WO2019018583A1 - 1,8-naphthyridinone compounds and uses thereof - Google Patents
1,8-naphthyridinone compounds and uses thereof Download PDFInfo
- Publication number
- WO2019018583A1 WO2019018583A1 PCT/US2018/042776 US2018042776W WO2019018583A1 WO 2019018583 A1 WO2019018583 A1 WO 2019018583A1 US 2018042776 W US2018042776 W US 2018042776W WO 2019018583 A1 WO2019018583 A1 WO 2019018583A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkylene
- compound
- halogen
- alkyl
- membered
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*(C)c1cc(cccc2)c2[n]1 Chemical compound C*(C)c1cc(cccc2)c2[n]1 0.000 description 8
- OSRARURJYPOUOV-UHFFFAOYSA-N Cc1cc2nccnc2cc1 Chemical compound Cc1cc2nccnc2cc1 OSRARURJYPOUOV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4709—Non-condensed quinolines and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- This disclosure relates generally to therapeutics for treatment mediated through a G- protein-coupled receptor (GPCR) signaling pathway and, more particularly, to compounds that inhibit an adenosine receptor (such as an A2A antagonist).
- GPCR G- protein-coupled receptor
- the disclosure also provides pharmaceutically acceptable compositions comprising such compounds and methods of using the compounds or compositions in the treatment of a disease associated with a GPCR signaling pathway.
- Adenosine receptors are distributed throughout the body and are responsible for numerous biological functions.
- the seven trans-membrane G -protein-coupled receptors (GPCRs) have been divided into four different subtypes: A;, A 2 A, A 2 B, and A3.
- the A 2 A and A 2B ARs stimulate activity of the adenylyl cyclase, inducing an increase of c AMP levels.
- a 2 AARS have a distinct tissue localization, different biochemical pathways, and specific pharmacological profiles.
- Adenosine is one of the human body's most important neuromodulators in both the central and the peripheral nervous systems. Adenosine is released from tumor cells and its concentration in the extracellular fluid of tumors can reach immunosuppressive levels (Blay et ai. (1997), Cancer Res., 57(13), pp. 2602-5). The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Id. This increase in adenosine concentration is a result of increases in CD 73 (ecto-5 '-nucleotidase) and CD39 (nucleoside triphosphate dephosphorylase) enzymes, which are responsible for directly catabolizing ATP into adenosine.
- CD 73 ecto-5 '-nucleotidase
- CD39 nucleoside triphosphate dephosphorylase
- upregulations are triggered by hypoxia and the generation of HIF- ⁇ .
- High levels of adenosine around tumor cells act to regulate multiple immune cells (e.g., CD4 " T-celis and cytotoxic CD8 " T-cells) via activation of multiple adenosine receptor subtypes, but particularly A 2 A receptors, resulting the suppressing of pro-inflammatory activities and upregulation of antiinflammatory molecules and immunoregulatory cells (Kumar et al. (2013), Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Piirinergic Signal., 9(2), pp
- CAR chimeric antigen receptor
- Blockade of striatal adenosine A 2 A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum, J. Neurosci, 22(5) pp. 1967-75, Gessi et al. (2011). Adenosine receptors and cancer. Biochim Biophys Acta, 1808(5), pp, 1400-12),
- a 2A and A 3 subtypes appear promising targets for therapeutic development.
- activation of A 2 A receptors leads to immunosuppressive effects, which decreases anti -tumoral immunity and thereby encourages tumor growth.
- the A 2B receptor is another potential target for therapeutic development.
- a 2S blockade may reduce tumor metastasis in an immune-independent manner (Beavis et ai. (2013). Blockade of A 2 A receptors potently suppresses the metabolism of CD73 " Tumors. Proc. Natl. Acad. Set., 110(36) pp. 14711-6). A 2B expression also correlates with relapse-free survival (RFS) in triple negative breast cancer suggesting that this pathway may be clinically relevant. A 2 B blockade also has the potential to modulate the
- tumor-associated immune cells including dendritic ceils and myeloid-derived suppressor cells (MDSCs) (Cekic et al. (2011). Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J. Immunol. 188(1), pp. 198-205;
- Blockade of A 2B adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia, 15(12), pp. 1400-9.
- the compound of the formula (I), or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing is of the formula (II) or (III), or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, as detailed herein.
- the compound of the formula (I), or a tautomer thereof, or a salt of any of the foregoing is of the formula (II).
- the compound of the formula (I), or a tautomer thereof, or a salt of any of the foregoing is of the formula (III).
- a disease such as a proliferative disease
- the compound of formula (I) or a tautomer thereof, or a salt of any of the foregoing is administered to the individual in combination with another therapeutic agent.
- the compound of fonnula (I) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing is administered to the individual in combination with another therapeutic agent.
- the compound of formula (I) or a salt thereof is a compound of the formula (II) or (III) or a tautomer thereof, or a or a salt of any of the foregoing.
- pharmaceutically acceptable salt of any of the foregoing is a compound of the formula (II) or (III), or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing.
- compositions comprising (A) a compound detailed herein, such as a compound of formula (I) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, or a compound of fonnula (II) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, or a compound of formula (III) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, and (B) a pharmaceutically acceptable carrier or excipient.
- a compound detailed herein such as a compound of formula (I) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, or a compound of fonnula (II) or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, or a compound of formula (III) or a tautomer or is
- compositions comprising (A) a compound detailed herein, such as a compound of formula (I) or a tautomer thereof, or a or a salt of any of the foregoing, or a compound of fonnula (II) or a tautomer thereof, or a or a salt of any of the foregoing, or a compound of fonnula (III) or a tautomer thereof, or a or a salt of any of the foregoing, and (B) a pharmaceutically acceptable earner or excipient.
- a compound detailed herein such as a compound of formula (I) or a tautomer thereof, or a or a salt of any of the foregoing, or a compound of fonnula (II) or a tautomer thereof, or a or a salt of any of the foregoing, or a compound of fonnula (III) or a tautomer thereof, or a or a salt of any
- Kits comprising a compound detailed herein or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing and instructions for use are also provided. Kits comprising a compound detailed herein or a salt thereof and instructions for use are also provided. A compound detailed herein or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing is also provided for the manufacture of a medicament for the treatment of cancer. Compounds as detailed herein or a pharmaceutically acceptable salt thereof are also provided for the manufacture of a medicament for the treatment of cancer. DETAILED DESCRIPTION OF THE INVENTION
- the alkenyl group may be in "cis” or “trans” configurations, or alternatively in "E” or "Z” configurations.
- alkenyl groups are those having 2 to 20 carbon atoms (a "C 2 -C 2 0 alkenyl"), having 2 to 8 carbon atoms (a “C 2 - Cg alkenyl”), having 2 to 6 carbon atoms (a “C ⁇ -Ce alkenyl”), or having 2 to 4 carbon atoms (a "C 2 -C4 alkenyl”).
- alkenyl examples include, but are not limited to, groups such as ethenyl (or vinyl), prop-l-enyl, prop-2-enyl (or aliyi), 2-methylprop-l-enyl, but-l-enyl, but-2-enyl, but- 3- enyl, buta ⁇ i,3 ⁇ dienyl, 2-methylbuta- 1 ,3 -dienyl, homologs and isomers thereof, and the like.
- groups such as ethenyl (or vinyl), prop-l-enyl, prop-2-enyl (or aliyi), 2-methylprop-l-enyl, but-l-enyl, but-2-enyl, but- 3- enyl, buta ⁇ i,3 ⁇ dienyl, 2-methylbuta- 1 ,3 -dienyl, homologs and isomers thereof, and the like.
- alkyl refers to and includes saturated linear and branched univalent hydrocarbon stractures and combination thereof, having the number of carbon atoms designated ⁇ i.e. , C 1 -C 10 means one to ten carbons). Particular alkyl groups are those having 1 to 20 carbon atoms (a "C 1 -C 2 0 alkyl").
- alkyl groups are those having 1 to 8 carbon atoms (a "Cj-Cg alkyl"), 3 to 8 carbon atoms (a “Cs-Cg alkyl”), 1 to 6 carbon atoms (a “Cj-Q alkyl”), 1 to 5 carbon atoms (a "C 1 -C5 alkyl”), or 1 to 4 carbon atoms (a "C 1 -C4 alkyl”).
- alkyl examples include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n -butyl, t-butyl, isobutyl, sec-butyl, homologs and isomers of, for example, n-pentyl, n-hexyl, n-heptyl, n-octyl, and the like.
- alkylene refers to the same residues as alkyl, but having bivalency. Particular alkylene groups are those having 1 to 6 carbon atoms (a "Cj-Ce alkylene"), 1 to 5 carbon atoms (a “C 1 -C5 alkylene”), 1 to 4 carbon atoms (a “C1-C4 alkylene”) or 1 to 3 carbon atoms (a "C 1 -C3 alkylene”). Examples of alkylene include, but are not limited to, groups such as methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), propylene (-CH 2 CH 2 CH 2 -), butylene
- Alkynyl refers to an unsaturated linear or branched univalent hydrocarbon chain or combination thereof, having at least one site of acetylenic unsaturation (i.e. , having at least one moiety of the formula ( ' C) and having the number of carbon atoms designated (i.e., C 2 -C 10 means two to ten carbon atoms).
- Particular alkynyl groups are those having 2 to 20 carbon atoms (a "C 2 -C 20 a kynyl"), having 2 to 8 carbon atoms (a "Ci-Cg aikynyi”), having 2 to 6 carbon atoms (a "C Ce alkynyl”), or having 2 to 4 carbon atoms (a "C 2 - C4 alkynyl”) .
- alkynyl examples include, but are not l imited to, groups such as ethynyl (or acetylenyl), prop-l-ynyl, prop-2-ynyl (or propargyl), but-l -ynyl, but-2-ynyl, but-3-ynyl, homologs and isomers thereof, and the like.
- aryl refers to and includes polyunsaturated aromatic hydrocarbon groups.
- Aryl may contain additional fused rings (e.g., from 1 to 3 rings), including additionally fused aryl, heteroaryl, cycloalkyl, and/or heterocyclyl rings.
- the aryl group contains from 6 to 14 annular carbon atoms. Examples of aryl groups include, but are not lim ited to, phenyl, naphthyl, biphenyl, and the like.
- cycloalkyl or “carbocycle” are used interchangeably and refer to and include cyclic univalent hydrocarbon structures, which may be fully saturated, mono- or polyunsaturated, but which are non-aromatic, having the number of carbon atoms designated (e.g. , Ci -Cio means one to ten carbons).
- Cycloalkyl or carbocycle groups can consist of one ring, such as cyclohexyl, or multiple rings, such as adamantyl, but excludes aryl groups.
- a cycloalkyl or carbocycle comprising more than one ring may be fused, spiro or bridged, or combinations thereof.
- a preferred cycloalkyl or carbocycle is a cyclic hydrocarbon having from 3 to 13 annular carbon atoms.
- a more preferred cycloalkyl or carbocycle is a cyclic hydrocarbon having from. 3 to 8 annular carbon atoms (a "Cs-Cs cycloalkyl").
- Examples of cycloalkyl or carbocycle groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3-cyclohexenyl, cycloheptyl, norbornyl, and the like.
- Halo or "halogen” refers to elements of the Group 17 series having atomic number 9 to 85.
- Preferred halo groups include fluoro, chloro, bromo and iodo. Where a residue is substituted with more than one halogen, it may be referred to by using a prefix corresponding to the number of halogen moieties attached, e.g., dihaloaryl, dihaloalkyl, trihaloaryl etc. refer to aryl and alkyl substituted with two ("di") or three ("tri") halo groups, which may be but are not necessarily the same halo; thus 4-chloro-3 -fluorophenyl is within the scope of dihaloaryl.
- An alkyl group in which each hydrogen is replaced with a halo group is referred to as a
- perhaloalkyl A preferred perhaloalkyl group is trifluoroalkyl (-CF 3 ).
- perhaloalkoxy refers to an alkoxy group in which a halogen takes the place of each H in the hydrocarbon making up the alkyl moiety of the alkoxy group.
- An example of a perhaloalkoxy group is trifluoromethoxy (-OCF3).
- heteroaryl refers to and includes unsaturated aromatic cyclic groups having from 1 to 10 annular carbon atoms and at least one annular heteroatom, including but not limited to heteroatonis such as nitrogen, oxygen and sulfur, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule at an annular carbon or at an annular heteroatom.
- Heteroaryl may contain additional fused rings (e.g., from 1 to 3 rings), including additionally fused aryl, heteroaryl, cycloalkyl, and/or heterocyclyl rings.
- heteroaryl groups include, but are not limited to, pyridyl, pyrimidy], thiophenyl, furanyl, thiazolyl, and the like.
- he teroaryl groups also include, but are not limited to, pyridyl, pyrimidyl, thiophenyl, furanyl, thiazolyl, oxazolyl, isoxazolyl, thiophenyl, pyrrolyl, pyrazolyl, 1,3,4- oxadiazolyl, imidazolyl, isothiazolyl, triazolyl, 1 ,3,4-thiadiazolyl, tetrazolyl, benzofuranyl, benzothiophenyl, pyrazolopyridinyl, indazolyl, benzothiazolyl, benzooxazoly] or
- a heteroaryl containing at least one additional fused ring that is nonaromatic is attached to the parent structure at an annular atom of the additional ring.
- a heteroaryl containing at least one additional ring that is nonaromatic is attached to the parent stmcture at an annular atom of the aromatic ring,
- heterocycle or “heteroeyclyf' refers to a saturated or an unsaturated nonaromatic group having from 1 to 10 annular carbon atoms and from. 1 to 4 annular heteroatonis, such as nitrogen, sulfur or oxygen, and the like, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heterocyclyl group may have a single ring or multiple condensed rings, but excludes heteroaryl groups.
- a heterocycle comprising more than one ring may be fused, spiro or bridged, or any combination thereof. In fused ring systems, one or more of the fused rings can be aryl, cycloalkyl or heterocyclyl. Examples of heterocyclyl groups include, but are not limited to,
- a heterocyclyl containing at least one additional ring (such as a fused additional ring) that does not contain a heteroatom is attached to the parent structure at an annular atom of the additional ring.
- a heterocyclyl containing at least one additional ring (such as a fused additional ring) that does not contain a heteroatom is attached to the parent stmcture at an annular atom of the ring containing a heteroatom.
- Optionally substituted unless otherwise specified means that a group may be unsubstituted or substituted by one or more (e.g., 1, 2, 3, 4 or 5) of the substituents listed for that group in which the substituents may be the same of different.
- an optionally substituted group has one substituent.
- an optionally substituted group has two substituents.
- an optionally substituted group has three substituents.
- an optionally substituted group has four substituents.
- an optionally substituted group has 1 to 2, 2 to 5, 3 to 5, 2 to 3, 2 to 4, 3 to 4, 1 to 3, 1 to 4 or 1 to 5 substituents.
- a "pharmaceutically acceptable earner” refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject.
- a pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excipient, stabilizer, or preservative.
- treatment is an approach for obtaining beneficial or desired results including clinical results.
- beneficial or desired results include, but are not limited to, one or more of the following: decreasing symptoms resulting from the disease, increasing the quality of life of those suffering from the disease, decreasing the dose of oilier medications required to treat the disease, delaying the progression of the disease, and/or prolonging survival of individuals.
- beneficial or desired results include shrinking a tumor (reducing tumor size); decreasing the growth rate of the tumor (such as to suppress tumor growth); reducing the number of cancer ceils; inhibiting, retarding or slowing to some extent and preferably stopping cancer cell infiltration into peripheral organs; inhibiting (slowing to some extent and preferably stopping) tumor metastasis; inhibiting tumor growth; preventing or delaying occurrence and/or recurrence of tumor; and/or relieving to some extent one or more of the symptoms associated with the cancer.
- beneficial or desired results include preventing or delaying occurrence and/or recurrence, such as of unwanted cell proliferation.
- delaying development of a disease means to defer, hinder, slow, retard, stabilize, and/or postpone development of the disease (such as cancer). This delay can be of varying lengths of time, depending on the history- of the disease and/or individual being treated. As is evident to one skilled in the art, a sufficient or significant delay can, in effect, encompass prevention, in that the individual does not develop the disease. For example, a late stage cancer, such as development of metastasis, may be delayed.
- an "effective dosage" or “effective amount” of compound or salt thereof or pharmaceutical composition is an amount sufficient to effect beneficial or desired results.
- beneficial or desired results include results such as eliminating or reducing the risk, lessening the severity of, or delaying the onset of the disease, including biochemical, histological and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease.
- beneficial or desired results include ameliorating, palliating, lessening, delaying or decreasing one or more symptoms resulting from the disease, increasing the quality of life of those suffering from the disease, decreasing the dose of other medications required to treat the disease, enhancing effect of another medication such as via targeting, delaying the progression of the disease, and/or prolonging survival.
- an effective amount comprises an amount sufficient to cause a tumor to shrink and/or to decrease the growth rate of the tumor (such as to suppress tumor growth) or to prevent or delay oilier unwanted cell proliferation. In some embodiments, an effective amount is an amount sufficient to delay development. In some embodiments, an effective amount is an amount sufficient to prevent or delay occurrence and/or recurrence.
- an effective amount can be administered in one or more administrations, in the case of cancer, the effective amount of the drug or composition may: (i) reduce the number of cancer cells; (ii) reduce tumor size; (iii) inhibit, retard, slow to some extent and preferably stop cancer cell infiltration into peripheral organs; (iv) inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; (v) inhibit tumor growth; (vi) prevent or delay occurrence and/or recurrence of tumor; and/or (vii) relieve to some extent one or more of the symptoms associated with the cancer.
- An effective dosage can be administered in one or more administrations.
- an effective dosage of compound or a salt thereof, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly. It is intended and understood that an effective dosage of a compound or salt thereof, or pharmaceutical
- composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition.
- an "effective dosage" may be considered in the context of administering one or more therapeutic agents, and a single agent may be considered to be given in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved.
- the term "individual” is a mammal, including humans.
- An individual includes, but is not limited to, human, bovine, horse, feline, canine, rodent, or primate.
- the individual is human.
- the individual (such as a human) may have advanced disease or lesser extent of disease, such as low tumor burden, in some embodiments, the individual is at an early stage of a proliferative disease (such as cancer). In some embodiments, the individual is at an advanced stage of a proliferati ve disease (such as an advanced cancer).
- Reference to "about” a value or parameter herein includes (and describes) embodiments that are directed to that value or parameter per se. For example, description referring to '"about X” includes description of "X”.
- R 1 is H or Cj-Ce alkyi wherein the Ci-Ce alkyl of R 1 is optionally substituted with oxo or
- R 2 and R 4 are each independently H, R D or oxo;
- R- and R " are each independently H or R c ;
- each R a , R , and R c is independently Ci -Ce alkyl, Cj-Ce aikenyl, Cj-Ce aikynyi, halogen,
- each R 3 , R b , and R ' is independently optionally substituted by halogen, oxo, -OR ! 1 , -NR n R 12 , -C(0)R n , -CN, -S(0)R n , -S(0) 2 R n ,
- R 1 is Ci-Q alkyl
- R is other tlian -NR 9 R l0 and R' is otlie than - C(0)R 8 :
- ⁇ is a single bond or a double bond, wherem when *n v is a double bond, R 4 is oxo;
- one of and » ⁇ is a double bond and the other is a single bond
- A is C6-C 12 aryl, 5- to 10-membered heteroaryl, 9- to 10-membered carbocycle, or 9- to 10-membered heterocycle, wherein the C & -Ci 2 aryl, 5- to 10-membered heteroaryl, 9- to 10- membered carbocycle, or 9- to 10-membered heterocycle of A is optionally further substituted with R 6 ;
- B is phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered carbocycle, 5- to 6- membered heterocycle, or 9 ⁇ to 10-membered heteroaryl, wherein the phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered carbocycle, 5- to 6-membered heterocycle, or 9- to 10-membered heteroaryl of B is optionally further substituted with R-';
- each R 6 and R 7 is independently oxo, Ci -Ce alkyl, Ci-Ce alkenyl, C 2 -Ce alkynyl, halogen,
- each R 8 is independently hydrogen, C Ce alkyl, C 2 -Ce alkenyl, C 2 -C'e alkynyl, C3-C6 cycloalkyi, Ce-Ci 4 aryl, 5-6-membered heteroaryl, 3-6-membered lieterocyclyl, -(Cr
- R and R'° are each independently hydrogen, Ci-Cg alkyl, C 2 -Ce alkenyl, C 2 -C6 alkynyl, Cs-Ce cycloalkyi, C6-C14 aryl, 5-6-membered heteroaryl, 3-6 membered lieterocyclyl, ⁇ (Cr C3 alkyleneJNR 1 'R 12 , -(C1-C3 alkylene)(C 3 -C6 cycloalkyi), -(C1-C3 alkylene)(3-6-membered heterocyely!), -(Ci-C 3 alkyiene)(5-6-membered heteroaryl) or -(Cj-C 3 alkylene)(C 6 aryl), wherein the C Ce alkyl, C 2 -Ce alkenyl, C 2 -Ce alkynyl, C 3 -Ce cycloalkyi, Ce-Ci4 aryl, 5-6-
- R 9 and R 10 are taken together with the atom to which they attached to form a 3- 6 membered lieterocyclyl optionally substituted by halogen, oxo, -OR 13 , -NR 3 R 14 or Ci- Ce alkyl optionally substituted by halogen, oxo or -OH;
- R J i and R are each independently hydrogen, Ci -Ce alkyl optionally substituted by halogen or oxo, ( ⁇ - ⁇ ' ,. alkenyl optionally substituted by halogen or oxo, or C 2 -Ce alkynyl optionally substituted by halogen or oxo;
- R' 1 and R 12 are taken together with the atom to which they attached to form a
- R" and R 14 are each independently hydrogen, Ci-C& alkyl optionally substituted by halogen or oxo, Cq-Ce alkenyl optionally substituted by halogen or oxo, or C -Ce alkynyl optionally substituted by halogen or oxo;
- R 1J and R 14 are taken together with the atom to which they attached to form a
- R 1 is H or Ci-Ce alkyl wherein tiie Cj-Ce alkyl of R 1 is optionally substituted witli oxo or
- R 2 and R 4 are each independently H, R b or oxo;
- R 3 and R 5 are each independently H or R°;
- each R a , R b , and R c is independently optionally substituted by halogen, oxo, -OR 11 , -NR n R 12 , -C(0)R 1 ! , -CN, -S(0)R n , -S(0) 2 R n ,
- R 1 is Ci-Ce alkyl
- R 4 is other than -NR 9 R'° and R 3 is other than - OC(0)R 8 ;
- R " is oxo
- ⁇ is a single bond or a double bond, wherein when ⁇ i s a double bond, R 4 is oxo;
- A is aryl, 5- to 10-membered heteroaiyl, 9 ⁇ to 10 ⁇ membered carbocycle, or 9- to 10-membered heterocycle, wherein the C6-C 12 aryl, 5- to 10-membered heteroaryl, 9- to 10- membered carbocycle, or 9- to 10-membered heterocycle of A is optionally further substituted with R 6 ;
- B is phenyl, 5- to 6 ⁇ membered heteroaryl, 5- to 6-membered carbocycle, 5- to 6- membered heterocycle, or 9- to 10-membered heteroaiyl, wherein the phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered carbocycle, 5- to 6-membered heterocycle, or 9- to 10-membered heteroaryl of B is optionally further substituted with R ';
- B is 5- to 6-membered heterocycle, A is other than phenyl or pyridyl optionally further substituted with R' ;
- each R 6 and R 7 is independently oxo, Ci -Ce alkyl, C 2 -Ce aikenyl, C 2 -Ce alkynyl, halogen,
- R 8 is independently hydrogen, Cj -Ce alkyl, C 2 -Ce aikenyl, C 2 -Ce alkynyl, C3-C6
- cycloalkyl C C 14 aryl, 5-6-membered heteroaryl or 3-6-membered heterocyclyi, wherein the C i -Ce alkyl, C 2 -Ce aikenyl, C 2 -Ce alkynyl, C 3 -C6 cycloalkyl, Ce-Cw aiyl, 5-6-membered heteroaiyl and 3-6-membered heterocyclyi are independently optionally substituted by halogen, oxo, -CN, -OR 13 , -NR 13 R 14 , -P(0)(OR 13 )(OR l4 ), phenyl optionally substituted by halogen, or Ci- C alkyl optionally substituted by halogen, -OH or oxo;
- R 9 and R 10 are each independently hydrogen, Cj-Ce alkyl, C 2 -C6 aikenyl, C 2 -Ce alkynyl, C3-C6 cycloalkyl, C6-C1 aryl, 5-6-membered heteroaryl or 3-6 membered heterocyclyi, wherein the CrCe alkyi, C 2 -C6 aikenyl, C 2 -Ce alkynyl, C 3 -Ce cycloalkyl, Q-Cu aryl, 5-6-membered heteroaiyl and 3-6 membered heterocyclyi are independently optionally substituted by halogen, oxo, -CN, -OR 13 , -NR 13 R 14 or Ci-Ce alkyl optionally substituted by halogen, -OH or oxo;
- R 9 and R 10 are taken together with the atom to which they attached to form a 3-
- Ce alkyl optionally substituted by halogen, oxo or -OH;
- R 11 and R l are each independently hydrogen, Ci -Ce alkyl optionally substituted by halogen or oxo, C 2 -Ce aikenyl optionally substituted by halogen or oxo, or Cj-Ce alkynyl optionally substituted by halogen or oxo;
- R 11 and R 12 are taken together with the atom to which they attached to form a
- R 13 and R 14 are each independently hydrogen, C i -Ce alkyl optionally substituted by halogen or oxo, C 2 -Ce aikenyl optionally substituted by halogen or oxo, or C 2 -Ce alkynyl optionally substituted by halogen or oxo; or R l3 and R 14 are taken together with the atom to which they attached to form a 3-6 membered heterocyclyl optionally substituted by halogen, oxo or Ci-Ce alkyl optionally substituted by oxo or halogen.
