WO2019062804A1 - 一种氧杂螺环类衍生物的制备方法及其中间体 - Google Patents
一种氧杂螺环类衍生物的制备方法及其中间体 Download PDFInfo
- Publication number
- WO2019062804A1 WO2019062804A1 PCT/CN2018/107901 CN2018107901W WO2019062804A1 WO 2019062804 A1 WO2019062804 A1 WO 2019062804A1 CN 2018107901 W CN2018107901 W CN 2018107901W WO 2019062804 A1 WO2019062804 A1 WO 2019062804A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- formula
- aryl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C1(CC(O)=O)CC2(CCCC2)OCC1 Chemical compound *C1(CC(O)=O)CC2(CCCC2)OCC1 0.000 description 7
- RQEUFEKYXDPUSK-ZETCQYMHSA-N C[C@@H](c1ccccc1)N Chemical compound C[C@@H](c1ccccc1)N RQEUFEKYXDPUSK-ZETCQYMHSA-N 0.000 description 1
- WOBPBSWVWCCPLE-HNNXBMFYSA-N OC(C[C@@]1(CC2(CCCC2)OCC1)c1ccccn1)=O Chemical compound OC(C[C@@]1(CC2(CCCC2)OCC1)c1ccccn1)=O WOBPBSWVWCCPLE-HNNXBMFYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4433—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/96—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings spiro-condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the invention relates to a preparation method of an oxaspirocyclic derivative and an intermediate thereof.
- Postoperative pain is the most common acute pain.
- Commonly used drugs are opioids, such as fentanyl, morphine, meperidine, oxycodone, etc.
- the pharmacological activity of analgesia is activated by activation in the central nervous system as well as the gastrointestinal tract.
- the G ⁇ i protein receptor ( ⁇ opioid receptor, MOR) on the cell membrane inhibits the hyperpolarization of nerve fibers to achieve.
- Opioid receptors are an important class of G protein-coupled receptors (GPCRs), which are targets for the binding of endogenous opioid peptides and opioids.
- GPCRs G protein-coupled receptors
- the opioid receptors activate the nervous system and the endocrine system. It has a regulating effect, and opioids are the strongest and commonly used central analgesics.
- Endogenous opioid peptides are naturally occurring opioid active substances in mammals.
- endogenous opioid peptides are broadly classified into enkephalins, endorphins, dynorphins and neomorphins (Pharmacol Rev 2007; 59:88-123).
- opioid receptors in the central nervous system namely ⁇ (MOR), ⁇ (DOR), ⁇ (KOR) receptors and the like.
- MOR is a target for endogenous enkephalins and opioid analgesics such as morphine.
- the method has the problems of small batch size, post-treatment method using chiral column separation and purification, thin layer chromatography purification, low yield, etc., wherein the reaction yield of preparing compound 19 is only 35%; the reduction used in the preparation of compound 5a
- the agent DIBAL is a dangerous flammable reagent, and large impurities are generated during the reaction, which is not conducive to industrial expansion, and it is necessary to improve the preparation method.
- the present invention provides a method for producing a compound represented by D1 or a salt thereof,
- the method comprises the steps of chiral resolution of a compound represented by formula D or a salt thereof, which is preferably a chromatographic resolution method (such as chiral HPLC) or chemical resolution (such as using a hand). Separation of sexual resolving agents),
- R is selected from aryl or heteroaryl, which is optionally selected from the group consisting of alkyl, haloalkyl, halogen, amino, nitro, cyano, oxo, alkenyl, haloalkoxy Base, hydroxyalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, OR 3 , C(O)R 3 , C(O)OR 3 , S(O) m R 3 or NR 4 R 5 Substituted by one or more substituents;
- R 3 is selected from a hydrogen atom, an alkyl group, a halogenated alkyl group, an amino group, an alkoxy group, a hydroxyalkyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the alkyl group, the cycloalkyl group , heterocyclyl, aryl and heteroaryl are optionally selected from alkyl, halo, hydroxy, amino, nitro, cyano, alkoxy, hydroxyalkyl, cycloalkyl, heterocyclyl, aryl or Substituting one or more substituents in the heteroaryl;
- R 4 and R 5 are each independently selected from a hydrogen atom, an alkyl group, an alkoxy group, a hydroxyalkyl group, a hydroxyl group, an amino group, a carboxylate group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group.
- the alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl groups are optionally selected from the group consisting of alkyl, halogen, hydroxy, amino, carboxylate, nitro, cyano, alkoxy, hydroxyalkane Substituted by one or more substituents in the group, cycloalkyl, heterocyclyl, aryl or heteroaryl;
- n 0, 1, or 2.
- the resolving agent used in the chiral resolution method is an alkaline chiral resolving agent, which may be S-phenylethylamine, quinidine, cinchonidine or arginine.
- the resolving agent used in the chiral resolution method is S-phenylethylamine.
- the method for producing the compound represented by D1 or a salt thereof further comprises the step of preparing a compound of the formula D, wherein the reaction condition is preferably from alkaline hydrolysis,
- R is as defined in the compound of formula D.
