WO2020210481A1 - Heterocyclic compounds as kinase inhibitors for therapeutic uses - Google Patents
Heterocyclic compounds as kinase inhibitors for therapeutic uses Download PDFInfo
- Publication number
- WO2020210481A1 WO2020210481A1 PCT/US2020/027453 US2020027453W WO2020210481A1 WO 2020210481 A1 WO2020210481 A1 WO 2020210481A1 US 2020027453 W US2020027453 W US 2020027453W WO 2020210481 A1 WO2020210481 A1 WO 2020210481A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cancer
- inhibitor
- alkyl
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- XDTMQSROBMDMFD-UHFFFAOYSA-N C1CCCCC1 Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- ORYMWWSJZRCUSV-UHFFFAOYSA-N CN(C)c(cc1)cc2c1c(Oc(cc1)ccc1NC(NCc1cnc(C(F)(F)F)cc1)=O)ncn2 Chemical compound CN(C)c(cc1)cc2c1c(Oc(cc1)ccc1NC(NCc1cnc(C(F)(F)F)cc1)=O)ncn2 ORYMWWSJZRCUSV-UHFFFAOYSA-N 0.000 description 1
- FFRKHRBQLZXWRV-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)cc(OC)c3NC(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)cc(OC)c3NC(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 FFRKHRBQLZXWRV-UHFFFAOYSA-N 0.000 description 1
- QPZIKJXOWYCEHE-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)cc(cc4)c3[n]4C(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)cc(cc4)c3[n]4C(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 QPZIKJXOWYCEHE-UHFFFAOYSA-N 0.000 description 1
- ASMLTJAJYAJQHR-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(NCc(cc3)ccc3OC)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(NCc(cc3)ccc3OC)=O)c2cc1 ASMLTJAJYAJQHR-UHFFFAOYSA-N 0.000 description 1
- BQRHGQMIXCBRQP-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(NCc(cc3)cnc3OC)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(NCc(cc3)cnc3OC)=O)c2cc1 BQRHGQMIXCBRQP-UHFFFAOYSA-N 0.000 description 1
- PNYUBWHIXNWCPO-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3Cl)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3Cl)=O)c2cc1 PNYUBWHIXNWCPO-UHFFFAOYSA-N 0.000 description 1
- RTWJZWFAMFOLDK-UHFFFAOYSA-N CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3OC)=O)c2cc1 Chemical compound CN(C)c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3OC)=O)c2cc1 RTWJZWFAMFOLDK-UHFFFAOYSA-N 0.000 description 1
- SPJRXTUKIPXWSO-UHFFFAOYSA-N CN(CC1)CCN1c(cc1)cc2c1c(Oc(cc1)ccc1NC(NCc1cnc(C(F)(F)F)cc1)=O)ncn2 Chemical compound CN(CC1)CCN1c(cc1)cc2c1c(Oc(cc1)ccc1NC(NCc1cnc(C(F)(F)F)cc1)=O)ncn2 SPJRXTUKIPXWSO-UHFFFAOYSA-N 0.000 description 1
- JQKCOTLZRULGMD-UHFFFAOYSA-N COC(CC1)CCN1c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3Cl)=O)c2cc1 Chemical compound COC(CC1)CCN1c1cc2ncnc(Oc(cc3)ccc3NC(Nc(cc3)ccc3Cl)=O)c2cc1 JQKCOTLZRULGMD-UHFFFAOYSA-N 0.000 description 1
- AHGLDKTYBGYJKP-UHFFFAOYSA-N COC(CC1)CCN1c1cc2ncnc(Oc(cc3)ccc3NC(Nc3ccccc3)=O)c2cc1 Chemical compound COC(CC1)CCN1c1cc2ncnc(Oc(cc3)ccc3NC(Nc3ccccc3)=O)c2cc1 AHGLDKTYBGYJKP-UHFFFAOYSA-N 0.000 description 1
- JDHLFXVNARQERV-HDICACEKSA-N C[C@H](C1)N[C@@H](C)CN1c1cc2ncnc(Oc(cc3)ccc3NC(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 Chemical compound C[C@H](C1)N[C@@H](C)CN1c1cc2ncnc(Oc(cc3)ccc3NC(NCc3cnc(C(F)(F)F)cc3)=O)c2cc1 JDHLFXVNARQERV-HDICACEKSA-N 0.000 description 1
- 0 Cc1c(*=I)nc(C)[o]1 Chemical compound Cc1c(*=I)nc(C)[o]1 0.000 description 1
- CVAQBRNPLXZEND-UHFFFAOYSA-N Cc1ncc(CNC(Nc(cc2)ccc2Oc2c(ccc(N(C)C)c3)c3ncn2)=O)cc1 Chemical compound Cc1ncc(CNC(Nc(cc2)ccc2Oc2c(ccc(N(C)C)c3)c3ncn2)=O)cc1 CVAQBRNPLXZEND-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/255—Esters, e.g. nitroglycerine, selenocyanates of sulfoxy acids or sulfur analogues thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/88—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Definitions
- Colony-stimulating factor-1 receptor (CSF1R) is a member of tyrosine kinase class III. It plays an important role in cell proliferation, differentiation, migration, and survival. See
- CSF1R is related to differentiation of tumor-associated macrophages (TAMs).
- TAMs express, on their surfaces, CSF1R, which forms a signaling axis with an active ligand, i.e., colony stimulating factor-1 (CSF1).
- CSF1R/CSF1 signaling axis promotes proliferation of monocytes, differentiation of the monocytes into TAMs, and survival of the TAMs.
- TAMs modify tumor microenvironment to render it more conducive to cancer cell growth, angiogenesis, and metastasis. Further, they can cause localized immunosuppression in tumor tissues, resulting in resistance to cancer therapy. As such, inhibiting the CSF1R/CSF1 signaling axis presents an attractive avenue for treating cancers associated with overexpression of CSF1.
- the present invention is based on unexpected discoveries that certain heterocyclic compounds effectively inhibit colony-stimulating factor-1 receptor (CSF1R).
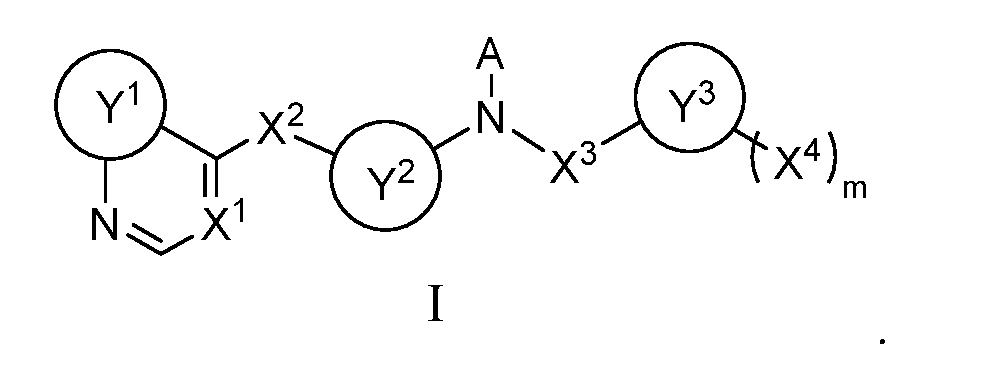
- this invention relates to these heterocyclic compounds and other heterocyclic compounds analogous thereto covered by formula I:
- R 1 in (R 1 )n, n being 0-4, is, independently, F, Cl, Br, NO 2 , CN, amino, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 8 cycloalkyl, C 5 -C 15 heterocycloalkyl, aryl, heteroaryl, carbonyl, thionyl, iminyl, or spiroamino; and R 2 in (R 2 )o, o
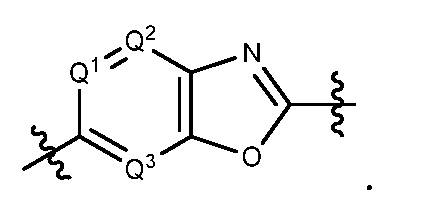
- each of Q 1 , Q 2 , Q 3 , Q 4 , Q 5 , Q 6 , Q 7 , and Q 8 is, independently, N or CR 4 , R 4 being H, F, Cl, Br, CN, amino, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, or C 1 -C 6 alkoxyl;
- Z 1 is O, S, or NRr;
- Z 2 is O, S, or NRr;
- G and H are, respectively, C or N and N or C;
- X 3 is deleted, CH 2 , ( CH 2 ) 2 , or
- Y 3 is C 1 -C 6 alkyl, aryl, heteroaryl, C 3 -C 8 cycloalkyl, or C 5 -C 6 heterocycloalkyl having one heteroatom, in which the one heteroatom is O or N; and
- alkyl refers to a straight or branched monovalent hydrocarbon moiety containing 1-20 carbon atoms, e.g., methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, and t-butyl.
- haloalkyl refers to an alkyl group substituted with one or more halogen atoms.
- alkenyl refers to a straight or branched monovalent or bivalent hydrocarbon containing 2- 20 carbon atoms and one or more double bonds, e.g., ethenyl, propenyl, propenylene, allyl, and 1,4-butadienyl.
- alkynyl refers to a straight or branched monovalent or bivalent hydrocarbon containing 2-20 carbon atoms and one or more triple bonds, e.g., ethynyl, ethynylene, 1-propynyl, 1- and 2-butynyl, and 1 -methyl-2-butynyl.
- aryl refers to a monovalent 6-carbon monocyclic, 10-carbon bicyclic, 14-carbon tricyclic aromatic ring system, e.g., phenyl, naphthyl, and anthracenyl.
- heteroaryl refers to a monovalent aromatic 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic ring system having one or more heteroatoms (such as O, N, S, or Se), e.g., imidazolyl, pyrazoyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyridyl, pyrazinyl, pyrimidyl, pyridazinyl, furyl, and thienyl.
- heteroatoms such as O, N, S, or Se
- cycloalkyl refers to a monovalent or bivalent saturated hydrocarbon ring system having 3 to 30 carbon atoms (e.g., C3-C12), e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, 1,4-cyclohexylene, cycloheptyl, and cyclooctyl.
- C3-C12 carbon atoms
- cycloalkenyl refers to a monovalent or bivalent non-aromatic hydrocarbon ring system having 3 to 30 carbons (e.g., C3-C12) and one or more double bonds, e.g., cyclopentenyl, cyclohexenyl, and cycloheptenyl.
- heterocycloalkyl refers to a monovalent or bivalent nonaromatic 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic ring system having one or more heteroatoms (such as O, N, S, or Se), e.g., piperidinyl, piperazinyl, pyrrolidinyl, dioxanyl, morpholinyl, tetrahydrofuranyl, and tetrahydropyranyl.
- heteroatoms such as O, N, S, or Se
- heterocycloalkenyl refers to a monovalent or bivalent nonaromatic 5-8 membered monocyclic, 8-12 membered bicyclic, or 11-14 membered tricyclic ring system having one or more heteroatoms (such as O, N, S, or Se) and one or more double bonds.
- amino refers to a -NRR' moiety, in which R and R' are, independently, H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, aryl, heteroaryl, aralkyl, or heteroarakyl.
- carbonyl refers to a -C(0)R moiety, in which R is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, alkoxyl, amino, aryl, or heteroaryl.
- thionyl refers to a -S(0)R moiety, in which R is H, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, alkoxyl, amino, aryl, or heteroaryl.
- aminospiroamino refers to a monovalent 7-11 membered bicyclic spiro moiety containing one N or a monovalent 10-16 membered tricyclic spiro moiety containing one N.
- alkyl, alkenyl, alkynyl, aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl, and alkoxyl can be substituted or unsubstituted.
- the compounds of formula I include the compounds themselves, as well as their salts, their stereoisomers, their solvates, their tautomers, their deuterated analogues, and their prodrugs, if applicable.
- a salt for example, can be formed between an anion and a positively charged group (e.g., ammonium) on a heterocyclic compound of this invention.
- Suitable anions include chloride, bromide, iodide, sulfate, bisulfate, sulfamate, nitrate, phosphate, citrate,
- a salt can also be formed between a cation and a negatively charged group (e.g., carboxylate) on a heterocyclic compound. Suitable cations include sodium ion, potassium ion, magnesium ion, calcium ion, and an ammonium cation such as tetramethylammonium ion.
- the salts of the heterocyclic compounds of this invention can also contain quaternary nitrogen atoms.
- prodrugs include esters and other pharmaceutically acceptable derivatives, which, upon administration to a subject, are capable of providing active heterocyclic compounds.
- a solvate refers to a complex formed between an active heterocyclic compound and a pharmaceutically acceptable solvent, e.g., water, ethanol, isopropanol, ethyl acetate, acetic acid, and ethanolamine.
- An additional aspect of this invention relates to a pharmaceutical composition containing one or more of the heterocyclic compounds covered by formula I.
- the pharmaceutical composition can be used for treatment of a CSF1R modulated condition.
- a method of treating a CSF1R modulated condition e.g., a cancer, an inflammatory disorder, a bone disorder, or an autoimmune disease.
- the method includes administering to a subject in need thereof an effective amount of one or more of the above-described heterocyclic compounds.
- treatment refers to administering one or more heterocyclic compounds of this invention to a subject who has a CSF1R modulated condition, a symptom of such a condition, or a predisposition toward it, with the purpose of conferring a therapeutic or prophylactic effect.
- An effective amount refers to the amount of an active compound that is required to confer such effect. Effective doses will vary, as recognized by those skilled in the art, depending on the types of disease treated, route of administration, excipient usage, and the possibility of co-usage with other therapeutic treatment.
- a pharmaceutical composition of this invention can be administered parenterally, orally, nasally, rec tally, topically, or buccally.
- parenteral refers to subcutaneous, intracutaneous, intravenous, intraperitoneal, intramuscular, intraarticular, intraarterial, intrasynovial, intrastemal, intrathecal, intralesional, or intracranial injection, as well as any suitable infusion technique.
- a sterile injectable composition can be a solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,3-butanediol.
- a non-toxic parenterally acceptable diluent or solvent such as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that can be employed are mannitol, water, Ringer’s solution, and isotonic sodium chloride solution.
- fixed oils are conventionally employed as a solvent or suspending medium (e.g., synthetic mono- or di-glycerides).
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in preparation of injectables, as are natural pharmaceutically acceptable oils, such as olive oil and castor oil, especially in their
- oil solutions or suspensions can also contain a long chain alcohol diluent or dispersant, carboxymethyl cellulose, or similar dispersing agents.
- Other commonly used surfactants such as Tweens and Spans or other similar emulsifying agents or bioavailability enhancers which are commonly used in the manufacture of pharmaceutically acceptable solid, liquid, or other dosage forms can also be used for the purpose of formulation.
- a composition for oral administration can be any orally acceptable dosage form including capsules, tablets, emulsions and aqueous suspensions, dispersions, and solutions.
- commonly used carriers include lactose and com starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried com starch.
- a nasal composition can be prepared according to techniques well known in the art of pharmaceutical formulation.
- such a composition can be prepared as a solution in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- a pharmaceutical composition of this invention can also be administered in the form of a suppository for rectal administration.
- the carrier in a pharmaceutical composition must be“acceptable” in the sense that it is compatible with the active ingredient of the composition and, preferably, capable of stabilizing the active ingredient, and not deleterious to the subject to be treated.
- One or more solubilizing agents can be utilized as pharmaceutical excipients for delivery of an active heterocyclic compound of this invention.
- examples of other carriers include colloidal silicon oxide, magnesium stearate, cellulose, sodium lauryl sulfate, and D&C Yellow #10.
- A, Y 1 , X 1 , X 2 , Y 2 , X 3 , Y 3 , X 4 , and m are defined in the SUMMARY section above.
- the compounds of formula I have Y 1 being phenyl substituted with (R1 )n, 5-membered heteroaryl substituted with (R 2 ) 0 , or 5-membered heterocycloalkenyl substituted with (R 2 )o, in which R 1 in (R 1 )n is, independently, F, Cl, Br, NO 2 , CN, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, C 2 -C 6 alkynyl, C 3 -C 8 cycloalkyl, C 5 -C 15 heterocycloalkyl, aryl, heteroaryl, carbonyl, thionyl, iminyl, or spiroamino; and R 2 in (R 2 ) 0 is, independently, F, Cl, Br, NO 2 , CN, amino, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, C 2 -C 6 alkynyl,
- the compounds of formula I have Y 2 being
- Z 2 is O or NRr.
- the compounds of formula I have Y 2 being Y 3 being pyridyl, and R 1 being amino.
- R 1 is amino or C 5 -C 15 heterocycloalkyl.
- the compounds of formula la have Y 2 being Y 3 being pyridyl; X 3 being CH 2 ; X 4 being CH 3 , CH 2 F, CHF 2 , CF 3 , or OCH 3 ; m being 1; and R 1 preferably being amino.
- the compounds have Y 2 being Y 3 being phenyl; X 3 being CH 2 ; each of X 4 being, independently, F, Cl, Br, CN, SO 2 NH 2 , CH 3 , CH 2 F, CHF 2 , CF 3 , OCF 3 , C 1 -C 6 alkoxyl, or amino; and m being 0-2.
- the compounds have Y 2 being Y 3 being phenyl; X 3 being deleted; each of X 4 being, independently, F, Cl, Br, CN, SO 2 NH 2 , CH 3 , CH 2 F, CHF 2 , CF 3 , OCF 3 , C 1 -C 6 alkoxyl, or amino; and m being 0-2.
- Formula la includes compounds in which Y 2 is Y 3
- Y 2 is
- Formula la further includes compounds in which Y 2 is
- Y 3 is phenyl or pyridyl, and X 3 is CH 2 .
- Y 2 is
- heterocyclic compounds of this invention are covered by formula lb:
- a subset of compounds of formula lb have Y 1 being Y 3 being phenyl or
- Y 3 being phenyl or pyridyl; and X 3 being deleted or CH 2 , in which Q 9 is N or CR 5 , R 5 being H, F, Cl, Br, CN, amino, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, or C 1 -C 6 alkoxyl.
- the compounds of formula lb have each of X 4 being, independently, CH 3 , CH 2 F, CHF 2 , CF 3 , or OCH 3 and m being 0-2.
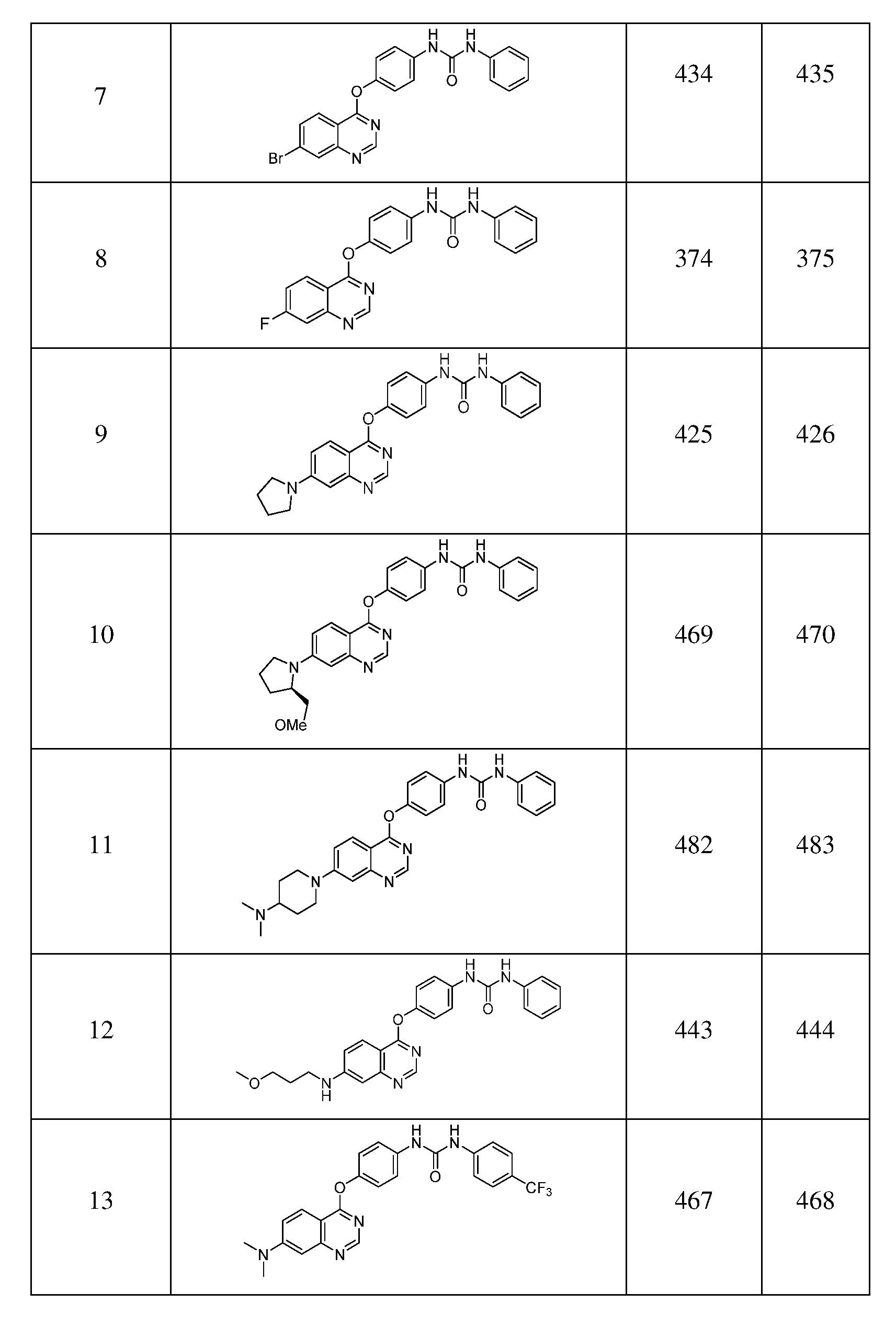
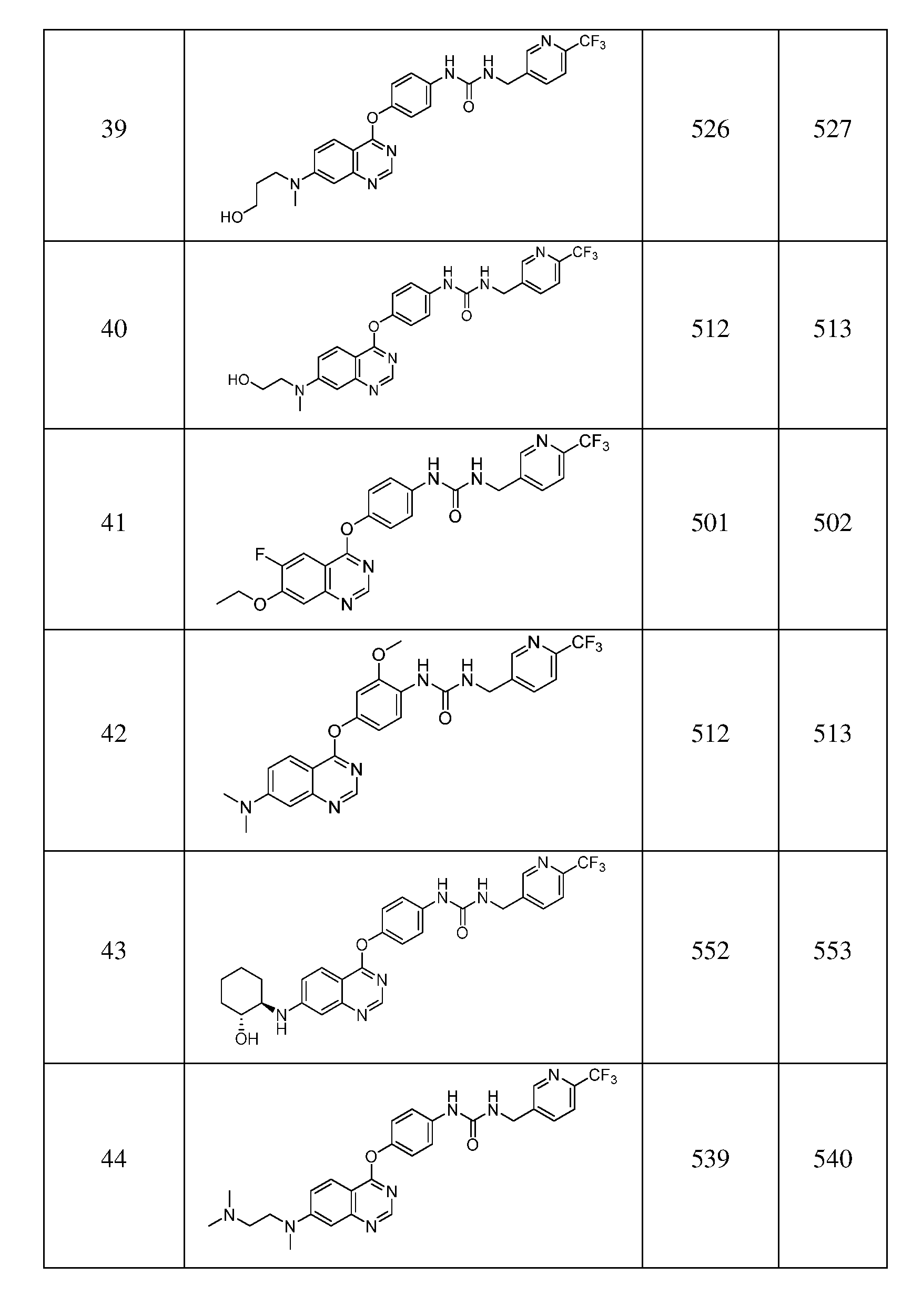
- Exemplary compounds of formula I include, but are not limited to, the following compounds:
- the compounds of formula I can be prepared according to methods well known in the field. See, for example, R. Larock, Comprehensive Organic Transformations (2nd Ed., VCH Publishers 1999); P. G. M. Wuts and T. W. Greene, Greene’s Protective Groups in Organic Synthesis (4th Ed., John Wiley and Sons 2007); L. Fieser and M. Fieser, Fieser and Fieser’s Reagents for Organic Synthesis (John Wiley and Sons 1994); L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis (2nd ed., John Wiley and Sons 2009); and G. J. Yu et al., J. Med. Chem. 2008, 51, 6044-6054.
- compositions containing one or more of the heterocyclic compounds of formula I are also within the scope of this invention.
- the pharmaceutical composition is used for treating a CSF1R modulated condition.
- the pharmaceutical composition further contains one of the following therapeutic agents: an anti-proliferative agent, an anti-inflammatory agent, an immunomodulatory agent, and an immunosuppressive agent.
- the pharmaceutical composition further contains one of the following therapeutic agents: an alkylating agent, e.g., adozelesin, altretamine, bizelesin, busulfan, carboplatin, carboquone, carmustine, chlorambucil, cisplatin, cyclophosphamide, dacarbazine, estramustine, fotemustine, hepsulfam, ifosfamide, improsulfan, irofulven, lomustine, mechlorethamine, melphalan, oxaliplatin, piposulfan, semustine, streptozocin, temozolomide, thiotepa, and treosulfan; an antibody, e.g., alemtuzumab, bevacizumab, cetuximab, galiximab, gemtuzumab, nivolumab, panitumumab, pembrolizum
- the condition can be a cancer, e.g., acute myeloid leukemia, bladder cancer, breast cancer, cervical cancer, colon cancer, gastric cancer, gastrointestinal stromal tumor, glioblastoma multiforme, hepatocellular carcinoma, Hodgkin’s lymphoma, kidney cancer, liver cancer, lung cancer, melanoma, metastatic tumor, ovarian cancer, pancreatic cancer, pigmented villondular synovitis, prostate cancer, tenosynovial giant cell tumors, endometrial cancer, multiple myeloma, myelocytic leukemia, bone cancer, renal cancer, brain cancer, myeloproliferative disorder, esophageal cancer, squamous cell carcinoma, uveal melanoma, follicular lymphoma, colorectal cancer, head
- a cancer e.g., acute myeloid leukemia, bladder cancer, breast cancer, cervical cancer, colon cancer, gastric cancer, gastrointestinal strom
- Table 1 Exemplary compounds of this invention, shown in Table 1 below, were prepared by procedures shown in Scheme 1, Scheme 2, Scheme 3, or Scheme 4.
- Table 1 includes mass spectral data for the compounds.
- LCMS Liquid chromatography mass spectrometry
- SOCk is thionylchloride
- DMF is dimethylformamide
- PhMe is toluene
- tBuOK is potassium tert-butoxide
- THF is tetrahydrofuran
- DMSO is dimethylsulfoxide
- CH 2 CI 2 is methylene chloride.
- Scheme 3 Among the listed reagents, solvents and catalysts in Scheme 3, SOCl 2 is thionylchloride, RuPhos Pd G3 is (2-dicyclohexylphosphino-2',6'-diisopropoxy-1, 1' -biphenyl) [2-2 , -amino-l,V- biphenyl]] palladium(II) methanesulfonate, RuPhos is 2-dicyclohexylphosphino-2',6'- diisopropoxybiphenyl, and Cs 2 CO 3 is cesium carbonate.
- POCl 3 is phosphoryl chloride
- Cul is copper iodide
- 1,10-phen is 1,10-phenanthroline
- K2CO3 is potassium carbonate
- fBuOH is tert-butanol.
- CSF1R Kinase-Glo assay Activity of CSF1R kinase was determined using a CSF1R Kinase-Glo assay.
- Recombinant N-terminal GST-CSF1R (CSF1R residues L534-C972) containing the CSFIR kinase domain was expressed in Sf9 insect cells and purified.
- the kinase assay was carried out in 96-well plates at 30 °C for 180 min with the tested compounds in a final volume of 50 ml including the following components: 25 mM Tris-HCl pH 7.4, 4 mM MnCl 2 , 10 mM MgCl 2 , 0.01% BSA, 0.5 mM Na 3 VO 4 , 0.02% Triton X-100, 40mM ATP, 2 mM DTT and 20mM poly(Glu.Tyr) 4:1 peptide, and 600 ng recombinant GST-CSF1R.
- the cell lines M-NFS-60 (ATOC ® CRL-1838TM) and BaF3-CSFlR-1600 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA).
- the stable BaF3- CSF1R-1600 cell line expresses ETV6-CSF1R fusion protein consisting of N-terminal ETS- variant gene 6 protein (ETV6 residues M1-G337) and CSF1R tyrosine kinase (CSF1R residues L533-C972).
- the M-NSF-60 and BaF3-CSFlR-1600 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 0.05 mM 2-ME, 10 U/ml penicillin, and 10 g/ml streptomycin at 37 °C and 5% CO 2 .
- M-NFS-60 and BaF3-CSFlR-1600 cells were seeded in 96-well plates at a density of 10000 cells/100 ml and 8000 cells/100 ml per well, respectively, for 16 h and treated with vehicle or various concentrations of test compounds in medium for 72 h.
- Viable cells were quantified using the MTS method (Promega, Madison, WI, USA) according to manufacturer’s recommended protocol. The results were determined by measuring absorbance at 490 nm using a plate reader (Victor 2).
- the GI50 value was defined as the amount of compound that caused 50% reduction in cell viability in comparison with DMSO-treated (vehicle) control and was calculated using Prism GraphPad Prism version 6 software (GraphPad).
- CSF1R kinase A study was conducted to determine kinase selectivity of compounds 27 and 67. More specifically, each compound was tested for inhibitory activity of CSF1R kinase, as compared to that of seven other kinases, i.e., Aurora A, Aurora B, tyrosine-protein kinase Kit (c-Kit), fins-like tyrosine kinase 3 (FLT3), platelet-derived growth factor receptor (PDGFR) A, PDGFR B, and discoidin domain receptor tyrosine kinase 1 (DDR1). Results from this study are shown in Table 3 below.
- c-Kit tyrosine-protein kinase Kit
- FLT3 fins-like tyrosine kinase 3
- PDGFR platelet-derived growth factor receptor
- DDR1 discoidin domain receptor tyrosine kinase 1
- kinase activity was determined as the percentage of the remaining kinase activity in a test sample compared to a vehicle (DMSO) reaction.
- IC50 value and a dose-response curve for each compound against each kinase were obtained using Prism (Graph Pad Software). Selectivity, expressed as an IC50 ratio, was determined by dividing the IC50 value of a kinase, e.g., Aurora A, by that of CSF1R.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Physical Education & Sports Medicine (AREA)
- Immunology (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
Abstract
Description











































Claims






















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20787959.4A EP3952865A4 (en) | 2019-04-12 | 2020-04-09 | HETEROCYCLIC COMPOUNDS AS KINASE INHIBITORS FOR THERAPEUTIC USES |
| CN202080024676.8A CN114025755B (en) | 2019-04-12 | 2020-04-09 | Therapeutic use of heterocyclic compounds as kinase inhibitors |
| US17/603,273 US20220213064A1 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic compounds as kinase inhibitors for therapeutic uses |
| JP2021560707A JP7568641B2 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic Compounds as Kinase Inhibitors for Therapeutic Use - Patent application |
| KR1020217032764A KR102913995B1 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic compounds as kinase inhibitors for therapeutic use |
| AU2020271855A AU2020271855B2 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic compounds as kinase inhibitors for therapeutic uses |
| CA3132855A CA3132855A1 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic compounds as kinase inhibitors for therapeutic uses |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962833364P | 2019-04-12 | 2019-04-12 | |
| US62/833,364 | 2019-04-12 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2020210481A1 true WO2020210481A1 (en) | 2020-10-15 |
| WO2020210481A8 WO2020210481A8 (en) | 2020-11-19 |
Family
ID=72751520
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2020/027453 Ceased WO2020210481A1 (en) | 2019-04-12 | 2020-04-09 | Heterocyclic compounds as kinase inhibitors for therapeutic uses |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20220213064A1 (en) |
| EP (1) | EP3952865A4 (en) |
| JP (1) | JP7568641B2 (en) |
| KR (1) | KR102913995B1 (en) |
| CN (1) | CN114025755B (en) |
| AU (1) | AU2020271855B2 (en) |
| CA (1) | CA3132855A1 (en) |
| TW (1) | TWI757722B (en) |
| WO (1) | WO2020210481A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114031561A (en) * | 2021-12-10 | 2022-02-11 | 江西科技师范大学 | 4-phenoxy-containing quinazoline compound and application thereof |
| US11691963B2 (en) | 2020-05-06 | 2023-07-04 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US11970494B2 (en) | 2021-11-09 | 2024-04-30 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12043632B2 (en) | 2020-12-23 | 2024-07-23 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12162881B2 (en) | 2021-11-09 | 2024-12-10 | Ajax Therapeutics, Inc. | Forms and compositions of inhibitors of JAK2 |
| JP2025507088A (en) * | 2022-03-08 | 2025-03-13 | 安徽中科拓苒▲薬▼物科学研究有限公司 | New uses for quinoline compounds |
| US12415816B2 (en) | 2018-11-07 | 2025-09-16 | Dana-Farber Cancer Institute, Inc. | Benzothiazole derivatives and 7-aza-benzothiazole derivatives as janus kinase 2 inhibitors and uses thereof |
| US12509455B2 (en) | 2018-11-07 | 2025-12-30 | Dana-Farber Cancer Institute, Inc. | Imidazopyridine derivatives and aza-imidazopyridine derivatives as Janus kinase 2 inhibitors and uses thereof |
| US12522583B2 (en) | 2018-11-07 | 2026-01-13 | Dana-Farber Cancer Institute, Inc. | Benzimidazole derivatives and aza-benzimidazole derivatives as Janus kinase 2 inhibitors and uses thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102910577B1 (en) * | 2022-02-17 | 2026-01-08 | 내셔날 헬스 리서치 인스티튜트 | The Method of the Preparation of Fused Multicyclic Compounds |
| TW202448857A (en) * | 2023-05-12 | 2024-12-16 | 大陸商正大天晴藥業集團股份有限公司 | Compounds containing aromatic dicyclic rings |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120053192A1 (en) * | 2009-05-05 | 2012-03-01 | Nanjing Luyesike Pharmaceutical Co., Ltd. | Heterocyclic Substituted Acardite Derivate and Application Thereof |
| WO2017161045A1 (en) | 2016-03-16 | 2017-09-21 | Plexxikon Inc. | Compounds and methods for kinase modulation and indications therefore |
| US20170355678A1 (en) * | 2003-09-26 | 2017-12-14 | Exelixis, Inc. | C-Met Modulators and Methods of Use |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100522934C (en) * | 1999-01-13 | 2009-08-05 | 拜尔有限公司 | Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| ATE538794T1 (en) * | 1999-01-13 | 2012-01-15 | Bayer Healthcare Llc | GAMMA CARBOXYARYL SUBSTITUTED DIPHENYL UREA COMPOUNDS AS P38 KINASE INHIBITORS |
| DE60137273D1 (en) * | 2000-10-20 | 2009-02-12 | Eisai R&D Man Co Ltd | Process for the preparation of 4-phenoxyquinoline derivatives |
| EA009994B1 (en) * | 2003-12-23 | 2008-06-30 | Пфайзер Инк. | Novel quinoline derivatives |
| GB0509224D0 (en) * | 2005-05-05 | 2005-06-15 | Chroma Therapeutics Ltd | Inhibitors of intracellular enzymatic activity |
| PE20080359A1 (en) * | 2006-04-19 | 2008-06-06 | Novartis Ag | BENZOXAZOLE AND BENZOTHIAZOLE 6-0-SUBSTITUTE COMPOUNDS AND METHODS OF INHIBITION OF CSF-1R SIGNALING |
| ES2452349T3 (en) * | 2007-05-21 | 2014-04-01 | Novartis Ag | CSF-1R inhibitors, compositions, and methods of use |
| CN101372475B (en) * | 2008-03-19 | 2012-01-04 | 南京工业大学 | Aromatic heterocyclic substituted diphenyl urea derivative and application thereof |
| JO3265B1 (en) * | 2008-12-09 | 2018-09-16 | Novartis Ag | Pyridyloxyindoles Inhibitors of VEGF-R2 and Use Thereof for Treatment of Disease |
| KR101106050B1 (en) * | 2009-03-25 | 2012-01-18 | 한국과학기술연구원 | Aminoquinoline compound, preparation method thereof and pharmaceutical composition containing same |
| US9358235B2 (en) * | 2012-03-19 | 2016-06-07 | Plexxikon Inc. | Kinase modulation, and indications therefor |
| CN104177346A (en) * | 2013-05-21 | 2014-12-03 | 苏州科捷生物医药有限公司 | Quinazoline compound and use thereof |
| WO2016161952A1 (en) * | 2015-04-07 | 2016-10-13 | 广东众生药业股份有限公司 | Tyrosine kinase inhibitor and pharmaceutical composition comprising same |
| CN108137585B (en) * | 2015-09-21 | 2021-10-22 | 普莱希科公司 | Heterocyclic compounds and their applications |
-
2020
- 2020-04-09 AU AU2020271855A patent/AU2020271855B2/en active Active
- 2020-04-09 CN CN202080024676.8A patent/CN114025755B/en active Active
- 2020-04-09 EP EP20787959.4A patent/EP3952865A4/en active Pending
- 2020-04-09 KR KR1020217032764A patent/KR102913995B1/en active Active
- 2020-04-09 JP JP2021560707A patent/JP7568641B2/en active Active
- 2020-04-09 CA CA3132855A patent/CA3132855A1/en active Pending
- 2020-04-09 US US17/603,273 patent/US20220213064A1/en active Pending
- 2020-04-09 WO PCT/US2020/027453 patent/WO2020210481A1/en not_active Ceased
- 2020-04-10 TW TW109112137A patent/TWI757722B/en active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170355678A1 (en) * | 2003-09-26 | 2017-12-14 | Exelixis, Inc. | C-Met Modulators and Methods of Use |
| US20120053192A1 (en) * | 2009-05-05 | 2012-03-01 | Nanjing Luyesike Pharmaceutical Co., Ltd. | Heterocyclic Substituted Acardite Derivate and Application Thereof |
| WO2017161045A1 (en) | 2016-03-16 | 2017-09-21 | Plexxikon Inc. | Compounds and methods for kinase modulation and indications therefore |
Non-Patent Citations (11)
| Title |
|---|
| "Encyclopedia of Reagents for Organic Synthesis", 2009, JOHN WILEY AND SONS |
| CANNARILE ET AL., J. IMMUNOTHER. CANCER, vol. 5, 2017, pages 53 |
| DATABASE PubChem compound 19 July 2005 (2005-07-19), "2-[2-({6-[(7-Chloro-4-quinazolinyl)oxy]-3-pyridinyl}amino)-2-oxoethoxy]acetic acid", XP055749486, retrieved from NCBI Database accession no. 2814130 * |
| DATABASE PubChem compound 30 November 2012 (2012-11-30), "[5-Chloro-6-(6,7-dimethoxyquinolin-4-yl)oxypyridin-3-yl] 2-amino-2-oxoacetate", XP055749483, retrieved from NCBI Database accession no. 67379395 * |
| EL-GAMAL ET AL., J. MED. CHEM., vol. 61, 2018, pages 5450 - 5466 |
| G. J. YU ET AL., J. MED. CHEM., vol. 51, 2008, pages 6044 - 6054 |
| L. FIESERM. FIESER: "Fieser and Fieser's Reagents for Organic Synthesis", 1994, JOHN WILEY AND SONS |
| MERCK KIESELGEL, vol. 60, no. 9385, pages 230 - 400 |
| P. G. M. WUTST. W. GREENE: "Greene's Protective Groups in Organic Synthesis", 2007, JOHN WILEY AND SONS |
| R. LAROCK: "Comprehensive Organic Transformations", 1999, VCH |
| See also references of EP3952865A4 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12415816B2 (en) | 2018-11-07 | 2025-09-16 | Dana-Farber Cancer Institute, Inc. | Benzothiazole derivatives and 7-aza-benzothiazole derivatives as janus kinase 2 inhibitors and uses thereof |
| US12509455B2 (en) | 2018-11-07 | 2025-12-30 | Dana-Farber Cancer Institute, Inc. | Imidazopyridine derivatives and aza-imidazopyridine derivatives as Janus kinase 2 inhibitors and uses thereof |
| US12522583B2 (en) | 2018-11-07 | 2026-01-13 | Dana-Farber Cancer Institute, Inc. | Benzimidazole derivatives and aza-benzimidazole derivatives as Janus kinase 2 inhibitors and uses thereof |
| US11691963B2 (en) | 2020-05-06 | 2023-07-04 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12275717B2 (en) | 2020-05-06 | 2025-04-15 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12043632B2 (en) | 2020-12-23 | 2024-07-23 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US11970494B2 (en) | 2021-11-09 | 2024-04-30 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12162881B2 (en) | 2021-11-09 | 2024-12-10 | Ajax Therapeutics, Inc. | Forms and compositions of inhibitors of JAK2 |
| CN114031561A (en) * | 2021-12-10 | 2022-02-11 | 江西科技师范大学 | 4-phenoxy-containing quinazoline compound and application thereof |
| JP2025507088A (en) * | 2022-03-08 | 2025-03-13 | 安徽中科拓苒▲薬▼物科学研究有限公司 | New uses for quinoline compounds |
| EP4491181A4 (en) * | 2022-03-08 | 2026-03-11 | Tarapeutics Science Inc | NEW USE OF A CHINOLINE COMPOUND |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3952865A1 (en) | 2022-02-16 |
| TWI757722B (en) | 2022-03-11 |
| AU2020271855B2 (en) | 2025-11-27 |
| CN114025755B (en) | 2024-11-29 |
| TW202104212A (en) | 2021-02-01 |
| WO2020210481A8 (en) | 2020-11-19 |
| EP3952865A4 (en) | 2023-05-03 |
| KR102913995B1 (en) | 2026-01-16 |
| AU2020271855A1 (en) | 2021-11-04 |
| KR20210151818A (en) | 2021-12-14 |
| JP7568641B2 (en) | 2024-10-16 |
| CA3132855A1 (en) | 2020-10-15 |
| JP2022528780A (en) | 2022-06-15 |
| US20220213064A1 (en) | 2022-07-07 |
| CN114025755A (en) | 2022-02-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020271855B2 (en) | Heterocyclic compounds as kinase inhibitors for therapeutic uses | |
| CN104125957B (en) | Substituted benzylpyrazoles | |
| US9102631B2 (en) | 1-(arylmethyl)-5,6,7,8-tetrahydroquinazoline-2,4-diones and analogs and the use thereof | |
| KR102388312B1 (en) | Aminopyrimidine compound, preparation method and use thereof | |
| EP3184521A1 (en) | Indazole compounds as fgfr kinase inhibitor, preparation and use thereof | |
| JP2019518059A (en) | Azabenzimidazole derivatives as PI3K beta inhibitors | |
| JP2011510010A (en) | 3H- [1,2,3] triazolo [4,5-d] pyrimidine compounds, their use as mTOR kinase and PI3 kinase inhibitors, and their synthesis | |
| CN103052627A (en) | Pyrimidine derivatives as fak inhibitors | |
| EP3459952B1 (en) | Pyrimidine derivative, method for preparing same and use thereof in medicine | |
| KR102372288B1 (en) | Aminothiazole compounds as protein kinase inhibitors | |
| AU2004283093A1 (en) | Compounds and compositions as protein kinase inhibitors | |
| CN116670134A (en) | Substituted imidazo[1,5-b]pyridazine compounds as kinase inhibitors and their use | |
| CN114269742B (en) | Derivatives of 4- (imidazo [1,2-a ] pyridin-3-yl) -N- (pyridinyl) pyrimidin-2-amine as therapeutic agents | |
| CN109280048A (en) | A kind of pyrimidine compound containing substituted phenylacrylamide structure and its application | |
| CN114502555A (en) | Derivatives of 4- (imidazo [1,2-a ] pyridin-3-yl) -N- (pyridin-3-yl) pyrimidin-2-amines for the treatment of proliferative diseases and disorders | |
| CN111499613B (en) | N-formamide derivative, its preparation method and its use in medicine | |
| WO2024061554A1 (en) | Pharmaceutical compound | |
| HK40061639A (en) | Heterocyclic compounds as kinase inhibitors for therapeutic uses | |
| HK40061639B (en) | Heterocyclic compounds as kinase inhibitors for therapeutic uses | |
| JP2024540004A (en) | Quinazoline derivative compounds and uses thereof | |
| WO2025215187A1 (en) | Therapeutic compounds and their use | |
| WO2025215188A1 (en) | Therapeutic compounds and their use as inhibitors of pkmyt1 | |
| WO2026057598A1 (en) | Therapeutic compounds as pkmyt1 inhibitors and their use | |
| HK40066516B (en) | Derivatives of 4-(imidazo[1,2-a]pyridin-3-yl)-n-(pyridinyl)pyrimidin-2-amine as therapeutic agents | |
| HK40066516A (en) | Derivatives of 4-(imidazo[1,2-a]pyridin-3-yl)-n-(pyridinyl)pyrimidin-2-amine as therapeutic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20787959 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3132855 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2021560707 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020271855 Country of ref document: AU Date of ref document: 20200409 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020787959 Country of ref document: EP Effective date: 20211112 |