WO2020204172A1 - 水溶性アジュバント - Google Patents
水溶性アジュバント Download PDFInfo
- Publication number
- WO2020204172A1 WO2020204172A1 PCT/JP2020/015359 JP2020015359W WO2020204172A1 WO 2020204172 A1 WO2020204172 A1 WO 2020204172A1 JP 2020015359 W JP2020015359 W JP 2020015359W WO 2020204172 A1 WO2020204172 A1 WO 2020204172A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- compound
- alkyl
- pharmaceutically acceptable
- vaccine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 Cc1c(Cc2c(*)cc(*)cc2)c(NC(*)*)nc(N)n1 Chemical compound Cc1c(Cc2c(*)cc(*)cc2)c(NC(*)*)nc(N)n1 0.000 description 4
- QMVQLEXPIXHOLU-UHFFFAOYSA-N CCCCCNc1nc(N)nc(C)c1Cc(cc(C=O)cc1)c1OC Chemical compound CCCCCNc1nc(N)nc(C)c1Cc(cc(C=O)cc1)c1OC QMVQLEXPIXHOLU-UHFFFAOYSA-N 0.000 description 1
- USVNKARHEBVFPD-UHFFFAOYSA-N CCCCCNc1nc(N)nc(C)c1Cc(cc(CNC)cc1)c1OC Chemical compound CCCCCNc1nc(N)nc(C)c1Cc(cc(CNC)cc1)c1OC USVNKARHEBVFPD-UHFFFAOYSA-N 0.000 description 1
- KYJMOHBCGHZCAF-UHFFFAOYSA-N CCCCCNc1nc(N)nc(C)c1Cc(ccc(COCCOCCOCCOCCOC)c1)c1OC Chemical compound CCCCCNc1nc(N)nc(C)c1Cc(ccc(COCCOCCOCCOCCOC)c1)c1OC KYJMOHBCGHZCAF-UHFFFAOYSA-N 0.000 description 1
Images


