WO2018135417A1 - 1,2,3,5,6-ペンタチエパンの製造方法 - Google Patents
1,2,3,5,6-ペンタチエパンの製造方法 Download PDFInfo
- Publication number
- WO2018135417A1 WO2018135417A1 PCT/JP2018/000737 JP2018000737W WO2018135417A1 WO 2018135417 A1 WO2018135417 A1 WO 2018135417A1 JP 2018000737 W JP2018000737 W JP 2018000737W WO 2018135417 A1 WO2018135417 A1 WO 2018135417A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- solvent
- tetrathiocarbonate
- ethanol
- toluene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D341/00—Heterocyclic compounds containing rings having three or more sulfur atoms as the only ring hetero atoms
Definitions
- the present invention relates to a method for producing high purity 1,2,3,5,6-pentathiepan.
- 1,2,3,5,6-pentathiepan (hereinafter sometimes referred to as “lenthionine”) is effective for optical materials (Patent Document 1) and medical applications (Patent Document 2), and is expected to be used in a wide range of applications. It is a compound.
- a method for synthesizing lenthionine for example, a method using dimethyl disulfide as a starting material is known (Non-patent Document 1). In this method, since an oily solution containing lenthionine is obtained after the reaction, it is necessary to purify using column chromatography, which is industrially disadvantageous. In addition, dimethyl disulfide, which is difficult to obtain industrially, must be used as a raw material.
- Non-patent Document 2 a method of reacting sodium sulfide as a starting material with diiodomethane or dibromomethane in an ethanol solvent is known (Non-patent Document 2). Even in this method, column chromatography is necessary for purification, which is industrially disadvantageous.
- An object of the present invention is to provide a method for easily producing high-purity lenthionine by suppressing the formation of insoluble polysulfide compounds.
- the present inventor has found that the above problem can be solved by reacting a reaction of tetrathiocarbonate with dihalogenated methane in a specific solvent. That is, the present invention is as follows.
- a method for producing 1,2,3,5,6-pentathiepan having Step A and Step B Step A: Step of synthesizing tetrathiocarbonate in a protic solvent
- Step B Reaction of tetrathiocarbonate with dihalogenated methane is carried out by mixing solvent (mass ratio of protic solvent to aprotic solvent is 13:87 to 38 : The process performed in 62)
- [2] The method for producing 1,2,3,5,6-pentathiepan according to [1], wherein the protic solvent contains an alcohol.
- [4] The method for producing 1,2,3,5,6-pentathiepan according to any one of [1] to [3], wherein the aprotic solvent contains an aromatic hydrocarbon.
- [5] The method for producing 1,2,3,5,6-pentathiepan according to any one of [1] to [3], wherein the aprotic solvent contains toluene.
- [6] The process for producing 1,2,3,5,6-pentathiepan according to any one of [1] to [5], wherein the tetrathiocarbonate is sodium tetrathiocarbonate.
- insoluble polysulfide compounds can be suppressed, and high-purity lenthionine can be easily produced.
- this high-purity lenthionine it can be suitably used for various applications such as improving the performance of optical materials.
- the method for producing lenthionine of the present invention has the following step A and step B.
- Step A Step of synthesizing tetrathiocarbonate in a protic solvent
- Step B Reaction of tetrathiocarbonate with dihalogenated methane is carried out by mixing solvent (mass ratio of protic solvent to aprotic solvent is 13:87 to 38 : 62) The process performed in the following. These A process and B process are demonstrated in detail hereafter.
- Step A Step of synthesizing tetrathiocarbonate in a protic solvent>
- the tetrathiocarbonate used in the present invention is a compound represented by M 2 CS 4 (M is a cationic species).
- M is a cationic species.
- Preferred examples include sodium tetrathiocarbonate, potassium tetrathiocarbonate, and lithium tetrathiocarbonate.
- Sodium tetrathiocarbonate represented by the following structural formula is more preferable because the raw materials are industrially easily available and the synthesis is simple. .
- the protic solvent in step A can be used as long as it is a solvent that can be mixed with the aprotic solvent used in step B.
- Specific examples include water, methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, t-butanol, n-pentanol, neopentanol, n-hexanol, n-heptanol, n-octanol, and n-nonanol.
- Methanol, ethanol and isopropanol are preferable because of high solubility of sulfides described later, and ethanol is particularly preferable. These can be used in combination of two or more.
- the amount of the protic solvent used in Step A is preferably in the range of 5 to 40 times by mass with respect to the sulfide, and more preferably in the range of 10 to 30 times by mass from the viewpoint of production efficiency and reactivity.
- Tetrathiocarbonate is obtained by reacting sulfide, carbon disulfide and sulfur in a protic solvent.
- the sulfide include sodium sulfide, potassium sulfide and lithium sulfide.
- sodium tetrathiocarbonate can be easily synthesized by reacting sodium sulfide and carbon disulfide in ethanol to synthesize disodium trithiocarbonate and adding sulfur thereto.
- the amount of carbon disulfide used is preferably in the range of 0.5 to 1.5 molar equivalents relative to the sulfide, and in the range of 0.8 to 1.2 molar equivalents because the side reaction can be suppressed. And more preferred.
- the amount of sulfur used is preferably in the range of 0.5 to 1.5 molar equivalents relative to the sulfide, and since it is possible to suppress the progress of side reactions, it is more preferable to be in the range of 0.8 to 1.2 molar equivalents. preferable.
