WO2020227100A1 - Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes - Google Patents
Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes Download PDFInfo
- Publication number
- WO2020227100A1 WO2020227100A1 PCT/US2020/031044 US2020031044W WO2020227100A1 WO 2020227100 A1 WO2020227100 A1 WO 2020227100A1 US 2020031044 W US2020031044 W US 2020031044W WO 2020227100 A1 WO2020227100 A1 WO 2020227100A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polymerase chain
- chain reaction
- nucleic acid
- sequence
- oligonucleotide primer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/178—Oligonucleotides characterized by their use miRNA, siRNA or ncRNA
Definitions
- FIELD [0003] The present application relates to methods and markers for identifying and quantifying nucleic acid sequence, mutation, expression, splice variant, translocation, copy number, and/or methylation changes using combinations of bisulfite treatment, nuclease, ligation, and polymerase reactions with carryover prevention.
- BACKGROUND [0004] Cancer is the leading cause of death in developed countries and the second leading cause of death in developing countries. Cancer kills 580,000 patients annually in the US, 1.3 million in Europe, and 2.8 million in China (Siegel et al.,“Cancer Statistics, 2016,” CA Cancer J. Clin.66(1):7-30 (2016)).
- Cancer is now the biggest cause of mortality worldwide, with an estimated 8.2 million deaths from cancer in 2012 (Torre et al.,“Global Cancer Statistics, 2012,” CA Cancer J. Clin.65(2):87-108 (2015)). Cancer cases worldwide are forecast to rise by 75% and reach close to 25 million over the next two decades. The lifetime risk of a woman dying from an invasive cancer is 19%, for a man it is 23%. With total annual costs of cancer care in the U.S. exceeding $400 billion, there is no other medical issue that so urgently needs intelligent solutions.
- lung cancer (27%), prostate cancer (8%), colorectal cancer (8%), and lung cancer (26%), breast cancer (14%), colorectal cancer (8%), for men and women, respectively.
- These cancers are driven by different biological processes, and while there have been exciting advancements in the treatment of some cancers, such as the emergence of targeted therapeutics and immunotherapy, most cancers are found at later stage, where survival is poor. Due to lack of reliable and inexpensive early detection tests, many cancer types are diagnosed at later stages, where survival rates for some cancers drop to below 10%.
- Cancer cells may undergo apoptosis (triggered cell death), which releases cell free DNA (cfDNA) into the patients’ blood (Salvi et al.,“Cell-free DNA as a Diagnostic Marker for Cancer: Current Insights,” OncoTargets and Therapy 9:6549-6559 (2016)).
- cfDNA cell free DNA
- the levels of cfDNA in serum from patients with cancer vary from vanishingly small to high, but do not correlate with cancer stage (Perlin et al.,“Serum DNA Levels in Patients With Malignant Disease,” American Journal of Clinical Pathology 58(5):601-602 (1972); Leon et al.,“Free DNA in the Serum of Cancer Patients and the Effect of Therapy,” Cancer Res.37(3):646-650 (1977)).
- exosomes lipid vesicles ranging from 30 to 100 nm
- exosomes can contain the same RNA molecules which serve as transcriptional signatures of the tumors.
- Exosomes, or tumor associated vesicles shield mRNA, lncRNA, ncRNA, and even mutant tumor DNA from exogenous nucleases, and, as such, the markers are in a protected state.
- Other protected states include, but are not limited to, DNA, RNA, and proteins within circulating tumor cells (CTCs), within other non-cellular membrane containing vesicles or particles, within nucleosomes, or within Argonaute or other protein complexes.
- cfDNA in particular, contains the same molecular aberrations as the solid tumors, such as mutations hyper/hypo methylation, copy number changes, or chromosomal rearrangements (Ignatiadis et al.,“Circulating Tumor Cells and Circulating Tumor DNA for Precision Medicine: Dream or Reality?” Ann. Oncol.25(12):2304-2313 (2014)).
- Methylation signatures have better specificity towards a particular cancer type likely because methylation patterns are highly tissue specific (Issa JP,“DNA Methylation as a Therapeutic Target in Cancer,” Clin. Cancer Res.13(6):1634-1637 (2007)).
- methylation at promoter regions of tumor suppressor genes has been detected in patients’ cfDNAs (Tang et al.,“Blood-based DNA Methylation as Biomarker for Breast Cancer: a Systematic Review,” Clinical Epigenetics 8:115 (2016)).
- a caveat for using methylation markers is that bisulfite conversion tends to destroy DNA, and thus decreases the overall signal that can be detected. Methylation detection techniques may also lead to false-positive signals due to incomplete conversion of unmethylated cytosines.
- methylation marker detection assays enable a higher level of multiplexing with single-molecule detection capabilities, which are predicted to allow for higher sensitivity and specificity across a broad spectrum of cancers.
- the challenge to develop reliable diagnostic and screening tests is to distinguish those markers emanating from the tumor that are indicative of disease (e.g., early cancer) vs. presence of the same markers emanating from normal tissue (which would lead to a false- positive signal).
- TCGA Cancer Genome Atlas Consortium
- CRC mutation markers such as those of KRAS and BRAF are found in late-stage primary cancers and metastases (Spindler et al.,“Circulating free DNA as Biomarker and Source for Mutation Detection in Metastatic Colorectal Cancer,” PloS One 10(4):e0108247 (2015); Gonzalez-Cao et al.,“BRAF Mutation Analysis in Circulating Free Tumor DNA of Melanoma Patients Treated with BRAF Inhibitors,” Melanoma Res.25(6):486-495 (2015); Sakai et al., “Extended RAS and BRAF Mutation Analysis Using Next-Generation Sequencing,” PloS One 10(5):e0121891 (2015)).
- the nucleic acid assay should serve primarily as a screening tool, requiring the availability of secondary diagnostic follow-up (e.g., colonoscopy for colorectal cancer).
- Compounding the biological problem is the need to reliably quantify mutation, CpG methylation, or DNA or RNA copy number from either a very small number of initial cells (i.e. from CTCs), or when the cancer signal is from cell-free DNA (cfDNA) in the blood and diluted by an excess of nucleic acid arising from normal cells, or inadvertently released from normal blood cells during sample processing (Mateo et al.,“The Promise of Circulating Tumor Cell Analysis in Cancer Management,” Genome Biol.15:448 (2014); Haque et al.,“Challenges in Using ctDNA to Achieve Early Detection of Cancer,” BioRxiv.237578 (2017)).
- DNA sequencing provides the ultimate ability to distinguish all nucleic acid changes associated with disease. However, the process still requires multiple up-front sample and template preparation, and consequently, DNA sequencing is not always cost-effective.
- DNA microarrays can provide substantial information about multiple sequence variants, such as SNPs or different RNA expression levels, and are less costly then sequencing; however, they are less suited for obtaining highly quantitative results, nor for detecting low abundance mutations.
- the TaqManTM reaction which provides real-time quantification of a known gene, but is less suitable for distinguishing multiple sequence variants or low abundance mutations.
- NGS requires substantial up-front sample preparation to polish ends and append linkers, and the current error rates of 0.7% are too high to identify 2-3 molecules of mutant sequence in a 10,000-fold excess of wild-tye molecules.“Deep sequencing” protocols have been developed to overcome this deficiency by appending unique molecular identifiers to both strands of an individual fragment.
- Deep sequencing of cfDNAs for 58 cancer-related genes at 30,000-fold coverage is capable of detecting Stage 1 or 2 cancer at moderately high sensitivity but missed 29% of CRC, 41% of breast, 41% of lung, and 32% of ovarian cancer, respectively (Phallen et al., “Direct Detection of Early-stage Cancers Using Circulating Tumor DNA,” Science Translational Medicine 9(403) (2017)).
- the Hopkins team very recently combined NGS with quantitation of serum protein markers (such as CA-125, CA19-9, CEA, HGF, Myeloperoxidase, OPN, Prolactin, TIMP-1) and improved detection of five cancer types (ovary, liver, stomach, pancreas, and esophagus) at sensitivities ranging from 69% to 98% (Cohen et al.“Detection and Localization of Surgically Resectable Cancers with a Multi-analyte Blood Test,” Science (2018).
- serum protein markers such as CA-125, CA19-9, CEA, HGF, Myeloperoxidase, OPN, Prolactin, TIMP-
- the patient samples comprised more than 20 types of cancer, including hormone receptor-negative breast, colorectal, esophageal, gallbladder, gastric, head and neck, lung, lymphoid leukemia, multiple myeloma, ovarian, and pancreatic cancer.
- the overall specificity was 99.4%, meaning only 0.6% of the results incorrectly indicated that cancer was present.
- the sensitivity of the assay for detecting a pre-specified high mortality cancer was 76%. Within this group, the sensitivity was 32% for patients with stage I cancer; 76% for those with stage II; 85% for stage III; and 93% for stage IV. Sensitivity across all cancer types was 55%, with similar increases in detection by stage.
- cancer-specific RNA markers may also be present in blood, either free of any compartment (Souza et al.,“Circulating mRNAs and miRNAs as Candidate Markers for the Diagnosis and Prognosis of Prostate Cancer,” PloS One 12(9):e0184094 (2017)), or contained in exosomes (Nedaeinia et al., “Circulating Exosomes and Exosomal microRNAs as Biomarkers in Gastrointestinal Cancer,” Cancer Gene Ther 24(2):48-56 (2017); Lai et al.,“A microRNA Signature in Circulating Exosomes is Superior to Exosomal Glypican-1 Levels for Diagnosing Pancreatic Cancer,” Cancer Lett 39:86-93 (2017)) or circulating tumor cells (“CTCs”), and have been tagged as potential indicators of early- stage cancers.
- CTCs circulating tumor cells
- nucleic acid detection Central to the concept of nucleic acid detection is the selective amplification or purification of the desired cancer-specific markers away from the same or closely similar markers from normal cells. These approaches include: (i) multiple primer binding regions for orthogonal amplification and detection, (ii) affinity selection of CTC’s or exosomes, and (iii) spatial dilution of the sample.
- PCR-LDR which uses 4 primer-binding regions to assure sensitivity and specificity, has previously been demonstrated. Desired regions are amplified using pairs or even tandem pairs of PCR primers, followed by orthogonal nested LDR primer pairs for detection.
- One advantage of using PCR-LDR is the ability to perform proportional PCR amplification of multiple fragments to enrich for low copy targets, and then use quantitative LDR to directly identify cancer-specific mutations.
- Biofire/bioMerieux has developed a similar technology termed“film array”; wherein initial multiplexed PCR reaction products are redistributed into individual wells, and then nested real-time PCR performed with SYBR Green Dye detection.
- the DNA may be amplified via PCR, and then detected via probe hybridization or TaqManTM reaction, giving in essence a 0/1 digital score.
- the approach is currently the most sensitive for finding point mutations in plasma, but it does require prior knowledge of the mutations being scored, as well as a separate digital dilution for each mutation, which would deplete the entire sample to score just a few mutations (Alcaide et al.,“A Novel Multiplex Droplet Digital PCR Assay to Identify and Quantify KRAS Mutations in Clinical Specimens,” J. Mol.
- a first aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences of other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules are then provided.
- One or more primary oligonucleotide primer sets are also provided.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the sample, the one or more first primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixtures and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising nucleotide sequences complementary to the target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more second primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixtures, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- dU deoxyuracil
- the method further comprises subjecting the one or more first polymerase chain reaction mixtures to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more first polymerase chain reaction products comprising the target nucleotide sequence or a complement thereof.
- One or more oligonucleotide probe sets are then provided.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence- specific portion and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on a complementary target nucleotide sequence of a secondary extension product.
- the one or more first polymerase chain reaction products are blended with a ligase, and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures.
- the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets are ligated together, when hybridized to complementary sequences, to form ligated product sequences in the ligation reaction mixtures wherein each ligated product sequence comprises the 5’ primer-specific portion, the target- specific portions, and the 3’ primer-specific portion.
- the method further includes providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more second polymerase chain reaction products.
- dU deoxyuracil
- the method further comprises detecting and distinguishing the one or more second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, one or more nucleases capable of digesting nucleic acid molecules not comprising modified nucleotides, and one or more first primary oligonucleotide primer(s) are provided.
- the one or more first primary oligonucleotide primer(s) comprise a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence.
- the sample, the one or more first primary oligonucleotide primers, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix that comprises one or more modified nucleotides that protect extension products but not target DNA from nuclease digestion, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixture and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the target nucleotide sequence.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a first 5’ primer-specific portion and a 3’ portion that is complementary to a portion of a primary extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a second 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary oligonucleotide primer sets, the one or more nucleases, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the first polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more first polymerase chain reaction products comprising the first 5’ primer-specific portion, a target-specific nucleotide sequence or a complement thereof, and a complement of the second 5’ primer-specific portion.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the first 5’ primer-specific portion of the one or more first polymerase chain reaction products and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the one or more first polymerase chain reaction products.
- the one or more first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU) containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more second polymerase chain reaction products.
- the method further involves detecting and distinguishing the one or more second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences of other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules and one or more nucleases capable of digesting nucleic acid molecules present not comprising modified nucleotides are provided.
- the method also involves providing one or more primary oligonucleotide primer sets.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the sample, the one or more first primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix that comprises one or more modified nucleotides that protect extension product but not target DNA from nuclease digestion, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixtures and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the target nucleotide sequence.
- the method further comprises blending the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more second primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more nucleases, a deoxynucleotide mix, and a DNA polymerase to form one or more first polymerase chain reaction mixtures.
- the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the target nucleotide sequence or a complement thereof.
- One or more secondary oligonucleotide primer sets are then provided.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the first polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues and subjecting the nucleic acid molecules in the sample to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite- treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite-treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- dU deoxyuracil
- the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the bisulfite-treated target nucleotide sequence or a complement thereof.
- the method further involves providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ bisulfite-treated target nucleotide sequence-specific or complement sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ bisulfite-treated target nucleotide sequence- specific or complement sequence-specific portion and a 3’ primer-specific portion, and wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on a complementary nucleotide sequence of a first polymerase chain reaction product.
- the first polymerase chain reaction products are blended with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures.
- the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets are ligated together, when hybridized to complementary sequences, to form ligated product sequences in the ligation reaction mixture wherein each ligated product sequence comprises the 5’ primer-specific portion, the bisulfite-treated target nucleotide sequence-specific or complement sequence- specific portions, and the 3’ primer-specific portion.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming a second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues.
- the nucleic acid molecules in the sample are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules are provided, and one or more first primary oligonucleotide primer(s) are provided.
- Each first primary oligonucleotide primer comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, to form primary extension products comprising the complement of the bisulfite-treated target nucleotide sequence.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of the polymerase extension reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)- containing nucleic acid molecules, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the first polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reactions products comprising a 5’ primer- specific portion of the first secondary oligonucleotide primer, the bisulfite-treated target nucleotide sequence-specific or complement sequence-specific portion, and a complement of the 5’ primer-specific portion of
- oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reactions product sequence.
- the first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further involves detecting and distinguishing the secondary polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues.
- the nucleic acid molecules in the sample are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures.
- the one or more polymerase extension reaction mixtures to are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixture, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- the one or more first polymerase chain reaction mixtures are sujected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the bisulfite- treated target nucleotide sequence or a complement thereof.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of a first polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a first polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the first polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues, and subjecting the nucleic acid molecules in the sample to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample are provided.
- One or more primary oligonucleotide primer sets are also provided. Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures.
- the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixture, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- the method further comprises subjecting the one or more first polymerase chain reaction mixtures to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reactions products comprising the bisulfite-treated target nucleotide sequence or a complement thereof.
- One or more secondary oligonucleotide primer sets are provided.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products or their complements and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the first polymerase chain reaction products or their complements.
- oligonucleotide primer sets the one or more enzymes capable of digesting deoxyuracil
- (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Another aspect of the present application is directed to a method for identifying in a sample, one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- the method involves providing a sample containing one or more parent ribonucleic acid molecules containing a target ribonucleic acid molecule potentially differing in sequence from other parent ribonucleic acid molecules and providing one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU containing nucleic acid molecules potentially present in the sample.
- One or more primary oligonucleotide primer sets are then provided.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to the RNA sequence in the parent ribonucleic acid molecule adjacent to the target ribonucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of the cDNA extension product formed from the first primary oligonucleotide primer.
- the contacted sample, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix including dUTP, a reverse transcriptase, and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target ribonucleic nucleic acid and to carry out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse
- the method further comprises providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on complementary portions of a reverse transcriptase/polymerase product corresponding to the target ribonucleic acid molecule sequence.
- the reverse transcriptase/polymerase products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures, and the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligase reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming first polymerase chain reaction products.
- the method further comprises detecting and distinguishing the first polymerase chain reaction products, thereby identifying the presence of one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- Another aspect of the present application is directed to a method for identifying in a sample, one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- the method involves providing a sample containing one or more parent ribonucleic acid molecules containing a target ribonucleic acid molecule potentially differing in sequence from other parent ribonucleic acid molecules and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU containing nucleic acid molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets, each primary oligonucleotide primer set comprising (a) a first primary
- oligonucleotide primer that comprises a nucleotide sequence that is complementary to the RNA sequence in the parent ribonucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of the cDNA extension product formed from the first primary oligonucleotide primer.
- the contacted sample, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse- transcription/polymerase chain reaction mixtures, and the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target RNA and to carry out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse-transcription/primary polymerase chain reaction products.
- cDNA complementary deoxyribonucleic acid
- the method futher comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of a reverse-transcription/primary polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a reverse-transcription/primary polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the reverse-transcription/primary polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming first polymerase chain reaction products.
- the method further involves detecting and distinguishing the first polymerase chain reaction products, thereby identifying the presence of one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequences differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- dU deoxyuracil
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample, and the contacted sample is blended with a ligase and one or more first oligonucleotide preliminary probes comprising a 5’ phosphate, a 5’ stem-loop portion, an internal primer-specific portion within the loop region, a blocking group, and a 3’ nucleotide sequence that is complementary to a 3’ portion of the target miRNA molecule sequence to form one or more first ligation reaction mixtures.
- the method further comprises ligating, in the one or more first ligation reaction mixtures, the one or more target miRNA molecules at their 3’end to the 5’ phosphate of the one or more first
- oligonucleotide preliminary probes to generate chimeric nucleic acid molecules comprising the target miRNA molecule sequence, if present in the sample, appended to the one or more first oligonucleotide preliminary probes.
- One or more primary oligonucleotide primer sets are then provided.
- Each primer set comprises (a) a first primary oligonucleotide primer comprising a nucleotide sequence that is complementary to the internal primer-specific portion of the first oligonucleotide preliminary probe, and (b) a second primary oligonucleotide primer comprising a 5’ primer-specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the one or more first ligation reaction mixtures comprising chimeric nucleic acid molecules, the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the sample, a deoxynucleotide mix including dUTP, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- dU deoxyuracil
- a deoxynucleotide mix including dUTP
- a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the reverse-transcription/polymerase chain reaction mixtures to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the chimeric nucleic acid molecules, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different primary reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence, and the complement of the internal primer-specific portion, and complements thereof.
- dU deoxyuracil
- cDNA complementary deoxyribonucleic acid
- the method further involves providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion, a portion complementary to a primary extension product, and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on complementary portions of a primary reverse-transcription/polymerase chain reaction product corresponding to the target miRNA molecule sequence, or complement thereof.
- the primary reverse- transcription/polymerase chain reaction products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more second ligation reaction mixtures, and the one or more second ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligation reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences and the one or more secondary oligonucleotide primer sets are blended with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further comprises detecting and distinguishing the secondary polymerase chain reaction products in the one or more reactions thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- dU deoxyuracil
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample, and the contacted sample is blended with a ligase and one or more first oligonucleotide probes comprising a 5’ phosphate, a 5’ stem-loop portion, an internal primer-specific portion within the loop region, a blocking group, and a 3’ nucleotide sequence that is complementary to a 3’ portion of the target miRNA molecule sequence to form one or more ligation reaction mixtures.
- the method further involves ligating, in the one or more ligation reaction mixtures, the one or more target miRNA molecules at their 3’end to the 5’ phosphate of the one or more first oligonucleotide probes to generate chimeric nucleic acid molecules comprising the target miRNA molecule sequence, if present in the sample, appended to the one or more first oligonucleotide probes.
- One or more primary oligonucleotide primer sets are then provided. Each primer set comprises (a) a first primary oligonucleotide primer comprising a nucleotide sequence that is complementary to the internal primer-specific portion of the first oligonucleotide probe, and (b) a second primary
- oligonucleotide primer comprising a 5’ primer-specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the one or more ligation reaction mixtures comprising chimeric nucleic acid molecules, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse- transcription/polymerase chain reaction mixtures.
- the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the chimeric nucleic acid molecules, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different primary reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence, and the complement of the internal primer-specific portion, and complements thereof.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the primary reverse- transcription/polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising a 5’ primer-specific portion of the first secondary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence or a complement thereof, and a complement of the other 5’ primer- specific portion second secondary oligonucleotide primer.
- the method further involves providing one or more tertiary oligonucleotide primer sets.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products or their complements and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reaction products or their complements.
- the first polymerase chain reaction process products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU) containing nucleic acid molecules present in the secondpolymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products, thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- dU deoxyuracil
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample.
- the contacted sample is blended with ATP and a Poly(A) polymerase to form a Poly(A) polymerase reaction mixture, and the Poly(A) polymerase reaction mixture is subjected to conditions suitable for appending homopolymer A to the 3’ ends of the one or more target miRNA molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets.
- Each primer set comprises (a) a first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising
- oligonucleotide primer may be the same or may differ from other first primary oligonucleotide primers in other sets, and (b) a second primary oligonucleotide primer comprising a 5’ primer- specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the Poly(A) polymerase reaction mixture, the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the sample, a deoxynucleotide mix including dUTP, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the reverse- transcription/polymerase chain reaction mixtures, then to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target miRNA sequences with 3’ polyA tails, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse-transcription/polymerase chain reaction products comprising the 5’ primer- specific portion of the second primary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence, a poly dA region, and the complement of the 5’ primer-specific portion of the first primary oligonucleotide primer, and complements thereof.
- dU deoxyuracil
- cDNA complementary deoxyribonucleic acid
- the method further comprises providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion, a portion complementary to the one or more reverse- transcription/polymerase chain reaction products, and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, to complementary portions of the one or more reverse-transcription/polymerase chain reaction products corresponding to the target miRNA molecule sequence, or complement thereof.
- the one or more reverse-transcription/polymerase chain reaction products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures, and the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligation reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences and the one or more secondary oligonucleotide primer sets are blended with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further comprises detecting and distinguishing the secondary polymerase chain reaction products, thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample.
- the contacted sample is blended with ATP and a Poly(A) polymerase to form a Poly(A) polymerase reaction mixture, and the Poly(A) polymerase reaction mixture is subjected to conditions suitable for appending a homopolymer A to the 3’ ends of the one or more target miRNA molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets. Each primer set comprises (a) a first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary
- oligonucleotide primer may be the same or may differ from other first primary oligonucleotide primers in other sets, and (b) a second primary oligonucleotide primer comprising a 5’ primer- specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the Poly(A) polymerase reaction mixture potentially comprising target miRNA sequences is blended with 3’ polyA tails, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse- transcriptase activity to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target miRNA sequences with 3’ polyA tails, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more different reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion of the second primary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence, a poly dA region, and the complement of the 5’ primer-specific portion of the first primary oligonucleotide primer, and complements thereof.
- cDNA complementary deoxyribonucleic acid
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of a reverse-transcription/polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a reverse- transcription/polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the reverse-transcription/polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising a 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence or a complement thereof, and a complement of the other 5’ primer-specific portion.
- the method further involves providing one or more tertiary oligonucleotide primer sets.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction product sequence and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reaction product sequence.
- the first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures, and one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products in the one or more reactions thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of cells or tissue based on identifying the presence or level of a plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers in a biological sample of an individual.
- the plurality of markers is in a set comprising from 6-12 markers, 12-24 markers, 24-36 markers, 36-48 markers, 48-72 markers, 72-96 markers, or > 96 markers.
- Each marker in a given set is selected by having any one or more of the following criteria: present, or above a cutoff level, in > 50% of biological samples of the disease cells or tissue from individuals diagnosed with the disease state; absent, or below a cutoff level, in > 95% of biological samples of the normal cells or tissue from individuals without the disease state; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with the disease state; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without the disease state; present with a z-value of > 1.65 in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65, comprise of one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with the disease state.
- the method involves obtaining a biological sample.
- the biological sample includes cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, and the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor-associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- Nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- At least two enrichment steps are carried out for 50% or more disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with the disease state if a minimum of 2 or 3 markers are present or are above a cutoff level in a marker set comprising from 6-12 markers; or a minimum of 3, 4, or 5 markers are present or are above a cutoff level in a marker set comprising from 12-24 markers; or a minimum of 3, 4, 5, or 6 markers are present or are above a cutoff level in a marker set comprising from 24-36 markers; or a minimum of 4, 5, 6, 7, or 8 markers are present or are above a cutoff level in a marker set comprising from 36
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of a solid tissue cancer including colorectal adenocarcinoma, stomach adenocarcinoma, esophageal carcinoma, breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, uterine carcinosarcoma, lung adenocarcinoma, lung squamous cell carcinoma, head & neck squamous cell carcinoma, prostate adenocarcinoma, invasive urothelial bladder cancer, liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma based on identifying the presence or level of a plurality of disease- specific and/or cell/tissue-specific DNA, RNA, and/or protein
- the plurality of markers is in a set comprising from 48-72 total cancer markers, 72-96 total cancer markers or 3 96 total cancer markers, wherein on average greater than one quarter such markers in a given set cover each of the aforementioned major cancers being tested.
- Each marker in a given set for a given solid tissue cancer is selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 50% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65, comprise of one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with a given solid tissue cancer.
- the method involves obtaining a biological sample including cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor-associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- the nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues. At least two enrichment steps are carried out for 50% or more of the given solid tissue cancer-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of cancer -specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with the a solid-tissue cancer if a minimum of 4 markers are present or are above a cutoff level in a marker set comprising from 48-72 total cancer markers; or a minimum of 5 markers are present or are above a cutoff level in a marker set comprising from 72-96 total cancer markers; or a minimum of 6 or “n”/18 markers are present or are above a cutoff level in a marker set comprising 96 to“n” total cancer markers, when“n” > 96 total cancer markers.
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of and identifying the most likely specific tissue(s) of origin of a solid tissue cancer in the following groups: Group 1 (colorectal adenocarcinoma, stomach adenocarcinoma, esophageal carcinoma); Group 2 (breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, uterine carcinosarcoma); Group 3 (lung adenocarcinoma, lung squamous cell carcinoma, head & neck squamous cell carcinoma); Group 4 (prostate adenocarcinoma, invasive urothelial bladder cancer); and/or Group 5 (liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adeno
- Each marker in a given set for a given solid tissue cancer is selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 50% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without that given solid tissue cancer; present with a z-value of > 1.65 in the biological sample comprising
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65 comprise one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with a given solid tissue cancer.
- the method involves obtaining the biological sample.
- the biological sample includes cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor- associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- the nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- At least two enrichment steps are carried out for 50% or more of the given solid tissue cancer-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of cancer-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with a solid-tissue cancer if a minimum of 4 markers are present or are above a cutoff level in a marker set comprising from 36-48 group- specific cancer markers; or a minimum of 5 markers are present or are above a cutoff level in a marker set comprising from 48-64 group-specific cancer markers; or a minimum of 6 or“n”/12 markers are present or are above a cutoff level in a marker set comprising 64 to“n” total cancer markers, when“n” > 64 group-specific cancer markers.
- the present application describes a number of approaches for detecting mutations, expression, splice variant, translocation, copy number, and/or methylation changes in target nucleic acid molecules using nuclease, ligase, and polymerase reactions.
- the present application solves the problems of carry over prevention, as well as allowing for spatial multiplexing to provide relative quantification, similar to digital PCR.
- Such technology may be utilized for non- invasive early detection of cancer, non-invasive prognosis of cancer, and monitoring for cancer recurrence from plasma or serum samples.
- the present application provides a comprehensive roadmap of nucleic acid methylation, miRNA, lncRNA, ncRNA, mRNA Exons, as well as cancer-associated protein markers that are specific for solid-tissue cancers and matched normal tissues.
- the present application teaches the art of selecting the desired number of markers and types of markers for both pan-oncology and specific cancers (i.e. colorectal cancer) to guide the physician to improve the treatment of the patient. Details on primer design and optimized primer sequences are provided to enable rapid validation of these tests for both pan-oncology and specific cancers.
- the two-step procedure is designed to cast a wide net to initially identify most of the individuals harboring an early cancer, followed by a more stringent second step to improve specificity and narrow the patients to those most likely to harbor a hidden cancer, who are then sent for imaging and followup.
- the advantage of this 2-step approach is that it not only identifies the potential tissue of origin, but it is designed to provide the highest positive predictive value (PPV).
- PSV positive predictive value
- FIG. 1A-B illustrates a conditional logic tree for an early detection colorectal cancer test based on analysis of a patient’s blood sample.
- Figure 1A illustrates a one-step colorectal cancer assay using 24 markers at average sensitivity of 50%.
- Figure 1B illustrates a two-step colorectal cancer assay using 24 markers in the first step at average sensitivity of 50%, and 48 markers in a second step.
- Figures 1C-D illustrate a conditional logic tree for a two-step assay for early detection pan-oncology pan-oncology cancer test based on analysis of a patient’s blood sample.
- Figure 1C illustrates a two-step pan-oncology assay using 96 group-specific markers at average sensitivity of 50% in the first step, followed by 1 or 2 groups of 64 type- specific markers each at average sensitivity of 50% in the second step.
- Figure 1D illustrates a two-step pan-oncology assay using 96 group-specific markers at average sensitivity of 66% in the first step, followed by 1 or 2 groups of 64 type-specific markers each at average sensitivity of 66% in the second step.
- Figure 2 illustrates exPCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 3 illustrates exPCR-LDR-qPCR carryover prevention reaction with UniTaq detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 4 illustrates exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 5 illustrates exPCR-qPCR carryover prevention reaction with UniTaq detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 6 illustrates a variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 7 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 8 illustrates a variation of exPCR-qPCR carryover prevention reaction with UniTaq detection to identify or relatively quantify target(s) and/or low-level mutations.
- Figure 9 illustrates exPCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 10 illustrates a variation of exPCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 11 illustrates exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 12 illustrates a variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 13 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 14 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 15 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 16 illustrates another variation of exPCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 17 illustrates another variation of exPCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 18 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 19 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 20 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 21 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 22 illustrates another variation of exPCR-qPCR carryover prevention reaction with TaqmanTM detection to identify or relatively quantify low-level methylation.
- Figure 23 illustrates RT-PCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate translocation events at the mRNA level.
- Figure 24 illustrates RT-PCR-qPCR carryover prevention reaction
- TaqmanTM detection to detect and enumerate translocation events at the mRNA level.
- Figure 25 illustrates RT-PCR-PCR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate translocation events at the mRNA level.
- Figure 26 illustrates RT-PCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate RNA copy number.
- Figure 27 illustrates RT-PCR-qPCR carryover prevention reaction
- TaqmanTM detection to detect and enumerate RNA copy number.
- Figure 28 illustrates RT-PCR-PCR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate RNA copy number.
- Figure 29 illustrates Ligation-RT-PCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate miRNA.
- Figure 30 illustrates Ligation-RT-PCR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate miRNA.
- Figure 31 illustrates RT-PCR-LDR-qPCR carryover prevention reaction with TaqmanTM detection to detect and enumerate miRNA.
- Figure 32 illustrates RT-PCR-qPCR carryover prevention reaction with
- TaqmanTM detection to detect and enumerate miRNA.
- Figures 33A-B illustrate results for calculated overall Sensitivity and Specificity for a 24-marker assay, where the average individual marker sensitivity is 50% ( Figure 33A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 33B).
- Figures 34A-B illustrate results for calculated overall Sensitivity and Specificity for a 36-marker assay, where the average individual marker sensitivity is 50% ( Figure 34A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 34B).
- Figures 35A-B illustrate results for calculated overall Sensitivity and Specificity for a 48-marker assay, where the average individual marker sensitivity is 50% ( Figure 35A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 35B).
- Figures 36A-B illustrate results for calculated overall Sensitivity and Specificity for a 96-marker assay, where the average individual marker sensitivity is 50% ( Figure 36A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 36B).
- Figures 37A-B illustrate the ROC curve for a 48-marker assay, where the average individual marker sensitivity is 50%, as well as the calculated AUC, when the average number of molecules per marker in the blood ranges from 150 to 600 molecules.
- the calculations are based on an average individual marker false-positive rate of 2% and 3%, respectively.
- Figures 38A-B illustrate the ROC curve for a 48-marker assay, where the average individual marker sensitivity is 50%, as well as the calculated AUC, when the average number of molecules per marker in the blood ranges from 150 to 600 molecules.
- the calculations are based on an average individual marker false-positive rate of 4% and 5%, respectively.
- Figures 39A-B provide a list of blood-based, colon cancer-specific microRNA markers derived through analysis of TCGA microRNA datasets, which may be present in exosomes or other protected state in the blood.
- Figures 40A-X provide a list of blood-based, colon cancer-specific ncRNA and lncRNA markers, which may be present in exosomes or other protected state in the blood.
