en 23 kDa que corresponde a otra forma de ANCUT2 producto de hidrólisis de la de 37 kDa.
La figura 17A se muestra un zimograma de la actividad de la cutinasa ANCUT1 a través del proceso de purificación en gel SDS-PAGE revelado con ot naftil acetato y Fast Red. Carril 1: Extracto crudo de P. pastoris X-33 después de 24 horas de inducción. Carril 2: Extracto crudo de la clona de P. pastoris con ANCUT1 después de 24 horas de inducción. Carril 3: Ultrafiltrado del extracto crudo de la clona de P. pastoris con ANCUT1 después de 24 horas de inducción. Carril 4: ANCUT1 pura. La única banda que se observa en cada carril en color obscuro corresponde a la cutinasa activa.
La figura 17B muestra un perfil de proteínas en un gel SDS- PAGE con tinción de plata del proceso de purificación de ANCUT1. Carril 5: Marcador de peso molecular (kaleidoscópico, BioRad) . Carril 6: ANCUT1 pura. Carril 7: ültrafiltrado del extracto crudo de la clona de P. pastoris con ANCUT1 después de 24 horas de inducción. Carril 8: Extracto crudo de la clona de P. pastoris con ANCUT1 después de 24 horas de inducción. Carril 9: Extracto crudo de P. pastoris X-33 después de 24 horas de inducción. En los carriles 6 a 8, los números al lado de cada banda observada indican los pesos moleculares de las proteínas expresadas.
La figura 17C muestra un Western blot para la identificación de ANCUT1 pura por la presencia del tallo de polihistidina en la enzima recombinante en membrana de PVDF. Carril 1: Marcador de peso molecular (kaleidoscópico, BioRad) . Carriles 3, 4 y 6: ANCUT1 pura. Carriles 2 y 5: Se dejaron vacíos. La figura 18A muestra muestra un Western blot para la identificación de ANCUT2 pura por la presencia del tallo de polihistidina en la enzima recombinante con anticuerpos específicos anti his-tag en membrana de PVDF. Carril MPM: marcador de peso molecular (Kaleidosdcopic de BioRad) . Carriles 1 y 2: ANCUT2 pura.
La figura 18B muestra la enzima ANCUT2 recombinante pura en gel SDS-PAGE, el cual se reveló con tinción de plata. Carril
MPM: marcador de peso molecular (Kaleidosdcopic de BioRad) . Carriles 1 y 2: ANCUT2 pura.
La figura 19 muestra los resultados de la evaluación preliminar en placa de degradación de poliésteres por ANCUT1 (izquierda) y ANCUT2 (derecha) después de 168 horas. En forma horizontal, Bloque 1: Hidrólisis de PLLA. Bloque 2: Hidrólisis de PBS. Bloque 3: Hidrólisis de PES. Bloque 4: Hidrólisis de PCL. La degradación se marca con flechas y se observa un halo de hidrólisis alrededor de las colonias de P. pastoris.
La figura 20 muestra un gráfico de los resultados del porcentaje de pérdida de peso de PBS después de 24, 48 y 72 horas de hidrólisis con las cutinasas ANCUT1 y ANCUT2. Para ambas cutinasas se observa una mayor pérdida de peso conforme aumenta el tiempo. La mayor hidrólisis se observa con ANCUT1 a las 72 horas con una pérdida de peso de 49.86%.
La figura 21 muestra un gráfico de los resultados del porcentaje de pérdida de peso de PES después de 24, 48 y 72 horas de hidrólisis con las cutinasas ANCUT1 y ANCUT2. Para ambas cutinasas se observa una mayor pérdida de peso conforme aumenta el tiempo. La mayor hidrólisis se observa con ANCUT1 a las 72 horas con una pérdida de peso de 87.39%.
La figura 22 muestra un gráfico de los resultados del porcentaje de pérdida de peso de PCL después de 24, 48 y 72
horas de hidrólisis con las cutinasas ANCUT1 y ANCUT2. Para ambas cutinasas se observa una mayor pérdida de peso conforme aumenta el tiempo. La mayor hidrólisis se observa con ANCUT1 a las 72 horas con una pérdida de peso de 71.99%.
La figura 23 muestra un gráfico de los resultados del porcentaje de pérdida de peso de PLLA después de 24, 48 y 72 horas de hidrólisis con las cutinasas ANCUT1 y ANCUT2. Para ambas cutinasas se observa una mayor pérdida de peso conforme aumenta el tiempo. La mayor hidrólisis se observa con ANCUT1 y ANCUT2 a las 72 horas.
La figura 24 muestra un gráfico de los resultados de la titulación de ácidos grasos libres después de la degradación de PBS por 24, 48 y 72 horas. En esta figura se observa un gráfico del volumen de NaOH gastado para el vire de coloración del indicador de pH (fenolftaleina) contra tiempo de reacción de hidrólisis enzimática. Ά las 24 horas de reacción se observa que la hidrólisis llevada a cabo por la ANCUT1 requirió un volumen mayor de NaOH para titular el medio de reacción (8.87 mi) que el requerido por la hidrólisis con ANCUT2 (1.47 mi). En una hidrólisis de 72 horas se observa que la realizada por la ANCUT1 requirió un volumen mayor de NaOH para titular el medio de reacción (19.72 mi) que el requerido por la hidrólisis con ANCUT2 (2.22 mi) con lo que se plantea que la concentración de
ácidos grasos liberados aumenta conforme aumenta el tiempo de reacción y que la enzima ANCUT1 lleva a cabo una mayor liberación de ácidos grasos que la ANCUT2.
La figura 25 muestra un gráfico de la titulación de ácidos grasos libres después de la degradación de PES.
La figura 26 muestra un gráfico de la titulación de ácidos grasos libres después de la degradación de PCL.
La figura 27 muestra un gráfico de la titulación de ácidos grasos libres después de la degradación de PLLA.
La figura 28 muestra un gráfico de la pérdida de peso del polimero PBS después de su hidrólisis con ANCUT1 a 40°C, 50°C y 60*C y ANCUT2 a 30°C, 40°C y 50°C y diferentes pH de 7.0, 8.0 y 9.0.
La figura 29 muestra un gráfico de la pérdida de peso del polimero PES después de su hidrólisis con ANCUT1 a 40°C, 50°C y 60°C y ANCUT2 a 30°C, 40°C y 50°C y diferentes pH de 7.0, 8.0 y 9.0.
La figura 30 muestra un gráfico de la pérdida de peso (%) del polimero PCL después de su hidrólisis con ANCÜT1 a 40°C, 50°C y 60°C y con ANCUT2 a 30°C, 40°C y 50°C. Ambas evaluadas también a diferente pH de 7.0, 8.0 y 9.0.
La figura 31 muestra un gráfico de la pérdida de peso del polimero PLLA después de su hidrólisis con ANCUT1 a 40°C,
50°C y 60°C y ANCUT2 a 30°C, 40°C y 50°C y diferentes pH de 7.0, 8.0 y 9.0.
La figura 32 muestra un gráfico de la pérdida de peso del polímero PBS después de su reacción de hidrólisis con hexano y tolueno (30%) con las enzimas ANCUT1 y ANCUT2.
La figura 33 muestra un gráfico de la pérdida de peso del polímero PES después de su reacción de hidrólisis con hexano y tolueno (30%) con las enzimas ANCUT1 y ANCUT2.
La figura 34 muestra un gráfico de la pérdida de peso del polímero PCL después de su reacción de hidrólisis con hexano y tolueno (30%) con las enzimas ANCUT1 y ANCUT2.
La figura 35 muestra las micrografías obtenidas por microscopía electrónica de barrido (SEM) de los poliésteres PBS y PCL después de 72 horas de degradación. Ά: control (cepa silvestre X-33) . B: ANCÜT1. C: ANCUT2.
La figura 36 muestra las micrografías obtenidas por microscopía electrónica de barrido (SEM) de los poliésteres PES, PLLA y PET después de 72 horas de degradación. A: control (cepa silvestre X-33). B: ANCUT1. C: ANCUT2.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Microorganisnos «aplaadoe
El microorganismo utilizado para la obtención de genes fue Aspergillus nidulans PW1 (biAl; argB2; methGl; veAl) , cepa auxótrofa a arginina adquirida del Fungal Genetics Stock Center (Kansas City, KS, USA) .
Las esporas de A. nidulans se cosecharon de placas con medio minimo (Kafer, 1977). Se colectaron en solución al 0.1% de NaCl y se conservaron a 4°C en silica gel (Kawasaki et al. 1995) .
Para la clonación en Escherichia coli con el fin de obtener la construcción con los genes de interés se utilizó una de las varias cepas hospederas disponibles de E. coli, la DH5a <F" 4>80dIacZDM15A (JacZYA-argF) U169 endhl recAl hsdR17 (rK- πΐκ+) deoR thi-l supE44 \-gyrhS6 relAl) , la cual fue adquirida en Novagen.
Para la clonación y expresión en Pichia pastoris se utilizó como hospedero a la cepa silvestre P. pastoris X-33 y el vector pPICZotB adquiridos de Invitrogen, esto no excluye que se puedan utilizar otras cepas disponibles comercialmente de P. pastoris como la GS115, KM71, MC100-3, SMD1163, SMD1165 y SMD1168.
Medio» y condiciones da cultivo.
A. nidulans se creció en medio optimizado CEHM y en medio con cutina como única fuente de carbono como ya se ha descrito
previamente inoculando 1 x 106 esporas/ml (Peña-Montes et al. 2008; Esqueda-Dominguez 2012) .
E. coli se creció a 37°C en medio Luria-Bertani (LB) (1% extracto de levadura-0.5% Bacto-triptona-1% NaCl) y LB bajo en sales (con 0.5% de NaCl) (Bertani, 1951). Para selección de clonas recombinantes, a las placas con LB o LB bajo en sales se les adicionó 100 μg ampicilina/ml ó 25 μg zeocina/ml respectivamente. P. pastoris se creció en medio YEPD que contiene 1% (p/v) extracto de levadura, 2% (ρ/v) peptona y 2% (p/v) dextrosa. Para la selección de clonas recombinantes, a las placas con YEPD se les adicionó 1 M sorbitol y 100 μg/ml zeocina (medio YEPS) .
Las clonas que secretaron la enzima extracelularmente se seleccionaron en medio YEPDT con 0.5% (p/v) de dextrosa, 1% (v/v) tributirina (sustrato para esterasas) y 100 μg I zeocina (medio YEPT) . La inducción de la enzima en cajas Petri se mantuvo adicionando 200 μl de metanol cada 24 horas. Las clonas positivas que generaron halos (debido a hidrólisis de la tributirina por la cutinasa) se crecieron durante 24horas en medio liquido BMGY (1% (p/v) extracto de levadura, 2% (p/v) peptona, 100 mM de fosfato de potasio, 1.34% (p/v)
bases nitrogenadas sin aminoácidos, 4x10-5 % (p/v) biotina y 1% (v/v) glicerol) con el pH ajustado a 6.0.
Para la inducción, las células crecidas en BMGY se centrifugaron (3000 x g, 10 minutos) y se resuspendieron en medio BMMY (BMGY con 0.5% (v/v) de metanol en lugar de glicerol) .
Alalamianto da MRNA y RT-PCR.
Se colectó por filtración el micelio de A. nidulans crecido en las condiciones de inducción para cada enzima (medio minimo optimizado para ANCUT2 (CEHM) y medio con inducción con cutina para ANCUT1) . El micelio se lavó con el amortiguador A (10 mM EDTA, 0.1 M NaCl, 40 mM Tris-HCl pH 8.0) y se pulverizó en un mortero en presencia de nitrógeno liquido hasta obtener un polvo fino. Se aisló el RNA total con el kit "RNeasy Plant Mini Kit" (Qiagen, Valencia, CA) y se purificó el mRNA con el kit "PolyATrack mRNA isolation system" (Promega, Madison, WI) de acuerdo las instrucciones del proveedor. Se sintetizó cDNA con el kit wcDNA cycle" (Invitrogen) y el cDNA obtenido se usó como templado para las reacciones posteriores de PCR.
PCR para amplificación da ganas ancutl y an.ca.t2.
