WO2020111030A1 - アルキレン骨格を有するジビニルエーテル化合物の製造方法 - Google Patents
アルキレン骨格を有するジビニルエーテル化合物の製造方法 Download PDFInfo
- Publication number
- WO2020111030A1 WO2020111030A1 PCT/JP2019/046071 JP2019046071W WO2020111030A1 WO 2020111030 A1 WO2020111030 A1 WO 2020111030A1 JP 2019046071 W JP2019046071 W JP 2019046071W WO 2020111030 A1 WO2020111030 A1 WO 2020111030A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- reaction
- group
- divinyl ether
- acetylene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/05—Preparation of ethers by addition of compounds to unsaturated compounds
- C07C41/06—Preparation of ethers by addition of compounds to unsaturated compounds by addition of organic compounds only
- C07C41/08—Preparation of ethers by addition of compounds to unsaturated compounds by addition of organic compounds only to carbon-to-carbon triple bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
- B01J23/04—Alkali metals
Definitions
- the present invention relates to a method for producing a divinyl ether compound having an alkylene skeleton.
- a Reppe method is known in which an alcohol (diol) is reacted with acetylene in the presence of an alkali metal catalyst.
- diethylene glycol and acetylene are reacted in the presence of diethylene glycol divinyl ether to allow the reaction to proceed until both diethylene glycol monovinyl ether and diethylene glycol divinyl ether are produced, and then extractive distillation is performed to obtain diethylene glycol.
- the technique of separating and recovering divinyl ether is described.
- any two of R 11 to R 16 represent a hydroxy group or a hydroxymethyl group, and the other represent a hydrogen atom.
- ⁇ 5> The production method according to any one of ⁇ 1> to ⁇ 4>, wherein the alkali metal catalyst is at least one selected from the group consisting of alkali metal hydroxides and alkali metal carbonates.
- the amount of the alkali metal catalyst used is in the range of 1 to 60 mol with respect to 100 mol of the compound represented by the formula (1).
- a divinyl ether compound having an alkylene skeleton can be produced from alkanediol and acetylene with a rapid production rate and a high reaction yield.
- the production method of the present invention is a method for producing a divinyl ether compound (2) by reacting an alkanediol (1) with acetylene in the absence of a solvent using an alkali metal catalyst.
- the alkylene group (alkanediyl group) represented by R 1 has 4 to 20 carbon atoms, but preferably 6 to 18 from the viewpoint of enhancing the desired effect of the present invention. is there.
- the alkylene group may be linear or branched.
- alkylene group examples include butane-1,1-diyl group, butane-1,2-diyl group, butane-1,3-diyl group, butane-1,4-diyl group, pentane-1,1- Diyl group, pentane-1,2-diyl group, pentane-1,3-diyl group, pentane-1,4-diyl group, pentane-1,5-diyl group, hexane-1,1-diyl group, hexane- 1,2-diyl group, hexane-1,3-diyl group, hexane-1,4-diyl group, hexane-1,5-diyl group, hexane-1,6-diyl group, 3-methyl-pentane-1 ,5-diyl group, heptane-1,7-diyl group, octan
- the alkanediol (1) used in the present invention preferably has a boiling point of 50° C. or higher under normal pressure, and more preferably 100-450° C. under normal pressure.
- those represented by the following formulas (3) to (5) are used in terms of enhancing the desired effects of the present invention and usefulness as a material of the corresponding divinyl ether compound. preferable.
- Examples of the alkanediol represented by the formula (3) include those represented by the formulas (3-1) to (3-3). According to the production method of the present invention, a corresponding divinyl ether compound can be obtained even from an alkanediol having such a structure.
- any two of R 11 to R 16 represent a hydroxy group or a hydroxymethyl group, and the other represent a hydrogen atom.
- R 11 to R 16 represent a hydroxy group or a hydroxymethyl group, they may be the same or different.
- any two of R 17 to R 19 represent a hydroxy group or a hydroxymethyl group, and the other represent a hydrogen atom.
- R 17 to R 19 represent a hydroxy group or a hydroxymethyl group, they may be the same or different.
- R 20 and R 21 each independently represent a hydroxy group or a hydroxymethyl group.
- any one of R 31 to R 40 represents a methyl group, and the other represent a hydrogen atom.
- R 41 ⁇ R 64 is a hydrogen atom or an alkyl group having 1 to 8 carbon atoms
- a combination of R 41 ⁇ R 64 is any one of R 41 ⁇ R 64 Carbon A combination in which all are alkyl groups of the formulas 1 to 8 and the others are hydrogen atoms, or a combination in which R 41 to R 64 are all hydrogen atoms.
- the alkali metal catalyst used in the present invention is not particularly limited, and conventionally known ones can be used. Examples thereof include hydroxides of alkali metals such as potassium hydroxide and sodium hydroxide; carbonates of alkali metals such as potassium carbonate and sodium carbonate.
- the alkali metal catalysts may be used alone or in combination of two or more. Of these, alkali metal hydroxides are preferable from the viewpoint of enhancing the desired effects of the present invention.
- the amount of the alkali metal catalyst used is preferably in the range of 1 to 60 mol, and more preferably 5 to 100 mol of the alkanediol (1), from the viewpoint of enhancing the desired effects of the present invention and manufacturing cost. It is in the range of up to 45 mol.
- the supply pressure of acetylene is not particularly limited, but from the viewpoint of safety and reaction progress, the gauge pressure is preferably 0.01 to 0.4 MPa, and more preferably the gauge pressure is 0. It is from 01 to 0.08 MPa.
- the reaction temperature of the reaction between the alkanediol (1) and acetylene is preferably in the range of 50 to 170° C., and more preferably in the range of 90 to 160° C. from the viewpoint of safety and reaction progress.
- the alkanediol (1) is previously treated with an alkali metal catalyst to be alkoxidized prior to the supply of acetylene, the alkoxidation reaction can be performed at the same reaction temperature.
- the order of contacting the alkanediol (1), acetylene and the alkali metal catalyst is arbitrary before and after, for example, after starting the supply of the alkanediol (1), acetylene and the alkali metal catalyst into the reactor,
- the reaction can be carried out by advancing the vinyl etherification reaction, or by preliminarily charging the alkanediol (1) and the alkali metal catalyst in the reactor and raising the acetylene pressure in the reactor after raising the temperature to a predetermined temperature.
- the method of starting is mentioned.
- the alkoxide reaction may be allowed to proceed prior to the supply of acetylene.
- the reaction pressure of the alkoxide reaction is preferably 1 to 101.3 kPa in absolute pressure.
- the obtained divinyl ether compound (2) can be treated and isolated by a known operation and treatment method.
- the target divinyl ether compound (2) can be isolated by separating the catalyst by filtration and then performing distillation.
- the divinyl ether compound (2) is derived from the alkanediol (1) and has a corresponding chemical structure.
- the divinyl ether compound (2) can be produced from the alkanediol (1) and acetylene with a rapid production rate and a high reaction yield. Further, since it is a solvent-free reaction, in addition to an aprotic polar solvent such as dimethyl sulfoxide, a divinyl ether compound corresponding to the alkanediol (1) used as a raw material (the same chemical structure as the target divinyl ether compound (2) The divinyl ether compound (2) can be easily obtained at low cost without adding a liquid compound other than the alkanediol (1) at room temperature other than the alkanediol (1).
- reaction process production rate measurement The following formula was used to calculate the reaction step production rate.
- r2 M/ ⁇ (t1+t2) ⁇ V ⁇ r2: reaction process production rate (g/L ⁇ hr) M: Divinyl ether production (g) t1: reaction time (hr) t2: Preparation time of potassium alkoxide (hr) V: Reaction liquid volume (L)
- Example 1 Synthesis of 1,12-octadecanediol divinyl ether (ODDVE)) 0.18 kg of potassium hydroxide (manufactured by Nippon Soda Co., Ltd.) in a pressure-resistant reaction vessel made of SUS316 having a capacity of 10 L equipped with an agitator, a pressure gauge, a thermometer, a gas introduction pipe, a gas purge line, a decompression line, and a liquid sampling line 3.3 mol) and 3.50 kg (12.2 mol) of 1,12-octadecanediol (manufactured by Ogura Gosei Kogyo Co., Ltd.) previously dissolved by heating were charged, and the inside of the container was replaced with nitrogen.
