WO2019194399A1 - 고흡수성 수지의 제조방법 - Google Patents
고흡수성 수지의 제조방법 Download PDFInfo
- Publication number
- WO2019194399A1 WO2019194399A1 PCT/KR2019/001109 KR2019001109W WO2019194399A1 WO 2019194399 A1 WO2019194399 A1 WO 2019194399A1 KR 2019001109 W KR2019001109 W KR 2019001109W WO 2019194399 A1 WO2019194399 A1 WO 2019194399A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fine powder
- water
- polymer
- reassembly
- super absorbent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/002—Methods
- B29B7/005—Methods for mixing in batches
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/80—Component parts, details or accessories; Auxiliary operations
- B29B7/88—Adding charges, i.e. additives
- B29B7/94—Liquid charges
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/16—Auxiliary treatment of granules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/04—Polymerisation in solution
- C08F2/10—Aqueous solvent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/48—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light
- C08F2/50—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light with sensitising agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
- B29B2009/125—Micropellets, microgranules, microparticles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/16—Auxiliary treatment of granules
- B29B2009/161—Absorbing, i.e. introducing a gas, a liquid or a solid material into the granules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Definitions
- the present invention relates to a method for preparing a superabsorbent polymer, and more particularly, by controlling the stirring speed in the preparation step of the fine powder reassembly during the preparation of the superabsorbent polymer, and by dividing water into the fine powder, the granulation strength of the fine granule assembly. It is possible to operate a stable fine powder reassembly manufacturing process while maintaining a, can reduce the fun fraction, and relates to a manufacturing method that can produce a super absorbent polymer with a fast initial absorption rate.
- Super Absorbent Polymer is from 500 to 500
- the absorption mechanism of the superabsorbent polymer is characterized by the interaction between the penetration pressure due to the difference in electrical attraction force of the charge of the polymer electrolyte, the affinity between water and the polymer electrolyte, the expansion of the molecule due to the repulsive force between the polymer electrolyte ions, and the expansion inhibition due to crosslinking. Is ruled by That is, the absorbency of the absorbent polymer depends on the affinity and molecular expansion described above, and the rate of absorption depends largely on the penetration pressure of the absorbent polymer itself.
- the fine powder is a fine powder reassembly process of mixing and coagulating fine powder and water.
- the present invention is to solve the problems of the prior art as described above, by adjusting the stirring speed in the manufacturing step of the fine powder reassembly during the production of superabsorbent polymer, and by adding water to the fine powder, the assembly strength of the fine granules It is possible to operate a stable fine powder reassembly manufacturing process while maintaining, to reduce the fun fraction, and to provide a method for producing a superabsorbent polymer having a fast initial absorption rate.
- Preparing a hydrogel polymer by thermally polymerizing or photopolymerizing a monomer composition including a water-soluble ethylenically unsaturated monomer and a polymerization initiator; Drying and pulverizing the hydrogel polymer and classifying into fine particles having a particle size of less than 150 /, and normal particles having a particle size of greater than 150 and less than 850 III; beauty
- the fine powder is mixed with water while stirring at a linear speed of 6.5 to 14. 3/4 1/3, and reassembled to prepare a fine powder reassembly;
- the water When mixing the fine powder and water, the water is divided into two times or more to the fine powder, provides a method for producing a super absorbent polymer.
- the method for preparing superabsorbent polymer according to the present invention enables the operation of a stable fine powder reassembly manufacturing process while maintaining the assembly strength of the fine powder assembly, and can reduce the fun fraction.
- 1 is a graph showing the result of comparing the secondary aggregation formation time according to the stirring linear velocity.
- Figure 2 is a photograph observing the internal state of the mixer and the shape of the prepared powder reassembly according to the stirring time in the preparation of the powder reassembled in Example 1-1.
- fine powder refers to particles having a particle size of 150 / year or less in a polymer including an acidic group and polymerizing a water-soluble ethylenically unsaturated monomer in which at least a part of the acidic group is neutralized, and wherein the fine powder is generated or the surface Irrespective of the crosslinking or the like, all of the processes of the super absorbent polymer, for example, the polymerization process, the drying process, the pulverization of the dried polymer, the surface crosslinking process and the like can be included.
- the "fine powder reassembly" may be reassembled by mixing water with respect to fine powder, or may be reassembled by further mixing a mixing stabilizer in addition to water.
- a hydrogel polymer by thermally polymerizing or photopolymerizing a monomer composition comprising a water-soluble ethylenically unsaturated monomer and a polymerization initiator (step 1);
- the hydrogel polymer was dried and pulverized to have fine powder having a particle size of 150_ or less, and more than 150 850 ash! Classifying into normal particles having the following particle diameters (step 2); And
- the water is divided into two or more times with respect to the fine powder.
- polymer or “polymer” means a water soluble ethylenically unsaturated monomer in a polymerized state and may cover all water content ranges, all particle size ranges, all surface crosslinking states, or processing states. Can be.
- a polymer having a water content (water content) of about 40% by weight or more after being dried after polymerization may be referred to as a 'functional gel polymer'.
- a polymer having a particle diameter of 150_ or less among the polymers may be referred to as "fine powder”.
- Superabsorbent polymer also means the polymer itself, depending on the context, or in a state suitable for commercialization by further processing such as surface crosslinking, fine powder reassembly, drying, grinding, classification, etc. for the polymer. It is used to encompass everything.
- step 1 is a step of preparing a hydrous gel polymer.
- the hydrogel polymer may be prepared by thermally polymerizing or photopolymerizing a monomer composition including a water-soluble ethylenically unsaturated monomer and a polymerization initiator.
- the monomer composition which is a raw material of the super absorbent polymer includes a water-soluble ethylenically unsaturated monomer and a polymerization initiator.
- the water-soluble ethylenically unsaturated monomer is usually used for the production of super absorbent polymers. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- Any monomer used may be used without particular limitation. Any one or more monomers selected from the group consisting of anionic monomers and salts thereof, nonionic hydrophilic-containing monomers and amino group-containing unsaturated monomers and quaternized compounds thereof can be used.
- an alkali metal salt such as acrylic acid or a salt thereof, for example acrylic acid or a sodium salt thereof can be used, and the use of such a monomer makes it possible to prepare a super absorbent polymer having better physical properties.
- acrylic acid can be neutralized with a basic compound such as caustic soda).
- the concentration of the water-soluble ethylenically unsaturated monomer may be an appropriate concentration in consideration of the polymerization time, reaction conditions and the like. However, when the concentration of the monomer is too low, the yield of the super absorbent polymer may be low and economic problems may occur. On the contrary, when the activity is excessively high, the grinding efficiency may be low during the pulverization of a hydrogel polymer polymer in which a part of the monomer is precipitated or polymerized. Since there may be a process problem such as appearing and the physical properties of the superabsorbent polymer may be lowered, the monomer composition comprising the raw material and the solvent of the superabsorbent polymer is about 20 to about 60% by weight, preferably about 40 To about 50% by weight.
- the polymerization initiator used in the polymerization in the superabsorbent polymer production method of the present invention is not particularly limited as long as it is generally used for the production of superabsorbent polymers. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the polymerization initiator may use a thermal polymerization initiator or a photopolymerization initiator according to UV irradiation depending on the polymerization method.
- a thermal polymerization initiator may be additionally included.
- the photopolymerization initiator may be used without any limitation as long as it is a compound capable of forming radicals by light such as ultraviolet rays.
- photopolymerization initiator examples include benzoin ether, dialkyl acetophenone, hydroxyl alkylketone, phenyl glyoxylate, and benzyl dimethyl ketal. ketal), acyl phosphine, and one or more selected from the group consisting of alpha -aminoketone.
- acylphosphine a commercially available lucirin TP0, that is, 2, 4, 6-trimethyl-benzoyl-trimethyl phosphine oxide ⁇ -trimethybbenzoyl trimethyl phosphine oxide
- lucirin TP0 a commercially available lucirin TP0, that is, 2, 4, 6-trimethyl-benzoyl-trimethyl phosphine oxide ⁇ -trimethybbenzoyl trimethyl phosphine oxide
- the photopolymerization initiator may be included in a concentration of about 0.01 to about 1.0 wt% based on the monomer composition. When the concentration of the photopolymerization initiator is too low, the polymerization rate may be slow. When the concentration of the photopolymerization initiator is too high, the molecular weight of the superabsorbent polymer may be low and the physical properties may be uneven.
- the thermal polymerization initiator may be used at least one selected from the group consisting of persulfate initiator, azo initiator, hydrogen peroxide and ascorbic acid.
- persulfate-based initiators include sodium persulfate (Na 2 S 2 3 ⁇ 4), potassium persulfate (K 2 S 2 O 8 ), and ammonium persulfate (Ammoni persulfate; (NH 4 ) 2 S 2 ( ) 8 )
- examples of azo initiators include 2, 2-azobis- (2-amidinopropane) dihydrochloride, 2,2-azobis- (N, N_dimethylene) isobutyramidine dihydrochloride (2,2-azobis_ (N, N- 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the thermal polymerization initiator may be included in a concentration of about 0.001 to about 0.5% by weight based on the monomer composition.
- concentration of the thermal polymerization initiator is too low, additional thermal polymerization hardly occurs, and thus the effect of the addition of the thermal polymerization initiator may be insignificant.
- concentration of the thermal polymerization initiator is too high, the molecular weight of the superabsorbent polymer becomes small and the physical properties become uneven. Can be.
- the monomer composition may further include an internal crosslinking agent as a raw material of the super absorbent polymer.
- the internal crosslinking agent include a crosslinking agent having one or more ethylenically unsaturated groups while having at least one functional group capable of reacting with the water-soluble substituent of the water-soluble ethylenically unsaturated monomer; Or the crosslinking agent which has 2 or more functional groups which can react with the water-soluble substituent of the said monomer, and / or the water-soluble substituent formed by hydrolysis of the monomer can be used.
- the internal crosslinking agent examples include bisacrylamide having 8 to 12 carbon atoms, bismethacrylamide, poly (meth) acrylate of polyol having 2 to 10 carbon atoms, poly (meth) allyl ether of polyol having 2 to 10 carbon atoms, and the like.
- One or more selected from the group consisting of triacrylate, trimethol triacrylate, triallylamine, triarylcyanurate, triallyl isocyanate, polyethylene glycol, diethylene glycol and propylene glycol can be used.
- Such an internal crosslinking agent may be included in a concentration of about 0.01 wt% to about 0.5 wt% based on the monomer composition to crosslink the polymerized polymer. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the monomer composition may further include additives such as thickeners, plasticizers, preservative stabilizers, antioxidants and the like as necessary.
- Raw materials such as the above-mentioned water-soluble ethylenically unsaturated monomers, photopolymerization initiators, thermal polymerization initiators, internal crosslinking agents and additives may be prepared in the form of a monomer composition solution dissolved in a solvent.
- the solvent that can be used at this time can be used without limitation of the composition as long as it can dissolve the above-mentioned components, for example, water, ethanol, ethylene glycol, diethylene glycol, triethylene glycol, 1,4-butanediol, Propylene glycol, ethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate, methyl ethyl ketone, acetone, methyl amyl ketone, cyclonucleanone, cyclopentanone, diethylene glycol monomethyl ether, diethylene glycol
- One or more selected from ethyl ether, toluene, xylene, butyrolactone, carbitol, methyl cellosolve acetate and N, N-dimethylacetamide can be used in combination.
- the solvent may be included in the remaining amount except for the above-described components with respect to the total content of the monomer composition.
- the method of forming a hydrogel polymer by thermally polymerizing or photopolymerizing such a monomer composition is not particularly limited as long as it is a commonly used polymerization method.
- the polymerization method is largely divided into thermal polymerization and photopolymerization according to the polymerization energy source, and when the thermal polymerization is usually carried out, it can be carried out in a reactor having a stirring shaft such as kneader, and when the polymerization is carried out, Although it can proceed in a reactor with a conveyor belt, the above-described polymerization method is an example, the present invention is not limited to the above-described polymerization method.
- the hydrogel polymer obtained by supplying hot air to the reactor such as a kneader having a stirring shaft or by heating the reactor as described above may be a reactor outlet according to the shape of the stirring shaft provided in the reactor.
- the hydrogel polymer discharged may be in the form of several centimeters to several millimeters.
- the size of the hydrogel polymer obtained is the size of the monomer composition to be injected. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- hydrogel polymer having a weight average particle diameter of about 2 mm to about 50 mm may be obtained.
- the form of the hydrogel polymer generally obtained may be a hydrogel gel polymer on a sheet having a width of the belt.
- the thickness of the polymer sheet depends on the concentration and the injection rate of the monomer composition to be injected, but it is preferable to supply the monomer composition so that a polymer on a sheet having a thickness of about 0.5 cm to about 5 cm can be obtained.
- the monomer composition When the monomer composition is supplied to such an extent that the thickness of the polymer on the sheet is less than 0.5 cm, the production efficiency is low, which is not preferable, and when the thickness of the polymer on the sheet exceeds 5 cm, due to the excessively thick thickness, the polymerization reaction is full thickness. It may not happen evenly over.
- the normal water content of the hydrogel polymer obtained by the above method may be 40 to 80% by weight.
- water content refers to the value of the moisture content of the total hydrogel polymer weight minus the weight of the polymer in the dry state. Specifically, it is defined as a value calculated by measuring the weight loss due to moisture evaporation in the polymer in the process of raising the temperature of the polymer through infrared heating and drying. At this time, the drying condition is to increase the temperature to 180 ° C at room temperature and then maintained at 180 ° C. The total drying time is set to 20 minutes, including 5 minutes of the temperature rise step, the moisture content is measured.
- the coarsely pulverizing process may optionally be further performed for the hydrogel polymer obtained above.
- the pulverizer used in the coarse grinding process is not limited in configuration, specifically, the vertical cutter (Vert ical pulver i zer), turbo cutter (Turbo cut ter), turbo grinder (Turbo gr inder), rotary cutting Rotary cutter mill, cutter mill, disc scissor, shred crusher, crusher, chopper and disc cutter It may include any one selected from the group of grinding devices consisting of a cutter), but is not limited to the above-described example. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the coarse grinding step may be pulverized so that the particle size of the hydrogel polymer is about 2ä to about 20ä.
- Coarse grinding of the particle diameter below 21 1 is not technically easy due to the high water content of the hydrogel polymer, and may also cause agglomeration between the crushed particles.
- the particle size is coarsely pulverized more than 20ä, the effect of increasing the efficiency of the subsequent drying step may be insignificant.
- step 2 is a step of drying and pulverizing the hydrous gel polymer prepared in step 1 and classifying the fine powder into normal particles.
- the drying process is carried out on the hydrogel polymer immediately after polymerization in step 1 or without polymerization.
- the drying temperature of the drying step may be about 150 to about 250 ° (: When the drying temperature is less than 150 °C, there is a fear that the drying time is too long and the physical properties of the final superabsorbent polymer is finally formed, When the drying temperature exceeds 250 ° ( :), only the surface of the polymer is dried excessively, fine powder may occur in a subsequent grinding process, and there is a possibility that the physical properties of the final superabsorbent polymer are lowered.
- drying time in consideration of the process efficiency, etc., it may proceed for about 20 minutes to about 90 minutes, but is not limited thereto.
- the drying method of the drying step is also commonly used as a drying step of the hydrogel polymer, it can be selected and used without limitation of the configuration. Specifically, the drying step may be performed by hot air supply, infrared irradiation, microwave irradiation, or ultraviolet irradiation.
- the water content of the polymer after such a drying step may be about 0.01% to about 10% by weight.
- the polymer powder obtained after the grinding step may have a particle diameter of about 150_ to about 850 // m.
- the grinder used to grind to such a particle size specifically includes a pin mill () 1 1 1 1), a hammer mill (113_la 1111 1 1), a screw mill (root 1) ⁇ 1 1), 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the polymer powder obtained after grinding is generally classified according to the particle size.
- the particles are classified into particles having a particle size of 150 / mi or less, and particles having a diameter of more than 150 and 850_ or less.
- fine particles having a particle size of less than or equal to a specific particle size, specifically 150 or less are referred to as superabsorbent polymer fine powder, SAP fine powder or fine powder (f ines, f ine powder), and have a particle diameter of more than 150 and less than 850_. Is referred to as normal particle.
- SAP fine powder or fine powder f ines, f ine powder
- the fine powder may be generated during the polymerization process, the drying process, or during the pulverization of the dried polymer. When the fine product is included in the final product, the fine powder is difficult to handle and exhibits a gel blocking phenomenon. It is desirable to exclude them from inclusion or to reuse them to be normal particles.
- the fine powder may be reassembled to agglomerate to a normal particle size.
- a reassembly process of coagulating fine particles in a wet state is performed.
- the higher the moisture content of the fine powder the higher the cohesive strength of the fine powder, but the reassembly lump may be too large during the reassembly process, which may cause problems during the operation of the process. After that, it is often broken into fine powder.
- the fine powder reassembly thus obtained has lower physical properties such as water-retaining capacity (CRC) and pressure-absorbing capacity (AUP) than normal particles, resulting in deterioration of the quality of the super absorbent polymer.
- CRC water-retaining capacity
- AUP pressure-absorbing capacity
- step 3 is a step of preparing a fine powder reassembly by reassembling the fine powder classified in the step 2.
- the fine powder classified in step 2 having a particle size of I50im or less is mixed with water, and the water is divided into two or more times for fine powder and reassembled to prepare a fine powder reassembly.
- the mixing of the fine powder and water is a mixing device that can add shear 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the stirring linear speed when mixing the fine powder and water in the present invention is 6.5 to 14.5111 / 3 3 ⁇ 4 number have.
- the stirring linear speed increases, the second agglomeration formation time of the powder becomes shorter, but the operation stability of the mixing apparatus or the mixer decreases.
- the stirring linear velocity is too slow, less than 6.5111 / 3, the second agglomeration formation time may be relatively long, so that the mixer operation may be stable, but there is a concern that the funnel generation rate of the reassembled body may be increased.
- the stirring linear velocity is excessively faster than 14.5 micro / 3
- the secondary coagulation formation time of the powder becomes excessively fast, and it may have sufficient assembly strength, but a reassembled mass is formed to increase the load on the mixer. Continuous operation is difficult.
- the agitation linear velocity may be 8.5 to 13 ⁇ 4 1/3 days.
- the water may be included in an amount such that the fine powder can be reassembled in a wet state, specifically, may be 80 to 150 parts by weight, preferably 80 to 110 parts by weight with respect to 100 parts by weight of the fine powder. .
- the water is evaporated during the redrying process after forming the fine powder reassembly, wherein if the water content is less than 80 parts by weight, the fine powder and water are dispersed evenly due to the fast absorption rate of fine powder in the mixing process Since it is difficult to reduce the uniformity of the fine powder reassembly, the amount of fun meal can be increased.
- the said water is divided into two or more times with respect to fine powder.
- the mixing stability may be lowered.
- the split-injected water after the second is dispersed and long-lived on the surface, thereby improving the mixing stability.
- the water is twice or three times with respect to the fine powder 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- Partial injection may be performed.
- the mixing step of the fine powder and water in the manufacturing method of the superabsorbent polymer composition according to the present invention is the primary mixing step of mixing the primary with water to the fine powder
- the 1 Secondary mixing may include a secondary mixing step of mixing the remaining water after the secondary mixing.
- the water input amount is not particularly limited for each input time, and may be divided into equal amounts within the above input amount range, or may be added in different amounts for each input time.
- the amount of water added firstly is more than 50% by weight to 90% by weight or less based on the total weight of water added, and the amount of water added secondly is 10 to 50% by weight. It may be less than weight percent.
- the second water is introduced in a smaller amount than the water at the time of the first injection, but when the amount of the water added at the second is 50% by weight or more, the aggregation time is increased and the mixing stability may be lowered. If less than 10% by weight, the improvement effect of the second input is not sufficient.
- the amount of water to be added firstly is 60 to 90% by weight based on the total weight of water added, and the amount of water to be added secondly may be 10 to 40% by weight, more specifically, to be added first.
- Amount of water is 65 to 80% by weight based on the total weight of water added,
- the amount of water added to the secondary may be 20 to 35% by weight, the balance.
- the first input is at the beginning of mixing with the fine powder
- the second and subsequent split inputs are 2 to 1 ⁇ seconds after the previous order of water input, more specifically, 2 to It may be performed at intervals of 5 seconds. If the second or more divided dose interval is less than 2 seconds, it is difficult to obtain a sufficient effect according to the divided dose, and when it is carried out for more than 10 seconds, the interval between inputs is too long, which is inefficient.
- the finely divided reassembly reassembled through the divided input described above is preferably quickly discharged out of the mixer. If the reassembly is secondary agglomerated and agglomerated, the load increases in the mixer, making the continuous process difficult.
- At least one of a water-soluble polymer and a surfactant as a mixing stabilizer for promoting the lubricating action of the fine powder reassembly in the mixing mixer when mixing the fine powder and water to improve the mixing stability and uniformity 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- One or more may optionally be further used.
- water-soluble polymer examples include polyethylene glycol, polyethylene oxide, polyvinylpyrrolidone, polyvinyl alcohol, polyacrylamide, polyacrylic acid, polystyrene sulfonic acid, polysilic acid, polyphosphoric acid, and polyethylene sulfonic acid.
- Synthetic polymers such as polyvinylphenol, polyvinylphenylsulfonic acid, polyethylenephosphoric acid, polyethyleneamide, polyamine or polyamideamine; Or non-synthetic polymers such as starch, rubber, cellulose, and the like, and any one or a mixture of two or more thereof may be used.
- polyethylene glycol may be more preferable because of chemical stability, harmlessness to human body, excellent dispersion / lubrication effect and economic characteristics.
- the polyethylene glycol when polyethylene glycol is used, the polyethylene glycol may have a weight average molecular weight of 2,000 to 200, 000g / mol, more preferably 4,000 to 100, 000g / mol. If the molecular weight of polyethylene glycol is less than 2,000 g / mol, the lubrication action may be lowered, and if it exceeds 200, 000 g / mol, the solubility in water may be lowered.
- the surfactant is specifically sodium dodecyl sulfate (SDS), diisooctyl sodium sulfosuccinate (DSS), sodium tetradecyl sulfate (sodium tetradecyl sulfate), sodium Nucleodecyl sulfate, sodium dodecylbenzene sulfonate, xylenesulfonate, sodium oleate, 4_n_ decylbenzenesulfonate ( 4-11- ( 160 15611761163111 £ 011 6), sodium laurate, 4-dodecylbenzenesul fonic acid, dodecylamine hydrochloride, dodecyltrimethylammoni chloride, 4_n_octylbenzene sulfonate (4-n-octylbenzene sulfonate), ethoxylated sulfonate (Et:
- dodecyl amine tetradecyltrimethylammonhim chloride
- dodecyl polysacchar ide glycoside cyclodextrin
- glycolipid glycol ipids lipoprotein-lipopeptides
- phosphol ipides phosphol ipides
- para-toluene sul foni c acid or trisiloxane
- any one or a mixture of two or more thereof may be used.
- commercially available products such as triton (tr i tonä X-100) may be used.
- sodium dodecyl sulfate may be more preferred due to its chemical stability and good dispersion / lubrication effect.
- the additives described above may be used in an amount of 0.001 to 1 part by weight, more preferably 0.001 to 0.5 part by weight, based on 100 parts by weight of fine powder. If the content of the additive is less than 0.001 parts by weight, it is difficult to obtain the effect of improving the mixing stability and uniformity of the fine powder reassembly according to the use of the additive, and if the content of the additive is more than 1 part by weight, the performance of the superabsorbent polymer may be degraded and the discoloration may be feared. There is.
- the fine powder reassembly prepared in step 3 mixed with the hydrogel polymer prepared in step 1, dried and pulverized, not more than 150 // m Reassembling fine powder having a particle size (hereinafter referred to as 'regrinding'), and classifying the reassembly normal particles having a particle size of greater than 150_ and not more than 850 n (step 4).
- step 4 is a step of mixing the fine powder reassembly prepared in step 3 with the hydrogel polymer prepared in step 1, drying and pulverizing, and classifying the reassembled fine powder and the reassembled normal particles.
- the drying process may be performed using a conventional drying apparatus, but according to one embodiment of the invention may be performed using a paddle type dryer or forced circulation dryer.
- the drying process is carried out in these dryers, the secondary granulated finely divided granules can be more easily primary granulated by the force generated during the flow, and as a result, the drying rate and the drying efficiency can be increased.
- the drying process may be performed at a temperature of about 12CTC to about 220 ° C. If the temperature during the drying process is less than 120 ° C, the drying time will be longer, 220 ° C 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the fine powder reassembly is preferably carried out so that the moisture content in the fine powder reassembly is 1% by weight or less.
- the initial temperature of the drying process specifically, the temperature of the dryer inlet is about 120 ° C to about 160 ° C
- the late drying process specifically, the temperature of the outlet of the rear end of the dryer is about 15 CTC to about 200 ° C to improve the drying efficiency. It can raise, and it is more preferable.
- the heating medium may be supplied or directly heated by means such as electricity, but the present invention is not limited to the above-described example. Specific heat sources that may be used include steam, electricity, ultraviolet rays, infrared rays, and the like, and a heated thermal fluid may be used.
- the drying process may be performed at the same time as the cutting process using the Spur. In this case, the drying efficiency can be increased and the drying time can be shortened.
- the drying process may be performed so that the moisture content in the dried fine powder reassembly is 1% by weight or more, more specifically 1 to 2% by weight.
- the moisture content in the dried fine powder reassembly is dried to less than 1% by weight, there is a fear of deterioration of physical properties of the fine powder reassembly.
- the secondary granulated fine powder reassembly is first granulated in the form of a single particle.
- the dried fine powder reassembly is pulverized and classified.
- the pulverization may be performed such that the dried fine powder reassembly has a particle diameter of more than 150 m and 850 / M or less.
- the grinder used to grind to such a particle size specifically includes a pin mill, a hammer mill, a screw mill, a roll mill, a disk mill. (di sc mi ll) or jog mill (j og mi ll) and the like can be used, but the present invention is not limited to the above examples.
- the polymer powder obtained after grinding is generally classified according to the particle size.
- fun powder having a particle size of less than 150 / / m, and 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the fine powder reassembly obtained through the process has a high cohesive strength due to a low rate of recrushing into fine powder after the grinding step.
- the fine powder reassembly may have a weight ratio of fine powder having a particle diameter of 150 / ® or less after grinding, about 50% by weight or less, and preferably about 30% by weight or less, based on the total weight of the fine powder reassembly. .
- step (step 5) to the surface cross-linking by introducing the re-assembly normal particles classified in step 4 into the surface cross-linking mixer and mixed with the surface cross-linking agent.
- the surface crosslinking is a step of increasing the crosslink density near the surface of the superabsorbent polymer particles with respect to the crosslink density inside the particles.
- the surface crosslinking agent is applied to the surface of the superabsorbent resin particles.
- this reaction takes place on the surface of the superabsorbent resin particles and improves the crosslinkability on the surface of the particles without substantially affecting the inside of the known particles.
- the surface crosslinked superabsorbent resin particles thus have a higher degree of crosslinking in the vicinity of the surface than in the interior.
- the surface crosslinking agent is not limited as long as it is a compound capable of reacting with the functional group of the polymer.
- the surface crosslinking agent may be a polyhydric alcohol compound; Epoxy compounds; Polyamine compounds; Haloepoxy compound; bamboo products of haloepoxy compounds; Oxazoline compounds; Mono-, di- or polyoxazolidinone compounds; Cyclic urea compounds; Polyvalent metal salts; And an alkylene carbonate compound may be used one or more selected from the group consisting of.
- examples of the polyhydric alcohol compound include mono-, di-, tri-, tetra_ or polyethylene glycol, monopropylene glycol, 1, 3-propanediol, dipropylene glycol, 2, 3, 4-trimethyl-1, 3 -Pentanediol, polypropylene glycol, glycerol, polyglycerol, 2-butene-1, 4-diol, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol, 1,6-nucleic acid diol, and 1 type selected from the group consisting of 1,2-cyclonucleodimethanol 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- Ethylene glycol diglycidyl ether and glycidol or poly (ethylene glycol) diglycidyl ether may be used as the epoxy compound, and as polyamine compounds, ethylene diamine, diethylene triamine, triethylene tetra
- polyamine compounds ethylene diamine, diethylene triamine, triethylene tetra
- amines, tetraethylenepentamine, pentaethylenenucleomine, polyethyleneimine and polyamide polyamine can be used.
- haloepoxy compound epichlorohydrin, epibromohydrin and methyl epichlorohydrin can be used.
- mono-, di- or polyoxazolidinone compound for example, 2-oxazolidinone can be used.
- an alkylene carbonate compound ethylene carbonate etc. can be used. These may be used alone or in combination with each other.
- it is preferable to use including at least 1 type of polyhydric alcohol compound among these surface crosslinking agents More preferably, C2-C10 polyhydric alcohol compounds can be used.
- the amount of the surface crosslinking agent to be added may be appropriately selected depending on the kind of the surface crosslinking agent to be added or the reaction conditions, but is generally about 0.001 to about 5 parts by weight, preferably about 0.01 to about 100 parts by weight of the polymer. 3 parts by weight, more preferably about 0.05 to about 2 parts by weight can be used.
- the surface crosslinking reaction When the content of the surface crosslinking agent is too small, the surface crosslinking reaction hardly occurs, and when 100 parts by weight of the polymer is more than 5 parts by weight, the excessive absorption of the surface crosslinking reaction may cause a phenomenon of deterioration of absorption capacity and physical properties. .
- the surface crosslinking reaction may be performed at the time of dry crosslinking reaction.
- the temperature raising means for surface crosslinking reaction is not specifically limited. It can be heated by supplying a heat medium or by directly supplying a heat source.
- the kind of heat medium that can be used may be a mild fluid such as steam, hot air, or hot oil, but the present invention is not limited thereto, and the temperature of the heat medium to be supplied is a means of heating medium, a temperature rising rate, and a temperature increase.
- the heat source directly supplied may be a heating method through electricity, a gas heating method, the present invention is not limited to the above-described example.
- the surface crosslinking fine powder having a particle size of 150 / cm or less, and the surface crosslinking fine powder having a particle size of more than 150_ 850 / L or less, and the surface crosslinking fine powder having a particle size of 150 // m or less Silver is reintroduced into a process for fine powder reassembly, and the surface crosslinked normal particles can be commercialized and used.
- the superabsorbent polymer prepared by the above-described method is a fine powder which is reassembled by mixing water with respect to fine powder having a particle size of 150 or less in a polymer containing an acidic group and polymerizing a water-soluble ethylenically unsaturated monomer in which at least a part of the acidic group is neutralized.
- Superabsorbent polymer with surface crosslinked reassembly, £ 0 ⁇ method The water holding capacity (03 ⁇ 4 :) measured according to 241.3 is 33.0 to 39.0 0.3 measured according to law Pressurized absorption capacity) is for 20.0 to 33.0 //; Vortex method (absorption rate by 0 6 may be 100 seconds or less.
- the polymer comprises an acidic group and polymerized a water-soluble ethylenically unsaturated monomer in which at least a part of the acidic group is neutralized. Same as described.
- Acrylic acid 100 Polyethyleneglycol diacrylate as crosslinking agent 0.3 Sodium persulfate as polymerization initiator 0.033 Melting, caustic soda 38.9 And a mixture of 103.9 sugars in water to prepare a monomer mixture having a monomer concentration of 50% by weight. Thereafter, the monomer mixture was placed on a continuously moving conveyor belt, and irradiated with ultraviolet rays (irradiation amount: 21 / ( 2 )) to proceed polymerization for 2 minutes to obtain a hydrous gel polymer.
- ultraviolet rays irradiation amount: 21 / ( 2 )
- the hydrogel polymer was pulverized by using a meat chopper (hole size 10 ⁇ ) 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- a hydrous gel polymer was obtained. 1701: dried for 1 hour in a hot air dryer at a temperature, and ground with a pin mill grinder It is classified into standard mesh of the standard and the normal particle which has the particle size of more than 150_ and less than 850 / L and 150 ⁇ ! Fine particles having the following particle diameters were obtained.
- Example 1-2 After the stirring, the resultant fine powder reassembly was put into a forced circulation dryer, and then dried at 1801: temperature for 60 minutes to recover the fine powder reassembly.
- Example 1-2
- Example 1-1 the fine powder reassembled was recovered in the same manner as in Example 1-1, except that the linear speed was 12.8111 / 3 when the powder was stirred. Secondary aggregation formation time was 7 seconds. Comparative Example 1-1
- Example 1-1 the fine powder reassembly was recovered in the same manner as in Example 1-1, except that the linear velocity at the time of differential stirring was 4.25111 / 3. Secondary aggregation formation time was 58 seconds. Comparative Example 1-2
- Example 1-1 the fine powder reassembly was recovered in the same manner as in Example 1-1, except that the linear speed of the stirring of the fine powder was 16.61 ⁇ / 3. Secondary aggregation formation time was 5 seconds. 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- Example 2-2 The resulting fine powder reassembly was put in a forced circulation dryer, and dried at 180 ° C. for 60 minutes to recover the fine powder reassembly.
- Example 2-2 The resulting fine powder reassembly was put in a forced circulation dryer, and dried at 180 ° C. for 60 minutes to recover the fine powder reassembly.
- Example 2-1 the fine powder reassembled was recovered in the same manner as in Example 2-1, except that the linear velocity was 12.801 / 3 when the powder was stirred. Secondary aggregation formation time was 10 seconds.
- Example 2-3 the fine powder reassembled was recovered in the same manner as in Example 2-1, except that the linear velocity was 12.801 / 3 when the powder was stirred. Secondary aggregation formation time was 10 seconds.
- Example 2-1 the fine powder was prepared in the same manner as in Example 2-1, except that 31.9 was used as the first input of water and 74.4 ⁇ was added secondly after 2 seconds. The reassembly was recovered. Secondary aggregation formation time was 18 seconds.
- Example 2-4 the fine powder was prepared in the same manner as in Example 2-1, except that 31.9 was used as the first input of water and 74.4 ⁇ was added secondly after 2 seconds. The reassembly was recovered. Secondary aggregation formation time was 18 seconds.
- Example 2-4 the fine powder was prepared in the same manner as in Example 2-1, except that 31.9 was used as the first input of water and 74.4 ⁇ was added secondly after 2 seconds. The reassembly was recovered. Secondary aggregation formation time was 18 seconds.
- Example 2-4 the fine powder was prepared in the same manner as in Example 2-1, except that 31.9 was used as the first input of water and 74.4 ⁇ was added secondly after 2 seconds. The reassembly was recovered. Secondary aggregat
- Example 2-1 35. was firstly added when the water was added, 35 was secondly added after 2 seconds, and 35.4 ⁇ was added secondly after 2 seconds.
- the fine powder reassembly was recovered in the same manner as in. Secondary aggregation formation time was 19 seconds.
- Example 1-1 the fine powder reassembly was carried out in the same manner as in Example 1-1, except that 56 dragons were firstly added when water was added, and 54 dragons were secondly added after 2 seconds. Was recovered. Secondary aggregation formation time was 13 seconds. Comparative Example 2-1
- Example 2-1 the fine powder reassembly was recovered in the same manner as in Example 2-1, except that the linear velocity was 4.23 ⁇ 4 / 3 when the powder was stirred. Secondary aggregation formation time was 85 seconds. Comparative Example 2-2
- Example 2-1 the fine powder reassembly was recovered in the same manner as in Example 2-1, except that the linear speed of the fine powder stirring was 16.6111 / 3. Secondary aggregation formation time was 8 seconds. . Comparative Example 2-3
- Example 2-1 except that the water in a batch, the fine powder reassembly was recovered in the same manner as in Example 2-1. Secondary aggregation formation time was 11 seconds. Comparative Example 3-1
- the fine powder 100 silver having a particle size of 150 / ⁇ 1 or less prepared in Additive Manufacturing Example 1 was placed in a mixer, and 111 ⁇ 2 of water was added in a batch while stirring at a linear speed of 4.23 ⁇ 4 / 3 . At this time, the time from the completion of the water input until the second agglomeration occurred was measured, and the mixing of the fine powder re-agglomeration was stopped immediately after the mixer load was increased, the mixer agitation was stopped. Secondary aggregation formation time was 28 seconds.
- Comparative Example 3-1 the fine powder reassembly was recovered in the same manner as in Comparative Example 3-1 except that the linear velocity during the stirring of the powder was 8. k8. Secondary aggregation formation time was 9 seconds. Comparative Example 3-3
- Comparative Example 3-1 the fine powder reassembly was recovered in the same manner as in Comparative Example 3-1, except that the linear velocity during the stirring of the powder was set to 12. 3/4. Secondary aggregation formation time was 4 seconds.
- the fine powder reassembly prepared in Example 1-1 was mixed with a hydrous gel polymer prepared in the fine powder preparation process of Preparation Example 1 in a weight ratio of 20:80, then chopped and dried, followed by 1 01 1 1 1 Crushed and classified into a standard mesh of Shoshone standard, and classified into fine particles of reassembly of 150_ or less and normal particles of reassembly having a particle diameter of more than 150 / L and 850_ or less.
- Classified reassembly normal particles 100 ⁇ poly (ethylene glycol) diglycidyl ether 0.2, methanol 5 ⁇ water 4 ⁇ silica aero 1105) After mixing with a surface crosslinking solution containing 0.03 ⁇ 4,
- Example 3-1 using the fine powder reassembly prepared in Example 1-2, and Example 2-1 to Example 2-5 instead of the fine powder reassembly prepared in Example 1-1 Except for the high-top water resin was prepared in the same manner as in Example 3-1.
- Example 3-1 instead of the fine powder reassembly prepared in Example 1-1 prepared in Comparative Examples 1-2, Comparative Examples 2-1 to 2-3, and Comparative Examples 3-1 to 3-3 A super absorbent polymer was prepared in the same manner as in Example 3-1, except that the fine powder reassembles were used.
- Experimental Example 1 A super absorbent polymer was prepared in the same manner as in Example 3-1, except that the fine powder reassembles were used.
- Examples 2-1 to 2-2 and Comparative Examples 2-1 to 2-2 are Examples 1-1 to 1-2 and Comparative Examples 1-1 to 2 under the same stirring linear velocity and water input conditions. Secondary aggregation formation time was slow compared to -1, respectively. This is because the mixing stability is increased by adding a mixing stabilizer when water is added to the mixed fine powder.
- Comparative Examples 1-1 and Examples 1-1 to 1-2 in which water was added in portions were compared with Comparative Examples 3-1 to 3-3, in which water was collectively added under conditions of the same stirring linear velocity. Secondary aggregation time slowed down. This is due to the increased mixing stability due to the dispersion of water and the presence of water on the surface for a long time.
- Example 1-1 In the same manner as in Example 1-1 to prepare a fine powder reassembly, the stirring time was changed to 5s, 10s and 20s, the stirring time was determined by observing the change in physical properties of the prepared fine powder assembly The effect on the assembly was confirmed.
- 0.2 g of a sample of fine powder reassembly having a particle diameter of more than 300 m and a particle size of 600 pm or less in the prepared fine powder reassembly is placed in a tea bag and precipitated in a 0.9% saline solution for 30 minutes. After dehydration for 3 minutes using a centrifugal force of 250G (gravi ty) was measured the amount W 2 (g) absorbed saline solution. In addition, the mass Kg at that time was measured after the same operation without using a powder reassembly.
- W 0 (g) is the initial weight (g) of the differential reassembly
- Wjg is the device weight measured after dehydration at 250G for 3 minutes using a centrifuge, without using a fine powder reassembly
- 3 ⁇ 4 (g) is absorbed by submerging the fine powder reassembly in a 0.9 wt% physiological saline at room temperature for 30 minutes and then dehydrated at 250G for 3 minutes using a centrifuge, 2019/194399 1 »(: 1 ⁇ 1 ⁇ 2019/001109
- the weight of the device including the powder assembly.
- BPKBase Polymer Index For the prepared powder reassembly, the sample having a particle diameter of more than 300 m and a particle size of 600 / L or less was used as the centrifugal support capacity (CRC, unit: g / g) by EDANA Test 441.2-02.
- CRC centrifugal support capacity
- AUP pressure-absorbing capacity
- the secondary flocculation formation time was 13 seconds or more, indicating the mixing stability.
- Example 2-4 when divided into three times (Example 2-4), the mixing stability was reduced compared to Example 2-1, but improved mixing stability compared to Comparative Example 3-2 in a batch Indicated.
- Diameter 90ä and thickness on the inside of the petri dish of diameter 150 _ A glass filter was placed so that physiological saline consisting of 0.9% by weight sodium chloride was at the same level as the top of the glass filter.
- One sheet of filter paper having a diameter of 90ä was loaded thereon. The measuring device was placed on the filter paper and the liquid was absorbed for 1 hour under load. After an hour, immediately lift the device, and its weight? 4 (Yo) was instantaneous.
- ⁇ 3 ⁇ 4 of the super absorbent polymer of Example 3-1 was 34.2 for / ⁇ , and 0.3? ⁇ was 28. 3 ⁇ 4 / for, and the absorption rate was 35 seconds.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
본 발명은 고흡수성 수지의 제조방법에 관한 것으로, 보다 자세하게는 고흡수성 수지의 제조시 미분 재조립체의 제조 단계에서 교반 속도를 조절하고, 또 미분에 대해 물을 분할 투입함으로써, 미분 조립체의 조립 강도를 유지하면서 안정적인 미분 재조립체 제조 공정의 운전이 가능하고, 재미분율을 감소시킬 수 있으며, 초기 흡수 속도가 빠른 고흡수성 수지를 제조할 수 있다.
Description
2019/194399 1»(:1^1{2019/001109
【발명의 명칭】
고흡수성 수지의 제조방법
【기술분야】
관련 출원 (들)과의 상호 인용
본 출원은 2018년 4월 3일자 한국 특허 출원 제 10-2018-0038632호에 기초한 우선권의 이익을주장하며, 해당 한국 특하출원의 문헌에 개시된 모든 내용은 본 명세서의 일부로서 포함된다.
본 발명은 고흡수성 수지의 제조방법에 관한 것으로, 보다 자세하게는 고흡수성 수지의 제조시 미분 재조립체의 제조 단계에서 교반 속도를 조절하고, 또 미분에 대해 물을 분할 투입함으로써, 미분 조립체의 조립 강도를 유지하면서 안정적인 미분 재조립체 제조 공정의 운전이 가능하고, 재미분율을 감소시킬 수 있으며, 초기 흡수 속도가 빠른 고흡수성 수지를 제조할 수 있는 제조방법에 관한 것이다.
【배경기술】
고흡수성 수지 (Super Absorbent Polymer , SAP)란 자체 무게의 5백 내지
1천 배 정도의 수분을 흡수할 수 있는 기능을 가진 합성 고분자 물질로, 생리용구로 실용화되기 시작해서, 현재는 어린이용 종이 기저귀 등 위생용품 외에 원예용 토양보수제, 토목, 건축용 지수재, 육묘용 시트, 식품 유통분야에서의 신선도 유지제, 및 찜질용등의 재료로 널리 사용되고 있다. 이러한 고흡수성 수지의 흡수 메카니즘은 고분자 전해질의 전하가 나타내는 전기적 흡인력의 차이에 의한 침투압, 물과 고분자 전해질 사이의 친화력, 고분자 전해질 이온 사이의 반발력에 의한 분자 팽창 및 가교 결합으로 인한 팽창 억제의 상호 작용에 의하여 지배된다. 즉, 흡수성 수지의 흡수성은 전술한 친화력과 분자 팽창에 의존하며, 흡수 속도는 흡수성 고분자 자체의 침투압에 크게 좌우되는 것이다.
통상 고흡수성 수지의 제조 공정 중 분쇄 및 이송 과정에서 약 20 내지 30%의 미분이 발생한다. 이러한 미분은 고흡수성 수지의 가압흡수능 (AUP)과 투수성 (Permeability)에 영향을 미치기 때문에, 두 물성을 동시에 높인 고흡수성 수지를 제조하는데 어려움이 있다.
또, 미분은 미분과 물을 혼합하고, 응집시키는 미분 재조립 공정을
\¥0 2019/194399 1»(:1'/¾3조2019/001109
통해 큰 입자가 된다. 그러나, 상기 미분과 물이 혼합되어 재조립되는 과정에서 과도한 응집이 발생할 경우, 장치 부하가 증가하게 되고, 이로 인해 연속운전이 어렵다. .
따라서, 고흡수성 수지의 연속 생산을 위해서는, 안정적인 미분 재조립 공정의 운전이 필요하다. 특히 초기 흡수속도가 빠른 흡수성 수지의 경우 안정적인 미분 재조립 운전이 필수적이다.
【발명의 상세한설명】
【기술적 과제】
본 발명은 상기와 같은 종래 기술의 문제점을 해결하기 위한 것으로, 고흡수성 수지의 제조시 미분 재조립체의 제조 단계에서 교반 속도를 조절하고, 또 미분에 대해 물을 분할 투입함으로써, 미분 조립체의 조립 강도를 유지하면서 안정적인 미분 재조립체 제조 공정의 운전이 가능하고, 재미분율을 감소시킬 수 있으며 , 초기 흡수 속도가 빠른 고흡수성 수지를 제조할 수 있는 방법을 제공하기 위한 것이다.
【기술적 해결방법】
상기 목적을 달성하기 위하여 본 발명은,
수용성 에틸렌계 불포화 단량체 및 중합개시제를 포함하는 모노머 조성물에 열중합또는 광중합을 진행하여 함수겔상중합체를 제조하는 단계; 상기 함수겔상 중합체를 건조 및 분쇄하고 150/ 이하의 입경을 갖는 미분, 및 150 초과 850 III 이하의 입경을 갖는 정상 입자로 분급하는 단계; 미
상기 미분을 선속도 6.5 내지 14. ¾1/3로 교반하면서 물과 혼합하고, 재조립하여 미분 재조립체를 제조하는 단계;를 포함하며 ,
상기 미분과 물의 혼합시, 상기 물을 미분에 대해 2회 이상 분할 투입하는, 고흡수성 수지의 제조방법을 제공한다.
【발명의 효과】
본 발명에 따른 고흡수성 수지의 제조방법은 미분 조립체의 조립 강도를 유지하면서 안정적인 미분 재조립체 제조 공정의 운전이 가능하며, 재미분율을 감소시킬 수 있다.
또, 상기 제조방법에 의해 초기 흡수 속도가 빠른 고흡수성 수지를
2019/194399 1»(:1^1{2019/001109
제조할수 있다.
【도면의 간단한설명】
도 1은 교반 선속도에 따른 2차 응집 형성 시간을 비교한 결과를 나타낸 그래프이다.
도 2는 실시예 1-1에서 미분 재조립체의 제조시 교반시간에 따른 믹서 내부상태 및 제조되는 미분 재조립체 형상을 관찰한사진이다.
【발명을 실시하기 위한구체적인 내용】
본 명세서에서 사용되는 용어는 단지 예시적인 실시예들을 설명하기 위해 사용된 것으로, 본 발명을 한정하려는 의도는 아니다. 단수의 표현은 문맥상 명백하게 다르게 뜻하지 않는 한, 복수의 표현을 포함한다. 본 명세서에서, "포함하다" , ’’구비하다’’ 또는 "가지다’’ 등의 용어는 실시된 특징, 단계, 구성 요소 또는 이들을 조합한 것이 존재함을 지정하려는 것이지, 하나 또는 그 이상의 다른 특징들이나 단계, 구성 요소, 또는 이들을 조합한 것들의 존재 또는부가가능성을 미리 배제하지 않는 것으로 이해되어야 한다.
본 발명은 다양한 변경을 가할 수 있고 여러 가지 형태를 가질 수 있는 바, 특정 실시예들을 예시하고 하기에서 상세하게 설명하고자 한다. 그러나, 이는 본 발명을 특정한 개시 형태에 대해 한정하려는 것이 아니며, 본 발명의 사상 및 기술 범위에 포함되는 모든 변경, 균등물 내지 대체물을 포함하는 것으로 이해되어야 한다.
본 발명에 있어서 ”미분"은 산성기를 포함하고 상기 산성기의 적어도 일부가 중화된 수용성 에틸렌계 불포화 단량체를 중합시킨 중합체 중 입경이 150/해 이하인 입자를 지칭하며, 상기 미분이 발생되는 단계 또는 표면 가교 여부 등에 상관없이, 고흡수성 수지의 모든 공정, 예를 들어, 중합 공정, 건조 공정, 건조된 중합체의 분쇄 공정, 또는 표면 가교 공정 등에서 발생한 것을 모두 포괄할수 있다.
또, 본 발명에 있어서, "미분 재조립체”는 미분에 대해, 물을 혼합하여 재조립되거나, 또는 물에 더하여 혼합안정화제를 더 혼합하여 재조립된 것일 수 있다.
이하, 발명의 구체적인 구현예에 따라 고흡수성 수지의 제조방법에 대해 보다상세히 설명하기로 한다.
2019/194399 1»(:1^1{2019/001109
본 발명의 일 구현예에 따른 고흡수성 수지의 제조방법은,
수용성 에틸렌계 불포화 단량체 및 중합개시제를 포함하는 모노머 조성물에 열중합 또는 광중합을 진행하여 함수겔상 중합체를 제조하는 단계(단계 1);
상기 함수겔상 중합체를 건조 및 분쇄하여 150_ 이하의 입경을 갖는 미분, 및 150 초과 850쌔! 이하의 입경을 갖는 정상 입자로 분급하는 단계(단계 2); 및
상기 미분을 선속도 6.5 내지 14.5111/3로 교반하면서 물과 혼합하고, 재조립하여 미분 재조립체를 제조하는 단계(단계 3);를 포함하며 ,
상기 미분과 물의 혼합시, 상기 물을 미분에 대해 2회 이상 분할 투입한다.
참고로, 본 발명의 명세서에서 "중합체", 또는 "고분자"는 수용성 에틸렌계 불포화 단량체가중합된 상태인 것을 의미하며, 모든 수분 함량 범위, 모든 입경 범위, 모든 표면 가교 상태 또는 가공 상태를 포괄할 수 있다. 상기 중합체 중, 중합 후 건조 전 상태의 것으로 함수율(수분 함량)이 약 40 중량% 이상의 중합체를 ’’함수겔상 중합체’’로 지칭할 수 있다. 또한, 상기 중합체 중 입경이 150_ 이하인 중합체를 "미분”으로 지칭할수 있다.
또한 "고흡수성 수지’’는 문맥에 따라 상기 중합체 자체를 의미하거나, 또는 상기 중합체에 대해 추가의 공정, 예를 들어 표면 가교, 미분 재조립, 건조, 분쇄, 분급 등을 거쳐 제품화에 적합한 상태로 한 것을 모두 포괄하는 것으로사용된다.
이하고흡수성 수지의 제조 단계 별로 상세히 설명한다.
본 발명의 고흡수성 수지의 제조방법에 있어서 단계 1은 함수겔상 중합체를 제조하는 단계이다.
상기 함수겔상 중합체는 수용성 에틸렌계 불포화 단량체 및 중합개시제를 포함하는 모노머 조성물에 열중합 또는 광중합을 진행함으로써 제조될 수 있다.
상기 고흡수성 수지의 원료 물질인 모노머 조성물은 수용성 에틸렌계 불포화 단량체 및 중합 개시제를 포함한다.
상기 수용성 에틸렌계 불포화 단량체는 고흡수성 수지의 제조에 통상
2019/194399 1»(:1^1{2019/001109
사용되는 임의의 단량체를 별다른 제한없이 사용할 수 있다. 여기에는 음이온성 단량체와 그 염, 비이온계 친수성 함유 단량체 및 아미노기 함유 불포화 단량체 및 그의 4급화물로 이루어진 군에서 선택되는 어느 하나 이상의 단량체를사용할수 있다.
구체적으로는 (메타)아크릴산, 무수말레인산, 푸말산, 크로톤산, 이타콘산, 2 -아크릴로일에탄 술폰산, 2 -메타아크릴로일에탄술폰산, 2 - (메타)아크릴로일프로판술폰산 또는 2-(메타)아크릴아미드- 2 -메틸 프로판 술폰산의 음이온성 단량체와 그 염; (메타)아크릴아미드, 치환(메타)아크릴레이트, 2 -히드록시에틸(메타)아크릴레이트, 2- 히드록시프로필(메타)아크릴레이트,
메톡시폴리에틸렌글리콜(메타)아크릴레이트 또는 폴리에틸렌 글리콜(메타)아크릴레이트의 비이온계 친수성 함유 단량체; 및 어 )_ 디메틸아미노에틸(메타) 아크릴레이트 또는 (比 - 디메틸아미노프로필(메타)아크릴아미드의 아미노기 함유 불포화 단량체 및 그의 4급화물로 이루어진 군에서 선택된 어느 하나 이상을사용할수 있다. 더욱 바람직하게는 아크릴산 또는 그 염, 예를 들어, 아크릴산 또는 그 나트륨염 등의 알칼리 금속염을 사용할 수 있는데, 이러한 단량체를 사용하여 보다 우수한 물성을 갖는 고흡수성 수지의 제조가 가능해 진다. 상기 아크릴산의 알칼리 금속염을 단량체로 사용하는 경우, 아크릴산을 가성소다어크細)와 같은 염기성 화합물로 중화시켜 사용할수 있다.
상기 수용성 에틸렌계 불포화 단량체의 농도는, 중합 시간 및 반응 조건 등을 고려해 적절한 농도로 될 수 있다. 다만, 상기 단량체의 농도가 지나치게 낮아지면 고흡수성 수지의 수율이 낮고 경제성에 문제가 생길 수 있고, 반대로 능도가 지나치게 높아지면 단량체의 일부가 석출되거나 중합된 함수겔상중합체의 분쇄 시 분쇄 효율이 낮게 나타나는 등 공정상 문제가 생길 수 있으며 고흡수성 수지의 물성이 저하될 우려가 있으므로, 상기 고흡수성 수지의 원료 물질 및 용매를 포함하는 단량체 조성물에 대해 약 20 내지 약 60 중량%, 바람직하게는 약 40 내지 약 50중량%로 될 수 있다.
본 발명의 고흡수성 수지 제조방법에서 중합시 사용되는 중합 개시제는 고흡수성 수지의 제조에 일반적으로 사용되는것이면 특별히 한정되지 않는다.
2019/194399 1»(:1^1{2019/001109
구체적으로, 상기 중합 개시제는 중합 방법에 따라 열중합 개시제 또는 UV 조사에 따른 광중합 개시제를 사용할 수 있다. 다만 광중합 방법에 의하더라도, 자외선 조사 등의 조사에 의해 일정량의 열이 발생하고, 또한 발열 반응인 중합 반응의 진행에 따라 어느 정도의 열이 발생하므로, 추가적으로 열중합 개시제를 포함할수도 있다.
상기 광중합 개시제는 자외선과 같은 광에 의해 라디칼을 형성할 수 있는 화합물이면 그 구성의 한정이 없이 사용될 수 있다.
상기 광중합 개시제로는 예를 들어, 벤조인 에테르(benzoin ether) , 디알킬아세토페논(dialkyl acetophenone) , 하이드록실 알킬케톤(hydroxyl alkylketone) , 페닐글리옥실레이트(phenyl glyoxylate) , 벤질디메틸케탈(Benzyl dimethyl ketal ) , 아실포스핀(acyl phosphine) 및 알파-아미노케톤 ( a -aminoketone)으로 이루어진 군에서 선택되는 하나 이상을 사용할 수 있다. 한편, 아실포스핀의 구체예로, 상용하는 lucirin TP0, 즉, 2 , 4, 6 -트리메틸-벤조일-트리메틸 포스핀 옥사이드 ^ ^^-trimethybbenzoyl- trimethyl phosphine oxide)를 사용할 수 있다. 보다 다양한 광개시제에 대해서는 Reinhold Schwalm 저서인 "UY Coatings: Basics, Recent
Developments and New Applicat ion(Elsevier 2007년V’ pll5에 잘 명시되어 있으며, 상술한 예에 한정되지 않는다.
상기 광중합 개시제는 상기 모노머 조성물에 대하여 약 0.01 내지 약 1.0 중량%의 농도로 포함될 수 있다. 이러한 광중합 개시제의 농도가 지나치게 낮을 경우 중합 속도가 느려질 수 있고, 광중합 개시제의 농도가 지나치게 높으면 고흡수성 수지의 분자량이 작고 물성이 불균일해질 수 있다.
또한, 상기 열중합 개시제로는 과황산염계 개시제, 아조계 개시제, 과산화수소 및 아스코르빈산으로 이루어진 개시제 군에서 선택되는 하나 이상을 사용할 수 있다. 구체적으로, 과황산염계 개시제의 예로는 과황산나트륨(Sodium persulfate; Na2S2¾) , 과황산칼륨(Potassium persulfate; K2S2O8) , 과황산암모늄 (Ammoni· persulfate; (NH4)2S2〔) 8) 등이 있으며, 아조 (Azo)계 개시제의 예로는 2, 2 -아조비스- (2 -아미디노프로판)이염산염 (2 , 2-azobis ( 2-amidinopropane ) dihydrochloride) , 2 , 2 -아조비스- (N, N_ 디메틸렌)이소부티라마이딘 디하이드로클로라이드 (2 , 2-azobis_(N, N-
2019/194399 1»(:1^1{2019/001109
dimethylene) isobutyramidine dihydrochloride) , 2 -
(카바모일아조)이소부티로니트릴(2-(carbamoyIazo)isobutyIonitril), 2 , 2 - 아조비스 [2-(2 -이미다졸린- 2 -일)프로판] 디하이드로클로라이드(2 , 2-azobis[2- (2-imidazol in-2-yl)propane] dihydrochloride) , 4 , 4 -아조비스-(4- 시아노발레릭 산)(4, 4-azobis_(4-cyanovaleric acid)) 등이 있다. 보다 다양한 열중합 개시제에 대해서는 Odian저서인 'Principle of Polymerizat ion(Wi ley, 1981)' , p203에 잘 명시되어 있으며 , 상술한 예에 한정되지 않는다.
상기 열중합 개시제는 상기 모노머 조성물에 대하여 약 0.001 내지 약 0.5 중량%의 농도로 포함될 수 있다. 이러한 열 중합 개시제의 농도가 지나치게 낮을 경우 추가적인 열중합이 거의 ·일어나지 않아 열중합 개시제의 추가에 따른 효과가 미미할수 있고, 열중합 개시제의 농도가 지나치게 높으면 고흡수성 수지의 분자량이 작고 물성이 불균일해질 수 있다.
본 발명의 일 실시예에 따르면, 상기 모노머 조성물은 고흡수성 수지의 원료 물질로서 내부 가교제를 더 포함할 수 있다. 상기 내부 가교제로는 상기 수용성 에틸렌계 불포화 단량체의 수용성 치환기와 반응할 수 있는 관능기를 1개 이상 가지면서, 에틸렌성 불포화기를 1개 이상 갖는 가교제; 혹은 상기 단량체의 수용성 치환기 및/또는 단량체의 가수분해에 의해 형성된 수용성 치환기와 반응할수 있는 관능기를 2개 이상 갖는 가교제를 사용할 수 있다. 상기 내부 가교제의 구체적인 예로는, 탄소수 8 내지 12의 비스아크릴아미드, 비스메타아크릴아미드, 탄소수 2 내지 10의 폴리올의 폴리(메타)아크릴레이트 또는 탄소수 2 내지 10의 폴리올의 폴리(메타)알릴에테르 등을 들 수 있고, 보다 구체적으로, N.N1 - 메틸렌비스(메타)아크릴레이트, 에틸렌옥시(메타)아크릴레이트, 폴리에틸렌옥시(메타)아크릴레이트, 프로필렌옥시(메타)아크릴레이트, 글리세린 디아크릴레이트, 글리세린 트리아크릴레이트 , 트리메티롤 트리아크릴레이트, 트리알릴아민 , 트리아릴시아누레이트, 트리알릴이소시아네이트 , 폴리에틸렌글리콜, 디에틸렌글리콜 및 프로필렌글리콜로 이루어진 군에서 선택된 하나 이상을사용할수 있다.
이러한 내부 가교제는 상기 모노머 조성물에 대하여 약 0.01 내지 약 0.5중량%의 농도로 포함되어, 중합된 고분자를 가교시킬 수 있다.
2019/194399 1»(:1^1{2019/001109
본 발명의 제조방법에서 , 상기 모노머 조성물은 필요에 따라 증점제 (thi ckener ) , 가소제, 보존안정제, 산화방지제 등의 첨가제를 더 포함할 수 있다.
상술한 수용성 에틸렌계 불포화 단량체, 광중합 개시제, 열중합 개시제, 내부 가교제 및 첨가제와 같은 원료 물질은 용매에 용해된 모노머 조성물 용액의 형태로 준비될 수 있다.
이 때 사용할 수 있는 상기 용매는 상술한 성분들을 용해할 수 있으면 그 구성의 한정이 없이 사용될 수 있으며, 예를 들어 물, 에탄올, 에틸렌글리콜, 디에틸렌글리콜, 트리에틸렌글리콜, 1,4 -부탄디올, 프로필렌글리콜, 에틸렌글리콜모노부틸에테르 , 프로필렌글리콜모노메틸에테르 , 프로필렌글리콜모노메틸에테르아세테이트, 메틸에틸케톤 , 아세톤, 메틸아밀케톤 , 시클로핵사논 , 시클로펜타논, 디에틸렌글리콜모노메틸에테르 , 디에틸렌글리콜에틸에테르, 톨루엔, 크실렌, 부틸로락톤, 카르비톨, 메틸셀로솔브아세테이트 및 N,N-디메틸아세트아미드등에서 선택된 1종 이상을 조합하여 사용할 수 있다.
상기 용매는 모노머 조성물의 총 함량에 대하여 상술한 성분을 제외한 잔량으로 포함될 수 있다.
한편, 이와 같은 모노머 조성물을 열중합 또는 광중합하여 함수겔상 중합체를 형성하는 방법 또한 통상 사용되는 중합 방법이면, 특별히 구성의 한정이 없다.
구체적으로, 중합 방법은 중합 에너지원에 따라 크게 열중합 및 광중합으로 나뉘며, 통상 열중합을 진행하는 경우, 니더 (kneader )와 같은 교반축을 가진 반응기에서 진행될 수 있으며, 광중합을 진행하는 경우, 이동 가능한 컨베이어 벨트를 구비한 반응기에서 진행될 수 있으나, 상술한 중합 방법은 일 예이며, 본 발명은 상술한 중합 방법에 한정되지는 않는다.
일 예로, 상술한 바와 같이 교반축을 구비한 니더 (kneader )와 같은 반응기에, 열풍을 공급하거나 반응기를 가열하여 열중합을 하여 얻어진 함수겔상 중합체는 반응기에 구비된 교반축의 형태에 따라, 반응기 배출구로 배출되는 함수겔상 중합체는 수 센티미터 내지 수 밀리미터 형태일 수 있다. 구체적으로, 얻어지는 함수겔상 중합체의 크기는 주입되는 모노머 조성물의
2019/194399 1»(:1^1{2019/001109
농도 및 주입속도 등에 따라 다양하게 나타날 수 있는데 , 통상 중량 평균 입경이 약 2mm내지 약 50mm인 함수겔상중합체가 얻어질 수 있다.
또한, 상술한 바와 같이 이동 가능한 컨베이어 벨트를 구비한 반응기에서 광중합을 진행하는 경우, 통상 얻어지는 함수겔상 중합체의 형태는 벨트의 너비를 가진 시트 상의 함수겔상 중합체일 수 있다. 이 때, 중합체 시트의 두께는 주입되는 단량체 조성물의 농도 및 주입속도에 따라 달라지나, 통상 약 0.5cm 내지 약 5cm의 두께를 가진 시트 상의 중합체가 얻어질 수 있도록 단량체 조성물을 공급하는 것이 바람직하다, 시트 상의 중합체의 두께가 0.5cm 미만으로 지나치게 얇을 정도로 단량체 조성물을 공급하는 경우, 생산 효율이 낮아 바람직하지 않으며, 시트 상의 중합체 두께가 5cm를 초과하는 경우에는 지나치게 두꺼운 두께로 인해, 중합 반응이 전 두께에 걸쳐 고르게 일어나지 않을수가 있다.
이때 이와 같은 방법으로 얻어진 함수겔상 중합체의 통상 함수율은 40 내지 80 중량%일 수 있다. 한편, 본 명세서 전체에서 "함수율"은 전체 함수겔상 중합체 중량에 대해 차지하는 수분의 함량으로 함수겔상 중합체의 중량에서 건조 상태의 중합체의 중량을 뺀 값을 의미한다. 구체적으로는, 적외선 가열을 통해 중합체의 온도를 올려 건조하는 과정에서 중합체 중의 수분증발에 따른 무게감소분을 측정하여 계산된 값으로 정의한다. 이때, 건조 조건은 상온에서 180°C까지 온도를 상승시킨 뒤 180°C에서 유지하는 방식으로 총 건조시간은 온도상승단계 5분을 포함하여 20분으로 설정하여, 함수율을 측정한다.
발명의 일 구현에에 따르면, 상기에서 수득한 함수겔상 중합체에 대해 조분쇄 공정이 선택적으로 더 수행될 수 있다.
이때, 조분쇄 공정에 사용되는 분쇄기는 구성의 한정은 없으나, 구체적으로, 수직형 절단기 (Vert ical pulver i zer ) , 터보 커터 (Turbo cut ter ) , 터보 글라인더 (Turbo gr inder) , 회전 절단식 분쇄기 (Rotary cutter mi l l ) , 절단식 분쇄기 (Cutter mi l l ) , 원판 분쇄기 (Di sc mi l l ) , 조각 파쇄기 (Shred crusher ) , 파쇄기 (Crusher ) , 초퍼 (chopper ) 및 원판식 절단기 (Di sc cutter)로 이루어진 분쇄 기기 군에서 선택되는 어느 하나를 포함할 수 있으나, 상술한 예에 한정되지는 않는다.
2019/194399 1»(:1^1{2019/001109
이때 조분쇄 단계는 함수겔상 중합체의 입경이 약 2ä 내지 약 20ä가 되도록 분쇄할수 있다.
입경이 21 1 미만으로 조분쇄하는 것은 함수겔상 중합체의 높은 함수율로 인해 기술적으로 용이하지 않으며, 또한 분쇄된 입자 간에 서로 응집되는 현상이 나타날 수도 있다. 한편, 입경이 20ä 초과로 조분쇄하는 경우, 추후 이루어지는 건조 단계의 효율 증대 효과가 미미할 수 있다.
다음으로, 발명의 일 구현예에 따른 고흡수성 수지의 제조방법에 있어서 단계 2는, 상기 단계 1에서 제조한 함수겔상 중합체를 건조 및 분쇄하고, 미분과 정상 입자로 분급하는 단계이다.
상기 건조 공정은 단계 1에서 조분쇄되거나, 혹은 조분쇄 단계를 거치지 않은 중합 직후의 함수겔상 중합체에 대해 수행된다. 이때 상기 건조 단계의 건조 온도는 약 150公 내지 약 250° (:일 수 있다. 건조 온도가 150ᄃ 미만인 경우, 건조 시간이 지나치게 길어지고 최종 형성되는 고흡수성 수지의 물성이 저하될 우려가 있고, 건조 온도가 250° (:를 초과하는 경우, 지나치게 중합체 표면만 건조되어, 추후 이루어지는 분쇄 공정에서 미분이 발생할 수도 있고, 최종 형성되는 고흡수성 수지의 물성이 저하될 우려가 있다. 따라서 바람직하게 상기 건조는 약 1501: 내지 약 2001:의 온도에서, 더욱 바람직하게는 약 160 내지 약 1801:의 온도에서 진행될 수 있다.
한편, 건조 시간의 경우에는 공정 효율 등을 고려하여, 약 20분 내지 약 90분 동안 진행될 수 있으나, 이에 한정되지는 않는다.
상기 건조 단계의 건조 방법 역시 함수겔상 중합체의 건조 공정으로 통상 사용되는 것이면, 그 구성의 한정이 없이 선택되어 사용될 수 있다. 구체적으로, 열풍 공급, 적외선 조사, 극초단파 조사, 또는 자외선 조사 등의 방법으로 건조 단계를 진행할 수 있다. 이와 같은 건조 단계 진행 후의 중합체의 함수율은 약 0. 1중량%내지 약 10중량%일 수 있다.
다음으로, 이와 같은 건조 단계를 거쳐 얻어진 건조된 중합체에 대한 분쇄 공정이 수행된다.
분쇄 단계 후 얻어지는 중합체 분말은 입경이 약 150_ 내지 약 850 // m일 수 있다. 이와 같은 입경으로 분쇄하기 위해 사용되는 분쇄기는 구체적으로, 핀 밀 ( ]1 1 1 1 ) , 해머 밀 (113_라 1111 1 1 ) , 스크류 밀 ( 근 1)^ 1 1 ) ,
2019/194399 1»(:1^1{2019/001109
롤 밀 (rol l mi l l ) , 디스크 밀 (di sc mi l l ) 또는 조그 밀 (jog mi l l ) 등을사용할 수 있으나, 상술한 예에 본 발명이 한정되는 것은 아니다.
이와 같은 분쇄 단계 이후 최종 제품화되는 고흡수성 수지 분말의 물성을 관리하기 위해, 일반적으로 분쇄 후 얻어지는 중합체 분말을 입경에 따라 분급한다. 바람직하게는 입경이 150/mi 이하인 입자와, 150 초과 850_ 이하인 입자로 분급하는 단계를 거친다.
본 발명의 명세서에서는 일정 입자 크기 이하, 구체적으로 150 이하의 입자 크기를 갖는 미분 입자를 고흡수성 중합체 미분, SAP 미분 또는 미분 ( f ines , f ine powder )으로 지칭하며, 입경이 150 초과 850_ 이하인 입자를 정상 입자로 지칭한다. 상기 미분은 중합공정 , 건조 공정 또는 건조된 중합체의 분쇄 단계 동안 발생될 수 있는데, 최종 제품에 미분이 포함될 경우 취급이 어렵고 겔 블로킹 (gel blocking) 현상을 나타내는 등 물성을 저하시키기 때문에 최종 수지 제품에 포함되지 않도록 배제하거나 정상 입자가 되도록 재사용하는 것이 바람직하다.
한 예로, 상기 미분들을 정상 입자 크기가 되도록 응집시키는 재조립 과정을 거칠 수 있다. 재조립 과정에서 일반적으로 응집 강도를 높이기 위해 미분 입자들을 습윤 .상태에서 응집시키는 재조립 공정을 진행한다. 이때 미분의 함수율이 높을수록 미분의 응집 강도가 높아지나 재조립 공정시 너무 큰 재조립체 덩어리가 생겨 공정 운전시 문제가 생길 수 있고, 함수율이 낮으면 재조립 공정은 용이하나 응집강도가 낮아 재조립 이후 다시 미분으로 파쇄되는 경우가 많다. 또한, 이렇게 얻어진 미분 재조립체는 정상 입자보다 보수능 (CRC)이나 가압흡수능 (AUP)과 같은 물성이 저하되어 고흡수성 수지의 품질 하락을 가져오기도 한다.
다음으로, 발명의 일 구현예에 따른 고흡수성 수지의 제조방법에 있어서 단계 3는, 상기 단계 2에서 분급한 미분을 재조립화하여 미분 재조립체를 제조하는 단계이다.
구체적으로, 상기 단계 2에서 분급한, I50im 이하의 입경을 갖는 미분을 물과 혼합하되, 상기 물을 미분에 대해 2회 이상 분할 투입하고 재조립하여 미분 재조립체를 제조한다.
이때 상기 미분과 물의 혼합은 전단력을 부가할 수 있는 혼합 장치나
2019/194399 1»(:1^1{2019/001109
믹서를 사용하여 수행될 수 있으며, 이때 전단력에 의한 교반 선속도 제어를 통해 2차 응집 시간을 제어할 수 있는데, 본 발명에서의 상기 미분과 물의 혼합시 교반 선속도는 6.5 내지 14.5111/3 ¾ 수 있다. 교반 선속도의 증가에 따라 미분의 2차 응집 형성 시간은 짧아지지만 혼합장치나 믹서의 운전 안정성은 저하되게 된다. 구체적으로 교반선속도가 6.5111/3 미만으로 지나치게 느릴 경우, 2차응집 형성 시간이 상대적으로 길어져 믹서 운전이 안정할 수는 있으나, 재조립체의 재미분 발생율이 증가될 우려가 있다. 또 교반 선속도가 14.5미/3를 초과하여 지나치게 빠를 경우 미분의 2차 응집 형성 시간이 지나치게 빨라지게 되고, 또 충분한 조립 강도를 가질 수 있으나 재조립체 덩어리가 생성되어 믹서에 부하가 증가되어 믹서의 연속 운전이 어렵다. 보다 바람직하게는 상기 교반 선속도는 8.5 내지 1¾1/3일 수 있다.
또, 상기 물은 미분이 습윤 상태에서 재조립될 수 있도록 하는 양으로 포함될 수 있는데 , 구체적으로는 상기 미분 100중량부에 대하여 , 80 내지 150 중량부, 바람직하게는 80 내지 110 중량부가 될 수 있다. 상기 물은 미분 재조립체를 형성한 뒤 재건조 과정에서 증발되게 되는데, 이때 물의 함량이 80중량부 미만이면, 미분과 물이 혼합 과정에서 미분의 빠른 흡수 속도로 인해 적은 양의 물을 고르게 분산하기 어려우므로 미분 재조립체의 균일성이 저하되고, 재미분 발생량이 증가될 수 있다. 또, 제조되는 미분 재조립체의 함수율이 감소함으로써 후속의 공정 동안에 단단한 덩어리가 형성되어 공정 운전 안정성이 저하될 우려가 있으며, 최종 제조된 고흡수성 수지의 흡수능이 저하될 수 있다. 또 물 함량이 150중량부를 초과할 경우 혼합 과정에서 미분재조립체의 끈적임이 증가해 정상적인 혼합이 이루어지지 못하며, 건조 과정에서 증발시켜야 할 물의 양이 증가하여 건조기의 부하가 증가하는 등의 우려가 있다.
또, 상기 물은 미분에 대해 2회 이상분할 투입된다.
상기 미분과 물의 혼합시, 미분에 대해 물이 일괄 투입될 경우, 투입된 물이 미분과 조립되어 2차 응집이 빠르게 발생하여 발생하기 때문에 혼합 안정성이 저하될 우려가 있으나, 상기와 같이 2회 이상으로 분할 투입될 경우, 2차 이후로 분할 투입되는 물이 분산되어 표면에 오래 존재하게 됨으로써 혼합 안정성이 향상될 수 있다. 구체적으로 상기 물은 미분에 대해 2회 또는 3회
2019/194399 1»(:1^1{2019/001109
분할투입될 수 있다.
일례로, 물이 2회로 분할 투입되는 경우, 본 발명에 따른 고흡수성 수지 조성물의 제조방법에 있어서 상기 미분과 물의 혼합 공정은 미분에 대해 물의 1차 투입하고 혼합하는 1차 혼합 공정과, 상기 1차 혼합 후 잔부의 물을 2차로 투입하여 혼합하는 2차혼합공정을 포함할수 있다.
또 물이 2회 이상으로 분할 투입되는 경우, 투입시기 별로 물 투입량이 특별히 한정되는 것은 아니며, 상기한 투입량 범위 내에서 동등 양으로 분할 투입될 수도, 또는투입 시기별로 다른 양으로 투입될 수도 있다.
일 례로, 2회 분할 투입의 경우, 1차로 투입되는 물의 양은 전체 투입되는 물 총 중량에 대해 50중량% 초과 내지 90중량% 이하이고, 2차로 투입되는 물의 양은 잔부인, 10중량% 이상 내지 50중량% 미만일 수 있다. 이와 같이 2차로 투입되는 물은 1차투입시의 물에 비해 적은 양으로 투입되는 것이 바람직한데 , 2차투입되는 물의 양이 50중량% 이상일 경우 응집 시간이 빨라져 혼합 안정성이 저하될 우려가 있고, 또 10중량% 미만일 경우 2차 투입에 따른 개선 효과가 충분하지 않다. 보다 구체적으로는 1차로 투입되는 물의 양은 전체 투입되는 물 총 중량에 대해 60 내지 90중량%이고, 2차로 투입되는 물의 양은 잔부인, 10 내지 40중량%일 수 있으며, 보다 구체적으로는 1차로 투입되는 물의 양은 전체 투입되는 물 총 중량에 대해 65 내지 80중량%이고,
2차로 투입되는 물의 양은 잔부인, 20 내지 35중량%일 수 있다.
또, 물이 2회 이상 분할 투입되는 경우, 1차 투입은 미분과의 혼합 초기에, 그리고 2차 이후의 분할투입은, 이전 차수의 물 투입 후 2 내지 1◦초, 보다 구체적으로는 2 내지 5초의 간격으로 수행될 수 있다. 2차 이상의 분할 투입 간격이 2초 미만이면, 분할 투입에 따른 충분한 효과를 얻기 어렵고, 10초를 초과하여 수행될 경우, 투입 간격이 지나치게 길어져 비효율적이다. 상기한 분할 투입을 통해 재조립된 미분 재조립체는 빠르게 혼합기 밖으로 배출되는 것이 바람직하다. 재조립체가 2차 응집되어 덩어리화될 경우 혼합기에 부하증가로 연속 공정이 어렵다.
또, 본 발명의 일 구현예에 따르면, 상기 미분과 물의 혼합시 혼합 믹서 내에서 미분 재조립체의 윤활 작용을 촉진시켜 혼합 안정성 및 균일성을 향상시키기 위한 혼합 안정화제로서 수용성 고분자 및 계면활성제 중 적어도
2019/194399 1»(:1^1{2019/001109
1종 이상이 선택적으로 더 사용될 수 있다.
상기 수용성 고분자로는 구체적으로 폴리에틸렌 글리콜, 폴리에틸렌 옥사이드, 폴리비닐피롤리돈, 폴리비닐알콜, 폴리아크릴아마이드, 폴리아크릴릭애시드, 폴리스타이렌설포닉애시드, 폴리실리식애시드, 폴리포스포릭애시드, 폴리에틸렌설포닉애시드, 폴리비닐페놀, 폴리비닐페닐설포닉애시드 , 폴리에틸렌포스포릭애시드, 폴리에틸렌아마이드 , 폴리아민 또는 폴리아마이드아민 등과 같은 합성 고분자 ; 또는 전분, 고무, 셀룰로오스 등과 같은 비합성 고분자를 들 수 있으며, 이들 중 어느 하나또는 둘 이상의 혼합물이 사용될 수 있다. 상기한 수용성 고분자 중에서도 폴리에틸렌 글리콜아화학적 안정성, 인체 무해성, 우수한 분산/윤활 효과 및 경제적인 특성으로 인해 보다 바람직할수 있다.
또, 폴리에틸렌 글리콜이 사용될 경우, 상기 폴리에틸렌 글리콜은 중량평균분자량이 2 , 000 내지 200 , 000g/mol, 보다 바람직하게는 4 , 000 내지 100 , 000g/mol 일 수 있다. 폴리에틸렌글리콜의 분자량이 2 , 000g/mol 미만이면 윤활 작용이 저하될 우려가 있고, 또 200 , 000g/mol 초과할 경우 물에 용해도가 저하될 우려가 있다.
한편, 상기 계면활성제로는 구체적으로 소둠 도데실 설페이트(sodium dodecyl sulfate, SDS) , 다이아이소옥틸 소듐 설포숙시네이트염 (di isooctyl sodium sulfosuccinate, DSS) , 소듐 테트라데실 설페이트(sodium tetradecyl sulfate) , 소듐 핵사데실 설페이트 (sodi· hexadecyl sulfate) , 소듐 도데실벤젠 설페이트 (sodi· dodecylbenzene sulfonate) , 크실렌 설포네이트 (xylenesulfonate) , 소듐 올레이트 (sodi· oleate) , 4_n_ 데실벤젠설포네이트(4-11-(160 15611761163111 £011 6), 소듐 라우레이트(sodium laurate) , 4-도데실벤젠설폰산 (4-dodecylbenzenesul fonic acid) , 도데실아민 하이드로클로라이드(dodecylamine hydrochloride) , 도데실트리메틸암모늄 클로라이드 (dodecyltrimethylammoni· chloride) , 4_n_옥틸벤젠 설포네이트 (4- n-octylbenzene sulfonate) , 에톡시화 설포네이트 (Et:hoxylated sulfonate) , 데실벤젠 설포네이트(decylbenzene sulfonate) , 포타슘 올레이트 (Potassi· oleate) , n_데실벤젠 설포네이트 (n-decylbenzene sulfonate) , 알킬트리메틸암모늄 브로마이드 (alkyltrimethylammoni· bromide, C10-C16
2019/194399 1»(:1^1{2019/001109
chains) , 도데실 아민 (dodecyl amine) , 테트라데실트리메틸암모늄 클로라이드 (tetradecyl tr imethyl ammonhim chlor ide) , 도데실 폴리사카라이드 글리코시드 (dodecyl polysacchar ide glycoside) , 사이클로덱스트린 (cyc lodextr ins), 글리코리피드 (glycol ipids) , 리포프로테인_ 리포펩타이드 ( l ipoprotein-ipopept ides),포스포리피드 (phosphol ipides) , para_ 톨루엔 설폰산 (para-toluene sul foni c acid) , 또는 트리실옥세인 (tr i si loxane) 등을 들 수 있으며, 이들 중 어느 하나 또는 둘 이상의 혼합물이 사용될 수 있다. 또 트리톤 (tr i tonä X-100)과 같은 상업적으로 입수가능한 상품이 사용될 수도 있다. 상기한 계면활성제 중에서도 소듐 도데실 설페이트가 화학적 안정성 및 우수한분산/윤활 효과로 인해 보다 바람직할수 있다.
상기한 첨가제들은 미분 100중량부에 대하여 0.001 내지 1중량부, 보다 바람직하게는 0.001 내지 0.5중량부로 사용될 수 있다. 첨가제의 함량이 0.001 중량부 미만이면, 첨가제 사용에 따른 미분 재조립체의 혼합 안정성 및 균일성 향상 효과를 얻기 어렵고, 또 첨가제의 함량이 1중량부를 초과할 경우 고흡수성 수지의 성능 저하 및 변색의 우려가 있다.
발명의 일 구현예에 따른 고흡수성 수지의 제조방법은, 상기 단계 3에서 제조한 미분 재조립체를, 상기 단계 1에서 제조한 함수겔상 중합체와 혼합하고, 건조 및 분쇄하여, 150//m 이하의 입경을 갖는 재조립체 미분 (이하 ’재미분'이라 함) , 및 150_ 초과 850 n 이하의 입경을 갖는 재조립체 정상 입자로 분급하는 단계 (단계 4)를 더 포함할수 있다.
구체적으로, 단계 4는 상기 단계 3에서 제조한 미분 재조립체를, 상기 단계 1에서 제조한 함수겔상 중합체와 혼합 후 건조 및 분쇄하고, 재조립체 미분 및 재조립체 정상 입자로 분급하는 단계이다.
상기 건조 공정은 통상의 건조 기기를 사용하여 수행될 수 있으나, 발명의 일 구현예에 따르면 패들형 건조기 또는 강제순환형 건조기를 이용하여 수행될 수 있다. 이들 건조기에서 건조 공정을 수행할 경우, 유동시 발생되는 힘에 의해 2차응집화된 미분 조립체가보다용이하게 1차 입자화될 수 있으며, 그 결과 건조 속도 및 건조 효율이 증가될 수 있다.
또, 상기 건조 공정은 약 12CTC 내지 약 220 °C의 온도에서 수행될 수 있다. 건조 공정시 온도가 120 °C 미만이면 건조 시간이 길어지고, 220 °C를
2019/194399 1»(:1^1{2019/001109
초과하면 미분 재조립체의 열화로 물성 저하의 우려가 있다. 보다 바람직하게는 약 150 °C 내지 약 20CTC의 온도에서, 미분 재조립체내 함수율이 1중량% 이하가되도록 수행될 수 있다.
또 상기한 온도 범위 내에서 건조 공정 초기에 비해 건조 공정 후기로 갈수록 온도를 높여 주는 것이 건조 효율을 더욱 높일 수 있어 바람직하다. 구체적으로는 건조 공정 초기, 구체적으로 건조기 투입구의 온도가 약 120°C 내지 약 160°C고, 건조 공정 후기, 구체적으로는 건조기 후단 배출구의 온도가 약 15CTC 내지 약 200°C인 것이 건조 효율을 높일 수 있어 보다 바람직하다. 또, 상기 건조 공정시 건조를 위한 승온 수단으로는, 그 구성의 한정이 없다. 구체적으로, 열 매체를 공급하거나, 전기 등의 수단으로 직접 가열할 수 있으나, 본 발명이 상술한 예에 한정되는 것은 아니다. 구체적으로 사용될 수 있는 열원으로는 스팀, 전기, 자외선, 적외선 등이 있으며, 가열된 열유체 등을사용할수도 있다.
또, 상기 건조 공정은 츠퍼를 이용한 절단 공정과 동시에 수행될 수도 있다. 이 경우 건조 효율을높이고, 건조 시간을 단축할수 있다.
상기 건조 공정은 건조된 미분 재조립체 내 함수율이 1중량% 이상, 보다 구체적으로는 1 내지 2중량%가 되도록 수행될 수 있다. 건조된 미분 재조립체내 함수율이 1중량% 미만으로 건조할 경우 미분 재조립체의 물성 저하의 우려가 있다.
상기와 같은 건조 공정시 2차 입자화된 미분 재조립체가 단일 입자 형태인 1차 입자화된다.
다음으로, 상기 건조된 미분 재조립체를 분쇄 및 분급한다.
상기 분쇄는 건조된 미분 재조립체를 입경이 150 m 초과 850/M 이하가 되도록 수행할 수 있다. 이와 같은 입경으로 분쇄하기 위해 사용되는 분쇄기는 구체적으로, 핀 밀 (pin mi l l ) , 해머 밀 (hammer mi l l ) , 스크류 밀 (screw mi 1 1 ) , 롤 밀 (rol l mi l l ) , 디스크 밀 (di sc mi l l ) 또는 조그 밀 ( j og mi l l ) 등을사용할 수 있으나, 상술한 예에 본 발명이 한정되는 것은 아니다.
이와 같은 분쇄 단계 이후 최종 제품화되는 고흡수성 수지 분말의 물성을 관리하기 위해, 일반적으로 분쇄 후 얻어지는 중합체 분말을 입경에 따라 분급한다. 바람직하게는 입경이 150//m 이하의 입경을 갖는 재미분, 및
2019/194399 1»(:1^1{2019/001109
150쌘!초과 850 이하의 입경을 갖는 재조립체 정상 입자로 분급하는 단계를 거친다.
상기 공정을 통해 수득되는 상기 미분 재조립체는, 분쇄 단계를 거친 후 다시 미분으로 재파쇄되는 비율이 낮아 높은 응집강도를 가진다. 구체적으로 상기 미분 재조립체는, 분쇄 후 입경이 150/® 이하인 미분이 생성된 중량 비율이, 전체 미분 재조립체의 중량에 대하여 약 50중량% 이하, 바람직하게는 약 30중량% 이하로 될 수 있다.
또, 발명의 일 구현예에 따르면, 상기 단계 4에 분급한 상기 재조립체 정상 입자를 표면가교 혼합기 내로 유입하여 표면 가교제와 혼합하여 표면 가교 하는 단계(단계 5)가선택적으로 더 수행될 수도 있다.
상기 표면 가교는 입자 내부의 가교결합 밀도와 관련하여 고흡수성 고분자 입자 표면 근처의 가교결합 밀도를 증가시키는 단계이다. 일반적으로, 표면 가교 제는 고흡수상 수지 입자의 표면에 도포된다. 따라서, 이 반응은 고흡수성 수지 입자의 표면 상에서 일어나며, 아는 입자 내부에는 실질적으로 영향을 미치지 않으면서 입자의 표면 상에서의 가교 결합성은 개선시킨다. 따라서 표면 가교 결합된 고흡수성 수지 입자는 내부에서보다 표면 부근에서 더 높은 가교 결합도를 갖는다.
이때 상기 표면 가교제로는 중합체가 갖는 관능기와 반응 가능한 화합물이라면 그 구성의 한정이 없다.
바람직하게는 생성되는 고흡수성 수지의 특성을 향상시키기 위해, 상기 표면 가교제로 다가 알콜 화합물; 에폭시 화합물; 폴리아민 화합물; 할로에폭시 화합물; 할로에폭시 화합물의 죽합 산물; 옥사졸린 화합물류; 모노-, 디-또는 폴리옥사졸리디논 화합물; 환상우레아 화합물; 다가금속염 ; 및 알킬렌 카보네이트 화합물로 이루어진 군에서 선택되는 1 종 이상을사용할 수 있다.
구체적으로, 다가 알콜 화합물의 예로는 모노- , 디-, 트리-, 테트라_ 또는 폴리에틸렌 글리콜, 모노프로필렌 글리콜, 1 , 3 -프로판디올, 디프로필렌 글리콜, 2 , 3 , 4 -트리메틸- 1 , 3 -펜탄디올, 폴리프로필렌 글리콜, 글리세롤, 폴리글리세롤, 2 -부텐- 1 , 4 -디올, 1, 4 -부탄디올, 1 , 3 -부탄디올, 1, 5 -펜탄디올, 1 , 6 -핵산디올, 및 1 , 2 -사이클로핵산디메탄올로 이루어진 군에서 선택되는 1 종
2019/194399 1»(:1^1{2019/001109
이상을사용할수 있다.
또한, 에폭시 화합물로는 에틸렌 글리콜 디글리시딜 에테르 및 글리시돌 또는 폴리(에틸렌글리콜)디글리시딜에테르 등을 사용할 수 있으며, 폴리아민 화합물류로는 에틸렌디아민 , 디에틸렌트리아민 , 트리에틸렌테트라아민 , 테트라에틸렌펜타민, 펜타에틸렌핵사민 , 폴리에틸렌이민 및 폴리아미드폴리아민로 이루어진 군에서 선택되는 1 종 이상을사용할수 있다.
그리고 할로에폭시 화합물로는 에피클로로히드린 , 에피브로모히드린 및 메틸에피클로로히드린을 사용할 수 있다. 한편, 모노-, 디- 또는 폴리옥사졸리디논 화합물로는 예를 들어 2 -옥사졸리디논 등을사용할 수 있다. 그리고, 알킬렌 카보네이트 화합물로는 에틸렌 카보네이트 등을 사용할 수 있다. 이들을 각각 단독으로 사용하거나 서로 조합하여 사용할 수도 있다. 한편, 표면 가교 공정의 효율을 높이기 위해, 이들 표면 가교제 중에서 1 종 이상의 다가 알코올 화합물을 포함하여 사용하는 것이 바람직하며, 더욱 바람직하게는 탄소수 2내지 10의 다가 알코올 화합물류를사용할수 있다. 상기 첨가되는 표면 가교제의 함량은 구체적으로 추가되는 표면 가교제의 종류나 반응 조건에 따라 적절히 선택될 수 있지만, 통상 중합체 100 중량부에 대해, 약 0.001 내지 약 5 중량부, 바람직하게는 약 0.01 내지 약 3 중량부, 더욱 바람직하게는 약 0.05 내지 약 2 중량부를사용할 수 있다.
표면 가교제의 함량이 지나치게 적으면, 표면 가교 반응이 거의 일어나지 않으며, 중합체 100 중량부에 대해, 5 중량부를 초과하는 경우, 과도한 표면 가교 반응의 진행으로 인해 흡수능력 및 물성의 저하 현상이 발생할수 있다.
표면 가교제가 첨가된 중합체 입자에 대해 가열시킴으로써 표면 가교 결합 반응 및 건조가동시에 이루어질 수 있다.
표면 가교 반응을 위한 승온 수단은 특별히 한정되지 않는다. 열매체를 공급하거나, 열원을 직접 공급하여 가열할 수 있다. 이때, 사용 가능한 열매체의 종류로는 스팀, 열풍, 뜨거운 기름과 같은 승은한 유체 등을 사용할 수 있으나, 본 발명이 이에 한정되는 것은 아니며, 또한 공급되는 열매체의 온도는 열매체의 수단, 승온 속도 및 승온 목표 온도를 고려하여 적절히
0 2019/194399 1»(:1^1{2019/001109
선택할 수 있다. 한편, 직접 공급되는 열원으로는 전기를 통한 가열, 가스를 통한 가열 방법을 들 수 있으나, 상술한 예에 본 발명이 한정되는 것은 아니다. 또, 상기 표면 가교 후, 150/께 이하의 입경을 갖는 표면가교 미분, 및 150_ 초과 850 /패 이하의 입경을 갖는 표면가교 정상 입자로 분급하고, 150 // m 이하의 입경을 갖는 표면가교 미분은 미분 재조립을 위한 공정으로 재투입하고, 표면 가교 정상 입자는 제품화하여 사용될 수 있다.
상술한 방법으로 제조된 고흡수성 수지는, 산성기를 포함하고 상기 산성기의 적어도 일부가 중화된 수용성 에틸렌계 불포화 단량체를 중합시킨 중합체 중 입경이 150 이하인 미분에 대해, 물을 혼합하여 재조립된 미분 재조립체를 표면 가교시킨 고흡수성 수지로, £0^ 법
 241.3에 따라 측정한 보수능(0¾:)이 33.0 내지 39.0
241.3에 따라 측정한 보수능(0¾:)이 33.0 내지 39.0
 법 으므 241.3에 따라 측정한 0.3
법 으므 241.3에 따라 측정한 0.3
 가압 흡수능 )이 20.0 내지 33.0 용/ 이고; 볼텍스 법( 0 6 에 의한흡수 속도가 100초 이하인 것일 수 있다.
가압 흡수능 )이 20.0 내지 33.0 용/ 이고; 볼텍스 법( 0 6 에 의한흡수 속도가 100초 이하인 것일 수 있다.
일 구현예의 고흡수성 수지에 있어, 상기 중합체는 산성기를 포함하고 상기 산성기의 적어도 일부가 중화된 수용성 에틸렌계 불포화 단량체를 중합시킨 것으로, 이에 사용되는 구체적인 물질 및 제조방법의 구체적인 설명은 앞서 예시적으로 설명한 바와동일하다.
이하 본 발명을 실시예에 기초하여 더욱 상세하게 설명한다. 단, 하기의 실시예는 본 발명을 예시하는 것일 뿐, 본 발명의 내용이 하기의 실시예에 의하여 한정되는 것은 아니다. 또한, 이하의 실시예 , 비교예에서 함유량을 나타내는 "V 및 ’’부’’는 특별히 언급하지 않는 한중량 기준이다.
<미분제조>
제조예 1
아크릴산 100
 가교제로 폴리에틸렌글리콜 디아크릴레이트 0.3 중합 개시제로 과황산나트륨 0.033 융, 가성소다(犯(犯) 38.9
가교제로 폴리에틸렌글리콜 디아크릴레이트 0.3 중합 개시제로 과황산나트륨 0.033 융, 가성소다(犯(犯) 38.9
 및 물 103.9 당의 비율로혼합하여, 단량체 농도가 50중량%인 단량체 혼합물을 준비하였다. 이후, 상기 단량체 혼합물을 연속 이동하는 콘베이어 벨트상에 투입하고 자외선을 조사(조사량: 21 /( 2)하여 2분 동안 중합을 진행하여 함수겔상중합체를 얻었다.
및 물 103.9 당의 비율로혼합하여, 단량체 농도가 50중량%인 단량체 혼합물을 준비하였다. 이후, 상기 단량체 혼합물을 연속 이동하는 콘베이어 벨트상에 투입하고 자외선을 조사(조사량: 21 /( 2)하여 2분 동안 중합을 진행하여 함수겔상중합체를 얻었다.
상기 함수겔상 중합체를 미트 초퍼(홀 크기 10 ■)로 분쇄하여 조분쇄
2019/194399 1»(:1^1{2019/001109
함수겔상 중합체를 얻었다. 이를 1701: 온도의 열풍 건조기에서 1시간 동안 건조하고, 핀밀 분쇄기로 분쇄한
 규격의 표준 망체로 분급하여 150_ 초과 850 /패 이하의 입자 크기를 갖는 정상 입자와, 150쌘! 이하의 입경을 갖는 미분 입자를 수득하였다.
규격의 표준 망체로 분급하여 150_ 초과 850 /패 이하의 입자 크기를 갖는 정상 입자와, 150쌘! 이하의 입경을 갖는 미분 입자를 수득하였다.
<미분재조립체의 제조>
실시예 1-1
상기 제조예 1에서 제조된, 150;_ 이하의 입경을 갖는 미분 10½을 믹서에 넣고, 선속도 8.5½八로 교반하면서, 물 7¾을 1차로 투입하고, 2초 뒤 물 33§을 2차 투입하고 교반하였다. 이때 상기 물의 2차 투입 완료 시점에서부터 2차 응집이 일어날 때까지의 시간을 측정하였으며, 미분 재조립체가 한 덩어리가 되어 믹서 부하가 증가된 후 바로 믹서 교반을 멈추었다. 2차응집 형성시간은 13초 이었다.
교반 후 결과로 수득한 미분 재조립체를 강제순환형 건조기에 투입한 후 1801: 온도에서 60분간 건조하여 미분 재조립체를 회수하였다. 실시예 1-2
상기 실시예 1-1에서, 미분 교반시 선속도를 12.8111/3로 하는 것을 제외하고는 상기 실시예 1-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 7초 이었다. 비교예 1-1
상기 실시예 1-1에서 , 미분 교반시 선속도를 4.25111/3로 하는 것을 제외하고는 상기 실시예 1-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 58초 이었다. 비교예 1-2
상기 실시예 1-1에서, 미분 교반시 선속도를 16.61^/3로 하는 것을 제외하고는 상기 실시예 1-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 5초 이었다.
2019/194399 1»(:1^1{2019/001109
실시예 2-1
상기 제조예 1에서 제조된, 150m 이하의 입경을 갖는 미분 ioog을 믹서에 넣고, 선속도 8.51m/s로 교반하면서, SDS( sodium dodecyl sul fate) 0. 1중량%를 첨가한 수용액 3.7g과, 물 74.4g을 1차로 투입하고, 2초 뒤 물 31 .9g을 2차 투입하고 교반하였다. 이때 상기 물의 2차 투입 완료 시점에서부터 2차 응집이 일어날 때까지의 시간을 측정하였으며, 미분 재조립체가 한 덩어리가 되어 믹서 부하가 증가된 후 바로 믹서 교반을 멈추었다. 2차 응집 형성시간은 23초 이었다.
결과로 수득한 미분 재조립체를 강제순환형 건조기에 투입한 후 180 °C 온도에서 60분간 건조하여 미분 재조립체를 회수하였다. 실시예 2-2
상기 실시예 2-1에서, 미분 교반시 선속도를 12.801/3로 하는 것을 제외하고는 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 10초 이었다. 실시예 2-3
상기 실시예 2-1에서, 물의 투입시 1차로 31 .9용을 투입하고, 2초 뒤 2차로 74.4§을 투입하는 것을 제외하고는, 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차 응집 형성시간은 18초 이었다. 실시예 2-4
상기 실시예 2-1에서, 물의 투입시 1차로 35. 을 투입하고, 2초 뒤 2차로 35 을 투입하였으며, 2초 뒤 3차로 35.4§을 투입하는 것을 제외하고는, 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차 응집 형성시간은 19초 이었다. 실시예 2-5
2019/194399 1»(:1^1{2019/001109
상기 실시예 1-1에서, 물의 투입시 1차로 56용을 투입하고, 2초 뒤 2차로 54용을 투입하는 것을 제외하고는, 상기 실시예 1-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차 응집 형성시간은 13초 이었다. 비교예 2-1
상기 실시예 2-1에서, 미분 교반시 선속도를 4.2¾/3로 하는 것을 제외하고는 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 85초 이었다. 비교예 2-2
상기 실시예 2-1에서, 미분 교반시 선속도를 16.6111/3로 하는 것을 제외하고는 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 8초 이었다. . 비교예 2-3
상기 실시예 2-1에서 , 물을 일괄로 투입하는 것을 제외하고는, 상기 실시예 2-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차 응집 형성시간은 11초이었다. 비교예 3-1
상가 제조예 1에서 제조된, 150/^1 이하의 입경을 갖는 미분 100은을 믹서에 넣고, 선속도 4.2¾/3로 교반하면서, 물 11½을 일괄 투입하고 교반하였다. 이때 상기 물 투입 완료 시점에서부터 2차 응집이 일어날 때까지의 시간을 측정하였으며, 미분 재조립체가 한 덩어리가 되어 믹서 부하가 증가된 후 바로 믹서 교반을 멈추었다. 2차 응집 형성시간은 28초 이었다.
결과로 수득한 미분 재조립체를 강제순환형 건조기에 투입한 후 1801: 온도에서 60분간 건조하여 미분 재조립체를 회수하였다.
2019/194399 1»(:1^1{2019/001109
비교예 3-2
상기 비교예 3-1에서, 미분 교반시 선속도를 8.크止八로 하는 것을 제외하고는 상기 비교예 3-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 9초 이었다. 비교예 3-3
상기 비교예 3-1에서, 미분 교반시 선속도를 12. ¾/3로 하는 것을 제외하고는 상기 비교예 3-1에서와 동일한 방법으로 수행하여 미분 재조립체를 회수하였다. 2차응집 형성시간은 4초 이었다.
<고흡수성 수지의 제조>
실시예 3-1
상기 실시예 1-1에서 제조한 미분 재조립체를, 상기 제조예 1의 미분 제조과정에서 중합 제조한 함수겔상 중합체와 20 : 80의 중량비로 혼합 후, 쵸핑하고, 건조한후, 1 01 1 1 1 분쇄하고, 쇼紅 규격의 표준 망체로 분급하여 150_ 이하인 재조립체 미분과, 150 /패 초과 850_ 이하의 입경을 갖는 재조립체 정상 입자로 분급하였다.
분급된 재조립체 정상 입자 100§에 폴리(에틸렌글리콜)디글리시딜에테르 0.2요, 메탄올 5§, 물 4§ 실리카 에어로
 1105사) 0.0¾을 포함하는 표면 가교 용액과 혼합한 후,
1105사) 0.0¾을 포함하는 표면 가교 용액과 혼합한 후,
180方의 온도에서 60분간 표면 가교 반응을 수행하여 최종 고흡수성 수지를 얻었다. 실시예 3-2내지 3-7
상기 실시예 3-1에서, 실시예 1-1에서 제조한 미분 재조립체 대신에 실시예 1-2, 및 실시예 2-1 내지 실시예 2-5에서 제조한 미분 재조립체를 각각 사용하는 것을 제외하고는 상기 실시예 3-1에서와 동일한 방법으로 수행하여 고톱수성 수지를 제조하였다. 비교예 4-1
2019/194399 1»(:1^1{2019/001109
상기 실시예 3-1에서, 실시예 1-1에서 제조한 미분 재조립체 대신에 비교예 1-1에서 제조한 미분 재조립체를 사용하는 것을 제외하고는, 상기 실시예 3-1과 동일한 방법으로 수행하여 고흡수성 수지를 얻었다. 비교예 4-2내지 4-8
상기 실시예 3-1에서 , 실시예 1-1에서 제조한 미분 재조립체 대신에 비교예 1-2 , 비교예 2-1 내지 2-3 , 및 비교예 3-1 내지 3-3에서 제조한 미분 재조립체를 각각 사용하는 것을 제외하고는 상기 실시예 3-1에서와 동일한 방법으로 수행하여 고흡수성 수지를 제조하였다. 실험예 1
상기 실시예 1-1 내지 2-2 , 그리고 비교예 1-1 내지 3-3에 따른 미분 재조립체의 제조시, 교반 선속도에 따른 2차응집 형성 시간을 비교하였다. 그 결과를 도 1에 나타내었다.
6=실시예 2-1 , =실시예 2-2 , 비교예 2-1 , 11=비교예 2-2 , 비교예 3-1 , 」_=비교예 3-2 , 뇨=비교예 3-3의 결과이다.
실험결과, 미분 재조립체의 제조시 물 투입 등 동일한 조건에서 교반 선속도가증가할수록 2차응집 형성시간이 빨라짐을 확인하였다.
또, 실시예 2-1 내지 2-2 , 및 비교예 2-1 내지 2-2는 동일 교반 선속도 및 물 투입 조건에서, 실시예 1-1 내지 1-2 및 비교예 1-1 내지 2-1과 각각 비교하여 2차 응집 형성시간이 느려졌다. 이는 혼합 미분에 대한 물 투입시 혼합 안정제가더 투입됨으로써 혼합 안정성이 증가되었기 때문이다.
또, 물을 분할 투입한 비교예 1-1 및 실시예 1-1 내지 1-2는, 동일 교반 선속도의 조건에서 .물을 일괄 투입한 비교예 3-1 내지 3-3과 각각 비교하여 2차 응집 시간이 느려졌다. 이는 물의 분할 투입으로 물이 분산되어 표면에 오래 존재함으로써 혼합 안정성이 증가되었기 때문이다. 실험예 2
상기 실시예 1-1에 따른 미분 재조립체의 제조시 교반 시간에 따른
2019/194399 1»(:1^1{2019/001109
믹서 내부 상태 및 미분 재조립체의 형상을 관찰하였다. 그 결과를 도 2에 나타내었다.
관찰결과, 2차 물 투입 완료 직후 믹서 내부에서 미분이 재조립되었으며 , 교반시간의 경과에 따라 2차 응집이 발생하여 재조립체가 한 덩어리가 되었다. 구체적으로, 교반 시간 13초일 때 2차 응집이 일어났으며, 교반 완료후 믹서에서 꺼내었을 때 한 덩어리로 되어 있었다. 실험예 3
상기 실시예 1-1에서와 동일한 방법으로 수행하여 미분 재조립체를 제조하되, 교반 시간을 5s , 10s 및 20s로 변화시켰으며, 제조되는 미분 재조립체에 대한 물성 변화 관찰을 통해 교반 시간이 미분 재조립체에 미치는 영향을 확인하였다.
미분 재조립체에 대한 물성은 하기 방법으로 측정하였으며, 그 결과는 하기 표 1에 나타내었다.
( 1) 보수능 (CRC, Centr i fugal Retent ion Capaci ty) : EDANA 법 WSP
241.3을 기준으로 하였다. 준비된 미분 재조립체 중 입경 300 m 초과 600 pm 이하의 입경을 갖는 미분 재조립체 시료 0.2 g을 티백에 넣고 0.9 % 염수 용액에 30분간 침전한다. 이후 250G (gravi ty)의 원심력으로 3분간 탈수한 후 염수 용액이 흡수된 양 W2(g)을 측정하였다. 또 미분 재조립체를 이용하지 않고 동일한조작을 한후에 그때의 질량 Kg)을 측정했다.
이렇게 얻어진 각 질량을 이용하여 다음의 수학식 1에 따라 CRC (g/g)를산출하여 보수능을 확인하였다.
[수학식 1]
CRC(g/g) = { [W2(g) - Wi(g) ] /Wo(g) > - 1
상기 수학식 1에서,
W0(g)는 미분 재조립체의 초기 무게 (g)이고,
Wjg)는 미분 재조립체를 사용하지 않고, 원심분리기를 사용하여 250G로 3분간 탈수한후에 측정한 장치 무게이고,
¾(g)는 상온에서 0.9 중량% 생리 식염수에 미분 재조립체를 30분 동안 침수하여 흡수시킨 다음, 원심분리기를 사용하여 250G로 3분간 탈수한 후에,
2019/194399 1»(:1^1{2019/001109
미분재조립체를포함하여 측정한장치 무게이다.
(2) BPKBase Polymer Index): 준비된 미분 재조립체 중 입경 300 m 초과 600 /패 이하의 입경을 갖는 미분 재조립체 시료에 대하여 EDANA 시험법 441.2-02에 의해 원심지지용량(CRC, 단위 : g/g), EDANA 시험법 270.2에 의해 수가용성분의 함량(단위: 중량%), 및 EDANA시험법 442.2-02에 의해 0.7 ps i 의 하중 하에서 가압흡수능(AUP , 단위: g/g)을 측정하고, 하기 수학식 2에 따라 BPI를 계산하였다.
[수학식 到
CRG + 8.7585
o of _
Oi l ~~ ..
in(수가용성분의 함량)
(3) 재미분율(중량%) - 준비된 미분 재조립체를 Hammer mi l l로 분쇄하고, 분급한 후, 150/im 이하의 입자 크기를 갖는 재미분의 함량을 측정하고, 그 결과로부터 미분 재조립체 총 양에 대한 재미분의 함량비(재미분율)를 계산하였다.
【표 1]

상기 실시예 1-1 내지 1-2 및 비교예 1-1에 따른 미분 재조립체의 제조시, 교반 선속도에 따른 2차 응집 형성 시간의 변화 및 제조되는 미분
2019/194399 1»(:1^1{2019/001109
재조립체의 물성 변화를 관찰하였다.
미분 재조립체의 물성은 상기 실험예 3에서와 동일한 방법으로 측정하였으며 , 그 결과는 하기 표 2에 나타내었다. 【표 2]
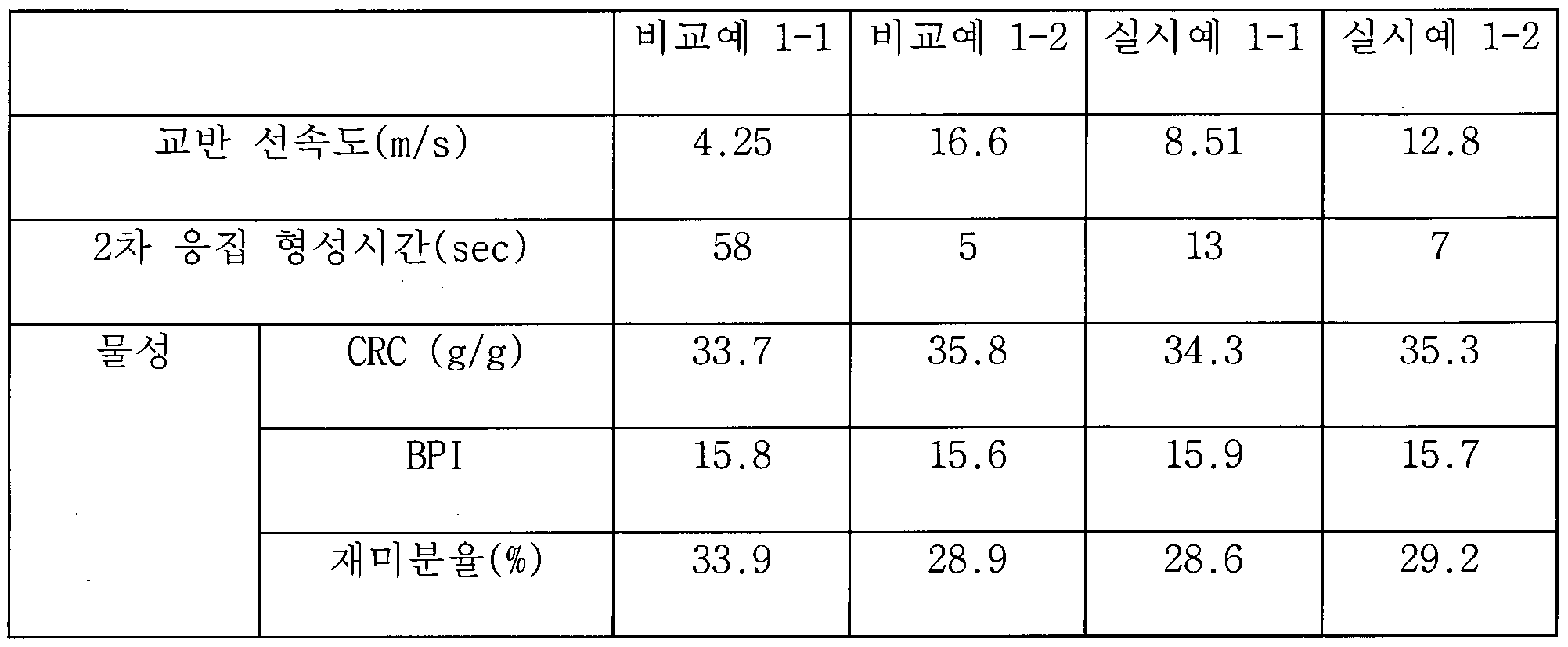
상기 실시예 1-1, 2-1, 2-3 , 2-4, 2-5 및 비교예 3-2에 따른 미분 재조립체의 제조시, 동일한 선속도 조건 하에 물의 분할 투입 및 투입량에 따른 2차 응집 형성 시간의 변화를 관찰하였다. 그 결과를 하기 표 3에 나타내었다. 【표 3】
2019/194399 1»(:1^1{2019/001109

한편, 초기 투입시에 비해 2차 투입시의 물 투입량이 더 크거나 (실시예
2-3) , 또는 3회 분할 투입한 경우 (실시예 2-4) , 실시예 2-1에 비해서는 혼합 안정성이 감소하였으나, 일괄 투입한 비교예 3-2에 비해서는 개선된 혼합 안정성을 나타내었다.
이로부터 미분과 물의 혼합시 물을 일괄 투입하는 것 보다는 분할 투입을 하는 것이, 그리고 2회 분할 투입시 1차투입시 물의 투입량이 더 높은 경우가 안정성 면에서 보다유리함을 알수 있다. 실험예 6
상기 실시예 3-1에서 제조한 고흡수성 수지에 대해, 보수능 (CRC), 0.3AUP 및 vortex를 각각측정하였다.
( 1) 보수능 (CRC) : 상기 실험예 3에서와동일한 방법으로 수행하였다.
2019/194399 1»(:1^1{2019/001109
(2) 0.3
 가압흡수능( !5): 표 법 ¾吸^ 242.3에 따라 측정하였다. 구체적으로, 내경 60 1페1의 플라스틱의 원통 바닥에 스테인레스제 400 016311 철망을 장착시켰다. 상온 및 습도 50%의 조건 하에서 철망상에 고흡수성 수지 (§) (0.90 을 균일하게 살포하고, 그 위에 0.3
가압흡수능( !5): 표 법 ¾吸^ 242.3에 따라 측정하였다. 구체적으로, 내경 60 1페1의 플라스틱의 원통 바닥에 스테인레스제 400 016311 철망을 장착시켰다. 상온 및 습도 50%의 조건 하에서 철망상에 고흡수성 수지 (§) (0.90 을 균일하게 살포하고, 그 위에 0.3
 하중을 균일하게 더 부여할수 있는 피스톤은 외경 60 1ä보다 약간 작고 원통의 내벽과툼이 없고 상하 움직임이 방해받지 않게 하였다. 이때 상기 장치의 중량 ¾(용)을 측정하였다.
하중을 균일하게 더 부여할수 있는 피스톤은 외경 60 1ä보다 약간 작고 원통의 내벽과툼이 없고 상하 움직임이 방해받지 않게 하였다. 이때 상기 장치의 중량 ¾(용)을 측정하였다.
직경 150 _의 페트로 접시의 내측에 직경 90ä 및 두께
 유리 필터를 두고, 0.9 중량% 염화나트륨으로 구성된 생리식염수를 유리 필터의 윗면과 동일 레벨이 되도록 하였다. 그 위에 직경 90ä의 여과지 1장을 실었다. 여과지 위에 상기 측정 장치를 싣고, 액을 하중 하에서 1시간 동안 흡수시켰다. 1시간후 즉정 장치를 들어올리고, 그 중량 ?4(요)을 즉정하였다.
유리 필터를 두고, 0.9 중량% 염화나트륨으로 구성된 생리식염수를 유리 필터의 윗면과 동일 레벨이 되도록 하였다. 그 위에 직경 90ä의 여과지 1장을 실었다. 여과지 위에 상기 측정 장치를 싣고, 액을 하중 하에서 1시간 동안 흡수시켰다. 1시간후 즉정 장치를 들어올리고, 그 중량 ?4(요)을 즉정하였다.
얻어진 각 질량을 이용하여 다음 수학식 3에 따라 가압 흡수능(£/용)을 산출하였다.
[수학식 3]
( ) = [、 ) - 13(용)]/ 0(용)
(3) 흡수 속도(\¾ 6)0
50 1111 식염수를 100 011 비커에 마그네틱 바와 함께 넣고, 교반기를 사용하여 교반 속도를 600 印미으로 지정하였다. 교반되고 있는 식염수에 상기 실시예 3-1에서 제조한 고흡수성 수지 2.0 §을 넣는 동시에 시간을 측정하였다. 비커 안에 소용돌이가 없어지는 시점에 시간측정을 종료하였다.
실험 결과, 실시예 3-1의 고흡수성 수지의 □¾:는 34.2용/§이었고, 0.3?^ 사 는 28. ¾/용이었으며, 흡수속도는 35초 이었다.
Claims
2019/194399 1»(:1^1{2019/001109
【청구범위】
【청구항 11
수용성 에틸렌계 불포화 단량체 및 중합개시제를 포함하는 모노머 조성물에 열중합또는 광중합을 진행하여 함수겔상 중합체를 제조하는 단계; 상기 함수겔상 중합체를 건조 및 분쇄하고 150 이하의 입경을 갖는 미분, 및 150 초과 850 /패 이하의 입경을 갖는 정상 입자로 분급하는 단계; 더
상기 미분을 선속도 6.5 내지 14. ¾1/3로 교반하면서 물과 혼합하고, 재조립하여 미분 재조립체를 제조하는 단계;를 포함하며,
상기 미분과 물의 혼합시, 상기 물을 미분에 대해 2회 이상 분할 투입하는, 고흡수성 수지의 제조방법.
【청구항 2]
제 1항에 있어서,
상기 미분과 물의 혼합시, 물은 2회 분할 투입되며, 투입되는 물의 총 양에 대하여 50중량%초과 내지 90중량% 이하가 1차로 투입되고, 잔부가 2차로 투입되는, 고흡수성 수지의 제조방법.
【청구항 3]
제 1항에 있어서,
상기 미분과 물의 혼합시, 물은 미분 100중량부에 대해 80 내지 150중량부로 혼합되는, 고흡수성 수지의 제조방법.
【청구항 4]
제 1항에 있어서,
상기 미분과 물의 혼합시, 수용성 고분자 및 계면활성제 중 1종 이상의 혼합 안정화제가더 첨가되는, 고흡수성 수지의 제조방법 .
【청구항 5]
제 4항에 있어서,
2019/194399 1»(:1^1{2019/001109
상기 혼합 안정화제는 미분 100중량부에 대하여 0.001 내지 1중량부로 투입되는, 고흡수성 수지의 제조방법.
【청구항 6]
제 4항에 있어서,
상기 수용성 고분자는 폴리에틸렌 글리콜, 폴리에틸렌 옥사이드, 폴리비닐피롤리돈, 폴리비닐알콜, 폴리아크릴아마이드, 폴리아크릴릭애시드, 폴리스타이렌설포닉애시드, 폴리실리식애시드 , 폴리포스포릭애시드, 폴리에틸렌설포닉애시드, 폴리비닐페놀 , 폴리비닐페닐설포닉애시드 , 폴리에틸렌포스포릭애시드, 폴리에틸렌아마이드, 폴리아민, 폴리아마이드아민, 전분, 고무 및 셀룰로오스로 이루어진 .군에서 선택되는 어느 하나 또는 둘 이상을포함하는, 고흡수성 수지의 제조방법 .
【청구항 7]
제 4항에 있어서,
상기 계면활성제는 소듐 도데실 설페이트, 다이아이소옥틸 소듐 설포숙시네이트염, 소듐 테트라데실 설페이트, 소듐 핵사데실 설페이트, 소듐 도데실벤젠 설페이트, 크실렌 설포네이트, 소듐 올레이트, 4-11- 데실벤젠설포네이트 , 소둠 라우레이트 , 4 -도데실벤젠설폰산, 도데실아민 하이드로클로라이드 , 도데실트리메틸암모늄 클로라이드, 4-11-옥틸벤젠 설포네이트, 에톡시화 설포네이트, 데실벤젠 설포네이트, 포타슘 올레이트, 11- 데실벤젠 설포네이트, 알킬트라메틸암모늄 브로마이드, 도데실 아민 , 테트라데실트리메틸암모늄 클로라이드 , 도데실 폴리사카라이드 글리코시드, 사이클로덱스트린, 글리코리피드, 리포프로테인-리포펩타이드, 포스포리피드, 파라-톨루엔 설폰산, 및 트리실옥세인으로 이루어진 군에서 선택되는 어느 하나또는 둘 이상을 포함하는, 고흡수성 수지의 제조방법 .
【청구항 8]
제 4항에 있어서,
상기 혼합 안정화제는 폴리에틸렌 글리콜 및 소둠 도데실 설페이트 중
2019/194399 1»(:1^1{2019/001109
적어도 1종을 포함하는, 고흡수성 수지의 제조방법 .
【청구항 9]
제 1항에 있어서,
상기 미분 재조립체를 상기 단계 1에서 제조한 함수겔상 중합체와 혼합 후, 건조 및 분쇄하여 150_ 이하의 입경을 갖는 재조립체 미분, 및 150 /페 초과 850_ 이하의 입경을 갖는 재조립체 정상 입자로 분급하는 단계;를 더 포함하는 고흡수성 수지의 제조방법 . 【청구항 10】
제 9항에 있어서,
상기 재조립체 정상 입자를 표면 가교제와 혼합하여 표면 가교하는 단계를 더 포함하는 고흡수성 수지의 제조방법 .
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019548022A JP7008718B2 (ja) | 2018-04-03 | 2019-01-25 | 高吸水性樹脂の製造方法 |
| US16/485,607 US11161942B2 (en) | 2018-04-03 | 2019-01-25 | Method for preparing superabsorbent polymer |
| EP19756315.8A EP3730539B1 (en) | 2018-04-03 | 2019-01-25 | Method for preparing superabsorbent polymer |
| CN201980001318.2A CN110557949B (zh) | 2018-04-03 | 2019-01-25 | 用于制备超吸收性聚合物的方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020180038632A KR102555381B1 (ko) | 2018-04-03 | 2018-04-03 | 고흡수성 수지의 제조방법 |
| KR10-2018-0038632 | 2018-04-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019194399A1 true WO2019194399A1 (ko) | 2019-10-10 |
Family
ID=68101059
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2019/001109 Ceased WO2019194399A1 (ko) | 2018-04-03 | 2019-01-25 | 고흡수성 수지의 제조방법 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11161942B2 (ko) |
| EP (1) | EP3730539B1 (ko) |
| JP (1) | JP7008718B2 (ko) |
| KR (1) | KR102555381B1 (ko) |
| CN (1) | CN110557949B (ko) |
| WO (1) | WO2019194399A1 (ko) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102566284B1 (ko) * | 2018-11-14 | 2023-08-10 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102417829B1 (ko) | 2018-12-12 | 2022-07-06 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| JP7793530B2 (ja) * | 2020-10-06 | 2026-01-05 | 住友精化株式会社 | 吸水性樹脂粒子の製造方法 |
| JP7794754B2 (ja) * | 2020-10-06 | 2026-01-06 | 住友精化株式会社 | 吸水性樹脂粒子の製造方法 |
| KR102909959B1 (ko) * | 2020-11-17 | 2026-01-07 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR102883895B1 (ko) * | 2020-11-27 | 2025-11-10 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조 방법 |
| KR20230097688A (ko) * | 2021-12-24 | 2023-07-03 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR20240056335A (ko) * | 2022-10-21 | 2024-04-30 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| CN116942882B (zh) * | 2023-07-27 | 2024-03-26 | 广东美登新材料科技有限公司 | 一种超薄芯体纸尿裤及其制备方法 |
| WO2026070548A1 (ja) * | 2024-09-30 | 2026-04-02 | 住友精化株式会社 | 吸水性樹脂粒子の製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20100126372A (ko) * | 2008-03-14 | 2010-12-01 | 코니카 미놀타 비지니스 테크놀로지즈 가부시키가이샤 | 관형 플로우 반응 장치, 고분자 수지 미립자의 제조 방법 |
| WO2014155419A1 (ja) * | 2013-03-29 | 2014-10-02 | パナソニック株式会社 | 導電性高分子微粒子分散体の製造方法およびその導電性高分子微粒子分散体を用いた電解コンデンサの製造方法 |
| KR20150068322A (ko) * | 2013-12-11 | 2015-06-19 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조방법 |
| KR20170106154A (ko) * | 2016-03-11 | 2017-09-20 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법, 및 고흡수성 수지 |
| KR20180003815A (ko) * | 2016-07-01 | 2018-01-10 | 한화케미칼 주식회사 | 고흡수성 수지 제조 방법 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5002986A (en) | 1989-02-28 | 1991-03-26 | Hoechst Celanese Corporation | Fluid absorbent compositions and process for their preparation |
| JP3979724B2 (ja) * | 1997-06-18 | 2007-09-19 | 株式会社日本触媒 | 吸水性樹脂造粒物の乾燥体の製造方法 |
| KR20070104663A (ko) * | 2005-02-15 | 2007-10-26 | 니폰 쇼쿠바이 컴파니 리미티드 | 수 흡수제, 수 흡수제품 및 수 흡수제의 제조방법 |
| US8648150B2 (en) | 2009-03-04 | 2014-02-11 | Nippon Shokubai Co., Ltd. | Method for producing water absorbent resin |
| JP5631866B2 (ja) | 2009-03-31 | 2014-11-26 | 株式会社日本触媒 | 粒子状吸水性樹脂の製造方法 |
| CN102666670B (zh) | 2009-10-09 | 2014-02-19 | 巴斯夫欧洲公司 | 再润湿表面后交联的吸水性聚合物颗粒的方法 |
| KR101559081B1 (ko) * | 2012-11-15 | 2015-10-08 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 이로부터 제조되는 고흡수성 수지 |
| KR101632058B1 (ko) | 2013-06-14 | 2016-06-20 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| KR20150061270A (ko) | 2013-11-27 | 2015-06-04 | 주식회사 엘지화학 | 고흡수성 수지 및 그 제조 방법 |
| JP6722507B2 (ja) | 2015-05-14 | 2020-07-15 | 株式会社日本触媒 | 吸水性樹脂の製造方法 |
| KR101848470B1 (ko) | 2015-07-10 | 2018-04-12 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 및 이로부터 제조된 고흡수성 수지 |
| KR101960043B1 (ko) * | 2015-11-03 | 2019-07-04 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
-
2018
- 2018-04-03 KR KR1020180038632A patent/KR102555381B1/ko active Active
-
2019
- 2019-01-25 CN CN201980001318.2A patent/CN110557949B/zh active Active
- 2019-01-25 US US16/485,607 patent/US11161942B2/en active Active
- 2019-01-25 EP EP19756315.8A patent/EP3730539B1/en active Active
- 2019-01-25 JP JP2019548022A patent/JP7008718B2/ja active Active
- 2019-01-25 WO PCT/KR2019/001109 patent/WO2019194399A1/ko not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20100126372A (ko) * | 2008-03-14 | 2010-12-01 | 코니카 미놀타 비지니스 테크놀로지즈 가부시키가이샤 | 관형 플로우 반응 장치, 고분자 수지 미립자의 제조 방법 |
| WO2014155419A1 (ja) * | 2013-03-29 | 2014-10-02 | パナソニック株式会社 | 導電性高分子微粒子分散体の製造方法およびその導電性高分子微粒子分散体を用いた電解コンデンサの製造方法 |
| KR20150068322A (ko) * | 2013-12-11 | 2015-06-19 | 주식회사 엘지화학 | 고흡수성 수지 및 이의 제조방법 |
| KR20170106154A (ko) * | 2016-03-11 | 2017-09-20 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법, 및 고흡수성 수지 |
| KR20180003815A (ko) * | 2016-07-01 | 2018-01-10 | 한화케미칼 주식회사 | 고흡수성 수지 제조 방법 |
Non-Patent Citations (3)
| Title |
|---|
| "Principle of Polymerization", 1981, WILEY, pages: 203 |
| REINHOLD SCHWALM: "Recent Developments and New Application", 2007, ELSEVIER, article "UV Coatings: Basics", pages: 115 |
| See also references of EP3730539A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102555381B1 (ko) | 2023-07-12 |
| CN110557949B (zh) | 2022-06-24 |
| KR20190115661A (ko) | 2019-10-14 |
| CN110557949A (zh) | 2019-12-10 |
| JP2020520390A (ja) | 2020-07-09 |
| EP3730539A1 (en) | 2020-10-28 |
| JP7008718B2 (ja) | 2022-01-25 |
| US11161942B2 (en) | 2021-11-02 |
| EP3730539B1 (en) | 2023-07-05 |
| EP3730539A4 (en) | 2021-04-21 |
| US20200247960A1 (en) | 2020-08-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019194399A1 (ko) | 고흡수성 수지의 제조방법 | |
| US9937482B2 (en) | Super absorbent polymer resin and method for preparing same | |
| US11492451B2 (en) | Manufacturing method of super absorbent polymer and super absorbent polymer | |
| KR102555379B1 (ko) | 고흡수성 수지의 제조 방법 | |
| US9701763B2 (en) | Method for preparing super absorbent polymer | |
| CN107207745B (zh) | 超吸收性聚合物的制备方法 | |
| US9433921B2 (en) | Method for preparing a super absorbent polymer | |
| KR101584719B1 (ko) | 고흡수성 수지의 제조 방법 | |
| KR102518937B1 (ko) | 고흡수성 수지의 제조 방법 | |
| US10711074B2 (en) | Super absorbent polymer and method for preparing same | |
| JP7039108B2 (ja) | 高吸水性樹脂の製造方法、および高吸水性樹脂 | |
| EP3159359B2 (en) | Method for producing a super absorbent polymer containing water-soluble salt | |
| KR20140036866A (ko) | 고흡수성 수지의 제조 방법 | |
| KR20120049004A (ko) | 고흡수성 수지의 제조 방법 | |
| US12023646B2 (en) | Preparation method of super absorbent polymer | |
| KR20150141424A (ko) | 고흡수성 수지의 제조용 냉각기 | |
| WO2018135928A9 (ko) | 고흡수성 수지 및 이의 제조 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2019548022 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |