WO2020189748A1 - Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ - Google Patents
Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ Download PDFInfo
- Publication number
- WO2020189748A1 WO2020189748A1 PCT/JP2020/012189 JP2020012189W WO2020189748A1 WO 2020189748 A1 WO2020189748 A1 WO 2020189748A1 JP 2020012189 W JP2020012189 W JP 2020012189W WO 2020189748 A1 WO2020189748 A1 WO 2020189748A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigen
- binding
- mta
- antibody
- binding domain
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/44—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material not provided for elsewhere, e.g. haptens, metals, DNA, RNA, amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1037—Screening libraries presented on the surface of microorganisms, e.g. phage display, E. coli display
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/005—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies constructed by phage libraries
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/244—Interleukins [IL]
- C07K16/248—IL-6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2866—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for cytokines, lymphokines, interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/42—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against immunoglobulins
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/06—Libraries containing nucleotides or polynucleotides, or derivatives thereof
- C40B40/08—Libraries containing RNA or DNA which encodes proteins, e.g. gene libraries
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/10—Libraries containing peptides or polypeptides, or derivatives thereof
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6845—Methods of identifying protein-protein interactions in protein mixtures
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6854—Immunoglobulins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/02—Screening involving studying the effect of compounds C on the interaction between interacting molecules A and B (e.g. A = enzyme and B = substrate for A, or A = receptor and B = ligand for the receptor)
Definitions
- the present disclosure describes an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in a Methylthioadenosine (MTA) -dependent manner, a method for producing or screening the antigen-binding domain or the antigen-binding molecule, the antigen-binding domain or the antigen.
- the present invention relates to a library for obtaining a binding molecule, a method for designing the same, and a pharmaceutical composition containing the antigen-binding molecule.
- the present disclosure also relates to a method for designing a library for efficiently acquiring an antigen-binding domain whose antigen-binding activity changes depending on a small molecule compound.
- the present disclosure relates to an antigen-binding molecule that specifically binds to MTA, a method for measuring MTA concentration using the antigen-binding molecule, and a method for diagnosing a disease.
- Non-Patent Document 1 Non-Patent Document 2
- Non-Patent Document 3 Rituxan for the CD20 antigen, cetuximab for the EGFR antigen, Herceptin for the HER2 antigen, etc. have been approved as cancer therapeutic agents using antibody drugs. These antibody molecules bind to antigens expressed in cancer cells and exert damaging activity against cancer cells by ADCC or the like. Since it is known that such cytotoxic activity by ADCC or the like depends on the number of antigens expressed in the target cells of the therapeutic antibody (Non-Patent Document 4), it is treated that the expression level of the target antigen is high. It is preferable from the viewpoint of the effect of the antibody for use.
- the antigen targeted by the therapeutic antibody as a cancer therapeutic agent is specifically expressed in cancer cells.
- Non-Patent Document 5 natural human A second-generation improved antibody molecule that exerts strong cellular cytotoxicity by enhancing ADCC activity by enhancing binding to Fc ⁇ RIIIa by substituting an amino acid in the Fc region of IgG1 (Non-Patent Document 6) has been reported.
- ADC Antibody Drug Conjugate
- Non-patent document in which a drug having strong cytotoxic activity is conjugated with an antibody as an antibody drug that exerts a damaging activity on cancer cells by a mechanism other than the above-mentioned ADCC activity mediated by NK cells.
- a drug having strong cytotoxic activity is conjugated with an antibody as an antibody drug that exerts a damaging activity on cancer cells by a mechanism other than the above-mentioned ADCC activity mediated by NK cells.
- improved antibody molecules that exert stronger cytotoxic activity such as small antibody (Non-Patent Document 8) that exerts damaging activity against cancer cells by recruiting T cells to cancer cells are also reported. Has been done.
- bivatuzumab mertansine which is an ADC in which mertansine is bound to an antibody against CD44v6 that is highly expressed in cancer cells, is clinically serious skin toxicity and hepatotoxicity because CD44v6 is also expressed in normal tissues. Is recognized (Non-Patent Document 10).
- the target antigen When an antibody capable of exerting strong cytotoxic activity against cancer cells in which the expression of the antigen is low is used, the target antigen must be expressed extremely cancer-specifically.
- the number of cancer antigens that are extremely cancer-specifically expressed is limited, as HER2, which is the target antigen of Herceptin, and EGFR, which is the target antigen of cetuximab, are also expressed in normal tissues. it is conceivable that. Therefore, although the cytotoxic activity against cancer can be enhanced, side effects due to the cytotoxic action on normal tissues can be a problem.
- Non-Patent Document 11 iprimumab, which enhances tumor immunity by inhibiting CTLA4, which contributes to immunosuppression in cancer, prolongs overall survival against metastatic melanoma.
- iprimumab inhibits CTLA4 systemically, it becomes a problem that while tumor immunity is enhanced, it shows serious side effects like autoimmune disease due to systemic immunity activation. (Non-Patent Document 12).
- Non-Patent Document 13 For example, for lesion sites such as cancer tissues and inflammatory tissues, pH-dependent antibodies utilizing the acidic condition of pH in these diseased tissues have been reported (Patent Documents 1 and 2).
- the decrease in pH that is, the increase in hydrogen ion concentration
- the increase in hydrogen ion concentration is slight as compared with normal tissue, and an antibody that acts by detecting a slight increase in hydrogen ion concentration with extremely small molecular weight
- the pH may be acidic even in normal tissues such as osteoclast bone resorption socket area and tissues other than the target lesion, and the pH condition is an environment specific to the lesion site. It was considered that there are still many problems to use as a factor.
- Patent Document 3 a method for producing an antibody that exhibits antigen-binding activity for the first time by cleaving with a protease expressed at a lesion site such as cancer tissue or inflammatory tissue has been reported.
- a protease expressed at a lesion site such as cancer tissue or inflammatory tissue.
- the cleavage of the antibody by protease is irreversible, it is a problem that the antibody cleaved at the lesion site can bind to the antigen even in the normal tissue by returning to the normal tissue in the bloodstream. it was thought.
- an antigen-binding molecule whose antigen-binding activity changes depending on the concentration of a compound specific to a disease tissue has been reported (Patent Documents 4 and 5).
- the present disclosure newly finds a low molecular weight compound that is specifically present or produced in a diseased tissue, and further, an antigen-binding molecule (low molecular weight compound switch antigen) in which binding to a target antigen is controlled depending on the low molecular weight compound.
- an antigen-binding molecule low molecular weight compound switch antigen
- One of the purposes is to provide a binding molecule) and further to provide a method for efficiently obtaining such an antigen-binding molecule in a short period of time.
- MTA Methylthioadenosine
- the present inventors have succeeded in creating a library capable of efficiently screening an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- the present inventors have also succeeded in creating a library capable of screening an antigen-binding domain in which the antigen-binding activity changes depending on the MTA and / or a small molecule compound other than MTA, and the small molecule other than MTA and / or MTA.
- the present inventors have also succeeded in obtaining an antigen-binding molecule that specifically binds to the MTA itself.
- MTA 5'-Methylthioadenosine
- the antigen-binding molecule described in [A1] to [A3], and the antigen-binding domain contained in the antigen-binding molecule has an antigen-binding activity of S- (5'-Adenosyl) -L-homocysteine ( An antigen-binding molecule that is substantially unaffected by SAH).
- [A7] The antigen-binding molecule described in [A1], [A2] or [A6], and the antigen-binding domain contained in the antigen-binding molecule has an antigen-binding activity of S- (5'-Adenosyl)-. An antigen-binding molecule that also changes in a dependent manner with respect to L-homocysteine (SAH).
- SAH L-homocysteine
- the antigen-binding molecule according to [A1], [A2], [A6] or [A7] has an antigen-binding activity of AMP, ADP and /. Or an antigen-binding molecule that also changes depending on ATP.
- [A13] The invention according to any one of [A1] to [A12], wherein the antigen-binding activity of the antigen-binding domain in the presence of MTA is stronger than the antigen-binding activity of the antigen-binding domain in the absence of MTA.
- Antigen binding molecule [A14] The invention according to any one of [A1] to [A12], wherein the antigen-binding activity of the antigen-binding domain in the presence of MTA is weaker than that of the antigen-binding domain in the absence of MTA.
- Antigen binding molecule [A15] The antigen-binding molecule according to any one of [A1] to [A14], wherein the antigen-binding domain has an amino acid residue that interacts with an MTA.
- the antigen-binding domain comprises an antibody variable region or monodomain antibody, and the amino acid residue that interacts with the MTA is located in the antibody variable region or the CDR of the monodomain antibody, [A15] or The antigen-binding molecule according to [A16].
- the antigen-binding domain is an antibody variable region, and the amino acid residues that interact with the MTA are the heavy chain positions 34, 35a, and 47 specified by Kabat numbering in the amino acid sequence of the antibody variable region.
- the antigen-binding molecule according to any one of [A15] to [A17], which is an amino acid residue located at at least one amino acid site.
- the antigen-binding domain is an antibody variable region, and the antibody variable region is a heavy chain W34, C35a, W47, F52, Y52e, E101, a light chain R32, S34, Y36, L46, Y49, S50, A89, G90.
- the antigen-binding molecule according to any one of [A15] to [A18], which comprises at least one amino acid.
- Antigen-binding molecule Any of A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 30th position of the heavy chain; Any of A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 31st position of the heavy chain; A located in the 32nd position of the heavy chain; Either A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 33rd position of the heavy chain; W located at the 34th position of the heavy chain; M located at the 35th position of the heavy chain; C located at the heavy chain 35a position; C located at the 50th position of the heavy chain; I located in the 51st position of the heavy chain; F located in the 52nd position of
- the antigen-binding domain is an antibody variable region, and the amino acid residues that interact with the MTA are heavy chains at positions 34, 47, and 50 specified by Kabat numbering in the amino acid sequence of the antibody variable region. At least one amino acid site selected from the group of amino acid sites at positions, 58, 95, 98, 99, 100a, light chain 28, 91, 95b, 95c and 96.
- the antigen-binding molecule according to any one of [A15] to [A17], which is a located amino acid residue.
- the antigen-binding domain is an antibody variable region, and the antibody variable region is heavy chain W34, W47, C50, Y58, E95, F98, G99, G100a, light chain Y28, T91, F95b, Y95c, and F96 (The antigen-binding molecule according to any one of [A15] to [A17], and [A21], which comprises at least one amino acid selected from (Kabat numbering).
- the antigen-binding domain is an antibody variable region, which comprises at least one amino acid selected from the group of amino acids below, any of [A1] to [A17] or [A21].
- Antigen-binding molecule (Kabat numbering) according to any one of [A22]: Any of A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 31st position of the heavy chain; Any of A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 32nd position of the heavy chain; Either A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 33rd position of the heavy chain; W located at the 34th position of the heavy chain; M located at the 35th position of the heavy chain; C located at the heavy chain 35a position; C located at the 50th position of the heavy chain; I located in the 51st position of the heavy chain; Either A, C, D, E,
- the antigen-binding domain is an antibody variable region, and amino acid residues that interact with the MTA are heavy chains at positions 33, 50, and 52 specified by Kabat numbering in the amino acid sequence of the antibody variable region. At least one or more amino acid sites selected from the group of amino acid sites at positions, 54, 56, 57, 58, 99, 100, 100a, light chain 91, 95c, and 96.
- the antigen-binding molecule according to any one of [A15] to [A17], which is an amino acid residue located in.
- the antigen-binding domain is an antibody variable region, which is a heavy chain A33, I50, G52, D54, S56, T57, W58, G99, Y100, T100a, light chain S91, Y95c, and N96 ( The antigen-binding molecule according to any one of [A15] to [A17], and [A24], which comprises at least one amino acid selected from (Kabat numbering).
- the antigen-binding domain is an antibody variable region, which comprises at least one amino acid selected from the group of amino acids below, either [A1] to [A17] or [A24].
- Antigen-binding molecule (Kabat numbering) according to any one of [A25]: Either A, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 26th position of the heavy chain; Either A, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 28th position of the heavy chain; Either A or L located at position 29 of the heavy chain; Any of A, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W or Y located at the 30th position of the heavy chain; Either A, D, E, F, G, H, I, K, L, N, Q, R, S, T, V, W or Y located at the 31st position of the heavy chain; Either D, E, F, H, N, P, R
- [A27] The antigen binding according to any one of [A1] to [A26], wherein the antigen is a molecule other than MTA, or a molecule other than MTA and having immunogenicity in the human body. molecule.
- [A28] The antigen-binding molecule according to any one of [A1] to [A27], wherein the antigen is any of a peptide, a polypeptide, and a protein.
- the antigen-binding molecule further has a second antigen-binding domain, and the second antigen-binding domain has a binding activity to a second antigen different from the antigen to which the antigen-binding domain binds.
- the antigen-binding molecule according to any one of A1] to [A28].
- the binding activity of the second antigen-binding domain to the second antigen in the presence of MTA is different from the binding activity of the second antigen-binding domain to the second antigen in the absence of MTA.
- the antigen-binding molecule is different from the binding activity of the second antigen-binding domain to the second antigen in the absence of MTA.
- the antigen expressed in the cancer tissue is an antigen expressed in cancer cells, or an antigen expressed in cancer stromal cells or immune tissue in cancer tissue [A35].
- [A37] The antigen-binding molecule according to [A35] or [A36], wherein the cancer tissue is a cancer tissue in which an MTA is accumulated.
- the cancer tissue is a cancer tissue in which the gene encoding MTA phospholylase (MTAP) is deficient or underexpressed, or has a mutation or splicing variant that reduces enzyme activity [A38].
- [A39] The antigen-binding molecule according to any one of [A35] to [A37], wherein the cancer tissue is a cancer tissue in which the activity of MTAP is deficient or decreased.
- [A40] The antigen-binding molecule according to any one of [A1] to [A39], wherein the antigen is a membrane-type molecule, and the antigen-binding molecule causes cytotoxicity to cells expressing the antigen.
- the antigen-binding molecule according to [A40] which exhibits at least one or more cytotoxic activity selected from ADCC activity, ADCP activity or CDC activity.
- [A42] The antigen-binding molecule according to [A1] to [A39], which has agonist activity against the antigen.
- One of the antigen and the second antigen is an antigen expressed in a target cell, and the other is an antigen expressed in an effector cell [A29] to [A32].
- the cancer cell encodes the MTAP or a cancer cell in which the gene encoding MTAP is deficient or underexpressed, or has a mutation or splicing variant that reduces enzyme activity.
- the target cells are other than cancer cells that are present around cancer cells in which the gene encoding MTAP is deficient or underexpressed, or have mutations or splicing variants that reduce enzyme activity.
- [A47] The antigen-binding molecule according to [A43], wherein the cells other than the cancer cells existing around the cancer cells are cancer-associated fibroblasts (CAF) or tumor associated macrophages (TAM).
- CAF cancer-associated fibroblasts
- TAM tumor associated macrophages
- [A48] The antigen-binding molecule according to any one of [A43] to [47], wherein the effector cell is a T cell.
- [A49] The antigen-binding molecule according to [A48], wherein the antigen expressed in the effector cell is a T cell receptor (TCR) complex.
- TCR T cell receptor
- [A50] The antigen-binding molecule according to either [A48] or [A49], wherein the antigen expressed in the effector cell is CD3.
- [A51] The antigen-binding molecule according to any one of [A48] to [A50], which induces cytotoxic activity against target cells by activating effector cells.
- [A52] The antigen-binding molecule according to any one of [A48] to [A51], which has TDCC activity.
- [A53] The antigen-binding molecule according to any one of [A1] to [A39], wherein the antigen is a soluble molecule, and the antigen-binding molecule exhibits neutralizing activity against the antigen. , Antigen binding molecule.
- [A54] The description in any one of [A1] to [A53], wherein the KD value for the antigen in the absence of the MTA of the antigen-binding domain and the KD value for the antigen in the presence of the MTA of the antigen-binding domain are different.
- Antigen-binding molecule A pharmaceutical composition comprising the antigen-binding molecule according to any one of [A1] to [A54].
- a pharmaceutical composition for treating cancer which comprises the antigen-binding molecule according to any one of [A1] to [A55] as an active ingredient.
- the cancer is a cancer in which the gene encoding MTAP is deficient or underexpressed, or has a mutation or splicing variant that reduces enzyme activity, [A56] to [A57].
- the pharmaceutical composition according to any one of the above.
- A59 The pharmaceutical composition according to any one of [A56] to [A58], wherein the cancer is a cancer in which the activity of MTAP is deficient or decreased.
- [A60] The method for producing an antigen-binding molecule according to any one of [A1] to [A54].
- [A61] The polynucleotide encoding the antigen-binding molecule according to any one of [A1] to [A52].
- [A62] A cell carrying the vector according to [A63] [A62] containing the polynucleotide according to [A61].
- [A64] An antigen-binding molecule recovered from the culture supernatant by culturing the cells according to [A63].
- [G2] A pharmaceutical formulation comprising the antigen-binding molecule described in [G1] and a pharmaceutically acceptable carrier.
- [G3] A method for producing an antigen-binding molecule having a high plasma retention and / or a low plasma antigen-accumulating ability as compared with a control antigen-binding molecule, wherein (a) the concentration of MTA is high.
- a library consisting of nucleic acids encoding antigen-binding molecules containing multiple antigen-binding domains having different sequences from each other and / and antigen-binding molecules containing multiple antigen-binding domains having different sequences from each other, and mutual with MTA.
- a library mainly composed of an antigen-binding molecule containing an antigen-binding domain having an amino acid residue of action, and / and a nucleic acid encoding the antigen-binding molecule.
- [B3] Multiple antibody variable region variants having amino acids different from those located at one or more amino acid sites in the antibody variable region unmodified product having binding activity to MTA, and / or having different sequences from each other. Includes nucleic acids encoding multiple antibody variable region variants having amino acids different from those located at one or more amino acid sites in the antibody variable region unmodified product having binding activity to MTA and having different sequences from each other.
- the amino acid site having an amino acid different from that of the antibody variable region unmodified form in the antibody variable region variant is one or more amino acid sites selected from the following group of amino acid sites, [B3].
- the amino acid site having an amino acid different from that of the antibody variable region unmodified form in the antibody variable region variant is one or more amino acid sites selected from the following group of amino acid sites, [B3].
- H-CDR1 containing XAXWMC SEQ ID NO: 65
- H-CDR2 containing CIFAXXXYXXSGGSTYYASWAKG SEQ ID NO: 66
- H-CDR3 containing GXGXXXGXXDEL SEQ ID NO: 67
- L-CDR1 containing QSSEXVXXXLS SEQ ID NO: 68
- L-CDR2 containing XAXTXPX SEQ ID NO: 69
- L-CDR3 containing AGLYXGNIPA SEQ ID NO: 70
- the library described in [B2], wherein X refers to an arbitrary amino acid, and X existing at different positions does not have to be an amino acid of the same type.
- H-CDR1 containing XAXWMC SEQ ID NO: 65
- H-CDR2 containing CIFAX 1 XXYXXSGGSTYYASWAKG SEQ ID NO: 71
- H-CDR3 containing GXGXXXGXXDEL SEQ ID NO: 67
- L-CDR1 containing QSSEXVXXX 1 XLS SEQ ID NO: 72
- L-CDR2 containing XAXTXPX SEQ ID NO: 69
- L-CDR3 containing AGLYXGNIPA SEQ ID NO: 70
- Is an antibody variable region containing X is any amino acid
- X 1 is an amino acid selected from A, D, E, G, H, I, K, L, M, N, P, Q, R, S, T and V
- H-CDR1 containing XXAXWMC (SEQ ID NO: 73); H-CDR2 containing CIFAX 1 XXYXXSGGSTYYASWAKG (SEQ ID NO: 71); H-CDR3 containing GXGXXXGXXDEL (SEQ ID NO: 67); L-CDR1 containing QSSEXVXXX 1 XLS (SEQ ID NO: 72); L-CDR2 containing XAXTXPX (SEQ ID NO: 69); and L-CDR3 containing AGLYXGNIPA (SEQ ID NO: 70); Is an antibody variable region containing X is any amino acid, X 1 is an amino acid selected from A, D, E, G, H, I, K, L, M, N, P, Q, R, S, T and V, and X or X 1 exists
- H-CDR1 containing XXXWMC SEQ ID NO: 74
- H-CDR2 containing CIXSXXXXTXYASWVNG SEQ ID NO: 75
- H-CDR3 containing EXXXXSGALNL SEQ ID NO: 76
- L-CDR1 containing HSSKXVXXXXLA SEQ ID NO: 77
- L-CDR2 containing XAXXLAS SEQ ID NO: 78
- L-CDR3 containing QGTYXXXXFYFA SEQ ID NO: 79
- the library described in [B2] wherein X refers to an arbitrary amino acid, and X existing at different positions does not have to be an amino acid of the same type.
- H-CDR1 containing X 2 X 3 X 4 X 5 G (SEQ ID NO: 80); H-CDR2 containing X 6 IGX 7 X 8 X 9 X 10 X 11 WX 12 PX 13 WVKX 14 (SEQ ID NO: 81); H-CDR3 including GX 15 X 16 X 17 X 18 X 19 X 20 NAX 21 DP (SEQ ID NO: 82); L-CDR1 containing QSSQSVX 22 X 23 NNX 24 LS (SEQ ID NO: 83); L-CDR2 containing DASTLAS (SEQ ID NO: 84); and L-CDR3 including HGX 25 X 26 X 27 X 28 X 29 X 30 X 31 X 32 DNX 33 (SEQ ID NO: 85); Is an antibody variable region containing X 2 is an amino acid selected from A, D, E, F, G, H, I, K, L
- X 3 is an amino acid selected from D, E, F, H, K, N, P, R and Y
- X 4 is an amino acid selected from A, I, P, T and V
- X 5 is an amino acid selected from A, E, F, H, I, K, L, M, N, Q, S, T, V, W and Y
- X 6 is an amino acid selected from D, I and V
- X 7 is an amino acid selected from A, D, E, G, I, K, Q and R
- X 8 is an amino acid selected from D, E, F, G, H, I, K, L, P, Q, R, S, T, V, W and Y.
- X 9 is an amino acid selected from A, D, E, F, G, H and S
- X 10 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, Q, R, S, T, V, W and Y
- X 11 is an amino acid selected from A, D, E, G, H, I, K, L, N, P, Q, R, S, T and V
- X 12 is an amino acid selected from A, D, E, F, G, H, I, K, L, Q, R, S, T, V, W and Y.
- X 13 is an amino acid selected from A, F, Q, R, S, T, V, W and Y
- X 14 is an amino acid selected from A, F and G
- X 15 is an amino acid selected from A, E, F, G, H, K, L, Q, R, S, T, W and Y
- X 16 is an amino acid selected from F, H, K, N, W and Y
- X 17 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 18 is an amino acid selected from A, D, E, G, H, Q and S
- X 19 is an amino acid selected from F and Y
- X 20 is an amino acid selected from N, T and V
- X 21 is an amino acid selected from F and W
- X 22 is an amino acid selected from A, E, F, H, I, K, L, N, R, S, T, V, W and Y
- X 23 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 24 is an amino acid selected from A, E, F, G, H, S and Y
- X 25 is an amino acid selected from A, S and T
- X 26 is an amino acid selected from A, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W and Y
- X 27 is an amino acid selected from A, D, E, F, G, H, L, N, Q, R, S, T, V and Y
- X 28 is an amino acid selected from A, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 29 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W, Y
- X 30 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, V, W and Y
- X 31 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 32 is an amino acid selected from A, F, H, I, K, L, N, P, Q, R, S, T, V, W and Y
- X 33 is an amino acid selected from A and G, the library described in [B2].
- [B16] From the library described in [B15], a library enriched with nucleic acids encoding an antigen-binding molecule containing an antigen-binding domain that binds to an MTA.
- [B17] The library according to [B16], wherein the binding of the antigen binding domain to the MTA is the binding to the MTA in the absence of the antigen.
- [B18] The concentration is performed in the following steps (1) to (2): (1) The step of contacting the antigen-binding domain displayed from the library described in [B15] with the MTA, and (2) The step of selecting the antigen-binding domain bound to the MTA in the step (1), The library described in [B16] or [B17], including. [B19] A library enriched with nucleic acids encoding an antigen-binding domain that binds to adenosine from the library described in [B15]. [B20] The library according to [B19], wherein the binding of the antigen-binding domain to adenosine is binding to adenosine in the absence of an antigen.
- the concentration is performed in the following steps (1) to (2): (1) A step of contacting the antigen-binding domain displayed from the library described in [B15] with adenosine, and (2) The step of selecting the antigen-binding domain bound to adenosine in the step (1).
- the amino acid modification in the step (b) satisfies at least one or more of the following (1) to (3): (1) When comparing the structure when the antigen-binding domain variant having the amino acid modification binds to the MTA and the structure when it does not bind to the MTA, the structural change rate of the amino acid site where the modified amino acid is located is large; (2) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the MTA and the structure when it is not bound to the MTA, the antigen-binding domain modification is caused by the presence of the modified amino acid.
- the antigen-binding domain variant having the amino acid modification does not significantly attenuate the binding activity to the MTA as compared with the antigen-binding domain unmodified form;
- [C3] The production method according to any one of [C1] to [C2], wherein the antigen-binding domain having an MTA-binding activity does not substantially bind to adenosine.
- [C4] The production method according to [C3], wherein the antigen-binding domain having MTA-binding activity does not bind to (5'-Adenosyl) -L-homocysteine (SAH), AMP, ADP or / and ATP.
- [C5] A library manufactured by the manufacturing method according to any one of [C1] to [C4].
- the amino acid modification in the step (b) satisfies at least one or more of the following (1) to (3): (1) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the low molecular weight compound and the structure when it is not bound to the low molecular weight compound, the amino acid in which the modified amino acid is located is compared. Large rate of structural change in the site; (2) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the low molecular weight compound and the structure when it is not bound to the low molecular weight compound, the modified amino acid is present.
- the antigen-binding domain variant having the amino acid modification does not significantly attenuate the binding activity to the low molecular weight compound as compared with the antigen-binding domain unmodified form; The manufacturing method described in [C5].
- the low molecular weight compound is selected from at least one selected from adenosine, adenosine triphosphate, adenosine diphosphate, adenosine monophosphate, and S- (5'-Adenosyl) -L-homocysteine (SAH).
- SAH S- (5'-Adenosyl) -L-homocysteine
- the present disclosure also includes aspects exemplified below.
- [D1] A method for screening an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- [D2] Comparing the antigen-binding activity of the antigen-binding domain in the presence of the first concentration of MTA with the antigen-binding activity at a concentration different from the first concentration (second concentration) in the presence of MTA [D1]. The method described in.
- step (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the above step (a) in the presence of a second concentration of MTA, and (c) The step of isolating the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b).
- the method described in [D1], including. [D7] The following steps (a) to (d); (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain has bound to the antigen in the step (a).
- step (c) A step of placing an antigen-binding molecule containing the antigen-binding domain bound to the antigen in the presence of a second concentration of MTA, and (d) A step of isolating an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b).
- the method described in [D1], including. [D8] The method according to [D6] or [D7], which confirms that the antigen-binding domain can bind to the MTA before performing the step (a) above.
- [D9] The following steps (a) to (d): (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding molecule containing an antigen-binding domain that does not bind to the antigen to the antigen in the presence of a second concentration of MTA, and (d) The step of isolating the antigen-binding molecule containing the antigen-binding domain bound to the antigen in the step (c).
- [D11] The following steps (a) to (d); (a) A step of contacting a library displaying an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain bound to an antigen in the step (a). (c) The step of placing the antigen-binding molecule containing the antigen-binding domain selected in the above step (b) in the presence of a second concentration of MTA, and (d) A step of isolating an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in step (b) in step (c).
- the library that comprises contact with the antigen in the presence of the first concentration of MTA in the step (a) is a library in which the antigen-binding molecule containing the antigen-binding domain selected in the steps (1) and (2) is displayed.
- [D13] The following steps (a) to (d): (a) A step of contacting an antigen with a library in which an antigen-binding molecule containing an antigen-binding domain is displayed in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain that does not bind to an antigen in the step (a). (c) The step of binding the antigen-binding molecule containing the antigen-binding domain selected in the above step (b) to the antigen in the presence of a second concentration of MTA, and (d) The step of isolating the antigen-binding molecule containing the antigen-binding domain bound to the antigen in the step (c).
- [D1] The method described in [D1], including. [D14] The method according to any one of [D2] to [D8], wherein the first concentration is a concentration higher than the second concentration.
- [D15] The method according to [D1], comprising comparing the antigen-binding activity of an antigen-binding domain in the presence of an MTA with that of an antigen-binding domain in the absence of an MTA.
- [D16] The method according to [D1] or [D15], comprising the step of selecting an antigen-binding domain in which the antigen-binding activity in the presence of MTA and the antigen-binding activity in the absence of MTA are different.
- [D17] The method according to [D1] or [D15], comprising selecting an antigen-binding domain whose antigen-binding activity in the presence of an MTA is higher than that in the absence of an MTA.
- [D18] The following steps (a) to (c): (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the above step (a) in the absence of MTA, and (c) The step of isolating the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b).
- the method described in [D1] including.
- [D19] The following steps (a) to (d); (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain has bound to the antigen in the step (a). (c) A step of placing an antigen-binding molecule containing the antigen-binding domain bound to the antigen in the absence of an MTA, and (d) A step of isolating an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b).
- step (c) A step of binding an antigen-binding molecule containing an antigen-binding domain that does not bind to the antigen to the antigen in the absence of MTA, and (d) The step of isolating the antigen-binding molecule containing the antigen-binding domain bound to the antigen in the step (c).
- the method described in [D1], including. [D22] The following steps (a) to (c): (a) A step of contacting a library displaying an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA.
- step (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the above step (a) in the absence of MTA, and (c) The step of isolating the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b).
- the method described in [D1], including. [D23] The following steps (a) to (d); (a) A step of contacting a library displaying an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain bound to an antigen in the step (a).
- step (c) The step of placing the antigen-binding molecule containing the antigen-binding domain selected in step (b) in the absence of MTA, and (d) A step of isolating an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in step (b) in step (c).
- the method described in [D1], including. [D24] Before the step (a), the following steps (1) to (2): (1) A step of contacting the displayed library with the antigen-binding molecule containing the antigen-binding domain with the MTA, and (2) The step of selecting an antigen-binding molecule containing an antigen-binding domain bound to MTA in the above step (1).
- the library to be contacted with the antigen in the presence of the MTA in the step (a) is a library in which the antigen-binding molecule containing the antigen-binding domain selected in the steps (1) and (2) is displayed.
- [D25] The following steps (a) to (d): (a) A step of contacting an antigen with a library in which an antigen-binding molecule containing an antigen-binding domain is displayed in the presence of an MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain that does not bind to an antigen in the step (a).
- step (c) The step of binding the antigen-binding molecule containing the antigen-binding domain selected in step (b) to the antigen in the absence of MTA, and (d) The step of isolating the antigen-binding molecule containing the antigen-binding domain bound to the antigen in the step (c).
- the method described in [D1], including. [D26] The library on which the antigen-binding molecule containing the antigen-binding domain is displayed, or the naive human antibody display library, or the synthetic human antibody display library, or the library according to any one of [B1] to [B14].
- the present disclosure also includes aspects exemplified below.
- [F1] A method for producing an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- [F2] Comparing the antigen-binding activity of the antigen-binding domain in the presence of the first concentration of MTA with the antigen-binding activity in the presence of a different concentration (second concentration) of MTA from the first concentration, [F1] ] The method described in.
- [F3] The method according to [F1] or [F2], which comprises a step of selecting an antigen-binding domain in which the antigen-binding activity in the presence of the first concentration of MTA and the antigen-binding activity in the presence of the second concentration of MTA are different. .. [F4] Any of [F2] to [F3], which comprises a step of selecting an antigen-binding domain whose antigen-binding activity in the presence of the first concentration of MTA is higher than that of the antigen-binding activity in the presence of the second concentration of MTA. The method described in one.
- Steps to recover antigen-binding molecules containing domains The method described in [F1], including. [F8] The following steps (a) to (e); (a) The step of contacting the antigen binding domain with the antigen in the presence of the first concentration of MTA, (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a).
- step (c) The step of placing the antigen-binding domain bound to the antigen in the presence of a second concentration of MTA, (d) A step of isolating an antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b), and (e) A cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium. Steps to recover antigen-binding molecules containing domains, The method described in [F1], including.
- step (d) The step of isolating the antigen-binding domain bound to the antigen in the above step (c), and (e) A cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium. Steps to recover antigen-binding molecules containing domains, The method described in [F1], including. [F11] The following steps (a) to (d): (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of a first concentration of MTA.
- [F12] The following steps (a) to (e); (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the presence of a second concentration of MTA, (d) Isolation of an antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in step (b) in step (c), and (e) A cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium.
- Steps to recover antigen-binding molecules containing domains The method described in [F1], including. [F13] Before the step (a), the following steps (1) to (2): (1) The process of contacting the library displaying the antigen-binding domain with the MTA, and (2) The step of selecting the antigen-binding domain bound to the MTA in the step (1), The library to be contacted with the antigen in the presence of the first concentration of MTA in the step (a) is a library in which the antigen-binding domain selected in the steps (1) and (2) is displayed [F11]. ] Or the method described in [F12].
- [F16] The method according to [F1], which comprises comparing the antigen-binding activity of an antigen-binding domain in the presence of an MTA with the antigen-binding activity in the absence of an MTA.
- [F17] The method according to [F1] or [F16], which comprises a step of selecting an antigen-binding domain in which the antigen-binding activity in the presence of MTA and the antigen-binding activity in the absence of MTA are different.
- [F18] The method according to [F1] or [F16], which comprises a step of selecting an antigen-binding domain having a high antigen-binding activity in the presence of MTA but a high antigen-binding activity in the absence of MTA.
- a cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing the selected antigen-binding domain has been operably linked is introduced is cultured, and the antigen-binding molecule containing the antigen-binding domain is cultivated from the cell culture medium.
- Steps to recover antigen-binding molecules containing domains The method described in [F1], including. [F21] The following steps (a) to (e); (a) The step of contacting the antigen binding domain with the antigen in the presence of the MTA, (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a).
- step (c) The step of placing the antigen-binding domain bound to the antigen in the absence of MTA, (d) A step of isolating an antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b), and (e) A cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium. Steps to recover antigen-binding molecules containing domains, The method described in [F1], including.
- [F22] The method according to [F20] or [F21], which confirms that the antigen-binding domain can bind to the MTA before performing the step (a).
- [F23] The following steps (a) to (e): (a) The step of contacting the antigen binding domain with the antigen in the presence of the MTA, (b) The step of confirming that the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding domain that does not bind to the antigen to the antigen in the absence of MTA.
- step (d) The step of isolating the antigen-binding domain bound to the antigen in the above step (c), and (e)
- a cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium. Steps to recover antigen-binding molecules containing domains, The method described in [F1], including.
- Steps to recover antigen-binding molecules containing domains The method described in [F1], including. [F25] The following steps (a) to (e); (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the absence of MTA, (d) Isolation of an antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in step (b) in step (c), and (e) A cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium.
- Steps to recover antigen-binding molecules containing domains The method described in [F1], including.
- the following steps (1) to (2) (1) The process of contacting the library displaying the antigen-binding domain with the MTA, and (2) The step of selecting the antigen-binding domain bound to the MTA in the step (1),
- the library to be contacted with the antigen in the presence of the MTA in step (a) is a library in which the antigen binding domain selected in steps (1) and (2) is displayed, [F24] or [ The method described in F25].
- step (b) The step of selecting an antigen-binding domain that does not bind to an antigen in the step (a).
- step (c) The step of binding the antigen-binding domain selected in step (b) to the antigen in the absence of MTA,
- step (d) The step of isolating the antigen-binding domain bound to the antigen in the above step (c), and
- a cell into which a vector into which a polynucleotide encoding an antigen-binding molecule containing an antigen-binding molecule isolated in the above step (d) is operably linked is introduced is cultured, and antigen-binding is performed from the cell culture medium. Steps to recover antigen-binding molecules containing domains, The method described in [F1], including.
- [F28] The library on which the antigen-binding domain is displayed, or the naive human antibody display library, or the synthetic human antibody display library, or the library according to any one of [B1] to [B14], or [C5].
- [F29] The method according to any one of [F1] to [F28], wherein the antigen-binding domain is an antibody variable region or a monodomain antibody.
- [E1] An antigen-binding molecule that specifically binds to MTA.
- [E2] The antigen-binding molecule according to [E1], which is an antigen-binding molecule that binds to MTA and the antigen-binding molecule does not substantially bind to adenosine.
- [E3] An antigen-binding molecule that binds to MTA, which does not substantially bind to (5'-Adenosyl) -L-homocysteine (SAH), AMP, ADP and / and ATP [E1] or The antigen-binding molecule according to [E2].
- [E4] A method for measuring MTA concentration using the antigen-binding molecule according to any one of [E1] to [E3].
- [E5] The measuring method according to [E4], wherein the MTA concentration is the MTA concentration in a tissue.
- [E6] The measuring method according to any one of [E4] and [E5], which measures the MTA concentration using an antigen-antibody reaction.
- [E7] The measuring method according to any one of [E4] to [E5], which uses an immunohistochemical staining method.
- [E8] The measurement method according to any one of [E4] to [E5] using the imaging method.
- [E9] The measurement method according to any one of [E4] to [E5], which uses an in vivo imaging method.
- [E10] A method for diagnosing a disease using the antigen-binding molecule according to any one of [E1] to [E3].
- [E11] The method according to [E10], wherein the diagnosis is to determine the presence or absence of a disease or to predict a therapeutic effect on a disease.
- [E12] The method according to any one of [E10] to [E11], wherein the disease is cancer.
- [E13] The method according to [E12], wherein the cancer is a cancer in which MTA is accumulated in cancer tissue.
- the cancer is a cancer tissue in which the gene encoding MTAP is deficient or underexpressed, or has a mutation or splicing variant that reduces enzyme activity [E12] or [E13].
- [E15] A disease diagnostic kit comprising the antigen-binding molecule according to any one of [E1] to [E3].
- [E16] The kit according to [E15], wherein the diagnosis determines the presence or absence of a disease or predicts a therapeutic effect on a disease.
- [E17] The kit according to any one of [E15] to [E16], wherein the disease is cancer.
- [E18] The kit according to [E17], wherein the cancer is a cancer in which MTA is accumulated in cancer tissue.
- the antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes depending on the MTA of the present disclosure, and the pharmaceutical composition containing the antigen-binding molecule do not act systemically in normal tissues or blood and are reversible in cancer. By acting on, it is possible to exert a medicinal effect while avoiding side effects and treat cancer. Furthermore, if a library containing a plurality of antigen-binding molecules having different sequences from each other and containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner is used, as described above, cancer tissue-specific. It is possible to efficiently obtain various antigen-binding molecules whose antigen-binding activity changes in an MTA-dependent manner, which is useful for treating diseases, in a short time.
- MTAP- means MTAP-deficient cell line and MTAP + means MTAP-free strain.
- MTA concentration in the culture medium in which each cell line was cultured The vertical axis is the MTA concentration in the culture medium, and the horizontal axis is the culture time, cell line name, and MTAP deficiency status.
- MTAP- means MTAP-deficient cell line and MTAP + means MTAP-free strain.
- the vertical axis is the MTA concentration in the culture solution
- the horizontal axis is the culture time.
- Each spot shows each measured data.
- the graph on the left shows the MTA concentration in the culture medium when HT-1376 of non-MTAP cells was cultured
- the graph on the right shows the MTA in the culture medium when SK-MES-1 of non-MTAP-deficient cells was cultured. It shows the concentration.
- the vertical axis is the MTA concentration in the tumor
- the horizontal axis is the name of the cell line transplanted into the mouse.
- Each spot shows each measured data. It is a figure which shows the relationship between the amount of MTAP DNA in a human clinical sample and the MTA concentration in a tissue.
- the graph on the left shows the results of measuring clinical samples of bladder cancer, and the graph on the right shows the results of measuring clinical samples of esophageal cancer.
- Each spot in the graph shows a clinical sample, and the light-colored spots show a sample in which the MTA concentration in the tissue was below the detection limit.
- the vertical axis is the MTA concentration in the tissue
- the horizontal axis is ⁇ Ct obtained by subtracting the Ct value of the ⁇ X4 gene from the Ct value of the MTAP gene. It is a figure which shows the extracellular MTA concentration in the tissue of a cancer-bearing mouse. The graph on the left shows the extracellular MTA concentration in the tumor, and the graph on the right shows the extracellular MTA concentration in the liver, which is a normal tissue.
- the vertical axis is the extracellular MTA concentration in the tissue
- the horizontal axis is the name of the cell line transplanted into the mouse. Each spot shows each measured data, and the hollow spot is the data below the lower limit of MTA quantification.
- the vertical axis shows the absorbance by phage ELISA when the MTA is not solid-phased
- the horizontal axis shows the absorbance by the phage ELISA when the MTA is solid-solidified. It is a figure which shows the binding amount to MTA of the clone group acquired after panning to MTA of the light chain variable region phage display library.
- the vertical axis shows the absorbance by phage ELISA when the MTA is not solid-phased
- the horizontal axis shows the absorbance by the phage ELISA when the MTA is solid-solidified.
- Amino acid residues that interact with the MTA are shown in a stick model.
- the dashed line and its value indicate the hydrogen bond distance between the amino acid residue and the MTA.
- the unit is indicated by ⁇ .
- the heavy chain of the antibody is shown in black, the light chain is shown in gray, and the MTA is shown in a ball-and-stick model.
- the heavy chain of the antibody is shown in black, the light chain is shown in gray, and the MTA is shown in a ball-and-stick model.
- Amino acid residues that interact with the MTA are shown in a stick model.
- the dashed lines and their numbers indicate the distance between hydrogen bonds, CH- ⁇ interactions, or ⁇ - ⁇ interactions between each amino acid residue and the MTA.
- the unit is indicated by ⁇ .
- the heavy chain of the antibody is shown in black, the light chain is shown in light gray, and the MTA is shown in a stick model.
- the C ⁇ atoms of isoleucine, leucine, valine, and alanine contained in the heavy chain of the antibody are indicated by dark gray spheres.
- the C ⁇ atoms of isoleucine, leucine, valine, and alanine contained in the light chain are indicated by white spheres. It is a figure of the crystal structure of the complex of MTA0330Fab and MTA. In the figure, the heavy chain of the antibody is shown in black, the light chain is shown in light gray, and the MTA is shown in a stick model. The C ⁇ atoms of isoleucine, leucine, valine, and alanine contained in the heavy chain of the antibody are indicated by dark gray spheres. The C ⁇ atoms of isoleucine, leucine, valine, and alanine contained in the light chain are indicated by white spheres.
- the superposition of the 1H-15N TROSY spectra of the MTA0303Fab in the MTA bound state and the unbound state is shown. It is a figure which shows the spectrum of the bonded state in black, and the unbonded state in gray.
- the superposition of the 1H-13C SOFAST-HMQC spectra of MTA0303Fab in the MTA bound state and the unbound state is shown. It is a figure which shows the spectrum of the bonded state in black, and the unbonded state in gray.
- the superposition of the 1H-15N TROSY spectra of the MTA0330Fab in the MTA bound state and the unbound state is shown. It is a figure which shows the spectrum of the bonded state in black, and the unbonded state in gray.
- the interaction between the antibody and the antigen is insufficient and the antibody cannot bind to the antigen, but in the presence of small molecules, the antibody binds to the antigen by being sandwiched between the antibody and the antigen. It becomes possible.
- Bispecific with an antigen-binding domain that binds to IL-6R and an antigen-binding domain that binds to CD3 in an MTA-dependent manner in the presence or absence of MTA or ADO tested using NFAT-RE-luc2-Jurkat cells The figure which shows the sex antibody T cell activation ability.
- the X-axis shows the antibody concentration ( ⁇ g / mL), and the Y-axis shows the relative luminescence amount (RLU).
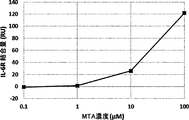
- FIG. 1 It is a figure which shows the binding amount to hIL-6R of the anti-IL-6R antibody which binds to IL-6R in an MTA-dependent manner under different MTA concentrations measured using BiacoreT200.
- the vertical axis shows the amount of hIL-6R bound per solid phase antibody amount, and the horizontal axis shows the concentration of MTA.
- It is a sensorgram showing the time course of the binding amount of the anti-IL-6R antibody that binds to IL-6R in an MTA-dependent manner to the antigen (hIL-6R), which was measured using the Octet RED384 system.
- the upper and lower segment diagrams show the measurement results with 100 ⁇ M and 10 ⁇ M MTA, respectively.
- Amino Acids In the present specification, for example, Ala / A, Leu / L, Arg / R, Lys / K, Asn / N, Met / M, Asp / D, Phe / F, Cys / C, Pro / P, Gln / Amino acids are single-letter codes or as represented by Q, Ser / S, Glu / E, Thr / T, Gly / G, Thrp / W, His / H, Tyr / Y, Ile / I, Val / V. It is written in 3-character code or both.
- Modification of Amino Acids For modification of amino acids in the amino acid sequence of an antigen-binding molecule, site-specific mutagenesis (Kunkel et al. (Proc. Natl. Acad. Sci. USA (1985) 82, 488-492)) and Overlap A known method such as extension PCR can be appropriately adopted. In addition, a plurality of known methods may also be adopted as methods for modifying amino acids other than natural amino acids (Annu. Rev. Biophys. Biomol. Struct. (2006) 35, 225-249, Proc. Natl. Acad. Sci. USA (2003) 100 (11), 6353-6357).
- a cell-free translation system (Clover Direct (Protein Express)) containing a tRNA in which an unnatural amino acid is bound to a complementary amber suppressor tRNA of a UAG codon (amber codon), which is one of the stop codons, is also preferable. Used.
- the meaning of the terms "and / or” used to describe the site of modification of an amino acid includes any combination of “and” and “or” as appropriate.
- “the amino acids at positions 33, 55, and / or 96 are substituted” includes the following variations of amino acid modifications; (a) 33rd, (b) 55th, (c) 96th, (d) 33rd and 55th, (e) 33rd and 96th, (f) 55th and 96th, (g) 33rd And 55th and 96th.
- an expression indicating the modification of an amino acid an expression in which the one-character code or the three-character code of the amino acid before and after the modification is written before and after the number indicating a specific position can be appropriately used.
- the modification N100bL or Asn100bLeu used when substituting the amino acid contained in the antibody variable region represents the substitution of Asn at position 100b with Leu represented by Kabat numbering. That is, the number represents the position of the amino acid represented by Kabat numbering, the one-letter code or three-letter code of the amino acid described before it is the amino acid before replacement, and the one-letter code or 3 of the amino acid described after that.
- the character code represents the amino acid after replacement.
- the modification P238D or Pro238Asp used when substituting an amino acid for the Fc region contained in the antibody constant region represents the substitution of Pro at position 238 with Asp, which is represented by EU numbering. That is, the number represents the position of the amino acid represented by the EU numbering, and the one-letter code or three-letter code of the amino acid described before it is the amino acid before replacement, and the one-letter code or 3 of the amino acid described after it.
- the character code represents the amino acid after replacement.
- the structure of an "antigen” is not limited to a specific structure as long as it contains an epitope to which an antigen-binding domain binds.
- the antigen is a peptide, or polypeptide, or protein of 4 or more amino acids.
- the antigens are the following molecules; 17-IA, 4-1BB, 4Dc, 6-keto-PGF1a, 8-iso-PGF2a, 8-oxo-dG, A1 adenosine receptor, A33, ACE, ACE-2, Activin, Activin A, Activin AB, Activin B, Activin C, Activin RIA, Activin RIA ALK-2, Activin RIB ALK-4, Activin RIIA, Activin RIIB, ADAM, ADAM10, ADAM12, ADAM15, ADAM17 / TACE, ADAM8, ADAM9 , ADAMTS, ADAMTS4, ADAMTS5, addressin, aFGF, ALCAM, ALK, ALK-1, ALK-7, alpha-1-antitrypsin, alpha-V / beta-1 antagonist, ANG, Ang, APAF-1, APE, APJ , APP, APRIL, AR, ARC,
- Receptors are also described in the above-mentioned examples of antigens, but even when these receptors are present in a soluble form in a biological fluid, antigen binding in which the antigen-binding activity changes depending on the MTA of the present disclosure. It can be used as an antigen to which an antigen-binding molecule containing a domain binds.
- soluble receptors for example, soluble IL-6R as described by Mullberg et al. (J. Immunol. (1994) 152 (10), 4958-4968).
- SEQ ID NO: 1 the protein consisting of amino acids 1 to 357 can be exemplified.
- antigens examples include membrane-type molecules expressed on cell membranes and soluble-type molecules secreted extracellularly from cells.
- an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes depending on the MTA of the present disclosure binds to a soluble molecule secreted from a cell, the antigen-binding molecule is described later. It is preferable that it has a neutralizing activity.
- the soluble molecule can be present in a biological fluid, that is, any liquid that fills between a vessel or a tissue / cell in the living body.
- the soluble molecule to which the antigen-binding molecule of the present disclosure binds can be present in extracellular fluid.
- extracellular fluid is a component in bone and cartilage such as plasma, intertissue fluid, lymph fluid, tight connective tissue, cerebrospinal fluid, cerebrospinal fluid, puncture fluid, or joint fluid, and alveolar fluid (aqueous humor).
- aqueous humor aqueous humor
- an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes depending on the MTA of the present disclosure binds to a membrane-type molecule expressed on a cell membrane
- a preferred example of the antigen-binding molecule will be described later.
- Preferred are antigen-binding molecules that have cytotoxic activity or the ability to bind or bind cytotoxic substances.
- an antigen having a neutralizing activity in place of or in addition to the property of having a cytotoxic activity or having the ability to bind or bind a cytotoxic substance.
- Bound molecules are also preferably cited as a non-limiting aspect.
- the "antigen-binding domain” can be a domain of any structure as long as it binds to the antigen of interest.
- a module called A domain of about 35 amino acids contained in Avimmer which is a cell membrane protein existing in the living body (International Publication WO2004 / 044011, WO2005 / 040229), Adnectin containing 10Fn3 domain, which is a domain that binds to a protein in fibronectin, which is a sugar protein expressed on the cell membrane (International Publication WO2002 / 032295), and a bundle of three helices consisting of 58 amino acids of Protein A Affibody (International Publication WO1995 / 001937) whose IgG binding domain constitutes scaffold, ankyrin repeat having a structure in which a turn containing 33 amino acid residues and two antiparallel helix and loop sub
- Anticalin et al. International Publication WO2003 / 029462
- LRR leucine-rich-repeat
- VLR variable lymphocyte receptor
- a recessed region of the parallel sheet structure International Publication WO2008 / 016854
- Preferable examples of the antigen-binding domains of the present disclosure include antigen-binding domains containing variable regions of the heavy and light chains of antibodies.
- antigen-binding domains are "scFv (single chain Fv)", “single chain antibody”, “Fv”, “scFv2 (single chain Fv 2)", “Fab” or “F ( ab') 2 ”and the like are preferably mentioned.
- antigen-binding molecules including an antigen-binding domain are used in the broadest sense, and specifically, as long as they include an antigen-binding domain, various molecular types are included.
- the antigen-binding molecule may be a molecule consisting only of the antigen-binding domain, or may be a molecule containing the antigen-binding domain and other domains.
- examples thereof include a complete antibody and an antibody fragment.
- Antibodies can include single monoclonal antibodies (including agonist and antagonist antibodies), human antibodies, humanized antibodies, chimeric antibodies and the like.
- a scaffold molecule in which a three-dimensional structure such as an existing stable ⁇ / ⁇ barrel protein structure is used as a scaffold (base) and only a part of the structure is made into a library for constructing an antigen-binding domain is also an antigen of the present disclosure. Can be included in the binding molecule.
- an antibody refers to an immunoglobulin that is natural or produced by partial or complete synthesis.
- Antibodies can be isolated from naturally occurring natural resources such as plasma and serum and culture supernatants of hybridoma cells that produce the antibodies, or partially or completely by using techniques such as genetic recombination. Can be synthesized. Suitable examples of antibodies include immunoglobulin isotypes and subclasses of those isotypes. Nine classes (isotypes) of IgG1, IgG2, IgG3, IgG4, IgA1, IgA2, IgD, IgE, and IgM are known as human immunoglobulins.
- the antibodies of the present disclosure may include IgG1, IgG2, IgG3, IgG4 among these isotypes.
- human IgG1, human IgG2, human IgG3, and human IgG4 constant regions multiple allotype sequences due to gene polymorphisms are described in Sequences of proteins of immunological interest, NIH Publication No. 91-3242, but in the present disclosure. Either of them may be used.
- sequence of human IgG1 the amino acid sequence at positions 356-358 represented by EU numbering may be DEL or EEM.
- the amino acid positions assigned to the CDRs and FRs of an antibody are defined according to Kabat (Sequences of Proteins of Immunological Interest (National Institute of Health, Bethesda, Md.). , 1987 and 1991).
- Kabat Sequences of Proteins of Immunological Interest (National Institute of Health, Bethesda, Md.). , 1987 and 1991).
- the amino acids in the variable region are numbered according to Kabat numbering
- the amino acids in the constant region are numbered EU according to the amino acid position of Kabat. It is expressed according to.
- variable region refers to the heavy or light chain domain of an antibody that is involved in binding the antibody to an antigen.
- the heavy and light chain variable domains of native antibodies are similar, with each domain usually containing four conserved framework regions (FR) and three hypervariable regions (HVR).
- FR conserved framework regions
- HVR hypervariable regions
- antibodies that bind to a particular antigen may be isolated by screening a complementary library of VL or VH domains using the VH or VL domains from the antibody that binds to that antigen, respectively. See, for example, Portolano et al., J. Immunol. 150: 880-887 (1993); Clarkson et al., Nature 352: 624-628 (1991).
- Hypervariable Regions As used herein, the terms “supervariable regions” or “HVRs” are hypervariable in sequence (“complementarity determining regions” or “CDRs” (complementarity determining regions)) and / or structurally. Refers to each region of the variable domain of an antibody that forms a defined loop (“supervariable loop”) and / or contains antigen contact residues (“antigen contact”). Usually, the antibody contains 6 HVRs: 3 for VH (H1, H2, H3) and 3 for VL (L1, L2, L3).
- Illustrative HVRs herein include: (a) At amino acid residues 26-32 (L1), 50-52 (L2), 91-96 (L3), 26-32 (H1), 53-55 (H2), and 96-101 (H3) The resulting hypervariable loop (Chothia and Lesk, J. Mol. Biol. 196: 901-917 (1987)); (b) At amino acid residues 24-34 (L1), 50-56 (L2), 89-97 (L3), 31-35b (H1), 50-65 (H2), and 95-102 (H3).
- the resulting CDR (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed.
- HVR residues and other residues in the variable domain are numbered herein according to Kabat et al., Supra.
- variable domain FR refers to variable domain residues other than hypervariable region (HVR) residues.
- a variable domain FR usually consists of four FR domains: FR1, FR2, FR3, and FR4.
- sequences of HVR and FR usually appear in VH (or VL) in the following order: FR1-H1 (L1) -FR2-H2 (L2) -FR3-H3 (L3) -FR4.
- the Fc region contains an amino acid sequence derived from the constant region of the antibody heavy chain.
- the Fc region is the portion of the heavy chain constant region of the antibody that contains the hinge, CH2 and CH3 domains from the N-terminus of the hinge region of the papain cleavage site at the amino acid at position 216 represented by EU numbering.
- the Fc region can be obtained from human IgG1, but is not limited to any particular subclass of IgG.
- a preferable example of the Fc region is an Fc region having FcRn-binding activity in an acidic pH range, as described later. Further, as a preferable example of the Fc region, there is an Fc region having an Fc ⁇ receptor-binding activity as described later.
- an Fc region As a non-limiting aspect of such an Fc region, it is represented by human IgG1 (SEQ ID NO: 5), IgG2 (SEQ ID NO: 6), IgG3 (SEQ ID NO: 7), or IgG4 (SEQ ID NO: 8).
- the Fc region is illustrated.
- the antibody used in the present disclosure is not limited to the full length molecule of the antibody, and may be a low molecular weight antibody or a modified product thereof.
- the low molecular weight antibody is not particularly limited as long as it contains an antibody fragment in which a part of a full-length antibody (for example, whole antibody such as whole IgG) is deleted and has an antigen-binding activity.
- the low molecular weight antibody of the present disclosure is not particularly limited as long as it is a part of the full-length antibody, but preferably contains a heavy chain variable region (VH) and / and a light chain variable region (VL).
- VH heavy chain variable region
- VL light chain variable region
- the amino acid sequence of VH or VL may be substituted, deleted, added and / or inserted.
- variable region may be chimerized or humanized.
- the antibody fragment include Fab, Fab', F (ab') 2, Fv and the like.
- Specific examples of low-molecular-weight antibodies include Fab, Fab', F (ab') 2, Fv, scFv (single chain Fv), Diabody, sc (Fv) 2 (single chain (Fv) 2). And so on. Multimers of these antibodies (eg, dimers, trimmers, tetramers, polymers) are also included in the low molecular weight antibodies of the present disclosure.
- Antibody fragments can be produced by treating the antibody with an enzyme such as papain, pepsin, or by constructing a gene encoding these antibody fragments, introducing it into an expression vector, and then expressing it in a suitable host cell.
- an enzyme such as papain, pepsin
- a gene encoding these antibody fragments introducing it into an expression vector, and then expressing it in a suitable host cell.
- Diabody refers to a bivalent low molecular weight antibody constructed by gene fusion (Holliger et al. (Proc. Natl. Acad. Sci. USA 90, 6444-6448 (1993), European Publication EP404097, and PCT). Publication WO 1993/011161 etc.). Diabody is a dimer composed of two polypeptide chains, and usually the polypeptide chains are so short that VL and VH cannot bind to each other in the same chain, for example. , It is bound by a linker of about 5 residues. VL and VH encoded on the same polypeptide chain cannot form a single-chain variable region fragment because the linker between them is short, and a dimer is formed. To form, the Diabody will have two antigen binding sites.
- ScFv is obtained by linking the H-chain variable region and the L-chain variable region of an antibody.
- the H-chain variable region and the L-chain variable region are linked via a linker, preferably a peptide linker (Huston et al. (Proc. Natl. Acad. Sci. USA (1988) 85, 5879-5883).
- the H-chain variable region and the L-chain variable region in scFv may be derived from any of the antibodies described herein as antibodies.
- the peptide linker that links the variable regions is not particularly limited, but for example. Any single-chain peptide consisting of about 3 to 25 residues, a peptide linker described later, or the like can be used.
- the above-mentioned PCR method can be used as a method for linking variable regions.
- the H chain of the antibody Or, of the DNA sequence encoding the H chain variable region and the DNA sequence encoding the L chain or the L chain variable region, the DNA portion encoding the entire or desired amino acid sequence is used as a template, and the sequences at both ends thereof are supported.
- the DNA encoding the scFv can be amplified by the PCR method using a pair of primers having the same sequence.
- the DNA encoding the peptide linker moiety and both ends thereof are designed to be linked to the H chain and the L chain, respectively.
- a DNA having a desired sequence can be obtained by performing a PCR reaction by combining a pair of primers having the same sequence.
- an expression vector containing them and the said DNA can be obtained according to a conventional method, and the scFv can be obtained by culturing the resulting recombinant cells to express the DNA encoding the scFv.
- sc (Fv) 2 is a low molecular weight antibody in which two VHs and two VLs are bound to each other with a linker or the like to form a single chain (Hudson et al. (J. Immunol. Methods (1999) 231, 177-189). . Sc (Fv) 2 can be produced, for example, by linking scFv with a linker.
- two VHs and two VLs are arranged in the order of VH, VL, VH, VL ([VH] linker [VL] linker [VH] linker [VL]) starting from the N-terminal side of the single-stranded polypeptide.
- Antibodies characterized by being present are preferred.
- the order of the two VHs and the two VLs is not particularly limited to the above arrangement, and may be arranged in any order. For example, the following arrangement can be mentioned.
- linker that binds the variable region of the antibody a linker similar to the linker described in the above-mentioned antigen-binding molecule section can be used.
- a linker similar to the linker described in the above-mentioned antigen-binding molecule section can be used.
- the following sc (Fv) 2 can be mentioned, for example.
- three linkers are usually required, but all may use the same linker or different linkers.
- the antibody is treated with an enzyme such as papain, pepsin, etc. to produce antibody fragments, or these antibody fragments or DNA encoding the low molecular weight antibody is constructed.
- an enzyme such as papain, pepsin, etc.
- This may be introduced into an expression vector and then expressed in an appropriate host cell (for example, Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976; Better, M. And Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515; Lamoyi, E., Methods Enzymol.
- a chimeric antigen in which a fusion of an antigen or a fragment thereof that recognizes an antigen instead of a T cell receptor and a signal domain of T cells is integrated into T cells.
- Chimeric antigen receptor and T cells incorporating the chimeric antigen receptor can also be mentioned, but are not limited to this.
- Single Domain Antibodies One of the preferred examples of the antigen binding domain of the present invention is a single domain antibody (sdAb).
- the structure of the term "monodomain antibody” in the present specification is not limited as long as the domain alone can exert antigen-binding activity.
- Normal antibodies such as IgG antibodies show antigen-binding activity when a variable region is formed by pairing VH and VL, whereas monodomain antibodies do not pair with other domains. It is known that the domain structure of the monodomain antibody itself can exert antigen-binding activity alone.
- Monodomain antibodies usually have a relatively low molecular weight and are present in monomeric form.
- monodomain antibodies include, but are not limited to, antigen-binding molecules that are congenitally lacking a light chain, such as VHH of camelids, VNAR of sharks, or all or part of the VH domain of an antibody. Included are antibody fragments containing all or part of the VL domain. Examples of monodomain antibodies that are antibody fragments that include all or part of the VH / VL domain of an antibody are, but are not limited to, human antibodies VH or human antibodies as described, for example, in US Pat. No. 6,248,516 B1. Examples include monodomain antibodies that are artificially produced starting from VL. In some embodiments of the invention, one monodomain antibody has three CDRs (CDR1, CDR2 and CDR3).
- the monodomain antibody can be obtained from an animal capable of producing a monodomain antibody or by immunizing an animal capable of producing a monodomain antibody.
- animals capable of producing a single domain antibody include, but are not limited to, camelids and transgenic animals into which a gene capable of producing a single domain antibody has been introduced.
- Camelids include camels, llamas, alpaca, dromedaries and guanaco.
- transgenic animals into which a gene capable of producing a monodomain antibody has been introduced include, but are not limited to, the transgenic animals described in International Publication WO2015 / 143414 and US Patent Publication US2011 / 0123527 A1.
- a humanized monodomain antibody can also be obtained by setting the framework sequence of the monodomain antibody obtained from an animal to a human germline sequence or a sequence similar thereto.
- a humanized single domain antibody (eg, humanized VHH) is also an embodiment of the single domain antibody of the present invention.
- Humanized monodomain antibody refers to a chimeric monodomain antibody that comprises an amino acid residue from a non-human CDR and an amino acid residue from a human FR.
- the humanized monodomain antibody corresponds to all or substantially all CDRs of a non-human antibody, and all or substantially all FRs correspond to those of a human antibody.
- FR In a humanized antibody, even if some of the residues in the FR do not correspond to those of the human antibody, substantially all FRs are considered as an example corresponding to those of the human antibody. For example, when humanizing VHH, which is an aspect of a single domain antibody, some of the residues in FR must be residues that do not correspond to those of human antibody (C Vincke et al., The Journal of Biological Chemistry). 284, 3273-3284.).
- the monodomain antibody can be obtained from a polypeptide library containing the monodomain antibody by ELISA, panning, or the like.
- polypeptide libraries containing monodomain antibodies include, but are not limited to, naive antibody libraries obtained from various animals or humans (eg Methods in Molecular Biology 2012 911 (65-78), Biochimica et Biophysica Acta-Proteins. and Proteomics 2006 1764: 8 (1307-1319)), antibody libraries obtained by immunizing various animals (eg Journal of Applied Microbiology 2014 117: 2 (528-536)), or antibody genes of various animals or humans.
- Synthetic antibody libraries prepared from the above eg, Journal of Biological Screening 2016 21: 1 (35-43), Journal of Biological Chemistry 2016 291: 24 (12641-12657), AIDS 2016 30:11 (1691-1701) Be done.
- Method for producing an antibody having a desired binding activity to an antigen A method for producing an antibody having a desired binding activity for an antigen, which is a molecule different from the low molecular weight compound and is not dependent on a small molecule compound containing MTA. It is known to those skilled in the art.
- the method for producing an antibody that binds to IL-6R is exemplified below.
- Antibodies that bind to antigens other than IL-6R can also be appropriately prepared according to the following examples.
- the anti-IL-6R antibody can be obtained as a polyclonal or monoclonal antibody using known means.
- a monoclonal antibody derived from a mammal can be preferably produced.
- Monoclonal antibodies derived from mammals include those produced by hybridomas, those produced by host cells transformed with an expression vector containing an antibody gene by genetic engineering techniques, and the like.
- the monoclonal antibody of the present invention includes a "humanized antibody” and a "chimeric antibody".
- the monoclonal antibody-producing hybridoma can be produced, for example, as follows by using a known technique. That is, the IL-6R protein is used as a sensitizing antigen to immunize mammals according to conventional immunization methods. The resulting immune cells are fused with known parent cells by conventional cell fusion methods. Hybridomas that produce anti-IL-6R antibodies can then be selected by screening monoclonal antibody-producing cells by conventional screening methods.
- the production of the monoclonal antibody is carried out as shown below, for example.
- the IL-6R gene whose nucleotide sequence is disclosed in SEQ ID NO: 2
- the IL-6R protein represented by SEQ ID NO: 1 used as a sensitizing antigen for antibody acquisition can be obtained. .. That is, an appropriate host cell is transformed by inserting the gene sequence encoding IL-6R into a known expression vector.
- the desired human IL-6R protein is purified from the host cell or culture supernatant by a known method.
- the soluble IL-6R as described by Mullberg et al. (J. Immunol.
- IL-6R polypeptide sequence represented by SEQ ID NO: 1 which is IL-6R
- a protein consisting of amino acids 1 to 357 is expressed in place of the IL-6R protein represented by SEQ ID NO: 1.
- Purified natural IL-6R protein can also be used as a sensitizing antigen as well.
- the purified IL-6R protein can be used as a sensitizing antigen used for immunization against mammals. Partial peptides of IL-6R can also be used as sensitizing antigens. At this time, the partial peptide can also be obtained by chemical synthesis from the amino acid sequence of human IL-6R. It can also be obtained by incorporating a part of the IL-6R gene into an expression vector and expressing it. Furthermore, it can be obtained by degrading the IL-6R protein using a proteolytic enzyme, but the region and size of the IL-6R peptide used as a partial peptide are not particularly limited to special embodiments.
- any sequence can be selected from the amino acid sequences corresponding to the 20th to 357th amino acids in the amino acid sequence of SEQ ID NO: 1.
- the number of amino acids constituting the peptide to be the sensitizing antigen is preferably at least 5, for example, 6 or more, or 7 or more. More specifically, peptides of 8 to 50, preferably 10 to 30 residues can be used as sensitizing antigens.
- a desired partial polypeptide of the IL-6R protein or a fusion protein obtained by fusing a peptide with a different polypeptide can be used as a sensitizing antigen.
- fusion proteins used as sensitizing antigens for example, Fc fragments of antibodies, peptide tags and the like can be preferably utilized.
- a vector expressing a fusion protein can be prepared by in-frame fusion of genes encoding two or more desired polypeptide fragments and inserting the fusion gene into an expression vector as described above. The method for producing the fusion protein is described in Molecular Cloning 2nd ed.
- the mammal immunized with the sensitizing antigen is not limited to a specific animal, but is preferably selected in consideration of compatibility with the parent cell used for cell fusion. Generally, rodent animals such as mice, rats, hamsters, rabbits, monkeys and the like are preferably used.
- the above animals are immunized with the sensitizing antigen according to a known method.
- immunization is performed by injecting a sensitizing antigen intraperitoneally or subcutaneously into a mammal.
- a sensitizing antigen diluted with PBS (Phosphate-Buffered Saline), physiological saline, or the like at an appropriate dilution ratio is optionally mixed with a usual adjuvant, for example, Freund's complete adjuvant, and emulsified.
- the sensitizing antigen is administered to the mammal several times every 4 to 21 days.
- suitable carriers may be used during immunization of the sensitizing antigen.
- a partial peptide having a small molecular weight when used as a sensitizing antigen, it may be desirable to immunize the sensitizing antigen peptide bound to a carrier protein such as albumin or keyhole limpet hemocyanin.
- a carrier protein such as albumin or keyhole limpet hemocyanin.
- Hybridomas that produce the desired antibody can also be produced using DNA immunity as follows.
- DNA immunization means that a sensitized antigen is expressed in the living body of the immunized animal in which the vector DNA constructed in such a manner that the gene encoding the antigen protein can be expressed in the immunized animal is administered. It is an immune method in which immune stimulation is given by being performed. Compared with the general immunization method in which a protein antigen is administered to an immunized animal, DNA immunization is expected to have the following advantages. -Immune stimulation can be given by maintaining the structure of membrane proteins such as IL-6R. -No need to purify immune antigens.
- DNA expressing the IL-6R protein is administered to an immunized animal.
- the DNA encoding IL-6R can be synthesized by known methods such as PCR.
- the resulting DNA is inserted into a suitable expression vector and administered to an immune animal.
- a commercially available expression vector such as pcDNA3.1 can be preferably used.
- a method for administering the vector to a living body a commonly used method can be used.
- DNA immunization is performed by introducing gold particles adsorbed by an expression vector into the cells of an immune animal individual with a gene gun.
- the production of an antibody that recognizes IL-6R can also be produced by using the method described in WO2003 / 104453.
- immune cells are collected from the mammal and subjected to cell fusion.
- splenocytes in particular can be used.
- Mammalian myeloma cells are used as cells fused with the immune cells.
- Myeloma cells preferably include a suitable selectable marker for screening.
- Selectable markers refer to traits that can (or cannot) survive under specific culture conditions.
- Known selectable markers include hypoxanthine-guanine-phosphoribosyltransferase deficiency (hereinafter abbreviated as HGPRT deficiency) or thymidine kinase deficiency (hereinafter abbreviated as TK deficiency).
- HGPRT deficiency hypoxanthine-guanine-phosphoribosyltransferase deficiency
- TK deficiency thymidine kinase deficiency
- Cells with HGPRT or TK deficiency have hypoxanthine-aminopterin-thymidine sensitivity (hereinafter abbreviated as HAT sensitivity).
- HAT-sensitive cells
- HGPRT-deficient and TK-deficient cells can be selected in a medium containing 6 thioguanine, 8 azaguanine (hereinafter abbreviated as 8AG), or 5'bromodeoxyuridine, respectively.
- 8AG 8 azaguanine
- 5'bromodeoxyuridine normal cells that incorporate these pyrimidine analogs into their DNA die.
- cells deficient in these enzymes that cannot uptake these pyrimidine analogs can survive in selective medium.
- G418 resistance confers resistance to 2-deoxystreptamine antibiotics (gentamicin analogs) by the neomycin resistance gene.
- gentamicin analogs gentamicin analogs
- myeloma cells suitable for cell fusion are known.
- Such myeloma cells include, for example, P3 (P3x63Ag8.653) (J.Immunol. (1979) 123 (4), 1548-1550), P3x63Ag8U.1 (Current Topics in Microbiology and Immunology (1978) 81,1- 7), NS-1 (C.Eur.J.Immunol. (1976) 6 (7), 511-519), MPC-11 (Cell (1976) 8 (3), 405-415), SP2 / 0 ( Nature (1978) 276 (5685), 269-270), FO (J. Immunol. Methods (1980) 35 (1-2), 1-21), S194 / 5.XX0.BU.1 (J. Exp. Med. (1978) 148 (1), 313-323), R210 (Nature (1979) 277 (5692), 131-133) and the like can be preferably used.
- P3 P3x63Ag8.653
- cell fusion between the immune cells and myeloma cells is performed according to a known method, for example, the method of Koehler and Milstein et al. (Methods Enzymol. (1981) 73, 3-46).
- the cell fusion can be carried out, for example, in the presence of a cell fusion promoter in a normal nutrient culture medium.
- a cell fusion promoter for example, polyethylene glycol (PEG), Sendai virus (HVJ) and the like are used, and an auxiliary agent such as dimethyl sulfoxide is optionally added and used in order to further increase the fusion efficiency.
- the usage ratio of immune cells and myeloma cells can be set arbitrarily. For example, it is preferable to increase the number of immune cells by 1 to 10 times that of myeloma cells.
- the culture medium used for the cell fusion for example, RPMI1640 culture medium suitable for the growth of the myeloma cell line, MEM culture medium, and other ordinary culture mediums used for this type of cell culture are used, and further, cows are used. Serum supplements such as fetal bovine serum (FCS) can be suitably added.
- FCS fetal bovine serum
- a predetermined amount of the immune cells and myeloma cells are well mixed in the culture medium, and a PEG solution preheated to about 37 ° C. (for example, an average molecular weight of about 1000 to 6000) is usually 30 to 60%. It is added at a concentration of (w / v).
- the desired fusion cells are formed by gently mixing the mixed solution. Then, the appropriate culture solution mentioned above is sequentially added, and by repeating the operation of centrifuging and removing the supernatant, a cell fusion agent or the like that is unfavorable for the growth of hybridoma can be removed.
- the hybridoma thus obtained can be selected by culturing in a normal selective culture medium, for example, a HAT culture medium (a culture medium containing hypoxanthine, aminopterin and thymidine). Culture using the above HAT culture medium can be continued for a sufficient time (usually, such a sufficient time is several days to several weeks) for cells other than the desired hybridoma (non-fusion cells) to die.
- a HAT culture medium a culture medium containing hypoxanthine, aminopterin and thymidine.
- Culture using the above HAT culture medium can be continued for a sufficient time (usually, such a sufficient time is several days to several weeks) for cells other than the desired hybridoma (non-fusion cells) to die.
- the hybridomas that produce the desired antibody are then screened and single cloned by conventional limiting dilution methods.
- the hybridoma thus obtained can be selected by using a selective culture solution according to the selectable marker possessed by the myeloma used for cell fusion.
- cells deficient in HGPRT or TK can be selected by culturing in HAT culture medium (culture medium containing hypoxanthine, aminopterin and thymidine). That is, when HAT-sensitive myeloma cells are used for cell fusion, cells that have succeeded in cell fusion with normal cells can selectively proliferate in the HAT culture medium. Culture using the above HAT culture medium is continued for a sufficient time for cells other than the desired hybridoma (non-fusion cells) to die.
- the desired hybridoma can be selected by culturing for several days to several weeks. Screening and single cloning of hybridomas producing the desired antibody can then be performed by conventional limiting dilution methods.
- Screening and single cloning of the desired antibody can be suitably performed by a screening method based on a known antigen-antibody reaction.
- a monoclonal antibody that binds to IL-6R can bind to IL-6R expressed on the cell surface.
- Such monoclonal antibodies can be screened, for example, by FACS (fluorescence activated cell sorting).
- FACS fluorescence activated cell sorting
- FACS is a system that makes it possible to measure the binding of an antibody to the cell surface by analyzing the cells in contact with a fluorescent antibody with laser light and measuring the fluorescence emitted by each cell.
- cells expressing IL-6R are prepared.
- Preferred cells for screening are mammalian cells that have been forced to express IL-6R.
- the binding activity of the antibody to IL-6R on the cell surface can be selectively detected. That is, a hybridoma that produces an IL-6R monoclonal antibody can be obtained by selecting a hybridoma that produces an antibody that does not bind to a host cell but binds to an IL-6R forced expression cell.
- the antibody binding activity to immobilized IL-6R-expressing cells can be evaluated based on the principle of ELISA.
- IL-6R-expressing cells are immobilized in wells of an ELISA plate.
- the hybridoma culture supernatant is brought into contact with the immobilized cells in the well, and an antibody that binds to the immobilized cells is detected.
- the monoclonal antibody is of mouse origin, the antibody bound to the cell can be detected by an anti-mouse immunoglobulin antibody.
- the hybridoma that produces the desired antibody having the ability to bind to the antigen selected by these screenings can be cloned by a limiting dilution method or the like.
- the hybridoma producing the monoclonal antibody thus produced can be subcultured in a normal culture medium.
- the hybridoma can be stored in liquid nitrogen for a long period of time.
- the hybridoma can be cultured according to a usual method, and a desired monoclonal antibody can be obtained from the culture supernatant.
- a hybridoma can be administered to a compatible mammal to proliferate and a monoclonal antibody can be obtained from the ascites.
- the former method is suitable for obtaining a high-purity antibody.
- An antibody encoded by an antibody gene cloned from an antibody-producing cell such as the hybridoma can also be preferably used.
- the cloned antibody gene By incorporating the cloned antibody gene into an appropriate vector and introducing it into a host, the antibody encoded by the gene is expressed.
- Methods for isolating antibody genes, introducing them into vectors, and transforming host cells have already been established, for example, by Vandamme et al. (Eur. J. Biochem. (1990) 192 (3), 767- 775). Methods for producing recombinant antibodies are also known, as described below.
- a cDNA encoding the variable region (V region) of an anti-IL-6R antibody is obtained from a hybridoma cell that produces an anti-IL-6R antibody.
- the total RNA is usually first extracted from the hybridoma.
- the following method can be used. -Guanidine ultracentrifugation (Biochemistry (1979) 18 (24), 5294-5299) -AGPC method (Anal. Biochem. (1987) 162 (1), 156-159)
- the extracted mRNA can be purified using mRNA Purification Kit (manufactured by GE Healthcare Bioscience) or the like.
- kits for extracting total mRNA directly from cells such as QuickPrep mRNA Purification Kit (manufactured by GE Healthcare Bioscience) are also commercially available.
- mRNA can be obtained from hybridomas.
- a cDNA encoding the antibody V region can be synthesized from the obtained mRNA using reverse transcriptase.
- the cDNA can be synthesized by AMV Reverse Transcriptase First-strand cDNA Synthesis Kit (manufactured by Seikagaku Corporation) or the like.
- the desired cDNA fragment is purified from the obtained PCR product and then ligated with the vector DNA.
- a desired recombinant vector can be prepared from the Escherichia coli that formed the colony. Then, whether or not the recombinant vector has the base sequence of the target cDNA is confirmed by a known method, for example, the dideoxynucleotide chain illumination method or the like.
- cDNA is synthesized using RNA extracted from hybridoma cells as a template, and a 5'-RACE cDNA library is obtained.
- kits such as SMART RACE cDNA amplification kit are appropriately used for the synthesis of 5'-RACE cDNA library.
- the antibody gene is amplified by the PCR method using the obtained 5'-RACE cDNA library as a template.
- Primers for mouse antibody gene amplification can be designed based on known antibody gene sequences. These primers have different base sequences for each subclass of immunoglobulin. Therefore, it is desirable to determine the subclass in advance using a commercially available kit such as the IsoStrip mouse monoclonal antibody isotyping kit (Roche Diagnostics).
- a primer capable of amplifying a gene encoding ⁇ 1, ⁇ 2a, ⁇ 2b, ⁇ 3 as a heavy chain and a gene encoding a ⁇ chain and a ⁇ chain as a light chain is used.
- a primer that anneals to a portion corresponding to a constant region close to the variable region is generally used as a primer on the 3'side.
- the primer attached to the 5'RACE cDNA library preparation kit is used as the primer on the 5'RACE cDNA library preparation kit.
- the PCR product thus amplified can be used to reconstitute an immunoglobulin consisting of a combination of heavy and light chains.
- the desired antibody can be screened using the binding activity of the reconstituted immunoglobulin to IL-6R as an index. For example, when the purpose is to obtain an antibody against IL-6R, it is more preferable that the binding of the antibody to IL-6R is specific.
- Antibodies that bind to IL-6R can be screened, for example, as follows; (1) A step of contacting an IL-6R-expressing cell with an antibody containing a V region encoded by cDNA obtained from a hybridoma. (2) A step of detecting the binding between IL-6R-expressing cells and an antibody, and (3) a step of selecting an antibody that binds to IL-6R-expressing cells.
- a method for detecting the binding between an antibody and IL-6R-expressing cells is known. Specifically, the binding between the antibody and IL-6R-expressing cells can be detected by a method such as FACS described above. Fixed specimens of IL-6R-expressing cells can be appropriately utilized to assess antibody binding activity.
- a panning method using a phage vector is also preferably used.
- an antibody gene is obtained as a library of heavy chain and light chain subclasses from a polyclonal antibody-expressing cell group
- a screening method using a phage vector is advantageous.
- Genes encoding the variable regions of the heavy and light chains can form a single chain Fv (scFv) by linking with the appropriate linker sequence.
- the DNA encoding scFv having the desired binding activity can be recovered. By repeating this operation as necessary, scFv having the desired binding activity can be concentrated.
- the cDNA is digested by a restriction enzyme that recognizes the restriction enzyme sites inserted at both ends of the cDNA.
- Preferred restriction enzymes recognize and digest base sequences that rarely appear in the base sequences that make up an antibody gene. Further, in order to insert one copy of the digested fragment into the vector in the correct direction, it is preferable to insert a restriction enzyme that gives an attachment end.
- An antibody expression vector can be obtained by inserting the cDNA encoding the V region of the anti-IL-6R antibody digested as described above into an appropriate expression vector.
- a chimeric antibody expression vector can be constructed by inserting the V region gene into an expression vector having a constant region in advance. Specifically, for example, a restriction enzyme recognition sequence of a restriction enzyme that digests the V region gene can be appropriately arranged on the 5'side of an expression vector holding a DNA encoding a desired antibody constant region.
- a chimeric antibody expression vector is constructed by in-frame fusion of both digested with the same combination of restriction enzymes.
- the antibody gene is incorporated into an expression vector so that it is expressed under the control of an expression control region.
- the expression control region for expressing an antibody includes, for example, an enhancer and a promoter.
- an appropriate signal sequence can be added to the amino terminus so that the expressed antibody is secreted extracellularly.
- a peptide having the amino acid sequence MGWSCIILFLVATATGVHS (SEQ ID NO: 3) is used as the signal sequence, but a suitable signal sequence is added in addition to this.
- the expressed polypeptide is cleaved at the carboxyl-terminal portion of the above sequence, and the cleaved polypeptide can be secreted extracellularly as a mature polypeptide.
- a suitable host cell is then transformed with this expression vector to obtain a recombinant cell expressing the DNA encoding the anti-IL-6R antibody.
- the DNA encoding the antibody heavy chain (H chain) and light chain (L chain) is integrated into separate expression vectors.
- An antibody molecule having an H chain and an L chain can be expressed by simultaneously co-transfecting the same host cell with a vector incorporating the H chain and the L chain.
- the host cell can be transformed by integrating the DNA encoding the H and L chains into a single expression vector (see WO 1994/011523).
- host cells and expression vectors are known for producing antibodies by introducing the isolated antibody gene into a suitable host. Any of these expression systems can be applied to isolate the antigen binding domain of the present invention.
- eukaryotic cells animal cells, plant cells, or fungal cells can be used as appropriate. Specifically, the following cells can be exemplified as animal cells.
- Mammalian cells : CHO (Chinese hamster ovary cell line), COS (Monkey kidney cell line), Mieroma (Sp2 / 0, NS0, etc.), BHK (baby hamster kidney cell line), Hela, Vero, HEK293 (human) embryonic kidney cell line with sheared adenovirus (Ad) 5 DNA), PER.C6 cell (human embryonic retinal cell line transformed with the Adenovirus Type 5 (Ad5) E1A and E1B genes), etc. (Current Protocols in Protein Science (May, 2001,) Unit 5.9, Table 5.9.1))
- Amphibian cells Xenopus oocytes, etc.
- Insect cells sf9, sf21, Tn5, etc.
- an expression system of an antibody gene by a cell derived from the genus Nicotiana such as Nicotiana tabacum is known.
- Callus-cultured cells can be appropriately used for transformation of plant cells.
- -Yeast The genus Saccharomyces such as Saccharomyces serevisiae and the genus Pichia such as methanol-utilizing yeast (Pichia pastoris).
- -Filamentous fungi Aspergillus genus such as Aspergillus niger.
- an antibody gene expression system using prokaryotic cells is also known.
- bacterial cells such as Escherichia coli (E. coli) and Bacillus subtilis can be appropriately used.
- An expression vector containing the antibody gene of interest is introduced into these cells by transformation. By culturing the transformed cells in vitro, the desired antibody can be obtained from the culture of the transformed cells.
- transgenic animals can also be used to produce recombinant antibodies. That is, the antibody can be obtained from an animal into which a gene encoding a desired antibody has been introduced.
- an antibody gene can be constructed as a fusion gene by in-frame insertion into a gene encoding a protein that is uniquely produced in milk.
- a protein secreted into milk for example, goat ⁇ casein and the like can be utilized.
- the DNA fragment containing the fusion gene into which the antibody gene is inserted is injected into a goat embryo, and the injected embryo is introduced into a female goat.
- a desired antibody can be obtained as a fusion protein with a milk protein from milk produced by a transgenic goat (or its offspring) born from a goat that has received an embryo. Hormones can also be administered to transgenic goats to increase the amount of milk containing the desired antibody produced by transgenic goats (Bio / Technology (1994), 12 (7), 699-702). ..
- the antigen-binding domain in the antigen-binding molecule is a recombinant type artificially modified for the purpose of reducing heterologous antigenicity to humans.
- Antigen-binding domains derived from antibodies can be appropriately adopted.
- Genetically modified antibodies include, for example, humanized antibodies and the like. These modified antibodies are appropriately produced using known methods.
- the method for producing an antibody having a desired binding activity for a specific low molecular weight compound As a method for producing an antibody having a desired binding activity for a specific low molecular weight compound, the method for producing an antibody that binds to a normal protein antigen has a desired binding activity for the low molecular weight compound by the same method as the method for producing an antibody. Antibodies can be obtained.
- a method for producing a sensitizing antigen used for obtaining an antibody against a low molecular weight compound a method of linking Mariculture Keyhole Limpet Hemocyanin (KLH) with a low molecular weight compound is exemplified.
- KLH Mariculture Keyhole Limpet Hemocyanin
- An example of a non-limiting sensitizing antigen produced to obtain an antibody against an MTA is 6'-MTA- Keyhole Limpet Hemocyanin (6'-MTA-KLH).
- Mariculture Keyhole Limpet Hemocyanin is a highly antigenic protein that can be recognized by T cell receptors expressed on helper T cells and is known to activate antibody production. Therefore, it should be linked with MTA. It is expected to enhance the production of antibodies against MTA.
- the low molecular weight compound to be linked to KLH is not limited to MTA, and it is possible to prepare a sensitizing antigen for various low molecular weight compounds that can be synthesized by the same method, and as a non-limiting example, Examples include AMP, ADP, ATP, adenosine, or SAH.
- the international publication WO2013 / 180200 also discloses the design of low molecular weight compound immunogens.
- the antigen linked to the low molecular weight compound is not limited to KLH, and for example, an antigen linked to biotin or the like may be used as a sensitizing antigen, and further, it can be linked to a low molecular weight compound other than KLH and biotin. If so, it can be used.
- the present disclosure also includes aspects exemplified below. [1] Biotinylated MTA. [2] Biotin-2'-MTA. [3] 6'-MTA-biotin. [4] Use of the biotinylated MTA according to [1] to [3] for screening an antigen-binding molecule that binds to MTA.
- a multispecific antigen-binding molecule or a multiparatopic antigen-binding molecule The at least one antigen-binding domain binds to the first epitope in the antigen molecule, and the at least one other antigen-binding domain is the second in the antigen molecule.
- An antigen-binding molecule containing at least two antigen-binding domains having a characteristic of binding to an epitope of is called a multispecific antigen-binding molecule from the viewpoint of the specificity of the reaction.
- the antigen-binding molecule binds to two different epitopes by two types of antigen-binding domains contained in one antigen-binding molecule, the antigen-binding molecule is called a bispecific antigen-binding molecule.
- the antigen-binding molecule binds to three different epitopes by three types of antigen-binding domains contained in one antigen-binding molecule, the antigen-binding molecule is called a trispecific antigen-binding
- the structure of the paratope in the antigen-binding domain that binds to the first epitope in the antigen molecule is different from that of the paratope in the antigen-binding domain that binds to the second epitope that has a different structure from the first epitope. Therefore, at least two of them have the characteristic that at least one antigen-binding domain binds to the first epitope in the antigen molecule and at least one other antigen-binding domain binds to the second epitope in the antigen molecule.
- An antigen-binding molecule containing an antigen-binding domain is called a multiparatopic antigen-binding molecule from the viewpoint of its structural specificity.
- the antigen-binding molecule When the antigen-binding molecule binds to two different epitopes by two types of antigen-binding domains contained in one antigen-binding molecule, the antigen-binding molecule is called a double paratopic antigen-binding molecule. When the antigen-binding molecule binds to three different epitopes by three types of antigen-binding domains contained in one antigen-binding molecule, the antigen-binding molecule is called a triple paratopic antigen-binding molecule.
- Bispecific antibody and its preparation method A multivalent multispecific or multiparatopic antigen-binding molecule containing one or more antigen-binding domains and its preparation method are described in Conrath et al. (J. Biol. Chem. (2001)). Non-patent literature such as 276 (10) 7346-7350), Muyldermans (Rev. Mol. Biotech. (2001) 74, 277-302) and Kontermann RE (2011) Bispecific Antibodies (Springer-Verlag), as well as international publication WO 1996 / It is also described in patent documents such as 034103 or WO1999 / 023221. By using the multiple specificity or multiple paratopic antigen-binding molecule described above and the method for preparing the same, it is possible to prepare the antigen-binding molecule of the present disclosure.
- a bispecific antibody and its preparation method are exemplified below.
- a bispecific antibody is an antibody that contains two types of variable regions that specifically bind to different epitopes.
- IgG-type bispecific antibodies can be secreted by hybrid hybridomas (quadromas) produced by fusing two hybridomas that produce IgG antibodies (Milstein et al. (Nature (1983) 305, 537-540)). ..
- genes encoding heavy chains containing the two variable regions of interest are introduced into cells and co-produced.
- a method of expression can be adopted.
- a pair of heavy chains containing a variable region that binds to the first epitope and a heavy chain containing a variable region that binds to the second epitope are paired.
- a combination of heavy chains with only a pair of chains is a mixture in which the number of molecules is 2: 1: 1. It is difficult to purify an antigen-binding molecule containing the desired heavy chain combination from a mixture of these three types of heavy chain combinations.
- a bispecific antibody containing a heterogeneous combination of heavy chains can be obtained by modifying the CH3 domain constituting the heavy chain with an appropriate amino acid substitution. It can be secreted preferentially. Specifically, the amino acid side chain existing in the CH3 domain of one heavy chain is replaced with a larger side chain (knob (meaning "protrusion")), and the amino acid side existing in the CH3 domain of the other heavy chain is replaced. Replacing the chain with a smaller side chain (hole (meaning "void")) allows protrusions to be placed in the void, causing promotion of heterogeneous heavy chain formation and inhibition of homologous heavy chain formation.
- the method International Publication WO 1996027011, Ridgway et al. (Protein Engineering (1996) 9, 617-621), Merchant et al. (Nat. Biotech. (1998) 16, 677-681)).
- carcinomas originating from epithelial tissues such as the gastrointestinal tract and skin include brain cancer, skin cancer, neck cancer, esophageal cancer, lung cancer, gastric cancer, duodenal cancer, breast cancer, prostate cancer, cervical cancer, etc.
- Examples thereof include endometrial cancer, pancreatic cancer, liver cancer, colon cancer, colon cancer, bladder cancer, and ovarian cancer.
- non-limiting examples of sarcomas originating from non-epithelial tissues (interstitium) such as muscles include osteosarcoma, chondrosarcoma, rhabdomyosarcoma, leiomyosarcoma, liposarcoma, and angiosarcoma. ..
- non-limiting examples of hematopoietic-derived blood cancers include malignant lymphomas, including Hodgkin's lymphoma and non-Hodgkin's lymphoma, acute myelocytic leukemia, or chronic myelocytic.
- Neoplasm means any newly developed pathogenic tissue tumor.
- neoplasms produce tumor formation, which is partially characterized by angiogenesis.
- Neoplasms are, for example, benign such as hemangiomas, gliomas, teratomas, or malignant such as carcinomas, sarcomas, gliomas, astrocytes, neuroblastomas, retinoblastomas, etc. sell.
- cancer tissue means a tissue containing at least one cancer cell. Thus, it refers to all cell types that contribute to the formation of tumor masses, including cancer cells and endothelial cells, such as cancer tissue containing cancer cells and blood vessels. In the present specification, the mass means a tumor tissue lesion (a foci of tumor tissue).
- tumor is commonly used to mean benign or malignant neoplasms.
- the "cancer tissue in which MTA is accumulated” means a cancer tissue in which a large amount of MTA is detected as compared with a normal tissue in the cancer tissue, and is a normal tissue to be compared.
- the normal tissue adjacent to the cancer tissue and the tissue of a healthy person can be exemplified.
- cancer tissues lacking methylthioadenosine phosphorylase (MTAP), which metabolizes MTA, and cancer tissues with reduced MTAP function are also aspects of cancer tissues in which MTA is accumulated. ..
- cancer tissues in which the gene encoding MTAP is deficient or underexpressed cancer tissues expressing mutations or splicing variants that reduce MTAP activity, or MTAP enzyme activity is reduced.
- the cancerous tissue is an aspect of the cancerous tissue in which MTA is accumulated. Decreased expression or function of MTAP can be determined by comparison with normal tissue, and non-limiting examples of normal tissue to be compared include normal tissue adjacent to cancer tissue and tissue of a healthy person. Can be done.
- Cancer-associated fibroblast In the present disclosure, "Cancer-associated fibroblast (CAF)” is a heterogeneous population having various origins such as endothelial cells existing around cancer tissue.
- the characteristics of non-limiting CAF include promoting the growth of cancer cells, promoting angiogenesis, promoting vascular infiltration of cancer cells, controlling the immune response, and constructing a microenvironment favorable for cancer progression. is there.
- ⁇ -smooth muscle actin ⁇ -SMA
- fibroblast activation protein FAP
- TN-C tenascin-C
- POSTN periostin
- NG2 chondroitin sulfate proteoglycan
- PDGFR Platelet derived growth factor receptor
- FSP1 fibroblast specific protein-1
- Tumor-associated macrophage refers to macrophages present in and around cancer tissue.
- TAM Tumor-associated macrophage
- a macrophage population that forms a cancer microenvironment together with fibroblasts, vascular endothelial cells, and the like is exemplified.
- An example of a non-limiting TAM feature is the induction of new blood vessels by the production of various angiogenic factors, such as the production of anti-inflammatory factors and the suppression of anti-tumor immunity by promoting the infiltration of regulatory T cells.
- cells expressing a marker selected from CD163, CD204, IL-10, TGF- ⁇ , and Prastagradin E2 are exemplified.
- effector cells are T cells (CD4 + (helper lymphocytes) T cells and / or CD8 + (cytotoxic) T cells), polynuclear leukocytes (neutrophils, eosinophils, etc.).
- Leukocytes such as eosinophils, obese cells), monospheres, macrophages, histospheres or natural killer cells (NK cells), NK-like T cells, Kupper cells, Langerhans cells, or lymphocain-activated killer cells (LAK cells), B It can be used in the broadest sense, including lymphocytes, or antigen-presenting cells such as dendritic cells or macrophages, but examples of suitable effector cells are CD8 + (cytotoxic) T cells, NK cells, or macrophages. Can be mentioned.
- a membrane-type molecule expressed on the cell membrane of an effector cell can be used as an antigen to which at least one antigen-binding domain contained in the antigen-binding molecule of the present disclosure binds, but a suitable membrane-type molecule comprises TCR.
- Polypeptides, CD3, CD2, CD28, CD44, CD16, CD32, CD64, or NKG2D or NK cell activating antigens can be exemplified as non-limiting examples.
- Methylthioadenosine As used herein, the term “MTA” refers to Methylthioadenosine, specifically a compound represented by the following chemical formula. MTA (CAS number: 2457-80-9)
- MTA analogs refers to low molecular weight compounds other than MTAs that have some common structure with MTAs.
- MTA analog a low molecular weight compound having adenosine as a common skeleton in the molecule can be exemplified, and a low molecular weight compound having a side chain containing a sulfur atom or an oxygen atom in the carbon at the 5-position in the adenosine can be exemplified.
- SAM S-adenosylmethionine
- SAH S-adenosylhomocystein
- S- (5'-) Metabolites of polyamine biosynthetic pathways such as Adenosyl) -L-homocysteine
- adenosine adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP)
- ATP adenosine triphosphate
- Adenosine Adenosine monophosphate (AMP) Adenosine diphosphate (ADP)
- Low-molecular-weight compounds refers to naturally-derived chemical substances or non-naturally-derived chemical substances other than "biopolymers" existing in the living body.
- low molecular weight compounds include, but are not limited to, naturally occurring or artificially synthesized compounds having a molecular weight of 10,000 or less, preferably compounds having a molecular weight of 1000 or less.
- Non-limiting aspects of low molecular weight compounds include, for example, cancer tissue-specific compounds, inflammatory tissue-specific compounds, and non-natural compounds.
- cancer tissue-specific compound is used in cancer tissue as compared to non-cancer tissue. Refers to a compound that exists as a target.
- the cancer tissue-specific compound is present in the cancerous tissue but not in the non-cancerous tissue, or not in the cancerous tissue but present in the non-cancerous tissue. It can be a compound defined by qualitative cancer tissue specificity such as.
- the cancer tissue specific compound is present in the cancer tissue at a different concentration (eg, high or low concentration) as compared to the non-cancer tissue. It can be a compound defined by specificity.
- cancer tissue-specific compounds are differentially present at any concentration. However, cancer tissue-specific compounds are generally at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%.
- Cancer tissue-specific compounds have p-values less than 0.05 and / or q-values, as determined using either Welch's t-test or Wilcoxon's rank-sum test, which are statistically significant. Is less than 0.10), preferably present differentially.
- One non-limiting aspect of the cancer tissue-specific compound is the cancer tissue-specific metabolism produced by the metabolic activity peculiar to cancer cells, immune cells, and stromal cells contained in the cancer tissue as follows. Compounds that are products (cancer tissue-specific metabolites; cancer cell-specific metabolites, immune cell-specific metabolites invading cancer tissue, cancer stromal cell-specific metabolites) can be exemplified. ..
- metabolism refers to chemical changes that occur within the tissues of an organism and includes “assimilation” and “catabolism.” Anabolism refers to the biosynthesis or accumulation of molecules, and catabolism refers to the decomposition of molecules.
- a “metabolite” is an intermediate or product resulting from the metabolism of a substance.
- Primary metabolites refer to metabolites that are directly involved in the growth or reproduction process of cells or organisms, and “secondary metabolites” are those that are directly involved in the growth or reproduction process of cells or organisms. It refers to products such as antibiotics and pigments that are produced as a result of metabolism that biosynthesizes substances that are not directly involved in common biological phenomena.
- the metabolite can be a "biopolymer” metabolite or a "small molecule” metabolite.
- a “biopolymer” is a polymer composed of one or more types of repeating units. Biopolymers are generally found in biological systems, and are molecules having a molecular weight of about 5000 or more, particularly polysaccharides (carbohydrates, etc.) that form structures such as cells that organize organisms and intercellular matrices and intertissue matrices that adhere to them. ) And peptides (the term is used to include polypeptides and proteins) and polynucleotides, as well as their analogs, eg, those compounds composed of or containing amino acid analogs or non-amino acid groups.
- a non-limiting aspect of the cancer tissue-specific metabolites described herein preferably includes cancer cell-specific small molecule metabolites (Eva Gottfried, Katrin Peter and Marina P. Kreutz, From. Molecular to Modular Tumor Therapy (2010) 3 (2), 111-132).
- cancer cell-specific small molecule metabolites Eva Gottfried, Katrin Peter and Marina P. Kreutz, From. Molecular to Modular Tumor Therapy (2010) 3 (2), 111-1312.
- high levels of metabolites produced by immune cells that infiltrate cancer tissue and stromal cells that infiltrate cancer tissue and stromal cells (cancer stromal cells or cancer stromal fibroblasts (CAF)) that support the survival and / or growth of cancer cells. It also includes the metabolites it produces.
- infiltrating immune cells include dendritic cells, suppressive dendritic cells, suppressive T cells, exhausted T cells, myeloma-derived suppressor cells, MDSCs, and the like.
- cancer cells when cells existing in cancer tissue (cancer cells, immune cells, stromal cells) die due to apoptosis, necrosis, etc., they are released from the inside of the cell to the outside of the cell.
- cancer cells cancer cells, immune cells, stromal cells
- stromal cells when cells existing in cancer tissue (cancer cells, immune cells, stromal cells) die due to apoptosis, necrosis, etc., they are released from the inside of the cell to the outside of the cell.
- Compounds are also included.
- inflammatory tissue-specific compound inflammatory tissue-specific compound
- inflammation tissue-specific compound is a compound that is differentially present in inflammatory tissue as compared to non-inflammatory tissue.
- inflamed tissue is exemplified as follows.
- Inflammatory tissue-specific metabolites are high-produced metabolites produced by immune cells infiltrating inflammatory tissue and high in injured normal cells in inflammatory tissue. It is a metabolite. Examples of infiltrating immune cells include effector T cells, mature dendritic cells, neutrophils, granule cells (mast cells), basophils and the like.
- the metabolites in the present invention also include compounds released from inside cells to outside cells when cells (immune cells, normal cells) existing in inflamed tissues die due to apoptosis, necrosis, or the like.
- non-natural compound refers to a non-naturally occurring chemical substance and its metabolites.
- One aspect of the invention is a non-naturally occurring chemical substance and its metabolite having the property of accumulating in a target tissue after being administered in vivo from outside the living body.
- unnatural compounds include (1) capecitabine (Xeloda) and its biotransformer 5-FU (fluorouracil), and (2) TH-302 and bromoisophosphamide mustard (Br-IPM). Illustrated.
- 5-FU is a metabolite of capecitabine (Xeloda) and is known to be metabolized by cancer tissue-specific metabolizing enzymes cytidine deaminase and thymidine phosphorylase (Desmoulin F. et al. Drug). Metab Dispos. 2002). It is also known that TH-302 is converted to Br-IPM by being reduced under hypoxic conditions, such as around cancer tissue (Duan JX, et al. J Med Chem. 2008). .. For example, when capecitabine (xeloda) is administered, it is metabolized to 5-FU by cancer-specific metabolic enzymes, resulting in a high concentration of 5-FU in the cancerous area (Desmoulin F.
- a 5-FU-switched antibody can selectively bind to the target antigen only locally in the cancer because of Drug Metab Dispos. 2002).
- metabolic enzymes it is also possible to use molecules produced in a cancer-specific hypoxic environment or acidic environment as switches.
- TH-302 Duan JX, et al. J Med Chem. 2008
- Br-IPM is metabolized to Br-IPM under hypoxic conditions, so it is selectively selected only locally in the cancer by an antibody that switches to Br-IPM. It is thought that it will be possible to bind to the target antigen.
- methods for administering an unnatural compound to a living body include, but are limited to, known administration methods such as oral administration, eye drop administration, transdermal administration, nasal administration, intravenous administration, and pulmonary administration. It's not a thing.
- Non-limiting aspects of the low molecular weight compound in the present disclosure include, for example, MTA, SAM, SAH, adenosine, adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and the like. Will be done.
- Glycolytic systems such as lactic acid, succinic acid, and citric acid, or primary metabolites of the Krebs cycle Low molecular weight compounds used in the present invention, or cancer tissue-specific compounds, particularly cancer cell-specific metabolites.
- primary metabolites produced as a result of glucose metabolism present in cancer tissues at a higher concentration than surrounding non-cancerous tissues such as lactic acid, succinic acid, and citric acid are preferably mentioned. Be done.
- glycolytic phenotype characterized as an upregulation of glycolytic (Embden-Myerhof pathway) enzymes such as pyruvate kinase, hexokinase, and lactate dehydrogenase (LDH), has a Warburg effect on solid tumors. It has been conventionally known to be a feature.
- M2 isotype pyruvate kinase required for erobic glycolysis under anaerobic conditions instead of M1 isotype in tumor cells is advantageous for the growth of tumor cells in vivo. It is believed to be working (Christofk et al. (Nature (2008) 452, 230-233). Pyruvate produced by pyruvate kinase is the result of equilibrium reaction by lactic acid glycolysis (LDH) under anaerobic conditions. The lactic acid produced causes feedback inhibition. The feedback inhibition promotes respiration (Krebs circuit) in mitochondria and suppresses cell proliferation, so that upregulation of LDH, hexokinskin, and glucose transporter (GLUT) causes tumor cells.
- LDH lactic acid glycolysis
- GLUT glucose transporter
- lactic acid, succinic acid, citric acid and the like which are primary metabolites produced by such glycolytic metabolism, are preferably mentioned, and by cell death. It is known that glycolysis, which is present in high concentrations in cells, leaks out of cells (Nature Immunology, (2008) 9, 1261-1269). Therefore, glycolysis occurs in tumor tissues where cell death occurs frequently. It is considered that the concentration of acid is increasing.
- Amino acids such as alanine, glutamic acid, and aspartic acid
- amino acids in tumor cells that require continuous supply of essential amino acids and non-essential amino acids necessary for biosynthesis of biopolymers under anaerobic conditions. It is known that metabolism is also changing.
- Glutamine is the most widely distributed amino acid in the body, acting as a nitrogen carrier with two nitrogens in its side chains. Tumor cells, whose intracellular uptake rate of glutamine is increased, are said to function as glutamine traps.
- glutaminolysis This increase in glutamine uptake and conversion to glutamic acid and lactic acid is called “glutaminolysis” and is thought to be characteristic of transformed (tumor) cells (Mazurek and Eigenbrodt (Anticancer). Res. (2003) 23, 1149-1154, and Mazurek et al. (J. Cell. Physiol. (1999) 181, 136-146)). As a result, cancer patients have decreased levels of glutamine in plasma. shows an increase in glutamate concentration (Droge et al. (Immunobiology (1987) 174, 473-479 ).
- 13 C-labeled succinic acid by metabolic studies of 13 C radiolabeled glucose lung cancer tissue, 13 C-labeled alanine, 13 C A correlation was observed between the concentrations of labeled glutamic acid and 13 C-labeled citric acid.
- glutamine degradation causes cancer.
- Preferable examples include alanine, glutamic acid, aspartic acid, etc., which accumulate in high concentrations in tissues.
- IDO Indoleamine 2,3 -dioxygenase
- a metabolite of amino acids such as kynurenine
- kidney cancer Uyttenhove et al. (Nat. Med. (2003) 9, 1269-127), two isoforms are known to exist (Lob et al. (CancerImmunol. Immunother. (2009) 58, 153-157)).
- IDO Is the first enzyme in the metabolite pathway of nicotine amide nucleotides (NADs) that catalyzes the conversion of tryptophan to kynurenine (represented by the formula below), and in gliomas that do not express IDO, tryptophan 2,3-di in the liver.
- Oxygenase (TDO) produces kynurenine from tryptophan (Opitz et al. (Nature (2011) 478, 7368, 197-203)), and IDO is also expressed in dendritic cells invading cancer tissue. , Tree cells also produce kynurenine (J. Immunol. (2008) 181, 5396-5404).
- IDO is also expressed in myeloid-derived inhibitory cells (MDSC) in cancer tissues, and MDSC also produces kynurenine. Produces (Yu et al. (J. Immunol. (2013) 190, 3783-3797)). Kynurenine
- Kynurenine is known to suppress allogeneic T cell responses (Frumento et al. (J. Exp. Med. (2002) 196, 459-468), through which tumor cells survive the antitumor immune response and A mechanism has been proposed in which glioma cell proliferation is promoted through an autocrine proliferation mechanism in which kynurenine acts as an endogenous ligand for the allyl hydrocarbon receptor expressed in glioma (Opitz et al. (Supra)). Kynurenine is a quinureninease. Converted to anthranilic acid (represented by the formula below) and to 3-hydroxyquinurenine (represented by the formula below) by kynurenine 3-hydroxylase. Both anthranic acid and 3-hydroxyquinurenine are precursors of NAD. It is converted to the body 3-hydroxyanthranilic acid.
- Kynurenine is converted to kynurenic acid (represented by the following formula) by kynurenine aminotransferase.
- kynurenine aminotransferase As a non-limiting aspect of the low molecular weight compounds, cancer tissue-specific compounds, particularly cancer cell-specific metabolites used in the present invention, such kynurenine and its metabolites, anthranilic acid and 3-hydroxyquinurenin. , And metabolites of amino acids such as kynurenic acid are preferred.
- Prostaglandin E2 a metabolite of arachidonic acid such as prostaglandin E2 is a prostaglandin synthesized by cyclooxygenase (COX) -1 / 2. And a metabolite of arachidonic acid called plastonoids, including thromboxane (Warner and Mitchell (FASEB J. (2004) 18, 790-804)). It promotes the proliferation of PGE2 colon cancer cells and suppresses their apoptosis (Sheng et al. (Cancer Res. (1998) 58, 362-366)). It is known that the expression of cyclooxygenase is altered in many cancer cells.
- COX-1 is constitutively expressed in almost all tissues
- COX-2 is mainly found to be induced in tumors by certain inflammatory cytokines and oncogenes. (Warner and Mitchell (supra)).
- Overexpression of COX-2 has a poor prognosis for breast cancer (Denkert et al. (Clin. Breast Cancer (2004) 4, 428-433), and rapid disease progression of ovarian cancer (Denker et al. (Mod. Pathol. (2006)). It has also been reported to be associated with 19, 1261-1269), and suppressive T cells invading cancer tissue also produce prostaglandin E2 (Curr. Med. Chem. (2011). ) 18, 5217-5223).
- Low molecular weight compounds such as prostaglandins and leukotrienes, which are metabolites of arachidonic acid, act as stimulators to control cancer autocline and / or paracrine growth.
- Known Non-limiting aspect of immune cell-specific metabolites infiltrating tissue is preferably a metabolite of arachidonic acid such as prostaglandin E2.
- thromboxane A2 TXA2 is upregulated in cancer tissues such as colon cancer (J. Lab. Clin. Med. (1993) 122, 518-523), a non-limiting aspect of the metabolite of arachidonic acid of the present invention. Is preferably mentioned.
- nucleoside cancer cells having a purine ring structure such as adenosine, adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) die, a large amount of intracellular ATP is generated. It is known to leak out of the cell. Therefore, the ATP concentration in cancer tissues is significantly higher than that in normal tissues (PLoS One. (2008) 3, e2599). Multiple types of cells release ATP, ADP and AMP types of adenine nucleotides. Metabolized by extracellular enzymes on the cell surface such as extracellular-5'-nucleotidase (CD73) (Resta and Thompson (Immunol. Rev.
- Adenosine is a purinucleotidase that is constitutively present in the extracellular environment at low concentrations, but extracellular adenosine concentrations in hypoxic tissues found in solid tumors. has been reported to be significantly increased (Blay and Hoskin (Cancer Res. (1997) 57, 2602-2605). CD73 is expressed on the surface of tumors and immune cells (Kobie et al. (J. Immunol. (2006)). ) 177, 6780-6786), breast cancer (Canbolat et al. (Breast Cancer Res. Treat.
- inhibitory T cells invading cancer tissue also express ATP-degrading enzymes and produce adenosine (Proc. Natl. Acad). Sci. (2006) 103 (35), 13132-13137, Curr. Med. Chem. (2011) 18, 5217-5223).
- Produced adenosine transmits cancer tissues via adenosine receptors such as A2A receptor. It is believed to create an immunosuppressive environment (Curr. Med. Chem. (2011), 18,5217-23).
- Non-limiting low molecular weight compounds and cancer tissue-specific compounds used in the present invention are examples of the present invention.
- ATP, ADP, AMP, adenosine and the like which accumulate in high concentrations in cancer tissues by metabolism of such purine nucleotides such as ATP, are preferably mentioned. Furthermore, adenosine is decomposed into inosine by adenosine deaminase, so that inosine accumulates in high concentration.
- ATP concentration is high in the alveoli inflamed due to bronchial asthma (Nat. Med. (2007) 13, 913-919). It is also known that ATP concentration is high in alveoli inflamed due to COPD (Am. J. Respir. Crit. Care Med. (2010) 181, 928-934). In addition, high adenosine concentrations have been observed in the joint fluid of patients with rheumatoid arthritis (Journal of Pharmaceutical and Biomedical Analysis (2004) 36 877-882). Furthermore, it is known that the ATP concentration is high in tissues in which GVHD causes rejection (Nat. Med. (2010) 16, 1434-1438).
- inflammatory tissue-specific compounds used in the present invention ATP, ADP, AMP, or adenosine, which accumulates in high concentrations in inflammatory tissues by metabolism of such purine nucleotides such as ATP. Etc. are preferably mentioned.
- adenosine is decomposed into inosine by adenosine deaminase, so that inosine accumulates at a high concentration.
- uric acid is a product of the metabolic pathway of purine nucleoside in vivo, it is liberated into the extracellular blood or interstitial ⁇ . In recent years, it has been clarified that it is released from dead cells existing at lesion sites such as cancer tissues (Nat. Med. (2007) 13, 851-856).
- uric acid which accumulates in a high concentration in the cancer tissue by metabolism of such purine nucleotides such as ATP is also preferably mentioned.
- uric acid released from cells undergoing necrosis promotes an inflammatory response (J. Clin. Invest. (2010) 120 (6), 1939-1949. ).
- uric acid which accumulates in a high concentration in the inflammatory tissue by metabolism of such purine nucleotides such as ATP is also preferably mentioned.
- 1-Methylnicotinamide It is known that the enzyme nicotinamide N-methyltransferase is highly expressed in a plurality of human cancer tissues. When this enzyme produces 1-methylnicotinamide, a stable metabolite from nicotinamide, it consumes the methyl group of S-adenosylmethionine (SAM), which is a methyl donor, so that SAM in cancer cells. It has been proposed that high expression of nicotinamide N-methyltransferase contributes to tumorigenesis through a mechanism that impairs the methylation ability of DNA with a decrease in concentration (Ulanovskaya et al. (Nat. Chem.). Biol. (2013) 9 (5) 300-306)).
- SAM S-adenosylmethionine
- 1-Methylnicotinamide a stable metabolite of this enzyme, is known to be secreted extracellularly of cancer cells (Yamada et al. (J. Nutr. Sci. Vitaminol. (2010) 56, 83). -86)), as a non-limiting aspect of the low molecular weight compound and cancer tissue-specific compound used in the present invention, 1-methylnicotinamide, which accumulates in high concentration in cancer tissue by metabolism of such nicotinamide, etc. Is also preferably mentioned.
- the low molecular weight compounds in the present disclosure can interact with antigen-binding molecules.
- Amino acid residues in the antigen-binding molecule capable of interacting with the low-molecular-weight compound may be present in the antigen-binding domain in the antigen-binding molecule, or may be present at a site other than the antigen-binding domain.
- An antigen-binding domain is exemplified, but is not limited to, as a site that interacts with a low molecular weight compound in an antigen-binding molecule.
- Antigen-binding domain that specifically binds to an antigen means that the antigen-binding domain refers to a specific epitope among a plurality of epitopes contained in an antigen. It is used when it is specific.
- the antigen-binding molecule having the antigen-binding domain can bind to various antigens including the epitope.
- the molecule does not substantially bind according to the method described in the section of the binding activity, and the binding activity of the specific binding molecule to the molecule other than the partner is the binding activity to the molecule of the partner. It means that it exhibits a binding activity of 80% or less, usually 50% or less, preferably 30% or less, and particularly preferably 15% or less.
- the binding of the antigen-binding domain to the antigen is a condition in which the antigen-binding activity of the antigen-binding domain is high. (For example, under a specific concentration of MTA or in the absence of MTA, under a specific concentration of a low molecular weight compound other than MTA, or in the absence of a low molecular weight compound other than MTA).
- binding activity refers to the term “binding activity” between one or more binding sites of a molecule (eg, an antibody) and a binding partner (eg, an antigen) of the molecule. Refers to the total strength of non-covalent interactions.
- binding activity is not strictly limited to a 1: 1 interaction between members of a binding pair (eg, antibody and antigen). For example, when the members of a binding pair reflect a 1: 1 interaction in monovalent, the binding activity refers to the inherent binding affinity (“affinity”). If the members of the binding pair are capable of both monovalent and multivalent binding, the binding activity is the sum of these binding forces.
- binding activity of the molecule X to its partner Y can generally be expressed by the dissociation constant (KD) or the "analyte binding amount per unit ligand amount". Binding activity can be measured by conventional methods known in the art, including those described herein. Conditions other than the concentration of the target tissue-specific compound can be appropriately determined by those skilled in the art. Specific examples and exemplary embodiments for measuring binding activity are described below.
- the antigen-binding molecule provided herein is an antibody, and the binding activity of the antibody is ⁇ 1 ⁇ M, ⁇ 100 nM, ⁇ 10 nM, ⁇ 1 nM, ⁇ 0.1.
- the dissociation constant (KD) of nM, ⁇ 0.01 nM or ⁇ 0.001 nM eg, 10 -8 M or less, eg 10 -8 M to 10 -13 M, eg 10 -9 M to 10 -13 M).
- the binding activity of the antibody is measured by surface plasmon resonance analysis, for example, ligand capture using BIACORE (registered trademark) T200 or BIACORE (registered trademark) 4000 (GE Healthcare, Uppsala, Sweden). The method is used. BIACORE (registered trademark) Control Software is used to operate the equipment.
- BIACORE registered trademark
- Control Software is used to operate the equipment.
- an amine coupling kit (GE Healthcare, Uppsala, Sweden) is used according to the supplier's instructions, and a ligand capture molecule, such as an anti-tag, is applied to a sensor chip (GE Healthcare, Uppsala, Sweden) coated with carboxymethyl dextran. Immobilize antibodies, anti-IgG antibodies, protein A, etc.
- the ligand trapping molecule is diluted with a 10 mM sodium acetate solution at the appropriate pH and injected at the appropriate flow rate and injection time.
- a buffer solution containing 0.05% polysorbate 20 (Tween (registered trademark) -20 as another name) is used as a buffer solution for measurement, the flow velocity is 10-30 ⁇ L / min, and the measurement temperature is preferably 25 ° C. Measured at 37 ° C.
- the antibody is captured as a ligand by a ligand-capturing molecule and the measurement is performed, the antibody is injected to capture the target amount, and then the antigen and / or Fc receptor stage prepared using the measurement buffer solution.
- a diluent is injected.
- the antigen and / or Fc receptor is captured as a ligand in the ligand capture molecule for measurement
- the antigen and / or Fc receptor is injected to capture the target amount, and then prepared using a buffer for measurement.
- a stepwise dilution (analyte) of the antibody is injected.
- the measurement results are analyzed using BIACORE® Evaluation Software.
- Kinetic parameter calculations are performed by simultaneously fitting sensorgrams of binding and dissociation using a 1: 1 Binding model, with binding rate (kon or ka) and dissociation rate (koff or kd).
- the equilibrium dissociation constant (KD) can be calculated. If the binding activity is weak, especially if the dissociation is fast and it is difficult to calculate the kinetic parameters, the equilibrium dissociation constant (KD) may be calculated using the Steady state model.
- the "analyte binding amount per unit ligand amount” can also be calculated by dividing the binding amount (RU) of a specific concentration of analyte by the ligand capture amount (RU).
- the antibody binding activity can be measured by, for example, the Octet RED 96e system or the Octet RED 384 system (Pall Forte Bio) based on the Bio-Layer Interferometry (BLI) method.
- Qualitative binding characteristic analysis and kinetic analysis of antigen-antibody reaction can be performed by using according to the instruction of the supplier.
- the specific measurement method after immobilizing an antibody on a Protein A (ProA) biosensor (Pall ForteBio), the antigen is allowed to interact as an analyst to change the amount of binding between antibody antigens. Can be measured.
- ProA Protein A
- the binding between the antibody and the antigen in the presence of MTA is reversibly dissociated in the absence of MTA over time. It is possible to observe.
- kd dissociation rate constant
- apparent kd Apparent dissociation rate constant
- the kd (dissociation rate constant) and the apparent kd (apparent dissociation rate constant) can be measured by a method known to those skilled in the art, and for example, Biacore (GE healthcare), a flow cytometer, or the like can be used. It is possible.
- the interaction between the test antigen-binding molecule and the antigen is measured under a specific concentration of the low-molecular-weight compound or It can be done in the absence.
- an antigen-binding molecule that does not exhibit antigen-binding activity in the absence of a specific low-molecular compound exhibits binding activity to the antigen in the presence of the low-molecular compound, the binding activity is evaluated in the presence of the low-molecular compound. can do.
- the binding activity is exhibited in the absence of the small molecule compound.
- the binding activity is under the condition that the low-molecular compound is present at a high concentration.
- the condition where the low-molecular-weight compound is present at a low concentration when the antigen-binding activity under the condition where the specific low-molecular-weight compound is present at a high concentration is lower than the antigen-binding activity under the condition where the low-molecular-weight compound is present at a low concentration, the condition where the low-molecular-weight compound is present at a low concentration.
- the binding activity can be evaluated in.
- the conditions that can affect the binding activity of the antigen and the antigen-binding molecule are not limited to the presence / absence of the low molecular weight compound and the concentration of the low molecular weight compound, and examples thereof include ion concentration, ion composition, and temperature. Is not limited to these. Furthermore, it is naturally possible to evaluate the binding activity under the condition that the above-mentioned different plurality of factors are combined.
- Epitope meaning an antigenic determinant present in an antigen means a site on the antigen to which the antigen-binding domain in the antigen-binding molecule disclosed herein binds.
- the site on the antigen to which the antigen-binding molecule binds in the present disclosure can be defined by evaluating the presence or absence of binding of the antigen-binding molecule.
- Epitopes can be defined by their structure.
- the epitope can also be defined by the binding activity to the antigen in the antigen-binding molecule that recognizes the epitope.
- the antigen is a peptide or polypeptide, it is also possible to identify the epitope by the amino acid residues constituting the epitope.
- the epitope is a sugar chain, it is also possible to specify the epitope by a specific sugar chain structure.
- a linear epitope is an epitope containing an epitope in which the primary amino acid sequence is recognized.
- Linear epitopes typically contain at least three, and most usually at least five, such as about 8 to about 10, 6 to 20 amino acids in their unique sequences.
- a three-dimensional structure epitope in contrast to a linear epitope, is an epitope in which the primary sequence of the amino acid containing the epitope is not a single defining component of the recognized epitope (eg, the primary sequence of the amino acid necessarily defines the epitope It is an epitope that is not recognized by the antibody).
- the conformational epitope may contain an increased number of amino acids relative to the linear epitope.
- an antibody recognizes the three-dimensional structure of a peptide or protein. For example, when a protein molecule is folded to form a three-dimensional structure, certain amino acid and / or polypeptide backbones that form the conformational epitope are parallel, allowing the antibody to recognize the epitope.
- Methods for determining the conformation of an epitope include, but are not limited to, for example, X-ray crystallography, two-dimensional nuclear magnetic resonance spectroscopy and site-specific spin labeling and electromagnetic paramagnetic resonance spectroscopy. For example, see Epitope Mapping Protocols in Methods in Molecular Biology (1996), Vol. 66, Morris (eds.).
- the structure of the antigen-binding domain that binds to an epitope is called a paratope.
- the epitope and paratope are stably bonded by hydrogen bonds, electrostatic forces, van der Waals forces, hydrophobic bonds, etc. that act between the epitope and the paratope.
- the binding force between this epitope and the paratope is called affinity.
- the sum of the binding forces when a plurality of antigens and a plurality of antigen-binding molecules bind to each other is called avidity.
- the avidity is higher than the affinity because the binding force (affinity) works synergistically.
- the method for confirming the binding to the epitope by the test antigen-binding molecule containing the antigen-binding domain for IL-6R is illustrated below, and the binding to the epitope by the test antigen-binding molecule containing the antigen-binding domain for an antigen other than IL-6R is illustrated below.
- the method for confirming the above can also be appropriately carried out according to the following examples.
- the test antigen-binding molecule containing the antigen-binding domain for IL-6R recognizes the linear epitope present in the IL-6R molecule, for example, as follows.
- a linear peptide consisting of the amino acid sequences constituting the extracellular domain of IL-6R is synthesized.
- the peptide can be chemically synthesized.
- it can be obtained by genetic engineering techniques using the region encoding the amino acid sequence corresponding to the extracellular domain in the cDNA of IL-6R.
- the binding activity between the linear peptide consisting of the amino acid sequence constituting the extracellular domain and the test antigen-binding molecule containing the antigen-binding domain for IL-6R is evaluated.
- ELISA using an immobilized linear peptide as an antigen can evaluate the binding activity of the antigen-binding molecule to the peptide.
- the binding activity to the linear peptide can be clarified based on the level of inhibition by the linear peptide in the binding of the antigen-binding molecule to IL-6R-expressing cells. These tests can reveal the binding activity of the antigen-binding molecule to the linear peptide.
- test antigen-binding molecule containing the antigen-binding domain for IL-6R recognizes the three-dimensional structure epitope as follows.
- Cells expressing IL-6R are prepared for the above purposes.
- a test antigen-binding molecule containing an antigen-binding domain for IL-6R comes into contact with an IL-6R-expressing cell, it binds strongly to the cell, while the extracellular domain of IL-6R on which the antigen-binding molecule is immobilized is formed.
- substantially non-binding means a binding activity of 80% or less, usually 50% or less, preferably 30% or less, particularly preferably 15% or less of the binding activity to human IL-6R expressing cells.
- a test antigen-binding molecule containing an antigen-binding domain to IL-6R to IL-6R-expressing cells for example, the method described in Antibodies A Laboratory Manual (Ed Harlow, David Lane, Cold Spring Harbor Laboratory (Ed Harlow, David Lane, Cold Spring Harbor Laboratory) 1988) 359-420). That is, it can be evaluated by the principle of ELISA or FACS (fluorescence activated cell sorting) using IL-6R-expressing cells as an antigen.
- the binding activity of a test antigen-binding molecule containing an antigen-binding domain to IL-6R to IL-6R-expressing cells is quantitatively evaluated by comparing the signal levels generated by the enzymatic reaction. That is, a test polypeptide aggregate is added to an ELISA plate on which IL-6R-expressing cells are immobilized, and a test antigen-binding molecule bound to the cell is detected using an enzyme-labeled antibody that recognizes the test antigen-binding molecule.
- the binding activity of the test antigen-binding molecule to the IL-6R-expressing cell is compared by creating a dilution series of the test antigen-binding molecule and determining the antibody-binding titer to the IL-6R-expressing cell. Can be done.
- the binding of the test antigen-binding molecule to the antigen expressed on the cell surface suspended in a buffer solution or the like can be detected by a flow cytometer.
- a flow cytometer for example, the following devices are known.
- FACSCanto TM II FACSAria TM FACSArray TM FACSVantage TM SE FACSCalibur TM both are brand names of BD Biosciences
- EPICS XL-MCL ADC EPICS XL ADC Cell Lab Quanta / Cell Lab Quanta SC (both are Beckman Coulter product names)
- the following method can be mentioned as an example of a preferable method for measuring the antigen-binding activity of a test antigen-binding molecule containing an antigen-binding domain for IL-6R.
- a test antigen-binding molecule containing an antigen-binding domain for IL-6R.
- the test antigen-binding molecule is prepared and used at a desired concentration by appropriately diluting the test antigen-binding molecule with a suitable buffer solution. For example, it can be used at any concentration between 10 ⁇ g / ml and 10 ng / ml.
- the fluorescence intensity and cell number are measured by FACSCalibur (BD).
- the amount of antibody bound to the cell is reflected in the fluorescence intensity obtained by analysis using CELLQUEST Software (BD), that is, the value of Geometric Mean. That is, by obtaining the value of the Geometric Mean, the binding activity of the test antigen-binding molecule represented by the binding amount of the test antigen-binding molecule can be measured.
- BD CELLQUEST Software
- test antigen-binding molecule containing an antigen-binding domain for antibody IL-6R that binds to the same epitope shares an epitope with an antigen-binding molecule can be confirmed by competition for the same epitope by both. Competition between antigen-binding molecules is detected by cross-blocking assays and the like. For example, a competitive ELISA assay is a preferred cross-blocking assay.
- the IL-6R protein coated on the wells of a microtiter plate is preincubated in the presence or absence of a candidate competing antigen-binding molecule before the test antigen.
- the binding molecule is added.
- the amount of test antigen-binding molecule bound to the IL-6R protein in the well indirectly correlates with the binding capacity of competing antigen-binding molecules that are candidates for binding to the same epitope. That is, the greater the affinity of the competing antigen-binding molecule for the same epitope, the lower the binding activity of the test antigen-binding molecule to the IL-6R protein-coated well.
- the amount of the test antigen-binding molecule bound to the well via the IL-6R protein can be easily measured by labeling the antigen-binding molecule in advance.
- a biotin-labeled antigen-binding molecule is measured by using an avidin peroxidase conjugate and a suitable substrate.
- Cross-blocking assays that utilize enzyme labels such as peroxidase are particularly referred to as competitive ELISA assays.
- the antigen-binding molecule can be labeled with other labeling substances that can be detected or measured. Specifically, a radioactive label or a fluorescent label is known.
- the competing antigen-binding molecule binds the test antigen-binding molecule containing the antigen-binding domain to IL-6R.
- the test antigen-binding molecule binds to or binds to substantially the same epitope as the competing antigen-binding molecule, provided that it can block at least 20%, preferably at least 20-50%, and even more preferably at least 50%. It is an antigen-binding molecule that competes with.
- binding activity for example, it can be measured by comparing the binding activity of the test antigen-binding molecule and the control antigen-binding molecule to the linear peptide into which the mutation has been introduced in the above-mentioned ELISA format.
- the binding activity to the mutant peptide bound to the column is quantified as the antigen-binding molecule eluted in the eluate after the test antigen-binding molecule and the control antigen-binding molecule are allowed to flow down the column. It can also be measured by A method of adsorbing a mutant peptide on a column as, for example, a fusion peptide with GST is known.
- the sharing of the epitope between the test antigen-binding molecule and the control antigen-binding molecule can be evaluated by the following method.
- IL-6R-expressing cells and IL-6R-expressing cells in which mutations have been introduced into the epitope are prepared.
- a test antigen-binding molecule and a control antigen-binding molecule are added to a cell suspension in which these cells are suspended in a suitable buffer such as PBS.
- a FITC-labeled antibody capable of recognizing the test antigen-binding molecule and the control antigen-binding molecule is added to the cell suspension appropriately washed with a buffer solution.
- the fluorescence intensity and cell number of cells stained with the labeled antibody are measured by FACSCalibur (BD).
- concentrations of the test antigen-binding molecule and the control antigen-binding molecule are adjusted to a desired concentration and used by appropriately diluting with a suitable buffer solution. For example, it is used at any concentration between 10 ⁇ g / ml and 10 ng / ml.
- the amount of labeled antibody bound to the cells is reflected in the fluorescence intensity obtained by analysis using CELLQUEST Software (BD), that is, the value of Geometric Mean. That is, by obtaining the Geometric Mean value, the binding activity of the test antigen-binding molecule and the control antigen-binding molecule represented by the binding amount of the labeled antibody can be measured.
- substantially not binding to mutant IL-6R-expressing cells can be determined by the following method.
- FACSCalibur is used as flow cytometry for fluorescence detection
- the obtained fluorescence intensity can be analyzed using CELLQUEST Software.
- Geometric Mean comparison values (mutated IL-6R molecule ⁇ Geo-Mean value) that reflect the amount of the test antigen-binding molecule bound to the mutant IL-6R-expressing cells obtained by the analysis are applied to the IL-6R-expressing cells of the test antigen-binding molecule. Compare with the ⁇ Geo-Mean comparison value that reflects the amount of binding.
- the concentrations of the test antigen-binding molecules used to obtain the ⁇ Geo-Mean comparative values for the mutant IL-6R-expressing cells and the IL-6R-expressing cells may be prepared to be the same or substantially the same as each other. Especially preferable.
- An antigen-binding molecule that has been confirmed to recognize an epitope in IL-6R in advance is used as a control antigen-binding molecule.
- the ⁇ Geo-Mean comparison value of the test antigen-binding molecule against the mutant IL-6R-expressing cells is at least 80%, preferably 50%, more preferably 30% of the ⁇ Geo-Mean comparison value of the test antigen-binding molecule against the IL-6R-expressing cells. %, Especially preferably less than 15%, "substantially does not bind to mutant IL-6R expressing cells".
- the calculation formula for obtaining the Geo-Mean value (Geometric Mean) is described in the CELL QUEST Software User's Guide (BD biosciences).
- the epitopes of the test antigen-binding molecule and the control antigen-binding molecule can be evaluated to be identical to the extent that they can be substantially equated by comparing the comparative values.
- Antigen-binding domain whose antigen-binding activity changes in a low-molecular-weight compound-dependent manner
- the "antigen-binding domain in which the antigen-binding activity changes in a low-molecular-weight compound-dependent manner" is different from that in the presence of a low-molecular-weight compound concentration.
- Antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner in the present disclosure is for an antigen which is a molecule different from MTA under conditions of different MTA concentrations.
- the antigen to which these antigen-binding domains bind may be a membrane-type molecule or a soluble-type molecule.
- the antigen to which these antigen-binding domains bind is expressed in an antigen expressed in a diseased tissue, more preferably an antigen expressed in a cancer tissue, and further preferably expressed in a cancer tissue in which MTA is accumulated. It is an antigen.
- the antigen expressed in the cancer tissue may be an antigen expressed in the cancer cell, or may be an antigen expressed in the cancer stromal cell or the immune tissue in the cancer tissue.
- an antigen-binding domain in which the antigen-binding activity in the presence of MTA is stronger than the antigen-binding activity in the absence of MTA, and the binding to the antigen in the presence of MTA can be exemplified.
- the antigen-binding activity in the presence of MTA of the present disclosure is stronger than the antigen-binding activity in the absence of MTA.
- the binding activity of the antigen-binding domain to the antigen in the absence of MTA is weaker than the binding activity to the antigen in the presence of MTA.
- the ratio of the binding activity to the antigen in the absence of MTA and the binding activity to the antigen in the presence of MTA is not particularly limited, it is preferably present as KD (Dissociation constant) in the absence of MTA to the antigen.
- the upper limit of the value of KD (without MTA) / KD (with MTA) is not particularly limited, and may be any value such as 400, 1000, 10000, etc. as long as it can be produced by a person skilled in the art. In the absence of MTA, if no antigen-binding activity is observed, this upper limit is infinite.
- Antigen-binding domains whose antigen-binding activity in the presence of MTA is stronger than that in the absence of MTA include antigen-binding domains that do not substantially bind to antigen in the absence of MTA.
- the antigen-binding activity in the presence of MTA of the present disclosure is weaker than the antigen-binding activity in the absence of MTA.
- the antigen-binding activity of the antigen-binding domain in the absence of MTA is stronger than the antigen-binding activity in the presence of MTA.
- KD Dissociation constant
- the value of KD (in the presence of MTA) / KD (in the absence of MTA), which is the ratio of KD below, is 2 or more, more preferably 10 or more, and further preferably 40 or more.
- the upper limit of the value of KD (in the presence of MTA) / KD (in the absence of MTA) is not particularly limited, and may be any value such as 400, 1000, 10000, etc. as long as it can be produced by a person skilled in the art. In the presence of MTA, if no antigen-binding activity is observed, this upper limit is an infinite number.
- the antigen-binding domain When the antigen-binding activity of the antigen-binding domain is weaker than that of the antigen-binding domain in the absence of MTA, the antigen-binding domain includes an antigen-binding domain that does not substantially bind to the antigen in the presence of MTA.
- the dissociation rate As another index showing the ratio of the antigen-binding activity of the antigen-binding domain (or the antigen-binding molecule containing the domain) of the present disclosure to the antigen in the absence of MTA and the antigen-binding activity in the presence of the MTA, for example, the dissociation rate.
- a constant kd Dissociation rate constant
- kd dissociation rate constant
- KD dissociation constant
- kd dissociation rate constant
- the value of kd (in the absence of MTA) / kd (in the presence of MTA), which is the ratio of (constant), is preferably 2 or more, more preferably 5 or more, still more preferably 10 or more, and more preferably. Is over 30.
- the upper limit of the value of Kd (without MTA) / kd (in the presence of MTA) is not particularly limited, and may be any value such as 50, 100, 200, etc. as long as it can be produced according to the common general technical knowledge of those skilled in the art. In the absence of MTA, no dissociation occurs when no antigen-binding activity is observed, so this upper limit is an infinite number.
- an appropriate MTA concentration can be set, and as a non-limiting example, for example, the condition in which 100 ⁇ M MTA is present can be regarded as the condition in the presence of MTA. Further, as a non-limiting aspect of the MTA concentration described as "in the presence of MTA" in the present disclosure, the concentration exemplified as the threshold value for distinguishing between the low concentration and the high concentration of MTA described later can be applied.
- Non-limiting examples of antigen-binding domains whose antigen-binding activity changes in an MTA-dependent manner include antigen-binding domains that are stronger than antigen-binding activity in the presence of low-concentration MTA, and high-concentration antigen-binding activity.
- An example is an antigen-binding domain in which the antigen-binding activity in the presence of the MTA is weaker than that in the presence of a low concentration of MTA.
- Antigen-binding activity in the presence of a high concentration of MTA of the present disclosure The binding activity of an antigen-binding domain stronger than the antigen-binding activity in the presence of a low concentration of MTA to an antigen in the presence of a low concentration of MTA is in the presence of a high concentration of MTA.
- the ratio of the binding activity to the antigen in the presence of a low concentration of MTA and the binding activity to the antigen in the presence of a high concentration of MTA is not particularly limited, but is preferably in the presence of a low concentration of MTA to the antigen.
- KD in the presence of low MTA concentration
- KD in the presence of high MTA concentration
- the value of KD (in the presence of low MTA concentration) / KD (in the presence of high MTA concentration) which is the ratio of KD (Dissociation constant) to KD in the presence of high concentration
- MTA in the presence of low concentration of MTA
- KD dissociation constant
- the upper limit of the value of KD (in the presence of low MTA concentration) / KD (in the presence of high concentration of MTA) is not particularly limited, and may be any value such as 400, 1000, 10000, etc. as long as it can be produced by a person skilled in the art. In the presence of low concentrations of MTA, this upper limit is infinite when no antigen-binding activity is observed.
- Antigen-binding activity in the presence of high concentrations of MTA Antigen-binding activity stronger than antigen-binding activity in the presence of low-concentration MTA includes antigen-binding domains that do not substantially bind to antigen in the presence of low-concentration MTA.
- Antigen-binding activity in the presence of a high concentration of MTA of the present disclosure The binding activity of an antigen-binding domain weaker than the antigen-binding activity in the presence of a low concentration of MTA to an antigen in the presence of a low concentration of MTA is in the presence of a high concentration of MTA.
- the ratio of the binding activity to the antigen in the presence of a low concentration of MTA and the binding activity to the antigen in the presence of a high concentration of MTA is not particularly limited, but is preferably in the presence of a high concentration of MTA to the antigen.
- the value of KD (in the presence of high MTA concentration) / KD (in the presence of low MTA concentration), which is the ratio of KD (Dissociation constant) to KD in the presence of low concentration, is 2 or more, more preferably 10 or more. It is more preferably 40 or more.
- the upper limit of the value of KD (in the presence of high MTA concentration) / KD (in the presence of low concentration of MTA) is not particularly limited, and may be any value such as 400, 1000, 10000, etc. as long as it can be produced by a person skilled in the art. In the presence of high concentrations of MTA, this upper limit is infinite when no antigen-binding activity is observed.
- Antigen-binding activity in the presence of a high concentration of MTA Antigen-binding activity weaker than that in the presence of a low concentration of MTA includes an antigen-binding domain that does not substantially bind to an antigen in the presence of a high concentration of MTA.
- Kd Dissociation rate constant
- kd dissociation rate constant
- KD dissociation constant
- kd dissociation rate constant
- the value of kd (in the presence of low MTA concentration) / kd (in the presence of high MTA concentration), which is the ratio of (dissociation rate constant) is preferably 2 or more, more preferably 5 or more, still more preferably. It is 10 or more, more preferably 30 or more.
- the upper limit of the value of Kd (in the presence of low MTA concentration) / kd (in the presence of high concentration of MTA) is not particularly limited, and may be any value such as 50, 100, 200, etc. as long as it can be produced according to the common general technical knowledge of those skilled in the art. In the presence of low concentrations of MTA, no dissociation occurs when no antigen-binding activity is observed, so this upper limit is an infinite number.
- a method for evaluating the relative binding activity can also be used in addition to the method for measuring the KD value described above.
- a method using a flow cytometer, ELISA, cavity electrophoresis, liquid chromatography, or the like is generally known, but the method is not limited thereto.
- the test molecule has stronger binding activity or weaker binding activity than the reference molecule even when the KD value cannot be calculated directly. It is also possible to evaluate the relative binding activity.
- the threshold can be appropriately set from a value of 10 nM, 1 nM, 100 pM, 10 pM, 1 pM or 0 M. ..
- high concentration conditions are at least 110%, at least 120%, at least 130%, at least 140%, at least 150%, at least 2x, at least 5x, at least 10x, at least 50x each threshold.
- at least 100-fold, at least 10 3 fold, at least 10 4 fold, at least 10 5 times it may appropriately be set at least 10 6 times the value.
- the antigen-binding activity of the antigen-binding domain is adenosine, S- (5'-Adenosyl) -L-. Included are antigen-binding domains that are substantially unaffected by one or more small molecule compounds selected from homocysteine (SAH), SAM, AMP, ADP, ATP.
- SAH homocysteine
- SAM SAM
- AMP AMP
- ADP ATP
- the adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH), SAM, AMP, ADP, and ATP have adenosine as a common skeleton in the molecule and are compounds similar to MTA. .. It is common to obtain antigen-binding molecules in which the antigen-binding activity of the antigen-binding domain in the present disclosure is not substantially affected by the presence of these similar molecules, and the binding activity is changed solely by the MTA. It is considered difficult. However, in the present disclosure, a library for obtaining an antibody that binds to an antibody in an MTA-dependent manner as exemplified in various examples described later is constructed, and such a library is screened.
- the antigen-binding activity of an antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is substantially unaffected by adenosine.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is substantially equivalent to S- (5'-Adenosyl) -L-homocysteine (SAH). Not affected.
- SAH S- (5'-Adenosyl) -L-homocysteine
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is substantially unaffected by SAM.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is substantially unaffected by any of adenosine, AMP, ADP, and ATP.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH). It is virtually unaffected by any of the above.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH).
- SAM is virtually unaffected.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH).
- SAM, AMP, ADP, ATP are virtually unaffected.
- the antigen-binding activity is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH), and the like.
- SAH S- (5'-Adenosyl) -L-homocysteine
- examples thereof include antigen-binding domains that change depending on one or more small molecule compounds selected from SAM, AMP, ADP, and ATP.
- the antigen-binding activity of an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure also changes in an adenosine-dependent manner.
- Antigen-binding domains whose binding activity changes in a dependent manner on both MTA and adenosine molecules are not bound by any particular theory, but by interacting with common structures in both MTA and adenosine molecules. It can be understood that the binding activity to the antigen changes. In that case, even if the concentration of MTA decreases in the solvent for evaluating the binding activity, the binding activity is maintained if the concentration of adenosine exceeds the degree of decrease in the binding activity due to the decrease in the concentration of MTA. It can be detected or strongly detected.
- the binding activity is maintained if the adenosine concentration decreases beyond the degree of increase in the binding activity due to the increase in the MTA concentration. , Or it can be detected low.
- the antigen-binding activity of an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure is S- (5'-Adenosyl) -L-homocysteine (SAH) -dependent. Also changes.
- the antigen-binding activity of an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure also changes in a SAM-dependent manner.
- the antigen-binding activity of an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure changes in an adenosine, AMP, ADP, or ATP-dependent manner. ..
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH). It changes depending on any of the above.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH).
- SAM changes depending on both.
- the antigen-binding activity of the antigen-binding domain whose MTA-dependently changing antigen-binding activity of the present disclosure is adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH). , SAM, AMP, ADP, and ATP.
- an antigen-binding domain containing an antibody variable region and / or a monodomain antibody is exemplified.
- the antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure is an antibody variable region, and the antibody variable region is at least one or more selected from the group of amino acids below.
- Antigen-binding molecules, including amino acids, can be exemplified.
- the antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure is an antibody variable region, and the antibody variable region is at least one selected from the group of amino acids below.
- An antigen-binding molecule containing the amino acids of can be exemplified.
- the antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner according to the present disclosure is an antibody variable region, and the antibody variable region is at least one selected from the group of amino acids below.
- An antigen-binding molecule containing the amino acids of can be exemplified.
- the antigen-binding domain in the present disclosure may have amino acid residues that interact with the MTA.
- the amino acid residue that interacts with the MTA may be present at the interface that directly interacts with the antigen in the antigen-binding domain, or may be present at other portions.
- the "interface that directly interacts with an antigen” refers to a site in which an antigen and an antigen-binding molecule are in close proximity to each other in the structure of a complex of an antigen and an antigen-binding domain analyzed by a method such as crystal structure analysis.
- a distance of 4 to 6 ⁇ can be exemplified as the distance between the antigen and the antigen-binding molecule, but in the structure of the complex of the antigen and the antigen-binding molecule, "what is the antigen and the antigen-binding molecule close to each other?" It refers to sites that are relatively close to each other in the complex, and is not limited to the above-exemplified distance.
- the amino acid residue that interacts with MTA may be a residue that interacts with MTA when the antigen-binding molecule is bound to the antigen, or a residue that interacts with MTA in the absence of the antigen. You may.
- amino acid residue that interacts with MTA can be a residue that interacts with MTA both in the state where the antigen-binding molecule is bound to the antigen and in the state where the antigen is absent. Further, the amino acid residue that interacts with the MTA may be only one residue or a plurality of amino acid residues in the antigen binding domain.
- the amino acid that interacts with the MTA may be present in the CDR in the antibody variable region or in the FR.
- the amino acid that interacts with the MTA may be present in CDR or FR in the monodomain antibody.
- the amino acid residue that interacts with MTA in the antigen-binding domain is a complex of two antigen-binding molecules containing MTA and the antigen-binding domain, or a tripartite complex of MTA, an antigen, and an antigen-binding molecule containing an antigen-binding domain. It can be identified by a method such as crystal structure analysis, three-dimensional structure analysis using NMR, or introduction of an amino acid mutation.
- the amino acid residue of the antigen-binding molecule that interacts with MTA can be identified from the crystal structure analysis of the complex of MTA and the antigen-binding molecule.
- "interacts with MTA” means that an atom of a side chain or a main chain of an amino acid forming an antigen-binding molecule and an atom of a low-molecular-weight compound can exert an effect on MTA-binding activity.
- a certain amino acid residue forms a three-dimensional structure such as a CDR loop in a state where an interaction between MTAs is formed, or in an embodiment in which the antigen-binding domain in the antigen-binding molecule contains an antibody heavy chain variable region and an antibody light chain variable region.
- a state that contributes to the binding of MTA including indirect effects such as stabilizing the conformation when MTA binds, and a state that satisfies both of them.
- the side chain or main chain of the amino acid forming the antigen-binding molecule is determined. It can be determined based on the interatomic distance between the non-hydrogen atoms that compose and the non-hydrogen atoms that compose MTA.
- the above interatomic distance is preferably within 3.0 ⁇ , 3.2 ⁇ , 3.4 ⁇ , 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ , 4.2 ⁇ , 4.4 ⁇ , 4.6 ⁇ , 4.8 ⁇ or 5.0 ⁇ , but is not limited to this. Absent. More preferably, the interatomic distance is within 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ or 4.2 ⁇ . More specifically, it is possible to determine the possibility of direct interaction based on the information on the interatomic distance on the three-dimensional structure, the type of intermolecular interaction formed, and the type of atom. More precisely, it can be judged from the effect of mutagenesis of amino acid residues such as modification to Ala or Gly on the activity of low molecular weight compounds, but it is not limited to this.
- the amino acid residue of the antigen-binding molecule that interacts with MTA can be identified from the crystal structure analysis of the complex of MTA, antigen, and antigen-binding molecule.
- interacting with MTA means the distance at which an atom of a side chain or a main chain and an atom of a low molecular weight compound in an amino acid forming an antigen-binding molecule can exert an effect on MTA-binding activity in the presence of an antigen.
- a certain amino acid residue is a CDR loop. It refers to a state that contributes to MTA binding, including indirect effects such as stabilizing the conformation when MTA binds in the presence of an antigen, and a state that satisfies both of them. ..
- the amino acids forming the antigen-binding molecule in the presence of an atom It can be determined based on the interatomic distance between the non-hydrogen atoms constituting the side chain or the main chain and the non-hydrogen atoms constituting the MTA.
- the above interatomic distance is preferably within 3.0 ⁇ , 3.2 ⁇ , 3.4 ⁇ , 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ , 4.2 ⁇ , 4.4 ⁇ , 4.6 ⁇ , 4.8 ⁇ or 5.0 ⁇ , but is not limited to this. Absent.
- the interatomic distance is within 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ or 4.2 ⁇ . More specifically, it is possible to determine the possibility of direct interaction based on the information on the interatomic distance on the three-dimensional structure, the type of intermolecular interaction formed, and the type of atom. More precisely, it can be judged from the effect of mutagenesis of amino acid residues such as modification to Ala or Gly on the activity of low molecular weight compounds, but it is not limited to this. Regarding the "indirectly influencing state" in the present specification, for example, from the three-dimensional structure of the complex of MTA, antigen, and antigen-binding molecule, the conformation of each amino acid residue and the molecule with surrounding residues.
- the antigen-binding domain is an antibody variable region
- the amino acid residue that interacts with the MTA is the amino acid sequence of the antibody variable region at position 34, 35a of the heavy chain identified by Kabat numbering.
- An antigen-binding domain, which is an amino acid residue located at at least one or more amino acid sites selected from the above, can be exemplified.
- the antigen-binding domain is an antibody variable region, which is a heavy chain W34, C35a, W47, F52, Y52e, E101, light chain R32, S34, Y36, L46, Y49, S50. , A89, G90, L91, and P96 (Kabat numbering) include antigen binding domains containing at least one amino acid selected from.
- the antigen-binding domain is an antibody variable region, and the amino acid residue that interacts with MTA is located at position 34 of the heavy chain specified by Kabat numbering in the amino acid sequence of the antibody variable region. At least one selected from the group of amino acid sites at positions 47, 50, 58, 95, 98, 99, 100a, light chain 28, 91, 95b, 95c and 96.
- An antigen-binding domain, which is an amino acid residue located at the amino acid site of, can be exemplified.
- the antigen-binding domain is an antibody variable region, which is a heavy chain W34, W47, C50, Y58, E95, F98, G99, G100a, light chain Y28, T91, F95b, Y95c.
- an antigen binding domain containing at least one amino acid selected from F96 is exemplified.
- the antigen-binding domain is an antibody variable region, and the amino acid residue that interacts with MTA is included in the amino acid sequence of the antibody variable region.
- An antigen-binding domain which is an amino acid residue located at at least one or more amino acid sites selected from the group of, can be exemplified.
- the antigen-binding domain is an antibody variable region, and the antibody variable region is heavy chain A33, I50, G52, D54, S56, T57, W58, G99, Y100, T100a, light chain S91, Y95c, and N96 (Kabat).
- An antigen-binding domain containing at least one or more amino acids selected from (numbering) can be exemplified.
- Antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner is described in the present disclosure. It is a molecule containing the antibody Fc region.
- the antibody Fc region contained in the antigen-binding molecule of the present disclosure may be a natural Fc region or a modified Fc region. Examples of the natural Fc region include an Fc region represented by human IgG1 (SEQ ID NO: 5), IgG2 (SEQ ID NO: 6), IgG3 (SEQ ID NO: 7), or IgG4 (SEQ ID NO: 8).
- the antigen-binding molecule of the present disclosure can include at least a portion of the Fc region that mediates binding to the Fc ⁇ receptor and / or binding to FcRn.
- the antigen binding molecule can be an antibody or an Fc fusion protein.
- a fusion protein is a chimeric polypeptide comprising a polypeptide comprising a first amino acid sequence linked to a polypeptide having a second amino acid sequence to which it is not naturally linked.
- the fusion protein contains a polypeptide comprising an amino acid sequence encoding at least a portion of the Fc region (eg, a portion of the Fc region that imparts binding to the Fc ⁇ receptor and / or a portion of the Fc region that imparts binding to FcRn).
- the amino acid sequences can be in separate proteins that are carried together to the fusion protein, or they can usually be in the same protein, but are put into a new rearrangement in the fusion polypeptide.
- the fusion protein can be made, for example, by chemical synthesis or by a genetic recombination technique that creates a polynucleotide in which the peptide region is encoded in the desired relationship and expresses it.
- Each domain in the antigen-binding molecule of the present disclosure can be directly linked by a polypeptide bond or can be linked via a linker.
- the linker any peptide linker that can be introduced by genetic engineering, or a linker disclosed in a synthetic compound linker (for example, Holliger et al. (Protein Engineering (1996) 9 (3), 299-305)) can be used.
- peptide linkers are preferred in the present disclosure.
- the length of the peptide linker is not particularly limited and can be appropriately selected by those skilled in the art depending on the intended purpose, but the preferred length is 5 amino acids or more (the upper limit is not particularly limited, but usually 30 amino acids or less is preferable. Is 20 amino acids or less), particularly preferably 15 amino acids.
- Synthetic chemical linkers are commonly used cross-linking agents for cross-linking peptides, such as N-hydroxysuccinimide (NHS), discicin-imidyl sverate (DSS), bis (sulfosuccinimidyl).
- NHS N-hydroxysuccinimide
- DSS discicin-imidyl sverate
- bis sulfosuccinimidyl
- BS3 Dithiobis (Succinimidyl Propionate) (DSP), Dithiobis (Sulfone Imidine Propionate) (DTSSP), Ethylene Glycolbis (Succinimidyl Succinate) (EGS), Ethylene Glycolbis (sulfosuccinimidyl succinate) (sulfo-EGS), dissuccinimidyl tartrate (DST), disulfosuccinimidyl tartrate (sulfo-DST), bis [2- (sulfinimideoxycarbonyloxy) ) Ethyl] sulfone (BSOCOES), bis [2- (sulfosuccinimideoxycarbonyloxy) ethyl] sulfone (sulfo-BSOCOES), etc., and these cross-linking agents are commercially available.
- linkers linking each domain When a plurality of linkers linking each domain are used, all the same kind of linkers can be used, and different kinds of linkers can also be used.
- linkers having peptide tags such as His tag, HA tag, myc tag, and FLAG tag can also be used as appropriate.
- the property of binding to each other by hydrogen bonds, disulfide bonds, covalent bonds, ionic interactions or combinations of these bonds can also be suitably utilized.
- the affinity between CH1 and CL of an antibody may be utilized, or the Fc region originating from the bispecific antibody described above may be used for association of hetero Fc regions.
- disulfide bonds formed between domains can also be suitably utilized.
- the polynucleotide encoding the domain is linked in frame.
- methods such as restriction fragment ligation, fusion PCR, and overlap PCR are known, and these methods may be used alone or in combination as appropriate for the production of the antigen-binding molecule of the present disclosure. Can be used.
- the terms "connected”, “fused”, “connected” or “fused” are used interchangeably. These terms refer to connecting elements or components such as two or more polypeptides so as to form one structure by all means including the above-mentioned chemical bonding means or recombination method.
- In-frame fusion means that when two or more elements or components are a polypeptide, two or more reading frames are used to form a continuous longer reading frame so as to maintain the correct reading frame for the polypeptide. Concatenation of units of.
- the antibody which is the antigen-binding molecule of the present disclosure in which the constant region containing the antigen-binding domain and the Fc region is linked in-frame by a peptide bond without a linker is used.
- the present disclosure provides antigen-binding molecules with high plasma retention.
- the antigen-binding molecule increases its antigen-binding activity as the concentration of MTA increases.
- the antigen-binding molecule has higher antigen-binding activity in the target tissue than in the non-target tissue.
- the antigen-binding molecule is an antibody.
- the antigen-dependent disappearance (clearance) of the antigen-binding molecule in tissues other than the target tissue is reduced.
- the reduction in antigen-dependent elimination (clearance) in most tissues in the body (tissues other than the target tissue) leads to high plasma retention of the antigen-binding molecule.
- the antigen-binding molecule in the present invention has high plasma retention can be determined by relative comparison with the control antigen-binding molecule.
- the antigen-binding molecule whose antigen-binding activity increases as the concentration of MTA increases has higher plasma retention as compared to the control antigen-binding molecule.
- control antigen-binding molecule is an antigen-binding molecule that does not have antigen-binding activity depending on the concentration of MTA.
- an antigen-binding molecule that does not have antigen-binding activity depending on the concentration of the compound has a difference in antigen-binding activity between the presence and absence of MTA, for example, less than 2-fold, more than 1.8-fold. It means an antigen-binding molecule that is smaller, less than 1.5 times, less than 1.3 times, less than 1.2 times, or less than 1.1 times. From a comparative point of view, it is desirable that the antigen-binding molecule of the present disclosure and the control antigen-binding molecule have substantially the same antigen-binding activity in the presence of a sufficient amount of MTA.
- the magnitude of the antigen-dependent disappearance of the antigen-binding molecule detected in vivo changes according to the quantitative balance between the antigen present in plasma and the antigen-binding molecule.
- the more antigens present in plasma / the less antigen-binding molecules the easier it is to detect antigen-dependent elimination of antigen-binding molecules, and conversely, the less antigens present in plasma / antigens.
- the more binding molecules there are the more difficult it is to detect the antigen-dependent disappearance of antigen-binding molecules.
- the antigen-binding molecule of the present disclosure does not have to exhibit high plasma retention under all conditions, and may exhibit high plasma retention under appropriate conditions such that sufficient antigen-dependent elimination is detected. .. When the amount of antigen in plasma is small, the amount of antigen may be increased by some artificial means and then the plasma retention may be evaluated.
- the present invention provides an antigen-binding molecule having a low plasma antigen-accumulating ability.
- the antigen-binding molecule increases its antigen-binding activity as the concentration of MTA increases.
- the MTA is a target tissue-specific compound.
- the antigen-binding molecule has higher antigen-binding activity in the target tissue than in the non-target tissue.
- the antigen-binding molecule is an antibody.
- the antigen-binding ability of the antigen-binding molecule in tissues other than the target tissue decreases.
- the ability of the antigen-binding molecule to form an antigen-antibody complex in a tissue other than the target tissue is reduced.
- an antigen-binding molecule such as an antibody binds to an antigen, the clearance of the antigen decreases and the concentration of the antigen in plasma increases (antigen accumulates).
- the decrease in the ability to form an antigen-antibody complex in most tissues in the body means that, overall, low antigen accumulation (in other words, low antigen of antigen-binding molecule) Accumulation capacity).
- Whether or not the antigen-binding molecule in the present invention has a low plasma antigen-accumulating ability can be determined by relative comparison with a control antigen-binding molecule.
- the antigen-binding molecule whose antigen-binding activity increases as the concentration of MTA increases has a lower plasma antigen-accumulating capacity as compared to the control antigen-binding molecule.
- control antigen-binding molecule is an antigen-binding molecule that does not have antigen-binding activity depending on the concentration of MTA.
- an antigen-binding molecule that does not have MTA concentration-dependent antigen-binding activity has a 1.8-fold difference in antigen-binding activity between the presence and absence of the compound, for example, less than 2-fold. It means an antigen-binding molecule that is smaller, less than 1.5 times, less than 1.3 times, less than 1.2 times, or less than 1.1 times. From the viewpoint of comparison, it is desirable that the antigen-binding molecule of the present invention and the control antigen-binding molecule have substantially the same antigen-binding activity in the presence of a sufficient amount of the compound.
- the amount of the antigen-antibody complex formed in the living body is considered to depend on the amount of the antigen and the antibody present in plasma. In general, as the amount of antigen / antibody in plasma increases, the amount of antigen-antibody complex formed also increases, and conversely, as the amount of antigen / antibody in plasma decreases, the amount of antigen-antibody formed increases. The amount of complex is also considered to decrease.
- the antigen-binding molecule of the present invention does not have to exhibit a low plasma antigen-accumulating ability under all conditions, but exhibits a low plasma antigen-accumulating ability under appropriate conditions such that a sufficient antigen-antibody complex is formed. Just do it. When the amount of antigen in plasma is small, the amount of antigen in plasma may be evaluated after increasing the amount of antigen by some artificial means.
- Fc ⁇ receptor An Fc ⁇ receptor (also referred to as Fc ⁇ R) is a receptor capable of binding to the Fc region of an IgG1, IgG2, IgG3, IgG4 monoclonal antibody, and is substantially any member of the family of proteins encoded by the Fc ⁇ receptor gene. means.
- this family includes Fc ⁇ RI (CD64) containing isoforms Fc ⁇ RIa, Fc ⁇ RIb and Fc ⁇ RIc; isoforms Fc ⁇ RIIa (including allotypes H131 and R131; i.e., Fc ⁇ RIIa (H) and Fc ⁇ RIIa (R)), Fc ⁇ RIIb (Fc ⁇ RIIb- Fc ⁇ RII (CD32) containing 1 and Fc ⁇ RIIb-2 and Fc ⁇ RIIc; and isoforms Fc ⁇ RIIIa (including allotypes V158 and F158; i.e., Fc ⁇ RIIIa (V) and Fc ⁇ RIIIa (F)) and Fc ⁇ RIIIb (allotypes Fc ⁇ RIIIb-NA1 and Fc ⁇ Includes, but is not limited to, Fc ⁇ RIII (CD16), including (including -NA2), as well as any undiscovered human Fc ⁇ Rs or Fc ⁇ R isoforms or allotypes.
- Fc ⁇ Rs can be of any biological origin, including but not limited to humans, mice, rats, rabbits and monkeys.
- Mouse Fc ⁇ Rs include Fc ⁇ RI (CD64), Fc ⁇ RII (CD32), Fc ⁇ RIII (CD16) and Fc ⁇ RIII-2 (Fc ⁇ RIV, CD16-2), as well as any undiscovered mouse Fc ⁇ Rs or Fc ⁇ R isoforms or allotypes. , Not limited to these.
- Preferable examples of such Fc ⁇ receptors include human Fc ⁇ RI (CD64), Fc ⁇ RIIa (CD32), Fc ⁇ RIIb (CD32), Fc ⁇ RIIIa (CD16) and / or Fc ⁇ RIIIb (CD16).
- the polynucleotide sequence and amino acid sequence of human Fc ⁇ RI are SEQ ID NO: 9 (NM_000566.3) and 10 (NP_000557.1), respectively, and the polynucleotide sequence and amino acid sequence of human Fc ⁇ RIIa (allotype H131) are SEQ ID NO: 11 (BC020823), respectively. .1) and 12 (AAH20823.1) (allotype R131 is the sequence in which the 166th amino acid of SEQ ID NO: 12 is replaced with Arg), the polynucleotide sequence and amino acid sequence of Fc ⁇ RIIb are SEQ ID NO: 13, respectively.
- polynucleotide and amino acid sequences of Fc ⁇ RIIIa are in SEQ ID NOs: 15 (BC033678.1) and 16 (AAH33678.1), respectively, and the polynucleotide and amino acids of Fc ⁇ RIIIb.
- sequences are described in SEQ ID NOs: 17 (BC128562.1) and 18 (AAI28563.1), respectively (the numbers in parentheses indicate database registration numbers such as RefSeq).
- Fc ⁇ receptor has binding activity in the Fc region of IgG1, IgG2, IgG3, and IgG4 monoclonal antibodies is determined by the FACS and ELISA formats described above, as well as the ALPHA screen (Amplified Luminescent Proximity Homogeneous Assay) and surface plasmon resonance. It can be confirmed by the BIACORE method using the resonance (SPR) phenomenon (Proc. Natl. Acad. Sci. USA (2006) 103 (11), 4005-4010).
- SPR resonance
- Fc ⁇ RI (CD64) containing Fc ⁇ RIa, Fc ⁇ RIb and Fc ⁇ RIc and Fc ⁇ RIIIa (including allotypes V158 and F158) and Fc ⁇ RIIIb (including allotypes Fc ⁇ RIIIb-NA1 and Fc ⁇ RIIIb-NA2) are the Fc regions of IgG.
- the ⁇ chain that binds and the common ⁇ chain that has ITAM that transmits the activation signal inside the cell are associated.
- ITAM is contained in the own cytoplasmic domain of isoforms Fc ⁇ RIIa (including allotypes H131 and R131) and Fc ⁇ RII (CD32) containing Fc ⁇ RIIc.
- Fc ⁇ receptor having the ability to transmit an activation signal as described above is referred to herein as an active Fc ⁇ receptor.
- the intracytoplasmic domain of Fc ⁇ RIIb contains ITIM that transmits an inhibitory signal.
- Fc ⁇ RIIb contains ITIM that transmits an inhibitory signal.
- B cells cross-linking between Fc ⁇ RIIb and B cell receptor (BCR) suppresses the activation signal from BCR, resulting in suppression of BCR antibody production.
- macrophages cross-linking between Fc ⁇ RIII and Fc ⁇ RIIb suppresses phagocytosis and inflammatory cytokine production.
- the Fc ⁇ receptor having the ability to transmit an inhibitory signal as described above is referred to herein as an inhibitory Fc ⁇ receptor.
- examples of the Fc region contained in the antigen-binding molecule of the present disclosure include an Fc region having Fc ⁇ receptor binding activity.
- an Fc region having Fc ⁇ receptor binding activity.
- it is represented by human IgG1 (SEQ ID NO: 5), IgG2 (SEQ ID NO: 6), IgG3 (SEQ ID NO: 7), or IgG4 (SEQ ID NO: 8). The Fc region is illustrated.
- Fc ⁇ receptor has binding activity in the Fc region of IgG1, IgG2, IgG3, and IgG4 monoclonal antibodies is determined by the FACS and ELISA formats described above, as well as the ALPHA screen (Amplified Luminescent Proximity Homogeneous Assay) and surface plasmon resonance. It can be confirmed by the BIACORE method using the resonance (SPR) phenomenon (Proc. Natl. Acad. Sci. USA (2006) 103 (11), 4005-4010).
- SPR resonance
- the ALPHA screen is carried out based on the following principle by ALPHA technology using two beads, donor and acceptor.
- the luminescent signal is detected only when the molecule bound to the donor bead biologically interacts with the molecule bound to the acceptor bead and the two beads are in close proximity.
- the photosensitizer in the donor beads excited by the laser converts the surrounding oxygen into excited singlet oxygen.
- Singlet oxygen diffuses around the donor beads, and when it reaches the adjacent acceptor beads, it causes a chemiluminescent reaction inside the beads, eventually emitting light.
- the chemiluminescent reaction does not occur because the singlet oxygen produced by the donor bead does not reach the acceptor bead.
- an antigen-binding molecule containing a biotin-labeled Fc region is bound to donor beads, and an Fc ⁇ receptor tagged with glutathione S transferase (GST) is bound to acceptor beads.
- GST glutathione S transferase
- antigen-binding molecules containing native Fc regions and Fc ⁇ receptors interact to produce a signal of 520-620 nm.
- Antigen-binding molecules containing untagged Fc region variants compete with interactions between antigen-binding molecules with native Fc regions and Fc ⁇ receptors. Relative binding affinity can be determined by quantifying the decrease in fluorescence that results from competition.
- an antigen-binding molecule such as an antibody is biotinylated using Sulfo-NHS-biotin or the like.
- a method for tagging the Fc ⁇ receptor with GST it is expressed in cells or the like in which a fusion gene in which a polynucleotide encoding the Fc ⁇ receptor and a polynucleotide encoding GST are fused in-frame is held in a vector operably linked.
- a method of purification using a glutathione column and the like can be appropriately adopted.
- the obtained signal is preferably analyzed by adapting it to a one-site competition model using nonlinear regression analysis using software such as GRAPHPAD PRISM (GraphPad, San Diego).
- the Biacore system takes the above-mentioned shift amount, that is, the change in mass on the surface of the sensor chip on the vertical axis, and displays the change in mass over time as measurement data (sensorgram).
- Kinetics binding rate constant (ka) and dissociation rate constant (kd) can be obtained from the curve of the sensorgram, and affinity (KD) can be obtained from the ratio of the constants.
- the inhibition measurement method is also preferably used. Examples of inhibition measures are described in Proc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010.
- Fc ⁇ receptor (Fc ⁇ R) binding modified Fc region included in the present disclosure includes human IgG1 (SEQ ID NO: 5), IgG2 (SEQ ID NO: 6), IgG3 (SEQ ID NO: 7), or IgG4 (SEQ ID NO: 8).
- an Fc ⁇ R-binding modified Fc region having a higher Fc ⁇ receptor-binding activity than the Fc ⁇ receptor-binding activity of the natural human IgG Fc region can also be appropriately used.
- the "Fc region of natural human IgG” is bound to the EU numbering 297 position of the Fc region of human IgG1, IgG2, IgG3 or IgG4 exemplified by SEQ ID NO: 5, 6, 7 or 8. It means the Fc region where the sugar chain is a fucose-containing sugar chain.
- Such an Fc ⁇ R-binding modified Fc region can be prepared by modifying the amino acids in the Fc region of native human IgG.
- Fc ⁇ R-binding activity of the Fc ⁇ R-binding modified Fc region is higher than the Fc ⁇ R-binding activity of the Fc region of natural human IgG can be appropriately carried out by using the method described in the above-mentioned binding activity section.
- amino acid modification or “amino acid modification” of the Fc region includes modification to an amino acid sequence different from the amino acid sequence of the starting Fc region.
- Any Fc region can be used as the starting Fc region as long as the modified variant of the starting Fc region can bind to the human Fc ⁇ receptor in the neutral pH range.
- the Fc region that has already been modified can be used as the starting Fc region, and the Fc region that has been further modified can also be preferably used as the Fc region of the present disclosure.
- the starting Fc region can mean the polypeptide itself, a composition containing the starting Fc region, or an amino acid sequence encoding the starting Fc region.
- the starting Fc region may include known Fc regions produced by recombination outlined in the section on antibodies.
- the origin of the starting Fc region can be obtained, but not limited to, from any non-human animal organism or human.
- any organism preferably includes an organism selected from mice, rats, guinea pigs, hamsters, gerbils, cats, rabbits, dogs, goats, sheep, cows, horses, camels, and non-human primates. ..
- the starting Fc region can also be obtained from cynomolgus monkeys, marmosets, rhesus monkeys, chimpanzees, or humans.
- the starting Fc region can be obtained from human IgG1, but is not limited to any particular class of IgG.
- the Fc region of human IgG1, IgG2, IgG3, or IgG4 can be appropriately used as the starting Fc region.
- the Fc region of any class or subclass of IgG from any of the aforementioned organisms can preferably be used as the starting Fc region. Examples of naturally occurring IgG variants or engineered types are available in the known literature (Curr. Opin. Biotechnol. (2009) 20 (6), 685-91, Curr. Opin. Immunol. (2008) 20 (4). ), 460-470, Protein Eng. Des. Sel. (2010) 23 (4), 195-202, International Publication WO2009 / 086320, WO2008 / 092117, WO2007 / 041635, and WO2006 / 105338) Not limited to.
- modifications include one or more mutations, eg, mutations substituted with amino acid residues different from the amino acids in the starting Fc region, or insertion of one or more amino acid residues into the amino acids in the starting Fc region or the starting Fc region. Includes deletion of one or more amino acids from the amino acids of.
- the modified amino acid sequence of the Fc region comprises an amino acid sequence that includes at least a portion of the non-naturally occurring Fc region.
- Such variants necessarily have less than 100% sequence identity or similarity to the starting Fc region.
- the variant has an amino acid sequence identity or similarity of about 75% to less than 100%, more preferably about 80% to less than 100%, more preferably about 85% to 100% with the amino acid sequence of the starting Fc region.
- the present disclosure has less than, more preferably about 90% to less than 100%, and most preferably about 95% to less than 100% amino acid sequences of identity or similarity.
- the difference in amino acids between the starting Fc region and the Fc ⁇ R-binding modified Fc region of the present disclosure can also be suitably identified by the difference in the amino acids in which the positions of the amino acid residues specified in the above-mentioned EU numbering are specified. Methods for making such variants are illustrated in the section "Modifying Amino Acids".
- the Fc ⁇ R-binding modified Fc region (Fc ⁇ R-binding modified Fc region) contained in the antigen-binding molecule of the present disclosure, which has a higher Fc ⁇ receptor-binding activity than the Fc-region-binding activity of the Fc region of natural human IgG, can be obtained by any method.
- the Fc ⁇ R-binding modified Fc region can be obtained by modifying the amino acid of the human IgG-type immunoglobulin used as the starting Fc region.
- Preferred IgG-type immunoglobulin Fc regions for modification include, for example, human IgG (IgG1, IgG2, IgG3, or IgG4, and variants thereof) exemplified by SEQ ID NO: 5, 6, 7 or 8.
- the Fc region can be mentioned.
- Modifications to other amino acids can modify amino acids at any position as long as the binding activity to the Fc ⁇ receptor is higher than the binding activity of the Fc region of natural human IgG to the Fc ⁇ receptor.
- the antigen-binding molecule contains the Fc region of human IgG1 as the human Fc region
- the sugar chain bound to EU numbering 297 is based on the Fc ⁇ receptor-binding activity of the Fc region of natural human IgG, which is a fucose-containing sugar chain. It is also preferable that a modification having a high binding activity to the Fc ⁇ receptor is included.
- Examples of such amino acid modifications include WO2007 / 024249, WO2007 / 021841, WO2006 / 031370, WO2000 / 042072, WO2004 / 029207, WO2004 / 099249, WO2006 / 105338, WO2007 / 041635, WO2008 / 092117, WO2005 / 070963, It has been reported in WO2006 / 020114, WO2006 / 116260 and WO2006 / 023403.
- the pH condition for measuring the binding activity between the Fc ⁇ receptor binding domain contained in the antigen-binding molecule of the present disclosure and the Fc ⁇ receptor conditions in the acidic pH range to the neutral pH range can be appropriately used.
- the pH acidic range or pH neutral range as a condition for measuring the binding activity between the Fc ⁇ receptor binding domain contained in the antigen-binding molecule of the present disclosure and the Fc ⁇ receptor usually means pH 5.8 to pH 8.0. It is preferably in the range indicated by any pH value from pH 6.0 to pH 7.4, preferably pH 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2.
- the binding affinity between the Fc ⁇ receptor binding domain and the human Fc ⁇ receptor can be evaluated at any temperature of 10 ° C to 50 ° C.
- a temperature of 15 ° C. to 40 ° C. is used to determine the binding affinity between the human Fc ⁇ receptor binding domain and the Fc ⁇ receptor. More preferably, from 20 ° C to 35 ° C, such as any one of 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, and 35 ° C.
- Any temperature of Fc ⁇ receptor is similarly used to determine the binding affinity between the Fc ⁇ receptor binding domain and the Fc ⁇ receptor.
- the temperature of 25 ° C. is a non-limiting example of aspects of the present disclosure.
- the binding activity of the Fc ⁇ R-binding modified Fc region to the Fc ⁇ receptor is higher than the binding activity of the natural Fc region to the Fc ⁇ receptor as Fc ⁇ RI, Fc ⁇ RIIa, Fc ⁇ RIIb, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb of the Fc ⁇ R-binding modified Fc region. It means that the binding activity to any of the human Fc ⁇ receptors is higher than the binding activity of the natural Fc region to these human Fc ⁇ receptors.
- the binding activity of the antigen-binding molecule containing the Fc ⁇ R-binding modified Fc region is 105% or more as compared with the binding activity of the antigen-binding molecule containing the natural Fc region of human IgG as a control.
- the natural Fc region a starting Fc region can also be used, and a natural Fc region of an antibody of the same subclass can also be used.
- the natural Fc region of human IgG as a control the Fc region of natural human IgG in which the sugar chain bound to the amino acid at position 297 represented by EU numbering is a fucose-containing sugar chain is preferably used. ..
- a known method can be used to determine whether or not the sugar chain bound to the amino acid at position 297 represented by EU numbering is a fucose-containing sugar chain (Non-fucosylated therapeutic antibodydies as next-generation therapeutic antibodies. Satoh M, Iida S, Shitara K., Expert Opin. Biol. Ther. (2006) 6 (11), 1161-1173).
- the sugar chain bound to the Fc region of natural human IgG is a fucose-containing sugar chain by the following method.
- N-Glycosidase F Fucose F
- sugar chains are released from the test natural human IgG (Weitzhandler et al. (J. Pharma. Sciences (1994) 83, 12, 1670-). 1675).
- the concentrated dry matter of the reaction solution Schoenk et al. (J. Clin. Investigation (2001) 108 (11) 1687-1695) from which the protein was removed by reacting with ethanol was prepared by 2-aminopyridine. Fluorescently labeled (Bigge et al. (Anal.
- an antigen-binding molecule having an Fc region of an IgG monoclonal antibody can be appropriately used.
- the structure of the Fc region is SEQ ID NO: 5 (A is added to the N end of database registration number AAC82527.1), SEQ ID NO: 6 (A is added to the N end of database registration number AAB59393.1), and SEQ ID NO: 7 (database registration). No. CAA27268.1) and SEQ ID NO: 8 (A is added to the N end of database registration number AAB59394.1).
- an antigen-binding molecule containing the Fc region of a specific isotype antibody When an antigen-binding molecule containing the Fc region of a specific isotype antibody is used as a test substance, an antigen-binding molecule having an Fc region of the specific isotype IgG monoclonal antibody is used as a control. The effect of the binding activity on the Fc ⁇ receptor by the antigen-binding molecule containing the test Fc region is verified. As described above, an antigen-binding molecule containing an Fc region whose binding activity to the Fc ⁇ receptor has been verified to be high is appropriately selected.
- Fc region having selective Fc ⁇ receptor-binding activity As an example of the Fc ⁇ receptor-binding domain preferably used in the present disclosure, the property that the binding activity to a specific Fc ⁇ receptor is higher than that to other Fc ⁇ receptors.
- Fc ⁇ receptor-binding domains having (Fc ⁇ receptor-binding domain having selective Fc ⁇ receptor-binding activity) are also preferably mentioned.
- an antibody is used as the antigen-binding molecule (the Fc region as the Fc ⁇ receptor-binding domain)
- one molecule of the antibody can bind to only one Fc ⁇ receptor, so that one molecule of the antigen-binding molecule becomes an inhibitory Fc ⁇ receptor. It cannot bind to other active Fc ⁇ Rs in the bound state, and cannot bind to other active Fc ⁇ receptors or inhibitory Fc ⁇ receptors in the bound state.
- the active Fc ⁇ receptor includes Fc ⁇ RIa, Fc ⁇ RIb and Fc ⁇ RI (CD64) containing Fc ⁇ RIc, Fc ⁇ RIIa and Fc ⁇ RIIIa (allotype V158). And Fc ⁇ RIII (including F158) and Fc ⁇ RIIIb (including allotypes Fc ⁇ RIIIb-NA1 and Fc ⁇ RIIIb-NA2) are preferred.
- Fc ⁇ RIIb (including Fc ⁇ RIIb-1 and Fc ⁇ RIIb-2) is a suitable example of the inhibitory Fc ⁇ receptor.
- the binding activity to a specific Fc ⁇ receptor is higher than the binding activity to other Fc ⁇ receptors
- the binding activity to the active Fc ⁇ receptor is higher than the binding activity to the inhibitory Fc ⁇ receptor.
- the binding activity of the antigen-binding molecule containing the Fc region to the human Fc ⁇ receptor of any of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb is preferably 105% or more of the binding activity to Fc ⁇ RIIb.
- an Fc region in which the binding activity to the active Fc ⁇ receptor is higher than the binding activity to the inhibitory Fc ⁇ receptor can be suitably included in the antigen-binding molecule of the present disclosure in which the antigen-binding domain binds to the membrane-type molecule. Since it is known that the IgG1 antibody containing such an Fc region has enhanced ADCC activity described later, the antigen-binding molecule containing the Fc region can be used as an antigen-binding molecule contained in the pharmaceutical composition of the present disclosure. Is also useful.
- the binding activity to the inhibitory Fc ⁇ receptor is higher than the binding activity to the active Fc ⁇ receptor
- the binding activity to a specific Fc ⁇ receptor is higher than the binding activity to other Fc ⁇ receptors
- the binding activity to the type Fc ⁇ receptor is higher than the binding activity to the active Fc ⁇ receptor.
- the binding activity of the Fc region to Fc ⁇ RIIb is higher than the binding activity of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb to any human Fc ⁇ receptor.
- the binding activity of the antigen-binding molecule containing the Fc region to Fc ⁇ RIIb is preferably 105% or more, preferably 105% or more of the binding activity of Fc ⁇ RIa, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb to the human Fc ⁇ receptor.
- An Fc region in which the binding activity to the inhibitory Fc ⁇ receptor is higher than the binding activity to the active Fc ⁇ receptor can be suitably included in the antigen-binding molecule of the present disclosure in which the antigen-binding domain binds to the soluble molecule.
- the Fc Among the amino acids in the region the Fc region in which 238 or 328 amino acids represented by EU numbering are modified to amino acids different from the natural Fc region is preferably mentioned.
- the Fc region which is an amino acid represented by the EU numbering of the Fc region and in which 238 amino acids represented by the EU numbering are modified to Asp or 328 amino acids are modified to any one or more of Glu.
- the Fc region described in US2009 / 0136485 or a modification can be appropriately selected.
- the amino acid represented by the EU numbering of the Fc region is Asp, or 328 amino acids are one or more of Glu.
- the modified Fc region is preferably mentioned.
- the substitution of Pro at position 238 represented by EU numbering with Asp and the amino acid at position 237 represented by EU numbering as exemplified by PCT / JP2012 / 054624 are Trp.
- the 237th amino acid represented by EU numbering is Phe
- the 267th amino acid represented by EU numbering is Val
- the 267th amino acid represented by EU numbering is Gln
- the 268th amino acid represented by EU numbering is Gln.
- the amino acid at position 271 represented by EU numbering is Gly
- the amino acid at position 326 represented by EU numbering is Leu
- the amino acid at position 326 represented by EU numbering is Gln
- the amino acid at position 326 represented by EU numbering is Gln
- position 326 represented by EU numbering is represented by EU numbering.
- the amino acid at position 326 is represented by Glu
- the amino acid at position 326 represented by EU numbering is Met
- the amino acid at position 239 represented by EU numbering is Asp
- the amino acid at position 267 represented by EU numbering is represented by Ala and EU numbering.
- the amino acid at position 234 is Trp, the amino acid at position 234 is represented by EU numbering, Tyr, the amino acid at position 237 represented by EU numbering is Ala, and the amino acid at position 237 represented by EU numbering is represented by Asp and EU numbering.
- the amino acid at position 237 is Glu, the amino acid at position 237 represented by EU numbering is Leu, the amino acid at position 237 represented by EU numbering is Met, and the amino acid at position 237 represented by EU numbering is Tyr, EU numbering.
- the amino acid at position 330 represented by is Lys
- the amino acid at position 330 represented by EU numbering is Arg
- the amino acid at position 233 represented by EU numbering is Asp
- the amino acid at position 268 represented by EU numbering is Asp
- the amino acid at position 268 represented by EU numbering is Glu
- the amino acid at position 326 represented by EU numbering is Asp
- the amino acid at position 326 represented by EU numbering is Ser
- the amino acid at position 326 represented by EU numbering is Ser.
- the amino acid at position 323 represented by EU numbering is Ile
- the amino acid at position 323 represented by EU numbering is Leu
- the amino acid at position 323 represented by EU numbering is Met
- the amino acid at position 323 represented by EU numbering is Met
- at position 296 represented by EU numbering is Asp
- amino acid at position 326 represented by EU numbering is Ala
- amino acid at position 326 represented by EU numbering is Asn
- EU numbering The Fc region in which the amino acid at position 330 represented by G is modified to one or more of Met is preferable.
- the composition of the sugar chain bound to the Fc region is such that the proportion of the Fc region bound to the fucose-deficient sugar chain is high.
- an Fc region modified to increase the proportion of the Fc region added with bisecting N-acetylglucosamine may also be included. It is known that the affinity for Fc ⁇ RIIIa is enhanced by removing the fucose residue from N-acetylglucosamine at the reducing end of the N-glycosidic bond complex sugar chain that binds to the Fc region of the antibody (Non-Patent Document 6).
- the antigen-binding molecule containing the Fc region can be used as an antigen-binding molecule contained in the pharmaceutical composition of the present disclosure. Is also useful.
- Antibodies for which fucose residues have been removed from N-acetylglucosamine at the reducing end of the N-glycosidic bond complex sugar chain that binds to the Fc region include, for example, the following antibodies; Antibodies with modified glycosylation (International Publication WO 1999/054342, etc.), Antibodies lacking fucose added to sugar chains (International Publication WO2000 / 061739, WO2002 / 031140, WO2006 / 067913, etc.),
- a fucose residue is removed from N-acetylglucosamine at the reducing end of an N-glycosidic bond complex sugar chain that binds to the antibody Fc region.
- a fucose residue is removed from N-acetylglucosamine at the reducing end of an N-glycosidic bond complex sugar chain that binds to the antibody Fc region.
- the antibody lacking fucose in the sugar chain can be recovered from the culture solution of the host cell.
- Fucose transferase EC 2.4.1.152
- fucose transporter SLC35C1
- GMD GDP-mannose 4,6-dehydrase
- Fx GDP- Selected from the group consisting of keto-6-deoxymannose 3,5-epimerase, 4-reductase
- GFP GDP- ⁇ -L-fucose pyrophosphorylase
- a protein capable of exerting these activities is referred to as a functional protein.
- One non-limiting aspect of the method of modifying these activities includes deletion of these activities.
- known methods such as a method of incapacitatingly disrupting the genes of these functional proteins can be appropriately adopted (International Publication WO2000 / 061739, WO2002 / 031140, WO 2006/067913 etc.).
- Host cells lacking such activity are CHO cells, BHK cells, NS0 cells, SP2 / 0 cells, YO myeloma cells, P3X63 mouse myeloma cells, PER cells, PER.C6 cells, HEK293 cells, or hybridomas. It can be produced by a method of incapacitatingly destroying the genes of these functional proteins that are endogenous to cells or the like.
- Antibodies having a sugar chain having bisecting GlcNAc are known.
- GnTIII ⁇ -1,4-mannosyl-glycoprotein, 4- ⁇ -N-acetylglucosaminyltransferase
- EC ⁇ -1,4-mannosyl-glycoprotein, 4- ⁇ -N-acetylglucosaminyltransferase
- a host cell expressing a gene encoding a functional protein having 2.4.1.144) activity or GalT ( ⁇ -1,4-galactosyltransferase) (EC 2.4.1.38) activity is generated.
- GnTI ⁇ -1,2-
- a gene encoding a functional protein having human ManII (mannosidase II) (3.2.1.114) activity, in addition to the functional protein described above.
- a gene encoding a protein, a gene encoding a functional protein having ManI (mannosidase) (EC 3.2.1.113) activity, and a host cell co-expressing with ⁇ -1,6-fucosyltransferase (International release WO2004 / 065540).
- an antibody Fc By transfecting an expression vector containing an antibody gene into a host cell having a low ability to add fucose to a sugar chain as described above and a host cell having an activity of forming a sugar chain containing a bisecting GlcNAc structure, an antibody Fc An antibody in which the fucose residue is removed from N-acetylglucosamine at the reducing end of the N-glycoside-linked complex sugar chain that binds to the region and an antibody having a sugar chain having a bisecting GlcNAc can be produced.
- the method for producing these antibodies is such that the composition of the sugar chain bound to the Fc region of the present disclosure increases the proportion of the Fc region bound to the fucose-deficient sugar chain, or the composition of the Fc region to which bisecting N-acetylglucosamine is added. It can also be applied to a method for producing an antigen-binding molecule containing a modified Fc region modified so as to have a high proportion.
- the composition of the sugar chain bound to the Fc region contained in the antigen-binding molecule of the present disclosure produced by such a production method can be confirmed by the method described in the above-mentioned "Fc ⁇ receptor (Fc ⁇ R) binding modified Fc region".
- the present disclosure is not bound by any particular theory, but by the formation of a heterocomplex containing the Fc region contained in the antigen-binding molecule, two molecules of FcRn, and one molecule of active Fc ⁇ receptor.
- the pharmacokinetics of the antigen-binding molecule in vivo (retention in plasma) when the molecule is administered in vivo, and the immune response (immunogenicity) to the administered antigen-binding molecule are as follows. It may have an impact.
- FcRn is expressed on immune cells in addition to various active Fc ⁇ receptors, and the formation of such a quaternary complex on immune cells by antigen-binding molecules improves the affinity for immune cells and further.
- associating intracellular domains enhances the internalization signal and promotes uptake into immune cells.
- antigen-presenting cells suggesting that the formation of a quaternary complex on the cell membrane of antigen-presenting cells may facilitate the uptake of antigen-binding molecules into antigen-presenting cells.
- the antigen-binding molecule taken up by an antigen-presenting cell is degraded in lysosomes in the antigen-presenting cell and presented to T cells.
- the formation of the above-mentioned quaternary complex on the cell membrane of the antigen-presenting cell may promote the uptake of the antigen-binding molecule into the antigen-presenting cell, thereby deteriorating the retention of the antigen-binding molecule in plasma.
- an immune response may be induced (exacerbated).
- an antigen-binding molecule having a reduced ability to form such a quaternary complex when administered to a living body, the plasma retention of the antigen-binding molecule is improved, and the induction of an immune response by the living body is suppressed. Can be considered.
- the following three types can be mentioned as preferable modes of the antigen-binding molecule that inhibits the formation of the complex on immune cells including such antigen-presenting cells.
- Antigen-binding molecule that inhibits the formation of heterocomplexes (Mode 1) An antigen-binding molecule containing an Fc region that has FcRn-binding activity under neutral pH conditions and whose binding activity to active Fc ⁇ R is lower than that of the natural Fc region to active Fc ⁇ R.
- the antigen-binding molecule of mode 1 forms a tripartite complex by binding to two FcRn molecules, but does not form a complex containing the active Fc ⁇ R.
- An Fc region in which the binding activity to the active Fc ⁇ R is lower than the binding activity to the active Fc ⁇ R in the natural Fc region can be prepared by modifying the amino acid in the natural Fc region as described above. Whether or not the binding activity of the modified Fc region to the active Fc ⁇ R is lower than the binding activity of the natural Fc region to the active Fc ⁇ R can be appropriately carried out by using the method described in the above-mentioned section of binding activity.
- Activated Fc ⁇ receptors include Fc ⁇ RIa, Fc ⁇ RIb and Fc ⁇ RIc containing Fc ⁇ RI (CD64), Fc ⁇ RIIa (including allotypes R131 and H131) and isoforms Fc ⁇ RIIIa (including allotypes V158 and F158) and Fc ⁇ RIIIb (allotypes Fc ⁇ RIIIb-NA1 and Fc).
- Fc ⁇ RIII (CD16) containing (including NA2) is preferably mentioned.
- the binding activity of the Fc region variant to the active Fc ⁇ receptor is lower than the binding activity of the native Fc region to the active Fc ⁇ receptor as Fc ⁇ RI, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb of the Fc region variant. It means that the binding activity to any of the human Fc ⁇ receptors is lower than the binding activity of the natural Fc region to these human Fc ⁇ receptors.
- the binding activity of the antigen-binding molecule containing the Fc region variant is 95% or less, preferably 90%, as compared with the binding activity of the antigen-binding molecule containing the natural Fc region as a control.
- % Or less 85% or less, 80% or less, 75% or less, particularly preferably 70% or less, 65% or less, 60% or less, 55% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, 25% or less, 20% or less, 15% or less, 10% or less, 9% or less, 8% or less, 7% or less, 6% or less, 5% or less, 4% or less, 3% or less, 2%
- the binding activity is 1% or less.
- a starting Fc region can also be used, or an Fc region of a different isotype of a wild-type antibody can also be used.
- the binding activity to the natural active Fc ⁇ R is preferably the binding activity of human IgG1 to the Fc ⁇ receptor, and in order to reduce the binding activity to the Fc ⁇ receptor, in addition to the above modifications, human IgG2, human IgG3, etc. It can also be achieved by changing the isotype to human IgG4.
- the Fc ⁇ receptor-binding activity can be reduced by expressing an antigen-binding molecule containing an Fc region having an Fc ⁇ receptor-binding activity in a host such as Escherichia coli to which no sugar chain is added. Can be done.
- an antigen-binding molecule having an Fc region of an IgG monoclonal antibody can be appropriately used as the antigen-binding molecule containing the Fc region as a control.
- the structure of the Fc region is SEQ ID NO: 5 (A is added to the N end of RefSeq registration number AAC82527.1), SEQ ID NO: 6 (A is added to the N end of RefSeq registration number AAB59393.1), and SEQ ID NO: 7 (. RefSeq registration number CAA27268.1), SEQ ID NO: 8 (A is added to the N end of RefSeq registration number AAB59394.1).
- an antigen-binding molecule containing the Fc region of a specific isotype antibody When an antigen-binding molecule containing the Fc region of a specific isotype antibody is used as a test substance, an antigen-binding molecule having an Fc region of the specific isotype IgG monoclonal antibody is used as a control. The effect of the binding activity on the Fc ⁇ receptor by the antigen-binding molecule containing the Fc region will be verified. As described above, an antigen-binding molecule containing an Fc region whose binding activity to the Fc ⁇ receptor has been verified to be high is appropriately selected.
- the amino acids in the Fc region are represented by EU numbering 234. , 235, 236, 237, 238, 239, 270, 297, 298, 325, 328, and 329, in which one or more amino acids are modified to different amino acids from the natural Fc region.
- the modification of the Fc region is not limited to the above modification, and for example, the dessaccharide chains (N297A, N297Q) and IgG1- described in Cur. Opin. In Biotech. (2009) 20 (6), 685-691.
- amino acid at position 234 can be Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Lys, Met, Phe, Pro, Ser, Thr or Trp.
- the amino acid at position 235 is either Ala, Asn, Asp, Gln, Glu, Gly, His, Ile, Lys, Met, Pro, Ser, Thr, Val or Arg.
- the amino acid at position 236 is either Arg, Asn, Gln, His, Leu, Lys, Met, Phe, Pro or Tyr.
- the amino acid at position 237 is either Ala, Asn, Asp, Gln, Glu, His, Ile, Leu, Lys, Met, Pro, Ser, Thr, Val, Tyr or Arg.
- the amino acid at position 238 is either Ala, Asn, Gln, Glu, Gly, His, Ile, Lys, Thr, Trp or Arg.
- the amino acid at position 239 is either Gln, His, Lys, Phe, Pro, Trp, Tyr or Arg.
- the amino acid at position 265 is either Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Met, Phe, Ser, Thr, Trp, Tyr or Val.
- Amino acid at position 266 can be Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Lys, Phe, Pro, Ser, Thr, Trp or Tyr.
- the amino acid at position 267 is either Arg, His, Lys, Phe, Pro, Trp or Tyr.
- the amino acid at position 269 is either Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr or Val.
- the amino acid at position 270 is either Ala, Arg, Asn, Gln, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr or Val.
- the amino acid at position 271 is either Arg, His, Phe, Ser, Thr, Trp or Tyr.
- the amino acid at position 295 is either Arg, Asn, Asp, Gly, His, Phe, Ser, Trp or Tyr.
- Amino acid at position 296 is either Arg, Gly, Lys or Pro, Ala at position 297
- the amino acid at position 298 is either Arg, Gly, Lys, Pro, Trp or Tyr,
- Amino acid at position 300 is either Arg, Lys or Pro, Amino acid at position 324 of either Lys or Pro,
- the amino acid at position 325 is either Ala, Arg, Gly, His, Ile, Lys, Phe, Pro, Thr, TrpTyr, or Val.
- the amino acid at position 327 is either Arg, Gln, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr or Val.
- Amino acid at position 328 is either Arg, Asn, Gly, His, Lys or Pro
- the amino acid at position 329 is either Asn, Asp, Gln, Glu, Gly, His, Ile, Leu, Lys, Met, Phe, Ser, Thr, Trp, Tyr, Val or Arg.
- Amino acid at position 330 of either Pro or Ser The amino acid at position 331 is either Arg, Gly or Lys, or the amino acid at position 332 is either Arg, Lys or Pro.
- the Fc region modified to any one or more of the above is preferably mentioned.
- Mode 2 An antigen-binding molecule containing an Fc region that has FcRn-binding activity under neutral pH conditions and has a higher Fc ⁇ R-binding activity than the active Fc ⁇ receptor.
- the antigen-binding molecule of mode 2 can form a complex containing these four molecules by binding to two molecules of FcRn and one molecule of inhibitory Fc ⁇ R.
- one molecule of antigen-binding molecule can bind to only one molecule of Fc ⁇ R, one molecule of antigen-binding molecule cannot bind to other active Fc ⁇ R in a state of being bound to inhibitory Fc ⁇ R.
- antigen-binding molecules that are incorporated into the cell while bound to the inhibitory Fc ⁇ R are recycled onto the cell membrane and avoid intracellular degradation (Immunity (2005) 23, 503-514). That is, it is considered that an antigen-binding molecule having selective binding activity to inhibitory Fc ⁇ R cannot form a heterocomplex including active Fc ⁇ R and two molecules of FcRn that cause an immune response.
- Activated Fc ⁇ receptors include Fc ⁇ RIa, Fc ⁇ RIb and Fc ⁇ RIc containing Fc ⁇ RI (CD64), Fc ⁇ RIIa (including allotypes R131 and H131) and isoforms Fc ⁇ RIIIa (including allotypes V158 and F158) and Fc ⁇ RIIIb (allotypes Fc ⁇ RIIIb-NA1 and Fc).
- Fc ⁇ RIII (CD16) containing (including NA2) is preferably mentioned.
- Fc ⁇ RIIb (including Fc ⁇ RIIb-1 and Fc ⁇ RIIb-2) is a suitable example of the inhibitory Fc ⁇ receptor.
- the binding activity to the inhibitory Fc ⁇ R is higher than the binding activity to the active Fc ⁇ receptor, which means that the binding activity of the Fc region variant to Fc ⁇ RIIb is one of Fc ⁇ RI, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb. It is higher than the binding activity to the Fc ⁇ receptor.
- the binding activity of the antigen-binding molecule containing the Fc region variant to Fc ⁇ RIIb is 105% or more of the binding activity of Fc ⁇ RI, Fc ⁇ RIIa, Fc ⁇ RIIIa and / or Fc ⁇ RIIIb to the human Fc ⁇ receptor.
- the binding activity to Fc ⁇ RIIb is all higher than that of Fc ⁇ RIa, Fc ⁇ RIIa (including allotypes R131 and H131) and Fc ⁇ RIIIa (including allotypes V158 and F158). Since Fc ⁇ RIa has an extremely high affinity for natural IgG1, it is considered that the binding is saturated by a large amount of endogenous IgG1 in vivo, so that the binding activity to Fc ⁇ RIIb is higher than that of Fc ⁇ RIIa and Fc ⁇ RIIIa and lower than that of Fc ⁇ RIa. However, it is considered possible to inhibit the formation of the complex.
- an antigen-binding molecule having an Fc region of an IgG monoclonal antibody can be appropriately used as the antigen-binding molecule containing the Fc region as a control.
- the structure of the Fc region is SEQ ID NO: 5 (A is added to the N end of RefSeq registration number AAC82527.1), SEQ ID NO: 6 (A is added to the N end of RefSeq registration number AAB59393.1), and SEQ ID NO: 7 (. RefSeq registration number CAA27268.1), SEQ ID NO: 8 (A is added to the N end of RefSeq registration number AAB59394.1).
- an antigen-binding molecule containing the Fc region of a specific isotype antibody When an antigen-binding molecule containing the Fc region of a specific isotype antibody is used as a test substance, an antigen-binding molecule having an Fc region of the specific isotype IgG monoclonal antibody is used as a control. The effect of the binding activity on the Fc ⁇ receptor by the antigen-binding molecule containing the Fc region will be verified. As described above, an antigen-binding molecule containing an Fc region whose binding activity to the Fc ⁇ receptor has been verified to be high is appropriately selected.
- an Fc region having selective binding activity to inhibitory Fc ⁇ R 238 or 328 amino acids represented by EU numbering among the amino acids in the Fc region are natural Fc regions.
- the Fc region modified to an amino acid different from the above is preferably mentioned.
- the Fc region described in US2009 / 0136485 or a modification can be appropriately selected.
- the amino acid represented by the EU numbering of the Fc region is Asp, or 328 amino acids are one or more of Glu.
- the modified Fc region is preferably mentioned.
- the substitution of Pro at position 238 represented by EU numbering with Asp, and the amino acid at position 237 represented by EU numbering are Trp, at position 237 represented by EU numbering.
- the amino acid is Phe
- the 267th amino acid represented by EU numbering is Val
- the 267th amino acid represented by EU numbering is Gln
- the 268th amino acid represented by EU numbering is Asn, 271 represented by EU numbering.
- the amino acid at position Gly, the amino acid at position 326 represented by EU numbering is Leu
- the amino acid at position 326 represented by EU numbering is Gln
- the amino acid at position 326 represented by EU numbering is represented by Glu and EU numbering.
- the 326th amino acid is Met, the 239th amino acid represented by EU numbering is Asp, the 267th amino acid represented by EU numbering is Ala, and the 234th amino acid represented by EU numbering is Trp and EU numbering.
- the 234th amino acid represented is Tyr, the 237th amino acid represented by EU numbering is Ala, the 237th amino acid represented by EU numbering is Asp, and the 237th amino acid represented by EU numbering is Glu, EU.
- the amino acid at position 237 represented by numbering is Leu, the amino acid at position 237 represented by EU numbering is Met, the amino acid at position 237 represented by EU numbering is Tyr, and the amino acid at position 330 represented by EU numbering is Lys.
- the amino acid at position 330 represented by EU numbering is Arg
- the amino acid at position 233 represented by EU numbering is Asp
- the amino acid at position 268 represented by EU numbering is Asp
- the amino acid at position 268 represented by EU numbering is Asp
- Is Glu the amino acid at position 326 represented by EU numbering is Asp
- the amino acid at position 326 represented by EU numbering is Ser
- the amino acid at position 326 represented by EU numbering is Thr
- position 323 represented by EU numbering position 323 represented by EU numbering.
- Amino acid is Ile
- amino acid at position 323 represented by EU numbering is Leu
- amino acid at position 323 represented by EU numbering is Met
- amino acid at position 296 represented by EU numbering is represented by Asp and EU numbering.
- the amino acid at position 326 is Ala
- the amino acid at position 326 represented by EU numbering is Asn
- the amino acid at position 330 represented by EU numbering is Met.
- the antigen-binding molecule of mode 3 can form a tripartite complex by binding to one molecule of FcRn and one molecule of Fc ⁇ R, but a heterocomplex containing two molecules of FcRn and one molecule of Fc ⁇ R is a heterocomplex. Does not form.
- One of the two polypeptides constituting the Fc region contained in the antigen-binding molecule of this mode 3 has FcRn-binding activity under the condition of neutral pH, and the other polypeptide has neutral pH.
- an Fc region originating from a bispecific antibody bispecific antibody
- Bispecific antibodies are two types of antibodies that have specificity for different antigens.
- IgG-type bispecific antibodies can be secreted by hybrid hybridomas (quadromas) produced by fusing two hybridomas that produce IgG antibodies (Milstein et al. (Nature (1983) 305, 537-540)). ..
- the gene encoding the polypeptide constituting the two target Fc regions is introduced into the cell.
- a method of co-expressing them can be adopted.
- one of the two polypeptides constituting the Fc region has a binding activity to FcRn under the condition of the neutral pH range, and the other polypeptide has the binding activity to the FcRn under the condition of the neutral pH range.
- Fc region that does not have FcRn-binding activity in FcRn and Fc region in which both of the two polypeptides that make up the Fc region have FcRn-binding activity under neutral pH conditions, and Fc region.
- Both of the two polypeptides form a mixture in which Fc regions having no binding activity to FcRn under neutral pH conditions are present in a ratio of 2: 1: 1 molecular number. It is difficult to purify an antigen-binding molecule containing the desired combination of Fc regions from three types of IgG.
- the antigen-binding molecule of mode 3 is prioritized by modifying the CH3 domain constituting the Fc region with an appropriate amino acid substitution.
- Yes International Publication WO 1996027011, Riggway et al. (Protein Engineering (1996) 9, 617-621), Merchant et al. (Nat. Biotech. (1998) 16, 677-681)).
- a technique for producing a bispecific antibody by utilizing a method for controlling the association of polypeptides or the association of heterologous multimers composed of polypeptides for the association of two polypeptides constituting the Fc region.
- a method for controlling the association of polypeptides or the association of heterologous multimers composed of polypeptides for the association of two polypeptides constituting the Fc region are known. That is, by modifying the amino acid residues forming the interface in the two polypeptides constituting the Fc region, the association of the polypeptides constituting the Fc region having the same sequence is inhibited, and the two Fc regions having different sequences are inhibited.
- a method of controlling the formation of the polypeptide aggregates constituting the above can be adopted for the production of bispecific antibodies (International Publication WO 2006/106905). Specifically, the method described in the section of the bispecific antibody and the method for producing the same can be adopted as a non-limiting aspect in producing the antigen-binding molecule of mode 3
- All of these antigen-binding molecules of Modes 1 to 3 are capable of lowering immunogenicity and improving plasma retention as compared with antigen-binding molecules capable of forming a quaternary complex. Be expected.
- FcRn Unlike Fc ⁇ receptors, which belong to the immunoglobulin superfamily, human FcRn is structurally similar to major histocompatibility complex (MHC) class I polypeptides and 22-29% sequence with class I MHC molecules. It has the same identity (Ghetie et al., Immunol. Today (1997) 18 (12), 592-598). FcRn is expressed as a heterodimer consisting of transmembrane ⁇ or heavy chains complexed with soluble ⁇ or light chain ( ⁇ 2 microglobulin). Like MHC, the ⁇ chain of FcRn consists of three extracellular domains ( ⁇ 1, ⁇ 2, ⁇ 3), and the short cytoplasmic domain anchors the protein to the cell surface. The ⁇ 1 and ⁇ 2 domains interact with the FcRn-binding domain in the Fc region of the antibody (Raghavan et al. (Immunity (1994) 1, 303-315).
- MHC major histocompatibility complex
- FcRn is expressed in the maternal placenta or yolk sac of mammals, which is involved in the transfer of IgG from the mother to the fetus.
- FcRn is involved in the transfer of FcRn across the brush border epithelium of maternal IgG from the ingested primary milk or milk.
- FcRn is expressed across many species in many other tissues, as well as in various endothelial cell lines. It is also expressed in human adult vascular endothelium, musculoskeletal system, and hepatic sinusoidal capillaries.
- FcRn is thought to play a role in maintaining the plasma concentration of IgG by binding to IgG and recycling it into serum. Binding of FcRn to IgG molecules is usually strictly pH dependent and optimal binding is found in the acidic pH range below 7.0.
- Human FcRn which is a precursor of a polypeptide containing the signal sequence represented by SEQ ID NO: 28, is a human ⁇ 2-microglobulin in vivo (the polypeptide containing the signal sequence is described in SEQ ID NO: 29). Form a complex with. Soluble human FcRn, which forms a complex with ⁇ 2-microglobulin, is produced by using conventional recombinant expression techniques. The binding activity of the Fc region of the present disclosure to soluble human FcRn forming a complex with such ⁇ 2-microglobulin can be evaluated.
- human FcRn refers to a form capable of binding to the Fc region of the present disclosure, and examples thereof include a complex of human FcRn and human ⁇ 2-microglobulin.
- FcRn bond-enhancing modified Fc technology at acidic pH (WO2002060919, WO2004035752, WO2000042072)
- FcRn binding enhancing modified Fc technology at neutral pH (WO2011122011, WO2012133782)
- inhibitory type Antibodies such as Fc ⁇ receptor selective binding enhancement technology (WO2012115241, WO2013125667), active Fc ⁇ receptor selective binding enhancement technology (ADCC activity enhancement technology) (WO2013002362), technology to reduce binding activity to Rheumatoid factor (WO2013046704), etc.
- a combination with a modification technique can be mentioned.
- variable region modification technique includes a combination with a modification technique such as a pH-dependent antibody (WO2009125825) and a calcium-dependent antibody (WO2012073992).
- the antigen-binding molecule in the present disclosure may have an amino acid residue that interacts with the MTA.
- the amino acid residue that interacts with the MTA may be present in the antigen-binding domain in the antigen-binding molecule, or may be present at a site other than the antigen-binding domain. Further, the amino acid residue that interacts with the MTA may be only one residue or a plurality of amino acid residues in the antigen-binding molecule.
- An antigen-binding domain is exemplified as a site where an amino acid residue that interacts with an MTA is located.
- an antibody is exemplified as a non-limiting aspect of an antigen-binding molecule containing an amino acid residue that interacts with an MTA.
- Amino acid residues that interact with the MTA can be present in the constant and / or variable regions of the antibody, and can also be present in CDRs, FRs in the antibody variable regions, but are not limited to either.
- a portion different from the antigen-binding domain of the antigen-binding molecule is exemplified.
- the structure of the antigen-binding domain is changed by the interaction of amino acid residues in a portion different from the antigen-binding domain of the antigen-binding molecule with the MTA, and the antigen-binding activity can be indirectly changed in an MTA-dependent manner.
- Amino acid residues that interact with MTA in the antigen-binding molecule are crystal structure analysis of the complex of MTA and the antigen-binding molecule or the tripartite complex of MTA, antigen, and antigen-binding molecule, and three-dimensional structure analysis using NMR. Or, it can be identified by a method such as introduction of an antigen mutation.
- An antibody is exemplified as a non-limiting aspect of an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner in the present disclosure.
- an antigen-binding molecule has two or more antigen-binding domains and can bind to a plurality of different antigens at the same time.
- the antigen-binding molecule of the present disclosure further has a second antigen-binding domain, and the second antigen-binding domain is for a second antigen different from the antigen to which the antigen-binding domain binds.
- all the antigen-binding domains contained in the antigen-binding molecule may be MTA or an antigen-binding domain in which the antigen-binding activity changes depending on a small molecule compound other than MTA.
- a plurality of antigen-binding domains may be antigen domains in which the antigen-binding activity changes depending on different low-molecular-weight compounds, or one antigen-binding molecule has a low-molecular-weight compound-dependent antigen-binding activity.
- the changing antigen-binding domain and the antigen-binding domain whose antigen-binding activity is not affected by the concentration of the small molecule compound may be present at the same time.
- the binding activity of the second antigen binding domain to the second antigen is substantially unaffected by the MTA. In a further aspect, the binding activity of the second antigen-binding domain to the second antigen changes in an MTA-dependent manner. In a more detailed aspect, the binding activity of the second antigen-binding domain to the second antigen in the presence of MTA is the binding activity of the second antigen-binding domain to the second antigen in the absence of MTA.
- Bispecific antibodies can be exemplified, but are limited to, as non-limiting examples of antigen-binding molecules having two or more antigen-binding domains and capable of simultaneously binding to a plurality of different antigens. It's not something.
- a membrane type that is different from each other and one of the paratopes binds to the cell membrane of a cancer cell or a cell invading cancer tissue.
- a Diabody or sc (Fv) 2 that binds to an epitope present in a molecule and the other paratope to an epitope present in a membrane-type molecule expressed on the cell membrane of effector cells can be exemplified.
- the binding activity of one paratope to an epitope present in a membrane-type molecule that binds to the cell membrane of a cancer cell or a cell invading cancer tissue changes in an MTA-dependent manner.
- the binding activity of one paratope to an epitope present in the membrane-type molecule that binds to the cell membrane of effector cells can change in an MTA-dependent manner, and the binding activity of both paratopes changes in an MTA-dependent manner.
- the antigen-binding domain in which the binding activity to the antigen does not change in an MTA-dependent manner is a low-molecular-weight compound other than MTA (for example, cancer tissue-specific). It may be an antigen-binding domain in which the antigen-binding activity changes depending on the compound (compound, non-natural small molecule compound).
- membrane-type molecules that bind to the cell membrane of effector cells include, but are not limited to, TCR complexes, CD3, and CD3e.
- One paratope binds to an epitope present in a membrane-type molecule that binds to the cell membrane of cancer cells, cells invading cancer tissue, etc. (also called target cells), and the other paratope binds to the cell membrane of effector cells.
- the double paratopic antigen-binding molecule binds to both the target cell and the effector cell. It can activate effector cells and induce cytotoxic activity against target cells. When the effector cells are T cells, the cytotoxic activity evoked is T-cell-dependent cytotoxicity (TDCC) activity.
- TDCC T-cell-dependent cytotoxicity
- the cytotoxic substance may be bound to the antigen-binding molecule.
- the cytotoxic substance may be a chemotherapeutic agent exemplified below, or a compound disclosed in Curr Opin Chem Biol (2010) 14, 529-37 or WO 2009/140242. Often, these compounds are bound to the antigen-binding molecule with a suitable linker or the like.
- the antigen-binding molecule of the present disclosure is used as a pharmaceutical composition, it is possible to bind these cytotoxic substances to a subject (subject, patient, etc.) before administering the antigen-binding molecule. It can also be administered before, after or at the same time as administration.
- a modified antigen-binding molecule-modified product to which a cytotoxic substance such as a chemotherapeutic agent, a toxic peptide or a radiochemical substance, which will be described later, is bound can also be suitably used as an antigen-binding molecule having cytotoxic activity of the present disclosure. ..
- a modified antigen-binding molecule (hereinafter, referred to as an antigen-binding molecule drug conjugate) can be obtained by chemically modifying the obtained antigen-binding molecule.
- a method for modifying the antigen-binding molecule a method already established in the field of antibody-drug conjugate or the like can be appropriately used.
- the modified antigen-binding molecule to which the toxic peptide is bound expresses a fusion gene in which the gene encoding the toxic peptide and the gene encoding the antigen-binding molecule of the present disclosure are linked in frame in an appropriate host cell. It can be obtained by isolating the cells from the culture medium after the cells are allowed to grow.
- Chemotherapeutic agents that bind to the antigen-binding molecules of the present disclosure can be exemplified: azaribine, anastrozole, azacytidine, bleomycin, bortezomib, briostatin-1 ( bryostatin-1), busulfan, camptothecin, 10-hydroxycamptothecin, carmustine, celebrex, chlorambucil, cisplatin, irinotecan , Carboplatin, cladribine, cyclophosphamide, cytarabine, dacarbazine, docetaxel, dactinomycin, daunomycin single chronide, daunomycin glucuronide daunorubicin, dexamethasone, diethylstilbestrol, doxorubicin, doxorubicin glucuronide, epirubicin, epirubicin, ethinyl estradiol (ethinyl estradi
- the preferred chemotherapeutic agent is a low molecular weight chemotherapeutic agent.
- Small molecule chemotherapeutic agents are less likely to interfere with the function of the antigen-binding molecule even after the antigen-binding molecule of the present disclosure has bound.
- low molecular weight chemotherapeutic agents typically have a molecular weight of 100-2000, preferably 200-1000.
- the chemotherapeutic agents exemplified here are all small molecule chemotherapeutic agents.
- These chemotherapeutic agents in the present disclosure include prodrugs that are converted into active chemotherapeutic agents in vivo. Activation of a prodrug can be an enzymatic or non-enzymatic conversion.
- toxic peptides such as Pseudomonas exotoxin A, Saporin-s6, Diphtheria toxin, Cnidarian toxin, Radioiodine, and Photosensitizer can also be exemplified.
- toxic peptides are preferably, for example: Diphtheria toxin A Chain (Langone et al. (Methods in Enzymology (1983) 93, 307-308)); Pseudomonas Exotoxin (Nature Medicine (1996) 2, 350-353); Ricin A Chain (Fulton et al. (J. Biol.
- cytotoxicity to cells containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner and expressing a membrane-type molecule on the cell membrane.
- An antigen-binding molecule having activity and a pharmaceutical composition containing the antigen-binding molecule as an active ingredient are provided.
- cellular cytotoxicity refers to, for example, antibody-dependent cell-mediated cytotoxicity (ADCC) activity, complement-dependent cytotoxicity (CDC) activity, and T cells. Examples include T-cell-dependent cytotoxicity (TDCC) activity and antibody-dependent cellular cytotoxicity (ADCP) activity.
- CDC activity means cytotoxic activity by the complement system.
- ADCC activity means that an immune cell or the like binds to the Fc region of an antigen-binding molecule containing an antigen-binding domain that binds to a membrane-type molecule expressed on the cell membrane of a target cell via an Fc ⁇ receptor expressed on the immune cell.
- TDCC activity is an antigen-binding domain that binds to a membrane-type molecule expressed on the cell membrane of a target cell, or an antigen-binding domain to any of the constituent subsystems of the T cell receptor (TCR) complex on T cells, particularly.
- T cells to damage the target cells by bringing the target cells closer to the T cells by using a bi-specific antibody containing an antigen-binding domain that binds to the CD3 epsilon chain.
- the antigen-binding molecule of interest has ADCC activity, CDC activity, TDCC activity or ADCP activity can be measured by a known method.
- the effector cells, complement solution, and target cells are prepared.
- Spleen cells are isolated from the spleen excised from CBA / N mice or the like in RPMI1640 medium (Invitrogen). Effector cells can be prepared by adjusting the concentration of the spleen cells washed in the same medium containing 10% fetal bovine serum (FBS, HyClone) to 5 ⁇ 10 6 / mL.
- FBS fetal bovine serum
- HyClone fetal bovine serum
- Target cells can be radioactively labeled by culturing antigen-expressing cells with 0.2 mCi of 51 Cr-sodium chromate (GE Healthcare Bioscience) in DMEM medium containing 10% FBS for 1 hour at 37 ° C. ..
- the target cells can be prepared by adjusting the concentration of cells washed three times in RPMI1640 medium containing 10% FBS after radiolabeling to 2 ⁇ 10 5 / mL.
- ADCC activity or CDC activity can be measured by the method described below.
- For the measurement of ADCC activity 50 ⁇ l of each target cell added to a 96-well U-bottom plate (Becton Dickinson) and an antigen-binding molecule are reacted at room temperature for 15 minutes. Then, the plate to which 100 ⁇ l of effector cells are added is allowed to stand in a carbon dioxide incubator for 4 hours.
- the final concentration of the antigen-binding molecule can be set to, for example, 0 or 10 ⁇ g / ml. After standing, the radioactivity of 100 ⁇ l of the supernatant collected from each well is measured using a gamma counter (COBRAII AUTO-GAMMA, MODEL D5005, Packard Instrument Company).
- the cytotoxic activity (%) can be calculated based on the formula (A-C) / (B-C) x100.
- A represents the radioactivity (cpm) in each sample
- B represents the radioactivity (cpm) in the sample to which 1% NP-40 (nacalai tesque) is added
- C represents the radioactivity (cpm) in the sample containing only the target cells.
- ADCP activity can be measured by the methods described below. Labeling of target cells expressing the antigen is performed using a PKH26 dye labeling kit (Sigma). The target cells are exfoliated with trypsin solution and washed with PBS. The detached cells were suspended so as to be 1x10 7 cells / ml in Diluent C, PKH26 dye stock to (1 mM) was diluted to 8 ⁇ M in Diluent C, immediately adding the cell suspension in an amount equal dye dilution. After standing at room temperature for 5 minutes, RPMI1640 containing 10% FBS is added and washed twice to prepare target cells.
- PKH26 dye labeling kit Sigma
- the target cells are exfoliated with trypsin solution and washed with PBS.
- the detached cells were suspended so as to be 1x10 7 cells / ml in Diluent C, PKH26 dye stock to (1 mM) was diluted to 8 ⁇ M in Diluent C, immediately adding the cell suspension in an amount
- the antigen-binding molecule which is the test substance, is diluted to 20 ⁇ g / ml using the medium of the target cells, and the target cells are dispensed and mixed so as to be 2 ⁇ 10 6 cells / 100 ⁇ l / tube.
- the final concentration of the antigen-binding molecule can be set to, for example, 0 or 10 ⁇ g / ml. After allowing to stand on ice for 30 minutes, the supernatant is discarded, washed twice with the culture solution, and suspended in 500 ⁇ L of the culture solution. The supernatant is removed from the effector cells, and the effector cells are added to the target cell suspension mixed with the antigen-binding molecule and mixed.
- the cells After culturing in a CO 2 incubator for 3 hours, the cells are detached with Trypsin-EDTA and the cells are collected. For example, FITC-labeled anti-CD11b antibody is added to the collected cells, and the cells are allowed to stand on ice for 30 minutes. After standing, the supernatant is discarded, and after washing twice with the culture solution, the collected cells are suspended in 300 ⁇ l of the culture solution, and measurement is performed with, for example, FACS Calibur (Becton Dickinson). In CD11b-positive macrophage cells, ADCP activity can be measured by evaluating the PKH26-positive fraction as phagocytosis-positive cells.
- TDCC activity can be measured by the methods described below.
- the TDCC activity of a test antibody can be measured using human peripheral blood mononucleosis (hereinafter referred to as human PBMC) as an effector cell.
- human PBMC human peripheral blood mononucleosis
- (1) Preparation of human PBMC solution Use a syringe in which 500 ⁇ l of 5000 units / 5 ml of heparin solution has been injected in advance, and collect 50 ml of peripheral blood from a healthy person.
- the peripheral blood diluted 2-fold with PBS is divided into 4 equal parts and added to a pre-injected and centrifuged Leucosep lymphocyte separation tube (GE Healthcare) with 15 ml Ficoll-Paque PLUS.
- TDCC activity can be evaluated by the LDH release method (LDH Cytotoxicity Detection Kit: TAKARA).
- LDH Cytotoxicity Detection Kit: TAKARA LDH Cytotoxicity Detection Kit
- each well of a 96-well U-bottom plate was prepared with an antibody solution of each concentration (0.000004, 0.00004, 0.0004, 0.004, 0.04, 0.4, 4, 40 ⁇ g / ml) diluted 4 times the final concentration in the culture medium of each target cell. Add 50 ⁇ l each.
- 50 ⁇ l of the target cells prepared in 2 x 10 5 cells / ml in the culture medium of each target cell is seeded (1 x 10 4 cells / well) and allowed to stand at room temperature for 15 minutes.
- a plate prepared by adding 100 ⁇ l (2 x 10 5 cells / well) of each human PBMC solution prepared in the culture medium of each target cell prepared in (1) to each well was placed in a 5% carbon dioxide incubator at 37 ° C. After leaving it to stand for about 24 hours, centrifuge. Transfer 100 ⁇ l of culture supernatant in each well of the plate to a 96-well flat bottom plate. Dissolve the catalyst solution of the LDH Cytotoxicity Detection Kit in 1 ml of H2O and mix with the dye solution at a ratio of 1:45.
- A is the absorbance of a mixture of target cells, effector cells, and antibodies
- B is the absorbance of effector cells
- C is the absorbance of target cells
- D is the absorbance of target cells supplemented with Triton X-100.
- a T cell receptor (T cell receptor on T cells) comprising an antigen binding domain in which the antigen binding activity changes in an MTA-dependent manner.
- An antigen-binding molecule capable of activating T cells by binding to any of the constituent subsystems of the TCR) complex, and a pharmaceutical composition containing the antigen-binding molecule as an active ingredient are provided. It binds to the antigen-binding domain that binds to the membrane-type molecule expressed on the cell membrane of the target cell, and to the antigen-binding domain to any of the constituent subsystems of the T cell receptor (TCR) complex on T cells, especially the CD3 epsilon chain.
- the activation of T cells can be induced by bringing the target cells and T cells closer to each other by using a bi-specific antibody containing an antigen-binding domain.
- the activation ability of T cells can be measured using, for example, GloResponse NFAT-RE-Luc2 Jurkat cells (Promega).
- the cells incorporate a luciferase reporter whose expression is induced by the NFAT response element (NFAT-RE), which is a Nuclear factor of activated T cells (NFAT) response sequence, and is a TCR complex expressed on the cell membrane of Jurkat. Activation can be detected by measuring the expression of the reporter protein.
- NFAT-RE NFAT response element
- NFAT Nuclear factor of activated T cells
- TCR ligands By using the cells, it is possible to measure the T cell activation ability of TCR ligands, anti-TCR antibodies, and anti-CD3 antibodies that can activate the TCR complex.
- the measurement of the activation ability of T cells is not limited to the method using the above cells, and the measurement is carried out by constructing and using cells of the T cell lineage into which the same NFAT response element (NFAT-RE) and reporter protein have been introduced. Is also possible.
- the measurement of the activation ability of T cells is not limited to the above method, and for example, the expression of T cell activation markers such as CD69 and CD25 may be confirmed by a flow cytokine method or a gene expression analysis method, or TNF.
- T cell activation may be performed by measuring cytokines secreted by activated T cells such as - ⁇ , IFN- ⁇ , IL-6 and IL-2, but it is not limited to these methods and T cell activation is measured. Any method that can be used can be used regardless of the cell line used or the measurement method.
- a bispecific antibody T cell having an antigen-binding domain that binds to the target antigen hIL-6R and an antigen-binding domain that binds to CD3
- a method for measuring the activation ability is illustrated below.
- NFAT-RE-luc2-Jurkat cells are used as the human T cell line, and CT26 / hIL-6R in which hIL-6R is forcibly expressed in CT26 is used as the hIL-6R expression cell line.
- NFAT-RE-luc2-Jurkat cells and CT26 / hIL-6R cells can be suspended in assay buffer to a cell density of 3 ⁇ 10 6 cells / mL and 1 ⁇ 10 6 cells / mL, respectively.
- the cell density and the mixing ratio of each cell may be increased or decreased depending on the cell type used for evaluation.
- To each well of the 384-well plate add 10 ⁇ l each of the prepared human T cell line and hIL-6R-expressing cell line suspensions, and further add 10 ⁇ l of the prepared antibody solution. After standing at 37 ° C. for 6 hours in a 5% CO 2 incubator, the activating ability of the human T cell line can be evaluated by measuring the luciferase activity of the sample.
- the measurement of luciferase activity can be carried out using a commercially available reagent for measuring luciferase activity (Bio-Glo luciferase assay system, Promega), but if the luciferase activity can be measured, the reagent can be used without limitation.
- an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner and having neutralizing activity for the membrane-type molecule.
- a pharmaceutical composition containing the above as an active ingredient is provided.
- a pharmaceutical composition containing an antigen-binding molecule having a neutralizing activity against a membrane-type molecule as an active ingredient.
- the neutralizing activity refers to an activity that inhibits the biological activity of a ligand having a biological activity on a cell, such as a virus or a toxin. That is, the substance having neutralizing activity refers to a substance that binds to the ligand or the receptor to which the ligand binds and inhibits the binding between the ligand and the receptor. A receptor whose binding to a ligand is blocked by neutralizing activity cannot exert biological activity through the receptor.
- the antigen-binding molecule is an antibody
- an antibody having such neutralizing activity is generally called a neutralizing antibody.
- the neutralizing activity of a test substance can be measured by comparing its biological activity in the presence of a ligand between conditions in the presence or absence of the test substance.
- IL-6 represented by SEQ ID NO: 27 is preferably used as the main ligand of the IL-6 receptor.
- the IL-6 receptor a type I membrane protein whose amino ends form an extracellular domain, forms a heterotetramer together with the gp130 receptor whose dimerization is induced by IL-6 (Heinrich et al. (Biochem.). J. (1998) 334, 297-314)).
- the formation of the heterotetramer activates Jak associated with the gp130 receptor. Jak performs autophosphorylation and receptor phosphorylation.
- Receptor and Jak phosphorylation sites act as binding sites for SH2-bearing Stat family molecules such as Stat3, MAP kinase, PI3 / Akt, and other SH2-bearing proteins and adapters.
- the Stat bound to the gp130 receptor is then phosphorylated by Jak.
- Phosphorylated Stats form dimers and translocate into the nucleus, regulating transcription of target genes. Jaks or Stats can also participate in the signal cascade via other classes of receptors.
- the decontrolled IL-6 signal cascade is observed in the pathophysiology and inflammation of autoimmune diseases and in cancers such as multiple myeloma and prostate cancer.
- Stat3, which can act as an oncogene, is constitutively activated in many cancers.
- a target molecule can be appropriately set for each target cell, and is not limited to the above factors.
- Neutralization activity can be evaluated by measuring the activation of in vivo signals.
- activation of in vivo signals can be detected by using the transcription-inducing action on a target gene existing downstream of the in vivo signal cascade as an index.
- Changes in the transcriptional activity of the target gene can be detected by the principle of the reporter assay. Specifically, a reporter gene such as GFP (Green Fluorescence Protein) or luciferase is placed downstream of the transcription factor or promoter region of the target gene, and the reporter activity is measured to measure the change in transcription activity as the reporter activity. can do.
- a commercially available measurement kit for in vivo signal activation can be appropriately used (for example, Mercury Pathway Profiling Luciferase System (Clontech), etc.).
- the antigen-binding molecule is measured by measuring the proliferation activity of the target cell. Can be evaluated for its neutralizing activity.
- the following method is preferable as a method for evaluating or measuring the inhibitory effect based on the neutralizing activity of an anti-HB-EGF antibody on the proliferation of cells whose proliferation is promoted by growth factors of the EGF family such as HB-EGF. used.
- a method for measuring the uptake of [ 3 H] -labeled thymidine added to the medium by living cells as an index of DNA replication ability is used.
- the dye elimination method for measuring the ability to eliminate dyes such as trypan blue extracellularly under a microscope and the MTT method are used.
- the latter utilizes the ability of living cells to convert the tetrazolium salt MTT (3- (4,5-dimethylthiazol-2-yl) -2,5-diphenyl tetrazolium bromide) to a blue formazan product.
- the MTT solution is added to the culture solution and allowed to stand for a certain period of time to allow the cells to take up MTT. ..
- the yellow compound MTT is converted to the blue compound by intracellular succinate dehydrogenase in the mitochondria. This blue product is lysed and colored, and then its absorbance is measured to use it as an index of the number of living cells.
- reagents such as MTS, XTT, WST-1, and WST-8 are also commercially available (such as nacalai tesque) and can be preferably used.
- an antibody having the same isotype as the anti-HB-EGF antibody but not having the cell growth inhibitory activity is used as a control antibody in the same manner as the anti-HB-EGF antibody, and the anti-HB-EGF is used.
- the activity can be determined by the antibody exhibiting stronger cell growth inhibitory activity than the control antibody.
- Examples of cells for evaluating activity include the RMG-1 cell line, which is an ovarian cancer cell whose proliferation is promoted by HB-EGF, and the extracellular domain of human EGFR and the mouse G-CSF receptor.
- Mouse Ba / F3 cells transformed with a vector bound so as to express a gene encoding hEGFR / mG-CSFR, which is a fusion protein in which the intracellular domains of the cells are fused in frame can also be preferably used.
- those skilled in the art can use the cells for measuring the cell proliferation activity by appropriately selecting the cells for evaluating the activity.
- the "antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner" in the present disclosure can bring about a positive pharmacological action when administered to a living body.
- the antigen-binding domain in the antigen-binding molecule is artificially used for the purpose of reducing heterologous antigenicity to humans.
- An antigen-binding domain derived from a modified recombinant antibody can be appropriately adopted.
- Genetically modified antibodies include, for example, humanized antibodies and the like. These modified antibodies are appropriately produced using known methods.
- variable regions of an antibody used to create the antigen-binding domain in the antigen-binding molecules described herein are usually three complementarity regions sandwiched between four framework regions (FRs). -determining region; CDR).
- CDRs are essentially regions that determine the binding specificity of an antibody.
- the amino acid sequence of CDR is rich in diversity.
- the amino acid sequences constituting FR often show high identity even among antibodies having different binding specificities. Therefore, it is generally said that the binding specificity of one antibody can be transplanted to another antibody by transplanting CDR.
- Humanized antibodies are also referred to as reshaped human antibodies. Specifically, as animals other than humans, humanized antibodies obtained by transplanting CDRs of antibodies such as mice and rabbits into human antibodies are known. Common gene recombination techniques for obtaining humanized antibodies are also known. Specifically, but not limited to this, for example, Overlap Extension PCR is known as a method for transplanting CDR of a mouse antibody into human FR. In Overlap Extension PCR, a nucleotide sequence encoding the CDR of a mouse antibody to be transplanted is added to a primer for synthesizing FR of a human antibody. Primers are prepared for each of the four FRs.
- human FRs having high identity with mouse FRs in maintaining the function of CDRs. That is, in general, it is preferable to use human FR having an amino acid sequence having a high identity with the amino acid sequence of FR adjacent to the mouse CDR to be transplanted.
- the origin of the CDR is not limited to mice and rabbits, but can be selected from antibodies of any non-human animal.
- the base sequences to be linked are designed to be linked to each other in frame.
- Human FR is individually synthesized by each primer.
- a product is obtained in which the DNA encoding the mouse CDR is added to each FR.
- the nucleotide sequences encoding the mouse CDRs of each product are designed to overlap each other.
- the overlapping CDR portions of the products synthesized using the human antibody gene as a template are annealed to each other to carry out a complementary strand synthesis reaction. By this reaction, human FR is linked via the sequence of mouse CDR.
- a human antibody expression vector can be prepared by inserting the DNA obtained as described above and the DNA encoding the human antibody C region into an expression vector so as to be fused in frame. After introducing the integrated vector into a host to establish recombinant cells, the recombinant cells are cultured and the DNA encoding the humanized antibody is expressed, so that the humanized antibody is a culture of the cultured cells. Produced in (see European Patent Publication EP239400, International Publication WO 1996/002576).
- the CDR forms a good antigen-binding site when linked via the CDR.
- the FR of such a human antibody can be preferably selected.
- the amino acid residues of FR can be substituted so that the CDRs of the reconstituted human antibody form the appropriate antigen binding site.
- the PCR method used for transplanting mouse CDRs into human FR can be applied to introduce mutations in the amino acid sequence into FR.
- a partial base sequence mutation can be introduced into the primer annealing to FR.
- a mutation in the base sequence is introduced into the FR synthesized by such a primer.
- a mutant FR sequence having desired properties can be selected by measuring and evaluating the antigen-binding activity of a mutant antibody substituted with an amino acid by the above method (Cancer Res., (1993) 53, 851-856). ..
- compositions containing antigen-binding molecules that do not act systemically in normal tissues or blood but act in cancer in order to exert drug efficacy while avoiding side effects. Since the antigen-binding molecule contained in the pharmaceutical composition of the present disclosure is controlled to bind to a target antigen in an MTA-dependent manner, for example, when the antigen-binding molecule targets an antigen in cancer tissue, Side effects due to cytotoxic effects and neutralizing effects on normal tissues because they cannot bind to antigens expressed in cancer cells, immune cells, stromal cells, etc. in cancer tissues or antigens secreted in cancer tissues.
- a bispecific antigen-binding molecule or biparatopic containing an antigen-binding domain that changes its binding activity to CD3 expressed on T cells in an MTA-dependent manner and an antigen-binding domain that binds to EGFR expressed on cancer cells.
- the antigen-binding molecule does not bind to EGFR expressed in normal tissues, but binds to EGFR expressed in cancer cells, so that it exerts a strong antitumor effect while avoiding side effects.
- antigen-binding molecules that bind to antigens in cancer tissues and do not bind to antigens in other normal tissues or blood exert their medicinal effects while avoiding side effects.
- An antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner (MTA switch antigen-binding molecule, an antigen-binding molecule that binds to an antigen using MTA as a switch) provided in the present disclosure. (Also known as) does not bind to the antigen in a normal environment in which MTA is absent, and can bind to an antigen in cancer tissues in which MTA is present at a high concentration.
- the structure of the paratope for the antigen of the antigen-binding molecule is changed.
- An example is an antigen-binding molecule whose antigen-binding activity changes in an MTA-dependent manner that functions as a switch. In the absence of MTA, the interaction between the paratope contained in the antigen-binding molecule of the present disclosure and the epitope contained in the antigen becomes insufficient, and the antigen-binding molecule of the present disclosure cannot bind to the antigen, but MTA is present.
- the binding molecule can exert a medicinal effect on cells expressing the antigen (FIGS. 26 and 27).
- an amino acid residue that interacts with MTA is present in the antigen-binding domain whose antigen-binding activity changes depending on the MTA of the present disclosure, the MTA is contained between the paratope contained in the antigen-binding molecule and the epitope contained in the antigen. It is easy to be pinched or the structure of the paratope is easily changed.
- the antigen-binding molecule of the present disclosure capable of binding to pathological cells such as cancer cells and immune cells in cancer tissue, or binding to an antigen secreted in cancer tissue to exert a medicinal effect.
- the pharmaceutical compositions of the present disclosure may include a pharmaceutically acceptable carrier.
- the pharmaceutical composition usually refers to a drug for treating or preventing a disease, or for testing / diagnosis.
- pharmaceutical composition containing an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner refers to "an antigen whose antigen-binding activity changes in an MTA-dependent manner". It can be paraphrased as "a method for treating a disease including administration of an antigen-binding molecule containing a binding domain to a therapeutic subject", or "MTA-dependent antigen-binding activity in the manufacture of a drug for treating a disease”.
- the pharmaceutical composition of the present disclosure can be formulated using a method known to those skilled in the art.
- it can be used parenterally in the form of sterile solutions with water or other pharmaceutically acceptable liquids, or injectable suspensions.
- pharmacologically acceptable carriers or vehicles specifically sterile water or saline, vegetable oils, emulsifiers, suspensions, surfactants, stabilizers, flavoring agents, excipients, vehicles, preservatives. , And the like, can be formulated by mixing in the generally accepted unit dose form required for pharmaceutical practice. The amount of active ingredient in these formulations is set to provide an appropriate volume in the indicated range.
- Aseptic compositions for injection can be formulated according to routine formulation practices using vehicles such as distilled water for injection.
- Aqueous solutions for injection include, for example, saline, isotonic solutions containing glucose and other adjuvants (eg, D-sorbitol, D-mannose, D-mannitol, sodium chloride).
- Suitable solubilizers such as alcohols (ethanol, etc.), polyalcohols (propylene glycol, polyethylene glycol, etc.), nonionic surfactants (polysorbate 80 (TM), HCO-50, etc.) can be used in combination.
- oily liquid examples include sesame oil and soybean oil, and benzyl benzoate and / or benzyl alcohol can also be used as a solubilizing agent. It can also be blended with buffers (eg, phosphate buffers and sodium acetate buffers), soothing agents (eg, prokine hydrochloride), stabilizers (eg, benzyl alcohol and phenol), antioxidants.
- buffers eg, phosphate buffers and sodium acetate buffers
- soothing agents eg, prokine hydrochloride
- stabilizers eg, benzyl alcohol and phenol
- antioxidants antioxidants.
- the prepared injection solution is usually filled in a suitable ampoule.
- the pharmaceutical composition of the present disclosure is preferably administered by parenteral administration.
- parenteral administration for example, an injection type, a nasal administration type, a pulmonary administration type, and a transdermal administration type composition are administered.
- it can be administered systemically or locally by intravenous injection, intramuscular injection, intraperitoneal injection, subcutaneous injection, or the like.
- the administration method can be appropriately selected depending on the patient's age and symptoms.
- the dose of the pharmaceutical composition containing the antigen-binding molecule can be set, for example, in the range of 0.0001 mg to 1000 mg per kg of body weight at a time. Alternatively, for example, a dose of 0.001 to 100,000 mg per patient can be set, but the disclosure is not necessarily limited to these numbers.
- the dose and method of administration vary depending on the weight, age, symptoms, etc. of the patient, but those skilled in the art can set an appropriate dose and method of administration in consideration of these conditions.
- MTA-dependently binding antigen-binding molecules in the present disclosure can be combined with various existing technologies for pharmaceutical applications.
- CAR-T cells chimeric antigen receptor T cells
- One of the non-limiting methods for producing CAR-T is to link an antigen-binding molecule that specifically binds to a tumor-related antigen to the extracellular domain of TCR, and to enhance T cell activation such as CD28.
- a nucleic acid encoding an antigen-binding molecule that binds in an MTA-dependent manner is incorporated into a living body using a virus vector or the like.
- a method of directly expressing the antigen-binding molecule include, but are not limited to, adenovirus.
- nucleic acid encoding an antigen-binding molecule that binds in an MTA-dependent manner into a living body by an electroporation method or a method of directly administering a nucleic acid without using a virus vector, and the antigen-binding molecule. It is also possible to continuously secrete the antigen-binding molecule in the living body by administering the cell genetically modified so as to secrete and express the nucleic acid.
- oligonucleotide refers to a synthetic polynucleotide that is usually single-stranded, usually less than about 200 nucleotides in length, but not necessarily.
- the terms “oligonucleotide” and “polynucleotide” are not mutually exclusive. The above description of polynucleotides is equally and fully applicable to oligonucleotides. In the present specification, “oligonucleotide” and “nucleic acid” are used interchangeably.
- An isolated polynucleotide encoding an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes MTA-dependently is an antigen-binding containing an antigen-binding domain whose antigen-binding activity changes MTA-dependently.
- the polynucleotide refers to one or more polynucleotide molecules encoding the heavy and light chains (or fragments thereof) of the antibody, one vector or separate vectors. Includes polynucleotide molecules on board and polynucleotide molecules present at one or more positions in the host cell.
- vector refers to a nucleic acid molecule to which it can augment another nucleic acid to which it is linked.
- the term includes a vector as a self-replicating nucleic acid structure and a vector that is incorporated into the genome of the host cell into which it has been introduced.
- Certain vectors can result in the expression of nucleic acids to which they are operably linked.
- Such vectors are also referred to herein as "expression vectors.”
- the expression vector can be introduced into the host cell by a method using a virus or an electroporation method, but the introduction of the expression vector is not limited to the introduction in vitro, and the vector can be directly introduced into the living body. It is possible.
- host cell refers to any suitable cell that uses foreign nucleic acid-introduced cells, including progeny of such cells.
- Host cells include “transformants” and “transformants”, which include primary transformants and progeny derived from those cells regardless of the number of passages.
- the offspring do not have to be exactly the same in the content of the parent cell and the nucleic acid and may contain mutations.
- Variant progeny with the same function or biological activity as those used when the original transformed cells were screened or selected are also included herein.
- Antigen-binding molecules can be prepared using recombinant methods and configurations, as described, for example, in US Pat. No. 4,816,567.
- an isolated nucleic acid is provided that encodes an antigen-binding molecule that comprises an antigen-binding domain that changes its antigen-binding activity in an MTA-dependent manner as described herein.
- an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner comprises an antibody variable region
- such nucleic acid is an amino acid sequence containing VL and / or VH of the antibody (eg, light of the antibody). Chains and / or heavy chains) may be encoded.
- one or more vectors containing such nucleic acids are provided.
- a host cell containing such a nucleic acid is provided.
- the host cell comprises (1) a vector containing a nucleic acid encoding an amino acid sequence containing the VL of the antigen binding molecule and an amino acid sequence containing the VH of the antigen binding molecule, or (2) antigen binding. It contains a first vector containing a nucleic acid encoding an amino acid sequence containing the VL of the molecule and a second vector containing a nucleic acid encoding the amino acid sequence containing the VH of the antigen binding molecule (eg, transformed).
- the host cell is eukaryotic (eg, Chinese hamster ovary (CHO) cells) or lymphoid cells (eg, Y0, NS0, Sp2 / 0 cells).
- a host cell containing a nucleic acid encoding the antigen-binding molecule is cultured as described above under conditions suitable for expressing an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- a method of making an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner which comprises recovering the antibody from a host cell (or host cell culture medium). Will be done.
- nucleic acids encoding antigen-binding molecules have been isolated, further cloned and / Alternatively, it is inserted into one or more vectors for expression in host cells.
- nucleic acids will be readily isolated and sequenced using conventional procedures (eg, in the case of antibodies, they can specifically bind to the genes encoding the heavy and light chains of the antibody. By using an oligonucleotide probe).
- Host cells suitable for cloning or expression of a vector encoding an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner include prokaryotic cells or eukaryotic cells described herein.
- the antibody may be produced in bacteria, especially if glycosylation and Fc effector functions are not required. See, for example, US Pat. Nos. 5,648,237, 5,789,199, and 5,840,523 for expression of antibody fragments and polypeptides in bacteria. (In addition, see Charlton, Methods in Molecular Biology, Vol.
- an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner may be isolated from the bacterial cell paste in a soluble fraction and can be further purified.
- filamentous fungi or yeasts including strains of fungi and yeasts whose glycosylation pathways are "humanized", resulting in the production of antigen-binding molecules with partial or complete human glycosylation patterns.
- Eukaryotic microorganisms are suitable cloning or expression hosts for antigen-binding molecular coding vectors. See Gerngross, Nat. Biotech. 22: 1409-1414 (2004) and Li et al., Nat. Biotech. 24: 210-215 (2006).
- invertebrates and vertebrates are also suitable host cells for the expression of glycosylated antigen-binding molecules.
- invertebrate cells include plant and insect cells. Numerous baculovirus strains have been identified for use in conjugation with insect cells, especially for transformation of Spodoptera frugiperda cells.
- Plant cell cultures can also be used as hosts. See, for example, U.S. Pat. Nos. 5,959,177, 6,040,498, 6,420,548, 7,125,978, and 6,417,429 (described PLANTIBODIES TM technology for producing antigen-binding molecules in transgenic plants). ..
- Vertebrate cells can also be used as hosts.
- a mammalian cell line adapted to grow in a suspended state would be useful.
- Another example of a useful mammalian host cell line is the SV40-transformed monkey kidney CV1 strain (COS-7); human fetal kidney strain (Graham et al., J. Gen Virol. 36:59 (1977). ); 293 or 293 cells described in, etc.); Mammal hamster kidney cells (BHK); Mouse cell line cells (TM4 cells described in Mather, Biol. Reprod.
- Monkey kidney cells (CV1) ); African green monkey kidney cells (VERO-76); Human cervical cancer cells (HELA); Canine kidney cells (MDCK); Buffalo rat hepatocytes (BRL 3A); Human lung cells (W138); Human hepatocytes (W138) Hep G2); Mammalian breast cancer (MMT 060562); TRI cells (for example, described in Mather et al., Annals NY Acad. Sci. 383: 44-68 (1982)); MRC5 cells; and FS4 cells.
- Other useful mammalian host cell lines include Chinese hamster ovary (CHO) cells, including DHFR-CHO cells (Urlaub et al., Proc. Natl.
- Y0 Includes myeloma cell lines such as NS0 and Sp2 / 0.
- myeloma cell lines such as NS0 and Sp2 / 0.
- Yazaki and Wu Methods in Molecular Biology, Vol. 248 (BKC Lo, ed., Humana Press, Totowa, NJ), pp. 255 See -268 (2003).
- the example of the host cell is not limited to the animal cell line.
- various cells such as effector cells extracted from a living body and mesenchymal stem cells (MSC) as host cells.
- a method for screening an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner The present disclosure describes a method for screening an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- an antigen-binding domain or an antigen-binding molecule containing the domain
- whose antigen-binding activity changes in an MTA-dependent manner can be obtained.
- some specific examples thereof are illustrated below.
- the binding activity of an antigen-binding domain to an antigen in the presence of a first concentration of MTA and an antigen-binding domain (or including the domain) to an antigen in the presence of a second concentration of MTA.
- the antigen-binding domain or the antigen-binding molecule containing the domain to the antigen in the presence of the first concentration of MTA and the second concentration of MTA. The binding activity of is compared.
- an antigen-binding domain (or an antigen-binding containing the domain) to an antigen in the presence of a first concentration of MTA and a second concentration of MTA.
- Antigen-binding domains with different binding activities (molecules) are selected.
- the first concentration and the second concentration are different concentrations, and either one of these concentrations can be set to 0 (that is, MTA is absent).
- the following acquisition method is exemplified.
- an antigen-binding domain or an antigen-binding molecule containing the domain in which the antigen-binding activity in the presence of a high concentration of MTA is higher than that in the presence of a low concentration of MTA
- the antigen-binding activity at the first concentration is required.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having high antigen-binding activity at the second concentration is selected.
- the second concentration is set to 0 and the first concentration is set.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having a high antigen-binding activity at the second concentration is selected.
- the antigen-binding activity in the presence of the first concentration of MTA is the second.
- the difference between the antigen-binding activity in the presence of the first concentration of MTA and the antigen-binding activity in the presence of the second concentration of MTA is not particularly limited, but is preferably the first.
- the antigen-binding activity in the presence of one concentration of MTA is twice or more, more preferably 10 times or more, and more preferably 40 times or more of the antigen-binding activity in the presence of a second concentration of MTA.
- the upper limit of the difference in the antigen-binding activity is not particularly limited, and may be any value such as 400 times, 1000 times, 10000 times, etc. as long as it can be produced by a person skilled in the art. If no antigen-binding activity is observed in the presence of a second concentration of MTA, this upper limit is an infinite number.
- an antigen-binding domain or an antigen-binding molecule containing the domain whose antigen-binding activity in the presence of a high concentration of MTA is lower than that in the presence of a low concentration of MTA, the antigen-binding activity at the first concentration is required.
- An antigen-binding domain or an antigen-binding molecule containing the domain having low antigen-binding activity at the second concentration is selected.
- the second concentration is set to 0 and the first concentration.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having a low antigen-binding activity at the second concentration is selected.
- the antigen-binding activity in the presence of the first concentration of MTA is the second.
- the difference between the antigen-binding activity in the presence of the first concentration of MTA and the antigen-binding activity in the presence of the second concentration of MTA is not particularly limited, but is preferably the first.
- the antigen-binding activity in the presence of two concentrations of MTA is twice or more, more preferably 10 times or more, and more preferably 40 times or more of the antigen-binding activity in the presence of the first concentration of MTA.
- the upper limit of the difference in the antigen-binding activity is not particularly limited, and may be any value such as 400 times, 1000 times, 10000 times, etc. as long as it can be produced by a person skilled in the art. If no antigen-binding activity is observed in the presence of the first concentration of MTA, this upper limit is an infinite number.
- the expression "the antigen-binding activity in the presence of MTA is higher than the antigen-binding activity in the absence of MTA” means "MTA of an antigen-binding domain (or an antigen-binding molecule containing the domain)".
- the binding activity to the antigen in the absence of MTA is lower than the binding activity to the antigen in the presence of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the absence of MTA is lower than the antigen-binding activity in the presence of MTA” is referred to as "antigen-binding domain".
- the antigen-binding activity of (or the antigen-binding molecule containing the domain) in the absence of MTA is weaker than the antigen-binding activity in the presence of MTA.
- the antigen-binding activity in the presence of the first concentration of MTA is higher than the antigen-binding activity in the presence of the second concentration of MTA
- the first concentration is higher than the second concentration.
- the expression “the antigen-binding activity in the presence of a high concentration of MTA is higher than the antigen-binding activity in the presence of a low concentration of MTA” or “an antigen-binding domain (or an antigen-binding molecule containing the domain)"
- the antigen-binding activity in the presence of a low concentration of MTA is lower than the antigen-binding activity in the presence of a high concentration of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the presence of a low concentration of MTA is lower than the antigen-binding activity in the presence of a high concentration of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the presence of a low concentration of MTA is weaker than the antigen-binding activity in the presence of a high concentration of MTA.
- Conditions other than the concentration of MTA when measuring the binding activity to the antigen can be appropriately selected by those skilled in the art, and are not particularly limited. For example, it can be measured under the condition of HEPES buffer and 37 ° C. For example, it can be measured using Biacore (GE Healthcare) or the like. In the measurement of the binding activity between the antigen-binding domain (or the antigen-binding molecule containing the domain) and the antigen, if the antigen is a soluble molecule, the antigen-binding domain (or the antigen-binding molecule containing the domain) is immobilized.
- low molecular weight compounds include, but are not limited to, S-adenosylmethionine (SAM: S-adenosylmethionine), S-adenosylhomocystein (SAH: S-adenosylhomocystein, S).
- SAM S-adenosylmethionine
- SAH S-adenosylhomocystein
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the presence of a second concentration of MTA, and (c) the antigen binding containing the antigen-binding domain dissociated in the step (b). The step of isolating the molecule, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below;
- the step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the criteria confirmed to bind to the antigen A method for screening an antigen-binding molecule, including
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain capable of binding to an MTA with an antigen in the presence of a first concentration of MTA. (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the presence of a second concentration of MTA, and (c) the antigen binding containing the antigen-binding domain dissociated in the step (b). The step of isolating the molecule, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below;
- the step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the criteria confirmed to bind to the antigen A method for screening an antigen-binding molecule, including
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding molecule containing an antigen-binding domain that does not bind to the antigen to the antigen in the presence of a second concentration of MTA, and (d) a step of binding the antigen-binding domain to the antigen in the step (c). The step of isolating the antigen-binding molecule, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting a library displaying an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the presence of a second concentration of MTA, and (c) the antigen binding containing the antigen-binding domain dissociated in the step (b). The step of isolating the molecule, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below;
- the present disclosure describes steps (1) to (c) below: (1) A step of contacting a library in which an antigen-binding molecule containing an antigen-binding domain is displayed with an MTA, and (2) a step of selecting an antigen-binding molecule containing an antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying an antigen-binding molecule containing the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of a first concentration of MTA.
- step (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the presence of a second concentration of MTA, and (c) the antigen binding containing the antigen-binding domain dissociated in the step (b).
- the step of isolating the molecule Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (1) to (d) below: (1) A step of contacting a library in which an antigen-binding molecule containing an antigen-binding domain is displayed with an MTA, and (2) a step of selecting an antigen-binding molecule containing an antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying an antigen-binding molecule containing the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain bound to an antigen in the step (a).
- step (c) The step of placing the antigen-binding molecule containing the antigen-binding domain selected in the step (b) in the presence of a second concentration of MTA, and (d) the step (d) of the antigen-binding activity in the step (b). ), The step of isolating the antigen-binding molecule containing an antigen-binding domain weaker than the criteria selected in.
- a method for screening an antigen-binding molecule containing The step of placing the antigen-binding molecule containing the antigen-binding domain selected in the step (b) in the presence of a second concentration of MTA, and (d) the step (d) of the antigen-binding activity in the step (b).
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen with a library in which an antigen-binding molecule containing an antigen-binding domain is displayed in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain that does not bind to an antigen in the step (a). (c) The step of binding the antigen-binding molecule containing the antigen-binding domain selected in the step (b) to the antigen in the presence of a second concentration of MTA, and (d) the antigen bound to the antigen in the step (c). The step of isolating the antigen-binding molecule containing the binding domain, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the absence of MTA, and (c) the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b).
- Isolation step A method for screening an antigen-binding molecule, including
- the present disclosure describes steps (a)-(d) below; (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain has bound to the antigen in the step (a). (c) The step of placing the antigen-binding molecule containing the antigen-binding domain bound to the antigen in the absence of MTA, and (d) the antigen-binding activity in the step (c) to the antigen in the step (b).
- a method for screening an antigen-binding molecule which comprises a step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the criteria confirmed to be bound.
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain capable of binding to an MTA with an antigen in the presence of an MTA. (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the absence of MTA, and (c) the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b). Isolation step, A method for screening an antigen-binding molecule, including
- the present disclosure describes steps (a)-(d) below;
- a method for screening an antigen-binding molecule which comprises a step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the criteria confirmed to be bound.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of confirming that the antigen-binding molecule containing the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding molecule containing an antigen-binding domain that does not bind to the antigen to the antigen in the absence of MTA, and (d) an antigen-binding containing the antigen-binding domain bound to the antigen in the step (c). The step of isolating the molecule, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(c) below: (a) A step of contacting a library displaying an antigen-binding molecule containing an antigen-binding domain with an antigen in the presence of an MTA. (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the absence of MTA, and (c) the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b). Isolation step, Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below;
- the step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the selected criteria Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (1) to (c) below: (1) A step of contacting a library in which an antigen-binding molecule containing an antigen-binding domain is displayed with an MTA, and (2) a step of selecting an antigen-binding molecule containing an antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying an antigen-binding molecule containing the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of an MTA.
- step (b) The step of placing the antigen-binding molecule containing the antigen-binding domain bound in the step (a) in the absence of MTA, and (c) the antigen-binding molecule containing the antigen-binding domain dissociated in the step (b).
- Isolation step Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (1) to (d) below: (1) A step of contacting a library in which an antigen-binding molecule containing an antigen-binding domain is displayed with an MTA, and (2) a step of selecting an antigen-binding molecule containing an antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying an antigen-binding molecule containing the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of an MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain bound to an antigen in the step (a).
- step (c) The step of placing the antigen-binding molecule containing the antigen-binding domain selected in the step (b) in the absence of MTA, and (d) the antigen-binding activity in the step (c) in the step (b).
- step of isolating an antigen-binding molecule containing an antigen-binding domain that is weaker than the selected criteria Provided is a method for screening an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen with a library in which an antigen-binding molecule containing an antigen-binding domain is displayed in the presence of an MTA. (b) The step of selecting an antigen-binding molecule containing an antigen-binding domain that does not bind to an antigen in the step (a). (c) The step of binding the antigen-binding molecule containing the antigen-binding domain selected in the step (b) to the antigen in the absence of MTA, and (d) the antigen-binding domain bound to the antigen in the step (c). The step of isolating the antigen-binding molecule containing Provided is a method for screening an antigen-binding molecule containing.
- a naive human antibody display library As a library in which an antigen-binding molecule containing an antigen-binding domain is displayed, a naive human antibody display library, a synthetic human antibody display library, or a library completed by the present disclosure described later can be used.
- the antigen-binding domain (or antigen-binding molecule containing the domain) of the present disclosure screened by the screening method may be any antigen-binding domain (or antigen-binding molecule), for example, the above-mentioned antigen-binding domain (or antigen-binding molecule). It is possible to screen. For example, an antigen-binding domain (or antigen-binding molecule) having a natural sequence may be screened, or an antigen-binding domain (or antigen-binding molecule) in which an amino acid sequence has been substituted may be screened.
- One aspect of the present disclosure is a method for producing an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner.
- a method for producing an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner will provide a.
- an antigen-binding domain (or an antigen-binding molecule containing the domain) whose antigen-binding activity changes in an MTA-dependent manner can be obtained.
- some specific examples thereof are illustrated below.
- the binding activity of an antigen-binding domain to an antigen in the presence of a first concentration of MTA and an antigen-binding domain (or including the domain) to an antigen in the presence of a second concentration of MTA.
- the antigen-binding domain or the antigen-binding molecule containing the domain to the antigen in the presence of the first concentration of MTA and the second concentration of MTA. The binding activity of is compared.
- an antigen-binding domain (or an antigen-binding containing the domain) to an antigen in the presence of a first concentration of MTA and a second concentration of MTA.
- Antigen-binding domains with different binding activities (molecules) are selected.
- the first concentration and the second concentration are different concentrations, and either one of these concentrations can be set to 0 (that is, MTA is absent).
- the following acquisition method is exemplified.
- an antigen-binding domain or an antigen-binding molecule containing the domain in which the antigen-binding activity in the presence of a high concentration of MTA is higher than that in the presence of a low concentration of MTA
- the antigen-binding activity at the first concentration is required.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having high antigen-binding activity at the second concentration is selected.
- the second concentration is set to 0 and the first concentration.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having a high antigen-binding activity at the second concentration is selected.
- the antigen-binding activity in the presence of the first concentration of MTA is the second.
- the difference between the antigen-binding activity in the presence of the first concentration of MTA and the antigen-binding activity in the presence of the second concentration of MTA is not particularly limited, but is preferably the first.
- the antigen-binding activity in the presence of one concentration of MTA is twice or more, more preferably 10 times or more, and more preferably 40 times or more of the antigen-binding activity in the presence of a second concentration of MTA.
- the upper limit of the difference in the antigen-binding activity is not particularly limited, and may be any value such as 400 times, 1000 times, 10000 times, etc. as long as it can be produced by a person skilled in the art. If no antigen-binding activity is observed in the presence of a second concentration of MTA, this upper limit is an infinite number.
- an antigen-binding domain or an antigen-binding molecule containing the domain whose antigen-binding activity in the presence of a high concentration of MTA is lower than that in the presence of a low concentration of MTA, the antigen-binding activity at the first concentration is required.
- An antigen-binding domain or an antigen-binding molecule containing the domain having low antigen-binding activity at the second concentration is selected.
- the second concentration is set to 0 and the first concentration.
- An antigen-binding domain (or an antigen-binding molecule containing the domain) having a low antigen-binding activity at the second concentration is selected.
- the antigen-binding activity in the presence of the first concentration of MTA is the second.
- the difference between the antigen-binding activity in the presence of the first concentration of MTA and the antigen-binding activity in the presence of the second concentration of MTA is not particularly limited, but is preferably the first.
- the antigen-binding activity in the presence of two concentrations of MTA is twice or more, more preferably 10 times or more, and more preferably 40 times or more of the antigen-binding activity in the presence of the first concentration of MTA.
- the upper limit of the difference in the antigen-binding activity is not particularly limited, and may be any value such as 400 times, 1000 times, 10000 times, etc. as long as it can be produced by a person skilled in the art. If no antigen-binding activity is observed in the presence of the first concentration of MTA, this upper limit is an infinite number.
- MTA-dependent by culturing cells into which a vector operably linked with a polynucleotide encoding an antigen-binding molecule containing a selected antigen-binding domain has been introduced and recovering the antigen-binding molecule from the cell culture medium. It is possible to produce an antigen-binding molecule containing an antigen-binding domain whose antigen-binding activity changes.
- the expression "the antigen-binding activity in the presence of MTA is higher than the antigen-binding activity in the absence of MTA” means "MTA of an antigen-binding domain (or an antigen-binding molecule containing the domain)".
- the binding activity to the antigen in the absence of MTA is lower than the binding activity to the antigen in the presence of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the absence of MTA is lower than the antigen-binding activity in the presence of MTA” is referred to as "antigen-binding domain".
- the antigen-binding activity of (or the antigen-binding molecule containing the domain) in the absence of MTA is weaker than the antigen-binding activity in the presence of MTA.
- the antigen-binding activity in the presence of the first concentration of MTA is higher than the antigen-binding activity in the presence of the second concentration of MTA
- the first concentration is higher than the second concentration.
- the expression “the antigen-binding activity in the presence of a high concentration of MTA is higher than the antigen-binding activity in the presence of a low concentration of MTA” or “an antigen-binding domain (or an antigen-binding molecule containing the domain)"
- the antigen-binding activity in the presence of a low concentration of MTA is lower than the antigen-binding activity in the presence of a high concentration of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the presence of a low concentration of MTA is lower than the antigen-binding activity in the presence of a high concentration of MTA.
- the antigen-binding activity of an antigen-binding domain (or an antigen-binding molecule containing the domain) in the presence of a low concentration of MTA is weaker than the antigen-binding activity in the presence of a high concentration of MTA.
- Conditions other than the concentration of MTA when measuring the binding activity to the antigen can be appropriately selected by those skilled in the art, and are not particularly limited. For example, it can be measured under the condition of HEPES buffer and 37 ° C. For example, it can be measured using Biacore (GE Healthcare) or the like. In the measurement of the binding activity between the antigen-binding domain (or the antigen-binding molecule containing the domain) and the antigen, if the antigen is a soluble molecule, the antigen-binding domain (or the antigen-binding molecule containing the domain) is immobilized.
- low molecular weight compounds include, but are not limited to, S-adenosylmethionine (SAM: S-adenosylmethionine), S-adenosylhomocystein (SAH: S-adenosylhomocystein, S).
- SAM S-adenosylmethionine
- SAH S-adenosylhomocystein
- the present disclosure describes steps (a)-(d) below: (a) The step of contacting the antigen binding domain with the antigen in the presence of the first concentration of MTA, (b) The step of placing the antigen-binding domain bound in the above step (a) in the presence of a second concentration of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated.
- a step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium.
- Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) The step of contacting the antigen binding domain with the antigen in the presence of the first concentration of MTA, (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a). (c) The step of placing the antigen-binding domain bound to the antigen in the presence of a second concentration of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b) in the step (c), and (e) in the step (d).
- Process Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen-binding domain capable of binding an MTA with an antigen in the presence of a first concentration of MTA. (b) The step of placing the antigen-binding domain bound in the above step (a) in the presence of a second concentration of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated. A step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium. Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) A step of contacting an antigen-binding domain capable of binding an MTA with an antigen in the presence of a first concentration of MTA. (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a). (c) The step of placing the antigen-binding domain bound to the antigen in the presence of a second concentration of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b) in the step (c), and (e) in the step (d).
- Process Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below: (a) The step of contacting the antigen binding domain with the antigen in the presence of the first concentration of MTA, (b) The step of confirming that the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding domain that does not bind to the antigen to the antigen in the presence of a second concentration of MTA. (D) The step of isolating the antigen-binding domain bound to the antigen in the step (c), and (e) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in the step (d).
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of placing the antigen-binding domain bound in the above step (a) in the presence of a second concentration of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated. A step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium. Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of a first concentration of MTA. (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the presence of a second concentration of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in the step (b) in the step (c), and (e) the antigen binding isolated in the step (d).
- a step of culturing a cell into which a vector in which a polynucleotide encoding an antigen-binding molecule containing a domain is operably linked has been introduced, and recovering the antigen-binding molecule containing the antigen-binding domain from the cell culture medium.
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes the following steps (1) to (d): (1) The step of contacting the library in which the antigen-binding domain is displayed with the MTA, and (2) the step of selecting the antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting the library displaying the antigen-binding domain selected in the steps (1) and (2) with the antigen in the presence of the first concentration of MTA.
- step (b) The step of placing the antigen-binding domain bound in the above step (a) in the presence of a second concentration of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated.
- a step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium.
- Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes the following steps (1) to (e): (1) The step of contacting the library in which the antigen-binding domain is displayed with the MTA, and (2) the step of selecting the antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting the library displaying the antigen-binding domain selected in the steps (1) and (2) with the antigen in the presence of the first concentration of MTA.
- step (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the presence of a second concentration of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in the step (b) in the step (c), and (e) the antigen binding isolated in the step (d).
- a step of culturing a cell into which a vector in which a polynucleotide encoding an antigen-binding molecule containing a domain is operably linked has been introduced, and recovering the antigen-binding molecule containing the antigen-binding domain from the cell culture medium.
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (a)-(e) below: (a) A step of contacting a library in which an antigen-binding domain is displayed in the presence of a first concentration of MTA with an antigen. (b) The step of selecting an antigen-binding domain that does not bind to an antigen in the step (a). (c) The step of binding the antigen-binding domain selected in step (b) to the antigen in the presence of a second concentration of MTA. (D) The step of isolating the antigen-binding domain bound to the antigen in the step (c), and (e) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in the step (d).
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (a)-(d) below: (a) The step of contacting the antigen binding domain with the antigen in the presence of the MTA, (b) The step of placing the antigen-binding domain bound in the above step (a) in the absence of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated.
- a step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium.
- Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) The step of contacting the antigen binding domain with the antigen in the presence of the MTA, (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a). (c) The step of placing the antigen-binding domain bound to the antigen in the absence of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b) in the step (c), and (e) in the step (d).
- Process Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting an antigen-binding domain capable of binding to an MTA with an antigen in the presence of an MTA, (b) The step of placing the antigen-binding domain bound in the above step (a) in the absence of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated. A step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium. Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) A step of contacting an antigen-binding domain capable of binding to an MTA with an antigen in the presence of an MTA, (b) The step of confirming that the antigen-binding domain is bound to the antigen in the step (a). (c) The step of placing the antigen-binding domain bound to the antigen in the absence of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criterion confirmed to be bound to the antigen in the step (b) in the step (c), and (e) in the step (d).
- Process Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below: (a) The step of contacting the antigen binding domain with the antigen in the presence of the MTA, (b) The step of confirming that the antigen-binding domain does not bind to the antigen in the step (a). (c) A step of binding an antigen-binding domain that does not bind to the antigen to the antigen in the absence of MTA. (D) The step of isolating the antigen-binding domain bound to the antigen in the step (c), and (e) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in the step (d).
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (a)-(d) below: (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of placing the antigen-binding domain bound in the above step (a) in the absence of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated. A step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium. Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (a)-(e) below; (a) A step of contacting a library displaying an antigen-binding domain with an antigen in the presence of an MTA, (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the absence of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in the step (b) in the step (c), and (e) the antigen binding isolated in the step (d).
- a step of culturing a cell into which a vector in which a polynucleotide encoding an antigen-binding molecule containing a domain is operably linked has been introduced, and recovering the antigen-binding molecule containing the antigen-binding domain from the cell culture medium.
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (1) to (d) below: (1) The step of contacting the library in which the antigen-binding domain is displayed with the MTA, and (2) the step of selecting the antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of an MTA.
- step (b) The step of placing the antigen-binding domain bound in the above step (a) in the absence of MTA, The steps of (c) isolating the antigen-binding domain dissociated in step (b) and (d) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in step (c) can be activated.
- a step of culturing a cell into which a vector linked to is introduced and recovering an antigen-binding molecule containing an antigen-binding domain from the cell culture medium.
- Provided is a method for producing an antigen-binding molecule containing.
- the present disclosure describes steps (1) to (e) below: (1) The step of contacting the library in which the antigen-binding domain is displayed with the MTA, and (2) the step of selecting the antigen-binding domain bound to the MTA in the above step (1). (a) A step of contacting a library displaying the antigen-binding domain selected in steps (1) and (2) with an antigen in the presence of an MTA.
- step (b) The step of selecting an antigen-binding domain bound to an antigen in the above step (a), (c) The step of placing the antigen-binding domain selected in step (b) in the absence of MTA, (d) The step of isolating the antigen-binding domain whose antigen-binding activity is weaker than the criteria selected in the step (b) in the step (c), and (e) the antigen binding isolated in the step (d).
- a step of culturing a cell into which a vector in which a polynucleotide encoding an antigen-binding molecule containing a domain is operably linked has been introduced, and recovering the antigen-binding molecule containing the antigen-binding domain from the cell culture medium.
- a method for producing an antigen-binding molecule containing is provided.
- the present disclosure describes steps (a)-(e) below: (a) A step of contacting a library in which an antigen-binding domain is displayed in the presence of an MTA with an antigen. (b) The step of selecting an antigen-binding domain that does not bind to an antigen in the step (a). (c) The step of binding the antigen-binding domain selected in step (b) to the antigen in the absence of MTA, (D) The step of isolating the antigen-binding domain bound to the antigen in the step (c), and (e) the polynucleotide encoding the antigen-binding molecule containing the antigen-binding domain isolated in the step (d).
- a method for producing an antigen-binding molecule containing is provided.
- a naive human antibody display library As the library in which the antigen-binding domain is displayed, a naive human antibody display library, a synthetic human antibody display library, or a library completed by the present disclosure described later can be used.
- the polynucleotide encoding the antigen-binding domain is derived from a virus such as a phage, as described in Examples described later. Isolated by normal gene amplification.
- the antigen-binding domain or antibody thus isolated is selected from the culture medium of a cell such as a hybridoma, the antibody gene or the like is a normal gene from the cell as shown in the above-mentioned antibody section. Isolated by amplification.
- the antigen-binding molecule of the present disclosure produced by the above-mentioned production method may be any antigen-binding molecule, for example, an antigen-binding molecule having a natural sequence may be produced, or an antigen-binding molecule in which an amino acid sequence is substituted may be produced. You may.
- a "library” is a nucleic acid or polynucleotide that encodes an antigen-binding molecule that contains multiple antigen-binding domains that are different in sequence from each other, and / and an antigen-binding molecule that contains multiple antigen-binding domains that are different in sequence from each other.
- the sequence of the nucleic acid encoding the antigen-binding molecule containing the antigen-binding domain or / and the antigen-binding molecule contained in the library is not a single sequence, but multiple antigen-binding molecules or / having different sequences from each other. And nucleic acids encoding multiple antigen-binding molecules with different sequences from each other.
- the antigen-binding domain of the present disclosure contains at least one amino acid residue that changes the antigen-binding activity in an MTA-dependent manner. It can be obtained from a library consisting primarily of nucleic acids encoding multiple antigen-binding molecules that are different in sequence from each other and / and antigen-binding molecules that contain multiple antigen-binding domains that are different in sequence from each other. In the following, a plurality of antigen-binding molecules having at least one amino acid residue that changes the antigen-binding activity in an MTA-dependent manner in the antigen-binding domain, or / and a plurality of antigen-binding molecules having different sequences from each other. Illustrated are libraries consisting primarily of nucleic acids encoding antigen-binding molecules containing domains.
- An antigen-binding molecule library containing an antigen-binding domain having an amino acid residue that changes the antigen-binding activity in an MTA-dependent manner includes an antigen-binding domain in which the antigen-binding activity changes in an MTA-dependent manner and such an antigen-binding domain. It is possible to efficiently obtain an antigen-binding molecule.
- an MTA switch antigen-binding domain that is, an antigen-binding molecule containing an antigen-binding domain that binds to a target antigen in the presence of MTA but not to the target antigen in the absence of MTA, or a high concentration.
- a library capable of efficiently obtaining an antigen-binding molecule containing an antigen-binding domain that binds to a target antigen in the presence of MTA but does not bind to the target antigen in the presence of a low concentration of MTA, but also MTA reverse switch antigen binding.
- a fusion polypeptide of the antigen-binding molecule of the present disclosure and a heterologous polypeptide can be prepared.
- the fusion polypeptide is fused with at least a portion of a virus coat protein selected from the group consisting of virus coat proteins such as pIII, pVIII, pVII, pIX, Soc, Hoc, gpD, pVI and variants thereof. obtain.
- the antigen-binding molecules of the present disclosure can be ScFv, Fab fragments, F (ab) 2 or F (ab') 2 , and thus in another embodiment they are heterologous to these antigen-binding molecules.
- these antigen-binding molecules and viral coat proteins such as at least a portion of the viral coat proteins selected from the group consisting of pIII, pVIII, pVII, pIX, Soc, Hoc, gpD, pVI and variants thereof.
- the antigen-binding molecules of the present disclosure may further comprise a dimerization domain.
- the dimerization domain may be present between the variable region of the heavy or light chain of the antibody and at least a portion of the viral coat protein.
- the dimerization domain may include at least one of the dimerization sequences and / or a sequence containing one or more cysteine residues. This dimerization domain can preferably be linked to the C-terminus of the heavy chain variable region or constant region.
- the dimerization domain is created by whether the antibody variable region is made as a fusion polypeptide component with the coat protein component of the virus (no amber stop codon after the dimerization domain), or It is possible to take various structures depending on whether the antibody variable region is prepared mainly containing no virus-coated protein component (for example, having an amber stop codon after the dimerization domain). ..
- the antibody variable region is made primarily as a fusion polypeptide with a viral coat protein component, divalent presentation is provided by one or more disulfide bonds and / or a single dimerized sequence.
- a library having a diversity of 1.2 ⁇ 10 8 or more that is, an antigen-binding molecule containing a plurality of antigen-binding domains having a diversity of 1.2 ⁇ 10 8 or more and different sequences from each other.
- the term "antigen-binding molecules containing a plurality of antigen-binding domains having different sequences from each other” as used in the description of "different sequences from each other” means that the sequences of individual antigen-binding molecules in the library are different from each other. Means that. That is, the number of sequences that differ from each other in the library reflects the number of independent clones of the sequences that differ from each other in the library, and is sometimes referred to as "library size". It is 10 6 to 10 12 in a normal phage display library, and the library size can be expanded to 10 14 by applying a known technique such as the ribosome display method.
- the actual number of phage particles used during the panning selection of the phage library is usually 10 to 10,000 times larger than the library size.
- This excess multiple also referred to as the "library equivalent” indicates that there can be 10 to 10,000 individual clones with the same amino acid sequence. Therefore, the term "different sequences from each other" in the present disclosure means that the sequences of individual antigen-binding molecules in the library excluding the library equivalent number are different from each other, and more specifically, antigen-binding molecules having different sequences from each other. It means that there are 10 6 to 10 14 molecules, preferably 10 7 to 10 12 molecules, more preferably 10 8 to 10 11 , and particularly preferably 10 8 to 10 10 .
- a library mainly composed of a nucleic acid encoding an antigen-binding molecule containing a plurality of antigen-binding domains having different sequences from each other and / or an antigen-binding molecule containing a plurality of antigen-binding domains having different sequences from each other.
- the term "plurality”, for example, an antigen-binding molecule, fusion polypeptide, polynucleotide molecule, vector or virus of the present disclosure usually refers to a set of two or more types of the substance. For example, if two or more substances differ from each other with respect to a particular trait, it means that there are two or more types of the substance.
- An example may be a variant in which an amino acid mutation is observed at a specific amino acid site in the amino acid sequence.
- an amino acid mutation is observed at a specific amino acid site in the amino acid sequence.
- there are two or more antigen-binding molecules having substantially the same, preferably the same sequence, except for the amino acid at a specific amino acid site there are a plurality of antigen-binding molecules.
- there are multiple polynucleotide molecules if there are two or more polynucleotide molecules that are substantially the same, preferably the same sequence, except for the base encoding the amino acid at a particular amino acid site.
- a library mainly composed of a nucleic acid encoding an antigen-binding molecule containing a plurality of antigen-binding domains having different sequences from each other and / and an antigen-binding molecule containing a plurality of antigen-binding domains having a different sequence from each other.
- the term "mainly from” reflects the number of antigen-binding molecules in the library that differ in their antigen-binding activity to the antigen in an MTA-dependent manner, out of the number of independent clones with different sequences.
- the antigen-binding molecules that exhibit such binding activity is present at least 10 4 molecules in the library.
- the antigen binding domains of this disclosure such antigen binding molecules exhibiting binding activity may be obtained from at least 10 5 molecules library found. More preferably, the antigen binding domains of this disclosure such antigen binding molecules exhibiting binding activity may be obtained from at least 10 6 molecules library found. Particularly preferably, the antigen-binding domain of the present disclosure can be obtained from a library in which at least 10 7 antigen-binding molecules exhibiting such binding activity are present. Also preferably, the antigen binding domains of this disclosure such antigen binding molecules exhibiting binding activity may be obtained from at least 10 8 molecules library found.
- the antigen-binding domains of the present disclosure are 10-6 % to 80%, preferably 10-5 %, of the number of independent clones in the library in which antigen-binding molecules exhibiting such binding activity have different sequences.
- the number of molecules or the ratio in the whole molecule can be expressed as described above.
- a virus in the case of a virus, as in the above, it can be expressed by the number of virus individuals or the ratio in the whole individual.
- the antigen-binding domain of the present disclosure can be obtained from a library in which at least 10 antigen-binding molecules exhibiting such binding activity are present.
- the antigen-binding domain of the present disclosure can be obtained from a library in which at least 100 antigen-binding molecules exhibiting such binding activity are present.
- the antigen-binding domain of the present disclosure can be obtained from a library in which at least 1000 antigen-binding molecules exhibiting such binding activity are present.
- the disclosure provides a library containing nucleic acids encoding multiple antibody variable regions and / and antibody variable regions.
- H-CDR1 containing XAXWMC (SEQ ID NO: 65); H-CDR2 containing CIFAXXXYXXSGGSTYYASWAKG (SEQ ID NO: 66); H-CDR3 containing GXGXXXGXXDEL (SEQ ID NO: 67); L-CDR1 containing QSSEXVXXXXLS (SEQ ID NO: 68); L-CDR2 containing XAXTXPX (SEQ ID NO: 69); and L-CDR3 containing AGLYXGNIPA (SEQ ID NO: 70); It is an antibody variable region containing.
- X refers to an arbitrary amino acid, and X existing at different positions may or may not be an amino acid of the same type.
- H-CDR1 containing XAXWMC (SEQ ID NO: 65); H-CDR2 containing CIFAX 1 XXYXXSGGSTYYASWAKG (SEQ ID NO: 71); H-CDR3 containing GXGXXXGXXDEL (SEQ ID NO: 67); L-CDR1 containing QSSEXVXXX 1 XLS (SEQ ID NO: 72); L-CDR2 containing XAXTXPX (SEQ ID NO: 69); and L-CDR3 containing AGLYXGNIPA (SEQ ID NO: 70); It is an antibody variable region containing.
- X is an arbitrary amino acid
- X 1 is an amino acid selected from A, D, E, G, H, I, K, L, M, N, P, Q, R, S, T and V.
- X or X 1 existing at different positions may be the same kind of amino acid, and may not be the same kind of amino acid.
- H-CDR1 containing XXAXWMC (SEQ ID NO: 73); H-CDR2 containing CIFAX 1 XXYXXSGGSTYYASWAKG (SEQ ID NO: 71); H-CDR3 containing GXGXXXGXXDEL (SEQ ID NO: 67); L-CDR1 containing QSSEXVXXX 1 XLS (SEQ ID NO: 72); L-CDR2 containing XAXTXPX (SEQ ID NO: 69); and L-CDR3 containing AGLYXGNIPA (SEQ ID NO: 70); It is an antibody variable region containing.
- X is an arbitrary amino acid
- X 1 is an amino acid selected from A, D, E, G, H, I, K, L, M, N, P, Q, R, S, T and V.
- X or X 1 existing at different positions may be the same kind of amino acid, and may not be the same kind of amino acid.
- H-CDR1 containing XXXWMC (SEQ ID NO: 74); H-CDR2 containing CIXSXXXXTXYASWVNG (SEQ ID NO: 75); H-CDR3 containing EXXXXSGALNL (SEQ ID NO: 76); L-CDR1 containing HSSKXVXXXXLA (SEQ ID NO: 77); L-CDR2 containing XAXXLAS (SEQ ID NO: 78); and L-CDR3 containing QGTYXXXXFYFA (SEQ ID NO: 79); It is an antibody variable region containing.
- X refers to an arbitrary amino acid, and X existing at different positions may or may not be an amino acid of the same type.
- H-CDR1 containing X 2 X 3 X 4 X 5 G (SEQ ID NO: 80); H-CDR2 containing X 6 IGX 7 X 8 X 9 X 10 X 11 WX 12 PX 13 WVKX 14 (SEQ ID NO: 81); H-CDR3 including GX 15 X 16 X 17 X 18 X 19 X 20 NAX 21 DP (SEQ ID NO: 82); L-CDR1 containing QSSQSVX 22 X 23 NNX 24 LS (SEQ ID NO: 83); L-CDR2 containing DASTLAS (SEQ ID NO: 84); and L-CDR3 containing HGX 25 X 26 X 27 X 28 X 29 X 30 X 31 X 32 DNX 33 (SEQ ID NO: 85); Is an antibody variable
- X 3 is an amino acid selected from D, E, F, H, K, N, P, R and Y
- X 4 is an amino acid selected from A, I, P, T and V
- X 5 is an amino acid selected from A, E, F, H, I, K, L, M, N, Q, S, T, V, W and Y
- X 6 is an amino acid selected from D, I and V
- X 7 is an amino acid selected from A, D, E, G, I, K, Q and R
- X 8 is an amino acid selected from D, E, F, G, H, I, K, L, P, Q, R, S, T, V, W and Y.
- X 9 is an amino acid selected from A, D, E, F, G, H and S
- X 10 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, Q, R, S, T, V, W and Y
- X 11 is an amino acid selected from A, D, E, G, H, I, K, L, N, P, Q, R, S, T and V
- X 12 is an amino acid selected from A, D, E, F, G, H, I, K, L, Q, R, S, T, V, W and Y.
- X 13 is an amino acid selected from A, F, Q, R, S, T, V, W and Y
- X 14 is an amino acid selected from A, F and G
- X 15 is an amino acid selected from A, E, F, G, H, K, L, Q, R, S, T, W and Y
- X 16 is an amino acid selected from F, H, K, N, W and Y
- X 17 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 18 is an amino acid selected from A, D, E, G, H, Q and S
- X 19 is an amino acid selected from F and Y
- X 20 is an amino acid selected from N, T and V
- X 21 is an amino acid selected from F and W
- X 22 is an amino acid selected from A, E, F, H, I, K, L, N, R, S, T, V, W and Y
- X 23 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 24 is an amino acid selected from A, E, F, G, H, S and Y
- X 25 is an amino acid selected from A, S and T
- X 26 is an amino acid selected from A, D, E, F, G, H, I, K, L, M, N, Q, R, S, T, V, W and Y
- X 27 is an amino acid selected from A, D, E, F, G, H, L, N, Q, R, S, T, V and Y
- X 28 is an amino acid selected from A, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y.
- X 29 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W, Y
- X 30 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, V, W and Y
- X 31 is an amino acid selected from A, D, E, F, G, H, I, K, L, N, P, Q, R, S, T, V, W and Y
- X 32 is an amino acid selected from A, F, H, I, K, L, N, P, Q, R, S, T, V, W and Y
- X 33 is an amino acid chosen from A and G.
- the antigen-binding domain in an antigen-binding molecule library containing an amino acid residue having an amino acid residue that changes the antigen-binding activity in an MTA-dependent manner is an antibody variable region and is included in the library.
- the antigen-binding domain contained in the "plurality of antigen-binding molecules" encoded by the nucleic acid contained in the library is an amino acid located at one or more amino acid sites in an antibody variable region unmodified body having MTA-binding activity. Contains multiple antibody variable region variants having different amino acids and different sequences from each other.
- the antibody variable region unmodified form is an antibody variable region having binding activity to MTA.
- the unmodified antibody variable region of the present disclosure may be an antibody variable region that does not substantially bind to adenosine and / and S- (5'-Adenosyl) -L-homocysteine (SAH).
- the antibody variable region unmodified form of the present disclosure may be an antibody variable region having binding activity to adenosine.
- the antibody variable region unmodified form of the present disclosure may be an antibody variable region having binding activity to (5'-Adenosyl) -L-homocysteine (SAH), AMP, ADP and / and ATP.
- the antibody variable region unmodified form having the binding activity to the MTA can be obtained from the antibody having the binding activity to the MTA.
- a "template” or “template” can be used as a starting molecule when designing an antigen-binding molecule library containing an antigen-binding domain having an amino acid residue that changes its antigen-binding activity in an MTA-dependent manner. It may be called.
- the antibody variable region unmodified product of the present disclosure may be any of the following: a) Antibody variable region containing the heavy chain variable region shown by SEQ ID NO: 46 and the light chain variable region shown by SEQ ID NO: 47; b) Antibody variable region containing the heavy chain variable region represented by SEQ ID NO: 50 and the light chain variable region represented by SEQ ID NO: 51; c) Antibody variable region containing the heavy chain variable region shown by SEQ ID NO: 48 and the light chain variable region shown by SEQ ID NO: 49; d) Antibody variable region containing the heavy chain variable region shown by SEQ ID NO: 52 and the light chain variable region shown by SEQ ID NO: 53; e) An antibody variable region having a heavy chain variable region shown by SEQ ID NO: 31 and a light chain variable region shown by SEQ ID NO: 32.
- these antigen-binding domains and the like may be prepared in any way, and the antigen-binding that exists in advance.
- Domains or antibodies, pre-existing libraries (such as phage libraries), antibodies or libraries made from hybridomas obtained from immunization of animals or B cells from immunized animals, such as, but not limited to. Made from immune cells such as animal B cells immunized with a conjugate with an adjuvanted agent such as a highly immunogenic T cell epitope peptide that is properly linked to a small molecule compound containing MTA. It is possible to use the antibody or library.
- T cell epitope peptide As a non-limiting example of the T cell epitope peptide, a p30 helper peptide derived from Tetanus toxin (represented by SEQ ID NO: 4 and also referred to as Fragment C (FrC)) and the like are preferably mentioned.
- Fragment C Fragment C
- an antigen-binding domain containing a variable region of a heavy chain and a light chain of an antibody can be mentioned.
- antigen-binding domains are "scFv (single chain Fv)", “single chain antibody”, “Fv”, “scFv2 (single chain Fv2)", “Fab” or "F (F)”.
- scFv single chain Fv
- Fv single chain antibody
- scFv2 single chain Fv2
- Fab or "F (F)
- “Ab') 2" or IgG is preferably mentioned, and a library composed of these can also be used.
- the above-mentioned library is used to have binding activity to a small molecule compound by panning.
- Techniques for preparing antigen-binding domains or antigen-binding molecules can be used.
- the library is not limited to this, but is limited to a phage display library, a ribosome display library, an mRNA display library, a cDNA display library, a CIS display library, an E. coli display library, a gram-positive bacterium display library, a yeast display library, and a mammalian cell display library.
- Virus display library, in vitro virus display library, etc. can be used.
- a low-molecular-weight compound immobilized on a carrier such as beads can be used.
- Immobilized low-molecular-weight compounds are a method in which a low-molecular-weight compound synthesized in a state of being chemically connected to biotin via a linker is brought into contact with beads or plates on which streptavidin or neutravidin is immobilized.
- a low molecular weight compound covalently linked to an adjuvant such as bovine serum albumin (BSA) can be produced by, but is not limited to, a method of adhering to a bead or a plate by a hydrophobic interaction. These methods are already known (J Immunol Methods. 2003 Sep; 280 (1-2): 139-55, BMC Biotechnol. 2009 Jan 29; 9: 6. doi: 10.1186 / 1472-6750-9-6). ..
- an antigen-binding domain or antigen-binding molecule having binding activity to the low-molecular-weight compound is prepared. It becomes possible to do.
- a fluorescent-labeled low-molecular compound or a biotin-labeled low-molecular compound can be used as another aspect of the method for preparing an antigen-binding domain or an antigen-binding molecule having binding activity to a low-molecular compound by the panning.
- Fluorescently labeled streptavidin (or neutravidin or avidin) can be used as another aspect of the method for preparing an antigen-binding domain or an antigen-binding molecule having binding activity to a low-molecular compound by the panning.
- FACS fluorescence activated cell sorting
- an antigen-binding domain having a binding activity to a pre-existing low-molecular-weight compound is used.
- an antigen-binding domain having an MTA-binding activity an antigen-binding domain in which the antigen-binding activity changes depending on the MTA, and an amino acid interacting with the MTA. It is possible to use an antigen-binding domain that has a residue.
- modifiers refers to variants of the parent sequence in which at least one or more amino acids have been substituted with respect to the unmodified form of the parent sequence.
- an antigen-binding molecule contained in the heavy chain variable region or light chain variable region sequence, or the CDR sequence or framework sequence of the antibody variable region unmodified form is described above.
- Known methods such as site-specific mutagenesis (Kunkel et al. (Proc. Natl. Acad. Sci. USA (1985) 82, 488-492)) and Overlap extension PCR can be appropriately adopted to prepare the drug.
- amino acid sites in which the antibody variable region variants contained in the library of the present disclosure have different amino acids from the antibody variable region unmodified variants are the heavy and / or light chain CDR1, CDR2, and /. Or located in CDR3.
- amino acid sites in which the antibody variable region variant has an amino acid different from that of the antibody variable region unmodified are located at FR1, FR2, FR3 and / or FR4 on the heavy and / or light chains. To do.
- the light chain and / or heavy chain variable region framework sequence of the antigen binding molecule can be any framework sequence as long as the amino acid that interacts with MTA is contained in the heavy chain and / or light chain antigen binding domain. Can be used.
- the origin of the framework sequence can be obtained from any non-human animal organism or human, without limitation.
- any organism preferably includes an organism selected from mice, rats, guinea pigs, hamsters, gerbils, cats, rabbits, dogs, goats, sheep, cows, horses, camels, and non-human primates. ..
- the framework sequences of the light and / or heavy chain variable regions of the antigen binding molecule preferably have a human germline framework sequence.
- the antigen-binding molecules of the present disclosure will have little or no immunogenic response when administered to humans (eg, treatment of disease). It is unlikely to cause it.
- "including a germline sequence" in the present disclosure means that a part of the framework sequence of the present disclosure is the same as a part of any human germline framework sequence. Means.
- the framework sequences of the present disclosure have at least 50% or more, 60% or more, 70% or more, 80% or more, 90% or more, or 100% or more homology with germline sequences. Is preferable.
- the heavy chain FR2 sequence of the antigen-binding molecule of the present disclosure is a combination of the heavy chain FR2 sequences of a plurality of different human germline framework sequences
- the "germline sequence" of the present disclosure is also used. Is an antigen-binding molecule.
- the framework sequence of the antigen-binding molecule of the present disclosure is a substituted sequence, it is also an antigen-binding molecule "containing a germline sequence" of the present disclosure.
- substituted sequences in particular, some amino acids in the human germline framework sequence alter the binding activity of the antigen-binding molecule to the antigen depending on the presence or absence of MTA and / or adenosine. Examples thereof include sequences that are substituted with amino acids that cause
- a framework is the currently known fully humanoid framework contained on websites such as V-Base (http://vbase.mrc-cpe.cam.ac.uk/). The arrangement of work areas is preferably mentioned.
- the sequences of these framework regions can be appropriately used as the sequences of germline lines contained in the antigen-binding molecules of the present disclosure.
- Germline sequences can be classified based on their similarities (Tomlinson et al. (J. Mol. Biol. (1992) 227, 776-798) Williams and Winter (Eur. J. Immunol. (1993) 23, 1456). -1461) and Cox et al. (Nat. Genetics (1994) 7, 162-168)).
- Suitable germline sequences can be appropriately selected from V ⁇ , which is classified into 7 subgroups, V ⁇ , which is classified into 10 subgroups, and VH, which is classified into 7 subgroups.
- VH1 subgroups eg, VH1-2, VH1-3, VH1-8, VH1-18, VH1-24, VH1-45).
- VH1-46, VH1-58, VH1-69) VH2 subgroup (eg VH2-5, VH2-26, VH2-70)
- VH3 subgroup VH3-7, VH3-9, VH3-11, VH3) -13, VH3-15, VH3-16, VH3-20, VH3-21, VH3-23, VH3-30, VH3-33, VH3-35, VH3-38, VH3-43, VH3-48, VH3-49 , VH3-53, VH3-64, VH3-66, VH3-72, VH3-73, VH3-74), VH4 subgroups (VH4-4, VH4-28, VH4-31, VH4-34, VH4-39, VH
- the fully human V ⁇ sequence is not limited to the following, but is, for example, A20, A30, L1, L4, L5, L8, L9, L11, L12, L14, L15, which are classified into the Vk1 subgroup.
- Fully human V ⁇ sequences are not limited to the following, but are, for example, classified into the VL1 subgroup V1-2, V1-3, V1-4, V1-5, V1-7, V1- 9, V1-11, V1-13, V1-16, V1-17, V1-18, V1-19, V1-20, V1-22, VL1 classified into subgroups V2-1, V2-6, V2 -7, V2-8, V2-11, V2-13, V2-14, V2-15, V2-17, V2-19, VL3 subgroups V3-2, V3-3, V3-4, V4-1, V4-2, V4-3, V4-4, V4-6 classified into VL4 subgroup, V5-1, V5-2, V5-4, V5-6 classified into VL5 subgroup, etc. (Kawasaki et al. (Genome Res. (1997) 7, 250-261)) is preferably mentioned.
- framework sequences differ from each other due to differences in one or more amino acid residues.
- Examples of fully human frameworks used in this disclosure include, but are not limited to, KOL, NEWM, REI, EU, TUR, TEI, LAY, POM, etc. (For example, Kabat et al. (1991) and Wu et al. (J. Exp. Med. (1970) 132, 211-250) mentioned above).
- amino acids at positions 1, 5, 10, 30, 48 and 58 can be exemplified. More specifically, Gln at 1st place, Gln at 5th place, Asp at 10th place, Asn at 30th place, Leu at 48th place, and Asn at 58th place can be exemplified. In order to improve the stability of the antibody, it is possible to replace these amino acids with the corresponding amino acids contained in the germline sequence.
- VH3-21 can be exemplified as a sequence of one non-limiting aspect of such germline.
- 1st place Gln is Glu
- 5th place Gln is Val
- 10th place Asp is Gly
- 30th place Asn is Ser
- 48th place Leu is Val
- 58th place Asn is Tyr.
- the amino acid site in which the antigen-binding domain variant has an amino acid different from that of the antigen-binding domain unmodified form is one or more selected from the following group of amino acid sites.
- the amino acid site in which the antigen-binding domain variant has an amino acid different from that of the unmodified antigen-binding domain is one or more amino acid sites selected from the group of amino acid sites below: 1) Amino acid sites corresponding to amino acid sites that are not involved in binding to MTA in the antigen-binding domain unmodified form, 2) Amino acid sites that do not significantly attenuate the binding of the antigen-binding domain variant to the MTA as compared to the antigen-binding domain variant.
- Amino acid sites that can be diversified Amino acid sites having an amino acid whose sequence of the unmodified antigen-binding domain is different from the sequence of the modified antigen-binding domain should be designed to contain various amino acids when designing the library. In the present disclosure, it is also referred to as a “diversifiable amino acid site”.
- Amino acid sites that can be diversified can include: (i) Amino acid sites exposed on the surface of the antigen-binding domain; (ii) Amino acid sites located in regions where the rate of structural change is large when comparing the structure when the antigen-binding domain binds to the MTA and the structure when it does not bind to the MTA; (iii) Amino acid sites not involved in binding to MTA; (iv) Amino acid site that does not significantly attenuate binding to MTA; (v) Amino acid sites with varying frequency of amino acid appearance in animal species to which the antigen-binding domain belongs; (vi) Amino acid sites that are not important for the formation of Canonical structure; (vii) An amino acid site that tends to contribute to MTA-dependent binding of an antigen-binding domain variant to an antigen.
- amino acid sites not involved in binding to MTA are crystal structure analysis of the complex of MTA and antigen-binding molecule, three-dimensional structure analysis using NMR, or amino acid mutation. It can be identified by a method such as introduction.
- the amino acid residue of the antigen-binding molecule that is not involved in the binding to the MTA, particularly the binding to the MTA is involved. Amino acid residues in no antigen-binding domain can be identified.
- involved in binding to MTA means that when the antigen-binding domain is an antibody variable region, the side chain or main chain atom and the MTA atom in the amino acid forming the H chain or L chain of the antibody variable region are used. However, in a state where an intermolecular interaction is formed at a distance that can affect the binding activity, or a certain amino acid residue stabilizes a conformation such as a CDR loop to the conformation when MTA binds. A state that contributes to MTA binding, including indirect effects, and a state that satisfies both.
- the determination of an amino acid site that is not involved in binding to the MTA is an amino acid site other than any one or more amino acid sites that are selected from among the amino acid sites that are involved in binding to the MTA.
- H of the antibody variable region It can be determined based on the interatomic distance between the non-hydrogen atom constituting the side chain or main chain of the amino acid forming the chain or L chain and the non-hydrogen atom constituting the MTA.
- the above interatomic distance is preferably within 3.0 ⁇ , 3.2 ⁇ , 3.4 ⁇ , 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ , 4.2 ⁇ , 4.4 ⁇ , 4.6 ⁇ , 4.8 ⁇ or 5.0 ⁇ , but is not limited to this. Absent. More preferably, the interatomic distance is within 3.6 ⁇ , 3.8 ⁇ , 4.0 ⁇ or 4.2 ⁇ . More specifically, it is possible to determine the possibility of direct interaction based on the information on the interatomic distance on the three-dimensional structure, the type of intermolecular interaction formed, and the type of atom. More precisely, it can be judged from the effect of mutagenesis of amino acid residues such as modification to Ala and Gly on the activity of MTA, but it is not limited to this.
- amino acid sites that do not significantly attenuate binding to MTA can be identified by the method of mutagenesis of amino acids.
- the amino acids in the variable region are comprehensively modified, and the binding of each variant to the MTA is measured by a known method using Biacore or the like, and the binding activity (affinity) of each variant to the MTA is determined. Calculated as a KD value.
- the KD value is compared with the KD value of the unmodified form which is the parent sequence, and the modified site showing a binding exceeding a certain standard is determined to be an "amino acid site that does not significantly attenuate the binding to the MTA".
- the binding activity (affinity) of each variant to MTA is calculated as a KD value, and the MTA binding ability of the heavy chain site is not modified by modification.
- the MTA binding ability is 1/100, 1/50, 1/10, 1/9, 1/8, 1/7, 1/6, 1/5 of the unmodified form due to modification.
- Those that do not fall below the binding ability of 1/4, 1/3, or 1/2 are determined to be amino acid sites that do not significantly attenuate the binding to the MTA, but are not limited to the above criteria.
- the binding activity (affinity) of each variant to the MTA is calculated as the KD value, not the comparison with the KD value of the unmodified variant, which is the parent sequence, and the heavy chain sites are 10 mM, 1 mM, 100 uM, and 10 uM.
- the measurement of the binding activity of the unmodified and modified products to the MTA can be carried out by appropriately selecting from methods known to those skilled in the art (Biacore, ELISA, ECL, etc.).
- the determination of an amino acid site that does not significantly attenuate the binding to the MTA is selected from among the amino acid sites involved in the binding to the MTA, other than any one or more amino acid sites. Can also be thought of as.
- amino acid sites that easily contribute to MTA-dependent binding are crystal structure analysis of the complex of MTA and antigen-binding molecule, three-dimensional structure analysis using NMR, or amino acids. It can be identified or estimated by a method such as introduction of mutations in.
- amino acid sites where amino acid residues exposed on the surface of antigen-binding molecular proteins are located or non-binding when MTA binds.
- An example is a method for identifying an amino acid site located in a region whose structure changes with respect to time.
- amino acid site exposed on the surface of an antigen-binding domain refers to an amino acid site located on the surface of a protein's three-dimensional structure. Amino acid residue located on the protein surface of the antigen-binding domain by a method such as crystal structure analysis or three-dimensional structure analysis using NMR for a single antigen-binding molecule containing an antigen-binding domain or a complex of MTA and an antigen-binding molecule. Group and amino acid sites can be identified. As a non-limiting method for identifying the amino acid site exposed on the surface of the antigen-binding domain, there is a method for identifying the amino acid site exposed on the surface by calculating the solvent exposed surface area (Accessible surface area).
- Amino acid site located in a region where the rate of structural change is large during MTA binding / non-binding of the antigen-binding domain The "region where the rate of structural change is large during MTA binding / non-binding of the antigen-binding domain" in the present disclosure is referred to by crystal structure analysis or NMR. Identify by identifying the region where the structure changes by comparing the structure of the antigen-binding molecule alone containing the antigen-binding domain and the complex of the MTA and the antigen-binding molecule using the method such as the three-dimensional structure analysis used. Can be done.
- Amino acid site located in the region where the structural change rate is large during MTA binding / non-binding of the antigen-binding domain compares the structure of the antigen-binding molecule alone including the antigen-binding domain and the structure of the complex of MTA and the antigen-binding molecule. It can also be identified directly by doing so.
- the antigen-binding domain is an antibody variable region, it is calculated when the structure of the antibody H chain and L chain when bound to MTA and the structure of the antibody H chain and L chain when not bound to MTA are superimposed.
- the amino acid site where the amino acid residue is located has a large rate of structural change during MTA binding / non-binding of the antigen-binding domain. It can be determined whether or not the amino acid site is located in the region. For example, the amino acid site where the amino acid residue where the distance between C ⁇ atoms at the time of MTA binding / unbonding is equal to or greater than the average value of the distance between C ⁇ atoms at the time of MTA binding / unbonding of all amino acid residues in the variable region is located.
- an amino acid site located in a region where the rate of structural change is large during MTA binding / non-binding of the antigen-binding domain It can be determined as "an amino acid site located in a region where the rate of structural change is large during MTA binding / non-binding of the antigen-binding domain", but the present invention is not limited to this. It is expected that MTA-dependent antigen-binding activity will be brought about by substituting various amino acids for amino acids at the amino acid site located in the region where the structural change rate is large during MTA binding / non-binding of the antigen-binding domain. To. This makes it possible to design a library in which various amino acids appear at such amino acid sites.
- a library mainly composed of a plurality of antigen-binding molecules is prepared so as to be a collection of antigen-binding molecules having different amino acids at amino acid sites located in regions where the structural change rate is large during MTA binding / non-binding of the antigen-binding domain. It is possible to design.
- amino acid sites with diverse amino acid appearance frequencies are found to exist at an occurrence frequency of 1% or more as a repertoire of antibodies of animal species to which the parent sequence belongs. It refers to an amino acid site in which two or more types of amino acids are recognized.
- amino acid sites having various amino acid appearance frequencies in the animal species to which the antigen-binding domain belongs is defined in the repertoire of antibody gene sequences found on the gene in the animal species from which the relevant unmodified product is derived. It refers to the amino acid site where diversity is recognized.
- the antibody repertoire of the animal species to which the unmodified product belongs is the repertoire of antibody gene sequences found on the human gene.
- the antibody repertoire of the animal species to which the unmodified product belongs refers to the repertoire of antibody gene sequences found on the rabbit gene.
- it does not include sequences that are not actually expressed as antibodies due to the presence of stop codons or frameshifts.
- the unmodified product is of non-human animal origin, it can be humanized according to conventional methods, and this method is widely known to those skilled in the art (European Patent Publication EP239400, International Publication WO 1996/002576). , WO1993 / 012227, WO1992 / 003918, WO1994 / 002602, WO1994 / 025585, WO1996 / 034096, WO1996 / 033735, WO1992 / 001047, WO1992 / 020791, WO1993 / 006213, WO1993 / 011236, WO1993 / 019172, WO1995 / 001438, WO1995 / 015388, Cancer Res., (1993) 53, 851-856, BBRC., (2013) 436 (3): 543-50, etc.).
- the antibody repertoire of the animal species to which the unmodified product belongs in the present disclosure includes the antigen-binding domain before humanization and humans. Both of the antigen-binding domains after conversion can be treated as antigen-binding domains, and accordingly, the repertoire of both humans and the animal species from which the antigen-binding domain before humanization was performed as the same animal species is applied. be able to.
- the antigen-binding domain prior to humanization is derived from rabbits
- the antibody repertoire of the animal species to which the unmodified belongs is human and / or rabbit. Refers to the repertoire of antibody gene sequences found on genes. However, even if it is present on the gene, it does not include sequences that are not actually expressed as antibodies due to the presence of stop codons or frameshifts.
- the antibody repertoire of the animal species to which the unmodified substance belongs is, for example, not limited to this, but can be investigated by referring to a known database. Sites with varying amino acid frequency appear generally in the CDR regions. In one aspect, provided by Kabat, Sequences of Proteins of Immunological Interest (National Institute of Health Bethesda Md.) (1987 and 1991) in determining the highly diverse positions of known and / or native antibodies. The data to be used is valid.
- the antibody repertoire of the animal species to which the unmodified substance belongs can be investigated by cloning the antibody gene obtained from the relevant species and analyzing the sequence.
- the human antibody repertoire is a naive library consisting of naive sequences, which are antibody sequences constructed from antibody genes derived from healthy human lymphocytes and without bias in the repertoire.
- Can be investigated by analyzing the sequence of (Gejima et al. (Human Antibodies (2002) 11,121-129) and Cardoso et al. (Scand. J. Immunol. (2000) 51, 337-344)).
- the repertoire of antibodies of the animal species to which the unmodified product belongs in the present disclosure is more preferably not limited to this, but it is desirable that a survey be conducted on the germline subgroup to which the unmodified product belongs.
- An example of a framework is the currently known fully humanoid framework contained on websites such as V-Base (http://vbase.mrc-cpe.cam.ac.uk/).
- the sequence of the work region can be appropriately used as a germline sequence contained in the antigen-binding molecule of the present disclosure.
- Germline sequences can be subgrouped based on their similarity (Tomlinson et al. (J. Mol. Biol. (1992) 227, 776-798) Williams and Winter (Eur. J. Immunol.
- the canonical structure of an antibody refers to the clustering of the three-dimensional structures of the antibody heavy and light chains, mainly CDR1 and 2. As shown, the structure can be categorized by antibody subgroup and CDR length or sequence. In each canonical structure, residues important for maintaining the structure are already known, and Chothia et al. (J. Mol. Biol. (1992) 227, 799-817), Al-Lazikani et al. (J. Mol. Biol. (J. Mol.).
- a library produced by the following steps (a) and (b) is provided.
- a library manufactured by a method that includes The amino acid modification in step (b) satisfies at least one or more of the following (1) to (3): (1) When comparing the structure when the antigen-binding domain variant having the amino acid modification binds to the MTA and the structure when it does not bind to the MTA, the structural change rate of the amino acid site where the modified amino acid is located is large; (2) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the MTA and the structure when it is not bound to the MTA, the antigen-binding domain modification is caused by the presence of the modified amino acid. Structural changes in the body are not inhibited; (3) The antigen-binding domain variant having the amino acid modification does not significantly attenuate the binding activity to the MTA as compared with the antigen-binding domain unmodified form.
- the “one or more amino acids” in the present disclosure is not particularly limited in the number of amino acids, and is defined as two or more kinds of amino acids, five or more kinds of amino acids, ten or more kinds of amino acids, 15 or more kinds of amino acids or 20 kinds. It may be a kind of amino acid.
- designing a library containing nucleic acids encoding a plurality of variants of the antigen-binding domain having different sequences from each other means, for example, NNK, TRIM Library, etc. (Gonzalez-Munoz A et al. MAbs 2012, Using the known library technology of Lee CV et al. J Mol Biol. 2004, Knappik A. et al. J Mol Biol. 2000, Tiller T et al. MAbs 2013), the amino acid at a specific site is converted to the desired amino acid.
- Modifier refers to a variant of the parent sequence in which at least one or more amino acids have been substituted with respect to the unmodified form of the parent sequence.
- the frequency of appearance in germline is 10% for the sites of CDR1 and CDR2.
- the modified product in the step of producing "a plurality of variants of the antigen-binding domain having different sequences from each other" in the present disclosure, the structure of the variant in which the amino acid of the specified amino acid site is substituted and the structure when bound to MTA and binding to MTA
- the modified product can be prepared so that the structural change rate of the amino acid site where the substituted amino acid is located is large, but the modification is not limited to this.
- the modified product in the step of producing "a plurality of variants of the antigen-binding domain having different sequences from each other" in the present disclosure, the structure of the variant in which the amino acid of the specified amino acid site is substituted and the structure when bound to the MTA and the binding to the MTA
- the modified product can be prepared so that the presence of the post-substitution amino acid does not inhibit the structural change due to the MTA binding of the modified product, but the modification is not limited to this. Absent.
- the binding activity of the modified amino acid-substituted variant at the specified amino acid site to the MTA is higher than that of the unmodified variant.
- Variants can be made without being significantly attenuated, but are not limited to this.
- the library of the present disclosure provides a library enriched with nucleic acids encoding an antigen-binding molecule that comprises an antigen-binding domain that binds to MTA.
- “Concentrating” in the present disclosure means increasing the abundance ratio of nucleic acid encoding a variant having the desired activity in comparison with the library before performing the concentration work.
- concentration The state in which the abundance ratio of the nucleic acid encoding the variant having the desired activity is increased is referred to as "concentrated”.
- enriching the nucleic acid encoding the antigen-binding molecule that binds to MTA means that the abundance ratio of the nucleic acid encoding the antigen-binding molecule having binding activity to MTA by panning is determined. It can be achieved by raising it. More specifically, as an example, a library containing a plurality of antigen-binding molecules brings a phage presented on the surface by a phage display method into contact with MTA, and presents a molecule having no binding activity by a washing operation.
- the antigen-binding molecule that has binding activity to MTA is obtained by panning by collecting only the phage that presents the antigen-binding molecule that maintains the binding. It is possible, but not limited to, to increase the abundance of the encoding nucleic acid.
- This method includes (1) contacting the antigen-binding domain displayed from the library with the MTA, and (2) selecting the antigen-binding domain bound to the MTA in the above step (1).
- the abundance ratio of the nucleic acid encoding the antigen-binding molecule having the desired activity is increased by 1.1 times or more as compared with the library before the concentration operation. More preferably, the abundance ratio of the nucleic acid encoding the antigen-binding molecule having the desired activity is increased 1.2 times or more, 1.5 times or more, 2 times or more, 4 times or more, 10 times or more, 25 times or more, 100 times or more. Thereby, it is possible to manufacture the library described in the present disclosure.
- the binding activity of an antigen-binding molecule in a library enriched in this way to an MTA is also demonstrated in the absence of an antigen, which is a different type of molecule than the MTA.
- the type of library to be enriched is not limited as long as it contains an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner. More specifically, examples include, but are not limited to, naive human antibody display libraries, synthetic human antibody display libraries, libraries designed according to the methods herein described below, and libraries described in the examples herein. ..
- enriching a nucleic acid encoding an antigen-binding molecule that binds to adenosine means the presence of a nucleic acid encoding an antigen-binding molecule that has binding activity to adenosine by panning. This can be achieved by increasing the ratio. More specifically, as an example, a library containing a plurality of antigen-binding molecules brings a phage presented on the surface by the phage display method into contact with adenosine and presents a molecule having no binding activity by a washing operation.
- the antigen-binding molecule that has binding activity to adenosine is obtained by panning by collecting only the phage that presents the antigen-binding molecule that maintains the binding. It is possible, but not limited to, to increase the abundance of the encoding nucleic acid.
- This method includes (1) contacting the antigen-binding domain displayed from the library with adenosine, and (2) selecting the antigen-binding domain bound to adenosine in the above step (1).
- the abundance ratio of the nucleic acid encoding the antigen-binding molecule having the desired activity is increased by 1.1 times or more as compared with the library before the concentration operation. More preferably, the abundance ratio of the nucleic acid encoding the antigen-binding molecule having the desired activity is increased 1.2 times or more, 1.5 times or more, 2 times or more, 4 times or more, 10 times or more, 25 times or more, 100 times or more. Thereby, it is possible to manufacture the library described in the present disclosure.
- the binding activity of an antigen-binding molecule in a library enriched in this way to adenosine is also demonstrated in the absence of an antigen, which is a different type of molecule than adenosine.
- the type of library to be enriched is not limited as long as it contains an antigen-binding domain whose antigen-binding activity changes in an adenosine-dependent manner. More specifically, examples thereof include, but are not limited to, a naive human antibody display library, a synthetic human antibody display library, a library manufactured according to the library manufacturing method of the present specification, and a library described in the present specification.
- the present disclosure also relates to methods of manufacturing the various aspects of the "library” included in the present disclosure described above.
- the "library manufacturing method” according to the present disclosure is not limited to any specific method as exemplified below, and any method that can manufacture the above-mentioned "library” of the present disclosure can be used. Methods are also included. Examples of the “library manufacturing method” according to the present disclosure include the methods illustrated below. It should be noted that each specific item in the "method for producing a library” illustrated below has the technical significance as described in detail above for "enriched” from the "library” of the present disclosure.
- the process of designing a library containing nucleic acids that encode each body The amino acid modification in step (b) satisfies at least one or more of the following (1) to (3): (1) When comparing the structure when the antigen-binding domain variant having the amino acid modification binds to the MTA and the structure when it does not bind to the MTA, the structural change rate of the amino acid site where the modified amino acid is located is large; (2) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the MTA and the structure when it is not bound to the MTA, the antigen-binding domain modification is caused by the presence of the modified amino acid.
- the antigen-binding domain variant having the amino acid modification does not significantly attenuate the binding activity to the MTA as compared with the antigen-binding domain unmodified form;
- a method for producing a library encoding an antigen-binding domain having an amino acid residue that interacts with MTA but also an antigen-binding domain having an amino acid residue that interacts with various small molecule compounds is encoded.
- a method of manufacturing the library to be used is also provided.
- Each specific matter in the method for producing the library illustrated below has the same technical significance as the method for producing the M library detailed above, and those skilled in the art should read "MTA" as "small molecule compound”. Can understand the meaning of various specific matters.
- the process of designing a library containing nucleic acids that encode each body A method of manufacturing a library including the
- the process of designing a library containing nucleic acids that encode each body The amino acid modification in step (b) satisfies at least one or more of the following (1) to (3): (1) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the low molecular weight compound and the structure when it is not bound to the low molecular weight compound, the amino acid in which the modified amino acid is located is compared. Large rate of structural change in the site; (2) When comparing the structure when the antigen-binding domain variant having the amino acid modification is bound to the low molecular weight compound and the structure when it is not bound to the low molecular weight compound, the modified amino acid is present.
- the antigen-binding domain variant having the amino acid modification does not significantly attenuate the binding activity to the low molecular weight compound as compared with the antigen-binding domain unmodified form;
- a method of manufacturing a library including the above is provided.
- MTA-Specifically Binding Antigen-Binding Molecules The present disclosure also relates to MTA-specifically binding antigen-binding molecules.
- an antigen-binding molecule that specifically binds to MTA one or more small molecules selected from S- (5'-Adenosyl) -L-homocysteine (SAH), SAM, AMP, ADP, and ATP.
- SAH S- (5'-Adenosyl) -L-homocysteine
- SAH S- (5'-Adenosyl) -L-homocysteine
- SAM SAM
- AMP AMP
- ADP ADP
- ATP ATP
- An example is an antigen-binding molecule that does not substantially bind to a compound.
- substantially no binding is determined according to the method described in the section of binding activity herein, and is 85% or less of the binding activity to the MTA, usually 50 or less, with respect to low molecular weight compounds other
- the antigen-binding molecule that specifically binds to the MTA of the present disclosure does not substantially bind to adenosine. In another non-limiting aspect, the antigen-binding molecule that specifically binds to the MTA of the present disclosure does not substantially bind to S- (5'-Adenosyl) -L-homocysteine (SAH). In another non-limiting aspect, the antigen-binding molecule that specifically binds to the MTA of the present disclosure does not substantially bind to SAM. In another non-limiting aspect, the antigen-binding molecule that specifically binds to the MTA of the present disclosure does not substantially bind to any of adenosine, AMP, ADP, ATP.
- the antigen-binding molecule that specifically binds to the MTA of the present disclosure substantially binds to either adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH). do not do.
- the antigen-binding molecule that specifically binds to the MTA of the present disclosure is substantially any of adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH), SAM. Does not combine with.
- the antigen-binding molecules that specifically bind to the MTA of the present disclosure are adenosine, S- (5'-Adenosyl) -L-homocysteine (SAH), SAM, AMP, ADP, ATP. Substantially does not bind to any of.
- an isolated antibody that binds to the MTA can be mentioned.
- the antigen-binding molecule that specifically binds to the MTA has been humanized.
- the antigen-binding molecule that specifically binds to the MTA comprises HVR in any of the embodiments described above, and further comprises an acceptor human framework (eg, a human immunoglobulin framework or a human consensus framework).
- the antigen-binding molecule that specifically binds to the above-mentioned MTA may be introduced with an amino acid modification for the purpose of enhancing the binding activity to the MTA, or a modification for further enhancing the specificity for a specific low-molecular-weight compound. May be introduced.
- the purpose of introducing the amino acid modification is not limited to the above, and the modification for any purpose can be introduced.
- the antigen-binding molecule that specifically binds to MTA is at least 90%, 91%, 92%, 93%, 94%, 95 with respect to the amino acid sequence of SEQ ID NO: 46, 48, 50, 52. Includes heavy chain variable region sequences with%, 96%, 97%, 98%, 99%, or 100% sequence identity. In certain embodiments, a heavy chain variable region sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity is a reference sequence.
- an antigen-binding molecule that comprises substitution (eg, conservative substitution), insertion, or deletion, but specifically binds to the MTA containing the sequence retains the ability to bind to the MTA. In certain embodiments, a total of 1 to 10 amino acids are substituted, inserted, and / or deleted at SEQ ID NOs: 46, 48, 50, 52.
- the antigen-binding molecule that specifically binds to MTA is at least 90%, 91%, 92%, 93%, 94%, 95 with respect to the amino acid sequence of SEQ ID NO: 47, 49, 51, 53. Includes light chain variable region sequences with%, 96%, 97%, 98%, 99%, or 100% sequence identity. In certain embodiments, a light chain variable region sequence having at least 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% identity is a reference sequence. In contrast, an antigen-binding molecule that comprises substitution (eg, conservative substitution), insertion, or deletion, but specifically binds to the MTA containing the sequence, retains the ability to bind to the MTA. In certain embodiments, a total of 1 to 10 amino acids are substituted, inserted, and / or deleted at SEQ ID NOs: 47, 49, 51, 53.
- the antibody is a post-translational modification of the heavy and light chain variable region sequences in SEQ ID NOs: 46, 48, 50, 52 and SEQ ID NOs: 47, 49, 51, 53, respectively. Includes, including those included. Post-translational modifications include, but are not limited to, modification of heavy or light chain N-terminal glutamine or glutamate to pyroglutamylation by pyroglutamylation.
- any of the antigen-binding molecules that specifically bind to an MTA provided herein are useful for detecting the presence of an MTA in a biological sample.
- the term "detection” includes quantitative or qualitative detection.
- the biological sample comprises cells or tissues, such as cancerous tissue.
- an antigen-binding molecule that specifically binds to an MTA for use in a diagnostic or detection method is provided.
- a method of detecting the presence of MTA in a biological sample is provided.
- the method comprises an antigen-binding molecule that specifically binds to the MTA described herein and biology under conditions that allow binding of the antigen-binding molecule that specifically binds to the MTA. Includes contacting a target sample and detecting whether a complex has been formed between the MTA and an antigen-binding molecule that specifically binds to the MTA.
- Such a method can be an in vitro method or an in vivo method.
- the antigen-binding molecule that specifically binds to MTA is an antigen-binding molecule that contains an antigen-binding domain whose antigen-binding activity changes in an MTA-dependent manner, for example, when MTA is a biomarker for selecting a patient.
- MTA is a biomarker for selecting a patient.
- it can also be used as a method for predicting a therapeutic effect on a patient.
- a non-limiting aspect of its use for selecting a subject is exemplified by determining whether an MTA is an accumulating cancer.
- MTAs are accumulated in cancer tissues, and by detecting MTAs in tissues and / and in peripheral areas, it is possible to diagnose whether or not they have those cancers. It is possible. Also, as a non-limiting aspect of its use for predicting therapeutic efficacy, detection of MTA in tissue and / and in peripherals can indirectly diagnose shrinkage or exacerbation of cancerous tissue.
- an antigen-binding molecule that specifically binds to a labeled MTA.
- Labels are detected indirectly through directly detected labels or moieties (eg, fluorescent labels, color-developing labels, high electron density labels, chemically luminescent labels, and radioactive labels) and, for example, enzymatic reactions or intermolecular interactions. Includes, but is not limited to, moieties such as enzymes or ligands.
- Exemplary labels include, but are not limited to: radioactive isotopes 32P, 14C, 125I, 3H and 131I, peroxidases such as rare earth chelate or luciferase and derivatives thereof, rhodamine and derivatives thereof.
- Luciferase Luciferin, 2,3-Dihydrophthalazinedione, Horseradish peroxidase (HRP), Alkaline phosphatase, ⁇ -galactosidase , Glucoamylase, luciferase, monosaccharide oxidase (eg glucose oxidase, galactose oxidase and glucose-6-phosphate dehydrogenase), heterocyclic oxidases such as uricase and xanthin oxidase, enzymes that oxidize dye precursors using hydrogen peroxide ( For example, HRP, lactoperoxidase, or microperoxidase) linked, biotin / avidin, spin-labeled, bacteriophage-labeled, stable free radicals, and the like.
- HRP Horseradish peroxidase
- Alkaline phosphatase Alkaline phosphatase
- ⁇ -galactosidase Gluco
- amino acids contained in the amino acid sequence described in the present disclosure may be modified after translation (for example, modification of N-terminal glutamine to pyroglutamylation by pyroglutamylation is a modification well known to those skilled in the art). However, even if the amino acid is modified after translation as such, it is naturally included in the amino acid sequence described in the present disclosure.
- Example 1 Analysis of MTA accumulation in tumor tissue and normal tissue As cell lines used in the experiment, ATCC to HCC827, HPAC, HT-1376, NCI-H2228, SK-LU-1, SK-MES-1, I got U-87 MG, Mia-PaCa2 from Dainippon, KM12 from NCI, KM12-Luc from JCRB, and PK-1 from RIKEN.
- the MTAP-deficient state was determined by referring to the SNPs array dataset of the cell lines published by Barretina J et al. Nature. (2012) 483: 603-7. Although KM12-Luc was not included in this dataset, it was considered to have the same MTAP-deficient state as KM12 because it is a derivative of KM12.
- the MTA solution was added to the culture solution so that the MTA concentration became 100 nM, and the cells were cultured for a predetermined time (MTA: Sigma, Catalog No. D5011-100MG).
- MTA Sigma, Catalog No. D5011-100MG
- the culture solution was collected at each time point of 0, 4, 24, 48, and 72 hours, and the MTA concentration in the collected culture solution was measured by LC-MS.
- the MTA concentration in the culture solution over time after the start of culture. It was observed to decrease to (Fig. 3). It was suggested that when cells can metabolize MTA using MTAP, extracellular MTA is absorbed by the cells and disappears rapidly.
- the SIL-MTA elimination rate constant from the blood was calculated, and the elimination rate constant was the blood volume per mouse body weight ( The total blood clearance calculated by multiplying (85 mL / kg) was estimated to be 46685 mL / h / kg. From this result, it was found that the MTA added to the mouse blood rapidly disappeared from the blood.
- MTA in vivo clearance example (1-4) in mice suggested that MTA blood clearance is large in vitro, so it is examined whether MTA clearance is actually large even in vivo.
- SIL-MTA was used as the test substance to distinguish it from the endogenous MTA.
- Plasma SIL-MTA concentration was measured by LC-MS / MS.
- the SIL-MTA systemic clearance calculated from the plasma SIL-MTA concentration 30 minutes after the start of administration and the administration rate of SIL-MTA is 38961 mL / h / kg or more, and the MTA disappears rapidly from the plasma even in vivo. It became clear to do.
- the supernatant was filtered, the filtrate was analyzed by LC-MS / MS to measure the MTA concentration, and the MTA concentration (pmol / g tissue) in the tumor was calculated from the amount of MTA in the filtrate and the tumor weight.
- MTA concentration pmol / g tissue
- the MTA concentration in the tissue interstitial fluid collected 90 to 120 minutes after the probe was inserted was quantified by the LC-MS / MS method.
- a standard solution was prepared by adding MTA to a BSA solution (0, 1, 10, 100 mg / mL) at a known concentration, and the MTA recovery rate was calculated from the ratio of the MTA concentration recovered from the microdilisys probe to the MTA concentration of the standard solution. Calculated.
- the extracellular MTA concentration in the tissue was calculated by correcting the MTA concentration in the collected tissue interstitial fluid with the MTA recovery rate (16.3%).
- Example 2 Acquisition of MTA-dependent antigen-binding antibody from naive or synthetic library (2-1) MTA-dependent manner from the library that mimic the naive antibody repertoire of panning person for obtaining an antibody that binds to the antigen binding activity to human IL-6 receptor (hIL-6R) under MTA presence Antibodies showing the above were screened.
- Naive human antibody phage consisting of multiple phages that present Fab domains of human antibody sequences different from each other according to a method known to those skilled in the art, using poly A RNA prepared from human PBMC or commercially available human poly A RNA as a template.
- a display library (called a naive library) was built.
- a synthetic human antibody phage display library consisting of a plurality of phages presenting Fab domains of different human antibody sequences according to a method known to those skilled in the art, based on a design artificially designed with reference to the human naive antibody repertoire.
- (Called a synthetic library) was constructed. Specifically, Escherichia coli carrying the constructed phagemid vector was infected with M13KO7TC (WO2015046554A1) or M13KO7 ⁇ pIII (called hyperphage) (PROGEN Biotechnik), and the supernatant was cultured overnight at 30 ° C. Phage were recovered. An antibody-presenting phage library solution was prepared by diluting a population of phages precipitated by adding 2.5 M NaCl / 10% PEG to the culture solution of Escherichia coli in which phages were produced, with TBS.
- Panning was carried out by the following method.
- BSA was added to the phage library solution to a final concentration of 4%. If necessary, in order to remove the antibody that binds to hIL-6R in the absence of MTA, magnetic beads on which 0.1 nmol of biotin-labeled hIL-6R was immobilized and the prepared phage library solution were mixed and reacted at room temperature for 60 minutes. Later, the phage solution was recovered from the beads separated using a magnetic stand. To 0.8 mL of the recovered phage library solution, 0.1 nmol of biotin-labeled hIL-6R and MTA having a final concentration of 100 ⁇ M were added, and the mixture was reacted at room temperature for 60 minutes.
- Magnetic beads NeuronalAvidin beads (TAMAGAWA SEIKI) or Dynabeads MyOne StreptAvidin T1 (Thermo Fisher Scientific) blocked by BSA were added to this reaction solution, and the mixture was reacted at room temperature for 15 minutes.
- the beads were washed 2 or 3 times with 0.5 mL TBS / 0.1% Tween 20 and 1 or 2 times with TBS.
- the beads to which TBS was added were then suspended at room temperature, and the phage solution was recovered from the beads separated using a magnetic stand. After repeating this operation two or three times, the eluted phage solution was mixed. A final concentration of 1 mg / mL trypsin was added to the recovered phage solution.
- the recovered phage was added to 20 mL of E. coli strain ER2738 in the logarithmic growth phase (OD600 0.4-0.7). Phage was infected with Escherichia coli by stirring and culturing the above E. coli at 37 ° C. for 1 hour. E. coli was inoculated on a 225 mm x 225 mm plate. This series of work was repeated three additional times.
- TBS or TBS-diluted TBS containing 100 ⁇ M final concentration M13 antibody (GE Healthcare) is added to the wells. It was left for a while.
- TMB single solution ZYMED
- the color reaction of the solution is stopped by the addition of sulfuric acid, and then the absorbance at 450 nm wavelength.
- 4 clones were confirmed that presented an antibody whose binding activity to hIL-6R was changed in the presence of 100 ⁇ M MTA and in the absence of MTA.
- Example 3 Designing a library using humanized SMB0002 as a template for obtaining antibodies that bind to antigens in an MTA-dependent manner (3-1) Evaluation of MTA binding of humanized SMB0002 Due to the structural similarity between MTA and adenosine, an antibody that binds to adenosine may cross-react with MTA, and an adenosine antibody that cross-reacts with MTA is MTA. It may be used as a template antibody when designing a library for obtaining an antibody that binds to an antigen in a dependent manner.
- Humanized SMB0002 (heavy chain variable region: SEQ ID NO: 31; light chain variable region: SEQ ID NO: 32), which was reported to bind to adenosine, designed a library for obtaining antibodies that bind to antigens in an MTA-dependent manner.
- Biacore T200 GE Healthcare was used to analyze the binding of humanized SMB0002 to the MTA to verify that it could be used as a template antibody.
- Capture Select TM Biotin Anti-IgG-CH1 Conjugate (Thermo fisher scientific), which is a biotinylated anti-human IgG CH1 molecule, is bound to Sensor chip SA (GE Healthcare) on which streptavidin is immobilized, and then humanized. SMB0002 was captured and MTA (sigma-aldrich) or adenosine (Wako) was allowed to interact with humanized SMB0002.
- MTA Sigma-aldrich
- adenosine Wide
- As a running buffer 50 mM Tris-HCl, 150 mM NaCl (Takara, T903), 0.02% (w / v) Tween20, 5% DMSO was used.
- the MTA or adenosine was allowed to interact with humanized SMB0002 for 42 seconds at a flow rate of 100 ⁇ L / min and then dissociated from the antibody in running buffer. The interaction was measured at 37 ° C. and the same buffer as the running buffer was used to dilute the MTA or adenosine.
- the dissociation constant K D (M) is calculated based on the binding rate constant ka (1 / Ms) and the dissociation rate constant kd (1 / s), which are kinetic parameters calculated from the sensorgrams obtained by the measurement. Was done.
- the dissociation constant K D (M) was calculated using the Steady state analysis. Biacore T200 Evaluation Software (GE Healthcare) was used to calculate each parameter. By this measurement, the binding activity of humanized SMB0002 to MTA and adenosine was determined as shown in Table 1, and it was confirmed that humanized SMB0002 has binding activity not only to adenosine but also to MTA.
- Crystallographic statistics are shown in Table 2.
- a molecular replacement method using Phaser J. Appl. Cryst. (2007) 40, 658-674 was carried out using the known crystal structure of Fab as a search model. The initial structure has been determined. After that, model construction and refinement by Coot (Acta Cryst. D66: 486-501 (2010)) and Refmac5 (Acta Cryst. D67: 355-467 (2011)) were repeated, and the final refined coordinates were obtained. It was. Crystallographic statistics are shown in Table 2.
- Crystallographic statistics are shown in Table 2.
- the initial structure was determined by the molecular replacement method using Phaser (J. Appl. Cryst. (2007) 40, 658-674) using the known crystal structure of Fab as a search model. It has been determined. After that, model construction and refinement by Coot (Acta Cryst. D66: 486-501 (2010)) and Refmac5 (Acta Cryst. D67: 355-467 (2011)) were repeated, and the final refined coordinates were obtained. It was. Crystallographic statistics are shown in Table 2.
- the adenine ring portion of adenosine is recognized by the light chains S91 and N96 of humanized SMB0002, the side chains of heavy chains A33, I50 and Y100, and the main chains of heavy chains T100a and G99. ..
- the 1-position N of the adenine ring and the side chain of the antibody light chain N96 between the NH2 at the 6-position of the adenine ring and the side chains of the antibody light chains N96 and S91, and the carbonyl oxygen of the main chain of the antibody heavy chain T100a.
- a hydrogen bond is formed between the 7-position N of the adenine ring and the amide NH of the main chain of the antibody heavy chain T100a.
- a weak hydrogen bond is formed between the 8-position CH of the adenine ring and the carbonyl oxygen of the main chain of the antibody heavy chain G99. Furthermore, an interaction is formed between the adenine ring and each side chain of the antibody heavy chains A33, I50, and Y100 using the pi-electrons of the adenine ring portion such as CH- ⁇ or ⁇ - ⁇ . In addition, the O at the 3'position of the ribose moiety forms a hydrogen bond with each side chain of the antibody heavy chains D54 and S56.
- adenosine is surrounded by antibody heavy chains T57, G52, W58, N100b, F100d, and antibody light chain Y95c, forming van der Waals interactions. From these interactions, it is inferred that adenosine is recognized by humanized SMB0002.
- the amino acid residue numbering of Fab is based on the Kabat numbering scheme.
- Example (3-1) it has been clarified that humanized SMB0002 binds not only to adenosine but also to MTA. Since there are parts where the molecular structures of adenosine and MTA are similar, it is possible to consider the binding mode when MTA binds to humanized SMB0002 from the crystal structures of humanized SMB0002 and adenosine. In the crystal structure of adenosine and humanized SMB0002, an intramolecular hydrogen bond is formed between OH at the ribose 5'position of adenosine and N at the 3-position of the adenine ring, but in MTA, the thiomethyl group is this interaction. Cannot be formed.
- the thiomethyl group of the MTA should move away from the adenine ring moiety. Must be located. As a result, it is presumed that the thiomethyl group is located closer to the heavy chain CDR2 and the light chain CDR3 than the ribose 5'position OH of adenosine and on the surface side of the antibody. Therefore, it is considered that MTA is more likely to form a direct interaction with antigen binding than adenosine.
- FIG. 9 shows a superposition of variable regions extracted from molecule 1 (SMB0002hFab_1) and molecule 2 (SMB0002hFab_2). As shown in FIG. 9, the structures of both are the structure of heavy chain CDR1 and the structure of heavy chain and light chain. It was shown that the twist angles between them were different. From FIG.
- FIG. 11 is a diagram showing the superposition of the crystal structure of the SMB0002hFab-adenosine complex and the crystal structure of SMB0002hFab_2.
- humanized SMB0002 may be fixed to one structure such as the SMB0002hFab-adenosine complex by adenosine binding. Specifically, the structure of the heavy chain CDR1 and the twist angle between the heavy chain and the light chain are changed by the binding of adenosine.
- the difference between the structure of humanized SMB0002 immobilized by adenosine and the structure of humanized SMB0002 in other states is important for adenosine-dependent antigen binding in antibodies that bind to antigen in an adenosine-dependent manner. Conceivable. That is, in an antibody that binds to an antigen in an adenosine-dependent manner, the antigen can bind to an antibody having a structure immobilized by adenosine, while in the absence of adenosine, the antigen binds to the antibody. It is assumed that it is difficult.
- Amino acid sites of the light chain that can be diversified based on consideration of interaction with adenosine (sites represented by Kabat numbering described as "Kabat” in the table) and amino acids before modification at the sites (in the table).
- sites represented by Kabat numbering described as "Kabat” in the table amino acids before modification at the sites (in the table).
- the amino acid appearing at the site of humanized SMB0002 described as “natural sequence” and the modified amino acid are shown in Table 4.
- Amino acid sites of the heavy chain that can be diversified based on consideration of interaction with MTA sites represented by Kabat numbering described as "Kabat” in the table
- amino acids before modification at the sites in the table.
- the amino acid appearing at the site of humanized SMB0002 described as “natural sequence") and the modified amino acid (amino acid described as "modified amino acid” in the table) are shown in Table 5.
- Amino acid sites of the light chain that can be diversified based on consideration of interaction with MTA (sites represented by Kabat numbering described as "Kabat” in the table) and amino acids before modification at the sites (in the table).
- sites represented by Kabat numbering described as "Kabat” in the table amino acids before modification at the sites (in the table).
- the amino acid appearing at the site of humanized SMB0002 described as “natural sequence") and the modified amino acid are shown in Table 6.
- Binding of humanized SMB0002 or its monosubstituted variants to adenosine or MTA was measured and analyzed by the method using Biacore T200 (GE Healthcare).
- the antibody to be analyzed was captured on the Sensor chip, and the interaction with adenosine or MTA was observed.
- Adenosine diluent / MTA diluent and a blank running buffer were added to the antibody captured on the sensor chip, and binding of adenosine / MTA to the antibody was observed.
- the running buffer was then flushed and dissociation of adenosine / MTA from the antibody was observed.
- Biacore T200 Evaluation Software (GE Healthcare) was used to calculate each parameter.
- K D values were modified K D values / heavy chain for adenosine humanized SMB0002 the ratio (parent antibody with a K D values for adenosine each variant with altered K D values and heavy chain for adenosine humanized SMB0002 a parent antibody Table 7 K D values) for adenosine in each variant, the ratio (parent antibody with a K D values for adenosine each variant with altered K D values and light for adenosine humanized SMB0002 a parent antibody human K D values) for adenosine each variant with altered K D values / light chain for adenosine reduction SMB0002 shown in Table 8.
- Table 9 for MTA for each variant, the human is the ratio (parent antibody with a K D values for MTA for each variant with altered K D values and light for MTA humanized SMB0002 a parent antibody K D values) for MTA for each variant with altered K D values / light chain for the MTA of SMB0002 shown in Table 10.
- Parent antibody K D values / the site the amino acid K D values and the sites for adenosine ratio parent antibody with a K D value for the adenosine heavy chain variants modified in the amino acids to adenosine (humanized SMB0002) the K D values / the sites that amino acid for adenosine K D values) of 0.4 or more modified amino acids to adenosine modified heavy chain variant, the ratio (the parent antibody with a K D value for the adenosine light chain variant in K D values) of 0.1 or more modified amino acids to adenosine altered light chain variant, it is determined that amino acids which are not significantly involved in binding to adenosine conditions 1), was selected as an amino acid usable diversification ..
- the amino acid at position 32 of the heavy chain Kabat numbering is D or E, or if the amino acid at position 33 is V or I or T, the amino acid is determined to be an amino acid that satisfies condition 2 and can be used for diversification. It was selected as an amino acid.
- a library for obtaining an antibody that binds to an antigen in an MTA or adenosine-dependent manner by designing a library in which at least one or more of the selected diversifiable amino acids appears (hereinafter, S02). Library) was built. The amino acid diversified amino acid sites in the heavy chains of the S02 library, as well as the amino acid repertoire at those sites, are shown in Table 11.
- the amino acid diversified amino acid sites in the light chain of the S02 library, as well as the amino acid repertoire at those sites, are shown in Table 12.
- the site represented by the Kabat numbering described as "Kabat” is the amino acid diversified amino acid site
- the amino acid described as "natural sequence” is the amino acid of the unmodified humanized SMB0002 at the site.
- the amino acids described as "libraryable amino acids” indicate the amino acid repertoire at the site.
- a library was designed in which at least one of the amino acids contained in the selected amino acid repertoire appears at each diversifiable amino acid site contained in the heavy chain.
- a library was designed in which at least one of the amino acids contained in the selected amino acid repertoire appears at each diversifiable amino acid site contained in the light chain.
- Example 4 Construction of a library for obtaining an antibody that binds to an antigen in an MTA-dependent manner and acquisition of an antibody that binds to an antigen in a library MTA-dependent manner.
- a heavy chain variable region phage display library for obtaining an antibody that binds to an antigen in an MTA or adenosine-dependent manner was constructed.
- the light chain variable region portion in the designed S02 library was gene-synthesized using TRIM technology (Tiller T et al. MAbs 2013) and combined with the heavy chain variable region sequence of humanized SMB0002 (SEQ ID NO: 64) in humans. It was introduced into a suitable phagemid vector having an IgG-derived CH1 sequence and a human IgG-derived light chain constant region sequence.
- Biotin-2'-MTA immobilized on magnetic beads was performed under two conditions.
- BSA was added to the antibody polyvalent presentation phage library solution to a final concentration of 4%.
- NeutrAvidin beads (TAMAGAWA SEIKI) or Dynabeads MyOne StreptAvidin T1 (Thermo Fisher Scientific) were used as the magnetic beads.
- Biotin-2'-MTA was immobilized on the magnetic beads by reacting the BSA-blocked magnetic beads with Biotin-2'-MTA at a final concentration of 10 ⁇ M at room temperature for 30 minutes.
- the magnetic beads on which Biotin-2'-MTA was immobilized were washed 3 times with TBST, and under the first condition, 0.4 mL of the prepared antibody multivalent-presented phage library solution was added to the washed magnetic beads. Under the second condition, 0.4 mL of the antibody polyvalent presenting phage library solution prepared in the washed magnetic beads was added in the presence of 1 mM adenosine, and the reaction was carried out at room temperature for 60 minutes. The beads were washed 3 times with 400 ⁇ L TBST and 2 times with TBS.
- the beads to which 0.5 mL of TBS containing trypsin at a final concentration of 1 mg / mL was added were suspended at room temperature for 15 minutes, and the phage was eluted. The beads were separated using a magnetic stand and the phage solution was recovered. The recovered phage was added to 20 mL of E. coli strain ER2738 in the logarithmic growth phase (OD600 0.4-0.7). Phage was infected with Escherichia coli by stirring and culturing the above E. coli at 37 ° C. for 1 hour. E. coli was inoculated on a 225 mm x 225 mm plate. This series of operations was repeated three additional times, and the antibody group that binds to the MTA was concentrated from each of the heavy chain variable region phage display library and the light chain variable region phage display library.
- Example (4-1-5) Construction of a library for obtaining an antibody that binds to an antibody in an MTA-dependent manner using panning for MTA
- the MTA-binding antibody group concentrated in Example (4-1-2) is concerned.
- the Fab-presented phage library prepared by combining the modified heavy chain and the modified light chain contained in the antibody group is also expected to contain a large number of antibodies that maintain binding to MTA, and antibodies that bind to antigens in an MTA-dependent manner are expected. It was thought that a library for acquisition could be built.
- the antibody gene was extracted from Escherichia coli infected with the phage presenting the MTA-binding antibody group concentrated in Example (4-1-2) by a method known to those skilled in the art.
- the heavy chain variable region gene was amplified using a primer (SEQ ID NO: 34, 35) capable of amplifying the heavy chain variable region against the antibody group concentrated from the heavy chain variable region phage library.
- a primer SEQ ID NO: 34, 35
- the heavy chain variable region sequence of humanized SMB0002 was removed by restriction enzyme treatment, and the light chain variable region library gene, human IgG-derived light chain constant region sequence, and human were removed.
- a phagemid fragment containing an IgG-derived CH1 sequence was prepared.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Physics & Mathematics (AREA)
- Biotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Cell Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Crystallography & Structural Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Plant Pathology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Peptides Or Proteins (AREA)
Abstract
本開示は、標的組織特異的な低分子化合物濃度に応じて抗原結合活性の変化する抗原結合分子、当該抗原結合分子をコードするポリヌクレオチド、当該ポリヌクレオチドを含むベクター、当該ベクターを保持する細胞、それぞれ異なる複数の該抗原結合分子を含むライブラリ、当該抗原結合分子を含む医薬組成物、当該抗原結合分子をスクリーニングする方法、およびそれを製造する方法等を提供することを課題とする。本発明者らは、腫瘍組織特異的な低分子化合物としてMethylthioadenosine(MTA)を見出し、MTAの濃度に依存して抗原に対する結合活性が変化する抗原結合ドメインまたは抗原結合ドメインを含む抗原結合分子を創作し、さらにはそれぞれ異なる複数の抗原結合ドメインまたは該抗原結合ドメインを含む抗原結合分子を含むライブラリを創作し、該ライブラリを用いることで、上記課題を解決できることを見出した。本開示の抗原結合分子を用ることにより、標的組織(例えば、腫瘍組織)に起因する各種疾患(例えば、癌)を標的組織特異的に治療することが可能となる。
Description
本開示は、Methylthioadenosine(MTA)依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子、当該抗原結合ドメインまたは当該抗原結合分子の製造方法およびスクリーニング方法、当該抗原結合ドメインまたは当該抗原結合分子を取得するためのライブラリとそのデザイン方法、ならびに当該抗原結合分子を含む医薬組成物に関する。また、本開示は低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを効率的に取得するためのライブラリの設計方法にも関する。更に、本開示はMTAに特異的に結合する抗原結合分子及び当該抗原結合分子を用いるMTA濃度測定方法や疾患の診断方法にも関する。
抗体は血漿中での安定性が高く、副作用も少ないことから医薬品として注目されている。中でもIgG型の抗体医薬は多数上市されており、現在も数多くの抗体医薬が開発されている(非特許文献1、および非特許文献2)。
抗体医薬を用いたがん治療薬として、これまでのCD20抗原に対するリツキサン、EGFR抗原に対するセツキシマブ、HER2抗原に対するハーセプチン等が承認されている(非特許文献3)。これらの抗体分子は、がん細胞に発現している抗原に対して結合し、ADCC等によってがん細胞に対する傷害活性を発揮する。こうしたADCC等による細胞傷害活性は、治療用抗体の標的細胞に発現する抗原の数に依存することが知られている(非特許文献4)ため、標的となる抗原の発現量が高いことが治療用抗体の効果の観点からは好ましい。しかし、抗原の発現量が高くても、正常組織に抗原が発現していると、正常細胞に対してADCC等の傷害活性を発揮してしまうため、副作用が大きな問題となる。そのため、がん治療薬として治療用抗体が標的とする抗原は、がん細胞に特異的に発現していることが好ましい。
ADCC活性による細胞傷害活性を発揮する抗体医薬の成功を受けて、天然型ヒトIgG1のFc領域のN型糖鎖のフコースを除去することによるADCC活性の増強(非特許文献5)、天然型ヒトIgG1のFc領域のアミノ酸置換によりFcγRIIIaへの結合を増強することによるADCC活性の増強(非特許文献6)等によって強力な細胞傷害活性を発揮する第二世代の改良抗体分子が報告されている。上述のNK細胞が介在するADCC活性以外のメカニズムでがん細胞に傷害活性を発揮する抗体医薬として、強力な細胞傷害活性のある薬物を抗体とコンジュゲートしたAntibody Drug Conjugate(ADC)(非特許文献7)、および、T細胞をがん細胞にリクルートすることによってがん細胞に対する傷害活性を発揮する低分子抗体(非特許文献8)等のより強力な細胞傷害活性を発揮する改良抗体分子も報告されている。
こうしたより強力な細胞傷害活性を発揮する抗体分子は、抗原の発現が多くはないがん細胞に対しても細胞傷害活性を発揮することが出来る一方で、抗原の発現が少ない正常組織に対しても同様に細胞傷害活性を発揮してしまう。実際、EGFR抗原に対する天然型ヒトIgG1であるセツキシマブと比較して、CD3とEGFRに対する二重特異性抗体であるEGFR-BiTEはT細胞をがん細胞にリクルートすることによってがん細胞に対して強力な細胞傷害活性を発揮し抗腫瘍効果を発揮することができる。その一方で、EGFRは正常組織においても発現しているため、EGFR-BiTEをカニクイザルに投与した際に深刻な副作用が現れることも認められている(非特許文献9)。また、がん細胞で高発現しているCD44v6に対する抗体にmertansineを結合させたADCであるbivatuzumab mertansineは、CD44v6が正常組織においても発現していることから、臨床において重篤な皮膚毒性&肝毒性が認められている(非特許文献10)。
このように抗原の発現が少ないようながん細胞に対しても強力な細胞傷害活性を発揮することが出来る抗体を用いた場合、標的抗原が極めてがん特異的に発現している必要があるが、ハーセプチンの標的抗原であるHER2やセツキシマブの標的抗原であるEGFRは正常組織にも発現しているように、極度にがん特異的に発現しているがん抗原の数は限られていると考えられる。そのため、がんに対する細胞傷害活性を強化することはできるものの、正常組織に対する細胞傷害作用による副作用が問題となり得る。
また、最近、がんにおける免疫抑制に寄与しているCTLA4を阻害することによって腫瘍免疫を増強するイプリムマブが転移性メラノーマに対してOverall survivalを延長させることが示された(非特許文献11)。しかしながら、イプリムマブはCTLA4を全身的に阻害するため、腫瘍免疫が増強される一方で、全身的に免疫が活性化されることによる自己免疫疾患様の重篤な副作用を示すことが問題となっている(非特許文献12)。
第二世代の抗体医薬に適用可能な技術として様々な技術が開発されており、エフェクター機能、抗原結合能、薬物動態、安定性を向上させる、あるいは、免疫原性リスクを低減させる技術等が報告されているが(非特許文献13)、上記のような副作用を解決するための、抗体医薬を疾患組織に特異的に作用可能とする技術はほとんど報告されていない。例えば、がん組織や炎症性組織のような病変部位については、これらの疾患組織におけるpHが酸性条件であることを利用したpH依存性抗体が報告されている(特許文献1、2)。しかしながら、がん組織や炎症性組織における正常組織と比較したpHの低下(すなわち水素イオン濃度の上昇)は僅かであり、分子量が極めて小さい水素イオン濃度の僅かな上昇を検知して作用する抗体を作製することは困難であると同時に、破骨細胞骨吸収窩領域等正常組織や対象とする病変以外の組織でもpHが酸性条件である場合もあり、pHの条件が病変部位に特異的な環境因子として利用するにはなお多くの課題があると考えられた。一方、がん組織や炎症性組織のような病変部位で発現するプロテアーゼで切断されることによって、初めて抗原結合活性を発揮する抗体を作製する方法が報告されている(特許文献3)。しかし、プロテアーゼによる抗体の切断は不可逆的であるため、当該病変部位で切断された抗体が、正常組織に血流に乗って戻ることで正常組織でも抗原に結合できてしまうことが課題であると考えられた。また、そのようなプロテアーゼのがん特異性にも課題があると考えられた。そのような課題を克服することを目的として、疾患組織特異体な化合物の濃度に応じて抗原に対する結合活性が変化する抗原結合分子が報告されている(特許文献4、5)。
Monoclonal antibody successes in the clinic. Janice M Reichert, Clark J Rosensweig, Laura B Faden & Matthew C Dewitz, Nat. Biotechnol. (2005) 23, 1073 - 1078
The therapeutic antibodies market to 2008. Pavlou AK, Belsey MJ., Eur. J. Pharm. Biopharm. (2005) 59 (3), 389-396
Monoclonal antibodies: versatile platforms for cancer immunotherapy. Weiner LM, Surana R, Wang S., Nat. Rev. Immunol. (2010) 10 (5), 317-327
Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, Shepard HM, Cancer Immunol. Immunotherapy (1993) 37, 255-263
Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Satoh M, Iida S, Shitara K., Expert Opin. Biol. Ther. (2006) 6 (11), 1161-1173
Optimizing engagement of the immune system by anti-tumor antibodies: an engineer's perspective. Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY., Drug Discov. Today (2007) 12 (21-22), 898-910
Antibody-drug conjugates: targeted drug delivery for cancer. Alley SC, Okeley NM, Senter PD., Curr. Opin. Chem. Biol. (2010) 14 (4), 529-537
BiTE: Teaching antibodies to engage T-cells for cancer therapy. Baeuerle PA, Kufer P, Bargou R., Curr. Opin. Mol. Ther. (2009) 11 (1), 22-30
T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, Friedrich M, Thomas O, Lorenczewski G, Rau D, Schaller E, Herrmann I, Wolf A, Urbig T, Baeuerle PA, Kufer P., Proc. Natl. Acad. Sci. U.S.A. (2010) 107 (28), 12605-12610
Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K, Erhardt T, Gronau S., Oral Oncol. (2008) 44 (9), 823-829
Ipilimumab in the treatment of melanoma. Trinh VA, Hwu WJ., Expert Opin. Biol. Ther., (2012) Apr 14 (doi:10.1517/14712598.2012.675325)
IPILIMUMAB - A NOVEL IMMUNOMODULATING THERAPY CAUSING AUTOIMMUNE HYPOPHYSITIS: A CASE REPORT AND REVIEW. Juszczak A, Gupta A, Karavitaki N, Middleton MR, Grossman A., Eur. J. Endocrinol. (2012) Apr 10 (doi: 10.1530/EJE-12-0167)
Antibody engineering for the development of therapeutic antibodies. Kim SJ, Park Y, Hong HJ., Mol. Cells. (2005) 20 (1), 17-29
本開示は、疾患組織において特異的に存在または産生される低分子化合物を新たに見出し、さらに当該低分子化合物に依存的に標的抗原への結合が制御される抗原結合分子(低分子化合物スイッチ抗原結合分子)を提供し、さらには斯かる抗原結合分子を短期間に効率的に取得する方法の提供を目的の一つとする。
本発明者らは、上記の目的を達成するために鋭意研究を進めたところ、がん組織に特異的な低分子化合物としてMethylthioadenosine(MTA)を新たに見出し、さらにMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を創作した。また、本発明者らは、当該抗原結合分子または当該抗原結合分子を含む医薬組成物が、がん治療に有用であることを見出すとともに、当該抗原結合分子を投与することを含むがん治療に有用であること、およびがん治療のための医薬の製造において当該抗原結合分子が有用であることを見出した。
本発明者らはまた、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインをスクリーニングおよび製造する方法を創作した。 また、本発明者らはMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを効率よくスクリーニングできるライブラリの作製に成功した。本発明者らはさらに、MTAおよび/またはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインをスクリーニングできるライブラリの作成に成功し、前記MTAおよび/またはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインをスクリーニングする方法および製造する方法も創作した。
さらに、本発明者らは、MTA自体に対して特異的に結合する抗原結合分子の取得にも成功した。
さらに、本発明者らは、MTA自体に対して特異的に結合する抗原結合分子の取得にも成功した。
本開示はこのような知見に基づくものであり、具体的には以下に例示的に記載する態様を包含するものである。
[A1] 5’-Methylthioadenosine(MTA)依存的に抗原に対する結合活性が 変化する抗原結合ドメインを含む抗原結合分子。
[A2] 前記抗原結合ドメインのMTA存在下における前記抗原に対する結合活性がMTA非存在下における前記抗原に対する結合活性と異なる、抗原結合分子。
[A3] [A1]または[A2]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はアデノシンに実質的に影響されない、抗原結合分子。
[A4] [A1]から[A3]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はS-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に影響されない、抗原結合分子。
[A5] [A1]から[A4]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はAMP、ADPまたはATPに実質的に影響されない、抗原結合分子。
[A6] [A1]または[A2]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はアデノシンに対しても依存的に変化する、抗原結合分子。
[A7] [A1]、[A2]または[A6]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はS-(5'-Adenosyl)-L-homocysteine (SAH)に対しても依存的に変化する、抗原結合分子。
[A8] [A1]、[A2]、[A6]または[A7]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はAMP、ADPおよび/または ATPに対しても依存的に変化する、抗原結合分子。
[A9] 前記抗原結合ドメインは抗体可変領域または/及び単ドメイン抗体を含む、[A1]から[A8]のいずれか一つに記載の抗原結合分子。
[A10] 前記抗原結合分子が抗体である、[A1]から[A9]のいずれか一つに記載の抗原結合分子。
[A11] 抗体Fc領域を含む[A1]から[A10]のいずれか一つに記載の抗原結合分子。
[A12] 前記抗体Fc領域が天然型Fc領域または改変Fc領域である、[A11]に記載の抗原結合分子。
[A13] 前記抗原結合ドメインのMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より強い、[A1]から[A12]のいずれか一つに記載の抗原結合分子。
[A14] 前記抗原結合ドメインのMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より弱い、[A1]から[A12]のいずれか一つに記載の抗原結合分子。
[A15] 前記抗原結合ドメインはMTAと相互作用するアミノ酸残基を有する、[A1]から[A14]いずれか一つに記載の抗原結合分子。
[A16] 前記MTAと相互作用するアミノ酸残基は、前記抗原結合ドメインの抗原と結合している状態においてMTAと相互作用する、[A15]に記載の抗原結合分子。
[A17] 前記抗原結合ドメインは抗体可変領域または単ドメイン抗体を含んでおり、前記MTAと相互作用するアミノ酸残基は前記抗体可変領域または前記単ドメイン抗体のCDR中に位置する、[A15]または[A16]に記載の抗原結合分子。
[A18] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、35a位、47位、52位、52e位、101位、軽鎖32位、34位、36位、46位、49位、50位、89位、90位、91位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A19] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、C35a、W47、F52、Y52e、E101、軽鎖R32、S34、Y36、L46、Y49、S50、A89、G90、L91、及びP96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む [A15]から[A18]のいずれか一つに記載の抗原結合分子。
[A20] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A19]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するF;
重鎖52a位に位置するA;
重鎖52b位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
重鎖52c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52d位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52e位に位置するY;
重鎖52f位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52g位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖53位に位置するS;
重鎖54位に位置するG;
重鎖55位に位置するG;
重鎖56位に位置するS;
重鎖57位に位置するT;
重鎖58位に位置するY;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するA;
重鎖64位に位置するK;
重鎖65位に位置するG;
重鎖95位に位置するG;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するG;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100a位に位置するG;
重鎖100b位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖101位に位置するE;
重鎖102位に位置するL;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するE;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖28位に位置するV;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するT;
軽鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖55位に位置するP;
軽鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖89位に位置するA;
軽鎖90位に位置するG;
軽鎖91位に位置するL;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するG;
軽鎖95位に位置するN;
軽鎖95a位に位置するI;
軽鎖96位に位置するP;
軽鎖97位に位置するA。
[A21] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、47位、50位、58位、95位、98位、99位、100a位、軽鎖28位、91位、95b位、95c位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A22] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、W47、C50、Y58、E95、F98、G99、G100a、軽鎖Y28、T91、F95b、Y95c、及びF96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む、[A15]から[A17]、および[A21]のいずれか一つに記載の抗原結合分子。
[A23] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A17]のいずれかまたは[A21]から[A22]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52a位に位置するS;
重鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するT;
重鎖58位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するN;
重鎖65位に位置するG;
重鎖95位に位置するE;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するS;
重鎖100a位に位置するG;
重鎖100b位に位置するA;
重鎖100c位に位置するL;
重鎖101位に位置するN;
重鎖102位に位置するL;
軽鎖24位に位置するH;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するK;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖27b位に位置するV;
軽鎖28位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するA;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するQ;
軽鎖90位に位置するG;
軽鎖91位に位置するT;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95b位に位置するF;
軽鎖95c位に位置するY;
軽鎖96位に位置するF;
軽鎖97位に位置するA。
[A24] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖33位、50位、52位、54位、56位、57位、58位、99位、100位、100a位、軽鎖91位、95c位、及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A25] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖A33、I50、G52、D54、S56、T57、W58、G99、Y100、T100a、軽鎖S91、Y95c、及びN96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む、 [A15]から[A17]、および[A24]のいずれか一つに記載の抗原結合分子。
[A26] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A17]のいずれかまたは[A24]から[A25]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖26位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖28位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖29位に位置するAまたはLのいずれか;
重鎖30位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するD、E、F、H、N、P、RまたはYのいずれか;
重鎖33位に位置するA、I、P、TまたはVのいずれか;
重鎖34位に位置するA、E、F、H、I、K、L、M、N、Q、S、T、V、WまたはYのいずれか;
重鎖35位に位置するG;
重鎖50位に位置するD、IまたはVのいずれか;
重鎖51位に位置するI;
重鎖52位に位置するG;
重鎖53位に位置するA、D、E、G,I、K、QまたはRのいずれか;
重鎖54位に位置するD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、D、E、F、GまたはHのいずれか;
重鎖56位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するA、D、E、G、H、I、K、L、N、P、Q、R、S、TまたはVのいずれか;
重鎖58位に位置するW;
重鎖59位に位置するA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WまたはYのいずれか;
重鎖60位に位置するP;
重鎖61位に位置するA、F、Q、R、S、T、V、WまたはYのいずれか;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するK;
重鎖65位に位置するA、FまたはG;
重鎖95位に位置するG;
重鎖96位に位置するA、E、F、G、H、K、L、Q、R、S、T、WまたはYのいずれか;
重鎖97位に位置するA、F、H、K、N、WまたはYのいずれか;
重鎖98位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、D、E、G、H、QまたはSのいずれか;
重鎖100位に位置するFまたはY;
重鎖100a位に位置するN、TまたはV
重鎖100b位に位置するN;
重鎖100c位に位置するA;
重鎖100d位に位置するFまたはW;
重鎖101位に位置するD;
重鎖102位に位置するP;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するQ;
軽鎖27e位に位置するS;
軽鎖27f位に位置するV;
軽鎖28位に位置するA、E、F、H、I、K、L、N、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するN;
軽鎖31位に位置するN ;
軽鎖32位に位置するA、E、F、G、H、SまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖50位に位置するD ;
軽鎖51位に位置するA;
軽鎖52位に位置するS ;
軽鎖53位に位置するT ;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するH;
軽鎖90位に位置するG;
軽鎖91位に位置するA、SまたはTのいずれか;
軽鎖92位に位置するA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WまたはYのいずれか;
軽鎖93位に位置するA、D、E、F、G、H、L、N、Q、R、S、T、VまたはYのいずれか;
軽鎖94位に位置するA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WまたはYのいずれか;
軽鎖95b位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95c位に位置するA、F、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖96d位に位置するD;
軽鎖96位に位置するN;
軽鎖97位に位置するAまたはG;
軽鎖98位に位置するA、F、I、LまたはV。
[A27] 前記抗原はMTA以外の分子である、またはMTA以外の分子でありヒトの生体内で免疫原性を有する分子である[A1]から[A26]のいずれか一つに記載の抗原結合分子。
[A28] 前記抗原はペプチド、ポリペプチド、タンパク質のいずれかである、[A1]から[A27]のいずれか一つに記載の抗原結合分子。
[A29] 前記抗原結合分子は更に第2の抗原結合ドメインを有し、前記第2の抗原結合ドメインは前記抗原結合ドメインが結合する抗原と異なる第2の抗原に対して結合活性を有する、[A1]から[A28]のいずれか一つに記載の抗原結合分子。
[A30] 前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTAに実質的に影響されない、[A29]に記載の抗原結合分子。
[A31] 前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTA依存的に変化する、[A29]に記載の抗原結合分子。
[A32] 前記第2の抗原結合ドメインのMTA存在下における前記第2の抗原に対する結合活性が、前記第2の抗原結合ドメインのMTA非存在下における前記第2の抗原に対する結合活性と異なる、[A31]に記載の抗原結合分子。
[A33] 前記抗原が膜型分子または可溶型分子である、[A1]から[A32]のいずれか一つに記載の抗原結合分子。
[A34] 前記抗原が膜型分子であり、且つ疾患組織で発現している抗原である[A33]に記載の抗原結合分子。
[A35] 前記抗原が可溶型分子であり、且つがん組織で発現している抗原である[A33]に記載の抗原結合分子。
[A36] 前記がん組織で発現している抗原ががん細胞に発現している抗原、もしくはがん組織中のがん間質細胞や免疫組織で発現している抗原である[A35]に記載の抗原結合分子。
[A37] 前記がん組織はMTAが蓄積しているがん組織である、[A35]または[A36]に記載の抗原結合分子。
[A38] 前記がん組織は、MTA phospholylase (MTAP)をコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん組織である[A35]から[A37]のいずれか一つに記載の抗原結合分子。
[A39] 前記がん組織は、MTAPの活性が欠損または低下しているがん組織である[A35]から[A37]のいずれか一つに記載の抗原結合分子。
[A40] [A1]から[A39]のいずれか一つに記載の抗原結合分子であって、前記抗原が膜型分子であり、前記抗原結合分子は前記抗原を発現する細胞に対して細胞傷害活性を示す、抗原結合分子。
[A41] ADCC活性、ADCP活性またはCDC活性の中から選択される少なくとも1つ以上の細胞傷害活性を示す[A40]に記載の抗原結合分子。
[A42] 前記抗原対してアゴニスト活性を有する[A1]から[A39]に記載の抗原結合分子。
[A43] 前記抗原と前記第2の抗原の一方は標的細胞において発現している抗原であり、他方はエフェクター細胞において発現している抗原である[A29]から[A32]のいずれか一つに記載の抗原結合分子。
[A44] 前記標的細胞はがん細胞である、[A43]に記載の抗原結合分子。
[A45] 前記がん細胞はMTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞、または前記MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞の周囲に存在するがん細胞である[A44]に記載の抗原結合分子。
[A46] 前記標的細胞はMTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞の周囲に存在するがん細胞以外の細胞である[A43]に記載の抗原結合分子。
[A47] 前記がん細胞の周囲に存在するがん細胞以外の細胞はcancer - associated fibroblasts (CAF) またはtumor associated macrophages (TAM)である[A43]に記載の抗原結合分子。
[A48] 前記エフェクター細胞がT細胞である、[A43]から[47]のいずれか一つに記載の抗原結合分子。
[A49] 前記エフェクター細胞において発現している抗原がT細胞レセプター(TCR)複合体である、[A48]に記載の抗原結合分子。
[A50] 前記エフェクター細胞において発現している抗原がCD3である、[A48]または[A49]のいずれか記載の抗原結合分子。
[A51] エフェクター細胞を活性化することで、標的細胞に対して細胞傷害活性を惹起する[A48]から[A50]のいずれか一つに記載の抗原結合分子。
[A52] TDCC活性を有する、[A48]から[A51]のいずれか一つに記載の抗原結合分子。
[A53] [A1]から[A39]のいずれか一つに記載の抗原結合分子であって、前記抗原が可溶型分子であり、前記抗原結合分子は前記抗原に対して中和活性を示す、抗原結合分子。
[A54] 前記抗原結合ドメインのMTA非存在下における前記抗原に対するKD値と前記抗原結合ドメインのMTA存在下における前記抗原に対するKD値が異なる[A1]から[A53]のいずれか一つに記載の抗原結合分子。
[A55] [A1]から[A54]のいずれか一つに記載の抗原結合分子を含む医薬組成物。
[A56] [A1]から[A55]のいずれか一つに記載の抗原結合分子を有効成分として含むがんの治療用医薬組成物。
[A57] 前記がんが、MTAが組織中に蓄積しているがんである[A56]に記載の医薬組成物。
[A58] 前記がんが、MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがんである、[A56]から[A57]のいずれか一つに記載の医薬組成物。
[A59] 前記がんが、MTAPの活性が欠損または低下しているであるがんある、[A56]から[A58]のいずれか一つに記載の医薬組成物。
[A60] [A1]から[A54]のいずれか一つに記載の抗原結合分子を製造する方法。
[A61] [A1]から[A52]のいずれか一つに記載の抗原結合分子コードするポリヌクレオチド。
[A62] [A61]に記載のポリヌクレオチドを含むベクター
[A63] [A62]に記載のベクターを保持する細胞。
[A64] [A63]に記載の細胞を培養し、培養上清から回収される抗原結合分子。
[G1] MTAの濃度依存的に結合しない対照の抗原結合分子と比較して、高い血漿中滞留性を有する、および/または低い血漿中抗原蓄積能を有する、[A1]から[A54]のいずれか一つに記載の抗原結合分子。
[G2] [G1]に記載の抗原結合分子および薬学的に許容される担体を含む、薬学的製剤。
[G3] 対照の抗原結合分子と比較して、高い血漿中滞留性を有する、および/または低い血漿中抗原蓄積能を有する抗原結合分子を製造する方法であって、(a) MTAの濃度が高まるにつれて抗原結合活性が増大する抗原結合分子を製造する工程、および(b) (a)で製造された抗原結合分子の血漿中滞留性、および/または血漿中抗原蓄積能を測定する工程を含む、方法。
[A1] 5’-Methylthioadenosine(MTA)依存的に抗原に対する結合活性が 変化する抗原結合ドメインを含む抗原結合分子。
[A2] 前記抗原結合ドメインのMTA存在下における前記抗原に対する結合活性がMTA非存在下における前記抗原に対する結合活性と異なる、抗原結合分子。
[A3] [A1]または[A2]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はアデノシンに実質的に影響されない、抗原結合分子。
[A4] [A1]から[A3]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はS-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に影響されない、抗原結合分子。
[A5] [A1]から[A4]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はAMP、ADPまたはATPに実質的に影響されない、抗原結合分子。
[A6] [A1]または[A2]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はアデノシンに対しても依存的に変化する、抗原結合分子。
[A7] [A1]、[A2]または[A6]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はS-(5'-Adenosyl)-L-homocysteine (SAH)に対しても依存的に変化する、抗原結合分子。
[A8] [A1]、[A2]、[A6]または[A7]に記載の抗原結合分子であって、当該抗原結合分子に含まれる抗原結合ドメインの前記抗原に対する結合活性はAMP、ADPおよび/または ATPに対しても依存的に変化する、抗原結合分子。
[A9] 前記抗原結合ドメインは抗体可変領域または/及び単ドメイン抗体を含む、[A1]から[A8]のいずれか一つに記載の抗原結合分子。
[A10] 前記抗原結合分子が抗体である、[A1]から[A9]のいずれか一つに記載の抗原結合分子。
[A11] 抗体Fc領域を含む[A1]から[A10]のいずれか一つに記載の抗原結合分子。
[A12] 前記抗体Fc領域が天然型Fc領域または改変Fc領域である、[A11]に記載の抗原結合分子。
[A13] 前記抗原結合ドメインのMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より強い、[A1]から[A12]のいずれか一つに記載の抗原結合分子。
[A14] 前記抗原結合ドメインのMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より弱い、[A1]から[A12]のいずれか一つに記載の抗原結合分子。
[A15] 前記抗原結合ドメインはMTAと相互作用するアミノ酸残基を有する、[A1]から[A14]いずれか一つに記載の抗原結合分子。
[A16] 前記MTAと相互作用するアミノ酸残基は、前記抗原結合ドメインの抗原と結合している状態においてMTAと相互作用する、[A15]に記載の抗原結合分子。
[A17] 前記抗原結合ドメインは抗体可変領域または単ドメイン抗体を含んでおり、前記MTAと相互作用するアミノ酸残基は前記抗体可変領域または前記単ドメイン抗体のCDR中に位置する、[A15]または[A16]に記載の抗原結合分子。
[A18] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、35a位、47位、52位、52e位、101位、軽鎖32位、34位、36位、46位、49位、50位、89位、90位、91位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A19] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、C35a、W47、F52、Y52e、E101、軽鎖R32、S34、Y36、L46、Y49、S50、A89、G90、L91、及びP96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む [A15]から[A18]のいずれか一つに記載の抗原結合分子。
[A20] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A19]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するF;
重鎖52a位に位置するA;
重鎖52b位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
重鎖52c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52d位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52e位に位置するY;
重鎖52f位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52g位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖53位に位置するS;
重鎖54位に位置するG;
重鎖55位に位置するG;
重鎖56位に位置するS;
重鎖57位に位置するT;
重鎖58位に位置するY;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するA;
重鎖64位に位置するK;
重鎖65位に位置するG;
重鎖95位に位置するG;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するG;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100a位に位置するG;
重鎖100b位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖101位に位置するE;
重鎖102位に位置するL;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するE;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖28位に位置するV;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するT;
軽鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖55位に位置するP;
軽鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖89位に位置するA;
軽鎖90位に位置するG;
軽鎖91位に位置するL;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するG;
軽鎖95位に位置するN;
軽鎖95a位に位置するI;
軽鎖96位に位置するP;
軽鎖97位に位置するA。
[A21] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、47位、50位、58位、95位、98位、99位、100a位、軽鎖28位、91位、95b位、95c位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A22] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、W47、C50、Y58、E95、F98、G99、G100a、軽鎖Y28、T91、F95b、Y95c、及びF96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む、[A15]から[A17]、および[A21]のいずれか一つに記載の抗原結合分子。
[A23] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A17]のいずれかまたは[A21]から[A22]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52a位に位置するS;
重鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するT;
重鎖58位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するN;
重鎖65位に位置するG;
重鎖95位に位置するE;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するS;
重鎖100a位に位置するG;
重鎖100b位に位置するA;
重鎖100c位に位置するL;
重鎖101位に位置するN;
重鎖102位に位置するL;
軽鎖24位に位置するH;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するK;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖27b位に位置するV;
軽鎖28位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するA;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するQ;
軽鎖90位に位置するG;
軽鎖91位に位置するT;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95b位に位置するF;
軽鎖95c位に位置するY;
軽鎖96位に位置するF;
軽鎖97位に位置するA。
[A24] 前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖33位、50位、52位、54位、56位、57位、58位、99位、100位、100a位、軽鎖91位、95c位、及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である、[A15]から[A17]のいずれか一つに記載の抗原結合分子。
[A25] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖A33、I50、G52、D54、S56、T57、W58、G99、Y100、T100a、軽鎖S91、Y95c、及びN96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む、 [A15]から[A17]、および[A24]のいずれか一つに記載の抗原結合分子。
[A26] 前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、[A1]から[A17]のいずれかまたは[A24]から[A25]のいずれか一つに記載の抗原結合分子(Kabatナンバリング):
重鎖26位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖28位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖29位に位置するAまたはLのいずれか;
重鎖30位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するD、E、F、H、N、P、RまたはYのいずれか;
重鎖33位に位置するA、I、P、TまたはVのいずれか;
重鎖34位に位置するA、E、F、H、I、K、L、M、N、Q、S、T、V、WまたはYのいずれか;
重鎖35位に位置するG;
重鎖50位に位置するD、IまたはVのいずれか;
重鎖51位に位置するI;
重鎖52位に位置するG;
重鎖53位に位置するA、D、E、G,I、K、QまたはRのいずれか;
重鎖54位に位置するD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、D、E、F、GまたはHのいずれか;
重鎖56位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するA、D、E、G、H、I、K、L、N、P、Q、R、S、TまたはVのいずれか;
重鎖58位に位置するW;
重鎖59位に位置するA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WまたはYのいずれか;
重鎖60位に位置するP;
重鎖61位に位置するA、F、Q、R、S、T、V、WまたはYのいずれか;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するK;
重鎖65位に位置するA、FまたはG;
重鎖95位に位置するG;
重鎖96位に位置するA、E、F、G、H、K、L、Q、R、S、T、WまたはYのいずれか;
重鎖97位に位置するA、F、H、K、N、WまたはYのいずれか;
重鎖98位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、D、E、G、H、QまたはSのいずれか;
重鎖100位に位置するFまたはY;
重鎖100a位に位置するN、TまたはV
重鎖100b位に位置するN;
重鎖100c位に位置するA;
重鎖100d位に位置するFまたはW;
重鎖101位に位置するD;
重鎖102位に位置するP;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するQ;
軽鎖27e位に位置するS;
軽鎖27f位に位置するV;
軽鎖28位に位置するA、E、F、H、I、K、L、N、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するN;
軽鎖31位に位置するN ;
軽鎖32位に位置するA、E、F、G、H、SまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖50位に位置するD ;
軽鎖51位に位置するA;
軽鎖52位に位置するS ;
軽鎖53位に位置するT ;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するH;
軽鎖90位に位置するG;
軽鎖91位に位置するA、SまたはTのいずれか;
軽鎖92位に位置するA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WまたはYのいずれか;
軽鎖93位に位置するA、D、E、F、G、H、L、N、Q、R、S、T、VまたはYのいずれか;
軽鎖94位に位置するA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WまたはYのいずれか;
軽鎖95b位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95c位に位置するA、F、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖96d位に位置するD;
軽鎖96位に位置するN;
軽鎖97位に位置するAまたはG;
軽鎖98位に位置するA、F、I、LまたはV。
[A27] 前記抗原はMTA以外の分子である、またはMTA以外の分子でありヒトの生体内で免疫原性を有する分子である[A1]から[A26]のいずれか一つに記載の抗原結合分子。
[A28] 前記抗原はペプチド、ポリペプチド、タンパク質のいずれかである、[A1]から[A27]のいずれか一つに記載の抗原結合分子。
[A29] 前記抗原結合分子は更に第2の抗原結合ドメインを有し、前記第2の抗原結合ドメインは前記抗原結合ドメインが結合する抗原と異なる第2の抗原に対して結合活性を有する、[A1]から[A28]のいずれか一つに記載の抗原結合分子。
[A30] 前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTAに実質的に影響されない、[A29]に記載の抗原結合分子。
[A31] 前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTA依存的に変化する、[A29]に記載の抗原結合分子。
[A32] 前記第2の抗原結合ドメインのMTA存在下における前記第2の抗原に対する結合活性が、前記第2の抗原結合ドメインのMTA非存在下における前記第2の抗原に対する結合活性と異なる、[A31]に記載の抗原結合分子。
[A33] 前記抗原が膜型分子または可溶型分子である、[A1]から[A32]のいずれか一つに記載の抗原結合分子。
[A34] 前記抗原が膜型分子であり、且つ疾患組織で発現している抗原である[A33]に記載の抗原結合分子。
[A35] 前記抗原が可溶型分子であり、且つがん組織で発現している抗原である[A33]に記載の抗原結合分子。
[A36] 前記がん組織で発現している抗原ががん細胞に発現している抗原、もしくはがん組織中のがん間質細胞や免疫組織で発現している抗原である[A35]に記載の抗原結合分子。
[A37] 前記がん組織はMTAが蓄積しているがん組織である、[A35]または[A36]に記載の抗原結合分子。
[A38] 前記がん組織は、MTA phospholylase (MTAP)をコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん組織である[A35]から[A37]のいずれか一つに記載の抗原結合分子。
[A39] 前記がん組織は、MTAPの活性が欠損または低下しているがん組織である[A35]から[A37]のいずれか一つに記載の抗原結合分子。
[A40] [A1]から[A39]のいずれか一つに記載の抗原結合分子であって、前記抗原が膜型分子であり、前記抗原結合分子は前記抗原を発現する細胞に対して細胞傷害活性を示す、抗原結合分子。
[A41] ADCC活性、ADCP活性またはCDC活性の中から選択される少なくとも1つ以上の細胞傷害活性を示す[A40]に記載の抗原結合分子。
[A42] 前記抗原対してアゴニスト活性を有する[A1]から[A39]に記載の抗原結合分子。
[A43] 前記抗原と前記第2の抗原の一方は標的細胞において発現している抗原であり、他方はエフェクター細胞において発現している抗原である[A29]から[A32]のいずれか一つに記載の抗原結合分子。
[A44] 前記標的細胞はがん細胞である、[A43]に記載の抗原結合分子。
[A45] 前記がん細胞はMTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞、または前記MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞の周囲に存在するがん細胞である[A44]に記載の抗原結合分子。
[A46] 前記標的細胞はMTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん細胞の周囲に存在するがん細胞以外の細胞である[A43]に記載の抗原結合分子。
[A47] 前記がん細胞の周囲に存在するがん細胞以外の細胞はcancer - associated fibroblasts (CAF) またはtumor associated macrophages (TAM)である[A43]に記載の抗原結合分子。
[A48] 前記エフェクター細胞がT細胞である、[A43]から[47]のいずれか一つに記載の抗原結合分子。
[A49] 前記エフェクター細胞において発現している抗原がT細胞レセプター(TCR)複合体である、[A48]に記載の抗原結合分子。
[A50] 前記エフェクター細胞において発現している抗原がCD3である、[A48]または[A49]のいずれか記載の抗原結合分子。
[A51] エフェクター細胞を活性化することで、標的細胞に対して細胞傷害活性を惹起する[A48]から[A50]のいずれか一つに記載の抗原結合分子。
[A52] TDCC活性を有する、[A48]から[A51]のいずれか一つに記載の抗原結合分子。
[A53] [A1]から[A39]のいずれか一つに記載の抗原結合分子であって、前記抗原が可溶型分子であり、前記抗原結合分子は前記抗原に対して中和活性を示す、抗原結合分子。
[A54] 前記抗原結合ドメインのMTA非存在下における前記抗原に対するKD値と前記抗原結合ドメインのMTA存在下における前記抗原に対するKD値が異なる[A1]から[A53]のいずれか一つに記載の抗原結合分子。
[A55] [A1]から[A54]のいずれか一つに記載の抗原結合分子を含む医薬組成物。
[A56] [A1]から[A55]のいずれか一つに記載の抗原結合分子を有効成分として含むがんの治療用医薬組成物。
[A57] 前記がんが、MTAが組織中に蓄積しているがんである[A56]に記載の医薬組成物。
[A58] 前記がんが、MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがんである、[A56]から[A57]のいずれか一つに記載の医薬組成物。
[A59] 前記がんが、MTAPの活性が欠損または低下しているであるがんある、[A56]から[A58]のいずれか一つに記載の医薬組成物。
[A60] [A1]から[A54]のいずれか一つに記載の抗原結合分子を製造する方法。
[A61] [A1]から[A52]のいずれか一つに記載の抗原結合分子コードするポリヌクレオチド。
[A62] [A61]に記載のポリヌクレオチドを含むベクター
[A63] [A62]に記載のベクターを保持する細胞。
[A64] [A63]に記載の細胞を培養し、培養上清から回収される抗原結合分子。
[G1] MTAの濃度依存的に結合しない対照の抗原結合分子と比較して、高い血漿中滞留性を有する、および/または低い血漿中抗原蓄積能を有する、[A1]から[A54]のいずれか一つに記載の抗原結合分子。
[G2] [G1]に記載の抗原結合分子および薬学的に許容される担体を含む、薬学的製剤。
[G3] 対照の抗原結合分子と比較して、高い血漿中滞留性を有する、および/または低い血漿中抗原蓄積能を有する抗原結合分子を製造する方法であって、(a) MTAの濃度が高まるにつれて抗原結合活性が増大する抗原結合分子を製造する工程、および(b) (a)で製造された抗原結合分子の血漿中滞留性、および/または血漿中抗原蓄積能を測定する工程を含む、方法。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[B1] お互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸からなるライブラリであって、MTAと相互作用するアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子、または/および前記抗原結合分子をコードする核酸から主としてなるライブラリ。
[B2] 前記抗原結合ドメインが抗体可変領域である、[B1]に記載のライブラリ。
[B3] MTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体、または/およびMTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体をそれぞれコードする核酸を含む[B2]に記載のライブラリ。
[B4] 前記抗体可変領域改変体中の、前記抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である、[B3]に記載のライブラリ:
1) 前記抗体可変領域未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗体可変領域改変体と比較して、前記抗体可変領域改変体のMTAとの結合を著しく減弱させないアミノ酸部位、及び
3) 前記抗体可変領域改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
[B5] 前記抗体可変領域改変体中の、前記抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である、[B3]に記載のライブラリ:
1) 前記抗体可変領域未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗体可変領域改変体と比較して、前記抗体可変領域改変体のMTAとの結合を著しく減弱させないアミノ酸部位、
3) 前記抗体可変領域未改変体の表面に露出するアミノ酸部位に対応するアミノ酸部位、及び
4) 前記抗体可変領域未改変体においてMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に対応するアミノ酸部位。
[B6] 前記抗体可変領域の未改変体はアデノシンまたは/及びS-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に結合しない、[B3]から[B5]のいずれかに記載のライブラリ。
[B7] 前記抗体可変領域の未改変体は下記いずれかである、[B3]から[B6]のいずれか一つに記載のライブラリ:
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域。
[B8] 前記複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAXXXYXXSGGSTYYASWAKG(配列番号:66)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXXXLS(配列番号:68)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B9] 前記複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、
Xは任意のアミノ酸、
X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であり、異なる位置に存在するXもしくはX1は同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B10] 前記複数の抗体可変領域は、下記:
XXAXWMC(配列番号:73)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、
Xは任意のアミノ酸、
X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であり、異なる位置に存在するXもしくはX1は同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B11] 前記複数の抗体可変領域は、下記:
XXXWMC(配列番号:74)を含むH-CDR1;
CIXSXXXXTXYASWVNG(配列番号:75)を含むH-CDR2;
EXXXXSGALNL(配列番号:76)を含むH-CDR3;
HSSKXVXXXXXLA(配列番号:77)を含むL-CDR1;
XAXXLAS(配列番号:78)を含むL-CDR2;及び
QGTYXXXXFYFA(配列番号:79)を含むL-CDR3;
を含む抗体可変領域であり、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B12] 前記抗体可変領域の未改変体はアデノシンに対しても結合活性を有する、[B3]から[B5]のいずれか一つに記載のライブラリ。
[B13] 前記抗体可変領域の未改変体は(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに対しても結合活性を有する[B12]に記載のライブラリ。
[B14] 前記抗体可変領域の未改変体は、配列番号:31で示す重鎖可変領域と配列番号:32で示す軽鎖可変領域を有する抗体可変領域である、[B3]から[B5]、[B12]のいずれか一つに記載のライブラリ。
[B15] 前記複数の抗体可変領域は、下記:
X2X3X4X5G(配列番号:80)を含むH-CDR1;
X6IGX7X8X9X10X11WX12PX13WVKX14(配列番号:81)を含むH-CDR2;
GX15X16X17X18X19X20NAX21DP(配列番号:82)を含むH-CDR3;
QSSQSVX22X23NNX24LS(配列番号:83)を含むL-CDR1;
DASTLAS(配列番号:84)を含むL-CDR2;及び
HGX25X26X27X28X29X30X31X32DNX33(配列番号:85)を含むL-CDR3;
を含む抗体可変領域であり、
X2はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X3はD、E、F、H、K、N、P、RおよびYから選ばれるアミノ酸、
X4はA、I、P、TおよびVから選ばれるアミノ酸、
X5はA、E、F、H、I、K、L、M、N、Q、S、T、V、WおよびYから選ばれるアミノ酸、
X6はD、IおよびVから選ばれるアミノ酸、
X7はA、D、E、G、I、K、QおよびRから選ばれるアミノ酸、
X8はD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X9はA、D、E、F、G、HおよびSから選ばれるアミノ酸、
X10はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X11はA、D、E、G、H、I、K、L、N、P、Q、R、S、TおよびVから選ばれるアミノ酸、
X12はA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X13はA、F、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X14はA、FおよびGから選ばれるアミノ酸、
X15はA、E、F、G、H、K、L、Q、R、S、T、WおよびYから選ばれるアミノ酸、
X16はF、H、K、N、WおよびYから選ばれるアミノ酸、
X17はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X18はA、D、E、G、H、QおよびSから選ばれるアミノ酸、
X19はFおよびYから選ばれるアミノ酸、
X20はN、TおよびVから選ばれるアミノ酸、
X21はFおよびWから選ばれるアミノ酸、
X22はA、E、F、H、I、K、L、N、R、S、T、V、WおよびYから選ばれるアミノ酸、
X23はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X24はA、E、F、G、H、SおよびYから選ばれるアミノ酸、
X25はA、SおよびTから選ばれるアミノ酸、
X26はA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X27はA、D、E、F、G、H、L、N、Q、R、S、T、VおよびYから選ばれるアミノ酸、
X28はA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X29はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、W、Yから選ばれるアミノ酸、
X30はA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WおよびYから選ばれるアミノ酸、
X31はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X32はA、F、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X33はAおよびGから選ばれるアミノ酸である、[B2]に記載のライブラリ。
[B16] [B15]に記載のライブラリから、MTAに結合する抗原結合ドメインを含む抗原結合分子をコードする核酸が濃縮されたライブラリ。
[B17] 前記抗原結合ドメインのMTAに対する結合は、抗原の非存在下におけるMTAに対する結合である、[B16]に記載のライブラリ。
[B18] 前記濃縮は、以下(1)~(2)の工程:
(1) [B15]に記載のライブラリからディスプレイされた抗原結合ドメインをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含む、[B16]または[B17]に記載のライブラリ。
[B19] [B15]に記載のライブラリから、アデノシンに結合する抗原結合ドメインをコードする核酸が濃縮されたライブラリ。
[B20] 前記抗原結合ドメインのアデノシンに対する結合は、抗原の非存在下におけるアデノシンに対する結合である、[B19]に記載のライブラリ。
[B21] 前記濃縮は、以下(1)~(2)の工程:
(1) [B15]に記載のライブラリからディスプレイされた抗原結合ドメインをアデノシンと接触させる工程、及び
(2) 前記工程(1)でアデノシンに結合した抗原結合ドメインを選択する工程、
を含む、[B19]または[B20]に記載のライブラリ。
[B1] お互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸からなるライブラリであって、MTAと相互作用するアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子、または/および前記抗原結合分子をコードする核酸から主としてなるライブラリ。
[B2] 前記抗原結合ドメインが抗体可変領域である、[B1]に記載のライブラリ。
[B3] MTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体、または/およびMTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体をそれぞれコードする核酸を含む[B2]に記載のライブラリ。
[B4] 前記抗体可変領域改変体中の、前記抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である、[B3]に記載のライブラリ:
1) 前記抗体可変領域未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗体可変領域改変体と比較して、前記抗体可変領域改変体のMTAとの結合を著しく減弱させないアミノ酸部位、及び
3) 前記抗体可変領域改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
[B5] 前記抗体可変領域改変体中の、前記抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である、[B3]に記載のライブラリ:
1) 前記抗体可変領域未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗体可変領域改変体と比較して、前記抗体可変領域改変体のMTAとの結合を著しく減弱させないアミノ酸部位、
3) 前記抗体可変領域未改変体の表面に露出するアミノ酸部位に対応するアミノ酸部位、及び
4) 前記抗体可変領域未改変体においてMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に対応するアミノ酸部位。
[B6] 前記抗体可変領域の未改変体はアデノシンまたは/及びS-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に結合しない、[B3]から[B5]のいずれかに記載のライブラリ。
[B7] 前記抗体可変領域の未改変体は下記いずれかである、[B3]から[B6]のいずれか一つに記載のライブラリ:
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域。
[B8] 前記複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAXXXYXXSGGSTYYASWAKG(配列番号:66)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXXXLS(配列番号:68)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B9] 前記複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、
Xは任意のアミノ酸、
X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であり、異なる位置に存在するXもしくはX1は同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B10] 前記複数の抗体可変領域は、下記:
XXAXWMC(配列番号:73)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域であり、
Xは任意のアミノ酸、
X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であり、異なる位置に存在するXもしくはX1は同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B11] 前記複数の抗体可変領域は、下記:
XXXWMC(配列番号:74)を含むH-CDR1;
CIXSXXXXTXYASWVNG(配列番号:75)を含むH-CDR2;
EXXXXSGALNL(配列番号:76)を含むH-CDR3;
HSSKXVXXXXXLA(配列番号:77)を含むL-CDR1;
XAXXLAS(配列番号:78)を含むL-CDR2;及び
QGTYXXXXFYFA(配列番号:79)を含むL-CDR3;
を含む抗体可変領域であり、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸でなくて良い、[B2]に記載のライブラリ。
[B12] 前記抗体可変領域の未改変体はアデノシンに対しても結合活性を有する、[B3]から[B5]のいずれか一つに記載のライブラリ。
[B13] 前記抗体可変領域の未改変体は(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに対しても結合活性を有する[B12]に記載のライブラリ。
[B14] 前記抗体可変領域の未改変体は、配列番号:31で示す重鎖可変領域と配列番号:32で示す軽鎖可変領域を有する抗体可変領域である、[B3]から[B5]、[B12]のいずれか一つに記載のライブラリ。
[B15] 前記複数の抗体可変領域は、下記:
X2X3X4X5G(配列番号:80)を含むH-CDR1;
X6IGX7X8X9X10X11WX12PX13WVKX14(配列番号:81)を含むH-CDR2;
GX15X16X17X18X19X20NAX21DP(配列番号:82)を含むH-CDR3;
QSSQSVX22X23NNX24LS(配列番号:83)を含むL-CDR1;
DASTLAS(配列番号:84)を含むL-CDR2;及び
HGX25X26X27X28X29X30X31X32DNX33(配列番号:85)を含むL-CDR3;
を含む抗体可変領域であり、
X2はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X3はD、E、F、H、K、N、P、RおよびYから選ばれるアミノ酸、
X4はA、I、P、TおよびVから選ばれるアミノ酸、
X5はA、E、F、H、I、K、L、M、N、Q、S、T、V、WおよびYから選ばれるアミノ酸、
X6はD、IおよびVから選ばれるアミノ酸、
X7はA、D、E、G、I、K、QおよびRから選ばれるアミノ酸、
X8はD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X9はA、D、E、F、G、HおよびSから選ばれるアミノ酸、
X10はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X11はA、D、E、G、H、I、K、L、N、P、Q、R、S、TおよびVから選ばれるアミノ酸、
X12はA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X13はA、F、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X14はA、FおよびGから選ばれるアミノ酸、
X15はA、E、F、G、H、K、L、Q、R、S、T、WおよびYから選ばれるアミノ酸、
X16はF、H、K、N、WおよびYから選ばれるアミノ酸、
X17はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X18はA、D、E、G、H、QおよびSから選ばれるアミノ酸、
X19はFおよびYから選ばれるアミノ酸、
X20はN、TおよびVから選ばれるアミノ酸、
X21はFおよびWから選ばれるアミノ酸、
X22はA、E、F、H、I、K、L、N、R、S、T、V、WおよびYから選ばれるアミノ酸、
X23はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X24はA、E、F、G、H、SおよびYから選ばれるアミノ酸、
X25はA、SおよびTから選ばれるアミノ酸、
X26はA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X27はA、D、E、F、G、H、L、N、Q、R、S、T、VおよびYから選ばれるアミノ酸、
X28はA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X29はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、W、Yから選ばれるアミノ酸、
X30はA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WおよびYから選ばれるアミノ酸、
X31はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X32はA、F、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X33はAおよびGから選ばれるアミノ酸である、[B2]に記載のライブラリ。
[B16] [B15]に記載のライブラリから、MTAに結合する抗原結合ドメインを含む抗原結合分子をコードする核酸が濃縮されたライブラリ。
[B17] 前記抗原結合ドメインのMTAに対する結合は、抗原の非存在下におけるMTAに対する結合である、[B16]に記載のライブラリ。
[B18] 前記濃縮は、以下(1)~(2)の工程:
(1) [B15]に記載のライブラリからディスプレイされた抗原結合ドメインをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含む、[B16]または[B17]に記載のライブラリ。
[B19] [B15]に記載のライブラリから、アデノシンに結合する抗原結合ドメインをコードする核酸が濃縮されたライブラリ。
[B20] 前記抗原結合ドメインのアデノシンに対する結合は、抗原の非存在下におけるアデノシンに対する結合である、[B19]に記載のライブラリ。
[B21] 前記濃縮は、以下(1)~(2)の工程:
(1) [B15]に記載のライブラリからディスプレイされた抗原結合ドメインをアデノシンと接触させる工程、及び
(2) 前記工程(1)でアデノシンに結合した抗原結合ドメインを選択する工程、
を含む、[B19]または[B20]に記載のライブラリ。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[C1] 以下(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法。
[C2] 前記工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない;
[C1]に記載の製造方法。
[C3] 前記MTAに対する結合活性を有する抗原結合ドメインは、アデノシンに実質的に結合しない、[C1]から[C2]のいずれか一つに記載の製造方法。
[C4] 前記MTAに対する結合活性を有する抗原結合ドメインは、(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに対して結合しない[C3]に記載の製造方法。
[C5] [C1]から[C4]のいずれか一つに記載の製造方法により製造されたライブラリ。
[C6] 以下(a)および(b)の工程:
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法。
[C7] 前記工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、前記低分子化合物に対する結合活性が著しく減弱されていない;
[C5]に記載の製造方法。
[C8] 以下(a)および(b)の工程:
(a) 低分子化合物に対して相互作用する抗原結合ドメインにおいて、以下(i)乃至(iv)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) 前記低分子化合物との結合に関与しないアミノ酸部位;
(iv) 前記低分子化合物との結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位に以下の(1)乃至(2)の少なくとも一つを満たすアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
を含むライブラリの製造方法。
[C9] 前記低分子化合物はアデノシン、アデノシン3リン酸、アデノシン2リン酸、アデノシン1リン酸、S-(5'-Adenosyl)-L-homocysteine (SAH)から選択される少なくとも一つから選ばれる、[C6]から[C8]のいずれか一つに記載の製造方法。
[C10] [C6]から[C8]のいずれか一つに記載の製造方法により製造されたライブラリ。
[C1] 以下(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法。
[C2] 前記工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない;
[C1]に記載の製造方法。
[C3] 前記MTAに対する結合活性を有する抗原結合ドメインは、アデノシンに実質的に結合しない、[C1]から[C2]のいずれか一つに記載の製造方法。
[C4] 前記MTAに対する結合活性を有する抗原結合ドメインは、(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに対して結合しない[C3]に記載の製造方法。
[C5] [C1]から[C4]のいずれか一つに記載の製造方法により製造されたライブラリ。
[C6] 以下(a)および(b)の工程:
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法。
[C7] 前記工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、前記低分子化合物に対する結合活性が著しく減弱されていない;
[C5]に記載の製造方法。
[C8] 以下(a)および(b)の工程:
(a) 低分子化合物に対して相互作用する抗原結合ドメインにおいて、以下(i)乃至(iv)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) 前記低分子化合物との結合に関与しないアミノ酸部位;
(iv) 前記低分子化合物との結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位に以下の(1)乃至(2)の少なくとも一つを満たすアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
を含むライブラリの製造方法。
[C9] 前記低分子化合物はアデノシン、アデノシン3リン酸、アデノシン2リン酸、アデノシン1リン酸、S-(5'-Adenosyl)-L-homocysteine (SAH)から選択される少なくとも一つから選ばれる、[C6]から[C8]のいずれか一つに記載の製造方法。
[C10] [C6]から[C8]のいずれか一つに記載の製造方法により製造されたライブラリ。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[D1] MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をスクリーニングする方法。
[D2] 抗原結合ドメインの第1濃度のMTA存在下における抗原に対する結合活性と第1濃度と異なる濃度(第2濃度)のMTA存在下における抗原に対する結合活性を比較することを含む、[D1]に記載の方法。
[D3] 第1濃度のMTA存在下における抗原に対する結合活性と第2濃度のMTA存在下における抗原に対する結合活性が異なる抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D1]または[D2]に記載の方法。
[D4] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D2]から[D3]のいずれか一つに記載の方法。
[D5] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より低い抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D2]から[D3]のいずれか一つに記載の方法。
[D6] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D7] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D8] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[D6]または[D7]に記載の方法。
[D9] 以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D10] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D11] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D12] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
を含み、前記工程(a)で第1濃度のMTA存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリである、[D10]または[D11]に記載の方法。
[D13] 以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D14] 前記第1濃度は前記第2濃度より高い濃度である、[D2]から[D8]のいずれか一つに記載の方法。
[D15] 抗原結合ドメインのMTA存在下における抗原に対する結合活性とMTA非存在における抗原に対する結合活性を比較することを含む、[D1]に記載の方法。
[D16] MTA存在下における抗原に対する結合活性とMTA非存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[D1]または[D15]に記載の方法。
[D17] MTA存在下における抗原に対する結合活性がMTA非存在下における抗原に対する結合活性より高い抗原結合ドメインを選択する工程含む、[D1]または[D15]に記載の方法。
[D18] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D19] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D20] 前記(a)工程を行う前に、抗原結合ドメインを含む抗原結合分子がMTAに結合可能であることを確認する、[D18]または[D19]に記載の方法。
[D21] 以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D22] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D23] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D24] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
を含み、前記工程(a)でMTAの存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリである、[D22]または[D23]に記載の方法。
[D25] 以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D26] 前記抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリは、またはナイーブヒト抗体ディスプレイライブラリ、または合成ヒト抗体ディスプレイライブラリ、または[B1]から[B14]のいずれか一つに記載のライブラリ、または[C5]に記載のライブラリである、[D10]から[D14]または[D22]から[D25]のいずれか一つに記載の方法。
[D27] 前記抗原結合ドメインが抗体可変領域もしくは単ドメイン抗体である、[D1]から[D26]のいずれか一つに記載の方法。
[D1] MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をスクリーニングする方法。
[D2] 抗原結合ドメインの第1濃度のMTA存在下における抗原に対する結合活性と第1濃度と異なる濃度(第2濃度)のMTA存在下における抗原に対する結合活性を比較することを含む、[D1]に記載の方法。
[D3] 第1濃度のMTA存在下における抗原に対する結合活性と第2濃度のMTA存在下における抗原に対する結合活性が異なる抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D1]または[D2]に記載の方法。
[D4] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D2]から[D3]のいずれか一つに記載の方法。
[D5] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より低い抗原結合ドメインを含む抗原結合分子を選択する工程含む、[D2]から[D3]のいずれか一つに記載の方法。
[D6] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D7] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D8] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[D6]または[D7]に記載の方法。
[D9] 以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D10] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D11] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D12] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
を含み、前記工程(a)で第1濃度のMTA存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリである、[D10]または[D11]に記載の方法。
[D13] 以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D14] 前記第1濃度は前記第2濃度より高い濃度である、[D2]から[D8]のいずれか一つに記載の方法。
[D15] 抗原結合ドメインのMTA存在下における抗原に対する結合活性とMTA非存在における抗原に対する結合活性を比較することを含む、[D1]に記載の方法。
[D16] MTA存在下における抗原に対する結合活性とMTA非存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[D1]または[D15]に記載の方法。
[D17] MTA存在下における抗原に対する結合活性がMTA非存在下における抗原に対する結合活性より高い抗原結合ドメインを選択する工程含む、[D1]または[D15]に記載の方法。
[D18] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D19] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D20] 前記(a)工程を行う前に、抗原結合ドメインを含む抗原結合分子がMTAに結合可能であることを確認する、[D18]または[D19]に記載の方法。
[D21] 以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D22] 以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D23] 以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D24] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
を含み、前記工程(a)でMTAの存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリである、[D22]または[D23]に記載の方法。
[D25] 以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、[D1]に記載の方法。
[D26] 前記抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリは、またはナイーブヒト抗体ディスプレイライブラリ、または合成ヒト抗体ディスプレイライブラリ、または[B1]から[B14]のいずれか一つに記載のライブラリ、または[C5]に記載のライブラリである、[D10]から[D14]または[D22]から[D25]のいずれか一つに記載の方法。
[D27] 前記抗原結合ドメインが抗体可変領域もしくは単ドメイン抗体である、[D1]から[D26]のいずれか一つに記載の方法。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[F1] MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を製造する方法。
[F2] 抗原結合ドメインの第1濃度のMTA存在下における抗原に対する結合活性と第1濃度とは異なる濃度(第2濃度)のMTA存在下における抗原に対する結合活性を比較することを含む、[F1]に記載の方法。
[F3] 第1濃度のMTA存在下における抗原に対する結合活性と第2濃度のMTA存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[F1]または[F2]に記載の方法。
[F4] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い抗原結合ドメインを選択する工程含む、[F2]から[F3]のいずれか一つに記載の方法。
[F5] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より低い抗原結合ドメインを選択する工程含む、[F2]から[F3]のいずれか一つに記載の方法。
[F6] 前記選択された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程を更に含む、[F3]から[F5]のいずれか一つに記載の方法。
[F7] 以下(a)~(d)の工程:
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F8] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F9] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[F7]または[F8]に記載の方法。
[F10] 以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F11] 以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F12] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F13] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含み、前記工程(a)で第1濃度のMTA存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリである、[F11]または[F12]に記載の方法。
[F14] 以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F15] 前記第1濃度は前記第2濃度より高い濃度である、[F2]から[F14]のいずれか一つに記載の方法。
[F16] 抗原結合ドメインのMTA存在下における抗原に対する結合活性とMTA非存在における抗原に対する結合活性を比較することを含む、[F1]に記載の方法。
[F17] MTA存在下における抗原に対する結合活性とMTA非存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[F1]または[F16]に記載の方法。
[F18] MTA存在下における抗原に対する結合活性がMTA非存在下における抗原に対する結合活性が高い抗原結合ドメインを選択する工程含む、[F1]または[F16]に記載の方法。
[F19] 前記選択された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程を更に含む、[F16]から[F18]のいずれか一つに記載の方法。
[F20] 以下(a)~(d)の工程:
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F21] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F22] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[F20]または[F21]に記載の方法。
[F23] 以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F24] 以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F25] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F26] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含み、前記工程(a)でMTAの存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリである、[F24]または[F25]に記載の方法。
[F27] 以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F28] 前記抗原結合ドメインがディスプレイされたライブラリは、またはナイーブヒト抗体ディスプレイライブラリ、または合成ヒト抗体ディスプレイライブラリ、または[B1]から[B14]のいずれか一つに記載のライブラリ、または[C5]に記載のライブラリである、[F11]から[F15]または[F24]から[F27]のいずれか一つに記載の方法。
[F29] 前記抗原結合ドメインが抗体可変領域もしくは単ドメイン抗体である、[F1]から[F28]のいずれか一つに記載の方法。
[F1] MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を製造する方法。
[F2] 抗原結合ドメインの第1濃度のMTA存在下における抗原に対する結合活性と第1濃度とは異なる濃度(第2濃度)のMTA存在下における抗原に対する結合活性を比較することを含む、[F1]に記載の方法。
[F3] 第1濃度のMTA存在下における抗原に対する結合活性と第2濃度のMTA存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[F1]または[F2]に記載の方法。
[F4] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い抗原結合ドメインを選択する工程含む、[F2]から[F3]のいずれか一つに記載の方法。
[F5] 前記第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より低い抗原結合ドメインを選択する工程含む、[F2]から[F3]のいずれか一つに記載の方法。
[F6] 前記選択された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程を更に含む、[F3]から[F5]のいずれか一つに記載の方法。
[F7] 以下(a)~(d)の工程:
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F8] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F9] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[F7]または[F8]に記載の方法。
[F10] 以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F11] 以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F12] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F13] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含み、前記工程(a)で第1濃度のMTA存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリである、[F11]または[F12]に記載の方法。
[F14] 以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F15] 前記第1濃度は前記第2濃度より高い濃度である、[F2]から[F14]のいずれか一つに記載の方法。
[F16] 抗原結合ドメインのMTA存在下における抗原に対する結合活性とMTA非存在における抗原に対する結合活性を比較することを含む、[F1]に記載の方法。
[F17] MTA存在下における抗原に対する結合活性とMTA非存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、[F1]または[F16]に記載の方法。
[F18] MTA存在下における抗原に対する結合活性がMTA非存在下における抗原に対する結合活性が高い抗原結合ドメインを選択する工程含む、[F1]または[F16]に記載の方法。
[F19] 前記選択された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程を更に含む、[F16]から[F18]のいずれか一つに記載の方法。
[F20] 以下(a)~(d)の工程:
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F21] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F22] 前記(a)工程を行う前に、抗原結合ドメインがMTAに結合可能であることを確認する、[F20]または[F21]に記載の方法。
[F23] 以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F24] 以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F25] 以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F26] 前記(a)工程の前に、以下(1)から(2)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
を含み、前記工程(a)でMTAの存在下で抗原と接触させるライブラリは、前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリである、[F24]または[F25]に記載の方法。
[F27] 以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む、[F1]に記載の方法。
[F28] 前記抗原結合ドメインがディスプレイされたライブラリは、またはナイーブヒト抗体ディスプレイライブラリ、または合成ヒト抗体ディスプレイライブラリ、または[B1]から[B14]のいずれか一つに記載のライブラリ、または[C5]に記載のライブラリである、[F11]から[F15]または[F24]から[F27]のいずれか一つに記載の方法。
[F29] 前記抗原結合ドメインが抗体可変領域もしくは単ドメイン抗体である、[F1]から[F28]のいずれか一つに記載の方法。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[E1] MTAに特異的に結合する抗原結合分子。
[E2] MTAに結合する抗原結合分子であって、当該抗原結合分子はアデノシンに実質的に結合しない[E1]に記載の抗原結合分子。
[E3] MTAに結合する抗原結合分子であって、当該抗原結合分子は(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに実質的に結合しない[E1]または[E2]に記載の抗原結合分子。
[E4] [E1]から[E3]のいずれか一つに記載の抗原結合分子を用いる、MTA濃度測定方法。
[E5] 前記MTA濃度は、組織中のMTA濃度である、[E4]に記載の測定方法。
[E6] 抗原抗体反応を利用してMTA濃度を測定する[E4]または[E5]のいずれか一つに記載の測定方法。
[E7] 免疫組織染色法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E8] imaging法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E9] in vivo imaging法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E10] [E1]から[E3]のいずれか一つに記載の抗原結合分子を用いる、疾患の診断方法。
[E11] 前記診断は、疾患の罹患有無を判断する、または疾患に対する治療効果を予測する、[E10]に記載の方法。
[E12] 前記疾患はがんである、[E10]から[E11]のいずれか一つに記載の方法。
[E13] 前記がんは、がん組織中にMTAが蓄積されているがんである、[E12]に記載の方法。
[E14] 前記がんは、MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん組織である[E12]または[E13]のいずれかに一つに記載の方法。
[E15] [E1]から[E3]のいずれか一つに記載の抗原結合分子を含む、疾患の診断キット。
[E16] 前記診断は、疾患の罹患有無を判断する、または疾患に対する治療効果を予測する、[E15]に記載のキット。
[E17] 前記疾患はがんである、[E15]から[E16]のいずれか一つに記載のキット。
[E18] 前記がんは、がん組織中にMTAが蓄積されているがんである、[E17]に記載のキット。
[E1] MTAに特異的に結合する抗原結合分子。
[E2] MTAに結合する抗原結合分子であって、当該抗原結合分子はアデノシンに実質的に結合しない[E1]に記載の抗原結合分子。
[E3] MTAに結合する抗原結合分子であって、当該抗原結合分子は(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに実質的に結合しない[E1]または[E2]に記載の抗原結合分子。
[E4] [E1]から[E3]のいずれか一つに記載の抗原結合分子を用いる、MTA濃度測定方法。
[E5] 前記MTA濃度は、組織中のMTA濃度である、[E4]に記載の測定方法。
[E6] 抗原抗体反応を利用してMTA濃度を測定する[E4]または[E5]のいずれか一つに記載の測定方法。
[E7] 免疫組織染色法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E8] imaging法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E9] in vivo imaging法を用いる、[E4]から[E5]のいずれか一つに記載の測定方法。
[E10] [E1]から[E3]のいずれか一つに記載の抗原結合分子を用いる、疾患の診断方法。
[E11] 前記診断は、疾患の罹患有無を判断する、または疾患に対する治療効果を予測する、[E10]に記載の方法。
[E12] 前記疾患はがんである、[E10]から[E11]のいずれか一つに記載の方法。
[E13] 前記がんは、がん組織中にMTAが蓄積されているがんである、[E12]に記載の方法。
[E14] 前記がんは、MTAPをコードしている遺伝子が欠損もしくは発現低下している、または酵素活性を低下させる変異もしくはスプライシングバリアントを有しているがん組織である[E12]または[E13]のいずれかに一つに記載の方法。
[E15] [E1]から[E3]のいずれか一つに記載の抗原結合分子を含む、疾患の診断キット。
[E16] 前記診断は、疾患の罹患有無を判断する、または疾患に対する治療効果を予測する、[E15]に記載のキット。
[E17] 前記疾患はがんである、[E15]から[E16]のいずれか一つに記載のキット。
[E18] 前記がんは、がん組織中にMTAが蓄積されているがんである、[E17]に記載のキット。
本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子、ならびにこれを含む医薬組成物は、正常組織や血液中において全身的に作用せず、がんにおいて可逆的に作用することにより、副作用を回避しつつ薬効を発揮し、がんを治療することができる。
さらに、本開示の、互いに配列の異なる複数の、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を含むライブラリを用いれば、上述したとおりの、がん組織特異的な疾患の治療に有用なMTA依存的に抗原に対する結合活性が変化する様々な抗原結合分子を、短時間で効率良く取得することが可能である。
さらに、本開示の、互いに配列の異なる複数の、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を含むライブラリを用いれば、上述したとおりの、がん組織特異的な疾患の治療に有用なMTA依存的に抗原に対する結合活性が変化する様々な抗原結合分子を、短時間で効率良く取得することが可能である。
以下の定義および詳細な説明は、本明細書において説明する本開示の理解を容易にするために提供される。
アミノ酸
本明細書において、たとえば、Ala/A、Leu/L、Arg/R、Lys/K、Asn/N、Met/M、Asp/D、Phe/F、Cys/C、Pro/P、Gln/Q、Ser/S、Glu/E、Thr/T、Gly/G、Trp/W、His/H、Tyr/Y、Ile/I、Val/Vと表されるように、アミノ酸は1文字コードまたは3文字コード、またはその両方で表記されている。
アミノ酸
本明細書において、たとえば、Ala/A、Leu/L、Arg/R、Lys/K、Asn/N、Met/M、Asp/D、Phe/F、Cys/C、Pro/P、Gln/Q、Ser/S、Glu/E、Thr/T、Gly/G、Trp/W、His/H、Tyr/Y、Ile/I、Val/Vと表されるように、アミノ酸は1文字コードまたは3文字コード、またはその両方で表記されている。
アミノ酸の改変
抗原結合分子のアミノ酸配列中のアミノ酸の改変のためには、部位特異的変異誘発法(Kunkelら(Proc. Natl. Acad. Sci. USA (1985) 82, 488-492))やOverlap extension PCR等の公知の方法が適宜採用され得る。また、天然のアミノ酸以外のアミノ酸に置換するアミノ酸の改変方法として、複数の公知の方法もまた採用され得る(Annu. Rev. Biophys. Biomol. Struct. (2006) 35, 225-249、Proc. Natl. Acad. Sci. U.S.A. (2003) 100 (11), 6353-6357)。例えば、終止コドンの1つであるUAGコドン(アンバーコドン)の相補的アンバーサプレッサーtRNAに非天然アミノ酸が結合されたtRNAが含まれる無細胞翻訳系システム(Clover Direct(Protein Express))等も好適に用いられる。
抗原結合分子のアミノ酸配列中のアミノ酸の改変のためには、部位特異的変異誘発法(Kunkelら(Proc. Natl. Acad. Sci. USA (1985) 82, 488-492))やOverlap extension PCR等の公知の方法が適宜採用され得る。また、天然のアミノ酸以外のアミノ酸に置換するアミノ酸の改変方法として、複数の公知の方法もまた採用され得る(Annu. Rev. Biophys. Biomol. Struct. (2006) 35, 225-249、Proc. Natl. Acad. Sci. U.S.A. (2003) 100 (11), 6353-6357)。例えば、終止コドンの1つであるUAGコドン(アンバーコドン)の相補的アンバーサプレッサーtRNAに非天然アミノ酸が結合されたtRNAが含まれる無細胞翻訳系システム(Clover Direct(Protein Express))等も好適に用いられる。
本明細書において、アミノ酸の改変部位を表す際に用いられる「および/または」の用語の意義は、「および」と「または」が適宜組み合わされたあらゆる組合せを含む。具体的には、例えば「33位、55位、および/または96位のアミノ酸が置換されている」とは以下のアミノ酸の改変のバリエーションが含まれる;
(a) 33位、(b)55位、(c)96位、(d)33位および55位、(e)33位および96位、(f)55位および96位、(g)33位および55位および96位。
(a) 33位、(b)55位、(c)96位、(d)33位および55位、(e)33位および96位、(f)55位および96位、(g)33位および55位および96位。
本明細書において、また、アミノ酸の改変を表す表現として、特定の位置を表す数字の前後に改変前と改変後のアミノ酸の1文字コードまたは3文字コードを併記した表現が適宜使用され得る。例えば、抗体可変領域に含まれるアミノ酸の置換を加える際に用いられるN100bLまたはAsn100bLeuという改変は、Kabatナンバリングで表される100b位のAsnのLeuへの置換を表す。すなわち、数字はKabatナンバリングで表されるアミノ酸の位置を表し、その前に記載されるアミノ酸の1文字コード又は3文字コードは置換前のアミノ酸、そのあとに記載されるアミノ酸の1文字コードまたは3文字コードは置換後のアミノ酸を表す。同様に、抗体定常領域に含まれるFc領域にアミノ酸の置換を加える際に用いられるP238DまたはPro238Aspという改変は、EUナンバリングで表される238位のProのAspへの置換を表す。すなわち、数字はEUナンバリングで表されるアミノ酸の位置を表し、その前に記載されるアミノ酸の1文字コードまたは3文字コードは置換前のアミノ酸、そのあとに記載されるアミノ酸の1文字コードまたは3文字コードは置換後のアミノ酸を表す。
抗原
本明細書において「抗原」は抗原結合ドメインが結合するエピトープを含む限りその構造は特定の構造に限定されない。ある態様において、抗原は4アミノ酸以上のペプチド、またはポリペプチド、またはタンパク質である。抗原としては下記のような分子;17-IA、4-1BB、4Dc、6-ケト-PGF1a、8-イソ-PGF2a、8-オキソ-dG、A1 アデノシン受容体、A33、ACE、ACE-2、アクチビン、アクチビンA、アクチビンAB、アクチビンB、アクチビンC、アクチビンRIA、アクチビンRIA ALK-2、アクチビンRIB ALK-4、アクチビンRIIA、アクチビンRIIB、ADAM、ADAM10、ADAM12、ADAM15、ADAM17/TACE、ADAM8、ADAM9、ADAMTS、ADAMTS4、ADAMTS5、アドレシン、aFGF、ALCAM、ALK、ALK-1、ALK-7、アルファ-1-アンチトリプシン、アルファ-V/ベータ-1アンタゴニスト、ANG、Ang、APAF-1、APE、APJ、APP、APRIL、AR、ARC、ART、アルテミン、抗Id、ASPARTIC、心房性ナトリウム利尿因子、av/b3インテグリン、Axl、b2M、B7-1、B7-2、B7-H、B-リンパ球刺激因子(BlyS)、BACE、BACE-1、Bad、BAFF、BAFF-R、Bag-1、BAK、Bax、BCA-1、BCAM、Bcl、BCMA、BDNF、b-ECGF、bFGF、BID、Bik、BIM、BLC、BL-CAM、BLK、BMP、BMP-2 BMP-2a、BMP-3 オステオゲニン(Osteogenin)、BMP-4 BMP-2b、BMP-5、BMP-6 Vgr-1、BMP-7(OP-1)、BMP-8(BMP-8a、OP-2)、BMPR、BMPR-IA(ALK-3)、BMPR-IB(ALK-6)、BRK-2、RPK-1、BMPR-II(BRK-3)、BMP、b-NGF、BOK、ボンベシン、骨由来神経栄養因子、BPDE、BPDE-DNA、BTC、補体因子3(C3)、C3a、C4、C5、C5a、C10、CA125、CAD-8、カルシトニン、cAMP、癌胎児性抗原(CEA)、癌関連抗原、カテプシンA、カテプシンB、カテプシンC/DPPI、カテプシンD、カテプシンE、カテプシンH、カテプシンL、カテプシンO、カテプシンS、カテプシンV、カテプシンX/Z/P、CBL、CCI、CCK2、CCL、CCL1、CCL11、CCL12、CCL13、CCL14、CCL15、CCL16、CCL17、CCL18、CCL19、CCL2、CCL20、CCL21、CCL22、CCL23、CCL24、CCL25、CCL26、CCL27、CCL28、CCL3、CCL4、CCL5、CCL6、CCL7、CCL8、CCL9/10、CCR、CCR1、CCR10、CCR10、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、CCR9、CD1、CD2、CD3、CD3E、CD4、CD5、CD6、CD7、CD8、CD10、CD11a、CD11b、CD11c、CD13、CD14、CD15、CD16、CD18、CD19、CD20、CD21、CD22、CD23、CD25、CD27L、CD28、CD29、CD30、CD30L、CD32、CD33(p67タンパク質)、CD34、CD38、CD40、CD40L、CD44、CD45、CD46、CD49a、CD52、CD54、CD55、CD56、CD61、CD64、CD66e、CD74、CD80(B7-1)、CD89、CD95、CD123、CD137、CD138、CD140a、CD146、CD147、CD148、CD152、CD164、CEACAM5、CFTR、cGMP、CINC、ボツリヌス菌毒素、ウェルシュ菌毒素、CKb8-1、CLC、CMV、CMV UL、CNTF、CNTN-1、COX、C-Ret、CRG-2、CT-1、CTACK、CTGF、CTLA-4、PD1、PDL1、LAG3、TIM3、galectin-9、CX3CL1、CX3CR1、CXCL、CXCL1、CXCL2、CXCL3、CXCL4、CXCL5、CXCL6、CXCL7、CXCL8、CXCL9、CXCL10、CXCL11、CXCL12、CXCL13、CXCL14、CXCL15、CXCL16、CXCR、CXCR1、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、サイトケラチン腫瘍関連抗原、DAN、DCC、DcR3、DC-SIGN、補体制御因子(Decay accelerating factor)、des(1-3)-IGF-I(脳IGF-1)、Dhh、ジゴキシン、DNAM-1、Dnase、Dpp、DPPIV/CD26、Dtk、ECAD、EDA、EDA-A1、EDA-A2、EDAR、EGF、EGFR(ErbB-1)、EMA、EMMPRIN、ENA、エンドセリン受容体、エンケファリナーゼ、eNOS、Eot、エオタキシン1、エフリンB2/EphB4、EPO、ERCC、E-セレクチン、ET-1、ファクターIIa、ファクターVII、ファクターVIIIc、ファクターIX、線維芽細胞活性化タンパク質(FAP)、Fas、FcR1、FEN-1、フェリチン、FGF、FGF-19、FGF-2、FGF3、FGF-8、FGFR、FGFR-3、フィブリン、FL、FLIP、Flt-3、Flt-4、卵胞刺激ホルモン、フラクタルカイン、FZD1、FZD2、FZD3、FZD4、FZD5、FZD6、FZD7、FZD8、FZD9、FZD10、G250、Gas6、GCP-2、GCSF、GD2、GD3、GDF、GDF-1、GDF-3(Vgr-2)、GDF-5(BMP-14、CDMP-1)、GDF-6(BMP-13、CDMP-2)、GDF-7(BMP-12、CDMP-3)、GDF-8(ミオスタチン)、GDF-9、GDF-15(MIC-1)、GDNF、GDNF、GFAP、GFRa-1、GFR-アルファ1、GFR-アルファ2、GFR-アルファ3、GITR、グルカゴン、Glut4、糖タンパク質IIb/IIIa(GPIIb/IIIa)、GM-CSF、gp130、gp72、GRO、成長ホルモン放出因子、ハプテン(NP-capまたはNIP-cap)、HB-EGF、HCC、HCMV gBエンベロープ糖タンパク質、HCMV gHエンベロープ糖タンパク質、HCMV UL、造血成長因子(HGF)、Hep B gp120、ヘパラナーゼ、Her2、Her2/neu(ErbB-2)、Her3(ErbB-3)、Her4(ErbB-4)、単純ヘルペスウイルス(HSV) gB糖タンパク質、HSV gD糖タンパク質、HGFA、高分子量黒色腫関連抗原(HMW-MAA)、HIV gp120、HIV IIIB gp 120 V3ループ、HLA、HLA-DR、HM1.24、HMFG PEM、HRG、Hrk、ヒト心臓ミオシン、ヒトサイトメガロウイルス(HCMV)、ヒト成長ホルモン(HGH)、HVEM、I-309、IAP、ICAM、ICAM-1、ICAM-3、ICE、ICOS、IFNg、Ig、IgA受容体、IgE、IGF、IGF結合タンパク質、IGF-1R、IGFBP、IGF-I、IGF-II、IL、IL-1、IL-1R、IL-2、IL-2R、IL-4、IL-4R、IL-5、IL-5R、IL-6、IL-6R、IL-8、IL-9、IL-10、IL-12、IL-13、IL-15、IL-18、IL-18R、IL-21、IL-23、IL-27、インターフェロン(INF)-アルファ、INF-ベータ、INF-ガンマ、インヒビン、iNOS、インスリンA鎖、インスリンB鎖、インスリン様増殖因子1、インテグリンアルファ2、インテグリンアルファ3、インテグリンアルファ4、インテグリンアルファ4/ベータ1、インテグリンアルファ4/ベータ7、インテグリンアルファ5(アルファV)、インテグリンアルファ5/ベータ1、インテグリンアルファ5/ベータ3、インテグリンアルファ6、インテグリンベータ1、インテグリンベータ2、インターフェロンガンマ、IP-10、I-TAC、JE、カリクレイン2、カリクレイン5、カリクレイン6、カリクレイン11、カリクレイン12、カリクレイン14、カリクレイン15、カリクレインL1、カリクレインL2、カリクレインL3、カリクレインL4、KC、KDR、ケラチノサイト増殖因子(KGF)、ラミニン5、LAMP、LAP、LAP(TGF-1)、潜在的TGF-1、潜在的TGF-1 bp1、LBP、LDGF、LECT2、レフティ、ルイス-Y抗原、ルイス-Y関連抗原、LFA-1、LFA-3、Lfo、LIF、LIGHT、リポタンパク質、LIX、LKN、Lptn、L-セレクチン、LT-a、LT-b、LTB4、LTBP-1、肺表面、黄体形成ホルモン、リンホトキシンベータ受容体、Mac-1、MAdCAM、MAG、MAP2、MARC、MCAM、MCAM、MCK-2、MCP、M-CSF、MDC、Mer、METALLOPROTEASES、MGDF受容体、MGMT、MHC(HLA-DR)、MIF、MIG、MIP、MIP-1-アルファ、MK、MMAC1、MMP、MMP-1、MMP-10、MMP-11、MMP-12、MMP-13、MMP-14、MMP-15、MMP-2、MMP-24、MMP-3、MMP-7、MMP-8、MMP-9、MPIF、Mpo、MSK、MSP、ムチン(Muc1)、MUC18、ミュラー管抑制物質、Mug、MuSK、NAIP、NAP、NCAD、N-Cアドヘリン、NCA 90、NCAM、NCAM、ネプリライシン、ニューロトロフィン-3、-4、または-6、ニュールツリン、神経成長因子(NGF)、NGFR、NGF-ベータ、nNOS、NO、NOS、Npn、NRG-3、NT、NTN、OB、OGG1、OPG、OPN、OSM、OX40L、OX40R、p150、p95、PADPr、副甲状腺ホルモン、PARC、PARP、PBR、PBSF、PCAD、P-カドヘリン、PCNA、PDGF、PDGF、PDK-1、PECAM、PEM、PF4、PGE、PGF、PGI2、PGJ2、PIN、PLA2、胎盤性アルカリホスファターゼ(PLAP)、PlGF、PLP、PP14、プロインスリン、プロレラキシン、プロテインC、PS、PSA、PSCA、前立腺特異的膜抗原(PSMA)、PTEN、PTHrp、Ptk、PTN、R51、RANK、RANKL、RANTES、RANTES、レラキシンA鎖、レラキシンB鎖、レニン、呼吸器多核体ウイルス(RSV)F、RSV Fgp、Ret、リウマイド因子、RLIP76、RPA2、RSK、S100、SCF/KL、SDF-1、SERINE、血清アルブミン、sFRP-3、Shh、SIGIRR、SK-1、SLAM、SLPI、SMAC、SMDF、SMOH、SOD、SPARC、Stat、STEAP、STEAP-II、TACE、TACI、TAG-72(腫瘍関連糖タンパク質-72)、TARC、TCA-3、T細胞受容体(例えば、T細胞受容体アルファ/ベータ)、TdT、TECK、TEM1、TEM5、TEM7、TEM8、TERT、睾丸PLAP様アルカリホスファターゼ、TfR、TGF、TGF-アルファ、TGF-ベータ、TGF-ベータ Pan Specific、TGF-ベータRI(ALK-5)、TGF-ベータRII、TGF-ベータRIIb、TGF-ベータRIII、TGF-ベータ1、TGF-ベータ2、TGF-ベータ3、TGF-ベータ4、TGF-ベータ5、トロンビン、胸腺Ck-1、甲状腺刺激ホルモン、Tie、TIMP、TIQ、組織因子、TMEFF2、Tmpo、TMPRSS2、TNF、TNF-アルファ、TNF-アルファベータ、TNF-ベータ2、TNFc、TNF-RI、TNF-RII、TNFRSF10A(TRAIL R1 Apo-2、DR4)、TNFRSF10B(TRAIL R2 DR5、KILLER、TRICK-2A、TRICK-B)、TNFRSF10C(TRAIL R3 DcR1、LIT、TRID)、TNFRSF10D(TRAIL R4 DcR2、TRUNDD)、TNFRSF11A(RANK ODF R、TRANCE R)、TNFRSF11B(OPG OCIF、TR1)、TNFRSF12(TWEAK R FN14)、TNFRSF13B(TACI)、TNFRSF13C(BAFF R)、TNFRSF14(HVEM ATAR、HveA、LIGHT R、TR2)、TNFRSF16(NGFR p75NTR)、TNFRSF17(BCMA)、TNFRSF18(GITR AITR)、TNFRSF19(TROY TAJ、TRADE)、TNFRSF19L(RELT)、TNFRSF1A(TNF RI CD120a、p55-60)、TNFRSF1B(TNF RII CD120b、p75-80)、TNFRSF26(TNFRH3)、TNFRSF3(LTbR TNF RIII、TNFC R)、TNFRSF4(OX40 ACT35、TXGP1 R)、TNFRSF5(CD40 p50)、TNFRSF6(Fas Apo-1、APT1、CD95)、TNFRSF6B(DcR3 M68、TR6)、TNFRSF7(CD27)、TNFRSF8(CD30)、TNFRSF9(4-1BB CD137、ILA)、TNFRSF21(DR6)、TNFRSF22(DcTRAIL R2 TNFRH2)、TNFRST23(DcTRAIL R1 TNFRH1)、TNFRSF25(DR3 Apo-3、LARD、TR-3、TRAMP、WSL-1)、TNFSF10(TRAIL Apo-2リガンド、TL2)、TNFSF11(TRANCE/RANKリガンド ODF、OPGリガンド)、TNFSF12(TWEAK Apo-3リガンド、DR3リガンド)、TNFSF13(APRIL TALL2)、TNFSF13B(BAFF BLYS、TALL1、THANK、TNFSF20)、TNFSF14(LIGHT HVEMリガンド、LTg)、TNFSF15(TL1A/VEGI)、TNFSF18(GITRリガンド AITRリガンド、TL6)、TNFSF1A(TNF-a コネクチン(Conectin)、DIF、TNFSF2)、TNFSF1B(TNF-b LTa、TNFSF1)、TNFSF3(LTb TNFC、p33)、TNFSF4(OX40リガンド gp34、TXGP1)、TNFSF5(CD40リガンド CD154、gp39、HIGM1、IMD3、TRAP)、TNFSF6(Fasリガンド Apo-1リガンド、APT1リガンド)、TNFSF7(CD27リガンド CD70)、TNFSF8(CD30リガンド CD153)、TNFSF9(4-1BBリガンド CD137リガンド)、TP-1、t-PA、Tpo、TRAIL、TRAIL R、TRAIL-R1、TRAIL-R2、TRANCE、トランスフェリン受容体、TRF、Trk、TROP-2、TLR(Toll-like receptor)1、TLR2、TLR3、TLR4、TLR5、TLR6、TLR7、TLR8、TLR9、TLR10、TSG、TSLP、腫瘍関連抗原CA125、腫瘍関連抗原発現ルイスY関連炭水化物、TWEAK、TXB2、Ung、uPAR、uPAR-1、ウロキナーゼ、VCAM、VCAM-1、VECAD、VE-Cadherin、VE-cadherin-2、VEFGR-1(flt-1)、VEGF、VEGFR、VEGFR-3(flt-4)、VEGI、VIM、ウイルス抗原、VLA、VLA-1、VLA-4、VNRインテグリン、フォン・ヴィレブランド因子、WIF-1、WNT1、WNT2、WNT2B/13、WNT3、WNT3A、WNT4、WNT5A、WNT5B、WNT6、WNT7A、WNT7B、WNT8A、WNT8B、WNT9A、WNT9A、WNT9B、WNT10A、WNT10B、WNT11、WNT16、XCL1、XCL2、XCR1、XCR1、XEDAR、XIAP、XPD、HMGB1、IgA、Aβ、CD81、CD97、CD98、DDR1、DKK1、EREG、Hsp90、IL-17/IL-17R、IL-20/IL-20R、酸化LDL、PCSK9、prekallikrein 、RON、TMEM16F、SOD1、Chromogranin A、Chromogranin B、tau、VAP1、高分子キニノーゲン、IL-31、IL-31R、Nav1.1、Nav1.2、Nav1.3、Nav1.4、Nav1.5、Nav1.6、Nav1.7、Nav1.8、Nav1.9、EPCR、C1、C1q、C1r、C1s、C2、C2a、C2b、C3、C3a、C3b、C4、C4a、C4b、C5、C5a、C5b、C6、C7、C8、C9、factor B、factor D、factor H、properdin、sclerostin、fibrinogen、fibrin、prothrombin、thrombin、組織因子、factor V、factor Va、factor VII、factor VIIa、factor VIII、factor VIIIa、factor IX、factor IXa、factor X、factor Xa、factor XI、factor XIa、factor XII、factor XIIa、factor XIII、factor XIIIa、TFPI、antithrombin III、EPCR、トロンボモデュリン、TAPI、tPA、plasminogen、plasmin、PAI-1、PAI-2、GPC3、Syndecan-1、Syndecan-2、Syndecan-3、Syndecan-4、LPA、S1Pならびにホルモンおよび成長因子のための受容体が例示され得る。抗原としてはがん組織におけるがん細胞・免疫細胞・ストローマ細胞等に発現する抗原
が好ましい。
本明細書において「抗原」は抗原結合ドメインが結合するエピトープを含む限りその構造は特定の構造に限定されない。ある態様において、抗原は4アミノ酸以上のペプチド、またはポリペプチド、またはタンパク質である。抗原としては下記のような分子;17-IA、4-1BB、4Dc、6-ケト-PGF1a、8-イソ-PGF2a、8-オキソ-dG、A1 アデノシン受容体、A33、ACE、ACE-2、アクチビン、アクチビンA、アクチビンAB、アクチビンB、アクチビンC、アクチビンRIA、アクチビンRIA ALK-2、アクチビンRIB ALK-4、アクチビンRIIA、アクチビンRIIB、ADAM、ADAM10、ADAM12、ADAM15、ADAM17/TACE、ADAM8、ADAM9、ADAMTS、ADAMTS4、ADAMTS5、アドレシン、aFGF、ALCAM、ALK、ALK-1、ALK-7、アルファ-1-アンチトリプシン、アルファ-V/ベータ-1アンタゴニスト、ANG、Ang、APAF-1、APE、APJ、APP、APRIL、AR、ARC、ART、アルテミン、抗Id、ASPARTIC、心房性ナトリウム利尿因子、av/b3インテグリン、Axl、b2M、B7-1、B7-2、B7-H、B-リンパ球刺激因子(BlyS)、BACE、BACE-1、Bad、BAFF、BAFF-R、Bag-1、BAK、Bax、BCA-1、BCAM、Bcl、BCMA、BDNF、b-ECGF、bFGF、BID、Bik、BIM、BLC、BL-CAM、BLK、BMP、BMP-2 BMP-2a、BMP-3 オステオゲニン(Osteogenin)、BMP-4 BMP-2b、BMP-5、BMP-6 Vgr-1、BMP-7(OP-1)、BMP-8(BMP-8a、OP-2)、BMPR、BMPR-IA(ALK-3)、BMPR-IB(ALK-6)、BRK-2、RPK-1、BMPR-II(BRK-3)、BMP、b-NGF、BOK、ボンベシン、骨由来神経栄養因子、BPDE、BPDE-DNA、BTC、補体因子3(C3)、C3a、C4、C5、C5a、C10、CA125、CAD-8、カルシトニン、cAMP、癌胎児性抗原(CEA)、癌関連抗原、カテプシンA、カテプシンB、カテプシンC/DPPI、カテプシンD、カテプシンE、カテプシンH、カテプシンL、カテプシンO、カテプシンS、カテプシンV、カテプシンX/Z/P、CBL、CCI、CCK2、CCL、CCL1、CCL11、CCL12、CCL13、CCL14、CCL15、CCL16、CCL17、CCL18、CCL19、CCL2、CCL20、CCL21、CCL22、CCL23、CCL24、CCL25、CCL26、CCL27、CCL28、CCL3、CCL4、CCL5、CCL6、CCL7、CCL8、CCL9/10、CCR、CCR1、CCR10、CCR10、CCR2、CCR3、CCR4、CCR5、CCR6、CCR7、CCR8、CCR9、CD1、CD2、CD3、CD3E、CD4、CD5、CD6、CD7、CD8、CD10、CD11a、CD11b、CD11c、CD13、CD14、CD15、CD16、CD18、CD19、CD20、CD21、CD22、CD23、CD25、CD27L、CD28、CD29、CD30、CD30L、CD32、CD33(p67タンパク質)、CD34、CD38、CD40、CD40L、CD44、CD45、CD46、CD49a、CD52、CD54、CD55、CD56、CD61、CD64、CD66e、CD74、CD80(B7-1)、CD89、CD95、CD123、CD137、CD138、CD140a、CD146、CD147、CD148、CD152、CD164、CEACAM5、CFTR、cGMP、CINC、ボツリヌス菌毒素、ウェルシュ菌毒素、CKb8-1、CLC、CMV、CMV UL、CNTF、CNTN-1、COX、C-Ret、CRG-2、CT-1、CTACK、CTGF、CTLA-4、PD1、PDL1、LAG3、TIM3、galectin-9、CX3CL1、CX3CR1、CXCL、CXCL1、CXCL2、CXCL3、CXCL4、CXCL5、CXCL6、CXCL7、CXCL8、CXCL9、CXCL10、CXCL11、CXCL12、CXCL13、CXCL14、CXCL15、CXCL16、CXCR、CXCR1、CXCR2、CXCR3、CXCR4、CXCR5、CXCR6、サイトケラチン腫瘍関連抗原、DAN、DCC、DcR3、DC-SIGN、補体制御因子(Decay accelerating factor)、des(1-3)-IGF-I(脳IGF-1)、Dhh、ジゴキシン、DNAM-1、Dnase、Dpp、DPPIV/CD26、Dtk、ECAD、EDA、EDA-A1、EDA-A2、EDAR、EGF、EGFR(ErbB-1)、EMA、EMMPRIN、ENA、エンドセリン受容体、エンケファリナーゼ、eNOS、Eot、エオタキシン1、エフリンB2/EphB4、EPO、ERCC、E-セレクチン、ET-1、ファクターIIa、ファクターVII、ファクターVIIIc、ファクターIX、線維芽細胞活性化タンパク質(FAP)、Fas、FcR1、FEN-1、フェリチン、FGF、FGF-19、FGF-2、FGF3、FGF-8、FGFR、FGFR-3、フィブリン、FL、FLIP、Flt-3、Flt-4、卵胞刺激ホルモン、フラクタルカイン、FZD1、FZD2、FZD3、FZD4、FZD5、FZD6、FZD7、FZD8、FZD9、FZD10、G250、Gas6、GCP-2、GCSF、GD2、GD3、GDF、GDF-1、GDF-3(Vgr-2)、GDF-5(BMP-14、CDMP-1)、GDF-6(BMP-13、CDMP-2)、GDF-7(BMP-12、CDMP-3)、GDF-8(ミオスタチン)、GDF-9、GDF-15(MIC-1)、GDNF、GDNF、GFAP、GFRa-1、GFR-アルファ1、GFR-アルファ2、GFR-アルファ3、GITR、グルカゴン、Glut4、糖タンパク質IIb/IIIa(GPIIb/IIIa)、GM-CSF、gp130、gp72、GRO、成長ホルモン放出因子、ハプテン(NP-capまたはNIP-cap)、HB-EGF、HCC、HCMV gBエンベロープ糖タンパク質、HCMV gHエンベロープ糖タンパク質、HCMV UL、造血成長因子(HGF)、Hep B gp120、ヘパラナーゼ、Her2、Her2/neu(ErbB-2)、Her3(ErbB-3)、Her4(ErbB-4)、単純ヘルペスウイルス(HSV) gB糖タンパク質、HSV gD糖タンパク質、HGFA、高分子量黒色腫関連抗原(HMW-MAA)、HIV gp120、HIV IIIB gp 120 V3ループ、HLA、HLA-DR、HM1.24、HMFG PEM、HRG、Hrk、ヒト心臓ミオシン、ヒトサイトメガロウイルス(HCMV)、ヒト成長ホルモン(HGH)、HVEM、I-309、IAP、ICAM、ICAM-1、ICAM-3、ICE、ICOS、IFNg、Ig、IgA受容体、IgE、IGF、IGF結合タンパク質、IGF-1R、IGFBP、IGF-I、IGF-II、IL、IL-1、IL-1R、IL-2、IL-2R、IL-4、IL-4R、IL-5、IL-5R、IL-6、IL-6R、IL-8、IL-9、IL-10、IL-12、IL-13、IL-15、IL-18、IL-18R、IL-21、IL-23、IL-27、インターフェロン(INF)-アルファ、INF-ベータ、INF-ガンマ、インヒビン、iNOS、インスリンA鎖、インスリンB鎖、インスリン様増殖因子1、インテグリンアルファ2、インテグリンアルファ3、インテグリンアルファ4、インテグリンアルファ4/ベータ1、インテグリンアルファ4/ベータ7、インテグリンアルファ5(アルファV)、インテグリンアルファ5/ベータ1、インテグリンアルファ5/ベータ3、インテグリンアルファ6、インテグリンベータ1、インテグリンベータ2、インターフェロンガンマ、IP-10、I-TAC、JE、カリクレイン2、カリクレイン5、カリクレイン6、カリクレイン11、カリクレイン12、カリクレイン14、カリクレイン15、カリクレインL1、カリクレインL2、カリクレインL3、カリクレインL4、KC、KDR、ケラチノサイト増殖因子(KGF)、ラミニン5、LAMP、LAP、LAP(TGF-1)、潜在的TGF-1、潜在的TGF-1 bp1、LBP、LDGF、LECT2、レフティ、ルイス-Y抗原、ルイス-Y関連抗原、LFA-1、LFA-3、Lfo、LIF、LIGHT、リポタンパク質、LIX、LKN、Lptn、L-セレクチン、LT-a、LT-b、LTB4、LTBP-1、肺表面、黄体形成ホルモン、リンホトキシンベータ受容体、Mac-1、MAdCAM、MAG、MAP2、MARC、MCAM、MCAM、MCK-2、MCP、M-CSF、MDC、Mer、METALLOPROTEASES、MGDF受容体、MGMT、MHC(HLA-DR)、MIF、MIG、MIP、MIP-1-アルファ、MK、MMAC1、MMP、MMP-1、MMP-10、MMP-11、MMP-12、MMP-13、MMP-14、MMP-15、MMP-2、MMP-24、MMP-3、MMP-7、MMP-8、MMP-9、MPIF、Mpo、MSK、MSP、ムチン(Muc1)、MUC18、ミュラー管抑制物質、Mug、MuSK、NAIP、NAP、NCAD、N-Cアドヘリン、NCA 90、NCAM、NCAM、ネプリライシン、ニューロトロフィン-3、-4、または-6、ニュールツリン、神経成長因子(NGF)、NGFR、NGF-ベータ、nNOS、NO、NOS、Npn、NRG-3、NT、NTN、OB、OGG1、OPG、OPN、OSM、OX40L、OX40R、p150、p95、PADPr、副甲状腺ホルモン、PARC、PARP、PBR、PBSF、PCAD、P-カドヘリン、PCNA、PDGF、PDGF、PDK-1、PECAM、PEM、PF4、PGE、PGF、PGI2、PGJ2、PIN、PLA2、胎盤性アルカリホスファターゼ(PLAP)、PlGF、PLP、PP14、プロインスリン、プロレラキシン、プロテインC、PS、PSA、PSCA、前立腺特異的膜抗原(PSMA)、PTEN、PTHrp、Ptk、PTN、R51、RANK、RANKL、RANTES、RANTES、レラキシンA鎖、レラキシンB鎖、レニン、呼吸器多核体ウイルス(RSV)F、RSV Fgp、Ret、リウマイド因子、RLIP76、RPA2、RSK、S100、SCF/KL、SDF-1、SERINE、血清アルブミン、sFRP-3、Shh、SIGIRR、SK-1、SLAM、SLPI、SMAC、SMDF、SMOH、SOD、SPARC、Stat、STEAP、STEAP-II、TACE、TACI、TAG-72(腫瘍関連糖タンパク質-72)、TARC、TCA-3、T細胞受容体(例えば、T細胞受容体アルファ/ベータ)、TdT、TECK、TEM1、TEM5、TEM7、TEM8、TERT、睾丸PLAP様アルカリホスファターゼ、TfR、TGF、TGF-アルファ、TGF-ベータ、TGF-ベータ Pan Specific、TGF-ベータRI(ALK-5)、TGF-ベータRII、TGF-ベータRIIb、TGF-ベータRIII、TGF-ベータ1、TGF-ベータ2、TGF-ベータ3、TGF-ベータ4、TGF-ベータ5、トロンビン、胸腺Ck-1、甲状腺刺激ホルモン、Tie、TIMP、TIQ、組織因子、TMEFF2、Tmpo、TMPRSS2、TNF、TNF-アルファ、TNF-アルファベータ、TNF-ベータ2、TNFc、TNF-RI、TNF-RII、TNFRSF10A(TRAIL R1 Apo-2、DR4)、TNFRSF10B(TRAIL R2 DR5、KILLER、TRICK-2A、TRICK-B)、TNFRSF10C(TRAIL R3 DcR1、LIT、TRID)、TNFRSF10D(TRAIL R4 DcR2、TRUNDD)、TNFRSF11A(RANK ODF R、TRANCE R)、TNFRSF11B(OPG OCIF、TR1)、TNFRSF12(TWEAK R FN14)、TNFRSF13B(TACI)、TNFRSF13C(BAFF R)、TNFRSF14(HVEM ATAR、HveA、LIGHT R、TR2)、TNFRSF16(NGFR p75NTR)、TNFRSF17(BCMA)、TNFRSF18(GITR AITR)、TNFRSF19(TROY TAJ、TRADE)、TNFRSF19L(RELT)、TNFRSF1A(TNF RI CD120a、p55-60)、TNFRSF1B(TNF RII CD120b、p75-80)、TNFRSF26(TNFRH3)、TNFRSF3(LTbR TNF RIII、TNFC R)、TNFRSF4(OX40 ACT35、TXGP1 R)、TNFRSF5(CD40 p50)、TNFRSF6(Fas Apo-1、APT1、CD95)、TNFRSF6B(DcR3 M68、TR6)、TNFRSF7(CD27)、TNFRSF8(CD30)、TNFRSF9(4-1BB CD137、ILA)、TNFRSF21(DR6)、TNFRSF22(DcTRAIL R2 TNFRH2)、TNFRST23(DcTRAIL R1 TNFRH1)、TNFRSF25(DR3 Apo-3、LARD、TR-3、TRAMP、WSL-1)、TNFSF10(TRAIL Apo-2リガンド、TL2)、TNFSF11(TRANCE/RANKリガンド ODF、OPGリガンド)、TNFSF12(TWEAK Apo-3リガンド、DR3リガンド)、TNFSF13(APRIL TALL2)、TNFSF13B(BAFF BLYS、TALL1、THANK、TNFSF20)、TNFSF14(LIGHT HVEMリガンド、LTg)、TNFSF15(TL1A/VEGI)、TNFSF18(GITRリガンド AITRリガンド、TL6)、TNFSF1A(TNF-a コネクチン(Conectin)、DIF、TNFSF2)、TNFSF1B(TNF-b LTa、TNFSF1)、TNFSF3(LTb TNFC、p33)、TNFSF4(OX40リガンド gp34、TXGP1)、TNFSF5(CD40リガンド CD154、gp39、HIGM1、IMD3、TRAP)、TNFSF6(Fasリガンド Apo-1リガンド、APT1リガンド)、TNFSF7(CD27リガンド CD70)、TNFSF8(CD30リガンド CD153)、TNFSF9(4-1BBリガンド CD137リガンド)、TP-1、t-PA、Tpo、TRAIL、TRAIL R、TRAIL-R1、TRAIL-R2、TRANCE、トランスフェリン受容体、TRF、Trk、TROP-2、TLR(Toll-like receptor)1、TLR2、TLR3、TLR4、TLR5、TLR6、TLR7、TLR8、TLR9、TLR10、TSG、TSLP、腫瘍関連抗原CA125、腫瘍関連抗原発現ルイスY関連炭水化物、TWEAK、TXB2、Ung、uPAR、uPAR-1、ウロキナーゼ、VCAM、VCAM-1、VECAD、VE-Cadherin、VE-cadherin-2、VEFGR-1(flt-1)、VEGF、VEGFR、VEGFR-3(flt-4)、VEGI、VIM、ウイルス抗原、VLA、VLA-1、VLA-4、VNRインテグリン、フォン・ヴィレブランド因子、WIF-1、WNT1、WNT2、WNT2B/13、WNT3、WNT3A、WNT4、WNT5A、WNT5B、WNT6、WNT7A、WNT7B、WNT8A、WNT8B、WNT9A、WNT9A、WNT9B、WNT10A、WNT10B、WNT11、WNT16、XCL1、XCL2、XCR1、XCR1、XEDAR、XIAP、XPD、HMGB1、IgA、Aβ、CD81、CD97、CD98、DDR1、DKK1、EREG、Hsp90、IL-17/IL-17R、IL-20/IL-20R、酸化LDL、PCSK9、prekallikrein 、RON、TMEM16F、SOD1、Chromogranin A、Chromogranin B、tau、VAP1、高分子キニノーゲン、IL-31、IL-31R、Nav1.1、Nav1.2、Nav1.3、Nav1.4、Nav1.5、Nav1.6、Nav1.7、Nav1.8、Nav1.9、EPCR、C1、C1q、C1r、C1s、C2、C2a、C2b、C3、C3a、C3b、C4、C4a、C4b、C5、C5a、C5b、C6、C7、C8、C9、factor B、factor D、factor H、properdin、sclerostin、fibrinogen、fibrin、prothrombin、thrombin、組織因子、factor V、factor Va、factor VII、factor VIIa、factor VIII、factor VIIIa、factor IX、factor IXa、factor X、factor Xa、factor XI、factor XIa、factor XII、factor XIIa、factor XIII、factor XIIIa、TFPI、antithrombin III、EPCR、トロンボモデュリン、TAPI、tPA、plasminogen、plasmin、PAI-1、PAI-2、GPC3、Syndecan-1、Syndecan-2、Syndecan-3、Syndecan-4、LPA、S1Pならびにホルモンおよび成長因子のための受容体が例示され得る。抗原としてはがん組織におけるがん細胞・免疫細胞・ストローマ細胞等に発現する抗原
が好ましい。
上記の抗原の例示には受容体も記載されるが、これらの受容体が生体液中に可溶型で存在する場合にも、本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子が結合する抗原として使用され得る。そのような可溶型受容体の非限定な一態様として、例えば、Mullbergら(J. Immunol. (1994) 152 (10), 4958-4968)によって記載されているような可溶型IL-6Rである、配列番号:1で表されるIL-6Rポリペプチド配列のうち、1から357番目のアミノ酸からなるタンパク質が例示され得る。
上記の抗原の例示には、細胞膜に発現する膜型分子、および細胞から細胞外に分泌される可溶型分子が含まれる。本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子が、細胞から分泌された可溶型分子に結合する場合、当該抗原結合分子としては、後述されるように中和活性を有していることが好適である。
可溶型分子が存在する溶液に限定はなく生体液、すなわち生体内の脈管又は組織・細胞の間を満たす全ての液体に本可溶型分子は存在し得る。非限定な一態様では、本開示の抗原結合分子が結合する可溶型分子は、細胞外液に存在することができる。細胞外液とは、脊椎動物では血漿、組織間液、リンパ液、密な結合組織、脳脊髄液、髄液、穿刺液、または関節液等の骨および軟骨中の成分、肺胞液(気管支肺胞洗浄液)、腹水、胸水、心嚢水、嚢胞液、または眼房水(房水)等の細胞透過液(細胞の能動輸送・分泌活動の結果生じた各種腺腔内の液、および消化管腔その他の体腔内液)の総称をいう。
本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子が、細胞膜に発現する膜型分子に結合する場合、当該抗原結合分子の好適な例として、後述されるように細胞傷害活性を有している、もしくは細胞傷害性物質を結合するまたは結合する能力を有している抗原結合分子が好適に挙げられる。また、細胞傷害活性を有している、もしくは細胞傷害性物質を結合するまたは結合する能力を有しているという性質に代えて、または当該性質に加えて、中和活性を有している抗原結合分子もまた非限定な一態様として好適に挙げられる。
抗原結合ドメイン
本明細書において、「抗原結合ドメイン」は目的とする抗原に結合するかぎりどのような構造のドメインも使用され得る。そのようなドメインの例として、例えば、抗体の重鎖および軽鎖の可変領域、生体内に存在する細胞膜タンパクであるAvimerに含まれる35アミノ酸程度のAドメインと呼ばれるモジュール(国際公開WO2004/044011、WO2005/040229)、細胞膜に発現する糖たんぱく質であるfibronectin中のタンパク質に結合するドメインである10Fn3ドメインを含むAdnectin(国際公開WO2002/032925)、ProteinAの58アミノ酸からなる3つのヘリックスの束(bundle)を構成するIgG結合ドメインをscaffoldとするAffibody(国際公開WO1995/001937)、33アミノ酸残基を含むターンと2つの逆並行ヘリックスおよびループのサブユニットが繰り返し積み重なった構造を有するアンキリン反復(ankyrin repeat:AR)の分子表面に露出する領域であるDARPins(Designed Ankyrin Repeat proteins)(国際公開WO2002/020565)、好中球ゲラチナーゼ結合リポカリン(neutrophil gelatinase-associated lipocalin(NGAL))等のリポカリン分子において高度に保存された8つの逆並行ストランドが中央方向にねじれたバレル構造の片側を支える4つのループ領域であるAnticalin等(国際公開WO2003/029462)、ヤツメウナギ、ヌタウナギなど無顎類の獲得免疫システムとしてイムノグロブリンの構造を有さない可変性リンパ球受容体(variable lymphocyte receptor(VLR))のロイシン残基に富んだリピート(leucine-rich-repeat(LRR))モジュールが繰り返し積み重なった馬てい形の構造の内部の並行型シート構造のくぼんだ領域(国際公開WO2008/016854)が好適に挙げられる。
本開示の抗原結合ドメインの好適な例として、抗体の重鎖および軽鎖の可変領域を含む抗原結合ドメインが挙げられる。こうした抗原結合ドメインの例としては、「scFv(single chain Fv)」、「単鎖抗体(single chain antibody)」、「Fv」、「scFv2(single chain Fv 2)」、「Fab」または「F(ab')2」等が好適に挙げられる。
本明細書において、「抗原結合ドメイン」は目的とする抗原に結合するかぎりどのような構造のドメインも使用され得る。そのようなドメインの例として、例えば、抗体の重鎖および軽鎖の可変領域、生体内に存在する細胞膜タンパクであるAvimerに含まれる35アミノ酸程度のAドメインと呼ばれるモジュール(国際公開WO2004/044011、WO2005/040229)、細胞膜に発現する糖たんぱく質であるfibronectin中のタンパク質に結合するドメインである10Fn3ドメインを含むAdnectin(国際公開WO2002/032925)、ProteinAの58アミノ酸からなる3つのヘリックスの束(bundle)を構成するIgG結合ドメインをscaffoldとするAffibody(国際公開WO1995/001937)、33アミノ酸残基を含むターンと2つの逆並行ヘリックスおよびループのサブユニットが繰り返し積み重なった構造を有するアンキリン反復(ankyrin repeat:AR)の分子表面に露出する領域であるDARPins(Designed Ankyrin Repeat proteins)(国際公開WO2002/020565)、好中球ゲラチナーゼ結合リポカリン(neutrophil gelatinase-associated lipocalin(NGAL))等のリポカリン分子において高度に保存された8つの逆並行ストランドが中央方向にねじれたバレル構造の片側を支える4つのループ領域であるAnticalin等(国際公開WO2003/029462)、ヤツメウナギ、ヌタウナギなど無顎類の獲得免疫システムとしてイムノグロブリンの構造を有さない可変性リンパ球受容体(variable lymphocyte receptor(VLR))のロイシン残基に富んだリピート(leucine-rich-repeat(LRR))モジュールが繰り返し積み重なった馬てい形の構造の内部の並行型シート構造のくぼんだ領域(国際公開WO2008/016854)が好適に挙げられる。
本開示の抗原結合ドメインの好適な例として、抗体の重鎖および軽鎖の可変領域を含む抗原結合ドメインが挙げられる。こうした抗原結合ドメインの例としては、「scFv(single chain Fv)」、「単鎖抗体(single chain antibody)」、「Fv」、「scFv2(single chain Fv 2)」、「Fab」または「F(ab')2」等が好適に挙げられる。
抗原結合分子
本開示において、抗原結合ドメインを含む抗原結合分子は最も広義な意味として使用されており、具体的には、それらが抗原結合ドメインを含む限り、様々な分子型が含まれる。抗原結合分子は、抗原結合ドメインのみからなる分子であっても良く、抗原結合ドメイン及び他のドメインを含む分子であっても良い。例えば、抗原結合分子が抗原結合ドメインとFc領域が結合した分子で有る場合、例として、完全抗体や抗体断片が挙げられる。抗体には、単一のモノクローナル抗体(アゴニストおよびアンタゴニスト抗体を含む)、ヒト抗体、ヒト化抗体、キメラ抗体等が含まれ得る。既存の安定なα/βバレルタンパク質構造等の立体構造が scaffold(土台)として用いられ、その一部分の構造のみが抗原結合ドメインの構築のためにライブラリ化されたスキャフォールド分子も、本開示の抗原結合分子に含まれ得る 。
本開示において、抗原結合ドメインを含む抗原結合分子は最も広義な意味として使用されており、具体的には、それらが抗原結合ドメインを含む限り、様々な分子型が含まれる。抗原結合分子は、抗原結合ドメインのみからなる分子であっても良く、抗原結合ドメイン及び他のドメインを含む分子であっても良い。例えば、抗原結合分子が抗原結合ドメインとFc領域が結合した分子で有る場合、例として、完全抗体や抗体断片が挙げられる。抗体には、単一のモノクローナル抗体(アゴニストおよびアンタゴニスト抗体を含む)、ヒト抗体、ヒト化抗体、キメラ抗体等が含まれ得る。既存の安定なα/βバレルタンパク質構造等の立体構造が scaffold(土台)として用いられ、その一部分の構造のみが抗原結合ドメインの構築のためにライブラリ化されたスキャフォールド分子も、本開示の抗原結合分子に含まれ得る 。
抗体
本明細書において、抗体とは、天然のものであるかまたは部分的もしくは完全合成により製造された免疫グロブリンをいう。抗体はそれが天然に存在する血漿や血清等の天然資源や抗体を産生するハイブリドーマ細胞の培養上清から単離され得るし、または遺伝子組換え等の手法を用いることによって部分的にもしくは完全に合成され得る。抗体の例としては免疫グロブリンのアイソタイプおよびそれらのアイソタイプのサブクラスが好適に挙げられる。ヒトの免疫グロブリンとして、IgG1、IgG2、IgG3、IgG4、IgA1、IgA2、IgD、IgE、IgMの9種類のクラス(アイソタイプ)が知られている。本開示の抗体には、これらのアイソタイプのうちIgG1、IgG2、IgG3、IgG4が含まれ得る。ヒトIgG1、ヒトIgG2、ヒトIgG3、ヒトIgG4定常領域としては、遺伝子多型による複数のアロタイプ配列がSequences of proteins of immunological interest, NIH Publication No.91-3242 に記載されているが、本開示においてはそのいずれであっても良い。特にヒトIgG1の配列としては、EUナンバリングで表される356-358位のアミノ酸配列がDELであってもEEMであってもよい。また、ヒトIgκ(Kappa)定常領域とヒトIgλ (Lambda)定常領域としては、遺伝子多型による複数のアロタイプ配列がSequences of proteins of immunological interest, NIH Publication No.91-3242に記載されているが、本開示においてはそのいずれであっても良い。
本明細書において、抗体とは、天然のものであるかまたは部分的もしくは完全合成により製造された免疫グロブリンをいう。抗体はそれが天然に存在する血漿や血清等の天然資源や抗体を産生するハイブリドーマ細胞の培養上清から単離され得るし、または遺伝子組換え等の手法を用いることによって部分的にもしくは完全に合成され得る。抗体の例としては免疫グロブリンのアイソタイプおよびそれらのアイソタイプのサブクラスが好適に挙げられる。ヒトの免疫グロブリンとして、IgG1、IgG2、IgG3、IgG4、IgA1、IgA2、IgD、IgE、IgMの9種類のクラス(アイソタイプ)が知られている。本開示の抗体には、これらのアイソタイプのうちIgG1、IgG2、IgG3、IgG4が含まれ得る。ヒトIgG1、ヒトIgG2、ヒトIgG3、ヒトIgG4定常領域としては、遺伝子多型による複数のアロタイプ配列がSequences of proteins of immunological interest, NIH Publication No.91-3242 に記載されているが、本開示においてはそのいずれであっても良い。特にヒトIgG1の配列としては、EUナンバリングで表される356-358位のアミノ酸配列がDELであってもEEMであってもよい。また、ヒトIgκ(Kappa)定常領域とヒトIgλ (Lambda)定常領域としては、遺伝子多型による複数のアロタイプ配列がSequences of proteins of immunological interest, NIH Publication No.91-3242に記載されているが、本開示においてはそのいずれであっても良い。
EUナンバリングおよびKabatナンバリング
本開示で使用されている方法によると、抗体のCDRとFRに割り当てられるアミノ酸位置はKabatにしたがって規定される(Sequences of Proteins of Immunological Interest(National Institute of Health, Bethesda, Md., 1987年および1991年)。本明細書において、抗原結合分子が抗体または抗原結合断片である場合、可変領域のアミノ酸はKabatナンバリングにしたがい、定常領域のアミノ酸はKabatのアミノ酸位置に準じたEUナンバリングにしたがって表される。
本開示で使用されている方法によると、抗体のCDRとFRに割り当てられるアミノ酸位置はKabatにしたがって規定される(Sequences of Proteins of Immunological Interest(National Institute of Health, Bethesda, Md., 1987年および1991年)。本明細書において、抗原結合分子が抗体または抗原結合断片である場合、可変領域のアミノ酸はKabatナンバリングにしたがい、定常領域のアミノ酸はKabatのアミノ酸位置に準じたEUナンバリングにしたがって表される。
可変領域
用語「可変領域」または「可変ドメイン」は、抗体を抗原へと結合させることに関与する、抗体の重鎖または軽鎖のドメインのことをいう。天然型抗体の重鎖および軽鎖の可変ドメイン(それぞれVHおよびVL)は、通常、各ドメインが4つの保存されたフレームワーク領域 (FR) および3つの超可変領域 (HVR) を含む、類似の構造を有する。(例えば、Kindt et al. Kuby Immunology, 6th ed., W.H. Freeman and Co., page 91 (2007) 参照。)1つのVHまたはVLドメインで、抗原結合特異性を与えるに充分であろう。さらに、ある特定の抗原に結合する抗体は、当該抗原に結合する抗体からのVHまたはVLドメインを使ってそれぞれVLまたはVHドメインの相補的ライブラリをスクリーニングして、単離されてもよい。例えばPortolano et al., J. Immunol. 150:880-887 (1993); Clarkson et al., Nature 352:624-628 (1991) 参照。
用語「可変領域」または「可変ドメイン」は、抗体を抗原へと結合させることに関与する、抗体の重鎖または軽鎖のドメインのことをいう。天然型抗体の重鎖および軽鎖の可変ドメイン(それぞれVHおよびVL)は、通常、各ドメインが4つの保存されたフレームワーク領域 (FR) および3つの超可変領域 (HVR) を含む、類似の構造を有する。(例えば、Kindt et al. Kuby Immunology, 6th ed., W.H. Freeman and Co., page 91 (2007) 参照。)1つのVHまたはVLドメインで、抗原結合特異性を与えるに充分であろう。さらに、ある特定の抗原に結合する抗体は、当該抗原に結合する抗体からのVHまたはVLドメインを使ってそれぞれVLまたはVHドメインの相補的ライブラリをスクリーニングして、単離されてもよい。例えばPortolano et al., J. Immunol. 150:880-887 (1993); Clarkson et al., Nature 352:624-628 (1991) 参照。
超可変領域
本明細書で用いられる用語「超可変領域」または「HVR」は、配列において超可変であり(「相補性決定領域」または「CDR」(complementarity determining region))、および/または構造的に定まったループ(「超可変ループ」)を形成し、および/または抗原接触残基(「抗原接触」)を含む、抗体の可変ドメインの各領域のことをいう。通常、抗体は6つのHVRを含む:VHに3つ(H1、H2、H3)、およびVLに3つ(L1、L2、L3)である。本明細書での例示的なHVRは、以下のものを含む:
(a) アミノ酸残基26-32 (L1)、50-52 (L2)、91-96 (L3)、26-32 (H1)、53-55 (H2)、および96-101 (H3)のところで生じる超可変ループ (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987));
(b) アミノ酸残基24-34 (L1)、50-56 (L2)、89-97 (L3)、31-35b (H1)、50-65 (H2)、 および95-102 (H3)のところで生じるCDR (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991));
(c) アミノ酸残基27c-36 (L1)、46-55 (L2)、89-96 (L3)、30-35b (H1)、47-58 (H2)、および93-101 (H3) のところで生じる抗原接触 (MacCallum et al. J. Mol. Biol. 262: 732-745 (1996));ならびに、
(d) HVRアミノ酸残基46-56 (L2)、47-56 (L2)、48-56 (L2)、49-56 (L2)、26-35 (H1)、26-35b (H1)、49-65 (H2)、93-102 (H3)、および94-102 (H3)を含む、(a)、(b)、および/または(c)の組合せ。
別段示さない限り、HVR残基および可変ドメイン中の他の残基(例えば、FR残基)は、本明細書では上記のKabatらにしたがって番号付けされる。
本明細書で用いられる用語「超可変領域」または「HVR」は、配列において超可変であり(「相補性決定領域」または「CDR」(complementarity determining region))、および/または構造的に定まったループ(「超可変ループ」)を形成し、および/または抗原接触残基(「抗原接触」)を含む、抗体の可変ドメインの各領域のことをいう。通常、抗体は6つのHVRを含む:VHに3つ(H1、H2、H3)、およびVLに3つ(L1、L2、L3)である。本明細書での例示的なHVRは、以下のものを含む:
(a) アミノ酸残基26-32 (L1)、50-52 (L2)、91-96 (L3)、26-32 (H1)、53-55 (H2)、および96-101 (H3)のところで生じる超可変ループ (Chothia and Lesk, J. Mol. Biol. 196:901-917 (1987));
(b) アミノ酸残基24-34 (L1)、50-56 (L2)、89-97 (L3)、31-35b (H1)、50-65 (H2)、 および95-102 (H3)のところで生じるCDR (Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991));
(c) アミノ酸残基27c-36 (L1)、46-55 (L2)、89-96 (L3)、30-35b (H1)、47-58 (H2)、および93-101 (H3) のところで生じる抗原接触 (MacCallum et al. J. Mol. Biol. 262: 732-745 (1996));ならびに、
(d) HVRアミノ酸残基46-56 (L2)、47-56 (L2)、48-56 (L2)、49-56 (L2)、26-35 (H1)、26-35b (H1)、49-65 (H2)、93-102 (H3)、および94-102 (H3)を含む、(a)、(b)、および/または(c)の組合せ。
別段示さない限り、HVR残基および可変ドメイン中の他の残基(例えば、FR残基)は、本明細書では上記のKabatらにしたがって番号付けされる。
フレームワーク
「フレームワーク」または「FR」は、超可変領域 (HVR) 残基以外の、可変ドメイン残基のことをいう。可変ドメインのFRは、通常4つのFRドメイン:FR1、FR2、FR3、およびFR4からなる。それに応じて、HVRおよびFRの配列は、通常次の順序でVH(またはVL)に現れる:FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4。
「フレームワーク」または「FR」は、超可変領域 (HVR) 残基以外の、可変ドメイン残基のことをいう。可変ドメインのFRは、通常4つのFRドメイン:FR1、FR2、FR3、およびFR4からなる。それに応じて、HVRおよびFRの配列は、通常次の順序でVH(またはVL)に現れる:FR1-H1(L1)-FR2-H2(L2)-FR3-H3(L3)-FR4。
Fc領域
Fc領域は、抗体重鎖の定常領域に由来するアミノ酸配列を含む。Fc領域は、EUナンバリングで表されるおよそ216位のアミノ酸における、パパイン切断部位のヒンジ領域のN末端から、当該ヒンジ、CH2およびCH3ドメインを含める抗体の重鎖定常領域の部分である。Fc領域は、ヒトIgG1から取得され得るが、IgGの特定のサブクラスに限定されるものでもない。当該Fc領域の好適な例として、後述されるようにpH酸性域におけるFcRnに対する結合活性を有するFc領域が挙げられる。また当該Fc領域の好適な例として、後述されるようにFcγレセプターに対する結合活性を有するFc領域が挙げられる。そのようなFc領域の非限定な一態様として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。
Fc領域は、抗体重鎖の定常領域に由来するアミノ酸配列を含む。Fc領域は、EUナンバリングで表されるおよそ216位のアミノ酸における、パパイン切断部位のヒンジ領域のN末端から、当該ヒンジ、CH2およびCH3ドメインを含める抗体の重鎖定常領域の部分である。Fc領域は、ヒトIgG1から取得され得るが、IgGの特定のサブクラスに限定されるものでもない。当該Fc領域の好適な例として、後述されるようにpH酸性域におけるFcRnに対する結合活性を有するFc領域が挙げられる。また当該Fc領域の好適な例として、後述されるようにFcγレセプターに対する結合活性を有するFc領域が挙げられる。そのようなFc領域の非限定な一態様として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。
低分子化抗体
本開示で使用される抗体は、抗体の全長分子に限られず、低分子化抗体またはその修飾物であってもよい。低分子化抗体は、全長抗体(例えば、whole IgG等のwhole antibody)の一部分が欠損している抗体断片を含み、抗原に対する結合活性を有していれば特に限定されない。本開示の低分子化抗体は、全長抗体の一部分であれば特に限定されないが、重鎖可変領域(VH)又は/及び軽鎖可変領域(VL)を含んでいることが好ましい。VHまたはVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入がされていてもよい。さらに抗原に対する結合活性を有する限り、VH又は/及びVLの一部を欠損させてもよい。又、可変領域はキメラ化やヒト化されていてもよい。抗体断片の具体例としては、例えば、Fab、Fab'、F(ab')2、Fvなどを挙げることができる。また、低分子化抗体の具体例としては、例えば、Fab、Fab'、F(ab')2、Fv、scFv(single chain Fv)、Diabody、sc(Fv)2(single chain (Fv)2)などを挙げることができる。これら抗体の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本開示の低分子化抗体に含まれる。
本開示で使用される抗体は、抗体の全長分子に限られず、低分子化抗体またはその修飾物であってもよい。低分子化抗体は、全長抗体(例えば、whole IgG等のwhole antibody)の一部分が欠損している抗体断片を含み、抗原に対する結合活性を有していれば特に限定されない。本開示の低分子化抗体は、全長抗体の一部分であれば特に限定されないが、重鎖可変領域(VH)又は/及び軽鎖可変領域(VL)を含んでいることが好ましい。VHまたはVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入がされていてもよい。さらに抗原に対する結合活性を有する限り、VH又は/及びVLの一部を欠損させてもよい。又、可変領域はキメラ化やヒト化されていてもよい。抗体断片の具体例としては、例えば、Fab、Fab'、F(ab')2、Fvなどを挙げることができる。また、低分子化抗体の具体例としては、例えば、Fab、Fab'、F(ab')2、Fv、scFv(single chain Fv)、Diabody、sc(Fv)2(single chain (Fv)2)などを挙げることができる。これら抗体の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本開示の低分子化抗体に含まれる。
抗体断片は、抗体を酵素、例えばパパイン、ペプシンで処理することによって生成され得るし、または、これら抗体断片をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現され得る(例えば、Coら(J. Immunol.(1994)152, 2968-2976)、BetterおよびHorwitz(Methods in Enzymology(1989)178, 476-496)、PlueckthunおよびSkerraら(Methods in Enzymology(1989)178, 476-496)、Lamoyi(Methods in Enzymology(1989)121, 652-663)、Rousseauxら(Methods in Enzymology(1989)121, 663-669)およびBirdら(TIBTECH(1991)9, 132-137)を参照)。
Diabodyは、遺伝子融合により構築された二価(bivalent)の低分子化抗体を指す(Holligerら(Proc. Natl. Acad. Sci. U.S.A. 90, 6444-6448 (1993)、欧州公開公報EP404097、およびPCT公開公報WO1993/011161等)。Diabodyは、2本のポリペプチド鎖から構成されるダイマーであり、通常、ポリペプチド鎖は各々、同じ鎖中でVL及びVHが、互いに結合できない位に短い、例えば、5残基程度のリンカーにより結合されている。同一ポリペプチド鎖上にコードされるVLとVHとは、その間のリンカーが短いため単鎖可変領域フラグメントを形成することが出来ず二量体を形成するため、Diabodyは2つの抗原結合部位を有することとなる。
scFvは、抗体のH鎖可変領域とL鎖可変領域とを連結することにより得られる。このscFvにおいて、H鎖可変領域とL鎖可変領域は、リンカー、好ましくはペプチドリンカーを介して連結される(Hustonら(Proc. Natl. Acad. Sci. U.S.A. (1988) 85, 5879-5883)。scFvにおけるH鎖可変領域およびL鎖可変領域は、本明細書に抗体として記載されたもののいずれの抗体由来であってもよい。可変領域を連結するペプチドリンカーとしては、特に制限はないが、例えば3から25残基程度からなる任意の一本鎖ペプチド、また、後述のペプチドリンカー等を用いることができる。可変領域の連結方法としては上記のようなPCR法が利用できる。前記抗体のH鎖またはH鎖可変領域をコードするDNA配列、およびL鎖またはL鎖可変領域をコードするDNA配列のうち、全部又は所望のアミノ酸配列をコードするDNA部分を鋳型として、及び、その両端の配列に対応する配列を有するプライマーの一対を用いたPCR法によってscFvをコードするDNAが増幅できる。次いで、ペプチドリンカー部分をコードするDNA、およびその両端が各々H鎖、L鎖と連結されるように設計された配列を有するプライマーの一対を組み合わせてPCR反応を行うことによって、所望の配列を有するDNAが取得できる。また、一旦scFvをコードするDNAが作製されると、それらを含有する発現ベクター、および当該発現ベクターにより形質転換された組換え細胞が常法に従って取得でき、また、その結果得られる組換え細胞を培養して当該scFvをコードするDNAを発現させることにより、当該scFvが取得できる。
sc(Fv)2は、2つのVH及び2つのVLをリンカー等で結合して一本鎖にした低分子化抗体である(Hudsonら(J. Immunol. Methods (1999) 231, 177-189)。sc(Fv)2は、例えば、scFvをリンカーで結ぶことによって作製できる。
また2つのVH及び2つのVLが、一本鎖ポリペプチドのN末端側を基点としてVH、VL、VH、VL([VH]リンカー[VL]リンカー[VH]リンカー[VL])の順に並んでいることを特徴とする抗体が好ましい。2つのVHと2つのVLの順序は特に上記配置に限定されず、どのような順序で並べられていてもよい。例えば以下のような配置も挙げることができる。
-[VL]リンカー[VH]リンカー[VH]リンカー[VL]
-[VH]リンカー[VL]リンカー[VL]リンカー[VH]
-[VH]リンカー[VH]リンカー[VL]リンカー[VL]
-[VL]リンカー[VL]リンカー[VH]リンカー[VH]
-[VL]リンカー[VH]リンカー[VL]リンカー[VH]
-[VL]リンカー[VH]リンカー[VH]リンカー[VL]
-[VH]リンカー[VL]リンカー[VL]リンカー[VH]
-[VH]リンカー[VH]リンカー[VL]リンカー[VL]
-[VL]リンカー[VL]リンカー[VH]リンカー[VH]
-[VL]リンカー[VH]リンカー[VL]リンカー[VH]
抗体の可変領域を結合するリンカーとしては、前記の抗原結合分子の項で記載されたリンカーと同様のリンカーが使用され得る。例えば、本開示において特に好ましいsc(Fv)2の態様としては、例えば、以下のsc(Fv)2を挙げることができる。
-[VH]ペプチドリンカー(15アミノ酸)[VL]ペプチドリンカー(15アミノ酸)[VH]ペプチドリンカー(15アミノ酸)[VL]
4つの抗体可変領域を結合する場合には、通常、3つのリンカーが必要となるが、全て同じリンカーを用いてもよいし、異なるリンカーを用いてもよい。
-[VH]ペプチドリンカー(15アミノ酸)[VL]ペプチドリンカー(15アミノ酸)[VH]ペプチドリンカー(15アミノ酸)[VL]
4つの抗体可変領域を結合する場合には、通常、3つのリンカーが必要となるが、全て同じリンカーを用いてもよいし、異なるリンカーを用いてもよい。
このような低分子化抗体を得るには、抗体を酵素、例えば、パパイン、ペプシンなどで処理し、抗体断片を生成させるか、もしくはこれらの抗体断片または低分子化抗体をコードするDNAを構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させればよい(例えば、Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976 ; Better, M. and Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496 ; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515 ; Lamoyi, E., Methods Enzymol. (1986) 121, 652-663 ; Rousseaux, J. et al., Methods Enzymol. (1986) 121, 663-669 ; Bird, R. E. and Walker, B. W., Trends Biotechnol. (1991) 9, 132-137参照)。
また、本開示における抗体の非限定の一態様としては、T細胞受容体に代えて抗原を認識する抗体またはそのフラグメントと、T細胞のシグナルドメインの融合体がT細胞に組み込まれたキメラ型抗原受容体(Chimeric antigen receptor)ならびに該キメラ型抗原受容体が組み込まれたT細胞も挙げることができるが,これに限定されることはない。
単ドメイン抗体
本発明の抗原結合ドメインの好適な例の一つとして、単ドメイン抗体(sdAb)が挙げられる。
本発明の抗原結合ドメインの好適な例の一つとして、単ドメイン抗体(sdAb)が挙げられる。
本明細書で用語「単ドメイン抗体」は、そのドメイン単独で抗原結合活性を発揮できるかぎりその構造は限定されない。IgG抗体等で例示される通常の抗体は、VHとVLのペアリングにより可変領域を形成された状態では抗原結合活性を示すのに対し、単ドメイン抗体は他のドメインとペアリングすることなく、単ドメイン抗体自身のドメイン構造単独で抗原結合活性を発揮できると知られている。単ドメイン抗体は通常比較的に低分子量を有し、単量体の形態で存在する。
単ドメイン抗体の例として、それだけに限定されないが、例えば、ラクダ科の動物のVHH、サメのVNARのような、先天的に軽鎖を欠如する抗原結合分子、または抗体のVHドメインのすべてもしくは一部分またはVLドメインのすべてもしくは一部分を含む抗体断片が挙げられる。抗体のVH/VLドメインのすべてもしくは一部分を含む抗体断片である単ドメイン抗体の例として、それだけに限定されないが、例えば、米国特許第6,248,516号B1等に記載されているようなヒト抗体VHまたはヒト抗体VLから出発して人工的に作製された単ドメイン抗体が挙げられる。本発明のいくつかの実施態様において、1つの単ドメイン抗体は3つのCDR(CDR1、CDR2及びCDR3)を有する。
単ドメイン抗体は、単ドメイン抗体を産生できる動物から、または単ドメイン抗体を産生できる動物を免疫することにより取得し得る。単ドメイン抗体を産生できる動物の例として、それだけに限定されないが、例えば、ラクダ科動物、単ドメイン抗体を産生できる遺伝子が導入された遺伝子導入動物(transgenic animals)が挙げられる。ラクダ科動物はラクダ、ラマ、アルパカ、ヒトコブラクダおよびグアナコ等を含む。単ドメイン抗体を産生できる遺伝子が導入された遺伝子導入動物の例として、それだけに限定されないが、国際公開WO2015/143414号、米国特許公開US2011/0123527号A1に記載の遺伝子導入動物が挙げられる。動物から取得した単ドメイン抗体のフレームワーク配列をヒトジャームライン配列あるいはそれに類似した配列とすることで、ヒト化した単ドメイン抗体を取得することも出来る。ヒト化した単ドメイン抗体(例えば、ヒト化VHH)はまた、本発明の単ドメイン抗体の一実施態様である。「ヒト化単ドメイン抗体」は、非ヒトCDRからのアミノ酸残基およびヒトFRからのアミノ酸残基を含む、キメラ単ドメイン抗体のことをいう。ある態様では、ヒト化単ドメイン抗体は、すべてのもしくは実質的にすべてのCDRは非ヒト抗体のものに対応し、かつ、すべてのもしくは実質的にすべてのFRはヒト抗体のものに対応する。ヒト化抗体において、FR中の残基の一部がヒト抗体のものと対応しない場合も、実質的にすべてのFRはヒト抗体のものに対応する一例として考えられる。たとえば、単ドメイン抗体の一態様であるVHHをヒト化する場合、FR中の残基の一部をヒト抗体のものと対応しない残基にする必要がある(C Vinckeら、The Journal of Biological Chemistry 284, 3273-3284.)。
また、単ドメイン抗体は、単ドメイン抗体を含むポリペプチドライブラリから、ELISA、パニング等により取得し得る。単ドメイン抗体を含むポリペプチドライブラリの例として、それだけに限定されないが、例えば、各種動物若しくはヒトから取得したナイーブ抗体ライブラリ(例:Methods in Molecular Biology 2012 911 (65-78)、Biochimica et Biophysica Acta - Proteins and Proteomics 2006 1764:8 (1307-1319))、各種動物を免疫することで取得した抗体ライブラリ(例:Journal of Applied Microbiology 2014 117:2 (528-536))、または各種動物若しくはヒトの抗体遺伝子より作製した合成抗体ライブラリ(例:Journal of Biomolecular Screening 2016 21:1 (35-43)、Journal of Biological Chemistry 2016 291:24 (12641-12657)、AIDS 2016 30:11 (1691-1701))が挙げられる。
抗原に対して所望の結合活性を有する抗体の作製方法
MTAを含む低分子化合物に依存的でなく、低分子化合物と異なる分子である抗原に対して所望の結合活性を有する抗体を作製する方法は当業者において公知である。以下に、IL-6Rに結合する抗体(抗IL-6R抗体)を作製する方法が例示される。IL-6R以外の抗原に結合する抗体も下記の例示に準じて適宜作製され得る。
MTAを含む低分子化合物に依存的でなく、低分子化合物と異なる分子である抗原に対して所望の結合活性を有する抗体を作製する方法は当業者において公知である。以下に、IL-6Rに結合する抗体(抗IL-6R抗体)を作製する方法が例示される。IL-6R以外の抗原に結合する抗体も下記の例示に準じて適宜作製され得る。
抗IL-6R抗体は、公知の手段を用いてポリクローナルまたはモノクローナル抗体として取得され得る。抗IL-6R抗体としては、哺乳動物由来のモノクローナル抗体が好適に作製され得る。哺乳動物由来のモノクローナル抗体には、ハイブリドーマにより産生されるもの、および遺伝子工学的手法により抗体遺伝子を含む発現ベクターで形質転換した宿主細胞によって産生されるもの等が含まれる。なお本願発明のモノクローナル抗体には、「ヒト化抗体」や「キメラ抗体」が含まれる。
モノクローナル抗体産生ハイブリドーマは、公知技術を使用することによって、例えば以下のように作製され得る。すなわち、IL-6Rタンパク質を感作抗原として使用して、通常の免疫方法にしたがって哺乳動物が免疫される。得られる免疫細胞が通常の細胞融合法によって公知の親細胞と融合される。次に、通常のスクリーニング法によって、モノクローナルな抗体産生細胞をスクリーニングすることによって抗IL-6R抗体を産生するハイブリドーマが選択され得る。
具体的には、モノクローナル抗体の作製は例えば以下に示すように行われる。まず、配列番号:2にそのヌクレオチド配列が開示されたIL-6R遺伝子を発現することによって、抗体取得の感作抗原として使用される配列番号:1で表されるIL-6Rタンパク質が取得され得る。すなわち、IL-6Rをコードする遺伝子配列を公知の発現ベクターに挿入することによって適当な宿主細胞が形質転換される。当該宿主細胞中または培養上清中から所望のヒトIL-6Rタンパク質が公知の方法で精製される。培養上清中から可溶型のIL-6Rを取得するためには、例えば、Mullbergら(J. Immunol. (1994) 152 (10), 4958-4968)によって記載されているような可溶型IL-6Rである、配列番号:1で表されるIL-6Rポリペプチド配列のうち、1から357番目のアミノ酸からなるタンパク質が、配列番号:1で表されるIL-6Rタンパク質の代わりに発現される。また、精製した天然のIL-6Rタンパク質もまた同様に感作抗原として使用され得る。
哺乳動物に対する免疫に使用する感作抗原として当該精製IL-6Rタンパク質が使用できる。IL-6Rの部分ペプチドもまた感作抗原として使用できる。この際、当該部分ペプチドはヒトIL-6Rのアミノ酸配列より化学合成によっても取得され得る。また、IL-6R遺伝子の一部を発現ベクターに組込んで発現させることによっても取得され得る。さらにはタンパク質分解酵素を用いてIL-6Rタンパク質を分解することによっても取得され得るが、部分ペプチドとして用いるIL-6Rペプチドの領域および大きさは特に特別の態様に限定されない。好ましい領域は配列番号:1のアミノ酸配列において20-357番目のアミノ酸に相当するアミノ酸配列から任意の配列が選択され得る。感作抗原とするペプチドを構成するアミノ酸の数は少なくとも5以上、例えば6以上、或いは7以上であることが好ましい。より具体的には8~50、好ましくは10~30残基のペプチドが感作抗原として使用され得る。
また、IL-6Rタンパク質の所望の部分ポリペプチドやペプチドを異なるポリペプチドと融合した融合タンパク質が感作抗原として利用され得る。感作抗原として使用される融合タンパク質を製造するために、例えば、抗体のFc断片やペプチドタグなどが好適に利用され得る。融合タンパク質を発現するベクターは、所望の二種類又はそれ以上のポリペプチド断片をコードする遺伝子がインフレームで融合され、当該融合遺伝子が前記のように発現ベクターに挿入されることにより作製され得る。融合タンパク質の作製方法はMolecular Cloning 2nd ed. (Sambrook, J et al., Molecular Cloning 2nd ed., 9.47-9.58(1989)Cold Spring Harbor Lab. press)に記載されている。感作抗原として用いられるIL-6Rの取得方法及びそれを用いた免疫方法は、国際公開WO2003/000883、WO2004/022754、WO2006/006693等にも具体的に記載されている。
当該感作抗原で免疫される哺乳動物としては、特定の動物に限定されるものではないが、細胞融合に使用する親細胞との適合性を考慮して選択するのが好ましい。一般的にはげっ歯類の動物、例えば、マウス、ラット、ハムスター、あるいはウサギ、サル等が好適に使用される。
公知の方法にしたがって上記の動物が感作抗原により免疫される。例えば、一般的な方法として、感作抗原が哺乳動物の腹腔内または皮下に注射によって投与されることにより免疫が実施される。具体的には、PBS(Phosphate-Buffered Saline)や生理食塩水等で適当な希釈倍率で希釈された感作抗原が、所望により通常のアジュバント、例えばフロイント完全アジュバントと混合され、乳化された後に、該感作抗原が哺乳動物に4から21日毎に数回投与される。また、感作抗原の免疫時には適当な担体が使用され得る。特に分子量の小さい部分ペプチドが感作抗原として用いられる場合には、アルブミン、キーホールリンペットヘモシアニン等の担体タンパク質と結合した該感作抗原ペプチドを免疫することが望ましい場合もある。
また、所望の抗体を産生するハイブリドーマは、DNA免疫を使用し、以下のようにしても作製され得る。DNA免疫とは、免疫動物中で抗原タンパク質をコードする遺伝子が発現され得るような態様で構築されたベクターDNAが投与された当該免疫動物中で、感作抗原が当該免疫動物の生体内で発現されることによって、免疫刺激が与えられる免疫方法である。蛋白質抗原が免疫動物に投与される一般的な免疫方法と比べて、DNA免疫には、次のような優位性が期待される。
-IL-6Rのような膜蛋白質の構造を維持して免疫刺激が与えられ得る。
-免疫抗原を精製する必要が無い。
-IL-6Rのような膜蛋白質の構造を維持して免疫刺激が与えられ得る。
-免疫抗原を精製する必要が無い。
DNA免疫によって本発明のモノクローナル抗体を得るために、まず、IL-6Rタンパク質を発現するDNAが免疫動物に投与される。IL-6RをコードするDNAは、PCRなどの公知の方法によって合成され得る。得られたDNAが適当な発現ベクターに挿入され、免疫動物に投与される。発現ベクターとしては、たとえばpcDNA3.1などの市販の発現ベクターが好適に利用され得る。ベクターを生体に投与する方法として、一般的に用いられている方法が利用され得る。たとえば、発現ベクターが吸着した金粒子が、gene gunで免疫動物個体の細胞内に導入されることによってDNA免疫が行われる。さらに、IL-6Rを認識する抗体の作製は国際公開WO2003/104453に記載された方法を用いても作製され得る。
このように哺乳動物が免疫され、血清中におけるIL-6Rに結合する抗体力価の上昇が確認された後に、哺乳動物から免疫細胞が採取され、細胞融合に供される。好ましい免疫細胞としては、特に脾細胞が使用され得る。
前記免疫細胞と融合される細胞として、哺乳動物のミエローマ細胞が用いられる。ミエローマ細胞は、スクリーニングのための適当な選択マーカーを備えていることが好ましい。選択マーカーとは、特定の培養条件の下で生存できる(あるいはできない)形質を指す。選択マーカーには、ヒポキサンチン-グアニン-ホスホリボシルトランスフェラーゼ欠損(以下HGPRT欠損と省略する)、あるいはチミジンキナーゼ欠損(以下TK欠損と省略する)などが公知である。HGPRTやTKの欠損を有する細胞は、ヒポキサンチン-アミノプテリン-チミジン感受性(以下HAT感受性と省略する)を有する。HAT感受性の細胞はHAT選択培地中でDNA合成を行うことができず死滅するが、正常な細胞と融合すると正常細胞のサルベージ回路を利用してDNAの合成を継続することができるためHAT選択培地中でも増殖するようになる。
HGPRT欠損やTK欠損の細胞は、それぞれ6チオグアニン、8アザグアニン(以下8AGと省略する)、あるいは5'ブロモデオキシウリジンを含む培地で選択され得る。これらのピリミジンアナログをDNA中に取り込む正常な細胞は死滅する。他方、これらのピリミジンアナログを取り込めないこれらの酵素を欠損した細胞は、選択培地の中で生存することができる。この他G418耐性と呼ばれる選択マーカーは、ネオマイシン耐性遺伝子によって2-デオキシストレプタミン系抗生物質(ゲンタマイシン類似体)に対する耐性を与える。細胞融合に好適な種々のミエローマ細胞が公知である。
このようなミエローマ細胞として、例えば、P3(P3x63Ag8.653)(J. Immunol.(1979)123 (4), 1548-1550)、P3x63Ag8U.1(Current Topics in Microbiology and Immunology(1978)81, 1-7)、NS-1(C. Eur. J. Immunol.(1976)6 (7), 511-519)、MPC-11(Cell(1976)8 (3), 405-415)、SP2/0(Nature(1978)276 (5685), 269-270)、FO(J. Immunol. Methods(1980)35 (1-2), 1-21)、S194/5.XX0.BU.1(J. Exp. Med.(1978)148 (1), 313-323)、R210(Nature(1979)277 (5692), 131-133)等が好適に使用され得る。
基本的には公知の方法、たとえば、ケーラーとミルステインらの方法(Methods Enzymol.(1981)73, 3-46)等に準じて、前記免疫細胞とミエローマ細胞との細胞融合が行われる。
より具体的には、例えば細胞融合促進剤の存在下で通常の栄養培養液中で、前記細胞融合が実施され得る。融合促進剤としては、例えばポリエチレングリコール(PEG)、センダイウイルス(HVJ)等が使用され、更に融合効率を高めるために所望によりジメチルスルホキシド等の補助剤が添加されて使用される。
免疫細胞とミエローマ細胞との使用割合は任意に設定され得る。例えば、ミエローマ細胞に対して免疫細胞を1から10倍とするのが好ましい。前記細胞融合に用いる培養液としては、例えば、前記ミエローマ細胞株の増殖に好適なRPMI1640培養液、MEM培養液、その他、この種の細胞培養に用いられる通常の培養液が使用され、さらに、牛胎児血清(FCS)等の血清補液が好適に添加され得る。
細胞融合は、前記免疫細胞とミエローマ細胞との所定量を前記培養液中でよく混合し、予め37℃程度に加温されたPEG溶液(例えば平均分子量1000から6000程度)が通常30から60%(w/v)の濃度で添加される。混合液が緩やかに混合されることによって所望の融合細胞(ハイブリドーマ)が形成される。次いで、上記に挙げた適当な培養液が逐次添加され、遠心して上清を除去する操作を繰り返すことによりハイブリドーマの生育に好ましくない細胞融合剤等が除去され得る。
このようにして得られたハイブリドーマは、通常の選択培養液、例えばHAT培養液(ヒポキサンチン、アミノプテリンおよびチミジンを含む培養液)で培養することにより選択され得る。所望のハイブリドーマ以外の細胞(非融合細胞)が死滅するのに十分な時間(通常、係る十分な時間は数日から数週間である)上記HAT培養液を用いた培養が継続され得る。次いで、通常の限界希釈法によって、所望の抗体を産生するハイブリドーマのスクリーニングおよび単一クローニングが実施される。
このようにして得られたハイブリドーマは、細胞融合に用いられたミエローマが有する選択マーカーに応じた選択培養液を利用することによって選択され得る。例えばHGPRTやTKの欠損を有する細胞は、HAT培養液(ヒポキサンチン、アミノプテリンおよびチミジンを含む培養液)で培養することにより選択され得る。すなわち、HAT感受性のミエローマ細胞を細胞融合に用いた場合、HAT培養液中で、正常細胞との細胞融合に成功した細胞が選択的に増殖し得る。所望のハイブリドーマ以外の細胞(非融合細胞)が死滅するのに十分な時間、上記HAT培養液を用いた培養が継続される。具体的には、一般に、数日から数週間の培養によって、所望のハイブリドーマが選択され得る。次いで、通常の限界希釈法によって、所望の抗体を産生するハイブリドーマのスクリーニングおよび単一クローニングが実施され得る。
所望の抗体のスクリーニングおよび単一クローニングが、公知の抗原抗体反応に基づくスクリーニング方法によって好適に実施され得る。例えば、IL-6Rに結合するモノクローナル抗体は、細胞表面に発現したIL-6Rに結合することができる。このようなモノクローナル抗体は、たとえば、FACS(fluorescence activated cell sorting)によってスクリーニングされ得る。FACSは、蛍光抗体と接触させた細胞をレーザー光で解析し、個々の細胞が発する蛍光を測定することによって細胞表面への抗体の結合を測定することを可能にするシステムである。
FACSによって本発明のモノクローナル抗体を産生するハイブリドーマをスクリーニングするためには、まずIL-6Rを発現する細胞を調製する。スクリーニングのための好ましい細胞は、IL-6Rを強制発現させた哺乳動物細胞である。宿主細胞として使用した形質転換されていない哺乳動物細胞を対照として用いることによって、細胞表面のIL-6Rに対する抗体の結合活性が選択的に検出され得る。すなわち、宿主細胞に結合せず、IL-6R強制発現細胞に結合する抗体を産生するハイブリドーマを選択することによって、IL-6Rモノクローナル抗体を産生するハイブリドーマが取得され得る。
あるいは固定化したIL-6R発現細胞に対する抗体の結合活性がELISAの原理にもとづいて評価され得る。たとえば、ELISAプレートのウェルにIL-6R発現細胞が固定化される。ハイブリドーマの培養上清をウェル内の固定化細胞に接触させ、固定化細胞に結合する抗体が検出される。モノクローナル抗体がマウス由来の場合、細胞に結合した抗体は、抗マウスイムノグロブリン抗体によって検出され得る。これらのスクリーニングによって選択された、抗原に対する結合能を有する所望の抗体を産生するハイブリドーマは、限界希釈法等によりクローニングされ得る。
このようにして作製されるモノクローナル抗体を産生するハイブリドーマは通常の培養液中で継代培養され得る。また、当該ハイブリドーマは液体窒素中で長期にわたって保存され得る。
当該ハイブリドーマを通常の方法に従い培養し、その培養上清から所望のモノクローナル抗体が取得され得る。あるいはハイブリドーマをこれと適合性がある哺乳動物に投与して増殖せしめ、その腹水からモノクローナル抗体が取得され得る。前者の方法は、高純度の抗体を得るのに好適なものである。
当該ハイブリドーマ等の抗体産生細胞からクローニングされる抗体遺伝子によってコードされる抗体も好適に利用され得る。クローニングした抗体遺伝子を適当なベクターに組み込んで宿主に導入することによって、当該遺伝子によってコードされる抗体が発現する。抗体遺伝子の単離と、ベクターへの導入、そして宿主細胞の形質転換のための方法は例えば、Vandammeらによって既に確立されている(Eur. J. Biochem.(1990)192 (3), 767-775)。下記に述べるように組換え抗体の製造方法もまた公知である。
たとえば、抗IL-6R抗体を産生するハイブリドーマ細胞から、抗IL-6R抗体の可変領域(V領域)をコードするcDNAが取得される。そのために、通常、まずハイブリドーマから全RNAが抽出される。細胞からmRNAを抽出するための方法として、たとえば次のような方法を利用することができる。
-グアニジン超遠心法(Biochemistry (1979) 18 (24), 5294-5299)
-AGPC法(Anal. Biochem. (1987) 162 (1), 156-159)
-グアニジン超遠心法(Biochemistry (1979) 18 (24), 5294-5299)
-AGPC法(Anal. Biochem. (1987) 162 (1), 156-159)
抽出されたmRNAは、mRNA Purification Kit (GEヘルスケアバイオサイエンス製)等を使用して精製され得る。あるいは、QuickPrep mRNA Purification Kit (GEヘルスケアバイオサイエンス製)などのように、細胞から直接全mRNAを抽出するためのキットも市販されている。このようなキットを用いて、ハイブリドーマからmRNAが取得され得る。得られたmRNAから逆転写酵素を用いて抗体V領域をコードするcDNAが合成され得る。cDNAは、AMV Reverse Transcriptase First-strand cDNA Synthesis Kit(生化学工業社製)等によって合成され得る。また、cDNAの合成および増幅のために、SMART RACE cDNA 増幅キット(Clontech製)およびPCRを用いた5'-RACE法(Proc. Natl. Acad. Sci. U.S.A. (1988) 85 (23), 8998-9002、Nucleic Acids Res. (1989) 17 (8), 2919-2932)が適宜利用され得る。更にこうしたcDNAの合成の過程においてcDNAの両末端に後述する適切な制限酵素サイトが導入され得る。
得られたPCR産物から目的とするcDNA断片が精製され、次いでベクターDNAと連結される。このように組換えベクターが作製され、大腸菌等に導入されコロニーが選択された後に、該コロニーを形成した大腸菌から所望の組換えベクターが調製され得る。そして、当該組換えベクターが目的とするcDNAの塩基配列を有しているか否かについて、公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法等により確認される。
可変領域をコードする遺伝子を取得するためには、可変領域遺伝子増幅用のプライマーを使った5'-RACE法を利用するのが簡便である。まずハイブリドーマ細胞より抽出されたRNAを鋳型としてcDNAが合成され、5'-RACE cDNAライブラリが得られる。5'-RACE cDNAライブラリの合成にはSMART RACE cDNA 増幅キットなど市販のキットが適宜用いられる。
得られた5'-RACE cDNAライブラリを鋳型として、PCR法によって抗体遺伝子が増幅される。公知の抗体遺伝子配列をもとにマウス抗体遺伝子増幅用のプライマーがデザインされ得る。これらのプライマーは、イムノグロブリンのサブクラスごとに異なる塩基配列である。したがって、サブクラスは予めIso Stripマウスモノクローナル抗体アイソタイピングキット(ロシュ・ダイアグノスティックス)などの市販キットを用いて決定しておくことが望ましい。
具体的には、たとえばマウスIgGをコードする遺伝子の取得を目的とするときには、重鎖としてγ1、γ2a、γ2b、γ3、軽鎖としてκ鎖とλ鎖をコードする遺伝子の増幅が可能なプライマーが利用され得る。IgGの可変領域遺伝子を増幅するためには、一般に3'側のプライマーには可変領域に近い定常領域に相当する部分にアニールするプライマーが利用される。一方5'側のプライマーには、5' RACE cDNAライブラリ作製キットに付属するプライマーが利用される。
こうして増幅されたPCR産物を利用して、重鎖と軽鎖の組み合せからなるイムノグロブリンが再構成され得る。再構成されたイムノグロブリンの、IL-6Rに対する結合活性を指標として、所望の抗体がスクリーニングされ得る。たとえばIL-6Rに対する抗体の取得を目的とするとき、抗体のIL-6Rへの結合は、特異的であることがさらに好ましい。IL-6Rに結合する抗体は、たとえば次のようにしてスクリーニングされ得る;
(1)ハイブリドーマから得られたcDNAによってコードされるV領域を含む抗体をIL-6R発現細胞に接触させる工程、
(2)IL-6R発現細胞と抗体との結合を検出する工程、および
(3)IL-6R発現細胞に結合する抗体を選択する工程。
(1)ハイブリドーマから得られたcDNAによってコードされるV領域を含む抗体をIL-6R発現細胞に接触させる工程、
(2)IL-6R発現細胞と抗体との結合を検出する工程、および
(3)IL-6R発現細胞に結合する抗体を選択する工程。
抗体とIL-6R発現細胞との結合を検出する方法は公知である。具体的には、先に述べたFACSなどの手法によって、抗体とIL-6R発現細胞との結合が検出され得る。抗体の結合活性を評価するためにIL-6R発現細胞の固定標本が適宜利用され得る。
結合活性を指標とする抗体のスクリーニング方法として、ファージベクターを利用したパニング法も好適に用いられる。ポリクローナルな抗体発現細胞群より抗体遺伝子を重鎖と軽鎖のサブクラスのライブラリとして取得した場合には、ファージベクターを利用したスクリーニング方法が有利である。重鎖と軽鎖の可変領域をコードする遺伝子は、適当なリンカー配列で連結することによってシングルチェインFv(scFv)を形成することができる。scFvをコードする遺伝子をファージベクターに挿入することにより、scFvを表面に発現するファージが取得され得る。このファージと所望の抗原との接触の後に、抗原に結合したファージを回収することによって、目的の結合活性を有するscFvをコードするDNAが回収され得る。この操作を必要に応じて繰り返すことにより、所望の結合活性を有するscFvが濃縮され得る。
目的とする抗IL-6R抗体のV領域をコードするcDNAが得られた後に、当該cDNAの両末端に挿入した制限酵素サイトを認識する制限酵素によって該cDNAが消化される。好ましい制限酵素は、抗体遺伝子を構成する塩基配列に出現する頻度が低い塩基配列を認識して消化する。更に1コピーの消化断片をベクターに正しい方向で挿入するためには、付着末端を与える制限酵素の挿入が好ましい。上記のように消化された抗IL-6R抗体のV領域をコードするcDNAを適当な発現ベクターに挿入することによって、抗体発現ベクターが取得され得る。このとき、抗体定常領域(C領域)をコードする遺伝子と、前記V領域をコードする遺伝子とがインフレームで融合されれば、キメラ抗体が取得される。ここで、キメラ抗体とは、定常領域と可変領域の由来が異なることをいう。したがって、マウス-ヒトなどの異種キメラ抗体に加え、ヒト-ヒト同種キメラ抗体も、本発明におけるキメラ抗体に含まれる。予め定常領域を有する発現ベクターに、前記V領域遺伝子を挿入することによって、キメラ抗体発現ベクターが構築され得る。具体的には、たとえば、所望の抗体定常領域をコードするDNAを保持した発現ベクターの5'側に、前記V領域遺伝子を消化する制限酵素の制限酵素認識配列が適宜配置され得る。同じ組み合わせの制限酵素で消化された両者がインフレームで融合されることによって、キメラ抗体発現ベクターが構築される。
抗IL-6Rモノクローナル抗体を製造するために、抗体遺伝子が発現制御領域による制御の下で発現するように発現ベクターに組み込まれる。抗体を発現するための発現制御領域とは、例えば、エンハンサーやプロモーターを含む。また、発現した抗体が細胞外に分泌されるように、適切なシグナル配列がアミノ末端に付加され得る。後に記載される実施例ではシグナル配列として、アミノ酸配列MGWSCIILFLVATATGVHS(配列番号:3)を有するペプチドが使用されているが、これ以外にも適したシグナル配列が付加される。発現されたポリペプチドは上記配列のカルボキシル末端部分で切断され、切断されたポリペプチドが成熟ポリペプチドとして細胞外に分泌され得る。次いで、この発現ベクターによって適当な宿主細胞が形質転換されることによって、抗IL-6R抗体をコードするDNAを発現する組換え細胞が取得され得る。
抗体遺伝子の発現のために、抗体重鎖(H鎖)および軽鎖(L鎖)をコードするDNAは、それぞれ別の発現ベクターに組み込まれる。H鎖とL鎖が組み込まれたベクターによって、同じ宿主細胞に同時に形質転換(co-transfect)されることによって、H鎖とL鎖を備えた抗体分子が発現され得る。あるいはH鎖およびL鎖をコードするDNAが単一の発現ベクターに組み込まれることによって宿主細胞が形質転換され得る(国際公開WO 1994/011523を参照のこと)。
単離された抗体遺伝子を適当な宿主に導入することによって抗体を作製するための宿主細胞と発現ベクターの多くの組み合わせが公知である。これらの発現系は、いずれも本発明の抗原結合ドメインを単離するのに応用され得る。真核細胞が宿主細胞として使用される場合、動物細胞、植物細胞、あるいは真菌細胞が適宜使用され得る。具体的には、動物細胞としては、次のような細胞が例示され得る。
(1)哺乳類細胞、:CHO(Chinese hamster ovary cell line)、COS(Monkey kidney cell line)、ミエローマ(Sp2/0、NS0等)、BHK (baby hamster kidney cell line)、Hela、Vero、HEK293(human embryonic kidney cell line with sheared adenovirus (Ad)5 DNA)、PER.C6 cell (human embryonic retinal cell line transformed with the Adenovirus Type 5 (Ad5) E1A and E1B genes)など(Current Protocols in Protein Science (May, 2001, Unit 5.9, Table 5.9.1))
(2)両生類細胞:アフリカツメガエル卵母細胞など
(3)昆虫細胞:sf9、sf21、Tn5など
(1)哺乳類細胞、:CHO(Chinese hamster ovary cell line)、COS(Monkey kidney cell line)、ミエローマ(Sp2/0、NS0等)、BHK (baby hamster kidney cell line)、Hela、Vero、HEK293(human embryonic kidney cell line with sheared adenovirus (Ad)5 DNA)、PER.C6 cell (human embryonic retinal cell line transformed with the Adenovirus Type 5 (Ad5) E1A and E1B genes)など(Current Protocols in Protein Science (May, 2001, Unit 5.9, Table 5.9.1))
(2)両生類細胞:アフリカツメガエル卵母細胞など
(3)昆虫細胞:sf9、sf21、Tn5など
あるいは植物細胞としては、ニコティアナ・タバカム(Nicotiana tabacum)などのニコティアナ(Nicotiana)属由来の細胞による抗体遺伝子の発現系が公知である。植物細胞の形質転換には、カルス培養した細胞が適宜利用され得る。
更に真菌細胞としては、次のような細胞を利用することができる。
-酵母:サッカロミセス・セレビシエ(Saccharomyces serevisiae)などのサッカロミセス(Saccharomyces )属、メタノール資化酵母(Pichia pastoris)などのPichia属。
-糸状菌:アスペスギルス・ニガー(Aspergillus niger)などのアスペルギルス(Aspergillus )属。
-酵母:サッカロミセス・セレビシエ(Saccharomyces serevisiae)などのサッカロミセス(Saccharomyces )属、メタノール資化酵母(Pichia pastoris)などのPichia属。
-糸状菌:アスペスギルス・ニガー(Aspergillus niger)などのアスペルギルス(Aspergillus )属。
また、原核細胞を利用した抗体遺伝子の発現系も公知である。たとえば、細菌細胞を用いる場合、大腸菌(E. coli )、枯草菌などの細菌細胞が適宜利用され得る。これらの細胞中に、目的とする抗体遺伝子を含む発現ベクターが形質転換によって導入される。形質転換された細胞をin vitroで培養することにより、当該形質転換細胞の培養物から所望の抗体が取得され得る。
組換え抗体の産生には、上記宿主細胞に加えて、トランスジェニック動物も利用され得る。すなわち所望の抗体をコードする遺伝子が導入された動物から、当該抗体を得ることができる。例えば、抗体遺伝子は、乳汁中に固有に産生されるタンパク質をコードする遺伝子の内部にインフレームで挿入することによって融合遺伝子として構築され得る。乳汁中に分泌されるタンパク質として、たとえば、ヤギβカゼインなどを利用され得る。抗体遺伝子が挿入された融合遺伝子を含むDNA断片はヤギの胚へ注入され、当該注入された胚が雌のヤギへ導入される。胚を受容したヤギから生まれるトランスジェニックヤギ(またはその子孫)が産生する乳汁からは、所望の抗体が乳汁タンパク質との融合タンパク質として取得され得る。また、トランスジェニックヤギから産生される所望の抗体を含む乳汁量を増加させるために、ホルモンがトランスジェニックヤギに対して投与され得る(Bio/Technology (1994), 12 (7), 699-702)。
本明細書において記載される抗原結合分子がヒトに投与される場合、当該抗原結合分子における抗原結合ドメインとして、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体由来の抗原結合ドメインが適宜採用され得る。遺伝子組換え型抗体には、例えば、ヒト化(Humanized)抗体等が含まれる。これらの改変抗体は、公知の方法を用いて適宜製造される。
特定の低分子化合物に対して所望の結合活性を有する抗体を作製する方法として、通常のタンパク抗原に結合する抗体を作製する方法と同様の方法により低分子化合物に対して所望の結合活性を有する抗体が取得され得る。低分子化合物に対する抗体を取得するために使用する感作抗原の作成法の一態様として、Mariculture Keyhole Limpet Hemocyanin(KLH)と低分子化合物を連結させる方法が例示される。MTAに対して抗体を取得するために作成される非限定な感作抗原の一例として、6’-MTA- Keyhole Limpet Hemocyanin(6’-MTA-KLH)が例示される。Mariculture Keyhole Limpet Hemocyanin(KLH)は、ヘルパーT細胞上に発現するT細胞受容体が認識できる抗原性の高い蛋白質であり、抗体産生を活性化することが知られているため、MTAと連結させることでMTAに対する抗体の産生を亢進させることが期待される。KLHに連結させる低分子化合物はMTAに限定されるものではなく、合成可能な様々な低分子化合物に対して同様の手法で感作抗原を作成することが可能であり、非限定な例として、AMP、ADP、ATP、アデノシン、またはSAHなどが例示される。国際公開WO2013/180200号においても低分子化合物免疫原のデザインについて開示している。
また、低分子化合物に連結させる抗原はKLHに限定されるものではなく、例えばビオチンなどと連結したものを感作抗原として使用してもよく、さらにKLH、ビオチン以外にも低分子化合物に連結可能であれば使用可能され得る。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[1] ビオチン化MTA。
[2] Biotin-2’-MTA。
[3] 6’-MTA-biotin。
[4]MTAに結合する抗原結合分子をスクリーニングするための[1]から[3]に記載のビオチン化MTAの使用。
[5] MTAに結合する抗原結合分子を取得するための免疫原としての[1]から[3]に記載のビオチン化MTAの使用。
[6] [1]から[3]に記載のビオチン化MTAを用いて、MTAに結合する抗原結合分子をスクリーニングする方法。
また、低分子化合物に連結させる抗原はKLHに限定されるものではなく、例えばビオチンなどと連結したものを感作抗原として使用してもよく、さらにKLH、ビオチン以外にも低分子化合物に連結可能であれば使用可能され得る。
本開示はまた、以下に例示的に記載する態様を包含するものである。
[1] ビオチン化MTA。
[2] Biotin-2’-MTA。
[3] 6’-MTA-biotin。
[4]MTAに結合する抗原結合分子をスクリーニングするための[1]から[3]に記載のビオチン化MTAの使用。
[5] MTAに結合する抗原結合分子を取得するための免疫原としての[1]から[3]に記載のビオチン化MTAの使用。
[6] [1]から[3]に記載のビオチン化MTAを用いて、MTAに結合する抗原結合分子をスクリーニングする方法。
多重特異性抗原結合分子または多重パラトピックな抗原結合分子
その少なくとも一つの抗原結合ドメインが抗原分子中の第一のエピトープに結合し、その少なくとも一つの別の抗原結合ドメインが抗原分子中の第二のエピトープに結合する特徴を有する、少なくとも二つの抗原結合ドメインを含む抗原結合分子は、その反応の特異性という観点から多重特異性抗原結合分子と呼ばれる。一分子の抗原結合分子に含まれる二種類の抗原結合ドメインによって当該抗原結合分子が、二つの異なるエピトープに結合する場合、当該抗原結合分子は二重特異性抗原結合分子と呼ばれる。また、一分子の抗原結合分子に含まれる三種類の抗原結合ドメインによって当該抗原結合分子が、三つの異なるエピトープに結合する場合、当該抗原結合分子は三重特異性抗原結合分子と呼ばれる。
その少なくとも一つの抗原結合ドメインが抗原分子中の第一のエピトープに結合し、その少なくとも一つの別の抗原結合ドメインが抗原分子中の第二のエピトープに結合する特徴を有する、少なくとも二つの抗原結合ドメインを含む抗原結合分子は、その反応の特異性という観点から多重特異性抗原結合分子と呼ばれる。一分子の抗原結合分子に含まれる二種類の抗原結合ドメインによって当該抗原結合分子が、二つの異なるエピトープに結合する場合、当該抗原結合分子は二重特異性抗原結合分子と呼ばれる。また、一分子の抗原結合分子に含まれる三種類の抗原結合ドメインによって当該抗原結合分子が、三つの異なるエピトープに結合する場合、当該抗原結合分子は三重特異性抗原結合分子と呼ばれる。
抗原分子中の第一のエピトープに結合する抗原結合ドメイン中のパラトープと、第一のエピトープと構造の異なる第二のエピトープに結合する抗原結合ドメイン中のパラトープとはその構造が互いに異なる。ゆえに、その少なくとも一つの抗原結合ドメインが抗原分子中の第一のエピトープに結合し、その少なくとも一つの別の抗原結合ドメインが抗原分子中の第二のエピトープに結合する特徴を有する、少なくとも二つの抗原結合ドメインを含む抗原結合分子は、その構造の特異性という観点から多重パラトピック抗原結合分子と呼ばれる。一分子の抗原結合分子に含まれる二種類の抗原結合ドメインによって当該抗原結合分子が、二つの異なるエピトープに結合する場合、当該抗原結合分子は二重パラトピック抗原結合分子と呼ばれる。また、一分子の抗原結合分子に含まれる三種類の抗原結合ドメインによって当該抗原結合分子が、三つの異なるエピトープに結合する場合、当該抗原結合分子は三重パラトピック抗原結合分子と呼ばれる。
二重特異性抗体とその作製方法
一つまたは複数の抗原結合ドメインを含む多価の多重特異性または多重パラトピック抗原結合分子とその調製方法は、Conrathら(J.Biol.Chem. (2001) 276 (10) 7346-7350)、Muyldermans(Rev. Mol. Biotech. (2001) 74, 277-302)およびKontermann R.E. (2011) Bispecific Antibodies(Springer-Verlag)等の非特許文献、ならびに国際公開WO1996/034103またはWO1999/023221等の特許文献等にも記載されている。これらに記載された多重特異性または多重パラトピック抗原結合分子とその調製方法を用いることによって、本開示の抗原結合分子を作製することが可能である
一つまたは複数の抗原結合ドメインを含む多価の多重特異性または多重パラトピック抗原結合分子とその調製方法は、Conrathら(J.Biol.Chem. (2001) 276 (10) 7346-7350)、Muyldermans(Rev. Mol. Biotech. (2001) 74, 277-302)およびKontermann R.E. (2011) Bispecific Antibodies(Springer-Verlag)等の非特許文献、ならびに国際公開WO1996/034103またはWO1999/023221等の特許文献等にも記載されている。これらに記載された多重特異性または多重パラトピック抗原結合分子とその調製方法を用いることによって、本開示の抗原結合分子を作製することが可能である
前記のような多重特異性または多重パラトピック抗原結合分子とその調製方法の一態様として、二重特異性抗体とその作製方法が下記に例示される。二重特異性抗体とは、異なるエピトープに対して特異的に結合する二種類の可変領域を含む抗体である。IgG型の二重特異性抗体はIgG抗体を産生するハイブリドーマ二種を融合することによって生じるhybrid hybridoma(quadroma)によって分泌させることが可能である(Milsteinら(Nature (1983) 305, 537-540)。
二重特異性抗体を前記の抗体の項で記載されたような組換え手法を用いて製造する場合、目的の二種の可変領域を含む重鎖をコードする遺伝子を細胞に導入しそれらを共発現させる方法が採用され得る。しかしながら、こうした共発現させる方法における重鎖の組合せを考慮するだけでも、(i) 第一のエピトープに結合する可変領域を含む重鎖と第二のエピトープに結合する可変領域を含む重鎖が一対となった重鎖の組合せ、(ii) 第一のエピトープに結合する可変領域を含む重鎖のみが一対となった重鎖の組合せ、(iii) 第二のエピトープに結合する可変領域を含む重鎖のみが一対となった重鎖の組合せが、2:1:1の分子数の割合で存在する混合物となる。これら三種類の重鎖の組合せの混合物から目的の重鎖の組合せを含む抗原結合分子を精製することは困難である。
こうした組換え手法を用いて二重特異性抗体を製造する際に、重鎖を構成するCH3ドメインに適当なアミノ酸置換の改変を加えることによってヘテロな組合せの重鎖を含む二重特異性抗体が優先的に分泌され得る。具体的には、一方の重鎖のCH3ドメインに存在するアミノ酸側鎖をより大きい側鎖(knob(「突起」の意))に置換し、もう一方の重鎖のCH3ドメインに存在するアミノ酸側鎖をより小さい側鎖(hole(「空隙」の意))に置換することによって、突起が空隙内に配置され得るようにして異種の重鎖形成の促進および同種の重鎖形成の阻害を引き起こす方法である(国際公開WO1996027011、Ridgwayら(Protein Engineering (1996) 9, 617-621)、Merchantら(Nat. Biotech. (1998) 16, 677-681))。
また、ポリペプチドの会合、またはポリペプチドによって構成される異種多量体の会合の制御方法を、重鎖の会合に利用することによって二重特異性抗体を作製する技術も知られている。即ち、重鎖内の界面を形成するアミノ酸残基を改変することによって、同一配列を有する重鎖の会合が阻害され、配列の異なる二つの重鎖が形成されるように制御する方法が二重特異性抗体の作製に採用され得る(国際公開WO2006/106905)。このような方法も二重特異性抗体を製造する際に、採用され得る。
がん
本明細書において、「がん」という用語は、一般に、悪性新生物を表すために用いられ、それは、転移性または非転移性であってよい。例えば、消化管や皮膚等の上皮組織から発生した癌腫の非限定な例として、脳腫瘍、皮膚癌、頸頭部癌、食道癌、肺癌、胃癌、十二指腸癌、乳癌、前立腺癌、子宮頸癌、子宮体癌、膵臓癌、肝臓癌、大腸癌、結腸癌、膀胱癌、および卵巣癌等が例示される。また、筋肉等の非上皮性組織(間質)から発生した肉腫の非限定な例として、骨肉腫、軟骨肉腫、横紋筋肉腫、平滑筋肉腫、脂肪肉腫、および血管肉腫等が例示される。さらに、造血器由来の血液がんの非限定な例として、ホジキンリンパ腫(Hodgkin's lymphoma)および非ホジキンリンパ腫(non Hodgkin's lymphoma)を含む悪性リンパ腫、急性(acute myelocytic leukemia)または慢性骨髄性白血病(chronic myelocytic leukemia)、および急性(acute lymphatic leukemia)または慢性リンパ性白血病(chronic lymphatic leukemia)を含む白血病、ならびに多発性骨髄腫(multiple myeloma)が例示される。本明細書で広く用いられる「新生物」という用語は、新たに生じたいかなる病的組織腫瘍をも意味する。本開示においては、新生物は腫瘍の形成を生じ、それは部分的に血管形成を特徴とする。新生物は、例えば、血管腫、神経膠腫、奇形腫等の良性、あるいは、例えば、癌腫、肉腫、膠細胞腫、星状膠細胞腫、神経芽細胞腫、網膜芽腫等の悪性でありうる。
本明細書において、「がん」という用語は、一般に、悪性新生物を表すために用いられ、それは、転移性または非転移性であってよい。例えば、消化管や皮膚等の上皮組織から発生した癌腫の非限定な例として、脳腫瘍、皮膚癌、頸頭部癌、食道癌、肺癌、胃癌、十二指腸癌、乳癌、前立腺癌、子宮頸癌、子宮体癌、膵臓癌、肝臓癌、大腸癌、結腸癌、膀胱癌、および卵巣癌等が例示される。また、筋肉等の非上皮性組織(間質)から発生した肉腫の非限定な例として、骨肉腫、軟骨肉腫、横紋筋肉腫、平滑筋肉腫、脂肪肉腫、および血管肉腫等が例示される。さらに、造血器由来の血液がんの非限定な例として、ホジキンリンパ腫(Hodgkin's lymphoma)および非ホジキンリンパ腫(non Hodgkin's lymphoma)を含む悪性リンパ腫、急性(acute myelocytic leukemia)または慢性骨髄性白血病(chronic myelocytic leukemia)、および急性(acute lymphatic leukemia)または慢性リンパ性白血病(chronic lymphatic leukemia)を含む白血病、ならびに多発性骨髄腫(multiple myeloma)が例示される。本明細書で広く用いられる「新生物」という用語は、新たに生じたいかなる病的組織腫瘍をも意味する。本開示においては、新生物は腫瘍の形成を生じ、それは部分的に血管形成を特徴とする。新生物は、例えば、血管腫、神経膠腫、奇形腫等の良性、あるいは、例えば、癌腫、肉腫、膠細胞腫、星状膠細胞腫、神経芽細胞腫、網膜芽腫等の悪性でありうる。
用語「がん組織」とは、少なくとも一つのがん細胞を含む組織を意味する。したがって、例えばがん組織ががん細胞と血管を含んでいるように、がん細胞および内皮細胞を含む腫瘤(tumor mass)の形成に寄与するすべての細胞型をいう。本明細書において、腫瘤とは腫瘍組織巣(a foci of tumor tissue)をいう。「腫瘍」という用語は、一般に、良性新生物または悪性新生物を意味するために用いられる。
本開示において、「MTAが蓄積しているがん組織」とは、前記がん組織中にMTAが正常組織と比較して多量に検出されるがん組織を意味し、比較対象となる正常組織の非限定な一例としては、がん組織の隣接正常組織や健常人の組織が例示され得る。
また、MTAを代謝するメチルチオアデノシンホスフォリラーゼ(MTAP:methylthioadenosine phosphorylase)が欠損したがん組織やMTAPの機能が低下しているがん組織もMTAが蓄積しているがん組織の一態様である。具体的には、MTAPをコードしている遺伝子が欠損もしくは発現低下しているがん組織、MTAPの活性を低下させる変異もしくはスプライシングバリアントを発現しているがん組織、またはMTAPの酵素活性が低下しているがん組織が、MTAが蓄積しているがん組織の一態様である。MTAPの発現または機能の低下は、正常組織と比較することで決定することができ、比較対象となる正常組織の非限定な一例としては、がん組織の隣接正常組織や健常人の組織が例示され得る。
また、MTAを代謝するメチルチオアデノシンホスフォリラーゼ(MTAP:methylthioadenosine phosphorylase)が欠損したがん組織やMTAPの機能が低下しているがん組織もMTAが蓄積しているがん組織の一態様である。具体的には、MTAPをコードしている遺伝子が欠損もしくは発現低下しているがん組織、MTAPの活性を低下させる変異もしくはスプライシングバリアントを発現しているがん組織、またはMTAPの酵素活性が低下しているがん組織が、MTAが蓄積しているがん組織の一態様である。MTAPの発現または機能の低下は、正常組織と比較することで決定することができ、比較対象となる正常組織の非限定な一例としては、がん組織の隣接正常組織や健常人の組織が例示され得る。
Cancer-associated fibroblast (CAF)
本開示において「Cancer-associated fibroblast (CAF)」とは、がん組織周辺部に存在す内皮細胞等,多様な起源を持つheterogeneous な集団である。非限定なCAFの特徴としては、がん細胞の増殖促進,血管新生の促進,がん細胞の脈管浸潤促進,免疫応答の制御等を通じてがんの進展に有利な微小環境を構築する等がある。さらに、非限定なCAFの一態様として、α-smooth muscle actin (α-SMA)、fibroblast activation protein (FAP)、tenascin-C (TN-C)、periostin (POSTN)、NG2 chondroitin sulfate proteoglycan (NG2)、platelet derived growth factor receptor (PDGFR)、vimentin、desmin、fibroblast specific protein-1 (FSP1)、fibronectinから選ばれるマーカーが発現している細胞が例示される。
本開示において「Cancer-associated fibroblast (CAF)」とは、がん組織周辺部に存在す内皮細胞等,多様な起源を持つheterogeneous な集団である。非限定なCAFの特徴としては、がん細胞の増殖促進,血管新生の促進,がん細胞の脈管浸潤促進,免疫応答の制御等を通じてがんの進展に有利な微小環境を構築する等がある。さらに、非限定なCAFの一態様として、α-smooth muscle actin (α-SMA)、fibroblast activation protein (FAP)、tenascin-C (TN-C)、periostin (POSTN)、NG2 chondroitin sulfate proteoglycan (NG2)、platelet derived growth factor receptor (PDGFR)、vimentin、desmin、fibroblast specific protein-1 (FSP1)、fibronectinから選ばれるマーカーが発現している細胞が例示される。
Tumor-associated macrophage(TAM)
本開示において「Tumor-associated macrophage(TAM)」とは、がん組織およびその周辺部に存在するマクロファージをいう。例えば、線維芽細胞や血管内皮細胞などとともに、がんの微小環境を形成するマクロファージ集団が例示される。非限定なTAMの特徴として、抗炎症性因子の産生や制御性T細胞の浸潤を促すことで抗腫瘍免疫を抑制するなど、種々の血管新生因子の産生によって新生血管を誘導することが例示される。さらに、非限定なTAMの一態様として、CD163、CD204、IL-10、TGF-β、Prastaglandin E2から選ばれるマーカーが発現している細胞が例示される。
本開示において「Tumor-associated macrophage(TAM)」とは、がん組織およびその周辺部に存在するマクロファージをいう。例えば、線維芽細胞や血管内皮細胞などとともに、がんの微小環境を形成するマクロファージ集団が例示される。非限定なTAMの特徴として、抗炎症性因子の産生や制御性T細胞の浸潤を促すことで抗腫瘍免疫を抑制するなど、種々の血管新生因子の産生によって新生血管を誘導することが例示される。さらに、非限定なTAMの一態様として、CD163、CD204、IL-10、TGF-β、Prastaglandin E2から選ばれるマーカーが発現している細胞が例示される。
エフェクター細胞
本開示において、「エフェクター細胞」とは、T細胞(CD4+(ヘルパーリンパ球)T細胞および/またはCD8+(細胞傷害性)T細胞)、多核白血球(好中球、好酸球、好塩基球、肥満細胞)、単球、マクロファージ、組織球またはナチュラルキラー細胞(NK細胞)、NK様T細胞、クッパー細胞、ランゲルハンス細胞、またはリンフォカイン活性化キラー細胞(LAK細胞)等の白血球、Bリンパ球、もしくは樹状細胞またはマクロファージ等の抗原提示細胞を含む最も広義な意味で使用され得るが、好適なエフェクター細胞の例としては、CD8+(細胞傷害性)T細胞、NK細胞、またはマクロファージが挙げられる。エフェクター細胞の細胞膜に発現する膜型分子であれば、本開示の抗原結合分子に含まれる少なくとも1つの抗原結合ドメインが結合する抗原として使用され得るが、好適な膜型分子としては、TCRを構成するポリペプチド、CD3、CD2、CD28、CD44、CD16、CD32、CD64、またはNKG2DもしくはNK細胞活性化リガンドが非限定な例として例示され得る。
本開示において、「エフェクター細胞」とは、T細胞(CD4+(ヘルパーリンパ球)T細胞および/またはCD8+(細胞傷害性)T細胞)、多核白血球(好中球、好酸球、好塩基球、肥満細胞)、単球、マクロファージ、組織球またはナチュラルキラー細胞(NK細胞)、NK様T細胞、クッパー細胞、ランゲルハンス細胞、またはリンフォカイン活性化キラー細胞(LAK細胞)等の白血球、Bリンパ球、もしくは樹状細胞またはマクロファージ等の抗原提示細胞を含む最も広義な意味で使用され得るが、好適なエフェクター細胞の例としては、CD8+(細胞傷害性)T細胞、NK細胞、またはマクロファージが挙げられる。エフェクター細胞の細胞膜に発現する膜型分子であれば、本開示の抗原結合分子に含まれる少なくとも1つの抗原結合ドメインが結合する抗原として使用され得るが、好適な膜型分子としては、TCRを構成するポリペプチド、CD3、CD2、CD28、CD44、CD16、CD32、CD64、またはNKG2DもしくはNK細胞活性化リガンドが非限定な例として例示され得る。
メチルチオアデノシン(MTA:Methylthioadenosine)
本明細書中で用いられる用語「MTA」は、メチルチオアデノシン(Methylthioadenosine)を指しており、具体的には、下記化学式で示される化合物を指す。