- Figures 41A-C provide a list of candidate blood-based colon cancer-specific exon transcripts that may be enriched in in exosomes or other protected state in the blood.
- Figures 42A-J provide a list of cancer proteins markers, identified through, mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from Colorectal tumors, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Figure 43 provides a list of protein markers that can be secreted by Colorectal tumors into the blood.
- Figures 44A-Y provide a list of primary CpG sites that are Colorectal Cancer and Colon-tissue specific markers, that may be used to identify the presence of colorectal cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 45A-P provide a list of chromosomal regions or sub-regions within which are primary CpG sites that are Colorectal Cancer and Colon-tissue specific markers, that may be used to identify the presence of colorectal cancer from cfDNA, or DNA within exosomes, or DNA in other protected state (such as within CTCs) within the blood.
- Figures 46A-B illustrate results for calculated overall Sensitivity and Specificity for a 24-marker assay, where the average individual marker sensitivity is 50%, and the average individual marker false-positive rate is from 2% to 5%; including one marker with a sensitivity at 90% ( Figure 46A) and a 10% (Figure 46B) false-positive rate.
- Figures 47A-B illustrate results for calculated overall Sensitivity and Specificity for a 24-marker assay, where the average individual marker sensitivity is 50%, and the average individual marker false-positive rate is from 2% to 5%; including two markers with a sensitivity at 90% ( Figure 47A) and a 10% (Figure 47B) false-positive rate.
- Figures 48A-B illustrate results for calculated overall Sensitivity and Specificity for a 48-marker assay, where the average individual marker sensitivity is 50%, and the average individual marker false-positive rate is from 2% to 5%; including one marker with a sensitivity at 90% ( Figure 48A) and a 10% (Figure 48B) false-positive rate.
- Figures 49A-B illustrate results for calculated overall Sensitivity and Specificity for a 48-marker assay, where the average individual marker sensitivity is 50%, and the average individual marker false-positive rate is from 2% to 5%; including two markers with a sensitivity at 90% ( Figure 49A) and a 10% (Figure 49B) false-positive rate.
- Figures 50A-B illustrate results for calculated overall Sensitivity and Specificity for a 24-marker assay, where the average individual marker sensitivity is 66% ( Figure 50A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 50B).
- Figures 51A-B illustrate results for calculated overall Sensitivity and Specificity for a 36-marker assay, where the average individual marker sensitivity is 66% ( Figure 51A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 51B).
- Figures 52A-B illustrate results for calculated overall Sensitivity and Specificity for a 48-marker assay, where the average individual marker sensitivity is 66% ( Figure 52A), and the average individual marker false-positive rate is from 2% to 5% ( Figure 52B).
- Figure 53 provides a list of blood-based, solid tumor-specific ncRNA and lncRNA markers, which may be present in exosomes or other protected state in the blood.
- Figures 54A-F provide a list of candidate blood-based solid tumor-specific exon transcripts that may be enriched in in exosomes or other protected state in the blood.
- Figures 55A-H provide a list of cancer proteins markers, identified through, mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from solid tumors, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Figures 56A-S provide a list of primary CpG sites that are solid-tumor and tissue- specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 57A-J provide a list of chromosomal regions or sub-regions within which are primary CpG sites that are solid-tumor and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 58 provide a list of cancer proteins markers, identified through, mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from colon adenocarcinoma, rectal adenocarcinoma, stomach adenocarcinoma, or esophageal carcinoma, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Figures 59A-S provide a list of primary CpG sites that are colon adenocarcinoma, rectal adenocarcinoma, stomach adenocarcinoma, or esophageal carcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 60A-J provide a list of chromosomal regions or sub-regions within which are primary CpG sites that are colon adenocarcinoma, rectal adenocarcinoma, stomach adenocarcinoma, or esophageal carcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 61A-C provide a list of primary CpG sites that are breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, or uterine carcinosarcoma and tissue- specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 62A-B provide a list of chromosomal regions or sub-regions within which are primary CpG sites that are breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, or uterine carcinosarcoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 63 provides a list of primary CpG sites that are lung adenocarcinoma, lung squamous cell carcinoma, or head & neck squamous cell carcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 64 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are lung adenocarcinoma, lung squamous cell carcinoma, or head & neck squamous cell carcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 65 provides a list of primary CpG sites that are prostate adenocarcinoma or invasive urothelial bladder cancer and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 66 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are prostate adenocarcinoma or invasive urothelial bladder cancer and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 67 provides a list of blood-based, liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma-specific ncRNA and lncRNA markers, which may be present in exosomes or other protected state in the blood.
- Figures 68A-E provide a list of candidate blood-based liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma-specific exon transcripts that may be enriched in exosomes or other protected state in the blood.
- Figures 69A-B provide a list of cancer proteins markers, identified through, mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Figures 70A-E provide a list of primary CpG sites that are liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figures 71A-C provide a list of chromosomal regions or sub-regions within which are primary CpG sites that are liver hepatoceullular carcinoma, pancreatic ductal
- adenocarcinoma or gallbladder adenocarcinoma and tissue-specific markers, that may be used to identify the presence of solid-tumor cancer from cfDNA, or DNA within exosomes, or DNA in another protected state (such as within CTCs) within the blood.
- Figure 72 illustrates the real-time PCR amplification plots obtained in the pixel Bisulfite-PCR-LDR-qPCR experiments to enumerate single molecules of methylated DNA in the presence of an excess of unmethylated DNA (Roche DNA).
- Figure 73 illustrates the real-time PCR amplification plots obtained in a multiplexed detection of 10 CRC methylation markers by Bisulfite-PCR-LDR-qPCR, using HT29 cell line DNA, with an average of 20 molecules each marker in 10,000 molecules of normal, e.g. unmethylated DNA (Roche DNA).
- Figure 74 illustrates the real-time PCR amplification plots obtained in a multiplexed detection of 7 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using HT29 cell line DNA, with an average of 30 molecules each marker in 3,000 molecules of normal, e.g. unmethylated DNA (Roche DNA).
- Figures 75A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 7 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using cfDNA isolated from CRC ( Figure 75A) and Normal ( Figure 75B) plasma.
- Figures 76A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 7 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using cfDNA isolated from CRC ( Figure 76A) and Normal ( Figure 76B) plasma.
- Figures 77A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 7 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using cfDNA isolated from CRC ( Figure 77A) and Normal ( Figure 77B) plasma.
- Figures 78A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 20 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using HT29 cell line DNA, with 1,500 genome equivalents of HT29 cell line DNA in 7,500 genome equivalents of normal, e.g. unmethylated DNA (Roche DNA; Figure 78A) compared with 7,500 genome equivalents of normal, e.g. unmethylated DNA ( Figure 78B).
- Figures 79A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 20 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using reverse primers with tails, using HT29 cell line DNA, with 200 genome equivalents of HT29 cell line DNA in 7,500 genome equivalents of normal, e.g. unmethylated DNA (Roche DNA; Figure 79A) compared with 7,500 genome equivalents of normal, e.g. unmethylated DNA ( Figure 79B).
- Figures 80A-B illustrate the real-time PCR amplification plots obtained in a multiplexed detection of 20 CRC methylation markers by Bisulfite-exPCR-LDR-qPCR, using reverse primers without tails, using HT29 cell line DNA, with 200 genome equivalents of HT29 cell line DNA in 7,500 genome equivalents of normal, e.g. unmethylated DNA (Roche DNA; Figure 80A) compared with 7,500 genome equivalents of normal, e.g. unmethylated DNA ( Figure 80B).
- DETAILED DESCRIPTION A Universal Design for Early Detection of Cancer Using“Cancer Marker Load”
- the most cost-effective early cancer detection test may combine an initial multiplexed coupled amplification and ligation assay to determine“cancer load”. For cancer detection, this would achieve > 95% sensitivity for all cancers (pan-oncology), at > 97% specificity.
- FIG. 1 Several flow charts for a cancer tumor load assay is illustrated in Figure 1.
- the assay would be a one-step assay to identify individuals with early colorectal cancer (CRC).
- CRC colorectal cancer
- a blood sample is fractionated into plasma and other components as needed, a set of 24 markers with average sensitivity of 50% are assayed, and the results are recorded ( Figure 1A).
- Figure 1A For example, an initial multiplexed PCR/LDR screening assay scoring for mutation, methylation, miRNA, mRNA, alternative splicing, and/or translocations identifies those samples with positive results.
- the physician is not concerned with which specific markers are positive but gives a simple directive. Those patients with 0-2 markers positive are told not to worry, go home, you are cancer-free.
- a two-step assay would be performed to identify if the patient has colorectal cancer.
- the rationale for a two-step test is to initially cast a wide net to maximize sensitivity in identifying the most individuals with potential cancer, followed by a second step only on the positive samples (which contain both true and false- positives) to maximize specificity, eliminate virtually all the false-positives, and hone in on those individuals most likely to have cancer.
- a blood sample is fractionated into plasma and other components as needed, followed by an assay to interrogate an initial set of 24 markers with an average sensitivity of 50% (Figure 1B).
- the first step assay can employ multiplexed PCR/LDR, or digital PCR screening to score for mutation, methylation, miRNA, mRNA, alternative splicing, and/or translocations events.
- patients with 0-2 markers positive are presumed to be cancer-free.
- patients with 3 3 markers positive will undergo a second step, wherein 48 (new) markers are assayed and scored as follows: 0-3 positive markers are considered cancer-free; 4-5 positive markers are advised to come back in 3-6 months for retesting; 3 6 positive markers are directed to go get a colonoscopy.
- the assay in the first step the assay would screen 96 markers, wherein on average 3 36 such markers would exhibit an average sensitivity of 50% for most major cancers (see Figure 1C). These cancers would cluster to certain groups, which include: Group 1 (Colorectal, Stomach, Esophagus); Group 2 (Breast, Endometrial, Ovarian, Cervical, Uterine); Group 3 (Lung, Head & Neck); Group 4 (Prostate, Bladder), and Group 5 (Liver, Pancreatic, Gall Bladder). Patients with 0-4 markers positive are presumed to be cancer- free, while patients with 3 5 markers positive will undergo a second step.
- Group 1 Cold, Stomach, Esophagus
- Group 2 Breast, Endometrial, Ovarian, Cervical, Uterine
- Group 3 Long, Head & Neck
- Group 4 Prostate, Bladder
- Group 5 Liver, Pancreatic, Gall Bladder
- Presumptive positive samples are then assayed in the second step, testing 1 or 2 groups, using 64 markers per group, wherein on average 3 36 such markers would exhibit an average sensitivity of 50% for each specific type of cancer within that group, including using tissue-specific markers to validate the initial result and to identify tissue of origin.
- Results are scored as follows: 0-3 positive markers are considered cancer-free; 4 positive markers are advised to come back in 3-6 months for retesting; 3 5 positive markers are directed to go to imaging that matches the type(s) of cancer most likely to be the tissue of origin.
- both the initial 96 markers in the first step, and the group-specific markers in the second step would have average sensitivity of 66% ( Figure 1D).
- the physician may then order targeted sequencing to further guide treatment decisions for the patient.
- the present application is directed to a universal diagnostic approach that seeks to combine the best features of digital polymerase chain reaction (PCR), or quantitative polymerase chain reaction (qPCR), with bisulfite conversion, ligation detection reaction (LDR), and quantitative detection of multiple disease markers, e.g., cancer markers. Multiplexing, avoiding False-Positives, and Carryover Protection
- the first theme is multiplexing. PCR works best when primer concentration is relatively high, from 50nM to 500nM, limiting multiplexing. Further, the more PCR primer pairs added, the chances of amplifying incorrect products or creating primer-dimers increase exponentially. In contrast, for LDR probes, low concentrations on the order of 4 nM to 20 nM are used, and probe-dimers are limited by the requirement for adjacent hybridization on the target to allow for a ligation event. Use of low concentrations of gene-specific PCR primers or LDR probes containing universal primer sequence“tails” allows for subsequent addition of higher concentrations of universal primers to achieve proportional amplification of the initial PCR or LDR products.
- PCR primers containing a few extra bases and a blocking group which is liberated to form a free 3’OH by cleavage with a nuclease only when hybridized to the target, e.g., a ribonucleotide base as the blocking group and RNase H2 as the cleaving nuclease.
- the second theme is fluctuations in signal due to low input target nucleic acids.
- the target nucleic acid originated from a few cells, either captured as CTCs, or from tumor cells that underwent apoptosis and released their DNA as small fragments (140– 160 bp) in the serum. Under such conditions, it is preferable to perform some level of proportional amplification to avoid missing the signal altogether or reporting inaccurate copy number due to fluctuations when distributing small numbers of starting molecules into individual wells (for real-time, or droplet PCR quantification).
- target-independent signal also known as“No Template Control” (NTC). This arises from either polymerase or ligase reactions that occur in the absence of the correct target. Some of this signal may be minimized by judicious primer design.
- the 5’ ® 3’ nuclease activity of polymerase may be used to liberate the 5’ phosphate of the downstream ligation primer (only when hybridized to the target), so it is suitable for ligation.
- Further specificity for distinguishing presence of a low-level mutation using LDR may be achieved by: (i) using upstream mutation-specific LDR probes containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild- type sequence that would reduce hybridization of mutation-specific LDR probes to wild-type sequences, (iii) using LDR probes to wild-type sequence that (optionally) ligate but do not undergo additional amplification, and (iv) using upstream LDR probes containing a few extra bases and a blocking group, which is liberated to form a free 3’OH by cleavage with a nuclease only when hybridized to the complementary target (e.g., RNase H
- Similar approaches for improving the specificity for distinguishing presence of a low-level mutation using PCR may be achieved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild- type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) using upstream PCR primers containing a few extra bases and a blocking group, which is liberated to form a free 3’OH by cleavage with a nuclease only when hybridized to the complementary target (e.g., RNase H2 and a ribonucleotide base).
- the complementary target e.g., RNase H2 and a ribonucleotide base
- the fourth theme is either suppressed (reduced) amplification or incorrect (false) amplification due to unused primers in the reaction.
- One approach to eliminate such unused primers is to capture genomic or target or amplified target DNA on a solid support, allow ligation probes to hybridize and ligate, and then remove probes or products that are not hybridized.
- Alternative solutions include pre-amplification, followed by subsequent nested LDR and/or PCR steps, such that there is a second level of selection in the process.
- the fifth theme is carryover prevention.
- Carryover signal may be eliminated by standard uracil incorporation during the universal PCR amplification step, and by using UDG (and optionally AP endonuclease) in the pre-amplification workup procedure. Incorporation of carryover prevention is central to the methods of the present application as described in more detail below.
- the initial PCR amplification is performed using incorporation of uracil.
- the LDR reaction is performed with LDR probes lacking uracil. Thus, when the LDR products are subjected to real-time PCR quantification, addition of UDG destroys the initial PCR products, but not the LDR products.
- LDR is a linear process and the tag primers use sequences absent from the human genome, accidental carryover of LDR products back to the original PCR will not cause template-independent amplification.
- Additional schemes to provide carryover prevention with methylated targets include use of restriction endonucleases to destroy unmethylated DNA prior to PCTR amplification, or capturing and enriching methylated DNA using methyl-specific DNA binding proteins or antibodies.
- the sixth theme is achieving even amplification of many mutation-specific or methylation-specific targets in the multiplexed reaction.
- One approach as already described above, is to perform limited initial PCR amplifications (8 to 12, or 12 to 20 cycles). However, sometimes different products amplify at different rates, especially when using mutation-or methylation-specific primers, or when using blocking LNA or PNA probes or other means to suppress amplification of wild-type DNA. This is because a regular PCR reaction has both forward and reverse primers working simultaneously. Although there may be preferential amplification using as an example a forward methylation-specific primer (i.e.
- the reverse primer will amplify both methylated and un-methylated DNA (again, after bisulfite treatment), and thus will magnify differences in initial rates of forward primer amplification. Further, and this also holds when using mutation-specific forward primers, the use of non-selecting reverse primers means that initial amplification products still contain substantial amounts of wild-type DNA sequence, which may lead to undesired false-positives in subsequent amplification steps.
- One approach is to perform an initial single-sided linear amplification, using primers that amplify only one strand of target DNA. This is particularly useful when amplifying bisulfite-treated DNA, where the two resultant strands are no longer complementary to each other.
- An important variation of this theme destroys the initial target DNA after the linear amplification step.
- This may be achieved by incorporating one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion.
- a-thio-dNTPs that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion.
- the original bisulfite- converted DNA may be destroyed using UDG.
- a first aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences of other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules are then provided.
- One or more primary oligonucleotide primer sets are also provided.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the sample, the one or more first primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixtures and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising nucleotide sequences complementary to the target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more second primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixtures, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- dU deoxyuracil
- the method further comprises subjecting the one or more first polymerase chain reaction mixtures to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more first polymerase chain reaction products comprising the target nucleotide sequence or a complement thereof.
- One or more oligonucleotide probe sets are then provided.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence- specific portion and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on a complementary target nucleotide sequence of a secondary extension product.
- the one or more first polymerase chain reaction products are blended with a ligase, and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures.
- the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets are ligated together, when hybridized to complementary sequences, to form ligated product sequences in the ligation reaction mixtures wherein each ligated product sequence comprises the 5’ primer-specific portion, the target- specific portions, and the 3’ primer-specific portion.
- the method further includes providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more second polymerase chain reaction products.
- dU deoxyuracil
- the method further comprises detecting and distinguishing the one or more second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- Figures 2 and 3 illustrate various embodiments of this aspect of the present application, abbreviated as exPCR-LDR-qPCR carryover prevention reaction to detect low-level mutations (exPCR is an abbreviation for one-sided extension using primers to one strand of a locus, followed by PCR– using either the same primers in the initial extension, or additional primers for the PCR step).
- Genomic or cfDNA is isolated ( Figure 2, step A), and the isolated DNA sample is treated with UDG to digest dU containing nucleic acid molecules that may be present in the sample ( Figure 2, step B).
- Suitable enzymes include, without limitation, E.
- UDG coli uracil DNA glycosylase
- hSMUG1 Human single-strand-selective monofunctional uracil-DNA Glycosylase
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising wild-type sequence, and a deoxynucleotide mix that includes dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H (star symbol) removes the RNA base to liberate a 3’OH group which is a few bases upstream of the mutation, and suitable for polymerase extension ( Figure 2 or 3, step B; see e.g., Dobosy et. al.“RNase H-Dependent PCR (rhPCR): Improved Specificity and Single Nucleotide Polymorphism Detection Using Blocked Cleavable Primers,” BMC Biotechnology 11(80):1011 (2011), which is hereby incorporated by reference in its entirety).
- a blocking LNA or PNA probe comprising wild-type sequence that partially overlaps with the upstream PCR primer will preferentially compete in binding to wild-type sequence over the upstream primer, but not as much to mutant DNA, and thus suppresses extension of wild- type DNA during each round of primer extension.
- Sample is optionally aliquoted into 12, 24, 36, 48, or 96 wells prior to the initial extension step. Subsequently, the locus-specific downstream primers are added, followed by limited (8 to 20 cycles) or full (20-40 cycles) PCR. Optionally, the downstream primers contain identical 8-11 base tails to prevent primer dimers.
- the initial extension products incorporate one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion.
- the down-stream locus-specific primers (optionally containing identical 8-11 base tails) are added, again followed by limited (8 to 20 cycles) or full (20-40 cycles) PCR.
- the amplified products contain dU as shown in Figure 2 or 3, step D, which allows for subsequent treatment with UDG or a similar enzyme for carryover prevention.
- step E target-specific oligonucleotide probes are hybridized to the amplified products and ligase (filled circle) covalently seals the two oligonucleotides together when hybridized to their complementary sequence.
- the upstream oligonucleotide probe having a sequence specific for detecting the mutation of interest further contains a 5’ primer-specific portion (Ai) to facilitate subsequent detection of the ligation product.
- the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses ligation to wild-type target sequence if present after the enrichment of mutant sequence during the PCR amplification step.
- the downstream oligonucleotide probe having a sequence common to both mutant and wild-type sequences contains a 3’ primer-specific portion (Ci’) that, together with the 5’ primer specific portion (Ai) of the upstream probe having a sequence specific for detecting the mutation, permit subsequent amplification and detection of only mutant ligation products.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- r RNA base
- step F target-specific oligonucleotide probes are hybridized to the amplified products and ligase (filled circle) covalently seals the two oligonucleotides together when hybridized to their complementary sequence.
- the upstream oligonucleotide probe contains a 5’ primer-specific portion (Ai) and the downstream
- oligonucleotide probe contains a 3’ primer-specific portion (Ci’) that permits subsequent amplification of the ligation product.
- the ligation products are aliquoted into separate wells, micro-pores or droplets containing one or more tag-specific primer pairs, each pair comprising matched primers Ai and Ci, treated with UDG or similar enzyme to remove dU containing amplification products or contaminants, PCR amplified, and detected.
- steps G & H detection of the ligation product can be carried out using traditional TaqManTM detection assay (see U.S. Patent No.6,270,967 to Whitcombe et al., and U.S.
- Patent No.7,601,821 to Anderson et al. which are hereby incorporated by reference in their entirety).
- an oligonucleotide probe spanning the ligation junction is used in conjunction with primers suitable for hybridization on the primer-specific portions of the ligation products for amplification and detection.
- the TaqManTM probe contains a fluorescent reporter group on one end (F1) and a quencher molecule (Q) on the other end that are in close enough proximity to each other in the intact probe that the quencher molecule quenches fluorescence of the reporter group.
- F1 fluorescent reporter group on one end
- Q quencher molecule
- the TaqManTM probe and upstream primer hybridize to their complementary regions of the ligation product.
- the 5’ ® 3’ nuclease activity of the polymerase extends the hybridized primer and liberates the fluorescent group of the TaqManTM probe to generate a detectable signal ( Figure 2, step H).
- the Taqman probe contains a second quencher group (ZEN) about 9 bases in from the fluorescent reporter group, and the probe is designed such that the ZEN group is at or adjacent to the mutant base.
- ZEN second quencher group
- target-specific oligonucleotide probes are hybridized to the amplified products and ligase (filled circle) covalently seals the two oligonucleotides together when hybridized to their complementary sequence.
- the upstream oligonucleotide probe having a sequence specific for detecting the mutation of interest further contains a 5’ primer-specific portion (Ai) to facilitate subsequent detection of the ligation product.
- the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses ligation to wild-type target sequence if present after the enrichment of mutant sequence during the PCR amplification step.
- the downstream oligonucleotide probe having a sequence common to both mutant and wild-type sequences contains a 3’ primer-specific portion (Bi-Ci’) that, together with the 5’ primer specific portion (Ai) of the upstream probe having a sequence specific for detecting the mutation, permit subsequent amplification and detection of only mutant ligation products.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- r RNA base
- the ligation probes are designed to contain UniTaq primer and tag sequences to facilitate detections.
- the UniTaq system is fully described in U.S. Patent Application Publication No.2011/0212846 to Spier, which is hereby incorporated by reference in its entirety.
- the UniTaq system involves the use of three unique“tag” sequences, where at least one of the unique tag sequences (Ai) is present in the first oligonucleotide probe, and the second and third unique tag portions (Bi’ and Ci’) are in the second oligonucleotide probe sequence as shown in Figure 3, step D & E.
- the resulting ligation product Upon ligation of oligonucleotide probes in a probe set, the resulting ligation product will contain the Ai sequence—target specific sequences—Bi’ sequence—Ci’ sequence.
- the essence of the UniTaq approach is that both oligonucleotide probes of a ligation probe set need to be correct in order to get a positive signal, which allows for highly multiplexed nucleic acid detection. For example, and as described herein, this is achieved by requiring hybridization of two parts, i.e., two of the tags, to each other.
- the sample Prior to detecting the ligation product, the sample is treated with UDG to destroy original target amplicons allowing only authentic ligation products to be detected. Following ligation, the ligation products are aliquoted into separate wells, micro-pores or droplets containing one or more tag-specific primer pairs.
- the ligation product containing Ai (a first primer-specific portion), Bi’ (a UniTaq detection portion), and Ci’ (a second primer-specific portion) is primed on both strands using a first oligonucleotide primer having the same nucleotide sequence as Ai, and a second oligonucleotide primer that is complementary to Ci’ (i.e., Ci).
- the first oligonucleotide primer also includes a UniTaq detection probe (Bi) that has a detectable label F1 on one end and a quencher molecule (Q) on the other end (F1-Bi-Q-Ai).
- a polymerase- blocking unit e.g., HEG, THF, Sp-18, ZEN, or any other blocker known in the art that is sufficient to stop polymerase extension.
- a ZEN quencher group is also positioned about 9 bases from the fluorescent reporter group to assure more complete quenching. PCR amplification results in the formation of double stranded products as shown in Figure 3, step G).
- a polymerase-blocking unit prevents a polymerase from copying the 5' portion (Bi) of the first universal primer, such that the bottom strand of product cannot form a hairpin when it becomes single-stranded. Formation of such a hairpin would result in the 3' end of the stem annealing to the amplicon such that polymerase extension of this 3' end would terminate the PCR reaction.
- the double stranded PCR products are denatured, and when the temperature is subsequently decreased, the upper strand of product forms a hairpin having a stem between the 5' portion (Bi) of the first oligonucleotide primer and portion Bi’ at the opposite end of the strand ( Figure 3, step H). Also, during this step, the second oligonucleotide primer anneals to the 5’- primer specific portion (Ci’) of the hairpinned product.
- 5' nuclease activity of the polymerase cleaves the detectable label D1 or the quencher molecule from the 5' end of the amplicon, thereby increasing the distance between the label and the quencher and permitting detection of the label.
- Ligases suitable for ligating oligonucleotide probes of a probe set together include, without limitation Thermus aquaticus ligase, E. coli ligase, T4 DNA ligase, T4 RNA ligase, Taq ligase, 9 N ligase, and Pyrococcus ligase, or any other thermostable ligase known in the art.
- the nuclease-ligation process of the present application can be carried out by employing an oligonucleotide ligation assay (OLA) reaction (see Landegren, et al., "A Ligase-Mediated Gene Detection Technique," Science 241:1077-80 (1988); Landegren, et al., “DNA Diagnostics -- Molecular Techniques and Automation,” Science 242:229-37 (1988); and U.S. Patent No.
- OVA oligonucleotide ligation assay
- LDR ligation detection reaction
- LCR ligation chain reaction
- oligonucleotide probes see e.g., WO 90/17239 to Barany et al, which is hereby incorporated by reference in its entirety).
- the oligonucleotide probes of a probe sets can be in the form of ribonucleotides, deoxynucleotides, modified ribonucleotides, modified deoxyribonucleotides, peptide nucleotide analogues, modified peptide nucleotide analogues, modified phosphate-sugar-backbone oligonucleotides, nucleotide analogs, and mixtures thereof.
- the hybridization step in the ligase detection reaction discriminates between nucleotide sequences based on a distinguishing nucleotide at the ligation junctions.
- the difference between the target nucleotide sequences can be, for example, a single nucleic acid base difference, a nucleic acid deletion, a nucleic acid insertion, or rearrangement. Such sequence differences involving more than one base can also be detected.
- the oligonucleotide probe sets have substantially the same length so that they hybridize to target nucleotide sequences at substantially similar hybridization conditions.
- Ligase discrimination can be further enhanced by employing various probe design features.
- an intentional mismatch or nucleotide analogue e.g., Inosine,
- Nitroindole or Nitropyrrole
- RNA bases that are cleaved by RNases can be incorporated into the oligonucleotide probes to ensure template-dependent product formation.
- This approach can be used to generate either ligation-competent 3’OH (for standard DNA ligases) or 5’-P, or both, in the latter case, provided a ligase that can ligate 5’-RNA base is utilized.
- abasic sites e.g., internal abasic furan or oxo-G. These abnormal“bases” are removed by specific enzymes to generate ligation- competent 3’-OH or 5’P sites.
- Endonuclease IV, Tth EndoIV (NEB) will remove abasic residues after the ligation oligonucleotides anneal to the target nucleic acid, but not from a single-stranded DNA.
- Tth EndoIV Tth EndoIV
- Ligation discrimination can also be enhanced by using the coupled nuclease- ligase reaction described in WO2013/123220 to Barany et al. or U.S. Patent Application Publication No.2006/0234252 to Anderson et al., which are hereby incorporated by reference in their entirety.
- the first oligonucleotide probe bears a ligation competent 3’ OH group while the second oligonucleotide probe bears a ligation incompetent 5’ end (i.e., an oligonucleotide probe without a 5’ phosphate).
- the oligonucleotide probes of a probe set are designed such that the 3’-most base of the first oligonucleotide probe is overlapped by the immediately flanking 5’-most base of the second oligonucleotide probe that is complementary to the target nucleic acid molecule.
- the overlapping nucleotide is referred to as a“flap”.
- the phosphodiester bond immediately upstream of the flap nucleotide of the second oligonucleotide probe is discriminatingly cleaved by an enzyme having flap endonuclease (FEN) or 5’ nuclease activity.
- FEN flap endonuclease
- flap endonucleases or 5’ nucleases that are suitable for cleaving the 5’ flap of the second oligonucleotide probe prior to ligation include, without limitation, polymerases with 5’ nuclease activity such as E.coli DNA polymerase and polymerases from Taq and T. thermophilus, as well as T4 RNase H and TaqExo.
- the second probe of the probe set has a 3 ⁇ primer-specific portion, a target specific portion, and a 5 ⁇ nucleotide sequence, where the 5 ⁇ nucleotide sequence is complementary to at least a portion of the 3 ⁇ primer-specific portion, and where the 5 ⁇ nucleotide sequence hybridizes to its complementary portion of the 3 ⁇ primer-specific portion to form a hair-pinned second oligonucleotide probe when the second probe is not hybridized to a target nucleotide sequence.
- incorporation of a matched base or nucleotide analogues e.g., -amino-dA or 5-propynyl-dC
- incorporation of a matched base or nucleotide analogues improves stability and may improve discrimination of such frameshift mutations from wild-type sequences.
- thiophosphate-modified nucleotides downstream from the desired scissile phosphate bond of the second oligonucleotide probe will prevent inappropriate cleavage by the 5’ nuclease enzyme when the probes are hybridized to wild-type DNA, and thus reduce false-positive ligation on wild-type target.
- thiophosphate-modified nucleotides upstream from the desired scissile phosphate bond of the second oligonucleotide probe will prevent inappropriate cleavage by the 5’ nuclease enzyme when the probes are hybridized to wild-type DNA, and thus reduce false-positive ligation on wild-type target.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, one or more nucleases capable of digesting nucleic acid molecules not comprising modified nucleotides, and one or more first primary oligonucleotide primer(s) are provided.
- the one or more first primary oligonucleotide primer(s) comprise a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence.
- the sample, the one or more first primary oligonucleotide primers, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix that comprises one or more modified nucleotides that protect extension products but not target DNA from nuclease digestion, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixture and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the target nucleotide sequence.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a first 5’ primer-specific portion and a 3’ portion that is complementary to a portion of a primary extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a second 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary oligonucleotide primer sets, the one or more nucleases, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the first polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more first polymerase chain reaction products comprising the first 5’ primer-specific portion, a target-specific nucleotide sequence or a complement thereof, and a complement of the second 5’ primer-specific portion.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the first 5’ primer-specific portion of the one or more first polymerase chain reaction products and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the one or more first polymerase chain reaction products.
- the one or more first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU) containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more second polymerase chain reaction products.
- the method further involves detecting and distinguishing the one or more second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- FIG. 4-8 illustrate various embodiments of this aspect of the present application.
- Figure 4 illustrates an exemplary exPCR-qPCR carryover prevention reaction to detect low-level mutations.
- Genomic or cfDNA is isolated ( Figure 4, step A), and the isolated DNA sample is treated with UDG to digest dU containing nucleic acid molecules that may be present in the sample ( Figure 4, step A).
- the sample is then subject to a linear amplification reaction, e.g., one or more polymerase extension reactions to generate complementary copies of mutation containing regions of interest.
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising wild-type sequence, and a deoxynucleotide mix that includes one of more modified nucleotides.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- a 3’ cleavable blocking group e.g. C3 spacer
- RNA base r
- RNase H star symbol
- a blocking LNA or PNA probe comprising wild-type sequence that partially overlaps with the upstream PCR primer will preferentially compete in binding to wild-type sequence over the upstream primer, but not as much to mutant DNA, and thus suppresses extension of wild-type DNA during each round of primer extension.
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the initial extension products incorporate one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion ( Figure 4, step C).
- a-thio-dNTPs that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion
- Using just upstream locus- specific primers in the presence of blocking LNA or PNA probes enriches for extension of mutation-containing products with each extension cycle.
- the exonuclease digestion destroys wild-type DNA present in the original genomic or cfDNA sample, and thus the enriched extension products will not be diluted by subsequent extension or amplification off original wild- type DNA (see step D below).
- step D mutation-specific and locus-specific oligonucleotide primers are added to then perform limited cycle nested PCR to amplify the mutation-containing sequence, if present in the sample.
- the upstream mutation-specific primer having a sequence specific for detecting the mutation of interest further contains a 5’ primer- specific portion (Ai) to facilitate subsequent detection of the nested PCR product.
- the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses extension of wild-type target sequence if present after the enrichment of mutant sequence during the initial extension step.
- the reverse locus-specific primer having a sequence common to both mutant and wild-type sequences contains a 5’ primer-specific portion (Ci) that, together with the 5’ primer specific portion (Ai) of the upstream primer having a sequence specific for detecting the mutation, permit subsequent amplification and detection of only mutant PCR products.
- another layer of specificity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the mutation-specific and locus-specific primers.
- Blk 3’ e.g. C3 spacer
- r RNA base
- the liberated 3’OH base is a few bases upstream from the mutation position, and thus would extend both wild-type and mutant sequences if cleaved (although the blocking LNA or PNA should limit cleavage of primer hybridized to wild-type sequence).
- the mutation-specific base of the primer is at the 3’OH base, such that extension on wild-type sequence would be less likely, since the base is mismatched.
- the specificity for polymerase extension of mutant over wild-type sequence may be further improved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild-type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) avoiding G:T or T:G mismatches between primer and wild-type sequence at the 3’OH base.
- nested PCR products comprise a 5’ primer-specific portion (Ai) target-specific sequence, and a 3’ primer-specific portion (Ci’) that permits subsequent amplification of the nested PCR product.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing one or more tag- specific primer pairs, each pair comprising matched primers Ai and Ci, treated with UDG or similar enzyme to remove dU containing amplification products or contaminants, PCR amplified, and detected.
- steps F & G detection of the ligation product can be carried out using traditional TaqManTM detection assay (see U.S.
- an oligonucleotide probe spanning the mutation-specific region is used in conjunction with primers suitable for hybridization on the primer-specific portions of the nested PCR products for amplification and detection.
- the TaqManTM probe contains a fluorescent reporter group on one end (F1) and a quencher molecule (Q) on the other end that are in close enough proximity to each other in the intact probe that the quencher molecule quenches fluorescence of the reporter group.
- the TaqManTM probe and upstream primer hybridize to their complementary regions of the nested PCR product.
- the 5’ ® 3’ nuclease activity of the polymerase extends the hybridized primer and liberates the fluorescent group of the TaqManTM probe to generate a detectable signal ( Figure 4, step G).
- the TaqManTM probe contains a second quencher group (ZEN) about 9 bases in from the fluorescent reporter group, and the probe is designed such that the ZEN group is at or adjacent to the mutant base.
- ZEN second quencher group
- Figure 5 illustrates an another exPCR-qPCR carryover prevention reaction to detect low-level mutations.
- Genomic or cfDNA is isolated ( Figure 5, step A), and the isolated DNA sample is treated with UDG to digest dU containing nucleic acid molecules that may be present in the sample ( Figure 5, step A).
- the sample is then subject to a linear amplification reaction, e.g., one or more polymerase extension reactions to generate complementary copies of mutation containing regions of interest.
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising wild-type sequence, and a deoxynucleotide mix that includes one of more modified nucleotides.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- a 3’ cleavable blocking group e.g. C3 spacer
- RNA base r
- RNase H star symbol
- a blocking LNA or PNA probe comprising wild-type sequence that partially overlaps with the upstream PCR primer will preferentially compete in binding to wild-type sequence over the upstream primer, but not as much to mutant DNA, and thus suppresses extension of wild-type DNA during each round of primer extension.
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the initial extension products incorporate one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion ( Figure 5, step C).
- a-thio-dNTPs that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion
- Using just upstream locus- specific primers in the presence of blocking LNA or PNA probes enriches for extension of mutation-containing products with each extension cycle.
- the exonuclease digestion destroys wild-type DNA present in the original genomic or cfDNA sample, and thus the enriched extension products will not be diluted by subsequent extension or amplification off original wild- type DNA (see step D below).
- step D mutation-specific and locus-specific oligonucleotide primers are added to then perform limited cycle nested PCR to amplify the mutation-containing sequence, if present in the sample.
- the upstream mutation-specific primer having a sequence specific for detecting the mutation of interest further contains a 5’ primer- specific portion (Ai) to facilitate subsequent detection of the nested PCR product.
- the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses extension of wild-type target sequence if present after the enrichment of mutant sequence during the initial extension step.
- the reverse locus-specific primer having a sequence common to both mutant and wild-type sequences contains a 3’ primer-specific portion (Bi-Ci) that, together with the 5’ primer specific portion (Ai) of the upstream primer having a sequence specific for detecting the mutation, permit subsequent amplification and detection of only mutant PCR products.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- r RNA base
- step D Upon target-specific hybridization, RNase H (star symbol) removes the RNA base to generate a polymerase extension competent 3’OH group ( Figure 5, step D).
- the liberated 3’OH base is a few bases upstream from the mutation position, and thus would extend both wild-type and mutant sequences if cleaved (although the blocking LNA or PNA should limit cleavage of primer hybridized to wild-type sequence).
- the mutation-specific base of the primer is at the 3’OH base, such that extension on wild-type sequence would be less likely, since the base is mismatched.
- the specificity for polymerase extension of mutant over wild-type sequence may be further improved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild-type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) avoiding G:T or T:G mismatches between primer and wild-type sequence at the 3’OH base.
- nested PCR products comprise a 5’ primer-specific portion (Ai) target-specific sequence, and a 3’ primer-specific portion (Bi’-Ci’) that permits subsequent amplification of the nested PCR product.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing one or more tag- specific primer pairs, each pair comprising matched primers F1-Bi-Q-Ai and Ci, treated with UDG or similar enzyme to remove dU containing amplification products or contaminants, PCR amplified ( Figure 5, step F), and detected.
- PCR amplification results in the formation of double stranded products as shown in Figure 5, step G.
- a polymerase-blocking unit prevents a polymerase from copying the 5' portion (Bi) of the first universal primer, such that the bottom strand of product cannot form a hairpin when it becomes single-stranded. Formation of such a hairpin would result in the 3' end of the stem annealing to the amplicon such that polymerase extension of this 3' end would terminate the PCR reaction.
- the double stranded PCR products are denatured, and when the temperature is subsequently decreased, the upper strand of product forms a hairpin having a stem between the 5' portion (Bi) of the first oligonucleotide primer and portion Bi’ at the opposite end of the strand ( Figure 5, step H). Also, during this step, the second oligonucleotide primer anneals to the 5’- primer specific portion (Ci’) of the hairpinned product.
- 5' nuclease activity of the polymerase cleaves the detectable label D1 or the quencher molecule from the 5' end of the amplicon, thereby increasing the distance between the label and the quencher and permitting detection of the label.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences of other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences of other parent nucleic acid molecules by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules and one or more nucleases capable of digesting nucleic acid molecules present not comprising modified nucleotides are provided.
- the method also involves providing one or more primary oligonucleotide primer sets.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the parent nucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the sample, the one or more first primary oligonucleotide primers of the primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix that comprises one or more modified nucleotides that protect extension product but not target DNA from nuclease digestion, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase extension reaction mixtures and for carrying out one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the target nucleotide sequence.
- the method further comprises blending the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more second primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more nucleases, a deoxynucleotide mix, and a DNA polymerase to form one or more first polymerase chain reaction mixtures.
- the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the target nucleotide sequence or a complement thereof.
- One or more secondary oligonucleotide primer sets are then provided.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the first polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more nucleotides, one or more copy numbers, one or more transcript sequences, and/or one or more methylated residues.
- FIGS 6, 7, and 8 illustrate various embodiments of this aspect of the present application.
- Figure 6 illustrates another exemplary exPCR-qPCR carryover prevention reaction to detect low-level mutations.
- Genomic or cfDNA is isolated ( Figure 6, step A), and the isolated DNA sample is treated with UDG to digest dU containing nucleic acid molecules that may be present in the sample ( Figure 6, step A).
- the sample is then subject to a linear amplification reaction, e.g., one or more polymerase extension reactions to generate complementary copies of mutation containing regions of interest.
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising wild-type sequence, and a deoxynucleotide mix that includes one of more modified nucleotides.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- a 3’ cleavable blocking group e.g. C3 spacer
- RNA base r
- RNase H star symbol
- a blocking LNA or PNA probe comprising wild-type sequence that partially overlaps with the upstream PCR primer will preferentially compete in binding to wild-type sequence over the upstream primer, but not as much to mutant DNA, and thus suppresses extension of wild-type DNA during each round of primer extension.
- the initial extension products incorporate one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion ( Figure 6, step B).
- modified nucleotides such as a-thio-dNTPs
- Figure 6, step B Optionally aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the locus-specific downstream primers are added, followed by limited cycle PCR (8 to 12 cycles, Figure 6, step C).
- the locus-specific downstream primers are approximately 20 to 40 bases downstream from the locus- specific upstream primers.
- the downstream primers contain identical 8-11 base tails to prevent primer dimers.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, mutation-specific, and locus- specific primers, to amplify the mutation-containing sequence, if present in the sample ( Figure 6, step D).
- Figure 6, step D the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses extension of wild-type target sequence if present after the enrichment of mutant sequence during the initial extension-amplification steps.
- another layer of specificity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g.
- RNA base in the mutation-specific and locus-specific primers.
- RNase H star symbol
- the liberated 3’OH is a few bases upstream from the mutation position, and thus would extend both wild-type and mutant sequences if cleaved (although the blocking LNA or PNA should limit cleavage of primer hybridized to wild-type sequence).
- the mutation-specific base of the primer is at the 3’OH, such that extension on wild-type sequence would be less likely, since the base is mismatched.
- the specificity for polymerase extension of mutant over wild-type sequence may be further improved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild-type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) avoiding G:T or T:G mismatches between primer and wild-type sequence at the 3’OH base.
- the TaqManTM probe spans the mutation region and contains a fluorescent reporter group on one end (F1) and a quencher molecule (Q) on the other end that are in close enough proximity to each other in the intact probe that the quencher molecule quenches fluorescence of the reporter group.
- F1 fluorescent reporter group on one end
- Q quencher molecule
- the TaqManTM probe and upstream primer hybridize to their complementary regions of the initial PCR product.
- the 5’ ® 3’ nuclease activity of the polymerase extends the hybridized primer and liberates the fluorescent group of the TaqManTM probe to generate a detectable signal (Figure 6, step E).
- the TaqmanTM probe contains a second quencher group (ZEN) about 9 bases in from the fluorescent reporter group, and the probe is designed such that the ZEN group is at or adjacent to the mutant base.
- ZEN second quencher group
- Figures 7 and 8 illustrate additional exemplary exPCR-qPCR carryover prevention reaction to detect low-level mutations.
- Genomic or cfDNA is isolated ( Figure 7 and 8, step A), and the isolated DNA sample is treated with UDG to digest dU containing nucleic acid molecules that may be present in the sample ( Figure 7, step A).
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising wild-type sequence, and a deoxynucleotide mix that includes one of more modified nucleotides.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g.
- RNA base in the upstream primer.
- RNase H star symbol
- a blocking LNA or PNA probe comprising wild- type sequence that partially overlaps with the upstream PCR primer will preferentially compete in binding to wild-type sequence over the upstream primer, but not as much to mutant DNA, and thus suppresses extension of wild-type DNA during each round of primer extension.
- the initial extension products incorporate one or more modified nucleotides, such as a-thio-dNTPs, that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion ( Figure 7 and 8, step B).
- modified nucleotides such as a-thio-dNTPs
- a-thio-dNTPs that protect the initial extension products (but not the original cfDNA or genomic DNA) from exonuclease I digestion
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, mutation-specific primers comprising 5’ primer-specific portions (Ai), locus-specific primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci. These primers combine to amplify the mutation-containing sequence, if present in the sample ( Figure 7, step C).
- the presence of blocking LNA or PNA probe comprising wild-type sequence suppresses extension of wild-type target sequence if present after the enrichment of mutant sequence during the initial extension-amplification steps ( Figure 7, step B).
- step C of this Figure another layer of specificity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the mutation- specific and locus-specific primers.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- RNA base r
- RNase H star symbol
- the liberated 3’OH is a few bases upstream from the mutation position, and thus would extend both wild-type and mutant sequences if cleaved (although the blocking LNA or PNA should limit cleavage of primer hybridized to wild-type sequence).
- the mutation-specific base of the upstream primer is at the 3’OH, such that extension on wild- type sequence would be less likely, since the base is mismatched.
- the products can be detected by the pairs of matched primers Ai and Ci, and TaqManTM probes that span the ligation junction as described supra for Figure 4 steps F-G (see Figure 7, steps E & F), or using other suitable means known in the art.
- the specificity for polymerase extension of mutant over wild-type sequence may be further improved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild-type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) avoiding G:T or T:G mismatches between primer and wild-type sequence at the 3’OH base.
- the longer target-specific primers are at a significantly lower concentration than the TaqmanTM probe and tag-specific primers (Ai, Ci), such that the longer mutation-specific primers are depleted, allowing the TaqmanTM probe and tag-specific primers to hybridize and enable target-dependent detection.
- the PCR products are aliquoted into separate wells, micro-pores, or droplets containing TaqmanTM probes, mutation-specific primers comprising 5’ primer-specific portions (Ai), locus-specific primers comprising 5’ primer-specific portions (Bi-Ci) and matching UniTaq primers F1-Bi-Q-Ai and Ci.
- These primers combine to amplify the mutation-containing sequence, if present in the sample ( Figure 8, step C).
- the presence of blocking LNA or PNA probe comprising wild- type sequence suppresses extension of wild-type target sequence if present after the enrichment of mutant sequence during the initial extension-amplification steps ( Figure 8, step B).
- step C of this Figure another layer of specificity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the mutation-specific and locus-specific primers.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- RNA base r
- RNase H star symbol
- the liberated 3’OH is a few bases upstream from the mutation position, and thus would extend both wild-type and mutant sequences if cleaved (although the blocking LNA or PNA should limit cleavage of primer hybridized to wild-type sequence).
- the mutation-specific base of the upstream primer is at the 3’OH, such that extension on wild-type sequence would be less likely, since the base is mismatched.
- the products can be detected by the pairs of matched UniTaq primers (i.e. F1-Bi-Q-Ai and Ci), as described supra for Figure 5 steps F-H (see Figure 8, steps E-G), or using other suitable means known in the art.
- the specificity for polymerase extension of mutant over wild-type sequence may be further improved by: (i) using mutation-specific PCR Primers containing a mismatch in the 2 nd or 3 rd position from the 3’OH base, (ii) using LNA or PNA probes to wild-type sequence that would reduce hybridization of mutation-specific PCR primers to wild-type sequences, (iii) using PCR primers to wild-type sequence that are blocked and do not undergo additional amplification, and (iv) avoiding G:T or T:G mismatches between primer and wild-type sequence at the 3’OH base.
- the longer target-specific primers are at a significantly lower concentration than the UniTaq primers (F1-Bi-Q-Ai, Ci), such that the longer mutation-specific primers are depleted, allowing the UniTaq primers to hybridize and enable target-dependent detection.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues and subjecting the nucleic acid molecules in the sample to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite- treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite-treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- dU deoxyuracil
- the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the bisulfite-treated target nucleotide sequence or a complement thereof.
- the method further involves providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ bisulfite-treated target nucleotide sequence-specific or complement sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ bisulfite-treated target nucleotide sequence- specific or complement sequence-specific portion and a 3’ primer-specific portion, and wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on a complementary nucleotide sequence of a first polymerase chain reaction product.
- the first polymerase chain reaction products are blended with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures.
- the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets are ligated together, when hybridized to complementary sequences, to form ligated product sequences in the ligation reaction mixture wherein each ligated product sequence comprises the 5’ primer-specific portion, the bisulfite-treated target nucleotide sequence-specific or complement sequence- specific portions, and the 3’ primer-specific portion.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming a second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reaction products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Figures 9 and 10 illustrate exPCR-LDR-qPCR carryover prevention reaction to detect low-level methylation in accordance with this aspect of the present application.
- the steps are similar to those steps described for Figure 2, with two key differentiators.
- Bisulfite converts unmethylated cytosines, but not 5- methyl cytosines (5meC) nor 5-hydroxymethyl cytosine (5hmC) into a uracil base, which base- pairs with A.
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- locus-specific downstream primers with optional identical 8-11 base tails
- a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is suitable for polymerase extension ( Figure 9, step B).
- UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the locus-specific upstream primers are added, followed by limited (8 to 20 cycles) or full (20-40 cycles) PCR using a deoxynucleotide mix that includes dUTP ( Figure 9, step C).
- RNase H Upon target-specific hybridization, RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension (Figure 9, step C).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- the downstream primers contain identical 8-11 base tails to prevent primer dimers.
- tails provide the option for asymmetric PCR at the end of the PCR cycles, by raising the hybridization temperature above that for the forward primers, but at or below that for the reverse primers– which at 8-11 bases longer will have higher Tm values. This generates more bottom strand products, which are suitable substrates for the subsequent LDR step.
- the amplified products contain dU as shown in Figure 9, step D, which allows for subsequent treatment with UDG or a similar enzyme for carryover prevention.
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement), and a deoxynucleotide mix that does NOT include dUTP.
- a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement)
- a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 10, step B).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- Add UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- RNA sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- locus-specific downstream primers are added, followed by limited (8 to 20 cycles) or full (20-40 cycles) PCR using a deoxynucleotide mix that includes dUTP ( Figure 14, step C).
- the downstream primers contain identical 8-11 base tails to prevent primer dimers.
- methylation-specific upstream and locus-specific downstream probes containing tails enable formation of a ligation product in the presence of bisulfite converted methylated base-containing PCR products.
- the ligation products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the ligation junction as described supra for Figure 2 (see Figure 9, steps E-H), or using other suitable means known in the art.
- methylation-specific upstream and locus-specific downstream probes containing tails enable formation of a ligation product in the presence of bisulfite converted methylated base-containing PCR products.
- the ligation products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 3, or using other suitable means known in the art.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues.
- the nucleic acid molecules in the sample are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules are provided, and one or more first primary oligonucleotide primer(s) are provided.
- Each first primary oligonucleotide primer comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures, and the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, to form primary extension products comprising the complement of the bisulfite-treated target nucleotide sequence.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of the polymerase extension reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)- containing nucleic acid molecules, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting nucleic acid molecules present in the first polymerase chain reaction mixtures, but not primary extension products comprising modified nucleotides and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reactions products comprising a 5’ primer- specific portion of the first secondary oligonucleotide primer, the bisulfite-treated target nucleotide sequence-specific or complement sequence-specific portion, and a complement of the 5’ primer-specific portion of
- oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reactions product sequence.
- the first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further involves detecting and distinguishing the secondary polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Figures 11, 12, 18, and 19 illustrate various embodiments of this aspect of the present application.
- Figure 11 illustrates an exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylations.
- Genomic or cfDNA is isolated and is optionally treated with a DNA repair kit prior to bisulfite conversion (Figure 11, Step A).
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H removes the RNA base to liberate a 3’OH group which is suitable for polymerase extension ( Figure 11, step B). If the locus-specific downstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence. After the extension cycles, add UDG, which destroys the bisulfite converted DNA (but not the primer extension products). Optionally aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement), and a deoxynucleotide mix that does not include dUTP.
- a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement)
- a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 12, step B).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- Add UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- step C bisulfite converted methylation base- specific primers (comprising 5’ primer-specific portions Ai) and bisulfite converted locus- specific primers (comprising 5’ primer-specific portions Ci) are added to then perform limited cycle nested PCR to amplify the bisulfite converted methylation-containing sequence, if present in the sample.
- Blocking LNA or PNA probes comprising the bisulfite converted unmethylated sequence (or its complement) enables amplification of originally methylated but not originally un-methylated allele. Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the bisulfite-converted methylation target regions as described supra for Figure 4 (see Figures 11 and 12, steps D-F), or using other suitable means known in the art.
- bisulfite converted methylation base-specific primers comprising 5’ primer-specific portions Ai
- bisulfite converted locus-specific primers comprising 5’ primer-specific portions Bi-Ci
- Blocking LNA or PNA probes comprising the bisulfite converted unmethylated sequence (or its complement) enables amplification of originally methylated but not originally un-methylated alleles.
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 5, or using other suitable means known in the art.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues.
- the nucleic acid molecules in the sample are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite-treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures.
- the one or more polymerase extension reaction mixtures to are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixture, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- the one or more first polymerase chain reaction mixtures are sujected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising the bisulfite- treated target nucleotide sequence or a complement thereof.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of a first polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a first polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the first polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- FIG. 13-15, 20, and 21 illustrate various embodiments of this aspect of the present application.
- Figure 13 illustrates another exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylation.
- Genomic or cfDNA is isolated, and optionally treated with a DNA repair kit prior to bisulfite conversion ( Figure 13, Step A).
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- RNase H removes the RNA base to liberate a 3’OH group which is suitable for polymerase extension ( Figure 13, step B). If the locus-specific downstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence. After the extension cycles, add UDG, which destroys the bisulfite converted DNA (but not the primer extension products). Optionally aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using locus-specific upstream primers, a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement), and a deoxynucleotide mix that does not include dUTP.
- a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement)
- a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 13, step C).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, bisulfite-converted, methylation base-specific, and bisulfite converted locus-specific primers, to amplify the bisulfite converted methylation-containing sequence, if present in the sample ( Figure 13, step D).
- the bisulfite converted methylation-containing products are amplified and detected as described supra for Figure 6 (see Figure 13, steps D-E), or using other suitable means known in the art.
- Figures 14 and 15 illustrate additional exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylation.
- Genomic or cfDNA is isolated, and optionally treated with a DNA repair kit prior to bisulfite conversion ( Figures 14 and 15, Step A).
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H removes the RNA base to liberate a 3’OH group which is suitable for polymerase extension ( Figure 14, step B). If the locus-specific downstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence. After the extension cycles, add UDG, which destroys the bisulfite converted DNA (but not the primer extension products). Optionally aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using locus- specific upstream primers, a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement), and a deoxynucleotide mix that does not include dUTP.
- a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement)
- a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 14, step C).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- the regions of interest are selectively extended using locus-specific upstream primers, a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement), and a deoxynucleotide mix that does not include dUTP.
- a blocking LNA or PNA probe comprising bisulfite converted unmethylated sequence (or its complement)
- a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- RNase H removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 15, step B).
- a blocking LNA or PNA probe comprising the bisulfite converted unmethylated sequence (or its complement) that partially overlaps with the upstream PCR primer will preferentially compete for binding to the bisulfite converted unmethylated sequence over the upstream primer, thus suppressing amplification of bisulfite converted unmethylated sequence DNA during each round of PCR.
- Add UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- locus-specific downstream primers are added, followed by limited cycle PCR (8 to 12 cycles, Figure 15, step C). If the locus-specific downstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- TaqmanTM probes bisulfite converted methylation base-specific primers comprising 5’ primer- specific portions (Ai), bisulfite converted locus-specific primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci. These primers combine to amplify the bisulfite converted methylation-containing sequence, if present in the sample ( Figures 14 and 15, step D). Blocking LNA or PNA probes comprising the bisulfite converted unmethylated sequence (or its complement) enables amplification of originally methylated but not originally un-methylated allele. Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the bisulfite-converted methylation target regions as described supra for Figure 4 (see Figure 14, steps E-G), or using other suitable means known in the art.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, bisulfite converted methylation base-specific primers comprising 5’ primer-specific portions (Ai), bisulfite converted locus-specific primers comprising 5’ primer-specific portions (Bi-Ci) and matching UniTaq primers F1-Bi-Q-Ai and Ci.
- Blocking LNA or PNA probes comprising the bisulfite converted unmethylated sequence (or its complement) enables amplification of originally methylated but not originally un-methylated alleles.
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 5, or using other suitable means known in the art.
- Figures 16 and 17 illustrate additional exemplary exPCR-LDR-qPCR carryover prevention reactions to detect low-level methylation.
- Genomic or cfDNA is isolated and then either treated with: (i) methyl-sensitive restriction endonucleases, e.g., Bsh1236I (CG ⁇ CG), to completely digest unmethylated DNA and prevent carryover, or (ii) capture and enrich for methylated DNA, (iii) followed by a DNA repair kit ( Figures 16 and 17, step A).
- the DNA is bisulfite-treated to convert unmethylated residues to uracil thereby rendering the double stranded DNA non-complementary.
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- locus-specific downstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using locus-specific upstream primers, and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- RNA base r
- RNase H star symbol
- the locus- specific upstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- the regions of interest are selectively extended using locus-specific upstream primers for bisulfite converted DNA, and a
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H star symbol
- the locus-specific upstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- the locus-specific downstream primers are added, followed by limited (8 to 20 cycles) or full (20-40 cycles) PCR using a deoxynucleotide mix that includes dUTP ( Figure 17, step C).
- the downstream primers contain identical 8-11 base tails to prevent primer dimers.
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the amplified products contain dU as shown in Figure 17, step D, which allows for subsequent treatment with UDG or a similar enzyme for carryover prevention.
- methylation-specific upstream and locus-specific downstream probes containing tails enable formation of a ligation product in the presence of bisulfite converted methylated base-containing PCR products.
- the ligation products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the ligation junction as described supra for Figure 2 (see Figure 16, steps E-H), or using other suitable means known in the art.
- methylation-specific upstream and locus-specific downstream probes containing tails enable formation of a ligation product in the presence of bisulfite converted methylated base-containing PCR products.
- the ligation products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 3, or using other suitable means known in the art.
- Figures 18 and 19 illustrate additional exemplary exPCR-LDR-qPCR carryover prevention reactions to detect low-level methylation.
- Genomic or cfDNA is isolated, and then either treated with: (i) methyl-sensitive restriction endonucleases, e.g., Bsh1236I (CG ⁇ CG), to completely digest unmethylated DNA and prevent carryover, or (ii) capture and enrich for methylated DNA, (iii) followed by a DNA repair kit ( Figures 18 and 19, step A).
- the DNA is bisulfite-treated to convert unmethylated residues to uracil thereby rendering the double stranded DNA non-complementary.
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does NOT include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- locus-specific downstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively extended using locus-specific upstream primers for bisulfite converted DNA, and a
- Blocking LNA or PNA probes comprising the bisulfite converted unmethylated sequence (or its complement) enables amplification of originally methylated but not originally un-methylated alleles.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H star symbol
- Add UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- step C bisulfite converted methylation base-specific primers (comprising 5’ primer-specific portions Ai) and bisulfite converted locus-specific primers (comprising 5’ primer-specific portions Ci) are added to then perform limited cycle nested PCR to amplify the bisulfite converted methylation-containing sequence, if present in the sample.
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the bisulfite-converted methylation target regions as described supra for Figure 4 (see Figure 18, steps D-F), or using other suitable means known in the art.
- bisulfite converted methylation base-specific primers comprising 5’ primer-specific portions Ai
- bisulfite converted locus-specific primers comprising 5’ primer-specific portions Bi-Ci
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 5, or using other suitable means known in the art.
- Figure 20 illustrates another exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylation.
- Genomic or cfDNA is isolated and then either treated with: (i) methyl-sensitive restriction endonucleases, e.g., Bsh1236I (CG ⁇ CG), to completely digest unmethylated DNA and prevent carryover, or (ii) capture and enrich for methylated DNA, (iii) followed by a DNA repair kit ( Figure 20, step A).
- the DNA is bisulfite-treated to convert unmethylated residues to uracil thereby rendering the double stranded DNA non-complementary.
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- locus-specific downstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite- converted unmethylated sequence.
- UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using locus-specific upstream primers, and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H star symbol
- the locus-specific upstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, bisulfite-converted, methylation base-specific, and bisulfite converted locus-specific primers, to amplify the bisulfite converted methylation-containing sequence, if present in the sample ( Figure 20, step D).
- the bisulfite converted methylation-containing products are amplified and detected as described supra for Figure 6 (see Figure 20, steps D-E), or using other suitable means known in the art.
- Figure 21 illustrate additional exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylation.
- Genomic or cfDNA is isolated, and then either treated with: (i) methyl-sensitive restriction endonucleases, e.g., Bsh1236I (CG ⁇ CG), to completely digest unmethylated DNA and prevent carryover, or (ii) capture and enrich for methylated DNA, (iii) followed by a DNA repair kit ( Figure 21, step A).
- the DNA is bisulfite-treated to convert unmethylated residues to uracil thereby rendering the double stranded DNA non-complementary.
- the regions of interest are selectively extended using locus-specific downstream primers (with optional identical 8-11 base tails), and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- locus-specific downstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite- converted unmethylated sequence.
- UDG which destroys the bisulfite converted DNA (but not the primer extension products) is added.
- samples are aliquoted into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using locus- specific upstream primers, and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- a 3’ cleavable blocking group Blk 3’, e.g. C3 spacer
- RNA base r
- RNase H star symbol
- the locus-specific upstream primer covers one or more methylation sites
- another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, bisulfite converted methylation base-specific primers comprising 5’ primer-specific portions (Ai), bisulfite converted locus-specific primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci.
- These primers combine to amplify the bisulfite converted methylation-containing sequence, if present in the sample ( Figure 21, step D).
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the bisulfite-converted methylation target regions as described supra for Figure 4 (see Figure 21, steps E-G), or using other suitable means known in the art.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes, bisulfite converted methylation base-specific primers comprising 5’ primer-specific portions (Ai), bisulfite converted locus-specific primers comprising 5’ primer-specific portions (Bi-Ci) and matching UniTaq primers F1-Bi-Q-Ai and Ci. Primers are unblocked with RNaseH2 only when bound to correct target. Following PCR, the products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 5, or using other suitable means known in the art.
- UniTaq-specific primers i.e., F1-Bi-Q-Ai, Ci
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more parent nucleic acid molecules containing a target nucleotide sequence differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- the method involves providing a sample containing one or more parent nucleic acid molecules potentially containing the target nucleotide sequence differing from the nucleotide sequences in other parent nucleic acid molecules by one or more methylated residues, and subjecting the nucleic acid molecules in the sample to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- One or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample are provided.
- One or more primary oligonucleotide primer sets are also provided. Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a sequence in the bisulfite-treated parent nucleic acid molecules adjacent to the bisulfite-treated target nucleotide sequence containing the one or more methylated residue and (b) a second primary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer.
- the bisulfite treated sample, the one or more first primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more polymerase extension reaction mixtures.
- the one or more polymerase extension reaction mixtures are subjected to conditions suitable for one or more polymerase extension reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming primary extension products comprising the complement of the bisulfite treated target nucleotide sequence.
- the one or more polymerase extension reaction mixtures comprising the primary extension products, the one or more secondary primary oligonucleotide primers of the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the reaction mixture, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures.
- the method further comprises subjecting the one or more first polymerase chain reaction mixtures to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reactions products comprising the bisulfite-treated target nucleotide sequence or a complement thereof.
- One or more secondary oligonucleotide primer sets are provided.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products or their complements and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer- specific portion of the first polymerase chain reaction products or their complements.
- oligonucleotide primer sets the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further involves detecting and distinguishing the second polymerase chain reactions products in the one or more second polymerase chain reaction mixtures to identify the presence of one or more parent nucleic acid molecules containing target nucleotide sequences differing from nucleotide sequences in other parent nucleic acid molecules in the sample by one or more methylated residues.
- Figure 22 illustrates an embodiment of this aspect of the present application.
- Figure 22 illustrates an additional exemplary exPCR-qPCR carryover prevention reaction to detect low-level methylation.
- Genomic or cfDNA is isolated, and then either treated with: (i) methyl-sensitive restriction endonucleases, e.g., Bsh1236I (CG ⁇ CG), to completely digest unmethylated DNA and prevent carryover, or (ii) capture and enrich for methylated DNA, (iii) followed by a DNA repair kit ( Figure 22, step A).
- the DNA is bisulfite-treated to convert unmethylated residues to uracil thereby rendering the double stranded DNA non-complementary.
- the regions of interest are selectively extended using bisulfite converted locus-specific downstream primers comprising 5’ primer-specific portions (Ci for Figure 22), and a deoxynucleotide mix that does not include dUTP.
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the downstream primer.
- Blk 3’ e.g. C3 spacer
- RNA base RNA base
- the locus-specific downstream primer covers one or more methylation sites, and another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- UDG which destroys the bisulfite converted DNA (but not the primer extension products).
- aliquot sample into 12, 24, 36, 48, or 96 wells prior to the initial extension step.
- the regions of interest are selectively amplified in a limited cycle PCR (8-20 cycles) using bisulfite converted methylation base-specific upstream primers comprising 5’ primer-specific portions (Ai), and a
- another layer of selectivity can be incorporated into the method by including a 3’ cleavable blocking group (Blk 3’, e.g. C3 spacer), and an RNA base (r), in the upstream primer.
- Blk 3 cleavable blocking group
- r RNA base
- RNase H star symbol
- RNA base removes the RNA base to liberate a 3’OH group which is a few bases upstream of the bisulfite converted methylated target base, and suitable for polymerase extension ( Figure 22, step C). Since the methylation base-specific upstream primer covers one or more methylation sites, another layer of specificity may be added by using blocking primers whose sequence corresponds to the bisulfite-converted unmethylated sequence.
- the limited cycle PCR products comprise of Ai tag sequence, methylation-specific sequence, and Ci’ tag sequence, and are distributed into wells, micro-pores, or droplets for TaqmanTM reactions.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the bisulfite- converted methylation target regions as described supra for Figure 4 (see Figure 22, steps D-F), or using other suitable means known in the art.
- the limited cycle PCR products comprise of Ai tag sequence, methylation-specific sequence, and Bi’-Ci’ tag sequence, and are distributed into wells, micro- pores, or droplets for TaqmanTM reactions.
- the products are amplified using UniTaq-specific primers (i.e., F1-Bi-Q-Ai, Ci) and detected as described supra for Figure 5, or using other suitable means known in the art.
- the method further comprises contacting the sample with DNA repair enzymes to repair damaged DNA, abasic sites, oxidized bases, or nicks in the DNA.
- the method further comprises contacting the sample with at least a first methylation sensitive enzyme to form a restriction enzyme reaction mixture prior to, or concurrent with, said blending to form one or more polymerase extension reaction mixtures, wherein said first methylation sensitive enzyme cleaves nucleic acid molecules in the sample that contain one or more unmethylated residues within at least one methylation sensitive enzyme recognition sequence, and whereby said detecting involves detection of one or more parent nucleic acid molecules containing the target nucleotide sequence, wherein said parent nucleic acid molecules originally contained one or more methylated residues.
- a“methylation sensitive enzyme” is an endonuclease that will not cleave or has reduced cleavage efficiency of its cognate recognition sequence in a nucleic acid molecule when the recognition sequence contains a methylated residue (i.e., it is sensitive to the presence of a methylated residue within its recognition sequence).
- A“methylation sensitive enzyme recognition sequence” is the cognate recognition sequence for a methylation sensitive enzyme.
- the methylated residue is a 5-methyl-C, within the sequence CpG (i.e., 5-methyl-CpG).
- methylation sensitive restriction endonuclease enzymes that are suitable for use in the methods of the present invention include, without limitation, AciI, HinP1I, Hpy99I, HpyCH4IV, BstUI, HpaII, HhaI, or any combination thereof.
- the method further comprises contacting the sample with an immobilized methylated nucleic acid binding protein or antibody to selectively bind and enrich for methylated nucleic acid in the sample.
- the primers from the one or more primary or secondary oligonucleotide primer sets comprise a portion that has no or one nucleotide sequence mismatch when hybridized in a base-specific manner to the target nucleic acid sequence or
- bisulfite-converted methylated nucleic acid sequence or complement sequence thereof but have one or more additional nucleotide sequence mismatches that interferes with polymerase extension when primers from said one or more primary or secondary oligonucleotide primer sets hybridize in a base-specific manner to a corresponding nucleotide sequence portion in wildtype nucleic acid sequence or bisulfite-converted unmethylated nucleic acid sequence or complement sequence thereof.
- oligonucleotide primer set may have a 3’ portion comprising a cleavable nucleotide or nucleotide analogue and a blocking group, such that the 3’ end of said primer or primers is unsuitable for polymerase extension.
- This embodiment of the method further comprises cleaving the cleavable nucleotide or nucleotide analog of one or both oligonucleotide primers during said hybridization treatment, thereby liberating free 3’OH ends on one or both oligonucleotide primers prior to said extension treatment.
- the cleavable nucleotide comprises one or more RNA bases.
- primers from the one or more primary or secondary oligonucleotide primer sets comprise a sequence that differs from the target nucleic acid sequence or bisulfite-converted methylated nucleic acid sequence or complement sequence thereof, said difference is located two or three nucleotide bases from the liberated free 3’OH end.
- the method further comprises providing one or more blocking oligonucleotide primers comprising one or more mismatched bases at the 3’ end or one or more nucleotide analogs and a blocking group at the 3’ end, such that the 3’ end of said blocking oligonucleotide primer is unsuitable for polymerase extension when hybridized in a base-specific manner to wildtype nucleic acid sequence or bisulfite-converted unmethylated nucleic acid sequence or complement sequence thereof, wherein said blocking oligonucleotide primer comprises a portion having a nucleotide sequence that is the same as a nucleotide sequence portion in the wildtype nucleic acid sequence or bisulfite-converted unmethylated nucleic acid sequence or complement sequence thereof to which the blocking oligonucleotide primer hybridizes but has one or more nucleotide sequence mismatches to a corresponding nucleotide sequence portion in the target nucleic acid sequence or bisulfite-con
- oligonucleotide primers are blended with the sample or subsequent products prior to a polymerase extension reaction, polymerase chain reaction, or ligation reaction, whereby during hybridization the one or more blocking oligonucleotide primers preferentially hybridize in a base-specific manner to a wildtype nucleic acid sequence or bisulfite-converted unmethylated nucleic acid sequence or complement sequence thereof, thereby interfering with polymerase extension or ligation during reaction of a primer or probes hybridized in a base-specific manner to the wildtype sequence or bisulfite-converted unmethylated sequence or complement sequence thereof.
- the first secondary oligonucleotide primer has a 5’ primer-specific portion and the second secondary oligonucleotide primer has a 5’ primer-specific portion
- said one or more secondary oligonucleotide primer sets further comprising a third secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer- specific portion of the first secondary oligonucleotide primer and (d) a fourth secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the second secondary oligonucleotide primer.
- Another aspect of the present application is directed to a method for identifying in a sample, one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- the method involves providing a sample containing one or more parent ribonucleic acid molecules containing a target ribonucleic acid molecule potentially differing in sequence from other parent ribonucleic acid molecules and providing one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU containing nucleic acid molecules potentially present in the sample.
- One or more primary oligonucleotide primer sets are then provided.
- Each primary oligonucleotide primer set comprises (a) a first primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to the RNA sequence in the parent ribonucleic acid molecule adjacent to the target ribonucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of the cDNA extension product formed from the first primary oligonucleotide primer.
- the contacted sample, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix including dUTP, a reverse transcriptase, and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target ribonucleic nucleic acid and to carry out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse
- the method further comprises providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on complementary portions of a reverse
- transcriptase/polymerase product corresponding to the target ribonucleic acid molecule sequence.
- the reverse transcriptase/polymerase products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures, and the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligase reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences, the one or more secondary oligonucleotide primer sets with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming first polymerase chain reaction products.
- the method further comprises detecting and distinguishing the first polymerase chain reaction products, thereby identifying the presence of one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- Figures 23 and 26 illustrate embodiments of this aspect of the present application.
- Figure 23 illustrates an exemplary RT-PCR-LDR-qPCR carryover prevention reaction to detect translocations at the mRNA level.
- fusion mRNA may be isolated from circulating tumor cells, exosomes or from other plasma fractions. For accurate enumeration, aliquot into 12, 24, 36, or 48 wells prior to PCR. For higher copy number, distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells.
- mRNA is isolated ( Figure 23, step A), and treated with UDG for carryover prevention ( Figure 23, step B).
- cDNA is generated using 3’ transcript-specific primers and reverse-transcriptase in the presence of dUTP.
- Suitable reverse transcriptases include but are not limited to Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, New England Biolabs), or Superscript II or III Reverse Transcriptase (Life Technologies).
- Taq polymerase is activated to perform limited cycle PCR amplification (12-20) to maintain relative ratios of different amplicons ( Figure 23, step B).
- the primers contain identical 8-11 base tails to prevent primer dimers.
- PCR products incorporate dUTP, allowing for carryover prevention (Figure 23, step C).
- step D exon junction-specific ligation oligonucleotide probes containing primer-specific portions (Ai, Ci’) suitable for subsequent PCR amplification, hybridize to their corresponding target sequence in a base-specific manner.
- Ligase covalently seals the two oligonucleotides together ( Figure 23, step D), and ligation products are aliquoted into separate wells for detection using tag-primers (Ai, Ci) and TaqManTM probe (F1-Q) which spans the ligation junction ( Figure 23, step E).
- Treat samples with UDG for carryover prevention, which also destroys original target amplicons Figure 23, step E).
- Another aspect of the present application is directed to a method for identifying in a sample, one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequence differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- the method involves providing a sample containing one or more parent ribonucleic acid molecules containing a target ribonucleic acid molecule potentially differing in sequence from other parent ribonucleic acid molecules and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU containing nucleic acid molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets, each primary oligonucleotide primer set comprising (a) a first primary
- oligonucleotide primer that comprises a nucleotide sequence that is complementary to the RNA sequence in the parent ribonucleic acid molecule adjacent to the target nucleotide sequence and (b) a second primary oligonucleotide primer that comprises a nucleotide sequence that is complementary to a portion of the cDNA extension product formed from the first primary oligonucleotide primer.
- the contacted sample, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse- transcription/polymerase chain reaction mixtures, and the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target RNA and to carry out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse-transcription/primary polymerase chain reaction products.
- cDNA complementary deoxyribonucleic acid
- the method futher comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 3’ portion that is complementary to a portion of a reverse-transcription/primary polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a reverse-transcription/primary polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the reverse-transcription/primary polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming first polymerase chain reaction products.
- the method further involves detecting and distinguishing the first polymerase chain reaction products, thereby identifying the presence of one or more parent ribonucleic acid molecules containing a target ribonucleic acid sequences differing from ribonucleic acid sequences of other parent ribonucleic acid molecules in the sample due to alternative splicing, alternative transcript, alternative start site, alternative coding sequence, alternative non-coding sequence, exon insertion, exon deletion, intron insertion, translocation, mutation, or other rearrangement at the genome level.
- FIGS 24, 25, 27, and 28 illustrate various embodiments of this aspect of the present application.
- Figures 24 and 25 illustrate additional exemplary RT-PCR-LDR-qPCR carryover prevention reaction to detect translocations at the mRNA level.
- mRNA is isolated ( Figures 24 and 25, step A), and treated with UDG for carryover prevention ( Figures 24 and 25, step B).
- cDNA is generated using 3’ transcript-specific primers and reverse-transcriptase.
- Taq polymerase is activated to perform limited cycle PCR amplification (8-20) to maintain relative ratios of different amplicons ( Figures 24 and 25, step B).
- the primers contain identical 8-11 base tails to prevent primer dimers and are added only to wells with the anticipated low-copy dilution.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the cDNA fusion junction, cDNA-specific primers, to amplify the junction sequence, if present in the sample ( Figure 24, step C).
- the fusion cDNA products are amplified and detected as described supra for Figure 6 (see Figure 24, steps C-D), or using other suitable means known in the art.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the cDNA fusion junction, cDNA-specific (forward) primers comprising 5’ primer-specific portions (Ai), cDNA-specific (reverse) primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci.
- These primers combine to amplify the fusion cDNA sequence, if present in the sample ( Figure 25, step C).
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the fusion cDNA regions as described supra for Figure 4 (see Figure 25, steps D-F), or using other suitable means known in the art.
- Figure 26 illustrates an exemplary RT-PCR-LDR-qPCR carryover prevention reaction to enumerate mRNA, ncRNA, or lncRNA copy number.
- RNA is isolated from whole blood cells, exosomes, CTCs or other plasma fractions. For accurate enumeration, aliquot into 12, 24, 36, or 48 wells prior to PCR. For higher copy number, distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells.
- mRNA is isolated ( Figure 26, step A), and treated with UDG for carryover prevention ( Figure 26, step B).
- cDNA is generated using 3’ transcript-specific primers and reverse-transcriptase in the presence of dUTP.
- Taq polymerase is activated to perform limited cycle PCR amplification (12-20) to maintain relative ratios of different amplicons (Figure 26, step B).
- the primers contain identical 8-11 base tails to prevent primer dimers.
- PCR products incorporate dUTP, allowing for carryover prevention (Figure 26, step C).
- exon junction-specific ligation oligonucleotide probes containing primer-specific portions (Ai, Ci’) suitable for subsequent PCR amplification hybridize to their corresponding target sequence in a base-specific manner.
- Ligase covalently seals the two oligonucleotides together ( Figure 26, step D), and ligation products are aliquoted into separate wells for detection using tag-primers (Ai, Ci) and TaqManTM probe (F1-Q) which spans the ligation junction ( Figure 26, step E).
- Treat samples with UDG for carryover prevention, which also destroys original target amplicons Figure 26, step E).
- Figures 27 illustrates an exemplary RT-PCR-LDR-qPCR carryover prevention reaction to enumerate mRNA, ncRNA, or lncRNA copy number.
- mRNA is isolated ( Figures 27, step A), and treated with UDG for carryover prevention ( Figures 27, step B).
- cDNA is generated using 3’ transcript-specific primers and reverse-transcriptase in the presence of dUTP.
- Taq polymerase is activated to perform limited cycle PCR amplification (8-20) to maintain relative ratios of different amplicons ( Figures 27, step B).
- the primers contain identical 8-11 base tails to prevent primer dimers.
- PCR products incorporate dUTP, allowing for carryover prevention ( Figures 27, step C).
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the cDNA target region, cDNA-specific primers, to amplify the target sequence, if present in the sample ( Figure 27, step C).
- the cDNA target products are amplified and detected as described supra for Figure 6 (see Figure 27, steps C-D), or using other suitable means known in the art.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the cDNA target region, cDNA-specific (forward) primers comprising 5’ primer- specific portions (Ai), cDNA-specific (reverse) primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci.
- These primers combine to amplify the target cDNA sequence, if present in the sample ( Figure 28, step C).
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the target cDNA regions as described supra for Figure 4 (see Figure 28, steps D-F), or using other suitable means known in the art.
- the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the cDNA target region, cDNA-specific (forward) primers comprising 5’ primer-specific portions (Ai), cDNA- specific (reverse) primers comprising 5’ primer-specific portions (Bi-Ci) and matching primers F1-Bi-Q-Ai and Ci ( Figure not shown).
- These primers combine to amplify the target cDNA sequence, if present in the sample.
- Primers are unblocked with RNaseH2 only when bound to correct target.
- the products can be detected using pairs of matched primers F1- Bi-Q-Ai and Ci, and TaqManTM probes that span the target cDNA regions as described supra for Figure 4, or using other suitable means known in the art.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- dU deoxyuracil
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample, and the contacted sample is blended with a ligase and one or more first oligonucleotide preliminary probes comprising a 5’ phosphate, a 5’ stem-loop portion, an internal primer-specific portion within the loop region, a blocking group, and a 3’ nucleotide sequence that is complementary to a 3’ portion of the target miRNA molecule sequence to form one or more first ligation reaction mixtures.
- the method further comprises ligating, in the one or more first ligation reaction mixtures, the one or more target miRNA molecules at their 3’end to the 5’ phosphate of the one or more first
- oligonucleotide preliminary probes to generate chimeric nucleic acid molecules comprising the target miRNA molecule sequence, if present in the sample, appended to the one or more first oligonucleotide preliminary probes.
- One or more primary oligonucleotide primer sets are then provided.
- Each primer set comprises (a) a first primary oligonucleotide primer comprising a nucleotide sequence that is complementary to the internal primer-specific portion of the first oligonucleotide preliminary probe, and (b) a second primary oligonucleotide primer comprising a 5’ primer-specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the one or more first ligation reaction mixtures comprising chimeric nucleic acid molecules, the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the sample, a deoxynucleotide mix including dUTP, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- dU deoxyuracil
- a deoxynucleotide mix including dUTP
- a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the reverse-transcription/polymerase chain reaction mixtures to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the chimeric nucleic acid molecules, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different primary reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence, and the complement of the internal primer-specific portion, and complements thereof.
- dU deoxyuracil
- cDNA complementary deoxyribonucleic acid
- the method further involves providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion, a portion complementary to a primary extension product, and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, on complementary portions of a primary reverse-transcription/polymerase chain reaction product corresponding to the target miRNA molecule sequence, or complement thereof.
- the primary reverse- transcription/polymerase chain reaction products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more second ligation reaction mixtures, and the one or more second ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligation reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences and the one or more secondary oligonucleotide primer sets are blended with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the second polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further comprises detecting and distinguishing the secondary polymerase chain reaction products in the one or more reactions thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Figure 29 illustrates an embodiment of this aspect of the present application.
- Figure 29 illustrates an exemplary Ligation-RT-PCR-LDR-qPCR carryover prevention reaction to quantify miRNA.
- aliquot into 12, 24, 36, or 48 wells prior to PCR.
- For higher copy number distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells.
- This method involves isolating miRNA from exosomes and treating with UDG for carryover prevention ( Figure 29, step B).
- An oligonucleotide probe having a portion that is complementary to the 3’ end of the target miRNA, and containing a stem-loop, tag (Di’), and blocking group (filled circle) is ligated at its 5’ end to the 3’ end of the target miRNA.
- the ligation product comprises the miRNA, Di’ tag, the blocking group, and a sequence complementary to the 3’ portion of the miRNA (Figure 29, step B).
- a reverse transcriptase such as Moloney Murine Leukemia Virus Reverse Transcriptase (M- MLV RT, New England Biolabs), or Superscript II or III Reverse Transcriptase (Life trademark).
- the reverse transcriptase undergoes strand switching and copies the Ei tag sequence.
- the PCR products incorporate dU, allowing for carryover prevention ( Figure 29, step C).
- step D miRNA sequence-specific ligation probes containing primer-specific portions (Ai, Ci’) suitable for subsequent PCR amplification, hybridize to their corresponding target sequence in a base-specific manner.
- the ligation products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the ligation junction as described supra for Figure 2 (see Figure 29, steps D-F), or using other suitable means known in the art.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- dU deoxyuracil
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample, and the contacted sample is blended with a ligase and one or more first oligonucleotide probes comprising a 5’ phosphate, a 5’ stem-loop portion, an internal primer-specific portion within the loop region, a blocking group, and a 3’ nucleotide sequence that is complementary to a 3’ portion of the target miRNA molecule sequence to form one or more ligation reaction mixtures.
- the method further involves ligating, in the one or more ligation reaction mixtures, the one or more target miRNA molecules at their 3’end to the 5’ phosphate of the one or more first oligonucleotide probes to generate chimeric nucleic acid molecules comprising the target miRNA molecule sequence, if present in the sample, appended to the one or more first oligonucleotide probes.
- One or more primary oligonucleotide primer sets are then provided.
- Each primer set comprises (a) a first primary oligonucleotide primer comprising a nucleotide sequence that is complementary to the internal primer-specific portion of the first oligonucleotide probe, and (b) a second primary oligonucleotide primer comprising a 5’ primer-specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the one or more ligation reaction mixtures comprising chimeric nucleic acid molecules, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse- transcription/polymerase chain reaction mixtures.
- the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the chimeric nucleic acid molecules, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different primary reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence, and the complement of the internal primer-specific portion, and complements thereof.
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of an extension product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of an extension product formed from the first secondary oligonucleotide primer.
- the primary reverse- transcription/polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising a 5’ primer-specific portion of the first secondary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence or a complement thereof, and a complement of the other 5’ primer- specific portion second secondary oligonucleotide primer.
- the method further involves providing one or more tertiary oligonucleotide primer sets.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction products or their complements and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reaction products or their complements.
- the first polymerase chain reaction process products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures, and the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU) containing nucleic acid molecules present in the secondpolymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products, thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Figure 30 illustrates an embodiment of this aspect of the present application.
- Figure 30 illustrates an exemplary Ligation-RT-PCR-qPCR carryover prevention reaction to quantify miRNA. For accurate enumeration, aliquot into 12, 24, 36, or 48 wells prior to PCR. For higher copy number, distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells. This method involves isolating miRNA from exosomes and treating with UDG for carryover prevention ( Figure 30, step B).
- oligonucleotide probe having a portion that is complementary to the 3’ end of the target miRNA, and containing a stem-loop, tag (Di’), and blocking group (filled circle) is ligated at its 5’ end to the 3’ end of the target miRNA.
- the ligation product comprises the miRNA, Di’ tag, the blocking group, and a sequence complementary to the 3’ portion of the miRNA (Figure 30, step B).
- a reverse transcriptase extends primer (Di) to make a full-length copy of the target and appends three C bases to the 3’ end of extended target sequence ( Figure 30, step B).
- the reverse transcriptase undergoes strand switching and copies the Ei tag sequence.
- step C following the limited cycle PCR, the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the miRNA target region, miRNA-specific (forward) primers comprising 5’ primer- specific portions (Ai), cDNA-specific (reverse) primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci. These primers combine to amplify the target miRNA sequence, if present in the sample ( Figure 30, step C). Primers are unblocked with RNaseH2 only when bound to correct target. Following PCR, the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the target miRNA regions as described supra for Figure 4 (see Figure 30, steps C-F), or using other suitable means known in the art.
- the 3’ portion of the second primary oligonucleotide primer comprises ribo-G and/or G nucleotide analogue, wherein the reverse transcriptase appends two or three cytosine nucleotides to the 3’ end of the complementary deoxyribonucleic acid products of the target miRNAs, enabling transient hybridization to the 3’ end of the second primary oligonucleotide primer, enabling the reverse transcriptase to undergo strand switching and to extend the complementary deoxyribonucleic acid products to include the complementary sequence of the 5’ primer-specific portion of the second primary oligonucleotide primer to form the one or more different first polymerase chain reaction products comprising a 5’ primer- specific portion, a nucleotide sequence portion corresponding to the target miRNA molecule sequence or a complement thereof, a further portion, and a complement of the other 5’ primer- specific portion.
- the 3’ portion of the second primary oligonucleotide primers contains from 6 to 14 bases comprising, from 5’ to 3’, three ribo-G or G bases, followed by additional bases that are the same as the 5’ end of the target miRNA sequences, wherein the reverse transcriptase appends two or three cytosine residues to the 3’ end of the initial complementary deoxyribonucleic acid extension products of the target miRNAs, and wherein subsequent to when the denaturation treatment of the polymerase chain reaction is initiated the conditions are adjusted to enable transient hybridization to the 3’ end of the second primary oligonucleotide primers to the 3’ end of the complementary deoxyribonucleic acid extension products, allowing for extension of either one or both the second primary oligonucleotide primers and the complementary deoxyribonucleic acid extension products to form the one or more different primary reverse-transcription/polymerase chain reaction products comprising a 5’ primer-specific portion, a
- the second oligonucleotide probe of the oligonucleotide probe set further comprises a unitaq detection portion, thereby forming ligated product sequences comprising the 5’ primer-specific portion, the target-specific portions, the unitaq detection portion, and the 3’ primer-specific portion.
- a unitaq detection portion thereby forming ligated product sequences comprising the 5’ primer-specific portion, the target-specific portions, the unitaq detection portion, and the 3’ primer-specific portion.
- one or more unitaq detection probes are provided, wherein each unitaq detection probe hybridizes to a complementary unitaq detection portion and said detection probe comprises a quencher molecule and a detectable label separated from the quencher molecule.
- the one or more unitaq detection probes are added to the second polymerase chain reaction mixture, and the one or more unitaq detection probes are hybridized to complementary unitaq detection portions on the ligated product sequence or complement thereof during said subjecting the second polymerase chain reaction mixture to conditions suitable for one or more polymerase chain reaction cycles, wherein the quencher molecule and the detectable label are cleaved from the one or more unitaq detection probes during the extension treatment and said detecting involves the detection of the cleaved detectable label.
- one primary oligonucleotide primer or one secondary oligonucleotide primer further comprises a unitaq detection portion, thereby forming extension product sequences comprising the 5’ primer-specific portion, the target-specific portions, the unitaq detection portion, and the complement of the other 5’ primer-specific portion, and complements thereof.
- one or more unitaq detection probes are provided, wherein each unitaq detection probe hybridizes to a complementary unitaq detection portion and said detection probe comprises a quencher molecule and a detectable label separated from the quencher molecule.
- the one or more unitaq detection probes are added to the one or more first or second polymerase chain reaction mixtures, and the one or more unitaq detection probes are hybridized to complementary unitaq detection portions on the ligated product sequence or complement thereof during polymerase chain reaction cycles after the first polymerization chain reaction, wherein the quencher molecule and the detectable label are cleaved from the one or more unitaq detection probes during the extension treatment and said detecting involves the detection of the cleaved detectable label.
- one or both oligonucleotide probes of the oligonucleotide probe set comprises a portion that has no or one nucleotide sequence mismatch when hybridized in a base-specific manner to the target nucleic acid sequence or bisulfite-converted methylated nucleic acid sequence or complement sequence thereof, but have one or more additional nucleotide sequence mismatches that interferes with ligation when said oligonucleotide probe hybridizes in a base-specific manner to a corresponding nucleotide sequence portion in the wildtype nucleic acid sequence or bisulfite-converted unmethylated nucleic acid sequence or complement sequence thereof.
- the 3’ portion of the first oligonucleotide probe of the oligonucleotide probe set comprises a cleavable nucleotide or nucleotide analogue and a blocking group, such that the 3’ end is unsuitable for polymerase extension or ligation.
- the cleavable nucleotide or nucleotide analog of the first oligonucleotide probe is cleaved when said probe is hybridized to it complementary target nucleotide sequence of the primary extension product, thereby liberating a 3’OH on the first oligonucleotide probe prior to the ligating.
- the one or more first oligonucleotide probe of the oligonucleotide probe set may comprises a sequence that differs from the target nucleic acid sequence or bisulfite-converted methylated nucleic acid sequence or complement sequence thereof, said difference is located two or three nucleotide bases from the liberated free 3’OH end.
- the second oligonucleotide probe has, at its 5’ end, an overlapping identical nucleotide with the 3’ end of the first oligonucleotide probe, and, upon hybridization of the first and second oligonucleotide probes of a probe set at adjacent positions on a complementary target nucleotide sequence of a primary extension product to form a junction, the overlapping identical nucleotide of the second oligonucleotide probe forms a flap at the junction with the first oligonucleotide probe.
- This embodiment further comprises cleaving the overlapping identical nucleotide of the second oligonucleotide probe with an enzyme having 5’ nuclease activity thereby liberating a phosphate at the 5’ end of the second oligonucleotide probe prior to said ligating.
- the one or more oligonucleotide probe sets further comprise a third oligonucleotide probe having a target-specific portion, wherein the second and third oligonucleotide probes of a probe set are configured to hybridize adjacent to one another on the target nucleotide sequence with a junction between them to allow ligation between the second and third oligonucleotide probes to form a ligated product sequence comprising the first, second, and third oligonucleotide probes of a probe set.
- the sample is selected from the group consisting of tissue, cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, cell-free circulating nucleic acids, cell-free circulating tumor nucleic acids, cell-free circulating fetal nucleic acids in pregnant woman, circulating tumor cells, tumor, tumor biopsy, and exosomes.
- the one or more target nucleotide sequences may be low-abundance nucleic acid molecules comprising one or more nucleotide base mutations, insertions, deletions,
- translocations splice variants, miRNA variants, alternative transcripts, alternative start sites, alternative coding sequences, alternative non-coding sequences, alternative splicings, exon insertions, exon deletions, intron insertions, or other rearrangement at the genome level and/or methylated nucleotide bases.
- low abundance nucleic acid molecule refers to a target nucleic acid molecule that is present at levels as low as 1% to 0.01% of the sample.
- a low abundance nucleic acid molecule with one or more nucleotide base mutations, insertions, deletions, translocations, splice variants, miRNA variants, alternative transcripts, alternative start sites, alternative coding sequences, alternative non-coding sequences, alternative splicings, exon insertions, exon deletions, intron insertions, other rearrangement at the genome level, and/or methylated nucleotide bases can be distinguished from a 100 to 10,000-fold excess of nucleic acid molecules in the sample (i.e., high abundance nucleic acid molecules) having a similar nucleotide sequence as the low abundance nucleic acid molecules but without the one or more nucleotide base mutations, insertions, deletions, translocations, splice variants, miRNA variants
- the copy number of one or more low abundance target nucleotide sequences are quantified relative to the copy number of high abundance nucleic acid molecules in the sample having a similar nucleotide sequence as the low abundance nucleic acid molecules.
- the one or more target nucleotide sequences are quantified relative to other nucleotide sequences in the sample.
- the relative copy number of one or more target nucleotide sequences is quantified. Methods of relative and absolute (i.e., copy number) quantitation are well known in the art.
- the low abundance target nucleic acid molecules to be detected can be present in any biological sample, including, without limitation, tissue, cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, cell-free circulating nucleic acids, cell-free circulating tumor nucleic acids, cell-free circulating fetal nucleic acids in pregnant woman, circulating tumor cells, tumor, tumor biopsy, and exosomes.
- the methods of the present invention are suitable for diagnosing or prognosing a disease state and/or distinguishing a genotype or disease predisposition.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample.
- the contacted sample is blended with ATP and a Poly(A) polymerase to form a Poly(A) polymerase reaction mixture, and the Poly(A) polymerase reaction mixture is subjected to conditions suitable for appending homopolymer A to the 3’ ends of the one or more target miRNA molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets.
- Each primer set comprises (a) a first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary
- oligonucleotide primer may be the same or may differ from other first primary oligonucleotide primers in other sets, and (b) a second primary oligonucleotide primer comprising a 5’ primer- specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the Poly(A) polymerase reaction mixture, the one or more primary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules in the sample, a deoxynucleotide mix including dUTP, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse-transcriptase activity are blended to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse- transcription/polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the reverse- transcription/polymerase chain reaction mixtures, then to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target miRNA sequences with 3’ polyA tails, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming one or more different reverse-transcription/polymerase chain reaction products comprising the 5’ primer- specific portion of the second primary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence, a poly dA region, and the complement of the 5’ primer-specific portion of the first primary oligonucleotide primer, and complements thereof.
- dU deoxyuracil
- cDNA complementary deoxyribonucleic acid
- the method further comprises providing one or more oligonucleotide probe sets.
- Each probe set comprises (a) a first oligonucleotide probe having a 5’ primer-specific portion and a 3’ target sequence-specific portion, and (b) a second oligonucleotide probe having a 5’ target sequence-specific portion, a portion complementary to the one or more reverse- transcription/polymerase chain reaction products, and a 3’ primer-specific portion, wherein the first and second oligonucleotide probes of a probe set are configured to hybridize, in a base specific manner, to complementary portions of the one or more reverse-transcription/polymerase chain reaction products corresponding to the target miRNA molecule sequence, or complement thereof.
- the one or more reverse-transcription/polymerase chain reaction products are contacted with a ligase and the one or more oligonucleotide probe sets to form one or more ligation reaction mixtures, and the one or more ligation reaction mixtures are subjected to one or more ligation reaction cycles whereby the first and second oligonucleotide probes of the one or more oligonucleotide probe sets, when hybridized to their complement, are ligated together to form ligated product sequences in the ligation reaction mixture, wherein each ligated product sequence comprises the 5’ primer-specific portion, the target-specific portions, and the 3’ primer-specific portion.
- the method further involves providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the ligated product sequence and (b) a second secondary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the ligated product sequence.
- the ligated product sequences and the one or more secondary oligonucleotide primer sets are blended with one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures and for carrying out one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming secondary polymerase chain reaction products.
- the method further comprises detecting and distinguishing the secondary polymerase chain reaction products, thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Figure 31 illustrates an embodiment of this aspect of the present application.
- Figure 31 illustrates an exemplary RT-PCR-LDR-qPCR carryover prevention reaction to quantify miRNA.
- For higher copy number distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells.
- This method involves isolating miRNA from exosomes and treating with UDG for carryover prevention ( Figure 31, step B).
- a reverse transcriptase extends primer comprising a Tag Di on the 5’ end, and a T30VN sequence at the 3’ end, to make a full-length copy of the target, and appends three C bases to the 3’ end of extended target sequence (Figure 31, step B).
- Tag oligonucleotide (Ei) having the 3’ rGrG+G hybridizes to the three C bases of the extended target sequence as shown in Figure 31, step B.
- the reverse transcriptase undergoes strand switching and copies the Ei tag sequence.
- the PCR products incorporate dU, allowing for carryover prevention (Figure 31, step C).
- step D miRNA sequence-specific ligation probes containing primer-specific portions (Ai, Ci’) suitable for subsequent PCR amplification, hybridize to their corresponding target sequence in a base-specific manner.
- the ligation products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the ligation junction as described supra for Figure 2 (see Figure 31, steps D-F), or using other suitable means known in the art.
- Another aspect of the present application is directed to a method for identifying, in a sample, one or more target micro-ribonucleic acid (miRNA) molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- the method involves providing a sample containing one or more target miRNA molecules potentially differing in sequence from other miRNA molecules in the sample by one or more bases and providing one or more enzymes capable of digesting deoxyuracil (dU)-containing nucleic acid molecules present in the sample.
- the sample is contacted with one or more enzymes capable of digesting dU-containing nucleic acid molecules potentially present in the sample.
- the contacted sample is blended with ATP and a Poly(A) polymerase to form a Poly(A) polymerase reaction mixture, and the Poly(A) polymerase reaction mixture is subjected to conditions suitable for appending a homopolymer A to the 3’ ends of the one or more target miRNA molecules potentially present in the sample.
- the method further involves providing one or more primary oligonucleotide primer sets. Each primer set comprises (a) a first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary oligonucleotide primer comprising a 5’ primer-specific portion, an internal poly dT portion, and a 3’ portion comprising from 1 to 10 bases complementary to the 3’ end of the target miRNA, wherein the first primary
- oligonucleotide primer may be the same or may differ from other first primary oligonucleotide primers in other sets, and (b) a second primary oligonucleotide primer comprising a 5’ primer- specific portion and a 3’ portion, wherein the second primary oligonucleotide primer may be the same or may differ from other second primary oligonucleotide primers in other sets.
- the Poly(A) polymerase reaction mixture potentially comprising target miRNA sequences is blended with 3’ polyA tails, the one or more primary oligonucleotide primer sets, a deoxynucleotide mix, and a reverse transcriptase and a DNA polymerase or a DNA polymerase with reverse- transcriptase activity to form one or more reverse-transcription/polymerase chain reaction mixtures.
- the one or more reverse-transcription/polymerase chain reaction mixtures are subjected to conditions suitable for generating complementary deoxyribonucleic acid (cDNA) molecules to the target miRNA sequences with 3’ polyA tails, and to one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming one or more different reverse-transcription/polymerase chain reaction products comprising the 5’ primer-specific portion of the second primary oligonucleotide primer, a nucleotide sequence corresponding to the target miRNA molecule sequence, a poly dA region, and the complement of the 5’ primer-specific portion of the first primary oligonucleotide primer, and complements thereof.
- cDNA complementary deoxyribonucleic acid
- the method further comprises providing one or more secondary oligonucleotide primer sets.
- Each secondary oligonucleotide primer set comprises (a) a first secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that is complementary to a portion of a reverse-transcription/polymerase chain reaction product formed from the first primary oligonucleotide primer and (b) a second secondary oligonucleotide primer having a 5’ primer-specific portion and a 3’ portion that comprises a nucleotide sequence that is complementary to a portion of a reverse- transcription/polymerase chain reaction product formed from the first secondary oligonucleotide primer.
- the reverse-transcription/polymerase chain reaction products, the one or more secondary oligonucleotide primer sets, a deoxynucleotide mix, and a DNA polymerase are blended to form one or more first polymerase chain reaction mixtures, and the one or more first polymerase chain reaction mixtures are subjected to conditions suitable for two or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment, thereby forming first polymerase chain reaction products comprising a 5’ primer-specific portion, a nucleotide sequence corresponding to the target miRNA molecule sequence or a complement thereof, and a complement of the other 5’ primer-specific portion.
- the method further involves providing one or more tertiary oligonucleotide primer sets.
- Each tertiary oligonucleotide primer set comprises (a) a first tertiary oligonucleotide primer comprising the same nucleotide sequence as the 5’ primer-specific portion of the first polymerase chain reaction product sequence and (b) a second tertiary oligonucleotide primer comprising a nucleotide sequence that is complementary to the 3’ primer-specific portion of the first polymerase chain reaction product sequence.
- the first polymerase chain reaction products, the one or more tertiary oligonucleotide primer sets, the one or more enzymes capable of digesting deoxyuracil (dU) containing nucleic acid molecules, a deoxynucleotide mix including dUTP, and a DNA polymerase are blended to form one or more second polymerase chain reaction mixtures.
- the one or more second polymerase chain reaction mixtures are subjected to conditions suitable for digesting deoxyuracil (dU)-containing nucleic acid molecules present in the first polymerase chain reaction mixtures, and one or more polymerase chain reaction cycles comprising a denaturation treatment, a hybridization treatment, and an extension treatment thereby forming second polymerase chain reaction products.
- the method further comprises detecting and distinguishing the second polymerase chain reaction products in the one or more reactions thereby identifying one or more target miRNA molecules differing in sequence from other miRNA molecules in the sample by one or more bases.
- Figure 32 illustrates an embodiment of this aspect of the present application.
- Figure 32 illustrates an exemplary RT-PCR-qPCR carryover prevention reaction to quantify miRNA. For accurate enumeration, aliquot into 12, 24, 36, or 48 wells prior to PCR. For higher copy number, distribute equally in 13 wells, dilute last well equally into 13 additional wells, and repeat for remaining wells. This method involves isolating miRNA from exosomes and treating with UDG for carryover prevention ( Figure 32, step B). PolyA tail miRNA with E. coli Poly(A) polymerase.
- a reverse transcriptase extends primer comprising a Tag Di on the 5’ end, and a T30VN sequence at the 3’ end, to make a full-length copy of the target, and appends three C bases to the 3’ end of extended target sequence (Figure 32, step B).
- Tag oligonucleotide (Ei) having the 3’ rGrG+G hybridizes to the three C bases of the extended target sequence as shown in Figure 32, step B.
- the reverse transcriptase undergoes strand switching and copies the Ei tag sequence.
- step C following the limited cycle PCR, the PCR products are aliquoted into separate wells, micro-pores or droplets containing TaqmanTM probes across the miRNA target region, miRNA-specific (forward) primers comprising 5’ primer- specific portions (Ai), cDNA-specific (reverse) primers comprising 5’ primer-specific portions (Ci) and matching primers Ai and Ci. These primers combine to amplify the target miRNA sequence, if present in the sample ( Figure 32, step C). Primers are unblocked with RNaseH2 only when bound to correct target. Following PCR, the products can be detected using pairs of matched primers Ai and Ci, and TaqManTM probes that span the target miRNA regions as described supra for Figure 6 (see Figure 32, steps C-F), or using other suitable means known in the art.
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of cells or tissue based on identifying the presence or level of a plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers in a biological sample of an individual.
- the plurality of markers is in a set comprising from 6-12 markers, 12-24 markers, 24-36 markers, 36-48 markers, 48-72 markers, 72-96 markers, or > 96 markers.
- Each marker in a given set is selected by having any one or more of the following criteria: present, or above a cutoff level, in > 50% of biological samples of the disease cells or tissue from individuals diagnosed with the disease state; absent, or below a cutoff level, in > 95% of biological samples of the normal cells or tissue from individuals without the disease state; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with the disease state; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without the disease state; present with a z-value of > 1.65 in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65, comprise of one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with the disease state.
- the method involves obtaining a biological sample.
- the biological sample includes cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, and the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor-associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- Nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- At least two enrichment steps are carried out for 50% or more disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with the disease state if a minimum of 2 or 3 markers are present or are above a cutoff level in a marker set comprising from 6-12 markers; or a minimum of 3, 4, or 5 markers are present or are above a cutoff level in a marker set comprising from 12-24 markers; or a minimum of 3, 4, 5, or 6 markers are present or are above a cutoff level in a marker set comprising from 24-36 markers; or a minimum of 4, 5, 6, 7, or 8 markers are present or are above a cutoff level in a marker set comprising from 36
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of a solid tissue cancer including colorectal adenocarcinoma, stomach adenocarcinoma, esophageal carcinoma, breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, uterine carcinosarcoma, lung adenocarcinoma, lung squamous cell carcinoma, head & neck squamous cell carcinoma, prostate adenocarcinoma, invasive urothelial bladder cancer, liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adenocarcinoma based on identifying the presence or level of a plurality of disease- specific and/or cell/tissue-specific DNA, RNA, and/or protein
- the plurality of markers is in a set comprising from 48-72 total cancer markers, 72-96 total cancer markers or 3 96 total cancer markers, wherein on average greater than one quarter such markers in a given set cover each of the aforementioned major cancers being tested.
- Each marker in a given set for a given solid tissue cancer is selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 50% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65, comprise of one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with a given solid tissue cancer.
- the method involves obtaining a biological sample including cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor-associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- the nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues. At least two enrichment steps are carried out for 50% or more of the given solid tissue cancer-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of cancer -specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with the a solid-tissue cancer if a minimum of 4 markers are present or are above a cutoff level in a marker set comprising from 48-72 total cancer markers; or a minimum of 5 markers are present or are above a cutoff level in a marker set comprising from 72-96 total cancer markers; or a minimum of 6 or “n”/18 markers are present or are above a cutoff level in a marker set comprising 96 to“n” total cancer markers, when“n” > 96 total cancer markers.
- each marker in a given set for a given solid tissue cancer may be selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 66% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 66% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without that given solid tissue cancer
- Another aspect of the present application is directed to a method of diagnosing or prognosing a disease state of and identifying the most likely specific tissue(s) of origin of a solid tissue cancer in the following groups: Group 1 (colorectal adenocarcinoma, stomach adenocarcinoma, esophageal carcinoma); Group 2 (breast lobular and ductal carcinoma, uterine corpus endometrial carcinoma, ovarian serous cystadenocarcinoma, cervical squamous cell carcinoma and adenocarcinoma, uterine carcinosarcoma); Group 3 (lung adenocarcinoma, lung squamous cell carcinoma, head & neck squamous cell carcinoma); Group 4 (prostate adenocarcinoma, invasive urothelial bladder cancer); and/or Group 5 (liver hepatoceullular carcinoma, pancreatic ductal adenocarcinoma, or gallbladder adeno
- Each marker in a given set for a given solid tissue cancer is selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 50% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 50% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without that given solid tissue cancer; present with a z-value of > 1.65 in the biological sample comprising
- At least 50% of the markers in a set each comprise one or more methylated residues, and/or at least 50% of the markers in a set that are present, or above a cutoff level, or present with a z-value of > 1.65 comprise one or more methylated residues, in the biological sample comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from at least 50% of individuals diagnosed with a given solid tissue cancer.
- the method involves obtaining the biological sample.
- the biological sample includes cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- the sample is fractionated into one or more fractions, wherein at least one fraction comprises exosomes, tumor- associated vesicles, other protected states, or cell-free DNA, RNA, and/or protein.
- the nucleic acid molecules in the one or more fractions are subjected to a bisulfite treatment under conditions suitable to convert unmethylated cytosine residues to uracil residues.
- At least two enrichment steps are carried out for 50% or more of the given solid tissue cancer-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers during either said fractionating and/or by carrying out a nucleic acid amplification step.
- the method further involves performing one or more assays to detect and distinguish the plurality of cancer -specific and/or cell/tissue-specific DNA, RNA, and/or protein markers, thereby identifying their presence or levels in the sample, wherein individuals are diagnosed or prognosed with a solid-tissue cancer if a minimum of 4 markers are present or are above a cutoff level in a marker set comprising from 36-48 group- specific cancer markers; or a minimum of 5 markers are present or are above a cutoff level in a marker set comprising from 48-64 group-specific cancer markers; or a minimum of 6 or“n”/12 markers are present or are above a cutoff level in a marker set comprising 64 to“n” total cancer markers, when“n” > 64 group-specific cancer markers
- each marker in a given set for a given solid tissue cancer may be selected by having any one or more of the following criteria for that solid tissue cancer: present, or above a cutoff level, in > 66% of biological samples of a given cancer tissue from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples of the normal tissue from individuals without that given solid tissue cancer; present, or above a cutoff level, in > 66% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals diagnosed with a given solid tissue cancer; absent, or below a cutoff level, in > 95% of biological samples comprising cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, bodily excretions, or fractions thereof, from individuals without that given solid tissue cancer; present
- the at least two enrichment steps comprise of two or more of the following steps: capturing or separating exosomes or extracellular vesicles or markers in other protected states; capturing or separating a platelet fraction; capturing or separating circulating tumor cells; capturing or separating RNA-containing complexes; capturing or separating cfDNA-nucleosome or differentially modified cfDNA-histone complexes; capturing or separating protein targets or protein target complexes; capturing or separating auto-antibodies; capturing or separating cytokines; capturing or separating methylated cfDNA; capturing or separating marker specific DNA, cDNA, miRNA, lncRNA, ncRNA, or mRNA, or amplified complements, by hybridization to complementary capture probes in solution, on magnetic beads, or on a microarray; amplifying miRNA markers, non-coding RNA markers (lncRNA & ncRNA markers), mRNA markers, ex
- the one or more assays to detect and distinguish the plurality of disease-specific and/or cell/tissue-specific DNA, RNA, or protein markers comprise one or more of the following: a quantitative real-time PCR method (qPCR); a reverse transcriptase-polymerase chain reaction (RTPCR) method; a bisulfite qPCR method; a digital PCR method (dPCR); a bisulfite dPCR method; a ligation detection method, a ligase chain reaction, a restriction endonuclease cleavage method; a DNA or RNA nuclease cleavage method; a micro-array hybridization method; a peptide-array binding method; an antibody-array method; a mass spectrometry method; a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method; a capillary or gel electrophoresis method; a chemiluminescence method; a fluor
- the one or more cutoff levels of the one or more assays to detect and distinguish the plurality of disease-specific and/or cell/tissue-specific DNA, RNA, or protein markers comprise one or more of the following calculations, comparisons, or determinations, in the one or more marker assays comparing samples from the disease vs.
- marker ⁇ Ct value is > 2; marker ⁇ Ct value is > 4; ratio of detected marker-specific signal is > 1.5; ratio of detected marker-specific signal is > 3; ratio of marker concentrations is > 1.5; ratio of marker concentrations is > 3; enumerated marker-specific signals differ by > 20%; enumerated marker-specific signals differ by > 50%; marker-specific signal from a given disease sample is > 85%; > 90%; > 95%; > 96%; > 97%; or > 98% of the same marker-specific signals from a set of normal samples; or marker-specific signal from a given disease sample has a z- score of > 1.03; > 1.28; > 1.65; > 1.75; > 1.88; or > 2.05 compared to the same marker-specific signals from a set of normal samples.
- Another aspect of the present application relates to a two-step method of diagnosing or prognosing a disease state of cells or tissue based on identifying the presence or level of a plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers in a biological sample of an individual.
- the method involves obtaining a biological sample, the biological sample including exosomes, tumor-associated vesicles, markers within other protected states, cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- a first step is applied to the biological samples with an overall sensitivity of > 80% and an overall specificity of > 90% or an overall Z-score of > 1.28 to identify individuals more likely to be diagnosed or prognosed with the disease state.
- a second step is then applied to biological samples from those individuals identified in the first step with an overall specificity of > 95% or an overall Z-score of > 1.65 to diagnose or prognose individuals with the disease state.
- the first step and the second step are carried out using a method of the present application.
- the first step uses markers to cover many cancers, where the aim is to obtain high sensitivity for early cancers where the number of marker molecules in the blood may be limited.
- the second step then would score for additional markers to verify that the initial result was a true positive, as well as to identify the likely tissue of origin.
- the second step may include the methods described herein, and/or additional methods such as next-generation sequencing. [0273] Fluorescent labeling.
- G 2 F2 300 FU KRAS
- ddPCR digital droplet PCR
- ddPCR comprises 10,000 droplets or micro-pores or micro- wells, on average, only 1 in 20 will actually comprise a PCR reaction; the chances of a given droplet having two amplicons that would compete with each other for resources would be about 1 in 400, or about 25 droplets would comprise 2 amplicons, which would be only 5% of the total number of droplets with only a single amplicon. Since there are 6 combinations of 2 different amplicons, on average, less than 2% of the droplets would contain two amplicons.
- the rare droplet comprising 2 or 3 or 4 colors would not need to be de-convoluted, they could simply be ignored as they represent approximately 4-6 droplets compared to about 48 droplets arising from a single molecule in the original sample. While it may be a bit difficult to distinguish 190 from 237 droplets, i.e. starting with 4 or 5 molecules of a given methylated target, it should be relatively straightforward to distinguish 95; 190; and 48 copies,
- a given chamber will comprise of 44; 88; 110; 22; 0; 22; 66; 44; 0; and 22 copies of the PCR products for methylated VIM, SEPT9, CLIP4, GSG1L, PP1R16B, KCNA3, GDF6, ZNF677, CCNA1, and STK32B, respectively.
- This is a total of about 418 of molecules that would be amplified with primers for the total PCR products for methylated VIM, SEPT9, CLIP4, GSG1L, PP1R16B, KCNA3, GDF6, ZNF677, CCNA1, and STK32B.
- the ddPCR comprises 10,000 droplets or micro-pores or micro-wells, on average, only 1 in 25 will actually comprise a PCR reaction; the chances of a given droplet having two amplicons that would compete with each other for resources would be about 1 in 625, or about 16 droplets would comprise 2 amplicons, which would be only 4% of the total number of droplets with only a single amplicon. Since there are 45 combinations of 2 different amplicons, on average, less than 0.1% of the droplets would contain a given two amplicons.
- the rare droplet comprising 2 or 3 or 4 colors would not need to be de-convoluted, they could simply be ignored as they represent one or two droplets compared to about 22 droplets arising from a single molecule in the original sample. While it may be a bit difficult to distinguish 88 from 110 droplets, i.e. starting with 4 or 5 molecules of a given methylated target, it should be relatively straightforward to distinguish 44, 88, and 22 copies, corresponding to 2, 4, and 1 target molecules in the original sample.
- Gene 10 F3 + F4 (ncRNA10) [0282]
- a given chamber will comprise of 25; 51; 192; 13; 0; 128; 28; 256; 0; and 13 copies of the PCR products for mRNA1-4 and ncRNA5-10, respectively. This is a total of about 706 of molecules that would be amplified with primers for the total PCR products for methylated mRNA1-4 and ncRNA5-10. If the ddPCR comprises 10,000 droplets or micro-pores or micro-wells, on average, only 1 in 14 will actually comprise a PCR reaction.
- RNA 3 and ncRNA5 would be present on average of 1 in 52 and 1 in 39, thus the chances of a given droplet having these two amplicons that would compete with each other for resources would be about 1 in 2028, or about 5 droplets would comprise 2 amplicons, which is still less than for a single molecule after amplification– which will generate 13 copies.
- the rare droplet comprising 2 or 3 or 4 colors would not need to be de-convoluted, they could simply be ignored as they represent from 1 to 5 droplets compared to at least 13 droplets arising from a single molecule in the original sample.
- RNA molecules are present in higher amounts, one can still de-convolute multiple signals arising from 2 amplicons in a given droplet, using the same approach of different color probes at different levels of FU (i.e.300 FU for products with a single color; 100 FU each for products using 2 colors) as articulated earlier.
- levels of FU i.e.300 FU for products with a single color; 100 FU each for products using 2 colors
- Another aspect of the present application relates to the ability to distinguish cancer at the earliest stages when analyzing markers within a blood sample.
- the average body contains about 6 liters (6,000 ml) of blood.
- a 10 ml sample will then comprise 1/600th of the sample.
- cancers i.e. lung cancer, melanoma
- other cancers i.e. breast, ovarian
- methylation changes in promoter regions i.e. methylation markers
- the mutation rate for gene K-ras is ⁇ 30% and > 90% for colorectal cancer and pancreatic cancer, respectively. While p53 is found mutated in about 50% of all cancers, more often than not, such a mutation is manifested in late-stage tumors.
- a given cancer during its earliest stage generates at least one detectable mutation.
- a more accurate prediction would be based on the Poisson distribution. If there are 200 objects (i.e. mutated molecules) distributed into 600 bins (i.e.600 aliquots of 10 ml representing the total blood volume of a patient), Poisson calculation would indicate that: 72% of wells will have 0 objects, 23.7% will have 1 object, 3.9% will have 2 objects, 0.4% will have 3 objects, etc. In other words, 28.1% of the aliquots would have at least one mutated molecule. If the assay is capable of detecting every single mutated molecule, then its sensitivity would be 28.1%. Likewise, if there were 300 objects (i.e.
- mutated molecules distributed into 600 bins (i.e.600 aliquots of 10 ml), then: 61% of wells will have 0 objects, 30.3% will have 1 object, 7.6% will have 2 objects, 1.3% will have 3 objects, etc. In other words, 39.4% of the aliquots would have at least one mutated molecule. If the assay is capable of detecting every single mutated molecule, its sensitivity is at 39.4%. Likewise, if there were 400 objects (i.e. mutated molecules) distributed into 600 bins (i.e.600 aliquots of 10 ml), then: 51% of wells will have 0 objects, 34.3% will have 1 object, 11.5% will have 2 objects, 2.5% will have 3 objects, etc.
- the overall early cancer detection sensitivity is a function of the average number of each marker in the blood, the average number of markers positive, the minimum number of markers required to call the sample positive, and the total number of markers scored. For example, if the test uses 12 methylation markers, that on average are methylated in > 50% of tumors for that cancer type, then on average, about 6 markers will be methylated for a given sample.
- 600 x 600 3,600 objects (i.e. methylated molecules) are distributed into 600 bins (i.e.600 aliquots of 10 ml).
- 600 bins i.e.600 aliquots of 10 ml.
- the distribution would be: 0.2% of wells will have 0 objects, 1.5% will have 1 object, 4.5% will have 2 objects, 8.9% will have 3 objects, 13.3% will have 4 objects, 16.0% will have 5 objects, 16.0% will have 6 objects, 13.8% will have 7 objects, 10.3% will have 8 objects etc.
- at least two markers need to be called positive.
- the overall early cancer detection specificity is a function of the average number of markers positive, the false-positive rate for each individual marker, the minimum number of markers required to call the sample positive, and the total number of markers scored.
- Figures 33, 34, 35, and 36 illustrate results for calculated overall Sensitivity and Specificity for 24, 36, 48, and 96 markers, respectively. These graphs are based on the assumption that the average individual marker sensitivity is 50%, and the average individual marker false-positive rate is from 2% to 5%.
- the sensitivity curves provide overall sensitivity as a function of the average number of molecules in the blood for each marker, with separate curves for each minimum number of markers needed to call a sample as positive.
- the specificity curves provide overall specificity as a function of individual marker false-positive rates, again with separate curves for each minimum number of markers needed to call a sample as positive.
- the calculated numbers for overall Sensitivity and Specificity for 12, 24, 36, 48, 72 and 96 markers, respectively are provided in the tables below. Table 1
- the receiver operating characteristic (ROC) curves may be calculated by plotting Sensitivity vs.1-Specificity. Since these are theoretical calculations, the curves were generated for different levels of average marker false-positive rates of 2%, 3%, 4%, and 5%. To assist in visualizing the graphs and calculating the AUC (Area under curve), the edges were set at 100% and 0%, respectively.
- the ROC curves for 24 marker, 3% & 4% average marker false-positives, 36 marker, 3% & 4% average marker false-positives, and 48 marker, 2%, 3%, 4% & 5% average marker false-positives are provided in Table 13 below and for 48 Markers illustrated in Figures 37 and 38, respectively.
- AUC values are at 95% with 24 markers, improve to 99% with 36 markers, and range from 98% to >99% with 48 markers, depending on average marker false-positive values.
- the test would miss 6.2%; i.e. for Stage I & II cancer the overall sensitivity would be 93.8% (See Figure 33A), e.g. the test would correctly identify 93.8% of individuals with disease, which would be 126,630 individuals (out of 135,000 new cases).
- a PPV of 17.5% is quite respectable, however, it would be achieved at the cost of missing 28.5% of early cancer.
- Another aspect of the present application relates to a two-step method of diagnosing or prognosing a disease state of cells or tissue based on identifying the presence or level of a plurality of disease-specific and/or cell/tissue-specific DNA, RNA, and/or protein markers in a biological sample of an individual.
- the method involves obtaining a biological sample, the biological sample including exosomes, tumor-associated vesicles, markers within other protected states, cell-free DNA, RNA, and/or protein originating from the cells or tissue and from one or more other tissues or cells, wherein the biological sample is selected from the group consisting of cells, serum, blood, plasma, amniotic fluid, sputum, urine, bodily fluids, bodily secretions, and bodily excretions, or fractions thereof.
- a first step is applied to the biological samples with an overall sensitivity of > 80% and an overall specificity of > 85% or an overall Z-score of > 1.03 to identify individuals more likely to be diagnosed or prognosed with the disease state.
- a second step is the applied to biological samples from those individuals identified in the first step with an overall specificity of > 95% or an overall Z-score of > 1.65 to diagnose or prognose individuals with the disease state.
- the first step and the second step are carried out using a method of the present application.
- the first step uses markers to cover many cancers, where the aim is to obtain high sensitivity for early cancers where the number of marker molecules in the blood may be limited.
- the second step then would score for additional markers to verify that the initial result was a true positive, as well as to identify the likely tissue of origin.
- the second step may include the methods described herein, and/or additional methods such as next-generation sequencing.
- the first step uses markers to cover many cancers, where the aim is to obtain high sensitivity for early cancers where the number of marker molecules in the blood may be limited.
- the second step then would score for additional markers to verify that the initial result was a true positive, as well as to identify the likely tissue of origin.
- the second step may include the methods described herein, and/or additional methods such as next-generation sequencing.
- the parameters may be adjusted to improve BOTH sensitivity and specificity.
- the aforementioned 24 marker test using 3 markers, for Stage I & II cancer (at about 300 molecules of each positive marker in the blood), the overall sensitivity would be 93.8%.
- Those samples that are scored as positives in the first step (24-markers specific to GI cancers)– including the false-positives would be retested in the second step with an expanded panel of 48 markers to provide coverage of colorectal cancers. If the individual marker FP rate is 3%, then if there is a 5-marker minimum, then overall FP rate is 4.2% for 48 markers, for a specificity of 95.8% (See Figure 35B).
- the test would miss 0.7%; i.e. for Stage I & 2 cancer the sensitivity would be 99.3% (See Figure 35A).
- the test would miss 1.9%; i.e.
- the first step would identify 5,778,000 individuals (out of 107,000,000 total adults over 50 in the U.S.) which would include at 93.8% sensitivity about 126,630 individuals with Stage I & II colorectal cancer (out of 135,000 total).
- plasma, urine include, but are not limited to plasma microRNAs (miRNA); mutations or methylation in cfDNA; exosomes with surface cancer-specific protein markers, or internal miRNA, ncRNA, lncRNA, mRNA, DNA; circulating cytokines, circulating proteins, or circulating antibodies against cancer-antigens; or nucleic-acid markers in whole blood (for review, see Nikolaou et al.,“Systematic Review of Blood
- miRNAs include, but are not limited to: miR-1290; miR-21; miR-24; miR-320a; miR-423-5p; miR-29a; miR-125b; miR-17-3p; miR-92a; miR-19a; miR-19b; miR-15b; mir23a; miR-150; miR-223; miR-1229; miR-1246; miR-612; miR-1296; miR-933; miR-937; miR-1207; miR-31; miR-141; miR-224-3p; miR-576-5p; miR-885-5p; miR-200c; miR-203 (Imaoka et al., “Circulating MicroRNA-1290
- Figure 39 provides a list of blood-based, colon cancer-specific microRNA markers derived through analysis of TCGA microRNA datasets, which may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- ncRNAs include but are not limited to: NEAT_v1; NEAT_v2; CCAT1; HOTAIR; CRNDE-h; H19; MALAT1; 91H; GAS5 (Wu et al.,“Nuclear-enriched Abundant Transcript 1 as a
- Figure 40 provides a list of blood-based, colon cancer-specific ncRNA and lncRNA markers derived through analysis of various publicly available Affymetrix Exon ST CEL data, which were aligned to GENCODE annotations to generate ncRNA and lncRNA transriptome datasets.
- Figure 40 Comparative analyses across these datasets (various cancer types, along with normal tissues, and peripheral blood) were conducted to generate the ncRNA and lncRNA markers list ( Figure 40). Such lncRNA and ncRNA may be enriched in exosomes or other protected states in the blood.
- Figure 41 provides a list of blood-based colon cancer-specific exon transcripts that may be enriched in exosomes, tumor-associated vesicles, or other protected states in the blood.
- OC-Light iFOB Test also called OC Light S FIT
- QuickVue iFOB manufactured by Quidel (91.9% : 74.9%)
- Hemosure One-Step iFOB Test manufactured by Hemosure, Inc.
- Cut- off values for FIT tests may range from 10 ug protein/gram stool to 300 ug protein/gram stool (See Robertson et al.,“Recommendations on Fecal Immunochemical Testing to Screen for Colorectal Neoplasia: a Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer,” Gastrointest. Endosc.85(1):2-21 (2017), which is hereby incorporated by reference in its entirety).
- a number of tumor-associated antigens elicit an immune response within the patient, and these may be identified as autoantibodies, or indirectly as increased cytokines in the serum. Some tumor antigens may be detected directly within the serum, or on the surface of cancer-associated exosomes or extracellular vesicles, while others may be detected indirectly, for example by an increase in mRNA within cancer-associated exosomes or extracellular vesicles.
- cancer-specific proteins markers may be identified through, mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product, and these markers include but are not limited to: RPH3AL; RPL36; SLP2; TP53;
- Figure 43 provides protein markers that can be secreted by Colorectal tumors into the blood.
- a comparative analysis was performed across various TCGA datasets (tumors, normals), followed by an additional bioinformatics filter (Meinken et al.,“Computational Prediction of Protein Subcellular Locations in Eukaryotes: an Experience Report,” Computational Molecular Biology 2(1):1-7 (2012), which is hereby incorporated by reference in its entirety), which predicts the likelihood that the translated protein is secreted by the cells.
- SEPT9 methylation is the basis for Epi proColon test, a CRC-detection assay by Epigenomics (Lofton-Day et al:,“DNA Methylation Biomarkers for Blood-based Colorectal Cancer Screening,” Clinical Chemistry 54(2):414-423 (2008), which is hereby incorporated by reference in its entirety).
- Epigenetic changes may mark not only the DNA (as methylation or hydroxy-methylation of promoter CpG sites) but also by appending methyl or acetyl groups on the histone proteins that bind to these promoters.
- Figure 44 provides a list of primary CpG sites that are Colorectal Cancer and Colon-tissue specific markers, that may be used to identify the presence of colorectal cancer from cfDNA, or DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 45 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are Colorectal Cancer and Colon-tissue specific markers, that may be used to identify the presence of colorectal cancer from cfDNA, or DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Mutation or methylation status may give a clear analytical cut-off, i.e. the assay either records a mutation or CpG methylation event, and false-positives are a consequence of biology, for example from age-related methylation.
- cut-offs may be defined by“Z-score”, 2 standard deviations above normal values, or by setting the false-positive rate at an arbitrary level, i.e.5% when evaluating a suitable set of age-matched normal samples.
- the set of age-matched normal should be suitably large enough to set cut-off of the marker-specific signal from a given disease sample at > 85%; > 90%; > 95%; > 96%; > 97%; or > 98% of the same marker-specific signals from the set of normal samples.
- the cut-off for marker-specific signal from a given disease sample may be set at a z-score of > 1.03; > 1.28; > 1.65; > 1.75; > 1.88; or > 2.05 compared to the same marker- specific signals from the set of normal samples
- marker levels i.e.
- DNA methylation levels for several gene promoter regions in plasma, or miRNA levels in urine are quantified in relation to another marker, either internal or externally added in a qPCR reaction, where the cut-off is determined as a ⁇ Ct value in the assay (Fackler et al.,“Novel Methylated Biomarkers and a Robust Assay to Detect Circulating Tumor DNA in Metastatic Breast Cancer,” Cancer Res.74(8):2160-70 (2014); United States Patent No.9,416,404 to Sukumar et al., which are hereby incorporated by reference in their entirety). Methylation status at defined promoter regions may also be determined using digital bisulfite genomic sequencing and digital
- MethyLight assays using bisulfite conversion and preferential amplification of converted methylated sequences by blocking primers that interfere with amplification of converted unmethylated sequences; or depletion of unmethylated DNA using methyl-sensitive restriction endonucleases, followed by PCR (see U.S. Patent No.9,290,803 to Laird et al.; U.S. Patent No. 9,476,100 to Frumkin, et al.; U.S. Patent No.9,765,397 to McEvoy et al.; U.S. Patent No.
- the genome-wide methylation profile of cfDNA (known as the methylome) can be determined using next-generation sequencing, and the methylation pattern may be used to identify the presence of fetal, tumor, or other tissue DNA in the plasma (Sun et al.,“Plasma DNA Tissue Mapping by Genome-wide Methylation Sequencing for Noninvasive Prenatal, Cancer, and Transplantation Assessments,” Proc. Natl. Acad. Sci. U S A 112(40):E5503-12 (2015); Lehmann-Werman et al.,“Identification of Tissue-specific Cell Death Using Methylation Patterns of Circulating DNA,” Proc. Natl. Acad. Sci.
- a protein marker at 90% sensitivity but with 10% false-positives
- B Average individual markers at 50% sensitivity and 2%-5% false-positives, with one marker at 80% sensitivity but with 15% false-positives
- C Average individual markers at 50% sensitivity and 2%-5% false-positives, with two markers at 90% sensitivity each but with 10% false-positives each
- D Average individual markers at 50% sensitivity and 2%-5% false-positives, with two markers at 80% sensitivity each but with 15% false-positives each.
- Figures 47-48 illustrate results for calculated overall Sensitivity and Specificity for 24 markers using conditions (A) and (C).
- the sensitivity curves provide overall sensitivity as a function of the average number of molecules in the blood for each marker, with separate curves for each minimum number of markers needed to call a sample as positive.
- the specificity curves provide overall specificity as a function of individual marker false-positive rates, again with separate curves for each minimum number of markers needed to call a sample as positive.
- the calculated numbers for overall Sensitivity and Specificity for 24 markers using the above 4 conditions are provided in the tables below.
- the test would miss 42.3%; i.e. for Stage I cancer the overall sensitivity would be 57.7% (See Figure 33A). However, if the individual marker FP rate is 5%, then if there is a 4-marker minimum, then overall FP rate is 6.6% for 24 markers, for a specificity of 93.4% (See Figure 33B). At 4 markers, for Stage I cancer (at about 150 molecules of each positive marker in the blood), the test would miss 64.7%; i.e. for Stage I cancer the sensitivity would be 35.3% (See Figure 33A).
- condition (B) the average individual markers are at 50% sensitivity and 2%- 5% false-positives, with one marker at 80% sensitivity but with 15% false-positives. Under these conditions, the specificity for 3 markers positive out of 24 markers would be at 87.9%, and thus would most likely not be used. Use of 4 markers positive out of 24 markers provides a sensitivity of 65.1% - still better than the original number of 57.7%, but now specificity improves to 95.7% with individual marker FP rates of 3%.
- condition (D) the average individual markers are at 50% sensitivity and 2%- 5% false-positives, with two markers at 80% sensitivity each but with 15% false-positives each. Under these conditions, the specificity for 3 markers positive out of 24 markers would be below 80%, and thus would not be used.
- Use of 4 markers positive out of 24 markers provides a sensitivity of 71.1% - still better than the original number of 57.7%, but now specificity is at 90.4% with individual marker FP rates of 2%. Should the individual marker FP rates rise to 3%, then 5 markers would be required, and while overall specificity would rise to 97.4%, the sensitivity would drop to 46.1%, which is worse than the original number of 57.7%.
- condition (C) provided the best improvement in overall sensitivity (71.9%) for detecting Stage I cancer, while still keeping overall specificity reasonable (95.7%) for the initial 24 marker screen, should it now include two markers with higher sensitivity (90%), but worse FP rate of 10% for each of these markers.
- the sensitivity curves provide overall sensitivity as a function of the average number of molecules in the blood for each marker, with separate curves for each minimum number of markers needed to call a sample as positive.
- the specificity curves provide overall specificity as a function of individual marker false-positive rates, again with separate curves for each minimum number of markers needed to call a sample as positive. The calculated numbers for overall Sensitivity and Specificity for 36 markers using the above two conditions are provided in the tables below.
- the test would miss 17.4%; i.e. for Stage I cancer the overall sensitivity would be 82.6% (See Figure 34A). However, if the individual marker FP rate is 3%, then if there is a 4-marker minimum, then overall FP rate is 4.8% for 36 markers, for a specificity of 95.2% (See Figure 34B).
- the test would miss 34.1%; i.e. for Stage I cancer the sensitivity would be 65.8% (See Figure 34A). While the specificity is reasonable, limiting the number of samples that would need to be retested in the second step of the assay, the assay would miss 1/3 rd of the earliest cancers.
- condition (C) the average individual markers are at 50% sensitivity and 2%- 5% false-positives, with two markers at 90% sensitivity each but with 10% false-positives each. Under these conditions, use of 5 markers positive out of 36 markers provides a sensitivity of 76.3% - still better than the original number of 65.8%, but now specificity is at 97.0% with individual marker FP rates of 2%. Should the individual marker FP rates rise to 3%, then overall specificity drops to 89.8%.
- condition (C) provided the best improvement in overall sensitivity (76.3%) for detecting Stage I cancer, while still keeping overall specificity reasonable (97.0%) for the initial 36 marker screen, should it now include two markers with higher sensitivity (90%), but worse FP rate of 10% for each of these markers.
- Figures 49-50 illustrate results for calculated overall Sensitivity and Specificity for 48 markers using the aforementioned 2 conditions: (A) Average individual markers at 50% sensitivity and 2%-5% false-positives, with one marker (i.e. a protein marker) at 90% sensitivity but with 10% false-positives; and (C) Average individual markers at 50% sensitivity and 2%-5% false-positives, with two markers at 90% sensitivity each but with 10% false-positives each.
- the sensitivity curves provide overall sensitivity as a function of the average number of molecules in the blood for each marker, with separate curves for each minimum number of markers needed to call a sample as positive.
- the specificity curves provide overall specificity as a function of individual marker false-positive rates, again with separate curves for each minimum number of markers needed to call a sample as positive.
- the calculated numbers for overall Sensitivity and Specificity for 48 markers using the above two conditions are provided in the tables below. Table 26.
- the test would miss 15.1%; i.e. for Stage I cancer the overall sensitivity would be 84.9% (See Figure 35A). However, if the individual marker FP rate is 3%, then if there is a 5-marker minimum, then overall FP rate is 4.2% for 48 markers, for a specificity of 95.8% (See Figure 35B). At 5 markers minimum, for Stage I cancer (at about 150 molecules of each positive marker in the blood), the test would miss 28.4%; i.e. for Stage I cancer the sensitivity would be 71.6% (See Figure 35A). While the specificity is reasonable, limiting the number of samples that would need to be retested in the second step of the assay, the assay would miss a little over 1/4 th of the earliest cancers.
- condition (C) For condition (C), with average individual markers at 50% sensitivity and 2%-5% false-positives, with two markers at 90% sensitivity each but with 10% false-positives each, see graph in Figure 49. Under these conditions, use of 5 markers positive out of 48 markers provides a sensitivity of 90.9% - still better than the original number of 71.6%, but now specificity is at 97.0% with individual marker FP rates of 2%. If the FP rate were 3%, this would require use of 6 markers positive out of 48 markers to provide a sensitivity of 81.0% - still better than the original number of 71.6%, but now specificity changes to 95.5% with individual marker FP rates of 3%.
- the receiver operating characteristic (ROC) curves may be calculated by plotting Sensitivity vs.1-Specificity. Since these are theoretical calculations, the curves were generated for different levels of average marker false-positive rates of 2%, 3%, 4%, and 5%.
- the AUC Ana under curve
- Sensitivity with 10% FP 24 markers, with average individual marker at 50% Sensitivity with 2%-3% FP, and two markers at 90% Sensitivity with 10% FP; 36 markers, with average individual marker at 50% Sensitivity with 2%-3% FP, and one marker at 90% Sensitivity with 10% FP; 36 markers, with average individual marker at 50% Sensitivity with 2%-3% FP, and two markers at 90% Sensitivity with 10% FP; 48 markers, with average individual marker at 50% Sensitivity with 2%-3% FP, and one marker at 90% Sensitivity with 10% FP; and 48 markers, with average individual marker at 50% Sensitivity with 2%-3% FP, and two markers at 90% Sensitivity with 10% FP; and are provided in the Tables below.
- AUC values are at 77% with 24 markers (average individual marker at 50% Sensitivity), improve to 91% with 24 markers (average individual marker at 50% Sensitivity, and one marker at 90% Sensitivity with 10% FP), but decrease to 83% with 24 markers (average individual marker at 50% Sensitivity, and two markers at 90% Sensitivity with 10% FP); AUC values are at 87% with 36 markers (average individual marker at 50%
- Sensitivity improve to 91% with 36 markers (average individual marker at 50% Sensitivity, and one marker at 90% Sensitivity with 10% FP), but decrease to 85% with 36 markers (average individual marker at 50% Sensitivity, and two markers at 90% Sensitivity with 10% FP); and AUC values are at 89% with 48 markers (average individual marker at 50% Sensitivity), improve to 91% with 48 markers (average individual marker at 50% Sensitivity, and one marker at 90% Sensitivity with 10% FP), and improve slightly to 92% with 48 markers (average individual marker at 50% Sensitivity, and two markers at 90% Sensitivity with 10% FP).
- Figures 51-53 illustrate results for calculated overall Sensitivity and Specificity for 24, 36, and 48 markers, respectively. These graphs are based on the assumption that the average individual marker sensitivity is 66%, and the average individual marker false-positive rate is from 2% to 5%.
- the sensitivity curves provide overall sensitivity as a function of the average number of molecules in the blood for each marker, with separate curves for each minimum number of markers needed to call a sample as positive.
- the specificity curves provide overall specificity as a function of individual marker false-positive rates, again with separate curves for each minimum number of markers needed to call a sample as positive.
- the calculated numbers for overall Sensitivity and Specificity for 24, 36, and 48 markers, respectively, where the average individual marker sensitivity is 50% (as described previously) or 66% are provided in the tables below. Table 32.
- Stage I cancer at about 150 molecules of each positive marker in the blood
- overall sensitivity improves from 35.3% to 56.7%, when the average individual marker sensitivity improves from 50% to 66% (See Figure 33A and Figure 50A).
- overall sensitivity improves from 82.6% to 93.8%, when the average individual marker sensitivity improves from 50% to 66%, for detecting Stage I cancer (at about 150 molecules of each positive marker in the blood, see Figures 34A and 52A).
- the receiver operating characteristic (ROC) curves may be calculated by plotting Sensitivity vs.1-Specificity. Since these are theoretical calculations, the curves were generated for different levels of average marker false-positive rates of 2%, 3%, 4%, and 5%.
- the AUC values, calculated for ROC curves for 24 markers, with average individual marker at 66% Sensitivity with 2%-3% FP; 36 markers, with average individual marker at 66% Sensitivity with 2%-3% FP; and 48 markers, with average individual marker at 66% Sensitivity with 2%-3% FP; are provided in the Table below.
- AUC values are at 77% with 24 markers (average individual marker at 50% Sensitivity), improve to 87% with 24 markers (average individual marker at 66% Sensitivity); AUC values are at 87% with 36 markers (average individual marker at 50% Sensitivity), improve to 95% with 36 markers (average individual marker at 66% Sensitivity); and AUC values are at 89% with 48 markers (average individual marker at 50% Sensitivity), improve to 97% with 48 markers (average individual marker at 66% Sensitivity).
- the test would miss 6.2%; i.e. for Stage I & II cancer the overall sensitivity would be 93.8% (See Figure 33A), e.g. the test would correctly identify 93.8% of individuals with disease, which would be 126,630 individuals (out of 135,000 new cases).
- the positive predictive value would be
- 126,630/(126,630 + 1,712,000) around 6.8%, in other words, only one in 14 individuals who tested positive would actually have colorectal cancer, the rest would be false-positives.
- the test would miss 1.4%; i.e. for Stage I & II cancer the overall sensitivity would be 98.6% (See Figure 50A), e.g. the test would correctly identify 98.6% of individuals with disease, which would be 133,110 individuals (out of 135,000 new cases).
- the positive predictive value would be
- 133,110/(133,110 + 1,712,000) around 7.2%, in other words, only one in 14 individuals who tested positive would actually have colorectal cancer, the rest would be false-positives.
- the FP is low, i.e.2%, then there is marginal benefit in going from an average marker sensitivity of 50% to an average marker sensitivity of 66%.
- 90,540/(90,540 + 428,000) around 17.5%, in other words, one in 5.7 individuals who tested positive would actually have colorectal cancer, the rest would be false-positives.
- a PPV of 17.5% is quite respectable, however, it would be achieved at the cost of missing 28.5% of early cancer.
- the test would miss 10.0%; i.e. for Stage I & II cancer the overall sensitivity would be 90.0% (See Figure 50A), e.g. the test would correctly identify 90.0% of individuals with disease, which would be 121,500 individuals (out of 135,000 new cases).
- a PPV of 22.1% is excellent, and further, it would be achieved at the cost of missing only 10% of early cancer.
- the FP is more realistic i.e.4%, then there is a significant benefit in going from an average marker sensitivity of 50% to an average marker sensitivity of 66%.
- Stage I within them, who are non- compliant to testing, for the purposes of this calculation, assume that the average late cancer was once the average early cancer, and thus individuals with Stage I cancer would be about 40,500 individuals. With the assumption of these samples containing at least 150 molecules with one mutation in the blood, such a test would find 8,910 individuals (out of 40,500 individuals with Stage I cancer) with colorectal cancer. However, with a specificity for sequencing at 98%, there would be about 2.1 million false-positives. The positive predictive value of such a test would be around 0.4%, in other words, only one in 236 individuals who tested positive would actually have Stage I colorectal cancer, the rest would be false-positives.
- the first step has 24 methylation markers specific to GI cancers
- the second step has 48 methylation markers specific to colorectal cancer.
- the average individual marker sensitivity is set at 66%.
- the calculations are done with the anticipation of an average of 150 methylated molecules per positive marker in the blood.
- the first step would identify 5,778,000 individuals (out of 107,000,000 total adults over 50 in the U.S.) which would include at 76.2% sensitivity or about 30,861 individuals with Stage I colorectal cancer (out of 40,500 individuals with Stage I cancer).
- the ultimate goal is to develop a high-throughput scalable test to detect the majority of cancers that occur worldwide.
- the solid tumor cancers have been grouped into the following subclasses, as listed below in Tables 42, 43, and 44 for both sexes, for men, and for women.
- liquid cancers does not include liquid cancers, nor some of the less common solid tumors.
- Worldwide incidence (numbers in thousands) of liquid tumors include Non-Hodgkin lymphoma (225), leukemia (187), multiple myeloma (70), and Hodgkin lymphoma (33). These would be detected in a separate test not discussed herein. Further, the list excludes melanoma (287) and brain tumors (134). Testing for these would be done with separate sets of markers, optimized as described above for colorectal cancer.
- cancers listed in the tables above are of extreme medical importance (e.g., mesothelioma, thyroid cancer, and the three different subcategories of kidney cancer), their biology is sufficiently different as to usually merit a separate set of markers, again, optimized as described above for colorectal cancer.
- a Pan-Oncology test is developed that would include the following major cancers by the following groupings: Group 1 (colorectal, stomach, and esophagus); Group 2 (breast, endometrial, ovarian, cervical, and uterine); Group 3 (lung and head & neck); Group 4 (prostate and bladder); and Group 5 (liver, pancreatic, or gall bladder). Note that some cancers within Group 3 may be tested as a sputum sample, and cancers in Group 4 may be tested as a urine sample.
- the first step is to identify markers that cover multiple cancers in one or more of the above groups.
- the markers should be sufficiently diverse as to cover cancers in all 5 groups.
- a first step of the assay would use a set of 96 markers that on average comprise of at least 36 markers with 50% sensitivity that covers each of the aforementioned 16 types of solid tumors (covered in the 5 Groups; see Figure 1E; for 66% sensitivity, see Figure 1C). If at least 5 markers are positive, the assay would then move to a second step that would be used to verify the initial results and identify the most probable tissue of origin. In most cases, more than 5 markers would be positive, and then pattern of distribution of these methylation markers would guide the choice of which groups to test in the second step.
- the second step of the assay would test, on average, 2 or more sets of the group-specific markers.
- the second step of the assay would use 2 or more sets of 64 group-specific markers that, on average, comprise of at least 36 markers with 50% sensitivity that covers each of the aforementioned types of solid tumors that may be present in that group (for 66% sensitivity, see Figure 1D).
- the physician can identify the most probable tissue of origin, and subsequently send the patient to the appropriate imaging.
- pan-oncology markers that meet the criteria for use in a set of 96 markers that on average comprise of at least 36 markers with 50% sensitivity that covers each of the aforementioned 16 types of solid tumors.
- pan- oncology markers include but are not limited to cancer-specific microRNA markers, cancer- specific ncRNA and lncRNA markers, cancer-specific exon transcripts, tumor-associated antigens, cancer protein markers, protein markers that can be secreted by solid tumors into the blood, common mutations, primary CpG sites that are solid tumor and tissue specific markers, chromosomal regions or sub-regions within which are primary CpG sites that are solid tumor and tissue specific markers, and primary and flanking CpG sites that are solid tumor and tissue specific markers. Methods for detecting said markers have been discussed supra, and these markers are listed below and in accompanying figures.
- Blood-based, solid tumor-specific microRNA markers derived through analysis of TCGA microRNA datasets includes, but is not limited to, the following markers: (mir ID , Gene ID); hsa-mir-21, MIR21; hsa-mir-182, MIR182; hsa-mir-454, MIR454; hsa-mir-96, MIR96; hsa- mir-183, MIR183; hsa-mir-549, MIR549; hsa-mir-301a, MIR301A; hsa-mir-548f-1, MIR548F1; hsa-mir-301b, MIR301B; hsa-mir-103-1, MIR1031; hsa-mir-18a, MIR18A; hsa-mir-147b, MIR147B; hsa-mir-4326, MIR4326; and hsa-mir-573, MIR573. These markers may be present in ex
- Figure 53 provides a list of blood-based, solid tumor-specific ncRNA and lncRNA markers derived through analysis of various publicly available Affymetrix Exon ST CEL data, which were aligned to GENCODE annotations to generate ncRNA and lncRNA transcriptome datasets. Comparative analyses across these datasets (various cancer types, along with normal tissues, and peripheral blood) were conducted to generate the ncRNA and lncRNA markers list. Such lncRNA and ncRNA may be enriched in exosomes or other protected states in the blood.
- Figure 54 provides a list of blood-based solid tumor-specific exon transcripts that may be enriched in exosomes, tumor-associated vesicles, or other protected states in the blood. Overexpressed oncogene transcripts, or transcripts of mutant oncogenes may be enriched in exosomes, as they may drive spread of the cancer.
- Figure 55 provides a list of cancer protein markers, identified through mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from solid tumors, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Protein markers that can be secreted by solid tumors into the blood include, but are not limited to: (Protein name, UniProt ID); Uncharacterized protein C19orf48, Q6RUI8; Protein FAM72B, Q86X60; Protein FAM72D, Q6L9T8; Hydroxyacylglutathione hydrolase-like protein, Q6PII5; Putative methyltransferase NSUN5, Q96P11; RNA pseudouridylate synthase domain-containing protein 1, Q9UJJ7; Collagen triple helix repeat-containing protein 1, Q96CG8; Interleukin-11, P20809; Stromelysin-2, P09238; Matrix metalloproteinase-9, P14780; Podocan-like protein 1, Q6PEZ8;Putative peptide YY-2, Q9NRI6; Osteopontin, P10451;
- TP53 tumor protein p53
- TTN titin
- MUC16 mucin 16
- KRAS Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- a deep analysis of the TCGA database of methylation markers that are absent in blood but on average are present in solid tumor types at 50% sensitivity show three general categories of clusters: (i) Markers that are present in colorectal cancers, and related GI cancer (stomach & esophagus), (ii) Markers that are present in colorectal cancers, and related GI cancer (stomach & esophagus), as well as other tumors, and (iii) Markers that are mostly absent in colorectal cancers, but present in other tumors.
- the first 48 markers comprised of about 12 markers that were strongly represented in Group 2 tumors, about 12 markers that were strongly represented in Group 3 tumors, about 12 markers that were strongly represented in Group 4 tumors, and about 12 markers that were strongly represented in Group 5 tumors.
- the remaining 48 markers comprised of about 12 markers that were strongly represented in Groups 1 & 2 tumors, about 12 markers that were strongly represented in Groups 1 & 3 tumors, about 12 markers that were strongly represented in Groups 1 & 4 tumors, and about 12 markers that were strongly represented in Groups 1 & 5 tumors.
- Figure 56 provides a list of primary CpG sites that are solid tumors and tissue- specific markers, that may be used to identify the presence of solid tumors from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 57 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are solid tumors and tissue-specific markers, that may be used to identify the presence of solid tumors from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Table 47 in the experimental section, provides simulations of the 96-marker assay, with average sensitivities of 50%, for identifying most probably group for tissue of origin, for both sexes. A set of 96 markers was assembled as above and the percentage of samples positive within each of the cancer patients in the TCGA and GEO databases was assessed.
- the columns reflect the total percent patients positive for each of the markers divided by the total number of markers used– for the first row of all cancers, that would be 96 markers.
- adenocarcinoma 45 marker equivalents
- lung squamous cell carcinoma 45 marker equivalents
- head & neck 48 marker equivalents
- bladder 53 marker equivalents
- pancreatic 39 marker equivalents
- gall bladder 69 marker equivalents
- markers were then re-ordered for each of the above cancer types such that the most prevalent markers were listed first. For example, with CRC, of the 96 markers, 54 markers gave scores above 55 (i.e. were positive in greater than 55% of the 395 patients) and 9 gave scores of between 25 and 54 (i.e. were positive for from 25% to 54% of the 395 patients). Half of the higher, and a third of the lower set, for a total of 30 markers were distributed into two marker test sets, designated“CRC1” and“CRC2” (Table 47, rows 2 & 3). These marker sets would reflect an ideal result if half the markers with the potential to be positive are detected in the assay.
- marker sets that are in the same range or higher than the number of positive markers for that cancer type are also shown with a light grey background.
- a patient with colorectal, stomach, or esophageal cancer will be scored as potentially positive with stomach cancer. This makes sense as the markers for these three cancers ovelap (i.e., they all bin to Group 1). They could be distinguished in step 2 of the assay on the group 1 markers, where these markers are more cancer types specific and tease out the most probable cancer type. Evaluation of the ST-Pt column shows that simulations for one of the two LUAD, BLAD, and both PANC also gave scores that might be interpreted as stomach cancer.
- the first step is not always able to pinpoint what Groups should be tested in the second step of the assay.
- most of the ambiguity is within group members (i.e. Group 2), and this makes sense, since the markers were chosen to maximize the ability to chose which groups to test in the second step.
- Tables 48 and 49 (see prophetic experimental section) takes the aforementioned results in the simulations in Table 47 and multiplies them by the percent incidence of the given cancer type for that gender (see tables 37 and 38 respectively), and the result is adjusted to the same order of magnitude (multiple by 10).
- the concept is for the physician to take into account that a lower score for a high incidence cancer (such as CRC) may be a more common tissue of origin for a higher score for a low incidence cancer (such as lung squamous cell carcinoma).
- Tables 48 and 49 show the level of ambiguity in identifying tissue of origin is higher among female patients than among male patients, as indicated by the number of cells highlighted in grey that are not on the diagonal. In all cases, the physician will need to incorporate all data, such as smoking history, and not just molecular data to determine the most likely tissue of origin before sending the patient to confirmatory imaging.
- each group with a set of 64 markers that on average comprise of at least 36 markers with 50% sensitivity that covers each of the aforementioned 16 types of solid tumors in the following groups: Group 1 (colorectal, stomach, and esophagus); Group 2 (breast, endometrial, ovarian, cervical, and uterine); Group 3 (lung and head & neck); Group 4 (prostate and bladder); and Group 5 (liver, pancreatic, or gall bladder).
- Group-specific and cancer type-specific markers include, but are not limited to, cancer-specific microRNA markers, cancer-specific ncRNA and lncRNA markers, cancer-specific exon transcripts, tumor-associated antigens, cancer protein markers, protein markers that can be secreted by solid tumors into the blood, common mutations, primary CpG sites that are solid tumor and tissue specific markers, chromosomal regions or sub-regions within which are primary CpG sites that are solid tumor and tissue specific markers, and primary and flanking CpG sites that are solid tumor and tissue specific markers. Methods for detecting said markers have been discussed supra, and listing of these markers are described for each of the groups below as well as in the corresponding figures.
- Group 1 (colorectal, stomach, and esophagus): Blood-based, colorectal, stomach, and esophageal cancer-specific microRNA markers that may be used to distinguish group 1 from other groups include, but are not limited to: (mir ID, Gene ID): hsa-mir-624, MIR624. This miRNA was identified through analysis of TCGA microRNA datasets, and may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- Blood-based, colorectal, stomach, and esophageal cancer-specific ncRNA and lncRNA markers that may be used to distinguish group 1 from other groups include, but are not limited to: [Gene ID, Coordinate (GRCh38)], ENSEMBL ID: LINC01558, chr6:167784537- 167796859, ENSG00000146521.8.
- This ncRNA was identified through comparative analysis of various publicly available Affymetrix Exon ST CEL data, which were aligned to GENCODE annotations to generate ncRNA and lncRNA transcriptome datasets. Such lncRNA and ncRNA may be enriched in exosomes or other protected states in the blood.
- Figure 58 provides a list of blood-based colorectal, stomach, and esophageal cancer-specific exon transcripts that may be enriched in exosomes, tumor-associated vesicles, or other protected states in the blood.
- Colorectal, stomach, and esophageal cancer protein encoding markers that may be used to distinguish group 1 from other groups include, but are not limited to: (Gene Symbol , Chromosome Band, Gene Title, UniProt ID): SELE, 1q22-q25, selectin E, P16581; OTUD4, 4q31.21, OTU domain containing 4, Q01804; BPI, 20q11.23, bactericidal/permeability- increasing protein, P17213; ASB4, 7q21-q22, ankyrin repeat and SOCS box containing 4, Q9Y574; C6orf123, 6q27, chromosome 6 open reading frame 123, Q9Y6Z2; KPNA3, 13q14.3, karyopherin alpha 3 (importin alpha 4), O00505; NUP98, 11p15, nucleoporin 98kDa , P52948, identified through mRNA sequences, protein expression levels, protein product concentrations
- Protein markers that can be secreted by colorectal, stomach, and esophageal cancer into the blood, and may be used to distinguish group 1 from other groups include, but are not limited to: (Protein name, UniProt ID); Bactericidal permeability-increasing protein (BPI) (CAP 57), P1721.
- BPI Bactericidal permeability-increasing protein
- a comparative analysis was performed across various TCGA datasets (tumors, normals), followed by an additional bioinformatics filter (Meinken et al.,
- Figure 59 provides a list of primary CpG sites that are colorectal, stomach, and esophageal cancer and tissue-specific markers, that may be used to identify the presence of colorectal, stomach, and esophageal cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 60 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are colorectal, stomach, and esophageal cancer and tissue-specific markers, that may be used to identify the presence of colorectal, stomach, and esophageal cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- These lists contain preferred primary CpG sites and their flanking sites, as well as alternative markers that are high in CRC, and alternative markers that are low to no-CRC, but high in stomach and/or esophageal cancers.
- Primer sets for exemplary preferred and alternate methylation markers are listed in Table 53 in the experimental section.
- Group 2 (breast, endometrial, ovarian, cervical, and uterine): Blood-based, breast, endometrial, ovarian, cervical, and uterine cancer-specific microRNA markers that may be used to distinguish group 2 from other groups include, but are not limited to: (mir ID, Gene ID): hsa- mir-1265, MIR1265. This marker was identified through analysis of TCGA microRNA datasets, which may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- Blood-based breast, endometrial, ovarian, cervical, and uterine cancer-specific exon transcripts that may be used to distinguish group 2 from other groups include, but are not limited to: (mir ID, Gene ID): hsa-mir-1265, MIR1265. This marker was identified through analysis of TCGA microRNA datasets, which may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- Breast, endometrial, ovarian, cervical, and uterine cancer protein markers that may be used to distinguish group 2 from other groups include, but are not limited to: (Gene Symbol, Chromosome Band, Gene Title, UniProt ID): RSPO2, 8q23.1, R-spondin 2, Q6UXX9; KLC4 , 6p21.1, kinesin light chain 4, Q9NSK0; GLRX, 5q14, glutaredoxin (thioltransferase), P35754.
- markers may be identified through mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from breast, endometrial, ovarian, cervical, and uterine cancers, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Protein markers that can be secreted by breast, endometrial, ovarian, cervical, and uterine cancer into the blood that may be used to distinguish group 2 from other groups include, but are not limited to: (Protein name, UniProt ID); R-spondin-2 (Roof plate-specific spondin-2) (hRspo2), Q6UXX9.
- R-spondin-2 Root plate-specific spondin-2)
- Q6UXX9 Q6UXX9.
- a comparative analysis was performed across various TCGA datasets (tumors, normals), followed by an additional bioinformatics filter (Meinken et al., 2012, as described above), which predicts the likelihood that the translated protein is secreted by the cells.
- PIK3CA phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- TTN titin
- Figure 61 provides a list of primary CpG sites that are breast, endometrial, ovarian, cervical, and uterine cancer and tissue-specific markers, that may be used to identify the presence of breast, endometrial, ovarian, cervical, and uterine cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 62 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are breast, endometrial, ovarian, cervical, and uterine cancer and tissue-specific markers, that may be used to identify the presence of breast, endometrial, ovarian, cervical, and uterine cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- These lists contain preferred primary CpG sites and their flanking sites, as well as alternative markers that may be used to distinguish breast, endometrial, ovarian, cervical, and uterine cancers.
- Primer sets for exemplary preferred and alternate methylation markers are listed in Table 54 in the experimental section.
- Group 3 lung adenocarcinoma, lung squamous cell carcinoma, and head & neck: Blood-based, lung, head, and neck cancer-specific microRNA markers that may be used to distinguish group 3 from other groups include, but are not limited to: (mir ID, Gene ID): hsa-mir- 28, MIR28. This marker was identified through analysis of TCGA microRNA datasets, and may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- Blood-based lung, head, and neck cancer-specific exon transcripts that may be used to distinguish group 3 from other groups include, but are not limited to: (Exon location, Gene); chr2: chr1:93307721-93309752:-, FAM69A; chr1:93312740-93312916:-, FAM69A; chr1:93316405-93316512:-, FAM69A; chr1:93341853-93342152:-, FAM69A; chr1:93426933- 93427079:-, FAM69A; chr7:40221554-40221627:+, C7orf10; chr7:40234539-40234659:+, C7orf10;chr8:22265823-22266009:+, SLC39A14; chr8:22272293-22272415:+, SLC39A14; chr14:39509936-395100
- Chromosome Band Gene Title, UniProt ID: STRN3, 14q13-q21, striatin, calmodulin binding protein 3, Q13033; LRRC17, 7q22.1, leucine rich repeat containing 17, Q8N6Y2; FAM69A, 1p22, family with sequence similarity 69, member A, Q5T7M9; ATF2 , 2q32, activating transcription factor 2, P15336; BHMT, 5q14.1, betaine--homocysteine S-methyltransferase, Q93088; ODZ3/TENM3, 4q34.3-q35.1, teneurin transmembrane protein 3, Q9P273; ZFHX4, 8q21.11, zinc finger homeobox 4, Q86UP3.
- markers may be identified through mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from lung, head, and neck cancers, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Protein markers that can be secreted by lung, head, and neck cancer into the blood may be used to distinguish group 3 from other groups include, but are not limited to: (Protein name, UniProt ID); Leucine-rich repeat-containing protein 17 (p37NB), Q8N6Y2.
- a comparative analysis was performed across various TCGA datasets (tumors, normals), followed by an additional bioinformatics filter (Meinken et al.,“Computational Prediction of Protein Subcellular Locations in Eukaryotes: an Experience Report,” Computational Molecular Biology 2(1):1-7 (2012), which is hereby incorporated by reference in its entirety), which predicts the likelihood that the translated protein is secreted by the cells.
- Figure 63 provides a list of primary CpG sites that are lung, head, and neck cancer and tissue-specific markers, that may be used to identify the presence of lung, head, and neck cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 64 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are lung, head, and neck cancer and tissue-specific markers, that may be used to identify the presence of lung, head, and neck from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Group 4 Blood or urine-based, prostate and bladder cancer-specific microRNA markers may be used to distinguish group 4 from other groups include, but are not limited to: (mir ID, Gene ID): hsa-mir-491, MIR491; hsa-mir-1468, MIR1468. These markers were identified through analysis of TCGA microRNA datasets, and may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood or urine.
- Blood or urine-based, prostate and bladder cancer-specific ncRNA and lncRNA markers that may be used to distinguish group 4 from other groups include, but are not limited to: [Gene ID, Coordinate (GRCh38), ENSEMBL ID]: AC007383.3, chr2:206084605- 206086564, ENSG00000227946.1; LINC00324, chr17:8220642-8224043,
- ENSG00000178977.3 These markers were identified through comparative analysis of various publicly available Affymetrix Exon ST CEL data, which were aligned to GENCODE annotations to generate ncRNA and lncRNA transcriptome datasets. Such lncRNA and ncRNA may be enriched in exosomes or other protected states in the blood or urine.
- Blood or urine-based prostate and bladder cancer-specific exon transcripts that may be used to distinguish group 4 from other groups include, but are not limited to: (Exon location, Gene); chr21:45555942-45556055:+ , C21orf33 and may be enriched in exosomes, tumor-associated vesicles, or other protected states in the blood or urine.
- Prostate and bladder cancer protein markers that may be used to distinguish group 4 from other groups include, but are not limited to: (Gene Symbol, Chromosome Band, Gene Title, UniProt ID): PMM1, 22q13, phosphomannomutase 1, Q92871. This marker may be identified through mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from lung, head, and neck cancers, which may be identified in the blood, either within exosomes, other protected states, tumor- associated vesicles, or free within the plasma, or within the urine.
- BAGE2 BAGE family member 2
- DNM1P47 dynamin 1 pseudogene 47
- FRG1BP region gene 1 family member B, pseudogene
- KRAS Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- RP11-156P1.3 TTN (titin)
- TUBB8P7 tubulin beta 8 class VIII pseudogene 7
- Figure 65 provides a list of primary CpG sites that are prostate and bladder cancer-specific markers, that may be used to identify the presence of prostate and bladder cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood or urine.
- Figure 66 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are prostate and bladder cancer specific markers, that may be used to identify the presence of prostate and bladder from cfDNA, or DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood or urine. These lists contain preferred primary CpG sites and their flanking sites that may be used to distinguish prostate and bladder cancers.
- Primer sets for exemplary methylation markers are listed in Table 56 in the experimental section.
- markers for example by increasing from 48 to 64 markers and including markers that were positive for both prostate and bladder, would rectify this situation.
- the markers were limited to those that were not methylated in normal prostate, bladder, or kidney tissue to minimize false-positive results from urine samples.
- Group 5 liver, pancreatic and gall-bladder: Blood-based, liver, pancreatic and gall-bladder cancer-specific microRNA markers that may be used to distinguish group 5 from other groups include, but are not limited to: (mir ID, Gene ID): hsa-mir-132, MIR132. This marker was identified through analysis of TCGA microRNA datasets, which may be present in exosomes, tumor-associated vesicles, Argonaute complexes, or other protected states in the blood.
- Figure 67 provides a list of blood-based, liver, pancreatic and gall-bladder cancer- specific ncRNA and lncRNA markers derived through comparative analysis of various publicly available Affymetrix Exon ST CEL data, which were aligned to GENCODE annotations to generate ncRNA and lncRNA transcriptome datasets. Such lncRNA and ncRNA may be enriched in exosomes or other protected states in the blood.
- Figure 68 provides a list of blood-based liver, pancreatic and gall- bladder cancer-specific exon transcripts that may be enriched in exosomes, tumor-associated vesicles, or other protected states in the blood.
- Figure 69 provides a list of liver, pancreatic and gall-bladder cancer protein markers, identified through mRNA sequences, protein expression levels, protein product concentrations, cytokines, or autoantibody to the protein product arising from liver, pancreatic and gall-bladder cancers, which may be identified in the blood, either within exosomes, other protected states, tumor-associated vesicles, or free within the plasma.
- Protein markers that can be secreted by liver, pancreatic and gall-bladder cancer into the blood that may be used to distinguish group 5 from other groups include, but are not limited to: (Protein name, UniProt ID); Gelsolin (AGEL) (Actin-depolymerizing factor) (ADF) (Brevin), P06396; Pro-neuregulin-2, O14511; CD59 glycoprotein (1F5 antigen) (20 kDa homologous restriction factor) (HRF-20) (HRF20) (MAC-inhibitory protein) (MAC-IP) (MEM43 antigen) (Membrane attack complex inhibition factor) (MACIF) (Membrane inhibitor of reactive lysis) (MIRL) (Protectin) (CD antigen CD59), P13987; Divergent protein kinase domain 2B (Deleted in autism-related protein 1), Q9H7Y0.
- GEL Actin-depolymerizing factor
- ADF Actin-depolymerizing factor
- Figure 70 provides a list of primary CpG sites that are liver, pancreatic and gall- bladder cancer and tissue-specific markers, that may be used to identify the presence of lung, head, and neck cancer from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- Figure 71 provides a list of chromosomal regions or sub-regions within which are primary CpG sites that are liver, pancreatic and gall-bladder cancer and tissue-specific markers, that may be used to identify the presence of liver, pancreatic and gall-bladder from cfDNA, DNA within exosomes, or DNA in other protected states (such as within CTCs) within the blood.
- liver and gall bladder were above the average 36-marker equivalents minimum, while pancreatic was below.
- the first step would identify 4,494,000 individuals (out of 107,000,000 total adults over 50 in the U.S.) which would include at 71.6% sensitivity or about 28,998 individuals with Stage I colorectal cancer (out of 40,500 individuals with Stage I cancer).
- the first step using 96 markers (48 markers for CRC) with average sensitivities of 50%, requiring a minimum of 5 markers positive, and an overall specificity of 95.8% the first step would identify 4,494,000 individuals (out of 107,000,000 total adults over 50 in the U.S.). This would include, at 90.1% sensitivity, or about 36,490 individuals with Stage I colorectal cancer (out of 40,500 individuals with Stage I cancer). However, those 4,494,000 presumptive positive individuals would be evaluated in a second step of 64 markers (48 markers for CRC) with average sensitivities of 50%, requiring a minimum of 5 markers positive.
- Stage I for the purposes of this calculation, assume that the stages are evenly divided. Thus, the number of individuals with Stage I ovarian cancer would be about 5,500 individuals. Assuming individual marker false-positive rates of 3%, the first step using 96 markers (36 markers for ovarian) with average sensitivities of 50%, requiring a minimum of 5 markers positive, and an overall specificity of 99.1%, the first step would identify 486,000 individuals (out of 54,000,000 total women ages 50-79 in the U.S.) with ovarian cancer. This would include, at 46.8% sensitivity, or about 2,574 individuals with Stage I ovarian cancer (out of 5,500 individuals with Stage I ovarian cancer).
- the first step using 96 markers (36 markers for ovarian) with average sensitivities of 50%, and requiring a minimum of 5 markers positive, with an overall specificity of 99.1% would identify 486,000 individuals (out of 54,000,000 total women ages 50-79 in the U.S.) with ovarian cancer. This would include at, 71.5% sensitivity, about 3,932 individuals with Stage I ovarian cancer (out of 5,500 individuals with Stage I ovarian cancer). However, those 486,000 presumptive positive individuals would be evaluated in a second step of 64 markers (36 markers for ovarian cancer) with average sensitivities of 50%, requiring a minimum of 5 markers positive.
- the first step using 96 markers (48 markers for CRC) with average sensitivities of 66%, and requiring a minimum of 5 markers positive then, with an overall specificity of 95.8%, the first step would identify 4,494,000 individuals with colorectal cancer (out of 107,000,000 total adults over 50 in the U.S.). This would include, at 98.0% sensitivity, about 39,690 individuals with Stage I colorectal cancer (out of 40,500 individuals with Stage I cancer). However, those 4,494,000 presumptive positive individuals would be evaluated in a second step of 64 markers (48 markers for CRC) with average sensitivities of 66%, requiring a minimum of 5 markers positive.
- the first step using 96 markers (36 markers for ovarian) with average sensitivities of 66%, and requiring a minimum of 5 markers positive then, with an overall specificity of 99.1%, the first step would identify 486,000 individuals (out of 54,000,000 total women ages 50-79 in the U.S). This would include, at 90.0% sensitivity, about 4,950 individuals with Stage I ovarian cancer (out of 5,500 individuals with Stage I ovarian cancer). However, those 486,000 presumptive positive individuals would be evaluated in a second step of 64 markers (36 markers for ovarian cancer) with average sensitivities of 66%, requiring a minimum of 5 markers positive.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Immunology (AREA)
- Analytical Chemistry (AREA)
- Genetics & Genomics (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description

























































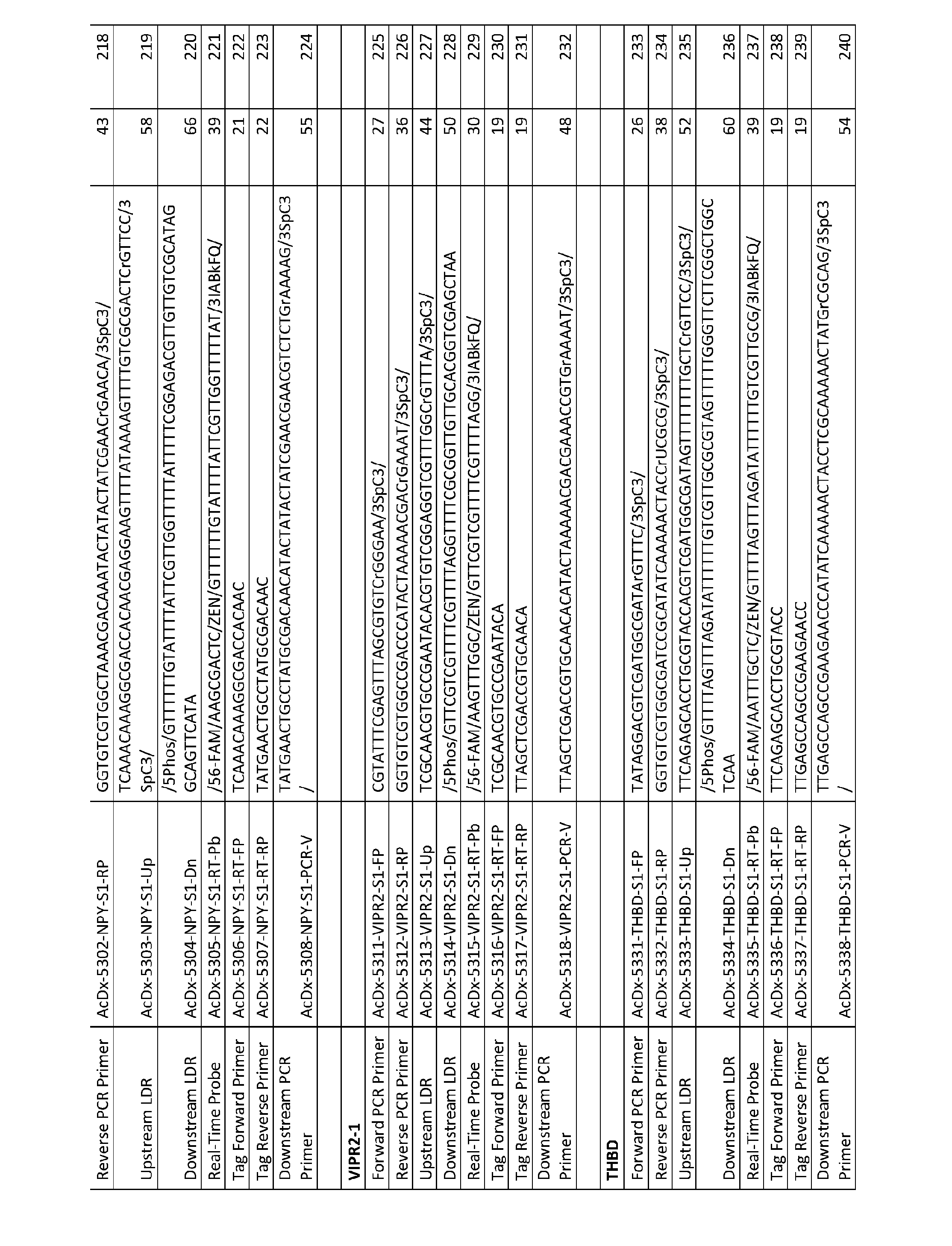
























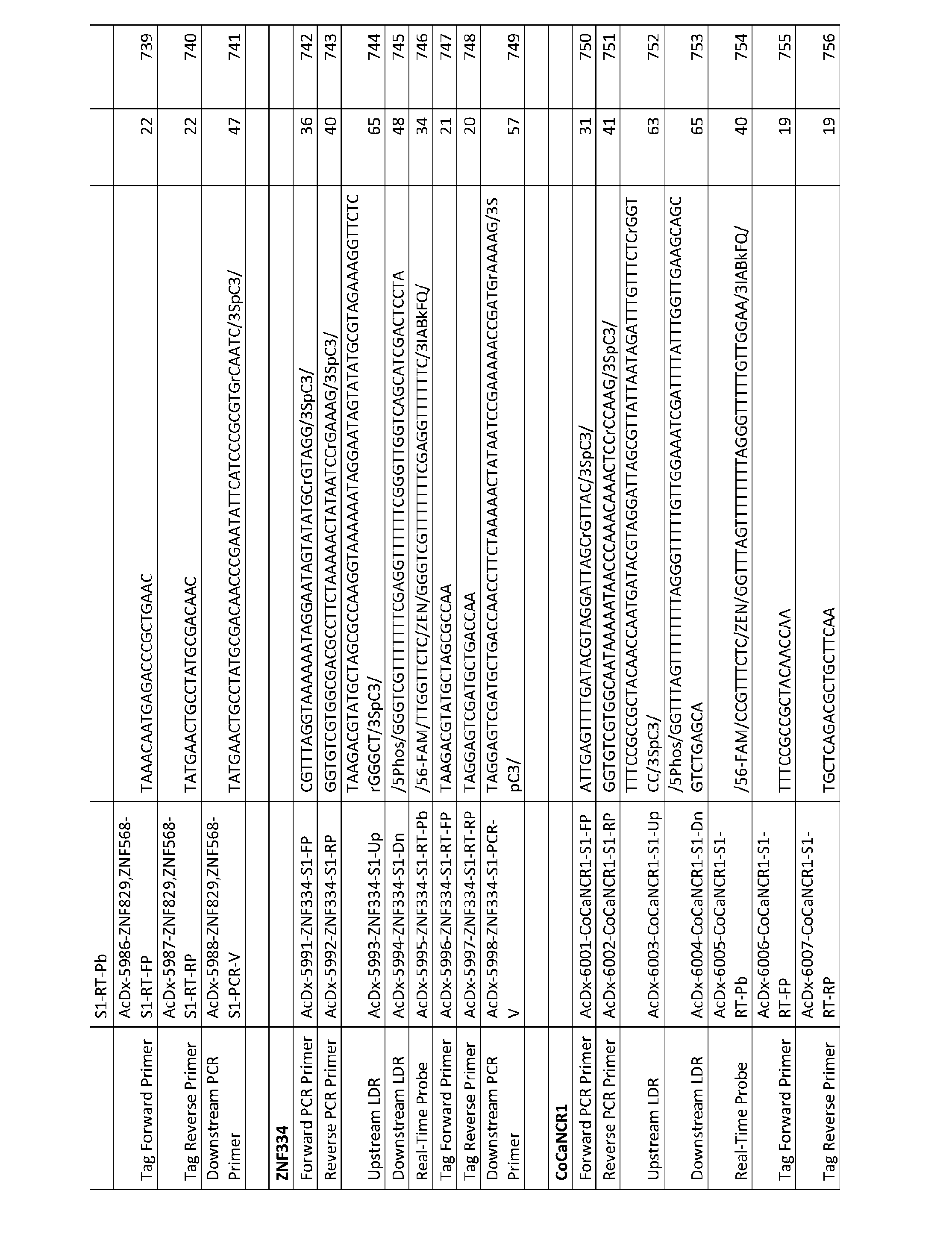

















































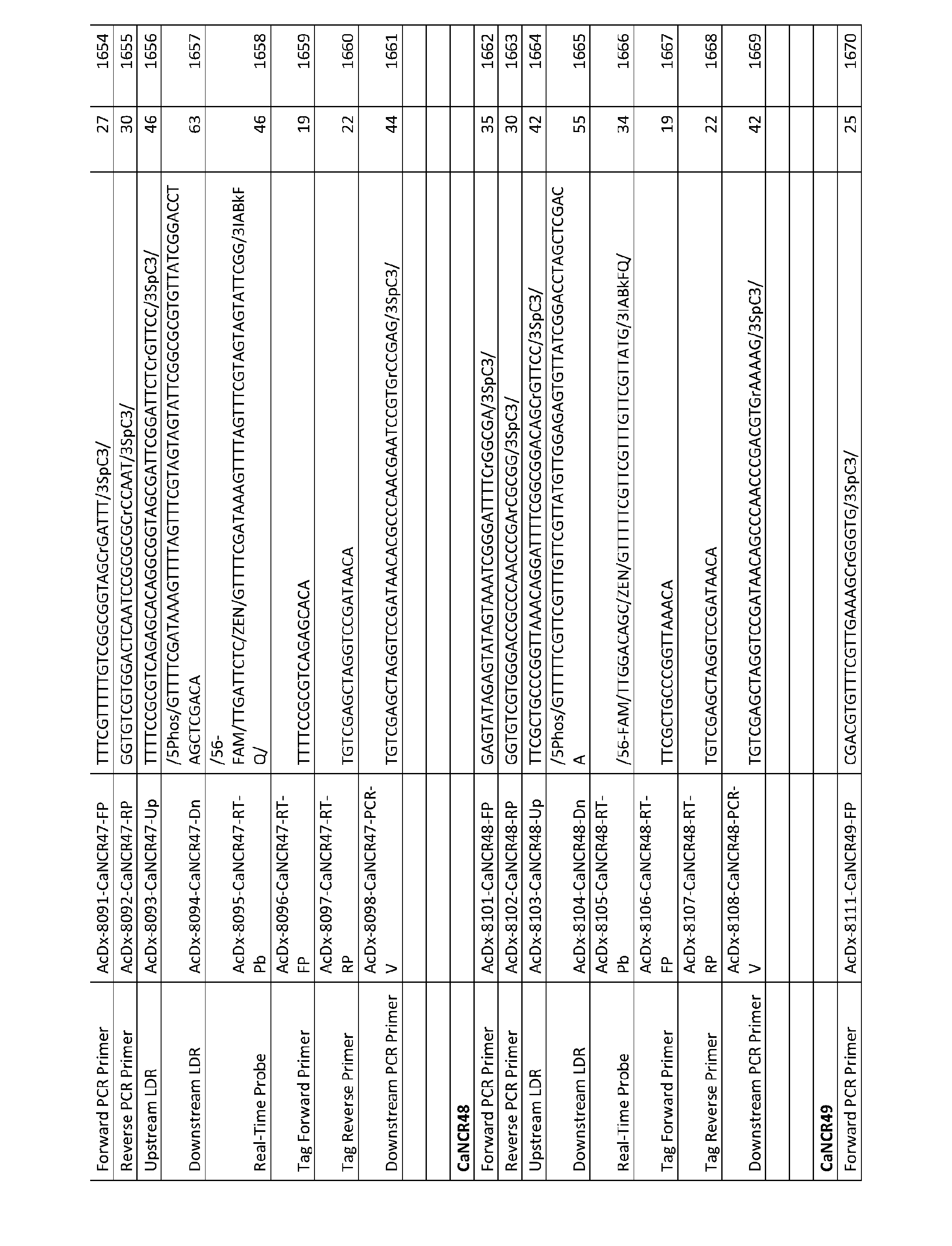






































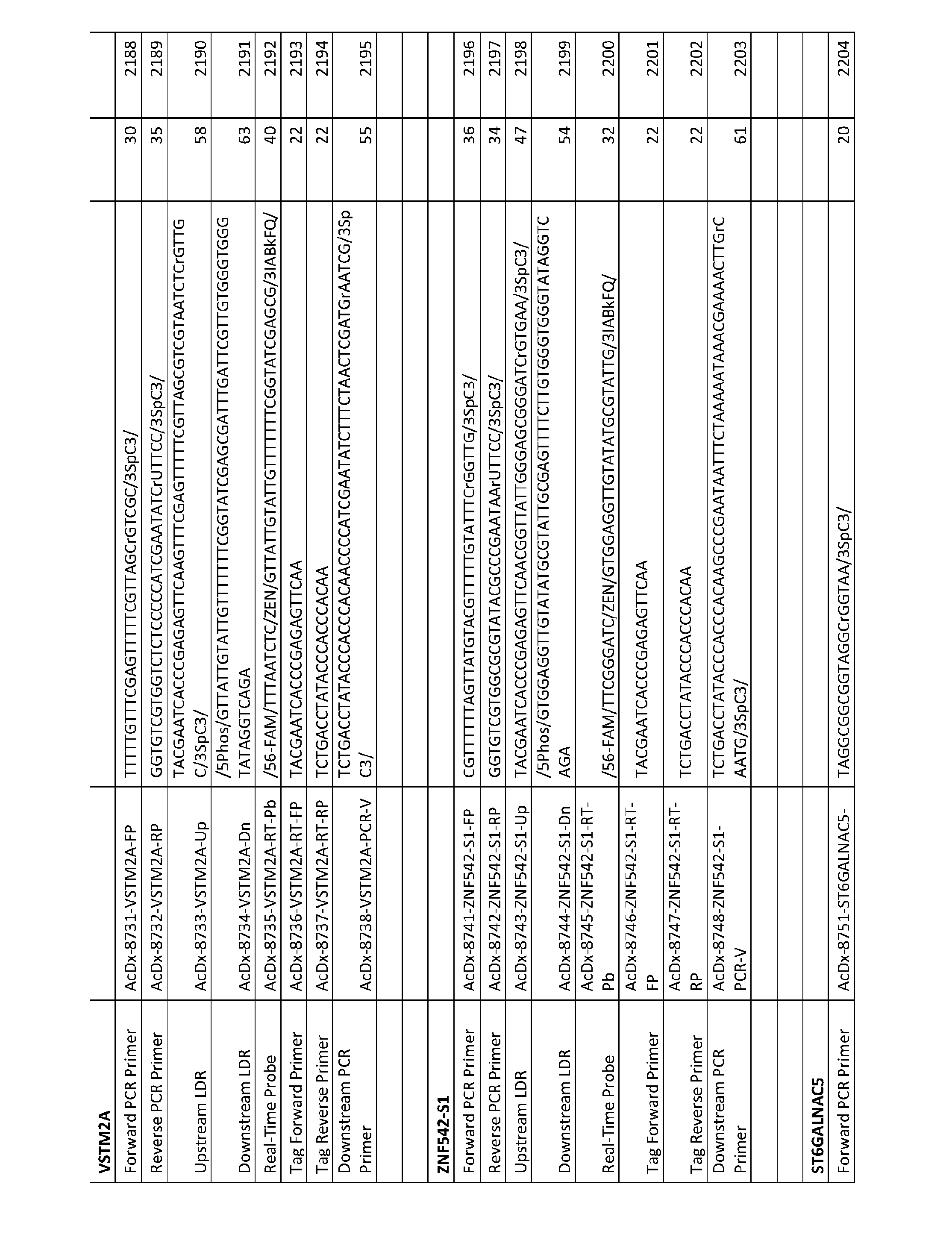










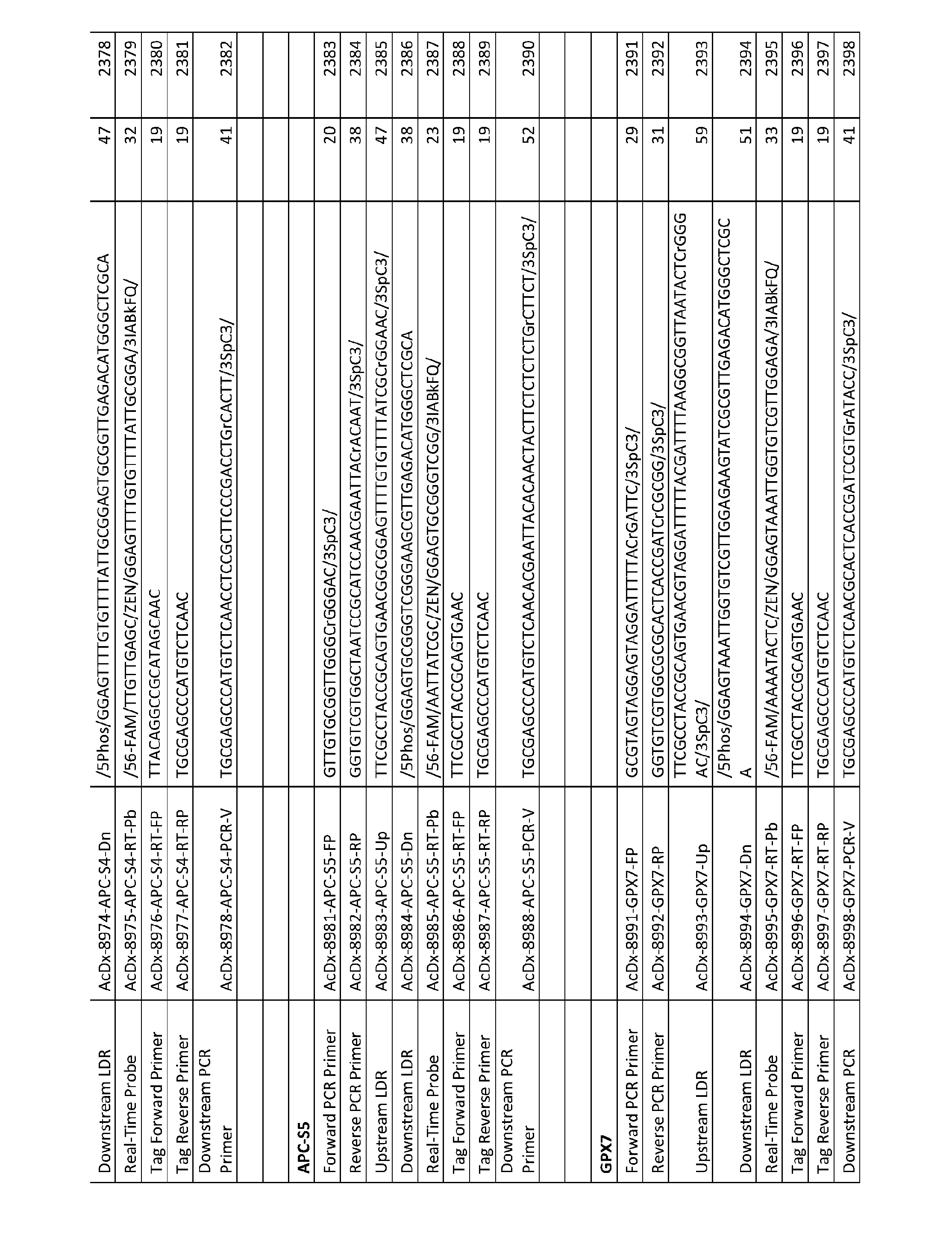




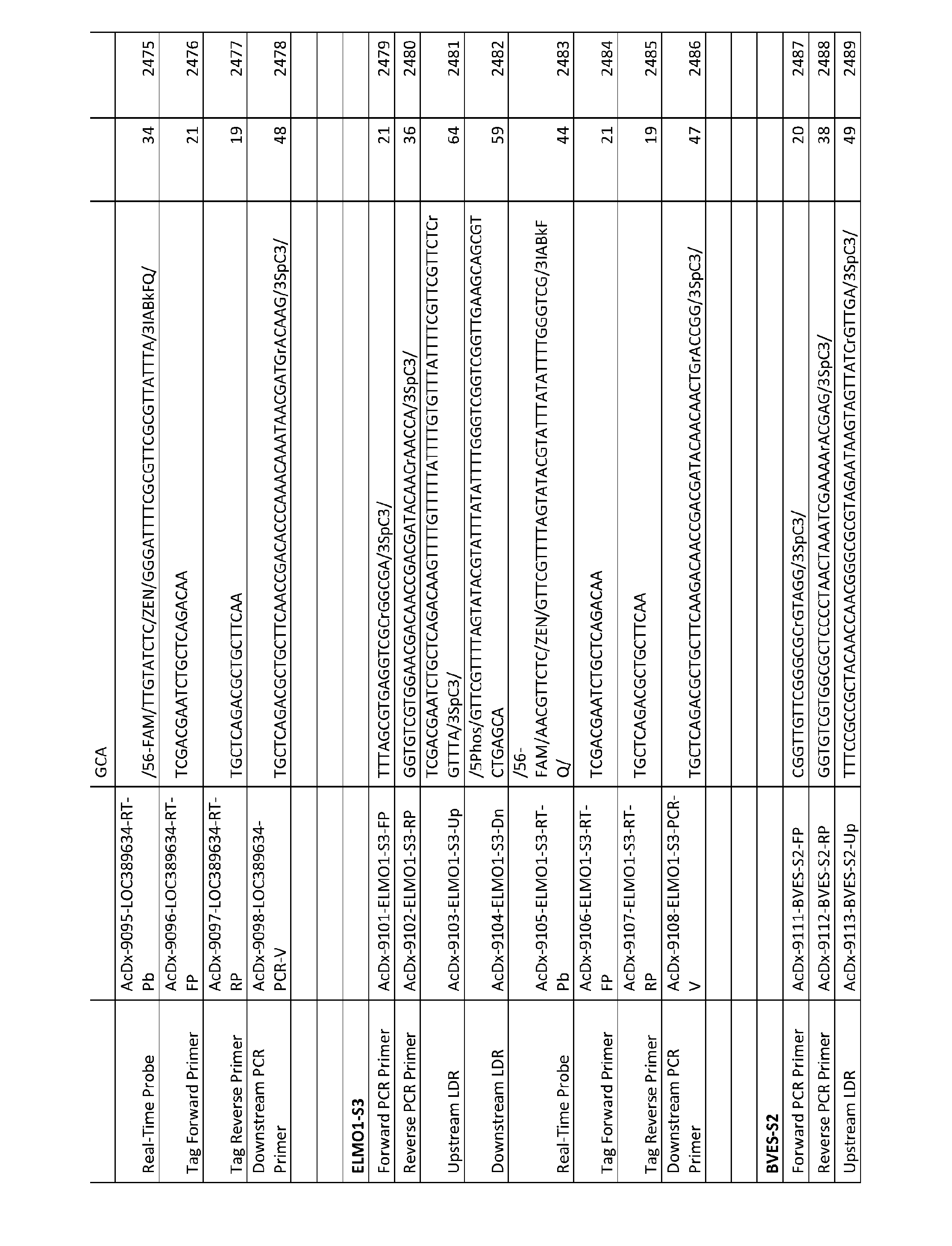



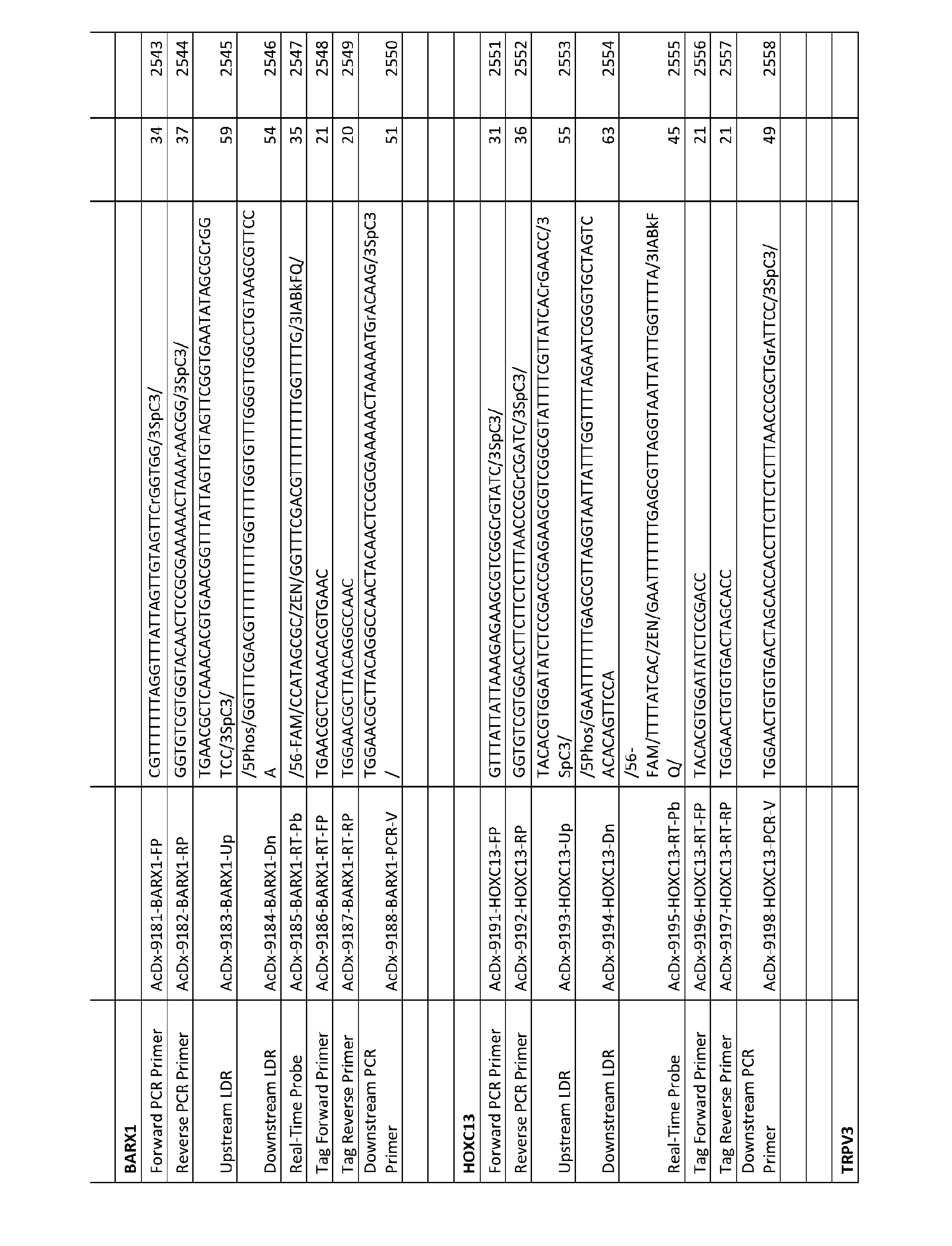
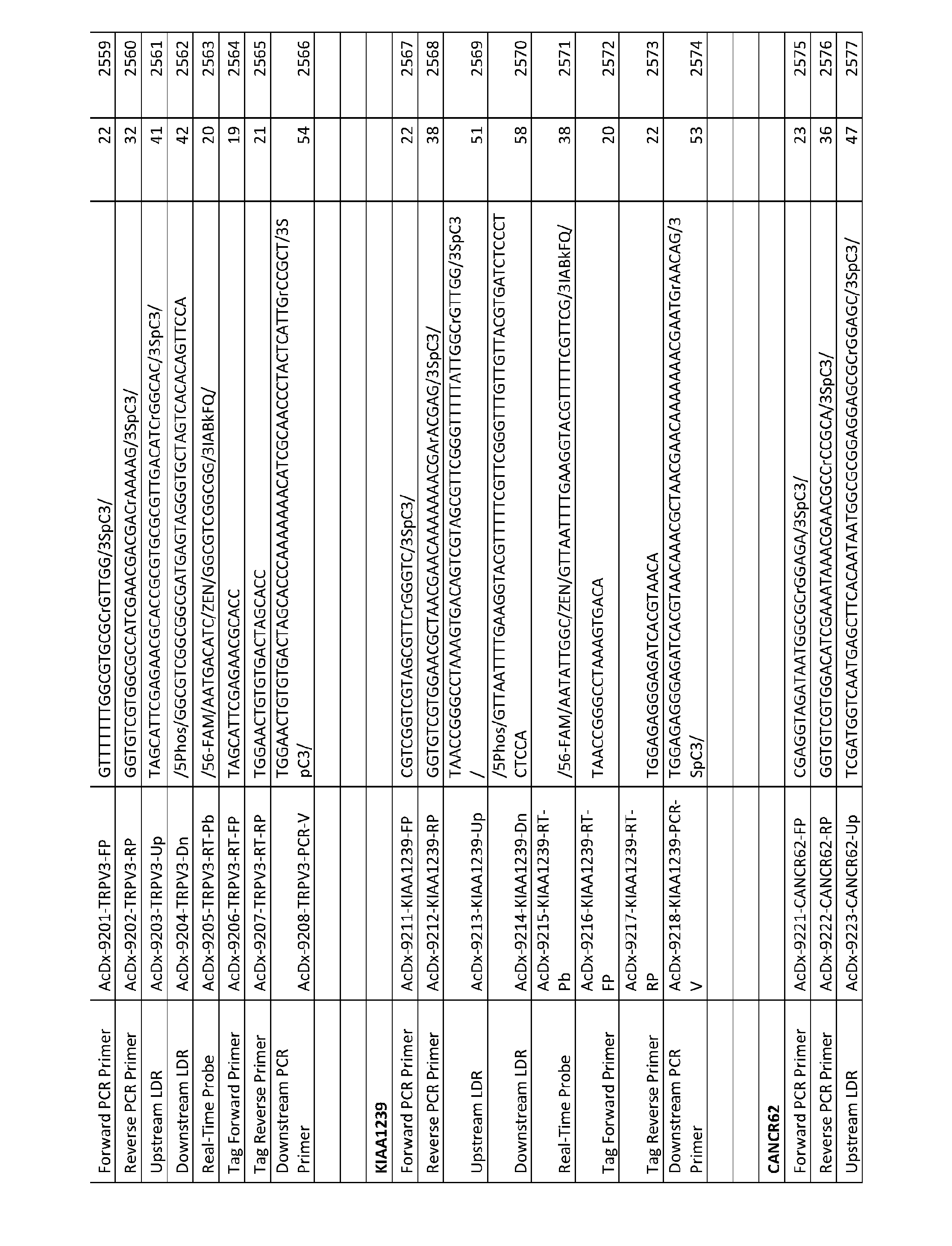













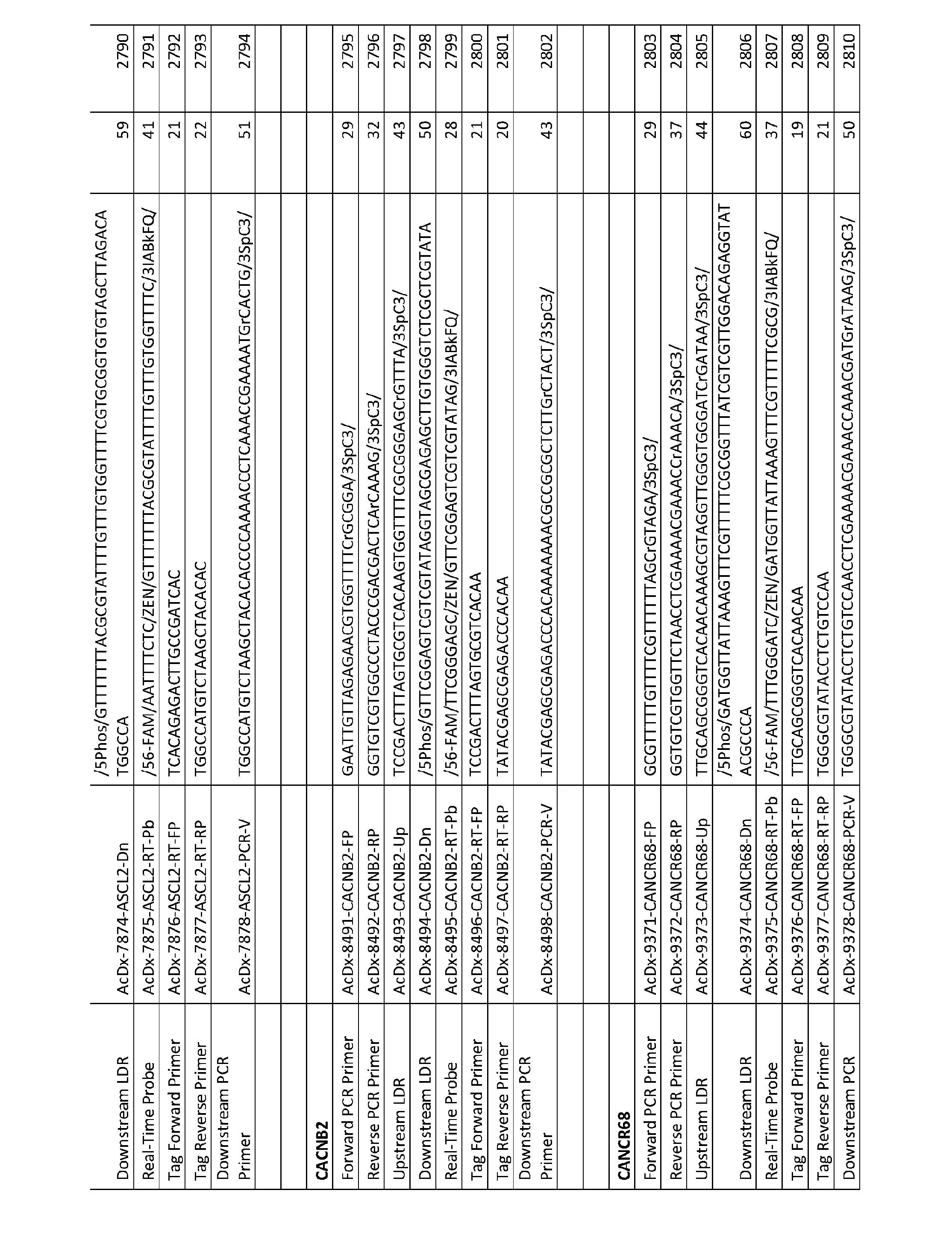





















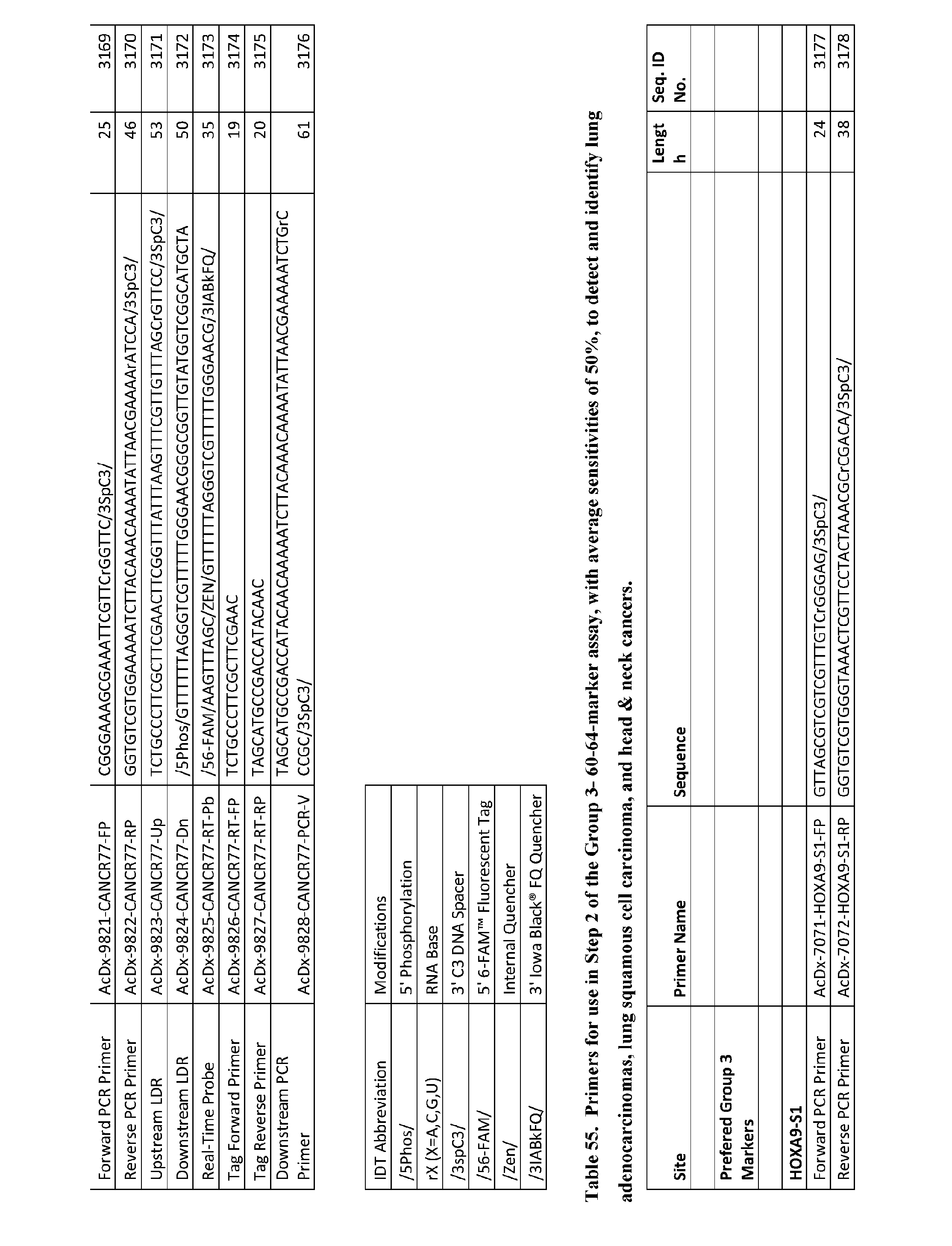







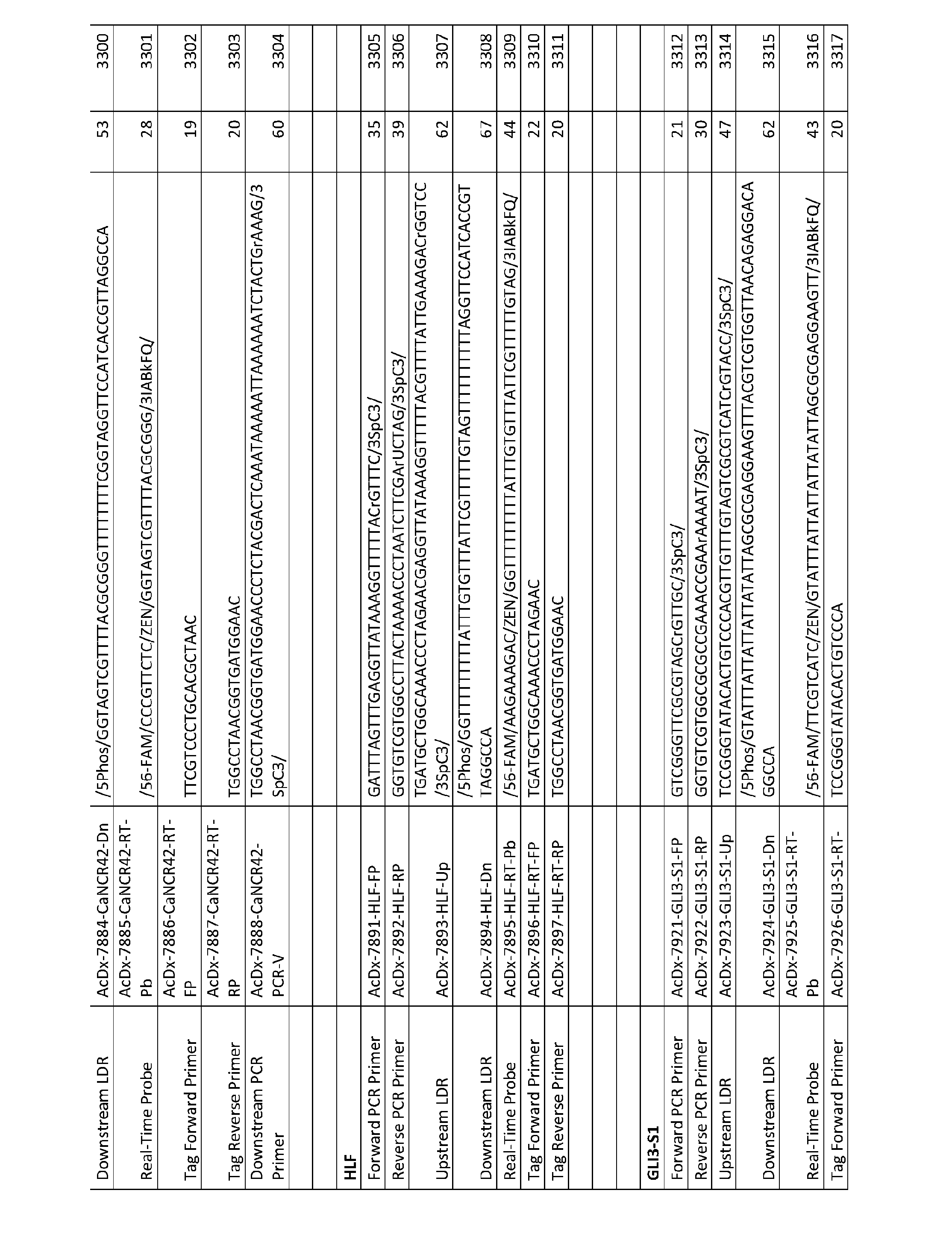









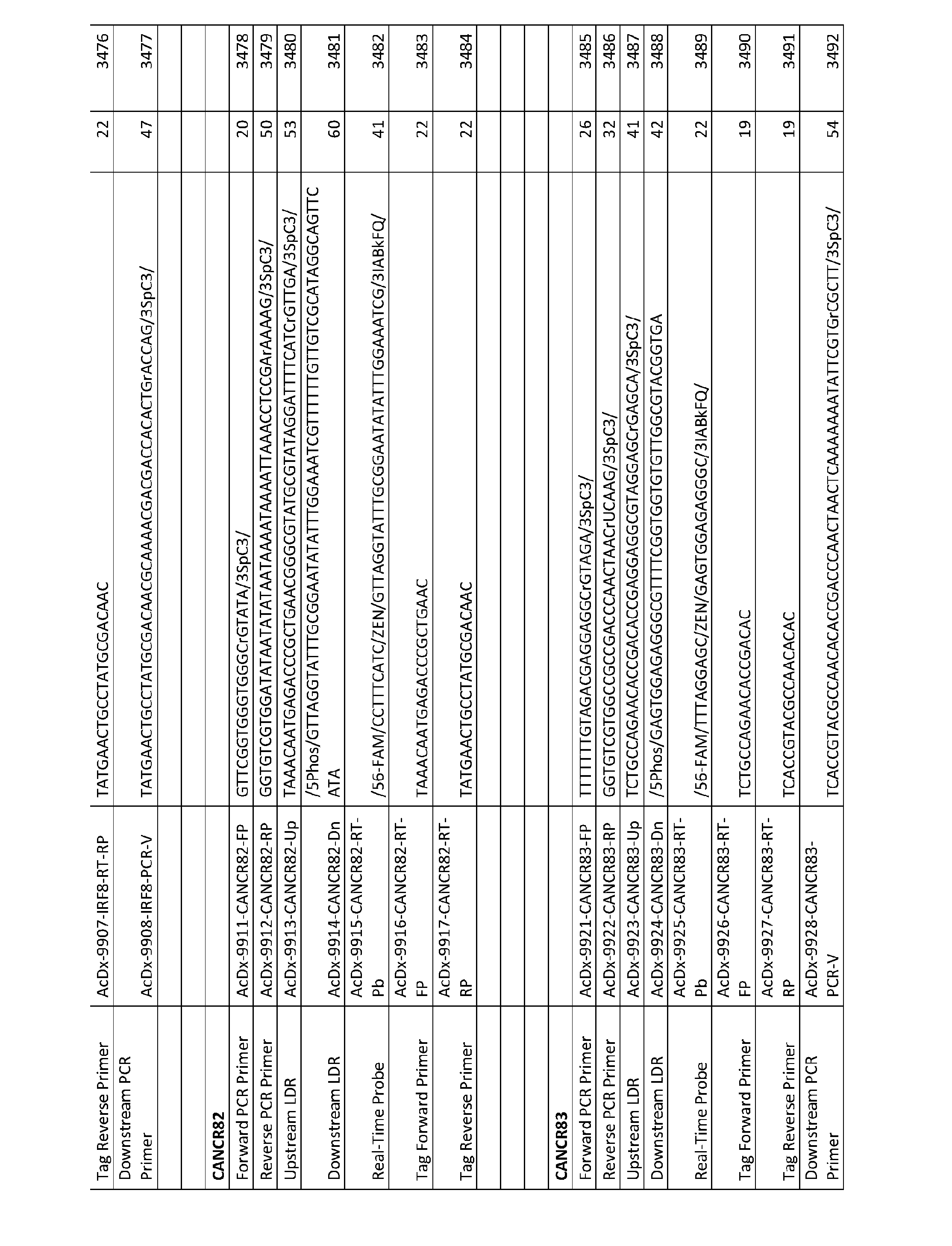













































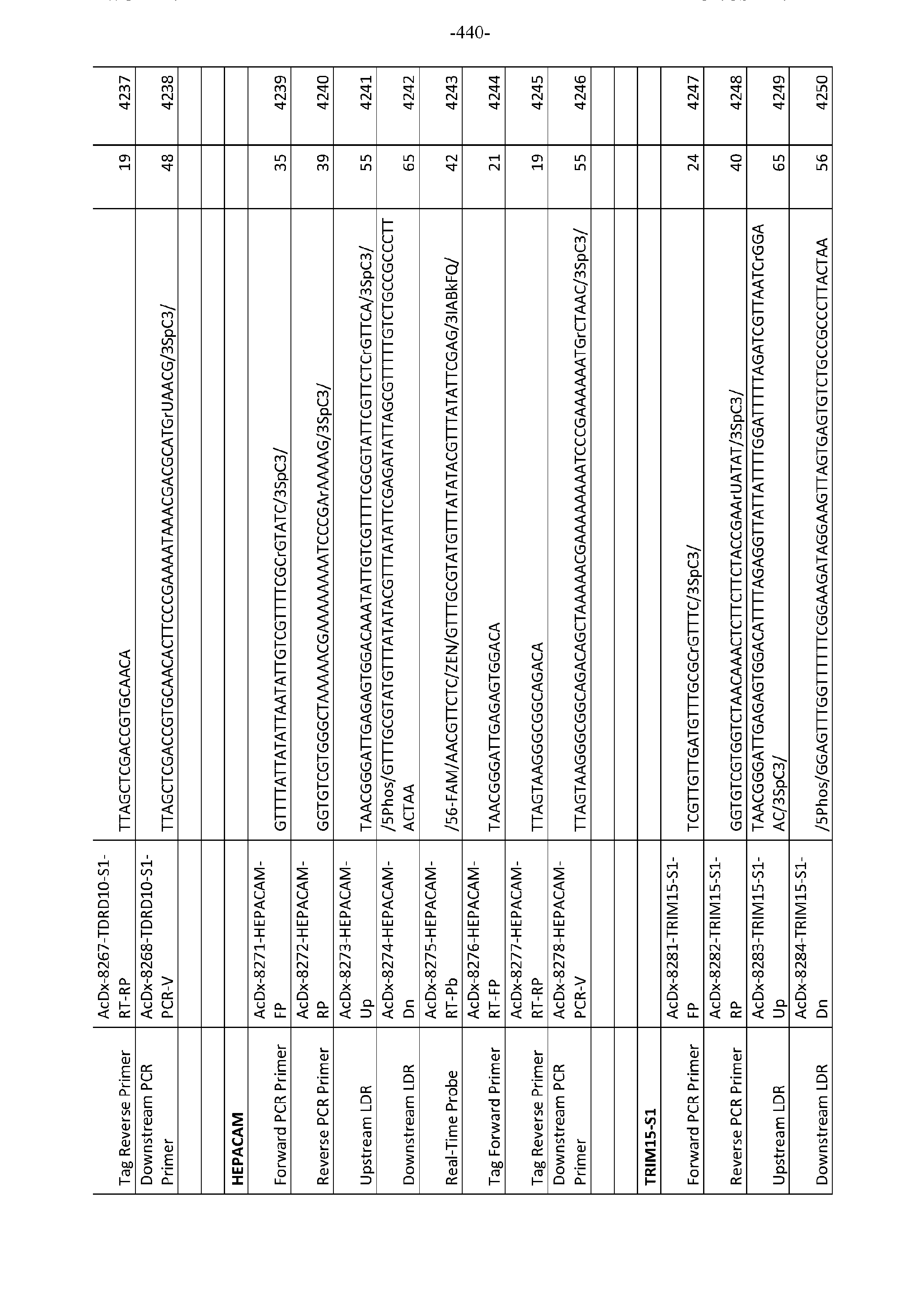

























Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3135726A CA3135726A1 (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes |
| SG11202112246UA SG11202112246UA (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes |
| EP20801778.0A EP3963095A4 (en) | 2019-05-03 | 2020-05-01 | MARKERS FOR THE IDENTIFICATION AND QUANTIFICATION OF A MUTATION, EXPRESSION, SPLICING VARIANT, TRANSLOCATION, COPY NUMBER OR NUCLEIC ACID SEQUENCE METHYLATION CHANGES |
| JP2021565022A JP2022530920A (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying mutations, expression, splice variants, translocations, copy counts, or methylation changes in nucleic acid sequences |
| AU2020268885A AU2020268885A1 (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes |
| US17/608,537 US20220243263A1 (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes |
| CN202080048135.9A CN115244188A (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying nucleic acid sequence mutations, expression, splice variants, translocations, copy number or methylation changes |
| BR112021022084A BR112021022084A2 (en) | 2019-05-03 | 2020-05-01 | Markers to identify and quantify nucleic acid sequence mutation, expression, splice variant, translocation, copy number or methylation changes |
| IL287804A IL287804A (en) | 2019-05-03 | 2021-11-02 | Markers for the detection and quantification of mutation, expression, fusion variant, copy number transition, or methylation changes of a nucleic acid sequence |
| JP2025091539A JP2026002789A (en) | 2019-05-03 | 2025-06-02 | Markers for identifying and quantifying mutations, expression, splice variants, translocations, copy number, or methylation changes in nucleic acid sequences |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962843032P | 2019-05-03 | 2019-05-03 | |
| US62/843,032 | 2019-05-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2020227100A1 true WO2020227100A1 (en) | 2020-11-12 |
| WO2020227100A8 WO2020227100A8 (en) | 2022-01-13 |
Family
ID=73050598
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2020/031044 Ceased WO2020227100A1 (en) | 2019-05-03 | 2020-05-01 | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20220243263A1 (en) |
| EP (1) | EP3963095A4 (en) |
| JP (2) | JP2022530920A (en) |
| CN (1) | CN115244188A (en) |
| AU (1) | AU2020268885A1 (en) |
| BR (1) | BR112021022084A2 (en) |
| CA (1) | CA3135726A1 (en) |
| IL (1) | IL287804A (en) |
| SG (1) | SG11202112246UA (en) |
| WO (1) | WO2020227100A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022140793A1 (en) * | 2020-12-24 | 2022-06-30 | Life Technologies Corporation | Compositions and methods for highly sensitive detection of target sequences in multiplex reactions |
| WO2022187695A1 (en) * | 2021-03-05 | 2022-09-09 | Mayo Foundation For Medical Education And Research | Detecting cervical cancer |
| WO2022246000A1 (en) * | 2021-05-19 | 2022-11-24 | City Of Hope | Compositions and methods for determining dna methylation level in cancer |
| WO2023017001A1 (en) * | 2021-08-09 | 2023-02-16 | Epigenomics Ag | Improved methods for detecting colorectal cancer |
| WO2024154075A1 (en) * | 2023-01-18 | 2024-07-25 | Pixelgen Technologies Ab | Gap-fill ligation and primer extension reactions employing exonuclease-resistant or capture moiety means |
| US12442043B2 (en) | 2019-10-31 | 2025-10-14 | Mayo Foundation For Medical Education And Research | Detecting ovarian cancer |
| US12584177B2 (en) | 2019-01-24 | 2026-03-24 | Mayo Foundation For Medical Education And Research | Detecting endometrial cancer |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3268032A1 (en) * | 2022-09-23 | 2024-03-28 | Flagship Pioneering Innovations Vi, Llc | Methods for amplifying nucleic acids |
| WO2024223698A1 (en) * | 2023-04-24 | 2024-10-31 | Saga Diagnostics Ab | Detection of variants |
| WO2024259135A1 (en) | 2023-06-13 | 2024-12-19 | Intellia Therapeutics, Inc. | Assays for analysis of ribonucleic acid (rna) molecules |
| CN117305312B (en) * | 2023-10-19 | 2024-08-20 | 西南大学 | A gene Zfh3 that determines silkworm's exclusive feeding on mulberry leaves and its application |
| US20250223652A1 (en) * | 2024-01-05 | 2025-07-10 | Harbinger Health, Inc. | Tiered testing for high risk populations |
| CN118986863A (en) * | 2024-10-18 | 2024-11-22 | 四川新泌学生物科技有限公司 | External body gel for vulva and preparation method thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000056929A2 (en) * | 1999-03-19 | 2000-09-28 | Cornell Research Foundation, Inc. | Coupled polymerase chain reaction-restriction endonuclease digestion-ligase detection reaction process |
| WO2013123220A1 (en) * | 2012-02-14 | 2013-08-22 | Cornell University | Method for relative quantification of nucleic acid sequence, expression, or copy changes, using combined nuclease, ligation, and polymerase reactions |
| US20180265917A1 (en) * | 2014-10-08 | 2018-09-20 | Cornell University | Method for identification and quantification of nucleic acid expression, splice variant, translocation, copy number, or methylation changes |
| US20180346973A1 (en) * | 2015-03-23 | 2018-12-06 | The University Of North Carolina At Chapel Hill | Method for identification and enumeration of nucleic acid sequences, expression, splice variant, translocation, copy, or dna methylation changes using combined nuclease, ligase, polymerase, terminal transferase, and sequencing reactions |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2446250T3 (en) * | 2005-05-02 | 2014-03-06 | University Of Southern California | DNA methylation markers associated with the CpG island methylation phenotype (CIMP) in human colorectal cancer |
| CN104846092A (en) * | 2007-02-21 | 2015-08-19 | 奥斯陆大学医院Hf | New markers for cancer |
| BR122021021823B1 (en) * | 2012-09-20 | 2023-04-25 | The Chinese University Of Hong Kong | METHOD TO DETERMINE A FIRST METYLATION PROFILE OF A BIOLOGICAL SAMPLE OF AN ORGANISM, AND MEMORY STORAGE MEDIA |
| WO2015023285A1 (en) * | 2013-08-15 | 2015-02-19 | Xia Xueliang James | Biomarkers for use in colorectal cancer |
| US10030272B2 (en) * | 2015-02-27 | 2018-07-24 | Mayo Foundation For Medical Education And Research | Detecting gastrointestinal neoplasms |
| RS61152B2 (en) * | 2015-05-12 | 2024-06-28 | Hoffmann La Roche | Therapeutic and diagnostic methods for cancer |
| GB201511152D0 (en) * | 2015-06-24 | 2015-08-05 | Ucl Business Plc | Method of diagnosing bladder cancer |
| WO2018087129A1 (en) * | 2016-11-08 | 2018-05-17 | Region Nordjylland, Aalborg University Hospital | Colorectal cancer methylation markers |
| WO2019068082A1 (en) * | 2017-09-29 | 2019-04-04 | Arizona Board Of Regents On Behalf Of The University Of Arizona | DNA METHYLATION BIOMARKERS FOR THE DIAGNOSIS OF CANCER |
-
2020
- 2020-05-01 WO PCT/US2020/031044 patent/WO2020227100A1/en not_active Ceased
- 2020-05-01 CA CA3135726A patent/CA3135726A1/en active Pending
- 2020-05-01 JP JP2021565022A patent/JP2022530920A/en active Pending
- 2020-05-01 EP EP20801778.0A patent/EP3963095A4/en active Pending
- 2020-05-01 BR BR112021022084A patent/BR112021022084A2/en unknown
- 2020-05-01 AU AU2020268885A patent/AU2020268885A1/en active Pending
- 2020-05-01 CN CN202080048135.9A patent/CN115244188A/en active Pending
- 2020-05-01 SG SG11202112246UA patent/SG11202112246UA/en unknown
- 2020-05-01 US US17/608,537 patent/US20220243263A1/en active Pending
-
2021
- 2021-11-02 IL IL287804A patent/IL287804A/en unknown
-
2025
- 2025-06-02 JP JP2025091539A patent/JP2026002789A/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000056929A2 (en) * | 1999-03-19 | 2000-09-28 | Cornell Research Foundation, Inc. | Coupled polymerase chain reaction-restriction endonuclease digestion-ligase detection reaction process |
| WO2013123220A1 (en) * | 2012-02-14 | 2013-08-22 | Cornell University | Method for relative quantification of nucleic acid sequence, expression, or copy changes, using combined nuclease, ligation, and polymerase reactions |
| US20180265917A1 (en) * | 2014-10-08 | 2018-09-20 | Cornell University | Method for identification and quantification of nucleic acid expression, splice variant, translocation, copy number, or methylation changes |
| US20180346973A1 (en) * | 2015-03-23 | 2018-12-06 | The University Of North Carolina At Chapel Hill | Method for identification and enumeration of nucleic acid sequences, expression, splice variant, translocation, copy, or dna methylation changes using combined nuclease, ligase, polymerase, terminal transferase, and sequencing reactions |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3963095A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12584177B2 (en) | 2019-01-24 | 2026-03-24 | Mayo Foundation For Medical Education And Research | Detecting endometrial cancer |
| US12442043B2 (en) | 2019-10-31 | 2025-10-14 | Mayo Foundation For Medical Education And Research | Detecting ovarian cancer |
| WO2022140793A1 (en) * | 2020-12-24 | 2022-06-30 | Life Technologies Corporation | Compositions and methods for highly sensitive detection of target sequences in multiplex reactions |
| WO2022187695A1 (en) * | 2021-03-05 | 2022-09-09 | Mayo Foundation For Medical Education And Research | Detecting cervical cancer |
| WO2022246000A1 (en) * | 2021-05-19 | 2022-11-24 | City Of Hope | Compositions and methods for determining dna methylation level in cancer |
| WO2023017001A1 (en) * | 2021-08-09 | 2023-02-16 | Epigenomics Ag | Improved methods for detecting colorectal cancer |
| WO2024154075A1 (en) * | 2023-01-18 | 2024-07-25 | Pixelgen Technologies Ab | Gap-fill ligation and primer extension reactions employing exonuclease-resistant or capture moiety means |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3135726A1 (en) | 2020-11-12 |
| BR112021022084A2 (en) | 2022-02-08 |
| US20220243263A1 (en) | 2022-08-04 |
| EP3963095A1 (en) | 2022-03-09 |
| JP2022530920A (en) | 2022-07-04 |
| WO2020227100A8 (en) | 2022-01-13 |
| CN115244188A (en) | 2022-10-25 |
| EP3963095A4 (en) | 2023-01-25 |
| SG11202112246UA (en) | 2021-12-30 |
| JP2026002789A (en) | 2026-01-08 |
| IL287804A (en) | 2022-01-01 |
| AU2020268885A1 (en) | 2021-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220243263A1 (en) | Markers for identifying and quantifying of nucleic acid sequence mutation, expression, splice variant, translocation, copy number, or methylation changes | |
| JP7757338B2 (en) | Breast cancer detection | |
| CN113557308B (en) | Detection of endometrial cancer | |
| JP2023040174A (en) | Methods for identifying and quantifying nucleic acid expression, splice variants, translocations, copy number, or methylation changes | |
| Radpour et al. | Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer | |
| US20220205050A1 (en) | Method and markers for identifying and quantifying of nucleic acid sequence, mutation, copy number, or methylation changes | |
| US20230287484A1 (en) | Method and markers for identification and relative quantification of nucleic acid sequence, mutation, copy number, or methylation changes using combinations of nuclease, ligation, deamination, dna repair, and polymerase reactions with carryover prevention | |
| Cheung et al. | The potential of circulating cell free RNA as a biomarker in cancer | |
| KR20210099044A (en) | Characterization of Methylated DNA, RNA, and Proteins in Pulmonary Neoplasia Detection | |
| KR20220092561A (en) | Ovarian Cancer Detection | |
| CN114096680A (en) | Methods and systems for detecting methylation changes in DNA samples | |
| US9328379B2 (en) | Hypermethylation biomarkers for detection of head and neck squamous cell cancer | |
| WO2009108917A2 (en) | Markers for improved detection of breast cancer | |
| US10697021B2 (en) | Methods of detecting breast cancer brain metastasis with genomic and epigenomic biomarkers | |
| WO2018069450A1 (en) | Methylation biomarkers for lung cancer | |
| CN109996891B (en) | Methods for performing early detection of colon cancer and/or colon cancer precursor cells and for monitoring colon cancer recurrence | |
| JP2021513858A (en) | Improved detection of microsatellite instability | |
| CN116219020A (en) | Methylation reference gene and application thereof | |
| US20090186360A1 (en) | Detection of GSTP1 hypermethylation in prostate cancer | |
| JPWO2020227100A5 (en) | ||
| US20240352537A1 (en) | Composition for diagnosing prostate cancer by using cpg methylation changes in specific genes, and use thereof | |
| Zhao et al. | The role of methylation-specific PCR and associated techniques in clinical diagnostics | |
| US20080213781A1 (en) | Methods of detecting methylation patterns within a CpG island | |
| Burgener | Multimodal Profiling of Cell-Free DNA for Detection and Characterization of Circulating Tumour DNA in Low Tumour Burden Settings | |
| Trapani | DNA hypomethylation and hypermethylation in colorectal cancer initiation. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20801778 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3135726 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2021565022 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112021022084 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2020268885 Country of ref document: AU Date of ref document: 20200501 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020801778 Country of ref document: EP Effective date: 20211203 |
|
| ENP | Entry into the national phase |
Ref document number: 112021022084 Country of ref document: BR Kind code of ref document: A2 Effective date: 20211103 |