WO2020262371A1 - 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、その組成物、ゴム架橋物及びタイヤ部材 - Google Patents
変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、その組成物、ゴム架橋物及びタイヤ部材 Download PDFInfo
- Publication number
- WO2020262371A1 WO2020262371A1 PCT/JP2020/024577 JP2020024577W WO2020262371A1 WO 2020262371 A1 WO2020262371 A1 WO 2020262371A1 JP 2020024577 W JP2020024577 W JP 2020024577W WO 2020262371 A1 WO2020262371 A1 WO 2020262371A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- copolymer
- structural unit
- modified
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C19/00—Chemical modification of rubber
- C08C19/25—Incorporating silicon atoms into the molecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/34—Monomers containing two or more unsaturated aliphatic radicals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/34—Monomers containing two or more unsaturated aliphatic radicals
- C08F212/36—Divinylbenzene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
- B60C1/0016—Compositions of the tread
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08C—TREATMENT OR CHEMICAL MODIFICATION OF RUBBERS
- C08C19/00—Chemical modification of rubber
- C08C19/22—Incorporating nitrogen atoms into the molecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/14—Monomers containing five or more carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/02—Monomers containing only one unsaturated aliphatic radical
- C08F212/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F212/06—Hydrocarbons
- C08F212/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F236/04—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F236/06—Butadiene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F36/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F36/02—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F36/04—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/42—Introducing metal atoms or metal-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
- C08G81/02—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers at least one of the polymers being obtained by reactions involving only carbon-to-carbon unsaturated bonds
- C08G81/021—Block or graft polymers containing only sequences of polymers of C08C or C08F
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G81/00—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers
- C08G81/02—Macromolecular compounds obtained by interreacting polymers in the absence of monomers, e.g. block polymers at least one of the polymers being obtained by reactions involving only carbon-to-carbon unsaturated bonds
- C08G81/021—Block or graft polymers containing only sequences of polymers of C08C or C08F
- C08G81/022—Block or graft polymers containing only sequences of polymers of C08C or C08F containing sequences of polymers of conjugated dienes and of polymers of alkenyl aromatic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/01—Use of inorganic substances as compounding ingredients characterized by their specific function
- C08K3/013—Fillers, pigments or reinforcing additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L53/00—Compositions of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L87/00—Compositions of unspecified macromolecular compounds, obtained otherwise than by polymerisation reactions only involving unsaturated carbon-to-carbon bonds
- C08L87/005—Block or graft polymers not provided for in groups C08L1/00 - C08L85/04
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L15/00—Compositions of rubber derivatives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/04—Homopolymers or copolymers of styrene
- C08L25/08—Copolymers of styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/04—Homopolymers or copolymers of styrene
- C08L25/08—Copolymers of styrene
- C08L25/10—Copolymers of styrene with conjugated dienes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/16—Homopolymers or copolymers of alkyl-substituted styrenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
- C08L9/06—Copolymers with styrene
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/80—Technologies aiming to reduce greenhouse gasses emissions common to all road transportation technologies
- Y02T10/86—Optimisation of rolling resistance, e.g. weight reduction
Definitions
- the present invention relates to a modified vinyl aromatic copolymer having excellent reactivity in a reaction with a living terminal of a conjugated diene polymer using an organic lithium compound as an initiator in a hydrocarbon solvent, and a method for producing the same.
- the present invention also relates to a modified vinyl aromatic copolymer having a branched structure and a method for producing the same.
- a modified conjugated diene-based copolymer composed of a modified conjugated diene-based copolymer having excellent workability, tensile strength and abrasion resistance, and a modified conjugated diene-based copolymer composed of the modified conjugated diene-based copolymer, and a crosslinked rubber product thereof.
- tire members are also arranged to a modified vinyl aromatic copolymer having excellent reactivity in a reaction with a living terminal of a conjugated diene polymer using an organic lithium compound as an initiator in a hydrocarbon solvent, and a method for producing the same.
- Conjugate diene rubbers such as SBR (styrene-butadiene rubber), BR (butadiene rubber), IR (isoprene rubber), and styrene-isoprene rubber have excellent wear resistance, elasticity, and water resistance, and are modified for molding materials and resins. It is used for various purposes such as agents.
- This conjugated diene rubber is automobile tires.
- the characteristics required for a tire include mechanical strength, wear resistance, wet grip property and the like (hereinafter, also referred to as strength and the like).
- strength and the like mechanical strength, wear resistance, wet grip property and the like.
- eco-tires fuel efficiency
- This eco-tire is required to have low rolling resistance in addition to strength and the like.
- Patent Document 1 an organolithium compound is used as an initiator in a non-polar solvent, and a block copolymer composed of ⁇ -methylstyrene block and butadiene block is synthesized by living anionic polymerization, and further, if necessary, polyfunctionality.
- S-SBR By reacting the coupling agent, S-SBR having both high temperature characteristics and rubber-like properties is obtained.
- Patent Document 2 discloses a star-block interpolymer having a random copolymer block of a conjugated diene and a monovinyl aromatic monomer, a polyconjugated diene block, and a functional group derived from a polyfunctional lithium-based initiator.
- Patent Documents 1 and 2 are considered to have the effect of ensuring the workability of rubber by introducing a branched structure into the rubber component.
- a rubber composition obtained by blending a predetermined amount of carbon black with a blended rubber containing a plurality of diene-based rubbers has a functional group at the end of the molecular chain that interacts with carbon black, and is diene-based.
- a rubber composition comprising a low molecular weight functional group-containing polymer having a polymer structure similar to the rubber component of rubber is disclosed.
- the amount of carbon black distributed into each diene-based rubber component can be controlled by blending a low molecular weight compound that interacts with carbon black into the rubber. Therefore, the characteristics of each rubber component can be effectively expressed, and it is possible to achieve both the rolling characteristics and the rubber characteristics having a contradictory relationship such as wet characteristics.
- Patent Document 4 a crosslinked rubber particle containing a conjugated diene unit, an aromatic vinyl unit and a unit having at least two polymerizable unsaturated groups, and a conjugated diene / aromatic containing a conjugated diene unit having a specific bonding structure.
- a rubber composition containing a group vinyl copolymerized rubber is disclosed, and it is disclosed that the crosslinked rubber particles may contain a monomer unit having a carboxylic acid group, a hydroxyl group and / or an epoxy group.
- the inorganic filler is excellent in dispersibility and processability.
- the substances disclosed as a monomer unit having at least two polymerizable unsaturated groups and a monomer unit having a carboxylic acid group, a hydroxyl group and / or an epoxy group are all small molecules. .. Therefore, the reactivity is excessively high, and gelation may proceed in the crosslinked rubber particles and the rubber composition.
- patent documents 6 and 7 and the like with respect to compounds similar to the polyfunctional vinyl aromatic copolymer compound disclosed in Patent Document 5.
- these analogs do not teach to be used as building blocks for copolymer rubbers.
- the polyfunctional vinyl aromatic copolymer disclosed in Patent Document 8 is a copolymer having a wide molecular weight distribution containing a large amount of high molecular weight compounds having a remarkably developed branched structure, it can be used as a constituent unit of the copolymer rubber. When used, there was a problem that a small amount of microgel was produced as a by-product.
- the present invention solves this problem and processes a modified vinyl aromatic copolymer having reactivity and solubility that can be used for producing a modified conjugated diene-based copolymer without a small amount of by-product of microgel.
- An object of the present invention is to provide a modified conjugated diene-based copolymer composition having properties, strength and homogeneity, a crosslinked rubber product obtained by cross-linking the same, and a tire member.
- the present inventors have made a vinyl aromatic having a branch containing a structural unit (a) derived from a specific divinyl aromatic compound and a structural unit (b) derived from a monovinyl aromatic compound.
- the present invention has been completed by finding that a copolymer in which the active end of the system copolymer is modified with an amino group, an alkoxysilyl group or a hydroxyl group can solve the above-mentioned problems.
- the modified vinyl aromatic copolymer in which the side chain or the terminal of the polyfunctional vinyl aromatic copolymer is modified with a silane compound has high reactivity in the production of the modified conjugated diene copolymer.
- the present invention has been completed by finding that the above problems can be solved by using this modified conjugated diene-based copolymer as a constituent unit of the modified conjugated diene-based copolymer composition.
- the present invention is a modified vinyl aromatic copolymer containing a structural unit (a) derived from a divinyl aromatic compound and a structural unit (b) derived from a monovinyl aromatic compound, wherein the structural unit (a) is At least a part of is the following crosslinked structural unit (a1), (Here, R 1 represents an aromatic hydrocarbon group having 6 to 30 carbon atoms.) It is a modified vinyl aromatic copolymer characterized in that one or more terminals are modified with a modifying group having at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group and a hydroxyl group. ..
- the present invention is a copolymer containing a structural unit (a) derived from a divinyl aromatic compound and a structural unit (b) derived from a monovinyl aromatic compound, wherein the structural unit (a) and the structural unit (a) The structural unit (a) was contained in an amount of 0.5 to 95.0 mol% and the structural unit (b) was contained in an amount of 5.0 to 99.5 mol% based on the total of b).
- the following formula (c1) or formula (c2) is attached to the side chain or terminal of the copolymer.
- R 2 is a divalent alkylene group having 1 to 6 carbon atoms
- R 3 , R 4 , R 5 , R 7 , R 8 and R 9 are independently methoxy group, ethoxy group and propoxy, respectively.
- a modified vinyl fragrance having a number average molecular weight Mn of 500 to 100,000 and a molecular weight distribution (Mw / Mn) represented by the ratio of the weight average molecular weight Mw to the number average molecular weight Mn of 30.0 or less. It is a group copolymer.
- the modified vinyl aromatic copolymer may contain one or more copolymers represented by any of the following formulas (2) to (13).
- R 2 is a divalent alkylene group having 1 to 6 carbon atoms
- R 3 , R 4 and R 5 agree with the above formula (c1)
- R 6 is an alkyl having 1 to 6 carbon atoms.
- Polymer is a copolymer chain.
- R 7 , R 8 and R 9 agree with the above formula (c2), except that the benzene ring in the formula is arbitrary. It can be an aromatic ring.
- Structural units (b) derived from the monovinyl aromatic compound include styrene, vinylnaphthalene, vinylbiphenyl, m-methylstyrene, p-methylstyrene, o, p-dimethylstyrene, m-ethylvinylbenzene, inden and p. Examples thereof include structural units derived from one or more monomers selected from the group consisting of ethylvinylbenzene.
- the present invention is a method for producing the above-mentioned modified vinyl aromatic copolymer from a polyfunctional vinyl aromatic copolymer and a silane compound.
- the polyfunctional vinyl aromatic copolymer contains 0.5 mol% or more and 95.0 mol% or less of the structural unit (a) derived from the divinyl aromatic compound, and the structural unit (b) derived from the monovinyl aromatic compound. ) Is contained in an amount of 5.0 mol% or more and 99.5 mol% or less.
- At least a part of the structural unit (a) is the crosslinked structural unit (a1) and the following vinyl group-containing structural unit (a2).
- R 1 represents an aromatic hydrocarbon group having 6 to 30 carbon atoms.
- the mole fraction of the crosslinked structural unit (a1) with respect to the structural unit (a) is in the range of 0.05 to 0.90.
- the molar fraction of the vinyl group-containing structural unit (a2) with respect to the sum of the structural units (a) and (b) is in the range of 0.001 to 0.80.
- the silane-based compound is a silane-based compound represented by the following formula (15) and / or formula (16).
- a modified vinyl aromatic copolymer characterized by dissolving the above-mentioned polyfunctional vinyl aromatic copolymer and a silane compound in a hydrocarbon solvent and adding them at a temperature of 0 to 150 ° C. in the presence of a catalyst. This is a method for producing a polymer.
- the present invention comprises a polymer of a conjugated diene compound (B) having an active terminal, or a copolymer of a conjugated diene compound (B) having an active terminal and an aromatic vinyl compound (C), and the above-mentioned modified vinyl aromatic. It is a modified conjugated diene polymer obtained by reacting the system copolymer (A) as a modifier.
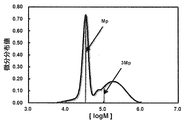
- the modified conjugated diene-based copolymer contains 0.001 to 6% by weight of the structural unit (A1) derived from the modified vinyl aromatic copolymer (A) and the structural unit (B) derived from the conjugated diene compound (B). It often contains 29 to 99.999% by weight of B1) and 0 to 70% by weight of the structural unit (C1) derived from the aromatic vinyl compound (C), and by gel permeation chromatography (GPC) measurement.
- GPC gel permeation chromatography
- At least one reinforcing filler selected from the group consisting of fillers or silica-based inorganic fillers, metal oxides, metal hydroxides and carbon black is blended with the modified conjugated diene-based copolymer. It is a modified conjugated diene-based copolymer composition characterized by the above, and this composition can further contain a cross-linking agent.
- the present invention is a rubber crosslinked product characterized in that the above-mentioned modified conjugated diene-based copolymer composition is crosslinked. Further, it is a tire member characterized by containing this rubber crosslinked product.
- the present invention is a modified vinyl aromatic copolymer containing a structural unit (a) derived from a divinyl aromatic compound and a structural unit (b) derived from a monovinyl aromatic compound. 95 mol% or more of a) is the crosslinked structural unit (a1).
- the above-mentioned modified vinyl aromatic copolymer characterized in that two or more terminals are modified with a modifying group having at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group and a hydroxyl group. Is.
- the modified vinyl aromatic copolymer contains 0.5 to 95.0 mol% of the structural unit (a) with respect to the total of the structural unit (a) and the structural unit (b), and the structural unit (b) Contains 5.0 to 99.5 mol%, the number average molecular weight Mn is 500 to 50,000, the molecular weight distribution (Mw / Mn) is 10.0 or less, and the average per molecule.
- the number of functional groups is in the range of 2 to 20, or the monovinyl aromatic compound is styrene, vinylnaphthalene, vinylbiphenyl, m-methylstyrene, p-methylstyrene, o, p-dimethylstyrene, m-ethylvinylbenzene. It is preferable to satisfy any one or more of the monomers selected from the group consisting of, inden and p-ethylvinylbenzene.
- an alkali metal compound or an alkaline earth metal compound is used as an anionic polymerization initiator, and a divinyl aromatic compound and a monovinyl aromatic compound, or a divinyl aromatic compound and a monovinyl aromatic compound can be anion-copolymerized with them.
- the above-mentioned modified vinyl aromatic system which comprises a compound having at least one functional group selected from the group consisting of the above, or a terminal modification step of reacting a precursor compound thereof to form a functional group.
- This is a method for producing a copolymer.
- the present invention is also characterized in that it is obtained by reacting a polymer of a conjugated diene compound or a copolymer of a conjugated diene compound and an aromatic vinyl compound with the above-mentioned modified vinyl aromatic copolymer. It is a diene-based copolymer.
- the modified conjugated diene-based copolymer contains 0.001 to 6% by weight of the structural unit (A1) derived from the modified vinyl aromatic copolymer and 29 to 99 by weight of the structural unit (B1) derived from the conjugated diene compound. Most when it contains 0 to 70% by weight of a structural unit (C1) derived from .999% by weight and an aromatic vinyl compound, or when the total area is 100% in the differential molecular weight distribution curve obtained by GPC measurement. It is preferable that the area of the region having a number average molecular weight (Mn) three times or more the peak on the low molecular weight side is 10% or more.
- 100 parts by weight of the above-mentioned modified conjugated diene-based copolymer is filled with at least one reinforcing material selected from the group consisting of silica-based inorganic fillers, metal oxides, metal hydroxides and carbon black.
- the rubber composition is characterized by containing 0.5 to 200 parts by weight of the agent, and the rubber composition can further contain a cross-linking agent.
- the present invention is a rubber crosslinked product characterized by cross-linking the rubber composition, and a tire member including the rubber cross-linked product.
- the modified vinyl aromatic copolymer of the present invention can be used as a raw material for the modified conjugated diene copolymer.
- the crosslinked rubber composition in which the modified conjugated diene-based copolymer contains a filler and is crosslinked is excellent in the dispersibility of the filler, and is excellent in mechanical strength and abrasion resistance. Therefore, tires (particularly tire treads). It is useful as an elastomer material such as seismic isolation rubber, rubber hose, rubber roller, and footwear material. It can also be applied to molding materials, resin modifiers, and the like. It can be provided as a dielectric material, an insulating material, a heat resistant material, a structural material, etc. in fields such as the electrical / electronic industry and the space / aircraft industry.
- the film and sheet coated with the curable resin composition containing the modified vinyl aromatic copolymer of the present invention are suitably used in plastic optical parts, touch panels, flat displays, film liquid crystal elements, and the like. It can also be used as a modifier for modifying properties such as heat resistance, dielectric properties and optical properties of a thermoplastic resin or curable resin composition used as a main material of films, sheets and prepregs. Further, the curable resin composition can be processed into a film, a sheet and a prepreg and used, and can also be used as various optical elements such as an optical waveguide and an optical lens.
- the modified vinyl aromatic copolymer of the present invention (hereinafter, also referred to as a modified copolymer or copolymer) has a structural unit (a) derived from a divinyl aromatic compound and a structure derived from a monovinyl aromatic compound.
- a modified vinyl aromatic copolymer containing the unit (b), at least a part of the structural unit (a) is the above-mentioned crosslinked structural unit (a1), and one or more of the ends of the copolymer.
- one or more are modified with a modifying group having at least one functional group selected from the group consisting of an amino group, a hydroxyl group and an alkoxysilyl group.
- a modified vinyl aromatic copolymer (hereinafter, also referred to as a modified copolymer or copolymer A) modified with a modifying group containing an alkoxysilyl group will be described.
- This modified vinyl aromatic copolymer contains 0.5 to 95.0 mol% of the structural unit (a) derived from the divinyl aromatic compound, and 5 structural units (b) derived from the monovinyl aromatic compound. Contains 0.0 to 99.5 mol% or less. Then, the crosslinked structural unit (a1) is included as at least a part of the structural unit (a). Further, the copolymer has an alkoxysilyl group represented by the above formula (c1) or (c2) at the side chain or the terminal.
- At least one in R 3 R 4 R 5 or at least one in R 7 R 8 R 9 is an alkoxy group.
- the alkoxy group is a group represented by RO-, and R is an alkyl group, an aralkyl group or an aryl group, preferably an alkyl group, a benzyl group or a phenyl group of C1-12. Since this alkoxysilyl group is a silane-based functional group, it is also referred to as a silane-based functional group (c) or a structural unit (c).
- the number average molecular weight Mn of the modified vinyl aromatic copolymer is 500 to 100,000, and Mw / Mn is 30.0 or less.
- the mole fraction of the crosslinked structural unit (a1) in the structural unit (a) is in the range of 0.05 to 0.90, and the number of the crosslinked structural unit (c) per molecule is in the range of 1 to 20.
- the number is preferably 2 to 20.
- the mole fraction (also referred to as the degree of cross-linking) of the cross-linked structural unit (a1) with respect to the structural unit (a) is in the range of 0.05 to 0.90, but the preferable lower limit is 0.06, which is more preferable. Is 0.07.
- the upper limit is preferably 0.80, and more preferably 0.70. Particularly preferably, it is 0.60, and most preferably 0.50. Optimal is in the range of 0.20 to 0.50. If the mole fraction is less than 0.05, the degree of cross-linking of the copolymer is small, and therefore, when the structural unit (c) is introduced, a cross-linking reaction in the molecule of the copolymer occurs.
- the modified vinyl aromatic copolymer of the present invention may be reacted with an unmodified polyfunctional vinyl aromatic copolymer and a silane compound represented by the above formulas (15) and / or (16). It can be obtained by the means of.
- R 2 is a divalent alkylene group having 1 to 6 carbon atoms
- R 3 , R 4 , R 5 , R 6 , R 7 and R 8 are independent of each other.
- R 3 , R 4 and R 5 may be a methoxy group, an ethoxy group, a propoxy group, a butoxy group, an octoxy group, a lauryloxy group, a phenoxy group, or a benzyloxy group.
- Examples of the silane compound represented by the above formula (15) include mercaptomethylenemethyldiethoxysilane, mercaptomethylenetriethoxysilane, 2-mercaptoethyltrimethoxysilane, 2-mercaptoethyltriethoxysilane, and 2-mercaptoethyl.
- the mercapto group (-SH) of the silane compound represented by the above formula (15) undergoes a radical addition reaction with a carbon-carbon unsaturated bond contained in the polyfunctional vinyl aromatic copolymer, whereby the silane compound
- the modified vinyl aromatic copolymer represented by the above formulas (2) to (7) having the structural unit (c) derived from the above can be obtained.
- Examples of the silane compound represented by the above formula (16) include trimethoxysilane, triethoxysilane, dimethoxymethylsilane, dimethoxychloromethylsilane, methoxydimethylsilane, dimethoxyphenylsilane, 1,3,3,5. Examples thereof include alkoxysilanes such as 5,7,7-heptamethyl-1,1-dimethoxytetrasiloxane. These silane compounds may be used alone or in combination of two or more. Among these, dimethoxymethylsilane, diethoxymethylsilane, trimethoxysilane, and triethoxysilane are preferable.
- the hydrosilyl group (-SiH) of the silane compound is a catalyst containing a Group 8 transition metal, a quinone compound having two or more organic groups having three or more carbon atoms, and an ester group, an ether group, a thioether group, and an amide.
- Modified vinyl aromatic copolymers represented by the above formulas (8) to (13) having a structural unit (c) derived from a silane compound by adding (hydrosilylating) to the compound. Is obtained.
- -R 2 -Si-R 3 R 4 R 5 or -Si-R 7 R 8 R 9 is referred to as a silane functional group (c) or a structural unit (c).
- Polymer is a copolymer chain, and the tertiary carbon-containing group and secondary carbon-containing group between this and the silane-based functional group (c) are the terminal, side chain, and / or center of the copolymer chain. Is a group that arises from.
- the benzene ring located between the silane functional group (c) and the copolymer chain is contained in the unit generated from the copolymer chain, it is also a part of the copolymer chain, and this benzene ring is also present. May be another aromatic ring.
- the average number of structural units (c) per molecule is 1 to 20, preferably 1 to 15, more preferably 1 to 10, and even more preferably 1 to 9.
- this number is less than 1, the rubber composition has a low affinity with the filler (C), and the filler dispersibility in the rubber composition cannot be improved.
- the wear resistance is improved.
- it exceeds 20 even the crosslinked product obtained from the rubber composition tends to deteriorate in physical properties such as wear resistance or wet grip performance.
- modified conjugated diene-based copolymer (hereinafter, also referred to as a diene-based copolymer) due to the introduction of an appropriate amount of functional groups into the modified vinyl aromatic copolymer. It is presumed that) is likely to be concentrated in the vicinity of the reinforcing filler, the reinforcing effect of the reinforcing filler is increased, and the wear resistance of the obtained crosslinked product is improved. Further, by using the modified vinyl aromatic copolymer, the affinity between the diene copolymer and the reinforcing filler is improved, and the dispersed state of each component such as the reinforcing filler in the rubber composition can be obtained.
- the copolymer adsorbed on the reinforcing filler is excessively strongly adsorbed, which lowers the dispersibility of the reinforcing filler and reduces the dispersibility of the modified vinyl. It is presumed that the aromatic copolymer does not contribute to the improvement of the characteristics (wear resistance, strength) of the crosslinked product composed of the diene copolymer and the reinforcing filler.
- the average number of functional groups per molecule of the copolymer of the present invention can be calculated by the following formula.
- (Average number of functional groups per molecule) [(number average degree of polymerization) * (average molecular weight per repeating unit)] / (equivalent of functional groups)
- the equivalent of the functional group means the mass of the divinyl aromatic compound unit and the monovinyl aromatic compound bonded to each functional group.
- the functional group equivalent can be calculated from the area ratio of the peak derived from the functional group to the peak derived from the polymer main chain using 1 H-NMR or 13 C-NMR.
- the peak derived from the functional group refers to the peak derived from the alkoxy group.
- the amount of the silane compound represented by the formula (15) and / or the formula (16) is 100% by mass of the polyfunctional vinyl aromatic copolymer. 1 to 200 parts by mass is preferable, 1 to 100 parts by mass is more preferable, 1 to 60 parts by mass is further preferable, 1 to 50 parts by mass is more preferable, and 1 to 40 parts by mass is further preferable.
- the amount used is more than 200 parts by mass, the dispersibility effect of the reinforcing filler is poor, the workability is deteriorated, and the wear resistance of the obtained crosslinked product tends to be lowered.
- the dispersibility effect of the reinforcing filler is poor, and it tends to be less than ideal for improving the physical properties of the crosslinked product in which a dispersed state such as the reinforcing filler can be obtained.
- the amount of the silane compound added to the modified vinyl aromatic copolymer can be determined by using various analytical instruments such as nuclear magnetic resonance spectroscopy.
- the method for adding the silane compound represented by the formula (15) to the polyfunctional vinyl aromatic copolymer is not particularly limited, and for example, the silane compound in the polyfunctional vinyl aromatic copolymer, and further. If necessary, a method of adding a radical catalyst and heating in the presence or absence of an organic solvent can be adopted.
- the radical generator to be used is not particularly limited, and commercially available organic peroxides, azo compounds, hydrogen peroxide and the like can be used.
- organic peroxide examples include methyl ethyl ketone peroxide, cyclohexanone peroxide, 3,3,5-trimethylcyclohexanone peroxide, methylcyclohexanone peroxide, acetylacetone peroxide, and 1,1-bis (t-butylperoxy).
- Examples of the azo compound include 2,2'-azobisisobutyronitrile, 1,1'-azobis (cyclohexane-1-carbonitrile), and 2,2'-azobis (2-methylbutyronitrile). , 2,2'-azobis (2,4-dimethylvaleronitrile), 2,2'-azobis (2,4-dimethyl-4-methoxyvaleronitrile), 2,2'-azobis (2- (2-imidazoline) -2-yl) propane), 2,2'-azobis (2,4,4-trimethylpentane), 2,2'-azobis (2-methylpropane), 2,2'-azobis (2-hydroxymethylpropion) Nitrile), 4,4'-azobis (4-cyanovaleric acid), dimethyl 2,2'-azobis (2-methylpropionate), 2-cyano-2-propylazoformamide, 2-phenylazo-4- Examples thereof include methoxy-2,4-dimethylvaleronitrile.
- Examples of the organic solvent used in the above method generally include hydrocarbon solvents and halogenated hydrocarbon solvents.
- hydrocarbon solvents such as n-butane, n-hexane, n-heptane, cyclohexane, methylcyclohexane, ethylcyclohexane, dimethylcyclohexane, decalin, benzene, toluene and xylene are preferable.
- an antiaging agent may be added from the viewpoint of suppressing side reactions.
- Preferred anti-aging agents used at this time include, for example, 2,6-dit-butyl-4-methylphenol (BHT), 2,2'-methylenebis (4-methyl-6-t-butylphenol), 4,4.
- the above-mentioned anti-aging agent may be used alone or in combination of two or more.
- the amount of the antiaging agent added is preferably 0 to 10 parts by mass, more preferably 0 to 5 parts by mass, based on 100 parts by mass of the unmodified polyfunctional vinyl aromatic copolymer.
- the molecular weight in the region of Mt ⁇ 1.45 or more is preferably in the range of 0 to 40%, more preferably in the range of 0 to 30%, and preferably in the range of 0 to 20%. Further preferably, it is particularly preferably in the range of 0 to 10%.
- the position where the functional group is introduced may be the polymerization terminal or the side chain of the polymer chain, but a plurality of functional groups can be easily introduced. From the viewpoint of introduction, it is preferably a side chain of the polymerized chain. Further, the functional group may be contained alone or in combination of two or more. Therefore, the copolymer may be modified with one modified compound, or may be modified with two or more modified compounds.
- the mixing ratio of the unmodified polyfunctional vinyl aromatic copolymer and the silane compound represented by the above formula (15) is such that, for example, the average number of functional groups per molecule of the copolymer becomes a desired value.
- the unmodified polyfunctional vinyl aromatic copolymer and the silane compound may be mixed so that the mass ratio (former / latter) is 0.3 to 100, for example. It may be mixed so as to be 0.3 to 50.
- the temperature in the reaction of adding a silane compound to the polyfunctional vinyl aromatic copolymer is preferably 10 to 200 ° C, more preferably 50 ° C to 180 ° C, still more preferably 50 ° C to 140 ° C.
- the reaction time is preferably 1 to 200 hours, more preferably 1 to 100 hours, even more preferably 1 to 50 hours, still more preferably 1 to 25 hours.
- the method for adding the silane compound represented by the above formula (16) to the polyfunctional vinyl aromatic copolymer is not particularly limited, and for example, the Group 8 transition in the polyfunctional vinyl aromatic copolymer. At least one selected from the group consisting of a catalyst containing a metal, a quinone compound having two or more organic groups having three or more carbon atoms, and an ester group, an ether group, a thioether group, an amide group, a urea group, and a urethane group.
- a method of adding (hydrosilylating) a silane compound to an unmodified vinyl aromatic copolymer having an unsaturated group in the presence of a phenolic compound having the same functional group as the above can be adopted. ..
- the silane compound represented by the formula (16) is added (hydrosilylated) to an unmodified vinyl aromatic copolymer having a carbon-carbon unsaturated bond, and is an unmodified vinyl aromatic copolymer. Responsible for introducing reactive silicon groups into the.
- the ratio (molar ratio) to the unsaturated group in the polyfunctional vinyl aromatic copolymer is important, and the molar ratio (in the above formula (16))
- the number of moles of the represented silane compound / the number of moles of the unsaturated group) is preferably 0.1 to 20, more preferably 0.5 to 3.
- the polyfunctional vinyl aromatic copolymer is a polyfunctional vinyl aromatic copolymer having at least one unsaturated group in the molecule capable of an addition reaction (hydrosilylation reaction) with a silicon hydride group. ..
- the unsaturated group may be referred to as an unsaturated group having a hydrosilylation activity.
- the number of hydrosilylation-active unsaturated groups present in one molecule of this polyfunctional vinyl aromatic copolymer is 1 to 20, preferably 1 to 15, and even more preferably 1 to 10. 1 to 9 are particularly preferable.
- the catalyst used when adding (hydrosilylating) the silane compound represented by the formula (16) to the polyfunctional vinyl aromatic copolymer having a carbon-carbon unsaturated bond is the eighth.
- Examples of the Group 8 transition metal include cobalt, nickel, ruthenium, rhodium, palladium, iridium, and platinum.
- the catalyst containing the Group 8 transition metal examples include a metal simple substance of the above-mentioned Group 8 transition metal, a metal salt, or a complex with an organic compound.
- platinum is supported on a carrier such as alumina, silica, or carbon black.
- dicarbonyldichloroplatinum the platinum-hydrocarbon complex described in Ashby's US Pat. Nos. 3159601 and 3159662, and the platinum-alcolate catalyst described in Lamoreaux's US Pat. No. 3220972.
- the platinum chloride-olefin complex described in Modic's US Pat. No. 3,516,946 is also preferred.
- platinum chloride acid, platinum-olefin complex, platinum-acetylacetonate complex, and platinum-vinylsiloxane complex are more preferable because they have relatively high reaction activity. Only one type of the catalyst may be added to the reaction system, or a plurality of types may be added in combination.
- the ratio of the amount of the catalyst added to the amount of the unmodified vinyl aromatic copolymer having a carbon-carbon unsaturated bond is important, and the molar ratio (number of moles of catalyst / number of moles of unsaturated group) is 10 -1 to 10 -8 is preferable, and 10 -3 to 10 -6 is more preferable. If the molar ratio (the number of moles of the catalyst / the number of moles of unsaturated groups) is less than 10-8 , the hydrosilylation reaction may not proceed sufficiently. In addition, if the molar ratio exceeds 10 -1 , problems such as an increase in raw material cost and a decrease in coloration and transparency of the product due to contamination of the catalyst residue may occur.
- a method for adding the catalyst a method in which the catalyst is dissolved in various solvents and added is preferable because the catalyst is stabilized and easy to handle.
- the solvent used when the catalyst is dissolved in a solvent and added is not particularly limited, and for example, hydrocarbon solvents such as benzene, toluene, and xylene; halogenated hydrocarbons; alcohols; glycols; ethers; esters. And so on.
- the quinone compound is used in combination with a catalyst and plays a role as a co-catalyst of the catalyst.
- the quinone compound is not particularly limited, and is 2,5-di-tert-butyl-1,4-benzoquinone, 2,6-di-tert-butyl-1,4-benzoquinone, 1,4-benzoquinone, 2-.
- Methyl-1,4-benzoquinone, 2,6-dimethyl-1,4-benzoquinone, 2,3,5,6, -tetramethyl-1,4-benzoquinone, 2-tert-butyl-1,4-benzoquinone examples include 1,4-naphthoquinone, 2-methyl-1,4-naphthoquinone (vitamin K3), among which 2,5-di-tert-butyl-1,4-benzoquinone, 2,6-di- tert-butyl-1,4-benzoquinone is preferred from the standpoint of reactivity, color suppression and toxicity.
- the quinone compound may be added as it is, but the quinone compound is used as a solvent because it is easy to handle and can be uniformly dispersed in the entire reaction solution. It is preferable to add the solution after dissolving it in a uniform diluted solution.
- any method of batch, division, and continuous may be used.
- the solvent used when the quinone compound is diluted and added to the solvent is not particularly limited, and is, for example, a hydrocarbon solvent such as benzene, toluene, xylene, hexane, or a halogenated hydrocarbon, alcohol, glycols, ether. Classes, esters and the like.
- the quinone compound (D) when an equimolar quinone compound (D) is added to the catalyst, the quinone compound with respect to the catalyst is added. Is added in a small amount, and the effect of the present invention may not be sufficiently achieved. Further, as the amount of the quinone compound added, the ratio to the amount of the unmodified vinyl aromatic copolymer having a carbon-carbon unsaturated bond added is also important, and the molar ratio (the number of moles of the quinone compound / unsaturated group) is important. The number of moles) is preferably 0.00001 to 100 times, more preferably 0.0001 to 10 times, and particularly preferably 0.001 to 1 times.
- a phenolic compound having at least one functional group selected from the group consisting of an ester group, an ether group, a thioether group, an amide group, a urea group and a urethane group can be used.
- Phenolic compounds are used in combination with catalysts and are thought to play a role as co-catalysts for catalysts.
- the hydrosilylation reaction can proceed under the condition of adding a small amount of oxygen at a better reaction rate as compared with the conventional system of the catalyst and the quinone compound alone.
- the generation of side reaction products can be suppressed, and the amount of the catalyst or quinone compound added can be reduced.
- the phenolic compound is not particularly limited as long as it has at least one functional group selected from the group consisting of an ester group, an ether group, a thioether group, an amide group, a urea group and a urethane group in its structure. It is preferable to have an alkyl group having 1 to 20 carbon atoms at the 2-position and the 6-position as a functional group with respect to the phenol hydroxyl group.
- Such compounds include, for example, ethylenebis (oxyethylene) bis [3- (5-tert-butyl-4-hydroxy-m-tolyl) propionate], pentaerythritol tetrakis [3- (3,5-).
- the phenolic compound may be added as it is, but since it can be uniformly dispersed in the entire reaction solution, the phenolic compound is heated and melted. It is preferable to add it as a liquid, or to dissolve a phenolic compound in a solvent to make a uniform diluted solution, and then add the compound. Moreover, as the addition method, any method of batch, division, and continuous may be used.
- the solvent used when the phenolic compound is diluted and added to the solvent is not particularly limited, and is, for example, a hydrocarbon solvent such as benzene, toluene, xylene, hexane, or a halogenated hydrocarbon, alcohol, glycols, etc. Examples include ethers and esters.
- the ratio to the amount of the organic polymer having an unsaturated group added is also important, and the molar ratio (number of moles of the phenolic compound / number of moles of unsaturated group) is 0.0002 to It is preferably 1 time, more preferably 0.001 to 0.5 times, and particularly preferably 0.005 to 0.1 times.
- the hydrosilylation reaction can be carried out in a solvent-free system or in the presence of a solvent.
- the solvent used in the hydrosilylation reaction is not particularly limited, and examples thereof include hydrocarbons; halogenated hydrocarbons; ethers; esters, etc. Among them, heptane, hexane, benzene, toluene, xylene and the like. Etc. are preferable.
- the hydrosilylation reaction is carried out under an inert gas such as nitrogen or helium in order to ensure safety in handling flammable substances. However, since it has an effect of promoting the hydrosilylation reaction, it is preferable to mix oxygen in the inert gas within a range that does not give an explosive mixed composition.
- the volume fraction of oxygen contained in the gas phase portion in the reaction vessel is preferably 0.1% or more, more preferably 0.5 to 10%. If the volume fraction of oxygen is less than 0.1%, the effect of promoting the hydrosilylation reaction may not be exhibited.
- the reaction temperature of the hydrosilylation reaction is preferably 30 ° C. or higher and 200 ° C. or lower, more preferably 50 ° C. or higher and 150 ° C. or lower.
- the reaction time is not particularly limited, but is preferably 30 minutes or more and 15 hours or less, more preferably 30 minutes or more and 10 hours or less, and further preferably 30 minutes or more and 6 hours or less.
- the modified vinyl aromatic copolymer of the present invention contains 0.5 to 95.0 mol% of the structural unit (a) derived from the divinyl aromatic compound.
- the mole fraction of the structural unit (a) is 0.005 to the sum of the structural units (a) and (b). It becomes 0.95. This mole fraction is calculated by the following formula (7).
- the lower limit of the molar fraction is preferably 0.006, and more preferably 0.007.
- the upper limit is preferably 0.80, more preferably 0.70.
- Optimal is 0.01 to 0.60.
- the lower limit of the preferable content is 0.2 mol%, more preferably 0.4 mol%, and further preferably 0. It is 6 mol%.
- the upper limit is preferably 70 mol%, more preferably 60 mol%, and even more preferably 50 mol%.
- the modified vinyl aromatic copolymer of the present invention contains 5.0 to 99.5 mol% of the structural unit (b) derived from the monovinyl aromatic compound. In terms of mole fraction, it is 0.05 to 0.995.
- the preferred lower limit is 0.20.
- a more preferable lower limit is 0.30.
- the upper limit is preferably 0.994, more preferably 0.993.
- Optimal is 0.40 to 0.99.
- the mole fraction of the structural unit (b) is calculated by the following formula (8) when it consists only of the structural units (a), (b) and (c). (B) / [(a) + (b)] (8) (Here, (a) and (b) are synonymous with equation (7).) Even when the structural unit (a) and the structural unit other than (b) are included, the preferable mole fraction of the structural unit (b) is in the above range.
- the structural unit (a) derived from the divinyl aromatic compound contains a vinyl group as a branching component for exhibiting the copolymerization reactivity with the conjugated diene compound, while the structural unit (a) derived from the monovinyl aromatic compound ( Since b) does not have a vinyl group involved in the curing reaction, it imparts moldability, compatibility, and the like. Further, if the molar fraction of the structural unit (a) is less than 0.005, the heat resistance of the cured product is insufficient, and if it exceeds 0.95, the molding processability is lowered. Further, when the molar fraction of the structural unit (b) exceeds 0.995, the heat resistance is lowered, and when it is less than 0.05, the molding processability is lowered.
- the copolymer of the present invention may contain other structural units in addition to the above structural units. Details of the other structural units are understood from the description of the manufacturing method.
- the Mn (number average molecular weight in terms of standard polystyrene measured using gel permeation chromatography) of the copolymer of the present invention is 500 to 100,000, preferably 600 to 50,000, and more preferably 700 to 700. It is 40,000. More preferably, it is 800 to 30,000, and most preferably 900 to 20,000. The optimum is 900 to 10,000. If Mn is less than 300, the amount of monofunctional copolymer contained in the copolymer increases, so that the copolymerization reactivity with the conjugated diene compound tends to decrease, and if it exceeds 100,000. In addition to facilitating the formation of gel, the moldability and tensile elongation at break tend to decrease.
- the molecular weight distribution is 30.0 or less, preferably 25.0 or less, and more preferably 1.3 to 20.0.
- the optimum value is 2.0 to 15.0.
- Mw / Mn exceeds 30.0, the processing characteristics of the copolymerized rubber tend to deteriorate and gel tends to be generated.
- the copolymer of the present invention is soluble in a solvent selected from toluene, xylene, tetrahydrofuran, dichloroethane or chloroform, but is advantageously soluble in any of the above solvents.
- a solvent-soluble and polyfunctional copolymer it is necessary that a part of the vinyl group of divinylbenzene is not crosslinked and has an appropriate degree of branching.
- Such a copolymer or a method for producing the same is known in the above patent documents and the like.
- an unmodified polyfunctional vinyl aromatic copolymer also referred to as an unmodified copolymer
- an unmodified copolymer which is a precursor of the modified vinyl aromatic copolymer of the present invention.
- the copolymer of the present invention can be advantageously produced.
- the presence of a Lewis acid catalyst in a uniform solvent in which a polymerization raw material containing a divinyl aromatic compound and a monovinyl aromatic compound is dissolved in a solvent having a dielectric constant of 2.0 to 15.0 Underneath, polymerize at a temperature of 0-120 ° C.
- the divinyl aromatic compound branches the copolymer to make it polyfunctional, and also plays an important role as a cross-linking component for forming a branch when the copolymer is copolymerized with the conjugated diene compound.
- divinyl aromatic compounds divinylbenzene (including each isomer), divinylnaphthalene (including each isomer), and divinylbiphenyl (including each isomer) are preferably used, but are limited thereto. is not. In addition, these can be used alone or in combination of two or more. From the viewpoint of moldability, divinylbenzene (m-form, p-form or a mixture thereof) is more preferable.
- the monovinyl aromatic compound improves the solvent solubility, compatibility, and processability of the copolymer.
- monovinyl aromatic compounds are vinyl aromatic compounds such as styrene, vinylnaphthalene, vinylbiphenyl, ⁇ -methylstyrene; o-methylstyrene, m-methylstyrene, p-methylstyrene, o, p-dimethylstyrene, Nuclear alkyl-substituted vinyl aromatic compounds such as o-ethylvinylbenzene, m-ethylvinylbenzene, and p-ethylvinylbenzene; cyclic vinyl aromatic compounds such as inden, acenaphthylene, benzothiophene, and kumaron can be mentioned.
- Ethylvinylnaphthalene (including each isomer) and indene are preferred and used from the standpoint of cost and availability. From the viewpoint of compatibility and cost, styrene, ethylvinylbenzene (m-form, p-form or a mixture thereof) and inden are more preferable.
- a trivinyl aromatic compound, a trivinyl aliphatic compound, a divinyl aliphatic compound and a monovinyl aliphatic compound are used as long as the effects of the present invention are not impaired.
- Cycloolefin compounds and other monomers can be used to introduce structural units (e) derived from these other monomers into the copolymer.
- the other monomer are preferably 1,3,5-trivinylbenzene, 1,3,5-trivinylnaphthalene, 1,2,4-trivinylcyclohexene, and ethylene glycol di.
- the structural unit (e) derived from the other monomer is within the range of less than 30 mol% with respect to the total amount of the structural unit in the copolymer.
- a divinyl aromatic compound, a monovinyl aromatic compound, or a monomer containing another monomer if necessary is polymerized in the presence of a Lewis acid catalyst to obtain a copolymer.
- a Lewis acid catalyst to obtain a copolymer.
- the ratio of each component serving as a monomer may be in the following range with respect to a total of 100 mol% of the divinyl aromatic compound and the monovinyl aromatic compound.
- Divinyl aromatic compounds 0.5-95 mol%, preferably 0.3-65 mol%, more preferably 0.4-55 mol%, optimally 5-50 mol%.
- Monovinyl aromatic compounds 7-99 mol%, preferably 10-95 mol%, more preferably 15-90 mol%.
- the Lewis acid catalyst used here is a compound composed of a metal ion (acid) and a ligand (base), and can be used without particular limitation as long as it can receive an electron pair.
- metal fluorides or complexes thereof are preferable from the viewpoint of thermal decomposition resistance of the obtained copolymer, and particularly B, Al, Ga, In, Si, Ge, Sn, Pb, Sb, Bi, Ti, A divalent to hexavalent metal fluoride such as W, Zn, Fe and V or a complex thereof is preferable.
- These catalysts can be used alone or in combination of two or more.
- an ether complex of boron trifluoride is most preferably used.
- examples of the ether of the ether complex include diethyl ether and dimethyl ether.
- the Lewis acid catalyst is preferably used in the range of 0.001 to 100 mol, more preferably 0.01 to 50 mol, based on 100 mol of the total monomer components. Most preferably 0.1 to 10 mol. If it exceeds 100 mol, the polymerization rate becomes too high, and it becomes difficult to control the molecular weight distribution. Further, if it is less than 0.001 mol, the polymerization rate becomes too small, which leads to an increase in cost and is not suitable for industrial implementation.
- one or more Lewis base compounds can be used as a co-catalyst, if desired.
- Specific examples of the Lewis base compound include the following compounds. 1) Ester compounds such as ethyl acetate, butyl acetate, phenyl acetate and methyl propionate, 2) Thioester compounds such as methyl mercaptopropionic acid and ethyl mercaptopropionic acid, 3) Ketone compounds such as methyl ethyl ketone, methyl isobutyl ketone, and benzophenone, 4) Amine-based compounds such as methylamine, ethylamine, propylamine, butylamine, cyclohexylamine, methylethylamine, dimethylamine, diethylamine, dipropylamine and dibutylamine, 5) Ether compounds such as diethyl ether and tetrahydropyran, 6) Thioether compounds such as diethyl sulfide
- ester compounds and ketone compounds are preferably used because they act synergistically with the Lewis acid catalyst to easily control the polymerization rate and the molecular weight distribution of the polymer.
- One type or two or more types of these Lewis base compounds can be used.
- the Lewis base compound which is a co-catalyst component, is linked to the growth reaction by controlling the interaction between the active species carbocation and the counter anion by coordinating with the Lewis acid catalyst, which is the counter anion, during the polymerization reaction. It regulates the frequency of reactions relative to the transfer reaction and contributes to volume control.
- a Lewis base compound by adding a Lewis base compound, the interaction between the active species carbocation and the counter anion is strengthened, so that the divinyl aromatic compound and the monovinyl aromatic compound are suppressed from causing an excessive insertion reaction, and the monomer is inserted.
- the chain transfer reaction after the reaction is likely to occur, the molecular weight is suppressed from being excessively increased, and the molecular weight can be easily controlled.
- the Lewis base compound as a co-catalyst is preferably 0.005 to 500 mol, more preferably 0.01 to 200 mol, more preferably 0.1 to 100 mol, based on 100 mol of the total of all the monomers.
- the polymerization rate is appropriately maintained, the selectivity of the reaction between the monomers is improved, the productivity is excellent, and the excessive increase or decrease of the molecular weight is suppressed, and the molding processability is suppressed.
- An excellent copolymer can be obtained.
- a polymerization raw material containing the mixture of the above monomers and a Lewis acid catalyst is cation copolymerized at a temperature of 20 to 120 ° C. in a uniform solvent dissolved in a solvent having a dielectric constant of 2.0 to 15.0.
- the solvent is a compound that does not essentially inhibit cationic polymerization, and dissolves a catalyst, a polymerization additive, a co-catalyst, a monomer, and a vinyl aromatic copolymer to be produced to form a uniform solution. Therefore, an organic solvent having a dielectric constant in the range of 2.0 to 15.0 is preferable, and it can be used alone or in combination of two or more. If the dielectric constant of the solvent is less than 2.0, the molecular weight distribution becomes wider, which is not preferable, and if it exceeds 15.0, the polymerization rate decreases.
- the organic solvent toluene, xylene, n-hexane, cyclohexane, methylcyclohexane or ethylcyclohexane is particularly preferable from the viewpoint of the balance between polymerization activity and solubility.
- the concentration of the copolymer in the polymerization solution at the end of the polymerization is 1 to 90 wt%, preferably 10 to 80 wt% in consideration of the viscosity of the obtained polymerization solution and the ease of heat removal. , Particularly preferably 20 to 70 wt%. If this concentration is less than 1 wt%, the cost is increased due to the low polymerization efficiency, and if it exceeds 90 wt%, the molecular weight and the molecular weight distribution are increased, and the molding processability is deteriorated.
- the polymerization temperature exceeds 120 ° C.
- the selectivity of the reaction decreases, which causes problems such as an increase in the molecular weight distribution and the generation of gel.
- the polymerization temperature is lower than 20 ° C., the catalytic activity is significantly reduced. It becomes necessary to add the catalyst of.
- the method for recovering the copolymer after the polymerization reaction is stopped is not particularly limited, and for example, a commonly used method such as a heat concentration method, a steam stripping method, or precipitation in a poor solvent may be used.
- the unmodified copolymer obtained by the above production method contains a structural unit (a) derived from a divinyl aromatic compound and a structural unit (b) derived from a monovinyl aromatic compound, and is derived from the divinyl aromatic compound. At least a part of the structural unit (a) is present as the vinyl group-containing structural unit (a1) represented by the above formula (1). And it is soluble in toluene, xylene, tetrahydrofuran, dichloroethane or chloroform.
- the copolymer of the present invention synthesized from the unmodified copolymer has a reactive silane-based functional group, it may be molded and cured independently, but other polymerizable resins may be modified and have a high molecular weight. It is good to use for multi-branched component synthesis.
- the modified vinyl aromatic copolymer of the present invention is used when obtaining a conjugated diene compound alone and / or a conjugated diene-based copolymer (rubber) obtained by copolymerizing a conjugated diene compound with another monomer. Used for modification and synthesis of high molecular weight multi-branched components.
- the modified vinyl aromatic copolymer of the present invention (hereinafter, also referred to as a modified copolymer or copolymer A') is a structural unit (a) derived from a divinyl aromatic compound and a monovinyl aromatic compound. It is a copolymer containing the derived structural unit (b), and 95 mol% or more of the structural unit (a) is the crosslinked structural unit (a1) represented by the above formula (1). Then, two or more of the terminals are modified by the modifying group having the above functional group.
- the divinyl aromatic compound unit (a) contained in the modified vinyl aromatic copolymer plays an important role as a cross-linking component that branches the copolymer to make it polyfunctional.
- a high molecular weight multi-branched component can be generated and the abrasion resistance can be improved.
- divinyl aromatic compounds divinylbenzene (including each isomer), divinylnaphthalene (including each isomer), and divinylbiphenyl (including each isomer) are preferably used, but are limited thereto. is not. In addition, these can be used alone or in combination of two or more. From the viewpoint of moldability, divinylbenzene (m-form, p-form or a mixture thereof) is more preferable.
- the monovinyl aromatic compound improves the solvent solubility, compatibility and processability of the copolymer.
- monovinyl aromatic compounds are vinyl aromatic compounds such as styrene, vinylnaphthalene, vinylbiphenyl, ⁇ -methylstyrene; o-methylstyrene, m-methylstyrene, p-methylstyrene, o, p-dimethylstyrene, Nuclear alkyl-substituted vinyl aromatic compounds such as o-ethylvinylbenzene, m-ethylvinylbenzene, and p-ethylvinylbenzene; cyclic vinyl aromatic compounds such as inden, acenaphthylene, benzothiophene, and kumaron can be mentioned.
- Ethylvinylnaphthalene (including each isomer) and indene are preferred and used from the standpoint of cost and availability. From the viewpoint of compatibility and cost, styrene, ethylvinylbenzene (m-form, p-form or a mixture thereof) and inden are more preferable.
- the modified copolymer can be produced by the production method of the present invention.
- an alkali metal compound or an alkaline earth metal compound is used as an anionic polymerization initiator, and a divinyl aromatic compound and a monovinyl aromatic compound, or a divinyl aromatic compound and a monovinyl aromatic compound and an anionic copolymerization thereof.
- a polymerization step of copolymerizing a possible monomer to obtain a vinyl aromatic copolymer having a branched structure and an active terminal, and forming the above functional group at the active terminal of the vinyl aromatic copolymer. Includes terminal transformation step.
- the anion copolymerizable monomer improves the toughness and compatibility of the copolymer.
- anion copolymerizable monomers examples include butadiene and isoprene; 2,3-dimethylbutadiene, 2-phenylbutadiene, 1,3-pentadiene, 2-methyl-1,3-pentadiene, 1,3-hexadiene, 1 , 3-Octadiene, 1,3-Cyclohexadiene, 2-methyl-1,3-octadien, 1,3,7-octatriene, Milsen, and conjugated diene (b1) other than butadiene and isoprene such as chloroprene. ..
- Butadiene and isoprene are preferable as the anion copolymerizable monomer contained in the modified vinyl aromatic copolymer of the present invention because of the ease of industrial implementation.
- the vinyl aromatic copolymer having the above-mentioned branched structure and active terminal is a multi-branched vinyl aromatic copolymer having an unmodified active terminal, and is also a precursor of the modified copolymer of the present invention.
- the method for producing a vinyl aromatic copolymer having a branched structure and an active terminal in addition to the divinyl aromatic compound and the monovinyl aromatic compound, other trivinyl aromatic compounds and the like are used as long as the effects of the present invention are not impaired.
- the structural unit (e) derived from these other monomers can be introduced into the copolymer by using the monomer of.
- the above-mentioned other monomers include, but are not limited to, 1,3,5-trivinylbenzene, 1,3,5-trivinylnaphthalene and the like. These can be used alone or in combination of two or more. Other monomers may be used in the range of less than 30 mol% of the total monomer. As a result, the structural unit (e) derived from the other monomer is within the range of less than 30 mol% with respect to the total amount of the structural unit in the copolymer.
- a divinyl aromatic compound, a monovinyl aromatic compound, or a monomer containing another monomer if necessary is used as an alkali metal compound or an alkaline earth metal.
- a copolymer is produced by polymerizing in the presence of a polymerization initiator composed of a compound.
- At least a part of the structural unit (a) is a crosslinked structural unit (a1) represented by the above formula (1), but the crosslinked structural unit (a1) derived from a divinyl aromatic compound with respect to the structural unit (a).
- the mole fraction also referred to as the degree of cross-linking
- the degree of cross-linking of the cross-linked structural unit (a1) is in the range of 0.95 or more, but the preferable lower limit is 0.98 or more, and more preferably 0.99 or more.
- the above degree of cross-linking is a parameter that can be arbitrarily controlled and changed, but when the degree of cross-linking is smaller than 0.95, a large amount of highly reactive pendant vinyl groups remain in the modified copolymer of the present invention.
- this polymer is used for the modification of the conjugated diene-based (co) polymer, the pendant vinyl group remaining in the copolymer is used as a starting point in the molecule due to the thermal history in the subsequent steps. Crosslinking reaction is likely to occur, and microgels tend to be formed during compounding and smelting.
- the method for producing the modified copolymer includes a polymerization step and a terminal modification step as described above.
- a polymerization initiator composed of an alkali metal compound or an alkaline earth metal compound used in the polymerization step will be described.
- the alkali metal compound used as the polymerization initiator is not particularly limited, but for example, an organic lithium compound is preferable.
- the organolithium compound may be either a low molecular weight organolithium compound or a solubilized oligomeric organolithium compound. Examples of the bonding mode between the organic group and lithium include a compound having a carbon-lithium bond, a compound having a nitrogen-lithium bond, and a compound having a tin-lithium bond.
- the organic lithium compound is not particularly limited, and examples thereof include an organic monolithium compound, an organic dilithium compound, and an organic polylithium compound.
- the organic group a hydrocarbon containing a functional group is preferable, and in that case, there is an advantage that the solubility in an organic solvent is excellent, and further, the starting speed is also excellent. Further, by using a compound having a nitrogen-lithium bond and a compound having a tin-lithium bond, a modifying group containing a functional group can be imparted to the starting end.
- the other organic alkali metal compound of the organic lithium compound is not particularly limited, and examples thereof include an organic sodium compound, an organic potassium compound, an organic rubidium compound, and an organic cesium compound. More specifically, sodium naphthalene, potassium naphthalene and the like can be mentioned. In addition, alcoholides such as lithium, sodium and potassium, sulfonates, carbonates, amides and the like can be mentioned. In addition, it may be used in combination with other organometallic compounds.
- alkaline earth metal compound examples include an organic magnesium compound, an organic calcium compound, and an organic strontium compound. Further, compounds such as alkali earth metal alcoxides, sulfonates, carbonates and amides may be used. These organometallic earth metal compounds may be used in combination with the above-mentioned alkali metal compounds and other organometallic compounds.
- the alkali metal compound having a functional group that can be used as a polymerization initiator is not particularly limited, but for example, a lithium amide compound obtained by lithiumizing hydrogen of a secondary amine, or an alkyl to which the functional group is bonded. There is lithium etc.
- a functional group can be imparted to the polymerization initiation terminal of the conjugated diene-based copolymer.
- the functional group is not particularly limited, but a functional group inert to an alkali metal is preferable, and for example, a disubstituted amino group, that is, a tertiary amine, a protected monosubstituted amino group, and a protected amino group are preferable. ..
- Examples of the protected monosubstituted amino group or the protected amino group include one hydrogen of a monosubstituted amino group or two hydrogens of an amino group substituted with a trialkylsilyl group. ..
- the organic lithium compound is not particularly limited, and for example, mono-organic lithium compounds such as n-butyllithium, sec-butyllithium, tert-butyllithium, n-propyllithium, iso-propyllithium, and benzyllithium; 1,4 -Dilithiobutane, 1,5-dilithiopentane, 1,6-dilithiohexane, 1,10-dilithiodecane, 1,1-dilithiodiphenylene, dilithiopolybutadiene, dilithiopolyisoprene, 1,4-dilithiobenzene, 1 , 2-Dilithio-1,2-diphenylethane, 1,4-dilithio-2-ethylcyclohexane, 1,3,5-trilithiobenzene, 1,3,5-trilithio-2,4,6-triethylbenzene, etc. Polyfunctional organic lithium compounds of.
- the polymerization initiator having a functional group is not particularly limited, and specific examples thereof include the compounds shown below.
- the types of functional groups that can be added to the polymer are described in parentheses.
- an oligomer initiator in which various lithium-based initiators and monomers are reacted can be used.
- a monomer having at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group, and a hydroxyl group can be used as the monomer.
- the oligomer initiator is not particularly limited, but a molecular weight of 1000 or less is preferable because it is industrially easy to handle.
- Polymerization or copolymerization is carried out using a polymerization initiator having a functional group, and at least one selected from the group consisting of an amino group, an alkoxysilyl group and a hydroxyl group at the active terminal of the vinyl aromatic copolymer in the next step. It is possible to introduce a compound having a functional group of the above, or a compound forming at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group and a hydroxyl group.
- the polyfunctional initiator is not particularly limited, but specifically, there are an organic dilithium compound and an organic polylithium compound.
- the organic group is not particularly limited, but a hydrocarbon is preferable. This has the advantage of excellent solubility in organic solvents, and also has an excellent starting speed.
- the method for preparing the polyfunctional initiator is not particularly limited, and examples thereof include a method by reacting a dispersion of metallic lithium with a polyhalogenated hydrocarbon compound.
- a polar compound may be added.
- a polar compound it is involved in the initiation reaction and the growth reaction, and is also effective in controlling the molecular weight and molecular weight distribution and promoting the polymerization reaction.
- polar compound examples include ethers such as tetrahydrofuran, diethyl ether, dioxane, ethylene glycol dimethyl ether, ethylene glycol dibutyl ether, diethylene glycol dimethyl ether, diethylene glycol dibutyl ether, dimethoxybenzene, and 2,2-bis (2-oxolanyl) propane; Tertiary amine compounds such as methylethylenediamine, dipiperidinoethane, trimethylamine, triethylamine, pyridine and quinuclidine; alkali metal alkoxides such as potassium-tert-amylate, potassium-tert-butyrate, sodium-tert-butyrate and sodium amylate.
- ethers such as tetrahydrofuran, diethyl ether, dioxane, ethylene glycol dimethyl ether, ethylene glycol dibutyl ether, diethylene glycol dimethyl ether, diethylene glycol dibutyl ether,
- Examples include phosphine compounds such as triphenylphosphine. These polar compounds may be used alone or in combination of two or more.
- the amount of the polar compound used is not particularly limited and can be selected according to the purpose and the like. Usually, it is preferably 0.01 to 100 mol with respect to 1 mol of the polymerization initiator or the polyfunctional initiator.
- Such a polar compound can be used in an appropriate amount as a regulator of the copolymer initiation reaction and the growth reaction, depending on the desired molecular weight and molecular weight distribution.
- Many polar compounds have a randomizing effect effective in copolymerizing a divinyl aromatic compound and a monovinyl aromatic compound at the same time, and can be used as an agent for adjusting the distribution of the aromatic vinyl compound or adjusting the amount of styrene block. it can.
- the copolymerization of the divinyl aromatic compound and the monomer containing the monovinyl aromatic compound is preferably carried out by solution polymerization in an inert solvent.
- the polymerization solvent is not particularly limited, and for example, hydrocarbon solvents such as saturated hydrocarbons and aromatic hydrocarbons are used.
- aliphatic hydrocarbons such as butane, pentane, hexane and heptane
- alicyclic hydrocarbons such as cyclopentane, cyclohexane, methylcyclopentane, methylcyclohexane, dimethylcyclohexane, ethylcyclohexane and decalin
- benzene, toluene examples thereof include hydrocarbon-based solvents composed of aromatic hydrocarbons such as xylene and mixtures thereof. It is preferable that the monomer and the polymerization solvent are treated alone or a mixture thereof is treated with an organometallic compound.
- the divinyl aromatic compound, the monomer such as the monovinyl aromatic compound, and the allenes and acetylenes contained in the polymerization solvent can be treated.
- a polymer having a high concentration of active terminals can be obtained, and a high denaturation rate can be achieved.
- the polymerization temperature at the time of copolymerization is not particularly limited as long as it is the temperature at which the living anionic polymerization proceeds, but from the viewpoint of productivity, it is preferably 0 ° C. or higher, and the active terminal is subjected to the terminal modification step after the completion of polymerization. From the viewpoint of ensuring a sufficient amount of reaction, the temperature is preferably 120 ° C. or lower. More preferably, it is 50 to 100 ° C.
- the mode of the polymerization reaction is not particularly limited, but it can be carried out by a polymerization mode such as a batch type (also referred to as "batch type") or a continuous type.
- a polymerization mode such as a batch type (also referred to as "batch type") or a continuous type.
- a continuous system one or two or more connected reactors can be used.
- the reactor a tank type with a stirrer, a tube type, or the like is used.
- the molecular weight distribution of the obtained polymer is generally narrow, and Mw / Mn tends to be 1.0 or more and less than 1.8.
- the continuous formula the molecular weight distribution is generally wide, and in Mw / Mn, it tends to be 1.8 or more and 3 or less.
- a vinyl aromatic copolymer having a branched structure and an active terminal (also referred to as an unmodified copolymer) is obtained in the polymerization step, and then an amino group and an alkoxysilyl group are added to the active terminal.
- a compound having at least one functional group selected from the group consisting of hydroxyl groups, or a compound forming at least one functional group selected from the group consisting of amino groups, alkoxysilyl groups, and hydroxyl groups is reacted to carry out the functionalization. It is subjected to a terminal modification step in which a group is introduced.
- the reaction temperature, reaction time, etc. when reacting the compound having the above functional group (including the precursor) at the active terminal are not particularly limited, but the reaction may be carried out at 0 to 120 ° C. for 30 seconds or more. preferable.
- the amount of the compound having a functional group added is not particularly limited, but the number of moles of the polymerization initiator or the polyfunctional initiator, that is, the total number of moles of the compound having a functional group with respect to the polymer molecule is 0.01. It is preferably in the range of about 6 times, more preferably in the range of 0.02 to 3 times, and further preferably in the range of 0.05 to 2 times.
- the addition amount is 0.05 times or more, it is preferable from the viewpoint of obtaining a sufficient modification rate in the target modified vinyl aromatic copolymer.
- the amount of the compound having a functional group added is preferably 0.7 times or less as the functional group equivalent with respect to the number of moles of the polymerization initiator or the polyfunctional initiator, that is, the number of polymer molecules.
- the terminal modification step may be carried out subsequently in the reactor used in the polymerization step, or may be transferred to the next reactor. If the polymerization step is continuous, it is transferred to the next reactor.
- the terminal denaturation step is preferably carried out immediately following the polymerization step, preferably within 5 minutes by mixing the denaturant to carry out the reaction.
- the reactor for the denaturation reaction is preferably one in which sufficient stirring is performed. Specifically, there are a static mixer type reactor, a row type reactor with a stirrer, and the like.
- a compound having at least one functional group selected from an amino group, an alkoxysilyl group, and a hydroxyl group at the active terminal of the unmodified copolymer (including a precursor thereof, which is also referred to as a modified compound).
- the functional group to be bonded to the active terminal of the conjugated diene polymer is not particularly limited, but specifically, a halogen group, a double bond, a ketone group, an ester group, an amide group, etc. Examples thereof include an epoxy group and an alkoxysilyl group.
- the compound having an amino group is not particularly limited, but specifically, a compound having an amino group and a functional group bonded to the active end of the polymer in the molecule, preferably having no active hydrogen.
- the amino group is not particularly limited, but specifically, a functional group inactive with respect to an alkali metal is preferable, and a disubstituted amino group, that is, a tertiary amine, a protected monosubstituted amino group, and two hydrogens. Amino groups protected by are preferred.
- the compound having an alkoxysilyl group is not particularly limited, but specifically, a compound having a plurality of alkoxysilyl groups in the molecule (including a compound having a silyl group in which a plurality of alkoxy groups are bonded), And compounds having an alkoxysilyl group in the molecule and a functional group that binds to the active terminal of the polymer. It is preferable that these are compounds having no active hydrogen.
- the compound that forms a hydroxyl group is not particularly limited, but specifically, it is a functional group that binds to the active terminal of the polymer, and a compound having a functional group that produces a hydroxyl group after the bonding reaction, or a compound that binds to the active terminal of the polymer.
- the non-functional group include a compound having a functional group in which a hydroxyl group is later generated by a reaction such as hydrolysis, and a compound having no active hydrogen is preferable.
- Examples of the compound having a functional group in which a hydroxyl group is generated after the binding reaction include a compound having a ketone group, an ester group, an amide group, an epoxy group and the like.
- examples of the compound having a functional group in which a hydroxyl group is generated by a reaction such as hydrolysis after the binding reaction include a compound having an alkoxysilyl group, an aminosilyl group and the like.
- the compound that forms an amino group and a hydroxyl group at the end of the copolymer is not particularly limited, but is limited to N, N, N', N'-tetramethyl-4,4'-diaminobenzophenone (Michler's ketone), N, N, Ketone compounds having an amino group such as N', N'-tetraethyl-4,4'-diaminobenzophenone; cyclic urea compounds such as N, N'-dimethylimidazolidinone and N-methylpyrrolidone; cyclic amides, that is, lactam compounds.
- Amino group-containing epoxy compounds such as N, N, N', N'-tetraglycidyl-1,3-bisaminomethylcyclohexane; and the epoxy compounds having a nitrogen-containing heterocyclic group described in JP2001-131227A are exemplified.
- the compound that forms an alkoxysilyl group at the end of the copolymer is not particularly limited, but is a halogenated alkoxysilane compound such as trimetokichlorosisilane, triethoxychlorosilane, and diphenoxydicrylorosilane; bis (trimethoxysilyl).
- Examples thereof include polyfunctional alkoxysilane compounds such as ethane and bis (3-triethoxysilylpropyl) ethane.
- the compound that forms an alkoxysilyl group and a hydroxyl group at the terminal of the copolymer is not particularly limited, but 3-glycidoxypropyltrimethoxysilane, 3-glycidoxypropyltriethoxysilane epoxy group and alkoxysilyl group are molecules.
- Examples thereof include polysiloxane compounds contained therein.
- the compound that forms an amino group and an alkoxysilyl group at the end of the copolymer is not particularly limited, but is limited to 3-dimethylaminopropyltrimethoxysilane, 3-dimethylaminopropyldimethoxymethylsilane, and 3-dimethylaminopropyltriethoxysilane.
- Alkoxysilane compound with an alkyl group having an amino substituent such as bis (3-trimethoxysilylpropyl) methylamine, bis (3-triethoxysilylpropyl) methylamine; N- [3- (triethoxysilyl) -Propyl] -N, N'-diethyl-N'-trimethylsilyl-ethane-1,2-diamine, 3- (4-trimethylsilyl-1-piperazinyl) propyltriethoxysilane, etc., as described in WO2007 / 034785.
- Alkoxysilane compound to which a mono-substituted amino group is bonded N- [2- (trimethoxysilanyl) -ethyl] -N, N', N'-trimethylethane-1,2-diamine, 1- [3-( Triethoxysilanyl) -propyl] -4-methylpiperazin, 2- (trimethoxysilanyl) -1,3-dimethylimidazolidine, bis- (3-dimethylaminopropyl) -dimethoxysilane, etc., as described in WO2008 / 013090A.
- the compound that forms a hydroxyl group at the end of the copolymer is not particularly limited, and examples thereof include epoxy compounds such as ethylene oxide and propylene oxide; and ketone compounds such as benzophenone.
- the modified copolymer of the present invention or the modified vinyl aromatic copolymer of the present invention obtained by the above-mentioned production method has a reactive functional group, it may be molded and cured alone, but other polymerizations.
- the sex resin may be used for functional group modification and high molecular weight polybranched component synthesis.
- this modified copolymer is used when the conjugated diene compound alone and / or the conjugated diene compound is copolymerized with another monomer to obtain a conjugated diene-based copolymer (also referred to as modified rubber). It is used for functional group modification and synthesis of high molecular weight multi-branched components.
- the modified copolymer of the present invention is excellent as a modifier for a conjugated diene-based (co) polymer (rubber).
- the above rubber is modified by introducing an appropriate amount of functional groups into the modified copolymer of the present invention, and the modified conjugated diene-based copolymer (modified rubber) is modified.
- the reinforcing effect of the reinforcing filler is increased, and the wear resistance of the obtained crosslinked product is improved.
- the affinity between the modified copolymer of the present invention and the reinforcing filler is improved, and the dispersed state of each component such as the reinforcing filler in the rubber composition can be obtained. It is estimated to be ideal for improving the physical properties of objects (for example, improving wear resistance, improving steering stability, dry grip performance, wet grip performance).
- the number of functional groups of the modified rubber becomes too large, the interaction between the modified rubbers adsorbed on the reinforcing filler is too strong, and the effect of dispersing the reinforcing filler becomes small. It is presumed that the modified copolymer of the present invention reduces the effect of contributing to the improvement of the affinity between the modified rubber and the reinforcing filler.
- the average number of functional groups per molecule of the modified vinyl aromatic copolymer is preferably in the range of 2 to 20.
- the amount of the modified compound added to the modified vinyl aromatic copolymer is preferably 1 to 200 parts by mass, more preferably 1 to 100 parts by mass, based on 100 parts by mass of the unmodified polyfunctional vinyl aromatic copolymer. It is more preferably 1 to 60 parts by mass, further preferably 1 to 50 parts by mass, and may be 1 to 40 parts by mass.
- the amount of the modified compound is more than 200 parts by mass, the dispersibility effect of the reinforcing filler is poor, the processability is deteriorated, and the wear resistance of the obtained crosslinked product tends to be lowered.
- the dispersibility effect of the reinforcing filler is poor, and it tends to be less than ideal for improving the physical properties of the crosslinked product in which a dispersed state such as the reinforcing filler can be obtained.
- the amount of the modified compound having a functional group selected from an amino group, an alkoxysilyl group, and a hydroxyl group added to the modified vinyl aromatic copolymer can be determined by, for example, various analytical instruments such as nuclear magnetic resonance spectroscopy. It can be obtained by using.
- the modified vinyl aromatic copolymer of the present invention contains 0.5 to 95.0 mol% of the structural unit (a) derived from the divinyl aromatic compound.
- the mole fraction of the structural unit (a) is 0.005 to the sum of the structural units (a) and (b). It becomes 0.95. This mole fraction is calculated by the following formula (7).
- the lower limit of the molar fraction is preferably 0.006, and more preferably 0.007.
- the upper limit is preferably 0.80, more preferably 0.70.
- Optimal is 0.01 to 0.60.
- the lower limit of the preferable content is 0.2 mol%, more preferably 0.4 mol%, and further preferably 0. It is 6 mol%.
- the upper limit is preferably 70 mol%, more preferably 60 mol%, and even more preferably 50 mol%.
- the modified vinyl aromatic copolymer of the present invention contains 5.0 to 99.5 mol% of the structural unit (b) derived from the monovinyl aromatic compound. In terms of mole fraction, it is 0.05 to 0.995.
- the preferred lower limit is 0.20.
- a more preferable lower limit is 0.30.
- the upper limit is preferably 0.994, more preferably 0.993.
- Optimal is 0.40 to 0.99.
- the mole fraction of the structural unit (b) is calculated by the following formula (8) when it consists only of the structural units (a), (b) and (c). (B) / [(a) + (b)] (8) (Here, (a) and (b) are synonymous with equation (7).) Even when the structural unit (a) and the structural unit other than (b) are included, the preferable mole fraction of the structural unit (b) is in the above range.
- the structural unit (a) derived from the divinyl aromatic compound contains a vinyl group as a branching component for exhibiting the copolymerization reactivity with the conjugated diene compound, while the structural unit (a) derived from the monovinyl aromatic compound ( Since b) does not have a vinyl group involved in the curing reaction, it imparts moldability, compatibility, and the like. Further, if the molar fraction of the structural unit (a) is less than 0.005, the heat resistance of the cured product is insufficient, and if it exceeds 0.95, the molding processability is lowered. Further, when the molar fraction of the structural unit (b) exceeds 0.995, the heat resistance is lowered, and when it is less than 0.05, the molding processability is lowered.
- the modified vinyl aromatic copolymer of the present invention may contain other structural units in addition to the above structural units. Details of the other structural units are understood from the description of the manufacturing method.
- the Mn (number average molecular weight in terms of standard polystyrene measured by gel permeation chromatography) of the modified vinyl aromatic copolymer of the present invention is 500 to 100,000, preferably 600 to 50,000, and more. It is preferably 700 to 40,000. More preferably, it is 800 to 30,000, and most preferably 900 to 20,000. The optimum is 900 to 10,000. If Mn is less than 300, the amount of functional groups contained in the copolymer decreases, so that the reactivity of the conjugated diene-based copolymer with the active terminal tends to decrease, and if it exceeds 100,000, it tends to decrease. In addition to facilitating the formation of gel, the moldability and tensile elongation at break tend to decrease.
- the molecular weight distribution is 30.0 or less, preferably 25.0 or less, and more preferably 1.3 to 20.0.
- the optimum value is 1.6 to 15.0.
- Mw / Mn exceeds 30.0, the processing characteristics of the copolymerized rubber tend to deteriorate and gel tends to be generated.
- the modified vinyl aromatic copolymer of the present invention is soluble in a solvent selected from toluene, xylene, tetrahydrofuran, dichloroethane or chloroform, but is advantageously soluble in any of the above solvents.
- a solvent selected from toluene, xylene, tetrahydrofuran, dichloroethane or chloroform
- Such a copolymer or a method for producing the same is known in the above patent documents and the like.
- it is preferable that 50 g or more is dissolved in 100 g of the solvent. More preferably, it dissolves 80 g or more.
- the modified vinyl aromatic copolymer of the present invention obtained by the above-mentioned production method has at least one functional group selected from a reactive amino group, alkoxysilyl group and hydroxyl group, it can be molded and cured independently.
- other polymerizable resins may be used for functional group modification and high molecular weight multi-branched component synthesis.
- the modified vinyl aromatic copolymer of the present invention is used when obtaining a conjugated diene compound alone and / or a conjugated diene-based copolymer (rubber) obtained by copolymerizing a conjugated diene compound with another monomer. , Used for functional modification and high molecular weight polybranched component synthesis.
- the modified conjugated diene-based polymer is a polymer of a conjugated diene compound (B) having an active terminal, or a copolymer of a conjugated diene compound (B) having an active terminal and an aromatic vinyl compound (C) of the present invention. It is obtained by reacting a modified vinyl aromatic copolymer (A) or (A') as a modifier.
- a modified vinyl aromatic copolymer (A) or (A') as a modifier.
- the polymer of the conjugated diene compound (B) or the copolymer of the conjugated diene compound (B) having an active terminal and the aromatic vinyl compound (C) are also collectively referred to as a diene-based polymer.
- modified vinyl aromatic copolymer (A) of the first aspect and the modified vinyl aromatic copolymer (A') of the second aspect can be used in the same manner, both are described below. It is also referred to as a modified vinyl aromatic copolymer (A).
- modified vinyl aromatic copolymer (A) of the present invention By copolymerizing the modified vinyl aromatic copolymer (A) of the present invention as a raw material with 1) a conjugated diene compound (B) or 2) a conjugated diene compound (B) and an aromatic vinyl compound (C).
- a modified conjugated diene-based copolymer having a branched polymer-type modifying group based on the modified vinyl aromatic copolymer of the present invention can be obtained.
- a modified diene-based rubber such as butadiene rubber or isoprene rubber
- a modified conjugated diene such as SBR can be obtained.
- a system copolymer can be obtained. Since these modified conjugated diene-based copolymers exhibit the characteristics of rubber, they are also referred to as modified copolymer rubbers.
- an alkali metal compound or an alkaline earth metal compound is used as a polymerization initiator, and the conjugated diene compound (B) is polymerized, or the conjugated diene compound (B) and A polymerization step of copolymerizing the aromatic vinyl compound (C) to obtain a conjugated diene-based copolymer having an active terminal, and introducing a branched polymer-type modifying group based on the modified vinyl aromatic-based copolymer. It consists of a terminal modification step.
- conjugated diene compound (B) examples include 1,3-butadiene, isoprene, 2,3-dimethyl-1,3-butadiene, 1,3-pentadiene, 3-methyl-1,3-pentadiene, 1,3. -Heptadiene, 1,3-hexadiene and the like can be mentioned. Of these, 1,3-butadiene and isoprene are preferable. These may be used alone or in combination of two or more.
- aromatic vinyl compound (C) examples include styrene, ⁇ -methylstyrene, 1-vinylnaphthalene, 3-vinyltoluene, ethylvinylbenzene, vinylxylene, 4-cyclohexylstyrene, 2,4,6-trimethylstyrene, and tert-.
- Butoxydimethylsilylstyrene, isopropoxydimethylsilylstyrene and the like can be used alone or in combination of two or more, and among these, styrene is particularly preferable.
- styrene-butadiene rubber SBR
- butadiene rubber BR
- isoprene is used as the conjugated diene compound (B) and there is no structural unit of the aromatic vinyl compound (C)
- IR isoprene rubber
- SBR styrene-butadiene rubber
- the alkali metal compound used as the polymerization initiator is not particularly limited, but for example, an organic lithium compound is preferable.
- the organolithium compound may be either a low molecular weight organolithium compound or a solubilized oligomeric organolithium compound.
- Examples of the bonding mode between the organic group and lithium include a compound having a carbon-lithium bond, a compound having a nitrogen-lithium bond, and a compound having a tin-lithium bond. When an organolithium compound is used, the starting efficiency is good and the living rate of the polymer is also good.
- the organic lithium compound is not particularly limited, and examples thereof include an organic monolithium compound, an organic dilithium compound, and an organic polylithium compound.
- the organic group a hydrocarbon containing a functional group is preferable, and in that case, there is an advantage that the solubility in an organic solvent is excellent, and further, the starting speed is also excellent. Further, by using a compound having a nitrogen-lithium bond and a compound having a tin-lithium bond, a modifying group containing a functional group can be imparted to the starting end.
- the other organic alkali metal compound of the organic lithium compound is not particularly limited, and examples thereof include an organic sodium compound, an organic potassium compound, an organic rubidium compound, and an organic cesium compound. More specifically, sodium naphthalene, potassium naphthalene and the like can be mentioned. In addition, alcoholides such as lithium, sodium and potassium, sulfonates, carbonates, amides and the like can be mentioned. In addition, it may be used in combination with other organometallic compounds. Examples of the alkaline earth metal compound used as the polymerization initiator include organic magnesium compounds, organic calcium compounds, and organic strontium compounds. Further, compounds such as alkali earth metal alcoxides, sulfonates, carbonates and amides may be used. These organometallic earth metal compounds may be used in combination with the above-mentioned alkali metal compounds and other organometallic compounds.
- the alkali metal compound having a functional group that can be used as a polymerization initiator is not particularly limited, but for example, a lithium amide compound obtained by lithiumizing hydrogen of a secondary amine, or an alkyl to which the functional group is bonded. There is lithium etc.
- a functional group can be imparted to the polymerization initiation terminal of the conjugated diene-based copolymer.
- the functional group is not particularly limited, but a functional group inert to an alkali metal is preferable, and for example, a disubstituted amino group, that is, a tertiary amine, a protected monosubstituted amino group, and a protected amino group are preferable. ..
- Examples of the protected monosubstituted amino group or the protected amino group include one hydrogen of a monosubstituted amino group or two hydrogens of an amino group substituted with a trialkylsilyl group. ..
- the organic lithium compound used as an initiator is not particularly limited, and for example, mono-organic lithium such as n-butyllithium, sec-butyllithium, tert-butyllithium, n-propyllithium, iso-propyllithium, and benzyllithium.
- the polymerization initiator having a functional group is not particularly limited, but specific examples thereof include the compounds shown below.
- the types of functional groups that can be added to the polymer are described in parentheses.
- an oligomer initiator in which various lithium-based initiators and monomers are reacted can be used.
- a monomer having at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group, and a hydroxyl group can be used as the monomer.
- the oligomer initiator is not particularly limited, but a molecular weight of 1000 or less is preferable because it is industrially easy to handle. It is possible to carry out polymerization or copolymerization using a polymerization initiator having a functional group, and to introduce a branched polymer type modifying group derived from the modified vinyl aromatic copolymer of the present invention at the terminal end in the next step. Is.
- the conjugated diene compound (B) is polymerized by using the polyfunctional initiator, or the conjugated diene compound (B) and the aromatic vinyl compound (C) are copolymerized.
- This is a step of polymerizing to obtain a conjugated diene-based copolymer having an active terminal.
- a modified vinyl aromatic copolymer is added to two or more active ends of the conjugated diene-based copolymer.
- the reaction is carried out to carry out a terminal modification step in which two or more branched polymer-type modifying groups (A) based on the modified vinyl aromatic copolymer are introduced.
- the polyfunctional initiator is not particularly limited, and specific examples thereof include an organic dilithium compound and an organic polylithium compound.
- the organic group is not particularly limited, but a hydrocarbon is preferable. This has the advantage of excellent solubility in organic solvents, and also has an excellent starting speed.
- the method for preparing the polyfunctional initiator is not particularly limited, but specifically, a method by a reaction between a dispersion of metallic lithium and a polyhalogenated hydrocarbon compound, a method by a reaction between metallic lithium and a diene compound, and an organic substance. Examples thereof include a method based on the reaction of a monolithium compound and a polyfunctional compound. Of these, the reaction of the organic monolithium compound and the polyfunctional compound is preferable.
- the polyfunctional compound is not particularly limited, but specifically, it is an aromatic compound and has a plurality of double bonds adjacent to an aromatic group, and a plurality of active hydrogens that can be replaced with lithium. Examples include compounds. When the organic monolithium compound is reacted with these polyfunctional compounds, a conjugated diene compound and an aromatic vinyl compound can be present to form an oligomer. Further, in order to enhance the reactivity of the organic monolithium compound, a polar compound such as an ether compound or a tertiary amine compound may be present.
- the aromatic compound having a plurality of double bonds adjacent to the aromatic group is not particularly limited, and examples thereof include diisopropenylbenzene and divinylbenzene.
- the compound having a plurality of active hydrogens that can be substituted with lithium is not particularly limited, and examples thereof include 1,3,5-trimethylbenzene.
- the above-mentioned hydrocarbon dilithium compound is not particularly limited, but for example, 1,3-bis (1-lithio-1,3-dimethylpentyl) obtained from the reaction of secondary or tertiary butyl lithium with diisopropenylbenzene.
- Benzene, 1,4-bis (1-lithio-1,3-dimethylpentyl) benzene, 1,3-bis (1-lithio-1,3,3-trimethylbutyl) benzene, and 1,4-bis (1) -Lithio-1,3,3-trimethylbutyl) benzene and the like can be mentioned.
- the hydrocarbon polylithium compound is not particularly limited, and is, for example, a reaction product of n-butyllithium, divinylbenzene and 1,3-butadiene, secondary or tertiary butyllithium and 1,3,5-trimethyl. Examples thereof include reaction products with benzene.
- the amount of these polyfunctional initiators used is adjusted by the molecular weight of the target butadiene polymer, and 0.01 to 0.2 parts by mass can be used with respect to 100 parts by mass of the monomer. It is preferably 0.02 to 0.15 parts by mass. At this time, it is preferable to consider deactivation due to moisture and impurities in the monomer and solvent, chain transfer due to impurities, deactivation due to formation of metal hydride at the polymerization terminal, and the like.
- a polar compound may be added when producing a conjugated diene-based copolymer.
- the vinyl aromatic compound can be randomly copolymerized with a conjugated diene compound such as 1,3-butadiene.
- the polar compound can also be used as a vinylizing agent for controlling the microstructure of the conjugated diene portion. It is also effective in promoting the polymerization reaction.
- polar compound examples include ethers such as tetrahydrofuran, diethyl ether, dioxane, ethylene glycol dimethyl ether, ethylene glycol dibutyl ether, diethylene glycol dimethyl ether, diethylene glycol dibutyl ether, dimethoxybenzene, and 2,2-bis (2-oxolanyl) propane; Tertiary amine compounds such as methylethylenediamine, dipiperidinoethane, trimethylamine, triethylamine, pyridine and quinuclidine; alkali metal alkoxides such as potassium-tert-amylate, potassium-tert-butyrate, sodium-tert-butyrate and sodium amylate.
- ethers such as tetrahydrofuran, diethyl ether, dioxane, ethylene glycol dimethyl ether, ethylene glycol dibutyl ether, diethylene glycol dimethyl ether, diethylene glycol dibutyl ether,
- Examples include phosphine compounds such as triphenylphosphine. These polar compounds may be used alone or in combination of two or more.
- the amount of the polar compound used is not particularly limited and can be selected according to the purpose and the like. Usually, it is preferably 0.01 to 100 mol with respect to 1 mol of the polymerization initiator or the polyfunctional initiator.
- Such a polar compound can be used in an appropriate amount as a regulator of the microstructure of the polymer-conjugated diene moiety, depending on the desired amount of vinyl bond.
- Many polar compounds have an effective randomizing effect in the copolymerization of the conjugated diene compound and the aromatic vinyl compound at the same time, and can be used as an agent for adjusting the distribution of the aromatic vinyl compound and adjusting the amount of styrene block.
- a method for randomizing the conjugated diene compound and the aromatic vinyl compound for example, the total amount of styrene and a part of the conjugated diene compound (B) as described in JP-A-59-140211 can be used.
- a method may be used in which the copolymerization reaction is started and the remaining conjugated diene compound (B) is continuously or intermittently added during the copolymerization reaction.
- the polymerization or copolymerization of the conjugated diene compound (B) is preferably carried out by solution polymerization in an inert solvent.
- the polymerization solvent is not particularly limited, and for example, hydrocarbon solvents such as saturated hydrocarbons and aromatic hydrocarbons are used.
- aliphatic hydrocarbons such as butane, pentane, hexane and heptane
- alicyclic hydrocarbons such as cyclopentane, cyclohexane, methylcyclopentane, methylcyclohexane, dimethylcyclohexane, ethylcyclohexane and decalin
- benzene, toluene examples thereof include hydrocarbon-based solvents composed of aromatic hydrocarbons such as xylene and mixtures thereof.
- the above-mentioned conjugated diene compound and the polymerization solvent are preferably used alone or a mixture thereof is preferably treated with an organometallic compound.
- the polymerization temperature at the time of copolymerizing the conjugated diene compound (B) or the conjugated diene compound (B) and the aromatic vinyl compound (C) is not particularly limited as long as the living anionic polymerization proceeds, but the productivity is limited.
- the temperature is preferably 0 ° C. or higher, and the structural unit (c) composed of the silane-based functional group represented by the above formula (2) and / or the formula (3) of the present invention with respect to the active terminal after the completion of polymerization.
- the polymerization mode for copolymerizing the conjugated diene compound (B) or the conjugated diene compound (B) and the aromatic vinyl compound (C) is not particularly limited, but is a batch type (also referred to as “batch type”). , It can be carried out by a polymerization mode such as a continuous type. In the continuous system, one or two or more connected reactors can be used. As the reactor, a tank type with a stirrer, a tube type, or the like is used. In the batch formula, the molecular weight distribution of the obtained polymer is generally narrow, and Mw / Mn tends to be 1.0 or more and less than 1.8. Further, in the continuous formula, the molecular weight distribution is generally wide, and in Mw / Mn, it tends to be 1.8 or more and 3 or less.
- the modified vinyl aromatic copolymer of the present invention is reacted with the active terminal of the conjugated diene copolymer to introduce the functional group and synthesize a multi-branched high molecular weight polymer. It is a process. This step is the same regardless of whether the polymerization initiator is used or the polyfunctional initiator is used.
- the reaction temperature, reaction time, etc. when reacting the modified vinyl aromatic copolymer with the active end of the polymer are not particularly limited, but it is preferable to react at 0 to 120 ° C. for 30 seconds or longer.
- the amount of the modified vinyl aromatic copolymer added is not particularly limited, but is that of the modified vinyl aromatic copolymer (A) with respect to the number of moles of the polymerization initiator or polyfunctional initiator, that is, the number of polymer molecules.
- the total number of moles is preferably in the range of 0.01 to 6 times, more preferably in the range of 0.02 to 3 times, and further preferably in the range of 0.05 to 2 times. preferable.
- the addition amount is 0.05 times or more, it is preferable from the viewpoint of obtaining a sufficient modification rate in the target modified conjugated diene polymer.
- the degree of branching and the amount of the branched polymer component in which the polymer ends are coupled to each other can be arbitrarily controlled by the structure of the modified vinyl aromatic copolymer (A).
- a modified vinyl aromatic copolymer (A) having a high functionality can be used as the modified vinyl aromatic copolymer (A).
- the amount of the modified vinyl aromatic copolymer (A) added is 0.7 times or less as the functional group equivalent with respect to the number of moles of the polymerization initiator or the polyfunctional initiator, that is, the number of polymer molecules. preferable.
- the modification reaction when the polymerization step is a batch type, the modification reaction may be subsequently carried out in the reactor used in the polymerization step, or may be transferred to the next reactor. If the polymerization step is continuous, it is transferred to the next reactor.
- the terminal denaturation step is preferably carried out immediately following the polymerization step, preferably within 5 minutes by mixing the denaturant to carry out the reaction.
- the reactor for the denaturation reaction is preferably one in which sufficient stirring is performed. Specifically, there are a static mixer type reactor, a row type reactor with a stirrer, and the like.
- the active terminal of the conjugated diene-based copolymer is used.
- One or more functional groups different from the modified vinyl aromatic copolymer can be introduced.
- the active terminal of the conjugated diene-based copolymer is composed of an amino group, an alkoxysilyl group, and a hydroxyl group.
- a modified compound having at least one functional group selected is reacted to form a bond, or a compound forming these functional groups is reacted to modify the terminal end.
- the functional group to be bonded to the active terminal of the conjugated diene polymer is not particularly limited, but specifically, a halogen group, a double bond, a ketone group, an ester group, an amide group, etc. Examples thereof include an epoxy group and an alkoxysilyl group.
- a modification compound having at least one functional group selected from an amino group, an alkoxysilyl group, and a hydroxyl group is reacted with the active terminal of the butadiene-based copolymer to form a bond, or a bond thereof is formed.
- the functional group to be bonded to the active terminal of the conjugated diene polymer is not particularly limited, but specifically, a halogen group, a double bond, a ketone group, an ester group, an amide group, etc. Examples thereof include an epoxy group and an alkoxysilyl group.
- the compound having an amino group is not particularly limited, but specifically, a compound having an amino group and a functional group bonded to the active end of the polymer in the molecule, preferably having no active hydrogen.
- the amino group is not particularly limited, but specifically, a functional group inactive with respect to an alkali metal is preferable, and a disubstituted amino group, that is, a tertiary amine, a protected monosubstituted amino group, and two hydrogens. Amino groups protected by are preferred.
- the compound having an alkoxysilyl group is not particularly limited, but specifically, a compound having a plurality of alkoxysilyl groups in the molecule (this has a silyl group in which a plurality of alkoxy groups are bonded). (Including compounds), and compounds having an alkoxysilyl group and a functional group bonded to the active terminal of the polymer in the molecule can be mentioned. It is preferable that these are compounds having no active hydrogen.
- the compound that forms a hydroxyl group is not particularly limited, but specifically, a compound having a functional group that binds to the active terminal of the polymer and having a functional group that forms a hydroxyl group after the binding reaction, heavy weight. Examples thereof include compounds having a functional group that does not bind to the combined active terminal and having a functional group in which a hydroxyl group is later generated by a reaction such as hydrolysis, and a compound having no active hydrogen is preferable.
- Examples of the compound having a functional group in which a hydroxyl group is generated after the above-mentioned bonding reaction include a compound having a ketone group, an ester group, an amide group, an epoxy group and the like.
- examples of the compound having a functional group in which a hydroxyl group is generated by a reaction such as hydrolysis after the binding reaction include a compound having an alkoxysilyl group, an aminosilyl group and the like.
- the compound that binds to the active end of the polymer to form an amino group and a hydroxyl group at the end of the polymer is not particularly limited, but is N, N, N', N'-tetramethyl-4,4'-diaminobenzophenone ( Michler's Ketone), N, N, N', N'-tetraethyl-4,4'-diaminobenzophenone and other ketone compounds; cyclic urea compounds such as N, N'-dimethylimidazolidinone and N-methylpyrrolidone.
- Cyclic amide that is, a lactam compound; an amino group-containing epoxy compound such as N, N, N', N'-tetraglycidyl-1,3-bisaminomethylcyclohexane; a nitrogen-containing heterocyclic compound described in JP-A-2001-131227.
- Examples thereof include epoxy compounds having a group.
- the compound that binds to the active terminal of the polymer to form an alkoxysilyl group at the end of the polymer is not particularly limited, but is a halogenated alkoxysilane compound such as trimetokichlorosisilane, triethoxychlorosilane, or diphenoxydicrylorosilane.
- Examples thereof include polyfunctional alkoxysilane compounds such as bis (trimethoxysilyl) ethane and bis (3-triethoxysilylpropyl) ethane.
- the compound that binds to the active end of the polymer to form an alkoxysilyl group and a hydroxyl group at the end of the polymer is not particularly limited, but 3-glycidoxypropyltrimethoxysilane and 3-glycidoxypropyltriethoxysilaneepoxy.
- Examples thereof include polysiloxane compounds having a group and an alkoxysilyl group in the molecule.
- the compound that binds to the active terminal of the polymer to form an amino group and an alkoxysilyl group at the end of the polymer is not particularly limited, but is limited to 3-dimethylaminopropyltrimethoxysilane, 3-dimethylaminopropyldimethoxymethylsilane, 3 -Alkoxysilane compound to which an alkyl group having an amino substituent such as dimethylaminopropyltriethoxysilane, bis (3-trimethoxysilylpropyl) methylamine, bis (3-triethoxysilylpropyl) methylamine is bonded; N-[ 3- (Triethoxysilyl) -propyl] -N, N'-diethyl-N'-trimethylsilyl-ethane-1,2-diamine, 3- (4-trimethylsilyl-1-piperazinyl) propyltriethoxysilane, etc.
- WO 2008/013090 Alkoxysilane compound to which a plurality of substituted amino groups are bonded; 1,4-bis [3- (trimethoxysilyl) propyl] piperazine, 1,4-bis [3- (triethoxysilyl) Alkoxysilane compound to which a nitrogen-containing heterocycle described in WO2011 / 040312 such as propyl] piperazine is bonded; 3-[N, N-bis (trimethylsilyl) amino] propyltrimethoxysilane, 3- [N, N-bis (trimethylsilyl) ) Amino] propylmethyldiethoxysilane, 2,2-dimethoxy-1- (3-trimethoxysilylpropyl) -1-aza-2-silacyclopentane, 2,2-diethoxy-1- (3-triethoxysilyl) Examples thereof include alkoxysilane compounds to which an azasilane group is bonded as described in WO2011 / 129425, such
- main chain modification step silicon having at least one functional group selected from the group consisting of a vinyl group of the main chain of the modified conjugated diene polymer having a modified terminal, an amino group, an alkoxysilyl group, and a hydroxyl group.
- This is a step of modifying the main chain by hydrosilylating a compound or a silicon compound forming at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group, or a hydroxyl group. This step is the same regardless of whether the polymerization initiator is used or the polyfunctional initiator is used.
- the hydrosilylation reaction can be carried out in an organic solvent solution or as a polymer (without solvent) in a kneader.
- a silicon compound such as a hydrosilane compound having a functional group can be reacted in the presence of a catalyst to modify the vinyl group of the main chain of the conjugated diene-based copolymer.
- a catalyst to modify the vinyl group of the main chain of the conjugated diene-based copolymer.
- the silicon compound is not particularly limited, and specific examples thereof include a hydrosilane compound.
- a hydrosilane compound is not particularly limited, and specifically, any hydrosilane compound having at least one functional group selected from the group consisting of an amino group, an alkoxysilyl group, and a hydroxyl group may be used.
- the general formula HSiR'' 3-n X n (R'' is not particularly limited, but represents a hydrocarbon group having 1 to 20 carbon atoms, and X represents an amino group, an alkoxy group, a hydroxyl group or an amino.
- a hydrocarbon or organosilane compound group having at least one functional group selected from the group consisting of a group, an alkoxysilyl group and a hydroxyl group is represented, and n is 1 to 3). More preferably, n is 1, and R ′′ preferably represents a hydrocarbon having 1 to 3 carbon atoms.
- the hydrosilane compound has such a structure, the yield of the hydrosilylation reaction is high. Further, it may be a silicon compound that forms the above-mentioned functional group by performing hydrolysis or the like after the hydrosilaneization reaction.
- Such a silicon compound is not particularly limited, and specifically, a hydrosilane compound having a functional group that forms at least one functional group of an amino group, an alkoxysilyl group, or a hydroxyl group by performing hydrolysis or the like. Can be mentioned. More specifically, a hydrosilane compound having a protected mono-substituted amino group, a protected di-substituted amino group, a protected hydroxyl group and the like can be mentioned.
- the silicon compound having an alkoxy group is not particularly limited, but specifically, dimethylmonomethoxysilane, dimethylmonoethoxysilane, dimethylmonopropoxysilane, dimethylmonobutoxysilane, methyldimethoxysilane, methyldiethoxysilane, and methyldi.
- organosiloxane compounds having an H—Si group and an alkoxysilyl group in the molecule such as propoxysilane, ethyldiethoxysilane, trimethoxysilane, and triethoxysilane.
- the silicon compound having a disubstituted amino group is not particularly limited, but specifically, dimethylaminodimethylsilane, diethylaminodimethylsilane, diethylaminodiethylsilane, 3-diethylaminopropyldimethylsilane, 4-dimethylaminobutyldimethylsilane, 6 -Diethylaminohexyldimethylsilane.
- the silicon compound having a protected monosubstituted amino group is not particularly limited, and specific examples thereof include N-methyl-N-trimethylsilylaminodimethylsilane and N-ethyl-N-trimethylsilylaminodiethylsilane.
- the silicon compound having a protected disubstituted amino group is not particularly limited, and specific examples thereof include N, N-bistrimethylsilylaminodimethylsilane and N, N-bistrimethylsilylaminodiethylsilane.
- the silicon compound having a hydroxyl group is not particularly limited, and specific examples thereof include dimethylhydroxysilane, diethylhydroxysilane, and dibutylhydroxysilane.
- the silicon compound that forms a hydroxyl group by hydrolysis is not particularly limited, but specifically, an alkoxysilane compound such as dimethylmonomethoxysilane, dimethylmonoethoxysilane, methyldimethoxysilane, trimethoxysilane, and triethoxysilane; dimethyl.
- alkoxysilane compound such as dimethylmonomethoxysilane, dimethylmonoethoxysilane, methyldimethoxysilane, trimethoxysilane, and triethoxysilane; dimethyl.
- silane compounds having an epoxy group such as glycidylsilane and diethylglycidylsilane.
- the amount of the hydrosilane compound to react is arbitrary depending on the purpose, but is preferably 1 to 10 per 1 mol of the main chain of the copolymer. It is a molar.
- 1 to 10 mol with respect to 1 mol of the main chain good affinity can be obtained when a modified diene copolymer and silica are mixed to obtain a rubber composition, as will be described later. , Excellent workability. More preferably, it is 2 to 5 mol with respect to 1 mol of the main chain.
- a predetermined catalyst may be used when carrying out the hydrosilylation reaction.
- the catalyst is not particularly limited, but for example, platinum or a platinum-containing catalyst is mainly used.
- a homogeneous platinum catalyst is preferably used, for example, a platinum chloride solution (ie, Spieer catalyst), a Pt 2 (divinyltetramethyldisiloxane) 3 solution (ie, Karstedt catalyst), dichloro ( ⁇ 4-cyclo-1, 5-Diene) Pt (II) and the like.
- the amount of platinum catalyst used in the reaction is preferably 0.01 to 10 mmol / mol, more preferably 0.1 to 1 mmol / mol per hydrosilane compound.
- examples of the catalyst used in carrying out the hydrosilylation reaction include metallocene compounds containing any of Ti, Zr, Hf, Ni, Co, Ru, and Rh, and in particular, titanocene compounds and organolithium or organoaluminum.
- the reaction product with is suitable.
- the hydrosilylation reaction is preferably carried out in the range of 20 to 150 ° C, more preferably in the range of 50 to 120 ° C. In this range, it can be carried out with an appropriate reaction time, and there are few side reactions such as gelation, which is practical.
- the polymerization solution is used as it is and the hydrosilylation reaction is carried out after the terminal denaturation reaction, it can be carried out at the same temperature as the polymerization temperature.
- the reaction time is preferably 10 minutes to 5 hours, more preferably 30 minutes to 2 hours.
- the weight average molecular weight (in terms of polystyrene) of the modified conjugated diene copolymer of the present invention is preferably 100,000 to 2,000,000, more preferably 150,000 to 1,000,000 in consideration of processability and physical properties.
- the weight average molecular weight can be determined by measuring a chromatogram using a GPC using a column using a polystyrene-based gel as a filler and using a calibration curve using standard polystyrene.
- the polyfunctional vinyl aromatic copolymer (A) and the conjugated diene compound (B) are used.
- the ratio is preferably in the following range.
- the structural unit (A1) derived from the modified vinyl aromatic copolymer (A) having the structural unit (c) composed of the silane-based functional group represented by the formula (2) and / or the formula (3) is It is 0.001 to 6% by weight, preferably 0.001 to 5% by weight, more preferably 0.005 to 5% by weight, still more preferably 0.01 to 5% by weight, and optimally 0.001 to 1%. By weight%.
- the structural unit (B1) derived from the conjugated diene compound (B) is 29 to 99.999% by weight, preferably 80 to 99.999% by weight, more preferably 90 to 99.995% by weight, and further. It is preferably 95 to 99.99% by weight.
- the structural unit (A1) has the same range as described above, and the structural unit (B1) is 30 to 97.999% by weight, preferably 45 to 94.995% by weight, and more preferably 55 to 89.99% by weight. %.
- the structural unit (C1) derived from the aromatic vinyl compound (C) is 2 to 50% by weight, preferably 5 to 45% by weight, and more preferably 10 to 40% by weight.
- the microstructure (cis, trans, vinyl bond amount) of the modified conjugated diene copolymer can be arbitrarily changed by using a polar compound or the like, but in the state before the terminal is modified, the conjugated diene unit is used.
- the content of the vinyl bond (1,2-bond) in the mixture is preferably 10 to 80 mol%.
- the modified conjugated diene-based copolymer of the present invention is used as a rubber composition described later and further crosslinked to be used as an automobile tire, 20 to 75 is required to highly balance rolling resistance performance and wear resistance. Mol% is preferred, 30-75 mol% is more preferred, and 40-70 mol% is even more preferred.
- a reaction terminator may be added to the polymer solution of the modified conjugated diene-based copolymer obtained by the above-mentioned polymerization method.
- the reaction terminator for example, alcohols such as methanol, ethanol and propanol; organic acids such as stearic acid, lauric acid and octanoic acid; water and the like can be used.
- the metals contained in the polymer may be decalcified as necessary.
- a decalcification method for example, a method is used in which an oxidizing agent such as water, an organic acid, an inorganic acid, or hydrogen peroxide is brought into contact with a polymer solution to extract metals, and then the aqueous layer is separated. ..
- the antioxidant is not particularly limited, and known ones can be used.
- examples of the antioxidant include phenol-based stabilizers, phosphorus-based stabilizers, sulfur-based stabilizers and the like.
- BHT 2,6-di-tert-butyl-4-hydroxytoluene
- Antioxidants such as propinate, 2-methyl-4,6-bis [(octylthio) methyl] phenol are preferred.
- alcohols such as water, methanol, ethanol, and isopropanol can be added as additives to remove or neutralize ionic substances, or stearic acid, oleic acid, myristic acid, lauric acid, and decane can be added.
- Carboxylic acids such as acid, citric acid and oleic acid, aqueous inorganic acid solutions, carbon dioxide and the like may be added.
- the modified conjugated diene copolymer of the present invention has a number average molecular weight (Mn) that is three times or more the peak on the lowest molecular weight side when the total area is 100% in the differential molecular weight distribution curve obtained by GPC measurement.
- the area of the region having the above is preferably 10% or more. This will be described with reference to FIG. FIG. 1 shows a typical example of a GPC elution curve and the positions of peak top Mp and a molecular weight of 3 Mp three times that of peak top.
- the area of the number average molecular weight 3Mp region (the region on the right side of 3Mp in FIG. 1), which is three times the number average molecular weight Mp of the peak on the lowest molecular weight side, should be 10 area% or more. This means that there are relatively many high molecular weight components.
- the modified vinyl aromatic copolymer (A) of the present invention itself can be used for modifying a rubber composition.
- this copolymer (A) By blending this copolymer (A) into the rubber composition, the processability of the rubber composition is improved, and the affinity of the reinforcing filler in the obtained rubber composition is improved. Therefore, the rubber composition Improving the physical properties of crosslinked products (for example, dry grip performance, wet grip performance), which tends to be present in the vicinity of the reinforcing filler when the rubber composition is produced, resulting in a dispersed state of the reinforcing filler in the rubber composition. ) Is estimated to be ideal. Further, as a result of the branched polymer-type modifying group based on the modified vinyl aromatic copolymer being likely to be present in the vicinity of the reinforcing filler, a crosslinked product having excellent wear resistance can be obtained.
- the modified conjugated diene-based copolymer composition of the present invention (hereinafter, also referred to as a rubber composition) will be described.
- the rubber composition contains the modified conjugated diene polymer and a filler.
- Another aspect of the rubber composition of the present invention contains 0.5 to 200 parts by mass of a reinforcing filler with respect to 100 parts by mass of the modified conjugated diene polymer.
- Another aspect of the rubber composition of the present invention contains 100 parts by mass of the raw material rubber containing 20 parts by mass or more of the modified conjugated diene polymer and 5 to 200 parts by mass of the filler.
- the content of the modified conjugated diene polymer in 100 parts by mass of the raw material rubber is 20 parts by mass or more, preferably 40 parts by mass or more, more preferably 50 parts by mass or more, and further preferably 60 parts by mass or more.
- the dispersibility that is the object of the present invention is excellent, and when the rubber composition of the present embodiment is a vulcanized composition, the performance such as tensile property and viscoelastic property is excellent, and it is used for tires.
- excellent fuel efficiency, grip performance, abrasion resistance, and rigidity can be obtained.
- the content of the modified conjugated diene polymer is preferably 90 parts by mass or less, more preferably 80 parts by mass or less. By setting the content to 90 parts by mass or less, the Mooney viscosity of the unvulcanized modified conjugated diene copolymer composition of the present invention is lowered and the processability is improved.
- the raw material rubber other than the modified butadiene polymer is not particularly limited, and for example, a conjugated diene polymer or a hydrogenated product thereof, a random copolymer of a conjugated diene compound and a vinyl aromatic compound or a hydrogenated product thereof. , Block copolymers of conjugated diene compounds and vinyl aromatic compounds or hydrogenated products thereof, other conjugated diene copolymers or hydrogenated products thereof, non-diene polymers, natural rubbers and the like.
- conjugated diene polymer or its hydrogenated product examples include butadiene rubber or its hydrogenated product, isoprene rubber or its hydrogenated product, and the like.
- random copolymer of the conjugated diene compound and the vinyl aromatic compound or the hydrogenated product thereof are not particularly limited, and examples thereof include styrene-butadiene copolymer rubber or the hydrogenated product thereof.
- block copolymer of the conjugated diene compound and the vinyl aromatic compound or the hydrogenated product thereof are not particularly limited, and for example, the styrene-butadiene block copolymer or the hydrogenated product thereof, the styrene-isoprene block.
- examples thereof include styrene-based elastomers such as a copolymer or a hydrogenated product thereof.
- conjugated diene-based copolymers or hydrogenated products thereof are not particularly limited, and examples thereof include acrylonitrile-butadiene rubber or hydrogenated products thereof.
- the non-diene polymer is not particularly limited, and is, for example, an olefin-based elastomer such as ethylene-propylene rubber, ethylene-propylene-diene rubber, ethylene-butene-diene rubber, ethylene-butene rubber, ethylene-hexene rubber, and ethylene-octene rubber.
- olefin-based elastomer such as ethylene-propylene rubber, ethylene-propylene-diene rubber, ethylene-butene-diene rubber, ethylene-butene rubber, ethylene-hexene rubber, and ethylene-octene rubber.
- the modified conjugated diene polymer is a modified styrene-butadiene rubber
- polybutadiene is preferable as the other rubber.
- modified butadiene polymer is modified polybutadiene, natural rubber or polyisoprene rubber is preferable as the other rubber.
- the weight average molecular weight of the various rubber-like polymers described above is preferably 2,000 to 2,000,000, preferably 5,000 to 1,500,000 from the viewpoint of the balance between performance and processing characteristics. Is more preferable. Further, so-called liquid rubber having a low molecular weight can also be used. These rubber-like polymers may be used alone or in combination of two or more.
- the weight average molecular weight referred to here is a polystyrene-equivalent weight average molecular weight (Mw) obtained by gel permeation chromatography (GPC) measurement.
- the amount of filler used with respect to the raw rubber is adjusted so that the hardness and modulus increase when a large amount of filler is used, and the desired physical properties are obtained according to the application. If it is within this range, the filler is well dispersed and the workability is good.
- the filler is preferably 5 to 150 parts by mass, and for footwear applications, preferably 30 to 200 parts by mass. Within the scope of this embodiment, it is possible to handle a wide range of materials from soft to hard.
- a silica-based inorganic filler In the rubber composition as another aspect of the present invention, a silica-based inorganic filler, a metal oxide, and metal water are used with respect to 100 parts by mass of the modified conjugated diene polymer, preferably 100 parts by mass of the raw material rubber. It contains 0.5 to 200 parts by mass of at least one reinforcing filler selected from the group consisting of oxides and carbon blacks.
- the silica-based inorganic filler contained in the rubber composition it is preferable to use solid particles containing SiO 2 or a silicate as a main component of the constituent unit.
- the main component means a component that occupies 50% by mass or more of the whole, preferably a component that occupies 70% by mass or more, and more preferably a component that occupies 90% by mass or more.
- the silica-based inorganic filler include inorganic fibrous substances such as silica, clay, talc, mica, diatomaceous earth, wallacenite, montmorillonite, zeolite, and glass fiber.
- the silica-based inorganic filler may be used alone or in combination of two or more.
- a silica-based inorganic filler having a hydrophobic surface, or a mixture of a silica-based inorganic filler and a non-silica-based inorganic filler can also be used.
- silica and glass fiber are preferable, and silica is more preferable.
- silica dry silica, wet silica, synthetic silicate silica and the like can be used, but among them, wet silica is preferable because it is more excellent in both improvement of fracture characteristics and wet skid resistance performance.
- silica used as the reinforcing filler silica having a BET specific surface area of 50 to 500 m 2 / g is used. By blending such silica, excellent fuel efficiency, wear resistance, wet skid performance and steering stability can be obtained.
- the dispersibility of silica is good, it is possible to disperse silica having a high specific surface area, that is, fine particle size silica.
- the silica-based inorganic filler can be fine particle size silica, and the fine particle size silica may contain silica having a CTAB specific surface area of 180 m 2 / g or more and a BET specific surface area of 185 m 2 / g or more. it can. By blending such fine particle size silica, a rubber composition having excellent fuel efficiency, wear resistance, wet skid performance and steering stability can be obtained.
- the CTAB (cetyltrimethylammonium bromide) specific surface area of the fine particle size silica is not particularly limited, and is preferably 190 m 2 / g or more, more preferably 195 m 2 / g or more, and further preferably 197 m 2 / g or more.
- the upper limit of the specific surface area is not particularly limited, and is preferably 500 m 2 / g or less, more preferably 300 m 2 / g or less, and further preferably 250 m 2 / g or less.
- the CTAB specific surface area is 500 m 2 / g or less, a rubber composition having excellent workability can be obtained. More preferably, the CTAB specific surface area is 190 to 300 m 2 / g.
- the CTAB specific surface area is measured according to ASTM D3765-92.
- the BET specific surface area of the fine particle size silica is not particularly limited, and is preferably 190 m 2 / g or more, more preferably 195 m 2 / g or more, and further preferably 210 m 2 / g or more. When the BET specific surface area is 185 m 2 / g or more, a rubber composition having excellent wear resistance can be obtained.
- the upper limit of the BET specific surface area is not particularly limited, and is preferably 500 m 2 / g or less, more preferably 300 m 2 / g or less, and further preferably 260 m 2 / g or less. When the BET specific surface area is 500 m 2 / g or less, a modified conjugated diene-based copolymer composition having excellent processability can be obtained.
- the BET specific surface area of silica is measured according to ASTM D3037-81.
- the aggregate size of the fine particle size silica is not particularly limited and can be 30 nm or more, preferably 35 nm or more, more preferably 40 nm or more, still more preferably 45 nm or more, still more preferably 50 nm or more, still more preferable. Is 55 nm or more.
- the aggregate size is not particularly limited, and is preferably 100 nm or less, more preferably 80 nm or less, still more preferably 70 nm or less, and particularly preferably 65 nm or less. By having such an aggregate size, it is possible to provide excellent reinforcing property, fuel efficiency, wear resistance, wet skid performance and steering stability while having good dispersibility.
- the aggregate size is also called the aggregate diameter or the maximum frequency Stokes equivalent diameter, and is the particle diameter when a silica aggregate composed of a plurality of primary particles is regarded as one particle. It is equivalent.
- the aggregate size can be measured using, for example, a disk centrifugal sedimentation type particle size distribution measuring device such as BI-XDC (manufactured by Brookhaven Instruments Corporation). Specifically, it can be measured by the method described in Japanese Patent Application Laid-Open No. 2011-132307.
- the average primary particle size of the fine particle size silica is not particularly limited, and is preferably 25 nm or less, more preferably 22 nm or less, still more preferably 17 nm or less, and particularly preferably 14 nm or less.
- the lower limit of the average primary particle size is not particularly limited, but is preferably 3 nm or more, more preferably 5 nm or more, and further preferably 7 nm or more. Within this range, dispersibility and reinforcing properties are excellent.
- the average primary particle size of the fine particle size silica can be determined by observing with a transmission type or scanning electron microscope, measuring 400 or more primary particles of silica observed in the visual field, and averaging them.
- the blending amount of the fine particle size silica in the rubber composition of the present invention is not particularly limited, and is preferably 5 parts by mass or more, more preferably 15 parts by mass or more, still more preferably 15 parts by mass or more with respect to 100 parts by mass of the rubber component. It is 20 parts by mass or more, more preferably 25 parts by mass or more, and even more preferably 30 parts by mass or more. If it is 5 parts by mass or more, the effect of blending the fine particle silica can be sufficiently obtained.
- the upper limit of the blending amount of the fine particle size silica is 200 parts by mass or less, preferably 100 parts by mass or less, more preferably 80 parts by mass or less, still more preferably 60 parts by mass or less, still more preferably 55 parts by mass or less. If it is 200 parts by mass or less, substantially good workability can be obtained.
- the rubber composition of the present invention preferably further contains 5 to 100 parts by mass of silica or carbon black having a BET specific surface area of less than 185 m 2 / g as a reinforcing filler.
- silica having a BET method nitrogen adsorption specific surface area (N2SA) of less than 185 m 2 / g is preferably used as the reinforcing filler, and silica having a BET method nitrogen adsorption specific surface area (N2SA) of less than 150 m 2 / g is preferably used, preferably 50 m 2 / g or more. Is used. In this range, the balance between reinforcement and dispersibility is good. Further, a particle having a suitable particle size is used depending on the application.
- Examples of the carbon black include N110, N220, N330, N339, N550, N660 and the like from the classification of carbon black for rubber by ASTM, and are selected according to the application. By using carbon black together, it is possible to improve the reinforcing property and improve the dry grip performance when used for tire tread applications.
- the carbon black used has a BET method nitrogen adsorption specific surface area (N2SA) of preferably less than 185 m 2 / g, preferably 30 m 2 / g or more, and more preferably in the range of 50 to 130 m 2 / g. .. In this range, the balance between reinforcement and dispersibility is good. When used for tire tread applications, it is more preferably N220, N330, N339.
- the reinforcing filler in addition to the above-mentioned silica and the like, other reinforcing fillers can be used.
- the other reinforcing filler is not particularly limited, but the metal oxide as the reinforcing filler has the chemical formula MxOy (M represents a metal atom, and x and y each represent an integer of 1 to 6). It is preferably a solid particle containing the component as a main component.
- the main component means a component that occupies 50% by mass or more of the whole, preferably a component that occupies 70% by mass or more, and more preferably a component that occupies 90% by mass or more.
- the metal oxide for example, alumina, titanium oxide, magnesium oxide, zinc oxide and the like can be used.
- metal hydroxide as the reinforcing filler examples include aluminum hydroxide, magnesium hydroxide, zirconium hydride and the like.
- metal oxides and metal hydroxides as the other reinforcing fillers may be used alone or in combination of two or more. Further, a mixture with an inorganic filler other than these can also be used.
- a silane coupling agent may be used.
- the silane coupling agent is not particularly limited, and is, for example, a compound having both a silica-affinitive portion and a polymer-affinitive portion in the molecule, and is, for example, a sulfide compound, a mercapto compound, a vinyl compound, an amino compound, and the like. Examples thereof include glycidoxy-based compounds, nitro-based compounds, and chloro-based compounds.
- sulfide compound examples include bis (3-triethoxysilylpropyl) tetrasulfide, bis (2-triethoxysilylethyl) tetrasulfide, bis (3-trimethoxysilylpropyl) tetrasulfide, and bis (2-trimethoxy).
- Cyrilethyl) tetrasulfide bis (3-triethoxysilylpropyl) trisulfide, bis (3-trimethoxysilylpropyl) trisulfide, bis (3-triethoxysilylpropyl) disulfide, bis (3-trimethoxysilylpropyl) Disulfide, 3-trimethoxysilylpropyl-N, N-dimethylthiocarbamoyltetrasulfide, 3-triethoxysilylpropyl-N, N-dimethylthiocarbamoyltetrasulfide, 2-trimethoxysilylethyl-N, N-dimethylthiocarbamoyl Tetrasulfide, 3-trimethoxysilylpropylbenzothiazole tetrasulfide, 3-triethoxysilylpropylbenzothiazole tetrasulfide, 3-triethoxysilyl
- Examples of the mercapto-based compound include 3-mercaptopropyltrimethoxysilane, 3-mercaptopropyltriethoxysilane, 2-mercaptoethyltrimethoxysilane, and 2-mercaptoethyltriethoxysilane.
- Examples of the vinyl compound include vinyltriethoxysilane and vinyltrimethoxysilane.
- Examples of amino compounds include 3-aminopropyltriethoxysilane, 3-aminopropyltrimethoxysilane, 3- (2-aminoethyl) aminopropyltriethoxysilane, and 3- (2-aminoethyl) aminopropyltrimethoxy. Examples include silane.
- Examples of the glycidoxy-based compound include ⁇ -glycidoxypropyltriethoxysilane, ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -glycidoxypropylmethyldiethoxysilane, and ⁇ -glycidoxypropylmethyldimethoxysilane. Can be mentioned.
- Examples of the nitro compound include 3-nitropropyltrimethoxysilane and 3-nitropropyltriethoxysilane.
- chloro compound examples include 3-chloropropyltrimethoxysilane, 3-chloropropyltriethoxysilane, 2-chloroethyltrimethoxysilane, and 2-chloroethyltriethoxysilane.
- examples of other compounds include octyltriethoxysilane, methyltriethoxysilane, methyltrimethoxysilane, hexadecyltrimethoxysilane and the like.
- silane coupling agents may be used alone or in combination of two or more.
- sulfur-containing silane coupling agents such as sulfide compounds and mercapto compounds are preferable from the viewpoint of large reinforcing effect, and bis (3-triethoxysilylpropyl) disulfide and bis (3-).
- Triethoxysilylpropyl) tetrasulfide and 3-mercaptopropyltrimethoxysilane are more preferred.
- the blending amount of the silane coupling agent is 1 to 20 parts by mass, preferably 2 to 15 parts by mass with respect to 100 parts by mass of silica.
- a silane coupling agent is blended in this range, the dispersibility of silica is further improved, the workability is improved, and the wear resistance is further improved, and the performance of the vulcanized rubber is improved.
- the hardness and modulus can be adjusted by using a plasticizer.
- the plasticizer is not particularly limited, but for example, the same oil as the above-mentioned extension oil can be used, and in addition, various natural oils, synthetic oils, low molecular weight polymers and the like can be used. Moreover, a known processing aid can be used.
- the rubber composition of the present invention may be a rubber composition further subjected to a cross-linking treatment by adding a cross-linking agent, a compounding agent and the like.
- the cross-linking agent is not particularly limited, and for example, a sulfur-based vulcanizing agent, an organic peroxide, or the like is used.
- the sulfur-based sulfide is not particularly limited, but for example, sulfur, morpholini disulfide and the like are used, and examples of the organic peroxide include benzoyl peroxide, dicumyl peroxide, di-t-butyl peroxide, and the like. t-Butyl cumyl peroxide, cumene hydroperoxide and the like are used.
- a vulcanization accelerator may be blended if necessary, and the vulcanization accelerator is not particularly limited, but for example, sulfenamide-based, thiazole-based, thiuram-based, thiourea-based, guadinin-based, and dithiocarbamic acid. Those containing at least one of a system, an aldehyde-amine system, an aldehyde-ammonia system, an imidazoline system, or a xantate system vulcanization accelerator can be used.
- a vulcanization aid may be blended as needed, and the vulcanization aid is not particularly limited, but for example, zinc oxide, stearic acid, or the like can be used. Furthermore, an anti-aging agent can be used.
- the modified conjugated diene-based copolymer composition of the present invention can be produced by mixing each of the above components.
- a modified conjugated diene-based copolymer at least one reinforcing filler selected from the group consisting of silica-based inorganic fillers, metal oxides, metal hydroxides and carbon black, and optionally a silane coupling agent.
- the method of mixing the above is not particularly limited. For example, a melt-kneading method using a general mixer such as an open roll, a Banbury mixer, a kneader, a single-screw screw extruder, a twin-screw screw extruder, or a multi-screw screw extruder. After melting and mixing each component, a solvent is added. Examples thereof include a method of removing by heating.
- melt-kneading method using a roll, a Banbury mixer, a kneader, or an extruder is preferable from the viewpoint of productivity and good kneading. Further, either a method of kneading the rubber component and various compounding agents at once or a method of mixing them in a plurality of times can be applied.
- the degree of polymer concentration on the surface of the filler can be expressed by the amount of bound rubber of the modified conjugated diene polymer at 25 ° C.
- the amount of bound rubber in the rubber composition after completion of the above-mentioned kneading is preferably 15% by mass or more, more preferably 20% by mass or more, from the viewpoint of improving wear resistance and breaking strength.
- the rubber composition may be a vulcanized composition that has been vulcanized with a vulcanizing agent.
- a radical generator such as an organic peroxide and an azo compound, an oxime compound, a nitroso compound, a polyamine compound, sulfur, and a sulfur compound can be used.
- Sulfur compounds include sulfur monochloride, sulfur dichloride, disulfide compounds, high molecular weight polysulfur compounds and the like.
- the amount of the vulcanizing agent used is not particularly limited, but is preferably 0.01 to 20 parts by mass and more preferably 0.1 to 15 parts by mass with respect to 100 parts by mass of the conjugated diene-based copolymer.
- the vulcanization method a conventionally known method can be applied, and the vulcanization temperature is preferably, for example, 120 ° C. to 200 ° C., more preferably 140 ° C. to 180 ° C.
- a vulcanization accelerator may be used if necessary.
- the vulcanization accelerator conventionally known materials can be used, for example, sulfenamide type, guanidine type, thiuram type, aldehyde-amine type, aldehyde-ammonia type, thiazole type, thiourea type, dithiocarbamate type.
- the vulcanization aid zinc oxide, stearic acid and the like can be used.
- the amount of the vulcanization accelerator used is not particularly limited, but is preferably 0.01 to 20 parts by mass and more preferably 0.1 to 15 parts by mass with respect to 100 parts by mass of the conjugated diene copolymer.
- the rubber composition of the present invention may contain a softening agent for rubber in order to improve workability.
- a softening agent for rubber mineral oil, liquid or synthetic softener having a low molecular weight is suitable.
- a mineral oil-based rubber softener called process oil or extender oil used to soften, increase the volume, and improve processability of rubber is a mixture of aromatic rings, naphthen rings, and paraffin chains, and is paraffin.
- a chain in which the number of carbon atoms accounts for 50% or more of the total carbon is called paraffin type, a chain having a naphthen ring carbon number of 30 to 45% is a naphthen type, and a chain having an aromatic carbon number of more than 30% is an aromatic type. It is called.
- the softening agent for rubber used in the present embodiment naphthenic and / or paraffin-based ones are preferable.
- the amount of the softening agent for rubber to be blended is not particularly limited, but is preferably 10 to 80 parts by mass, more preferably 20 to 50 parts by mass, based on 100 parts by mass of the conjugated diene copolymer.
- the rubber composition contains softeners and fillers other than those described above, as well as heat-resistant stabilizers, antistatic agents, weather-resistant stabilizers, anti-aging agents, colorants, and lubricants, as long as the object of the present embodiment is not impaired.
- Various additives such as and the like may be used.
- Specific examples of the filler include calcium carbonate, magnesium carbonate, aluminum sulfate, barium sulfate and the like.
- the softening agent to be blended as necessary in order to adjust the hardness and fluidity of the target product include liquid paraffin, castor oil, linseed oil and the like.
- Known materials can be applied as the heat-resistant stabilizer, antistatic agent, weather-resistant stabilizer, anti-aging agent, colorant, and lubricant.
- the crosslinked rubber of the present invention can be obtained by crosslinking a rubber composition containing the crosslinking agent of the present invention.
- the tire member of the present invention is obtained from a crosslinked rubber product.
- a tire is manufactured by extruding the above rubber composition according to the shape of the tire (specifically, the shape of the tread), forming the tire, and heating and pressurizing the tread in a vulcanizer.
- the desired tire can be manufactured.
- the rubber composition of the present invention is excellent in mechanical strength and wear resistance when it is a crosslinked rubber product. Therefore, as described above, it can be suitably applied to treads of tires such as fuel-efficient tires, large tires, and high-performance tires, and tire members such as sidewall members. Further, in addition to the tire member, it can be suitably used for a rubber belt, a rubber hose, a material for footwear and the like.
- GPC Molecular weight and molecular weight distribution
- HLC-8220GPC tetrahydrofuran
- Mooney viscosity (ML (1 + 4) 100 ° C.) It was determined according to JIS K6300-1 with an L-shaped rotor, preheating 1 minute, rotor operating time 4 minutes, and temperature 100 ° C.
- Haze A sample in which 0.5 g of a copolymer is dissolved in 100 g of toluene is placed in a quartz cell, and the Haze (turbidity) of the sample is an integrating sphere type light transmittance measuring device (manufactured by Nippon Denshoku Co., Ltd., SZ) using toluene as a reference sample. The Haze value was measured using ⁇ 90).
- Tg Glass transition temperature
- a solution of a polyfunctional vinyl aromatic copolymer dissolved in toluene is uniformly applied to a glass substrate so that the thickness after drying becomes 20 ⁇ m, and the mixture is heated for 30 minutes at 90 minutes using a hot plate and dried. I let you.
- the resin film obtained together with the glass substrate is set in a TMA (thermomechanical analyzer), heated to 220 ° C. at a heating rate of 10 ° C./min under a nitrogen stream, and further heat-treated at 220 ° C. for 20 minutes. The remaining solvent was removed and the polyfunctional vinyl aromatic copolymer was cured.
- TMA thermomechanical analyzer
- the analytical probe After allowing the glass substrate to cool to room temperature, the analytical probe is brought into contact with the sample in the TMA measuring device, and scan measurement is performed from 30 ° C to 360 ° C at a temperature rise rate of 10 ° C / min under a nitrogen stream, and the tangential method is used. The softening temperature was determined.
- Solvent-soluble copolymer was soluble in toluene, xylene, THF, dichloroethane, dichloromethane, and chloroform, and 100 g or more of the copolymer was dissolved in 100 g of each of the above solvents, and gel formation was not observed. The case was solvent-soluble ⁇ .
- Bound rubber amount (%) 100 parts by mass of the modified conjugated diene-based polymer and 60 parts by mass of wet silica (BET method specific surface area: 205 ⁇ 10 m 2 / g, manufactured by Tosou Silica Co., Ltd., trade name "Nipcil AQ”) are kneaded.
- ⁇ (volume of the rubber composition being kneaded / volume in the kneading chamber) ⁇ 100 ⁇ is kneaded at 60% and the maximum kneading temperature reaches 160 ° C.
- the rubber kneaded product is discharged to obtain a rubber composition M for measuring the amount of bound rubber.
- this rubber composition M is cut into 1 mm squares, the mass is measured, the mixture is added to 25 mL of toluene, left at 25 ° C. for 48 hours, and then filtered through an ADVANTEC glass fiber filter to insoluble toluene. After separating the above, the separated toluene insoluble matter is vacuum dried at 25 ° C. and then weighed, and the amount of bound rubber is determined by the following formula.
- Bound rubber amount (%) ⁇ (mass of toluene insoluble matter-mass of wet silica in rubber composition M) / (mass of rubber composition M-mass of wet silica in rubber composition M) ⁇ ⁇ 100
- the specific surface area of silica by the BET method is measured in accordance with ISO 5794/1.
- Example 10 Abrasion resistance Using a method using a Lambourn type abrasion tester conforming to JIS K6264, the amount of abrasion with a slip ratio of 30% was measured, and the measured value of the crosslinked rubber obtained in Example 10 was set to 100. It was indexed. The larger the index value, the better the wear resistance.
- DVB-630 mixture of divinylbenzene component (DVB) and ethylvinylbenzene component (EVB), DVB and EVB (mixture of m-form and p-form); DVB content 63.0 wt%, Nittetsu Chemical & Materials Made) BHT; 2,6-di-tert-butyl-p-cresol BTESPA; bis (3-trimethoxysilylpropyl) methylamine
- Synthesis example 1 DVB-630 31.00 g (DVB 0.150 mol, EVB 0.088 mol), styrene 65.72 g (0.631 mol), diisobutylene 70.80 g (0.631 mol), n-propyl acetate 60 mmol ( 6.90 mL) and 48.55 g (0.527 mol) of toluene were placed in a 500 mL reactor, and at 70 ° C., 15.2 mmol of boron trifluoride diethyl ether complex (1.91 mL) was added. The reaction was carried out for 2.0 hours.
- the Mn of the copolymer A-1 was 941, the Mw was 2850, and the Mw / Mn was 3.03.
- the structural unit derived from DVB is 19.0 mol% (21.8 wt%)
- the structural unit derived from EVB is 8.3 mol% (9.6 wt%)
- the structural unit derived from styrene is 48.6 mol%. It contained (44.6 wt%) and 24.2 mol% (24.0 wt%) of structural units derived from diisobutylene.
- the crosslinked structural unit (a1) was 4.6 mol% (5.2 wt%), and the degree of crosslinking (a1 / a) was 0.24.
- the structural unit (a2) having a residual vinyl group is 14.4 mol% (16.5 wt%), which is the structural unit (a2) with respect to the sum of the structural units (a), (b), and (c). ) was 0.144.
- the Tg of the cured product was 167 ° C, and the softening temperature was 280 ° C or higher.
- the weight loss at 350 ° C. was 1.41 wt%.
- a sample in which 0.5 g of a polyfunctional vinyl aromatic copolymer (A-1) is dissolved in 100 g of toluene is placed in a quartz cell, and the Haze (turbidity) thereof is measured by integrating sphere light transmittance using toluene as a reference sample.
- the Haze value when measured using the rate measuring device was 0.02.
- Example 1 50.0 g of the above-mentioned copolymer A-1 and 50.0 g of ethylcyclohexane were placed in a 300 mL three-necked flask, and nitrogen was degassed while stirring at 60 ° C. for 3 hours. 0.032 g of t-butylperoxy2-ethylhexanoate and 30.6 g of (3-mercaptopropyl) trimethoxysilane (MPTMS) were added and reacted at 90 ° C. for 8 hours to carry out a modified vinyl aromatic copolymer. 80.6 g of a coalescence (copolymer A-2) was obtained in terms of solid content.
- MPTMS (3-mercaptopropyl) trimethoxysilane
- Example 2 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Synthesis example 2 DVB-630 46.49 g (DVB 0.225 mol, EVB 0.132 mol), styrene 53.33 g (0.512 mol), diisobutylene 70.80 g (0.631 mol), n-propyl acetate 60 mmol ( 6.90 mL) and 48.55 g (0.527 mol) of toluene were placed in a 500 mL reactor, and 10.0 mmol of boron trifluoride diethyl ether complex (1.26 mL) was added at 70 ° C. The reaction was carried out for 2.0 hours.
- the Mn of the copolymer B-1 was 1240, the Mw was 4980, and the Mw / Mn was 4.02.
- the structural unit derived from DVB is 28.2 mol% (31.5 wt%)
- the structural unit derived from EVB is 12.2 mol% (13.8 wt%)
- the structural unit derived from styrene is 37.6 mol%. It contained (33.6 wt%) and 22.0 mol% (21.1 wt%) of structural units derived from diisobutylene.
- the crosslinked structural unit (a1) was 7.6 mol% (8.5 wt%), and the degree of crosslinking was 0.27.
- the structural unit (a2) was 20.6 mol% (23.0 wt%), and the molar fraction of the structural unit (a2) was 0.206.
- the Tg of the cured product was 176 ° C.
- the softening temperature was 280 ° C. or higher
- the weight loss at 350 ° C. was 1.32 wt%.
- the Haze value was 0.03.
- Example 3 50.0 g of the copolymer B-1 and 50.0 g of ethylcyclohexane obtained in Synthesis Example 2 were placed in a 300 mL three-necked flask, and nitrogen degassed while stirring at 60 ° C. for 3 hours. 0.036 g of t-butylperoxy2-ethylhexanoate and 34.5 g of (3-mercaptopropyl) trimethoxysilane were added and reacted at 90 ° C. for 8 hours to obtain a modified vinyl aromatic copolymer (co-polymer). 84.5 g of the polymer B-2) was obtained in terms of solid content.
- Example 4 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Synthesis example 3 DVB-630 62.00 g (DVB 0.300 mol, EVB 0.176 mol), styrene 40.92 g (0.393 mol), diisobutylene 70.80 g (0.631 mol), n-propyl acetate 60 mmol ( 6.90 mL) and 48.55 g (0.527 mol) of toluene were placed in a 500 mL reactor, and at 70 ° C., 8.2 mmol of boron trifluoride diethyl ether complex (1.03 mL) was added. The reaction was carried out for 1.5 hours.
- the obtained copolymer C-1 had a Mn of 1430, a Mw of 5490, and a Mw / Mn of 3.84.
- the structural unit derived from DVB is 39.4 mol% (42.6 wt%)
- the structural unit derived from EVB is 15.9 mol% (17.4 wt%)
- the structural unit derived from styrene is 25.7 mol%. It contained (22.2 wt%) and 19.1 mol% (17.8 wt%) of structural units derived from diisobutylene.
- the crosslinked structural unit (a1) was 11.4 mol% (12.3 wt%), and the degree of crosslinking was 0.29.
- the structural unit (a2) was 28.0 mol% (30.2 wt%), and its molar fraction was 0.280.
- the Tg of the cured product was 183 ° C, and the softening temperature was 280 ° C or higher.
- the weight loss at 350 ° C. was 1.28 wt%.
- the Haze value was 0.05.
- Example 5 50.0 g of the copolymer C-1 and 50.0 g of ethylcyclohexane obtained in Synthesis Example 3 were placed in a 300 mL three-necked flask, and nitrogen degassed while stirring at 60 ° C. for 3 hours. 0.044 g of t-butylperoxy2-ethylhexanoate and 42.3 g of (3-mercaptopropyl) trimethoxysilane (MPTMC) were added and reacted at 90 ° C. for 8 hours to carry out a modified vinyl aromatic copolymer. A coalescence (copolymer C-2) was obtained in an amount of 92.3 g in terms of solid content.
- MPTMC (3-mercaptopropyl) trimethoxysilane
- Example 6 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Comparative Example 1 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Comparative Example 2 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Example 7 The copolymer A-3 (copolymer rubber) obtained in Example 2, process oil, carbon black, zinc oxide, stearic acid and an antiaging agent were blended, and using a laboplast mill, 4 at 155 ° C. and 60 rpm. Kneaded for minutes. Sulfur and a vulcanization accelerator were added to the kneaded product obtained by the above kneading, and the mixture was kneaded at 70 ° C. and 60 rpm for 1 minute using a laboplast mill, and vulcanized to obtain crosslinked rubber A. The mixing ratio (part by weight) is shown in Table 3. Table 4 shows the physical characteristics of the crosslinked rubber A.
- Process oil Idemitsu Kosan Dyna process oil
- AC-12 Silica ULTRASIL VN3 manufactured by Degusa Carbon Black: Niteron # 300 made by Shin Nikka Carbon Zinc oxide: Mitsui Mining & Smelting Co., Ltd.
- Zinc oxide No. 1 stearic acid NOF Corporation
- Anti-aging agent Ouchi Shinko Kagaku Kogyo Noxeller NS Sulfur: Tsurumi Kagaku Kogyo Co., Ltd.
- Powdered sulfur vulcanization accelerator N-tert-butylbenzothiazole-2-sulfenamide
- the rubber crosslinked product using the copolymer of the present invention imparts the same or better dispersibility of silica and adhesion at the interface as compared with the case of using divinylbenzene which is a known branching agent. From this, it can be seen that the vulcanized rubber is excellent in tensile strength and abrasion resistance.
- Example 11 5 g of a cyclohexane solution containing 270 ml (210.3 g) of cyclohexane and 1.05 g (9.0 mmol) of N, N, N', N'-tetramethylethylenediamine was charged, and n-butyllithium was purely contained at 50 ° C.
- DVB-630 4.73 g (DVB 22.9 mmol, EVB 13.4 mmol), styrene 27.96 g (269 mmol), after adding 50 g of a cyclohexane solution containing 2.88 g (45.0 mmol). was added to initiate polymerization.
- the Mn of the copolymer A-11 was 4520, the Mw was 8870, and the Mw / Mn was 1.96.
- the structural unit derived from DVB is 6.55 mol% (6.88 wt%)
- the structural unit derived from EVB is 3.79 mol% (4.04 wt%)
- the structural unit derived from styrene is 76.8 mol%. It contained (64.5 wt%) and 12.9 mol% (24.5 wt%) of structural units derived from 3-glycidoxypropyltrimethoxysilane.
- the crosslinked structural unit (a1) was 6.55 mol% (6.88 wt%), and the degree of crosslinking (a1 / a) was 1.00.
- the structural unit (a2) having a residual vinyl group was 0.0 mol%.
- the weight loss at 350 ° C. was 1.25 wt%, and the Haze value was 0.02.
- Example 12 5 g of a cyclohexane solution containing 270 ml (210.3 g) of cyclohexane and 1.05 g (9.0 mmol) of N, N, N', N'-tetramethylethylenediamine was charged, and n-butyllithium was purely contained at 50 ° C.
- the Mn of the copolymer B-11 was 2190, the Mw was 3620, and the Mw / Mn was 1.65.
- the structural unit derived from DVB is 6.56 mol% (6.10 wt%), the structural unit derived from EVB is 3.78 mol% (3.58 wt%), and the structural unit derived from styrene is 76.8 mol%. It contained (57.3 wt%) and 12.8 mol% (33.1 wt%) of structural units derived from 3-glycidoxypropyltrimethoxysilane.
- the crosslinked structural unit (a1) was 6.55 mol% (6.09 wt%), and the degree of crosslinking was 1.00.
- the structural unit (a2) was 0.0 mol%.
- the weight loss at 350 ° C. was 1.39 wt%, and the Haze value was 0.03.
- the analysis results of the copolymers of Examples 11 and 12 are shown in Table 5.
- Example 13 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Example 14 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- the obtained copolymer C-11 had a Mn of 941, a Mw of 2850, and a Mw / Mn of 3.03.
- the structural unit derived from DVB is 19.0 mol% (21.8 wt%)
- the structural unit derived from EVB is 8.3 mol% (9.6 wt%)
- the structural unit derived from styrene is 48.6 mol%. It contained (44.6 wt%) and 24.2 mol% (24.0 wt%) of structural units derived from diisobutylene.
- the crosslinked structural unit (a1) was 4.6 mol% (5.2 wt%), and the degree of crosslinking was 0.24.
- the structural unit (a2) was 14.4 mol% (16.5 wt%), and its mole fraction was 0.144.
- the Tg of the cured product was 167 ° C, and the softening temperature was 280 ° C or higher.
- the weight loss at 350 ° C. was 1.41 wt%.
- the Haze value was 0.02.
- Comparative Example 11 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 81 ° C.
- Comparative Example 12 A modified conjugated diene-based copolymer (copolymer D-12) was obtained in the same manner as in Comparative Example 2. The analysis results of the obtained copolymer are shown in Table 6.
- Example 15 The copolymer A-12 (copolymer rubber) obtained in Example 13, process oil, carbon black, zinc oxide, stearic acid and an antiaging agent were blended, and using a laboplast mill, 4 at 155 ° C. and 60 rpm. Kneaded for minutes. Sulfur and a vulcanization accelerator were added to the kneaded product obtained by the above kneading, and the mixture was kneaded at 70 ° C. and 60 rpm for 1 minute using a laboplast mill, and vulcanized to obtain crosslinked rubber A.
- the blending ratio and the additives used are the same as in Example 7 and Table 3.
- Crosslinked rubbers B to D were obtained in the same manner as in 15.
- Table 7 shows the types of modified conjugated diene-based copolymers used and the physical properties of the obtained crosslinked rubbers B to D.
- Example 17 The reactor was charged with 240 ml (186.96 g) of ethylcyclohexane and 2.31 g (12.0 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and at 50 ° C., n-butyllithium as a pure content 3 After adding 37.5 g of an n-hexane solution containing .84 g (60.0 mmol), 6.37 g (DVB 30.0 mmol, EVB 18.9 mmol) of DVB-630 with impurities removed in advance, 38.65 g of styrene (371). .1 mmol) was added to initiate polymerization.
- the copolymer F-2 contains 6.24 mol% (6.30 wt%) of divinylbenzene-derived structural units, 3.93 mol% (3.98 wt%) of ethylvinylbenzene-derived structural units, and styrene. It contained 77.17 mol% (62.38 wt%) of structural units derived from it and 12.66 mol% (27.34 wt%) of structural units derived from GPTES.
- the crosslinked structural unit (a1) was 6.05 mol% (6.11 wt%), and the degree of crosslinking (a1 / a) was 0.97.
- the structural unit (a2) derived from divinylbenzene having a residual vinyl group is 0.19 mol% (0.19 wt%), it is a structural unit with respect to the sum of the structural units (a) and (b).
- the mole fraction of (a2) was 0.0021.
- Example 18 240 ml (186.96 g) of ethylcyclohexane and 2.31 g (12.0 mmol) of 2,2-di (2-tetrahydrofuryl) propane were charged, and at 50 ° C., 3.84 g (pure content) of n-butyllithium was charged. After adding 37.5 g of an n-hexane solution containing (60.0 mmol), DVB-630 8.92 g (DVB 42.0 mmol, EV 26.5 mmol) and styrene 42.86 g (411.5 mmol) from which impurities have been removed in advance. , Was added to initiate polymerization.
- the copolymer G-2 contains 7.77 mol% (7.96 wt%) of DVB-derived structural units, 4.90 mol% (5.02 wt%) of EVB-derived structural units, and a styrene-derived structure. It contained 76.09 mol% (62.37 wt%) of units and 11.25 mol% (24.66 wt%) of structural units derived from GPTES.
- the crosslinked structural unit (a1) was 7.45 mol% (7.64 wt%), and the degree of crosslinking (a1 / a) was 0.96.
- the structural unit (a2) is 0.31 mol% (0.32 wt%), and the molar fraction of the structural unit (a2) with respect to the sum of the structural unit (a) and (b) is 0. It was 0035.
- Example 19 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 76 ° C.
- Example 20 A nitrogen-substituted autoclave reactor was charged with 200 g of cyclohexane and 5 g of a cyclohexane solution containing 6.2 mg (0.032 mmol) of 2,2-di (2-tetrahydrofuryl) propane, and n-butyllithium at 50 ° C. After adding 5 g of a cyclohexane solution containing 10 mg (0.16 mmol) as a pure content, 10 g of styrene and 40 g of 1,3-butadiene from which impurities had been removed in advance were added to initiate polymerization. The temperature of the reaction solution rose due to the heat of polymerization, and the maximum temperature reached 77 ° C.
- Examples 21-22 Crosslinked rubbers F to G were obtained in the same manner as in Example 15 except that the copolymer F-3 or G-3 was used.
- Table 10 shows the types of the modified conjugated diene-based copolymer used and the physical properties of the obtained crosslinked rubbers FG.
- the modified vinyl aromatic copolymer of the present invention can be used as a raw material for a conjugated diene-based copolymer.
- the crosslinked rubber composition in which the conjugated diene-based copolymer contains a filler and is crosslinked is excellent in dispersibility of the filler, mechanical strength, and abrasion resistance. Therefore, tires (particularly tire treads), It is useful as an elastomer material such as seismic isolation rubber, rubber hose, rubber roller, and footwear material.
- the polymer or a composition containing the same is useful as a molding material, a resin modifier, a dielectric material, an insulating material, a heat resistant material, a structural material, an optical material and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
また、特許文献2では、共役ジエン及びモノビニル芳香族モノマーのランダムコポリマーブロックと、ポリ共役ジエンブロックと、多官能性リチウム系開始剤由来の官能基とを有する、星形-ブロックインターポリマーが開示され、転がり抵抗の低減やトラクション特性の改善といった優れた特性を有するタイヤトレッドの作製におけるゴムとして、広く使用することができることが開示されている。
特許文献1及び2の技術は、ゴム成分に分岐構造を導入することで、ゴムの加工性を担保する効果があると考えられる。しかし、強度を担保するためのフィラーとの相互作用については、特段の工夫はなく、強度に対する寄与は十分ではない。
また、特許文献4では、共役ジエン単位、芳香族ビニル単位及び少なくとも2個の重合性不飽和基を有する単位を含む架橋ゴム粒子、並びに特定の結合構造を有する共役ジエン単位を含む共役ジエン/芳香族ビニル共重合ゴムを含有するゴム組成物が開示され、この架橋ゴム粒子はカルボン酸基、ヒドロキシル基及び/又はエポキシ基を有する単量体単位を含んでも良いと開示されている。この技術は、シリカ等の無機充填剤(フィラー)との適度な相互作用を有することから、無機充填剤の分散性や加工性に優れる。しかし、少なくとも2個の重合性不飽和基を有する単量体単位や、カルボン酸基、ヒドロキシル基及び/又はエポキシ基を有する単量体単位として開示されている物質は、いずれも低分子である。そのため、反応性が過剰に高く、架橋ゴム粒子及びゴム組成物においてゲル化が進行する恐れがあった。また、この技術は共役ジエン/芳香族ビニル共重合ゴムとは別途架橋ゴムを合成した上で、その架橋ゴムを共役ジエン/芳香族ビニル共重合ゴムと配合することが必須であり、工程の簡易性の観点で、改善が必要である。
しかし、この多官能ビニル芳香族共重合体化合物においては、ゲル化については大幅に改善されるものの、依然として、ミクロゲルが少量副生するといった問題があった。
また、特許文献8で開示した多官能ビニル芳香族共重合体は、分岐構造が著しく発達した高分子量体を多く含む分子量分布の広い共重合体であるために、共重合体ゴムの構成単位として使用した場合、ミクロゲルが少量副生するといった問題があった。

(ここで、R1は炭素数6~30の芳香族炭化水素基を示す。)
末端の1個以上がアミノ基、アルコキシシリル基及び水酸基からなる群より選ばれる少なくとも1種の官能基を有する変性基により変性されていることを特徴とする変性ビニル芳香族系共重合体である。
共重合体の側鎖又は末端に、下記式(c1)又は式(c2)

(ここで、R2は炭素数1~6の2価のアルキレン基であり、R3、R4、R5、R7、R8及びR9はそれぞれ独立に、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、ベンジルオキシ基、メチル基、エチル基、プロピル基、ブチル基、シクロヘキシル基、フェニル基、又はベンジル基を示す。但し、R3、R4及びR5の少なくとも1つ、又はR7、R8及びR9の少なくとも1つはメトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、又はベンジルオキシ基である。)
で表されるシラン系官能基(c)を1種類以上含有し、
構造単位(a)に対する架橋構造単位(a1)のモル分率が、0.05~0.90の範囲であり、
シラン系官能基(c)を、一分子当たり1~20個有し、
数平均分子量Mnが500~100,000であり、重量平均分子量Mwと数平均分子量Mnの比で表される分子量分布(Mw/Mn)が30.0以下であることを特徴とする変性ビニル芳香族系共重合体である。
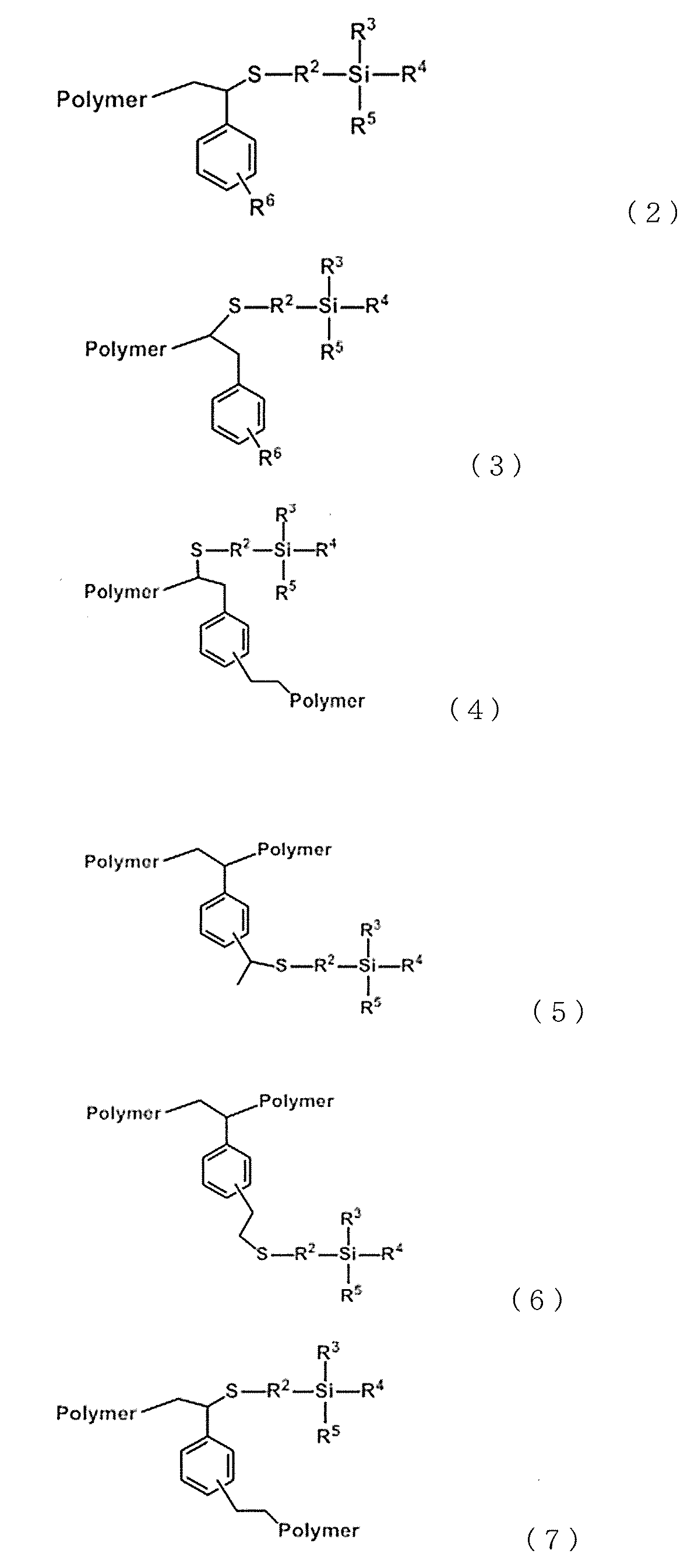

(式中、R2は炭素数1~6の2価のアルキレン基であり、R3、R4及びR5は上記式(c1)と同意であり、R6は炭素数1~6のアルキル基、ビニル基、又は水素である。Polymerは、共重合体鎖である。R7、R8及びR9は上記式(c2)と同意である。但し、式中のベンゼン環は、任意の芳香族環であることができる。)
多官能ビニル芳香族共重合体が、ジビニル芳香族化合物に由来する構造単位(a)を0.5モル%以上、95.0モル%以下含有し、モノビニル芳香族化合物に由来する構造単位(b)を5.0モル%以上、99.5モル%以下含有し、
構造単位(a)の少なくとも一部は上記架橋構造単位(a1)と、下記ビニル基含有構造単位(a2)であり、

(式中、R1は炭素数6~30の芳香族炭化水素基を示す。)
構造単位(a)に対する架橋構造単位(a1)のモル分率が、0.05~0.90の範囲であり、
構造単位(a)及び(b)の総和に対する上記ビニル基含有構造単位(a2)のモル分率が、0.001~0.80の範囲であり、
Mnが300~50,000であり、Mw/Mnが20.0以下である多官能ビニル芳香族共重合体であり、
シラン系化合物が、下記式(15)及び/又は式(16)で表されるシラン系化合物であり、

(ここで、R2、R3、R4及びR5は上記式(c1)と同意であり、R6、R7及びR8は上記式(c2)のR7、R8及びR9と同意である。)
上記多官能ビニル芳香族共重合体と、シラン系化合物を、炭化水素系溶剤に溶解させ、0~150℃の温度で、触媒の存在下で付加させることを特徴とする変性ビニル芳香族系共重合体の製造方法である。
末端の2個以上がアミノ基、アルコキシシリル基及び水酸基からなる群より選ばれる少なくとも1種の官能基を有する変性基により変性されていることを特徴とする上記の変性ビニル芳香族系共重合体である。
また、本発明の変性ビニル芳香族系共重合体を含有する硬化性樹脂組成物をコーティングしたフィルム及びシートは、プラスチック光学部品、タッチパネル、フラットディスプレイ、フィルム液晶素子などで好適に使用される他、フィルム、シート及びプリプレグの主材として使用される熱可塑性樹脂又は硬化性樹脂組成物の耐熱性、誘電特性及び光学特性等の特性を改質する改質剤として使用することもできる。また、上記硬化性樹脂組成物を、フィルム、シート及びプリプレグに加工して使用することもできる他、光導波路や光学レンズを始めとする各種光学素子として使用することもできる。
この変性ビニル芳香族系共重合体は、ジビニル芳香族化合物に由来する構造単位(a)を0.5~95.0モル%含有し、モノビニル芳香族化合物に由来する構造単位(b)を5.0~99.5モル%以下含有する。そして、構造単位(a)の少なくとも一部として、上記架橋構造単位(a1)を含む。
更に、この共重合体の側鎖もしくは末端に、上記式(c1)又は(c2)で表されるアルコキシシリル基を有する。ここで、R3R4R5中の少なくとも1つ又はR7R8R9中の少なくとも1つは、アルコキシ基である。ここで、アルコキシ基は、RO-で表される基であり、Rはアルキル基、アラルキル基又はアリール基であるが、好ましくはC1~12のアルキル基、ベンジル基、又はフェニル基である。
このアルコキシシリル基は、シラン系官能基であるので、シラン系官能基(c)又は構造単位(c)ともいう。
変性ビニル芳香族系共重合体の数平均分子量Mnが500~100,000であり、Mw/Mnが30.0以下である。
式(15)、式(16)式中、R2は炭素数1~6の2価のアルキレン基であり、R3、R4、R5、R6、R7及びR8はそれぞれ独立に、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、ベンジルオキシ基、メチル基、エチル基、プロピル基、ブチル基、シクロヘキシル基、フェニル基、又はベンジル基を示す。但し、R3、R4及びR5の少なくとも1つはメトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、又はベンジルオキシ基であることがよい。
上記式(15)で表されるシラン系化合物のメルカプト基(-SH)が、多官能ビニル芳香族系共重合体に含まれる炭素-炭素不飽和結合にラジカル付加反応することにより、シラン系化合物に由来する構造単位(c)を有する上記式(2)~(7)で表される変性ビニル芳香族系共重合体が得られる。
上記シラン系化合物のヒドロシリル基(-SiH)が、第8族遷移金属を含む触媒、炭素数が3個以上の有機基を2個以上有するキノン化合物、及びエステル基、エーテル基、チオエーテル基、アミド基、ウレア基、ウレタン基からなる群から選択される少なくとも1種の官能基を構造に有するフェノール系化合物の存在下で、炭素-炭素不飽和結合を有する未変性のビニル芳香族系共重合体に、付加(ヒドロシリル化)することなどにより、具体的にはシラン系化合物に由来する構造単位(c)を有する上記式(8)~(13)で表される変性ビニル芳香族系共重合体が得られる。
(一分子当たりの平均官能基数)=[(数平均重合度)*(1繰返し単位の平均分子量)]/(官能基の当量)
なお、官能基の当量は、官能基1個当たりに結合しているジビニル芳香族化合物単位及びモノビニル芳香族化合物の質量を意味する。官能基の当量は、 1H-NMR又は 13C-NMRを用いて官能基由来のピークと重合体主鎖に由来するピークの面積比から算出することができる。なお、官能基由来のピークとは、アルコキシ基由来のピークを指す。
この使用量が200質量部より多い場合には、補強性充填剤の分散性効果に乏しく、加工性が悪化し、得られる架橋物の耐摩耗性も低下する傾向にある。1質量部より低い場合には、補強性充填剤の分散性効果に乏しく、補強性充填剤などの分散状態が得られる架橋物の物性向上のためには理想的にならない傾向にある。
なお、変性ビニル芳香族系共重合体中に付加されたシラン系化合物の量は、例えば核磁気共鳴分光法等の各種分析機器を用いて求めることができる。
この時に用いる好ましい老化防止剤としては、例えば、2,6-ジt-ブチル-4-メチルフェノール(BHT)、2,2’-メチレンビス(4-メチル-6-t-ブチルフェノール)、4,4’-チオビス(3-メチル-6-t-ブチルフェノール)、4,4’-ブチリデンビス(3-メチル-6-t-ブチルフェノール)(AO-40)、3,9-ビス[1,1-ジメチル-2-[3-(3-t-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオニルオキシ]エチル]-2,4,8,10-テトラオキサスピロ[5.5]ウンデカン(AO-80)、2,4-ビス[(オクチルチオ)メチル]-6-メチルフェノール(Irganox1520L)、2,4-ビス[(ドデシルチオ)メチル]-6-メチルフェノール(Irganox1726)、2-[1-(2-ヒドロキシ-3,5-ジt-ペンチルフェニル)エチル]-4,6-ジt-ペンチルフェニルアクリレート(SumilizerGS)、2-t-ブチル-6-(3-t-ブチル-2-ヒドロキシ-5-メチルベンジル)-4-メチルフェニルアクリレート(SumilizerGM)、6-t-ブチル-4-[3-(2,4,8,10-テトラ-t-ブチルジベンゾ[d,f][1,3,2]ジオキサホスフェピン-6-イルオキシ)プロピル]-2-メチルフェノール(SumilizerGP)、亜リン酸トリス(2,4-ジt-ブチルフェニル)(Irgafos168)、ジオクタデシル3,3’-ジチオビスプロピオネート、ヒドロキノン、p-メトキシフェノール、N-フェニル-N’-(1,3-ジメチルブチル)-p-フェニレンジアミン(ノクラック6C)、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート(LA-77Y)、N,N-ジオクタデシルヒドロキシルアミン(IrgastabFS042)、ビス(4-t-オクチルフェニル)アミン(Irganox5057)などが挙げられる。上記老化防止剤は、1種単独で用いてもよく、2種以上を併用してもよい。
老化防止剤の添加量は、未変性の多官能ビニル芳香族共重合体100質量部に対して0~10質量部が好ましく、0~5質量部がより好ましい。
式(16)で表されるシラン系化合物は、炭素-炭素不飽和結合を有する未変性のビニル芳香族系共重合体に、付加(ヒドロシリル化)され、未変性のビニル芳香族系共重合体に反応性ケイ素基を導入する役割を担う。
多官能ビニル芳香族共重合体は、水素化ケイ素基と付加反応(ヒドロシリル化反応)が可能な不飽和基を、分子中に少なくとも1個有している多官能ビニル芳香族共重合体である。なお、前記不飽和基を、ヒドロシリル化活性のある不飽和基と呼ぶ場合もある。この多官能ビニル芳香族共重合体1分子中に存在するヒドロシリル化活性のある不飽和基の数は、1~20個であり、1~15個が好ましく、1~10個がさらにより好ましく、1~9個が特に好ましい。
また、ジカルボニルジクロロ白金や、Ashbyの米国特許第3159601および3159662号明細書中に記載の白金-炭化水素複合体、Lamoreauxの米国特許第3220972号明細書中に記載の白金-アルコラート触媒も好ましい。
さらに、Modicの米国特許第3516946号明細書中に記載の塩化白金-オレフィン複合体も好ましい。
前記の第8族遷移金属を含む触媒のなかでも、塩化白金酸、白金-オレフィン錯体、白金-アセチルアセトナート錯体、白金-ビニルシロキサン錯体は、比較的反応活性が高いためより好ましい。
前記の触媒は反応系中に1種類のみを添加してもよく、複数種を組み合わせて添加しても良い。
触媒の添加方法としては、触媒を安定化すること、取扱が容易なこと、などにより、触媒を種々の溶媒に溶解して添加する方法が好ましい。触媒を溶媒に溶解して添加する場合に用いられる溶媒としては、特に限定されず、例えばベンゼン、トルエン、キシレンなどの炭化水素系溶媒;ハロゲン化炭化水素;アルコール;グリコール類;エーテル類;エステル類などがあげられる。
キノン化合物としては、特に限定されず、2,5-ジ-tert-ブチル-1,4-ベンゾキノン、2,6-ジ-tert-ブチル-1,4-ベンゾキノン、1,4-ベンゾキノン、2-メチル-1,4-ベンゾキノン、2,6-ジメチル-1,4-ベンゾキノン、2,3,5,6,-テトラメチル-1,4-ベンゾキノン、2-tert-ブチル-1,4-ベンゾキノン、1,4-ナフトキノン、2-メチル-1,4-ナフトキノン(ビタミンK3)などが挙げられ、この中では、2,5-ジ-tert-ブチル-1,4-ベンゾキノン、2,6-ジ-tert-ブチル-1,4-ベンゾキノンが反応性、着色抑制、毒性の面から好ましい。
キノン化合物を溶媒に希釈して添加する場合に用いられる溶媒としては、特に限定されず、例えばベンゼン、トルエン、キシレン、ヘキサン等の炭化水素系溶媒、あるいはハロゲン化炭化水素、アルコール、グリコール類、エーテル類、エステル類等が挙げられる。
なお、Jean Fisherらが(Chem.Eur.J.,4,2008-2017,(1998))開示しているように、触媒と等モルのキノン化合物(D)を添加した場合、触媒に対するキノン化合物の添加量が少なく、本発明の効果が十分に達成されない場合がある。
また、キノン化合物の添加量としては、炭素-炭素不飽和結合を有する未変性のビニル芳香族系共重合体の添加量に対する比率も重要となり、モル比(キノン化合物のモル数/不飽和基のモル数)としては、0.00001~100倍が好ましく、0.0001~10倍がより好ましく、0.001~1倍が特に好ましい。
フェノール系化合物は触媒と併用して用いられ、触媒の助触媒としての働きを担うと考えられる。フェノール系化合物を添加することにより、ヒドロシリル化反応を少量の酸素の添加条件下で、触媒とキノン化合物単独の従来の系に比較し、良好な反応速度で進行させることができる。さらには、副反応生成物の発生が抑制され、また、触媒やキノン化合物の添加量を減少させることが可能となる。
フェノール系化合物は、単独で用いてもよいが、2種もしくは3種の混合物であってもよい。
フェノール系化合物を溶媒に希釈して添加する場合に用いられる溶媒としては、特に限定されず、例えばベンゼン、トルエン、キシレン、ヘキサン等の炭化水素系溶媒、あるいはハロゲン化炭化水素、アルコール、グリコール類、エーテル類、エステル類等が挙げられる。
反応容器中の気相部に含まれる酸素の体積分率としては、0.1%以上が好ましく、0.5~10%がより好ましい。酸素の体積分率が0.1%未満の場合、ヒドロシリル化反応の促進効果が発現しない場合がある。ヒドロシリル化反応の反応温度は、30℃以上200℃以下が好ましく、50℃以上150℃以下がより好ましい。反応時間は特に限定されないが、30分以上15時間以下が好ましく、30分以上10時間以下がより好ましく、さらには30分以上6時間以下が最も好ましい。
構造単位が、構造単位(a)、及び(b)だけからなる場合は、構造単位(a)のモル分率は、構造単位(a)、及び(b)の総和に対して0.005~0.95となる。このモル分率は、下記式(7)で計算される。
(a)/[(a)+(b)] (7)
(ここで、(a):ジビニル芳香族化合物に由来する構造単位(a)のモル分率、及び(b):モノビニル芳香族化合物に由来する構造単位(b)のモル分率である。)
上記モル分率は、好ましい下限は0.006、より好ましくは0.007である。また、好ましい上限は、0.80、より好ましくは0.70である。最適には、0.01~0.60である。
なお、構造単位(a)、及び(b)以外の構造単位を含む場合においては、好ましい含有率の下限は0.2モル%、より好ましくは0.4モル%であり、更に好ましくは0.6モル%である。また、好ましい上限は、70モル%、より好ましくは60モル%であり、更に好ましくは50モル%である。
(b)/[(a)+(b)] (8)
(ここで、(a)、及び(b)は、式(7)と同義である。)
なお、構造単位(a)、及び(b)以外の構造単位を含む場合においても、構造単位(b)の好ましいモル分率は、上記の範囲である。
さらに、構造単位(a)のモル分率が0.005に満たないと硬化物の耐熱性が不足し、0.95を超えると成形加工性が低下する。また、構造単位(b)のモル分率が0.995を超えると耐熱性が低下し、0.05に満たないと成形加工性が低下する。
上記未変性共重合体の製造方法では、ジビニル芳香族化合物、及びモノビニル芳香族化合物を含む重合原料を誘電率2.0~15.0の溶媒に溶解させた均一溶媒中、ルイス酸触媒の存在下で、0~120℃の温度で重合させる。
ジビニル芳香族化合物;0.5~95モル%、好ましくは0.3~65モル%、より好ましくは0.4~55モル%、最適には5~50モル%。
モノビニル芳香族化合物;7~99モル%、好ましくは10~95モル%、より好ましくは15~90モル%。
上記ルイス塩基化合物の具体例としては、次の化合物が挙げられる。
1)酢酸エチル、酢酸ブチル、酢酸フェニル、プロピオン酸メチル等のエステル系化合物、
2)メチルメルカプトプロピオン酸、エチルメルカプトプロピオン酸等のチオエステル系化合物、
3)メチルエチルケトン、メチルイソブチルケトン、ベンゾフェノン等のケトン系化合物、
4)メチルアミン、エチルアミン、プロピルアミン、ブチルアミン、シクロヘキシルアミン、メチルエチルアミン、ジメチルアミン、ジエチルアミン、ジプロピルアミン、ジブチルアミン等のアミン系化合物、
5)ジエチルエーテル、テトラヒドロラン等のエーテル系化合物、
6)ジエチルスルフィド、ジフェニルスルフィド等のチオエーテル系化合物、及び
7)トリプロピルホスフィン、トリブチルホスフィン、トリヘキシルホスフィン、トリシクロヘキシルホスフィン、トリオクチルホスフィン、ビニルホスフィン、プロペニルホスフィン、シクロヘキセニルホスフィン、ジアルケニルホスフィン、トリアルケニルホスフィンなどのホスフィン系化合物。
これらの中でも、ルイス酸触媒と相乗的に作用して、重合速度及び重合体の分子量分布を容易に制御できる点からエステル系化合物及びケトン系化合物が好ましく使用される。
これらのルイス塩基化合物は、1種又は2種以上を使用することができる。
未変性共重合体から合成される本発明の共重合体は、反応性のシラン系官能基を有するので、単独で成形、硬化させてもよいが、他の重合性樹脂を変性及び高分子量の多分岐成分合成に利用することがよい。特に、本発明の変性ビニル芳香族共重合体は、共役ジエン化合物単独、及び/又は共役ジエン化合物と他の単量体と共重合させた共役ジエン系共重合体(ゴム)を得る際に、変性及び高分子量の多分岐成分合成のために使用される。
上記本発明の変性ビニル芳香族系共重合体(以下、変性共重合体又は共重合体A'ともいう。)は、ジビニル芳香族化合物に由来する構造単位(a)と、モノビニル芳香族化合物に由来する構造単位(b)を含有する共重合体であって、構造単位(a)の95モル%以上が上記式(1)で表される架橋構造単位(a1)となっている。そして、この末端の2個以上が、上記官能基を有する変性基により変性されている。
ジビニル芳香族化合物の例としては、ジビニルベンゼン(各異性体含む)、ジビニルナフタレン(各異性体を含む)、ジビニルビフェニル(各異性体を含む)が好ましく使用されるが、これらに限定されるものではない。また、これらは単独又は2種以上を組み合わせて用いることができる。成形加工性の観点から、より好ましくはジビニルベンゼン(m-体、p-体又はこれらの異性体混合物)である。
本発明の製造方法は、アルカリ金属化合物又はアルカリ土類金属化合物をアニオン重合開始剤として用い、ジビニル芳香族化合物及びモノビニル芳香族化合物、又はジビニル芳香族化合物及びモノビニル芳香族化合物とこれらとアニオン共重合可能な単量体を共重合して、分岐構造と活性末端を有するビニル芳香族系共重合体を得る重合工程と、このビニル芳香族系共重合体の活性末端に、上記官能基を形成する末端変成工程を含む。
ここで、アニオン共重合可能な単量体は、共重合体の靱性、相溶性を改善する。アニオン共重合可能な単量体としては、ブタジエン、イソプレン;2,3-ジメチルブタジエン、2-フェニルブタジエン、1,3-ペンタジエン、2-メチル-1,3-ペンタジエン、1,3-ヘキサジエン、1,3-オクタジエン、1,3-シクロヘキサジエン、2-メチル-1,3-オクタジエン、1,3,7-オクタトリエン、ミルセン、及びクロロプレン等のブタジエン及びイソプレン以外の共役ジエン(b1)が挙げられる。本発明の変性ビニル芳香族系共重合体に含まれるアニオン共重合可能な単量体としては、工業的実施の容易さから、ブタジエン及びイソプレンが好ましい。
架橋構造単位(a1)の上記架橋度は、0.95以上の範囲であるが、好ましい下限は、0.98以上であり、より好ましくは、0.99以上である。
重合工程で使用されるアルカリ金属化合物又はアルカリ土類金属化合物からなる重合開始剤について、説明する。
重合開始剤として用いるアルカリ金属化合物は、特に制限されないが、例えば、有機リチウム化合物が好ましい。有機リチウム化合物としては、低分子量有機リチウム化合物、可溶化したオリゴマーの有機リチウム化合物のうちいずれでもよい。有機基とリチウムの結合様式において炭素-リチウム結合を有する化合物、窒素-リチウム結合を有する化合物、錫-リチウム結合を有する化合物等が挙げられる。有機リチウム化合物を用いると、開始効率がよく、重合体のリビング率もよい。有機リチウム化合物としては、特に制限されないが、例えば、有機モノリチウム化合物、有機ジリチウム化合物、有機ポリリチウム化合物が挙げられる。有機基としては官能基を含む炭化水素が好適であり、その場合は有機溶剤への溶解性が優れる利点があり、さらに開始速度も優れる。また、窒素-リチウム結合を有する化合物、錫-リチウム結合を有する化合物を用いることにより、開始末端に官能基を含む変性基を付与することもできる。
有機リチウム化合物の他の有機アルカリ金属化合物としては、特に限定されないが、例えば、有機ナトリウム化合物、有機カリウム化合物、有機ルビジウム化合物、有機セシウム化合物等が挙げられる。より具体的には、ナトリウムナフタレン、カリウムナフタレン等が挙げられる。その他にも、リチウム、ナトリウム及びカリウム等のアルコキサイド、スルフォネート、カーボネート、アミド等が挙げられる。また、他の有機金属化合物と併用してもよい。
が好ましい。
例えば、ジプロピルアミノリチウム、ジイソプロピルアミノリチウム、ジブチルアミノリチウム、テトラメチレンイミノリチウム、ペンタメチレンイミノリチウム、ヘキサメチレンイミノリチウム、ヘプタメチレンイミノリチウム、2-ジメチルアミノエチルリチウム、3-ジメチルアミノプロピルリチウム、3-ジエチルアミノプロピルリチウム、4-ジメチルアミノブチルリチウム(以上は2置換アミノ基)、2-トリメチルシリルエチルアミノエチルリチウム、3-トリメチルシリルメチルアミノプロピルリチウム(以上は1置換アミノ基)、2-ビストリメチルシリルアミノエチルリチウム、3-ビストリメチルシリルアミノプロピルリチウム(以上はアミノ基)がある。
極性化合物としては、例えば、テトラヒドロフラン、ジエチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジブチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールジブチルエーテル、ジメトキシベンゼン、2,2-ビス(2-オキソラニル)プロパン等のエーテル類;テトラメチルエチレンジアミン、ジピペリジノエタン、トリメチルアミン、トリエチルアミン、ピリジン、キヌクリジン等の第3級アミン化合物;カリウム-tert-アミラート、カリウム-tert-ブチラート、ナトリウム-tert-ブチラート、ナトリウムアミラート等のアルカリ金属アルコキシド化合物;トリフェニルホスフィン等のホスフィン化合物等が挙げられる。これらの極性化合物は、単独で用いてもよく、2種以上組み合わせて用いてもよい。
極性化合物の使用量は、特に限定されず、目的等に応じて選択することができる。通常、重合開始剤又は多官能開始剤1モルに対して0.01~100モルであることが好ましい。このような極性化合物は共重合体の開始反応と成長反応の調節剤として、所望の分子量、分子量分布に応じて、適量用いることができる。多くの極性化合物は、同時にジビニル芳香族化合物と、モノビニル芳香族化合物との共重合において有効なランダム化効果を有し、芳香族ビニル化合物の分布の調整やスチレンブロック量の調整剤として用いることができる。
上記単量体及び重合溶媒は、それぞれ単独であるいはこれらの混合液を、有機金属化合物を用いて処理しておくことが好ましい。これにより、ジビニル芳香族化合物と、モノビニル芳香族化合物等の単量体や、重合溶媒に含まれているアレン類やアセチレン類を処理できる。その結果、高濃度の活性末端を有する重合体が得られるようになり、高い変性率を達成できるようになる。
本発明の製造方法では、重合工程で分岐構造と活性末端を有するビニル芳香族系共重合体(未変性共重合体ともいう。)を得た後、この活性末端に、アミノ基、アルコキシシリル基、水酸基からなる群より選ばれる少なくとも1種の官能基を有する化合物、又はアミノ基、アルコキシシリル基、水酸基からなる群より選ばれる少なくとも1種の官能基を形成する化合物を反応させて、前記官能基を導入する末端変性工程に付す。
上記官能基を有する化合物の添加量は、特に限定されないが、重合開始剤又は多官能開始剤のモル数、すなわち重合体分子に対して、官能基を有する化合物の合計モル数が、0.01~6倍となる範囲であることが好ましく、0.02~3倍となる範囲であることがより好ましく、0.05~2倍となる範囲であることがさらに好ましい。添加量が0.05倍以上であれば、目的とする変性ビニル芳香族共重合体において十分な変性率を得る観点から好ましい。
このような場合、上記官能基を有する化合物の添加量は、重合開始剤又は多官能開始剤のモル数すなわち重合体分子数に対して、官能基当量として、0.7倍以下が好ましい。
上記結合反応後に水酸基が生成する官能基を有する化合物としては、ケトン基、エステル基、アミド基、エポキシ基等を有する化合物が挙げられる。また、結合反応後に加水分解等の反応によって水酸基が生成する官能基を有する化合物としては、アルコキシシリル基、アミノシリル基等を有する化合物が挙げられる。
共重合体の末端にアミノ基及び水酸基を形成する化合物としては、特に限定されないが、N,N,N’,N’-テトラメチル-4,4’-ジアミノベンゾフェノン(ミヒラーズケトン)、N,N,N’,N’-テトラエチル-4,4’-ジアミノベンゾフェノン等のアミノ基を有するケトン化合物;N,N’-ジメチルイミダゾリジノン、N-メチルピロリドン等の環状尿素化合物;環状アミド、すなわちラクタム化合物;N,N,N’,N’-テトラグリシジル-1,3-ビスアミノメチルシクロヘキサン等のアミノ基含有エポキシ化合物;JP2001-131227Aに記載の含窒素複素環式基を有するエポキシ化合物等が例示される。
共重合体の末端にアルコキシシリル基を形成する化合物としては、特に限定されないが、トリメトキクロロシシラン、トリエトキシクロロシラン、ジフェノキシジクリロロシラン等のハロゲン化アルコキシシラン化合物;ビス(トリメトキシシリル)エタン、ビス(3-トリエトキシシリルプロピル)エタン等の多官能アルコキシシラン化合物等が例示される。
共重合体の末端にアルコキシシリル基及び水酸基を形成する化合物としては、特に限定されないが、3-グリシドキシプロピルトリメトキシシラン、3-グリシドキシプロピルトリエトキシシランエポキシ基及びアルコキシシリル基を分子内に有するポリシロキサン化合物等が例示される。
共重合体の末端に水酸基を形成する化合物としては、特に限定されないが、エチレンオキサイド、プロピレンオキサイド等のエポキシ化合物;ベンゾフェノン等のケトン化合物等が例示される。
また、上記変性ゴムを介することで、本発明の変性共重合体と補強性充填剤との親和性が向上し、ゴム組成物中の補強性充填剤等各成分の分散状態が、得られる架橋物の物性向上(例えば、耐摩耗性の向上、操縦安定性の向上、ドライグリップ性能、ウェットグリップ性能)のためには理想的になると推定される。
一方、変性ゴムとしてその官能基数が大きくなり過ぎると、補強性充填剤に吸着した変性ゴム同士の相互作用が強すぎるために、補強性充填剤を分散させる効果が小さくなってしまうこととなり、本発明の変性共重合体が、変性ゴムと補強性充填剤との親和性向上に寄与する効果が低下してしまうものと推定される。
構造単位が、構造単位(a)、及び(b)だけからなる場合は、構造単位(a)のモル分率は、構造単位(a)、及び(b)の総和に対して0.005~0.95となる。このモル分率は、下記式(7)で計算される。
(a)/[(a)+(b)] (7)
(ここで、(a):ジビニル芳香族化合物に由来する構造単位(a)のモル分率、及び(b):モノビニル芳香族化合物に由来する構造単位(b)のモル分率である。)
上記モル分率は、好ましい下限は0.006、より好ましくは0.007である。また、好ましい上限は、0.80、より好ましくは0.70である。最適には、0.01~0.60である。
なお、構造単位(a)、及び(b)以外の構造単位を含む場合においては、好ましい含有率の下限は0.2モル%、より好ましくは0.4モル%であり、更に好ましくは0.6モル%である。また、好ましい上限は、70モル%、より好ましくは60モル%であり、更に好ましくは50モル%である。
(b)/[(a)+(b)] (8)
(ここで、(a)、及び(b)は、式(7)と同義である。)
なお、構造単位(a)、及び(b)以外の構造単位を含む場合においても、構造単位(b)の好ましいモル分率は、上記の範囲である。
さらに、構造単位(a)のモル分率が0.005に満たないと硬化物の耐熱性が不足し、0.95を超えると成形加工性が低下する。また、構造単位(b)のモル分率が0.995を超えると耐熱性が低下し、0.05に満たないと成形加工性が低下する。
変性共役ジエン系重合体は、活性末端を有する共役ジエン化合物(B)の重合体、又は活性末端を有する共役ジエン化合物(B)及び芳香族ビニル化合物(C)の共重合体に、本発明の変性ビニル芳香族系共重合体(A)又は(A’)を変性剤として反応させて得られる。以下、共役ジエン化合物(B)の重合体、又は活性末端を有する共役ジエン化合物(B)及び芳香族ビニル化合物(C)の共重合体を、まとめてジエン系重合体ともいう。
第一の態様の変性ビニル芳香族系共重合体(A)と、第二の態様の変性ビニル芳香族系共重合体(A’)は、同様にして使用することができるので、以下両者を変性ビニル芳香族系共重合体(A)ともいう。
重合開始剤として用いるアルカリ金属化合物は、特に制限されないが、例えば、有機リチウム化合物が好ましい。有機リチウム化合物としては、低分子量有機リチウム化合物、可溶化したオリゴマーの有機リチウム化合物のうちいずれでもよい。有機基とリチウムの結合様式において炭素-リチウム結合を有する化合物、窒素-リチウム結合を有する化合物、錫-リチウム結合を有する化合物等が挙げられる。有機リチウム化合物を用いると、開始効率がよく、重合体のリビング率もよい。有機リチウム化合物としては、特に制限されないが、例えば、有機モノリチウム化合物、有機ジリチウム化合物、有機ポリリチウム化合物が挙げられる。有機基としては官能基を含む炭化水素が好適であり、その場合は有機溶剤への溶解性が優れる利点があり、さらに開始速度も優れる。また、窒素-リチウム結合を有する化合物、錫-リチウム結合を有する化合物を用いることにより、開始末端に官能基を含む変性基を付与することもできる。
重合開始剤として用いるアルカリ土類金属化合物としては、有機マグネシウム化合物、有機カルシウム化合物、有機ストロンチウム化合物等が挙げられる。また、アルカリ土類金属のアルコキサイド、スルフォネート、カーボネート、アミド等の化合物を用いてもよい。これらの有機アルカリ土類金属化合物は、前記アルカリ金属化合物や、その他有機金属化合物と併用してもよい。
その後、活性末端を有する共役ジエン系共重合体に、2以上の末端に官能基を導入する方法として、前記共役ジエン系共重合体の2以上の活性末端に変性ビニル芳香族系共重合体を反応させて、前記変性ビニル芳香族系共重合体に基づく分岐高分子型変性基(A)を2以上導入する末端変性工程を行う。
多官能開始剤としては、特に限定されないが、具体的には、有機ジリチウム化合物、有機ポリリチウム化合物がある。有機基としては特に限定されないが、炭化水素が好適である。これにより、有機溶剤への溶解性が優れる利点があり、さらに開始速度も優れる。
また、多官能開始剤の調製方法としては、特に限定されないが、具体的には、金属リチウムのディスパージョンとポリハロゲン化炭化水素化合物の反応による方法、金属リチウムとジエン化合物の反応による方法、有機モノリチウム化合物と多官能性化合物の反応による方法が挙げられる。この中でも、有機モノリチウム化合物と多官能性化合物の反応によるものが好ましい。多官能性化合物としては、特に限定されないが、具体的には、芳香族化合物であって芳香族基に隣接する二重結合を複数個有する化合物、リチウムに置換し得る活性な水素を複数個有する化合物が挙げられる。有機モノリチウム化合物とこれらの多官能性化合物を反応させる際に、共役ジエン化合物、芳香族ビニル化合物を存在させて、オリゴマーとすることも可能である。また、有機モノリチウム化合物の反応性を高めるため、エーテル化合物、第3級アミン化合物等の極性化合物を存在させてもよい。芳香族化合物であって芳香族基に隣接する二重結合を複数有する化合物としては、特に限定されないが、例えば、ジイソプロペニルベンゼン、ジビニルベンゼン等がある。リチウムに置換し得る活性な水素を複数有する化合物としては、特に限定されないが、例えば、1,3,5-トリメチルベンゼン等がある。
また、上記炭化水素ポリリチウム化合物としては、特に限定されないが、例えば、n-ブチルリチウム、ジビニルベンゼン及び1,3-ブタジエンの反応生成物、セカンダリー又はターシャリーブチルリチウムと1,3,5-トリメチルベンゼンとの反応生成物等が挙げられる。
これらの多官能開始剤の使用量は、目的とするブタジエン系重合体の分子量により調節され、単量体100質量部に対し、0.01~0.2質量部を用いることができる。好ましくは0.02~0.15質量部である。この際に、単量体及び溶媒中の水分及び不純物による失活、不純物による連鎖移動、重合末端の金属ハイドライドの生成による失活等を考慮することが好ましい。
極性化合物としては、例えば、テトラヒドロフラン、ジエチルエーテル、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジブチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールジブチルエーテル、ジメトキシベンゼン、2,2-ビス(2-オキソラニル)プロパン等のエーテル類;テトラメチルエチレンジアミン、ジピペリジノエタン、トリメチルアミン、トリエチルアミン、ピリジン、キヌクリジン等の第3級アミン化合物;カリウム-tert-アミラート、カリウム-tert-ブチラート、ナトリウム-tert-ブチラート、ナトリウムアミラート等のアルカリ金属アルコキシド化合物;トリフェニルホスフィン等のホスフィン化合物等が挙げられる。これらの極性化合物は、単独で用いてもよく、2種以上組み合わせて用いてもよい。
極性化合物の使用量は、特に限定されず、目的等に応じて選択することができる。通常、重合開始剤又は多官能開始剤1モルに対して0.01~100モルであることが好ましい。このような極性化合物は重合体共役ジエン部分のミクロ構造の調節剤として、所望のビニル結合量に応じて、適量用いることができる。多くの極性化合物は、同時に共役ジエン化合物と芳香族ビニル化合物との共重合において有効なランダム化効果を有し、芳香族ビニル化合物の分布の調整やスチレンブロック量の調整剤として用いることができる。共役ジエン化合物と芳香族ビニル化合物とをランダム化する方法としては、例えば、特開昭59-140211号公報に記載されているような、スチレンの全量と共役ジエン化合物(B)の一部とで共重合反応を開始させ、共重合反応の途中に残りの共役ジエン化合物(B)を連続的又は断続的に添加する方法を用いてもよい。
上記した共役ジエン系化合物及び重合溶媒は、それぞれ単独であるいはこれらの混合液を、有機金属化合物を用いて処理しておくことが好ましい。これにより、共役ジエン化合物や重合溶媒に含まれているアレン類やアセチレン類を処理できる。その結果、高濃度の活性末端を有する重合体が得られるようになり、高い変性率を達成できるようになる。
共役ジエン化合物(B)又は、共役ジエン化合物(B)と芳香族ビニル化合物(C)とを共重合する際の重合様式としては、特に限定されないが、回分式(「バッチ式」ともいう。)、連続式等の重合様式で行うことができる。連続式においては、1個又は2個以上の連結された反応器を用いることができる。反応器は、撹拌機付きの槽型、管型等のものが用いられる。回分式では、得られる重合体の分子量分布が一般に狭く、Mw/Mnでは1.0以上、1.8未満となりやすい。また、連続式では一般に分子量分布が広く、Mw/Mnでは1.8以上、3以下となりやすい。
末端変性工程は、前記共役ジエン系共重合体の活性末端に、本発明の変性ビニル芳香族系共重合体を反応させて、前記官能基を導入すると共に、多分岐の高分子量体を合成する工程である。この工程は、重合開始剤を用いる場合も、前記多官能開始剤を用いる場合も同様である。
上記変性ビニル芳香族系共重合体を、重合体活性末端に反応させる際の、反応温度、反応時間等については、特に限定されないが、0~120℃で、30秒以上反応させることが好ましい。変性ビニル芳香族系共重合体の添加量は、特に限定されないが、重合開始剤又は多官能開始剤のモル数すなわち重合体分子数に対して、変性ビニル芳香族系共重合体(A)の合計モル数が、0.01~6倍となる範囲であることが好ましく、0.02~3倍となる範囲であることがより好ましく、0.05~2倍となる範囲であることがさらに好ましい。添加量が0.05倍以上であれば、目的とする変性共役ジエン系重合体において十分な変性率を得る観点から好ましい。また、加工性改良のために重合体末端同士がカップリングした分岐状重合体成分の分岐度と生成量を任意に変性ビニル芳香族系共重合体(A)の構造によってコントロールとすることもでき、その場合には、変性ビニル芳香族系共重合体(A)として官能基度の高い変性ビニル芳香族系共重合体(A)を用いることができる。このような変性ビニル芳香族系共重合体(A)の添加量は、重合開始剤又は多官能開始剤のモル数すなわち重合体分子数に対して、官能基当量として、0.7倍以下が好ましい。
また、変性ビニル芳香族系共重合体の官能基同士が結合する副反応を防止する目的や、異なった種類のフィラーとの親和性の向上を目的として、共役ジエン系共重合体の活性末端に、変性ビニル芳香族系共重合体とは異なる官能基を1種以上導入することもできる。
末端変性工程は、前記ブタジエン系共重合体の活性末端に、アミノ基、アルコキシシリル基、水酸基から選ばれる少なくとも1種の官能基を有する変性化合物を反応させて結合を形成するか、またはこれらの官能基を形成する化合物を反応させ、終了末端を変性する工程である。この工程で用いる変性化合物において、共役ジエン系重合体の活性末端と結合する官能基としては、特に限定されないが、具体的には、ハロゲン基、二重結合、ケトン基、エステル基、アミド基、エポキシ基、アルコキシシリル基等が挙げられる。
変性剤のうち、水酸基を形成する化合物としては、特に限定されないが、具体的には、重合体活性末端と結合する官能基であって、結合反応後に水酸基が生成する官能基を有する化合物、重合体活性末端と結合しない官能基であって、後に加水分解等の反応によって水酸基が生成する官能基を有する化合物が挙げられ、活性水素を有しない化合物であることが好ましい。
重合体活性末端と結合して重合体の末端にアミノ基及び水酸基を形成する化合物としては、特に限定されないが、N,N,N’,N’-テトラメチル-4,4’-ジアミノベンゾフェノン(ミヒラーズケトン)、N,N,N’,N’-テトラエチル-4,4’-ジアミノベンゾフェノン等のアミノ基を有するケトン化合物;N,N’-ジメチルイミダゾリジノン、N-メチルピロリドン等の環状尿素化合物;環状アミド、すなわちラクタム化合物;N,N,N’,N’-テトラグリシジル-1,3-ビスアミノメチルシクロヘキサン等のアミノ基含有エポキシ化合物;特開2001-131227に記載の含窒素複素環式基を有するエポキシ化合物等が例示される。
重合体活性末端と結合して重合体の末端にアルコキシシリル基を形成する化合物としては、特に限定されないが、トリメトキクロロシシラン、トリエトキシクロロシラン、ジフェノキシジクリロロシラン等のハロゲン化アルコキシシラン化合物;ビス(トリメトキシシリル)エタン、ビス(3-トリエトキシシリルプロピル)エタン等の多官能アルコキシシラン化合物等が例示される。
重合体活性末端と結合して重合体の末端にアルコキシシリル基及び水酸基を形成する化合物としては、特に限定されないが、3-グリシドキシプロピルトリメトキシシラン、3-グリシドキシプロピルトリエトキシシランエポキシ基及びアルコキシシリル基を分子内に有するポリシロキサン化合物等が例示される。
重合体活性末端と結合して重合体の末端に水酸基を形成する化合物としては、特に限定されないが、エチレンオキサイド、プロピレンオキサイド等のエポキシ化合物;ベンゾフェノン等のケトン化合物等が例示される。
主鎖変性工程は、上記の末端が変性された変性共役ジエン系重合体の主鎖のビニル基と、アミノ基、アルコキシシリル基、水酸基からなる群より選ばれる少なくとも1種の官能基を有する珪素化合物、又はアミノ基、アルコキシシリル基、又は水酸基からなる群より選ばれる少なくとも1種の官能基を形成する珪素化合物とをヒドロシリル化反応させることにより、主鎖を変性する工程である。この工程は、重合開始剤を用いる場合も、前記多官能開始剤を用いる場合も同様である。
ヒドロシリル化反応は、有機溶剤溶液中又は重合体のまま(溶剤なしの状態)で、混練機中で、行なうことができる。具体的には、前記官能基を有するヒドロシラン化合物等の珪素化合物を触媒存在下に反応させ、共役ジエン系共重合体の主鎖のビニル基の変性を行ことができる。溶液重合における重合工程後に、さらに上述のように重合体の末端部を変性した重合体溶液をそのまま用いることが好ましい。
また、ヒドロシラン化反応の後に加水分解等を行うことで、上記の官能基を形成する珪素化合物であってもよい。このような珪素化合物としては、特に限定されず、具体的には加水分解等を行うことで、アミノ基、アルコキシシリル基、又は水酸基の少なくとも1種の官能基を形成する官能基を有するヒドロシラン化合物を挙げることができる。より具体的には、保護化1置換アミノ基、保護化2置換アミノ基、保護化水酸基などを有するヒドロシラン化合物を挙げることができる。
2置換アミノ基を有する珪素化合物としては、特に限定されないが、具体的には、ジメチルアミノジメチルシラン、ジエチルアミノジメチルシラン、ジエチルアミノジエチルシラン、3-ジエチルアミノプロピルジメチルシラン、4-ジメチルアミノブチルジメチルシラン、6-ジエチルアミノヘキシルジメチルシランが挙げられる。
保護化1置換アミノ基を有する珪素化合物としては、特に限定されないが、具体的には、N-メチル-N-トリメチルシリルアミノジメチルシラン、N-エチル-N-トリメチルシリルアミノジエチルシランが挙げられる。
前記式(2)及び/又は式(3)で表されるシラン系官能基からなる構造単位(c)を有する変性ビニル芳香族系共重合体(A)に由来する構造単位(A1)は、0.001~6重量%であり、好ましくは0.001~5重量%、より好ましくは0.005~5重量%、更に好ましくは0.01~5重量%、最適には0.001~1重量%である。共役ジエン化合物(B)に由来する構造単位(B1)は、29~99.999重量%であり、好ましくは80~99.999重量%であり、より好ましくは90~99.995重量%、更に好ましくは95~99.99重量%である。
上述のようにして、末端変性及び主鎖変性がなされた、変性共役ジエン系重合体は、溶液として得られる場合、必要に応じて酸化防止剤、添加剤を加えた後、通常の方法で溶媒の除去、乾燥を行うことができる。これにより、後述するゴム組成物の原料とすることができる。具体的には、スチームストリッピング及び脱水乾燥による方法、ドラムドライヤー、フラッシング及びベント押出し機による直脱法などである。
これを、図1を参照して説明する。
図1は、GPC溶出曲線の典型例と、ピークトップMp及びピークトップの3倍の分子量3Mpの位置を示す。最も低分子量側のピークの数平均分子量Mpの3倍の数平均分子量3Mp領域(図1では、3Mpより右側の領域)の面積が10面積%以上であることがよい。これは比較的高分子量成分が多いことを意味する。
上記ゴム組成物は、前記変性共役ジエン系重合体とフィラーを含有する。
本発明のゴム組成物の他の態様は、前記変性共役ジエン系重合体100質量部に対し、補強性充填剤を0.5~200質量部含有する。
本発明のゴム組成物の他の態様は、前記変性共役ジエン系重合体を20質量部以上含む原料ゴム100質量部と、フィラー5~200質量部を含有する。
原料ゴム100質量部中の変性共役ジエン系重合体含有量は、20質量部以上であり、好ましくは40質量部以上、より好ましくは50質量部以上、さらに好ましくは60質量部以上である。20質量部以上であると、本発明の目的とする分散性が優れ、本実施形態のゴム組成物を加硫化組成物とした場合、引っ張り特性、粘弾性特性などの性能が優れ、タイヤ用の材料として用いた場合、優れた燃費性能、グリップ性能、耐摩耗性、剛性が得られる。また、変性共役ジエン系重合体の含有量は、好ましくは90質量部以下、より好ましくは80質量部以下である。90質量部以下とすることにより、本発明の未加硫の変性共役ジエン系共重合体組成物のムーニー粘度を下げて加工性が向上する。
本発明の他の態様としてのゴム組成物では、前記変性共役ジエン系重合体100質量部に対し、好ましくは上記原料ゴム100質量部に対して、シリカ系無機充填剤、金属酸化物、金属水酸化物及びカーボンブラックからなる群より選ばれる少なくとも1種の補強性充填剤を0.5~200質量部含む。
金属酸化物としては、例えば、アルミナ、酸化チタン、酸化マグネシウム、酸化亜鉛等を用いることができる。
ビニル系化合物としては、例えばビニルトリエトキシシラン、ビニルトリメトキシシランなどが挙げられる。
アミノ系化合物としては、例えば、3-アミノプロピルトリエトキシシラン、3-アミノプロピルトリメトキシシラン、3-(2-アミノエチル)アミノプロピルトリエトキシシラン、3-(2-アミノエチル)アミノプロピルトリメトキシシランなどが挙げられる。
ニトロ系化合物としては、例えば、3-ニトロプロピルトリメトキシシラン、3-ニトロプロピルトリエトキシシラン等が挙げられる。
その他の化合物としては、例えば、オクチルトリエトキシシラン、メチルトリエトキシシラン、メチルトリメトキシシラン、ヘキサデシルトリメトキシシランなどが挙げられる。
本発明のゴム組成物は、架橋剤、配合剤等を加えて、架橋処理をさらに施したゴム組成物としてもよい。このような架橋剤としては、特に制限されないが、例えば、硫黄系加硫剤、有機過酸化物等が用いられる。
目的とする製品の硬さや流動性を調節するために、必要に応じて配合する軟化剤としては、例えば、流動パラフィン、ヒマシ油、アマニ油等が挙げられる。耐熱安定剤、帯電防止剤、耐候安定剤、老化防止剤、着色剤、滑剤としては、公知の材料を適用できる。
分子量及び分子量分布測定は、GPC(東ソー製、HLC-8220GPC)を使用し、溶媒にテトラヒドロフラン(THF)、流量1.0ml/min、カラム温度38℃、単分散ポリスチレンによる検量線を用いて行った。
日本電子製JNM-LA600型核磁気共鳴分光装置を用い、13C-NMR及び1H-NMR分析により決定した。溶媒としてクロロホルム-d1を使用し、テトラメチルシランの共鳴線を内部標準として使用した。
JIS K6300‐1に従って、L形ローター、予熱1分、ローター作動時間4分、温度100℃ で求めた。
共重合体 0.5gをトルエン 100gに溶解させたサンプルを石英セルに入れ、そのHaze(濁り度)を、トルエンを基準サンプルとして、積分球式光線透過率測定装置(日本電色社製、SZ-Σ90)を用いHaze値を測定した。
乾燥後の厚さが20μmになるように、ガラス基板に多官能ビニル芳香族共重合体をトルエンに溶解させた溶液を均一に塗布し、ホットプレートを用いて90分で30分間加熱し、乾燥させた。ガラス基板とともに得られた樹脂膜はTMA(熱機械分析装置)にセットし、窒素気流下、昇温速度10℃/分で220℃まで昇温し、更に220℃で20分間加熱処理することにより残存する溶媒を除去するとともに多官能ビニル芳香族共重合体を硬化させた。ガラス基板を室温まで放冷した後、TMA測定装置中の試料に分析用プローブを接触させ、窒素気流下、昇温速度10℃/分で30℃から360℃までスキャン測定を行い、接線法で軟化温度を求めた。
共重合体が、トルエン、キシレン、THF、ジクロロエタン、ジクロロメタン、およびクロロホルムに可溶で、前記各溶媒100gに対し、共重合体が100g以上溶解し、ゲルの生成は認められなかった場合を、溶剤可溶性〇とした。
試料を二硫化炭素溶液とし、溶液セルを用いて、赤外線スペクトルを600~1000cm-1の範囲で測定し、所定の波数における吸光度によりハンプトン(スチレン-ブタジエン共重合体)の方法の計算式に従いビニル結合量を求めた。装置は、パーキンエルマー社製のSpectrum100を用いた。
変性共役ジエン系重合体100質量部と湿式シリカ(BET法比表面積:205±10m2/g、東ソウ・シリカ株式会社製、商品名「ニプシルAQ」)60質量部とを混練室内容積2リットルの密閉式混練装置を使用して、{(混練されているゴム組成物の体積/混練室内容積)×100}が60%にて混練りし、最高混練温度160℃に達した時点で、当該ゴム混練り物を排出して、バウンドラバー量測定用ゴム組成物Mを得る。
このゴム組成物M0.2gを1mm角に裁断して質量を測定した後、トルエン25mL中に加えて、25℃にて48時間放置した後、ADVANTEC社製ガラス繊維フィルターでろ過し、トルエン不溶分を分離した後、分離したトルエン不溶分を25℃にて真空乾燥した後秤量し、下記式によりバウンドラバー量を求める。
バウンドラバー量(%)={(トルエン不溶分の質量―ゴム組成物M中の湿式シリカの質量)/(ゴム組成物Mの質量―ゴム組成物M中の湿式シリカの質量)}×100
なお、シリカのBET法比表面積は、ISO 5794/1に準拠して測定する。
JIS K6251の引張試験法により300%モジュラスを測定し、実施例7で得られた架橋ゴムの測定値を100として、指数化した。指数値が大きいほど引張り強度に優れることを示す。
JIS K6264に準拠したランボーン型摩耗試験機を使用した方法を用い、スリップ率が30%の摩耗量を測定し、実施例10で得られた架橋ゴムの測定値を100として、指数化した。指数値が大きいほど耐摩耗性は良好である。
DVB-630;ジビニルベンゼン成分(DVB)とエチルビニルベンゼン成分(EVB)の混合物、DVBとEVBは(m-体とp-体の混合物);DVB含有率63.0wt%、日鉄ケミカル&マテリアル製)
BHT;2,6-ジ-tert-ブチル-p-クレゾール
BTESPA;ビス(3-トリメトキシシリルプロピル)メチルアミン
DVB-630 31.00g(DVB 0.150モル、EVB 0.088モル)、スチレン 65.72g(0.631モル)、ジイソブチレン 70.80g(0.631モル)、酢酸n-プロピル 60ミリモル(6.90mL)、トルエン 48.55g(0.527モル)を500mLの反応器内に投入し、70℃で、15.2ミリモルの三フッ化ホウ素ジエチルエーテル錯体(1.91mL)を添加し、2.0時間反応させた。重合溶液を炭酸水素ナトリウム水溶液で停止させた後、純水で3回油層を洗浄し、60℃で減圧脱揮し、共重合体を回収し、多官能ビニル芳香族共重合体(共重合体A-1) 60.6gを得た。
また、硬化物のTgは167℃で、軟化温度は280℃以上であった。350℃における重量減少は1.41wt%であった。
また多官能ビニル芳香族共重合体(A-1) 0.5gをトルエン100gに溶解させたサンプルを石英セルに入れ、そのHaze(濁り度)を、トルエンを基準サンプルとして、積分球式光線透過率測定装置を用い測定したときのHaze値は、0.02であった。
上記共重合体A-1 50.0g、エチルシクロヘキサン 50.0gを300mLの三口フラスコに仕込み、60℃で3時間撹拌をしながら窒素脱気をした。t-ブチルパーオキシ2-エチルヘキサノエート 0.032gと(3-メルカプトプロピル)トリメトキシシラン(MPTMS) 30.6gを添加し、90℃で8時間反応させて、変性ビニル芳香族系共重合体(共重合体A-2)を固形分換算で80.6g得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。
重合反応終了後、反応器に変性剤として実施例1で得られた共重合体A-2 0.031gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体A-3)を得た。
DVB-630 46.49g(DVB 0.225モル、EVB 0.132モル)、スチレン 53.33g(0.512モル)、ジイソブチレン 70.80g(0.631モル)、酢酸n-プロピル 60ミリモル(6.90mL)、トルエン 48.55g(0.527モル)を500mLの反応器内に投入し、70℃で、10.0ミリモルの三フッ化ホウ素ジエチルエーテル錯体(1.26mL)を添加し、2.0時間反応させた。重合溶液を炭酸水素ナトリウム水溶液で停止させた後、純水で3回油層を洗浄し、60℃で減圧脱揮し、共重合体を回収し、多官能ビニル芳香族共重合体(共重合体B-1) 66.4gを得た。
また、硬化物のTgは176℃であり、軟化温度は280℃以上であり、350℃における重量減少は1.32wt%であった。また、上記Haze値は、0.03であった。
合成例2で得た共重合体B-1 50.0g、エチルシクロヘキサン 50.0gを300mLの三口フラスコに仕込み、60℃で3時間撹拌をしながら窒素脱気をした。t-ブチルパーオキシ2-エチルヘキサノエート 0.036gと(3-メルカプトプロピル)トリメトキシシラン 34.5gを添加し、90℃で8時間反応させて、変性ビニル芳香族系共重合体(共重合体B-2)を固形分換算で84.5g得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤として実施例3で得られた共重合体B-2 0.027gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体B-3)を得た。
DVB-630 62.00g(DVB 0.300モル、EVB 0.176モル)、スチレン 40.92g(0.393モル)、ジイソブチレン 70.80g(0.631モル)、酢酸n-プロピル 60ミリモル(6.90mL)、トルエン 48.55g(0.527モル)を500mLの反応器内に投入し、70℃で、8.2ミリモルの三フッ化ホウ素ジエチルエーテル錯体(1.03mL)を添加し、1.5時間反応させた。重合溶液を炭酸水素ナトリウム水溶液で停止させた後、純水で3回油層を洗浄し、60℃で減圧脱揮し、共重合体を回収し、多官能ビニル芳香族共重合体(共重合体C-1) 56.3gを得た。
また、硬化物のTgは183℃であり、軟化温度は280℃以上であった。350℃における重量減少は1.28wt%であった。また、Haze値は、0.05であった。
合成例3で得た共重合体C-1 50.0g、エチルシクロヘキサン 50.0gを300mLの三口フラスコに仕込み、60℃で3時間撹拌をしながら窒素脱気をした。t-ブチルパーオキシ2-エチルヘキサノエート 0.044gと(3-メルカプトプロピル)トリメトキシシラン(MPTMC) 42.3gを添加し、90℃で8時間反応させて、変性ビニル芳香族系共重合体(共重合体C-2)を固形分換算で92.3g得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤として実施例5で得られた共重合体C-2 0.023gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体C-3)を得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に分岐剤として合成例3で得られた共重合体(C-1) 0.021gを含むシクロヘキサン溶液5gを添加し、分岐反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.16mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体D-3)を得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に、BTESPAを0.16mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体E-3を得た。
各共重合体の分析結果を表1及び表2に示す。

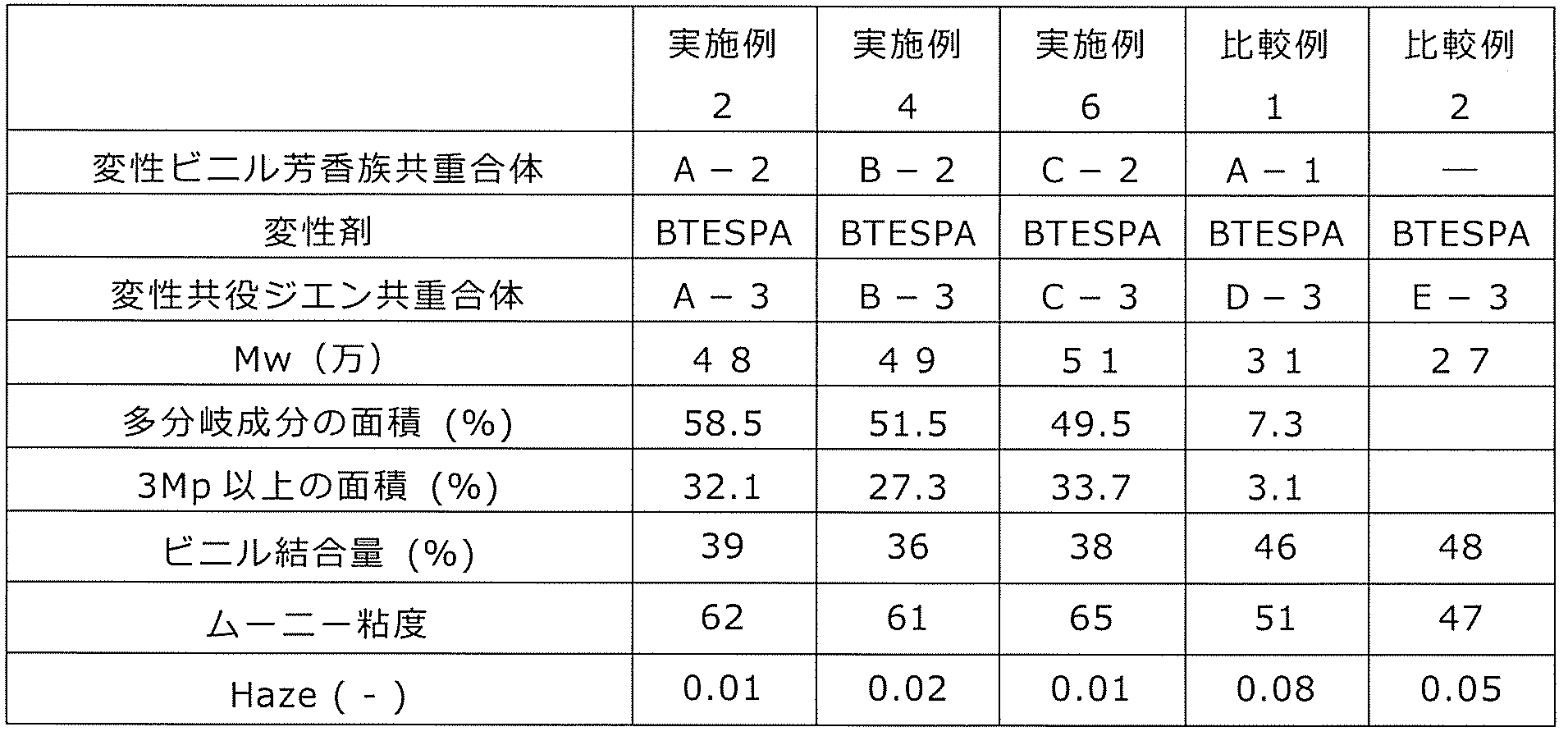
実施例2で得た共重合体A-3(共重合体ゴム)、プロセスオイル、カーボンブラック、酸化亜鉛、ステアリン酸及び老化防止剤を配合し、ラボプラストミルを用い、155℃、60rpmで4分間混練した。
上記混練で得られた混練物に、硫黄と加硫促進剤を加え、ラボプラストミルを用い、70℃、60rpmで1分間混練し、加硫して架橋ゴムAを得た。
配合割合(重量部)を表3に示す。また、架橋ゴムAの物性を表4に示す。

プロセスオイル:出光興産製 ダイナプロセスオイル AC-12
シリカ:デグサ社製、ULTRASIL VN3
カーボンブラック:新日化カーボン製 ニテロン#300
酸化亜鉛:三井金属鉱業製 亜鉛華1号
ステアリン酸:日油製
老化防止剤:大内新興化学工業製 ノクセラーNS
硫黄:鶴見化学工業製 粉末硫黄
加硫促進剤:N-tert-ブチルベンゾチアゾール-2-スルフェンアミド
変性共役ジエン系共重合体A-3の代わりに、上記実施例4及び実施例6、又は比較例1~2で合成した変性共役ジエン系共重合体B-3、C-3、D-3又はE-3を使用した以外は、実施例7と同様の手法で架橋ゴムB~Fを得た。
使用した変性共役ジエン系共重合体の種類と、得られた架橋ゴムB~Eの物性を表4に示す。
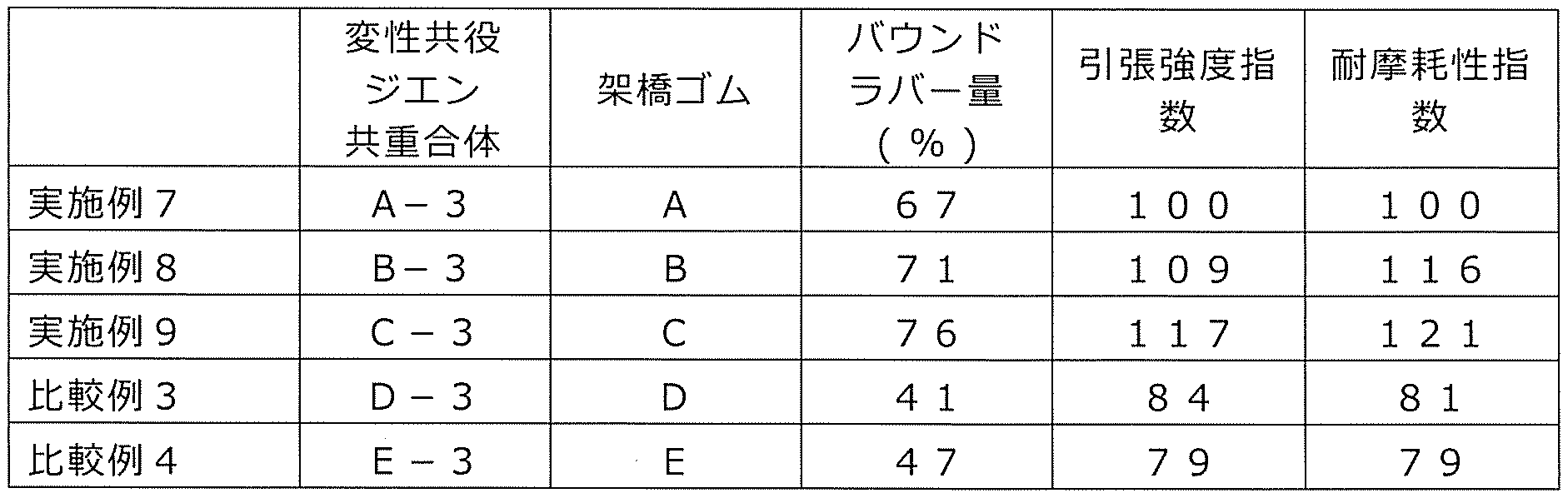
シクロヘキサン 270ml(210.3g)、N,N,N’,N’-テトラメチルエチレンジアミン 1.05g(9.0mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として2.88g(45.0mmol)を含むシクロヘキサン溶液50gを添加した後、予め不純物を除去したDVB-630 4.73g(DVB 22.9mmol、EVB 13.4mmol)、スチレン 27.96g(269mmol)、を添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤として3-グリシドキシプロピルトリメトキシシラン(GPTMS) 10.63g(45.0mmol)を添加して30分間変性反応させ、変性ビニル芳香族系共重合体含有ポリマー溶液を得た。
得られた重合溶液を脱揮することにより濃縮し、変性ビニル芳香族系共重合体(共重合体A-11)を固形分換算の収量で42.89g(収率:99.0wt%)を得た。
また、350℃における重量減少は1.25wt%で、上記Haze値は、0.02であった。
シクロヘキサン 270ml(210.3g)、N,N,N’,N’-テトラメチルエチレンジアミン 1.05g(9.0mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として2.88g(45.0mmol)を含むシクロヘキサン溶液50gを添加した後、予め不純物を除去したDVB-630 4.73g(ジビニルベンゼン(m-体とp-体の混合物)成分22.9mmol、エチルビニルベンゼン(m-体とp-体の混合物)成分13.4mmol)、スチレン 27.96g(269mmol)、を添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤としてトリエトキシリルチオプロピルトリメトキシシラン(TESTPTMS) 16.14g(45.0mmol)を添加して30分間変性反応させ、変性ビニル芳香族系共重合体(共重合体B-11)含有ポリマー溶液を得た。
得られた重合溶液を脱揮することにより濃縮し、変性ビニル芳香族系共重合体B-11を固形分換算の収量で48.34g(収率:99.0wt%)を得た。
また、350℃における重量減少は1.39wt%で、Haze値は、0.03であった。
実施例11及び12の共重合体の分析結果を表5に示す。
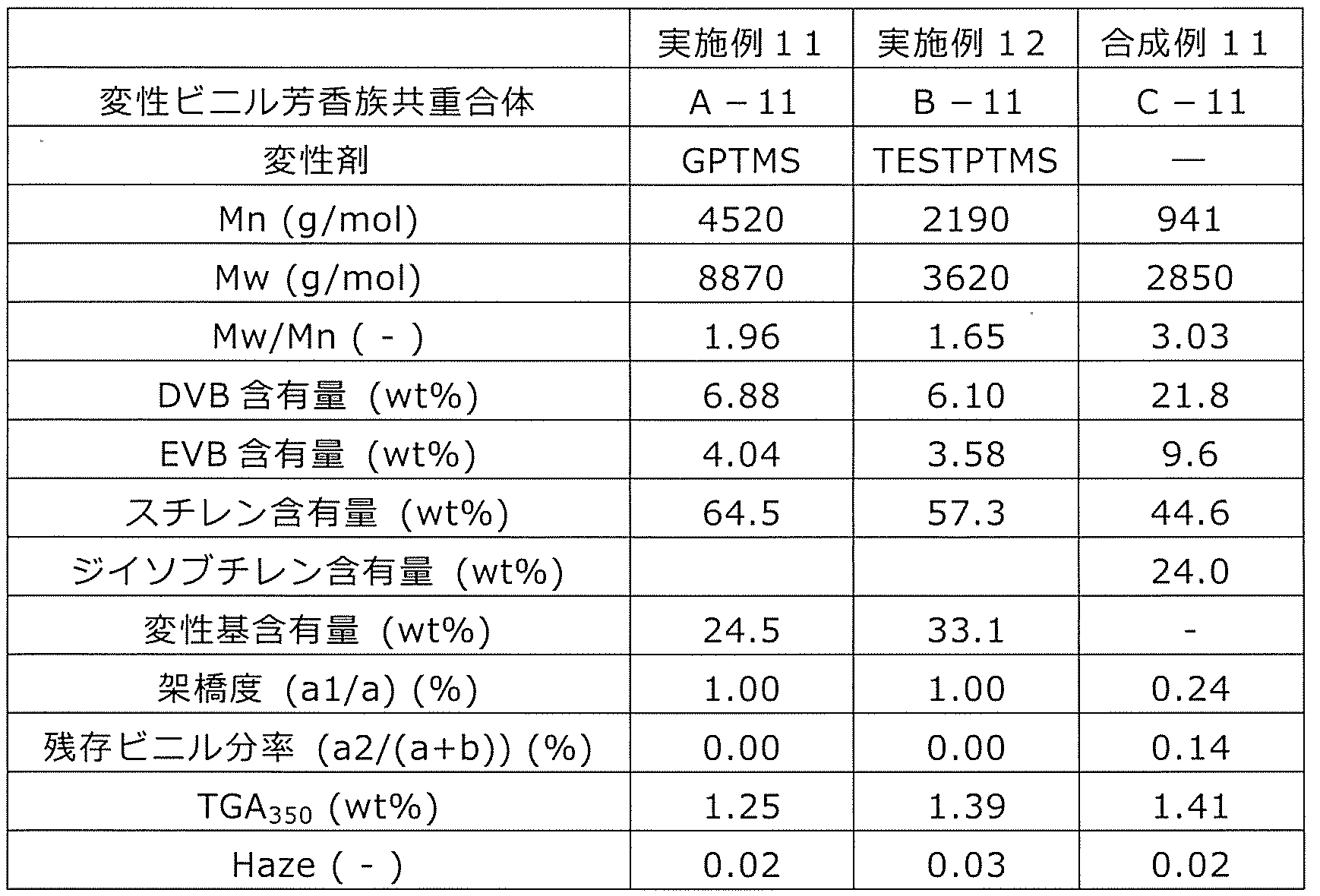
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤として実施例11で得られた変性ビニル芳香族系共重合体(A-11) 0.076gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体A-12)を得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に変性剤として実施例12で得られた共重合体B-11 0.085gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体B-12)を得た。
DVB-630 31.00g(DVB 0.150モル、EVB 0.088モル)、スチレン 65.72g(0.631モル)、ジイソブチレン 70.80g(0.631モル)、酢酸n-プロピル 60ミリモル(6.90mL)、トルエン 48.55g(0.527モル)を500mLの反応器内に投入し、70℃で、15.2ミリモルの三フッ化ホウ素ジエチルエーテル錯体(1.91mL)を添加し、2.0時間反応させた。重合溶液を炭酸水素ナトリウム水溶液で停止させた後、純水で3回油層を洗浄し、60℃で減圧脱揮し、共重合体を回収し、ビニル芳香族共重合体(共重合体C-11) 60.6gを得た。
また、硬化物のTgは167℃であり、軟化温度は280℃以上であった。350℃における重量減少は1.41wt%であった。Haze値は、0.02であった。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は81℃に達した。重合反応終了後、反応器に分岐剤として合成例11で得られた共重合体C-11 0.021gを含むシクロヘキサン溶液5gを添加し、分岐反応を実施した、50℃の温度条件で30分間の変性反応を実施して重合体溶液を得た。
さらにBTESPAを0.16mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体C-12)を得た。
比較例2と同様にして変性共役ジエン系共重合体(共重合体D-12)を得た。
得られた共重合体の分析結果を表6に示す。

実施例13で得た共重合体A-12(共重合体ゴム)、プロセスオイル、カーボンブラック、酸化亜鉛、ステアリン酸及び老化防止剤を配合し、ラボプラストミルを用い、155℃、60rpmで4分間混練した。
上記混練で得られた混練物に、硫黄と加硫促進剤を加え、ラボプラストミルを用い、70℃、60rpmで1分間混練し、加硫して架橋ゴムAを得た。
配合割合及び使用した添加剤は、実施例7及び表3と同じである。
変性共役ジエン系共重合体A-12の代わりに、上記実施例14、又は比較例11~12で得た共重合体B-12、C-12又はD-12を使用した以外は、実施例15と同様の手法で架橋ゴムB~Dを得た。
使用した変性共役ジエン系共重合体の種類と、得られた架橋ゴムB~Dの物性を表7に示す。

反応器にエチルシクロヘキサン 240ml(186.96g)、2,2-ジ(2-テトラヒドロフリル)プロパン 2.31g(12.0mmol)を装入し、50℃において、n-ブチルリチウムを純分として3.84g(60.0mmol)を含むn-ヘキサン溶液37.5gを添加した後、予め不純物を除去したDVB-630 6.37g(DVB 30.0mmol、EVB 18.9mmol)、スチレン 38.65g(371.1mmol)、を添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は67℃に達した。重合反応終了後、反応器に変性剤として3-グリシドキシプロピルトリエトキシシラン(GPTES) 16.94g(60.9mmol)を添加して60分間変性反応させ、官能基変性ビニル芳香族系共重合体含有ポリマー溶液を得た。得られた重合溶液を、コハク酸を使用して中和した後、ろ過を行った。
得られた重合溶液を脱揮することにより濃縮し、官能基変性ビニル芳香族系共重合体(共重合体F-2)を固形分換算の収量で61.34g(収率:99.0wt%)を得た。
共重合体F-2の分析結果を表8に示す。
エチルシクロヘキサン 240ml(186.96g)、2,2-ジ(2-テトラヒドロフリル)プロパン 2.31g(12.0mmol)を装入し、50℃において、n-ブチルリチウムを純分として3.84g(60.0mmol)を含むn-ヘキサン溶液37.5gを添加した後、予め不純物を除去したDVB-630 8.92g(DVB 42.0mmol、EV 26.5mmol)、スチレン 42.86g(411.5mmol)、を添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は65℃に達した。重合反応終了後、反応器に変性剤としてGPTES 16.94g(60.9mmol)を添加して60分間変性反応させ、官能基変性ビニル芳香族系共重合体含有ポリマー溶液を得た。得られた重合溶液を、コハク酸を使用して中和した後、ろ過を行った。
得られた重合溶液を脱揮することにより濃縮し、変性ビニル芳香族系共重合体(共重合体G-2)を固形分換算の収量で68.31g(収率:99.4wt%)を得た。
共重合体G-2の分析結果を表8に示す。
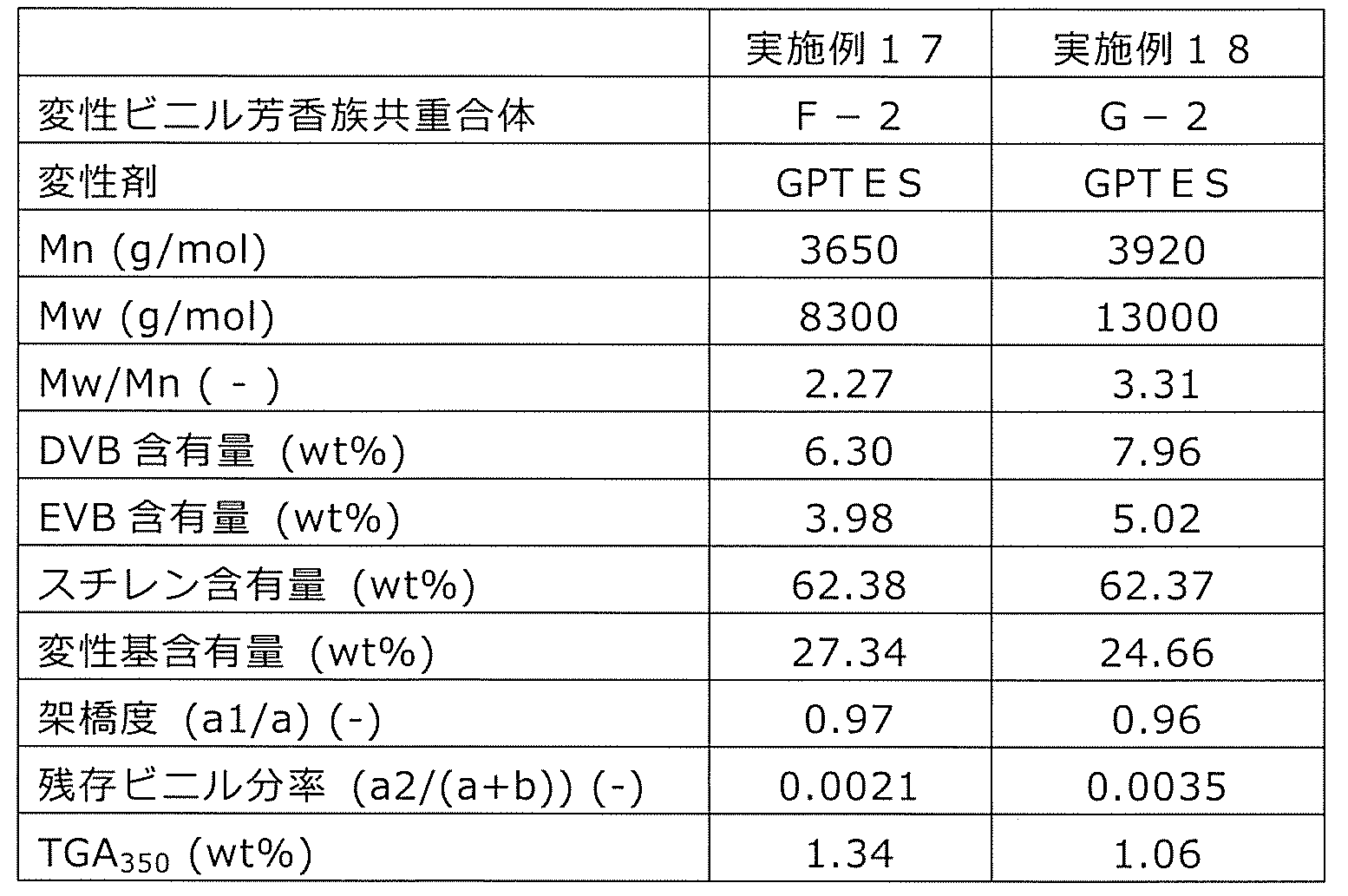
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は76℃に達した。重合反応終了後、反応器に変性剤として実施例17で得られた共重合体F-2 0.081gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で60分間の変性反応を実施して重合体溶液を得た。
さらにBTESPABTESPAを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体F-3)を得た。
窒素置換されたオートクレーブ反応器に、シクロヘキサン200g、2,2-ジ(2-テトラヒドロフリル)プロパン6.2mg(0.032mmol)を含むシクロヘキサン溶液5gを装入し、50℃において、n-ブチルリチウムを純分として10mg(0.16mmol)を含むシクロヘキサン溶液5gを添加した後、予め不純物を除去したスチレン10g、1,3-ブタジエン40gを添加して重合を開始した。重合熱により反応溶液の温度が上昇し、最高温度は77℃に達した。重合反応終了後、反応器に変性剤として実施例18で得られた共重合体G-2 0.090gを含むシクロヘキサン溶液5gを添加し、変性反応を実施した、50℃の温度条件で60分間の変性反応を実施して重合体溶液を得た。
さらにビス(3-トリエトキシシリルプロピル)メチルアミンを0.08mmol添加して30分間変性反応させ、変性共役ジエン系共重合体含有ポリマー溶液を得た。
得られた重合溶液に、酸化防止剤として2,6-ジ-tert-4-ヒドロキシトルエンを0.045g添加した後、スチームストリッピングにより溶媒を除去し、真空乾燥を経て、変性共役ジエン系共重合体(共重合体G-3)を得た。
得られた共重合体の分析結果を表9に示す。

共重合体F-3又はG-3を使用した以外は、実施例15と同様の手法で架橋ゴムF~Gを得た。
使用した変性共役ジエン系共重合体の種類と、得られた架橋ゴムF~Gの物性を表10に示す。
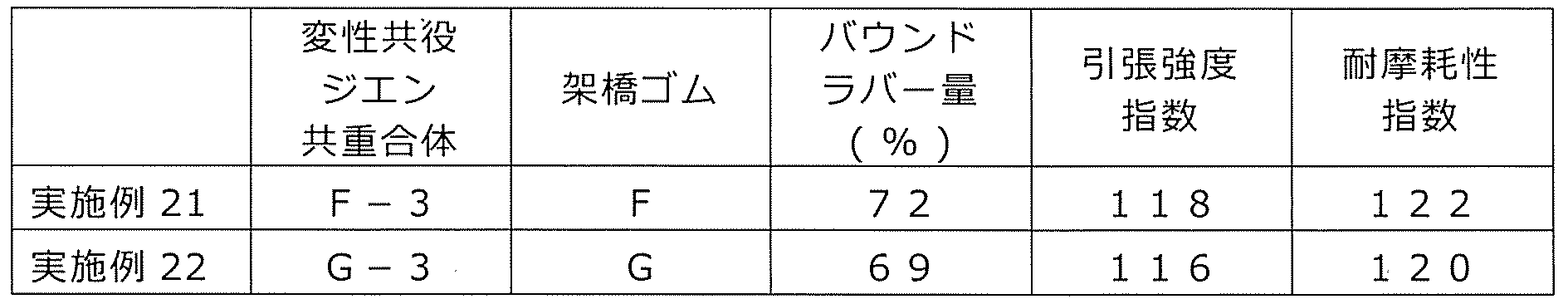
Claims (25)
- ジビニル芳香族化合物に由来する構造単位(a)と、モノビニル芳香族化合物に由来する構造単位(b)を含有する変性ビニル芳香族系共重合体であって、構造単位(a)の少なくとも一部は下記の架橋構造単位(a1)であり、

(ここで、R1は炭素数6~30の芳香族炭化水素基を示す。)
末端の1個以上がアミノ基、アルコキシシリル基及び水酸基からなる群より選ばれる少なくとも1種の官能基を有する変性基により変性されていることを特徴とする変性ビニル芳香族系共重合体。 - ジビニル芳香族化合物に由来する構造単位(a)と、モノビニル芳香族化合物に由来する構造単位(b)を含む共重合体であって、構造単位(a)と構造単位(b)の合計に対し、構造単位(a)を0.5モル%~95.0モル%含有し、構造単位(b)を5.0モル%~99.5モル%含有し、
共重合体の側鎖又は末端に、下記式(c1)又は式(c2)
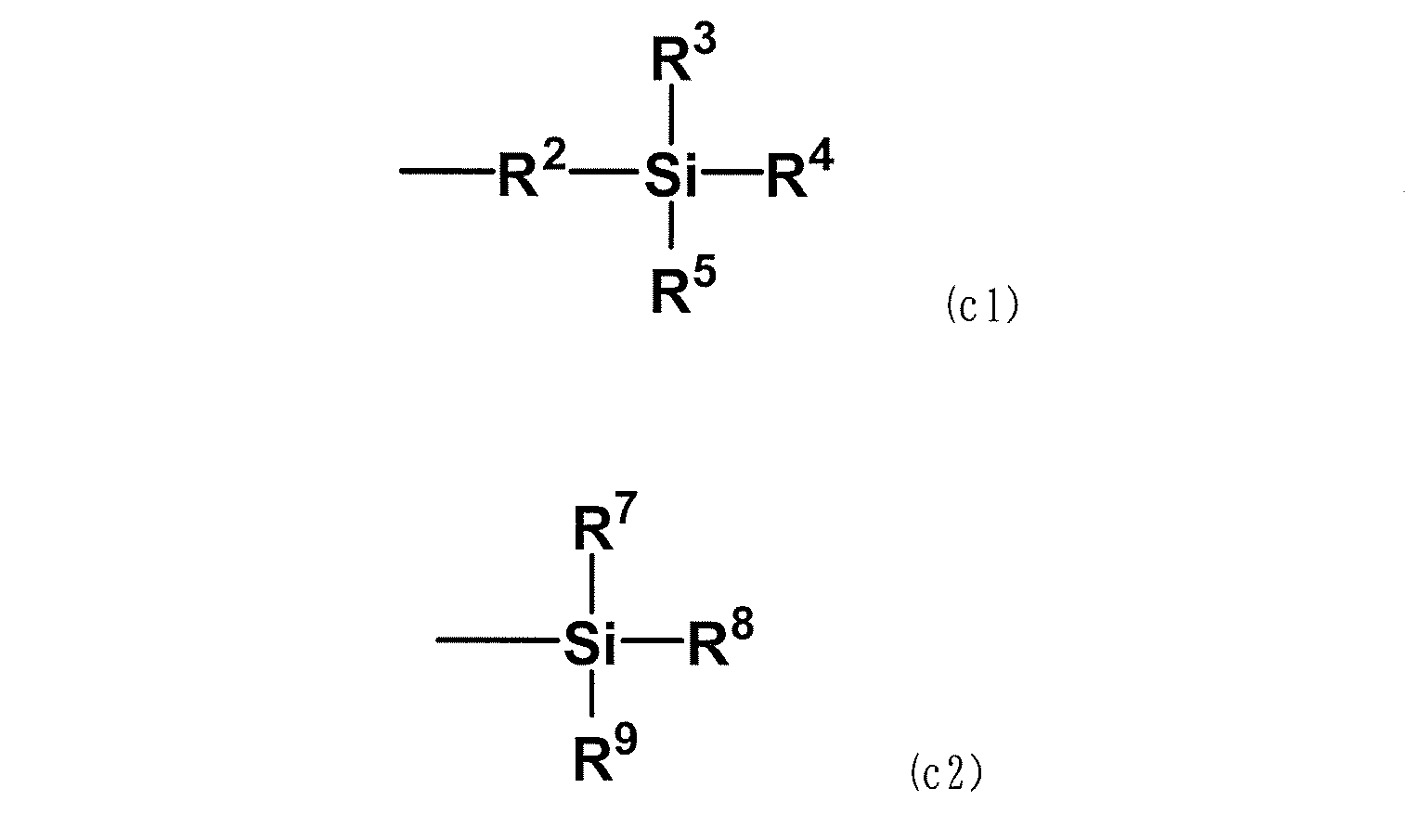
(ここで、R2は炭素数1~6の2価のアルキレン基であり、R3、R4、R5、R7、R8及びR9はそれぞれ独立に、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、ベンジルオキシ基、メチル基、エチル基、プロピル基、ブチル基、シクロヘキシル基、フェニル基、又はベンジル基を示す。但し、R3、R4及びR5の少なくとも1つ、又はR7、R8及びR9の少なくとも1つはメトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、又はベンジルオキシ基である。)
で表されるシラン系官能基(c)を1種類以上含有し、
構造単位(a)に対する架橋構造単位(a1)のモル分率が、0.05~0.90の範囲であり、
シラン系官能基(c)を、一分子当たり1~20個有し、
数平均分子量Mnが500~100,000であり、重量平均分子量Mwと数平均分子量Mnの比で表される分子量分布(Mw/Mn)が30.0以下であることを特徴とする請求項1に記載の変性ビニル芳香族系共重合体。 - 下記式(2)~(13)のいずれかで表される共重合体を1種以上含むことを特徴とする請求項2に記載の変性ビニル芳香族系共重合体。

(式中、R2は炭素数1~6の2価のアルキレン基であり、R3、R4及びR5はそれぞれ独立に、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、ベンジルオキシ基、メチル基、エチル基、プロピル基、ブチル基、シクロヘキシル基、フェニル基、又はベンジル基を示す。但し、R3、R4及びR5の少なくとも1つはメトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、又はベンジルオキシ基である。R6は炭素数1~6のアルキル基、ビニル基、又は水素である。Polymerは、共重合体鎖である。但し、式中のベンゼン環は、任意の芳香族環であることができる。)

(式中、R7、R8及びR9はそれぞれ独立に、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、ベンジルオキシ基、メチル基、エチル基、プロピル基、ブチル基、シクロヘキシル基、フェニル基、又はベンジル基を示す。但し、R7、R8及びR9の少なくとも1つはメトキシ基、エトキシ基、プロポキシ基、ブトキシ基、オクトキシ基、ラウリルオキシ基、フェノキシ基、又はベンジルオキシ基である。R6は炭素数1~6のアルキル基、ビニル基、又は水素である。また、Polymerは、共重合体鎖である。) - モノビニル芳香族化合物に由来する構造単位(b)が、スチレン、ビニルナフタレン、ビニルビフェニル、m-メチルスチレン、p-メチルスチレン、o,p-ジメチルスチレン、m-エチルビニルベンゼン、インデン及びp-エチルビニルベンゼンからなる群から選ばれる1種以上の単量体に由来する構造単位である請求項2又は3に記載の変性ビニル芳香族系共重合体。
- 多官能ビニル芳香族共重合体と、シラン系化合物とから請求項2~4のいずれか一項に記載の変性ビニル芳香族系共重合体を製造する方法であって、
多官能ビニル芳香族共重合体が、ジビニル芳香族化合物に由来する構造単位(a)を0.5モル%以上、95.0モル%以下含有し、モノビニル芳香族化合物に由来する構造単位(b)を5.0モル%以上、99.5モル%以下含有し、
構造単位(a)の少なくとも一部は上記架橋構造単位(a1)と、下記ビニル基含有構造単位(a2)であり、

(式中、R1は炭素数6~30の芳香族炭化水素基を示す。)
構造単位(a)に対する架橋構造単位(a1)のモル分率が、0.05~0.90の範囲であり、
構造単位(a)及び(b)の総和に対する上記ビニル基含有構造単位(a2)のモル分率が、0.001~0.80の範囲であり、
Mnが300~50,000であり、Mw/Mnが20.0以下である多官能ビニル芳香族共重合体であり、
シラン系化合物が、下記式(5)及び/又は式(6)で表されるシラン系化合物であり、
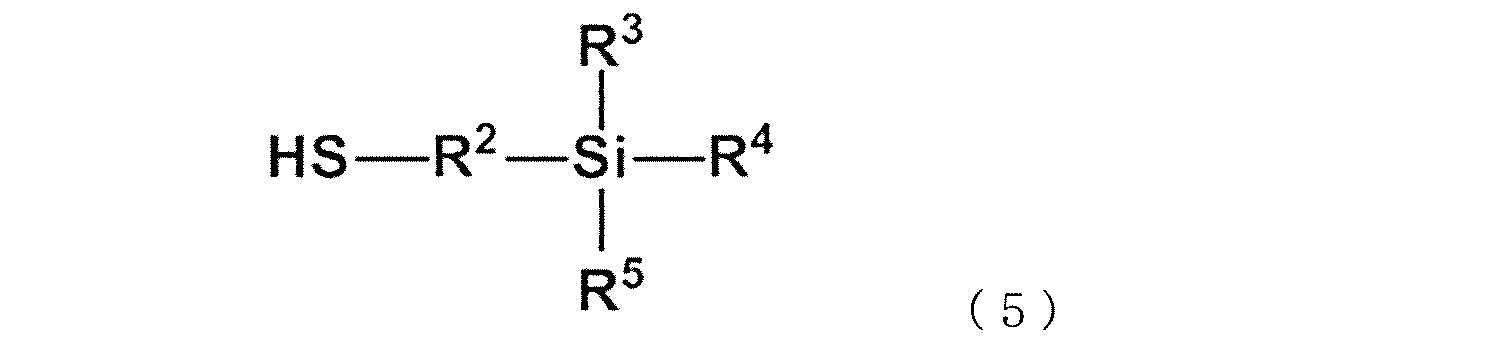
(式中、R2、R3、R4及びR5は上記式(c1)と同じである。)
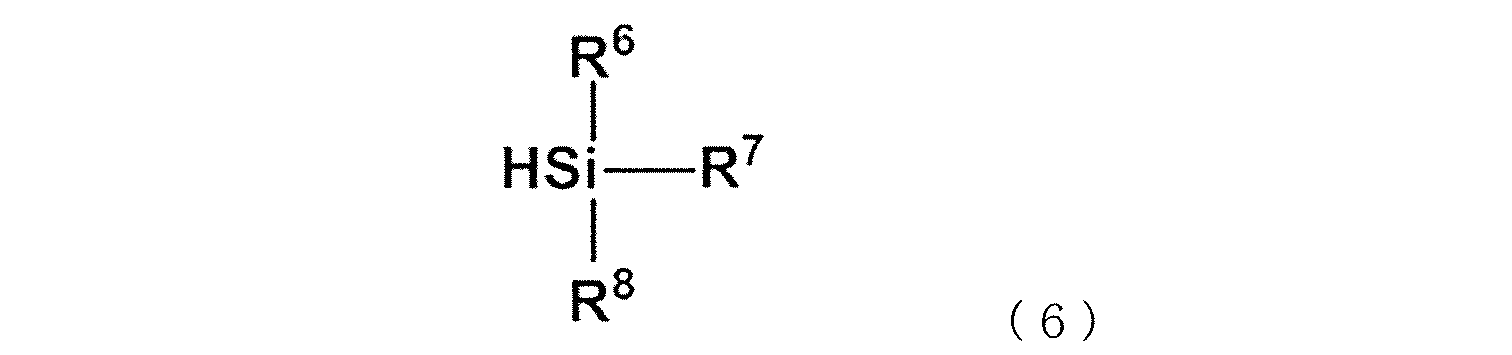
(式中、R6、R7及びR8は上記式(c2)のR7、R8及びR9と同じである。)
上記多官能ビニル芳香族共重合体と、シラン系化合物を、炭化水素系溶剤に溶解させ、0~150℃の温度で、触媒の存在下で付加させることを特徴とする変性ビニル芳香族系共重合体の製造方法。 - 活性末端を有する共役ジエン化合物(B)の重合体、又は活性末端を有する共役ジエン化合物(B)及び芳香族ビニル化合物(C)の共重合体に、請求項2~4のいずれか一項に記載の変性ビニル芳香族系共重合体(A)を変性剤として反応させて得られることを特徴とする変性共役ジエン系重合体。
- 変性ビニル芳香族系共重合体(A)に由来する構造単位(A1)を0.001~6重量%、共役ジエン化合物(B)に由来する構造単位(B1)を29~99.999重量%及び芳香族ビニル化合物(C)に由来する構造単位(C1)を0~70重量%含有する請求項6に記載の変性共役ジエン系共重合体。
- ゲルパーミエーションクロマトグラフィー(GPC)測定によって得られる微分分子量分布曲線において、全面積を100%とした場合に、最も低分子量側のピークの3倍以上の数平均分子量(Mn)を有する領域の面積が10%以上である請求項6又は7に記載の変性共役ジエン系共重合体。
- 請求項6~8のいずれか一項に記載の変性共役ジエン系共重合体に、フィラー又はシリカ系無機充填剤、金属酸化物、金属水酸化物及びカーボンブラックからなる群より選ばれる少なくとも1種の補強性充填剤を配合したことを特徴とする変性共役ジエン系共重合体組成物。
- 架橋剤を更に含有する請求項9に記載の変性共役ジエン系共重合体組成物。
- 請求項10に記載の変性共役ジエン系共重合体組成物を架橋してなることを特徴とするゴム架橋物。
- 請求項11に記載のゴム架橋物を含むことを特徴とするタイヤ部材。
- 構造単位(a)の95モル%以上が上記架橋構造単位(a1)であり、
末端の2個以上がアミノ基、アルコキシシリル基及び水酸基からなる群より選ばれる少なくとも1種の官能基を有する変性基により変性されていることを特徴とする請求項1に記載の変性ビニル芳香族系共重合体。 - 構造単位(a)と構造単位(b)の合計に対し、構造単位(a)を0.5モル%~95.0モル%含有し、構造単位(b)を5.0~99.5モル%含有している請求項13に記載の変性ビニル芳香族系共重合体。
- 数平均分子量Mnが500~50,000であり、分子量分布(Mw/Mn)が10.0以下である請求項13又は14に記載の変性ビニル芳香族系共重合体。
- 一分子当たりの平均官能基数は2~20個の範囲である請求項13~15のいずれか一項に記載の変性ビニル芳香族系共重合体。
- モノビニル芳香族化合物が、スチレン、ビニルナフタレン、ビニルビフェニル、m-メチルスチレン、p-メチルスチレン、o,p-ジメチルスチレン、m-エチルビニルベンゼン、インデン及びp-エチルビニルベンゼンからなる群から選ばれる1種以上の単量体である請求項13~16のいずれか一項に記載の変性ビニル芳香族系共重合体。
- アルカリ金属化合物又はアルカリ土類金属化合物をアニオン重合開始剤として用い、ジビニル芳香族化合物及びモノビニル芳香族化合物、又はジビニル芳香族化合物及びモノビニル芳香族化合物とこれらとアニオン共重合可能な単量体を共重合して、分岐構造と活性末端を有するビニル芳香族系共重合体を得る重合工程と、
前記ビニル芳香族系共重合体の活性末端に、アミノ基、アルコキシシリル基、水酸基からなる群より選ばれる少なくとも1種の官能基を有する化合物、又はそれらの前駆体化合物を反応させて官能基を形成する末端変成工程と、を含むことを特徴とする請求項13~17のいずれかに記載の変性ビニル芳香族系共重合体の製造方法。 - 共役ジエン化合物の重合体、又は共役ジエン化合物と芳香族ビニル化合物の共重合体に、請求項13~17のいずれか一項に記載の変性ビニル芳香族系共重合体を反応させて得られることを特徴とする変性共役ジエン系共重合体。
- 変性ビニル芳香族系共重合体に由来する構造単位(A1)を0.001~6重量%、共役ジエン化合物に由来する構造単位(B1)を29~99.999重量%及び芳香族ビニル化合物に由来する構造単位(C1)を0~70重量%含有する請求項19に記載の変性共役ジエン系共重合体。
- ゲルパーミエーションクロマトグラフィー(GPC)測定によって得られる微分分子量分布曲線において、全面積を100%とした場合に、最も低分子量側のピークの3倍以上の数平均分子量(Mn)を有する領域の面積が10%以上である請求項19又は20に記載の変性共役ジエン系共重合体。
- 請求項19~21のいずれか一項に記載の変性共役ジエン系共重合体100重量部に対し、シリカ系無機充填剤、金属酸化物、金属水酸化物及びカーボンブラックからなる群より選ばれる少なくとも1種の補強性充填剤を0.5~200重量部を含有することを特徴とするゴム組成物。
- 架橋剤を更に含有する請求項22に記載のゴム組成物。
- 請求項23に記載のゴム組成物を架橋してなることを特徴とするゴム架橋物。
- 請求項24に記載のゴム架橋物を含むことを特徴とするタイヤ部材。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20831000.3A EP4005819A4 (en) | 2019-06-25 | 2020-06-23 | Modified vinylaromatic copolymer, production method therefor, modified conjugated-diene copolymer obtained therefrom and composition thereof, crosslinked rubber object, and tire member |
| KR1020227000275A KR20220025802A (ko) | 2019-06-25 | 2020-06-23 | 변성 비닐 방향족계 공중합체 및 그 제조 방법, 그것으로부터 얻어지는 변성 공역 디엔계 공중합체, 그 조성물, 고무 가교물 및 타이어 부재 |
| JP2021527638A JP7636324B2 (ja) | 2019-06-25 | 2020-06-23 | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、その組成物、ゴム架橋物及びタイヤ部材 |
| US17/622,699 US12173098B2 (en) | 2019-06-25 | 2020-06-23 | Modified vinylaromatic copolymer, production method therefor, modified conjugated diene copolymer obtained therefrom and composition thereof, crosslinked rubber object, and tire member |
| CN202080043545.4A CN113966350B (zh) | 2019-06-25 | 2020-06-23 | 改性乙烯基芳香族系共聚物及其制造方法、改性共轭二烯系共聚物、及这些的应用 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019117636 | 2019-06-25 | ||
| JP2019117688 | 2019-06-25 | ||
| JP2019-117636 | 2019-06-25 | ||
| JP2019-117688 | 2019-06-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020262371A1 true WO2020262371A1 (ja) | 2020-12-30 |
Family
ID=74061645
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/024577 Ceased WO2020262371A1 (ja) | 2019-06-25 | 2020-06-23 | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、その組成物、ゴム架橋物及びタイヤ部材 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US12173098B2 (ja) |
| EP (1) | EP4005819A4 (ja) |
| JP (1) | JP7636324B2 (ja) |
| KR (1) | KR20220025802A (ja) |
| CN (1) | CN113966350B (ja) |
| TW (1) | TWI855098B (ja) |
| WO (1) | WO2020262371A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021054428A1 (ja) * | 2019-09-20 | 2021-03-25 | ||
| WO2023100993A1 (ja) * | 2021-12-03 | 2023-06-08 | 日鉄ケミカル&マテリアル株式会社 | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、樹脂組成物、樹脂架橋物及び構造部材 |
| WO2023145935A1 (ja) * | 2022-01-31 | 2023-08-03 | 日鉄ケミカル&マテリアル株式会社 | 樹脂組成物、及びそれから得られる成形体、硬化物、フィルム、複合材料、硬化複合材料、積層体、樹脂付き金属箔、並びに回路基板材料用ワニス |
| JPWO2023189949A1 (ja) * | 2022-03-28 | 2023-10-05 | ||
| WO2025258655A1 (ja) * | 2024-06-13 | 2025-12-18 | デンカ株式会社 | オレフィン-芳香族ビニル化合物-芳香族ポリエン共重合体及びその製造方法、当該共重合体を含有する組成物、並びにこれらの硬化体 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113725725B (zh) | 2017-09-28 | 2025-05-02 | 苹果公司 | 使用量子阱混合技术的激光架构 |
| US11171464B1 (en) | 2018-12-14 | 2021-11-09 | Apple Inc. | Laser integration techniques |
| US12426139B1 (en) | 2022-06-27 | 2025-09-23 | Apple Inc. | Feedback control of a diode element |
Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3159662A (en) | 1962-07-02 | 1964-12-01 | Gen Electric | Addition reaction |
| US3159601A (en) | 1962-07-02 | 1964-12-01 | Gen Electric | Platinum-olefin complex catalyzed addition of hydrogen- and alkenyl-substituted siloxanes |
| US3220972A (en) | 1962-07-02 | 1965-11-30 | Gen Electric | Organosilicon process using a chloroplatinic acid reaction product as the catalyst |
| US3516946A (en) | 1967-09-29 | 1970-06-23 | Gen Electric | Platinum catalyst composition for hydrosilation reactions |
| JPS59140211A (ja) | 1983-02-01 | 1984-08-11 | Nippon Erasutomaa Kk | スチレン−ブタジエン共重合体の製造方法 |
| JPH07207029A (ja) * | 1994-01-19 | 1995-08-08 | Japan Synthetic Rubber Co Ltd | 架橋ポリマー粒子およびその製造方法 |
| JPH08259584A (ja) * | 1995-02-24 | 1996-10-08 | Ciba Geigy Ag | シリル化フェロセニルジホスフィン、無機又はポリマー性有機支持体に結合したシリル化フェロセニルジホスフィン、及びそれらの金属錯体、並びにそれらの製造法及び用途 |
| JP2001131227A (ja) | 1999-10-29 | 2001-05-15 | Bridgestone Corp | 変性共役ジエン系重合体、それを用いたゴム組成物及び空気入りタイヤ |
| WO2002000779A1 (en) | 2000-06-29 | 2002-01-03 | Jsr Corporation | Rubber composition |
| JP2003073434A (ja) | 2001-06-22 | 2003-03-12 | Kuraray Co Ltd | ブロック共重合体の製造方法 |
| JP2004123873A (ja) | 2002-10-01 | 2004-04-22 | Nippon Steel Chem Co Ltd | 可溶性多官能ビニル芳香族共重合体及びその重合方法 |
| JP2004517202A (ja) | 2000-10-19 | 2004-06-10 | チャイナ・ペトロリアム・アンド・ケミカル・コーポレーション | 星形−ブロックインターポリマー及びそれらの製造 |
| JP2005213381A (ja) | 2004-01-29 | 2005-08-11 | Toyo Tire & Rubber Co Ltd | ゴム組成物 |
| WO2007034785A1 (ja) | 2005-09-22 | 2007-03-29 | Asahi Kasei Chemicals Corporation | 共役ジエン系重合体およびその製造方法 |
| WO2008013090A1 (en) | 2006-07-24 | 2008-01-31 | Asahi Kasei Chemicals Corporation | Modified conjugated diene polymer and method for producing the same |
| WO2011040312A1 (ja) | 2009-10-02 | 2011-04-07 | 旭化成ケミカルズ株式会社 | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、及び変性共役ジエン系重合体組成物 |
| WO2011129425A1 (ja) | 2010-04-16 | 2011-10-20 | 旭化成ケミカルズ株式会社 | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、及び変性共役ジエン系重合体組成物 |
| JP2013155268A (ja) * | 2012-01-30 | 2013-08-15 | Jsr Corp | 共役ジエン系ゴムおよびその製造方法、ゴム組成物、ゴム弾性体並びにタイヤ |
| JP2016530361A (ja) * | 2013-07-22 | 2016-09-29 | トリンゼオ ヨーロッパ ゲゼルシャフト ミット ベシュレンクテル ハフツング | 重合反応開始剤 |
| JP2018039995A (ja) | 2016-08-31 | 2018-03-15 | 新日鉄住金化学株式会社 | 可溶性多官能ビニル芳香族共重合体、その製造方法、硬化性樹脂組成物及びその硬化物 |
| WO2018084128A1 (ja) | 2016-11-01 | 2018-05-11 | 新日鉄住金化学株式会社 | 共重合体ゴム及びその製造方法、並びに架橋ゴム組成物 |
| WO2018181842A1 (ja) | 2017-03-30 | 2018-10-04 | 新日鉄住金化学株式会社 | 可溶性多官能ビニル芳香族共重合体、その製造方法並びに硬化性樹脂組成物及びその硬化物 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6311794B1 (en) | 1994-05-27 | 2001-11-06 | Deka Products Limited Partneship | System and method for stair climbing in a cluster-wheel vehicle |
| KR101445405B1 (ko) * | 2006-07-28 | 2014-09-26 | 가부시키가이샤 브리지스톤 | 상간 영역을 갖는 중합체성 코어-쉘 나노입자 |
| CN105175582B (zh) * | 2008-10-14 | 2017-10-17 | 旭化成株式会社 | 改性共轭二烯系聚合物、其制造方法、改性共轭二烯系聚合物组合物及轮胎 |
| JP2012092197A (ja) | 2010-10-26 | 2012-05-17 | Denki Kagaku Kogyo Kk | 樹脂及びシート |
| CN110256742A (zh) * | 2014-03-14 | 2019-09-20 | 株式会社可乐丽 | 橡胶组合物 |
| CN105175659B (zh) * | 2015-07-23 | 2017-11-03 | 湖南博瑞康新材料有限公司 | 一种透明型氢化聚苯乙烯‑b‑无规共聚共轭二烯/苯乙烯树脂及其制备方法 |
| JP6411313B2 (ja) | 2015-11-26 | 2018-10-24 | ヤンマー株式会社 | 燃料噴射ポンプ |
| DE102017003296B3 (de) | 2017-04-04 | 2018-05-30 | Te Connectivity Germany Gmbh | Steckverbinder und Verfahren zur Herstellung einer Steckverbindung |
| KR102818943B1 (ko) * | 2018-09-28 | 2025-06-11 | 닛테츠 케미컬 앤드 머티리얼 가부시키가이샤 | 다관능 비닐 방향족 공중합체 및 그 제조 방법, 그것으로부터 얻어지는 공중합체 고무, 고무 조성물, 고무 가교물, 및 타이어 부재 |
-
2020
- 2020-06-23 KR KR1020227000275A patent/KR20220025802A/ko active Pending
- 2020-06-23 JP JP2021527638A patent/JP7636324B2/ja active Active
- 2020-06-23 WO PCT/JP2020/024577 patent/WO2020262371A1/ja not_active Ceased
- 2020-06-23 EP EP20831000.3A patent/EP4005819A4/en active Pending
- 2020-06-23 CN CN202080043545.4A patent/CN113966350B/zh active Active
- 2020-06-23 US US17/622,699 patent/US12173098B2/en active Active
- 2020-06-24 TW TW109121556A patent/TWI855098B/zh active
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3159601A (en) | 1962-07-02 | 1964-12-01 | Gen Electric | Platinum-olefin complex catalyzed addition of hydrogen- and alkenyl-substituted siloxanes |
| US3220972A (en) | 1962-07-02 | 1965-11-30 | Gen Electric | Organosilicon process using a chloroplatinic acid reaction product as the catalyst |
| US3159662A (en) | 1962-07-02 | 1964-12-01 | Gen Electric | Addition reaction |
| US3516946A (en) | 1967-09-29 | 1970-06-23 | Gen Electric | Platinum catalyst composition for hydrosilation reactions |
| JPS59140211A (ja) | 1983-02-01 | 1984-08-11 | Nippon Erasutomaa Kk | スチレン−ブタジエン共重合体の製造方法 |
| JPH07207029A (ja) * | 1994-01-19 | 1995-08-08 | Japan Synthetic Rubber Co Ltd | 架橋ポリマー粒子およびその製造方法 |
| JPH08259584A (ja) * | 1995-02-24 | 1996-10-08 | Ciba Geigy Ag | シリル化フェロセニルジホスフィン、無機又はポリマー性有機支持体に結合したシリル化フェロセニルジホスフィン、及びそれらの金属錯体、並びにそれらの製造法及び用途 |
| JP2001131227A (ja) | 1999-10-29 | 2001-05-15 | Bridgestone Corp | 変性共役ジエン系重合体、それを用いたゴム組成物及び空気入りタイヤ |
| WO2002000779A1 (en) | 2000-06-29 | 2002-01-03 | Jsr Corporation | Rubber composition |
| JP2004517202A (ja) | 2000-10-19 | 2004-06-10 | チャイナ・ペトロリアム・アンド・ケミカル・コーポレーション | 星形−ブロックインターポリマー及びそれらの製造 |
| JP2003073434A (ja) | 2001-06-22 | 2003-03-12 | Kuraray Co Ltd | ブロック共重合体の製造方法 |
| JP2004123873A (ja) | 2002-10-01 | 2004-04-22 | Nippon Steel Chem Co Ltd | 可溶性多官能ビニル芳香族共重合体及びその重合方法 |
| JP2005213381A (ja) | 2004-01-29 | 2005-08-11 | Toyo Tire & Rubber Co Ltd | ゴム組成物 |
| WO2007034785A1 (ja) | 2005-09-22 | 2007-03-29 | Asahi Kasei Chemicals Corporation | 共役ジエン系重合体およびその製造方法 |
| WO2008013090A1 (en) | 2006-07-24 | 2008-01-31 | Asahi Kasei Chemicals Corporation | Modified conjugated diene polymer and method for producing the same |
| WO2011040312A1 (ja) | 2009-10-02 | 2011-04-07 | 旭化成ケミカルズ株式会社 | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、及び変性共役ジエン系重合体組成物 |
| WO2011129425A1 (ja) | 2010-04-16 | 2011-10-20 | 旭化成ケミカルズ株式会社 | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、及び変性共役ジエン系重合体組成物 |
| JP2013155268A (ja) * | 2012-01-30 | 2013-08-15 | Jsr Corp | 共役ジエン系ゴムおよびその製造方法、ゴム組成物、ゴム弾性体並びにタイヤ |
| JP2016530361A (ja) * | 2013-07-22 | 2016-09-29 | トリンゼオ ヨーロッパ ゲゼルシャフト ミット ベシュレンクテル ハフツング | 重合反応開始剤 |
| JP2018039995A (ja) | 2016-08-31 | 2018-03-15 | 新日鉄住金化学株式会社 | 可溶性多官能ビニル芳香族共重合体、その製造方法、硬化性樹脂組成物及びその硬化物 |
| WO2018084128A1 (ja) | 2016-11-01 | 2018-05-11 | 新日鉄住金化学株式会社 | 共重合体ゴム及びその製造方法、並びに架橋ゴム組成物 |
| WO2018181842A1 (ja) | 2017-03-30 | 2018-10-04 | 新日鉄住金化学株式会社 | 可溶性多官能ビニル芳香族共重合体、その製造方法並びに硬化性樹脂組成物及びその硬化物 |
Non-Patent Citations (2)
| Title |
|---|
| JEAN FISHER ET AL., CHEM. EUR. J., vol. 4, 1998, pages 2008 - 2017 |
| See also references of EP4005819A4 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021054428A1 (ja) * | 2019-09-20 | 2021-03-25 | ||
| JP7273982B2 (ja) | 2019-09-20 | 2023-05-15 | 株式会社クラレ | 共役ジエン系グラフト重合体、およびその製造方法 |
| WO2023100993A1 (ja) * | 2021-12-03 | 2023-06-08 | 日鉄ケミカル&マテリアル株式会社 | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、樹脂組成物、樹脂架橋物及び構造部材 |
| WO2023145935A1 (ja) * | 2022-01-31 | 2023-08-03 | 日鉄ケミカル&マテリアル株式会社 | 樹脂組成物、及びそれから得られる成形体、硬化物、フィルム、複合材料、硬化複合材料、積層体、樹脂付き金属箔、並びに回路基板材料用ワニス |
| JPWO2023189949A1 (ja) * | 2022-03-28 | 2023-10-05 | ||
| WO2023189949A1 (ja) * | 2022-03-28 | 2023-10-05 | 旭化成株式会社 | 多官能ビニル芳香族共重合体 |
| WO2025258655A1 (ja) * | 2024-06-13 | 2025-12-18 | デンカ株式会社 | オレフィン-芳香族ビニル化合物-芳香族ポリエン共重合体及びその製造方法、当該共重合体を含有する組成物、並びにこれらの硬化体 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI855098B (zh) | 2024-09-11 |
| TW202108640A (zh) | 2021-03-01 |
| US20230102867A1 (en) | 2023-03-30 |
| EP4005819A4 (en) | 2023-09-27 |
| KR20220025802A (ko) | 2022-03-03 |
| JPWO2020262371A1 (ja) | 2020-12-30 |
| US12173098B2 (en) | 2024-12-24 |
| EP4005819A1 (en) | 2022-06-01 |
| CN113966350A (zh) | 2022-01-21 |
| JP7636324B2 (ja) | 2025-02-26 |
| CN113966350B (zh) | 2025-03-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7636324B2 (ja) | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、その組成物、ゴム架橋物及びタイヤ部材 | |
| JP5520829B2 (ja) | 変性共役ジエン系重合体、その製造方法、変性共役ジエン系重合体組成物、及びタイヤ | |
| JP6032880B2 (ja) | 変性共役ジエン系重合体組成物、トレッド用組成物、サイドウォール用組成物及びタイヤ | |
| WO2018034194A1 (ja) | 変性共役ジエン系重合体、ゴム組成物、及びタイヤ | |
| JP6158480B2 (ja) | ゴム組成物、ゴム組成物の製造方法 | |
| WO2011129425A1 (ja) | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、及び変性共役ジエン系重合体組成物 | |
| WO2016199779A1 (ja) | 変性共役ジエン系重合体及びその製造方法、ゴム組成物、並びにタイヤ | |
| WO2013031599A1 (ja) | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、変性共役ジエン系重合体組成物、ゴム組成物、及びタイヤ | |
| KR20160097262A (ko) | 실란 개질된 엘라스토머 중합체 | |
| KR102818943B1 (ko) | 다관능 비닐 방향족 공중합체 및 그 제조 방법, 그것으로부터 얻어지는 공중합체 고무, 고무 조성물, 고무 가교물, 및 타이어 부재 | |
| WO2018034195A1 (ja) | 変性共役ジエン系重合体及びそのゴム組成物、並びにタイヤ | |
| CN108026222A (zh) | 聚合物化合物、使用其制备改性共轭二烯类聚合物的方法以及改性共轭二烯类聚合物 | |
| JP5534913B2 (ja) | 変性共役ジエン系ゴム組成物及び変性共役ジエン系ゴム組成物の製造方法 | |
| JP5844575B2 (ja) | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、変性共役ジエン系重合体組成物、及びタイヤ | |
| US10294314B2 (en) | Functionalized elastomeric polymer compositions, their preparation methods and crosslinked rubber compositions thereof | |
| JP5866161B2 (ja) | 変性共役ジエン系重合体の製造方法、変性共役ジエン系重合体、変性共役ジエン系重合体組成物、及びタイヤ | |
| EP4442713A1 (en) | Modified vinyl aromatic copolymer, method for producing same, modified conjugated diene copolymer obtained from same, resin composition, crosslinked resin and structural member | |
| WO2023243644A1 (ja) | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、樹脂組成物、樹脂架橋物及び構造部材 | |
| JP2025109590A (ja) | 変性ビニル芳香族系共重合体及びその製造方法、それから得られる変性共役ジエン系共重合体、樹脂組成物、樹脂架橋物及び構造部材 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20831000 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2021527638 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20227000275 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020831000 Country of ref document: EP Effective date: 20220125 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 202080043545.4 Country of ref document: CN |