- ODDVE 1,12-octadecanediol divinyl ether
- the temperature inside the container was raised to 150° C. and the mixture was stirred at 250 rpm.
- the pressure in the container was gradually reduced to 3 kPaA (A indicates an absolute pressure), and bubbling with nitrogen of 0.1 NL/min was performed for 3 hours from the liquid sampling line.
- 0.06 kg of a distillate whose main component was water was extracted. In this way, potassium alkoxide derived from 1,12-octadecanediol was obtained.
- the inside of the container was replaced with acetylene while stirring at 420 rpm.
- the potassium concentration in the filtrate was 0.4% by mass. Further, the filtrate was subjected to simple distillation under reduced pressure of 0.02 kPaA to obtain 3.37 kg (10.0 mol) of 1,12-octadecanediol divinyl ether (hereinafter referred to as ODDVE). The obtained ODDVE had a purity of 99% or higher and a yield of 81%. The results are shown in Table 1.
- Example 2 Synthesis of 2,4-diethyl-1,5-pentanediol divinyl ether (DEPDVE)
- DEPDVE 2,4-diethyl-1,5-pentanediol divinyl ether
- Example 3 Synthesis of 1,12-dodecanediol divinyl ether (3DVE)
- the reaction rate at this time was calculated to be 52 g/L ⁇ hr, and the reaction step production rate was calculated to be 44 g/L ⁇ hr.
- the catalyst was separated by pressure-filtering this reaction liquid at 120° C. and a nitrogen pressure of 0.1 MPaG using the same filter cloth as in Example 1.
- the potassium concentration in the filtrate was 0.2% by mass.
- the filtrate was subjected to simple distillation under reduced pressure of 0.06 kPaA to obtain 3.34 kg (13.1 mol) of 1,12-dodecanediol divinyl ether (hereinafter referred to as 3DVE).
- 3DVE 1,12-dodecanediol divinyl ether
- Example 4 Synthesis of 3-methyl-1,5-pentanediol divinyl ether (MPDVE)
- MPDVE 3-methyl-1,5-pentanediol divinyl ether
- the conversion rate was 99% or more and the selectivity was 91%.
- the reaction rate at this time was calculated to be 62 g/L ⁇ hr, and the reaction step production rate was calculated to be 51 g/L ⁇ hr.
- the catalyst was separated by pressure-filtering this reaction liquid at 120° C. and a nitrogen pressure of 0.1 MPaG using the same filter cloth as in Example 1.
- the potassium concentration in the filtrate was 0.2% by mass.
- the filtrate was subjected to simple distillation under reduced pressure of 1.3 kPaA to obtain 3.41 kg (20.0 mol) of 3-methyl-1,5-pentanediol divinyl ether (hereinafter referred to as MPDVE).
- the obtained MPDVE had a purity of 99% or more and a yield of 70%.
- the results are shown in Table 1.
- ODDVE ODDVE
- the obtained ODDVE had a purity of 99% or higher and a yield of 72%.
- Table 1 The results are shown in Table 1.
- DEPDVE 0.87 kg (4.1 mol) was obtained under conditions of a pressure of 1.3 kPaA and a reflux ratio of 10, DEPDVE 0.87 kg (4.1 mol) was obtained.
- the obtained DEPDVE had a purity of 99% or more and a yield of 65%.
- the results are shown in Table 1.
- reaction rate r2 reaction step production rate
- ODDVE 1,12-octadecanediol divinyl ether
- DEPDVE 2,4-diethyl-1,5-pentanediol divinyl ether
- 3DVE 1,12-dodecanediol divinyl ether
- MPDVE 3- Methyl-1,5-pentanediol divinyl ether
- DMSO dimethyl sulfoxide
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
アルキレン骨格を有するジビニルエーテル化合物を、速やかな生産速度で且つ高い反応収率でアルカンジオール及びアセチレンから製造できる方法を提供すること。 式(1)で表される化合物とアセチレンとをアルカリ金属触媒を用いて反応させ、式(2)で表される化合物を製造する方法であって、前記反応を無溶媒で行う、製造方法。 〔式(1)中、R1は、炭素数4~20のアルキレン基を示す。〕 〔式(2)中、R1は、式(1)中のR1と同義である。〕
Description
本発明は、アルキレン骨格を有するジビニルエーテル化合物の製造方法に関するものである。
2つのビニルエーテル構造を有する化合物は、ジビニルエーテル化合物と呼ばれ、重合組成物原料の架橋成分、硬化成分として用いられる。中でも、アルキレン骨格を有するジビニルエーテル化合物は、低毒性、低刺激性、低収縮性等といった優れた特徴から、接着剤、塗料、印刷用インク、レジスト材料等への応用が期待されている。
ジビニルエーテル化合物の製造方法としては、アルカリ金属触媒の存在下でアルコール(ジオール)とアセチレンとを反応させるレッペ法が知られている。例えば、特許文献1には、ジエチレングリコールとアセチレンをジエチレングリコールジビニルエーテル存在下で反応させてジエチレングリコールモノビニルエーテルとジエチレングリコールジビニルエーテルの両方が生成した状態まで反応を進行させた後、抽出蒸留を行うことにより、ジエチレングリコールジビニルエーテルを分離回収するという手法が記載されている。
しかしながら、特許文献1に記載の製造方法では、ジエチレングリコールジビニルエーテルを高純度で回収できるものの、ジエチレングリコールジビニルエーテルまで完全には反応を進行させずに、ジエチレングリコールモノビニルエーテルを高い割合(約30質量%)で生成させるため、反応1回当たりのジエチレングリコールジビニルエーテルの反応収率が低く、実用化した場合に生産効率を損なうものと考えられる。また、この方法ではアルキレン骨格を有するジビニルエーテル化合物の製造を実現するに至っていなかった。
しかしながら、特許文献1に記載の製造方法では、ジエチレングリコールジビニルエーテルを高純度で回収できるものの、ジエチレングリコールジビニルエーテルまで完全には反応を進行させずに、ジエチレングリコールモノビニルエーテルを高い割合(約30質量%)で生成させるため、反応1回当たりのジエチレングリコールジビニルエーテルの反応収率が低く、実用化した場合に生産効率を損なうものと考えられる。また、この方法ではアルキレン骨格を有するジビニルエーテル化合物の製造を実現するに至っていなかった。
また、特許文献2には、ジメチルスルホキシドを非プロトン性極性溶媒として使用し、アルカンジオール及びアセチレンからレッペ法でアルキレン骨格を有するジビニルエーテル化合物を製造する手法が記載されているが、ジビニルエーテル化合物の生成速度(生産速度)が低いという問題があった。
一方、レッペ法とは異なる方法によるアルキレン骨格を有するジビニルエーテル化合物の製造方法として、ジオールと酢酸ビニルを用いてビニル基交換反応を行う方法が報告されている(特許文献3)。しかしながら、この手法においても、ジビニルエーテル化合物の生成速度が低く、また、高価なイリジウム触媒が反応に用いられるため、製造コストの増大という問題もあった。
本発明の課題は、アルキレン骨格を有するジビニルエーテル化合物を、速やかな生産速度で且つ高い反応収率でアルカンジオール及びアセチレンから製造できる方法を提供することにある。
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、アルカンジオールとアセチレンとの反応を無溶媒で行うことによって、アルキレン骨格を有するジビニルエーテル化合物を、速やかな生産速度で且つ高い反応収率で製造できることを見出し、本発明の完成に至った。
すなわち、本発明は、以下の<1>~<6>を提供するものである。
<1> 式(1)で表される化合物(以下、アルカンジオール(1)ともいう)とアセチレンとをアルカリ金属触媒を用いて反応させ、式(2)で表される化合物(以下、ジビニルエーテル化合物(2)ともいう)を製造する方法であって、前記反応を無溶媒で行う、製造方法(以下、本発明の製造方法とも称する)。

〔式(1)中、R1は、炭素数4~20のアルキレン基を示す。〕
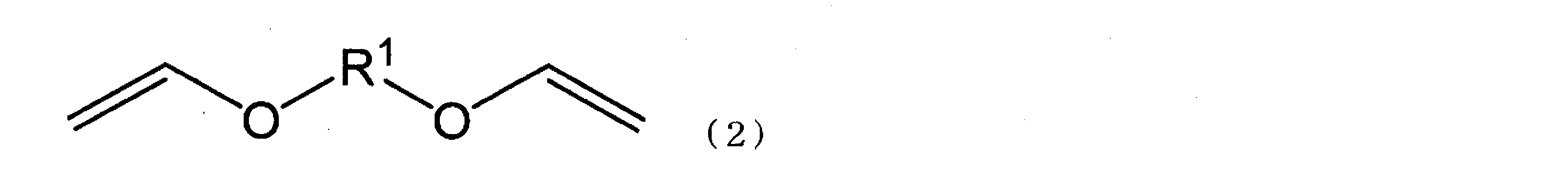
〔式(2)中、R1は、式(1)中のR1と同義である。〕
<2> 式(1)で表される化合物が、式(3)、(4)又は(5)で表されるものである、<1>に記載の製造方法。

〔式(3)中、R11~R16のうちいずれか2つはヒドロキシ基又はヒドロキシメチル基を示し、その他は水素原子を示す。〕

〔式(4)中、R31~R40のうちいずれか1つはメチル基を示し、その他は水素原子を示す。〕
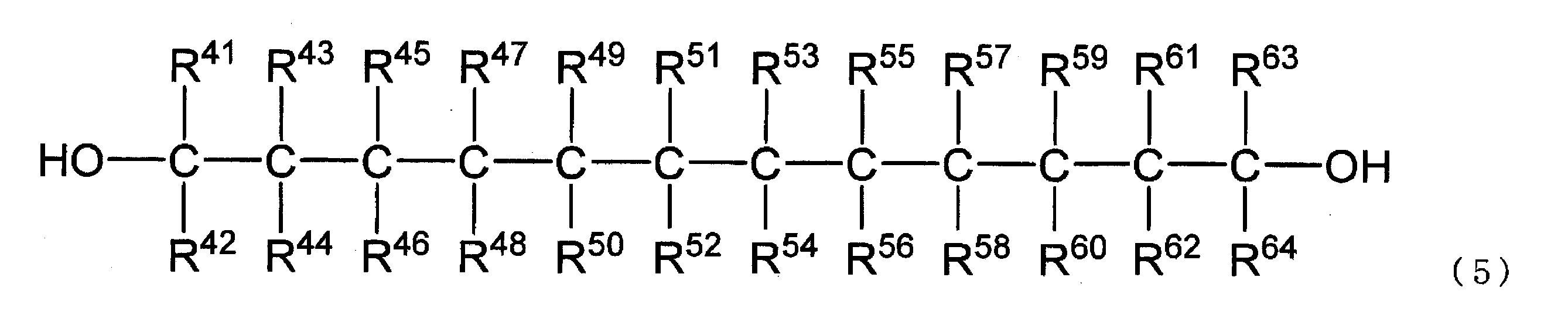
〔式(5)中、R41~R64は、水素原子又は炭素数1~8のアルキル基を示すが、R41~R64の組み合わせは、R41~R64のうちいずれか1つが炭素数1~8のアルキル基であり、その他は水素原子である組み合わせ、又はR41~R64がいずれも水素原子である組み合わせである。〕
<3> 式(1)で表される化合物が、3-メチル-1,5-ペンタンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-ブチル-2-エチル-1,3-プロパンジオール、1,12-ドデカンジオール及び1,12-オクタデカンジオールからなる群から選択される少なくとも1種である、<1>又は<2>に記載の製造方法。
<4> 前記反応を50~170℃の範囲で行う、<1>~<3>のいずれかに記載の製造方法。
<5> 前記アルカリ金属触媒が、アルカリ金属の水酸化物及びアルカリ金属の炭酸塩からなる群から選択される少なくとも1種である、<1>~<4>のいずれかに記載の製造方法。
<6> 前記アルカリ金属触媒の使用量が、式(1)で表される化合物100モルに対して、1~60モルの範囲である、<1>~<5>のいずれかに記載の製造方法。
<6> 前記アルカリ金属触媒の使用量が、式(1)で表される化合物100モルに対して、1~60モルの範囲である、<1>~<5>のいずれかに記載の製造方法。
本発明によれば、アルキレン骨格を有するジビニルエーテル化合物を、速やかな生産速度で且つ高い反応収率でアルカンジオール及びアセチレンから製造できる。
本発明の製造方法は、アルカンジオール(1)とアセチレンとを、アルカリ金属触媒を用いて無溶媒で反応させ、ジビニルエーテル化合物(2)を製造する方法である。
式(1)、(2)中、R1で示されるアルキレン基(アルカンジイル基)の炭素数は4~20であるが、本発明の所望の効果を高める点で、好ましくは6~18である。また、アルキレン基は直鎖状でも分岐鎖状でもよい。
アルキレン基の具体例としては、ブタン-1,1-ジイル基、ブタン-1,2-ジイル基、ブタン-1,3-ジイル基、ブタン-1,4-ジイル基、ペンタン-1,1-ジイル基、ペンタン-1,2-ジイル基、ペンタン-1,3-ジイル基、ペンタン-1,4-ジイル基、ペンタン-1,5-ジイル基、ヘキサン-1,1-ジイル基、ヘキサン-1,2-ジイル基、ヘキサン-1,3-ジイル基、ヘキサン-1,4-ジイル基、ヘキサン-1,5-ジイル基、ヘキサン-1,6-ジイル基、3-メチル-ペンタン-1,5-ジイル基、ヘプタン-1,7-ジイル基、オクタン-1,8-ジイル基、ノナン-1,9-ジイル基、2,4-ジエチル-ペンタン-1,5-ジイル基、2-ブチル-2-エチル-プロパン-1,3-ジイル基、デカン-1,10-ジイル基、ウンデカン-1,11-ジイル基、ドデカン-1,12-ジイル基、トリデカン-1,13-ジイル基、テトラデカン-1,14-ジイル基、ペンタデカン-1,15-ジイル基、ヘキサデカン-1,16-ジイル基、ヘプタデカン-1,17-ジイル基、オクタデカン-1,12-ジイル基、オクタデカン-1,18-ジイル基等が挙げられる。
アルキレン基の具体例としては、ブタン-1,1-ジイル基、ブタン-1,2-ジイル基、ブタン-1,3-ジイル基、ブタン-1,4-ジイル基、ペンタン-1,1-ジイル基、ペンタン-1,2-ジイル基、ペンタン-1,3-ジイル基、ペンタン-1,4-ジイル基、ペンタン-1,5-ジイル基、ヘキサン-1,1-ジイル基、ヘキサン-1,2-ジイル基、ヘキサン-1,3-ジイル基、ヘキサン-1,4-ジイル基、ヘキサン-1,5-ジイル基、ヘキサン-1,6-ジイル基、3-メチル-ペンタン-1,5-ジイル基、ヘプタン-1,7-ジイル基、オクタン-1,8-ジイル基、ノナン-1,9-ジイル基、2,4-ジエチル-ペンタン-1,5-ジイル基、2-ブチル-2-エチル-プロパン-1,3-ジイル基、デカン-1,10-ジイル基、ウンデカン-1,11-ジイル基、ドデカン-1,12-ジイル基、トリデカン-1,13-ジイル基、テトラデカン-1,14-ジイル基、ペンタデカン-1,15-ジイル基、ヘキサデカン-1,16-ジイル基、ヘプタデカン-1,17-ジイル基、オクタデカン-1,12-ジイル基、オクタデカン-1,18-ジイル基等が挙げられる。
本発明で用いるアルカンジオール(1)としては、常圧での沸点が50℃以上のものが好ましく、常圧での沸点が100~450℃のものがより好ましい。
また、アルカンジオール(1)としては、本発明の所望の効果を高める点や対応するジビニルエーテル化合物の材料としての有用性の点で、下記式(3)~(5)で表されるものが好ましい。また、式(3)で表されるアルカンジオールとしては、式(3-1)~(3-3)で表されるものが挙げられる。本発明の製造方法によれば、このような構造のアルカンジオールからでも、対応するジビニルエーテル化合物を得ることができる。
また、アルカンジオール(1)としては、本発明の所望の効果を高める点や対応するジビニルエーテル化合物の材料としての有用性の点で、下記式(3)~(5)で表されるものが好ましい。また、式(3)で表されるアルカンジオールとしては、式(3-1)~(3-3)で表されるものが挙げられる。本発明の製造方法によれば、このような構造のアルカンジオールからでも、対応するジビニルエーテル化合物を得ることができる。

〔式(3)中、R11~R16のうちいずれか2つはヒドロキシ基又はヒドロキシメチル基を示し、その他は水素原子を示す。〕
なお、R11~R16は、ヒドロキシ基又はヒドロキシメチル基を示す場合、同種であっても異種であってもよい。
なお、R11~R16は、ヒドロキシ基又はヒドロキシメチル基を示す場合、同種であっても異種であってもよい。

〔式(3-1)中、R17~R19のうちいずれか2つはヒドロキシ基又はヒドロキシメチル基を示し、その他は水素原子を示す。〕
なお、R17~R19は、ヒドロキシ基又はヒドロキシメチル基を示す場合、同種であっても異種であってもよい。
なお、R17~R19は、ヒドロキシ基又はヒドロキシメチル基を示す場合、同種であっても異種であってもよい。
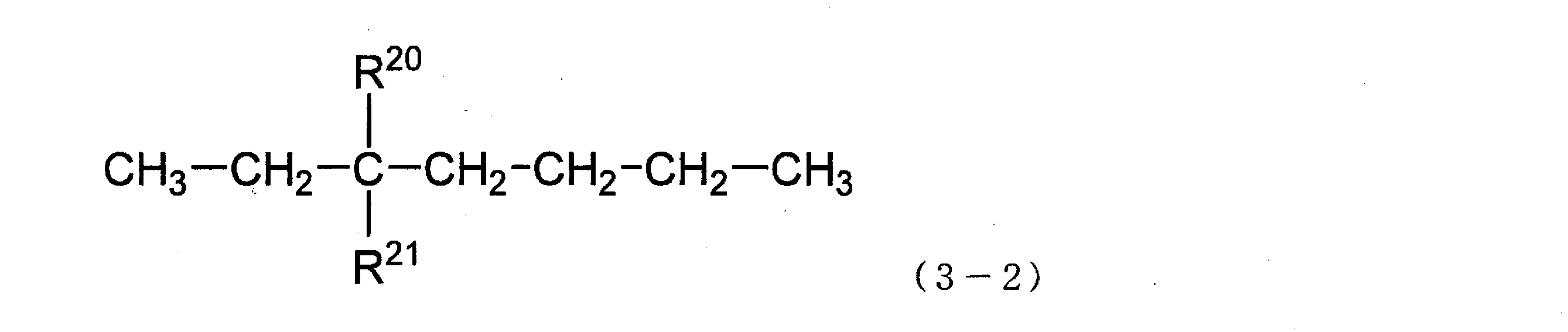
〔式(3-2)中、R20及びR21は、それぞれ独立して、ヒドロキシ基又はヒドロキシメチル基を示す。〕

〔式(3-3)中、R22及びR23は、それぞれ独立して、ヒドロキシ基又はヒドロキシメチル基を示す。〕

〔式(4)中、R31~R40のうちいずれか1つはメチル基を示し、その他は水素原子を示す。〕

〔式(5)中、R41~R64は、水素原子又は炭素数1~8のアルキル基を示すが、R41~R64の組み合わせは、R41~R64のうちいずれか1つが炭素数1~8のアルキル基であり、その他は水素原子である組み合わせ、又はR41~R64がいずれも水素原子である組み合わせである。〕
R41~R64で示されるアルキル基は直鎖状でも分岐鎖状でもよく、例えば、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、ネオペンチル基、n-ヘキシル基、イソヘキシル基、n-ヘプチル基、n-オクチル基等が挙げられる。