- the reaction temperature is in the range of ⁇ 10 to 60 ° C., and preferably in the range of 20 to 40 ° C. from the viewpoint of reaction time and reaction yield.
- Step B Step of performing reaction between tetrathiocarbonate and dihalogenated methane in mixed solvent (mass ratio of protic solvent to aprotic solvent 13:87 to 38:62)> [Tetrathiocarbonate]
- the tetrathiocarbonate in Step B is obtained in Step A, and can be used sequentially from the Step A together with the reaction solution.
- the dihalogenated methane used in the present invention is dichloromethane, dibromomethane, diiodomethane, chlorobromomethane, chloroiodomethane, and bromoiodomethane. From the viewpoint of reactivity, dibromomethane and diiodomethane are preferable, and dibromomethane is particularly preferable. .
- the amount of dihalogenated methane used is preferably in the range of 0.5 to 1.5 molar equivalents relative to tetrathiocarbonate, and the progress of side reactions can be suppressed, so that it is in the range of 0.8 to 1.2 molar equivalents. More preferably.
- the protic solvent in Step B can be selected from the protic solvents used in Step A. Alternatively, the reaction can be carried out sequentially using the protic solvent in step A as it is.
- the amount of the protic solvent used in Step B is preferably in the range of 5 to 40 times by mass with respect to tetrathiocarbonate, and more preferably in the range of 10 to 30 times by mass from the viewpoint of production efficiency and reactivity.
- aprotic solvent examples include hydrocarbons, aromatic hydrocarbons, ethers, esters, nitriles, ketones, amides, and halogen-based solvents. Of these, hydrocarbons, aromatic hydrocarbons and ether solvents are preferred, cyclic compounds are more preferred, and benzene, toluene and tetrahydrofuran are preferred because the reaction yield of lenthionine is high and the solvent removal after the reaction is easy. Particularly preferred, toluene is most preferred from the viewpoint of yield. These can be used in combination of two or more.
- reaction of tetrathiocarbonate with dihalogenated methane The reaction between tetrathiocarbonate and dihalogenated methane is performed in a mixed solvent of a protic solvent and an aprotic solvent.
- the mass ratio of the protic solvent to the aprotic solvent is 13:87 to 38:62, and preferably 15:85 to 30:70. If there is too much protic solvent, an insoluble polysulfide compound is produced, and if there is too much aprotic solvent, the reaction does not proceed.
- the amount of the mixed solvent used is preferably in the range of 5 to 40 times by mass with respect to tetrathiocarbonate, and more preferably from 10 to 30 times by mass from the viewpoint of production efficiency and reactivity.
- the reaction temperature of tetrathiocarbonate and dihalogenated methane is preferably in the range of ⁇ 10 to 60 ° C., more preferably in the range of 20 to 40 ° C. When the temperature is low, the reaction is slow, and when the temperature is high, the side reaction tends to proceed.
- an aqueous acid solution is added to quench the reaction.
- Any acidic aqueous solution can be used as long as it is an acidic aqueous solution, but industrially inexpensive sulfuric acid, hydrochloric acid, nitric acid and phosphoric acid can be suitably used.
- the ethanol solution of sodium tetrathiocarbonate prepared previously was dropped into a toluene solution of dibromomethane, and the reaction was carried out at 35 ° C.
- the production yield of lenthionine was 33 mol% after 3 hours and 43 mol% after 20 hours.
- 100 g of 1N sulfuric acid aqueous solution was added to quench. By this operation, the aqueous layer and the organic layer were separated into two layers. The organic layer was washed 3 times with 100 ml of ion exchange water. No polysulfide compound was confirmed in each step.
- Diethanolmethane (13.2 g, 76 mmol) was diluted by adding 18 g of ethanol.
- An ethanol solution of dibromomethane was added dropwise to the ethanol solution of sodium tetrathiocarbonate prepared previously, and the reaction was performed at 35 ° C.
- the production yield of lenthionine was 5 mol% after 3 hours and 4 mol% after 20 hours.
- 100 g of 1N sulfuric acid aqueous solution was added to quench, and washing was performed 3 times with 100 ml of ion-exchanged water.
- the residue was a yellow oily compound, and as a result of adding 100 g of toluene, a large amount of insoluble components were generated.
- the ethanol solution of sodium tetrathiocarbonate prepared above was added dropwise to the ethanol solution of dibromomethane and reacted at 35 ° C.
- the yield of lenthionine was 23 mol% after 3 hours and 23 mol% after 20 hours.
- Met. After 20 hours, 100 g of 1N sulfuric acid aqueous solution was added for quenching, 100 g of toluene was added for extraction, and washing was performed 3 times with 100 ml of ion-exchanged water. After concentrating the toluene solution to 12 g, the same crystallization operation as in Example 6 was performed. As a result, 1.1 g of crude crystals (purity 95%) was obtained, but 2.3 g of polysulfide compound was present in the crude crystals. It was contained and a high purity product could not be obtained by crystallization.
- the production yield of lenthionine was 19 mol% after 3 hours and 13 mol% after 20 hours.
- the polysulfide compound was contained in the crude crystals, and a high-purity product could not be obtained by crystallization.
- the polysulfide compound was 4.0 g.
- the production yield of lenthionine was 33 mol% after 3 hours and 22 mol% after 20 hours.
- the polysulfide compound was contained in the crude crystals, and a high-purity product could not be obtained by crystallization.
- the polysulfide compound was 2.8 g.
- the production yield of lenthionine was 41 mol% after 3 hours and 33 mol% after 20 hours.
