WO2018182014A1 - 腫瘍溶解性ウイルスの増殖方法及び抗腫瘍剤 - Google Patents
腫瘍溶解性ウイルスの増殖方法及び抗腫瘍剤 Download PDFInfo
- Publication number
- WO2018182014A1 WO2018182014A1 PCT/JP2018/013974 JP2018013974W WO2018182014A1 WO 2018182014 A1 WO2018182014 A1 WO 2018182014A1 JP 2018013974 W JP2018013974 W JP 2018013974W WO 2018182014 A1 WO2018182014 A1 WO 2018182014A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxaliplatin
- type
- virus
- antitumor
- cva11
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images










Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
- A61K35/768—Oncolytic viruses not provided for in groups A61K35/761 - A61K35/766
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/28—Compounds containing heavy metals
- A61K31/282—Platinum compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/555—Heterocyclic compounds containing heavy metals, e.g. hemin, hematin, melarsoprol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2750/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssDNA viruses
- C12N2750/00011—Details
- C12N2750/14011—Parvoviridae
- C12N2750/14111—Dependovirus, e.g. adenoassociated viruses
- C12N2750/14132—Use of virus as therapeutic agent, other than vaccine, e.g. as cytolytic agent
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/32011—Picornaviridae
- C12N2770/32311—Enterovirus
- C12N2770/32332—Use of virus as therapeutic agent, other than vaccine, e.g. as cytolytic agent
Definitions
- the present invention relates to a novel antitumor therapy using an oncolytic virus.
- Malignant tumor is the first cause of Japanese death, and statistically one in three people die from malignant tumors. Through years of efforts, advances in surgical treatment for malignant tumors, radiation therapy, and chemotherapy including molecular targeted drugs have been remarkable, and treatment results have improved. However, the mortality rate due to malignant tumor is still high, and a new treatment method effective for malignant tumor is desired.
- oncolytic virus therapy has attracted attention because it has a direct cytocidal effect.
- oncolytic adenoviruses and herpes simplex viruses which are DNA viruses, have been conducted on brain tumors and breast cancer, and results suggesting safety and effectiveness have been reported.
- RNA virus Picornaviridae enterovirus
- the RNA virus has no integration into the host cell genome after infection and has a low risk of canceration due to genetic mutation.
- the growth rate in the cell is fast, a rapid and high antitumor effect can be expected.
- enterovirus coxsackievirus (CV) A21 type, echovirus (EV) type 6, EV7 type, EV11 type, EV12 type, EV13 type, EV29 type Patent Document 1
- CVA13 type Oncolytic virus therapy using CVA15 type, CVA18 type, CVA21 type, EV1 type, EV7 type, EV8 type, EV22 type Patent Document 2 has been reported.
- Coxsackievirus A11 type referred to as “CVA11 type”
- Echovirus type 4 EV4 type
- the present invention relates to providing an antitumor therapy using an oncolytic virus that exhibits an excellent antitumor effect and has few side effects.
- the present inventor has promoted the growth of the virus by using a specific anticancer agent in combination with Coxsackie virus and the like, and the antitumor caused by the virus without enhancing side effects It has been found that the effect is remarkably enhanced.
- the present invention includes the following 1) to 18).
- An antitumor agent comprising a combination of an oncolytic virus and an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite.
- the antitumor agent according to 1) wherein the oncolytic virus is Coxsackie virus or adenovirus.
- the antitumor agent according to 2) wherein the Coxsackie virus is A11 type or B3 type.
- An antitumor agent comprising an oncolytic virus and an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolite agents.
- the antitumor agent according to any one of 6) to 8), wherein the plant alkaloid anticancer agent comprises one or more selected from SN-38, irinotecan and salts thereof.
- any one of 1) to 5 which is a kit comprising a drug comprising an oncolytic virus and a drug comprising an anticancer agent selected from oxaliplatin and plant alkaloid anticancer agents.
- the antitumor agent as described in. 12 An antitumor effect enhancer for an oncolytic virus comprising as an active ingredient an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolites.
- a method for promoting the growth of an oncolytic virus by culturing an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite together with an oncolytic virus.
- a virus receptor expression enhancer for cancer cells comprising as an active ingredient an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolites. 16) The virus receptor expression enhancer according to 15), wherein the virus receptor is DAF and / or ICAM-1. 17) Use of an oncolytic virus and an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolite agents for producing an antitumor agent.
- an antitumor therapy that exhibits an excellent antitumor effect and is highly safe for humans.
- A The figure at the time of oxaliplatin and CVA11 combined use.
- B The figure at the time of cisplatin and CVA11 combined use. The figure which shows that the proliferation of CVB3 type
- An oncolytic virus is a virus that infects cancer cells to lyse and kill the cancer cells.
- the oncolytic virus of the present invention is not particularly limited as long as it is a virus that lyses cancer cells and causes cell death.
- enteroviruses such as CVA11 type, CVB3 type (Coxsackie virus), EV4 type (echovirus), adeno AAV which is a virus, HF10 which is a mutant of a simple perpes virus, and the like are mentioned, and CVA11 type, CVB3 type and AAV are particularly preferable.
- CVA11 type and CVB3 type are Coxsackie virus, which is a kind of enteric virus (Enterovirus) belonging to the Picornaviridae family.
- Coxsackie viruses are divided into two groups, A and B. Group A is further divided into type 24 and group B is divided into type 6.
- the CVA11 type of the present invention is a group A 11 type coxsackie virus
- the CVB3 type is a group B type 3 coxsackie virus.
- Oncolytic viruses can infect cells by binding to viral receptors on the cell surface. Examples of the virus receptor include decay accelerating factor (DAF or CD55), intercellular adhesion molecule-1 (ICAM-1 or CD54), integrin ⁇ 2 ⁇ 1 (CD49b), and the like. The oncolytic virus interacts with the viral receptor to destabilize the capsid, which induces the oncolytic virus shed.
- DAF decay accelerating factor
- IAM-1 intercellular adhesion molecule-1
- CD49b integrin ⁇ 2 ⁇ 1
- the oncolytic virus can be isolated from a specimen or the like by a known virus isolation method such as a centrifugation method or a virus propagation method using cultured cells.
- the oncolytic virus of the present invention can be selected from organisms by culturing naturally occurring viruses in cell lines over many passages so as to acquire high infectivity to cancer cells. Good.
- Cell lines suitable for biological selection are preferably those having viral receptors such as DAF, ICAM-1, integrin ⁇ 2 ⁇ 1 , HEK293 cells, H1299 cells, A549 cells, LK-87 cells, PC-9 cells, H460 cell etc. are mentioned.
- the oncolytic virus of the present invention may be naturally occurring, modified, or partially mutated.
- a vector virus may be used.
- CVA11 type variants include those from which the capsid has been removed.
- the capsid can be removed, for example, by treatment with a protease such as chymotrypsin or trypsin.
- the capsid can be removed by treating CVA11 type with chymotrypsin in the presence of a surfactant such as an alkyl sulfate.
- the protein present in the capsid is a major activator of humoral immunity and cellular immunity of the host, the host immune response can be reduced by removing the CVA11 type capsid. As a result, the infectivity to CVA11 type cancer cells, and thus the cytotoxicity of the pharmaceutical composition to cancer cells can be improved.
- the oncolytic virus includes a nucleic acid derived from an oncolytic virus that infects cancer cells.
- Nucleic acids derived from oncolytic viruses include viral RNA isolated directly from oncolytic viruses, synthetic RNA, and cDNA corresponding to the base sequence of isolated viral RNA. To isolate viral RNA, any method such as phenol / chloroform extraction or magnetic bead isolation can be used.
- the nucleic acid may be a virus plasmid or expression vector into which a nucleic acid for generating a virus is incorporated.
- Expression vectors include, for example, plasmids that can express DNA encoding viral proteins required to generate viruses.
- the expression vector may include a transcriptional regulatory control sequence to which the inserted nucleic acid is operably linked.
- the transcriptional regulatory control sequence here is, for example, a promoter for initiating transcription, an expression control element for enabling ribosome binding to the transcribed mRNA, and the like.
- the nucleic acid derived from the CVA11 type specifically includes a nucleic acid having the base sequence shown in SEQ ID NO: 1.
- Specific examples of the nucleic acid derived from CVB3 type include a nucleic acid having the base sequence shown in SEQ ID NO: 2.
- Examples of expression vectors include pSV2neo, pEF-PGk. puro, pTk2, non-replicating adenovirus shuttle vector, cytomegalovirus promoter, and the like can be used.
- a cDNA encoding a viral protein necessary to give rise to a virus can be prepared by reverse transcription of viral RNA or a fragment thereof.
- the nucleic acid derived from AAV specifically includes a nucleic acid having the base sequence shown in SEQ ID NO: 3.