- the base used in the hydrolysis reaction is selected from the group consisting of sodium hydroxide, potassium hydroxide or lithium hydroxide, and the like.
- the compound of Formula D1 is:
- the preparation method comprises the following steps:
- a method of preparing a compound of D1 or a salt thereof comprising:
- R is as defined in the compound of formula D.
- a method of preparing a compound of the formula D2 or a salt thereof comprising:
- the present invention also provides a compound of the formula D1 or a salt thereof,
- the compound of Formula D1 is:
- the compound salt of formula D1 is:
- M is selected from the group consisting of S-phenylethylamine, quinidine, cinchonidine or arginine.
- the salt of the compound of formula D1 is
- the invention also provides a process for the preparation of a compound of formula D1-1, comprising the steps of reacting a compound of formula D with a chiral resolving agent M to give a compound of formula D1-1, said chiral resolution
- the agent is preferably an alkaline chiral resolving agent, more preferably S-phenylethylamine, quinidine, cinchonidine or arginine.
- the method for preparing the compound represented by the formula D1-1 further comprises:
- R is as defined in formula D1.
- R is M is S-phenethylamine
- the preparation method is as follows:
- the present invention also provides a process for the preparation of a compound of the formula B or a stereoisomer thereof, which comprises: the compound of the formula D or a stereoisomer thereof is prepared by a one-step or one-step reaction to obtain a compound of the formula B or The steps of its stereoisomers,
- R is as defined in the compound represented by formula D1, preferably from
- the compound of Formula B is:
- the preparation method comprises the following:
- a compound containing an aldehyde group is usually produced by a one-step reaction, a two-step reaction or a two-step reaction.
- the preferred two-step reaction of the present invention is preferred.
- a compound containing an aldehyde group is prepared.
- a method of preparing a compound of Formula B or a stereoisomer thereof comprises:
- R is as defined in the compound represented by formula D1, preferably from
- the compound of formula B or a stereoisomer thereof is
- the preparation method comprises the following:
- the compound of Formula B or a stereoisomer thereof is
- the preparation method comprises the following:
- a method of preparing a compound of Formula B or a stereoisomer thereof comprises:
- R is as defined in the compound represented by formula D1.
- the method of preparing a compound of Formula B or a stereoisomer thereof further comprises the method steps described above for the preparation of a compound of Formula D1 or a salt thereof.
- the compound of Formula B or a stereoisomer thereof is
- the preparation method comprises the following:
- R is as defined in the compound represented by formula D1.
- the compound of Formula B or a stereoisomer thereof is
- the preparation method comprises the following:
- the invention also provides a compound of formula C,
- R is as defined in the compound represented by formula D1, preferably from
- the compound of Formula C is:
- R is as defined in the compound represented by formula D1, preferably from
- the compound of Formula C is:
- the invention also provides a process for the preparation of a compound of formula C, which comprises:
- R is as defined in the compound represented by formula D1.
- the compound of Formula C is:
- the preparation method comprises the following:
- the method of preparing a compound of Formula C1 further comprises the steps of the foregoing method of preparing a compound of Formula D1.
- a method of preparing a compound of Formula C1 includes:
- R is as defined in the compound represented by formula D1.
- the compound of Formula C is:
- the preparation method comprises the following:
- the method of preparing a compound of Formula C2 further comprises the steps of the foregoing method of preparing a compound of Formula D-1.
- a method of preparing a compound of Formula C2 includes:
- the present invention also provides a process for the preparation of a compound of the formula I or a salt thereof, which comprises reacting a compound of the formula D or a stereoisomer thereof to give a compound of the formula B or a stereoisomer thereof, the formula B A step of reacting a compound or a stereoisomer thereof with a compound of formula A or a stereoisomer thereof to give a compound of formula I,
- R1 is selected from a hydrogen atom or an alkyl group
- R2 is selected from an optionally substituted aryl or heteroaryl group selected from a hydrogen atom, an alkyl group, a halogenated alkyl group, a halogen, an amino group, a nitro group, a cyano group, Oxo, alkenyl, haloalkoxy, hydroxyalkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, -OR3, -C(O)R3, -C(O)OR3, -S( O) mR3 or -NR4R5, wherein the alkyl, alkoxy, alkenyl, haloalkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl groups are optionally selected from the group consisting of a halogen atom, an alkyl group, an alkyl halide Substituted by one or more substituents in the group, hal
- the method for the preparation of a compound of Formula I or a salt thereof further comprises:
- R is as defined in the compound represented by formula D1.
- the method for preparing the compound of the formula I or a salt thereof further comprises:
- the method of preparing a compound of Formula I or a salt thereof further comprises the method steps described above for the preparation of a compound of Formula D1 or a salt thereof.
- the method of preparing a compound of Formula I or a salt thereof further comprises the aforementioned method steps of preparing a compound of Formula B or a stereoisomer thereof.