- the polysulfide compound was contained in the crude crystals, and a high-purity product could not be obtained by crystallization.
- the polysulfide compound was 0.5 g.
- the yield of lenthionine was 5 mol% after 3 hours and 15 mol% after 20 hours. After 20 hours, the same operation as in Comparative Example 1 was performed. As a result, no polysulfide compound insoluble in toluene was confirmed. A crystallization operation similar to that of Example 6 was performed, but since the yield was low, a high-purity product could not be obtained by crystallization.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
レンチオニンの合成方法としては、例えばジメチルジスルフィドを出発原料とした方法が知られている(非特許文献1)。この方法では、反応後にレンチオニンを含むオイル状の溶液となるため、カラムクロマトグラフィーを用いて精製する必要があり工業的に不利である。また、工業的に入手しにくいジメチルジスルフィドを原料に使用しなければならない。
また、硫化ナトリウムを出発原料に、エタノール溶媒中でジヨードメタン、ジブロモメタンと反応させる方法が知られている(非特許文献2)。この方法でも、精製にはカラムクロマトグラフィーが必要であり、工業的に不利である。
反応系内に不溶性のポリスルフィド化合物が多量に生成すると、洗浄が容易でなく工業化が困難になるだけでなく、最終的にレンチオニンに混入するため、高純度のレンチオニンを得ることが困難になる。このポリスルフィドの生成が公知のレンチオニン合成における精製を困難にしている。
すなわち、本発明は以下の通りである。
A工程:テトラチオ炭酸塩をプロトン性溶媒中で合成する工程
B工程:テトラチオ炭酸塩とジハロゲン化メタンとの反応を、混合溶媒(プロトン性溶媒と非プロトン性溶媒の質量比が13:87~38:62)中で行う工程
[2] 前記プロトン性溶媒がアルコールを含有する、[1]に記載の1,2,3,5,6-ペンタチエパンの製造方法。
[3] 前記プロトン性溶媒がエタノールを含有する、[1]に記載の1,2,3,5,6-ペンタチエパンの製造方法。
[4] 前記非プロトン性溶媒が芳香族炭化水素を含有する、[1]~[3]のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
[5] 前記非プロトン性溶媒がトルエンを含有する、[1]~[3]のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
[6] 前記テトラチオ炭酸塩がテトラチオ炭酸ナトリウムである、[1]~[5]のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
[7] 前記ジハロゲン化メタンがジブロモメタン又はジヨードメタンを含有する、[1]~[6]のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
[8] 前記A工程及びB工程を逐次的に行う、[1]~[7]のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
A工程:テトラチオ炭酸塩をプロトン性溶媒中で合成する工程
B工程:テトラチオ炭酸塩とジハロゲン化メタンとの反応を、混合溶媒(プロトン性溶媒と非プロトン性溶媒の質量比が13:87~38:62)中で行う工程
以下、これらA工程、B工程について詳細に説明する。
[テトラチオ炭酸塩]
本発明で使用されるテトラチオ炭酸塩は、M2CS4(Mはカチオン種)で表される化合物である。好ましい具体例として、テトラチオ炭酸ナトリウム、テトラチオ炭酸カリウム及びテトラチオ炭酸リチウムが挙げられ、原料が工業的に入手しやすく、また合成が簡便である理由から下記構造式で表されるテトラチオ炭酸ナトリウムがより好ましい。

A工程におけるプロトン性溶媒は、B工程で使用される非プロトン性溶媒と混合可能な溶媒であれば使用可能である。
具体例として水、メタノール、エタノール、n-プロパノール、イソプロパノール、n-ブタノール、イソブタノール、t-ブタノール、n-ペンタノール、ネオペンタノール、n-ヘキサノール、n-ヘプタノール、n-オクタノール、n-ノナノール、n-デカノール、エチレングリコール及びプロピレングリコールが挙げられ、メタノール、エタノール及びイソプロパノールが後述する硫化塩の溶解性が高いため好ましく、エタノールが特に好ましい。これらは2種以上を組み合わせて用いることができる。
A工程におけるプロトン性溶媒の使用量は、硫化塩に対して5~40質量倍の範囲が好ましく、生産効率と反応性の観点から10~30質量倍の範囲にあるとより好ましい。
テトラチオ炭酸塩は硫化塩、二硫化炭素及び硫黄をプロトン性溶媒中で反応させることで得られる。硫化塩としては、具体例として硫化ナトリウム、硫化カリウム及び硫化リチウムが挙げられる。
例えば、硫化ナトリウム及び二硫化炭素をエタノール中で反応させて、トリチオ炭酸ジナトリウムを合成し、これに硫黄を添加することで容易にテトラチオ炭酸ナトリウムが合成可能である。
二硫化炭素の使用量は、硫化塩に対して0.5~1.5モル当量の範囲が好ましく、副反応の進行を抑えることができるため0.8~1.2モル当量の範囲にあるとより好ましい。
硫黄の使用量は、硫化塩に対して0.5~1.5モル当量の範囲が好ましく、副反応の進行を抑えることができるため0.8~1.2モル当量の範囲にあるとより好ましい。
反応温度は-10~60℃の範囲であり、反応時間と反応収率観点から20~40℃の範囲にあると好ましい。
[テトラチオ炭酸塩]
B工程におけるテトラチオ炭酸塩はA工程で得られるものであり、A工程から逐次的に反応液ごと用いることもできる。