- A is selected from the group consisting of e-Cn aryl and 5- to 10-membered heteroaryl, wherein the Ce-Cn aryl and 5- to 10-membered heteroaryl of A is optionally further substituted with R u .
- B is selected from the group consisting of phenyl and 5- to 6- membered heteroaryl, wherein the phenyl and 5- to 6-membered heteroaryl of B is optionally further substituted with R' .
- A is selected from the group consisting of Ce-Cn aryl and 5- to 10-membered heteroaryl, wherein the Ce-Cn aryl and 5- to 10-membered heteroaryl of A is optionally further substituted with R D and B is selected from the group consisting of phenyl and 5- to 6-membered heteroaryl, wherein the phenyl and 5- to 6-membered heteroaryl of B is optionally further substituted with R'.
- R 3 , R b , and R c are independently Ci-Ce alkyl, C 2 -C 6 alkenyl, C 2 ⁇ C 6 alkynyl, halogen, -CN, -OR 8 , -SR.
- R", R b , and R c are independently Cj-Ce alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, halogen, -CN, -OR 8 , -SR 8 , -NR 9 R 10 , -C(0)NR 9 R 10 , -NR 8 C(0)R 9 , C3-C6 cycloalkyl, 3-12-membered heterocyclyl, 5- to 10-membered heteroaryl or Ce-Ci 4 aryl.
- R 3 , R b , and R c are independently Ci-Ce alkyl, halogen, -CN, -OR 8 , -SR 8 or -NR 9 9R formula10
- R", R , and R" are independently -CH 3 , halogen, -CN or -OCH 3 .
- R 1 is H or methyl .
- R s is H, Ci-Ce alkyl, halogen,
- R 1 , R 2 , R 3 , and ⁇ ⁇ are each H and R 4 is oxo. In some embodiments of the compound of Formula (I), R 1 , R 3 , R and R 3 are each H and R is oxo.
- At least one of R 3 , R 4 , and R ' is not H.
- at least one of R 3 , R 4 , and R 5 is Cj -Ce alkyl, halogen, C6-C14 aryl, -CN, or -OR 8 .
- R R 3 , R 4 , and R 5 are each H.
- At least one of R 2 , R ! . and R 5 is not H.
- at least one of R 2 , R 3 , and R J is C ⁇ ⁇ Ce alkyl, halogen, Ce ⁇ Ci 4 aryl, -CN, or -OR 8 .
- R R 2 , R J , and R 5 are each H.
- R 1 is Ci-C 6 alkyl, ( ⁇ .-( ' ,. alkenyl, C 2 -C 6 alkynyl, halogen, -CN, -OR s , -SR 8 , ⁇ NR 9 R !0 , -C(O)NR 9 R !0 , -NR 8 C(0)R 9 , -NR 8 C(0)NR 9 R 10 , -S(0)R 8 , -S(0) 2 R 8 , -NR 8 S(0)R 9 , -NR 8 S(0) 2 R 9 , -S(O)NR 9 R i0 , -S(0) 2 NR 9 R 10 , C3-C6 cycloalkyi, 3-12-membered heterocyclyl, 5- to 10-membered heteroaryl or C6-C14 aryl.
- R 1 is Ci-Ce alkyl, C 2 -Ce alkenyl, C 2 -C 6 alkynyl, halogen, -CN, -OR 8 , -SR 8 , -NR 9 R !0 , -C(O)NR 9 R !0 , -NR 8 C(0)R 9 , C 3 - Ce cycloalkyi, 3- 12-membered heterocyclyl, 5- to 10-membered heteroaryl or C6-C14 aryl.
- R 1 is Ci -Ce alkyl, halogen, -CN, -OR 8 , -SR 8 or -NR R 10 .
- R 1 is - CH 3 , halogen, -CN or -OCH 3 .
- R 1 is H or methyl.
- R 1 is H, Ci -Ce alkyl, halogen, -CN, or -OR 8 .
- R ! is H or C 1 -C & alkyl.
- R 1 is H or methyl. In some embodiments, R 1 is H.
- R 2 is Ci-Cc, alkyl, C 2 -C 6 alkenyl, C 2 ⁇ C 6 alkynyl, halogen, -CN, -OR 8 , -SR.
- R 2 is Ci-Ce alkyl, C 2 -Ce alkenyl, C 2 ⁇ C 6 alkynyl, halogen, -CN, -OR 8 , -SR. 8 , -NR 9 R !0 , -C(O)NR 9 R !0 , -NR 8 C(0)R 9 , C 3 - Ce cycloalkyl, 3- 12-membered heterocyclyl, 5- to 10-membered heteroaryl or C6-C14 aryl.
- R 2 is Ci -Ce alkyl, halogen, -CN, -OR * , -SR" or -NR R .
- R ' is - CH3, halogen, -CN or -OCH3.
- R 2 is H or methyl.
- R 2 is H, Ci-C & alkyl, halogen, -CN, or -OR 8 .
- R 2 is oxo.
- R J is Ci-C 6 alkyl, ( ⁇ .-( ' ,. alkenyl, C 2 -C 6 alkynyl, halogen, -CN, -OR 8 , -SR.*.
- R 3 is C .
- R 3 is Ci -Ce alkyl, halogen, -CN, -OR * , -SR e or -NR R .
- R is - CH 3 , halogen, -CN or -OCH 3 .
- R 3 is H or methyl.
- R J is H, Ci-C 6 alkyl, halogen, -CN, or -OR 8 .
- R 4 is CpCe alkyl
- R 4 is Ci-Ce aikyl, QrG aikenyl, C 2 -C 6 alkynyl, halogen, -CN, -OR 8 , -SR 8 , -NR 9 R i0 , -C(O)NR 9 R i0 , -NR 8 C(0)R 9 , C 3 - Ce cycloaikyl, 3-12-membered heterocyciyi, 5- to 10-nienibered heteroaiyl or Ce-Ci 4 aryl.
- R 4 is Ci-Ce aikyl, halogen, -CN, -OR 8 . -SR 8 or -NR 9 R 10 .
- R is - CH3, halogen, -CN or -OCH3.
- ir is H or methyl.
- R is H, Ci-Ce aikyl, halogen, -CN, or -OR 8 .
- R 4 is oxo.
- R 5 is Ci-Ce aikyl, (X V. aikenyl, ( >-C profession alkynyl, halogen, -CN, -OR 8 , -SR 8 , -NR 9 R 10 , -C(O)NR 9 R i0 , -NR 8 C(0)R 9 , -NR 8 C(0)NR 9 R 10 , -S(0)R 8 , -S(0) 2 R 8 , -NR 8 S(0)R 9 , -NR 8 S(0) 2 R 9 , -S(O)NR 9 R !0 , -S(0) 2 NR 9 R 10 , C 3 -C6 cycloaikyl, 3-12-membered heterocyciyi, 5- to 10-membered heteroaiyl or C6-C14 aryl.
- R 3 is Ci-Cr, aikyl, C 2 -C & aikenyl, C 2 -C6 alkynyl, halogen, -CN, -OR 8 , -SR 8 , -NR 9 R i0 , -C(O)NR 9 R i0 , -NR 8 C(0)R 9 , C 3 ⁇
- R 5 is Ci-Ce aikyl, halogen, -CN, -OR 8 . -SR 8 or -NR 9 R 10 .
- R 3 is - CH 3 , halogen, -CN or -OCH 3 .
- R 3 is H or methyl.
- R 5 is H, Ci-Ce aikyl, halogen, -CN, or -OR 8 . In some embodiments of a compound of Formula (I), (II), or (III), R 5 is H, Ci-Ce aikyl, halogen, -CN, or -OR 8 . In some embodiments, R 5 is H. In particular embodiments of a compound of Formula (I), (II), or (III), R 1 is H or Ci-Ce aikyl (such as methyl) and R 5 is H, ( ' ⁇ . -( " (, aikyl, halogen, -CN, or -OR 8 .
- A is Ce-C ⁇ . aryl optionally further substituted with R 6 .
- A is phenyl or naphthyl, optionally substituted with R D .
- A is phenyl.
- A is naphthyl.
- A is phenyl or naphthyl, substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR 8 , -NR 9 R 10 , -N() 2 , -C(0)R 8 , -C(0)OR 8 , -C(0)NR 9 R 10 ,
- A is phenyl, substituted with one or more groups selected from halogen, -CN, -OH, -OC Ce aikyl, -NH 2 , -N0 2 , C 3 -C 6 cycloaikyl and C C 6 aikyl optionally substituted by halogen.
- A is phenyl, substituted with one or more groups selected from halogen, -OH, and Ci-Ce alkyl.
- A is 5- to 10- membered heteroaryl optionally further substituted with R b .
- A is selected from the group consisting of pyridyl, quinolinyl, isoquinolinyl, quinoxalinyl, cinnolinyl, quinazolinyl, naphthyridinyl, benzoxazolyl, benzothiazolyl, benzoimidazoyl, pyrrolyl, pyrazolvl, imidazolyi, triazolyl, tetrazolyl, furanyl, isoxazolyl, oxazolvl, oxadiazolyl, thioplienyl, isothiazolyl, thiazolyl, thiadiazolyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, te
- the optional substitution with R 6 provides a moiety that is unsubstituted.
- the optional substitution with R D provides a moiety that is substituted with 1-3 R 6 , which may be the same or different.
- A is a 10-membered heteroaryl optionally further substituted with R D , wherein the 10-membered heteroaryl is a 6/6-ring fused system (i.e., a ring system formed by fusing a 6-membered ring with a 6-membered ring).
- A is a 9- membered heteroaryl, wherein the 9-membered heteroaryl is a 6/5 -ring fused system (i.e., a ring system formed by fusing a 6-membered ring with a 5-membered ring).
- the 6/5 -ring fused system of A is attached to the rest of the compound via the 6-membered ring.
- the 6/5-ring fused system of A is attached to the rest of the compound via the 5-membered ring.
- A is selected from the group consisting of:
- such groups are not further substituted with R 6 .
- A is selected from the group consisting of:
- A is 5- to 10-membered heteroaryl optionally further substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR S , -NR 9 R 10 , -N0 2 , -C(0)R 8 , -C(0)OR 8 , -C(())NR 9 R i0 , -C(0)NR 8 S(0) 2 R 9 , -OC(())R 8 , -OC(O)NR 9 R !0 , -NR 8 C(0)R 9 , -NR 8 C(O)NR 9 R i0 , -S(0)R 8 , -S(0) 2 R 8 , C3-C6 cycloaikyl and Ci-Ce alkyl optionally substituted by halogen.
- A is 5- to 10-membere
- A is a 9- to 10- membered carbocycle optionally further substituted with R 6 .
- A is a 10- membered carbocycle, wherein the 10-membered carbocycle is a 6/6-ring fused system (i.e., a ring system formed by fusing a 6-membered ring with a 6-membered ring).
- A is a 9-membered carbocycle, wherein the 9 ⁇ membered carbocycle is a 6/5-ring fused system (i.e., a ring system formed by fusing a 6-membered ring with a 5 -membered ring).
- the 6/5-ring fused system of A is attached to the rest of the compound via the 6-membered ring.
- the 6/5-ring fused system of A is attached to the rest of the compound via the 5 -me inhered ring.
- A is a fully saturated 9- to 10-membered carbocycle.
- A is a partially saturated 9- to 10- membered carbocycle.
- A is selected from the group consisting of decahydronaphthalenyl, octahydroindenyl, 1,2,3,4- tetrahydronaphthalenyl, and 2,3-dihydroindenyl, each optionally substituted with R 6 .
- A is a 9- to 10-membered carbocycle optionally further substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR.
- A is a 9- to 10-membered carbocy cle optionally further substituted with one or more groups selected from Ci -Ce alkyl, halogen, -CN, -OH, and -OCi-Ce alkyl.
- A is a 9- to 10-membered heterocycle optionally further substituted with R b .
- A is a 10-membered heterocycle optionally further substituted with R D , wherein the 10-membered heterocycle is a 6/6-ring fused system, (i.e., a ring system formed by fusing a 6-membered ring with a 6-membered ring).
- A is a 9-membered heterocycle, wherein the 9-membered heterocycle is a 6/5-ring fused system (i.e., a ring system formed by fusing a 6-membered ring with a 5-membered ring).
- the 6/5-ring fused system of A is attached to the rest of the compound via the 6- membered ring. In other embodiments, the 6/5-ring fused system of A is attached to the rest of the compound via the 5-membered ring. In some embodiments, A is a fully saturated 9- to 10- membered heterocycle. In some embodiments, A is a partially saturated 9- to 10-membered heterocycle.
- A is selected from the group consisting of teirahydroquinolinvl, tetrahydroisoquinolinyl, decahydroquinolinyl, decahydroisoquinolinyl, indolinyl, isomdolinyl, tetrahydronaphthyridinyl and hexahydrobenzoimidazolyl, each optionally further substituted with R b .
- A is a 9- to 10-membered heterocycle optionally further substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR 8 , -NR 9 R 10 , -NO 2 , -C(0)R 8 , -C(())()R 8 , -C(O)NR 9 R !0 , -C(0)NR 8 S(0) 2 R 9 , -OC(0)R 8 , -OC(O)NR 9 R i0 , -NR 8 C(0)R 9 , -NR 8 C(O)NR 9 R !0 , -S(0)R 8 , -S(0) 2 R 8 , C 3 -C 6 cycloaikyl and d- Cf 3 alkyl optionally substituted by halogen.
- A is a 9- to 10-membered heterocycle optionally further substituted with one or more groups selected from Ci-Ce alkyl, halogen, -CN, -QCi-Ce alkyl. In some embodiments, A is selected from the group
- R D each optionally substituted with R D .
- such groups are not further substituted with R b .
- such groups are further substituted with 1-3 R 6 , which may be the same or different.
- R" is independently selected from the group consisting of halogen, -CN, -OR 8 , -SR 8 , -NR 9 R !0 , -N0 2 , -C(0)R 8 , -C(())()R 8 , -C(0)NR 9 R 10 , -C(0)NR 8 S(0) 2 R 9 , -OC(0)R 8 , -OC(O)NR 9 R i0 , -NR 8 C(0)R 9 , -NR 8 C(0)NR 9 R 10 , -S(Q)R 8 , -S(0) 2 R 8 , C 3 -C 6 cycloaikyl and C C 6 alkyl optionally substituted by halogen.
- R 6 is independently selected from the group consisting of halogen, -CN
- each R 6 is independently selected from the group consisting of halogen, -CN, -OR 8 , -SR 8 , -NR V R 10 , -NO 2 , -C(())R 8 , -C(0)OR 8 , -C(O)NR 9 R !0 , -C(0)NR 8 S(0) 2 R 9 , -OC(0)R 8 , -OC(0)NR 9 R 10 ,
- R D is independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OR 8 .
- A is selected from group consisting of:
- each description of A may be combined with each description of R 1 - R 5 the same as if each and every combination were specifically and individually listed. It is similarly understood that each description of A may be combined with each description of B (and furtlier with each description of R 1 -R 5 ) the same as if each and every combination were specifically and individually listed.
- each description of A may be combined in one aspect with a variation in which R 1 , R J , R 5 are each hydrogen and one of R 2 and R 4 is hydrogen and one of R 2 and R 4 is oxo. In one such variation, each description of A is combined in one aspect with a variation in which R !
- each description of A is combined in one aspect with a variation in which R ! , R 3 , R 4 , R 3 are each hydrogen and R is oxo.
- Such embodiments may furher be combined with each description of B.
- B is phenyl, optionally further substituted with R' .
- B is 5- to 6-membered heteroaryl optionally further substituted with R' .
- B is pyrrolyl, pyrazolyl,
- imidazolyl triazolyl, tetrazolyl, furanyl, isoxazolyi, oxazolyl, oxadiazolyl, thiophenyl,
- B is furanyl, pyridinyl, oxazoyi, or oxadiazoyl, each optionally substituted with R 7 .
- B is a 5- to 6- membered carbocycle optionally further substituted with R 7 .
- B is a fully saturated 5- to 6-membered carbocycle optionally further substituted with R .
- B is cyclopentyl or cyclohexyl, optionally further substituted with R' .
- B is a 5- to 6-membered carbocycle optionally substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR 8 , -NR 9 R 10 , -NC) 2 , -C(0)R 8 , -C(0)OR 8 ,
- B is a 5- to 6-membered carbocycle optionally substituted with halogen.
- B is a 5- to 6- membered heterocycJe optionally further substituted with R' .
- B is a fully saturated 5- to 6-membered heterocycle optionally further substituted with R 7 .
- B is pyrrolidinyl, pyrazolidinyl, imidazolidinyl, tetrahydrofuranyl, 1,3-dioxolanyl, tetrahydrothiophenyl, oxathiolanyl, sulfolanyl, piperidinyl, piperazinyl, tetrahydropyranyl, dioxanyl, thianyl, dithianyi, trithianyl, morpholinyl, thiomorpholinyl optionally further substituted with R' .
- B is a 5- to 6-membered heterocycle optionally substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR 8 , ⁇ NR 9 R 10 , ⁇ N0 2 , -C(0)R 8 , -C(0)OR 8 , -C(0)NR 9 R 10 , -C(0)NR 8 S(0) 2 R 9 , -OC(0)R 8 , -OC(0)NR 9 R 10 ,
- B is a 5- to 6-membered heterocycle optionally substituted with halogen.
- B is a 9- to 10- membered heteroaryl optionally further substituted with R 7 .
- B is selected from the group consisting of pyridyl, quinolinyl, isoquinolinyl, quinoxalinyl, cinnolinyl.
- B is a 9- to 10-membered heteroaryi optionally substituted with one or more groups selected from halogen, -CN, -OR 8 , -SR S , -NR 9 R 10 , -N0 2 , -C(0)R 8 , -C(0)OR 8 , -C(O)NR 9 R 10 5 -C(0)NR 8 S(0) 2 R 9 , -OC(0)R 8 , -OC(O)NR 9 R !0 , -NR 8 C(0)R 9 , -NR 8 C(0)NR 9 R 10 , -S(0)R 8 , -S(0) 2 R 8 , C 3 -C 6 cycloalkyl and CVC 6 alkyl optionally substituted by halogen.
- B is a 9- to 10-membered heteroaryi optionally substituted with halogen.
- R 7 is
- R 7 is halogen.
- B is selected from the group consisting of:
- B is selected from the group consisting of:
- B is selected from
- A is C 6 -Ci 2 aryl or 5- to 10-rnembered heteroaryl, each optionally further substituted with R 6
- B is phenyl or 5- to 6-membered heteroaryl, each optionally further substituted with R .
- A is Ce-Cii aryl, optionaliy further substituted with R 6
- B is phenyl, optionally further substituted with R ' .
- A is Ce-Cu aryl, optionally further substituted with R 6 , and B is 5- to 6-membered heteroaryl, optionally further substituted with R ' .
- A is 5- to 10-membered heteroaryl, optionaliy further substituted with R 6
- B is phenyl, optionally further substituted with R' .
- A is 5- to 10-membered heteroaryl, optionally further substituted with R 6
- B is 5- to 6-membered heteroaryl, optionally further substituted with R 7 .
- A is 9- to 10- membered carbocycle or 9- to 10-membered heterocycle, each optionally further substituted with R°, and B is phenyl, 5- to 6-membered heteroaryl, 5- to 6-membered carbocycle, or 5- to 6- membered heterocycle, each optionally further substituted with R ' .
- A is 9- to 10-membered carbocycle, optionally further substituted with R 6
- B is phenyl, optionally further substituted with R ? .
- A is 9- to 10-membered carbocycle, optionaliy further substituted with R 6 , and B is 5- to 6-membered heteroaryl, optionally further substituted with R' .
- A is 9- to 10-membered carbocycle, optionally further substituted with R 6 , and B is 5- to 6-membered carbocycle, optionally further substituted with R' .
- A is 9- to 10-membered carbocycle, optionally further substituted with R 6 , and B is 5- to 6-membered heterocycle, optionally further substituted with R 7 .
- A is Ce-Cu aryl or
- A is Ce-C] -, aryl, optionally further substituted with R 6 , and B is 5- to
- A is C & - Ci2 aryl, optionally further substituted with R 6 , and B is 5- to 6-membered heterocycle, optionally further substituted with R 7 .