- the expression vector include pSV2neo, pEF-PGk. puro, pTk2, non-replicating adenovirus shuttle vector, cytomegalovirus promoter, and the like can be used.
- a cDNA encoding a viral protein necessary to give rise to a virus can be prepared by reverse transcription of viral RNA or a fragment thereof.
- the anticancer agent is selected from oxaliplatin, plant alkaloid anticancer agents, and antimetabolites, and examples of plant alkaloid anticancer agents include vincristine, vinblastine, vindesine, vinorelbine. , Etoposide, irinotecan or an active metabolite thereof or a salt thereof, nogitecan, sobuzoxane, docetaxel, paclitaxel, paclitaxel injection, eribulin and the like, and irinotecan, SN-38 and a salt thereof are preferable.
- Antimetabolites include 5-fluorouracil (5-FU), 5-FU prodrugs (eg, tegafur or a salt thereof), capecitabine or a salt thereof, TS-1 (also referred to as S-1, a modulator for tegafur).
- 5-FU 5-fluorouracil
- 5-FU prodrugs eg, tegafur or a salt thereof
- capecitabine or a salt thereof
- TS-1 also referred to as S-1, a modulator for tegafur
- fluoropyrimidine anticancer agents such as carmofur and doxyfluridine, gemcitabine, cytarabine, enositabine, mercaptopurine, fludarabine, cladribine, methotrexate, pemetrexed, hydroxycarbamide, nelarabine, pentostatin and their pros Drugs and the like can be mentioned, and fluorinated pyrimidine-based anticancer agents that allow 5-fluorouracil to exist in vivo are more preferable, and 5-FU or a salt thereof is particularly preferable.
- Oxaliplatin is a third generation platinum complex anticancer agent also known as L-OHP.
- oxaliplatin means cis-oxaloto (trans-l-1,2-diaminocyclohexane) platinum (II), and its optical enantiomer, cis-oxaloto (trans-d-1,2-diaminocyclohexane).
- Platinum (II) and mixtures thereof are included.
- Irinotecan is a derivative of camptothecin, which is an antitumor alkaloid derived from Cannaboku, and has a topoisomerase I inhibitory action.
- SN-38 (7-ethyl-10-hydroxycamptothecin) is an active metabolite of irinotecan and has stronger antitumor activity than irinotecan.
- Examples of the salt of irinotecan and SN-38 include salts with inorganic acids or organic acids, and hydrochlorides are preferable.
- 5-FU is a fluorinated pyrimidine-based antimetabolite and is an anticancer agent that exerts an antitumor effect by inhibiting nucleic acid synthesis.
- oxaliplatin is preferred.
- Oxaliplatin and SN-38, 5-FU have antitumor effects by themselves, but as shown in the examples described later, oxaliplatin has an action of promoting the growth of oncolytic viruses, particularly coxsackie viruses, In addition, it has an action of promoting the expression of viral receptors (DAF, ICAM-1) of cancer cells.
- DAF viral receptors
- plant alkaloid anticancer agents such as oxaliplatin and SN-38 and antimetabolites such as 5-FU, when coxsackie virus and oxaliplatin are used in combination, prevent coxsackie virus against cancer cells resistant to oxaliplatin. It exhibits much stronger cytotoxicity than when applied as a single agent.
- an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite is oncolytic.
- an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite can be an expression enhancer of a cancer cell virus receptor.
- the growth promoting effect of oncolytic virus by an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agent and antimetabolite is from oxaliplatin, plant alkaloid anticancer agent and antimetabolite It is obtained by culturing the selected anticancer agent and oncolytic virus together.
- a known method such as a virus propagation method using cultured cells can be used.
- the growth promoting effect can be evaluated by using a known method for calculating the infectious titer (MOI) of a virus.
- the antitumor effect (cytotoxicity against cancer cells) by the anticancer drug selected from the oncolytic virus and oxaliplatin of the present invention, plant alkaloid anticancer drug and antimetabolite is in the presence of the anticancer drug.
- Methods for testing cell line survival include, for example, a method of quantifying the number of viable stained cells by staining fixed cells with a staining solution, a method of quantifying a crystal violet method, an apoptosis-specific marker, etc. There is. Using these methods, cancer cells that have been incubated with oncolytic virus in the presence of anti-cancer drugs are quantified after lapse of time, resulting in tumor lysis. The dead cancer cells can be quantified by the cytotoxicity of the sex virus and the anticancer agent.
- the cancer type targeted by the anti-tumor therapy of the present invention is not particularly limited as long as it is an oncolytic virus, particularly an oncolytic virus that infects cancer cells and exhibits cytotoxicity.
- Including cancer examples of cancer cells in which particularly strong cytotoxicity is induced in solid cancer include, for example, small cell lung cancer, non-small cell lung cancer, lung squamous cell carcinoma, malignant mesothelioma, colorectal cancer, colorectal cancer, gastric cancer, esophageal cancer, Examples include cancer cells such as hypopharyngeal cancer, breast cancer, cervical cancer, ovarian cancer, prostate cancer, and bladder cancer.
- cancer cells such as non-Hodgkin lymphoma, lymphocytic leukemia, human B lymphoma and the like in addition to the solid cancer, and cancer cells of colorectal cancer and colorectal cancer are preferably used. Particularly preferred.
- the antitumor therapy of the present invention can also be used for the treatment of cancers resistant to oxaliplatin, plant alkaloid anticancer agents or antimetabolites, so-called refractory cancers.
- an oxaliplatin-resistant cancer for example, a cancer in which a decrease in tumor volume, suppression of increase, or improvement of a condition related to the cancer is not observed even when a dose of oxaliplatin that is clinically effective is administered
- an anticancer agent selected from an oncolytic virus and oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite is contained in an appropriate combination ratio, and Even a combination drug formulated into a dosage form (single-drug form) can be formulated separately with drugs containing effective amounts of oncolytic virus and anticancer drug so that they can be used separately at the same time or at intervals. It may be a combination (two-drug form, referred to as a kit).
- the combination drug may contain a carrier, a diluent, an adjuvant and a carrier.
- a carrier for example, liposome, micelle and the like are preferable.
- Liposomes contain a combination of lipids and steroids or steroid precursors that contribute to membrane stability.
- examples of the lipid include phosphatidyl compounds such as phosphatidylglycerol, phosphatidylcholine, phosphatidylserine, sphingolipid, phosphatidylethanolamine, cerebroside, and ganglioside.
- Oncolytic viruses covered with liposomes or micelles can reduce the host immune response.
- the diluent include demineralized water, distilled water, and physiological saline.
- the adjuvant include plant oils, cellulose derivatives, polyethylene glycol, and fatty acid esters.
- the carrier various types commonly used for ordinary drugs, such as excipients, binders, disintegrants, lubricants, diluents, solubilizers, suspending agents, isotonic agents, pH adjusting agents. , Buffers, stabilizers, colorants, flavoring agents, flavoring agents and the like.
- a compounding agent can be administered in combination with drugs other than a compounding agent.
- the kit is administered in combination with a drug containing an oncolytic virus and a drug containing an anticancer drug selected from oxaliplatin, plant alkaloid anticancer drugs and antimetabolite drugs. can do.
- the above-mentioned preparation containing an oncolytic virus may contain a carrier, a diluent or an adjuvant in addition to the oncolytic virus.
- a carrier for example, liposome, micelle and the like are preferable.
- Liposomes contain a combination of lipids and steroids or steroid precursors that contribute to membrane stability.
- examples of the lipid include phosphatidyl compounds such as phosphatidylglycerol, phosphatidylcholine, phosphatidylserine, sphingolipid, phosphatidylethanolamine, cerebroside, and ganglioside.
- Oncolytic viruses covered with liposomes or micelles can reduce the host immune response.
- the diluent include demineralized water, distilled water, and physiological saline.
- the adjuvant include plant oils, cellulose derivatives, polyethylene glycol, and fatty acid esters.
- a preparation containing an anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolites can be prepared by a generally known method using a pharmacologically acceptable carrier.
- a pharmacologically acceptable carrier include various types commonly used for ordinary drugs, such as excipients, binders, disintegrants, lubricants, diluents, solubilizers, suspending agents, isotonic agents, pH. Examples include regulators, buffers, stabilizers, colorants, flavoring agents, and flavoring agents.
- the amount of oncolytic virus in the preparation is, for example, 1 ⁇ 10 2 to 1 ⁇ 10 10 plaque forming units per 1 mL of solution, and 1 ⁇ 10 5 plaque forming units or more. preferable.
- the compounding amount of the anticancer agent selected from oxaliplatin, plant alkaloid anticancer agents and antimetabolites is, for example, preferably 1 to 1000 mg in the preparation.
- an oncolytic virus and an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite are used in various ways, that is, oral, muscle, subcutaneous, rectal, vagina. It can be administered to cancer patients via the nasal cavity or the like, but is preferably administered intratumorally, intravenously, or intraperitoneally depending on the type of cancer.