アルカンジオール(1)の具体例としては、例えば、1,2-ブタンジオール、1,3-ブタンジオール、1,4-ブタンジオール、2,3-ブタンジオール、2-メチル-1,3-プロパンジオール、1,2-ペンタンジオール、1,4-ペンタンジオール、1,5-ペンタンジオール、2,4-ペンタンジオール、2-メチル-1,3-ブタンジオール、3-メチル-1,3-ブタンジオール、2-メチル-2,3-ブタンジオール、2,2-ジメチル-1,3-プロパンジオール、1,2-ヘキサンジオール、1,5-ヘキサンジオール、1,6-ヘキサンジオール、2,4-ヘキサンジオール、2,5-ヘキサンジオール、3-メチル-1,5-ペンタンジオール、4-メチル-2,3-ペンタンジオール、ヘキシレングリコール、3,3-ジメチル-1,2-ブタンジオール、2,2-ジメチル-1,3-ブタンジオール、ピナコール、2-エチル-2-メチル-1,3-プロパンジオール、1,2-ヘプタンジオール、1,4-ヘプタンジオール、1,7-ヘプタンジオール、3,3-ヘプタンジオール、3,4-ヘプタンジオール、3,5-ヘプタンジオール、4,4-ヘプタンジオール、5-メチル-2,4-ヘキサンジオール、3,3-ジメチル-1,5-ペンタンジオール、2,4-ジメチル-2,4-ペンタンジオール、2,2-ジエチル-1,3-プロパンジオール、2-メチル-2-プロピル-1,3-プロパンジオール、1,2-オクタンジオール、1,8-オクタンジオール、4,5-オクタンジオール、3-メチル-2,4-ヘプタンジオール、2-エチル-1,2-ヘキサンジオール、2-エチル-1,3-ヘキサンジオール、2-エチル-1,4-ヘキサンジオール、2,5-ジメチル-2,5-ヘキサンジオール、2-プロピル-1,2-ペンタンジオール、2,4,4-トリメチル-1,2-ペンタンジオール、2-プロピル-1,3-ペンタンジオール、2-メチル-2-(1-メチルプロピル)-1,3-プロパンジオール、2,2,4-トリメチル-1,3-ペンタンジオール、2,4,4-トリメチル-2,3-ペンタンジオール、1,2-ノナンジオール、1,9-ノナンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-エチル-3-プロピル-1,4-ブタンジオール、2-ブチル-2-エチル-1,3-プロパンジオール、1,2-デカンジオール、1,10-デカンジオール、2,7-ジメチル-2,7-オクタンジオール、3,6-ジメチル-3,6-オクタンジオール、2,3,4,5-テトラメチル-3,4-ヘキサンジオール、2,2-ジブチル-1,3-プロパンジオール、2,2-ジイソブチル-1,3-プロパンジオール、1,2-ドデカンジオール、1,12-ドデカンジオール、5-エチル-3-メチル-2,4-ノナンジオール、7-エチル-2-メチル-4,6-ノナンジオール、2-ブチル-1,3-オクタンジオール、2-メチル-2,3-ドデカンジオール、2,2-ジイソアミル-1,3-プロパンジオール、2-(4,4-ジメチルペンチル)-2-プロピル-1,3-プロパンジオール、1,2-テトラデカンジオール、1,14-テトラデカンジオール、2,2,9,9-テトラメチル-1,10-デカンジオール、2-オクチル-2-プロピル-1,3-プロパンジオール、1,2-ヘキサデカンジオール、1,16-ヘキサデカンジオール、2-デシル-2-プロピル-1,3-プロパンジオール、2-(2-メチルプロピル)-2-ノニル-1,3-プロパンジオール、5,13-ヘプタデカンジオール、2-ドデシル-2-エチル-1,3-プロパンジオール、1,12-オクタデカンジオール、2,9-ジメチル-2,9-ジプロピル-1,10-デカンジオール、2-ドデシル-2-プロピル-1,3-プロパンジオール、2,2-ジオクチル-1,3-プロパンジオール、8-エチル-1,18-オクタデカンジオールが挙げられる。
これらの中では、3-メチル-1,5-ペンタンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-ブチル-2-エチル-1,3-プロパンジオール、1,12-ドデカンジオール及び1,12-オクタデカンジオールからなる群から選択される少なくとも1種が好ましい。
これらの中では、3-メチル-1,5-ペンタンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-ブチル-2-エチル-1,3-プロパンジオール、1,12-ドデカンジオール及び1,12-オクタデカンジオールからなる群から選択される少なくとも1種が好ましい。
アルカンジオール(1)の使用量は、製造コストや生成するジビニルエーテル化合物の高純度化の点で、反応系内の液相(生成物を除く)の総量に対して、好ましくは85~99質量%、より好ましくは90~99質量%である。
また、アルカンジオール(1)とアルカリ金属触媒の合計の仕込み割合は、仕込み成分(アセチレンを除く)の合計に対して、好ましくは95~100質量%、より好ましくは97.5~100質量%である。
また、アルカンジオール(1)とアルカリ金属触媒の合計の仕込み割合は、仕込み成分(アセチレンを除く)の合計に対して、好ましくは95~100質量%、より好ましくは97.5~100質量%である。
本発明で用いるアルカリ金属触媒は特に限定されるものでなく従来公知のものを用いることができる。例えば、水酸化カリウム、水酸化ナトリウム等のアルカリ金属の水酸化物;炭酸カリウム、炭酸ナトリウム等のアルカリ金属の炭酸塩等が挙げられる。なお、アルカリ金属触媒は1種を単独で用いても2種以上を組み合わせて用いてもよい。これらの中では、本発明の所望の効果を高める点で、アルカリ金属の水酸化物が好ましい。
アルカリ金属触媒の使用量は、本発明の所望の効果を高める点や製造コストの点で、アルカンジオール(1)100モルに対して、好ましくは1~60モルの範囲であり、より好ましくは5~45モルの範囲である。
アルカリ金属触媒の使用量は、本発明の所望の効果を高める点や製造コストの点で、アルカンジオール(1)100モルに対して、好ましくは1~60モルの範囲であり、より好ましくは5~45モルの範囲である。
本発明の製造方法における、アセチレンの供給圧力は特に限定されないが、安全性及び反応進行性の観点から、好ましくはゲージ圧で0.01~0.4MPaであり、より好ましくはゲージ圧で0.01~0.08MPaである。
アルカンジオール(1)とアセチレンの反応の反応温度は、安全性及び反応進行性の観点から、好ましくは50~170℃の範囲であり、より好ましくは90~160℃の範囲である。なお、アセチレン供給に先立ちアルカンジオール(1)をアルカリ金属触媒で予め処理してアルコキシド化する場合、当該アルコキシド化反応も同様の反応温度で行うことができる。
アルカンジオール(1)とアセチレンの反応の反応時間は、アルカンジオール(1)の種類等に応じて調整すればよいが、通常1~72時間であり、好ましくは1.5~48時間である。
また、アルカンジオール(1)とアセチレンの反応は、バッチ法、半連続法又は連続法で実施することができる。
アルカンジオール(1)とアセチレンの反応の反応温度は、安全性及び反応進行性の観点から、好ましくは50~170℃の範囲であり、より好ましくは90~160℃の範囲である。なお、アセチレン供給に先立ちアルカンジオール(1)をアルカリ金属触媒で予め処理してアルコキシド化する場合、当該アルコキシド化反応も同様の反応温度で行うことができる。
アルカンジオール(1)とアセチレンの反応の反応時間は、アルカンジオール(1)の種類等に応じて調整すればよいが、通常1~72時間であり、好ましくは1.5~48時間である。
また、アルカンジオール(1)とアセチレンの反応は、バッチ法、半連続法又は連続法で実施することができる。
また、アルカンジオール(1)、アセチレン及びアルカリ金属触媒の接触の順番の前後は任意であり、例えば、アルカンジオール(1)、アセチレン及びアルカリ金属触媒の反応器内への供給を開始してからジビニルエーテル化反応を進行させる手法や、アルカンジオール(1)とアルカリ金属触媒を反応器内に予め仕込んでおき、所定の温度に昇温後、反応器内のアセチレン圧力を昇圧することによって、反応を開始させる手法が挙げられる。アルカンジオール(1)とアルカリ金属触媒を反応器内に予め仕込んでおく場合において、アセチレン供給に先立ちアルコキシド化反応を進行させてもよい。アルコキシド化反応の反応圧力は、好ましくは絶対圧力で1~101.3kPaである。
アルカンジオール(1)とアセチレンの反応の反応終了後、得られたジビニルエーテル化合物(2)は、公知の操作、処理方法により処理し、単離することができる。例えば、ろ過によって触媒を分離した後、蒸留を行うことで、目的とするジビニルエーテル化合物(2)を単離することができる。なお、ジビニルエーテル化合物(2)は、アルカンジオール(1)に由来するものであり、対応した化学構造を有する。
本発明によれば、ジビニルエーテル化合物(2)を、速やかな生産速度で且つ高い反応収率でアルカンジオール(1)及びアセチレンから製造できる。また、無溶媒反応であるため、ジメチルスルホキシド等の非プロトン性極性溶媒の他、原料として使用したアルカンジオール(1)に対応するジビニルエーテル化合物(目的とするジビニルエーテル化合物(2)と同じ化学構造のジビニルエーテル化合物)などといった、アルカンジオール(1)以外の常温で液状化合物を添加することなく、ジビニルエーテル化合物(2)を低コストで且つ簡便に得ることができる。
以下、実施例を挙げて本発明を詳細に説明するが、本発明はこれら実施例に限定されるものではない。なお、以下の実施例における測定は、次の測定方法に従った。
(反応液の組成分析)
反応液の組成分析にはガスクロマトグラフィーを用いた。分析条件は以下のとおりである。
装置:製品名「GC-2010Plus」(島津製作所製)
検出器:FID
カラム:DB-5(30m、0.25mmID、1.0μm、Agilent社製)
反応液の組成分析にはガスクロマトグラフィーを用いた。分析条件は以下のとおりである。
装置:製品名「GC-2010Plus」(島津製作所製)
検出器:FID
カラム:DB-5(30m、0.25mmID、1.0μm、Agilent社製)