- A is 5- to 10-membered heteroaryi, optionally further substituted with R 6 , and B is 5- to 6-membered carbocycle, optionally further substituted with R 7 .
- A is 5- to 10-membered heteroaryi, optionally further substituted with R b
- B is 5- to 6-membered heterocycle, optionally further substituted with R' .
- B is not a saturated heterocycle.
- A is C 6 -C !2 aryl or 5- to 10-membered heteroaiyl, each optionally further substituted with R 6 , and B is 9- to 10- membered carbocycle, optionally further substituted with R' .
- A is Ce- Ci 2 aryl, optionally further substituted with R", and B is 9- to 10-membered carbocycle, optionally further substituted with R' .
- A is 5- to 10-membered heteroaryi, optionally fuither substituted with R 6 , and B is 9- to 10-membered carbocycle, optionally further substituted with R' .
- A is
- R 6a , R 6 , R 6c , R 6d , R 6e , and R 6f are each independently H, C, -C 6 alkyl, C 2 -C 6 alkenyl, C 2 ⁇ C 6 alkynyl, halogen, -CN, -OR 8 , -SR.
- R 6a , R 6b , R 6e , R 6d , R 6e , and R 6f are each independently H, C C 6 alkyi, halogen, -CN, or -OC, -C 6 alkyi.
- R 63 , R 6b , R 6c , R 6d , R 6e , and R 6f are each H,
- one of R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f is CI, F, Br, or I.
- one of R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f is CI.
- one of R 6a , R 6b , R DC , R 6d , R ue , and R ui is halogen and the others are each H.
- one of R 6a , R 6 , R 6c , R 6d , R 6e , and R 6f is halogen and one of R 6a , R 6b , R & , R 6d , R 6e , and R 6f is C,-C 6 alkyi.
- R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f is CI and one of R 6a , R 6b , R 6c , R 6d , R 6e , and R Di is methyl.
- R 6a is CpCe alkyi.
- R 6b is C ⁇ - Ce alkyi.
- R bc is Cj-Q alkyi.
- R 6d is Ci-Ce alkyi.
- R 6e is C ⁇ ⁇ alkyi.
- R of is C e alkyi.
- R 6a is methyl, ethyl, n-propyl, isopropyi, n-buty], isobutyl, secbutyl, or tertbutyl.
- R 6 ° is methyl, ethyl, n-propyl, isopropyi, n-buty 1, isobutyl, secbutyl, or teitbutyl.
- R 6c is methyl, ethyl, n-propyl, isopropyi, n-buty 1, isobutyl, secbutyl, or tertbutyl.
- R 6a is methyl, ethyl, n-propyl, isopropyi, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R 68 is methyl, ethyl, n-propyl, isopropyi, n-butyl, isobutyl, secbutyl, or tertbutyi.
- R 6f is methyl, ethyl, n-propyl, isopropyi, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R D3 is Cj-Ce alkyi and R bb is halogen.
- R 6a is Ci-C'e alkyi and R bC is halogen.
- R 6d is Ci-Ce alkyi and R Dd is halogen.
- R ud is Ci-Ce alkyi and R 6e is halogen.
- R 6a is Ci -Ce alkyi and R 6f is halogen.
- R 6b is Ci-C & alkyi and R b3 is halogen.
- R 6b is Ci-C & alkyi and R 6c is halogen .
- R 6b is Ci-Ce alkyi and R 6a is halogen.
- R ob is Cj-Ce alkyi and R 6e is halogen.
- R 6b is Ci-Ce alkyi and R" f is halogen.
- R" is Cj-Ce alkyi and R 6a is halogen.
- R 6c is Ci-Ce alkvl and R 6b is halogen.
- R 6c is Ci-Ce alkvl and R 6u is halogen.
- R 6c is Cj -Ce alkyi and R 6e is halogen.
- R 6c is Ci-Ce alkvl and R of is halogen.
- R od is Cj -Ce alkyl and R 6a is halogen.
- R 6d is Ci-Ce alkyl and R 6b is halogen.
- R 6a is Ci-Ce alkyl and R 6'" is halogen.
- R 6a is Ci-Ce alkyl and R De is halogen.
- R 6d is Ci-Ce alkyl and R Di is halogen.
- R oe is Ci-Ce alkyl and R 6d is halogen.
- R 6e is Ci-C& alkyl and R 6b is halogen. In some embodiments, R 6e is Ci-Cr, alkyl and R 6c is halogen. In some embodiments, R 6e is Ci-Ce alkyl and R Dd is halogen. In some embodiments, R ue is Ci-Ce alkyl and R 61 is halogen. In some embodiments, R 61 is Cj -Ce alkyl and R 6a is halogen. In some embodiments, R 61 is Ci-Ce alkyl and R 6b is halogen. In some embodiments, oi is C Cc, alkyl and R 6c is halogen .
- R 6 ' is Ci-Ce alkyl and R 6d is halogen.
- R Di is C i-Ce alkyl and R De is halogen.
- two of R ud , R Db , R 6c , R 6 , R 6e , and R 6f are halogen.
- two of R 63 , R 6b , R 6c , R 6d , R 6e , and R 6f are Ci-Ce alkyl ,
- A is
- R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f are independently selected from the group consisting of Cj -Ce alkyl, halogen, -CN, and -OCi-Ce alkyl, and the remainder of R 63 , R 6b , R 6c , R 6d , R 6e , and R bf are each H; and B is phenyl, optionally substituted with R 7 .
- A is
- R 6a , R 6b , R 6c , R 6a , R 6e , and R 6 ' are independently selected from the group consisting of C pCe alkyl, halogen, -CN, and -OCj -Ce alkyl, and the remainder of R 6a , R 6b , R DC , R bd , R 6e , and R 61 are each H: and B is a 5- to 6-membered heteroaryl, optionally substituted with R 7 .
- R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f are independently selected from the group consisting of Ci-Cr, alkvl, halogen, -CN, and -Od-Ce alkyl, and the remainder of R 6 ", R Db , R 6t , R ud , R 6s , and R 6i are each H; and B is a 5- to 6-membered carbocycle, optionally substituted with R 7 .
- A is
- R 6a , R 6 , R 6c , R 6d , R 6e , and R 6f are independently selected from the group consisting of d-Ce alkyl, halogen, -CN, and -OCi-Ce alkyl, and the remainder of R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f are each H: and B is a 5- to 6-membered
- R 6a , R 6b , R 6c , R 6d , R 6e , and R 6f are independently selected from the group consisting of Ci-Cr, alkyl, halogen, -CN, and -Od-Ce alkyl, and remainder of R 6a , R Db , R 6t , R ud , R 6s , and R 6i are each H; and B is a 9- to 10-membered
- heteroaryi optionally substituted with R .
- A is
- R 6a , R 6 , R 6c , R 6d , R 6e , and R 6f are independently lected from the group consisting of d-Ce alkyl, halogen, -CN, and -OCi ⁇ Ce alkyl, and th
- R b3 , R 6b , R" 1' , R od , R 6s , and R 61 are each H: and B is selected from the group consisting of:
- A is
- R 6a , R 6b , R 6c , R 6a , R 6e , and R 6 ' are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCj -Ce aikyl, and the remainder of R b3 , R 6b , R" 1' , R od , R 6s , and R 61 are each H: and B is selected from the group consisting of
- A is
- R 6a , R , R 6a , R 6e , and R are independent! selected from the group consisting of Ci ⁇ C 6 alkyl, halogen, -CN, and -OCi-C 6 alkyl, and the
- R 6b , R 6c , R bd , R 6e , R 6f , and R 6g are each independently H, C C 6 alkyl, halogen, ⁇ CN, or -OCi-Ce alkyl.
- R 6D , R 6'" , R 6d , R 6e , R 6f , and R 6 ⁇ are each H.
- one of R 6b , R uC , R Dd , R 6e , R ui , and R 6g is CI, F, Br, or I.
- one of R* R 6c , R 6d , R 6e , R 6f , and R 6g is CI.
- one of R* R 6c , R 6d , R 6e , R 61 , and R Dg is halogen and the others are each H.
- one of R 6b , R 6c , R 6d , R 6e , R 6f , and R 6g is halogen and one of R 6b , R 6c , R 6d , R 6e , R 6f , and R 6g is C C 6 alkyl.
- R 6b , R 6c , R 6d , R 6e , R 6f , and R 6 is CI and one of R 6b , R 6c , R 6d , R 68 , R 6i , and R Dg is methyl.
- R 6g is Ci-Ce alkyl.
- R bb is Cr C-6 alkyl.
- R D£ is Ci-Ce alkyl.
- R 6d is C 5 ⁇ C & alkyl.
- R 6e is Cj-Ce alkyl .
- R 6f is Cj -Ce alkyl.
- R 6s is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or tertbutyl.
- R b0 is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or terthuty!.
- R 6c is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or tertbutyl.
- R ba is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or tertbutyl.
- R 6e is metliyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or tertbutyl.
- R 6t is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyi, secbutyl, or tertbutyl.
- R 6g is C -.
- R 6 is Ci-Ce alkyl and R 6c is halogen.
- R 6g is Ci -Ce alkyl and R 6d is halogen.
- R 6g is Cj-Ce alkyl and R 6e is halogen .
- R 6 is d-Ce alkyl and R 61 is halogen.
- R bb is Ci-C-6 alkyl and R 6 is halogen.
- R 6b is Ci-Ce alkyl and R 6 is halogen.
- R 6b is Ci-Ce alkyl and R DC is halogen.
- R 6b is Ci-Ce alkyl and R DC1 is halogen.
- R" b is C . -Ce alkyl and R 6s is halogen.
- R Db is Ci -Ce alkyl and R 6f is halogen.
- R bL is Ci-Ce alkyl and R bg is halogen.
- R 6c is Ci-Ce alkyl and R Db is halogen.
- R uC is Ci-Ce alkyl and R 6d is halogen.
- R bc is Cj -Ce alkyl and R 6e is halogen.
- R 6c is Ci ⁇ Ce alkyl and R" 1 is halogen .
- R od is Ci-Ce alkyl and R 6g is halogen. In some embodiments, R 6d is Ci-Ce alkyl and R 6b is halogen. In some embodiments, R DC1 is Ci-C-6 alkyl and R 6 " is halogen. In some embodiments, R bC! is Ci-Ce alkyl and R oe is halogen. In some embodiments, R 6d is Ci-Ce alkyl and R of is halogen. In some embodiments, R 6e is G-G alkyl and R 6g is halogen. In some embodiments, R 6e is G-G alkyl and R 6b is halogen.
- R 6s is G-G alkyl and R 6c is halogen.
- R 6e is G ⁇ G alkyl and R od is halogen.
- R 6e is G-G alkyl and R 61 is halogen.
- R 6i is G ⁇ G alkyl and R 6g is halogen.
- R 6i is G-G alkyl and R 6b is halogen.
- R 6f is G ⁇ G alkyl and R DC is halogen.
- R 6x is G-G alkyl and R 6d is halogen.
- R of is G-G alkyl and R oe is halogen.
- two of R 6b , R w' , R 6d , R be , R 6 , and R 6g are halogen.
- two of R 6b , R 6c , R 6d , R be , R 6i , and R 6g are d-Ce alkyl .
- A is
- R Dg are independently selected from the group consisting of Cj-Ce alkyl, halogen, -CN, and ⁇ OG ⁇ G alkyl, and the remainder of R 6D , R 6" , R 6d , R 6e , R 6f , and R 6g are each H; and B is phenyl, optionally substituted with R .
- R 6b , R DC , R 6d , R 6t , R DI , and R 6g are independently selected from the group consisting of Ci-CV, alkyl, halogen, -CN, and -OG-Ce alkyl, and the remainder of R 6b , R 6c , R 6d , R 6e , R 61 , and R 6g are each H; and B is a 5- to 6-membered heteroaryl, optionally substituted with R 7 .
- R Dg are independently selected from the group consisting of G-Ce alkyl, halogen, -CN, and -QG-G alkyl, and the remainder of R 6b , R 6c , R 6d , R 6e , R 61 , and R 6g are each H; and B is a 5- to 6-membered carbocycle, optionally substituted with R ? .
- R Dg are independently selected from the group consisting of Cj-Ce alkyl, halogen, -CN, and -OC ⁇ ⁇ C ⁇ , alkvl, and the remainder of R 6 °, R 6" , R 6d , R 6e , R 6f , and R 6g are each H; and B is a 5- to 6-membered heterocycle, optionally substituted with R'.
- R 6b , R DC , R 6d , R & , R DI , and R 6g are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCj-Ce alkyl, and the remainder of R 6b , R 6c , R 6d , R 6e , R 61 , and R 6g are each H; and B is a 9- to 10-membered heteroaryl, optionally substituted with R 7 .
- R Dg are independently selected from the group consisting of Cj-Ce alkyl, halogen, -CN, and -OC ⁇ ⁇ C ⁇ , alkyl, and the remainder of 6b , R 6'" , R 6d , R 6e , R 6f , and R 6g are each H; and B is selected from the group
- R 6 °, R 6c , R 6d , R Dt , R 6f , and R 6 are each H; and B is selected from the group consisting of:
- R , R 6c , R , R , R , and R 6g are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCj-Ce alkyl, and the remainder of R 6b , R 6c , R 6d , R 6e , R 6 , and R 6g are each H; and B is selected from the group
- R 6b , R 6c , R 6d , R 6s , R 6f , and R 6g are independently selected from the group consisting of Cj-Ce alkyl, halogen, -CN, and -OCi-Ce alkyl, and the
- R 6b , R 6c , R 6d , R 6e , R 6 , and R 6g are each H; and B is selected
- X is selected from the group consisting of N, C, and CH; X is selected from the group consisting of NH, O, and S; and R 6a , R 6b , and R bC are each
- each R Da , R 6 °, and R 6c is independently optionally substituted by halogen, oxo, -OR 11 , -NR l l R 12 , -C(0)R n , -CN, -8(0)1 ' -S(Q) 2 R 1 !
- A is , wherein X 1 is selected from the group consisting of N, C, and CH; X 2 is selected from the group consisting of NH, O, and S; and R Da , R 6 °, and R 6c are each
- R ⁇ a , R , and R 6c are each H.
- one of R 6 , R D0 , and R 6c is CI, F, Br, or I.
- one of R ba , R 6 °, and R 6c is CI.
- one of R ba , R 6 °, and R 6c is halogen and the others are each H.
- one of R 6a , R ub , and R 6c is halogen and one of R 6a , R 6b , and R 6c is C C 6 alkyi.
- one of R 63 , R 6b , and R bc is CI and one of R 6 , R b0 , and R 6c is methyl.
- R 6a is Ci-Ce alkyi.
- R 6b is C i -Ce alkyi.
- R 6c is Ci-Ce alkyi. in some
- R 6d is methyl, ethyl, n-propyl, isopropvl, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R b0 is methyl, ethyl, n-propyl, isopropvl, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R 6c is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R ba is Cj-Ce alkyi and R 6b is halogen.
- R oa is Cj-Ce alkyi and R 6c is halogen.
- R 6 is Cj -Ce aikyl and R"' 1 is halogen.
- R 6b is C Ce, alkyi and R" is halogen.
- R 6c is Ci-Ce alkyi and R 6a is halogen .
- R 6c is Ci-Ce alkyi and R 6b is halogen.
- two of R 6a , R ub , and R 6c are halogen.
- two of R 6a , R" b , and R DC are C Ce alkyi.
- A is , wherein one or two of R 6a , R 6b , and R bC are independently selected from the group consisting of C Ce alkyi, halogen, -CN, and -OCpCe alkyi, and the remainder of R Da , R 6b , and R 6c are each H; and B is phenyl, optionally substituted with R' .
- A is , wherein one or two of R 6a , R 6D , and R are independently selected from the group consisting of C-.
- A is , wherein one or two of R 6a , R 60 , and R 61' are independently selected from the group consisting of C -.
- R 6a , R 6 and R° c are each H; and B is a 5- to 6-membered carbocycle, optionally substituted with R'. 0104] In some embodiments of a compound of Formula (I), (II), or ( I I I )..
- A is , wherein one or two of R 6a , R 6B , and R 6c are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6a , R* and R 6c are each H; and B is a 5- to 6-membered heterocycle, optionally substituted with R' .
- A is , wh ⁇ erei ⁇ n one or two of - R-- 6a a , R,6 °, and T Rj6 ' c c are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6a , R*, and R 6c are each H; and B is a 9- to 10-membered heteroaryl, optionally substituted with R 7 .
- R DC are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6a , R*,
- R 6d , R 6 °, and R are independently selected from the group consisting of Ci-Ce alkyl, halogen, ⁇ CN, and -OCi -Ce alkyl, and the remainder of R 6a , R* and R 6c are each H; and B is selected from the group consisting of:
- A is and R DC are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi-C & alkyl, a nndd tthhee rreemmaaiinnddeerr of R 'a , R b , and R DC are each H; and B is selected from the group consisting of
- R a , R " utilizaton and R' c are independently selected from tlie group consisting of Ci-Ce alkyl, halogen, -CN, and -OC i -Ce alkyl, and the remainder of R ba , R 6b ,
- R "' are each 1 1, and B is selected from
- X is selected from the group consisting of N, C, and CH; X is selected from the group consisting of NH, O, and S; and R 6g , R Db , and R DC are each
- X 1 is selected from the group consisting of N, C, and CH
- X 2 is selected from the group consisting of NH, O, and S
- R bg , R 6b , and R 6c are each
- R 6G , R 6B , and R 6C is CI, F, Br, or I. In some embodiments, one of R 6G , R 6B , and R 6C is CI. In some embodiments, one of R 6G , R 6B , and R DC is halogen and the others are each H. In some embodiments, one of R 6g , R 6B , and R 6C is halogen and one of R 6G , R B0 , and R 6T is Ci-Ce alkyl. In some embodiments, one of R BG , R 6B , and R 6l is CI and one of R 6G , R 6B , and R 6'" is methyl. In some embodiments, R 6S is Ci -Ce alkyl. In some embodiments, R 6 ° is C Ce alkyl. In some embodiments, R 6 ° is C Ce alkyl. In some embodiments, R 6 ° is C Ce alkyl. In some embodiments, R 6 ° is
- R 6C is Ci-Ce alkyl.
- R BG is methyl, ethyl, n-propyl, isopropvi, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R B0 is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R 6C is methyl, ethyl, n- propyl, isopropyl, n-butyl, isobutyl, secbutyl, or tertbutyl.
- R 6G is Cj- Ce alkyl and R* is halogen. In some embodiments, R 6G is C 5 ⁇ C & alkyl and R 6C is halogen. In some embodiments, R 6B is C ⁇ ⁇ Ce alkyl and R 6G is halogen. In some embodiments, R 6 ° is Cr C-6 alkyl and R UC is halogen. In some embodiments, R DC is Cj-Ce alkyl and R 6G is halogen. In
- R DC is Cj -Ce alkyl and R° is halogen.
- R og , R ! and R bC are halogen.
- R bg , R 61 , and R" c are Ci-Ce alkyl.
- R bg , R 6b , and R 6c are independently selected from the
- R 6b group consisting of C Ce alkyl, halogen, -CN, and -OCpCe alkyl, and the remainder of R Dg , R* and R 6c are each H; and B is phenyl, optionally substituted with R' .
- B is phenyl, optionally substituted with R' .
- R 6g , R 6b , and R 6c are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6g , R 6b , and R 6c are each H; and B is a 5- to 6-membered heteroaryl, optionally substituted with R 7 .
- A is
- R 6b , and R 6t are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -QCi -C& alkyl, and the remainder of R 6g , R 6b , and R 6c are each H; and B is a 5- to 6-membered carbocycle, optionally substituted with R ' .
- R Gg , R 6 , and R ⁇ c are independently selected from the
- R 6b group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R Dg , R* and R 6c are each H; and B is a 5- to 6-membered heterocycle, optionally substituted with R'
- R 6 , R 6 , and R" 1' are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OC i -Ce alkyl, and the remainder of R bg , R 6b , and R 6c are each H; and B is a 9- to lO-mernbered heteroaryl, optionally substituted with R' .
- R R , and R Dt' are independently selected from the group consisting of Ci -Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6g , R 6b ,
- R 6b , and R 6t are independently selected from the group consisting of Ci-Ce alkyl, halogen, -CN, and -OCi -Ce alkyl, and the remainder of R 6g , R 6! and R 6c are each H; and B is selected from the group consisting of:
- A is herein one or two of R Dg , R 6b , and R 6t are independently selected from the group consisting of C Ce alkyl, halogen, -CN, and -OCi-Ce alkyl, and the remainder of R 6 ' g R D 6b and R D are each H; and B is selected from the group consisting
- A is , wherein one or two of R bg , R 6b , and R 6c are independently selected from the 6b group consisting of Ci-Ce alkyl, hal remainder of R og , R*
- R "' are each 1 1, and B is selecte rom
- salts of compounds referred to herein such as pharmaceutically acceptable salts.
- the invention also includes any or all of the stereochemical forms, including any enantiomeric or diastereomeric forms, and any tautomers or other forms of the compounds described.
- a compound as detailed herein may in one aspect be in a purified form and compositions comprising a compound in purified forms are detailed herein.
- Compositions comprising a compound as detailed herein or a salt thereof are provided, such as compositions of substantially pure compounds.
- a composition containing a compound as detailed herein or a salt thereof is in substantially pure form.
- substantially pure intends a composition that contains no more than 35 % impurity, wherein the impurity denotes a compound other than the compound comprising the majority of the composition or a salt thereof.
- a composition of substantially pure compound or a salt thereof is provided wherein the composition con tains no more than 25 %, 20%, 15%, 10%, or 5% impurity.
- a composition of substantially pure compound or a salt thereof is provided wherein the composition contains or no more than 3 %, 2%, 1% or 0.5% impurity,
- the compounds depicted herein may be present as salts even if salts are not depicted and it is understood that the present disclosure embraces all salts and solvates of the compounds depicted here, as well as the non-salt and non-so!vate form of the compound, as is well understood by the skilled artisan.
- the salts of the compounds provided herein are pharmaceutically acceptable salts. Where one or more tertiary amine moiety is present in the compound, the N-oxides are also provided and described.
- tautomeric forms may be present for any of the compounds described herein, each and every tautomeric form is intended even though only one or some of the tautomeric forms may be explicitly depicted .
- the tautomeric forms specifically depicted may or may not be the predominant forms in solution or when used according to the methods described herein.
- the present disclosure also includes any or all of the stereochemical forms, including any enantiomeric or diastereomeric forms of the compounds described. The structure or name is intended to embrace all possible isomers of a compound depicted.
- compositions comprising a compound of the invention are also intended, such as a composition of substantially pure compound, including a specific stereochemical form thereof, or a composition comprising mixtures of compounds of the invention in any ratio, including two or more stereochemical forms, such as in a racemic or non-racemic mixture.
- the invention also intends isotopically-labeled and/or isotopically-enriched forms of compounds described herein.
- the compounds herein may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the compound is isotopically-labeled, such as an isotopically-labeled compound of the formula (I) or v ariations thereof described herein, where a fraction of one or more atoms are replaced by an isotope of the same element.