MTA (CAS番号:2457-80-9)
本明細書中で用いられる用語「MTA」は、メチルチオアデノシン(Methylthioadenosine)を指しており、具体的には、下記化学式で示される化合物を指す。
MTA類縁体
本明細書中で用いられる用語「MTA類縁体」は、MTAと一部共通の構造を有するMTA以外の低分子化合物を指す。MTA類縁体として、分子中にアデノシンを共通骨格として有する低分子化合物が例示されうるし、またアデノシン中の5位の炭素に硫黄原子や酸素原子を含む側鎖を有する低分子化合物が例示される。さらにMTA類縁体として、これらに限定されるものではないが、例えば、S-アデノシルメチオニン(SAM:S-adenoshylmethionine)、S-アデノシルホモシステイン(SAH:S-adenosylhomocystein、S-(5'-Adenosyl)-L-homocysteine)等のポリアミン生合成経路の代謝物(Stevensら(J Chromatogr A. 2010 May 7;1217(19):3282-8))が好適に挙げられる。

SAM

SAH
また、MTA類縁体の非限定的な一態様として、アデノシン、アデノシン3リン酸(ATP)、アデノシン2リン酸(ADP)、アデノシン1リン酸(AMP)が挙げられる。

アデノシン

アデノシン1リン酸(AMP)

アデノシン2リン酸(ADP)

アデノシン3リン酸(ATP)
本明細書中で用いられる用語「MTA類縁体」は、MTAと一部共通の構造を有するMTA以外の低分子化合物を指す。MTA類縁体として、分子中にアデノシンを共通骨格として有する低分子化合物が例示されうるし、またアデノシン中の5位の炭素に硫黄原子や酸素原子を含む側鎖を有する低分子化合物が例示される。さらにMTA類縁体として、これらに限定されるものではないが、例えば、S-アデノシルメチオニン(SAM:S-adenoshylmethionine)、S-アデノシルホモシステイン(SAH:S-adenosylhomocystein、S-(5'-Adenosyl)-L-homocysteine)等のポリアミン生合成経路の代謝物(Stevensら(J Chromatogr A. 2010 May 7;1217(19):3282-8))が好適に挙げられる。
また、MTA類縁体の非限定的な一態様として、アデノシン、アデノシン3リン酸(ATP)、アデノシン2リン酸(ADP)、アデノシン1リン酸(AMP)が挙げられる。
低分子化合物
本開示中で用いられる用語「低分子化合物」は、生体に存在する「生体高分子」以外の天然由来の化学物質又は非天然由来の化学物質をいう。低分子化合物として、これらに限定されるものではないが、例えば、天然に存在する、もしくは人工的に合成される分子量が10000以下の化合物、好ましくは分子量が1000以下の化合物が列挙される。低分子化合物の非限定的な態様として、例えば、がん組織特異的化合物、炎症組織特異的化合物、非天然化合物が例示される。
本開示中で用いられる用語「低分子化合物」は、生体に存在する「生体高分子」以外の天然由来の化学物質又は非天然由来の化学物質をいう。低分子化合物として、これらに限定されるものではないが、例えば、天然に存在する、もしくは人工的に合成される分子量が10000以下の化合物、好ましくは分子量が1000以下の化合物が列挙される。低分子化合物の非限定的な態様として、例えば、がん組織特異的化合物、炎症組織特異的化合物、非天然化合物が例示される。
がん組織特異的化合物
本明細書中で用いられる、用語「がん組織特異的な化合物(がん組織特異的化合物)」とは、非がん組織と比較してがん組織中に差示的に存在する化合物をいう。
本明細書中で用いられる、用語「がん組織特異的な化合物(がん組織特異的化合物)」とは、非がん組織と比較してがん組織中に差示的に存在する化合物をいう。
例えば、いくつかの実施形態では、がん組織特異的化合物は、がん組織に存在するが非がん組織には存在しない、またはがん組織には存在しないが非がん組織には存在している等の定性的ながん組織特異性で規定される化合物であり得る。別の実施形態では、がん組織特的化合物は、非がん組織と比較して異なる濃度(例えば、高濃度または低濃度)でがん組織に存在している等の定量的ながん組織特異性で規定される化合物であり得る。例えば、がん組織特異的化合物は任意の濃度で差示的に存在する。しかし、一般にがん組織特異的化合物は、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%、少なくとも50%、少なくとも55%、少なくとも60%、少なくとも65%、少なくとも70%、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、少なくとも95%、少なくとも100%、少なくとも110%、少なくとも120%、少なくとも130%、少なくとも140%、少なくとも150%、少なくとも2倍、少なくとも5倍、少なくとも10倍、少なくとも50倍、少なくとも100倍、少なくとも103倍、少なくとも104倍、少なくとも105倍、少なくとも106倍、またはそれ以上であって、無限大(すなわち非がん組織に不存在である場合)までの増加する濃度で、存在することが可能であり、あるいは一般に、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%、少なくとも50%、少なくとも60%、少なくとも65%、少なくとも70%、少なくとも75%、少なくとも80%、少なくとも85%、少なくとも90%、少なくとも95%、または少なくとも100%(すなわち、不存在を表す)まで減少する濃度で、存在することが可能である。がん組織特異的化合物は、統計的に有意である濃度(すなわち、ウェルチのt検定またはウィルコクソンの順位和検定のいずれかを用いて決定されるように、p値は0.05未満および/またはq値は0.10未満)で、好ましくは差示的に存在する。がん組織特異的化合物の非限定な一態様としては、以下のようながん組織に含まれるがん細胞、免疫細胞、ストローマ細胞に特有の代謝活性によって産生されたがん組織特異的な代謝産物(がん組織特異的代謝産物;がん細胞特異的代謝産物、がん組織に浸潤している免疫細胞特異的な代謝産物、がんストローマ細胞特異的代謝産物)である化合物が例示され得る。
がん組織特異的代謝産物
用語「代謝」は、生物の組織内で生ずる化学変化のことをいい、「同化」および「異化」が含まれる。同化とは、分子の生合成または蓄積のことをいい、異化は分子の分解のことをいう。「代謝産物」は、物質代謝に起因する中間体または生成物である。「一次代謝産物」とは、細胞または生物の成長もしくは繁殖の過程に直接関わる代謝産物を指し、「二次代謝産物」とはそれらの成長もしくは繁殖の過程には直接関わらず、細胞または生物に共通の生命現象に直接関与しない物質を生合成する代謝の結果生じる抗生物質や色素等の生産物をいう。代謝産物は、「生体高分子」の代謝産物でもあり得るし、「低分子」の代謝産物でもあり得る。「生体高分子」は、一種類以上の反復単位からなる高分子である。生体高分子は、一般に生物系で見出され、生物を組織する細胞およびそれに付着する細胞間マトリックス、組織間マトリックス等の構造物を形成する分子量がおよそ5000以上の分子、特に多糖類(炭水化物等)およびペプチド(この用語はポリペプチドおよびタンパク質を含むようにして用いられる)およびポリヌクレオチド、同様にそれらの類似体、例えばアミノ酸類似体もしくは非アミノ酸基から構成もしくは含むそれらの化合物が挙げられる。
用語「代謝」は、生物の組織内で生ずる化学変化のことをいい、「同化」および「異化」が含まれる。同化とは、分子の生合成または蓄積のことをいい、異化は分子の分解のことをいう。「代謝産物」は、物質代謝に起因する中間体または生成物である。「一次代謝産物」とは、細胞または生物の成長もしくは繁殖の過程に直接関わる代謝産物を指し、「二次代謝産物」とはそれらの成長もしくは繁殖の過程には直接関わらず、細胞または生物に共通の生命現象に直接関与しない物質を生合成する代謝の結果生じる抗生物質や色素等の生産物をいう。代謝産物は、「生体高分子」の代謝産物でもあり得るし、「低分子」の代謝産物でもあり得る。「生体高分子」は、一種類以上の反復単位からなる高分子である。生体高分子は、一般に生物系で見出され、生物を組織する細胞およびそれに付着する細胞間マトリックス、組織間マトリックス等の構造物を形成する分子量がおよそ5000以上の分子、特に多糖類(炭水化物等)およびペプチド(この用語はポリペプチドおよびタンパク質を含むようにして用いられる)およびポリヌクレオチド、同様にそれらの類似体、例えばアミノ酸類似体もしくは非アミノ酸基から構成もしくは含むそれらの化合物が挙げられる。
本明細書に記載される非限定な一態様のがん組織特異的代謝産物として、がん細胞特異的な低分子代謝産物が好適に挙げられる(Eva Gottfried, Katrin Peter and Marina P. Kreutz, From Molecular to Modular Tumor Therapy (2010) 3 (2), 111-132)。さらには、がん組織に浸潤する免疫細胞が高く産生する代謝産物やがん細胞の生存および/または成長をサポートするストローマ細胞(がんストローマ細胞または癌間質線維芽細胞(CAF))が高く産生する代謝産物も含まれる。浸潤する免疫細胞としては、樹状細胞、抑制性樹状細胞、抑制性T細胞、疲弊T細胞(exhausted T cell)、骨髄系由来抑制細胞(myeloma derived suppressor cell、MDSC)等が例示される。また、本発明における代謝産物には、がん組織に存在する細胞(がん細胞、免疫細胞、ストローマ細胞)が、アポトーシスやネクローシス等によって細胞死した際に、細胞内から細胞外に放出される化合物も含まれる。
がん細胞特異的代謝産物を同定するため、トランスクリプトーム・レベルでの解析、(例えば、Dhanasekaranら(Nature (2001) 412, 822-826)、Lapointeら(Proc. Natl. Acad. Sci. U.S.A. (2004) 101, 811-816またはPerouら(Nature (2000) 406, 747-752等が例示される)もしくはプロテオーム・レベルでの解析(例えば、Ahramら(Mol. Carcinog. (2002) 33, 9-15、Hoodら(Mol. Cell. Proteomics (2005) 4, 1741-1753)のほか、代謝学的プロファイリングを中心とする代謝学(メタボロミックス)解析が、適宜使用される。すなわち、被験試料中の代謝産物を同定するために高圧液体クロトグラフィ(HPLC)、核磁気共鳴(NMR)(Brindleら(J. Mol. Recognit. (1997) 10, 182-187)、質量分析法(GatesおよびSweeley(Clin. Chem. (1978) 24, 1663-1673)(GC/MSおよび LC/MS))およびELISA等を単独でおよび/または組み合わせて用いる代謝学的プロファイリングが適宜使用され得る。
これらの研究によって、がん細胞が低い酸素圧条件下で成育することを可能にする代謝産物(例えばブドウ糖または酸素)および生長因子の濃度勾配を変えることによって構成された腫瘍内の異質性が明らかにされた(DangおよびSemenza(Trends Biochem. Sci. (1999) 24, 68-72))。これらの研究においては、腫瘍の悪性度の異なる程度によるエネルギー利用経路の変化を理解するために細胞株モデルも使用されている(Vizanら(Cancer Res. (2005) 65, 5512-5515)。代謝学プラットフォームの技術的構成要素の非限定な一態様として、Lawtonら(Pharmacogenomics (2008) 9, 383)に記載された、試料抽出、分離、検出、分光分析、データ正規化、クラス特異的代謝産物の描写、経路マッピング、確認、および候補代謝産物の機能的特徴付けが例示される。これらの方法によって所望のがん組織におけるがん細胞特異的代謝産物を同定することが可能である。
炎症組織特異的化合物
本明細書中で用いられる、用語「炎症組織特異的な化合物(炎症組織特異的化合物)」とは、非炎症組織と比較して炎症組織中に差示的に存在する化合物をいう。本明細書において、「炎症組織」とは、例えば、以下が例示的に挙げられる。
・関節リウマチや変形性関節症における関節
・気管支喘息やCOPDにおける肺(肺胞)
・炎症性腸疾患やクローン病や潰瘍性大腸炎における消化器官
・肝臓、腎臓、肺における線維化症における線維化組織
・臓器移植における拒絶反応が起こっている組織
・動脈硬化や心不全における血管、心臓(心筋)
・メタボリック症候群における内臓脂肪
・アトピー性皮膚炎その他皮膚炎における皮膚組織
・椎間板ヘルニアや慢性腰痛における脊髄神経
本明細書中で用いられる、用語「炎症組織特異的な化合物(炎症組織特異的化合物)」とは、非炎症組織と比較して炎症組織中に差示的に存在する化合物をいう。本明細書において、「炎症組織」とは、例えば、以下が例示的に挙げられる。
・関節リウマチや変形性関節症における関節
・気管支喘息やCOPDにおける肺(肺胞)
・炎症性腸疾患やクローン病や潰瘍性大腸炎における消化器官
・肝臓、腎臓、肺における線維化症における線維化組織
・臓器移植における拒絶反応が起こっている組織
・動脈硬化や心不全における血管、心臓(心筋)
・メタボリック症候群における内臓脂肪
・アトピー性皮膚炎その他皮膚炎における皮膚組織
・椎間板ヘルニアや慢性腰痛における脊髄神経
炎症組織特異的代謝産物
炎症組織特異的代謝産物とは、炎症性組織に浸潤にしている免疫細胞が高く産生する代謝産物、および、炎症組織において傷害を受けている正常細胞特異的が高く産生する代謝産物である。浸潤する免疫細胞としては、エフェクターT細胞、成熟樹状細胞、好中球、顆粒細胞(肥満細胞)、好塩基球等が例示される。また、本発明における代謝産物には、炎症組織に存在する細胞(免疫細胞、正常細胞)が、アポトーシスやネクローシス等によって細胞死した際に、細胞内から細胞外に放出される化合物も含まれる。
炎症組織特異的代謝産物とは、炎症性組織に浸潤にしている免疫細胞が高く産生する代謝産物、および、炎症組織において傷害を受けている正常細胞特異的が高く産生する代謝産物である。浸潤する免疫細胞としては、エフェクターT細胞、成熟樹状細胞、好中球、顆粒細胞(肥満細胞)、好塩基球等が例示される。また、本発明における代謝産物には、炎症組織に存在する細胞(免疫細胞、正常細胞)が、アポトーシスやネクローシス等によって細胞死した際に、細胞内から細胞外に放出される化合物も含まれる。
本明細書中で用いられる用語「非天然化合物」とは、非天然由来の化学物質及びその代謝物をいう。発明の一態様としては、生体外から生体内に投与された後に、標的組織に集積する性質をもつ非天然由来の化学物質及びその代謝物である。非天然化合物としては、(1)カペシタビン(Capecitabine (Xeloda))とその代謝物である5-FU(fluorouracil)、並びに(2)TH-302とブロモイソホスファミドマスタード(Br-IPM)等が例示される。5-FUはカペシタビン(Capecitabine (Xeloda))の代謝物であり、がん組織特異的な代謝酵素であるcytidine deaminaseと thymidine phosphorylaseによって代謝されることが知られている(Desmoulin F. et al. Drug Metab Dispos. 2002)。またTH-302はがん組織周辺のように、低酸素の条件下で還元されることでBr-IPMに変換されることが知られている(Duan JX, et al. J Med Chem. 2008)。例えば、カペシタビン(capecitabine (xeloda))を投与すると、がん特異的な代謝酵素等によって5-FUに代謝されるため、がん局所における5-FUの濃度が高くなる(Desmoulin F. et al. Drug Metab Dispos. 2002)ため、5-FUをスイッチとする抗体によって、がん局所でのみ選択的に標的抗原に結合することが可能となると考えられる。また、代謝酵素以外でも、がんに特異的な低酸素環境や酸性環境下によって生成される分子をスイッチとして用いることも可能であると考えられる。例えば、TH-302(Duan JX, et al. J Med Chem. 2008)は低酸素条件下においてBr-IPMに代謝されるため、Br-IPMをスイッチとする抗体によって、がん局所でのみ選択的に標的抗原に結合することが可能となると考えられる。例えば、非天然化合物を生体に投与する方法としては、経口投与、点眼投与、経皮投与、経鼻投与、経静脈投与、経肺投与など公知の投与方法が挙げられるが、これらに限定されるものではない。
本開示における低分子化合物の非限定的な態様として、例えば、MTA、SAM、SAH、アデノシン、アデノシン3リン酸(ATP)、アデノシン2リン酸(ADP)、アデノシン1リン酸(AMP)等が例示される。
本開示における低分子化合物の非限定的な態様として、更に以下の化合物が例示される:
(1)乳酸、コハク酸、クエン酸等の解糖系、またはクレブス回路の一次代謝産物
本発明で使用される低分子化合物、またはがん組織特異的化合物、とくにがん細胞特異的代謝産物の非限定な一態様として、乳酸、コハク酸、クエン酸等の周囲に存在する非がん部組織よりもがん組織において高濃度に存在するグルコース代謝の結果生成される一次代謝産物が好適に挙げられる。ピルビン酸キナーゼ、ヘキソキナーゼ、および乳酸脱水素酵素(LDH)等の解糖系(Embden-Myerhof経路)酵素の上方調節(アップレギュレーション)として特徴付けられる解糖系表現型は、Warburg効果として固形腫瘍の特徴であることが従来から知られている。
本発明で使用される低分子化合物、またはがん組織特異的化合物、とくにがん細胞特異的代謝産物の非限定な一態様として、乳酸、コハク酸、クエン酸等の周囲に存在する非がん部組織よりもがん組織において高濃度に存在するグルコース代謝の結果生成される一次代謝産物が好適に挙げられる。ピルビン酸キナーゼ、ヘキソキナーゼ、および乳酸脱水素酵素(LDH)等の解糖系(Embden-Myerhof経路)酵素の上方調節(アップレギュレーション)として特徴付けられる解糖系表現型は、Warburg効果として固形腫瘍の特徴であることが従来から知られている。
すなわち、腫瘍細胞ではM1アイソ型ではなく嫌気条件下での解糖(erobic glycolysis)に必要なM2アイソ型のピルビン酸キナーゼが高発現していることが、生体内における腫瘍細胞の生育に有利に働いていると考えられている(Christofkら(Nature (2008) 452, 230-233)。ピルビン酸キナーゼによって生成されたピルビン酸は、嫌気条件下における乳酸脱水素酵素(LDH)による平衡反応の結果生成される、乳酸によってフィードバック阻害を受ける。当該フィードバック阻害によってミトコンドリアにおける呼吸(クレブス回路)の促進、および細胞増殖抑制が生じるため、LDH、ヘキソキナーゼ、およびグルコーストランスポーター(GLUT)の上方調節が腫瘍細胞の増殖に重要な役割を果たすといわれている(Fantinら(Cancer Cell (2006) 9, 425-434))。グルコースは解糖系で代謝され、その最終代謝産物である乳酸が腫瘍の周囲にプロトンとともに共輸送される結果、腫瘍の周辺組織のpHは酸性条件に変化するといわれている。解糖系の最終産物である乳酸、ミトコンドリアにおける呼吸の促進によって生成されるコハク酸およびクエン酸が、癌組織において蓄積していることが知られている(Teresaら(Mol. Cancer (2009) 8, 41-59))。本発明で使用される低分し化合物、がん組織特異的化合物、とくにがん細胞特異的代謝産物の非限定な一態様として、こうした解糖系の代謝によって生成される一次代謝産物である、乳酸、コハク酸、クエン酸等が好適に挙げられる。また、細胞死により細胞内に高濃度で存在するコハク酸が細胞外に漏出することが知られている(Nature Immunology, (2008) 9, 1261-1269)。そのため、細胞死が頻繁に起こっている癌組織においてコハク酸の濃度が上昇していると考えられる。
(2)アラニン、グルタミン酸、アスパラギン酸等のアミノ酸
上述されたグルコース代謝以外にも、嫌気条件下における生体高分子の生合成に必要な必須アミノ酸および非必須アミノ酸の連続供給が必要な腫瘍細胞ではアミノ酸代謝も変化していることが知られている。グルタミンはその側鎖に二つの窒素を含む窒素運搬体として作用する、生体においてもっとも広範に分布するアミノ酸である。グルタミンの細胞内への取込み速度が上昇している腫瘍細胞はグルタミントラップ(glutamine trap)として機能しているといわれている。こうしたグルタミンの取込みとグルタミン酸および乳酸へ変換される活性の上昇は「グルタミン分解(glutaminolysis)」と呼ばれ、形質転換された(腫瘍)細胞の特徴であると思われている(MazurekおよびEigenbrodt(Anticancer Res. (2003) 23, 1149-1154、ならびにMazurekら(J. Cell. Physiol. (1999) 181, 136-146))。その結果、がん患者は血漿中のグルタミンのレベルの減少の一方でグルタミン酸濃度の増大を示す(Drogeら(Immunobiology (1987) 174, 473-479)。そして、肺癌組織の13C放射標識されたグルコースの代謝研究によって13C標識コハク酸、 13C 標識アラニン、13C 標識グルタミン酸、および13C 標識クエン酸の濃度間で相関が観察された。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたグルタミン分解等によってがん組織において高濃度に蓄積する、アラニン、グルタミン酸、アスパラギン酸等が好適に挙げられる。
上述されたグルコース代謝以外にも、嫌気条件下における生体高分子の生合成に必要な必須アミノ酸および非必須アミノ酸の連続供給が必要な腫瘍細胞ではアミノ酸代謝も変化していることが知られている。グルタミンはその側鎖に二つの窒素を含む窒素運搬体として作用する、生体においてもっとも広範に分布するアミノ酸である。グルタミンの細胞内への取込み速度が上昇している腫瘍細胞はグルタミントラップ(glutamine trap)として機能しているといわれている。こうしたグルタミンの取込みとグルタミン酸および乳酸へ変換される活性の上昇は「グルタミン分解(glutaminolysis)」と呼ばれ、形質転換された(腫瘍)細胞の特徴であると思われている(MazurekおよびEigenbrodt(Anticancer Res. (2003) 23, 1149-1154、ならびにMazurekら(J. Cell. Physiol. (1999) 181, 136-146))。その結果、がん患者は血漿中のグルタミンのレベルの減少の一方でグルタミン酸濃度の増大を示す(Drogeら(Immunobiology (1987) 174, 473-479)。そして、肺癌組織の13C放射標識されたグルコースの代謝研究によって13C標識コハク酸、 13C 標識アラニン、13C 標識グルタミン酸、および13C 標識クエン酸の濃度間で相関が観察された。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたグルタミン分解等によってがん組織において高濃度に蓄積する、アラニン、グルタミン酸、アスパラギン酸等が好適に挙げられる。
(3)キヌレニン(kynurenine)等のアミノ酸の代謝産物
インドールアミン2, 3-ジオキシゲナーゼ(IDO)はメラノーマ、結腸癌、および腎臓癌等の多くのがんで高発現しているトリプトファン代謝酵素であり(Uyttenhoveら(Nat. Med. (2003) 9, 1269-127)、二つのアイソフォームが存在することが知られている(Lobら(CancerImmunol. Immunother. (2009) 58, 153-157))。IDOはトリプトファンのキヌレニン(下記式で表される)への変換を触媒しニコチンアミドヌクレオチド(NAD)の新生経路の最初の酵素である。また、IDOを発現しないグリオーマでは肝臓のトリプトファン2, 3-ジオキシゲナーゼ(TDO)によって、トリプトファンからキヌレニンが生成する(Opitzら(Nature (2011) 478, 7368, 197-203))。またIDOはがん組織に浸潤している樹状細胞にも発現しており、樹状細胞もキヌレニンを産生する(J. Immunol. (2008) 181, 5396-5404)。またIDOはがん組織の 骨髄系由来抑制細胞(MDSC)にも発現しており、MDSCもキヌレニンを産生する(Yuら(J. Immunol. (2013) 190, 3783-3797))。

キヌレニン
インドールアミン2, 3-ジオキシゲナーゼ(IDO)はメラノーマ、結腸癌、および腎臓癌等の多くのがんで高発現しているトリプトファン代謝酵素であり(Uyttenhoveら(Nat. Med. (2003) 9, 1269-127)、二つのアイソフォームが存在することが知られている(Lobら(CancerImmunol. Immunother. (2009) 58, 153-157))。IDOはトリプトファンのキヌレニン(下記式で表される)への変換を触媒しニコチンアミドヌクレオチド(NAD)の新生経路の最初の酵素である。また、IDOを発現しないグリオーマでは肝臓のトリプトファン2, 3-ジオキシゲナーゼ(TDO)によって、トリプトファンからキヌレニンが生成する(Opitzら(Nature (2011) 478, 7368, 197-203))。またIDOはがん組織に浸潤している樹状細胞にも発現しており、樹状細胞もキヌレニンを産生する(J. Immunol. (2008) 181, 5396-5404)。またIDOはがん組織の 骨髄系由来抑制細胞(MDSC)にも発現しており、MDSCもキヌレニンを産生する(Yuら(J. Immunol. (2013) 190, 3783-3797))。
キヌレニンは同種T細胞応答を抑制することが知られており(Frumentoら(J. Exp. Med. (2002) 196, 459-468)、こうした抑制を通じて腫瘍細胞が抗腫瘍免疫応答を潜り抜けるとともに、グリオーマに発現するアリル炭化水素受容体の内因性リガンドとしてキヌレニンが作用するオートクライン増殖機構を通じて、グリオーマ細胞の増殖が促進されるメカニズムが提唱されている(Opitzら(上掲))。キヌレニンはキヌレニダーゼによってアントラニル酸(下記式で表される)に、およびキヌレニン3-ヒドロキシラーゼによって3-ヒドロキシキヌレニン(下記式で表される)に変換される。アントラニル酸、および3-ヒドロキシキヌレニンはともにNADの前駆体となる3-ヒドロキシアントラニル酸に変換される。

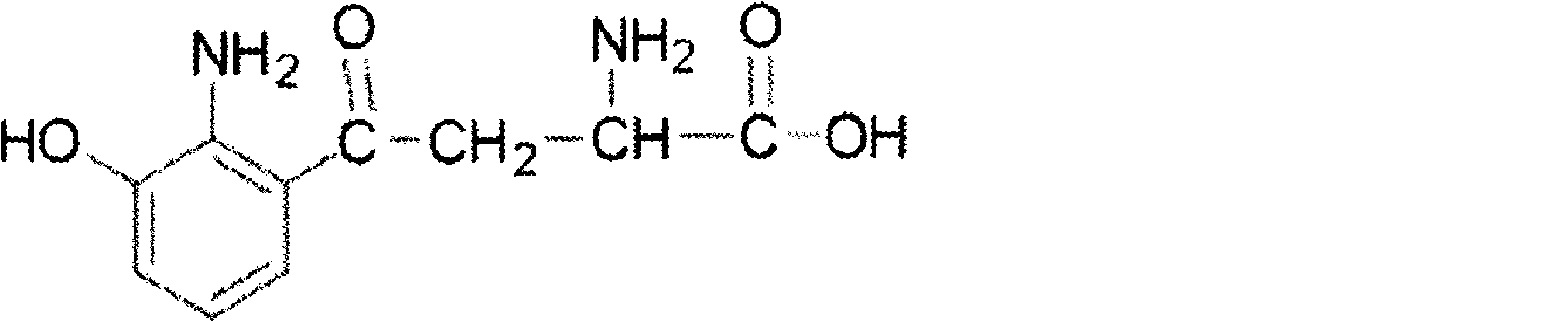
キヌレニンはキヌレニンアミノトランスフェラーゼによってキヌレン酸(下記式で表される)に変換される。本発明で使用される低分子化合物、がん組織特異的化合物、とくにがん細胞特異的代謝産物の非限定な一態様として、こうしたキヌレニン、およびその代謝産物である、アントラニル酸、3-ヒドロキシキヌレニン、およびキヌレン酸等のアミノ酸の代謝産物が好適に挙げられる。

(4)プロスタグランジンE2(Prostaglandin E2)等のアラキドン酸の代謝産物
プロスタグランジンE2(PGE2)(下記式で表される)は、シクロオキシゲナーゼ(COX)-1/2によって合成されるプロスタグランジンおよびトロンボキサンを含むプラストノイドと呼ばれるアラキドン酸の代謝物である(WarnerおよびMitchell(FASEB J. (2004) 18, 790-804))。PGE2結腸癌細胞の増殖を促進し、そのアポトーシスを抑制する(Shengら(Cancer Res. (1998) 58, 362-366))。 多くのがん細胞ではシクロオキシゲナーゼの発現が変化していることが知られている。すなわち、COX-1はほぼすべての組織において構成的に発現しているのに対して、COX-2は腫瘍においてある種の炎症性サイトカインおよびがん遺伝子によって誘導されることが主に見出されている(WarnerおよびMitchell(前掲))。COX-2の過剰発現は乳癌の予後の悪さ(Denkertら(Clin. Breast Cancer (2004) 4, 428-433)、および卵巣癌の急速な疾患の進行(Denkerら(Mod. Pathol. (2006) 19, 1261-1269)と関連性があることも報告されている。またがん組織に浸潤している抑制性T細胞もプロスタグランジンE2を産生している(Curr. Med. Chem. (2011) 18, 5217-5223)。アラキドン酸の代謝物のプロスタグランジン、ロイコトリエン等の低分子化合物ががんのオートクライン、および/またはパラクラインな増殖を制御する刺激因子として作用していることが知られている(Nat. Rev. Cancer (2012) 12 (11) 782-792)。本発明で使用される低分子化合物、がん組織特異的化合物、とくにがん細胞特異的代謝産物やがん組織に浸潤している免疫細胞特異的代謝物の非限定な一態様として、こうしたプロスタグランジンE2等のアラキドン酸の代謝産物が好適に挙げられる。プロスタグランジンE2以外にも、トロンボキサンA2 (TXA2)が大腸癌等のがん組織で産生が亢進しており(J. Lab. Clin. Med. (1993) 122, 518-523)、本発明のアラキドン酸の代謝産物の非限定な一態様として好適に挙げられる。
プロスタグランジンE2(PGE2)(下記式で表される)は、シクロオキシゲナーゼ(COX)-1/2によって合成されるプロスタグランジンおよびトロンボキサンを含むプラストノイドと呼ばれるアラキドン酸の代謝物である(WarnerおよびMitchell(FASEB J. (2004) 18, 790-804))。PGE2結腸癌細胞の増殖を促進し、そのアポトーシスを抑制する(Shengら(Cancer Res. (1998) 58, 362-366))。 多くのがん細胞ではシクロオキシゲナーゼの発現が変化していることが知られている。すなわち、COX-1はほぼすべての組織において構成的に発現しているのに対して、COX-2は腫瘍においてある種の炎症性サイトカインおよびがん遺伝子によって誘導されることが主に見出されている(WarnerおよびMitchell(前掲))。COX-2の過剰発現は乳癌の予後の悪さ(Denkertら(Clin. Breast Cancer (2004) 4, 428-433)、および卵巣癌の急速な疾患の進行(Denkerら(Mod. Pathol. (2006) 19, 1261-1269)と関連性があることも報告されている。またがん組織に浸潤している抑制性T細胞もプロスタグランジンE2を産生している(Curr. Med. Chem. (2011) 18, 5217-5223)。アラキドン酸の代謝物のプロスタグランジン、ロイコトリエン等の低分子化合物ががんのオートクライン、および/またはパラクラインな増殖を制御する刺激因子として作用していることが知られている(Nat. Rev. Cancer (2012) 12 (11) 782-792)。本発明で使用される低分子化合物、がん組織特異的化合物、とくにがん細胞特異的代謝産物やがん組織に浸潤している免疫細胞特異的代謝物の非限定な一態様として、こうしたプロスタグランジンE2等のアラキドン酸の代謝産物が好適に挙げられる。プロスタグランジンE2以外にも、トロンボキサンA2 (TXA2)が大腸癌等のがん組織で産生が亢進しており(J. Lab. Clin. Med. (1993) 122, 518-523)、本発明のアラキドン酸の代謝産物の非限定な一態様として好適に挙げられる。

また、関節リウマチや変形性関節症においてPGE2濃度が高いことが知られている(Eur. J. Clin. Pharmacol. (1994) 46, 3-7.、Clin. Exp. Rheumatol. (1999) 17, 151-160、Am. J. Vet. Res. (2004) 65, 1269-1275.)。本発明で使用される低分子化合物、炎症組織特異的化合物、とくに炎症細胞特異的代謝産物や炎症組織に浸潤する免疫細胞特異的代謝物の非限定な一態様として、こうしたプロスタグランジンE2等のアラキドン酸の代謝産物が好適に挙げられる。
(5)アデノシン、アデノシン3リン酸(ATP)、アデノシン2リン酸(ADP)、アデノシン1リン酸(AMP)等のプリン環構造を有するヌクレオシド
がん細胞が細胞死すると細胞内の大量のATPが細胞外に漏出することが知られている。そのため、がん組織におけるATP濃度は正常組織と比較して著しく高い(PLoS One. (2008) 3, e2599)。複数の型の細胞がATP、ADPおよびAMPの型のアデニンヌクレオチドを遊離する。細胞外-5'-ヌクレオチダーゼ(eco-5'-nucleotidase)(CD73)のような細胞表面の細胞外酵素によって代謝される(RestaおよびThompson(Immunol. Rev. (1998) 161, 95-109)ならびにSadejら(Melanoma Res. (2006) 16, 213-222)。アデノシンは低濃度で細胞外環境に構成的に存在するプリンヌクレオシドであるが、固形癌で見出される低酸素組織では細胞外アデノシン濃度の顕著な増加が報告されている(BlayおよびHoskin(Cancer Res. (1997) 57, 2602-2605)。CD73は腫瘍および免疫細胞の表面に発現しており(Kobieら(J. Immunol. (2006) 177, 6780-6786)、乳癌(Canbolatら(Breast Cancer Res. Treat. (1996) 37, 189-193)、胃癌(Durakら(Cancer Lett. (1994) 84, 199-202)、膵臓癌(FlockeおよびMannherz(Biochim. Biophys. Acta (1991) 1076, 273-281)およびグリオブラストーマ(Bardotら(Br. J. Cancer (1994) 70, 212-218))において活性の上昇が見出されている。がん組織におけるアデノシンの蓄積は、細胞質の5'-ヌクレオチダーゼによるAMPの脱リン酸によって細胞内アデノシン生成が増加することに起因している可能性が提唱されている(HeadrickおよびWillis(Biochem. J. (1989) 261, 541-550)。さらにがん組織に浸潤している抑制性T細胞等もATP分解酵素を発現しており、アデノシンを産生している(Proc. Natl. Acad. Sci. (2006) 103 (35), 13132-13137、Curr. Med. Chem. (2011) 18, 5217-5223)。産生されたアデノシンは、A2Aレセプター等のアデノシンレセプターを介してがん組織を免疫抑制的な環境にしていると考えられている(Curr. Med. Chem. (2011),18,5217-23)。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によってがん組織において高濃度に蓄積する、ATP、ADP、AMP、またはアデノシン等が好適に挙げられる。さらにアデノシンは、adenosine deaminaseによってイノシンに分解されるため、イノシンが高濃度に蓄積する。
がん細胞が細胞死すると細胞内の大量のATPが細胞外に漏出することが知られている。そのため、がん組織におけるATP濃度は正常組織と比較して著しく高い(PLoS One. (2008) 3, e2599)。複数の型の細胞がATP、ADPおよびAMPの型のアデニンヌクレオチドを遊離する。細胞外-5'-ヌクレオチダーゼ(eco-5'-nucleotidase)(CD73)のような細胞表面の細胞外酵素によって代謝される(RestaおよびThompson(Immunol. Rev. (1998) 161, 95-109)ならびにSadejら(Melanoma Res. (2006) 16, 213-222)。アデノシンは低濃度で細胞外環境に構成的に存在するプリンヌクレオシドであるが、固形癌で見出される低酸素組織では細胞外アデノシン濃度の顕著な増加が報告されている(BlayおよびHoskin(Cancer Res. (1997) 57, 2602-2605)。CD73は腫瘍および免疫細胞の表面に発現しており(Kobieら(J. Immunol. (2006) 177, 6780-6786)、乳癌(Canbolatら(Breast Cancer Res. Treat. (1996) 37, 189-193)、胃癌(Durakら(Cancer Lett. (1994) 84, 199-202)、膵臓癌(FlockeおよびMannherz(Biochim. Biophys. Acta (1991) 1076, 273-281)およびグリオブラストーマ(Bardotら(Br. J. Cancer (1994) 70, 212-218))において活性の上昇が見出されている。がん組織におけるアデノシンの蓄積は、細胞質の5'-ヌクレオチダーゼによるAMPの脱リン酸によって細胞内アデノシン生成が増加することに起因している可能性が提唱されている(HeadrickおよびWillis(Biochem. J. (1989) 261, 541-550)。さらにがん組織に浸潤している抑制性T細胞等もATP分解酵素を発現しており、アデノシンを産生している(Proc. Natl. Acad. Sci. (2006) 103 (35), 13132-13137、Curr. Med. Chem. (2011) 18, 5217-5223)。産生されたアデノシンは、A2Aレセプター等のアデノシンレセプターを介してがん組織を免疫抑制的な環境にしていると考えられている(Curr. Med. Chem. (2011),18,5217-23)。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によってがん組織において高濃度に蓄積する、ATP、ADP、AMP、またはアデノシン等が好適に挙げられる。さらにアデノシンは、adenosine deaminaseによってイノシンに分解されるため、イノシンが高濃度に蓄積する。
また、気管支喘息に起因する炎症が起こっている肺胞においてATP濃度が高いことが知られている(Nat. Med. (2007) 13, 913-919)。また、COPDに起因する炎症が起こっている肺胞においてATP濃度が高いこともまた知られている(Am. J. Respir. Crit. Care Med. (2010) 181, 928-934)。また、関節リウマチ患者の関節液中でアデノシン濃度が高いことが観察されている(Journal of Pharmaceutical and Biomedical Analysis (2004) 36 877-882)。さらにGVHDにより拒絶反応が起こっている組織においてATP濃度が高いことが知られている(Nat. Med. (2010) 16, 1434-1438)。また、肺、肝臓、腎臓における線維化組織においてアデノシン濃度が亢進していることも知られている(FASEB J. (2008) 22, 2263-2272、J. Immunol. (2006) 176, 4449-4458、J. Am. Soc. Nephrol. (2011) 22 (5), 890-901、PLoS ONE J. (2010) 5 (2), e9242)。また肺線維症患者の線維化組織においてATP濃度が上昇していることが観察されている(Am. J. Respir. Crit. Care Med. (2010) 182, 774-783)。本発明で使用される低分子化合物、炎症性組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によって炎症組織において高濃度に蓄積する、ATP、ADP、AMP、またはアデノシン等が好適に挙げられる。さらにアデノシンは、adenosine deaminaseによってイノシンに分解されるため、イノシンが高濃度に蓄積する。
(6)尿酸
尿酸は生体内におけるプリンヌクレオシドの代謝経路の産物であり、血液または間質腔等の細胞外に遊離される。また、近年では、がん組織等の病変部位に存在する死細胞から遊離されることが明らかとなっている(Nat. Med. (2007) 13, 851-856)。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によってがん組織において高濃度に蓄積する尿酸も好適に挙げられる。
尿酸は生体内におけるプリンヌクレオシドの代謝経路の産物であり、血液または間質腔等の細胞外に遊離される。また、近年では、がん組織等の病変部位に存在する死細胞から遊離されることが明らかとなっている(Nat. Med. (2007) 13, 851-856)。本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によってがん組織において高濃度に蓄積する尿酸も好適に挙げられる。
また、近年では、壊死(necrosis)を進行する細胞から遊離される尿酸が炎症性応答を促進することが明らかとなっている(J. Clin. Invest. (2010) 120 (6), 1939-1949)。本発明で使用される低分子化合物、炎症組織特異的化合物の非限定な一態様として、こうしたATP等のプリンヌクレオチドの代謝によって炎症性組織において高濃度に蓄積する尿酸も好適に挙げられる。
(7)1-メチルニコチンアミド
複数のヒトがん組織において酵素ニコチンアミドN-メチルトランスフェラーゼが高発現していることが知られている。本酵素がニコチンアミドから安定的な代謝物である1-メチルニコチンアミドを産生する際、メチル供与体となるS-アデノシルメチオニン(SAM)のメチル基を消費するために、がん細胞におけるSAM濃度の減少に伴ったDNAのメチル化能を損ねる機構を通じて、ニコチンアミドN-メチルトランスフェラーゼの高発現が腫瘍化(tumorigenesis)に寄与していることが提唱されている(Ulanovskayaら(Nat. Chem. Biol. (2013) 9 (5) 300-306))。本酵素の安定的な代謝産物である1-メチルニコチンアミドは、がん細胞の細胞外に分泌することが知られており(Yamadaら(J. Nutr. Sci. Vitaminol. (2010) 56, 83-86))、本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたニコチンアミドの代謝によってがん組織において高濃度に蓄積する1-メチルニコチンアミド等も好適に挙げられる。
本開示における低分子化合物は抗原結合分子と相互作用し得る。低分子化合物と相互作用し得る抗原結合分子中のアミノ酸残基は、抗原結合分子中の抗原結合ドメインに存在しても良いし、抗原結合ドメイン以外の部位に存在しても良い。抗原結合分子中の低分子化合物と相互作用する部位として、抗原結合ドメインが例示されるがこれに限定されるものではない。
複数のヒトがん組織において酵素ニコチンアミドN-メチルトランスフェラーゼが高発現していることが知られている。本酵素がニコチンアミドから安定的な代謝物である1-メチルニコチンアミドを産生する際、メチル供与体となるS-アデノシルメチオニン(SAM)のメチル基を消費するために、がん細胞におけるSAM濃度の減少に伴ったDNAのメチル化能を損ねる機構を通じて、ニコチンアミドN-メチルトランスフェラーゼの高発現が腫瘍化(tumorigenesis)に寄与していることが提唱されている(Ulanovskayaら(Nat. Chem. Biol. (2013) 9 (5) 300-306))。本酵素の安定的な代謝産物である1-メチルニコチンアミドは、がん細胞の細胞外に分泌することが知られており(Yamadaら(J. Nutr. Sci. Vitaminol. (2010) 56, 83-86))、本発明で使用される低分子化合物、がん組織特異的化合物の非限定な一態様として、こうしたニコチンアミドの代謝によってがん組織において高濃度に蓄積する1-メチルニコチンアミド等も好適に挙げられる。
本開示における低分子化合物は抗原結合分子と相互作用し得る。低分子化合物と相互作用し得る抗原結合分子中のアミノ酸残基は、抗原結合分子中の抗原結合ドメインに存在しても良いし、抗原結合ドメイン以外の部位に存在しても良い。抗原結合分子中の低分子化合物と相互作用する部位として、抗原結合ドメインが例示されるがこれに限定されるものではない。
抗原に特異的に結合する抗原結合ドメイン
本明細書において、「抗原に特異的に結合する抗原結合ドメイン」は、抗原結合ドメインが、ある抗原中に含まれる複数のエピトープのうち特定のエピトープに対して特異的である場合に用いられる。また、抗原結合ドメインが結合するエピトープが複数の異なる抗原に含まれる場合には、当該抗原結合ドメインを有する抗原結合分子は当該エピトープを含む様々な抗原と結合することができる。ここで、実質的に結合しないとは上記結合活性の項で記載される方法に準じて決定され、前記相手方以外の分子に対する特異的結合分子の結合活性が、前記相手方の分子に対するの結合活性の80%以下、通常50%以下、好ましくは30%以下、特に好ましくは15%以下の結合活性を示すことをいう。抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインの場合、抗原に対する当該抗原結合ドメインの結合は、当該抗原結合ドメインの抗原に対する結合活性が高い条件(例えば、MTAの特定濃度下またはMTA非存在下、MTA以外の低分子化合物の特定濃度下またはMTA以外の低分子化合物非存在下)にて測定される。
本明細書において、「抗原に特異的に結合する抗原結合ドメイン」は、抗原結合ドメインが、ある抗原中に含まれる複数のエピトープのうち特定のエピトープに対して特異的である場合に用いられる。また、抗原結合ドメインが結合するエピトープが複数の異なる抗原に含まれる場合には、当該抗原結合ドメインを有する抗原結合分子は当該エピトープを含む様々な抗原と結合することができる。ここで、実質的に結合しないとは上記結合活性の項で記載される方法に準じて決定され、前記相手方以外の分子に対する特異的結合分子の結合活性が、前記相手方の分子に対するの結合活性の80%以下、通常50%以下、好ましくは30%以下、特に好ましくは15%以下の結合活性を示すことをいう。抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインの場合、抗原に対する当該抗原結合ドメインの結合は、当該抗原結合ドメインの抗原に対する結合活性が高い条件(例えば、MTAの特定濃度下またはMTA非存在下、MTA以外の低分子化合物の特定濃度下またはMTA以外の低分子化合物非存在下)にて測定される。
抗原結合活性及び抗原結合活性の確認方法
用語「結合活性(binding activity)」は、分子(例えば、抗体)の1個またはそれ以上の結合部位と、分子の結合パートナー(例えば、抗原)との間の、非共有結合的な相互作用の合計の強度のことをいう。ここで、「結合活性(binding activity)」は、ある結合対のメンバー(例えば、抗体と抗原)の間の1:1相互作用に厳密に限定されない。例えば、結合対のメンバーが1価での1:1相互作用を反映する場合、結合活性は固有の結合アフィニティ(「アフィニティ」)のことをいう。結合対のメンバーが、1価での結合および多価での結合の両方が可能である場合、結合活性は、これらの結合力の総和となる。分子XのそのパートナーYに対する結合活性は、一般的に、解離定数 (KD) または「単位リガンド量当たりのアナライト結合量」により表すことができる。結合活性は、本明細書に記載のものを含む、当該技術分野において知られた通常の方法によって測定され得る。標的組織特異的な化合物の濃度以外の条件については当業者が適宜決定することが可能である。
結合活性を測定するための具体的な実例となるおよび例示的な態様については、下で述べる。
用語「結合活性(binding activity)」は、分子(例えば、抗体)の1個またはそれ以上の結合部位と、分子の結合パートナー(例えば、抗原)との間の、非共有結合的な相互作用の合計の強度のことをいう。ここで、「結合活性(binding activity)」は、ある結合対のメンバー(例えば、抗体と抗原)の間の1:1相互作用に厳密に限定されない。例えば、結合対のメンバーが1価での1:1相互作用を反映する場合、結合活性は固有の結合アフィニティ(「アフィニティ」)のことをいう。結合対のメンバーが、1価での結合および多価での結合の両方が可能である場合、結合活性は、これらの結合力の総和となる。分子XのそのパートナーYに対する結合活性は、一般的に、解離定数 (KD) または「単位リガンド量当たりのアナライト結合量」により表すことができる。結合活性は、本明細書に記載のものを含む、当該技術分野において知られた通常の方法によって測定され得る。標的組織特異的な化合物の濃度以外の条件については当業者が適宜決定することが可能である。
結合活性を測定するための具体的な実例となるおよび例示的な態様については、下で述べる。
抗原結合分子の結合活性
特定の態様において、本明細書で提供される抗原結合分子は抗体であり、抗体の結合活性(binding activity)は、≦1μM、≦100nM、≦10nM、≦1nM、≦0.1nM、≦0.01nMまたは≦0.001nM(例えば、10-8M以下、例えば10-8M~10-13M、例えば10-9M~10-13M)の解離定数 (KD) である。
特定の態様において、本明細書で提供される抗原結合分子は抗体であり、抗体の結合活性(binding activity)は、≦1μM、≦100nM、≦10nM、≦1nM、≦0.1nM、≦0.01nMまたは≦0.001nM(例えば、10-8M以下、例えば10-8M~10-13M、例えば10-9M~10-13M)の解離定数 (KD) である。
一態様において、抗体の結合活性(binding activity)は表面プラズモン共鳴分析法を測定原理とする例えばBIACORE(商標登録)T200またはBIACORE(商標登録)4000(GE Healthcare, Uppsala, Sweden)を用いたリガンド捕捉法が用いられる。機器操作にはBIACORE(商標登録)Control Softwareが用いられる。一態様においてアミンカップリングキット(GE Healthcare, Uppsala, Sweden)を供給元の指示にしたがって使用し、カルボキシメチルデキストランをコーティングしたセンサーチップ(GE Healthcare, Uppsala, Sweden)にリガンド捕捉用分子、たとえば抗タグ抗体、抗IgG抗体、プロテインAなど、を固相化する。リガンド捕捉分子は適切なpHの10 mM酢酸ナトリウム溶液を用いて希釈され、適切な流速および注入時間で注入される。結合活性測定は0.05%ポリソルベート20(その他の名称としてTween(商標登録)-20)含有緩衝液を測定用緩衝液として使用し、流速は10- 30 μL/分、測定温度は好ましくは25℃や37℃で測定される。リガンド捕捉用分子に抗体をリガンドとして捕捉させて測定を実施する場合は、抗体を注入して目的量を捕捉させたのち、測定用緩衝液を用いて調製された抗原およびまたはFc受容体の段階希釈物(アナライト)が注入される。リガンド捕捉用分子に抗原およびまたはFc受容体をリガンドとして捕捉させて測定を実施する場合は、抗原およびまたはFc受容体を注入して目的量を捕捉させたのち、測定用緩衝液を用いて調製された抗体の段階希釈物(アナライト)が注入される。
一態様において、測定結果はBIACORE(登録商標)Evaluation Softwareを用いて解析される。速度論的パラメータ(kinetics parameter)算出は1:1 Bindingのモデルを用いて、結合および解離のセンサーグラムを同時にフィッティングすることによって実施され、結合速度 (konもしくはka) 、解離速度 (koffもしくはkd) 、平衡解離定数 (KD)が計算され得る。結合活性が弱い、特に解離が早く速度論的パラメータ算出が困難な場合はSteady stateモデルを用いて平衡解離定数 (KD)を計算しても良い。結合活性の他のパラメータとしては、特定の濃度のアナライトの結合量(RU)をリガンドの捕捉量(RU)で除して「単位リガンド量当たりのアナライト結合量」も算出され得る。
一態様において、抗体の結合活性はバイオレイヤー干渉(Bio-Layer Interferometry, BLI)法を測定原理とする例えばOctet RED96eシステムまたはOctet RED 384システム(Pall ForteBio)等により測定することができる。供給元の指示に従い使用することで抗原抗体反応の定性的結合特性解析やカイネティクス解析を行うことができる。具体的測定手法の非限定な一態様として、Protein A (ProA)バイオセンサー(Pall ForteBio)に抗体を固相化した後、抗原をアナライトとして相互作用させることで抗体抗原間の結合量の変化を測定することができる。例えば、MTA存在下で抗原に結合する抗体と抗原との結合量を測定する場合、結合相としてMTAが終濃度0, 10, 100 μMの濃度で添加された20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4で希釈された3000nMのアナライトを含むバッファー中で抗体とアナライトの結合反応を測定することが可能であり、解離相としてアナライトを含まない結合相と同様のバッファーを用いることで抗体とアナライトの解離反応を測定することが可能である。さらに、解離相として結合相と同濃度のアナライトを含み、MTAを含まないバッファーを用いることで、MTA存在下における抗体と抗原の結合がMTA非存在下において可逆的に解離することを経時的に観察することが可能である。
抗原結合活性の値として、抗原が可溶型分子の場合はkd(解離速度定数)を用いることが可能であり、抗原が膜型分子の場合は見かけのkd(Apparent dissociation rate constant:見かけの解離速度定数)を用いることが可能である。kd(解離速度定数)、および、見かけのkd(見かけの解離速度定数)は、当業者公知の方法で測定することが可能であり、例えばBiacore(GE healthcare)、フローサイトメーター等を用いることが可能である。
低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む被験抗原結合分子の抗原に対する結合活性を測定するとき、被験抗原結合分子と抗原の相互作用を低分子化合物の特定濃度下もしくは非存在下で行うことが出来る。
特定の低分子化合物が非存在の条件において抗原結合活性を示さない抗原結合分子が、当該低分子化合物存在下において当該抗原に対する結合活性を示す場合は、当該低分子化合物存在下において結合活性を評価することができる。同様に、特定の低分子化合物存在下において抗原に対する結合活性を示さず、当該低分子化合物の非存在下で当該抗原に対する結合活性を示す場合は、当該低分子化合物の非存在下において結合活性を評価することができる。特定の低分子化合物が低濃度で存在する条件における抗原結合活性が当該低分子化合物が高濃度で存在する条件における抗原結合活性より高い場合、当該低分子化合物が高濃度で存在する条件において結合活性を評価することができる。同様に、特定の低分子化合物が高濃度で存在する条件における抗原結合活性が当該低分子化合物が低濃度で存在する条件における抗原結合活性より低い場合、当該低分子化合物が低濃度で存在する条件において結合活性を評価することができる。抗原と抗原結合分子の結合活性に影響を与えうる条件は、低分子化合物の存在下/非存在下やその低分子化合物の濃度に限定されず、イオン濃度、イオン組成または温度などが例示されるがこれらに限定されるものではない。さらに前記の異なる複数の因子が組み合わされた条件下で結合活性を評価することも当然あり得る。
特定の低分子化合物が非存在の条件において抗原結合活性を示さない抗原結合分子が、当該低分子化合物存在下において当該抗原に対する結合活性を示す場合は、当該低分子化合物存在下において結合活性を評価することができる。同様に、特定の低分子化合物存在下において抗原に対する結合活性を示さず、当該低分子化合物の非存在下で当該抗原に対する結合活性を示す場合は、当該低分子化合物の非存在下において結合活性を評価することができる。特定の低分子化合物が低濃度で存在する条件における抗原結合活性が当該低分子化合物が高濃度で存在する条件における抗原結合活性より高い場合、当該低分子化合物が高濃度で存在する条件において結合活性を評価することができる。同様に、特定の低分子化合物が高濃度で存在する条件における抗原結合活性が当該低分子化合物が低濃度で存在する条件における抗原結合活性より低い場合、当該低分子化合物が低濃度で存在する条件において結合活性を評価することができる。抗原と抗原結合分子の結合活性に影響を与えうる条件は、低分子化合物の存在下/非存在下やその低分子化合物の濃度に限定されず、イオン濃度、イオン組成または温度などが例示されるがこれらに限定されるものではない。さらに前記の異なる複数の因子が組み合わされた条件下で結合活性を評価することも当然あり得る。
エピトープ
抗原中に存在する抗原決定基を意味するエピトープは、本明細書において開示される抗原結合分子中の抗原結合ドメインが結合する抗原上の部位を意味する。本開示における抗原結合分子が結合する抗原上の部位は、当該抗原結合分子の結合の有無を評価することによって定義されうる。
抗原中に存在する抗原決定基を意味するエピトープは、本明細書において開示される抗原結合分子中の抗原結合ドメインが結合する抗原上の部位を意味する。本開示における抗原結合分子が結合する抗原上の部位は、当該抗原結合分子の結合の有無を評価することによって定義されうる。
エピトープは、その構造によって定義され得る。また、当該エピトープを認識する抗原結合分子中の抗原に対する結合活性によっても当該エピトープが定義され得る。抗原がペプチド又はポリペプチドである場合には、エピトープを構成するアミノ酸残基によってエピトープを特定することも可能である。また、エピトープが糖鎖である場合には、特定の糖鎖構造によってエピトープを特定することも可能である。
直線状エピトープは、アミノ酸一次配列が認識されたエピトープを含むエピトープである。直線状エピトープは、典型的には、少なくとも3つ、および最も普通には少なくとも5つ、例えば約8ないし約10個、6ないし20個のアミノ酸が固有の配列において含まれる。
立体構造エピトープは、直線状エピトープとは対照的に、エピトープを含むアミノ酸の一次配列が、認識されたエピトープの単一の規定成分ではないエピトープ(例えば、アミノ酸の一次配列が、必ずしもエピトープを規定する抗体により認識されないエピトープ)である。立体構造エピトープは、直線状エピトープに対して増大した数のアミノ酸を包含するかもしれない。立体構造エピトープの認識に関して、抗体は、ペプチドまたはタンパク質の三次元構造を認識する。例えば、タンパク質分子が折り畳まれて三次元構造を形成する場合には、立体構造エピトープを形成するあるアミノ酸および/またはポリペプチド主鎖は、並列となり、抗体がエピトープを認識するのを可能にする。エピトープの立体構造を決定する方法には、例えばX線結晶学、二次元核磁気共鳴分光学並びに部位特異的なスピン標識および電磁常磁性共鳴分光学が含まれるが、これらには限定されない。例えば、Epitope Mapping Protocols in Methods in Molecular Biology (1996)、第66巻、Morris(編)を参照。
エピトープに結合する抗原結合ドメインの構造はパラトープと呼ばれる。エピトープとパラトープの間に作用する、水素結合、静電気力、ファンデルワールス力、疎水結合等によりエピトープとパラトープは安定して結合する。このエピトープとパラトープの間の結合力はアフィニティー(affinity)と呼ばれる。複数の抗原と複数の抗原結合分子が結合するときの結合力の総和はアビディティ(avidity)と呼ばれる。複数の抗原結合ドメインを含む(すなわち多価の)抗体等が複数のエピトープに結合する際には、結合力(affinity)が相乗的に働くため、アビディティはアフィニティーよりも高くなる。
以下にIL-6Rに対する抗原結合ドメインを含む被験抗原結合分子によるエピトープへの結合の確認方法が例示されるが、IL-6R以外の抗原に対する抗原結合ドメインを含む被験抗原結合分子によるエピトープへの結合の確認方法も以下の例示に準じて適宜実施され得る。
例えば、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子が、IL-6R分子中に存在する線状エピトープを認識することは、たとえば次のようにして確認することができる。上記の目的のためにIL-6Rの細胞外ドメインを構成するアミノ酸配列からなる線状のペプチドが合成される。当該ペプチドは、化学的に合成され得る。あるいは、IL-6RのcDNA中の、細胞外ドメインに相当するアミノ酸配列をコードする領域を利用して、遺伝子工学的手法により得られる。次に、細胞外ドメインを構成するアミノ酸配列からなる線状ペプチドと、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子との結合活性が評価される。たとえば、固定化された線状ペプチドを抗原とするELISAによって、当該ペプチドに対する当該抗原結合分子の結合活性が評価され得る。あるいは、IL-6R発現細胞に対する当該抗原結合分子の結合における、線状ペプチドによる阻害のレベルに基づいて、線状ペプチドに対する結合活性が明らかにされ得る。これらの試験によって、線状ペプチドに対する当該抗原結合分子の結合活性が明らかにされ得る。
また、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子が立体構造エピトープを認識することは、次のようにして確認され得る。上記の目的のために、IL-6Rを発現する細胞が調製される。IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子がIL-6R発現細胞に接触した際に当該細胞に強く結合する一方で、当該抗原結合分子が固定化されたIL-6Rの細胞外ドメインを構成するアミノ酸配列からなる線状ペプチドに対して実質的に結合しないとき等が挙げられる。ここで、実質的に結合しないとは、ヒトIL-6R発現細胞に対する結合活性の80%以下、通常50%以下、好ましくは30%以下、特に好ましくは15%以下の結合活性をいう。
IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子のIL-6R発現細胞に対する結合活性を測定する方法としては、例えば、Antibodies A Laboratory Manual記載の方法(Ed Harlow, David Lane, Cold Spring Harbor Laboratory (1988) 359-420)が挙げられる。即ちIL-6R発現細胞を抗原とするELISAやFACS(fluorescence activated cell sorting)の原理によって評価され得る。
ELISAフォーマットにおいて、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子のIL-6R発現細胞に対する結合活性は、酵素反応によって生成するシグナルレベルを比較することによって定量的に評価される。すなわち、IL-6R発現細胞を固定化したELISAプレートに被験ポリペプチド会合体を加え、細胞に結合した被験抗原結合分子が、被験抗原結合分子を認識する酵素標識抗体を利用して検出される。あるいはFACSにおいては、被験抗原結合分子の希釈系列を作成し、IL-6R発現細胞に対する抗体結合力価(titer)を決定することにより、IL-6R発現細胞に対する被験抗原結合分子の結合活性が比較され得る。
緩衝液等に懸濁した細胞表面上に発現している抗原に対する被験抗原結合分子の結合は、フローサイトメーターによって検出することができる。フローサイトメーターとしては、例えば、次のような装置が知られている。
FACSCantoTM II
FACSAriaTM
FACSArrayTM
FACSVantageTM SE
FACSCaliburTM (いずれもBD Biosciences社の商品名)
EPICS ALTRA HyPerSort
Cytomics FC 500
EPICS XL-MCL ADC EPICS XL ADC
Cell Lab Quanta / Cell Lab Quanta SC(いずれもBeckman Coulter社の商品名)
FACSCantoTM II
FACSAriaTM
FACSArrayTM
FACSVantageTM SE
FACSCaliburTM (いずれもBD Biosciences社の商品名)
EPICS ALTRA HyPerSort
Cytomics FC 500
EPICS XL-MCL ADC EPICS XL ADC
Cell Lab Quanta / Cell Lab Quanta SC(いずれもBeckman Coulter社の商品名)
例えば、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子の抗原に対する結合活性の好適な測定方法の一例として、次の方法が挙げられる。まず、IL-6Rを発現する細胞と反応させた被験抗原結合分子を認識するFITC標識した二次抗体で染色する。被験抗原結合分子を適宜好適な緩衝液によって希釈することによって、当該抗原結合分子が所望の濃度に調製して用いられる。例えば、10μg/mlから10 ng/mlまでの間のいずれかの濃度で使用され得る。次に、FACSCalibur(BD社)により蛍光強度と細胞数が測定される。当該細胞に対する抗体の結合量は、CELL QUEST Software(BD社)を用いて解析することにより得られた蛍光強度、すなわちGeometric Meanの値に反映される。すなわち、当該Geometric Meanの値を得ることにより、被験抗原結合分子の結合量によって表される被験抗原結合分子の結合活性が測定され得る。
同じエピトープに結合する抗体
IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子が、ある抗原結合分子とエピトープを共有することは、両者の同じエピトープに対する競合によって確認され得る。抗原結合分子間の競合は、交叉ブロッキングアッセイなどによって検出される。例えば競合ELISAアッセイは、好ましい交叉ブロッキングアッセイである。
IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子が、ある抗原結合分子とエピトープを共有することは、両者の同じエピトープに対する競合によって確認され得る。抗原結合分子間の競合は、交叉ブロッキングアッセイなどによって検出される。例えば競合ELISAアッセイは、好ましい交叉ブロッキングアッセイである。
具体的には、交叉ブロッキングアッセイにおいては、マイクロタイタープレートのウェル上にコートしたIL-6Rタンパク質が、候補となる競合抗原結合分子の存在下、または非存在下でプレインキュベートされた後に、被験抗原結合分子が添加される。ウェル中のIL-6Rタンパク質に結合した被験抗原結合分子の量は、同じエピトープへの結合に対して競合する候補となる競合抗原結合分子の結合能に間接的に相関している。すなわち同一エピトープに対する競合抗原結合分子の親和性が大きくなればなる程、被験抗原結合分子のIL-6Rタンパク質をコートしたウェルへの結合活性は低下する。
IL-6Rタンパク質を介してウェルに結合した被験抗原結合分子の量は、予め抗原結合分子を標識しておくことによって、容易に測定され得る。たとえば、ビオチン標識された抗原結合分子は、アビジンペルオキシダーゼコンジュゲートと適切な基質を使用することにより測定される。ペルオキシダーゼなどの酵素標識を利用した交叉ブロッキングアッセイは、特に競合ELISAアッセイといわれる。抗原結合分子は、検出あるいは測定が可能な他の標識物質で標識され得る。具体的には、放射標識あるいは蛍光標識などが公知である。
候補の競合抗原結合分子会合体の非存在下で実施されるコントロール試験において得られる結合活性と比較して、競合抗原結合分子が、IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子の結合を少なくとも20%、好ましくは少なくとも20-50%、さらに好ましくは少なくとも50%ブロックできるならば、当該被験抗原結合分子は競合抗原結合分子と実質的に同じエピトープに結合するか、又は同じエピトープへの結合に対して競合する抗原結合分子である。
IL-6Rに対する抗原結合ドメインを含む被験抗原結合分子が結合するエピトープの構造が同定されている場合には、被験抗原結合分子と対照抗原結合分子とがエピトープを共有することは、当該エピトープを構成するペプチドにアミノ酸変異を導入したペプチドに対する両者の抗原結合分子の結合活性を比較することによって評価され得る。
こうした結合活性を測定する方法としては、例えば、前記のELISAフォーマットにおいて変異を導入した線状のペプチドに対する被験抗原結合分子及び対照抗原結合分子の結合活性を比較することによって測定され得る。ELISA以外の方法としては、カラムに結合した当該変異ペプチドに対する結合活性を、当該カラムに被検抗原結合分子と対照抗原結合分子を流下させた後に溶出液中に溶出される抗原結合分子を定量することによっても測定され得る。変異ペプチドを例えばGSTとの融合ペプチドとしてカラムに吸着させる方法は公知である。
また、同定されたエピトープが立体エピトープの場合には、被験抗原結合分子と対照抗原結合分子とがエピトープを共有することは、次の方法で評価され得る。まず、IL-6Rを発現する細胞とエピトープに変異が導入されたIL-6Rを発現する細胞が調製される。これらの細胞がPBS等の適切な緩衝液に懸濁された細胞懸濁液に対して被験抗原結合分子と対照抗原結合分子が添加される。次いで、適宜緩衝液で洗浄された細胞懸濁液に対して、被験抗原結合分子と対照抗原結合分子を認識することができるFITC標識された抗体が添加される。標識抗体によって染色された細胞の蛍光強度と細胞数がFACSCalibur(BD社)によって測定される。被験抗原結合分子と対照抗原結合分子の濃度は好適な緩衝液によって適宜希釈することによって所望の濃度に調製して用いられる。例えば、10μg/mlから10 ng/mlまでの間のいずれかの濃度で使用される。当該細胞に対する標識抗体の結合量は、CELL QUEST Software(BD社)を用いて解析することにより得られた蛍光強度、すなわちGeometric Meanの値に反映される。すなわち、当該Geometric Meanの値を得ることにより、標識抗体の結合量によって表される被験抗原結合分子と対照抗原結合分子の結合活性を測定することができる。
本方法において、例えば「変異IL-6R発現細胞に実質的に結合しない」ことは、以下の方法によって判断することができる。まず、変異IL-6Rを発現する細胞に対して結合した被験抗原結合分子と対照抗原結合分子が、標識抗体で染色される。次いで細胞の蛍光強度が検出される。蛍光検出にフローサイトメトリーとしてFACSCaliburを用いた場合、得られた蛍光強度はCELL QUEST Softwareを用いて解析され得る。ポリペプチド会合体存在下および非存在下でのGeometric Meanの値から、この比較値(ΔGeo-Mean)を下記の式1に基づいて算出することにより、抗原結合分子の結合による蛍光強度の増加割合を求めることができる。
(式1)
ΔGeo-Mean=Geo-Mean(ポリペプチド会合体存在下)/Geo-Mean(ポリペプチド会合体非存在下)
ΔGeo-Mean=Geo-Mean(ポリペプチド会合体存在下)/Geo-Mean(ポリペプチド会合体非存在下)
解析によって得られる被験抗原結合分子の変異IL-6R発現細胞に対する結合量が反映されたGeometric Mean比較値(変異IL-6R分子ΔGeo-Mean値)を、被験抗原結合分子のIL-6R発現細胞に対する結合量が反映されたΔGeo-Mean比較値と比較する。この場合において、変異IL-6R発現細胞及びIL-6R発現細胞に対するΔGeo-Mean比較値を求める際に使用する被験抗原結合分子の濃度は互いに同一又は実質的に同一の濃度で調製されることが特に好ましい。予めIL-6R中のエピトープを認識していることが確認された抗原結合分子が、対照抗原結合分子として利用される。
被験抗原結合分子の変異IL-6R発現細胞に対するΔGeo-Mean比較値が、被験抗原結合分子のIL-6R発現細胞に対するΔGeo-Mean比較値の、少なくとも80%、好ましくは50%、更に好ましくは30%、特に好ましくは15%より小さければ、「変異IL-6R発現細胞に実質的に結合しない」ものとする。Geo-Mean値(Geometric Mean)を求める計算式は、CELL QUEST Software User's Guide(BD biosciences社)に記載されている。比較値を比較することによってそれが実質的に同視し得る程度であれば、被験抗原結合分子と対照抗原結合分子のエピトープは同一であると評価され得る。
低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む被験抗原結合分子間の競合/同一エピトープへ結合するか否かを評価するとき、被験抗原結合分子と抗原の相互作用を低分子化合物の特定濃度下もしくは非存在下で行うことができ、被験抗原結合分子間では低分子化合物の濃度条件を同一にすることが好ましい。
低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメイン
本明細書において、「低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメイン」とは、異なる低分子化合物濃度存在下において、前記低分子化合物とは異なる分子である抗原に対する結合活性が変化する抗原結合ドメインを意味する。
本明細書において、「低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメイン」とは、異なる低分子化合物濃度存在下において、前記低分子化合物とは異なる分子である抗原に対する結合活性が変化する抗原結合ドメインを意味する。
MTA依存的に抗原に対する結合活性の変化する抗原結合ドメイン
本開示におけるMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインは、MTA濃度の異なる条件下において、MTAとは異なる分子である抗原に対する結合活性が異なる抗原結合ドメインである。これらの抗原結合ドメインが結合する抗原は、膜型分子であっても良く、可溶型分子であっても良い。また、これらの抗原結合ドメインが結合する抗原は、疾患組織で発現している抗原、より好ましくはがん組織で発現している抗原、更に好ましくはMTAが蓄積しているがん組織で発現する抗原である。がん組織で発現する抗原は、がん細胞に発現している抗原でもよく、がん組織中のがん間質細胞や免疫組織で発現している抗原でもよい。
本開示におけるMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインは、MTA濃度の異なる条件下において、MTAとは異なる分子である抗原に対する結合活性が異なる抗原結合ドメインである。これらの抗原結合ドメインが結合する抗原は、膜型分子であっても良く、可溶型分子であっても良い。また、これらの抗原結合ドメインが結合する抗原は、疾患組織で発現している抗原、より好ましくはがん組織で発現している抗原、更に好ましくはMTAが蓄積しているがん組織で発現する抗原である。がん組織で発現する抗原は、がん細胞に発現している抗原でもよく、がん組織中のがん間質細胞や免疫組織で発現している抗原でもよい。
MTA依存的に抗原に対する結合活性を変化する非限定な一例として、MTA存在下における抗原に対する結合活性が、MTA非存在下における抗原に対する結合活性より強い抗原結合ドメイン、またMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より弱い抗原結合ドメインが例示され得る。
本開示のMTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より強い抗原結合ドメインの、MTAの非存在下における抗原に対する結合活性が、MTAの存在下における抗原に対する結合活性よりも弱い限り、MTAの非存在下における抗原に対する結合活性とMTAの存在下における抗原に対する結合活性の比は特に限定されないが、好ましくは抗原に対するMTAの非存在下におけるKD(Dissociation constant:解離定数)と存在下におけるKDの比であるKD(MTA非存在下)/KD(MTA存在下)の値が2以上であり、さらに好ましくはKD(MTA非存在下)/KD(MTA存在下)の値が10以上であり、さらに好ましくはKD(MTA非存在下)/KD(MTA存在下)の値が40以上である。KD(MTA非存在下)/KD(MTA存在下)の値の上限は特に限定されず、当業者の技術において作製可能な限り、400、1000、10000等、いかなる値でもよい。MTAの非存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
MTA存在下における抗原に対する結合活性が、MTA非存在下における抗原に対する結合活性より強い抗原結合ドメインは、MTA非存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
MTA存在下における抗原に対する結合活性が、MTA非存在下における抗原に対する結合活性より強い抗原結合ドメインは、MTA非存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
本開示のMTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より弱い抗原結合ドメインの、MTAの非存在下における抗原に対する結合活性が、MTAの存在下における抗原に対する結合活性よりも強い限り、MTAの非存在下における抗原に対する結合活性とMTAの存在下における抗原に対する結合活性の比は特に限定されないが、好ましくは抗原に対するMTAの存在下におけるKD(Dissociation constant:解離定数)と非存在下におけるKDの比であるKD(MTA存在下)/KD(MTA非存在下)の値が2以上であり、さらに好ましくは10以上であり、さらに好ましくは40以上である。KD(MTA存在下)/KD(MTA非存在下)の値の上限は特に限定されず、当業者の技術において作製可能な限り、400、1000、10000等、いかなる値でもよい。MTAの存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
MTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より弱い抗原結合ドメインの場合、MTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
MTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より弱い抗原結合ドメインの場合、MTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
また、本開示の抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性と存在下における抗原に対する結合活性の比を示す他の指標として、例えば、解離速度定数であるkd(Dissociation rate constant:解離速度定数)もまた好適に用いられ得る。結合活性の比を示す指標としてKD(解離定数)の代わりにkd(解離速度定数)を用いる場合、MTAの非存在下における抗原に対するkd(解離速度定数)とMTAの存在下におけるkd(解離速度定数)の比である、kd(MTA非存在下)/kd(MTA存在下)の値は、好ましくは2以上であり、さらに好ましくは5以上であり、さらに好ましくは10以上であり、より好ましくは30以上である。Kd(MTA非存在下)/kd(MTA存在下)の値の上限は特に限定されず、当業者の技術常識において作製可能な限り、50、100、200等、いかなる値でもよい。MTAの非存在下において、抗原に対する結合活性が観察されない場合には解離も生じないため、この上限は無限大の数値となる。
MTAの存在下条件は、適宜なMTA濃度を設定することができ、非限定的な一例として、例えば、100μMのMTAが存在する条件をMTAの存在下条件と見なすことができる。また、本開示における「MTA存在下」と記載されるのMTA濃度の非限定な一態様としては、後記MTAの低濃度及び高濃度を区別する閾値として例示された濃度が適用されうる。
MTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの非限定な一例として、高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より強い抗原結合ドメインや、高濃度のMTA存在下における抗原結合活性が低濃度のMTA存在下における抗原結合活性より弱い抗原結合ドメインが例示される。
本開示の高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より強い抗原結合ドメインの、MTAの低濃度存在下における抗原に対する結合活性が、MTAの高濃度存在下における抗原に対する結合活性よりも弱い限り、MTAの低濃度存在下における抗原に対する結合活性とMTAの高濃度存在下における抗原に対する結合活性の比は特に限定されないが、好ましくは抗原に対するMTAの低濃度存在下におけるKD(Dissociation constant:解離定数)と高濃度存在下におけるKDの比であるKD(MTA低濃度存在下)/KD(MTA高濃度存在下)の値が2以上であり、さらに好ましくはKD(MTA低濃度存在下)/KD(MTA高濃度存在下)の値が10以上であり、さらに好ましくはKD(MTA低濃度存在下)/KD(MTA高濃度存在下)の値が40以上である。KD(MTA低濃度存在下)/KD(MTA高濃度存在下)の値の上限は特に限定されず、当業者の技術において作製可能な限り、400、1000、10000等、いかなる値でもよい。MTAの低濃度存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より強い抗原結合ドメインは、低濃度のMTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より強い抗原結合ドメインは、低濃度のMTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
本開示の高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より弱い抗原結合ドメインの、MTAの低濃度存在下における抗原に対する結合活性が、MTAの高濃度存在下における抗原に対する結合活性よりも強い限り、MTAの低濃度存在下における抗原に対する結合活性とMTAの高濃度存在下における抗原に対する結合活性の比は特に限定されないが、好ましくは抗原に対するMTAの高濃度存在下におけるKD(Dissociation constant:解離定数)と低濃度存在下におけるKDの比であるKD(MTA高濃度存在下)/KD(MTA低濃度存在下)の値が2以上であり、さらに好ましくは10以上であり、さらに好ましくは40以上である。KD(MTA高濃度存在下)/KD(MTA低濃度存在下)の値の上限は特に限定されず、当業者の技術において作製可能な限り、400、1000、10000等、いかなる値でもよい。MTAの高濃度存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より弱い抗原結合ドメインは、高濃度のMTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
高濃度のMTA存在下における抗原結合活性低濃度のMTA存在下における抗原結合活性より弱い抗原結合ドメインは、高濃度のMTA存在下において抗原に実質的に結合しない抗原結合ドメインを含む。
また、本開示の抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性と高濃度存在下における抗原に対する結合活性の比を示す他の指標として、例えば、解離速度定数であるkd(Dissociation rate constant:解離速度定数)もまた好適に用いられ得る。結合活性の比を示す指標としてKD(解離定数)の代わりにkd(解離速度定数)を用いる場合、MTAの低濃度存在下における抗原に対するkd(解離速度定数)とMTAの高濃度存在下におけるkd(解離速度定数)の比である、kd(MTA低濃度存在下)/kd(MTA高濃度存在下)の値は、好ましくは2以上であり、さらに好まくは5以上であり、さらに好ましくは10以上であり、より好ましくは30以上である。Kd(MTA低濃度存在下)/kd(MTA高濃度存在下)の値の上限は特に限定されず、当業者の技術常識において作製可能な限り、50、100、200等、いかなる値でもよい。MTAの低濃度存在下において、抗原に対する結合活性が観察されない場合には解離も生じないため、この上限は無限大の数値となる。
結合活性を評価する方法として、上述したKD値を測定する方法以外にも相対的な結合活性を評価する方法を用いることもできる。そのような非限定な方法の一例として、フローサイトメーター、ELISA、キャビラリー電気泳動、または液体クロマトグラフィー等を用いる手法が一般に知られているが、これらに限定されない。また例えばBiacoreによりKD値を算出した抗原結合結合分子を参照分子として比較することで、KD値を直接算出できない場合であっても被験分子が参照分子よりも結合活性が強い、もしくは結合活性が弱いという相対的な結合活性の評価も可能である。
MTAの低濃度及び高濃度を区別する閾値の非限定な一態様では、閾値として、低濃度条件は10 nM、1 nM、100 pM、10 pM、1 pMまたは0 Mの値から適宜設定され得る。設定された閾値に応じて、高濃度条件は各閾値の少なくとも110%、少なくとも120%、少なくとも130%、少なくとも140%、少なくとも150%、少なくとも2倍、少なくとも5倍、少なくとも10倍、少なくとも50倍、少なくとも100倍、少なくとも103倍、少なくとも104倍、少なくとも105倍、少なくとも106倍の値から適宜設定され得る。
本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの非限定的な一態様としては、当該抗原結合ドメインの抗原に対する結合活性がアデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPから選ばれる一つまたは複数の低分子化合物に実質的に影響されない抗原結合ドメインが挙げられる。ここで、前記アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPは分子中にアデノシンを共通骨格として有しており、MTAに類似する化合物である。本開示における抗原結合ドメインの抗原に対する結合活性が、これらの類似する分子の存在に実質的に影響されず、MTAのみに依存的に結合活性を変化させる抗原結合分子を取得することは一般的に困難であると考えられる。しかしながら、本開示においては、後述する各種の実施例に例示的に記載されるとおりのMTA依存的に抗体に結合する抗体を取得するためのライブラリを構築し、斯かるライブラリをスクリーニングすることにより、MTA特異的に依存して抗原に結合し、MTAと類似した構造を示す低分子化合物に対して依存的に抗原に対する結合を示さない、優れたMTA依存特異性を有する抗原結合ドメインを取得することに成功したものである。
本明細書において、「抗原に対する結合活性が低分子化合物に実質的に影響されない」は、当該低分子化合物存在下における抗原結合ドメインの抗原結合活性と当該低分子化合物非存在下における抗原結合ドメインの抗原結合活性が、お互いの少なくとも0.5倍~2倍であることをいう。
本明細書において、「抗原に対する結合活性が低分子化合物に実質的に影響されない」は、当該低分子化合物存在下における抗原結合ドメインの抗原結合活性と当該低分子化合物非存在下における抗原結合ドメインの抗原結合活性が、お互いの少なくとも0.5倍~2倍であることをいう。
非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシンに実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、S-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、SAMに実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、AMP、ADP、ATPのいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、S-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、SAMに実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、AMP、ADP、ATPのいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも実質的に影響されない。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも実質的に影響されない。
本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの非限定的な一態様としては、抗原に対する結合活性がアデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPから選ばれる一つまたは複数の低分子化合物に依存的にも変化する抗原結合ドメインが挙げられる。
非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン依存的にも変化する。MTAとアデノシンの両方の分子に依存的に結合活性が変化する抗原結合ドメインは、特定の理論に拘束されるものではないが、MTAとアデノシンの両方の分子において共通の構造と相互作用することにより抗原に対する結合活性が変化すると理解することができる。その場合、結合活性を評価する溶媒中において仮にMTAの濃度が減少したとしても、アデノシンの濃度がMTAの濃度の減少による結合活性の低下の程度を上回る濃度で存在する場合は、結合活性が維持される、もしくは強く検出されることはあり得る。また、結合活性を評価する溶媒中において仮にMTAの濃度が増加したとしても、MTAの濃度の増加による結合活性の増加の程度を越えてアデノシンの濃度が低下する場合は、結合活性が維持される、もしくは低く検出されることはあり得る。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、S-(5'-Adenosyl)-L-homocysteine (SAH)依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、SAM依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、AMP、ADP、ATPのいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも依存的にも変化する。
非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン依存的にも変化する。MTAとアデノシンの両方の分子に依存的に結合活性が変化する抗原結合ドメインは、特定の理論に拘束されるものではないが、MTAとアデノシンの両方の分子において共通の構造と相互作用することにより抗原に対する結合活性が変化すると理解することができる。その場合、結合活性を評価する溶媒中において仮にMTAの濃度が減少したとしても、アデノシンの濃度がMTAの濃度の減少による結合活性の低下の程度を上回る濃度で存在する場合は、結合活性が維持される、もしくは強く検出されることはあり得る。また、結合活性を評価する溶媒中において仮にMTAの濃度が増加したとしても、MTAの濃度の増加による結合活性の増加の程度を越えてアデノシンの濃度が低下する場合は、結合活性が維持される、もしくは低く検出されることはあり得る。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、S-(5'-Adenosyl)-L-homocysteine (SAH)依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、SAM依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、AMP、ADP、ATPのいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも依存的にも変化する。
別の非限定的な一態様において、本開示のMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインの抗原結合活性は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも依存的にも変化する。
本開示におけるMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインの非限定な一態様としては、抗体可変領域または/及び単ドメイン抗体を含む抗原結合ドメインが例示される。
非限定的な一態様として、本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む、抗原結合分子が例示されうる。(Kabatナンバリング):
重鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するF;
重鎖52a位に位置するA;
重鎖52b位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
重鎖52c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52d位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52e位に位置するY;
重鎖52f位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52g位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖53位に位置するS;
重鎖54位に位置するG;
重鎖55位に位置するG;
重鎖56位に位置するS;
重鎖57位に位置するT;
重鎖58位に位置するY;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するA;
重鎖64位に位置するK;
重鎖65位に位置するG;
重鎖95位に位置するG;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するG;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100a位に位置するG;
重鎖100b位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖101位に位置するE;
重鎖102位に位置するL;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するE;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖28位に位置するV;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するT;
軽鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖55位に位置するP;
軽鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖89位に位置するA;
軽鎖90位に位置するG;
軽鎖91位に位置するL;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するG;
軽鎖95位に位置するN;
軽鎖95a位に位置するI;
軽鎖96位に位置するP;
軽鎖97位に位置するA。
重鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するF;
重鎖52a位に位置するA;
重鎖52b位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
重鎖52c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52d位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52e位に位置するY;
重鎖52f位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52g位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖53位に位置するS;
重鎖54位に位置するG;
重鎖55位に位置するG;
重鎖56位に位置するS;
重鎖57位に位置するT;
重鎖58位に位置するY;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するA;
重鎖64位に位置するK;
重鎖65位に位置するG;
重鎖95位に位置するG;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するG;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100a位に位置するG;
重鎖100b位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100c位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖101位に位置するE;
重鎖102位に位置するL;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するE;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖28位に位置するV;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TまたはVのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するT;
軽鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖55位に位置するP;
軽鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖89位に位置するA;
軽鎖90位に位置するG;
軽鎖91位に位置するL;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するG;
軽鎖95位に位置するN;
軽鎖95a位に位置するI;
軽鎖96位に位置するP;
軽鎖97位に位置するA。
非限定的な別の態様として、本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む抗原結合分子が例示されうる。(Kabatナンバリング):
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52a位に位置するS;
重鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するT;
重鎖58位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するN;
重鎖65位に位置するG;
重鎖95位に位置するE;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するS;
重鎖100a位に位置するG;
重鎖100b位に位置するA;
重鎖100c位に位置するL;
重鎖101位に位置するN;
重鎖102位に位置するL;
軽鎖24位に位置するH;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するK;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖27b位に位置するV;
軽鎖28位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するA;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するQ;
軽鎖90位に位置するG;
軽鎖91位に位置するT;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95b位に位置するF;
軽鎖95c位に位置するY;
軽鎖96位に位置するF;
軽鎖97位に位置するA。
重鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖33位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖34位に位置するW;
重鎖35位に位置するM;
重鎖35a位に位置するC;
重鎖50位に位置するC;
重鎖51位に位置するI;
重鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖52a位に位置するS;
重鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖54位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖56位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するT;
重鎖58位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖59位に位置するY;
重鎖60位に位置するA;
重鎖61位に位置するS;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するN;
重鎖65位に位置するG;
重鎖95位に位置するE;
重鎖96位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖97位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖98位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖100位に位置するS;
重鎖100a位に位置するG;
重鎖100b位に位置するA;
重鎖100c位に位置するL;
重鎖101位に位置するN;
重鎖102位に位置するL;
軽鎖24位に位置するH;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するK;
軽鎖27a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖27b位に位置するV;
軽鎖28位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖31位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖32位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するA;
軽鎖49位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖50位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖51位に位置するA;
軽鎖52位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖53位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するQ;
軽鎖90位に位置するG;
軽鎖91位に位置するT;
軽鎖92位に位置するY;
軽鎖93位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖94位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95b位に位置するF;
軽鎖95c位に位置するY;
軽鎖96位に位置するF;
軽鎖97位に位置するA。
非限定的な更なる態様として、本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は下記アミノ酸の群から選択される少なくとも一つ以上のアミノ酸を含む抗原結合分子が例示されうる。(Kabatナンバリング):
重鎖26位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖28位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖29位に位置するAまたはLのいずれか;
重鎖30位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するD、E、F、H、N、P、RまたはYのいずれか;
重鎖33位に位置するA、I、P、TまたはVのいずれか;
重鎖34位に位置するA、E、F、H、I、K、L、M、N、Q、S、T、V、WまたはYのいずれか;
重鎖35位に位置するG;
重鎖50位に位置するD、IまたはVのいずれか;
重鎖51位に位置するI;
重鎖52位に位置するG;
重鎖53位に位置するA、D、E、G,I、K、QまたはRのいずれか;
重鎖54位に位置するD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、D、E、F、GまたはHのいずれか;
重鎖56位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するA、D、E、G、H、I、K、L、N、P、Q、R、S、TまたはVのいずれか;
重鎖58位に位置するW;
重鎖59位に位置するA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WまたはYのいずれか;
重鎖60位に位置するP;
重鎖61位に位置するA、F、Q、R、S、T、V、WまたはYのいずれか;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するK;
重鎖65位に位置するA、FまたはG;
重鎖95位に位置するG;
重鎖96位に位置するA、E、F、G、H、K、L、Q、R、S、T、WまたはYのいずれか;
重鎖97位に位置するA、F、H、K、N、WまたはYのいずれか;
重鎖98位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、D、E、G、H、QまたはSのいずれか;
重鎖100位に位置するFまたはY;
重鎖100a位に位置するN、TまたはV
重鎖100b位に位置するN;
重鎖100c位に位置するA;
重鎖100d位に位置するFまたはW;
重鎖101位に位置するD;
重鎖102位に位置するP;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するQ;
軽鎖27e位に位置するS;
軽鎖27f位に位置するV;
軽鎖28位に位置するA、E、F、H、I、K、L、N、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するN;
軽鎖31位に位置するN ;
軽鎖32位に位置するA、E、F、G、H、SまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖50位に位置するD ;
軽鎖51位に位置するA;
軽鎖52位に位置するS ;
軽鎖53位に位置するT ;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するH;
軽鎖90位に位置するG;
軽鎖91位に位置するA、SまたはTのいずれか;
軽鎖92位に位置するA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WまたはYのいずれか;
軽鎖93位に位置するA、D、E、F、G、H、L、N、Q、R、S、T、VまたはYのいずれか;
軽鎖94位に位置するA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WまたはYのいずれか;
軽鎖95b位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95c位に位置するA、F、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖96d位に位置するD;
軽鎖96位に位置するN;
軽鎖97位に位置するAまたはG;
軽鎖98位に位置するA、F、I、LまたはV。
重鎖26位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖28位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖29位に位置するAまたはLのいずれか;
重鎖30位に位置するA、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖31位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖32位に位置するD、E、F、H、N、P、RまたはYのいずれか;
重鎖33位に位置するA、I、P、TまたはVのいずれか;
重鎖34位に位置するA、E、F、H、I、K、L、M、N、Q、S、T、V、WまたはYのいずれか;
重鎖35位に位置するG;
重鎖50位に位置するD、IまたはVのいずれか;
重鎖51位に位置するI;
重鎖52位に位置するG;
重鎖53位に位置するA、D、E、G,I、K、QまたはRのいずれか;
重鎖54位に位置するD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖55位に位置するA、D、E、F、GまたはHのいずれか;
重鎖56位に位置するA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WまたはYのいずれか;
重鎖57位に位置するA、D、E、G、H、I、K、L、N、P、Q、R、S、TまたはVのいずれか;
重鎖58位に位置するW;
重鎖59位に位置するA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WまたはYのいずれか;
重鎖60位に位置するP;
重鎖61位に位置するA、F、Q、R、S、T、V、WまたはYのいずれか;
重鎖62位に位置するW;
重鎖63位に位置するV;
重鎖64位に位置するK;
重鎖65位に位置するA、FまたはG;
重鎖95位に位置するG;
重鎖96位に位置するA、E、F、G、H、K、L、Q、R、S、T、WまたはYのいずれか;
重鎖97位に位置するA、F、H、K、N、WまたはYのいずれか;
重鎖98位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
重鎖99位に位置するA、D、E、G、H、QまたはSのいずれか;
重鎖100位に位置するFまたはY;
重鎖100a位に位置するN、TまたはV
重鎖100b位に位置するN;
重鎖100c位に位置するA;
重鎖100d位に位置するFまたはW;
重鎖101位に位置するD;
重鎖102位に位置するP;
軽鎖24位に位置するQ;
軽鎖25位に位置するS;
軽鎖26位に位置するS;
軽鎖27位に位置するQ;
軽鎖27e位に位置するS;
軽鎖27f位に位置するV;
軽鎖28位に位置するA、E、F、H、I、K、L、N、R、S、T、V、WまたはYのいずれか;
軽鎖29位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖30位に位置するN;
軽鎖31位に位置するN ;
軽鎖32位に位置するA、E、F、G、H、SまたはYのいずれか;
軽鎖33位に位置するL;
軽鎖34位に位置するS;
軽鎖50位に位置するD ;
軽鎖51位に位置するA;
軽鎖52位に位置するS ;
軽鎖53位に位置するT ;
軽鎖54位に位置するL;
軽鎖55位に位置するA;
軽鎖56位に位置するS;
軽鎖89位に位置するH;
軽鎖90位に位置するG;
軽鎖91位に位置するA、SまたはTのいずれか;
軽鎖92位に位置するA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WまたはYのいずれか;
軽鎖93位に位置するA、D、E、F、G、H、L、N、Q、R、S、T、VまたはYのいずれか;
軽鎖94位に位置するA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95a位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WまたはYのいずれか;
軽鎖95b位に位置するA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖95c位に位置するA、F、H、I、K、L、N、P、Q、R、S、T、V、WまたはYのいずれか;
軽鎖96d位に位置するD;
軽鎖96位に位置するN;
軽鎖97位に位置するAまたはG;
軽鎖98位に位置するA、F、I、LまたはV。
本開示における抗原結合ドメインはMTAと相互作用するアミノ酸残基を有し得る。MTAと相互作用するアミノ酸残基は抗原結合ドメイン中の抗原と直接相互作用する界面に存在しても良いし、それ以外の部分に存在しても良い。「抗原と直接相互作用する界面」とは結晶構造解析等の手法により解析された抗原と抗原結合ドメインの複合体の構造において、抗原と抗原結合分子が相互に近接している部位をいう。前記抗原と抗原結合分子が相互に近接する距離として、4~6Åの距離が例示され得るが、抗原と抗原結合分子の複合体の構造において、「抗原と抗原結合分子が相互に近接するとは」複合体の中で相対的に近接しる部位を指し、前記例示した距離に限定されるものではない。MTAと相互作用するアミノ酸残基は、抗原結合分子が抗原と結合している状態においてMTAと相互作用する残基であっても良く、抗原非存在の状態においてMTAと相互作用する残基であってもよい。さらに、MTAと相互作用するアミノ酸残基は、抗原結合分子が抗原と結合している状態においても、抗原非存在の状態においてもMTAと相互作用する残基であり得る。またMTAと相互作用するアミノ酸残基は、抗原結合ドメイン中に1残基のみでもよく、複数存在してもよい。
抗原結合ドメインが抗体重鎖可変領域と抗体軽鎖可変領域を含む態様においては、MTAと相互作用するアミノ酸は抗体可変領域中CDRに存在してもよく、FRに存在してもよい。抗原結合ドメインが単ドメイン抗体を含む態様においては、MTAと相互作用するアミノ酸は単ドメイン抗体中CDRに存在してもよく、FRに存在してもよい。
抗原結合ドメイン中のMTAと相互作用するアミノ酸残基は、MTAと抗原結合ドメインを含む抗原結合分子二者の複合体、またはMTA、抗原、抗原結合ドメインを含む抗原結合分子の三者複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定することができる。
本開示の非限定の一態様として、MTAと抗原結合分子二者の複合体の結晶構造解析から、MTAと相互作用する抗原結合分子のアミノ酸残基を同定することができる。ここで、「MTAと相互作用する」とは、抗原結合分子を形成するアミノ酸における側鎖又は主鎖の原子と低分子化合物の原子が、MTA結合活性に効果を及ぼしうる距離で抗原結合分子-MTA間相互作用を形成している状態、又は、抗原結合分子中の抗原結合ドメインが抗体重鎖可変領域および抗体軽鎖可変領域を含む態様において、あるアミノ酸残基がCDRループ等の立体構造をMTAが結合する際のコンフォメーションに安定化させる等の間接的な影響を含む、MTAの結合に寄与している状態、ならびにその双方を満たす状態のことをいう。
本明細書における「分子間相互作用を形成している状態」については,例えばMTAと抗原結合分子二者の複合体の結晶構造解析から、抗原結合分子を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入が低分子化合物の活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTAと抗原結合分子二者の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、MTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
本明細書における「分子間相互作用を形成している状態」については,例えばMTAと抗原結合分子二者の複合体の結晶構造解析から、抗原結合分子を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入が低分子化合物の活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTAと抗原結合分子二者の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、MTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
本開示の非限定の一態様として、MTA、抗原、抗原結合分子三者の複合体の結晶構造解析から、MTAと相互作用する抗原結合分子のアミノ酸残基を同定することができる。ここで、「MTAと相互作用する」とは、抗原結合分子を形成するアミノ酸における側鎖又は主鎖の原子と低分子化合物の原子が、抗原の存在下でMTA結合活性に効果を及ぼしうる距離で抗原結合分子-MTA間相互作用を形成している状態、又は、抗原結合分子中の抗原結合ドメインが抗体重鎖可変領域および抗体軽鎖可変領域を含む態様において、あるアミノ酸残基がCDRループ等の立体構造を抗原存在下でMTAが結合する際のコンフォメーションに安定化させる等の間接的な影響を含む、MTAの結合に寄与している状態、ならびにその双方を満たす状態のことをいう。
本明細書における「分子間相互作用を形成している状態」については,例えばMTA、抗原、抗原結合分子三者の複合体の結晶構造解析から、抗原存在下で抗原結合分子を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入が低分子化合物の活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTA、抗原、抗原結合分子三者の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、抗原存在下でのMTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
本明細書における「分子間相互作用を形成している状態」については,例えばMTA、抗原、抗原結合分子三者の複合体の結晶構造解析から、抗原存在下で抗原結合分子を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入が低分子化合物の活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTA、抗原、抗原結合分子三者の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、抗原存在下でのMTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
非限定な一態様として、抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、35a位、47位、52位、52e位、101位、軽鎖32位、34位、36位、46位、49位、50位、89位、90位、91位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である抗原結合ドメインが例示されうる。
非限定な一態様として、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、C35a、W47、F52、Y52e、E101、軽鎖R32、S34、Y36、L46、Y49、S50、A89、G90、L91、及びP96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示される。
非限定な一態様として、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、C35a、W47、F52、Y52e、E101、軽鎖R32、S34、Y36、L46、Y49、S50、A89、G90、L91、及びP96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示される。
また、非限定な一態様として、抗原結合ドメインは抗体可変領域であり、MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖34位、47位、50位、58位、95位、98位、99位、100a位、軽鎖28位、91位、95b位、95c位及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である抗原結合ドメインが例示されうる。
非限定な一態様として、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、W47、C50、Y58、E95、F98、G99、G100a、軽鎖Y28、T91、F95b、Y95c、及びF96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示される。
非限定な一態様として、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖W34、W47、C50、Y58、E95、F98、G99、G100a、軽鎖Y28、T91、F95b、Y95c、及びF96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示される。
また、MTAと相互作用する抗原結合ドメインの非限定な一態様として、前記抗原結合ドメインは抗体可変領域であり、前記MTAと相互作用するアミノ酸残基は、前記抗体可変領域のアミノ酸配列の内、Kabatナンバリングで特定される重鎖33位、50位、52位、54位、56位、57位、58位、99位、100位、100a位、91位、95c位、及び96位のアミノ酸部位の群から選択される少なくとも一つ以上のアミノ酸部位に位置するアミノ酸残基である抗原結合ドメインが例示されうる。
また、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖A33、I50、G52、D54、S56、T57、W58、G99、Y100、T100a、軽鎖S91、Y95c、及びN96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示されうる。
また、前記抗原結合ドメインは抗体可変領域であり、当該抗体可変領域は重鎖A33、I50、G52、D54、S56、T57、W58、G99、Y100、T100a、軽鎖S91、Y95c、及びN96(Kabatナンバリング)から選択される少なくとも一つ以上のアミノ酸を含む抗原結合ドメインが例示されうる。
MTA依存的に抗原に対する結合活性の変化する抗原結合ドメインを含む抗原結合分子
非限定な一態様においては、本開示にMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインを含む抗原結合分子は抗体Fc領域を含む分子である。本開示の抗原結合分子に含まれる抗体Fc領域は、天然型Fc領域であっても改変Fc領域であっても良い。天然型Fc領域として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。
非限定な一態様においては、本開示にMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインを含む抗原結合分子は抗体Fc領域を含む分子である。本開示の抗原結合分子に含まれる抗体Fc領域は、天然型Fc領域であっても改変Fc領域であっても良い。天然型Fc領域として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。
本開示の抗原結合分子は、Fcγレセプターに対する結合、および/またはFcRnに対する結合を媒介するFc領域の少なくとも部分を含むことができる。例えば、非限定な一態様では、抗原結合分子は抗体またはFc融合タンパク質であり得る。融合タンパク質とは、天然ではそれが自然に連結しない第二のアミノ酸配列を有するポリペプチドに連結された第一のアミノ酸配列を含むポリペプチドを含むキメラポリペプチドをいう。例えば、融合タンパク質は、Fc領域の少なくとも部分(例えば、Fcγレセプターに対する結合を付与するFc領域の部分および/またはFcRnに対する結合を付与するFc領域の部分)をコードするアミノ酸配列を含むポリペプチド、を含むことができる。アミノ酸配列は、一緒に融合タンパク質に運ばれる別々のタンパク質に存在できるか、あるいはそれらは通常は同一タンパク質に存在できるが、融合ポリペプチド中の新しい再編成に入れられる。融合タンパク質は、例えば、化学合成によって、またはペプチド領域が所望の関係でコードされたポリヌクレオチドを作成し、それを発現する遺伝子組換えの手法によって作製され得る。
本開示の抗原結合分子中の各ドメインはポリペプチド結合によって直接連結され得るし、リンカーを介して連結され得る。リンカーとしては、遺伝子工学により導入し得る任意のペプチドリンカー、又は合成化合物リンカー(例えば、Holligerら(Protein Engineering (1996) 9 (3), 299-305))に開示されるリンカー等が使用され得るが、本開示においてはペプチドリンカーが好ましい。ペプチドリンカーの長さは特に限定されず、目的に応じて当業者が適宜選択することが可能であるが、好ましい長さは5アミノ酸以上(上限は特に限定されないが、通常、30アミノ酸以下、好ましくは20アミノ酸以下)であり、特に好ましくは15アミノ酸である。
例えば、ペプチドリンカーの場合:
Ser
Gly・Ser
Gly・Gly・Ser
Ser・Gly・Gly
Gly・Gly・Gly・Ser(配列番号:19)
Ser・Gly・Gly・Gly(配列番号:20)
Gly・Gly・Gly・Gly・Ser(配列番号:21)
Ser・Gly・Gly・Gly・Gly(配列番号:22)
Gly・Gly・Gly・Gly・Gly・Ser(配列番号:23)
Ser・Gly・Gly・Gly・Gly・Gly(配列番号:24)
Gly・Gly・Gly・Gly・Gly・Gly・Ser(配列番号:25)
Ser・Gly・Gly・Gly・Gly・Gly・Gly(配列番号:26)
(Gly・Gly・Gly・Gly・Ser(配列番号:21))n
(Ser・Gly・Gly・Gly・Gly(配列番号22))n
[nは1以上の整数である]等が好適に挙げられる。但し、ペプチドリンカーの長さや配列は目的に応じて当業者が適宜選択することができる。
Ser
Gly・Ser
Gly・Gly・Ser
Ser・Gly・Gly
Gly・Gly・Gly・Ser(配列番号:19)
Ser・Gly・Gly・Gly(配列番号:20)
Gly・Gly・Gly・Gly・Ser(配列番号:21)
Ser・Gly・Gly・Gly・Gly(配列番号:22)
Gly・Gly・Gly・Gly・Gly・Ser(配列番号:23)
Ser・Gly・Gly・Gly・Gly・Gly(配列番号:24)
Gly・Gly・Gly・Gly・Gly・Gly・Ser(配列番号:25)
Ser・Gly・Gly・Gly・Gly・Gly・Gly(配列番号:26)
(Gly・Gly・Gly・Gly・Ser(配列番号:21))n
(Ser・Gly・Gly・Gly・Gly(配列番号22))n
[nは1以上の整数である]等が好適に挙げられる。但し、ペプチドリンカーの長さや配列は目的に応じて当業者が適宜選択することができる。
合成化学物リンカー(化学架橋剤)は、ペプチドの架橋に通常用いられている架橋剤、例えばN-ヒドロキシスクシンイミド(NHS)、ジスクシンイミジルスベレート(DSS)、ビス(スルホスクシンイミジル)スベレート(BS3)、ジチオビス(スクシンイミジルプロピオネート)(DSP)、ジチオビス(スルホスクシンイミジルプロピオネート)(DTSSP)、エチレングリコールビス(スクシンイミジルスクシネート)(EGS)、エチレングリコールビス(スルホスクシンイミジルスクシネート)(スルホ-EGS)、ジスクシンイミジル酒石酸塩(DST)、ジスルホスクシンイミジル酒石酸塩(スルホ-DST)、ビス[2-(スクシンイミドオキシカルボニルオキシ)エチル]スルホン(BSOCOES)、ビス[2-(スルホスクシンイミドオキシカルボニルオキシ)エチル]スルホン(スルホ-BSOCOES)等であり、これらの架橋剤は市販されている。
各ドメインを連結するリンカーが複数用いられる場合には、全て同種のリンカーが用いられ得るし、異種のリンカーも用いられ得る。また、上記記載で例示されるリンカーのほか、例えばHisタグ、HAタグ、mycタグ、FLAGタグ等のペプチドタグを有するリンカーも適宜使用され得る。また、水素結合、ジスルフィド結合、共有結合、イオン性相互作用またはこれらの結合の組合せにより互いに結合する性質もまた好適に利用され得る。例えば、抗体のCH1とCL間の親和性が利用されたり、ヘテロFc領域の会合に際して前述の二重特異性抗体を起源とするFc領域が用いられたりする。さらに、ドメイン間に形成されるジスルフィド結合もまた好適に利用され得る。
各ドメインをペプチド結合で連結するために、当該ドメインをコードするポリヌクレオチドがインフレームで連結される。ポリヌクレオチドをインフレームで連結する方法としては、制限断片のライゲーションやフュージョンPCR、オーバーラップPCR等の手法が公知であり、本開示の抗原結合分子の作製にも適宜これらの方法が単独または組合せで使用され得る。本開示では、用語「連結され」、「融合され」、「連結」または「融合」は相互交換的に用いられる。これらの用語は、上記の化学結合手段または組換え手法を含めた全ての手段によって、二以上のポリペプチド等のエレメントまたは成分を一つの構造を形成するように連結することをいう。インフレームで融合するとは、二以上のエレメントまたは成分がポリペプチドである場合に、当該ポリペプチドのの正しい読み取り枠を維持するように連続したより長い読み取り枠を形成するための二以上の読取り枠の単位の連結をいう。二分子のFabが抗原結合ドメインとして用いられた場合、当該抗原結合ドメインとFc領域を含む定常領域がリンカーを介することなくペプチド結合によってインフレームで連結された本開示の抗原結合分子である抗体は、本開示の好適な抗原結合分子として使用され得る。
別の局面において、本開示は、高い血漿中滞留性を有する抗原結合分子を提供する。ある局面において、当該抗原結合分子は、MTAの濃度が高まるにつれて抗原結合活性が増大する。ある態様において、当該抗原結合分子は、非標的組織における抗原結合活性に比べて、標的組織における抗原結合活性がより高い。いくつかの局面において、抗原結合分子は抗体である。特定の理論に拘束されるわけではないが、上述の血漿中における動態の変化は以下のように解釈することができる。抗原結合分子のMTAの濃度に依存した抗原結合活性が高まるにつれて、当該抗原結合分子の標的組織以外の組織における抗原結合能が低下する。その結果、当該抗原結合分子の標的組織以外の組織における抗原依存的な消失(クリアランス)が低下することになる。生体内の大部分の組織(標的組織以外の組織)において、抗原依存的な消失(クリアランス)が低下することは、総合的に見れば、当該抗原結合分子の高い血漿中滞留性につながる。
本発明における抗原結合分子が高い血漿中滞留性を有しているかどうかの判断は、対照となる抗原結合分子との相対的な比較によって行うことができる。いくつかの態様において、MTAの濃度が高まるにつれて抗原結合活性が増大する抗原結合分子は、対照となる抗原結合分子と比べて、高い血漿中滞留性を有する。一態様において、対照となる抗原結合分子は、MTAの濃度に依存した抗原結合活性を有していない抗原結合分子である。特定の態様において、化合物の濃度に依存した抗原結合活性を有していない抗原結合分子とは、MTAの存在下と非存在下における抗原結合活性の差が、例えば2倍より小さい、1.8倍より小さい、1.5倍より小さい、1.3倍より小さい、1.2倍より小さい、または1.1倍より小さい抗原結合分子を意味する。比較の観点からすると、本開示の抗原結合分子と、対照となる抗原結合分子とでは、十分量のMTAの存在下における抗原結合活性が互いに実質的に等しいことが望ましい。
本発明における抗原結合分子が高い血漿中滞留性を有しているかどうかの判断は、対照となる抗原結合分子との相対的な比較によって行うことができる。いくつかの態様において、MTAの濃度が高まるにつれて抗原結合活性が増大する抗原結合分子は、対照となる抗原結合分子と比べて、高い血漿中滞留性を有する。一態様において、対照となる抗原結合分子は、MTAの濃度に依存した抗原結合活性を有していない抗原結合分子である。特定の態様において、化合物の濃度に依存した抗原結合活性を有していない抗原結合分子とは、MTAの存在下と非存在下における抗原結合活性の差が、例えば2倍より小さい、1.8倍より小さい、1.5倍より小さい、1.3倍より小さい、1.2倍より小さい、または1.1倍より小さい抗原結合分子を意味する。比較の観点からすると、本開示の抗原結合分子と、対照となる抗原結合分子とでは、十分量のMTAの存在下における抗原結合活性が互いに実質的に等しいことが望ましい。
ここで、生体内で検出される抗原結合分子の抗原依存的な消失の大きさは、血漿中に存在する抗原と抗原結合分子の量的なバランスに応じて変化すると考えられる。一般的に、血漿中に存在する抗原が多いほど/抗原結合分子が少ないほど、抗原結合分子の抗原依存的な消失は検出されやすくなり、逆に、血漿中に存在する抗原が少ないほど/抗原結合分子が多いほど、抗原結合分子の抗原依存的な消失は検出されにくくなると考えられる。本開示の抗原結合分子は、全ての条件で高い血漿中滞留性を示す必要はなく、十分な抗原依存的な消失が検出されるような適切な条件下において高い血漿中滞留性を示せばよい。血漿中の抗原量が少ない場合は、何らかの人為的な手段によって抗原量を増やしてから血漿中滞留性を評価してもよい。
別の局面において、本発明は、低い血漿中抗原蓄積能を有する抗原結合分子を提供する。さらなる局面において、当該抗原結合分子は、MTAの濃度が高まるにつれて抗原結合活性が増大する。特定の態様において、MTAは、標的組織特異的化合物である。さらなる態様において、当該抗原結合分子は、非標的組織における抗原結合活性に比べて、標的組織における抗原結合活性がより高い。いくつかの局面において、抗原結合分子は抗体である。特定の理論に拘束されるわけではないが、上述の血漿中における動態の変化は以下のように解釈することができる。抗原結合分子のMTAの濃度に依存した抗原結合活性が高まるにつれて、当該抗原結合分子の標的組織以外の組織における抗原結合能が低下する。その結果、当該抗原結合分子の標的組織以外の組織における抗原-抗体複合体の形成能が低下することになる。一般的に、抗体などの抗原結合分子が抗原と結合すると、抗原のクリアランスが低下して血漿中の抗原濃度が上昇(抗原が蓄積)することが知られている。生体内の大部分の組織(標的組織以外の組織)において、抗原-抗体複合体の形成能が低下することは、総合的に見れば、抗原の低い蓄積(言い換えると、抗原結合分子の低い抗原蓄積能)につながる。本発明における抗原結合分子が低い血漿中抗原蓄積能を有しているかどうかの判断は、対照となる抗原結合分子との相対的な比較によって行うことができる。いくつかの態様において、MTAの濃度が高まるにつれて抗原結合活性が増大する抗原結合分子は、対照となる抗原結合分子と比べて、低い血漿中抗原蓄積能を有する。一態様において、対照となる抗原結合分子は、MTAの濃度に依存した抗原結合活性を有していない抗原結合分子である。特定の態様において、MTAの濃度に依存した抗原結合活性を有していない抗原結合分子とは、当該化合物の存在下と非存在下における抗原結合活性の差が、例えば2倍より小さい、1.8倍より小さい、1.5倍より小さい、1.3倍より小さい、1.2倍より小さい、または1.1倍より小さい抗原結合分子を意味する。比較の観点からすると、本発明の抗原結合分子と、対照となる抗原結合分子とでは、十分量の化合物の存在下における抗原結合活性が互いに実質的に等しいことが望ましい。
ここで、生体内で形成される抗原-抗体複合体の量は、血漿中に存在する抗原および抗体の量に依存すると考えられる。一般的に、血漿中の抗原・抗体量が増加するほど、形成される抗原-抗体複合体量も増加し、逆に、血漿中の抗原・抗体量が減少するほど、形成される抗原-抗体複合体量も減少すると考えられる。本発明の抗原結合分子は、全ての条件で低い血漿中抗原蓄積能を示す必要はなく、十分な抗原-抗体複合体が形成されるような適切な条件下において低い血漿中抗原蓄積能を示せばよい。血漿中の抗原量が少ない場合は、何らかの人為的な手段によって抗原量を増やしてから血漿中抗原蓄積能を評価して
もよい。
もよい。
Fcγレセプター(FcγR)
Fcγレセプター(FcγRとも記載される)とは、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合し得るレセプターをいい、実質的にFcγレセプター遺伝子にコードされるタンパク質のファミリーのいかなるメンバーをも意味する。ヒトでは、このファミリーには、アイソフォームFcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64);アイソフォームFcγRIIa(アロタイプH131およびR131を含む。即ち、FcγRIIa (H)およびFcγRIIa (R))、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)およびFcγRIIcを含むFcγRII(CD32);およびアイソフォームFcγRIIIa(アロタイプV158およびF158を含む。即ち、FcγRIIIa (V)およびFcγRIIIa (F))およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)、並びにいかなる未発見のヒトFcγR類またはFcγRアイソフォームまたはアロタイプも含まれるが、これらに限定されるものではない。FcγRは、ヒト、マウス、ラット、ウサギおよびサルを含むが、これらに限定されるものではない、いかなる生物由来でもよい。マウスFcγR類には、FcγRI(CD64)、FcγRII(CD32)、FcγRIII(CD16)およびFcγRIII-2(FcγRIV、CD16-2)、並びにいかなる未発見のマウスFcγR類またはFcγRアイソフォームまたはアロタイプも含まれるが、これらに限定されない。こうしたFcγレセプターの好適な例としてはヒトFcγRI(CD64)、FcγRIIa(CD32)、FcγRIIb(CD32)、FcγRIIIa(CD16)及び/又はFcγRIIIb(CD16)が挙げられる。ヒトFcγRIのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:9(NM_000566.3)及び10(NP_000557.1)に、ヒトFcγRIIa(アロタイプH131)のポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:11(BC020823.1)及び12(AAH20823.1)に(アロタイプR131は配列番号:12の166番目のアミノ酸がArgに置換されている配列である)、FcγRIIbのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:13(BC146678.1)及び14(AAI46679.1)に、FcγRIIIaのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:15(BC033678.1)及び16(AAH33678.1)に、及びFcγRIIIbのポリヌクレオチド配列及びアミノ酸配列は、それぞれ配列番号:17(BC128562.1)及び18(AAI28563.1)に記載されている(カッコ内はRefSeq等のデータベース登録番号を示す)。Fcγレセプターが、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合活性を有するか否かは、上記に記載されるFACSやELISAフォーマットのほか、ALPHAスクリーン(Amplified Luminescent Proximity Homogeneous Assay)や表面プラズモン共鳴(SPR)現象を利用したBIACORE法等によって確認され得る(Proc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010)。
Fcγレセプター(FcγRとも記載される)とは、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合し得るレセプターをいい、実質的にFcγレセプター遺伝子にコードされるタンパク質のファミリーのいかなるメンバーをも意味する。ヒトでは、このファミリーには、アイソフォームFcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64);アイソフォームFcγRIIa(アロタイプH131およびR131を含む。即ち、FcγRIIa (H)およびFcγRIIa (R))、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)およびFcγRIIcを含むFcγRII(CD32);およびアイソフォームFcγRIIIa(アロタイプV158およびF158を含む。即ち、FcγRIIIa (V)およびFcγRIIIa (F))およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)、並びにいかなる未発見のヒトFcγR類またはFcγRアイソフォームまたはアロタイプも含まれるが、これらに限定されるものではない。FcγRは、ヒト、マウス、ラット、ウサギおよびサルを含むが、これらに限定されるものではない、いかなる生物由来でもよい。マウスFcγR類には、FcγRI(CD64)、FcγRII(CD32)、FcγRIII(CD16)およびFcγRIII-2(FcγRIV、CD16-2)、並びにいかなる未発見のマウスFcγR類またはFcγRアイソフォームまたはアロタイプも含まれるが、これらに限定されない。こうしたFcγレセプターの好適な例としてはヒトFcγRI(CD64)、FcγRIIa(CD32)、FcγRIIb(CD32)、FcγRIIIa(CD16)及び/又はFcγRIIIb(CD16)が挙げられる。ヒトFcγRIのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:9(NM_000566.3)及び10(NP_000557.1)に、ヒトFcγRIIa(アロタイプH131)のポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:11(BC020823.1)及び12(AAH20823.1)に(アロタイプR131は配列番号:12の166番目のアミノ酸がArgに置換されている配列である)、FcγRIIbのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:13(BC146678.1)及び14(AAI46679.1)に、FcγRIIIaのポリヌクレオチド配列及びアミノ酸配列はそれぞれ配列番号:15(BC033678.1)及び16(AAH33678.1)に、及びFcγRIIIbのポリヌクレオチド配列及びアミノ酸配列は、それぞれ配列番号:17(BC128562.1)及び18(AAI28563.1)に記載されている(カッコ内はRefSeq等のデータベース登録番号を示す)。Fcγレセプターが、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合活性を有するか否かは、上記に記載されるFACSやELISAフォーマットのほか、ALPHAスクリーン(Amplified Luminescent Proximity Homogeneous Assay)や表面プラズモン共鳴(SPR)現象を利用したBIACORE法等によって確認され得る(Proc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010)。
FcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64)ならびにアイソフォームFcγRIIIa(アロタイプV158およびF158を含む)およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)は、IgGのFc領域と結合するα鎖と細胞内に活性化シグナルを伝達するITAMを有する共通γ鎖が会合する。一方、アイソフォームFcγRIIa(アロタイプH131およびR131を含む)およびFcγRIIcを含むFcγRII(CD32)の自身の細胞質ドメインにはITAMが含まれている。これらのレセプターは、マクロファージやマスト細胞、抗原提示細胞等の多くの免疫細胞に発現している。これらのレセプターがIgGのFc領域に結合することによって伝達される活性化シグナルによって、マクロファージの貪食能や炎症性サイトカインの産生、マスト細胞の脱顆粒、抗原提示細胞の機能亢進が促進される。上記のように活性化シグナルを伝達する能力を有するFcγレセプターは、本明細書において活性型Fcγレセプターと呼ばれる。
一方、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)の自身の細胞質内ドメインには抑制型シグナルを伝達するITIMが含まれている。B細胞ではFcγRIIbとB細胞レセプター(BCR)との架橋によってBCRからの活性化シグナルが抑制される結果BCRの抗体産生が抑制される。マクロファージでは、FcγRIIIとFcγRIIbとの架橋によって貪食能や炎症性サイトカインの産生能が抑制される。上記のように抑制化シグナルを伝達する能力を有するFcγレセプターは、本明細書において抑制型Fcγレセプターと呼ばれる。
FcγRに対するFc領域の結合活性
前述されるように、本開示の抗原結合分子に含まれるFc領域として、Fcγレセプターに対する結合活性を有するFc領域が挙げられる。そのようなFc領域の非限定な一態様として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。Fcγレセプターが、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合活性を有するか否かは、上記に記載されるFACSやELISAフォーマットのほか、ALPHAスクリーン(Amplified Luminescent Proximity Homogeneous Assay)や表面プラズモン共鳴(SPR)現象を利用したBIACORE法等によって確認され得る(Proc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010)。
前述されるように、本開示の抗原結合分子に含まれるFc領域として、Fcγレセプターに対する結合活性を有するFc領域が挙げられる。そのようなFc領域の非限定な一態様として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域が例示される。Fcγレセプターが、IgG1、IgG2、IgG3、IgG4モノクローナル抗体のFc領域に結合活性を有するか否かは、上記に記載されるFACSやELISAフォーマットのほか、ALPHAスクリーン(Amplified Luminescent Proximity Homogeneous Assay)や表面プラズモン共鳴(SPR)現象を利用したBIACORE法等によって確認され得る(Proc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010)。
ALPHAスクリーンは、ドナーとアクセプターの2つのビーズを使用するALPHAテクノロジーによって下記の原理に基づいて実施される。ドナービーズに結合した分子が、アクセプタービーズに結合した分子と生物学的に相互作用し、2つのビーズが近接した状態の時にのみ、発光シグナルを検出される。レーザーによって励起されたドナービーズ内のフォトセンシタイザーは、周辺の酸素を励起状態の一重項酸素に変換する。一重項酸素はドナービーズ周辺に拡散し、近接しているアクセプタービーズに到達するとビーズ内の化学発光反応を引き起こし、最終的に光が放出される。ドナービーズに結合した分子とアクセプタービーズに結合した分子が相互作用しないときは、ドナービーズの産生する一重項酸素がアクセプタービーズに到達しないため、化学発光反応は起きない。
例えば、ドナービーズにビオチン標識されたFc領域を含む抗原結合分子が結合され、アクセプタービーズにはグルタチオンSトランスフェラーゼ(GST)でタグ化されたFcγレセプターが結合される。競合するFc領域改変体を含む抗原結合分子の非存在下では、天然型Fc領域を有する抗原結合分子とFcγレセプターは相互作用し520-620 nmのシグナルを生ずる。タグ化されていないFc領域改変体を含む抗原結合分子は、天然型Fc領域を有する抗原結合分子とFcγレセプター間の相互作用と競合する。競合の結果表れる蛍光の減少を定量することによって相対的な結合親和性が決定され得る。抗体等の抗原結合分子をSulfo-NHS-ビオチン等を用いてビオチン化することは公知である。FcγレセプターをGSTでタグ化する方法としては、FcγレセプターをコードするポリヌクレオチドとGSTをコードするポリヌクレオチドをインフレームで融合した融合遺伝子が作動可能に連結されたベクターに保持した細胞等において発現し、グルタチオンカラムを用いて精製する方法等が適宜採用され得る。得られたシグナルは例えばGRAPHPAD PRISM(GraphPad社、San Diego)等のソフトウェアを用いて非線形回帰解析を利用する一部位競合(one-site competition)モデルに適合させることにより好適に解析される。
相互作用を観察する物質の一方(リガンド)をセンサーチップの金薄膜上に固定し、センサーチップの裏側から金薄膜とガラスの境界面で全反射するように光を当てると、反射光の一部に反射強度が低下した部分(SPRシグナル)が形成される。相互作用を観察する物質の他方(アナライト)をセンサーチップの表面に流しリガンドとアナライトが結合すると、固定化されているリガンド分子の質量が増加し、センサーチップ表面の溶媒の屈折率が変化する。この屈折率の変化により、SPRシグナルの位置がシフトする(逆に結合が解離するとシグナルの位置は戻る)。Biacoreシステムは上記のシフトする量、すなわちセンサーチップ表面での質量変化を縦軸にとり、質量の時間変化を測定データとして表示する(センサーグラム)。センサーグラムのカーブからカイネティクス:結合速度定数(ka)と解離速度定数(kd)が、当該定数の比からアフィニティー(KD)が求められる。BIACORE法では阻害測定法も好適に用いられる。阻害測定法の例はProc. Natl. Acad. Sci. U.S.A. (2006) 103 (11), 4005-4010において記載されている。
Fcγレセプター(FcγR)結合改変Fc領域
本開示が含むFc領域として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域のほかに、天然型ヒトIgGのFc領域のFcγレセプターに対する結合活性よりもFcγレセプターに対する結合活性が高いFcγR結合改変Fc領域も適宜使用され得る。本明細書において、「天然型ヒトIgGのFc領域」とは、配列番号:5、6、7または8で例示されるヒトIgG1、IgG2、IgG3またはIgG4のFc領域のEUナンバリング297位に結合した糖鎖がフコース含有糖鎖であるFc領域を意味する。そのようなFcγR結合改変Fc領域は、天然型ヒトIgGのFc領域のアミノ酸を改変することによって作製され得る。FcγR結合改変Fc領域のFcγRに対する結合活性が、天然型ヒトIgGのFc領域のFcγRに対する結合活性より高いか否かは、前記の結合活性の項で記載された方法を用いて適宜実施され得る。
本開示が含むFc領域として、ヒトIgG1(配列番号:5)、IgG2(配列番号:6)、IgG3(配列番号:7)、またはIgG4(配列番号:8)で表されるFc領域のほかに、天然型ヒトIgGのFc領域のFcγレセプターに対する結合活性よりもFcγレセプターに対する結合活性が高いFcγR結合改変Fc領域も適宜使用され得る。本明細書において、「天然型ヒトIgGのFc領域」とは、配列番号:5、6、7または8で例示されるヒトIgG1、IgG2、IgG3またはIgG4のFc領域のEUナンバリング297位に結合した糖鎖がフコース含有糖鎖であるFc領域を意味する。そのようなFcγR結合改変Fc領域は、天然型ヒトIgGのFc領域のアミノ酸を改変することによって作製され得る。FcγR結合改変Fc領域のFcγRに対する結合活性が、天然型ヒトIgGのFc領域のFcγRに対する結合活性より高いか否かは、前記の結合活性の項で記載された方法を用いて適宜実施され得る。
本開示において、Fc領域の「アミノ酸の改変」または「アミノ酸改変」とは、出発Fc領域のアミノ酸配列とは異なるアミノ酸配列に改変することを含む。出発Fc領域の修飾改変体がpH中性域においてヒトFcγレセプターに結合することができる限り、いずれのFc領域も出発Fc領域として使用され得る。また、既に改変が加えられたFc領域を出発Fc領域としてさらなる改変が加えられたFc領域も本開示のFc領域として好適に使用され得る。出発Fc領域とは、ポリペプチドそのもの、出発Fc領域を含む組成物、または出発Fc領域をコードするアミノ酸配列を意味し得る。出発Fc領域には、抗体の項で概説された組換えによって産生された公知のFc領域が含まれ得る。出発Fc領域の起源は、限定されないが非ヒト動物の任意の生物またはヒトから取得され得る。好ましくは、任意の生物としては、マウス、ラット、モルモット、ハムスター、アレチネズミ、ネコ、ウサギ、イヌ、ヤギ、ヒツジ、ウシ、ウマ、ラクダ、および非ヒト霊長類から選択される生物が好適に挙げられる。別の態様において、出発Fc領域はまた、カニクイザル、マーモセット、アカゲザル、チンパンジー、またはヒトから取得され得る。好ましくは、出発Fc領域は、ヒトIgG1から取得され得るが、IgGの特定のクラスに限定されるものでもない。このことは、ヒトIgG1、IgG2、IgG3、またはIgG4のFc領域を出発Fc領域として適宜用いることができることを意味する。同様に、本明細書において、前記の任意の生物からのIgGの任意のクラスまたはサブクラスのFc領域を、好ましくは出発Fc領域として用いることができることを意味する。天然に存在するIgGのバリアントまたは操作された型の例は、公知の文献(Curr. Opin. Biotechnol. (2009) 20 (6), 685-91、Curr. Opin. Immunol. (2008) 20 (4), 460-470、Protein Eng. Des. Sel. (2010) 23 (4), 195-202、国際公開WO2009/086320、WO2008/092117、WO2007/041635、およびWO2006/105338)に記載されるがそれらに限定されない。
改変の例としては一以上の変異、例えば、出発Fc領域のアミノ酸とは異なるアミノ酸残基に置換された変異、あるいは出発Fc領域のアミノ酸に対して一以上のアミノ酸残基の挿入または出発Fc領域のアミノ酸から一以上のアミノ酸の欠失等が含まれる。好ましくは、改変後のFc領域のアミノ酸配列には、天然に生じないFc領域の少なくとも部分を含むアミノ酸配列を含む。そのような変種は必然的に出発Fc領域と100%未満の配列同一性または類似性を有する。好ましい実施形態において、変種は出発Fc領域のアミノ酸配列と約75%~100%未満のアミノ酸配列同一性または類似性、より好ましくは約80%~100%未満、より好ましくは約85%~100%未満の、より好ましくは約90%~100%未満、最も好ましくは約95%~100%未満の同一性または類似性のアミノ酸配列を有する。本開示の非限定の一態様において、出発Fc領域および本開示のFcγR結合改変Fc領域の間には少なくとも1つのアミノ酸の差がある。出発Fc領域と本開示のFcγR結合改変Fc領域のアミノ酸の違いは、特に前述のEUナンバリングで特定されるアミノ酸残基の位置の特定されたアミノ酸の違いによっても好適に特定可能である。そのような変種の作製方法は「アミノ酸の改変」の項に例示されている。
本開示の抗原結合分子に含まれる、天然型ヒトIgGのFc領域のFcγレセプターに対する結合活性よりもFcγレセプターに対する結合活性が高いFcγR結合改変Fc領域(FcγR結合改変Fc領域)はいかなる方法によっても取得され得るが、具体的には、出発Fc領域として用いられるヒトIgG型免疫グロブリンのアミノ酸の改変によって当該FcγR結合改変Fc領域が取得され得る。改変のための好ましいIgG型免疫グロブリンのFc領域としては、例えば、配列番号:5、6、7または8で例示されるヒトIgG(IgG1、IgG2、IgG3、またはIgG4、およびそれらの改変体)のFc領域が挙げられる。
他のアミノ酸への改変は、天然型ヒトIgGのFc領域のFcγレセプターに対する結合活性よりもFcγレセプターに対する結合活性が高いかぎり、いかなる位置のアミノ酸も改変され得る。抗原結合分子が、ヒトFc領域としてヒトIgG1のFc領域を含んでいる場合、EUナンバリング297位に結合した糖鎖がフコース含有糖鎖である天然型ヒトIgGのFc領域のFcγレセプターに対する結合活性よりもFcγレセプターに対する結合活性が高い効果をもたらす改変が含まれていることが好ましい。こうしたアミノ酸の改変としては、例えば国際公開WO2007/024249、WO2007/021841、WO2006/031370、WO2000/042072、WO2004/029207、WO2004/099249、WO2006/105338、WO2007/041635、WO2008/092117、WO2005/070963、WO2006/020114、WO2006/116260およびWO2006/023403などにおいて報告されている。
本開示の抗原結合分子に含まれるFcγレセプター結合ドメインとFcγレセプターとの結合活性を測定するpHの条件はpH酸性域乃至pH中性域の条件が適宜使用され得る。本開示の抗原結合分子に含まれるFcγレセプター結合ドメインとFcγレセプターとの結合活性を測定する条件としてのpH酸性域乃至pH中性域とは、通常pH5.8~pH8.0を意味する。好ましくはpH6.0 ~pH7.4の任意のpH値によって示される範囲であり、好ましくはpH6.0、6.1、6.2、6.3、6.4、6.5、6.6、6.7、6.8、6.9、7.0、7.1、7.2、7.3、および7.4から選択され、特に好ましくは癌組織のpHに近いpH6.15~7.4である(Vaupelら(Cancer Res. (1989) 49, 6449-6665))。測定条件に使用される温度として、Fcγレセプター結合ドメインとヒトFcγレセプターとの結合アフィニティーは、10℃~50℃の任意の温度で評価され得る。好ましくは、ヒトFcγレセプター結合ドメインとFcγレセプターとの結合アフィニティーを決定するために、15℃~40℃の温度が使用される。より好ましくは、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、および35℃のいずれか1つのような20℃から35℃までの任意の温度も同様に、Fcγレセプター結合ドメインとFcγレセプターとの結合アフィニティーを決定するために使用される。25℃という温度は本開示の態様の非限定な一例である。
本明細書において、FcγR結合改変Fc領域のFcγレセプターに対する結合活性が天然型Fc領域のFcγレセプターに対する結合活性よりも高いとは、FcγR結合改変Fc領域のFcγRI、FcγRIIa、FcγRIIb、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性が、これらのヒトFcγレセプターに対する天然型Fc領域の結合活性よりも高いことをいう。例えば、上記の解析方法にもとづいて、対照とするヒトIgGの天然型Fc領域を含む抗原結合分子の結合活性に比較してFcγR結合改変Fc領域を含む抗原結合分子の結合活性が、105%以上、好ましくは110%以上、115%以上、120%以上、125%以上、特に好ましくは130%以上、135%以上、140%以上、145%以上、150%以上、155%以上、160%以上、165%以上、170%以上、175%以上、180%以上、185%以上、190%以上、195%以上、2倍以上、2.5倍以上、3倍以上、3.5倍以上、4倍以上、4.5倍以上、5倍以上、7.5倍以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍以上、60倍以上、70倍以上、80倍以上、90倍以上、100倍以上の結合活性を示すことをいう。天然型Fc領域としては、出発Fc領域も使用され得るし、同じサブクラスの抗体の天然型Fc領域も使用され得る。
本開示では、対照とするヒトIgGの天然型Fc領域として、EUナンバリングで表される297位のアミノ酸に結合した糖鎖がフコース含有糖鎖である天然型ヒトIgGのFc領域が好適に用いられる。EUナンバリングで表される297位のアミノ酸に結合した糖鎖がフコース含有糖鎖であるか否かは、既知の手法が用いられ得る(Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Satoh M, Iida S, Shitara K., Expert Opin. Biol. Ther. (2006) 6 (11), 1161-1173)。例えば、下記のような方法によって、天然型ヒトIgGのFc領域に結合した糖鎖がフコース含有糖鎖であるか否かを判定することが可能である。被験天然型ヒトIgGにN-Glycosidase F(Roche diagnostics)を反応させることによって、被験天然型ヒトIgGから糖鎖が遊離される(Weitzhandlerら(J. Pharma. Sciences (1994) 83, 12, 1670-1675)。次に、エタノールを反応させてタンパク質が除かれた反応液(Schenkら(J. Clin. Investigation (2001) 108 (11) 1687-1695)の濃縮乾固物が、2-アミノピリジンによって蛍光標識される(Biggeら(Anal. Biochem. (1995) 230 (2) 229-238)。セルロースカートリッジを用いた固相抽出により脱試薬された、蛍光標識された2-AB化糖鎖が、順相クロマトグラフィによって解析される。検出されるクロマトグラムのピークを観察することによって、ヒトIgGの天然型Fc領域に結合した糖鎖がフコース含有糖鎖であるか否かを判定することが可能である。
対照とする同じサブクラスの抗体の天然型Fc領域を含む抗原結合分子としては、IgGモノクローナル抗体のFc領域を有する抗原結合分子が適宜使用され得る。当該Fc領域の構造は、配列番号:5(データベース登録番号AAC82527.1のN末にA付加)、配列番号6(データベース登録番号AAB59393.1のN末にA付加)、配列番号7(データベース登録番号CAA27268.1)、および配列番号8(データベース登録番号AAB59394.1のN末にA付加)に記載する。また、ある特定のアイソタイプの抗体のFc領域を含む抗原結合分子を被検物質として使用する場合には、当該特定のアイソタイプのIgGモノクローナル抗体のFc領域を有する抗原結合分子を対照として用いることによって、被験Fc領域を含む抗原結合分子によるFcγレセプターに対する結合活性の効果が検証される。上記のようにして、Fcγレセプターに対する結合活性が高いことが検証されたFc領域を含む抗原結合分子が適宜選択される。
選択的なFcγレセプターに対する結合活性を有するFc領域
また、本開示において好適に用いられる、Fcγレセプター結合ドメインの例として、特定のFcγレセプターに対する結合活性がそのほかのFcγレセプターに対する結合活性よりも高い性質を有するFcγレセプター結合ドメイン(選択的なFcγレセプターに対する結合活性を有するFcγレセプター結合ドメイン)もまた好適に挙げられる。抗原結合分子として抗体が(Fcγレセプター結合ドメインとしてFc領域が)用いられる場合には、一分子の抗体は一分子のFcγレセプターとしか結合できないため、一分子の抗原結合分子は抑制型Fcγレセプターに結合した状態で他の活性型FcγRに結合することはできないし、活性型Fcγレセプターに結合した状態で他の活性型Fcγレセプターや抑制型Fcγレセプターに結合することはできない。
また、本開示において好適に用いられる、Fcγレセプター結合ドメインの例として、特定のFcγレセプターに対する結合活性がそのほかのFcγレセプターに対する結合活性よりも高い性質を有するFcγレセプター結合ドメイン(選択的なFcγレセプターに対する結合活性を有するFcγレセプター結合ドメイン)もまた好適に挙げられる。抗原結合分子として抗体が(Fcγレセプター結合ドメインとしてFc領域が)用いられる場合には、一分子の抗体は一分子のFcγレセプターとしか結合できないため、一分子の抗原結合分子は抑制型Fcγレセプターに結合した状態で他の活性型FcγRに結合することはできないし、活性型Fcγレセプターに結合した状態で他の活性型Fcγレセプターや抑制型Fcγレセプターに結合することはできない。
活性型Fcγレセプターに対する結合活性が抑制型Fcγレセプターに対する結合活性よりも高いFc領域
前記したように、活性型Fcγレセプターとしては、FcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64)、FcγRIIaならびにFcγRIIIa(アロタイプV158およびF158を含む)およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)が好適に挙げられる。また、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)が抑制型Fcγレセプターの好適な例として挙げられる。
前記したように、活性型Fcγレセプターとしては、FcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64)、FcγRIIaならびにFcγRIIIa(アロタイプV158およびF158を含む)およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)が好適に挙げられる。また、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)が抑制型Fcγレセプターの好適な例として挙げられる。
本明細書において、特定のFcγレセプターに対する結合活性がそれ以外のFcγレセプターに対する結合活性よりも高い例として、例えば、活性型Fcγレセプターに対する結合活性が抑制型Fcγレセプターに対する結合活性よりも高い場合が挙げられる。この場合、Fc領域のFcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性が、FcγRIIbに対する結合活性よりも高いことをいう。例えば、上記の解析方法にもとづいて、Fc領域を含む抗原結合分子のFcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性が、FcγRIIbに対する結合活性の、105%以上、好ましくは110%以上、120%以上、130%以上、140%以上、特に好ましくは150%以上、160%以上、170%以上、180%以上、190%以上、200%%以上、250%以上、300%以上、350%以上、400%以上、450%以上、500%以上、750%以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍、60倍、70倍、80倍、90倍、100倍以上の結合活性を示すことをいう。活性型Fcγレセプターに対する結合活性が抑制型Fcγレセプターに対する結合活性よりも高いFc領域は、抗原結合ドメインが膜型分子に結合する本開示の抗原結合分子に好適に含まれ得る。こうしたFc領域を含むIgG1抗体は、後述するADCC活性が増強されていることが知られていることから、当該Fc領域を含む抗原結合分子は、本開示の医薬組成物に含まれる抗原結合分子としても有用である。
本開示の非限定な一態様では、活性型Fcγレセプターに対する結合活性が抑制型Fcγレセプターに対する結合活性よりも高い(抑制型Fcγレセプターに対する選択的な結合活性を有する)Fc領域の例として、前述されたEUナンバリングで表される221位、222位、223位、224位、225位、227位、228位、230位、231位、232位、233位、234位、235位、236位、237位、238位、239位、240位、241位、243位、244位、245位、246位、247位、249位、250位、251位、254位、255位、256位、258位、260位、262位、263位、264位、265位、266位、267位、268位、269位、270位、271位、272位、273位、274位、275位、276位、278位、279位、280位、281位、282位、283位、284位、285位、286位、288位、290位、291位、292位、293位、294位、295位、296位、297位、298位、299位、300位、301位、302位、303位、304位、305位、311位、313位、315位、317位、318位、320位、322位、323位、324位、325位、326位、327位、328位、329位、330位、331位、332位、333位、334位、335位、336位、337位、339位、376位、377位、378位、379位、380位、382位、385位、392位、396位、421位、427位、428位、429位、434位、436位および440位の群から選択される少なくとも一つ以上のアミノ酸が天然型Fc領域と異なるアミノ酸に改変されているFc領域が好適に挙げられる。
抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高いFc領域
本明細書において、特定のFcγレセプターに対する結合活性がそれ以外のFcγレセプターに対する結合活性よりも高い例として、例えば、抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高い場合が挙げられる。この場合、Fc領域のFcγRIIbに対する結合活性が、FcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性よりも高いことをいう。例えば、上記の解析方法にもとづいて、Fc領域を含む抗原結合分子のFcγRIIbに対する結合活性が、FcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性の、105%以上、好ましくは110%以上、120%以上、130%以上、140%以上、特に好ましくは150%以上、160%以上、170%以上、180%以上、190%以上、200%%以上、250%以上、300%以上、350%以上、400%以上、450%以上、500%以上、750%以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍、60倍、70倍、80倍、90倍、100倍以上の結合活性を示すことをいう。抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高いFc領域は、抗原結合ドメインが可溶型分子に結合する本開示の抗原結合分子に好適に含まれ得る。
本明細書において、特定のFcγレセプターに対する結合活性がそれ以外のFcγレセプターに対する結合活性よりも高い例として、例えば、抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高い場合が挙げられる。この場合、Fc領域のFcγRIIbに対する結合活性が、FcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性よりも高いことをいう。例えば、上記の解析方法にもとづいて、Fc領域を含む抗原結合分子のFcγRIIbに対する結合活性が、FcγRIa、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性の、105%以上、好ましくは110%以上、120%以上、130%以上、140%以上、特に好ましくは150%以上、160%以上、170%以上、180%以上、190%以上、200%%以上、250%以上、300%以上、350%以上、400%以上、450%以上、500%以上、750%以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍、60倍、70倍、80倍、90倍、100倍以上の結合活性を示すことをいう。抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高いFc領域は、抗原結合ドメインが可溶型分子に結合する本開示の抗原結合分子に好適に含まれ得る。
本開示の非限定な一態様では、抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高い(抑制型Fcγレセプターに対する選択的な結合活性を有する)Fc領域の例として、前記Fc領域のアミノ酸のうちEUナンバリングで表される238または328のアミノ酸が天然型Fc領域と異なるアミノ酸に改変されているFc領域が好適に挙げられる。
また本開示の非限定な一態様では、抑制型Fcγレセプターに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高い(抑制型Fcγレセプターに対する選択的な結合活性を有する)Fc領域の例として、前記Fc領域のEUナンバリングで表されるアミノ酸であってEUナンバリングで表される238のアミノ酸がAsp、または328のアミノ酸がGluのいずれか一つ以上に改変されているFc領域が好適に挙げられる。また、抑制型Fcγレセプターに対する選択的な結合活性を有するFc領域として、US2009/0136485に記載されているFc領域あるいは改変も適宜選択することができる。
また本開示の非限定の一態様では、前記Fc領域のEUナンバリングで表されるアミノ酸であってEUナンバリングで表される238のアミノ酸がAsp、または328のアミノ酸がGluのいずれか一つ以上に改変されているFc領域が好適に挙げられる。
さらに本開示の非限定の一態様では、PCT/JP2012/054624で例示される、EUナンバリングで表される238位のProのAspへの置換、およびEUナンバリングで表される237位のアミノ酸がTrp、EUナンバリングで表される237位のアミノ酸がPhe、EUナンバリングで表される267位のアミノ酸がVal、EUナンバリングで表される267位のアミノ酸がGln、EUナンバリングで表される268位のアミノ酸がAsn、EUナンバリングで表される271位のアミノ酸がGly、EUナンバリングで表される326位のアミノ酸がLeu、EUナンバリングで表される326位のアミノ酸がGln、EUナンバリングで表される326位のアミノ酸がGlu、EUナンバリングで表される326位のアミノ酸がMet、EUナンバリングで表される239位のアミノ酸がAsp、EUナンバリングで表される267位のアミノ酸がAla、EUナンバリングで表される234位のアミノ酸がTrp、EUナンバリングで表される234位のアミノ酸がTyr、EUナンバリングで表される237位のアミノ酸がAla、EUナンバリングで表される237位のアミノ酸がAsp、EUナンバリングで表される237位のアミノ酸がGlu、EUナンバリングで表される237位のアミノ酸がLeu、EUナンバリングで表される237位のアミノ酸がMet、EUナンバリングで表される237位のアミノ酸がTyr、EUナンバリングで表される330位のアミノ酸がLys、EUナンバリングで表される330位のアミノ酸がArg、EUナンバリングで表される233位のアミノ酸がAsp、EUナンバリングで表される268位のアミノ酸がAsp、EUナンバリングで表される268位のアミノ酸がGlu、EUナンバリングで表される326位のアミノ酸がAsp、EUナンバリングで表される326位のアミノ酸がSer、EUナンバリングで表される326位のアミノ酸がThr、EUナンバリングで表される323位のアミノ酸がIle、EUナンバリングで表される323位のアミノ酸がLeu、EUナンバリングで表される323位のアミノ酸がMet、EUナンバリングで表される296位のアミノ酸がAsp、EUナンバリングで表される326位のアミノ酸がAla、EUナンバリングで表される326位のアミノ酸がAsn、EUナンバリングで表される330位のアミノ酸がMet、のいずれか一つ以上に改変されているFc領域が好適に挙げられる。
糖鎖が修飾されたFc領域
本開示が提供する抗原結合分子に含まれるFc領域として、Fc領域に結合した糖鎖の組成がフコース欠損糖鎖を結合したFc領域の割合が高くなるように、またはバイセクティングN-アセチルグルコサミンが付加したFc領域の割合が高くなるように修飾されたFc領域も含まれ得る。抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基を除去すると、FcγRIIIaに対する親和性が増強されることが知られている(非特許文献6)。こうしたFc領域を含むIgG1抗体は、後述するADCC活性が増強されていることが知られていることから、当該Fc領域を含む抗原結合分子は、本開示の医薬組成物に含まれる抗原結合分子としても有用である。抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基が除去された抗体としては、例えば、次のような抗体;
グリコシル化が修飾された抗体(国際公開WO1999/054342等)、
糖鎖に付加するフコースが欠損した抗体(国際公開WO2000/061739、WO2002/031140、WO2006/067913等)、
本開示が提供する抗原結合分子に含まれるFc領域として、Fc領域に結合した糖鎖の組成がフコース欠損糖鎖を結合したFc領域の割合が高くなるように、またはバイセクティングN-アセチルグルコサミンが付加したFc領域の割合が高くなるように修飾されたFc領域も含まれ得る。抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基を除去すると、FcγRIIIaに対する親和性が増強されることが知られている(非特許文献6)。こうしたFc領域を含むIgG1抗体は、後述するADCC活性が増強されていることが知られていることから、当該Fc領域を含む抗原結合分子は、本開示の医薬組成物に含まれる抗原結合分子としても有用である。抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基が除去された抗体としては、例えば、次のような抗体;
グリコシル化が修飾された抗体(国際公開WO1999/054342等)、
糖鎖に付加するフコースが欠損した抗体(国際公開WO2000/061739、WO2002/031140、WO2006/067913等)、
より具体的には、抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基が除去された抗体の異なる非限定の一態様として、糖鎖に付加するフコースが欠損した抗体(国際公開WO2000/061739、WO2002/031140、WO2006/067913等)を作製するために、糖鎖修飾を受けるポリペプチドの糖鎖構造を形成する活性が改変された結果、糖鎖にフコースを付加する能力が低い宿主細胞が作製される。当該宿主細胞において所望の抗体遺伝子を発現することによって、当該宿主細胞の培養液からその糖鎖中のフコースが欠損した当該抗体が回収され得る。ポリペプチドの糖鎖構造を形成する活性として、フコシルトランスフェラーゼ(EC 2.4.1.152)、フコーストランスポーター(SLC35C1)、GMD(GDP-マンノース4,6-デヒドラターゼ)(EC 4.2.1.47)、Fx(GDP-ケト-6-デオキシマンノース3,5-エピメラーゼ,4-レダクターゼ)(EC 1.1.1.271)、およびGFPP(GDP-β-L-フコースピロフォスフォリラーゼ)(EC 2.7.7.30)からなる群から選択される酵素またはトランスポーターの活性が非限定の好適な例として挙げられ得る。これらの酵素またはトランスポーターは、その活性を発揮することができれば必ずしもその構造は特定されない。本明細書においては、これらの活性を発揮することが可能なタンパク質を機能性タンパク質という。これらの活性を改変する方法の非限定の一態様として、これらの活性の欠失が挙げられる。これらの活性が欠失した宿主細胞を作製するために、これらの機能性タンパク質の遺伝子を機能不能に破壊する方法等公知の方法が適宜採用され得る(国際公開WO2000/061739、WO2002/031140、WO2006/067913等)。そのような活性が欠失した宿主細胞は、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞、YO骨髄腫細胞、P3X63マウス骨髄腫細胞、PER細胞、PER.C6細胞、HEK293細胞、またはハイブリドーマ細胞等に内在性であるこれらの機能性タンパク質の遺伝子を機能不能に破壊する方法等によって作製され得る。
バイセクティングGlcNAcを有する糖鎖を有する抗体(国際公開WO2002/079255等)が公知である。非限定な一態様では、バイセクティングGlcNAcを有する糖鎖を有する抗体を作製するために、GnTIII(β-1,4-マンノシル-グリコプロテイン,4-β-N-アセチルグルコサミニルトランスフェラーゼ)(EC 2.4.1.144)活性またはGalT(β-1,4-ガラクトシルトランスフェラーゼ)(EC 2.4.1.38)活性を有する機能性タンパク質をコードする遺伝子を発現する宿主細胞が作製される。別の非限定の好適な一態様では、前記の機能性タンパク質に加えて、ヒトManII(マンノシダーゼII)(3.2.1.114)活性を有する機能性タンパク質をコードする遺伝子、GnTI(β-1,2-アセチルグルコサミニルトランスフェラーゼI)(EC 2.4.1.94)活性を有する機能性タンパク質をコードする遺伝子、GnTII(β-1,2-アセチルグルコサミニルトランスフェラーゼII)(EC 2.4.1.143)活性を有する機能性タンパク質をコードする遺伝子、ManI(マンノシダーゼ)(EC 3.2.1.113)活性を有する機能性タンパク質をコードする遺伝子、およびα-1,6-フコシルトランスフェラーゼ(EC 2.4.1.68)と共発現する宿主細胞が作製される(国際公開WO2004/065540)。
前記のような糖鎖にフコースを付加する能力が低い宿主細胞、およびバイセクティングGlcNAc構造を含む糖鎖を形成する活性を有する宿主細胞に抗体遺伝子を含む発現ベクターを形質導入することによって、抗体Fc領域に結合するN -グリコシド結合複合型糖鎖還元末端のN -アセチルグルコサミンからフコース残基が除去された抗体、およびバイセクティングGlcNAcを有する糖鎖を有する抗体がそれぞれ作製され得る。これらの抗体の製造方法は本開示のFc領域に結合した糖鎖の組成がフコース欠損糖鎖を結合したFc領域の割合が高くなるように、またはバイセクティングN-アセチルグルコサミンが付加したFc領域の割合が高くなるように修飾された改変Fc領域を含む抗原結合分子の製造方法にも適用することが可能である。こうした製造方法によって作製された本開示の抗原結合分子に含まれるFc領域に結合した糖鎖の組成は、前記の「Fcγレセプター(FcγR)結合改変Fc領域」で記載された方法によって確認され得る。
二分子のFcRnおよび一分子の活性型Fcγレセプターの四者を含むヘテロ複合体
FcRnとIgG抗体との結晶学的研究によって、FcRn-IgG複合体は、二分子のFcRnに対して一分子のIgGから構成され、IgGのFc領域の両側に位置するCH2およびCH3ドメインの接触面付近において、二分子の結合が起こると考えられている(Burmeisterら(Nature (1994) 372, 336-343)。一方、PCT/JP2012/058603の実施例3において確認されたように、抗体のFc領域が二分子のFcRnおよび一分子の活性型Fcγレセプターの四者を含む複合体を形成できることが明らかとなった(PCT/JP2012/058603)。このヘテロ複合体の形成は、pH中性域の条件下でFcRnに対する結合活性を有するFc領域を含む抗原結合分子の性質について解析を進めた結果明らかとなった現象である。
FcRnとIgG抗体との結晶学的研究によって、FcRn-IgG複合体は、二分子のFcRnに対して一分子のIgGから構成され、IgGのFc領域の両側に位置するCH2およびCH3ドメインの接触面付近において、二分子の結合が起こると考えられている(Burmeisterら(Nature (1994) 372, 336-343)。一方、PCT/JP2012/058603の実施例3において確認されたように、抗体のFc領域が二分子のFcRnおよび一分子の活性型Fcγレセプターの四者を含む複合体を形成できることが明らかとなった(PCT/JP2012/058603)。このヘテロ複合体の形成は、pH中性域の条件下でFcRnに対する結合活性を有するFc領域を含む抗原結合分子の性質について解析を進めた結果明らかとなった現象である。
本開示は特定の理論に拘束されるわけではないが、抗原結合分子に含まれるFc領域と二分子のFcRnおよび一分子の活性型Fcγレセプターの四者を含むヘテロ複合体の形成によって、抗原結合分子が生体内に投与されたときの抗原結合分子の当該生体内における薬物動態(血漿中滞留性)、および、投与された抗原結合分子に対する免疫応答(免疫原性)に対して以下のような影響がもたらされることも考えられる。免疫細胞上には各種活性型Fcγレセプターに加えFcRnが発現しており、抗原結合分子が免疫細胞上でこのような四者複合体を形成することは、免疫細胞に対する親和性を向上させ、さらに細胞内ドメインを会合化させることにより内在化シグナルを増強させ、免疫細胞への取り込みが促進されることが示唆される。抗原提示細胞においても同様であり、抗原提示細胞の細胞膜上で四者複合体を形成することにより、抗原結合分子が抗原提示細胞へ取り込まれやすくなる可能性が示唆される。一般的に、抗原提示細胞に取り込まれた抗原結合分子は、抗原提示細胞内のリソソームにおいて分解され、T細胞へと提示される。結果として、抗原提示細胞の細胞膜上で上記の四者複合体を形成することにより、抗原結合分子に対する抗原提示細胞への取り込みが促進されことによって抗原結合分子の血漿中滞留性が悪化する可能性もある。また、同様にして、免疫応答が誘起される(増悪する)可能性がある。
そのため、このような四者複合体を形成する能力が低下した抗原結合分子が生体に投与された場合、当該抗原結合分子の血漿中滞留性が向上し、当該生体による免疫応答の誘起が抑制されると考えられ得る。このような抗原提示細胞を含む免疫細胞上における当該複合体の形成を阻害する抗原結合分子の好ましい様態として、以下の三種類が挙げられ得る。
ヘテロ複合体の形成を阻害する抗原結合分子
(様態1) pH中性域の条件下でのFcRnに対する結合活性を有し、活性型FcγRに対する結合活性が天然型Fc領域の活性型FcγRに対する結合活性より低いFc領域を含む抗原結合分子
(様態1) pH中性域の条件下でのFcRnに対する結合活性を有し、活性型FcγRに対する結合活性が天然型Fc領域の活性型FcγRに対する結合活性より低いFc領域を含む抗原結合分子
様態1の抗原結合分子は、二分子のFcRnに結合することによって三者複合体を形成するが、活性型FcγRを含めた複合体は形成しない。活性型FcγRに対する結合活性が天然型Fc領域の活性型FcγRに対する結合活性より低いFc領域は、前記のように天然型Fc領域のアミノ酸を改変することによって作製され得る。改変Fc領域の活性型FcγRに対する結合活性が、天然型Fc領域の活性型FcγRに対する結合活性より低いか否かは、前記の結合活性の項で記載された方法を用いて適宜実施され得る。
活性型Fcγレセプターとしては、FcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64)、FcγRIIa(アロタイプR131およびH131を含む)ならびにアイソフォームFcγRIIIa(アロタイプV158およびF158を含む)およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)が好適に挙げられる。
本明細書において、Fc領域改変体の活性型Fcγレセプターに対する結合活性が天然型Fc領域の活性型Fcγレセプターに対する結合活性よりも低いとは、Fc領域改変体のFcγRI、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性が、これらのヒトFcγレセプターに対する天然型Fc領域の結合活性よりも低いことをいう。例えば、上記の解析方法にもとづいて、対照とする天然型Fc領域を含む抗原結合分子の結合活性に比較してFc領域改変体を含む抗原結合分子の結合活性が、95%以下、好ましくは90%以下、85%以下、80%以下、75%以下、特に好ましくは70%以下、65%以下、60%以下、55%以下、50%以下、45%以下、40%以下、35%以下、30%以下、25%以下、20%以下、15%以下、10%以下、9%以下、8%以下、7%以下、6%以下、5%以下、4%以下、3%以下、2%以下、1%以下の結合活性を示すことをいう。天然型Fc領域としては、出発Fc領域も使用され得るし、野生型抗体の異なるアイソタイプのFc領域も使用され得る。
また、天然型の活性型FcγRに対する結合活性とは、ヒトIgG1のFcγレセプターに対する結合活性であることが好ましく、Fcγレセプターに対する結合活性を低減させるには上記改変以外にも、ヒトIgG2、ヒトIgG3、ヒトIgG4にアイソタイプを変更することでも達成し得る。また、Fcγレセプターに対する結合活性を低下させるのは上記改変以外にも、Fcγレセプターに対する結合活性を有するFc領域を含む抗原結合分子を大腸菌等の糖鎖を付加しない宿主で発現させることによっても得ることができる。
対照とするFc領域を含む抗原結合分子としては、IgGモノクローナル抗体のFc領域を有する抗原結合分子が適宜使用され得る。当該Fc領域の構造は、配列番号:5(RefSeq登録番号AAC82527.1のN末にA付加)、配列番号:6(RefSeq登録番号AAB59393.1のN末にA付加)、配列番号:7(RefSeq登録番号CAA27268.1)、配列番号:8(RefSeq登録番号AAB59394.1のN末にA付加)に記載されている。また、ある特定のアイソタイプの抗体のFc領域を含む抗原結合分子を被検物質として使用する場合には、当該特定のアイソタイプのIgGモノクローナル抗体のFc領域を有する抗原結合分子を対照として用いることによって、当該Fc領域を含む抗原結合分子によるFcγレセプターに対する結合活性の効果が検証される。上記のようにして、Fcγレセプターに対する結合活性が高いことが検証されたFc領域を含む抗原結合分子が適宜選択される。
本開示の非限定の一態様では、活性型FcγRに対する結合活性が天然型Fc領域の活性型FcγRに対する結合活性より低いFc領域の例として、前記Fc領域のアミノ酸のうちEUナンバリングで表される234、235、236、237、238、239、270、297、298、325、328、および329のいずれか一つ以上のアミノ酸が天然型Fc領域と異なるアミノ酸に改変されているFc領域が好適に挙げられるが、Fc領域の改変は上記改変に限定されず、例えばCur. Opin. in Biotech. (2009) 20 (6), 685-691に記載されている脱糖鎖(N297A, N297Q)、IgG1-L234A/L235A、IgG1-A325A/A330S/P331S、IgG1-C226S/C229S、IgG1-C226S/C229S/E233P/L234V/L235A、IgG1-L234F/L235E/P331S、IgG1-S267E/L328F、IgG2-V234A/G237A、IgG2-H268Q/V309L/A330S/A331S、IgG4-L235A/G237A/E318A、IgG4-L236E等の改変、および、国際公開WO2008/092117に記載されているG236R/L328R、L235G/G236R、N325A/L328R、N325L/L328R等の改変、および、EUナンバリング233位、234位、235位、237位におけるアミノ酸の挿入、国際公開WO2000/042072に記載されている個所の改変であってもよい。
また本開示の非限定の一態様では、前記Fc領域のEUナンバリングで表されるアミノ酸であって;
234位のアミノ酸をAla、Arg、Asn、Asp、Gln、Glu、Gly、His、Lys、Met、Phe、Pro、Ser、ThrまたはTrpのいずれか、
235位のアミノ酸をAla、Asn、Asp、Gln、Glu、Gly、His、Ile、Lys、Met、Pro、Ser、Thr、ValまたはArgのいずれか、
236位のアミノ酸をArg、Asn、Gln、His、Leu、Lys、Met、Phe、ProまたはTyrのいずれか、
237位のアミノ酸をAla、Asn、Asp、Gln、Glu、His、Ile、Leu、Lys、Met、Pro、Ser、Thr、Val、TyrまたはArgのいずれか、
238位のアミノ酸をAla、Asn、Gln、Glu、Gly、His、Ile、Lys、Thr、TrpまたはArgのいずれか、
239位のアミノ酸をGln、His、Lys、Phe、Pro、Trp、TyrまたはArgのいずれか、
265位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Ser、Thr、Trp、TyrまたはValのいずれか、
266位のアミノ酸をAla、Arg、Asn、Asp、Gln、Glu、Gly、His、Lys、Phe、Pro、Ser、Thr、TrpまたはTyrのいずれか、
267位のアミノ酸をArg、His、Lys、Phe、Pro、TrpまたはTyrのいずれか、
269位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
270位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
271位のアミノ酸をArg、His、Phe、Ser、Thr、TrpまたはTyrのいずれか、
295位のアミノ酸をArg、Asn、Asp、Gly、His、Phe、Ser、TrpまたはTyrのいずれか、
296位のアミノ酸をArg、Gly、LysまたはProのいずれか、
297位のアミノ酸をAla、
298位のアミノ酸をArg、Gly、Lys、Pro、TrpまたはTyrのいずれか、
300位のアミノ酸をArg、LysまたはProのいずれか、
324位のアミノ酸をLysまたはProのいずれか、
325位のアミノ酸をAla、Arg、Gly、His、Ile、Lys、Phe、Pro、Thr、TrpTyr、もしくはValのいずれか、
327位のアミノ酸をArg、Gln、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
328位のアミノ酸をArg、Asn、Gly、His、LysまたはProのいずれか、
329位のアミノ酸をAsn、Asp、Gln、Glu、Gly、His、Ile、Leu、Lys、Met、Phe、Ser、Thr、Trp、Tyr、ValまたはArgのいずれか、
330位のアミノ酸をProまたはSerのいずれか、
331位のアミノ酸をArg、GlyまたはLysのいずれか、もしくは
332位のアミノ酸をArg、LysまたはProのいずれか、
のいずれか一つ以上に改変されているFc領域が好適に挙げられる。
234位のアミノ酸をAla、Arg、Asn、Asp、Gln、Glu、Gly、His、Lys、Met、Phe、Pro、Ser、ThrまたはTrpのいずれか、
235位のアミノ酸をAla、Asn、Asp、Gln、Glu、Gly、His、Ile、Lys、Met、Pro、Ser、Thr、ValまたはArgのいずれか、
236位のアミノ酸をArg、Asn、Gln、His、Leu、Lys、Met、Phe、ProまたはTyrのいずれか、
237位のアミノ酸をAla、Asn、Asp、Gln、Glu、His、Ile、Leu、Lys、Met、Pro、Ser、Thr、Val、TyrまたはArgのいずれか、
238位のアミノ酸をAla、Asn、Gln、Glu、Gly、His、Ile、Lys、Thr、TrpまたはArgのいずれか、
239位のアミノ酸をGln、His、Lys、Phe、Pro、Trp、TyrまたはArgのいずれか、
265位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Ser、Thr、Trp、TyrまたはValのいずれか、
266位のアミノ酸をAla、Arg、Asn、Asp、Gln、Glu、Gly、His、Lys、Phe、Pro、Ser、Thr、TrpまたはTyrのいずれか、
267位のアミノ酸をArg、His、Lys、Phe、Pro、TrpまたはTyrのいずれか、
269位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
270位のアミノ酸をAla、Arg、Asn、Gln、Gly、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
271位のアミノ酸をArg、His、Phe、Ser、Thr、TrpまたはTyrのいずれか、
295位のアミノ酸をArg、Asn、Asp、Gly、His、Phe、Ser、TrpまたはTyrのいずれか、
296位のアミノ酸をArg、Gly、LysまたはProのいずれか、
297位のアミノ酸をAla、
298位のアミノ酸をArg、Gly、Lys、Pro、TrpまたはTyrのいずれか、
300位のアミノ酸をArg、LysまたはProのいずれか、
324位のアミノ酸をLysまたはProのいずれか、
325位のアミノ酸をAla、Arg、Gly、His、Ile、Lys、Phe、Pro、Thr、TrpTyr、もしくはValのいずれか、
327位のアミノ酸をArg、Gln、His、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、TyrまたはValのいずれか、
328位のアミノ酸をArg、Asn、Gly、His、LysまたはProのいずれか、
329位のアミノ酸をAsn、Asp、Gln、Glu、Gly、His、Ile、Leu、Lys、Met、Phe、Ser、Thr、Trp、Tyr、ValまたはArgのいずれか、
330位のアミノ酸をProまたはSerのいずれか、
331位のアミノ酸をArg、GlyまたはLysのいずれか、もしくは
332位のアミノ酸をArg、LysまたはProのいずれか、
のいずれか一つ以上に改変されているFc領域が好適に挙げられる。
(様態2) pH中性域の条件下でのFcRnに対する結合活性を有し、抑制型FcγRに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高いFc領域を含む抗原結合分子
様態2の抗原結合分子は、二分子のFcRnと一分子の抑制型FcγRに結合することによってこれら四者を含む複合体を形成し得る。しかしながら、一分子の抗原結合分子は一分子のFcγRとしか結合できないため、一分子の抗原結合分子は抑制型FcγRに結合した状態で他の活性型FcγRに結合することはできない。さらに、抑制型FcγRに結合した状態で細胞内へと取り込まれた抗原結合分子は、細胞膜上へとリサイクルされ、細胞内での分解を回避することが報告されている(Immunity (2005) 23, 503-514)。すなわち、抑制型FcγRに対する選択的結合活性を有する抗原結合分子は、免疫応答の原因となる活性型FcγRおよび二分子のFcRnを含めたヘテロ複合体を形成することができないと考えられる。
活性型Fcγレセプターとしては、FcγRIa、FcγRIbおよびFcγRIcを含むFcγRI(CD64)、FcγRIIa(アロタイプR131およびH131を含む)ならびにアイソフォームFcγRIIIa(アロタイプV158およびF158を含む)およびFcγRIIIb(アロタイプFcγRIIIb-NA1およびFcγRIIIb-NA2を含む)を含むFcγRIII(CD16)が好適に挙げられる。また、FcγRIIb(FcγRIIb-1およびFcγRIIb-2を含む)が抑制型Fcγレセプターの好適な例として挙げられる。
本明細書において、抑制型FcγRに対する結合活性が活性型Fcγレセプターに対する結合活性よりも高いとは、Fc領域改変体のFcγRIIbに対する結合活性が、FcγRI、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性よりも高いことをいう。例えば、上記の解析方法にもとづいて、Fc領域改変体を含む抗原結合分子のFcγRIIbに対する結合活性が、FcγRI、FcγRIIa、FcγRIIIa及び/又はFcγRIIIbのいずれかのヒトFcγレセプターに対する結合活性の、105%以上、好ましくは110%以上、120%以上、130%以上、140%以上、特に好ましくは150%以上、160%以上、170%以上、180%以上、190%以上、200%%以上、250%以上、300%以上、350%以上、400%以上、450%以上、500%以上、750%以上、10倍以上、20倍以上、30倍以上、40倍以上、50倍以上の結合活性を示すことをいう。
FcγRIIbに対する結合活性が、FcγRIa、FcγRIIa(アロタイプR131およびH131を含む)およびFcγRIIIa(アロタイプV158およびF158を含む)よりもすべて高いことがもっとも好ましい。FcγRIa は天然型IgG1に対するアフィニティーが極めて高いことから、生体内においては大量の内因性IgG1によって結合が飽和されていると考えられるため、FcγRIIbに対する結合活性はFcγRIIaおよびFcγRIIIaよりも高く、FcγRIaよりは低くても当該複合体の形成阻害は可能であると考えられる。
対照とするFc領域を含む抗原結合分子としては、IgGモノクローナル抗体のFc領域を有する抗原結合分子が適宜使用され得る。当該Fc領域の構造は、配列番号:5(RefSeq登録番号AAC82527.1のN末にA付加)、配列番号:6(RefSeq登録番号AAB59393.1のN末にA付加)、配列番号:7(RefSeq登録番号CAA27268.1)、配列番号:8(RefSeq登録番号AAB59394.1のN末にA付加)に記載されている。また、ある特定のアイソタイプの抗体のFc領域を含む抗原結合分子を被検物質として使用する場合には、当該特定のアイソタイプのIgGモノクローナル抗体のFc領域を有する抗原結合分子を対照として用いることによって、当該Fc領域を含む抗原結合分子によるFcγレセプターに対する結合活性の効果が検証される。上記のようにして、Fcγレセプターに対する結合活性が高いことが検証されたFc領域を含む抗原結合分子が適宜選択される。
本開示の非限定の一態様では、抑制型FcγRに対する選択的な結合活性を有するFc領域の例として、前記Fc領域のアミノ酸のうちEUナンバリングで表される238または328のアミノ酸が天然型Fc領域と異なるアミノ酸に改変されているFc領域が好適に挙げられる。また、抑制型Fcγレセプターに対する選択的な結合活性を有するFc領域として、US2009/0136485に記載されているFc領域あるいは改変も適宜選択することができる。
また本開示の非限定の一態様では、前記Fc領域のEUナンバリングで表されるアミノ酸であってEUナンバリングで表される238のアミノ酸がAsp、または328のアミノ酸がGluのいずれか一つ以上に改変されているFc領域が好適に挙げられる。
さらに本開示の非限定の一態様では、EUナンバリングで表される238位のProのAspへの置換、およびEUナンバリングで表される237位のアミノ酸がTrp、EUナンバリングで表される237位のアミノ酸がPhe、EUナンバリングで表される267位のアミノ酸がVal、EUナンバリングで表される267位のアミノ酸がGln、EUナンバリングで表される268位のアミノ酸がAsn、EUナンバリングで表される271位のアミノ酸がGly、EUナンバリングで表される326位のアミノ酸がLeu、EUナンバリングで表される326位のアミノ酸がGln、EUナンバリングで表される326位のアミノ酸がGlu、EUナンバリングで表される326位のアミノ酸がMet、EUナンバリングで表される239位のアミノ酸がAsp、EUナンバリングで表される267位のアミノ酸がAla、EUナンバリングで表される234位のアミノ酸がTrp、EUナンバリングで表される234位のアミノ酸がTyr、EUナンバリングで表される237位のアミノ酸がAla、EUナンバリングで表される237位のアミノ酸がAsp、EUナンバリングで表される237位のアミノ酸がGlu、EUナンバリングで表される237位のアミノ酸がLeu、EUナンバリングで表される237位のアミノ酸がMet、EUナンバリングで表される237位のアミノ酸がTyr、EUナンバリングで表される330位のアミノ酸がLys、EUナンバリングで表される330位のアミノ酸がArg、EUナンバリングで表される233位のアミノ酸がAsp、EUナンバリングで表される268位のアミノ酸がAsp、EUナンバリングで表される268位のアミノ酸がGlu、EUナンバリングで表される326位のアミノ酸がAsp、EUナンバリングで表される326位のアミノ酸がSer、EUナンバリングで表される326位のアミノ酸がThr、EUナンバリングで表される323位のアミノ酸がIle、EUナンバリングで表される323位のアミノ酸がLeu、EUナンバリングで表される323位のアミノ酸がMet、EUナンバリングで表される296位のアミノ酸がAsp、EUナンバリングで表される326位のアミノ酸がAla、EUナンバリングで表される326位のアミノ酸がAsn、EUナンバリングで表される330位のアミノ酸がMet、のいずれか一つ以上に改変されているFc領域が好適に挙げられる。
(様態3) Fc領域を構成する二つのポリペプチドの一方がpH中性域の条件下でのFcRnに対する結合活性を有し、他方がpH中性域の条件下でのFcRnに対する結合能活性を有しないFc領域を含む抗原結合分子
様態3の抗原結合分子は、一分子のFcRnと一分子のFcγRに結合することによって三者複合体を形成しうるが、二分子のFcRnと一分子のFcγRの四者を含むヘテロ複合体は形成しない。本様態3の抗原結合分子に含まれる、Fc領域を構成する二つのポリペプチドの一方がpH中性域の条件下でのFcRnに対する結合活性を有し、他方のポリペプチドがpH中性域の条件下でのFcRnに対する結合能活性を有しないFc領域として、二重特異性抗体(bispecific抗体)を起源とするFc領域も適宜使用され得る。二重特異性抗体とは、異なる抗原に対して特異性を有する二種類の抗体である。IgG型の二重特異性抗体はIgG抗体を産生するハイブリドーマ二種を融合することによって生じるhybrid hybridoma(quadroma)によって分泌させることが可能である(Milsteinら(Nature (1983) 305, 537-540)。
上記の様態3の抗原結合分子を前記の抗体の項で記載されたような組換え手法を用いて製造する場合、目的の二種のFc領域を構成するポリペプチドをコードする遺伝子を細胞に導入しそれらを共発現させる方法が採用され得る。しかしながら、製造されるFc領域は、Fc領域を構成する二つのポリペプチドの一方がpH中性域の条件下でのFcRnに対する結合活性を有し、他方のポリペプチドがpH中性域の条件下でのFcRnに対する結合能活性を有しないFc領域と、Fc領域を構成する二つのポリペプチドの双方がpH中性域の条件下でのFcRnに対する結合活性を有するFc領域と、Fc領域を構成する二つのポリペプチドの双方がpH中性域の条件下でのFcRnに対する結合活性を有しないFc領域が、2:1:1の分子数の割合で存在する混合物となる。3種類のIgGから目的の組合せのFc領域を含む抗原結合分子を精製することは困難である。
こうした組換え手法を用いて様態3の抗原結合分子を製造する際に、Fc領域を構成するCH3ドメインに適当なアミノ酸置換の改変を加えることによってヘテロな組合せのFc領域を含む抗原結合分子が優先的に分泌され得る。具体的には、一方の重鎖のCH3ドメインに存在するアミノ酸側鎖をより大きい側鎖(knob(「突起」の意))に置換し、もう一方の重鎖のCH3ドメインに存在するアミノ酸側鎖をより小さい側鎖(hole(「空隙」の意))に置換することによって、突起が空隙内に配置され得るようにして異種H鎖形成の促進および同種H鎖形成の阻害を引き起こす方法である(国際公開WO1996027011、Ridgwayら(Protein Engineering (1996) 9, 617-621)、Merchantら(Nat. Biotech. (1998) 16, 677-681))。
また、ポリペプチドの会合、またはポリペプチドによって構成される異種多量体の会合の制御方法を、Fc領域を構成する二つのポリペプチドの会合に利用することによって二重特異性抗体を作製する技術も知られている。即ち、Fc領域を構成する二つのポリペプチド内の界面を形成するアミノ酸残基を改変することによって、同一配列を有するFc領域を構成するポリペプチドの会合が阻害され、配列の異なる二つのFc領域を構成するポリペプチド会合体が形成されるように制御する方法が二重特異性抗体の作製に採用され得る(国際公開WO2006/106905)。具体的には前記の二重特異性抗体とその作製方法の項で記載された方法が本開示の様態3の抗原結合分子を製造する際に、非限定な一態様として採用され得る。
これら様態1~3の抗原結合分子は、四者複合体を形成しうる抗原結合分子に比較して、いずれも免疫原性を低下させ、また血漿中滞留性を向上させることが可能であると期待される。
FcRn
免疫グロブリンスーパーファミリーに属するFcγレセプターと異なり、ヒトFcRnは構造的には主要組織不適合性複合体(MHC)クラスIのポリペプチドに構造的に類似しクラスIのMHC分子と22から29%の配列同一性を有する(Ghetieら,Immunol. Today (1997) 18 (12), 592-598)。FcRnは、可溶性βまたは軽鎖(β2マイクログロブリン)と複合体化された膜貫通αまたは重鎖よりなるヘテロダイマーとして発現される。MHCのように、FcRnのα鎖は3つの細胞外ドメイン(α1、α2、α3)よりなり、短い細胞質ドメインはタンパク質を細胞表面に繋留する。α1およびα2ドメインが抗体のFc領域中のFcRn結合ドメインと相互作用する(Raghavanら(Immunity (1994) 1, 303-315)。
免疫グロブリンスーパーファミリーに属するFcγレセプターと異なり、ヒトFcRnは構造的には主要組織不適合性複合体(MHC)クラスIのポリペプチドに構造的に類似しクラスIのMHC分子と22から29%の配列同一性を有する(Ghetieら,Immunol. Today (1997) 18 (12), 592-598)。FcRnは、可溶性βまたは軽鎖(β2マイクログロブリン)と複合体化された膜貫通αまたは重鎖よりなるヘテロダイマーとして発現される。MHCのように、FcRnのα鎖は3つの細胞外ドメイン(α1、α2、α3)よりなり、短い細胞質ドメインはタンパク質を細胞表面に繋留する。α1およびα2ドメインが抗体のFc領域中のFcRn結合ドメインと相互作用する(Raghavanら(Immunity (1994) 1, 303-315)。
FcRnは、哺乳動物の母性胎盤または卵黄嚢で発現され、それは母親から胎児へのIgGの移動に関与する。加えてFcRnが発現するげっ歯類新生児の小腸では、FcRnが摂取された初乳または乳から母性IgGの刷子縁上皮を横切る移動に関与する。FcRnは多数の種にわたって多数の他の組織、並びに種々の内皮細胞系において発現している。それはヒト成人血管内皮、筋肉血管系、および肝臓洞様毛細血管でも発現される。FcRnは、IgGに結合し、それを血清にリサイクルすることによって、IgGの血漿中濃度を維持する役割を演じていると考えられている。FcRnのIgG分子への結合は、通常、厳格にpHに依存的であり、最適結合は7.0未満のpH酸性域において認められる。
配列番号:28で表されたシグナル配列を含むポリペプチドを前駆体とするヒトFcRnは、生体内で(配列番号:29にシグナル配列を含むそのポリペプチドが記載されている)ヒトβ2-ミクログロブリンとの複合体を形成する。β2-ミクログロブリンと複合体を形成している可溶型ヒトFcRnが通常の組換え発現手法を用いることによって製造される。このようなβ2-ミクログロブリンと複合体を形成している可溶型ヒトFcRnに対する本開示のFc領域の結合活性が評価され得る。本開示において、特に記載のない場合は、ヒトFcRnは本開示のFc領域に結合し得る形態であるものを指し、例としてヒトFcRnとヒトβ2-ミクログロブリンとの複合体が挙げられる。
本開示と定常領域改変技術の組み合わせの態様として、酸性pHでのFcRn結合増強改変Fc技術(WO2002060919、WO2004035752、WO2000042072)、中性pHでのFcRn結合増強改変Fc技術(WO2011122011、WO2012133782)、抑制型Fcγ受容体選択的結合増強技術(WO2012115241、WO2013125667)、活性型Fcγ受容体選択的結合増強技術(ADCC活性増強技術)(WO2013002362)、Rheumatoid factorへの結合活性を低下させる技術(WO2013046704) 等の抗体改変技術との組み合わせが挙げられる。
本開示と可変領域改変技術との非限定の組み合わせの態様としては、pH依存性抗体(WO2009125825)、カルシウム依存性抗体(WO2012073992)等の改変技術との組み合わせが挙げられる。
本開示における抗原結合分子はMTAと相互作用するアミノ酸残基を有し得る。MTAと相互作用するアミノ酸残基は、抗原結合分子中の抗原結合ドメインに存在しても良いし、抗原結合ドメイン以外の部位に存在しても良い。またMTAと相互作用するアミノ酸残基は、抗原結合分子中に1残基のみでもよく、複数存在してもよい。
MTAと相互作用するアミノ酸残基が位置する部位として、抗原結合ドメインが例示される。さらに、MTAと相互作用するアミノ酸残基を含む抗原結合分子の非限定な一態様としては抗体が例示される。MTAと相互作用するアミノ酸残基は抗体の定常領域および/または可変領域に存在し得るし、さらに抗体可変領域中のCDR、FRに存在し得るがいずれかに限定されるものではない。
MTAと相互作用するアミノ酸残基が位置する部位として、抗原結合分子の抗原結合ドメインとは異なる部分が例示される。抗原結合分子の抗原結合ドメインとは異なる部分のアミノ酸残基がMTAと相互作用することにより抗原結合ドメインの構造が変化し、間接的にMTA依存的に抗原結合活性が変化し得る。
MTAと相互作用するアミノ酸残基が位置する部位として、抗原結合分子の抗原結合ドメインとは異なる部分が例示される。抗原結合分子の抗原結合ドメインとは異なる部分のアミノ酸残基がMTAと相互作用することにより抗原結合ドメインの構造が変化し、間接的にMTA依存的に抗原結合活性が変化し得る。
抗原結合分子中のMTAと相互作用するアミノ酸残基は、MTAと抗原結合分子二者の複合体またはMTA、抗原、抗原結合分子の三者複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定することができる。
本開示におけるMTA依存的に抗原に対する結合活性の変化する抗原結合ドメインを含む抗原結合分子の非限定な一態様として抗体が例示される。
本開示において、抗原結合分子が2つ以上の複数の抗原結合ドメインを有し、さらに複数の異なる抗原に対して同時に結合し得る。
非限定的な一態様として、本開示の抗原結合分子は更に第2の抗原結合ドメインを有し、前記第2の抗原結合ドメインは前記抗原結合ドメインが結合する抗原と異なる第2の抗原に対して結合活性を有する。
抗原結合分子が異なる複数の抗原結合ドメインを有する場合、その内の少なくとも一つの抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを有しても良いし、抗原結合分子が含む全ての抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインであっても良い。また複数の抗原結合ドメインがそれぞれ異なる低分子化合物依存的に抗原に対する結合活性が変化する抗原ドメインであっても良いし、一つの抗原結合分子の中に低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインと低分子化合物の濃度に抗原に対する結合活性が影響されない抗原結合ドメインが同時に存在しても良い。
更なる一態様において、前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTAに実質的に影響されない。
更なる一態様において、前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTA依存的に変化する。より詳細な一態様において、前記第2の抗原結合ドメインのMTA存在下における前記第2の抗原に対する結合活性が、前記第2の抗原結合ドメインのMTA非存在下における前記第2の抗原に対する結合活性と異なる。
2つ以上の複数の抗原結合ドメインを有し、さらに複数の異なる抗原に対して同時に結合し得る抗原結合分子の非限定な例示として、二重特異性抗体が例示され得るが、これに限定されるものではない。
非限定的な一態様として、本開示の抗原結合分子は更に第2の抗原結合ドメインを有し、前記第2の抗原結合ドメインは前記抗原結合ドメインが結合する抗原と異なる第2の抗原に対して結合活性を有する。
抗原結合分子が異なる複数の抗原結合ドメインを有する場合、その内の少なくとも一つの抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを有しても良いし、抗原結合分子が含む全ての抗原結合ドメインがMTAまたはMTA以外の低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインであっても良い。また複数の抗原結合ドメインがそれぞれ異なる低分子化合物依存的に抗原に対する結合活性が変化する抗原ドメインであっても良いし、一つの抗原結合分子の中に低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインと低分子化合物の濃度に抗原に対する結合活性が影響されない抗原結合ドメインが同時に存在しても良い。
更なる一態様において、前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTAに実質的に影響されない。
更なる一態様において、前記第2の抗原結合ドメインの前記第2の抗原に対する結合活性はMTA依存的に変化する。より詳細な一態様において、前記第2の抗原結合ドメインのMTA存在下における前記第2の抗原に対する結合活性が、前記第2の抗原結合ドメインのMTA非存在下における前記第2の抗原に対する結合活性と異なる。
2つ以上の複数の抗原結合ドメインを有し、さらに複数の異なる抗原に対して同時に結合し得る抗原結合分子の非限定な例示として、二重特異性抗体が例示され得るが、これに限定されるものではない。
本開示において非限定な二重パラトピック抗原結合分子の一態様として、互いに異なるパラトープであって、一方のパラトープががん細胞やがん組織に浸潤している細胞等の細胞膜に結合する膜型分子に存在するエピトープに結合し、もう一方のパラトープがエフェクター細胞の細胞膜に発現する膜型分子中に存在するエピトープに結合するDiabody又はsc(Fv)2が例示され得る。上記のDiabody又はsc(Fv)2では、がん細胞やがん組織に浸潤している細胞等の細胞膜に結合する膜型分子に存在するエピトープに対する一方のパラトープの結合活性がMTA依存的に変化し得るし、エフェクター細胞の細胞膜に結合する膜型分子に存在するエピトープに対する一方のパラトープの結合活性がMTA依存的に変化し得るし、また、双方のパラトープの結合活性がMTA依存的に変化し得る。がん細胞やがん組織に浸潤している細胞等の細胞膜に結合する膜型分子に存在するエピトープに対する一方のパラトープの結合活性と、エフェクター細胞の細胞膜に結合する膜型分子に存在するエピトープに対する一方のパラトープの結合活性のどちらか片方のみがMTA依存的に変化する場合、MTA依存的に抗原に対する結合活性が変化しない抗原結合ドメインは、MTA以外の低分子化合物(例えば、がん組織特異的化合物、非天然低分子化合物)依存的に抗原に対する結合活性が変化する抗原結合ドメインであっても良い。
エフェクター細胞の細胞膜に結合する膜型分子として、これらに限るものではないが、TCR複合体、CD3、CD3eが例示される。
一方のパラトープががん細胞やがん組織に浸潤している細胞等(標的細胞とも呼ぶ)の細胞膜に結合する膜型分子に存在するエピトープに結合し、もう一方のパラトープがエフェクター細胞の細胞膜に発現する膜型分子中に存在するエピトープに結合する二重パラトピック抗原結合分子の一態様いおいては、当該二重パラトピック抗原結合分子が標的細胞とエフェクター細胞の両方に結合することで、エフェクター細胞を活性化され、標的細胞に対して細胞傷害活性を惹起することが出来る。エフェクター細胞がT細胞である場合、惹起される細胞傷害活性はT細胞による細胞傷害(T-cell-dependent cytotoxicity: TDCC)活性である。
エフェクター細胞の細胞膜に結合する膜型分子として、これらに限るものではないが、TCR複合体、CD3、CD3eが例示される。
一方のパラトープががん細胞やがん組織に浸潤している細胞等(標的細胞とも呼ぶ)の細胞膜に結合する膜型分子に存在するエピトープに結合し、もう一方のパラトープがエフェクター細胞の細胞膜に発現する膜型分子中に存在するエピトープに結合する二重パラトピック抗原結合分子の一態様いおいては、当該二重パラトピック抗原結合分子が標的細胞とエフェクター細胞の両方に結合することで、エフェクター細胞を活性化され、標的細胞に対して細胞傷害活性を惹起することが出来る。エフェクター細胞がT細胞である場合、惹起される細胞傷害活性はT細胞による細胞傷害(T-cell-dependent cytotoxicity: TDCC)活性である。
細胞傷害性物質
本開示の抗原結合分子ががん細胞に結合し、細胞傷害活性を発揮するために、抗原結合分子に細胞傷害性物質が結合されていてもよい。細胞傷害性物質としては、以下に例示される化学療法剤であってもよく、またCurr Opin Chem Biol (2010) 14, 529-37や国際公開2009/140242に開示されている化合物であってもよく、これらの化合物が適切なリンカー等で抗原結合分子に結合される。本開示の抗原結合分子が医薬組成物として使用される場合、対象(被験者、患者、等)に当該抗原結合分子を投与する前にこれらの細胞傷害性物質を結合させることも可能であるし、投与の前後または同時に投与することも可能である。
本開示の抗原結合分子ががん細胞に結合し、細胞傷害活性を発揮するために、抗原結合分子に細胞傷害性物質が結合されていてもよい。細胞傷害性物質としては、以下に例示される化学療法剤であってもよく、またCurr Opin Chem Biol (2010) 14, 529-37や国際公開2009/140242に開示されている化合物であってもよく、これらの化合物が適切なリンカー等で抗原結合分子に結合される。本開示の抗原結合分子が医薬組成物として使用される場合、対象(被験者、患者、等)に当該抗原結合分子を投与する前にこれらの細胞傷害性物質を結合させることも可能であるし、投与の前後または同時に投与することも可能である。
また、後述される、化学療法剤、毒性ペプチド或いは放射性化学物質などの細胞傷害性物質が結合された修飾抗原結合分子修飾物も本開示の細胞傷害活性を有する抗原結合分子として好適に使用され得る。このような修飾抗原結合分子(以下、抗原結合分子薬物コンジュゲートと称する。)は、得られた抗原結合分子を化学的に修飾することによって取得され得る。なお、抗原結合分子の修飾方法として、抗体薬物コンジュゲート等の分野においてすでに確立されている方法が適宜使用され得る。また、毒性ペプチドが結合された修飾抗原結合分子は、当該毒性ペプチドをコードする遺伝子と本開示の抗原結合分子をコードする遺伝子がインフレームで連結された融合遺伝子を、適切な宿主細胞中で発現させた後に、当該細胞の培養液から単離することによって、取得され得る。
本開示の抗原結合分子に結合される化学療法剤が例示され得る:アザリビン(azaribine)、アナストロゾール(anastrozole)、アザシチジン(azacytidine)、ブレオマイシン(bleomycin)、ボルテゾミブ(bortezomib)、ブリオスタチン-1(bryostatin-1)、ブスルファン(busulfan)、カンプトテシン(camptothecin)、10-ヒドロキシカンプトテシン(10-hydroxycamptothecin)、カルムスチン(carmustine)、セレブレックス(celebrex)、クロラムブシル(chlorambucil)、シスプラチン(cisplatin)、イリノテカン(irinotecan)、カルボプラチン(carboplatin)、クラドリビン(cladribine)、シクロホスファミド(cyclophosphamide)、シタラビン(cytarabine)、ダカルバジン(dacarbazine)、ドセタキセル(docetaxel)、ダクチノマイシン(dactinomycin)、ダウノマイシングルクロニド(daunomycin glucuronide)、ダウノルビシン(daunorubicin)、デキサメタゾン(dexamethasone)、ジエチルスチルベストロール(diethylstilbestrol)、ドキソルビシン(doxorubicin)、ドキソルビシンブルクロニド(doxorubicin glucuronide)、エピルビシン(epirubicin)、エチニルエストラジオール(ethinyl estradiol)、エストラムスチン(estramustine)、エトポシド(etoposide)、エトポシドグルクロニド(etoposide glucuronide)、フロキシウリジン(floxuridine)、フルダラビン(fludarabine)、フルタミド(flutamide)、フルオロウラシル(fluorouracil)、フルオキシメステロン(fluoxymesterone)、ゲムシタビン(gemcitabine)、ヒドロキシプロゲステロンカプロエート(hydroxyprogesterone caproate)、ヒドロキシウレア(hydroxyurea)、イダルビシン(idarubicin)、イフォスファミド(ifosfamide)、ロイコボリン(leucovorin)、ロムスチン(lomustine)、マイタンシノイド(maytansinoid)、メクロレタミン(mechlorethamine)、メドロキシプロゲステロンアセテート(medroxyprogesterone acetate)、メゲストロールアセテート(megestrol acetate)、メルファラン(melphalan)、メルカプトプリン(mercaptopurine)、メトトレキセート(methotrexate)、ミトキサントロン(mitoxantrone)、ミトラマイシン(mithramycin)、ミトマイシン(mitomycin)、ミトタン(mitotane)、フェニルブチレート(phenylbutyrate)、プレドニゾン(prednisone)、プロカルバジン(procarbazine)、パクリタキセル(paclitaxel)、ペントスタチン(pentostatin)、セムスチン(semustine)、ストレプトゾシン(streptozocin)、タモキシフェン(tamoxifen)、タキサン類(taxanes)、タキソール(taxol)、テストステロンプロピオネート(testosterone propionate)、サリドマイド(thalidomide)、チオグアニン(thioguanine)、チオテパ(thiotepa)、テニポシド(teniposide)、トポテカン(topotecan)、ウラシルマスタード(uracil mustard)、ビンブラスチン(vinblastine)、ビノレルビン(vinorelbine)、ビンクリスチン(vincristine)。
本開示において、好ましい化学療法剤は、低分子の化学療法剤である。低分子の化学療法剤は、本開示の抗原結合分子が結合した後も、抗原結合分子の機能に干渉する可能性が低い。本開示において、低分子の化学療法剤は、通常100~2000、好ましくは200~1000の分子量を有する。ここに例示した化学療法剤は、いずれも低分子の化学療法剤である。これらの本開示における化学療法剤は、生体内で活性な化学療法剤に変換されるプロドラッグを含む。プロドラッグの活性化は酵素的な変換であり得るし、非酵素的な変換でもあり得る。
また、本開示の抗原結合分子に結合される細胞傷害物質としては、Pseudomonas exotoxin A、Saporin-s6、Diphtheria toxin、Cnidarian toxin等の毒性ペプチド(トキシン)やRadioiodine、Photosensitizerも例示され得る。毒性ペプチドの例としては、例えば、次のものが好適に挙げられる。
ジフテリアトキシンA鎖(Diphtheria toxin A Chain)(Langoneら(Methods in Enzymology (1983) 93, 307-308));
シュードモナスエンドトキシン(Pseudomonas Exotoxin)(Nature Medicine (1996) 2, 350-353);
リシン鎖(Ricin A Chain)(Fultonら(J. Biol. Chem. (1986) 261, 5314-5319)、Sivamら(Cancer Res. (1987) 47, 3169-3173)、Cumberら、(J. Immunol. Methods (1990) 135,15-24、Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、およびGheeite ら(J. Immunol.Methods (1991) 142,223-230));
無糖鎖リシンA鎖(Deglicosylated Ricin A Chain)(Thorpeら(Cancer Res. (1987) 47, 5924-5931));
アブリンA鎖(Abrin A Chain)(Wawrzynczakら(Br. J. Cancer (1992) 66, 361-366)、Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、Sivamら(Cancer Res. (1987) 47, 3169-3173)、およびThorpeら(Cancer Res. (1987) 47, 5924-5931));
ゲロニン(Gelonin)(Sivamら(Cancer Res. (1987) 47, 3169-3173)、Cumberら(J. Immunol. Methods (1990) 135, 15-24)、Wawrzynczakら(Cancer Res., (1990) 50, 7519-7562)、およびBolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ポークウイード抗ウィルス蛋白(PAP-s; Pokeweed anti-viral protein fromseeds)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ブリオジン(Briodin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
サポリン(Saporin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
モモルジン(Momordin)(Cumberら(J. Immunol. Methods (1990) 135, 15-24);Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、およびBolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
モモルコキン(Momorcochin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ジアンシン32(Dianthin 32)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ジアンシン30(Dianthin 30)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
モデッシン(Modeccin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ビスカミン(Viscumin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ボルケシン(Volkesin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ドデカンドリン(Dodecandrin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
トリチン(Tritin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ルフィン(Luffin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));および
トリコキリン(Trichokirin)(Casellasら(Eur. J. Biochem. (1988) 176, 581-588)、およびBolognesiら(Clin. exp. Immunol., (1992) 89, 341-346))。
ジフテリアトキシンA鎖(Diphtheria toxin A Chain)(Langoneら(Methods in Enzymology (1983) 93, 307-308));
シュードモナスエンドトキシン(Pseudomonas Exotoxin)(Nature Medicine (1996) 2, 350-353);
リシン鎖(Ricin A Chain)(Fultonら(J. Biol. Chem. (1986) 261, 5314-5319)、Sivamら(Cancer Res. (1987) 47, 3169-3173)、Cumberら、(J. Immunol. Methods (1990) 135,15-24、Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、およびGheeite ら(J. Immunol.Methods (1991) 142,223-230));
無糖鎖リシンA鎖(Deglicosylated Ricin A Chain)(Thorpeら(Cancer Res. (1987) 47, 5924-5931));
アブリンA鎖(Abrin A Chain)(Wawrzynczakら(Br. J. Cancer (1992) 66, 361-366)、Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、Sivamら(Cancer Res. (1987) 47, 3169-3173)、およびThorpeら(Cancer Res. (1987) 47, 5924-5931));
ゲロニン(Gelonin)(Sivamら(Cancer Res. (1987) 47, 3169-3173)、Cumberら(J. Immunol. Methods (1990) 135, 15-24)、Wawrzynczakら(Cancer Res., (1990) 50, 7519-7562)、およびBolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ポークウイード抗ウィルス蛋白(PAP-s; Pokeweed anti-viral protein fromseeds)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ブリオジン(Briodin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
サポリン(Saporin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
モモルジン(Momordin)(Cumberら(J. Immunol. Methods (1990) 135, 15-24);Wawrzynczakら(Cancer Res. (1990) 50, 7519-7562)、およびBolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
モモルコキン(Momorcochin)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ジアンシン32(Dianthin 32)(Bolognesiら(Clin. exp. Immunol. (1992) 89, 341-346));
ジアンシン30(Dianthin 30)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
モデッシン(Modeccin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ビスカミン(Viscumin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ボルケシン(Volkesin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ドデカンドリン(Dodecandrin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
トリチン(Tritin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));
ルフィン(Luffin)(Stirpe F., Barbieri L.(FEBS letter (1986) 195, 1-8));および
トリコキリン(Trichokirin)(Casellasら(Eur. J. Biochem. (1988) 176, 581-588)、およびBolognesiら(Clin. exp. Immunol., (1992) 89, 341-346))。
細胞傷害活性及び細胞傷害活性の測定方法
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、膜型分子をその細胞膜に発現する細胞に対する細胞傷害活性を有する抗原結合分子、および当該抗原結合分子を有効成分として含む医薬組成物が提供される。本開示において細胞傷害活性とは、例えば抗体依存性細胞介在性細胞傷害(antibody-dependent cell-mediated cytotoxicity:ADCC)活性、補体依存性細胞傷害(complement-dependent cytotoxicity:CDC)活性、T細胞による細胞傷害(T-cell-dependent cytotoxicity: TDCC)活性および抗体依存性細胞貪食(Antibody-Dependent Cellular Phagocytosis:ADCP)活性等が挙げられる。本開示において、CDC活性とは補体系による細胞傷害活性を意味する。またADCC活性とは、標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメインを含む抗原結合分子のFc領域に、免疫細胞等が当該免疫細胞に発現したFcγレセプターを介して結合し、当該免疫細胞が標的細胞に傷害を与える活性を意味する。またTDCC活性とは、標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメイン、およびT細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに対する抗原結合ドメイン、特にCD3 epsilon鎖に結合する抗原結合ドメインを含むbi-specific抗体を用いることで標的細胞とT細胞を接近させることにより、T細胞が標的細胞に障害を与える活性を意味する。目的の抗原結合分子がADCC活性、CDC活性、TDCC活性またはADCP活性を有するか否かは公知の方法により測定され得る。
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、膜型分子をその細胞膜に発現する細胞に対する細胞傷害活性を有する抗原結合分子、および当該抗原結合分子を有効成分として含む医薬組成物が提供される。本開示において細胞傷害活性とは、例えば抗体依存性細胞介在性細胞傷害(antibody-dependent cell-mediated cytotoxicity:ADCC)活性、補体依存性細胞傷害(complement-dependent cytotoxicity:CDC)活性、T細胞による細胞傷害(T-cell-dependent cytotoxicity: TDCC)活性および抗体依存性細胞貪食(Antibody-Dependent Cellular Phagocytosis:ADCP)活性等が挙げられる。本開示において、CDC活性とは補体系による細胞傷害活性を意味する。またADCC活性とは、標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメインを含む抗原結合分子のFc領域に、免疫細胞等が当該免疫細胞に発現したFcγレセプターを介して結合し、当該免疫細胞が標的細胞に傷害を与える活性を意味する。またTDCC活性とは、標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメイン、およびT細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに対する抗原結合ドメイン、特にCD3 epsilon鎖に結合する抗原結合ドメインを含むbi-specific抗体を用いることで標的細胞とT細胞を接近させることにより、T細胞が標的細胞に障害を与える活性を意味する。目的の抗原結合分子がADCC活性、CDC活性、TDCC活性またはADCP活性を有するか否かは公知の方法により測定され得る。
具体的には、まず、エフェクター細胞、補体溶液、標的細胞の調製が実施される。
(1)エフェクター細胞の調製
CBA/Nマウスなどから摘出された脾臓から、RPMI1640培地(Invitrogen)中で脾臓細胞が分離される。10%ウシ胎児血清(FBS、HyClone)を含む同培地で洗浄された当該脾臓細胞の濃度を5×106/mLに調製することによって、エフェクター細胞が調製され得る。
(2)補体溶液の調製
10% FBS含有培地(Invitrogen)によってBaby Rabbit Complement(CEDARLANE)を10倍に希釈することによって、補体溶液が調製され得る。
(3)標的細胞の調製
(1)エフェクター細胞の調製
CBA/Nマウスなどから摘出された脾臓から、RPMI1640培地(Invitrogen)中で脾臓細胞が分離される。10%ウシ胎児血清(FBS、HyClone)を含む同培地で洗浄された当該脾臓細胞の濃度を5×106/mLに調製することによって、エフェクター細胞が調製され得る。
(2)補体溶液の調製
10% FBS含有培地(Invitrogen)によってBaby Rabbit Complement(CEDARLANE)を10倍に希釈することによって、補体溶液が調製され得る。
(3)標的細胞の調製
抗原を発現する細胞を0.2 mCiの51Cr-クロム酸ナトリウム(GEヘルスケアバイオサイエンス)とともに、10% FBS含有DMEM培地中で37℃にて1時間培養することにより該標的細胞が放射性標識され得る。放射性標識後、10% FBS含有RPMI1640培地にて3回洗浄された細胞の濃度を2×105/mLに調製することによって、当該標的細胞が調製され得る。
ADCC活性、又はCDC活性は下記に述べる方法により測定され得る。ADCC活性の測定の場合は、96ウェルU底プレート(Becton Dickinson)に加えられた各50μlずつの標的細胞と抗原結合分子が室温にて15分間反応させられる。その後、エフェクター細胞100μlが加えられた当該プレートが、炭酸ガスインキュベーター内で4時間静置される。抗原結合分子の終濃度は例えば0または10μg/ml等の濃度が設定され得る。静置後、各ウェルから回収された100μlの上清の放射活性が、ガンマカウンター(COBRAII AUTO-GAMMA、MODEL D5005、Packard Instrument Company)を用いて測定される。測定値を用いて細胞傷害活性(%)が(A-C) / (B-C) x 100の計算式に基づいて計算され得る。Aは各試料における放射活性(cpm)、Bは1% NP-40(nacalai tesque)を加えた試料における放射活性(cpm)、Cは標的細胞のみを含む試料の放射活性(cpm)を表す。
一方、CDC活性の測定の場合は、96ウェル平底プレート(Becton Dickinson)に加えられた各50μlずつの標的細胞と抗原結合分子が氷上にて15分間反応させられる。その後、補体溶液100μlが加えられた当該プレートが、炭酸ガスインキュベーター内で4時間静置される。抗原結合分子の終濃度は例えば0または3μg/mL等の濃度が設定され得る。静置後、各ウェルから回収された100μlの上清の放射活性が、ガンマカウンターを用いて測定される。細胞傷害活性はADCC活性の測定と同様に計算され得る。
ADCP活性は下記に述べる方法により測定され得る。抗原を発現する標的細胞のラベリングをPKH26 dye labeling kit(Sigma社)を用いて行う。標的細胞をトリプシン溶液で剥離し、PBSで洗浄する。剥離した細胞をDiluent Cで1x107 cells/mlになるよう懸濁し、PKH26 dye stock(1mM)をDiluent Cで8μMに希釈し、すぐに細胞浮遊液と等量のdye希釈液を添加する。室温に5分間放置した後、10% FBS含有RPMI1640を加え2回洗浄を行うことで標的細胞を調製する。 被験物質である抗原結合分子を標的細胞の培地を用いて20μg/mlに希釈し、前記標的細胞を2x106 cells/100μl/tubeとなるように分注し、混合する。抗原結合分子の終濃度は例えば0または10μg/ml等の濃度が設定され得る。氷上で30分間静置した後、上清を捨て、培養液で2回洗浄し、培養液500μLに懸濁する。エフェクター細胞から上清を除き、前記抗原結合分子と混合した標的細胞懸濁液にエフェクター細胞を加え混合する。CO2インキュベータ内で3時間培養した後、Trypsin-EDTAで細胞を剥離し、細胞を回収する。回収した細胞に例えばFITC標識抗CD11b抗体を加え、氷上で30分間静置する。静置後に上清を捨て、培養液で2回洗浄した後に、回収した細胞を300μlの培養液で懸濁し、例えばFACS Calibur(ベクトンディッキンソン社)にて測定を行う。CD11b陽性のマクロファージ細胞において、PKH26陽性画分を貪食陽性細胞として評価することで、ADCP活性が測定されうる。
TDCC活性は下記に述べる方法により測定され得る。ヒト末梢血単核球(以下、ヒトPBMCと指称する。)をエフェクター細胞として用いて被験抗体のTDCC活性が測定され得る。
(1)ヒトPBMC溶液の調製
5000単位/5mlのヘパリン溶液が予め500μl注入された注射器を用い、健常人より末梢血50 mlを採取する。PBSを用いて2倍に希釈された当該末梢血を4等分し、15 mlのFicoll-Paque PLUSが予め注入されて遠心操作が行なわれたLeucosepリンパ球分離管(GE Healthcare)に加える。当該末梢血が分注された分離管に1000 gの速度によって10分間室温にて遠心分離の操作をした後、単核球画分層を分取する。10%FBSを含むRPMI-1640(10%FBS/RPMI-1640)によって1回当該各分層に含まれる細胞を洗浄した後、当該細胞を各標的細胞の培養液中にその細胞密度が2 x 106 細胞/ mlとなるように懸濁する。当該細胞懸濁液をエフェクター細胞として以後の実験に供する。
(1)ヒトPBMC溶液の調製
5000単位/5mlのヘパリン溶液が予め500μl注入された注射器を用い、健常人より末梢血50 mlを採取する。PBSを用いて2倍に希釈された当該末梢血を4等分し、15 mlのFicoll-Paque PLUSが予め注入されて遠心操作が行なわれたLeucosepリンパ球分離管(GE Healthcare)に加える。当該末梢血が分注された分離管に1000 gの速度によって10分間室温にて遠心分離の操作をした後、単核球画分層を分取する。10%FBSを含むRPMI-1640(10%FBS/RPMI-1640)によって1回当該各分層に含まれる細胞を洗浄した後、当該細胞を各標的細胞の培養液中にその細胞密度が2 x 106 細胞/ mlとなるように懸濁する。当該細胞懸濁液をエフェクター細胞として以後の実験に供する。
(2)LDH遊離試験(TDCC活性)
TDCC活性はLDHリリース法(LDH Cytotoxicity Detection Kit: TAKARA)にて評価され得る。まず、各標的細胞の培養液で終濃度の4倍に希釈した各濃度(0.000004、0.00004、0.0004、0.004、0.04、0.4、4、40μg/ml)の抗体溶液を96ウェルU底プレートの各ウェル中に50μlずつ添加する。次に、各標的細胞の培養液で2 x 105細胞/mlに調製した標的細胞を50μlずつ播種し(1 x 104細胞/ウェル)室温にて15分間静置する。各ウェル中に(1)で調製した各標的細胞の培養液中で調製したヒトPBMC溶液各100μl(2 x 105細胞/ウェル)を加えた当該プレートを、5%炭酸ガスインキュベータ中において37℃で約24時間静置した後に、遠心操作する。当該プレートの各ウェル中の100μlの培養上清を96ウェル平底プレートに移す。前記LDH Cytotoxicity Detection Kit のcatalyst溶液を1 mlのH2Oに溶解し、dye溶液と1:45で混合する。catalyst溶液およびdye溶液の混合溶液を100μl/ウェルで培養上清を移した96ウェル平底プレートに分注し、15-30分、室温で静置した。490-492nmの吸光度をプレートリーダーで測定する。対照波長は600-620nmとして、490-492nmの吸光度から引いた。培養液のみのウェル(ブランク)の平均値を引いた値を以下の式に当てはめる。
下式:
Cytotoxicty (TDCC) (%) = ((A-B)-C))×100 / (D-C)
に基づいて細胞傷害活性を求めることができる。 ここでAは標的細胞、エフェクター細胞、抗体の混合の吸光度、Bはエフェクター細胞の吸光度、Cは標的細胞の吸光度、Dは標的細胞にTriton X-100を添加した吸光度とする。
TDCC活性はLDHリリース法(LDH Cytotoxicity Detection Kit: TAKARA)にて評価され得る。まず、各標的細胞の培養液で終濃度の4倍に希釈した各濃度(0.000004、0.00004、0.0004、0.004、0.04、0.4、4、40μg/ml)の抗体溶液を96ウェルU底プレートの各ウェル中に50μlずつ添加する。次に、各標的細胞の培養液で2 x 105細胞/mlに調製した標的細胞を50μlずつ播種し(1 x 104細胞/ウェル)室温にて15分間静置する。各ウェル中に(1)で調製した各標的細胞の培養液中で調製したヒトPBMC溶液各100μl(2 x 105細胞/ウェル)を加えた当該プレートを、5%炭酸ガスインキュベータ中において37℃で約24時間静置した後に、遠心操作する。当該プレートの各ウェル中の100μlの培養上清を96ウェル平底プレートに移す。前記LDH Cytotoxicity Detection Kit のcatalyst溶液を1 mlのH2Oに溶解し、dye溶液と1:45で混合する。catalyst溶液およびdye溶液の混合溶液を100μl/ウェルで培養上清を移した96ウェル平底プレートに分注し、15-30分、室温で静置した。490-492nmの吸光度をプレートリーダーで測定する。対照波長は600-620nmとして、490-492nmの吸光度から引いた。培養液のみのウェル(ブランク)の平均値を引いた値を以下の式に当てはめる。
下式:
Cytotoxicty (TDCC) (%) = ((A-B)-C))×100 / (D-C)
に基づいて細胞傷害活性を求めることができる。 ここでAは標的細胞、エフェクター細胞、抗体の混合の吸光度、Bはエフェクター細胞の吸光度、Cは標的細胞の吸光度、Dは標的細胞にTriton X-100を添加した吸光度とする。
T細胞の活性化及びT細胞の活性化能の測定方法
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、T細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに結合することでT細胞の活性化能を有する抗原結合分子、および当該抗原結合分子を有効成分として含む医薬組成物が提供される。標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメイン、およびT細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに対する抗原結合ドメイン、特にCD3 epsilon鎖に結合する抗原結合ドメインを含むbi-specific抗体を用いることで標的細胞とT細胞を接近させることにより、T細胞の活性化を誘導することができる。T細胞の活性化能の測定は、例えばGloResponse NFAT-RE-Luc2 Jurkat細胞(Promega)を用いて測定することができる。前記細胞にはNuclear factor of activated T cells(NFAT)応答配列であるNFAT response element(NFAT-RE)により発現が誘導されるルシフェラーゼレポーターが組み込まれており、Jurkatの細胞膜に発現しているTCR複合体の活性化を、レポータータンパク質の発現を測定することにより検出することが可能である。当該細胞を用いることでTCR複合体を活性化可能なTCRリガンドや抗TCR抗体、抗CD3抗体によるT細胞活性化能を測定することが可能である。T細胞の活性化能の測定は、前記細胞を用いた方法に限定されず同様のNFAT response element(NFAT-RE)およびレポータータンパク質を導入したT細胞系の細胞を構築して用いて測定することも可能である。また、T細胞の活性化能の測定は前記手法に限定されず、例えばCD69やCD25等のT細胞活性化マーカーの発現確認をフローサイトメトリー法や遺伝子発現解析法により行っても良いし、TNF-α、IFN-γ、IL-6 及びIL-2等の活性化したT細胞が分泌するサイトカインを測定することで行っても良いが、これらの手法に限定されずT細胞の活性化を測定できる方法であれば使用する細胞株や測定方法によらず使用することができる。
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、T細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに結合することでT細胞の活性化能を有する抗原結合分子、および当該抗原結合分子を有効成分として含む医薬組成物が提供される。標的細胞の細胞膜に発現された膜型分子に結合する抗原結合ドメイン、およびT細胞上のT細胞レセプター(TCR)複合体の構成サブユニットのいずれかに対する抗原結合ドメイン、特にCD3 epsilon鎖に結合する抗原結合ドメインを含むbi-specific抗体を用いることで標的細胞とT細胞を接近させることにより、T細胞の活性化を誘導することができる。T細胞の活性化能の測定は、例えばGloResponse NFAT-RE-Luc2 Jurkat細胞(Promega)を用いて測定することができる。前記細胞にはNuclear factor of activated T cells(NFAT)応答配列であるNFAT response element(NFAT-RE)により発現が誘導されるルシフェラーゼレポーターが組み込まれており、Jurkatの細胞膜に発現しているTCR複合体の活性化を、レポータータンパク質の発現を測定することにより検出することが可能である。当該細胞を用いることでTCR複合体を活性化可能なTCRリガンドや抗TCR抗体、抗CD3抗体によるT細胞活性化能を測定することが可能である。T細胞の活性化能の測定は、前記細胞を用いた方法に限定されず同様のNFAT response element(NFAT-RE)およびレポータータンパク質を導入したT細胞系の細胞を構築して用いて測定することも可能である。また、T細胞の活性化能の測定は前記手法に限定されず、例えばCD69やCD25等のT細胞活性化マーカーの発現確認をフローサイトメトリー法や遺伝子発現解析法により行っても良いし、TNF-α、IFN-γ、IL-6 及びIL-2等の活性化したT細胞が分泌するサイトカインを測定することで行っても良いが、これらの手法に限定されずT細胞の活性化を測定できる方法であれば使用する細胞株や測定方法によらず使用することができる。
具体的なT細胞の活性化能の測定方法の非限定な一態様として、標的抗原hIL-6Rに結合する抗原結合ドメインとCD3に結合する抗原結合ドメインを有する二重特異性抗体のT細胞の活性化能の測定方法を以下に例示する。ヒトT細胞株としてNFAT-RE-luc2-Jurkat細胞を使用し、hIL-6R発現細胞株としてCT26にhIL-6Rを強制発現させたCT26/hIL-6Rを用いる。NFAT-RE-luc2-Jurkat細胞およびCT26/hIL-6R細胞は細胞密度がそれぞれ3×106 cells/mL、1×106 cells/mLとなるようにアッセイバッファーで懸濁して使用することができるが、細胞密度や各細胞の混合比は評価に用いる細胞種に応じて増減させて用いても良い。384ウェルプレートの各ウェルに調製したヒトT細胞株およびhIL-6R発現細胞株の懸濁液をそれぞれ10 μlずつ添加し、さらに調製した抗体溶液10 μlを添加する。5% CO2インキュベーターにおいて37℃で6時間静置した後、当該サンプルのルシフェラーゼ活性を測定することでヒトT細胞株の活性化能を評価することができる。ルシフェラーゼ活性の測定には市販のルシフェラーゼ活性測定試薬(Bio-Glo luciferase assay system、Promega)を用いて実施することができるが、ルシフェラーゼ活性を測定できれば用いる試薬に限定されず使用することができる。
中和活性及び中和活性の測定方法
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、当該膜型分子に対する中和活性を有する抗原結合分子を有効成分として含む医薬組成物が提供される。本開示の非限定な別の一態様では、低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、膜型分子をその細胞膜に発現する細胞に対する細胞傷害活性に加えて、当該膜型分子に対する中和活性を有する抗原結合分子を有効成分として含む医薬組成物が提供される。一般的に、中和活性とは、ウイルスや毒素など、細胞に対して生物学的活性を有するリガンドの当該生物学的活性を阻害する活性をいう。即ち、中和活性を有する物質とは、当該リガンド又は当該リガンドが結合するレセプターに結合し、当該リガンドとレセプターの結合を阻害する物質をさす。中和活性によりリガンドとの結合を阻止されたレセプターは、当該レセプターを通じた生物学的活性を発揮することができなくなる。抗原結合分子が抗体である場合、このような中和活性を有する抗体は一般に中和抗体と呼ばれる。ある被検物質の中和活性は、リガンドの存在下における生物学的活性をその被検物質の存在又は非存在下の条件の間で比較することにより測定され得る。
本開示の非限定な一態様では、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、当該膜型分子に対する中和活性を有する抗原結合分子を有効成分として含む医薬組成物が提供される。本開示の非限定な別の一態様では、低分子化合物依存的に抗原に対する結合活性が変化する抗原結合ドメインを含み、膜型分子をその細胞膜に発現する細胞に対する細胞傷害活性に加えて、当該膜型分子に対する中和活性を有する抗原結合分子を有効成分として含む医薬組成物が提供される。一般的に、中和活性とは、ウイルスや毒素など、細胞に対して生物学的活性を有するリガンドの当該生物学的活性を阻害する活性をいう。即ち、中和活性を有する物質とは、当該リガンド又は当該リガンドが結合するレセプターに結合し、当該リガンドとレセプターの結合を阻害する物質をさす。中和活性によりリガンドとの結合を阻止されたレセプターは、当該レセプターを通じた生物学的活性を発揮することができなくなる。抗原結合分子が抗体である場合、このような中和活性を有する抗体は一般に中和抗体と呼ばれる。ある被検物質の中和活性は、リガンドの存在下における生物学的活性をその被検物質の存在又は非存在下の条件の間で比較することにより測定され得る。
例えば、IL-6レセプターの主要なリガンドとして考えられているものは配列番号:27で表されるIL-6が好適に挙げられる。そのアミノ末端が細胞外ドメインを形成するI型膜タンパク質であるIL-6レセプターは、IL-6によって二量体化が誘導されたgp130レセプターとともにヘテロ四量体を形成する(Heinrichら(Biochem. J. (1998) 334, 297-314))。当該ヘテロ四量体の形成によって、gp130レセプターに会合しているJakが活性化される。Jakは自己リン酸化とレセプターのリン酸化を行う。受容体及びJakのリン酸化部位は、Stat3のようなSH2を持つStatファミリーに属する分子や、MAPキナーゼ、PI3/Akt、そのほかのSH2を持つタンパク質やアダプターに対して、結合部位の役割を果たす。次に、gp130レセプターに結合したStatが、Jakによってリン酸化される。リン酸化されたStatは二量体を形成して核内に移行し、標的遺伝子の転写を調節する。JakまたはStatは他のクラスのレセプターを介してシグナルカスケードに関与することもできる。脱制御されたIL-6のシグナルカスケードは、自己免疫疾患の病態や炎症、多発性骨髄腫や前立腺癌などの癌で観察される。癌遺伝子として作用し得るStat3は、多くのがんにおいて恒常的に活性化している。前立腺癌と多発性骨髄腫では、IL-6レセプターからのシグナルカスケードと、上皮成長因子受容体 (EGFR) ファミリーメンバーからのシグナルカスケードとの間にクロストークがある(Ishikawaら(J. Clin. Exp. Hematopathol. (2006) 46 (2), 55-66))。
こうした細胞内のシグナルカスケードは細胞種毎に異なるため、目的とする標的細胞毎に適宜標的分子を設定することができ、上記の因子に限定されるものではない。生体内シグナルの活性化を測定することにより、中和活性を評価することができる。また、生体内シグナルカスケードの下流に存在する標的遺伝子に対する転写誘導作用を指標として、生体内シグナルの活性化を検出することもできる。標的遺伝子の転写活性の変化は、レポーターアッセイの原理によって検出することができる。具体的には、標的遺伝子の転写因子又はプロモーター領域の下流にGFP(Green Fluorescence Protein)やルシフェラーゼなどのレポーター遺伝子を配し、そのレポーター活性を測定することにより、転写活性の変化をレポーター活性として測定することができる。生体内シグナルの活性化の測定キットは市販のものを適宜使用することができる(例えば、Mercury Pathway Profiling Luciferase System(Clontech)等)。
更に、通常は細胞増殖を促進する方向に働くシグナルカスケードに作用するEGFレセプターファミリー等のレセプターリガンドの中和活性を測定する方法として、標的とする細胞の増殖活性を測定することによって、抗原結合分子の中和活性を評価することができる。例えば、HB-EGF等その増殖がEGFファミリーの成長因子によって促進される細胞の増殖に対する、抗HB-EGF抗体の中和活性に基づく抑制効果を評価又は測定する方法として、以下の方法が好適に使用される。試験管内において当該細胞増殖抑制活性を評価又は測定する方法としては、培地中に添加した[3H]ラベルしたチミジンの生細胞による取り込みをDNA複製能力の指標として測定する方法が用いられる。より簡便な方法としてトリパンブルー等の色素を細胞外に排除する能力を顕微鏡下で計測する色素排除法や、MTT法が用いられる。後者は、生細胞がテトラゾリウム塩であるMTT(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide)を青色のホルマザン産物へ転換する能力を有することを利用している。
より具体的には、被検細胞の培養液にリガンドと共に被検抗体を添加して一定時間を経過した後に、MTT溶液を培養液に加えて一定時間静置することによりMTTを細胞に取り込ませる。その結果、黄色の化合物であるMTTが細胞内のミトコンドリア内のコハク酸脱水素酵素により青色の化合物に変換される。この青色生成物を溶解し呈色させた後にその吸光度を測定することにより生細胞数の指標とするものである。MTT以外に、MTS、XTT、WST-1、WST-8等の試薬も市販されており(nacalai tesqueなど)好適に使用することができる。活性の測定に際しては、対照抗体として抗HB-EGF抗体と同一のアイソタイプを有する抗体で当該細胞増殖抑制活性を有しない結合抗体を、抗HB-EGF抗体と同様に使用して、抗HB-EGF抗体が対照抗体よりも強い細胞増殖抑制活性を示すことにより活性を判定することができる。
活性を評価するための細胞として、例えば、その増殖がHB-EGFによって促進される細胞である、卵巣癌細胞であるRMG-1細胞株や、ヒトEGFRの細胞外ドメインとマウスG-CSF受容体の細胞内ドメインをインフレームで融合した融合タンパク質であるhEGFR/mG-CSFRをコードする遺伝子を発現する様に結合したベクターによって形質転換されたマウスBa/F3細胞等も好適に使用され得る。このように、当業者は、活性を評価するための細胞を適宜選択することによって前記の細胞増殖活性の測定に使用することが可能である。
本開示における「MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子」は、生体に投与されたときに正の薬理作用をもたらし得る。
ヒト化抗体の製造方法
本明細書において記載される抗原結合分子がヒトに投与される場合、当該抗原結合分子における抗原結合ドメインとして、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体由来の抗原結合ドメインが適宜採用され得る。遺伝子組換え型抗体には、例えば、ヒト化(Humanized)抗体等が含まれる。これらの改変抗体は、公知の方法を用いて適宜製造される。
本明細書において記載される抗原結合分子がヒトに投与される場合、当該抗原結合分子における抗原結合ドメインとして、ヒトに対する異種抗原性を低下させること等を目的として人為的に改変した遺伝子組換え型抗体由来の抗原結合ドメインが適宜採用され得る。遺伝子組換え型抗体には、例えば、ヒト化(Humanized)抗体等が含まれる。これらの改変抗体は、公知の方法を用いて適宜製造される。
本明細書において記載される抗原結合分子における抗原結合ドメインを作製するために用いられる抗体の可変領域は、通常、4つのフレームワーク領域(FR)にはさまれた3つの相補性決定領域(complementarity-determining region ; CDR)で構成されている。CDRは、実質的に、抗体の結合特異性を決定している領域である。CDRのアミノ酸配列は多様性に富む。一方FRを構成するアミノ酸配列は、異なる結合特異性を有する抗体の間でも、高い同一性を示すことが多い。そのため、一般に、CDRの移植によって、ある抗体の結合特異性を、他の抗体に移植することができるとされている。
ヒト化抗体は、再構成(reshaped)ヒト抗体とも称される。具体的には、ヒト以外の動物として、マウス、ウサギなどの抗体のCDRをヒト抗体に移植したヒト化抗体などが公知である。ヒト化抗体を得るための一般的な遺伝子組換え手法も知られている。具体的には、これに限定されるわけではないが、マウスの抗体のCDRをヒトのFRに移植するための方法として、たとえばOverlap Extension PCRが公知である。Overlap Extension PCRにおいては、ヒト抗体のFRを合成するためのプライマーに、移植すべきマウス抗体のCDRをコードする塩基配列が付加される。プライマーは4つのFRのそれぞれについて用意される。一般に、マウスCDRのヒトFRへの移植においては、マウスのFRと同一性の高いヒトFRを選択するのが、CDRの機能の維持において有利であるとされている。すなわち、一般に、移植すべきマウスCDRに隣接しているFRのアミノ酸配列と同一性の高いアミノ酸配列からなるヒトFRを利用するのが好ましい。CDRの由来は、マウスやウサギに限定されず、あらゆる非ヒト動物の抗体から選択され得る。
また連結される塩基配列は、互いにインフレームで接続されるようにデザインされる。それぞれのプライマーによってヒトFRが個別に合成される。その結果、各FRにマウスCDRをコードするDNAが付加された産物が得られる。各産物のマウスCDRをコードする塩基配列は、互いにオーバーラップするようにデザインされている。続いて、ヒト抗体遺伝子を鋳型として合成された産物のオーバーラップしたCDR部分を互いにアニールさせて相補鎖合成反応が行われる。この反応によって、ヒトFRがマウスCDRの配列を介して連結される。
最終的に3つのCDRと4つのFRが連結された可変領域遺伝子は、その5'末端と3'末端にアニールし適当な制限酵素認識配列を付加されたプライマーによってその全長が増幅される。上記のように得られたDNAとヒト抗体C領域をコードするDNAとをインフレームで融合するように発現ベクター中に挿入することによって、ヒト型抗体発現用ベクターが作成できる。当該組込みベクターを宿主に導入して組換え細胞を樹立した後に、当該組換え細胞を培養し、当該ヒト化抗体をコードするDNAを発現させることによって、当該ヒト化抗体が当該培養細胞の培養物中に産生される(欧州特許公開EP239400、国際公開WO1996/002576参照)。
上記のように作製されたヒト化抗体の抗原への結合活性を定性的又は定量的に測定し、評価することによって、CDRを介して連結されたときに該CDRが良好な抗原結合部位を形成するようなヒト抗体のFRが好適に選択できる。必要に応じ、再構成ヒト抗体のCDRが適切な抗原結合部位を形成するようにFRのアミノ酸残基を置換することもできる。たとえば、マウスCDRのヒトFRへの移植に用いたPCR法を応用して、FRにアミノ酸配列の変異を導入することができる。具体的には、FRにアニーリングするプライマーに部分的な塩基配列の変異を導入することができる。このようなプライマーによって合成されたFRには、塩基配列の変異が導入される。アミノ酸を置換した変異型抗体の抗原への結合活性を上記の方法で測定し評価することによって所望の性質を有する変異FR配列が選択され得る(Cancer Res., (1993) 53, 851-856)。
医薬組成物
本開示によって、副作用を回避しつつ薬効を発揮するために、正常組織や血液中において全身的に作用せず、がんにおいて作用する抗原結合分子を含む医薬組成物が提供される。本開示の医薬組成物に含まれる抗原結合分子は、MTA依存的に標的抗原への結合が制御されることから、例えば、該抗原結合分子が、がん組織における抗原を標的とする場合、がん組織におけるがん細胞・免疫細胞・ストローマ細胞等に発現する抗原、あるいはがん組織に分泌されている抗原に結合することが出来ないため、正常組織に対する細胞傷害作用や中和作用等による副作用を回避しつつ、がんに対する強力な細胞傷害作用や増殖抑制作用、免疫亢進作用等を発揮する。例えば、MTA依存的にT細胞に発現するCD3に対する結合活性が変化する抗原結合ドメインと、がん細胞に発現するEGFRに結合する抗原結合ドメインを含む二重特異性抗原結合分子または二重パラトピックな抗原結合分子は、正常組織に発現するEGFRに結合せず、がん細胞に発現しているEGFRに結合するため、副作用を回避しつつ強力な抗腫瘍効果を発揮する。すなわち、がん細胞近傍にいるT細胞に発現しているCD3にはMTAに依存的に結合するが、がん細胞近傍以外にいるT細胞に発現しているCD3に結合しないため、がん細胞近傍にいるT細胞を活性化し副作用を回避しつつ強力な抗腫瘍効果を発揮する。
本開示によって、副作用を回避しつつ薬効を発揮するために、正常組織や血液中において全身的に作用せず、がんにおいて作用する抗原結合分子を含む医薬組成物が提供される。本開示の医薬組成物に含まれる抗原結合分子は、MTA依存的に標的抗原への結合が制御されることから、例えば、該抗原結合分子が、がん組織における抗原を標的とする場合、がん組織におけるがん細胞・免疫細胞・ストローマ細胞等に発現する抗原、あるいはがん組織に分泌されている抗原に結合することが出来ないため、正常組織に対する細胞傷害作用や中和作用等による副作用を回避しつつ、がんに対する強力な細胞傷害作用や増殖抑制作用、免疫亢進作用等を発揮する。例えば、MTA依存的にT細胞に発現するCD3に対する結合活性が変化する抗原結合ドメインと、がん細胞に発現するEGFRに結合する抗原結合ドメインを含む二重特異性抗原結合分子または二重パラトピックな抗原結合分子は、正常組織に発現するEGFRに結合せず、がん細胞に発現しているEGFRに結合するため、副作用を回避しつつ強力な抗腫瘍効果を発揮する。すなわち、がん細胞近傍にいるT細胞に発現しているCD3にはMTAに依存的に結合するが、がん細胞近傍以外にいるT細胞に発現しているCD3に結合しないため、がん細胞近傍にいるT細胞を活性化し副作用を回避しつつ強力な抗腫瘍効果を発揮する。
このようにがん組織において抗原に結合し、それ以外の正常組織や血液中では抗原に結合しない抗原結合分子は、副作用を回避しつつ薬効を発揮する。本開示が提供する、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子(MTAをスイッチとして抗原に結合する抗原結合分子、MTAスイッチ抗原結合分子(MTA switch antigen binding molecule)ともいう)は、MTAが存在しない正常の環境では抗原に結合せず、MTAが高濃度で存在するがん組織では抗原に結合することが可能である。
本開示の抗原結合分子(に含まれるパラトープ)と抗原(に含まれるエピトープ)に挟まれて、もしくは本開示の抗原結合分子に結合することで抗原結合分子の抗原に対するパラトープの構造に変化を起こしてスイッチ機能を果たすMTA依存的に抗原に対する結合活性が変化する抗原結合分子が例示される。MTAが存在しなければ本開示の抗原結合分子に含まれるパラトープと抗原に含まれるエピトープとの相互作用が不十分となり本開示の抗原結合分子は抗原に結合することができないが、MTAが存在すれば本開示の抗原結合分子に含まれるパラトープと抗原に含まれるエピトープとの間に挟まる、もしくはパラトープの構造を変化させることによって、MTAが高濃度で存在するがん組織において抗原に結合した当該抗原結合分子が、当該抗原を発現する細胞に対して薬効を発揮することができる(図26、図27)。本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインにMTAと相互作用するアミノ酸残基が存在する場合、MTAが抗原結合分子に含まれるパラトープと抗原に含まれるエピトープとの間に挟まれやすい、もしくはパラトープの構造を変化させやすい。また、このスイッチとなるMTAへの結合は可逆的であるため、これらのMTAのスイッチによる本開示の抗原結合分子の抗原に対する結合の制御は可逆的であると考えられる。このようにがん組織におけるがん細胞や免疫細胞等の病的細胞に結合し、あるいは、がん組織において分泌された抗原に結合し、薬効を発揮することが可能な本開示の抗原結合分子は医薬組成物として有用である。本開示の医薬組成物には医薬的に許容される担体が含まれ得る。
本開示において医薬組成物とは、通常、疾患の治療もしくは予防、あるいは検査・診断のための薬剤をいう。また、本開示において、「MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を含む医薬組成物」との用語は、「MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を治療対象に投与することを含む疾患の治療方法」と言い換えることも可能であるし、「疾患を治療するための医薬の製造におけるMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の使用」と言い換えることも可能である。また、「MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を含む医薬組成物」との用語を、「疾患を治療するためのMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の使用」と言い換えることも可能である。
本開示の医薬組成物は、当業者に公知の方法を用いて製剤化され得る。例えば、水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、又は懸濁液剤の注射剤の形で非経口的に使用され得る。例えば、薬理学上許容される担体もしくは媒体、具体的には、滅菌水や生理食塩水、植物油、乳化剤、懸濁剤、界面活性剤、安定剤、香味剤、賦形剤、ベヒクル、防腐剤、結合剤等と適宜組み合わせて、一般に認められた製薬実施に要求される単位用量形態で混和することによって製剤化され得る。これら製剤における有効成分量は、指示された範囲の適当な容量が得られるように設定される。
注射のための無菌組成物は注射用蒸留水のようなベヒクルを用いて通常の製剤実施にしたがって処方され得る。注射用の水溶液としては、例えば生理食塩水、ブドウ糖やその他の補助薬(例えばD-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウム)を含む等張液が挙げられる。適切な溶解補助剤、例えばアルコール(エタノール等)、ポリアルコール(プロピレングリコール、ポリエチレングリコール等)、非イオン性界面活性剤(ポリソルベート80(TM)、HCO-50等)が併用され得る。
油性液としてはゴマ油、大豆油が挙げられ、溶解補助剤として安息香酸ベンジル及び/またはベンジルアルコールも併用され得る。また、緩衝剤(例えば、リン酸塩緩衝液及び酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩酸プロカイン)、安定剤(例えば、ベンジルアルコール及びフェノール)、酸化防止剤と配合され得る。調製された注射液は通常、適切なアンプルに充填される。
本開示の医薬組成物は、好ましくは非経口投与により投与される。例えば、注射剤型、経鼻投与剤型、経肺投与剤型、経皮投与型の組成物が投与される。例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などにより全身または局部的に投与され得る。
投与方法は、患者の年齢、症状により適宜選択され得る。抗原結合分子を含有する医薬組成物の投与量は、例えば、一回につき体重1 kgあたり0.0001 mgから1000 mgの範囲に設定され得る。または、例えば、患者あたり0.001~100000 mgの投与量が設定され得るが、本開示はこれらの数値に必ずしも制限されるものではない。投与量及び投与方法は、患者の体重、年齢、症状などにより変動するが、当業者であればそれらの条件を考慮し適当な投与量及び投与方法を設定することが可能である。
MTA依存的に結合する抗原結合分子の利用法
本開示におけるMTA依存的に結合する抗原結合分子は、様々な既存の医薬用途の技術と組み合わることが可能である。そのような技術の組み合わせの非限定な一態様として、MTA依存的に結合する抗原結合分子を利用したキメラ抗原受容体 T細胞(CAR-T細胞)の作成が例示される。CAR-Tの非限定な作成手法の一つとしては、TCRの細胞外ドメインに腫瘍関連抗原に特異的に結合する抗原結合分子を連結し、またT細胞の活性化を増強する目的でCD28等の共刺激分子のシグナルドメインを細胞内に連結したキメラ受容体を遺伝子改変技術によりT細胞を主とするエフェクター細胞に導入する手法が挙げられる。前記腫瘍関連抗原に特異的に結合する抗原結合分子として、本開示におけるMTA依存的に結合する抗原結合分子を使用することで、MTAが蓄積した腫瘍組織特異的にCAR-T細胞が抗原に結合することが可能となる。
また、本開示のMTA依存的に結合する抗原結合分子と組み合わせ可能な技術の非限定な例として、ウィルスベクター等を用いてMTA依存的に結合する抗原結合分子をコードする核酸を生体内に組み込み、当該抗原結合分子を直接発現させる方法が挙げられる。ウィルスベクターとして、アデノウィルスが例示されるがこれに限定されるものではない。またウィルスベクターを用いることなくエレクトロポレーション法や核酸を直接投与する手法等によりMTA依存的に結合する抗原結合分子をコードする核酸を直接生体内に組み込むことも可能であるし、当該抗原結合分子を分泌発現させるように遺伝子改変した細胞を生体に投与することで、当該抗原結合分子を継続的に生体内で分泌させることも可能である。
本開示におけるMTA依存的に結合する抗原結合分子は、様々な既存の医薬用途の技術と組み合わることが可能である。そのような技術の組み合わせの非限定な一態様として、MTA依存的に結合する抗原結合分子を利用したキメラ抗原受容体 T細胞(CAR-T細胞)の作成が例示される。CAR-Tの非限定な作成手法の一つとしては、TCRの細胞外ドメインに腫瘍関連抗原に特異的に結合する抗原結合分子を連結し、またT細胞の活性化を増強する目的でCD28等の共刺激分子のシグナルドメインを細胞内に連結したキメラ受容体を遺伝子改変技術によりT細胞を主とするエフェクター細胞に導入する手法が挙げられる。前記腫瘍関連抗原に特異的に結合する抗原結合分子として、本開示におけるMTA依存的に結合する抗原結合分子を使用することで、MTAが蓄積した腫瘍組織特異的にCAR-T細胞が抗原に結合することが可能となる。
また、本開示のMTA依存的に結合する抗原結合分子と組み合わせ可能な技術の非限定な例として、ウィルスベクター等を用いてMTA依存的に結合する抗原結合分子をコードする核酸を生体内に組み込み、当該抗原結合分子を直接発現させる方法が挙げられる。ウィルスベクターとして、アデノウィルスが例示されるがこれに限定されるものではない。またウィルスベクターを用いることなくエレクトロポレーション法や核酸を直接投与する手法等によりMTA依存的に結合する抗原結合分子をコードする核酸を直接生体内に組み込むことも可能であるし、当該抗原結合分子を分泌発現させるように遺伝子改変した細胞を生体に投与することで、当該抗原結合分子を継続的に生体内で分泌させることも可能である。
オリゴヌクレオチド
本明細書で用いられる「オリゴヌクレオチド」は、通常約200ヌクレオチド長未満であるがそうである必要はない、通常一本鎖である合成ポリヌクレオチドのことをいう。用語「オリゴヌクレオチド」および「ポリヌクレオチド」は、相互に排他的ではない。ポリヌクレオチドに関する上記の説明は、オリゴヌクレオチドに等しくかつ完全に適用可能である。本明細書において、「オリゴヌクレオチド」及び「核酸」は互換的に使用される。
本明細書で用いられる「オリゴヌクレオチド」は、通常約200ヌクレオチド長未満であるがそうである必要はない、通常一本鎖である合成ポリヌクレオチドのことをいう。用語「オリゴヌクレオチド」および「ポリヌクレオチド」は、相互に排他的ではない。ポリヌクレオチドに関する上記の説明は、オリゴヌクレオチドに等しくかつ完全に適用可能である。本明細書において、「オリゴヌクレオチド」及び「核酸」は互換的に使用される。
「MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をコードする単離されたポリヌクレオチド」は、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をコードする1つまたは複数のポリヌクレオチド分子のことをいい、1つのベクターまたは別々のベクターに乗っているポリヌクレオチド分子、および、宿主細胞中の1つまたは複数の位置に存在しているポリヌクレオチド分子を含む。当該抗原結合分子が抗体である場合、前記ポリヌクレオチドは、抗体の重鎖および軽鎖(またはその断片)をコードする1つまたは複数のポリヌクレオチド分子のことをいい、1つのベクターまたは別々のベクターに乗っているポリヌクレオチド分子、および、宿主細胞中の1つまたは複数の位置に存在しているポリヌクレオチド分子を含む。
ベクター
本明細書で用いられる用語「ベクター」は、それが連結されたもう1つの核酸を増やすことができる、核酸分子のことをいう。この用語は、自己複製核酸構造としてのベクター、および、それが導入された宿主細胞のゲノム中に組み入れられるベクターを含む。あるベクターは、自身が動作的に連結された核酸の、発現をもたらすことができる。そのようなベクターは、本明細書では「発現ベクター」とも称される。発現ベクターはウィルスを用いる方法やエレクトロポレーション法などにより宿主細胞に導入することができるが、発現ベクターの導入は生体外での導入に限られるものではなく、ベクターを直接生体に導入することも可能である。
本明細書で用いられる用語「ベクター」は、それが連結されたもう1つの核酸を増やすことができる、核酸分子のことをいう。この用語は、自己複製核酸構造としてのベクター、および、それが導入された宿主細胞のゲノム中に組み入れられるベクターを含む。あるベクターは、自身が動作的に連結された核酸の、発現をもたらすことができる。そのようなベクターは、本明細書では「発現ベクター」とも称される。発現ベクターはウィルスを用いる方法やエレクトロポレーション法などにより宿主細胞に導入することができるが、発現ベクターの導入は生体外での導入に限られるものではなく、ベクターを直接生体に導入することも可能である。
宿主細胞
用語「宿主細胞」、「宿主細胞株」、および「宿主細胞培養物」は、相互に交換可能に用いられ、外来核酸を導入された細胞(そのような細胞の子孫を含む)のことをいう。宿主細胞は「形質転換体」および「形質転換細胞」を含み、これには初代の形質転換細胞および継代数によらずその細胞に由来する子孫を含む。子孫は、親細胞と核酸の内容において完全に同一でなくてもよく、変異を含んでいてもよい。オリジナルの形質転換細胞がスクリーニングされたまたは選択された際に用いられたものと同じ機能または生物学的活性を有する変異体子孫も、本明細書では含まれる。
用語「宿主細胞」、「宿主細胞株」、および「宿主細胞培養物」は、相互に交換可能に用いられ、外来核酸を導入された細胞(そのような細胞の子孫を含む)のことをいう。宿主細胞は「形質転換体」および「形質転換細胞」を含み、これには初代の形質転換細胞および継代数によらずその細胞に由来する子孫を含む。子孫は、親細胞と核酸の内容において完全に同一でなくてもよく、変異を含んでいてもよい。オリジナルの形質転換細胞がスクリーニングされたまたは選択された際に用いられたものと同じ機能または生物学的活性を有する変異体子孫も、本明細書では含まれる。
組み換えの方法および構成
例えば、米国特許第4,816,567号に記載されるとおり、抗原結合分子は組み換えの方法や構成を用いて製造することができる。一態様において、本明細書に記載のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をコードする、単離された核酸が提供される。MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインが抗体可変領域を含む態様において、そのような核酸は、抗体のVLを含むアミノ酸配列および/またはVHを含むアミノ酸配列(例えば、抗体の軽鎖および/または重鎖)をコードしてもよい。さらなる態様において、このような核酸を含む1つまたは複数のベクター(例えば、発現ベクター)が提供される。さらなる態様において、このような核酸を含む宿主細胞が提供される。このような態様の1つでは、宿主細胞は、(1)抗原結合分子のVLを含むアミノ酸配列および抗原結合分子のVHを含むアミノ酸配列をコードする核酸を含むベクター、または、(2)抗原結合分子のVLを含むアミノ酸配列をコードする核酸を含む第一のベクターと抗原結合分子のVHを含むアミノ酸配列をコードする核酸を含む第二のベクターを含む(例えば、形質転換されている)。一態様において、宿主細胞は、真核性である(例えば、チャイニーズハムスター卵巣 (CHO) 細胞)またはリンパ系の細胞(例えば、Y0、NS0、Sp2/0細胞))。一態様において、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の発現に好適な条件下で、上述のとおり当該抗原結合分子をコードする核酸を含む宿主細胞を培養すること、および任意で、当該抗体を宿主細胞(または宿主細胞培養培地)から回収することを含む、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を作製する方法が提供される。
例えば、米国特許第4,816,567号に記載されるとおり、抗原結合分子は組み換えの方法や構成を用いて製造することができる。一態様において、本明細書に記載のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をコードする、単離された核酸が提供される。MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインが抗体可変領域を含む態様において、そのような核酸は、抗体のVLを含むアミノ酸配列および/またはVHを含むアミノ酸配列(例えば、抗体の軽鎖および/または重鎖)をコードしてもよい。さらなる態様において、このような核酸を含む1つまたは複数のベクター(例えば、発現ベクター)が提供される。さらなる態様において、このような核酸を含む宿主細胞が提供される。このような態様の1つでは、宿主細胞は、(1)抗原結合分子のVLを含むアミノ酸配列および抗原結合分子のVHを含むアミノ酸配列をコードする核酸を含むベクター、または、(2)抗原結合分子のVLを含むアミノ酸配列をコードする核酸を含む第一のベクターと抗原結合分子のVHを含むアミノ酸配列をコードする核酸を含む第二のベクターを含む(例えば、形質転換されている)。一態様において、宿主細胞は、真核性である(例えば、チャイニーズハムスター卵巣 (CHO) 細胞)またはリンパ系の細胞(例えば、Y0、NS0、Sp2/0細胞))。一態様において、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の発現に好適な条件下で、上述のとおり当該抗原結合分子をコードする核酸を含む宿主細胞を培養すること、および任意で、当該抗体を宿主細胞(または宿主細胞培養培地)から回収することを含む、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を作製する方法が提供される。
MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の組み換え製造のために、(例えば、上述したものなどの)抗原結合分子をコードする核酸を単離し、さらなるクローニングおよび/または宿主細胞中での発現のために、1つまたは複数のベクターに挿入する。そのような核酸は、従来の手順を用いて容易に単離および配列決定されるだろう(例えば、抗体の場合、抗体の重鎖および軽鎖をコードする遺伝子に特異的に結合することができるオリゴヌクレオチドプローブを用いることで)。
MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をコードするベクターのクローニングまたは発現に好適な宿主細胞は、本明細書に記載の原核細胞または真核細胞を含む。例えば、抗体は、特にグリコシル化およびFcエフェクター機能が必要とされない場合は、細菌で製造してもよい。細菌での抗体断片およびポリペプチドの発現に関して、例えば、米国特許第5,648,237号、第5,789,199号、および第5,840,523号を参照のこと。(加えて、大腸菌における抗体断片の発現について記載したCharlton, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ, 2003), pp.245-254も参照のこと。)発現後、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子は細菌細胞ペーストから可溶性フラクション中に単離されてもよく、またさらに精製することができる。
原核生物に加え、部分的なまたは完全なヒトのグリコシル化パターンを伴う抗原結合分子の産生をもたらす、グリコシル化経路が「ヒト化」されている菌類および酵母の株を含む、糸状菌または酵母などの真核性の微生物は、抗原結合分子コードベクターの好適なクローニングまたは発現宿主である。Gerngross, Nat. Biotech. 22:1409-1414 (2004)および Li et al., Nat. Biotech. 24:210-215 (2006) を参照のこと。
多細胞生物(無脊椎生物および脊椎生物)に由来するものもまた、グリコシル化された抗原結合分子の発現のために好適な宿主細胞である。無脊椎生物細胞の例は、植物および昆虫細胞を含む。昆虫細胞との接合、特にSpodoptera frugiperda細胞の形質転換に用いられる、数多くのバキュロウイルス株が同定されている。
植物細胞培養物も、宿主として利用することができる。例えば、米国特許第5,959,177号、第6,040,498号、第6,420,548号、第7,125,978号、および第6,417,429号(トランスジェニック植物で抗原結合分子を産生するための、PLANTIBODIES(商標)技術を記載)を参照のこと。
脊椎動物細胞もまた宿主として使用できる。例えば、浮遊状態で増殖するように適応された哺乳動物細胞株は、有用であろう。有用な哺乳動物宿主細胞株の他の例は、SV40で形質転換されたサル腎CV1株 (COS-7);ヒト胎児性腎株(Graham et al., J. Gen Virol. 36:59 (1977) などに記載の293または293細胞);仔ハムスター腎細胞 (BHK);マウスセルトリ細胞(Mather, Biol. Reprod. 23:243-251 (1980) などに記載のTM4細胞);サル腎細胞 (CV1);アフリカミドリザル腎細胞 (VERO-76);ヒト子宮頸部癌細胞 (HELA);イヌ腎細胞 (MDCK);Buffalo系ラット肝細胞 (BRL 3A);ヒト肺細胞 (W138);ヒト肝細胞 (Hep G2);マウス乳癌 (MMT 060562);TRI細胞(例えば、Mather et al., Annals N.Y. Acad. Sci. 383:44-68 (1982) に記載);MRC5細胞;および、FS4細胞などである。他の有用な哺乳動物宿主細胞株は、DHFR- CHO細胞 (Urlaub et al., Proc. Natl. Acad. Sci. USA 77:4216 (1980)) を含むチャイニーズハムスター卵巣 (CHO) 細胞;およびY0、NS0、およびSp2/0などの骨髄腫細胞株を含む。抗原結合分子産生に好適な特定の哺乳動物宿主細胞株の総説として、例えば、Yazaki and Wu, Methods in Molecular Biology, Vol. 248 (B.K.C. Lo, ed., Humana Press, Totowa, NJ), pp. 255-268 (2003) を参照のこと。また、宿主細胞の例は動物細胞株に限られるものではない。生体から抽出したエフェクター細胞やmesenchymal stem cell (MSC)をはじめとする各種細胞を宿主細胞として使用することも当然可能である。
MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子のスクリーニング方法
本開示は、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をスクリーニングする方法を提供する。 本開示の記載事項に従いMTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が取得され得る。取得する方法の非限定な一態様として、以下にその具体例がいくつか例示される。
例えば、第1濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性と第2濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性の方が異なることを確認するためには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が比較される。MTA依存的に抗原に対する結合活性がの変化する抗原結合ドメインを選択するには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が異なる抗原結合ドメインが選択される。第1濃度と第2濃度は異なる濃度であり、これらの濃度のどちらか一方を0(即ち、MTAが非存在する)とすることが出来る。
本開示は、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をスクリーニングする方法を提供する。 本開示の記載事項に従いMTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が取得され得る。取得する方法の非限定な一態様として、以下にその具体例がいくつか例示される。
例えば、第1濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性と第2濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性の方が異なることを確認するためには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が比較される。MTA依存的に抗原に対する結合活性がの変化する抗原結合ドメインを選択するには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が異なる抗原結合ドメインが選択される。第1濃度と第2濃度は異なる濃度であり、これらの濃度のどちらか一方を0(即ち、MTAが非存在する)とすることが出来る。
第1濃度を第2濃度より高い濃度に設定する場合、以下の取得方法が例示される。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
MTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第2濃度を0と設定した上、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性が高い抗原結合ドメインを選択するには、第1濃度のMTA存在下における抗原に対する結合活性が、第2濃度のMTA存在下における抗原に対する結合活性よりも高い限り、第1濃度のMTA存在下における抗原結合活性と第2濃度のMTA存在下における抗原結合活性の差は特に限定されないが、好ましくは、第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性の2倍以上であり、さらに好ましくは10倍以上であり、より好ましくは40倍以上である。当該抗原結合活性の差の上限は特に限定されず、当業者の技術において作製可能な限り、400倍、1000倍、10000倍等、いかなる値でもよい。第2濃度のMTA存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が低い抗原結合ドメイ(または当該ドメインを含む抗原結合分子)ンが選択される。
MTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第2濃度を0と設定した上、第1濃度における抗原結合活性が第2濃度における抗原結合活性が低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性が低い抗原結合ドメインを選択するには、第1濃度のMTA存在下における抗原に対する結合活性が、第2濃度のMTA存在下における抗原に対する結合活性よりも低い限り、第1濃度のMTA存在下における抗原結合活性と第2濃度のMTA存在下における抗原結合活性の差は特に限定されないが、好ましくは、第2濃度のMTA存在下における抗原結合活性が第1濃度のMTA存在下における抗原結合活性の2倍以上であり、さらに好ましくは10倍以上であり、より好ましくは40倍以上である。当該抗原結合活性の差の上限は特に限定されず、当業者の技術において作製可能な限り、400倍、1000倍、10000倍等、いかなる値でもよい。第1濃度のMTA存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
さらに本開示において、「MTAの存在下における抗原に対する結合活性が、MTAの非存在下における抗原に対する結合活性より高い」という表現は、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも低い」と表現することもできる。なお本開示においては、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも低い」を「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも弱い」と記載する場合もある。
さらに本開示において、「第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い」および「第1濃度は前記第2濃度より高い濃度である」という表現は、「MTAの高濃度存在下における抗原に対する結合活性が、MTAの低濃度存在下における抗原に対する結合活性よりも高い」、または「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも低い」と表現することもできる。なお本開示においては、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも低い」を「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも弱い」と記載する場合もある。
抗原に対する結合活性を測定する際のMTAの濃度以外の条件は、当業者が適宜選択することが可能であり、特に限定されない。例えば、HEPESバッファー、37℃の条件において測定することが可能である。例えば、Biacore(GE Healthcare)などを用いて測定することが可能である。抗原結合ドメイン(または当該ドメインを含む抗原結合分子)と抗原との結合活性の測定は、抗原が可溶型分子である場合は、抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を固定化したチップへ、抗原をアナライトとして流すことで可溶型分子に対する結合活性を評価することが可能であり、抗原が膜型分子である場合は、抗原を固定化したチップへ、抗原結合ドメインを(または当該ドメインを含む抗原結合分子)アナライトとして流すことで膜型分子に対する結合活性を評価することが可能である。
上記のMTAと同様にMTA類縁体を含む他の低分子化合物に対しても、抗原結合ドメイン(または抗原結合分子)の異なる低分子化合物濃度における抗原結合活性を比較することが可能である。そのような低分子化合物の具体例として、これらに限定されるものではないが、例えば、S-アデノシルメチオニン(SAM:S-adenoshylmethionine)、S-アデノシルホモシステイン(SAH:S-adenosylhomocystein、S-(5'-Adenosyl)-L-homocysteine)等のポリアミン生合成経路の代謝物(Stevensら(J Chromatogr A. 2010 May 7;1217(19):3282-8))、またプリンヌクレオチド代謝経路中の代謝物であるAMP、ADP、ATPもしくはアデノシン等が例示され得る。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子を第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(c)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(d)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 第1濃度のMTA存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子を第2濃度のMTA存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む、抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、を含む抗原結合分子のスクリーニング方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを含む抗原結合分子をMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子を抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインを含む抗原結合分子が抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(c)の工程:
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(d)の工程;
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(c)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(c) 前記工程(b)で解離した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(d)の工程:
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(1) 抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを含む抗原結合分子をMTAの非存在下に置く工程、及び
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
(a) MTAの存在下で抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを含む抗原結合分子を選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを含む抗原結合分子をMTAの非存在下で抗原に結合させる工程、及び
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを含む抗原結合分子を単離する工程、
を含む抗原結合分子のスクリーニング方法を提供する。
抗原結合ドメインを含む抗原結合分子がディスプレイされたライブラリとして、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、または後述する本開示により完成されたライブラリを使用することが出来る。
前記のスクリーニング方法によりスクリーニングされる本開示の抗原結合ドメイン(または当該ドメインを含む抗原結合分子)はいかなる抗原結合ドメイン(または抗原結合分子)でもよく、例えば上述の抗原結合ドメイン(または抗原結合分子)をスクリーニングすることが可能である。例えば、天然の配列を有する抗原結合ドメイン(または抗原結合分子)をスクリーニングしてもよいし、アミノ酸配列が置換された抗原結合ドメイン(または抗原結合分子)をスクリーニングしてもよい。
MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の製造方法
本開示の一側面はMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の製造方法を提供する。
本開示の記載事項に従いMTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が取得され得る。取得する方法の非限定な一態様として、以下にその具体例がいくつか例示される。
例えば、第1濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性と第2濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性の方が異なることを確認するためには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が比較される。MTA依存的に抗原に対する結合活性がの変化する抗原結合ドメインを選択するには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が異なる抗原結合ドメインが選択される。第1濃度と第2濃度は異なる濃度であり、これらの濃度のどちらか一方を0(即ち、MTAが非存在する)とすることが出来る。
本開示の一側面はMTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子の製造方法を提供する。
本開示の記載事項に従いMTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が取得され得る。取得する方法の非限定な一態様として、以下にその具体例がいくつか例示される。
例えば、第1濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性と第2濃度のMTAの存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性の方が異なることを確認するためには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が比較される。MTA依存的に抗原に対する結合活性がの変化する抗原結合ドメインを選択するには、第1濃度のMTA存在下および第2濃度のMTA存在下における抗原に対する抗原結合ドメイン(または当該ドメインを含む抗原結合分子)の結合活性が異なる抗原結合ドメインが選択される。第1濃度と第2濃度は異なる濃度であり、これらの濃度のどちらか一方を0(即ち、MTAが非存在する)とすることが出来る。
第1濃度を第2濃度より高い濃度に設定する場合、以下の取得方法が例示される。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
MTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第2濃度を0と設定した上、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
MTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第2濃度を0と設定した上、第1濃度における抗原結合活性が第2濃度における抗原結合活性が高い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性が高い抗原結合ドメインを選択するには、第1濃度のMTA存在下における抗原に対する結合活性が、第2濃度のMTA存在下における抗原に対する結合活性よりも高い限り、第1濃度のMTA存在下における抗原結合活性と第2濃度のMTA存在下における抗原結合活性の差は特に限定されないが、好ましくは、第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性の2倍以上であり、さらに好ましくは10倍以上であり、より好ましくは40倍以上である。当該抗原結合活性の差の上限は特に限定されず、当業者の技術において作製可能な限り、400倍、1000倍、10000倍等、いかなる値でもよい。第2濃度のMTA存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
MTAの高濃度存在下における抗原結合活性がMTAの低濃度存在下における抗原結合活性より低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第1濃度における抗原結合活性が第2濃度における抗原結合活性が低い抗原結合ドメイ(または当該ドメインを含む抗原結合分子)ンが選択される。
MTA存在下における抗原結合活性がMTA非存在下における抗原結合活性より低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を取得するには、第2濃度を0と設定した上、第1濃度における抗原結合活性が第2濃度における抗原結合活性が低い抗原結合ドメイン(または当該ドメインを含む抗原結合分子)が選択される。
第1濃度のMTA存在下における抗原結合活性が第2濃度のMTA存在下における抗原結合活性が低い抗原結合ドメインを選択するには、第1濃度のMTA存在下における抗原に対する結合活性が、第2濃度のMTA存在下における抗原に対する結合活性よりも低い限り、第1濃度のMTA存在下における抗原結合活性と第2濃度のMTA存在下における抗原結合活性の差は特に限定されないが、好ましくは、第2濃度のMTA存在下における抗原結合活性が第1濃度のMTA存在下における抗原結合活性の2倍以上であり、さらに好ましくは10倍以上であり、より好ましくは40倍以上である。当該抗原結合活性の差の上限は特に限定されず、当業者の技術において作製可能な限り、400倍、1000倍、10000倍等、いかなる値でもよい。第1濃度のMTA存在下において、抗原に対する結合活性が観察されない場合には、この上限は無限大の数値となる。
選択された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合分子を回収することにより、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を製造することが可能である。
さらに本開示において、「MTAの存在下における抗原に対する結合活性が、MTAの非存在下における抗原に対する結合活性より高い」という表現は、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも低い」と表現することもできる。なお本開示においては、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも低い」を「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの非存在下における抗原に対する結合活性がMTAの存在下における抗原に対する結合活性よりも弱い」と記載する場合もある。
さらに本開示において、「第1濃度のMTA存在下における抗原に対する結合活性が前記第2濃度のMTA存在下における抗原に対する結合活性より高い」および「第1濃度は前記第2濃度より高い濃度である」という表現は、「MTAの高濃度存在下における抗原に対する結合活性が、MTAの低濃度存在下における抗原に対する結合活性よりも高い」、または「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも低い」と表現することもできる。なお本開示においては、「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも低い」を「抗原結合ドメイン(または当該ドメインを含む抗原結合分子)のMTAの低濃度存在下における抗原に対する結合活性がMTAの高濃度存在下における抗原に対する結合活性よりも弱い」と記載する場合もある。
抗原に対する結合活性を測定する際のMTAの濃度以外の条件は、当業者が適宜選択することが可能であり、特に限定されない。例えば、HEPESバッファー、37℃の条件において測定することが可能である。例えば、Biacore(GE Healthcare)などを用いて測定することが可能である。抗原結合ドメイン(または当該ドメインを含む抗原結合分子)と抗原との結合活性の測定は、抗原が可溶型分子である場合は、抗原結合ドメイン(または当該ドメインを含む抗原結合分子)を固定化したチップへ、抗原をアナライトとして流すことで可溶型分子に対する結合活性を評価することが可能であり、抗原が膜型分子である場合は、抗原を固定化したチップへ、抗原結合ドメインを(または当該ドメインを含む抗原結合分子)アナライトとして流すことで膜型分子に対する結合活性を評価することが可能である。
上記のMTAと同様にMTA類縁体を含む他の低分子化合物に対しても、抗原結合ドメイン(または抗原結合分子)の異なる低分子化合物濃度における抗原結合活性を比較することが可能である。そのような低分子化合物の具体例として、これらに限定されるものではないが、例えば、S-アデノシルメチオニン(SAM:S-adenoshylmethionine)、S-アデノシルホモシステイン(SAH:S-adenosylhomocystein、S-(5'-Adenosyl)-L-homocysteine)等のポリアミン生合成経路の代謝物(Stevensら(J Chromatogr A. 2010 May 7;1217(19):3282-8))、またプリンヌクレオチド代謝経路中の代謝物であるAMP、ADP、ATPもしくはアデノシン等が例示され得る。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) MTAに結合可能な抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) MTAに結合可能な抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAに結合可能な抗原結合ドメインを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 第1濃度のMTA存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(1)~(d)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(1)~(e)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリを第1濃度のMTA存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインを第2濃度のMTA存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(e)の工程:
(a) 第1濃度のMTA存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 第1濃度のMTA存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインを第2濃度のMTA存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) MTAに結合可能な抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAに結合可能な抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) MTAに結合可能な抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAに結合可能な抗原結合ドメインをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合したことを確認する工程、
(c) 前記抗原に結合した前記抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で抗原に結合したことを確認した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAの存在下で抗原結合ドメインを抗原に接触させる工程、
(b) 前記工程(a)で前記抗原結合ドメインが抗原に結合しないことを確認する工程、
(c) 前記抗原に結合しない抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(d)の工程:
(a) 抗原結合ドメインがディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインがディスプレイされたライブラリを MTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下の(a)~(e)の工程;
(a) 抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) 抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(d)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で結合した抗原結合ドメインをMTAの非存在下に置く工程、
(c) 前記工程(b)で解離した抗原結合ドメインを単離する工程、及び
(d) 前記工程(c)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(1)から(e)の工程:
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(1) 抗原結合ドメインがディスプレイされたライブラリをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、
(a) 前記工程(1)及び(2)で選択された抗原結合ドメインがディスプレイされたライブラリをMTAの存在下で抗原と接触させる工程、
(b) 前記工程(a)で抗原に結合した抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択した抗原結合ドメインをMTAの非存在下に置く工程、
(d) 前記工程(c)で抗原結合活性が、前記工程(b)で選択した基準より弱い抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
非限定的な一態様において、本開示は、以下(a)~(e)の工程:
(a) MTAの存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
(a) MTAの存在下で抗原結合ドメインがディスプレイされたライブラリを抗原に接触させる工程、
(b) 前記工程(a)で抗原に結合しない抗原結合ドメインを選択する工程、
(c) 前記工程(b)で選択された抗原結合ドメインをMTAの非存在下で抗原に結合させる工程、
(d) 前記工程(c)で抗原に結合した抗原結合ドメインを単離する工程、及び
(e) 前記工程(d)で単離された抗原結合ドメインを含む抗原結合分子をコードするポリヌクレオチドが作動可能に連結されたベクターが導入された細胞を培養し、細胞の培養液から抗原結合ドメインを含む抗原結合分子を回収する工程、
を含む抗原結合分子の製造方法を提供する。
抗原結合ドメインがディスプレイされたライブラリとして、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、または後述する本開示により完成されたライブラリを使用することが出来る。
本開示の製造方法において単離された抗原結合ドメインがライブラリから選択された場合には、後述する実施例に記載されているように、当該抗原結合ドメインをコードするポリヌクレオチドはファージ等のウイルスから通常の遺伝子増幅によって単離される。また、このように単離された抗原結合ドメインまたは抗体がハイブリドーマ等の細胞の培養液から選択された場合には、前記の抗体の項で示したように当該細胞から抗体遺伝子等が通常の遺伝子増幅によって単離される。
前記の製造方法により製造される本開示の抗原結合分子はいかなる抗原結合分子でもよく、例えば天然の配列を有する抗原結合分子を製造してもよいし、アミノ酸配列が置換された抗原結合分子を製造してもよい。
ライブラリ
本明細書において「ライブラリ」とは互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸若しくはポリヌクレオチドをいう。ライブラリ中に含まれる抗原結合ドメインを含む抗原結合分子、または/および抗原結合ドメインを含む抗原結合分子をコードする核酸の配列は単一の配列ではなく、互いに配列の異なる複数の抗原結合分子または/および互いに配列の異なる複数の抗原結合分子をコードする核酸である。
本明細書において「ライブラリ」とは互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸若しくはポリヌクレオチドをいう。ライブラリ中に含まれる抗原結合ドメインを含む抗原結合分子、または/および抗原結合ドメインを含む抗原結合分子をコードする核酸の配列は単一の配列ではなく、互いに配列の異なる複数の抗原結合分子または/および互いに配列の異なる複数の抗原結合分子をコードする核酸である。
ある一態様によれば、本開示の抗原結合ドメイン(または当該ドメインを含む抗原結合分子)は、MTA依存的に抗原に対する結合活性を変化させる少なくとも一つのアミノ酸残基が抗原結合ドメインに含まれている互いに配列の異なる複数の抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸から主としてなるライブラリから取得され得る。以下では、MTA依存的に抗原に対する結合活性を変化させる少なくとも一つのアミノ酸残基が抗原結合ドメインに含まれている互いに配列の異なる複数の抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸から主としてなるライブラリについて例示される。
MTA依存的に抗原に対する結合活性を変化させるアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子ライブラリは、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインおよびそのような抗原結合ドメインを含む抗原結合分子を効率的に取得することが可能である。本明細書における「ライブラリ」の態様として、MTAスイッチ抗原結合ドメイン、即ちMTA存在下で標的抗原に結合するがMTA非存在下では標的抗原に結合しない抗原結合ドメインを含む抗原結合分子、または高濃度のMTA存在下で標的抗原に結合するが低濃度のMTA存在下で標的抗原に結合しない抗原結合ドメインを含む抗原結合分子を効率的に取得できるライブラリを提供するだけではなく、MTA逆スイッチ抗原結合ドメイン、即ちMTA非存在化で標的抗原に結合するかMTA存在下では標的抗原に結合しない抗原結合ドメインを含む抗原結合分子、および低濃度のMTA存在下で標的抗原に結合するが高濃度のMTA存在下で標的抗原に結合しない抗原結合ドメインを含む抗原結合分子を効率的に取得できるライブラリも提供することができる。
本開示における一つの実施形態では、本開示の抗原結合分子と異種ポリペプチドとの融合ポリペプチドが作製され得る。ある実施形態では、融合ポリペプチドはウイルスコートタンパク質、例えばpIII、pVIII、pVII、pIX、Soc、Hoc、gpD、pVIおよびその変異体からなる群から選択されるウイルスコートタンパク質の少なくとも一部と融合され得る。
ある実施形態では、本開示の抗原結合分子は、ScFv、Fab断片、F(ab)2またはF(ab')2であり得るため、別の一つの実施形態では、これらの抗原結合分子と異種ポリペプチドとの融合ポリペプチドであって互いに配列の異なる複数の融合ポリペプチドから主としてなるライブラリが提供される。具体的には、これらの抗原結合分子とウイルスコートタンパク質、例えばpIII、pVIII、pVII、pIX、Soc、Hoc、gpD、pVIおよびその変異体からなる群から選択されるウイルスコートタンパク質の少なくとも一部と融合された融合ポリペプチドであって互いに配列の異なる複数の融合ポリペプチドから主としてなるライブラリが提供される。本開示の抗原結合分子はさらに二量体化ドメインを含み得る。ある実施形態では、前記二量体化ドメインは抗体の重鎖または軽鎖の可変領域とウイルスコートタンパク質の少なくとも一部との間に存在し得る。この二量体化ドメインには、二量体化配列の少なくとも1つ、および/または1つまたは複数のシステイン残基を含む配列が含まれ得る。この二量体化ドメインは、好ましくは重鎖可変領域または定常領域のC末端と連結され得る。二量体化ドメインは、前記抗体可変領域がウイルスのコートタンパク質成分との融合ポリペプチド成分として作製されている(二量体化ドメインの後ろにアンバー終止コドンを有さない)かどうかによって、または、前記抗体可変領域が主にウイルスコートタンパク質成分を含まずに作製されている(例えば、二量体化ドメインの後にアンバー終止コドンを有する)かどうかによって、様々な構造をとることが可能である。前記抗体可変領域が主にウイルスのコートタンパク質成分との融合ポリペプチドとして作製されるときは、1つまたは複数のジスルフィド結合および/または単一の二量体化配列によって二価提示がもたらされる。
本開示のライブラリの非限定的な一態様としては、1.2×108以上の多様性を有するライブラリが、即ち、1.2×108以上のお互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸を含むライブラリが挙げられる。
本明細書においては、「互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子」という記載における「互いに配列の異なる」との用語は、ライブラリ中の個々の抗原結合分子の配列が相互に異なることを意味する。すなわち、ライブラリ中における互いに異なる配列の数は、ライブラリ中の配列の異なる独立クローンの数が反映され、「ライブラリサイズ」と指称される場合もある。通常のファージディスプレイライブラリでは106から1012であり、リボゾームディスプレイ法等の公知の技術を適用することによってライブラリサイズを1014まで拡大することが可能である。しかしながら、ファージライブラリのパンニング選択時に使用されるファージ粒子の実際の数は、通常、ライブラリサイズよりも10ないし10,000倍大きい。この過剰倍数は、「ライブラリ当量数」とも呼ばれるが、同じアミノ酸配列を有する個々のクローンが10ないし10,000存在し得ることを表す。よって本開示における「互いに配列の異なる」との用語はライブラリ当量数が除外されたライブラリ中の個々の抗原結合分子の配列が相互に異なること、より具体的には互いに配列の異なる抗原結合分子が106から1014分子、好ましくは107から1012分子、さらに好ましくは108から1011、特に好ましくは108から1010存在することを意味する。
また、本開示の、互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸から主としてなるライブラリという記載における「複数の」との用語は、例えば本開示の抗原結合分子、融合ポリペプチド、ポリヌクレオチド分子、ベクターまたはウイルスは、通常、その物質の2つ以上の種類の集合を指す。例えば、ある2つ以上の物質が特定の形質に関して互いに異なるならば、その物質には2種類以上が存在することを表す。例としては、アミノ酸配列中の特定のアミノ酸部位でアミノ酸変異が観察される変異体が挙げられ得る。例えば、特定のアミノ酸部位のアミノ酸以外は実質的に同じ、好ましくは同一の配列である2つ以上の抗原結合分子がある場合、抗原結合分子は複数個存在する。他の例では、特定のアミノ酸部位のアミノ酸をコードする塩基以外は実質的に同じ、好ましくは同一の配列である2つ以上のポリヌクレオチド分子があるならば、ポリヌクレオチド分子は複数個存在する。
さらに、本開示の、互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸から主としてなるライブラリという記載における「から主としてなる」との用語は、ライブラリ中の配列の異なる独立クローンの数のうち、MTA依存的に抗原に対する抗原結合分子の結合活性が異なっている抗原結合分子の数が反映される。具体的には、そのような結合活性を示す抗原結合分子がライブラリ中に少なくとも104分子存在することが好ましい。また、より好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも105分子存在するライブラリから取得され得る。さらに好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも106分子存在するライブラリから取得され得る。特に好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも107分子存在するライブラリから取得され得る。また、好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも108分子存在するライブラリから取得され得る。別の表現では、ライブラリ中の配列の異なる独立クローンの数のうち、MTA依存的に抗原に対する抗原結合ドメインの結合活性が変化する抗原結合分子の割合としても好適に表現され得る。具体的には、本開示の抗原結合ドメインは、そのような結合活性を示す抗原結合分子がライブラリ中の配列の異なる独立クローンの数の10-6%%から80%、好ましくは10-5%から60%、より好ましくは10-4%から40%含まれるライブラリから取得され得る。融合ポリペプチド、ポリヌクレオチド分子またはベクターの場合も、上記と同様、分子の数や分子全体における割合で表現され得る。また、ウイルスの場合も、上記と同様、ウイルス個体の数や個体全体における割合で表現され得る。本開示の非限定の一態様として、複数の抗原結合分子が結合する抗原が1種類の場合、そのような結合活性を示す抗原結合分子がライブラリ中に少なくとも10分子, 100分子, 1000分子, 104分子,105分子,106分子,107分子又は108分子存在することが好ましい。また、より好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも10分子存在するライブラリから取得され得る。さらに好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも100分子存在するライブラリから取得され得る。特に好ましくは、本開示の抗原結合ドメインはそのような結合活性を示す抗原結合分子が少なくとも1000分子存在するライブラリから取得され得る。
一側面において、本開示は複数の抗体可変領域または/および抗体可変領域をコードする核酸を含むライブラリを提供する。
非限定的な一態様において、本開示のライブラリに含まれるもしくは本開示のライブラリに含まれる核酸にコードされる複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAXXXYXXSGGSTYYASWAKG(配列番号:66)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXXXLS(配列番号:68)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAXXXYXXSGGSTYYASWAKG(配列番号:66)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXXXLS(配列番号:68)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
非限定的な一態様において、本開示のライブラリに含まれるもしくは本開示のライブラリに含まれる核酸にコードされる複数の抗体可変領域は、下記:
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸、X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸である。更に、異なる位置に存在するXもしくはX1は同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
XAXWMC(配列番号:65)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸、X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸である。更に、異なる位置に存在するXもしくはX1は同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
非限定的な一態様において、本開示のライブラリに含まれるもしくは本開示のライブラリに含まれる核酸にコードされる複数の抗体可変領域は、下記:
XXAXWMC(配列番号:73)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸、X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であ。更に、異なる位置に存在するXもしくはX1は同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
XXAXWMC(配列番号:73)を含むH-CDR1;
CIFAX1XXYXXSGGSTYYASWAKG(配列番号:71)を含むH-CDR2;
GXGXXXGXXDEL(配列番号:67)を含むH-CDR3;
QSSEXVXXX1XLS(配列番号:72)を含むL-CDR1;
XAXTXPX(配列番号:69)を含むL-CDR2;及び
AGLYXGNIPA(配列番号:70)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸、X1はA、D、E、G、H、I、K、L、M、N、P、Q、R、S、TおよびVから選ばれるアミノ酸であ。更に、異なる位置に存在するXもしくはX1は同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
非限定的な一態様において、本開示のライブラリに含まれるもしくは本開示のライブラリに含まれる核酸にコードされる複数の抗体可変領域は、下記:
XXXWMC(配列番号:74)を含むH-CDR1;
CIXSXXXXTXYASWVNG(配列番号:75)を含むH-CDR2;
EXXXXSGALNL(配列番号:76)を含むH-CDR3;
HSSKXVXXXXXLA(配列番号:77)を含むL-CDR1;
XAXXLAS(配列番号:78)を含むL-CDR2;及び
QGTYXXXXFYFA(配列番号:79)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
XXXWMC(配列番号:74)を含むH-CDR1;
CIXSXXXXTXYASWVNG(配列番号:75)を含むH-CDR2;
EXXXXSGALNL(配列番号:76)を含むH-CDR3;
HSSKXVXXXXXLA(配列番号:77)を含むL-CDR1;
XAXXLAS(配列番号:78)を含むL-CDR2;及び
QGTYXXXXFYFA(配列番号:79)を含むL-CDR3;
を含む抗体可変領域である。ここで、Xは任意のアミノ酸を指し、異なる位置に存在するXは同種のアミノ酸であっても良く、同種のアミノ酸でなくても良い。
非限定的な一態様において、本開示のライブラリに含まれるもしくは本開示のライブラリに含まれる核酸にコードされる複数の抗体可変領域は、下記:
X2X3X4X5G(配列番号:80)を含むH-CDR1;
X6IGX7X8X9X10X11WX12PX13WVKX14(配列番号:81)を含むH-CDR2;
GX15X16X17X18X19X20NAX21DP(配列番号:82)を含むH-CDR3;
QSSQSVX22X23NNX24LS(配列番号:83)を含むL-CDR1;
DASTLAS(配列番号:84)を含むL-CDR2;及び
HGX25X26X27X28X29X30X31X32DNX33(配列番号:85)を含むL-CDR3;
を含む抗体可変領域であり、
X2はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X3はD、E、F、H、K、N、P、RおよびYから選ばれるアミノ酸、
X4はA、I、P、TおよびVから選ばれるアミノ酸、
X5はA、E、F、H、I、K、L、M、N、Q、S、T、V、WおよびYから選ばれるアミノ酸、
X6はD、IおよびVから選ばれるアミノ酸、
X7はA、D、E、G、I、K、QおよびRから選ばれるアミノ酸、
X8はD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X9はA、D、E、F、G、HおよびSから選ばれるアミノ酸、
X10はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X11はA、D、E、G、H、I、K、L、N、P、Q、R、S、TおよびVから選ばれるアミノ酸、
X12はA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X13はA、F、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X14はA、FおよびGから選ばれるアミノ酸、
X15はA、E、F、G、H、K、L、Q、R、S、T、WおよびYから選ばれるアミノ酸、
X16はF、H、K、N、WおよびYから選ばれるアミノ酸、
X17はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X18はA、D、E、G、H、QおよびSから選ばれるアミノ酸、
X19はFおよびYから選ばれるアミノ酸、
X20はN、TおよびVから選ばれるアミノ酸、
X21はFおよびWから選ばれるアミノ酸、
X22はA、E、F、H、I、K、L、N、R、S、T、V、WおよびYから選ばれるアミノ酸、
X23はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X24はA、E、F、G、H、SおよびYから選ばれるアミノ酸、
X25はA、SおよびTから選ばれるアミノ酸、
X26はA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X27はA、D、E、F、G、H、L、N、Q、R、S、T、VおよびYから選ばれるアミノ酸、
X28はA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X29はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、W、Yから選ばれるアミノ酸、
X30はA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WおよびYから選ばれるアミノ酸、
X31はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X32はA、F、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X33はAおよびGから選ばれるアミノ酸である。
X2X3X4X5G(配列番号:80)を含むH-CDR1;
X6IGX7X8X9X10X11WX12PX13WVKX14(配列番号:81)を含むH-CDR2;
GX15X16X17X18X19X20NAX21DP(配列番号:82)を含むH-CDR3;
QSSQSVX22X23NNX24LS(配列番号:83)を含むL-CDR1;
DASTLAS(配列番号:84)を含むL-CDR2;及び
HGX25X26X27X28X29X30X31X32DNX33(配列番号:85)を含むL-CDR3;
を含む抗体可変領域であり、
X2はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X3はD、E、F、H、K、N、P、RおよびYから選ばれるアミノ酸、
X4はA、I、P、TおよびVから選ばれるアミノ酸、
X5はA、E、F、H、I、K、L、M、N、Q、S、T、V、WおよびYから選ばれるアミノ酸、
X6はD、IおよびVから選ばれるアミノ酸、
X7はA、D、E、G、I、K、QおよびRから選ばれるアミノ酸、
X8はD、E、F、G、H、I、K、L、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X9はA、D、E、F、G、HおよびSから選ばれるアミノ酸、
X10はA、D、E、F、G、H、I、K、L、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X11はA、D、E、G、H、I、K、L、N、P、Q、R、S、TおよびVから選ばれるアミノ酸、
X12はA、D、E、F、G、H、I、K、L、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X13はA、F、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X14はA、FおよびGから選ばれるアミノ酸、
X15はA、E、F、G、H、K、L、Q、R、S、T、WおよびYから選ばれるアミノ酸、
X16はF、H、K、N、WおよびYから選ばれるアミノ酸、
X17はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X18はA、D、E、G、H、QおよびSから選ばれるアミノ酸、
X19はFおよびYから選ばれるアミノ酸、
X20はN、TおよびVから選ばれるアミノ酸、
X21はFおよびWから選ばれるアミノ酸、
X22はA、E、F、H、I、K、L、N、R、S、T、V、WおよびYから選ばれるアミノ酸、
X23はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X24はA、E、F、G、H、SおよびYから選ばれるアミノ酸、
X25はA、SおよびTから選ばれるアミノ酸、
X26はA、D、E、F、G、H、I、K、L、M、N、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X27はA、D、E、F、G、H、L、N、Q、R、S、T、VおよびYから選ばれるアミノ酸、
X28はA、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X29はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、W、Yから選ばれるアミノ酸、
X30はA、D、E、F、G、H、I、K、L、N、P、Q、R、V、WおよびYから選ばれるアミノ酸、
X31はA、D、E、F、G、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X32はA、F、H、I、K、L、N、P、Q、R、S、T、V、WおよびYから選ばれるアミノ酸、
X33はAおよびGから選ばれるアミノ酸である。
未改変体(鋳型、テンプレート)
本開示の一態様において、MTA依存的に抗原に対する結合活性を変化させるアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子ライブラリ中の抗原結合ドメインは抗体可変領域であり、当該ライブラリに含まれるもしくは当該ライブラリに含まれる核酸にコードされる「複数の抗原結合分子」に含まれる抗原結合ドメインは、MTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体を含む。
本開示の一態様において、MTA依存的に抗原に対する結合活性を変化させるアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子ライブラリ中の抗原結合ドメインは抗体可変領域であり、当該ライブラリに含まれるもしくは当該ライブラリに含まれる核酸にコードされる「複数の抗原結合分子」に含まれる抗原結合ドメインは、MTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体を含む。
非限定的な一態様において、前記抗体可変領域未改変体はMTAに対する結合活性を有する抗体可変領域である。詳細な一態様において、本開示の抗体可変領域未改変体は、アデノシンまたは/及びS-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に結合しない抗体可変領域であってもい。詳細な別態様において、本開示の抗体可変領域未改変体は、アデノシンに対しても結合活性を有する抗体可変領域であっても良い。また、本開示の抗体可変領域未改変体は、(5'-Adenosyl)-L-homocysteine (SAH)、AMP,ADPまたは/及びATPに対しても結合活性を有する抗体可変領域であって良い。
MTAに対する結合活性を有する抗体可変領域未改変体は、MTAに対する結合活性を有する抗体から取得することができる。
MTA依存的に抗原に対する結合活性を変化させるアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子ライブラリをデザインするときに出発分子として使用することが出来るため、本明細書で「鋳型」または「テンプレート」と称する場合もある。
MTA依存的に抗原に対する結合活性を変化させるアミノ酸残基を有する抗原結合ドメインを含む抗原結合分子ライブラリをデザインするときに出発分子として使用することが出来るため、本明細書で「鋳型」または「テンプレート」と称する場合もある。
非限定的な一態様において、本開示の抗体可変領域未改変体は下記いずれかであっても良い:
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域;
e) 配列番号:31で示す重鎖可変領域と配列番号:32で示す軽鎖可変領域を有する抗体可変領域。
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域;
e) 配列番号:31で示す重鎖可変領域と配列番号:32で示す軽鎖可変領域を有する抗体可変領域。
本開示においてライブラリを製造する際に用いられる出発分子(鋳型、テンプレート)の取得方法(スクリーニング方法)に関し、これらの抗原結合ドメイン等はどのように調製されてもよく、あらかじめ存在している抗原結合ドメイン又は抗体、あらかじめ存在しているライブラリ(ファージライブラリ等)、動物への免疫から得られたハイブリドーマや免疫動物からのB細胞から作製された抗体又はライブラリ、例えばこれに限定されるものではないが、MTAを含む低分子化合物と適切に連結された免疫原性が高いT細胞エピトープペプチドのようなアジュバント作用剤との連結剤(conjugate)によって免疫された動物のB細胞等の免疫細胞から作製された抗体またはライブラリ等を用いることが可能である 。当該T細胞エピトープペプチドの非限定な一例として、Tetanus toxin由来のp30ヘルパーペプチド(配列番号:4で表され、Fragment C(FrC)とも指称される)等が好適に挙げられる。
前記のスクリーニング方法によって取得される本発明の抗原結合ドメインの非限定的な一態様として、抗体の重鎖および軽鎖の可変領域を含む抗原結合ドメインが挙げられる。こうした抗原結合ドメインの例としては、「scFv(single chain Fv)」、「単鎖抗体(single chain antibody)」、「Fv」、「scFv2(single chain Fv 2)」、「Fab」または「F(ab')2」またはIgG等が好適に挙げられ、これらより成るライブラリを用いることも可能である。
さらに、前記のスクリーニング方法によって取得される本発明の抗原結合ドメイン又は抗原結合分子の調製法の非限定の1態様として、前記のライブラリを用いて、パンニングにより低分子化合物に対して結合活性を有する抗原結合ドメインまたは抗原結合分子を調製する手法を用いることが可能である。ライブラリとしては、これに限定されないが、ファージディスプレイライブラリ、リボゾームディスプレイライブラリ、mRNAディスプレイライブラリ、cDNAディスプレイライブラリ、CISディスプレイライブラリ、E. coliディスプレイライブラリ、グラム陽性菌ディスプレイライブラリ、酵母ディスプレイライブラリ、哺乳類細胞ディスプレイライブラリ、ウイルスディスプレイライブラリ、インビトロウイルスディスプレイライブラリ等を用いることが可能である。
前記パンニングにより低分子化合物に対して結合活性を有する抗原結合ドメイン又は抗原結合分子を調製する手法の一態様として、ビーズ等の担体に固相化された低分子化合物を使用することが可能である。固相化された低分子化合物は、化学的にリンカーを介してビオチンと接続された状態で合成された低分子化合物を、ストレプトアビジンやニュートラアビジンが固相化されたビーズもしくはプレートと接触させる手法や、ウシ血清アルブミン(BSA)等のアジュバントと共有結合により接続された低分子化合物を、疎水相互作用によりビーズもしくはプレートに接着させる手法等により作製することが可能だがこれに限定されない。これらの方法は既に公知である(J Immunol Methods. 2003 Sep;280(1-2):139-55、BMC Biotechnol. 2009 Jan 29;9:6. doi: 10.1186/1472-6750-9-6)。これらの、固相化された低分子化合物に対して結合活性を有する抗原結合ドメイン又は抗原結合分子を回収することにより、低分子化合物に対して結合活性を有する抗原結合ドメイン又は抗原結合分子を調製することが可能となる。
または、前記パンニングにより低分子化合物に対して結合活性を有する抗原結合ドメイン又は抗原結合分子を調製する手法の別の一態様として、蛍光標識された低分子化合物や、ビオチン標識された低分子化合物と蛍光標識されたストレプトアビジン(もしくはニュートラアビジンまたはアビジン)を使用することが可能である。細胞等の表面に提示されたライブラリに対して当該蛍光標識された低分子化合物、もしくはビオチン標識された低分子化合物と蛍光標識されたストレプトアビジン(もしくはニュートラアビジンまたはアビジン)を接触させたのちに、fluorescence activated cell sorting(FACS)法を使用することによって、当該低分子化合物に対して結合活性を有する抗原結合ドメイン又は抗原結合分子を調製することが可能となる。これらの方法は既に公知である(Proc Natl Acad Sci U S A. 2000 Sep 26;97(20):10701-5.)。
また、前記のスクリーニング方法によって取得される本開示の抗原結合ドメイン又は抗原結合分子の調製法の非限定の一態様として、あらかじめ存在している低分子化合物に結合活性を有する抗原結合ドメインを用いることが可能である。例えば、これに限定されるわけではないが、MTAを例にした場合、MTAに結合活性を有する抗原結合ドメイン、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン、MTAと相互作用するアミノ酸残基を有する抗原結合ドメインを用いることが可能である。
また、前記のスクリーニング方法によって取得される本開示の抗原結合ドメイン又は抗原結合分子の調製法の非限定の一態様として、あらかじめ存在している低分子化合物に結合活性を有する抗原結合ドメインを用いることが可能である。例えば、これに限定されるわけではないが、MTAを例にした場合、MTAに結合活性を有する抗原結合ドメイン、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン、MTAと相互作用するアミノ酸残基を有する抗原結合ドメインを用いることが可能である。
改変体
本開示における「改変体」とは、親配列の未改変体に対して、少なくとも1又はそれ以上のアミノ酸が置換されている親配列の各々異なる改変体をいう。
一態様において本開示の抗体可変領域改変体を作製するため、前記の抗体可変領域未改変体の重鎖可変領域または軽鎖可変領域の配列、もしくはCDR配列またはフレームワーク配列に含まれる抗原結合分子を作製するために部位特異的変異誘発法(Kunkelら(Proc. Natl. Acad. Sci. U.S.A. (1985) 82, 488-492))やOverlap extension PCR等の公知の方法が適宜採用され得る。
本開示における「改変体」とは、親配列の未改変体に対して、少なくとも1又はそれ以上のアミノ酸が置換されている親配列の各々異なる改変体をいう。
一態様において本開示の抗体可変領域改変体を作製するため、前記の抗体可変領域未改変体の重鎖可変領域または軽鎖可変領域の配列、もしくはCDR配列またはフレームワーク配列に含まれる抗原結合分子を作製するために部位特異的変異誘発法(Kunkelら(Proc. Natl. Acad. Sci. U.S.A. (1985) 82, 488-492))やOverlap extension PCR等の公知の方法が適宜採用され得る。
非限定的な一態様において、本開示のライブラリに含まれる抗体可変領域改変体が抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、重鎖および/または軽鎖のCDR1、CDR2、および/またはCDR3に位置する。非限定な別の一態様では、前記抗体可変領域改変体が抗体可変領域未改変体と異なるアミノ酸を有するアミノ酸部位は、重鎖および/または軽鎖のFR1、FR2、FR3および/またはFR4に位置する。
抗原結合分子の軽鎖および/または重鎖可変領域のフレームワーク配列はMTAと相互作用するアミノ酸が重鎖および/または軽鎖の抗原結合ドメインに含まれている限り、どのようなフレームワーク配列も使用され得る。フレームワーク配列の起源は、限定されないが非ヒト動物の任意の生物またはヒトから取得され得る。好ましくは、任意の生物としては、マウス、ラット、モルモット、ハムスター、アレチネズミ、ネコ、ウサギ、イヌ、ヤギ、ヒツジ、ウシ、ウマ、ラクダ、および非ヒト霊長類から選択される生物が好適に挙げられる。特に好適な実施形態では、抗原結合分子の軽鎖および/または重鎖可変領域のフレームワーク配列は、ヒトの生殖細胞系フレームワーク配列を有していることが望ましい。したがって、本開示の一態様においてフレームワーク配列が完全にヒトの配列であるならば、ヒトに投与(例えば疾病の治療)された場合、本開示の抗原結合分子は免疫原性反応を殆どあるいは全く引き起こさないと考えられる。上記の意味から、本開示の「生殖細胞系列の配列を含む 」とは、本開示のフレームワーク配列の一部が、いずれかのヒトの生殖細胞系フレームワーク配列の一部と同一であることを意味する。具体的には、本開示のフレームワーク配列は、生殖細胞系列の配列と少なくとも50%以上、60%以上、70%以上、80%以上、90%以上、又は100%以上の相同性を有することが好ましい。例えば、本開示の抗原結合分子の重鎖FR2の配列が複数の異なるヒトの生殖細胞系フレームワーク配列の重鎖FR2配列が組み合わされた配列である場合も、本開示の「生殖細胞系列の配列を含む 」抗原結合分子である。また、本開示の抗原結合分子のフレームワーク配列が、置換されている配列である場合も、本開示の「生殖細胞系列の配列を含む 」抗原結合分子である。そのような置換されている配列の例として、とくに、ヒトの生殖細胞系フレームワーク配列の一部のアミノ酸が、MTA及び/またはアデノシンの存在もしくは非存在によって抗原に対する抗原結合分子の結合活性を変化させるアミノ酸に置換されている配列が挙げられる。
フレームワークの例としては、例えばV-Base(http://vbase.mrc-cpe.cam.ac.uk/)等のウェブサイトに含まれている、現在知られている完全にヒト型のフレームワーク領域の配列が好適に挙げられる。これらのフレームワーク領域の配列が本開示の抗原結合分子に含まれる生殖細胞系列の配列として適宜使用され得る。生殖細胞系列の配列はその類似性にもとづいて分類され得る(Tomlinsonら(J. Mol. Biol. (1992) 227, 776-798)WilliamsおよびWinter(Eur. J. Immunol. (1993) 23, 1456-1461)およびCoxら(Nat. Genetics (1994) 7, 162-168))。 7つのサブグループに分類されるVκ、10のサブグループに分類されるVλ、7つのサブグループに分類されるVHから好適な生殖細胞系列の配列が適宜選択され得る。
完全にヒト型のVH配列は、下記のみに限定されるものではないが、例えばVH1サブグループ(例えば、VH1-2、VH1-3、VH1-8、VH1-18、VH1-24、VH1-45、VH1-46、VH1-58、VH1-69)、VH2サブグループ(例えば、VH2-5、VH2-26、VH2-70)、VH3サブグループ(VH3-7、VH3-9、VH3-11、VH3-13、VH3-15、VH3-16、VH3-20、VH3-21、VH3-23、VH3-30、VH3-33、VH3-35、VH3-38、VH3-43、VH3-48、VH3-49、VH3-53、VH3-64、VH3-66、VH3-72、VH3-73、VH3-74)、VH4サブグループ(VH4-4、VH4-28、VH4-31、VH4-34、VH4-39、VH4-59、VH4-61)、VH5サブグループ(VH5-51)、VH6サブグループ(VH6-1)、VH7サブグループ(VH7-4、VH7-81)のVH配列等が好適に挙げられる。これらは公知文献(Matsudaら(J. Exp. Med. (1998) 188, 1973-1975))等にも記載されており、当業者はこれらの配列情報をもとに本開示の抗原結合分子を適宜設計することが可能である。これら以外の完全にヒト型のフレームワークまたはフレームワークの準領域も好適に使用され得る。
完全にヒト型のVκ配列は、下記のみに限定されるものではないが、例えばVk1サブグループに分類されるA20、A30、L1、L4、L5、L8、L9、L11、L12、L14、L15、L18、L19、L22、L23、L24、O2、O4、O8、O12、O14、O18、Vk2サブグループに分類されるA1、A2、A3、A5、A7、A17、A18、A19、A23、O1、O11、Vk3サブグループに分類されるA11、A27、L2、L6、L10、L16、L20、L25、Vk4サブグループに分類されるB3、Vk5サブグループに分類されるB2(本明細書においてはVk5-2とも指称される))、Vk6サブグループに分類されるA10、A14、A26等(Kawasakiら(Eur. J. Immunol. (2001) 31, 1017-1028)、SchableおよびZachau(Biol. Chem. Hoppe Seyler (1993) 374, 1001-1022)およびBrensing-Kuppersら(Gene (1997) 191, 173-181))が好適に挙げられる。
完全にヒト型のVλ配列は、下記のみに限定されるものではないが、例えばVL1サブグループに分類されるV1-2、V1-3、V1-4、V1-5、V1-7、V1-9、V1-11、V1-13、V1-16、V1-17、V1-18、V1-19、V1-20、V1-22、VL1サブグループに分類されるV2-1、V2-6、V2-7、V2-8、V2-11、V2-13、V2-14、V2-15、V2-17、V2-19、VL3サブグループに分類されるV3-2、V3-3、V3-4、VL4サブグループに分類されるV4-1、V4-2、V4-3、V4-4、V4-6、VL5サブグループに分類されるV5-1、V5-2、V5-4、V5-6等(Kawasakiら(Genome Res. (1997) 7, 250-261))が好適に挙げられる。
通常これらのフレームワーク配列は一またはそれ以上のアミノ酸残基の相違により互いに異なっている。本開示に使用される完全にヒト型のフレームワークの例としては、これだけに限定されるわけではないが、ほかにもKOL、NEWM、REI、EU、TUR、TEI、LAY、POM等が挙げられる(例えば、前記のKabatら (1991)およびWuら(J. Exp. Med. (1970) 132, 211-250))。
本開示は特定の理論に拘束されるものではないが、生殖細胞系の配列の使用がほとんどの個人において有害な免疫反応を排除すると期待されている一つの理由は、以下の通りであると考えられている。通常の免疫反応中に生じる親和性成熟ステップの結果、免疫グロブリンの可変領域に体細胞の突然変異が頻繁に生じる。これらの突然変異は主にその配列が超可変的であるCDRの周辺に生じるが、フレームワーク領域の残基にも影響を及ぼす。これらのフレームワークの突然変異は生殖細胞系の遺伝子には存在せず、また患者の免疫原性になる可能性は少ない。一方、通常のヒトの集団は生殖細胞系の遺伝子によって発現されるフレームワーク配列の大多数にさらされており、免疫寛容の結果、これらの生殖細胞系のフレームワークは患者において免疫原性が低いあるいは非免疫原性であると予想される。免疫寛容の可能性を最大にするため、可変領域をコード化する遺伝子が普通に存在する機能的な生殖細胞系遺伝子の集合から選択され得る。
また、本開示の非限定な一態様では、抗原結合分子の安定性を向上させるために、CDR領域および/またはフレームワーク領域を含む可変領域のアミノ酸を適宜改変することも可能である。そのようなアミノ酸の非限定の一態様として、1位、5位、10位、30位、48位、58位のアミノ酸が例示され得る。より具体的には1位のGln、5位のGln、10位のAsp、30位のAsn、48位のLeu、58位のAsnが例示され得る。抗体の安定性を向上させるために、これらのアミノ酸を生殖細胞系列の配列に含まれている対応するアミノ酸に置換することが可能である。そのような生殖細胞系列の非限定な一態様の配列としてVH3-21の配列が例示され得る。この場合において、1位のGlnがGluに、5位のGlnがValに、10位のAspがGlyに、30位のAsnがSerに、48位のLeuがValに、58位のAsnがTyrに置換され得る。
改変体中のアミノ酸改変部位
非限定的な一態様において、前記抗原結合ドメイン改変体が抗原結合ドメイン未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である:
1) 前記抗原結合ドメイン未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗原結合ドメイン改変体と比較して、前記抗原結合ドメイン改変体のMTAとの結合を著しく減弱させないアミノ酸部位、及び
3) 前記抗原結合ドメイン改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
非限定的な一態様において、前記抗原結合ドメイン改変体が抗原結合ドメイン未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である:
1) 前記抗原結合ドメイン未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗原結合ドメイン改変体と比較して、前記抗原結合ドメイン改変体のMTAとの結合を著しく減弱させないアミノ酸部位、及び
3) 前記抗原結合ドメイン改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
非限定的な一態様において、前記抗原結合ドメイン改変体が抗原結合ドメイン未改変体と異なるアミノ酸を有するアミノ酸部位は、下記のアミノ酸部位の群から選ばれる一つまたは複数のアミノ酸部位である:
1) 前記抗原結合ドメイン未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗原結合ドメイン改変体と比較して、前記抗原結合ドメイン改変体のMTAとの結合を著しく減弱させないアミノ酸部位、
3) 前記抗原結合ドメイン未改変体の表面に露出するアミノ酸部位に対応するアミノ酸部位、及び
4) 前記抗原結合ドメイン未改変体においてMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に対応するアミノ酸部位。
1) 前記抗原結合ドメイン未改変体中のMTAとの結合に関与しないアミノ酸部位に対応するアミノ酸部位、
2) 前記抗原結合ドメイン改変体と比較して、前記抗原結合ドメイン改変体のMTAとの結合を著しく減弱させないアミノ酸部位、
3) 前記抗原結合ドメイン未改変体の表面に露出するアミノ酸部位に対応するアミノ酸部位、及び
4) 前記抗原結合ドメイン未改変体においてMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に対応するアミノ酸部位。
多様化可能なアミノ酸部位
前記抗原結合ドメイン未改変体の配列が抗原結合ドメイン改変体の配列と異なるアミノ酸を有するアミノ酸部位は、ライブラリを設計するときに、多様なアミノ酸が含まれるように設計することが可能な部位とし、本開示において、「多様化可能なアミノ酸部位」とも呼ばれる。多様化可能なアミノ酸部位は、以下を含むことが出来る:
(i) 抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(vii) 抗原結合ドメイン改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
前記抗原結合ドメイン未改変体の配列が抗原結合ドメイン改変体の配列と異なるアミノ酸を有するアミノ酸部位は、ライブラリを設計するときに、多様なアミノ酸が含まれるように設計することが可能な部位とし、本開示において、「多様化可能なアミノ酸部位」とも呼ばれる。多様化可能なアミノ酸部位は、以下を含むことが出来る:
(i) 抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(vii) 抗原結合ドメイン改変体の抗原に対するMTA依存的結合に寄与しやすいアミノ酸部位。
MTAとの結合に関与しないアミノ酸部位
本開示における「MTAとの結合に関与しないアミノ酸部位」は、MTAと抗原結合分子の複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定することができる。本開示の非限定の一態様として、MTAと抗原結合分子の複合体の結晶構造解析から、MTAとの結合に関与していない抗原結合分子のアミノ酸残基、特にMTAとの結合に関与していない抗原結合ドメインのアミノ酸残基を同定することができる。ここで、「MTAとの結合に関与」とは、抗原結合ドメインが抗体可変領域である場合、抗体可変領域のH鎖又はL鎖を形成するアミノ酸における側鎖又は主鎖の原子とMTAの原子が、結合活性に効果を及ぼしうる距離で分子間相互作用を形成している状態、又は、あるアミノ酸残基がCDRループ等の立体構造をMTAが結合する際のコンフォメーションに安定化させる等の間接的な影響を含む、MTAの結合に寄与している状態、ならびにその双方を満たす状態のことをいう。
別の一態様では、MTAに対する結合に関与しないアミノ酸部位の判定は、MTAとの結合に関与しているアミノ酸部位のうちから選択される、いずれか一つ以上のアミノ酸部位以外のアミノ酸部位であるとも考えることができる。
本明細書における「分子間相互作用を形成している状態」については、抗原結合ドメインが抗体可変領域である場合、例えばMTAと抗原結合分子の複合体の結晶構造解析から、抗体可変領域のH鎖又はL鎖を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTAと抗原結合分子の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、MTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
本開示における「MTAとの結合に関与しないアミノ酸部位」は、MTAと抗原結合分子の複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定することができる。本開示の非限定の一態様として、MTAと抗原結合分子の複合体の結晶構造解析から、MTAとの結合に関与していない抗原結合分子のアミノ酸残基、特にMTAとの結合に関与していない抗原結合ドメインのアミノ酸残基を同定することができる。ここで、「MTAとの結合に関与」とは、抗原結合ドメインが抗体可変領域である場合、抗体可変領域のH鎖又はL鎖を形成するアミノ酸における側鎖又は主鎖の原子とMTAの原子が、結合活性に効果を及ぼしうる距離で分子間相互作用を形成している状態、又は、あるアミノ酸残基がCDRループ等の立体構造をMTAが結合する際のコンフォメーションに安定化させる等の間接的な影響を含む、MTAの結合に寄与している状態、ならびにその双方を満たす状態のことをいう。
別の一態様では、MTAに対する結合に関与しないアミノ酸部位の判定は、MTAとの結合に関与しているアミノ酸部位のうちから選択される、いずれか一つ以上のアミノ酸部位以外のアミノ酸部位であるとも考えることができる。
本明細書における「分子間相互作用を形成している状態」については、抗原結合ドメインが抗体可変領域である場合、例えばMTAと抗原結合分子の複合体の結晶構造解析から、抗体可変領域のH鎖又はL鎖を形成するアミノ酸の側鎖又は主鎖を構成する非水素原子とMTAを構成する非水素原子との原子間距離をもとに判定できる。たとえば,上記原子間距離は3.0Å、3.2Å、3.4Å、3.6Å、3.8Å、4.0Å、4.2Å、4.4Å、4.6Å、4.8Å又は5.0Å以内が好ましいがこれに限定されることはない。更に好ましい上記原子間距離は3.6Å、3.8Å、4.0Å又は4.2Å以内である。
より詳しくは、立体構造上での原子間距離と形成される分子間相互作用の種類ならびに原子の種類の情報をもとに直接的に相互作用している可能性が判定できる。さらに正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができるが、これに限定されるわけではない。
本明細書における「間接的に影響している状態」についても,例えば、MTAと抗原結合分子の複合体の立体構造から各アミノ酸残基のコンフォメーションや周囲の残基との分子間相互作用の状況を詳細に解析することで、MTAの結合に間接的に影響しているかどうかを推定できるが、より正確にはAlaやGlyへの改変といったアミノ酸残基の変異導入がMTAの活性に与える影響から判断することができる。
本開示の一態様として、MTAの結合に関与していないと同定された残基を他のアミノ酸に置換しても、MTAへの結合を適切な程度に維持できるアミノ酸を選択することができる。これにより、選択された残基において選択されたアミノ酸が出現するライブラリを設計することができる。この場合において、MTAの結合に関与していないと同定された残基が互いに異なるアミノ酸に置換された抗原結合分子の集合となるように、複数の抗原結合分子から主としてなるライブラリを設計することが可能である。
MTAとの結合を著しく減弱させないアミノ酸部位
本開示における非限定の一態様として、「MTAとの結合を著しく減弱させないアミノ酸部位」は、アミノ酸の変異導入の方法によって同定することができる。例えば、可変領域のアミノ酸を網羅的に改変して、個々の改変体のMTAへの結合が、Biacore等を用いた公知の方法で測定され、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出される。当該KD値が親配列である未改変体のKD値と比較され、一定の基準を上回る結合を示す改変箇所が、「MTAとの結合を著しく減弱させないアミノ酸部位」と判定される。例えば、Biacore等の公知の方法を用いた測定の結果、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出され、重鎖の部位としては改変によってMTA結合能が未改変体の1/100、1/50、1/10、1/9、1/8、1/7、1/6、1/5、1/4、1/3又は1/2の結合能を下回らないもの、および軽鎖の部位としては改変によってMTA結合能が未改変体の1/100、1/50、1/10、1/9、1/8、1/7、1/6、1/5、1/4、1/3又は1/2の結合能を下回らないものがMTAとの結合を著しく減弱させないアミノ酸部位と判定されるが、上記基準に限定されるものではない。あるいは、親配列である未改変体のKD値との比較ではなく、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出され、重鎖の部位としては10mM、1mM、100uM、10uM、1uM、100nM、10nM、1nM、100pM、10pM、又は1pMの結合能を下回らないもの、および軽鎖の部位としては10mM、1mM、100uM、10uM、1uM、100nM、10nM、1nM、100pM、10pM、又は1pMの結合能を下回らないものがMTAとの結合を著しく減弱させないアミノ酸部位と判定されるが、上記基準に限定されるものではない。未改変及び改変体のMTAに対する結合活性の測定は、当業者公知の方法(Biacore、ELISA、ECL等)から適宜選択して実施することができる。
別の一態様では、MTAとの結合を著しく減弱させないアミノ酸部位の判定は、MTAとの結合に関与しているアミノ酸部位のうちから選択される、いずれか一つ以上のアミノ酸部位以外のアミノ酸部位であるとも考えることができる。
本開示における非限定の一態様として、「MTAとの結合を著しく減弱させないアミノ酸部位」は、アミノ酸の変異導入の方法によって同定することができる。例えば、可変領域のアミノ酸を網羅的に改変して、個々の改変体のMTAへの結合が、Biacore等を用いた公知の方法で測定され、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出される。当該KD値が親配列である未改変体のKD値と比較され、一定の基準を上回る結合を示す改変箇所が、「MTAとの結合を著しく減弱させないアミノ酸部位」と判定される。例えば、Biacore等の公知の方法を用いた測定の結果、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出され、重鎖の部位としては改変によってMTA結合能が未改変体の1/100、1/50、1/10、1/9、1/8、1/7、1/6、1/5、1/4、1/3又は1/2の結合能を下回らないもの、および軽鎖の部位としては改変によってMTA結合能が未改変体の1/100、1/50、1/10、1/9、1/8、1/7、1/6、1/5、1/4、1/3又は1/2の結合能を下回らないものがMTAとの結合を著しく減弱させないアミノ酸部位と判定されるが、上記基準に限定されるものではない。あるいは、親配列である未改変体のKD値との比較ではなく、個々の改変体のMTAに対する結合活性(アフィニティ)がKD値として算出され、重鎖の部位としては10mM、1mM、100uM、10uM、1uM、100nM、10nM、1nM、100pM、10pM、又は1pMの結合能を下回らないもの、および軽鎖の部位としては10mM、1mM、100uM、10uM、1uM、100nM、10nM、1nM、100pM、10pM、又は1pMの結合能を下回らないものがMTAとの結合を著しく減弱させないアミノ酸部位と判定されるが、上記基準に限定されるものではない。未改変及び改変体のMTAに対する結合活性の測定は、当業者公知の方法(Biacore、ELISA、ECL等)から適宜選択して実施することができる。
別の一態様では、MTAとの結合を著しく減弱させないアミノ酸部位の判定は、MTAとの結合に関与しているアミノ酸部位のうちから選択される、いずれか一つ以上のアミノ酸部位以外のアミノ酸部位であるとも考えることができる。
MTA依存的結合に寄与しやすいアミノ酸部位
本開示における「MTA依存的結合に寄与しやすいアミノ酸部位」は、MTAと抗原結合分子の複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定または推定することができる。「MTA依存的結合に寄与しやすいアミノ酸部位」を推定する非限定な手法として、抗原結合分子蛋白の表面に露出しているアミノ酸残基が位置するアミノ酸部位、もしくはMTAが結合したときに非結合時と比較して構造変化する領域に位置するアミノ酸部位を同定する手法が例示される。
本開示における「MTA依存的結合に寄与しやすいアミノ酸部位」は、MTAと抗原結合分子の複合体の結晶構造解析、NMRを用いた立体構造解析、又はアミノ酸の変異導入等の手法によって同定または推定することができる。「MTA依存的結合に寄与しやすいアミノ酸部位」を推定する非限定な手法として、抗原結合分子蛋白の表面に露出しているアミノ酸残基が位置するアミノ酸部位、もしくはMTAが結合したときに非結合時と比較して構造変化する領域に位置するアミノ酸部位を同定する手法が例示される。
抗原結合ドメインの表面に露出するアミノ酸部位
本開示における「抗原結合ドメインの表面に露出するアミノ酸部位」は、タンパク質の立体構造上の表面に位置するアミノ酸部位をいう。抗原結合ドメインを含む抗原結合分子単体、またはMTAと抗原結合分子の複合体に対して、結晶構造解析やNMRを用いた立体構造解析等の手法によって、抗原結合ドメインのタンパク質表面に位置するアミノ酸残基およびアミノ酸部位を同定することができる。抗原結合ドメインの表面に露出するアミノ酸部位を同定する非限定な一手法として、溶媒露出表面積(Accessible surface area)を算出することで、表面に露出するアミノ酸部位を同定する手法が挙げられる。
本開示における「抗原結合ドメインの表面に露出するアミノ酸部位」は、タンパク質の立体構造上の表面に位置するアミノ酸部位をいう。抗原結合ドメインを含む抗原結合分子単体、またはMTAと抗原結合分子の複合体に対して、結晶構造解析やNMRを用いた立体構造解析等の手法によって、抗原結合ドメインのタンパク質表面に位置するアミノ酸残基およびアミノ酸部位を同定することができる。抗原結合ドメインの表面に露出するアミノ酸部位を同定する非限定な一手法として、溶媒露出表面積(Accessible surface area)を算出することで、表面に露出するアミノ酸部位を同定する手法が挙げられる。
このような、抗原結合ドメインの表面に露出するアミノ酸部位に位置するアミノ酸を多様なアミノ酸に置換することにより、様々な抗原分子への結合活性をもたらすことが期待される。これにより、このようなアミノ酸部位において多様なアミノ酸が出現するライブラリを設計することができる。この場合において、抗原結合ドメインの表面に露出するアミノ酸部位に異なるアミノ酸を有する抗原結合分子の集合となるように、複数の抗原結合分子から主としてなるライブラリを設計することが可能である。
抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位
本開示における「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域」は、結晶構造解析やNMRを用いた立体構造解析等の手法を用いて、抗原結合ドメインを含む抗原結合分子単体、及びMTAと抗原結合分子の複合体の構造を比較することにより、構造変化する領域を同定することにより同定することが出来る。
また、「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」は、抗原結合ドメインを含む抗原結合分子単体、及びMTAと抗原結合分子の複合体の構造を比較することにより直接同定することもできる。抗原結合ドメインが抗体可変領域の場合、MTAに結合した時の抗体H鎖およびL鎖の構造と、MTAに結合していない時の抗体H鎖、L鎖構造を重ね合わせたときに計算される、MTA結合時/非結合時の各アミノ酸残基のCα原子間の距離をもとに、当該アミノ酸残基が位置するアミノ酸部位が「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」であるか否かを判定できる。例えば、 MTA結合時/非結合時のCα原子間の距離が可変領域全アミノ酸残基のMTA結合時/非結合時のCα原子間距離の平均値以上であるアミノ酸残基が位置するアミノ酸部位を「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」と判定することが出来るが、これに限定されることはない。
このような、抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位のアミノ酸を多様なアミノ酸に置換することにより、MTA依存的な抗原結合活性をもたらすことが期待される。これにより、このようなアミノ酸部位において多様なアミノ酸が出現するライブラリを設計することができる。この場合において、抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に異なるアミノ酸を有する抗原結合分子の集合となるように、複数の抗原結合分子から主としてなるライブラリを設計することが可能である。
本開示における「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域」は、結晶構造解析やNMRを用いた立体構造解析等の手法を用いて、抗原結合ドメインを含む抗原結合分子単体、及びMTAと抗原結合分子の複合体の構造を比較することにより、構造変化する領域を同定することにより同定することが出来る。
また、「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」は、抗原結合ドメインを含む抗原結合分子単体、及びMTAと抗原結合分子の複合体の構造を比較することにより直接同定することもできる。抗原結合ドメインが抗体可変領域の場合、MTAに結合した時の抗体H鎖およびL鎖の構造と、MTAに結合していない時の抗体H鎖、L鎖構造を重ね合わせたときに計算される、MTA結合時/非結合時の各アミノ酸残基のCα原子間の距離をもとに、当該アミノ酸残基が位置するアミノ酸部位が「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」であるか否かを判定できる。例えば、 MTA結合時/非結合時のCα原子間の距離が可変領域全アミノ酸残基のMTA結合時/非結合時のCα原子間距離の平均値以上であるアミノ酸残基が位置するアミノ酸部位を「抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位」と判定することが出来るが、これに限定されることはない。
このような、抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位のアミノ酸を多様なアミノ酸に置換することにより、MTA依存的な抗原結合活性をもたらすことが期待される。これにより、このようなアミノ酸部位において多様なアミノ酸が出現するライブラリを設計することができる。この場合において、抗原結合ドメインのMTA結合/非結合時に構造変化率が大きい領域に位置するアミノ酸部位に異なるアミノ酸を有する抗原結合分子の集合となるように、複数の抗原結合分子から主としてなるライブラリを設計することが可能である。
アミノ酸出現頻度の多様性のあるアミノ酸部位
本開示における「アミノ酸出現頻度の多様性のあるアミノ酸部位」とは、親配列が属する動物種の抗体のレパートリーとして、1%以上の出現頻度で存在が認められるアミノ酸の種類として2種以上が認められるアミノ酸部位のことをいう。
本開示における「アミノ酸出現頻度の多様性のあるアミノ酸部位」とは、親配列が属する動物種の抗体のレパートリーとして、1%以上の出現頻度で存在が認められるアミノ酸の種類として2種以上が認められるアミノ酸部位のことをいう。
本開示における「抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位」とは、該当する未改変体が由来する動物種において遺伝子上で認められる抗体遺伝子配列のレパートリー中で多様性が認められるアミノ酸部位のことを言う。一例として、これに限定されるわけではないが、該当する未改変体がヒト由来の場合、未改変体が属する動物種の抗体のレパートリーとは、ヒトの遺伝子上において認められる抗体遺伝子配列のレパートリーのことを指し、該当する未改変体がウサギ由来の場合は、未改変体が属する動物種の抗体のレパートリーとは、ウサギの遺伝子上において認められる抗体遺伝子配列のレパートリーのことを指す。ただし、遺伝子上に存在したとしても、終始コドンの存在やフレームシフトによって実際に抗体して発現されることのない配列は含まれない。
該当する未改変体が非ヒト動物由来である場合、常法に従いこれをヒト化することが可能であり、この手法については当業者に広く公知である(欧州特許公開EP239400、国際公開WO1996/002576、WO1993/012227、WO1992/003918、WO1994/002602、WO1994/025585、WO1996/034096、WO1996/033735、WO1992/001047、WO1992/020791、WO1993/006213、WO1993/011236、WO1993/019172、WO1995/001438、WO1995/015388、Cancer Res., (1993) 53, 851-856、BBRC., (2013)436(3):543-50等)。該当する未改変体を常法に従ってヒト化した後にライブラリ化を実施する場合、本開示における未改変体が属する動物種の抗体のレパートリーにおいては、ヒト化を実施する前の抗原結合ドメインと、ヒト化した後の抗原結合ドメインの双方を抗原結合ドメインとして扱うことができ、それに伴い同一動物種としてヒト化を実施する前の抗原結合ドメインが由来する動物種と、ヒトの双方のレパートリーを適用することができる。一例として、これに限定されるわけではないが、ヒト化を実施する前の抗原結合ドメインがウサギに由来する場合、未改変体が属する動物種の抗体のレパートリーとは、ヒトおよび/またはウサギの遺伝子上において認められる抗体遺伝子配列のレパートリーのことを指す。ただし、遺伝子上に存在したとしても、終始コドンの存在やフレームシフトによって実際に抗体して発現されることのない配列は含まれない。
未改変体の属する動物種の抗体のレパートリーは、一例として、これに限定されるわけではないが、既知のデータベースを参照することによって調査が可能である。アミノ酸出現頻度の多様性のある部位は一般的にCDR領域に存在する。一態様では、公知のおよび/または天然抗体の非常に多様な位置を決定する際には、Kabat, Sequences of Proteins of Immunological Interest (National Institute of Health Bethesda Md.) (1987年および1991年)が提供するデータが有効である。また、インターネット上の複数のデータベース(http://vbase.mrc-cpe.cam.ac.uk/、http://www.bioinf.org.uk/abs/index.html)では収集された多数のヒト軽鎖および重鎖の配列とその配置が提供されており、これらの配列とその配置の情報は本開示における非常に多様な位置の決定に有用である。
また、別の一態様としては、未改変体の属する動物種の抗体のレパートリーは、該当する生物種から取得した抗体遺伝子をクローニングし、配列を解析することによっても調査が可能である。一例として、これに限定されるわけではないが、ヒトの抗体レパートリーについては、健常人のリンパ球由来の抗体遺伝子から構築され、そのレパートリーにバイアスを含まない抗体配列であるナイーブ配列からなるナイーブライブラリの配列を解析することによって調査され得る(Gejimaら(Human Antibodies (2002) 11,121-129)およびCardosoら(Scand. J. Immunol. (2000) 51, 337-344))。レパートリーの調査の際には、少なくとも100種類、好ましくは200種類、さらに好ましくは400種類以上の配列について解析が行われることが望ましい。
本開示における未改変体の属する動物種の抗体のレパートリーは、より好ましくは、これに限定されるわけではないが、未改変体の属するジャームラインのサブグループについて調査が実施されることが望ましい。フレームワークの例としては、例えばV-Base(http://vbase.mrc-cpe.cam.ac.uk/)等のウェブサイトに含まれている、現在知られている完全にヒト型のフレームワーク領域の配列が本開示の抗原結合分子に含まれる生殖細胞系列の配列として適宜使用され得る。生殖細胞系列の配列はその類似性に基づいてサブグループに分類され得る(Tomlinsonら(J. Mol. Biol. (1992)227, 776-798)WilliamsおよびWinter(Eur. J. Immunol.(1993)23, 1456-1461)およびCoxら(Nat. Genetics(1994)7, 162-168)。一例として、ヒトの抗体においては重鎖可変領域については7つのサブグループ、Vκについては7つのサブグループ、Vλについては10種類のサブグループが報告されており、各アミノ酸部位について、未改変体が属しているサブグループ内におけるアミノ酸のレパートリーを解析することによって調査され得るが、特にこの態様に限定されるものではない。
Canonical structureの形成に重要でないアミノ酸部位
本開示における「Canonical structureの形成に重要でないアミノ酸部位」において、抗体のcanonical structureとは、抗体重鎖及び軽鎖の主にCDR1、2の立体構造のクラスタリングを示しており、構造は抗体のサブグループや、CDRの長さや配列によって分類されることができる。各canonical structureにおいて、構造の維持に重要な残基は既に公知であり、Chothiaら(J. Mol. Biol. (1992) 227, 799-817)、Al-Lazikaniら(J. Mol. Biol. (1997) 273, 927-948)、Tomlinsonら(J. Mol. Biol.(1992) 227, 776-798)等の報告を参照することにより、該当する親抗原結合分子が分類されるcanonical structure、及びその構造に重要な残基を特定することが可能である。
また、抗体以外の抗原結合ドメインにおいても、構造の維持に重要な残基が存在していることは公知であり、これに限定されるわけではないが、作製した変異体の構造解析等により、個々の抗原結合ドメインにおいてcanonical structureの形成に重要でないアミノ酸部位を同定することが可能である。
本開示における「Canonical structureの形成に重要でないアミノ酸部位」において、抗体のcanonical structureとは、抗体重鎖及び軽鎖の主にCDR1、2の立体構造のクラスタリングを示しており、構造は抗体のサブグループや、CDRの長さや配列によって分類されることができる。各canonical structureにおいて、構造の維持に重要な残基は既に公知であり、Chothiaら(J. Mol. Biol. (1992) 227, 799-817)、Al-Lazikaniら(J. Mol. Biol. (1997) 273, 927-948)、Tomlinsonら(J. Mol. Biol.(1992) 227, 776-798)等の報告を参照することにより、該当する親抗原結合分子が分類されるcanonical structure、及びその構造に重要な残基を特定することが可能である。
また、抗体以外の抗原結合ドメインにおいても、構造の維持に重要な残基が存在していることは公知であり、これに限定されるわけではないが、作製した変異体の構造解析等により、個々の抗原結合ドメインにおいてcanonical structureの形成に重要でないアミノ酸部位を同定することが可能である。
製造されたライブラリ
本開示の一態様として、以下(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含む方法によって製造されるライブラリが提供される。
本開示の一態様として、以下(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含む方法によって製造されるライブラリが提供される。
本開示の一態様として、 以下の(a)および(b)の工程により製造されるライブラリが提供される。
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含む方法により製造されたライブラリであって、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない。
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含む方法により製造されたライブラリであって、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない。
本開示における「1または複数のアミノ酸」とは、特にアミノ酸の数を限定されることはなく、2種類以上のアミノ酸、5種類以上のアミノ酸、10種類以上のアミノ酸、15種類以上のアミノ酸又は20種類のアミノ酸であってもよい。
本開示における「互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する」とは、例えばNNKやTRIM Library等(Gonzalez-Munoz A et al. MAbs 2012, Lee CV et al. J Mol Biol. 2004, Knappik A. et al. J Mol Biol. 2000, Tiller T et al. MAbs 2013)の公知のライブラリ技術を利用して、特定部位のアミノ酸を所望のアミノ酸に改変されている抗原結合ドメインまたは抗原結合ドメインを含む抗原結合分子の複数の改変体を含むライブラリを設計することが包含されるが、特にこの態様に限定されるものではない。本開示における「改変体」とは、親配列の未改変体に対して、少なくとも1又はそれ以上のアミノ酸が置換されている親配列の各々異なる改変体をいう。
本開示における「互いに配列が異なる、前記抗原結合ドメインの複数の改変体」を作製する工程において、特定されたアミノ酸部位のうち、CDR1及びCDR2の部位に関しては生殖細胞系列での出現頻度が10%以上、9%以上、8%以上、7%以上、6%以上、5%以上、4%以上、3%以上、2%以上又は1%以上のアミノ酸に置換することができ、CDR3の部位に関しては生殖細胞系列での出現頻度が10%以上、9%以上、8%以上、7%以上、6%以上、5%以上、4%以上、3%以上、2%以上又は1%以上のアミノ酸に置換して、個々の改変体を作製することができるが、これに限定されるものではない。
本開示における「互いに配列が異なる、前記抗原結合ドメインの複数の改変体」を作製する工程において、特定されたアミノ酸部位のアミノ酸を置換した改変体の、MTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該置換後アミノ酸が位置するアミノ酸部位の構造変化率が大きいように改変体を作製することができるが、これに限定されるものではない。
本開示における「互いに配列が異なる、前記抗原結合ドメインの複数の改変体」を作製する工程において、特定されたアミノ酸部位のアミノ酸を置換した改変体の、MTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該置換後アミノ酸が存在することにより改変体のMTA結合による構造変化が阻害されないように改変体を作製することができるが、これに限定されるものではない。
本開示における「互いに配列が異なる、前記抗原結合ドメインの複数の改変体」を作製する工程において、特定されたアミノ酸部位のアミノ酸を置換した改変体のMTAに対する結合活性が、未改変体に比べて著しく減弱されないように改変体を作製することが出来るが、これに限定されるものではない。
濃縮されたライブラリ
非限定的な一態様において、本開示のライブラリはMTAに結合する抗原結合ドメインを含む抗原結合分子をコードする核酸が濃縮されたライブラリが提供される。
本開示における「濃縮する」とは、濃縮作業実施前のライブラリとの比較において、目的の活性を有する改変体をコードする核酸の存在比率を上昇させることを言う。目的の活性を有する改変体をコードする核酸の存在比率が上昇した状態を、「濃縮された」と言う。
一例として、これに限定されるわけではないが、MTAに結合する抗原結合分子をコードする核酸を濃縮するとは、パンニングによりMTAに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることによって達成可能である。より具体的には、一例として、複数の抗原結合分子を含むライブラリがファージディスプレイ法により表面に提示されたファージをMTAと接触させ、洗浄作業により結合活性を有していない分子を提示しているファージ及び分子を提示していないファージを除去したのち、結合を維持している抗原結合分子を提示しているファージのみを回収することによって、パンニングによりMTAに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることが可能であるが、これに限定されることはない。このような方法は、即ち、以下(1)~(2)の工程:
(1) ライブラリからディスプレイされた抗原結合ドメインをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、を含む方法である。
目的の活性を有する抗原結合分子コードする核酸の存在比率が、濃縮作業実施前のライブラリとの比較において、1.1倍以上上昇することが好ましい。より好ましくは、目的の活性を有する抗原結合分子をコードする核酸の存在比率を、1.2倍以上、1.5倍以上、2倍以上、4倍以上、10倍以上、25倍以上、100倍以上上昇させることにより、本開示に記載のライブラリを製造することが可能である。非限定的な一態様において、このような方法で濃縮されたライブラリ中の抗原結合分子がMTAに対する結合活性は、MTAとは異なる種類の分子である抗原の非存在下でも示される。
濃縮対象となるライブラリは、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含んでいる限りその種類は限定されない。より具体的に、これらに限定されないが、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、後述する本明細書の方法に沿って設計されるライブラリ、本明細書実施例に記載のライブラリが挙げられる。
非限定的な一態様において、本開示のライブラリはMTAに結合する抗原結合ドメインを含む抗原結合分子をコードする核酸が濃縮されたライブラリが提供される。
本開示における「濃縮する」とは、濃縮作業実施前のライブラリとの比較において、目的の活性を有する改変体をコードする核酸の存在比率を上昇させることを言う。目的の活性を有する改変体をコードする核酸の存在比率が上昇した状態を、「濃縮された」と言う。
一例として、これに限定されるわけではないが、MTAに結合する抗原結合分子をコードする核酸を濃縮するとは、パンニングによりMTAに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることによって達成可能である。より具体的には、一例として、複数の抗原結合分子を含むライブラリがファージディスプレイ法により表面に提示されたファージをMTAと接触させ、洗浄作業により結合活性を有していない分子を提示しているファージ及び分子を提示していないファージを除去したのち、結合を維持している抗原結合分子を提示しているファージのみを回収することによって、パンニングによりMTAに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることが可能であるが、これに限定されることはない。このような方法は、即ち、以下(1)~(2)の工程:
(1) ライブラリからディスプレイされた抗原結合ドメインをMTAと接触させる工程、及び
(2) 前記工程(1)でMTAに結合した抗原結合ドメインを選択する工程、を含む方法である。
目的の活性を有する抗原結合分子コードする核酸の存在比率が、濃縮作業実施前のライブラリとの比較において、1.1倍以上上昇することが好ましい。より好ましくは、目的の活性を有する抗原結合分子をコードする核酸の存在比率を、1.2倍以上、1.5倍以上、2倍以上、4倍以上、10倍以上、25倍以上、100倍以上上昇させることにより、本開示に記載のライブラリを製造することが可能である。非限定的な一態様において、このような方法で濃縮されたライブラリ中の抗原結合分子がMTAに対する結合活性は、MTAとは異なる種類の分子である抗原の非存在下でも示される。
濃縮対象となるライブラリは、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含んでいる限りその種類は限定されない。より具体的に、これらに限定されないが、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、後述する本明細書の方法に沿って設計されるライブラリ、本明細書実施例に記載のライブラリが挙げられる。
別の一例として、これに限定されるわけではないが、アデノシンに結合する抗原結合分子をコードする核酸を濃縮するとは、パンニングによりアデノシンに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることによって達成可能である。より具体的には、一例として、複数の抗原結合分子を含むライブラリがファージディスプレイ法により表面に提示されたファージをアデノシンと接触させ、洗浄作業により結合活性を有していない分子を提示しているファージ及び分子を提示していないファージを除去したのち、結合を維持している抗原結合分子を提示しているファージのみを回収することによって、パンニングによりアデノシンに対して結合活性を有する抗原結合分子をコードする核酸の存在比率を上昇させることが可能であるが、これに限定されることはない。このような方法は、即ち、以下(1)~(2)の工程:
(1) ライブラリからディスプレイされた抗原結合ドメインをアデノシンと接触させる工程、及び
(2) 前記工程(1)でアデノシンに結合した抗原結合ドメインを選択する工程、を含む方法である。
目的の活性を有する抗原結合分子コードする核酸の存在比率が、濃縮作業実施前のライブラリとの比較において、1.1倍以上上昇することが好ましい。より好ましくは、目的の活性を有する抗原結合分子をコードする核酸の存在比率を、1.2倍以上、1.5倍以上、2倍以上、4倍以上、10倍以上、25倍以上、100倍以上上昇させることにより、本開示に記載のライブラリを製造することが可能である。非限定的な一態様において、このような方法で濃縮されたライブラリ中の抗原結合分子がアデノシンに対する結合活性は、アデノシンとは異なる種類の分子である抗原の非存在下でも示される。
濃縮対象となるライブラリは、アデノシン依存的に抗原に対する結合活性が変化する抗原結合ドメインを含んでいる限りその種類は限定されない。より具体的に、これらに限定されないが、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、本明細書のライブラリ製造方法に沿って製造されるライブラリ、本明細書に記載のライブラリが挙げられる。
(1) ライブラリからディスプレイされた抗原結合ドメインをアデノシンと接触させる工程、及び
(2) 前記工程(1)でアデノシンに結合した抗原結合ドメインを選択する工程、を含む方法である。
目的の活性を有する抗原結合分子コードする核酸の存在比率が、濃縮作業実施前のライブラリとの比較において、1.1倍以上上昇することが好ましい。より好ましくは、目的の活性を有する抗原結合分子をコードする核酸の存在比率を、1.2倍以上、1.5倍以上、2倍以上、4倍以上、10倍以上、25倍以上、100倍以上上昇させることにより、本開示に記載のライブラリを製造することが可能である。非限定的な一態様において、このような方法で濃縮されたライブラリ中の抗原結合分子がアデノシンに対する結合活性は、アデノシンとは異なる種類の分子である抗原の非存在下でも示される。
濃縮対象となるライブラリは、アデノシン依存的に抗原に対する結合活性が変化する抗原結合ドメインを含んでいる限りその種類は限定されない。より具体的に、これらに限定されないが、ナイーブヒト抗体ディスプレイライブラリ、合成ヒト抗体ディスプレイライブラリ、本明細書のライブラリ製造方法に沿って製造されるライブラリ、本明細書に記載のライブラリが挙げられる。
ライブラリの製造方法
本開示はまた、上記に説明した本開示に包含される様々な態様の「ライブラリ」を製造する方法に関する。
本開示に係る「ライブラリの製造方法」は、以下に例示するようないずれかの特定の方法に限定されるものではなく、上述した本開示の「ライブラリ」を製造し得る方法であればいずれの方法も包含される。
本開示に係る「ライブラリの製造方法」としては、例えば、以下に例示するような方法を挙げることができる。
なお、以下に例示する「ライブラリの製造方法」における各特定事項は、上記で本開示の「ライブラリ」から「濃縮された」について詳述したとおりの技術的意義を有する。
本開示はまた、上記に説明した本開示に包含される様々な態様の「ライブラリ」を製造する方法に関する。
本開示に係る「ライブラリの製造方法」は、以下に例示するようないずれかの特定の方法に限定されるものではなく、上述した本開示の「ライブラリ」を製造し得る方法であればいずれの方法も包含される。
本開示に係る「ライブラリの製造方法」としては、例えば、以下に例示するような方法を挙げることができる。
なお、以下に例示する「ライブラリの製造方法」における各特定事項は、上記で本開示の「ライブラリ」から「濃縮された」について詳述したとおりの技術的意義を有する。
本開示の一態様として、 以下の(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法が提供される。
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法が提供される。
本開示の一態様として、 以下の(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない;
を含むライブラリの製造方法が提供される。
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体がMTAに結合したときの構造とMTAに結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、MTAに対する結合活性が著しく減弱されていない;
を含むライブラリの製造方法が提供される。
本開示の一側面において、MTAと相互作用するアミノ酸残基を有する抗原結合ドメインをコードするライブラリの製造方法に限らず、様々な低分子化合物と相互作用するアミノ酸残基を有する抗原結合ドメインをコードするライブラリの製造方法も提供される。
以下に例示するライブラリの製造方法における各特定事項は、上記で詳述したMライブラリの製造方法と同様な技術的意義を有し、当業者は、「MTA」を「低分子化合物」と読み替えることにより様々な特定事項の意味を理解できる。
以下に例示するライブラリの製造方法における各特定事項は、上記で詳述したMライブラリの製造方法と同様な技術的意義を有し、当業者は、「MTA」を「低分子化合物」と読み替えることにより様々な特定事項の意味を理解できる。
一態様として、以下(a)および(b)の工程:
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法が提供される。
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法が提供される。
一の態様において、以下(a)および(b)の工程:
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、前記低分子化合物に対する結合活性が著しく減弱されていない;
を含むライブラリの製造方法が提供される。
(a) 低分子化合物に対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(ii)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
工程(b)のアミノ酸改変は、以下(1)乃至(3)の少なくとも一つ以上を満たす:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
(3) 当該アミノ酸改変を有する抗原結合ドメイン改変体が、前記抗原結合ドメイン未改変体と比べて、前記低分子化合物に対する結合活性が著しく減弱されていない;
を含むライブラリの製造方法が提供される。
一態様として、以下(a)および(b)の工程:
(a) 低分子化合物に対して相互作用する抗原結合ドメインにおいて、以下(i)乃至(iv)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) 前記低分子化合物との結合に関与しないアミノ酸部位;
(iv) 前記低分子化合物との結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位に以下の(1)乃至(2)の少なくとも一つを満たすアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
を含むライブラリの製造方法が提供される。
(a) 低分子化合物に対して相互作用する抗原結合ドメインにおいて、以下(i)乃至(iv)の少なくとも一つを満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインが前記低分子化合物に結合したときの構造と前記低分子化合物に結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) 前記低分子化合物との結合に関与しないアミノ酸部位;
(iv) 前記低分子化合物との結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位に以下の(1)乃至(2)の少なくとも一つを満たすアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程:
(1) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が位置するアミノ酸部位の構造変化率が大きい;
(2) 当該アミノ酸改変を有する抗原結合ドメイン改変体が前記低分子化合物に結合したときの構造と前記低分子化合物に結合していないときの構造を比較する場合、当該改変後アミノ酸が存在することにより前記抗原結合ドメイン改変体の構造変化が阻害されない;
を含むライブラリの製造方法が提供される。
MTA特異的に結合する抗原結合分子
本開示はまた、MTAに特異的に結合する抗原結合分子に関する。MTAに特異的に結合する抗原結合分子の非限定な一例として、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPから選ばれる一つまたは複数の低分子化合物に実質的に結合しない抗原結合分子が例示される。
ここで、実質的に結合しないとは本明細書結合活性の項で記載される方法に準じて決定され、前記MTA以外の低分子化合物に対し、前記MTAに対する結合活性の85%以下、通常50%以下、好ましくは30%以下、特に好ましくは20%以下の結合活性を示すことをいう。
本開示はまた、MTAに特異的に結合する抗原結合分子に関する。MTAに特異的に結合する抗原結合分子の非限定な一例として、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPから選ばれる一つまたは複数の低分子化合物に実質的に結合しない抗原結合分子が例示される。
ここで、実質的に結合しないとは本明細書結合活性の項で記載される方法に準じて決定され、前記MTA以外の低分子化合物に対し、前記MTAに対する結合活性の85%以下、通常50%以下、好ましくは30%以下、特に好ましくは20%以下の結合活性を示すことをいう。
非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシンに実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、S-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、SAMに実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、AMP、ADP、ATPのいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、S-(5'-Adenosyl)-L-homocysteine (SAH)に実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、SAMに実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、AMP、ADP、ATPのいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)のいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAMのいずれにも実質的に結合しない。
別の非限定的な一態様において、本開示のMTAに特異的に結合する抗原結合分子は、アデノシン、S-(5'-Adenosyl)-L-homocysteine (SAH)、SAM、AMP、ADP、ATPのいずれにも実質的に結合しない。
本開示におけるMTAに特異的に結合する抗原結合分子の非限定な一態様として、
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域をのいずれか一つ含む抗原結合分子が挙げられる。
a) 配列番号:46で示す重鎖可変領域と配列番号:47で示す軽鎖可変領域を含む抗体可変領域;
b) 配列番号:50で示す重鎖可変領域と配列番号:51で示す軽鎖可変領域を含む抗体可変領域;
c) 配列番号:48で示す重鎖可変領域と配列番号:49で示す軽鎖可変領域を含む抗体可変領域;
d) 配列番号:52で示す重鎖可変領域と配列番号:53で示す軽鎖可変領域を含む抗体可変領域をのいずれか一つ含む抗原結合分子が挙げられる。
本開示におけるMTAに特異的に結合する抗原結合分子の非限定な一態様として、MTAに結合する単離された抗体が挙げられる。
上述の態様の任意のものにおいて、MTAに特異的に結合する抗原結合分子は、ヒト化されている。一態様において、MTAに特異的に結合する抗原結合分子は、上述の態様の任意のものにおけるHVRを含み、かつさらに、アクセプターヒトフレームワーク(例えば、ヒト免疫グロブリンフレームワークまたはヒトコンセンサスフレームワーク)を含む。さらに上記のMTAに特異的に結合する抗原結合分子は、MTAに対する結合活性を増強する目的でアミノ酸改変を導入しても良いし、特定の低分子化合物に対して特異性をさらに高めるための改変を導入しても良い。アミノ酸改変を導入する目的は上記に限定されず、任意の目的のための改変を導入し得る。
別の局面において、MTAに特異的に結合する抗原結合分子は、配列番号:46、48、50、52のアミノ酸配列に対して少なくとも90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、または100%の配列同一性を有する重鎖可変領域配列を含む。特定の態様において、少なくとも90%、91%、92%、93%、94%、95%、96%、97%、98%、または99%の同一性を有する重鎖可変領域配列は、参照配列に対して、置換(例えば、保存的置換)、挿入、または欠失を含むが、当該配列を含むMTAに特異的に結合する抗原結合分子は、MTAに結合する能力を保持する。特定の態様において、合計1個から10個のアミノ酸が、配列番号:46、48、50、52において、置換、挿入、および/または欠失される。
別の局面において、MTAに特異的に結合する抗原結合分子は、配列番号:47、49、51、53のアミノ酸配列に対して少なくとも90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、または100%の配列同一性を有する軽鎖可変領域配列を含む。特定の態様において、少なくとも90%、91%、92%、93%、94%、95%、96%、97%、98%、または99%の同一性を有する軽鎖可変領域配列は、参照配列に対して、置換(例えば、保存的置換)、挿入、または欠失を含むが、当該配列を含むMTAに特異的に結合する抗原結合分子は、MTAに結合する能力を保持する。特定の態様において、合計1個から10個のアミノ酸が、配列番号:47、49、51、53において、置換、挿入、および/または欠失される。
別の局面において、上述の態様の任意のものにおける重鎖可変領域、および上述の態様の任意のものにおける軽鎖可変領域を含む、MTAに特異的に結合する抗原結合分子が提供される。一態様において、抗体はそれぞれ配列番号:46、48、50、52および配列番号:47、49、51、53中の重鎖可変領域配列および軽鎖可変領域配列を、当該配列の翻訳後修飾を含んだものも含めて、含む。翻訳後修飾は、重鎖又は軽鎖N末端のグルタミン又はグルタミン酸のピログルタミル化によるピログルタミン酸への修飾を含むが、これに限定されない。
診断ならびに検出のための方法および組成物
特定の態様において、本明細書で提供されるMTAに特異的に結合する抗原結合分子のいずれも、生物学的サンプルにおけるMTAの存在を検出するのに有用である。本明細書で用いられる用語「検出」は、定量的または定性的な検出を包含する。特定の態様において、生物学的サンプルは、細胞または組織、例えば、がん組織を含む。
特定の態様において、本明細書で提供されるMTAに特異的に結合する抗原結合分子のいずれも、生物学的サンプルにおけるMTAの存在を検出するのに有用である。本明細書で用いられる用語「検出」は、定量的または定性的な検出を包含する。特定の態様において、生物学的サンプルは、細胞または組織、例えば、がん組織を含む。
一態様において、診断方法または検出方法において使用するためのMTAに特異的に結合する抗原結合分子が提供される。さらなる局面において、生物学的サンプル中のMTAの存在を検出する方法が提供される。特定の態様において、この方法は、MTAへのMTAに特異的に結合する抗原結合分子の結合が許容される条件下で本明細書に記載のMTAに特異的に結合する抗原結合分子と生物学的サンプルを接触させること、およびMTAに特異的に結合する抗原結合分子とMTAの間で複合体が形成されたかどうかを検出することを含む。そのような方法は、インビトロの方法またはインビボの方法であり得る。一態様において、MTAに特異的に結合する抗原結合分子は、例えばMTAが患者を選択するためのバイオマーカーである場合、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を用いる治療に適合する被験体を選択するために使用されうる。また別の一態様として、患者に対する治療効果を予測する方法にも使用されうる。
被験体を選択するための使用の非限定な一態様として、MTAが蓄積しているがんであるか否かの判断が例示される。本開示において、MTAががん組織にて蓄積されていることが検証されたことにより、組織中または/および末梢中のMTAを検出することによりそれらのがんに罹患しているかどうかの診断が可能である。
また治療効果を予測するための使用の非限定な一態様として、組織中または/および抹消中のMTAを検出することにより、がん組織の縮小や増悪を間接的に診断し得る。
被験体を選択するための使用の非限定な一態様として、MTAが蓄積しているがんであるか否かの判断が例示される。本開示において、MTAががん組織にて蓄積されていることが検証されたことにより、組織中または/および末梢中のMTAを検出することによりそれらのがんに罹患しているかどうかの診断が可能である。
また治療効果を予測するための使用の非限定な一態様として、組織中または/および抹消中のMTAを検出することにより、がん組織の縮小や増悪を間接的に診断し得る。
特定の態様において、標識されたMTAに特異的に結合する抗原結合分子が提供される。標識は、直接的に検出される標識または部分(例えば、蛍光標識、発色標識、高電子密度標識、化学発光標識、および放射性標識)ならびに、例えば酵素反応または分子間相互作用を通じて間接的に検出される部分(例えば酵素またはリガンド)を含むが、これらに限定されない。例示的な標識は、これらに限定されるものではないが、以下を含む:放射性同位体32P、14C、125I、3Hおよび131I、希土類キレートなどの発蛍光団またはフルオレセインおよびその誘導体、ローダミンおよびその誘導体、ダンシル、ウンベリフェロン、ホタルルシフェラーゼおよび細菌ルシフェラーゼ(米国特許第4,737,456号)などのルシフェラーゼ、ルシフェリン、2,3-ジヒドロフタラジンジオン、西洋ワサビペルオキシダーゼ (horseradish peroxidase: HRP)、アルカリホスファターゼ、β-ガラクトシダーゼ、グルコアミラーゼ、リゾチーム、単糖オキシダーゼ(例えばグルコースオキシダーゼ、ガラクトースオキシダーゼおよびグルコース-6-リン酸デヒドロゲナーゼ)、ウリカーゼおよびキサンチンオキシダーゼなどの複素環オキシダーゼ、過酸化水素を用いて色素前駆体を酸化する酵素(例えばHRP、ラクトペルオキシダーゼ、またはミクロペルオキシダーゼ)と連結されたもの、ビオチン/アビジン、スピン標識、バクテリオファージ標識、安定なフリーラジカル類、ならびにこれらに類するもの。
なお、本開示に記載するアミノ酸配列に含まれるアミノ酸は翻訳後に修飾(例えば、N末端のグルタミンのピログルタミル化によるピログルタミン酸への修飾は当業者によく知られた修飾である)を受ける場合もあるが、そのようにアミノ酸が翻訳後修飾された場合であっても当然のことながら本開示に記載するアミノ酸配列に含まれる。
本明細書に記載の1又は複数の態様を任意に組み合わせたものも、当業者の技術常識に基づいて技術的に矛盾しない限り、本開示に含まれることが当業者には当然に理解される。
なお本明細書において引用されたすべての先行技術文献は、参照として本明細書に組み入れられる。
以下本開示を実施例により具体的に説明するが、本開示はこれら実施例に制限されるものではない。
[実施例1] 腫瘍組織及び正常組織におけるMTA蓄積量の分析
実験に使用する細胞株として、ATCCからHCC827, HPAC, HT-1376, NCI-H2228, SK-LU-1, SK-MES-1, U-87 MGを、DainipponからMia-PaCa2を、NCIからKM12を、JCRBからKM12-Lucを、RIKENからPK-1を入手した。KM12-Luc以外の細胞株について、Barretina JらNature. (2012)483:603-7.が公開した細胞株のSNPsアレイデータセットを参照して、MTAP欠損状態を判定した。KM12-Lucはこのデータセットに含まれていないが、KM12の派生株であることから、MTAP欠損状態はKM12と同じであるとみなした。
実験に使用する細胞株として、ATCCからHCC827, HPAC, HT-1376, NCI-H2228, SK-LU-1, SK-MES-1, U-87 MGを、DainipponからMia-PaCa2を、NCIからKM12を、JCRBからKM12-Lucを、RIKENからPK-1を入手した。KM12-Luc以外の細胞株について、Barretina JらNature. (2012)483:603-7.が公開した細胞株のSNPsアレイデータセットを参照して、MTAP欠損状態を判定した。KM12-Lucはこのデータセットに含まれていないが、KM12の派生株であることから、MTAP欠損状態はKM12と同じであるとみなした。
(1-1)MTAP欠損/非欠損細胞の細胞内MTA濃度の測定
MTAP欠損細胞のMia-PaCa2, NCI-H2228, SK-LU-1, U-87 MG、およびMTAP非欠損細胞のHCC827, HPAC, KM12-Lucを75cm2 bottleに106個撒き、48時間培養した。細胞をTrypsinで剥がして回収し、回収された細胞にH2Oを加え、Sonicationによる細胞破砕を実施して細胞破砕液を得た。細胞破砕液に、細胞破砕液の3倍量のMeOHを加えてVortexし、更に遠心でDebrisを除去し、遠心後上清中のMTA濃度をLC-MS(TSQ vantage, ThermoFisher)を用いて測定した。Reinoso RF et al., Drug Metab Dispos. 2001 Apr;29(4 Pt 1):453-9.の論文報告を参照し、細胞体積を106細胞あたり4.0uLと仮定し、遠心後上清中のMTA濃度から細胞内MTA濃度を算出した。図1で示すように、MTAP非欠損細胞に比べ、MTAP欠損細胞のいずれも高い細胞内MTA濃度を有することが分かった。
MTAP欠損細胞のMia-PaCa2, NCI-H2228, SK-LU-1, U-87 MG、およびMTAP非欠損細胞のHCC827, HPAC, KM12-Lucを75cm2 bottleに106個撒き、48時間培養した。細胞をTrypsinで剥がして回収し、回収された細胞にH2Oを加え、Sonicationによる細胞破砕を実施して細胞破砕液を得た。細胞破砕液に、細胞破砕液の3倍量のMeOHを加えてVortexし、更に遠心でDebrisを除去し、遠心後上清中のMTA濃度をLC-MS(TSQ vantage, ThermoFisher)を用いて測定した。Reinoso RF et al., Drug Metab Dispos. 2001 Apr;29(4 Pt 1):453-9.の論文報告を参照し、細胞体積を106細胞あたり4.0uLと仮定し、遠心後上清中のMTA濃度から細胞内MTA濃度を算出した。図1で示すように、MTAP非欠損細胞に比べ、MTAP欠損細胞のいずれも高い細胞内MTA濃度を有することが分かった。
(1-2)MTAP欠損/非欠損細胞の培養液中MTA濃度の測定
MTAP欠損細胞のMia-PaCa2, NCI-H2228, SK-LU-1, U-87 MGおよびMTAP非欠損細胞のHCC827, KM12-Lucを75cm2 bottleに106個撒き、培地を14mL加え培養した。24, 48時間の各タイムポイントで培養液を回収し、回収された培養液に3倍量のMeOHを加え、遠心してDebrisを除去したのちに上清を取得し、取得された培養液上清中のMTA濃度をLC-MS(LTQ VelosおよびTSQ vantage, ThermoFisher)で測定した。その結果、MTAP欠損細胞の培養液中ではMTA濃度の経時的上昇(MTAの蓄積)が観察された。一方、MTAP非欠損細胞のHCC827の培養液中では、いずれのタイムポイントにおいてもMTAは検出限界の5nMを下回り、MTAP非欠損細胞のKM12-Lucの培養液中のMTA濃度も、5nMをやや上回る程度であった(図2)。
MTAP欠損細胞のMia-PaCa2, NCI-H2228, SK-LU-1, U-87 MGおよびMTAP非欠損細胞のHCC827, KM12-Lucを75cm2 bottleに106個撒き、培地を14mL加え培養した。24, 48時間の各タイムポイントで培養液を回収し、回収された培養液に3倍量のMeOHを加え、遠心してDebrisを除去したのちに上清を取得し、取得された培養液上清中のMTA濃度をLC-MS(LTQ VelosおよびTSQ vantage, ThermoFisher)で測定した。その結果、MTAP欠損細胞の培養液中ではMTA濃度の経時的上昇(MTAの蓄積)が観察された。一方、MTAP非欠損細胞のHCC827の培養液中では、いずれのタイムポイントにおいてもMTAは検出限界の5nMを下回り、MTAP非欠損細胞のKM12-Lucの培養液中のMTA濃度も、5nMをやや上回る程度であった(図2)。
(1-3)MTAP非欠損細胞による細胞外MTAの取り込み
MTAP欠損細胞の培養液中のMTA濃度が経時的に増加することから、細胞内から細胞外へのMTA移行経路があることが推測される。しかし、MTAP非欠損細胞では、細胞内に~1uMのMTAがあると予想される(図1)にも関わらず、培養液中のMTA濃度が非常に低かった(図2)。MTAP非欠損細胞培養液中のMTA蓄積が見られない理由に関する手がかりを得るため、MTAP非欠損細胞のHT-1376、SK-MES-1を6-wells plateに3 x 105個撒き、培地を3mL加え、24hrインキュベーションして細胞を接着させた。その後、MTA濃度が100nMになるようにMTA溶液を培養液に加え、所定の時間培養した(MTA: Sigma, Catalog No. D5011-100MG)。0, 4, 24, 48, 72時間の各タイムポイントで培養液を回収し、回収した培養液中のMTA濃度をLC-MSで測定したところ、培養開始後に培養液中のMTA濃度が経時的に減少することが観察された(図3)。細胞がMTAPを使ってMTAを代謝できる場合、細胞外のMTAが細胞に吸収され、速やかに消失することが示唆された。
MTAP欠損細胞の培養液中のMTA濃度が経時的に増加することから、細胞内から細胞外へのMTA移行経路があることが推測される。しかし、MTAP非欠損細胞では、細胞内に~1uMのMTAがあると予想される(図1)にも関わらず、培養液中のMTA濃度が非常に低かった(図2)。MTAP非欠損細胞培養液中のMTA蓄積が見られない理由に関する手がかりを得るため、MTAP非欠損細胞のHT-1376、SK-MES-1を6-wells plateに3 x 105個撒き、培地を3mL加え、24hrインキュベーションして細胞を接着させた。その後、MTA濃度が100nMになるようにMTA溶液を培養液に加え、所定の時間培養した(MTA: Sigma, Catalog No. D5011-100MG)。0, 4, 24, 48, 72時間の各タイムポイントで培養液を回収し、回収した培養液中のMTA濃度をLC-MSで測定したところ、培養開始後に培養液中のMTA濃度が経時的に減少することが観察された(図3)。細胞がMTAPを使ってMTAを代謝できる場合、細胞外のMTAが細胞に吸収され、速やかに消失することが示唆された。
(1-4)MTAの血液クリアランス
MTA添加培地でMTAP非欠損細胞を培養するとき、培養液中MTAが速やかに消失したことを受け、正常組織でもMTAが細胞に吸収され速やかに消失するかを検討した。
マウスから新鮮血液を採取した。予め37℃で加温しておいたマウス血液に終濃度5 ng/mLになるようにMTAの安定同位体(SIL-MTA)を添加し、5秒後に等量のアセトニトリルを添加した。アセトニトリルで除タンパクした血液試料にメタノールならびに内標準物質を添加し、遠心濾過後の濾液をLC-MS/MSで分析し、SIL-MTA添加5秒後のマウス血液中SIL-MTA濃度を測定した。マウス血液とアセトニトリルを等量混合した溶液に終濃度5 ng/mLとなるようにSIL-MTAを添加したサンプルを用いて測定したSIL-MTA濃度をSIL-MTA濃度の初期値とした。SIL-MTA濃度の初期値とSIL-MTA添加5秒後のマウス血液中SIL-MTA濃度から、血液からのSIL-MTA消失速度定数を算出し、消失速度定数にマウス単位体重当たりの血液量(85 mL/kg)を乗じて算出した全血液クリアランスは46685 mL/h/kgと推定された。この結果から、マウス血液中に添加されたMTAは、血液から速やかに消失することが分かった。
MTA添加培地でMTAP非欠損細胞を培養するとき、培養液中MTAが速やかに消失したことを受け、正常組織でもMTAが細胞に吸収され速やかに消失するかを検討した。
マウスから新鮮血液を採取した。予め37℃で加温しておいたマウス血液に終濃度5 ng/mLになるようにMTAの安定同位体(SIL-MTA)を添加し、5秒後に等量のアセトニトリルを添加した。アセトニトリルで除タンパクした血液試料にメタノールならびに内標準物質を添加し、遠心濾過後の濾液をLC-MS/MSで分析し、SIL-MTA添加5秒後のマウス血液中SIL-MTA濃度を測定した。マウス血液とアセトニトリルを等量混合した溶液に終濃度5 ng/mLとなるようにSIL-MTAを添加したサンプルを用いて測定したSIL-MTA濃度をSIL-MTA濃度の初期値とした。SIL-MTA濃度の初期値とSIL-MTA添加5秒後のマウス血液中SIL-MTA濃度から、血液からのSIL-MTA消失速度定数を算出し、消失速度定数にマウス単位体重当たりの血液量(85 mL/kg)を乗じて算出した全血液クリアランスは46685 mL/h/kgと推定された。この結果から、マウス血液中に添加されたMTAは、血液から速やかに消失することが分かった。
(1-5)マウスにおけるMTAのin vivoクリアランス
実施例(1-4)の検討から、in vitroではMTA血液クリアランスが大きいことが示唆されたため、実際にin vivoでもMTAのクリアランスが大きいかを検討するためにMTAの薬物動態試験を実施した。内因性MTAと区別するために被験物質にSIL-MTAを用いた。NOD-scidマウスにSIL-MTAを0.1~100 μg/min/kg、25 mL/kg(n=3)で定速静脈内投与して、投与開始後2, 5, 10, 30分に血液を採取した。血液採取後、12000rpm、5分間、4℃で遠心して細胞を除去し、血漿を得た。血漿中SIL-MTA濃度はLC-MS/MSで測定した。投与開始後30分の血漿中SIL-MTA濃度とSIL-MTAの投与速度から算出されたSIL-MTA全身クリアランスは38961 mL/h/kg以上であり、in vivoにおいてもMTAは速やかに血漿から消失することが明らかとなった。
実施例(1-4)の検討から、in vitroではMTA血液クリアランスが大きいことが示唆されたため、実際にin vivoでもMTAのクリアランスが大きいかを検討するためにMTAの薬物動態試験を実施した。内因性MTAと区別するために被験物質にSIL-MTAを用いた。NOD-scidマウスにSIL-MTAを0.1~100 μg/min/kg、25 mL/kg(n=3)で定速静脈内投与して、投与開始後2, 5, 10, 30分に血液を採取した。血液採取後、12000rpm、5分間、4℃で遠心して細胞を除去し、血漿を得た。血漿中SIL-MTA濃度はLC-MS/MSで測定した。投与開始後30分の血漿中SIL-MTA濃度とSIL-MTAの投与速度から算出されたSIL-MTA全身クリアランスは38961 mL/h/kg以上であり、in vivoにおいてもMTAは速やかに血漿から消失することが明らかとなった。
(1-6)担癌マウスモデルの腫瘍中MTA濃度測定
MTAP欠損細胞のMia-PaCa2とMTAP非欠損細胞のPK-1をNOD SCIDマウスの皮下に移植して担癌マウスを作製し、腫瘍体積が188~347mm3の時点で腫瘍を採取した(n=3)。腫瘍重量の3倍量のメタノールを添加してステンレスビーズを用いてホモジェナイズし、12000rpm、10分間遠心分離して上清を得た。得られた上清を75%メタノールで10倍希釈し、内標準物質を含む水溶液を添加して10000rpm、10分間遠心分離した。上清をフィルターで濾過し、濾液をLC-MS/MS分析してMTA濃度を測定し、濾液中のMTA量と腫瘍重量から腫瘍中のMTA濃度(pmol/g tissue)を計算した。その結果、MTAP欠損細胞のMia-PaCa2を移植したNOD SCIDマウスにおいて高い腫瘍中MTA濃度が検出された(図4)。
MTAP欠損細胞のMia-PaCa2とMTAP非欠損細胞のPK-1をNOD SCIDマウスの皮下に移植して担癌マウスを作製し、腫瘍体積が188~347mm3の時点で腫瘍を採取した(n=3)。腫瘍重量の3倍量のメタノールを添加してステンレスビーズを用いてホモジェナイズし、12000rpm、10分間遠心分離して上清を得た。得られた上清を75%メタノールで10倍希釈し、内標準物質を含む水溶液を添加して10000rpm、10分間遠心分離した。上清をフィルターで濾過し、濾液をLC-MS/MS分析してMTA濃度を測定し、濾液中のMTA量と腫瘍重量から腫瘍中のMTA濃度(pmol/g tissue)を計算した。その結果、MTAP欠損細胞のMia-PaCa2を移植したNOD SCIDマウスにおいて高い腫瘍中MTA濃度が検出された(図4)。
(1-7)ヒト臨床サンプルにおけるMTAP DNA量およびMTA濃度の測定
Asterand社およびILS社から購入した膀胱がん8例と、食道がん10例のFresh frozen臨床サンプルにおいてMTAP 欠損に伴うMTA蓄積が見られるかを調べた。
Asterand社およびILS社から購入した膀胱がん8例と、食道がん10例のFresh frozen臨床サンプルにおいてMTAP 欠損に伴うMTA蓄積が見られるかを調べた。
(1-7-1)ヒト臨床サンプルにおけるMTAP DNA量分析
臨床サンプルからQIAamp Fast DNA Tissue Kit [QIAGEN, cat.no. 51404] を用いてDNAを抽出した。抽出されたDNAを鋳型にして、RealTime-PCR法でMTAPの遺伝子領域の増幅を行って、MTAP遺伝子のCt値を求めた。Ct値は増幅DNA量が閾値に達するまでのPCRサイクル数であり、初期DNA量が少ないと大きい値となる。実験間の増幅効率の差を補正するため、M’soka TJら(Leukemia (2000) 14, 935-940)に倣い、別染色体上にあるMTAP偽遺伝子のΨX4のCt値も求めた。各サンプルについて、MTAP遺伝子のCt値からΨX4遺伝子のCt値を引いてΔCtを求めた。
臨床サンプルからQIAamp Fast DNA Tissue Kit [QIAGEN, cat.no. 51404] を用いてDNAを抽出した。抽出されたDNAを鋳型にして、RealTime-PCR法でMTAPの遺伝子領域の増幅を行って、MTAP遺伝子のCt値を求めた。Ct値は増幅DNA量が閾値に達するまでのPCRサイクル数であり、初期DNA量が少ないと大きい値となる。実験間の増幅効率の差を補正するため、M’soka TJら(Leukemia (2000) 14, 935-940)に倣い、別染色体上にあるMTAP偽遺伝子のΨX4のCt値も求めた。各サンプルについて、MTAP遺伝子のCt値からΨX4遺伝子のCt値を引いてΔCtを求めた。
(1-7-2)ヒト臨床サンプルにおけるMTA濃度分析
臨床サンプル重量の3倍量のメタノールを添加してステンレスビーズを用いてホモジェナイズし、12000rpm、10分間遠心分離して上清を得た。得られた上清を75%メタノールで10倍希釈し、内標準物質を含む水溶液を添加して10000rpm、10分間遠心分離した。上清をフィルターで濾過し、濾液をLC-MS/MS分析してMTA濃度を測定し、濾液中のMTA量と臨床サンプル重量から各サンプルの組織中MTA濃度(pmol/g tissue)を計算した。検出限界(30 pmol/g tissue)を下回ったサンプルに関しては、検出下限の値を測定値とした。
臨床サンプル重量の3倍量のメタノールを添加してステンレスビーズを用いてホモジェナイズし、12000rpm、10分間遠心分離して上清を得た。得られた上清を75%メタノールで10倍希釈し、内標準物質を含む水溶液を添加して10000rpm、10分間遠心分離した。上清をフィルターで濾過し、濾液をLC-MS/MS分析してMTA濃度を測定し、濾液中のMTA量と臨床サンプル重量から各サンプルの組織中MTA濃度(pmol/g tissue)を計算した。検出限界(30 pmol/g tissue)を下回ったサンプルに関しては、検出下限の値を測定値とした。
(1-7-3)ヒト臨床サンプルにおけるMTAP DNA量とMTA濃度の相関性分析
ΔCt値を横軸に、組織中のMTA量を縦軸に作図し、MTAP DNA量とMTA濃度の相関性を分析した(図5)。膀胱がん臨床サンプルと食道がん臨床サンプルのいずれにおいても、ΔCtが大きくなる、すなわち組織中MTAP DNA量が少なくなるにつれ、MTA濃度が大きくなることが観察された。
ΔCt値を横軸に、組織中のMTA量を縦軸に作図し、MTAP DNA量とMTA濃度の相関性を分析した(図5)。膀胱がん臨床サンプルと食道がん臨床サンプルのいずれにおいても、ΔCtが大きくなる、すなわち組織中MTAP DNA量が少なくなるにつれ、MTA濃度が大きくなることが観察された。
(1-8)マイクロダイアリシスによる担癌マウスモデル組織中MTA濃度の測定
MTAP欠損細胞のMia-PaCa2とMTAP非欠損細胞のKM12をBALB/c nu/nuマウスの皮下に移植し、担癌マウス(以下、Mia-PaCa2モデル、KM12モデル)を作成した。移植した腫瘍体積が200mm3以上になった時点でイソフルラン麻酔下、マイクロダイアリスプローブを腫瘍および肝臓(対照としての正常組織)に挿入し、組織間質液を採取した。プローブ挿入後90-120分に採取された組織間質液中のMTA濃度をLC-MS/MS法で定量した。BSA溶液(0, 1, 10,100mg/mL)にMTAを既知濃度添加した標準溶液を作製し、マイクロダイリシスのプローブから回収されたMTA濃度と標準溶液のMTA濃度の比から、MTA回収率を算出した。採取された組織間質液中MTA濃度に対して、MTA回収率(16.3%)で補正し、組織中の細胞外MTA濃度が算出された。
腫瘍中細胞外MTA濃度については、Mia-PaCa2モデルの方がKM12モデルより高く、MTAP欠損細胞からMTAが間質液に排出されていることが示唆された(図6)。一方、肝臓中細胞外MTA濃度については、両担癌マウスモデル間では差が見られなかった。以上結果から、MTAP欠損細胞からMTAが排出されているが、MTAP欠損細胞由来のMTAは速やかに体内から消失するため正常組織中の細胞外MTA濃度は低く保たれていると推察された。
MTAP欠損細胞のMia-PaCa2とMTAP非欠損細胞のKM12をBALB/c nu/nuマウスの皮下に移植し、担癌マウス(以下、Mia-PaCa2モデル、KM12モデル)を作成した。移植した腫瘍体積が200mm3以上になった時点でイソフルラン麻酔下、マイクロダイアリスプローブを腫瘍および肝臓(対照としての正常組織)に挿入し、組織間質液を採取した。プローブ挿入後90-120分に採取された組織間質液中のMTA濃度をLC-MS/MS法で定量した。BSA溶液(0, 1, 10,100mg/mL)にMTAを既知濃度添加した標準溶液を作製し、マイクロダイリシスのプローブから回収されたMTA濃度と標準溶液のMTA濃度の比から、MTA回収率を算出した。採取された組織間質液中MTA濃度に対して、MTA回収率(16.3%)で補正し、組織中の細胞外MTA濃度が算出された。
腫瘍中細胞外MTA濃度については、Mia-PaCa2モデルの方がKM12モデルより高く、MTAP欠損細胞からMTAが間質液に排出されていることが示唆された(図6)。一方、肝臓中細胞外MTA濃度については、両担癌マウスモデル間では差が見られなかった。以上結果から、MTAP欠損細胞からMTAが排出されているが、MTAP欠損細胞由来のMTAは速やかに体内から消失するため正常組織中の細胞外MTA濃度は低く保たれていると推察された。
[実施例2]
ナイーブライブラリもしくは合成ライブラリからのMTA依存的に抗原に結合する抗体の取得
(2-1)MTA依存的に抗原に結合する抗体を取得するためのパニング
ヒトのナイーブ抗体レパートリーを模したライブラリから、MTA存在下でヒトIL-6レセプター(hIL-6R)に対して結合活性を示す抗体がスクリーニングされた。
ヒトPBMCから調製したポリA RNAや、市販されているヒトポリA RNAなどを鋳型として、当業者に公知な方法に従い、互いに異なるヒト抗体配列のFabドメインを提示する複数のファージからなるナイーブヒト抗体ファージディスプレイライブラリ(ナイーブライブラリと呼称する)が構築された。
またヒトのナイーブ抗体レパートリーを参考に人為的に設計したデザインをもとに、当業者に公知な方法に従い、互いに異なるヒト抗体配列のFabドメインを提示する複数のファージからなる合成ヒト抗体ファージディスプレイライブラリ(合成ライブラリと呼称する)が構築された。
具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7TC(WO2015046554A1)もしくはM13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、30℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団を、TBSにて希釈することによって抗体提示ファージライブラリ液が調製された。
ナイーブライブラリもしくは合成ライブラリからのMTA依存的に抗原に結合する抗体の取得
(2-1)MTA依存的に抗原に結合する抗体を取得するためのパニング
ヒトのナイーブ抗体レパートリーを模したライブラリから、MTA存在下でヒトIL-6レセプター(hIL-6R)に対して結合活性を示す抗体がスクリーニングされた。
ヒトPBMCから調製したポリA RNAや、市販されているヒトポリA RNAなどを鋳型として、当業者に公知な方法に従い、互いに異なるヒト抗体配列のFabドメインを提示する複数のファージからなるナイーブヒト抗体ファージディスプレイライブラリ(ナイーブライブラリと呼称する)が構築された。
またヒトのナイーブ抗体レパートリーを参考に人為的に設計したデザインをもとに、当業者に公知な方法に従い、互いに異なるヒト抗体配列のFabドメインを提示する複数のファージからなる合成ヒト抗体ファージディスプレイライブラリ(合成ライブラリと呼称する)が構築された。
具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7TC(WO2015046554A1)もしくはM13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、30℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団を、TBSにて希釈することによって抗体提示ファージライブラリ液が調製された。
パニングが以下の方法により実施された。当該ファージライブラリ液に終濃度4%となるようにBSAが添加された。必要に応じてMTA非存在下でhIL-6Rに結合する抗体を除去するため、0.1 nmolのビオチン標識hIL-6Rを固相した磁気ビーズと調製されたファージライブラリ液を混合し室温で60分間反応後、磁気スタンドを用いて分離されたビーズからファージ溶液が回収された。回収されたファージライブラリ液0.8 mLに0.1 nmolのビオチン標識hIL-6Rと終濃度100 μMのMTAを加え、室温で60分間反応させた。この反応液に、BSAでブロッキングされた磁気ビーズ(NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific))を加え室温で15分反応させた。ビーズは0.5 mLのTBS/0.1%Tween20にて2回もしくは3回、TBSにて1回もしくは2回洗浄された。その後TBSが加えられたビーズが室温で懸濁され、磁気スタンドを用いて分離されたビーズからファージ溶液が回収された。この作業が2回または3回繰り返された後、溶出されたファージ溶液が混合された。回収されたファージ溶液に最終濃度1mg/mLのトリプシンが加えられた。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった20mLの大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。大腸菌は225 mm x 225 mmのプレートへ播種された。この一連の作業を追加で3回繰り返した。
(2-2)ファージELISAによるパニング後取得ファージ群のMTA存在下でのhIL-6R結合活性の評価
パニングで得られた大腸菌のシングルコロニーらから、それぞれ常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収された。NucleoFast96(MACHERY-NAGEL)を用いて、回収されたファージ培養上清が限外濾過された。
TBSまたは終濃度100 μMのMTAを含むTBSに希釈されたファージが以下の手順でELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、ビオチン標識hIL-6Rを含むTBSが10 μl添加され1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80uLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、当該ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示する抗体をMTA非存在下もしくは存在下においてビオチン標識hIL-6Rに結合させた。当該ウェルをTBSTもしくは終濃度100 μMのMTAを含むTBSTで洗浄した後、当該ウェルにTBSまたは終濃度100 μMのMTAを含むTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。当該ウェルをTBSTもしくは終濃度100 μMのMTAを含むTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、評価した384クローン中、100μMのMTA存在下とMTAの非存在下でhIL-6Rに対する結合活性が変化する抗体を提示するファージが4クローン確認された。
パニングで得られた大腸菌のシングルコロニーらから、それぞれ常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収された。NucleoFast96(MACHERY-NAGEL)を用いて、回収されたファージ培養上清が限外濾過された。
TBSまたは終濃度100 μMのMTAを含むTBSに希釈されたファージが以下の手順でELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、ビオチン標識hIL-6Rを含むTBSが10 μl添加され1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80uLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、当該ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示する抗体をMTA非存在下もしくは存在下においてビオチン標識hIL-6Rに結合させた。当該ウェルをTBSTもしくは終濃度100 μMのMTAを含むTBSTで洗浄した後、当該ウェルにTBSまたは終濃度100 μMのMTAを含むTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。当該ウェルをTBSTもしくは終濃度100 μMのMTAを含むTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、評価した384クローン中、100μMのMTA存在下とMTAの非存在下でhIL-6Rに対する結合活性が変化する抗体を提示するファージが4クローン確認された。
(2-3)MTA依存的に抗原に結合する抗体の発現と精製
実施例(2-2)において確認された、100μMのMTA存在下とMTAの非存在下でhIL-6Rに対する結合活性が変化する抗体の重鎖および軽鎖の可変領域配列は、重鎖抗体定常領域配列(配列番号:56)もしくは軽鎖kappa定常領域配列(配列番号:39)を有する動物発現用プラスミドへとそれぞれ挿入された。プライマー(配列番号:44および45)を用いて増幅された抗体遺伝子の塩基配列を解析したところ、一種類の抗体可変領域配列(重鎖可変領域:配列番号:54、軽鎖可変領域:配列番号:55)が同定された。参考実施例1の方法を用いて抗体(C03H- BH076N17/C03L-KT0)が発現された。
実施例(2-2)において確認された、100μMのMTA存在下とMTAの非存在下でhIL-6Rに対する結合活性が変化する抗体の重鎖および軽鎖の可変領域配列は、重鎖抗体定常領域配列(配列番号:56)もしくは軽鎖kappa定常領域配列(配列番号:39)を有する動物発現用プラスミドへとそれぞれ挿入された。プライマー(配列番号:44および45)を用いて増幅された抗体遺伝子の塩基配列を解析したところ、一種類の抗体可変領域配列(重鎖可変領域:配列番号:54、軽鎖可変領域:配列番号:55)が同定された。参考実施例1の方法を用いて抗体(C03H- BH076N17/C03L-KT0)が発現された。
(2-4)表面プラズモン共鳴を用いたMTA存在下での抗原結合活性の評価
Biacore T200(GE Healthcare)を用いて、C03H- BH076N17/C03L-KT0とhIL-6Rとの抗原抗体反応におけるMTAとアデノシンの影響が評価された。ランニングバッファーとして、TBS、0.02%(w/v)Tween20、pH7.4が用いられた。センサーチップCM5(GE Healthcare)上にアミンカップリングによりProA/G(Pierce)を固定化し、その上に抗体を固相化した後、各種抗原をアナライトとして相互作用させることで、抗原抗体間の結合量の変化が観察された。抗原の希釈には、ランニングバッファーおよびランニングバッファーにMTA、またはアデノシンのいずれかがそれぞれ添加されたバッファーが使用され、数段階の濃度系列で調製された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。
MTAもしくはアデノシンの異なる濃度下での、C03H- BH076N17/C03L-KT0のhIL-6Rに対する結合量を図7に示した。C03H- BH076N17/C03L-KT0はMTA濃度が高いほどhIL-6Rに対して強い結合活性を示すことが確認された。またアデノシン存在下ではいかなる濃度においてもhIL-6Rへの結合は観察されなかった。したがってC03H- BH076N17/C03L-KT0はMTA特異的に依存的にhIL-6Rに結合する抗体であることが示された。
Biacore T200(GE Healthcare)を用いて、C03H- BH076N17/C03L-KT0とhIL-6Rとの抗原抗体反応におけるMTAとアデノシンの影響が評価された。ランニングバッファーとして、TBS、0.02%(w/v)Tween20、pH7.4が用いられた。センサーチップCM5(GE Healthcare)上にアミンカップリングによりProA/G(Pierce)を固定化し、その上に抗体を固相化した後、各種抗原をアナライトとして相互作用させることで、抗原抗体間の結合量の変化が観察された。抗原の希釈には、ランニングバッファーおよびランニングバッファーにMTA、またはアデノシンのいずれかがそれぞれ添加されたバッファーが使用され、数段階の濃度系列で調製された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。
MTAもしくはアデノシンの異なる濃度下での、C03H- BH076N17/C03L-KT0のhIL-6Rに対する結合量を図7に示した。C03H- BH076N17/C03L-KT0はMTA濃度が高いほどhIL-6Rに対して強い結合活性を示すことが確認された。またアデノシン存在下ではいかなる濃度においてもhIL-6Rへの結合は観察されなかった。したがってC03H- BH076N17/C03L-KT0はMTA特異的に依存的にhIL-6Rに結合する抗体であることが示された。
[実施例3]
MTA依存的に抗原に結合する抗体を取得するための、ヒト化SMB0002を鋳型としたライブラリのデザイン
(3-1)ヒト化SMB0002のMTA結合性評価
MTAとアデノシンの構造の類似性から、アデノシンに結合する抗体は、MTAに交差反応する可能性があり、MTAに交差反応するアデノシン抗体は、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインするときの鋳型抗体として使用できる可能性がある。アデノシンに結合すると報告されたヒト化SMB0002(重鎖可変領域:配列番号:31;軽鎖可変領域:配列番号:32)が、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインするときの鋳型抗体として利用できるか検証するため、Biacore T200 (GE Healthcare)を用いて、ヒト化SMB0002のMTAに対する結合性が解析された。
MTA依存的に抗原に結合する抗体を取得するための、ヒト化SMB0002を鋳型としたライブラリのデザイン
(3-1)ヒト化SMB0002のMTA結合性評価
MTAとアデノシンの構造の類似性から、アデノシンに結合する抗体は、MTAに交差反応する可能性があり、MTAに交差反応するアデノシン抗体は、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインするときの鋳型抗体として使用できる可能性がある。アデノシンに結合すると報告されたヒト化SMB0002(重鎖可変領域:配列番号:31;軽鎖可変領域:配列番号:32)が、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインするときの鋳型抗体として利用できるか検証するため、Biacore T200 (GE Healthcare)を用いて、ヒト化SMB0002のMTAに対する結合性が解析された。
ストレプトアビジンがあらかじめ固定化されたSensor chip SA(GE Healthcare)に、ビオチン化された抗ヒトIgG CH1分子であるCaptureSelectTM Biotin Anti-IgG-CH1 Conjugate (Thermo fisher scientific) を結合させ、そののちヒト化SMB0002をキャプチャーさせ、MTA (sigma-aldrich)またはアデノシン (Wako)をヒト化SMB0002と相互作用させた。ランニングバッファーとして、50 mM Tris-HCl, 150mM NaCl (Takara, T903), 0.02% (w/v) Tween20, 5% DMSOが用いられた。MTAまたはアデノシンは流速100μL/minで、ヒト化SMB0002と42秒間相互作用させ、その後ランニングバッファーにて抗体から解離させた。相互作用は37℃で測定され、MTAまたはアデノシンの希釈にはランニングバッファーと同じバッファーが使用された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、ヒト化SMB0002のMTAおよびアデノシンに対する結合活性が表1のように決定され、ヒト化SMB0002はアデノシンだけでなくMTAに対しても結合活性を有すると確認された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、ヒト化SMB0002のMTAおよびアデノシンに対する結合活性が表1のように決定され、ヒト化SMB0002はアデノシンだけでなくMTAに対しても結合活性を有すると確認された。

(3-2)ヒト化SMB0002のX線結晶構造解析
ヒト化SMB0002のFab断片(SMB0002hFab)単独、およびSMB0002hFabとアデノシンの複合体の結晶構造が解析された。
ヒト化SMB0002のFab断片(SMB0002hFab)単独、およびSMB0002hFabとアデノシンの複合体の結晶構造が解析された。
(3-2-1)結晶化用ヒト化SMB0002全長抗体の調製
結晶化用ヒト化SMB0002全長抗体の調製及び精製は当業者公知の方法により行われた。
結晶化用ヒト化SMB0002全長抗体の調製及び精製は当業者公知の方法により行われた。
(3-2-2)SMB0002hFabの結晶構造解析のためのFab断片の調製
SMB0002hFabは、ヒト化SMB0002全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)による制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex200 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
SMB0002hFabは、ヒト化SMB0002全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)による制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex200 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
(3-2-3)SMB0002hFabとアデノシン複合体の結晶の作製
結晶化は、当業者公知の方法で精製され、約13mg/mLまで濃縮されたSMB0002hFabを用いて、シッティングドロップ蒸気拡散法により5℃で行われた。リザーバー溶液は、0.1 M リン酸水素二ナトリウム・クエン酸 pH 4.2、40 %w/v ポリエチレングリコール200から成るものであった。
結晶化は、当業者公知の方法で精製され、約13mg/mLまで濃縮されたSMB0002hFabを用いて、シッティングドロップ蒸気拡散法により5℃で行われた。リザーバー溶液は、0.1 M リン酸水素二ナトリウム・クエン酸 pH 4.2、40 %w/v ポリエチレングリコール200から成るものであった。
(3-2-4)SMB0002hFabとアデノシンの複合体の結晶からのX線回折データの収集および構造決定
得られた結晶は、液体窒素で凍結され、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-17AでX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計720枚が収集された。得られた回折画像は、Xia2 (J. Appl. Cryst. (2010). 43, 186-190)を用いて処理され、分解能2.24 Åまでの回折強度データが取得された。結晶学的統計値が表2に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法を実施し、初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表2に示される。
得られた結晶は、液体窒素で凍結され、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-17AでX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計720枚が収集された。得られた回折画像は、Xia2 (J. Appl. Cryst. (2010). 43, 186-190)を用いて処理され、分解能2.24 Åまでの回折強度データが取得された。結晶学的統計値が表2に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法を実施し、初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表2に示される。
(3-2-5)SMB0002hFabの結晶の作製
当業者公知の方法で精製されたSMB0002hFabは、尿素により変性された後、透析法によってリフォールディングされ、続いて、ゲル濾過クロマトグラフィー(Superdex200 10/300 increase、GE Healthcare)により精製された。結晶化は、約13mg/mLまで濃縮されたSMB0002hFabを用いて、シッティングドロップ蒸気拡散法により20℃で行われた。リザーバー溶液は、0.1 M Morpheus buffer 2 pH 7.5、37.5% w/v M1K3350、0.1M Morpheus Carboxylic acids(Morpheus、Molecular Dimensions)から成るものであった。
当業者公知の方法で精製されたSMB0002hFabは、尿素により変性された後、透析法によってリフォールディングされ、続いて、ゲル濾過クロマトグラフィー(Superdex200 10/300 increase、GE Healthcare)により精製された。結晶化は、約13mg/mLまで濃縮されたSMB0002hFabを用いて、シッティングドロップ蒸気拡散法により20℃で行われた。リザーバー溶液は、0.1 M Morpheus buffer 2 pH 7.5、37.5% w/v M1K3350、0.1M Morpheus Carboxylic acids(Morpheus、Molecular Dimensions)から成るものであった。
(3-2-6)SMB0002hFabの結晶からのX線回折データの収集および構造決定
得られた結晶は、液体窒素で凍結され、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-17AでX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計4320枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.83 Åまでの回折強度データが取得された。結晶学的統計値が表2に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法により初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表2に示される。
得られた結晶は、液体窒素で凍結され、高エネルギー加速器研究機構の放射光施設フォトンファクトリーのBL-17AでX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計4320枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.83 Åまでの回折強度データが取得された。結晶学的統計値が表2に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法により初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表2に示される。

(3-2-7)SMB0002hFabとアデノシンの相互作用部位の同定
SMB0002hFabとアデノシンの複合体(SMB0002hFab-アデノシン複合体)の結晶構造の非対称単位は、2つのSMB0002hFabとアデノシンの複合体を含んでいた。両者の構造は良好に重ね合わせることができる。以下で説明する図8は、一方の分子のみを用いて作成された。
SMB0002hFab-アデノシン複合体の結晶構造より、アデノシンは、主に当該抗体Fab断片の重鎖と軽鎖の間に形成されるポケットに、アデニン環部分がポケットの奥に向かう形で結合することが明らかになった。
図8に示すように、アデノシンのアデニン環部分は、ヒト化SMB0002の軽鎖S91、N96及び重鎖A33、I50、Y100の各側鎖ならびに、重鎖T100a、G99の各主鎖により認識される。特に、アデニン環の1位Nと抗体軽鎖N96の側鎖の間および、アデニン環の6位のNH2と抗体軽鎖N96およびS91の側鎖、抗体重鎖T100aの主鎖のカルボニル酸素の間、並びにアデニン環の7位Nと抗体重鎖T100aの主鎖のアミドNHの間には水素結合が形成されている。また、アデニン環の8位CHと抗体重鎖G99の主鎖のカルボニル酸素の間には、弱い水素結合が形成されている。さらに、アデニン環と抗体重鎖A33、I50、Y100の各側鎖の間には、CH-πまたはπ-πなどといったアデニン環部分のパイ電子を利用した相互作用が形成されている。また、リボース部分の3’位のOは、抗体重鎖D54、S56の各側鎖と水素結合を形成している。さらにこれらの相互作用に加えて、アデノシンは、抗体重鎖T57、G52、W58、N100b、F100d、抗体軽鎖Y95cにより取り囲まれており、ファンデルワールス相互作用が形成されている。これらの相互作用によって、アデノシンはヒト化SMB0002に認識されていることが推察される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
SMB0002hFabとアデノシンの複合体(SMB0002hFab-アデノシン複合体)の結晶構造の非対称単位は、2つのSMB0002hFabとアデノシンの複合体を含んでいた。両者の構造は良好に重ね合わせることができる。以下で説明する図8は、一方の分子のみを用いて作成された。
SMB0002hFab-アデノシン複合体の結晶構造より、アデノシンは、主に当該抗体Fab断片の重鎖と軽鎖の間に形成されるポケットに、アデニン環部分がポケットの奥に向かう形で結合することが明らかになった。
図8に示すように、アデノシンのアデニン環部分は、ヒト化SMB0002の軽鎖S91、N96及び重鎖A33、I50、Y100の各側鎖ならびに、重鎖T100a、G99の各主鎖により認識される。特に、アデニン環の1位Nと抗体軽鎖N96の側鎖の間および、アデニン環の6位のNH2と抗体軽鎖N96およびS91の側鎖、抗体重鎖T100aの主鎖のカルボニル酸素の間、並びにアデニン環の7位Nと抗体重鎖T100aの主鎖のアミドNHの間には水素結合が形成されている。また、アデニン環の8位CHと抗体重鎖G99の主鎖のカルボニル酸素の間には、弱い水素結合が形成されている。さらに、アデニン環と抗体重鎖A33、I50、Y100の各側鎖の間には、CH-πまたはπ-πなどといったアデニン環部分のパイ電子を利用した相互作用が形成されている。また、リボース部分の3’位のOは、抗体重鎖D54、S56の各側鎖と水素結合を形成している。さらにこれらの相互作用に加えて、アデノシンは、抗体重鎖T57、G52、W58、N100b、F100d、抗体軽鎖Y95cにより取り囲まれており、ファンデルワールス相互作用が形成されている。これらの相互作用によって、アデノシンはヒト化SMB0002に認識されていることが推察される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
また、実施例(3―1)で示されるように、ヒト化SMB0002はアデノシンだけではなく、MTAにも結合することが明らかになっている。アデノシンとMTAの分子構造が類似している部分があるため、ヒト化SMB0002とアデノシンの結晶構造から、MTAがヒト化SMB0002に結合した場合の結合様式を考察することが可能である。アデノシンとヒト化SMB0002の結晶構造では、アデノシンのリボース5’位OHとアデニン環部分3位のNの間で分子内水素結合が形成されているが、MTAにおいては、そのチオメチル基は本相互作用を形成することはできない。アデニン環部分または、リボース部分と、ヒト化SMB0002の間で形成される相互作用を維持しつつ、MTAがヒト化SMB0002に結合するためには、MTAのチオメチル基は、アデニン環部分から離れる方向に位置する必要がある。その結果、チオメチル基は、アデノシンのリボース5’位OHよりも重鎖CDR2および軽鎖CDR3の近傍かつ、抗体の表面側に存在すると推測される。そのため、アデノシンよりもMTAの方が、抗原の結合に、直接的な相互作用を形成しやすいと考えられる。
(3-2-8)SMB0002hFabと、SMB0002hFab-アデノシン複合体の結晶構造の比較
SMB0002hFabの結晶構造の非対称単位は、2つのSMB0002hFab分子(分子1、分子2)を含んでいた。図9は、分子1(SMB0002hFab_1)と分子2(SMB0002hFab_2)から可変領域を抽出した重ね合わせを示す図9で示されるように、両者の構造は、重鎖CDR1の構造と重鎖と軽鎖の間のねじれ角が異なっていることが示された。
SMB0002hFab-アデノシン複合体の結晶構造と、SMB0002hFab_1の重ね合わせを示す図10から、両者の構造は良好に重ね合わせることができることが明らかになり、SMB0002hFab_1は、アデノシン結合時と同様な構造を取っているのではないかと推察される。一方で、SMB0002hFab_2の構造は、アデノシンが結合した複合体の構造とは異なっており、アデノシンが結合していない時の構造であることが推察される。
図11は、SMB0002hFab-アデノシン複合体の結晶構造とSMB0002hFab_2の結晶構造の重ね合わせを示す図である。ヒト化SMB0002の構造は、アデノシンの結合によって、SMB0002hFab-アデノシン複合体のような1つの構造に固定される可能性が示唆される。具体的には重鎖CDR1の構造及び、重鎖と軽鎖の間のねじれ角がアデノシンの結合により変化する。
SMB0002hFabの結晶構造の非対称単位は、2つのSMB0002hFab分子(分子1、分子2)を含んでいた。図9は、分子1(SMB0002hFab_1)と分子2(SMB0002hFab_2)から可変領域を抽出した重ね合わせを示す図9で示されるように、両者の構造は、重鎖CDR1の構造と重鎖と軽鎖の間のねじれ角が異なっていることが示された。
SMB0002hFab-アデノシン複合体の結晶構造と、SMB0002hFab_1の重ね合わせを示す図10から、両者の構造は良好に重ね合わせることができることが明らかになり、SMB0002hFab_1は、アデノシン結合時と同様な構造を取っているのではないかと推察される。一方で、SMB0002hFab_2の構造は、アデノシンが結合した複合体の構造とは異なっており、アデノシンが結合していない時の構造であることが推察される。
図11は、SMB0002hFab-アデノシン複合体の結晶構造とSMB0002hFab_2の結晶構造の重ね合わせを示す図である。ヒト化SMB0002の構造は、アデノシンの結合によって、SMB0002hFab-アデノシン複合体のような1つの構造に固定される可能性が示唆される。具体的には重鎖CDR1の構造及び、重鎖と軽鎖の間のねじれ角がアデノシンの結合により変化する。
アデノシンにより固定化されたヒト化SMB0002の構造と、それ以外の状態におけるヒト化SMB0002の構造が異なることは、アデノシン依存的に抗原に結合する抗体において、アデノシン依存的な抗原結合に重要であると考えられる。
つまり、アデノシン依存的に抗原に結合する抗体中では、抗原は、アデノシンにより固定化された構造を取っている抗体に結合することができる一方で、アデノシン非存在下では、抗原は抗体に結合しにくいということが想定される。従って、アデノシンの結合に伴い構造変化が生じる領域の利用はアデノシン依存的な抗原結合能の発揮に有効であると期待される。アデノシンの結合により固定される抗体の抗原結合界面を利用することが、アデノシン依存的な抗原結合能の発揮に重要であると考えられる。
以上のヒト化SMB0002の結晶構造解析の結果より、アデノシンの結合に大きく関与していないアミノ酸部位、もしくは抗原とのアデノシン依存的な結合に関与しやすいと想定されるアミノ酸部位(抗体蛋白の表面に露出しているアミノ酸残基が位置するアミノ酸部位、もしくはアデノシンが結合したときに構造変化する抗体領域に位置するアミノ酸部位)が選抜された。
つまり、アデノシン依存的に抗原に結合する抗体中では、抗原は、アデノシンにより固定化された構造を取っている抗体に結合することができる一方で、アデノシン非存在下では、抗原は抗体に結合しにくいということが想定される。従って、アデノシンの結合に伴い構造変化が生じる領域の利用はアデノシン依存的な抗原結合能の発揮に有効であると期待される。アデノシンの結合により固定される抗体の抗原結合界面を利用することが、アデノシン依存的な抗原結合能の発揮に重要であると考えられる。
以上のヒト化SMB0002の結晶構造解析の結果より、アデノシンの結合に大きく関与していないアミノ酸部位、もしくは抗原とのアデノシン依存的な結合に関与しやすいと想定されるアミノ酸部位(抗体蛋白の表面に露出しているアミノ酸残基が位置するアミノ酸部位、もしくはアデノシンが結合したときに構造変化する抗体領域に位置するアミノ酸部位)が選抜された。
(3-3)ヒト化SMB0002を鋳型としたライブラリの設計のための一置換改変体の網羅的評価
結晶構造解析の結果から、ヒト化SMB0002中の、ヒト化SMB0002のアデノシンもしくはMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのアデノシン依存的結合もしくは抗原とのMTA依存的結合に関与しやすいと想定されるアミノ部位が推定された。これらの部位のアミノ酸をヒト化SMB0002と異なるアミノ酸に改変しても、MTAもしくはアデノシンとの相互作用を保持する可能性が高いと考えられるため、ライブラリ設計のための多様化可能なアミノ酸部位となると考えられた。多様化可能なアミノ酸部位において、配置しても抗体とアデノシンもしくはMTAと相互作用に影響しないアミノ酸の種類を同定するため、ヒト化SMB0002のこれらの多様化可能なアミノ酸部位のアミノ酸を一つ改変した、ヒト化SMB0002一置換改変体が網羅的に作製された。
重鎖のうちアデノシンとの相互作用に関する考察に基づいた多様化可能なアミノ酸部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載される、ヒト化SMB0002の当該部位に出現するアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表3に示した。
結晶構造解析の結果から、ヒト化SMB0002中の、ヒト化SMB0002のアデノシンもしくはMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのアデノシン依存的結合もしくは抗原とのMTA依存的結合に関与しやすいと想定されるアミノ部位が推定された。これらの部位のアミノ酸をヒト化SMB0002と異なるアミノ酸に改変しても、MTAもしくはアデノシンとの相互作用を保持する可能性が高いと考えられるため、ライブラリ設計のための多様化可能なアミノ酸部位となると考えられた。多様化可能なアミノ酸部位において、配置しても抗体とアデノシンもしくはMTAと相互作用に影響しないアミノ酸の種類を同定するため、ヒト化SMB0002のこれらの多様化可能なアミノ酸部位のアミノ酸を一つ改変した、ヒト化SMB0002一置換改変体が網羅的に作製された。
重鎖のうちアデノシンとの相互作用に関する考察に基づいた多様化可能なアミノ酸部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載される、ヒト化SMB0002の当該部位に出現するアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表3に示した。

軽鎖のうちアデノシンとの相互作用に関する考察に基づいた多様化可能なアミノ酸部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載される、ヒト化SMB0002の当該部位に出現するアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表4に示した。


軽鎖のうちMTAとの相互作用に関する考察に基づいた多様化可能なアミノ酸部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載される、ヒト化SMB0002の当該部位に出現するアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表6に示した。

ヒト化SMB0002またはその一置換改変体のアデノシンまたはMTAに対する結合は、Biacore T200 (GE Healthcare)を用いた方法で測定され、解析された。Sensor chip上に解析対象の抗体がキャプチャーされ、アデノシンまたはMTAとの相互作用が観察された。
センサーチップ上にキャプチャーされた抗体に対して、アデノシン希釈液/MTA希釈液とブランクであるランニングバッファーが添加され、アデノシン/MTAと抗体の結合が観察された。その後ランニングバッファーが流され、アデノシン/MTAの抗体からの解離が観察された。各パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。
センサーチップ上にキャプチャーされた抗体に対して、アデノシン希釈液/MTA希釈液とブランクであるランニングバッファーが添加され、アデノシン/MTAと抗体の結合が観察された。その後ランニングバッファーが流され、アデノシン/MTAの抗体からの解離が観察された。各パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。
測定の結果、各改変体のアデノシンとMTAに対するAffinityがKD値として算出された。親抗体であるヒト化SMB0002のアデノシンに対するKD値と重鎖を改変した各改変体のアデノシンに対するKD値の比(親抗体であるヒト化SMB0002のアデノシンに対するKD値/重鎖を改変した各改変体のアデノシンに対するKD値)を表7、親抗体であるヒト化SMB0002のアデノシンに対するKD値と軽鎖を改変した各改変体のアデノシンに対するKD値の比(親抗体であるヒト化SMB0002のアデノシンに対するKD値/軽鎖を改変した各改変体のアデノシンに対するKD値)を表8に示した。親抗体であるヒト化SMB0002のMTAに対するKD値と重鎖を改変した各改変体のMTAに対するKD値の比(親抗体であるヒト化SMB0002のMTAに対するKD値/重鎖を改変した各改変体のMTAに対するKD値)を表9、親抗体であるヒト化SMB0002のMTAに対するKD値と軽鎖を改変した各改変体のMTAに対するKD値の比(親抗体であるヒト化SMB0002のMTAに対するKD値/軽鎖を改変した各改変体のMTAに対するKD値)を表10に示した。




(3-4)ヒト化SMB0002を鋳型としたライブラリの設計
ライブラリの設計にあたり、一置換改変体の網羅的評価で得られた情報をもとに、以下の条件のうち少なくとも一つを満たすアミノ酸が多様化に使用可能なアミノ酸として選定された。
条件1)MTAあるいはアデノシンに対する結合に大きく関与しないアミノ酸;
条件2)アデノシン結合時と非結合時でヒト化SMB0002の構造変化率が大きいと予想されるアミノ酸。
ライブラリの設計にあたり、一置換改変体の網羅的評価で得られた情報をもとに、以下の条件のうち少なくとも一つを満たすアミノ酸が多様化に使用可能なアミノ酸として選定された。
条件1)MTAあるいはアデノシンに対する結合に大きく関与しないアミノ酸;
条件2)アデノシン結合時と非結合時でヒト化SMB0002の構造変化率が大きいと予想されるアミノ酸。
親抗体(ヒト化SMB0002)のMTAに対するKD値と当該部位が当該アミノ酸に改変された重鎖改変体のMTAに対するKD値の比(親抗体のMTAに対するKD値/当該部位が当該アミノ酸に改変された重鎖改変体のMTAに対するKD値)が0.4以上の改変アミノ酸、軽鎖改変体のMTAに対するKD値の比(親抗体のMTAに対するKD値/当該部位が当該アミノ酸に改変された軽鎖改変体のMTAに対するKD値)が0.1以上の改変アミノ酸が、条件1)中のMTAに対する結合に大きく関与しないアミノ酸と判定され、多様化に使用可能なアミノ酸として選定された。
親抗体(ヒト化SMB0002)のアデノシンに対するKD値と当該部位が当該アミノ酸に改変された重鎖改変体のアデノシンに対するKD値の比(親抗体のアデノシンに対するKD値/当該部位が当該アミノ酸に改変された重鎖改変体のアデノシンに対するKD値)が0.4以上の改変アミノ酸、軽鎖改変体のアデノシンに対するKD値の比(親抗体のアデノシンに対するKD値/当該部位が当該アミノ酸に改変された軽鎖改変体のアデノシンに対するKD値)が0.1以上の改変アミノ酸が、条件1)中のアデノシンに対する結合に大きく関与しないアミノ酸と判定され、多様化に使用可能なアミノ酸として選定された。
重鎖のKabatナンバリング32位のアミノ酸がDもしくは Eである場合、または33位のアミノ酸がVもしくは IもしくはTである場合、当該アミノ酸が、条件2を満たすアミノ酸と判定され、多様化に使用可能なアミノ酸として選定された。
選定された多様化可能なアミノ酸のうち、少なくともいずれか一つ以上のアミノ酸が出現するライブラリを設計することによって、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するためのライブラリ(以下、S02ライブラリ)が構築された。S02ライブラリの重鎖におけるアミノ酸多様化されたアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表11に示した。S02ライブラリの軽鎖におけるアミノ酸多様化されたアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表12に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位はアミノ酸多様化されたアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位における未改変体ヒト化SMB0002のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸は当該部位におけるアミノ酸レパートリーを、それぞれ示す。重鎖に含まれる各多様化可能なアミノ酸部位において、選出されたアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。また、軽鎖に含まれる各多様化可能なアミノ酸部位において、選出されたアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。
選定された多様化可能なアミノ酸のうち、少なくともいずれか一つ以上のアミノ酸が出現するライブラリを設計することによって、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するためのライブラリ(以下、S02ライブラリ)が構築された。S02ライブラリの重鎖におけるアミノ酸多様化されたアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表11に示した。S02ライブラリの軽鎖におけるアミノ酸多様化されたアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表12に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位はアミノ酸多様化されたアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位における未改変体ヒト化SMB0002のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸は当該部位におけるアミノ酸レパートリーを、それぞれ示す。重鎖に含まれる各多様化可能なアミノ酸部位において、選出されたアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。また、軽鎖に含まれる各多様化可能なアミノ酸部位において、選出されたアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。
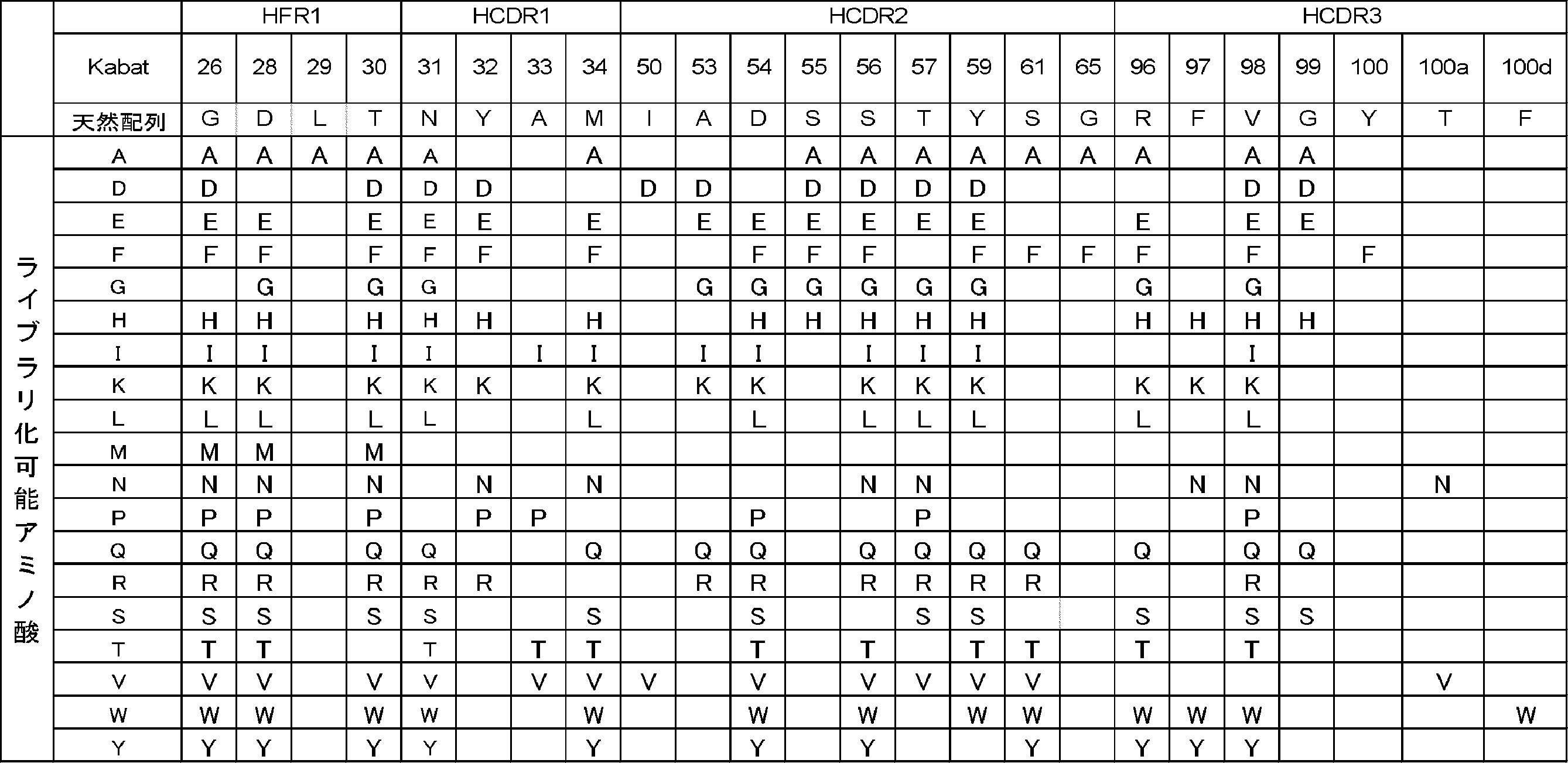

[実施例4]
MTA依存的に抗原に結合する抗体を取得するためのライブラリの構築及び当該ライブラリMTA依存的に抗原に結合する抗体の取得
MTA依存的に抗原に結合する抗体を取得するためのライブラリの構築及び当該ライブラリMTA依存的に抗原に結合する抗体の取得
(4-1)MTA依存的に抗原に結合する抗体を取得するためのライブラリの構築
(4-1-1)重鎖もしくは軽鎖可変領域ファージディスプレイライブラリの構築
設計されたS02ライブラリ中の重鎖可変領域部分はTRIM技術(Tiller T et al. MAbs 2013)を利用して遺伝子合成され、ヒト化SMB0002の軽鎖可変領域配列(配列番号:32)と組み合わせヒトIgG由来CH1配列およびヒトIgG由来軽鎖定常領域配列を有する適切なファージミドベクターに導入された。このファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するための重鎖可変領域ファージディスプレイライブラリが構築された。
設計されたS02ライブラリ中の軽鎖可変領域部分はTRIM技術(Tiller T et al. MAbs 2013)を利用して遺伝子合成され、ヒト化SMB0002の重鎖可変領域配列(配列番号:64)と組み合わせヒトIgG由来CH1配列およびヒトIgG由来軽鎖定常領域配列を有する適切なファージミドベクターに導入された。このファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するための軽鎖可変領域ファージディスプレイライブラリが構築された。
(4-1-1)重鎖もしくは軽鎖可変領域ファージディスプレイライブラリの構築
設計されたS02ライブラリ中の重鎖可変領域部分はTRIM技術(Tiller T et al. MAbs 2013)を利用して遺伝子合成され、ヒト化SMB0002の軽鎖可変領域配列(配列番号:32)と組み合わせヒトIgG由来CH1配列およびヒトIgG由来軽鎖定常領域配列を有する適切なファージミドベクターに導入された。このファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するための重鎖可変領域ファージディスプレイライブラリが構築された。
設計されたS02ライブラリ中の軽鎖可変領域部分はTRIM技術(Tiller T et al. MAbs 2013)を利用して遺伝子合成され、ヒト化SMB0002の重鎖可変領域配列(配列番号:64)と組み合わせヒトIgG由来CH1配列およびヒトIgG由来軽鎖定常領域配列を有する適切なファージミドベクターに導入された。このファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTAもしくはアデノシン依存的に抗原に結合する抗体を取得するための軽鎖可変領域ファージディスプレイライブラリが構築された。
(4-1-2)重鎖および軽鎖可変領域ファージディスプレイライブラリを用いたMTAに結合する抗体群の濃縮
実施例(4-1-1)で構築された重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、MTAに結合する抗体群を濃縮するために、ビオチン化標識MTA(Biotin-2’-MTA)を用いたパニングが実施された。
具体的には、構築された重鎖可変領域ファージディスプレイライブラリまたは軽鎖可変領域ファージディスプレイライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
実施例(4-1-1)で構築された重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、MTAに結合する抗体群を濃縮するために、ビオチン化標識MTA(Biotin-2’-MTA)を用いたパニングが実施された。
具体的には、構築された重鎖可変領域ファージディスプレイライブラリまたは軽鎖可変領域ファージディスプレイライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
磁気ビーズに固定化されたBiotin-2’-MTAを用いたパニングが2つの条件で実施された。抗体多価提示ファージライブラリ液に終濃度4%となるようにBSAが添加された。磁気ビーズとして、NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific)が用いられた。BSAでブロッキングされた磁気ビーズと終濃度10 μMのBiotin-2’-MTAを室温で30分間反応させることによって、磁気ビーズに対してBiotin-2’-MTAが固相化された。Biotin-2’-MTAが固相化された磁気ビーズがTBSTで3回洗浄され、一つ目の条件では洗浄された磁気ビーズに調製された抗体多価提示ファージライブラリ液0.4mLが加えられ、二つ目の条件では洗浄された磁気ビーズに調製された抗体多価提示ファージライブラリ液0.4mLが1 mMのアデノシンが共存する状態で加えられ、室温で60分間反応させた。ビーズは400μLのTBSTにて3回、TBSにて2回洗浄された。その後終濃度1mg/mLのトリプシンを含むTBS 0.5 mLが加えられたビーズが室温で15分懸濁され、ファージが溶出された。磁気スタンドを用いてビーズが分離され、ファージ溶液が回収された。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった20mLの大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。大腸菌は225 mm x 225 mmのプレートへ播種された。この一連の作業を追加で3回繰り返し、重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、MTAに結合する抗体群が濃縮された。
(4-1-3)ファージELISAによるパニング後取得クローン群のビオチン化MTAに対する結合活性の評価
実施例(4-1-2)で濃縮されたMTAに結合する抗体群中の抗体を提示するファージを感染させた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収され、ELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、Biotin-2’-MTAを含むTBSが10 μL添加され、1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80μLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示される抗体をBiotin-2’-MTAに結合させた。各ウェルをTBSTで洗浄した後、各ウェルにTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。各ウェルをTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、Biotin-2’-MTAに結合する抗体を提示するファージが複数確認された。ファージELISAの結果を図12(重鎖可変領域ファージディスプレイライブラリから濃縮された抗体群)、図13(軽鎖可変領域ファージディスプレイライブラリから濃縮された抗体群)、表13に示した。ELISAの結果から、実施例(4-1-2)のパニングにより濃縮された抗体群中にMTA結合抗体が多数含まれていることが示された。
実施例(4-1-2)で濃縮されたMTAに結合する抗体群中の抗体を提示するファージを感染させた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収され、ELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、Biotin-2’-MTAを含むTBSが10 μL添加され、1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80μLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示される抗体をBiotin-2’-MTAに結合させた。各ウェルをTBSTで洗浄した後、各ウェルにTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。各ウェルをTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、Biotin-2’-MTAに結合する抗体を提示するファージが複数確認された。ファージELISAの結果を図12(重鎖可変領域ファージディスプレイライブラリから濃縮された抗体群)、図13(軽鎖可変領域ファージディスプレイライブラリから濃縮された抗体群)、表13に示した。ELISAの結果から、実施例(4-1-2)のパニングにより濃縮された抗体群中にMTA結合抗体が多数含まれていることが示された。

(4-1-4)ファージELISAで評価されたクローンの抗体配列解析
ファージELISAで評価されたクローンについて、プライマー(配列番号:33および34)を用いて増幅された抗体遺伝子の塩基配列が解析された。解析の結果、ファージELISAで評価された192クローンの中での抗体配列重複サンプル2クローン以下であり、実施例(4-1-2)の濃縮を経てもライブラリが高い配列多様性を保持していることが示された。
ファージELISAで評価されたクローンについて、プライマー(配列番号:33および34)を用いて増幅された抗体遺伝子の塩基配列が解析された。解析の結果、ファージELISAで評価された192クローンの中での抗体配列重複サンプル2クローン以下であり、実施例(4-1-2)の濃縮を経てもライブラリが高い配列多様性を保持していることが示された。
(4-1-5)MTAに対するパニングを利用したMTA依存的に抗体に結合する抗体を取得するためのライブラリの構築
実施例(4-1-2)で濃縮されたMTA結合抗体群について、当該抗体群に含まれる改変重鎖と改変軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもMTAに対する結合を維持する抗体が多数含まれることが予想され、MTA依存的に抗原に結合する抗体を取得するためのライブラリが構築できると考えられた。
実施例(4-1-2)で濃縮されたMTA結合抗体群を提示するファージが感染した大腸菌から、当業者公知の方法で抗体遺伝子が抽出された。重鎖可変領域ファージライブラリから濃縮された抗体群に対して、重鎖可変領域を増幅できるプライマー(配列番号:34, 35)を用いて重鎖可変領域遺伝子が増幅された。軽鎖可変領域ファージライブラリから濃縮された抗体群に対して、ヒト化SMB0002の重鎖可変領域配列を制限酵素処理により除去し、軽鎖可変領域ライブラリ遺伝子とヒトIgG由来軽鎖定常領域配列とヒトIgG由来CH1配列を含むファージミド断片が調製された。前記ファージミドベクター断片に重鎖可変領域ライブラリから増幅された重鎖可変領域遺伝子を挿入することで、重鎖/軽鎖可変領域ライブラリ遺伝子が導入されたファージミドベクターが構築された。このベクターをエレクトロポレーションにより大腸菌へと導入することで、ヒト抗体可変領域と定常領域からなるFabドメインを提示し、MTA依存的に抗原に結合する抗体を取得するためのライブラリ(以下、MTA濃縮S02ライブラリ)が構築された。
実施例(4-1-2)で濃縮されたMTA結合抗体群について、当該抗体群に含まれる改変重鎖と改変軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもMTAに対する結合を維持する抗体が多数含まれることが予想され、MTA依存的に抗原に結合する抗体を取得するためのライブラリが構築できると考えられた。
実施例(4-1-2)で濃縮されたMTA結合抗体群を提示するファージが感染した大腸菌から、当業者公知の方法で抗体遺伝子が抽出された。重鎖可変領域ファージライブラリから濃縮された抗体群に対して、重鎖可変領域を増幅できるプライマー(配列番号:34, 35)を用いて重鎖可変領域遺伝子が増幅された。軽鎖可変領域ファージライブラリから濃縮された抗体群に対して、ヒト化SMB0002の重鎖可変領域配列を制限酵素処理により除去し、軽鎖可変領域ライブラリ遺伝子とヒトIgG由来軽鎖定常領域配列とヒトIgG由来CH1配列を含むファージミド断片が調製された。前記ファージミドベクター断片に重鎖可変領域ライブラリから増幅された重鎖可変領域遺伝子を挿入することで、重鎖/軽鎖可変領域ライブラリ遺伝子が導入されたファージミドベクターが構築された。このベクターをエレクトロポレーションにより大腸菌へと導入することで、ヒト抗体可変領域と定常領域からなるFabドメインを提示し、MTA依存的に抗原に結合する抗体を取得するためのライブラリ(以下、MTA濃縮S02ライブラリ)が構築された。
(4-2)MTA依存的に抗原に結合する抗体の取得
実施例(4-1-5)で構築されたMTA濃縮S02ライブラリから、MTA存在下でヒトIL-6レセプター(hIL-6R)、ヒトIL-6(hIL-6)、ヒトIgA(hIgA)のそれぞれに対して結合活性を示す抗体がスクリーニングされた。
具体的には、構築されたMTA濃縮S02ライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
実施例(4-1-5)で構築されたMTA濃縮S02ライブラリから、MTA存在下でヒトIL-6レセプター(hIL-6R)、ヒトIL-6(hIL-6)、ヒトIgA(hIgA)のそれぞれに対して結合活性を示す抗体がスクリーニングされた。
具体的には、構築されたMTA濃縮S02ライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
磁気ビーズに固定化されたhIL-6R、hIL-6、hIgAを用いたパニングがそれぞれ実施された。抗体多価提示ファージライブラリ液に終濃度4%となるようにBSAが添加された。BSAが添加された抗体多価提示ファージライブラリ液0.8 mLに0.1 nmolのビオチン標識抗原と終濃度100 μMのMTAを加え、室温で60分間反応させ、BSAでブロッキングされた磁気ビーズを加え室温で15分反応させた。磁気ビーズとして、NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific)が用いられた。磁気ビーズは0.8 mLの終濃度100 μMのMTAを含むTBSTにて2回もしくは3回、終濃度100 μMのMTAを含むTBSにて1回もしくは2回洗浄された。その後0.25 mLのTBSが加えられたビーズが室温で懸濁され、磁気スタンドを用いてビーズが分離され、ファージ溶液が溶出された。分離されたビーズに再度0.25 mLのTBSを加えてビーズを室温で懸濁され、磁気スタンドを用いてビーズが分離され、ファージ溶液が溶出された。2回の溶出されたファージ溶液が混合された。回収されたファージ溶液に最終濃度1mg/mLのトリプシンが加えられた。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった20mLの大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。大腸菌は225 mm x 225 mmのプレートへ播種された。この一連の作業を追加で3回繰り返した。
(4-3)ファージELISAによるパニング後取得クローン群のMTA存在下での抗原結合活性の評価
実施例(4-2)のパニングで得られた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収された。NucleoFast96(MACHERY-NAGEL)を用いて、回収されたファージ培養上清が限外濾過された。具体的には培養上清各0.2 mLがNucleoFast96の各ウェルに添加され、6000 g、40分間遠心分離を行いフロースルーが除去された。その後H2O 0.2 mLが各ウェルに添加され、6000 g、20分間遠心分離を行いフロースルーが除去された。その後TBS 0.2 mLが各ウェルに添加され、室温で5分間静置された後、抗体多価提示ファージ溶液が回収された。
実施例(4-2)のパニングで得られた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収された。NucleoFast96(MACHERY-NAGEL)を用いて、回収されたファージ培養上清が限外濾過された。具体的には培養上清各0.2 mLがNucleoFast96の各ウェルに添加され、6000 g、40分間遠心分離を行いフロースルーが除去された。その後H2O 0.2 mLが各ウェルに添加され、6000 g、20分間遠心分離を行いフロースルーが除去された。その後TBS 0.2 mLが各ウェルに添加され、室温で5分間静置された後、抗体多価提示ファージ溶液が回収された。
MTA非存在下のファージELISAは以下のように行われた。TBSに希釈されたファージが以下の手順でELISAに供された。384 Well Microplates Streptavidin-coated(greiner)にビオチン標識抗原を含むTBSが10 μl添加され1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80μLの0.02%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに回収された抗体多価提示ファージが加えられ一時間静置することで、ファージに提示する抗体をMTA非存在下においてビオチン標識抗原に結合させた。各ウェルをTBSTで洗浄した後、各ウェルにTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。当該ウェルをTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。
MTA存在下のファージELISAは以下のように行われた。終濃度100 μMのMTAを含むTBSに希釈されたファージが以下の手順でELISAに供された。384 Well Microplates Streptavidin-coated(greiner)にビオチン標識抗原を含むTBSが10 μl添加され1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80μLの0.02%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに回収された抗体多価提示ファージが加えられ一時間静置することで、ファージに提示する抗体をMTA存在下においてビオチン標識抗原に結合させた。各ウェルを終濃度100 μMのMTAを含むTBSTで洗浄した後、各ウェルに終濃度100 μMのMTAを含むTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。当該ウェルを終濃度100 μMのMTAを含むTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。
解析の結果、ビオチン標識ヒトIL-6レセプター(hIL-6R)、ヒトIL-6(hIL-6)、ヒトIgA(hIgA)のそれぞれを用いたパニングから、MTA依存的に抗原に結合する抗体が複数確認された。ファージELISAの結果を表14に示した。どの抗原に対してもMTA存在下で抗原に結合でき、MTA非存在下で抗原に結合できないクローンが複数取得された。それらのクローンの中に、アデノシンとMTA両方に対して依存的に抗原に結合する抗体もあるが、アデノシン存在下では抗原に対する結合を示さなかったクローン(MTA特異的に依存的に抗原に結合する抗体)も取得できた。したがってMTA濃縮S02ライブラリから、MTA特異的に依存して抗原に結合し、MTAと類似した構造を示す低分子化合物に対して依存的に抗原に対する結合を示さない、優れたMTA依存特異性を有する抗体を取得できることが示された。

(4-4)MTA依存的に抗原に結合する抗体の配列解析
ファージELISAで評価された抗体群のいくつかについて、プライマー(配列番号:33および34)を用いて増幅された抗体遺伝子の塩基配列が解析された。
ファージELISAで評価された抗体群のいくつかについて、プライマー(配列番号:33および34)を用いて増幅された抗体遺伝子の塩基配列が解析された。
(4-5)MTA依存的に抗原に結合する抗体の発現と精製
抗体の重鎖可変領域と軽鎖可変領域配列はそれぞれ、重鎖抗体定常領域配列(配列番号:38)もしくは軽鎖kappa定常領域配列(配列番号:39)を有する動物発現用プラスミドへとそれぞれ挿入された。表15で示す抗体を参考実施例1の方法を用いて発現させ、精製した。
抗体の重鎖可変領域と軽鎖可変領域配列はそれぞれ、重鎖抗体定常領域配列(配列番号:38)もしくは軽鎖kappa定常領域配列(配列番号:39)を有する動物発現用プラスミドへとそれぞれ挿入された。表15で示す抗体を参考実施例1の方法を用いて発現させ、精製した。

(4-6)表面プラズモン共鳴を用いたMTA存在下での抗体抗原結合活性の評価
Biacore T200(GE Healthcare)を用いて、実施例(4-5)で精製製された抗体と標的抗原との抗原抗体反応におけるMTAとアデノシンの影響が評価された。ランニングバッファーとして、20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4が用いられ、25℃で測定された。センサーチップCM4上にアミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、各種抗原をアナライトとして相互作用させることで、抗原抗体間の結合量の変化が観察された。抗原の希釈には、ランニングバッファーおよびランニングバッファーにMTAまたはアデノシンのいずれかがそれぞれ終濃度100 μMの濃度で添加されたバッファーが使用された。また抗原は数段階の濃度系列で調製され、各抗原濃度に対するセンサーグラムからシングルサイクルカイネティクス解析により各クローンの解離定数KDが算出された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。100 μM MTA存在下での各クローンの解離定数KDを表16に示した。また100 μMアデノシン存在下では抗原への結合は観察されなかった。
Biacore T200(GE Healthcare)を用いて、実施例(4-5)で精製製された抗体と標的抗原との抗原抗体反応におけるMTAとアデノシンの影響が評価された。ランニングバッファーとして、20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4が用いられ、25℃で測定された。センサーチップCM4上にアミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、各種抗原をアナライトとして相互作用させることで、抗原抗体間の結合量の変化が観察された。抗原の希釈には、ランニングバッファーおよびランニングバッファーにMTAまたはアデノシンのいずれかがそれぞれ終濃度100 μMの濃度で添加されたバッファーが使用された。また抗原は数段階の濃度系列で調製され、各抗原濃度に対するセンサーグラムからシングルサイクルカイネティクス解析により各クローンの解離定数KDが算出された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。100 μM MTA存在下での各クローンの解離定数KDを表16に示した。また100 μMアデノシン存在下では抗原への結合は観察されなかった。

100 μM MTAもしくは100 μM アデノシン存在下と非存在下での各クローンの抗原に対する結合量を図14に示した。図14に示されたように、各クローンは100 μM MTA存在下では抗原に結合するが、MTA非存在下では抗原に結合しなかった。このことから、精製された各抗体がMTA依存的に抗原に結合する性質を有することが確認された。
(4-7)MTA依存的に結合する抗体のMTA依存的なT細胞活性化能の評価
MTA依存的に結合する抗体がMTA依存的に機能することを確認するために、MTA依存的に結合する抗体を用いてMTAの存在下および非存在下におけるT細胞の活性化能を評価した。評価には、実施例(4-5)でクローニングされたMTA依存性抗hIL-6R抗体(6RS2T007H-F760mnN17/6RS2T007L-KT0)、および抗CD3抗体(重鎖可変領域:配列番号:40、重鎖定常領域:配列番号:42、軽鎖可変領域:配列番号:41、軽鎖定常領域:配列番号:39)からなる抗hIL-6R/hCD3二重特異性抗体を用いた。
上記MTA依存性抗hIL-6R抗体および抗CD3抗体はそれぞれ参考実施例1の方法に従い調製した。取得されたMTA依存性抗IL-6R抗体および抗CD3抗体を混合し25 mM 2-MEA(2-メルカプトエチルアミン)存在下で37℃、90分間反応させることで、両抗体の重鎖および軽鎖を有し、MTA依存的にhIL-6Rに結合する抗原結合ドメインとCD3に結合する抗原結合ドメインを有する二重特異性抗体を作製した。作製した二重特異性抗体は透析によりPBSへ溶媒置換した後、分光光度計を用いて280nmでの吸光度を測定した。得られた吸光度の測定値から参考実施例1に記載のPACE法により算出された吸光係数を用いて精製抗体の濃度が算出された。抗体は10% FBSを含むRPMI1640培地(アッセイバッファー)またはMTAもしくはアデノシンを含むアッセイバッファーで希釈してT細胞活性化能の評価に用いた。
MTA依存的に結合する抗体がMTA依存的に機能することを確認するために、MTA依存的に結合する抗体を用いてMTAの存在下および非存在下におけるT細胞の活性化能を評価した。評価には、実施例(4-5)でクローニングされたMTA依存性抗hIL-6R抗体(6RS2T007H-F760mnN17/6RS2T007L-KT0)、および抗CD3抗体(重鎖可変領域:配列番号:40、重鎖定常領域:配列番号:42、軽鎖可変領域:配列番号:41、軽鎖定常領域:配列番号:39)からなる抗hIL-6R/hCD3二重特異性抗体を用いた。
上記MTA依存性抗hIL-6R抗体および抗CD3抗体はそれぞれ参考実施例1の方法に従い調製した。取得されたMTA依存性抗IL-6R抗体および抗CD3抗体を混合し25 mM 2-MEA(2-メルカプトエチルアミン)存在下で37℃、90分間反応させることで、両抗体の重鎖および軽鎖を有し、MTA依存的にhIL-6Rに結合する抗原結合ドメインとCD3に結合する抗原結合ドメインを有する二重特異性抗体を作製した。作製した二重特異性抗体は透析によりPBSへ溶媒置換した後、分光光度計を用いて280nmでの吸光度を測定した。得られた吸光度の測定値から参考実施例1に記載のPACE法により算出された吸光係数を用いて精製抗体の濃度が算出された。抗体は10% FBSを含むRPMI1640培地(アッセイバッファー)またはMTAもしくはアデノシンを含むアッセイバッファーで希釈してT細胞活性化能の評価に用いた。
T細胞の活性化能の評価にはヒトT細胞株としてNFAT-RE-luc2-Jurkat細胞を使用し、hIL-6R発現細胞株としてCT26にhIL-6Rを強制発現させたCT26/hIL-6Rを用いた。NFAT-RE-luc2-Jurkat細胞およびCT26/hIL-6R細胞は細胞密度がそれぞれ3×106 cells/mL、1×106 cells/mLとなるようにアッセイバッファーで懸濁して使用した。384ウェルプレートの各ウェルに調製したヒトT細胞株およびhIL-6R発現細胞株の懸濁液をそれぞれ10 μlずつ添加し、さらに調製した抗体溶液10 μlを添加した。5% CO2インキュベーターにおいて37℃で6時間静置した。静置後当該サンプルのルシフェラーゼ活性を測定することでヒトT細胞株の活性化能を評価した。ルシフェラーゼ活性の測定には市販のルシフェラーゼ活性測定試薬(Bio-Glo luciferase assay system、Promega)を用いて実施した。ルシフェラーゼ発光基質30 μlを静置後のプレートのウェルに添加し、さらに10分間静置後、ウェルの発光量をEnvision(PerkinElmer)により測定した。各ウェルの測定値を抗体未添加のコントロールウェルの測定値で割った値をLuminescence foldとしてT細胞活性化能を評価する指標とした。
MTAもしくはアデノシンの存在下、非存在下における作製した二重特異性抗体のT細胞活性化能を評価した結果を図28に示した。図28に示されたように、作製した二重特異性抗体はMTA非存在下と比較して300 μM MTA存在下ではT細胞を強く活性化することが確認された。さらに、300 μMアデノシン存在下と比較し300 μM MTA存在下ではT細胞を強く活性化することが確認されたことから、作成したMTA依存的にhIL-6Rに結合する抗原結合ドメインとCD3に結合する抗原結合ドメインを有する二重特異性抗体がMTA依存的、かつMTA特異的にT細胞を活性化することが確認された。
(4-8)MTA依存的に結合する抗体のMTA依存的な結合の検証
実施例(4-5)でクローニングされたMTA依存的に結合する抗hIL-6R抗体(6RS2T007H-F760mnN17/6RS2T007L-KT0)について、MTA濃度依存的な抗原結合量の変化をBiacore T200(GE Healthcare)を用いて検証した。測定はランニングバッファーとして、MTAが終濃度0.1、 1、 10、 100 μMの濃度で添加された20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4を用いて、25℃で測定した。センサーチップとしてSeries S Sensor Chip CM4を使用し、アミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、hIL-6Rをアナライトとして相互作用させることで、抗原抗体間の結合量の変化を測定した。抗原の希釈には、ランニングバッファーを用いた。各MTA濃度における抗体の抗原結合量変化を図29に示した。この結果からMTA依存的に結合する抗体がMTA濃度に応じて抗原への結合量が段階的に変化することが確認され、MTAの濃度依存的に結合していることが確認された。
実施例(4-5)でクローニングされたMTA依存的に結合する抗hIL-6R抗体(6RS2T007H-F760mnN17/6RS2T007L-KT0)について、MTA濃度依存的な抗原結合量の変化をBiacore T200(GE Healthcare)を用いて検証した。測定はランニングバッファーとして、MTAが終濃度0.1、 1、 10、 100 μMの濃度で添加された20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4を用いて、25℃で測定した。センサーチップとしてSeries S Sensor Chip CM4を使用し、アミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、hIL-6Rをアナライトとして相互作用させることで、抗原抗体間の結合量の変化を測定した。抗原の希釈には、ランニングバッファーを用いた。各MTA濃度における抗体の抗原結合量変化を図29に示した。この結果からMTA依存的に結合する抗体がMTA濃度に応じて抗原への結合量が段階的に変化することが確認され、MTAの濃度依存的に結合していることが確認された。
また、Octet RED384システム(Forte Bio)を用いて、MTA依存的に結合した抗体がMTA濃度の低下に応じて可逆的に抗原から解離することを検証した。Protein Aバイオセンサー(Forte Bio)に抗体を固相化した後、結合相においてhIL-6Rをアナライトとして相互作用させることで、抗原抗体間の結合量の変化を測定した。結合相としてMTAが終濃度10, 100 μMの濃度で添加された20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4で希釈された3000nMのアナライトを含むバッファーを使用した。解離相においては、結合相と同濃度のアナライトを含み、MTAを結合相と同濃度添加した溶液とMTAを添加していないバッファーを使用した。結合相から解離相までの抗原に対する結合量の経時的変化を示すセンサーグラムを図30に示した。図中のw/MTAは解離相において結合相と同濃度のMTAを含むことを示し、w/o MTAは解離相においてMTAを含まないことを示している。この結果からMTAを含む溶液中では解離相においてアナライトとの結合が維持されているのに対し、MTAを含まない溶液中ではアナライトとの解離速度が増加していることが確認され、MTA依存的に結合する抗体と抗原との結合はMTA存在下と非存在下で可逆的に結合活性を変化させることができることが確認された。
[実施例5]ウサギB細胞クローニングによる抗MTA抗体の取得及び評価
(5-1)ウサギB細胞クローニングによる抗MTA抗体の取得
(5-1-1)抗MTA抗体取得用の免疫原のデザイン
ウサギに免疫する免疫原として6’-MTA- Keyhole Limpet Hemocyanin(6’-MTA-KLH)が用いられた。Mariculture Keyhole Limpet Hemocyanin(KLH)は、ヘルパーT細胞上に発現するT細胞受容体が認識できる抗原性の高い蛋白質であり、抗体産生を活性化することが知られているため、MTAと連結させることでMTAに対する抗体の産生を亢進させることが期待される。
また、KLHの代わりにビオチンをMTAに連結させた6’-MTA-biotinが作製された。
(5-1)ウサギB細胞クローニングによる抗MTA抗体の取得
(5-1-1)抗MTA抗体取得用の免疫原のデザイン
ウサギに免疫する免疫原として6’-MTA- Keyhole Limpet Hemocyanin(6’-MTA-KLH)が用いられた。Mariculture Keyhole Limpet Hemocyanin(KLH)は、ヘルパーT細胞上に発現するT細胞受容体が認識できる抗原性の高い蛋白質であり、抗体産生を活性化することが知られているため、MTAと連結させることでMTAに対する抗体の産生を亢進させることが期待される。
また、KLHの代わりにビオチンをMTAに連結させた6’-MTA-biotinが作製された。
(5-1-2)抗MTA抗体取得用の免疫原の合成
(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-(クロロメチル)オキソラン-3,4-ジオール

(2R,3R,4S,5R)-2-(6-アミノ-9H-プリン-9-イル)-5-(ヒドロキシメチル)オキソラン-3,4-ジオール (16.08g,60.17mmol,1等量) のアセトニトリル溶液(210 mL)にピリジン(9.78 mL,2.0等量)を窒素雰囲気下加えた。この溶液にチオニルクロリド(22.2mL,5.0等量)を0℃で撹拌しながらゆっくり滴下し,5℃で6時間次いで25℃で終夜撹拌した。溶液をメタノール:水=5:1(v/v,360mL)で希釈し,更に28%アンモニア水(30mL)で中和した。析出した固体をろ集,乾燥し(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-(クロロメチル)オキソラン-3,4-ジオールを白色固体として得た(12.0g,収率70%)。
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.35(s,1H),8.17(s,1H),7.30(s,2H),5.94(d,J=5.6Hz,1H),5.60 (d,J=6.0Hz,1H),5.46(d,J=5.2Hz,1H),4.76(q,J =5.6Hz,1H),4.23(q,J=4.8Hz,1H),4.14-4.06(m, 1H),3.96(dd,J=11.6,5.1Hz,1H),3.85(dd,J=11.6, 6.4Hz,1H).
(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-(クロロメチル)オキソラン-3,4-ジオール
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.35(s,1H),8.17(s,1H),7.30(s,2H),5.94(d,J=5.6Hz,1H),5.60 (d,J=6.0Hz,1H),5.46(d,J=5.2Hz,1H),4.76(q,J =5.6Hz,1H),4.23(q,J=4.8Hz,1H),4.14-4.06(m, 1H),3.96(dd,J=11.6,5.1Hz,1H),3.85(dd,J=11.6, 6.4Hz,1H).
(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-[(メチルスルファニル)メチル]オキソラン-3,4-ジオール

(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-(クロロメチル)オキソラン-3,4-ジオール(10.0g,35.0mmol,1等量)N,N-ジメチルホルムアミド溶液(300mL)にナトリウムチオメトキシド(14.73g,210mmol,3.0等量)を加え,窒素雰囲気下にて50℃で2時間撹拌した。反応溶液を減圧下濃縮し,得られた残渣を水(500mL)で希釈した。溶液のpHを4N塩酸で7に調整し析出した固体をろ集し(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-[(メチルスルファニル)メチル]オキソラン-3,4-ジオールを白色固体として得た(7.0g,収率67%)。
LC-MS(ES,m/z):298[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.36(s,1H),8.16(s,1H),7.28(s,2H),5.90(d,J=5.7Hz,1H),5.52 (d,J=5.7Hz,1H),5.37-5.31(m,1H),4.76(q,J=5.0 Hz,1H),4.16(q,J=3.6Hz,1H),4.04(td,J=6.3,3.6 Hz,1H),2.94-2.71(m,2H),2.06(s,3H).
LC-MS(ES,m/z):298[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.36(s,1H),8.16(s,1H),7.28(s,2H),5.90(d,J=5.7Hz,1H),5.52 (d,J=5.7Hz,1H),5.37-5.31(m,1H),4.76(q,J=5.0 Hz,1H),4.16(q,J=3.6Hz,1H),4.04(td,J=6.3,3.6 Hz,1H),2.94-2.71(m,2H),2.06(s,3H).
9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-アミン

(2R,3R,4S,5S)-2-(6-アミノ-9H-プリン-9-イル)-5-[(メチルスルファニル)メチル]オキソラン-3,4-ジオール(7.0g,23.54mmol, 1.0等量)のアセトン溶液(500mL)に2,2-ジメトキシプロパン(19.55g,187.71mmol,3.0等量)およびp-トルエンスルホン酸(7.91g,45.93 mmol,2.0等量)を0℃にて加え,25℃にて4時間撹拌した。1Nアンモニア水で溶液のpHを7に調節したのち,溶液を減圧下濃縮した。残渣を酢酸エチル(200mL)に溶解し蒸留水(100mL)で2回洗浄し,有機層を減圧下濃縮した。残渣をシリカゲルカラム(ジクロロメタン:メタノール=10:1)で精製し,9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-アミンを白色固体として得た(6.0g,収率76%)。
LC-MS(ES,m/z):338[M+H]+
1H-NMR(300MHz,Chloroform-d,ppm) δ 8.32(s, 1H),7.91(s,1H),6.06(d,J=2.2Hz,1H),5.89(s,2H),5.49(dd,J=6.4,2.2Hz,1H),5.04(dd,J=6.4,3.2 Hz,1H),4.45-4.33(m,1H),2.75(qt,J=18.4,9.8 Hz,2H),2.08(s,3H),1.59(s,3H),1.38(s,3H).
LC-MS(ES,m/z):338[M+H]+
1H-NMR(300MHz,Chloroform-d,ppm) δ 8.32(s, 1H),7.91(s,1H),6.06(d,J=2.2Hz,1H),5.89(s,2H),5.49(dd,J=6.4,2.2Hz,1H),5.04(dd,J=6.4,3.2 Hz,1H),4.45-4.33(m,1H),2.75(qt,J=18.4,9.8 Hz,2H),2.08(s,3H),1.59(s,3H),1.38(s,3H).
6-([9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸エチル

9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-アミン (2.0g,5.93mmol,1等量)のN,N-ジメチルホルムアミド溶液(17mL)に水素化ナトリウム(60%,248mg,6.2mmol,1.05等量)を25℃にて加え窒素雰囲気下で2時間撹拌した。6-ブロモヘキサン酸エチル(1.38g,6.19mmol,1.05等量)のN,N-ジメチルホルムアミド溶液(4mL)をゆっくり滴下し2時間60℃にて撹拌した。反応混合物を100mLの氷水に注ぎ,酢酸エチル(100mL)にて抽出した。有機層を減圧下濃縮し得られた残渣をシリカゲルカラム(酢酸エチル:石油エーテル=1:1)にて精製し6-([9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸エチルを白色固体として得た(1.4g,収率49%)。
LC-MS(ES,m/z):480[M+H]+
LC-MS(ES,m/z):480[M+H]+
6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸

6-([9-[(3aR,4R,6S,6aS)-2,2-ジメチル-6-[(メチルスルファニル)メチル]-テトラヒドロ-2H-フロ[3,4-d][1,3]ジオキソール-4-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸エチル(450mg,0.94mmol,1等量)のテトラヒドロフラン溶液(7mL)に1N塩酸(3.5mL)を加え6時間50℃にて撹拌した。反応液を減圧下濃縮した残渣を,蒸留水(8mL)にて希釈し,析出した固体をろ集した。得られた粗生成物をプレパラティブHPLC(島津HPLC-10,XBridge Shield RP18 OBDカラム,5μm,19mmx150mm,移動相 水(0.05%アンモニア水含有)およびアセトニトリル,グラジエント5.0-20.0%,検出UV254nm)にて精製し,6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸を白色固体として得た(159.5mg,41%)。
LC-MS(ES,m/z):412[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 11.81(s,1H),8.35(s,1H),8.22(s,1H),7.82(s,1H),5.90(d,J=5.7 Hz,1H),5.49(s,1H),5.31(s,1H),4.75(t,J=5.3Hz,1H),4.16(t,J=4.3Hz,1H),4.04(ddd,J=6.9,5.8, 3.7Hz,1H),3.47(s,2H),2.94-2.74(m,2H),2.20(t, J=7.3Hz,2H),2.07(s,3H),1.56(dp,J=25.6,7.4Hz,4H),1.33(qd,J=8.4,6.0Hz,2H).
LC-MS(ES,m/z):412[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 11.81(s,1H),8.35(s,1H),8.22(s,1H),7.82(s,1H),5.90(d,J=5.7 Hz,1H),5.49(s,1H),5.31(s,1H),4.75(t,J=5.3Hz,1H),4.16(t,J=4.3Hz,1H),4.04(ddd,J=6.9,5.8, 3.7Hz,1H),3.47(s,2H),2.94-2.74(m,2H),2.20(t, J=7.3Hz,2H),2.07(s,3H),1.56(dp,J=25.6,7.4Hz,4H),1.33(qd,J=8.4,6.0Hz,2H).
6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)-N-(プロパ-2-イン-1-イル)ヘキサンアミド

6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸(580mg,1.41mmol,1等量)のジクロロメタン溶液(20mL)に1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(542mg,2.83mmol, 2.0等量),ジイソプロピルエチルアミン(910mg,7.04mmol,5.0等量)およびプロパ-2-イン-1-アミン(388mg,7.04mmol,5.0等量)を加え25℃にて3日間撹拌した。溶液を100mLの蒸留水にて希釈し,析出した固体をろ集し乾燥し6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)-N-(プロパ-2-イン-1-イル)ヘキサンアミドを白色固体として取得し、これを更に精製することなく次工程に使用した(420mg,収率66%)。
N-[[1-(2-[2-[2-(2-[5-[(3aS,4S,6aR)-2-オキソ-ヘキサヒドロ-1H-チエノ[3,4-d]イミダゾリジン-4-イル]ペンタンアミド]エトキシ)エトキシ]エトキシ]エチル)-1H-1,2,3-トリアゾール-4-イル]メチル]-6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサンアミド

6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)-N-(プロパ-2-イン-1-イル)ヘキサンアミド(100mg,0.22mmol,1等量)のテトラヒドロフラン(3mL)およびtert-ブタノール(3mL)の溶液にN-(2-(2-(2-(2-アジドエトキシ)エトキシ)エトキシ)エチル)-5-((3aS,4S,6aR)-2-オキソ-ヘキサヒドロ-1H-チエノ[3,4-d]イミダゾール-4-イル)ペンタンアミド(99.1mg,0.22mmol,1.0等量), CuSO4 5H2O(55.5mg,0.22mmol,1.0等量),アスコルビン酸ナトリウム(1M水溶液、7滴)を加え20℃にて16時間撹拌した。溶液を20mLの酢酸エチルにて希釈し、15mLの蒸留水にて分配した。有機層を減圧下濃縮し得られた粗生成物をプレパラティブHPLC(島津HPLC-10,XBridge Shield RP18 OBDカラム,5um,19mmx150mm,移動相 水(10mmol/L NH4HCO3+0.1%アンモニア水含有)およびアセトニトリル,グラジエント16.0-27.0%,検出UV220nm)にて精製し、N-[[1-(2-[2-[2-(2-[5-[(3aS,4S,6aR)-2-オキソ-ヘキサヒドロ-1H-チエノ[3,4-d]イミダゾリジン-4-イル]ペンタンアミド]エトキシ)エトキシ]エトキシ]エチル)-1H-1,2,3-トリアゾール-4-イル]メチル]-6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサンアミドを白色固体として得た。(57.4mg,収率29%)。
LC-MS(ES,m/z):893[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.35(s,1H), 8.25(dd,J=13.4,7.7Hz,2H),7.89-7.78(m,3H),6.41(s,1H),6.35(s,1H),5.90(d,J=5.7Hz, 1H),5.48(d,J=6.0Hz,1H),5.31(d,J=5.0Hz, 1H),4.75(q,J=5.6Hz,1H),4.49(t,J=5.3Hz, 2H),4.29(dd,J=14.3,6.7Hz,3H),4.20-4.08(m, 2H),4.04(td,J=6.3,3.7Hz,1H),3.80(t,J=5.3Hz,2H),3.55-3.27(m,14H),3.23-3.04(m,3H), 2.94-2.74(m,3H),2.58(d,J=12.4Hz,1H),2.14-2.02(m,7H),1.53(ddtd,J=35.3,28.2,13.8,7.6Hz,8H),1.38-1.22(m,4H)
LC-MS(ES,m/z):893[M+H]+
1H-NMR(400MHz,DMSO-d6,ppm) δ 8.35(s,1H), 8.25(dd,J=13.4,7.7Hz,2H),7.89-7.78(m,3H),6.41(s,1H),6.35(s,1H),5.90(d,J=5.7Hz, 1H),5.48(d,J=6.0Hz,1H),5.31(d,J=5.0Hz, 1H),4.75(q,J=5.6Hz,1H),4.49(t,J=5.3Hz, 2H),4.29(dd,J=14.3,6.7Hz,3H),4.20-4.08(m, 2H),4.04(td,J=6.3,3.7Hz,1H),3.80(t,J=5.3Hz,2H),3.55-3.27(m,14H),3.23-3.04(m,3H), 2.94-2.74(m,3H),2.58(d,J=12.4Hz,1H),2.14-2.02(m,7H),1.53(ddtd,J=35.3,28.2,13.8,7.6Hz,8H),1.38-1.22(m,4H)
6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸のKeyhole Limpet Hemocyanin(KLH)コンジュゲート(6’-MTA-KLH)
6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸(4.94mg,0.012mmol,1等量))のDMSO (200μl)溶液に1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(2.30mg,0.012mmol, 1.0等量),ヒドロキシ-2,5-ジオキソピロリジン-3-スルホン酸 ナトリウム塩(2.61mg,0.012mmol,1.0等量)を加え20℃にて2時間撹拌した。この溶液を50μl取り、KLH(10mg,ThermoFisher Scientific社,77600)のリン酸バッファー溶液(pH8.4,0.95μL)を4℃にて加え緩やかに撹拌し4℃にて24時間静置した。得られたコンジュゲートを含む溶液を、そのまま動物免疫に用いた。
6-([9-[(2R,3R,4S,5S)-3,4-ジヒドロキシ-5-[(メチルスルファニル)メチル]オキソラン-2-イル]-9H-プリン-6-イル]アミノ)ヘキサン酸(4.94mg,0.012mmol,1等量))のDMSO (200μl)溶液に1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(2.30mg,0.012mmol, 1.0等量),ヒドロキシ-2,5-ジオキソピロリジン-3-スルホン酸 ナトリウム塩(2.61mg,0.012mmol,1.0等量)を加え20℃にて2時間撹拌した。この溶液を50μl取り、KLH(10mg,ThermoFisher Scientific社,77600)のリン酸バッファー溶液(pH8.4,0.95μL)を4℃にて加え緩やかに撹拌し4℃にて24時間静置した。得られたコンジュゲートを含む溶液を、そのまま動物免疫に用いた。
(5-1-3)ウサギB細胞クローニングによる抗MTA抗体のスクリーニング
実施例(5-1-2)で合成された6’-MTA-KLHを用いて、ウサギが当業者公知の方法で免疫された。免疫されたウサギの血液または脾臓から採取された細胞懸濁液から、autoMACS Pro SeparatorとFACSAria(BD)を用いた未標識アデノシン共存下でもMTA結合活性を有する細胞の候補が選抜された。次に選抜された細胞の培養上清中に分泌された抗体を用いて細胞がスクリーニングされた。具体的には、ELISA法により、6’-MTA-biotinに対する結合活性の有無、及び未標識MTA共存下における6’-MTA-biotinへの結合活性の有無が評価された。6’-MTA-biotinに結合し、未標識MTA共存下においてアデノシンの共存有無にかかわらず6’-MTA-biotinへの結合が抑制される抗体を分泌することを指標に選抜された細胞からPCR法を用いて重鎖可変領域遺伝子および軽鎖可変領域遺伝子が取得された。取得された可変領域遺伝子から、ヒトIgG1重鎖定常領域、およびヒト軽鎖定常領域と組み合わせて抗体を発現させた。
実施例(5-1-2)で合成された6’-MTA-KLHを用いて、ウサギが当業者公知の方法で免疫された。免疫されたウサギの血液または脾臓から採取された細胞懸濁液から、autoMACS Pro SeparatorとFACSAria(BD)を用いた未標識アデノシン共存下でもMTA結合活性を有する細胞の候補が選抜された。次に選抜された細胞の培養上清中に分泌された抗体を用いて細胞がスクリーニングされた。具体的には、ELISA法により、6’-MTA-biotinに対する結合活性の有無、及び未標識MTA共存下における6’-MTA-biotinへの結合活性の有無が評価された。6’-MTA-biotinに結合し、未標識MTA共存下においてアデノシンの共存有無にかかわらず6’-MTA-biotinへの結合が抑制される抗体を分泌することを指標に選抜された細胞からPCR法を用いて重鎖可変領域遺伝子および軽鎖可変領域遺伝子が取得された。取得された可変領域遺伝子から、ヒトIgG1重鎖定常領域、およびヒト軽鎖定常領域と組み合わせて抗体を発現させた。
(5-1-4)ウサギB細胞クローニングから得られたクローンの配列解析
実施例(5-1-3)で取得された抗体について、プライマー(配列番号:44および45)を用いて増幅された抗体遺伝子の塩基配列が解析された。配列を以下の表17に示す。
実施例(5-1-3)で取得された抗体について、プライマー(配列番号:44および45)を用いて増幅された抗体遺伝子の塩基配列が解析された。配列を以下の表17に示す。

(5-2)ウサギB細胞クローニングから得られたクローンのMTAとその類縁体に対する結合活性の評価
ウサギB細胞クローニングから得られたクローンがMTA特異的に結合するか否かを検証するため、Biacore T200 (GE Healthcare)を用いて、MTAおよびその類縁体に対する結合性が解析された。
Protein Aがあらかじめ固定化されたSensor chip Protein A (GE healthcare)、またはストレプトアビジンがあらかじめ固定化されたSensor chip SA(GE Healthcare)にビオチン化された抗ヒトIgG CH1分子であるCaptureSelect Biotin Anti-IgG-Fc (Hu) (Thermo fisher scientific) を結合させた固定化表面に対し、目的の抗体をキャプチャーさせ、MTA (sigma-aldrich)またはMTA類縁体を相互作用させた。MTA類縁体として、MTAとの構造類似性及びMTAP欠損細胞株の担癌マウスの腫瘍組織内濃度から、アデノシン (Wako)(腫瘍組織内測定濃度:182 nM)およびS-(5'-Adenosyl)-L-homocysteine, (SAH) (sigma-aldrich)(腫瘍組織内測定濃度:30 nM)が選抜された。ランニングバッファーには20mM ACES-NaOH, 150mM NaCl, 0.05% Tween20, 0.05%DMSOが用いられた。MTAまたはアデノシンまたはSAHは流速30μL/minで、120秒間相互作用させ、その後ランニングバッファーにて解離させた。相互作用は25℃で測定され、MTAおよびその類縁体の希釈にはランニングバッファーと同じバッファーが使用された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、各抗体のMTAおよびその類縁体に対する結合活性が表18のように決定され、選択された抗体のいずれもMTAに対して高い選択性を有することが確認された。
ウサギB細胞クローニングから得られたクローンがMTA特異的に結合するか否かを検証するため、Biacore T200 (GE Healthcare)を用いて、MTAおよびその類縁体に対する結合性が解析された。
Protein Aがあらかじめ固定化されたSensor chip Protein A (GE healthcare)、またはストレプトアビジンがあらかじめ固定化されたSensor chip SA(GE Healthcare)にビオチン化された抗ヒトIgG CH1分子であるCaptureSelect Biotin Anti-IgG-Fc (Hu) (Thermo fisher scientific) を結合させた固定化表面に対し、目的の抗体をキャプチャーさせ、MTA (sigma-aldrich)またはMTA類縁体を相互作用させた。MTA類縁体として、MTAとの構造類似性及びMTAP欠損細胞株の担癌マウスの腫瘍組織内濃度から、アデノシン (Wako)(腫瘍組織内測定濃度:182 nM)およびS-(5'-Adenosyl)-L-homocysteine, (SAH) (sigma-aldrich)(腫瘍組織内測定濃度:30 nM)が選抜された。ランニングバッファーには20mM ACES-NaOH, 150mM NaCl, 0.05% Tween20, 0.05%DMSOが用いられた。MTAまたはアデノシンまたはSAHは流速30μL/minで、120秒間相互作用させ、その後ランニングバッファーにて解離させた。相互作用は25℃で測定され、MTAおよびその類縁体の希釈にはランニングバッファーと同じバッファーが使用された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、各抗体のMTAおよびその類縁体に対する結合活性が表18のように決定され、選択された抗体のいずれもMTAに対して高い選択性を有することが確認された。

(5-3)抗MTA抗体MTA0303のX線結晶構造解析
MTA0303の Fab断片(MTA0303Fab)とメチルチオアデノシン(MTA)の複合体(MTA0303Fab-MTA)の結晶構造が解析された。
MTA0303の Fab断片(MTA0303Fab)とメチルチオアデノシン(MTA)の複合体(MTA0303Fab-MTA)の結晶構造が解析された。
(5-3-1)結晶化用MTA0303全長抗体の調製
結晶化用MTA0303全長抗体の調製及び精製は当業者公知の方法により行われた。
結晶化用MTA0303全長抗体の調製及び精製は当業者公知の方法により行われた。
(5-3-2)MTA0303Fabの結晶構造解析のためのFab断片の調製
MTA0303Fabは、MTA0303全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)により制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex75 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
MTA0303Fabは、MTA0303全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)により制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex75 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
(5-3-3)MTA0303FabとMTAの複合体(MTA0303Fab-MTA複合体)の結晶の作製
結晶化は、当業者公知の方法で精製され、約16 mg/mLまで濃縮されたMAT0303Fabに終濃度2mMになるようにMTAを混合した溶液を用いて、シーディング法と、株式会社 創晶において行われたフェムト秒レーザーの照射(J. Appl. Phys. 42: L798-L800 (2003)) を組み合わせたシッティングドロップ蒸気拡散法により5℃で行われた。リザーバー溶液は、0.1 M HEPES pH 7.5、70 % v/v (+/-)-2-Methyl-2,4-pentanediolから成るものであった。
結晶化は、当業者公知の方法で精製され、約16 mg/mLまで濃縮されたMAT0303Fabに終濃度2mMになるようにMTAを混合した溶液を用いて、シーディング法と、株式会社 創晶において行われたフェムト秒レーザーの照射(J. Appl. Phys. 42: L798-L800 (2003)) を組み合わせたシッティングドロップ蒸気拡散法により5℃で行われた。リザーバー溶液は、0.1 M HEPES pH 7.5、70 % v/v (+/-)-2-Methyl-2,4-pentanediolから成るものであった。
(5-3-4)MTA0303Fab-MTA複合体の結晶からのX線回折データの収集および構造決定
得られた結晶は、液体窒素で凍結され、ポール・シェラー研究所の放射光施設Swiss Light Source X10SAにてX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計720枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.95Åまでの回折強度データが取得された。結晶学的統計値が表19に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法を実施し、初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表19に示される。
得られた結晶は、液体窒素で凍結され、ポール・シェラー研究所の放射光施設Swiss Light Source X10SAにてX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計720枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.95Åまでの回折強度データが取得された。結晶学的統計値が表19に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法を実施し、初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表19に示される。
(5-3-5)MTA0303FabとMTAの相互作用部位の同定
図20に示すMTA0303Fab-MTA複合体の結晶構造より、MTAは、MTA0303Fabの重鎖と軽鎖の間に形成されるポケットにチオメチル基部分がポケットの奥に向かう形で結合することが明らかになった。また、図15に示されるように、MTAは、重鎖CDR2に覆いかぶさられるように、奥深くに埋もれるように結合していた。
図20に示すMTA0303Fab-MTA複合体の結晶構造より、MTAは、MTA0303Fabの重鎖と軽鎖の間に形成されるポケットにチオメチル基部分がポケットの奥に向かう形で結合することが明らかになった。また、図15に示されるように、MTAは、重鎖CDR2に覆いかぶさられるように、奥深くに埋もれるように結合していた。
図16に示すように、MTAのアデニン環部分は、当該抗体の軽鎖R32、S50、L91、重鎖W34、Y52eの各側鎖により認識される。特に、アデニン環の7位Nと重鎖Y52eの側鎖の間および、アデニン環の2位CHと軽鎖S50の側鎖の間には水素結合が形成されている。さらに、アデニン環と重鎖W34および、軽鎖R32、L91の各側鎖の間には、CH-πやπ-πなどといったアデニン環部分のパイ電子を利用した相互作用が形成されている。また、リボース部分の3’位のOと軽鎖Y36の側鎖の間および、リボース部分の2’位のOと軽鎖S34および、重鎖E101の各側鎖の間には水素結合が形成されている。チオメチル基部分は、重鎖W34、W47、F52の各側鎖との間に、CH-π相互作用や、硫黄原子とパイ電子の間に形成される相互作用を利用して認識されている。さらにこれらの相互作用に加えて、MTAは、重鎖C35a、軽鎖L46、Y49、A89、G90、P96により取り囲まれており、ファンデルワールス相互作用が形成されている。これらの相互作用によって、MTAは当該抗体により強く認識されていることが推察される。
また、当該抗体の重鎖CDR2は図17に示されるように、特徴的なへリックス構造を形成している。この2次構造中に含まれる重鎖Y52eの側鎖は、MTAのアデニン環部分と水素結合を形成することが結晶構造から明らかになっている。この相互作用の形成が、へリックス構造を形作る引き金となっているのではないかと考えられる。従って、当該抗体は、MTAの結合に伴い構造が変化する可能性が考えられる。
MTA非存在下における当該抗体の構造と、MTA存在下における当該抗体の構造が異なることは、MTA依存的な抗原への結合に重要であると考えられる。つまり、蛋白質抗原は、MTAの結合に伴い構造が変化した抗体のみに結合することができる一方で、MTA非存在下では、当該抗体の構造は、MTA存在下における構造とは異なるため、MTAが存在しない状態では、蛋白質抗原は抗体に結合することができないということが想定される。従って、MTAの結合に伴い構造変化を生じる領域の利用はMTA依存的な抗原結合能の発揮に有効であると期待される。当該抗体においては、MTAの結合に伴う重鎖CDR2の構造変化が考えられるため、重鎖CDR2の利用によるMTA依存的に抗原に結合する抗体の取得が期待される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
MTA非存在下における当該抗体の構造と、MTA存在下における当該抗体の構造が異なることは、MTA依存的な抗原への結合に重要であると考えられる。つまり、蛋白質抗原は、MTAの結合に伴い構造が変化した抗体のみに結合することができる一方で、MTA非存在下では、当該抗体の構造は、MTA存在下における構造とは異なるため、MTAが存在しない状態では、蛋白質抗原は抗体に結合することができないということが想定される。従って、MTAの結合に伴い構造変化を生じる領域の利用はMTA依存的な抗原結合能の発揮に有効であると期待される。当該抗体においては、MTAの結合に伴う重鎖CDR2の構造変化が考えられるため、重鎖CDR2の利用によるMTA依存的に抗原に結合する抗体の取得が期待される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
(5-4)抗MTA抗体MTA0330のX線結晶構造解析
MTA0330のFab断片(MTA0330Fab)とMTAの複合体(MTA0330Fab-MTA)の結晶構造が解析された。
MTA0330のFab断片(MTA0330Fab)とMTAの複合体(MTA0330Fab-MTA)の結晶構造が解析された。
(5-4-1)結晶化用MTA0330全長抗体の調製
結晶化用MTA0330全長抗体の調製及び精製は当業者公知の方法により行われた。
結晶化用MTA0330全長抗体の調製及び精製は当業者公知の方法により行われた。
(5-4-2)MTA0330Fabの結晶構造解析のためのFab断片の調製
MTA0330Fabは、MTA0330全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)により制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex75 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
MTA0330Fabは、MTA0330全長抗体をEndoproteinase Lys-C(Roche、カタログ番号11047825001)により制限消化し、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)、カチオン交換カラム(HiTrap SP HP、GE Healthcare)、およびゲル濾過カラム(Superdex75 16/60、GE Healthcare)へのローディングを用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、-80℃で保存された。
(5-4-3)MTA0330FabとMTAの複合体(MTA0330Fab-MTA複合体)の結晶の作製
結晶化は、当業者公知の方法で精製され、約13.3 mg/mLまで濃縮されたMAT0330 Fabに終濃度2mMになるようにMTAを混合した溶液を用いて、シッティングドロップ蒸気拡散法により20℃で行われた。リザーバー溶液は、30.0 %w/v P550MME_P20K、 0.1 M Morpheus buffer 1 pH 6.5、60 mM Morpheus Divalents (Morpheus、Molecular Dimensions)から成るものであった。
結晶化は、当業者公知の方法で精製され、約13.3 mg/mLまで濃縮されたMAT0330 Fabに終濃度2mMになるようにMTAを混合した溶液を用いて、シッティングドロップ蒸気拡散法により20℃で行われた。リザーバー溶液は、30.0 %w/v P550MME_P20K、 0.1 M Morpheus buffer 1 pH 6.5、60 mM Morpheus Divalents (Morpheus、Molecular Dimensions)から成るものであった。
(5-4-4)MTA0330Fab-MTA複合体の結晶からのX線回折データの収集および構造決定
得られた結晶は、液体窒素で凍結され、ポール・シェラー研究所の放射光施設Swiss Light Source X10SAにてX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計1440枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.46Åまでの回折強度データが取得された。結晶学的統計値が表19に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法により初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表19に示される。
得られた結晶は、液体窒素で凍結され、ポール・シェラー研究所の放射光施設Swiss Light Source X10SAにてX線回折データが測定された。測定中、結晶は常に-178℃の窒素流下に置おくことで凍結状態が維持された。X線回折画像は、ビームラインに接続されたPilatus 6M検出器(DECTRIS)を用いて、結晶を1回に0.25°回転させながら、合計1440枚が収集された。得られた回折画像は、autoPROC(Acta Cryst. D67: 293-302 (2011))を用いて処理され、分解能1.46Åまでの回折強度データが取得された。結晶学的統計値が表19に示される。
得られたX線回折強度データを用いて、既知のFabの結晶構造をサーチモデルとして、Phaser(J. Appl. Cryst. (2007) 40, 658-674)を用いた分子置換法により初期構造が決定された。その後、Coot(Acta Cryst. D66: 486-501 (2010))とRefmac5(Acta Cryst. D67: 355-467 (2011))によるモデル構築と精密化が繰り返され、最終的な精密化座標が得られた。結晶学的統計値は、表19に示される。

(5-4-5)MTA0330 FabとMTAの相互作用部位の同定
図21に示すMTA0330Fab-MTA複合体の結晶構造より、MTAは、MTA0330Fabの重鎖と軽鎖の間に形成されるポケットにチオメチル基部分がポケットの奥に向かう形で、また、図18に示すようにMTAのアデニン環部分は当該抗体の表面から露出するように結合することが明らかになった。
図19に示すように、MTAのアデニン環部分は、当該抗体の軽鎖F95bおよび、重鎖F98の各側鎖の間で、π-π相互作用が形成されることで認識される。また、リボース部分は、2’位のOおよび3’位のOと重鎖E95の側鎖と水素結合を形成することで認識される。チオメチル基部分は、軽鎖F96の側鎖との間でCH-π相互作用が形成されている。さらにこれらの相互作用に加えて、MTAは、重鎖W34、W47、C50、Y58、G99、G100aおよび、軽鎖Y28、T91、Y95cにより取り囲まれており、ファンデルワールス相互作用が形成されている。これらの相互作用によって、MTAは当該抗体により強く認識されていることが推察される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
図21に示すMTA0330Fab-MTA複合体の結晶構造より、MTAは、MTA0330Fabの重鎖と軽鎖の間に形成されるポケットにチオメチル基部分がポケットの奥に向かう形で、また、図18に示すようにMTAのアデニン環部分は当該抗体の表面から露出するように結合することが明らかになった。
図19に示すように、MTAのアデニン環部分は、当該抗体の軽鎖F95bおよび、重鎖F98の各側鎖の間で、π-π相互作用が形成されることで認識される。また、リボース部分は、2’位のOおよび3’位のOと重鎖E95の側鎖と水素結合を形成することで認識される。チオメチル基部分は、軽鎖F96の側鎖との間でCH-π相互作用が形成されている。さらにこれらの相互作用に加えて、MTAは、重鎖W34、W47、C50、Y58、G99、G100aおよび、軽鎖Y28、T91、Y95cにより取り囲まれており、ファンデルワールス相互作用が形成されている。これらの相互作用によって、MTAは当該抗体により強く認識されていることが推察される。なお、Fabのアミノ酸の残基番号付けは、Kabat番号付けスキームに基づく。
(5-5)抗MTA抗体MTA0303およびMTA0330のNMR解析
(5-5-1)NMR用MTA0303およびMTA0330全長抗体の調製
NMR用MTA0303およびMTA0330全長抗体の調製及び精製は当業者公知の方法により行われた。但し、アミノ酸の安定同位体標識を実施するため、培地中に200mg/L [d-13CH3, 15N, 2H]Isoleucine, 300mg/L [d2-13CH3, 2H, 15N]Leucine, 190mg/L [g1-13CH3, 2H, 15N]Valine, 360mg/L [b-13CH3, 2H, 15N]Alanine, 60mg/L β-chloro-L-Alanineを添加した。
(5-5-1)NMR用MTA0303およびMTA0330全長抗体の調製
NMR用MTA0303およびMTA0330全長抗体の調製及び精製は当業者公知の方法により行われた。但し、アミノ酸の安定同位体標識を実施するため、培地中に200mg/L [d-13CH3, 15N, 2H]Isoleucine, 300mg/L [d2-13CH3, 2H, 15N]Leucine, 190mg/L [g1-13CH3, 2H, 15N]Valine, 360mg/L [b-13CH3, 2H, 15N]Alanine, 60mg/L β-chloro-L-Alanineを添加した。
(5-5-2)MTA0303FabおよびMTA0330FabのNMR解析のためのFab断片の調製
MTA0303のFab断片(MTA0303Fab)およびMTA0330のFab断片(MTA0330Fab)を、Lys-C(Roche、カタログ番号11 047 825 001)による制限消化、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)を用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、5mM d-citrate, pH6.5, 20mM NaCl, 5% D2Oに置換し、0.15mM~0.3mMとなるまで濃縮した。
MTA0303のFab断片(MTA0303Fab)およびMTA0330のFab断片(MTA0330Fab)を、Lys-C(Roche、カタログ番号11 047 825 001)による制限消化、続いて、Fc断片を除去するためのプロテインAカラム(MabSlect SuRe、GE Healthcare)を用いるという、従来の方法により調製された。Fab断片を含む画分はプールされ、5mM d-citrate, pH6.5, 20mM NaCl, 5% D2Oに置換し、0.15mM~0.3mMとなるまで濃縮した。
(5-5-3)MTA0303FabおよびMTA0330FabのNMRスペクトル測定
実施例(5-5-2)で調製した試料を用いて、AvanceIII 600 (Bruker社)でNMRスペクトルを測定した。測定温度を305Kとした。1H-15N TROSYスペクトルおよび1H-13C SOFAST-HMQCスペクトルを取得するため、それぞれtrosyetf3gpiasi、sfhmqcf2gpphパルスプログラム(Bruker社標準パルスプログラム)を用いた。スペクトルのフーリエ変換には、TopSpin (Bruker社)を使用した。
実施例(5-5-2)で調製した試料を用いて、AvanceIII 600 (Bruker社)でNMRスペクトルを測定した。測定温度を305Kとした。1H-15N TROSYスペクトルおよび1H-13C SOFAST-HMQCスペクトルを取得するため、それぞれtrosyetf3gpiasi、sfhmqcf2gpphパルスプログラム(Bruker社標準パルスプログラム)を用いた。スペクトルのフーリエ変換には、TopSpin (Bruker社)を使用した。
(5-5-4)MTA0303FabとMTAの複合体(MTA0303Fab-MTA複合体)およびMTA0330FabとMTAの複合体(MTA0330Fab-MTA複合体)のNMRスペクトル測定
実施例(5-5-2)で調製した資料に、終濃度が0.4 mMになるようにMTAを添加し、Avance III 600でNMRスペクトルを測定した。測定温度を305Kとした。1H-15N TROSYスペクトルおよび1H-13C SOFAST-HMQCスペクトルを取得するため、それぞれtrosyetf3gpiasi、sfhmqcf2gpphパルスプログラム(Bruker社標準パルスプログラム)を用いた。スペクトルのフーリエ変換には、TopSpin (Bruker社)を使用した。
実施例(5-5-2)で調製した資料に、終濃度が0.4 mMになるようにMTAを添加し、Avance III 600でNMRスペクトルを測定した。測定温度を305Kとした。1H-15N TROSYスペクトルおよび1H-13C SOFAST-HMQCスペクトルを取得するため、それぞれtrosyetf3gpiasi、sfhmqcf2gpphパルスプログラム(Bruker社標準パルスプログラム)を用いた。スペクトルのフーリエ変換には、TopSpin (Bruker社)を使用した。
(5-5-5)MTA0303FabとMTA0303Fab-MTA複合体の1H-15N TROSYスペクトル比較
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0303FabのNMRスペクトルとMTA0303Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図22に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは17個あった。
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0303FabのNMRスペクトルとMTA0303Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図22に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは17個あった。
(5-5-6)MTA0303FabとMTA0303Fab-MTA複合体の1H-13C SOFAST-HMQCスペクトル比較
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0303FabのNMRスペクトルとMTA0303Fab-MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図23に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは20個あった。
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0303FabのNMRスペクトルとMTA0303Fab-MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図23に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは20個あった。
(5-5-7)MTA0330FabとMTA0330Fab-MTA複合体の1H-15N TROSYスペクトル比較
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0330FabのNMRスペクトルとMTA0330Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図24に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは7個あった。
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0330FabのNMRスペクトルとMTA0330Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図24に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは7個あった。
(5-5-8)MTA0330FabとMTA0330Fab-MTA複合体の1H-13C SOFAST-HMQCスペクトル比較
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0330FabのNMRスペクトルとMTA0330Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図25に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは7個あった。
NMRスペクトル表示ソフトウエアであるSparky(UCSF)を用いて、MTA0330FabのNMRスペクトルとMTA0330Fab―MTA複合体のNMRスペクトルを重ね合わせた後、スペクトルの比較を行った。結果を図25に示す。MTA結合状態と非結合状態で化学シフトがピーク1個分以上、変化しているシグナルは7個あった。
[実施例6]ウサギB細胞クローニングから得られた抗MTA抗体を鋳型としたライブラリのデザイン
実施例5で取得した抗MTA抗体のMTA0303、MTA0330を鋳型として、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインした。
実施例5で取得した抗MTA抗体のMTA0303、MTA0330を鋳型として、MTA依存的に抗原に結合する抗体を取得するためのライブラリをデザインした。
(6-1)ウサギB細胞クローニングから得られた抗体MTA0303とMTA0330のヒト化
MTA0303とMTA0330のヒト化が当業者公知の方法で実施された(欧州特許公開EP239400、国際公開WO1996/00257、WO1993/012227、WO1992/003918、WO1994/002602、WO1994/025585、WO1996/034096、WO1996/033735、WO1992/001047、WO1992/020791、WO1993/006213、WO1993/011236、WO1993/019172、WO1995/001438、WO1995/015388、Cancer Res.,(1993)53, 851-856, BBRC.,(2013)436(3):543-50等)。ヒト化抗体の配列を以下の表20に示す。
MTA0303とMTA0330のヒト化が当業者公知の方法で実施された(欧州特許公開EP239400、国際公開WO1996/00257、WO1993/012227、WO1992/003918、WO1994/002602、WO1994/025585、WO1996/034096、WO1996/033735、WO1992/001047、WO1992/020791、WO1993/006213、WO1993/011236、WO1993/019172、WO1995/001438、WO1995/015388、Cancer Res.,(1993)53, 851-856, BBRC.,(2013)436(3):543-50等)。ヒト化抗体の配列を以下の表20に示す。

(6-2)表面プラズモン共鳴を用いたヒト化MTA0303とヒト化MTA0330のMTA結合活性評価
ヒト化MTA0303、ヒト化MTA0330がMTAに特異的に結合するか否かを検証するため、Biacore T200 (GE Healthcare)を用いて、MTAおよびMTA類縁体(アデノシン、SAH)に対する結合性が解析された。
Protein Aがあらかじめ固定化されたSensor chip Protein A (GE healthcare)、またはストレプトアビジンがあらかじめ固定化されたSensor chip SA(GE Healthcare)にビオチン化された抗ヒトIgG CH1分子であるCaptureSelect Biotin Anti-IgG-Fc (Hu) (Thermo fisher scientific) を結合させた固定化表面に対し、目的の抗体をキャプチャーさせ、MTA (sigma-aldrich)、またはアデノシン (Wako)、またはS-(5'-Adenosyl)-L-homocysteine (SAH) (sigma-aldrich) を相互作用させた。ランニングバッファーには20mM ACES-NaOH, 150mM NaCl, 0.05% Tween20, 0.05%DMSOが用いられた。MTAまたはアデノシンまたはSAHは流速30μL/minで、120秒間相互作用させ、その後ランニングバッファーにて解離させた。相互作用は25℃で測定され、MTAまたはアデノシンまたはSAHの希釈にはランニングバッファーと同じバッファーが使用された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、各抗体のMTAおよびMTA類縁体に対する結合活性が表21のように決定され、ヒト化抗体のいずれもMTAに対して高い選択性を有することが確認された。
ヒト化MTA0303、ヒト化MTA0330がMTAに特異的に結合するか否かを検証するため、Biacore T200 (GE Healthcare)を用いて、MTAおよびMTA類縁体(アデノシン、SAH)に対する結合性が解析された。
Protein Aがあらかじめ固定化されたSensor chip Protein A (GE healthcare)、またはストレプトアビジンがあらかじめ固定化されたSensor chip SA(GE Healthcare)にビオチン化された抗ヒトIgG CH1分子であるCaptureSelect Biotin Anti-IgG-Fc (Hu) (Thermo fisher scientific) を結合させた固定化表面に対し、目的の抗体をキャプチャーさせ、MTA (sigma-aldrich)、またはアデノシン (Wako)、またはS-(5'-Adenosyl)-L-homocysteine (SAH) (sigma-aldrich) を相互作用させた。ランニングバッファーには20mM ACES-NaOH, 150mM NaCl, 0.05% Tween20, 0.05%DMSOが用いられた。MTAまたはアデノシンまたはSAHは流速30μL/minで、120秒間相互作用させ、その後ランニングバッファーにて解離させた。相互作用は25℃で測定され、MTAまたはアデノシンまたはSAHの希釈にはランニングバッファーと同じバッファーが使用された。
測定で得られたセンサーグラムから算出されたカイネティクスパラメーターである結合速度定数 ka(1/Ms)、および解離速度定数 kd(1/s)をもとに、解離定数KD(M)が算出された。あるいは、衡状態解析法 (Steady state analysis) を用いて、解離定数KD(M)が算出された。各パラメーターの算出には Biacore T200 Evaluation Software(GE Healthcare)が用いられた。
この測定によって、各抗体のMTAおよびMTA類縁体に対する結合活性が表21のように決定され、ヒト化抗体のいずれもMTAに対して高い選択性を有することが確認された。

(6-3)ヒト化MTA0303改変体の網羅的評価
実施例(5-3)のMTA0303結晶構造解析の結果から、ヒト化MTA0303中のMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのMTA依存的な結合に関与しやすいと想定される抗体可変領域のアミノ酸部位が推定された。これらの部位は多様化してもMTAに対する結合性を保持する可能性が高いと考えられるため、ライブラリのための多様化部位の候補となると考えられた。推定した部位の中で実際にMTAへの結合に大きく関与しない部位を同定するため、各部位のアミノ酸に対するAla/Val置換改変体が作製された。
各MTA0303重鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)および当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表22に示した。
実施例(5-3)のMTA0303結晶構造解析の結果から、ヒト化MTA0303中のMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのMTA依存的な結合に関与しやすいと想定される抗体可変領域のアミノ酸部位が推定された。これらの部位は多様化してもMTAに対する結合性を保持する可能性が高いと考えられるため、ライブラリのための多様化部位の候補となると考えられた。推定した部位の中で実際にMTAへの結合に大きく関与しない部位を同定するため、各部位のアミノ酸に対するAla/Val置換改変体が作製された。
各MTA0303重鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)および当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表22に示した。

各MTA0303軽鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表23に示した。

Biacore T200(GE Healthcare)を用いて、調製された改変体とMTAとの結合活性が評価された。ランニングバッファーとして、20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4が用いられ、25℃で測定された。センサーチップCM4上にアミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、MTAをアナライトとして相互作用させることで、MTAと抗体間の結合量の変化が観察された。MTAの希釈には、ランニングバッファーが使用され、数段階の濃度系列で調製され、MTA濃度に対するセンサーグラムからシングルサイクルカイネティクス解析により各クローンの解離定数KDが算出された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。ヒト化MTA0303のMTAに対する解離定数KDと各改変体のMTAに対する解離定数KDの比(ヒト化MTA0303のMTAに対するKD/各改変体のMTAに対するKD)を表22(MTA0303重鎖改変体)、表23(MTA0303軽鎖改変体)、)に示した。
(6-4)ヒト化MTA0303を鋳型としたライブラリ設計
ライブラリの設計にあたり、得られた情報をもとに、結合評価した改変体の中で、以下の条件を満たす部位を多様化可能なアミノ酸部位として選定された:
改変体の網羅的評価の結果から、MTAに対する結合に大きく関与しないアミノ酸部位。
ライブラリの設計にあたり、得られた情報をもとに、結合評価した改変体の中で、以下の条件を満たす部位を多様化可能なアミノ酸部位として選定された:
改変体の網羅的評価の結果から、MTAに対する結合に大きく関与しないアミノ酸部位。
重鎖に位置するアミノ酸部位の内、アミノ酸残基が抗体の表面側に存在し、その側鎖が抗体表面に露出し、想定される抗原結合面にそのアミノ酸残基が位置していること、または小分子との結合に大きくは関与していないと推定されるアミノ酸残基であり、親抗体(ヒト化MTA0303)のMTAに対するKD値が当該部位のアミノ酸が改変されたMTA0303改変体のMTAに対するKD値の50%以上であるアミノ酸部位、または親抗体(ヒト化MTA0303)のMTAに対するKD値が当該部位のアミノ酸が改変されたMTA0303改変体のMTAに対するKD値の70%以上であるアミノ酸部位が、条件1を満たす多様化可能なアミノ酸部位と判定された。
判定された多様化可能なアミノ酸部位に、少なくともいずれか一つ以上のライブラリ化可能なアミノ酸が出現するライブラリを設計することによって、MTA依存的に抗原に結合する抗体取得用のライブラリが構築された。ライブラリ化可能なアミノ酸として全種類のアミノ酸を採用し、X線結晶構造解析の結果から、重鎖S52b、軽鎖N31についてのみ、MTAが結合した際の抗体の構造変化を抑制すると予想されるアミノ酸を除いた。
ヒト化MTA0303の重鎖における多様化可能なアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表24に示した。ヒト化MTA0303の軽鎖における多様化可能なアミノ酸位、ならびに当該部位におけるアミノ酸レパートリーは表25に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位は多様化可能なアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位におけるヒト化MTA0303のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸)は当該部位における多様化可可能アミノ酸を、それぞれ示す。重鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。軽鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが構築された。
判定された多様化可能なアミノ酸部位に、少なくともいずれか一つ以上のライブラリ化可能なアミノ酸が出現するライブラリを設計することによって、MTA依存的に抗原に結合する抗体取得用のライブラリが構築された。ライブラリ化可能なアミノ酸として全種類のアミノ酸を採用し、X線結晶構造解析の結果から、重鎖S52b、軽鎖N31についてのみ、MTAが結合した際の抗体の構造変化を抑制すると予想されるアミノ酸を除いた。
ヒト化MTA0303の重鎖における多様化可能なアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表24に示した。ヒト化MTA0303の軽鎖における多様化可能なアミノ酸位、ならびに当該部位におけるアミノ酸レパートリーは表25に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位は多様化可能なアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位におけるヒト化MTA0303のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸)は当該部位における多様化可可能アミノ酸を、それぞれ示す。重鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。軽鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが構築された。


(6-5)ヒト化MTA0330改変体の網羅的評価
実施例(5-4)のMTA0330の結晶構造解析の結果から、ヒト化MTA0330中のMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのMTA依存的な結合に関与しやすいと想定される抗体可変領域のアミノ酸部位が推定された。これらの部位は多様化してもMTAに対する結合性を保持する可能性が高いと考えられるため、ライブラリのための多様化部位の候補となると考えられた。推定した部位の中で実際にMTAへの結合に大きく関与しない部位を同定するため、各部位のアミノ酸に対するAla置換改変体が作製された。
各MTA0330重鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)および当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表26に示した。
実施例(5-4)のMTA0330の結晶構造解析の結果から、ヒト化MTA0330中のMTAへの結合に大きく関与していないアミノ酸部位、もしくは抗原とのMTA依存的な結合に関与しやすいと想定される抗体可変領域のアミノ酸部位が推定された。これらの部位は多様化してもMTAに対する結合性を保持する可能性が高いと考えられるため、ライブラリのための多様化部位の候補となると考えられた。推定した部位の中で実際にMTAへの結合に大きく関与しない部位を同定するため、各部位のアミノ酸に対するAla置換改変体が作製された。
各MTA0330重鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)および当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表26に示した。

各MTA0330軽鎖改変体の、改変された部位(表中、「Kabat」と記載されるKabatナンバリングで表される部位)ならびに当該部位における改変前のアミノ酸(表中、「天然配列」と記載されるアミノ酸)および改変後のアミノ酸(表中、「改変アミノ酸」と記載されるアミノ酸)は表27に示した。

Biacore T200(GE Healthcare)を用いて、調製された改変体とMTAとの結合活性が評価された。ランニングバッファーとして、20 mM ACES、150 mM NaCl、0.05%(w/v)Tween20、pH7.4が用いられ、25℃で測定された。センサーチップCM4上にアミンカップリングによりProA/Gを固定化し、その上に抗体を固相化した後、MTAをアナライトとして相互作用させることで、MTAと抗体間の結合量の変化が観察された。MTAの希釈には、ランニングバッファーが使用され、数段階の濃度系列で調製され、MTA濃度に対するセンサーグラムからシングルサイクルカイネティクス解析により各クローンの解離定数KDが算出された。パラメーターの算出にはBiacore T200 Evaluation Software(GE Healthcare)が用いられた。ヒト化MTA0330のMTAに対する解離定数KDと各改変体のMTAに対する解離定数KDとの比(ヒト化MTA0330のMTAに対するKD/各改変体のMTAに対するKD)を表26(MTA0330重鎖改変体)、表27(MTA0330軽鎖改変体)に示した。
(6-6)ヒト化MTA0303とヒト化MTA0330を鋳型としたライブラリ設計
ライブラリの設計にあたり、得られた情報をもとに、結合評価した改変体の中で、以下の条件を満たす部位を多様化可能なアミノ酸部位として選定された:
改変体の網羅的評価の結果から、MTAに対する結合に大きく関与しないアミノ酸部位。
ライブラリの設計にあたり、得られた情報をもとに、結合評価した改変体の中で、以下の条件を満たす部位を多様化可能なアミノ酸部位として選定された:
改変体の網羅的評価の結果から、MTAに対する結合に大きく関与しないアミノ酸部位。
重鎖に位置し、親抗体(ヒト化MTA0330)のMTAに対するKD値が当該部位のアミノ酸が改変されたMTA0330改変体のMTAに対するKD値の40%以上であるアミノ酸部位、または軽鎖に位置し、親抗体(ヒト化MTA0330)のMTAに対するKD値が当該部位のアミノ酸が改変されたMTA0330改変体のMTAに対するKD値の70%以上であるアミノ酸部位が、条件1満たす多様化可能なアミノ酸部位と判定された。
判定された多様化可能なアミノ酸部位に、少なくともいずれか一つ以上のライブラリ化可能なアミノ酸が出現するライブラリを設計することによって、MTA依存的に抗原に結合する抗体取得用のライブラリが構築された。ライブラリ化可能なアミノ酸として全種類のアミノ酸を採用した。
ヒト化MTA0330の重鎖における多様化可能なアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表28に示した。ヒト化MTA0330の軽鎖における多様化可能なアミノ酸位、ならびに当該部位におけるアミノ酸レパートリーは表29に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位は多様化可能なアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位におけるヒト化MTA0330のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸)は当該部位における多様化可可能アミノ酸を、それぞれ示す。重鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。軽鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが構築された。
ヒト化MTA0330の重鎖における多様化可能なアミノ酸部位、ならびに当該部位におけるアミノ酸レパートリーは表28に示した。ヒト化MTA0330の軽鎖における多様化可能なアミノ酸位、ならびに当該部位におけるアミノ酸レパートリーは表29に示した。表中、「Kabat」と記載されるKabatナンバリングで表される部位は多様化可能なアミノ酸部位を、「天然配列」と記載されるアミノ酸は当該部位におけるヒト化MTA0330のアミノ酸を、「ライブラリ化可能アミノ酸」と記載されるアミノ酸)は当該部位における多様化可可能アミノ酸を、それぞれ示す。重鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが設計された。軽鎖の各多様化可能アミノ酸部位において、表中のアミノ酸レパートリーに含まれるアミノ酸のうち、少なくともいずれか一つが出現するライブラリが構築された。

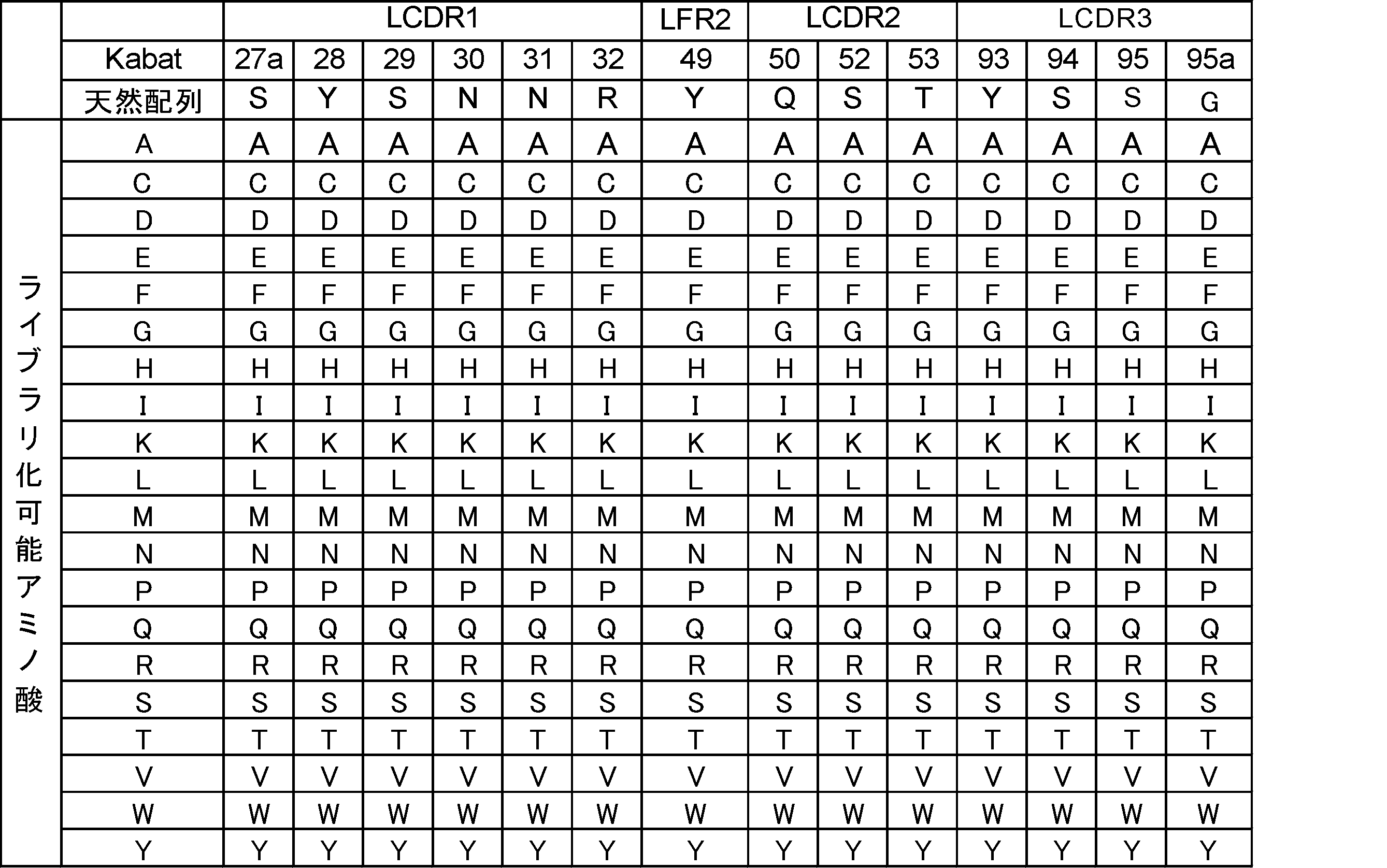
(6-7)ヒト化MTA0330を鋳型としたライブラリからのMTA依存性抗体の取得
(6-7-1)ヒト化MTA0330重鎖もしくは軽鎖可変領域ファージディスプレイライブラリの構築
実施例(6-6)で構築したヒト化MTA0330を鋳型として設計した重鎖可変領域ライブラリを遺伝子合成し、合成したヒト化MTA0330の重鎖可変領域をヒト化MTA0330の軽鎖可変領域、およびヒトIgG由来重鎖CH1配列およびヒトIgG由来軽鎖定常領域配列含むファージミドベクターに導入した。同様の手法を用いてヒト化MTA0330を鋳型として設計された軽鎖可変領域ライブラリを遺伝子合成し、合成したヒト化MTA0330の軽鎖可変領域をヒト化MTA0330の重鎖可変領域、およびヒトIgG由来重鎖CH1配列およびヒトIgG由来軽鎖定常領域配列を含むファージミドベクターに導入した。作成した上記ファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTA依存的に抗原に結合できる重鎖・軽鎖可変領域を取得可能なファージディスプレイライブラリが構築された。
(6-7-1)ヒト化MTA0330重鎖もしくは軽鎖可変領域ファージディスプレイライブラリの構築
実施例(6-6)で構築したヒト化MTA0330を鋳型として設計した重鎖可変領域ライブラリを遺伝子合成し、合成したヒト化MTA0330の重鎖可変領域をヒト化MTA0330の軽鎖可変領域、およびヒトIgG由来重鎖CH1配列およびヒトIgG由来軽鎖定常領域配列含むファージミドベクターに導入した。同様の手法を用いてヒト化MTA0330を鋳型として設計された軽鎖可変領域ライブラリを遺伝子合成し、合成したヒト化MTA0330の軽鎖可変領域をヒト化MTA0330の重鎖可変領域、およびヒトIgG由来重鎖CH1配列およびヒトIgG由来軽鎖定常領域配列を含むファージミドベクターに導入した。作成した上記ファージミドベクターをエレクトロポレーションにより大腸菌へと導入することで、MTA依存的に抗原に結合できる重鎖・軽鎖可変領域を取得可能なファージディスプレイライブラリが構築された。
(6-7-2)重鎖および軽鎖可変領域ファージディスプレイライブラリを用いたパニングによるMTAに結合する抗体の取得
実施例(6-7-1)において構築された重鎖可変領域、軽鎖可変領域のそれぞれのファージディスプレイライブラリから、MTAに結合する抗体可変領域を有するファージの集団を取得するために、ビオチン化標識MTAを用いたパニングを実施した。具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液をTBSに溶媒置換し抗体多価提示ファージライブラリ液が調製された。
実施例(6-7-1)において構築された重鎖可変領域、軽鎖可変領域のそれぞれのファージディスプレイライブラリから、MTAに結合する抗体可変領域を有するファージの集団を取得するために、ビオチン化標識MTAを用いたパニングを実施した。具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液をTBSに溶媒置換し抗体多価提示ファージライブラリ液が調製された。
磁気ビーズに固定化された抗原を用いてパニングが実施された。当該ファージライブラリ液に12%のBSAが添加された。磁気ビーズとして、NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific)が用いられた。BSAでブロッキングされた磁気ビーズと終濃度5.7 μMのBiotin-6’-MTAと0.91 mMのAdenosineを調製されたファージライブラリ液0.3mLに加え、室温で60分間反応させた。ビーズは400μLのTBSTにて2回、TBSにて1回洗浄された。その後最終濃度1mg/mLのトリプシン入りのTBS 0.5 mLが加えられたビーズが室温で15分懸濁された。磁気スタンドを用いて分離されたビーズからファージ溶液が回収された。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。この一連の作業を追加でさらに1回繰り返した。
(6-7-3)MTAに対するパンニングを利用したMTA依存性抗体取得用ライブラリの構築
重鎖、軽鎖可変領域ファージディスプレイライブラリそれぞれから取得されたMTAに対して結合する重鎖および軽鎖の集団について、その集団の中の重鎖と軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもMTAに対する結合を維持するクローンが多数含まれることが予想され、より効率よくMTA依存性抗体を取得できるライブラリが構築できると考えられた。
MTAに対するパニングで取得された重鎖、軽鎖それぞれのファージライブラリが感染した大腸菌から当業者公知の方法で遺伝子が抽出された。パンニング後重鎖可変領域ファージライブラリにおいては、重鎖可変領域遺伝子が重鎖可変領域を増幅できるプライマー(配列番号:88、89)を用いて増幅された。
重鎖、軽鎖可変領域ファージディスプレイライブラリそれぞれから取得されたMTAに対して結合する重鎖および軽鎖の集団について、その集団の中の重鎖と軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもMTAに対する結合を維持するクローンが多数含まれることが予想され、より効率よくMTA依存性抗体を取得できるライブラリが構築できると考えられた。
MTAに対するパニングで取得された重鎖、軽鎖それぞれのファージライブラリが感染した大腸菌から当業者公知の方法で遺伝子が抽出された。パンニング後重鎖可変領域ファージライブラリにおいては、重鎖可変領域遺伝子が重鎖可変領域を増幅できるプライマー(配列番号:88、89)を用いて増幅された。
パンニング後軽鎖可変領域ファージライブラリにおいてはヒト化MTA0330の重鎖可変領域配列を制限酵素処理により抜き取り、軽鎖可変領域ライブラリ遺伝子、ヒトIgG由来軽鎖定常領域配列およびヒトIgG由来CH1配列を含むファージミド断片が調製された。調製された軽鎖可変領域ライブラリ遺伝子を含むファージミドベクター断片に重鎖可変領域ライブラリ遺伝子を挿入することで、重鎖/軽鎖可変領域ライブラリ遺伝子が導入されたファージミドベクターが構築された。このベクターをエレクトロポレーションにより大腸菌へと導入することで、ヒト抗体可変領域と定常領域からなるFabドメインを提示し、MTA依存的に抗原に結合できる抗体を取得できるライブラリが構築された。
(6-7-4)M30ライブラリからのMTA依存性抗体取得パンニング
構築されたMTA依存性抗体取得用ファージディスプレイライブラリ(M30ライブラリと呼称する)から、MTA存在下でヒトIL-6レセプター(hIL-6R)、ヒトIL-6(hIL-6)、ヒトIgA(hIgA)のそれぞれに対して結合活性を示す抗体がスクリーニングされた。
具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液をTBSに溶媒置換し抗体多価提示ファージライブラリ液が調製された。
構築されたMTA依存性抗体取得用ファージディスプレイライブラリ(M30ライブラリと呼称する)から、MTA存在下でヒトIL-6レセプター(hIL-6R)、ヒトIL-6(hIL-6)、ヒトIgA(hIgA)のそれぞれに対して結合活性を示す抗体がスクリーニングされた。
具体的には、構築されたファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液をTBSに溶媒置換し抗体多価提示ファージライブラリ液が調製された。
磁気ビーズに固定化されたそれぞれの抗原を別条件として用いてパンニングが実施された。当該ファージライブラリ液に10%のBSAが添加された。調製されたファージライブラリ液0.8 mLに0.1 nmolのビオチン標識抗原と終濃度100 μMのMTAを加え、室温で60分間反応させた。磁気ビーズとして、NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific)が用いられた。BSAでブロッキングされた磁気ビーズをファージと抗原の反応液に加え室温で15分反応させた。ビーズは0.8 mLのTBSTにて2回もしくは3回、TBSにて1回もしくは2回洗浄された。その後0.25 mLのTBSが加えられたビーズが室温で懸濁され、磁気スタンドを用いて分離されたビーズからファージ溶液が回収された。この作業が再度繰り返された後、2回に分けて溶出されたファージ溶液が混合された。回収されたファージ溶液に最終濃度1mg/mLのトリプシンが加えられた。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。大腸菌は225 mm x 225 mmのプレートへ播種された。この一連の作業を追加で2回繰り返した。
(6-7-5)フローサイトメトリーによるMTA存在下での抗原結合活性の評価
パンニングで得られた重鎖および軽鎖の可変領域配列は、重鎖抗体定常領域配列(配列番号:86)もしくは軽鎖kappa定常領域配列(配列番号:87)を有する動物発現用プラスミドへと挿入された。播種されたExpi293株(Thermo Fisher Scientific)に、調製されたプラスミドがリポフェクション法により導入された。CO2インキュベーター(37 ℃、8% CO2、1000 rpm)で4日間培養された培養上清を回収した。
培養上清は以下の手順でフローサイトメトリーを利用した抗原結合評価に供された。384ウェルマイクロプレート(greiner)に各ビオチン標識抗原を固相した磁気ビーズ、培養上清、0.1% BSA/5mM Mg2CL2/PBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSが添加され30分間以上静置された。当該プレートのビーズを0.1% BSA/5mM Mg2CL2/PBSまたは100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで洗浄した後、当該ウェルにPBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで希釈されたDyLight488標識抗ヒトFc抗体(invitrogen)が加えられ30分間以上静置された。当該ウェルを0.1% BSA/5mM Mg2CL2/PBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで洗浄した後、iQue screener(Intellicyt)でサンプルの蛍光強度を測定した。解析の結果、ビオチン標識hIL-6R、hIL-6、hIgAのそれぞれを用いてパンニングされた各条件について、MTA依存的に抗原に結合する抗体が複数確認された。結果を表31に示した。
パンニングで得られた重鎖および軽鎖の可変領域配列は、重鎖抗体定常領域配列(配列番号:86)もしくは軽鎖kappa定常領域配列(配列番号:87)を有する動物発現用プラスミドへと挿入された。播種されたExpi293株(Thermo Fisher Scientific)に、調製されたプラスミドがリポフェクション法により導入された。CO2インキュベーター(37 ℃、8% CO2、1000 rpm)で4日間培養された培養上清を回収した。
培養上清は以下の手順でフローサイトメトリーを利用した抗原結合評価に供された。384ウェルマイクロプレート(greiner)に各ビオチン標識抗原を固相した磁気ビーズ、培養上清、0.1% BSA/5mM Mg2CL2/PBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSが添加され30分間以上静置された。当該プレートのビーズを0.1% BSA/5mM Mg2CL2/PBSまたは100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで洗浄した後、当該ウェルにPBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで希釈されたDyLight488標識抗ヒトFc抗体(invitrogen)が加えられ30分間以上静置された。当該ウェルを0.1% BSA/5mM Mg2CL2/PBSもしくは終濃度100 μMのMTAを含む0.1% BSA/5mM Mg2CL2/PBSで洗浄した後、iQue screener(Intellicyt)でサンプルの蛍光強度を測定した。解析の結果、ビオチン標識hIL-6R、hIL-6、hIgAのそれぞれを用いてパンニングされた各条件について、MTA依存的に抗原に結合する抗体が複数確認された。結果を表31に示した。

この結果から、配列も構造も異なる複数の抗原に対してMTA存在下で抗原に結合でき、MTA非存在下で抗原に結合できないクローンが複数取得された。また取得されたクローンは全てアデノシン存在下では抗原に対する結合を示さなかったことから、MTA特異的かつMTA濃度依存的に抗原に結合する抗体を取得できることが示された。
[実施例7] アデノシン依存的に抗原に結合する抗体を取得するためのライブラリの構築
(7-1)重鎖および軽鎖可変領域ファージディスプレイライブラリを用いたアデノシンに結合する抗体群の濃縮
実施例(4-1-1)で構築された重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、アデノシンに結合する抗体群を濃縮するために、Biotin-2’-アデノシンとBiotin-5’-アデノシンの混合物(Biotin-アデノシン混合物)を用いたパニングが実施された。
具体的には、構築された重鎖可変領域ファージディスプレイライブラリまたは軽鎖可変領域ファージディスプレイライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
(7-1)重鎖および軽鎖可変領域ファージディスプレイライブラリを用いたアデノシンに結合する抗体群の濃縮
実施例(4-1-1)で構築された重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、アデノシンに結合する抗体群を濃縮するために、Biotin-2’-アデノシンとBiotin-5’-アデノシンの混合物(Biotin-アデノシン混合物)を用いたパニングが実施された。
具体的には、構築された重鎖可変領域ファージディスプレイライブラリまたは軽鎖可変領域ファージディスプレイライブラリのファージミドベクターを保持する大腸菌に対して、M13KO7ΔpIII(ハイパーファージと呼称される)(PROGEN Biotechnik)を感染させ、25℃で一晩培養した上清からファージが回収された。ファージ産生が行われた大腸菌の培養液に2.5M NaCl/10%PEGを添加することによって沈殿させたファージの集団をTBSにて希釈することによって抗体多価提示ファージライブラリ液が調製された。
磁気ビーズに固定化されたBiotin-アデノシン混合物を用いたパニングが実施された。抗体多価提示ファージライブラリ液に終濃度4%となるようにBSAが添加された。磁気ビーズとして、NeutrAvidin beads(TAMAGAWA SEIKI)もしくはDynabeads MyOne StreptAvidin T1(Thermo Fisher Scientific)が用いられた。BSAでブロッキングされた磁気ビーズと終濃度10 μMのBiotin-アデノシン混合物を室温で30分間反応させることによって、磁気ビーズに対してBiotin-アデノシン混合物が固相化された。TBSTで3回洗浄されたBiotin-アデノシン混合物が固相化された磁気ビーズに、調製された抗体多価提示ファージライブラリ液0.8mLを加え、室温で60分間反応させた。ビーズは400μLのTBSTにて2回、TBSにて1回洗浄された。その後終濃度1mg/mLのトリプシンを含むTBS 0.5 mLが加えられたビーズが室温で15分懸濁され、ファージが溶出された。磁気スタンドを用いてビーズが分離され、ファージ溶液が回収された。回収されたファージが、対数増殖期(OD600が0.4-0.7)となった20mLの大腸菌株ER2738に添加された。37℃で1時間上記大腸菌の撹拌培養を行うことによって、ファージを大腸菌に感染させた。大腸菌は225 mm x 225 mmのプレートへ播種された。この一連の作業を追加で1回繰り返し、重鎖可変領域ファージディスプレイライブラリ、軽鎖可変領域ファージディスプレイライブラリのそれぞれから、アデノシンに結合する抗体群が濃縮された。
(7-2)ファージELISAによるパニング後取得クローン群のビオチン化アデノシンに対する結合活性の評価
実施例(7-1)で濃縮されたアデノシンに結合する抗体群中の抗体を提示するファージを感染させた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収され、ELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、Biotin-アデノシン混合物を含むTBSが10 μL添加され、1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80uLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示される抗体をBiotin-アデノシン混合物に結合させた。各ウェルをTBSTで洗浄した後、各ウェルにTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。各ウェルをTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、Biotin-2’-アデノシンに結合する抗体を提示するファージが複数確認された。ファージELISAの結果を表30に示した。ELISAの結果から、実施例(7-1)のパニングにより濃縮された抗体群中にアデノシン結合抗体が多数含まれていることが示された。
実施例(7-1)で濃縮されたアデノシンに結合する抗体群中の抗体を提示するファージを感染させた大腸菌のシングルコロニーから、常法(Methods mol. Biol. (2002) 178, 133-145)に倣い、ファージ含有培養上清が回収された。ヘルパーファージとしてハイパーファージを用い、抗体多価提示ファージが回収され、ELISAに供された。384 Well Microplates Streptavidin-coated(greiner)に、Biotin-アデノシン混合物を含むTBSが10 μL添加され、1時間以上静置された。当該プレートの各ウェルをTBSTで洗浄した後、各ウェルは80uLの0.2%スキムミルク-TBSで1時間以上ブロッキングされた。各ウェルをTBSTで洗浄した後、各ウェルに調製されたファージが加えられ一時間静置することで、ファージに提示される抗体をBiotin-アデノシン混合物に結合させた。各ウェルをTBSTで洗浄した後、各ウェルにTBSで希釈されたHRP結合抗M13抗体(GE Healthcare)が加えられ一時間静置された。各ウェルをTBSTで洗浄した後、TMB single溶液(ZYMED)が加えられ、一定時間後溶液の発色反応が硫酸の添加により停止された後、450nm波長の吸光度が測定された。解析の結果、Biotin-2’-アデノシンに結合する抗体を提示するファージが複数確認された。ファージELISAの結果を表30に示した。ELISAの結果から、実施例(7-1)のパニングにより濃縮された抗体群中にアデノシン結合抗体が多数含まれていることが示された。

(7-3)アデノシンに対するパニングを利用したアデノシン依存的に抗体に結合する抗体を取得するためのライブラリの構築
実施例(7-2)で濃縮されたアデノシン結合抗体群について、当該抗体群に含まれる改変重鎖と改変軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもアデノシンに対する結合を維持する抗体が多数含まれることが予想され、アデノシン依存的に抗原に結合する抗体を取得するためのライブラリが構築できると考えられた。
実施例(7-2)で濃縮されたアデノシン結合抗体群を提示するファージが感染した大腸菌から、当業者公知の方法で抗体遺伝子が抽出された。重鎖可変領域ファージライブラリから濃縮された抗体群に対して、重鎖可変領域を増幅できるプライマー(配列番号:34、35)を用いて重鎖可変領域遺伝子が増幅された。軽鎖可変領域ファージライブラリおよびそのライブラリから濃縮された抗体群に対して、ヒト化SMB0002の重鎖可変領域配列を制限酵素処理により除去し、軽鎖可変領域ライブラリ遺伝子とヒトIgG由来軽鎖定常領域配列とヒトIgG由来CH1配列を含むファージミド断片が調製された。前記ファージミドベクター断片に重鎖可変領域ライブラリから増幅された重鎖可変領域遺伝子を挿入することで、重鎖/軽鎖可変領域ライブラリ遺伝子が導入されたファージミドベクターが構築された。このベクターをエレクトロポレーションにより大腸菌へと導入することで、ヒト抗体可変領域と定常領域からなるFabドメインを提示し、アデノシン依存的に抗原に結合する抗体を取得するためのライブラリ(アデノシン濃縮S02ライブラリ)が構築された。
実施例(7-2)で濃縮されたアデノシン結合抗体群について、当該抗体群に含まれる改変重鎖と改変軽鎖を組み合わせることにより作製されるFab提示ファージライブラリもアデノシンに対する結合を維持する抗体が多数含まれることが予想され、アデノシン依存的に抗原に結合する抗体を取得するためのライブラリが構築できると考えられた。
実施例(7-2)で濃縮されたアデノシン結合抗体群を提示するファージが感染した大腸菌から、当業者公知の方法で抗体遺伝子が抽出された。重鎖可変領域ファージライブラリから濃縮された抗体群に対して、重鎖可変領域を増幅できるプライマー(配列番号:34、35)を用いて重鎖可変領域遺伝子が増幅された。軽鎖可変領域ファージライブラリおよびそのライブラリから濃縮された抗体群に対して、ヒト化SMB0002の重鎖可変領域配列を制限酵素処理により除去し、軽鎖可変領域ライブラリ遺伝子とヒトIgG由来軽鎖定常領域配列とヒトIgG由来CH1配列を含むファージミド断片が調製された。前記ファージミドベクター断片に重鎖可変領域ライブラリから増幅された重鎖可変領域遺伝子を挿入することで、重鎖/軽鎖可変領域ライブラリ遺伝子が導入されたファージミドベクターが構築された。このベクターをエレクトロポレーションにより大腸菌へと導入することで、ヒト抗体可変領域と定常領域からなるFabドメインを提示し、アデノシン依存的に抗原に結合する抗体を取得するためのライブラリ(アデノシン濃縮S02ライブラリ)が構築された。
[参考実施例1] 抗体の発現と精製
以下の方法を用いて抗体蛋白が発現された。播種されたヒト胎児腎細胞由来FreeStyle293株またはExpi293株(Thermo Fisher Scientific)に、調製されたプラスミドがリポフェクション法により導入された。CO2インキュベーター(37 ℃、8% CO2、1000 rpm)で4日間培養された培養上清から、rProtein A Sepharose Fast Flow(Amersham Biosciences)またはMonoSpin ProA(ジーエルサイエンス)を用いて当業者公知の方法で抗体が精製された。分光光度計を用いて、精製された抗体溶液の280nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製抗体の濃度が算出された(Protein Science(1995) 4、2411-2423)。
以下の方法を用いて抗体蛋白が発現された。播種されたヒト胎児腎細胞由来FreeStyle293株またはExpi293株(Thermo Fisher Scientific)に、調製されたプラスミドがリポフェクション法により導入された。CO2インキュベーター(37 ℃、8% CO2、1000 rpm)で4日間培養された培養上清から、rProtein A Sepharose Fast Flow(Amersham Biosciences)またはMonoSpin ProA(ジーエルサイエンス)を用いて当業者公知の方法で抗体が精製された。分光光度計を用いて、精製された抗体溶液の280nmでの吸光度が測定された。得られた測定値からPACE法により算出された吸光係数を用いて精製抗体の濃度が算出された(Protein Science(1995) 4、2411-2423)。
本開示のMTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン及び当該抗原結合ドメインを含む抗原結合分子、ならびにこれを含む医薬組成物は、正常組織や血液中において全身的に作用せず、がんにおいて標的抗原と結合し作用することにより、副作用を回避しつつ薬効を発揮し、がんを治療することができる。
さらに、本開示の、互いに配列の異なる複数の、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインまたは抗原結合ドメインを含む抗原結合分子を含むライブラリを用いれば、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン及び当該抗原結合ドメインを含む抗原結合分子を、短時間で効率良く取得することが可能である。
さらに、本開示の、互いに配列の異なる複数の、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインまたは抗原結合ドメインを含む抗原結合分子を含むライブラリを用いれば、MTA依存的に抗原に対する結合活性が変化する抗原結合ドメイン及び当該抗原結合ドメインを含む抗原結合分子を、短時間で効率良く取得することが可能である。
Claims (15)
- 5’-Methylthioadenosine(MTA)依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子。
- 前記抗原結合ドメインのMTA存在下における前記抗原に対する結合活性が、MTA非存在下における前記抗原に対する結合活性と異なる、請求項1記載の抗原結合分子。
- 前記抗原結合ドメインが、抗体可変領域または/及び単ドメイン抗体を含む、請求項1また2記載の抗原結合分子。
- 前記抗原結合ドメインのMTA存在下における抗原に対する結合活性が、前記抗原結合ドメインのMTA非存在下における抗原に対する結合活性より強い、請求項1乃至3のいずれか一項記載の抗原結合分子。
- 前記抗原が、膜型分子であり、且つがん組織で発現している抗原である、請求項1乃至4のいずれか一項記載の抗原結合分子。
- 請求項1乃至5のいずれか一項記載の抗原結合分子を含む医薬組成物。
- お互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子、または/および互いに配列の異なる複数の抗原結合ドメインを含む抗原結合分子をコードする核酸から主としてなるライブラリであって、前記抗原結合ドメインはMTAと相互作用する抗原結合ドメインである、ライブラリ。
- 前記抗原結合ドメインが、抗体可変領域である、請求項7記載のライブラリ。
- MTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体、または/およびMTAに対する結合活性を有する抗体可変領域未改変体中の1つまたは複数のアミノ酸部位に位置するアミノ酸と異なるアミノ酸を有し、互いに配列が異なる複数の抗体可変領域改変体をそれぞれコードする核酸を含む、請求項8記載のライブラリ。
- 以下(a)および(b)の工程:
(a) MTAに対する結合活性を有する抗原結合ドメインにおいて、以下(i)乃至(vi)の少なくとも一つ以上を満たすアミノ酸部位を特定する工程:
(i) 前記抗原結合ドメインの表面に露出するアミノ酸部位;
(ii) 前記抗原結合ドメインがMTAに結合したときの構造とMTAに結合していない時の構造を比較するときに構造変化率が大きい領域に位置するアミノ酸部位;
(iii) MTAとの結合に関与しないアミノ酸部位;
(iv) MTAとの結合を著しく減弱させないアミノ酸部位;
(v) 前記抗原結合ドメインが属する動物種において、アミノ酸出現頻度の多様性のあるアミノ酸部位;または
(vi) Canonical structureの形成に重要でないアミノ酸部位;
(b) 前記抗原結合ドメインにおいて、未改変体をコードする核酸及び工程(a)で特定された1または複数のアミノ酸部位にアミノ酸改変を有する、互いに配列が異なる、前記抗原結合ドメインの複数の改変体をそれぞれコードする核酸を含むライブラリを設計する工程、
を含むライブラリの製造方法。 - MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子をスクリーニングする方法。
- 第1濃度のMTA存在下における抗原に対する結合活性と第2濃度のMTA存在下における抗原に対する結合活性が異なる抗原結合ドメインを選択する工程含む、請求項11記載の方法。
- MTA依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子を製造する方法。
- MTAに特異的に結合する抗原結合分子。
- 請求項14記載の抗原結合分子を含む、疾患の診断キット。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20773939.2A EP3943108A4 (en) | 2019-03-19 | 2020-03-19 | ANTIGEN-BINDING MOLECULE WITH ANTIGEN-BINDING DOMAIN WITH VARIABLE BINDING ACTIVITY TO ANTIGEN DEPENDING ON MTA AND LIBRARY TO OBTAIN THE ANTIGEN-BINDING DOMAIN |
| CN202511354577.7A CN121159688A (zh) | 2019-03-19 | 2020-03-19 | 包含对抗原的结合活性因mta而变化的抗原结合结构域的抗原结合分子及用于获得该抗原结合结构域的文库 |
| CN202080021856.0A CN113613676A (zh) | 2019-03-19 | 2020-03-19 | 包含对抗原的结合活性因mta而变化的抗原结合结构域的抗原结合分子及用于获得该抗原结合结构域的文库 |
| US17/438,993 US20220153875A1 (en) | 2019-03-19 | 2020-03-19 | Antigen-binding molecule containing antigen-binding domain of which binding activity to antigen is changed depending on mta, and library for obtaining said antigen-binding domain |
| JP2021507411A JP7601758B2 (ja) | 2019-03-19 | 2020-03-19 | Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ |
| JP2024211901A JP2025038000A (ja) | 2019-03-19 | 2024-12-05 | Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-051965 | 2019-03-19 | ||
| JP2019051965 | 2019-03-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020189748A1 true WO2020189748A1 (ja) | 2020-09-24 |
Family
ID=72520304
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/012189 Ceased WO2020189748A1 (ja) | 2019-03-19 | 2020-03-19 | Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220153875A1 (ja) |
| EP (1) | EP3943108A4 (ja) |
| JP (2) | JP7601758B2 (ja) |
| CN (2) | CN113613676A (ja) |
| WO (1) | WO2020189748A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11673947B2 (en) | 2012-05-30 | 2023-06-13 | Chugai Seiyaku Kabushiki Kaisha | Target tissue-specific antigen-binding molecule |
| US11912989B2 (en) | 2013-12-04 | 2024-02-27 | Chugai Seiyaku Kabushiki Kaisha | Antigen-binding molecules, the antigen-binding activity of which varies according to the concentration of compounds, and libraries of said molecules |
| US12304960B2 (en) | 2018-08-10 | 2025-05-20 | Chugai Seiyaku Kabushiki Kaisha | Anti-CD137 antigen-binding molecule and utilization thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117130035A (zh) * | 2017-08-18 | 2023-11-28 | 南京中硼联康医疗科技有限公司 | 生物剂量计及具有其的中子捕获治疗系统 |
| IL297697A (en) * | 2020-06-17 | 2022-12-01 | Tiziana Life Sciences Plc | Preparations and methods for improving chimeric antigen receptor t cell therapies |
Citations (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0239400A2 (en) | 1986-03-27 | 1987-09-30 | Medical Research Council | Recombinant antibodies and methods for their production |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| EP0404097A2 (de) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispezifische und oligospezifische, mono- und oligovalente Rezeptoren, ihre Herstellung und Verwendung |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992003918A1 (en) | 1990-08-29 | 1992-03-19 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| WO1993012227A1 (en) | 1991-12-17 | 1993-06-24 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1994002602A1 (en) | 1992-07-24 | 1994-02-03 | Cell Genesys, Inc. | Generation of xenogeneic antibodies |
| WO1994011523A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Fully impaired consensus kozac sequences for mammalian expression |
| WO1994025585A1 (en) | 1993-04-26 | 1994-11-10 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995001937A1 (fr) | 1993-07-09 | 1995-01-19 | Association Gradient | Procede de traitement de residus de combustion et installation de mise en ×uvre dudit procede |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| WO1996000257A1 (en) | 1994-06-24 | 1996-01-04 | Imperial Chemical Industries Plc | Foaming method |
| WO1996002576A1 (en) | 1994-07-13 | 1996-02-01 | Chugai Seiyaku Kabushiki Kaisha | Reconstituted human antibody against human interleukin-8 |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| WO1996033735A1 (en) | 1995-04-27 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1996034103A1 (en) | 1995-04-25 | 1996-10-31 | Vrije Universiteit Brussel | Variable fragments of immunoglobulins - use for therapeutic or veterinary purposes |
| WO1996034096A1 (en) | 1995-04-28 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1999023221A2 (en) | 1997-10-27 | 1999-05-14 | Unilever Plc | Multivalent antigen-binding proteins |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1999054342A1 (en) | 1998-04-20 | 1999-10-28 | Pablo Umana | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (fr) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Methode de regulation de l'activite d'une molecule immunologiquement fonctionnelle |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| WO2002020565A2 (en) | 2000-09-08 | 2002-03-14 | Universität Zürich | Collections of repeat proteins comprising repeat modules |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| WO2002032925A2 (en) | 2000-10-16 | 2002-04-25 | Phylos, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| WO2002060919A2 (en) | 2000-12-12 | 2002-08-08 | Medimmune, Inc. | Molecules with extended half-lives, compositions and uses thereof |
| WO2002079255A1 (en) | 2001-04-02 | 2002-10-10 | Idec Pharmaceuticals Corporation | RECOMBINANT ANTIBODIES COEXPRESSED WITH GnTIII |
| WO2003000883A1 (fr) | 2001-06-22 | 2003-01-03 | Chugai Seiyaku Kabushiki Kaisha | Inhibiteurs de proliferation cellulaire renfermant un anticorps anti-glypicane 3 |
| WO2003029462A1 (en) | 2001-09-27 | 2003-04-10 | Pieris Proteolab Ag | Muteins of human neutrophil gelatinase-associated lipocalin and related proteins |
| WO2003104453A1 (ja) | 2002-06-05 | 2003-12-18 | 中外製薬株式会社 | 抗体作製方法 |
| WO2004022754A1 (ja) | 2002-09-04 | 2004-03-18 | Chugai Seiyaku Kabushiki Kaisha | MRL/lprマウスを用いた抗体の作製 |
| WO2004029207A2 (en) | 2002-09-27 | 2004-04-08 | Xencor Inc. | Optimized fc variants and methods for their generation |
| WO2004035752A2 (en) | 2002-10-15 | 2004-04-29 | Protein Design Labs, Inc. | ALTERATION OF FcRn BINDING AFFINITIES OR SERUM HALF-LIVES OF ANTIBODIES BY MUTAGENESIS |
| WO2004044011A2 (en) | 2002-11-06 | 2004-05-27 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| WO2004065540A2 (en) | 2003-01-22 | 2004-08-05 | Glycart Biotechnology Ag | Fusion constructs and use of same to produce antibodies with increased fc receptor binding affinity and effector function |
| WO2004099249A2 (en) | 2003-05-02 | 2004-11-18 | Xencor, Inc. | Optimized fc variants and methods for their generation |
| WO2005040229A2 (en) | 2003-10-24 | 2005-05-06 | Avidia, Inc. | Ldl receptor class a and egf domain monomers and multimers |
| WO2005070963A1 (en) | 2004-01-12 | 2005-08-04 | Applied Molecular Evolution, Inc | Fc region variants |
| WO2006006693A1 (ja) | 2004-07-09 | 2006-01-19 | Chugai Seiyaku Kabushiki Kaisha | 抗グリピカン3抗体 |
| WO2006020114A2 (en) | 2004-08-04 | 2006-02-23 | Applied Molecular Evolution, Inc. | Variant fc regions |
| WO2006023403A2 (en) | 2004-08-16 | 2006-03-02 | Medimmune, Inc. | Eph receptor fc variants with enhanced antibody dependent cell-mediated cytotoxicity activity |
| WO2006031370A2 (en) | 2004-08-19 | 2006-03-23 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2006067913A1 (ja) | 2004-12-22 | 2006-06-29 | Chugai Seiyaku Kabushiki Kaisha | フコーストランスポーターの機能が阻害された細胞を用いた抗体の作製方法 |
| WO2006105338A2 (en) | 2005-03-31 | 2006-10-05 | Xencor, Inc. | Fc VARIANTS WITH OPTIMIZED PROPERTIES |
| WO2006106905A1 (ja) | 2005-03-31 | 2006-10-12 | Chugai Seiyaku Kabushiki Kaisha | 会合制御によるポリペプチド製造方法 |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| WO2006116260A2 (en) | 2005-04-26 | 2006-11-02 | Medimmune, Inc. | Modulation of antibody effector function by hinge domain engineering |
| WO2007021841A2 (en) | 2005-08-10 | 2007-02-22 | Macrogenics, Inc. | Identification and engineering of antibodies with variant fc regions and methods of using same |
| WO2007024249A2 (en) | 2004-11-10 | 2007-03-01 | Macrogenics, Inc. | Engineering fc antibody regions to confer effector function |
| WO2007041635A2 (en) | 2005-10-03 | 2007-04-12 | Xencor, Inc. | Fc variants with optimized fc receptor binding properties |
| JP2007531724A (ja) * | 2004-04-05 | 2007-11-08 | クアーク・ファーマスーティカルス、インコーポレイテッド | 5’−デオキシ−5’−メチルチオアデノシンに対する抗体及びその使用 |
| WO2008016854A2 (en) | 2006-08-02 | 2008-02-07 | The Uab Research Foundation | Methods and compositions related to soluble monoclonal variable lymphocyte receptors of defined antigen specificity |
| WO2008092117A2 (en) | 2007-01-25 | 2008-07-31 | Xencor, Inc. | Immunoglobulins with modifications in the fcr binding region |
| US20090136485A1 (en) | 2007-05-30 | 2009-05-28 | Xencor, Inc. | Methods and compositions for inhibiting CD32B expressing cells |
| WO2009086320A1 (en) | 2007-12-26 | 2009-07-09 | Xencor, Inc | Fc variants with altered binding to fcrn |
| WO2009125825A1 (ja) | 2008-04-11 | 2009-10-15 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2009140242A1 (en) | 2008-05-13 | 2009-11-19 | Genentech, Inc. | Analysis of antibody drug conjugates by bead-based affinity capture and mass spectrometry |
| US20110123527A1 (en) | 2008-05-23 | 2011-05-26 | Hiroaki Shizuya | Method of generating single vl domain antibodies in transgenic animals |
| WO2011122011A2 (en) | 2010-03-30 | 2011-10-06 | Chugai Seiyaku Kabushiki Kaisha | Antibodies with modified affinity to fcrn that promote antigen clearance |
| WO2012073992A1 (ja) | 2010-11-30 | 2012-06-07 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2012115241A1 (ja) | 2011-02-25 | 2012-08-30 | 中外製薬株式会社 | FcγRIIb特異的Fc抗体 |
| WO2012133782A1 (ja) | 2011-03-30 | 2012-10-04 | 中外製薬株式会社 | 抗原結合分子の血漿中滞留性と免疫原性を改変する方法 |
| WO2013002362A1 (ja) | 2011-06-30 | 2013-01-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2013046704A2 (en) | 2011-09-30 | 2013-04-04 | Chugai Seiyaku Kabushiki Kaisha | THERAPEUTIC ANTIGEN-BINDING MOLECULE WITH A FcRn-BINDING DOMAIN THAT PROMOTES ANTIGEN CLEARANCE |
| WO2013125667A1 (ja) | 2012-02-24 | 2013-08-29 | 中外製薬株式会社 | FcγRIIBを介して抗原の消失を促進する抗原結合分子 |
| WO2013180200A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 標的組織特異的抗原結合分子 |
| WO2015046554A1 (ja) | 2013-09-30 | 2015-04-02 | 中外製薬株式会社 | 改変されたヘルパーファージを用いて抗原結合分子を作製する方法 |
| WO2015083764A1 (ja) * | 2013-12-04 | 2015-06-11 | 中外製薬株式会社 | 化合物の濃度に応じて抗原結合能の変化する抗原結合分子及びそのライブラリ |
| WO2015143414A2 (en) | 2014-03-21 | 2015-09-24 | Regeneron Pharmaceuticals, Inc. | Non-human animals that make single domain binding proteins |
| JP2018537473A (ja) * | 2015-12-03 | 2018-12-20 | アジオス ファーマシューティカルズ, インコーポレイテッド | Mtapヌル癌を処置するためのmat2a阻害剤 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110059101A9 (en) * | 2006-03-13 | 2011-03-10 | Ablynx N.V. | Amino Acid Sequences Directed Against Il-6 And Polypetides Comprising The Same For The Treatment Of Diseases And Disorders Associated With Il-6 Mediated Signalling |
-
2020
- 2020-03-19 EP EP20773939.2A patent/EP3943108A4/en active Pending
- 2020-03-19 CN CN202080021856.0A patent/CN113613676A/zh active Pending
- 2020-03-19 US US17/438,993 patent/US20220153875A1/en active Pending
- 2020-03-19 CN CN202511354577.7A patent/CN121159688A/zh active Pending
- 2020-03-19 WO PCT/JP2020/012189 patent/WO2020189748A1/ja not_active Ceased
- 2020-03-19 JP JP2021507411A patent/JP7601758B2/ja active Active
-
2024
- 2024-12-05 JP JP2024211901A patent/JP2025038000A/ja active Pending
Patent Citations (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4737456A (en) | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| EP0239400A2 (en) | 1986-03-27 | 1987-09-30 | Medical Research Council | Recombinant antibodies and methods for their production |
| US6248516B1 (en) | 1988-11-11 | 2001-06-19 | Medical Research Council | Single domain ligands, receptors comprising said ligands methods for their production, and use of said ligands and receptors |
| EP0404097A2 (de) | 1989-06-22 | 1990-12-27 | BEHRINGWERKE Aktiengesellschaft | Bispezifische und oligospezifische, mono- und oligovalente Rezeptoren, ihre Herstellung und Verwendung |
| US6417429B1 (en) | 1989-10-27 | 2002-07-09 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| US5959177A (en) | 1989-10-27 | 1999-09-28 | The Scripps Research Institute | Transgenic plants expressing assembled secretory antibodies |
| WO1992001047A1 (en) | 1990-07-10 | 1992-01-23 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992020791A1 (en) | 1990-07-10 | 1992-11-26 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1992003918A1 (en) | 1990-08-29 | 1992-03-19 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5648237A (en) | 1991-09-19 | 1997-07-15 | Genentech, Inc. | Expression of functional antibody fragments |
| WO1993006213A1 (en) | 1991-09-23 | 1993-04-01 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| WO1993011161A1 (en) | 1991-11-25 | 1993-06-10 | Enzon, Inc. | Multivalent antigen-binding proteins |
| WO1993011236A1 (en) | 1991-12-02 | 1993-06-10 | Medical Research Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| WO1993012227A1 (en) | 1991-12-17 | 1993-06-24 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| WO1993019172A1 (en) | 1992-03-24 | 1993-09-30 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1994002602A1 (en) | 1992-07-24 | 1994-02-03 | Cell Genesys, Inc. | Generation of xenogeneic antibodies |
| WO1994011523A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Fully impaired consensus kozac sequences for mammalian expression |
| WO1994025585A1 (en) | 1993-04-26 | 1994-11-10 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| WO1995001438A1 (en) | 1993-06-30 | 1995-01-12 | Medical Research Council | Sbp members with a chemical moiety covalently bound within the binding site; production and selection thereof |
| WO1995001937A1 (fr) | 1993-07-09 | 1995-01-19 | Association Gradient | Procede de traitement de residus de combustion et installation de mise en ×uvre dudit procede |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| WO1996000257A1 (en) | 1994-06-24 | 1996-01-04 | Imperial Chemical Industries Plc | Foaming method |
| WO1996002576A1 (en) | 1994-07-13 | 1996-02-01 | Chugai Seiyaku Kabushiki Kaisha | Reconstituted human antibody against human interleukin-8 |
| US5789199A (en) | 1994-11-03 | 1998-08-04 | Genentech, Inc. | Process for bacterial production of polypeptides |
| WO1996027011A1 (en) | 1995-03-01 | 1996-09-06 | Genentech, Inc. | A method for making heteromultimeric polypeptides |
| US5840523A (en) | 1995-03-01 | 1998-11-24 | Genetech, Inc. | Methods and compositions for secretion of heterologous polypeptides |
| WO1996034103A1 (en) | 1995-04-25 | 1996-10-31 | Vrije Universiteit Brussel | Variable fragments of immunoglobulins - use for therapeutic or veterinary purposes |
| WO1996033735A1 (en) | 1995-04-27 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1996034096A1 (en) | 1995-04-28 | 1996-10-31 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| WO1999023221A2 (en) | 1997-10-27 | 1999-05-14 | Unilever Plc | Multivalent antigen-binding proteins |
| WO1999054342A1 (en) | 1998-04-20 | 1999-10-28 | Pablo Umana | Glycosylation engineering of antibodies for improving antibody-dependent cellular cytotoxicity |
| US6040498A (en) | 1998-08-11 | 2000-03-21 | North Caroline State University | Genetically engineered duckweed |
| WO2000042072A2 (en) | 1999-01-15 | 2000-07-20 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2000061739A1 (fr) | 1999-04-09 | 2000-10-19 | Kyowa Hakko Kogyo Co., Ltd. | Methode de regulation de l'activite d'une molecule immunologiquement fonctionnelle |
| US6420548B1 (en) | 1999-10-04 | 2002-07-16 | Medicago Inc. | Method for regulating transcription of foreign genes |
| US7125978B1 (en) | 1999-10-04 | 2006-10-24 | Medicago Inc. | Promoter for regulating expression of foreign genes |
| WO2002020565A2 (en) | 2000-09-08 | 2002-03-14 | Universität Zürich | Collections of repeat proteins comprising repeat modules |
| WO2002031140A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions |
| WO2002032925A2 (en) | 2000-10-16 | 2002-04-25 | Phylos, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| WO2002060919A2 (en) | 2000-12-12 | 2002-08-08 | Medimmune, Inc. | Molecules with extended half-lives, compositions and uses thereof |
| WO2002079255A1 (en) | 2001-04-02 | 2002-10-10 | Idec Pharmaceuticals Corporation | RECOMBINANT ANTIBODIES COEXPRESSED WITH GnTIII |
| WO2003000883A1 (fr) | 2001-06-22 | 2003-01-03 | Chugai Seiyaku Kabushiki Kaisha | Inhibiteurs de proliferation cellulaire renfermant un anticorps anti-glypicane 3 |
| WO2003029462A1 (en) | 2001-09-27 | 2003-04-10 | Pieris Proteolab Ag | Muteins of human neutrophil gelatinase-associated lipocalin and related proteins |
| WO2003104453A1 (ja) | 2002-06-05 | 2003-12-18 | 中外製薬株式会社 | 抗体作製方法 |
| WO2004022754A1 (ja) | 2002-09-04 | 2004-03-18 | Chugai Seiyaku Kabushiki Kaisha | MRL/lprマウスを用いた抗体の作製 |
| WO2004029207A2 (en) | 2002-09-27 | 2004-04-08 | Xencor Inc. | Optimized fc variants and methods for their generation |
| WO2004035752A2 (en) | 2002-10-15 | 2004-04-29 | Protein Design Labs, Inc. | ALTERATION OF FcRn BINDING AFFINITIES OR SERUM HALF-LIVES OF ANTIBODIES BY MUTAGENESIS |
| WO2004044011A2 (en) | 2002-11-06 | 2004-05-27 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| WO2004065540A2 (en) | 2003-01-22 | 2004-08-05 | Glycart Biotechnology Ag | Fusion constructs and use of same to produce antibodies with increased fc receptor binding affinity and effector function |
| WO2004099249A2 (en) | 2003-05-02 | 2004-11-18 | Xencor, Inc. | Optimized fc variants and methods for their generation |
| WO2005040229A2 (en) | 2003-10-24 | 2005-05-06 | Avidia, Inc. | Ldl receptor class a and egf domain monomers and multimers |
| WO2005070963A1 (en) | 2004-01-12 | 2005-08-04 | Applied Molecular Evolution, Inc | Fc region variants |
| JP2007531724A (ja) * | 2004-04-05 | 2007-11-08 | クアーク・ファーマスーティカルス、インコーポレイテッド | 5’−デオキシ−5’−メチルチオアデノシンに対する抗体及びその使用 |
| WO2006006693A1 (ja) | 2004-07-09 | 2006-01-19 | Chugai Seiyaku Kabushiki Kaisha | 抗グリピカン3抗体 |
| WO2006020114A2 (en) | 2004-08-04 | 2006-02-23 | Applied Molecular Evolution, Inc. | Variant fc regions |
| WO2006023403A2 (en) | 2004-08-16 | 2006-03-02 | Medimmune, Inc. | Eph receptor fc variants with enhanced antibody dependent cell-mediated cytotoxicity activity |
| WO2006031370A2 (en) | 2004-08-19 | 2006-03-23 | Genentech, Inc. | Polypeptide variants with altered effector function |
| WO2007024249A2 (en) | 2004-11-10 | 2007-03-01 | Macrogenics, Inc. | Engineering fc antibody regions to confer effector function |
| WO2006067913A1 (ja) | 2004-12-22 | 2006-06-29 | Chugai Seiyaku Kabushiki Kaisha | フコーストランスポーターの機能が阻害された細胞を用いた抗体の作製方法 |
| WO2006105338A2 (en) | 2005-03-31 | 2006-10-05 | Xencor, Inc. | Fc VARIANTS WITH OPTIMIZED PROPERTIES |
| WO2006106905A1 (ja) | 2005-03-31 | 2006-10-12 | Chugai Seiyaku Kabushiki Kaisha | 会合制御によるポリペプチド製造方法 |
| WO2006116260A2 (en) | 2005-04-26 | 2006-11-02 | Medimmune, Inc. | Modulation of antibody effector function by hinge domain engineering |
| WO2007021841A2 (en) | 2005-08-10 | 2007-02-22 | Macrogenics, Inc. | Identification and engineering of antibodies with variant fc regions and methods of using same |
| WO2007041635A2 (en) | 2005-10-03 | 2007-04-12 | Xencor, Inc. | Fc variants with optimized fc receptor binding properties |
| WO2008016854A2 (en) | 2006-08-02 | 2008-02-07 | The Uab Research Foundation | Methods and compositions related to soluble monoclonal variable lymphocyte receptors of defined antigen specificity |
| WO2008092117A2 (en) | 2007-01-25 | 2008-07-31 | Xencor, Inc. | Immunoglobulins with modifications in the fcr binding region |
| US20090136485A1 (en) | 2007-05-30 | 2009-05-28 | Xencor, Inc. | Methods and compositions for inhibiting CD32B expressing cells |
| WO2009086320A1 (en) | 2007-12-26 | 2009-07-09 | Xencor, Inc | Fc variants with altered binding to fcrn |
| WO2009125825A1 (ja) | 2008-04-11 | 2009-10-15 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2009140242A1 (en) | 2008-05-13 | 2009-11-19 | Genentech, Inc. | Analysis of antibody drug conjugates by bead-based affinity capture and mass spectrometry |
| US20110123527A1 (en) | 2008-05-23 | 2011-05-26 | Hiroaki Shizuya | Method of generating single vl domain antibodies in transgenic animals |
| WO2011122011A2 (en) | 2010-03-30 | 2011-10-06 | Chugai Seiyaku Kabushiki Kaisha | Antibodies with modified affinity to fcrn that promote antigen clearance |
| WO2012073992A1 (ja) | 2010-11-30 | 2012-06-07 | 中外製薬株式会社 | 複数分子の抗原に繰り返し結合する抗原結合分子 |
| WO2012115241A1 (ja) | 2011-02-25 | 2012-08-30 | 中外製薬株式会社 | FcγRIIb特異的Fc抗体 |
| WO2012133782A1 (ja) | 2011-03-30 | 2012-10-04 | 中外製薬株式会社 | 抗原結合分子の血漿中滞留性と免疫原性を改変する方法 |
| WO2013002362A1 (ja) | 2011-06-30 | 2013-01-03 | 中外製薬株式会社 | ヘテロ二量化ポリペプチド |
| WO2013046704A2 (en) | 2011-09-30 | 2013-04-04 | Chugai Seiyaku Kabushiki Kaisha | THERAPEUTIC ANTIGEN-BINDING MOLECULE WITH A FcRn-BINDING DOMAIN THAT PROMOTES ANTIGEN CLEARANCE |
| WO2013125667A1 (ja) | 2012-02-24 | 2013-08-29 | 中外製薬株式会社 | FcγRIIBを介して抗原の消失を促進する抗原結合分子 |
| WO2013180200A1 (ja) | 2012-05-30 | 2013-12-05 | 中外製薬株式会社 | 標的組織特異的抗原結合分子 |
| WO2015046554A1 (ja) | 2013-09-30 | 2015-04-02 | 中外製薬株式会社 | 改変されたヘルパーファージを用いて抗原結合分子を作製する方法 |
| WO2015083764A1 (ja) * | 2013-12-04 | 2015-06-11 | 中外製薬株式会社 | 化合物の濃度に応じて抗原結合能の変化する抗原結合分子及びそのライブラリ |
| WO2015143414A2 (en) | 2014-03-21 | 2015-09-24 | Regeneron Pharmaceuticals, Inc. | Non-human animals that make single domain binding proteins |
| JP2018537473A (ja) * | 2015-12-03 | 2018-12-20 | アジオス ファーマシューティカルズ, インコーポレイテッド | Mtapヌル癌を処置するためのmat2a阻害剤 |
Non-Patent Citations (181)
| Title |
|---|
| ACTA CRYST, vol. D66, 2010, pages 486 - 501 |
| ACTA CRYST, vol. D67, 2011, pages 293 - 302 |
| AHRAM ET AL., MOL. CARCINOG., vol. 33, 2002, pages 9 - 15 |
| AIDS, vol. 30, no. 11, 2016, pages 1691 - 1701 |
| AL-LAZIKANI ET AL., J. MOL. BIOL., vol. 273, 1997, pages 927 - 948 |
| ALLEY SCOKELEY NMSENTER PD.: "Antibody-drug conjugates: targeted drug delivery for cancer", CURR. OPIN. CHEM. BIOL., vol. 14, no. 4, 2010, pages 529 - 537, XP055042125, DOI: 10.1016/j.cbpa.2010.06.170 |
| AM. J. RESPIR. CRIT. CARE MED., vol. 182, 2010, pages 774 - 783 |
| AM. J. VET. RES., vol. 65, 2004, pages 1269 - 1275 |
| ANAL. BIOCHEM., vol. 162, no. 1, 1987, pages 156 - 159 |
| ANNU. REV. BIOPHYS. BIOMOL. STRUCT., vol. 35, 2006, pages 225 - 249 |
| BAEUERLE PAKUFER PBARGOU R.: "BiTE: Teaching antibodies to engage T-cells for cancer therapy", CURR. OPIN. MOL. THER., vol. 11, no. 1, 2009, pages 22 - 30, XP009151509 |
| BARDOT ET AL., BR. J. CANCER, vol. 70, 1994, pages 212 - 218 |
| BARRETINA J ET AL., NATURE, vol. 483, 2012, pages 603 - 7 |
| BBRC, vol. 436, no. 3, 2013, pages 543 - 50 |
| BETTER, M.HORWITZ, A. H., METHODS ENZYMOL., vol. 178, 1989, pages 497 - 515 |
| BIGGE ET AL., ANAL. BIOCHEM., vol. 230, no. 2, 1995, pages 229 - 238 |
| BIO/TECHNOLOGY, vol. 12, no. 7, 1994, pages 699 - 702 |
| BIOCHEMISTRY, vol. 18, no. 24, 1979, pages 5294 - 5299 |
| BIOCHIMICA ET BIOPHYSICA ACTA-PROTEINS AND PROTEOMICS, vol. 1764, no. 8, 2006, pages 1307 - 1319 |
| BIRD ET AL., TIBTECH, vol. 9, 1991, pages 132 - 137 |
| BIRD, R. E.WALKER, B. W., BIOTECHNOL, vol. 9, 1991, pages 132 - 137 |
| BLAYHOSKIN, CANCER RES., vol. 57, 1997, pages 2602 - 2605 |
| BMC BIOTECHNOL, vol. 9, 29 January 2009 (2009-01-29), pages 6 |
| BOLOGNESI ET AL., CLIN. EXP. IMMUNOL., vol. 89, 1992, pages 341 - 346 |
| BRENSING-KUPPERS ET AL., GENE, vol. 191, 1997, pages 173 - 181 |
| BRINDLE ET AL., J. MOL. RECOGNIT., vol. 10, 1997, pages 182 - 187 |
| BURMEISTER ET AL., NATURE, vol. 372, 1994, pages 336 - 343 |
| C VINCKE ET AL., THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 284, pages 3273 - 3284 |
| C. EUR. J. IMMUNOL., vol. 6, no. 7, 1976, pages 511 - 519 |
| CANBOLAT ET AL., BREAST CANCER RES. TREAT., vol. 37, 1996, pages 189 - 193 |
| CANCER RES., vol. 53, 1993, pages 851 - 856 |
| CARDOSO ET AL., SCAND. J. IMMUNOL., vol. 51, 2000, pages 337 - 344 |
| CASELLAS ET AL., EUR. J. BIOCHEM., vol. 176, 1988, pages 581 - 588 |
| CELL, vol. 8, no. 3, 1976, pages 405 - 415 |
| CHOTHIA AND LESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| CHRISTOFK ET AL., NATURE, vol. 452, 2008, pages 230 - 233 |
| CLARKSON ET AL., NATURE, vol. 352, 1991, pages 624 - 628 |
| CLIN. EXP. RHEUMATOL., vol. 17, 1999, pages 151 - 160 |
| CO, M. S. ET AL., J. IMMUNOL., vol. 152, no. 10, 1994, pages 2968 - 2976 |
| CONRATH ET AL., J. BIOL. CHEM., vol. 276, no. 10, 2001, pages 7346 - 7350 |
| COX ET AL., NAT. GENETICS, vol. 7, 1994, pages 162 - 168 |
| CUMBER ET AL., J. IMMUNOL. METHODS, vol. 135, 1990, pages 15 - 24 |
| CUR. OPIN. IN BIOTECH., vol. 20, no. 6, 2009, pages 685 - 691 |
| CURR OPIN CHEM BIOL, vol. 14, 2010, pages 529 - 37 |
| CURR. MED. CHEM., vol. 18, 2011, pages 5217 - 5223 |
| CURR. OPIN. BIOTECHNOL., vol. 20, no. 6, 2009, pages 685 - 91 |
| CURR. OPIN. IMMUNOL., vol. 20, no. 4, 2008, pages 460 - 470 |
| CURRENT PROTOCOLS IN PROTEIN SCIENCE, May 2001 (2001-05-01) |
| CURRENT TOPICS IN MICROBIOLOGY AND IMMUNOLOGY, vol. 81, 1978, pages 1 - 7 |
| DANGSEMENZA, TRENDS BIOCHEM. SCI., vol. 24, 1999, pages 68 - 72 |
| DENKER ET AL., MOD. PATHOL., vol. 19, 2006, pages 1261 - 1269 |
| DENKERT ET AL., CLIN. BREAST CANCER, vol. 4, 2004, pages 428 - 433 |
| DESJARLAIS JRLAZAR GAZHUKOVSKY EACHU SY.: "Optimizing engagement of the immune system by anti-tumor antibodies: an engineer's perspective", DRUG DISCOV. TODAY, vol. 12, 2007, pages 898 - 910, XP022338218, DOI: 10.1016/j.drudis.2007.08.009 |
| DESMOULIN F ET AL., DRUG METAB DISPOS, 2002 |
| DHANASEKARAN ET AL., NATURE, vol. 412, 2001, pages 822 - 826 |
| DROGE, IMMUNOBIOLOGY, vol. 174, 1987, pages 473 - 479 |
| DUAN JX ET AL., J MED CHEM., 2008 |
| DURAK ET AL., CANCER LETT., vol. 84, 1994, pages 199 - 202 |
| ENOMOTO K. ET AL.: "Development of high-throughput spermidine synthase activity assay using homogeneous time-resolved fluorescence", ANALYTICAL BIOCHEMISTRY, vol. 351, 2006, pages 229 - 240, XP024942404, DOI: 10.1016/j.ab.2006.01.012 * |
| EPITOPE MAPPING PROTOCOLS IN METHODS IN MOLECULAR BIOLOGY, vol. 66, 1996 |
| EUR. J. CLIN. PHARMACOL., vol. 46, 1994, pages 3 - 7 |
| EVA GOTTFRIEDKATRIN PETERMARINA P. KREUTZ, FROM MOLECULAR TO MODULAR TUMOR THERAPY, vol. 3, no. 2, 2010, pages 111 - 132 |
| FANTIN ET AL., CANCER CELL, vol. 9, 2006, pages 425 - 434 |
| FASEB J., vol. 22, 2008, pages 2263 - 2272 |
| FLOCKEMANNHERZ, BIOCHIM. BIOPHYS. ACTA, vol. 1076, 1991, pages 273 - 281 |
| FRUMENTO ET AL., J. EXP. MED., vol. 196, 2002, pages 459 - 468 |
| FULTON ET AL., J. BIOL. CHEM., vol. 261, 1986, pages 5314 - 5319 |
| GATESSWEELEY, CLIN. CHEM., vol. 24, 1978, pages 1663 - 1673 |
| GEJIMA ET AL., HUMAN ANTIBODIES, vol. 11, 2002, pages 121 - 129 |
| GERNGROSS, NAT. BIOTECH., vol. 22, 2004, pages 1409 - 1414 |
| GHEEITE ET AL., J. IMMUNOL. METHODS, vol. 142, 1991, pages 223 - 230 |
| GONZALEZ-MUNOZ A ET AL., MABS, 2012 |
| GRAHAM ET AL., J. GEN VIROL., vol. 36, 1977, pages 59 |
| HEADRICKWILLIS, BIOCHEM. J., vol. 261, 1989, pages 541 - 550 |
| HEINRICH ET AL., BIOCHEM. J., vol. 334, 1998, pages 297 - 314 |
| HOLLIGER ET AL., PROTEIN ENGINEERING, vol. 9, no. 3, 1996, pages 299 - 305 |
| HOLLINGER ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6444 - 6448 |
| HOOD ET AL., MOL. CELL. PROTEOMICS, vol. 4, 2005, pages 1741 - 1753 |
| HUDSON ET AL., J. IMMUNOL. METHODS, vol. 231, 1999, pages 177 - 189 |
| HUSTON ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 85, no. 23, 1988, pages 8998 - 9002 |
| IMMUNITY, vol. 23, 2005, pages 503 - 514 |
| ISHIKAWA ET AL., J. CLIN. EXP. HEMATOPATHOL., vol. 46, no. 2, 2006, pages 55 - 66 |
| J. AM. SOC. NEPHROL., vol. 22, no. 5, 2011, pages 890 - 901 |
| J. APPL. CRYST., vol. 40, 2007, pages 658 - 674 |
| J. APPL. CRYST., vol. 43, 2010, pages 186 - 190 |
| J. APPL. PHYS., vol. 248, 2003, pages L798 - L800 |
| J. CLIN. INVEST., vol. 120, no. 6, 2010, pages 1939 - 1949 |
| J. EXP. MED., vol. 148, no. 1, 1978, pages 313 - 323 |
| J. IMMUNOL. METHODS, vol. 35, no. 1-2, 1980, pages 1 - 21 |
| J. IMMUNOL. METHODS., 28 September 2003 (2003-09-28), pages 139 - 55 |
| J. IMMUNOL., vol. 123, no. 4, 1979, pages 1548 - 1550 |
| J. IMMUNOL., vol. 181, 2008, pages 5396 - 5404 |
| J. LAB. CLIN. MED., vol. 122, 1993, pages 518 - 523 |
| JANICE M REICHERTCLARK J ROSENSWEIGLAURA B FADENMATTHEW C DEWITZ: "Monoclonal antibody successes in the clinic", NAT. BIOTECHNOL., vol. 23, 2005, pages 1073 - 1078, XP002555770, DOI: 10.1038/nbt0905-1073 |
| JOURNAL OF APPLIED MICROBIOLOGY, vol. 117, no. 2, 2014, pages 528 - 536 |
| JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 291, no. 24, 2016, pages 12641 - 12657 |
| JOURNAL OF BIOMOLECULAR SCREENING, vol. 21, no. 1, 2016, pages 35 - 43 |
| JOURNAL OF PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, vol. 36, 2004, pages 877 - 882 |
| KATHARINA LIMM, WALLNER SUSANNE, MILENKOVIC VLADIMIR M., WETZEL CHRISTIAN H., BOSSERHOFF ANJA-KATRIN: "The metabolite 5'-methylthioadenosine signals through the adenosine receptor A2B in melanoma", EUROPEAN JOURNAL OF CANCER, vol. 50, no. 15, 30 July 2014 (2014-07-30) - October 2014 (2014-10-01), pages 2714 - 2724, XP055741192 * |
| KAWASAKI ET AL., EUR. J. IMMUNOL., vol. 31, 2001, pages 1017 - 1028 |
| KAWASAKI ET AL., GENOME RES, vol. 7, 1997, pages 250 - 261 |
| KIM SJPARK YHONG HJ.: "Antibody engineering for the development of therapeutic antibodies", MOL. CELLS., vol. 20, no. 1, 2005, pages 17 - 29 |
| KNAPPIK A. ET AL., J MOL BIOL., 2000 |
| KOBIE ET AL., J. IMMUNOL., vol. 176, 2006, pages 4449 - 4458 |
| KOHLERMILSTEIN ET AL., METHODS ENZYMOL., vol. 73, 1981, pages 3 - 46 |
| KUNKEL ET AL., PROC. NATL. ACAD. SCI. USA, vol. 82, 1985, pages 488 - 492 |
| LANGONE ET AL., METHODS IN ENZYMOLOGY, vol. 93, 1983, pages 307 - 308 |
| LAPOINTE ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 101, 2004, pages 811 - 816 |
| LAWTON ET AL., PHARMACOGENOMICS, vol. 9, 2008, pages 383 |
| LEE CV ET AL., J MOL BIOL., 2004 |
| LEWIS GDFIGARI IFENDLY BWONG WLCARTER PGORMAN CSHEPARD HM: "Differential responses of human tumor cell lines to anti-pl85HER2 monoclonal antibodies", CANCER IMMUNOL. IMMUNOTHERAPY, vol. 37, 1993, pages 255 - 263, XP002961547, DOI: 10.1007/BF01518520 |
| LI ET AL., NAT. BIOTECH., vol. 24, 2006, pages 210 - 215 |
| LOB ET AL., CANCER IMMUNOL. IMMUNOTHER., vol. 58, 2009, pages 153 - 157 |
| LUTTERBUESE RRAUM TKISCHEL RHOFFMANN PMANGOLD SRATTEL BFRIEDRICH MTHOMAS OLORENCZEWSKI GRAU D: "T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells", PROC. NATL. ACAD. SCI. U.S.A., vol. 107, no. 28, 2010, pages 12605 - 12610, XP055067703, DOI: 10.1073/pnas.1000976107 |
| MACCALLUM ET AL., J. MOL. BIOL., vol. 262, 1996, pages 732 - 745 |
| MATHER ET AL., ANNALS N.Y. ACAD. SCI., vol. 383, 1982, pages 44 - 68 |
| MATHER, BIOL. REPROD., vol. 23, 1980, pages 243 - 251 |
| MATSUDA ET AL., J. EXP. MED., vol. 188, 1998, pages 1973 - 1975 |
| MAZUREK ET AL., J. CELL. PHYSIOL., vol. 181, 1999, pages 136 - 146 |
| MAZUREKEIGENBRODT, ANTICANCER RES, vol. 23, 2003, pages 1149 - 1154 |
| MERCHANT ET AL., NATURE BIOTECHNOLOGY, vol. 16, 1998, pages 677 - 681 |
| METHODS IN MOLECULAR BIOLOGY, vol. 911, 2012, pages 65 - 78 |
| METHODS MOL. BIOL., vol. 178, 2002, pages 133 - 145 |
| MILSTEIN ET AL., NATURE, vol. 305, 1983, pages 537 - 540 |
| MUYLDERMANS, REV. MOL. BIOTECH., vol. 74, 2001, pages 277 - 302 |
| NAT. MED., vol. 13, 2007, pages 851 - 856 |
| NAT. MED., vol. 16, 2010, pages 1434 - 1438 |
| NAT. REV. CANCER, vol. 12, no. 11, 2012, pages 782 - 792 |
| NATURE IMMUNOLOGY, vol. 9, 2008, pages 1261 - 1269 |
| NATURE MEDICINE, vol. 2, 1996, pages 350 - 353 |
| NATURE, vol. 276, no. 5685, 1978, pages 269 - 270 |
| NATURE, vol. 277, no. 5692, 1979, pages 131 - 133 |
| NUCLEIC ACIDS RES., vol. 17, no. 8, 1989, pages 2919 - 2932 |
| OPITZ ET AL., NATURE, vol. 478, no. 7368, 2011, pages 197 - 203 |
| PAVLOU AKBELSEY MJ.: "The therapeutic antibodies market to 2008", EUR. J. PHARM. BIOPHARM., vol. 59, no. 3, 2005, pages 389 - 396, XP025317626, DOI: 10.1016/j.ejpb.2004.11.007 |
| PEROU ET AL., NATURE, vol. 406, 2000, pages 747 - 752 |
| PLOS ONE J, vol. 5, no. 2, 2010, pages e9242 |
| PLOS ONE, vol. 3, 2008, pages e2599 |
| PLUECKTHUN AND SKERRA, METHODS IN ENZYMOLOGY, vol. 121, 1989, pages 663 - 669 |
| PORTOLANO ET AL., J. IMMUNOL., vol. 150, 1993, pages 880 - 887 |
| PROC NATL ACAD SCI USA., vol. 97, no. 20, 26 September 2000 (2000-09-26), pages 10701 - 5 |
| PROC. NATL. ACAD. SCI. U.S.A., vol. 100, no. 11, 2003, pages 6353 - 6357 |
| PROC. NATL. ACAD. SCI. U.S.A., vol. 103, no. 11, 2006, pages 4005 - 4010 |
| PROC. NATL. ACAD. SCI., vol. 103, no. 35, 2006, pages 13132 - 13137 |
| PROTEIN ENG. DES. SEL., vol. 23, no. 4, 2010, pages 195 - 202 |
| PROTEIN SCIENCE, vol. 4, 1995, pages 2411 - 2423 |
| RAGHAVAN ET AL., IMMUNITY, vol. 1, 1994, pages 303 - 315 |
| REINOSO RF ET AL., DRUG METAB DISPOS, vol. 29, April 2001 (2001-04-01), pages 453 - 9 |
| RESTATHOMPSON, IMMUNOL. REV., vol. 161, 1998, pages 95 - 109 |
| RIECHELMANN HSAUTER AGOLZE WHANFT GSCHROEN CHOERMANN KERHARDT TGRONAU S: "Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma", ORAL ONCOL, vol. 44, no. 9, 2008, pages 823 - 829, XP024341160, DOI: 10.1016/j.oraloncology.2007.10.009 |
| ROUSSEAUX, J. ET AL., METHODS ENZYMOL., vol. 121, 1986, pages 663 - 669 |
| SADEJ, MELANOMA RES, vol. 16, 2006, pages 213 - 222 |
| SATOH MIIDA SSHITARA K., EXPERT OPIN. BIOL. THER., vol. 6, no. 11, 2006, pages 1161 - 1173 |
| SATOH MIIDA SSHITARA K.: "Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies", EXPERT OPIN. BIOL. THER., vol. 6, no. 11, 2006, pages 1161 - 1173, XP008078583, DOI: 10.1517/14712598.6.11.1161 |
| SCHABLEZACHAU, BIOL. CHEM. HOPPE SEYLER, vol. 374, 1993, pages 1001 - 1022 |
| SCHENK ET AL., J. CLIN. INVESTIGATION, vol. 108, no. 11, 2001, pages 1687 - 1695 |
| See also references of EP3943108A4 |
| SHENG ET AL., CANCER RES., vol. 58, 1998, pages 362 - 366 |
| STEVENS A. P. ET AL.: "Quantitative analysis of 5'-deoxy-5'- methylthioadenosine in melanoma cells by liquid chromatography-stable isotope ratio tandem mass spectrometry", JOURNAL OF CHROMATOGRAPHY B, vol. 876, 2008, pages 123 - 128, XP025681507, DOI: 10.1016/j.jchromb.2008.10.038 * |
| STEVENS ET AL., J CHROMATOGR A., vol. 1217, no. 19, 7 May 2010 (2010-05-07), pages 3282 - 8 |
| STIRPE F.BARBIERI L., FEBS LETTER, vol. 195, 1986, pages 1 - 8 |
| TERESA ET AL., MOL. CANCER, vol. 8, 2009, pages 41 - 59 |
| THORPE ET AL., CANCER RES., vol. 47, 1987, pages 3169 - 3173 |
| TILLER T ET AL., MABS, 2013 |
| TOMLINSON ET AL., J. MOL. BIOL., vol. 227, 1992, pages 799 - 817 |
| TRINH VAHWU WJ.: "Ipilimumab in the treatment of melanoma", EXPERT OPIN. BIOL. THER., 2012 |
| ULANOVSKAYA ET AL., CHEM. BIOL., vol. 9, no. 5, 2013, pages 300 - 306 |
| URLAUB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 77, 1980, pages 4216 |
| UYTTENHOVE ET AL., NAT. MED., vol. 9, 2003, pages 1269 - 127 |
| VANDAMME ET AL., EUR. J. BIOCHEM., vol. 192, no. 3, 1990, pages 767 - 775 |
| VAUPEL ET AL., CANCER RES., vol. 49, 1989, pages 6449 - 6665 |
| VIZAN ET AL., CANCER RES., vol. 65, 2005, pages 5512 - 5515 |
| WARNERMITCHELL, FASEB J, vol. 18, 2004, pages 790 - 804 |
| WAWRZYNCZAK ET AL., BR. J. CANCER, vol. 66, 1992, pages 361 - 366 |
| WAWRZYNCZAK ET AL., CANCER RES., vol. 50, 1990, pages 7519 - 7562 |
| WEINER LMSURANA RWANG S.: "Monoclonal antibodies: versatile platforms for cancer immunotherapy", NAT. REV. IMMUNOL., vol. 10, no. 5, 2010, pages 317 - 327, XP055217481, DOI: 10.1038/nri2744 |
| WEITZHANDLER ET AL., J. PHARMA. SCIENCES, vol. 83, no. 12, 1994, pages 1670 - 1675 |
| WILLIAMSWINTER, EUR. J. IMMUNOL., vol. 23, 1993, pages 1456 - 1461 |
| WU ET AL., J. EXP. MED., vol. 132, 1970, pages 211 - 250 |
| YAMADA ET AL., J. NUTR. SCI. VITAMINOL., vol. 56, 2010, pages 83 - 86 |
| YU ET AL., J. IMMUNOL., vol. 190, 2013, pages 3783 - 3797 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11673947B2 (en) | 2012-05-30 | 2023-06-13 | Chugai Seiyaku Kabushiki Kaisha | Target tissue-specific antigen-binding molecule |
| US11912989B2 (en) | 2013-12-04 | 2024-02-27 | Chugai Seiyaku Kabushiki Kaisha | Antigen-binding molecules, the antigen-binding activity of which varies according to the concentration of compounds, and libraries of said molecules |
| US12304960B2 (en) | 2018-08-10 | 2025-05-20 | Chugai Seiyaku Kabushiki Kaisha | Anti-CD137 antigen-binding molecule and utilization thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2020189748A1 (ja) | 2020-09-24 |
| EP3943108A4 (en) | 2023-01-04 |
| US20220153875A1 (en) | 2022-05-19 |
| JP2025038000A (ja) | 2025-03-18 |
| CN121159688A (zh) | 2025-12-19 |
| JP7601758B2 (ja) | 2024-12-17 |
| CN113613676A (zh) | 2021-11-05 |
| EP3943108A1 (en) | 2022-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7488843B2 (ja) | 化合物の濃度に応じて抗原結合能の変化する抗原結合分子及びそのライブラリ | |
| JP6931034B2 (ja) | 標的組織特異的抗原結合分子 | |
| JP7601758B2 (ja) | Mta依存的に抗原に対する結合活性が変化する抗原結合ドメインを含む抗原結合分子及び当該抗原結合ドメイン取得用ライブラリ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20773939 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021507411 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020773939 Country of ref document: EP Effective date: 20211019 |