- many digestive organ cancers such as esophageal cancer and colon cancer can be directly injected into the tumor tissue while visually confirming the tumor tissue with an endoscope or the like. In this case, since the injected site can be confirmed with an endoscope or the like, there is also an advantage that it is easy to cope with bleeding.
- the oncolytic virus and the anticancer agent selected from oxaliplatin, plant alkaloid anticancer agent and antimetabolite should only be administered in an amount sufficient to treat cancer,
- the dose is determined based on the patient's weight, age, sex, tumor tissue size, and the like. For example, per adult day, oncolytic viruses can be exemplified by 1 ⁇ 10 2 to 1 ⁇ 10 10 plaque forming units, and anticancer agents can be exemplified by 1-1000 mg.
- the administration method may be single administration or multiple administration, and may be continuously administered as a sustained release preparation.
- the administration sequence and the administration interval are not particularly limited as long as the effect of the combination of an oncolytic virus and an anticancer agent selected from oxaliplatin, a plant alkaloid anticancer agent and an antimetabolite is obtained.
- Example 1 Antitumor effect by combined use of oxaliplatin and CVA11 type
- Method (a) Preparation of CVA11 type was obtained from National Institute of Infectious Diseases. CVA11 type was grown using HELA cells (purchased from ATCC).
- MOI Dulbecco's modified Eagle medium
- the collected HELA cells were frozen and thawed three times using liquid nitrogen, and then centrifuged at 4 ° C. and 3000 rpm for 15 minutes to collect the supernatant.
- the collected supernatant (virus solution) was stored at ⁇ 80 ° C.
- C Examination of antitumor effect using CVA11 type crystal violet method The antitumor effect (cytotoxicity) by CVA11 type was evaluated by the crystal violet method.
- Oxaliplatin-resistant colorectal cancer cell line (WiDr) was seeded in a 24-well plate at a density (3 ⁇ 10 4 cells / well) that became confluent after 72 hours.
- the medium was removed from the plate, 200 ⁇ L of CVA11 dilution was added to each well, and the plate was maintained at 37 ° C., 5% CO 2 for 1 hour.
- the CVA11 type diluent was removed, 1 mL of each cell culture medium was added to each well, and cultured for 72 hours. After 72 hours, the cells were gently washed with phosphate buffered saline (PBS), 300 ⁇ L of PBS containing 0.5% glutaraldehyde was added to each well, and then allowed to stand at room temperature for 15 minutes to fix viable adherent cells. .
- PBS phosphate buffered saline
- the glutaraldehyde-containing PBS is removed, washed with PBS, 300 ⁇ L of sterilized water containing 2% ethanol and 0.1% crystal violet is added to each well, and the cells are allowed to stand at room temperature for 10 minutes. Was stained. Each well of the stained plate was washed twice with 500 ⁇ L of sterilized water, and staining was recorded using a scanner, thereby confirming the antitumor effect.
- A Oxaliplatin and CVA11 type not added
- B Oxaliplatin only added (50 ⁇ M)
- C Oxaliplatin added (50 ⁇ M)
- CVA11 type added (MOI 0.01)
- FIG. 1 shows the results of the crystal violet method.
- B No antitumor effect was observed with WiDr to which only oxaliplatin was added and D: CVA11 type alone was added.
- D CVA11 type alone was added.
- MOI 0.01
- Example 1 (1) the infectious titer of the virus was measured by the method described in Example 1 (1) (b).
- A: Oxaliplatin and CVA11 type not added, B: Oxaliplatin not added, only CVA11 type added, C: Oxaliplatin added (50 ⁇ M), then CVA11 type added (MOI 0.01)
- C Oxaliplatin added (50 ⁇ M)
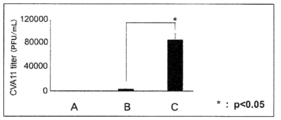
- FIG. 2 shows the CVA11 virus titer results. When oxaliplatin was added, a significant increase in the amount of CVA11 virus was observed. It was confirmed that oxaliplatin promotes the growth of CVA11 type.
- Example 3 Viral Receptor Expression Promoting Action by Oxaliplatin
- the cultured oxaliplatin-resistant colorectal cancer cell line (WiDr) was suspended in DMEM medium so as to be 3 ⁇ 10 6 cells / mL. 100 ⁇ L of the obtained cell suspension was dispensed into each well of a 96-well plate and seeded at 3 ⁇ 10 5 cells / well. After leaving the plate at 37 ° C., 5% CO 2 for about 8 hours, oxaliplatin was added to a final concentration of 50 ⁇ M. Thereafter, the plate was allowed to stand for about 42 hours at 37 ° C. and 5% CO 2 , and then mRNA was collected to prepare cDNA.
- FIG. 3 shows the results of real-time PCR.
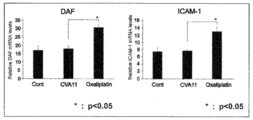
- the addition of oxaliplatin significantly increased the expression of viral receptors DAF and ICAM-1.
- the addition of CVA11 virus did not increase the expression of DAF and ICAM-1.
- Some of the receptors are known to affect the growth of the virus. From this result, the antitumor effect of CVA11 against WiDr was increased by pretreatment with oxaliplatin because DAF and ICAM-1 This was thought to be due to the growth of CVA11 virus.
- Example 4 In vivo antitumor effect 1 by combined use of oxaliplatin and CVA11 type 1 (1) Method The antitumor effect on CVA11 type cancer cells confirmed in Example 1 was examined using tumor-bearing nude mice with an oxaliplatin-resistant colorectal cancer cell line WiDr. WiDr was washed with PBS and suspended in OPTI-MEM I to 5.0 ⁇ 10 7 cells / mL. Suspensions containing WiDr were injected subcutaneously into the right flank of 6-8 week old BALB / c nude mice using a 27G needle.
- mice were divided into 4 groups: 1) non-administration group, 2) oxaliplatin single administration group, 3) CVA11 type single administration group, and 4) CVA11 type administration group after oxaliplatin administration.
- Oxaliplatin 100 ⁇ g was administered intraperitoneally to day1.
- Type CVA11 was locally injected into subcutaneous tumors on days 2, 4, 6, 8, 10 with 5 ⁇ 10 7 plaque forming units (PFU).
- PFU plaque forming units
- OPTI-MEM I not containing CVA11 type was administered to the right flank in the same amount as in the CVA11 type administered group.
- the tumor volume and body weight of each group were measured. The tumor volume was calculated by major axis ⁇ minor axis ⁇ minor axis ⁇ 0.5.
- the test was performed using 5 mice in each group, and the t-test was used for the test.
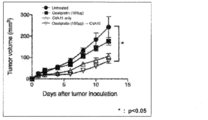
- FIG. 4 shows the tumor volume of the non-administered group and the tumor volume of the group administered with oxaliplatin alone, the group administered with CVA11 alone, and the group administered with CVA11 after administration of oxaliplatin (combination group).
- mice administered with CVA11 after administration of oxaliplatin an increase in tumor volume was significantly suppressed as compared to the non-administered group.
- the combination group had a high antitumor effect because the increase in tumor volume was suppressed as compared with the oxaliplatin single administration group and the CVA11 type single administration group.
- FIG. 5 shows changes in body weight of these four groups.
- mice administered with oxaliplatin alone As compared with the non-administration group, no significant weight loss was observed in mice administered with oxaliplatin alone, mice administered with CVA11 type alone, and mice administered with CVA11 type after oxaliplatin administration. Since weight loss at this point suggests an adverse event, it can be said that no weight loss was observed in mice administered CVA type 11 after oxaliplatin administration, and the antitumor therapy of the present invention was It can be said that safety is high.
- Example 5 In vivo antitumor effect 2 by combined use of oxaliplatin and CVA11 type 2 (1) Method The survival rate of tumor-bearing nude mice subcutaneously transplanted with the oxaliplatin-resistant colorectal cancer cell line WiDr in Example 4 was compared. E. (Hematoxylin eosin) staining was evaluated.
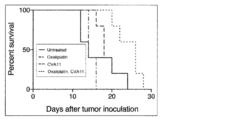
- FIG. 6 shows data comparing survival rates in cancer-bearing nude mice.
- the survival rate of mice administered with CVA11 after administration of oxaliplatin was improved as compared with mice in the untreated group, mice in the group administered with oxaliplatin alone, and mice in the group administered with CVA11 alone.
- 7 shows a pathological tissue (HE staining) of the tumor tissue.
- Example 6 Antitumor effect by combined use of oxaliplatin and CVB3 type
- DMEM Dulbecco's modified Eagle medium
- OPTI-MEM I After removing the medium, 1 mL of OPTI-MEM I was added to the petri dish for culture, and the cells were peeled and collected using a cell scraper.
- CVB3 type and HELA cells were cultured in an incubator at 37 ° C. and 5% CO 2 .
- the collected HELA cells were frozen and thawed three times using liquid nitrogen, and then centrifuged at 4 ° C. and 3000 rpm for 15 minutes to collect the supernatant.
- the collected supernatant (virus solution) was stored at ⁇ 80 ° C.
- FIG. 8 shows the results of the crystal violet method. No anti-tumor effect was observed in the case where only oxaliplatin was added (CVB3 type MOI in FIG. 8 in a horizontal row with 0), and in the case where only CVB3 type was added (OXA in FIG. 8 was in a vertical row). On the other hand, in the group to which CVA11 type was added after the addition of oxaliplatin (group in the frame), a strong antitumor effect was observed depending on the amount of MOI and oxaliplatin added.
- Example 7 CVB3 Type Growth Promotion Action by Oxaliplatin (1)
- Method CVB3 type was prepared in the same manner as in Example 6 (1) (a).
- the MOI was calculated by the same method as in Example 1 (1) (b).
- the cultured oxaliplatin-resistant colorectal cancer cell line (WiDr) was suspended in DMEM medium to 3 ⁇ 10 6 cells / mL. 100 ⁇ L of the obtained cell suspension was dispensed into each well of a 96-well plate, and seeded at 3 ⁇ 10 5 cells / well. After leaving the plate at 37 ° C. under 5% CO 2 for about 8 hours, oxaliplatin was added to a final concentration of 0 (no addition), 0.5, and 1.0 ⁇ M.
- Example 1 (1) the infectious titer of the virus was measured by the method described in Example 1 (1) (b). Comparison was made by dividing into three groups: 1: no oxaliplatin added, 2: oxaliplatin added at 0.5 ⁇ M, 3: oxaliplatin added at 1.0 ⁇ M. The test was performed 6 times, and t-test was used for the test.
- FIG. 9 shows the results of virus titer of CVB3 type.
- oxaliplatin When oxaliplatin was added, an increase in the amount of CVB3 virus was observed. In particular, a significant increase was observed when 1 ⁇ M oxaliplatin was added. Thus, it was confirmed that oxaliplatin promotes not only CVA11 type but also CVB3 type proliferation.
- Example 8 AAV Growth-Promoting Action by Oxaliplatin
- Method (a) Preparation of AAV As AAV pAAV-CMV Vector (manufactured by Takara Bio Inc.) was used.
- pAAV-CMV Vector was prepared using AAVpro® Helper Free System (manufactured by Takara Bio Inc.). The collected supernatant (virus solution) was stored at ⁇ 80 ° C.
- AAVpro® Titration Kit for Real Time PCR Ver. 2 (manufactured by Takara Bio Inc.) was used to measure the copy number of the virus. The t-test was used for the test.
- FIG. 10 shows the results of AAV copy numbers.
- oxaliplatin was added, a significant increase in the AAV copy number was observed.
- the amount of oxaliplatin added was between 0.25 and 1.0 ⁇ M. This confirmed that oxaliplatin promoted AAV proliferation.
- Example 9 Growth Promotion Action of CVA11 Type by SN-38
- Method Preparation of CVA11 type and calculation of MOI were carried out in the same manner as in Example 1 (1) (a) and (b).
- the cultured oxaliplatin-resistant colorectal cancer cell line (WiDr) was suspended in DMEM medium to 3 ⁇ 10 6 cells / mL. 100 ⁇ L of the obtained cell suspension was dispensed into each well of a 96-well plate, and seeded at 3 ⁇ 10 5 cells / well. After leaving the plate at 37 ° C., 5% CO 2 for about 8 hours, SN-38 was added to a final concentration of 0 (no addition), 1.0, 5.0, 50 ⁇ M.
- Example 1 (1) the infectious titer of the virus was measured by the method described in Example 1 (1) (b). Comparison was made in four groups: 1: SN-38 not added, 2: SN-38 added at 1.0 ⁇ M, 3: SN-38 added at 5.0 ⁇ M, 4: SN-38 added at 50 ⁇ M. The test was performed 6 times, and t-test was used for the test.
- FIG. 11 shows the results of virus titer of CVA11 type when SN-38 was added. When SN-38 was added, a significant increase in the amount of CVA11 virus was observed. It was confirmed that CVA11 type proliferation was promoted not only in oxaliplatin but also in SN-38.
- Example 10 Growth Promotion Action of CVA11 Type by 5-FU (1) Method Preparation of CVA11 type and calculation of MOI were performed in the same manner as in Example 1 (1) (a) and (b).
- the cultured oxaliplatin-resistant colorectal cancer cell line (WiDr) was suspended in DMEM medium to 3 ⁇ 10 6 cells / mL. 100 ⁇ L of the obtained cell suspension was dispensed into each well of a 96-well plate, and seeded at 3 ⁇ 10 5 cells / well. After leaving the plate at 37 ° C., 5% CO 2 for about 8 hours, 5-FU was added to a final concentration of 0 (no addition) and 50 ⁇ M. Thereafter, the plate was allowed to stand for about 12 hours at 37 ° C.
- Example 1 (1) the infectious titer of the virus was measured by the method described in Example 1 (1) (b). Comparison was made by dividing into two groups of 1: 5-FU not added and 2: 5-FU added at 50 ⁇ M. The test was performed 6 times, and t-test was used for the test.
- FIG. 12 shows the results of CVA11 type virus titer when 5-FU was added. With the addition of 5-FU, a significant increase in the amount of CVA11 virus was observed. It was confirmed that not only oxaliplatin and SN-38 but also 5-FU promotes the growth of CVA11 type.
- Example 11 Antitumor effect by combined use of oxaliplatin and CVA11 type when using brain tumor cell line U-87
- Method (a) Preparation of CVA11 type CVA11 type was prepared in Example 1 (1) (a ).
- C Examination of antitumor effect using crystal violet method of CVA11 type Antitumor effect (cytotoxicity) by combined use of CVA11 type and oxaliplatin when brain tumor cell line U-87 is used by crystal violet method evaluated.
- the CVA11 type diluent was removed, 1 mL of each cell culture medium was added to each well, and cultured for 72 hours. After 72 hours, the cells were gently washed with phosphate buffered saline (PBS), 300 ⁇ L of PBS containing 0.5% glutaraldehyde was added to each well, and then allowed to stand at room temperature for 15 minutes to fix viable adherent cells. . Thereafter, the glutaraldehyde-containing PBS is removed, washed with PBS, 300 ⁇ L of sterilized water containing 2% ethanol and 0.1% crystal violet is added to each well, and the cells are allowed to stand at room temperature for 10 minutes. Was stained. Each well of the stained plate was washed twice with 500 ⁇ L of sterilized water, and staining was recorded using a scanner, thereby confirming the antitumor effect.
- PBS phosphate buffered saline
- FIG. 13 shows the results of the crystal violet method.
- the anti-tumor effect was confirmed only with CVA11 type, but the enhancement of the anti-tumor effect was confirmed in combination with oxaliplatin 50 ⁇ M.
- the antitumor therapy is effective for brain tumors which are cancer types other than colorectal cancer.
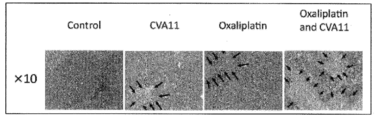
- FIG. 14A shows the results of the crystal violet method when oxaliplatin is added
- FIG. 14B shows the results of the crystal violet method when cisplatin is added.
- the combination of CVA11 type and oxaliplatin showed a strong antitumor effect compared to the combination of CVA11 type and cisplatin. From these results, it was shown that the combination of CVA11 type and oxaliplatin, which is an oncolytic virus, is particularly useful as compared with the combination of CVA11 type and cisplatin already reported (International Publication No. 2013-157648). .
- FIG. 15 shows the results of virus titer of CVB3 type. Unlike the case where oxaliplatin was added (FIG. 9), no increase in the amount of CVB3 virus was observed even when cisplatin was added. From this result, it was shown that oxaliplatin is more useful in combination with oncolytic virus than cisplatin.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Microbiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Oncology (AREA)
- Mycology (AREA)
- Biotechnology (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description
1)腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を組み合わせてなる抗腫瘍剤。
2)腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである1)記載の抗腫瘍剤。
3)コクサッキーウイルスが、A11型又はB3型である2)記載の抗腫瘍剤。
4)植物アルカロイド系抗がん剤が、SN-38、イリノテカン及びそれらの塩から選ばれる1以上からなる1)~3)のいずれかに記載の抗腫瘍剤。
5)代謝拮抗剤が、5-FU又はその塩である1)~3)のいずれかに記載の抗腫瘍剤。
6)腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を含む抗腫瘍剤。
7)腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである6)記載の抗腫瘍剤。
8)コクサッキーウイルスが、A11型又はB3型である7)記載の抗腫瘍剤。
9)植物アルカロイド系抗がん剤が、SN-38、イリノテカン及びそれらの塩から選ばれる1以上からなる6)~8)のいずれかに記載の抗腫瘍剤。
10)代謝拮抗剤が、5-FU又はその塩である6)~8)のいずれかに記載の抗腫瘍剤。
11)腫瘍溶解性ウイルスを含有してなる薬剤と、オキサリプラチン及び植物アルカロイド系抗がん剤から選ばれる抗がん剤を含有してなる薬剤からなるキットである1)~5)のいずれかに記載の抗腫瘍剤。
12)オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を有効成分とする腫瘍溶解性ウイルスの抗腫瘍効果増強剤。
13)腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである12)記載の抗腫瘍効果増強剤。
14)オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤と、腫瘍溶解性ウイルスを共に培養することによる腫瘍溶解性ウイルスの増殖促進方法。
15)オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を有効成分とする癌細胞のウイルス受容体発現増強剤。
16)ウイルス受容体が、DAF及び/又はICAM-1である15)記載のウイルス受容体発現増強剤。
17)抗腫瘍剤を製造するための、腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の使用。
18)抗腫瘍療法に使用される、腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の組み合わせ。
19)腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を患者に投与する、抗腫瘍療法。
腫瘍溶解性ウイルスは、細胞表面のウイルス受容体に結合することによって、当該細胞に感染することができる。ウイルス受容体としては、例えば、崩壊促進因子(DAF又はCD55)、細胞間接着分子‐1(ICAM‐1又はCD54)、インテグリンα2β1(CD49b)等が挙げられる。腫瘍溶解性ウイルスがウイルス受容体と相互作用することでカプシドが脱安定化され、これにより腫瘍溶解性ウイルスの脱外皮が誘導される。
ウイルスRNAを単離するには、フェノール/クロロホルム抽出法や磁気ビーズによる単離等の任意の方法を使用することができる。
また、上記核酸は、ウイルスを生じさせるための核酸を組み込んだウイルスプラスミドや発現ベクターであってもよい。発現ベクターには、例えば、ウイルスを生じさせるために必要なウイルスタンパク質をコードするDNAを発現することができるプラスミドが含まれる。発現ベクターには、挿入された核酸が機能的に連結された転写調節制御配列が含まれてもよい。ここでの転写調節制御配列は、例えば、転写を開始させるためのプロモーター、転写されたmRNAに対するリボソームの結合を可能にするための発現制御エレメント等である。
オキサリプラチンは、L-OHPとしても知られる第三世代の白金錯体系抗がん剤である。本発明において、「オキサリプラチン」は、cis-オキサロト(trans-l-1,2-ジアミノシクロヘキサン)白金(II)、その光学エナンチオマーであるcis-オキサロト(trans-d-1,2-ジアミノシクロヘキサン)白金(II)及びこれらの混合物が包含される。
イリノテカンは、カンレンボク由来の抗腫瘍性アルカロイドであるカンプトテシンの誘導体で、トポイソメラーゼI阻害作用を有する。SN-38(7-ethyl-10-hydroxycamptothecin)は、イリノテカンの活性代謝物であり、イリノテカンに比べ強い抗腫瘍活性を有する。
イリノテカン及びSN-38の塩としては、無機酸又は有機酸との塩が挙げられるが、好ましくは塩酸塩である。
5-FUは、フッ化ピリミジン系の代謝拮抗剤で、核酸の合成を阻害することにより抗腫瘍効果を発揮する抗がん剤である。
すなわち、オキサリプラチン、植物アルカロイド系抗がん剤又は代謝拮抗剤と、腫瘍溶解性ウイルスの併用において、オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤は腫瘍溶解性ウイルスの増殖促進剤、腫瘍溶解性ウイルスの抗腫瘍効果増強剤となり得、腫瘍溶解性ウイルスとオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の組み合わせは抗腫瘍剤となり得る(以下、これらを纏めて本発明の「抗腫瘍療法」とも称する)。また、オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤は癌細胞のウイルス受容体の発現増強剤となり得る。
固形癌において特に強い細胞傷害性が誘導される癌細胞としては、例えば、小細胞肺癌、非小細胞肺癌、肺扁平上皮癌、悪性中皮腫、大腸癌、結腸直腸癌、胃癌、食道癌、下咽頭癌、乳癌、子宮頸癌、卵巣癌、前立腺癌、膀胱癌等の癌細胞が挙げられる。したがって、本発明の抗腫瘍療法では、前記固形癌に加え、非ホジキンリンパ腫やリンパ性白血病、ヒトBリンパ腫等の癌細胞を対象に用いられるのが好ましく、大腸癌、結腸直腸癌の癌細胞が特に好ましい。
また、キットは、腫瘍溶解性ウイルスを含有する薬剤とオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を含有する薬剤以外に、さらに他の薬剤を組み合わせて投与することができる。
希釈剤としては、例えば、脱塩水、蒸留水及び生理的食塩水等が挙げられる、また、補助剤としては、植物系オイル、セルロース誘導体、ポリエチレングリコール及び脂肪酸エステル等が挙げられる。
投与方法は、単回投与でも複数回投与でもよく、徐放製剤として持続的に投与してもよい。また、投与順序や投与間隔は、腫瘍溶解性ウイルスと、オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の組み合わせの効果が得られる範囲であれば特に制限されないが、オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の投与後に腫瘍溶解性ウイルスを投与することがより好ましい。また、キットとする場合は、それぞれ単独の製剤を、同時に或いは間隔を空けて投与してもよい。
実施例1 オキサリプラチンとCVA11型の併用による抗腫瘍効果
(1)方法
(a)CVA11型の調製
CVA11型は、国立感染症研究所から入手した。CVA11型は、HELA細胞(ATCCから購入)を用いて増殖した。Dulbecco’s modified Eagle medium(DMEM)(Sigma-Aldrich製)を10mL用いて継代培養したHELA細胞(約2x106cells/mL)にCVA11型(播種量:MOI=0.1~1.0)を1時間孵置した後、培地をDMEMに置換し、細胞変性効果が始まるまで静置した。培地を除去後、培養用シャーレにOPTI-MEM I 1mLを添加し、セルスクレーパーを用いて細胞を剥離回収した。なお、CVA11型及びHELA細胞は、インキュベーター内で37℃、5%CO2下で培養した。液体窒素を用いて、回収したHELA細胞の凍結、融解を3回繰り返した後、4℃、3000rpmで15分間遠心を行い、上清を回収した。回収した上清(ウイルス溶液)は、-80℃で保存した。
MOIは、特許文献3に記載の以下の方法で算出した。
オキサリプラチン抵抗性大腸癌細胞株(WiDr)(ATCCから入手)を96穴のプレートに5×103cells/100μL/ウェルで播種し、37℃、5%CO2下で5時間維持した。ウイルスは、OPTI-MEM Iで100倍又は1000倍希釈して、これをMOI測定用のウイルス原液とした(ここでの希釈倍率の常用対数をLとする)。ウイルス原液を10倍ずつ段階希釈し(ここでの希釈倍率の常用対数をdとする)、希釈系列液を調製した。次に、各ウェルに希釈系列液を0.05mLずつ添加した(添加した希釈系列液の体積をvとする)。120時間後に50%以上の細胞変性効果が認められたウェル数の合計を8で除算した値Sを算出し、MOIを以下の式で算出した。
(数1)
log10(MOI)=L+d(S-0.5)+log10(1/v)
クリスタルバイオレット法により、CVA11型による抗腫瘍効果(細胞傷害性)を評価した。
オキサリプラチン抵抗性大腸癌細胞株(WiDr)を72時間後にコンフルエントになる密度(3×104cells/ウェル)で24穴のプレートに播種した。適切な感染力価(MOI=0.001、0.01、0.1)になるように、OPTI-MEM IでCVA11型を希釈し、CVA11型の希釈液を調製した。約6時間後、プレートから培地を除去し、CVA11型の希釈液を各ウェルに200μL添加し、37℃、5%CO2下にプレートを1時間維持した。次に、CVA11型の希釈液を除去し、各細胞用培地を各ウェルに1mL加え、72時間培養した。72時間後、リン酸緩衝食塩水(PBS)で緩やかに洗浄し、0.5%グルタルアルデヒド含有PBSを各ウェルに300μL添加した後、15分間室温に静置することで生存接着細胞を固定した。その後、グルタルアルデヒド含有PBSを除去し、PBSで洗浄後、2%エタノール及び0.1%クリスタルバイオレットを含有する滅菌水を、各ウェルに300μL添加し、室温で10分間静置することで生細胞を染色した。染色後のプレートの各ウェルを滅菌水500μLで2回洗浄し、スキャナを用いて染色を記録することで、抗腫瘍効果を確認した。