Se amplificaron los genes utilizando el cDNA obtenido de acuerdo a lo descrito anteriormente. El gen ancut2 se amplificó a partir del cDNA obtenido micelio del medio optimizado CEHM (Peña-Montes et al; 2008) y el gen ancutl del cDNA obtenido del micelio del medio inducido con cutina {Esqueda-Domínguez, 2012) . Se utilizaron los siguientes cebadores específicos (secuencias en dirección 5'a 3'):
Para amplificar el gen ancutl:
Cebador directo ancutlF:
SEQ ID NO 1: cctgaattctgaatccaatccgtctcgatc
Cebador reverso ancutlR:
SEQ ID NO 2: ccttctagagcaagcttcccaacaagaaagtc
Para amplificar el gen ancut2:
Cebador directo ancut2F:
SEQ ID NO 3: cctgaattctgagtcctttgaatcttgacgag
Cebador reverso ancut2R:
S*q ID NO 4: ccttctagagcgttaccaaggccagagaaccag
Es importante mencionar que para el diseño de los cebadores que amplifican cada gen se tomaron en cuenta los siguientes criterios:
Para el cebador directo de cada gen:
1.- Se diseñaron para amplificar cada gen justo después de las secuencias señal identificadas por Castro-Ochoa para
ANCUT2 y Esqueda-Domínguez para ANCUT1, es decir el amplicón de cada gen no tiene la secuencia señal.
2. - Se adicionaron los nucleótidos necesarios para que no se presente un cambio en el marco de lectura del gen justo después de la secuencia de reconocimiento por la enzima de restricción y antes de la secuencia del gen.
3. - Se adicionaron 3 nucleótidos previos a la secuencia del sitio de restricción (EcoRl) para permitir el anclaje de la enzima de restricción.
4.- Parte de la secuencia es complementaria al extremo 5 'de cada gen de cutinasa sin secuencia señal, ya sea ANCUT1 (AN05309) o ANCUT2 (AN07541) .
Para el cebador inverso de cada gen:
1. - En los cebadores inversos no se adicionó un codón de paro con el fin de que en ambas enzimas se adicione una cola de histidinas en el C-terminal.
2. - Se adicionaron los nucleótidos necesarios para que no se presente un cambio en el marco de lectura del gen justo después de la secuencia de reconocimiento por la enzima de restricción y antes de la secuencia del gen.
3. - Se adicionaron 3 nucleótidos previos a la secuencia del sitio de restricción (Xbal) para permitir el anclaje de la enzima de restricción.
4. -Parte de la secuencia es complementaria al extremo 3' de cada gen de cutinasa, ya sea ANCUT1 (AN05309) o ANCUT2(AN07541) . Se utilizaron las siguientes condiciones de amplificación con la enzima Pfu polimerasa (Fermentas) :
Tabla 2. Condiciones de amplificación para gen ancutl.

Tabla 3. Condiciones de amplificación para gen ancut2.

Clonación y Expresión da loa ganas ancutl y ancut2 an Plchim pmatoria.
Construcción da loa vetorea pPICZoüWCDTl y pVXCZcANCUT2.
Para la clonación de los genes en el vector pPICZaB, se introdujeron a cada amplicón por PCR los sitios de corte para
las enzimas de restricción EcoRl y Xbal. Cada gen de cutinasa completo se amplificó de cDNA con los cebadores específicos para cada gen (ancutlF-ancutlR o ancut2F-ancut2R) . Los amplicones de PCR y el vector se digirieron con las enzimas EcoRl y Xbal y se ligaron con una ligasa de acuerdo al protocolo del proveedor (Fermentas) . Los insertos se introdujeron en los sitios respectivos del vector pPICZaB generando los vectores pPICZotancutl y pPICZctancut2. Clonación en ¾. eoli.
Se realizaron células electrocompetentes de DH5ot y se transformaron por electroporación ya sea con el vector pPICZarancutl o pPICZaancut2. Las clonas recombinantes se crecieron a 37°C en 50 mi de medio LB con zeocina para la extracción de plásmidos con el kit "Plasmid midi" (Qiagen) . Se verificó la presencia de cada gen en ambas construcciones (pPICZora/icutl y pPICZaancut2) por amplificación de cada gen tanto con cebadores específicos como con universales (AOX) contenidos en el vector, asi como por la secuenciación de los amplicones obtenidos.
.Expresión de lom qmamm mncutl y *ncut2 ·η P. Pastoría.
Se prepararon células electrocompetentes de P. pastoría X-33 y se transformaron por electroporación ya sea con el vector
p?ICZccancutl o con pPICZotancut2. Todas las incubaciones se realizaron de 28°C a 30°C. Las clonas recombinantes se seleccionaron en medio YEPS con zeocina y se seleccionaron posteriormente las clonas productoras de halo (positivas) en medio YEPDT, ambos medios descritos anteriormente. Las transformantes positivas se crecieron en medio liquido BMGY hasta alcanzar una OD6oo■ 2 a 6, se centrifugaron (4,000 x g, 10 min) y el botón celular se transfirió al medio BMMY para inducir la expresión de la proteina. Se adicionó metanol (0.5% v/v) diariamente para la inducción. Los extractos inducidos se centrifugaron (4,000 x g, 10 min) y se colectó el sobrenadante.
Obtención da inóculoa de células da P. pastoría productora» da las aminas recombinante» ANCUT1 y ANCUT2.
Se tomaron las células transformantes con ANCUT1 ó ANCÜT2 y se inoculó un tubo Falcon de 50 mi estéril con 5 mi del medio YEPD y se incubó entre 16 horas a 18 horas de 28°C a 30°C de 200 a 250 rpm. Se centrifugó durante 5 min y el botón se resuspendió en 1 mi de medio YEPD-Glicerol 1%. Se tomaron alicuotas de 100 μl y almacenaron en tubos de 200 μl a -70°C.
Inducción da las enzimas recombinantes .
Se sembraron células transformantes con ANCUT1 ó ANCUT2 de P. pastoris en un matraz de 250 mi con 25 mi de medio BMGY estéril, el cual se incubó entre 16 y 18 horas a 28°C a 30°C a 200-250-300 rpm (O.D. de 2 a 6) . El contenido del matraz se colocó en un tubo falcon de 50 mi y centrifugó a 10000 rpm por 5 min.
Se resuspendió el botón celular en 100 mi de medio BMMY (BMGY con 0.5% de metanol en lugar del glicerol) y se tomó una alícuota del medio (2 a 4 mi) como una muestra del tiempo cero de inducción. Se incubó a 28°C a 30°C a 200-300 rpm. Se agregaron 0.5 mi de metanol absoluto cada 24 horas y se tomaron muestras a cada tiempo de inducción (2 a 4 mi cada 24 horas hasta llegar a las 120 h) para determinar el tiempo óptimo de producción de cada enzima. Al final de la inducción se centrifugó a 14000 rpm por 15 min a 4°C.
El sobrenadante con la enzima se utilizó posteriormente para la purificación de la cutinasa y el botón celular de cada uno de los tubos se almacenó a -70°C para su posterior uso en extracción de DNA y verificación de la presencia del gen de cutinasa en el genoma de P. pastoris por amplificación por PCR y secuenciación.
Concentración dm enzimas raconbinantas .
Una vez pasado el tiempo de incubación óptimo, se transfirió el cultivo a tubos falcon de 50 mi y se centrifugaron a 8500 rpm por 10 min para separar las células del sobrenadante (extracto enzimático) . Una vez centrifugados, se trabajó con el sobrenadante, midiendo el volumen obtenido. Se determinó la cantidad de proteína, actividad de esterasa, perfil de proteínas y zimograma. Posteriormente, se realizó una ultrafiltración del extracto crudo en un sistema Amicon (Millipore) con membrana de 10 kDa como limite de corte nominal para concentrar la muestra (1:5).
Evaluación da degradación en-.imática da poliéataras.
Se emplearon como modelo los polímeros de poli (1, 4-butulen succinato) (PBS) , policaprolactona (PCL) , poli (etilensuccinato) (PES) , poli (L-láctico) (PLLA) y polietilentereftalato (PET) .
Emulsión da polimaroa.
Se procedió a realizar emulsiones por separado de PBS, PCL, PES y PLLA (Sigma-Aldrich, Missouri, USA) . Para disolver el poliéster se añadieron 200 mg de polímero en 5 mi de cloroformo. Se preparó una solución de SDS al 0.1% en agua desionizada a la cual se le añadió el polímero disuelto en cloroformo. Se procedió a emulsificar homogenizando la
solución, hasta obtener una fase estable. La emulsión se evaporó a 40°C para remover el cloroformo (Hiraishi, 2008) .
Preparación da medio» en placas.
Se preparó el medio BMGY 2X y agar 1.5%, se realizó una dilución 1:1 agregando la emulsión del polímero. Una vez obtenida la placa, se sembró en ellas las clonas de P. pastoris que expresan las enzimas ANCUT1, ANCÜT2 y se utilizó también la cepa de P. pastoris X-33 como control negativo. Se incubaron a 29°C, añadiendo 100 μl de Metanol 20X cada 24 horas en la tapa de la caja Petri. La concentración final del polímero en las cajas es de 0.4%.
Preparación de películas da PBS, PES, PET, PCL y PUA.
Se prepararon películas circulares de aproximadamente 3 mm de diámetro y 1 mm de grosor, con un peso aproximado de 10 mg. El PBS y PCL se fundieron aproximadamente a 80°C y se comprimieron en un molde para obtener películas pequeñas (Sulaiman, 2012) .
Para el caso de PES y PLLA, se tomó una muestra directa del polímero para realizar la reacción de hidrólisis y para PET se tomaron muestras de botellas de agua comerciales (Bonafont y CIEL) , de las cuales se cortaron pequeñas películas de aproximadamente lcm x lcm. El proveedor de agua Bonafont,
indica en la etiqueta que su botella está hecha de PET y el proveedor de CIEL, menciona que es Bio-PET.
Reacción de hidrólisis de películas de PBS, PES, PCL y PLLA.
La reacción de hidrólisis enzimática se realizó colocando en un vial:
Aproximadamente 50 mg de películas, 1 mi de extracto crudo con enzima ANCÜT1 (1500 ü) y 2 mi de buffer de fosfatos 50 mM pH 7.0, y para la enzima ANCUT2 se agregó 100 μl (1500 ü) . En el caso para los controles, en el primero se utilizó 100 μl del extracto crudo de la cepa de P. pastoris X-33 y en el segundo 1 mi de buffer de fosfatos 50 mM, en lugar de la solución enzimática. Los viales se incubaron a 37*C a 225 rpm durante 3 dias, tomando muestra cada 24 horas. Para el caso de la enzima ANCUT2, se agregó 100 μl del extracto cada 24 horas .
Se determinó la degradación por pérdida de peso, acidez titulable y microscopía electrónica de barrido (SEM) .
Pérdida de peso de películas.
Una vez trascurrido el tiempo de reacción de degradación enzimática, las películas se lavaron con etanol y se secaron en estufa a 37°C durante toda la noche.
Se pesaron en balanza analítica y se registró el peso. La pérdida de peso se determina restado al peso inicial el peso final. Acide., titulable.
El sobrenadante de la reacción de degradación se tituló con NaOH valorada 0.01N y se utilizó como indicador fenolftaleina. Se registró el gasto de mi de NaOH. Interminación da condiciona» da reacción: Temperatura y pH.
Se procedió a realizar la reacción de hidrólisis anteriormente descrita, cambiando las condiciones de pH y temperatura, que se han observado son las óptimas para la expresión de la actividad de las enzimas recombinantes de P. pastoris ANCUT1 y ANCUT2 (Morales, 2015, Solís, 2015) .
Para el caso de la enzima ANCUT1, se procede a trabajar con las temperaturas de 40°C, 50°C y 60°C y pH de 7.0, 8.0 y 9.0 (Solis, 2015) y para la enzima ANCÜT2, se trabajan las temperaturas de 30°C, 40°C y 50°C y pH de 7.0, 8.0 y 9.0 (Morales, 2015) .
Los viales se incubaron a las temperaturas anteriormente descritas y a 225 rpm durante 24 horas. Ya que en la reacción de hidrólisis se demostró que hay una pérdida significativa
de peso. Para el caso de la enzima ANCUT2, se agregó 100 μl del extracto cada 24 horas.
Los buffers utilizados fueron buffer de fosfatos 0.1M (pH 7.2), buffer Tris-HCl 0.1M (pH 8.0) y buffer Tris-HCl 0.1M (pH 9.0) .
Sólo para el caso de PLLA se trabajó a condiciones de 60°C y pH de 7.0, 8.0 y 9.0 para a enzima ANCUT1 y para la enzima ANCUT2, a 50°C y pH de 7.0, 8.0 y 9.0.
Se determinó la degradación por medio de la pérdida de peso después de la reacción de hidrólisis.
Determinación de condiciones de reacción : Solventes y ti «arpo .
Se utilizaron como solventes hexano y tolueno al 30% para llevar a cabo la reacción de hidrólisis a 48 horas, tomando muestras cada 24 horas.
Para la reacción con la enzima ANCUT1, en un vial se colocó aproximadamente 50 mg de películas, 0.1 mi de enzima, 0.9 mi de solvente, y 2 mi de buffer de fosfatos 50 inM. Para la enzima ANCUT2, aproximadamente 50 mg de películas, 1 mi de enzima, 0.9 mi de solvente y 1.1 mi de buffer de fosfatos 50 mM. También se realizó un blanco solo con buffer de fosfatos 50 mM y el solvente.
La reacción se llevó a cabo por 96 horas a 37°C y 225 rpm, tomando muestras cada 24 horas. Después de la reacción, las
películas se lavaron con etanol y se secaron en estufa a 37°C durante toda la noche.
Para el caso de las películas de PCL con solvente tolueno, después de la reacción de hidrólisis, se colocaron en un baño a 50°C para evaporar el solvente, después se lavaron las películas con etanol y se secaron en estufa a 37°C durante toda la noche.
Se determinó la degradación por medio de la cuantificación de la pérdida de peso después de la reacción de hidrólisis.
Aplicación da hidrólisis enzimática en muestras comerciales
(PET) .
Para la reacción de hidrólisis de PET se extendió el ensayo a 3 semanas, agregando enzima ANCUT1 cada 72 horas y enzima ANCUT2 cada 24 horas, tomando muestras cada 72 horas. Se determinó la degradación por la pérdida de peso y microscopía electrónica de barrido (SEM) .
Morfología de películas.
La morfología de las películas antes y después de la reacción de hidrólisis se observaron mediante microscopía electrónica de barrido (SEM) (JSM-5900LV de JEOL) a alto vacío a un voltaje de aceleración de 20 kV. Las películas fueron
recubiertas con una capa fina de oro (10 Á) antes de ser observadas .
EJEMPLOS DE LAS MODALIDADES PREFERIDAS DE LA INVENCIÓN
Técnicas Bioquímicas utilizada».
1) Determinación de proteina por el método Bradford en microplaca (Bradford, 1976; Morales-Garcia, 2015) .
2) Determinación de actividad cualitativa de esterasa (CEH)con acetato de α-Naftilo (Mastropaolo, 1981).
3) Determinación de actividad cuantitativa de esterasa (CEH) con acetato de p-nitrofenilo (p-NPA) (y otros p-NP ésteres (p-NPE) ) (Ejima,2004; Nawani, 2006; Bornscheuer et al., 2005. 4) Determinación de perfil electroforético por preparación de geles SDS-PAGE (Laemmli, 1970) .
5) Determinación de actividad de esterasa por medio de zimogramas (Karpushova, 2004} .
6) Las técnicas moleculares utilizadas fueron las descritas por Sambrook et al. (1989).
7) Geles de agarosa. Se preparó agarosa al 1% en buffer TAE IX, para disolver la agarosa se deben calentar dichas preparaciones . Se agregó en una probeta 30 mi de agarosa y se agregan 3 μl de bromuro de etidio (10 mg/ml) en una
concentración final de 0.1 mg de bromuro/ml de agarosa. Se vertió en la sección de la cámara de electroforesis correspondiente, se colocó el peine y se dejó polimerizar. Cuando polimerizó se retiró el peine, se agregó buffer TAE IX y se cargó la muestra con buffer de carga 6 X de Thermo Scientific. Se utilizó el marcador de peso molecular (MPM) 1 kb de Thermo Scientific. Se corrió a 80 mV y se observaron los resultados en un transiluminador de rayos UV. EJEMPLO 1; Aislamiento da RMA y amplificación de ganas antnitl y antmt2 por RT-PCR.
En la figura 1 se observa el RNA obtenido de cada uno de los micelios de A. nidulans crecidos en condiciones de inducción para cada enzima, es decir, en medio mínimo optimizado para ANCUT2 (CEHM) (carril A) y medio con inducción con cutina para ANCUT1 (carril B) . En la figura 2 se observa el producto de PCR (amplicón) de cada gen de las cutinasas ancutl (carril A) y ancut2 (Carril B) obtenido por RT-PCR. En el caso de la cutinasa ANCUT1 se obtuvo un amplicón de 650 bp y para la cutinasa ANCUT2 de 750 bp, de acuerdo a lo esperado teóricamente.
EJEMPLO 2: Verificación de la presencia del gen ANCUT1 6 ANCUT2 en al gancma da P. pastoris y da la expresión da las entinases . Extracción da DNA.
Se centrifugó una muestra de un cultivo de células inducidas de P. pastoris para la producción de las enzimas recombinantes ANCUT1 y ANCUT2, tal como se indició en el apartado referente a la obtención de inóculos e inducción de enzimas recombinantes mencionados anteriormente, a 14,000 rpm durante 10 minutos a 4°C. Se eliminó el sobrenadante y se añadieron 500 μl de buffer de lisis (buffer Tris 2 M, pH 8.0, NaCl 250 mM, SDS 25 mM y EDTA 0.5%) y 200 μl del equivalente en perlas de vidrio, para ayudar a lisar a la levadura. Se agitó en un vortex a máxima velocidad durante 3 minutos. Se incubó en un baño de agua a 65°C durante 30 minutos. Se añadieron 20μ1 de RNAsa (20 μg/ml) y se incubó a 37°C durante 30 minutos. Se centrifugó a 14,000 rpm durante 10 minutos. Se transfirió el sobrenadante a un tubo nuevo y estéril. En la figura 3 se observa el DNA de la clona de P. pastoris X-33 con el gen ANCÜT1 (carriles 2 y 3) o ANCUT2 (carril 4) y de la cepa silvestre X-33 (carril 5) , se midió la concentración de DNA dando como resultado 3,782.3 ng/μl con una relación 260/280 de 1.89, lo que nos indica una pureza aceptable. Por
medio de una electroforesis en un gel de agarosa se verificó la presencia de DNA. En la figura 3 (carril 2, 3, 4 y 5) se muestra una banda por arriba de 10,000 pb que corresponden al DNA extraído. También se observa la presencia de una banda por debajo de 250 pb que corresponden a productos de degradación de RNA.
PCR con cébmdor»a especificacos y universal»».
Una vez obtenida la muestra de DNA de P. pastoris se realizó una PCR con cebadores específicos para cada gen de cutinasa (SEQ ID NO 1, 2, 3 y 4) en las condiciones anteriormente descritas en las tablas 2 y 3. También se realizó una PCR con cebadores universales para la región promotora y terminadora (AOX) descritos en el manual de expresión de P. pastoris al igual que las condiciones de reacción (Easy Select Pichia Manual, Invitrogen, 2009) .
En la figura 4A se aprecia de una banda cercana a 650 pb que corresponde al peso molecular teórico del gen ancutl. Se realizó adicionalmente un PCR con cebadores universales para la región promotora y terminadora del gen aox. En la figura 4B, en los carriles 2 a 4, se observa la presencia de una banda cercana al peso molecular teórico de 1200 pb que corresponde al fragmento de DNA insertado a partir de la construcción en el plásmido pPICZa (desde la región promotora
aox a la región terminadora aox) que codifica para: 1) la región promotora del gen aox, 2) la secuencia señal del cc- factor de Sacharomyces Cerevisiae, 3) el gen ANCÜT1 de la cutinasa, 4) un Epítope myc, 5) tallo de histidinas y 6) la región terminadora del gen aox. La banda adicional a la esperada que se muestra por arriba de 1500 pb puede corresponder a la amplificación del gen aox2 dado que los cebadores pueden hibridar con la región promotora y terminadora de la copia del gen aox (aox2) en el DNA de Pichia pastoris (Cerenghino y Cregg, 2000) . La otra banda por arriba de 6000 pb puede corresponder a productos de degradación de DNA o de amplificación inespecifica. Sin embargo, se realizó una purificación para obtener únicamente la banda de interés (1200 pb) . Por otro lado, la presencia de la banda de 592 pb en el carril 5 (figura 4B) corresponde a la amplificación de la misma región a partir del vector pPICZotB sin cutinasa (promotor aox-terminador aox) , el cual fue utilizado como control negativo.
En la figura 5 se observa la amplificación del gen ANCUT2 con cebadores específicos y universales AOX. Se observa que el amplicón de PCR con cebadores específicos tiene un peso aproximado de 700 pb (carriles 1 y 3) que coincide con lo esperado. Por otra parte, el amplicón obtenido con los cebadores universales de AOX muestra una banda que tiene un
peso aproximado de 1290 pb (carril 2) que corresponde al valor teórico. El proveedor del vector de clonación en Pichia pastoris indica que la región AOX tiene un tamaño de 592 pb y se considera adicionalmente el peso del gen insertado en el vector (700 pb) dando un tamaño total teórico de 1292 pb.
Purificación de anplicon>a y «acunciacién.
La purificación de los amplicones se realizó con el kit GenElute Minus EtBr spin columns de acuerdo al protocolo del proveedor (Sigma-Aldrich) . Al final se resuspendió el pellet en 50 μl de buffer TE. Se prepararon diluciones del amplicón puro obtenido de 50 ng/μl y se enviaron para una primera secuenciación a Macrogen, Corea.
Posteriormente, se realizó un duplicado de dicha secuenciación de la purificación de los productos de PCR con cebadores especificos y universales AOX, se midió la concentración de DNA y se realizó dicha segunda secuenciación también en Macrogen para verificar y comprobar la presencia del gen de cutinasa ANCÜT1 ó ANCUT2 insertado en el genoma de P. pastoris.
Para el caso de la ANCUT1, al analizar las secuencias se identificó que pertenecían al gen de la cutinasa ANCUT1 (AN05309) y no presentaba cambios en las bases nitrogenadas con respecto a las secuencias reportadas en las
bases de datos (figuras 6A y 6B) . Se observó la adición de 117 aa a la secuencia de la enzima nativa, utilizados como una estrategia del proceso de clonación, 94 residuos en la parte del N- terminal y 23 residuos en la parte del C- terminal. Sin embargo, es importante considerar que la región N- terminal, 89 residuos son eliminados debido a que pertenecen a la secuencia señal del factor a, quedando sólo 5 aa adicionales en el N- terminal de la proteina nativa.
Se realizó adicionalmente una identificación de péptidos de la cutinasa pura (como se explica adelante) por la técnica de espectrometría de masas (en el IBT y BioUSAII de la UNAM) , en las cuales se lograron identificar 9 péptidos con un 76.06% de cobertura. Todos los péptidos pertenecieron a la secuencia de aminoácidos de la cutinasa ANCUT1. Utilizando herramientas bioinformáticas ExPASy se determinó que la proteina heteróloga posee un peso molecular teórico de 23.34 kDa y un pl de 5.96. La secuencia de la proteina recombinante verificada con la secuencia de nucleótidos correspondiente se observa en la figura 7, así como los péptidos identificados. Para el caso de la ANCUT2, se realizaron los mismos procedimiento de secuenciación y verificación mencionados anteriormente para ANCUT1 (figuras 6C y 6D) .
Al analizar las secuencias se identificó que pertenecían al gen de la cutinasa ANCUT2 (ANO7541) y no presentaba cambios en
las bases nitrogenadas con respecto a las secuencias reportadas por Castro-Ochoa, 2012 y Bermúdez, 2014 (figura 8) . Se observó la adición de 116 aa a la secuencia de la enzima nativa, utilizados como una estrategia del proceso de clonación, 93 residuos en la parte del N- terminal y 23 residuos en la parte del C- terminal. Sin embargo, es importante considerar que la región N- terminal, 89 residuos son eliminados debido a que pertenecen a la secuencia señal del factor o, quedando sólo 4 aa adicionales en el N- terminal de la proteina nativa.
Se realizó adicionalmente una identificación de péptidos de la cutinasa pura (como se explica adelante) por la técnica de espectrometría de masas (en el IBT y BioUSAII de la UNAM) , en las cuales se lograron identificar 14 péptidos con un 69.8% de cobertura. Todos los péptidos pertenecieron a la secuencia de aminoácidos de la cutinasa ANCUT2. Utilizando herramientas bioinformáticas ExPASy se determinó que la proteina heteróloga posee un peso molecular teórico de 27.4 kDa y un pl de 4.7. La secuencia de la proteina recombinante verificada con la secuencia de nucleótidos correspondiente se observa en la figura 8, asi como los péptidos identificados.
EJEMPLO 3: Producción de las anglmas ANCUT1 y ANCUT2 recoadainantaa.
Se observó por medio de microscopía el microorganismo con el que llevó a cabo el trabajo experimental (P. pastoris) , el cual presentó las características típicas en cuanto a forma y tamaño de las levaduras.
Se realizó el crecimiento de las clonas y la inducción con metanol para la producción de la enzima de interés ANCUT1 y ANCUT2 durante varios días, se midió la actividad de esterasa a las alícuotas tomadas del extracto crudo cada 24 horas (0, 24, 48, 72, 96 y 120 h) durante el tiempo de inducción. Mediante la prueba de actividad cualitativa sobre acetato de α-naftilo como sustrato y Fast red como revelador, se comprobó que las fracciones de los diferentes tiempos de inducción mostraron actividad de esterasa desde las 24 horas para ambas enzimas (figura 9 fila Ά, y 10), debido a la presencia de coloración rojo/marrón que se observó, causada por la hidrólisis de acetato de oc-naftilo liberando el a- naftol que forma un compuesto cromógeno de color rojo/marrón al reaccionar con Fast Red.
Al mismo tiempo como control negativo se llevó a cabo la inducción de la cepa silvestre P. pastoris X-33 y se comprobó la ausencia de actividad esterasa durante el periodo de 0 a
48 horas de inducción, ya que a partir 72 horas se observó una coloración tenue que se puede atribuir a las enzimas intracelulares de Pichia pastoris liberadas durante la lisis por muerte celular (Figura 9 fila B) (Castro-Ochoa et al., 2013).
EJEMPLO 4: Cinética da la producción da las «Mimas racoabinantaa .
Con el fin de determinar el tiempo de inducción óptimo, se monitoreó la actividad en todos los tiempos de inducción. Se cuantificó por medio de hidrólisis de acetato de p- nitrofenilo. Se realizó la prueba de cuantificación de proteína del extracto crudo a cada tiempo de inducción y se determinó la actividad específica.
En la figura 11 se observan los resultados obtenidos para la ANCUT1, se detectó un pico máximo de actividad a las 24 horas de inducción, se estableció por lo tanto este tiempo como el óptimo de inducción. Se logra apreciar como aumenta la concentración de proteína a lo largo del tiempo de inducción. En este caso se observó para la ANCÜT1 una actividad específica de 750 LJ/mg a las 24 horas.
En el caso de la ANCÜT2, se puede observar en la figura 12 que el tiempo en el que se presenta mayor actividad es a las 48 horas de inducción, sin embargo, la actividad no varía
mucho respecto a la actividad a las 24 horas, por lo que se decidió llevar a cabo la inducción 24 horas con el fin de economizar en gastos de producción y reducir el tiempo de cosecha. Se observó para la ANCUT2 una actividad especifica de 549 U/mg a las 24 horas.
EJEMPLO 5: Determinación del perfil de proteínas y «laogramee .
Se determinó el perfil de proteínas en geles SDS-PAGE para observar la presencia de las enzimas de interés a lo largo de la inducción en el extracto crudo, cargando 10 mg de proteina por cada tiempo de inducción (figuras 13 y 14) .
En la figura 13 se puede observar para ANCUT1 que a partir de 24 horas de inducción (carril 3) hay dos proteínas a 22.7 kDa y a 24.8 KDa, respectivamente, cuya presencia se incrementa a lo largo del periodo de inducción. Es importante mencionar que la proteina de 22.7 kDa se encuentra cercana al peso molecular teórico de la cutinasa ANCUT1 recombinante (23.34 kDa) . Más adelante se describe que ambas bandas corresponden a la ANCUT1.
En la figura 14 para ANCUT2, se observa en el gel teñido con coomassie un aumento en la intensidad de las bandas que migraron a un peso equivalente a 37, 24 y 21kDa, conforme aumenta el tiempo de inducción. La banda de 37 kDa es la que
se observó con mayor intensidad, se describe más adelante que las tres bandas corresponden a la ANCUT2.
Para observar la presencia de actividad esterasa en el gel SDS-PAGE realizado con el extracto crudo de ANCUT1 a diferentes tiempos de inducción, se realizó un gel de actividad nln sltu", en el que no se utilizó β- mercaptoetanol, ni se aplicó calor a la muestra. Se cargaron 10 mg de proteina por cada tiempo de inducción. En la figura 15 en el carril número 2 (24 horas de inducción) se aprecia una banda gruesa que representa la actividad esterasa entre 20-25 kDa, la cual tiene una aparición constante a lo largo del tiempo de inducción. En el carril 6 (240 horas de inducción) se observaron 2 bandas adicionales con actividad de esterasa, una cercana a 15 kDa y otra entre 50 y 100 kDa. Este resultado puede deberse a la lisis celular de P. pastoris, la cual libera enzimas intracelulares con actividad de esterasa (Castro-Ochoa et al., 2013). Los resultados obtenidos en el perfil proteico y el zimograma muestran una correlación ya que durante el transcurso del tiempo de inducción la presencia de la que puede ser la proteina de interés ANCUT1 (22.7 kDa) va en aumento, al igual que la presencia de la actividad de esterasa para esa misma. Sin embargo, es importante mencionar que este comportamiento también se observó para la otra banda de 24.8 kDa.
En la figura 16 para ANCUT2, se observa que, de las tres bandas, tienen actividad las bandas de 37 y 24 kDa, observándose mayor actividad en la banda de 37 kDa, por lo que se supuso que se trata de la cutinasa recombinante mientras que las bandas de 24 y 21 kDa son productos de hidrólisis de la ANCUT2, se considera que el sitio activo se encuentra presente en la fracción de 24 kDa por lo que ésta conserva actividad, este comportamiento también se observó en la enzima nativa (Castro-Ochoa, 2014) . En la aparición de bandas de menor peso al esperado se descarta una hidrólisis por parte de proteasas expresadas por P. pastoris ya que al tratarse de un sistema de expresión heteróloga tiene deletados los genes para proteasas secretadas al medio. Este comportamiento debido, al parecer, a una autohidrólisis de la enzima ha sido observado anteriormente por Castro-Ochoa y colaboradores (2012), quienes reportan dos bandas con actividad de esterasa en los pesos de 29 y 24 kDa en extractos crudos, las cuales, al ser identificadas por técnicas de espectrometría de masas, se vio que corresponden a la ANCUT2. La banda de 29 kDa se identificó como la ANCUT2 de A. nidulans nativa y la de 24 kDa un producto de su autohidrólisis, por lo que se considera puede tratarse de un comportamiento propio de la enzima nativa que está repitiéndose en la enzima recombinante (Castro-Ochoa, 2012) .
EJEMPLO 6: Purificación da las enzimas recombinantes e identificación. Precipitación con sulfato da amonio.
Para el caso de la concentración de proteína del extracto crudo se llevó a cabo una precipitación con sulfato de amonio. Se filtraron los sobrenadantes de los cultivos y se colocaron en un baño de hielo sobre una parrilla en agitación. Se añadió lentamente sulfato de amonio hasta una concentración de 65% . Conforme se iba acercando el punto de saturación se añadió más lentamente la sal. Se continuó agitando 30 min después de que toda la sal sé hubo añadido y posteriormente se centrifugó la solución a 8500 rpm durante 10 min.
Después se decantó el sobrenadante y se resuspendió el botón en 1 a 2 volúmenes de regulador pH 7.0. ültrafiltraclón.
Se realizó una ultrafiltración del extracto crudo previa a la purificación para concentrar la proteína recombinante en Amicon (Millipore'") con membranas YM de límite de exclusión de 10 kDa. Se partió de 250 mi de extracto y se utilizó una
membrana de 10 kDa para concentrar la muestra reduciendo su volumen hasta 15 mi.
Purificación con columna da afinidad.
Se realizó la purificación del extracto ultrafiltrado con la columna Clontech** His Talón Gravity column. Se utilizaron los siguientes amortiguadores:
❖ Amortiguador de equilibrio (50 mM fosfato de sodio, 300 raM NaCl, pH 7.4, 1L)
· 7.089 g de Na2HP04
• 17.52 g NaCl
❖ Amortiguador de lavado (50 mM fosfato de sodio, 300 mM NaCl, 10 mM imidazol, pH 7.4, 1L)
• 7.089 g de Na2HPO<
· 17.52 g NaCl
• 0.681 g imidazol
❖ Amortiguador de elución (50 mM fosfato de sodio, 300 mM NaCl, 150 mM imidazol, pH 7.4, 1L)
• 7.089 g de Na2HP04
· 17.52 g NaCl
• 10.21 g imidazol
❖ Amortiguador de regeneración (20 mM MES, 0.3 mM NaCl, pH 5.0, 200 mi)
• 0.781 g de MES
• 3.50 g NaCl
Todas las soluciones se filtraron con membrana de 0.45 μπι. Se equilibró la columna con 5 a 10 volúmenes de amortiguador de equilibrio, se cargó la muestra clarificada, tomando en consideración que la capacidad de la columna era de hasta 3 mg de proteína y se colectaron fracciones de 1 mi. Se lavó la columna con 8 volúmenes amortiguador de equilibrio, seguido de 7 volúmenes de amortiguador de lavado. La cutinasa recombinante se eluyó con 5 a 8 volúmenes de amortiguador de elución y se colectaron fracciones de 1 mi. Se midió la absorbancia a 280 nm a las fracciones de elución. La columna se regeneró después de cada purificación con 20 mi de amortiguador de equilibrio o lavando con 10 mi de amortiguador de regeneración. La columna se almacenó con etanol al 20% a 4°C.
La ANCUT1 recombinante pura presentó una de actividad específica de 14,823.86 ü/mg, valor que se encuentra arriba de los reportados en la literatura para cutinasas puras recombinantes como la CUTAB1 de Alternaría brassicicola con un valor de 4,394 ü/mg o la cutinasa de F. solaní con una actividad específica de 3, 990 U/mg ambas expresadas en P. pastoris (Koschorreck et al., Kwon et al., 2009).
El rendimiento y factor de purificación de la enzima pura es 17.3 % y 2.4 respectivamente. El rendimiento puede mejorar si se optimiza el proceso de purificación de la enzima por medio de la columna de afinidad de cobalto. En la figura 17B se observó después de la purificación de la ANCUT1, la presencia de la banda de 22.7 kDa y la de 24.8 kDa. La primera con un peso cercano al peso teórico de la enzima ANCUT1 heteróloga 23.34 kDa. Este resultado nos indica que ambas proteínas corresponden a la ANCUT1 recombinante con el tallo de histidinas. La presencia de las dos bandas después de purificar que corresponden a la ANCUT1 recombinante puede explicarse por modificaciones post traduccionales durante la expresión de la proteina en P. Pastoris. Estos resultados coinciden con los de otros autores que han observado que se obtienen dos formas de una misma proteina después de ser inducida en P. pastoris. Estos autores han concluido que la presencia del tallo de polihistidina y Epitope myc en el C- terminal fusionados a la cutinasa tiene un efecto negativo significante en el proceso celular para la sintesis, plegamiento y secreción adecuado de la enzima en P. pastoris (Kwon et al., 2009) .
