WO2019126576A1 - Release segments and binding compositions comprising same - Google Patents
Release segments and binding compositions comprising same Download PDFInfo
- Publication number
- WO2019126576A1 WO2019126576A1 PCT/US2018/066939 US2018066939W WO2019126576A1 WO 2019126576 A1 WO2019126576 A1 WO 2019126576A1 US 2018066939 W US2018066939 W US 2018066939W WO 2019126576 A1 WO2019126576 A1 WO 2019126576A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cell
- recombinant polypeptide
- cancer
- antigen
- fbm
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*C(C(*)C(*)C(*)C*)(C(CC(*C1*)O)C1OC)O Chemical compound C*C(C(*)C(*)C(*)C*)(C(CC(*C1*)O)C1OC)O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/80—Vaccine for a specifically defined cancer
- A61K2039/812—Breast
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/54—F(ab')2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/50—Fusion polypeptide containing protease site
Definitions
- a primary goal of cancer therapy is to specifically destroy tumor cells, while leaving healthy cells and tissues as undamaged as possible.
- An approach that has recently generated interest is to induce an immune response against the tumor in which immune effector cells such as natural killer (NK) cells or cytotoxic T lymphocytes (CTLs) are induced to attack and destroy tumor cells.
- immune effector cells such as natural killer (NK) cells or cytotoxic T lymphocytes (CTLs) are induced to attack and destroy tumor cells.
- MAb monoclonal antibodies
- scFv single chain fragments
- bispecific antibodies combine the benefits of different binding specificities derived from two monoclonal antibodies into a single composition, enabling approaches or combinations of coverages that are not possible with monospecific antibodies.
- This approach relies on binding of one arm of the bispecific antibody to a tumor-associated antigen or marker, while the other arm, upon binding the marker of an effector cell (e.g., a CD3 molecule on T cells), triggers their cytotoxic activity by the release of effector molecules such as such as TNF -alpha, IFN-gamma, interleukins 2, 4 and 10, perforin, and granzymes.
- an effector cell e.g., a CD3 molecule on T cells
- effector molecules e.g., a CD3 molecule on T cells
- BiTEs function by recruiting and activating polyclonal populations of T-cells at tumor sites, and do so without the need for co-stimulation or conventional MHC recognition.
- cytokine storm or “cytokine release syndrome” (Lee DW et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014 124(2): 188-195) mediated by the release of TNF- alpha and IFN-gamma, amongst other cytokines, in addition to the fact that BiTE compositions have a very short half-life, necessitating continuous infusions of four to eight weeks in order to maintain BiTE within the therapeutic window for sufficient time to achieve a therapeutic effect.
- Proteases are enzymes that are capable of cleaving proteins and peptides by hydrolysis of peptide bonds. Proteases are involved in a diversity of functions, regulate the fate and activity of many proteins, create or inactivate bioactive molecules, affect cell proliferation and differentiation, tissue morphogenesis and remodeling, contribute to the processing of protein, and even are involved in molecular signaling. As a result of the action of proteases and protein responses, they play a role in angiogenesis, wound repair, hemostasis, blood coagulation, inflammation, immunity, necrosis, apoptosis, and the progression or amelioration of diseases, including cancers.
- matriptase as a prognostic marker in various human cancers.
- matriptase mRNA and protein are up-regulated in cancerous lesions compared with normal tissue, and there is a positive correlation between matriptase expression and histopathological grade of the tumor (Lee JW, et al. Increased expression of matriptase is associated with histopathologic grades of cervical neoplasia. Hum Pathol. (2005) 36(6):626-33).
- matriptase While matriptase is expressed at low levels in the normal ovary, it becomes highly expressed in early-stage ovarian carcinoma (Tanimoto H., et al., Transmembrane serine protease TADG-15 (STl4/Matriptase/MT-SPl): expression and prognostic value in ovarian cancer. Br J Cancer. (2005) 92(2):278-83). Similarly, matrix metalloproteinases (MMPs) are important cancer markers in that they are present in nearly all human cancers.
- MMPs matrix metalloproteinases
- MMPs can be expressed by healthy fibroblasts in the stroma adjacent to tumors, cancer-associated fibroblasts, or by non-fibroblastic cancer cells where they can influence the tumor environment by promoting angiogenesis, tumor growth, and metastasis (Bhowmick, N.A., Stromal fibroblasts in cancer initiation and progression. Nature, 432 (2004), pp. 332-337).
- legumain is overexpressed in the majority of human solid tumors (Liu, C, et al.
- tumor proteases An essential function of tumor proteases is to dissolve the extracellular matrix to allow the tumor cells to invade, and grow in an infiltrative manner in, normal tissue. These proteases also protect the tumor from the defense mechanisms of the body by cleaving and inactivating, for example, antibodies, cytokines, growth factors, complement factors, coagulation factors and mediators that would limit otherwise inhibit the tumor.
- protease-sensitive peptides can be incorporated into therapeutic biologies to confer certain properties on the intact and/or the product of a protease-treated drug or biologic, there exists a need to identify new peptide substrates for proteases associated with diseased tissues and to incorporate these peptide substrates in a variety of prodrug therapeutic, diagnostic and prophylactic compositions as a key mechanism to activate such compositions, improving the therapeutic index and outcome.
- the present disclosure provides recombinant polypeptides comprising cleavable release segments (RS) that are useful in the treatment or prevention of diseases, including but not limited to cancers, autoimmune, and inflammatory disorders.
- RS cleavable release segments
- the recombinant polypeptides comprising release segments described herein may address an unmet need and are superior in one or more aspects, including tailored designs that result in beneficial properties described herein.
- the disclosure provides recombinant polypeptides comprising a first release segment (RS1), wherein the RS1 is a substrate for cleavage by a mammalian protease.
- the RS1 comprises an amino acid sequence having at least 88%, or at least 89%, or at least 90%, or at least 91%, or at least 92%, or at least 93%, or at least 94%, or at least 95%, or at least 100% sequence identity to a sequence selected from the sequences set forth in Table 1, wherein the RS1 is a substrate for one or more mammalian proteases.
- the RS1 comprises an amino acid sequence having at least 88%, or at least 89%, or at least 90%, or at least 91%, or at least 92%, or at least 93%, or at least 94%, or at least 95%, or at least 96%, or at least 97%, or at least 100% sequence identity to a sequence selected from the sequences set forth in Table 2, wherein the RS1 is a substrate for one or more mammalian proteases.
- the RS1 comprises an amino acid sequence selected from the sequences of Table 1, wherein the RS1 is a substrate for one or more mammalian proteases.
- the RS1 comprises an amino acid sequence selected from the sequences of Table 2, wherein the RS1 is a substrate for one or more mammalian proteases.
- the present disclosure provides recombinant polypeptides comprising an RS1 and further comprising a first binding moiety (FBM) having binding affinity for a target cell marker on a target tissue or cell.
- FBM is an antibody, a cytokine, a cell receptor, or a fragment thereof.
- the RS1 is a substrate for cleavage by a mammalian protease wherein the mammalian protease is produced by or is co-localized with the target tissue or cell.
- the RS1 is a substrate for cleavage by multiple mammalian proteases wherein the mammalian proteases are produced by or are co-localized with the target tissue or cell.
- the RS1 of the subject compositions can be a substrate for a serine protease and/or a cysteine protease and/or a metalloproteinase.
- the RS1 is a substrate for a protease selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, and matriptase.
- the RS1 is a substrate for a protease set forth in Table 3.
- the RS1 of the embodiments is designed for cleavage by multiple proteases at one, two, or three cleavage sites in the RS1 sequence.
- the RS1 is a substrate for cleavage at two or more cleavage sites by two or more proteases selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, and matriptase.
- the RS1 is a substrate for cleavage at three or more cleavage sites by three or more proteases selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, and matriptase.
- the release segments of the subject compositions can be designed to have different rates of cleavage by the mammalian proteases at each of the cleavage sites.
- the rates of cleavage were determined relative to a control release segment having the amino acid sequence EAGRSANHEPLGLVAT, which can be cleaved by serine, cysteine and metalloproteinases, as described in Example 43.
- the disclosure provides recombinant polypeptides comprising an RS1 and a FBM, wherein the rate of cleavage of the RS1 by legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase is at least two-fold faster compared to the rate of cleavage of the control sequence having the sequence EAGRSANHEPLGLVAT by the same protease when assayed in vitro under equivalent molar concentrations.
- the disclosure provides recombinant polypeptides comprising an RS1 and a FBM, wherein the rate of cleavage of the RS1 by legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, or matriptase is at least two-fold slower compared to the rate of cleavage of the control sequence having the sequence EAGRSANHEPLGLVAT by the same protease when assayed in vitro under equivalent molar concentrations.
- the disclosure provides recombinant polypeptides comprising an RS1 and a FBM, wherein the RS1 is a substrate for cleavage by a protease selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase and wherein the RS1 has at least a 0.2 log 2 , or 0.4 log2, or 0.8 log2, or 1.0 log 2 higher cleavage efficiency in an in vitro biochemical competitive assay compared to the cleavage by the same protease of a control sequence having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase
- the RS1 has at least a 0.2 log 2 , or 0.4 log2, or 0.8 log2, or 1.0 log 2 higher cleavage efficiency in an in
- the disclosure provides recombinant polypeptides comprising an RS1 and a FBM, wherein the RS1 is a substrate for cleavage by a protease selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, or matriptase and wherein the RS1 has at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 lower cleavage efficiency in an in vitro biochemical competitive assay compared to the cleavage by the same protease of a control sequence having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, or matriptase
- the RS1 has at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 lower cle
- the disclosure relates to recombinant polypeptides comprising an RS1, a FBM, and at least a first bulking moiety.
- recombinant polypeptide compositions comprising an RS1, a FBM, and at least a first bulking moiety.
- the FBM when the recombinant polypeptide is in an intact, uncleaved state, has lower binding affinity for its ligand due to the shielding effect of the bulking moiety.
- the FBM Upon its release via cleavage of the release segment by a mammalian protease co-localized in a target tissue, for example, a tumor tissue, the FBM regains its full potential to bind the target cell marker as it is no longer being shielded by the bulking moiety.
- the bulking moiety is a first extended recombinant polypeptide (XTEN1).
- the XTEN1 comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from the sequences set forth in Table 8 or Table 10.
- the XTEN1 comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from AE l44_l A, AE 144_2A, AEE144_2B, AE 144_3A, AE144_3B, AE 144_4A, AE 144 4B, AE 144_5A, AE 144_6B, AE284, AE288_l, AE288_2, AE288_3, AE576, AE864, AE864 2, AE865, AE866, AE867, AE867 2, and AE868.
- the recombinant polypeptide comprising an RS1, a FBM, and an XTEN1 has, in an uncleaved state, a structural arrangement from N -terminus to C-terminus of FBM-RS 1 -XTEN 1.
- the recombinant polypeptide comprising an RS1, a FBM, and an XTEN1 has, in an uncleaved state, a structural arrangement from N-terminus to C-terminus of XTEN1-RS1-FBM.
- recombinant polypeptides comprising an RS1, a FBM, and an XTEN1
- the XTEN1 and the FBM are released from the recombinant polypeptide.
- the disclosure relates to recombinant polypeptides comprising an RS1, a FBM, and an XTEN1 wherein the FBM is an antibody fragment.
- the FBM is an antibody fragment selected from the group consisting of Fv, Fab, Fab', Fab'-SH, linear antibody, and single-chain variable fragment (scFv).
- the FBM antibody fragment has binding affinity for an effector cell antigen expressed on the surface of an effector cell selected from a plasma cell, a T cell, a B cell, a cytokine induced killer cell (CD cell), a mast cell, a dendritic cell, a regulatory T cell (RegT cell), a helper T cell, a myeloid cell, and a NK cell.
- an effector cell antigen expressed on the surface of an effector cell selected from a plasma cell, a T cell, a B cell, a cytokine induced killer cell (CD cell), a mast cell, a dendritic cell, a regulatory T cell (RegT cell), a helper T cell, a myeloid cell, and a NK cell.
- the FBM antibody fragment has binding affinity for an effector cell antigen expressed on the surface of a T cell.
- FBM antibody fragment has binding affinity for CD3.
- the antibody fragment comprises a VL and VH derived from a monoclonal antibody having
- the antibody fragment comprises a VL and VH selected from the sequences set forth in Table 4.
- the antibody fragment comprises complementarity- determining regions (CDR) derived from a monoclonal antibody having binding specificity to CD3.
- the antibody fragment comprises a CDR-H1 region, a CDR-H2 region, a CDR-H3 region, a CDR-L1 region, a CDR-L2 region, and a CDR-H3 region, wherein each is derived from a monoclonal antibody of Table 4.
- the disclosure relates to recombinant polypeptides comprising an RS1, an XTEN1, a FBM and a second binding moiety (SBM) wherein the SBM is an antibody fragment having binding affinity for a target cell marker.
- the FBM and the SBM are each an antibody fragment selected from the group consisting of Fv, Fab, Fab', Fab'-SH, linear antibody, and single-chain variable fragment (scFv) or the VL and VH of the FBM and SBM are configured as a single chain diabody.
- the SBM antibody fragment has binding affinity for a target cell marker on a tumor cell or a cancer cell.
- the SBM antibody fragment has binding affinity for a target cell marker selected from the target cell markers set forth in Table 5.
- the SBM antibody fragment has binding affinity for a target cell marker selected from A33 antigen, alpha- fetoprotein (AFP), alpha 4 integrin, Ang2, B7-H3, B7-H6, B-cell maturation antigen (BCMA), cancer antigen 19-9 (CA19-9), cancer antigen 125 (CA-125), Carbonic Anhydrase 6 (CA6), carbonic anhydrase IX (CALX), CEACAM5, cMET, CTLA4, C-C Motif Chemokine Receptor 1 (CCR1), C-C Motif Chemokine Receptor 2 (CCR2), C-C Motif Chemokine Receptor 3 (CCR3), C-C Motif Chemokine Receptor 4 (CCR4), C-C Motif Chemokine Receptor 5 (CCR5), C-C Motif Chemokine Receptor
- CD7 Differentiation 7
- globohexaosylceramide globo-H
- GD2, Glypican 3 Glypican 3
- GCC guanylyl cyclase C
- HER2, HER2 neu HER3, HER4, HER1, ELl3Ra2, insulin-like growth factor I receptor (IGF-IR ), Lysosomal Associated Membrane Protein 1 (LAMP1), Ll Cell Adhesion Molecule (L1CAM), lymphocyte antigen 6 (Ly-6), melanoma chondroitin sulfate proteoglycan (MCSP), Membrane- type metalloproteinase (MT-MMP), mesothelin, mucin 1 (MUC1), MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16, Muellerian inhibitory substance receptor type II (MISIIR), nectin cell adhesion molecule 4 (Nectin-4), 6-transmembrane epithelial
- the SBM antibody fragment comprises a VL and VH derived from a monoclonal antibody having binding affinity to the target cell marker.
- the SBM antibody fragment comprises a VL and VH derived from a monoclonal antibody, wherein the VL and VH are selected from the sequences set forth in Table 5.
- the SBM antibody fragment comprises a CDR-H1 region, a CDR-H2 region, a CDR-H3 region, a CDR-L1 region, a CDR-L2 region, and a CDR-H3 region, wherein each is derived from a monoclonal antibody set forth in Table 5.
- the recombinant polypeptide comprises the FBM, the SBM, the RS1 and the XTEN1, in an uncleaved state
- the recombinant polypeptide has a structural arrangement from N-terminus to C-terminus of SBM-FBM-RS1-XTEN1, FBM- SBM-RS 1 -XTEN 1 , XTEN 1 -RS 1 -SBM-FBM, XTEN 1 -RS 1 -FBM- SBM, or diabody-RSl- XTEN1, or XTENl-RSl-diabody, wherein the diabody comprises VL and VH of the FBM and SBM.
- the disclosure provides a recombinant polypeptide comprising a FBM, SBM, RS1, and an XTEN1, wherein the recombinant polypeptide comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from the group of sequences set forth in Table 14.
- the FBM and SBM upon cleavage of the RS1 by the mammalian protease and release of the FBM and SBM from the recombinant polypeptide, the FBM and SBM remain fused and are capable of binding to and linking together a T cell bearing the CD3 antigen and a tumor cell bearing the target cell marker in an in vitro assay comprising both the T cells and the tumor cells.
- the binding together of the T cell and the tumor cell results in cytotoxic activity against the tumor cell in the in vitro assay, as determined by quantitation of cell lysis or release of intracellular components.
- the released, fused FBM and SBM are capable of effecting a greater amount of cell lysis of the tumor cell compared to the cell lysis effected by the uncleaved recombinant polypeptide in in vitro assays performed under equivalent molar concentrations, as determined by quantitation of cell lysis or release of intracellular components.
- the amount of cell lysis effected by the released FBM and SBM of the recombinant polypeptide is at least 10-fold greater, or at least 30-fold, or at least 100-fold, or at least 300-fold, or at least 1000-fold, or at least 10,000-fold greater compared to the cell lysis effected by the uncleaved recombinant polypeptide in the in vitro assays performed under equivalent molar concentrations, as determined by quantitation of cell lysis or release of intracellular components.
- the cytotoxic activity and/or cell lysis of the tumor cell may be mediated by target specific activation of the T cell.
- the amount of activation of the T cell effected by the released FBM and SBM is at least lO-fold greater, or at least 30-fold, or at least lOO-fold, or at least 300-fold, or at least 1000-fold greater, or at least 10,000-fold greater compared to the activation effected by the uncleaved recombinant polypeptide, as determined by quantitation of T cell -derived effector molecules in in vitro assays performed under equivalent molar concentrations.
- the FBM and SBM upon cleavage of the RS1 by the mammalian protease and release of the FBM and SBM from the recombinant polypeptide, the FBM and SBM remain fused and exhibit increased binding affinity to the CD3 antigen and/or the target cell marker in an in vitro assay comprising CD3 antigen or target cell marker compared the binding affinity of the intact, uncleaved recombinant polypeptide to the CD3 antigen or to the target cell marker, when assayed under equivalent molar concentrations.
- the binding affinity of the released FBM to the CD3 antigen or the released SBM to the target cell marker is at least 10- fold greater, or at least 30-fold, or at least 100-fold, or at least 300-fold, or at least 1000-fold greater, as determined as a IQ constant in the in vitro assay, compared to the binding affinity of the intact, uncleaved recombinant polypeptide to the CD3 antigen or to the target cell marker, when assayed under equivalent molar concentrations.
- the I constant of the binding of the released FBM of the recombinant polypeptide to the CD3 antigen is between 10 5 to 10 9 M and the IQ of the binding of the released SBM to the target specific marker is between 10 5 to 10 9 M.
- the binding affinity of the released SBM to the target cell marker is at least one order of magnitude greater compared to the lower binding affinity of the released FBM to the CD3 antigen, as determined as IQ constants in the in vitro assay, when assayed under equivalent molar concentrations.
- the in vitro assay utilized can be selected from cell membrane integrity assay, mixed cell culture assay, FACS based propidium Iodide assay, trypan Blue influx assay, photometric enzyme release assay, radiometric 5lCr release assay, fluorometric Europium release assay, CalceinAM release assay, photometric MTT assay, XTT assay, WST-l assay, alamar blue assay, radiometric 3H-Thd incorporation assay, clonogenic assay measuring cell division activity, fluorometric rhodaminel23 assay measuring mitochondrial transmembrane gradient, apoptosis assay monitored by FACS-based
- phosphatidylserine exposure ELISA-based TUNEL test assay
- sandwich ELISA sandwich ELISA
- caspase activity assay cell-based LDH release assay
- cell morphology assay or any combination thereof.
- the disclosure relates to recombinant polypeptides comprising an RS1, FBM, SBM, XTEN1 having the elements described in the embodiments, above, and further comprising a second release segment (RS2) that is a substrate for cleavage by a mammalian protease, and a second XTEN (XTEN2).
- RS2 second release segment
- XTEN2 second XTEN
- the disclosure contemplates different configurations of the recombinant polypeptides, wherein in an uncleaved state, the recombinant polypeptide has a structural arrangement from N-terminus to C-terminus as follows: XTEN1-RS1-SBM-FBM- RS2-XTEN2, XTEN1 -RS 1 -FBM-SBM-RS2-XTEN2, XTEN2-RS2-SBM-FBM-RS 1 -XTEN1 , XTEN2-RS2-FBM-SBM-RS 1 -XTEN 1 , XTEN2-RS2-diabody-RSl-XTENl, wherein the diabody comprises VL and VH of the FBM and SBM, or XTENl-RSl-diabody-RS2-XTEN2, wherein the diabody comprises VL and VH of the FBM and SBM.
- the XTEN2 of the recombinant polypeptide comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from the group of sequences set forth in Table 8 or Table 10.
- the XTEN2 of the recombinant polypeptide comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from AE 144 1 A, AE 144 2A, AEE144 2B, AE 144_3A, AE144 3B, AE 144_4A, AE 144_4B, AE 144_5A, AE 144_6B, AE284, AE288_l, AE288_2, AE288_3, AE576, AE864, AE864_2, AE865, AE866, AE867, AE867_2, and AE868.
- the RS2 sequence is identical compared to the RS1 sequence.
- the RS2 sequence is different compared to the RS1 sequence and each comprise an amino acid sequence having at least 88%, or at least 89%, or at least 90%, or at least 91%, or at least 92%, or at least 93%, or at least 94%, or at least 95% sequence identity to sequences selected from the sequences of Table 1 or Table 2.
- the RS2 sequence is different compared to the RS1 sequence and each comprises a sequence selected from the sequences of Table 1 or Table 2.
- the disclosure provides a recombinant polypeptide comprising an XTEN1, RS1, SBM, FBM,
- recombinant polypeptide comprises an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a sequence selected from the group of sequences set forth in Table 15 or Table 18.
- the disclosure provides a recombinant polypeptide comprising an RS1, RS2, FBM, SBM, XTEN1, and XTEN2, wherein i) the RS1 and RS2, wherein the RS1 and RS2 are each a substrate for cleavage by a mammalian protease and each comprise an amino acid sequence having at least 90%, at least 93%, at least 97%, or 100% sequence identity to a sequence selected from the sequences of Table 2; ii) the FBM is an antibody fragment comprising a VL and VH derived from a monoclonal antibody having binding specificity to an effector cell; iii) the SBM is an antibody fragment comprising a VL and VH derived from a monoclonal antibody having binding affinity to a target cell marker; iv) the XTEN1 and XTEN2 each comprise an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%
- the effector cell is a T cell and the target cell marker is selected from A33 antigen, alpha-fetoprotein (AFP), alpha 4 integrin, Ang2, B7-H3, B7-H6, B-cell maturation antigen (BCMA), cancer antigen 19-9 (CA19-9), cancer antigen 125 (CA-125), Carbonic Anhydrase 6 (CA6), carbonic anhydrase IX (CALX),
- Phosphodiesterase 3 (ENPP3), EGFR, EGFRvIII, EpCAM, endosialin (CD248), epidermal growth factor receptor variant III (EGFRvIII), EphA2, F19 antigen, fetal acetylcholine receptor (fiiAChR), fibroblast activation antigen (FAP), Fos-related antigen 1 (FRA1), Folate Receptor 1 (FOLR1), fucosyl GM1, G250, ganglioside GD3, glypican-3 (GPC3), 9-0- Acetyl-GD3, GM2, Glucocorticoid induced TNF receptor (GITR), globohexaosylceramide (globo-H), GD2,
- Glypican 3 Glypican 3 (GPC3), guanylyl cyclase C (GCC), HER2, HER2 neu, HER3, HER4, HER1, ILl3Ra2, insulin-like growth factor I receptor (IGF-IR ), Lysosomal Associated Membrane Protein 1 (LAMP1), Ll Cell Adhesion Molecule (L1CAM), lymphocyte antigen 6 (Ly-6), melanoma chondroitin sulfate proteoglycan (MCSP), Membrane-type metalloproteinase (MT- MMP), mesothelin, mucin 1 (MUC1), MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16, Muellerian inhibitory substance receptor type II (MISIIR), nectin cell adhesion molecule 4 (Nectin-4), 6-transmembrane epithelial antigen of prostate (STEAP), plasma cell antigen 1, prostate stem cell antigen
- the RS1 and the RS2 sequences can be identical or they can be different sequences selected from Table 2.
- the RS2 sequence is different compared to the RS1 sequence and each is a substrate for a different protease set forth in Table 3.
- the RS1 and the RS2 sequences are identical each is a substrate for two or more proteases selected from legumain, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, and matriptase.
- the FBM and SBM upon cleavage of the RS1 and the RS2 by the mammalian protease(s) and release of the FBM and SBM from the recombinant polypeptide, the FBM and SBM remain fused and are capable of binding to and linking together a T cell bearing the CD3 antigen and a tumor cell bearing the target cell marker in an in vitro assay comprising both the T cells and the tumor cells.
- the lower ability of the recombinant polypeptide in an uncleaved state to induce lysis of the tumor cell bearing the target cell marker antigen in an in vitro assay comprising both T cells and tumor cells is at least two orders of magnitude less, or at least three orders of magnitude less, or at least four orders of magnitude less compared to the greater amount of lysis induced by the FBM or the SBM that have been released from the recombinant polypeptide by cleavage of the RS1 and RS2, as determined by quantitation of cell lysis or release of intracellular components when assayed under equivalent molar concentrations.
- the binding affinity of the uncleaved recombinant polypeptide to the CD3 antigen or to the target cell marker in an in vitro assay comprising CD3 antigen or target cell marker is at least one order of magnitude less, as determined as a I constant, compared to binding affinity to the CD3 antigen or to the target cell marker of an uncleaved recombinant polypeptide comprising an RS1, RS2, FBM, SBM, XTEN1 but not comprising a second release segment and a second XTEN, when assayed under equivalent molar concentrations.
- the binding affinity of the uncleaved recombinant polypeptide comprising an RS1, RS2, FBM, SBM, XTEN1, and XTEN2 to the CD3 antigen or to the target cell marker in an in vitro assay comprising CD3 antigen or target cell marker is at least two orders of magnitude less, or at least three orders of magnitude less, or at least four orders of magnitude less, as determined as a I constant in the in vitro assay, compared to the binding affinity to CD3 antigen or target cell marker of the FBM or the SBM that have been released from the recombinant polypeptide by cleavage of the RS1 and the RS2, when assayed under equivalent molar concentrations.
- the in vitro assay utilized can be selected from cell membrane integrity assay, mixed cell culture assay, FACS based propidium Iodide assay, trypan Blue influx assay, photometric enzyme release assay, radiometric 5lCr release assay, fluorometric Europium release assay, CalceinAM release assay, photometric MTT assay, XTT assay, WST-l assay, alamar blue assay, radiometric 3H-Thd incorporation assay, clonogenic assay measuring cell division activity, fluorometric rhodaminel23 assay measuring mitochondrial transmembrane gradient, apoptosis assay monitored by FACS-based
- phosphatidylserine exposure ELISA-based TUNEL test assay
- sandwich ELISA sandwich ELISA
- caspase activity assay cell-based LDH release assay
- cell morphology assay or any combination thereof.
- the recombinant polypeptide compositions provided herein can be useful for a variety of purposes including therapeutics and diagnostics.
- the disclosure relates to recombinant polypeptide compositions administered to a subject.
- administration of a recombinant polypeptide having the elements described in the embodiments, above, to a subject having a target cell, such as a tumor the release segment(s) of the recombinant polypeptide are capable of being cleaved when in proximity to the tumor, wherein the tumor or surrounding tissue is expressing one or more proteases for which the release segment(s) are a substrate.
- the fused FBM and SBM are capable of binding to and linking together a T cell bearing the CD3 antigen and a tumor cell bearing a tumor specific marker that is a ligand for the SBM in the subject.
- the binding results in the release of one or more T cell-derived effector molecules by the T cell.
- the one or more effector molecules are selected from TNF-alpha, IFN-gamma, interleukin 2, perforin, and granzymes.
- TNF-alpha TNF-alpha
- IFN-gamma interleukin 2
- perforin perforin
- granzymes Upon the binding together of the T cell bearing the CD3 antigen and the tumor cell bearing the tumor specific marker, lysis of the tumor cell in the subject is effected by the T cell-derived effector molecules.
- the subject is selected from the group consisting of mouse, rat, monkey, dog, and human.
- the disclosure relates to the pharmacokinetic properties of the subject recombinant polypeptides and the released components after administrations to a subject.
- the uncleaved recombinant polypeptide exhibits a terminal half-life following administration of a single dose to a subject that is at least five-fold, lO-fold, 20-fold, 30-fold, 40- fold, 50-fold, or lOO-fold greater compared to the terminal half-life of the fused FBM and SBM not linked to the recombinant polypeptide when the uncleaved recombinant polypeptide and the fused FBM and SBM are each administered to a subject at a equivalent molar dose.
- the fused FBM and SBM cleaved and released from the recombinant polypeptide exhibit a terminal half-life that is at least five-fold, lO-fold, or 20-fold, or 30-fold, or 50-fold, or lOO-fold less compared to the terminal half-life of the corresponding recombinant polypeptide that is not cleaved in the subject.
- the plasma Cmax concentration of the released fused FBM and SBM does not exceed about 0.01 ng/ml, or about 0.1 ng/ml, or about 1 ng/ml, or about 10 ng/ml, or about 100 ng/ml. In another embodiment, following the
- the plasma area under the curve of the released FBM and SBM is at least lO-fold lower, or at least 30-fold lower, or at least lOO-fold lower compared to the plasma area under the curve of the uncleaved recombinant polypeptide in the subject.
- the subject is selected from the group consisting of mouse, rat, monkey, dog, and human.
- compositions comprising any of the recombinant polypeptides described herein, together with one or more pharmaceutically suitable excipients.
- the pharmaceutical composition is formulated for intradermal, subcutaneous, intravenous, intra-arterial, intraabdominal, intraperitoneal, intrathecal, or intramuscular administration.
- the pharmaceutical composition is in a liquid form.
- the pharmaceutical composition is in a pre-filled syringe for a single injection.
- the pharmaceutical composition is formulated as a lyophilized powder to be reconstituted prior to administration.
- the present disclosure contemplates use of the recombinant polypeptide of any one of embodiments described herein in the preparation of a medicament for the treatment of a disease in a subject.
- the disease to be treated by the medicament is selected from the group consisting of carcinoma, Hodgkin's lymphoma, and non-Hodgkin's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, ER/PR+ breast cancer, Her2+ breast cancer, triple-negative breast cancer, colon cancer, colon cancer with malignant ascites, mucinous tumors, prostate cancer, head and neck cancer, skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervix cancer, colorectal, uterine cancer, meso
- the disclosure relates to methods of treating a disease in a subject.
- the disclosure provides a method of treating a disease in a subject, comprising administering to the subject in need thereof one or more therapeutically effective doses of the recombinant polypeptide or a pharmaceutical composition comprising the recombinant polypeptide any one of the embodiments described herein.
- the disease to be treated by the method is selected from the group consisting of carcinomas, Hodgkin's lymphoma, non-Hodgkin's lymphoma, B cell lymphoma, T-cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, colon cancer, prostate cancer, head and neck cancer, any form of skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervical cancer, colorectal cancer, an epithelia intraperitoneal malignancy with malignant ascites, uterine cancer, mesothelioma in the peritoneum kidney cancers, lung cancer, small-cell lung cancer, non-small cell lung cancer, gastric cancer, esophageal cancer, stomach cancer, small intestine cancer, liver cancer, hepatocar
- the disclosure provides a method of treatment wherein the pharmaceutical composition or
- the recombinant polypeptide is administered to the subject as one or more therapeutically effective doses administered twice weekly, once a week, every two weeks, every three weeks, or monthly.
- the pharmaceutical composition or recombinant polypeptide is administered to the subject as one or more therapeutically effective doses over a period of at least two weeks, or at least one month, or at least two months, or at least three months, or at least four months, or at least five months, or at least six months.
- the dose can be administered intradermally, subcutaneously, intravenously, intra-arterially, intra-abdominally, intraperitoneally, intrathecally, or intramuscularly.
- the pharmaceutical composition or recombinant polypeptide dose is administered as a bolus dose or by infusion of 5 minutes to 96 hours as tolerated for maximal safety and efficacy.
- the dose to be administered is selected from the group consisting of at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg, at least about 0.12 mg/kg, at least about 0.14 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg/kg, at least 0.4 mg/kg, at least about 0.5 mg/kg, at least about 0.6 mg
- an initial dose is selected from the group consisting of at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg
- a subsequent dose is selected from the group consisting of at least about 0.1 mg/kg, at least about 0.12 mg/kg, at least about 0.14 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg/kg, at least 0.4.
- the administration to the subject results in a plasma concentration of the recombinant polypeptide of at least about 0.1 ng/mL to at least about 2 ng/mL or more in the subject for at least about 3 days, at least about 7 days, at least about 10 days, at least about 14 days, or at least about 21 days.
- the subject is selected from the group consisting of mouse, rat, monkey, and human.
- the disclosure relates to treatment regimens.
- the treatment regimen uses a recombinant polypeptide or pharmaceutical composition described herein for use in a method for the treatment of a disease, the method comprising administering the pharmaceutical composition or the recombinant polypeptide to a subject with the disease, optionally according to a treatment regimen comprising two or more consecutive doses using a therapeutically effective dose.
- the disease to be treated by the regimen is selected from the group consisting of carcinomas, Hodgkin's lymphoma, non-Hodgkin's lymphoma, B cell lymphoma, T-cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, colon cancer, prostate cancer, head and neck cancer, any form of skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervical cancer, colorectal cancer, an epithelia intraperitoneal malignancy with malignant ascites, uterine cancer, mesothelioma in the peritoneum kidney cancers, lung cancer, small-cell lung cancer, non-small cell lung cancer, gastric cancer, esophageal cancer, stomach cancer, small intestine cancer, liver cancer, hepatocarcinoma
- the pharmaceutical composition or the recombinant polypeptide for the use in the treatment regimen is part of a specified treatment cycle.
- the treatment cycle can comprise administration of the pharmaceutical composition or the recombinant polypeptide twice a week, every week, every 10 days, every two weeks, every three weeks, or every month per each treatment cycle.
- the treatment regimen results in the improvement of a clinical parameter or endpoint associated with the disease in the subject.
- the clinical parameter or endpoint associated with the disease in the subject can be one or any combination of the group consisting of tumor shrinkage as a complete, partial or incomplete response; time-to-progression, time to treatment failure, biomarker response; progression-free survival; disease free-survival; time to recurrence; time to metastasis; time of overall survival; improvement of quality of life; and improvement of symptoms.
- the disclosure provides kits.
- the disclosure provides a kit comprising the pharmaceutical composition of any one of the embodiments described herein, together with a container and a label or package insert on or associated with the container.
- the disclosure provides one or more isolated nucleic acids, the nucleic acid comprising (a) a polynucleotide encoding a recombinant polypeptide of any one of the embodiments described herein; or (b) the complement of the polynucleotide of (a).
- the disclosure also provides an expression vector comprising the polynucleotide sequences encoding the recombinant polypeptide of any one of the embodiments described herein and a recombinant regulatory sequence operably linked to the polynucleotide sequence.
- the disclosure also provides an isolated host cell, comprising the foregoing expression vector. In one embodiment the host cell is a prokaryote. In another embodiment, the host cell is E. coli.
- the disclosure relates to methods of manufacturing an activatable recombinant polypeptide.
- the disclosure provides a method of
- an activatable recombinant polypeptide composition comprising: a) culturing a host cell comprising a nucleic acid construct that encodes the activatable recombinant polypeptide under conditions that lead to expression of the activatable recombinant polypeptide, wherein the activatable recombinant polypeptide comprises an RS1, RS2, FBM, SBM, XTEN1, and XTEN2, wherein: i) the RS1 and RS2, wherein the RS1 and RS2 are each substrates for cleavage by a mammalian protease and each comprise an amino acid sequence having at least 88%, or at least 89%, or at least 90%, or at least 91%, or at least 92%, or at least 93%, or at least 94%, or at least 95%, or 100% sequence identity to a sequence selected from the sequences of Table 1 or Table 2; ii) the FBM is an antibody fragment comprising a VL and VH derived from a
- Phosphodiesterase 3 (ENPP3), EGFR, EGFRvIII, EpCAM, endosialin (CD248), epidermal growth factor receptor variant III (EGFRvIII), EphA2, F19 antigen, fetal acetylcholine receptor (fiiAChR), fibroblast activation antigen (FAP), Fos-related antigen 1 (FRA1), Folate Receptor 1 (FOLR1), fucosyl GM1, G250, ganglioside GD3, glypican-3 (GPC3), 9-0- Acetyl-GD3, GM2, Glucocorticoid induced TNF receptor (GITR), globohexaosylceramide (globo-H), GD2,
- Glypican 3 Glypican 3 (GPC3), guanylyl cyclase C (GCC), HER2, HER2 neu, HER3, HER4, HER1, ILl3Ra2, insulin-like growth factor I receptor (IGF-IR ), Lysosomal Associated Membrane Protein 1 (LAMP1), Ll Cell Adhesion Molecule (L1CAM), lymphocyte antigen 6 (Ly-6), melanoma chondroitin sulfate proteoglycan (MCSP), Membrane-type metalloproteinase (MT- MMP), mesothelin, mucin 1 (MUC1), MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16, Muellerian inhibitory substance receptor type II (MISIIR), nectin cell adhesion molecule 4 (Nectin-4), 6-transmembrane epithelial antigen of prostate (STEAP), plasma cell antigen 1, prostate stem cell antigen
- the activatable recombinant polypeptide is activated by cleavage of the RS1 and RS2 by one or more proteases capable of cleaving the RS1 and RS2, resulting in the release of the FBM and SBM from the composition, wherein the FBM and SBM remain fused.
- the XTEN1 and XTEN2 of the activatable recombinant polypeptide in an uncleaved state interfere with specific binding of the FBM to the CD3 and the SBM to the target cell marker such that the dissociation constant (K d ) of the FBM of the activatable recombinant polypeptide in an uncleaved state towards CD3 or the SBM to the target cell marker is at least 100 times greater compared to the FBM or the SBM released from the activatable recombinant polypeptide by cleavage of the RS1 and RS2, when measured in in vitro assays comprising the target cell marker under equivalent molar concentrations.
- K d dissociation constant
- FIG. 1 depicts the various schematic figures used in various drawings, together with descriptions of what they represent.
- FIG. 2 depicts a ProTIA composition (a form of recombinant polypeptide composition described herein) that is in the uncleaved,“pro” form and in the cleaved state after being acted on by a tumor associated protease.
- the figure also describes some of the non-limiting properties of both forms of the compositions.
- FIG. 3 shows the uncleaved“pro” form of ProTIA in FIG. 3 A and the cleaved form in FIG. 3B in which the uncleaved form is depicted in proximity to an effector cell and a tumor associated cell, each with cell-surface antigens; however the uncleaved form in FIG. 3A is unable to concurrently bind the two cells because of the steric hindrance and shielding effects of the XTEN on the binding moieties, while the cleaved form in FIG. 3B, with the released binding moieties, permits the concurrent binding of the two cells and allows and immune activation by the effector cell against the target tumor associated cell.
- FIG. 4 shows schematic representations of two configurations of the ProTIA
- compositions illustrating that the Release Segment and the XTEN can be attached to either the effector cell binding moiety or the tumor antigen binding moiety.
- FIG. 5 shows schematic representations of two configurations of the ProTIA
- compositions in which two Release Segments and two XTEN are linked to the binding moieties are linked to the binding moieties.
- one RS and XTEN is linked to the effector cell binding moiety and the other RS and XTEN is linked to the tumor antigen binding moiety, and the composition would be in a scFv configuration.
- both RS and XTEN are attached to either the effector cell binding moiety (on the left) or the tumor antigen binding moiety (on the right), and the binding moieties would be in a diabody configuration (thus permitting the composition to be produced in recombinant form).
- FIG. 6 shows schematic representations of two configurations of the ProTIA
- FIGS. 6A-D show alternative N- and C-terminal configurations for the binding moieties.