図1にクリスタルバイオレット法の結果を示す。B:オキサリプラチンのみを添加したもの、D:CVA11型のみを添加したWiDrでは抗腫瘍効果は認められなかった。一方、C:オキサリプラチン添加後CVA11型を添加(MOI=0.01)した群で強い抗腫瘍効果が認められた。
(1)方法
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、オキサリプラチンを最終濃度50μMとなるように添加した。その後、37℃、5%CO2下にプレートを約12時間静置した後、CVA11型をMOI=0.01となるように添加した。37℃、5%CO2下にプレートを約30時間静置した後、実施例1(1)(b)に記載の方法でウイルスの感染力価を測定した。A:オキサリプラチン、CVA11型ともに不添加、B:オキサリプラチンを添加せず、CVA11型のみを添加、C:オキサリプラチン添加(50μM)後、CVA11型添加(MOI=0.01)の3群に分けて比較をした。検定はt検定を用いた。
図2にCVA11型のウイルス力価の結果を示す。オキサリプラチン添加を行ったもので、有意なCVA11型ウイルス量の増加がみとめられた。オキサリプラチンがCVA11型の増殖を促進することが確認された。
(1)方法
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、オキサリプラチンを最終濃度50μMとなるように添加した。その後、37℃、5%CO2下にプレートを約42時間静置した後、mRNAを採取しcDNAを作成した。細胞播種後、約20時間後にCVA11型を添加したものと比較した。DAF(decay accelerating factor)、ICAM-1(intercellular adhesion molecule 1)の発現をreal-time PCRにより比較した。検定はt検定を用いた。
図3にreal-time PCRの結果を示す。
オキサリプラチン抵抗性大腸癌細胞株(WiDr)において、オキサリプラチン添加により、ウイルス受容体あるDAFとICAM-1の発現が有意に増加した。一方、CVA11型ウイルスを添加したものは、DAFとICAM-1の発現は増加しなかった。受容体の中にはウイルスの増殖に影響するものも知られており、この結果より、CVA11型によるWiDrに対する抗腫瘍効果がオキサリプラチンによる前処理によって増加したのは、DAFやICAM-1がウイルスの増殖に関与し、CVA11型ウイルスが増殖したためと考えられた。
(1)方法
実施例1で確認したCVA11型の癌細胞に対する抗腫瘍効果を、オキサリプラチン抵抗性大腸癌細胞株WiDrによる担癌ヌードマウスを用いて検討した。WiDrをPBS で洗浄し、5.0×107cells/mLになるようにOPTI-MEM Iに懸濁した。 6-8週齢のBALB/cヌードマウスの右側腹部にWiDrを含む懸濁液を、100μLずつ27G針を用いて皮下注射した。マウスは、1)非投与群、2)オキサリプラチン単独投与群、3)CVA11型単独投与群、4)オキサリプラチン投与後にCVA11型を投与した群の4つに分けた。オキサリプラチン100μgは、day1に腹腔内投与された。CVA11型は5×107プラーク形成ユニット(PFU)をday2,4,6,8,10に皮下腫瘍内に局所注射された。非投与群に対しては、右側腹部にCVA11型を含まないOPTI-MEM Iを、CVA11型投与群と同量投与した。CVA11型投与後、各群の腫瘍体積及び体重を測定した。なお、腫瘍体積は、長径×短径×短径×0.5で算出した。また、試験は各群5匹のマウスを用いて行い、検定はt検定を用いた。
図4は、非投与群の腫瘍体積およびオキサリプラチン単独投与群、CVA11型単独投与群、オキサリプラチン投与後にCVA11型を投与した群(併用群)の腫瘍体積を示す。オキサリプラチン投与後にCVA11型を投与したマウスでは、非投与群と比較して、腫瘍体積の増加が有意に抑制された。また、併用群は、オキサリプラチン単独投与群、CVA11型単独投与群と比較しても腫瘍体積の増加が抑制されており、高い抗腫瘍効果を有していることが確認された。
また、図5は、これら4群の体重の変化を示す。この結果、非投与群と比べて、オキサリプラチン単独投与したマウス、CVA11型を単独投与したマウス、オキサリプラチン投与後にCVA11型を投与したマウスにおいて有意な体重減少を認めなかった。この時点での体重減少は、有害事象を示唆するため、体重減少を認めないことは、オキサリプラチン投与後にCVA11型を投与したマウスにおいても有害事象を認めなかったといえ、本発明の抗腫瘍療法は安全性が高いといえる。
(1)方法
実施例4におけるオキサリプラチン抵抗性大腸癌細胞株WiDrを皮下移植された担癌ヌードマウスにおける生存率を比較し、皮下移植後day 40の腫瘍の病理組織をH.E.(hematoxylin eosin)染色にて評価した。
図6に担癌ヌードマウスにおける生存率を比較したデータを示す。オキサリプラチン投与後にCVA11型を投与したマウスの生存率は、未治療群のマウス、オキサリプラチン単独投与群のマウス、CVA11型単独投与群のマウスと比べて改善が見られた。また、図7に腫瘍組織の病理組織(H.E.染色)を示す。H.E.染色された腫瘍の病理組織においては、オキサリプラチン投与後にCVA11型を投与したマウスの腫瘍の病理組織において、より広範囲に細胞死をみとめた。これにより、生存率や病理組織の観察からも本発明が高い抗腫瘍効果を有することが確認された。
(1)方法
(a)CVB3型の調製
CVB3型は、国立感染症研究所から入手した。CVB3型は、HELA細胞(ATCCから購入)を用いて増殖した。Dulbecco’s modified Eagle medium(DMEM)(Sigma-Aldrich製)を10mL用いて継代培養したHELA細胞(約2x106cells/mL)にCVA11型(播種量:MOI=0.1~1.0)を1時間孵置した後、培地をDMEMに置換し、細胞変性効果が始まるまで静置した。培地を除去後、培養用シャーレにOPTI-MEM I 1mLを添加し、セルスクレーパーを用いて細胞を剥離回収した。なお、CVB3型及びHELA細胞は、インキュベーター内で37℃、5%CO2下で培養した。液体窒素を用いて、回収したHELA細胞の凍結、融解を3回繰り返した後、4℃、3000rpmで15分間遠心を行い、上清を回収した。回収した上清(ウイルス溶液)は、-80℃で保存した。
MOIの算出は、実施例1(1)(b)と同様の方法で行った。
ウイルスとしてCVB3型を使用した。また、CVB3型の感染力価(MOI)を0(不添加)、0.001、0.01、0.1とし、オキサリプラチンの添加量を0μM(不添加)、0.5μM、1μM、5μMとした以外は、実施例1(1)(c)と同様の方法で行った。
図8にクリスタルバイオレット法の結果を示す。オキサリプラチンのみを添加したもの(図8のCVB3型のMOIが0の横一列)、CVB3型のみを添加したもの(図8のOXAが0の縦一列)では抗腫瘍効果は認められなかった。一方、オキサリプラチン添加後CVA11型を添加した群(枠内の群)では、MOI及びオキサリプラチン添加量の量に依存して強い抗腫瘍効果が認められた。
(1)方法
CVB3型の調製は、実施例6(1)(a)と同様の方法で行った。また、MOIの算出は、実施例1(1)(b)と同様の方法で行った。
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、オキサリプラチンを最終濃度が0(不添加)、0.5、1.0μMとなるように添加した。その後、37℃、5%CO2下にプレートを約12時間静置した後、CVB3型をMOI=0.01となるように添加した。37℃、5%CO2下にプレートを約30時間静置した後、実施例1(1)(b)に記載の方法でウイルスの感染力価を測定した。1:オキサリプラチン不添加、2:オキサリプラチン 0.5μM添加、3:オキサリプラチン 1.0μM添加の3群に分けて比較した。試験は6回試験とし、検定はt検定を用いた。
図9にCVB3型のウイルス力価の結果を示す。オキサリプラチン添加を行ったもので、CVB3型ウイルス量の増加がみとめられた。特にオキサリプラチン1μM添加時には有意な増加がみとめられた。これにより、オキサリプラチンがCVA11型だけでなく、CVB3型の増殖も促進することが確認された。
(1)方法
(a)AAVの調製
AAVとしてpAAV-CMV Vector(タカラバイオ(株)製)を使用した。pAAV-CMV Vectorは、AAVpro(R) Helper Free System(タカラバイオ(株)製)を用いて調製を行った。回収した上清(ウイルス溶液)は、-80℃で保存した。
MOIの算出は、実施例1(1)(b)と同様の方法で行った。
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、オキサリプラチンを最終濃度が0(無添加)、0.25、0.5、1.0、2.5μMとなるように添加した。その後、37℃、5%CO2下にプレートを約24時間静置した後、AAVをMOI=0.01となるように添加した。37℃、5%CO2下にプレートを約30時間静置した後、AAVpro(R) Titration Kit(for Real Time PCR)Ver.2(タカラバイオ(株)製)を用いてウイルスのコピー数を測定した。検定はt検定を用いた。
図10にAAVのコピー数の結果を示す。オキサリプラチン添加を行ったもので、有意なAAVのコピー数の増加がみとめられた。特に、オキサリプラチンの添加量が0.25~1.0μMの間で顕著な増加が見られた。これにより、オキサリプラチンがAAVの増殖を促進することが確認された。
(1)方法
CVA11型の調製及びMOIの算出は、実施例1(1)(a)及び(b)と同様の方法で行った。
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、SN-38を最終濃度が0(不添加)、1.0、5.0、50μMとなるように添加した。その後、37℃、5%CO2下にプレートを約12時間静置した後、CVA11型をMOI=0.01となるように添加した。37℃、5%CO2下にプレートを約30時間静置した後、実施例1(1)(b)に記載の方法でウイルスの感染力価を測定した。1:SN-38不添加、2:SN-38 1.0μM添加、3:SN-38 5.0μM添加、4:SN-38 50μM添加の4群に分けて比較をした。試験は6回試験とし、検定はt検定を用いた。
図11にSN-38添加時のCVA11型のウイルス力価の結果を示す。SN-38添加を行ったもので、有意なCVA11型ウイルス量の増加がみとめられた。オキサリプラチンだけでなく、SN-38においてもCVA11型の増殖を促進することが確認された。
(1)方法
CVA11型の調製及びMOIの算出は、実施例1(1)(a)及び(b)と同様の方法で行った。
培養したオキサリプラチン抵抗性大腸癌細胞株(WiDr)を3×106cells/mLになるようにDMEM培地に懸濁した。96穴のプレートの各ウェルに、得られた細胞懸濁液を100μLずつ分注し、3×105cells/ウェルとなるように播種した。37℃、5%CO2下にプレートを約8時間静置した後、5-FUを最終濃度が0(不添加)、50μMとなるように添加した。その後、37℃、5%CO2下にプレートを約12時間静置した後、CVA11型をMOI=0.01となるように添加した。37℃、5%CO2下にプレートを約30時間静置した後、実施例1(1)(b)に記載の方法でウイルスの感染力価を測定した。1:5-FU不添加、2:5-FU 50μM添加の2群に分けて比較をした。試験は6回試験とし、検定はt検定を用いた。
図12に5-FU添加時のCVA11型のウイルス力価の結果を示す。5-FU添加を行ったもので、有意なCVA11型ウイルス量の増加がみとめられた。オキサリプラチン、SN-38だけでなく、5-FUにおいてもCVA11型の増殖を促進することが確認された。
(1)方法
(a)CVA11型の調製
CVA11型の調製は、実施例1(1)(a)と同様の方法で行った。
(b)MOIの算出
MOIの算出は、オキサリプラチン抵抗性大腸癌細胞株(WiDr)の代わりに脳腫瘍細胞株U-87を使用したこと以外は実施例1(1)(b)と同様の方法で行った。
(c)CVA11型のクリスタルバイオレット法を用いた抗腫瘍効果の検討
クリスタルバイオレット法により、脳腫瘍細胞株U-87を使用した際のCVA11型及びオキサリプラチンの併用による抗腫瘍効果(細胞傷害性)を評価した。
脳腫瘍細胞株U-87を72時間後にコンフルエントになる密度(3×104 cells/ウェル)で24穴のプレートに播種した。その後、オキサリプラチンを0(不添加)又は50μM添加した。感染力価がMOI=0.001になるように、OPTI-MEM IでCVA11型を希釈し、CVA11型の希釈液を調製した。約6時間後、プレートから培地を除去し、CVA11型の希釈液を各ウェルに200μL添加し、37℃、5%CO2下にプレートを1時間維持した。次に、CVA11型の希釈液を除去し、各細胞用培地を各ウェルに1mL加え、72時間培養した。72時間後、リン酸緩衝食塩水(PBS)で緩やかに洗浄し、0.5%グルタルアルデヒド含有PBSを各ウェルに300μL添加した後、15分間室温に静置することで生存接着細胞を固定した。その後、グルタルアルデヒド含有PBSを除去し、PBSで洗浄後、2%エタノール及び0.1%クリスタルバイオレットを含有する滅菌水を、各ウェルに300μL添加し、室温で10分間静置することで生細胞を染色した。染色後のプレートの各ウェルを滅菌水500μLで2回洗浄し、スキャナを用いて染色を記録することで、抗腫瘍効果を確認した。
図13にクリスタルバイオレット法の結果を示す。CVA11型のみにおいても抗腫瘍効果が確認されたが、オキサリプラチン50μMとの併用で抗腫瘍効果の増強が確認された。これにより大腸癌以外の癌種である脳腫瘍に対しても当該抗腫瘍療法が効果的であることが示された。
(1)方法
(a)CVA11型の調製及びMOIの算出
CVA11型の調製及びMOIの算出は、実施例1(1)(a)及び(b)と同様の方法で行った。
CVA11型の感染力価(MOI)を0(不添加)、0.001、0.01、0.1とし、オキサリプラチン又はシスプラチンの添加量を0μM(不添加)、0.5μM、1μM、5μMとした。それ以外は、実施例1(1)(c)と同様の方法で行った。
図14(a)にオキサリプラチン添加時、図14(b)にシスプラチン添加時のクリスタルバイオレット法の結果を示す。CVA11型とオキサリプラチンの併用は、CVA11型とシスプラチンの併用と比較して、強い抗腫瘍効果を示した。この結果より、腫瘍溶解性ウイルスであるCVA11型とオキサリプラチンの組み合わせは、既報(国際公開第2013-157648号)のCVA11型とシスプラチンとの組み合わせと比較して特に有用であることが示された。
(1)方法
オキサリプラチンの代わりにシスプラチンを使用した以外は、実施例7と同様の方法で行った。1:シスプラチン不添加、2:シスプラチン 0.5μM添加、3:シスプラチン 1.0μM添加の3群に分けて比較をした。
図15にCVB3型のウイルス力価の結果を示す。オキサリプラチンを添加した場合(図9)と異なり、シスプラチンを添加してもCVB3型ウイルス量の増加はみとめられなかった。この結果より、オキサリプラチンはシスプラチンと比較して、腫瘍溶解性ウイルスとの併用において有用であることが示された。
Claims (19)
- 腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を組み合わせてなる抗腫瘍剤。
- 腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである請求項1記載の抗腫瘍剤。
- コクサッキーウイルスが、A11型又はB3型である請求項2記載の抗腫瘍剤。
- 植物アルカロイド系抗がん剤が、SN-38、イリノテカン及びそれらの塩から選ばれる1以上からなる請求項1~3のいずれか1項記載の抗腫瘍剤。
- 代謝拮抗剤が、5-FU又はその塩である請求項1~3のいずれか1項記載の抗腫瘍剤。
- 腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を含む抗腫瘍剤。
- 腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである請求項6記載の抗腫瘍剤。
- コクサッキーウイルスが、A11型又はB3型である請求項7記載の抗腫瘍剤。
- 植物アルカロイド系抗がん剤が、SN-38、イリノテカン及びそれらの塩から選ばれる1以上からなる請求項6~8のいずれか1項記載の抗腫瘍剤。
- 代謝拮抗剤が、5-FU又はその塩である請求項6~8のいずれか1項記載の抗腫瘍剤。
- 腫瘍溶解性ウイルスを含有してなる薬剤と、オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を含有してなる薬剤からなるキットである請求項1~5のいずれか1項記載の抗腫瘍剤。
- オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を有効成分とする腫瘍溶解性ウイルスの抗腫瘍効果増強剤。
- 腫瘍溶解性ウイルスがコクサッキーウイルス又はアデノウイルスである請求項12記載の抗腫瘍効果増強剤。
- オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤と、腫瘍溶解性ウイルスを共に培養することによる腫瘍溶解性ウイルスの増殖促進方法。
- オキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を有効成分とする癌細胞のウイルス受容体発現増強剤。
- ウイルス受容体が、DAF及び/又はICAM-1である請求項15記載のウイルス受容体発現増強剤。
- 抗腫瘍剤を製造するための、腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の使用。
- 抗腫瘍療法に使用される、腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤の組み合わせ。
- 腫瘍溶解性ウイルス、並びにオキサリプラチン、植物アルカロイド系抗がん剤及び代謝拮抗剤から選ばれる抗がん剤を患者に投与する、抗腫瘍療法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18778015.0A EP3610870A4 (en) | 2017-03-31 | 2018-03-30 | METHOD OF EXPANSION OF AN ONCOLYTIC VIRUS AND ANTI-TUMOR AGENT |
| JP2019509428A JP7114571B2 (ja) | 2017-03-31 | 2018-03-30 | 腫瘍溶解性ウイルスの増殖方法及び抗腫瘍剤 |
| CN201880020318.2A CN110461323A (zh) | 2017-03-31 | 2018-03-30 | 溶瘤病毒的增殖方法和抗肿瘤剂 |
| US16/497,705 US11857584B2 (en) | 2017-03-31 | 2018-03-30 | Oncolytic virus growth method and antitumor agent |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017-071296 | 2017-03-31 | ||
| JP2017071296 | 2017-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018182014A1 true WO2018182014A1 (ja) | 2018-10-04 |
Family
ID=63676197
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/013974 Ceased WO2018182014A1 (ja) | 2017-03-31 | 2018-03-30 | 腫瘍溶解性ウイルスの増殖方法及び抗腫瘍剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11857584B2 (ja) |
| EP (1) | EP3610870A4 (ja) |
| JP (1) | JP7114571B2 (ja) |
| CN (1) | CN110461323A (ja) |
| WO (1) | WO2018182014A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1878351A3 (en) * | 2006-07-14 | 2008-10-01 | Daisey Machinery Co., Ltd. | Method for removing bean sprout both end portions and bean sprouts treating devices therefor |
| CN119868519A (zh) * | 2023-10-23 | 2025-04-25 | 中山大学 | Stt3a在溶瘤病毒治疗疗效评估中的应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007527719A (ja) | 2004-03-11 | 2007-10-04 | ヴィロターグ ピーティワイ リミテッド | 改変された腫瘍崩壊性ウイルス |
| JP2012046489A (ja) | 2002-12-18 | 2012-03-08 | Viralytics Ltd | 直接的なピコルナウイルス媒介腫瘍崩壊による対象における悪性腫瘍の治療方法 |
| WO2013157648A1 (ja) | 2012-04-19 | 2013-10-24 | 国立大学法人九州大学 | 医薬組成物 |
| WO2014171526A1 (ja) * | 2013-04-17 | 2014-10-23 | 国立大学法人九州大学 | 遺伝子改変コクサッキーウイルス |
| JP2016526531A (ja) * | 2013-06-14 | 2016-09-05 | サイオクサス セラピューティクス リミテッド | B型アデノウイルスのための投与計画および製剤 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG158133A1 (en) * | 2004-12-31 | 2010-01-29 | Sonne Holm | Method for reversing multiple resistance in animal cells |
| EP2073823A1 (en) | 2006-10-13 | 2009-07-01 | Medigene AG | Use of oncolytic viruses and antiangiogenic agents in the treatment of cancer |
| US20100297072A1 (en) | 2009-05-19 | 2010-11-25 | Depinho Ronald A | Combinations of Immunostimulatory Agents, Oncolytic Virus, and Additional Anticancer Therapy |
| CA2689707A1 (en) * | 2009-11-16 | 2011-05-16 | Jean-Simon Diallo | Identification of the novel small molecule viral sensitizer vse1 using high-throughput screening |
| WO2014201492A1 (en) * | 2013-06-17 | 2014-12-24 | Darren Raymond Shafren | Methods for the treatment of bladder cancer |
| WO2015155263A1 (en) * | 2014-04-10 | 2015-10-15 | Transgene S.A. | Poxviral oncolytic vectors |
| WO2017043815A1 (en) * | 2015-09-08 | 2017-03-16 | Sillajen, Inc. | Modified oncolytic vaccinia viruses expressing a cytokine and a car- boxylesterase and methods of use thereof |
| FI127460B (en) * | 2016-01-15 | 2018-06-29 | Targovax Oy | Combining adenovirus and chemotherapeutic agents for treating cancer |
-
2018
- 2018-03-30 EP EP18778015.0A patent/EP3610870A4/en not_active Withdrawn
- 2018-03-30 CN CN201880020318.2A patent/CN110461323A/zh active Pending
- 2018-03-30 WO PCT/JP2018/013974 patent/WO2018182014A1/ja not_active Ceased
- 2018-03-30 JP JP2019509428A patent/JP7114571B2/ja active Active
- 2018-03-30 US US16/497,705 patent/US11857584B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012046489A (ja) | 2002-12-18 | 2012-03-08 | Viralytics Ltd | 直接的なピコルナウイルス媒介腫瘍崩壊による対象における悪性腫瘍の治療方法 |
| JP2007527719A (ja) | 2004-03-11 | 2007-10-04 | ヴィロターグ ピーティワイ リミテッド | 改変された腫瘍崩壊性ウイルス |
| WO2013157648A1 (ja) | 2012-04-19 | 2013-10-24 | 国立大学法人九州大学 | 医薬組成物 |
| WO2014171526A1 (ja) * | 2013-04-17 | 2014-10-23 | 国立大学法人九州大学 | 遺伝子改変コクサッキーウイルス |
| JP2016526531A (ja) * | 2013-06-14 | 2016-09-05 | サイオクサス セラピューティクス リミテッド | B型アデノウイルスのための投与計画および製剤 |
Non-Patent Citations (3)
| Title |
|---|
| OTTOLINO-PERRY, K. ET AL.: "Oncolytic vaccinia virus synergizes with irinotecan in colorectal cancer", MOLECULAR ONCOLOGY, vol. 9, no. 8, 6 May 2015 (2015-05-06), pages 1539 - 1552, XP055201104 * |
| See also references of EP3610870A4 |
| TANI, KENZABURO: "Chemotherapy and gene therapy", CLINIC ALL-ROUND, vol. 50, no. 2, 2001, pages 210 - 215, XP009516812, ISSN: 0371-1900 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110461323A (zh) | 2019-11-15 |
| EP3610870A4 (en) | 2020-12-09 |
| EP3610870A1 (en) | 2020-02-19 |
| JP7114571B2 (ja) | 2022-08-08 |
| US11857584B2 (en) | 2024-01-02 |
| JPWO2018182014A1 (ja) | 2020-02-20 |
| US20200023023A1 (en) | 2020-01-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Gong et al. | A nanodrug combining CD47 and sonodynamic therapy efficiently inhibits osteosarcoma deterioration | |
| JP7564572B2 (ja) | 単離された組換え体腫瘍溶解性ワクシニアウイルスとnk細胞を含む治療薬、その使用およびそれを適用したキット | |
| Hoffman | Tumor-targeting Salmonella typhimurium A1-R: an overview | |
| JP7114571B2 (ja) | 腫瘍溶解性ウイルスの増殖方法及び抗腫瘍剤 | |
| JPWO2005095613A1 (ja) | Rad51の発現抑制剤、該発現抑制剤を有効成分として含む医薬、及びその使用 | |
| He et al. | Reconstructing tumor microenvironment using photoresponsive Cyanobacteria to reversal chemoresistance for robust chemotherapy | |
| RU2671857C1 (ru) | Новый способ производства липоплекса для местного введения и противоопухолевое средство, в котором используется такой липоплекс | |
| CN111939262B (zh) | 一种治疗肿瘤或癌症的药物组合物和其应用 | |
| JP7699838B2 (ja) | マイクロrnaで調節及び制御可能な単離された組換え腫瘍溶解性ポックスウイルス及びその使用 | |
| TW202409281A (zh) | 表現副孢菌素(parasporin)之載體及醫藥組成物 | |
| JP6093757B2 (ja) | 医薬組成物 | |
| JP5038309B2 (ja) | 悪性中皮腫治療剤 | |
| ES2847214T3 (es) | 8-oxo-dGTP para la prevención y tratamiento de tumores y aplicaciones del mismo | |
| WO2021024897A1 (ja) | 悪性腫瘍を治療するための併用薬、悪性腫瘍を治療するための医薬組成物、および、悪性腫瘍治療用医薬組成物 | |
| EP3011964A1 (en) | Compounds and associations for treating pancreatic cancer | |
| HK40056184A (en) | Isolated recombinant oncolytic poxvirus capable of being regulated and controlled by microrna and use thereof | |
| HK40056184B (zh) | 可受微小rna调控的分离的重组溶瘤痘病毒及其应用 | |
| HK40106046A (zh) | 治疗剂、药盒、分离的重组溶瘤痘病毒在治疗肿瘤和/或癌症的药物中的用途 | |
| WO2022232944A1 (en) | Pharmaceutical compositions comprising vanadium salts | |
| CN120549940A (zh) | 一种具有fgfr3-tacc3融合基因的肿瘤的联合用药物及其应用 | |
| HK1227705A1 (en) | New production method of lipoplex for local administration and antitumor drug using lipoplex | |
| HK1227705B (en) | New production method of lipoplex for local administration and antitumor drug using lipoplex | |
| JP2015020963A (ja) | 放射線治療増強薬及び放射線抵抗性癌の治療方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18778015 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019509428 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018778015 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2018778015 Country of ref document: EP Effective date: 20191031 |