- the compound of formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the preparation method comprises the following:
- a method of preparing a compound of Formula II includes:
- the compound of formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the preparation method comprises the following:
- a method for preparing a compound of Formula III includes:
- the method for preparing a compound of formula III comprises:
- the compound of the formula (E) is reacted with a base in an organic solvent to be hydrolyzed to a compound of the formula D-1;
- the basic condition is preferably sodium hydroxide, potassium hydroxide or aqueous ammonia.
- the compound of the formula D-1 is reacted with a chiral resolving reagent in an alcohol solvent to form a compound of the formula D2-1;
- the chiral resolving agent is preferably an alkaline chiral resolving agent, more preferably S-phenylethyl An amine, quinidine, cinchonidine or arginine;
- the alcohol solvent is preferably methanol, ethanol or isopropanol.
- the compound of the formula D2-1 is freed under basic conditions to give a compound of the formula D2; the basic condition is preferably sodium hydroxide, potassium hydroxide or aqueous ammonia.
- the compound of the formula C2 is reacted with a reducing agent to give a compound of the formula B2; the reducing agent is preferably red aluminum.
- the compound of formula B2 is reacted with a compound of formula A1 to give a compound of formula III.
- the compound of formula I is N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl-N-(2-aminoethyl)-2-aminoethyl
- the preparation method comprises the following:
- methods for preparing a compound of Formula IV include:
- the present invention also provides the use of the compound of the formula D1, the compound of the formula C or a salt thereof for the preparation of the compound of the formula I,
- the compound of Formula I is:
- the compound of Formula I is:
- the present invention also provides a process for the preparation of a pharmaceutically acceptable salt of a compound of Formula I, Formula II, Formula III, Formula IV, including the steps of the foregoing schemes, and by Formula I, Formula II, Formula III, Formula a step of reacting a compound of IV with an acid to prepare a pharmaceutically acceptable salt thereof, the acid being selected from the group consisting of an organic acid or a mineral acid, preferably an organic acid; the organic acid being selected from the group consisting of acetic acid, maleic acid, and fumaric acid.
- Tartaric acid citric acid, methanesulfonic acid, benzenesulfonic acid or p-toluenesulfonic acid, preferably fumaric acid; the inorganic acid being selected from the group consisting of hydrochloric acid, hydrobromic acid, sulfuric acid or phosphoric acid.
- halogen or halogen atom means a fluorine atom, a chlorine atom, a bromine atom, an iodine atom or the like.
- alkyl group in the present invention means a linear or branched alkyl group having 1 to 20 carbon atoms, and includes, for example, "C 1-6 alkyl group", “C 1-4 alkyl group”, etc., specific examples Including but not limited to: methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, 2-methylbutyl, Neopentyl, 1-ethylpropyl, n-hexyl, isohexyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3,3-dimethylbutyl, 2, 2-dimethylbutyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 2- Ethyl butyl,
- alkenyl group as used in the present invention means a straight or branched chain group having at least one double bond and having 2 to 20 carbon atoms, and includes, for example, "C 2-6 alkenyl group, C 2-4 alkenyl group”. Wait. Examples thereof include, but are not limited to, ethenyl, propenyl, 2-butenyl, 2-pentenyl, 3-pentenyl, 2-hexenyl, 3-hexenyl, and the like.
- haloalkyl refers to a group derived by substituting one or more "halogen atoms” for one or more hydrogen atoms on an "alkyl group” as described above. Defined.
- hydroxyalkyl or hydroxyalkyl refers to a radical derived from one or more "hydroxy” groups substituted by one or more hydrogen atoms on an "alkyl” group as previously described. definition.
- alkylsulfonylamino or alkylsulfonyl means alkyl-O-, haloalkyl-O-, alkyl-C(O)-, alkyl-OC(O)-, C(O)-alkane -O-, alkyl-C(O)-NH-, alkyl-NH-C(O)-, alkyl-NH-, (alkyl) 2 -N-, alkyl-S(O) 2 a group to which -NH- or alkyl-S(O) 2 - is attached, wherein "alkyl, haloalkyl” is as defined above.
- cycloalkyl refers to a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent which includes from 3 to 20 carbon atoms, preferably from 3 to 12 carbon atoms, more preferably cycloalkyl.
- the ring contains from 3 to 10 carbon atoms, and most preferably the cycloalkyl ring contains from 3 to 6 carbon atoms.
- Non-limiting examples of monocyclic cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, cycloheptyl, cycloheptatriene
- the alkenyl group, the cyclooctyl group and the like are preferably a cyclopropyl group or a cyclohexenyl group.
- Polycyclic cycloalkyl groups include spiro, fused, and bridged cycloalkyl groups.
- the "aryl group” as used in the present invention means a 6 to 14 membered all-carbon monocyclic or fused polycyclic ring (that is, a ring sharing a pair of adjacent carbon atoms) having a conjugated ⁇ -electron system, preferably 6 to 10
- the aryl group is more preferably a phenyl group, a naphthyl group, a dihydronaphthyl group, a tetrahydronaphthyl group, an anthracenyl group or a fluorenyl group, and most preferably a phenyl group.