- Exemplary isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, chlorine, such as 2 H, H, i !
- isotope labeled compounds e.g. 3 ⁇ 4 and 14 C
- isotope labeled compounds are useful in compound or substrate tissue distribution study. Incorporation of heavier isotopes such as deuterium ( 2 H) can afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life, or reduced dosage requirements and, hence may be preferred in some instances.
- Isotopically-labeled compounds of the present invention can generally be prepared by standard methods and techniques known to those skilled in the art or by procedures similar to those described in the accompanying Examples substituting appropriate isotopically-labeled reagents in place of the corresponding non-labeled reagent.
- the invention also includes any or all metabolites of any of the compounds described.
- the metabolites may include any chemical species generated by a biotransformation of any of the compounds described, such as intermediates and products of metabolism of the compound, such as would be generated in vivo following administration to a human.
- Articles of manufacture comprising a compound described herein, or a salt or solvate thereof, in a suitable container are provided.
- the container may be a vial, jar, ampoule, preloaded syringe, i.v. bag, and the like.
- the compounds detailed herein are orally bioavailable.
- the compounds may also be formulated for parenteral (e.g., intravenous) administration.
- One or several compounds described herein can be used in the preparation of a medicament by combining the compound or compounds as an active ingredient with a pharmacologically acceptable carrier, which are known in the art.
- a pharmacologically acceptable carrier which are known in the art.
- the carrier may be in various forms.
- the manufacture of a medicament is for use in any of the methods disclosed herein, e.g., for the treatment of cancer.
- the compounds of the invention may be prepared by a number of processes as generally described below and more specifically in the Examples hereinafter (such as the schemes provided in the Examples below).
- the symbols when used in the formulae depicted are to be understood to represent those groups described above in relation to the formulae herein.
- enantiomer of a compound may be accomplished from a corresponding mixture of enantiomers using any suitable conventional procedure for separating or resolving enantiomers.
- diastereomeric derivatives may be produced by reaction of a mixture of enantiomers, e.g.. a racernate, and an appropriate chiral compound.
- Tire diastereomers may then he separated by any convenient means, for example by crystallization and the desired enantiomer recovered.
- a racemate may be separated using chiral High Performance Liquid
- Chromatography, recrystallization and other conventional separation procedures may also be used with intermediates or final products where it is desired to obtain a particular isomer of a compound or to otherwise purify a product of a reaction.
- Solvates and/or polymorphs of a compound provided herein or a pharmaceutically acceptable salt thereof are also contemplated.
- Solvates contain either stoichiometric or non- stoichiometric amounts of a solvent, and are often formed during the process of crystallization. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol.
- Polymorphs include the different crystal packing arrangements of the same elemental composition of a compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and/or solubility. Various factors such as the recrystallization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate
- compounds of the formula (1) may be synthesized according to Scheme 1, 2, 3, 4, or 5.
- compositions of any of the compounds detailed herein are embraced by this disclosure.
- the present disclosure includes pharmaceutical compositions comprising a compound as detailed herein or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
- the pharmaceutically acceptable salt is an acid addition salt, such as a salt formed with an inorganic or organic acid.
- compositions may take a form, suitable for oral, buccal, parenteral, nasal, topical, or rectal administration or a form suitable for administration by inhalation.
- a compound as detailed herein may in one aspect be in a purified form and compositions comprising a compound in purified forms are detailed herein.
- Compositions comprising a compound as detailed herein or a salt thereof are provided, such as compositions of substantially pure compounds.
- a composition containing a compound as detailed herein or a salt thereof is in substantially pure form.
- the compounds herein are synthetic compounds prepared for administration to an individual.
- compositions are provided containing a compound in substantially pure form.
- the present disclosure embraces pharmaceutical compositions comprising a compound detailed herein and a pharmaceutically acceptable carrier.
- methods of administering a compound are provided. The purified forms, pharmaceutical compositions and methods of administering the compounds are suitable for any compound or form thereof detailed herein.
- a compound detailed herein or salt thereof may be formulated for any available deliver ⁇ 7 route, including an oral, mucosal, (e.g., nasal, sublingual, vaginal, buccal or rectal), parenteral (e.g., intramuscular, subcutaneous or intravenous), topical or transdermal delivery form.
- an oral, mucosal e.g., nasal, sublingual, vaginal, buccal or rectal
- parenteral e.g., intramuscular, subcutaneous or intravenous
- topical or transdermal delivery form e.g., topical or transdermal delivery form.
- a compound or salt thereof may be formulated with suitable carriers to provide delivery forms that include, but are not limited to, tablets, capiets, capsules (such as hard gelatin capsules or soft elastic gelatin capsules), cachets, troches, lozenges, gums, dispersions, suppositories, ointments, cataplasms (poultices), pastes, powders, dressings, creams, solutions, patches, aerosols ⁇ e.g., nasal spray or inhalers), gels, suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions or water-in-oil liquid emulsions), solutions and elixirs.
- suitable carriers include, but are not limited to, tablets, capiets, capsules (such as hard gelatin capsules or soft elastic gelatin capsules), cachets, troches, lozenges, gums, dispersions, suppositories, ointments, cataplasm
- One or several compounds described herein or a salt thereof can be used in the preparation of a formulation, such as a pharmaceutical formulation, by combining the compound or compounds, or a salt thereof, as an active ingredient with a pharmaceutically acceptable carrier, such as those mentioned above.
- a pharmaceutically acceptable carrier such as those mentioned above.
- the carrier may be in various forms.
- the carrier may be in various forms.
- compositions may contain preservatives, solubilizers, stabilizers, re-wetting agents, emuigators, sweeteners, dyes, adjusters, and salts for the adjustment of osmotic pressure, buffers, coating agents or antioxidants.
- Formulations comprising the compound may also contain other substances which have valuable therapeutic properties.
- Pharmaceutical formulations may be prepared by known pharmaceutical methods. Suitable formulations can be found, e.g., in Remington 's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, PA, 20* ed. (2000), which is incorporated herein by reference.
- Compounds as described herein may be administered to individuals in a form of generally accepted oral compositions, such as tablets, coated tablets, and gel capsules in a hard or in soft shell, emulsions or suspensions.
- carriers which may be used for the preparation of such compositions, are lactose, com starch or its derivatives, talc, stearate or its salts, etc.
- Acceptable carriers for gel capsules with soft, shell are, for instance, plant oils, wax, fats, semisolid and liquid poly-ols, and so on.
- pharmaceutical formulations may contain preservatives, solubilizers, stabilizers, re-wetting agents, emuigators, sweeteners, dyes, adjusters, and salts for the adjustment of osmotic pressure, buffers, coating agents or antioxidants.
- any of the compounds described herein can be formulated in a tablet in any dosage form described, for example, a compound as described herein or a pharmaceutically acceptable salt thereof can be formulated as a 10 mg tablet.
- compositions comprising a compound provided herein are also described.
- the composition comprises a compound or salt thereof and a pharmaceutically acceptable carrier or excipient.
- a composition of substantially pure compound is provided.
- Compounds and compositions detailed herein such as a pharmaceutical composition containing a compound of any formula provided herein or a salt thereof and a pharmaceutically acceptable carrier or excipient, may be used in methods of administration and treatment as provided herein.
- the compounds and compositions may also be used in in vitro methods, such as in vitro methods of administering a compound or composition to cells for screening purposes and/or for conducting quality control assays.
- Provided herein is a method of treating a disease in an individual comprising administering an effective amount of a compound of formula (I) or any embodiment, variation or aspect thereof (collectively, a compound of formula (I) or the present compounds or the compounds detailed or described herein) or a pharmaceutically acceptable salt thereof, to the individual.
- a method of treating a disease mediated by a G protein coupled receptor signaling pathway in an individual comprising administering an effecti ve amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, to the individual.
- the disease is mediated by a class A G protein coupled receptor.
- the disease is mediated by a class B G protein coupled receptor.
- the disease is mediated by a class C G protein coupled receptor.
- the G protein coupled receptor is a purinergic G protein receptor.
- the G protein coupled receptor is an adenosine receptor, such as any of the Aj, A 2 A, A 2 B, and A 3 receptors.
- the present compounds or salts thereof are believed to be effective for treating a variety of diseases and disorders.
- the present compositions may be used to treat a proliferative disease, such as cancer.
- the cancer is a solid tumor.
- the cancer is any of adult and pediatric oncology, myxoid and round cell carcinoma, locally advanced tumors, metastatic cancer, human soft tissue sarcomas, including E wing's sarcoma, cancer metastases, including lymphatic metastases, squamous cell carcinoma, particularly of the head and neck, esophageal squamous cell carcinoma, oral carcinoma, blood cell malignancies, including multiple myeloma, leukemias, including acute lymphocytic leukemia, acute nonlymphocytic leukemia, chronic lymphocytic leukemia, chronic myelocytic leukemia, and hairy cell leukemia, effusion lymphomas (body cavity based lymphomas), thymic lymphoma lung cancer, including small cell carcinoma, cutaneous T cell lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, cancer of the adrenal cortex, ACTH-producing tumors, nonsmall cell cancers, breast cancer
- the present compounds or salts thereof are used in treatment of tumors which produce high levels of ATP and/or adenosine.
- the extracellular concentration of adenosine is 10-20 times higher in the tumor compared to adjacent tissue.
- the present compounds or salts thereof are used in treatment of tumors that express high levels of an ectonucleotidase.
- the ectonucleotidase is CD39.
- the ectonucleotidase is CD73.
- ⁇ 55 Also provided herein is a method of enhancing an immune response in an individual in need thereof comprising administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, to the individual.
- Adenosine receptors are known to play an immunosuppressive role in cancer biology. High levels of adenosine present in the tumor microenvironment bind to adenosine receptors on immune cells to provide an
- a receptor provides an immunosuppressive signal that inhibits T cell proliferation, cytokine production and cytotoxicity.
- the AIA receptor signaling has been implicated in adenosine-mediated inhibition of NK cell cytotoxicity, NKT cell cytokine production and CD40L upregulation. Therefore, use of an A 2 A receptor antagonist, such as those provided herein, may reverse the
- the immune response is enhanced by a compound of formula (I) or a salt thereof enhancing activity of natural killer (NK) cells.
- NK natural killer
- the present compounds or salts thereof increase NK cell-meditated cytotoxicity.
- the immune response is enhanced by enhancing the activity of CD8T cells.
- the present compounds or salts thereof cause an inflammatory response in the tumor microenvironment.
- the present disclosure further provides a method of increasing the activity of a natural killer cell in an individual comprising administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, to the individual.
- the present compounds or salts thereof increase NK cell-meditated cytotoxicity.
- a compound of formula (I) or a salt thereof increases the number of NK cells.
- a compound of formula (I) or a salt thereof may be useful for modulating the activity of G protein receptor coupled signaling pathway proteins.
- a compound of formula (I) or a salt thereof activates a G protein receptor coupled signaling pathway protein (i.e. is an agonist of a G protein receptor).
- a compound of formula (I) or a salt thereof inhibits a G protein receptor coupled Signaling pathway protein (i.e., is a G protein receptor antagonist).
- a compound of formula (I) or a salt thereof is an adenosine receptor antagonist.
- a compound of formula (I) or a salt thereof is an antagonist of any of the A ( , A 2 A, A 2B , and A3 receptors.
- a method of modulating the activity of an A 2 A receptor in an individual comprising administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof to an individual.
- a compound of formula (I) or a salt thereof is an A 2 A receptor antagonist.
- a compound of formula (I) or a salt thereof reduces A 2 A receptor signaling by at least 10%, 2.0%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%j, 98%, or 99%.
- a compound of formula (I) or a salt thereof reduces A 2A receptor signaling by 40-99%, 50-99%, 60-99%, 70-99%, 80-99%, 90-99%, or 95-99%.
- a compound of formula (I) or a salt thereof binds to the A 2 A receptor with an IC50 of less than 1 ⁇ , less than 900 nM, less than 800 nM, less than 700 nM, less than 600 nM, less than 500 nM, less than 400 nM, less than 300 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM or less than 100 pM.
- [compound x] binds to the A 2 A receptor with an IC 50 of 500 nM to 100 pM, 400 nM to 100 pM, 300 nM to 100 pM, 200 nM to 100 pM, or 100 nM to 100 pM.
- Also provided herein is a method of modulating the activity of an A 2B receptor in an individual comprising administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof to an individual .
- a compound of formula (I) or a salt thereof is an A?B receptor antagonist.
- a compound of formula (1) or a salt thereof reduces A 2 B receptor signaling by at least 10%, 20%, 30%, 40%,
- a compound of formula (I) or a salt thereof reduces A 2B receptor signaling by 40-
- a compound of formula (I) or a salt thereof binds to the A 2B receptor with an IC 50 of less than 1 ⁇ , less than 900 nM, less than 800 nM, less than 700 nM, less than 600 nM, less than 500 nM, less than 400 nM, less than 300 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM or less than 100 pM.
- a compound of formula (I) or a salt thereof binds to the A 2B receptor with an IC 50 of 500 nM to 100 pM, 400 nM to 100 pM, 300 nM to 100 pM, 200 nM to 100 pM, or 100 nM to 100 pM.
- a method of modulating the activity of an A3 receptor in an individual comprising administering an effective amount of a compound of formula (1), or a pharmaceutically acceptable salt thereof to an individual , in some embodiments a compound of formula (I) or a salt thereof is an A 3 receptor antagonist.
- a compound of formula (1) or a salt thereof reduces A 3 receptor signaling by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99%.
- a compound of formula (I) or a salt thereof reduces A3 receptor signaling by 40- 99%, 50-99%, 60-99%, 70-99%, 80-99%, 90-99%, or 95-99%.
- a compound of formula (I) or a salt thereof binds to the A3 receptor with an IC JO of less than 1 ⁇ , less than 900 nM, less than 800 nM, less than 700 nM, less than 600 nM, less than 500 nM, less than 400 nM, less than 300 nM, less than 200 nM, less than 100 nM, less than 10 nM, less than 1 nM or less than 100 pM.
- a compound of formula (I) or a salt thereof binds to the A 3 receptor with an IC 50 of 500 nM to 100 pM, 400 nM to 100 pM, 300 nM to 100 pM, 200 nM to 100 pM, or 100 nM to 100 pM.
- the present invention comprises a method of inhibiting tumor metastasis in an individual in need thereof comprising administering a compound of formula (I), or a pharmaceutically acceptable salt thereof, to the individual .
- the metastasis is to the lung, liver, lymph node, bone, adrenal gland, brain, peritoneum, muscle, or vagina.
- a compound of formula (I) or a salt thereof inhibits metastasis of melanoma cells.
- the present disclosure includes a method of delaying tumor metastasis comprising administering a compound of formula (I), or a pharmaceutically acceptable salt thereof, to the individual.
- the time to metastatic is delayed by 1 month, 2 months 3 months, 4 months, 5 months, 6 months, 12 months, or more, upon treatment with the compounds of the present invention.
- a compound of formula (I) or a salt thereof is used to treat an individual having a proliferative disease, such as cancer as described herein.
- the individual is at risk of developing a proliferative disease, such as cancer.
- the individual is determined to be at risk of developing cancer based upon one or more risk factors.
- the risk factor is a family history and/or gene associated with cancer.
- the individual has a cancer that expresses a high level of a nucleotide metabolizing enzyme.
- the nucleotide metabolizing enzyme is a nucleotidase, such as CD73 (ecto-5 '-nucleotidase, EctoS'NTase).
- the individual has a cancer that expresses a high level of a nucleotidase, such as CD73.
- the nucleotide metabolizing enzyme is a nucleotidase, such as CD73 (ecto-5 '-nucleotidase, EctoS'NTase).
- CD73 ecto-5 '-nucleotidase, EctoS'NTase
- the individual has a cancer that expresses a high level of a nucleotidase, such as CD73.
- the nucleotide metabolizing enzyme is a nucleotidase, such as CD73 (ecto-5 '-nucleotidase, EctoS'NTase).
- the metabolizing enzyme is an ecto-nucleotidase.
- the ecto-nucleotidase degrades adenosine monophosphate.
- the nucleotide metabolizing enzyme is CD 39 (ecto-nucleoside triphosphate diphosphohydrolase 1, E-NTPDasel).
- the individual has a cancer that expresses a high level of CD39.
- the individual has a cancer that expresses a high level of an adenosine receptor, such as the A 2A receptor.
- the presently disclosed compounds or a salt thereof may activate the immune system by modulating the activity of a G protein coupled receptor signaling pathway, for example acting as an A 2A receptor antagonist, which results in significant antitumor effects. Accordingly, the present compounds or a salt thereof may be used in combination with other anti-cancer agents to enhance tumor immunotherapy.
- a method of treating a disease mediated by a G protein coupled receptor signaling pathway in an individual comprising administering an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and an additional therapeutic agent to the individual.
- the disease mediated by a G protein coupled receptor signaling pathway is a proliferative disease such as cancer.
- the additional therapeutic agent is a cancer immunotherapy. In some embodiments, the additional therapeutic agent is an immunostimuiatory agent. In some embodiments, the additional therapeutic agent targets a checkpoint protein. In some embodiments, the additional therapeutic agent is effective to stimulate, enhance or improve an immune response against a tumor.
- a combination therapy in which a compound of formula (I) is coadministered (which may be separately or simultaneously) with one or more additional agents that are effective in stimulating immune responses to thereby further enhance, stimulate or upregulate immune responses in a subject.
- a method for stimulating an immune response in a subject comprising administering to the subject a compound of formula (I) or a salt thereof and one or more immunostimuiatory- antibodies, such as an anti-PD-l antibody, an anti-PD-Ll antibody and/or an anti-CTLA-4 antibody, such that an immune response is stimulated in the subject, for example to inhibit tumor growth.
- a method for stimulating an immune response in a subject comprising administering to the subject a compound of formula (I) or a salt thereof and one or more immunostimulatory antibodies or immunotherapy like Chimeric antigen receptor (CAR) T-cell therapy: immunostimulatory antibodies, such as an anti-PD-l antibody, an anti-PD-Ll antibody and/or an anti-CTLA-4 antibody, such that an immune response is stimulated in the subject, for example to inhibit tumor growth.
- the subject is administered a compound of formula (I) or a salt thereof and an anti-PD-l antibody.
- the subject is administered a compound of formula (I) or a salt thereof and an anti-PD-Ll antibody.
- the subject is administered a compound of formula (I) or a salt thereof and an anti-CTLA-4 antibody.
- the immunostimulatory antibody e.g., anti- PD-l, anti-PD-Ll and/or anti-CTLA-4 antibody
- the immunostimulatory antibody is a human antibody.
- the immunostimulatory antibody can be, for example, a chimeric or humanized antibody (e.g., prepared from, a mouse anti-PD-l, anti-PD-Ll and/or anti-CTLA-4 antibody).
- the subject is administered a compound of formula (I) or a salt thereof and CAR T- ceils (genetically modified T cells).
- the present disclosure provides a method for treating a proliferative disease (e.g., cancer), comprising administering a compound of formula (I) or a salt thereof and an anti-PD-l antibody to a subject.
- a compound of formula (I) or a salt thereof is administered at a subtherapeutic dose
- the anti-PD-l antibody is administered at a subtherapeutic dose
- the present disclosure provides a method for altering an adverse event associated with treatment of a hyperproliferative disease with an immunostimulatory agent, comprising administering a compound of formula (I) or a salt thereof and a subtherapeutic dose of anti-PD-l antibody to a subject.
- the subject is human.
- the anti-PD- 1 antibody is a human sequence monoclonal antibody
- the present invention provides a method for treating a hyperproliferative disease (e.g., cancer), comprising administering a compound of formula (I) or a salt thereof and an anti-PD-Ll antibody to a subject.
- a compound of formula (I) or a salt thereof is administered at a subtherapeutic dose
- the anti-PD-Ll antibody is administered at a subtherapeutic dose
- both are administered at a subtherapeutic dose.
- the present invention provides a method for altering an adverse event associated with treatment of a hyperproliferative disease with an immunostimulatory agent, comprising administering a compound of formula (I) or a salt thereof and a subtherapeutic dose of anti-PD-Ll antibody to a subject.
- the subject is human.
- the anti-PD-Ll antibody is a human sequence monoclonal antibody.
- the combination of therapeutic agents discussed herein can be administered concurrently as a single composition in a pharmaceutically acceptable carrier, or concurrently as separate compositions each in a pharmaceutically acceptable carrier.
- the combination of therapeutic agents can be administered sequentially.
- an anti-CTLA-4 antibody and a compound of formula (I) or a salt thereof can be administered sequentially, such as anti-CTLA-4 antibody being administered first and a compound of fonnula (I) or a salt thereof second, or a compound of formula (I) or a salt thereof being administered first and anti-CTLA-4 antibody second.
- an anti-PD-1 antibody and a compound of formula (I) or a salt thereof can be administered sequentially, such as anti-PD-1 antibody being administered first and a compound of formula (I) or a salt the eof second, or a compound of formula (I) or a salt thereof being administered first and anti-PD-1 antibody second.
- an anti-PD-Ll antibody and a compound of formula (I) or a salt thereof can be administered sequentially, such as anti-PD-Ll antibody being administered fi rst and a compound of formula (I) or a salt thereof second, or a compound of formula (I) or a salt thereof being administered first and anti-PD-L l antibody second.
- the combination of a compound of formula (I) or a salt thereof can be further combined with an immunogenic agent, such as cancerous cells, purified tumor antigens (including recombinant proteins, peptides, and carbohydrate molecules), cells, and cells transfected with genes encoding immune stimulating cytokines.
- an immunogenic agent such as cancerous cells, purified tumor antigens (including recombinant proteins, peptides, and carbohydrate molecules), cells, and cells transfected with genes encoding immune stimulating cytokines.
- a compound of formula (1) or a salt thereof can also be further combined with standard cancer treatments.
- a compound of formula (I) or a salt thereof can be effectively combined with chemotherapeutic regimes.
- Other combination therapies with a compound of fonnula (I) or a salt thereof include radiation, surgery, or hormone deprivation.
- Angiogenesis inhibitors can also be combined with a compound of formula (I) or a salt tliereof. Inhibition of angiogenesis leads to tumor cell death, which can be a source of tumor antigen fed into host antigen presentation pathways.
- a compound of formula (I) or a salt thereof can be used in conjunction with anti -neoplastic antibodies.
- treatment with an anti-cancer antibody or an anti-cancer antibody conjugated to a toxin can lead to cancer cell death (e.g., tumor cells) which would potentiate an immune response mediated by CTLA-4, PD-1, PD-L1 or a compound of formula (I) or a salt thereof.
- a treatment of a hyperpiOliferative disease can include an anti-cancer antibody in combination with a compound of formula (I) or a salt tliereof and anti-CTLA-4 and/or anti -PD-1 and/or anti -PD -LI antibodies, concurrently or sequentially or any combination thereof, which can potentiate anti-tumor immune responses by the host.
- Other antibodies that can be used to activate host immune responsiveness can be further used in combination with a compound of formula (I) or a salt thereof.
- a compound of formula (I) or a salt thereof can be combined with an anti-CD73 therapy, such as an anti ⁇ CD73 antibody.