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/001152—Transcription factors, e.g. SOX or c-MYC
- A61K39/001153—Wilms tumor 1 [WT1]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
- C07D239/49—Two nitrogen atoms with an aralkyl radical, or substituted aralkyl radical, attached in position 5, e.g. trimethoprim
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55566—Emulsions, e.g. Freund's adjuvant, MF59
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/572—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 cytotoxic response
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/28—Steroids, e.g. cholesterol, bile acids or glycyrrhetinic acid
Definitions
- the present invention relates to a compound useful as a vaccine adjuvant for a vaccine (cancer vaccine or infectious disease vaccine), a method for producing the same, a pharmaceutical composition containing the compound, and a vaccine adjuvant for a vaccine (cancer vaccine or infectious disease vaccine) of the compound. It is about use as.
- Cancer vaccine therapy generally aims to treat cancer by activating tumor-specific immune cells using proteins and peptides derived from tumor antigens.
- the therapy using a tumor antigen peptide as an antigen is called a cancer peptide vaccine therapy.
- tumor antigen peptides alone generally have low immunogenicity
- vaccine adjuvants are used in combination for the purpose of inducing cytotoxic T cells (CTL), which are important for antitumor immunity.
- CTL cytotoxic T cells
- the aqueous phase of the W / O emulsion is the internal phase and it is easy to retain the peptide as an antigen in the internal phase, it is possible to show efficient induction of CTL by using the W / O emulsion as a vaccine adjuvant. It has been reported (Patent Document 1).
- Examples of the W / O emulsion used as a vaccine adjuvant for tumor antigen peptides include emulsion compositions for dilution (Patent Document 1), Incomplete Freund's Adjuvant (IFA), and Montanide (registered trademark) (Non-Patent Documents). 1, 2). Furthermore, Complete Freund's Adjuvant (CFA), which is a W / O emulsion supplemented with inactivated Mycobacterium Tuberculosis, is known. However, CFA has not been approved for human administration due to its high toxicity (Non-Patent Document 2).
- CFA Complete Freund's Adjuvant
- TLR7 Toll-like receptor 7
- Non-Patent Document 3 It is known that some easy-to-manufacture low-molecular-weight compounds act as ligands for TLR7, and it has been reported that compounds having a pyrimidine skeleton act as TLR7 agonists in addition to the marketing agent imiquimod. (Patent Document 2).
- the TLR7 agonist has been improved, and a compound having physical properties suitable for an adjuvant is being searched for.
- TLR7 agonists having a structure in which a phosphate group is compounded so as to bind to insoluble metal particles such as Alum adjuvant Patent Document 3, Non-Patent Documents 4 and 5.
- An object of the present invention is to provide a complex TLR7 agonist that enhances adjuvant activity.
- a TLR7 agonist that enhances adjuvant activity As a result, it was found that the TLR7 agonist having a pyrimidine skeleton and polyethylene glycol (PEG) were combined to impart water solubility, and the TLR7 agonist had excellent adjuvant activity, and the present invention was completed.
- a pyrimidine derivative represented by the following formula (1) hereinafter, may be referred to as “compound of the present invention”.
- the present invention is as follows.
- Equation (1) [In the formula, X represents methylene, oxygen atom, sulfur atom, SO, SO 2 , or NR 5 (R 5 represents hydrogen atom or C 1-6 alkyl).
- R 1 is an alkyl of C 1-6 (the alkyl may be substituted with 1-5 substituents independently selected from the group consisting of halogen, hydroxy, and C 1-6 alkoxy).
- R 2 and R 3 are each independently selected from a hydrogen atom or a C 1-6 alkyl (the alkyl is independently selected from the group consisting of halogen, hydroxy, C 1-6 alkoxy).
- R 4 is a hydrogen atom, halogen, hydroxy, C 1-6 alkyl (the alkyl may be substituted with 1 to 3 identical or different halogens), C 1-6 alkoxy (the alkoxy is 1).
- L represents a linker
- Y 1 represents ⁇ (CH 2 CH 2 O) m ⁇ R 6 (R 6 represents a hydrogen atom or C 1-6 alkyl, and m represents an integer from 3 to 100).
- Y 1 represents ⁇ (CH 2 CH 2 O) m ⁇ R 6 (R 6 represents a hydrogen atom or C 1-6 alkyl, and m represents an integer from 3 to 100).
- Item 2 The compound according to Item 1, or a pharmaceutically acceptable salt thereof, wherein X is methylene.
- Item 2 The compound according to Item 1 or 2, wherein R 1 is C 1-3 alkyl (the alkyl may be substituted with 1 to 3 identical or different halogens) or pharmaceutically acceptable thereof. Salt.
- Item 3 The compound according to Item 3 or a pharmaceutically acceptable salt thereof, wherein R 1 is methyl.
- R 4 is a hydrogen atom, hydroxy, C 1-3 alkyl or C 1-3 alkoxy, which is a compound or a pharmaceutically acceptable according to any one of claims 1-4.
- Item 5 The compound according to Item 5, or a pharmaceutically acceptable salt thereof, wherein R 4 is a hydrogen atom, hydroxy, or methoxy.
- Salts R 2 is C 1-6 alkyl, which is a compound or a pharmaceutically acceptable according to any one of claims 1-6.
- Item 6 The compound according to any one of Items 1 to 7, wherein R 3 is a hydrogen atom or C 1-3 alkyl (the alkyl may be substituted with 1 to 3 hydroxy groups) or a compound thereof. Pharmaceutically acceptable salt.
- L is -O-, -NR Y- , -C (O)-, -C (O) O-, -OC (O)-, -C (O) NR Y- , -NR YC (O) -, -CH 2 NR Y- , -CH 2 O-, -OC (O) O-, -NR 7 C (O) O-, -OC (O) NR Y- , -NR 7 C (O) NR Y- , -OC (S) NR Y- , or -NR 7 C (S) NR Y- (where R 7 represents a hydrogen atom or an alkyl having 1 to 6 carbon atoms, and RY is a hydrogen atom.
- Item 9 The compound according to Item 9 or pharmaceutically acceptable thereof, wherein L is -C (O) NR Y- , -CH 2 NR Y- , -C (O) O- or -CH 2 O-. salt.
- Item 9 The compound according to Item 9, or a pharmaceutically acceptable salt thereof, wherein L is -C (O) NR Y- or -CH 2 NR Y- .
- Item 9 The compound according to Item 9, or a pharmaceutically acceptable salt thereof, wherein L is -CH 2 NR Y- and RY is a hydrogen atom, an alkyl having 1 to 6 carbon atoms or Y 2 .
- Item 9 The compound according to Item 9, or a pharmaceutically acceptable salt thereof, wherein L is -CH 2 NR Y- and RY is a hydrogen atom or an alkyl having 1 to 6 carbon atoms.
- Item 15 Item 24. Item 14, wherein Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6 , R 6 is a hydrogen atom or C 1-6 alkyl, and m is an integer of 3 to 20. A compound or a pharmaceutically acceptable salt thereof.
- Equation (2) Or equation (3): [In the formula, R 2 is C 1-6 alkyl, R 3 is a hydrogen atom, or C 1-3 alkyl, which may be substituted with 1 to 3 hydroxys. R 4 is hydrogen atom, hydroxy or methoxy, L is -CH 2 NR Y- , -C (O) NR Y- , -C (O) O- or -CH 2 O-.
- RY is a hydrogen atom, C 1-6 alkyl or Y 2 and Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6 and Y 2 is ⁇ (CH 2 CH 2 O) n ⁇ R 8 and R 6 is a hydrogen atom or C 1-6 alkyl and is R 8 is a hydrogen atom or C 1-6 alkyl and is m and n are independently integers of 3 to 40.
- Item 2 The compound according to Item 1 or a pharmaceutically acceptable salt thereof.
- Equation (2) Or equation (3): [In the formula, R 2 is C 1-6 alkyl, R 3 is a hydrogen atom, or C 1-3 alkyl, which may be substituted with 1 to 3 hydroxys. R 4 is hydrogen atom, hydroxy or methoxy, L is -CH 2 NR Y- RY is a hydrogen atom, or C 1-6 alkyl, Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6 and R 6 is a hydrogen atom or C 1-6 alkyl and is m is an integer of 3 to 20. ] Item 2. The compound according to Item 1 or a pharmaceutically acceptable salt thereof.
- Equation (2) [In the formula, R 2 is C 1-6 alkyl, R 3 is a hydrogen atom, or C 1-3 alkyl, which alkyl may be substituted with one hydroxy.
- R 4 is hydrogen atom or methoxy
- L is -CH 2 NR Y- RY is a hydrogen atom, or C 1-6 alkyl
- Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6
- R 6 is a hydrogen atom or C 1-6 alkyl and is m is an integer of 3 to 40.
- Item 2 The compound according to Item 1 or a pharmaceutically acceptable salt thereof.
- Item 19 The compound according to Item 1 or a pharmaceutically acceptable salt thereof selected from the following compound group: 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14-tetraoxa- 2-Azahexadecane-16-all (Example 1), 1- ⁇ 4-[(2-Amino-4- ⁇ [(3S) -1-hydroxyhexane-3-yl] amino ⁇ -6-methylpyrimidine-5-yl) methyl] -3-methoxyphenyl ⁇ -2 -Methyl-5,8,11,14-Tetraoxa-2-azahexadecane-16-ol (Example 2), 1-(3- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -4-methoxyphenyl)
- Item 20 The compound according to Item 1 or a pharmaceutically acceptable salt thereof selected from the following compound group: 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14-tetraoxa- 2-Azahexadecane-16-all (Example 1), 1- ⁇ 4-[(2-Amino-4- ⁇ [(3S) -1-hydroxyhexane-3-yl] amino ⁇ -6-methylpyrimidine-5-yl) methyl] -3-methoxyphenyl ⁇ -2 -Methyl-5,8,11,14-Tetraoxa-2-azahexadecane-16-ol (Example 2), 1-(3- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -4-methoxyphenyl)
- Item 22 The pharmaceutical composition according to Item 21, which is an emulsion preparation, an oil-based suspension, a hydrogel preparation, or a lipid preparation.
- Item 23 The pharmaceutical composition according to Item 21, which is an emulsion preparation.
- Item 23 The pharmaceutical composition according to Item 23, wherein the emulsion preparation is a water-in-oil emulsion.
- Emulsion formulations include (1) ethyl oleate, octyldodecyl myristate, sorbitan monooleate, glycerin monooleate, polyoxyethylene hydrogenated castor oil 20, glycerin, and sodium dihydrogen phosphate, or (2) Montanede ISA 51VG Item 24.
- Item 26 The pharmaceutical composition according to Item 21, which is a lipid preparation.
- Item 28 The pharmaceutical composition according to Item 26, wherein the lipid preparation is a liposome preparation containing a phospholipid.
- Item 6 The pharmaceutical composition according to Item 26 or 27, wherein the lipid preparation is a liposome preparation containing sterols.
- Item 28 The pharmaceutical composition according to Item 28, wherein the sterols are cholesterol.
- the liposome preparation is a liposome preparation containing one or more additives selected from the group consisting of inorganic acids, inorganic acid salts, organic acids, organic acid salts, sugars, buffers, antioxidants, and polymers.
- the pharmaceutical composition according to any one of 29 to 29.
- Item 3 The pharmaceutical composition according to Item 31, wherein the antigen is a pathogen-derived antigen or a tumor antigen.
- Item 33 The pharmaceutical composition according to Item 31, wherein the antigen is a tumor antigen.
- Item 34 The pharmaceutical composition according to Item 33, wherein the tumor antigen is a tumor antigen peptide.
- Tumor antigen peptide RMFPNAPYL (SEQ ID NO: 1), ALLPAVPSL (SEQ ID NO: 8), SLGEQQYSV (SEQ ID NO: 9), RVPPGVAPTL (SEQ ID NO: 10), VLDFAPPGA (SEQ ID NO: 4), CMTWNQMNL (SEQ ID NO: 11), CYTWNQMNL (SEQ ID NO: 2), TYAGCLSQIF (SEQ ID NO: 18), Equation (4): [In the formula, the bond between C and C represents a disulfide bond. ], And equation (5): [In the formula, the bond between C and C represents a disulfide bond.
- the pharmaceutical composition according to Item 34 which comprises one or more peptides represented by an amino acid sequence selected from the group consisting of or a pharmaceutically acceptable salt thereof.
- Tumor antigen peptide RMFPNAPYL (SEQ ID NO: 1), ALLPAVPSL (SEQ ID NO: 8), SLGEQQYSV (SEQ ID NO: 9), RVPPGVAPTL (SEQ ID NO: 10), VLDFAPPGA (SEQ ID NO: 4), CMTWNQMNL (SEQ ID NO: 11), CYTWNQMNL (SEQ ID NO: 2), and Equation (4):
- the bond between C and C represents a disulfide bond.
- CWAPVLDFAPPGASAYGSL (SEQ ID NO: 12), WAPVLDFAPPGASAYGSLC (SEQ ID NO: 13), CNKRYFKLSHLQMHSRKHTG (SEQ ID NO: 14), CNKRYFKLSHLQMHSRKH (SEQ ID NO: 15), CNKRYFKLSHLQMHSRK (SEQ ID NO: 16), and KRYFKLSHLQMHSRKH (SEQ ID NO: 17), 34.
- the pharmaceutical composition according to Item 34 which comprises one or more peptides represented by an amino acid sequence selected from the group consisting of or a pharmaceutically acceptable salt thereof.
- the tumor antigen peptide is expressed in formula (4): [In the formula, the bond between C and C represents a disulfide bond. ]
- the pharmaceutical composition according to Item 34 which is a combination of a peptide represented by the amino acid sequence of the above or a pharmaceutically acceptable salt thereof.
- a vaccine adjuvant comprising the compound according to any one of Items 1 to 20 or a pharmaceutically acceptable salt thereof.
- Item 38 The vaccine adjuvant according to Item 38, which is a vaccine adjuvant for a cancer vaccine.
- Item 40 The compound according to any one of Items 1 to 20, or a pharmaceutically acceptable salt thereof, which is used as a vaccine adjuvant.
- Item 4 The compound according to any one of Items 1 to 20, or a pharmaceutically acceptable salt thereof, which is used as a vaccine adjuvant for a cancer vaccine.
- a method for inducing CTL in a mammal which comprises administering to the mammal the compound according to any one of Items 1 to 20 or a pharmaceutically acceptable salt thereof.
- a method for enhancing CTL induction in a mammal which comprises administering to the mammal the compound according to any one of Items 1 to 20 or a pharmaceutically acceptable salt thereof.
- a method for enhancing a specific immune response against an antigen in a mammal which comprises administering to the mammal the compound according to any one of Items 1 to 20 or a pharmaceutically acceptable salt thereof.
- [Item 50] a) A pharmaceutical composition containing the compound represented by the formula (1) of Item 1 or a pharmaceutically acceptable salt thereof, or the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof; and b) a tumor.
- [Item 51] a) A pharmaceutical composition containing the compound represented by the formula (1) of Item 1 or a pharmaceutically acceptable salt thereof, or the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof; and b) a pathogen.
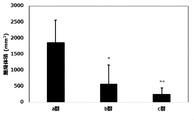
- FIG. 1 shows that in Test Example 3, the compound represented by Formula No. 4 and the compound represented by SEQ ID NO: 3 and the peptide represented by SEQ ID NO: 3 were mixed with Montanide ISA 51 VG to add the compound synthesized in Example 1 or Reference Example 12. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 2 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 24 : 02 gene transfer mouse.
- FIG. 2 shows the compound represented by Formula No. 4 and SEQ ID NO: 3 in Test Example 5 7 days before and 7 days after transplantation of MCA-A24 / Kb-WT1 tumor cells into HLA-A * 24: 02 gene-introduced mice.
- FIG. 3 shows CTL induction with respect to SEQ ID NO: 6 in vivo for a vaccine obtained by adding the compound synthesized in Example 1 to a vaccine in which the peptide represented by SEQ ID NO: 6 and Montanide ISA 51 VG were mixed in Test Example 6. It is a figure which shows the result of having tested the ability by IFN ⁇ ELISPOT assay using the HLA-A * 02 : 01 gene transfer mouse.
- FIG. 4 shows CTL induction with respect to SEQ ID NO: 5 in vivo for a vaccine obtained by adding the compound synthesized in Example 1 to a vaccine obtained by mixing the peptide represented by SEQ ID NO: 5 and Montanide ISA 51 VG in Test Example 7. It is a figure which shows the result of having tested the ability by IFN ⁇ ELISPOT assay using the HLA-A * 02 : 01 gene transfer mouse.
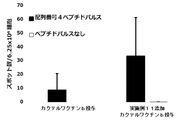
- FIG. 5 shows CTL induction on SEQ ID NO: 18 in vivo for a vaccine obtained by adding the compound synthesized in Example 1 to a vaccine obtained by mixing the peptide represented by SEQ ID NO: 18 and Montanide ISA 51 VG in Test Example 8.
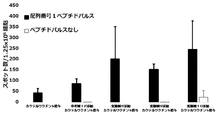
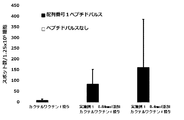
- FIG. 6 shows Example 8, 9, or 10 or Reference Example 12 in a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanede ISA 51 VG were mixed in Test Example 9. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by the IFN ⁇ ELISPOT assay using the HLA-A * 02 : 01 transgenic mouse for the vaccine to which the synthesized compound was added.
- FIG. 6 shows Example 8, 9, or 10 or Reference Example 12 in a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanede ISA 51 VG were mixed in Test Example 9. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by the IFN ⁇ ELISPOT assay using the HLA-A * 02 : 01 transgenic mouse for the vaccine to which the synthesized compound was
- Example 7 shows Example 8, 9, or 10 or Reference Example 12 in a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanede ISA 51 VG were mixed in Test Example 9. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by the IFN ⁇ ELISPOT assay using the HLA-A * 02 : 01 transgenic mouse for the vaccine to which the synthesized compound was added.
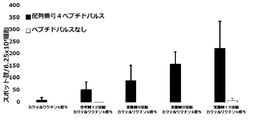
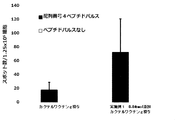
- FIG. 8 shows a vaccine obtained by adding the compound synthesized in Example 11 to a cocktail vaccine obtained by mixing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanide ISA 51 VG in Test Example 10.
- FIG. 9 shows a vaccine obtained by adding the compound synthesized in Example 11 to a cocktail vaccine obtained by mixing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanede ISA 51 VG in Test Example 10. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene transfer mouse.
- FIG. 9 shows a vaccine obtained by adding the compound synthesized in Example 11 to a cocktail vaccine obtained by mixing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 and Montanede ISA 51 VG in Test Example 10. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene transfer mouse.
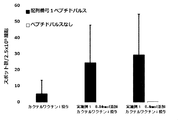
- FIG. 9 shows a vaccine obtained by adding the compound synthesized in
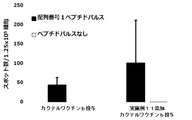
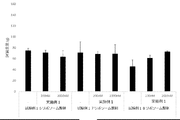
- FIG. 10 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine containing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 as the emulsifying composition 1 in Test Example 11. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene transfer mouse.
- FIG. 11 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine containing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 as the emulsifying composition 1 in Test Example 11.
- FIG. 12 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 are emulsified composition 2 in Test Example 12. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene transfer mouse.
- FIG. 13 shows the vaccine in which the compound represented by the formula No.
- FIG. 14 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 are emulsified composition 3 in Test Example 13.
- FIG. 15 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine using the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 as an oily suspension in Test Example 14. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- FIG. 15 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine using the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 as an oily suspension in Test Example 14. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- FIG. 16 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine using the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 as an oily suspension in Test Example 14. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- FIG. 17 shows the vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 were used as a hydrogel preparation in Test Example 15.
- FIG. 18 shows the vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 were used as a hydrogel preparation in Test Example 15. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene transfer mouse.
- FIG. 19 shows a vaccine in which the compound represented by the formula No.
- FIG. 20 shows the vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 were used as the liposome preparation 2 in Test Example 17 and the compound synthesized in Example 1 was added to the vaccine.
- FIG. 21 shows the vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 were used as the liposome preparation 2 in Test Example 17 and the compound synthesized in Example 1 was added to the vaccine. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- FIG. 21 shows the vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 were used as the liposome preparation 2 in Test Example 17 and the compound synthesized in Example 1 was added to the vaccine. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 4 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- Test Example 18 shows in Test Example 18 a vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 are used as the liposome preparation 3 and the compound synthesized in Example 1 is added to the vaccine. It is a figure which shows the result of having tested the CTL induction ability with respect to SEQ ID NO: 1 in vivo by IFN ⁇ ELISPOT assay using HLA-A * 02 : 01 gene-introduced mouse.
- FIG. 23 shows a vaccine in which the compound synthesized in Example 1 was added to a cocktail vaccine in which the peptide represented by SEQ ID NO: 1 and the peptide represented by SEQ ID NO: 17 and a pre-emulsification composition were mixed in Test Example 19.
- FIG. 24 shows a vaccine obtained by adding the compound synthesized in Example 1 to a cocktail vaccine obtained by mixing the peptide represented by SEQ ID NO: 1 and the peptide represented by SEQ ID NO: 17 and a pre-emulsification composition in Test Example 19.
- the ability to induce helper T cells for SEQ ID NO: 17 is shown by IFN ⁇ ELISPOT assay using HLA-A * 02: 01 / HLA-DRB1 * 01: 01 gene-introduced mice.
- FIG. 25 shows the CTL for SEQ ID NO: 2 in vivo for a vaccine obtained by adding the compound synthesized in Example 1 to a vaccine obtained by mixing the compound represented by the formula No. 5 and the pre-emulsification composition in Test Example 20. It is a figure which shows the result of having tested the inducibility by IFN ⁇ ELISPOT assay using the HLA-A * 24 : 02 gene transfer mouse.
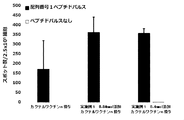
- FIG. 26 shows that a cocktail vaccine containing the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 in Test Examples 11 to 13 as an emulsified composition was administered with a vaccine obtained by adding the compound synthesized in Example 1. It is a figure which shows the result of having measured the spleen weight of a mouse.
- FIG. 27 shows that the compound synthesized in Example 1 was added to a cocktail vaccine in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 in Test Examples 14 and 15 were used as an oil suspension and a hydrogel preparation. It is a figure which shows the result of having measured the spleen weight of the mouse which administered the said vaccine.
- FIG. 28 shows mice administered with a cocktail vaccine prepared by using the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 in Test Examples 16 to 18 as a liposome preparation, to which the compound synthesized in Example 1 was added. It is a figure which shows the result of having measured the spleen weight of.
- the number of substituents in the groups defined as “may be substituted” or “substituted” is not particularly limited as long as it can be substituted. Unless otherwise indicated, the description of each group also applies when the group is part of another group or a substituent.
- halogen examples include fluorine, chlorine, bromine, and iodine. Fluorine or chlorine is preferable, and fluorine is more preferable.
- C 1-6 alkyl means a linear or branched saturated hydrocarbon group having 1 to 6 carbon atoms.
- C 1-4 alkyl is preferable, and “C 1-3 alkyl” is more preferable.
- Specific examples of “C 1-6 alkyl” include, for example, methyl, ethyl, propyl, 1-methylethyl, butyl, 2-methylpropyl, 1-methylpropyl, 1,1-dimethylethyl, pentyl, 3-methylbutyl, Examples thereof include 2-methylbutyl, 2,2-dimethylpropyl, 1-ethylpropyl, 1,1-dimethylpropyl, hexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl and 1-methylpentyl, and "C.
- Specific examples of " 1-4 alkyl” include examples of “C 1-6 alkyl” having 1 to 4 carbon atoms.
- Specific examples of “C 1-3 alkyl” include examples of "C
- C 1-6 alkoxy means “C 1-6 alkyloxy”, and the “C 1-6 alkyl” moiety is synonymous with the “C 1-6 alkyl”.
- C 1-6 alkoxy preferably, “C 1-4 alkoxy” is mentioned, and more preferably, “C 1-3 alkoxy” is mentioned.
- Specific examples of “C 1-6 alkoxy” include, for example, methoxy, ethoxy, propoxy, 1-methylethoxy, butoxy, 2-methylpropoxy, 1-methylpropoxy, 1,1-dimethylethoxy, pentyroxy, 3-methyl.
- Linker means a divalent group having two bonds within a functional group.
- the divalent group include C 1-6 alkylene, C 2-7 alkenylene, C 2-7 alkinylene, C 3-10 cycloalkylene, C 6-10 arylene, C 5-10 heteroarylene, ether, amine, and the like.
- Examples include carbonyl, ester, amide, carbonate, carbamate, thiocarbamate, or thiourea. Further, from these examples, they can be appropriately combined and used as a divalent group.
- the linker is preferably -O-, -NR Y- , -C (O)-, -C (O) O-, -OC (O)-, -C (O) NR Y- , -NR Y C ( O)-, -CH 2 NR Y- , -CH 2 O-, -OC (O) O-, -NR 7 C (O) O-, -OC (O) NR Y- , -NR 7 C (O) ) NR Y- , -OC (S) NR Y- , or -NR 7 C (S) NR Y- (in the formula, RY and R 7 are as defined in Item 9), more preferably-.
- C 1-6 alkylene means a linear or branched saturated hydrocarbon having 1 to 6 carbon atoms.
- Specific examples of “C 1-6 alkylene” include, for example, methylene, ethylene, propylene, 1-methylethylene, butylene, 2-methylpropylene, 1-methylpropylene, 1,1-dimethylethylene, pentylene, 3-methylbutylene.
- C 2-7 alkenylene means a linear or branched unsaturated hydrocarbon having 2 to 7 carbon atoms and containing 1 to 3 double bonds.
- Specific examples of “C 2-7 alkenylene” include, for example, vinylene, propenylene, methylpropenilen, butenylene, methylbutenylene, pentenylene, hexenylene, heptenylene, and the like, preferably vinylene or propenylene.
- C 2-7 alkynylene means a linear or branched unsaturated hydrocarbon having 2 to 7 carbon atoms and containing one triple bond.
- Specific examples of “C 2-7 alkynylene” include, for example, ethynylene, propinilen, methylpropinylene, butinirene, methylbutinirene, penterene, hexinylene, heptinylene, and the like, preferably ethynylene or propinylene.
- C 3-10 cycloalkylene means a cyclic alkylene having 3 to 10 carbon atoms, and a partially crosslinked structure is also included.
- C 3-10 cycloalkylene For example, cyclopropylene, cyclobutylene, cyclopentylene, cyclohexylene, cycloheptyrene, cyclooctylene, adamantylene and the like can be mentioned, with preference given to cyclopropylene or cyclobutylene.
- C 6-10 arylene means an aromatic hydrocarbon having 6 to 10 carbon atoms.
- Specific examples of “C 6-10 arylene” include, for example, phenylene, 1-naphthylene, 2-naphthylene, and the like, preferably phenylene.
- C 5-10 heteroarylene is a monocyclic 5- to 7-membered aromatic heterocycle containing 1 to 4 atoms independently selected from the group consisting of nitrogen, oxygen and sulfur atoms. , Or a two-ring 8- to 10-membered aromatic heterocycle.
- C 5-10 heteroarylene examples include pyridylene, pyridadinylene, isothiazolylen, pyrrolylene, furylene, thienylene, thiazolilen, imidazolylene, pyrimidinylene, thiadiazolilen, pyrazolylen, oxazolylen, isooxazolilen, pyrazilylene, triadylene , Imidazolidinylene, oxadiazolylene, triazolylen, tetrazolilen, indolylene, indazolylen, quinolylene, isoquinolylene, benzofuranilen, benzothienylene, benzoxazolylen, benzothiazolylen, benzisooxazolilen, benzisothiazolilen , Benzimidazolylene, or 6,11-dihydrodibenzo [b, e] thiepinylene and the
- Preferred examples of X include methylene, an oxygen atom, or NR 5 (R 5 represents a hydrogen atom or an alkyl of C 1-6 ), and more preferably methylene.
- Examples of Y 1 include ⁇ (CH 2 CH 2 O) m ⁇ R 6 .
- Examples of Y 2 include ⁇ (CH 2 CH 2 O) n ⁇ R 8 .
- R 1 is C 1-6 alkyl, which may be substituted with 1 to 3 identical or different halogens. More preferably, methyl, ethyl, propyl, 1-methylethyl, butyl, 2-methylpropyl, 1-methylpropyl, 1,1-dimethylethyl which are C 1-6 alkyls are mentioned, and more preferably, methyl is used. Can be mentioned.
- R 2 Hydrogen atom or (2) C 1-6 alkyl (the alkyl may be substituted with 1 to 3 substituents independently selected from halogen and hydroxy). Can be mentioned. More preferably, it includes a hydrogen atom or a C 1-6 alkyl, more preferably, include alkyl C 1-6, even more preferably include alkyl C 3-4.
- R 3 Hydrogen atom or (2) C 1-6 alkyl (the alkyl may be substituted with 1 to 3 substituents independently selected from halogen and hydroxy). Can be mentioned.
- R 3 More preferred as R 3, (1) Hydrogen atom, or (2) C 1-3 alkyl (the alkyl may be substituted with 1 to 3 hydroxys) Can be mentioned.
- R 3 More preferably the R 3, Examples thereof include (1) a hydrogen atom or (2) a C 1-3 alkyl (the alkyl may be substituted with one hydroxy).
- R 3 examples thereof include (1) a hydrogen atom or (2) a C 1-2 alkyl (the alkyl may be substituted with one hydroxy).
- R 4 Hydrogen atom (2) Halogen (3) Hydroxy (4) C 1-6 alkyl (The alkyl may be substituted with 1 to 3 identical or different halogens) Examples thereof include (5) C 1-6 alkoxy (the alkoxy may be substituted with 1 to 3 identical or different halogens), or (6) cyano.
- R 4 More preferred as R 4, (1) Hydrogen atom (2) Halogen (3) Hydroxy (4) C 1-6 alkyl (the alkyl may be substituted with 1 to 3 identical or different halogens), or (5) C 1 -6 Alkoxy (The alkoxy may be substituted with 1 to 3 identical or different halogens) Can be mentioned.
- R 4 includes hydrogen atom, hydroxy, C 1-3 alkyl, or C 1-3 alkoxy.
- R 4 Even more preferably as R 4, a hydrogen atom, hydroxy or methoxy and the like, and even more preferably include hydroxy or methoxy, most preferably, be mentioned a hydrogen atom or methoxy.
- R 5 is a hydrogen atom, or a C 1-3 includes alkyl. More preferably, a hydrogen atom, methyl, ethyl, or propyl can be mentioned.
- R 6 and R 8 preferably each independently include a hydrogen atom or C 1-3 alkyl, and more preferably each independently include a hydrogen atom, methyl, ethyl, or propyl. More preferably, a hydrogen atom or methyl can be mentioned.
- R 7 include a hydrogen atom or an alkyl of C 1-3 . More preferably, a hydrogen atom, methyl, ethyl, or propyl can be mentioned.
- the most preferable L is -CH 2 NR Y- .
- substitution positions of X, L, and R 4 on the benzene ring are preferably the following (1a) or (1aa).
- RY is preferably a hydrogen atom, C 1-6 alkyl or Y 2 , more preferably a hydrogen atom or C 1-6 alkyl, and even more preferably a hydrogen atom, methyl, ethyl, etc. Or propyl can be mentioned.
- RY is preferably a hydrogen atom, C 1-6 alkyl or Y 2 , and more preferably a hydrogen atom, methyl, Y 2 .
- the m and n are preferably 3 to 40 integers, more preferably 4 to 40 integers, and even more preferably 4 to 36 integers, respectively.
- an integer of 3 to 40 can be mentioned independently, an integer of 3 to 20 can be mentioned, and an integer of 5 to 20 can be mentioned more preferably.
- preferred examples of the compound include the following compounds or pharmaceutically acceptable salts thereof.
- (A) is mentioned as a preferable embodiment.
- (A) X is methylene, oxygen atom, sulfur atom, SO, SO 2 , or NR 5 ;
- R 1 is C 1-6 alkyl (the alkyl may be substituted with 1-5 substituents independently selected from the group consisting of halogen, hydroxy, C 1-6 alkoxy). Yes;
- R 2 and R 3 are independent of each other (1) Hydrogen atom, or (2) C 1-6 alkyl (the alkyl is substituted with 1-5 substituents independently selected from the group consisting of halogen, hydroxy, C 1-6 alkoxy.
- R 4 (1) Hydrogen atom (2) Halogen (3) Hydroxy (4) C 1-6 alkyl (The alkyl may be substituted with 1 to 3 identical or different halogens) (5) C 1-6 alkoxy (the alkoxy may be substituted with 1 to 3 identical or different halogens), or (6) cyano;
- R 5 It is (1) a hydrogen atom or (2) a C 1-6 alkyl;
- R 6 and R 8 are independent of each other It is (1) a hydrogen atom or (2) a C 1-6 alkyl;
- R 7 is It is (1) a hydrogen atom or (2) a C 1-6 alkyl;
- L is (1) -O- (2) -NR Y- (3) -C (O)- (4) -C (O) O- (5) -OC (O)- (6) -C (O) NR Y - (7) -NR Y C (O ) - (8) -CH 2 NR Y- (9) -CH 2 O- (10) -OC (O)
- X is methylene, oxygen atom, or NR 5 ;
- R 1 is a C 1-6 alkyl, which may be substituted with 1 to 3 identical or different halogens;
- R 2 is (1) a hydrogen atom or (2) a C 1-6 alkyl;
- R 3 It is (1) a hydrogen atom, or (2) a C 1-6 alkyl, which may be substituted with 1 to 3 hydroxys;
- R 4 (1) Hydrogen atom (2) Halogen (3) Hydroxy (4) C 1-6 alkyl (the alkyl may be substituted with 1 to 3 identical or different halogens), or (5) C 1 -6 Alkoxy, which may be substituted with 1 to 3 identical or different halogens;
- R 5 It is (1) a hydrogen atom or (2) C 1-3 alkyl;
- R 6 and R 8 are independent of each other It is (1) a hydrogen atom or (2) C 1-3 alkyl;
- L is (1) -O- (2) -NR 5 ;
- R 1 is a C
- (C) is methylene;
- R 1 is methyl, ethyl, propyl, 1-methylethyl, butyl, 2-methylpropyl, 1-methylpropyl or 1,1-dimethylethyl;
- R 2 is located at C 1-6 alkyl;
- R 3 It is (1) a hydrogen atom, or (2) a C 1-3 alkyl, which may be substituted with a single hydroxy;
- R 4 (1) hydrogen atom (2) hydroxy (3) C 1-3 alkyl, or (4) C 1-3 alkoxy;
- R 6 and R 8 are each independently a hydrogen atom, methyl, ethyl or propyl;
- L is (1) -C (O) NR Y -, (2) -CH 2 NR Y- , (3) -C (O) O- or (4) -CH 2 O- Is;
- RY is a hydrogen atom, methyl, ethyl, propyl
- Another aspect of the compound represented by the formula (1) is the following (D).
- (D) A compound represented by the formula (2) or (3) or a pharmaceutically acceptable salt thereof; [In the formula, R 2 is C 1-6 alkyl, R 3 is a hydrogen atom, or C 1-3 alkyl, which may be substituted with 1 to 3 hydroxys. R 4 is hydrogen atom, hydroxy or methoxy, L is -CH 2 NR Y- , -C (O) NR Y- , -C (O) O- or -CH 2 O-.
- RY is a hydrogen atom, methyl, ethyl, propyl or Y 2 and Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6 and Y 2 is ⁇ (CH 2 CH 2 O) n ⁇ R 8 and R 6 and R 8 are independently hydrogen atoms, methyl, ethyl or propyl, respectively.
- m and n are each independently an integer of 3 to 40.
- Another embodiment of the compound represented by the formula (1) includes the following (E).
- R 2 is C 1-6 alkyl
- R 3 is a hydrogen atom, or C 1-3 alkyl, which alkyl may be substituted with one hydroxy.
- R 4 is hydrogen atom or methoxy
- L is -CH 2 NR Y- RY is a hydrogen atom, methyl, ethyl or propyl
- Y 1 is ⁇ (CH 2 CH 2 O) m ⁇ R 6
- R 6 is a hydrogen atom, methyl, ethyl or propyl
- m is an integer of 4 to 36.
- the method for producing the compound of the present invention will be described below.
- the compound of the present invention represented by the formula (1) or a pharmaceutically acceptable salt thereof can be produced, for example, by the following production method.
- Manufacturing method A-1 Of the formula (1) compounds represented by or a pharmaceutically acceptable salt of the present invention -CR A1 R A2 NR Y - , or a compound with a linker represented by -CR A1 R A2 O- (a1-2) is produced by the following method.
- R A1 and R A2 each independently represent a hydrogen atom
- C represents an alkyl group of 1-6
- LG a1 represents a leaving group
- Nu a1 represents a nucleophilic agent
- L a1 represents a linker generated by the process
- This step is a substitution reaction between LG a1 which is a leaving group and Nu a1- Y 1 which is a nucleophile. Presence or absence of a suitable base, in a suitable solvent, and from Nu a1 -Y 1 compound (a1-1), a step to give compound (a1-2).
- the leaving group is not particularly limited, but preferably includes fluorine, chlorine, bromine, iodine, methanesulfonate, ethanesulfonate, p-toluenesulfonate, and the like, and more preferably chlorine, bromine, and methanesulfonate. Can be mentioned.
- the nucleophile is not particularly limited, but preferably an amine (the amine may be substituted with RY as defined in Item 9), alcohol, thiol and the like, and more preferably an amine (the amine). Amines may be substituted with RY as defined in Item 9), alcohols and the like.
- the base used in this step is selected from the bases exemplified below, and preferred examples include sodium hydride and potassium hydride.
- the solvent used in this step is selected from the solvents exemplified below, and DMF is preferable.
- the reaction time is usually from about 5 minutes to about 48 hours, preferably from about 10 minutes to about 24 hours.
- the reaction temperature is usually about ⁇ 78 ° C. to about 100 ° C., preferably about 0 ° C. to about 100 ° C.
- This step is a substitution reaction between LG a2 , which is a leaving group, and Nu a2 , which is a nucleophile. Presence or absence of a suitable base, in a suitable solvent, from LG a2 -Y 1 and the compound (a2-1), a step to give compound (a2-2).
- LG a2 , Nu a2 and La 2 are the same as the leaving group, nucleophile and linker described in Production Method A-1, respectively.
- the various reaction conditions are the conditions according to the description of the production method A-1.
- the method for producing the compound of the present invention will be described below.
- the compound of the present invention represented by the formula (1) can be produced, for example, by the following production method.
- Manufacturing method B-1 Among the compound represented by the formula (1) of the present invention or a pharmaceutically acceptable salt thereof, the compound (b1-2) having a linker represented by ⁇ CH 2 NR Y ⁇ is prepared by the following method. Manufactured. (In the equation, R 1 , R 2 , R 3 , R 4 , R 5 , X, and Y 1 are as defined in Item 1, RY is as defined in Item 9, and L b1. Represents the linker produced by this step)
- This step is a reductive amination reaction between aldehyde and amine.
- a suitable reducing agent in a suitable solvent, and the R Y -NH-Y 1 compound (b1-1), a step of obtaining a compound (b1-2).
- the reducing agent used in this step is not particularly limited, but preferably sodium borohydride, triacetoxyborohydride, and picoline borane.
- the solvent used in this step is selected from the solvents exemplified below, and preferred examples include THF and chloroform.
- the reaction time is usually from about 5 minutes to about 48 hours, preferably from about 10 minutes to about 24 hours.
- the reaction temperature is usually about ⁇ 78 ° C. to about 100 ° C., preferably about 0 ° C. to about 100 ° C.
- Manufacturing method B-2 Among the compounds represented by the formula (1) of the present invention or pharmaceutically acceptable salts thereof, the compound having a linker represented by -NR Y- or -CH 2 NR Y- (b2-2). Is manufactured by the following method. (In the equation, R 1 , R 2 , R 3 , R 4 , R 5 , X, and Y 1 are as defined in Item 1, RY is as defined in Item 9, and n b. Is 0 or 1 and L b2 represents the linker produced by this step)
- This step is a reductive amination reaction between aldehyde and amine. Presence of a suitable reducing agent, in a suitable solvent, and a Y 1 -CHO compound (b2-1), a step of obtaining a compound b2-2.
- the various reaction conditions are the conditions according to the description of the production method B-1.
- the method for producing the compound of the present invention will be described below.
- the compound of the present invention represented by the formula (1) can be produced, for example, by the following production method.
- Manufacturing method C-1 Of the formula (1) compounds represented by or a pharmaceutically acceptable salt of the present invention, -O -, - NR Y - , or -NR Y C (O) - with a linker represented by Compound (c1-2) is produced by the following method.
- R 1 , R 2 , R 3 , R 4 , R 5 , X, and Y 1 are as defined in Item 1, LG c 1 represents a leaving group, and Nu c 1 is a nucleophile.
- L c1 represents the linker produced by this step
- This step is a coupling reaction between LG c1 which is a leaving group and Nu c1- Y 1 which is a nucleophile.
- the compound (c1-2) is obtained from the compound (c1-1) and the nucleophile Nu c1- Y 1 in the presence and absence of the appropriate base in the presence of the appropriate catalyst and in the appropriate solvent. It is a process.
- examples of the catalyst include those supported on a carrier such as a transition metal such as palladium, a salt thereof, a complex thereof, and a polymer.
- the leaving group in this step is not particularly limited, but preferably includes boronic acid, boronic acid ester, halogen, trifluoromethanesulfonate, etc., and more preferably boronic acid, boronic acid ester, bromine atom, iodine. Examples include the atom, trifluoromethanesulfonate.
- the nucleophile in this step is not particularly limited, and examples thereof include amines (the amines may be substituted with RY defined in Item 9), alcohols, alkylmagnesiums, alkylzincs, alkyllithiums, and the like.
- amines the amines may be substituted with alkyl groups having 1 to 6 carbon atoms
- alcohols The solvent used in this step is selected from the solvents exemplified below, and preferably a dioxane-water mixed solvent or the like.
- the reaction time is usually from about 5 minutes to about 48 hours, preferably from about 10 minutes to about 24 hours.
- the reaction temperature is usually about ⁇ 78 ° C. to about 100 ° C., preferably about 0 ° C. to about 100 ° C.
- the method for producing the compound of the present invention will be described below.
- the compound of the present invention represented by the formula (1) can be produced, for example, by the following production method.
- This step is a condensation reaction of a compound having a carboxylic acid and (d1-1) and Nu d1 -Y 1.
- the nucleophile is not particularly limited, but preferably an amine (the amine may be substituted with RY defined in Item 9), alcohol, thiol and the like, and more preferably. Examples include amines (the amines may be substituted with one alkyl group having 1 to 6 carbon atoms) and alcohols.
- the condensing agent is selected from the condensing agents used in the conventional method, and preferably HBTU, HATU, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (including hydrochloride).
- the base is selected from the bases exemplified below, preferably a tertiary alkylamine, and more preferably DIPEA or triethylamine.
- the solvent used in this step is selected from the solvents exemplified below, and preferably DMF, dichloromethane, chloroform, THF and the like can be mentioned.
- the reaction time is usually from about 5 minutes to about 48 hours, preferably from about 10 minutes to about 24 hours.
- the reaction temperature is usually about ⁇ 78 ° C. to about 100 ° C., preferably about 0 ° C. to about 100 ° C.
- This step is a condensation reaction with a compound having a Y 1 -COOH and nucleophile Nu d2 having a carboxylic acid (d2-1).
- a suitable condensing agent in the presence and absence of a suitable base, in a suitable solvent, compounds containing a nucleophilic agent from Y 1 -COOH is electrophilic agent (d2-1), compound (d2 -2) is the process of obtaining.
- the various reaction conditions are the same as those described in Step D-1.
- the starting materials (a1-1, a2-1, b1-1, b2-1, c1-1, d1-1, d2-1) used in the production methods A to D are described in, for example, WO2009 / 067081. It can be manufactured according to the method.
- the compound of the present invention represented by the formula (1) can be produced, for example, by the following production method.
- Manufacturing method E-1 (Wherein, R 1, R 2, R 3, R 4, R 5, L, and Y 1 are as defined in claim 1, LG e1 and LG e2 represents a leaving group, R e is Represents a C 1-6 alkyl group)
- the compound (e1-1) which is the starting material of the step e-1 can be produced by using a commercially available product or by using a corresponding raw material compound and a reaction according to the methods described in the production methods A to D.
- Steps e-1 to e-4 are methods according to, for example, the manufacturing method described in WO2009 / 067081.
- the base used in each step of each of the above production methods should be appropriately selected depending on the reaction, the type of the starting compound, etc., and for example, alkali hydroxides such as sodium hydroxide and potassium carbonate, sodium carbonate, etc. , Alkali carbonates such as potassium carbonate, sodium hydride, metal hydrides such as potassium hydride, sodium hydroxide, alkali metal hydroxides such as potassium hydroxide, sodium methoxydo, sodium t-butoxide Alkali metal alkoxides such as, organic metal bases such as butyllithium, lithium diisopropylamide, triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo [5.4.0]-. Included are organic bases such as 7-undecene (DBU).
- DBU 7-undecene
- Examples of the condensing agent used in each process of each of the above manufacturing methods include those described in Volume 22 of the Experimental Chemistry Course (edited by the Chemical Society of Japan, Maruzen).
- phosphate esters such as diethyl cyanophosphate and diphenylphosphoryl azide
- carbodiimides such as 1-ethyl-3- (3-dimethylaminopropyl) -carbodiimide hydrochloride (WSC ⁇ HCl), dicyclohexylcarbodiimide (DCC);
- a combination of disulfides such as 2'-dipyridyldisulfide and phosphines such as triphenylphosphine; phosphorus halides such as N, N'-bis (2-oxo-3-oxazolidinyl) phosphinic chloride (BOPCl); azo Combination of azodicarboxylic acid diester such as diethyl dicarboxylic acid and phosphine such as
- the solvent used in each step of each of the above production methods should be appropriately selected depending on the reaction, the type of raw material compound, etc., and for example, alcohols such as methanol, ethanol and isopropanol, acetone, methyl ketone and the like. Ketones, halogenated hydrocarbons such as methylene chloride and chloroform, ethers such as tetrahydrofuran (THF) and dioxane, aromatic hydrocarbons such as toluene and benzene, aliphatic hydrocarbons such as hexane and heptane.
- alcohols such as methanol, ethanol and isopropanol
- acetone methyl ketone and the like.
- Ketones halogenated hydrocarbons
- ethers such as tetrahydrofuran (THF) and dioxane
- aromatic hydrocarbons such as toluene and benzene
- aliphatic hydrocarbons such as hex
- esters such as ethyl acetate, propyl acetate, amides such as N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone, sulfoxides such as dimethyl sulfoxide (DMSO), such as acetonitrile.
- DMF N, N-dimethylformamide
- sulfoxides such as dimethyl sulfoxide (DMSO)
- Examples of the “pharmaceutically acceptable salt” include acid addition salts and base addition salts.
- an inorganic acid salt such as hydrochloride, hydrobromide, sulfate, hydroiodide, nitrate, phosphate, or citrate, oxalate, phthalate, Fumarate, maleate, succinate, malate, acetate, formate, propionate, benzoate, trifluoroacetate, methanesulfonate, benzenesulfonate, para-toluenesulfonic acid
- examples thereof include organic acid salts such as salts and camphor sulfonates
- examples of the base addition salt include inorganic base salts such as sodium salt, potassium salt, calcium salt, magnesium salt, barium salt and aluminum salt, or trimethylamine, triethylamine and pyridine.
- Picolin 2,6-rutidine, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, dicyclohexylamine, N, N-dibenzylethylamine, organic bases
- salts include salts, and further examples include amino acid salts such as basic or acidic amino acids such as arginine, lysine, ornithine, aspartic acid, or glutamic acid.
- Suitable and pharmaceutically acceptable salts of the starting and target compounds are conventional non-toxic salts, such as organic acid salts (eg, acetates, trifluoroacetates, maleates, fumarates).
- the compound of the present invention when it is desired to obtain a salt of the compound of the present invention, if the compound of the present invention is obtained in the form of a salt, it may be purified as it is, and if it is obtained in the free form, it may be dissolved or suspended in an appropriate organic solvent. It may be turbid and an acid or base may be added to form a salt by a usual method. Further, the compound of the present invention and a pharmaceutically acceptable salt thereof may exist in the form of a solvate with water or various solvents, and these adducts are also included in the present invention.
- a deuterium converter obtained by converting any one or two or more H of the compound represented by the formula (1) into 2 H (D) is also included in the compound represented by the formula (1).
- the present invention includes a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof. It also includes solvates such as these hydrates or ethanol solvates. Furthermore, the present invention also includes all tautomers of the compound (1) of the present invention, all stereoisomers present, and crystalline forms of any mode.
- the compounds (1) of the present invention are optical isomers based on optically active centers, atropisomers based on axial or planar chirality generated by binding of intramolecular rotation, other stereoisomers, and remutability. All possible isomers and mixtures thereof, including these, are included within the scope of the present invention, although there may be conformations, geometric isomers and the like.
- a mixture of optical isomers of the compound of the present invention can be produced according to a usual method.
- the production method include a method using a raw material having an asymmetric point, or a method of introducing an asymmetry in an intermediate stage.
- the optical isomer can be obtained by using an optically active raw material or performing optical resolution at an appropriate stage in the manufacturing process.
- an optical division method for example, when the compound represented by the formula (1) or an intermediate thereof has a basic functional group, it is contained in an inert solvent (for example, an alcohol solvent such as methanol, ethanol or 2-propanol).
- Ether solvent such as diethyl ether, ester solvent such as ethyl acetate, hydrocarbon solvent such as toluene, aproton solvent such as acetonitrile, or a mixed solvent of two or more kinds selected from the above solvents
- optically active Acids eg, monocarboxylic acids such as mandelic acid, N-benzyloxyalanine, lactic acid, tartrate, dicarboxylic acids such as o-diisopropyridene tartrate, malic acid, sulfonic acids such as camphorsulfonic acid and bromocamparsulfonic acid
- a salt is formed using.
- an optically active amine for example, 1-phenylethylamine, quinine, quinidine, cinchonidine, cinchonine, strikinine
- Optical resolution can also be performed by forming a salt using an organic amine such as
- the compound of the present invention represented by the formula (1) or an intermediate thereof can be separated and purified by a method known to those skilled in the art.
- extraction, partitioning, reprecipitation, column chromatography (eg, silica gel column chromatography, ion exchange column chromatography or preparative liquid chromatography) or recrystallization can be mentioned.
- the recrystallizing solvent include alcohol solvents such as methanol, ethanol or 2-propanol, ether solvents such as diethyl ether, ester solvents such as ethyl acetate, aromatic hydrocarbon solvents such as benzene or toluene, acetone and the like.
- a ketone solvent, a halogen solvent such as dichloromethane or chloroform, a hydrocarbon solvent such as hexane, an aproton solvent such as dimethylformamide or acetonitrile, water, or a mixed solvent thereof can be used.
- a halogen solvent such as dichloromethane or chloroform
- a hydrocarbon solvent such as hexane
- an aproton solvent such as dimethylformamide or acetonitrile
- water or a mixed solvent thereof
- the intermediate or final product in the above production method is appropriately converted from its functional group, and in particular, various side chains are extended by using an amino group, a hydroxyl group, a carbonyl group, a halogen group, etc. as a foothold, and At that time, by performing the above protection and deprotection as necessary, it is possible to lead to another compound contained in the present invention.
- Functional group conversion and side chain extension can be performed by conventional methods (see, for example, Comprehensive Organic Transformations, R.C. Larock, John Wiley & Sons Inc. (1999), etc.).
- the temperature at which the salt is formed is selected from the range from -50 ° C to the boiling point of the solvent, preferably from 0 ° C to the boiling point, and more preferably from room temperature to the boiling point of the solvent. In order to improve the optical purity, it is desirable to raise the temperature to near the boiling point of the solvent. When the precipitated salt is collected by filtration, it can be cooled if necessary to improve the yield.
- the amount of the optically active acid or amine used is appropriately in the range of about 0.5 to about 2.0 equivalents, preferably about 1 equivalent with respect to the substrate.
- the crystals are placed in an inert solvent (for example, an alcohol solvent such as methanol, ethanol, 2-propanol, an ether solvent such as diethyl ether, an ester solvent such as ethyl acetate, a hydrocarbon solvent such as toluene, acetonitrile and the like. It is also possible to recrystallize in an aproton solvent of (or a mixed solvent of two or more kinds selected from the above solvents) to obtain a highly pure optically active salt. Further, if necessary, the optically resolved salt can be treated with an acid or a base by a usual method to obtain a free form.
- an inert solvent for example, an alcohol solvent such as methanol, ethanol, 2-propanol, an ether solvent such as diethyl ether, an ester solvent such as ethyl acetate, a hydrocarbon solvent such as toluene, acetonitrile and the like. It is also possible to recrystallize in an
- the present invention provides a compound represented by the above-defined formula (1) or a pharmaceutically acceptable salt thereof, which is useful as a vaccine adjuvant, preferably a vaccine adjuvant for a cancer vaccine.
- the present invention also combines a pharmaceutically acceptable diluent or carrier to form a pharmaceutical composition containing the compound represented by the formula (1) defined above or a pharmaceutically acceptable salt thereof (hereinafter referred to as pharmaceutically acceptable salt). ,
- a pharmaceutical composition of the present invention containing the compound represented by the formula (1) defined above or a pharmaceutically acceptable salt thereof (hereinafter referred to as pharmaceutically acceptable salt).
- the compound of the present invention or a pharmaceutically acceptable salt thereof can be used as an adjuvant for maintaining or enhancing the immunostimulatory activity of an active ingredient having immunostimulatory activity. That is, the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of inducing or enhancing an antigen-specific antibody, specifically, an antigen-specific IgG, and more particularly, a Th1-type antigen-specific IgG (for example, IgG2c). In addition, the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of increasing cytotoxic T cells (CTL).
- CTL cytotoxic T cells
- the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of inducing CTL in a mammal or an activity of enhancing CTL induction in a mammal.
- the compounds of the invention or pharmaceutically acceptable salts thereof have the activity of increasing CD4 positive (ie, MHC class II binding) and / or CD8 positive (ie, MHC class I binding) T cells.
- the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of increasing antigen-specific T cells.
- the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of increasing memory T cells, specifically CD8-positive effector memory T cells.
- the compound of the present invention or a pharmaceutically acceptable salt thereof is said to have a higher effect of increasing CTL when administered to mammals than when the same number of moles of a compound containing no PEG structure is administered. It has characteristics.
- the compound of the present invention or a pharmaceutically acceptable salt thereof has an activity of activating immunocompetent cells.
- the pharmaceutical composition of the present invention may contain a tumor antigen.
- a tumor antigen protein or a tumor antigen peptide derived from the tumor antigen protein can be used.
- the antigen peptide described later can be preferably used, but more preferably, a tumor antigen peptide derived from NY-ESO-1, MAGE-3, WT1, OR7C1 or Her2 / neu can be used.
- a tumor antigen peptide derived from WT1 is preferable.
- peptides derived from neoantigens produced as a result of genetic abnormalities in tumors can also be used with the compounds of the invention or pharmaceutically acceptable salts thereof.
- the compound of the present invention or a pharmaceutical composition containing a pharmaceutically acceptable salt thereof and a tumor antigen has an action of inhibiting the growth of a tumor expressing the antigen or suppressing the development of a tumor expressing the antigen. Has a preventive effect. Therefore, the compound of the present invention or a pharmaceutically acceptable salt thereof is useful as a drug for treating or preventing cancer by using it as a pharmaceutical composition in combination with the tumor antigens shown below.
- the tumor antigen peptide is not particularly limited, but the peptides described in the internationally published WO2014 / 157692 pamphlet or the internationally published WO2014 / 157704A1 pamphlet can be used.
- One embodiment of the tumor antigen peptide includes, for example, a tumor antigen peptide consisting of a peptide consisting of the following amino acid sequences or a pharmaceutically acceptable salt thereof: RMFPNAPYL (SEQ ID NO: 1), ALLPAVPSL (SEQ ID NO: 8), SLGEQQYSV (SEQ ID NO: 9), RVPPGVAPTL (SEQ ID NO: 10), VLDFAPPGA (SEQ ID NO: 4), CMTWNQMNL (SEQ ID NO: 11), CYTWNQMNL (SEQ ID NO: 2), WAPVLDFAPPGASAYGSL (SEQ ID NO: 3), CWAPVLDFAPPGASAYGSL (SEQ ID NO: 12), WAPVLDFAPPGASAYGSLC (SEQ
- equation (4) [In the formula, the bond between C and C represents a disulfide bond.
- equation (5) [In the formula, the bond between C and C represents a disulfide bond.
- a peptide consisting of the amino acid sequence represented by or a pharmaceutically acceptable salt thereof can also be used as the tumor antigen peptide in the present invention.
- tumor antigen peptide can be performed according to the method used in ordinary peptide chemistry.
- the synthesis method is described in the literature (Peptide Synthesis, Interscience, New York, 1966; The Proteins, Vol. 2, Academic Press Inc., New York, 1976) and the like. There is a method that is used.
- the pharmaceutical composition of the present invention may contain an antigen.
- the antigen include pathogen-derived antigens, for example, proteins derived from pathogens such as viruses and bacteria, or partial peptides thereof, and complexes of these antigens and carriers are also included in the category of antigens in the present specification. ..
- Such complexes include those in which an antigen (including, but not limited to, proteins and peptides) is chemically crosslinked with a protein as a carrier via a linker known to those skilled in the art, and the antigen is a virus-like particle. Examples include those included in (Virus-like Particle; VLP). Therefore, the compound of the present invention or a pharmaceutically acceptable salt thereof is useful as a drug for treating or preventing infectious diseases such as viruses and bacteria when used in combination with the antigen.
- the route of administration of the pharmaceutical composition of the present invention includes, for example, parenteral administration, specifically, intravascular (for example, intravenous), subcutaneous, intradermal, intramuscular, intratumoral, lymph node, and transdermal administration. ..
- parenteral administration specifically, intravascular (for example, intravenous), subcutaneous, intradermal, intramuscular, intratumoral, lymph node, and transdermal administration. ..
- the pharmaceutical composition of the present invention may contain a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable diluent or carrier.
- Examples of the form of the pharmaceutical composition of the present invention include liquid preparations and the like.
- liquid preparation of the present invention examples include an aqueous solution preparation or an aqueous suspension, an oily solution preparation or an oily suspension, a hydrogel preparation, a lipid preparation, an emulsion preparation and the like.
- an aqueous solution preparation or an aqueous suspension for example, an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof is dissolved or dispersed in water.
- an antigen tumor antigen or pathogen-derived antigen
- a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof is dissolved or dispersed in water.
- an antigen for example, an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof is dissolved or dispersed in an oily component.
- Formulations can be mentioned.
- the hydrogel preparation for example, an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof was dissolved or dispersed in water to impart consistency.
- the lipid preparation include a liposome preparation containing an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof.
- Emulsion preparations include, for example, preparations containing an aqueous solution and an oily composition containing an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof. Can be mentioned.
- an aqueous solution preparation or an aqueous suspension prepared by dissolving or dispersing a tumor antigen and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof in water.
- Additives used in the aqueous solution preparations or suspensions of the present invention include, for example, purified water, water for injection, buffers, pH regulators, stabilizers, tonicity agents, solubilizers or solubilizers. And so on.
- Additives used in the oily solution preparations or oily suspensions of the present invention include, for example, buffering agents, pH adjusters, stabilizers, tonicity agents, animal and vegetable fats and oils, hydrocarbons, fatty acids, fatty acid esters. , Solubilizers, solubilizers and the like.
- Additives used in the hydrogel formulation of the present invention include, for example, purified water, water for injection, buffers, pH regulators, stabilizers, tonicity agents, solubilizers, solubilizers, or thickeners. And so on.
- additives used in the liposome preparation of the present invention include purified water, water for injection, buffers, pH regulators, stabilizers, tonicity agents, solubilizers, solubilizers, lipids and the like. Can be mentioned.
- the emulsion preparation of the present invention includes an oil-in-water emulsion (also referred to as O / W emulsion), a water-in-oil emulsion (also referred to as W / O emulsion), and a water-in-oil oil-in-water emulsion (W / O / W emulsion). (Also described as) or an oil-in-oil-in-oil emulsion (also described as O / W / O emulsion) can be used.
- the emulsion preparation of the present invention preferably includes a water-in-oil emulsion (W / O emulsion).
- the emulsion preparation of the present invention can be produced by emulsifying the aqueous phase and the oil phase by a known method.
- the antigen (tumor antigen or pathogen-derived antigen) and / or the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof is contained in either or both of the oil phase and the aqueous phase in the emulsion. be able to.
- Additives used in the emulsion preparation of the present invention include, for example, water, buffers, pH adjusters, stabilizers, tonicity agents, animal and vegetable fats and oils, hydrocarbons, fatty acids, fatty acid esters, glycerin fatty acids. Examples thereof include esters, hydrophilic surfactants, lipophilic surfactants and the like.
- examples of water include purified water, water for injection and the like
- examples of buffering agents include phosphates and organic acid salts
- examples of pH adjusting agents include hydrochloric acids and sodium hydroxide
- examples of stabilizers include hydrochloric acids and sodium hydroxide.
- examples include glycerin, propylene glycol, sulfite, etc., as isotonic agents include salt, glucose, sucrose, mannitol, etc., and animal and vegetable fats and oils include olive oil, soybean oil, liver oil, etc.
- Examples include liquid paraffin, squalane, squalane and the like, fatty acids include oleic acid and myristic acid, and fatty acid esters include ethyl oleate, octyldodecyl myristate, cetyl 2-ethylhexanoate and myristic acid.
- Examples include isopropyl and the like, examples of glycerin fatty acid esters include medium-chain fatty acid triglyceride, medium-chain fatty acid diglyceride, and medium-chain fatty acid monoglyceride, and examples of hydrophilic surfactants include polyoxyethylene castor oil and polyoxyethylene hydrogenated castor oil.
- lipophilic surfactants include glycerin monooleate, glycerin dioleate, sorbitan monooleate (Span 80 (registered trademark)), sorbitan sesquioleate, and the like. Examples thereof include sorbitan dioleate, sorbitan trioleate (Span 85 (registered trademark)), PEG-30 dipolyhydroxystearate, and plant-derived surfactants (saponin, etc.).
- the emulsion composition for dilution described in International Publication WO2006 / 078059 Montanide ISA 51 VG (Seppic), Montanide ISA 720 VG (Seppic), Incomplete Freund's Adjuvant (IFA) and the like can be used, but the present invention is not limited thereto.
- the W / O emulsion preparation of the present invention includes a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof, ethyl oleate, octyldodecyl myristate, sorbitan monooleate, glycerin monooleate, polyoxy.
- the liposome means a microvesicle having an internal phase composed of a lipid multilayer such as a bilayer membrane (lipid bilayer) of an amphipathic lipid molecule.
- a lipid multilayer such as a bilayer membrane (lipid bilayer) of an amphipathic lipid molecule.
- the lipid multilayer is preferably a lipid bilayer.
- the liposome preparation of the present invention contains an amphipathic lipid molecule.
- the amphipathic lipid molecule preferably comprises one or more "phospholipids".
- the "phospholipid” include phosphatidylcholine, phosphatidylglycerol, phosphatidylate, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, sphingomyelin and the like.
- Preferred examples of the "phospholipid” include phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, sphingomyelin, and phosphatidylserine.
- the "phospholipid” includes phosphatidylcholine, sphingomyelin, and phosphatidylserine.
- the fatty acid residue in the "phospholipid” is not particularly limited, and examples thereof include saturated or unsaturated fatty acid residues having 14 to 18 carbon atoms, and specifically, myristic acid, palmitic acid, stearic acid, and oleic acid. , Acyl groups derived from fatty acids such as linoleic acid.
- phospholipids derived from natural products such as egg yolk lecithin and soy lecithin, and hydrogenated egg yolk lecithin and hydrogenated soy lecithin (hydrogenated soy phospholipid or hydrogenated soy phosphatidylcholine) obtained by hydrogenating the unsaturated fatty acid residue thereof ) Etc.
- the blending amount (molar fraction) of the phospholipid with respect to the entire liposome membrane component is not particularly limited, but is preferably 30 to 80%, and more preferably 40 to 70%.
- Liposomes containing the compounds of the present invention can contain sterols.
- sterols include cholesterol, ⁇ -citosterol, stigmasterol, campesterol, brushcastellol, ergosterol, fucostellol and the like.
- Preferable examples of sterols include cholesterol.
- the blending amount (molar fraction) of the sterols with respect to the entire liposome membrane component is not particularly limited, but is preferably 0 to 60%, more preferably 10 to 50%, still more preferably 30 to 50%. Can be mentioned.
- the liposome containing the compound of the present invention can contain a polymer-modified lipid.
- the polymer-modified lipid means a lipid modified with a polymer.
- Polymer-modified lipids are represented by "lipids"-"polymers".
- a hydrophilic polymer is preferable, and more preferably, a hydrophilic polymer in which the end of the polymer not bonded to the lipid is alkoxylated is preferable. More preferably, the polymer portion of the polymer-modified lipid includes a hydrophilic polymer in which the end of the polymer not bound to the lipid is methoxylated, ethoxylated or propoxylated.
- the most preferable polymer portion of the polymer-modified lipid is a hydrophilic polymer in which the end of the polymer not bound to the lipid is methoxylated.
- the polymer portion of the polymer-modified lipid is not particularly limited, but for example, polyethylene glycol, polypropylene glycol, polyvinyl alcohol, polyvinylpyrrolidone, methoxypolyethylene glycol, methoxypolypropylene glycol, methoxypolyvinyl alcohol, methoxypolyvinylpyrrolidone, ethoxy.
- polyethylene glycol examples thereof include polyethylene glycol, ethoxypolypropylene glycol, ethoxypolyvinyl alcohol, ethoxypolyvinylpyrrolidone, propoxypolyethylene glycol, propoxypolypropylene glycol, propoxypolyvinyl alcohol, and propoxypolyvinylpyrrolidone.
- the polymer portion of the polymer-modified lipid preferably includes polyethylene glycol, methoxypolyethylene glycol, methoxypolypropylene glycol, ethoxypolyethylene glycol, ethoxypolypropylene glycol, propoxypolyethylene glycol, and propoxypolypropylene glycol.
- the polymer portion of the polymer-modified lipid includes polyethylene glycol, methoxypolyethylene glycol, ethoxypolyethylene glycol, ethoxypolypropylene glycol, and propoxypolyethylene glycol.
- the polymer portion of the polymer-modified lipid is even more preferably polyethylene glycol and methoxypolyethylene glycol.
- the polymer portion of the polymer-modified lipid is most preferably methoxypolyethylene glycol.
- the molecular weight of the polymer portion of the polymer-modified lipid is not particularly limited, but for example, 100 to 10000 daltons, preferably 500 to 8000 daltons, more preferably 1000 to 7000 daltons, and further preferably 1500 to 1500 daltons.
- the lipid portion of the polymer-modified lipid is not particularly limited, and examples thereof include phosphatidylethanolamine and diacylglycerol. Phosphatidylethanolamine having a saturated or unsaturated fatty acid residue having 14 to 18 carbon atoms and diacylglycerol having a saturated or unsaturated fatty acid residue having 14 to 18 carbon atoms are preferable as the lipid portion of the polymer-modified lipid.
- Examples thereof include phosphatidylethanolamine having a saturated fatty acid residue having 14 to 18 carbon atoms and diacylglycerol having a saturated fatty acid residue having 14 to 18 carbon atoms, and more preferably a palmitoyl group or a stearoyl group.
- Examples include phosphatidylethanolamine having a palmitoyl group or diacylglycerol having a palmitoyl group or stearoyl group.
- the most preferred lipid portion of the polymer modified lipid is distearoylphosphatidylethanolamine.
- the blending amount (molar fraction) of the polymer-modified lipid with respect to the entire liposome membrane component is not particularly limited, but is preferably 0 to 20%, more preferably 1 to 10%, still more preferably 2 to 6%. Can be mentioned.
- the liposome containing the compound of the present invention can contain a pharmaceutically acceptable additive.
- Additives include, for example, inorganic acids, inorganic acid salts, organic acids, organic acid salts, sugars, buffers, antioxidants, and polymers.
- the inorganic acid include phosphoric acid, hydrochloric acid, and sulfuric acid.
- the inorganic acid salt include sodium hydrogen phosphate, sodium chloride, ammonium sulfate, and magnesium sulfate.
- organic acids include citric acid, acetic acid, succinic acid, and tartaric acid.
- Examples of the organic acid salt include sodium citrate, sodium acetate, disodium succinate, and sodium tartrate.
- sugars include glucose, sucrose, mannitol, sorbitol, and trehalose.
- Buffering agents include, for example, L-arginine, L-histidine, tromethamole (trishydroxymethylaminomethane, Tris) and salts thereof.
- Antioxidants include, for example, sodium sulfite, L-cysteine, sodium thioglycolate, sodium thiosulfate, ascorbic acid, and tocopherol.
- the polymers include polyvinyl alcohol, polyvinylpyrrolidone, carboxyvinyl polymer, and sodium carboxymethyl cellulose.
- the oily suspension preparation of the present invention dissolves or disperses an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof in an oily component. It can be contained in one or both states.
- Additives used in the oily suspension of the present invention include, for example, buffering agents, pH regulators, stabilizers, isotonic agents, animal and vegetable fats and oils, hydrocarbons, fatty acids, fatty acid esters, solubilizers. Alternatively, a dissolution aid and the like can be mentioned.
- examples of the buffering agent include phosphates and organic acid salts
- examples of the pH adjusting agent include hydrochloric acid and sodium hydroxide
- examples of the stabilizer include glycerin, propylene glycol and myristic acid salt.
- examples of the tonicity agent include salt, glucose, sucrose, mannitol and the like
- examples of animal and vegetable fats and oils include olive oil, soybean oil, liver oil and the like
- examples of hydrocarbons include liquid paraffin, squalene and squalane.
- fatty acids include oleic acid and myristic acid
- fatty acid esters include ethyl oleate, octyldodecyl myristate, cetyl 2-ethylhexanoate, isopropyl myristic acid, sucrose fatty acid ester, glycerin fatty acid ester, and sorbitan fatty acid.
- esters and propylene glycol fatty acid esters examples of the solubilizer or solubilizing agent include glycerin, propylene glycol, macrogol, ethanol and the like.
- an antigen for example, an antigen (tumor antigen or pathogen-derived antigen) and / or a compound represented by the formula (1) or a pharmaceutically acceptable salt thereof is dissolved or dispersed in water to increase the viscosity.
- antigen tumor antigen or pathogen-derived antigen
- Additives used in the hydrogel formulation of the present invention include, for example, purified water, water for injection, buffers, pH regulators, stabilizers, tonicity agents, solubilizers, solubilizers, or thickeners. And so on.
- examples of the buffering agent include phosphates and organic acid salts
- examples of the pH adjusting agent include hydrochloric acid and sodium hydroxide
- examples of the stabilizer include glycerin, propylene glycol and sulfite.
- examples of the tonicity agent include salt, glucose, sucrose, mannitol and the like
- examples of the solubilizing agent or solubilizing agent include glycerin, propylene glycol, macrogol, ethanol and the like
- examples of the thickening agent include carme. Examples thereof include loin sodium, poroxymers, popidone and the like.
- the compound represented by the formula (1) described in the present specification or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of the present invention is an additional drug in addition to the above-mentioned tumor antigen (the present specification). It can also be used in combination with a concomitant drug).
- the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof, or the pharmaceutical composition of the present invention is further used in combination with an "immunomodulator" in addition to the above-mentioned tumor antigen.
- an "immunomodulator” refers to a co-stimulatory signal by interacting with a molecule involved in the transmission of the co-stimulatory signal on the antigen-presenting cell and / or the T cell in T cell activation by the antigen-presenting cell. It refers to anything that controls the transmission of cells or controls the function of molecules that are directly or indirectly involved in the establishment of immune tolerance (immunosuppression) in the immune system.
- tumor antigenic peptides are agents that increase tumor-reactive CTLs in tumors
- their combined use with immunomodulators may reduce the dose of immunomodulators and reduce adverse events. That is, the combined use of the WT1 antigen peptide and the immunomodulator can provide the patient with a treatment method having both higher effect and higher safety.
- the “immunomodulator” can be, but is not limited to, a drug selected from antibodies, nucleic acids, proteins, peptides and small molecule compounds.
- the term “antibody” also includes an antibody fragment. Examples of the antibody fragment include heavy and light chain variable regions (VH and VL), F (ab') 2, Fab', Fab, Fv, Fd, sdFv, scFV and the like of the antibody.
- protein means any protein except antibody.
- Immunune regulators include, for example, immune checkpoint inhibitors, costimulatory molecular agonists, immunostimulators, and small molecule inhibitors.
- Immune checkpoint inhibitors inhibits the immunosuppressive action of cancer cells and antigen-presenting cells.
- Immune checkpoint inhibitors include, but are not limited to, agents for molecules selected from the group consisting of: (1) CTLA-4 (ipilimumab, tremelimumab, etc.); (2) PD-1 (nivolumab, Pembrolizumab, AMP-224, AMP-514 (MEDI0680), pidilizumab (CT-011), etc.); (3) LAG-3 (IMP-321, BMS-986016, etc.); (4) BTLA; (5) KIR (IPH2101) Etc.); (6) TIM-3 (LY3321367, CA-327, etc.); (7) PD-L1 (Durvalumab (MEDI4736), MPDL3280A, BMS-936559, Avelumab (MSB0010718C), BMS-1001, BMS-1116, CA -170, CA-327, etc.); (8) PD-L2
- Co-stimulatory molecule agonists activate T cells by transmitting co-stimulatory molecules on T cells and antigen-presenting cells, and attenuate the immunosuppressive effect of cancer cells and antigen-presenting cells.
- the co-stimulatory molecule agonist agent is not particularly limited, and examples thereof include agents for molecules selected from the following group: (1) 4-1BB (2) 4-1BB-L; (3) OX40 (4) OX40. -L; (5) GITR; (6) CD28; (7) CD40; (8) CD40-L (9) ICOS; (10) ICOS-L; (11) LIGHT; (12) CD27; and (13) DNAM-1.
- Immunoactivators efficiently stimulates killer T cells in the lymph nodes by directly or indirectly activating immune cells such as T cells and dendritic cells.
- Immunoactivators include, but are not limited to, Toll-like receptor (TLR) agonists, interferon gene stimulator (STING) agonists, cytokines, or agents against heat shock proteins (HSPs).
- TLR Toll-like receptor
- STING interferon gene stimulator
- cytokines cytokines
- HSPs heat shock proteins
- the "Toll-like receptor (TLR) agonist” is not particularly limited, but is, for example, a TLR1 / 2 agonist, a TLR2 agonist, a TLR3 agonist (PolyI: C, etc.), a TLR4 agonist (S-type lipopolysaccharide, etc.).
- TLR5 agonists Flangerin, etc.
- TLR6 / 2 agonists MALP-2, etc.
- TLR7 agonists TLR7 / 8 agonists
- TLR7 / 9 agonists hydroxychloroquine sulfate, etc.
- TLR8 agonists Motrimod (VTX-2337), etc.
- TLR9 agonists CpG-ODN, etc.
- TLR11 agonists profillin
- cytokine is not particularly limited, but is, for example, IL-1 ⁇ , IL-1 ⁇ , IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL. -9, IL-10, IL-11, IL-12, IL-13, IL-14, IL-15, IL-16, IL-17, IL-18, interferon (INF) - ⁇ , INF- ⁇ , Examples thereof include INF- ⁇ , SCF, GM-CSF, G-CSF, M-CSF, erythropoetin, tronpopoetin, MIP (macrophage inflammatory protein) and MCP (monocyte chemoattractant protein).
- INF interferon
- HSP heat shock protein
- HSP90 HSP90 inhibitors
- tanespimycin 17-AAG
- luminespimycin AUY-922, NVP-AUY922
- alvespimycin 17-DMAG hydrochloride
- Gnetespib STA-9090
- BIIB021 Tanespimycin (AT13387), Gerdanamycin, NVP-BEP800, SNX-2112 (PF-04928473), PF-4929113 (SNX-5422), KW-2478, XL888, VER155008, VER-50589, CH5138303, VER-49009 , NMS-E973, PU-H71, HSP990 (NVP-HSP990) or KNK437 and the like.
- the "small molecule inhibitor” is not particularly limited, and is, for example, a histone deacetylation inhibitor, a histone demethylation inhibitor, a histone acetylase inhibitor, a histone methylase inhibitor, and a DNA methyltransferase inhibitor.
- Agents anthracycline antibiotics, platinum preparations, MAPK inhibitors, ⁇ -catenin inhibitors, STAT3 inhibitors, NF-kB inhibitors, JAK inhibitors, mTOR inhibitors, IDO inhibitors, COX-2 inhibitors CXCR4 inhibitors Agents and arginase inhibitors and the like.
- histone deacetylase inhibitor is not particularly limited, and is, for example, vorinostat (SAHA, MK0683), entinostat (MS-275), panobinostat (LBH589), tricostatin A (TSA), mocetinostat (MGCD0103).
- histone demethylation inhibitor is not particularly limited, but for example, GSKJ4HCl, OG-L002, JIB-04, IOX1, SP2509, ORY-1001 (RG-6016), GSKJ1, ML324, GSK- Examples include LSD1 and 2HCl.
- histone acetylase inhibitor is not particularly limited, and examples thereof include C646, MG149, Remodelin, and Anacardic Acid.
- histone methylase inhibitor is not particularly limited, and is, for example, Pinometostat (EPZ5676), EPZ005678, GSK343, BIX01294, Tazemetostat (EPZ6438), 3-deazaneplanocin A (DZNeP) HCl, UNC1999. , Entacapone, EPZ015666, UNC0379, EI1, MI-2 (Menin-MLL Inhibitor), MI-3 (Menin-MLL Inhibitor), PFI-2, GSK126, EPZ04777, BRD4770, GSK-2816126 and UNC0631.
- the "DNA methyltransferase inhibitor” is not particularly limited, and examples thereof include decitabine, azatidine, RG108, thioguanine, zebularine, SGI-110, CC-486, SGI-1027, romeguatrib and procainamide hydrochloride. ..
- anthracycline antibiotics inhibit the unwinding of DNA by insertion between DNA strands.
- the anthracycline antibiotic is not particularly limited, and examples thereof include doxorubicin, liposomal doxorubicin, daunorubicin, pirarubicin, epirubicin, idarubicin, aclarubicin, amrubicin, aloin, and mitoxantrone.
- platinum preparation is not particularly limited, and examples thereof include cisplatin, carboplatin, miboplatin, nedaplatin, satraplatin (JM-126), oxaliplatin (ELOXATIN), tryplatin tetranitrate, and DDS preparations thereof.
- the "MAPK inhibitor” is not particularly limited, but is, for example, SB203580, Dramapimod (BIRB796), SB202190 (FHPI), LY2228820, VX-702, SB239063, Pexmetinib (ARRY-614), PH-779804, VX-745 or.
- Examples include TAK-715.
- ⁇ -catenin inhibitor is not particularly limited, but for example, XAV-939, ICG-001, IWR-1-endo, Wnt-C59 (C59), LGK-974, KY02111, IWP-2, IWP- Examples include L6, WIKI4 and FH535.
- STAT3 inhibitor is not particularly limited, but is, for example, S3I-201, Stattic, niclosamide, niclosamide, napabucasin (BBI608), cryptotansinone, HO-3867, WHI-P154, FLLL32, STA-21, WP1066 or SH-4-54 and the like can be mentioned.
- NF-kB inhibitor is not particularly limited, and examples thereof include QNZ (EVP4593), sodium 4-aminosalicylate, JSH-23, phenethyl caffeate, sodium salicylate, androglaholide, and SC75741.
- the "JAK inhibitor” is not particularly limited, but is, for example, ruxolitinib (INCB018424), tofacitinib (CP-6900550) citrate, AZD1480, fedratinib (SAR302503, TG101348), AT9283, tyrohostin B42 (AG-490), momerotinib.
- CYT387 tofacitinib (CP-690550, tasocitinib), WP1066, TG101209, gandtinib (LY2784544), NVP-BSK805 2HCl, baricitinib (LY3009104, INCB0280), AZ960, CEP-31549, AZ960, CEP-317 , S-ruxolitinib (INCB018424), ZM39923HCl, desernotinib (VX-509), Cerdulatinib (PRT062070, PRT2070), filgotinib (GLPG0634), FLLL32, FLLL32, peficitinib (ASP015K) Be done.
- the “mTOR inhibitor” is not particularly limited, and is, for example, silolimus (rapamycin), defololimus (AP23573, MK-8669), everolimus (RAD-001), temsirolimus (CCI-779, NSC683864), zotarolimus (ABT-578). ), And biolimus A9 (umilolimus), AZD8055, KU-0063794, Voctalisib (XL765, SAR245409), MHY1485, ductoric (BEZ235, NVP-BEZ235) or PI-103, Torkinib (PP242) and the like.
- the "IDO inhibitor” is not particularly limited, and examples thereof include NLG919, INCB024360 analog, indoxymod (NLG-8189), and Epacadostat (INCB024360).
- COX2 inhibitor is not particularly limited, and is, for example, valdecoxib, rofecoxib, carprofen, celecoxib, lumiracoxib, tolfenamic acid, nimesulide, niflumic acid, aspirinide, lornoxicum, sodium meclofenacate, diclofenac sodium hydrate, diclofenac hydrate.
- Examples include sodium, ketoprofen, ketoprofen, naproxene sodium, indomethacin, ibuprofen, aspirin, mephenamic acid, bromfenac sodium, oxaprozin, saltprofen and nepafenac.
- CXCR4 inhibitor is not particularly limited, and examples thereof include WZ811, Plerixafor (AMD3100) and Plerixafor 8HCl (AMD3100 8HCl).
- the compounds represented by the formula (1) described herein, and their pharmaceutically acceptable salts, and compositions are "hormonal therapeutic agents,” “immunotherapeutic agents,” and “biologics.” , "Cell Growth Factor”, “Cell Growth Factor Inhibitor”, “Cell Growth Factor Receptor Inhibitor”, “Radiotherapy Agent”, “Auxiliary Agent” or “Chemotherapy Agent” 1 Alternatively, it can be used in combination with a plurality of drugs.
- the peptides, compounds, and pharmaceutically acceptable salts thereof, and combinations thereof can be used in combination with 1 to 5, 1 to 3, or 1 drug selected from the above group. ..
- “Hormonal therapies” include corticosteroids (eg, steroidal anti-inflammatory agents, estrogen preparations, progesterone preparations, androgen preparations, etc.), anti-estrogen agents, estrogen regulators, estrogen synthesis inhibitors, anti-androgen agents, etc. Examples include androgen regulators, androgen synthesis inhibitors, LH-RH agonist preparations, LH-RH antagonist preparations, aromatase inhibitors, steroid lactase inhibitors, pill preparations, or agents that slow the metabolism of retinoids and retinoids.
- corticosteroids eg, steroidal anti-inflammatory agents, estrogen preparations, progesterone preparations, androgen preparations, etc.
- anti-estrogen agents eg., anti-estrogen agents, estrogen regulators, estrogen synthesis inhibitors, anti-androgen agents, etc.
- Examples include androgen regulators, androgen synthesis inhibitors, LH-RH agonist preparations, LH-
- hormone therapeutic agent examples include phosfestol, diethylstilbestrol, flutamidemesterol, chlorotrianicene, methyltestosterone, medroxyprogesterone acetate, megestrol acetate, chlormaginone acetate, ciproterone acetate, danazole, and allyl.
- Estrenol guestlinone, mepartricin, raloxifene, ormeroxyphene, levolmeroxyphene, tamoxyphene citrate, tremiphen citrate, iodoxifene, pills, mepitiostane, testrolactone, aminoglutetiimide, goseleline acetate, busererin, Leuprorelin, leuprorelin, raloxifene, epithiostanol, ethinyl estradiol sulfonate, estramustin, fadrosol hydrochloride, anastrozole, terrorazole, ketoconazole, retrozole, exemestane, borozole, formestane, exemestane, flutamide, bicalutamide, Examples thereof include niltamide, enzalutamide, mifepriston, finasterode, dexamethasone, prednisolone, betamethasone, triam
- immunotherapeutic agent examples include pisibanil, crestin, cisophyllan, lentinan, ubenimex, interferon (IL) - ⁇ , interferon (IL) - ⁇ , interferon (IL) - ⁇ , interleukin, macrophage colony stimulating factor, and granules.
- the “biological agent” is not particularly limited, and is, for example, interleukin-2 (Aldesleukin), interferon- ⁇ , interferon- ⁇ , interferon- ⁇ , erythropoietin (EPO), granulocyte colony stimulating factor (fill). Glastin), Granulocyte Macrophage Colony Stimulator (Sargramostim), IL13-PE38QQR, Bacillus Calmet-Gelan, Revamizol, Octreotide, CPG7909, Antibody, GVAX, Myvax, Favld, Lenalide, Lenalide, Trastuzumab, Ritsumib.
- interleukin-2 Aldesleukin
- interferon- ⁇ interferon- ⁇
- interferon- ⁇ interferon- ⁇
- erythropoietin EPO
- fill granulocyte colony stimulating factor
- Glastin Granulocyte Macrophage Colony Stimulator
- the cell growth factor in the "cell growth factor”, “cell growth factor inhibitor” and “cell growth factor receptor inhibitor” may be any substance as long as it promotes cell growth, for example, molecular weight. Peptides with a value of 20,000 or less include factors that exert their effects at low concentrations by binding to the receptor.
- the "cell growth factor” is not particularly limited, but is, for example, epidermal growth factor (EGF), insulin-Like Growth Factor: IGF (for example, insulin, IGF-1, IGF-2, etc.). )), Transforming Growth Factor: TGF (eg, TGF-alpha, TGF-beta), Nerve Growth Factor: NGF, Brain-derived Neurotrophic Factor: BDNF ), Vasicular Endothelial Growth Factor (VEGF), Colony Stimulating Factor: CSF (for example, Granulocyte-Colony Stimulating Factor: G-CSF), Granulocyte macrophage colony Stimulator (Granulocyte-Macrophage-Colony Stimulating Factor: GM-CSF), Platelet-Derived Growth Factor (PDGF), Erythropoietin (EPO), Fibroblast Growth Factor: Factor (For example, acidic FGF, basic FGF, KGK (Keratinocyte Growth Factor
- the "cell growth factor inhibitor” is not particularly limited, but for example, an epithelial growth factor inhibitor (EGF inhibitor), an insulin-like growth factor inhibitor (IGF inhibitor), a nerve growth factor inhibitor (NGF inhibitor). , Brain-derived neuronutrient factor inhibitor (NGF inhibitor), vascular endothelial cell growth factor inhibitor (VEGF inhibitor), colony stimulator factor inhibitor (CSF inhibitor), platelet-derived growth factor inhibitor (PDGF inhibitor), Examples thereof include erythropoetin inhibitor (EPO inhibitor), fibroblast growth factor inhibitor (FGF inhibitor), hepatocellular growth factor inhibitor (HGF inhibitor), herregulin inhibitor, and angiopoetin inhibitor.
- a cell growth factor inhibitor is synonymous with a growth factor inhibitor.
- the "cell growth factor receptor inhibitor” is not particularly limited, but for example, an epithelial growth factor receptor inhibitor (EGFR inhibitor), an insulin-like growth factor receptor inhibitor (IGFR inhibitor), and a nerve growth factor receptor.
- Body inhibitor NGFR inhibitor
- brain-derived neurotrophic factor receptor inhibitor NGFR inhibitor
- VEGF inhibitor vascular endothelial cell growth factor inhibitor
- CSF inhibitor colony stimulator factor inhibitor
- PDGFR inhibitor platelet-derived Growth factor receptor inhibitor
- EPOR inhibitor erythropoetin receptor inhibitor
- FGFR inhibitor fibroblast growth factor receptor inhibitor
- HGFR inhibitor hepatocellular growth factor receptor inhibitor
- Helegrine receptor inhibitor angiopoetin receptor inhibitor, and the like.
- the cell growth factor receptor inhibitor is synonymous with a growth factor receptor inhibitor.
- the “radiotherapy agent” is not particularly limited, and examples thereof include radioactive substances and radioactive sensitizers.
- auxiliary agent is used to suppress side effects and vomiting caused by anticancer agents, and is not particularly limited, and is, for example, aprepitant, ondansetron, lorazepam, dexamethasone, diphenhydramine, ranitidine, cimetidine, ranitidine, famotidine, cimetidine. , Procrit, epoetin alfa, filgrastim, oprelbekin, leucovorin and granulocyte macrophage colony stimulating factor (GM-CSF) and the like.
- GM-CSF granulocyte macrophage colony stimulating factor
- chemotherapeutic agent is not particularly limited, and includes, for example, an alkylating agent, a platinum preparation, an antimetabolite, a topoisomerase inhibitor, a DNA intercalator, an anti-thread fission agent, an anticancer antibiotic, and a plant-derived anticancer agent.
- Epigenomic drugs, immunomodulators, molecular targeted therapies, new angiogenesis inhibitors and other chemotherapeutic agents are used. A typical example is described below.
- alkylating agent is not particularly limited, but for example, nitrogen mustard, nitrogen mustard hydrochloride-N-oxide, chlorambucil, cyclophosphamide, ifosfamide, thiotepa, carbocon, improsulfan tosylate, busulfan, hydrochloric acid.
- platinum preparation is not particularly limited, and examples thereof include cisplatin, carboplatin, miboplatin, nedaplatin, satraplatin, oxaliplatin, triplatin tetranitrate, and their DDS preparations.
- antimetabolite is not particularly limited, and examples thereof include an antimetabolite, a pyrimidine metabolism inhibitor, a purine metabolism inhibitor, a ribonucleotide reductase inhibitor, and a nucleotide analog.
- the "antimetabolite” is not particularly limited, and is, for example, mercaptopurine, 6-mercaptopurine riboside, thioinosin, methotrexate, pemetrexed, eocitabine, enocitabine, cytarabine, cytarabine ocphosphate, ancitabine hydrochloride, 5-FU drug.
- topoisomerase inhibitor is not particularly limited, and is, for example, doxorubicin, daunorubicin, epirubicin, idarubicin, anthracendion, mitoxantrone, mitomycin C, bleomycin, dactinomycin, precatomycin, irinotecan, camptothecin, rubitecan, verotecan. , Etoposide, teniposide, topotecan, amsacrine and their DDS preparations.
- the "DNA intercalator” is not particularly limited, and examples thereof include proflavine, doxorubicin (adriamycin), daunorubicin, dactinomycin, thalidomide, and DDS preparations thereof.
- the "anti-thread-dividing agent” is not particularly limited, but is, for example, paclitaxel, paclitaxel derivative (eg, DHA paclitaxel, polyglutamated paclitaxel, nab-paclitaxel, paclitaxel micelle, 7 ⁇ -glucosyloxyacetylpaclitaxel, BMS-275183, etc.).
- paclitaxel paclitaxel derivative
- paclitaxel derivative eg, DHA paclitaxel, polyglutamated paclitaxel, nab-paclitaxel, paclitaxel micelle, 7 ⁇ -glucosyloxyacetylpaclitaxel, BMS-275183, etc.
- anticancer antibiotic is not particularly limited, and is, for example, actinomycin D, actinomycin C, mitomycin C, chromomycin A3, mitomycin A, bleomycin hydrochloride, bleomycin sulfate, pepromycin sulfate, daunorubicin hydrochloride, and doxorubicin hydrochloride.
- the "plant-derived anticancer agent” is not particularly limited, and is, for example, irinotecan, nogitecan, etoposide, etoposide phosphate, elibrin, sobzoxane, vinblastine sulfate, vincristine sulfate, bindecine sulfate, teniposide, paclitaxel, paclitaxel injection, docetaxel, DJ- 927, vinorelbine, topotecan and their DDS preparations and the like.
- the "epigenome drug” is not particularly limited, and is, for example, a DNA methylation inhibitor, a histone deacetylase (HDAC) inhibitor, a DNA methyltransferase (DNMT) inhibitor, and a histone deacetylase activator. , Histone demethylase inhibitors and methylated nucleotides and the like.
- the "epigenome drug” is not particularly limited, and is, for example, vorinostat, verinostat, mocetinostat (MGCD0103), entinostat (SNDX-275), romidepsin, azacitidine, decitabine, GSK28795522Hl, SGC707, ORY-1001 (RG). ), PFI-4, SirReal2, GSK2801, CPI-360, GSK503, AMI-1, CPI-169 and their DDS preparations.
- the "immunomodulator” is not particularly limited, and examples thereof include thalidomide, lenalidomide, pomalidomide, and their DDS preparations.
- the “molecular-targeted therapeutic agent” may be a low-molecular-weight compound or an antibody.
- the “molecular-targeted therapeutic agent” is not particularly limited, and examples thereof include kinase inhibitors, proteasome inhibitors, monoclonal antibodies, mTOR inhibitors, TNF inhibitors, and T cell inhibitors.
- the "kinase inhibitor” is not particularly limited, and includes, for example, a tyrosine kinase inhibitor, a serine / threonine kinase inhibitor, a Raf kinase inhibitor, a CDK (cyclin-dependent kinase) inhibitor, and MEK (mitogen-activated factor activation). Protein kinase) inhibitors and the like.
- the "kinase inhibitor” is not particularly limited, but is, for example, imatinib, gefitinib, ellotinib, afatinib, dasatinib, bostinib, bandetanib, snitinib, axitinib, pazopanib, lembatinib, lapatinib, nilotinib, lapatinib, nintedanib, lapatinib.
- proteasome inhibitor is not particularly limited, and examples thereof include bortezomib, carfilzomib, and their DDS preparations.
- the "monoclonal antibody” is not particularly limited, and is, for example, an anti-CD22 antibody, an anti-CD20 antibody, an anti-CD25 antibody, an anti-CD30 antibody, an anti-CD33 antibody, an anti-CD5 antibody, an anti-CD52 antibody, and an anti-epithelial growth factor receptor antibody ( EGFR antibody), anti-vascular endothelial cell proliferation factor antibody (VEGF antibody), anti-TNF- ⁇ antibody, anti-IL-1 receptor antibody, anti-IL-2 receptor antibody, anti-IL-5 receptor antibody, anti-IL-6 receptor antibody, Anti-HER2 antibody, anti-IgE antibody, anti-IgG antibody, anti-RS virus antibody, anti-CCR4 antibody, anti-CTLA-4 (cytotoxic T lymphocyte-related antigen 4, CD152) antibody, anti-PD-1 antibody, anti-RANKL (receptor) Activator of nuclear factor ⁇ B ligand) antibody, anti-c-Met antibody, anti-CXCR4 antibody and the like.
- EGFR antibody anti-vascular endo
- the "monoclonal antibody” is not particularly limited. , Ranibizumab, Celtrizumab, Oclerisumab, Mogamurizumab, Eculizumab, Pertuzumab, Alemtuzumab, Inotzumab, Panitumumab, Ofatumumab, Golimumab, Adalimumab, Ofatumumab, Golimumab, Adalimumab, Ramsilumab, Nibolumab, DDS preparations and the like.
- the “mTOR inhibitor” is not particularly limited, but is, for example, everolimus (RAD001), rapamycin (sirolimus), AZD8055, tacrolimus (CCI-779, NSC683864) KU-0063794, Voctalisib (XL-765, SAR245409), MHY14.
- the "TNF inhibitor” is not particularly limited, and examples thereof include etanercept, lenalidomide (CC-5013), pomalidomide, thalidomide, necrostatin-1 or QNZ (EVP4593).
- T cell inhibitor is not particularly limited, and examples thereof include abatacept.
- the "new angiogenesis inhibitor” is not particularly limited, and is, for example, CM101, IFN- ⁇ , IL-12, platelet factor-4, slamin, semaxanib, thrombospondin, VEGFR antagonist, new angiogenesis inhibitor steroid plus. Heparin, cartilage-derived new angiogenesis inhibitor, matrix metalloproteinase inhibitor, batimastat, marimastat, angiostatin, endostatin, 2-methoxyestradiol, tecogalane, thrombospondin, ⁇ V ⁇ 3 inhibitor, linomid, ADH-1, E7820 And their DDS preparations and the like.
- the "other chemotherapeutic agent” is not particularly limited, and examples thereof include finasteride, sobzoxane, obatocrax, ephaproxilal, tipifarnib, and lonafarnib.
- the pharmaceutical composition of the present invention may further contain other additives, and examples of the additives include surfactants, antioxidants, preservatives, and soothing agents.
- the compound of formula (1) or a pharmaceutically acceptable salt thereof can be administered simultaneously with the antigenic substance (immunogen) or at a time lag, and the dose is usually 5 to 5000 mg / m for warm-blooded animals. 2 (body surface area), i.e., a unit dose in the range of about 0.1 ng / kg to 100 mg / kg, which usually results in a dose effective as a vaccine adjuvant.
- Unit dosage forms such as injectables usually contain, for example, 1 ng to 250 mg of active ingredient. Preferably, it is used at a dose in the range of 1 ng to 50 mg / kg per day. However, this daily dose may also need to vary depending on the host being treated, the specific route of administration and the severity of the disease being treated. Therefore, the optimal dose can also be determined by the individual patient or the practitioner treating the warm-blooded animal.
- treatment is used to include alleviating any, some or all of the symptoms of a disease, in whole or in part, or blocking or delaying the progression of a condition. ..
- prevention refers to primary prevention (preventing the onset of a disease) or secondary prevention (preventing recurrence in patients whose symptoms have been alleviated or cured after the onset of the disease). , Prevention of recurrence).
- the compound of the present invention or a pharmaceutically acceptable salt thereof has immunoadjuvant activity in vitro or in vivo, it maintains the immunogenicity of the antigen (tumor antigen or pathogen-derived antigen) as a vaccine adjuvant. It is useful for enhancing.
- the compound of the present invention or a pharmaceutically acceptable salt thereof has an adjuvant activity of cell-mediated immunity in vitro or in vivo, as a vaccine adjuvant, in order to maintain or enhance the immunogenicity of the tumor antigen. It is useful.
- the compound of the present invention or a pharmaceutically acceptable salt thereof is an immunostimulatory substance (immunostimulator) that is a therapeutic or prophylactic agent for a disease, that is, a substance that induces an antigen (tumor antigen or pathogen-derived antigen) -specific immune response. It can be used to maintain or improve the action of.
- a pharmaceutical composition containing the compound of the present invention or a pharmaceutically acceptable salt thereof, and a substance (also referred to as a tumor antigen or a pathogen-derived antigen) that enhances a tumor antigen-specific immune response or a pathogen-specific immune response is also the present invention. This is one aspect.
- the tumor antigen is not particularly limited, but includes an antigen protein or an antigen peptide (partial peptide) derived from the antigen protein, a tumor antigen protein or a tumor antigen peptide derived from the tumor antigen protein (partial peptide), and these. Examples include a complex with a carrier.
- the compounds of the invention or pharmaceutically acceptable salts thereof treat or prevent cancer by administration in combination with a tumor antigen protein or tumor antigen peptide for cancer immunotherapy.
- cancer include leukemia, myelodysplastic syndrome, multiple myeloma, malignant lymphoma, gastric cancer, colon cancer, lung cancer, breast cancer, germ cell cancer, liver cancer, skin cancer, bladder cancer, prostate cancer, uterine cancer, and cervix.
- Cancer ovarian cancer, brain tumor, bone cancer, pancreatic cancer, head and neck cancer, skin or orbital malignant melanoma, rectal cancer, anal cancer, testicular cancer, oviductal carcinoma, endometrial carcinoma, cervical carcinoma, vaginal carcinoma, External pudendal calcinoma, Hodgkin's disease, non-hodgkin's lymphoma, esophageal cancer, small intestinal cancer, endocrine cancer, thyroid cancer, parathyroid cancer, adrenal cancer, soft tissue sarcoma, urinary tract cancer, penis cancer, acute myeloid leukemia, chronic myeloid leukemia , Acute lymphoblastic leukemia, Chronic or acute leukemia including chronic lymphocytic leukemia, Pediatric solid tumor, Lymphocytic lymphoma, Kidney or urinary tract cancer, Renal calcinoma, Central nervous system (CNS) tumor, Primary CNS Lymphoma, tumor neovascularization, spinal tumor, brain stem glioum
- the compounds of the invention or pharmaceutically acceptable salts thereof are administered in combination with the active ingredient of an infectious disease preventive vaccine for various infectious diseases such as varicella, vulgaris.
- Varicella plantar wart, hepatitis B, hepatitis C, simple herpesvirus, molluscum contagiosum, natural pox, human immunodeficiency virus (HIV), human papillomavirus (HPV), RS virus, norovirus, cytomegalo Viral diseases such as virus (CMV), varicella-zoster virus (VZV), rhinovirus, adenovirus, coronavirus, influenza, parainfluenza; bacterial diseases such as tuberculosis, mycobacterium abium, Hansen's disease; fungal disease, chlamydia , Candida, Aspergillus, Cryptococcus meningitis, Pneumocystis carini, Cryptospolidium disease, Histoplasmosis, Toxoplasm
- infectious diseases such as
- the active ingredient of the infectious disease preventive vaccine is not particularly limited, and examples thereof include substances derived from microorganisms / pathogens such as bacteria, fungi, protozoa, and viruses that cause infectious diseases, and examples thereof include antigen proteins and substances derived from the proteins. Examples thereof include antigen peptides (partial peptides), polysaccharides, lipids, and combinations thereof, or combinations of substances and carriers derived from the microorganism / pathogen.
- the viral antigen peptide derived from the viral antigen is not particularly limited, but for example, influenza matrix protein peptide 58-66 (Jager E et al., Int. J. Cancer 67: 54 (1996)), HPV16 E7 peptide 86. -93 (van Driel WJ et al., Eur. J. Cancer 35: 946 (1999)), HPV E7 peptide 12-20 (Scheibenbogen C et al., J. Immunother 23: 275 (2000)), HPV16 E7 peptide 11-20 (Smith JWI et al., J. Clin. Oncol.
- the carrier used in the present invention is a substance that chemically and / or physically binds an antigen protein or an antigen peptide, and examples thereof include proteins and lipids.
- the carrier is not particularly limited, but is, for example, CRM197 (Vaccine. 2013). Oct 1; 31 (42): 4827-33), KLH (Cancer Immunol Immunother. 2003 Oct; 52 (10): 608-16), virus-like particles (PLoS ONE 5 (3): e9809) and liposomes (J Liposome) Res. 2004; 14 (3-4): 175-89).
- the antigen protein can be prepared by cloning the cDNA encoding the antigen protein and expressing it in the host cell according to a basic book such as Molecular Cloning 2nd Edt., Cold Spring Harbor Laboratoy Press (1989).
- the synthesis of antigenic peptides can be performed according to the method used in ordinary peptide chemistry.
- the synthesis method is described in the literature (Peptide Synthesis, Interscience, New York, 1966; The Proteins, Vol. 2, Academic Press Inc., New York, 1976) and the like. There is a method that is used.
- kits containing the following: a) A pharmaceutical composition containing the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof, or the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof; b) A pharmaceutical composition containing an antigen (tumor antigen or pathogen-derived antigen) or antigen (tumor antigen or pathogen-derived antigen).
- the antigen is not particularly limited as long as it is an antigen that can be used as an active ingredient of a vaccine, but the above-mentioned antigen protein or an antigen peptide (partial peptide) derived from the antigen protein, etc. Examples include a complex and the like.
- the tumor antigen is not particularly limited as long as it is a tumor antigen that can be used as an active ingredient of a cancer vaccine, but the above-mentioned tumor antigen protein or a tumor antigen peptide (partial peptide) derived from the tumor antigen protein and the like can be used. Further, a complex of these and a carrier can be mentioned.
- kits containing the following: a) A pharmaceutical composition containing the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof, or the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof; b) A pathogen-derived antigen or a pharmaceutical composition containing a pathogen-derived antigen.
- the pathogen-derived antigen is not particularly limited as long as it is a pathogen-derived antigen that can be used as an active ingredient of an infectious disease vaccine, but is the above-mentioned pathogen-derived antigen protein or a pathogen-derived antigen peptide derived from the pathogen-derived antigen protein. (Partial peptide) and the like, further include a complex of these and a carrier and the like.
- a compound represented by the formula (1) for the production of a vaccine vaccine adjuvant, or a pharmaceutically acceptable salt thereof.
- a compound represented by the above-defined formula (I) as a vaccine adjuvant in the production of a vaccine for the treatment of cancer or infectious disease, or a pharmaceutically acceptable salt thereof Provide use.
- a compound represented by the formula (1) for producing a vaccine adjuvant for a cancer vaccine or a pharmaceutically acceptable salt thereof.
- the use of the compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof as a vaccine adjuvant in the production of a cancer vaccine for the treatment of cancer is used. provide.
- a compound represented by the formula (1) for producing a vaccine adjuvant for an infectious disease vaccine, or a pharmaceutically acceptable salt thereof.
- a compound represented by the above-defined formula (I) as a vaccine adjuvant in the production of an infectious disease vaccine for the treatment of infectious diseases, or a pharmaceutically acceptable salt thereof Provide use.
- a step of administering to a patient the compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof together with an antigen is included.
- an antigen tumor antigen or pathogen-derived antigen
- treatment of cancer, prevention of progression, or administration of a compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof to a patient together with a tumor antigen is included.
- a compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof is included.
- treatment and progression of an infectious disease which comprises a step of administering to a patient a compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof together with a pathogen-derived antigen.
- a compound represented by the above-defined formula (I) or a pharmaceutically acceptable salt thereof together with a pathogen-derived antigen.
- Fmoc 9-fluorenylmethyloxycarbonyl Boc: tert-butoxycarbonyl Alko: p-alkoxybenzyl alcohol
- PEG polyethylene glycol tBu: tert-butyl HBTU: O- (benzotriazole-1-yl) -N, N, N ', N'-Tetramethyluronium hexafluorophosphate
- DIPEA N, N-diisopropylethylamine
- DMF N, N-dimethylformamide
- TFA Trifluoroacetic acid
- TIS Triisopropylsilane
- THF tetrahydrofuran
- TBS tert-butyldimethylsilyl group
- TBDPS tert-butyldiphenylsilyl group
- LCMS liquid chromatography mass spectrometry
- a CS336X peptide synthesizer manufactured by CS Bio was used for solid phase synthesis, and deprotection of the Fmoc group was carried out by treating with a DMF solution of 20% piperidine for 5 minutes and 20 minutes. Coupling of the protected amino acids was carried out by reacting with a DMF solution of 1.05 mmol of protected amino acids, 1 mmol of HBTU and 2 mmol of DIPEA for 1 hour. The obtained resin was washed with DMF and ether and dried under reduced pressure to obtain a peptide resin.
- the obtained crude peptide was dissolved in a mixed solution of 20% aqueous acetic acid and acetonitrile (volume ratio 1/1), purified under the conditions shown below, and RMPNAPYL (Arg-Met-Phe-Pro-Asn-Ala-Pro-). 0.16 g of the trifluoroacetate of Tyr-Leu) (SEQ ID NO: 1) was obtained. From the obtained trifluoroacetate, an acetate was prepared according to a conventional method and used for evaluation.
- Triethylamine (4.90 mL) and tert-butyldimethylchlorosilane (3.54 g) were added to a DMF (20 mL) solution of the compound (3.91 g) obtained in Step 1 of Reference Example 9, and the mixture was stirred at room temperature for 2 hours. ..
- the reaction mixture was diluted with ethyl acetate, washed with water and saturated brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- Reference example 14 Synthesis of 3- ⁇ [2-amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -4-hydroxybenzaldehyde
- dichloromethane 5 mL
- boron tribromide 1.0 M, 0.8 mL
- the mixture was stirred at room temperature for 6 hours.
- saturated aqueous sodium hydrogen carbonate was added to the reaction solution, the mixture was extracted with chloroform, the organic layer was dried over sodium sulfate, filtered, and then concentrated.
- Example 1 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14-tetraoxa- Synthesis of 2-azahexadecane-16-ol trifluoroacetate
- pentyl-2,4-diamine 75mg
- ⁇ [4- (chloromethyl) -2-methoxyphenyl] methyl ⁇ -6-methyl -N 4 9 76 mg
- potassium carbonate 65 mg
- potassium iodide 67 mg
- Example 3 1-(3- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -4-methoxyphenyl) -2-methyl-5,8,11,14-tetraoxa- Synthesis of 2-azahexadecane-16-ol trifluoroacetate
- Reference Example 9 101 mg
- acetic acid 5.4 ⁇ L
- sodium triacetoxyborohydride 199 mg
- Example 5 4-[(2-Amino-4- ⁇ [(2S) -1-hydroxypentane-2-yl] amino ⁇ -6-methylpyrimidine-5-yl) methyl] -N- (20-hydroxy-3,6) , 9, 12, 15, 18-Hexaoxaicosan-1-yl) -3-methoxybenzamide synthesis
- a known compound 4-[(2-amino-4- ⁇ [(2S) -hydroxypentane-2-yl] amino ⁇ -6-methylpyrimidine-5-yl) methyl] -3-methoxybenzoic acid ((30 mg) )
- DMF diisopropylethylamine
- 20-amino-3,6,9,12,15,18-hexaoxaicosan-1-ol 31.3 mg
- HATU 33.
- Example 7 5- ⁇ [2-Methoxy-4- (2,5,8,11,14-pentaoxapentadecane-1-yl) phenyl] methyl ⁇ -6-methyl-N 4 -pentylpyrimidine-2,4-diamine Synthetic Sodium hydride (7.2 mg, content> 55%) was added to 2,5,8,11-tetraoxatridecane-13-ol and stirred at room temperature for 1 hour, and then the known compound 5- ⁇ [[ 4- (Chloromethyl) -2-methoxyphenyl] methyl ⁇ -6-methyl-N 4 -pentylpyrimidine-2,4-diamine (20 mg) was added, and the mixture was stirred at 60 ° C. for 3 hours.
- Example 8 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14,17, Synthesis of 20,23,26,29-Nonaoxa-2-azahentriacontane-3-1-ol Known compound 5- ⁇ [4- (chloromethyl) -2-methoxyphenyl] methyl ⁇ -6-methyl-N 4 -pentylpyrimidine-2,4-diamine (40 mg), Reference Example 17 (64.6 mg) , Potassium iodide (36.6 mg) and potassium carbonate (30.5 mg) were mixed with acetonitrile (1 mL), and the mixture was stirred at 80 ° C.
- Example 9 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14,17, Synthesis of 20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71-tricosaoxa-2-azatriheptacontane-73-ol
- the title compound (31.6 mg) was obtained by carrying out the reaction in the same manner as in Example 8 using Reference Example 18 (83 mg).
- Example 10 1-(4- ⁇ [2-Amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) -2-methyl-5,8,11,14,17, 20, 23, 26, 29, 32, 35, 38, 41, 44, 47, 50, 53, 56, 59, 62, 65, 68, 71, 74, 77, 80, 83, 86, 89, 92, Synthesis of 95,98,101,104,107-pentatriacontaoxa-2-azanonahexane-109-ol
- the title compound (26.3 mg) was obtained by carrying out the reaction in the same manner as in Example 8 using Reference Example 19 (95 mg).
- Example 11 12-[(4- ⁇ [2-amino-4-methyl-6- (pentylamino) pyrimidin-5-yl] methyl ⁇ -3-methoxyphenyl) methyl] -3,6,9,15,18,21 -Synthesis of hexaoxa-12-azatricosan-1,23-diol Using 3,6,9,15,18,21-hexaoxa-12-azatricosan-1,23-diol (40.7 mg), the title compound (13.1 mg) was obtained by carrying out the reaction in the same manner as in Example 1. Obtained.
- Test Example 1 Human TLR7 Reporter Gene Assay The TLR7 / NF- ⁇ B / SEAPorter TM HEK293 cell line (Imenex Corporation) is a stable co-expression that expresses full-length human TLR7 and secretory alkaline phosphatase (SEAP) reporter genes under transcriptional control of NF- ⁇ B response elements. Transfect cell line. TLR7 expression in this cell line has been tested by flow cytometry. Stablely expressed transformants were selected using the antibiotics Blasticidin and Genetisin. TLR signaling leads to NF- ⁇ B translocation and promoter activation results in SEAP gene expression. The cells are incubated with the compounds synthesized in Examples and References at 37 ° C.
- SEAP secretory alkaline phosphatase
- TLR7-specific activation was evaluated.
- the degree of human TLR7 activation of the compounds of the invention was evaluated using the human TLR7 reporter gene assay and is shown in Tables 3 and 4 as the concentration of the compound (EC 50 ) resulting in half the maximum level of SEAP.
- Test Example 2 The Mouse TLR7 Reporter Gene Assay HEK-Blue TM mTLR7 Cell Line (Invivogen) is a stable cotransfected cell line that expresses full-length mouse TLR7 and the secretory SEAP reporter gene under transcriptional control of the NF- ⁇ B response element. TLR7 expression in this cell line has been tested by RT-PCR. Stablely expressed transformants were selected using the antibiotics Blasticidin and Zeocin. TLR signaling leads to NF- ⁇ B translocation and promoter activation results in SEAP gene expression. The cells were incubated with the compounds synthesized in Examples and References at 37 ° C.
- TLR7 specific activation was evaluated.
- the degree of mouse TLR7 activation of the compounds of the invention was evaluated using the mouse TLR7 reporter gene assay and is shown in Tables 5 and 6 as the concentration of the compound (EC 50 ) resulting in half the maximum level of SEAP.
- the adjuvant activity was evaluated by an antigen-specific cytotoxic T cell (CTL) induction test.
- CTL cytotoxic T cell
- CYTWNQMNL SEQ ID NO: 2 contained in the compound represented by the formula No. 4 is an HLA-A * 24:02 restrictive WT1 protein-derived antigen peptide.
- HLA-A * 24:02 transgenic mice C57BL / 6CrHLA-A2402 / K b are mice expressing chimeric HLA of H-2K b are HLA-A * 24:02 and murine MHC is a human MHC Therefore, by using this mouse, it is possible to induce CTL with a peptide capable of inducing CTL in HLA-A * 24 : 02 positive humans. (Int J Cancer. 2002; 100: 565-70).
- cocktail vaccine b induces CTL against the target peptide (SEQ ID NO: 2) produces IFN ⁇ when the above mouse-derived splenocytes are restimulated with the peptide (SEQ ID NO: 2). It was judged by measuring whether to do it. Furthermore, whether or not the compound synthesized in Example 1 or the compound synthesized in Reference Example 12 exerts adjuvant activity in vivo depends on the number of CTLs induced by the administration of cocktail vaccine b and the cocktail vaccine b. The number of CTLs induced by administering a vaccine prepared by adding the compound synthesized in Example 1 or the compound synthesized in Reference Example 12 was compared, and it was judged whether or not the amount was increased.
- the concentration after dilution of the compound represented by the formula No. 4 is 6 mg / mL, SEQ ID NO: 3.
- the peptide represented by (1) was mixed with water for injection so as to be 4.5 mg / mL.
- Cocktail vaccine b prepared by mixing this peptide diluent with an equal amount of Montanede ISA 51 VG and emulsifying it, 300 ⁇ g / animal of the compound represented by the formula No. 4 and 225 ⁇ g of the peptide represented by SEQ ID NO: 3. /
- the mice were administered intradermally to the ridge.
- a vaccine prepared by adding the compound synthesized in Example 1 to the peptide diluent used for preparing cocktail vaccine b is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 225 ⁇ g / animal and the amount of the compound synthesized in Example 1 was 100 ng / animal.
- a vaccine prepared by adding the compound represented by Reference Example 12 to the peptide diluent used for preparing the cocktail vaccine b is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 225 ⁇ g / animal and the amount of the compound synthesized in Reference Example 12 was 69 ng / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- the IFN ⁇ ELISPOT assay kit was used for the measurement of IFN ⁇ production.
- the day before splenocyte preparation, ELISPOT plates were treated with anti-mouse IFN ⁇ antibody and blocked on the day with RPMI 1640 medium containing 10% fetal bovine serum (FBS).
- FBS fetal bovine serum
- the prepared HLA-A * 24 02 transgenic mouse-derived splenocytes were seeded at 2.5 ⁇ 10 5 cells / well on a blocked ELISPOT plate.
- the peptide (SEQ ID NO: 2) was added to splenocytes at a final concentration of 10 ⁇ g / mL in the presence of 0.1% (v / v).
- In vitro peptide restimulation was applied by culturing the peptide-added splenocytes overnight at 37 ° C. and 5% CO 2 . After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured by Immuno Spot Analyzer (manufactured by CTL).
- FIG. 1 The results of the IFN ⁇ ELISPOT assay using HLA-A * 24 : 02 transgenic mice are shown in FIG.
- the vertical axis shows the number of cells that produced IFN ⁇ in response to a stimulus in the seeded cells, and the horizontal axis shows the vaccine administered to mice.
- the black and white bars in FIG. 1 show the results of culturing HLA-A * 24 : 02 transgenic mouse-derived splenocytes in the presence and absence of the peptide represented by SEQ ID NO: 2.
- the difference between the values of the black bar and the white bar indicates the number of IFN ⁇ -producing cells, that is, CTLs, which are specific to the peptide represented by SEQ ID NO: 2 induced in the mouse body by the administration of the vaccine.
- the value of the white bar is hardly recognized. This indicates that mouse splenocytes hardly responded in the absence of the peptide of interest.
- the induction of peptide-reactive CTL represented by SEQ ID NO: 2 was confirmed in HLA-A * 24 : 02 transgenic mice to which cocktail vaccine b was administered.
- the number of peptide-reactive CTLs represented by SEQ ID NO: 2 was increased by adding the compound synthesized in Example 1 or the compound synthesized in Reference Example 12 to the cocktail vaccine b. Furthermore, an increase in the number of CTLs was observed more when the compound synthesized in Example 1 was added to the cocktail vaccine b than when the compound synthesized in Reference Example 12 was added to the cocktail vaccine b. It was.
- Test Example 4 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mice Regarding the in vivo adjuvant activity of the compound synthesized in Example 1 and the compound synthesized in Reference Example 12, it was synthesized in Reference Example 8.
- Examples were added to a cocktail vaccine (hereinafter referred to as “cocktail vaccine a”) in which the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 were mixed with a preliminary emulsion composition.
- a vaccine was prepared by adding the compound synthesized in 1 or the compound synthesized in Reference Example 12, and the vaccine was administered to HLA-A * 02 : 01 gene-introduced mice to obtain adjuvant activity by an antigen-specific CTL induction test. evaluated.
- Equation number 4 (In the formula, the bond between C and C represents a disulfide bond.)
- RMFPNAPYL SEQ ID NO: 1 contained in Formula No. 4
- VLDFAPPGA SEQ ID NO: 4 contained in the peptide represented by SEQ ID NO: 3 are HLA-A * 02:01 restrictive WT1 protein-derived antigen peptides.
- HLA-A * 02:01 transgenic mice C57BL / 6CrHLA-A2.1DR1 lacked mouse MHC and were of human MHC HLA-A * 02:01 and mouse MHC H-2D b .
- it is possible to induce CTL with a peptide that can induce CTL in HLA-A * 02 : 01 positive humans (Eur J Immunol. 2, 004; 34: 3, 060-9). ).
- cocktail vaccine a induces CTL against an antigenic peptide (SEQ ID NO: 1 or SEQ ID NO: 4) produces IFN ⁇ when the above-mentioned mouse-derived splenocytes are restimulated with the target peptide. It was judged by measuring whether to do it. Furthermore, whether or not the compound synthesized in Example 1 or the compound synthesized in Reference Example 12 exerts adjuvant activity in vivo depends on the number of CTLs induced by the administration of cocktail vaccine a and the cocktail vaccine a. The number of CTLs induced by administration of the vaccine prepared by adding the compound synthesized in Example 1 or the compound synthesized in Reference Example 12 was compared, and it was judged whether or not the number was increased.
- the in vivo adjuvant activity was evaluated as follows. 0.312 g of sodium dihydrogen phosphate dihydrate was dissolved in 80 g of water for injection. Ethyl oleate 14.0 g, octyldodecyl myristate 14.0 g, sorbitan monooleate 2.0 g, glycerin monooleate 2.8 g, polyoxyethylene hydrogenated castor oil 200.4 g, and glycerin 0.4 g were mixed.
- Cocktail vaccine a prepared by mixing an equal amount of the peptide diluted solution with the above-mentioned preliminary emulsifying composition and emulsifying it, and containing 300 ⁇ g / animal of the compound represented by the formula No. 4 and the peptide represented by SEQ ID NO: 3 It was administered intradermally to the ridge of mice so as to be 225 ⁇ g / animal.
- a vaccine prepared by adding the compound synthesized in Example 1 to a peptide diluent used for preparing cocktail vaccine a is represented by 300 ⁇ g / animal of the compound represented by the formula No.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide to be produced was 225 ⁇ g / animal and the amount of the compound synthesized in Example 1 was 32.5 ng / animal.
- a vaccine prepared by adding the compound synthesized in Reference Example 12 to a peptide diluent for preparing cocktail vaccine a is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide to be produced was 225 ⁇ g / animal and the amount of the compound synthesized in Reference Example 12 was 22.5 ng / animal.
- mice were euthanized with CO 2 gas and then the spleen was removed to prepare splenocytes.
- the IFN ⁇ ELISPOT assay kit was used for the measurement of IFN ⁇ production.
- ELISPOT plates were treated with anti-mouse IFN ⁇ antibody and blocked on the day with RPMI 1640 medium containing 10% FBS.
- prepared splenocytes 1.25 ⁇ 10 5 cells / well, were seeded into the blocked ELISPOT plates.
- the peptide (SEQ ID NO: 1 or SEQ ID NO: 4) was added to splenocytes at a final concentration of 10 ⁇ g / mL in the presence of 0.1% (v / v) DMSO.
- In vitro peptide restimulation was applied by culturing the peptide-added splenocytes overnight at 37 ° C. and 5% CO 2 . After culturing, the supernatant was removed and the ELISPOT plate was colored according to the attached protocol. The number of colored spots was measured by the ImmunoSpot Analyzer.
- Test Example 5 Enhancement effect of vaccine on in vivo tumor growth inhibitory effect HLA-A * 24: 02 3-methylcholanthrene suspended in corn oil was administered into the ventral skin of gene-introduced mice, and HLA-A-from the tumor that developed at the administration site. A * 2402 / Kb-expressing tumor cells were obtained.
- a cell line in which the WT1 antigen peptide (SEQ ID NO: 2) was stably expressed in the cells (also referred to as MCA-A24 / Kb-WT1 tumor cells in the present specification) was suspended in a Hanks balanced salt solution, and HLA- It was transplanted into the ventral intradermal of a * 24:02 transgenic mice (per mouse, 5 ⁇ 10 5 cells).
- a group to administer the vehicle (group a), a group to administer the vaccine (group b), and a group to administer the vaccine to which the compound synthesized in Example 1 was added (group c) were set.
- Six mice were used for each group. Seven days before and seven days after the tumor cells were transplanted, the mice in group a were mixed with an equal amount of Montanede ISA 51 VG to emulsify the composition containing water for injection, and then administered intradermally to the ridge (1). 0.1 mL / animal per dose). Mice in group b were mixed with an equal amount of Montanede ISA 51 VG to emulsify a composition containing the compound represented by the formula No.
- the average value of the tumor volume 27 days after the tumor transplantation of 6 mice in each group is shown in FIG.
- the vaccine (group b) significantly suppressed the growth of tumor cells relative to the vehicle (group a) (nonparametric Dunnett's multiplex test, *: p ⁇ 0.05). Furthermore, by adding the compound synthesized in Example 1 to the vaccine, the growth of tumor cells was more significantly suppressed (group c, **: p ⁇ 0.01).
- Example 6 Evaluation of in vivo adjuvant activity using HLA-A * 02:01 gene-introduced mice
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by SEQ ID NO: 6 synthesized in Reference Example 6.
- a vaccine was prepared by adding the compound synthesized in Example 1 to a vaccine prepared by mixing a peptide with Montanede ISA 51 VG (hereinafter referred to as "vaccine c"), and this was HLA-A * 02:01. It was administered to transgenic mice and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the peptide GLYDGMEHL represented by SEQ ID NO: 6 is an antigen peptide derived from the HLA-A * 02 : 01 restrictive MAGE-A10 protein.
- Whether or not administration of vaccine c induces CTL against the antigenic peptide is to measure whether IFN ⁇ is produced when the above-mentioned mouse-derived splenocytes are restimulated with the target peptide. Judged by. Furthermore, whether or not the compound synthesized in Example 1 exerts adjuvant activity in vivo depends on the number of CTLs induced by the administration of vaccine c and the compound synthesized in Example 1 is added to vaccine c. The number of CTLs induced by administration of the vaccine prepared in the above was compared and judged whether or not the number was increased.
- the peptide represented by SEQ ID NO: 6 was dissolved in DMSO and then mixed with water for injection so that the diluted concentration was 0.1 mg / mL.
- Vaccine c prepared by mixing this peptide diluent with an equal amount of Montanede ISA 51 VG and emulsifying it was administered into the ridge skin of mice so that the peptide represented by SEQ ID NO: 6 was 10 ⁇ g / animal. did.
- a vaccine prepared by adding the compound synthesized in Example 1 to the peptide diluent used for preparing the vaccine c is synthesized in Example 1 with 10 ⁇ g / animal of the peptide represented by SEQ ID NO: 6.
- the compound was administered intradermally to the ridge of the mouse so that the amount of the compound was 0.4 nmol / animal. Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes. Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 6) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 4 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- SEQ ID NO: 6 a peptide having a final concentration of 10 ⁇ g / mL
- the vertical axis represents the average number of cells that produced IFN ⁇ in response to stimulation in seeded cells of 3 mice in each group, and the horizontal axis represents the vaccine administered to the mice.
- the black and white bars show the results of culturing HLA-A * 02 : 01 transgenic mouse-derived splenocytes in the presence and absence of the peptide represented by SEQ ID NO: 6.
- the number of peptide-reactive CTLs represented by SEQ ID NO: 6 was higher due to the addition of the compound synthesized in Example 1 to vaccine c than when the compound was not added. Was done.
- Example 7 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mice
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by SEQ ID NO: 5 synthesized in Reference Example 5.
- a vaccine was prepared by adding the compound synthesized in Example 1 to a vaccine prepared by mixing a peptide with Montanede ISA 51 VG (hereinafter referred to as "vaccine d"), and this was HLA-A * 02:01. It was administered to transgenic mice and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the peptide VLQELNVTV represented by SEQ ID NO: 5 is an antigen peptide derived from the HLA-A * 02 : 01 restrictive Proteinase-3 protein.
- Whether or not administration of vaccine d induces CTL against the antigenic peptide is to measure whether IFN ⁇ is produced when the above-mentioned mouse-derived splenocytes are restimulated with the target peptide. Judged by. Furthermore, whether or not the compound synthesized in Example 1 exerts adjuvant activity in vivo depends on the number of CTLs induced by the administration of vaccine d and the compound synthesized in Example 1 is added to vaccine d. The number of CTLs induced by administration of the vaccine prepared in the above was compared and judged whether or not the number was increased.
- the peptide represented by SEQ ID NO: 5 was dissolved in DMSO and then mixed with water for injection so that the diluted concentration was 2 mg / mL.
- Vaccine d prepared by mixing this peptide diluent with an equal amount of Montanede ISA 51 VG and emulsifying it was administered into the ridge skin of mice so that the peptide represented by SEQ ID NO: 5 was 100 ⁇ g / animal. did.
- a vaccine prepared by adding the compound synthesized in Example 1 to a peptide diluent used for preparing vaccine d is synthesized in Example 1 with 100 ⁇ g / animal of the peptide represented by SEQ ID NO: 5.
- the compound was administered intradermally to the ridge of the mouse so that the amount of the compound was 330 ng / animal.
- the mice were euthanized with CO 2 gas and then the spleen was removed to prepare splenocytes.
- Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 5) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 4 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- the vertical axis represents the average number of cells that produced IFN ⁇ in response to stimulation in seeded cells of 3 mice in each group, and the horizontal axis represents the vaccine administered to the mice.
- the black and white bars show the results of culturing HLA-A * 02 : 01 transgenic mouse-derived splenocytes in the presence and absence of the peptide represented by SEQ ID NO: 5.
- the number of peptide-reactive CTLs represented by SEQ ID NO: 5 was higher due to the addition of the compound synthesized in Example 1 to vaccine d as compared with the case where no compound was added.
- Example 1 the compound synthesized in Example 1
- Example 1 the compound synthesized in Example 1
- Test Example 8 Evaluation of in vivo adjuvant activity using HLA-A * 24 : 02 gene-introduced mice
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by SEQ ID NO: 18 synthesized in Reference Example 20.
- a vaccine was prepared by adding the compound synthesized in Example 1 to a vaccine in which a peptide was mixed with Montanede ISA 51 VG (hereinafter referred to as "vaccine e"), and this was used as an HLA-A * 24:02 gene-introduced mouse.
- the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the peptide TYAGCLSQIF represented by SEQ ID NO: 18 is an antigen peptide derived from the HLA-A * 24: 02 restrictive Or7c1 protein.
- Whether or not administration of vaccine e induces CTL against the antigenic peptide is to measure whether IFN ⁇ is produced when the above-mentioned mouse-derived splenocytes are restimulated with the target peptide. Judged by. Furthermore, whether or not the compound synthesized in Example 1 exerts adjuvant activity in vivo depends on the number of CTLs induced by the administration of vaccine e and the compound synthesized in Example 1 is added to vaccine e. The number of CTLs induced by administration of the vaccine prepared in the above was compared and judged whether or not the number was increased.
- the peptide represented by SEQ ID NO: 18 was dissolved in DMSO and then mixed with water for injection so that the diluted concentration was 3 mg / mL.
- Vaccine e prepared by mixing this peptide diluent with an equal amount of Montanede ISA 51 VG and emulsifying it was administered into the ridge skin of mice so that the peptide represented by SEQ ID NO: 18 was 300 ⁇ g / animal. did.
- a vaccine prepared by adding the compound synthesized in Example 1 to the peptide diluent used for preparing the vaccine e is synthesized in Example 1 with 300 ⁇ g / animal of the peptide represented by SEQ ID NO: 18.
- the compound was administered intradermally to the ridge of the mouse so that the amount of the compound was 0.4 nmol / animal. Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes. Peptide in vitro by seeding splenocytes to which a peptide (SEQ ID NO: 18) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 3 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2 Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- SEQ ID NO: 18 a peptide having a final concentration of 10 ⁇ g / mL
- the vertical axis represents the average number of cells that produced IFN ⁇ in response to stimulation in seeded cells of 3 mice in each group, and the horizontal axis represents the vaccine administered to the mice.
- the black and white bars show the results of culturing HLA-A * 24 : 02 transgenic mouse-derived splenocytes in the presence and absence of the peptide represented by SEQ ID NO: 18.
- the number of peptide-reactive CTLs represented by SEQ ID NO: 18 was higher due to the addition of the compound synthesized in Example 1 to the vaccine e than when the compound was not added. Was done.
- Test Example 9 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mice Regarding the in vivo adjuvant activity of the compounds synthesized in Examples 8, 9 and 10 and the compound synthesized in Reference Example 12, reference Example 8, 9, or 10 was added to a cocktail vaccine b in which the compound represented by the formula No. 4 synthesized in Example 8 and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 were mixed with Montanide ISA51 VG.
- a vaccine was prepared by adding the compound synthesized in (1) or the compound synthesized in Reference Example 12, and the vaccine was administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test. did.
- the concentration after dilution of the compound represented by the formula No. 4 is 3 mg / mL, SEQ ID NO: 3.
- the peptide represented by (2) was mixed with water for injection so as to be 2.25 mg / mL.
- Cocktail vaccine b prepared by mixing this peptide diluent with an equal amount of Montanede ISA51 VG and emulsifying it, 300 ⁇ g / animal of the compound represented by the formula No. 4 and 225 ⁇ g / g of the peptide represented by SEQ ID NO: 3.
- the mice were administered intradermally to the ridge.
- a vaccine prepared by adding the compound synthesized in Example 8, 9, or 10 or the compound synthesized in Reference Example 12 to the peptide diluent for preparing the cocktail vaccine b is prepared by formula No. 4.
- the compound represented by 300 ⁇ g / animal, the peptide represented by SEQ ID NO: 3 is 225 ⁇ g / animal, the compound synthesized in Examples 8, 9, or 10 or the compound synthesized in Reference Example 12 is 0.4 nmol / animal. It was administered intradermally to the ridge of the mouse. Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 . In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- the number of peptide-reactive CTLs represented by SEQ ID NO: 1 or SEQ ID NO: 4 is the cocktail vaccine b of the compound synthesized in Example 8, 9, or 10 or the compound synthesized in Reference Example 12.
- the addition to more was observed compared to the case without the addition of the compound.
- the addition of the compound synthesized in Examples 8, 9 or 10 to Cocktail Vaccine b resulted in more peptide reactions.
- Sexual CTL was observed.
- Test Example 10 Evaluation of in vivo adjuvant activity using HLA-A * 02:01 gene-introduced mice Regarding the in vivo adjuvant activity of the compound synthesized in Example 11, the same cocktail vaccine b as in Test Example 9 was applied to Example 11.
- a vaccine was prepared by adding the compound synthesized in (1), administered to HLA-A * 02:01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the concentration after dilution of the compound represented by the formula No. 4 is 3 mg / mL, SEQ ID NO: 3.
- the peptide represented by (2) was mixed with water for injection so as to be 2.25 mg / mL.
- Cocktail vaccine b prepared by mixing this peptide diluent with an equal amount of Montanede ISA51 VG and emulsifying it, 300 ⁇ g / animal of the compound represented by the formula No. 4 and 225 ⁇ g / g of the peptide represented by SEQ ID NO: 3.
- the mice were administered intradermally to the ridge.
- a vaccine prepared by adding the compound synthesized in Example 11 to the peptide diluent used for preparing the cocktail vaccine b is represented by the compound represented by the formula No. 4 in an amount of 300 ⁇ g / animal and SEQ ID NO: 3.
- the peptide to be produced was 225 ⁇ g / animal, and the compound synthesized in Example 11 was 0.4 nmol / animal, which was administered intradermally to the ridge of the mouse. Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 . In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 11 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- the compound synthesized in Example 1 was added to a cocktail vaccine (hereinafter referred to as "cocktail vaccine f") containing the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 as the emulsified composition 1.
- a vaccine was prepared, administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the emulsified composition 1 was prepared as follows. Soybean oil 95.8% (w / w), dipolyhydroxystearate PEG-30 3% (w / w), and polysorbate 80 1.2% (w / w) were mixed to prepare an oil phase mixture. The amount prepared was increased or decreased as needed.
- the compound represented by the formula No. 4 was mixed with a pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) so that the compound represented by the formula No. 4 was 3 mg / mL and the peptide represented by the SEQ ID NO: 3 was 2.25 mg / mL.
- This peptide diluent was mixed with an equal amount of an oil phase mixture and emulsified to obtain a cocktail vaccine f prepared. This was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 300 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 225 ⁇ g / animal.
- a vaccine prepared by adding the compound synthesized in Example 1 to the peptide diluent used for preparing the cocktail vaccine f is represented by 300 ⁇ g / animal of the compound represented by the formula No.
- the peptide was administered intradermally to the ridge of the mouse so that the peptide to be produced was 225 ⁇ g / animal and the compound synthesized in Example 1 was 0.04 nmol / animal or 0.4 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 12 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- the compound synthesized in Example 1 was added to a cocktail vaccine (hereinafter referred to as "cocktail vaccine g") containing the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 as the emulsified composition 2.
- cocktail vaccine g cocktail vaccine
- a vaccine was prepared, administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the emulsified composition 2 was prepared as follows. Medium-chain fatty acid triglyceride (Miguriol 812) 95.8% (w / w), dipolyhydroxystearate PEG-30 3% (w / w), polysorbate 80 1.2% (w / w) are mixed and oiled. It was a phase mixture. The amount prepared was increased or decreased as needed.
- the compound represented by the formula No. 4 was mixed with a pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) so that the compound represented by the formula No. 4 was 3 mg / mL and the peptide represented by the SEQ ID NO: 3 was 2.25 mg / mL.
- This peptide diluent was mixed with an equal amount of an oil phase mixture and emulsified to obtain a cocktail vaccine g prepared. This was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 300 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 225 ⁇ g / animal.
- a vaccine prepared by adding the compound synthesized in Example 1 to a peptide diluent used for preparing cocktail vaccine g is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the peptide to be produced was 225 ⁇ g / animal and the compound synthesized in Example 1 was 0.04 nmol / animal or 0.4 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 13 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- the compound synthesized in Example 1 was added to a cocktail vaccine (hereinafter referred to as "cocktail vaccine h") using the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 as the emulsified composition 3.
- a vaccine was prepared, administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the emulsified composition 3 was prepared as follows. Isopropyl myristate 95.8% (w / w), PEG-30 dipolyhydroxystearate (w / w), and polysorbate 80 1.2% (w / w) were mixed to prepare an oil phase mixture. The amount prepared was increased or decreased as needed.
- the compound represented by the formula No. 4 was mixed with a pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) so that the compound represented by the formula No. 4 was 3 mg / mL and the peptide represented by the SEQ ID NO: 3 was 2.25 mg / mL.
- This peptide diluent was mixed with an equal amount of an oil phase mixture and emulsified to obtain a cocktail vaccine h prepared. This was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 300 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 225 ⁇ g / animal.
- a vaccine prepared by adding the compound synthesized in Example 1 to the peptide diluent used for preparing the cocktail vaccine h is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide to be produced was 225 ⁇ g / animal and the amount of the compound synthesized in Example 1 was 0.04 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 1) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 4 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Example 1 The results of measuring the spleen weight of the mice in Test Examples 11 to 13 are shown in FIG. As a result of this test, it was found that the addition of Example 1 did not cause a significant increase in spleen weight, and the compound synthesized in Example 1 exhibited adjuvant activity in vivo without causing splenomegaly. It was suggested.
- Test Example 14 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- the compound synthesized in Example 1 was added to a cocktail vaccine (hereinafter referred to as "cocktail vaccine i") using the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 as an oily suspension.
- a vaccine was prepared, administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the oily suspension is prepared by dissolving sucrose fatty acid ester (Ryoto sugar ester L-195) in cyclohexane so as to have a concentration of 12.5 mg / mL, and a compound represented by the formula No. 4. Was 0.75 mg / mL, and the peptide represented by SEQ ID NO: 3 was added to 0.5625 mg / mL to prepare an oil phase mixture.
- sucrose fatty acid ester Rosulfate
- a compound represented by the formula No. 4. Was 0.75 mg / mL
- the peptide represented by SEQ ID NO: 3 was added to 0.5625 mg / mL to prepare an oil phase mixture.
- pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) was added, dispersed with a homogenizer, and freeze-dried.
- cocktail vaccine i which is an oily suspension.
- the amount prepared was increased or decreased as needed.
- Cocktail vaccine i was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 300 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 225 ⁇ g / animal.
- a vaccine obtained by adding the compound synthesized in Example 1 to a pH 2.5 buffer solution used for preparing cocktail vaccine i is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 225 ⁇ g / animal and the compound synthesized in Example 1 was 0.04 nmol / animal or 0.4 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 15 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- a vaccine is prepared by adding the compound synthesized in Example 1 to a vaccine (hereinafter referred to as "cocktail vaccine j") in which a compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 are used as an adjuvant. It was prepared and administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the preparation of the hydrogel preparation includes 1.5 mg / mL of the compound represented by the formula No. 4, 1.125 mg / mL of the peptide represented by SEQ ID NO: 3, and polyoxyethylene (196).
- Polyoxypropylene (67) glycol was mixed with a pH 2.5 buffer (10 mM tartaric acid) and ice-cooled so as to have a concentration of 200 mg / mL, and the mixture was ice-cooled to prepare a cocktail vaccine j which is a hydrogel preparation. The amount prepared was increased or decreased as needed.
- the cocktail vaccine j was administered intradermally to the ridge of the mouse so that the compound represented by the formula No.
- a vaccine obtained by adding the compound synthesized in Example 1 to a pH 2.5 buffer solution used for preparing cocktail vaccine j is represented by 300 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 225 ⁇ g / animal and the amount of the compound synthesized in Example 1 was 0.4 nmol / animal. Administration was performed twice at weekly intervals.
- mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Example 1 The results of measuring the spleen weight of the mice in Test Examples 14 and 15 are shown in FIG. 27. As a result of this test, it was found that the addition of Example 1 did not cause a significant increase in spleen weight, and the compound synthesized in Example 1 exhibited adjuvant activity in vivo without causing splenomegaly. It was suggested.
- Test Example 16 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- a vaccine is prepared by adding the compound synthesized in Example 1 to a vaccine (hereinafter referred to as "cocktail vaccine k") in which the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 are used as the liposome preparation 1. It was prepared and administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the liposome preparation 1 is prepared by dissolving 47.1 mg of hydrogenated soybean phosphatidylcholine and 15.47 mg of cholesterol in t-butyl alcohol, then freeze-drying, and represented by the formula No. 4. Add 2 mL of an aqueous solution mixed with pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) so that the compound is 2.5 mg / mL and the peptide represented by SEQ ID NO: 3 is 1.875 mg / mL, and the temperature is adjusted to about 65 ° C.
- a buffer vaccine k which is a liposome preparation 1.
- the amount prepared was increased or decreased as needed.
- the cocktail vaccine k was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 500 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 375 ⁇ g / animal.
- a vaccine obtained by adding the compound synthesized in Example 1 to a pH 2.5 buffer solution for preparing a cocktail vaccine k is represented by 500 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of mice so that the amount of the peptide was 375 ⁇ g / animal and the compound synthesized in Example 1 was 0.4 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 1) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 4 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 17 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- a vaccine is prepared by adding the compound synthesized in Example 1 to a vaccine (hereinafter referred to as "cocktail vaccine l") in which the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 are used as the liposome preparation 2. It was prepared and administered to HLA-A * 02 : 01 gene-introduced mice, and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the liposome preparation 2 was prepared by dissolving 42.66 mg of sphingomyelin and 15.47 mg of cholesterol in t-butyl alcohol and then freeze-drying. Add a pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) to the lyophilized product so that the compound represented by formula No. 4 is 2.5 mg / mL and the peptide represented by SEQ ID NO: 3 is 1.875 mg / mL. 2 mL of the mixed aqueous solution was added, and an extruder (Mini-Extruder, manufactured by Avanti Polar Lipids) heated to about 65 ° C.
- a buffer vaccine l which is a liposome preparation 2.
- the amount prepared was increased or decreased as needed.
- the cocktail vaccine l was administered intradermally into the ridge of the mouse so that the compound represented by the formula No. 4 was 500 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 375 ⁇ g / animal.
- a vaccine obtained by adding the compound synthesized in Example 1 to a pH 2.5 buffer solution for preparing a cocktail vaccine l is represented by 500 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 375 ⁇ g / animal and the compound synthesized in Example 1 was 0.4 nmol / animal.
- Administration was performed twice at weekly intervals.
- the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 4) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 18 Evaluation of in vivo adjuvant activity using HLA-A * 02 : 01 gene-introduced mouse
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 4 synthesized in Reference Example 8.
- a vaccine is prepared by adding the compound synthesized in Example 1 to a vaccine (hereinafter referred to as "cocktail vaccine m") in which the compound and the peptide represented by SEQ ID NO: 3 synthesized in Reference Example 3 are used as liposome preparation 3. It was prepared and administered to HLA-A * 02 : 01 gene-introduced mice and evaluated by an antigen-specific CTL induction test.
- the preparation of the liposome preparation 3 includes 1,2-dipalmitoyl-sn-glycero-3-phosphocholine 36.7 mg and 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine. 7.58 mg of sodium salt and 15.47 mg of cholesterol were dissolved in a t-butyl alcohol / water mixture and then freeze-dried. Add a pH 2.5 buffer (10 mM tartaric acid, 10% trehalose) to the lyophilized product so that the compound represented by formula No. 4 is 2.5 mg / mL and the peptide represented by SEQ ID NO: 3 is 1.875 mg / mL.
- a buffer vaccine m which is a liposome preparation 3.
- the amount prepared was increased or decreased as needed.
- the cocktail vaccine m was administered intradermally to the ridge of the mouse so that the compound represented by the formula No. 4 was 500 ⁇ g / animal and the peptide represented by SEQ ID NO: 3 was 375 ⁇ g / animal.
- a vaccine obtained by adding the compound synthesized in Example 1 to a pH 2.5 buffer solution for preparing a cocktail vaccine m is represented by 500 ⁇ g / animal of the compound represented by the formula No. 4 and SEQ ID NO: 3.
- the peptide was administered intradermally to the ridge of the mouse so that the amount of the peptide was 375 ⁇ g / animal and the compound synthesized in Example 1 was 0.4 nmol / animal.
- Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 1) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 4 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- SEQ ID NO: 1 a peptide having a final concentration of 10 ⁇ g / mL
- Example 1 The results of measuring the spleen weight of the mice in Test Examples 16 to 18 are shown in FIG. 28. As a result of this test, it was found that the addition of Example 1 did not cause a significant increase in spleen weight, and the compound synthesized in Example 1 exhibited adjuvant activity in vivo without causing splenomegaly. It was suggested.
- Test Example 19 Evaluation of in vivo adjuvant activity using HLA-A * 02: 01 / HLA-DRB1 * 01: 01 gene-introduced mice Regarding the in vivo adjuvant activity of the compound synthesized in Example 1, it was synthesized in Reference Example 1.
- a cocktail vaccine (hereinafter referred to as “cocktail vaccine n”) prepared by mixing the peptide represented by SEQ ID NO: 1 and the peptide represented by SEQ ID NO: 17 synthesized in Reference Example 20 with a pre-emulsification composition.
- a vaccine was prepared by adding the compound synthesized in Example 1, and this vaccine was administered to HLA-A * 02: 01 / HLA-DRB1 * 01: 01 gene-introduced mice (C57BL / 6CrHLA-A2.1DR1). Evaluated by antigen-specific CTL and antigen-specific helper T cell induction tests.
- the peptide represented by SEQ ID NO: 17 is a helper peptide derived from the WT1 protein.
- the concentration after dilution of the peptide represented by SEQ ID NO: 1 is 2 mg / mL
- SEQ ID NO: 17 The peptide represented by (1) was mixed with water for injection so as to be 4 mg / mL.
- a cocktail vaccine n prepared by mixing this peptide diluent with an equal amount of the pre-emulsification composition prepared in the same manner as in Test Example 4 and emulsifying it, wherein the peptide represented by SEQ ID NO: 1 is 200 ⁇ g / animal, SEQ ID NO: The peptide represented by 17 was administered intradermally to the ridge of the mouse so as to be 400 ⁇ g / animal.
- a vaccine obtained by adding the compound synthesized in Example 1 to a peptide diluent used for preparing cocktail vaccine n is 200 ⁇ g / animal of the peptide represented by SEQ ID NO: 1 and the peptide represented by SEQ ID NO: 17.
- mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes.
- splenocytes supplemented with a peptide having a final concentration of 10 ⁇ g / mL (SEQ ID NO: 1 or SEQ ID NO: 17) were seeded on an ELISPOT plate and cultured overnight at 37 ° C. and 5% CO 2 .
- In vitro peptide restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- Test Example 20 Evaluation of in vivo adjuvant activity using HLA-A * 24 : 02 gene-introduced mice
- the in vivo adjuvant activity of the compound synthesized in Example 1 is represented by Formula No. 5 synthesized in Reference Example 22.
- a vaccine was prepared by adding the compound synthesized in Example 1 to a vaccine prepared by mixing a compound with a preliminary emulsion composition (hereinafter referred to as "vaccine o"), and this was HLA-A * 24 : 02. It was administered to transgenic mice and the adjuvant activity was evaluated by an antigen-specific CTL induction test.
- the compound represented by the formula No. 5 was dissolved in DMSO and then mixed with water for injection so that the diluted concentration of the compound represented by the formula No. 5 was 3 mg / mL.
- Vaccine o prepared by mixing this compound diluent with an equal amount of the pre-emulsification composition prepared in the same manner as in Test Example 4 and emulsifying it so that the compound represented by the formula No. 5 is 300 ⁇ g / animal. It was administered intradermally to the ridge of mice.
- a vaccine obtained by adding the compound synthesized in Example 1 to the compound diluent used for preparing the vaccine o is obtained by adding 300 ⁇ g / animal of the compound represented by the formula No. 5 and the compound synthesized in Example 1.
- mice were administered intradermally to the ridge of mice so as to be 0.4 nmol / animal. Administration was performed twice at weekly intervals. One week after the final administration, the mice were euthanized with CO 2 gas, and then the spleen was removed to prepare splenocytes. Peptides in vitro were obtained by seeding splenocytes to which a peptide (SEQ ID NO: 2) having a final concentration of 10 ⁇ g / mL was added in the same manner as in Test Example 3 on an ELISPOT plate and culturing overnight at 37 ° C. and 5% CO 2. Restimulation was added. After culturing, the supernatant was removed, and the number of spots on the colored ELISPOT plate was measured.
- SEQ ID NO: 2 a peptide having a final concentration of 10 ⁇ g / mL
- Test Example 21 A human TLR7 reporter gene assay was performed according to the method of Test Example 1 and the results shown in the table below were obtained.
- Test Example 22 A human TLR7 reporter gene assay was performed according to the method of Test Example 1 and the results shown in the table below were obtained. At this time, a HEK-Blue TM hTLR7 cell line (Invivogen) was used as the cells.
- the HEK-Blue TM hTLR7 cell line is a stable cotransfected cell line that expresses full-length human TLR7 and the secretory SEAP reporter gene under transcriptional control of the NF- ⁇ B response element. TLR7 expression in this cell line has been tested by RT-PCR. Stablely expressed transformants were selected using the antibiotics Blasticidin and Zeocin.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Immunology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Toxicology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Dispersion Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description
式(1):
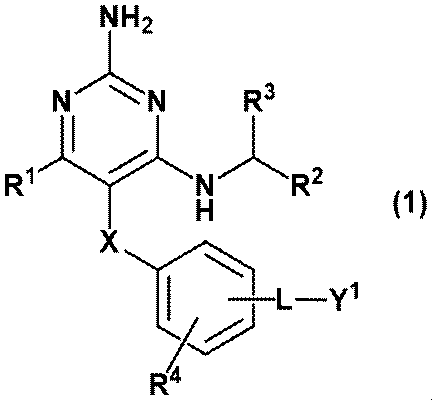
R1は、C1-6のアルキル(該アルキルは、ハロゲン、ヒドロキシ、およびC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
R2及びR3は、それぞれ独立して、水素原子、またはC1-6アルキル(該アルキルは、ハロゲン、ヒドロキシ、C1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
R4は、水素原子、ハロゲン、ヒドロキシ、C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)、C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)またはシアノを表し、
Lはリンカーを表し、
Y1は、-(CH2CH2O)m-R6(R6は、水素原子またはC1-6アルキルを表し、mは、3~100の整数を表す)を表す。]で示される化合物またはその製薬学的に許容される塩。
Xが、メチレンである、項1に記載の化合物またはその製薬学的に許容される塩。
R1が、C1-3アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)である、項1または2に記載の化合物またはその製薬学的に許容される塩。
R1が、メチルである、項3に記載の化合物またはその製薬学的に許容される塩。
R4が、水素原子、ヒドロキシ、C1-3アルキル、またはC1-3アルコキシである、項1~4のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
R4が、水素原子、ヒドロキシ、またはメトキシである、項5に記載の化合物またはその製薬学的に許容される塩。
R2が、C1-6アルキルである、項1~6のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
R3が、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)である、項1~7のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
Lが、-O-、-NRY-、-C(O)-、-C(O)O-、-OC(O)-、-C(O)NRY-、-NRYC(O)-、-CH2NRY-、-CH2O-、-OC(O)O-、-NR7C(O)O-、-OC(O)NRY-、-NR7C(O)NRY-、-OC(S)NRY-、または-NR7C(S)NRY-(ここで、R7は、水素原子または炭素数1~6のアルキルを表し、RYは、水素原子、炭素数1~6のアルキル、またはY2(Y2は、-(CH2CH2O)n-R8(R8は、水素原子または炭素数1~6のアルキルを表し、nは、3~100の整数を表す)を表す)を表す)である、項1~8のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
Lが、-C(O)NRY-、-CH2NRY-、-C(O)O-または-CH2O-である、項9に記載の化合物またはその製薬学的に許容される塩。
Lが、-C(O)NRY-、または-CH2NRY-である、項9に記載の化合物またはその製薬学的に許容される塩。
Lが、-CH2NRY-であり、RYが、水素原子、炭素数1~6のアルキルまたはY2である、項9に記載の化合物またはその製薬学的に許容される塩。
Lが、-CH2NRY-であり、RYが、水素原子、または炭素数1~6のアルキルである、項9に記載の化合物またはその製薬学的に許容される塩。
Y1が、-(CH2CH2O)m-R6であり、R6が、水素原子またはC1-6アルキルであり、mが、3~40の整数である、項1~13のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
Y1が、-(CH2CH2O)m-R6であり、R6が、水素原子またはC1-6アルキルであり、mが、3~20の整数である、項14に記載の化合物またはその製薬学的に許容される塩。
式(2):
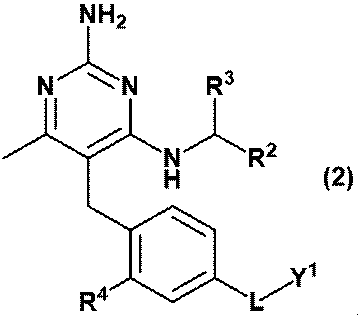
式(3):
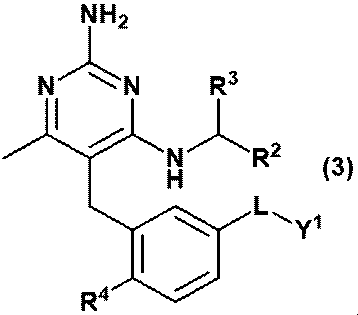
R3は、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、ヒドロキシ、またはメトキシであり、
Lは、-CH2NRY-、-C(O)NRY-、-C(O)O-または-CH2O-であり、
RYは、水素原子、C1-6アルキルまたはY2であり、
Y1は、-(CH2CH2O)m-R6であり、
Y2は、-(CH2CH2O)n-R8であり、
R6は、水素原子またはC1-6アルキルであり、
R8は、水素原子またはC1-6アルキルであり、
mおよびnは、各々独立して、3~40の整数である。]
で表される、項1に記載の化合物またはその製薬学的に許容される塩。
式(2):

式(3):
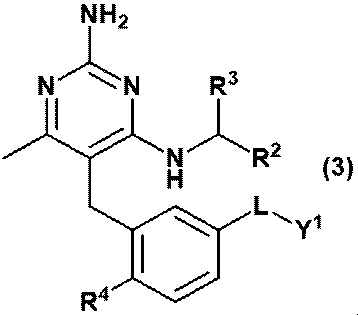
R3は、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、ヒドロキシ、またはメトキシであり、
Lは、-CH2NRY-であり、
RYは、水素原子、またはC1-6アルキルであり、
Y1は、-(CH2CH2O)m-R6であり、
R6は、水素原子またはC1-6アルキルであり、
mは、3~20の整数である。]
で表される、項1に記載の化合物またはその製薬学的に許容される塩。
式(2):
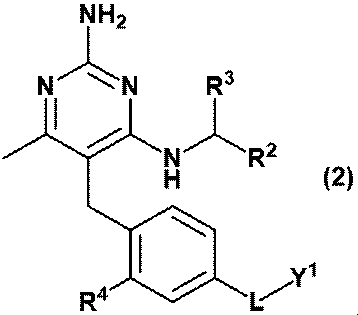
R3は、水素原子、またはC1-3アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、またはメトキシであり、
Lは、-CH2NRY-であり、
RYは、水素原子、またはC1-6アルキルであり、
Y1は、-(CH2CH2O)m-R6であり、
R6は、水素原子またはC1-6アルキルであり、
mは、3~40の整数である。]
で表される、項1に記載の化合物またはその製薬学的に許容される塩。
以下の化合物群から選択される、項1に記載の化合物又はその製薬学的に許容される塩:
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例1)、
1-{4-[(2-アミノ-4-{[(3S)-1-ヒドロキシヘキサン-3-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシフェニル}-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例2)、
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例3)、
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-ヒドロキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例4)、
4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-N-(20-ヒドロキシ-3,6,9,12,15,18-ヘキサオキサイコサン-1-イル)-3-メトキシベンズアミド(実施例5)、
2,5,8,11-テトラオキサトリデカン-13-イル 4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシベンゾエート(実施例6)、
5-{[2-メトキシ-4-(2,5,8,11,14-ペンタオキサペンタデカン-1-イル)フェニル]メチル}-6-メチル-N4-ペンチルピリミジン-2,4-ジアミン(実施例7)、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29-ノナオキサ-2-アザヘントリアコンタン-31-オール(実施例8)、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71-トリコサオキサ-2-アザトリヘプタコンタン-73-オール(実施例9)、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98,101,104,107-ペンタトリアコンタオキサ-2-アザノナヘクタン-109-オール(実施例10)、および
12-[(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)メチル]-3,6,9,15,18,21-ヘキサオキサ-12-アザトリコサン-1,23-ジオール(実施例11)。
以下の化合物群から選択される、項1に記載の化合物又はその製薬学的に許容される塩:
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例1)、
1-{4-[(2-アミノ-4-{[(3S)-1-ヒドロキシヘキサン-3-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシフェニル}-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例2)、
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例3)、および
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-ヒドロキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール(実施例4)。
項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩を含有する医薬組成物。
エマルション製剤、油性懸濁剤、ハイドロゲル製剤、又は脂質製剤である項21に記載の医薬組成物。
エマルション製剤である項21に記載の医薬組成物。
エマルション製剤が油中水型エマルションである項23に記載の医薬組成物。
エマルション製剤が、(1)オレイン酸エチル、ミリスチン酸オクチルドデシル、モノオレイン酸ソルビタン、モノオレイン酸グリセリン、ポリオキシエチレン硬化ヒマシ油20、グリセリン、及びリン酸二水素ナトリウム、又は(2)Montanide ISA 51VGを含有する項24に記載の医薬組成物。
脂質製剤である項21に記載の医薬組成物。
脂質製剤が、リン脂質を含むリポソーム製剤である項26に記載の医薬組成物。
脂質製剤が、ステロール類を含むリポソーム製剤である項26または27に記載の医薬組成物。
ステロール類が、コレステロールである項28に記載の医薬組成物。
リポソーム製剤が、無機酸、無機酸塩、有機酸、有機酸塩、糖類、緩衝剤、酸化防止剤、及びポリマー類からなる群から選択される1以上の添加物を含むリポソーム製剤である項27~29のいずれか一項に記載の医薬組成物。
更に、抗原を含む、項21~30のいずれか一項に記載の医薬組成物。
抗原が、病原体由来抗原又は腫瘍抗原である、項31に記載の医薬組成物。
抗原が、腫瘍抗原である、項31に記載の医薬組成物。
腫瘍抗原が、腫瘍抗原ペプチドである、項33に記載の医薬組成物。
腫瘍抗原ペプチドが、
RMFPNAPYL(配列番号:1)、
ALLPAVPSL(配列番号:8)、
SLGEQQYSV(配列番号:9)、
RVPGVAPTL(配列番号:10)、
VLDFAPPGA(配列番号:4)、
CMTWNQMNL(配列番号:11)、
CYTWNQMNL(配列番号:2)、
TYAGCLSQIF(配列番号:18)、
式(4):
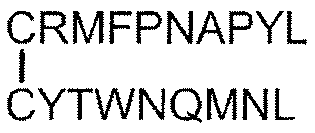
式(5):

からなる群から選択されるアミノ酸配列で表される一つ以上のペプチドまたはその製薬学的に許容される塩、並びに
WAPVLDFAPPGASAYGSL(配列番号:3)、
CWAPVLDFAPPGASAYGSL(配列番号:12)、
WAPVLDFAPPGASAYGSLC(配列番号:13)、
CNKRYFKLSHLQMHSRKHTG(配列番号:14)、
CNKRYFKLSHLQMHSRKH(配列番号:15)、
CNKRYFKLSHLQMHSRK(配列番号:16)、および
KRYFKLSHLQMHSRKH(配列番号:17)
からなる群から選択されるアミノ酸配列で表される一つ以上のペプチドまたはその製薬学的に許容される塩を含む、項34に記載の医薬組成物。
腫瘍抗原ペプチドが、
RMFPNAPYL(配列番号:1)、
ALLPAVPSL(配列番号:8)、
SLGEQQYSV(配列番号:9)、
RVPGVAPTL(配列番号:10)、
VLDFAPPGA(配列番号:4)、
CMTWNQMNL(配列番号:11)、
CYTWNQMNL(配列番号:2)、および
式(4):

からなる群から選択されるアミノ酸配列で表される一つ以上のペプチドまたはその製薬学的に許容される塩、並びに
WAPVLDFAPPGASAYGSL(配列番号:3)、
CWAPVLDFAPPGASAYGSL(配列番号:12)、
WAPVLDFAPPGASAYGSLC(配列番号:13)、
CNKRYFKLSHLQMHSRKHTG(配列番号:14)、
CNKRYFKLSHLQMHSRKH(配列番号:15)、
CNKRYFKLSHLQMHSRK(配列番号:16)、および
KRYFKLSHLQMHSRKH(配列番号:17)、
からなる群から選択されるアミノ酸配列で表される一つ以上のペプチドまたはその製薬学的に許容される塩を含む、項34に記載の医薬組成物。
腫瘍抗原ペプチドが、式(4):
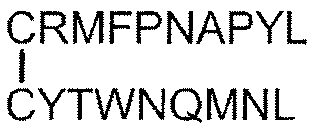
のアミノ酸配列で表されるペプチドまたはその製薬学的に許容される塩、および
配列番号3:WAPVLDFAPPGASAYGSL
のアミノ酸配列で表されるペプチドまたはその製薬学的に許容される塩の組み合わせである、項34に記載の医薬組成物。
項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩を含む、ワクチンアジュバント。
癌ワクチンのワクチンアジュバントである、項38に記載のワクチンアジュバント。
ワクチンアジュバントとして用いられる、項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
癌ワクチンのワクチンアジュバントとして用いられる、項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩。
項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩を含むCTL誘導剤。
項1~20のいずれか一項に記載の化合物またはその製薬学的に許容される塩を含む免疫賦活剤。
項1~20のいずれか一項に記載の化合物またはその薬学上許容される塩を哺乳動物に投与することを含む、哺乳動物におけるCTLを誘導する方法。
項1~20のいずれか一項に記載の化合物またはその薬学上許容される塩を哺乳動物に投与することを含む、哺乳動物におけるCTL誘導を増強する方法。
項1~20のいずれか一項に記載の化合物またはその薬学上許容される塩を哺乳動物に投与することを含む、哺乳動物における抗原に対する特異的免疫応答を増強する方法。
項1~20のいずれか一項に記載の化合物もしくはその薬学上許容される塩の、ワクチンアジュバントを製造するための使用。
項1~20のいずれか一項に記載の化合物もしくはその薬学上許容される塩の、癌ワクチンのワクチンアジュバントを製造するための使用。
a)項1~20のいずれか一項に記載の化合物またはその薬学上許容し得る塩、もしくは項1~20のいずれか一項に記載の化合物又はその薬学上許容し得る塩を含有する医薬組成物;および
b)抗原または抗原を含有する医薬組成物
を含有するキット。
a)項1の式(1)で示される化合物又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;および
b)腫瘍抗原または腫瘍抗原を含有する医薬組成物
を含有するキット。
a)項1の式(1)で示される化合物又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;および
b)病原体由来抗原または病原体由来抗原を含有する医薬組成物
を含有するキット。

(1)水素原子、または
(2)C1-6アルキル(該アルキルは、ハロゲン、ヒドロキシから独立して選択される1~3個の置換基で置換されていてもよい)
が挙げられる。より好ましくは、水素原子またはC1-6アルキルが挙げられ、更に好ましくは、C1-6のアルキルが挙げられ、更により好ましくは、C3-4のアルキルが挙げられる。
(1)水素原子、または
(2)C1-6アルキル(該アルキルは、ハロゲン、ヒドロキシから独立して選択される1~3個の置換基で置換されていてもよい)
が挙げられる。
(1)水素原子、または
(2)C1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)
が挙げられる。
(1)水素原子、または
(2)C1-3アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)が挙げられる。
(1)水素原子、または
(2)C1-2アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)が挙げられる。
(1)水素原子
(2)ハロゲン
(3)ヒドロキシ
(4)C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)
(5)C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)、または
(6)シアノ
が挙げられる。
(1)水素原子
(2)ハロゲン
(3)ヒドロキシ
(4)C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)、または
(5)C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)
が挙げられる。
(1)-O-
(2)-NRY-
(3)-C(O)-
(4)-C(O)O-
(5)-OC(O)-
(6)-C(O)NRY-
(7)-NRYC(O)-
(8)-CH2NRY-
(9)-CH2O-
(10)-OC(O)O-
(11)-NR7C(O)O-
(12)-OC(O)NRY-
(13)-NR7C(O)NRY-
(14)-OC(S)NRY-、または
(15)-NR7C(S)NRY-
が挙げられる。
(1)-O-
(2)-NRY-
(3)-C(O)-
(4)-C(O)O-
(5)-OC(O)-
(6)-C(O)NRY-
(7)-NRYC(O)-
(8)-CH2NRY-、または
(9)-CH2O-
が挙げられる。
(1)-C(O)NRY-、
(2)-CH2NRY-、
(3)-C(O)O-、または
(4)-CH2O-
が挙げられる。
(1)-C(O)NRY-、または
(2)-CH2NRY-
が挙げられる。

(A)
Xが、メチレン、酸素原子、硫黄原子、SO、SO2、またはNR5であり;
R1が、C1-6アルキル(該アルキルは、ハロゲン、ヒドロキシ、C1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい。)であり;
R2及びR3が、各々独立して、
(1)水素原子、または
(2)C1-6アルキル(該アルキルは、ハロゲン、ヒドロキシ、C1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)であり;
R4が、
(1)水素原子
(2)ハロゲン
(3)ヒドロキシ
(4)C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)
(5)C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)、または
(6)シアノ
であり;
R5が、
(1)水素原子、または
(2)C1-6アルキル
であり;
R6及びR8が、各々独立して、
(1)水素原子、または
(2)C1-6アルキル
であり;
R7が、
(1)水素原子、または
(2)C1-6アルキル
であり;
Lが、
(1)-O-
(2)-NRY-
(3)-C(O)-
(4)-C(O)O-
(5)-OC(O)-
(6)-C(O)NRY-
(7)-NRYC(O)-
(8)-CH2NRY-
(9)-CH2O-
(10)-OC(O)O-
(11)-NR7C(O)O-
(12)-OC(O)NRY-
(13)-NR7C(O)NRY-
(14)-OC(S)NRY-、または
(15)-NR7C(S)NRY-
であり;
RYが、
(1)水素原子
(2)C1-6アルキル、または
(3)Y2であり;
Y1が、-(CH2CH2O)m-R6であり;
Y2が、-(CH2CH2O)n-R8であり;
m及びnが、各々独立して、3~100で表される整数;
で表される化合物または製薬学的に許容される塩。
(B)
Xが、メチレン、酸素原子、またはNR5であり;
R1が、C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい。)であり;
R2が
(1)水素原子、または
(2)C1-6アルキルであり;
R3が、
(1)水素原子、または
(2)C1-6アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)であり;
R4が、
(1)水素原子
(2)ハロゲン
(3)ヒドロキシ
(4)C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)、または
(5)C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)、であり;
R5が、
(1)水素原子、または
(2)C1-3アルキル
であり;
R6及びR8が、各々独立して、
(1)水素原子、または
(2)C1-3アルキル
であり;
Lが、
(1)-O-
(2)-NRY-
(3)-C(O)-
(4)-C(O)O-
(5)-OC(O)-
(6)-C(O)NRY-
(7)-NRYC(O)-
(8)-CH2NRY-、または
(9)-CH2O-
であり;
RYが、
(1)水素原子、
(2)C1-6アルキル、または
(3)Y2であり;
Y1が、-(CH2CH2O)m-R6であり;
Y2が、-(CH2CH2O)n-R8であり;
m及びnが、各々独立して、3~40で表される整数;
で表される化合物または製薬学的に許容される塩。
(C)
Xが、メチレンであり;
R1が、メチル、エチル、プロピル、1-メチルエチル、ブチル、2-メチルプロピル、1-メチルプロピルまたは1,1-ジメチルエチルであり;
R2が、C1-6アルキルであり;
R3が、
(1)水素原子、または
(2)C1-3アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)であり;
R4が、
(1)水素原子
(2)ヒドロキシ
(3)C1-3アルキル、または
(4)C1-3アルコキシであり;
R6及びR8が、各々独立して水素原子、メチル、エチルまたはプロピルであり;
Lが、
(1)-C(O)NRY-、
(2)-CH2NRY-、
(3)-C(O)O-、または
(4)-CH2O-
であり;
RYが、水素原子、メチル、エチル、プロピルまたはY2であり;
Y1が、-(CH2CH2O)m-R6であり;
Y2が、-(CH2CH2O)n-R8であり;
m及びnが、各々独立して、3~40で表される整数;
で表される化合物または製薬学的に許容される塩。
(D)
式(2)または(3)で表される化合物またはその製薬学的に許容される塩;
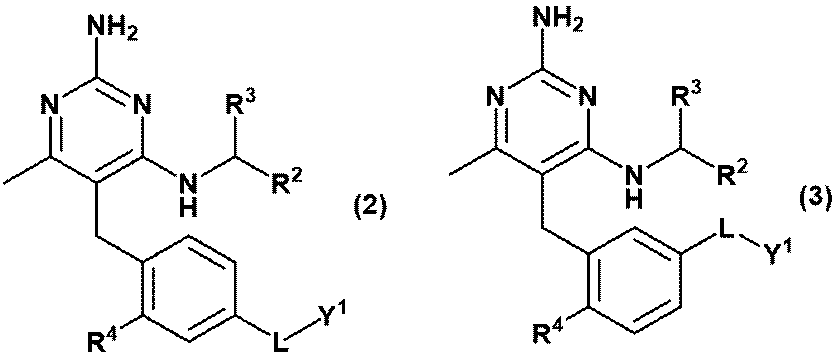
R3は、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、ヒドロキシ、またはメトキシであり、
Lは、-CH2NRY-、-C(O)NRY-、-C(O)O-または-CH2O-であり、
RYは、水素原子、メチル、エチル、プロピルまたはY2であり、
Y1は、-(CH2CH2O)m-R6であり、
Y2が、-(CH2CH2O)n-R8であり、
R6及びR8は、各々独立して水素原子、メチル、エチルまたはプロピルであり、
m及びnは、各々独立して3~40の整数である。]
(E)
式(2)で表される化合物またはその製薬学的に許容される塩;

R3は、水素原子、またはC1-3アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、またはメトキシであり、
Lは、-CH2NRY-であり、
RYは、水素原子、メチル、エチルまたはプロピルであり、
Y1は、-(CH2CH2O)m-R6であり、
R6は、水素原子、メチル、エチルまたはプロピルであり、
mは、4~36の整数である。]
製造法A-1
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-CRA1RA2NRY-、または-CRA1RA2O-で表されるリンカーを有する化合物(a1-2)は、以下の方法により製造される。
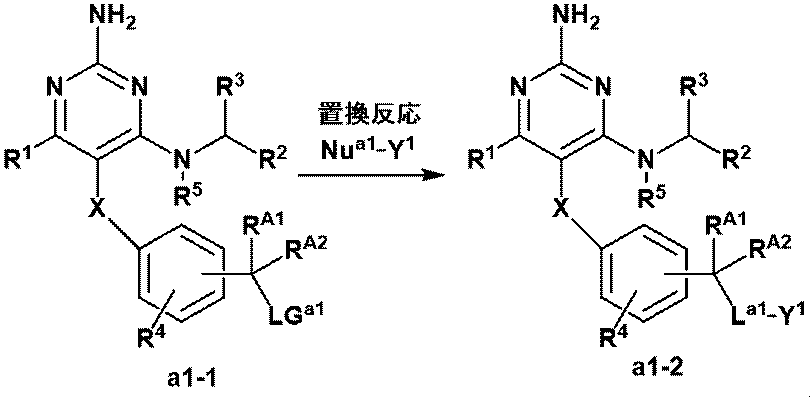
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-O-、-NRY-、-C(O)O-、-CH2NRY-、または-CH2O-で表されるリンカーを有する化合物(a2-2)は、以下の方法により製造される。
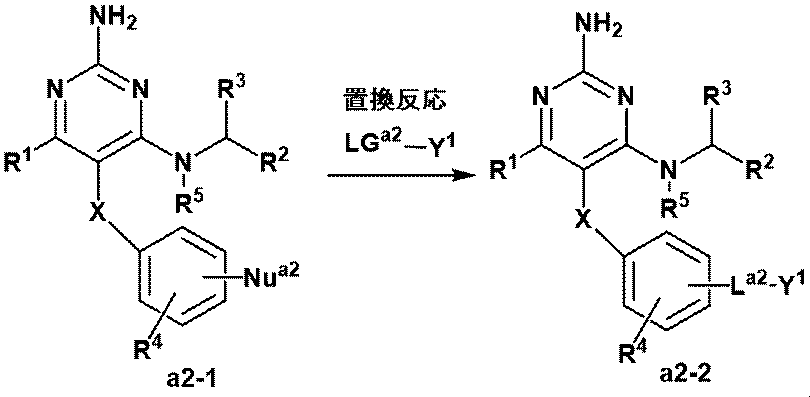
製造法B-1
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-CH2NRY-で表されるリンカーを有する化合物(b1-2)は、以下の方法により製造される。
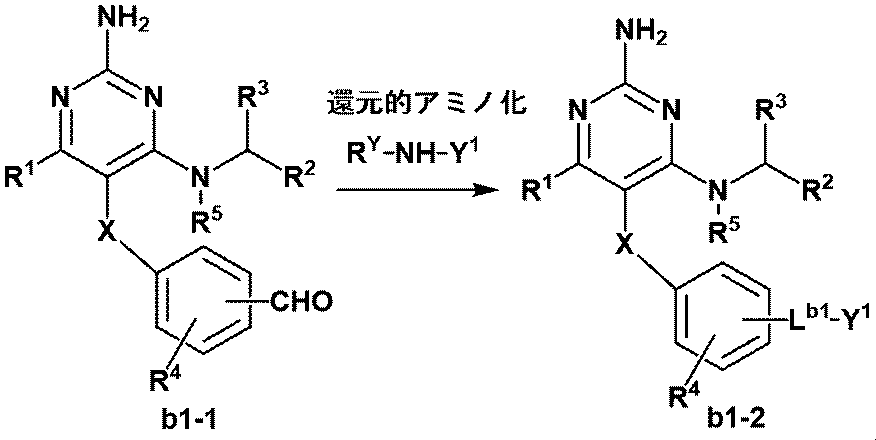
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-NRY-、または-CH2NRY-で表されるリンカーを有する化合物(b2-2)は、以下の方法により製造される。

製造法C-1
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-O-、-NRY-、または-NRYC(O)-で表されるリンカーを有する化合物(c1-2)は、以下の方法により製造される。
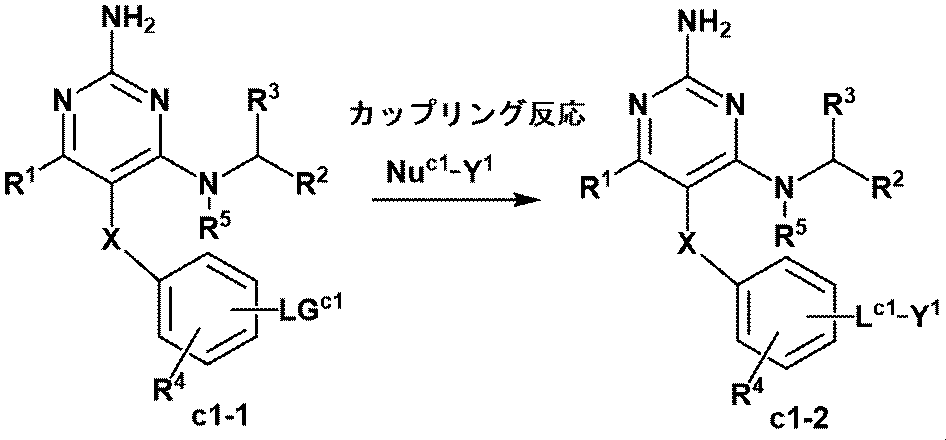
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-C(O)O-、または-C(O)NRY-リンカーを有する化合物(d1-2)は、以下の方法により製造される。
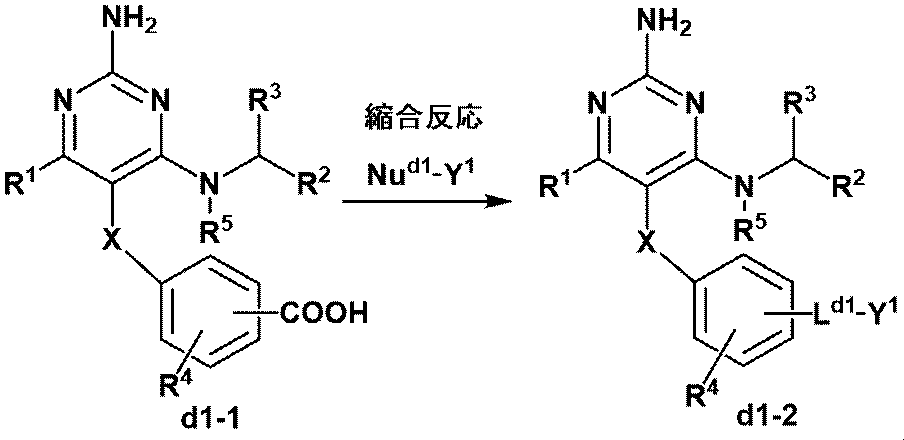
本発明の式(1)で表される化合物またはその製薬学的に許容される塩のうち、-OC(O)-、または-NRYC(O)-リンカーを有する化合物(d2-2)は、以下の方法により製造される。
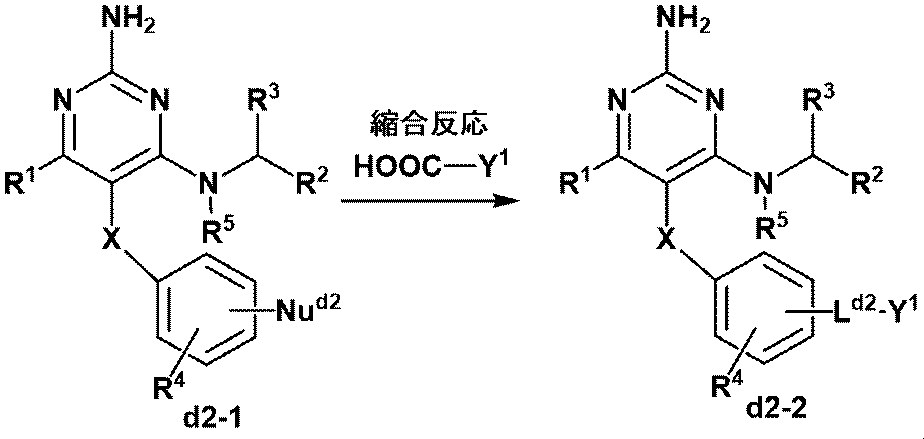
製造法E-1

出発化合物及び目的化合物の好適な塩及び医薬として許容しうる塩は、慣用の無毒性塩であり、それらとしては、有機酸塩(例えば酢酸塩、トリフルオロ酢酸塩、マレイン酸塩、フマル酸塩、クエン酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、ギ酸塩又はpara-トルエンスルホン酸塩など)及び無機酸塩(例えば塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩又はリン酸塩など)のような酸付加塩、アミノ酸(例えばアルギニン、アスパラギン酸又はグルタミン酸など)との塩、アルカリ金属塩(例えばナトリウム塩又はカリウム塩など)及びアルカリ土類金属塩(例えばカルシウム塩又はマグネシウム塩など)などの金属塩、アンモニウム塩、又は有機塩基塩(例えばトリメチルアミン塩、トリエチルアミン塩、ピリジン塩、ピコリン塩、ジシクロヘキシルアミン塩又はN,N’-ジベンジルエチレンジアミン塩など)などの他、当業者が適宜選択することができる。
また、本発明化合物及びその製薬学的に許容される塩は、水あるいは各種溶媒との溶媒和物の形で存在することもあるが、これらの付加物も本発明に包含される。
再結晶溶媒としては例えば、メタノール、エタノールもしくは2-プロパノールなどのアルコール系溶媒、ジエチルエーテルなどのエーテル系溶媒、酢酸エチルなどのエステル系溶媒、ベンゼンもしくはトルエンなどの芳香族炭化水素系溶媒、アセトンなどのケトン系溶媒、ジクロロメタンもしくはクロロホルムなどのハロゲン系溶媒、ヘキサンなどの炭化水素系溶媒、ジメチルホルムアミドもしくはアセトニトリルなどの非プロトン系溶媒、水、又はこれらの混合溶媒などを用いることができる。その他の精製方法としては、実験化学講座(日本化学会編、丸善)1巻などに記載された方法などを用いることができる。また、本発明化合物の分子構造の決定は、それぞれの原料化合物に由来する構造を参照して、核磁気共鳴法、赤外吸収法、円二色性スペクトル分析法などの分光学的手法、及び質量分析法により容易に行える。
すなわち、本発明の化合物又はその薬学上許容される塩は、抗原特異的抗体、詳しくは抗原特異的IgG、さらに詳しくはTh1型抗原特異的IgG(例えばIgG2c)を誘導又は亢進する活性を有する。
また、本発明の化合物又はその製薬学的に許容される塩は、細胞傷害性T細胞(CTL)を増加させる活性を有する。または、本発明の化合物又はその製薬学的に許容される塩は、哺乳動物におけるCTLを誘導する活性、もしくは哺乳動物におけるCTL誘導を増強する活性を有する。
また、本発明の化合物又はその製薬学的に許容される塩は、CD4陽性(すなわち、MHC class II拘束性)及び/又はCD8陽性(すなわち、MHC Class I拘束性)T細胞を増加させる活性を有する。
また、本発明の化合物又はその製薬学的に許容される塩は、抗原特異的なT細胞を増加させる活性を有する。
また、本発明の化合物又はその製薬学的に許容される塩は、メモリーT細胞、詳しくはCD8陽性エフェクターメモリーT細胞を増加させる活性を有する。
また、本発明の化合物又はその製薬学的に許容される塩は、哺乳動物に投与した場合、CTLを増加させる作用が、同モル数のPEG構造を含まない化合物を投与した場合よりも高いという特徴を有する。
また、本発明の化合物またはその製薬学的に許容される塩は、免疫担当細胞を活性化させる活性を有する。
本発明の医薬組成物は腫瘍抗原を含有し得る。当該腫瘍抗原としては、腫瘍抗原タンパク質、又は当該腫瘍抗原タンパク質由来の腫瘍抗原ペプチドを用いることができる。腫瘍抗原ペプチドとしては、好ましくは後記にある抗原ペプチドを用いることができるが、より好ましくはNY-ESO-1、MAGE-3、WT1、OR7C1またはHer2/neu由来の腫瘍抗原ペプチドが挙げられ、より好ましくはWT1由来の腫瘍抗原ペプチドが挙げられる。更には、腫瘍の遺伝子異常の結果、生成するネオアンチゲン由来のペプチドも本発明の化合物またはその製薬学的に許容される塩と共に用いることができる。
また、本発明の化合物又はその製薬学的に許容される塩と腫瘍抗原を含む医薬組成物は、当該抗原を発現する腫瘍の増殖を阻害する作用、または当該抗原を発現する腫瘍の発生を抑制する予防作用を有する。
従って、本発明の化合物又はその製薬学的に許容される塩は、以下に示す腫瘍抗原と組合せた医薬組成物として用いることで、癌の治療又は予防のための薬剤として有用である。
腫瘍抗原ペプチドの一つの態様として、例えば、以下のアミノ酸配列からなるペプチドまたはそれらの製薬学的に許容される塩である腫瘍抗原ペプチドが挙げられる:
RMFPNAPYL(配列番号:1)、
ALLPAVPSL(配列番号:8)、
SLGEQQYSV(配列番号:9)、
RVPGVAPTL(配列番号:10)、
VLDFAPPGA(配列番号:4)、
CMTWNQMNL(配列番号:11)、
CYTWNQMNL(配列番号:2)、
WAPVLDFAPPGASAYGSL(配列番号:3)、
CWAPVLDFAPPGASAYGSL(配列番号:12)、
WAPVLDFAPPGASAYGSLC(配列番号:13)、
CNKRYFKLSHLQMHSRKHTG(配列番号:14)、
CNKRYFKLSHLQMHSRKH(配列番号:15)、
CNKRYFKLSHLQMHSRK(配列番号:16)、
KRYFKLSHLQMHSRKH(配列番号:17)または
TYAGCLSQIF(配列番号:18)。
また、式(4):

式(5):

で表されるアミノ酸配列からなるペプチドまたはその製薬学的に許容される塩も本発明における腫瘍抗原ペプチドとして用いることができる。
水性溶液製剤もしくは水性懸濁液剤としては、例えば抗原(腫瘍抗原または病原体由来抗原)、及び/又は式(1)で表される化合物又はその薬学上許容される塩を水に溶解又は分散させた製剤が挙げられる。
油性溶液製剤もしくは油性懸濁液剤としては、例えば抗原(腫瘍抗原または病原体由来抗原)、及び/又は式(1)で表される化合物又はその薬学上許容される塩を油性成分に溶解又は分散させた製剤が挙げられる。
ハイドロゲル製剤としては、例えば抗原(腫瘍抗原または病原体由来抗原)、及び/又は式(1)で表される化合物又はその薬学上許容される塩を水に溶解又は分散させ粘稠性を付与した製剤が挙げられる。
脂質製剤としては、例えば抗原(腫瘍抗原または病原体由来抗原)、及び/又は式(1)で表される化合物又はその薬学上許容される塩を含有するリポソーム製剤が挙げられる。
エマルション製剤としては、例えば抗原(腫瘍抗原または病原体由来抗原)、及び/又は式(1)で表される化合物又はその薬学上許容される塩を含有する水性の溶液及び油状組成物を含む製剤が挙げられる。
本発明の液剤の別の態様としては、腫瘍抗原、及び/又は式(1)で表される化合物又はその薬学上許容される塩を水に溶解又は分散させた水性溶液製剤もしくは水性懸濁液剤;腫瘍抗原、及び/又は式(1)で表される化合物又はその薬学上許容される塩を油性成分に溶解又は分散させた油性溶液製剤もしくは油性懸濁液剤;水性の溶液及び油状組成物を含むエマルション製剤等が挙げられる。
本発明の水性溶液製剤もしくは水性懸濁液剤に使用される添加剤としては、例えば精製水、注射用水、緩衝剤、pH調節剤、安定化剤、等張化剤、可溶化剤又は溶解補助剤等が挙げられる。
本発明の油性溶液製剤もしくは油性懸濁液剤に使用される添加剤としては、例えば緩衝剤、pH調節剤、安定化剤、等張化剤、動植物性油脂、炭化水素類、脂肪酸、脂肪酸エステル類、可溶化剤又は溶解補助剤等が挙げられる。
本発明のハイドロゲル製剤に使用される添加剤としては、例えば精製水、注射用水、緩衝剤、pH調節剤、安定化剤、等張化剤、可溶化剤、溶解補助剤、又は粘稠剤等が挙げられる。
本発明のリポソーム製剤に使用される添加剤としては、例えば精製水、注射用水、緩衝剤、pH調節剤、安定化剤、等張化剤、可溶化剤、溶解補助剤、又は脂質類等が挙げられる。
本発明のエマルション製剤に使用される添加剤としては、例えば、水、緩衝剤、pH調節剤、安定化剤、等張化剤、動植物性油脂、炭化水素類、脂肪酸、脂肪酸エステル類、グリセリン脂肪酸エステル類、親水性界面活性剤、及び親油性界面活性剤等が挙げられる。ここで水としては精製水、注射用水等が挙げられ、緩衝剤としてはリン酸塩、有機酸塩等が挙げられ、pH調節剤としては塩酸、水酸化ナトリウム等が挙げられ、安定化剤としてはグリセリン、プロピレングリコール、亜硫酸塩等が挙げられ、等張化剤としては食塩、ブドウ糖、ショ糖、マンニトール等が挙げられ、動植物性油脂としてはオリーブ油、大豆油、肝油等が挙げられ、炭化水素類としては流動パラフィン、スクワレン、スクワラン等が挙げられ、脂肪酸としてはオレイン酸、ミリスチン酸等が挙げられ、脂肪酸エステル類としてはオレイン酸エチル、ミリスチン酸オクチルドデシル、2-エチルヘキサン酸セチル、ミリスチン酸イソプロピル等が挙げられ、グリセリン脂肪酸エステル類としては中鎖脂肪酸トリグリセリド、中鎖脂肪酸ジグリセリド、中鎖脂肪酸モノグリセリド等が挙げられ、親水性界面活性剤としてはポリオキシエチレンヒマシ油、ポリオキシエチレン硬化ヒマシ油、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリソルベート類等が挙げられ、親油性界面活性剤としてはモノオレイン酸グリセリン、ジオレイン酸グリセリン、モノオレイン酸ソルビタン(Span 80(登録商標))、セスキオレイン酸ソルビタン、ジオレイン酸ソルビタン、トリオレイン酸ソルビタン(Span 85(登録商標))、ジポリヒドロキシステアリン酸PEG-30、植物由来界面活性剤(サポニン等)等が挙げられる。
本発明のW/Oエマルション製剤としては、式(1)で表される化合物又はその薬学上許容される塩、オレイン酸エチル、ミリスチン酸オクチルドデシル、モノオレイン酸ソルビタン、モノオレイン酸グリセリン、ポリオキシエチレン硬化ヒマシ油20、グリセリン、及びリン酸二水素ナトリウムを含有する製剤;又は式(1)で表される化合物又はその薬学上許容される塩、及びMontanide ISA 51 VGを含有する製剤が挙げられる。
「リン脂質」における脂肪酸残基は特に限定されないが、例えば、炭素数14~18の飽和または不飽和の脂肪酸残基が挙げられ、具体的には、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸、リノール酸等の脂肪酸由来のアシル基が挙げられる。また、卵黄レシチン、大豆レシチン等の天然物由来のリン脂質、およびその不飽和脂肪酸残基に水素添加した水素添加卵黄レシチン、水素添加大豆レシチン(水素添加大豆リン脂質、または水素添加大豆ホスファチジルコリンともいう)等を用いることもできる。
リポソーム膜成分全体に対するリン脂質の配合量(モル分率)は特に限定されないが、好ましくは30~80%が挙げられ、より好ましくは40~70%が挙げられる。
ステロール類としては、コレステロール、β-シトステロ-ル,スチグマステロ-ル,カンペステロ-ル,ブラシカステロ-ル、エルゴステロ-ル、及びフコステロ-ル等が挙げられる。ステロール類として好ましくは、コレステロールが挙げられる。リポソーム膜成分全体に対するステロール類の配合量(モル分率)は特に限定されないが、好ましくは0~60%が挙げられ、より好ましくは10~50%が挙げられ、さらに好ましくは30~50%が挙げられる。
本発明の化合物もしくはその薬学上許容される塩、及び腫瘍抗原特異的免疫反応または病原体特異的免疫反応を増強させる物質(腫瘍抗原または病原体由来抗原ともいう)を含む医薬組成物も、本発明の一態様である。当該腫瘍抗原としては、特に限定はないが、抗原タンパク質又は該抗原タンパク質に由来する抗原ペプチド(部分ペプチド)、腫瘍抗原タンパク質又は該腫瘍抗原タンパク質に由来する腫瘍抗原ペプチド(部分ペプチド)、さらにこれらと担体との複合体が挙げられる。
a)式(1)で示される化合物、又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;
b)抗原(腫瘍抗原または病原体由来抗原)または抗原(腫瘍抗原または病原体由来抗原)を含有する医薬組成物。
ここで、抗原としては、ワクチンの有効成分として用いられ得る抗原であれば特に限定はないが、上述の抗原タンパク質又は該抗原タンパク質に由来する抗原ペプチド(部分ペプチド)等、さらにこれらと担体との複合体等が挙げられる。
a)式(1)で示される化合物又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;
b)腫瘍抗原または腫瘍抗原を含有する医薬組成物。
ここで、腫瘍抗原としては、癌ワクチンの有効成分として用いられ得る腫瘍抗原であれば特に限定はないが、上述の腫瘍抗原タンパク質又は該腫瘍抗原タンパク質に由来する腫瘍抗原ペプチド(部分ペプチド)等、さらにこれらと担体との複合体等が挙げられる。
a)式(1)で示される化合物又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;
b)病原体由来抗原または病原体由来抗原を含有する医薬組成物。
ここで、病原体由来抗原としては、感染症ワクチンの有効成分として用いられ得る病原体由来抗原であれば特に限定はないが、上述の病原体由来抗原タンパク質又は該病原体由来抗原タンパク質に由来する病原体由来抗原ペプチド(部分ペプチド)等、さらにこれらと担体との複合体等が挙げられる。
また、本発明の一態様として、癌または感染症の処置のためのワクチンの製造における、ワクチンアジュバントとしての、上で定義した式(I)で示される化合物、又はその薬学上許容し得る塩の使用を提供する。
また、本発明の一態様として、癌の処置のための癌ワクチンの製造における、ワクチンアジュバントとしての、上で定義した式(I)で示される化合物、又はその薬学上許容し得る塩の使用を提供する。
また、本発明の一態様として、感染症の処置のための感染症ワクチンの製造における、ワクチンアジュバントとしての、上で定義した式(I)で示される化合物、又はその薬学上許容し得る塩の使用を提供する。
Boc:tert-ブトキシカルボニル
Alko:p-アルコキシベンジルアルコール
PEG:ポリエチレングリコール
tBu:tert-ブチル
HBTU:O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスファート
DIPEA:N,N-ジイソプロピルエチルアミン
DMF:N,N-ジメチルホルムアミド
TFA:トリフルオロ酢酸
TIS:トリイソプロピルシラン
THF:テトラヒドロフラン
TBS:tert-ブチルジメチルシリル基
TBDPS:tert-ブチルジフェニルシリル基
MS検出器:LCMS-IT-TOF
HPLC:Shimadzu Nexera X2 LC 30AD
カラム:Kinetex 1.7μ C18 100A New column 50×2.1mm
流速:1.2ml/min
測定波長:254/220nm
移動相:A液;0.1%ギ酸水溶液
B液;アセトニトリル
タイムプログラム:
ステップ 時間(分)
1 0.01-1.40 A液:B液=90:10~5:95
2 1.40-1.60 A液:B液=5:95
3 1.61-2.00 A液:B液=99:1
MS検出器:ACQUITY(登録商標)SQdetecter (Waters社)
HPLC:ACQUITY(登録商標)system
カラム:Waters ACQUITY UPLC(登録商標)BEH C18(1.7μm,2.1mm×30mm)
流速:0.8ml/min
測定波長:254/220nm
移動相:A液:0.06%ギ酸/アセトニトリル、
B液:0.06%ギ酸水溶液
タイムプログラム:0.0-1.30 A液:B液=2:98~96:4
カラム温度:25℃
アミノ酸配列:RMFPNAPYL(Arg-Met-Phe-Pro-Asn-Ala-Pro-Tyr-Leu)(配列番号1)からなるペプチドの合成
(Fmoc-Lys(Boc)-Alko-PEG樹脂)1.00g(渡辺化学製;0.23mmol/g、0.23mmol)を出発原料とし、Fmoc/tBu法による固相合成によりペプチド鎖の組上げを行った。固相合成にはCS Bio社製CS336X型ペプチド合成機を用い、Fmoc基の脱保護は20%ピペリジンのDMF溶液で5分間及び20分間処理することにより行った。保護アミノ酸のカップリングは、1.05mmolの保護アミノ酸、1mmolのHBTU、2mmolのDIPEAのDMF溶液と1時間反応させることにより行った。得られた樹脂をDMF及びエーテル洗浄後減圧乾燥することにより、ペプチド樹脂得た。このペプチド樹脂にTFA/水/TIS(体積比:94/2.5/2.5)の混合液10mLを加え、室温にて2時間振とうした。樹脂を濾去後、反応液を減圧濃縮した。反応液を氷冷しジエチルエーテル(50mL)を加えた。生じた沈殿物を濾取し、エーテルで洗浄後減圧乾燥することにより粗ペプチドを得た。得られた粗ペプチドを20%酢酸水とアセトニトリルの混合液(体積比1/1)に溶解し、以下に示す条件で精製し、RMFPNAPYL(Arg-Met-Phe-Pro-Asn-Ala-Pro-Tyr-Leu)(配列番号1)のトリフルオロ酢酸塩を0.16g得た。得られたトリフルオロ酢酸塩から、定法に従い酢酸塩を作成し、評価に用いた。
質量分析: m/z = 554.73 [M+2H]+2、保持時間: 0.82min (LCMS条件A)
精製条件
HPLCシステム:Gilson社製ハイスループットHPLC分取システム
カラム:YMC ODS-A 3cmφ×25cm、10μm
溶出液1:0.1%TFA水
溶出液2:0.035%TFAアセトニトリル
流速:20mL/min
グラジエントメソッド:


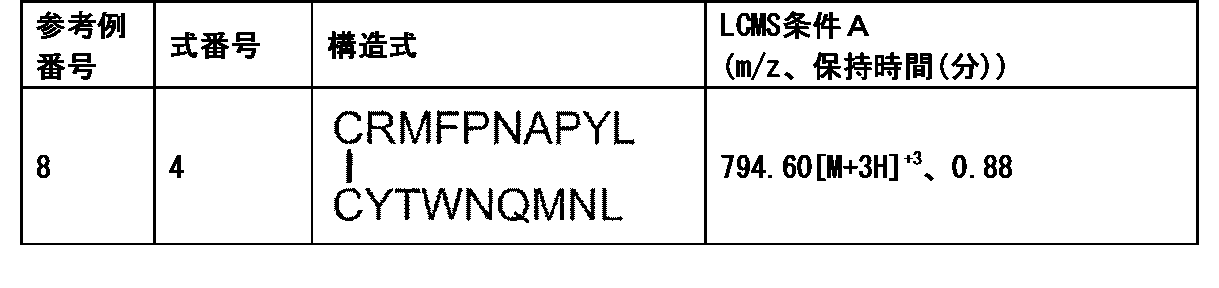
N-2,2,3,3-ペンタメチル-4,7,10,13,16-ペンタオキサ-3-シラオクタデカン-18-アミンの合成

m/z = 334 [M+H]+、Rt = 0.507 (LCMS条件B)

m/z = 448 [M+H]+、Rt = 1.153 (LCMS条件B)
m/z = 463 [M+H]+、Rt = 1.210 (LCMS条件B)
m/z = 366 [M+H+]、Rt = 0.718 (LCMS条件B)
N,2,2-トリメチル-3,3-ジフェニル-4,7,10,13,16-ペンタオキサ-3-シラオクタデカン-18-アミンの合成
m/z = 490 [M+H]+、Rt = 0.953 (LCMS条件B)
(3S)-3-{[2-アミノ-5-({2-メトキシ-4-[(メチルアミノ)メチル]フェニル}メチル)-6-メチルピリミジン-4-イル]アミノ}ヘキサン-1-オールの合成

m/z = 194 [M+2H]+2、Rt = 0.552 (LCMS条件B)
5-({2-メトキシ-4-[(メチルアミノ)メチル]フェニル}メチル)-6-メチル-N 4 -ペンチルピリミジン-2,4-ジアミンの合成
m/z = 179 [M+2H]+2、Rt = 0.475 (LCMS条件B)
3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-メトキシベンズアルデヒドの合成
m/z = 179 [M+2H]+2、Rt = 0.475 (LCMS条件B)
3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-ヒドロキシベンズアルデヒドの合成

m/z = 329 [M+H]+、Rt = 0.748 (LCMS条件B)
5-({2-メトキシ-5-[(メチルアミノ)メチル]フェニル}メチル)-6-メチル-N 4 -ペンチルピリミジン-2,4-ジアミンの合成
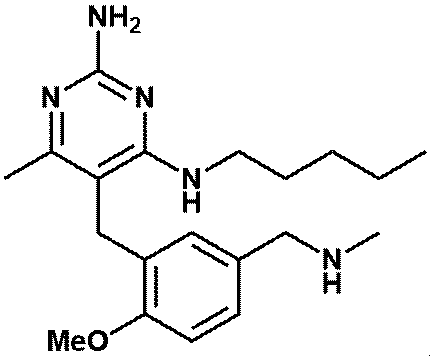
m/z = 179 [M+2H]+2、Rt = 0.599 (LCMS条件B)
2-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-[(メチルアミノ)メチル]フェノールの合成

m/z = 173 [M+2H]+2、Rt = 0.562 (LCMS条件B)
N,2,2,3,3-ペンタメチル-4,7,10,13,16,19,22,25,28,31-デカオキサ-3-シラトリトリアコンタン-33-アミン
m/z = 587 [M+H]+、Rt = 0.865 (LCMS条件B)
N,2,2,3,3-ペンタメチル-4,7,10,13,16,19,22,25,28,31,34,37,40,46,49,52,55,58,61,64,67,70,73-テトラコサオキサ-3-シラペンタヘプタコンタン-75-アミン
m/z = 402 [M+3H]+3、Rt = 0.946 (LCMS条件B)
N,2,2,3,3-ペンタメチル-4,7,10,13,16,19,22,25,28,31,34,37,40,46,49,52,55,58,61,64,67,70,73,76,79,82,85,88,91,94,97,100,103,106,109-ヘキサトリアコンタオキサ-3-シラヘンデカヘクタン-111-アミン

m/z = 866 [M+2H]+2、Rt = 0.948 (LCMS条件B)
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール・トリフルオロ酢酸塩の合成

m/z = 290 [M+2H]+2、Rt = 0.623 (LCMS条件B)
1H-NMR (CDCl3): δ 7.34 (s, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.08 (t, J = 5.6 Hz, 1H), 3.96 (s, 3H), 3.88 (m, 2H), 3.70 (m, 2H), 3.64-3.55 (m, 18H), 3.47 (s, 1H), 3.34 (dd, J = 6.8, 12 Hz, 2H), 2.81 (s, 3H), 2.47 (s, 3H), 1.49-1.42 (m, 2H), 1.30-1.23 (m, 2H), 1.21-1.13 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H)
1-{4-[(2-アミノ-4-{[(3S)-1-ヒドロキシヘキサン-3-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシフェニル}-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール・トリフルオロ酢酸塩の合成
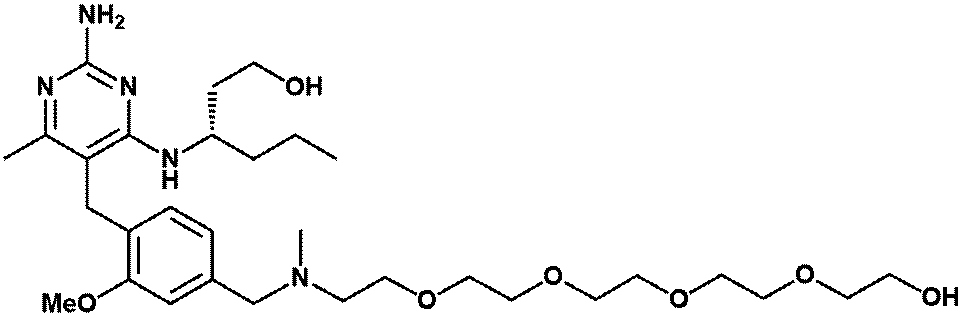
m/z = 305 [M+2H]+2、Rt = 0.527 (LCMS条件B)
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール・トリフルオロ酢酸塩の合成
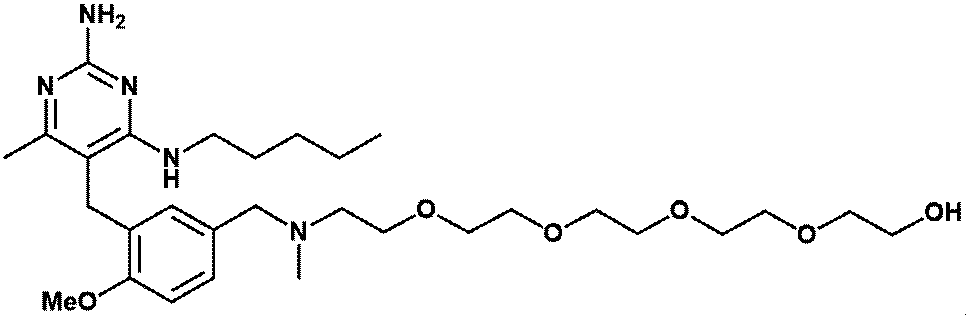
m/z = 290 [M+2H]+2、Rt = 0.661 (LCMS条件B)
1H-NMR (CDCl3): δ 7.32 (dd, J = 1.6, 8.4 Hz, 1H), 7.13 (d, J = 1.6 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.24 (t, J = 4.8 Hz, 1H), 4.26 (m, 2H), 3.86 (s, 3H), 3.81 (m, 2H), 3.64-3.49 (m, 18H), 3.29 (dd, J = 6.8, 12.8 Hz, 2H), 2.70 (s, 3H), 2.43 (s, 3H), 1.44-1.36 (m, 2H), 1.24-1.17 (m, 2H), 1.13-1.07 (m, 2H), 0.80 (t, J = 7.2 Hz, 3H)
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-ヒドロキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール・トリフルオロ酢酸塩の合成
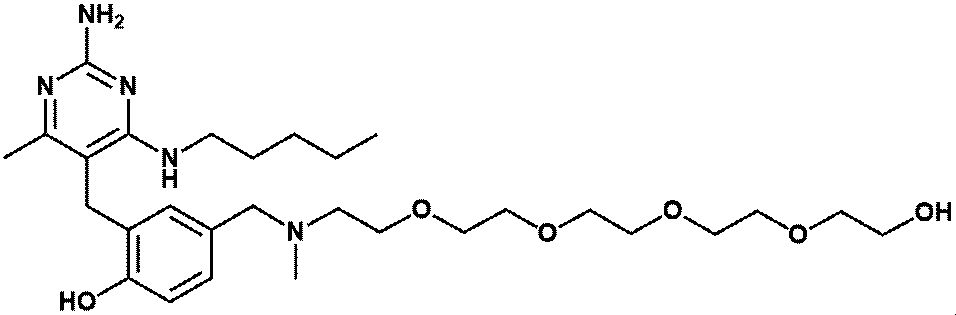
m/z = 290 [M+2H]+2、Rt = 0.661 (LCMS条件B)
1H-NMR (CDCl3): δ 7.30 (t, J = 5.2 Hz, 1H), 7.07 (d, J = 2.0 Hz, 1H), 6.97 (dd, J = 1.6, 8.0 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.10 (m, 2H), 3.70 (t, J = 5.6 Hz, 2H), 3.63 (d, J = 5.6 Hz, 2H), 3.55-3.49 (m, 18H), 3.26 (dd, J = 7.2, 13.2 Hz, 2H), 2.68 (s, 3H), 2.43 (s, 3H), 1.47-1.40 (m, 2H), 1.24-1.15 (m, 2H), 1.13-1.05 (m, 2H), 0.76 (t, J = 6.8 Hz, 3H)
4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-N-(20-ヒドロキシ-3,6,9,12,15,18-ヘキサオキサイコサン-1-イル)-3-メトキシベンズアミドの合成
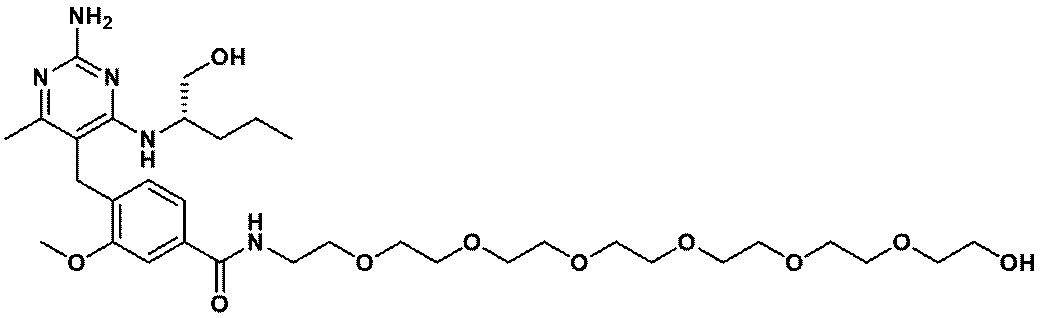
m/z = 683 [M+H]+、Rt = 0.558 (LCMS条件B)
1H-NMR (CDCl3): δ 7.49 (1H, s), 7.21 (1H, d, J = 7.9 Hz), 7.05 (1H, s),6.95 (1H, d, J = 7.9 Hz), 4.77 (1H, d, J = 6.7 Hz), 4.68 (2H, s), 4.01-3.92 (1H, m), 3.94 (3H, s), 3.75-3.68 (4H, m), 3.67-3.51 (26H, m), 3.42-3.35 (1H, m), 2.28 (3H, s), 1.45-1.36 (1H, m), 1.29-1.20 (1H, m), 1.16-1.06 (2H, m), 0.80 (3H, t, J = 7.3 Hz).
2,5,8,11-テトラオキサトリデカン-13-イル 4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシベンゾエートの合成

m/z = 565 [M+H]+、Rt = 0.665 (LCMS条件B)
1H-NMR (CDCl3):δ 7.56 (1H, d, J = 7.9 Hz), 7.51 (1H, s), 6.99 (1H, d, J = 7.9 Hz), 6.12 (1H, d, J = 7.9 Hz), 4.41 (2H, t, J = 4.6 Hz), 4.20-4.10 (1H, m), 3.93 (3H, s), 3.86-3.41 (18H, m), 3.29 (3H, s), 3.02 (1H, q, J = 7.5 Hz), 2.44 (3H, s), 1.50-1.22 (2H, m), 1.12-0.91 (2H, m), 0.74 (3H, t, J = 7.0 Hz).
5-{[2-メトキシ-4-(2,5,8,11,14-ペンタオキサペンタデカン-1-イル)フェニル]メチル}-6-メチル-N 4 -ペンチルピリミジン-2,4-ジアミンの合成
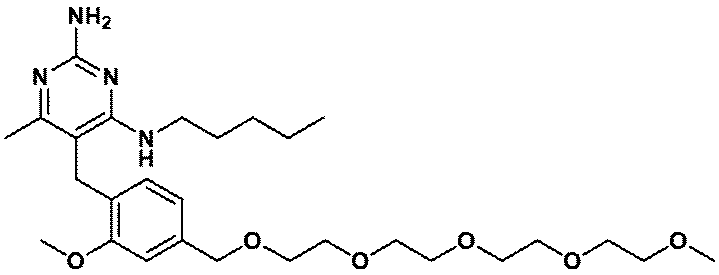
m/z = 536 [M+H]+、Rt = 0.850 (LCMS条件B)
1H-NMR (CDCl3):δ 6.88 (1H, s), 6.84 (1H, d, J = 7.3 Hz), 6.78 (1H, d, J = 7.3 Hz), 5.80-5.40 (2H, br), 4.46 (2H, s), 3.86 (3H, s), 3.67-3.46 (19H, m), 3.30 (3H, s), 3.24 (2H, td, J = 7.0, 5.5 Hz), 2.35 (3H, s), 1.36 (2H, m), 1.23-1.05 (4H, m), 0.78 (3H, t, J = 7.0 Hz).
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29-ノナオキサ-2-アザヘントリアコンタン-31-オールの合成
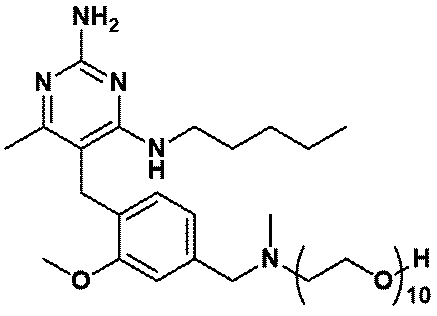
m/z = 400 [M+2H]+2、Rt = 0.583 (LCMS条件B)
1H-NMR (CDCl3):δ 6.86 (1H, s), 6.81 (1H, d, J = 7.9 Hz), 6.73 (1H, d, J = 7.9 Hz), 6.00-5.00 (2H, br), 3.85 (3H, s), 3.68 (2H, t, J = 4.6 Hz), 3.63-3.50 (40H, m), 3.44 (2H, s), 3.24 (2H, q, J = 6.5 Hz), 2.54 (2H, t, J = 6.1 Hz), 2.32 (3H, s), 2.19 (3H, s), 1.41-1.34 (2H, m), 1.23-1.16 (2H, m), 1.13-1.05 (2H, m), 0.78 (3H, t, J = 7.3 Hz).
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71-トリコサオキサ-2-アザトリヘプタコンタン-73-オールの合成

m/z = 708 [M+2H]+2、Rt = 0.672 (LCMS条件B)
1H-NMR (CDCl3):δ 6.83 (1H, s), 6.79 (1H, d, J = 7.9 Hz), 6.69 (1H, d, J = 7.3 Hz), 4.79 (1H, s), 4.56 (2H, s), 3.83 (3H, s), 3.76-3.40 (99H, m), 3.21 (2H, dd, J = 12.5, 7.0 Hz), 2.53 (2H, t, J = 6.1 Hz), 2.24 (3H, s), 2.18 (3H, s), 1.39-1.32 (2H, m), 1.22-1.14 (2H, m), 1.12-1.04 (2H, m), 0.77 (3H, t, J = 7.0 Hz).
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98,101,104,107-ペンタトリアコンタオキサ-2-アザノナヘクタン-109-オールの合成
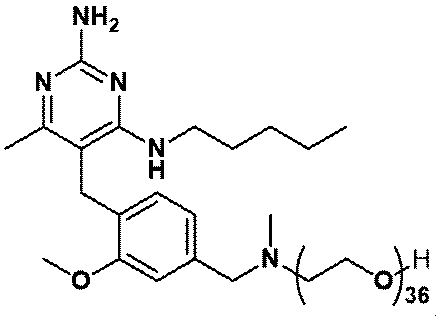
m/z = 487 [M+4H]+4、Rt = 0.693 (LCMS条件B)
1H-NMR (CDCl3):δ 6.83 (1H, s), 6.79 (1H, d, J = 7.9 Hz), 6.69 (1H, d, J = 9.2 Hz), 4.81 (1H, s), 4.60 (2H, s), 3.83 (3H, s), 3.76-3.38 (147H, m), 3.21 (2H, dd, J = 12.5, 7.0 Hz), 2.53 (2H, t, J = 5.8 Hz), 2.24 (3H, s), 2.18 (3H, s), 1.39-1.32 (2H, m), 1.21-1.14 (2H, m), 1.12-1.06 (2H, m), 0.77 (3H, t, J = 7.3 Hz).
12-[(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)メチル]-3,6,9,15,18,21-ヘキサオキサ-12-アザトリコサン-1,23-ジオールの合成
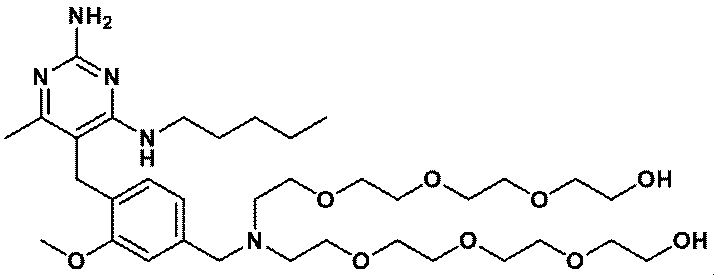
m/z = 349 [M+2H]+2、Rt = 0.554 (LCMS条件B)
1H-NMR (CDCl3):δ 6.85 (1H, s), 6.79 (1H, d, J = 7.9 Hz), 6.73 (1H, d, J = 7.9 Hz), 4.85 (1H, t, J = 5.5 Hz), 4.57 (2H, s), 3.83 (3H, s), 3.68-3.42 (34H, m), 3.21 (2H, td, J = 7.2, 5.3 Hz), 2.67 (4H, t, J = 6.1 Hz), 2.24 (3H, s), 1.41-1.33 (2H, m), 1.23-1.09 (4H, m), 0.78 (3H, t, J = 7.0 Hz).
ヒトTLR7レポータージーンアッセイ
TLR7/NF-κB/SEAPorterTM HEK293細胞株(Imgenex Corporation)は、NF-κB応答エレメントの転写制御下で全長ヒトTLR7と分泌型アルカリフォスファターゼ(SEAP)レポーター遺伝子を発現する安定なコトランスフェクト細胞株である。この細胞株でのTLR7発現はフローサイトメトリーにより試験済みである。抗生物質ブラストサイジンおよびジェネティシンを使用して、安定に発現する形質転換体を選択した。TLRシグナル伝達はNF-κBの転位を導き、プロモーターの活性化はSEAP遺伝子の発現をもたらす。この細胞を0.1%(v/v)ジメチルスルホキシド(DMSO)存在下で37℃にて実施例及び参考例で合成された化合物と24時間インキュベーションした後、生成したSEAPのレベルを測定することにより、TLR7特異的な活性化を評価した。本発明化合物のヒトTLR7活性化の程度を、ヒトTLR7レポータージーンアッセイを用いて評価し、SEAPの最大レベルの半分をもたらす化合物の濃度(EC50)として表3及び表4に示した。
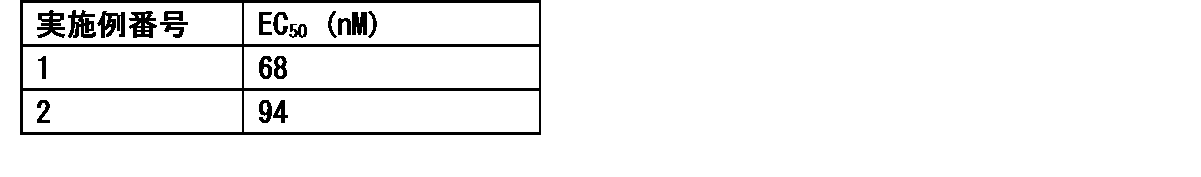

マウスTLR7レポータージーンアッセイ
HEK-BlueTM mTLR7細胞株(Invivogen)は、NF-κB応答エレメントの転写制御下で全長マウスTLR7と分泌型SEAPレポーター遺伝子を発現する安定なコトランスフェクト細胞株である。この細胞株でのTLR7発現はRT-PCRにより試験済みである。抗生物質ブラストサイジンおよびゼオシンを使用して、安定に発現する形質転換体を選択した。TLRシグナル伝達はNF-κBの転位を導き、プロモーターの活性化はSEAP遺伝子の発現をもたらす。この細胞を0.1%(v/v)DMSO存在下で37℃にて実施例及び参考例で合成された化合物と20-24時間インキュベーションした後、生成したSEAPのレベルを測定することにより、TLR7特異的な活性化を評価した。本発明化合物のマウスTLR7活性化の程度を、マウスTLR7レポータージーンアッセイを用いて評価し、SEAPの最大レベルの半分をもたらす化合物の濃度(EC50)として表5及び表6に示した。
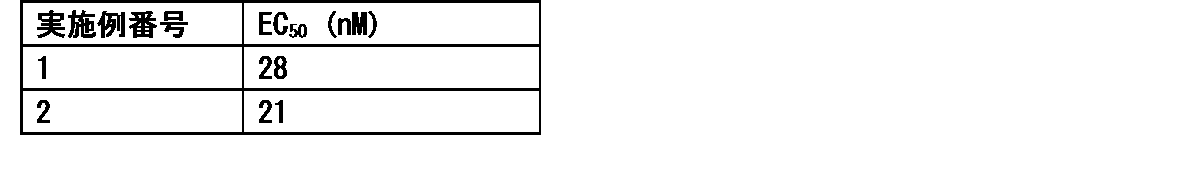
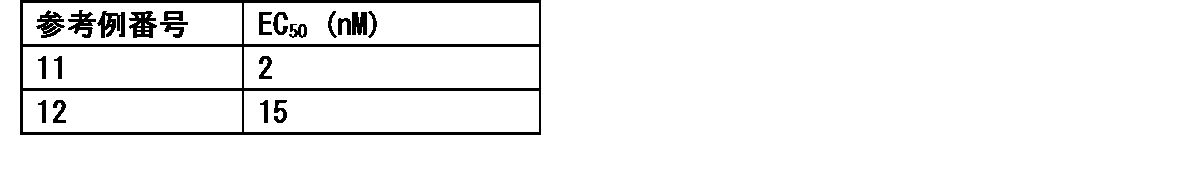
HLA-A * 24:02遺伝子導入マウスを用いた、in vivoでのCTL誘導能の評価
実施例1で合成された化合物および参考例12で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをMontanide ISA 51 VGと混合したカクテルワクチン(以下、「カクテルワクチンb」という)に実施例1で合成された化合物を添加したワクチンおよび、カクテルワクチンbに参考例12で合成された化合物を添加してワクチンを調製し、これをHLA-A*24:02遺伝子導入マウスに投与し、抗原特異的細胞傷害性T細胞(CTL)誘導試験によってアジュバント活性を評価した。
式番号4で表される化合物に含まれるCYTWNQMNL(配列番号2)はHLA-A*24:02拘束性WT1タンパク質由来抗原ペプチドである。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物および参考例12で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドを予備乳化組成物と混合したカクテルワクチン(以下、「カクテルワクチンa」という)に、実施例1で合成された化合物あるいは参考例12で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
式番号4:
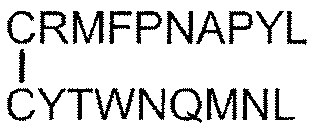
式番号4に含まれるRMFPNAPYL(配列番号1)および配列番号3で表されるペプチドに含まれるVLDFAPPGA(配列番号4)はHLA-A*02:01拘束性WT1タンパク質由来抗原ペプチドである。
リン酸二水素ナトリウム二水和物0.312gを注射用水80gに溶解した。オレイン酸エチル14.0g、ミリスチン酸オクチルドデシル14.0g、モノオレイン酸ソルビタン2.0g、モノオレイン酸グリセリン2.8g、ポリオキシエチレン硬化ヒマシ油200.4g、グリセリン0.4gを混合した。このうち2.354mL(2.077g相当量)を試験管にとり、ミキサー(ウルトラタラックスT10、IKA社、またはタッチミキサーMT-51、ヤマト科学)で撹拌しながらリン酸二水素ナトリウム水溶液0.396mL(0.396g相当量)を徐々に加えて乳化し、予備乳化組成物とした。調製量は必要に応じて増減した。
式番号4で表される化合物および配列番号3で表されるペプチドをDMSOで溶解した後、希釈後の濃度が式番号4で表される化合物が3mg/mL、配列番号3で表されるペプチドが2.25mg/mLになるよう注射用水と混合した。このペプチド希釈液を等量の上記予備乳化組成物と混合してエマルション化させて調製したカクテルワクチンaを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンaを調製する際のペプチド希釈液中に実施例1で合成された化合物を添加して調製したワクチンを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹、実施例1で合成された化合物が32.5ng/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンaを調製する際のペプチド希釈液中に参考例12で合成された化合物を添加して調製したワクチンを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹、参考例12で合成された化合物が22.5ng/匹となるよう、マウスの尾根部皮内に投与した。1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。IFNγ産生の測定には、IFNγ ELISPOT assay kitを用いた。脾細胞調製の前日に、ELISPOTプレートを抗マウスIFNγ抗体で処理し、当日に10%FBSを含むRPMI1640培地でブロッキングした。調製した脾細胞を1.25×105cells/wellで、ブロッキングしたELISPOTプレートに播種した。ペプチド(配列番号1あるいは配列番号4)を0.1%(v/v)DMSO存在下、最終濃度10μg/mLで脾細胞に添加した。ペプチドを添加した脾細胞を37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、ELISPOTプレートを、添付のプロトコールに従って発色させた。発色したスポット数は、ImmunoSpot Analyzerによって測定した。
ワクチンによるin vivo腫瘍増殖抑制効果に対する増強効果
HLA-A*24:02遺伝子導入マウスの腹側部皮内にコーン油に懸濁した3-methylcholanthreneを投与し、投与部位に発生した腫瘍からHLA-A*2402/Kb発現腫瘍細胞を取得した。本細胞にWT1抗原ペプチド(配列番号2)を安定発現させた細胞株(本明細書中、MCA-A24/Kb-WT1腫瘍細胞とも記載する)を、ハンクス平衡塩溶液中に浮遊させ、HLA-A*24:02遺伝子導入マウスの腹側部皮内に移植した(マウス1匹につき、5×105個)。ビークルを投与する群(a群)、ワクチンを投与する群(b群)、実施例1で合成された化合物を添加したワクチンを投与する群(c群)を設定した。それぞれの群について6匹のマウスを使用した。腫瘍細胞を移植した7日前および7日後に、a群のマウスには注射用水を含む組成物を等量のMontanide ISA 51 VGと混合しエマルション化させたのち、尾根部皮内に投与した(1回あたり、0.1mL/匹を投与)。b群のマウスには、式番号4で示される化合物および配列番号3で示されるペプチドを含む組成物を等量のMontanide ISA 51 VGと混合しエマルション化させたのち、尾根皮内に投与した(1回あたり、式番号4で示される化合物を300μg/匹および配列番号3で示されるペプチドを225μg/匹投与)。c群のマウスには、式番号4で示される化合物と配列番号3で示されるペプチドおよび実施例1で合成された化合物を含む組成物を等量のMontanide ISA 51 VGと混合しエマルション化させたのち、尾根皮内に投与した(1回あたり、式番号4で示される化合物を300μg/匹、配列番号3で示されるペプチドを225μg/匹および実施例1で合成された化合物を100ng/匹投与)。腫瘍移植27日後に腫瘍径を測定し、腫瘍体積を算出した。


HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例6で合成された配列番号6で表されるペプチドをMontanide ISA 51 VGと混合して調製したワクチン(以下、「ワクチンc」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。配列番号6で表されるペプチドGLYDGMEHLはHLA-A*02:01拘束性MAGE-A10タンパク質由来抗原ペプチドである。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例5で合成された配列番号5で表されるペプチドをMontanide ISA 51 VGと混合して調製したワクチン(以下、「ワクチンd」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。配列番号5で表されるペプチドVLQELNVTVはHLA-A*02:01拘束性Proteinase-3タンパク質由来抗原ペプチドである。
HLA-A * 24:02遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例20で合成された配列番号18で表されるペプチドをMontanide ISA 51 VGと混合したワクチン(以下、「ワクチンe」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*24:02遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。配列番号18で表されるペプチドTYAGCLSQIFはHLA-A*24:02拘束性Or7c1タンパク質由来抗原ペプチドである。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例8、9、10で合成された化合物および参考例12で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをMontanide ISA51 VGと混合したカクテルワクチンbに、実施例8、9、あるいは10で合成された化合物あるいは参考例12で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例11で合成された化合物のin vivoにおけるアジュバント活性に関しては、試験例9と同様のカクテルワクチンbに、実施例11で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドを乳化組成物1としたカクテルワクチン(以下、「カクテルワクチンf」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドを乳化組成物2としたカクテルワクチン(以下、「カクテルワクチンg」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドを乳化組成物3としたカクテルワクチン(以下、「カクテルワクチンh」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドを油性懸濁液剤としたカクテルワクチン(以下、「カクテルワクチンi」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
カクテルワクチンiを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンiを調製する際のpH2.5緩衝液中に実施例1で合成された化合物を添加したワクチンを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹、実施例1で合成された化合物が0.04nmol/匹あるいは0.4nmol/匹になるよう、マウスの尾根部皮内に投与した。投与は1週間間隔で2回行った。最終投与の1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。試験例4と同様に最終濃度10μg/mLのペプチド(配列番号1あるいは配列番号4)を添加した脾細胞をELISPOTプレートに播種して37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、発色させたELISPOTプレートのスポット数を測定した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをハイドロゲル製剤としたワクチン(以下、「カクテルワクチンj」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
カクテルワクチンjを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンjを調製する際のpH2.5緩衝液中に実施例1で合成された化合物を添加したワクチンを、式番号4で表される化合物が300μg/匹、配列番号3で表されるペプチドが225μg/匹、実施例1で合成された化合物が0.4nmol/匹になるよう、マウスの尾根部皮内に投与した。投与は1週間間隔で2回行った。最終投与の1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。試験例4と同様に最終濃度10μg/mLのペプチド(配列番号1あるいは配列番号4)を添加した脾細胞をELISPOTプレートに播種して37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、発色させたELISPOTプレートのスポット数を測定した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをリポソーム製剤1としたワクチン(以下、「カクテルワクチンk」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
カクテルワクチンkを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンkを調製する際のpH2.5緩衝液中に実施例1で合成された化合物を添加したワクチンを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹、実施例1で合成された化合物が0.4nmol/匹になるよう、マウスの尾根部皮内に投与した。投与は1週間間隔で2回行った。最終投与の1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。試験例4と同様に最終濃度10μg/mLのペプチド(配列番号1)を添加した脾細胞をELISPOTプレートに播種して37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、発色させたELISPOTプレートのスポット数を測定した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをリポソーム製剤2としたワクチン(以下、「カクテルワクチンl」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
カクテルワクチンlを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンlを調製する際のpH2.5緩衝液中に実施例1で合成された化合物を添加したワクチンを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹、実施例1で合成された化合物が0.4nmol/匹になるよう、マウスの尾根部皮内に投与した。投与は1週間間隔で2回行った。最終投与の1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。試験例4と同様に最終濃度10μg/mLのペプチド(配列番号1あるいは配列番号4)を添加した脾細胞をELISPOTプレートに播種して37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、発色させたELISPOTプレートのスポット数を測定した。
HLA-A * 02:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例8で合成された式番号4で表される化合物と参考例3で合成された配列番号3で表されるペプチドをリポソーム製剤3としたワクチン(以下、「カクテルワクチンm」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によって評価した。
カクテルワクチンmを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹になるよう、マウスの尾根部皮内に投与した。または、カクテルワクチンmを調製する際のpH2.5緩衝液中に実施例1で合成された化合物を添加したワクチンを、式番号4で表される化合物が500μg/匹、配列番号3で表されるペプチドが375μg/匹、実施例1で合成された化合物が0.4nmol/匹になるよう、マウスの尾根部皮内に投与した。投与は1週間間隔で2回行った。最終投与の1週間後に、マウスをCO2ガスにより安楽死させたのち脾臓を摘出し、脾細胞を調製した。試験例4と同様に最終濃度10μg/mLのペプチド(配列番号1)を添加した脾細胞をELISPOTプレートに播種して37℃、5% CO2下で一晩培養することで、in vitroにおけるペプチド再刺激を加えた。培養後に上清を除き、発色させたELISPOTプレートのスポット数を測定した。
HLA-A * 02:01/HLA-DRB1 * 01:01遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例1で合成された配列番号1で表されるペプチドと参考例20で合成された配列番号17で表されるペプチドを予備乳化組成物と混合して調製したカクテルワクチン(以下、「カクテルワクチンn」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*02:01/HLA-DRB1*01:01遺伝子導入マウス(C57BL/6CrHLA-A2.1DR1)に投与し、抗原特異的CTLおよび抗原特異的ヘルパーT細胞誘導試験によって評価した。配列番号17で表されるペプチドは、WT1タンパク質由来ヘルパーペプチドである。
HLA-A * 24:02遺伝子導入マウスを用いたin vivoアジュバント活性の評価
実施例1で合成された化合物のin vivoにおけるアジュバント活性に関しては、参考例22で合成された式番号5で表される化合物を予備乳化組成物と混合して調製したワクチン(以下、「ワクチンo」という)に、実施例1で合成された化合物を添加してワクチンを調製し、これをHLA-A*24:02遺伝子導入マウスに投与し、抗原特異的CTL誘導試験によってアジュバント活性を評価した。
試験例1の方法に準じ、ヒトのTLR7レポータージーンアッセイを実施し、以下の表の結果を得た。

試験例1の方法に準じ、ヒトのTLR7レポータージーンアッセイを実施し、以下の表の結果を得た。この時、細胞としてはHEK-BlueTM hTLR7細胞株(Invivogen)を用いた。HEK-BlueTM hTLR7細胞株は、NF-κB応答エレメントの転写制御下で全長ヒトTLR7と分泌型SEAPレポーター遺伝子を発現する安定なコトランスフェクト細胞株である。この細胞株でのTLR7発現はRT-PCRにより試験済みである。抗生物質ブラストサイジンおよびゼオシンを使用して、安定に発現する形質転換体を選択した。
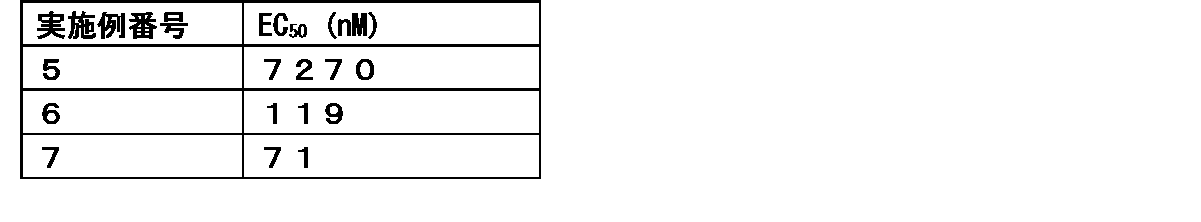
Claims (31)
- 式(1):

R1は、C1-6のアルキル(該アルキルは、ハロゲン、ヒドロキシ、およびC1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
R2及びR3は、それぞれ独立して、水素原子、またはC1-6アルキル(該アルキルは、ハロゲン、ヒドロキシ、C1-6アルコキシからなる群から独立して選択される1~5個の置換基で置換されていてもよい)を表し、
R4は、水素原子、ハロゲン、ヒドロキシ、C1-6アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)、C1-6アルコキシ(該アルコキシは、1~3個の同一または異なるハロゲンで置換されていてもよい)またはシアノを表し、
Lはリンカーを表し、
Y1は、-(CH2CH2O)m-R6(R6は、水素原子またはC1-6アルキルを表し、mは、3~100の整数を表す)を表す。]で示される化合物またはその製薬学的に許容される塩。 - Xが、メチレンである、請求項1に記載の化合物またはその製薬学的に許容される塩。
- R1が、C1-3アルキル(該アルキルは、1~3個の同一または異なるハロゲンで置換されていてもよい)である、請求項1または2に記載の化合物またはその製薬学的に許容される塩。
- R1が、メチルである、請求項3に記載の化合物またはその製薬学的に許容される塩。
- R4が、水素原子、ヒドロキシ、C1-3アルキル、またはC1-3アルコキシである、請求項1~4のいずれかに記載の化合物またはその製薬学的に許容される塩。
- R4が、水素原子、ヒドロキシ、またはメトキシである、請求項5に記載の化合物またはその製薬学的に許容される塩。
- R2が、C1-6アルキルである、請求項1~6のいずれかに記載の化合物またはその製薬学的に許容される塩。
- R3が、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)である、請求項1~7のいずれかに記載の化合物またはその製薬学的に許容される塩。
- Lが、-O-、-NRY-、-C(O)-、-C(O)O-、-OC(O)-、-C(O)NRY-、-NRYC(O)-、-CH2NRY-、-CH2O-、-OC(O)O-、-NR7C(O)O-、-OC(O)NRY-、-NR7C(O)NRY-、-OC(S)NRY-、または-NR7C(S)NRY-(ここで、R7は、水素原子または炭素数1~6のアルキルを表し、RYは、水素原子、炭素数1~6のアルキル、またはY2(Y2は、-(CH2CH2O)n-R8(R8は、水素原子または炭素数1~6のアルキルを表し、nは、3~100の整数を表す)を表す)を表す)である、請求項1~8のいずれかに記載の化合物またはその製薬学的に許容される塩。
- Lが、-C(O)NRY-、-CH2NRY-、-C(O)O-または-CH2O-である、請求項9に記載の化合物またはその製薬学的に許容される塩。
- Lが、-CH2NRY-であり、RYが、水素原子、炭素数1~6のアルキルまたはY2である、請求項9に記載の化合物またはその製薬学的に許容される塩。
- Y1が、-(CH2CH2O)m-R6であり、R6が、水素原子またはC1-6アルキルであり、mが、3~40の整数である、請求項1~11のいずれかに記載の化合物またはその製薬学的に許容される塩。
- 式(2):

式(3):

R3は、水素原子、またはC1-3アルキル(該アルキルは、1~3個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、ヒドロキシ、またはメトキシであり、
Lは、-CH2NRY-であり、
RYは、水素原子またはC1-6アルキルであり、
Y1は、-(CH2CH2O)m-R6であり、
R6は、水素原子またはC1-6アルキルであり、
mは、3~40の整数である。]
で表される、請求項1に記載の化合物またはその製薬学的に許容される塩。 - 式(2):
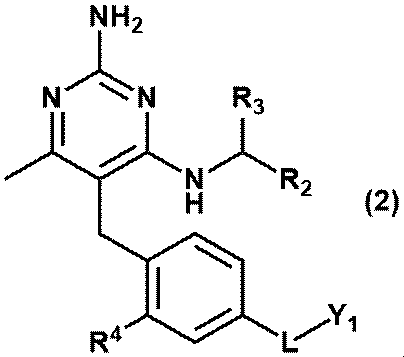
R3は、水素原子、またはC1-3アルキル(該アルキルは、1個のヒドロキシで置換されていてもよい)であり、
R4は、水素原子、またはメトキシであり、
Lは、-CH2NRY-であり、
RYは、水素原子またはC1-6アルキルであり、
Y1は、-(CH2CH2O)m-R6であり、
R6は、水素原子またはC1-6アルキルであり、
mは、3~40の整数である。]
で表される、請求項1に記載の化合物またはその製薬学的に許容される塩。 - 以下の化合物群から選択される、請求項1に記載の化合物又はその製薬学的に許容される塩:
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール、
1-{4-[(2-アミノ-4-{[(3S)-1-ヒドロキシヘキサン-3-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3-メトキシフェニル}-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール、
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール、
1-(3-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-4-ヒドロキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール、
4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-N-(20-ヒドロキシ-3,6,9,12,15,18-ヘキサオキサイコサン-1-イル)-3-メトキシベンズアミド、
2,5,8,11-テトラオキサトリデカン-13-イル 4-[(2-アミノ-4-{[(2S)-1-ヒドロキシペンタン-2-イル]アミノ}-6-メチルピリミジン-5-イル)メチル]-3メトキシベンゾエート、
5-{[2-メトキシ-4-(2,5,8,11,14-ペンタオキサペンタデカン-1-イル)フェニル]メチル}-6-メチル-N4-ペンチルピリミジン-2,4-ジアミン、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29-ノナオキサ-2-アザヘントリアコンタン-31-オール、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71-トリコサオキサ-2-アザトリヘプタコンタン-73-オール、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98,101,104,107-ペンタトリアコンタオキサ-2-アザノナヘクタン-109-オール、および
12-[(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)メチル]-3,6,9,15,18,21-ヘキサオキサ-12-アザトリコサン-1,23-ジオール。 - 以下の化合物群から選択される、請求項1に記載の化合物又はその製薬学的に許容される塩:
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14-テトラオキサ-2-アザヘキサデカン-16-オール、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29-ノナオキサ-2-アザヘントリアコンタン-31-オール、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71-トリコサオキサ-2-アザトリヘプタコンタン-73-オール、
1-(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)-2-メチル-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47,50,53,56,59,62,65,68,71,74,77,80,83,86,89,92,95,98,101,104,107-ペンタトリアコンタオキサ-2-アザノナヘクタン-109-オール、および
12-[(4-{[2-アミノ-4-メチル-6-(ペンチルアミノ)ピリミジン-5-イル]メチル}-3-メトキシフェニル)メチル]-3,6,9,15,18,21-ヘキサオキサ-12-アザトリコサン-1,23-ジオール。 - 請求項1~16のいずれかに記載の化合物またはその製薬学的に許容される塩を含有する医薬組成物。
- エマルション製剤、油性懸濁剤、ハイドロゲル製剤、又は脂質製剤である請求項17に記載の医薬組成物。
- エマルション製剤である請求項18に記載の医薬組成物。
- エマルション製剤が油中水型エマルションである請求項19に記載の医薬組成物。
- エマルション製剤が、(1)オレイン酸エチル、ミリスチン酸オクチルドデシル、モノオレイン酸ソルビタン、モノオレイン酸グリセリン、ポリオキシエチレン硬化ヒマシ油20、グリセリン、及びリン酸二水素ナトリウム、又は(2)Montanide ISA 51VGを含有する請求項20に記載の医薬組成物。
- 脂質製剤である請求項18に記載の医薬組成物。
- 脂質製剤が、リン脂質を含むリポソーム製剤である請求項22に記載の医薬組成物。
- 脂質製剤が、ステロール類を含むリポソーム製剤である請求項22または23に記載の医薬組成物。
- ステロール類が、コレステロールである請求項24に記載の医薬組成物。
- リポソーム製剤が、無機酸、無機酸塩、有機酸、有機酸塩、糖類、緩衝剤、酸化防止剤、及びポリマー類からなる群から選択される1以上の添加物を含むリポソーム製剤である項23~25のいずれか一項に記載の医薬組成物。
- 更に、腫瘍抗原を含む、請求項17~26のいずれかに記載の医薬組成物。
- 腫瘍抗原が、腫瘍抗原ペプチドである、請求項27に記載の医薬組成物。
- 腫瘍抗原ペプチドが、式(4):
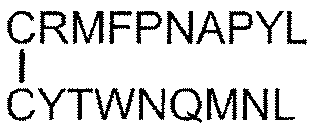
のアミノ酸配列で表されるペプチドまたはその製薬学的に許容される塩、および
配列番号3:WAPVLDFAPPGASAYGSL
のアミノ酸配列で表されるペプチドまたはその製薬学的に許容される塩の組み合わせである、請求項28に記載の医薬組成物。 - 請求項1~16のいずれかに記載の化合物またはその製薬学的に許容される塩を含む、癌ワクチンのワクチンアジュバント。
- a)請求項1の式(1)で示される化合物又はその薬学上許容し得る塩、もしくは式(1)で示される化合物又はその薬学上許容し得る塩を含有する医薬組成物;および
b)腫瘍抗原または腫瘍抗原を含有する医薬組成物
を含有するキット。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20784003.4A EP3949983A4 (en) | 2019-04-05 | 2020-04-03 | WATER SOLUBLE ADJUVANT |
| MX2021012201A MX2021012201A (es) | 2019-04-05 | 2020-04-03 | Adyuvante soluble en agua. |
| CA3136182A CA3136182A1 (en) | 2019-04-05 | 2020-04-03 | Water soluble adjuvant |
| AU2020251582A AU2020251582B2 (en) | 2019-04-05 | 2020-04-03 | Water soluble adjuvant |
| CN202080041623.7A CN113966326A (zh) | 2019-04-05 | 2020-04-03 | 水溶性佐剂 |
| JP2021512327A JP7539867B2 (ja) | 2019-04-05 | 2020-04-03 | 水溶性アジュバント |
| KR1020217033615A KR102921677B1 (ko) | 2019-04-05 | 2020-04-03 | 수용성 보조제 |
| US17/600,919 US12427192B2 (en) | 2019-04-05 | 2020-04-03 | Water soluble adjuvant and composition containing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-072910 | 2019-04-05 | ||
| JP2019072910 | 2019-04-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020204172A1 true WO2020204172A1 (ja) | 2020-10-08 |
Family
ID=72668226
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/015359 Ceased WO2020204172A1 (ja) | 2019-04-05 | 2020-04-03 | 水溶性アジュバント |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US12427192B2 (ja) |
| EP (1) | EP3949983A4 (ja) |
| JP (1) | JP7539867B2 (ja) |
| KR (1) | KR102921677B1 (ja) |
| CN (1) | CN113966326A (ja) |
| CA (1) | CA3136182A1 (ja) |
| MX (1) | MX2021012201A (ja) |
| WO (1) | WO2020204172A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023017836A1 (ja) * | 2021-08-12 | 2023-02-16 | 株式会社癌免疫研究所 | 癌を処置または予防するための医薬組成物および方法 |
| WO2023091879A1 (en) * | 2021-11-17 | 2023-05-25 | Lipella Pharmaceuticals Inc. | Intravesical delivery of hydrophilic therapeutic agents using liposomes |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12036318B2 (en) * | 2022-02-16 | 2024-07-16 | Chemical & Schutz High Performance Lubricants, S.A. De C.V. | Self-emulsionable mineral oil as a vehicle in vaccines for poultry |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006078059A1 (ja) | 2005-01-19 | 2006-07-27 | Dainippon Sumitomo Pharma Co., Ltd. | 希釈用乳化組成物および癌ワクチン組成物 |
| WO2007063903A1 (ja) | 2005-11-30 | 2007-06-07 | International Institute Of Cancer Immunology, Inc. | 新規ペプチド化合物 |
| WO2009067081A1 (en) | 2007-11-22 | 2009-05-28 | Astrazeneca Ab | Pyrimidine derivatives for the treatment of asthma, copd, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, cancer, hepatitis b, hepatitis c, hiv, hpv, bacterial infections and dermatosis |
| WO2012031140A1 (en) | 2010-09-01 | 2012-03-08 | Novartis Ag | Adsorption of immunopotentiators to insoluble metal salts |
| WO2012066336A1 (en) | 2010-11-19 | 2012-05-24 | Astrazeneca Ab | Benzylamine compounds as toll -like receptor 7 agonists |
| WO2014157704A1 (ja) | 2013-03-29 | 2014-10-02 | 大日本住友製薬株式会社 | Erap1によるトリミング機能をきっかけとしたコンジュゲートワクチン |
| WO2014157692A1 (ja) | 2013-03-29 | 2014-10-02 | 大日本住友製薬株式会社 | Wt1抗原ペプチドコンジュゲートワクチン |
| WO2017061532A1 (ja) | 2015-10-07 | 2017-04-13 | 大日本住友製薬株式会社 | ピリミジン化合物 |
| WO2018046460A1 (en) * | 2016-09-07 | 2018-03-15 | Glaxosmithkline Biologicals S.A. | Imidazoquinoline derivatives and their use in therapy |
| WO2018181420A1 (ja) * | 2017-03-29 | 2018-10-04 | 大日本住友製薬株式会社 | ワクチンアジュバント製剤 |
| JP2019500360A (ja) * | 2015-12-14 | 2019-01-10 | グラクソスミスクライン バイオロジカルズ ソシエテ アノニム | Tlr7及びtlr8アゴニストとしてのpeg化されたイミダゾキノリン |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2004268625B2 (en) * | 2003-08-27 | 2011-03-31 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
| KR20110117705A (ko) * | 2009-02-11 | 2011-10-27 | 더 리전트 오브 더 유니버시티 오브 캘리포니아 | 톨-유사 수용체 조정제 및 질병의 치료 |
| JO3257B1 (ar) * | 2009-09-02 | 2018-09-16 | Novartis Ag | مركبات وتركيبات كمعدلات لفاعلية tlr |
| KR102619747B1 (ko) * | 2017-01-10 | 2023-12-29 | 넥타르 테라퓨틱스 | Tlr 효현제 화합물의 다중-아암 중합체 컨쥬게이트 및 관련 면역 요법적 치료 방법 |
-
2020
- 2020-04-03 CN CN202080041623.7A patent/CN113966326A/zh active Pending
- 2020-04-03 KR KR1020217033615A patent/KR102921677B1/ko active Active
- 2020-04-03 MX MX2021012201A patent/MX2021012201A/es unknown
- 2020-04-03 JP JP2021512327A patent/JP7539867B2/ja active Active
- 2020-04-03 US US17/600,919 patent/US12427192B2/en active Active
- 2020-04-03 EP EP20784003.4A patent/EP3949983A4/en active Pending
- 2020-04-03 WO PCT/JP2020/015359 patent/WO2020204172A1/ja not_active Ceased
- 2020-04-03 CA CA3136182A patent/CA3136182A1/en active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006078059A1 (ja) | 2005-01-19 | 2006-07-27 | Dainippon Sumitomo Pharma Co., Ltd. | 希釈用乳化組成物および癌ワクチン組成物 |
| WO2007063903A1 (ja) | 2005-11-30 | 2007-06-07 | International Institute Of Cancer Immunology, Inc. | 新規ペプチド化合物 |
| WO2009067081A1 (en) | 2007-11-22 | 2009-05-28 | Astrazeneca Ab | Pyrimidine derivatives for the treatment of asthma, copd, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, cancer, hepatitis b, hepatitis c, hiv, hpv, bacterial infections and dermatosis |
| WO2012031140A1 (en) | 2010-09-01 | 2012-03-08 | Novartis Ag | Adsorption of immunopotentiators to insoluble metal salts |
| WO2012066336A1 (en) | 2010-11-19 | 2012-05-24 | Astrazeneca Ab | Benzylamine compounds as toll -like receptor 7 agonists |
| WO2014157704A1 (ja) | 2013-03-29 | 2014-10-02 | 大日本住友製薬株式会社 | Erap1によるトリミング機能をきっかけとしたコンジュゲートワクチン |
| WO2014157692A1 (ja) | 2013-03-29 | 2014-10-02 | 大日本住友製薬株式会社 | Wt1抗原ペプチドコンジュゲートワクチン |
| WO2017061532A1 (ja) | 2015-10-07 | 2017-04-13 | 大日本住友製薬株式会社 | ピリミジン化合物 |
| JP2019500360A (ja) * | 2015-12-14 | 2019-01-10 | グラクソスミスクライン バイオロジカルズ ソシエテ アノニム | Tlr7及びtlr8アゴニストとしてのpeg化されたイミダゾキノリン |
| WO2018046460A1 (en) * | 2016-09-07 | 2018-03-15 | Glaxosmithkline Biologicals S.A. | Imidazoquinoline derivatives and their use in therapy |
| WO2018181420A1 (ja) * | 2017-03-29 | 2018-10-04 | 大日本住友製薬株式会社 | ワクチンアジュバント製剤 |
Non-Patent Citations (22)
| Title |
|---|
| "Peptide Synthesis", 1966, INTERSCIENCE |
| "The Proteins", vol. 2, 1976, ACADEMIC PRESS INC. |
| BERMAN PW ET AL., SCIENCE, vol. 227, 1985, pages 1490 |
| CANCER IMMUNOL IMMUNOTHER, vol. 52, no. 10, October 2003 (2003-10-01), pages 608 - 16 |
| FREY SE ET AL., INFECT DIS, vol. 180, 1999, pages 1700 |
| GONCZOL E. ET AL., EXP. OPIN. BIOL. THER., vol. 1, 2001, pages 401 |
| INT J CANCER, vol. 100, 2002, pages 565 - 70 |
| J IMMUNOTHER CANCER., vol. 4, 20 September 2016 (2016-09-20), pages 56 |
| J. MED. CHEM., vol. 59, no. 12, 2016, pages 5868 - 5878 |
| JAGER E ET AL., INT. J. CANCER, vol. 67, 1996, pages 54 |
| NAT REV DRUG DISCOV, vol. 12, 2013, pages 130 - 146 |
| NAT REV IMMUNOL, vol. 14, 2014, pages 559 - 69 |
| NIKKEI, MEDICAL CANCER REVIEW, 2014, pages 9 |
| ROSA CL ET AL., BLOOD, vol. 100, 2002, pages 3681 |
| SCHEIBENBOGEN C ET AL., J. IMMUNOTHER, vol. 23, 2000, pages 275 |
| SCI TRANSL MED, vol. 6, 19 November 2014 (2014-11-19), pages 263 |
| See also references of EP3949983A4 |
| SEMIN IMMUNOL, vol. 22, no. 3, pages 155 - 61 |
| SMITH JWI ET AL., J. CLIN. ONCOL., vol. 21, pages 1562 |
| VACCINE, vol. 29, no. 17, 12 April 2011 (2011-04-12), pages 3341 - 55 |
| VACCINE, vol. 31, no. 42, 1 October 2013 (2013-10-01), pages 4827 - 33 |
| VAN DRIEL WJ ET AL., EUR. J. CANCER, vol. 35, 1999, pages 946 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023017836A1 (ja) * | 2021-08-12 | 2023-02-16 | 株式会社癌免疫研究所 | 癌を処置または予防するための医薬組成物および方法 |
| WO2023091879A1 (en) * | 2021-11-17 | 2023-05-25 | Lipella Pharmaceuticals Inc. | Intravesical delivery of hydrophilic therapeutic agents using liposomes |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3949983A4 (en) | 2022-12-21 |
| KR102921677B1 (ko) | 2026-02-02 |
| JP7539867B2 (ja) | 2024-08-26 |
| AU2020251582A1 (en) | 2021-12-02 |
| KR20220004637A (ko) | 2022-01-11 |
| US20220211844A1 (en) | 2022-07-07 |
| CN113966326A (zh) | 2022-01-21 |
| US12427192B2 (en) | 2025-09-30 |
| CA3136182A1 (en) | 2020-10-08 |
| JPWO2020204172A1 (ja) | 2020-10-08 |
| EP3949983A1 (en) | 2022-02-09 |
| MX2021012201A (es) | 2022-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6956967B2 (ja) | Wt1抗原ペプチドおよび免疫調節剤の併用 | |
| CN108290845B (zh) | 嘧啶化合物 | |
| JP7539867B2 (ja) | 水溶性アジュバント | |
| WO2018181648A1 (ja) | Wt1癌抗原ペプチドおよびこれを含むペプチドコンジュゲート体 | |
| WO2021230247A1 (ja) | 癌を処置するための医薬組成物 | |
| CN105658673B (zh) | 新型的具有4个连接的ctl表位的肽 | |
| CN105008399B (zh) | 新型的具有5个连接的ctl表位的肽 | |
| JP7209963B2 (ja) | Wt1ヘルパーペプチド及びこれと癌抗原ペプチドコンジュゲート体との組合せ | |
| JP7573516B2 (ja) | 水溶性アジュバントおよびそれを含有する組成物 | |
| JPWO2019131722A1 (ja) | Wt1由来ペプチドのコンジュゲート体およびこれを含む組成物 | |
| AU2020251582B2 (en) | Water soluble adjuvant | |
| HK40068431A (en) | Water soluble adjuvant | |
| HK40068589A (en) | Water soluble adjuvant and composition containing same | |
| WO2025263617A1 (ja) | コレステロールとtlr7アゴニストのコンジュゲート体およびそれを含む医薬組成物 | |
| HK40060502A (en) | Crystalline forms and salt forms of a kinase inhibitor | |
| HK40032856A (en) | Conjugate of wt1-derived peptides and composition comprising the same | |
| HK1224680A1 (en) | Novel four-ctl epitope-joined peptide | |
| HK1224680B (zh) | 新型的具有4个连接的ctl表位的肽 | |
| HK1250213B (en) | Combination of wt1 antigen peptide, immunomodulator and wt1 helper peptide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20784003 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2021512327 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 3136182 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020784003 Country of ref document: EP Effective date: 20211105 |
|
| ENP | Entry into the national phase |
Ref document number: 2020251582 Country of ref document: AU Date of ref document: 20200403 Kind code of ref document: A |
|
| WWG | Wipo information: grant in national office |
Ref document number: 17600919 Country of ref document: US |