- heterocyclyl refers to a saturated or partially unsaturated monocyclic or polycyclic cyclic hydrocarbon substituent comprising 3 to 20 ring atoms, wherein at least one of the ring atoms is a hetero atom such as a nitrogen atom or oxygen.
- An atom or a sulfur atom, and the remaining ring atoms are carbon; optionally, a ring atom (for example, a carbon atom, a nitrogen atom or a sulfur atom) in the cyclic structure may be oxidized.
- it comprises 3 to 12 ring atoms or 5 to 12 ring atoms, wherein 1 to 4 are hetero atoms, more preferably the heterocyclic ring contains 3 to 8 ring atoms, and more preferably the heterocyclic ring contains 5 to 6 Ring atom.
- monocyclic heterocyclic groups include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, homopiperazinyl, pyranyl, dihydrofuranyl, tetrahydrofuranyl Wait.
- Polycyclic heterocyclic groups include spiro, fused, and bridged heterocyclic groups.
- heteroaryl refers to a 5 to 14 membered aryl group having from 1 to 4 heteroatoms as ring atoms and the remainder of the ring atoms being carbon, wherein the heteroatoms include oxygen, sulfur or nitrogen.
- the "alcohol solvent” as used in the present invention means a group derived from one or more "hydroxyl groups” substituted with one or more hydrogen atoms on the "C 1-6 alkyl group", said "hydroxyl group” and "C” 1-6 alkyl” is as defined above, and specific examples include, but are not limited to, methanol, ethanol, isopropanol, n-propanol, isoamyl alcohol or trifluoroethanol.
- the “stereoisomers” described in the present invention are classified into conformational and conformational isomers, and the configurational isomerization is further classified into cis-trans isomerization and optical isomerization (or enantiomeric).
- Conformational isomerism refers to a stereoisomerism in which organic molecules of a certain configuration cause different arrangement of atoms or groups of molecules in space due to the rotation or distortion of carbon and carbon single bonds. Common alkanes and rings are common. The structure of an alkane compound, such as the chair conformation and the ship conformation that appear in the cyclohexane structure.
- optical isomer means that when the compound of the invention contains one or more asymmetric centers, it can be used as a racemate and a racemic mixture, a single enantiomer, or a non- Enantiomeric mixtures and single diastereomers.
- the compounds of the invention have asymmetric centers, each of which will independently produce two optical isomers, the scope of the invention including all possible optical isomers and mixtures of diastereomers and pure or partially Pure compound. If the compound of the present invention contains an olefinic double bond, the present invention includes a cis isomer and a trans isomer unless otherwise specified.
- the compounds of the present invention may exist in tautomeric forms which have different hydrogen attachment points by displacement of one or more double bonds.
- a ketone and its enol form are keto-enol tautomers.
- Each tautomer and mixtures thereof are included in the present invention.
- the structure of the compound is determined by nuclear magnetic resonance (NMR) or/and mass spectrometry (MS).
- NMR nuclear magnetic resonance
- MS mass spectrometry
- the NMR shift is given in units of 10-6 (ppm).
- the NMR measurement was carried out using a Bruker AVANCE-400 nuclear magnetic apparatus, and the solvent was determined to be a deuterated reagent, and the internal standard was tetramethylsilane (TMS).
- the measurement of the MS was carried out using a FINNIGAN LCQAd (ESI) mass spectrometer (manufacturer: Thermo, model: Finnigan LCQ advantage MAX).
- ESI FINNIGAN LCQAd
- HPLC measurements were performed using an Agilent 1200 DAD high pressure liquid chromatograph and a Waters 2695-2996 high pressure liquid chromatograph with octadecylsilane bonded silica as the column packing.
- the compound (25 g) represented by the formula (E1), potassium hydroxide (22.4 g) and ethylene glycol (150 mL) were mixed, and stirred at 150 ° C for 16 hours to terminate the reaction.
- the reaction solution was cooled to room temperature, diluted with water (150 mL), extracted with dichloromethane (150 mL ⁇ 2), the aqueous phase was adjusted to pH 6 to 7 with 3M hydrochloric acid, and extracted with dichloromethane (200 mL ⁇ 4) The mixture was washed with EtOAc EtOAc (EtOAc m.
- the compound of the formula (D-1) (28 g) was dissolved in absolute ethanol (100 mL), and the temperature was raised to 50 ° C.
- the resolving reagent S-phenylethylamine (6.2 g) was dissolved in anhydrous ethanol (100 mL).
- the S-phenylethylamine solution was dropped into the above solution at 50 ° C, and the mixture was heated to reflux and stirred for 2 hours.
- the compound of formula (C2) (334.4 g) was dissolved in toluene (2.2 kg) in a reaction flask, cooled to -45 ° C to -35 ° C, argon gas was protected, and the dropping temperature was controlled from -45 to -35 ° C.
- Red aluminum (348.76g) was added dropwise, and the reaction was stirred at -45 to -35 ° C for 3 to 4 hours.
- 10% aqueous citric acid solution (1 kg) was added dropwise to the reaction solution at -45 to -35 ° C, and then added.
- Concentrated hydrochloric acid solution was adjusted to pH 2-3, ethyl acetate (1.8 kg) was added, stirred, and allowed to stand for separation.
- the aqueous phase was adjusted to pH 11-13 with 5N sodium hydroxide solution and extracted with dichloromethane (3.3 kg ⁇ 2), the methylene chloride layer was combined, washed with a saturated sodium chloride solution (2.7 kg), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. Light red oil, directly into the next reaction.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Pain & Pain Management (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description































































Claims (21)
- 用于制备式D1所示化合物或其盐的方法,
 其包括:式D所示化合物或其盐经手性拆分的步骤,所述手性拆分方法优选自色谱拆分法或化学拆分法,
其包括:式D所示化合物或其盐经手性拆分的步骤,所述手性拆分方法优选自色谱拆分法或化学拆分法, 其中,R选自芳基或杂芳基,所述芳基或杂芳基任选被选自烷基、卤代烷基、卤素、氨基、硝基、氰基、氧代基、烯基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基、杂芳基、OR 3、C(O)R 3、C(O)OR 3、S(O) mR 3或NR 4R 5中的一个或多个取代基所取代;R 3选自氢原子、烷基、氘代烷基、氨基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基,其中所述的烷基、环烷基、杂环基、芳基和杂芳基任选被选自烷基、卤素、羟基、氨基、硝基、氰基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;R 4和R 5各自独立地选自氢原子、烷基、烷氧基、羟烷基、羟基、氨基、羧酸酯基、环烷基、杂环基、芳基或杂芳基,其中所述的烷基、环烷基、杂环基、芳基和杂芳基任选被选自烷基、卤素、羟基、氨基、羧酸酯基、硝基、氰基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;m为0、1或2。
其中,R选自芳基或杂芳基,所述芳基或杂芳基任选被选自烷基、卤代烷基、卤素、氨基、硝基、氰基、氧代基、烯基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基、杂芳基、OR 3、C(O)R 3、C(O)OR 3、S(O) mR 3或NR 4R 5中的一个或多个取代基所取代;R 3选自氢原子、烷基、氘代烷基、氨基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基,其中所述的烷基、环烷基、杂环基、芳基和杂芳基任选被选自烷基、卤素、羟基、氨基、硝基、氰基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;R 4和R 5各自独立地选自氢原子、烷基、烷氧基、羟烷基、羟基、氨基、羧酸酯基、环烷基、杂环基、芳基或杂芳基,其中所述的烷基、环烷基、杂环基、芳基和杂芳基任选被选自烷基、卤素、羟基、氨基、羧酸酯基、硝基、氰基、烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;m为0、1或2。 - 如权利要求1所述的方法,其中所述化学拆分法所用拆分剂为碱性手性拆分剂,优选自S-苯乙胺、奎尼丁、辛可尼丁或精氨酸。
- 如权利要求1或2所述的方法,其中还包括:式E所示化合物制备得式D所示化合物的步骤,所述反应条件优选自碱性水解,
 其中,R如权利要求1所述。
其中,R如权利要求1所述。 - 如权利要求1-3任一项所述方法,其中所述式D1所示化合物为:

- 如权利要求1-4任一项所述的方法,其为:

- 式D1所示化合物或其盐,
 其中,R如权利要求1所述。
其中,R如权利要求1所述。 - 如权利要求6所述的化合物,其为

- 如权利要求6所述的化合物,其盐为:
 其中,所述M选自S-苯乙胺、奎尼丁、辛可尼丁或精氨酸,优选自S-苯乙胺。
其中,所述M选自S-苯乙胺、奎尼丁、辛可尼丁或精氨酸,优选自S-苯乙胺。 - 用于制备式B所示化合物或其立体异构体的方法,
 其包括:式D所示化合物或其立体异构体通过一步或一步以上反应制备得式B所示化合物或其立体异构体的步骤,
其包括:式D所示化合物或其立体异构体通过一步或一步以上反应制备得式B所示化合物或其立体异构体的步骤, 其中,R如权利要求1所述,优选自
其中,R如权利要求1所述,优选自
- 如权利要求9所述的方法,其为:
 其中,R如权利要求1所述,优选自
其中,R如权利要求1所述,优选自
- 如权利要求9或10所述的方法,其中所述式B所示化合物为:

- 如权利要9-11任一项所述的方法,其中还包括权利要求1-5任一项所述的方法步骤。
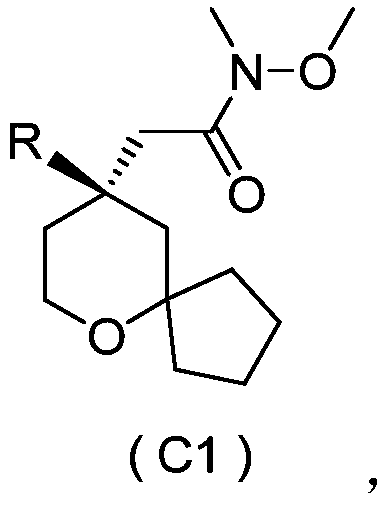
- 式C所示化合物,
 其中,R如权利要求1所述,优选自
其中,R如权利要求1所述,优选自
- 如权利要求13所述的化合物,其为

- 用于制备式I所示化合物或其盐的方法,其包括式D所示化合物或其立体异构体反应后得到式B所示化合物或其立体异构体,式B所示化合物或其立体异构体与式A所示化合物或其立体异构体反应得到式I所示化合物的步骤,
 其中,R 1选自氢原子或烷基;R 2选自任选取代的芳基或杂芳基,所述取代基选自氢 原子、烷基、卤代烷基、卤素、氨基、硝基、氰基、氧代基、烯基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基、杂芳基、-OR 3、-C(O)R 3、-C(O)OR 3、-S(O)mR 3或-NR 4R 5,其中所述的烷基、烷氧基、烯基、卤代烷基、环烷基、杂环基、芳基和杂芳基任选被选自氘原子、烷基、卤代烷基、卤素、氨基、硝基、氰基、羟基、烷氧基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;n选自0、1、2或3;R、R 3、R 4、R 5、m如权利要1所述。
其中,R 1选自氢原子或烷基;R 2选自任选取代的芳基或杂芳基,所述取代基选自氢 原子、烷基、卤代烷基、卤素、氨基、硝基、氰基、氧代基、烯基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基、杂芳基、-OR 3、-C(O)R 3、-C(O)OR 3、-S(O)mR 3或-NR 4R 5,其中所述的烷基、烷氧基、烯基、卤代烷基、环烷基、杂环基、芳基和杂芳基任选被选自氘原子、烷基、卤代烷基、卤素、氨基、硝基、氰基、羟基、烷氧基、卤代烷氧基、羟烷基、环烷基、杂环基、芳基或杂芳基中的一个或多个取代基所取代;n选自0、1、2或3;R、R 3、R 4、R 5、m如权利要1所述。 - 如权利要求15所述的方法,其还包括权利要求1-5任一项所述的方法步骤或权利要求9-12任一项所述的方法步骤。
- 如权利要求15或16所述的方法,其中所述式I所示化合物为

- 如权利要求15或16所述的方法,其中所述式I所示化合物为:

- 用于制备式III所示化合物的方法,其包括:

- 用于制备式IV所示化合物的方法,其包括:

- 权利要求6-8、13或14任一项所述的化合物或其盐在制备式I所示化合物中的用途,
 其中,R、R 1、R 2、n如权利要求15所述。
其中,R、R 1、R 2、n如权利要求15所述。
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2018341782A AU2018341782A1 (en) | 2017-09-28 | 2018-09-27 | Method for preparing oxaspirocycle derivative, and intermediate thereof |
| RU2020113709A RU2777983C2 (ru) | 2017-09-28 | 2018-09-27 | Способ получения оксаспироциклического производного и его промежуточного соединения |
| CA3076395A CA3076395A1 (en) | 2017-09-28 | 2018-09-27 | Method for preparing oxaspirocycle derivative, and intermediate thereof |
| JP2020517594A JP2020535177A (ja) | 2017-09-28 | 2018-09-27 | オキサスピロサイクル誘導体の調製方法およびその中間体 |
| BR112020005858-0A BR112020005858A2 (pt) | 2017-09-28 | 2018-09-27 | compostos derivados oxaspirocíclicos e intermediários do mesmo, métodos de preparação e uso dos referidos compostos |
| CN201880035013.9A CN110678453B (zh) | 2017-09-28 | 2018-09-27 | 一种氧杂螺环类衍生物的制备方法及其中间体 |
| EP18862192.4A EP3689859A4 (en) | 2017-09-28 | 2018-09-27 | PROCESS FOR MANUFACTURING OXASPIROCYCLE DERIVATIVES AND INTERMEDIATE THEREOF |
| KR1020207012222A KR20200060750A (ko) | 2017-09-28 | 2018-09-27 | 옥사스피로사이클 유도체의 제조 방법 및 이의 중간체 |
| US16/651,922 US11111236B2 (en) | 2017-09-28 | 2018-09-27 | Method for preparing oxaspirocycle derivative, and intermediate thereof |
| MX2020003784A MX395589B (es) | 2017-09-28 | 2018-09-27 | Método para preparar derivado de oxaespirociclo e intermediario del mismo. |
| ZA2020/02138A ZA202002138B (en) | 2017-09-28 | 2020-05-04 | Method for preparing oxaspirocycle derivative, and intermediate thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710896555.2 | 2017-09-28 | ||
| CN201710896555 | 2017-09-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019062804A1 true WO2019062804A1 (zh) | 2019-04-04 |
Family
ID=65900832
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/107901 Ceased WO2019062804A1 (zh) | 2017-09-28 | 2018-09-27 | 一种氧杂螺环类衍生物的制备方法及其中间体 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US11111236B2 (zh) |
| EP (1) | EP3689859A4 (zh) |
| JP (1) | JP2020535177A (zh) |
| KR (1) | KR20200060750A (zh) |
| CN (1) | CN110678453B (zh) |
| AU (1) | AU2018341782A1 (zh) |
| BR (1) | BR112020005858A2 (zh) |
| CA (1) | CA3076395A1 (zh) |
| MX (1) | MX395589B (zh) |
| TW (1) | TWI690524B (zh) |
| WO (1) | WO2019062804A1 (zh) |
| ZA (1) | ZA202002138B (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019205983A1 (zh) * | 2018-04-28 | 2019-10-31 | 四川科伦博泰生物医药股份有限公司 | 氧杂螺环类化合物及其制备方法和用途 |
| WO2022143715A1 (zh) * | 2020-12-29 | 2022-07-07 | 上海海雁医药科技有限公司 | 氧杂螺环取代的吡咯并吡唑衍生物及其中间体和制备方法 |
| CN117624138A (zh) * | 2022-08-29 | 2024-03-01 | 江苏恒瑞医药股份有限公司 | 一种氧杂螺环类衍生物的可药用盐、晶型及制备方法 |
| WO2024153159A1 (zh) | 2023-01-18 | 2024-07-25 | 上海森辉医药有限公司 | 一种四氢-1-萘胺及其衍生物的制备方法 |
| CN118638109A (zh) * | 2024-04-30 | 2024-09-13 | 中山大学 | 一种阿片类药物-奥赛利定的制备方法 |
| CN118994123A (zh) * | 2024-10-23 | 2024-11-22 | 江苏恒瑞医药股份有限公司 | 制备9-(吡啶-2-基)-6-氧杂螺[4.5]癸烷衍生物的方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20240317761A1 (en) * | 2021-07-13 | 2024-09-26 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Pharmaceutically acceptable salt of mor receptor agonist, and polymorph thereof and use thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012129495A1 (en) | 2011-03-23 | 2012-09-27 | Trevena, Inc. | Opioid receptor ligands and methods of using and making same |
| WO2017063509A1 (zh) | 2015-10-15 | 2017-04-20 | 江苏恒瑞医药股份有限公司 | 氧杂螺环类衍生物、其制备方法及其在医药上的应用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106588899B (zh) * | 2015-10-15 | 2019-11-15 | 江苏恒瑞医药股份有限公司 | 吡啶基取代的6-氧杂螺[4.5]癸烷类衍生物、其制备方法及其在医药上的应用 |
-
2018
- 2018-09-27 AU AU2018341782A patent/AU2018341782A1/en not_active Abandoned
- 2018-09-27 KR KR1020207012222A patent/KR20200060750A/ko not_active Withdrawn
- 2018-09-27 WO PCT/CN2018/107901 patent/WO2019062804A1/zh not_active Ceased
- 2018-09-27 JP JP2020517594A patent/JP2020535177A/ja not_active Ceased
- 2018-09-27 US US16/651,922 patent/US11111236B2/en not_active Expired - Fee Related
- 2018-09-27 BR BR112020005858-0A patent/BR112020005858A2/pt not_active Application Discontinuation
- 2018-09-27 TW TW107134070A patent/TWI690524B/zh not_active IP Right Cessation
- 2018-09-27 CA CA3076395A patent/CA3076395A1/en active Pending
- 2018-09-27 MX MX2020003784A patent/MX395589B/es unknown
- 2018-09-27 EP EP18862192.4A patent/EP3689859A4/en not_active Withdrawn
- 2018-09-27 CN CN201880035013.9A patent/CN110678453B/zh active Active
-
2020
- 2020-05-04 ZA ZA2020/02138A patent/ZA202002138B/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012129495A1 (en) | 2011-03-23 | 2012-09-27 | Trevena, Inc. | Opioid receptor ligands and methods of using and making same |
| WO2017063509A1 (zh) | 2015-10-15 | 2017-04-20 | 江苏恒瑞医药股份有限公司 | 氧杂螺环类衍生物、其制备方法及其在医药上的应用 |
Non-Patent Citations (3)
| Title |
|---|
| J. MED. CHEM., vol. 56, 2013, pages 8019 - 8031 |
| PHARMACOL. REV., vol. 59, 2007, pages 88 - 123 |
| See also references of EP3689859A4 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019205983A1 (zh) * | 2018-04-28 | 2019-10-31 | 四川科伦博泰生物医药股份有限公司 | 氧杂螺环类化合物及其制备方法和用途 |
| WO2022143715A1 (zh) * | 2020-12-29 | 2022-07-07 | 上海海雁医药科技有限公司 | 氧杂螺环取代的吡咯并吡唑衍生物及其中间体和制备方法 |
| CN116368138A (zh) * | 2020-12-29 | 2023-06-30 | 上海海雁医药科技有限公司 | 氧杂螺环取代的吡咯并吡唑衍生物及其中间体和制备方法 |
| CN116368138B (zh) * | 2020-12-29 | 2026-03-03 | 上海海雁医药科技有限公司 | 氧杂螺环取代的吡咯并吡唑衍生物及其中间体和制备方法 |
| CN117624138A (zh) * | 2022-08-29 | 2024-03-01 | 江苏恒瑞医药股份有限公司 | 一种氧杂螺环类衍生物的可药用盐、晶型及制备方法 |
| WO2024153159A1 (zh) | 2023-01-18 | 2024-07-25 | 上海森辉医药有限公司 | 一种四氢-1-萘胺及其衍生物的制备方法 |
| EP4653414A1 (en) | 2023-01-18 | 2025-11-26 | Shanghai Senhui Medicine Co., Ltd. | Method for preparing tetrahydro-1-naphthylamine and derivatives thereof |
| CN118638109A (zh) * | 2024-04-30 | 2024-09-13 | 中山大学 | 一种阿片类药物-奥赛利定的制备方法 |
| CN118994123A (zh) * | 2024-10-23 | 2024-11-22 | 江苏恒瑞医药股份有限公司 | 制备9-(吡啶-2-基)-6-氧杂螺[4.5]癸烷衍生物的方法 |
| CN118994123B (zh) * | 2024-10-23 | 2025-02-14 | 江苏恒瑞医药股份有限公司 | 制备9-(吡啶-2-基)-6-氧杂螺[4.5]癸烷衍生物的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2020003784A (es) | 2020-08-03 |
| EP3689859A4 (en) | 2021-08-25 |
| CN110678453A (zh) | 2020-01-10 |
| AU2018341782A1 (en) | 2020-04-16 |
| US20200308151A1 (en) | 2020-10-01 |
| US11111236B2 (en) | 2021-09-07 |
| RU2020113709A3 (zh) | 2022-01-25 |
| EP3689859A1 (en) | 2020-08-05 |
| JP2020535177A (ja) | 2020-12-03 |
| ZA202002138B (en) | 2021-05-26 |
| TW201914997A (zh) | 2019-04-16 |
| MX395589B (es) | 2025-03-25 |
| CN110678453B (zh) | 2023-03-10 |
| RU2020113709A (ru) | 2021-10-18 |
| CA3076395A1 (en) | 2019-04-04 |
| KR20200060750A (ko) | 2020-06-01 |
| TWI690524B (zh) | 2020-04-11 |
| BR112020005858A2 (pt) | 2020-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019062804A1 (zh) | 一种氧杂螺环类衍生物的制备方法及其中间体 | |
| US11897843B2 (en) | Process for the preparation of enantiomerically enriched 3-aminopiperidine | |
| EP2397141A1 (en) | Process for the synthesis of beta-amino acids and derivatives thereof | |
| KR20200144546A (ko) | 피롤로아미노피리다지논 화합물 및 이의 중간체의 제조 방법 | |
| BR112015015880B1 (pt) | Processos de produção de um composto e compostos | |
| WO2024167810A1 (en) | Ligand-enabled transannular c-h functionalization of cycloalkane carboxylic acids | |
| EP3081554B1 (en) | Method for preparing silodosin and intermediate thereof | |
| RU2777983C2 (ru) | Способ получения оксаспироциклического производного и его промежуточного соединения | |
| CN108239040B (zh) | 硝酸2-(4-甲基噻唑-5-基)乙酯盐酸盐的制备方法 | |
| CN111808040B (zh) | 多构型2-氧代噁唑烷-4-羧酸类化合物的合成方法 | |
| HK40019965A (zh) | 一种氧杂螺环类衍生物的制备方法及其中间体 | |
| CN110573495B (zh) | 反式异构杂环化合物及其制备方法 | |
| WO2024109801A1 (zh) | 一种手性吡咯衍生物的制备方法及其中间体 | |
| JP2006028154A (ja) | 光学活性化合物の製造方法 | |
| JP2008531487A (ja) | カンプトセシンサブユニットの新規の合成法 | |
| S'kof et al. | Stereoselective Amination of 5‐Substituted γ‐Lactones and γ‐Lactams–A Convenient Route for the Preparation of 5‐Substituted (3S, 5S)‐3‐Acetylaminotetrahydrofuran‐2‐ones and (3S, 5S)‐3‐Acetylaminopyrrolidin‐2‐ones | |
| TW200909434A (en) | Production process of coumarin dimer compound | |
| US20060241299A1 (en) | Process for making spirolactone compounds | |
| JP6169721B2 (ja) | パピローマウイルスの治療で用いることができるヒドラジンの合成方法 | |
| JP2010195688A (ja) | Npyy5受容体拮抗作用を有するアミド及びウレア誘導体 | |
| JPH0859581A (ja) | シクロヘキサジエン誘導体 | |
| CN102858741A (zh) | 制备p2x7r拮抗剂的新方法 | |
| JP2003192651A (ja) | 3−アミノカルボン酸およびそのエステルの製法 | |
| JPS6183161A (ja) | ピリジン誘導体 | |
| JP2003327568A (ja) | 光学活性β−アミノ酸誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18862192 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3076395 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020517594 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018341782 Country of ref document: AU Date of ref document: 20180927 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20207012222 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018862192 Country of ref document: EP Effective date: 20200428 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020005858 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112020005858 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200324 |