- a compound of formula (I) or a salt thereof can be combined with an anti-CD39 therapy, such as an anti-CD 39 antibody.
- a compound of formula (I) or a salt thereof is administered in combination another G protein receptor antagonist, such as an adenosine A ; and/or A 3 antagonist.
- the dose of a compound administered to an individual may vary with the particular compound or salt thereof, the method of administration, and the particular disease, such as type and stage of cancer, being treated.
- the amount of the compound or salt thereof is a therapeutically effecti e amount.
- Hie effective amount of the compound may in one aspect be a dose of between about 0.01 and about 100 mg/kg.
- Effective amounts or doses of the compounds of the invention may be ascertained by routine methods, such as modeling, dose escalation, or clinical trials, taking into account routine factors, e.g., the mode or route of administration or drug delivery, the pharmacokinetics of the agent, the severity and course of the disease to be treated, the subject's health status, condition, and weight.
- An exemplary dose is in the range of about from about 0.7 mg to 7 g daily, or about 7 mg to 350 mg daily, or about 350 mg to 1 .75 g daily, or about 1.75 to 7 g daily.
- Any of the m ethods provided herein may in one aspect comprise administering to an individual a pharmaceutical composition that contains an effective amount of a compound provided herein or a salt thereof and a pharmaceutically acceptable excipient.
- a compound or composition of the invention may be administered to an individual in accordance with an effective dosing regimen for a desired period of time or duration, such as at least about one month, at least about 2 months, at least about 3 months, at least about 6 months, or at least about 12 months or longer, which in some variations may be for the duration of the individual's life.
- the compound is administered on a daily or intermittent schedule.
- the compound can be administered to an individual continuously (for example, at least once daily) over a period of time.
- the dosing frequency can also be less than once daily, e.g. , about a once weekly dosing.
- the dosing frequency can be more than once daily, e.g. , twice or three times daily.
- the dosing frequency can also be intermittent, including a 'drag holiday ' (e.g. , once daily dosing for 7 days followed by no doses for 7 days, repeated for any 14 day time period, such as about 2 months, about 4 months, about 6 months or more). Any of the dosing frequencies can employ any of the compounds described herein together with any of the dosages described herein.
- a 'drag holiday ' e.g. , once daily dosing for 7 days followed by no doses for 7 days, repeated for any 14 day time period, such as about 2 months, about 4 months, about 6 months or more.
- the compounds provided herein or a salt thereof may be administered to an individual via various routes, including, e.g., intravenous, intramuscular, subcutaneous, oral and transdermal.
- a compound provided herein can be administered frequently at low doses, known as 'metronomic therapy,' or as part of a maintenance therapy using compound alone or in combination with one or more additional drugs.
- Metronomic therapy or maintenance therapy can comprise administration of a compound provided herein in cycles.
- Metronomic therapy or maintenance therapy can comprise intra-tumoral administration of a compound provided herein.
- the invention provides a method of treating cancer in an individual by parenterally administering to the individual (e.g., a human) an effective amount of a compound or salt thereof.
- the route of administration is intravenous, intra-arterial, intramuscular, or subcutaneous.
- the route of administration is oral .
- the route of administration is transdermal.
- compositions including pharmaceutical compositions as described herein for the use in treating, preventing, and/or delaying the onset and/or development of cancer and other methods described herein.
- the composition comprises a pharmaceutical formulation which is present in a unit dosage form.
- articles of manufacture comprising a compound of the disclosure or a salt thereof, composition, and unit dosages described herein in suitable packaging for use in the methods described herein.
- suitable packaging is known in the art and includes, for example, vials, vessels, ampules, bottles, jars, flexible packaging and the like.
- An article of manufacture may further be sterilized and/or sealed.
- kits for carrying out the methods of the invention which comprises one or more compounds described herein or a composition comprising a compound described herein .
- the kits may employ any of the compounds disclosed herein.
- the kit employs a compound described herein or a pharmaceutically acceptabie salt thereof.
- the kits may be used for any one or more of the uses described herein, and, accordingly, may contain instructions for the treatment of cancer.
- Kits generally comprise suitable packaging.
- Hie kits may comprise one or more containers comprising any compound described herein.
- Each component if there is more than one component
- kits may be in unit dosage forms, bulk packages (e.g. , multi-dose packages) or sub-unit doses.
- kits may be provided that contain sufficient dosages of a compound as disclosed herein and/or a second pharmaceutically active compound useful for a disease detailed herein (e.g., hypertension) to pro vide effecti ve treatment of an individual for an extended period, such as any of a week, 2 weeks, 3 weeks, 4 weeks, 6 w eeks. 8 weeks, 3 months, 4 months, 5 months, 7 months, 8 months, 9 months, or more.
- Kits may also include multiple unit doses of the compounds and instructions for use and be packaged in quantities sufficient for storage and use in pharmacies (e.g., hospital pharmacies and compounding pharmacies).
- kits may optionally include a set of instructions, generally written instructions, although electronic storage media (e.g. , magnetic diskette or optical disk) containing instructions are also acceptable, relating to the use of component(s) of the methods of the present invention.
- the instructions included with the kit generally include information as to the components and their administration to an individual.
- Step 1 Synthesis of (E ⁇ -3 ⁇ (dimethyIanimo)-l-phe5iySprop-2-en-1-one: A mixture of DMF ' DMA (21.9 g, 300 mmol, 3.6 equiv) and acetophenone (10.0 g, 1.0 equiv, 83 mmol) was heated at 100 °C for 16h. The reaction mixture was then cooled to RT and diluted with cold water. The yellow precipitates were filtered under vacuum and washed with excess water followed by hexane. Tire yellow solid (6 g) was used as such for next step without further purification. LCMS: 176 [M+l
- Step 2 Synthesis of ethyl 2-cyanoacetimidate: To a solution of malononitrile (10 g, 151.52 mmol) in diethyl ether (60 mL) was cooled to 0 °C. To this reaction mixture was ethanol (7.0 g, 151.52 mmol, 1 equiv) added 2M HQ in diethyl ether (40 mL) and the reaction mixture was allowed to stir at 10 °C for 16h. The precipitates formed were filtered and the solid was washed with ether and dried to give the desired product (6 g, 36%).
- Step 3 Synthesis of 2-amino-6-phenylnicotinonitrile: To a solution of (E)-3-
- Step 4 Synthesis of 2-amino-5-bromo-6-phenylnicotinonitrile: To a stirred solution of 2-amino-6-phenyl-pyridine-3-carbonitrile (2.80 g, 14.36 mmol, 1.0 equiv) in DMF (40 mL) was added NBS portion wise (2.56 g, 14.36 mmol, 1.0 equiv). The resulting solution was poured over ice to get the precipitates which were filtered under vacuum, washed with excess water and vacuum dried to get the desired product (3 g) which was used as such for next step without further purification.
- Step 5 Synthesis of 2-amino-5-bromo-6-phenylnicotinaldehyde: To a stirred solution of 2-amino-5-bromo-6-phenyl-pyridine-3-carbonitrile ( 1.0 g, 3.64 mmol, 1.0 equiv) in THF (30 mL) was added 1M solution of DIBAL-H in toluene (12.7 mL, 12.7 mmol, 3.5 eq) and the reaction mixture was allowed to stir at 0 °C for 45 min. Progress of the reaction was monitored by TLC and ⁇ NMR.
- reaction mixture was added 2M HC1 in water (16 mL) dropwise at 0 °C and the reaction mixture was allowed to stir at the same temperature for 10 min.
- the reaction mixture was basified with saturated sodium carbonate solution (20 mL) and extracted with ethyl acetate (3 ⁇ 75 mL). Combined organic layers were washed (brine), dried (anhydrous a 2 SC ) and concentrated under vacuum to get the yellow solid (0.98 g) which was used as such for next step without further purification.
- Step 6 Synthesis of ethyl (E)-3-(2-amino-5-bromo-6-phenylpyridin-3- yl)acrylate: To a solution of ethyl 2-diethoxyphosphorylacetate (0.98 g, 4.38 rnmol, 1.0 equiv) m THF was added NaH (0.174 g, 4.38 mmol, 1.1 equiv) at 0 °C. To this mixture was added 2- amino-5-bromo-6-phenyl-pyridine-3-carbaldehyde ( 1.10 g, 3,97 mmol, 1 .0 equiv). Progress of the reaction was monitored by TLC.
- reaction mixture was quenched by adding cold water and extracted by using ethyl acetate.
- the combined organic layers were washed (brine), dried ( anhydrous Na 2 S0 4 ) and concentrated under vacuum to get the desired product as yellow solid (0.210 g) which was used as such for next step without further purification.
- Step 8 Synthesis of 6-(3-chloro-4-hydroxyphenyl)-7-phenyl-l,8-naphthyridin- 2(lH)-one: To a stirred solution of 6-bromo-7-phenyl-lH-l,8-naph&yridin-2-one (0.140g, 0.46 mmol, 1.0 eq) and (4-chloro-3-hydroxy-phenyl)boronic acid (0.104 g, 0.60 mmol, 1.3 eq) in dioxane (3 mL) was added 2M aq. NaiCCh (0.107 g, 1 .0 mmol, 2.2 eq, 0.5 mL).
- Step ! Synthesis of l-(2 ⁇ ammo ⁇ 5 ⁇ bromo ⁇ 0-phenyl ⁇ 3 ⁇ pyridyi)ethanone: To a stirred solution of 2-amino-5-bromo-6-phenyl-pyridine-3-carbonitrile (2.0 g, 7.30 mmol, 1.0 eq) m THF (60 mL) was added 3M MeMgBr in diethyl ether (11.0 g, 146.9 mmol, 20.0 equiv) at 0 °C. The resulting reaction mixture was stirred at 50 °C for 16h. Reaction mixture was then cooled to 0 °C and quenched by adding dilute HCI.
- the acidic reaction mixture was neutralized by using aq. sodium bicarbonate solution and extracted by using ethyl acetate (2 ⁇ 75 mL). The combined organic layers were washed (brine), dried (anhydrous a2S0 4 ) and concentrated under vacuum to get the light green solid (2.0 g, 94%) which was used as such for next step without further purification
- Step 2 Synthesis of 6-bromo-7-phenyl-lH-l,8-naphthyridin-4-one: To a solution of l-(2-amino-5-bromo-6-phenyl-3-pyridyl)ethanone (2.0 g, 6.89 mmol, 1.0 eq) in DMF (10 mL) was added DMF DMA (1.5 g, 20.68 mmol, 3.0 equiv). The reaction mixture was warmed to 100 °C. After stirring for 3h, CS 2 CO3 (4.5 g, 13.78 mmol, 2.0 equiv) was added and the reaction was stirred for 16 h at 100 °C.
- the reaction mixture was cooled to 0 °C and diluted with cold water.
- the resulting precipitates were filtered, washed with excess water and vacuum dried.
- the solid obtained was purified by normal phase silica gel flash chromatography to get the yellow solid (0.250 g, 12%)
- Step 3 Synthesis of 6-(3-chloro-4-hydroxy-phenyl)-7-phenyl-lH-l,8- naphthyridin-4-one: To a stirred solution of 6-bromo-7-phenyl-lH-l,8-naphthyridin-4-one (0.120g, 0.40 mmol, 1.0 eq) and 2-ch]oro-5-(4,4,5,5-tetramethy]-l,3,2-dioxaborolan-2-yl)phenol
- 6-bromo-7-phenyl-l ,8-naphthyridin-2(IH)-one (0.120 g, 0.40 mmol, 1.0 equiv) in dioxane (4 mL) was added 2M aqueous Na 2 CC>3 (0.084 g, 0.80 mmol, 2.0 equiv, 0.4 mL).
- the reaction was purged with 2 for 5 min.
- Pd(dppf)Cl 2 *DCM (0,016 g, 5 mol%) and N 2 was purged again for another 5 min.
- the reaction mixture was heated at 90 °C for 4 h.
- reaction mixture was allowed to cool to RT and extracted using ethyl acetate (2 ⁇ 35 mL), The combined organic layers were washed (brine), dried (anhydrous Na ⁇ SC ⁇ ) and concentrated under vacuum to get the solid residue which was purified by reverse phase column chromatography to get the desired product as off white solid (0.020 g, 14 %).
- the reaction was monitored by 3 ⁇ 4 NMR and LCMS.
- the reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 50 mL). The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure.
- the crude product was purified by reverse phase column chromatography to afford (8 mg, 8%) as desired product (8 mg, 8%).
- Step 1 Synthesis of 6-bromo-l-methyl-7-phenyl-l,8-naphthyridin-4(lH)-one:
- Step 2 Synthesis of l-methy! ⁇ 7-p enyl-6 ⁇ (quinol!5i-6 ⁇ yl ⁇ -l,8-naphthyridin- 4(lH)-one: To a solution of 6-bromo-l-methyl-7-phenyl-l,8-naphthyridin-4(lH)-one (140 rng, 0.44 mmol, 1 eq.) in 1,4 dioxane (8 mL): water (2 mL) was added quinolin-6-yl boronic acid (84 mg, 0.49 mmol, 1 , 1 eq.), Na 2 C0 3 (94 mg, 0.89 mmol, 2 eq.), and PdCl 2 (dppf) ⁇ ⁇ I complex ( 18 mg, 0,022 mmol, 0.05 eq).
- the reaction mixture was deoxygenated using N 2 atmosphere followed by heating at 80 °C for 48 h.
- the reaction was monitored by TLC and LCMS.
- the reaction mixture was diluted with water (30 mL) and extracted using ethyl acetate (2 ⁇ 50 mL). The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure. Hie crude product was purified by reverse phase column chromatography to afford the desired product (25 mg, 15%).
- the reaction was monitored by NMR and LCMS.
- the reaction mixture was diluted with water (30 mL) and extracted using ethyl acetate (2 ⁇ 50 mL). The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure.
- the crude product was purified by reverse phase column chromatography to afford the desired product as a TFA salt (8 mg, 11 %).
- reaction mixture was heated at 90 °C for 18 h .
- the reaction mixture was allowed to cool to RT and extracted using ethyl acetate (2 x 35 mL).
- the combined organic layers were washed (brine), dried (anhydrous NaiSCX- and concentrated under vacuum to get the solid residue which was purified by reversed phase column chromatography to get the desired product as off white solid (0.006 g, 4 %).
- Step 1 Synthesis of (l,8-naphthyridin-3-yl)boronic acid: To a solution of 3- bromo-l ,8-naphthyridine (0.160 g, 0.76 mrnol, 1 eq.) in 1,4-dioxane (3 mL) was added 5- (4,4,5,5- bis(pinacolato)diboron (0.289 mg, 1.14 mmol, 1.5 eq.), KOAc (0.224g, 2.28 mrnol, 3 eq.), and Pd(PPh.3)Cl 2 complex ( 0.026 g, 0.038 mmol, 0.05 eq.).
- reaction mixture was deoxygenated with N 2 allowed stir at 80 °C for 16 h.
- the reaction mixture was cooled to RT and filtered through Celiie. Tlie filtrate was concentrated (0.120 g, 90%) and used for next step without further purification.
- Step 2 Synthesis of 2-phenyl-[3,3'-bi(l,8-naphthyridin)]-5(8H)-one: To a stirred solution of (l,8-naphthyridin-3-yl)boronic acid (0.068 g, 0.39 mmol, 1.2 equiv) and 6-bromo-7- phenyl-l,8-naphthyridin-4( lH)-one (0.100 g, 0.33 nmol, 1.0 equiv) in dioxane (3 mL) was added Na 2 C03 (0.069 g, 0.66 mmol, 2.0 equiv) and 1 mL water.
- reaction was purged with N 2 for 5 min.
- Pd(dppf)Cl 2 *DCM complex 0.14 g, 5 mol%) and 2 was purged again for another 5 min.
- Tlie reaction mixture was heated at 90 °C for 18 h.
- the reaction mixture was allowed to cool to RT and extracted using ethyl acetate (2 ⁇ 35 mL).
- the combined organic layers were washed (brine), dried (anhydrous Na 2 S0 4 ) and concentrated under vacuum to get the solid residue which was purified by reversed phase column chromatography to get the desired product as off white solid (0.040 g, 29 %).
- Step 1 Synthesis of 3-(dimethylamino)-l-(furan-2-yl)prop-2-en-l-one: A mixture of DMF-DMA (21 .9 g, 297.17 mmol, 3 ,6 eq) and l-(furan ⁇ 2 ⁇ yl)e ⁇ hanone (10.0 g, 90.81 mmol, 1.0 eq,) was heated at 100 °C for 16 h. The reaction mixture was then cooled to RT and diluted with cold water. The yellow precipitates were filtered under vacuum and washed with excess water followed by hexane. The yellow solid (13.4 g, 88%) was used as such for next step without further purification.
- Step 2 Synthesis of 2-amino-6-(furan-2-yl)nicotinonitriIe: To a solution of 3- (dimethylamino)-l-(furan-2-yl)prop-2-en-l-one (9.0 g, 54.54 mmol. 1.00 eq) in ethanol (200 mL) was added ethyl 2-cyanoethanimidate hydrochloride (9.68 g, 1.2 eq, 65.45 mmol) and ammonium acetate (48.50 g, 54.5 mmol, 10 eq). The reaction mixture was allowed to stir at 85 °C for 16 h. Progress of the reaction was monitored by LCMS. Reaction mixture was cooled to RT, diluted with cold water (250 mL) and the solid was filtered. The solid was washed with hexane and dried under vacuum to afford the desired product (5.7 g, 57 %).
- Step 3 Synthesis of 2-ammo-5 ⁇ bromo-6 ⁇ (furan-2-yi) cotinonitriie: To a stirred solution of 2-amino-6-(furan-2-yl)nicotinonitrile (3.0 g 16.21 rnmol, 1.0 equiv) in DMF (120 mL) was added NBS portion wise (2.88 g, 16.21 mmol, 1.0 equiv). The reaction mixture was allowed to stir at RT. The progress of the reaction was monitored by T ' LC. After completion of the reaction the reaction mixture was poured over ice to get the precipitates which were filtered under vacuum, washed with excess water and vacuum dried to get the desired product (2.2 g, 52 %) which was used as such for next step without further purification.
- Step 4 Synthesis of l-(2-amino-5-bromo-6-(furan-2-yl)pyridin-3-yl)ethanone:
- Step 5 Synthesis of l-(2-amino-5-bromo-6-(furan-2-yl)pyridin-3-yl)-3-
- Step 6 Synthesis of 6-bromo-7-(furan-2-yl)-l,8-naphthyridin-4(lH)-one: To a stirred solution of l-(2-amino-5-bromo-6-(mran-2-yl)pyridin-3-yl)-3-(dimethylamino)prop-2- en-l-one (1.0 g, 2.98 mmol, 1.0 eq) in DMF (10,0 mL) was added Cs 2 C0 3 (1.94 g, 2.0 eq, 5.97 mmol) and allowed to stir at 100 °C for 16 h. Progress of reaction was monitored by TLC. The reaction mixture was diluted with ice cold water and the resulting precipitates were filtered and vacuum dried to get the desired product (0.400 g, 50 %).
- Step 7 Synthesis of 7-(furan-2-yl)-6-(quinolin-6-yl)-l,8-naphthyridin-4(lH)-one:
- the reaction mixture was diluted with water (20 mL) and extracted using ethyl acetate (2 ⁇ 20 mL). The separated organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by reverse phase column chromatography to afford the desired product (15 mg, 13 %),
- Example SI 6 Synthesis of 6-(naphthalen-2-yl)- 7 -phenyl- L 8-naphthyridm-4( lH)-one.
- the reaction mixture was diluted with water (20 mL) and extracted using ethyl acetate (2 ⁇ 20 mL). The separated organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by reverse phase column chromatography to afford the desired product (12 mg, 9 %).
- Example SI 8 Synthesis of 7-(pyridin-4-yl)-6-(qiiinolin-6-yl) ' -l, 8-naphthyridin-4(lH) ' -one (Compound No. 1.18)
- Step 6 Synthesis of (E ⁇ -3 ⁇ (dimethyIammo)-l-(pyrid!n-4 ⁇ yl)prop ⁇ 2-e5i-l ⁇ one: A mixture of DMF ' DMA (21.9 g, 297.17 mmol, 3.6 eq) and l-(pyridin-4-yl)ethan-l-one (10.0 g, 1.0 eq, 83 mmol) was heated at 100 °C for 16 h. The reaction mixture was then cooled to RT and diluted with cold water. The yellow precipitates were filtered under vacuum and washed with excess water followed by hexane. The yellow solid (14.8 g) was used as such for next step without further purification.
- Step 2 Synthesis of 6-aminG- 2,4'-bipyridineJ-5-c3 ⁇ 4rbooitriIe: To a solution of (E)-3-(dimethylamino)-l -phenyl-prop-2-en-l-one (1 1.0 g, 62.5 mmol) in ethanol (200 mL) was added ethyl 2-cyanoethanimidate hydrochloride 11.1 g, 1.2 equiv, 75.0 mmol) and ammonium acetate (55.62g, 625.0 mmol, 10 equiv) and the reaction mixture was allowed to stir at 85 °C for 16 h. Progress of the reaction was monitored by ⁇ NMR. Reaction mixture was cooled to RT, diluted with cold water (250 mL) and the solid was filtered. The solid was washed with hexane and dried under vacuum to afford the desired product (9.8 g, 80%)
- Step 3 Synthesis of 6-aini!io-3-bronio-[2,4 , -bipyridine]-5-carboniir0e: To a stirred solution of 6 ⁇ amino- 2,4 , -bipyridine
- Step 4 Synthesis of l-(6-amino-3-bromo-[2,4'-bipyridin]-5-yl)ethan-l-one: To a stirred solution of 6-amino-3-bromo-[2,4'-bipyridine
- Reaction mixture was then cooled to 0 °C and quenched by adding dilute HCI.
- the acidic reaction mixture was neutralized by using aq. sodium bicarbonate solution and extracted by using ethyl acetate (2 ⁇ 75 mL).
- the combined organic layers were washed (brine), dried (anhydrous and concentrated under vacuum to get the light green solid (2.27 g, 51%) which was used as such for next step without further purification.
- Step 5 Synthesis of 6-bromo-7-(pyridin-4-yl)-l,8-naphthyridin-4(lH)-one: To a solution of l-(6-amino-3-bromo-[2,4'-bipyridin]-5-yl)ethan-l-one (1.3 g, 4.46 mmol, 1.0 eq) in DMF (10 mL) was added DMF » DMA (1.06 g, 8.92 mmol, 2.0 equiv). The reaction mixture was warmed to 100 °C.
- Step 6 Synthesis of 7-(pyridin-4-yl)-6-(quinolin-6-yl)-l,8-naphthyridin-4(lH)- one: To a stirred solution of 6-bromo-7-(pyridin-4-yl)-l,8-naphthyridin-4(lH)-one (0.100 g, 0.33 mmol, 1.0 eq) and quinolin-6-ylboronic acid (0.068 g, 0.39 mmol, 1.2 eq) in dioxane (5 mL) was added 0.4 mL 2M aq.
- Example S20 Synthesis of 7-(py dm-3-yl)-6-(quinolin-6-yl)-l, 8-naphthyridin-4(lH)-one.
- Step 1 l-(3-aniino-5-pheny!-6-(quinoIin-6-y!)p razin-2 ⁇ yS)ethan ⁇ l-one : To a stirred solution of 3-amino-5-phenyl-6-(quinolin-6-yl)pyrazine-2-carbonitrile (0.100 g, 0.32 mmol, 1.0 eq) in THF (5 mL) was added 3M MeMgBr in diethyl ether (1 mL, 0.360 g, 10.0 eq, 3.0 mmol) at 0 °C. The resulting reaction mixture was stirred at 50 °C for 16 h.
- reaction mixture was then cool to RT and acidified slowly with dilute HCL The acidified reaction mixture was stirred for lh at 50 °C. The reaction mixture was again allowed to cool to RT and extracted by using ethyl acetate (2 ⁇ 25 mL). The combined organic layers were washed (brine), dried (anhydrous NaiSO,]) and concentrated under vacuum to get the desired product as light yellow solid (0.090 g, 97%)
- Step 2 Synthesis of 7-(pyridin-3-yl)-6-(quinolin-6-yl)-l,8-naphthyridin-4(lH)- one: To a solution of l-(6-ainino-3-(quinolin-6-yl)-2,3'-bipyridin-5-yl)ethanone (0.090 g, 0.31 mmol, 1.00 eq.), in 1,4 dioxane (5 mL), was added DMF:DMA (0.044 g, 0.37 mmol, 1.2 eq.). The reaction mixture was heated at 90 °C for 30 minute. The reaction was monitored by TLC and LCMS. The reaction solvent was evaporated under reduced pressure.
- Step 1 Synthesis of 4-bromo-2-nitrophenol: To a solution of 6-bromophenol (5g, 28,90 mniol, 1 eq.) in acetic acid (10 mL) was added nitric acid (1 mL) drop wise. Reaction mixture was stirred at RT for Smin.The reaction was monitored by LCMS and found to be complete after 5 min. The reaction mixture was added with ice, the precipitated solid was filtered and dried to afford 4-bromo-2-nitrophenol (6 g, 95.23%) as yellow solid.
- Step 2 Synthesis of 2 ⁇ an3 ⁇ 4ino-4"hromophenoI: To a solution of 4-bromo-2- nitrophenol (3g, 13.76 mmol, 1 eq.) in ethanol -water (50mL) was added ammonium chloride (2.1 g, 41.28 mmol, 3 eq.) and iron( 2.3g, 41.28mmol. The reaction was stirred at 90 °C for 2 h. The reaction was monitored by LCMS and found to be complete after 2h. The reaction mixture was cooled to RT, evaporated under reduced pressure to remove the solvent diluted with water (20 mL) and extracted with ethyl acetate (2 ⁇ 50 mL).
- Step 3 Synthesis of 5-bromobenzo[d]oxazoIe: A solution of 2-amino-4- bromophenol (1.5, 7.90 mmol, 1 eq.) in methylorthoformate (10 mL) was stirred atl50 °C for 4- 6 h. The reaction was monitored by LCMS and found to be complete after 6 h. The reaction mixture was cooled to RT, evaporated under reduced pressure to remove the solvent diluted with water (20 mL) and extracted with ethyl acetate (2 x 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium, sulfate. Removal of solvent under reduced pressure gave crude which was purified by Combi-Flash to obtain the 5-bromobenzo[d]oxazole (1.2 g, 80.0%) as yellow solid.
- Step 4 Synthesis of 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzo[d]oxazole: To a solution of 5-bromobenzo[d]oxazole (1.2 g, 6.06 nimol, 1 eq.) in DMF (10 mL) was added 5-(4,4,5,5- bis(pinacolato)diboron (1.68 g, 1.66 mmol, 1 .1 eq.), KOAc (1.7g, 18.09 mmol, 3eq.), PdCl 2 (dppf DCM ( 247 mg, 0.23 mmol, 0.05 eq.).
- reaction mixture was deoxygenated with N 2 and the reaction mixture was stirred at 80 °C overnight.
- the reaction was monitored by LCMS and found to be complete after 18 h.
- the reaction mixture was cooled to RT, diluted with water (50 mL) and extracted with ethyl acetate (2 ⁇ 50 mL).
- Step 5 Synthesis of 6-(benzo[d]o.xazol-5-yl)-7-phenyI-l,8-naphthyridin-4(lH)- one: To a solution of 6-bromo-7-phenylquinolin-4(lH)-one (100 mg, 0.40 mmol, 1 eq.) in DME-water (2 mL) was added 5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzo[d]oxazole (97 mg, 0.39 mmol, 1 ,2 eq,), Sa 0 - (70 mg, 0.66 mmol, 2.0 eq.), PdC12(dppf)*DCM ( 13.86 mg, 0.01 mmol, 0.05 eq.).
- reaction mixture was deoxygenated with N 2 and the reaction mixture was stirred at 100 °C for 12 h.
- the reaction was monitored by LCMS and found to be complete after 12h.
- the reaction mixture was cooled to RT, diluted with water (20 mL) and extracted with ethyl acetate (2 ⁇ 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude which was purified by reverse phase column chromatography to afford the desired product (10 mg, 9%) as off white solid.
- Step 1 Synthesis of l-methyl-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)- lH-benzo[d]imidazole: To a solution of 5-bromo-l-methyl-lH-benzofd]imidazole (200 mg, 0.33 mmol, 1 eq.) in 1,4-dioxane (10 mL) was added 5-(4,4,5,5-bis(pinacolato)diboron (287 mg, 1.13 mmo! .
- Step 2 Synthesis of 6-(l-methyl-lH-benzo[d]imidazol-5-yl)-7-phenyl-l,8- naphthyridin-4(lH)-one: To a solution of 6-bromo-7-phenyl-l ,8-iiaphthyridin-4(lH)-one
- reaction was monitored by LCMS and found to be complete after 30 min.
- the reaction mixture was cooled to RT, diluted with water (20 mL) and extracted with ethyl acetate (2 x 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude which was purified by reverse phase column chromatography to afford the desired product (10 mg, 10%) as off white solid.
- Step 1 Synthesis of 8-methyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)quinoline: To a solution of 6-bromo-8-methylquinoline (100 mg, 0.45 mmol, 1 eq.) in 1,4- dioxane (10 mL) was added 5-(4,4,5,5-bis(pinacolato)diboron (127 mg, 0.49 mmol, 1.1 eq.), KOAc (88 mg, 0.90 mmol, 2 eq.), PdCl 2 (PPh 3 ) 2 ( 15.7 mg, 0.02 mmol, 0.05 eq.).
- reaction mixture was deoxygenated with N 2 and the reaction mixture was heated under microwave irradiation for 30 min at 120 °C. The reaction was monitored by LCMS and found to be complete after 30 min.
- the reaction mixture was cooled to RT, diluted with water (10 mL) and extracted with ethyl acetate (2 20 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude ( 150 mg) which was carried to next step without any further purification.
- Step 2 Synthesis of 6-(8-methylquinolin-6-yl)-7-phenyl-l,8-naphthyridin-4(lH)- one: To a solution of 6-bromo-7-phenyl-l,8-naphthyridin-4(lH)-one (100 mg, 0.33 mmol, 1 eq.) in DME-Water (2 mL) was added 8-methyl-6-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)quinoline (107 mg, 0.39 mmol, 1.2 eq.), Na 2 CC (70 rng, 0.66 mmol, 2.0 eq.),
- reaction mixture was cooled to RT, diluted with water (20 mL) and extracted with ethyl acetate (2 ⁇ 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude which was purified by reverse phase column chromatography to afford the desired product (3 mg 10%) as a light brown solid.
- Step I Synthesis of 2-amino-6-chloro-N-methoxy-N-methylnicotinamide: To a stirred solution of 2-amino-6-chloronicotinic acid ( 1.136 g, 6.56 mmol, 1.0 eq) and N,0- dimethylhydroxylamine hydrochloride (0.965 g, 9.85 mmol, 1.5 eq) in DMF (20 mL) was added EDC (1.9 g, 9.85 mmol, 1.5 eq), HOBt (1.5 g, 9.85 mmol, 1.5 eq), and DIPEA (2.58 g, 20.0 mmol, 3.0 eq). The reaction was allowed to stir at RT for 16 h.
- reaction mixture was then extracted using ethyl acetate (2 ⁇ 100 mL). the combined organic layer was washed (brine), dried (anhydrous sodium sulphate) and concentrated under vacuum to get the solid which was purified by normal phase flash column chromatography to get the desired product (1 .0 g)
- Step 2 Synthesis of l - ⁇ 2 ⁇ ammo ⁇ 6 ⁇ eh!eropyridin-3 ⁇ y!etlha5i05ie: To a solution of 2-amino-6-chloro-N-methoxy-N-methylnicotinamide (500 mg, 2.32 mmol, 1.00 eq) in THF (20 mL) was cooled to 0 °C and 3M MeMgBr in THF (2.713 mL, 3.5 mmol) was added at the same temperature. The reaction mixture was stirred at the same temperature for 30 min.
- reaction mixture was quenched using freshly prepared saturated solution of NH CI (50 mL) and extracted using ethyl acetate (2 x 50 mL). The combined organic layer was dried over sodium sulfate and concentrated under reduced pressure to afford the desired product (300 mg, 47 %).
- Step 3 Synthesis of l- ⁇ 2 ⁇ am!no-5 ⁇ bromo ⁇ 6-ch!oropyridin-3-yS)ethanone: To a solution of l-(2-amino-6-chloropyridin-3-yl)ethanone (1.5 g, 8.82 mmol, 1 eq.) in mixture of ACN (50 mL) was added N- bromosuccinimide (1.895 g, 10.58 mmol, 1.2 eq.). The reaction mixture was stirred at room temperature for 1 h. The reaction was monitored by TLC and NMR. The reaction solvent was evaporated under reduced pressure.
- reaction mixture was diluted with water ( 100 mL) and extracted by ethyl acetate (2 ⁇ 50 mL). The organic layer was separated, washed water (5 ⁇ 20 mL) and brine and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to afford (1.800 g, 85 %) of 1 -(2-amino-5- bromo-6-chloropyridin-3 -yl)ethanone .
- Step 4 Synthesis of l-(2-amino-6-chloro-5-(quinolin-6-yl)pyridin-3-yl)ethanone:
- the reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 x 50 mL), The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography to afford the desired product (350 mg, 26 %).
- Step 5 Synthesis of l-(2-amino-6-(5-methylfuran-2-yl)-5-(quinolin-6-yl)pyridin- 3-yl)ethanone: To a solution of l-(2-amino-6-chloro-5-(quinolin-6-yl)pyridin-3-yl)ethanone
- Step 6 Synthesis of (E)-N'-(3-acetyl-6-(5-methylfuran-2-yl)-5-(quinolin-6- yl)pyridin-2-yl)-N,N-dimethylformimidamide: To a solution of l-(2-amino-6-(5- methylfliran-2-yl)-5-(qumolm-6-yl)pyridin-3-yl)ethanone (100 mg, 0.29 mmol, 1 eq), in 1,4- dioxane (5 mL), was added DMF:DMA (346 mg, 2.91 mmol, 2.5 eq.). The reaction mixture was heated at 90° C for 30 min. The reaction was monitored by TLC and LCMS. The reaction solvent was evaporated under reduced pressure to afford the desired product ( 100 mg, 86 %).
- Step 7 Synthesis of 7-(5-methylfuran-2-yl)-6-(quinolin-6-yl)-l,8-naphthyridin- 4(lH)-one: To a solution ofN'-(3-a ⁇ tyl-6-(5-memyl uran-2-yl)-5-(quinolin-6-yl)pyridm-2-yi)- N,N-dimethylformimidamide (100 mg, 0.25 mmol, 1 eq), in DMF (1 mL), was added Cs 2 C0 3 (122 mg, 0.37 mmol, 1.5 eq). The reaction mixture was heated at 90° C for 18 h. The reaction was monitored by LCMS. The reaction mixture was diluted with ice cold water (25 mL). The precipitation of product was observed and filtered as sold. The crude product was submitted to reverse phase column chromatography to afford the desired product (10 mg, 11 %).
- Step 1 Synthesis of 8-methoxyquinolin-6-ylboronic acid: To a solution of 6- 6- bromo-8-methoxyquinoline (200 mg, 084 mmol, 1 eq.) in 1,4-dioxane (10 mL) was added 5- (4,4,5,5- bis(pinacolato)diboron (256 mg, l .OOmmol, 1.2 eq.), KOAc (164 mg, 1.68 mmol, 2eq.), PdCl 2 (PPh 3 ) 2 ( 68 mg, 0.168 mmol, 0.2 eq.). The reaction mixture was deoxygenated with 2 and the reaction mixture was heated at 80 °C for 12h.
- the reaction was monitored by LCMS and found to be complete after ! 2h.
- the reaction mixture was cooled to RT, diluted with water (10 mL) and extracted with ethyl acetate (2 20 niL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude which was carried to next step without any purification to afford 8-methox quinolin-6- ylboronic acid. (120 mg) which was used directly to the next step.
- Step 2 Synthesis of 6-(8-methoxyquinolin-6-yl)-7-phenyl-l,8-naphthyridin- 4(lH)-one: To a solution of 6-bromo-7-phenyl-l ,8-naphthyridin-4(l H)-one ( 100 mg, 0.33 mmol, 1 eq.) in DME-water (2 mL) was added 8-methoxyquinolin-6 ⁇ yiboronic acid (80.9 mg, 0.39 mmol 1.2 eq,), K 2 C0 3 (136.62 mg, 0,99 mmol, 3.0 eq.), PdCl 2 (dppf DCM (13.4 mg, 0.01 mmol, 0.05 eq.).
- reaction mixture was deoxygenated with N 2 and the reaction mixture was stirred at 120 °C for 30 min in microwave. Hie reaction was monitored by LCMS and found to be complete after 30 min.
- the reaction mixture was cooled to RT, diluted with water (20 mL) and extracted with ethyl acetate (2 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate. Removal of solvent under reduced pressure gave crude which was purified by reversed phase column chromatography to afford the desired product (10 mg, 8%) as off white solid,
- reaction mixture was deoxygenated with N 2 and the reaction mixture was stirred at 120 °C for 30 min under microwave irradiation. The reaction was monitored by LCMS and found to be complete after 30 min.
- the reaction mixture was cooled to room temperature, diluted with water (20 mL) and extracted with ethyl acetate (2 ⁇ 50 mL). Combined organic layer was washed with brine (20 mL) and dried over sodium sulfate . Removal of solvent under reduced pressure gave crude which was purified by reverse phase column chromatography to afford the desired product (3 mg, 8%) as off-white solid.
- HW!R 400 MHz, DMSO-de
- Step 1 Synthesis of l-(6-amino-3-(quinolin-6-yl)-2,3'-bipyridin-5-yl)ethanone:
- reaction was monitored by TLC and LCMS.
- the reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 x 50 mL).
- the separated organic layer was dried over sodium sulfate and concentrated under reduced pressure, to get the desired product (200 mg) which was used as such for next step without further purification,
- Step 2 Synthesis of 7-(pyridin-3-yl)-6-(quinolin-6-yI)-l,8-naphthyridin-4(lH)- one: To a solution of l-(6-amino-3-(quinolin-6-yl)-2,3'-bipyridin-5-yl)ethanone (200 mg, 0.58 mmol, 1.00 eq.), in 1,4 dioxane (5 mL), was added DMF:DMA (346 mg, 2.91 mmol, 2.5 eq.).
- the reaction mixture was heated at 90 °C for 30 min. The reaction was monitored by TLC and LCMS. The reaction solvent was evaporated under reduced pressure. The semisolid crude material obtained was dissolved in DMF (2 mL) and CS2CO3 (246 mg, 0.75 mmol, 1.5 eq.) was added. The reaction mixture was again heated at 90 °C for 18 h. The progress of the reaction was monitored by LCMS. The reaction mixture was diluted with ice cold water (25 mL) and the precipitates obtained were filtered under vacuum. The residue was purified by reversed phase column chromatography to afford the desired product (10 mg, 6 %).
- Step 1 Synthesis of (2-amino-6-chloropyridin-3-yl)methanol: To a stirred solution of 2-amino-6-chloro-N-methoxy-N-methylnicotinamide (1.0 g, 4.6 mmol, 1.0 eq) in THF (20 mL) was added 1M solution of DIBAL-H in toluene (23.14 mL, 5.0 eq) dropwise at 0 °C. Progress of the reaction was monitored by TLC, To the reaction mixture was added aqueous NH 4 CI and extracted using ethyl acetate (3 75 mL). Combined organic layers were washed (brine), dried (anhydrous ⁇ ⁇ ⁇ >,() ⁇ ) and concentrated under vacuum to get the solid which was further triturated with pentane to give the desired product (0,330 g, 45%)
- Step 2 Synthesis of 2-amino-6-chloronicotinaldehyde: To a solution of (2- amino-6-chloropyridin-3-yl)methanol (2.67 g, 16.83 mmol, 1.0 eq) in DCM (40 mL) was added PCC (7.26 g, 33.6 mmol, 2.0 eq) at 0°C, the reaction was allowed to stir at RT, progress of the reaction was monitored by TLC. After completion of the reaction the reaction was filtered through Celite. The filtrate were washed with brine and dried over sodium sulphate and concentrated under vacuum to get the desired product (1 .92g, 73%),
- Step 3 Synthesis of 2-amino-5-foromo-6-cMoroiikotinaIdelhyde: To a stirred solution of 2-amino-6-chloronicotinaldehyde (1.8 g, 1.0 eq, 1 1.49 mmol) in ACN (20 niL) were added NBS (2.0 g, 1.0 eq, 11.49 mmol) at 0°C. The reaction mixture was allowed to stir at RT. The progress of the reaction was monitored by TLC. After complete conversion of the starting material the reaction mixture was concentrated under vacuum.
- Step 4 Synthesis of ethyl (E)-3-(2-amino-5-bromo-6-chloropyridin-3- yl)acrylate: In a 20 mL THF, NaH (1.8 g, 1.0 eq, 7.6mmol) and TEPA (1.98 mL, 9.9 mmol, 1.3 eq) was added at 0°C. Followinged by addition of 2-amino-5-bromo-6-chloronicotinaldehyde (1.8 g, 1.0 eq, 7.6 mmol) and the reaction mixture was allowed to stir at RT. The progress of the reaction was monitored by TLC. The reaction was quenched by adding water extracted by using ethyl acetate (3 100 mL). The combined organics were washed by brine and dried over sodium sulphate and concentrated under vacuum to give the desired product (1.035 g, 34%)
- Step 5 Synthesis of 6-bromo-7-chloro-l,8-naphthyridin-2(lH)-one: To a freshly prepared sodium ethoxide (0.105 g Na metal in 10 mL ethanol) at 0°C was added 20 mL solution of ethyl (E)-3-(2-amino-5-bromo-6-chloropyridin-3-yl)acrylate (1.035 g, 4.56 mmol, 1.0 eq) in ethanol and the reaction mixture was allowed to stir at RT for 16h. The progress of the reaction was monitored by T ' LC. The reaction mixture was concentrated under vacuum and extracted with ethyl acetate (3 ⁇ 75 mL), the combined organic layers were washed with brine and dried over sodium sulphate and concentrated under vacuum to get the desired product (0.650 g, 55%)
- Step 6 Synthesis of 7-chloro-6-(quinolin-6-yl)-l,8-naphthyridin-2(lH)-one: To a stirred solution of 6-bromo-7-chloro-l ,8-naphthyridin-2(lH)-one (0, 120 g, 0,46 mmol, 1 .0 equiv) and quinolin-6-ylboronic acid (0.096 g, 0.56 mmol, 1.2 equiv) in dioxane (3 mL) was added a 2 C0 3 (0.097 g, 0.66 mmol, 2.0 equiv) and 1 mL water.
- Step 7 Synthesis of 7-(5-methylfuran-2-yl)-6-(quinolin-6-yl)-l,8-naphthyridin- 2(lH)-one: To a stirred solution of 7-ch3oro ⁇ 6-(quinolin-6-yl)-l,8-naphthyridin-2(lH)-one (0.100 g, 0.32 mmol, 1.0 equiv) and (5-methylfuran-2-yl)boronic acid (0.050 g, 0.38 mmol, 1.2 equiv) in dioxane (3 mL) was added Na 2 C0 3 (0.062 g, 0.64 mmol, 2.0 equiv) and 1 mL water. The reaction was purged with N 2 for 5 mm. To this reaction mixture was added
- Step 1 Synthesis of 7-chloro-6-(quinolin-6-yl)-l,8-naphthyridin-4(lH)-one
- l -(2-ammo-5-(quinolin-6-y])pyridm-3-yl)ethanone 297 mg, 0.83 mmol, 1 eq
- 1,4 dioxane 10 mL
- DMF:DMA 150 rng, 1.25 mmol, 1.5 eq.
- the reaction mixture was heated at 100 °C for 30 min.
- the reaction was monitored by TLC and LCMS.
- the reaction mixture was concentrated under reduced pressure.
- Step-2 Synthesis of 7-(3-methyl-lH-pyrazol-l-yl)-6-(quinolin-6-yl)-l,8- naphthyridin-4(lH)-one: To a stirred solution of 7-chloro-6-(quinolin-6-yl)-l,8-naphthyridin- 4(lH)-one (120 mg, 0.39 mmol, leq.) in DMF (2 mL) was added 3-methyl-lH-pyrazole (320 mg, 3.9 mmol, 10 eq.) and CS 2 CO 3 (380 mg, 1.16 mmol, 3 eq.).
- reaction mixture was heated at 90 C for 48 h, progress of the reaction was monitored by TLC and LCMS.
- the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate (50 mL x 2). Combined organic layers were washed with water (100 mL ⁇ 2), dried with anhydrous Na 2 S0 4 and concentrated under vacuum to get the solid residue which was purified by reversed phase column chromatography to get the desired product as off white solid (2 mg, 1%).
- Step 1 Synthesis of l-(2-3 ⁇ 4mino-6- ⁇ 5-methyIfur3 ⁇ 4n-2- S)pyridin-3-yl)ethanone: To a solution of l-(2-amino-6-chloropyridin-3-yl)ethanone (600 mg, 3.52 mmol, 1 eq.) in DME (15 mL); water (3 mL) was added 5-methylfuran-2-ylboronic acid (485 mg, 3.88 mmol, 1 .1 eq.), K 2 C0 3 (731 mg, 5.29 mmol, 1.5 eq.), PdCl 2 (dppf DCM ( 144 mg, 0.017 mmol, 0.05 eq.).
- the reaction mixture was deoxygenated using N 2 atmosphere and the reaction mixture was heated at 80 °C for 18h. The reaction was monitored by TLC and LCMS. The reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 ⁇ 50 mL). The separated organic layer was dned over sodium sulfate and concentrated under reduced pressure. The crude product was purified using Combi-flash column chromatography to afford the desired product (150 mg, 20 %).
- Step 2 Synthesis of l-(2-amino-5-bromo-6-(5-methylfuran-2-yl)pyridin-3- yI)ethanone: To a solution of l -(2-amino-6-(5-methylfuran-2-yl)pyridin-3-yl)ethanorie (150 mg, 0.69 mmol, 1 eq.) in mixture of ACN (10 mL) was added N- bromosuccimmide ( 160 mg, 0.89 mmol, 1.3 eq.). The reaction mixture was stirred at room temperature for 1 h. The reaction was monitored by TLC and NMR. The reaction solvent was evaporated under reduced pressure.
- reaction mixture was diluted with water (100 mL) and extracted by ethyl acetate (2 ⁇ 50 mL). The organic layer was separated, washed water (5 ⁇ 20 mL) and brine and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to afford the desired product (200 mg, 98 %).
- Step 4 Synthesis of 6-bromo-7-(5-methylfuran-2-yl)-l,8-naphthyridin-4(lH)- one: To a solution of N'-(3-acetyl-5-bromo-6-(5-metiiylfuran-2-yl)pyrid
- Step 5 Synthesis of 6-(benzo[d]thiazol-6-yl)-7-(5-methylfuran-2-yl)-l,8- naphthyridin-4(lH)-one: To a solution of 6-bromo-7-(5-methylfuran-2-yl)-l,8-naphtiiyridiri- 4(lH)-one (100 mg, 0,33 mmol, 1 eq.) in 1 ,4 dioxane (8 mL): water (2 mL) was added benzo[d]thiazol-6-ylboronic acid (64 mg, 0.36 mmol, 1.1 eq.), 2 CO 3 (68 mg, 0.49 mmol, 1.5 eq.), Pd(PPh 3 ) 4 ( 19 mg, 0.016 mmol, 0.05 eq.).
- the reaction mixture was deoxygenated using N 2 atmosphere and the reaction mixture was heated at 80°C for 18h. The reaction was monitored by TLC and LCMS. The reaction mixture was diluted with water (50 ml.) and extracted using ethyl acetate (2 ⁇ 50 mL). The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified using reverse phase column chromatography to afford the desired product as off-white solid(14 mg, 1 1 %).
- reaction mixture was deoxygenated again & heated at 100 °C for 16 h.
- the reaction mixture was allowed to cool to RT and extracted using ethyl acetate (3 ⁇ 25 mL).
- the combined organic layers were washed (brine), dried (anhydrous Na 2 S0 4 ) and concentrated under vacuum to get the solid residue which was purified by reversed phase column chromatography to get the desired product as off white solid (0.015g, 13%)
- Example S33 Synthesis of 7-(lH-pyrazol-l-yl)-6-(quinolin-6-yl)-l, 8-naphthyridin-4(lH)-one ( ompound No. 1. / 00)
- Step 1 To a stirred solution of 7-chloro-6-(qiiinolin-6-yl)-l,8-naphthyridin-4(lH)- one(90mg, 0.29 mmol, 1.0 equiv) & hydrazine hydrate (22mg, 0.43 mmol, 1.5 equiv) in dioxane (5 mL). The resulting mixture was heated at 90 °C for 16 h . The reaction mixture was allowed to cool to RT and concentrated under vacuum to get the solid residue (90 mg cmde) which was used for next reaction without purification.
- Step 2 To a stirred solution of 7-hydrazinyl-6-(quino]in-6-yl)-l ,8-naphthyridin- 4(lH)-one (90 mg, 0.29 mmol, 1.0 equiv) & 1,1,3, 3-tetraethoxypropane (97 mg, 0.44 mrnoi, 1.5 equiv) in dioxane (5 mL) was added 20% HCi in dioxane (0.3 ml). The reaction mixture was heated at 90 °C for 16 h. Progress of the reaction was monitored by LCMS.
- reaction mixture was allowed to cool to RT & basified with solution of NaHCOs (lOmL) and extracted with ethyl acetate i20mL 2 ). The combined organic layers were washed with water (50mL) and brine solution (50mL), dried (anhydrous Na2S0 4 ) & concentrated under vacuum to get the solid residue which was purified by reversed phase column chromatography to get the desired product (13 mg, 13 %).
- Reaction mixture was allowed to cool to RT & quenched by- adding aq. NaOH & extracted using ethyl acetate (3 x 100 mL) The combined organic layers were washed (brine), dried (anhydrous Na 2 S(> 4 ) & concentrated under vacuum to get the solid which was purified by normal phase column chromatography to get the desired product (0.0092 g, 8%).
- the progress of the reaction was monitored by LCMS.
- the reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 x 50 mL). The separated organic layer was dried over sodium sulfate and concentrated under reduced pressure.
- the crude product was purified by reverse phase column chromatography to afford the desired product (25 mg, 23%)
- the reaction mixture was deoxygenated using N 2 atmosphere and the reaction mixture was heated at 80 °C for 18 h. The reaction was monitored by NMR and LCMS. The reaction mixture was diluted with water (50 mL) and extracted using ethyl acetate (2 ⁇ 50 mL). The separated organic layer was dried over sodium sulfate and
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description


























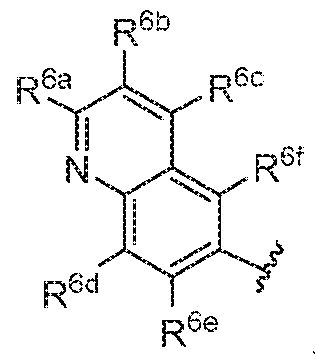



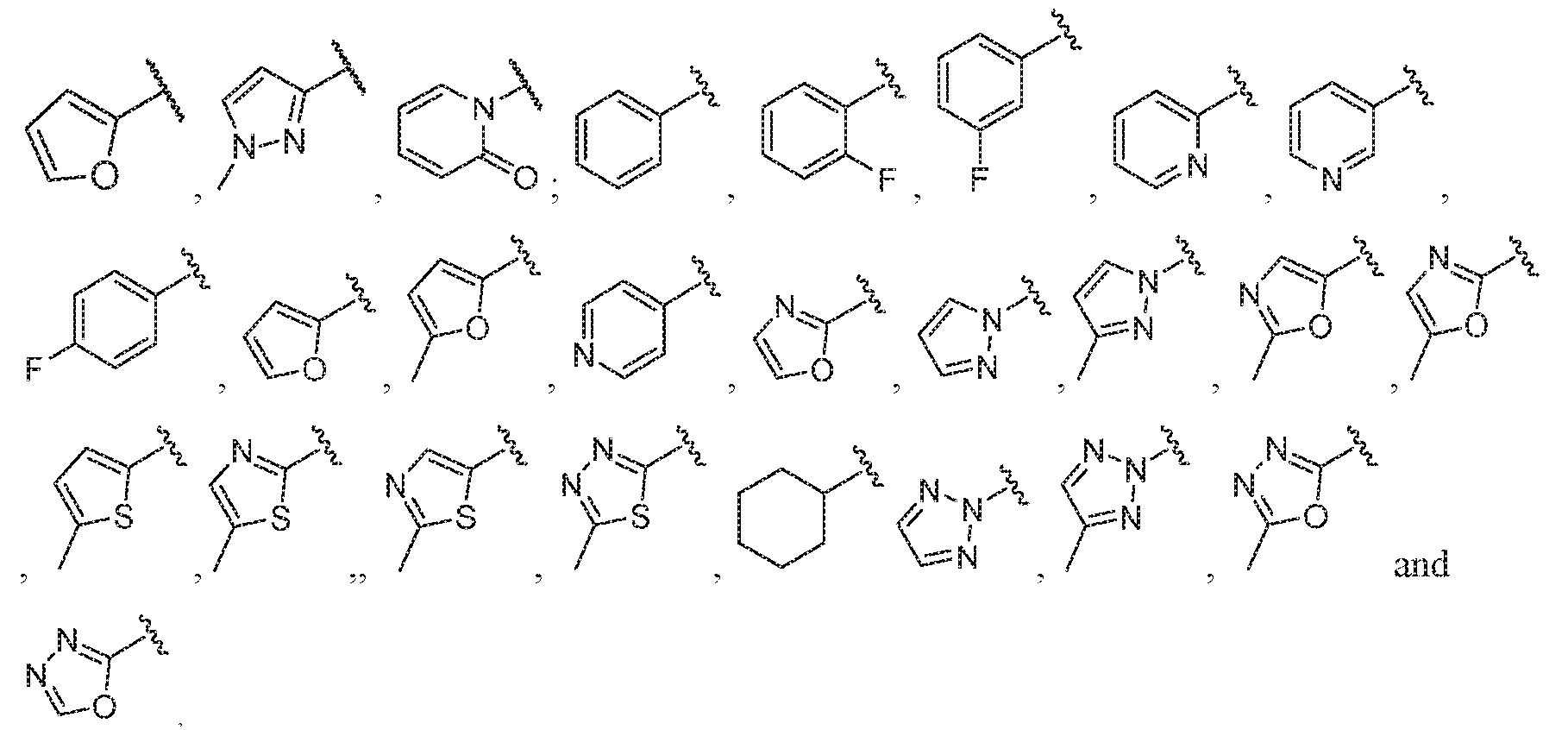






























































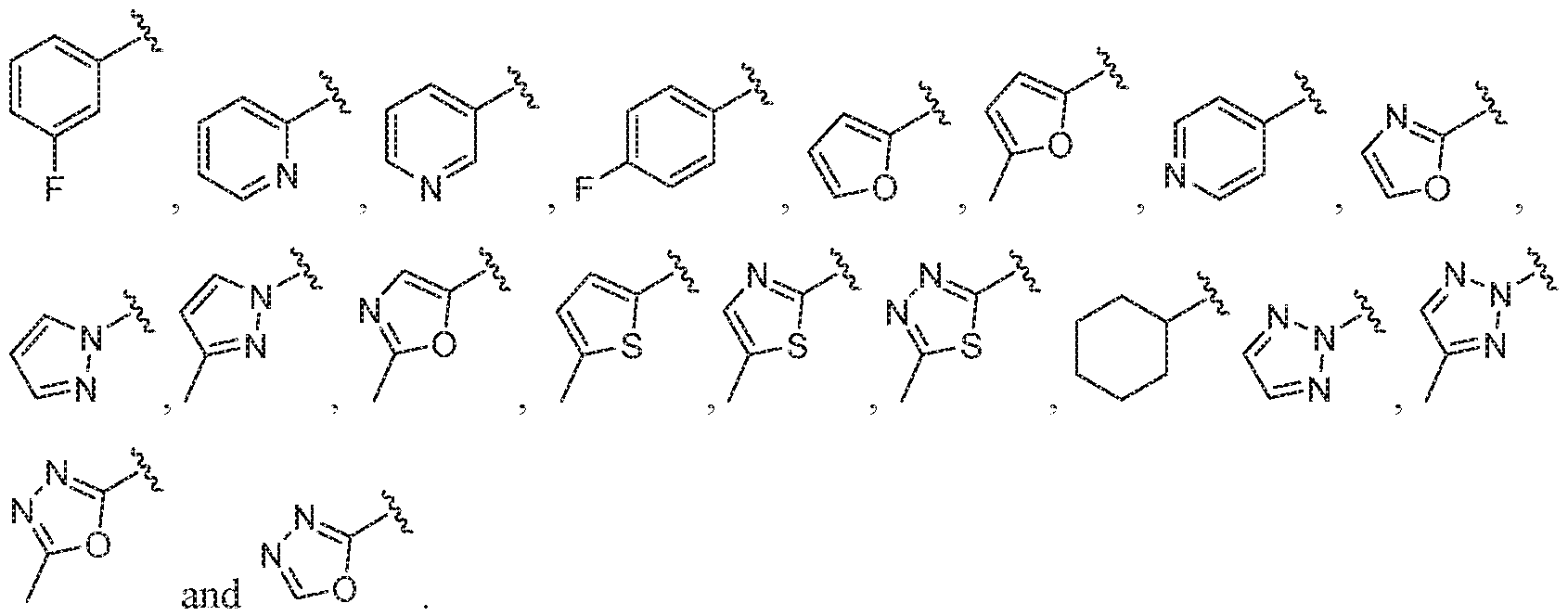






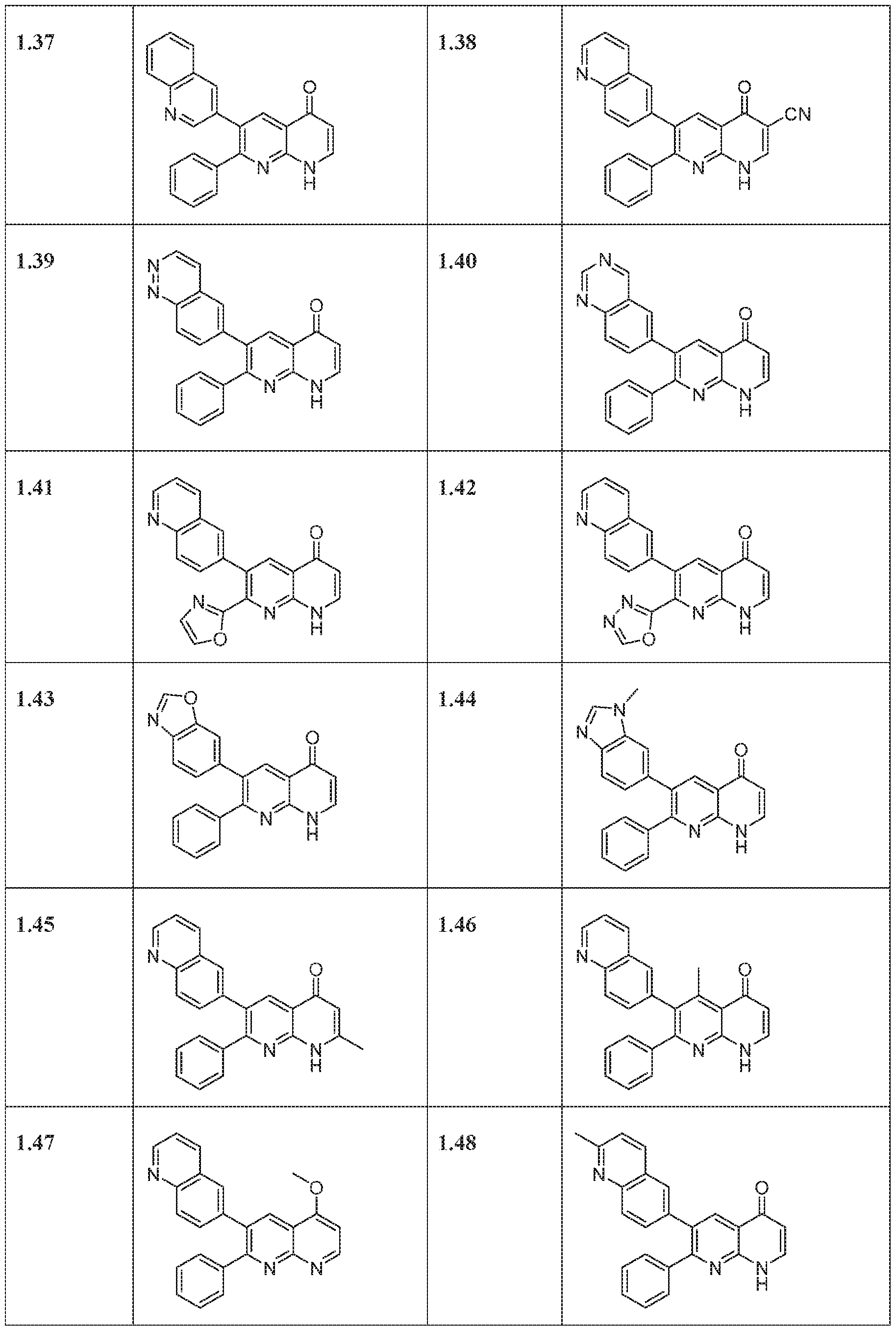
























































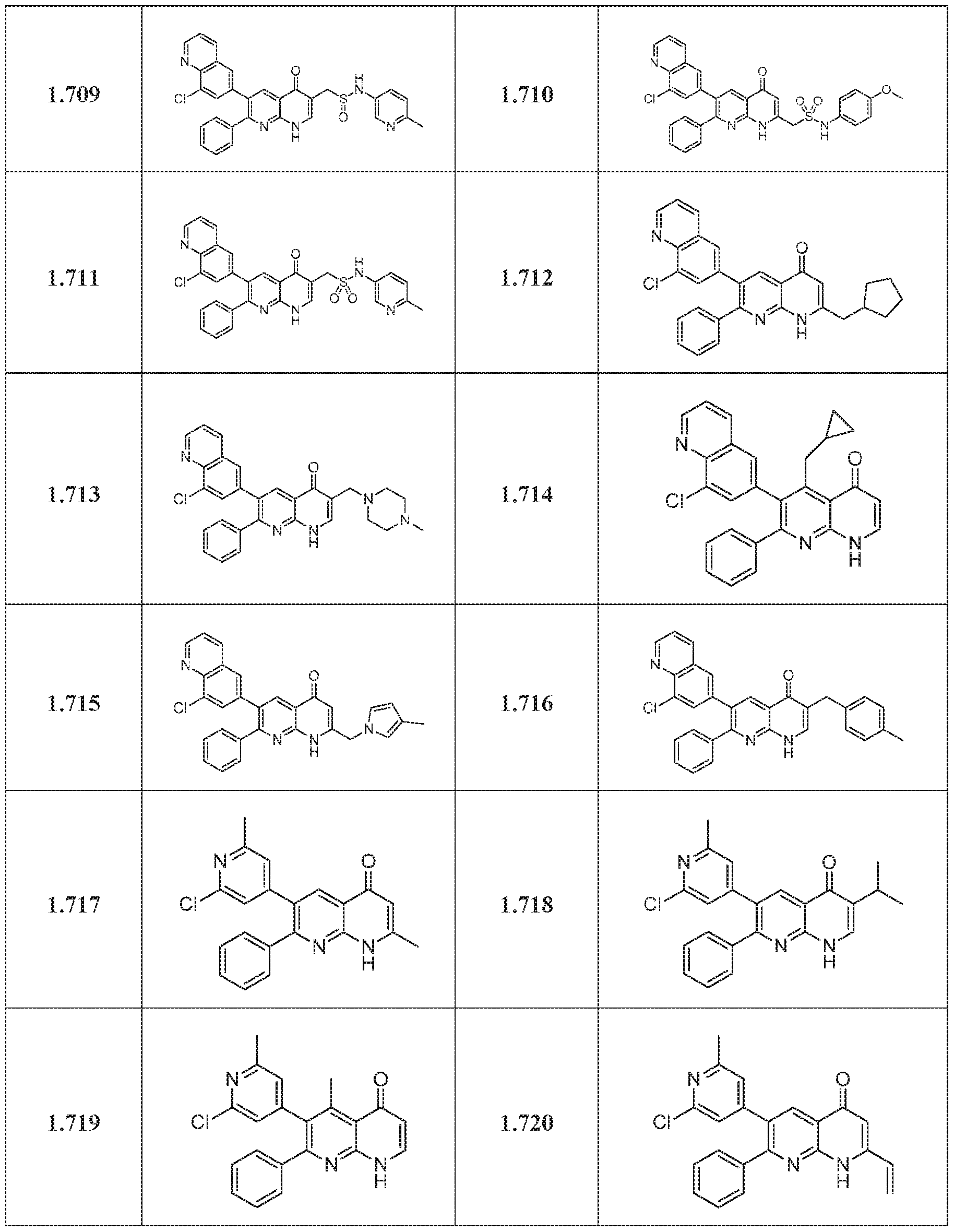
































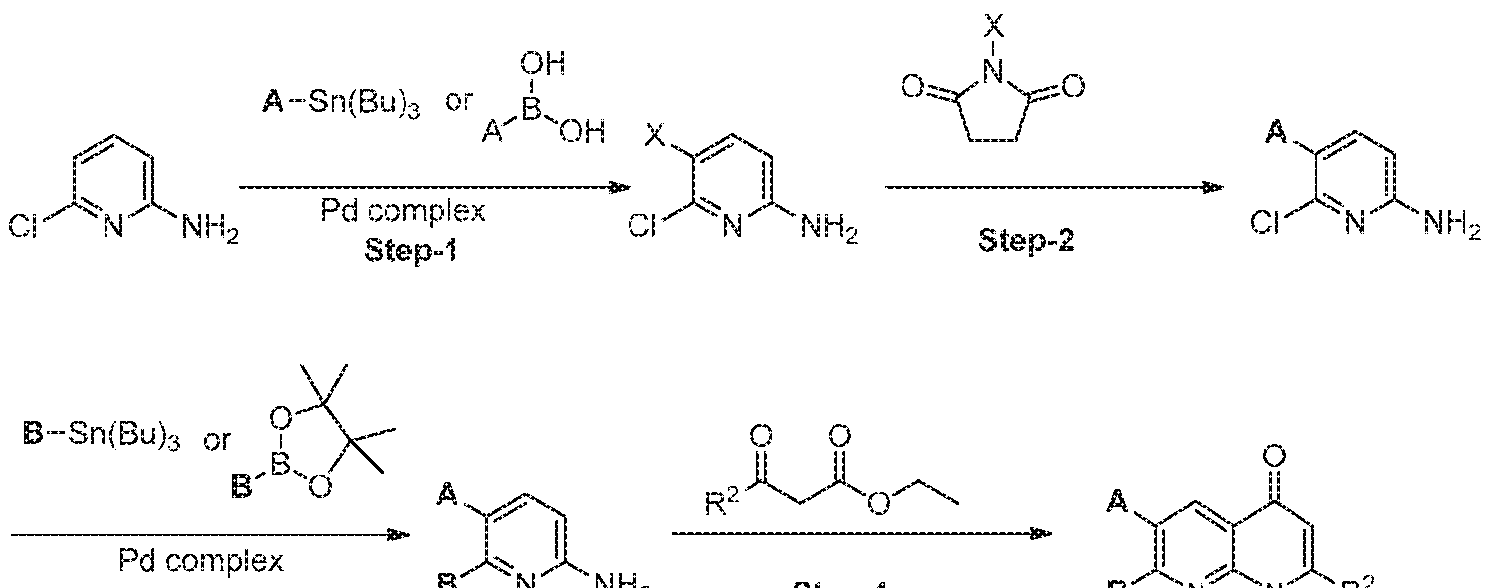



















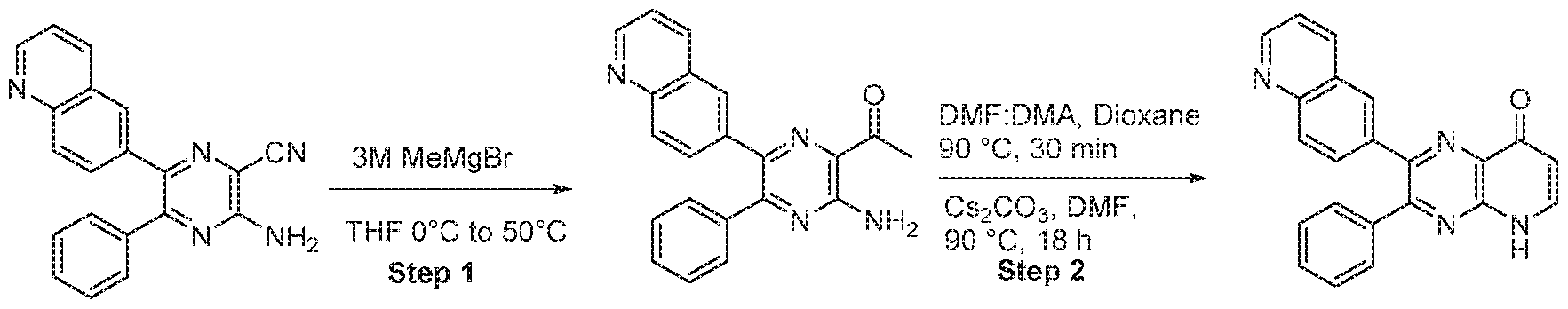





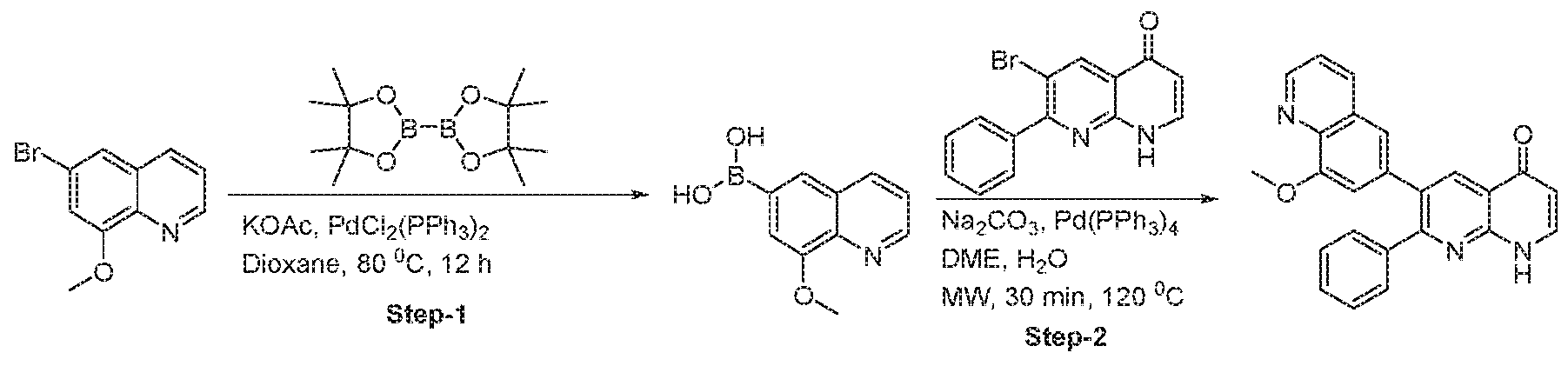

















Claims











Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2018302178A AU2018302178A1 (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof |
| CA3070073A CA3070073A1 (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof |
| KR1020207004250A KR20200040764A (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compound and use thereof |
| BR112020000962-8A BR112020000962A2 (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof |
| JP2020503017A JP2020527593A (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthylidinone compounds and their use |
| EP18835580.4A EP3654982A4 (en) | 2017-07-18 | 2018-07-18 | 1,8-NAPHTHYRIDINONE COMPOUNDS AND THEIR USES |
| MX2020000693A MX2020000693A (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof. |
| CN201880059414.8A CN111093666A (en) | 2017-07-18 | 2018-07-18 | 1,8-Naphthyridinone compound and use thereof |
| SG11202000431PA SG11202000431PA (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof |
| IL272055A IL272055A (en) | 2017-07-18 | 2020-01-15 | 1,8-naphthyridinone compounds and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762534185P | 2017-07-18 | 2017-07-18 | |
| US62/534,185 | 2017-07-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019018583A1 true WO2019018583A1 (en) | 2019-01-24 |
Family
ID=65014549
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2018/042776 Ceased WO2019018583A1 (en) | 2017-07-18 | 2018-07-18 | 1,8-naphthyridinone compounds and uses thereof |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US10793561B2 (en) |
| EP (1) | EP3654982A4 (en) |
| JP (1) | JP2020527593A (en) |
| KR (1) | KR20200040764A (en) |
| CN (1) | CN111093666A (en) |
| AU (1) | AU2018302178A1 (en) |
| BR (1) | BR112020000962A2 (en) |
| CA (1) | CA3070073A1 (en) |
| IL (1) | IL272055A (en) |
| MX (1) | MX2020000693A (en) |
| SG (1) | SG11202000431PA (en) |
| WO (1) | WO2019018583A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020150676A1 (en) * | 2019-01-18 | 2020-07-23 | Nuvation Bio Inc. | 1,8-naphthyridinone compounds and uses thereof |
| US11306071B2 (en) | 2019-01-18 | 2022-04-19 | Nuvation Bio Inc. | Heterocyclic compounds as adenosine antagonists |
| CN114507231A (en) * | 2020-11-17 | 2022-05-17 | 江苏先声药业有限公司 | Lactam compound and preparation method thereof |
| EP3911322A4 (en) * | 2019-01-18 | 2022-08-17 | Nuvation Bio Inc. | COMPOUNDS AND THEIR USES |
| US11564928B1 (en) | 2020-05-05 | 2023-01-31 | Teon Therapeutics, Inc. | Cannabinoid receptor type 2 (CB2) modulators and uses thereof |
| EP4276100A4 (en) * | 2020-12-29 | 2025-06-25 | Txinno Bioscience Inc. | Novel naphthyridinone derivative with inhibiting activity against ectonucleotide pyrophosphatase phosphodiesterase and use thereof |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019018584A1 (en) | 2017-07-18 | 2019-01-24 | GiraFpharma LLC | Heterocyclic compounds as adenosine antagonists |
| EP3654982A4 (en) * | 2017-07-18 | 2021-04-14 | Nuvation Bio Inc. | 1,8-NAPHTHYRIDINONE COMPOUNDS AND THEIR USES |
| WO2022184116A1 (en) * | 2021-03-05 | 2022-09-09 | 江苏先声药业有限公司 | New sos1 inhibitor, preparation method therefor and use thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140243346A1 (en) * | 2010-07-14 | 2014-08-28 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
Family Cites Families (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE47351E1 (en) | 1999-06-22 | 2019-04-16 | Gilead Sciences, Inc. | 2-(N-pyrazolo)adenosines with application as adenosine A2A receptor agonists |
| WO2003030909A1 (en) | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| US6992087B2 (en) | 2001-11-21 | 2006-01-31 | Pfizer Inc | Substituted aryl 1,4-pyrazine derivatives |
| CA2517256C (en) | 2003-02-26 | 2013-04-30 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| WO2004084824A2 (en) | 2003-03-24 | 2004-10-07 | Merck & Co., Inc. | Biaryl substituted 6-membered heterocyles as sodium channel blockers |
| CA2543644A1 (en) | 2003-10-27 | 2005-05-06 | Astellas Pharma Inc. | Pyrazine derivatives and pharmaceutical use thereof |
| AU2004289638B2 (en) * | 2003-11-04 | 2009-10-01 | Merck Sharp & Dohme Corp. | Substituted naphthyridinone derivatives |
| ES2270715B1 (en) * | 2005-07-29 | 2008-04-01 | Laboratorios Almirall S.A. | NEW DERIVATIVES OF PIRAZINA. |
| GB0800741D0 (en) | 2008-01-16 | 2008-02-20 | Univ Greenwich | Cyclic triazo and diazo sodium channel blockers |
| ES2480994T3 (en) | 2008-03-31 | 2014-07-29 | Genentech, Inc. | Compounds of benzopyran and benzoxepine PI3K inhibitors and methods of use |
| TW201102065A (en) * | 2009-05-29 | 2011-01-16 | Astrazeneca Ab | Heterocyclic urea derivatives and methods of use thereof |
| SI2531492T1 (en) | 2010-02-05 | 2016-08-31 | Heptares Therapeutics Limited | 1,2,4-triazine-4-amine derivatives |
| CN103261202B (en) * | 2010-09-24 | 2016-01-20 | 阿迪维纳斯疗法有限公司 | As the fused tricyclic compounds of adenosine receptor antagonists |
| US9142781B2 (en) | 2011-06-09 | 2015-09-22 | Novaled Ag | Compound for organic electronic device |
| AR089698A1 (en) | 2012-01-13 | 2014-09-10 | Novartis Ag | ANTAGONIST HETEROCICLIC COMPOUNDS OF THE IP RECEIVER |
| US9266891B2 (en) | 2012-11-16 | 2016-02-23 | Boehringer Ingelheim International Gmbh | Substituted [1,2,4]triazolo[4,3-A]pyrazines that are BRD4 inhibitors |
| EP2970312B1 (en) | 2013-03-11 | 2017-11-15 | The Regents of The University of Michigan | Bet bromodomain inhibitors and therapeutic methods using the same |
| EP3842424B1 (en) | 2013-03-15 | 2024-12-11 | The Trustees of Columbia University in the City of New York | Map kinase modulators and uses thereof in the tretament of tauopathies |
| CA2915838C (en) | 2013-06-21 | 2023-04-18 | Zenith Epigenetics Corp. | Bicyclic bromodomain inhibitors |
| CN104341386A (en) | 2013-07-23 | 2015-02-11 | 中国科学院上海药物研究所 | Aryl heterocycle micromolecule compounds, derivatives thereof, and preparing methods and uses of the compounds and the derivatives |
| EP3027616B1 (en) | 2013-07-30 | 2018-01-10 | Boehringer Ingelheim International GmbH | Azaindole compounds as modulators of rorc |
| JP6553632B2 (en) | 2013-11-18 | 2019-07-31 | フォーマ セラピューティクス,インコーポレイテッド | Tetrahydroquinoline compositions as BET bromodomain inhibitors |
| CN106029076B (en) | 2013-11-18 | 2019-06-07 | 福马疗法公司 | Benzo piperazine composition as BET bromine domain inhibitor |
| US9580430B2 (en) | 2014-02-28 | 2017-02-28 | The Regents Of The University Of Michigan | 9H-pyrimido[4,5-B]indoles and related analogs as BET bromodomain inhibitors |
| EA034972B1 (en) | 2014-04-23 | 2020-04-13 | Инсайт Корпорейшн | 1H-PYRROLO[2,3-c]PYRIDIN-7(6H)-ONES AS INHIBITORS OF BET PROTEINS |
| CA2961807A1 (en) * | 2014-09-19 | 2016-03-24 | Forma Therapeutics, Inc. | Phenyl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| CA2967944C (en) | 2014-11-18 | 2020-11-17 | Merck Sharp & Dohme Corp. | Aminopyrazine compounds with a2a antagonist properties |
| EP3230277B1 (en) | 2014-12-11 | 2019-09-18 | Zenith Epigenetics Ltd. | Substituted heterocycles as bromodomain inhibitors |
| EP3262045A1 (en) | 2015-02-27 | 2018-01-03 | The Regents of The University of Michigan | 9h-pyrimido [4,5-b]indoles as bet bromodomain inhibitors |
| JP2018527293A (en) | 2015-06-16 | 2018-09-20 | オリオン コーポレーション | Spiro [cyclobutane-1,3'-indoline] -2'-one derivatives as bromodomain inhibitors |
| SI3334431T1 (en) | 2015-08-11 | 2020-01-31 | Novartis Ag | 5-bromo-2,6-di-(1h-pyrazol-l-yl)pyrimidin-4-amine for use in the treatment of cancer |
| CN106478651B (en) * | 2015-08-31 | 2019-07-09 | 广东东阳光药业有限公司 | Substituted heteroaryl compound and combinations thereof and purposes |
| CN108137585B (en) | 2015-09-21 | 2021-10-22 | 普莱希科公司 | Heterocyclic compounds and their applications |
| US10370356B2 (en) | 2015-09-22 | 2019-08-06 | Glaxosmithkline Intellectual Property (No. 2) Limited | Pyridinone dicarboxamide for use as bromodomain inhibitors |
| JP6847958B2 (en) | 2015-12-24 | 2021-03-24 | コーバス・ファーマシューティカルズ・インコーポレイテッド | How to treat cancer |
| WO2018005533A1 (en) | 2016-07-01 | 2018-01-04 | G1 Therapeutics, Inc. | Antiproliferative pyrimidine-based compounds |
| EP3858835A1 (en) | 2016-07-01 | 2021-08-04 | G1 Therapeutics, Inc. | Pyrimidine-based antiproliferative agents |
| JP7106462B2 (en) | 2016-07-01 | 2022-07-26 | ジー1 セラピューティクス, インコーポレイテッド | Synthesis of N-(heteroaryl)-pyrrolo[3,2-D]pyrimidin-2-amines |
| GB201612092D0 (en) | 2016-07-12 | 2016-08-24 | Almac Discovery Ltd | Pharmaceutical compounds |
| WO2018081863A1 (en) | 2016-11-04 | 2018-05-11 | University Of Wollongong | 6-SUBSTITUTED DERIVATIVES OF HEXAMETHYLENE AMILORIDE AS INHIBITORS OF uPA AND USES THEREOF |
| CN110214012B (en) | 2017-01-20 | 2023-05-09 | 艾库斯生物科学有限公司 | Azolopyrimidines for the treatment of cancer related disorders |
| DK3601296T3 (en) | 2017-03-30 | 2022-08-29 | iTeos Belgium SA | 2-OXO-THIAZOLE DERIVATIVES AS A2A INHIBITORS AND COMPOUNDS FOR USE IN THE TREATMENT OF CANCER |
| WO2018178338A1 (en) | 2017-03-30 | 2018-10-04 | Iteos Therapeutics | 2-oxo-thiazole derivatives as a2a inhibitors and compounds for use in the treatment of cancers |
| CN107221611B (en) | 2017-06-15 | 2019-02-05 | 江西冠能光电材料有限公司 | A kind of stable easy processing organic semiconducting materials and its organic luminescent device application |
| JP2020525472A (en) | 2017-06-30 | 2020-08-27 | ライブ・セラピューティクス・スプウカ・アクツィーナRyvu Therapeutics S.A. | Imidazo[1,2-A]pyrazine modulator of adenosine A2A receptor |
| WO2019018584A1 (en) | 2017-07-18 | 2019-01-24 | GiraFpharma LLC | Heterocyclic compounds as adenosine antagonists |
| EP3654982A4 (en) * | 2017-07-18 | 2021-04-14 | Nuvation Bio Inc. | 1,8-NAPHTHYRIDINONE COMPOUNDS AND THEIR USES |
| CN111565722A (en) | 2017-11-06 | 2020-08-21 | 科尔沃斯制药股份有限公司 | Adenosine pathway inhibitors for cancer therapy |
| WO2019096322A1 (en) | 2017-11-20 | 2019-05-23 | 上海医药集团股份有限公司 | Pyrazolone-pyrimidine compound, preparation method therefor and application thereof |
| EP3723754A4 (en) | 2017-12-13 | 2021-05-19 | Merck Sharp & Dohme Corp. | IMIDAZO [1,2-C] QUINAZOLIN-5-AMINE COMPOUNDS WITH A2A ANTAGONIST PROPERTIES |
| WO2019120234A2 (en) | 2017-12-20 | 2019-06-27 | 贝达药业股份有限公司 | Compound functioning as bromodomain protein inhibitor, and composition |
| CN111315747B (en) | 2018-01-05 | 2023-05-02 | 四川科伦博泰生物医药股份有限公司 | Dihydropyrazolopyrimidine compounds and their preparation methods and uses |
| EP3741758A1 (en) | 2018-01-16 | 2020-11-25 | Shanghai Institute of Materia Medica, Chinese Academy of Sciences | Bromodomain inhibitor compound and use thereof |
| CA3088381A1 (en) | 2018-01-29 | 2019-08-01 | Beta Pharma, Inc. | 2h-indazole derivatives as cdk4 and cdk6 inhibitors and therapeutic uses thereof |
| CA3089159A1 (en) | 2018-02-06 | 2019-08-15 | Jiangsu Hengrui Medicine Co., Ltd. | Pyrazolo[1,5-a][1,3,5]triazine-2-amine derivative, preparation method therefor and medical use thereof |
| CA3090795A1 (en) | 2018-02-13 | 2019-08-22 | Iovance Biotherapeutics, Inc. | Expansion of tumor infiltrating lymphocytes (tils) with adenosine a2a receptor antagonists and therapeutic combinations of tils and adenosine a2a receptor antagonists |
| WO2019158070A1 (en) | 2018-02-15 | 2019-08-22 | 杭州阿诺生物医药科技有限公司 | A2a and/or a2b receptor antagonist |
| US11479555B2 (en) | 2018-02-23 | 2022-10-25 | Newave Pharmaceutical Inc. | Substituted 1,2-dihydro-3H-pyrazolo[3,4-D]pyrimidin-3-ones as inhibitors of WEE-1 kinase |
| BR122023024273A2 (en) | 2018-02-27 | 2024-02-20 | Incyte Corporation | IMIDAZOPIRIMIDINE AND TRIAZOLOPYRIMIDIINE COMPOUNDS, THEIR USES, METHOD FOR INHIBITING AN ADENOSINE RECEPTOR ACTIVITY AND PHARMACEUTICAL COMPOSITION THEREOF |
| EP3758706A4 (en) | 2018-02-28 | 2021-11-24 | The Regents Of The University Of Colorado | KINASE WEE1 INHIBITORS AND METHODS OF CANCER TREATMENT USING THESE INHIBITORS |
| MX2020009372A (en) | 2018-03-09 | 2020-10-14 | Recurium Ip Holdings Llc | Substituted 1,2-dihydro-3h-pyrazolo[3,4-d]pyrimidin-3-ones. |
| US11220510B2 (en) | 2018-04-09 | 2022-01-11 | Incyte Corporation | Pyrrole tricyclic compounds as A2A / A2B inhibitors |
-
2018
- 2018-07-18 EP EP18835580.4A patent/EP3654982A4/en not_active Withdrawn
- 2018-07-18 KR KR1020207004250A patent/KR20200040764A/en not_active Withdrawn
- 2018-07-18 US US16/039,301 patent/US10793561B2/en not_active Expired - Fee Related
- 2018-07-18 CA CA3070073A patent/CA3070073A1/en active Pending
- 2018-07-18 WO PCT/US2018/042776 patent/WO2019018583A1/en not_active Ceased
- 2018-07-18 AU AU2018302178A patent/AU2018302178A1/en not_active Abandoned
- 2018-07-18 MX MX2020000693A patent/MX2020000693A/en unknown
- 2018-07-18 BR BR112020000962-8A patent/BR112020000962A2/en not_active Application Discontinuation
- 2018-07-18 JP JP2020503017A patent/JP2020527593A/en active Pending
- 2018-07-18 SG SG11202000431PA patent/SG11202000431PA/en unknown
- 2018-07-18 CN CN201880059414.8A patent/CN111093666A/en active Pending
-
2020
- 2020-01-15 IL IL272055A patent/IL272055A/en unknown
-
2021
- 2021-12-13 US US17/644,052 patent/US20220220111A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140243346A1 (en) * | 2010-07-14 | 2014-08-28 | Novartis Ag | Ip receptor agonist heterocyclic compounds |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE PUBCHEM. [o] 30 November 2012 (2012-11-30), Database accession no. 66665902 * |
| See also references of EP3654982A4 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020150676A1 (en) * | 2019-01-18 | 2020-07-23 | Nuvation Bio Inc. | 1,8-naphthyridinone compounds and uses thereof |
| US11254670B2 (en) | 2019-01-18 | 2022-02-22 | Nuvation Bio Inc. | 1,8-naphthyridinone compounds and uses thereof |
| US11306071B2 (en) | 2019-01-18 | 2022-04-19 | Nuvation Bio Inc. | Heterocyclic compounds as adenosine antagonists |
| EP3911322A4 (en) * | 2019-01-18 | 2022-08-17 | Nuvation Bio Inc. | COMPOUNDS AND THEIR USES |
| EP3911324A4 (en) * | 2019-01-18 | 2022-08-17 | Nuvation Bio Inc. | 1,8-NAPHTHYRIDINONE COMPOUNDS AND THEIR USES |
| US11564928B1 (en) | 2020-05-05 | 2023-01-31 | Teon Therapeutics, Inc. | Cannabinoid receptor type 2 (CB2) modulators and uses thereof |
| US11957689B2 (en) | 2020-05-05 | 2024-04-16 | Teon Therapeutics, Inc. | Cannabinoid receptor type 2 (CB2) modulators and uses thereof |
| CN114507231A (en) * | 2020-11-17 | 2022-05-17 | 江苏先声药业有限公司 | Lactam compound and preparation method thereof |
| EP4276100A4 (en) * | 2020-12-29 | 2025-06-25 | Txinno Bioscience Inc. | Novel naphthyridinone derivative with inhibiting activity against ectonucleotide pyrophosphatase phosphodiesterase and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CN111093666A (en) | 2020-05-01 |
| CA3070073A1 (en) | 2019-01-24 |
| EP3654982A1 (en) | 2020-05-27 |
| EP3654982A4 (en) | 2021-04-14 |
| US20190023702A1 (en) | 2019-01-24 |
| JP2020527593A (en) | 2020-09-10 |
| MX2020000693A (en) | 2020-07-29 |
| IL272055A (en) | 2020-03-31 |
| US10793561B2 (en) | 2020-10-06 |
| BR112020000962A2 (en) | 2020-07-14 |
| AU2018302178A1 (en) | 2020-02-13 |
| US20220220111A1 (en) | 2022-07-14 |
| SG11202000431PA (en) | 2020-02-27 |
| KR20200040764A (en) | 2020-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019018583A1 (en) | 1,8-naphthyridinone compounds and uses thereof | |
| US11028058B2 (en) | Heterocyclic compounds as adenosine antagonists | |
| WO2019074979A1 (en) | Heterocyclic compounds and uses thereof | |
| US11306071B2 (en) | Heterocyclic compounds as adenosine antagonists | |
| US20220169648A1 (en) | 1,8-naphthyridinone compounds and uses thereof | |
| AU2020210013A1 (en) | Compounds and uses thereof | |
| WO2020150677A1 (en) | Heterocyclic compounds as adenosine antagonists | |
| WO2021146631A1 (en) | Heterocyclic compounds as adenosine antagonists | |
| WO2022082174A1 (en) | Heterocyclic compounds and uses thereof | |
| WO2021146629A1 (en) | Heterocyclic compounds as adenosine antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18835580 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3070073 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020503017 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020000962 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2018302178 Country of ref document: AU Date of ref document: 20180718 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018835580 Country of ref document: EP Effective date: 20200218 |
|
| ENP | Entry into the national phase |
Ref document number: 112020000962 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200116 |