・1,12-オクタデカンジオールジビニルエーテル(ODDVE)
INJ温度:250℃
DET温度:250℃
スプリット比:50
カラム温度:180℃で3min保持→10℃/minで250℃まで昇温→250℃で20min保持(計30min)
INJ温度:250℃
DET温度:250℃
スプリット比:50
カラム温度:180℃で3min保持→10℃/minで250℃まで昇温→250℃で20min保持(計30min)
・2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル(DEPDVE)
INJ温度:250℃
DET温度:300℃
スプリット比:50
カラム温度:100℃から160℃まで4℃/minで昇温→160℃で10min保持→20℃/minで300℃まで昇温→300℃で13min保持(計45min)
INJ温度:250℃
DET温度:300℃
スプリット比:50
カラム温度:100℃から160℃まで4℃/minで昇温→160℃で10min保持→20℃/minで300℃まで昇温→300℃で13min保持(計45min)
・1,12-ドデカンジオールジビニルエーテル(3DVE)
INJ温度:250℃
DET温度:270℃
スプリット比:50
カラム温度:180℃で3min保持→5℃/minで250℃まで昇温→250℃で13min保持(計30min)
INJ温度:250℃
DET温度:270℃
スプリット比:50
カラム温度:180℃で3min保持→5℃/minで250℃まで昇温→250℃で13min保持(計30min)
・3-メチル-1,5ペンタンジオールジビニルエーテル(MPDVE)
INJ温度:250℃
DET温度:300℃
スプリット比:50
カラム温度:100℃で5min保持→10℃/minで280℃まで昇温→280℃で7min保持(計30min)
INJ温度:250℃
DET温度:300℃
スプリット比:50
カラム温度:100℃で5min保持→10℃/minで280℃まで昇温→280℃で7min保持(計30min)
(カリウム濃度測定)
反応液のカリウム濃度測定には以下の装置を用いた。
装置:製品名「自動滴定装置COM-1700A」(平沼産業製)
滴定液:0.2mol/L 塩酸、エタノール
反応液のカリウム濃度測定には以下の装置を用いた。
装置:製品名「自動滴定装置COM-1700A」(平沼産業製)
滴定液:0.2mol/L 塩酸、エタノール
(留出液の水分測定)
留出液の水分測定には以下の装置を用いた。
装置:製品名「自動水分測定装置AQV-300」(平沼産業製)
滴定液:ハイドナール コンポジット5K(林純薬製)
滴定溶媒:カールフィッシャー試薬 ハヤシソルベント CE(林純薬製)
留出液の水分測定には以下の装置を用いた。
装置:製品名「自動水分測定装置AQV-300」(平沼産業製)
滴定液:ハイドナール コンポジット5K(林純薬製)
滴定溶媒:カールフィッシャー試薬 ハヤシソルベント CE(林純薬製)
(反応速度測定)
反応速度の計算には、以下の式を用いた。
r1=M/{(t1)×V}
r1:反応速度(g/L・hr)
M:ジビニルエーテル生成量(g)
t1:反応時間(hr)
V:反応液体積(L)
反応速度の計算には、以下の式を用いた。
r1=M/{(t1)×V}
r1:反応速度(g/L・hr)
M:ジビニルエーテル生成量(g)
t1:反応時間(hr)
V:反応液体積(L)
(反応工程生産速度測定)
反応工程生産速度の計算には、以下の式を用いた。
r2=M/{(t1+t2)×V}
r2:反応工程生産速度(g/L・hr)
M:ジビニルエーテル生成量(g)
t1:反応時間(hr)
t2:カリウムアルコキシド調製時間(hr)
V:反応液体積(L)
反応工程生産速度の計算には、以下の式を用いた。
r2=M/{(t1+t2)×V}
r2:反応工程生産速度(g/L・hr)
M:ジビニルエーテル生成量(g)
t1:反応時間(hr)
t2:カリウムアルコキシド調製時間(hr)
V:反応液体積(L)
(実施例1:1,12-オクタデカンジオールジビニルエーテル(ODDVE)の合成)
撹拌機、圧力計、温度計、ガス導入管、ガスパージライン、減圧ライン、液サンプリングラインを備えた容量10LのSUS316製耐圧反応容器に、水酸化カリウム(日本曹達(株)製)0.18kg(3.3mol)と、加温することで予め溶解させておいた1,12-オクタデカンジオール(小倉合成工業(株)製)3.50kg(12.2mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を150℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に3kPaA(Aは絶対圧力を示す)まで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.06kg抜き出した。このようにすることで、1,12-オクタデカンジオール由来のカリウムアルコキシドを得た。
その後、420rpmで撹拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG(Gはゲージ圧力を示す)、150℃に保持し、11.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は97%であった。このときの反応速度は61g/L・hr、反応工程生産速度は49g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、ろ布(通気度:0.6~1.8cc/cm2・sec、ポリフェニレンサルファイド樹脂製)を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.4質量%であった。さらに、0.02kPaA減圧下、ろ液の単蒸留を行い、1,12-オクタデカンジオールジビニルエーテル(以下、ODDVEと称す)3.37kg(10.0mol)を得た。得られたODDVEは、純度99%以上、収率81%であった。結果を表1に示す。
撹拌機、圧力計、温度計、ガス導入管、ガスパージライン、減圧ライン、液サンプリングラインを備えた容量10LのSUS316製耐圧反応容器に、水酸化カリウム(日本曹達(株)製)0.18kg(3.3mol)と、加温することで予め溶解させておいた1,12-オクタデカンジオール(小倉合成工業(株)製)3.50kg(12.2mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を150℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に3kPaA(Aは絶対圧力を示す)まで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.06kg抜き出した。このようにすることで、1,12-オクタデカンジオール由来のカリウムアルコキシドを得た。
その後、420rpmで撹拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG(Gはゲージ圧力を示す)、150℃に保持し、11.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は97%であった。このときの反応速度は61g/L・hr、反応工程生産速度は49g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、ろ布(通気度:0.6~1.8cc/cm2・sec、ポリフェニレンサルファイド樹脂製)を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.4質量%であった。さらに、0.02kPaA減圧下、ろ液の単蒸留を行い、1,12-オクタデカンジオールジビニルエーテル(以下、ODDVEと称す)3.37kg(10.0mol)を得た。得られたODDVEは、純度99%以上、収率81%であった。結果を表1に示す。
(実施例2:2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル(DEPDVE)の合成)
実施例1と同じ反応容器に、2,4-ジエチル-1,5-ペンタンジオール(KHネオケム(株)製)3.80kg(23.7mol)と、水酸化カリウム0.29kg(5.2mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を150℃まで昇温し、250rpmで撹拌した。液サンプリングラインから1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.09kg抜き出した。このようにすることで、2,4-ジエチル-1,5-ペンタンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、11.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は97%であった。このときの反応速度は60g/L・hr、反応工程生産速度は47g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、理論段数5の桐山蒸留塔を用いて、圧力1.3kPaA、還流比1の条件でろ液の精密蒸留を行い、2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル(以下、DEPDVEと称す)3.50kg(16.5mol)を得た。得られたDEPDVEは、純度99%以上、収率70%であった。結果を表1に示す。
