WO2017222402A1 - Polybenzimidazole polymer with e'unctionalized spacer chain and its method of preparation for removal of genotoxic impurities - Google Patents
Polybenzimidazole polymer with e'unctionalized spacer chain and its method of preparation for removal of genotoxic impurities Download PDFInfo
- Publication number
- WO2017222402A1 WO2017222402A1 PCT/PT2017/000008 PT2017000008W WO2017222402A1 WO 2017222402 A1 WO2017222402 A1 WO 2017222402A1 PT 2017000008 W PT2017000008 W PT 2017000008W WO 2017222402 A1 WO2017222402 A1 WO 2017222402A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymer
- adenine
- solution
- gti
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/264—Synthetic macromolecular compounds derived from different types of monomers, e.g. linear or branched copolymers, block copolymers, graft copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/18—Polybenzimidazoles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3202—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the carrier, support or substrate used for impregnation or coating
- B01J20/3206—Organic carriers, supports or substrates
- B01J20/3208—Polymeric carriers, supports or substrates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3214—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the method for obtaining this coating or impregnating
- B01J20/3217—Resulting in a chemical bond between the coating or impregnating layer and the carrier, support or substrate, e.g. a covalent bond
- B01J20/3219—Resulting in a chemical bond between the coating or impregnating layer and the carrier, support or substrate, e.g. a covalent bond involving a particular spacer or linking group, e.g. for attaching an active group
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3231—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the coating or impregnating layer
- B01J20/3242—Layers with a functional group, e.g. an affinity material, a ligand, a reactant or a complexing group
- B01J20/3244—Non-macromolecular compounds
- B01J20/3246—Non-macromolecular compounds having a well defined chemical structure
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3231—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the coating or impregnating layer
- B01J20/3242—Layers with a functional group, e.g. an affinity material, a ligand, a reactant or a complexing group
- B01J20/3268—Macromolecular compounds
- B01J20/3272—Polymers obtained by reactions otherwise than involving only carbon to carbon unsaturated bonds
- B01J20/3274—Proteins, nucleic acids, polysaccharides, antibodies or antigens
Definitions
- the present invention relates to a polybenziinidazoie (FBI) polymer with a functionalized spacer chain and its structural functional ization method with deoxyribonucleic acid (DNA) bases, namely adenine, thymine, cytosine or guanine, or carboxyiic acid groups (-COOK; ,
- FBI polybenziinidazoie
- GTI genotoxic impurities
- GTIs typically are elect rophi lie species that either- formed during the manufacturing process of the API or generated in vivo may chemically attack the nucleophilic centers present in the DNA molecule and may induce single- or double-strand breaks and damages whose repair mechanisms may not be able to reverse. Ail these anomalies compromise the DNA replication process, which may lead to genetic mutations, implying an increased risk to the health of the patients to whom the API is administered [A. Teasdale et al. Org. Process Res. Dev. 17 (2013) 221-230].
- the aromatic amine- type compounds constitute another group of GTIs, which although not Inherently genotoxic, during their metabolization in vivo, give rise to electrophilic species. Generally, when they undergo oxidation, N-hydroxy1 compounds are formed which are conjugated as acetates, sulfates or glucuronides . Further deconj ugation of these compounds results in nitrenium ions (ArN*H) , which are considered active genotox.i.ns that bind to DMA [D. J. Snodin, Org. Process Res. Dev. 14 (2010) 960- 976] .
- the polymeric material used in this invention is a linear polybenzimidazole (PBI) polymer belonging to a class of heterocyclic polymers, the structure of which is shown in Figure 1.
- the FBI is a polymer compatible with most organic solvents, with accessible functional! zable groups.
- Modification of PBI with a ONA base is intended to mimic the interaction occurring in vivo between a GTI, for example of the sulfonate type (electrophilic species), and DNA (nucleophiiic centers) .
- the final material obtained due to its robustness and versatility, allows it to be exploited as a highly specific adsorbent material for the GTI class, in particular the suiphonate fami ly .
- the polymer modified with -COOH groups allows the selective elimination of GTI from the aromatic amine family.
- the removal of electrophilic species is ensured, which would result from the in vivo raetabolization of these aromatic compounds , if they were present in the API when administered to the patient.
- the functionalization chemistry of the PBI polymer has been exploited to obtain highly cross-linked PBI membranes by reaction of the polymer -with dihaiogenated compounds at reflux (80°C) in acetonitriie (MeCN) to give them greater physical robustness in the formulation of membranes for nanof.titration resistant to organic solvents, for example [I. B . Vaitcheva et al . , J. Membrane Sci. 457 (2014) 62-72; I. B. Vaitcheva et al. , J . Membrane Sci. 493 (2015) 568- 579] .
- US 2012035333&1 document discloses a method for obtaining PBI with carboxyl groups by reaction of the polyraer with cyclic acid anhydrides. In this way a polymer with amide groups directly attached to the PBI rings and to the free carboxylic acid groups is obtained.
- a halogenated carboxylic acid with a spacer chain there is no amide bond formation and the carboxylic acid function is separated from the structural backbone of the PBI by two carbon atoms or more. This aspect is important to avoid stereochemical impediments that could hinder the binding to the GTIs that are intended to be removed.
- the relevance of the present invention is that it is possible to modify the structure of the relatively inert PBI polymer to contain a DNA base or a carboxylic acid type functionality by means of a spacer chain between the nitrogen from the imidazole rings of PBI and the new chemical functionality inserted.
- this spacer chain presents a great advantage, since it facilitates the interaction between the functional groups present in the adsorbent material and the GTI, in which the latter can present several spatial geometries, more or less bulky. In this way, the adsorbent presents a great versatility against several geometries of the different GTI molecules.
- the present invention relates to a poiybenzimidazole polymer with a functionalized spacer chain and its structural functionalization method with DNA bases or carboxylic acids, chemically compatible with organic solvents, to be applied in the removal of genotoxic impurities.
- the invention focuses on obtaining a material, which establishes specific interactions with genotoxic impurities of various chemical families, to be exploited as a selective adsorbent thereof.
- the present invention relates to a poiybenzimidazole polymer with a functionalized spacer chain and its structural functional ization method with deoxyribonucleic acid.
- (DNA) bases namely adenine, thymine, cytosine or guanine, or carboxylic acid groups.
- Poiybenzimidazole polymer with functionalized spacer chain general formula of poiybenzimidazole polymer functionalized with a spacer chain R and functional group 2 , R represents a spacer chain composed of aromatic or aliphatic hydrocarbons containing from 2 to 11 carbon atoms and 2 represents a DNA base, or a carboxylic acid group.
- the present invention relates to a polybenz imidazole polymer with a functionalized spacer chain and its structural functionaiization method with DMA bases or carboxylic acids, chemically compatible with organic solvents, to be applied in the removal of genotoxic impurities.
- the invention focuses on obtaining a material, which establishes specific interactions with genotoxic impurities of various chemical families, to be exploited as a selective adsorbent, thereof.
- the present invention relates to a polybenzimidazole polymer with a functionalized. spacer chain and a structural functionaiization method with deoxyribonucleic acid (DNA) bases, namely adenine, thymine, cytosine or guanine, or carboxylic acid groups.
- DNA deoxyribonucleic acid
- the spacer chain is composed of aromatic or aliphatic hydrocarbons containing from 2 to 11 carbon atoms.
- the most nucleophilic positions of the DNA bases are known to be the endocyclic nitrogens of the N3 and N7 positions of guanine and adenine. These sites are preferably reactive to the presence of eiectrophilic species, such as su1 fonate type GT I .
- the functional ization of the P3I polymer of the present invention results in obtaining a specific material for binding to certain genotoxic impurities, allowing the elimination thereof and consequently the production of active pharmaceutical, ingredients with high purity.
- genotoxic impurities refer to families of compounds of the sulfonate or aromatic amine type .
- the method of obtaining the polybenz imidazole polymer with the functional! zed spacer chain is a versatile method, in that it can be included in an industrial process, in particular the pharmaceutical industry, for elimination or reduction of levels of genotoxic impurities. It may also be included in any synthesis based industry, which exploits compounds structurally similar to GTIs, for their disposal of the respective final products.
- step b) Addition to the solution obtained in step b) of a base at atmospheric pressure;
- step d) Addition of the haiogenated compound obtained in step a) to the solution, obtained in step cj at atmospheric pressure ;
- X will have to be a good leaving group, selected from: F, CI, Br, 1; and R may be a chain with aromatic groups of the type ⁇ R-Ph-R 5 or a linear chain containing from 2 to 11 carbon atoms.
- the DNA molecule with a spacer chain which in the present invention by way of example, relates to the 9- (3- bromopropy1 ⁇ adenine compound, is described in the literature being obtained in yields between 8% and 38% (N . J . Leonard et al., J. Org, Chem. 34 (1969) 3240-3243; WO 2013151663] .
- yields between 8% and 38% (N . J . Leonard et al., J. Org, Chem. 34 (1969) 3240-3243; WO 2013151663] .
- For these low yield values contribute the formation of by-products, such as the adenine-R-adenine dimer and a tricyclic derivative.
- the present invention further describes the synthesis of this compound, the optimization of which allowed it to be obtained in a yield higher than 60%.
- the development of this synthesis has been achieved iteratively by introducing a set of innovations described below and allowing improved performance of a synthesis which is not. trivial.
- the step of obtaining an aromatic or aliphatic hydrocarbon compound containing at its ends a halogen atom and a DNA base, or a carboxylic acid group comprises the following steps:
- step b) Reaction of the solution obtained in step b) at room temperature, up to a period of 16 hours, obtaining the final solution;
- step f) Purification by column chromatography of the solid obtained in step f) with a DCM/MeOH mixture.
- the modified adenine base with a bromine atom at its end was incorporated into the PBI polymer. Briefly, the polymer is dissolved in dimethylsulfoxide (DMSO) at 100 °C to 180°C, and thereafter a basic compound is added, in this case the base (KsCCb) , to deprotonate the nitrogen atoms of the imidazole rings of the polymer, and finally the DNA base is added (e.g. modified adenine, In this nuclecphilic
- substitution compounds of the type HX e.g. HBr
- substitution compounds of the type HX e.g. HBr
- the bond between the DNA base (i.e. adenine) and the PBI takes place by the spacer chain present in the first, as shown schematically in Figure 4,
- the PBI initial solution concentration was increased to 15% and left stirring for 3 hours at 170 °C to achieve complete solubilization of the polymer. After this time, the mixture was allowed to cool to room temperature and 2. eq. of K 2 CG 3 and 1 eq. of S- ( 3 -bromopropyl ) adenine were added. At this stage, the mixture presented itself as a viscous gel that hindered the efficient homogeni zation of the reactionai mixture. For this reason, it was necessary to increase the temperature to 50°C and left stirring for 24 hours at this temperature. At the end of the reaction water (40 mL) was added giving a solid in the form of small particles which was washed multiple times with water and then dried under vacuum .
- This polymer ⁇ 50 ing was placed in contact with 1 ruL of GTI solution at a concentration of 100 ppro in DCM at room temperature for 24 hours and the percentage of GTI bound to the polymer was quantified at the end. of this time. With this polymer, a preliminary GTI binding value of 14% was obtained. Based on this result, we tried to optimize the reaction using 3 eq. of sodium hydroxide (NaOH) to replace K 2 CO 3 . In this case a final product of high hardness was obtained, impossible to be processed for use in the GTI removal trials. In a further approach, to the 15% PBI solution was added
- the PBI polymer can be solubilized in a suitable solvent including DMSO, dymethylacetami.de ( DMAc ) , dimethyl formamide (DMF) or N ⁇ methyI-2 ⁇ pyrrolidone (NMP) .
- a suitable solvent including DMSO, dymethylacetami.de ( DMAc ) , dimethyl formamide (DMF) or N ⁇ methyI-2 ⁇ pyrrolidone (NMP) .
- DMAc dymethylacetami.de
- DMF dimethyl formamide
- NMP N ⁇ methyI-2 ⁇ pyrrolidone
- K2CO3 can be used for this purpose.
- the DNA base may contain a spacer chain composed of an aromatic and/or aliphatic hydrocarbon containing from 2 to 11 carbon atoms, with a terminal halogenated function.
- the DNA molecule with a spacer chain [adenine- (R) -X] , wherein R is independently selected from aromatic and/or aliphatic hydrocarbon chains containing up to 11 carbon atoms, and X is selected from the following: F, Ci, Br or I.
- the residue obtained was purified by CC using DCM/MeOH 10:1 (v/v) as eluent and 0.55 g of a white solid were obtained in 61% yield.
- the product was characterized by i H and 13 C nuclear magnetic resonance (NMR) using a Bruker 300 MHz spectrometer for the *H.
- a PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of p.oly-2, 2' - (m-phenylene) -5, 5' - bibenzimidazole ⁇ commercially available) in 1.3 ml, of DMSO.
- a magnetic stirrer was introduced and a reflux condenser was coupled.
- the system was heated and stirred at atmospheric pressure up to 3 hours in a temperature range of 10Q ⁇ 18G°C. After, the mixture was cooled to about 5Q°C ana 1 eq. of was added.
- a PBI solution is prepared in a 25 rnL round-bottom flask by solubilizing 2 g of
- bibenzimidazole (commercially available) in 13 mL of DMSO.
- a magnetic stirrer is introduced and a reflux condenser is coupled.
- the system is heated and stirred at atmospheric pressure up to 3 hours in a temperature range of 1G0-180°C.
- the mixture is cooled to about 50°C and 1 eq. of K 2 C0 3 (1.7 g, 12.5 mmoi; is added followed by the addition of 0,25 eq. of 1- (3- broraopropyl ) thymine (3.2 mmoi, 0.79 g) and the mixture is left stirring for 24 hours at a temperature between 100°C to 150°C.
- the polymer is separated from the mother liquor by filtration with a 3G porous plate funnel.
- the solid obtained is allowed to air dry or under reduced pressure .
- the final product is characterized by MMR in DM30 -de and can be compared with the one obtained for the initial PBI in the same deuterated solvent, represented in Figure 6.
- ft P31 solution is prepared in a 25 mL round-bottom flask by solubilizing 2 g of poiy-2, 2' - (.m-phenyiene) -5, 5' - bifoenziraidazole (commercially available) in. 13 mL of DMSO.
- a magnetic stirrer is introduced and a reflux condenser is coupled.
- the system is heated and stirred at. atmospheric pressure up to 3 hours in a temperature range of 100-180oC.
- the mixture is cooled to about 50°C and 1 eq. of K2CO3 (1.7 g, 12.5 mmoi) is added followed by the addition of Q.25 eq.
- the solution is filtered and applied the same procedure using SO mL of OC ' M.
- the solid is filtered and dried to obtain the final polymer. Between each wash lug the polymer is separated from trie mother liquor t>y filtration with a 3G porous plate funnel. The solid, obtained is allowed to air dry or under reduced pressure.
- the final product is characterized by d; N.MR in DdiC-o. and can be compared with the one obtained for the initial PBI, in the same deaterated solvent represented in Figure 6.
- a oh I solution was prepared in a 25 ml, round-bot t om flask by solufoilising 2 g. of poIy-2, 2' - (m-pheny iene ) -5, 5 ' - bifoenzimidazole (commercially available) in 13 mX of DlxiSO, To the rouna-bo r. torn flask a magnetic stirrer was introduced and a reflux condenser was coupled. The system was beared and stirred at atmospheric pressure up to 3 hours in a temperature range of 100-180 °C . After, the mixture was cooled to about 50°C and .1. eq. of R:iCCb ( 1.
- a PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of
- a PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of
- the solution was filtered aod applied the same procedure using 80 mX of OCH.
- the solid. Vae filtered, and dried to obtain the final polymer. Between each washing the polymer was separated from the mother liquor, by filtration with a 3G porous plate funnel.. The collected solid was dried under vacuum.
- the final product was characterized by 1 H NflR in XgfSO-de and the spectrum is represented in Figure 10.
- example Z 4- (dimethylanino)pyridine a representative compound of the aromatic amine type GTIs, was used,
- the GTI binding percentage was deterinined by difference between the initial concentration and the equilibrium, concentration present in. the resulting test solution according to the equation: wherein Co (mg/L) is the initial concentration of GTI and C e (mg/L) is the equilibrium concentration, of GTI in solution.
- the adsorption capacity of each polymer was determined according to the following equation: wherein. is the amount of GTI bound to the polymer,
- V (L) is the volume of solution used
- M (g) is the mass of polymer used in the test.
- binding to GTI ranged from 591-87%.
- the polymer obtained .in example 1.2 was used.
- the GTI used was methyl metnanesulfonate (MMS) , a compound belonging to the sulfonate type GTIs.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Abstract
The invention relates to a polybenzimidazole polymer with a functionalized spacer chain, according to the general formula (I) and its method of preparation, with deoxyribonucleic acid bases or carboxylic acids, chemically compatible with organic solvents, to be applied in the remova1 of genotoxic impurities. In particular, the invention focuses on obtaining a material, which establishes specific interactions with genotoxic impurities of various chemical families, to be exploited as a selective adsorbent.
Description
DESCRIPTION OP THE INVENTION
"POLYBENZIMIDAZOLE POLYMER WITH FUNCTIONALIZED SPACER CHAIN AND ITS METHOD OF PREPARATION FOR REMOVAL OF GENOTOXIC IMPURITIES"
TECHNICAL FIELD OF THE INVENTION The present invention relates to a polybenziinidazoie (FBI) polymer with a functionalized spacer chain and its structural functional ization method with deoxyribonucleic acid (DNA) bases, namely adenine, thymine, cytosine or guanine, or carboxyiic acid groups (-COOK; ,
BACKGROUND OF THE INVENTION