En el caso del gel de actividad win situ", donde no se utilizó β-mercaptoetanol, ni calor (figura 17A) se puede observar, la presencia de una banda de actividad esterasa
gruesa en 20 a 25 kDa en el carril 4 que corresponde a la enzima pura.
Se realizó el ensayo inmonublot ("Western blot") para verificar si las dos proteínas que se encontraron después de la purificación tenían tallo de polihistidina (Figura 17C) . Se realizó un gel de electroforesis SDS-PAGE con 1.5 μg de proteina por carril y posteriormente la transferencia a una membrana de PVDF. Se obtuvo como resultado que las 2 bandas con pesos moleculares de 22.7 y 24.8 kDa, que se encontraron en los geles SDS-PAGE de purificación fueron detectadas en el "Western blot" con los anticuerpos contra el tallo de histidinas (anti-histag) , el resultado nos confirma que la enzima ANCUT1 recombinante posee 2 formas con tallo de histidinas.
En cuanto a la purificación de la cutinasa ANCUT2 , en la figura 18B se observa que el peso de la proteina pura corresponde a 37 kDa. Después de la purificación, ya no se observan las bandas de 24 y 21 kDa que se presentaban en el perfil proteico de inducción, esto podria indicar que estas bandas son fragmentos de la enzima recombinante que al hidrolizarse pierden la cola de histidinas y por lo tanto no se logran adherir a la columna.
Se realizó un ensayo inmunológico de detección anti HisTAG "Western blot" para confirmar que la enzima pura correspondia
efectivamente a la ANCUT2 recombinante a pesar de la diferencia que presentó con el peso molecular teórico (figura 18A) . Se usó un anticuerpo que reconocía al tallo de histidinas anexado en el carboxilo terminal de la enzima. En la figura 18A se pueden observar las bandas que confirman que la enzima es la ANCUT2 recombinante, ya que los anticuerpos dieron resultado positivo al ensayo de "Western blot". Es importante resaltar que ya no se observan las bandas correspondientes a los pesos de 21 y 24 kDa anteriormente mencionadas, con lo que se confirma que ambas carecían del tallo de histidinas y no fueron adheridas a la columna de afinidad.
Posteriormente, se llevó a cabo la identificación de las bandas que presentaron actividad en el zimograma de inducción de ambas enzimas recombinantes ANCUT1 y ANCUT2.
Se realizó una identificación de péptidos por la técnica de espectrometría de masas (en el IBT y BioUSAII de la UNAM) de la ANCUT1, en la cual se lograron identificar 9 péptidos con un 76.06% de cobertura de la banda de 22.7 kDa. Todos los péptidos pertenecieron a la secuencia de aminoácidos de la cutinasa ANCUT1 (AN05309) . Utilizando herramientas bioinformáticas ExPASy se determinó que la proteina heteróloga posee un peso molecular teórico de 23.34 kDa y un pl de 5.96. La secuencia de la proteina recombinante
verificada se observa en la figura 7, así como los péptidos identificados.
Para las ANCÜT2 se cortaron las bandas correspondientes a los pesos de 37 y 24 kDa del gel teñido con Coomassie Blue y fueron identificadas mediante el método LC-MS/MS en el Instituto de Biotecnología de la ÜNAM en Cuernavaca, Morelos. El análisis de las bandas mostró como resultado la secuencia de varios péptidos. Ambas bandas (37 y 24 kDa) se identificaron como la cutinasa 2 de Emericella nidulans (A. nidulans) con código AN7541 en el Gene Bank (tabla 10) . Una segunda identificación se mandó a realizar en la Bio Unidad de Servicios de Apoyo a la Investigación (BioUSAI) en la Facultad de Química de la UNAM para la enzima recombinante pura en solución bajo el método descrito anteriormente. Se confirmó de nuevo la presencia de la cutinasa 2 de Emericella nidulans (A. nidulans) . El péptido encontrado en ambas secuenciaciones se muestran en la figura 8.
EJEMPLO 7: Caracterización da las en«imaa puras (AKCUT1 y ANCUT2) .
Debido a la concentración de las enzimas, se realizó una dilución de las enzimas puras 1:100 con amortiguador de fosfatos pH 8.0 (pH óptimo de ensayo) que fue considerada para determinar actividad volumétrica y específica en todos
los ensayos de caracterización. En la tabla 4 se resumen las propiedades encontradas para cada enzima recombinante pura:
Tabla 4. Caracterización de las cutinasas recombinantes (Solis, 2014; Morales 2014) .

Fue necesario determinar primero el pH óptimo de ensayo de las enzimas puras para poder llevar a cabo las siguientes caracterizaciones bioquímicas al pH óptimo de ensayo. Para determinarlo, la actividad enzimática fue medida utilizando p-NPA como sustrato con la misma técnica reportada en el apartado de cuantificación de actividad enzimática en el rango de valores de pH de 5.0 a 11.0. Los amortiguadores empleados fueron: amortiguador de acetatos 50 mM para los diferentes valores de pH de 4.0 y 5.0, amortiguador de fosfatos 50 mM para los pH 6.0 y 7.0, amortiguador Tris-HCl 50 mM para los pH 8.0 y 9.0, y amortiguador CAPS 50 mM para los pH 10.0 y 11.0.
Efacto da la temperatura sobra la ANCUT2 raconbinanta.
Para evaluar el efecto de la temperatura sobre la enzima recombinante, la actividad enzimática fue medida por triplicado espectrofotométricamente en microplaca durante 10 min cada minuto a 30°C, 40°C, 50°C, 60°C y 70°C a pH 8.0 corriendo a la par siempre un blanco de autohidrólisis en cada condición. Se incubaron a cada temperatura 10 μl de
enzima en 170 μl de amortiguador y se añadieron 20 μl de sustrato p-NPA 1 mM. Se monitoreó la reacción cada 2 min durante 10 min. Datarminación da la aatabilidad térmica.
Se incubó la enzima pura sin sustrato a temperaturas de 30°C, 40°C, 50°C, 60°C y 70°C durante intervalos de tiempo de 15, 30 y 60 min. Posteriormente se dejó que la solución alcanzara la temperatura ambiente y se monitoreó su actividad frente a p-NPA a la temperatura óptima de trabajo.
Datarminación da la aatabilidad a pH.
Se determinó la estabilidad de la actividad enzimática frente al pH utilizando los mismos amortiguadores que en el punto anterior. La enzima suspendida en cada amortiguador se incubó durante 1 y 3 horas. Posteriormente se determinó su actividad frente a p-NPA en microplaca como en los ensayos anteriores al pH óptimo de trabajo. Bapacificldad fruta a «uatratoa.
Se determinó la actividad especifica de la enzima frente a p- NP ésteres de cadena corta, media y larga cubriendo un largo de cadena de 2 a 18 carbonos (C2-C18) . Los ensayos se llevaron a cabo a pH 8.0 y a 30°C para favorecer la
disolución de los sustratos sin que la autohidrólisis fuera excesiva. Los p-NP ésteres probados fueron p-NPAcetato (p- NPA), p-NPButirato (p-NPB) , p-NPMiristato <p-NPM) , p- NPLaurato (p-NPL) , p-NPPalmitato (p-NPP) y p-NPeStearato (p- NPS) .
Efecto de Iones frente a la actividad enzimatica.
Para determinar si algún ion actuaba como co-factor de la enzima, se determinó la actividad de la cutinasa recombinante en presencia de varios iones metálicos (Bermúdez, 2013) . Los iones utilizados (Ca2*, Mg2*' K*, Na* y Cu2*) se añadieron a la enzima resuspendida en amortiguador Tris-HCl 50 mM pH 8.0 a una concentración de 1 y 10 mM, y la solución se incubó durante 24 horas a 4°C. Transcurrido este tiempo, se midió la actividad con p-NPA como sustrato en ensayos de microplaca igual que en las otras determinaciones. La actividad residual se calculó utilizando como 100% la actividad de la enzima en amortiguador pH 9.0 sin iones añadidos con el quelante EDTA a una concentración 1 mM para eliminar el ruido que pudieran presentar los posibles iones en solución.
Efecto da poaiblaa de«naturali¾antes a inactivadoraa sobra la enzima recombinante.
Se evaluó el efecto de compuestos desnaturalizantes reportados para cutlnasas (Chen et al., 2010) sobre la actividad enzimática de la cutinasa recombinante de A. nidulans. Los compuestos utilizados (SDS, PMSF y Tween 80) se añadieron en concentraciones de 1 y 10 mM a la enzima resuspendida en amortiguador Tris-HCl pH 8.0 y se incubaron durante 24 horas a 4°C. Posteriormente se cuantificó la actividad con p-NPA y se reportó la actividad residual considerando como 100% la actividad obtenida sin la adición de ningún surfactante.
Efecto da solvnta» an la actividad•nrimátlca.
Se evaluó el efecto de diferentes solventes en la actividad enzimática de la cutinasa recombinante frente a p-NPA. Los solventes utilizados, acetona, etanol, metanol, hexano, isopropanol y DMSO se añadieron en concentraciones de 30% a la cutinasa diluida en amortiguador Tris-HCl pH 8.0 50 mM y se incubaron de tres diferentes formas: durante 24 horas a 4°C, 24 horas a 40°C y 3 horas a 40°C (40°C fue la temperatura óptima de ensayo de la enzima recombinante) antes de medir la actividad residual espectrofotométricamente. Se consideró como 100% de actividad a la actividad presentada por la enzima incubada en el mismo amortiguador sin la adición de solvente alguno.
Determinación do parámetros cinéticos.
Se determinaron las constantes cinéticas Km, kcat y km / kcat para la cutinasa recombinante pura. Primero se determinó la actividad especifica con p-NPA a diferentes concentraciones de sustrato utilizando el ensayo espectrofotométrico descrito anteriormente. La velocidad inicial fue calculada utilizando la región lineal de la curva de reacción (5 min) . Las constantes cinéticas aparentes fueron calculadas a partir de una gráfica de dobles inversos (1/v vs. l/[pNPA]) y son el resultado del promedio de tres mediciones independientes.
Determinación de sitio» de glicoailación.
Se realizó un ensayo para detectar posibles sitios de glicosilación en la cutinasa de 29 kDa utilizando el protocolo descrito en el kit DIG Glycan Differentiation (Roche) . El fundamento de este ensayo es el mismo que el de una réplica en Western blot, ya que utiliza diferentes lectinas acopladas a un anticuerpo. La lectina reconoce un único tipo de azúcar por lo que una reacción positiva indica a qué azúcar está unida la proteina de interés; es necesario utilizar controles positivos para cada una de las lectinas.
Ensayo de inmunodetección contra HiaTAS.
Se utilizaron los siguientes amortiguadores para llevar a cabo el ensayo inmunoreplicativo de "western blot":
❖ Amortiguador de transferencia (pH 7.5, 100 mi)
• 10 mi de solución 10X de glicina/ Tris-HCl
Para 400 mi de solución 10X: 72 g de glicina, 15.15 g Tris-HCl
• Metanol 10 mi (éste se agregó en el momento del uso del amortiguador de transferencia)
• 80 ra. de agua
❖ Amortiguador TBS 10X (pH, 7.5, 500 mi)
• 400 mi de solución NaCl/Tris-HCl (7.089 g Tris-HCl, 43.8 g NaCl)
• 2.5 m. Tween 20%
• Cbp agua
Para la solución IX:
• 15 mi de solución 10X se llevó a 150 mi
• Para Amortiguador TBS-Skin milk 3%: 0.9 g de skin milk en 30 mi de la solución IX.
Se corrió gel SDS-PAGE con la proteina pura y marcador de peso molecular Kaleidoscopic™ BioRad. Se enjuagó con agua desionizada y se incubó con amortiguador de transferencia 1 hora. Se llevó a cabo la transferencia semihúmeda empaquetando 2 papeles filtro especiales, la membrana de nitrocelulosa (humedecida con metanol), el gel y otros dos
papeles filtro en el equipo Transfer™ de BioRad a 15V durante 75 min. Una vez transferidas las proteínas a la membrana de transferencia se cortó el marcador de la membrana. Se incubó una hora en amortiguador TBS-skin milk 3%, y se realizaron dos lavados de 30 s cada uno con amortiguador TBS. Posteriormente se incubó con anticuerpo Anti-His (C-term) /AP Ab (novex™) durante 90 min, se volvieron a realizar dos lavados de 30 s cada uno con amortiguador TBS y finalmente se reveló con BCIP/NBT.