- FIG. 7 shows schematic representations of two configurations of the ProTIA
- compositions in which two Release Segments and two XTEN are linked to the binding moieties In the case of FIG. 7 A, one RS and one XTEN is linked to the effector cell binding moiety and the other RS and XTEN is linked to the tumor antigen binding moiety, and the composition would be in a scFv configuration. In the case of FIG. 7B, both RS and XTEN are attached to either the effector cell binding moiety (on the right) or the tumor antigen binding moiety (on the left), and the binding moieties would be in a diabody configuration (thus permitting the composition to be produced in recombinant form).
- FIG. 8 shows schematic representations of two configurations of the ProTIA
- FIG. 8 A depicts the binding moieties as XTEN.
- FIG. 8B depicts the binding moieties as albumin.
- FIG. 8C depicts the binding moieties as an Fc fragment.
- FIG. 9 shows schematic representations of configurations of the ProTIA compositions in which two Release Segments and two XTEN are linked to the binding moieties.
- FIG. 9 A depicts three configurations in which the two RS and XTEN are linked to both the effector cell binding moiety and the tumor antigen binding moiety (on the left), to the tumor antigen binding moiety (the center) or to the effector cell binding moiety (on the right).
- FIG. 9 shows schematic representations of configurations of the ProTIA compositions in which two Release Segments and two XTEN are linked to the binding moieties.
- FIG. 9 A depicts three configurations in which the two RS and XTEN are linked to both the effector cell binding moiety and the tumor antigen binding moiety (on the left), to the tumor antigen binding moiety (the center) or to the effector cell binding moiety (on the right).
- FIG. 9 A depicts three configurations in which the two RS and XTEN are linked to both the effector cell binding moiety and the tumor anti
- FIG. 9B depicts four configurations in which the one RS and XTEN are linked to the effector cell binding moiety and one RS and albumin are linked to the tumor antigen binding moiety (on the upper left), one RS and an XTEN are linked to the tumor antigen binding moiety and one RS and albumin are linked to the effector cell binding moiety (on the upper right), both the RS and an XTEN and the RS and albumin are linked to the tumor antigen binding moiety (on the lower left) and both the RS and an XTEN and the RS and albumin are linked to the effector cell binding moiety (on the lower right).
- 9C depicts four configurations in which the one RS and XTEN are linked to the effector cell binding moiety and one RS and Fc are linked to the tumor antigen binding moiety (on the upper left), one RS and an XTEN are linked to the tumor antigen binding moiety and one RS and Fc are linked to the effector cell binding moiety (on the upper right), both the RS and an XTEN and the RS and Fc are linked to the tumor antigen binding moiety (on the lower left) and both the RS and an XTEN and the RS and Fc are linked to the effector cell binding moiety (on the lower right).
- FIG. 10 shows schematic representations of a ProTIA in proximity to tumor tissue (on the left) and normal tissue (on the right) in which the more permeable vasculature in the tumor tissue permits the ProTIA to extravasate into the tissue where the tumor-associated proteases can act on the RS, cleaving it and releasing the binding moieties, which in turn can bind to and link together the effector cell and the tumor associated cell.
- the extravasation is either blocked by the tighter vasculature barriers or, in the case where the ProTIA does extravasate, the ProTIA remains in the“pro” form and while able to bind the effector cell, no tumor cells are present or, if present, insufficient proteases are present to release the binding moieties, with the net effect that an immunological synapse is not formed.
- FIG. 11 shows a schematic representation of an scFv configuration of the effector cell binding moiety the tumor antigen binding moiety, each with VH/VL pairs joined by linkers, and in a tandem format.
- FIG. 12 shows a schematic representation of a single chain diabody configuration of the effector cell binding moiety the tumor antigen binding moiety, each with VH/VL pairs joined by linkers.
- FIG. 13 shows schematic representations of constructs.
- FIG. 13A shows a schematic representation of a generic construct design.
- FIGS. 13B and 13C show schematic representations of ProTIA compositions in which the effector cell binding moiety and the tumor antigen binding moiety are in various permutations in scFv configurations (FIG. 13B) [with variable heavy (VH) and variable light (VL) domains linked either by intramolecular long linker (L) or intermolecular shorter linker (1)] and in single chain diabody configurations (FIG. 13C) [with the VH and VL domains linked either by long linker (L) or intermolecular shorter linker (1).
- FIG. 14 shows the purification of uncleaved AC 1278 from fermentation media, as described in Example 2.
- FIG. 14A shows exemplary SDS-PAGE of IMAC capture of AC1278 from fermentation media;
- FIG. 14B shows SDS-PAGE analysis of fractions in HIC polishing step;
- FIG. 14C shows SDS-PAGE analysis of fractions in ImpRes-Q polishing step.
- FIG. 15 shows the lot release analytics of uncleaved AC1278, as described in Example 2.
- FIG. 15A shows the lot release analytical SEC chromatography of uncleaved AC 1278 (in solid line) against XTEN length standard (in dashed line);
- FIG. 15B shows the lot release SDS- PAGE of uncleaved AC1278.
- FIG. 16 shows the preparation of cleaved ProTIA-A using uncleaved AC1278, as described in Example 2.
- FIG. 16A shows SDS-PAGE analysis of MMP-9 digestion reaction mixture;
- FIG. 16B show SDS-PAGE analysis of IMAC purification of MMP-9 digestion mixture to remove cleaved XTEN segment.
- FIG. 17 shows the lot release analytics of cleaved AC1278, as described in Example 2.
- FIG. 17A shows the lot release analytical SEC chromatography of cleaved AC1278 (in solid line) against globular protein standard (in dashed line);
- FIG. 17B shows the lot release SDS- PAGE of cleaved AC1278.
- FIG. 18 shows the purification of uncleaved AC 1476 from fermentation media, as described in Example 3.
- FIG. 18A shows exemplary SDS-PAGE of IMAC capture of AC1476 from fermentation media;
- FIG. 18B shows SDS-PAGE analysis of fractions in HIC polishing step;
- FIG. 18C shows SDS-PAGE analysis of fractions in ImpRes-Q polishing step.
- FIG. 19 shows the lot release analytics of uncleaved AC 1476 as described in Example 3.
- FIG. 19A shows the lot release analytical SEC chromatography of uncleaved AC1476 (in solid line) against XTEN length standard (in dashed line);
- FIG. 19B shows the lot release SDS-PAGE of uncleaved AC1476 with Coomassie staining;
- FIG. 19C shows the lot release SDS-PAGE of uncleaved AC 1476 with silver staining.
- FIG. 20 shows additional lot release analytics of uncleaved AC 1476 as described in Example 3.
- FIG. 20A shows the lot release ESI-MS of uncleaved AC 1476;
- FIG. 20B shows the lot release cation exchange chromatography of uncleaved AC1476.
- FIG. 21 shows the preparation of cleaved ProTIA-A using uncleaved AC 1476 as described in Example 3.
- FIG. 21 A shows the SDS-PAGE analysis of MMP-9 digestion reaction mixture;
- FIG. 21B shows the SDS-PAGE analysis of anion exchange fractions of MMP-9 digestion mixture to remove uncleaved substrate, as well as cleaved XTEN segment.
- FIG. 22 shows the lot release analytics of cleaved AC 1476 as described in Example 3.
- FIG. 22A shows the lot release analytical SEC of cleaved AC1476 (in solid line) against globular protein standard (in dashed line);
- FIG. 22B shows the lot release SDS-PAGE of cleaved AC1476 with Coomassie staining;
- FIG. 22C shows the lot release SDS-PAGE of cleaved AC1476 with silver staining.
- FIG. 23 shows the additional lot release analytics of cleaved AC1476 as described in Example 3.
- FIG. 23A shows the lot release ESI-MS of cleaved AC1476;
- FIG. 23B shows the lot release cation exchange chromatography of cleaved AC1476.
- FIG. 24 shows binding of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA for its ligand, as described in Example 4.
- FIG. 25 depicts results from the experiment to determine the in vitro activity of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 6.
- FIG. 26 depicts results from the experiment to determine the in vitro specificity of anti- EpCAM x anti-CD3 ProTIA, as described in Example 6.
- FIG. 27 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-uncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 6.
- FIG. 28 depicts results from the experiment to determine the PK of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 9.
- FIG. 29 shows schematic representations of the alternate N- to C-terminus
- FIG. 29 A shows the configuration of the effector cell binding moiety (ECBM) followed by release site segment (RS) and XTEN while FIG. 29B shows the configuration of XTEN followed by the RS segment and then ECBM.
- ECBM effector cell binding moiety
- RS release site segment
- XTEN XTEN
- FIG. 30 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-noncleavable anti-EpCAM x anti-CD3 ProTIA in SK-OV-3 as described in Example 6.
- FIG. 31 depicts tumor volume results from experiment to determine the anti-tumor effect of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 10.
- FIG. 32 depicts body weight results from an experiment to determine the anti-tumor effect of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 10.
- FIG. 33 depicts results from an experiment to determine the cytokine profile of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 12.
- FIG. 33A shows the results of the assay to detect IL-2 and
- FIG. 33B shows the results to detect IL-4.
- FIG. 34 depicts results from an experiment to determine the cytokine profile of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 12.
- FIG. 34A shows the results of the assay to detect IL-6 and
- FIG. 34B shows the results to detect IL-10.
- FIG. 35 depicts results from an experiment to determine the cytokine profile of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA, as described in Example 12.
- FIG. 35 A shows the results of the assay to detect IFN-gamma and
- FIG. 35B shows the results to detect TNF -alpha.
- FIG. 36 depicts the amino acid sequence of the release segment RSR-1517 and the location of the three cleavage sites where the listed proteases are able to cleave the peptide.
- FIG. 37 depicts results from a cytotoxicity assay against huEp-CHO 4-12B measuring released caspase 3/7 in culture supernatants, as described in Example 55.
- FIG. 38 depicts HCT-l 16 tumor volume results from experiment to determine the anti- tumor effect of anti-EpCAM x anti-CD3 ProTIA, protease-treated anti-EpCAM x anti-CD3 ProTIA and non-cleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 13.
- FIG. 39 depicts body weight results from experiment to determine the anti-HCT-l 16 tumor effect of anti-EpCAM x anti-CD3 ProTIA, protease-treated anti-EpCAM x anti-CD3 ProTIA and non-cleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 13.
- FIG. 40 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-non cleavable anti-EpCAM x anti-CD3 ProTIA in SK-OV-3 with human purified CD3 positive T cells as described in Example 14.
- FIG. 41 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-non cleavable anti-EpCAM x anti-CD3 ProTIA in OVCAR-3 with human purified CD3 positive T cells as described in Example 14.
- FIG. 42 depicts results from the experiment to measure activation of CD69 on CD8 and CD4 cells in co-culture of PBMC and SK-OV-3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 42A depicts the activation of CD69 on CD8 cells
- FIG. 42B depicts the activation of CD69 on CD4 cells.
- FIG. 43 depicts results from the experiment to measure activation of both CD69 and CD25 on CD8 and CD4 cells in co-culture of PBMC and SK-OV-3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 43A depicts the activation of both CD69 and CD25 on CD8 cells
- FIG. 43B depicts the activation of both CD69 and CD25 on CD4 cells.
- FIG. 44 depicts results from the experiment to measure activation of CD69 on CD8 and CD4 cells in co-culture of purified CD3+ cells and SK-OV-3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 44A depicts the activation of CD69 on CD8 cells
- FIG. 44B depicts the activation of CD69 on CD4 cells.
- FIG. 45 depicts results from the experiment to measure activation of both CD69 and CD25 on CD8 and CD4 cells in co-culture of purified CD3+ cells and SK-OV-3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 45A depicts the activation of both CD69 and CD25 on CD8 cells
- FIG. 45B depicts the activation of both CD69 and CD25 on CD4 cells.
- FIG. 46 depicts results from the experiment to measure activation of CD69 on CD8 and CD4 cells in co-culture of purified CD3+ cells and OVCAR3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 46A depicts the activation of CD69 on CD8 cells
- FIG. 46B depicts the activation of CD69 on CD4 cells.
- FIG. 47 depicts results from the experiment to measure activation of both CD69 and CD25 on CD8 and CD4 cells in co-culture of purified CD3+ cells and OVCAR3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 47A depicts the activation of both CD69 and CD25 on CD8 cells
- FIG. 47B depicts the activation of both CD69 and CD25 on CD4 cells.
- FIG. 48 depicts results from the experiment to measure activation of CD69 on CD8 and CD4 cells in co-culture of PBMC and OVCAR3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 48A depicts the activation of CD69 on CD8 cells
- FIG. 48B depicts the activation of CD69 on CD4 cells.
- FIG. 49 depicts results from the experiment to measure activation of both CD69 and granzyme B in CD8 and CD4 cells in co-culture of PBMC and OVCAR3 cells with protease- treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 8.
- FIG. 49A depicts the activation of both CD69 and granzyme B in CD8 cells
- FIG. 49B depicts the activation of both CD69 and granzyme B in CD4 cells.
- FIG. 50 depicts results from the experiment to measure release of cytokines IL-2 and IL-4 in co-culture of purified CD3+ cells and SK-OV-3 cells with protease-treated, protease- untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 15.
- FIG. 50A depicts the concentration of released IL-2
- FIG. 50B depicts the
- FIG. 51 depicts results from the experiment to measure release of cytokines IL-6 and IL-10 in co-culture of purified CD3+ cells and SK-OV-3 cells with protease-treated, protease- untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 15.
- FIG. 51 A depicts the concentration of released IL-6
- FIG. 51B depicts the
- FIG. 52 depicts results from the experiment to measure release of cytokines TNF-alpha and IFN-gamma in co-culture of purified CD3+ cells and SK-OV-3 cells with protease-treated, protease-untreated and protease noncleavable anti-EpCAM x anti-CD3 ProTIA, as described in Example 15.
- FIG. 52 A depicts the concentration of released TNF -alpha
- FIG. 52B depicts the concentration of released IFN-gamma.
- FIG. 53 shows the binding curves of protease-treated, protease-untreated.
- FIG. 54 shows binding specificity of protease treated antiEpCAM x antiCD3 ProTIA for rhEpCAM ligand, as described in Example 17.
- FIG. 55 depicts SW480 tumor volume results from the experiment to determine the antitumor effect of antiEpCAM x antiCD3 ProTIA, protease treated antiEpCAM x antiCD3 ProTIA and noncleavable antiEpCAM x antiCD3 ProTIA, as described in Example 18.
- FIG. 56 depicts body weight results from the experiment to determine the antiSW480 tumor effect of antiEpCAM x antiCD3 ProTIA, protease-treated antiEpCAM x antiCD3 ProTIA and noncleavable antiEpCAM x antiCD3 ProTIA, as described in Example 18.
- FIG. 57 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-noncleavable antiEpCAM x antiCD3 ProTIA in SKOV3 with human PBMC as described in Example 23.
- FIG. 58 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-noncleavable antiEpCAM x antiCD3 ProTIA in OVCAR3 with human PBMC as described in Example 23.
- FIG. 59 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-noncleavable antiEpCAM x antiCD3 ProTIA in HCT116 with human PBMC as described in Example 23.
- FIG. 60 depicts results from the experiment to determine the in vitro activity of protease-treated, protease-untreated and protease-noncleavable antiEpCAM x antiCD3 ProTIA in SW480 with human PBMC as described in Example 23.
- FIG. 61 depicts HCT-l 16 tumor volume results from experiment to determine the antitumor effect of protease-treated, protease-untreated, and non-cleavable anti-EpCAM x anti- CD3 ProTIAs, as described in Example 25
- FIG. 62 depicts human CA125 levels in control Group 1 bearing OVCAR-3 and PBMC, Group 8 bearing PBMC only and Group 9 bearing OVCAR-3 only, as described in Example 26.
- 64 depicts human CA125 levels from experiment to determine the antitumor effect of high dose protease-treated anti-EpCAM x anti-CD3 ProTIA (Group 3), protease-untreated anti-EpCAM x anti-CD3 ProTIA (Group 5), and non-cleavable anti-EpCAM x anti-CD3 ProTIA (Group 7), as described in Example 26.
- FIG. 65 depicts human CA125 levels from experiment to determine the antitumor effect of protease-untreated anti-EpCAM x anti-CD3 ProTIA administered intraperitoneally versus intravenously in mice bearing OVCAR-3 tumor, as described in Example 27.
- FIG. 66 depicts total tumor volume from experiment to determine the antitumor effect of protease-untreated anti-EpCAM x anti-CD3 ProTIA administered intraperitoneally versus intravenously in mice bearing OVCAR-3 tumor, as described in Example 27.
- FIG. 67 depicts total tumor volume from experiment to determine the antitumor effect of protease-untreated anti-EpCAM x anti-CD3 ProTIA versus bevacizumab in mice bearing OVCAR-3 tumor, as described in Example 27.
- FIG. 68 depicts binding of protease-untreated anti-EpCAM x anti-CD3 variants for CD3 epsilon/delta ligand, as described in Example 28.
- FIG. 69 depicts (FIG. 69 A) plasma and (FIG. 69B) ascites pharmacokinetics results of intravenously administered protease-treated, protease-untreated, and non-cleavable anti-EpCAM x anti-CD3 ProTIAs, as described in Example 30.
- FIG. 70 depicts (FIG. 70A) plasma and (FIG. 70B) ascites pharmacokinetics results of intraperitoneally administered protease-treated, protease-untreated, and non-cleavable anti- EpCAM x anti-CD3 ProTIAs, as described in Example 30.
- FIGS. 71A-F shows the results from cytokine assays of samples from an in vivo toxicity assessment of the intact, cleaved and uncleavable ProTIA constructs compared to a construct configured as a BiTE, as described Example 33.
- FIG. 72 shows the results from an experiment to determine the maximum tolerated dose of an intact ProTIA compared to the cleaved, activated form, graphed as a Kaplan-Meier plot, as described in Example 34.
- FIGS. 73A-F shows the results from an experiment to determine the maximum tolerated dose of an intact AC 1553 ProTIA compared to the cleaved, activated form, graphed as body weight of the dosed mice over time, as described in Example 34.
- FIG. 74 shows SDS-PAGE gels from the production of release segment-XTEN variants, as described in Example 41.
- FIG. 74A is a titer analysis of RS-XTEN variant expression.
- FIGS. 74(B)-(D) show the single-step IMAC purification of RS-XTEN variants AC1602, AC1609, AC1610, AC 1604, AC1608, AC1611, AC1612, AC1649, AC1650.
- FIG. 74E is the gel from the lot release of the purified RS-XTEN variants.
- FIG. 75 shows an SDS-PAGE gel of the cleavage profile of AC1611 when subject to seven human proteases implicated in cancer, as described in Example 42.
- FIG. 76 shows an SDS-PAGE gel of the uPA digestion of RS-XTEN variants with AC1611 as the reference, as described in Example 43.
- FIG. 77 shows results of body weight determinations in the vehicle and treatment groups, as described in Example 56.
- FIG. 78 shows results of body weight determinations in the treatment groups, as described in Example 57.
- FIG. 79 shows results of tumor volume in vehicle and treatment groups, as described in Example 60.
- FIG. 79A shows results of animals dosed with 0.5 mg/kg and
- FIG. 79B shows results of animals dosed with 0.1 mg/kg.
- FIG. 80 shows results of redirected cellular cytotoxicity assays of protease-untreated anti-EGFR x anti-CD3 ProTIA compositions compared to protease-treated anti-EGFR x anti- CD3 ProTIA and protease-non-cleavable as described in Example 61.
- FIG. 80A shows results of the in vitro caspase 3/7 assay of AC1955 and AC1958 against HCT-l 16 cells with human PBMC.
- FIG. 80B shows results of the in vitro caspase 3/7 assay of AC1955 and AC1958 against HT-29 cells with human PBMC.
- FIG. 81 shows results from redirected cellular cytotoxicity assays of protease-untreated anti-Her2 x anti-CD3 ProTIA compositions AC2038 and AC2040 compared to protease-treated anti-Her2 x anti-CD3 ProTIA and protease-non-cleavable AC2039), assessed in an in vitro cell- based assay of caspase 3/7 activities of apoptotic cells as described in Example 62.
- FIG. 81 A shows results with BT474 with human PBMC.
- FIG. 81B shows results with SK-OV-3 and human PBMC.
- FIG, 81C shows results with JIMT-l with human PBMC.
- FIG. 81D shows results with MDA-MB-231 with human PBMC.
- FIG. 82 shows results from in vivo experiments to determine to determine the anti- tumor effect of protease-treated and protease-untreated anti-EGFR x anti-CD3 ProTIA against Cetuximab as described in Example 63.
- FIG. 82 A depicts tumor volume results from animals with HT-29 tumor cells.
- FIG. 82B depicts body weight results from animals with HT-29 tumor cells.
- FIG. 83 shows results from in vivo experiments to determine the anti-tumor effect of protease-treated and protease-untreated anti-EGFR x anti-CD3 ProTIA in an established breast tumor model, as described in Example 64.
- FIG. 83A depicts depicts tumor volume results from animals with BT-474 tumor cells.
- FIG. 83B depicts body weight results from animals with BT- 474 tumor cells.
- FIG. 84 shows an SDS-PAGE of the lot release analysis of formulated drug substance, as described in Example 46.
- FIG. 85 shows lot release HPLC analyses of formulated drug substance, as described in Example 46.
- FIG. 85A shows the SE-HPLC analysis and
- FIG. 85B shows the HI-HPLC analysis.
- FIG. 86 shows lot release analyses of formulated drug substance, as described in Example 47.
- FIG. 86A shows an SDS-PAGE of the lot release analysis of formulated drug substance.
- FIG. 86B shows an ESI-MS of the lot release analysis of formulated drug substance.
- FIG. 87 shows lot release HPLC analyses of formulated drug substance, as described in Example 46.
- FIG. 87A shows the SE-HPLC analysis and
- FIG. 87B shows the HI-HPLC analysis.
- a“cleavage sequence”, as used herein, means“at least a first cleavage sequence” but includes a plurality of cleavage sequences.
- polypeptide “peptide”, and“protein” are used interchangeably herein to refer to polymers of amino acids of any length.
- the polymer may be linear or branched, it may comprise modified amino acids, and it may be interrupted by non-amino acids.
- the terms also encompass an amino acid polymer that has been modified, for example, by disulfide bond formation, glycosylation, lipidation, acetylation, phosphorylation, or any other manipulation, such as conjugation with a labeling component.
- N-terminus or“amino terminus”
- C-terminus or“carboxyl terminus”
- the term“monomeric” as applied to a polypeptide refers to the state of the polypeptide as being a single continuous amino acid sequence substantially unassociated with one or more additional polypeptide of the same or different sequence.
- the monomeric state of the polypeptide can be ascertained as a single proteinaceous entity of the same molecular weight by size exclusion chromatography.
- amino acid refers to either natural and/or unnatural or synthetic amino acids, including but not limited to both the D or L optical isomers, and amino acid analogs and peptidomimetics. Standard single or three letter codes may be used to designate amino acids.
- L-amino acid or“L-amino acid” means the L optical isomer forms of glycine (G), proline (P), alanine (A), valine (V), leucine (L), isoleucine (I), methionine (M), cysteine (C), phenylalanine (F), tyrosine (Y), tryptophan (W), histidine (H), lysine (K), arginine (R), glutamine (Q), asparagine (N), glutamic acid (E), aspartic acid (D), serine (S), and threonine (T).
- non-naturally occurring means polypeptide or polynucleotide sequences that do not have a counterpart to, are not
- a non-naturally occurring polypeptide or fragment may share no more than 99%, 98%, 95%, 90%, 80%, 70%, 60%, 50% or even less amino acid sequence identity as compared to a natural sequence when suitably aligned.
- the terms“hydrophilic” and“hydrophobic” refer to the degree of affinity that a substance has with water.
- a hydrophilic substance has a strong affinity for water, tending to dissolve in, mix with, or be wetted by water, while a hydrophobic substance substantially lacks affinity for water, tending to repel and not absorb water and tending not to dissolve in or mix with or be wetted by water.
- Amino acids can be characterized based on their hydrophobicity.
- a number of scales have been developed. An example is a scale developed by Levitt, M, et al., J Mol Biol (1976) 104:59, which is listed in Hopp, TP, et al., Proc Natl Acad Sci U S A (1981) 78:3824.
- hydrophilic amino acids are arginine, lysine, threonine, alanine, asparagine, and glutamine. Of particular interest are the hydrophilic amino acids aspartate, glutamate, and serine, and glycine.
- hydrophilic amino acids aspartate, glutamate, and serine, and glycine.
- hydrophobic amino acids are tryptophan, tyrosine, phenylalanine, methionine, leucine, isoleucine, and valine.
- A“fragment” when applied to a biologically active protein is a truncated form of a the biologically active protein that retains at least a portion of the therapeutic and/or biological activity.
- A“variant,” when applied to a biologically active protein is a protein with sequence homology to the native biologically active protein that retains at least a portion of the therapeutic and/or biological activity of the biologically active protein.
- a variant protein may share at least 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98% or 99% amino acid sequence identity compared with the reference biologically active protein.
- the term“biologically active protein variant” includes proteins modified deliberately, as for example, by site directed mutagenesis, synthesis of the encoding gene, insertions, or accidentally through mutations and that retain activity.
- sequence variant means polypeptides that have been modified compared to their native or original sequence by one or more amino acid insertions, deletions, or substitutions. Insertions may be located at either or both termini of the protein, and/or may be positioned within internal regions of the amino acid sequence.
- a non-limiting example is substitution of an amino acid in an XTEN with a different amino acid.
- deletion variants one or more amino acid residues in a polypeptide as described herein are removed. Deletion variants, therefore, include all fragments of a described polypeptide sequence.
- substitution variants one or more amino acid residues of a polypeptide are removed and replaced with alternative residues.
- substitutions are conservative in nature and conservative substitutions of this type are well known in the art.
- a sequence variant would retain at least a portion of the binding affinity or biological activity, respectively, of the unmodified polypeptide.
- the term“moiety” means a component of a larger composition or that is intended to be incorporated into a larger composition, such as a proteinaceous portion joined to a larger polypeptide as a contiguous or non-contiguous sequence.
- a moiety of a larger composition can confer a desired functionality.
- an antibody fragment may retain the ability to bind its ligand yet have a smaller molecular size and be in a single-chain format.
- XTEN may confer the functionality of increasing molecular weight and/or half-life of a resulting larger composition with which the XTEN is associated.
- RS refers to a peptide with one or more cleavage sites in the sequence that can be recognized and cleaved by one or more proteases.
- mimmalian protease means a protease that normally exists in the body fluids, cells, tissues, and may be found in higher levels in certain target tissues or cells, e.g., in diseased tissues (e.g., tumor) of a mammal.
- RS sequences can be engineered to be cleaved by various mammalian proteases or multiple mammalian proteases that are present in or proximal to target tissues in a subject or are introduced in an in vitro assay.
- proteases endogenous or exogenous
- RS sequence can be adjusted and tailored to the protease utilized and can incorporate linker amino acids to join to adjacent polypeptides of the composition; e.g., the binding moieties and the XTEN.
- an RS component when linked“within” an recombinant polypeptide, the RS may be linked to the N-terminus, the C-terminus, or may be inserted between any two amino acids of an XTEN polypeptide.
- “Activity” as applied to form(s) of a composition provided herein refers to an action or effect, including but not limited to receptor binding, antagonist activity, agonist activity, a cellular or physiologic response, cell lysis, cell death, or an effect generally known in the art for the effector component of the composition, whether measured by an in vitro, ex vivo or in vivo assay or a clinical effect.
- an effect cell includes any eukaryotic cells capable of conferring an effect on a target cell.
- an effect cell can induce loss of membrane integrity, pyknosis, karyorrhexis, apoptosis, lysis, and/or death of a target cell.
- an effector cell can induce division, growth, differentiation of a target cell or otherwise altering signal transduction of a target cell.
- Non-limiting examples of effector cell include plasma cell, T cell, CD4 cell, CD8 cell, B cell, cytokine induced killer cell (CD cell), master cell, dendritic cell, regulatory T cell (RegT cell), helper T cell, myeloid cell, macrophage, and NK cell.
- effector cell antigen refers to molecules expressed by an effector cell, including without limitation cell surface molecules such as proteins, glycoproteins or lipoproteins.
- effector cell antigens include proteins of the CD3 complex or the T cell receptor (TCR), CD4, CD8, CD25, CD38, CD69, CD45RO, CD57, CD95, CD107, and CD154, as well as effector molecules such as cytokines in association with, bound to, expressed within, or expressed and released by, an effector cell.
- TCR T cell receptor
- An effector cell antigen can serve as the binding counterpart of a binding moiety of the subject recombinant polypeptide.
- Non-limiting examples of effector cell antigens to which the subject composition may bind include antigens on the cell surface such as CD3, CD4, CD8, CD25, CD38, CD69, CD45RO, CD57, CD95, CD107, and CD154 as well as Thl cytokines selected from IL2, IL10, IL12, IFN-gamma, and TNF-alpha.
- ELISA refers to an enzyme-linked immunosorbent assay as described herein or as otherwise known in the art.
- A“host cell” includes an individual cell or cell culture which can be or has been a recipient for the subject vectors into which exogenous nucleic acid has been introduced, such as those described herein.
- Host cells include progeny of a single host cell. The progeny may not necessarily be completely identical (in morphology or in genomic of total DNA complement) to the original parent cell due to natural, accidental, or deliberate mutation.
- a host cell includes cells transfected in vivo with a vector of this disclosure.
- isolated when used to describe the various polypeptides disclosed herein, means polypeptide that has been identified and separated and/or recovered from a component of its natural environment or from a more complex mixture (such as during protein purification).
- Contaminant components of its natural environment are materials that would typically interfere with diagnostic or therapeutic uses for the polypeptide, and may include enzymes, hormones, and other proteinaceous or non-proteinaceous solutes.
- a non-naturally occurring polynucleotide, peptide, polypeptide, protein, antibody, or fragments thereof does not require“isolation” to distinguish it from its naturally occurring counterpart.
- a“concentrated”,“separated” or“diluted” polynucleotide, peptide, polypeptide, protein, antibody, or fragments thereof is distinguishable from its naturally occurring counterpart in that the concentration or number of molecules per volume is generally greater than that of its naturally occurring counterpart.
- an“isolated nucleic acid” is a nucleic acid molecule that is identified and separated from at least one contaminant nucleic acid molecule with which it is ordinarily associated in the natural source of the polypeptide-encoding nucleic acid.
- an isolated polypeptide- encoding nucleic acid molecule is other than in the form or setting in which it is found in nature. Isolated polypeptide-encoding nucleic acid molecules therefore are distinguished from the specific polypeptide-encoding nucleic acid molecule as it exists in natural cells.
- an isolated polypeptide-encoding nucleic acid molecule includes polypeptide-encoding nucleic acid molecules contained in cells that ordinarily express the polypeptide where, for example, the nucleic acid molecule is in a chromosomal or extra-chromosomal location different from that of natural cells.
- A“chimeric” protein or polypeptide contains at least one fusion polypeptide comprising at least one region in a different position in the sequence than that which occurs in nature.
- the regions may normally exist in separate proteins and are brought together in the fusion
- a chimeric protein may be created, for example, by chemical synthesis, or by recombinantly creating and translating a polynucleotide in which the peptide regions are encoded in the desired relationship.
- fusion protein or “chimeric protein” comprises a first amino acid sequence linked to a second amino acid sequence with which it is not naturally linked in nature.
- “Uncleaved” and“uncleaved state” are used interchangeably herein, and refers to a polypeptide that has not been cleaved or digested by a protease such that the polypeptide remains intact.
- XTENylated is used to denote a peptide or polypeptide that has been modified by the linking or fusion of one or more XTEN polypeptides (described, below) to the peptide or polypeptide, whether by recombinant or chemical cross-linking means.
- “Operably linked” means that the DNA sequences being linked are contiguous, and in reading phase or in-frame.
- An“in-frame fusion” refers to the joining of two or more open reading frames (ORFs) to form a continuous longer ORF, in a manner that maintains the correct reading frame of the original ORFs.
- ORFs open reading frames
- a promoter or enhancer is operably linked to a coding sequence for a polypeptide if it affects the transcription of the polypeptide sequence.
- the resulting recombinant fusion protein is a single protein containing two or more segments that correspond to polypeptides encoded by the original ORFs (which segments are not normally so joined in nature).
- Crosslinking and“conjugating,” are used interchangeably herein, and refer to the covalent joining of two different molecules by a chemical reaction.
- the crosslinking can occur in one or more chemical reactions, as known in the art.
- a“linear sequence” or a“sequence” is an order of amino acids in a polypeptide in an amino to carboxyl terminus (N- to C-terminus) direction in which residues that neighbor each other in the sequence are contiguous in the primary structure of the polypeptide.
- A“partial sequence” is a linear sequence of part of a polypeptide that is known to comprise additional residues in one or both directions.
- Heterologous means derived from a genotypically distinct entity from the rest of the entity to which it is being compared.
- a glycine rich sequence removed from its native coding sequence and operatively linked to a coding sequence other than the native sequence is a heterologous glycine rich sequence.
- the term“heterologous” as applied to a polynucleotide, a polypeptide means that the polynucleotide or polypeptide is derived from a genotypically distinct entity from that of the rest of the entity to which it is being compared.
- polynucleotides refer to nucleotides of any length, encompassing a singular nucleic acid as well as plural nucleic acids, either deoxyribonucleotides or ribonucleotides, or analogs thereof.
- Polynucleotides may have any three-dimensional structure, and may perform any function, known or unknown.
- polynucleotides coding or non-coding regions of a gene or gene fragment, loci (locus) defined from linkage analysis, exons, introns, messenger RNA (mRNA), transfer RNA, ribosomal RNA, ribozymes, cDNA, recombinant polynucleotides, branched polynucleotides, plasmids, vectors, isolated DNA of any sequence, isolated RNA of any sequence, nucleic acid probes, and primers.
- loci locus
- polynucleotide may comprise modified nucleotides, such as methylated nucleotides and nucleotide analogs. If present, modifications to the nucleotide structure may be imparted before or after assembly of the polymer.
- the sequence of nucleotides may be interrupted by non- nucleotide components.
- a polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component.
- polynucleotide denotes a polynucleotide molecule having a complementary base sequence and reverse orientation as compared to a reference sequence, such that it could hybridize with a reference sequence with complete fidelity.
- “Recombinant” as applied to a polynucleotide means that the polynucleotide is the product of various combinations of recombination steps which may include cloning, restriction and/or ligation steps, and other procedures that result in expression of a recombinant protein in a host cell.
- the terms“gene” and“gene fragment” are used interchangeably herein. They refer to a polynucleotide containing at least one open reading frame that is capable of encoding a particular protein after being transcribed and translated.
- a gene or gene fragment may be genomic or cDNA, as long as the polynucleotide contains at least one open reading frame, which may cover the entire coding region or a segment thereof.
- A“fusion gene” is a gene composed of at least two heterologous polynucleotides that are linked together.
- a "coding region” or “coding sequence” is a portion of polynucleotide which consists of codons translatable into amino acids.
- a “stop codon” (TAG, TGA, or TAA) is typically not translated into an amino acid, it may be considered to be part of a coding region, but any flanking sequences, for example promoters, ribosome binding sites,
- transcriptional terminators are not part of a coding region.
- the boundaries of a coding region are typically determined by a start codon at the 5' terminus, encoding the amino terminus of the resultant polypeptide, and a translation stop codon at the 3' terminus, encoding the carboxyl terminus of the resulting polypeptide.
- Two or more coding regions of the present disclosure can be present in a single polynucleotide construct, e.g., on a single vector, or in separate polynucleotide constructs, e.g., on separate (different) vectors.
- a single vector can contain just a single coding region, or comprise two or more coding regions, e.g., a single vector can separately encode a binding moiety-A and a binding moiety-B as described below.
- a vector, polynucleotide, or nucleic acid of the disclosure can encode heterologous coding regions, either fused or unfused to a nucleic acid encoding a binding moiety of the disclosure.
- Heterologous coding regions include without limitation specialized elements or motifs, such as a secretory signal peptide or a heterologous functional domain.
- downstream refers to a nucleotide sequence that is located 3' to a reference nucleotide sequence.
- downstream nucleotide sequences relate to sequences that follow the starting point of transcription. For example, the translation initiation codon of a gene is located downstream of the start site of transcription.
- upstream refers to a nucleotide sequence that is located 5' to a reference nucleotide sequence.
- upstream nucleotide sequences relate to sequences that are located on the 5' side of a coding region or starting point of transcription. For example, most promoters are located upstream of the start site of transcription.
- “Homology” or“homologous” or“Identity” interchangably refers to sequence similarity between two or more polynucleotide sequences or between two or more polypeptide sequences.
- polynucleotides that are homologous are those which hybridize under stringent conditions as defined herein and have at least 70%, preferably at least 80%, more preferably at least 90%, more preferably 95%, more preferably 97%, more preferably 98%, and even more preferably 99% sequence identity, when optimally aligned, compared to those sequences.
- Polypeptides that are homologous preferably have sequence identities that are at least 70%, preferably at least 80%, even more preferably at least 90%, even more preferably at least 95-99% identical when optimally aligned over sequences of comparable length.
- the ends of the DNA must be compatible with each other. In some cases, the ends will be directly compatible after endonuclease digestion. However, it may be necessary to first convert the staggered ends commonly produced after endonuclease digestion to blunt ends to make them compatible for ligation.
- stringent conditions or“stringent hybridization conditions” includes reference to conditions under which a polynucleotide will hybridize to its target sequence, to a detectably greater degree than other sequences (e.g., at least 2-fold over background). Generally, stringency of hybridization is expressed, in part, with reference to the temperature and salt concentration under which the wash step is carried out.
- stringent conditions will be those in which the salt concentration is less than about 1.5 M Na ion, typically about 0.01 to 1.0 M Na ion concentration (or other salts) at pH 7.0 to 8.3 and the temperature is at least about 30°C for short polynucleotides (e.g., 10 to 50 nucleotides) and at least about 60°C for long polynucleotides (e.g., greater than 50 nucleotides)— for example,“stringent conditions” can include hybridization in 50% formamide, 1 M NaCl, 1% SDS at 37°C, and three washes for 15 min each in 0.lxSSC/l% SDS at 60°C to 65°C.
- temperatures of about 65°C, 60°C, 55°C, or 42°C may be used.
- SSC concentration may be varied from about 0.1 to 2xSSC, with SDS being present at about 0.1%.
- wash temperatures are typically selected to be about 5°C to 20°C lower than the thermal melting point for the specific sequence at a defined ionic strength and pH.
- the Tm is the temperature (under defined ionic strength and pH) at which 50% of the target sequence hybridizes to a perfectly matched probe.
- blocking reagents are used to block non-specific hybridization.
- blocking reagents include, for instance, sheared and denatured salmon sperm DNA at about 100-200 pg/ml.
- Organic solvent such as formamide at a concentration of about 35-50% v/v, may also be used under particular circumstances, such as for RNA:DNA hybridizations. Useful variations on these wash conditions will be readily apparent to those of ordinary skill in the art.
- the terms“percent identity,” percentage of sequence identity,” and“% identity,” as applied to polynucleotide sequences, refer to the percentage of residue matches between at least two polynucleotide sequences aligned using a standardized algorithm. Such an algorithm may insert, in a standardized and reproducible way, gaps in the sequences being compared in order to optimize alignment between two sequences, and therefore achieve a more meaningful
- Percent identity may be measured over the length of an entire defined polynucleotide sequence, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined polynucleotide sequence, for instance, a fragment of at least 45, at least 60, at least 90, at least 120, at least 150, at least 210 or at least 450 contiguous residues.
- Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures or Sequence Listing, may be used to describe a length over which percentage identity may be measured.
- the percentage of sequence identity is calculated by comparing two optimally aligned sequences over the window of comparison, determining the number of matched positions (at which identical residues occur in both polypeptide sequences), dividing the number of matched positions by the total number of positions in the window of comparison (i.e., the window size), and multiplying the result by 100 to yield the percentage of sequence identity.
- the shortest sequence defines the length of the window of comparison. Conservative substitutions are not considered when calculating sequence identity.