The industrial production of active pharmaceutical ingredients (API) often occurs in organic phase using highly reactive species, which sometimes also originate in the reaction medium itself, and which present a great diversity of functional chemical groups ["Genotoxic Impurities: Strategies For Identification and Control, Andrew Teasdale, 2010, John Wiley & Sons, Chapter 9, 221-247; Guidance for Industry Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) , December 2008]. These compounds, generally referred to as genotoxic impurities (GTI), may persist in the formulation of the final compound, A recent, and exhaustive survey of GTI associated with various manufacturing processes of different APIs can be found in the literature [G. Szekely et al . , Chem. Rev. 115 (2015) 8182-82291.
GTIs typically are elect rophi lie species that either- formed during the manufacturing process of the API or generated in vivo may chemically attack the nucleophilic centers present in the DNA molecule and may induce single- or double-strand breaks and damages whose repair mechanisms may not be able to reverse. Ail these anomalies compromise the DNA replication process, which may lead to genetic mutations, implying an increased risk to the health of the patients to whom the API is administered [A. Teasdale et al. Org. Process Res. Dev. 17 (2013) 221-230].
Sometimes, some reactive species may interact with solvents, or other reagents, present in the production of the API and also generate genotoxic compounds. Sulfonic acids are an example of such compounds [A. Teasdale et al., Org. Process Res. Dev. 14 (2010) 999-1007] . When in the presence of alcohols {used in the manufacturing process in sterns of recrystallization, or even in the washing of equipment) lead to the formation of sulfonate esters, which are substances known to be genotoxic. An example of this kind occurred in 2007 with the production of Viracept anti-viral drug, whose detailed description of the incident can be found in the literature [C. Gerber et al., Toxicol, Lett. 190 (2009) 248- 253] .
On the other hand, the aromatic amine- type compounds constitute another group of GTIs, which although not Inherently genotoxic, during their metabolization in vivo, give rise to electrophilic species. Generally, when they undergo oxidation, N-hydroxy1 compounds are formed which are conjugated as acetates, sulfates or glucuronides . Further deconj ugation of these compounds results in nitrenium ions (ArN*H) , which are considered active genotox.i.ns that bind to
DMA [D. J. Snodin, Org. Process Res. Dev. 14 (2010) 960- 976] .
Due to their high toxicity and persistence, the maximum permissible levels for GTIs in API formulations are strictly regulated by the competent authorities with a reference value of 1.5 tig / day for the toxicological risk limit ["European Medicines Agency Evaluation of Medicines for Human Use, London, 28 June 2006,
For the elimination of these impurities it is necessary to resort to intermediate and final steps of purification in the manufacturing process of the API. The use of conventional purification techniques, such as recrystailization or distillation for example, entails high energy costs. Furthermore, since these processes are not selective for GTI removal, significant losses of API also occur, with enormous economic impact on their production [N. V. V. s. S. Raman et ai., J . Pharmaceut. Biomed. Anal. 55 (2011) 662-667] .
Therefore, it is necessary to develop versatile platforms for the different purification steps directed to the selective removal of GTI thus promoting the maximum recovery of the API with high purity. For this reason, obtaining a material that is robust in organic media capable of selectively removing a GTI, or a chemically related GTI family, presents itself as a major technological development with high potential for application in an industrial enviroriment .
The polymeric material used in this invention is a linear polybenzimidazole (PBI) polymer belonging to a class of heterocyclic polymers, the structure of which is shown in Figure 1. The FBI is a polymer compatible with most organic solvents, with accessible functional! zable groups. For these reasons it was chosen as a platform that, after a structural modification, allows to be explored in a purification stage in an industrial production process of an API, as am adsorbent material for selective removal of GTI, which has not yet been described and whose capacity found in terms of GTI removal and selection efficiency over API was surprisingly high for a stoichiometry of 0.25 moles of deoxyribonucleic acid base per PBI monomer.
Modification of PBI with a ONA base is intended to mimic the interaction occurring in vivo between a GTI, for example of the sulfonate type (electrophilic species), and DNA (nucleophiiic centers) .
To date there are no similar approaches described in the literature or in industrial application. For this reason, the invention described herein contains a strong innovation component which opens up numerous possibilities with regard to selective purification methodologies of APIs.
The final material obtained, due to its robustness and versatility, allows it to be exploited as a highly specific adsorbent material for the GTI class, in particular the suiphonate fami ly .
On the other hand, the polymer modified with -COOH groups allows the selective elimination of GTI from the aromatic amine family. The removal of electrophilic species is ensured, which would result from the in vivo
raetabolization of these aromatic compounds , if they were present in the API when administered to the patient.
The functionalization chemistry of the PBI polymer has been exploited to obtain highly cross-linked PBI membranes by reaction of the polymer -with dihaiogenated compounds at reflux (80°C) in acetonitriie (MeCN) to give them greater physical robustness in the formulation of membranes for nanof.titration resistant to organic solvents, for example [I. B . Vaitcheva et al . , J. Membrane Sci. 457 (2014) 62-72; I. B. Vaitcheva et al. , J . Membrane Sci. 493 (2015) 568- 579] .
US 7,259,230 and US 8,129,498 documents disclose processes in which hybrid organic-inorganic units or amide functions are inserted into the PBI structure, respectively. In both cases the ionization of the nitrogen atoms of the imidazole rings of the PBI with an alkali metal hydride (NaH) is carried out first and subsequent reaction, with compounds containing a halogenated group of the B.-C.1 type. In these cases, it is intended to favor the solubility of the PBI in organic medium in order to allow easier handling in the processing of the resulting polymers. In these two examples the chemical functionalities introduced into the PBI molecule are directly attached to the nitrogen atoms in the imidazole rings of the polymer.
In US 4,814,400 document the synthesis of modified PBI with carboxylic acid functions is described. This is achieved by first reacting the PBI with a halogenated ester of the type X-R-CQOR1. It is only after the basic hydrolysis of the ester functions that the functionalized polymer is obtained with -COOH groups.
In the present invention a direct method for obtaining modified PBI with -COGH functions in a single reaction step, in the presence of a basic compound is described, for example, potassium carbonate (K2CG3) with this approach being described for the first time, with obtained surprisingly positive results using this weaker base,
US 2012035333&1 document discloses a method for obtaining PBI with carboxyl groups by reaction of the polyraer with cyclic acid anhydrides. In this way a polymer with amide groups directly attached to the PBI rings and to the free carboxylic acid groups is obtained. In the present invention, by using a halogenated carboxylic acid with a spacer chain, there is no amide bond formation and the carboxylic acid function is separated from the structural backbone of the PBI by two carbon atoms or more. This aspect is important to avoid stereochemical impediments that could hinder the binding to the GTIs that are intended to be removed. The relevance of the present invention is that it is possible to modify the structure of the relatively inert PBI polymer to contain a DNA base or a carboxylic acid type functionality by means of a spacer chain between the nitrogen from the imidazole rings of PBI and the new chemical functionality inserted.
The presence of this spacer chain presents a great advantage, since it facilitates the interaction between the functional groups present in the adsorbent material and the GTI, in which the latter can present several spatial geometries, more or less bulky. In this way, the adsorbent presents a great versatility against several geometries of the different GTI molecules.
SUMMARY OP THE INVENTION
The present invention relates to a poiybenzimidazole polymer with a functionalized spacer chain and its structural functionalization method with DNA bases or carboxylic acids, chemically compatible with organic solvents, to be applied in the removal of genotoxic impurities. The invention focuses on obtaining a material, which establishes specific interactions with genotoxic impurities of various chemical families, to be exploited as a selective adsorbent thereof.
In. one embodiment, the present invention relates to a poiybenzimidazole polymer with a functionalized spacer chain and its structural functional ization method with deoxyribonucleic acid. (DNA) bases, namely adenine, thymine, cytosine or guanine, or carboxylic acid groups.
DESCRIPTION OF THE DRAWINGS
'figure 1 - Chemical structure of the poiybenzimidazole polymer (PBI) .
Figure 2 - Poiybenzimidazole polymer with functionalized spacer chain: general formula of poiybenzimidazole polymer functionalized with a spacer chain R and functional group 2 , R represents a spacer chain composed of aromatic or aliphatic hydrocarbons containing from 2 to 11 carbon atoms and 2 represents a DNA base, or a carboxylic acid group.
Figure 3 - .9- ( 3-bromopropyl 5 adenine formation reaction: DMF, at room temperature,

Figure 4 ~ PBI formation reaction with adenine groups : i) DMSO,

C
Figure 6 - ¾ NMR spectrum of PBI in DMSO-d5.
Figure 7 - !H NMR spectrum of PBI with adenine groups in DMSO-ds.
Figure 8 - lH NMR spectrum of PBI with carboxylic acid groups of 3-bromapropionic acid in DMSO-de .
Figure 9 - 1H NMR spectrum of PBI with carboxylic acid groups of 5-bromovaieric acid in DMSO-de,.
Figure 10 - *H NMR spectrum of FBI with 11-bromoundecanoic acid carboxylic acid groups in DMSO-de .
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to a polybenz imidazole polymer with a functionalized spacer chain and its structural functionaiization method with DMA bases or carboxylic acids, chemically compatible with organic solvents, to be applied in the removal of genotoxic impurities. The invention focuses on obtaining a material, which establishes specific interactions with genotoxic impurities of various chemical families, to be exploited as a selective adsorbent, thereof.
In one embodiment, the present invention relates to a polybenzimidazole polymer with a functionalized. spacer chain and a structural functionaiization method with deoxyribonucleic acid (DNA) bases, namely adenine, thymine, cytosine or guanine, or carboxylic acid groups.
The spacer chain is composed of aromatic or aliphatic hydrocarbons containing from 2 to 11 carbon atoms.
The most nucleophilic positions of the DNA bases are known to be the endocyclic nitrogens of the N3 and N7 positions of guanine and adenine. These sites are preferably reactive to the presence of eiectrophilic species, such as su1 fonate type GT I .
Surprisingly it was found that the functional ization of the P3I polymer of the present invention results in obtaining a specific material for binding to certain genotoxic impurities, allowing the elimination thereof and consequently the production of active pharmaceutical, ingredients with high purity.
In the present invention, genotoxic impurities refer to families of compounds of the sulfonate or aromatic amine type .
In addition, the method of obtaining the polybenz imidazole polymer with the functional! zed spacer chain is a versatile method, in that it can be included in an industrial process, in particular the pharmaceutical industry, for elimination or reduction of levels of genotoxic impurities. It may also be included in any synthesis based industry, which exploits compounds structurally similar to GTIs, for their disposal of the respective final products.
It should be noted once again that the industrial processes of synthesis to obtain active compounds in the pharmaceutical industry take place in reaction media essentially consisting of organic solvents. Curiently, adsorbent materials such as resins are designed to perform well in aqueous media and are inefficient in organic media. The fact that the functional!zed PSI polymers disclosed herein are organic solvent resistant adsorbent materials
gives them an exceptional property to be applied in the context of the treatment of post-synthesis solutions of these pharmaceutical active compounds for their purification, The method of obtaining the polybenzimidazole polymer with a functionalized spacer chain for the removal of genotoxic impurities has the following steps:
a) Obtention of an arom.at.ic or aliphatic hydrocarbon compound containing at its ends a halogen atom and. a DNA base, or a carboxylic acid group;
b) Dissolution of the polymer in a solvent at atmospheric pressure;
c) Addition to the solution obtained in step b) of a base at atmospheric pressure;
d) Addition of the haiogenated compound obtained in step a) to the solution, obtained in step cj at atmospheric pressure ;
e) Reaction, of the solution obtained in step d) up to a period of 24 h, obtaining the final solution;
f } Precipitation of the final solution with a co- solvent, followed by successive washing/ filtration steps.
Preparation of a haiogenated DNA base with a spacer chain composed of an aliphatic hydrocarbon.
Based on what is described in the literature, the introduction of molecules into the structure of the PBI polymer occurs by reaction of the heterocyclic nitrogen atoms of the polymer with organic compounds containing a. haiogenated terminal functional group, of the type R-X in which R may correspond to a diversity of organic compounds and possible combinations thereof with inorganic compounds and X to a halogen.
DNA bases, for example, adenine do not have a halogenated group in their molecular structure. Therefore, it is first necessary to modify these molecules so as to contain this terminal -X functional group, which will allow binding to the FBI, keeping intact the positions that will be used for GTI recognition. This is accomplished by reacting the DNA base, for example adenine, with a dibromoalkane, of the type X-R-X, under controlled conditions so as to have at the end the DNA base, e.g. adenine, with a spacer chain containing a terminal halogenated group as exemplified in Figure 3. X will have to be a good leaving group, selected from: F, CI, Br, 1; and R may be a chain with aromatic groups of the type {R-Ph-R 5 or a linear chain containing from 2 to 11 carbon atoms.
This same methodology can be applied to ail pyrimidine and purine bases by reaction with their more nucleophilic nitrogen atoms . In the DNA molecule, the bases form hydrogen bonds with their complementary base. In the example of adenine and its cornpiertieritary base, thymine, these hydrogen bonds lie between the 7 position nitrogen atom and one of the two hydrogen atoms of the -NHa group of the 6 position of adenine (Figure 3) . On the other hand, the binding of the DNA bases to the ribose is done by groups not involved in the interactions with complementary bases, in the case of adenine by the nitrogen atom of the 9 position. For this reason, this was the position in which the spacer chain of the forcmoalkane type, R~X, was introduced after reaction of the adenine with a halogenated compound.
The DNA molecule with a spacer chain, which in the present invention by way of example, relates to the 9- (3-
bromopropy1 } adenine compound, is described in the literature being obtained in yields between 8% and 38% (N . J . Leonard et al., J. Org, Chem. 34 (1969) 3240-3243; WO 2013151663] . For these low yield values contribute the formation of by-products, such as the adenine-R-adenine dimer and a tricyclic derivative.
The present invention further describes the synthesis of this compound, the optimization of which allowed it to be obtained in a yield higher than 60%. The development of this synthesis has been achieved iteratively by introducing a set of innovations described below and allowing improved performance of a synthesis which is not. trivial.
For the synthesis of
 were initially used 2 equivalents (eq.) of 1 , 3-dibromopropane relatively to adenine (in a 1 molar solution of adenine in DMF) at room temperature in the presence of 2 eq. of potassium carbonate (K2CO3) for 16 hours. The insolubility of K2CC3 in DMF did not allow to observe the complete dissolution of adenine in the 3.7 mL of solvent used. However, after the reaction, the reacticnai mixture was filtered and, both the filtrate as the solid, were analyzed by thin-layer chromatography (TLC) , to confirm that, both phases had ths same chromatographic pattern in TLC,
were initially used 2 equivalents (eq.) of 1 , 3-dibromopropane relatively to adenine (in a 1 molar solution of adenine in DMF) at room temperature in the presence of 2 eq. of potassium carbonate (K2CO3) for 16 hours. The insolubility of K2CC3 in DMF did not allow to observe the complete dissolution of adenine in the 3.7 mL of solvent used. However, after the reaction, the reacticnai mixture was filtered and, both the filtrate as the solid, were analyzed by thin-layer chromatography (TLC) , to confirm that, both phases had ths same chromatographic pattern in TLC,
To avoid final product loss, it was chosen to avoid the filtration step and, instead, totally evaporate the organic solvent. To remove K2CO3 the formed solid was washed with acetone, filtered and evaporated. The obtained solid was again washed with methanol (MeOH, 10 mL) , obtaining the final product in 16% yield.
In order to improve the reaction yield, the product wa s extracted with a brine solution and dichloromethane (DMC) . However, the formation of a precipitate in the organic phase wa s observed that interfered in phases separation. The organic phases were collected and, to avoid possible product losses, acetone was added to dissolve the precipitate formed. The mixture was filtered, evaporated and the solid was purified by column chromatography (CC) , obtaining the finai compound with 27% yield. posteriorly, the reaction temperature wa s raised to 50 'JC, but the degradation of the product after its formation and consequent production of secondary products was observed by TLC, comparatively to the reaction performed at room temperature. Thereby, it was chosen to maintain the reaction at room temperature to favor the formation of a more stable final product and also to avoid the presence of undesirable secondary products. At this point, it was important, to study the solubility of 9- ( 3-bromopropyl ) adenine in several solvents and it wa s verified that it was soluble in DMF, DMSO and in mixtures of DCM/MeOH. Therefore, a reaction with 3 eq. of 1 , 3-dibrom.opropane relatively to the adenine (solution of 1 M of adenine in DMF) at room temperature in the presence of 2 eq. of potassium carbonate { K2CO3) for 16 hours was performed. After, the solvent was evaporated, the solid was washed with a DCM/MeOH 10:1 to 4:1 solution (40 mL) , filtered, the filtrate was concentrated and purified by CC, obtaining the product in 35% yield. Following this methodology, using 5 eq. of 1 , 3-dibromopropane relatively to adenine, in DMF, was obtained a 50% yield for the same 16 hours of reaction.
In order to improve the yield of this synthesis, 5 eq. of 1 , 3-dibromopropane were used and the concentration of the
initial adenine solution was increased from. 1 M to 1.2 M, in DMF. In this case, 3 rnL of this solution were used in a reaction, followed by TLC, that underwent for only 3 hours, instead of the previous 16 hours. The resulting product was purified by CC, using a solvent gradient starting with 100% of DCM and followed by a DCM/MeOH 10:1 (v/v) mixture. With this procedure, the product was obtained in a 61% yield.
According to the present invention, in the case where the DNA. base is adenine, the step of obtaining an aromatic or aliphatic hydrocarbon compound containing at its ends a halogen atom and a DNA base, or a carboxylic acid group comprises the following steps:
a) Dissolution of the adenine in DMF, at room temperature and subsequent addition of K2CO3;
b) Slow addition of an aromatic or aliphatic dihalogenated hydrocarbon;
c) Reaction of the solution obtained in step b) at room temperature, up to a period of 16 hours, obtaining the final solution;
d) Evaporation of the DMT from the final solution to obtain a solid;
e) Addition of a 10:1 to 4:1 (v/v) solution of DCM/MeOH to the solid obtained in step d) and filtration;
f) Evaporation of the solvent from the solution obtained in step e) to obtain a solid;
g) Purification by column chromatography of the solid obtained in step f) with a DCM/MeOH mixture.
Functionalization of PBI with a halogenated DNA base with a spacer chain composed of aliphatic hydrocarbon.
After obtaining the modified adenine base with a bromine atom at its end, this unit was incorporated into the PBI
polymer. Briefly, the polymer is dissolved in dimethylsulfoxide (DMSO) at 100 °C to 180°C, and thereafter a basic compound is added, in this case the base (KsCCb) , to deprotonate the nitrogen atoms of the imidazole rings of the polymer, and finally the DNA base is added (e.g. modified adenine, In this nuclecphilic