Gel de iaoelectroenfoque.
Para determinar el punto isoeléctrico de la proteína se corrió un gel prefabricado con gradiente de pH de 3.0 a 9.0 en el equipo Phast System Pharmacia de Mod LKB. El gel almacenado a 4°C fue atemperado durante 20 min antes de cargar las muestras por capilaridad y se corrió con el protocolo 2 del equipo (70 V 20 min, 15 V 1 h) .
Hidrólisis ám cutiría.
Obtención cUi cutícula.
Para la obtención de cutícula se lavaron y pelaron 14 kg de manzana de la variedad Golden Delicious. La cáscara se disolvió en 32 g de ácido oxálico (4 g/1) y 128 g de oxalato
de amonio (16 g/1) en 8 litros de agua destilada. Al comenzar a hervir se contaron 30 min, al finalizar el tiempo se lavó y filtró la cáscara hasta quedar libre de residuos y se puso a secar en estufa a 50°C durante una noche. Una vez seca se molió hasta obtener un polvo fino el cual se sometió a una destilación por Soxlet con Hexano durante 24 horas. Se dejó secar el polvo resultante en campana de extracción durante 24 horas y posteriormente se sometió a una hidrólisis enzimática con celulasa y amilasa durante 24 horas obteniendo asi cutina pura {Esqueda-Domínguez, 2012) .
-Bvaluación da la actividad hidrolltica sobre cutina.
Se resuspendieron 5 mg de cutina en 300 μl de tolueno hasta su disolución y se añadieron 700 μl de una dilución 2.5 de la enzima pura para un volumen final de reacción de 1 mi con 50U de enzima. Además, se hizo un blanco de reacción en el que no se añadió la enzima. Se incubaron las reacciones durante 72 horas a 37 °C y 100 rpm en viales sellados.
Posteriormente se revelaron los productos de hidrólisis por Cromatografía en Capa Fina (CCF) utilizando como fase móvil éter de petróleo tdietil éter: ácido acético (60:40:1 v/v) Los productos se visualizaron en una cámara de UV utilizando como agente revelador diclorofluoresceina .
EJEMPLO 8: DEGRADACIÓN DE POLIÉSTERES. Evaluación praliminar da dagradación.
Se realizó un ensayo preliminar para ver si las cepas recombinantes que contenían las cutinasas ANCÜT1 y ANCÜT2 podían degradar el PBS, PES, PCL y PLLA. El crecimiento e inducción con metanol en las cajas Petri que contenían a PBS, PES, PCL y PLLA emulsificadas (0.4%), presentaron halos de hidrólisis después de 168 horas (figura 19) .
Pérdida da paao y acida., titulabla.
Los extractos crudos concentrados (1:5) de ANCUT1 y ANCUT2 se utilizaron para la reacción de hidrólisis de las películas de PBS, PES, PCL y PLLA, encontrándose por la determinación de la pérdida de peso, que la enzima ANCUT1 degrada a las 24 horas casi el 24% de PBS y a las 72 horas de reacción degrada casi el 50% de este mismo polímero (figura 20) . Para el caso de PES llega a degradarse el 62.43% después de 24 horas de reacción y el 87.39% a las 72 horas (figura 21). Con PCL, la degradación llevada a las 24 horas presentó un 43.89% de degradación y un 71.99% a las 72 horas (figura 22). PLLA a las 24 horas logró degradarse un 5% y a las 72 horas un 14.94% (figura 23). Para este ensayo el que presentó mayor
porcentaje de degradación fue el PES ya que después de 72 horas de degradación perdió casi el 90% de su peso inicial. La cutinasa ANCUT2, por el contrario, únicamente pudo degradar 2.02% de PBS, el 3.55 % de PES, el 5.28% de PCL y el 13,47% de PLLA después de 72 horas de reacción, lo cual se observa en las figuras 20 a 23.
Alternativamente se evaluó la hidrólisis por titulación de ácidos grasos. En las figuras 24 a 27 se observan que los resultados son congruentes con los obtenidos por el método de pérdida de peso, es decir, la cutinasa ANCUT1 es la que hidroliza más eficientemente a los polímeros PBS, PES, PCL y PLLA. Observándose un aumento con respecto al tiempo de mililitros de NaOH.
La titulación de ácidos grasos se hace de manera indirecta, ya que inicialmente no se tiene conocimiento que cuantos mi de "grasa o aceite" tenemos pero, sin embargo, sabemos que en la reacción de hidrólisis de las cutinasas con actividad de esterasas y los poliésteres, se realiza una fragmentación de polímero, liberando cadenas de ácidos grasos, cuya presencia se trató de determinar por la titulación de ácidos grasos, observándose una relación en cuanto a que, entre más es la pérdida de peso de los poliésteres más aumenta el gasto de mililitros de NaOH, por lo que se da a entender que si se
lleva a cabo hidrólisis, por consecuencia si se lleva a cabo una degradación con las cutinasas.
Después de analizar las pérdidas de peso de los poliésteres, se observa que el PES es el polímero que muestra mayor pérdida de peso después de 72 horas de reacción, seguido de la PCL con la enzima ANCUT1, mientras los demás presentan menos del 50% de degradación.
Por lo que se sabe de la ANCUT1 y ANCUT2, estas enzimas tienen mayor especificidad a los esteres de cadena corta (C2 y C4) . Pero se presenta una mayor afinidad de los poliésteres con la enzima ANCUT1 que con la enzima ANCUT2, pues el máximo de degradación que se observó fue con el PLLA con un 13.47% después de 72 horas de reacción.
Probablemente la degradación que se lleva acabo con los poliésteres y las enzimas se deba a la cristalización que obtuvo cada uno, pues se ha observado que la forma en que se llevó la cristalización de los polímeros tendrá diferencia en sus características físicas.
Determinación de condiciones de pH y tugara tura.
Para optimizar las condiciones de degradación con las enzimas ANCÜT1 y ANCUT2, se procedió a trabajar con diferentes rangos de temperatura y pH. La elección de trabajar con rangos de
temperatura y pH seleccionados, fue a partir de los resultados obtenidos de la caracterización descrita anteriormente (tabla 4), en donde se observó que la enzima tiene su máxima actividad a las 24 horas (figuras 11 y 12) . A partir de estas condiciones, se buscó optimizar la reacción de hidrólisis y aumentar el grado de degradación de los poliésteres.
Una vez obtenidos los resultados se encontró que en el PBS se presentó una mayor degradación (65.6%) a pH 8.0 y 60 °C, para el PES las condiciones en donde se presentó una mayor degradación (76.6%) fueron a pH 9.0 y a 60°C, para el PCL se presentó una mayor degradación (63.6%) en las condiciones de pH 8.0 a 60°C con la enzima ANCUT1, lo cual se muestra en las figuras 28, 29 y 30. Para el caso de la enzima ANCUT2 no se presenta un cambio en la degradación considerable en cada uno de los poliésteres. En el caso del PLLA sólo se realizó las condiciones de hidrólisis mediante el análisis previo de los otros polímeros, eligiéndose la temperatura de 60°C para ANCUT1 y 50°C para la ANCUT2, en donde se presentó mayor degradación a pH 7.0 para la enzima ANCÜT1, pero no presenta una pérdida significativa a comparación de la reacción de hidrólisis sin cambios de pH y temperatura (figura 31) .
A partir del análisis de los resultados obtenidos con la variación de pH y temperatura, se observó un aumento
significativo de degradación con PBS y en menor grado con PES Y PCL, con PLLA no se logró aumentar la degradación. Todos estos resultados se observaron en mayor grado para la enzima ANCUT1. Después de 24 horas de reacción se logró aumentas casi 3 veces (41.67%) la degradación del PBS a condiciones de pH 8.0 y a 60°C. En el caso de la PES sólo se logró aumentar un 14 a 17% (pH 9.0 a 60°C) y con PCL el 19.71% (pH 8.0 a 60°C) . Ρ·terminación de condicionas da solventas y timpo da reacción.
Se busca encontrar la estabilidad enzimática en solventes ya que es importante a nivel industrial, pues el uso de solventes orgánicos es esencial, pero pueden modificar la estabilidad de su actividad. Del tal manera que ahora se busco variar las condiciones de degradación de los poliésteres variando el solvente y alargando el ensayo a 96 horas obteniéndose los siguientes resultados:
Para el caso del PBS se observa un aumento en la perdida de peso con respecto al tiempo, pero sólo se logró degradar un 13.79% después de 96 horas de reacción con tolueno en el caso de la enzima ANCUT1. Por el contrario, para la enzima ANCUT2 no se presentó degradación (figura 32) . El PES al contrario del PBS, presenta una mayor degradación del 73.22% a las 96
horas con hexano con la enzima ANCUT1 (figura 33) . Con la enzima ANCUT2 sólo se logró degradar el 26.74% con hexano después de 96 horas de degradación. En el caso de la PCL, con enzima ANCUT1 se obtuvo un 48.97% de pérdida de peso después de 96 horas de reacción con tolueno, mientras que con ANCUT2 sólo el 12.64% (figura 34). Las pérdidas de peso obtenidas variando el tiempo de reacción y el solvente, no demostró aumentos significativos de pérdida de peso de los poliésteres, ya que a las 24 horas de reacción sólo se logra degradar la mitad de lo que se degradó en la reacción de hidrólisis sin cambian condiciones para el caso de la enzima ANCUT1. Para la enzima ANCUT2, se observó un ligero aumento en la pérdida de peso. Aplicación de hidrólisis enzimatica en nuestras conerclalsa de PKT.
Como al principio se mencionó, se trabajaron con dos diferentes muestras de PET, a pesar de que la degradación de PET es mínima y a que se extendió el ensayo hasta 15 días, no se observó una pérdida significativa después de pasados 3 días de reacción. Se presentó una mejor degradación del PET con la enzima ANCUT2 (Tabla 5) .
Tabla 5. Capacidad de degradación de PET después de la reacción de hidrólisis de las enzimas ANCUT1 y ANCÜT2.
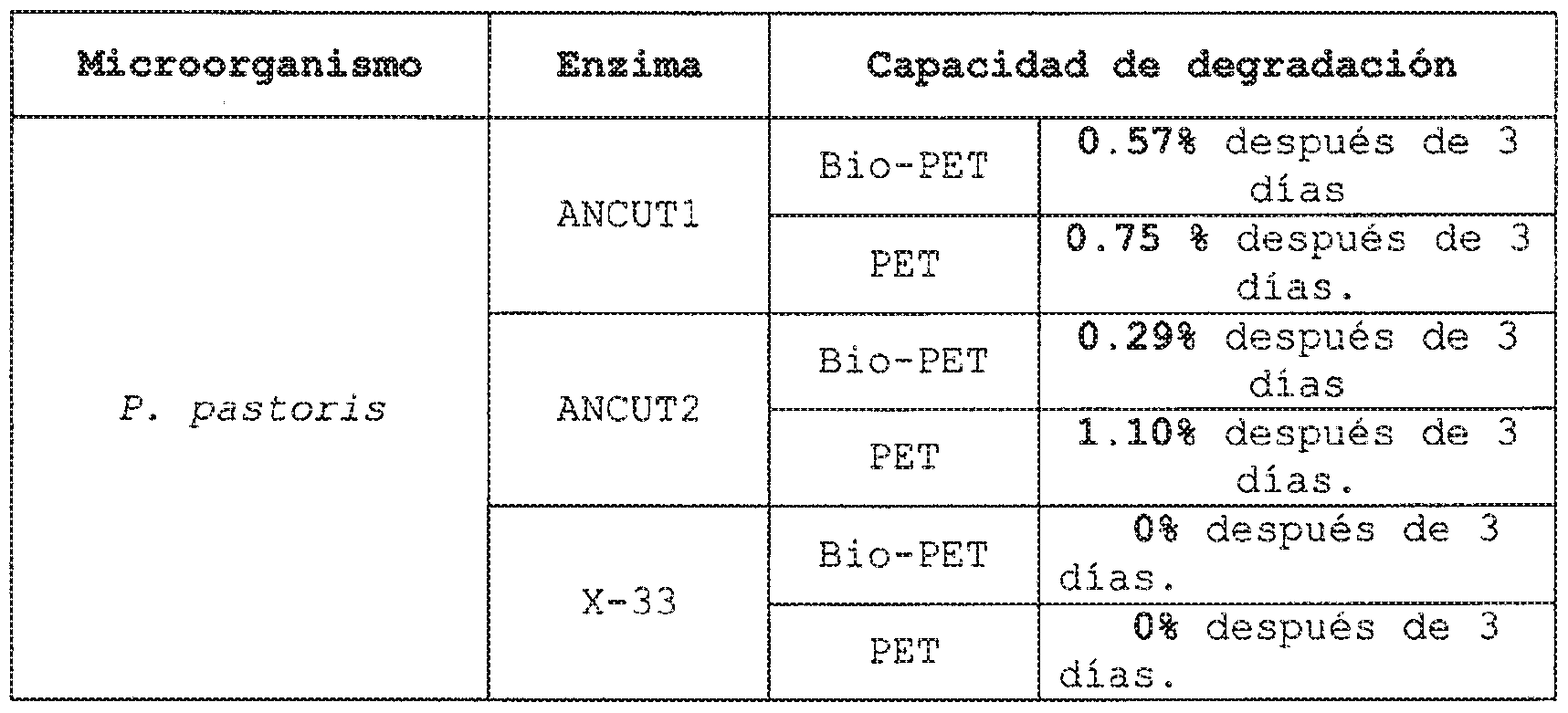
Condiciona» óptimas da degradación da poliéataraa para la» enzimas ANCÜT1 y AKCDT2.
A partir de los resultado obtenidos, en las tablas 6 y 7 se resumen las condiciones óptimas de degradación de poliésteres para las enzimas ANCUT1 y ANCUT2.
Tabla 6. Condiciones óptimas cié degradación en poliésteres con enzima ANCUT1 después de 24 h de reacción.

Tabla 7. Condiciones óptimas de degradación en poliésteres con enzima ANCUT2 después de 24 h de reacción.

Morfología de los poliésteres PBS, PCL, PES, PUA y PET después de 72h de degradación.
Las muestras de PBS, PCL, PES, PLLA y PET después de 72 horas de degradación por reacción de hidrólisis, fueron enviadas a la USAI y se obtuvieron imágenes por microscopía electrónica de barrido (SEM) con la finalidad de verificar su grado de degradación. Del análisis de dichas imágenes pudimos comprobar de manera visual y perceptible que, efectivamente, si se está llevando a cabo una degradación por hidrólisis con las enzimas ANCUT1 y ANCUT2. Ya que en las imágenes de las figura 35 y 36 se muestra una gran diferencia en la degradación de las películas. En particular, cabe señalar que las películas control que fueron sometidas a reacción con el extracto crudo de la cepa silvestre X-33 (la cual no contiene enzimas recombinantes) no presentan degradación aparente, mientras que resulta ser clara la degradación y el desgaste de las películas que estuvieron sometidas a la reacción con las enzimas ANCÜT1 y ANCUT2.
REFERANCIAS .
1. Ahmed Al-Tammar, K. , Ornar, 0., Murad, A., Muñir, A., Bakar, A., & Diba, F. (2016). Expression and characterization of a cutinase (AnCUT2) from Aspergillus niger. Open Life Sciences, 11(1), 29-38.
2. Bermúdez, E. P. (2013). Bases moleculares y bioquímicas para el estudio del sistema cutinolitica de A.
nidulans. Tesis de Maestría. Universidad Nacional Autónoma de México.
Bertani, G. (1951). STÜDIES ON LYSOGENESIS I.: The Mode of Phage Liberation by Lysogenic Escherichla coli 1. Journal of bacteriology, 62(3), 293.
Bornscheuer, U.T, et al. (2005). Selectivity of lipase and esterases towards phenol esters. J Mol Catal B: Enzimatic, 36'. 8-13.
Bradford, M.M. (1976) . Ά rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principie of protein-dye binding. Anal Biochem, 72: 248-254.
Carvalho, C.M.L., Aires-Barros, M.R., Cabral, J.M.S. (1998) . Cutinase structure, function and biocatalytic applications . EJB Electronic Journal of
Biotechnology, 1(3).
Carvalho, C.M.L., Aires-Barros, M.R., Cabral, J.M.S. 1999. Cutinase: from molecular level to bioprocess development. Biotechnol Bioeng. 66(1): 17-34.
Castro-Ochoa D, Peña-Montes C, González-Canto A, Alva- Gasca A, Esquivel-Bautista R, Navarro-Ocaña A & Farrés A (2012), ANCUT2, an extracellular cutinase from Aspergillus nidulans induced by olive oil, Appl Biochem Biotechnol 166(5) :1275-1290.
9. Castro-Ochoa, D. , Peña-Montes, C. & Farrés, A. (2013). Evaluation of Strategies to Improve the Production of Alkaline Protease PrtA from Aspergillus nidulans. Applied biochemistry and biotechnology, 169(5), 1672- 1682.
10. Castro-Ochoa, L. D. Estudio de la producción de actividad carboxiesterasa en Aspergillus nidulans. (2014) . Tesis de doctorado. Universidad Nacional Autónoma de México.
11. Cerenghino, J. L. & Cregg, J. M. 2000. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS microbiology reviews, 24(1), 45-66.
12. Chen, S., Su, L., Billig, S., Zimmermann, W., Chen, J.
& Wu, J. (2010) . Biochemical characterization of the cutinases from Thermobifida fusca. Journal of Molecular
Catalysis B: Enzymatic, 63(3), 121-127.
13. Chevallet, M., Luche, S. & Rabilloud, T. (2006). Silver staining of proteins in polyacrylamide gels. Nature protocols, 2(4), 1852-1858.
14. Cregg, J. M., Cerenghino, J. L., Shi, J., & Higgins, D.
R. (2000) . Recombinant protein expression in Pichia pastoris. Molecular biotechnology, 16(1), 23-52.
15. Dimarogona, M., Nikolaivits, E., Kanelli, M., Christakopoulos, P., Sandgren, M., & Topakas, E.
(2015) . Structural and functional studies of a Fusarium oxysporum cutinase with polyethylene terephthalate modification potential. Biochimica et Biophysica Acta (BBA) -General Subjects, 1850{11), 2308-2317.
16. Ejima, K. , Liu, J., Oshima, Y., Hirooka, K. , Shimanuki, S., Yokota, Y. and Nishino, T. (2004). Molecular cloning and characterization of a thermostable carboxylesterase from an archaeon, Sulfolobus shibatae DSM5389: non-linear kinetic behavior of a hormone- sensitive lipase family enzyrae. Journal of bioscience and bioengineering, 98{6), 445-451.
17. Esqueda-Dominguez K. (2012). Producción, identificación y caracterización de una cutinasa de Aspergillus nidulans PW1. Tesis de maestría en ciencias bioquímicas. Facultad de Química, UNAM.
18. Galagan, J.E., Calvo, et al. (2005). Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 438 (7071): 1105-1115.
19. Herrero-Acero, E., Ribitsch, D., et al., (2011).
Enzymatic surface hydrolysis of PET: Effect of structural diversity on kinetic properties of cutinases from Thermobifida. Macromol, 44(12): 4632-4640.
20. Hiraishi, N., Sadek F.T., et al; (2008). Susceptibility of a polycaprolactone-base root canal filling material
to degradation using an agar-well difussion assay. Am J Dent. April, 21(2): 119-123.
21. Hiraishi, N. , Yau, J.Y.Y, Loushine, R.J., Armstron, S.R., Weller, R.N., King, N.M., Pashley, D.H., Tay, F.R. (2007) . Susceptibility of a polycaprolactone-base root canal filling material to degradation. III. Turbidimetric evaluation of enzymatic hydrolysis. JOE. Aug; 33(8) : 952-956.
22. INEGI. Censo Nacional de Gobiernos Municipales y Delegacionales, (2013) . Módulo 6. Residuos Sólidos
Urbanos .
23. Invitrogen, E. S. T. (2009) . Pichia expression kit. A manual of methods for expression of recombinant proteins using pPICZ and PICZa in Pichia pastoris. Catalog, (K1740-01) .
24. Kafer, E. (1977). Meiotic and Mitotic Recombination in Aspergíllus and its Chromosomal Aberrations. Adv. Genet. 19: 33-131.
25. Karpushova A; Brümmer F; Barth S.; Lange, S; Schmid R.
D. (2005) Cloning, recombinant expression and biochemical characterization of novel esterases from Bacillus sp. associated with the marine sponge Aplysina aerophoba. Appl Microbiol Biotechnol 67: 59-69.
26. Kawasaki, L., Farrés, A., & Aguirre, J. (1995). Aspergillus nidulans mutants affected in acétate metabolism isolated as lipid nonutilizers. Experimental mycology, 19(1), 81-85.
27. Koschorreck, K. , Liu, D.r Kazenwadel, C, Schmid, R. D.
& Hauer, B. (2010) . Heterologous expression, characterization and site-directed mutagenesis of cutinase CUTAB1 from Alternaría brassicicola. Applied microbiology and biotechnology, 57(3), 991-997.
28. Kwon, M. A. , Kim, H. S., Yang, T. H. , Song, B. K. & Song, J. K. (2009) . High-level expression and characterization of Fusarium solani cutinase in Pichia pastoris. Protein expression and purification, 68(1), 104-109.
29. Laemmli, UK, (1970) . Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Wature 227(5259) : 680-685
30. Mastropaolo, W., & Yourno, J. (1981). An ultraviolet spectrophotometric assay for α-naphthyl acétate and a- naphthyl butyrate esterases. Analytical biochemistry,
225(1), 188-193.
31. Morales, S. (2015). Purificación y caracterización de la enzima recombinante ANCUT2 de Aspergillus nidulans.
Tesis de Licenciatura. Universidad Nacional Autónoma de México.
32. Nawani, N. , Dosanjh, N; Kaur, J. (1998). A novel thermostable lipase from a thermophilic Bacillus sp. : characterization and esterification studies. Biotechnol
Lett. 20 (10): 997-1000.
33. Nyyssóla, A., Pihlajaniemi, V., HSkkinen, M., Kontkanen, H. , Saloheimo, M., & Nakari-Setálá, T. (2014) . Cloning and characterization of a novel acidic cutinase from Sirococcus conigenus. Applied microbiology and biotechnology, 98(8), 3639-3650.
34. Peña-Montes, C. (2001). Producción, purificación y caracterización bioquimica de una enzima con actividad politica en Aspergillus nidulans. Tesis de Maestría. Universidad Nacional Autónoma de México.
35. Peña-Montes, C, González, A., Castro-Ochoa, D., Farrés, A. (2008) . Purification and biochemical characterization of a broad substrate specificity thermostable alkaline protease from Aspergillus nidulans. Appl Microbiol Biotechnol. 78 (4): 603-612.
36. Peña-Montes, C, Langa, S., Flores, I., Castro-Ochoa, D., Schmid, R., Cruz-Garcia, F., & Farrés, A. (2009). Molecular characterization of Stcl esterase from
Aspergillus nidulans. Applied microbiology and biotechnology, 84(5), 917-926.
37. Purdy, R. E. , & Kolattukudy, P. E. (1973).
Depolymerization of a hydroxy fatty acid biopolymer, cutin, by an extracellular enzyme from Fusarium solani f. pisi: isolation and some properties of the enzyme. Archives of Biochemistry and
Biophysics, 159(1), 61-69.
38. Ronkvist, A.M., Xie, W., Lu, W., Gross, R.A. (2009).
Cutinase-catalyzed hydrolysis of poly (ethylene terephthalate) . Macromol, 42 (14): 5128-5138.
39. Sambrook, J; E. F. Fritsch, y T. Maniatis. (1989). In Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, NY, Vol. 1, 2, 3.
40. Solis, I. (2015). Producción, purificación y caracterización de la cutinasa recombinante ANCUT1 producida en Pichia pastoris. Tesis de Licenciatura. Universidad Nacional Autónoma de México.
41. Sulaiman, S., et al. (2012). Isolation of a novel cutinase homolog with polyethylene terephthalate- degrading activity from leaf-branch compost by using a metagenomics approach. Appl. Environ. Microbiol, 78(5) : 1556-62.
42. Vega, F. (2013). Purificación y caracterización de la cutinasa ANCUT1 de Aspergillus nidulans. Tesis de Licenciatura. Universidad Nacional Autónoma de México.
43. Vertommen, M; et al. (2005) . Enzymatic surface modification of poly (ethylene terephthalate) . J
Biotech, Jun; 120: 376-386.
Claims
1. Un polinucleótido recombinante aislado que codifica para un polipéptido funcional que es capaz de degradar poliésteres, caracterizado porque consiste de la SEQ ID NO: 5.
2. Un polinucleótido recombinante aislado que codifica para un polipéptido funcional que es capaz de degradar poliésteres, caracterizado porque consiste de la SEQ ID NO: 6.
3. Un polinucleótido recombinante aislado que codifica para un polipéptido funcional que es capaz de degradar poliésteres de conformidad con las reivindicaciones 1 y 2, en donde dicho polinucleótido comprende una secuencia de nucleótidos que tiene una homología de al menos 70% con las secuencias SEQ ID NO: 5 o SEQ ID NO: 6.
4. Un polinucleótido caracterizado porque comprende el polinucleótido recombinante que codifica para un polipéptido funcional que es capaz de degradar poliésteres reclamado en las reivindicaciones 1, 2 o 3.
5. Un vector de expresión caracterizado porque comprende el polinucleótido recombinante que codifica para un polipéptido funcional que es capaz de degradar poliésteres reclamado en las reivindicaciones 1, 2 , 3 o 4.
6. Un cebador directo sintético para amplificar el gen ancutl que codifica para una cutinasa recombinante de Aspergillus nidulans, caracterizado porque consiste de la SEQ ID NO: 1.
7. Un cebador reverso sintético para amplificar el gen ancutl que codifica para una cutinasa recombinante de Aspergillus nidulans, caracterizado porque consiste de la SEQ ID NO: 2.
8. Un cebador directo sintético para amplificar el gen ancut2 que codifica para una cutinasa recombinante de Aspergillus nidulans, caracterizado porque consiste de la SEQ ID NO: 3.
9. Un cebador reverso sintético para amplificar el gen ancut2 que codifica para una cutinasa recombinante de Aspergillus nidulans, caracterizado porque consiste de la SEQ ID NO:
4.
10. Un microorganismo recombinante capaz de degradar poliésteres/ caracterizado porque comprende el vector de expresión reclamado en la reivindicación 5.
11. Un microorganismo recombinante de conformidad con la reivindicación 10, caracterizado porque es un microorganismo eucariótico o procariótico.
12. Un microorganismo recombinante de conformidad con la reivindicación 10 y 11, caracterizado porque es una levadura o una bacteria.
13. Un microorganismo recombinante de conformidad con las reivindicaciones 10 a 12, caracterizado porque la levadura es una cepa recombinante de Pichia pastoris.
14. Un microorganismo recombinante de conformidad con las reivindicaciones 10 a 12, caracterizado porque la bacteria es una cepa recombinante de Escherichia coli.
15. Un polipéptido recombinante funcional que es capaz de degradar poliésteres, caracterizado porque consiste de la SEQ ID NO: 7.
16. Un polipéptido recombinante funcional que es capaz de degradar poliésteres, caracterizado porque consiste de la SEQ ID NO: 8.
17. Un polipéptido recombinante funcional que es capaz de degradar poliésteres de conformidad con las reivindicaciones 15 y 16, en donde dicho polipéptido comprende una secuencia de aminoácidos derivada de la SEQ ID NO: 7 o SEQ ID NO: 8 por deleción de 89 aminoácidos en el extremo N-terminal.
18. Un polipéptido recombinante funcional que es capaz de degradar poliésteres de conformidad con las reivindicaciones 15 y 16, en donde dicho polipéptido
comprende una secuencia de aminoácidos que tiene una homología de al menos 70% con las secuencias SEQ ID NO: 7 o SEQ ID NO: 8.
19. Un polinucleótido caracterizado porque que codifica para un polipéptido recombinante funcional que es capaz de degradar poliésteres reclamado en las reivindicaciones 17 y 18.
20. Un método para producir un polipéptido recombinante funcional que es capaz de degradar poliésteres, caracterizado porque comprende los pasos de:
(a) cultivar un microorganismo recombinante de conformidad con las reivindicaciones 10 a 14 en las condiciones óptimas para el crecimiento del microorganismo y para la producción del polipéptido,
(b) separar el polipéptido recombinante del extracto crudo del microorganismo recombinante, y
(c) purificar dicho polipéptido recombinante.
21. Un método para producir una enzima recombinante funcional que es capaz de degradar poliésteres, caracterizado porque comprende los pasos de:
(a) cultivar un microorganismo recombinante de conformidad con las reivindicaciones 10 a 14 en las condiciones óptimas para el crecimiento del microorganismo y para la producción de la enzima,
(b) separar la enzima del extracto crudo del microorganismo recombinante, y
(c) purificar dicha enzima recombinante.
22. Un método para degradar plásticos de poliésteres, caracterizado porque comprende el paso de:
(a) poner en contacto el plástico de poliéster con al menos un polipéptido recombinante funcional que es capaz de degradar poliésteres de conformidad con las reivindicaciones 15 a 18.
23. Un método para degradar plásticos de poliésteres de conformidad con la reivindicación 22, en donde el plástico de poliéster se pone en contacto con una mezcla polipéptidos recombinantes funcionales que son capaces de degradar poliésteres de conformidad con las reivindicaciones 15 a 18.
24. Un método para degradar plásticos de poliésteres, caracterizado porque comprende el paso de:
(a) poner en contacto el plástico de poliéster con al menos un microorganismo recombinante capaz de degradar poliésteres de conformidad con las reivindicaciones 10 a
14.
25. Un método para degradar plásticos de poliésteres de conformidad con la reivindicación 24, en donde el plástico de poliéster se pone en contacto con una mezcla de
microorganismos recombinantes capaces de degradar poliésteres de conformidad con las reivindicaciones 10 a 14.
26. El uso de al menos un polipéptido recombinante funcional que es capaz de degradar poliésteres de conformidad con las reivindicaciones 15 a 18, para la manufactura de una solución para degradar plásticos de poliésteres.
27. El uso de conformidad con la reivindicación 26, en donde la solución para degradar plásticos de poliésteres es manufacturada con una mezcla polipéptidos recombinantes funcionales que son capaces de degradar poliésteres de conformidad con las reivindicaciones 15 a 18.
28. El uso de al menos un microorganismo recombinante capaz de degradar poliésteres de conformidad con las reivindicaciones 10 a 14, para la manufactura de una solución para degradar plásticos de poliésteres.
29. El uso de conformidad con la reivindicación 28, en donde la solución para degradar plásticos de poliésteres es manufacturada con una mezcla de microorganismos recombinantes capaces de degradar poliésteres de conformidad con las reivindicaciones 10 a 14.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2016006869A MX380946B (es) | 2016-05-26 | 2016-05-26 | Cutinasas recombinantes de aspergillus nidulans para biodegradacion de poliesteres. |
| MXMX/A/2016/006869 | 2016-05-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2017204615A2 true WO2017204615A2 (es) | 2017-11-30 |
| WO2017204615A3 WO2017204615A3 (es) | 2018-02-22 |
Family
ID=60411492
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/MX2017/000056 Ceased WO2017204615A2 (es) | 2016-05-26 | 2017-05-26 | Cutinasas recombinantes de aspergillus nidulans para biodegradación de poliésteres |
Country Status (2)
| Country | Link |
|---|---|
| MX (1) | MX380946B (es) |
| WO (1) | WO2017204615A2 (es) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3636768A1 (en) * | 2018-10-08 | 2020-04-15 | B4Plastics BVBA | Method and means for determining the biodegradability of polymeric materials |
| KR20250043611A (ko) | 2023-09-21 | 2025-03-31 | 경북대학교 산학협력단 | 아스퍼질러스 푸미가티아피니스 유래 신규 pet 가수분해효소 및 이의 변이체 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4273504B2 (ja) * | 2002-10-23 | 2009-06-03 | 株式会社 東北テクノアーチ | プラスチックの分解法及びそれを利用した有用物質の製造方法 |
-
2016
- 2016-05-26 MX MX2016006869A patent/MX380946B/es unknown
-
2017
- 2017-05-26 WO PCT/MX2017/000056 patent/WO2017204615A2/es not_active Ceased
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3636768A1 (en) * | 2018-10-08 | 2020-04-15 | B4Plastics BVBA | Method and means for determining the biodegradability of polymeric materials |
| KR20250043611A (ko) | 2023-09-21 | 2025-03-31 | 경북대학교 산학협력단 | 아스퍼질러스 푸미가티아피니스 유래 신규 pet 가수분해효소 및 이의 변이체 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017204615A3 (es) | 2018-02-22 |
| MX380946B (es) | 2025-03-12 |
| MX2016006869A (es) | 2017-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Takahashi et al. | Extracellular production of active Rhizopus oryzae lipase by Saccharomyces cerevisiae | |
| JP6537076B2 (ja) | 分泌シグナルペプチドならびにそれを利用したタンパク質の分泌および細胞表層提示 | |
| US7247463B2 (en) | Enzymes with lipase/acyltransferase activity, nucleic acids encoding the same and methods of use thereof | |
| WO2011023009A1 (en) | Recombinant expression of carboxylesterases | |
| ES2345973T3 (es) | Lipasa/aciltransferasa a partir de candida parapsilosis. | |
| Jiang et al. | Cell surface display of functionally active lipases from Yarrowia lipolytica in Pichia pastoris | |
| Seok et al. | Construction of an expression system for the secretory production of recombinant α-agarase in yeast | |
| US20240084244A1 (en) | Recombinant yeast host cell having enhanced growth rate | |
| KR101887732B1 (ko) | 리파아제 활성이 증대된 양친매성 펩타이드-리파아제 결합체 및 이의 용도 | |
| WO2017204615A2 (es) | Cutinasas recombinantes de aspergillus nidulans para biodegradación de poliésteres | |
| Kim et al. | Purification, characterization, and cloning of a cold-adapted protease from Antarctic Janthinobacterium lividum | |
| Iguch et al. | Cloning of a protopectinase gene of Trichosporon penicillatum and its expression in Saccharomyces cerevisiae | |
| WO2004055177A2 (en) | Artificial esterases | |
| Wirajana et al. | Secretion of Geobacillus thermoleovorans IT-08 α-L-arabinofuranosidase (AbfA) in Saccharomyces cerevisiae by Fusion with HM-1 Signal Peptide | |
| KR101920094B1 (ko) | 효모에서 엔테로키나아제를 생산하는 조성물과 방법 | |
| CN111088240A (zh) | 一种酯酶及其编码基因和应用 | |
| Ghasemian et al. | Production of recombinant microbial thermostable lipases | |
| RU2728240C1 (ru) | Способ получения секретируемой полностью функциональной фосфолипазы А2 в дрожжах Saccharomyces cerevisiae, белок-предшественник для осуществления этого способа (варианты) | |
| Yang et al. | Purification and characterization of a β-1, 3-glucomannanase expressed in Pichia pastoris | |
| KR101713885B1 (ko) | sn-1 위치 특이적 리파아제 및 그 생산방법 | |
| Ahmadpour et al. | Cloning and expression of an indigenous mesophile lipase and evaluation of Bacillus codon translation in Pichia pastoris under control of two different promoters | |
| ES2347398B1 (es) | Esterasa termofila de thermus thermophilus. | |
| JP7194112B2 (ja) | 細胞結合型異種タンパク質を発現する組換え酵母宿主細胞 | |
| ES2354326B1 (es) | Lipasa termófila. | |
| KR100354344B1 (ko) | 한세눌라 폴리모르파 유전자 및 이들 유전자가 결핍된 균주 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17803132 Country of ref document: EP Kind code of ref document: A2 |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17803132 Country of ref document: EP Kind code of ref document: A2 |