本発明で使用されるジハロゲン化メタンとは、ジクロロメタン、ジブロモメタン、ジヨードメタン、クロロブロモメタン、クロロヨードメタン及びブロモヨードメタンであり、反応性の観点からジブロモメタン及びジヨードメタンが好ましく、ジブロモメタンが特に好ましい。
ジハロゲン化メタンの使用量は、テトラチオ炭酸塩に対して0.5~1.5モル当量の範囲が好ましく、副反応の進行を抑えることができるため0.8~1.2モル当量の範囲にあるとより好ましい。
B工程におけるプロトン性溶媒は、A工程で用いられるプロトン性溶媒から選択することができる。また、A工程のプロトン性溶媒をそのまま用いて逐次的に反応を行うこともできる。
B工程におけるプロトン性溶媒の使用量は、テトラチオ炭酸塩に対して5~40質量倍の範囲が好ましく、生産効率と反応性の観点からから10~30質量倍の範囲にあるとより好ましい。
非プロトン性溶媒は、炭化水素、芳香族炭化水素、エーテル、エステル、ニトリル、ケトン、アミド及びハロゲン系溶媒が挙げられる。これらのうち、レンチオニンの反応収率が高く、かつ反応終了後の溶媒除去が容易であるため、炭化水素、芳香族炭化水素及びエーテル溶媒が好ましく、環状化合物が更に好ましく、ベンゼン、トルエン及びテトラヒドロフランが特に好ましく、収率の観点からトルエンが最も好ましい。これらは2種以上を組み合わせて用いることができる。
テトラチオ炭酸塩とジハロゲン化メタンとの反応は、プロトン性溶媒と非プロトン性溶媒の混合溶媒中で行う。プロトン性溶媒と非プロトン性溶媒との質量比は13:87~38:62であり、15:85~30:70が好ましい。プロトン性溶媒が多すぎると不溶性のポリスルフィド化合物が生成し、非プロトン性溶媒が多すぎると反応が進行しない。
混合溶媒の使用量は、テトラチオ炭酸塩に対して5~40質量倍の範囲が好ましく、生産効率と反応性の観点から10~30質量倍であることがより好ましい。
分析は液体クロマトグラフを使用し、ODSカラム(カラム:一般財団法人科学物質評価研究機構VP-ODS、カラムサイズ4.6φ×150mm)を使用した。
RI検出器を用いて原料の臭化メチレンのモル比を基準としたレンチオニンの生成収率を算出した。
[液体クロマトグラフ条件]
オーブン温度:40℃
溶離液:アセトニトリル/蒸留水(容積比)=50/50
溶液調製:サンプル5mgを、0.1%ギ酸溶液(アセトニトリル溶媒)10mlで希釈し分析試料とした。
レンチオニンはトルエンに易溶であり、ポリスルフィド化合物はトルエンに不溶である。そのため、クエンチ後に系内に確認された固体をろ過により回収し、トルエン50mlを添加し、更に水洗を実施後、トルエン中の不溶物をろ過で回収して乾燥した後、質量測定を行った。
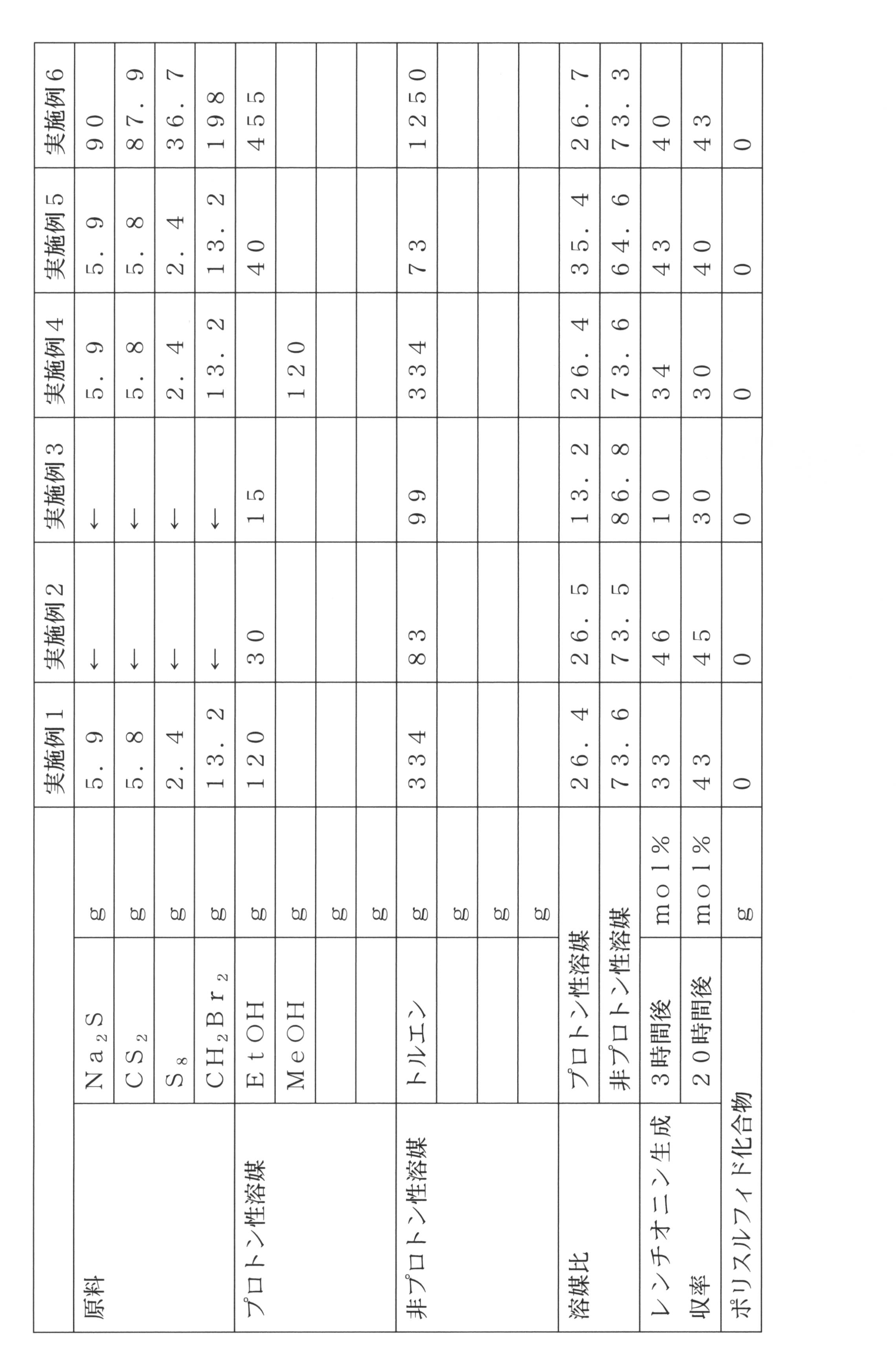
[溶媒:エタノール:トルエン=26.4:73.6]
硫化ナトリウム5.9g(76mmol)をエタノール120gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で20分反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行うことで、テトラチオ炭酸ナトリウムのエタノール溶液を調製した。
次に、ジブロモメタン13.2g(76mmol)にトルエン334gを添加して希釈した。先に調製したテトラチオ炭酸ナトリウムのエタノール溶液をジブロモメタンのトルエン溶液に滴下し、35℃で反応を行った。
レンチオニンの生成収率は3時間経過後で33mol%、20時間経過後で43mol%であった。20時間経過後、1N硫酸水溶液100gを添加しクエンチを行った。この操作で水層と有機層の2層に分離した。有機層を100mlのイオン交換水で3回洗浄を行った。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:エタノール:トルエン=26.5:73.5]
硫化ナトリウム5.9g(76mmol)にエタノール30gを添加し、二硫化炭素5.8g(76mmol)を滴下して、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。得られた黄色のスラリー溶液にトルエン83gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。
レンチオニンの生成収率は3時間経過後で46mol%、20時間経過後で45mol%であった。20時間経過後、実施例1と同様の操作を行った。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:エタノール:トルエン=13.2:86.8]
使用した溶媒をエタノールが15g、トルエンを99gとした以外は実施例2と同様の条件で反応を行った。
レンチオニンの生成収率は3時間経過後で10mol%、20時間経過後で30mol%であった。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:メタノール:トルエン=26.4:73.6]
使用した溶媒をメタノール120g、トルエン334gとした以外は実施例1と同様の条件で反応を行った。
レンチオニンの生成収率は3時間経過後で34mol%、20時間経過後で30mol%であった。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:エタノール:トルエン=35.4:64.6]
硫化ナトリウム5.9g(76mmol)にエタノール40gを添加し、二硫化炭素5.8g(76mmol)を滴下して、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。得られた黄色のスラリー溶液にトルエン73gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。
レンチオニンの生成収率は3時間経過後で43mol%、20時間経過後で40mol%であった。20時間経過後、実施例1と同様の操作を行った。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:エタノール:トルエン=26.7:73.3]
硫化ナトリウム90g(1.15mol)にエタノール455gを添加し、30℃で1時間撹拌した。更に二硫化炭素87.9g(1.15mol)を添加し、35℃で1時間反応を行った。得られた反応液に硫黄36.7g(1.14mol)を添加し、更に35℃で1時間反応を行った。得られた黄色のスラリー溶液にトルエン1250gを添加した後、ジブロモメタン198g(1.15mmol)を滴下し、35℃で20時間反応を行った。