実施例1と同じ反応容器に、2,4-ジエチル-1,5-ペンタンジオール(KHネオケム(株)製)3.80kg(23.7mol)と、水酸化カリウム0.29kg(5.2mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を150℃まで昇温し、250rpmで撹拌した。液サンプリングラインから1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.09kg抜き出した。このようにすることで、2,4-ジエチル-1,5-ペンタンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、11.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は97%であった。このときの反応速度は60g/L・hr、反応工程生産速度は47g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、理論段数5の桐山蒸留塔を用いて、圧力1.3kPaA、還流比1の条件でろ液の精密蒸留を行い、2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル(以下、DEPDVEと称す)3.50kg(16.5mol)を得た。得られたDEPDVEは、純度99%以上、収率70%であった。結果を表1に示す。
(実施例3:1,12-ドデカンジオールジビニルエーテル(3DVE)の合成)
実施例1と同じ反応容器に、水酸化カリウム0.23kg(4.1mol)と、加温することで予め溶解させておいた1,12-ドデカンジオール(富士フィルムワコーケミカル(株)製)3.96kg(19.6mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を135℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に20kPaAまで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.09kg抜き出した。このようにすることで、1,12-ドデカンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、17hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は94%であった。このときの反応速度は52g/L・hr、反応工程生産速度は44g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、0.06kPaA減圧下、ろ液の単蒸留を行い、1,12-ドデカンジオールジビニルエーテル(以下、3DVEと称す)3.34kg(13.1mol)を得た。得られた3DVEは、純度99%以上、収率67%であった。結果を表1に示す。
実施例1と同じ反応容器に、水酸化カリウム0.23kg(4.1mol)と、加温することで予め溶解させておいた1,12-ドデカンジオール(富士フィルムワコーケミカル(株)製)3.96kg(19.6mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を135℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に20kPaAまで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.09kg抜き出した。このようにすることで、1,12-ドデカンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、17hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は94%であった。このときの反応速度は52g/L・hr、反応工程生産速度は44g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、0.06kPaA減圧下、ろ液の単蒸留を行い、1,12-ドデカンジオールジビニルエーテル(以下、3DVEと称す)3.34kg(13.1mol)を得た。得られた3DVEは、純度99%以上、収率67%であった。結果を表1に示す。
(実施例4:3-メチル-1,5-ペンタンジオールジビニルエーテル(MPDVE)の合成)
実施例1と同じ反応容器に、3-メチル-1,5-ペンタンジオール(富士フィルムワコーケミカル(株)製)3.40kg(28.8mol)と、水酸化カリウム0.34kg(6.0mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を110℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に5.5kPaAまで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.11kg抜き出した。このようにすることで、3-メチル-1,5-ペンタンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、10hr反応させた。その後、容器内温度を135℃まで降温し、さらに3.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は91%であった。このときの反応速度は62g/L・hr、反応工程生産速度は51g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、1.3kPaA減圧下、ろ液の単蒸留を行い、3-メチル-1,5-ペンタンジオールジビニルエーテル(以下、MPDVEと称す)3.41kg(20.0mol)を得た。得られたMPDVEは、純度99%以上、収率70%であった。結果を表1に示す。
実施例1と同じ反応容器に、3-メチル-1,5-ペンタンジオール(富士フィルムワコーケミカル(株)製)3.40kg(28.8mol)と、水酸化カリウム0.34kg(6.0mol)とを仕込み、容器内を窒素置換した。
窒素置換後、容器内温度を110℃まで昇温し、250rpmで撹拌した。容器内圧力を徐々に5.5kPaAまで減圧し、液サンプリングラインから0.1NL/minの窒素によるバブリングを3hr行った。同操作により、主成分が水である留出液を0.11kg抜き出した。このようにすることで、3-メチル-1,5-ペンタンジオール由来のカリウムアルコキシドを得た。
その後、容器内温度を155℃まで昇温した後、420rpmで攪拌しながら、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内を0.03MPaG、155℃に保持し、10hr反応させた。その後、容器内温度を135℃まで降温し、さらに3.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は91%であった。このときの反応速度は62g/L・hr、反応工程生産速度は51g/L・hrと計算された。
この反応液を、120℃、窒素圧力0.1MPaGにて、実施例1と同じろ布を用いて加圧ろ過することで、触媒を分離した。ろ液中のカリウム濃度は0.2質量%であった。さらに、1.3kPaA減圧下、ろ液の単蒸留を行い、3-メチル-1,5-ペンタンジオールジビニルエーテル(以下、MPDVEと称す)3.41kg(20.0mol)を得た。得られたMPDVEは、純度99%以上、収率70%であった。結果を表1に示す。
(比較例1:1,12-オクタデカンジオールジビニルエーテル(ODDVE)の合成)
実施例1と同じ反応容器に、水酸化カリウム0.17kg(3.0mol)と、加温することで予め溶解させておいた1,12-オクタデカンジオール1.50kg(5.2mol)と、ジメチルスルホキシド(日本リファイン(株)製)3.50kgとを仕込み、容器内を窒素置換した。
容器内を420rpmで攪拌しながら、80℃まで昇温した後、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内圧力を0.03MPaGに保持し、容器内温度80℃で13.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は89%であった。このときの反応速度は22g/L・hr、反応工程生産速度は22g/L・hrと計算された。
この反応液を10Lポリ容器に移し静置して分液させた後、上層を抜き出した。上層液中のカリウム濃度は0.5質量%であった。さらに、0.02kPaA減圧下、上層液の単蒸留を行い、ODDVE1.28kg(3.8mol)を得た。得られたODDVEは、純度99%以上、収率72%であった。結果を表1に示す。
実施例1と同じ反応容器に、水酸化カリウム0.17kg(3.0mol)と、加温することで予め溶解させておいた1,12-オクタデカンジオール1.50kg(5.2mol)と、ジメチルスルホキシド(日本リファイン(株)製)3.50kgとを仕込み、容器内を窒素置換した。
容器内を420rpmで攪拌しながら、80℃まで昇温した後、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内圧力を0.03MPaGに保持し、容器内温度80℃で13.5hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は89%であった。このときの反応速度は22g/L・hr、反応工程生産速度は22g/L・hrと計算された。
この反応液を10Lポリ容器に移し静置して分液させた後、上層を抜き出した。上層液中のカリウム濃度は0.5質量%であった。さらに、0.02kPaA減圧下、上層液の単蒸留を行い、ODDVE1.28kg(3.8mol)を得た。得られたODDVEは、純度99%以上、収率72%であった。結果を表1に示す。
(比較例2:2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル(DEPDVE)の合成)
実施例1と同じ反応容器に、2,4-ジエチル-1,5-ペンタンジオール1.00kg(6.3mol)と、水酸化カリウム0.10kg(1.8mol)と、ジメチルスルホキシド4.00kgとを仕込み、容器内を窒素置換した。
容器内を420rpmで攪拌しながら、80℃まで昇温した後、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内圧力を0.03MPaGに保持し、容器内温度80℃で7hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は90%であった。このときの反応速度は32g/L・hr、反応工程生産速度は32g/L・hrと計算された。
この反応液を20Lポリ容器に移し、ヘキサン5.20kgを加えて振とうした後、静置した。静置後、上層を抜き出し、減圧濃縮した。さらに理論段数10の桐山蒸留塔を用いて、濃縮液の精密蒸留を行った。圧力1.3kPaA、還流比10の条件で、DEPDVE0.87kg(4.1mol)を得た。得られたDEPDVEは、純度99%以上、収率65%であった。結果を表1に示す。
実施例1と同じ反応容器に、2,4-ジエチル-1,5-ペンタンジオール1.00kg(6.3mol)と、水酸化カリウム0.10kg(1.8mol)と、ジメチルスルホキシド4.00kgとを仕込み、容器内を窒素置換した。
容器内を420rpmで攪拌しながら、80℃まで昇温した後、容器内をアセチレン置換した。そのままアセチレンを連続的に供給して容器内圧力を0.03MPaGに保持し、容器内温度80℃で7hr反応させた。反応液のガスクロマトグラフィー分析の結果、転化率は99%以上であり、選択率は90%であった。このときの反応速度は32g/L・hr、反応工程生産速度は32g/L・hrと計算された。
この反応液を20Lポリ容器に移し、ヘキサン5.20kgを加えて振とうした後、静置した。静置後、上層を抜き出し、減圧濃縮した。さらに理論段数10の桐山蒸留塔を用いて、濃縮液の精密蒸留を行った。圧力1.3kPaA、還流比10の条件で、DEPDVE0.87kg(4.1mol)を得た。得られたDEPDVEは、純度99%以上、収率65%であった。結果を表1に示す。

表中の記号は、それぞれ以下を示す。
r1:反応速度
r2:反応工程生産速度
ODDVE:1,12-オクタデカンジオールジビニルエーテル
DEPDVE:2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル
3DVE:1,12-ドデカンジオールジビニルエーテル
MPDVE:3-メチル-1,5-ペンタンジオールジビニルエーテル
DMSO:ジメチルスルホキシド
r1:反応速度
r2:反応工程生産速度
ODDVE:1,12-オクタデカンジオールジビニルエーテル
DEPDVE:2,4-ジエチル-1,5-ペンタンジオールジビニルエーテル
3DVE:1,12-ドデカンジオールジビニルエーテル
MPDVE:3-メチル-1,5-ペンタンジオールジビニルエーテル
DMSO:ジメチルスルホキシド
表1に示すとおり、上記のアルカンジオールとアセチレンとを無溶媒で反応させることにより、反応速度と反応工程生産速度が大幅に大きくなった。さらに、収率も高くなることが確認された。なお、レッペ法は、反応系中のアセチレン溶解度にその反応速度が影響されるものであるため、無溶媒で行うよりも溶媒存在下で行った方が、アセチレン溶解度が増加し、反応速度は大きくなるものと通常は考えられるものであるところ、上記のようにして無溶媒条件で反応速度が大きくなることは、意外なことである。
Claims (6)
- 式(1)で表される化合物とアセチレンとをアルカリ金属触媒を用いて反応させ、式(2)で表される化合物を製造する方法であって、前記反応を無溶媒で行う、製造方法。


- 式(1)で表される化合物が、式(3)、(4)又は(5)で表されるものである、請求項1に記載の製造方法。



- 式(1)で表される化合物が、3-メチル-1,5-ペンタンジオール、2,4-ジエチル-1,5-ペンタンジオール、2-ブチル-2-エチル-1,3-プロパンジオール、1,12-ドデカンジオール及び1,12-オクタデカンジオールからなる群から選択される少なくとも1種である、請求項1又は2に記載の製造方法。
- 前記反応を50~170℃の範囲で行う、請求項1~3のいずれか1項に記載の製造方法。
- 前記アルカリ金属触媒が、アルカリ金属の水酸化物及びアルカリ金属の炭酸塩からなる群から選択される少なくとも1種である、請求項1~4のいずれか1項に記載の製造方法。
- 前記アルカリ金属触媒の使用量が、式(1)で表される化合物100モルに対して、1~60モルの範囲である、請求項1~5のいずれか1項に記載の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020557720A JP7458323B2 (ja) | 2018-11-27 | 2019-11-26 | アルキレン骨格を有するジビニルエーテル化合物の製造方法 |
| EP19889569.0A EP3889125A4 (en) | 2018-11-27 | 2019-11-26 | METHOD FOR PRODUCING A DIVINE ETHER COMPOUND HAVING AN ALKYLENE SKELETON |
| US17/297,170 US20220033335A1 (en) | 2018-11-27 | 2019-11-26 | Method for producing divinyl ether compound having alkylene skeleton |
| CN201980077572.0A CN113166016A (zh) | 2018-11-27 | 2019-11-26 | 具有亚烷基骨架的二乙烯基醚化合物的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018-221035 | 2018-11-27 | ||
| JP2018221035 | 2018-11-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020111030A1 true WO2020111030A1 (ja) | 2020-06-04 |
Family
ID=70853945
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/046071 Ceased WO2020111030A1 (ja) | 2018-11-27 | 2019-11-26 | アルキレン骨格を有するジビニルエーテル化合物の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220033335A1 (ja) |
| EP (1) | EP3889125A4 (ja) |
| JP (1) | JP7458323B2 (ja) |
| CN (1) | CN113166016A (ja) |
| WO (1) | WO2020111030A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020111031A1 (ja) * | 2018-11-27 | 2020-06-04 | 丸善石油化学株式会社 | アルキレン骨格を有するジビニルエーテル化合物の製造方法 |
| CN114560756B (zh) * | 2022-03-16 | 2022-09-09 | 齐翔华利新材料有限公司 | 一种二乙二醇二乙烯基醚的制备方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2628408A1 (de) * | 1976-06-24 | 1978-01-12 | Basf Ag | Vinylaether des 3-methylpentandiol- 1,5 |
| JPH04500520A (ja) * | 1988-09-28 | 1992-01-30 | エクスフルアー・リサーチ・コーポレーシヨン | 液相フツ素置換 |
| JPH05279284A (ja) * | 1991-05-08 | 1993-10-26 | Nippon Carbide Ind Co Inc | 1,9−ノナンジオールモノビニルエーテル及び1,9−ノナンジオールジビニルエーテル |
| JPH08225479A (ja) * | 1994-10-29 | 1996-09-03 | Basf Ag | モノビニルエーテルの製法 |
| DE19924159A1 (de) * | 1999-05-26 | 2000-11-30 | Basf Ag | Verfahren zur Herstellung von Alken-Verbindungen |
| JP2014513048A (ja) | 2011-02-17 | 2014-05-29 | ビーエーエスエフ ソシエタス・ヨーロピア | ジビニルエーテル類の製造方法 |
| WO2015190376A1 (ja) | 2014-06-13 | 2015-12-17 | 丸善石油化学株式会社 | 新規なジビニルエーテル化合物およびその製造法 |
| JP2017068246A (ja) | 2015-09-30 | 2017-04-06 | キヤノン株式会社 | 硬化型液体現像剤 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE33211E (en) * | 1986-08-19 | 1990-05-08 | Allied-Signal Inc. | Vinyl ether terminated urethane resins |
| DE59608335D1 (de) * | 1995-03-24 | 2002-01-17 | Basf Ag | Verfahren zur Durchführung von Gas/Flüssigreaktionen unter Vermeidung einer zusammenhängenden Gasphase |
| DE10017222A1 (de) * | 2000-04-06 | 2001-10-11 | Basf Ag | Verfahren zur Herstellung von Alkenyl-ethern |
| DE10048696A1 (de) * | 2000-09-30 | 2002-04-11 | Basf Ag | Tertiäre Divinylether, Verfahren zu deren Herstellung und deren Verwendung |
| DE10255437B4 (de) * | 2001-12-05 | 2017-02-09 | Basf Se | Kontinuierliches Verfahren zur Herstellung von Alkenylverbindungen |
| US8946487B2 (en) * | 2011-02-17 | 2015-02-03 | Basf Se | Process for preparing divinyl ethers |
| CN108187682B (zh) * | 2018-01-03 | 2020-01-03 | 山西大学 | 一种合成4-羟丁基乙烯基醚的固体碱催化剂及其制备方法和应用 |
-
2019
- 2019-11-26 WO PCT/JP2019/046071 patent/WO2020111030A1/ja not_active Ceased
- 2019-11-26 JP JP2020557720A patent/JP7458323B2/ja active Active
- 2019-11-26 US US17/297,170 patent/US20220033335A1/en not_active Abandoned
- 2019-11-26 CN CN201980077572.0A patent/CN113166016A/zh active Pending
- 2019-11-26 EP EP19889569.0A patent/EP3889125A4/en not_active Withdrawn
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2628408A1 (de) * | 1976-06-24 | 1978-01-12 | Basf Ag | Vinylaether des 3-methylpentandiol- 1,5 |
| JPH04500520A (ja) * | 1988-09-28 | 1992-01-30 | エクスフルアー・リサーチ・コーポレーシヨン | 液相フツ素置換 |
| JPH05279284A (ja) * | 1991-05-08 | 1993-10-26 | Nippon Carbide Ind Co Inc | 1,9−ノナンジオールモノビニルエーテル及び1,9−ノナンジオールジビニルエーテル |
| JPH08225479A (ja) * | 1994-10-29 | 1996-09-03 | Basf Ag | モノビニルエーテルの製法 |
| DE19924159A1 (de) * | 1999-05-26 | 2000-11-30 | Basf Ag | Verfahren zur Herstellung von Alken-Verbindungen |
| JP2014513048A (ja) | 2011-02-17 | 2014-05-29 | ビーエーエスエフ ソシエタス・ヨーロピア | ジビニルエーテル類の製造方法 |
| WO2015190376A1 (ja) | 2014-06-13 | 2015-12-17 | 丸善石油化学株式会社 | 新規なジビニルエーテル化合物およびその製造法 |
| JP2017068246A (ja) | 2015-09-30 | 2017-04-06 | キヤノン株式会社 | 硬化型液体現像剤 |
Non-Patent Citations (3)
| Title |
|---|
| CRIVELLO, JAMES V. ET AL.: "Photoinitiated alternating copolymerization of dialkyl maleates and fumarates with vinyl ethers", JOURNAL OF POLYMER SCIENCE , PART A: POLYMER CHEMISTRY, vol. 48, no. 21, 7 September 2010 (2010-09-07), pages 4726 - 4736, XP055477965, DOI: 10.1002/pola.24264 * |
| KUNICHIKA, SANGO ET AL.: "Preparation and Polymerization of 1,4-Butanediol- Monovinyl Ether", THE JOURNAL OF THE SOCIETY OF CHEMICAL INDUSTRY, vol. 60, no. 6, 1957, Japan, pages 761 - 763, XP055713364 * |
| See also references of EP3889125A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220033335A1 (en) | 2022-02-03 |
| CN113166016A (zh) | 2021-07-23 |
| EP3889125A4 (en) | 2022-08-17 |
| JPWO2020111030A1 (ja) | 2021-10-14 |
| EP3889125A1 (en) | 2021-10-06 |
| JP7458323B2 (ja) | 2024-03-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111792985B (zh) | 一种含氟传热流体及其制备方法和应用 | |
| JP7458323B2 (ja) | アルキレン骨格を有するジビニルエーテル化合物の製造方法 | |
| EP2612859A1 (en) | Method for producing 1-triazole-2-butanol derivative | |
| JP5952384B2 (ja) | ジアルキルカーボネートの製造方法 | |
| EP3395813B1 (en) | Voriconazole intermediate and voriconazole synthesis method | |
| EP2818465A1 (en) | 2-Oxo-1,3-dioxolane-4-acyl halides, their preparation and use | |
| EP2436665B1 (en) | Process for producing vinyl ether | |
| WO2020111031A1 (ja) | アルキレン骨格を有するジビニルエーテル化合物の製造方法 | |
| JP2008201740A (ja) | エダラボンの精製方法及び高純度エダラボン | |
| WO2025006693A1 (en) | Cannabidiol-like compounds and methods for the selective preparation of the same | |
| JPWO2017119458A1 (ja) | ヒドロキシピバルアルデヒドの製造方法 | |
| CN110621680A (zh) | 具有烷氧基烷基的异氰脲酸衍生物及其制造方法 | |
| JP6269384B2 (ja) | テトラヒドロフラン化合物の製造方法 | |
| JP5893008B2 (ja) | 4,4−ビス[(エテニロキシ)メチル]シクロヘキセンおよびその製造方法 | |
| HK1209751A1 (en) | Process for the synthesis of substituted gamma lactams | |
| JP5115762B2 (ja) | 4−ホルミルピペリジンアセタール誘導体の製造方法 | |
| JP6543824B2 (ja) | デヒドロリナリルアセテートの製造方法(i) | |
| KR100972427B1 (ko) | 트리페닐메탄 보호기 제거 방법 | |
| KR102071898B1 (ko) | 높은 광학순도의 키랄성 진저롤 화합물의 제조 방법 | |
| JP2009143850A (ja) | ピラゾリノン誘導体の製造法 | |
| JPH0617348B2 (ja) | N−ビニルホルムアミドの回収法 | |
| CN106397268A (zh) | 二氰基化合物的制造方法 | |
| JP5082340B2 (ja) | 3−アミノ−6−クロロピリダジンの製造方法 | |
| JP2009501140A (ja) | ヒドロキノン誘導体の合成 | |
| JP2021520382A (ja) | 2,2−ジメチルピペラジンの調製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19889569 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020557720 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019889569 Country of ref document: EP Effective date: 20210628 |