- “Percent (%) sequence identity” and“percent (%) identity” with respect to the polypeptide sequences identified herein, is defined as the percentage of amino acid residues in a query sequence that are identical with the amino acid residues of a second, reference polypeptide sequence of comparable length or a portion thereof, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity, thereby resulting in optimal alignment. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software.
- Percent identity may be measured over the length of an entire defined polypeptide sequence, or may be measured over a shorter length, for example, over the length of a fragment taken from a larger, defined polypeptide sequence, for instance, a fragment of at least 15, at least 20, at least 30, at least 40, at least 50, at least 70 or at least 150 contiguous residues.
- Such lengths are exemplary only, and it is understood that any fragment length supported by the sequences shown herein, in the tables, figures or Sequence Listing, may be used to describe a length over which percentage identity may be measured.
- “Repetitiveness” used in the context of polynucleotide sequences refers to the degree of internal homology in the sequence such as, for example, the frequency of identical nucleotide sequences of a given length. Repetitiveness can, for example, be measured by analyzing the frequency of identical sequences.
- RNA messenger RNA
- tRNA transfer RNA
- shRNA small hairpin RNA
- siRNA small interfering RNA
- expression produces a "gene product.”
- a gene product can be either a nucleic acid, e.g., a messenger RNA produced by transcription of a gene, or a polypeptide which is translated from a transcript.
- Gene products described herein further include nucleic acids with post transcriptional modifications, e.g., polyadenylation or splicing, or polypeptides with post translational modifications, e.g., methylation, glycosylation, the addition of lipids, association with other protein subunits, or proteolytic cleavage.
- post transcriptional modifications e.g., polyadenylation or splicing
- polypeptides with post translational modifications e.g., methylation, glycosylation, the addition of lipids, association with other protein subunits, or proteolytic cleavage.
- A“vector” or“expression vector” are used interchangeably and refers to a nucleic acid molecule, preferably self-replicating in an appropriate host, which transfers an inserted nucleic acid molecule into and/or between host cells.
- the term includes vectors that function primarily for insertion of DNA or RNA into a cell, replication of vectors that function primarily for the replication of DNA or RNA, and expression vectors that function for transcription and/or translation of the DNA or RNA. Also included are vectors that provide more than one of the above functions.
- An“expression vector” is a polynucleotide which, when introduced into an appropriate host cell, can be transcribed and translated into a polypeptide(s).
- An“expression system” usually connotes a suitable host cell comprised of an expression vector that can function to yield a desired expression product.
- “Serum degradation resistance,” as applied to a polypeptide, refers to the ability of the polypeptides to withstand degradation in blood or components thereof, which typically involves proteases in the serum or plasma.
- the serum degradation resistance can be measured by combining the protein with human (or mouse, rat, dog, monkey, as appropriate) serum or plasma, typically for a range of days (e.g. 0.25, 0.5, 1, 2, 4, 8, 16 days), typically at about 37°C.
- the samples for these time points can be run on a Western blot assay and the protein is detected with an antibody.
- the antibody can be to a tag in the protein. If the protein shows a single band on the western, where the protein’s size is identical to that of the injected protein, then no degradation has occurred.
- the time point where 50% of the protein is degraded is the serum degradation half-life or“serum half-life” of the protein.
- the terms“fr / 2”,“half-life”,“terminal half-life”,“elimination half-life” and“circulating half-life” are used interchangeably herein and, as used herein means the terminal half-life calculated as Ih /K ⁇ . 3 ⁇ 4 is the terminal elimination rate constant calculated by linear regression of the terminal linear portion of the log concentration vs. time curve.
- Half-life typically refers to the time required for half the quantity of an administered substance deposited in a living organism to be metabolized or eliminated by normal biological processes. When a clearance curve of a given polypeptide is constructed as a function of time, the curve is usually biphasic with a rapid a-phase and longer beta-phase.
- the typical beta-phase half-life of a human antibody in humans is 21 days. Half-life can be measured using timed samples from any body fluid, but is most typically measured in plasma samples.
- molecular weight generally refers to the sum of atomic weights of the constituent atoms in a molecule. Molecular weight can be determined theoretically by summing the atomic masses of the constituent atoms in a molecule. When applied in the context of a polypeptide, the molecular weight is calculated by adding, based on amino acid composition, the molecular weight of each type of amino acid in the composition or by estimation from
- the calculated molecular weight of a molecule can differ from the apparent molecular weight of a molecule, which generally refers to the molecular weight of a molecule as determined by one or more analytical techniques.“Apparent molecular weight factor” and“apparent molecular weight” are related terms and when used in the context of a polypeptide, the terms refer to a measure of the relative increase or decrease in apparent molecular weight exhibited by a particular amino acid or polypeptide sequence.
- the apparent molecular weight can be determined, for example, using size exclusion chromatography (SEC) or similar methods by comparing to globular protein standards, as measured in“apparent kD” units.
- SEC size exclusion chromatography
- the apparent molecular weight factor is the ratio between the apparent molecular weight and the“molecular weight”; the latter is calculated by adding, based on amino acid composition as described above, or by estimation from comparison to molecular weight standards in an SDS electrophoresis gel. The determination of apparent molecular weight and apparent molecular weight factor is described in US patent number 8,673,860.
- the terms“hydrodynamic radius” or“Stokes radius” is the effective radius (R h in nm) of a molecule in a solution measured by assuming that it is a body moving through the solution and resisted by the solution’s viscosity.
- the hydrodynamic radius measurements of the XTEN polypeptides correlate with the“apparent molecular weight factor” which is a more intuitive measure.
- The“hydrodynamic radius” of a protein affects its rate of diffusion in aqueous solution as well as its ability to migrate in gels of macromolecules.
- the hydrodynamic radius of a protein is determined by its molecular weight as well as by its structure, including shape and compactness.
- “Diffusion coefficient” means the magnitude of the molar flux through a surface per unit concentration gradient out-of-plane. In dilute species transport, the flux due to diffusion is given by Fick's first law, which only depends on a single property of the solute's interaction with the solvent: the diffusion coefficient.
- “Physiological conditions” refers to a set of conditions in a living host as well as in vitro conditions, including temperature, salt concentration, pH, that mimic those conditions of a living subject.
- a host of physiologically relevant conditions for use in in vitro assays have been established.
- a physiological buffer contains a physiological concentration of salt and is adjusted to a neutral pH ranging from about 6.5 to about 7.8, and preferably from about 7.0 to about 7.5.
- a variety of physiological buffers are listed in Sambrook et al. (2001).
- Physiologically relevant temperature ranges from about 25°C to about 38°C, and preferably from about 35°C to about 37°C.
- binding moiety is used herein in the broadest sense, and is specifically intended to include the categories of cytokines, cell receptors, antibodies or antibody fragments that have specific affinity for an antigen or ligand such as cell-surface receptors, target cell markers, or antigens or glycoproteins, oligonucleotides, enzymatic substrates, antigenic determinants, or binding sites that may be present in or on the surface of a tissue or cell.
- antibody is used herein in the broadest sense and encompasses various antibody structures, including but not limited to monoclonal antibodies, polyclonal antibodies, multispecific antibodies (e.g., bispecific antibodies), and antibody fragments so long as they exhibit the desired antigen-binding activity.
- the full-length antibodies may be for example monoclonal, recombinant, chimeric, deimmunized, humanized and human antibodies.
- the term“monoclonal antibody” as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical and/or bind the same epitope, except for possible variant antibodies, e.g., containing naturally occurring mutations or arising during production of a monoclonal antibody preparation, such variants generally being present in minor amounts.
- polyclonal antibody preparations typically include different antibodies directed against different determinants (epitopes)
- each monoclonal antibody of a monoclonal antibody preparation is directed against a single determinant on an antigen.
- the modifier “monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by a variety of techniques, including but not limited to the hybridoma method, recombinant DNA methods, phage-display methods, and methods utilizing transgenic animals containing all or part of the human immunoglobulin loci, such methods and other exemplary methods for making monoclonal antibodies being known in the art or described herein.
- an“antibody fragment” refers to a molecule other than an intact antibody that comprises a portion of an intact antibody and that binds the antigen to which the intact antibody binds.
- antibody fragments include but are not limited to Fv, Fab, Fab', Fab'-SH, F(ab')2, diabodies, single chain diabodies, linear antibodies, a single domain antibody, a single domain camelid antibody, single-chain variable fragment (scFv) antibody molecules, and multispecific antibodies formed from antibody fragments.
- “scFv” or“single chain fragment variable” are used interchangeably herein to refer to an antibody fragment format comprising regions of variable heavy (“VH”) and variable light (“VL”) chains or two copies of a VH or VL chain, which are joined together by a short flexible peptide linker.
- the scFv is not actually a fragment of an antibody, but is a fusion protein of the variable regions of the heavy (VH) and light chains (VL) of immunoglobulins, and can be easily expressed in functional form in E. coli in either N- to C-termnus orientation; VL-VH or VH-VL.
- antigen “antigen”,“target cell marker” and“ligand” are used interchangeably herein to refer to the structure or binding determinant that a binding moiety, an antibody, antibody fragment or an antibody fragment-based molecule binds to or has binding specificity against.
- epitope refers to the particular site on an antigen molecule to which an antibody, antibody fragment, or binding moiety binds.
- An epitope is a ligand of an antibody, antibody fragment, or a binding moiety.
- “CD3” or“cluster of differentiation 3” means the T cell surface antigen CD3 complex, which includes in individual form or independently combined form all known CD3 subunits, for example CD3 epsilon, CD3 delta, CD3 gamma, CD3 zeta, CD3 alpha and CD3 beta.
- CD3 epsilon, CD3 delta, CD3 gamma, CD3 zeta, CD3 alpha and CD3 beta The extracellular domains of CD3 epsilon, gamma and delta contain an
- immunoglobulin-like domain so are therefore considered part of the immunoglobulin superfamily.
- the terms“specific binding” or“specifically bind” or“binding specificity” are used interchangeably herein to refer to the high degree of binding affinity of a binding moiety to its corresponding target. Typically, specific binding as measured by one or more of the assays disclosed herein would have a dissociation constant or IQ of less than about 10 6 M; e.g, 10 7 M - 10 12 M.
- “Affinity” refers to the strength of the sum total of noncovalent interactions between a single binding site of a molecule (e.g., an antibody) and its binding partner (e.g., an antigen). Unless indicated otherwise, as used herein,“binding affinity” refers to intrinsic binding affinity which reflects a 1 : 1 interaction between members of a binding pair (e.g., antibody and antigen).
- the affinity of a molecule X for its partner Y can generally be represented by the dissociation constant (IQ).
- a greater binding affinity” or“increased binding affinity” means a lower IQ value; e.g., 1 x 10 9 M is a greater binding affinity than 1 x 10 8 M, while a“lower binding affinity” means a greater IQ value; e.g., 1 x 10 7 M is a lower binding affinity than 1 x 10 8 M.
- “Inhibition constant”, or“K” are used interchangeably and mean the dissociation constant of the enzyme-inhibitor complex, or the reciprocal of the binding affinity of the inhibitor to the enzyme.
- the term“kon”, as used herein, is intended to refer to the on rate constant for association of an antibody to the antigen to form the antibody/antigen complex as is known in the art.
- the term“k 0ff ”, as used herein, is intended to refer to the off rate constant for dissociation of an antibody from the antibody/antigen complex as is known in the art.
- Techniques such as flow cytometry or surface plasmon resonance can be used to detect binding events.
- the assays may comprise soluble antigens or receptor molecules, or may determine the binding to cell-expressed receptors.
- Such assays may include cell-based assays, including assays for proliferation, cell death, apoptosis and cell migration.
- binding affinity of the subject compositions for the target ligands can be assayed using binding or competitive binding assays, such as Biacore assays with chip-bound receptors or binding proteins or ELISA assays, as described in US Patent 5,534,617, assays described in the
- radio-receptor assays or other assays known in the art.
- the binding affinity constant can then be determined using standard methods, such as Scatchard analysis, as described by van Zoelen, et al., Trends Pharmacol Sciences (1998) 19)12):487, or other methods known in the art.
- antagonists includes any molecule that partially or fully blocks, inhibits, or neutralizes a biological activity of a native polypeptide disclosed herein.
- Methods for identifying antagonists of a polypeptide may comprise contacting a native polypeptide with a candidate antagonist molecule and measuring a detectable change in one or more biological activities normally associated with the native polypeptide.
- antagonists may include proteins, nucleic acids, carbohydrates, antibodies or any other molecules that decrease the effect of a biologically active protein.
- A“target cell marker” refers to a molecule expressed by a target cell including but not limited to cell-surface receptors, cytokine receptors, antigens, tumor-associated antigens, glycoproteins, oligonucleotides, enzymatic substrates, antigenic determinants, or binding sites that may be present in the on the surface of a target tissue or cell that may serve as ligands for a binding moiety.
- target cell markers include the target markers of Table 5.
- a "target tissue” refers to a tissue that is the cause of or is part of a disease condition such as, but not limited to cancer or inflammatory conditions.
- Sources of diseased target tissue include a body organ, a tumor, a cancerous cell or population of cancerous cells or cells that form a matrix or are found in association with a population of cancerous cells, bone, skin, cells that produce cytokines or factors contributing to a disease condition.
- A“defined medium” refers to a medium comprising nutritional and hormonal requirements necessary for the survival and/or growth of the cells in culture such that the components of the medium are known. Traditionally, the defined medium has been formulated by the addition of nutritional and growth factors necessary for growth and/or survival.
- the defined medium provides at least one component from one or more of the following categories: a) all essential amino acids, and usually the basic set of twenty amino acids plus cysteine; b) an energy source, usually in the form of a carbohydrate such as glucose; c) vitamins and/or other organic compounds required at low concentrations; d) free fatty acids; and e) trace elements, where trace elements are defined as inorganic compounds or naturally occurring elements that are typically required at very low concentrations, usually in the micromolar range.
- the defined medium may also optionally be supplemented with one or more components from any of the following categories: a) one or more mitogenic agents; b) salts and buffers as, for example, calcium, magnesium, and phosphate; c) nucleosides and bases such as, for example, adenosine and thymidine, hypoxanthine; and d) protein and tissue hydrolysates.
- agonist is used in the broadest sense and includes any molecule that mimics a biological activity of a native polypeptide disclosed herein. Suitable agonist molecules specifically include agonist antibodies or antibody fragments, fragments or amino acid sequence variants of native polypeptides, peptides, small organic molecules, etc. Methods for identifying agonists of a native polypeptide may comprise contacting a native polypeptide with a candidate agonist molecule and measuring a detectable change in one or more biological activities normally associated with the native polypeptide.
- treatment or“treating,” or“palliating” or“ameliorating” is used interchangeably herein.
- therapeutic benefit is meant eradication or amelioration of the underlying disorder being treated.
- a therapeutic benefit is achieved with the eradication or amelioration of one or more of the physiological symptoms or improvement in one or more clinical parameters associated with the underlying disorder such that an improvement is observed in the subject, notwithstanding that the subject may still be afflicted with the underlying disorder.
- compositions may be administered to a subject at risk of developing a particular disease, or to a subject reporting one or more of the physiological symptoms of a disease, even though a diagnosis of this disease may not have been made.
- A“therapeutic effect” or“therapeutic benefit,” as used herein, refers to a physiologic effect, including but not limited to the mitigation, amelioration, or prevention of disease or an improvement in one or more clinical parameters associated with the underlying disorder in humans or other animals, or to otherwise enhance physical or mental wellbeing of humans or animals, resulting from administration of a polypeptide of the disclosure other than the ability to induce the production of an antibody against an antigenic epitope possessed by the biologically active protein.
- compositions may be administered to a subject at risk of developing a particular disease, a recurrence of a former disease, condition or symptom of the disease, or to a subject reporting one or more of the physiological symptoms of a disease, even though a diagnosis of this disease may not have been made.
- therapeutically effective amount and“therapeutically effective dose”, as used herein, refer to an amount of a drug or a biologically active protein, either alone or as a part of a polypeptide composition, that is capable of having any detectable, beneficial effect on any symptom, aspect, measured parameter or characteristics of a disease state or condition when administered in one or repeated doses to a subject. Such effect need not be absolute to be beneficial. Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein.
- the term“equivalent molar dose” means that the amounts of materials administered to a subject have an equivalent amount of moles, based on the molecular weight of the material used in the dose.
- therapeutically effective and non-toxic dose refers to a tolerable dose of the compositions as defined herein that is high enough to cause depletion of tumor or cancer cells, tumor elimination, tumor shrinkage or stabilization of disease without or essentially without major toxic effects in the subject.
- therapeutically effective and non-toxic doses may be determined by dose escalation studies described in the art and should be below the dose inducing severe adverse side effects.
- dose regimen refers to a schedule for consecutively administered multiple doses (i.e., at least two or more) of a composition, wherein the doses are given in therapeutically effective amounts to result in sustained beneficial effect on any symptom, aspect, measured parameter, endpoint, or characteristic of a disease state or condition.
- cancer refers to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth/proliferation.
- cancer include, but are not limited to, carcinomas, Hodgkin's lymphoma, non-Hodgkin's lymphoma, B cell lymphoma, T-cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, colon cancer, prostate cancer, head and neck cancer, any form of skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervical cancer, colorectal cancer, an epithelia intraperitoneal malignancy with malignant ascites, uterine cancer, mesothelioma in the peritoneum kidney cancers, lung cancer, small-cell lung cancer, non-small cell lung
- adenocarcinoma adenocarcinoma, sarcomas of any origin, primary hematologic malignancies including acute or chronic lymphocytic leukemias, acute or chronic myelogenous leukemias, myeloproliferative neoplastic disorders, or myelodysplastic disorders, myasthenia gravis, Morbus Basedow,
- Tumor-specific marker refers to an antigen that is found on or in a cancer cell.
- “Target cell” refers to a cell that has the ligand of a binding moiety, an antibody or antibody fragment of the subject compositions and is associated with or causes a disease or pathologic condition, including cancer cells, tumor cells, and inflammatory cells.
- the ligand of a target cell is referred to herein as a“target cell marker” or“target cell antigen” and includes, but is not limited to, cell surface receptors or antigens, cytokines, cytokine receptors, MHC proteins, and cytosol proteins or peptides that are exogenously presented.
- “target cell” would not include an effector cell.
- Host cells can be cultured in a variety of media.
- Commercially available media such as Ham's F10 (Sigma), Minimal Essential Medium (MEM, Sigma), RPMI-1640 (Sigma), and Dulbecco's Modified Eagle's Medium (DMEM, Sigma) are suitable for culturing eukaryotic cells.
- animal cells can be grown in a defined medium that lacks serum but is supplemented with hormones, growth factors or any other factors necessary for the survival and/or growth of a particular cell type.
- a defined medium supporting cell survival maintains the viability, morphology, capacity to metabolize and potentially, capacity of the cell to differentiate
- a defined medium promoting cell growth provides all chemicals necessary for cell proliferation or multiplication.
- the general parameters governing mammalian cell survival and growth in vitro are well established in the art. Physicochemical parameters which may be controlled in different cell culture systems are, e.g., pH, p0 2 , temperature, and osmolarity.
- the nutritional requirements of cells are usually provided in standard media formulations developed to provide an optimal environment. Nutrients can be divided into several categories: amino acids and their derivatives, carbohydrates, sugars, fatty acids, complex lipids, nucleic acid derivatives and vitamins.
- cells Apart from nutrients for maintaining cell metabolism, most cells also require one or more hormones from at least one of the following groups: steroids, prostaglandins, growth factors, pituitary hormones, and peptide hormones to proliferate in serum-free media (Sato, G. H., et al. in “Growth of Cells in Hormonally Defined Media”, Cold Spring Harbor Press, N.Y., 1982).
- hormones from at least one of the following groups: steroids, prostaglandins, growth factors, pituitary hormones, and peptide hormones to proliferate in serum-free media (Sato, G. H., et al. in “Growth of Cells in Hormonally Defined Media”, Cold Spring Harbor Press, N.Y., 1982).
- cells may require transport proteins such as transferrin (plasma iron transport protein), ceruloplasmin (a copper transport protein), and high-density lipoprotein (a lipid carrier) for survival and growth in vitro.
- transferrin plasm
- Growth media for growth of prokaryotic host cells include nutrient broths (liquid nutrient medium) or LB medium (Luria Bertani). Suitable media include defined and undefined media. In general, media contains a carbon source such as glucose needed for bacterial growth, water, and salts. Media may also include a source of amino acids and nitrogen, for example beef or yeast extract (in an undefined medium) or known quantities of amino acids (in a defined medium).
- the growth medium is LB broth, for example LB Miller broth or LB Lennox broth. LB broth comprises peptone (enzymatic digestion product of casein), yeast extract and sodium chloride.
- a selective medium is used which comprises an antibiotic. In this medium, only the desired cells possessing resistance to the antibiotic will grow.
- the present disclosure provides recombinant polypeptides comprising at least three categories of components; binding moieties, release segments (RS) and bulking moieties; each of which are described more fully herein.
- the disclosure also provides configurations of recombinant polypeptides that are specifically designed to confer pharmaceutical and therapeutic advantageous properties on the compositions in comparison to conventional antibody- and cytokine-based therapeutics.
- the disclosure provides recombinant polypeptide compositions having a first binding moiety (FBM) designed to bind a target ligand, a release segment that is a substrate for a mammalian protease, and a bulking moiety such as an XTEN, wherein the FBM is an antibody, a cytokine, a cell receptor, or a fragment thereof.
- FBM first binding moiety
- the recombinant polypeptides comprising a single binding moiety may be designed to confer a prodrug property on the composition in order to render it less reactive when in the circulation or when exposed to healthy tissues, but when in proximity to diseased tissues or cells that produce or have co-localized proteases that are capable of cleaving the RS incorporated into the recombinant polypeptide, the FBM and XTEN are released such that the XTEN no longer shields the FBM and the FBM regains its full potential for binding affinity for its ligand.
- FBM suitable for incorporation into the subject compositions include cytokines, chemokines and interleukins (such as but not limited to interleukin- 1 (IL-l), IL-12, and IL-18, tumor necrosis factor (TNF), interferon gamma (IFN-gamma), granulocyte-macrophage colony stimulating factor, C-C chemokines (RANTES, monocyte chemoattractant protein or MCP-l, monocyte inflammatory protein or MIP-la, and MP b), C-X-C chemokines (IL-8 also called growth related oncogene or GRO/KC), C chemokines (lymphotactin), and CXXXC chemokines (ffactalkine).
- IL-l interleukin- 1
- IL-12 tumor necrosis factor
- IFN-gamma interferon gamma
- granulocyte-macrophage colony stimulating factor granulocyte-macrophage colony
- FBM suitable for incorporation into the subject compositions include antibody fragments that have binding affinity tumor associated antigens, including but not limited to the target cell markers of Table 5.
- the recombinant polypeptides further comprise RS1 of Tables 1 or 2 and XTEN of Tables 8 or 10, or sequence variants thereof (as described more fully, below).
- the disclosure provides recombinant polypeptide compositions comprising two antibody fragments, one or more release segments, and one or more XTEN that are activatable by cleavage of the release segments such that the antibody fragments are released from the composition and regain their full potential for binding affinity for their respective ligands.
- Such recombinant polypeptides having two antibody fragments are also referred to herein as activatable antibody compositions (AAC).
- AAC activatable antibody compositions
- an AAC having a first binding moiety (FBM) fused to a second binding moiety (SBM) in which the FBM and SBM are both antibody fragments further comprises at least a first release segment and at least a first XTEN.
- the AAC constructs described herein confer multiple therapeutic advantages over traditional monoclonal antibodies and other smaller bispecific molecules.
- conditional activation of the AAC of the present disclosure The intact, uncleaved AAC have a reduced ability to bind their intended target cell markers due to the shielding effect of the bulky, unstructured XTEN tethered to the AAC by the release segment.
- the specific activity to non-diseased, normal tissue of the exemplary compositions of the disclosure is significantly reduced when compared to that of analogous antibodies and antibody fragments.
- AAC polypeptides to activate at their desired site of action (e.g., the proximity of a diseased tissue such as a tumor or cancer cell) while remaining essentially inactive during their progress to this site is an advance in the field of immune-oncologic therapeutics, offering the promise of potent and specific therapeutics with improved therapeutic index, as well as a readily designable and manufacturable format that can be applied to multiple target cells such as those disclosed herein.
- the AAC described herein with a FBM and SBM antibody fragment are designed to allow specific targeting and killing of cells expressing a target cell marker by recruiting cytotoxic effector cells, e.g., T cells.
- the intact, uncleaved AAC is in a prodrug form in that the XTEN shields the binding moieties, reducing their binding affinity towards their ligands until released from the composition by protease cleavage of any of the protease cleavage sites located within the RS. This improves the specificity of the composition towards diseased tissues or cells compared to bispecific T-cell engager therapeutics that are not in a prodrug format.
- the bispecific binding moieties and XTEN of the AAC constructs are released upon cleavage of the RS and the fused FBM and SBM antibody fragments can crosslink cytotoxic effector cells with cells expressing a target cell marker in a highly specific fashion, thereby directing the cytotoxic potential of the T cell towards the target cell.
- the fused FBM-SBM antibody fragment having a much smaller size compared to the uncleaved AAC, is then free to permeate the target sites, e.g., a tumor mass, in order to reach and bind to and link together the target cell and cytotoxic T cell. Additionally, the entire process is not dependent upon internalization of the composition.
- the AAC constructs described herein engage cytotoxic T cells via binding to the surface-expressed CD3, which forms part of the T cell receptor complex, causing T cell activation that mediates the subsequent lysis of the cell expressing the particular target cell marker.
- AAC are contemplated to display strong, specific and efficient target cell killing.
- the AAC described herein stimulate target cell killing by cytotoxic effector cells to eliminate the cells expressing the particular target cell marker bound by the target-specific targeting moiety of the AAC in protease-rich
- proteases known to be associated with diseased cells or tissues include but are not limited to serine proteases, cysteine proteases, aspartate proteases, and metalloproteases.
- the AAC of the disclosure may confer further therapeutic and pharmaceutical advantages over recognized monoclonal antibodies and other smaller bispecific molecules.
- bi-specific molecules are designed to bind to a target cell having a cell-specific marker associated with a pathogenic cell. Toxicity and undesirable side effects are possible when, in some cases, healthy cells or tissues express the same marker as the target cell.
- One benefit to an AAC of the disclosure is that binding to CD3 and the target cells is enhanced upon the release of the binding moieties by a protease expressed by, or in association with, the disease tissue harboring the target cell, such as a tumor cell, permitting the binding of the released fused FBM and SBM antibody fragments to the target cell marker and the effector cell, creating an immunologic synapse.
- the two antibody fragment binding moieties of the AAC are fused to each other by a short linker, and are, in turn, connected to the XTEN by the release segment having one or more cleavage sites to allow the release of the fused FBM and SBM antibody fragments from the XTEN upon cleavage by one or more proteases co-localized with the diseased tissue.
- the binding moieties of the AAC may be in any format of a single chain binding moiety including Fv, Fab, Fab', Fab'-SH, F(ab')2, linear antibodies, a single domain antibody, a single domain antibody, and single-chain variable fragment antibody molecules (scFv).
- the two fused antibody fragments FBM and SBM can also be configured in a single chain diabody format by the selective arrangement of the VL and VH and the linkers that join them.
- Polypeptide compositions capable of binding diseased tissues such as tumors have an optimal size for enhanced tissue penetration and distribution of the therapeutic. However, this is counterbalanced by the desire to have reduced first pass renal clearance as well as reduced extravasation from the circulation in normal tissue. Because the kidney generally filters out molecules below about 50 kDa, efforts to reduce clearance in the design of protein therapeutics have focused on increasing molecular size through fusions with proteins like albumin or the addition of polyethylene glycol polymers. However, while increasing the size of a protein therapeutic may prevent renal clearance and extravasation, the larger size also hinders penetration of the molecule into the target tissues.
- Exemplary AAC described herein avoid this by fusion of the binding moieties with release segments and bulking moieties such as XTEN, which greatly increase the apparent molecular weight of the composition (described more fully, below), and will prevent rapid renal clearance and extravasation in normal vasculature while having the ability to have the XTEN be released by the action of target tissue associated proteases on the RS, resulting in the release of the binding moieties having a small size, allowing for enhanced tissue penetration and distribution and optimal efficacy.
- the XTEN confers a number of favorable properties on the AAC embodiments, including but not limited to increased half-life, reduced extravasation in normal vasculature, increased solubility, reduced binding to healthy tissues, increased therapeutic index, and a prodrug format.
- the present disclosure provides AAC having a single chain binding moiety polypeptide directed to a ligand of a target cell and another single chain binding moiety polypeptide directed to an effector cell ligand, such as a CD3 antigen, making the configuration of this component of the AAC similar to bifunctional binding compositions such as blinatumomab (referred to as a BiTE ® composition).
- a representative target cell marker is an antigen found on the surface of a cancer cell, e.g., EGFR, EpCAM, HER2, or any of the target markers of Table 5.
- the AAC polypeptides comprise a FBM and a SBM, which can be scFv linked through a flexible linker such as those of Table 7, or can be configured as a single chain diabody. While each of the FBM and SBM have binding affinity for their respective ligands comparable to typical single chain binding moieties, the XTEN of the intact, uncleaved AAC composition serves to greatly reduce the ability of both scFv of the intact composition to bind their respective ligands by steric hindrance due to the ability of the flexible, unstructured XTEN to surround the binding moieties of the composition.
- the fused binding moieties separate from the XTEN, allowing the fused anti-target binding moiety and the anti-CD3 binding moiety to cooperatively bind their respective ligands and form an immunologic synapse between the target cell and the effector T cell.
- the recombinant polypeptide contains a single anti-target binding moiety, such as a cytokine or anti-cytokine
- the released binding moiety would similarly have an enhanced ability to bind its ligand upon release from the intact composition by action of a protease on the RS.
- the disclosure provides release segment (RS) peptides that are substrates for one or more mammalian proteases associated with or produced by disease tissues or cells found in proximity to disease tissues.
- proteases can include, but not be limited to the classes of proteases such as metalloproteinases, cysteine proteases, aspartate proteases, and serine proteases, including, but not limited to, the proteases of Table 3.
- the RS are useful for, amongst other things, incorporation into the subject recombinant polypeptides, conferring a prodrug format that can be activated by the cleavage of the RS by mammalian proteases.
- the RS are incorporated into the subject recombinant polypeptide compositions, linking the incorporated binding moieties to the XTEN (the configurations of which are described more fully, below) such that upon cleavage of the RS by action of the one or more proteases for which the RS are substrates, the binding moieties and XTEN are released from the composition and the binding moieties, no longer shielded by the XTEN, regain their full potential to bind their ligands.
- the compositions are also referred to herein as activatable antibody compositions (AAC).
- the disclosure provides activatable recombinant polypeptides comprising a first release segment (RS1) sequence having at least 88%, or at least 94%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 1, wherein the RS1 is a substrate for one or more mammalian proteases.
- RS1 first release segment
- the disclosure provides activatable recombinant polypeptides comprising a RS1 and a second release segment (RS2) sequence, each having at least 88%, or at least 94%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 1, wherein the RS1 and the RS2 each are a substrate for one or more mammalian proteases.
- RS1 and the RS2 each are a substrate for one or more mammalian proteases.
- disclosure provides activatable recombinant polypeptides comprising a first RS (RS1) sequence having at least 90%, at least 93%, at least 97%, or 100% identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 2, wherein the RS is a substrate for one or more mammalian proteases.
- RS1 RS1 sequence having at least 90%, at least 93%, at least 97%, or 100% identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 2, wherein the RS is a substrate for one or more mammalian proteases.
- the disclosure provides activatable recombinant polypeptides comprising a RS1 and a second release segment (RS2) sequence, each having at least 88%, or at least 94%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 2, wherein the RS1 and the RS2 are each a substrate for one or more mammalian proteases.
- the two release segments can be identical or the sequences can be different.
- the present disclosure contemplates release segments that are substrates for one, two or three different classes of proteases selected from metalloproteinases, cysteine proteases, aspartate proteases, and serine proteases, including the proteases of Table 3.
- the RS serve as substrates for proteases found in close association with or are co-localized with disease tissues or cells, such as but not limited to tumors, cancer cells, and inflammatory tissues, and upon cleavage of the RS, the binding moieties that are otherwise shielded by the XTEN of the subject recombinant polypeptide compositions (and thus have a lower binding affinity for their respective ligands) are released from the composition and regain their full potential to bind the target and/or effector cell ligands.
- disease tissues or cells such as but not limited to tumors, cancer cells, and inflammatory tissues
- the RS of the subject recombinant polypeptide compositions comprises an amino acid sequence that is a substrate for a cellular protease located within a targeted cell, including but not limited to the proteases of Table 3.
- the RS that are substrates for two or three classes of proteases were designed with sequences that are capable of being cleaved in different locations of the RS sequence by the different proteases, with a representative example depicted in FIG. 36.
- the RS that are substrates for two, three, or more classes of proteases have two, three, or a plurality of distinct cleavage sites in the RS sequence, but cleavage by a single protease nevertheless results in the release of the binding moieties and the XTEN from the recombinant polypeptide composition comprising the RS.
- the RS of the disclosure for incorporation into the subject recombinant polypeptide compositions is a substrate for one or more proteases selected from the group consisting of meprin, neprilysin (CD 10), PSMA, BMP-1, A disintegrin and
- ADAMs metalloproteinases
- ADAM8 ADAM9 ADAM 10
- ADAM 12 AD AMI 5
- AD AMI 7 TACE
- ADAM 19 ADAM28 (MDC-L)
- ADAM with thrombospondin motifs ADAMTS
- ADAMTSl ADAMTS4
- MMP-1 collagenase 1
- MMP-1 matrix metalloproteinase- 1
- MMP-2 matrix metalloproteinase-2
- MMP-3 matrix metalloproteinase-3
- MMP-7 matrix metalloproteinase-7
- Matrilysin 1 matrix metalloproteinases
- MMP-8 collagenase 2
- MMP-9 matrix metalloproteinase-9
- MMP-9 gelatinase B
- MMP-10 matrix metalloproteinase- 10
- MMP-11 matrix metalloproteinase- 11
- MMP-12 matrix metalloproteinase- 12
- MMP-13 collagenase 3
- matrix metalloproteinase- 14 matrix metalloproteinase- 14
- matrix metalloproteinase- 15 matrix metalloproteinase- 15
- MMP-15 matrix metalloproteinase- 15
- MMP-19 matrix metalloproteinase-23
- MMP-23 CA-MMP
- matrix metalloproteinase-24 MMP-24, MT5-MMP
- MMP-26 matrilysin 2
- MMP-27 metalloproteinase-27
- CMMP metalloproteinase-27
- legumain cathepsin B, cathepsin C, cathepsin K, cathepsin L, cathepsin S, cathepsin X, cathepsin D, cathepsin E, secretase, urokinase (uPA), tissue-type plasminogen activator (tPA), plasmin, thrombin, prostate-specific antigen (PSA, KLK3), human neutrophil elastase (HNE), elastase, tryptase, Type II transmembrane serine proteases (TTSPs), DESC1, hepsin (HPN), matriptase, matriptase-2, TMPRSS2, TMPRSS3, TMPRSS4 (CAP2), fibroblast activation protein (FAP), kallikrein-related peptidase (K
- the RS is a substrate for ADAM 17. In one embodiment, the RS is a substrate for BMP-l. In one embodiment, the RS is a substrate for cathepsin. In one embodiment, the RS is a substrate for HtrAl. In one embodiment, the RS is a substrate for legumain. In one embodiment, the RS is a substrate for MMP-l. In one embodiment, the RS is a substrate for MMP-2. In one embodiment, the RS is a substrate for MMP-7. In one embodiment, the RS is a substrate for MMP-9. In one embodiment, the RS is a substrate for MMP-l 1. In one embodiment, the RS is a substrate for MMP-l 4.
- the RS is a substrate for uPA. In one embodiment, the RS is a substrate for matriptase. In one embodiment, the RS is a substrate for MT-SP1. In one embodiment, the RS is a substrate for neutrophil elastase. In one embodiment, the RS is a substrate for thrombin. In one embodiment RS is a substrate for TMPRSS3. In one embodiment, the RS is a substrate for TMPRSS4.
- the RS of the subject recombinant polypeptide compositions is a substrate for at least two proteases selected from the group consisting of legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, and matriptase.
- the RS of the subject recombinant polypeptide compositions is a substrate for legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, and matriptase.
- the RS for incorporation into the subject recombinant polypeptides can be designed to be selectively sensitive in order to have different rates of cleavage and different cleavage efficiencies to the various proteases for which they are substrates.
- the disclosure provides RS that have had the individual amino acid sequences engineered to have a higher or lower cleavage efficiency for a given protease in order to ensure that the recombinant polypeptide is preferentially converted from the prodrug form to the active form (i.e., by the separation and release of the binding moieties and XTEN from the recombinant polypeptide after cleavage of the RS) when in proximity to the target cell or tissue and its co-localized proteases compared to the rate of cleavage of the RS in healthy tissue or the circulation such that the released antibody fragment binding moieties have a greater ability to bind to ligands in the diseased tissues compared to the prodrug form that remains in circulation.
- the therapeutic index of the resulting compositions can be
- cleavage efficiency is defined as the log 2 value of the ratio of the percentage of the test substrate comprising the RS cleaved to the percentage of the control substrate AC1611 cleaved when each is subjected to the protease enzyme in biochemical assays (further detailed in the Examples) in which reaction in conducted wherein the initial substrate concentration is 6 mM, the reactions are incubated at 37°C for 2 hours before being stopped by adding EDTA, with the amount of digestion products and uncleaved substrate analyzed by non- reducing SDS-PAGE to establish the ratio of the percentage cleaved.
- the cleavage efficiency is calculated as follows:
- a cleavage efficiency of -1 means that the amount of test substrate cleaved was 50% compared to that of the control substrate, while a cleavage efficiency of +1 means that the amount of test substrate cleaved was 200% compared to that of the control substrate.
- a higher rate of cleavage by the test protease relative to the control would result in a higher cleavage efficiency, and a slower rate of cleavage by the test protease relative to the control would result in a lower cleavage efficiency.
- a control RS sequence AC1611 (RSR-1517), having the amino acid sequence EAGRSANHEPLGLVAT, was established as having an appropriate baseline cleavage efficiency by the proteases legumain, MMP-2, MMP-7, MMP-9, MMP-14, uPA, and matriptase, when tested in in vitro biochemical assays for rates of cleavage by the individual proteases.
- RSR-1517 a control RS sequence AC1611 (RSR-1517), having the amino acid sequence EAGRSANHEPLGLVAT, was established as having an appropriate baseline cleavage efficiency by the proteases legumain, MMP-2, MMP-7, MMP-9, MMP-14, uPA, and matriptase, when tested in in vitro biochemical assays for rates of cleavage by the individual proteases.
- libraries of RS were created and evaluated against the panel of the 7 proteases (detailed more fully in the Examples), resulting in profiles that were used to establish guidelines for appropriate amino
- substitutions using the hydrophilic amino acids A, E, G, P, S, and T are preferred, however other L-amino acids can be substituted at given positions in order to adjust the cleavage efficiency so long as the RS retains at least some susceptibility to cleavage by a protease.
- Conservative substitutions of amino acids in a peptide to retain or effect activity is well within the knowledge and capabilities of a person within skill in the art.
- the disclosure provides RS in which the RS is cleaved by a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, or matriptase with at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 higher cleavage efficiency in an in vitro biochemical competitive assay compared to the cleavage by the same protease of a control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-14, uPA, or matriptase with at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 higher cleavage efficiency in an in vitro biochemical competitive assay compared to the cleavage by
- the disclosure provides RS in which the RS is cleaved by a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase with at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 lower cleavage efficiency in an in vitro biochemical competitive assay compared to the cleavage by the same protease of a control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase with at least a 0.2 log 2 , or 0.4 log 2 , or 0.8 log 2 , or 1.0 log 2 lower cleavage efficiency in an in vitro biochemical competitive assay compared to the clea
- the disclosure provides RS in which the rate of cleavage of the RS by a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase is at least 2-fold, or at least 4-fold, or at least 8 fold, or at least 16-fold faster compared to the control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase is at least 2-fold, or at least 4-fold, or at least 8 fold, or at least 16-fold faster compared to the control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- the disclosure provides RS in which the rate of cleavage of the RS by a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase is at least 2-fold, or at least 4-fold, or at least 8 fold, or at least 16-fold slower compared to the control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- a protease selected from legumain, MMP-l, MMP-2, MMP-7, MMP-9, MMP-l 1, MMP-l 4, uPA, or matriptase is at least 2-fold, or at least 4-fold, or at least 8 fold, or at least 16-fold slower compared to the control sequence RSR-1517 having the sequence EAGRSANHEPLGLVAT.
- the disclosure provides AAC comprising multiple RS wherein each RS sequence is selected from the group of sequences set forth in Table 1 and the RS are linked to each other by 1 to 6 amino acids selected from glycine, serine, alanine, and threonine.
- the AAC comprises a first RS and a second RS different from the first RS wherein each RS sequence is selected from the group of sequences set forth in Table 1 and the RS are linked to each other by 1 to 6 amino acids selected from glycine, serine, alanine, and threonine.
- the AAC comprises a first RS, a second RS different from the first RS, and a third RS different from the first and the second RS wherein each sequence is selected from the group of sequences set forth in Table 1 and the first and the second and the third RS are linked to each other by 1 to 6 amino acids selected from glycine, serine, alanine, and threonine. It is specifically intended that the multiple RS of the AAC can be concatenated to form a sequence that can be cleaved by multiple proteases at different rates or efficiency of cleavage.
- the disclosure provides AAC comprising an RS1 and an RS2 selected from the group of sequences set forth in Tables 1 and 2 and an XTEN 1 and XTEN 2 selected from the group of sequences set forth in Tables 8 and 10 wherein the RS1 is fused between the XTEN1 and the binding moieties and the RS2 is fused between the XTEN2 and the binding moieties.
- compositions would be more readily cleaved by diseased target tissues that express multiple proteases, compared with healthy tissues or when in the normal circulation, with the result that the resulting fragments bearing the binding moieties would more readily penetrate the target tissue; e.g., a tumor, and have an enhanced ability to bind and link the target cell and the effector cell (or just the target cell in the case of AAC designed with a single binding moiety.
- the RS of the disclosure are useful for inclusion in recombinant polypeptides as therapeutics for treatment of cancers, autoimmune diseases, inflammatory diseases and other conditions where localized activation of the recombinant polypeptide is desirable.
- the subject compositions address an unmet need and are superior in one or more aspects including enhanced terminal half-life, targeted delivery, and improved therapeutic ratio with reduced toxicity to healthy tissues compared to conventional antibody therapeutics or bispecific antibody
- the disclosure provides recombinant polypeptides comprising a first binding moiety (FBM) having specific binding affinity to a ligand.
- FBM first binding moiety
- the binding moiety is selected from an antibody, a cytokine, an interleukin, a chemokine, or a fragment thereof.
- the binding moiety is a cell receptor or a fragment thereof.
- the binding moiety is an antibody fragment having binding affinity to a cell receptor or target cell marker.
- the disclosure provides recombinant polypeptides that are AAC comprising a first binding moiety (FBM) and a second binding moiety (SBM), each having specific binding affinity to a their respective ligands.
- the AAC comprise a first and a second binding moiety, each of which are antibody fragments.
- a binding moiety directed against a target cell marker of a disease tissue is used in combination with a second binding moiety directed towards an effector cell marker; thus it is bifunctional.
- the antibody fragment is an antibody fragment containing an antigen binding domain that is capable of binding, especially specific binding, to a target ligand of interest.
- the antibody fragment can be, but is not limited to, variable or hypervariable regions of light and/or heavy chains of an antibody (VL, VH), variable fragments (Fv), Fab' fragments, F(ab')2 fragments, Fab fragments, single chain antibodies (scAb), single chain variable fragment (scFv), linear antibodies, a single domain antibody, complementarity determining regions (CDR), domain antibodies (dAbs), single domain heavy chain
- the immunoglobulins of the BHH or BNAR type single domain light chain immunoglobulins, or other polypeptides known in the art containing an antibody fragment capable of binding target proteins or epitopes on target proteins associated with a target or effector cell.
- the VL and VH of the antibody fragments can also be configured in a single chain diabody configuration.
- the first of the two binding moieties of the polypeptide contains an antibody fragment targeted to an effector cell ligand (such as, but not limited to CD3, CD 16, TCRa, TCRp, CD28 and the like) and the second binding moiety contains an antibody fragment that has a disease targeting domain (e.g., a target cell marker produced by a disease tissue or cell).
- the origin of the antibody fragments contemplated by the disclosure can be derived from a naturally occurring antibody or fragment thereof, a non-naturally occurring antibody or fragment thereof, a humanized antibody or fragment thereof, a synthetic antibody or fragment thereof, a hybrid antibody or fragment thereof, or an engineered antibody or fragment thereof.
- Methods for generating an antibody for a given target marker are well known in the art.
- the monoclonal antibodies may be made using the hybridoma method first described by Kohler et al., Nature, 256:495 (1975), or may be made by recombinant DNA methods (U.S. Pat. No. 4,816,567).
- VH and VL variable regions of heavy and light chains of an antibody
- scFv single chain variable regions
- CDR complementarity determining regions
- dAbs domain antibodies
- Therapeutic monoclonal antibodies from which VL and VH and CDR domains can be derived for the subject compositions are known in the art.
- Such therapeutic antibodies include, but are not limited to, rituximab, IDEC/Genentech/Roche (see, e.g., U.S. Pat. No.
- a chimeric anti-CD20 antibody used in the treatment of many lymphomas, leukemias, and some autoimmune disorders; ofatumumab, an anti-CD20 antibody approved for use for chronic lymphocytic leukemia, and under development for follicular non-Hodgkin’s lymphoma, diffuse large B cell lymphoma, rheumatoid arthritis and relapsing remitting multiple sclerosis, being developed by GlaxoSmithKline; lucatumumab (HCD122), an anti-CD40 antibody developed by Novartis for Non-Hodgkin's or Hodgkin's Lymphoma (see, for example, U.S. Pat. No.
- AME-133 an antibody developed by Applied Molecular Evolution which binds to cells expressing CD20 to treat non-Hodgkin's lymphoma
- veltuzumab hA20
- Immunomedics, Inc. which binds to cells expressing CD20 to treat immune thrombocytopenic purpura
- HumaLYM developed by Intracel for the treatment of low-grade B- cell lymphoma
- ocrelizumab developed by Genentech which is an anti-CD20 monoclonal antibody for treatment of rheumatoid arthritis (see, e.g., U.S.
- trastuzumab see, e.g., U.S. Pat. No. 5,677,171
- trastuzumab a humanized anti-HER2/neu antibody approved to treat breast cancer developed by Genentech
- pertuzumab an anti-HER2 dimerization inhibitor antibody developed by Genentech in treatment of in prostate, breast, and ovarian cancers
- cetuximab an anti-EGFR antibody used to treat epidermal growth factor receptor (EGFR)-expressing, KRAS wild-type metastatic colorectal cancer and head and neck cancer, developed by Imclone and BMS (see U.S. Pat. No.
- panitumumab a fully human monoclonal antibody specific to the epidermal growth factor receptor (also known as EGF receptor, EGFR, ErbB-l and HER1, currently marketed by Amgen for treatment of metastatic colorectal cancer (see U.S. Pat. No. 6,235,883); zalutumumab, a fully human IgGl monoclonal antibody developed by Genmab that is directed towards the epidermal growth factor receptor (EGFR) for the treatment of squamous cell carcinoma of the head and neck (see, e.g., U.S. Pat. No.
- EGFR epidermal growth factor receptor
- nimotuzumab a chimeric antibody to EGFR developed by Biocon, YM Biosciences, Cuba, and Oncosciences, Europe) in the treatment of squamous cell carcinomas of the head and neck, nasopharyngeal cancer and glioma (see, e.g., U.S. Pat. No. 5,891,996; U.S. Pat. No. 6,506,883); matuzumab, a humanized monoclonal that is directed towards the epidermal growth factor receptor (EGFR) that was developed by Takeda
- EGFR epidermal growth factor receptor
- compositions for the treatment of colorectal, lung, esophageal and stomach cancer see, e.g., U.S. Patent Application 20090175858A1; cetuximab, a chimeric (mouse/human) monoclonal antibody that is directed to epidermal growth factor receptor (EGFR) used for the treatment of metastatic colorectal cancer, metastatic non-small cell lung cancer and head and neck cancer that was developed by Bristol-Myers Squibb and Merck KGaA (see, e.g.,, U.S. Patent No.
- EGFR epidermal growth factor receptor
- alemtuzumab a humanized monoclonal antibody to CD52 marketed by Bayer Schering Pharma for the treatment of chronic lymphocytic leukemia (CLL), cutaneous T-cell lymphoma (CTCL) and T-cell lymphoma; muromonab-CD3, an anti-CD3 antibody developed by Ortho Biotech/Johnson & Johnson used as an immunosuppressant biologic given to reduce acute rejection in patients with organ transplants; ibritumomab tiuxetan, an anti-CD20 monoclonal antibody developed by IDEC/Schering AG as treatment for some forms of B cell non-Hodgkin's lymphoma; gemtuzumab ozogamicin, an anti-CD33 (p67 protein) antibody linked to a cytotoxic chelator tiuxetan, to which a radioactive isotope is attached, developed by Celltech/Wyeth used to treat acute myelogenous leukemia; ABX
- MDX-010 an anti-CTLA4 antibody developed by Medarex
- MDX-060 an anti- CD30 antibody developed by Medarex
- MDX-070 developed by Medarex
- MDX-018 developed by Medarex
- OSIDEM IDM-l
- an anti-HER2 antibody developed by Medarex and Immuno- Designed Molecules
- HuMax®-CD4 an anti-CD4 antibody developed by Medarex and Genmab
- HuMax-ILl5 an anti-ILl5 antibody developed by Medarex and Genmab
- ICM-l anti-intercellular adhesion molecule- 1 (ICAM-l) (CD54) antibodies developed by MorphoSys, MOR201;
- tremelimumab an anti-CTLA-4 antibody developed by Pfizer
- visilizumab an anti-CD3 antibody developed by Protein Design Labs
- Anti-a 5b1 Integrin developed by Protein Design Labs
- anti-IL-l2 developed by Protein Design Labs
- ING-l an anti-Ep-CAM antibody developed by Xoma
- MLN01 an anti-Beta2 integrin antibody developed by Xoma
- the complementary determining regions of the heavy chain and/or the light chain for the antibody fragment directed to the effector cells to be incorporated into the subject AAC compositions are derived from known anti-CD3 antibodies, such as, for example, muromonab-CD3 (OKT3), otelixizumab (TRX4), teplizumab (MGA031), visilizumab (Nuvion), SP-34 or I2C, TR-66 or X35-3, VIT3, BMA030 (BW264/56), CLB-T3/3, CRIS7, YTH12.5, Fl 11-409, CLB-T3.4.2, TR-66, WT32, SPv-T3b, 11D8, XIII-141, XIII-46, XIII-87, 12F6,
- known anti-CD3 antibodies such as, for example, muromonab-CD3 (OKT3), otelixizumab (TRX4), teplizumab (MGA031), visili
- the effector cell binding moiety of the subject AAC is a single chain antibody fragment comprising a paired VL and VH sequence as set forth in Table 4.
- the VL and VH are linked by long linkers of hydrophilic amino acids selected from the sequences set forth in Table 6 and the scFv are linked together by a short linker of hydrophilic amino acids selected from the group of sequences set forth in Table 7.
- the long linker used to link the VL and VH is L7 of Table 6 and the intermolecular linker that fuses the two scFv is S-l or S-2 of Table 7.
- the disclosure provides AAC compositions comprising a single chain diabody in which after folding, the first domain (VL or VH) is paired with the last domain (VH or VL) to form one scFv and the two domains in the middle are paired to form the other scFv in which the first and second domains, as well as the third and last domains, are fused together by a short linker of hydrophilic amino acids selected from the sequences set forth in Table 7 and the second and the third variable domains are fused by a long linker selected from Table 6.
- the selection of the short linker and long linker is to prevent the incorrect pairing of adjacent variable domains, thereby facilitating the formation of the single chain diabody configuration comprising the VL and VH of the first binding moiety and the second binding moiety.
- Table 4 Anti-CD3 Monoclonal Antibodies and Sequences
- the disclosure relates to recombinant polypeptides comprising at least a first bulking moiety that are incorporated into the subject compositions both in order to increase the mass and size of the construct, but that also serve to greatly reduce the ability of the binding moieties to bind their ligands when the molecule is in the intact, uncleaved state, described more fully, below.
- the disclosure provides a recombinant polypeptide comprising a single bulking moiety fused to the N- or C-terminus of the RS that is located between the binding moiety and the bulking moiety.
- bulking moieties include extended recombinant polypeptide (XTEN, as described herein, below);
- albumin binding domain albumin; IgG binding domain; polypeptides of at least 350 amino acid residues consisting of proline, serine, and alanine; fatty acid; elastin-like protein (ELP) (the individual subunit or building blocks of ELPs are derived from a five amino acid motif found in human protein elastin that is repeated multiple times to form the ELP biopolymer, as described in WO20l608l884),Fc domain, polyethylene glycol (PEG), PLGA, and hydoxylethyl starch.
- ELP elastin-like protein
- the disclosure provides a recombinant polypeptide comprising at least a first XTEN fused to the N- or C-terminus of the RS, which, in turn, is fused to the adjacent binding moiety.
- the recombinant polypeptide comprises two different XTEN sequences, wherein the two XTEN are each linked to two RS of the composition that, in turn, are linked to the binding moieties.
- recombinant polypeptide compositions comprise a first XTEN sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to an XTEN sequence of comparable length selected from the group of sequences set forth in Table 8 or Table 10.
- the recombinant polypeptide comprises a first and a second XTEN sequence (XTEN1 and XTEN2), each sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from sequences set forth in Table 8.
- the recombinant polypeptide comprises a first and a second XTEN sequence (XTEN1 and XTEN2), each sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from sequences set forth in Table 10.
- compositions to confer certain properties; 1) provide recombinant polypeptide compositions with a bulking moiety XTEN that shields the binding moieties and reduces binding affinity for the target cell markers and effector cell antigens when the composition is in its intact, prodrug form; ii) provide recombinant polypeptide compositions with a bulking moiety XTEN that provides enhanced half-life when administered to a subject, iii) contribute to the solubility and stability of the intact composition, thereby enhancing the pharmaceutical properties of the subject compositions; and iv) provide recombinant polypeptide compositions with a bulking moiety XTEN that reduces extravasation in normal tissues and organs yet permits a degree of extravasation in diseased tissues (e.g., a tumor) with larger pore sizes in the vasculature, yet could be released from the composition by action of certain mammalian proteases, thereby permitting the binding moieties of the composition to more readily penetrate into the disease
- compositions comprising one or more XTEN in which the XTEN provides increased mass and hydrodynamic radius to the resulting composition.
- the XTEN polypeptides of the embodiments provide certain advantages in the design of the subject compositions in that is provides not only provides increased mass and hydrodynamic radius, but its flexible, unstructured characteristics provides a shielding effect over the binding moieties of the composition, thereby reducing the likelihood of binding to antigens in normal tissues or the vasculature of normal tissues that don’t express or express reduced levels of target cell markers and/or effector cell antigens, and enhances solubility and proper folding of the single chain antibody fragment binding moieties during their expression and recovery.
- XTEN are polypeptides with non-naturally occurring, substantially non-repetitive sequences having a low degree or no secondary or tertiary structure under physiologic conditions, as well as additional properties described in the paragraphs that follow.
- XTEN typically have from at least about 100 to at least about 1000 or more amino acids, and more preferably at least about 200 to at least about 900 amino acids, of which the majority or the entirety are small hydrophilic amino acids selected from glycine, serine, threonine, glutamate, and proline.
- XTEN specifically excludes whole antibodies or antibody fragments (e.g. single- chain antibodies and Fc fragments).
- XTEN polypeptides have utility as fusion partners in that they serve in various roles, conferring certain desirable properties when linked to a composition comprising, for example, the bispecific binding moieties of the subject AAC compositions described herein.
- the resulting compositions have enhanced properties, such as enhanced pharmacokinetic, physicochemical, pharmacologic, and improved toxicological and
- binding moieties are known in the art to be used.
- the unstructured characteristic and physicochemical properties of the XTEN result, in part, from the overall amino acid composition that is disproportionately limited to 4-6 types of hydrophilic amino acids, the sequence of the amino acids in a quantifiable, substantially non- repetitive design, and from the resulting length of the XTEN polypeptide.
- the properties of XTEN disclosed herein are not tied to an absolute primary amino acid sequence, as evidenced by the diversity of the exemplary sequences of Tables 8 and 10 that, within varying ranges of length, possess similar properties and confer enhanced properties on the compositions to which they are linked, many of which are documented in the Examples.
- compositions of the disclosure not be limited to those XTEN specifically enumerated in Tables 8 or 10, but, rather, the embodiments include sequences having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity, when optimally aligned, to the sequences of Table 8 or Table 10 as they exhibit the properties of XTEN described herein. It has been established that such XTEN have properties more like non-proteinaceous, hydrophilic polymers (such as polyethylene glycol, or“PEG”) than they do proteins.
- PEG polyethylene glycol
- the XTEN of the present disclosure exhibit one or more of the following advantageous properties: defined and uniform length (for a given sequence), conformational flexibility, reduced or lack of secondary structure, high degree of random coil formation, high degree of aqueous solubility, high degree of protease resistance, low immunogenicity, low binding to mammalian receptors, a defined degree of charge, and increased hydrodynamic (or Stokes) radii; properties that are similar to certain hydrophilic polymers (e.g., polyethylene glycol) that make them particularly useful as fusion partners.
- hydrophilic polymers e.g., polyethylene glycol
- the XTEN component(s) of the subject recombinant polypeptides and AAC are designed to behave like denatured peptide sequences under physiological conditions, despite the extended length of the polymer.
- “Denatured” describes the state of a peptide in solution that is characterized by a large conformational freedom of the peptide backbone. Most peptides and proteins adopt a denatured conformation in the presence of high concentrations of denaturants or at elevated temperature. Peptides in denatured conformation have, for example, characteristic circular dichroism (CD) spectra and are characterized by a lack of long-range interactions as determined by NMR.“Denatured conformation” and“unstructured conformation” are used synonymously herein.
- CD characteristic circular dichroism
- the methods to measure such properties include analytical centrifugation, EPR, HPLC-ion exchange, HPLC-size exclusion chromatography (SEC), HPLC-reverse phase, light scattering, capillary electrophoresis, circular dichroism, differential scanning calorimetry, fluorescence, HPLC-ion exchange, HPLC-size exclusion, IR, NMR, Raman spectroscopy, reffactometry, and UV/Visible spectroscopy.
- secondary structure can be measured
- Secondary structure elements such as alpha-helix and beta-sheet, each give rise to a characteristic shape and magnitude of CD spectra, as does the lack of these structure elements. Secondary structure can also be predicted for a polypeptide sequence via certain computer programs or algorithms, such as the well-known Chou-Fasman algorithm (Chou, P. Y., et al. (1974) Biochemistry, 13: 222-45) and the Gamier-Osguthorpe-Robson algorithm (“GOR IV algorithm”) (Gamier J, Gibrat JF, Robson B.
- Chou-Fasman algorithm Chou, P. Y., et al. (1974) Biochemistry, 13: 222-45
- GOR IV algorithm Gamier-Osguthorpe-Robson algorithm
- the XTEN sequences of the subject compositions have an alpha- helix percentage ranging from 0% to less than about 5% and a beta-sheet percentage ranging from 0% to less than about 5% as determined by the Chou-Fasman algorithm and at least about 90%, or at least about 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% random coil formation as determined by the GOR IV algorithm.
- the XTEN sequences of the disclosed compositions have an alpha-helix percentage less than about 2% and a beta-sheet percentage less than about 2% as determined by the Chou-Fasman algorithm and at least about 90% random coil formation as determined by the GOR IV algorithm.
- the XTEN sequences of the compositions are substantially lacking secondary structure as measured by circular dichroism.
- the XTEN sequence used in the subject compositions of the disclosure is at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% identical to a sequence selected from the group consisting of AE 144 1A, AE 144 2A, AEE144 2B, AE 144_3A, AE144 3B, AE 144_4A, AE 144_4B, AE 144_5A, AE 144_6B, AE288_l, AE288_2, AE288_3, AE284, AE292, AE576, AE864, AE864_2, AE865, AE866, AE867, AE867_2, and AE868.
- amino acids of an XTEN in the subject compositions are selected from glycine (G), alanine (A), serine (S), threonine (T), glutamate (E) and proline (P), or wherein less than 100% of the sequence consists of the XTEN sequences of Table 8 or Table 10, the remaining amino acid residues of the XTEN are selected from any of the other 14 natural L-amino acids, but are preferentially selected from hydrophilic amino acids such that the XTEN sequence contains at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or at least about 99% hydrophilic amino acids.
- the content of hydrophobic amino acids in the XTEN utilized in the subject compositions can be less than 5%, or less than 2%, or less than 1% hydrophobic amino acid content.
- Hydrophobic residues that are less favored in construction of XTEN include tryptophan, phenylalanine, tyrosine, leucine, isoleucine, valine, and methionine.
- XTEN sequences can contain less than 5% or less than 4% or less than 3% or less than 2% or less than 1% or none of the following amino acids: methionine (for example, to avoid oxidation), or asparagine and glutamine (to avoid desamidation).
- amino acid sequences for certain XTEN utilized in the AAC embodiments of the disclosure are shown in Table 8.
- compositions comprising XTEN of intermediate lengths to those of Table 8, as well as XTEN of longers lengths in which motifs of 12 amino acids are added to the N- or C- terminus of an XTEN of Table 8 incorporated into the composition.
- a subject composition comprises an XTEN of Table 8 with the addition of one or more copies of one or more motifs selected from the group of motifs set forth in Table 9.
- the amino acid sequences for certain XTEN utilized in the embodiments of the disclosure are shown in Table 10.
- the AAC comprises a first XTEN (XTEN1) comprising an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 10.
- the AAC comprises an XTEN1 and a second XTEN (XTEN2) comprising an amino acid sequence having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Table 10.
- XTEN1 and XTEN2 are identical.
- the XTEN1 and XTEN2 are different.
- the AAC comprises an XTEN1 and an XTEN2 comprising amino acid sequences having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Tables 8 and 10.
- the AAC comprises an XTEN1 and an XTEN2 comprising amino acid sequences having at least about 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity, when optimally aligned, to a sequence selected from the sequences set forth in Tables 8 and 10 and further comprising a His tag of HHHHHH or HHHHHHHH at the N-terminus or C-terminus of the composition.
- the binding moieties have reduced ability to bind their ligands until the XTEN component of the recombinant polypeptides, which shields the binding moieties and reduces their binding affinity to their ligands, is released from the composition by cleavage of the release segment that fuses the binding moieties to the XTEN.
- compositions having a first binding moiety were driven by consideration of at least three properties: 1) compositions having a binding moiety with the capability to bind the desired target cell marker(s) on a target cell; 2) compositions with one or more XTEN that i) shields the binding moiety and reduces binding affinity for the target cell marker when the composition is in an intact form (thus rendering a prodrug form), ii) provides enhanced half-life when administered to a subject, iii) reduces extravasation of the intact composition from the circulation in normal tissues and organs compared to diseased tissues (e.g., tumor), and iv) confers an increased safety profile compared to conventional antibody therapeutics; and 3) is activated when the RS is cleaved by one or more mammalian proteases in proximity of or co-localized with diseased tissues or cells, thereby releasing the binding moiety such that the binding moiety regains its full binding affinity potential for the target ligand.
- the design of the subject composition is activated when the
- the recombinant polypeptides can have, in an uncleaved state, a structural arrangement from N-terminus to C-terminus of FBM-RS1-XTEN1 or XTEN1-RS1- FBM.
- the disclosure provides recombinant polypeptides having two binding moieties that are antibody fragments and are activatable (referred to as“AAC”) with a first binding moiety that targets an effector cell and a second binding moiety that targets a cell marker associated with a disease tissue or cell; both of which have specific binding affinity for their respective ligands.
- AAC activatable
- compositions having a first and a second binding moiety were driven by consideration of at least three properties: 1) compositions having bispecific binding moieties with the capability to bind to and link together an effector cell and a target cell with the resultant formation of an immunological synapse; 2) compositions with a XTEN that i) shields both of the binding moieties and reduces binding affinity for the target and effector cell ligands when the composition is in an intact prodrug form, ii) provides enhanced half-life when administered to a subject, iii) reduces extravasation of the intact composition from the circulation in normal tissues and organs compared to diseased tissues (e.g., tumor), and iv) confers an increased safety profile compared to conventional bispecific cytotoxic antibody therapeutics; and 3) is activated when the RS is cleaved by one or more mammalian proteases in proximity of diseased tissues, thereby releasing the bispecific binding moieties such that
- the two binding moieties of the AAC are connected to each other by a short linker, and are, in turn, connected to the XTEN by the release segment (RS) peptide that includes up to three different cleavage sites designed to allow separation and release of the binding moieties from the XTEN upon cleavage of any one or all of the cleavage sites.
- RS release segment
- the AAC can have, in an uncleaved state, a structural arrangement from N-terminus to C-terminus of SBM-FBM-RS 1 -XTEN 1 , FBM-SBM-RS 1 -XTEN 1 , XTEN1- RS1-SBM-FBM, XTEN 1 -RS 1 -FBM-SBM, or diabody-RSl-XTENl, or XTENl-RSl-diabody, wherein the diabody comprises VL and VH of the FBM and SBM.
- the FBM has VL and VH derived from antibodies having binding affinity to effector cells, including the antibodies of Table 4, the SBM VL and VH are derived from antibodies having binding affinity to target cell markers, including but not limited to the antibodies of Table 5, the release segments have sequences having 88-100% identity to the sequences of Table 1 or Table 2, and the XTEN have sequences having 90-100% identity to the sequences of Table 8 or Table 10.
- the disclosure provides AAC having two binding moieties, two RS, and two XTEN.
- the design of these AAC was driven by considerations of further reducing the binding affinity of the uncleaved compositions to the respective ligands of the FBM and SBM antibody fragments by the addition of the second XTEN in order to reduce the unintended binding of the AAC to healthy tissues or cells when administered to a subject, thereby further improving the therapeutic index of the subject compositions compared to AAC having only one RS and one XTEN.
- the addition of the second RS and second XTEN resulted in a suprising reduction of binding affinity of the intact, uncleaved AAC to the respective ligands of the FBM and SBM antibody fragments relative to those AAC having a single RS and XTEN, when assayed in vitro, and also resulted in redued toxicity in animal models of disease.
- the AAC can have, in an uncleaved state, a structural arrangement from N-terminus to C- terminus of XTEN1 -RS 1 -SBM-FBM-RS2-XTEN2, XTEN1 -RS 1 -FBM-SBM-RS2-XTEN2, XTEN2-RS2-SBM-FBM-RS 1 -XTEN1 , XTEN2-RS2-FBM-SBM-RS 1 -XTEN1 , XTEN2-RS2- diabody-RSl-XTENl, wherein the diabody comprises VL and VH of the FBM and SBM, or XTENl-RSl-diabody-RS2-XTEN2, wherein the diabody comprises VL and VH of the FBM and SBM.
- the FBM has VL and VH derived from antibodies having binding affinity to effector cells, including the antibodies of Table 4, the SBM VL and VH are derived from antibodies having binding affinity to target cell markers, including but not limited to the antibodies of Table 5, the release segments have sequences having 88-100% identity to the sequences of Table 1 or Table 2, and the XTEN have sequences having 90-100% identity to the sequences of Table 8 or Table 10.
- the binding affinity of each binding moiety released from the AAC is greater for the respective target ligands compared to the binding moieties of the intact composition that has not been cleaved, such as when assayed in an in vitro binding assay as described herein.
- the binding affinity of the effector cell binding moiety released from the composition by cleavage of the RS by a protease is at least 3 -fold, or at least 4- fold, or at least 5-fold, or at least 6-fold, or at least 7-fold, or at least 8-fold, or at least 9-fold, or at least lO-fold, or at least lOO-fold, or at least 1000-fold, or at least 10,000-fold greater for the released effector cell antigen compared to the effector cell binding moiety of the intact AAC, as measured in an in vitro cell assay with an effector cell having said effector cell antigen on the cell surface of said cell or in an ELISA with bound effector cell antigen, when assayed under comparable conditions, e.g., equivalent molar concentrations.
- the effector cell antigen is CD3.
- the binding affinity of the target cell binding moiety released from the composition by cleavage of the RS by a protease is at least 2 -fold, or at least 3- fold, or at least 4-fold, or at least 5-fold, or at least 6-fold, or at least 7-fold, or at least 8-fold, or at least 9-fold, or at least lO-fold, or at least lOO-fold, or at least 1000-fold or at least 10,000-fold greater for the target marker or target cell antigen compared to the target cell binding moiety of the intact AAC, as measured in an in vitro cell assay with an tumor cell having said antigen on the cell surface of said cell or in an ELISA with bound effector cell antigen, when assayed under comparable conditions, e.g., equivalent molar concentrations.
- the target marker or an antigen of a target cell is selected from the group consisting of alpha 4 integrin, Ang2, B7-H3, B7-H6, CEACAM5, cMET, CTLA4, FOLRl,EpCAM, CCR5, CD 19, HER2, HER2 neu, HER3, HER4, HER1 (EGFR), PD-L1, PSMA, CEA, MUC1 (mucin), MUC-2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16 hCG, Lewis-Y, CD20, CD33, CD38, CD30, CD56 (NCAM), CD133, ganglioside GD3; 9-0- Acetyl-GD3, GM2, Globo H, fucosyl GM1, GD2, carbonicanhydrase DC, CD44v6, Sonic Hedgehog (Shh), Wue-l, plasma cell antigen 1, melanoma chondroitin sulfate proteoglycan
- the shielding effect of the XTEN applies to both binding moieties of the foregoing embodiments of the intact, prodrug form of the activatable recombinant polypeptide composition, and that upon release of the XTEN from the AAC by cleavage of the RS, the full binding potential of the respective binding moieties is restored.
- the released fused FBM and SBM antibody fragments are capable of killing target cells by recruitment of cytotoxic effector cells without any need for pre- and/or co-stimulation. Further, the independence from pre- and/or co- stimulation of the effector cell may substantially contribute to the exceptionally high cytotoxicity mediated by the released binding moieties.
- the released FBM is designed with binding specificities such that it has the capability to bind and link together cytotoxic effector cells (e.g., T cells, NK cells, cytokine induced killer cell (CD cell)), to preselected target cell markers by the SBM (that remains linked to the FBM by a linker peptide) that has binding specificity to target cell markers associated with tumor cells or cancer cells, thereby effecting an immunological synapse and a selective, directed, and localized effect of released cytokines and effector molecules against the target tumor or cancer cell, with the result that tumor or cancer cells are damaged or destroyed, resulting in therapeutic benefit to a subject.
- cytotoxic effector cells e.g., T cells, NK cells, cytokine induced killer cell (CD cell)
- CD cell cytokine induced killer cell
- the released FBM that binds to an effector cell antigen is capable of modulating one or more functions of an effector cell, resulting in or contributed to the cytolytic effect on the target tumor cell.
- the effector cell antigen can by expressed by the effector cell or other cells.
- the effector cell antigen is expressed on cell surface of the effector cell.
- Non-limiting examples are CD3, CD4, CD8, CD16, CD25, CD38, CD45RO,
- the configurations of the subject compositions are intended to selectively or disproportionately deliver the active form of the composition to the target tumor tissue or cancer cell, compared to healthy tissue or healthy cells in a subject in which the composition is administered, with resultant therapeutic benefit.
- the disclosure provides a large family of polypeptides in designed configurations to effect the desired properties.
- the design of the AAC, with the shielding XTEN of the intact AAC and the concomitant reduction in binding to T cells and target tissues results in reduced production of Thl T-cell associated cytokines or other proinflammatory mediators during systemic exposure when administered to a subject such that the overall side-effect and safety profile is improved compared to bispecific binding compositions not linked to a bulking moiety such as XTEN.
- Thl T-cell associated cytokines or other proinflammatory mediators results in reduced production of Thl T-cell associated cytokines or other proinflammatory mediators during systemic exposure when administered to a subject such that the overall side-effect and safety profile is improved compared to bispecific binding compositions not linked to a bulking moiety such as XTEN.
- IL-2, TNF-alpha, and IFN-gamma are hallmarks of a Thl response (Romagnani S. T-cell subsets (Thl versus Th2). Ann Allergy Asthma Immunol. 2000.
- an intact, uncleaved AAC exhibits at least 3 -fold, or at least 4-fold, or at least 5 -fold, or at least 6-fold, or at least 7- fold, or at least 8-fold, or at least 9-fold, or at least 10-fold, or at least 20-fold, or at least 30-fold, or at least 50-fold, or at least 100-fold, or at least 1000-fold reduced potential to result in the production of Thl and/or proinflammatory cytokines when the intact, uncleaved AAC is in contact with the effector cell and a target cell in an in vitro cell-based cytokine stimulation assay (such as described in the Examples, below) compared to the cytokine levels stimulated by the corresponding released first and second binding moieties (which remain linked together after release) of a protease-treated AAC in the in vitro cell-based stimulation cytokine assay
- Thl and/or proinflammatory cytokines are IL-2, IL-4, IL-6, IL-10, TNF-alpha and IFN-gamma.
- the production of the Thl cytokine is assayed in an in vitro assay comprising effector cells such as PBMC or CD3+ T cells and target cells having a tumor specific marker antigen selected from the group consisting of A33 antigen, alpha- fetoprotein (AFP), alpha 4 integrin, Ang2, B7-H3, B7-H6, B-cell maturation antigen (BCMA), cancer antigen 19-9 (CA19-9), cancer antigen 125 (CA-125), Carbonic Anhydrase 6 (CA6), carbonic anhydrase IX (CALX), CEACAM5, cMET, CTLA4, C-C Motif Chemokine Receptor 1 (CCR1), C-C Motif Chemokine Receptor 2 (CCR2), C-C Motif Chemokine Receptor 3 (CCR3), C-C Motif Chemokine Receptor 4 (CCR4), C-C Motif Chemokine Receptor 1 (CCR1),
- CD7 Differentiation 7
- globohexaosylceramide globo-H
- GD2, Glypican 3 Glypican 3
- GCC guanylyl cyclase C
- HER2, HER2 neu HER3, HER4, HER1, ELl3Ra2, insulin-like growth factor I receptor (IGF-IR ), Lysosomal Associated Membrane Protein 1 (LAMP1), Ll Cell Adhesion Molecule (L1CAM), lymphocyte antigen 6 (Ly-6), melanoma chondroitin sulfate proteoglycan (MCSP), Membrane- type metalloproteinase (MT-MMP), mesothelin, mucin 1 (MUC1), MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16, Muellerian inhibitory substance receptor type II (MISIIR), nectin cell adhesion molecule 4 (Nectin-4), 6-transmembrane epithelial
- the assayed cytokine is IL-2. In another embodiment of the foregoing, the assayed cytokine is TNF-alpha. In another embodiment of the foregoing, the assayed cytokine is IFN-gamma.
- an intact, uncleaved AAC administered to a subject having a tumor with target cell marker that can be bound by the released binding moiety of the AAC exhibits at least 2 -fold, at least 3 -fold, at least 4-fold, at least 5 -fold, at least 6-fold, at least 7-fold, at least 8-fold, at least 9-fold, at least lO-fold, at least 20-fold, at least 30-fold, at least 50- fold, at least lOO-fold, or at least 1000-fold reduced potential to result in the systemic production of Thl and/or proinflammatory cytokines in the subject compared to the cytokine levels produced by the corresponding released binding moieties of a protease-treated AAC in a comparable subject with a tumor dosed with an equivalent molar concentration.
- the cytokines can be assessed from a blood, fluid, or tissue sample removed from the subject.
- the subject can be mouse, rat, monkey, and human.
- the cytolytic properties of the compositions do not require prestimulation by cytokines; that formation of the
- the production of proinflammatory cytokines are useful markers to assess the potency or the effects of the subject AACs; whether by in vitro assay or in the monitoring of treatment of a subject with a tumor.
- the binding site recognizing the target cell marker antigen has a high binding affinity in order to capture the target cells to be destroyed with high efficiency.
- the AACs of the disclosure have the advantage that they may be used a number of times for killing tumour cells since, in preferred embodiments, the target cell binding moiety of the released target cell binding moiety has an affinity with a K d value in the range of 10 7 to 10 -10 M, as determined in an vitro binding assay. If the affinity of a bispecific binding moiety for binding a target cell marker is too high, the composition binds the expressing target cell and remains on its surface, making it unable to release and bind to another cell.
- the released effector cell binding moiety of a subject AAC has a binding constant of between 10 5 and 10 8 M, as determined in an vitro binding assay, detailed examples of which are described in the Examples, below.
- the released effector cell binding moiety (FBM) of a subject AAC has a lower binding affinity to the effector cell ligand of at least one order of magnitude lower compared to the greater binding affinity of the SBM to the target cell marker, as determined as a I constant in an in vitro assay.
- the fused FBM and SBM bind to and link together an effector cell (e.g., a T cell bearing CD3) and a tumor or cancer cell bearing the target cell marker of a target cell targeted by the SBM, whereupon the effector cell is activated.
- an effector cell e.g., a T cell bearing CD3
- the subsequent concurrent binding of the effector cell and the target cell results in at least a 3 -fold, or a 10-fold, or a 30-fold, or a 100-fold, or a 300-fold, or a 1000-fold activation of the effector cell, wherein the activation is assessed by the production of cytokines, cytolytic proteins, or lysis of the target cell, assessed in an in vitro cell-based assay.
- the concurrent binding of a T cell bearing the CD3 antigen and a tumor cell bearing the target cell marker of a target cell by the released binding moieties forms an immunologic synapse, wherein the binding results in the release of T cell -derived effector molecules capable of lysing the tumor cell.
- the in vitro assay for measuring effector cell activation and/or cytolysis include cell membrane integrity assay, mixed cell culture assay,
- FACS based propidium Iodide assay trypan Blue influx assay, photometric enzyme release assay, ELISA, radiometric 5lCr release assay, fluorometric Europium release assay, CalceinAM release assay, photometric MTT assay, XTT assay, WST-l assay, alamarBlue assay, radiometric 3H-Thd incorporation assay, clonogenic assay measuring cell division activity, fluorometric Rhodaminel23 assay measuring mitochondrial transmembrane gradient, apoptosis assay monitored by FACS-based phosphatidylserine exposure, ELISA-based TUNEL test assay, caspase activity assay, and cell morphology assay, or other assays known in the art for the assay of cytokines, cytolytic proteins, or lysis of cells, or the methods of the Examples, below.
- the AAC are present in a prodrug form and are converted to a more active form when entering a certain cellular environment by the action of proteases co-localized with the disease tissue or cell.
- the first binding moiety with binding specificity to an effector cell antigen and the linked second binding moiety with binding specificity to a tumor- specific marker or an antigen of a target cell regain their full capability to bind to and link together the effector cell to the target cell, forming an immunological synapse.
- the formation of the immuological synapse causes the effector cell to become activated, with various signal pathways turning on new gene transcription and the release, by exocytosis, the effector molecule contents of its vesicles.
- different cytokines and lymphokines are released; e.g., Type 1 helper T cells (Thl) release cytokines like IFN-gamma, IL-2 and TNF-alpha while Type 2 helper T cells (Th2) release cytokines like IL-4, IL-5, IL-10, and IL-13 that stimulate B cells, and cytotoxic T Lymphocytes (CTLs) release cytotoxic molecules like perforin and granzymes that kill the target (collectively,“effector molecules”).
- Thl Type 1 helper T cells
- Th2 Type 2 helper T cells
- CTLs cytotoxic T Lymphocytes
- the tumor cell upon the concurrent binding to and linking together the effector cell to the target tumor cell by the released bispecific binding moieties of the AAC, at very low effector to target (E:T) ratios the tumor cell is acted upon by the effector molecules released by the effector cell into the immunological synapse between the cells, resulting in damage, perforin- mediated lysis, granzyme B-induced cell death and/or apoptosis of the tumor cell.
- E:T effector to target
- the prodrug form when the activatable recombinant polypeptide composition is administered to a subject with a tumor, the prodrug form remains in the circulatory system in normal tissue but is able to extravasate in the more permeable vasculature of the tumor such that the prodrug form of the assembly is activated by the proteases co-localized with the tumor and that the released binding moieties bind together and link an effector cell (e.g., a T cell) and a tumor cell expressing the target cell marker targeted by the SBM of the composition, whereupon the effector cell is activated and lysis of the tumor cell is effected.
- an effector cell e.g., a T cell
- the released binding moiety in the tumor of the subject bound to both a tumor cell and an effector cell exhibits an increased ability to activate effector cells of at least 10-fold, or at least 30-fold, or at least 100-fold, or at least 200-fold, or at least 300-fold, or at least 400-fold, or at least 500-fold, or at least 1000-fold compared to the corresponding intact, uncleaved AAC.
- the released binding moieties in the tumor of the subject bound to both a tumor cell and an effector cell exhibits an increased ability to lyse the tumor cell of at least 10-fold, or at least 30-fold, or at least 100-fold, or at least 200-fold, or at least 300-fold, or at least 400-fold, or at least 500-fold, or at least 1000-fold compared to the corresponding intact AAC that has not been cleaved.
- the effector cell activation and/or the cytotoxicity can be assayed by conventional methods known in the art, such as cytometric measurement of activated effector cells, assay of cytokines, measurement of tumor size, or by histopathology.
- the subject can be mouse, rat, dog, monkey, and human.
- the subject compositions are designed such that they have an enhanced therapeutic index and reduced toxicity or side effects, achieved by a combination of the shielding effect and steric hindrance of XTEN on binding affinity over the binding moieties in the prodrug form, yet are able to release the bispecific binding moieties (achieved by inclusion of the cleavage sequences in the RS) in proximity to or within a target tissue (e.g., a tumor) that produces a protease for which the RS is a substrate.
- a target tissue e.g., a tumor
- XTEN with cumulative lengths longer that about 400 residues incorporated into the compositions result in increased hydrodynamic radii, increased apparent molecular weight, and increased apparent molecular weight factor of a recombinant protein compared to a protein not linked to an XTEN.
- incorporation of the XTEN can effectively enlarge the hydrodynamic radius of the subject compositions beyond the glomerular pore size of approximately 3-5 nm (corresponding to an apparent molecular weight of about 70 kDa)
- the subject AAC were designed to take advantage of this differential in pore size by the addition of the XTEN, such that extravasation of the intact AAC in normal tissue is reduced, but in the leaky environment of the tumor vasculature or other areas of inflammation, the intact assembly can extravasate and be activated by the proteases in the tumor environment, releasing the binding moieties to the effector and target cells.
- the design takes advantage of the circumstance that when an AAC is in proximity to diseased tissues; e.g., a tumor, that elaborates one or more proteases, the RS sequences that are susceptible to the one or more proteases expressed by the tumor are capable of being cleaved by the proteases (described more fully, above).
- the action of the protease cleaves the release segment (RS) of the composition, separating the binding moieties from the XTEN, resulting in components with reduced molecular weight and hydrodynamic radii, particularly for the released binding moieties.
- the decrease in molecular weight and hydrodynamic radius of the composition also confers the property that the released binding moieties are able to more freely move in solution, move through smaller pore spaces in tissue and tumors, and extravsate more readily from the larger pores of the tumor vasculature and more readily penetrate into the tumor, resulting in an increased ability to attacht and link together the effector cell and the tumor cell.
- Such property can be measured by different assays.
- the binding moieties upon cleavage and release of the bispecific binding moieties and the XTEN from the AAC, have a diffusion coefficient in phosphate buffered saline that is at least 2 -fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, 10-fold, 20-fold, 50-fold, or 100-fold greater compared to the intact AAC composition.
- the apparent molecular weight of the intact AAC composition is at least 2-fold, at least 3 -fold, at least 4-fold, or at least 5 -fold, or at least 10-fold greater than the binding moieties released by cleavage of the RS by a mammalian protease, when the apparent molecular weight is determined by size exclusion chromatography (SEC).
- the hydrodynamic radius of the intact AAC composition is at least 2-fold, or at least 3 -fold, or at least 4-fold, or at least 5-fold, or at least 10- fold greater than the binding moieties released by cleavage of the RS by a mammalian protease, when the hydrodynamic radius is determined by size exclusion chromatography (SEC).
- the disclosure provides an AAC, wherein upon cleavage of the RS to release the binding moieties and the XTEN from the AAC, the hydrodynamic radius of the released binding moieties is less than about 30%, or less than about 40%, or less than about 50% of the hydrodynamic radius of the intact AAC, when hydrodynamic radius is assessed by size exclusion chromatography.
- the disclosure provides an AAC, wherein upon cleavage of the RS to release the binding moieties and the XTEN from the AAC, the hydrodynamic radius of the released binding moieties is less than about 5 nm, or less than about 4 nm, or less than about 3 nm when hydrodynamic radius is determined by size exclusion chromatography.
- the disclosure provides an AAC, wherein upon cleavage of the RS to release the binding moieties and the XTEN from the AAC, the released binding moieties having a hydrodynamic radius of less than about 5 nm, or less than about 4 nm, or less than about 3 nm, when hydrodynamic radius is determined by size exclusion
- the disclosure provides an AAC, wherein the hydrodynamic radius of the intact, uncleaved AAC is greater than about 8 nm, or greater than about 9 nm, or greater than about 10 nm, or greater than about 12 mm when hydrodynamic radius is determined by size exclusion chromatography.
- compositions will, by their design and linkage to XTEN, have enhanced pharmacokinetic properties when administered to a subject compared to the corresponding binding moieties not linked to XTEN.
- an AAC adenosine triphosphate
- composition administered to a subject using a therapeutically-effective dose exhibits a terminal half-life in a subject that is increased, upon or following administration to a subject, in comparison to the corresponding binding moieties not linked to the composition, by at least about 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, lO-fold, 20-fold, 30-fold, 40- fold, or 100-fold greater.
- an AAC composition administered to a subject using a therapeutically-effective dose exhibits increased area under the curve (AUC), upon or following administration to a subject, in comparison to the corresponding binding moieties not linked to the composition, of at least 25%, 50%, 100%, 200%, or at least 300% or more.
- an AAC composition administered to a subject using a therapeutically- effective dose exhibits a lower volume of distribution, upon or following administration to a subject, in comparison to the corresponding binding moieties not linked to the composition, of at least 25% lower, or 50%, or 100%, or 200%, or at least 300% lower.
- an AAC composition administered to a subject using a therapeutically-effective dose exhibits a terminal half-life of at least about 20 h, or at least about 30 h, or at least about 32 h, or at least about 48 h, or at least about 72 h, or at least about 96 h, or at least about 120 h, or at least about 144 h, or at least about 7 days, or at least about 10 days, or at least about 14 days following administration to a subject.
- the concentration of the released binding moieties in the circulation of a subject will be low, thereby contributing to the improved safety profile and lower incidence of side effects compared to bispecific compositions not having the protective XTEN and release segment.
- the disclosure provides an AAC, wherein the plasma Cmax concentration of the binding moieties released from the AAC by cleavage of the RS by a protease capable of cleaving the RS of the composition upon or following a single administration of the chimeric polypeptide composition to a subject does not exceed about 0.01 ng/ml, or about 0.03 ng/ml, or about 0.1 ng/ml, or about 0.3 ng/ml, or about 1 ng/ml, or about 10 ng/ml, or about 100 ng/ml.
- the disclosure provides an AAC, wherein the plasma Cmax concentration of the binding moieties released from the AAC following a single administration of the chimeric polypeptide composition to a subject in which the RS has been cleaved by a protease capable of cleaving the RS of the composition is a least lO-fold lower, or at least 30-fold lower, or at least lOO-fold, or at least 1000-fold lower than the plasma levels of the intact AAC in the same subject.
- the subject is a mouse, or a rat, or a dog, or a monkey, or a human.
- the AAC constructs described herein confer multiple therapeutic advantages over traditional monoclonal antibodies and other smaller bispecific molecules. Of particular note is the conditional activation of the AAC of the present disclosure.
- the constructs have a reduced ability to bind their intended target cell markers due to the shielding effect of the bulky, unstructured XTEN tethered to the AAC by the release segment.
- the specific activity to non-diseased, normal tissue of the exemplary compositions of the disclosure is significantly reduced when compared to that of analogous antibodies and antibody fragments.
- polypeptides to activate at their desired site of action (e.g., the proximity of a diseased tissue such as a tumor or cancer cell) while remaining inactive during their progress to this site is an advance in the field of immune-oncologic therapeutics, offering the promise of potent and specific therapeutics with improved therapeutic index, as well as a readily designable and manufacturable format.
- desired site of action e.g., the proximity of a diseased tissue such as a tumor or cancer cell
- the present disclosure provides cleavable recombinant polypeptide compositions and activatable antibody compositions and pharmaceutical compositions comprising a recombinant polypeptide or an activatable antibody that are particularly useful in medical settings; for example in the prevention, treatment and/or the amelioration of certain cancers, tumors or inflammatory diseases.
- a number of therapeutic strategies have been used to design the recombinant polypeptide compositions for use in methods of treatment of a subject with a cancerous disease, including the modulation of T cell responses by targeting TcR signaling, particularly using VL and VH portions of anti-human CD3 monoclonal antibodies that are widely used clinically in immunosuppressive regimes.
- the CD3 -specific monoclonal OKT3 was the first such monoclonal approved for use in humans (Sgro, Toxicology 105 (1995), 23-29) and is widely used clinically as an immunosuppressive agent in transplantation (Chatenoud L: Immunologic monitoring during OKT3 therapy. Clin Transplant 7:422 430, 1993).
- anti-CD3 monoclonals can induce partial T cell signaling and clonal anergy (Smith, J. Exp. Med. 185 (1997), 1413-1422).
- the OKT3 reacts with and blocks the function of the CD3 complex in the membrane of T cells; the CD3 complex being associated with the antigen recognition structure of T cells (TCR), which is essential for signal transduction.
- TCR T cells
- CD3 specific antibodies are able to induce various T cell responses, including cytokine production (Von Wussow, Human gamma interferon production by leukocytes induced with monoclonal antibodies recognizing T cells. J. Immunol. 127:1197-1200 (1981)), proliferation and suppressor T-cell induction.
- cytotoxic T cells In cancer, attempts have been made to utilize cytotoxic T cells to lyse cancer cells. Without being bound by theory, to effect target cell lysis, cytotoxic T cells are believed to require direct cell-to-cell contact; the TCR on the cytotoxic T cell must recognize and engage the appropriate antigen on the target cell. This creates the immunologic synapse that, in turn initiates a signaling cascade within the cytotoxic T cell, causing T-cell activation and the production of a variety of cytotoxic cytokines and effector molecules. Perforin and granzymes are highly toxic molecules that are stored in preformed granules that reside in activated cytotoxic T cells.
- the cytoplasmic granules of the engaged cytotoxic T cells migrate toward the cytotoxic T-cell membrane, ultimately fusing with it and releasing their contents in directed fashion into the immunological synapse to form a pore within the membrane of the target cell, disrupting the tumor cell plasma membrane.
- the created pore acts as a point of entry for granzymes; a family of serine proteases that that induce apoptosis of the tumor cells.
- the disclosure contemplates methods of use of AAC that are engineered to target a range of malignant cells, such as tumors, in addition to the effector cells, in order to initiate target cell lysis and to effect a beneficial therapeutic outcome in that the AAC are designed such that one binding moiety binds and engages CD3 to activate the cytotoxic T cell while the second binding moiety can be designed to target a variety of different target cell markers that are characteristic of specific malignancies; bridging them together for the creation of the immunological synapse.
- the physical binding of the cytotoxic effector cell and the cancer cell eliminates the need for antigen processing, MHCI/B2 -microglobulin, as well as co- stimulatory molecules.
- important tumor cell markers include, but are not limited to the markers of Table 5. Because of the range of tumor-specific markers (more extensively described, above) that can be engineered into the various embodiments of the subject compositions AAC, it will be appreciated that the resulting compositions will have utility against a variety of cancers, including solid and hematological tumors.
- the disclosure provides a method of treatment of a subject with a tumor.
- the tumor being treated can comprise tumor cells arising from a cell selected from the group consisting of stromal cell, fibroblasts, myofibroblasts, glial cells, epithelial cells, fat cells, lymphocytic cells, vascular cells, smooth muscle cells, mesenchymal cells, breast tissue cells, prostate cells, kidney cells, brain cells, colon cells, ovarian cells, uterine cells, bladder cells, skin cells, stomach cells, genito- urinary tract cells, cervix cells, uterine cells, small intestine cells, liver cells, pancreatic cells, gall bladder cells, bile duct cells, esophageal cells, salivary gland cells, lung cells, and thyroid cells.
- stromal cell fibroblasts, myofibroblasts, glial cells, epithelial cells, fat cells, lymphocytic cells, vascular cells, smooth muscle cells, mesenchymal cells, breast tissue cells, prostate cells, kidney cells, brain cells, colon cells, ovarian cells, uterine cells, bladder
- an activated effector cell can release and move on through the local tissue towards other target cancer cells, bind the target antigen, and initiate additional cell lysis.
- effector cell molecules such as perforin and granzymes will result in damage to tumor cells that are adjacent but not bound by a given molecule of the bispecific binding domains, resulting in stasis of growth or regression of the tumor.
- composition comprising an AAC described herein to a subject with a cancer or tumor having the target cell marker
- the composition can be acted upon by proteases in association with or co-localized with the cancer or tumor cells, releasing the fused FBM and SBM such that an immunological synapse can be created by the linking of the target cell and a effector cell, with the result that effector cell-derived effector molecules capable of lysing the target cell are released into the synapse, leading to apoptosis, cytolysis, or death of the target cancer or tumor cell.
- the disclosure relates to methods of treating a disease in a subject, such as a cancer or an inflammatory disorder.
- the disclosure provides a method of treating a disease in a subject, comprising administering to the subject in need thereof a therapeutically effective amount of a pharmaceutical composition comprising a recombinant polypeptide or AAC described herein.
- a therapeutically effective amount of the pharmaceutical composition may vary according to factors such as the disease state, age, sex, and weight of the individual, and the ability of the antibody or antibody portion to elicit a desired response in the individual.
- a therapeutically effective amount is also one in which any toxic or detrimental effects of the subject compositions are outweighed by the therapeutically beneficial effects.
- a prophylactically effective amount refers to an amount of pharmaceutical composition required for the period of time necessary to achieve the desired prophylactic result.
- the disease for treatment can be carcinomas, Hodgkin's lymphoma, non-Hodgkin's lymphoma, B cell lymphoma, T-cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, colon cancer, prostate cancer, head and neck cancer, any form of skin cancer, melanoma, genito- urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal
- carcinomatosis uterine serous carcinoma, endometrial cancer, cervical cancer, colorectal cancer, an epithelia intraperitoneal malignancy with malignant ascites, uterine cancer, mesothelioma in the peritoneum kidney cancers, lung cancer, small-cell lung cancer, non-small cell lung cancer, gastric cancer, esophageal cancer, stomach cancer, small intestine cancer, liver cancer, hepatocarcinoma, hepatoblastoma, liposarcoma, pancreatic cancer, gall bladder cancer, cancers of the bile duct, salivary gland carcinoma, thyroid cancer, epithelial cancer, adenocarcinoma, sarcomas of any origin, primary hematologic malignancies including acute or chronic
- the therapeutically effective amount can produce a beneficial effect in helping to treat (e.g., cure or reduce the severity) or prevent (e.g., reduce the likelihood of recurrence) of a cancer or a tumor.
- the pharmaceutical composition is administered to the subject as two or more therapeutically effective doses administered twice weekly, once a week, every two weeks, every three weeks, or monthly.
- the pharmaceutical composition is administered to the subject as two or more therapeutically effective doses over a period of at least two weeks, or at least one month, or at least two months, or at least three months, or at least four months, or at least five months, or at least six months.
- a first low priming dose is administered to the subject, followed by one or more higher maintenance doses over the dosing schedule of at least two weeks, or at least one month, or at least two months, or at least three months, or at least four months, or at least five months, or at least six months.
- the initial priming dose administered is selected from the group consisting of at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg, and one or more subsequent maintenance dose(s) administered is selected from the group consisting of at least about 0.02 mg/kg, at least about 0.05 mg/kg, at least about 0.1 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg/kg, at least 0.4.
- the pharmaceutical composition is administered to the subject intradermally, subcutaneously, intravenously, intra-arterially, intra-abdominally, intraperitoneally, intrathecally, or
- the pharmaceutical composition is administered to the subject as one or more therapeutically effective bolus doses or by infusion of 5 minutes to 96 hours as tolerated for maximal safety and efficacy.
- the pharmaceutical composition is administered to the subject as one or more therapeutically effective bolus doses or by infusion of 5 minutes to 96 hours, wherein the dose is selected from the group consisting of at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg, at least about 0.12 mg/kg, at least about 0.14 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg
- mg/kg at least about 0.5 mg/kg, at least about 0.6 mg/kg, at least about 0.7 mg/kg, at least about 0.8 mg/kg, at least about 0.9 mg/kg, at least about 1.0 mg/kg, at least about 1.5 mg/kg, or at least about 2.0 mg/kg.
- the pharmaceutical composition is administered to the subject as one or more therapeutically effective bolus doses or by infusion over a period of 5 minutes to 96 hours, wherein the administration to the subject results in a Cmax plasma concentration of the intact, uncleaved AAC of at least about 0.1 ng/mL to at least about 2 pg/mL or more in the subject that is maintained for at least about 3 days, at least about 7 days, at least about 10 days, at least about 14 days, or at least about 21 days.
- the therapeutically effective dose is at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg, at least about 0.12 mg/kg, at least about 0.14 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg/kg, at least 0.4 mg/kg, at least about 0.5 mg/kg, at least about 0.6 mg/kg, at least about 0.7 mg/kg, at least about 0.8 mg/kg, at least about 0.9 mg/kg, at least about 1.0 mg/kg, at least about 1.5 mg/kg, or at least about 2.0 mg/kg.
- an initial dose is selected from the group consisting of at least about 0.005 mg/kg, at least about 0.01 mg/kg, at least about 0.02 mg/kg, at least about 0.04 mg/kg, at least about 0.08 mg/kg, at least about 0.1 mg/kg
- a subsequent dose is selected from the group consisting of at least about 0.1 mg/kg, at least about 0.12 mg/kg, at least about 0.14 mg/kg, at least about 0.16 mg/kg, at least about 0.18 mg/kg, at least about 0.20 mg/kg, at least about 0.22 mg/kg, at least about 0.24 mg/kg, at least about 0.26 mg/kg, at least about 0.27 mg/kg, at least about 0.28 mg/kg, at least 0.3 mg/kg, at least 0.4.
- the administration to the subject results in a plasma concentration of the recombinant polypeptide of at least about 0.1 ng/mL to at least about 2 ng/mL or more in the subject for at least about 3 days, at least about 7 days, at least about 10 days, at least about 14 days, or at least about 21 days.
- the subject can be mouse, rat, monkey, and human.
- the pharmaceutical compositions can be used for the treatment of epithelial cancer, preferably adenocarcinomas, or minimal residual disease, more preferably early solid tumor, advanced solid tumor or metastatic solid tumor.
- epithelial cancer preferably adenocarcinomas, or minimal residual disease, more preferably early solid tumor, advanced solid tumor or metastatic solid tumor.
- the pharmaceutical compositions can be used for the treatment of epithelial cancer, preferably adenocarcinomas, or minimal residual disease, more preferably early solid tumor, advanced solid tumor or metastatic solid tumor.
- epithelial cancer preferably adenocarcinomas, or minimal residual disease, more preferably early solid tumor, advanced solid tumor or metastatic solid tumor.
- compositions provided in this disclosure are useful in the treatment of sarcomas.
- pharmaceutical compositions comprising a recombinant polypeptide provided in this disclosure are useful in the treatment of lymphomas and leukemias, including primary hematologic malignancies including acute or chronic lymphocytic leukemias, acute or chronic myelogenous leukemias, myeloproliferative neoplastic disorders, or myelodysplastic disorders, B-cell disorders such as B-cell lymphoma, Hodgkin's lymphoma, and non-Hodgkin's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, B-cell derived chronic lymphatic leukemia (B-CLL) and/or having a B-cell related autoimmune disease such as myasthenia gravis, Morbus Basedow, Hashimoto thyroiditis, or Goodpasture syndrome.
- B-CLL B-cell related
- compositions comprising a recombinant polypeptide provided in this disclosure are useful in the treatment of cancers leading to ascites, including genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervix cancer, colorectal, uterine cancer, mesothelioma in the peritoneum, pancreatic cancer, colon cancer, colon cancer with malignant ascites, and gastric cancer.
- the disclosure provides a method of for achieving a beneficial effect in a cancer or tumor mediated by administration of pharmaceutical compositions comprising recombinant polypeptide or AAC compositions.
- the disclosure provides the use of a pharmaceutical composition in a method of treatment of a cancer or tumor in a subject in need thereof by administration of a therapeutically effective amount of the pharmaceutical composition in which one binding moiety of the composition is derived from a parental antibody that binds to an effector cell CD3 antigen and a second binding domain is derived from a parental antibody that binds to a target cell marker antigen selected from the group consisting of A33 antigen, alpha-fetoprotein (AFP), alpha 4 integrin, Ang2, B7-H3, B7-H6, B-cell maturation antigen (BCMA), cancer antigen 19-9 (CA19-9), cancer antigen 125 (CA-125), Carbonic Anhydrase 6 (CA6), carbonic anhydrase IX (CAI
- Chemokine Receptor 5 CCR5
- Chemokine Receptor 7 CCR7
- Chemokine Receptor 9 CCR9, Cluster of Differentiation 7 (CD7), CD22, CD70, CD79a, CD79b, CD 19, CCR8, CEA, hCG, Lewis-Y, CA19-9, CA-125, CD20, CD22, CD25, CD33, CD38, CD30, CD44v6, CD47, CD56 (NCAM), CD63, CD79b, CD123, CD133, CD138, CD166, claudin-l, claudin 18.2, C-type lectin-like molecule-l (CLL-l), C-type lectin domain family 12 (CLEC12), Cora antigen, delta like canonical notch ligand 3 (DDL3), desmoglein 4, delta like non-xanonical notch ligand 1 (DLK1), Ectonucleotide Pyrophosphatase/ Phosphodiesterase 3 (ENPP3), EGFR, EGFRvIII, EpCAM, endosialin (CD248), epiderma
- the administration of the therapeutically effective amount of the pharmaceutical composition leads to the eradication or amelioration of the underlying cancer or tumor disorder such that an improvement is observed in the subject, notwithstanding that the subject may still be afflicted with the underlying disorder.
- the disclosure provides use of a pharmaceutical composition comprising an AAC in a method of treatment of a cancer or tumor in a subject by administration of a therapeutically effective amount of the pharmaceutical composition in which one binding moiety of the composition is derived from a parental antibody directed to an effector cell selected from the group consisting of the antibodies of Table 4 and a second binding moiety is derived from a parental antibody that binds to an target cell target antigen selected from the group consisting of the antibodies of Table 5 and further comprising one or more RS of Table 1 or Table 2 and one or more XTEN of Table 8 or Table 10, the AAC having a configuration as described herein.
- the pharmaceutical composition doses of the method are administered as a bolus dose.
- the pharmaceutical composition doses of the method are each administered by intravenous infusion. In another embodiment, the pharmaceutical composition doses of the method are each administered by intraabdominal infusion. In another embodiment, the pharmaceutical composition doses of the method are each administered by intra-arterial infusion. In another embodiment, the pharmaceutical composition doses of the method are each administered by subcutaneous injection. In another embodiment, the pharmaceutical composition doses of the method are each administered by intramuscular injection.
- the subject is selected from the group consisting of mouse, rat, dog, monkey, and human.
- the disclosure relates to a method of treating a cancer or a tumor in a subject according to a treatment regimen.
- the disclosure provides a method of treating a cancer or a tumor in a subject comprising administering to the subject with the disease according to a treatment regimen comprising two or more consecutive doses of a therapeutically effective amount of a pharmaceutical composition comprising a recombinant polypeptide or AAC composition disclosed herein.
- the disclosure provides a method of treating a cancer or a tumor in a subject comprising administering to the subject with the disease according to a treatment regimen comprising two or more consecutive doses of a therapeutically effective amount of the pharmaceutical composition wherein the administration of the therapeutically effective amount of a pharmaceutical composition to the subject achieves a beneficial therapeutic effect including.
- the disclosure provides a method of treating a cancer or a tumor in a subject comprising administering to the subject with the disease according to a treatment regimen comprising two or more consecutive doses of a therapeutically effective amount of a pharmaceutical composition disclosed herein wherein the treatment regimen results in the improvement of a clinical parameter or endpoint associated with the disease in the subject.
- the clinical parameter or endpoint is selected from one or any combination of the group consisting of tumor shrinkage as a complete, partial or incomplete response; time-to-progression; time to treatment failure; biomarker response;
- progression-free survival disease free-survival; time to recurrence; time to metastasis; time of overall survival; improvement of quality of life; and improvement of symptoms.
- the disclosure relates to a method of use in which the treatment regimen is part of a specified treatment cycle.
- the specified treatment cycle of the treatment regimen comprises administration of a pharmaceutical composition comprising a recombinant polypeptide or AAC disclosed herein twice a week, every week, every 10 days, every two weeks, every three weeks, or every month per each treatment cycle.
- the treatment regimen is used in treatment of a disease, wherein the disease is selected from the group consisting of carcinomas, Hodgkin's lymphoma, non-Hodgkin's lymphoma, B cell lymphoma, T-cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, colon cancer, prostate cancer, head and neck cancer, any form of skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervical cancer, colorectal cancer, an epithelia intraperitoneal malignancy with malignant ascites, uterine cancer, mesothelioma in the peritoneum kidney cancers, lung cancer, small-cell lung cancer, non-small cell lung cancer, gastric cancer, esophageal cancer, stomach cancer, small intestin
- myeloproliferative neoplastic disorders or myelodysplastic disorders, myasthenia gravis,
- the disclosure relates to improved methods of inducing death of a target cell, such as a cancer cell, utilizing the subject recombinant polypeptide or AAC compositions disclosed herein, wherein the method effects death or induces apoptosis in the target cell or tissue, but with reduced toxicity and side effects.
- a target cell such as a cancer cell
- the method effects death or induces apoptosis in the target cell or tissue, but with reduced toxicity and side effects.
- the enhanced properties of the compositions permit lower-dose
- the subject compositions can have superior efficacy and safety compared to the corresponding binding moieties not linked to the recombinant polypeptides or AAC because of the ability of the attached bulking moiety to reduce the non-specific binding to healthy tissues and to prevent extravasation from the circulatory system in healthy tissue, while permitting enhanced penetration and binding into the cancer or tumor tissue upon the cleavage of the RS and release of the binding moieties; thus resulting in a differential compartmentalization of the prodrug form versus the released binding moieties upon cleavage of the composition.
- the disclosure provides a method of inducing death of a target cell, the method comprising contacting the target cell and an effector cell with an AAC described herein, wherein the contact results in an effect in the target cell selected from the group consisting of loss of membrane integrity, pyknosis, karyorrhexis, inducement of the intrinsic pathway of apoptosis, inducement of the extrinsic pathway of apoptosis, apoptosis, cell lysis, and cell death.
- the effect can be determined in an in vitro cell-based assay comprising a mixed population of the target cells and the effector cells, and an effective amount of the recombinant polypeptide having binding affinity for the target cell marker and the effector cell.
- the disclosure provides methods of inducing death of a target cell in a subject having a cancer comprising a population of the target cell.
- the method comprises administering a therapeutically effective amount of a
- the method comprises administering the pharmaceutical composition as one or more consecutively administered therapeutically effective doses.
- the method comprises determining the amount of a pharmaceutical composition needed to achieve a therapeutic effect in the subject having the cancer and administering the amount as two or more consecutive doses to the subject.
- the cancer is selected from the group consisting of carcinoma, Hodgkin's lymphoma, and non-Hodgkin's lymphoma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma, blastoma, breast cancer, ER/PR+ breast cancer, Her2+ breast cancer, triple-negative breast cancer, colon cancer, colon cancer with malignant ascites, mucinous tumors, prostate cancer, head and neck cancer, skin cancer, melanoma, genito-urinary tract cancer, ovarian cancer, ovarian cancer with malignant ascites, peritoneal carcinomatosis, uterine serous carcinoma, endometrial cancer, cervix cancer, colorectal, uterine cancer, mesothelioma in the peritoneum, kidney cancer, Wilm's tumor, lung cancer, small-cell lung cancer, non-small cell lung cancer, gastric cancer, stomach cancer, small intestine cancer, liver cancer
- the method comprises administering a therapeutically effective amount of the pharmaceutical composition to the subject wherein the method results in an improvement of a clinical parameter or endpoint.
- Exemplary clinical parameters or endpoints can be overall survival, symptom endpoints, disease-free survival, objective response rate, complete response, duration of response, progression-free survival, time to progression, time-to- treatment failure, tumor measurement, tumor size, tumor response rate, time to metastasis, and biomarker concentration.
- the method comprises administering a therapeutically effective amount of the pharmaceutical composition to the subject wherein the method results in a reduction in the frequency, duration, or severity in diagnostically associated side effects in the subject compared to administration of a comparable dose, in mmoles/kg, to a comparable subject of a composition comprising the FBM and SBM of the AAC, wherein the side effects are selected from the group consisting of increased plasma levels of IL-2, increased plasma levels of TNF-alpha, increased plasma levels of IFN-gamma, sepsis, febrile neutropenia, neurotoxicity, convulsions, encephalopathy, cytokine release syndrome, speech disturbance, equilibrium disturbance, fever, headache, confusion, hypotension, neutropenia, nausea, impaired consciousness, disorientation, and increased liver enzymes.
- the side effects are selected from the group consisting of increased plasma levels of IL-2, increased plasma levels of TNF-alpha, increased plasma levels of IFN-gamma, sepsis, febrile neutropenia, neurotoxicity, convulsions
- the methods of the disclosure may include administration of consecutive doses of a therapeutically effective amount of the pharmaceutical composition for a period of time sufficient to achieve and/or maintain the desired parameter or clinical effect, and such
- consecutive doses of a therapeutically effective amount establishes the therapeutically effective dose regimen for the pharmaceutical composition; i.e., the schedule for consecutively
- the doses are given in therapeutically effective amounts to result in a sustained beneficial effect on any clinical sign or symptom, aspect, measured parameter or characteristic of a cancer disease state or condition, including, but not limited to, those cancers and tumors described herein.
- a method of treatment comprises administration of a therapeutically effective dose of a pharmaceutical composition comprising the recombinant polypeptide or A AC to a subject in need thereof that results in a gain in time spent within a therapeutic window established for the binding moiety components of the pharmaceutical composition compared to the corresponding binding moiety components not linked to the fusion protein and administered at a comparable molar dose to a subject.
- the gain in time spent within the therapeutic window is at least about three-fold, or at least about four-fold, or at least about five-fold, or at least about six- fold, or at least about eight-fold, or at least about 10-fold, or at least about 20-fold, or at least about 40-fold, or at least about 50-fold, or at least about 100-fold greater compared to the corresponding binding moiety components not linked to the recombinant protein or AAC and administered at a comparable molar dose to a subject.
- the methods further provide that administration of multiple consecutive doses of a pharmaceutical composition administered using a therapeutically effective dose regimen to a subject in need thereof can result in a gain in time between consecutive Cmax peaks and/or Cmin troughs for blood levels of the composition compared to the corresponding binding moiety components not linked to the fusion protein.
- the gain in time spent between consecutive Cmax peaks and/or Cmin troughs can be at least about three-fold, or at least about four-fold, or at least about five-fold, or at least about six-fold, or at least about eight-fold, or at least about 10-fold, or at least about 20- fold, or at least about 40-fold, or at least about 50-fold, or at least about 100-fold longer compared to the corresponding binding moiety component(s) not linked to the fusion protein and administered using a comparable molar dose regimen established for the targeting components.
- composition can result in an improvement in at least one parameter known to be useful for assessing the subject cancer or tumor using a lower unit dose in moles of recombinant polypeptide or AAC compared to the corresponding binding moiety components not linked to the recombinant polypeptide or AAC and administered at a comparable molar dose or dose regimen to a subject.
- the disclosure provides methods of manufacturing an AAC.
- the method comprises culturing a host cell comprising a nucleic acid construct that encodes an activatable recombinant polypeptide under conditions that lead to expression of the activatable recombinant polypeptide, wherein the activatable recombinant polypeptide comprises an RS1, RS2, FBM, SBM, XTEN1, and XTEN2, wherein: i) the RS1 and RS2, wherein the RS1 and RS2 are each substrates for cleavage by a mammalian protease and each comprise an amino acid sequence having at least 88% or at least 94%, or 100% sequence identity to a sequence selected from the sequences of Table 1; ii) the FBM is an antibody fragment comprising a VL and VH derived from a monoclonal antibody having binding specificity to CD3; iii) the SBM is an antibody fragment comprising a VL and VH derived
- Chemokine Receptor 4 CCR4
- C-C Motif Chemokine Receptor 5
- CCR5 CCR5
- Chemokine Receptor 6 CCR6
- C-C Motif Chemokine Receptor 7
- CCR7 C-C Motif
- Chemokine Receptor 8 CCR8
- C-C Motif Chemokine Receptor 9 CCR9
- CD7 Differentiation 7
- globohexaosylceramide globo-H
- GD2, Glypican 3 Glypican 3
- GCC guanylyl cyclase C
- HER2, HER2 neu HER3, HER4, HER1, ILl3Ra2, insulin-like growth factor I receptor (IGF-IR ), Lysosomal Associated Membrane Protein 1 (LAMP1), Ll Cell Adhesion Molecule (L1CAM), lymphocyte antigen 6 (Ly-6), melanoma chondroitin sulfate proteoglycan (MCSP), Membrane- type metalloproteinase (MT-MMP), mesothelin, mucin 1 (MUC1), MUC2, MUC3, MUC4, MUC5AC, MUC5B, MUC7, MUC16, Muellerian inhibitory substance receptor type II (MISIIR), nectin cell adhesion molecule 4 (Nectin-4), 6-transmembrane epithelial
- the activatable recombinant polypeptide is activated by cleavage of the RS1 and RS2 by one or more proteases capable of cleaving the RS1 and RS2, resulting in the release of the FBM and SBM from the composition, wherein the FBM and SBM remain fused and the XTEN1 and XTEN2 of the activatable recombinant polypeptide in an uncleaved state interfere with specific binding of the FBM to the CD3 and the SBM to the target cell marker such that the dissociation constant (IQ) of the FBM of the activatable recombinant polypeptide in an uncleaved state towards CD3 or the SBM to the target cell marker is at least 100 times greater compared to the FBM or the SBM released from the activatable recombinant polypeptide by cleavage of the RS1 and RS2, when measured in in vitro assays comprising the target cell marker under comparable conditions, e.g., equivalent molar
- the present disclosure relates to isolated polynucleotide sequences encoding the recombinant polypeptide or AAC compositions and sequences complementary to polynucleotide molecules encoding the recombinant polypeptide or AAC compositions.
- the disclosure provides polynucleotides encoding the
- the disclosure provides an isolated polynucleotide sequence encoding a recombinant polypeptide consisting of an amino acid sequence with at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to an amino acid sequence set forth in Table 11, Tables 14-16, or Table 18, or the complement of the polynucleotide sequence.
- the disclosure provides an isolated polynucleotide sequence encoding an AAC composition wherein the polynucleotide sequence has at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% sequence identity to a polynucleotide sequence set forth in Table 11, Table 16 or Table 18.
- the disclosure relates to methods to produce polynucleotide sequences encoding the recombinant polypeptide composition or AAC embodiments, or sequences complementary to the polynucleotide sequences, including homologous variants thereof, as well as methods to express the proteins expressed by the polynucleotide sequences.
- the methods include producing a polynucleotide sequence coding for the proteinaceous recombinant polypeptide or AAC composition and incorporating the encoding gene into an expression vector appropriate for a host cell.
- the method includes transforming an appropriate host cell with the expression vector, and culturing the host cell under conditions causing or permitting the resulting recombinant polypeptide or AAC to be expressed in the transformed host cell, thereby producing the recombinant polypeptide or AAC, which is recovered by methods described herein or by standard protein purification methods known in the art. Standard recombinant techniques in molecular biology are used to make the polynucleotides and expression vectors of the present disclosure.
- nucleic acid sequences that encode recombinant polypeptide compositions are used to generate recombinant DNA molecules that direct the expression in appropriate host cells.
- Several cloning strategies are suitable for performing the present disclosure, many of which are used to generate a construct that comprises a gene coding for a composition of the present disclosure, or its complement. In one
- the cloning strategy is used to create a gene that encodes a recombinant
- the genes can comprise nucleotides encoding the binding moieties, release segments, and the bulking moieties in the configurations disclosed herein.
- a construct is first prepared containing the DNA sequence
- recombinant polypeptide construct corresponding to recombinant polypeptide construct.
- exemplary methods for the preparation of such constructs are described in the Examples.
- the construct is then used to create an expression vector suitable for transforming a host cell, such as a prokaryotic host cell for the expression and recovery of the recombinant polypeptide construct.
- the host cell is an E. coli.
- Exemplary methods for the creation of expression vectors, the transformation of host cells and the expression and recovery of XTEN are described in the Examples.
- the gene encoding for the recombinant polypeptide construct can be made in one or more steps, either fully synthetically or by synthesis combined with enzymatic processes, such as restriction enzyme-mediated cloning, PCR and overlap extension, including methods more fully described in the Examples.
- the methods disclosed herein can be used, for example, to ligate sequences of polynucleotides encoding the various components (e.g., binding domains, linkers, release segments, and XTEN) genes of a desired length and sequence.
- Genes encoding recombinant polypeptide compositions are assembled from oligonucleotides using standard techniques of gene synthesis. The gene design can be performed using algorithms that optimize codon usage and amino acid composition appropriate for the E.
- coli host cell utilized in the production of the recombinant polypeptide.
- a library of polynucleotides encoding the components of the constructs is created and then assembled, as described above.
- the resulting genes are then assembled and the resulting genes used to transform a host cell and produce and recover the recombinant polypeptide compositions for evaluation of its properties, as described herein.
- the resulting polynucleotides encoding the recombinant polypeptide sequences can then be individually cloned into an expression vector.
- the nucleic acid sequence is inserted into the vector by a variety of procedures.
- DNA is inserted into an appropriate restriction endonuclease site(s) using techniques known in the art.
- Vector components generally include, but are not limited to, one or more of a signal sequence, an origin of replication, one or more marker genes, an enhancer element, a promoter, and a transcription termination sequence.
- the vector may, for example, be in the form of a plasmid, cosmid, viral particle, or phage that may conveniently be subjected to recombinant DNA procedures, and the choice of vector will often depend on the host cell into which it is to be introduced.
- the vector may be an autonomously replicating vector, i.e., a vector, which exists as an extrachromosomal entity, the replication of which is independent of chromosomal replication, e.g., a plasmid.
- the vector may be one which, when introduced into a host cell, is integrated into the host cell genome and replicated together with the chromosome(s) into which it has been integrated.
- the disclosure provides for the use of plasmid expression vectors containing replication and control sequences that are compatible with and recognized by the host cell, and are operably linked to the gene encoding the polypeptide for controlled expression of the polypeptide.
- the vector ordinarily carries a replication site, as well as sequences that encode proteins that are capable of providing phenotypic selection in transformed cells.
- Such vector sequences are well known for a variety of bacteria, yeast, and viruses.
- Useful expression vectors that can be used include, for example, segments of chromosomal, non-chromosomal and synthetic DNA sequences.
- “Expression vector” refers to a DNA construct containing a DNA sequence that is operably linked to a suitable control sequence capable of effecting the expression of the DNA encoding the polypeptide in a suitable host. The requirements are that the vectors are replicable and viable in the host cell of choice. Low- or high-copy number vectors may be used as desired.
- Suitable vectors include, but are not limited to, derivatives of SV40 and pcDNA and known bacterial plasmids such as col El, pCRl, pBR322, pMal-C2, pET, pGEX as described by Smith, et al., Gene 57:31-40 (1988), pMB9 and derivatives thereof, plasmids such as RP4, phage DNAs such as the numerous derivatives of phage I such as NM98 9, as well as other phage DNA such as Ml 3 and filamentous single stranded phage DNA; yeast plasmids such as the 2 micron plasmid or derivatives of the 2m plasmid, as well as centomeric and integrative yeast shuttle vectors; vectors useful in eukaryotic cells such as vectors useful in insect or mammalian cells; vectors derived from combinations of plasmids and phage DNAs, such as plasmids that have been modified to employ phage
- Yeast expression systems that can also be used in the present disclosure include, but are not limited to, the non-fusion pYES2 vector (Invitrogen), the fusion pYESHisA, B, C (Invitrogen), pRS vectors and the like.
- the control sequences of the vector include a promoter to effect transcription, an optional operator sequence to control such transcription, a sequence encoding suitable mRNA ribosome binding sites, and sequences that control termination of transcription and translation.
- the promoter may be any DNA sequence, which shows transcriptional activity in the host cell of choice and may be derived from genes encoding proteins either homologous or heterologous to the host cell.
- Promoters suitable for use in expression vectors with prokaryotic hosts include the b-lactamase and lactose promoter systems [Chang et al., Nature, 275:615 (1978); Goeddel et al., Nature, 281:544 (1979)], alkaline phosphatase, a tryptophan (trp) promoter system [Goeddel, Nucleic Acids Res., 8:4057 (1980); EP 36,776], and hybrid promoters such as the tac promoter [deBoer et al., Proc. Natl. Acad. Sci. USA, 80:21-25 (1983)], all is operably linked to the DNA encoding CFXTEN polypeptides. Promoters for use in bacterial systems can also contain a Shine-Dalgamo (S.D.) sequence, operably linked to the DNA encoding recombinant polypeptide polypeptides.
- S.D. Shine-Dalgamo
- the disclosure relates to methods of making the recombinant polypeptide compositions at high fermentation expression levels of functional protein using an E. coli host cell, as well as providing expression vectors encoding the constructs useful in methods to produce the cytotoxically active polypeptide construct compositions at high expression levels.
- the method comprises the steps of 1) preparing the polynucleotide encoding the recombinant polypeptide or AAC of any of the embodiments disclosed herein, 2) cloning the polynucleotide into an expression vector, which can be a plasmid or other vector under control of appropriate transcription and translation sequences for high level protein expression in a biological system, 3) transforming an appropriate E.
- the expression of the recombinant polypeptide or AAC results in fermentation titers of at least 0.1 g/L, or at least 0.2 g/L, or at least 0.3 g/L, or at least 0.5 g/L, or at least 0.6 g/L, or at least 0.7 g/L, or at least 0.8 g/L, or at least 0.9 g/L, or at least 1 g/L of the expression product of the host cell and wherein at least 70%, or at least 80%, or at least 90%, or at least 95%, or at least 97%, or at least 99% of the expressed protein are correctly folded.
- the term“correctly folded” means that the binding moiety component of the composition has the ability to specifically bind its target ligand.
- the disclosure provides a method for producing a recombinant polypeptide or AAC composition, the method comprising culturing in a fermentation reaction a host cell that comprises a vector encoding a polypeptide comprising the recombinant polypeptide or AAC composition under conditions effective to express the polypeptide product at a concentration of more than about 10 milligrams/gram of dry weight host cell (mg/g), or at least about 250 mg/g, or about 300 mg/g, or about 350 mg/g, or about 400 mg/g, or about 450 mg/g, or about 500 mg/g of said polypeptide when the fermentation reaction reaches an optical density of at least 130 at a wavelength of 600 nm, and wherein the binding moieties of the expressed protein are correctly folded.
- mg/g milligrams/gram of dry weight host cell
- the disclosure provides a method for producing a recombinant polypeptide composition or AAC, the method comprising culturing in a fermentation reaction a host cell that comprises a vector encoding a polypeptide comprising the recombinant polypeptide or AAC composition under conditions effective to express the polypeptide product at a concentration of more than about 10 milligrams/gram of dry weight host cell (mg/g), or at least about 250 mg/g, or about 300 mg/g, or about 350 mg/g, or about 400 mg/g, or about 450 mg/g, or about 500 mg/g of said polypeptide when the fermentation reaction reaches an optical density of at least 130 at a wavelength of 600 nm, and wherein the expressed polypeptide product is soluble.
- mg/g milligrams/gram of dry weight host cell
- Example 1 Construction of ProTIA construct with anti-EpCAM-anti-CD3-XTEN with
- Transformants were screened by DNA miniprep and the desired construct was confirmed by DNA sequencing.
- the final vector encodes the ProTIA molecule with the components (in the N- to C-terminus) of anti-EpCAM-anti-CD3 bispecific tandem scFv with BSRS-l as release segment fused to XTEN 864 gene under the control of a PhoA promoter and STII secretion leader.
- the resulting construct is AC 1278, with the DNA sequence and encoded amino acid sequence provided in Table 11.
- Another anti-EpCAM anti-CD3-XTEN with Release Segment designated AC1476 and with the DNA sequence and encoded amino acid sequence provided in Table 11 as well, was constructed in a similar manner into base vector pYS0044-XTEN864-H6 base vector.
- the underscored sequence represents signal peptide, which is cleaved off during secretion and is absent in the final mature protein.
- Table 11 DNA and amino acid sequence of AC1278 and AC1476 anti-EpCAM-anti-CD3- XTEN with Release Segment
- Example 2 Production of uncleaved and cleaved His8-aEnCAM-aCD3-BSRSl- XTEN864 from E. coli fermentation culture
- the fusion protein AC1278 (MKKNIAFLLASMFVFSIATNAYA-His(8)-aEpCAM- aCD3-BSRSl-XTEN_AE864) was expressed in a proprietary coli AmE098 strain.
- a 10L fermentation culture was grown at 37°C and temperature shifted to 26°C following depletion of the salt feed. During harvest, fermentation whole broth was centrifuged to pellet the cells. The supernatant was collected, and acid flocculation was then used to reduce endotoxin and host cell protein contamination. Using 1 M acetic acid, the supernatant pH was gradually lowered to pH
- Triton Wash buffer (20 mM Tris, 100 mM NaCl, 0.l%Triton X-l 14, pH 7.5
- Wash 2 buffer (20 mM Tris, 100 mM NaCl, pH 7.5) were prepared in advance, stored at 4°C, and kept on ice during use. After column equilibration, the supernatant was loaded to the column.
- Hydrophobic interaction chromatography was chosen as the subsequent polishing step.
- Two 20-mL RedisepRf 25G column housings (Teledyne Isco) were packed with 20 mL of Toyopearl-Phenyl-650M resin (TOSOH Biosciences). The columns were sanitized with 0.5M NaOH, thoroughly rinsed with distilled water, and equilibrated with 5 CVs of Buffer A (20 mM Tris, 1M (NH 4 ) 2 S0 4 , pH 7.5). Elution buffers at 75% Buffer A, 50% Buffer A, and 25% Buffer A were prepared in advance by mixing appropriate volumes of Buffer A and Buffer B (20 mM Tris, pH 7.5).
- IMAC elutions CV1 and 2 were pooled together from the previous column step, and ammonium sulfate was added to a final concentration of 1M before loading to the pre- equilibrated Phenyl columns. After loading and chasing with 3 CVs of Buffer A, the column was eluted with 3 CVs each of 75% Buffer A, 50% Buffer A, 25% Buffer A, and 0% Buffer A. The load, flowthrough, and elutions were analyzed by non-reducing 4-12% Bis-Tris SDS-PAGE and Coomassie staining. Based on the gel, wash and elutions CV1-2 at 750 mM (NH 4 ) 2 S0 4 (boxed) were pooled for further processing (FIG. 14B).
- anion exchange chromatography was chosen as the final polishing step.
- a XK16 column housing on AKTApurifier was packed with 5 mL of Capto Q Impress resin (GE Healthcare), sanitized with 0.5M NaOH, thoroughly rinsed with distilled water, stripped with 2 CVs of Buffer B (20 mM Tris, 500 mM NaCl, pH 7.5), and equilibrated with 5 CVs of Buffer A (20 mM Tris pH 7.5). The HIC elution pool was diluted 4 fold before loading to the column.
- Desired elution fractions (boxed in FIG. 14C) were concentrated and buffer exchanged into 50 mM Tris, 150 mM NaCl, pH 7.5. Formulated product was 0.2 pm sterile filtered. Lot release to determine product quality involved size exclusion chromatography analysis and SDS- PAGE analysis.
- SEC analysis 10 pg of formulated product was injected to an analytical SEC column, confirming >95% monomeric product. (FIG. 15 A). SDS-PAGE analysis was conducted by loading 5 pg of formulated product to a 4-12% Bis-Tris gel and staining with Coomassie Blue. The product purity was >90% (FIG. 15B).
- Recombinant mouse MMP-9 was supplied as zymogen from R&D Systems and required activation by 4-aminophenylmercuric acetate (APMA).
- APMA 4-aminophenylmercuric acetate
- APMA was first dissolved in 0.1M NaOH to a final concentration of 10 mM before the pH was readjusted to neutral using 0.1N HC1. Further dilution of the APMA stock to 2.5 mM was done in 50 mM Tris, 150 mM NaCl, 10 mM CaCl 2 pH 7.5.
- pro-MMP 1 mM APMA and 100 ug/mL of pro-MMP-9 were incubated at 37 °C for 3 hours.
- Activated enzyme added to a final concentration of 50% glycerol could then be stored at -20°C for several weeks.
- Coomassie Blue staining allowed visualization of the full- length His8-aEpCAM-aCD3-BSRS 1 -XTEN864 (ProTIA-X) before MMP-9 digestion and the His8-aEpCAM-aCD3 cleaved fragment (ProTIA-A) after MMP-9 digestion (FIG. 16A).
- Desired elutions (boxed in FIG. 16B) were concentrated and buffer exchanged into 50 mM Tris, 150 mM NaCl, pH 7.5. Lot release to determine product quality involved size exclusion chromatography analysis and SDS-PAGE analysis.
- SEC analysis 10 pg of product was injected to an analytical SEC column, confirming >95% monomeric product (FIG. 17A).
- SDS-PAGE analysis 5 pg of product was loaded on a 4-12% Bis-Tris gel, confirming >90% product purity (FIG. 17B).
- Example 3 Production of uncleaved and cleaved AC 1476 aEpCAM-aCD3-BSRSl- XTEN AE864-His(6) fromE. coli fermentation culture
- the fusion protein AC1476 (MKKNIAFLLASMFVFSIATNAYA-aEpCAM-aCD3- BSRSl-XTEN_AE864-His(6)) was expressed in a proprietary E. coli AmE097 strain.
- a 10L fermentation culture was grown at 37°C and temperature shifted to 28°C after depletion of the salt feed. During harvest, fermentation whole broth was centrifuged to pellet the cells. The supernatant was 0.20 pm filtered using a 3M LifeAssure filter capsule.
- a XK50 housing column was packed with 100 mL of Toyopearl-AF-Chelate-650M resin (TOSOH Biosciences) and connected to a peristaltic pump at 4°C.
- the column was sanitized with 0.5M NaOH, thoroughly rinsed with distilled water, charged with 0.1M NiS0 4 , and equilibrated with 5 CVs of equilibration buffer (20 mM Tris, 250 mM NaCl, pH 7.5). After column equilibration, the supernatant was loaded to the column, followed by Triton Wash, Wash 2, and elution similar to the process described above in Example 2-1. Elutions were collected in 1 ⁇ 4 CV (25 mL) fractions and EDTA was added to a final concentration of 5 mM to chelate free nickel.
- Hydrophobic interaction chromatography was chosen as the subsequent polishing step.
- a XK24 housing column on AKTApurifier was packed with 50 mL of Toyopearl-Phenyl- 650M resin (TOSOH Biosciences). The column was sanitized with 0.5M NaOH, thoroughly rinsed with distilled water, and equilibrated with 5 CVs of Buffer A (20 mM Tris, 1M
- Anion exchange chromatography was chosen as the final polishing step.
- a XK24 housing column was packed with 30 mL Capto Q Impress resin (GE Healthcare), sanitized with 0.5M NaOH, thoroughly rinsed with distilled water, stripped with 2 CVs of Buffer B (20 mM Tris, 500 mM NaCl, pH 7.5), and equilibrated with 5 CVs of Buffer A (20 mM Tris, pH 7.5).
- the elution pool was buffer exchanged through a Pellicon XL Ultrafiltration module Biomax 10 kDa into 20 mM Tris pH 7.5 until the permeate had a conductivity of 8 ms/cm.
- the permeate was loaded to the Capto Q Impress column, and the column was then washed with 3 CVs of 10% and 20 % Buffer B.
- Elutions were collected in 1 ⁇ 4 CV (7.5 mL) fractions in a gradient from 20% to 70% Buffer B over 10 CVs.
- the load, flowthrough, and elutions were analyzed by non- reducing 4-12% Bis-Tris SDS-PAGE and Coomassie staining. Based on the gel, selected elutions (boxed) were pooled for formulation (FIG. 18C).
- Desired elutions were concentrated and buffer exchanged into 50 mM Tris, 150 mM NaCl, pH 7.5. Lot release to determine product quality was performed following protocol established in Example 2 for SEC analysis (FIG. 19A) and SDS-PAGE (FIG. 19B). Additionally, 2 pg was loaded to a 4-12% Bis-Tris non-reducing SDS-PAGE gel, with subsequent silver staining (FIG. 19C). The results of SEC were also used to determine the apparent molecular weight and apparent molecular weight factor (relative to actual molecular weight) and the hydrodynamic radius of the aEpCAM-aCD3-BSRSl-XTEN864-His(6). The apparent molecular weight determined was 1.7 MDa, which would result in an apparent molecular weight factor of 12.3 and a calculated hydrodynamic radius of 10.8 nm.
- electrospray ionization mass spectrometry was performed and the experimental mass was determined to be 138,652 Da, with AMass of +1 Da when compared to theoretical molecular weight of 138,651 Da (FIG. 20A).
- ESI-MS electrospray ionization mass spectrometry
- 10 pg of sample was loaded onto Agilent Bio SCX NP3 with mobile phase A 20 mM sodium acetate, pH 4.5 and mobile phase B 20 mM sodium acetate, 1 M sodium chloride, pH 4.5.
- a linear gradient of 0-100% B was applied during the course of 20 minutes and only one single major peak was detected (FIG. 20B).
- the column was eluted with 2 CVs each of 150 mM, 200 mM, 250 mM, 300 mM, and 500 mM NaCl.
- the load, flowthrough, and elutions were analyzed by 4-12% Bis-Tris SDS-PAGE and Coomassie straining to determine fractions containing ProTIA-A (FIG. 21B).
- the apparent molecular weight determined was 39.8 kDa (the latter being about 23 -fold less than that of the intact construct, above), which would give apparent molecular weight factor of 0.7 (the latter being about l7-fold less than that of the intact construct, above) and a hydrodynamic radius of 2.3 nm (the latter being about 5 -fold less than that of the intact construct, above).
- electrospray ionization mass spectrometry (ESI-MS) was performed and the experimental mass was determined to be 58,071 Da, with AMass of +4 Da when compared to theoretical molecular weight of 58,067 Da (FIG. 23 A).
- Analytical cation exchange chromatography (FIG. 23B) using a protocol previously described in 2) also confirmed the homogeneity of the sample.
- Example 4 EpCAM binding assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIAI composition
- the binding capability of anti-EpCAM x anti-CD3 ProTIA composition was verified with an EpCAM/peroxidase-conjugated protein-L sandwich ELISA.
- recombinant human EpCAM rhEpCAM
- rhEpCAM recombinant human EpCAM
- the assay plate was washed and blocked with 3 % bovine serum albumin (BSA) for 1 h at room temperature.
- BSA bovine serum albumin
- the plate was washed again followed by the introduction of a dose range of non-cleavable anti-EpCAM x anti-CD3 ProTIA (i.e., a ProTIA without the release segment cleavage sequence and AC 1484, a ProTIA chimeric polypeptide assembly composition) and protease-treated and protease-untreated anti-EpCAM x anti-CD3 ProTIA (AC1476).
- the dose range utilized for non-cleavable and protease-treated and untreated ProTIA was 0.0006 to 5 nM, achieved with a 1 :6 fold serial dilution scheme from a starting concentration of 5 nM.
- the plate was allowed to incubate with shaking for 1 h at room
- TMB tetramethylbenzidine
- ProTIA that gave half-maximal response was derived with a 4-parameter logistic regression equation using GraphPad prism software.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA has a binding activity similar to that of protease-untreated anti-EpCAM x anti-CD3 bispecific ProTIA molecule each bearing an EC50 of 320 pM and 280 pM respectively.
- the protease-treated anti-EpCAM x anti-CD3 ProTIA has a binding activity similar to that of protease-untreated anti-EpCAM x anti-CD3 bispecific ProTIA molecule each bearing an EC50 of 320 pM and 280 pM respectively.
- ProTIA has the strongest binding activity at EC50 of 120 pM for the rhEpCAM ligand compared to the intact protease-untreated bispecific molecule or the non-cleavable ProTIA molecule.
- the data suggest that the presence of XTEN864 hindered the binding of the anti-EpCAM moiety for its ligand by at least 2.3 -fold.
- Example 5 Cell binding assessed by flow cytometry
- Bispecific binding of the anti-EpCAM x anti-CD3 ProTIA composition is also evaluated by fluorescence-activated cell sorting (FACS)-based assays utilizing CD3 positive human Jurkat cells and EpCAM positive human cells selected from SW480, HCT-l 16, Kato III, MDA-MB-453, MCF-7, MT3, SK-Br-3, SK-OV-3, OVCAR-3, BT-474, HPAF-II, JIMT-l, MDA-MB-436, NCI-H322, NCI-H660, NCI-H69 and PC3.
- FACS fluorescence-activated cell sorting
- CD3 + and EpCAM + cells are incubated with a dose range of untreated anti-EpCAM x anti-CD3 ProTIA, protease-treated anti- EpCAM x anti-CD3 ProTIA, and anti-CD3 scFv and anti-EpCAM scFv positive controls for 30 min at 4°C in FACS buffer containing PBS with 1% BSA and 0.05% sodium azide. After several washes in FACS buffer to remove unbound test material, cells are incubated with FITC- conjugated anti-His tag antibody (Abeam cat #abl206) for 30 min at 4°C.
- FITC- conjugated anti-His tag antibody Abeam cat #abl206
- Unbound FITC- conjugated antibody is removed by several washes with FACS buffer and cells resuspended in FACS buffer for acquisition on a FACS Calibur flow cytometer (Becton Dickerson) or equivalent instrument. All flow cytometry data are analyzed with FlowJo software (FlowJo LLC) or equivalent.
- anti-EpCAM scFv While anti-EpCAM scFv is not expected to bind to Jurkat cells, anti-CD3 scFv, untreated anti-EpCAM x anti-CD3 ProTIA and protease-treated anti-EpCAM x anti-CD3 ProTIA are all expected to bind to Jurkat cells as indicated by an increase in fluorescence intensity when compared to Jurkat cells incubated with FITC-conjugated anti-His tag antibody alone. Similarly, anti-EpCAM scFv, protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA are all expected to bind to EpCAM positive cells, while anti-CD3 scFv is not expected to bind to EpCAM positive cells.
- Example 6 Cytotoxicity assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA! composition
- PBMC peripheral blood mononuclear cells
- EpCAM positive human carcinoma cells such as SW480 colon cells (or selected from HCT-l 16, Kato III, NCI-N87, MKN45, MDA-MB-231, MDA-MB-453, MCF-7, MT3, SK-Br-3, SK-OV-3, OVCAR3, BT-474, HPAF-II, JIMT-l, MDA-MB-436, NCI-H322, NCI-H660, NCI-H69 and PC3) as targets.
- PBMC peripheral blood mononuclear cells
- PBMC peripheral blood mononuclear cells
- cytotoxicity assays Three different types of cytotoxicity assays are used for the determination of the cytolytic activity of non-cleavable anti-EpCAM x anti-CD3 composition (AC 1484), protease-treated and untreated anti-EpCAM x anti-CD3 cleavable ProTIA compositions (AC1278 & AC1476), namely lactate dehydrogenase (LDH) release assay, caspase 3/7 assay and FACS-based analysis.
- LDH lactate dehydrogenase
- the LDH release assay quantitatively measures the stable cytosolic enzyme LDH that is released upon cell lysis in much the same way as 51 Cr is released in radioactive assays. Released LDH in culture supernatants is measured by an enzymatic assay that converts a tetrazolium salt into a red formazan product; the amount of color formed being proportional to the number of lysed cells.
- composition samples were diluted in assay medium to the desired dose concentration and added in 20 microL to the respective experimental wells bringing the total assay volume to 200 microL.
- the protease-cleaved ProTIA was evaluated as a 12 -point, 5x serial diluted dose concentration starting at 440 nM to obtain a final dose range of 0.000005 to 44 nM.
- the untreated non-cleaved ProTIA composition was analyzed as a 12 point, 5x serial diluted dose concentration starting at 184 nM to derive at a final dose range of 0.000002 to 18.4 nM.
- Assay controls that included spontaneous LDH released by effector and target cells; target cell maximum LDH released; volume correction control due to the addition of lysis solution and culture medium background were also set up at this time.
- target spontaneous LDH released SW480 cells were incubated in 200 microL of assay medium in the absence of any protease-treated or untreated composition.
- effector spontaneous LDH released PBMC were incubated in 200 microL of assay medium in the absence of any protease-treated or untreated composition.
- Target cell maximum LDH released was determined by the addition of 20 microL of lOx lysis solution to SW480 (220 microL total volume) and incubating the target cells in the presence of lysis solution for 45 min prior to harvesting the supernatant for LDH measurement.
- Volume correction control was achieved by adding 20 microL of lOx lysis solution to 200 microL of assay media, while culture medium background was obtained by incubating 200 microL of assay medium.
- the amount of LDH released into the supernatant as a result of cell lysis was measured using the Promega CytoTox Assay kit and following manufacturer’s instructions. Briefly, 50 microL of the supernatant from each well of the assay plate was transferred to the corresponding well of a flat-bottomed enzymatic plate. To each well in the enzymatic plate, 50 microL of the reconstituted substrate was added. The plate was then covered, protected from light and allowed to incubate at room temperature for 30 min. After the desired incubation period, 50 microL of stop solution was added to each well and absorbance recorded at 490 nm.
- the specificity of the anti-EpCAM x anti-CD3 ProTIA was further evaluated by comparing the cytotoxic activity of protease-treated and protease-untreated ProTIA to that of unconjugated monospecific anti-EpCAM scFv and monospecific anti-CD3 scFv in the LDH assay. Briefly, PBMC and SW480 cells were co-cultured in an effector to target ratio of 5:1 in assay medium in a 96-well round-bottom plate as described above.
- Protease-treated anti- EpCAM x anti-CD3 ProTIA, protease-untreated anti-EpCAM x anti-CD3 ProTIA, and unconjugated monospecific anti-EpCAM scFv plus monospecific anti-CD3 scFv samples were all evaluated as a 12-point, 5x serial dilution of a final dose range of 0.00005 to 45 nM in a total assay volume to 200 microL. Together with experimental wells, all relevant assay controls as described above were also included in the assay plate and the plate was incubated overnight in a 37°C, 5% C0 2 humidified incubator.
- the release segment cleavage sequence present in the anti- EpCAM x anti-CD3 ProTIA may by itself be susceptible to cleavage by proteases released by the tumor cells or by activated CD3 positive T cells (e.g. granzymes).
- a non-cleavable anti-EpCAM x anti-CD3 ProTIA without the release segment (AC 1357) was constructed and evaluated in conjugation with the protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA (AC 1278). All three ProTIA were analyzed in the LDH assay using a 5:1 PBMC to SW480 ratio and tested in a 12-point dose concentration range of 0.00005 to 45 nM achieved with a 5x serial dilution scheme.
- untreated anti-EpCAM x anti-CD3 ProTIA is 32-fold less active than protease-treated ProTIA (EC50 of 288 pM vs. 8.9 pM).
- the non-cleavable anti- EpCAM x anti-CD3 ProTIA i.e., ProTIA without the release segment cleavage sequence
- the release segment contained within the cleavable anti-EpCAM x anti-CD3 ProTIA molecule is susceptible to some cleavage by proteases likely released from the tumor cells and/or activated CD3 positive T cells.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA without the release segment (AC1484) and protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA (AC1476) were also evaluated in human cell line of ovarian origin.
- PBMC was mixed with SK-OV-3 ovarian cells in a ratio of 5:1 and all three ProTIA molecules were tested as a 12-point, 5x serial dilution dose curve in the LDH assay as described above.
- the activity trend of the three ProTIA molecules profiled in SK-OV-3 ovarian cell line was found to be similar to that observed in the SW480 colorectal cell line.
- untreated anti- EpCAM x anti-CD3 ProTIA was 45 -fold less active than protease-treated ProTIA (EC50 of 136 pM vs. 3 pM); and the non-cleavable anti-EpCAM x anti-CD3 ProTIA was 600-fold less active than the protease-cleaved ProTIA (EC50 of 1793 pM vs. 3 pM) (FIG. 30).
- Example 7 Cell lysis assessed by flow cytometry
- EpCAM positive SK-OV-3 target cells (or target cells selected from HCT-l 16, Kato III, MDA-MB-453, MCF-7, MKN45, MT3, NCI-N87, SK-Br-3, SW480, OVCAR3, BT-474, HPAF-II, JIMT-l, MDA-MB-436, NCI- H322, NCI-H660, NCI-H69 and PC3 cell lines) are labeled with the fluorescent membrane dye CellVue Maroon dye (Affymetrix/eBioscience, cat #88-0870-16) according to manufacturer’s instructions.
- CellVue Maroon dye Affymetrix/eBioscience, cat #88-0870-16
- PKH26 (Sigma, cat #MINI26 and PKH26GL) can also be used.
- SK-OV-3 cells are washed twice with PBS followed by resuspension of 2 x 10 6 cells in 0.1 mL diluent C provided with the CellVue Maroon labeling kit.
- 2 mircoL of CellVue Maroon dye is mixed with 0.5 mL diluent C, and then 0.1 mL added to the SK-OV-3 cell suspension.
- the cell suspension and CellVue Maroon dye are mixed and incubated for 2 min at room temperature.
- the labeling reaction is then quenched by the addition of 0.2 mL of FCS.
- Labeled cells are washed twice with complete cell culture medium (RPMI-1640 containing 10% FCS) and total number of viable cells determined by trypan blue exclusion.
- RPMI-1640 containing 10% FCS complete cell culture medium
- total number of viable cells determined by trypan blue exclusion.
- lxl 0 5 PBMC are co-cultured with 2 x 10 4 CellVue Maroon-labeled SK-OV-3 cells per well in a 96-well round-bottom plate in the absence or presence of the indicated dose range concentration of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA samples.
- After 24 h, cells are harvested with Accutase
- Cytotoxicity results utilizing flow cytometry are expected to be in line with results obtained with the LDH assay. Exposure of SK-OV-3 cells to protease-cleaved and uncleaved anti-EpCAM x anti-CD3 ProTIA compositions in the absence of PBMC are expected to have no effect. Similarly, PBMC are not expected to be activated in the presence of ProTIA without target cells. These results are expected to indicate that ProTIA compositions need to be clustered on the surface of target cells in order to stimulate PBMC for cytotoxicity activity.
- CD 19 and HER2 positive target cells will be used instead of EpCAM positive cells.
- Example cell lines for CD 19 expressing cells will include but not limited to NAML-6, Blin-l, SKW6.4, Raji, Daudi and BJAB.
- HER2 positive cell lines such as SK-BR-3, BT474, HCC-1954, MDA-MB-453, SK-OV-3, NCI- N87, JIMT-l, HCT-l 16 will be used.
- Example 8 T-cell activation marker assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition
- SK-OV-3 cells were used as target cells, and activation of CD69 on CD8 and CD4 populations of purified CD3+ cells by untreated anti-EpCAM x anti-CD3 ProTIA (AC 1476) was ⁇ lOO-fold less active than protease- treated ProTIA (EC50 of 260 pM vs. 2.4 pM for CD8+, EC 50 of 240 pM vs.
- PBMC (1 X 10 5 ) were co-cultured with 2 X 10 4 OVCAR3 cells per assay well (i.e., effector to target ratio of 5:1) in the presence of anti-EpCAM x anti-CD3 ProTIA in a 96-well round-bottom plate with total final volume of 200 microL.
- CD69 and granzyme B are expressed in ProTIA-activated T cells in the presence of OVCAR3 cells. Additionally, a greater fraction of CD8+ cells express granzyme B compared to CD4+ cells (FIGS. 48 and 49).
- Example 9 Pharmacokinetic properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition
- Plasma concentration of protease-treated ProTIA was quantified by a rhEpCAM/biotinylated-anti-His tag sandwich ELISA with the protease-cleaved ProTIA as standard; while plasma concentration of untreated ProTIA was quantified by a rhEpCAM/biotinylated-anti-XTEN sandwich ELISA with the uncleaved ProTIA as standard.
- ELISA plate (Nunc Maxisorp cat# 442404) was coated with 0.1 mircog/lOO microL per well of rhEpCAM (R&D Systems, cat# EHH104111). After overnight incubation at 4°C, the ELISA plate was washed and blocked with 3% BSA for 1 h at room temperature. The plate was washed again followed by the appropriate addition of a dose range of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA standards, appropriate quality controls and plasma test samples. The plate was allowed to incubate with shaking for 1 h at room temperature to allow the ProTIA standards, quality controls and test samples to bind to rhEpCAM coated on the plate.
- biotinylated anti-His tag antibody R&D Systems, cat# BAM050
- biotinylated anti-XTEN antibody a proprietary antibody
- streptavidin-HRP (Thermo Scientific cat# 21130) was added at 1 : 30,000 dilution and plate incubated at room temperature for 1 h. After several washes, TMB substrate was added to each well. Once desired color intensity was reached, 0.2 N sulfuric acid was added to stop the reaction and absorbance (OD) was measured at 450 nm using a spectrophotometer. The intensity of the color is proportional to the concentration of protease-treated and untreated ProTIA captured by the respective rhEpCAM/biotinylated-anti-His tag and rhEpCAM/biotinylated-anti-XTEN sandwich ELISA.
- the concentration of ProTIA present in the plasma samples was determined against the appropriate protease-treated or untreated ProTIA standard curve using SoftMax Pro software. Pharmacokinetic calculations of terminal half-life (Ti /2 ) of the protease-cleaved and uncleaved anti-EpCAM x anti-CD3 ProTIA were performed with GraphPad Prism.
- the protease-treated anti-EpCAM x anti-CD3 ProTIA has a short terminal elimination half-life (Ti /2 ) of about 3.5 h, whereas the protease-untreated ProTIA (with attached XTEN) has an extended T 2 of 32 h (FIG. 28), confirming that the intact ProTIA molecule has significantly longer half-life (at least 9-fold longer) than the cleaved molecule.
- Example 10 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition in early treatment SW480 model
- Tumors were measured twice per week for a projected 35 days with a caliper in two perpendicular dimensions and tumor volumes were calculated by applying the (width 2 X length) / 2 formula.
- Body weight, general appearance and clinical observations such as seizures, tremors, lethargy, hyper-reactivity, pilo-erection, labored/rapid breathing, coloration and ulceration of tumor and death were also closely monitored as a measure of treatment related toxicity.
- Study endpoint was defined as a tumor volume of 2000 mm 3 or survival to 36 days, whichever comes first.
- Percent tumor growth inhibition index was calculated for each of the treatment group by applying the formula: ((Mean tumor volume of PBS vehicle control - Mean tumor volume of ProTIA treatment)/mean tumor volume of PBSvehicle control) x 100. Treatment group with %TGI >60% is considered therapeutically active.
- protease-untreated anti-EpCAM x anti-CD3 ProTIA variants is performed in SW480/PBMC inoculated NOD/SCID mice much like the study described above but with eight mice per treatment group.
- early treatment with PBS vehicle control non-cleavable anti-EpCAM x anti-CD3 ProTIA (AC 1357 or AC 1484), a bispecific negative control ProTIA (having the binding activity for CD3 but not for EpCAM), protease-untreated anti-EpCAM x anti-CD3 ProTIA (e.g.
- protease-treated anti-EpCAM x anti-CD3 ProTIA is initiated an hour after SW480/PBMC inoculation.
- the 1 mg/kg dose concentration of protease-untreated anti-EpCAM x anti-CD3 ProTIA as determined in the above study is used in this study and the bispecific negative control ProTIA, non-cleavable and protease-treated anti- EpCAM x anti-CD3 ProTIA test articles are all intravenously administered at equimolar concentration. Tumor volume, body weight and clinical observations are monitored two times per week for 35 days.
- Treatment with equimolar concentration of the non-cleavable ProTIA is expected to minimally retard tumor growth as it does not contain the substrate for protease cleavage.
- Example 11 Anti-tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition in established colorectal tumor model
- SW480 or HCT-l 16 tumor cells are independently implanted into NOG (NOD/Shi-scid/IL-2Rg nu11 ) or NSG (NOD.Cg- Prkdc scld .IL2rg tmlwjl /SzJ) mice on day 0.
- NOG or NSG mice are NOD/SCID mice bearing IL-2Rg mutation resulting in the mice lacking T, B and NK cells, dysfunctional macrophage, dysfunctional dendritic cells and reduced complement activity.
- Human PBMC are then intravenously or intraperitoneally introduced sometime between days 3 to 10.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA (e.g. AC1484) is expected to minimally retard tumor growth as it does not contain the substrate sequence for protease cleavage within the tumor environment.
- Example 12 Cytometric bead array analysis for human Thl/Th2 cytokines using stimulated normal healthy human PBMCs and intact and protease-treated anti-EpCAM x anti- CD3 ProTIA
- OKT3 (0, 10 nM, 100 nM and 1000 nM) and protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA (AC 1278 at 10 nM, 100 nM, 1000 nM and 2000 nM) were dry coated onto a 96-well flat bottomed plate by allowing the wells to evaporate overnight in the biosafety hood. Wells were then washed once gently with PBS and 1X10 6 PBMC in 200 microL were added to each well.
- the plate was then incubated at 37°C, 5% C0 2 for 24 h, after which tissue culture supernatant was collected from each well and analyzed for cytokine released using the validated commercial CBA kit (BD CBA human Thl/Th2 cytokine kit, cat # 551809) by flow cytometry following manufacturer’s instructions.
- BD CBA human Thl/Th2 cytokine kit cat # 551809
- Example 13 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered
- Immune Activator composition in early treatment HCT-l 16 model.
- ProTIA without the release segment cleavage sequence and an example of which being AC 1484) was evaluated using the human HCT-l 16 colorectal carcinoma xenograft model. Briefly, on day 0, four cohorts of 5 NOD/SCID mice per group were subcutaneously injected in the right flank with 5 X 10 6 human PBMC mixed with 5 x 10 6 HCT-l 16 cells.
- cohort 1 was injected with vehicle (PBS+0.05% Tween 80), cohort 2 with 0.21 mg/kg protease-treated anti-EpCAM x anti-CD3 ProTIA, cohort 3 with 0.5 mg/kg protease-untreated anti-EpCAM x anti-CD3 ProTIA and cohort 4 with 0.49 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA.
- cohort 1 to 4 were all subjected to four additional doses administered daily from day 1 to 4.
- Tumors were measured twice per week for a projected 35 days with a caliper in two perpendicular dimensions and tumor volumes were calculated by applying the (width 2 X length)
- %TGI Percent tumor growth inhibition index
- the non- cleavable anti-EpCAM x anti-CD3 ProTIA is considered therapeutically inactive.
- Example 14 Cytotoxicity assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition in the presence of purified CD3 positive T cells
- untreated anti- EpCAM x anti-CD3 ProTIA is 56-fold less active than protease-treated ProTIA (EC50 of 134 pM vs. 2.4 pM); and the non-cleavable anti-EpCAM x anti-CD3 ProTIA is >1000-fold less active than the protease-cleaved ProTIA (EC50 of 2660 pM vs. 2.4 pM) (FIG. 40).
- untreated anti-EpCAM x anti- CD3 ProTIA is only 2-fold less active than protease-treated ProTIA (EC50 of 0.7 pM vs. 0.3 pM); and the non-cleavable anti-EpCAM x anti-CD3 ProTIA is 287-fold less active than the protease-cleaved ProTIA (EC50 of 86 pM vs. 0.3 pM) (FIG. 41).
- Example 15 T-cell activation marker and cvtokine release assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition
- This assay can also be performed with other target cells selected from HCT-l 16, Kato III, MDA- MB-453, MCF-7, MKN45, MT3, NCI-N87, SK-Br-3, SW480, OVCAR3 and PC3 cell lines as well as PBMC in place of purified CD3+ cells.
- Cytokine analysis of interleukin (IL)-2, IL-4, IL-6, IL-10, tumor necrosis factor (TNF)- alpha and interferon (IFN)-gamma secreted into the cell culture supernatant was quantitated using the Human Thl/Th2 Cytokine Cytometric Bead Array (CBA) kit (BD Biosciences cat #550749) following manufacturer’s instruction. In the absence of ProTIA, no cytokine secretion above background is expected from purified CD3+ cells.
- CBA Human Thl/Th2 Cytokine Cytometric Bead Array
- ProTIA in the presence of EpCAM- positive target cells and purified CD3+ cells is expected to activate T cells and secrete a pattern of T cell cytokines with a high proportion of Thl cytokines such as IFN-gamma and TNF -alpha.
- anti-EpCAM x anti-CD3 ProTIA induced robust secretion of all cytokines (IL-2, IL-4, IL-6, IL-10, TNF-alpha, IFN-gamma) evaluated (see FIGS. 50-52).
- Stimulation of purified CD3+ cells with SK-OV-3 cells and protease-treated anti-EpCAM x anti-CD3 ProTIA (MMP-9 treated AC 1476) triggered significant cytokine expression, especially at concentrations higher than 20 pM for all of the cytokines tested.
- baseline levels of IL-2, IL-4, IL-6, IL-10, TNF -alpha and IFN-gamma were detected when the intact non-cleaved anti-EpCAM x anti-CD3 ProTIA molecule (AC1476) was used at a concentration range of 8 to 200 pM (EC 50 of 4.3 nM). Additionally, baseline levels of all cytokines tested were detected when the non- cleavable anti-EpCAM x anti-CD3 ProTIA molecule (AC 1484) was used at a concentration range of 40 pM to 1 nM.
- Example 16 CD3 binding specificity of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition
- ProTIA is a bispecific-targeting composition
- the binding capability of anti-EpCAM x anti-CD3 ProTIA composition was also evaluated for binding affinity to human CD3. This was determined with a CD3e & d/peroxidase-conjugated protein-L sandwich ELISA.
- recombinant human CD3 rhCD3e & d
- CD3E&CD3D-219H recombinant human CD3
- the assay plate was washed and blocked with 3 % bovine serum albumin (BSA) for 1 h at room temperature.
- BSA bovine serum albumin
- the plate was washed again followed by the introduction of dose ranges of non-cleavable anti-EpCAM x anti-CD3 ProTIA (AC 1484), protease-treated and protease-untreated anti-EpCAM x anti-CD3 ProTIA (AC1476).
- the dose range utilized for all three versions of ProTIA was 0.002 to 100 nM, achieved with a 1 :6 fold serial dilution scheme from a starting concentration of 100 nM.
- the plate was allowed to incubate with shaking for 1 h at room temperature to allow the non-cleavable, protease-cleaved and protease-untreated ProTIA to bind to the rhCD3e & d coated on the plate. Unbound components were removed with a wash step and a peroxidase-conjugated protein L
- any unbound reagent was removed by a wash step followed by the addition of tetramethylbenzidine (TMB) substrate to each well. After desired color intensity was reached, 0.2 N sulfuric acid was added to stop the reaction and absorbance (OD) was measured at 450 nm using a spectrophotometer. The intensity of the color is proportional to the concentration of non-cleavable, protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA captured by the rhCD3e & d/protein-L sandwich ELISA.
- the intensity of the color produced was ploted against protein concentration; and the concentration of non-cleavable, protease-cleaved and uncleaved anti-EpCAM x anti-CD3 ProTIA that gave half-maximal response (EC50) was derived with a 4-parameter logistic regression equation using GraphPad prism software.
- ProTIA molecule each bearing an EC50 of 1800 pM and 2200 pM respectively.
- the protease- treated ProTIA had the strongest binding activity at EC50 of 310 pM for the rhCD3e & d ligand compared to the intact protease-untreated bispecific molecule or the non-cleavable ProTIA molecule.
- the XTEN864 blocking moiety As the XTEN864 blocking moiety is located right after the anti-CD3scFv moiety, the XTEN864 results in hindrance in the binding of the non-cleaved anti-CD3 entity for its ligand by ⁇ 5.8 fold as compared to the cleaved and released anti-CD3scFv portion of the ProTIA binding to the CD3 ligand.
- Example 17 Binding specificity of anti-EpCAM x anti-CD3 Protease Triggered
- the binding specificity of an anti-EpCAM x anti-CD3 ProTIA was evaluated in conjunction with the control ProTIA compositions anti-CEA x anti-CD3 ProTIA (AC1432) and anti-HER2 x anti-CD3 ProTIA (AC 1408), in a target cell marker/biotin-conjugated protein-L sandwich ELISA. Both the anti-CEA x anti-CD3 ProTIA (AC1432) and the anti-HER2 x anti- CD3 ProTIA (AC 1408) bear the same anti-CD3 scFv component as the anti-EpCAM x anti-CD3 ProTIA (AC1476) albeit with different targeting component.
- recombinant human EpCAM (rhEpCAM) (R&D Systems cat # 960-EP-50), recombinant human CEA (Abeam cat # ab742) and recombinant human HER2 (AcroBiosystems cat# HE2-H525) were coated on a 96-well, flat-botomed plate at a concentration of 0.1 microg/lOO microL.
- the assay plate was washed and blocked with 3 % bovine serum albumin (BSA) for 1 h at room temperature.
- BSA bovine serum albumin
- the plate was washed again followed by the introduction of a dose range (0.0007 to 0.5 nM, achieved with a 1 :3 fold serial dilution scheme from a starting concentration of 0.5 nM) of protease-treated anti-EpCAM x anti-CD3 ProTIA (AC1476) to EpCAM-coated wells, CEA-coated wells and HER2 -coated wells.
- protease-treated anti-CEA x anti-CD3 ProTIA (AC1432) was introduced at a similar dose range onto CEA-coated wells, and protease-treated anti-HER2 x anti-CD3 ProTIA
- the intensity of the color is proportional to the concentration of the respective protease-treated ProTIAs captured by the appropriate antigen coated on the plate.
- the intensity of the color produced was plotted against ProTIA concentration; and the respective dose curve derived with a 4-parameter logistic regression equation using
- protease-treated anti-EpCAM x anti-CD3 ProTIA binds to rhEpCAM coated on the plate in a dose- dependent manner to yield an EC50 of 110 pM.
- protease-treated anti-CEA x anti-CD3 ProTIA binds to the CEA antigen coated on the plate in a dose-dependent manner to yield an EC50 of 70 pM; and protease-treated anti-HER2 x anti-CD3 ProTIA binds to the HER2 antigen coated on the plate in a dose-dependent manner to yield an EC50 of 47 pM.
- Example 18 Anti -tumor properties of intact anti-EpCAM x anti-CD3 ProTIA versus non-cleavable anti-EpCAM x anti-CD3 ProTIA in early treatment SW480 model
- protease susceptibility of the Release Segment (RS) as engineered into the anti- EpCAM x anti-CD3 ProTIA molecule (AC 1476) in tumor environment was also evaluated in vivo together with non-cleavable anti-EpCAM x anti-CD3 ProTIA (AC 1484), protease-treated and protease-untreated anti-EpCAM x anti-CD3 ProTIA (AC1476) in the SW480/PBMC inoculated NOD/SCID xenograft model.
- cohort 1 mice was injected with vehicle (PBS_0.05% Tween 80), cohort 2 with 0.21 mg/kg protease-treated anti-EpCAM x anti- CD3 ProTIA, cohort 3 with 0.5 mg/kg intact anti-EpCAM x anti-CD3 ProTIA and cohort 4 with 0.49 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA. All cohorts (i.e. 1 to 4) were further treated with four additional doses administered daily from day 1 to day 4. Tumor volume, body weight and clinical observations are monitored two times per week for a targeted 35 days. [00387] As shown in FIG.
- protease-treated anti-EpCAM x anti-CD3 ProTIA at 0.21 mg/kg (cohort 2), intact anti-EpCAM x anti-CD3 ProTIA at 0.5 mg/kg (cohort 3) and non-cleavable anti-EpCAM x anti-CD3 ProTIA at 0.49 mg/kg (cohort 4) are all determined to be
- %TGI tumor growth inhibition index
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA is less effective than intact anti- EpCAM x anti-CD3 ProTIA indicating that the presence of the release segment improved therapeutic efficacy of the composition by permitting the release of the anti-EpCAM x anti-CD3 binding domains.
- Example 19 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in OVCAR-3 ovarian model.
- ProTIA Protease Triggered Immune Activator
- NOG and NSG mice are characterized by the deficiency of T, B and NK cells, as well as the dysfunction of macrophages, dendritic cell and complement system. Briefly, on day 0, seven cohorts of 5 NOG or NSG mice per group are implanted intraperitoneally with 5-10 X 10 6 OVCAR-3 cells, followed by the intravenous introduction of 5-10 X 10 6 of PBMC on day 14.
- cohort 1 injected with vehicle (PBS+0.05% Tween 80) daily for 5 doses (qdx5)
- cohort 2 with 0.21 mg/kg protease-treated anti-EpCAM x anti-CD3 ProTIA qdx5
- cohort 3 with 1.05 mg/kg protease-treated anti-EpCAM x anti-CD3 ProTIA once per week (qw)
- cohort 4 with 0.5 mg/kg with protease-untreated anti-EpCAM x anti-CD3 ProTIA qdx5
- cohort 5 with 2.5 mg/kg with protease-untreated anti-EpCAM x anti-CD3 ProTIA qw
- cohort 6 with 0.49 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA qdx5
- cohort 7 with 2.45 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA qw.
- mice are monitored daily for behavior and survival, and twice weekly for body weight and abdomen distention. Blood are collected on day 30, day 40, day 50 and day 60 for CA125 determination as sign of tumor development. When weight of animals has increased by 30% from day 0, the animal is defined as having met study endpoint and is sacrificed and autopsied.
- Example 20 PK properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in OVCAR-3 ovarian model.
- ProTIA Protease Triggered Immune Activator
- Protease-cleaved, protease-untreated and non-cleavable anti-EpCAM x anti-CD3 ProTIAs’ PK and bio-distribution profile is evaluated as a mixture of independently metal- labeled molecules in the OVCAR-3 tumor bearing BALB/c nude mice.
- ten million OVCAR-3 cells are injected intraperitoneally on day 0.
- OVCAR-3 tumor bearing mice 18 are selected and randomized according to their individual body weight into 2 groups of 9 animals per group.
- One group of 9 mice is intravenously injected with 1.5 mg/kg of the mixture comprising of equimolar concentration of metal 1 -labeled protease-cleaved anti-EpCAM x anti- CD3 ProTIA, metal 2 -labeled protease-untreated anti-EpCAM x anti-CD3 ProTIA and metal 3- labeled non-cleavable anti-EpCAM x anti-CD3 ProTIA.
- the other group of 9 animals is administered intraperitoneally with 1.5 mg/kg of the same ProTIA mixture.
- blood is collected by jugular/mandibular vein puncture into lithium heparin tubes at 0.5 h, 4 h, 8 h, 24 h, 48 h, day 3, day 5 and day 7 post-test article administration.
- Blood is processed into plasma by centrifugation at 1300 g for 10 minutes at 4°C and stored at - 80°C till analysis.
- Ascites is collected from both intravenously and intraperitoneal administered groups at 4 h, 8 h, 24 h, 48 h, day 3, day 5 and day 7 post-test article administrations by alternating between animals in the same group. Ascites samples are immediately centrifuged at 300 g for 10 minutes at 4°C and fluid component frozen down at -80°C until analysis.
- mice from each group will be terminated on day 3, day 5 and day 7.
- Organs (brain, heart, liver, lung, spleen, and pancreas) and tumor nodules in the peritoneal cavity are harvested, weighed, flash frozen and stored at -80°C until analysis is performed.
- all 3 ProTIA versions are expected to be detectable in the ascites. Due to the presence of tumor in the intraperitoneal space, it is unknown if metal 2-labeled protease-untreated anti-EpCAM x anti- CD3 ProTIA and metal 3 -labeled non-cleavable anti-EpCAM x anti-CD3 ProTIA will have a longer retention time in the peritoneal cavity as compared to metal 1 -labeled protease-cleaved anti-EpCAM x anti-CD3 ProTIA. All 3 ProTIA versions are expected to be detected in plasma at a delayed time and at a lower concentration as compared to the intravenous route. All 3 ProTIA versions are expected to be present at higher concentration in tumor nodules extracted from the peritoneal cavity but minimally or none in normal organs.
- Example 21 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in SK-OV-3 ovarian model.
- ProTIA Protease Triggered Immune Activator
- the in vivo efficacy of anti-EpCAM x anti-CD3 ProTIA is also evaluated using the human ovarian SK-OV-3 cell line implanted intraperitoneally into the severely immunodeficient NSG (NOD.Cg-Prkdc scid .IL2rg tmlwjl /SzJ) or NOG (NOD/Shi-scid/IL-2Rg nu11 ) mice.
- NOG and NSG mice are characterized by the deficiency of T, B and NK cells, as well as the dysfunction of macrophages, dendritic cell and complement system.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA is also expected to retard tumor growth but to a much lesser magnitude than that exhibited by the release segment bearing protease-untreated ProTIA and the protease-treated ProTIA.
- Example 22 Performance of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA! composition in human malignant ascites samples.
- Human malignant ascites are collected from patients with primary intraperitoneal EpCAM positive epithelial malignancies which includes but not limited to advanced, relapsed and refractory ovarian (adenocarcinoma and mucinous), colorectal, gastric, bile
- duct/cholangiocarcinoma, Ampulla of Vater, pancreatic and non-clear renal cell carcinoma patients Patients who are receiving chemotherapy, immunological therapy, biologies and/or corticosteroid therapy within the last 30 days prior to sample collection are excluded. Malignant ascites are centrifuged at 300-400 g for 10 min at room temperature and the fluid and pellet component harvested.
- the concentration of human proteases including but not limited to MMP- 9, MMP-2, matriptase and uPA are quantitated in the fluid component using commercially available ELISA kits (human MMP-9, Invitrogen cat # KHC3061 or equivalent; human MMP-2, Invitrogen cat # KHC3081 or equivalent; human matriptase, Enzo cat # ADI-900-221; and human uPA, Abeam cat # 119611) following manufacturer’s instructions.
- the rate of intact anti- EpCAM x anti-CD3 e.g. AC1684, AC1685, AC1686, AC1693, AC1695, AC1714, and
- cleavage by protease found in the ascites fluid is determined by spiking a known concentration of the ProTIA into the ascites fluid component and incubating mixture at 37°C, with an aliquot withdrawn at indicated time points of 0.5 h, 2h, 4 h, 8 h, 24 h, 48 h, 3 day, 4 day, 5 day and 7 day.
- the amount of intact anti-EpCAM x anti-CD3 ProTIA present at the respective time points are then analyzed on a rhEpCAM/biotinylated-anti-XTEN sandwich ELISA with the corresponding intact anti-EpCAM x anti-CD3 as standard.
- ELISA plate (Nunc Maxisorp cat# 442404) is coated with 0.1 mircog/100 microL per well of rhEpCAM (R&D Systems, cat# EHH104111). After overnight incubation at 4°C, the ELISA plate is washed and blocked with 3% BSA for 1 h at room temperature. The plate is washed again followed by the appropriate addition of a dose range of intact, protease- untreated anti-EpCAM x anti-CD3 ProTIA standards, appropriate quality controls and ProTIA- spiked ascites test samples. The plate is allowed to incubate with shaking for 1 h at room temperature to allow the ProTIA standards, quality controls and test samples to bind to rhEpCAM coated on the plate. Unbound components are removed with several washes.
- Biotinylated anti-XTEN antibody (a proprietary antibody) is added at 0.1 microg/100 microL and the plate allowed to incubate at room temperature for 1 h. After washing away unbound biotinylated reagent, streptavidin-HRP (ThermoFisher Scientific cat # 21130) is added at 1 : 30,000 dilution and plate incubated at room temperature for 1 h. After several washes, TMB substrate is added to each well. Once desired color intensity is reached, 0.2 N sulfuric acid is added to stop the reaction and absorbance (OD) is measured at 450 nm using a
- the intensity of the color is proportional to the concentration of intact ProTIA captured by the rhEpCAM/biotinylated-anti-XTEN sandwich ELISA.
- concentration of intact ProTIA present in the ascites test samples is determined against the intact ProTIA standard curve using the SoftMax Pro software.
- the rate of decrease of intact ProTIA as detected in the rhEpCAM/biotinylated-anti-XTEN sandwich ELISA i.e. half-life
- GraphPad Prism It is postulated that differences in Release Segments are likely to play a role in the metabolism rate among the protease-untreated ProTIAs
- the ascites pellet is phenotyped for EpCAM, CD3, CD4, CD8, CA125 and CD56 expression.
- Malignant ascites samples tested positive for EpCAM and CD3 are used for cytotoxic analysis with protease-treated and protease-untreated ProTIA. Briefly, 1X10 5 ascites cells are reconstituted with appropriate amount of ascites fluid and allowed to adhere on a 24- well plate for 24 h in triplicate.
- concentration of protease-treated and untreated anti-EpCAM x anti-CD3 ProTIA is then plotted against luminescence signal and the concentration of protein that give half maximal response (EC50) is derived with a 4-parameter logistic regression equation using GraphPad prism software. It is expected that the human malignant ascites derived from advanced, relapsed and refractory EpCAM positive cancer patients will contain all necessary components for the cleavage and subsequent activation of intact anti-EpCAM x anti-CD3 ProTIA to the unXTENylated anti- EpCAM x anti-CD3 moiety that exert strong cytotoxic activity. A decrease in number of
- EpCAM positive cells as a sign of tumor elimination; and an increase in T cell activation markers such as CD69 and granzymes as reflective of T cell activation are also expected,
- Example 23 Caspase 3/7 assay of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition
- Redirected cellular cytotoxicity of anti-EpCAM x anti-CD3 ProTIA compositions was also assessed via caspase 3/7 activities of apoptotic cells. Similar to the LDH cytotoxicity assay described above, PBMC or purified CD3 positive T cells were mixed with EpCAM positive tumor target cells such as SW480, SK-OV-3 and OVAR-3 cells in a ratio of 5 effectors to 1 target, HCT-l 16 at a ratio of 10:1; and all three ProTIA versions were tested as a 12-point, 5x serial dilution dose concentrations as in the LDH assay described above.
- protease-uncleaved anti-EpCAM x anti- CD3 ProTIA is 4-fold less active than protease-treated ProTIA (EC50 of 9.8 pM vs.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA is 420-fold less active than the protease- cleaved ProTIA (EC50 of 1043 pM vs. 2.5 pM) (FIG. 58).
- protease-treated and intact protease-untreated anti-EpCAM x anti- CD3 ProTIA have almost similar activity (EC50 of 1.8 pM vs.
- protease-cleaved ProTIA EC50 of 240 pM vs. 1.8 pM
- protease-treated and protease-uncleaved anti-EpCAM x anti-CD3 ProTIA also demonstrated similar activity (EC50 of 2 pM vs.
- non-cleavable anti-EpCAM x anti-CD3 ProTIA is 70-fold less active than the protease-cleaved ProTIA (EC50 of 148 pM vs. 2 pM) (FIG. 60). Results demonstrated that non-cleavable ProTIA is consistently less active than the unXTENylated anti-EpCAM x anti-CD3 moiety.
- protease-untreated ProTIA ranged from similar to 12-fold less active as compared to protease-cleaved ProTIA, suggesting a difference in degree of susceptibility of the release segment to proteases postulated to be released from the tumor cells and/or activated CD3 positive T cells in the assay mixture.
- a panel of enzymes was used to digest the AC 1476 aEpCAM-aCD3-BSRSl- XTEN_AE864-His(6) ProTIA composition. 10 mM of the substrate composition was incubated individually with each enzyme in the following enzyme-to-substrate molar ratios: MMP-2 (1:200), MMP-9 (1: 2000), matriptase (1:12.5), and neutrophil elastase (1:1000). Reactions were incubated at 37°C for two hours before stopping digestion by gel loading dye and heating at 80°C.
- Example 25 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in established colorectal tumor model
- HCT-l 16 tumor cells were independently implanted into NOG (NOD/Shi-scid/IL-2Rg nu11 ) mice on day 0.
- NOG mice are NOD/SCID mice bearing IL-2Rg mutation resulting in the mice lacking T, B and NK cells, dysfunctional macrophage, dysfunctional dendritic cells and reduced complement activity.
- Human PBMC were then intravenously introduced on day 4.
- treatment with anti-EpCAM x anti-CD3 ProTIAs were initiated 3x per week for 4 weeks at equimolar concentration of 21.6 nmol/kg. This is equivalent to 1.26 mg/kg of protease-cleaved and 3.0 mg/kg of protease-untreated and non-cleavable anti-EpCAM x anti- CD3 ProTIA.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA e.g.
- Example 26 Anti-tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in OVCAR-3 ovarian model.
- ProTIA Protease Triggered Immune Activator
- Group 8 comprising of 5 NOG mice, was also set up on day 0 with no OVCAR-3 inoculation.
- tumor cells were observed to have progressed as reflected by an increase in human CA125 level from baseline (below limit of detection) to 300-400 U/mL on day 20, 10 X 10 6 of PBMC were intraperitoneally introduced to Groups 1-8.
- Group 9 did not received any PBMC.
- Treatments were initiated on day of PBMC inoculation with Groups 1, 8 and 9 injected with vehicle (PBS+0.05% Tween 80), Group 2 with 0.21 mg/kg protease-treated anti-EpCAM x anti- CD3 ProTIA, Group 3 with 1.05 mg/kg protease-treated anti-EpCAM x anti-CD3 ProTIA,
- Group 4 with 0.5 mg/kg with protease-untreated anti-EpCAM x anti-CD3 ProTIA, Group 5 with 2.5 mg/kg with protease-untreated anti-EpCAM x anti-CD3 ProTIA, Group 6 with 0.49 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA, and Group 7 with 2.46 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA. All cohorts were treated twice per week for 4 weeks. Mice were monitored daily for behavior and survival, and twice weekly for body weight and abdomen distention. Blood were collected on day 28, day 42 and day 48 for CA125 determination as sign of tumor development.
- the non-cleavable anti-EpCAM x anti-CD3 ProTIA treated groups demonstrated an increased in CA125 levels over time.
- Group 6 (0.49 mg/kg non-cleavable anti-EpCAM x anti- CD3 ProTIA) saw an increased in CA125 from 344 ⁇ 118 U/mL on day 20 to 3426 ⁇ 4170 U/mL on day 48; and
- Group 7 (2.46 mg/kg non-cleavable anti-EpCAM x anti-CD3 ProTIA) demonstrated an increased in CA125 from 351 ⁇ 113 U/mL on day 20 to 1905 ⁇ 2534 U/mL on day 48.
- Example 27 Anti -tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition in OVCAR-3 ovarian model versus standard of care.
- ProTIA Protease Triggered Immune Activator
- Groups 1-7 and Group 9 Eight cohorts (Groups 1-7 and Group 9) of 6 NOG mice per group were implanted intraperitoneally with 10 X 10 6 OVCAR-3 cells. Group 8, comprising of 6 NOG mice, was also set up on day 0 with no OVCAR-3 inoculation. When tumor cells were observed to have progressed as reflected by an increase in human CA125 level from baseline (below limit of detection) to approximately 650 U/mL on day 21, lOxlO 6 of PBMC were intravenously introduced to Groups 1-8. Group 9 did not received any PBMC. Treatments were initiated on day of PBMC inoculation with Groups 1,
- protease-untreated anti-EpCAM x anti-CD3 ProTIA e.g.
- CA125 level in Group 2 (0.5 mg/kg protease-untreated anti-EpCAM x anti-CD3 ProTIA, IP) increased slightly from 679 ⁇ 242 U/mL on day 21 to 891 ⁇ 897 U/mL at sacrifice;
- Group 3 (2.5 mg/kg protease-untreated anti-EpCAM x anti-CD3 ProTIA, IP) decreased from 677 ⁇ 241 U/mL on day 21 to 228 ⁇ 269 U/mL at sacrifice;
- Group 4 (0.5 mg/kg protease-untreated anti- EpCAM x anti-CD3 ProTIA, IV) remained relatively unchanged from 661 ⁇ 216 U/mL on day 21 to 661 ⁇ 861 U/mL at sacrifice; and
- Group 5 (2.5 mg/kg protease-untreated anti-EpCAM x anti-CD3 ProTIA, IV) decreased from 658 ⁇ 200 U/mL
- Example 28 CD3 binding assay of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition
- rhCD3 recombinant human CD3 (Creative Biomart cat # CD3E&CD3D-219H) was coated on a 96-well, flat-bottomed plate at a concentration of 0.25 microg/lOO microL. After overnight incubation at 4°C, the assay plate was washed and blocked with 3 % bovine serum albumin (BSA) for 1 h at room temperature.
- BSA bovine serum albumin
- non-cleavable anti-EpCAM x anti-CD3 ProTIA i.e., a ProTIA without the release segment cleavage sequence and AC 1484, a ProTIA chimeric polypeptide assembly composition
- protease-untreated anti-EpCAM x anti-CD3 ProTIA e.g. AC1684, AC1685, AC1686, AC1693, AC1695, AC1714, AC1715.
- the dose range utilized for non-cleavable and protease-untreated ProTIA was 3,600 to 0.077 ng/mL, achieved with a 1 :6 fold serial dilution scheme from a starting concentration of 3,600 ng/mL.
- the plate was allowed to incubate with shaking for 1 h at room temperature to allow the non-cleavable, protease-uncleaved ProTIA to bind to the rhCD3e & d coated on the plate. Unbound components were removed with a wash step and a proprietary biotinylated anti-XTEN monoclonal antibody was added. After an appropriate incubation period that allowed the anti-XTEN antibody to bind to the XTEN polypeptide on the ProTIAs, any unbound reagent was removed by a wash step followed by the addition of tetramethylbenzidine (TMB) substrate to each well. TMB is a chromogenic substrate of peroxidase.
- TMB tetramethylbenzidine
- the intensity of the color produced was plotted against protein concentration; and the concentration of non-cleavable and protease-uncleaved anti-EpCAM x anti-CD3 ProTIA that gave half-maximal response (EC50) was derived with a 4-parameter logistic regression equation using GraphPad prism software.
- AC 1685 is 29 ng/mL
- AC1686 is 26 ng/mL
- AC1695 is 28 ng/mL
- AC1714 is 30 ng/mL
- AC1715 is 34 ng/mL.
- Only AC 1693 with an EC50 of 9 ng/mL has a 3 -fold more active binding than the non-cleavable AC 1484 for the rhCD3e & d ligand.
- the data suggest that differences in Release Segment composition can influence the binding of ProTIA to the CD3 antigen found on T cells.
- Example 29 Pharmacokinetic properties of anti-EpCAM x anti-CD3 Protease
- protease-untreated anti-EpCAM x anti-CD3 ProTIA variants e.g. AC1684, AC1685, AC1686, AC1693, AC1695, AC1714, and AC1715
- non-cleavable anti-EpCAM x anti-CD3 ProTIA e.g. AC1484
- Each ProTIA would be evaluated with three mice per group at an intravenous dose of 4 mg/kg.
- blood would be collected into lithium heparinized tubes and processed into plasma.
- Plasma concentration of ProTIAs would be quantified by a rhEpCAM/biotinylated-anti-XTEN sandwich ELISA with the protease-untreated ProTIA as standard.
- ELISA plate (Nunc Maxisorp cat# 442404) would be coated with 0.1 mircog/100 microL per well of rhEpCAM (R&D Systems, cat# EHH104111). After overnight incubation at 4°C, the ELISA plate would be washed and blocked with 3% BSA for 1 h at room temperature. The plate would then be washed again followed by the appropriate addition of a dose range of protease-untreated and non-cleavable anti-EpCAM x anti-CD3 ProTIA standards, appropriate quality controls and plasma test samples.
- the plate would be allowed to incubate with shaking for 1 h at room temperature to allow the ProTIA standards, quality controls and test samples to bind to rhEpCAM coated on the plate. Unbound components would be removed with several washes.
- biotinylated anti-XTEN antibody would be added at 0.1 microg/lOO microL and the plate allowed to incubate at room temperature for 1 h.
- streptavidin-HRP streptavidin-HRP (Thermo Scientific cat# 21130) would be added at 1 : 30,000 dilution and plate incubated at room temperature for 1 h. After several washes, TMB substrate would be added to each well.
- 0.2 N sulfuric acid is added to stop the reaction and absorbance (OD) measured at 450 nm using a spectrophotometer.
- the intensity of the color is proportional to the concentration of protease- untreated and non-cleavable ProTIA captured by the rhEpCAM/biotinylated-anti-XTEN sandwich ELISA.
- the concentration of ProTIA present in the plasma samples is determined against the appropriate protease-untreated or non-cleavable ProTIA standard curve using
- Example 30 PK properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA! composition in OVCAR-3 ovarian model.
- Protease-cleaved, protease-untreated and non-cleavable anti-EpCAM x anti-CD3 ProTIAs’ PK profile was evaluated as a mixture of independently metal -labeled molecules in the OVCAR-3 tumor bearing BALB/c nude mice.
- OVCAR-3 tumor bearing BALB/c nude mice To each irradiated BALB/c nude mice, ten million OVCAR-3 cells are injected intraperitoneally on day 0. Treatment was initiated when abdominal distention was visibly observed and/or when animal body weight had increased by 10-15% over day 0. Out of twenty OVCAR-3 tumor bearing mice, 18 were selected and randomized according to their individual body weight into 2 groups of 9 animals per group.
- mice One group of 9 mice was intravenously injected with 1.5 mg/kg of the mixture comprising of equimolar concentration of Lutetium (Lu)-labeled protease-cleaved anti-EpCAM x anti-CD3 ProTIA, Holmium (Ho)-labeled protease-untreated anti-EpCAM x anti-CD3 ProTIA and
- Tb Terbium-labeled non-cleavable anti-EpCAM x anti-CD3 ProTIA.
- the other group of 9 animals is administered intraperitoneally with 1.5 mg/kg of the same ProTIA mixture.
- blood was collected by jugular/mandibular vein puncture into lithium heparin tubes at 0.5 h, 4 h, 8 h, 24 h, 48 h, day 3, day 5 and day 7 post-test article administration. Blood was processed into plasma by centrifugation at 1300 g for 10 minutes at 4°C and stored at -80°C till analysis.
- Ascites was collected from both intravenously and intraperitoneal administered groups at 4 h, 8 h, 24 h, 48 h, day 3, day 5 and day 7 post-test article administrations by alternating between animals within the same group. Ascites samples were immediately centrifuged at 300 g for 10 minutes at 4°C and fluid component frozen down at -80°C until analysis. All samples (blood and ascites) were analyzed by ICP-MS (inductively coupled plasma mass spectrometry).
- Ho-labeled protease- untreated anti-EpCAM x anti-CD3 ProTIA and Tb-labeled non-cleavable anti-EpCAM x anti- CD3 ProTIA demonstrated similar half-life of 19.5 h.
- Ho-labeled protease- untreated anti-EpCAM x anti-CD3 ProTIA had a longer systemic half-life compared to the Lu- labeled protease-cleaved anti-EpCAM x anti-CD3 ProTIA (19.5 h vs. 2 h) (FIG 69 A).
- Ho-labeled protease-untreated anti-EpCAM x anti-CD3 ProTIA and Tb-labeled non-cleavable anti-EpCAM x anti-CD3 ProTIA reached Cmax at -8 h, and demonstrated equivalent half-life of 21.4 and 22.9 h, respectively.
- the Ho-labeled protease-untreated anti-EpCAM x anti-CD3 ProTIA exhibited a longer systemic half-life compared to the Lu-labeled protease-cleaved anti-EpCAM x anti-CD3 ProTIA (21.4 h vs. 6.5 h) (FIG. 70A).
- Example 31 Casnase 3/7 assay of anti-EnCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition
- PBMC peripheral blood mononuclear cells
- EpCAM positive tumor target cells such as HPAF-II (human pancreatic tumor cell line), HCT-l 16 (human colorectal tumor cell line) and MDA-MB-231 (human triple negative breast cell line) in a ratio of 10 effectors to 1 target; and all ProTIA variants were tested as either a 8-point or a 12-point, 5x serial dilution dose concentrations as in the caspase assay described above.
- the three cell lines were selected to represent high, mid and low EpCAM antigen expressing cells with HPAF-II expressing 1.1 million EpCAM antigen per cell, HCT-l 16 500,000 per cell and MDA-MB-231 13,000 per cell.
- the activity of the non-cleavable ProTIA is consistently poorer as compared to all the protease-untreated ProTIAs (AC 1684, AC1685, AC1686, AC1693, AC1695, AC1714, and AC1715) in all three high, mid and low EpCAM expressing cell lines tested. (The only exception being AC 1684 and AC 1484 having equivalent activity in MDA-MB-231 cell line.)
- the fold difference in activity between protease-untreated versus non-cleavable in the EpCAM high expressing HPAF-II is
- Results demonstrated that EpCAM expression of approximately >500,000 per target cell is sufficient to provide strong cytotoxic activity, while EpCAM expression of approximately ⁇ 13,000 per target cell will induced much poorer cytotoxic activity. Differences in ProTIA Release Segment composition as represented by AC1684, AC1685, AC1686, AC1693, AC1695, AC1714, and AC1715 can influence the cytotoxic activity of ProTIAs to kill a specific target cell in the presence of effector PBMC. Results also demonstrated that non-cleavable ProTIA is consistently less active than the protease-untreated anti-EpCAM x anti-CD3 variants in all three high, mid and low EpCAM expressing cell lines evaluated.
- Example 32 Anti-tumor properties of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition in OVCAR-3 ovarian model versus standard of care.
- ProTIA Protease Triggered Immune Activator
- 10 X 10 6 of PBMC would be intravenously introduced to Groups 1-9.
- Treatments would be initiated on day of PBMC inoculation with Group 1 intravenously injected with vehicle (PBS+0.05% Tween 80); Groups 2-8, each intravenously administered with one protease-untreated anti-EpCAM x anti-CD3 ProTIA variants (e.g.
- CA125 level in Group 1 vehicle is expected to increase significantly over time.
- CA125 level in Group 9 bevacizumab is also expected to increase over time but to a lesser extent than that observed in Group 1 vehicle.
- CA125 levels in Groups 2-7 are expected to decrease over time due to efficacy imparted by the various protease-untreated anti-EpCAM x anti-CD3 ProTIA (e.g. AC1684, AC1685, AC1686, AC1693, AC1695, AC1714, and AC1715). As these variants bear different release segment, there is a good likely hood that differences in degree of efficacy (i.e. magnitude of CA125 decrease) would be observed among the protease- untreated variants.
- Example 33 In vivo toxicity assessment of ProTIA vs. BiTE equivalent.
- Toxicity of ProTIA was assessed by using surrogate molecules that bind to mouse EpCAM and mouse CD3e proteins.
- the main toxicity attributed to this molecule is cytokine release syndrome due to expression of EpCAM positive cells in the mouse lymphocytes.
- the test articles were AC1553X, AC1553A, and AC1476A.
- AC1553X is a l38kDa recombinant molecule consisting of anti-mouse EpCAM scFv fused to an anti-mouseCD3 scFv linked to an 864-amino acid XTEN protein (AE864), described more fully in Example 24.
- a tumor- associated protease-sensitive cleavage site was engineered between the aCD3 scFv and adjoining XTEN. Insertion of the unique protease cleavage site enables selective cleavage of AC1553X by tumor-associated proteases such as MMP-9, MMP-2, and matriptase to release the fused anti- mouseEpCAM and anti-mouse CD3 scFv cytotoxic moiety.
- AC1553A is configured in a format equivalent to a BiTE molecule that can be generated by cleavage of the AC1553X by the tumor- associated proteases, and it recognizes mouse EpCAM and mouse CD36/e receptors.
- AC1476A is a bispecific molecule that recognizes human EpCAM and human CD36/e receptor, and it is used as a negative control in the toxicity assessment because it does not recognize the mouse EpCAM nor mouse CD3 molecules.
- results Except for interferon-gamma (IFN-g; FIG. 71E), all of the cytokines for AC1553A were significantly higher than that of AC1553X at the corresponding dose (see results, FIG. 71).
- the max cytokine induction for IL-2, IL-4, IL-6, and TNF-a for AC1553A was at ⁇ 4h. post-dosing, and for IL-10 and INF-g was at ⁇ l0h post-dosing.
- the induction of cytokines for AC1553X in general was delayed to ⁇ 6 - lOh. post-dosing, and the magnitude of induction was much lower than that of AC1553A.
- mice treated with control AC 1476 A are in light circle, with 50ug/kg of AC1553A are in open diamond dotted line, with l50ug/kg of AC1553A are in open diamond dashed line, and with 500ug/kg of AC 1553 A are in open diamond solid line.
- results from mice treated with 120 ug/kg of AC1553X are in black triangle dotted line, with 360 ug/kg of AC1553X are in black triangle dashed line, and with 1,200 pg/kg of AC1553X are in black triangle solid line.
- Cytokine concentrations are shown in picogram per mL of serum plotted against time of blood collection.
- Example 34 Determination of the maximum tolerated dose of ProTIA in C57BL/6 mice.
- Toxicity of ProTIA was assessed by using a surrogate molecule that binds to mouse EpCAM and mouse CD3e proteins.
- the main toxicity attributed to this molecule is cytokine release syndrome due to expression of EpCAM positive cells in the mouse lymphocytes.
- the test articles were AC1553X and AC1553A, described in Example 24.
- Normal C57BL/6 mice were dosed with varying amount of AC1553A (50, 150, and 500 pg/kg), and matching molar amounts of AC1553X (120, 360, and 1,200 pg/kg), and health and body weight of the mice were monitored for 14 days post-dosing.
- FIG. 72 depicts a Kaplan-Meier plot of AC1553X and AC1553A treatment of C57BL/6 mice.
- mice per group were treated with 120 pg/kg of AC1553X (black triangle dotted line), 360 pg/kg of AC1553X (black triangle dashed line), and 1,200 pg/kg of AC1553X (black triangle solid line), and matching molar amounts of AC1553A.
- 50 pg/kg of AC1553A shown in an open diamond dotted line 150 pg/kg of AC 1553 A shown in an open diamond dashed line, and 500 pg/kg of
- mice that were treated with high dose of AC1553A exhibited greater than 10% body weight loss after 2 days of treatment, and all of the mice died within 3 days (FIG. 73A).
- Mice that were treated with mid dose of AC1553A displayed 10 - 20 % body weight loss within 2-4 days post dosing, and 3 mice died while the body weight of the remaining two mice recovered to pre-dosing body weight levels (FIG. 73B)
- Mice treated with low dose of AC1553A (50 pg/kg) displayed a temporary weight loss within 10% at 2 days following treatment, and all of the body weights of the mice recovered to equal or above pre- dosing levels (FIG 73C).
- mice For AC1553X, the high dose (1,200 pg/kg) mice displayed body weight loss of greater than 10 % after 2 -3 days post-dosing, but all of the body weights recovered to normal levels (FIG. 73F) For mid and low dosing of AC1553X, the body weight loss is insignificant (FIG. 73D, E) Percent weight change is plotted against time post drug dosing. Percent weight is calculated by taking the weight of the mice at times post-drug dosing and divided by the original pre-drug dosing weight and multiply by 100 [(post-drug dose weight/pre-drug dose weight) x 100]. 5 mice per group were treated with 120 pg/kg of
- AC1553X black triangle dotted lines
- 360 pg/kg of AC1553X black triangle dashed lines
- 1,200 pg/kg of AC1553X black triangle solid lines
- matching molar amounts of AC1553A 50 pg/kg of AC1553A shown in open diamond dotted lines
- 150 pg/kg of AC1553A shown in open diamond dashed lines 150 pg/kg of AC1553A shown in open diamond dashed lines
- 500 pg/kg of AC1553A in open diamond solid lines Each line represents weight changes of one mouse.
- Example 35 Binding affinity of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition.
- anti-EpCAM x anti- CD3 to the EpCAM chip was performed at 5-6 difference concentrations for full kinetic analysis and K d determination.
- the I for binding to human EpCAM was determined to be 11.4 nM for untreated anti-EpCAM x anti-CD3 ProTIA, 1.15 nM for protease-treated ProTIA, and 10.0 nM for non-cleavable ProTIA.
- binding constants for anti-EpCAM x anti-CD3 ProTIA binding to EpCAM- expressing and CD3 -expressing cells were determined by competition binding with a
- the fluorescently-labeled, protease-treated ProTIA was made by conjugation of Alexa Fluor 647 C2 maleimide (Thermo Fisher,
- Binding experiments were performed on 10,000 cells at 4 °C for 1 hour in a total volume of 100 microL of binding buffer (1% bovine serum albumin in phosphate-buffered saline). Cells were washed once with cold binding buffer, then re-suspended in 2% formaldehyde in phosphate- buffered saline and immediately analyzed on a Millipore Guava easyCyte flow cytometer.
- binding buffer 1% bovine serum albumin in phosphate-buffered saline.
- Binding of the fluorescently-labeled, protease-treated ProTIA revealed an apparent I of 0.85 nM to CHO cells stably transfected with human EpCAM (EpCAM-CHO) and 2.6 nM to CD3+ Jurkat cells. Competition binding of the fluorescently-labeled, protease-treated ProTIA to
- EpCAM-CHO cells resulted in apparent binding constants of 2.9 nM for untreated anti-EpCAM x anti-CD3 ProTIA (e.g. AC1476) and 0.29 nM for protease-treated ProTIA (e.g. MMP-9 treated AC1476).
- Competition binding of the fluorescently-labeled, protease-treated ProTIA to CD3+ Jurkat cells resulted in apparent binding constants of 31 nM for untreated anti-EpCAM x anti- CD3 ProTIA (e.g. AC1476) and 1.1 nM for protease-treated ProTIA (e.g. MMP-9 treated
- the binding affinity to EpCAM for the protease-treated ProTIA was about 10-fold stronger than untreated and non-cleavable ProTIA by both SPR and flow cytometry.
- the binding affinity to CD3 for the protease-treated ProTIA was stronger than untreated and non-cleavable ProTIA: about 10-fold by SPR and about 30-fold by flow cytometry.
- Example 36 T-cell activation marker assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition.
- 1 X 105 PBMC or purified CD3+ cells would be co-cultured in RPMI-1640 containing 10% FCS with 2 X 104 SK-OV-3 or OVCAR3 cells per assay well (i.e., effector to target ratio of 5:1) in the presence of anti-EpCAM x anti-CD3 ProTIA (e.g. AC 1695) in a 96- well round-bottom plate with total final volume of 200 microL.
- anti-EpCAM x anti-CD3 ProTIA e.g. AC 1695
- T-cell activation marker expression is expected to have a similar trend for the three ProTIA molecules [untreated (e.g. AC1695), protease-treated (e.g. MMP-9 treated AC1695), and non-cleavable (e.g. AC1484) anti-EpCAM x anti-CD3] as was observed by caspase 3/7 cytotoxicity assay.
- untreated anti-EpCAM x anti-CD3 ProTIA e.g. AC1695
- protease-treated ProTIA e.g. MMP-9 treated AC1695
- non-cleavable e.g. AC1484
- Example 37 Cvtokine release assays of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA] composition.
- This assay can also be performed with other target cells selected from HCT-116, Kato III, MDA-MB-453, MCF-7, MKN45, MT3, NCI-N87, SK-Br-3, SW480, OVCAR3 and PC3 cell lines as well as PBMC in place of purified CD3+ cells.
- Cytokine analysis of interleukin (IL)-2, IL-4, IL-6, IL-10, tumor necrosis factor (TNF)- alpha and interferon (IFN)-gamma secreted into the cell culture supernatant would be quantitated using the Human Thl/Th2 Cytokine Cytometric Bead Array (CBA) kit (BD Biosciences cat #550749) following manufacturer’s instruction. In the absence of ProTIA, no cytokine secretion above background is expected from purified CD3+ cells.
- CBA Human Thl/Th2 Cytokine Cytometric Bead Array
- ProTIA in the presence of EpCAM- positive target cells and purified CD3+ cells is expected to activate T cells and secrete a pattern of T cell cytokines with a high proportion of Thl cytokines such as IFN-gamma and TNF -alpha.
- Anti-EpCAM x anti-CD3 ProTIA is expected to induce robust secretion of all cytokines (IL-2, IL-4, IL-6, IL-10, TNF-alpha, IFN-gamma) that would be evaluated (see FIGS. 50-52).
- Stimulation of purified CD3+ cells with SK-OV-3 cells and protease-treated anti-EpCAM x anti- CD3 ProTIA e.g. MMP-9 treated AC1695
- MMP-9 treated AC1695 is expected to trigger significant cytokine expression, especially at concentrations higher than 20 pM for all of the cytokines that would be tested.
- baseline levels of IL-2, IL-4, IL-6, IL-10, TNF-alpha and IFN-gamma are expected when the intact non-cleaved anti-EpCAM x anti-CD3 ProTIA molecule (e.g. AC1695) is used at a concentration up to about 100 pM. Additionally, baseline levels of all cytokines that would be tested are expected for the non-cleavable anti-EpCAM x anti-CD3 ProTIA molecule (e.g.
- Example 38 Binding affinity of anti-EpCAM x anti-CD3 Protease Triggered Immune Activator (ProTIA) composition.
- the binding constants for anti-EpCAM x anti-CD3 ProTIA binding to EpCAM- expressing and CD3 -expressing cells would be measured by competition binding with a fluorescently-labeled, protease-treated ProTIA.
- the fluorescently-labeled, protease-treated ProTIA was made by conjugation of Alexa Fluor 647 C2 maleimide (Thermo Fisher,
- Binding experiments would be performed on 10,000 cells at 4°C for 1 hour in a total volume of 100 microL of binding buffer (1% bovine serum albumin in phosphate-buffered saline). Cells would be washed once with cold binding buffer, then re-suspended in 2% formaldehyde in phosphate-buffered saline and immediately analyzed on a Millipore Guava easyCyte flow cytometer.
- Binding of the fluorescently-labeled, protease-treated ProTIA would be expected to have an apparent I value of approximately 1 nM to CHO cells stably transfected with human EpCAM (EpCAM-CHO) and approximately 3 nM to CD3+ Jurkat cells.
- Competition binding of the fluorescently-labeled, protease-treated ProTIA to EpCAM-CHO cells is expected to result in apparent binding constants of single-digit nM for untreated anti-EpCAM x anti-CD3 ProTIA (e.g. AC1695) and sub-nM for protease-treated ProTIA (e.g. MMP-9 treated AC1695).
- the XTEN base vector AC 1611 with Release Segment RSR-1517 was built from modifying pNL0356 by PCR to encode the protein of HD2-V5-XTEN144-RSR-1517-XTEN712-H8 under the control of T71ac promoter, where HD2 sequence is MKNPEQAEEQAEEQREET and V5 is GKPIPNPLLGLDST.
- the coding sequence of release segment RSR-1517 on AC1611 is flanked by unique Nhel and Bsal restriction sites to enable replacement with another release segments. After ligation reaction, transformants were screened by DNA miniprep and the desired construct was confirmed by DNA sequencing. The resulting construct is AC1611, with the DNA sequence and encoded amino acid sequence provided in Table 16.
- Example 40 Shake flask expression of Release Sesment-XTEN variants
- BL21 DE3 strain New England Biolabs
- Starter cultures were prepared by inoculating glycerol stocks of E. coli BL21 DE3 carrying the corresponding plasmids into 6 mL of LB Broth media containing 50 pg/mL kanamycin and incubated overnight at 37 °C at 200 min-l. Overnight starter cultures were inoculated 1:50 into approximately 250 mL of ZY auto-induction media and grown at 26°C for 26 hours, at 200 min- 1. Cells were then harvested by centrifugation at 10,000 rpm for 30 mins for immediate use or frozen at -80°C until use.
- the ZY auto-induction media were made by mixing the components as follows: 928 mL of ZY media (10 g bacto tryptone, 5 g yeast extract and 925 mL of water; autoclaved), 1 mL 1M MgS04, 20 mL 50x 5052 solution (25 g glycerol, 2.5 g glucose (Fisher FLBP350-1), 10 g a- lactose (Sigma L3625), and 73 ml water; sterile-filtered), 50 ml 20x NPS solution (to make 100 mL, dissolve 6.6g (NH4)2S04, 13.6 g KH2P04, 14.2 g Na2HP04 in 90 mL water; sterile- filtered) and 1 mL kanamycin (50 mg/mL).
- Example 41 Purification of Release Segment-XTEN variants from E. coli shake flask cultures
- FIG. 74A is an exemplary titer analysis of 3 RS variants and the arrow indicates where the products migrate, with apparent molecular weight roughly around the 160 kDa molecular weight marker.
- Immobilized-metal affinity chromatography was used as the capture step.
- a 10- mL polypropylene column (Thermo Scientific) was packed with 5 mL of ToyoPearl-AF-Chelate 650M resin (TOSOH Biosciences).
- the column was equilibrated with 5 column volumes (CVs) of equilibration buffer (20 mM Tris, 100 mM NaCl, pH 7.5).
- the pH of the clarified cell lysates was adjusted to 7.5 before loading to the columns.
- Example 42 Enzyme activation, storage and digestion of RSR-l5l7-containing XTEN AC1611
- RSR-l5l7-containing XTEN constructs AC1611 can be cleaved by various tumor-associated proteases including recombinant human uPA, matriptase, legumain, MMP-2, MMP-7, MMP-9, and MMP-14, in test tubes.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicinal Preparation (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description






























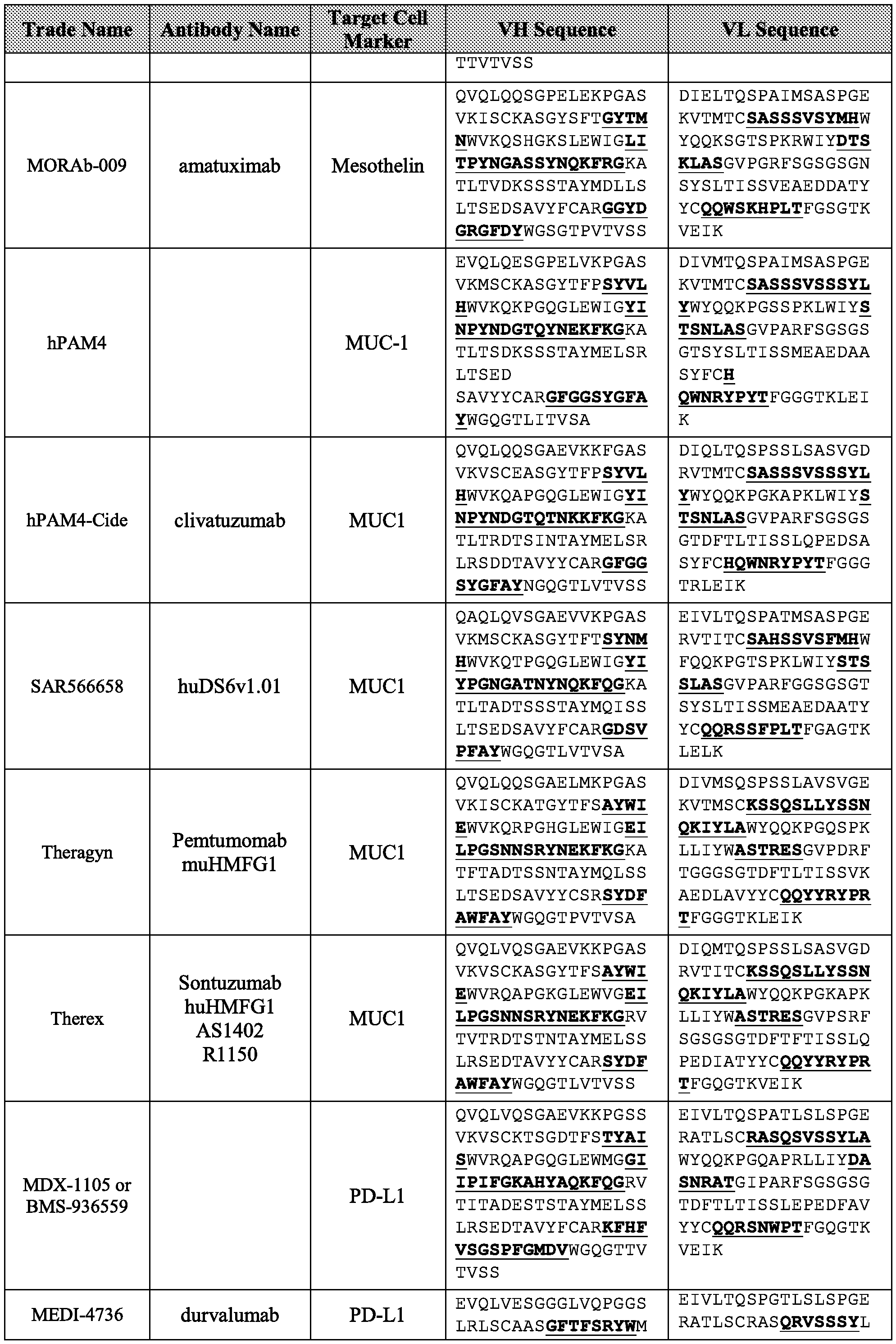



































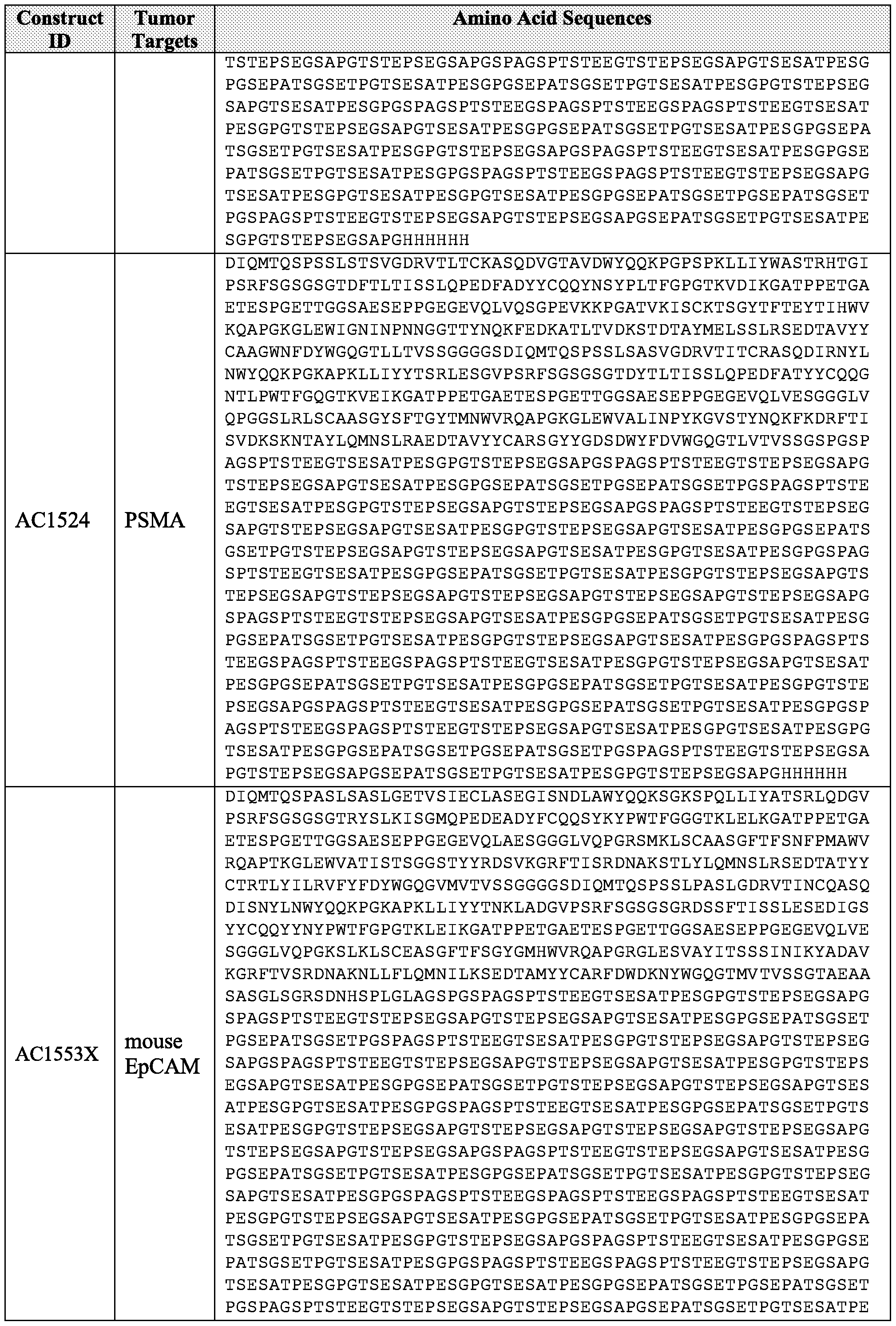













































Claims
Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201880090095.7A CN111819202A (en) | 2017-12-21 | 2018-12-20 | Release segment and binding composition comprising the same |
| IL323008A IL323008A (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| US16/954,145 US12060424B2 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| KR1020207020363A KR102935647B1 (en) | 2017-12-21 | 2018-12-20 | Glass segment and bonding composition comprising the same |
| MX2020006619A MX2020006619A (en) | 2017-12-21 | 2018-12-20 | RELEASE SEGMENTS AND BINDING COMPOSITIONS COMPRISING THEM. |
| AU2018393111A AU2018393111B2 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| CA3085950A CA3085950A1 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| JP2020534301A JP2021507706A (en) | 2017-12-21 | 2018-12-20 | Release segment and binding composition containing it |
| EP18890575.6A EP3728326A4 (en) | 2017-12-21 | 2018-12-20 | RELEASE SEGMENTS AND LIAISON COMPOSITIONS INCLUDING THEM |
| BR112020012220-3A BR112020012220A2 (en) | 2017-12-21 | 2018-12-20 | release segments and connection compositions comprising the same |
| KR1020267006604A KR20260039804A (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| EA202091326A EA202091326A1 (en) | 2018-12-17 | 2018-12-20 | RELEASING SEGMENTS AND BINDING COMPOSITIONS CONTAINING THEM |
| SG11202005674XA SG11202005674XA (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| IL317237A IL317237B2 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| IL275395A IL275395A (en) | 2017-12-21 | 2020-06-15 | Release segments and binding compositions comprising same |
| JP2024098514A JP2024123120A (en) | 2017-12-21 | 2024-06-19 | Release segments and binding compositions containing same |
| US18/765,614 US20250163153A1 (en) | 2017-12-21 | 2024-07-08 | Release segments and binding compositions comprising same |
| AU2025242242A AU2025242242A1 (en) | 2017-12-21 | 2025-10-03 | Release segments and binding compositions comprising same |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762609296P | 2017-12-21 | 2017-12-21 | |
| US62/609,296 | 2017-12-21 | ||
| US201862780719P | 2018-12-17 | 2018-12-17 | |
| US62/780,719 | 2018-12-17 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/954,145 A-371-Of-International US12060424B2 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
| US18/765,614 Division US20250163153A1 (en) | 2017-12-21 | 2024-07-08 | Release segments and binding compositions comprising same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019126576A1 true WO2019126576A1 (en) | 2019-06-27 |
Family
ID=66992850
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2018/066939 Ceased WO2019126576A1 (en) | 2017-12-21 | 2018-12-20 | Release segments and binding compositions comprising same |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US12060424B2 (en) |
| EP (1) | EP3728326A4 (en) |
| JP (2) | JP2021507706A (en) |
| KR (2) | KR20260039804A (en) |
| CN (1) | CN111819202A (en) |
| AU (2) | AU2018393111B2 (en) |
| BR (1) | BR112020012220A2 (en) |
| CA (1) | CA3085950A1 (en) |
| IL (3) | IL323008A (en) |
| MX (1) | MX2020006619A (en) |
| SG (1) | SG11202005674XA (en) |
| TW (1) | TW201930341A (en) |
| WO (1) | WO2019126576A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020260309A1 (en) * | 2019-06-24 | 2020-12-30 | Urteste Sp. Z O.O. | Novel diagnostic marker for pancreatic cancer |
| WO2020264200A1 (en) * | 2019-06-26 | 2020-12-30 | Amunix Pharmceuticals, Inc. | Cd3 antigen binding fragments and compositions comprising same |
| WO2021097186A1 (en) * | 2019-11-13 | 2021-05-20 | Amunix Pharmaceuticals, Inc. | Barcoded xten polypeptides and compositions thereof, and methods for making and using the same |
| EP3845664A1 (en) * | 2020-01-02 | 2021-07-07 | Urteste Sp. z o.o. | Novel diagnostic marker for prostate cancer |
| WO2021262985A1 (en) * | 2020-06-25 | 2021-12-30 | Amunix Pharmaceuticals, Inc. | Cytokine conjugates |
| WO2021263058A1 (en) * | 2020-06-25 | 2021-12-30 | Amunix Pharmaceuticals, Inc. | Her-2 targeted bispecific compositions and methods for making and using the same |
| CN114651012A (en) * | 2019-09-04 | 2022-06-21 | 苏黎世大学 | Bispecific binding agents that bind to CD117/c-KIT and CD3 |
| JP2023511376A (en) * | 2020-01-23 | 2023-03-17 | アダジーン アーゲー | Heterodimeric protein with FC mutation |
| JP2024508671A (en) * | 2021-02-11 | 2024-02-28 | アダジーン プライベート リミテッド | Anti-CD3 antibody and method of use thereof |
| WO2024150172A1 (en) * | 2023-01-11 | 2024-07-18 | Bright Peak Therapeutics Ag | Cleavable peptides and methods of use thereof |
| US12060424B2 (en) | 2017-12-21 | 2024-08-13 | Amunix Pharmaceuticals, Inc. | Release segments and binding compositions comprising same |
| EP4259202A4 (en) * | 2020-12-09 | 2025-01-15 | Janux Therapeutics, Inc. | Compositions and methods related to tumor activated antibodies targeting trop2 and effector cell antigens |
| EP4259193A4 (en) * | 2020-12-09 | 2025-01-15 | Janux Therapeutics, Inc. | COMPOSITIONS AND METHODS RELATED TO TUMOR-ACTIVATED ANTIBODIES AGAINST PSMA AND EFFECTOR CELL ANTIGENS |
| EP4352218A4 (en) * | 2021-06-07 | 2025-04-16 | Flagship Pioneering Innovations VII, LLC | Compositions and methods for targeted delivery of therapeutic agents |
| WO2025122957A1 (en) | 2023-12-08 | 2025-06-12 | Amunix Pharmaceuticals, Inc. | Protease activatable cytokines and methods for making and using the same |
| RU2851152C1 (en) * | 2020-06-25 | 2025-11-19 | Амуникс Фармасьютикалз, Инк. | Her-2-targeted bisspecific compositions and methods for their preparation and use |
| EP4403572A4 (en) * | 2021-09-13 | 2025-12-24 | Shandong Simcere Biopharmaceutical Co Ltd | ANTI-HUMAN CD3 ANTIBODIES AND USE OF THEM |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY204234A (en) | 2018-08-27 | 2024-08-16 | Regeneron Pharma | Use of raman spectroscopy in downstream purification |
| CN115197325A (en) * | 2021-04-14 | 2022-10-18 | 康方药业有限公司 | Use of Antibodies in Antitumor Therapy |
| WO2023161853A1 (en) | 2022-02-23 | 2023-08-31 | Bright Peak Therapeutics Ag | Activatable il-18 polypeptides |
| WO2024150175A1 (en) | 2023-01-11 | 2024-07-18 | Bright Peak Therapeutics Ag | Conditionally activated proteins and methods of use |
| US20240417436A1 (en) | 2023-01-11 | 2024-12-19 | Bright Peak Therapeutics Ag | Conditionally activated immunocytokines and methods of use |
| WO2025041101A1 (en) | 2023-08-23 | 2025-02-27 | Bright Peak Therapeutics Ag | Activatable il-18 immunocytokines and uses thereof |
| CN121517565A (en) * | 2023-09-25 | 2026-02-13 | 深圳湾实验室 | It can activate membrane-penetrating constructs and degradable molecules. |
| CN117169518B (en) * | 2023-11-03 | 2024-01-19 | 赛德特(北京)生物工程有限公司 | Method and kit for detecting CD3 antibody residues in T lymphocyte preparation |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120178691A1 (en) * | 2010-08-19 | 2012-07-12 | Amunix Operating Inc. | Factor viii compositions and methods of making and using same |
| US20120239554A1 (en) * | 2011-03-14 | 2012-09-20 | Christopher Primbas | System And Method To Eliminate Receiving Coins As Cents Due Less Than One Dollar |
| WO2013130683A2 (en) * | 2012-02-27 | 2013-09-06 | Amunix Operating Inc. | Xten conjugate compositions and methods of making same |
| US20150266943A1 (en) * | 2012-07-11 | 2015-09-24 | Amunix Operating Inc. | Factor viii complex with xten and von willebrand factor protein, and uses thereof |
| WO2016077505A2 (en) * | 2014-11-11 | 2016-05-19 | Amunix Operating Inc. | Targeted xten conjugate compositions and methods of making same |
| WO2017040344A2 (en) * | 2015-08-28 | 2017-03-09 | Amunix Operating Inc. | Chimeric polypeptide assembly and methods of making and using the same |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6225449B1 (en) | 1991-10-04 | 2001-05-01 | Washington University | Hormone analogs with multiple CTP extensions |
| US20030228309A1 (en) | 2000-11-08 | 2003-12-11 | Theodora Salcedo | Antibodies that immunospecifically bind to TRAIL receptors |
| PE20020908A1 (en) | 2001-03-21 | 2002-10-26 | Cell Therapeutics Inc | RECOMBINANT PRODUCTION OF POLYANIONIC POLYMERS AND USE OF THE SAME |
| MXPA05012201A (en) | 2003-05-14 | 2006-02-10 | Dow Corning | CONJUGATES OF ACTIVE PROTEIN POLYMER AGENTS OF REPETITION SEQUENCE, METHODS AND USES. |
| WO2006081249A2 (en) | 2005-01-25 | 2006-08-03 | Cell Therapeutics, Inc. | Conjugates of biologically active proteins having a modified in vivo half-life |
| KR100727466B1 (en) | 2005-02-07 | 2007-06-13 | 주식회사 잉크테크 | Organic silver complex compound, preparation method thereof and thin film formation method using the same |
| US7855279B2 (en) | 2005-09-27 | 2010-12-21 | Amunix Operating, Inc. | Unstructured recombinant polymers and uses thereof |
| US7846445B2 (en) | 2005-09-27 | 2010-12-07 | Amunix Operating, Inc. | Methods for production of unstructured recombinant polymers and uses thereof |
| AU2006301492B2 (en) | 2005-10-11 | 2011-06-09 | Amgen Research (Munich) Gmbh | Compositions comprising cross-species-specific antibodies and uses thereof |
| CN101384272B (en) | 2005-12-20 | 2013-05-01 | 杜克大学 | Methods and compositions for delivering active agents with enhanced pharmacological properties |
| WO2008049711A1 (en) | 2006-10-27 | 2008-05-02 | Novo Nordisk A/S | Peptide extended insulins |
| WO2008119566A2 (en) | 2007-04-03 | 2008-10-09 | Micromet Ag | Cross-species-specific bispecific binders |
| ATE502114T1 (en) | 2007-06-21 | 2011-04-15 | Univ Muenchen Tech | BIOLOGICALLY ACTIVE PROTEINS WITH INCREASED IN-VIVO AND/OR IN-VITRO STABILITY |
| US8933197B2 (en) | 2007-08-15 | 2015-01-13 | Amunix Operating Inc. | Compositions comprising modified biologically active polypeptides |
| US20100189651A1 (en) | 2009-01-12 | 2010-07-29 | Cytomx Therapeutics, Llc | Modified antibody compositions, methods of making and using thereof |
| US8680050B2 (en) | 2009-02-03 | 2014-03-25 | Amunix Operating Inc. | Growth hormone polypeptides fused to extended recombinant polypeptides and methods of making and using same |
| CA2748314C (en) | 2009-02-03 | 2018-10-02 | Amunix Operating Inc. | Extended recombinant polypeptides and compositions comprising same |
| US8703717B2 (en) | 2009-02-03 | 2014-04-22 | Amunix Operating Inc. | Growth hormone polypeptides and methods of making and using same |
| SI2440241T1 (en) | 2009-06-08 | 2017-11-30 | Amunix Operating Inc. | Growth hormone polypeptides and methods of making and using same |
| JP5839597B2 (en) | 2009-06-08 | 2016-01-06 | アムニクス オペレーティング インコーポレイテッド | Glucose-regulating polypeptide and methods for making and using the same |
| US9849188B2 (en) | 2009-06-08 | 2017-12-26 | Amunix Operating Inc. | Growth hormone polypeptides and methods of making and using same |
| AU2010290131C1 (en) | 2009-08-24 | 2015-12-03 | Amunix Operating Inc. | Coagulation factor VII compositions and methods of making and using same |
| WO2011028344A2 (en) | 2009-08-25 | 2011-03-10 | Amunix Operating Inc. | Interleukin-1 receptor antagonist compositions and methods of making and using same |
| US20110312881A1 (en) | 2009-12-21 | 2011-12-22 | Amunix, Inc. | Bifunctional polypeptide compositions and methods for treatment of metabolic and cardiovascular diseases |
| WO2011123830A2 (en) | 2010-04-02 | 2011-10-06 | Amunix Operating Inc. | Alpha 1-antitrypsin compositions and methods of making and using same |
| WO2012158818A2 (en) * | 2011-05-16 | 2012-11-22 | Fabion Pharmaceuticals, Inc. | Multi-specific fab fusion proteins and methods of use |
| EP2755675B1 (en) | 2011-09-12 | 2018-06-06 | Amunix Operating Inc. | Glucagon-like peptide-2 compositions and methods of making and using same |
| BR112014020694A2 (en) | 2012-02-15 | 2018-05-08 | Amunix Operating Inc. | factor viii fusion protein comprising extended recombinant polypeptide (xten) fusion factor polypeptide and its method of manufacture, nucleic acid, vectors, host cell, as well as pharmaceutical composition and its use in the treatment of coagulopathy, bleeding episode and hemophilia a |
| KR20150021072A (en) | 2012-06-05 | 2015-02-27 | 아뮤닉스 오퍼레이팅 인코포레이티드 | Hgh-xten fusion protein and its use in the treatment of growth hormone deficiency |
| HK1216617A1 (en) | 2013-03-11 | 2016-11-25 | Amunix Operating Inc. | Treatment of pediatric growth hormone deficiency with human growth hormone analogues |
| US10548953B2 (en) | 2013-08-14 | 2020-02-04 | Bioverativ Therapeutics Inc. | Factor VIII-XTEN fusions and uses thereof |
| AU2015323313B2 (en) | 2014-09-25 | 2021-04-01 | Amgen Inc. | Protease-activatable bispecific proteins |
| WO2016109823A1 (en) | 2015-01-02 | 2016-07-07 | Amunix Operation Inc. | Treatment of pediatric growth hormone deficiency with human growth hormone analogues |
| EP3778640A1 (en) | 2015-05-01 | 2021-02-17 | Genentech, Inc. | Masked anti-cd3 antibodies and methods of use |
| CR20180453A (en) | 2016-03-22 | 2018-12-05 | Hoffmann La Roche | Bispecific MOLECULES OF T-CELLS ACTIVATED BY PROTEASES |
| JP2019525931A (en) | 2016-07-27 | 2019-09-12 | アムニクス ファーマシューティカルズ, インコーポレイテッド | Treatment of adult growth hormone deficiency with human growth hormone analogs |
| EP3728326A4 (en) | 2017-12-21 | 2021-12-15 | Amunix Pharmaceuticals, Inc. | RELEASE SEGMENTS AND LIAISON COMPOSITIONS INCLUDING THEM |
| US20230121775A1 (en) * | 2019-06-26 | 2023-04-20 | Amunix Pharmaceuticals, Inc. | Cd3 antigen binding fragments and compositions comprising same |
-
2018
- 2018-12-20 EP EP18890575.6A patent/EP3728326A4/en active Pending
- 2018-12-20 KR KR1020267006604A patent/KR20260039804A/en active Pending
- 2018-12-20 WO PCT/US2018/066939 patent/WO2019126576A1/en not_active Ceased
- 2018-12-20 CA CA3085950A patent/CA3085950A1/en active Pending
- 2018-12-20 AU AU2018393111A patent/AU2018393111B2/en active Active
- 2018-12-20 JP JP2020534301A patent/JP2021507706A/en active Pending
- 2018-12-20 MX MX2020006619A patent/MX2020006619A/en unknown
- 2018-12-20 KR KR1020207020363A patent/KR102935647B1/en active Active
- 2018-12-20 TW TW107146132A patent/TW201930341A/en unknown
- 2018-12-20 IL IL323008A patent/IL323008A/en unknown
- 2018-12-20 IL IL317237A patent/IL317237B2/en unknown
- 2018-12-20 SG SG11202005674XA patent/SG11202005674XA/en unknown
- 2018-12-20 BR BR112020012220-3A patent/BR112020012220A2/en unknown
- 2018-12-20 US US16/954,145 patent/US12060424B2/en active Active
- 2018-12-20 CN CN201880090095.7A patent/CN111819202A/en active Pending
-
2020
- 2020-06-15 IL IL275395A patent/IL275395A/en unknown
-
2024
- 2024-06-19 JP JP2024098514A patent/JP2024123120A/en active Pending
- 2024-07-08 US US18/765,614 patent/US20250163153A1/en active Pending
-
2025
- 2025-10-03 AU AU2025242242A patent/AU2025242242A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120178691A1 (en) * | 2010-08-19 | 2012-07-12 | Amunix Operating Inc. | Factor viii compositions and methods of making and using same |
| US20120239554A1 (en) * | 2011-03-14 | 2012-09-20 | Christopher Primbas | System And Method To Eliminate Receiving Coins As Cents Due Less Than One Dollar |
| WO2013130683A2 (en) * | 2012-02-27 | 2013-09-06 | Amunix Operating Inc. | Xten conjugate compositions and methods of making same |
| US20150266943A1 (en) * | 2012-07-11 | 2015-09-24 | Amunix Operating Inc. | Factor viii complex with xten and von willebrand factor protein, and uses thereof |
| WO2016077505A2 (en) * | 2014-11-11 | 2016-05-19 | Amunix Operating Inc. | Targeted xten conjugate compositions and methods of making same |
| WO2017040344A2 (en) * | 2015-08-28 | 2017-03-09 | Amunix Operating Inc. | Chimeric polypeptide assembly and methods of making and using the same |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3728326A4 * |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12060424B2 (en) | 2017-12-21 | 2024-08-13 | Amunix Pharmaceuticals, Inc. | Release segments and binding compositions comprising same |
| JP2022538862A (en) * | 2019-06-24 | 2022-09-06 | ユルテステ・ソシエテ・アノニム | Novel diagnostic marker for pancreatic cancer |
| KR102798031B1 (en) | 2019-06-24 | 2025-04-17 | 우르테스테 에스.에이. | A new pancreatic cancer diagnostic marker |
| CN114096555A (en) * | 2019-06-24 | 2022-02-25 | 尔特斯特公司 | Novel pancreatic cancer diagnostic marker |
| KR20220024867A (en) * | 2019-06-24 | 2022-03-03 | 우르테스테 에스.에이. | New pancreatic cancer diagnostic markers |
| WO2020260309A1 (en) * | 2019-06-24 | 2020-12-30 | Urteste Sp. Z O.O. | Novel diagnostic marker for pancreatic cancer |
| JP7601804B2 (en) | 2019-06-24 | 2024-12-17 | ユルテステ・ソシエテ・アノニム | New diagnostic markers for pancreatic cancer |
| WO2020264200A1 (en) * | 2019-06-26 | 2020-12-30 | Amunix Pharmceuticals, Inc. | Cd3 antigen binding fragments and compositions comprising same |
| CN114651012A (en) * | 2019-09-04 | 2022-06-21 | 苏黎世大学 | Bispecific binding agents that bind to CD117/c-KIT and CD3 |
| WO2021097186A1 (en) * | 2019-11-13 | 2021-05-20 | Amunix Pharmaceuticals, Inc. | Barcoded xten polypeptides and compositions thereof, and methods for making and using the same |
| EP4058466A1 (en) * | 2019-11-13 | 2022-09-21 | Amunix Pharmaceuticals, Inc. | Barcoded xten polypeptides and compositions thereof, and methods for making and using the same |
| EP3845664A1 (en) * | 2020-01-02 | 2021-07-07 | Urteste Sp. z o.o. | Novel diagnostic marker for prostate cancer |
| JP7710450B2 (en) | 2020-01-23 | 2025-07-18 | アダジーン アーゲー | Heterodimeric proteins having FC mutations |
| JP2023511376A (en) * | 2020-01-23 | 2023-03-17 | アダジーン アーゲー | Heterodimeric protein with FC mutation |
| US12215156B2 (en) | 2020-06-25 | 2025-02-04 | Amunix Pharmaceuticals, Inc. | HER-2 targeted bispecific compositions and methods for making and using the same |
| CN116096730A (en) * | 2020-06-25 | 2023-05-09 | 阿穆尼克斯制药公司 | cytokine conjugates |
| RU2851152C1 (en) * | 2020-06-25 | 2025-11-19 | Амуникс Фармасьютикалз, Инк. | Her-2-targeted bisspecific compositions and methods for their preparation and use |
| EP4172179A4 (en) * | 2020-06-25 | 2024-11-20 | Amunix Pharmaceuticals, Inc. | Her-2 targeted bispecific compositions and methods for making and using the same |
| JP2023531496A (en) * | 2020-06-25 | 2023-07-24 | アムニクス ファーマシューティカルズ, インコーポレイテッド | Cytokine conjugate |
| WO2021262985A1 (en) * | 2020-06-25 | 2021-12-30 | Amunix Pharmaceuticals, Inc. | Cytokine conjugates |
| WO2021263058A1 (en) * | 2020-06-25 | 2021-12-30 | Amunix Pharmaceuticals, Inc. | Her-2 targeted bispecific compositions and methods for making and using the same |
| EP4259193A4 (en) * | 2020-12-09 | 2025-01-15 | Janux Therapeutics, Inc. | COMPOSITIONS AND METHODS RELATED TO TUMOR-ACTIVATED ANTIBODIES AGAINST PSMA AND EFFECTOR CELL ANTIGENS |
| EP4259202A4 (en) * | 2020-12-09 | 2025-01-15 | Janux Therapeutics, Inc. | Compositions and methods related to tumor activated antibodies targeting trop2 and effector cell antigens |
| JP2024508671A (en) * | 2021-02-11 | 2024-02-28 | アダジーン プライベート リミテッド | Anti-CD3 antibody and method of use thereof |
| EP4352218A4 (en) * | 2021-06-07 | 2025-04-16 | Flagship Pioneering Innovations VII, LLC | Compositions and methods for targeted delivery of therapeutic agents |
| EP4403572A4 (en) * | 2021-09-13 | 2025-12-24 | Shandong Simcere Biopharmaceutical Co Ltd | ANTI-HUMAN CD3 ANTIBODIES AND USE OF THEM |
| WO2024150172A1 (en) * | 2023-01-11 | 2024-07-18 | Bright Peak Therapeutics Ag | Cleavable peptides and methods of use thereof |
| WO2025122957A1 (en) | 2023-12-08 | 2025-06-12 | Amunix Pharmaceuticals, Inc. | Protease activatable cytokines and methods for making and using the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021507706A (en) | 2021-02-25 |
| BR112020012220A2 (en) | 2021-01-26 |
| IL323008A (en) | 2025-10-01 |
| US20200385469A1 (en) | 2020-12-10 |
| US12060424B2 (en) | 2024-08-13 |
| EP3728326A1 (en) | 2020-10-28 |
| IL317237B2 (en) | 2026-02-01 |
| AU2018393111B2 (en) | 2025-07-10 |
| AU2018393111A1 (en) | 2020-07-23 |
| KR20260039804A (en) | 2026-03-20 |
| IL275395A (en) | 2020-07-30 |
| SG11202005674XA (en) | 2020-07-29 |
| TW201930341A (en) | 2019-08-01 |
| AU2025242242A1 (en) | 2025-10-23 |
| JP2024123120A (en) | 2024-09-10 |
| EP3728326A4 (en) | 2021-12-15 |
| CA3085950A1 (en) | 2019-06-27 |
| MX2020006619A (en) | 2020-09-14 |
| IL317237A (en) | 2025-01-01 |
| IL317237B1 (en) | 2025-10-01 |
| KR102935647B1 (en) | 2026-03-06 |
| CN111819202A (en) | 2020-10-23 |
| KR20200111176A (en) | 2020-09-28 |
| US20250163153A1 (en) | 2025-05-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250163153A1 (en) | Release segments and binding compositions comprising same | |
| AU2026200378C1 (en) | Chimeric polypeptide assembly and methods of making and using the same | |
| EP3990497A1 (en) | Cd3 antigen binding fragments and compositions comprising same | |
| BR122026001258A2 (en) | METHOD OF MANUFACTURING A RECOMBINANT POLYPEPTIDE AND USE OF A RECOMBINANT POLYPEPTIDE OR A COMPOSITION COMPRISING THE SAME | |
| HK40081669A (en) | Chimeric polypeptide assembly and methods of making and using the same | |
| BR112018004045B1 (en) | CHIMERIC POLYPEPTIDE ASSEMBLY CONSTRUCTS AND THEIR USE | |
| BR122025010451A2 (en) | CHIMERIC POLYPEPTIDE SET, ITS USE, PHARMACEUTICAL COMPOSITION AND ISOLATED NUCLEIC ACID |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18890575 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3085950 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020534301 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20207020363 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018393111 Country of ref document: AU Date of ref document: 20181220 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018890575 Country of ref document: EP Effective date: 20200721 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020012220 Country of ref document: BR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112020012220 Country of ref document: BR Free format text: APRESENTAR, EM ATE 60 (SESSENTA) DIAS, TRADUCAO COMPLETA DO PEDIDO, ADAPTADA A NORMA VIGENTE, CONFORME CONSTA NO DEPOSITO INTERNACIONAL INICIAL PCT/US2018/066936 DE 20/12/2018, POIS A MESMA NAO FOI APRESENTADA ATE O MOMENTO. |
|
| ENP | Entry into the national phase |
Ref document number: 112020012220 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200617 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 275395 Country of ref document: IL |
|
| WWD | Wipo information: divisional of initial pct application |
Ref document number: 323008 Country of ref document: IL |