レンチオニンの生成収率は3時間経過後で40mol%、20時間経過後で43mol%であった。20時間経過後、1N硫酸水溶液750gを添加しクエンチを行い、水層を除去し更にイオン交換水350mlで2回洗浄を行った。トルエン層を分液し、エバポレーターを用いて30~40torr、40℃の条件で100mlになるまで濃縮を行った。撹拌しながら0~-1℃まで冷却し晶析操作を行った。得られた固体をろ過し、乾燥することで純度99%のレンチオニン31.9g(単離収率29.4mol%)を取得した。各工程においてポリスルフィド化合物は確認されなかった。
[溶媒:エタノール100%条件]
中国文献(中国調味品(CHINA CONDIMENT)2005年9月、No9、p25)に従い、レンチオニンの合成を行った。
即ち、硫化ナトリウム5.9g(76mmol)をエタノール67gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で20分反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行うことで、テトラチオ炭酸ナトリウムのエタノール溶液を調製した。
ジブロモメタン13.2g(76mmol)にエタノール18gを添加して希釈した。先に調製したテトラチオ炭酸ナトリウムのエタノール溶液にジブロモメタンのエタノール溶液を滴下し、35℃で反応を行った。
レンチオニンの生成収率は3時間経過後で5mol%、20時間経過後で4mol%であった。20時間経過後、1N硫酸水溶液100gを添加しクエンチを行い、100mlのイオン交換水で3回洗浄を行った。残差物は黄色のオイル状化合物であり、トルエン100gを添加した結果、不溶性の成分が多量に発生した。質量を測定した結果、2.1gであった。IR測定によりポリスルフィド化合物であることを確認した。
実施例6と同様の晶析操作を実施した結果、粗結晶中にポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。
[溶媒:エタノール100%条件]
硫化ナトリウム5.9g(76mmol)をエタノール120gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で20分反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行うことで、テトラチオ炭酸ナトリウムのエタノール溶液を調製した。
ジブロモメタン13.2g(76mmol)にエタノール302gを添加し希釈した。先に調製したテトラチオ炭酸ナトリウムのエタノール溶液をジブロモメタンのエタノール溶液に滴下し、35℃で反応を行った結果、レンチオニンの生成収率は3時間経過後で23mol%、20時間経過後で23mol%であった。20時間経過後、1N硫酸水溶液100gを添加しクエンチを行い、トルエン100gを添加して抽出し、100mlのイオン交換水で3回洗浄を行った。
トルエン溶液を12gまで濃縮後、実施例6と同様の晶析操作を実施した結果、粗結晶1.1g(純度95%)を得られたが、粗結晶中には2.3gのポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。
[溶媒:メタノール100%条件]
使用した溶媒をエタノールからメタノールに変えた以外は比較例2と同様の操作を行った。レンチオニンの生成収率は3時間経過後で17mol%、20時間経過後で9mol%であった。実施例6と同様の晶析操作を実施した結果、粗結晶中にポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。ポリスルフィド化合物は1.6gであった。
[溶媒:トルエン100%条件]
テトラチオ炭酸ナトリウムのエタノール溶液を比較例2と同様の手法で調合し、溶媒であるエタノールをエバポレーターで除去した。トルエン120gを添加し、テトラチオ炭酸ナトリウムの懸濁液を調合した。ジブロモメタン13.2g(76mmol)をトルエン334gで希釈し、テトラチオ炭酸ナトリウムのトルエン溶液に添加して35℃で反応を行った。3時間後および20時間後でレンチオニンの生成は確認されなかった。テトラチオ炭酸ナトリウムがトルエンに溶解しないため反応が進行しなかったものと考えられる。
[溶媒:エタノール:トルエン=67.4:32.6]
硫化ナトリウム5.9g(76mmol)をエタノール87gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。この溶液にトルエン42gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。レンチオニンの生成収率は3時間経過後で19mol%、20時間経過後で13mol%であった。実施例6と同様の晶析操作を実施した結果、粗結晶中にポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。ポリスルフィド化合物は4.0gであった。
[溶媒:エタノール:トルエン=50:50]
硫化ナトリウム5.9g(76mmol)をエタノール57gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。この溶液にトルエン57gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。レンチオニンの生成収率は3時間経過後で33mol%、20時間経過後で22mol%であった。実施例6と同様の晶析操作を実施した結果、粗結晶中にポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。ポリスルフィド化合物は2.8gであった。
[溶媒:エタノール:トルエン=40:60]
硫化ナトリウム5.9g(76mmol)をエタノール48gに溶解し、二硫化炭素5.8g(76mmol)を添加し、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。この溶液にトルエン72gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。レンチオニンの生成収率は3時間経過後で41mol%、20時間経過後で33mol%であった。実施例6と同様の晶析操作を実施した結果、粗結晶中にポリスルフィド化合物が含まれており、晶析により高純度品を得ることはできなかった。ポリスルフィド化合物は0.5gであった。