substitution compounds of the type HX (e.g. HBr) are formed, which are released into the reaction medium, and the bond between the DNA base (i.e. adenine) and the PBI takes place by the spacer chain present in the first, as shown schematically in Figure 4,
After this reaction, water (40 mL) was added to the reaction medium to promote the precipitation of the modified polymer and also to remove the base in solution and subsequently washing with MeOH (40 mL) and DCM (40 mL) to remove secondary products that have formed as well as modified adenine.
In the first attempt, a 1% (wt/v) PBI solution in DMSO in the presence of 2.2 eq. of K2CO3 and 1 eq. of 9- (3- bromopropyl) adenine at room temperature for 24 hours was used. At the end of this time water was added (40 mL) but no precipitation of the polymer was observed. Acetone (SO mL) was added to the DMSO and water mixture and left in the freezer {-20 °C) for 16 hours, obtaining a thin powder that was filtered and dried.
To favor the formation of the product the PBI initial solution concentration was increased to 15% and left stirring for 3 hours at 170 °C to achieve complete solubilization of the polymer. After this time, the mixture was allowed to cool to room temperature and 2. eq. of K2CG3 and 1 eq. of S- ( 3 -bromopropyl ) adenine were added. At this stage, the mixture presented itself as a viscous gel that hindered the
efficient homogeni zation of the reactionai mixture. For this reason, it was necessary to increase the temperature to 50°C and left stirring for 24 hours at this temperature. At the end of the reaction water (40 mL) was added giving a solid in the form of small particles which was washed multiple times with water and then dried under vacuum .
This polymer {50 ing) was placed in contact with 1 ruL of GTI solution at a concentration of 100 ppro in DCM at room temperature for 24 hours and the percentage of GTI bound to the polymer was quantified at the end. of this time. With this polymer, a preliminary GTI binding value of 14% was obtained. Based on this result, we tried to optimize the reaction using 3 eq. of sodium hydroxide (NaOH) to replace K2CO3. In this case a final product of high hardness was obtained, impossible to be processed for use in the GTI removal trials. In a further approach, to the 15% PBI solution was added
2 eq. of K2CO3 and 2 eq. of 9- { 3~broraopropyl} adenine, following the reaction for another 24 hours at 8Q°C. The final product had a GTI binding of about 24%. At this point, it was tried to make the addition of the reactants and the reaction itself at 100°C. A hard solid was obtained again, which could not be processed for use in the GTI removal assays .
It was hypothesized that the addition temperature of the reactants was very high and it was chosen to make this step at 50 °C and raise the temperature to 100eC for the course of the reaction. Under these latter conditions it has been found that in the presence of 1 or 2 eq. of K2CO3 the polymer obtained had a GTI removal value of 59%, similar for
both equivalents used. In this way we chose to use only 1 eq. of K2CO3 in the reaction for obtaining the adenine- modified FBI polymer. In the GTI assays, impurities from the reaction were observed, which persisted if the polymer was washed with water alone or with DCM. However, after further washing with MeOH it was found that these impurities were efficiently removed from the polymer without interfering with its binding to the GTI .
In the binding reaction of the halogenated adenine (9~ (3-bromopropyl } adenine ) to the FBI, different equivalents of halogenated adenine against. FBI were evaluated to select the ratio that originates the adenine-modified polymer with the best adsorption capacity for the sulfonate GTI in organic medium. Values for GTI removal were obtained between 16%- 97%, higher than the starting PBI which showed only about 10% binding to the GTI.
Functionalization of PBI with carboxylic acid with spacer chain composed of aliphatic hydrocarbon.
The reaction conditions optimized to obtain the polymer PBI with halogenated adenine (9- {3-bromopropyl) adenine) were applied in the synthesis of the polymer modified with carboxylic acids (Figure 5) . In this case, the reaction was carried out between the FBI and the halogen compound which consisted of a carboxylic acid with a halogenated function for attachment to the polymer.
The influence of several halogenated carboxylic acid equivalents against the PIB was also determined to select the ratio that causes a GTI removal of the aromatic amine by
the carboxylic acid modified PBI. GTI binding values were obtained between 59%-99%, higher than the starting PBI which showed only about 10% GTI binding. For a better understanding of the invention, some examples of the application of the present invention are described below by way of illustration and not limitation.
Examples
As an example, the PBI polymer can be solubilized in a suitable solvent including DMSO, dymethylacetami.de ( DMAc ) , dimethyl formamide (DMF) or N~methyI-2~pyrrolidone (NMP) . A solution of 15% P3I in DMSO was found to be suitable for performing the functionalization described in this invention .
Ionization of the nitrogen atoms of the imidazole rings by deprotonation with a base was then carried out. K2CO3 can be used for this purpose.
The DNA base, or the carboxylic group, may contain a spacer chain composed of an aromatic and/or aliphatic hydrocarbon containing from 2 to 11 carbon atoms, with a terminal halogenated function.
By way of example, the DNA molecule with a spacer chain, [adenine- (R) -X] , wherein R is independently selected from aromatic and/or aliphatic hydrocarbon chains containing up to 11 carbon atoms, and X is selected from the following: F, Ci, Br or I.
For the reaction to occur a minimum of 0.1 eq. of modified adenine or carboxylic acid derivative, relative to the imidazole ring nitrogens, are necessary.
Preferably for GTI removal the reaction requires about
0.25 eq. of the modified DNA base or 1.00 eq. of halogenated carboxylic acid. Both ionization of the imidazole groups and alkyiation steps can occur at temperatures between 50°C and 100 °C at atmospheric pressure.
Example 1
1.1 Synthesis of 9- (3-bromopropyi) adenine
To a solution of 0.5 g of adenine (3.7 romol) in 3 mL of DMF, in a 10 mL round-bottom flask, 2 eq. of K2CO3 were added (1 g, 7.2 mmol) . The mixture was left stirring for, approximately, 15 minutes at room temperature. After that time, 5 eq. of 1 , 3~dibromcpropane were slowly added (1.9 mL, 19 mmol) and the mixture was left stirring for 3 hours at room temperature. The solvent was removed under reduced pressure and. the solid was left stirring for 10 minutes in a solution of 4:1 (V/V) of DCM/MeOH. This mixture was filtered in a 3G porous plate and the solvent was removed under reduced pressure. The residue obtained was purified by CC using DCM/MeOH 10:1 (v/v) as eluent and 0.55 g of a white solid were obtained in 61% yield. The product was characterized by iH and 13C nuclear magnetic resonance (NMR) using a Bruker 300 MHz spectrometer for the *H.
1.2 Reaction between FBI polymer and 0.25 eq. of 9- (3- bromopropyl) adenine
A PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of p.oly-2, 2' - (m-phenylene) -5, 5' - bibenzimidazole {commercially available) in 1.3 ml, of DMSO. To the round-bottom flask a magnetic stirrer was introduced and a reflux condenser was coupled. The system was heated and stirred at atmospheric pressure up to 3 hours in a
temperature range of 10Q~18G°C. After, the mixture was cooled to about 5Q°C ana 1 eq. of
 was added. In this step, at 30°C the mixture was too viscous to continue the procedure and at 100°C the obtained final polymer was too hard to be easily handled. During this process the mixture kept the initial dark brown color. It was added 0.25 eq. of 9- ( 3-bromopropyl) adenine {3.2 mmol, 0.82 g) , prepared in example 1.1, and the mixture was left stirring for 24 hours at a temperature between 100°C to 150°C. After completion, 80 m.L of distilled water were added as co-solvent and the polymer precipitated immediately as a gold brown solid that was manually broken with the help of a spatula. This mixture was left stirring for some minutes and filtered in a 3G poraus plate and the solid was transferred to a glass becker (100 mL) and about 80 mL of MeOH were added and left stirring for some minutes. The solution was filtered and applied the same procedure using 80 mL of DCM. The solid was filtered and dried to obtain the final polymer. The final product was characterized by 1H NMR in DMSO-de and the spectrum is presented in Figure 7. 1H NMR (300 MHz, DMSO- de) : δ 2.03 is), 3.40 (s) , 4.01 ( s ) , 7.96-7.64 (m, 7H) , 8.35 (d, J=6 Hz, 2Hi, 9.16 and 8.66 (.:;, 1H) . This spectrum could be compared with the one obtained for the initial FBI in the same deuterated solvent, represented in Figure 6: NMR 4-ϊ (300
was added. In this step, at 30°C the mixture was too viscous to continue the procedure and at 100°C the obtained final polymer was too hard to be easily handled. During this process the mixture kept the initial dark brown color. It was added 0.25 eq. of 9- ( 3-bromopropyl) adenine {3.2 mmol, 0.82 g) , prepared in example 1.1, and the mixture was left stirring for 24 hours at a temperature between 100°C to 150°C. After completion, 80 m.L of distilled water were added as co-solvent and the polymer precipitated immediately as a gold brown solid that was manually broken with the help of a spatula. This mixture was left stirring for some minutes and filtered in a 3G poraus plate and the solid was transferred to a glass becker (100 mL) and about 80 mL of MeOH were added and left stirring for some minutes. The solution was filtered and applied the same procedure using 80 mL of DCM. The solid was filtered and dried to obtain the final polymer. The final product was characterized by 1H NMR in DMSO-de and the spectrum is presented in Figure 7. 1H NMR (300 MHz, DMSO- de) : δ 2.03 is), 3.40 (s) , 4.01 ( s ) , 7.96-7.64 (m, 7H) , 8.35 (d, J=6 Hz, 2Hi, 9.16 and 8.66 (.:;, 1H) . This spectrum could be compared with the one obtained for the initial FBI in the same deuterated solvent, represented in Figure 6: NMR 4-ϊ (300

1.3 Reaction between PBI polymer and 0.25 eq. of
A PBI solution is prepared in a 25 rnL round-bottom flask by solubilizing 2 g of

bibenzimidazole (commercially available) in 13 mL of DMSO. To the round-bottom flask a magnetic stirrer is introduced and a reflux condenser is coupled. The system is heated and
stirred at atmospheric pressure up to 3 hours in a temperature range of 1G0-180°C. After, the mixture is cooled to about 50°C and 1 eq. of K2C03 (1.7 g, 12.5 mmoi; is added followed by the addition of 0,25 eq. of 1- (3- broraopropyl ) thymine (3.2 mmoi, 0.79 g) and the mixture is left stirring for 24 hours at a temperature between 100°C to 150°C. After completion, 80 mL of distilled water are added as co-solvent and the polymer precipitates immediately as a gold brown solid that is manually broken -with the help of a spatula. This mixture is left stirring for some minutes and filtered in a 3G porous plate and the solid is transferred to a glass becker (100 mL) and about 80 mL of MeOH are added and left stirring for some minutes. The solution is filtered and applied the same procedure using 80 mL of DCM. The solid is filtered and dried to obtain the final polymer.
Between each washing the polymer is separated from the mother liquor by filtration with a 3G porous plate funnel. The solid obtained is allowed to air dry or under reduced pressure .
The final product is characterized by MMR in DM30 -de and can be compared with the one obtained for the initial PBI in the same deuterated solvent, represented in Figure 6.
1.4 Reaction between PBI polymer and 0.25 eq. of l-(3- brontopropyl) cytosina
ft P31 solution is prepared in a 25 mL round-bottom flask by solubilizing 2 g of poiy-2, 2' - (.m-phenyiene) -5, 5' - bifoenziraidazole (commercially available) in. 13 mL of DMSO. To the round-bottom flask a magnetic stirrer is introduced and a reflux condenser is coupled. The system is heated and stirred at. atmospheric pressure up to 3 hours in a temperature range of 100-180ºC. After, the mixture is cooled to about 50°C and 1 eq. of K2CO3 (1.7 g, 12.5 mmoi) is added followed by the addition of Q.25 eq. of 1- (3-
bromopropyi) cy tosine (3.2 mmol, 0.74 q) and. the mixture is If:: ft stirring for 24 hours at a temperature between 100°C to 150°C, After completion, 80 m.L of distilled xyster are ahded as co-solvent and the polymer precipitates iKumeftbat e l y as a gold brown solid that is manually broken with the help or" a spatula. This mixture is left stirring for some minutes and filtered in a 3G porous plate and the solid is transferred. :: u a glass becker (100 mid and about 80 mL of 'MeOH are added and left stirring for some minutes. The solution is filtered and applied the same procedure using SO mL of OC'M. The solid is filtered and dried to obtain the final polymer. Between each wash lug the polymer is separated from trie mother liquor t>y filtration with a 3G porous plate funnel. The solid, obtained is allowed to air dry or under reduced pressure. The final product is characterized by d; N.MR in DdiC-o. and can be compared with the one obtained for the initial PBI, in the same deaterated solvent represented in Figure 6.
Example 2
Reaction between FBI polymer and 1.00 eq. of 3-broiaopropi.onic acid
A oh I solution was prepared in a 25 ml, round-bot t om flask by solufoilising 2 g. of poIy-2, 2' - (m-pheny iene ) -5, 5 ' - bifoenzimidazole (commercially available) in 13 mX of DlxiSO, To the rouna-bo r. torn flask a magnetic stirrer was introduced and a reflux condenser was coupled. The system was beared and stirred at atmospheric pressure up to 3 hours in a temperature range of 100-180 °C . After, the mixture was cooled to about 50°C and .1. eq. of R:iCCb ( 1.7 g, 1.2.5 mmoi) was added followed by the addition of 1 eq, of 3-hro:i!:opropi ormc acid (11 mmol, I g) and the mixture was left stirring for 24 hours at a temperature out ween 100°C to 150°C, After completion, 30 tvL of distilled water were added as co-solvent and the polymer precipitated immediately as a gold brown sella that
was manually broken with the help of a spatula. This mixture was left stirring for some minutes and filtered in a 3G porous plate and the solid was transferred to a glass becker (100 mL) and about 80 ml. of MeOH were added and left stirring for sorae minutes. The solution was filtered and applied the same procedure using 80 m,L of DCM. The solid was filtered and dried to obtain the final polymer. Between each washing the polymer was separated from the mother liquor by filtration with a 3G porous plate funnel. The collected solid was dried under vacuum. The final product was characterized by lH NMR in DMSO-ds and the spectrum is represented in Figure
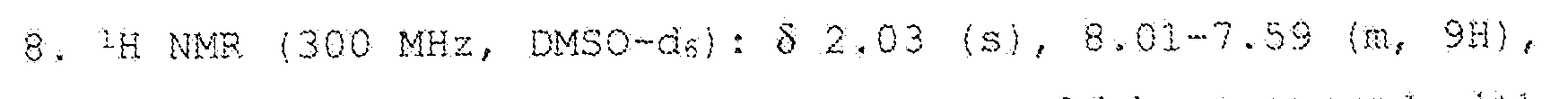
9.21 and 8.81 (s, 1H) . This spectrum could be compared with the one obtained for the initial PBI in the same deuterated solvent, represented in Figure 6.
Example 3
Reaction between PBI polymer and 1.00 eq. of 5-bromovaleric acid
A PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of

bibenzimidazcle (commercially available) in 13 mL of DMSO. To the round-bottom, flask a magnetic stirrer was introduced and a reflux condenser was coupled. The system was heated and stirred at atmospheric pressure up to 3 hours in a temperature range of 100-180 °C. After, the mixture was cooled to about 50°C and 1 eq. of K2CO3 (1/7 g, 12.5 mmol) was added. In this step at 30°C the mixture is too viscous to continue the procedure and at 100°C the obtained final polymer is too hard to be easily handled. During this process, the mixture kept the initial dark brown color. 1 eq. of 5-bromovaleric acid (13 mmol, 2.5 g) was added and the mixture was left stirring for 24 hours at a temperature between 100°C to 150°C. After completion, 80 mL of distilled water were added
as co-solvent and the polymer precipitated immediately a s a gold brown solid that was manually broken with the help of a spatula. This mixture was left stirring for some minutes and filtered in a 3G porous plate and the solid was transferred to a glass foecker (100 mL) and about 80 mL of MeOH were added and left stirring for some minutes. The solution was filtered and applied the same procedure using 80 mL of DCM. The solid was filtered and dried, to obtain the final polymer. Between each washing the polymer "wa s separated from the mother liquor by filtration with a 3G porous plate funnel. The collected solid was dried under vacuum. The final product was characterized by 3·Η NMR in BMSO-de and the spectrum is represented in Figure 9.

7H) , 8.35 (s, 2H) , 9.18 i s , 1H) . This spectrum could be compared with the one obtained for the initial PBI. in the same deuterated solvent, represented in Figure 6.
Example 4
Reaction between PBI polymer and 1.00 eq, of 11- bromoundecananoic acid
A PBI solution was prepared in a 25 mL round-bottom flask by solubilizing 2 g of

bibenzimidazole (commercially available) in 13 mL of DM30. To the round-bottom flask a -magnetic stirrer was introduced and a reflux condenser 'was coupled. The system was heated and stirred at atmospheric pressure up to 3 hours in a temperature range of 1Q0-18Q°C. After, the mixture was cooled to about 50°C and 1 eg. of K2CO3 (1.7 g, 12.5 mmol) was added. In this step at 30°C the mixture is too viscous to continue the procedure and at 100°C the obtained, final polymer is too hard to be easily handled. During this process, the mixture kept the initial dark brown color. 1 eq„ of 11- bromoundecanoic acid (13 mmol, 3. A g) was added and the
mixture was left stirring for 24 hours* at a temperature between 10D°C to 150°C» After completion, fiO mL of diet il led water were eddied as co-solvent and the polymer precipitated immediately aa a gold brown eolid that was *nahu$lly broken with- the help of a spatula r This mixture Was- left stirring for aome minutes and fil tered in a 36 porous plate and the solid was transferred to a glass becker (100 mL) and about 80" QtL Of MeOH were added and lef t stirring for some minutes . The solution was filtered aod applied the same procedure using 80 mX of OCH. The solid. Vae filtered, and dried to obtain the final polymer. Between each washing the polymer was separated from the mother liquor, by filtration with a 3G porous plate funnel.. The collected solid was dried under vacuum. The final product was characterized by 1H NflR in XgfSO-de and the spectrum is represented in Figure 10.


This spectrum could be

compared with the one obtained for the initial FBI in the same deuterated Solvent, represented in Figure 6.
Example 5
In these tests the polymers obtained in examples
sulfonate { A representative compound of the sulfonate

type GTXs. For the polymer with carboxylic. a-cid groups, example Z, 4- (dimethylanino)pyridine
 a representative compound of the aromatic amine type GTIs, was used,
a representative compound of the aromatic amine type GTIs, was used,
To 50 rag of each polymer va« added 1 ml of this respective GTI solution, with a concentration of 100 ppm, prepared in □CM. This suspension was allowed to stir for 24 hoars at
room temperature and 200 rpm. After this time the suspensions were centrifuged (13, 900 rpm, 35 minutes; and the supernatant was filtered with syringe filters and analyzed by HPLC (High Pressure Liquid Chromatography) . These analyzes were performed on a Hitachi Merck equipment coupled to an L--24Q0 UV detector with a Macherey-Nagel CIS reverse phase analytical column (Nucleosil 100-10, 250 x 4.6 mm) .
The GTI binding percentage was deterinined by difference between the initial concentration and the equilibrium, concentration present in. the resulting test solution according to the equation:
 wherein Co (mg/L) is the initial concentration of GTI and Ce (mg/L) is the equilibrium concentration, of GTI in solution.
wherein Co (mg/L) is the initial concentration of GTI and Ce (mg/L) is the equilibrium concentration, of GTI in solution.
The adsorption capacity of each polymer was determined according to the following equation:
 wherein. is the amount of GTI bound to the polymer,
wherein. is the amount of GTI bound to the polymer,

Co (mg/L) is the initial concentration of is

the equilibrium concentration of GTI in solution, V (L) is the volume of solution used and M (g) is the mass of polymer used in the test.
From the results shown in Table 1 it was found that the polymer obtained from the reaction with 0,25 eq. of 9- ( 3- broruopropyl) adenine shows the best percentage of removal of MPTS, corresponding to about 96%. For the remaining polymers the percentage of GTI removal was between
 In any case the GTI removal was always higher than that, observed
for the starting PBI polymer, used as a control, with only 10% GTI removal . Controls were also performed with GTI solutions processed identically, but in the absence of polymer, to detect GTI losses by adsorption on the walls of vials, filters or evaporation, in which case a negligible removal was observed.
In any case the GTI removal was always higher than that, observed
for the starting PBI polymer, used as a control, with only 10% GTI removal . Controls were also performed with GTI solutions processed identically, but in the absence of polymer, to detect GTI losses by adsorption on the walls of vials, filters or evaporation, in which case a negligible removal was observed.
Table 1 - Percentage removal and adsorption capacity of FBI polymers with adenine for the methyl p-toluenesulfonate GTI of a solution with a concentration of 100 ppm in DCM.

According to the data presented in Table 2, it is verified that for DM&P the use of 1 eq. of the carboxylic acid derivative in the reaction with the PBI results in the adsorbent having the best GTI removal characteristics, having the highest GTI removal values at about 99%.
For the remaining polymers, formed in the presence of different carboxylic acid equivalents, binding to GTI ranged from 591-87%.
The results show that polymers containing carboxylic acid groups have a higher affinity for the GTI as compared to the
startiaq FBI, used as a control, which exhibits a GTI re:novaI of only 10%.
Table 2 - Percentage removal and adsorption capacity of: FBI polymers with carboxylic acid for 4 - (dimethyiamino; pyridine GTI of a solution with a concentration ot 100 ppra in DOM.

Example 6
Removal efficiency, binding percentage of adenine- £unctionalized FBI polymer to methyl methanesulfonate GTI in "batch" and column chroxtiatography.
In this example the polymer obtained .in example 1.2 was used. The GTI used was methyl metnanesulfonate (MMS) , a compound belonging to the sulfonate type GTIs.
For the batch procedure; to 50 mg of the polymer was added 1 icL of the 0ΓΓ ; solution, at a concentration of 100 ρρπρ prepared in DCM. The suspension was stirred for 2·;! hours at rcoffi temperature and 200 rose After that time the mixture was centxifuged (13, 900 rpm, 35 minotes; and the supernatant was filtered with syringe filters and analyzed by K0LC as described in example 5.
For the chromatographic column procedure, 100 nig of polymer were conditioned on a glass column, through which 2 ir,L of the GTI solution, at a concentration of 100 ppm, prepared in DCM was passed. The resulting eiuate from the column was analyzed by HPLC as described in example 5.
A percentage of GTI removal of about 96% was determined in both the batch and chromatographic column assays.
Claims
1. Poiybenzimidazole polymer with funct.io.nalized. spacer chain characterized in that it presents the following general formula (I) :

a; R represents a spacer chain composed of aromatic o aliphatic hydrocarbons, containing from 2 to 11 carbon atoms b) Z represents a DNA base, or a carboxylic acid group..
2. Folybenzimidazole polymer according to claim 1, characterized in that the DNA base is selected from, the group: adenine, thymine, cytosine or guanine.
3. Method of obtaining the polybenzimidazole polymer with functionaiized spacer chain for the removal of genotoxic impurities, as defined in claims 1 and 2, characterized in that it presents the following steps:
a) Obtention of an aromatic or aliphatic hydrocarbon compound, containing at. its ends a halogen atom and a DNA base, or a carboxylic acid group;
b) Dissolution of the polymer in a solvent at atmospheric pressure ;
c) Addition to the solution obtained in step b) of a base at atmospheric pressure;
d) Addition of the haiogenated compound obtained in step a) to the solution obtained in step c) at atmospheric pressure; e} Reaction of the solution obtained in step d) to a period of 24 h, obtaining the final solution;
f) Precipitation of the final solution with a co-solvent, followed by successive washing/filtration steps.
4. Method according to claim 3, characterized in that, in the case where the DNA base is adenine, step a) comprises the following steps:
a) Dissolution of the adenine in DMF, at room temperature and subsequent addition of K2CO3;
b) Slow addition of an aromatic or aliphatic dihaiogenated hydrocarbon;
c) Reaction of the solution obtained in step b) at room, temperature, up to a period of 16 hours, obtaining the final solution;
d) Evaporation of the DMF from the final solution to give a solid;
e) Addition of a 10:1 to 4:1 (v/v) solution of DCM/MeOH to the solid obtained in step dS and filtration;
f) Evaporation of the solvent from the solution obtained in step e) to give a solid;
g) Purification by column chromatography of the solid obtained in step f) with a DCM/MeOH mixture.
5. Method according to claim. 3, characterized in that the solvent used for the dissolution of the FBI in step b) is selected from the group of: dimethylsulfoxide (DMSO) , N- methyl-2-pyrrol idone (NMP) , dymethylacetamide (DMAc) .
6. Method according to claim 3, characterized in that step b) is carried out at a temperature between 100 and 180°C.
7. Method according to claim 3, characterized in that step b) is carried out for up to 3 hours.
8. Method according to claim 3, characterized in that step c) is carried out at a temperature between 30 and 100CC
9. Method according to claim 3, characterized in that the base used in step c) is a carbonate.
10. Method according to claim 3, characterized in that step d) is carried out at a temperature between 30 and. lOO'C.
11. Method according to claim. 3, characterized in that step e) is carried out at a temperature between 100 and 150oC.
12. Method according to claim 3, characterized in that the co-solvent used in step f) is water.
13. Method according to claim 3, characterized in that the washing solvents used in the successive washing/filtration steps are water, methanol and dichloromethane, respectively.
14. Ose of the polybenzimidazole polymer, as defined in claims 1 and 2, characterized in. that it is employed in the removal of genotoxic impurities of the sulfonate or aromatic amine type .

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PT109480A PT109480B (en) | 2016-06-22 | 2016-06-22 | POLYMENZIMIDAZOLE POLYMER WITH FUNCTIONED SPACER CHAIN AND ITS METHOD OF OBTAINING TO REMOVE GENOToxic IMPURITIES |
| PT109480 | 2016-06-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017222402A1 true WO2017222402A1 (en) | 2017-12-28 |
Family
ID=59091554
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/PT2017/000008 Ceased WO2017222402A1 (en) | 2016-06-22 | 2017-05-11 | Polybenzimidazole polymer with e'unctionalized spacer chain and its method of preparation for removal of genotoxic impurities |
Country Status (2)
| Country | Link |
|---|---|
| PT (1) | PT109480B (en) |
| WO (1) | WO2017222402A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115326937A (en) * | 2021-05-11 | 2022-11-11 | 山东省食品药品检验研究院 | Solid-phase probe for capturing genotoxic impurities and using method and application thereof |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4814400A (en) | 1987-04-13 | 1989-03-21 | Hoechst Celanese Corporation | Polybenzimidazole ester and carboxylic acid |
| US7259230B2 (en) | 2004-06-07 | 2007-08-21 | Battelle Energy Alliance, Llc | Polybenzimidazole compounds, polymeric media, and methods of post-polymerization modifications |
| US20120035333A1 (en) | 2010-08-04 | 2012-02-09 | Chang Gung University | Carboxylic polybenzimidazole |
| US8129498B2 (en) | 2004-06-07 | 2012-03-06 | Battelle Energy Alliance, Llc | Polymeric medium |
| WO2012035556A1 (en) * | 2010-09-14 | 2012-03-22 | Council Of Scientific & Industrial Research | Quaternised polybenzimidazole |
| WO2013151663A1 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of membrane proteins |
| US20130344567A1 (en) * | 2010-12-30 | 2013-12-26 | Lfb Biotechnologies | Method for immobilising nucleic ligands |
-
2016
- 2016-06-22 PT PT109480A patent/PT109480B/en active IP Right Grant
-
2017
- 2017-05-11 WO PCT/PT2017/000008 patent/WO2017222402A1/en not_active Ceased
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4814400A (en) | 1987-04-13 | 1989-03-21 | Hoechst Celanese Corporation | Polybenzimidazole ester and carboxylic acid |
| US7259230B2 (en) | 2004-06-07 | 2007-08-21 | Battelle Energy Alliance, Llc | Polybenzimidazole compounds, polymeric media, and methods of post-polymerization modifications |
| US8129498B2 (en) | 2004-06-07 | 2012-03-06 | Battelle Energy Alliance, Llc | Polymeric medium |
| US20120035333A1 (en) | 2010-08-04 | 2012-02-09 | Chang Gung University | Carboxylic polybenzimidazole |
| WO2012035556A1 (en) * | 2010-09-14 | 2012-03-22 | Council Of Scientific & Industrial Research | Quaternised polybenzimidazole |
| US20130344567A1 (en) * | 2010-12-30 | 2013-12-26 | Lfb Biotechnologies | Method for immobilising nucleic ligands |
| WO2013151663A1 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of membrane proteins |
Non-Patent Citations (12)
| Title |
|---|
| "Food and Drug Administration, Center for Drug Evaluation and Research (CDER", December 2008, U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES, article "Guidance for Industry Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches" |
| A. TEASDALE ET AL., ORG. PROCESS RES. DEV, vol. 17, 2013, pages 221 - 230 |
| A. TEASDALE ET AL., ORG. PROCESS RES. DEV., vol. 14, 2010, pages 999 - 1007 |
| ANDREW TEASDALE: "Genotoxic Impurities-, Strategies For Identification and Control", 2010, JOHN WILEY & SONS, pages: 221 - 247 |
| C. GERBER ET AL., TOXICOL. LETT., vol. 190, 2009, pages 248 - 253 |
| CPMP/SWP/5199/02, EMEA/CHMP/QWP/25134 4/2006, COMMITTEE ON MEDICINAL PRODUCTS FOR HUMAN USE (CHMP), GUIDELINE ON THE LIMITS OF GENOTOXIC IMPURITIES, 28 June 2006 (2006-06-28) |
| D. J. SNODIN, ORG. PROCESS RES. DEV., vol. 14, 2010, pages 960 - 976 |
| G. SZEKELY ET AL., CHEM. REV., vol. 115, 2015, pages 8182 - 8229 |
| I. B. VALTCHEVA ET AL., J. MEMBRANE SCI., vol. 457, 2014, pages 62 - 72 |
| I. B. VALTCHEVA ET AL., J. MEMBRANE SCI., vol. 493, 2015, pages 568 - 5,791 |
| N. J. LEONARD ET AL., J. ORG. CHEM., vol. 34, 1969, pages 3240 - 3248 |
| N. V. V. S. S. RAMAN ET AL., J. PHARMACEUT. BIOMED. ANAL, vol. 55, 2011, pages 662 - 667 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115326937A (en) * | 2021-05-11 | 2022-11-11 | 山东省食品药品检验研究院 | Solid-phase probe for capturing genotoxic impurities and using method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| PT109480B (en) | 2020-03-13 |
| PT109480A (en) | 2017-12-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10577433B2 (en) | Process for the preparation of sugammadex | |
| CN100378140C (en) | Block copolymers with reduced impurity levels, polymeric carriers, pharmaceutical formulations in polymeric form and methods for their preparation | |
| JP6460366B2 (en) | Method for purifying polyethylene glycol having one amino group | |
| EP2137206B1 (en) | Process for the preparation of tauroursodesoxycholic acid | |
| CN104086691B (en) | A kind of preparation method of the polymer containing Cucurbituril structure | |
| US20180339286A1 (en) | Polymer-supported chelating agent | |
| WO2001060799A1 (en) | Resolution of the enantiomers of amlodipine | |
| Zhang et al. | Efficient synthesis of secondary amines by reductive amination of curdlan Staudinger ylides | |
| KR102054228B1 (en) | Process for Preparing Sugammadex Sodium | |
| WO2017222402A1 (en) | Polybenzimidazole polymer with e'unctionalized spacer chain and its method of preparation for removal of genotoxic impurities | |
| EP2488497B1 (en) | Process for manufacturing 2-[(3,5-difluoro-3'-methoxy-1,1'-biphenyl-4-yl)amino]nicotinic acid | |
| JP5424205B2 (en) | Polymer, method for producing the same, and method for recovering noble metal | |
| WO2023176809A1 (en) | Method for producing polyethylene glycol derivative | |
| Vicente et al. | Solvent compatible polymer functionalization with adenine, a DNA base, for api degenotoxification: Preparation and characterization | |
| CN114716663B (en) | Method for preparing polyethylene glycol modified lysine | |
| CN116096724B (en) | Method for preparing oligonucleotides | |
| US11866520B2 (en) | Functionalised compounds | |
| CA2394580A1 (en) | Macrocyclic compounds and their use | |
| KR20210077677A (en) | Methods of making releasable polymeric reagents | |
| CN119119000A (en) | Aromatic oligoamide folded body with protected aminoalkoxy side chain and preparation method and application thereof | |
| CN111196800B (en) | Method for preparing lenalidomide | |
| Zhang et al. | Efficient Synthesis of Secondary Amines by Reductive | |
| HK40108064A (en) | Method for producing oligonucleic acid compound | |
| AU2022424485A1 (en) | Method for producing oligonucleic acid compound | |
| CN119039201A (en) | Synthesis method of (S) -1-tert-butoxycarbonyl-3-aminopyrrolidine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17731657 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 17731657 Country of ref document: EP Kind code of ref document: A1 |