WO2020035880A1 - Inhibitors to target hiv-1 nef-cd80/cd86 interactions for therapeutic intervention - Google Patents
Inhibitors to target hiv-1 nef-cd80/cd86 interactions for therapeutic intervention Download PDFInfo
- Publication number
- WO2020035880A1 WO2020035880A1 PCT/IN2019/050594 IN2019050594W WO2020035880A1 WO 2020035880 A1 WO2020035880 A1 WO 2020035880A1 IN 2019050594 W IN2019050594 W IN 2019050594W WO 2020035880 A1 WO2020035880 A1 WO 2020035880A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- independently selected
- heterocyclyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- RUXXPHXYDKYWRL-LSHDLFTRSA-N COc(cc1)ccc1-c1cnc(N/N=C/c(cc2)ccc2F)nc1 Chemical compound COc(cc1)ccc1-c1cnc(N/N=C/c(cc2)ccc2F)nc1 RUXXPHXYDKYWRL-LSHDLFTRSA-N 0.000 description 1
- WYDVFOMCPMHKSM-UHFFFAOYSA-N COc(cc1)ccc1OC(N(c1ccccc1)c1ccccc1)=O Chemical compound COc(cc1)ccc1OC(N(c1ccccc1)c1ccccc1)=O WYDVFOMCPMHKSM-UHFFFAOYSA-N 0.000 description 1
- ZOOIAOPIDPIFMM-ZVBGSRNCSA-N [O-][N+](c(cccc1)c1-c1cnc(N/N=C/c(cc2)ccc2Br)nc1)=O Chemical compound [O-][N+](c(cccc1)c1-c1cnc(N/N=C/c(cc2)ccc2Br)nc1)=O ZOOIAOPIDPIFMM-ZVBGSRNCSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/17—Amides, e.g. hydroxamic acids having the group >N—C(O)—N< or >N—C(S)—N<, e.g. urea, thiourea, carmustine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/235—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/215—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids
- A61K31/235—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group
- A61K31/24—Esters, e.g. nitroglycerine, selenocyanates of carboxylic acids having an aromatic ring attached to a carboxyl group having an amino or nitro group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/421—1,3-Oxazoles, e.g. pemoline, trimethadione
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- the present disclosure relates to prevention/ treatment of immune evasion by human immunodeficiency virus (HIV) related infections.
- HIV human immunodeficiency virus
- the compounds of the present disclosure are useful as medicaments and their use in the manufacture of medicaments for treatment, prevention or suppression of diseases, and conditions mediated by HIV.
- HAART highly active antiretroviral therapy
- the anti-retro viral drugs have different mechanism of action, such as nucleoside/nucleotide reverse transcriptase inhibitors, non- nucleoside retroviral treatment, maturation and cellular inhibitors, integrase inhibitors, protease inhibitors and immune-based therapy.
- nucleoside/nucleotide reverse transcriptase inhibitors such as nucleoside/nucleotide reverse transcriptase inhibitors, non- nucleoside retroviral treatment, maturation and cellular inhibitors, integrase inhibitors, protease inhibitors and immune-based therapy.
- Nef protein, a HIV-l auxiliary protein, and inhibition of its interaction and complexes comprising Nef, with other cellular components have been studied in detail. Nef interaction with Src protein-tyrosine kinase family (series of signaling molecules) is required for the onset and progression of AIDS in HIV-l -infected persons. (Lugari et. al, Bioorganic & Medicinal Chemistry 19 (2011) 7401-7406; Emert-Sedlak et ah, ACS Chem Biol. 2009 4(11), 939-947; US 8541415). [0004] Interestingly, the virus uses the Nef protein to evade killing of its host cell by cytotoxic T-lymphocytes. Inhibiting /Ve/-mediated functions thus presents a different strategy for increasing CTL activity against HIV infected cells by making the latter more visible to the immune system.
- the present disclosure relates to compounds of Formula I, II, and III that restore immune activation in case of infections.
- the present disclosure specifically relates to a method for the prevention or treatment of an HIV infection or a disease associated with an HIV infection in a subject in need thereof, comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and amino, wherein C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and C 6 -io aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy, and C 2-10 heterocyclyl;
- X and Y are N;
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C2 10 heterocyclyl, C 5- 10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 1-10 alkyl, C 5- 10 aryl or C2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5;
- R 1 is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R 4 is independently selected from hydrogen or C 1-10 alkyl
- R 3 is selected from C 1-10 alkyl or halogen; and n is selected from 1 to 5.
- the present disclosure further relates to a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for prophylaxis and/or treatment of an HIV infection or a disease associated with an HIV infection
- the present disclosure further relates to use of a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, towards restoration of immune signaling via T cell activation through inhibiting Nef- CD80/86 interactions.
- the present disclosure further relates to a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in treating a disease or condition in a patient wherein said disease or condition is caused by cancers including chronic lymphocytic leukemia, colon carcinoma, multiple myeloma or viral infections including HIV, HPV, herpes and the like.
- the present disclosure further relates to use of a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in treating disease or condition in a subject, wherein said disease or condition is caused by HIV.
- the subject may be a mammal including humans.
- Figure 1 illustrates the biochemical screening of the compounds in accordance with an implementation of the present disclosure.
- Figure 2 illustrates the schematic representation of the cell-based assay, in accordance with an implementation of the present disclosure.
- Figure 3 illustrates the inhibition of Nef mediated internalization of surface
- CD80/86 as measured by flow cytometry by compounds from Formula I, II and III in accordance with an implementation of the present disclosure.
- Figure 4 illustrates the schematic representation of the cell-based T cell activation assay in accordance with an implementation of the present disclosure.
- Figure 5 illustrates the inhibition of Nef mediated T cell inactivation in an
- APC in accordance with an implementation of the present disclosure.
- the articles“a”,“an” and“the” are used to refer to one or to more than one (i.e., to at least one) of the grammatical object of the article.
- the term“therapeutic dose” refers to providing any compound of the present disclosure (drug) in a dose per unit time over an extended time to a subject in need thereof.
- the therapeutic dose according to the present disclosure may be in a range of 50 mM to 1000 pM.
- the term“relevant dose” refers to providing any compound of the present disclosure (drug) in a dose per unit time over an extended time to a subject for preventing HIV.
- low levels of CD80/86 receptors can be defined as any condition which leads to a decrease in levels of CD80/86 receptors on the T cells.
- alkyl refers to a monoradical branched or unbranched saturated hydrocarbon chain having from 1 to 10 carbon atoms. This term is exemplified by groups such as n-butyl, iso-butyl, t-butyl, n-hexyl, n-decyl, and the like. The groups may be optionally substituted.
- alkenyl refers to a monoradical of a branched or unbranched unsaturated hydrocarbon group preferably having from 4, 5, 6, 7, 8, 9, or 10 carbon atoms and having 1, 2, 3, 4, 5 or 6 double bond (vinyl), preferably 1 double bond. The groups may be optionally substituted.
- alkynyl refers to a monoradical of an unsaturated hydrocarbon, preferably having from 4, 5, 6, 7, 8, 9, or lOcarbon atoms and having 1, 2, 3, 4, 5 or 6 sites of acetylene (triple bond) unsaturation, preferably 1 triple bond.
- the groups may be optionally substituted.
- Halo or“Halogen”, alone or in combination with any other term means halogens such as chloro (Cl), fluoro (F), bromo (Br) and iodo (I).
- aryl refers to an aromatic carbocyclic group of 5 to 18 carbon atoms having a single ring (e.g. phenyl) or multiple rings (e.g. biphenyl), or multiple condensed (fused) rings (e.g. naphthyl or anthranyl).
- Preferred aryls include phenyl, naphthyl and the like.
- the groups may be optionally substituted.
- heteroaryl refers to a heteroaromatic carbocyclic group of 2 to 10 carbon atoms having a single ring or multiple rings, or multiple condensed rings.
- Preferred heteroaryls include pyrazole, thiazole,oxazole, benzoxazole, pyridine and the like.
- the groups may be optionally substituted.
- cycloalkyl refers to carbocyclic groups of from 3 to 12 carbon atoms having a single cyclic ring or multiple condensed rings, which may be partially unsaturated.
- Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclohexyl, cyclohexenyl, and the like, or multiple ring structures or carbocyclic groups to which is fused an aryl group.
- the groups may be optionally substituted.
- heterocyclyl refers to a saturated or partially unsaturated group or unsaturated group having a single ring or multiple condensed rings, having from 2 to 10 carbon atoms and from 1 to 10 hetero atoms, preferably 1, 2, or 3 heteroatoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen within the ring.
- Preferred heterocyclyls include morpholine, piperidine, and the like. The groups may be optionally substituted.
- substituted is contemplated to include all permissible substituents of organic compounds.
- permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds.
- the term "effective amount” means an amount of a compound or composition, which is sufficient enough to significantly and positively modify the symptoms and/or conditions to be treated.
- the effective amount will vary depending on various conditions and is within the knowledge and expertise of the attending physician.
- the compounds described herein may contain one or more chiral centers and/or double bonds and therefore, may exist as stereoisomers. Accordingly, the chemical structures depicted herein encompass all possible enantiomers and stereoisomers of the illustrated or identified compounds including the stereoisomerically pure form and enantiomeric and stereoisomeric mixtures. The compounds may also exist in several tautomeric forms including the enol form, the keto form and mixtures thereof. Accordingly, the chemical structures depicted herein encompass all possible tautomeric forms of the illustrated or identified compounds.
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms, which are suitable for use in contact with the tissues of human beings and animals and is understood by a person skilled in the art.
- “Pharmaceutically acceptable salt” embraces salts with a pharmaceutically acceptable acid or base.
- Pharmaceutically acceptable acids include both inorganic acids, and organic bases.
- polymorphs refers to crystal forms of the same molecule, and different polymorphs may have different physical properties.
- solvate refers to a crystal form of a substance, which contains solvent.
- hydrate refers to a solvate wherein the solvent is water.
- Ratios, concentrations, amounts, and other numerical data may be presented herein in a range format. It is to be understood that such range format is used merely for convenience and brevity and should be interpreted flexibly to include not only the numerical values explicitly recited as the limits of the range, but also to include all the individual numerical values or sub-ranges encompassed within that range as if each numerical value and sub range is explicitly recited.
- a concentration range of about 50 to 1000 m M should be interpreted to include not only the explicitly recited limits of about 50 mM to about 1000 mM, but also to include sub-ranges, such as 75-875 mM, 80-500 mM, and so forth, as well as individual amounts, including fractional amounts, within the specified ranges, such as 100 mM, and 155 mM, for example.
- the present disclosure relates to compounds for treatment of HIV infection.
- the target is novel interaction of Nef protein from the HIV-l virus with the host immune receptors CD80/CD86 responsible for T cell stimulation.
- Nef protein An indispensable factor for HIV pathogenicity is the Nef protein. It allows HIV to evade the immune response, maintain high viral load and is needed for replication and dissemination, down-regulate cell surface receptors involved in the generation of immune response, including MHC-l and MHC-2. It has been surprising found that in addition to relocation of MHC, Nef also down-regulates surface expression of the B-7 family of co-stimulatory proteins, namely CD80 and CD86 expressed on antigen presenting cells, leading to impaired T cell stimulation in vitro. Nef binds directly to the cytoplasmic tails of CD80 and CD86 and prone to reversal by introduction of peptides, making it a suitable target for therapeutic intervention.
- Targeting /Ve/-CD80/86 interactions has a unique mechanism of action: prevention of immune evasion of infected cells.
- the disclosure revolves around the inhibition of /Ve/-mediated functions, which is a distinct strategy since it targets a key function of Nef in its ability to directly modulate infected immune cell (macrophage) capacity to generate HIV -specific T cells from naive T cell, potentially bringing forth a different class of drugs.
- the disclosure presents a new line of antiviral therapy through immune signaling restoration.
- the host-virus interface is also less amenable to drug resistance than purely viral targets.
- the present disclosure discloses a method for preventing/ treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I. Formula II, and Formula III as described herein, wherein the therapeutic dose of the compound is in a range of 50 to 1000 m M.
- the present disclosure is intended to cover all the sub-ranges and individual values.
- the range can be 50 to 100 mM, or 100 to 1000 mM, or 150 to 900 mM, or 125 to 500 mM, or 500 to 900 mM, or 750 to 950 mM, or 75 to 150 mM, or 100 to 200 mM.
- the present disclosure provides a method for preventing/ treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and amino, wherein C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and C 5-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy, and C 2-10 heterocyclyl;
- R 3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl, and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2- 10 heterocyclyl;
- X and Y are N;
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C2-10 heterocyclyl, C 5- 10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- Ar is selected from C 1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
- R3 is absent or is selected from hydroxy, C 1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5;
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R4 is independently selected from hydrogen or C 1-10 alkyl
- R 3 is selected from C 1-10 alkyl or halogen
- n is selected from 1 to 5.
- the therapeutic dose of the compound is in a range of 50 to 1000 m M. In yet another embodiment of the present disclosure, the therapeutic dose of the compound is in a range of 50 to 100 mM. In an alternate embodiment of the present disclosure, the therapeutic dose of the compound is in a range of 100 to 1000 mM. In one another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 150 to 900 mM. In an alternate embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 125 to 500 mM. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 500 to 900 mM.
- the therapeutic dose of the compound is in the range of 750 to 950 m M. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 75 to 150 m M. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 100 to 200 mM.
- the present disclosure provides a method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, and amino, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy, and C 2-10 heterocyclyl;
- R 3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and Ci- io alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-i o heterocyclyl;
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, and C 1-10 alkyl, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, and C 1-10 alkoxy;
- R3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-i o heterocyclyl;
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 aryl, and Ci-5 alkyl, wherein C1-5 alkyl, and C 6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C 1-5 alkyl or C 1-5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from C 1-5 alkoxy, nitro, halogen or C 1-5 alkyl, wherein C 1-5 alkyl and C 1-5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-5 alkyl, C 1-5 alkoxy or C 2 10 heterocyclyl;
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 aryl, and CF 3 , wherein C 6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, C 1-5 alkyl or C 1-5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from -OCH 3 , -OCF 3 , nitro, fluorine, CF 3 ;
- the present disclosure provides a method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and amino, wherein C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl or C 1-10 alkoxy, C 2-10 heterocyclyl;
- R3 is C 6 -io aryl, wherein C 6 -io aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- the present disclosure provides a compound of Formula I or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:
- the present disclosure provides a compound of Formula I or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:
- the present disclosure provides a method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-i o heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-i o heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Riand R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein Ci-10 alkyl, C2 10 heterocyclyl, C 5- 10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-i o aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 5-i oaryl or C 2 i oheteroaryl
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is C 5-10 aryl
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano,
- n is selected from 0-5 .
- the present disclosure provides a compound of Formula II or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R 4 is independently selected from hydrogen or C 1-10 alkyl
- R 3 is selected from C 1-10 alkyl or halogen
- n is selected from 1 to 5.
- the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci- 5 alkyl;
- R 2 and R4 is independently selected from hydrogen or C 1-5 alkyl
- R3 is selected from Ci- 5 alkyl or chlorine; and n is selected from 1 to 2.
- the present disclosure provides a compound of Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 -io aryl, C 1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C 1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- R 3 is C 6 -io aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy, C 2 10 heterocyclyl;
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5- 10 aryl, C 1-10 alkoxy, halogen,
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, and
- n is selected from 0-5;
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R 4 is independently selected from hydrogen or C 1-10 alkyl
- R 3 is selected from C 1-10 alkyl or halogen
- n is selected froml to 5.
- the therapeutic dose of the compound is in the range of 50 to 1000 m M.
- the therapeutic dose of the compound is in the range of 50 to 100 m M.
- the therapeutic dose of the compound is in the range of 100 to 1000 mM.
- the therapeutic dose of the compound is in the range of 150 to 900 mM.
- the therapeutic dose of the compound is in the range of 125 to 500 mM.
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, and amino, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C2-10 heterocyclyl;
- R3 is C 6 -io aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, and C 1-10 alkyl, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl or C 1-10 alkoxy;
- R 3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen, C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-i o heterocyclyl;
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 aryl, and Ci- 5 alkyl, wherein C 1-5 alkyl, and C 6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C 1-5 alkyl or C 1-5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from Ci- 5 alkoxy, nitro, halogen, C 1-5 alkyl, wherein C 1-5 alkyl and C 1-5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-5 alkyl, C 1-5 alkoxy or C 2- 10 heterocyclyl;
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 aryl, and CF 3 , wherein C 6 aryl is optionally substituted with one to four substituents independently selected from hydroxyl, C1-5 alkyl or C 1 -5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from -OCH3, -OCF3, nitro, fluorine or CF3;
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C 1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- R3 is C 6 -io aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro or C 1-10 alkyl, wherein C 1-10 alkyl and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2 10 heterocyclyl;
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5- 10 aryl, C2 10 heteroaryl, C2 10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CPhPhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-i o heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5- 10 aryl, C 1-10 alkoxy, hal
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 5- 10 aryl or C2-10 heteroaryl
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C2-10 heterocyclyl, C 5- 10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-i o aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH
- R3 is absent or is selected from hydroxy, C 1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5 .
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci-io alkyl;
- R 2 and R 4 is independently selected from hydrogen or Ci-io alkyl
- R3 is selected from Ci-io alkyl or halogen; and n is selected from 1 to 5.
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-5 alkyl;
- R 2 and R 4 is independently selected from hydrogen or C 1-5 alkyl
- R 3 is selected from C 1-5 alkyl or chlorine; and n is selected from 1 to 2.
- the present disclosure provides a method for treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-i o alkenyl, C 2-i o alkynyl, and amino, wherein C 1-10 alkyl, C 2-i o alkenyl, C 2-i o alkynyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C 2-i o heterocyclyl;
- R 3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl, and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2 10 heterocyclyl
- X and Y are N;
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C 5- 10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5;
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R 4 is independently selected from hydrogen or C 1-10 alkyl
- R 3 is selected from C 1-10 alkyl or halogen
- n is selected from 1 to 5.
- the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, and amino, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C 2-i o heterocyclyl;
- R3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, and C 1-10 alkyl, wherein C 1-10 alkyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl or C 1-10 alkoxy;
- R 3 is C 6 -io aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R2 are independently selected from the group consisting of hydrogen, C 6 aryl, and Ci- 5 alkyl, wherein C 1-5 alkyl, and C 6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C 1-5 alkyl or C 1-5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from Ci- 5 alkoxy, nitro, halogen or Ci- 5 alkyl, wherein Ci- 5 alkyl and Ci- 5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, Ci- 5 alkyl, Ci- 5 alkoxy or C 2 10 heterocyclyl;
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6 aryl, and CF 3 , wherein C 6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, C 1-5 alkyl or C 1-5 alkoxy;
- R 3 is C 6 aryl, wherein C 6 aryl is optionally substituted with one to four substituents independently selected from -OCH 3 , -OCF 3 , nitro, fluorine or CF 3 ;
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-i o alkenyl, C 2-10 alkynyl, and amino, wherein C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and C 6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- R3 is C 6 -io aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro or C 1-10 alkyl, wherein C 1-10 alkyl and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2-10 heterocyclyl;
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and n is selected from 0-5 .
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and n is selected from 0-5 .
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is O
- Ar is selected from C 5-10 aryl or C 2-10 heteroaryl
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and n is selected from 0-5 .
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein Ci-10 alkyl, C2 10 heterocyclyl, C5-10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, Ci-10 alkyl, C5-10 aryl, Ci-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2 -Ph
- X is O
- Ar is C 5-10 aryl
- R 3 is absent or is selected from hydroxy, Ci-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, Ci-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and n is selected from 0-5 .
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by qf CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
- R 2 and R4 is independently selected from hydrogen or C1-10 alkyl
- R3 is selected from C1-10 alkyl or halogen; and n is selected from 1 to 5.
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula III
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci -5 alkyl;
- R 2 and R4 is independently selected from hydrogen or C1-5 alkyl
- R3 is selected from C1-5 alkyl or chlorine; and n is selected from 1 to 2.
- the present disclosure provides a compound of
- Formula I, II, or III or its or its stereoisomers pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, which is selected from a group consisting of 2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-(4-nitrophenyl)pyrimidine (Iia),
- the compounds for Formula I, II, or III causes restoration of immune signaling via T cell activation through inhibiting M?/-CD80/86 interactions.
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula II, wherein compound of Formula II is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II, wherein compound of Formula II is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula III, wherein compound of Formula III is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula III, wherein compound of Formula III is selected from a group consisting of:
- the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,
- Ri and R 2 are independently selected from the group consisting of hydrogen, C 6-10 aryl, C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and amino, wherein C 1-10 alkyl, C 2-10 alkenyl, C 2-10 alkynyl, and C 5-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C 1-10 alkyl, C 1-10 alkoxy, and C 2-10 heterocyclyl;
- R3 is C 6-10 aryl, wherein C 6-10 aryl is optionally substituted with one to four substituents independently selected from C 1-10 alkoxy, nitro, halogen or C 1-10 alkyl, wherein C 1-10 alkyl, and C 1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C 1-10 alkyl, C 1-10 alkoxy or C 2- 10 heterocyclyl;
- X and Y are N;
- Ri, and R 2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R 2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C 1-10 alkyl, C 2-10 heterocyclyl, C 5-10 aryl, C 2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH 2
- X is selected from O;
- Ar is selected from C 1-10 alkyl, C 5-10 aryl or C 2-10 heteroaryl;
- R 3 is absent or is selected from hydroxy, C 1-10 alkyl, C 5-10 aryl, C 2-10 heteroaryl, C 2-10 heterocyclyl, C 1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH 2 PhNHCOOPh-Me, NHCONHPh-(Cl) 2 or CONHNHS0 2 Ph-Me; and
- n is selected from 0-5;
- Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-10 alkyl;
- R 2 and R4 is independently selected from hydrogen or C 1-10 alkyl
- R3 is selected from C 1-10 alkyl or halogen
- n is selected from 1 to 5.
- the relevant dose is in the range of 50 to 1000 mM.
- the present disclosure provides a method of treating or preventing or treating HIV infection in a subject by immune evasion as described herein, wherein the compounds causes restoration of immune signaling via T cell activation through inhibiting Nef- CD80/86 interactions.
- the present disclosure provides a method for treating/preventing infections which have low levels of CD80/86 receptors compared to non-infected state, selected from the including chronic lymphocytic leukemia, colon carcinoma, multiple myeloma, viral infections including HIV, HPV, herpes.
- the present disclosure relates to a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (I 3 b), naphthalen-2-yl (2-fluorophenyl)carbamate (II3), and 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilka).
- the relevant dose is in the range of 50 to 1000 m M.
- the present disclosure relates to a method for preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4- (5-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (kb), naphthalen-2-yl (2- fluorophenyl)carbamate (Ik), 4-[2-(3,5-dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilka) and combinations thereof.
- the therapeutic dose is in the range of 50 to 1000 mM.
- the compound is a combination of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2- yl)morpholine (13b), naphthalen-2-yl (2-fluorophenyl)carbamate (II 3 ), 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilha).
- the present disclosure relates to method for treating
- HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4-(5-(4- (trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (hb), naphthalen-2-yl (2- fluorophenyljcarbamate (II3), and 4-[2-(3,5-dimethylphenoxy)acetamido]-2- hydroxybenzoic acid (IIHa).
- the relevant dose is in the range of 50 to 1000 mM.
- the present disclosure relates to method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4-(5-(4- (trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (Hb), naphthalen-2-yl (2- fluorophenyljcarbamate (II3), 4-[2-(3,5-dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (III 2 a), and combinations thereof.
- the therapeutic dose is in the range of 50 to 1000 mM.
- the compound is a combination of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2- yljmorpholine (Hb), naphthalen-2-yl (2-fluorophenyl)carbamate (II3), 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid ( 11 Ha).
- the present disclosure relates to a method for preventing or treating HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from 4-(5-(4-
- the relevant dose is in the range of 50 to 1000 mM.
- the present disclosure relates to a method for treating/preventing infections which have low levels of CD80/86 receptors compared to non-infected state, selected from the including cancers like chronic lymphocytic leukemia, colon carcinoma, multiple myeloma, viral infections including HIV, HPV, herpes using the compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof.
- cancers like chronic lymphocytic leukemia, colon carcinoma, multiple myeloma
- viral infections including HIV, HPV, herpes using the compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof.
- the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in killing or inhibiting the growth virus.
- the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in killing or inhibiting the growth of HIV.
- the present disclosure relates to use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in killing or inhibiting the growth of HIV.
- the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in treating a disease or condition in a patient wherein said disease or condition is caused by HIV.
- the present disclosure relates to use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in treating disease or condition in a patient, wherein said disease or condition is caused by HIV.
- the patient is a typically a mammal, preferably a human.
- the present disclosure relates to a method of treating a disease or condition in a patent, said method comprising administering to a patient a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, wherein said disease or condition is caused by HIV.
- the present disclosure relates to a compound of
- Formula I, Formula II, and Formula III or its stereoisomers pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for useas a medicament.
- the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for usein the preparation of medicaments for inhibiting viral growth.
- the present disclosure relates to medicaments that include a compound of Formula I, Formula II, and Formula III, or an addition salt of the compound of Formula I, Formula II, and Formula III with a pharmaceutically acceptable acid or base. These medicaments find their use in therapeutics, especially in the treatment of viral infection caused by HIV.
- the present disclosure relates to the use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in the manufacture of a medicament for the production of an antiviral effect in a warm-blooded animal such as man.
- the present disclosure relates to a method for producing an antiviral effect in a warm-blooded animal such as man, said method including administering to said animal an effective amount of a compound of Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof.
- the present disclosure relates to a compound of Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prophylaxis of viral infections in a warm-blooded animal, such as man.
- the present disclosure relates to a compound of
- Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof, for the therapeutic and prophylactic treatment of mammals including humans, in particular in treating viral infections caused by HIV, is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
- the present disclosure relates to a pharmaceutical composition
- a pharmaceutical composition including a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, and at least one pharmaceutically acceptable carrier, diluent, or excipient.
- pharmaceutically acceptable includes compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- the compounds of Formula I, Formula II, and Formula III may form stable pharmaceutically acceptable acid or base salts, and in such cases administration of a compound as a salt may be appropriate.
- the salts may be formed by conventional means.
- the compositions of the disclosure may be in a form suitable for oral use, for topical use, for administration by inhalation, for administration by insufflation or for parenteral administration.
- compositions of the present disclosure may be obtained by conventional procedures using conventional pharmaceutical excipients well known in the art.
- the compounds disclosed herein may be applied as a sole therapy or may involve, in addition to a compound of the disclosure, one or more other substances and/or treatments.
- Reagents and conditions (a) 2° amines, K 2 C0 3 , MeOH, rt, 12 h; (b) Pd(PPh 3 ) 4 , K 2 C0 3 , dioxane, 110 °C, 6h.
- the mixture was heated 180 °C on microwave for lh.
- the reaction mixture was diluted with EtOAc, filtered through celite, and washed with water.
- the combined organic layer was dried over anhydrous Na 2 S0 4 and concentrated under reduced pressure.
- the crude products were purified on a silica gel column using hexane/EtOAc to furnish compound I 4 as a white solid.
- CD80, CD86 and CD74 are coated on a microplate, while CD80 and CD86 has higher binding capacity with the Nef protein, CD74 is non-specific peptide and is a peptide negative control for Nef binding.
- the coated plates are incubated overnight at 4 °C. The plates were washed 5 times with PBS (0.02 %Tween-20) before every step. After washing, the wells were blocked with 5% blotto and incubated at RT with shaking (650 rpm). The plates were washed 5 times with PBS (0.02 % -tween20) after every step. Primary antibody mouse anti-Nef raised in rabbit dissolved in 2.5% blotto (1: 1000) were added and incubated on shaker for 1 hour at RT.
- a throughput screen was performed to pull out the actives.
- Standard quality control metrics like Z’ were used to qualify plates and Z’ scores were used to identify actives.
- the IC 50 value of the compounds were further determined to evaluate the efficacy of the test compounds.
- IC50 values of the compounds have been illustrated in Table 1. Eighteen compounds were found to show HIV inhibiting activity and increasing the expression of CD80 and CD86 proteins. Actives predominantly belong to compounds from Formula I, II and III with IC50S in nano or subnanomolar ranges were selected. Selected compounds were then screened in the cell-based assays systems.
- Table 1 pIC 5 o values of representative actives from Primary Screen
- Table 1 illustrates the efficacy of individual compounds in inhibiting the Nef mediated downregulation of CD80 and CD86 proteins. Out of the total 19 compounds of Formula I, II, and III, the compounds which were found effective in inhibiting Nef-COSO binding, in terms of higher pICso values (ICsos in nano or
- CD80 and CD86 are two major co-stimulatory cell surface proteins that are present on antigen presenting cells (APCs) and provide a co-stimulatory signal necessary for activation and survival of the naive T cells. Both these proteins get down regulated during HIV infection via a Nef mediated pathway. It has been speculated that Nef ability to reduce CD80 and CD86 surface expression in infected cells can prevent the activation of naive T cells, necessary for an efficient recognition and elimination of the target cells.
- Figure 2 illustrates that in the presence of the compounds of Formula I, Formula II or Formula III, the binding of Nef with CD80 and CD86 is inhibited thus increasing their expression. The increased expression would activate naive T cells and aid in eliminating the infection.
- monocyte / B cell lines were either delivered with Nef protein or exposed to replication deficient retrovirus and the levels of surface CD80/86 was evaluated by flow cytometry with/without compound treatment. The compounds were qualified based on the ability to reverse the down regulation of CD 80/86 caused by the Nef protein. Similar quality checks were used in the secondary screen as in primary screen. Results from representative compounds were tested in the phenotypic assay for downregulation is presented in the Figure 3. Briefly, B/ monocyte cells were preexposed to 100 m M of the 4 compounds for 2 hrs and then infected with non -replicative reterovirus containing NEF. The internalization of surface receptors were measured by flow cytometry after optimal infection (48-72 hrs). Representative compounds from each scaffold (I 3 b, II 3 and 1112 a ) showed reversal of Nef mediated internalization of CD80/86.
- FIG. 4 represents the principle behind the activation of naive T cells by the antigen presenting cells (APC), including dendritic cells, B cells, monocytes, macrophages.
- APC antigen presenting cells
- the activation is based on CD80/86 co-stimulation along with CD3 activation by major histocompatibility complex (MHC) as evidenced by an increase in IL-2 release from the activated T cells.
- MHC major histocompatibility complex
- Figure 4(B) is a schematic representation that displays the addition of Nef in the APC causes down regulation of the CD80/86 and hence loss of T cell activation, which leads to a reduction in IL-2 release.
- Figure 4(C) displays the rescue of internalization of CD80/86 back to the surface of the APC by means of inhibitiors to the Nef protein delivered to the APC cell line, which restores CD80/86 co-stimulation and hence results in IL-2 release from the T cells.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Emergency Medicine (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The compounds of Formula I, II, and III along with their stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof are described in the present disclosure. The said compounds restore immune activation in case of infections or a disease associated with an HIV infection in a subject in need thereof.
Description
INHIBITORS TO TARGET HIV-l ZVEE-CD80/CD86 INTERACTIONS FOR
THERAPEUTIC INTERVENTION
FIELD OF INVENTION [0001] The present disclosure relates to prevention/ treatment of immune evasion by human immunodeficiency virus (HIV) related infections. The compounds of the present disclosure are useful as medicaments and their use in the manufacture of medicaments for treatment, prevention or suppression of diseases, and conditions mediated by HIV.
BACKGROUND
[0002] In recent years, there has been considerable progress in the treatment of HIV- related illness through different approaches. One of them being use of highly active antiretroviral therapy (HAART), which involves the use of different kinds of anti retroviral agents that act on different stages of the HIV life cycle. The anti-retro viral drugs have different mechanism of action, such as nucleoside/nucleotide reverse transcriptase inhibitors, non- nucleoside retroviral treatment, maturation and cellular inhibitors, integrase inhibitors, protease inhibitors and immune-based therapy. Despite the rapid development of the HIV therapy, the key issue for eradication of virus reservoir composed of latently infected memory CD4+ T cells carrying integrated pro-virus remains. The resting memory CD4+ T cells can persist for months to years carrying replication competent viral genome thus posing problems for patients on HAART. The current therapy does not eradicate the provirus and needs more research to address this issue.
[0003] Nef protein, a HIV-l auxiliary protein, and inhibition of its interaction and complexes comprising Nef, with other cellular components have been studied in detail. Nef interaction with Src protein-tyrosine kinase family (series of signaling molecules) is required for the onset and progression of AIDS in HIV-l -infected persons. (Lugari et. al, Bioorganic & Medicinal Chemistry 19 (2011) 7401-7406; Emert-Sedlak et ah, ACS Chem Biol. 2009 4(11), 939-947; US 8541415).
[0004] Interestingly, the virus uses the Nef protein to evade killing of its host cell by cytotoxic T-lymphocytes. Inhibiting /Ve/-mediated functions thus presents a different strategy for increasing CTL activity against HIV infected cells by making the latter more visible to the immune system.
SUMMARY
[0005] The present disclosure relates to compounds of Formula I, II, and III that restore immune activation in case of infections. The present disclosure specifically relates to a method for the prevention or treatment of an HIV infection or a disease associated with an HIV infection in a subject in need thereof, comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-io aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy, and C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl, is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;
Formula II,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2 10 heterocyclyl, C5- 10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5- 10 aryl or C2-10 heteroaryl;
R3is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and
Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
R1 is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and n is selected from 1 to 5.
[0006] The present disclosure further relates to a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for prophylaxis and/or treatment of an HIV infection or a disease associated with an HIV infection
[0007] The present disclosure further relates to use of a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, towards restoration of immune signaling via T cell activation through inhibiting Nef- CD80/86 interactions.
[0008] The present disclosure further relates to a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof,
for use in treating a disease or condition in a patient wherein said disease or condition is caused by cancers including chronic lymphocytic leukemia, colon carcinoma, multiple myeloma or viral infections including HIV, HPV, herpes and the like.
[0009] The present disclosure further relates to use of a compound selected from the group consisting of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in treating disease or condition in a subject, wherein said disease or condition is caused by HIV. The subject may be a mammal including humans. These and other features, aspects, and advantages of the present subject matter will become better understood with reference to the following description. This summary is provided to introduce a selection of concepts in a simplified form. This summary is not intended to identify key features or essential features of the disclosure, nor is it intended to be used to limit the scope of the subject matter. BRIEF DESCRIPTION OF DRAWINGS
[00010] Figure 1 illustrates the biochemical screening of the compounds in accordance with an implementation of the present disclosure.
[00011] Figure 2 illustrates the schematic representation of the cell-based assay, in accordance with an implementation of the present disclosure.
[00012] Figure 3 illustrates the inhibition of Nef mediated internalization of surface
CD80/86 as measured by flow cytometry by compounds from Formula I, II and III in accordance with an implementation of the present disclosure.
[00013] Figure 4 illustrates the schematic representation of the cell-based T cell activation assay in accordance with an implementation of the present disclosure.
[00014] Figure 5 illustrates the inhibition of Nef mediated T cell inactivation in an
APC in accordance with an implementation of the present disclosure.
DETAILED DESCRIPTION
[00015] Those skilled in the art will be aware that the present disclosure is subject to variations and modifications other than those specifically described. It is to be
understood that the present disclosure includes all such variations and modifications. The disclosure also includes all such steps, features, compositions and compounds referred to or indicated in this specification, individually or collectively, and any and all combinations of any or more of such steps or features.
Definitions
[00016] For convenience, before further description of the present disclosure, certain terms employed in the specification, and examples are collected here. These definitions should be read in the light of the remainder of the disclosure and understood as by a person of skill in the art. The terms used herein have the meanings recognized and known to those of skill in the art, however, for convenience and completeness, particular terms and their meanings are set forth below.
[00017] The articles“a”,“an” and“the” are used to refer to one or to more than one (i.e., to at least one) of the grammatical object of the article.
[00018] The terms“comprise” and“comprising” are used in the inclusive, open sense, meaning that additional elements may be included. Throughout this specification, unless the context requires otherwise the word“comprise”, and variations, such as “comprises” and“comprising”, will be understood to imply the inclusion of a stated element or step or group of element or steps but not the exclusion of any other element or step or group of element or steps.
[00019] The term“including” is used to mean“including but not limited to”.
“Including” and“including but not limited to” are used interchangeably.
[00020] In the structural formulae given herein and throughout the present disclosure, the following terms have been indicated meaning, unless specifically stated otherwise.
[00021] The term“therapeutic dose” refers to providing any compound of the present disclosure (drug) in a dose per unit time over an extended time to a subject in need thereof. The therapeutic dose according to the present disclosure may be in a range of 50 mM to 1000 pM.
[00022] The term“relevant dose” refers to providing any compound of the present disclosure (drug) in a dose per unit time over an extended time to a subject for preventing HIV.
[00023] The term “low levels of CD80/86 receptors” can be defined as any condition which leads to a decrease in levels of CD80/86 receptors on the T cells.
[00024] The term "alkyl" refers to a monoradical branched or unbranched saturated hydrocarbon chain having from 1 to 10 carbon atoms. This term is exemplified by groups such as n-butyl, iso-butyl, t-butyl, n-hexyl, n-decyl, and the like. The groups may be optionally substituted. [00025] The term "alkenyl" refers to a monoradical of a branched or unbranched unsaturated hydrocarbon group preferably having from 4, 5, 6, 7, 8, 9, or 10 carbon atoms and having 1, 2, 3, 4, 5 or 6 double bond (vinyl), preferably 1 double bond. The groups may be optionally substituted.
[00026] The term "alkynyl" refers to a monoradical of an unsaturated hydrocarbon, preferably having from 4, 5, 6, 7, 8, 9, or lOcarbon atoms and having 1, 2, 3, 4, 5 or 6 sites of acetylene (triple bond) unsaturation, preferably 1 triple bond. The groups may be optionally substituted.
[00027] ‘Halo” or“Halogen”, alone or in combination with any other term means halogens such as chloro (Cl), fluoro (F), bromo (Br) and iodo (I).
[00028] The term "aryl" refers to an aromatic carbocyclic group of 5 to 18 carbon atoms having a single ring (e.g. phenyl) or multiple rings (e.g. biphenyl), or multiple condensed (fused) rings (e.g. naphthyl or anthranyl). Preferred aryls include phenyl, naphthyl and the like. The groups may be optionally substituted.
[00029] The term "heteroaryl" refers to a heteroaromatic carbocyclic group of 2 to 10 carbon atoms having a single ring or multiple rings, or multiple condensed rings.
Preferred heteroaryls include pyrazole, thiazole,oxazole, benzoxazole, pyridine and the like. The groups may be optionally substituted.
[00030] The term "cycloalkyl" refers to carbocyclic groups of from 3 to 12 carbon atoms having a single cyclic ring or multiple condensed rings, which may be partially
unsaturated. Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclohexyl, cyclohexenyl, and the like, or multiple ring structures or carbocyclic groups to which is fused an aryl group. The groups may be optionally substituted.
[00031] The term "heterocyclyl" refers to a saturated or partially unsaturated group or unsaturated group having a single ring or multiple condensed rings, having from 2 to 10 carbon atoms and from 1 to 10 hetero atoms, preferably 1, 2, or 3 heteroatoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen within the ring. Preferred heterocyclyls include morpholine, piperidine, and the like. The groups may be optionally substituted.
[00032] As used herein, the term "substituted" is contemplated to include all permissible substituents of organic compounds. In a broad aspect, the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and nonaromatic substituents of organic compounds.
[00033] The term "effective amount" means an amount of a compound or composition, which is sufficient enough to significantly and positively modify the symptoms and/or conditions to be treated. The effective amount will vary depending on various conditions and is within the knowledge and expertise of the attending physician.
[00034] The compounds described herein may contain one or more chiral centers and/or double bonds and therefore, may exist as stereoisomers. Accordingly, the chemical structures depicted herein encompass all possible enantiomers and stereoisomers of the illustrated or identified compounds including the stereoisomerically pure form and enantiomeric and stereoisomeric mixtures. The compounds may also exist in several tautomeric forms including the enol form, the keto form and mixtures thereof. Accordingly, the chemical structures depicted herein encompass all possible tautomeric forms of the illustrated or identified compounds.
[00035] The term "pharmaceutically acceptable" refers to those compounds, materials, compositions, and/or dosage forms, which are suitable for use in contact with the tissues of human beings and animals and is understood by a person skilled in the art.
[00036] “Pharmaceutically acceptable salt” embraces salts with a pharmaceutically acceptable acid or base. Pharmaceutically acceptable acids include both inorganic acids, and organic bases.
[00037] The term“polymorphs” refers to crystal forms of the same molecule, and different polymorphs may have different physical properties.
[00038] The term“solvate”, as used herein, refers to a crystal form of a substance, which contains solvent.
[00039] The term“hydrate” refers to a solvate wherein the solvent is water. Ratios, concentrations, amounts, and other numerical data may be presented herein in a range format. It is to be understood that such range format is used merely for convenience and brevity and should be interpreted flexibly to include not only the numerical values explicitly recited as the limits of the range, but also to include all the individual numerical values or sub-ranges encompassed within that range as if each numerical value and sub range is explicitly recited. For example, a concentration range of about 50 to 1000 m M should be interpreted to include not only the explicitly recited limits of about 50 mM to about 1000 mM, but also to include sub-ranges, such as 75-875 mM, 80-500 mM, and so forth, as well as individual amounts, including fractional amounts, within the specified ranges, such as 100 mM, and 155 mM, for example.
[00040] The present disclosure relates to compounds for treatment of HIV infection. The target is novel interaction of Nef protein from the HIV-l virus with the host immune receptors CD80/CD86 responsible for T cell stimulation.
[00041] An indispensable factor for HIV pathogenicity is the Nef protein. It allows HIV to evade the immune response, maintain high viral load and is needed for replication and dissemination, down-regulate cell surface receptors involved in the generation of immune response, including MHC-l and MHC-2. It has been surprising found that in addition to relocation of MHC, Nef also down-regulates surface expression of the B-7 family of co-stimulatory proteins, namely CD80 and CD86 expressed on antigen presenting cells, leading to impaired T cell stimulation in vitro. Nef binds directly to the cytoplasmic tails of CD80 and CD86 and prone to reversal by introduction of peptides,
making it a suitable target for therapeutic intervention. Targeting /Ve/-CD80/86 interactions has a unique mechanism of action: prevention of immune evasion of infected cells. The disclosure revolves around the inhibition of /Ve/-mediated functions, which is a distinct strategy since it targets a key function of Nef in its ability to directly modulate infected immune cell (macrophage) capacity to generate HIV -specific T cells from naive T cell, potentially bringing forth a different class of drugs. The disclosure presents a new line of antiviral therapy through immune signaling restoration.
[00042] The host-virus interface is also less amenable to drug resistance than purely viral targets. The present disclosure discloses a method for preventing/ treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I. Formula II, and Formula III as described herein, wherein the therapeutic dose of the compound is in a range of 50 to 1000 m M. The present disclosure is intended to cover all the sub-ranges and individual values. The range can be 50 to 100 mM, or 100 to 1000 mM, or 150 to 900 mM, or 125 to 500 mM, or 500 to 900 mM, or 750 to 950 mM, or 75 to 150 mM, or 100 to 200 mM.
[00043] In an embodiment, the present disclosure provides a method for preventing/ treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C5-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy, and C2-10 heterocyclyl;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl, and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2- 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5- 10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is selected from O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5. In another embodiment of the present disclosure, the therapeutic dose of the compound is in a range of 50 to 1000 m M. In yet another embodiment of the present disclosure, the therapeutic dose of the compound is in a range of 50 to 100 mM. In an alternate embodiment of the present disclosure, the therapeutic dose of the compound is in a range of 100 to 1000 mM. In one another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 150 to 900 mM. In an alternate embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 125 to 500 mM. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 500 to 900 mM. In another embodiment of the
present disclosure, the therapeutic dose of the compound is in the range of 750 to 950 m M. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 75 to 150 m M. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 100 to 200 mM.
[00044] In an embodiment, the present disclosure provides a method for treating
HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, and amino, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy, and C2-10heterocyclyl;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and Ci- io alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-io heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00045] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, and C1-10 alkyl, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, and C1-10 alkoxy;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-io heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00046] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and Ci-5 alkyl, wherein C1-5 alkyl, and C6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C1-5 alkyl or C1-5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from C1-5 alkoxy, nitro, halogen or C1-5 alkyl, wherein C1-5 alkyl and C1-5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-5 alkyl, C1-5 alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-5 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2 10 heteroaryl, wherein C2 10 heterocyclyl or C2 10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-5 alkyl; and X and Y are N.
[00047] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and CF3, wherein C6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, C1-5 alkyl or C1-5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from -OCH3, -OCF3, nitro, fluorine, CF3;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-5 alkyl, and -N=CHC6 aryl, wherein N=CHC6 aryl is further substituted with one to four substituents selected from fluorine and bromine; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-5 alkyl; and X and Y are N.
[00048] In an embodiment, the present disclosure provides a method for treating
HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents
independently selected from hydroxyl, halogen, C1-10 alkyl or C1-10 alkoxy, C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-io aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00049] In an embodiment, the present disclosure provides a compound of Formula I or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:
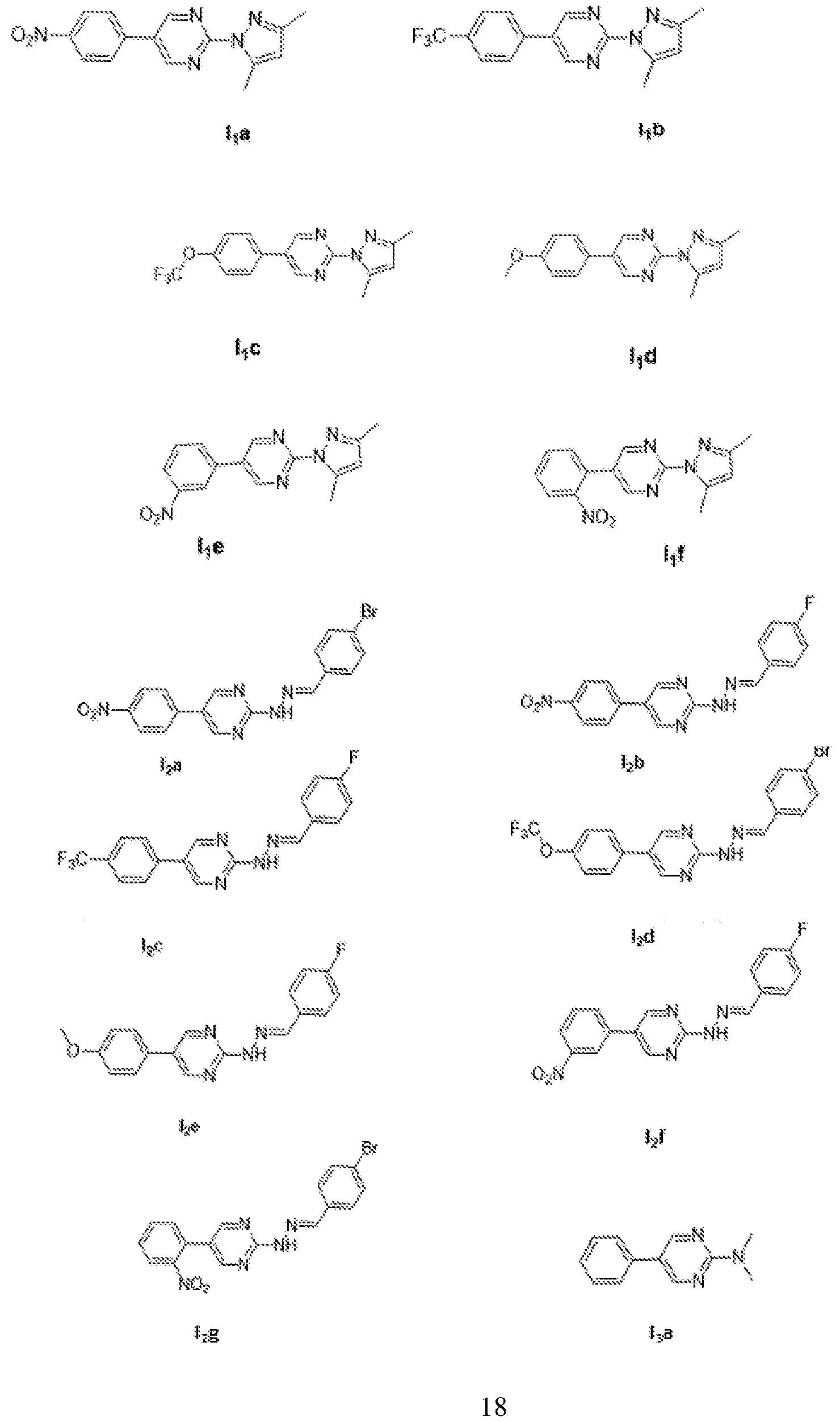

[00050] In an embodiment, the present disclosure provides a compound of Formula I or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:

HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-io heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00052] In an embodiment, the present disclosure provides a method for treating
HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-io heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Riand R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00053] In an embodiment, the present disclosure provides a method for treating
HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II

Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein Ci-10 alkyl, C2 10 heterocyclyl, C5- 10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-io aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C5-ioaryl or C2 ioheteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00054] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula II

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or
Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is C5-10 aryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano,
CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00055] In an embodiment, the present disclosure provides a compound of Formula II or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:

[00056] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5.
[00057] In an embodiment, the present disclosure provides a method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound of Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci-5alkyl;
R2 and R4 is independently selected from hydrogen or C1-5 alkyl;
R3 is selected from Ci-5alkyl or chlorine; and n is selected from 1 to 2.
[00058] In an embodiment, the present disclosure provides a compound of Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, which is selected from a group consisting of:

[00059] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-io aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy, C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-ioheterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N;

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5- 10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and

X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, and
n is selected from 0-5; and

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected froml to 5. In another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 50 to 1000 m M. In yet another embodiment of the
present disclosure, the therapeutic dose of the compound is in the range of 50 to 100 m M. In an another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 100 to 1000 mM. In an alternate embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 150 to 900 mM. In one another embodiment of the present disclosure, the therapeutic dose of the compound is in the range of 125 to 500 mM. In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, and amino, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10
heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00060] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, and C1-10 alkyl, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl or C1-10 alkoxy;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen, C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-io heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen, or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00061] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and Ci-5 alkyl, wherein C1-5 alkyl, and C6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C1-5 alkyl or C1-5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from Ci-5alkoxy, nitro, halogen, C1-5 alkyl, wherein C1-5 alkyl and C1-5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-5 alkyl, C1-5 alkoxy or C2- 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-5 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2 10 heteroaryl, wherein C2 10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-5 alkyl; and X and Y are N.
[00062] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and CF3, wherein C6 aryl is optionally substituted with one to four substituents independently selected from hydroxyl, C1-5 alkyl or C1 -5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from -OCH3, -OCF3, nitro, fluorine or CF3;
R4 and R5 are independently selected from the group consisting of hydrogen, C 1-5 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four substituents selected from fluorine and bromine; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2 10 heterocyclyl or C2 10 heteroaryl is optionally substituted with one to four substituents independently selected from C 1-5 alkyl; and X and Y are N.
[00063] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro or C1-10 alkyl, wherein C1-10 alkyl and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen, or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00064] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
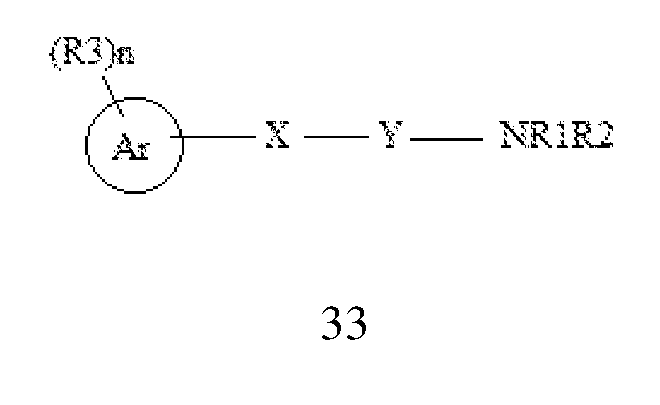
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5- 10 aryl, C2 10 heteroaryl, C2 10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CPhPhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00065] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-io heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5- 10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00066] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring,
wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C5- 10 aryl or C2-10 heteroaryl;
R3is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00067] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula II
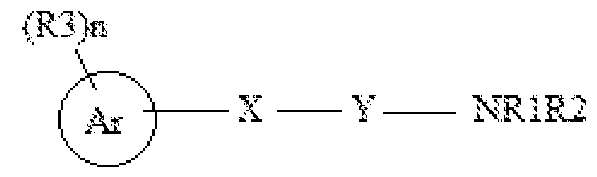
Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5- 10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-io aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;

R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5.
[00068] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci-io alkyl;
R2 and R4 is independently selected from hydrogen or Ci-io alkyl;
R3 is selected from Ci-io alkyl or halogen; and n is selected from 1 to 5.
[00069] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C 1-5 alkyl;
R2 and R4 is independently selected from hydrogen or C1-5 alkyl;
R3 is selected from C1-5 alkyl or chlorine; and n is selected from 1 to 2.
[00070] In an embodiment, the present disclosure provides a method for treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-io alkenyl, C2-io alkynyl, and amino, wherein C1-10 alkyl, C2-io alkenyl, C2-io alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-io heterocyclyl;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl, and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;
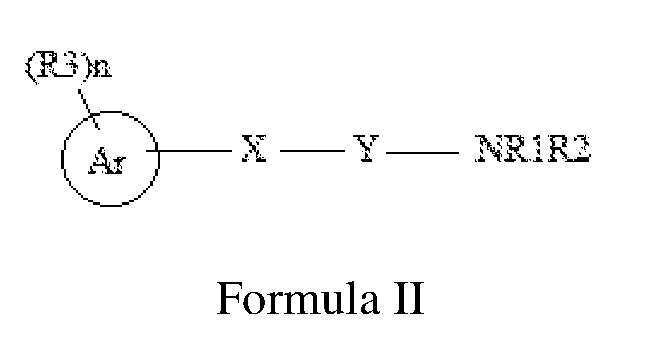
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5- 10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and

Formula III
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5.
[00071] In an embodiment, the present disclosure provides a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I,

Formula I
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, and amino, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-io heterocyclyl;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00072] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, and C1-10 alkyl, wherein C1-10 alkyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl or C1-10 alkoxy;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00073] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and Ci-5 alkyl, wherein C1-5 alkyl, and C6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, fluorine, C1-5 alkyl or C1-5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from Ci-5 alkoxy, nitro, halogen or Ci-5 alkyl, wherein Ci-5 alkyl and Ci-5 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, Ci-5 alkyl, Ci-5 alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-5 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2 10 heteroaryl, wherein C2 10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-5 alkyl; and X and Y are N.
[00074] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6 aryl, and CF3, wherein C6 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, C1-5 alkyl or C1-5 alkoxy;
R3 is C6 aryl, wherein C6 aryl is optionally substituted with one to four substituents independently selected from -OCH3, -OCF3, nitro, fluorine or CF3;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-5 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four
substituents selected from fluorine and bromine; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-5 alkyl; and X and Y are N.
[00075] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-io alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro or C1-10 alkyl, wherein C1-10 alkyl and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10
heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and X and Y are N.
[00076] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -
X is O;

Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and n is selected from 0-5.
[00077] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of
CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and n is selected from 0-5.
[00078] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and n is selected from 0-5.
[00079] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula II

Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein Ci-10 alkyl, C2 10 heterocyclyl, C5-10 aryl, C2 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, Ci-10 alkyl, C5-10 aryl, Ci-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is C5-10 aryl;
R3is absent or is selected from hydroxy, Ci-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, Ci-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and n is selected from 0-5.
[00080] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by qf CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula III

Formula III
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and n is selected from 1 to 5.
[00081] In an embodiment, the present disclosure provides a method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from the group consisting of Formula III

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is Ci-5 alkyl;
R2 and R4 is independently selected from hydrogen or C1-5 alkyl;
R3 is selected from C1-5 alkyl or chlorine; and n is selected from 1 to 2.
[00082] In an embodiment, the present disclosure provides a compound of
Formula I, II, or III or its or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, which is selected from a group consisting of 2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-(4-nitrophenyl)pyrimidine (Iia),
2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-[4-(trifluoromethyl)phenyl]pyrimidine(Iib),
2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-[4-(trifluoromethoxy)phenyl]pyrimidine (lie),
2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-(4-methoxyphenyl)pyrimidine (lid),
2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-(3-nitrophenyl)pyrimidine (lie),
2-(3, 5-Dimethyl- lif-pyrazol-l-yl)-5-(2-nitrophenyl)pyrimidine (Iif),
2-[2-(4-Bromobenzylidene)hydrazinyl)-5-(4-nitrophenyl)]pyrimidine (ha),
2-(2-(4-Fluorobenzylidene)hydrazinyl)-5-(4-nitrophenyl)pyrimidine (hb),
(£)-2-(2-(4-Fluorobenzylidene)hydrazinyl)-5-(4-(trifluoromethyl)phenyl)pyrimidine (he),
(h)-2-[2-(4-Bromobenzylidene)hydrazinyl)-5-(4-(trifluoromethoxy)phenyl] pyrimidine
(hd),
(£)-2-[2-(4-Fluorobenzylidene)hydrazinyl)-5-(4-methoxyphenyl]pyrimidine (he), (£)-2-(2-(4-Fluorobenzylidene)hydrazinyl)-5-(3-nitrophenyl)pyrimidine (hf), (£)-2-[(2-(4-Bromobenzylidene)hydrazinyl)-5-(2-nitrophenyl)]pyrimidine (hg), /V,/V-Dimethyl-5-phenylpyrimidin-2-amine (ha),
4-(5-(4-(Trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (hb),
5-(4-Fluorophenyl)-N,N-dimethylpyrimidin-2-amine (he),
4-(5-(4-Fluorophenyl)pyrimidin-2-yl)morpholine (hd),
2-(2-amino-5-(4-methoxyphenyl)-6-(trifluoromethyl)pyrimidin-4-yl)-4-ethyl-5- methoxyphenol (h),
Phenyl 2-th ioxobenzo[d]oxazole-3(2//)-carbox yl ate (Ih),
p-Tolyl (3,4-dichlorophenyl)carbamate (Iha),
[ 1, 1 '-biphenyl] -4-yl (3,4-dichlorophenyl)carbamate (Ihb),
4-Methoxyphenyl diphenylcarbamate (Ihc),
Di-p-tolyl [methylenebis(4,l-phenylene)]dicarbamate (Ihd),
Naphthalen-2-yl (2-fluorophenyl)carbamate (Ih),
2-[3-(3,4-dichlorophenyl)ureido]-4-nitrophenyl (3,4-dichlorophenyl)carbamate (Ih),
3-[(l,3,4-oxadiazol-2-yl)phenyl] diphenylcarbamate (Ih),
2,4-di-tert-butyl-6-methoxyphenyl phenylcarbamate (II6),
2-[(2-Tosylhydrazine-l-carbonyl)phenyl] phenylcarbamate (Ih),
Ethyl 4-[2-(4-ethylphenoxy)acetamido]benzoate (Ilha),
Methyl 4-[2-(2-isopropylphenoxy)acetamido]benzoate (Illib),
Propyl 4-[2-(4-isopropylphenoxy)acetamido]benzoate (IIIic),
Isopropyl 4-[2-(4-isopropylphenoxy)acetamido]benzoate (Illid),
Propyl 4-[2-(m-tolyloxy)acetamido]benzoate (Illie),
Isobutyl 2-hydroxy-4-[2-(4-isopropylphenoxy)acetamido]benzoate(IIIif),
4- [2-(3 ,5 -dime thy lphenoxy)acetamido] -2-hydroxybenzoic acid (IIFa) ,
4-(2-Phenoxypropanamido)benzoic acid ( 11 lob),
Ethyl 4-[2-(4-chlorophenoxy)-2-methylpropanamido]benzoate (IIPc), and
Methyl 4-[2-(o-tolyloxy)propanamido]benzoate (IIFd).
[00083] The compounds for Formula I, II, or III causes restoration of immune signaling via T cell activation through inhibiting M?/-CD80/86 interactions.
[00084] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:

[00085] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:

[00086] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV
infected subject a therapeutic dose of a compound selected the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:

[00087] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:

[00088] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of
Formula I, wherein compound of Formula I is selected from a group consisting of:

[00089] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of Formula I, wherein compound of Formula I is selected from a group consisting of:

[00090] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected the group consisting of Formula II, wherein compound of Formula II is selected from a group consisting of:

[00091] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the
group consisting of Formula II, wherein compound of Formula II is selected from a group consisting of:


[00093] In an embodiment, the present disclosure provides a method of treating or preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the
group consisting of Formula III, wherein compound of Formula III is selected from a group consisting of:


Formula I
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C5-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy, and C2-10 heterocyclyl;
R3 is C6-10 aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl, and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2- 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein -N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, and halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is selected from O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl;
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and

Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5. In another embodiment of the present disclosure, the relevant dose is in the range of 50 to 1000 mM.
[00095] In an embodiment, the present disclosure provides a method of treating or preventing or treating HIV infection in a subject by immune evasion as described herein, wherein the compounds causes restoration of immune signaling via T cell activation through inhibiting Nef- CD80/86 interactions.
[00096] In an embodiment, the present disclosure provides a method for treating/preventing infections which have low levels of CD80/86 receptors compared to non-infected state, selected from the including chronic lymphocytic leukemia, colon carcinoma, multiple myeloma, viral infections including HIV, HPV, herpes.
[00097] In an embodiment, the present disclosure relates to a method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from the group consisting of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (I3b), naphthalen-2-yl (2-fluorophenyl)carbamate (II3), and 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilka). In another embodiment of the present disclosure, the relevant dose is in the range of 50 to 1000 m M.
[00098] In an embodiment, the present disclosure relates to a method for preventing HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4- (5-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (kb), naphthalen-2-yl (2- fluorophenyl)carbamate (Ik), 4-[2-(3,5-dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilka) and combinations thereof. In another embodiment of the present disclosure,
the therapeutic dose is in the range of 50 to 1000 mM. In yet another embodiment, the compound is a combination of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2- yl)morpholine (13b), naphthalen-2-yl (2-fluorophenyl)carbamate (II3), 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (Ilha).
[00099] In an embodiment, the present disclosure relates to method for treating
HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4-(5-(4- (trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (hb), naphthalen-2-yl (2- fluorophenyljcarbamate (II3), and 4-[2-(3,5-dimethylphenoxy)acetamido]-2- hydroxybenzoic acid (IIHa). In another embodiment of the present disclosure, the relevant dose is in the range of 50 to 1000 mM.
[000100] In an embodiment, the present disclosure relates to method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of 4-(5-(4- (trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (Hb), naphthalen-2-yl (2- fluorophenyljcarbamate (II3), 4-[2-(3,5-dimethylphenoxy)acetamido]-2-hydroxybenzoic acid (III2a), and combinations thereof. In another embodiment of the present disclosure, the therapeutic dose is in the range of 50 to 1000 mM. In yet another embodiment, the compound is a combination of 4-(5-(4-(trifluoromethyl)phenyl)pyrimidin-2- yljmorpholine (Hb), naphthalen-2-yl (2-fluorophenyl)carbamate (II3), 4-[2-(3,5- dimethylphenoxy)acetamido]-2-hydroxybenzoic acid ( 11 Ha).
[000101] In an embodiment, the present disclosure relates to a method for preventing or treating HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising: administering to a subject a relevant dose of a compound selected from 4-(5-(4-
(trifluoromethyl)phenyl)pyrimidin-2-yl)morpholine (Hb), naphthalen-2-yl (2- fluorophenyljcarbamate (II3), or 4-[2-(3,5-dimethylphenoxy)acetamido]-2- hydroxybenzoic acid (III2a). In another embodiment of the present disclosure, the relevant dose is in the range of 50 to 1000 mM.
[000102] In an embodiment, the present disclosure relates to a method for treating/preventing infections which have low levels of CD80/86 receptors compared to non-infected state, selected from the including cancers like chronic lymphocytic leukemia, colon carcinoma, multiple myeloma, viral infections including HIV, HPV, herpes using the compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof.
[000103] The infections in bovine, feline, simian that act through Nef superfamily pathways.
[000104] In an embodiment, the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in killing or inhibiting the growth virus.
[000105] In an embodiment, the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for use in killing or inhibiting the growth of HIV.
[000106] In an embodiment, the present disclosure relates to use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in killing or inhibiting the growth of HIV.
[000107] In an embodiment, the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically
active forms and pharmaceutically active derivative thereof, for use in treating a disease or condition in a patient wherein said disease or condition is caused by HIV.
[000108] In an embodiment, the present disclosure relates to use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in treating disease or condition in a patient, wherein said disease or condition is caused by HIV. The patient is a typically a mammal, preferably a human.
[000109] In an embodiment, the present disclosure relates toa method of treating a disease or condition in a patent, said method comprising administering to a patient a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, wherein said disease or condition is caused by HIV.
[000110] In an embodiment, the present disclosure relates to a compound of
Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for useas a medicament.
[000111] In an embodiment, the present disclosure relates to a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, for usein the preparation of medicaments for inhibiting viral growth.
[000112] In an embodiment, the present disclosure relates to medicaments that include a compound of Formula I, Formula II, and Formula III, or an addition salt of the compound of Formula I, Formula II, and Formula III with a pharmaceutically acceptable acid or base. These medicaments find their use in therapeutics, especially in the treatment of viral infection caused by HIV.
[000113] In an embodiment, the present disclosure relates to the use of a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, in the manufacture of a medicament for the production of an antiviral effect in a warm-blooded animal such as man.
[000114] In an embodiment, the present disclosure relates to a method for producing an antiviral effect in a warm-blooded animal such as man, said method including administering to said animal an effective amount of a compound of Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof.
[000115] In an embodiment, the present disclosure relates to a compound of Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof, for use in the treatment and/or prophylaxis of viral infections in a warm-blooded animal, such as man.
[000116] In an embodiment, the present disclosure relates to a compound of
Formula I, Formula II, and Formula III or a pharmaceutically acceptable salt thereof, for the therapeutic and prophylactic treatment of mammals including humans, in particular in treating viral infections caused by HIV, is normally formulated in accordance with standard pharmaceutical practice as a pharmaceutical composition.
[000117] In an embodiment, the present disclosure relates to a pharmaceutical composition including a compound of Formula I, Formula II, and Formula III or its stereoisomers, pharmaceutically acceptable salts, complexes, hydrates, solvates, tautomers, polymorphs, racemic mixtures, optically active forms and pharmaceutically active derivative thereof, and at least one pharmaceutically acceptable carrier, diluent, or excipient.
[000118] The term,“pharmaceutically acceptable” includes compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals
without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
[000119] The compounds of Formula I, Formula II, and Formula III may form stable pharmaceutically acceptable acid or base salts, and in such cases administration of a compound as a salt may be appropriate. The salts may be formed by conventional means.The compositions of the disclosure may be in a form suitable for oral use, for topical use, for administration by inhalation, for administration by insufflation or for parenteral administration.
[000120] The compositions of the present disclosure may be obtained by conventional procedures using conventional pharmaceutical excipients well known in the art.
[000121] The compounds disclosed herein may be applied as a sole therapy or may involve, in addition to a compound of the disclosure, one or more other substances and/or treatments.
PROCESS AND CHARACTERIZATION DATA
[000122] There is also provided a process as shown in the following Schemes 1-7, for the preparation of compounds of the Formula I, Formula II, and Formula III, wherein all the groups are as defined earlier. The examples given below are provided by the way of illustration only and therefore should not be construed to limit the scope of the invention.
Scheme-1

General procedure for the synthesis of Iia-f of Scheme- 1
[000123] To a solution of compound 7 (3.4 mmol) in methanol was added acetyl acetone 8 (5.19 mmol) and catalytic amounts of acetic acid. The resulting mixture was stirred at 60° C- 70° C for 5 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound Iia-f.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-(4-nitrophenyl)pyrimidine (Iia, Scheme 1):
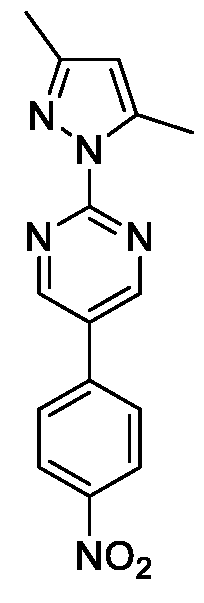
[000124]
 9.28 (m, 2H), 8.38 - 8.24 (m, 2H), 7.94 -
9.28 (m, 2H), 8.38 - 8.24 (m, 2H), 7.94 -
7.80 (m, 2H), 6.14 (s, 1H), 2.38 (s, 6H); HRMS (ESI-TOF) calcd for C
296.1147, found 296.1147.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-[4-(trifluoromethyl)phenyl]pyrimidine(Iib, Scheme 1 ):

[000125]
 9.24 (s, 2H), 7.70 - 7.67 (m, 2H), 7.61 - 7.58 (m, 2H), 6.13 (s, 1H), 2.36 (m, 6H); HRMS (ESI-TOF) calcd for C16H13F3N4
9.24 (s, 2H), 7.70 - 7.67 (m, 2H), 7.61 - 7.58 (m, 2H), 6.13 (s, 1H), 2.36 (m, 6H); HRMS (ESI-TOF) calcd for C16H13F3N4
[M+H]+ 318.1092, found 318.1083.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-[4-(trifluoromethoxy)phenyl]pyrimidine (lie, Scheme 1):

[000126]
 9.36 (s, 2H), 7.68 - 7.48 (m, 2H), 7.19 -
9.36 (s, 2H), 7.68 - 7.48 (m, 2H), 7.19 -
6.96 (m, 2H), 6.12 (s, 1H), 2.44 - 2.25 (m, 6H).; HRMS (ESI-TOF) calcd for C16H13F3N4O [M+H]+ 334.1041, found 334.1062.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-(4-methoxyphenyl)pyrimidine (lid, Scheme 1):

[000127]
 9.21 (s, 2H), 7.63 - 7.55 (m, 2H), 7.12 -
9.21 (s, 2H), 7.63 - 7.55 (m, 2H), 7.12 -
6.96 (m, 2H), 6.12 (s, 1H), 3.81 (s, 3H), 2.36 (s, 6H); HRMS (ESI-TOF) calcd for CieHieNrO [M+H]+ 280.1324, found 280.1348.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-(3-nitrophenyl)pyrimidine (lie, Scheme 1):

[000128]
 9.26 (s, 2H), 8.56 (s, 1H), 8.22-8.19 (m,
9.26 (s, 2H), 8.56 (s, 1H), 8.22-8.19 (m,
1H), 7.99-7.91 (m, 1H), 7.68-7.59 (m, 1H), 6.13 (s, 1H), 2.37 (s, 6H); HRMS (ESI-TOF) calcd for C15H13N5O2 [M+H]+ 295.1069, found 295.1054.
2-(3,5-Dimethyl-l//-pyrazol-l-yl)-5-(2-nitrophenyl)pyrimidine (Iif, Scheme 1):

(d, / = 29.8 Hz, 1H), 7.60 (s, 1H), 6.13 (s, 1H), 2.36 (m, 6H); HRMS (ESI-TOF) calcd for C15H13N5O2 [M+H]+ 295.1069, found 295.1047.
General procedure for the synthesis of Ha-g of Scheme 1:
[000130] To a solution of compound 7 (0.4 mmol) in methanol/ethanol was added substituted benzaldehyde 9 (0.51 mmol) and catalytic amounts of acetic acid. The resulting mixture was stirred at room temperature for 5 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCE and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound Ha-g.
2

[000131]
 8.74 (s, 1H), 8.71 (s, 2H), 8.28 (d, / = 8.8
8.74 (s, 1H), 8.71 (s, 2H), 8.28 (d, / = 8.8
Hz, 2H), 7.86 (s, 1H), 7.61 (dd, / = 15.3, 8.6 Hz, 5H), 7.48 (d, / = 8.5 Hz, 3H); HRMS (ESI-TOF) calcd for Ci7Hi3BrN502: [M+H] 399.2280 found 399.2282.


[000132]
 8.74 (s, 1H), 8.71 (s, 2H), 8.28 (d, / = 8.8
8.74 (s, 1H), 8.71 (s, 2H), 8.28 (d, / = 8.8
Hz, 2H), 7.86 (s, 1H), 7.61 (dd, / = 15.3, 8.6 Hz, 5H), 7.48 (d, / = 8.5 Hz, 3H); HRMS (ESI-TOF) calcd for C17H13FN5O2: [M+H] 338.1053 found 338.1055.
(£,)-2-(2-(4-Fluorobenzylidene)hydrazinyl)-5-(4-(trifluoromethyl)phenyl)pyrimidine (I2C, Scheme 1):

[000133]
 9.03 (s, 1H), 8.86 (m, 2H), 7.69 - 7.64 (m,
9.03 (s, 1H), 8.86 (m, 2H), 7.69 - 7.64 (m,
2H), 7.63 - 7.58 (m, 2H), 7.56 (s, 1H), 7.54 - 7.48 (m, 2H), 7.30 - 7.23 (m, 2H); HRMS (ESI-TOF) calcd for C18H12F4N4: [M+H] 360.0998 found 360.0986.
(£')-2-[2-(4-Bromobenzylidene) hydrazinyl)-5-(4— (trifluoromethoxy) phenyl] pyrimidine (Hd, Scheme 1):

[000134]
 9.81 (s, 1H), 8.87 (s, 2H), 7.67 - 7.57 (m,
9.81 (s, 1H), 8.87 (s, 2H), 7.67 - 7.57 (m,
4H), 7.32 - 7.24 (m, 2H), 7.15 - 7.09 (m, 2H), 7.00 (s, 1H); HRMS (ESI-TOF) calcd for Ci8Hi2BrF3N40: [M+H] 436.0147 found 436.0126.
(£')-2-[2-(4-Fluorobenzylidene)hydrazinyl)-5-(4-methoxyphenyl]pyrimidine (He, Scheme 1):

[000135]
 9.01 (s, 1H), 8.84 - 8.77 (m, 2H), 7.63 -
9.01 (s, 1H), 8.84 - 8.77 (m, 2H), 7.63 -
7.59 (m, 2H), 7.56 (s, 1H), 7.54 - 7.49 (m, 2H), 7.29 - 7.25 (m, 2H), 7.08 - 7.04 (m, 2H), 3.91 - 3.72 (m, 3H); HRMS (ESI-TOF) calcd for C18H15FN4O: [M+H] 322.1230 found
322.1242.
(£,)-2-(2-(4-Fluorobenzylidene)hydrazinyl)-5-(3-nitrophenyl)pyrimidine (Hf, Scheme 1):

[000136]
 9.04 (s, 1H), 8.89 - 8.83 (m, 2H), 8.59 (s,
9.04 (s, 1H), 8.89 - 8.83 (m, 2H), 8.59 (s,
1H), 8.25 (s, 1H), 8.01 (s, 1H), 7.71 (s, 1H), 7.57 (s, 1H), 7.54 - 7.48 (m, 2H), 7.32 - 7.23 (m, 2H); HRMS (ESI-TOF) calcd for C17H12FN5O2: [M+H] 337.0975 found
337.0954.


[000137]
 9.07 (s, 1H), 8.60 - 8.55 (m, 2H), 8.30 (s,
9.07 (s, 1H), 8.60 - 8.55 (m, 2H), 8.30 (s,
1H), 7.85 (s, 1H), 7.80 (s, 1H), 7.61 (s, 1H), 7.60 - 7.55 (m, 2H), 7.53 (s, 1H), 7.40 - 7.34 (m, 2H); HRMS (ESI-TOF) calcd for C17H12FN5O2: [M+H] 337.0975 found
337.0968.
Scheme-2

Reagents and conditions: (a) 2° amines, K2C03, MeOH, rt, 12 h; (b) Pd(PPh3)4, K2C03, dioxane, 110 °C, 6h.
General procedure for the synthesis of Ha-d of Scheme 2:
[000138] To a solution of 11 (8.2 mmol) in dioxane (3 mL) under nitrogen atmosphere were added tetrakis(triphenylphosphine)palladium (0.047 g, 0.41 mmol), substituted phenylboronic acid 12 (0.98 mmol), and K2CO3 dissolved in 2 mL of water. The mixture was stirred under reflux for 6 h. The reaction mixture was diluted with EtOAc, filtered through celite, and washed with water. The combined organic layer was dried over anhydrous NaoSCL and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound Ha-d.
AyV-Dimethyl-5-phenylpyrimidin-2-amine (La, Scheme 2):
[000139]
 8.49 (s, 2H), 7.38 (dt, J = 15.2, 7.5 Hz,
8.49 (s, 2H), 7.38 (dt, J = 15.2, 7.5 Hz,
4H), 7.26 (t, / = 7.1 Hz, 1H), 3.17 (s, 6H); HRMS (ESI-TOF) calcd for C12H14N3: [M+H] 200.1188 found 200.1187.
4-[5-(4-(Trifluoromethyl)phenyl)pyrimidin-2-yl]morpholine (I3I1, Scheme 2):

[000140]
 8.58 (s, 1H), 7.70 (d, / = 8.2 Hz, 1H), 7.59
8.58 (s, 1H), 7.70 (d, / = 8.2 Hz, 1H), 7.59
(d, / = 8. Hz, 2H), 3.87 (m, 4H), 3.83 - 3.77 (m, 4H); HRMS (ESI-TOF) calcd for C15H15F3N3O: [M+H]+ 310.1167 found 310.1170.
5-(4-Fluorophenyl)-N,N-dimethylpyrimidin-2-amine (I3C, Scheme 2):
[000141]
 8.65 (s, 2H), 7.68 - 7.51 (m, 2H), 7.29 -
8.65 (s, 2H), 7.68 - 7.51 (m, 2H), 7.29 -
7.12 (m, 2H), 2.97 (s, 6H); HRMS (ESI-TOF) calcd for C15H12FN3: [M+H]+ 217.1015 found 217.1036.
4-[5-(4-Fluorophenyl)pyrimidin-2-yl]morpholine (fed, Scheme 2):

General procedure for the synthesis of I4 of Scheme 3
[000143] To a solution of 13 (8.2 mmol) and 14 in dioxane (3 mL) were refluxed for 12 h. The reaction mixture was diluted with EtOAc, filtered through celite, and washed with water. The combined organic layer was dried over anhydrous NaoSCU and concentrated under reduced pressure. The crude product was further used without any purification. The compound 15 (8.2 mmol) was dissolved in acetonitrile (3 mL) under nitrogen atmosphere and then added tetrakis(triphenylphosphine)palladium (0.41 mmol), substituted compound 16 (0.98 mmol), and K2CO3 solution (dissolved in 2 mF of water). The mixture was heated 180 °C on microwave for lh. The reaction mixture was diluted with EtOAc, filtered through celite, and washed with water. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound I4 as a white solid.
2-[2-amino-5-(4-methoxyphenyl)-6-(trifluoromethyl)pyrimidin-4-yl]-4-ethyl-5- methoxyphenol (I4 , Scheme 3):

[000144]
 7.61 - 7.55 (d, 2H), 7.31 (s, 1H), 7.07 -
7.61 - 7.55 (d, 2H), 7.31 (s, 1H), 7.07 -
7.04 (d, 2H), 6.61 (s, 1H), 6.55 (s, 1H), 3.86 - 3.79 (m, 6H), 2.61 - 2.52 (q, 2H), 1.68 (s, 2H), 1.31 - 1.25 (t, 3H); HRMS (ESI-TOF) calcd for C21H20F3N3O3: [M+H] 419.1457 found 419. 1479.
Scheme-4

General procedure for the synthesis of III of Scheme 4:
[000145] To a solution of benzo[d]oxazole-2(3//)-thione 18 (2.6 mmol) in ethylacetate was added phenyl chloroformate 17 slowly at 0 °C (4.00 mmol) and catalytic amounts of pyridine. The resulting mixture was stirred at rt for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound Hi.
Phenyl 2-thioxobenzo[d]oxazole-3(2//)-carboxylate (Hi, Scheme 4):

[000146]

7.33 - 7.25 (m, 6H). LC-MS (ESI+): m\z 272.0303[M + H]+.
Scheme 5

General procedure for the synthesis of IHa-d of Scheme 5:
[000147] To a solution of substituted anilines 20 (2.6 mmol) in ethylacetate was added substituted phenyl chloroformate 19 slowly at 0 °C (4.00 mmol) and catalytic amounts of pyridine. The resulting mixture was stirred at rt for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCE and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound IHa-d.
p-Tolyl (3,4-dichlorophenyl)carbamate (lira, Scheme 5):

[000148]
 10.11 (s, 1H), 7.60 (s, 1H), 7.35 (d, / = 2.2
10.11 (s, 1H), 7.60 (s, 1H), 7.35 (d, / = 2.2
Hz, 2H), 7.15 - 7.03 (m, 2H), 7.03 - 6.90 (m, 2H), 2.41 - 2.18 (m, 3H). LC-MS (ESI+): mV 296.0240 [M + H]+.
[l,l'-biphenyl]-4-yl (3,4-dichlorophenyl)carbamate (Ibb, Scheme 5):

[000149] 510.11 (s, 1H), 7.83 (s, 1H), 7.76 - 7.47 (m,

4H), 7.47 - 7.41 (m, 2H), 7.35 (d, / = 3.4 Hz, 2H), 7.26 - 7.04 (m, 3H). LC-MS (ESI+): mV 358.0396[M + H]+.
4-Methoxyphenyl diphenylcarbamate (II2C, Scheme 5):

[000150]

7.03 - 6.95 (m, 1H), 6.88 - 6.81 (m, 1H), 6.81 - 6.75 (m, 1H), 6.75 - 6.67 (m, 1H), 3.74 (s, 1H). LC-MS (ESI+): m\z 320.1281 [M + H]+.
Di-p-tolyl [methylenebis(4,l-phenylene)]dicarbamate (II2d, Scheme 5):

[000151]
 -
-
7.29 (m, 4H), 7.19 - 7.07 (m, 4H), 7.04 - 6.95 (m, 4H), 3.91 - 3.57 (s, 2H), 2.50 - 2.17 (s, 6H). LC-MS (ESI+): mV 467.l965[M + H]+.
Scheme 6

General procedure for the synthesis of II3 of Scheme 6:
[000152] To a solution of substituted aniline 22 (2.6 mmol) in ethylacetate was added substituted phenyl chloroformate 21 slowly at 0 °C (4.00 mmol) and catalytic amounts of pyridine. The resulting mixture was stirred at rt for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCA and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound II3.
Naphthalen-2-yl (2-fluorophenyl)carbamate (II3, Scheme 6):

[000153]
 9.86 (s, 1H), 7.78 - 7.68 (m, 1H), 7.52 (s,
9.86 (s, 1H), 7.78 - 7.68 (m, 1H), 7.52 (s,
1H), 7.34 (t, / = 5.1 Hz, 1H), 7.10 (dd, / = 15.8, 3.3 Hz, 1H). LC-MS (ESI+): mSz 282.0295 [M + H]+.
Scheme 7

General procedure for the synthesis of II4 of Scheme 7:
[000154] To a solution of phenylisocyante 23(4.2mmol) in benzene was added potassium tert-butoxide (lOmol %) slowly and then 2-amino-4-nitrophenol 24 (4.2mmol) was added. The resulting mixture was stirred at 45-50 °C for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCA and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound II4.
2-[3-(3,4-dichlorophenyl)ureido]-4-nitrophenyl (3,4-dichlorophenyl)carbamate (II4, Scheme 7):

8.05 (s, 1H), 7.81 (s, 1H), 7.55 (d, J = 34.9 Hz, 2H), 7.46 - 7.32 (m, 3H), 7.28 (s, 1H), 7.01 (s, 1H). LC-MS (ESI+): mSz 528.9635[M + H]+.
Scheme 8

General procedure for the synthesis of IIs of Scheme 8:
[000156] To a solution of substituted aniline 20d (2.6 mmol) in ethylacetate was added substituted phenyl chloroformate 25 slowly at 0 °C (4.00 mmol)and catalytic amounts of pyridine. The resulting mixture was stirred at rt for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCA and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound IIs.
3-[(l,3,4-oxadiazol-2-yl)phenyl] diphenylcarbamate (IIs , Scheme 8):

[000157]
 9.45 (s, 1H), 7.82 (s, 1H), 7.51 (s, 1H),
9.45 (s, 1H), 7.82 (s, 1H), 7.51 (s, 1H),
7.42 - 7.26 (m, 10H), 7.18 (m, 1H), 7.14 - 7.03 (m, 2H). ). LC-MS (ESI+): mSz 358. l l86[M + H]+.
Scheme 9

General procedure for the synthesis of He of Scheme 9:
[000158] To a solution of substituted aniline 27 (2.6 mmol) in ethyl acetate was added substituted phenyl chloroformate 26 slowly at 0 °C (4.00 mmol) and catalytic amounts of pyridine. The resulting mixture was stirred at rt for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCA and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound He.
2,4-di-tert-butyl-6-methoxyphenyl phenylcarbamate (lie, Scheme 9):

[000159]
 9.86(s, 1H), 7.60 - 7.34 (m, 4H), 7.16 (m,
9.86(s, 1H), 7.60 - 7.34 (m, 4H), 7.16 (m,
1H), 7.03 (s, 1H), 6.79 (s, 1H), 3.81 - 3.74 (m, 3H), 1.43 - 1.36 (m, 9H), 1.35 - 1.29 (m, 9H); LC-MS (ESI+): mSz 356.2220[M + H]+.
Scheme 10

General procedure for the synthesis of II7 of Scheme 10):
[000160] To a solution of acid 29 (2 mmol) in benzene (5 mL) was added thionyl chloride (6 mmol). The reaction mixture was stirred under reflux for 2 hrs and concentrated to give the crude acid chlorides 30 as yellow liquids. This liquid was dissolved in dry THF (5 ml) added drop wise to p-tosylhydrazine 31 (1 mmol). After being refluxed for 2-4 hrs the reaction mixture was concentrated. Reaction completion monitored by TLC. The resulting mixture was partition between ethyl acetate and water and the organic layer was separated and finally washed with brine, dried over anhydrous sodium sulphate, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography by using gradient mixture of ethyl acetate and hexane to afford compounds II7.
2-[(2-Tosylhydrazine-l-carbonyl)phenyl] phenylcarbamate (II7, Scheme 10):

[000161]
 8.99 (s, 1H), 8.12 - 7.99 (m, 1H), 7.85 (s,
8.99 (s, 1H), 8.12 - 7.99 (m, 1H), 7.85 (s,
2H), 7.69 - 7.46 (m, 4H), 7.46 - 7.33 (m, 5H), 7.30 (s, 1H), 7.11 (s, 1H), 4.56 (s, 1H), 2.41 - 2.35 (m, 3H); LC-MS (ESI+): m\z 426.1 H8[M + H]+.
Scheme 11

General procedure for the synthesis of IHia-g of Scheme 11:
[000162] To a solution of substituted anilines 32 (5 mmol) in DCM (5ml) and triethylamine (10 mmol) was added chloroacetyl chloride (7.5 mmol). The reaction was stirred at room temperature for 6 hours then diluted with water and extract with DCM, the organic layer was separated and washed with brine, dried over anhydrous sodium sulphate and filtered. The filtrate was concentrated to give the crude 2-chloro amides 33 as a yellow liquid. This yellow liquid was dissolved in dimethyl formamide (5 ml) was added K2CO3 (15 mmol) and corresponding substituted phenols 34 at room temperature. After being stirred at room temperature for l2h, the reaction mixture was diluted with ice water and extract with ethyl acetate, the organic layer was separated and washed with brine, dried over anhydrous sodium sulphate and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using EtO Ac/Hexane as eluents to get compounds Illia-f.
Ethyl 4-[2-(4-ethylphenoxy)acetamido]benzoate (Illia, Scheme 11):

[000163]

 (q, / = 7.1 Hz, 2H), 2.62 (q, / = 7.6 Hz, 2H), 1.39 (t, / = 7.1 Hz, 3H), 1.22 (t, / = 7.6 Hz, 3H);HRMS (ESI-TOF) calcd for C19H21NO4 [M + H]+ 328.1549, found 328.1567.
(q, / = 7.1 Hz, 2H), 2.62 (q, / = 7.6 Hz, 2H), 1.39 (t, / = 7.1 Hz, 3H), 1.22 (t, / = 7.6 Hz, 3H);HRMS (ESI-TOF) calcd for C19H21NO4 [M + H]+ 328.1549, found 328.1567.
Methyl 4-[2-(2-isopropylphenoxy)acetamido]benzoate (Illib, Scheme 11):

[000164]
 8.49 (s, 1H), 8.03 (d, / = 8.65 Hz, 2H),
8.49 (s, 1H), 8.03 (d, / = 8.65 Hz, 2H),
7.64 (d, / = 8.65 Hz, 2H), 7.24 (d, / = 7.4 Hz, 1H), 7.19 (t, / = 8.02 Hz, 1H), 7.06 (t, / = 7.38 Hz, 1H), 6.84(d, J = 8.03Hz, 1H), 4.64 (s, 2H), 3.91 (s, 3H), 3.34 - 3.28 (m, 1H), 1.32 (d, / = 7.38 Hz, 6H); ; HRMS (ESI-TOF) calcd for C19H21NO4 [M + H]+ 328.1549, found 328.1565.
Propyl 4-[2-(4-isopropylphenoxy)acetamido]benzoate (IIIic, Scheme 11):

[000165]
 8.47 (s, 1H), 8.07 (d, J = 8.7 Hz, 2H), 7.71
8.47 (s, 1H), 8.07 (d, J = 8.7 Hz, 2H), 7.71
(d, J = 8.7 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.6 Hz, 2H), 4.63 (s, 2H), 4.30 (t, J = 6.7 Hz, 2H), 2.95 - 2.88 (m, 1H), 1.88 - l.76(m, 2H), 1.26 (d, J = 6.9 Hz, 6H), 1.05 (t, / = 7.4 Hz, 3H);;HRMS (ESI-TOF) calcd for C21H25NO4 [M + Na]+ 378.1682, found 378.1681.
Isopropyl 4-[2-(4-isopropylphenoxy)acetamido]benzoate (Illid, Scheme 11):

(d, J = 8.7 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 5.30 - 5.23(m, 1H), 4.63 (s, 2H), 2.95 - 2.88 (m, 1H), 1.39 (d, / = 6.3 Hz, 6H), 1.26 (d, / = 6.9 Hz, 6H); HRMS (ESI-TOF) calcd for C21H25NO4 [M + Na]+ 378.1682, found 378.1645.
Propyl 4-[2-(m-tolyloxy)acetamido]benzoate (Illie, Scheme 11):

[000167]
 8.44 (s, 1H), 8.05 (d, / = 8.7 Hz, 2H), 7.69
8.44 (s, 1H), 8.05 (d, / = 8.7 Hz, 2H), 7.69
(d, / = 8.7 Hz, 2H), 7.23 (t, / = 7.8 Hz, 1H), 6.89 (d, / = 7.5 Hz, 1H), 6.84 - 6.77 (m, 2H), 4.62 (s, 2H), 4.27 (t, / = 6.7 Hz, 2H), 2.37 (s, 3H), 1.85 - 1.74 (m, 2H), 1.03 (t, / = 7.4 Hz, 3H); HRMS (ESI-TOF) calcd for C19H21NO4 [M + H]+ 328.1549, found 328.1541.
Isobutyl 2-hydroxy-4-[2-(4-isopropylphenoxy)acetamido]benzoate(IIIif, Scheme 11):

[000168]
 7.85 (s, 4H), 7.36 - 7.15 (m, 8H), 7.12 (s,
7.85 (s, 4H), 7.36 - 7.15 (m, 8H), 7.12 (s,
4H), 7.04 (s, 4H), 7.01 - 6.90 (m, 12H), 4.93 - 4.87 (m, 8H), 4.37 (s, 4H), 4.28 - 4.03 (m, 8H), 3.03 (s, 3H), 2.27 (s, 3H), 1.42 - 1.20 (m, 24H), 1.13 - 0.90 (m, 24H). LC-MS
(ESI+): m\z 386.1962 [M + H]+.
Scheme 12

General procedure for the synthesis of IIHa-d, Scheme 12):
[000169] To a suspension of substituted phenoxyalkyl acids 38 (2 mmol) in benzene
(5 mL) was added thionyl chloride (6 mmol). The reaction mixture was stirred under reflux for 5 h and concentrated to give the crude acid chlorides 39 as yellow liquids. This liquid was dissolved in dry DCM (5 ml) added drop wise to the substituted anilines 40 ( 1 mmol) in triethylamine (3 mmol) and DCM (5 ml) at 0°C. After being stirred at room temperature for 4h the reaction mixture was concentrated. The resulting mixture was partition between ethyl acetate and water and the organic layer was separated. The organic layer washed with saturated bicarbonate solution and washed with 2N HC1 and finally washed with brine, dried over anhydrous sodium sulphate, and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography by using gradient mixture of ethyl acetate and hexane to afford compounds IIl2a-d.

[000170]
 9.88 (s, 1H), 8.05 (s, 1H), 7.49 (s, 1H),
9.88 (s, 1H), 8.05 (s, 1H), 7.49 (s, 1H),
7.10 (s, 1H), 6.78 - 6.68 (m, 3H), 4.84 - 4.78 (m, 2H), 2.38 - 2.32 (m, 6H). LC-MS (ESI+): m\z 316.1179[M + H]+.
4-(2-Phenoxypropanamido)benzoic acid (IIHb, Scheme 12):

[000171]
 Acetone-d6) d 9.72 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H),
Acetone-d6) d 9.72 (s, 1H), 7.98 (d, J = 8.7 Hz, 2H),
7.86 (d, J = 8.7 Hz, 2H), 7.36- 7.27 (m, 2H), 7.03 (d, J = 7.9 Hz, 2H), 6.99 (t, J = 7.4 Hz, 1H), 4.92 (q, J = 6.7 Hz, 1H), 1.63 (d, J = 6.7 Hz, 3H);HRMS (ESI-TOF) calcd for CieHisNOr [M + H]+ 286.1079, found 286.1074.
Ethyl 4-[2-(4-chlorophenoxy)-2-methylpropanamido]benzoate (III2C, Scheme 12):

[000172]
 8.73 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.69
8.73 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.69
(d, 7 = 8.6 Hz, 2H), 7.29 (d, / = 8.4 Hz, 2H), 6.95 (d, / = 8.6 Hz, 2H), 4.38 (q, J = 7.1 Hz, 2H), 1.59 (s, 6H), 1.41 (t, 7 = 7.1 Hz, 3H); HRMS (ESI-TOF) calcd for Ci8Hi8ClN04 [M
+ H]+ 348.1002, found 348.0999.
Methyl 4-[2-(o-tolyloxy)propanamido]benzoate (IIEd, Scheme 12):

[000173]
 8.52 (s, 1H), 8.04 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 7.27 - 7.16 (m, 2H), 6.99 (t, J = 7.4 Hz, 1H), 6.87 (d, J = 8.2 Hz,
8.52 (s, 1H), 8.04 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 7.27 - 7.16 (m, 2H), 6.99 (t, J = 7.4 Hz, 1H), 6.87 (d, J = 8.2 Hz,
1H), 4.82 (q, J = 6.7 Hz, 1H), 3.92 (s, 3H), 2.38 (s, 3H), 1.69 (d, J = 6.8 Hz, 3H); HRMS (ESI-TOF) calcd for C18H19NO4 [M + H]+ 314.1392, found 314.1428.
Synthesis of the Intermediates
General procedure for the synthesis of intermediate 3 of Iia-f (Scheme 1):
[000174] To a solution of compound 1 (1.93 mg, 10 mmol) in dry benzene at 0°C was added ethane thiol 2 (0.93 g, 1.5 mmol), followed by NaH (0.36 mg, 1.5 mmol) in
portion wise. The resulting mixture was stirred at room temperature for 3 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, it was quenched with ice-cold water and extracted with ethylacetate. The combined organic layer was dried over anhydrous NaoSCU and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to give compound 3 as a white liquid (1.9 g, 88%). 1 H NMR (500 MHz, CDCE) d 8.90 - 8.73 (s, 2H), 3.16 - 3.00 (q, 2H), 1.41 - 1.23 (t, 3H); Mass (ESI+) 217.9513 [M+H]
General procedure for the synthesis of intermediate 5a-f of Iia-f (Scheme 1)
[000175] To a solution of 3 (8.7 mmol) in dioxane (4 mL) under nitrogen atmosphere were added tetrakis(triphenylphosphine)palladium (0.44 mmol), substituted phenylboronic acid 4 (10.5 mmol), and K2CO3 (8 mmol), dissolved in 4 mL of water. The mixture was stirred under reflux for 6 h. The reaction mixture was diluted with EtOAc, filtered through celite, and washed with water. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to furnish compound 5.
2-(Ethylthio)-5-(4-nitrophenyl)pyrimidine 5a of Ii (Scheme 1)
[000176]
 8.87 - 8.58 (s, 2H), 8.46 - 8.20 (d, 2H),
8.87 - 8.58 (s, 2H), 8.46 - 8.20 (d, 2H),
7.93 - 7.64 (d, 2H), 3.18 - 3.05 (q, 2H), 1.38 - 1.29 (t, 3H); Mass (ESI+) 261.05 [M+H] 2-(Ethylthio)-5-(4-trifluoromethylphenyl)pyrimidine 5b of Ii (Scheme 1):
[000177]
 8.51 - 8.45 (s, 2H), 8.21 - 8.13 (d, 2H),
8.51 - 8.45 (s, 2H), 8.21 - 8.13 (d, 2H),
7.73 - 7.54 (d, 2H), 3.03 - 2.96 (q, 2H), 1.31 - 1.22 (t, 3H); Mass (ESI+) 255.09 [M+H]
2-(Ethylthio)-5-(4-trifluoromethoxyphenyl)pyrimidine 5c of Ii (Scheme 1):
[000178]
 8.57 - 8.49 (s, 2H), 8.21 - 8.17 (d, 2H), 7.29 - 7.13 (d, 2H), 3.03 - 2.96 (q, 2H), 1.31 - 1.22 (t, 3H); Mass (ESI+) 271.16 [M+H]
8.57 - 8.49 (s, 2H), 8.21 - 8.17 (d, 2H), 7.29 - 7.13 (d, 2H), 3.03 - 2.96 (q, 2H), 1.31 - 1.22 (t, 3H); Mass (ESI+) 271.16 [M+H]
2-(Ethylthio)-5-(4-methoxyphenyl)pyrimidine 5d of Ii (Scheme 1):
[000179]
 8.51 - 8.45 (s, 2H), 8.19 - 8.14 (d, 2H),
8.51 - 8.45 (s, 2H), 8.19 - 8.14 (d, 2H),
7.09 - 6.94 (d, 2H), 3.21 (s, 3H), 3.03 - 2.96 (q, 2H), 1.31 - 1.22 (t, 3H); Mass (ESI+) 217.12 [M+H]
2-(Ethylthio)-5-(3-nitrophenyl)pyrimidine 5e of Ii (Scheme 1):
[000180]
 8.87 - 8.58 (s, 2H), 8.48 - 8.23 (m, 4H),
8.87 - 8.58 (s, 2H), 8.48 - 8.23 (m, 4H),
3.18 - 3.05 (q, 2H), 1.38 - 1.29 (t, 3H); Mass (ESI+) 261.05 [M+H]
2-(Ethylthio)-5-(2-nitrophenyl)pyrimidine 5f of Ii (Scheme 1):
[000181]
 8.87 - 8.58 (s, 2H), 8.46 - 8.10 (m, 4H),
8.87 - 8.58 (s, 2H), 8.46 - 8.10 (m, 4H),
3.18 - 3.05 (q, 2H), 1.38 - 1.29 (t, 3H); Mass (ESI+) 261.05 [M+H]
General procedure for the synthesis of intermediate 6a-f of Ii (Scheme 1)
[000182] To a solution of compound 5 (7.6 mmol) in DCM at 0° C was added 3- chloroperbenzoic acid (15.3 mmol) in portion wise. The resulting mixture was stirred at room temperature for 3 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, saturated bicarbonate solution (10 mL) was added, and the reaction mixture was extracted with EtOAc. The combined organic layer was dried over anhydrous NaoSCU and concentrated under reduced pressure to give compound 6. The crude products were further used without any purification.
General procedure for the synthesis of intermediate 7a-f of Ii (Scheme 1)
[000183] To a solution of compound 6 (5.1 mmol) in THF was added hydrazine hydrate (0.5 g, 10.2 mmol). The resulting mixture was stirred at 60° C- 70° C for 3 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the reaction mixture was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure to give compound 7. The crude products were further used without any purification. General procedure for the synthesis of intermediate lla-b of Ha-d (Scheme 2)
[000184] To a solution of compound 10 (1 mmol) in methanol was added 2° amine (1.5 mmol), and K2CO3 (1 mmol). The resulting mixture was stirred at room temperature for 1 h. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to get compound 11.
General procedure for the synthesis of intermediate 29 of II7 (Scheme 10):
[000185] To a solution of phenylisocyante 23 (4.2mmol) in benzene was added potassium ferz-butoxide (lOmol %) slowly and then salicylic acid 28 (4.2mmol) was added. The resulting mixture was stirred at 45-50 °C for lh. After completion of the reaction (reaction monitored by TLC) the solvent was removed under vacuum, and the compound was extracted with EtOAc. The combined organic layer was dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude products were purified on a silica gel column using hexane/EtOAc to afford compound 29.
General procedure for the synthesis of intermediate 38a-d of IIHa-d (Scheme 12):
[000186] To a solution of substituted phenols 35 (10 mmol) in DMF (10 ml) was added K2CO3 (20 mmol) and substituted 2-chloroethylacetate 36 (12 mmol) at room temperature. The reaction mixture was stirred at room temperature for 12 h, and then the reaction mixture was poured in ice water, extracted with ethyl acetate twice. Combined the organic layers washed with brine solution and dried over anhydrous NaoSCU, and concentrated to get the crude compounds 37. These crude compounds 37 were dissolved in methanol (10 ml) was added 2N NaOH (6 ml) at room temperature and stirred for l2h. Solvent was evaporated under vaccum and the resulting mixture was acidified with 2N HC1 and then solids were filtered off. The solids were washed with water and hexane and dried for some time under vaccum to get the pure substituted 2-phenoxyacetic acids 38a- d in 80-90%.
2-(3,5-Dimethylphenoxy)acetic acid (38a, Scheme 12):
[000187]
 7.01 (s, 1H), 6.77 - 6.67 (m, 2H), 4.94 -
7.01 (s, 1H), 6.77 - 6.67 (m, 2H), 4.94 -
4.87 (s, 2H), 2.39 - 2.32 (m, 6H); LC-MS (ESI+): mV 386.1962 [M + H]+.
2-Phenoxypropanoic acid (38b, Scheme 12):
[000188]
 7.34 (t, / = 8.0 Hz, 2H), 7.01 (t, / = 7.4
7.34 (t, / = 8.0 Hz, 2H), 7.01 (t, / = 7.4
Hz, 1H), 6.97 (d, / = 7.9 Hz, 2H), 4.75 (q, / = 6.75 Hz, 1H), 1.64 (d, / = 6.75 Hz, 3H); LC-MS (ESI+): mV 167.08 [M + H]+.
2-(4-Chlorophenoxy)-2-methylpropanoic acid (38c, Scheme 12):
[000189]

Hz, 2H), 1.59 (s, 6H);LC-MS (ESI+): m\z 215.12[M + H]+.
2-(o-Tolyloxy (propanoic acid (38d, Scheme 12):
lH NMR (400 MHz, CD3OD) d 7.28 - 7.15 (m, 2H), 6.99 (t, / = 7.4 Hz, 1H), 6.87 (d, / = 8.2 Hz, 1H), 4.82 (q, J = 6.7 Hz, 1H), 1.69 (d, J = 6.8 Hz, 3H); LC-MS (ESI+): mSz
181.09[M + H]+.
EXAMPLES
[000190] The following examples provide the details about the, applications of the compounds of the present disclosure. It should be understood the following is representative only, and that the invention is not limited by the details set forth in these examples.
Example 1
Biological Activity/Assay
[000191] Through an in vitro biochemistry screen, small molecules were identified which can target Nef interaction with CD80 and CD86. Out of the 1061 compounds, three scaffolds were identified. Compounds from these scaffolds (Formula I, Formula II, and Formula III) were validated in (a.) Primary biochemistry screen; and (b.) Cell-based screen.
Validation of compounds in Primary Screen
[000192] Standard EFISA assay was used to validate the compounds as a primary screen. The basic steps of the assay are outlined in Figure 1. The cytosolic peptides
CD80, CD86 and CD74 are coated on a microplate, while CD80 and CD86 has higher binding capacity with the Nef protein, CD74 is non-specific peptide and is a peptide negative control for Nef binding. The coated plates are incubated overnight at 4 °C. The plates were washed 5 times with PBS (0.02 %Tween-20) before every step. After washing, the wells were blocked with 5% blotto and incubated at RT with shaking (650 rpm). The plates were washed 5 times with PBS (0.02 % -tween20) after every step. Primary antibody mouse anti-Nef raised in rabbit dissolved in 2.5% blotto (1: 1000) were added and incubated on shaker for 1 hour at RT. Plate was washed and IOOmE anti rabbit
raised in donkey dissolved in 2.5% blotto (1: 11000) was added and incubated at RT for 45 min. Plate was washed with PBS and TMB substrate solution was added and incubated in dark for 5-10 min. The reaction was stopped with 1N H2SO4 and the absorbance was measured at 450 nm. Positive control (Nef) and Negative control (No peptide with Nef) were analysed for highest binding of Nef with peptide. The absorbance of the plates was measured at 450 nm.
[000193] A throughput screen was performed to pull out the actives. Standard quality control metrics like Z’ were used to qualify plates and Z’ scores were used to identify actives. The IC50 value of the compounds were further determined to evaluate the efficacy of the test compounds.
[000194] The IC50 values of the compounds have been illustrated in Table 1. Eighteen compounds were found to show HIV inhibiting activity and increasing the expression of CD80 and CD86 proteins. Actives predominantly belong to compounds from Formula I, II and III with IC50S in nano or subnanomolar ranges were selected. Selected compounds were then screened in the cell-based assays systems.
Table 1: pIC5o values of representative actives from Primary Screen


[000195] Table 1 illustrates the efficacy of individual compounds in inhibiting the Nef mediated downregulation of CD80 and CD86 proteins. Out of the total 19 compounds of Formula I, II, and III, the compounds which were found effective in inhibiting Nef-COSO binding, in terms of higher pICso values (ICsos in nano or

which were active in inhibiting interactions of Nef protein with both CD80 and CD86 proteins were I2b, II4 and II2d respectively.
Example 2
Cell-Based Screen: Phenotypic screen for downregulation
[000196] The schematic of the cell-based assay is as shown in the Figure 2. CD80 and CD86 are two major co-stimulatory cell surface proteins that are present on antigen presenting cells (APCs) and provide a co-stimulatory signal necessary for activation and survival of the naive T cells. Both these proteins get down regulated during HIV infection via a Nef mediated pathway. It has been speculated that Nef ability to reduce CD80 and CD86 surface expression in infected cells can prevent the activation of naive T cells, necessary for an efficient recognition and elimination of the target cells. Figure 2 illustrates that in the presence of the compounds of Formula I, Formula II or Formula III, the binding of Nef with CD80 and CD86 is inhibited thus increasing their expression. The increased expression would activate naive T cells and aid in eliminating the infection.
[000197] Briefly monocyte / B cell lines were either delivered with Nef protein or exposed to replication deficient retrovirus and the levels of surface CD80/86 was evaluated by flow cytometry with/without compound treatment. The compounds were qualified based on the ability to reverse the down regulation of CD 80/86 caused by the Nef protein. Similar quality checks were used in the secondary screen as in primary
screen. Results from representative compounds were tested in the phenotypic assay for downregulation is presented in the Figure 3. Briefly, B/ monocyte cells were preexposed to 100 m M of the 4 compounds for 2 hrs and then infected with non -replicative reterovirus containing NEF. The internalization of surface receptors were measured by flow cytometry after optimal infection (48-72 hrs). Representative compounds from each scaffold (I3b, II3 and 1112 a ) showed reversal of Nef mediated internalization of CD80/86.
Example 3
Efficacy assay system to test T cell activation:
[000198] The appropriate cell system was designed to mimic the biology of the said interactions. The schematic representation of the cell-based T cell activation assay is as shown in Figure 4. Figure 4 (A) represents the principle behind the activation of naive T cells by the antigen presenting cells (APC), including dendritic cells, B cells, monocytes, macrophages. The activation is based on CD80/86 co-stimulation along with CD3 activation by major histocompatibility complex (MHC) as evidenced by an increase in IL-2 release from the activated T cells.
[000199] Figure 4(B) is a schematic representation that displays the addition of Nef in the APC causes down regulation of the CD80/86 and hence loss of T cell activation, which leads to a reduction in IL-2 release.
[000200] Figure 4(C) displays the rescue of internalization of CD80/86 back to the surface of the APC by means of inhibitiors to the Nef protein delivered to the APC cell line, which restores CD80/86 co-stimulation and hence results in IL-2 release from the T cells.
[000201] Representative compounds from the Lormula I, II and III were tested in the functional T cell assay and the results are as presented in Ligure 5. B-cell line was pre-exposed to 100 mM of the 4 compounds for 1 hr. The cells were washed and then Nef was introduced via protein delivery agent. The cells were washed again and co-cultured with T cell line. The IL-2 release (as an indicator of activation) was measured by ELISA after 3 hrs. Addition of Nef reduced the T cell activation by more than 50%. Addition of compounds (Lb, II3 and IIFa) reversed the Nef mediated inactivation.
ADVANTAGE
[000202] The method of treatment of HIV infection by targeting M?/-CD80/86 interactions has a very distinct and unique mechanism of action: prevention of immune evasion of infected cells. None of the potential or existing therapy has targeted the viral strategy of immune evasion and the present disclosure presents a new class of drugs. Also, targeting host-virus interface is also less amenable to drug resistance than pure viral targets. This, hence, would provide hope to class of people where existing retroviral treatments have failed and also a profound impact on pre-exposure prophylaxis to the hot-spot population.
Claims
1. A method for treating HIV subtypes causing disease in a subject comprising: administering to a HIV infected subject a therapeutic dose of a compound selected from a group consisting of Formula I,

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-10 aryl, C1-10 alkyl, C2-io alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-io aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro, halogen or C1-10 alkyl, wherein C1-10 alkyl, and C1-10 alkoxy is optionally substituted with one to four substituents selected from halogen, hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2- 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl,
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and

Formula III
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10alkyl or halogen; and
n is selected from 1 to 5.
2. A method for preventing HIV subtypes causing disease in a subject in need thereof comprising: administering to a HIV high risk subject a relevant dose of a compound selected from a group consisting of

Formula I
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-io aryl, C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro or C1-10 alkyl, wherein C1-10 alkyl, and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen; hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10
heterocyclyl or C2-io heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;

Formula II
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C 1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2- 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CH2-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from Ci-io alkyl, C5-10 aryl or C2-10 heteroaryl,
R3 is absent or is selected from hydroxy, Ci-io alkyl, C5-10 aryl, C2-10 heteroaryl, C2-10 heterocyclyl, Ci-io alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano,
CH2PhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and
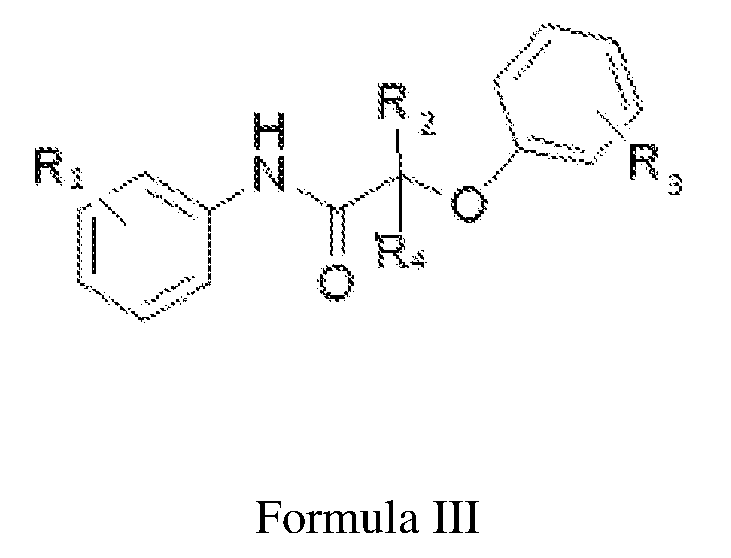
or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5.
3. The method according to claim 1 or 2, wherein compound of Formula I is selected from a group consisting of:


4. The method according to claim 1 or 2, wherein compound of Formula I is selected from a group consisting of:

5. The method according to claims 1 or 2, wherein compound of Formula II is selected from a group consisting of:

6. The method according to claims 1 or 2, wherein compound of Formula III is selected from a group consisting of:

7. A method of treating or preventing HIV infection in a subject by immune evasion mediated by internalization of CD80/86 receptors by its Nef protein comprising:
administering to a subject a relevant dose of a compound selected from a group consisting of

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein
Ri and R2 are independently selected from the group consisting of hydrogen, C6-io aryl, C1-10 alkyl, C2-io alkenyl, C2-10 alkynyl, and amino, wherein C1-10 alkyl, C2-10 alkenyl, C2-10 alkynyl, and C6-10 aryl are optionally substituted with one to four substituents independently selected from hydroxyl, halogen, C1-10 alkyl, C1-10 alkoxy or C2-10 heterocyclyl;
R3 is C6-io aryl, wherein C6-10 aryl is optionally substituted with one to four substituents independently selected from C1-10 alkoxy, nitro or C1-10 alkyl, wherein C1-10 alkyl, and Ci- 10 alkoxy is optionally substituted with one to four substituents selected from halogen; hydrogen, hydroxyl, C1-10 alkyl, C1-10 alkoxy or C2 10 heterocyclyl;
R4 and R5 are independently selected from the group consisting of hydrogen, C1-10 alkyl, and -N=CHC6 aryl, wherein N=CHC6 aryl is further substituted with one to four halogen; or R4 and R5 are joined to form a C2-10 heterocyclyl or a C2-10 heteroaryl, wherein C2-10 heterocyclyl or C2-10 heteroaryl is optionally substituted with one to four substituents independently selected from C1-10 alkyl; and
X and Y are N;

or its stereoisomers, pharmaceutically acceptable salts, polymorphs, solvates and hydrates thereof, wherein,
Ri, and R2 are independently selected from the group consisting of hydrogen, C1-10 alkyl, hydroxy, thio, amino, C2-10 heterocyclyl, C5-10 aryl, C2-10 heteroaryl, acyl, amido, imido, sulfinyl, sulfonyl, carboxaldehyde, cyano, isocyano, azido, hydrazino, nitro, halo; or Ri and R2 are joined to form an optionally substituted monocyclic or a bicyclic ring, wherein C1-10 alkyl, C2-10 heterocyclyl, C5-10 aryl, C2- 10 heteroaryl or monocyclic or a bicyclic ring are optionally substituted with one four substituents independently selected from hydroxy, C1-10 alkyl, C5-10 aryl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, cyano, thio, and -CFh-Ph-NH-COO-Ph-Me;
X is O;
Y is C=0;
Ar is selected from C1-10 alkyl, C5-10 aryl or C2-10 heteroaryl,
R3 is absent or is selected from hydroxy, C1-10 alkyl, C5- 10 aryl, C2 10 heteroaryl, C2 10 heterocyclyl, C1-10 alkoxy, halogen, haloalkyl, perhaloalkyl, nitro, cyano, CPhPhNHCOOPh-Me, NHCONHPh-(Cl)2 or CONHNHS02Ph-Me; and
n is selected from 0-5; and

Ri is independently selected from the group consisting of OH, COOH, and COORa, wherein Ra is C1-10 alkyl;
R2 and R4 is independently selected from hydrogen or C1-10 alkyl;
R3 is selected from C1-10 alkyl or halogen; and
n is selected from 1 to 5.
8. The method as claimed in claim 7, wherein the compounds causes restoration of immune signaling via T cell activation through inhibiting Nef- CD80/86 interactions.
9. A method for treating/preventing infections characterized by low levels of
CD80/86 receptors compared to non-infected state, including chronic lymphocytic leukemia, colon carcinoma, multiple myeloma, viral infections including HIV, HPV, herpes.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021532550A JP2021535200A (en) | 2018-08-13 | 2019-08-13 | Inhibitors targeting HIV-1 Nef-CD80 / CD86 interactions for therapeutic intervention |
| US17/268,440 US20220062281A1 (en) | 2018-08-13 | 2019-08-13 | Inhibitors to target hiv-1 nef-cd80/cd86 interactions for therapeutic intervention |
| EP19849238.1A EP3836933A4 (en) | 2018-08-13 | 2019-08-13 | INHIBITORS INTENDED TO TARGETING HIV-1 NEF-CD80/CD86 INTERACTIONS FOR THERAPEUTIC INTERVENTION |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201841005491 | 2018-08-13 | ||
| IN201841005491 | 2018-08-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020035880A1 true WO2020035880A1 (en) | 2020-02-20 |
Family
ID=69525270
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2019/050594 Ceased WO2020035880A1 (en) | 2018-08-13 | 2019-08-13 | Inhibitors to target hiv-1 nef-cd80/cd86 interactions for therapeutic intervention |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20220062281A1 (en) |
| EP (1) | EP3836933A4 (en) |
| JP (1) | JP2021535200A (en) |
| WO (1) | WO2020035880A1 (en) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8975254B2 (en) * | 2009-10-27 | 2015-03-10 | Orion Corporation | Androgen receptor modulating compounds |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03505579A (en) * | 1988-06-30 | 1991-12-05 | デービス,マイクル,エイチ | Method for inhibiting in vivo activity of human immunodeficiency virus (HIV) |
| JP2009126852A (en) * | 2007-11-27 | 2009-06-11 | Meiji Milk Prod Co Ltd | Pharmaceutical composition containing a pyrimidine derivative as an active ingredient |
| US8188131B2 (en) * | 2008-02-07 | 2012-05-29 | Massachusetts Eye & Ear Infirmary | Compounds that enhance Atoh1 expression |
-
2019
- 2019-08-13 EP EP19849238.1A patent/EP3836933A4/en not_active Withdrawn
- 2019-08-13 JP JP2021532550A patent/JP2021535200A/en active Pending
- 2019-08-13 WO PCT/IN2019/050594 patent/WO2020035880A1/en not_active Ceased
- 2019-08-13 US US17/268,440 patent/US20220062281A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8975254B2 (en) * | 2009-10-27 | 2015-03-10 | Orion Corporation | Androgen receptor modulating compounds |
Non-Patent Citations (3)
| Title |
|---|
| A. LUGARI ET AL.: "A specific protein disorder catalyzer of HIV-1 Nef", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 19, no. 24, 15 December 2011 (2011-12-15), pages 7401 - 7406, XP028597157, Retrieved from the Internet <URL:10.1016/j.bmc.2011.10.051> DOI: 10.1016/j.bmc.2011.10.051 * |
| S. BETZI ET AL.: "Protein-protein interaction inhibition (2P2I) combining high throughput and virtual screening: application to the HIV-1 Nef protein", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, vol. 104, no. 49, 4 December 2007 (2007-12-04), pages 19256 - 19261, XP008090298, DOI: 10.1073/pnas.0707130104 * |
| See also references of EP3836933A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3836933A1 (en) | 2021-06-23 |
| US20220062281A1 (en) | 2022-03-03 |
| JP2021535200A (en) | 2021-12-16 |
| EP3836933A4 (en) | 2022-08-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2942533C (en) | Hepatitis b core protein allosteric modulators | |
| JPWO2008023847A1 (en) | P2X4 receptor antagonist | |
| CN102015606B (en) | IRE-1α inhibitor | |
| JP5592278B2 (en) | Diazepinedione derivatives | |
| JP5843771B2 (en) | P2X4 receptor antagonist | |
| CN104955456A (en) | N-Substituted-5-Substituted Anthramoylbenzoic Acids as Sortilin Inhibitors | |
| AU2005286701A1 (en) | Compounds for inflammation and immune-related uses | |
| AU2008263166A1 (en) | Heteroaryl-substituted urea modulators of fatty acid amide hydrolase | |
| CN109824756B (en) | Phenylalanine derivative containing 4- (benzenesulfonyl) piperazine-2-ketone and preparation method and application thereof | |
| US11447501B2 (en) | Biphenyl-containing diarylpyrimido compounds, pharmaceutically-acceptable salts thereof, composition and preparation thereof | |
| JPWO2006059744A1 (en) | Activator of peroxisome proliferator activated receptor δ | |
| JPWO2012017876A1 (en) | P2X4 receptor antagonist | |
| WO2010090300A1 (en) | Diazepinedione derivatives | |
| ES2907746T3 (en) | Amide compounds and use thereof | |
| TW200812976A (en) | Compounds which inhibit the glycine transporter and uses thereof | |
| JP2009511559A (en) | Benzoxazoles useful for the treatment of inflammation | |
| NZ566722A (en) | Novel pyrimidine carboxamides | |
| WO2004069788A1 (en) | Carboxylic acid compounds | |
| WO2020035880A1 (en) | Inhibitors to target hiv-1 nef-cd80/cd86 interactions for therapeutic intervention | |
| CN110041272B (en) | 2- (2-chlorphenyl) quinazoline-4 (3H) -ketone derivative and preparation method and application thereof | |
| JP2009062278A (en) | P2X4 receptor antagonist | |
| WO2021049628A1 (en) | P2x4 receptor antagonist | |
| Hashimoto et al. | 4-Aryl/cycloalkyl-5-phenyloxazole derivatives as selective COX-2 inhibitors | |
| TW200800905A (en) | Substituted 1-amino-4-phenyldihydroisoquinolines, process for their preparation, their use as medicament, and medicament comprising them | |
| CN101362737A (en) | 2-(4-substituted phenyl-1,2,3-thiadiazole-5-mercapto)-N-substituted phenylacetamide derivatives and their preparation method and application |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19849238 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021532550 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019849238 Country of ref document: EP Effective date: 20210315 |