[溶媒:エタノール:トルエン=6:94]
硫化ナトリウム5.9g(76mmol)にエタノール7gを添加し、二硫化炭素5.8g(76mmol)を滴下して、35℃で1時間反応を行った。得られた反応液に硫黄2.4g(76mmol)を添加し、更に35℃で1時間反応を行った。反応後の黄色のスラリー溶液にトルエン109gを添加し、ジブロモメタン13.2g(76mmol)を滴下して35℃で反応を行った。レンチオニンの収率は3時間経過後で5mol%、20時間経過後で15mol%であった。20時間経過後、比較例1と同様の操作を実施した結果、トルエンに不溶のポリスルフィド化合物は全く確認されなかった。実施例6と同様の晶析操作を実施したが、収率が低かったため晶析により高純度品を得ることはできなかった。

Claims (8)
- A工程及びB工程を有する1,2,3,5,6-ペンタチエパンの製造方法。
A工程:テトラチオ炭酸塩をプロトン性溶媒中で合成する工程
B工程:テトラチオ炭酸塩とジハロゲン化メタンとの反応を、混合溶媒(プロトン性溶媒と非プロトン性溶媒の質量比が13:87~38:62)中で行う工程 - 前記プロトン性溶媒がアルコールを含有する、請求項1に記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記プロトン性溶媒がエタノールを含有する、請求項1に記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記非プロトン性溶媒が芳香族炭化水素を含有する、請求項1~3のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記非プロトン性溶媒がトルエンを含有する、請求項1~3のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記テトラチオ炭酸塩がテトラチオ炭酸ナトリウムである、請求項1~5のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記ジハロゲン化メタンがジブロモメタン又はジヨードメタンを含有する、請求項1~6のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
- 前記A工程及びB工程を逐次的に行う、請求項1~7のいずれかに記載の1,2,3,5,6-ペンタチエパンの製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18741370.3A EP3398943B1 (en) | 2017-01-23 | 2018-01-15 | Method for producing 1,2,3,5,6-pentathiepane |
| JP2018522701A JP6399257B1 (ja) | 2017-01-23 | 2018-01-15 | 1,2,3,5,6−ペンタチエパンの製造方法 |
| CN201880000898.9A CN108633276B (zh) | 2017-01-23 | 2018-01-15 | 1,2,3,5,6-五硫杂环庚烷的制造方法 |
| KR1020187022100A KR101942059B1 (ko) | 2017-01-23 | 2018-01-15 | 1,2,3,5,6-펜타티에판의 제조방법 |
| US16/073,609 US10472343B2 (en) | 2017-01-23 | 2018-01-15 | Method for producing 1,2,3,5,6-pentathiepane |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017009257 | 2017-01-23 | ||
| JP2017-009257 | 2017-01-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018135417A1 true WO2018135417A1 (ja) | 2018-07-26 |
Family
ID=62908645
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/000737 Ceased WO2018135417A1 (ja) | 2017-01-23 | 2018-01-15 | 1,2,3,5,6-ペンタチエパンの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US10472343B2 (ja) |
| EP (1) | EP3398943B1 (ja) |
| JP (1) | JP6399257B1 (ja) |
| KR (1) | KR101942059B1 (ja) |
| CN (1) | CN108633276B (ja) |
| TW (1) | TWI656119B (ja) |
| WO (1) | WO2018135417A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019181484A1 (ja) * | 2018-03-22 | 2019-09-26 | 三菱瓦斯化学株式会社 | 1,2,3,5,6-ペンタチエパンの製造方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112322569B (zh) * | 2020-09-29 | 2022-08-05 | 南京农业大学 | 干旱诱导香菇体内风味物质香菇精合成的方法 |
| CN119039269B (zh) * | 2024-09-03 | 2025-09-19 | 江苏视科新材料股份有限公司 | 一种高折射率硫醚型环硫化合物及其制备方法和应用 |
| CN119661497A (zh) * | 2024-12-13 | 2025-03-21 | 中山大学·深圳 | 一种环状硫化物的制备方法及环状硫化物的应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3503758A (en) * | 1965-10-14 | 1970-03-31 | Takeda Chemical Industries Ltd | Flavoring composition |
| JP2002040201A (ja) * | 2000-07-21 | 2002-02-06 | Mitsubishi Gas Chem Co Inc | 光学材料用組成物 |
| JP2002293783A (ja) * | 2000-12-08 | 2002-10-09 | Yokohama Rubber Co Ltd:The | 環状ポリスルフィド化合物の製造方法及びそれを含むゴム組成物 |
| WO2005034974A1 (ja) | 2003-10-09 | 2005-04-21 | Nihon University | 血栓症の予防または改善のための健康食品及び血栓症の予防または治療用医薬組成物 |
| CN102260240A (zh) * | 2011-08-08 | 2011-11-30 | 天津市化学试剂研究所 | 食用香料1,2,3,5,6-五硫环庚烷的合成方法 |
| JP2017165950A (ja) * | 2016-03-10 | 2017-09-21 | 三菱瓦斯化学株式会社 | 光学材料用高純度1,2,3,5,6−ペンタチエパン、およびその精製方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1292664B (de) * | 1965-10-13 | 1969-04-17 | Takeda Chemical Industries Ltd | 1, 2, 3, 5, 6-Pentathiacycloheptan, Verfahren zu dessen Herstellung und Verwendung |
| US6984711B2 (en) | 2000-12-08 | 2006-01-10 | The Yokohama Rubber Co., Ltd. | Method for production of cyclic polysulfide compound and rubber composition containing the same |
| CN101914087A (zh) | 2010-07-17 | 2010-12-15 | 天津市化学试剂研究所 | 食用香料香菇精的合成方法 |
| CN101897419A (zh) | 2010-07-17 | 2010-12-01 | 天津市化学试剂研究所 | 一种食用香料香菇精 |
| CN103141799A (zh) * | 2013-03-28 | 2013-06-12 | 天津春发生物科技集团有限公司 | 一种香菇香精及其制备方法 |
-
2018
- 2018-01-15 CN CN201880000898.9A patent/CN108633276B/zh active Active
- 2018-01-15 US US16/073,609 patent/US10472343B2/en active Active
- 2018-01-15 JP JP2018522701A patent/JP6399257B1/ja not_active Expired - Fee Related
- 2018-01-15 EP EP18741370.3A patent/EP3398943B1/en active Active
- 2018-01-15 WO PCT/JP2018/000737 patent/WO2018135417A1/ja not_active Ceased
- 2018-01-15 KR KR1020187022100A patent/KR101942059B1/ko active Active
- 2018-01-19 TW TW107101985A patent/TWI656119B/zh active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3503758A (en) * | 1965-10-14 | 1970-03-31 | Takeda Chemical Industries Ltd | Flavoring composition |
| JP2002040201A (ja) * | 2000-07-21 | 2002-02-06 | Mitsubishi Gas Chem Co Inc | 光学材料用組成物 |
| JP4573148B2 (ja) | 2000-07-21 | 2010-11-04 | 三菱瓦斯化学株式会社 | 光学材料用組成物 |
| JP2002293783A (ja) * | 2000-12-08 | 2002-10-09 | Yokohama Rubber Co Ltd:The | 環状ポリスルフィド化合物の製造方法及びそれを含むゴム組成物 |
| WO2005034974A1 (ja) | 2003-10-09 | 2005-04-21 | Nihon University | 血栓症の予防または改善のための健康食品及び血栓症の予防または治療用医薬組成物 |
| CN102260240A (zh) * | 2011-08-08 | 2011-11-30 | 天津市化学试剂研究所 | 食用香料1,2,3,5,6-五硫环庚烷的合成方法 |
| JP2017165950A (ja) * | 2016-03-10 | 2017-09-21 | 三菱瓦斯化学株式会社 | 光学材料用高純度1,2,3,5,6−ペンタチエパン、およびその精製方法 |
Non-Patent Citations (6)
| Title |
|---|
| CHINA CONDIMENT, September 2005 (2005-09-01), pages 25 |
| HANSEN, H. C. ET AL.: "Synthesis, structure, and reactions of thiocarbonic acid derivatives new pentathiodipercarbonates, (RSS)"2 C@?S, @a,@a,@a-tris(disulfides), and the first @a,@a,@a-tris(trisulfide)", TETRAHEDRON, vol. 41, no. 22, 1985, pages 5145 - 58, XP026645726 * |
| LIU, H. ET AL.: "Improvement of synthesis of lenthionine", SPECIALITY PETROCHEMICALS, 2005, pages 21 - 22, XP009508983, ISSN: 1003-9384 * |
| LIU, H. ET AL.: "Synthesis and thermal analysis of lenthionine", CHINA CONDIMENT, vol. 9, 2005, pages 25 - 27, 24, XP009509221, ISSN: 1000-9973 * |
| SPECIALITY PETROCHEMICALS, 2005, pages 22 |
| TETRAHEDRON, LETT 1981-22 1939 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019181484A1 (ja) * | 2018-03-22 | 2019-09-26 | 三菱瓦斯化学株式会社 | 1,2,3,5,6-ペンタチエパンの製造方法 |
| JPWO2019181484A1 (ja) * | 2018-03-22 | 2021-04-08 | 三菱瓦斯化学株式会社 | 1,2,3,5,6−ペンタチエパンの製造方法 |
| JP7226430B2 (ja) | 2018-03-22 | 2023-02-21 | 三菱瓦斯化学株式会社 | 1,2,3,5,6-ペンタチエパンの製造方法 |
| US11919877B2 (en) | 2018-03-22 | 2024-03-05 | Mitsubishi Gas Chemical Company, Inc. | Production method for 1,2,3,5,6-pentathiepane |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3398943A1 (en) | 2018-11-07 |
| EP3398943A4 (en) | 2018-12-26 |
| US10472343B2 (en) | 2019-11-12 |
| KR101942059B1 (ko) | 2019-01-24 |
| TW201838980A (zh) | 2018-11-01 |
| EP3398943B1 (en) | 2020-03-18 |
| CN108633276A (zh) | 2018-10-09 |
| KR20180090901A (ko) | 2018-08-13 |
| US20190040035A1 (en) | 2019-02-07 |
| CN108633276B (zh) | 2019-05-31 |
| JP6399257B1 (ja) | 2018-10-03 |
| JPWO2018135417A1 (ja) | 2019-01-24 |
| TWI656119B (zh) | 2019-04-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6399257B1 (ja) | 1,2,3,5,6−ペンタチエパンの製造方法 | |
| CN118772081B (zh) | 一种制备n-甲基-2,3-二氢苯并噻嗪-4-酮的方法 | |
| WO2015064712A1 (ja) | メチレンジスルホン酸化合物の製造方法 | |
| JP4901174B2 (ja) | 2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニルの製造方法 | |
| KR102513651B1 (ko) | 에테르 화합물 제조 방법 | |
| US8304580B2 (en) | Method for producing tris(perfluoroalkanesulfonyl)methide acid salt | |
| CN116410218B (zh) | 一种单氟烯基硅的合成方法 | |
| CN108456193B (zh) | 1,2,3,5,6-五硫杂环庚烷的制造方法 | |
| JP4738345B2 (ja) | 2,2’−ビス(トリフルオロメチル)−4,4’−ジアミノビフェニルの製造方法 | |
| JP2014156493A (ja) | テトラヒドロピラン化合物の製造中間体 | |
| JP7226430B2 (ja) | 1,2,3,5,6-ペンタチエパンの製造方法 | |
| CN109721565B (zh) | 一种重要的氟中间体合成工艺 | |
| JP7619730B2 (ja) | アルキルシリルオキシ置換ベンジルアミン化合物の製造方法 | |
| CN116102551B (zh) | 一种吡啶稠合的喹啉酮类化合物的制备方法 | |
| JP4258606B2 (ja) | 含リン有機ゼオライト類縁体及びその製造法 | |
| JP5940418B2 (ja) | 3−ハロゲン化アニリンの製造方法 | |
| JP2014122182A (ja) | 3−ハロゲン化アニリンの製造方法 | |
| WO2015064711A1 (ja) | メチレンジスルホニルクロライド化合物の製造方法 | |
| JP2007055928A (ja) | マレオニトリル類の製造方法 | |
| CN101096330A (zh) | 从1,4-二卤代-1,4-二(三甲基硅基)-1,3-丁二烯衍生物制备1,1,4,4-四卤代-1,3-丁二烯衍生物的方法 | |
| KR20110041306A (ko) | 아릴케톤의 α-할로겐화 방법 | |
| JP2011105672A (ja) | 2−ヒドロキシ−6−ビニルナフタレンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018522701 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187022100 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018741370 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2018741370 Country of ref document: EP Effective date: 20180802 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18741370 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |