WO2021002473A1 - Nrf2活性化化合物 - Google Patents
Nrf2活性化化合物 Download PDFInfo
- Publication number
- WO2021002473A1 WO2021002473A1 PCT/JP2020/026309 JP2020026309W WO2021002473A1 WO 2021002473 A1 WO2021002473 A1 WO 2021002473A1 JP 2020026309 W JP2020026309 W JP 2020026309W WO 2021002473 A1 WO2021002473 A1 WO 2021002473A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- group
- methyl
- ethyl
- reference example
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 Cc1ccc(*)[s]1 Chemical compound Cc1ccc(*)[s]1 0.000 description 4
- XQQBUAPQHNYYRS-UHFFFAOYSA-N Cc1ccc[s]1 Chemical compound Cc1ccc[s]1 XQQBUAPQHNYYRS-UHFFFAOYSA-N 0.000 description 2
- QYCHWMYWJNORMU-UHFFFAOYSA-N CC(C(c1cc(OC)c(C(C)=O)[s]1)c1ccc(C)c(CCl)c1)C(OCC[Si](C)(C)C)=O Chemical compound CC(C(c1cc(OC)c(C(C)=O)[s]1)c1ccc(C)c(CCl)c1)C(OCC[Si](C)(C)C)=O QYCHWMYWJNORMU-UHFFFAOYSA-N 0.000 description 1
- HXVLWNKFMNRJED-UHFFFAOYSA-N CC(c1cc(Br)c[s]1)=O Chemical compound CC(c1cc(Br)c[s]1)=O HXVLWNKFMNRJED-UHFFFAOYSA-N 0.000 description 1
- JOJMDXDMUDWEJV-SNAWJCMRSA-N CC(c1cc(C)c(/C=C/C(OC)=O)[s]1)=O Chemical compound CC(c1cc(C)c(/C=C/C(OC)=O)[s]1)=O JOJMDXDMUDWEJV-SNAWJCMRSA-N 0.000 description 1
- HMZLIPBWXKAIPL-GQCTYLIASA-N CC(c1ccc(/C=C/C(OC)=O)[s]1)=O Chemical compound CC(c1ccc(/C=C/C(OC)=O)[s]1)=O HMZLIPBWXKAIPL-GQCTYLIASA-N 0.000 description 1
- QSONDMOXTRFZLB-UHFFFAOYSA-N CC1(c2ccc(C(CC(OC)=O)c3ccc(C)c(COCc(cc4)ccc4OC)c3)[s]2)OCCO1 Chemical compound CC1(c2ccc(C(CC(OC)=O)c3ccc(C)c(COCc(cc4)ccc4OC)c3)[s]2)OCCO1 QSONDMOXTRFZLB-UHFFFAOYSA-N 0.000 description 1
- IHBTUTIFJGQDSO-UHFFFAOYSA-N CCC(CN1Cc2cc(C(CC(O)=O)c3c[s]c(C(NC)=O)c3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CCC(CN1Cc2cc(C(CC(O)=O)c3c[s]c(C(NC)=O)c3)ccc2C)Oc(cccc2)c2S1(=O)=O IHBTUTIFJGQDSO-UHFFFAOYSA-N 0.000 description 1
- XPSKIQNUJSROKK-ZMFCMNQTSA-N CC[C@H](CN1Cc2c(C)ccc(C(CC(OC)=O)c3cc(F)c(C(C)=O)[s]3)c2)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2c(C)ccc(C(CC(OC)=O)c3cc(F)c(C(C)=O)[s]3)c2)Oc(cccc2)c2S1(=O)=O XPSKIQNUJSROKK-ZMFCMNQTSA-N 0.000 description 1
- CFEPSLMWMXECSB-MIHMCVIASA-N CC[C@H](CN1Cc2c(C)ccc(C(CC(OC)=O)c3ccc(C(NC(C)C)=O)[o]3)c2)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2c(C)ccc(C(CC(OC)=O)c3ccc(C(NC(C)C)=O)[o]3)c2)Oc(cccc2)c2S1(=O)=O CFEPSLMWMXECSB-MIHMCVIASA-N 0.000 description 1
- WVVGHSXCLRAFRO-MIHMCVIASA-N CC[C@H](CN1Cc2cc(C(CC(O)=O)c3ccc(C(C)=O)[n]3CC)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(O)=O)c3ccc(C(C)=O)[n]3CC)ccc2C)Oc(cccc2)c2S1(=O)=O WVVGHSXCLRAFRO-MIHMCVIASA-N 0.000 description 1
- GIDBZEVBCINPSG-ZMFCMNQTSA-N CC[C@H](CN1Cc2cc(C(CC(O)=O)c3ccc(C(C)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(O)=O)c3ccc(C(C)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O GIDBZEVBCINPSG-ZMFCMNQTSA-N 0.000 description 1
- LNBJPTSUDVYMSI-MIHMCVIASA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(C(C)=O)c[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(C(C)=O)c[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O LNBJPTSUDVYMSI-MIHMCVIASA-N 0.000 description 1
- IIFYGJFQPDPULT-WTQRLHSKSA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(C)c(C(NC)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(C)c(C(NC)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O IIFYGJFQPDPULT-WTQRLHSKSA-N 0.000 description 1
- DCLUHYXGTCEIRB-ZMFCMNQTSA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(CNC4=O)c4[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3cc(CNC4=O)c4[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O DCLUHYXGTCEIRB-ZMFCMNQTSA-N 0.000 description 1
- QMRWPMGPLQKYNZ-ZMFCMNQTSA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(C)=O)[s]3)ccc2C)Oc(ccnc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(C)=O)[s]3)ccc2C)Oc(ccnc2)c2S1(=O)=O QMRWPMGPLQKYNZ-ZMFCMNQTSA-N 0.000 description 1
- XVHZDWXPZQWACY-ZMFCMNQTSA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(NC)=O)[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(NC)=O)[s]3)ccc2C)Oc(cccc2)c2S1(=O)=O XVHZDWXPZQWACY-ZMFCMNQTSA-N 0.000 description 1
- DEHUBGOUXSMCAK-MIHMCVIASA-N CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(NCCOC)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O Chemical compound CC[C@H](CN1Cc2cc(C(CC(OC)=O)c3ccc(C(NCCOC)=O)[o]3)ccc2C)Oc(cccc2)c2S1(=O)=O DEHUBGOUXSMCAK-MIHMCVIASA-N 0.000 description 1
- YLIWKIYVWPKWHH-SOFGYWHQSA-N CC[n]1c(C(C)=O)ccc1/C=C/C(OC)=O Chemical compound CC[n]1c(C(C)=O)ccc1/C=C/C(OC)=O YLIWKIYVWPKWHH-SOFGYWHQSA-N 0.000 description 1
- BEEILQXFNUFFLY-NSCUHMNNSA-N COC(/C=C/c1cc(CNC2=O)c2[s]1)=O Chemical compound COC(/C=C/c1cc(CNC2=O)c2[s]1)=O BEEILQXFNUFFLY-NSCUHMNNSA-N 0.000 description 1
- ZGUDNMKZVWJARD-ONEGZZNKSA-N C[n]1c(C#N)cc(/C=C/C(OC)=O)c1 Chemical compound C[n]1c(C#N)cc(/C=C/C(OC)=O)c1 ZGUDNMKZVWJARD-ONEGZZNKSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D291/00—Heterocyclic compounds containing rings having nitrogen, oxygen and sulfur atoms as the only ring hetero atoms
- C07D291/08—Heterocyclic compounds containing rings having nitrogen, oxygen and sulfur atoms as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/04—Artificial tears; Irrigation solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D419/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms
- C07D419/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D419/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D419/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms
- C07D419/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D515/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D515/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen, oxygen, and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D515/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/027—Organoboranes and organoborohydrides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
- C07F7/0812—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te comprising a heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Definitions
- the present invention relates to Nrf2 activating compounds and the like.
- Nrf2 (NF-E2-related factor 2) is a transcription factor that promotes the expression of oxidative stress protection genes. Nrf2 is ubiquitinated by interaction with Keap1 (Kelch-like ECH-associated protein 1) and undergoes degradation in the proteasome. Therefore, inhibition of the interaction between Nrf2 and Keap1 promotes the translocation of Nrf2 into the nucleus, and can activate the expression of the oxidative stress protection gene.
- the Nrf2 activator is used in the systemic area for multiple sclerosis, psoriasis, diabetic nephropathy, etc., and in the ophthalmic area, the Nrf2 activator is used for dry eye, diabetic retinopathy, retinitis pigmentosa, glaucoma, etc. It is expected as a therapeutic drug for various diseases.
- Patent Document 1 discloses an Nrf2 activator mainly for the treatment of COPD (chronic obstructive pulmonary disease).
- An object of the present invention is to provide a compound having an Nrf2 activating action.
- the present invention includes the following aspects.
- R 1a and R 1b are the same or different and represent a hydrogen atom, an alkyl group or a halogen atom.
- R 2 represents a heterocyclic group that may be substituted, and said heterocycle represents a fused ring containing a thiophene, furan, pyrrole, or thiazole, or any of these heterocycles.
- R 6 indicates an optionally substituted alkyl group.
- a 1 , A 2 , A 3 and A 4 are the same or different and indicate CH or N (provided that N is one or less).
- Z represents a hydrogen atom or a halogen atom.
- Item 2 The compound according to Item 1, a salt thereof, or a solvate thereof, which is a thienyl group in which R 2 may be substituted.
- Item 3 The compound according to Item 2, a salt thereof, or a solvate thereof, wherein the substituent that the thienyl group may have includes an electron attractant group.
- Item 4. The compound according to Item 2 or 3, a salt thereof, or a solvate thereof, wherein the substituent that the thienyl group may have includes an acetyl group, an aminosulfonyl group, a cyano group or a cycloalkylcarbonyl group.
- R 2 is a general formula (R2A):
- R 3 represents an acetyl group, an aminosulfonyl group, a cyano group or a cycloalkylcarbonyl group.
- Y represents a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group.
- Item 4 The compound according to any one of Items 1 to 4, a salt thereof, or a solvate thereof, which is a group represented by.
- R 2 is the general formula (R2Aa) or (R2Ab):
- R 3 represents an acetyl group, an aminosulfonyl group, a cyano group or a cycloalkylcarbonyl group.
- Y represents a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group.
- Item 5 The compound according to any one of Items 1 to 5, a salt thereof, or a solvate thereof, which is a group represented by.
- Item 7 The compound according to Item 5 or 6, a salt thereof, or a solvate thereof, wherein R 3 is an acetyl group.
- Item 8 The compound according to any one of Items 5 to 7, wherein R 4 and R 5 are the same or different and are hydrogen atoms or alkyl groups, salts thereof, or solvates thereof.
- Item 9 The compound according to any one of Items 5 to 8, wherein R 1a and R 1b are the same or different, and are hydrogen atoms or alkyl groups, salts thereof, or solvates thereof.
- the R 1a and the R 1b are the same or different and are hydrogen atoms or alkyl groups.
- the R 3 is an acetyl group
- the R 4 and the R 5 are the same or different and are hydrogen atoms or alkyl groups.
- the R 6 is an optionally substituted ethyl group.
- a 1 , A 2 and A 4 are all CH, and A 3 is CH or N.
- Y is a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group
- Z is a hydrogen atom.
- Item 5 The compound according to any one of Items 5 to 9, a salt thereof, or a solvate thereof.
- R 1a and R 1b are alkyl groups and the other is a hydrogen atom, or both R 1a and R 1b are alkyl groups, preferably the alkyl group has 1 to 3 carbon atoms.
- R 2 is a general formula (R2Aa):
- R 3 represents an acetyl group.
- Y represents a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group.
- the alkyl group has 1 to 3 carbon atoms
- the halogen atom is a fluorine atom
- the alkoxy group has 1 to 3 carbon atoms.
- the R 4 is a hydrogen atom
- the R 5 is an alkyl group, preferably the alkyl group has 1 to 3 carbon atoms.
- the R 6 may be an ethyl group substituted, preferably an arylalkoxy group as a substituent which the ethyl group may have, and particularly preferably a benzyloxy group.
- a 1 , A 2 and A 4 are all CH
- a 3 is CH or N
- Z is a hydrogen atom.
- Item 5 The compound according to any one of Items 5 to 10, a salt thereof, or a solvate thereof.
- Item 13 A pharmaceutical agent containing at least one selected from the group consisting of the compound according to any one of Items 1 to 12, a salt thereof, and a solvate thereof.
- Item 14 The medicament according to Item 13, which is an Nrf2 (NF-E2-related factor 2) activator.
- Item 15. The medicament according to Item 13 or 14, which is a locally administered preparation.
- Item 16 The drug according to any one of Items 13 to 15, which is an ophthalmic preparation, an otolaryngological preparation, a respiratory preparation, or a dermatological preparation.
- Item 17. The medicament according to Item 13 or 14, which is an orally administered preparation.
- Item 19 The medicament according to Item 13, 14, or 18, which is an intravenous administration preparation, a nasal administration preparation, or a transdermal administration preparation.
- Item 20 The drug according to any one of Items 11 to 19, which is a drug for preventing or treating brain disease, lung disease, skin disease, otolaryngology disease, renal disease or ophthalmic disease.
- Item 21 A method for preventing or treating a disease or symptom related to Nrf2 (NF-E2-related factor 2) activity regulation in a subject suffering from a disease related to Nrf2 (NF-E2-related factor 2) activity regulation. , The method comprising administering to the subject at least one selected from the group consisting of the compound according to any one of Items 1 to 13, a salt thereof, and a solvate thereof.
- Item 22 A method for preventing or treating brain disease, lung disease, skin disease, otolaryngological disease, renal disease or ophthalmic disease in a subject suffering from brain disease, lung disease, skin disease, otolaryngological disease, renal disease or ophthalmic disease. , The method comprising administering to the subject at least one selected from the group consisting of the compound according to any one of Items 1 to 13, a salt thereof, and a solvate thereof.
- Item 23 At least one selected from the group consisting of the compound according to any one of Items 1 to 13 for use as a medicine, a salt thereof, and a solvate thereof.
- Item 24 The compound according to any one of Items 1 to 13, a salt thereof, and a solvent thereof for use in the prevention or treatment of diseases or symptoms related to the regulation of Nrf2 (NF-E2-related factor 2) activity. At least one selected from the group consisting of Japanese products.
- Item 25 The compound according to any one of Items 1 to 13, a salt thereof, and a solvent sum thereof for use in the prevention or treatment of brain disease, lung disease, skin disease, otolaryngological disease, renal disease or ophthalmic disease. At least one selected from the group of things.
- Item 26 Use of at least one selected from the group consisting of the compounds according to any one of Items 1 to 13, salts thereof, and solvates thereof for the production of pharmaceuticals.
- Nrf2 activating action a compound having an Nrf2 activating action
- a salt thereof a compound having an Nrf2 activating action
- a solvate thereof a medicine containing at least one of these, more specifically, for example, Nrf2 activator, topical preparation, brain disease, lung disease, skin disease, otolaryngology disease, renal disease, or ophthalmology. It is possible to provide medicines for the prevention or treatment of diseases.
- compound (I) of the present invention It relates to a compound represented by (1), a salt thereof, or a solvate thereof (hereinafter, may be referred to as "compound (I) of the present invention”. This will be described below.
- R 1a , R 1b > R 1a and R 1b are the same or different and represent a hydrogen atom, an alkyl group or a halogen atom.
- the alkyl group represented by R 1a or R 1b includes both linear and branched chain (preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 4, preferably 1 to 3, more preferably 1 to 2, and even more preferably 1.
- Specific examples of the alkyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert-butyl group, a sec-butyl group and the like.
- halogen atom represented by R 1a or R 1b examples include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like, preferably a fluorine atom, a chlorine atom and the like, and more preferably a fluorine atom. ..
- R 1a and R 1b are the same or different, hydrogen atoms or alkyl groups. Particularly from the viewpoint of Nrf2 activation action, more preferably one of R 1a and R 1b is an alkyl group and the other is a hydrogen atom or an alkyl group, and more preferably one of R 1a and R 1b is an alkyl group. And the other is a hydrogen atom.
- R 2 > R 2 represents a heterocyclic group that may be substituted, and said heterocycle represents a fused ring containing a thiophene, furan, pyrrole, or thiazole, or any of these heterocycles.
- fused ring examples include a fused ring (bicyclic type) of thiophene, furan, pyrrole, or thiazole and a 5- to 7-membered ring.
- fused ring bicyclic type
- 5- to 7-membered ring forming the fused ring examples include a ring having or not having an oxo group having, for example, 0 to 3 heteroatoms (for example, sulfur atom, oxygen atom, nitrogen atom, etc.).
- fused ring More specific examples of the fused ring include indole, hydroindole, oxohydroindole, thienopyrrole, hydrothienopyrrole, oxohydrothienopyrrole and the like, and oxohydroindole (more specifically, for example 4- Oxo-1,5,6,7-tetrahydro-1H-indole), oxohydrothienopyrrole (more specifically, for example, 6-oxo-4,5-dihydro-6H-thieno [2,3-c] pyrrole) And so on.
- oxohydroindole more specifically, for example 4- Oxo-1,5,6,7-tetrahydro-1H-indole
- oxohydrothienopyrrole More specifically, for example, 6-oxo-4,5-dihydro-6H-thieno [2,3-c] pyrrole
- heterocyclic-derived group examples include a thienyl group (for example, 2-thienyl group, 3-thienyl group, etc., preferably 2-thienyl group), a frill group (for example, 2-furyl group, 3-furyl group, etc., preferably 2).
- thienyl group for example, 2-thienyl group, 3-thienyl group, etc., preferably 2-thienyl group
- frill group for example, 2-furyl group, 3-furyl group, etc., preferably 2.
- pyrolyl group for example, 1-pyrrolyl group, 2-pyrrolill group, 3-pyrrolill group
- thiazolyl group for example, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl
- Examples of the substituent that the heterocyclic-derived group represented by R 2 may have include an acyl group such as an acetyl group and a cycloalkylcarbonyl group, an aminosulfonyl group, a cyano group, and ⁇ R 21 ⁇ NR 22 R 23.
- R 21 indicates a single bond, carbonyl group or alkylene group.
- R 22 and R 23 are the same or different, hydrogen atom, optionally substituted alkyl group or R 22 and R 23 are bonded to each other and alkylene group.
- an alkoxy group, a halogen atom, an alkyl group and the like To form a ring together with an adjacent nitrogen atom), an alkoxy group, a halogen atom, an alkyl group and the like.
- the position of the substituent is not particularly limited, and is, for example, any of ring-constituting atoms (for example, carbon atom, sulfur atom, oxygen atom, nitrogen atom, etc.) on the hetero ring, preferably a carbon atom on the hetero ring. is there.
- the number of the above-mentioned substituents is not particularly limited, and is, for example, 0 to 4, preferably 0 to 2, and more preferably 1 to 2.
- the acyl group as a substituent that the group derived from the heterocycle may have is not particularly limited as long as it is a group obtained by removing the hydroxy group from the carboxylic acid, but is preferably ⁇ CO-R 24 (R 24).
- R 24 a group represented by an alkyl group or a cycloalkyl group (preferably an alkyl group).
- the alkyl group represented by R 24 includes both linear and branched chain (preferably linear) groups.
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- alkyl group examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n. -Hexyl group, 3-methylpentyl group and the like can be mentioned.
- the carbon number of the cycloalkyl group represented by R 24 is not particularly limited, but is, for example, 3 to 8, preferably 3 to 6, more preferably 3 to 4, and even more preferably 3.
- the acyl group as the substituent include an acetyl group and a cycloalkylcarbonyl group, and particularly preferably an acetyl group from the viewpoint of Nrf2 activating action.
- R 21 constituting -R 21- NR 22 R 23 as a substituent which the group derived from the heterocycle may have indicates a single bond, a carbonyl group or an alkylene group (preferably a carbonyl group).
- the alkylene group represented by R 21 includes both linear and branched chain (preferably straight chain) groups.
- the number of carbon atoms of the alkylene group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkylene group include a methylene group, an ethylene group, a propylene group, a butylene group and the like.
- R 22 and R 23 constituting -R 21- NR 22 R 23 as the above substituents are the same or different, and hydrogen atoms, optionally substituted alkyl groups or R 22 and R 23 are bonded to each other and alkylene.
- Form a group to form a ring with adjacent nitrogen atoms preferably R 22 and R 23 are the same or different, hydrogen or alkyl groups, more preferably one of R 22 and R 23 is a hydrogen atom.
- the other is an alkyl group, more preferably both R 22 and R 23 are hydrogen atoms).
- the alkyl group represented by R 22 or R 23 includes both linear and branched chain (preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkyl group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n. -Hexyl group, 3-methylpentyl group and the like can be mentioned.
- Examples of the substituent that the alkyl group may have include an alkoxy group.
- the alkoxy group includes either a linear group or a branched chain (preferably linear).
- the number of carbon atoms of the alkoxy group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkoxy group include a methoxy group, an ethoxy group, an n-propoxy group, an isopropoxy group, an n-butoxy group, an isobutoxy group, a sec-butoxy group, a tert-butoxy group and the like.
- the alkoxy group as a substituent that the heterocyclic group may have includes either a linear group or a branched chain (preferably linear).
- the number of carbon atoms of the alkoxy group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkoxy group include a methoxy group, an ethoxy group, an n-propoxy group, an isopropoxy group, an n-butoxy group, an isobutoxy group, a sec-butoxy group, a tert-butoxy group and the like.
- halogen atom as a substituent that the group derived from the hetero ring may have include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like, and preferably a fluorine atom, a chlorine atom and the like. More preferably, a fluorine atom is mentioned.
- the alkyl group as a substituent that the heterocyclic group may have includes either a linear group or a branched chain (preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 4, preferably 1 to 3, more preferably 1 to 2, and even more preferably 1.
- Specific examples of the alkyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert-butyl group, a sec-butyl group and the like.
- R 2 is preferably a optionally substituted thienyl group or optionally substituted frill group, more preferably from the viewpoint of being more easily metabolized.
- R 2 is a optionally substituted thienyl group.
- R 2 is a optionally substituted thienyl group or a optionally substituted frill group (preferably a optionally substituted thienyl group)
- the compound is a compound which the heteroring-derived group may have contains an electron-withdrawing group (that is, when there is one substituent, the substituent is an electron-withdrawing group. If there are two or more substituents, at least one substituent is an electron-withdrawing group), and more preferably, a substituent derived from a heterocycle may have an acyl group, an aminosulfonyl.
- a group or cyano group (more preferably an acetyl group, an aminosulfonyl group, a cyano group, or a cycloalkylcarbonyl group, particularly preferably an acetyl group from the viewpoint of Nrf2 activating activity) (ie, one substituent).
- the substituent is one of these groups, and in the case of two or more substituents, at least one substituent is one of these groups.
- the heterocyclic group preferably has 0 to 2 (preferably 1 to 2) substituents, and when the number of the substituents is 2 or more, at least the substituents.
- One is an alkyl group, a halogen atom or an alkoxy group.
- R 2 is a general formula (R2X1) or a general formula (R2X2):
- X indicates S, O or NR 31 .
- Y represents a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group.
- R 3 indicates an acyl group, a hydrogen atom, an aminosulfonyl group, a cyano group, or -R 21- NR 22 R 23 .
- R 31 represents a hydrogen atom or an alkyl group.
- It is a group represented by.
- X is preferably S or O, and particularly preferably S.
- R 3 examples include an acyl group (preferably ⁇ CO-R 24 (R 24 indicates an alkyl group or a cycloalkyl group)), an aminosulfonyl group, a cyano group and the like, and more preferably an acetyl group and an amino.
- R 3 examples include a sulfonyl group, a cyano group, a cycloalkylcarbonyl group and the like, and particularly preferably an acetyl group from the viewpoint of Nrf2 activating action.
- R 31 is preferably an alkyl group.
- the alkyl group represented by R 31 includes both linear and branched chain (preferably linear) groups.
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkyl group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n. -Hexyl group, 3-methylpentyl group and the like can be mentioned.
- R 2 represented by the general formula examples include a group represented by the general formula (R2X1), and more preferably the general formula (R2A):
- the alkyl group represented by R 4 or R 5 includes both linear and branched chain (preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 4, preferably 1 to 3, more preferably 1 to 2, and even more preferably 1.
- Specific examples of the alkyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert-butyl group, a sec-butyl group and the like.
- Examples of the substituent that the alkyl group represented by R 4 or R 5 may have include a halogen atom.
- the halogen atom include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like.
- the number of the substituents is not particularly limited, and is, for example, 0 to 3, preferably 0.
- R 4 and R 5 are the same or different and are hydrogen atoms or alkyl groups, more preferably one of R 4 and R 5 is an alkyl group and the other. It is a hydrogen atom, more preferably R 4 is a hydrogen atom and R 5 is an alkyl group.
- R 6 > R 6 indicates an optionally substituted alkyl group.
- the alkyl group represented by R 6 includes both linear and branched (preferably linear) groups.
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 2.
- Specific examples of the alkyl group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n. -Hexyl group, 3-methylpentyl group and the like can be mentioned.
- Substituents that the alkyl group represented by R 6 may have include, for example, a hydroxyl group, an alkoxy group, an arylalkoxy group, an aryloxy group, and -NR 61 R 62 (R 61 and R 62 are the same or different. It indicates a hydrogen atom or an alkyl group.) And the like.
- the position of the substituent is not particularly limited, but in one aspect of the present invention, it is preferably the terminal of the alkyl group.
- the number of the above-mentioned substituents is not particularly limited, and is, for example, 0 to 3, preferably 0 to 1.
- the alkoxy group as the substituent includes either a linear or branched chain (preferably linear).
- the number of carbon atoms of the alkoxy group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 1.
- Specific examples of the alkoxy group include a methoxy group, an ethoxy group, an n-propoxy group, an isopropoxy group, an n-butoxy group, an isobutoxy group, a sec-butoxy group, a tert-butoxy group and the like.
- the arylalkoxy group as the substituent is not particularly limited as long as it is the alkoxy group substituted with the aryl group.
- the number of carbon atoms of the aryl group is not particularly limited, but is, for example, 6 to 14, preferably 6 to 8.
- Specific examples of such an aryl group include a phenyl group, a naphthyl group, a biphenyl group, a pentarenyl group, an indenyl group, an anthryl group, a tetrasenyl group, a pentasenyl group, a pyrenyl group, a peryleneyl group, a fluorenyl group, a phenanthryl group and the like.
- a phenyl group and the like are particularly preferable.
- the substitution position of the aryl group in the alkoxy group is not particularly limited, but in one aspect of the present invention, it is preferably the terminal of the alkoxy group.
- Examples of the arylalkoxy group include a benzyloxy group and a phenethyloxy group.
- the number of carbon atoms of the aryloxy group as the above-mentioned substituent is not particularly limited, but is, for example, 6 to 14, preferably 6 to 8.
- Specific examples of the aryl group constituting the aryloxy group include a phenyl group, a naphthyl group, a biphenyl group, a pentarenyl group, an indenyl group, an anthranyl group, a tetrasenyl group, a pentasenyl group, a pyrenyl group, a perylenel group and a fluorenyl group. Examples thereof include a phenanthryl group, and particularly preferably a phenyl group and the like. Examples of the aryloxy group include a phenyloxy group and the like.
- R 61 and R 62 constituting -NR 61 R 62 as the above-mentioned substituents are the same or different and indicate a hydrogen atom or an alkyl group.
- the alkyl group represented by R 61 or R 62 includes both linear and branched chain (preferably linear).
- the number of carbon atoms of the alkyl group is not particularly limited, but is, for example, 1 to 8, preferably 1 to 6, more preferably 1 to 4, still more preferably 1 to 2, and particularly preferably 2.
- alkyl group examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, tert-butyl group, sec-butyl group, n-pentyl group, neopentyl group, n. -Hexyl group, 3-methylpentyl group and the like can be mentioned.
- R 6 is preferably an alkyl group optionally substituted with a hydroxyl group, an alkoxy group, or an arylalkoxy group, and more preferably substituted with an arylalkoxy group. It is an alkyl group, more preferably an alkyl group.
- a 1 , A 2 , A 3 , A 4 > A 1 , A 2 , A 3 and A 4 are the same or different and indicate CH or N (provided that N is one or less).
- a 1 or A 3 is preferably N, and more preferably A 3 is N.
- a 1 , A 2 and A 4 are all CH and A 3 is CH or N, more preferably A 1 , A 2 , A 3 and A 4 All are CH.
- Z > Z represents a hydrogen atom or a halogen atom.
- halogen atom represented by Z examples include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like, preferably a fluorine atom, a chlorine atom and the like, and more preferably a fluorine atom.
- Z When Z is a halogen atom, its bond position may be any of A 1 , A 2 , A 3 and A 4 , for example A 4 .
- Z is preferably a hydrogen atom.
- R 1a and R 1b are the same or different and are hydrogen atoms or alkyl groups.
- R 3 is an acetyl group
- R 4 and R 5 are the same or different, hydrogen atoms or alkyl groups
- R 6 is an optionally substituted ethyl group
- a 1 , A 2 , and A 4 are all CH
- a 3 is CH or N
- Y is a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group
- Z is a hydrogen atom, Is preferable.
- R 1a and R 1b are alkyl group and the other is a hydrogen atom, or both R 1a and R 1b are alkyl groups, preferably the carbon of the alkyl group.
- the number is 1-3,
- R 2 is a general formula (R2Aa):
- R 3 represents an acetyl group.
- Y represents a hydrogen atom, an alkyl group, a halogen atom or an alkoxy group.
- the alkyl group has 1 to 3 carbon atoms
- the halogen atom is a fluorine atom
- the alkoxy group has 1 to 3 carbon atoms.
- the R 4 is a hydrogen atom
- the R 5 is an alkyl group, preferably the alkyl group has 1 to 3 carbon atoms.
- the R 6 may be an ethyl group substituted, preferably an arylalkoxy group as a substituent which the ethyl group may have, and particularly preferably a benzyloxy group.
- a 1 , A 2 and A 4 are all CH
- a 3 is CH or N
- Z is a hydrogen atom. Is preferable.
- the following compounds are preferably mentioned as the compound represented by the general formula (I).
- the salt of the compound represented by the general formula (I) is not particularly limited as long as it is a pharmaceutically acceptable salt.
- an acidic salt or a basic salt can be adopted.
- acidic salts are inorganic acid salts such as hydrochloride, hydrobromide, sulfate, nitrate, phosphate; acetate, propionate, tartrate, fumarate, maleate, malic acid.
- Organic salts such as salts, citrates, methanesulfonates and paratoluenesulfonates are mentioned, and examples of basic salts are alkali metal salts such as sodium salts and potassium salts; as well as calcium salts and magnesium.
- Alkaline earth metal salts such as salts; Salts with ammonia; Morphorine, piperidine, pyrrolidine, monoalkylamine, dialkylamine, trialkylamine, mono (hydroxyalkyl) amine, di (hydroxyalkyl) amine, tri (hydroxyalkyl) Examples thereof include salts with organic amines such as amines.
- the solvate of the compound represented by the general formula (I) is not particularly limited.
- the solvent constituting the solvent field include water, a pharmaceutically acceptable organic solvent (for example, ethanol, glycerol, acetic acid, etc.) and the like.
- the compound (I) of the present invention can also be produced by modifying it into a compound in which B is independently C, CH, or N in the following general formula.
- B is independently C, CH, or N in the following general formula.
- the double line between the dotted line and the solid line indicates a single bond or a double bond.
- the number of N in the 6-membered ring containing B is 1 to 3.
- R 1a , R 1b , R 2 , R 4 , R 5 , R 6 , A 1 , A 2 , A 3 , A 4 , Z are the same as above.
- the compound (I) of the present invention can be produced by various methods.
- the compound (I) of the present invention can be produced, for example, by the method shown in Scheme 1 below or a method similar thereto.
- step a the synthetic intermediate 2 and the synthetic intermediate 3 are reacted in the presence of a catalyst and a base to produce the synthetic intermediate 4.
- the synthetic intermediate 3 can be used, for example, 1 to 5 equivalents, preferably 1.2 to 3 equivalents
- the catalyst can be used, for example, 0.05 to 0.5 equivalents, preferably 0.05 to 0.2 equivalents, relative to the synthetic intermediate 2.
- 1.5 to 5 equivalents, preferably 1.5 to 3 equivalents of the base can be used.
- the catalyst include a rhodium complex, preferably [RhCl (cod)] 2 .
- the base examples include N, N-dicyclohexylmethylamine, triethylamine, pyridine, N, N-diisopropylethylamine and the like, and triethylamine is preferable.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include 1,4-dioxane, 1,2-dimethoxyethane, tetrahydrofuran and the like, and 1,4-dioxane is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 18 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 150 ° C, preferably 25 ° C to 100 ° C.
- step b synthetic intermediate 4 is reacted with iodomethane in the presence of a base to produce synthetic intermediate 5.
- Lithium diisopropylamide can be used, for example, 1 to 2 equivalents, preferably 1 to 1.8 equivalents, and iodomethane can be used, for example, 1 to 20 equivalents, preferably 1 to 15 equivalents, relative to the synthetic intermediate 4.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 5 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually ⁇ 78 ° C. to 40 ° C., preferably ⁇ 78 ° C. to 25 ° C.
- step c the acetal of synthetic intermediate 5 is deprotected.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include methanol, dichloromethane, tetrahydrofuran, 1,4-dioxane and the like, preferably methanol or tetrahydrofuran.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 18 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 25 ° C to 100 ° C.
- the compound (I) of the present invention is produced by a hydrolysis reaction of synthetic intermediate 4 or an ester equivalent thereto.
- synthetic intermediate 4 for example, 5 to 20 equivalents, preferably 10 to 20 equivalents of the base can be used with respect to the synthetic intermediate 4.
- the base include lithium hydroxide, sodium hydroxide, potassium hydroxide and the like, and preferred examples include sodium hydroxide and lithium hydroxide.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include methanol, dichloromethane, tetrahydrofuran and the like, preferably methanol and tetrahydrofuran.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 18 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 25 ° C to 140 ° C.
- the compound (I) of the present invention can also be produced, for example, according to the method shown in Scheme 2 below or a method similar thereto.
- step e synthetic intermediate 8 is produced from synthetic intermediate 6 and synthetic intermediate 7.
- the synthetic intermediate 7 can be used, for example, 1 to 1.5 equivalents, preferably 1 to 1.2 equivalents
- n-butyllithium can be used, for example, 1 to 2 equivalents, preferably 1 to 1.5 equivalents.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 10 hours, preferably 1 hour to 4 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually ⁇ 80 ° C. to 25 ° C., preferably ⁇ 78 ° C. to 25 ° C.
- step f synthetic intermediate 9 is produced from synthetic intermediate 8.
- synthetic intermediate 8 trichloroacetamide can be used, for example 1-3 equivalents, preferably 1-2 equivalents, and 1,8-diazabicyclo [5,4,0] -7-undecene, eg 0.01-0.1.
- Equivalents preferably 0.01-0.05 equivalents, can be used, for example 1-3 equivalents, preferably 1-2.5 equivalents of dimethylketenmethyltrimethylsilylacetal, and bis (trifluoromethanesulfonyl) imide, for example 0.01-0.1.
- An equivalent, preferably 0.05 to 0.1 equivalent, can be used.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, tetrahydrofuran, acetonitrile and the like, preferably acetonitrile.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 10 hours, preferably 1 hour to 5 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 25 ° C, preferably 10 ° C to 25 ° C.
- step g synthetic intermediate 10 is produced from synthetic intermediate 9.
- synthetic intermediate 9 2,3-dichloro-5,6-dicyano-p-benzoquinone can be used, for example, 1 to 3 equivalents, preferably 1 to 2.5 equivalents.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include a mixed solvent of dichloromethane and water.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 5 hours, preferably 1 hour to 3 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 25 ° C, preferably 10 ° C to 25 ° C.
- synthetic intermediate 12 is produced from synthetic intermediate 10 and synthetic intermediate 11.
- the synthetic intermediate 11 can be used, for example, 1 to 2 equivalents, preferably 1 to 1.2 equivalents
- triphenylphosphine can be used, for example, 1 to 2 equivalents, preferably 1 to 1.2 equivalents.
- diethylazodicarboxylate can be used, for example, 1 to 1.5 equivalents, preferably 1 to 1.2 equivalents.
- diethyl azodicarboxylate bis (2-methoxyethyl) azodicarboxylate, diisopropylazodicarboxylate, cyanomethylenetributylphosphorane and the like can also be used.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, 1,4-dioxane, tetrahydrofuran, toluene, N, N-dimethylformamide and the like, and tetrahydrofuran is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 5 to 8 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 40 ° C, preferably 0 ° C to 25 ° C.
- the synthetic intermediates 2, synthetic intermediates 6, and synthetic intermediates 11 of Shcheme 1 and 2 are prepared by known methods, for example, the methods described in Patent Document 1 (WO2015 / 092713), WO2016 / 202253, or similar methods. For example, it can be manufactured according to Scheme 3 below.
- step i the synthetic intermediate 14 is produced by the amination reaction of the synthetic intermediate 13.
- Ammonia water is used in excess of the synthetic intermediate 13.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 18 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 25 ° C to 40 ° C.
- step j the synthetic intermediate 14 and the synthetic intermediate 15 are reacted to produce the synthetic intermediate 11 via the synthetic intermediate 16.
- the synthetic intermediate 14 and the synthetic intermediate 15 are reacted in the presence of a base.
- the base can be used, for example, 1 to 2 equivalents, preferably 1 to 1.5 equivalents
- the synthetic intermediate 15 can be used, for example, 0.8 to 1.5 equivalents, preferably 0.8 to 1.2 equivalents, relative to the synthetic intermediate 14.
- Examples of the base include potassium carbonate, sodium carbonate and the like, and preferably potassium carbonate can be mentioned.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include a mixed solvent of water and an organic solvent such as tetrahydrofuran or dimethylformamide, and preferably a mixed solvent of tetrahydrofuran and water. ..
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 3 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 25 ° C to 40 ° C.
- the synthetic intermediate 11 is produced from the synthetic intermediate 16 in the presence of a base.
- a base for example, 1 to 5 equivalents, preferably 2 to 3 equivalents of the base can be used with respect to the synthetic intermediate 16.
- the base include potassium tert-butoxide, potassium carbonate, potassium hydroxide and the like, and preferably potassium tert-butoxide.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dimethyl sulfoxide and dimethylformamide, and dimethyl sulfoxide is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 3 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 60 ° C to 100 ° C.
- the synthetic intermediate 18 is produced by the reduction reaction of the synthetic intermediate 17.
- 1 to 2 equivalents, preferably 1 to 1.5 equivalents of the reducing agent can be used with respect to the synthetic intermediate 17.
- the reducing agent include borane-tetrahydrofuran complex and borane-dimethyl sulfide complex.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and tetrahydrofuran can be used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 40 hours, preferably 1 hour to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 25 ° C to 150 ° C, preferably 25 ° C to 40 ° C.
- step l the synthetic intermediate 19 is reacted with bispinacolat diboron in the presence of a base and a catalyst to produce the synthetic intermediate 2.
- the base can be used, for example, 1-3 equivalents, preferably 1-2.5 equivalents
- the catalyst can be used, for example 0.05-0.1 equivalents, preferably 0.05-0.08 equivalents, bispina.
- 1 to 2 equivalents, preferably 1 to 1.5 equivalents of korat diboron can be used.
- the base for example, potassium acetate or the like can be used.
- the catalyst examples include palladium catalysts such as PdCl 2 (dppf) 2 ⁇ CH 2 Cl 2 and Pd 2 (dba) 3 , and preferably PdCl 2 (dppf) 2 ⁇ CH 2 Cl 2 can be mentioned.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include 1,4-dioxane, tetrahydrofuran, dimethyl sulfoxide, N, N-dimethylformamide and the like, and 1,4-dioxane is preferable. ..
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 5 to 8 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 60 ° C to 120 ° C, preferably 60 ° C to 100 ° C.
- step m synthetic intermediate 20 is produced from synthetic intermediate 18.
- Sodium hydride can be used, for example 1-2 equivalents, preferably 1-1.5 equivalents
- p-methoxybenzyl chloride can be used, for example 1-2 equivalents, preferably 1-1.2 equivalents, relative to the synthetic intermediate 20. be able to.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran, N, N-dimethylformamide and the like, and preferably N, N-dimethylformamide.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 5 hours to 15 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 60 ° C, preferably 0 ° C to 25 ° C.
- step n synthetic intermediate 6 is produced from synthetic intermediate 20.
- synthetic intermediate 20 n-butyllithium can be used, for example, 1 to 1.5 equivalents, preferably 1 to 1.2 equivalents, and N, N-dimethylformamide can be used, for example, 1 to 2 equivalents, preferably 1 to 1.5 equivalents.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 10 hours, preferably 1 hour to 3 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually ⁇ 80 ° C. to 25 ° C., preferably ⁇ 78 ° C. to 25 ° C.
- Synthetic intermediate 3 can be obtained, for example, by the method shown in Schemes 4 to 7 below or a method similar thereto.
- step o the synthetic intermediate 21 and N, O-dimethylhydroxylammonium hydrochloride are reacted in the presence of a base, a condensing agent and an additive to produce the synthetic intermediate 22.
- a base a condensing agent
- an additive to produce the synthetic intermediate 22.
- N, O-dimethylhydroxylamine hydrochloride can be usually used in an amount of 1 to 3 equivalents, preferably 1.5 to 2 equivalents.
- the condensing agent can be usually used in an amount of 1 to 3 equivalents, preferably 1 to 2 equivalents
- the additive can be used in an amount of usually 0.1 to 2 equivalents, preferably 1 to 2 equivalents, relative to the synthetic intermediate 21.
- the base can usually be used in an amount of 0.5 to 5 equivalents, preferably 2 to 4 equivalents.
- condensing agent examples include N, N'-dicyclohexylcarbodiimide, 1,1'-carbonyldiimidazole, 1-ethyl-3- [3- (dimethylamino) propyl] carbodiimide hydrochloride, and benzotriazole-1-yloxy.
- Tripyrolidinophosphonium hexafluorophosphate O- (benzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate, O- (7-azabenzotriazole-) 1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate, diethyl cyanophosphate, diphenylphosphoryl azide, pentafluoroacetyl trifluoroacetate, isopropylchloroformate and the like can be mentioned.
- 1-ethyl-3- [3- (dimethylamino) propyl] carbodiimide hydrochloride or O- (7-azabenzotriazole-1-yl) -N, N, N', N'-tetramethyluronium Hexafluorophosphate can be mentioned.
- the additive include 1-hydroxybenzotriazole, 1-hydroxy-7-azabenzotriazole, N-hydroxysuccinimide and the like, and 1-hydroxybenzotriazole is preferable.
- Examples of the base include triethylamine, pyridine, N, N-diisopropylethylamine, 4-dimethylaminopyridine and the like, and preferably triethylamine, N, N-diisopropylethylamine or 4-dimethylaminopyridine.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether, acetonitrile, N, N-dimethylformamide and the like.
- reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 25 ° C to 40 ° C.
- synthetic intermediate 23 or synthetic intermediate 25 is produced from synthetic intermediate 22 or synthetic intermediate 24 by a bromoization reaction.
- N-bromosuccinimide can usually be used in an amount of usually 1-5 equivalents, preferably 2-3 equivalents, and trifluoroacetic acid is usually 1-3 equivalents, preferably 1-to. 2 equivalents can be used.
- trifluoroacetic acid instead of trifluoroacetic acid, concentrated sulfuric acid, concentrated hydrochloric acid, acetic acid and the like can also be used.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, carbon tetrachloride, 1,2-dimethoxyethane, N, N-dimethylformamide and the like, and N, N-dimethyl is preferable.
- Formamide can be mentioned.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 25 ° C to 40 ° C.
- step q synthetic intermediate 25 is produced from synthetic intermediate 23.
- 1 to 3 equivalents, preferably 1 to 1.5 equivalents of methylmagnesium bromide can be used with respect to the synthetic intermediate 23.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether and the like, and tetrahydrofuran is preferable. it can.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 0 ° C to 40 ° C.
- step r the synthetic intermediate 25 is reacted with ethylene glycol in the presence of an acid catalyst to produce the synthetic intermediate 26.
- the acid catalyst can be used, for example, 0.1 to 5 equivalents, preferably 0.1 to 0.5 equivalents, and ethylene glycol can be used, for example, 5 to 20 equivalents, preferably 10 to 15 equivalents, relative to the synthetic intermediate 25.
- the acid catalyst include p-toluenesulfonic acid monohydrate, hydrochloric acid, trifluoroacetic acid and the like, and p-toluenesulfonic acid monohydrate is preferable.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include toluene, benzene and the like, and toluene is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 40 ° C to 150 ° C, preferably 100 ° C to 130 ° C.
- the synthetic intermediate 25 or the synthetic intermediate 26 is reacted with methyl acrylate in the presence of a catalyst, a base, and an additive to produce the synthetic intermediate 27.
- the catalyst can be used, for example, 0.05 to 0.2 equivalents, preferably 0.05 to 0.1 equivalents
- the base can be used, for example, 1.5 to 5 equivalents, preferably 1.5 to 3 equivalents.
- the additive can be used, for example, 0.05 to 0.2 equivalents, preferably 0.05 to 0.1 equivalents, and methyl acrylate can be used, for example, 1.5 to 3 equivalents, preferably 1.5 to 2 equivalents.
- Examples of the catalyst include palladium catalysts such as PdCl 2 (dppf) 2 , CHCl 3 , Pd (OAc) 2 , PdCl 2 (PPh 3 ) 2 , Pd (PPh 3 ) 4, and the like, preferably PdCl 2 (dppf). 2.
- CHCl 3 can be mentioned.
- Examples of the base include N, N-dicyclohexylmethylamine, triethylamine, pyridine, N, N-diisopropylethylamine and the like, and preferably N, N-dicyclohexylmethylamine.
- the additive examples include tetrabutylammonium chloride, tetrabutylammonium iodide and the like, and preferably tetrabutylammonium chloride and the like.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dimethylformamide and dimethylacetamide, and dimethylacetamide is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 1 hour to 8 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 40 ° C. to 150 ° C., preferably 100 ° C. to 140 ° C. using microwaves.
- Scheme 5 is performed according to steps o and s of Scheme 4, and in step o, the corresponding amine is used instead of N, O-dimethylhydroxylamine hydrochloride.
- step t synthetic intermediate 29 is produced from synthetic intermediate 28.
- 1 to 3 equivalents, preferably 1 to 1.5 equivalents of methylmagnesium bromide can be used with respect to the synthetic intermediate 28.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether and the like, and tetrahydrofuran is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 0 ° C to 40 ° C.
- the synthetic intermediate 29 is oxidized by the sulfur trioxide / pyridine complex in the presence of a base to produce the synthetic intermediate 30.
- a base for example, 1 to 5 equivalents, preferably 1 to 3 equivalents of the base can be used with respect to the synthetic intermediate 29, and 1 to 3 equivalents, preferably 1 to 2.5 equivalents of the sulfur trioxide / pyridine complex can be used.
- the base include triethylamine, pyridine, N, N-diisopropylethylamine, 4-dimethylaminopyridine and the like, and preferably triethylamine.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and dimethyl sulfoxide can be preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 0 ° C to 40 ° C.
- the synthetic intermediate 3 can be produced according to a known method, for example, the method described in JP2008 / 273924, or a method similar thereto, for example, Scheme 7 below.
- step v the synthetic intermediate 31 is reacted with tert-butylamine and acetic acid to produce the synthetic intermediate 32.
- tert-butylamine can be used, for example, 1 to 3 equivalents, preferably 1 to 1.5 equivalents
- acetic acid can be used, for example, 1 to 20 equivalents, preferably 2.5 to 5 equivalents.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, toluene and the like, preferably dichloromethane.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 5 hours, preferably 2 to 4 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 40 ° C, preferably 10 to 30 ° C.
- step w synthetic intermediate 34 is produced by the deprotection reaction of tert-butylamine of synthetic intermediate 33.
- a mixed solvent of trifluoroacetic acid and water can be preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 5 hours, preferably 2 to 4 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 150 ° C, preferably 90 ° C to 110 ° C.
- the compound (I) of the present invention can also be produced in Scheme 2 using synthetic intermediate 38 instead of synthetic intermediate 10.
- the synthetic intermediate 38 can also be produced, for example, according to the method shown in Scheme 8 below or a method similar thereto.
- the compound (I) of the present invention can also be produced in Scheme 1 using synthetic intermediate 47 instead of synthetic intermediate 2.
- the synthetic intermediate 47 can also be produced, for example, according to the method shown in Scheme 9 below or a method similar thereto.
- step x synthetic intermediate 40 is produced from synthetic intermediate 39.
- 1 to 2 equivalents, preferably 1 to 1.2 equivalents of bromine can be used with respect to the synthetic intermediate 39.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include acetic acid and trifluoroacetic acid, and acetic acid is preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 0.5 to 3 hours, preferably 1 to 2 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 40 ° C, preferably 0 ° C to 25 ° C.
- step y synthetic intermediate 41 is produced from synthetic intermediate 40.
- 1 to 10 equivalents, preferably 2 to 5 equivalents of tin (II) chloride can be used with respect to the synthetic intermediate 40.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include methanol, ethanol, propanol and isopropanol, and methanol is preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 6 hours, preferably 1.5 to 4 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 60 ° C to 80 ° C.
- step z the synthetic intermediate 42 is produced from the synthetic intermediate 41. Excess formic acid is used in excess of synthetic intermediate 41.
- the reaction time is usually 0.5 to 5 hours, preferably 1 to 2 hours.
- the reaction temperature is usually 25 ° C to 150 ° C, preferably 90 ° C to 110 ° C.
- step a-1 the synthetic intermediate 43 is produced from the synthetic intermediate 42. Trityl chloride can be used, for example, 1 to 3 equivalents, preferably 1.1 to 1.3 equivalents, and the base can be used, for example, 1 to 3 equivalents, preferably 1.1 to 1.7 equivalents, relative to the synthetic intermediate 42.
- Examples of the base include triethylamine, N, N-diisopropylethylamine, pyridine, 4-dimethylaminopyridine, N-methylmorpholine and the like, and preferably N-methylmorpholine or triethylamine.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether, acetonitrile, N, N-dimethylformamide and the like. And preferably dichloromethane can be mentioned.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 12 hours, preferably 1 to 2 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 40 ° C to 60 ° C.
- Step a-2 manufactures synthetic intermediate 44 from synthetic intermediate 43.
- 1 to 5 equivalents, preferably 1.5 to 3 equivalents of the reducing agent can be used with respect to the synthetic intermediate 43.
- the reducing agent include lithium borohydride, lithium aluminum hydride, lithium borohydride, borane-tetrahydrofuran complex and the like, and lithium borohydride is preferable.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran, diethyl ether, 1,2-dimethoxyethane and the like, and tetrahydrofuran is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 0.2 to 3 hours, preferably 0.5 to 1.5 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 60 ° C, preferably 0 ° C to 30 ° C.
- step a-3 methanesulfonyl chloride and lithium chloride are reacted in the presence of a base to produce synthetic intermediate 45 from synthetic intermediate 44.
- a base for the synthetic intermediate 44, methanesulfonyl chloride can be used, for example, 1-5 equivalents, preferably 1.5-2.5 equivalents, and lithium chloride can be used, for example, 1-10 equivalents, preferably 3-5 equivalents.
- 1 to 10 equivalents, preferably 3 to 8 equivalents of the base can be used.
- Examples of the base include triethylamine, N, N-diisopropylethylamine, pyridine, 4-dimethylaminopyridine, N-methylmorpholine and the like, and N, N-diisopropylethylamine is preferable.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, N, N-dimethylformamide and the like, and dichloromethane is preferable.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 1 to 12 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 40 ° C, preferably 0 ° C to 25 ° C.
- step a-4 synthetic intermediate 45 is reacted with synthetic intermediate 11 and tetrabutylammonium iodide in the presence of a base to produce synthetic intermediate 46.
- Synthetic intermediate 11 can be used, for example, 1 to 3 equivalents, preferably 1 to 1.2 equivalents, and tetrabutylammonium iodide, for example, 0.1 to 1 equivalents, preferably 0.1 to 0.2 equivalents, relative to synthetic intermediate 45.
- the base can be used, for example, 1 to 5 equivalents, preferably 1 to 3 equivalents. Examples of the base include potassium carbonate, sodium carbonate, sodium hydrogencarbonate and the like, and potassium carbonate is preferable.
- the reaction is usually carried out in a solvent.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, tetrahydrofuran, 1,4-dioxane, N, N-dimethylformamide, N, N-dimethylacetamide and the like, preferably N, N-. Dimethylformamide can be mentioned.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 1 to 18 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 60 ° C, preferably 25 ° C to 40 ° C.
- the compound (I) of the present invention can also be produced, for example, according to the method shown in Scheme 10 below or a method similar thereto.
- Processes a-3 and a-4 of Scheme 10 are performed according to Scheme 9, and process d is performed according to Scheme 1.
- the compound (I) of the present invention can also be produced, for example, according to the method shown in Scheme 11 below or a method similar thereto.
- Processes c and d of Scheme 11 are performed according to Scheme 1, process g is performed according to Scheme 2, and steps a-3 and a-4 are performed according to Scheme 9.
- step a-5 compound 49 is produced from compound 9.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include tetrahydrofuran, N, N-dimethylformamide, and tetrahydrofuran is preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 1 to 3 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 25 ° C to 40 ° C.
- step a-6 compound 50 is produced from compound 49.
- Manganese dioxide is used in an amount of 10 to 50 equivalents, preferably 25 to 50 equivalents, relative to compound 49.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, acetone, benzene, toluene, tetrahydrofuran, 1,4-dioxane and the like, and dichloromethane is preferably used.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 to 24 hours, preferably 2 to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 25 ° C to 40 ° C.
- step a-7 compound 52 is produced from compound 51.
- TMS ethanol is used in an amount of 1-5 equivalents, preferably 3-5 equivalents, relative to compound 51.
- the condensing agent is usually 1 to 3 equivalents, preferably 1.5 to 2 equivalents, and the base is usually 1 to 5 equivalents, preferably 2 to 5 equivalents.
- Examples of the condensing agent include N, N'-dicyclohexylcarbodiimide, 1,1'-carbonyldiimidazole, 1-ethyl-3- [3- (dimethylamino) propyl] carbodiimide hydrochloride, and benzotriazole-1-yloxy.
- Tripyrolidinophosphonium hexafluorophosphate O- (benzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate, O- (7-azabenzotriazole-) 1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate, diethyl cyanophosphate, diphenylphosphoryl azide, pentafluoroacetate pentafluorophenyl ester, isopropylchloroformate and the like can be mentioned.
- O- (benzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate or O- (7-azabenzotriazole-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate can be mentioned.
- the base include triethylamine, pyridine, N, N-diisopropylethylamine, 4-dimethylaminopyridine and the like, and preferably triethylamine, N, N-diisopropylethylamine or 4-dimethylaminopyridine.
- the solvent is not particularly limited as long as it does not adversely affect the reaction, and examples thereof include dichloromethane, chloroform, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, diethyl ether, acetonitrile, N, N-dimethylformamide and the like. And preferably, dichloromethane or N, N-dimethylformamide can be mentioned.
- the reaction time varies depending on the reagent or solvent used, but is usually 1 hour to 24 hours, preferably 3 hours to 24 hours.
- the reaction temperature varies depending on the reagent or solvent used, but is usually 0 ° C to 100 ° C, preferably 25 ° C to 40 ° C.
- the produced compound (I) of the present invention can be isolated or purified as a free form or as a salt thereof by subjecting it to a salt forming treatment by a conventional method.
- the isolation or purification method is not particularly limited, and for example, commonly used methods such as crystallization, recrystallization, distillation, liquid separation, and chromatography can be appropriately selected and combined. If a plurality of isomers are present, purification can give a single isomer or a mixture of the plurality of isomers. When a plurality of single isomers or a plurality of isomer mixtures are obtained by purification, each of them can be used as it is or a mixture thereof can be used as the compound (I) of the present invention.
- the solvate of the compound represented by the general formula (I) can be obtained according to a method known per se.
- the structure of the product can be identified by elemental analysis, MS (ESI-MS) analysis, IR analysis, 1 H-NMR, 13 C-NMR and the like.
- Compound (I) of the present invention has an Nrf2 activating action. Therefore, at least one selected from the group consisting of the compound represented by the general formula (I), a salt thereof, and a solvate thereof (in the present specification, it may be referred to as "the active ingredient of the present invention”. ) Can be used, for example, as a medicine, a reagent, or the like. From this point of view, the present invention relates to, in one aspect, a pharmaceutical product (sometimes referred to as "the pharmaceutical product of the present invention” in the present specification) containing the active ingredient of the present invention.
- the medicament of the present invention can be used as an Nrf2 activator, an oxidative stress protection gene expression activator, etc. based on the action of the active ingredient of the present invention.
- the medicament of the present invention can be used for the prevention or treatment of various diseases.
- the target disease is not particularly limited as long as it can be prevented or treated based on the above-mentioned action of the active ingredient of the present invention.
- the medicament of the present invention can be used for the prevention or treatment of diseases or symptoms related to the regulation of Nrf2 activity.
- Treatment means preventing the deterioration of the condition when a disorder or disease develops, delaying the progression, and maintaining, alleviating or eliminating the condition, and "prevention” means the disorder or disease. It means to prevent the onset of the disease before the onset of the disease.
- Target diseases include, for example, in ophthalmic diseases, for example, dry eye, diabetic retinopathy, retinal pigment degeneration, glaucoma, cataract, age-related luteal degeneration, proliferative vitreous retinopathy, retinal artery occlusion, retinal vein occlusion. , Vinitis, Label's disease, premature infant retinopathy, retinal detachment, retinal pigment epithelial detachment, retinal neuropathy caused by these diseases, optic neuropathy caused by glaucoma, ischemic optic neuropathy, postoperative inflammation suppression, pain suppression And so on.
- ophthalmic diseases for example, dry eye, diabetic retinopathy, retinal pigment degeneration, glaucoma, cataract, age-related luteal degeneration, proliferative vitreous retinopathy, retinal artery occlusion, retinal vein occlusion.
- Vinitis Label's disease, premature infant retinopathy, retinal detachment, retinal pigment epi
- skin diseases for example, UV-responsive skin damage, epidermal foam disease, etc., psoriasis, atopic dermatitis, strong skin disease, etc.
- otolaryngology diseases for example, noise-induced hearing loss, sensory hearing loss, etc.
- lung diseases such as Alzheimer's disease, Parkinson's disease, Huntington's disease, Batten's disease, dementia, epilepsy, etc., for example, asthma, pulmonary bronchitis, chronic obstructive pulmonary disease, etc.
- renal diseases for example, diabetic kidney.
- systemic diseases such as disease, Alport syndrome, autosomal dominant multiple cystic kidney, focal segmental glomerulosclerosis, IgA nephropathy, etc.
- arteriosclerosis hypertension, cancer, cardiac arrest, Friedrich ataxia Disease, muscular atrophic sclerosis, hepatitis, rheumatoid arthritis, pancreatitis, vasculitis, esophagitis, ulcerative colitis, neutrophilia, cellular immunity, diabetes, mitochondrial myopathy, sickle red anemia
- ophthalmic diseases such as multiple sclerosis, psoriasis, Alzheimer's disease, retinitis pigmentosa, glaucoma, and cataract are preferable.
- the administration target of the medicament of the present invention is not particularly limited, and examples thereof include mammals (for example, humans, mice, rats, hamsters, rabbits, cats, dogs, cows, sheep, monkeys, etc.).
- the route of administration of the medicament of the present invention is not particularly limited.
- enteral administration such as oral administration, tube feeding, and enema administration
- parenteral administration such as intravenous administration, transarterial administration, intramuscular administration, intracardiac administration, subcutaneous administration, intradermal administration, and intraperitoneal administration
- Topical ocular administration eg, eye drops, intravitreal administration, subconjunctival administration, subcapsular administration, etc.
- intradermal administration inhalation administration, enema administration, ear drops, nasal administration, intravaginal administration, etc.
- topical administration and parenteral administration are preferable.
- the medicament of the present invention preferably takes a formulation suitable for these preferred administration routes, and from this viewpoint, topical administration preparations and parenteral administration preparations are preferable.
- the medicament of the present invention can be an orally administered preparation or the like.
- the medicament of the present invention can be an ophthalmic preparation, an otolaryngological preparation, a respiratory preparation, a dermatological preparation, or the like.
- the medicament of the present invention can be an intravenous administration preparation, a nasal administration preparation, a transdermal administration preparation, or the like.
- the drug of the present invention can take a dosage form suitable for the route of administration.
- a dosage form suitable for the route of administration For example, eye drops, eye ointments, eye wash, injections, patches, lotions, creams, powders, granules, tablets, capsules, syrups, liquids, ointments, gels, liniments, suppositories, sprays. , Inhalants, sprays, nasal drops and the like.
- the active ingredient of the present invention can also be a preparation for intraocular implants, a preparation in the form of a DDS (drug delivery system) such as microspheres, and the like.
- DDS drug delivery system
- the medicament of the present invention may consist of only the active ingredient of the present invention, or may be referred to simply as an "additive" in the present specification as a pharmaceutically acceptable additive, if necessary. ) Can also be a composition containing.
- the pharmaceutical product of the present invention can be produced by mixing the active ingredient of the present invention with at least one additive or the like and following a method known per se in the technical field of pharmaceutical preparations.
- the additive can be appropriately selected depending on the form of the preparation preferable for administration.
- the content of the active ingredient of the present invention in the medicine varies depending on the dosage form, dosage and the like, and can be appropriately selected. For example, it can be usually 0.01 to 99.9% by mass, preferably 0.1 to 80% by mass of the whole drug.
- Additives include, for example, bases, carriers, solvents, dispersants, emulsifiers, buffers, stabilizers, excipients, binders, disintegrants, lubricants, thickeners, moisturizers, colorants, fragrances, etc. And a chelating agent and the like. These can be appropriately selected depending on the administration route, dosage form and the like.
- water purified or sterile water, distilled water for injection, etc.
- physiological saline physiological saline
- glucose solution glucose solution
- water-soluble organic solvent ethanol, isopropanol, etc.
- Lower aliphatic alcohols ethylene glycol, diethylene glycol, polyalkylene glycols such as polyethylene glycol), animal and plant oils (vegetable oils such as jojoba oil, olive oil, palm oil, cottonseed oil; animal oils such as squalane)
- Mineral oils liquid paraffin, silicone oil, etc.
- waxes honey wax, carnauba wax, lanolin, paraffin, vaseline, etc.
- long-chain fatty acid esters saturated or unsaturated fatty acid alkyl esters, fatty acids and polyhydric alcohols (poly C2-) 4 Esters with alkylene glycols, glycerin or polyglycerin), hardened oils, higher alcohols (saturated aliphatic alcohols such as stearyl alcohols, unsaturated aliphatic alcohols such as oleyl alcohols), higher fatty acids (stearic acid, olein, etc.) Acids, etc.) can also be added.
- preservatives or preservatives parabens such as methylparaben and butylparaben
- deodorants or fragrances fragments such as menthol
- refreshing agents painkillers (local anesthetics such as lidocaine), etc.
- Active ingredients for the treatment of infectious diseases and inflammation can also be blended.
- the amount of these additives added varies depending on the type of additive to be added, the intended use, and the like, but a concentration that can achieve the purpose of the additive may be added.
- examples of the solvent include purified water, ethanol, isopropanol, dipropylene glycol and the like, and one or more of these may be selected.
- animal and vegetable fats and oils such as olive oil, soybean oil, camellia oil, sesame oil, peanut oil, cacao butter, beef tallow, and pork fat; waxes such as carnauba wax and honey wax; fatty acids such as octyldodecanol, cetanol, and stearyl alcohol.
- Alcohols such as oleic acid, palmitic acid, stearic acid; oily bases such as hydrocarbons such as squalane, white petrolatum, liquid paraffin, selecin, microcrystalin wax, and hydrophilic bases such as gelatin and macrogol. , One or more of these can be selected and used.
- excipients such as light anhydrous silicic acid, crystalline cellulose, and dextrin; solubilizers such as diisopropyl adipate, capric acid, crotamitone, and propylene carbonate; propylene glycol alginate, sodium dioctyl sulfosuccinate, Sustaining agents for soy lecithin, povidone, etc .; polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan monolaurate, polyoxyethylene sorbitan monostearate, polyoxyethylene sorbitan monooleate, glycerin monostearate, sucrose fatty acid Surfactants or emulsifiers such as esters; viscous agents such as carboxyvinyl polymers, xanthan gum, carboxymethyl cellulose, hydroxypropyl cellulose, polyvinyl alcohol (partially saponified); plastic agents such as triacetin, isopropyl myristate
- the amount of these additives added varies depending on the type of additive to be added, the intended use, and the like, but a concentration that can achieve the purpose of the additive may be added.
- a concentration that can achieve the purpose of the additive may be added.
- stabilizers for example, sodium hydrogen sulfite, sodium thiosulfate, sodium edetate, etc.
- the drug of the present invention When used as an otolaryngological composition such as an ear drop or a nasal drop, it may be manufactured according to a method usually used in the field of formulation.
- a method usually used in the field of formulation For example, the 17th revised Japanese Pharmacopoeia, General Rules for Formulation, " It can be produced according to the method described in the section "Preparations to be administered to the ear” or “Preparations to be applied to the nose".
- the drug of the present invention When used as a dermatological composition such as an external preparation for skin, it may be produced according to a method usually used in the field of preparation. For example, the 17th revised Japanese Pharmacopoeia, General Rules for Preparation, "Applicable to skin, etc.” It can be produced according to the method described in the section "Preparation to be prepared”.
- the medicament of the present invention When the medicament of the present invention is used as an ophthalmic composition such as an eye drop or an eye ointment, it may be produced according to a method usually used in the field of formulation.
- the 17th revised Japanese Pharmacopoeia, General Rules for Formulation, "Eyes” can be produced based on the method described in the section of "drugs to be administered” (for example, the section of eye drops and the section of eye ointment).
- the dose of the medicament of the present invention varies depending on the target disease and cannot be unequivocally determined, but the concentration of the active ingredient of the present invention in the target tissue exerting the effect is, for example, 0.001 nM to 100 ⁇ M, preferably 0.01 nM. It can be set to ⁇ 100 ⁇ M.
- a drug containing the active ingredient of the present invention of 0.01 nM to 1000 ⁇ M, preferably 0.1 nM to 1000 ⁇ M, is preferably applied 1 to 8 times a day. Should be applied 1 to 5 times.
- the applicable amount can be appropriately set according to the concentration and dosage form of the active ingredient of the present invention.
- the medicament of the present invention may contain one or more of any other medicinal ingredients as long as the effects of the present invention are not impaired.
- Reference Examples and Examples were usually carried out in an anhydrous solvent under an argon atmosphere.
- the reaction was analyzed by TLC (Thin Layer Chromatography) and judged by the consumption of the starting material and terminated.
- TLC Thin Layer Chromatography
- silica gel 60F254 Merck was used, developed with an appropriate solvent, displayed at an appropriate position, and a UV detector was used for detection. Unless otherwise specified, elution in column chromatography of Reference Examples and Examples was performed under observation by TLC.
- Silica gel for column chromatography includes SI silica gel (particle size 30-50 ⁇ m) or NH silica gel (particle size 60 ⁇ m) manufactured by Fuji Silicia Chemical Co., Ltd., silica gel (particle size 40 ⁇ m) manufactured by Yamazen Co., Ltd., or amino silica gel (particle size 40 ⁇ m). A particle size of 40 ⁇ m) was used. Room temperature usually means a temperature of about 10 ° C to 35 ° C.
- the reaction solution was cooled to room temperature, ethyl acetate was added, the mixture was washed with water and saturated brine, the organic layer was dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- Tin (II) chloride (13.6 g, 72 mmol) was added to a solution of methyl 2-amino-5-bromo-3-nitrobenzoate (3.97 g, 14 mmol) in methanol (100 mL), and the mixture was stirred at 75 ° C. for 1.5 hours. .. The reaction solution was returned to room temperature, the reaction solution was concentrated under reduced pressure, water was added, 1 M aqueous sodium hydroxide solution (100 mL) and solid sodium hydrogen carbonate were added, the pH was adjusted to 9, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- the reaction solution was returned to room temperature, ethyl acetate was added, the mixture was washed with saturated aqueous sodium carbonate solution and saturated brine, dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- the obtained residue was dissolved in THF (12 mL), a 2M lithium borohydride THF solution (3.4 mL, 6.8 mmol) was added, and the mixture was stirred at room temperature for 1 hour. Water was added to the reaction solution to stop the reaction, and the mixture was extracted with ethyl acetate. The obtained organic layer was dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- N-Bromosuccinimide (1.17 g, 6.6 mmol) and TFA (1.0 mL, 13 mmol) in a DMF (6.0 mL) solution of 3-fluoro-N-methoxy-N-methylthiophen-2-carboxamide (620 mg, 3.3 mmol). ) was added.
- the reaction mixture was stirred at room temperature for 24 hours, 1N aqueous sodium hydroxide solution was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with water, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Methylmagnesium bromide (3M, 2-methyltetrahydrofuran solution) (0.3 mL, 0.9) in a THF (2.0 mL) solution of 2-bromo-N-methoxy-N-4-dimethylthiazole-5-carboxamide (209.0 mg, 0.8 mmol). The mmol) solution was added at 0 ° C. and stirred at room temperature for 10 minutes. The reaction was stopped by adding a saturated aqueous solution of ammonium chloride to the reaction solution, extraction was performed with ethyl acetate, the organic layer was washed with saturated brine, dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- Methylmagnesium bromide (3M, 2-methyltetrahydrofuran solution) (907 ⁇ L, 2.7 mol) was added to a solution of 5-bromothiophene-3-carbaldehyde (200 ⁇ L, 1.8 mmol) in THF (8.0 mL) at room temperature. Stirred for 1.5 hours. A saturated aqueous solution of ammonium chloride was added to this mixture to stop the reaction, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated aqueous ammonium chloride solution, dried over sodium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (503.8 mg, quant.) As a white solid.
- Example 123 The same procedure as in Example 123 was carried out to give the title compound (138.0 mg, 50%) as a pale yellow oily substance.
- Example 130 The same procedure as in Example 130 was carried out to give the title compound (22.2 mg, 21%) as a white solid.
- TEA 66 ⁇ L, 0.47 mmol
- the reaction solution was returned to room temperature, diluted with ethyl acetate, washed with water, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 130 The same procedure as in Example 130 was carried out to give the title compound (29.9 mg, 22%) as a colorless oil.
- Example 130 The same procedure as in Example 130 was carried out to give the title compound (52.0 mg, 29%) as a pale yellow oily substance.
- Example 130 The same procedure as in Example 130 was carried out to give the title compound (24.6 mg, 19%) as a light brown oily substance.
- a solution of diisopropylamine (30 ⁇ L, 0.21 mmol) in THF (200 ⁇ L) is cooled to -78 ° C, stirred for 10 minutes, and a 1.6 M solution of n-butyllithium hexane (110 ⁇ L, 0.18 mmol) at -78 ° C.
- the temperature was raised to -40 ° C, and then the mixture was stirred for 30 minutes.
- the reaction solution was cooled to room temperature, ethyl acetate was added, the mixture was washed with water and saturated brine, the organic layer was dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Reference example 173 3- (3- ⁇ [(R) -4-Ethyl-1,1-dioxide-3,4-dihydro-2H-benzo [b] [1,4,5] oxathiazepine-2-yl] methyl ⁇ -4 -Methylphenyl) -3- [4-fluoro-5- (1-hydroxyethyl) thiophen-2-yl] -2,2-Dimethylpropanoic acid production
- the reaction mixture was stirred at room temperature for 20 hours.
- the mixture was diluted with water and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (48.1 mg, 87%) as a colorless oil.
- the reaction solution was adjusted to be basic with saturated aqueous sodium hydrogen carbonate solution, extracted with ethyl acetate, dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
- reaction solution was diluted with ethyl acetate, washed with water and saturated brine, dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure to give the title compound (26.7 mg, 92%) as a brown transparent oily substance. ..
- the reaction solution was cooled to room temperature, ethyl acetate was added, the mixture was washed with water and saturated brine, the organic layer was dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Diabetes (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pulmonology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description

で表される化合物、その塩、又はそれらの溶媒和物。

で表される基である、項1~4のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
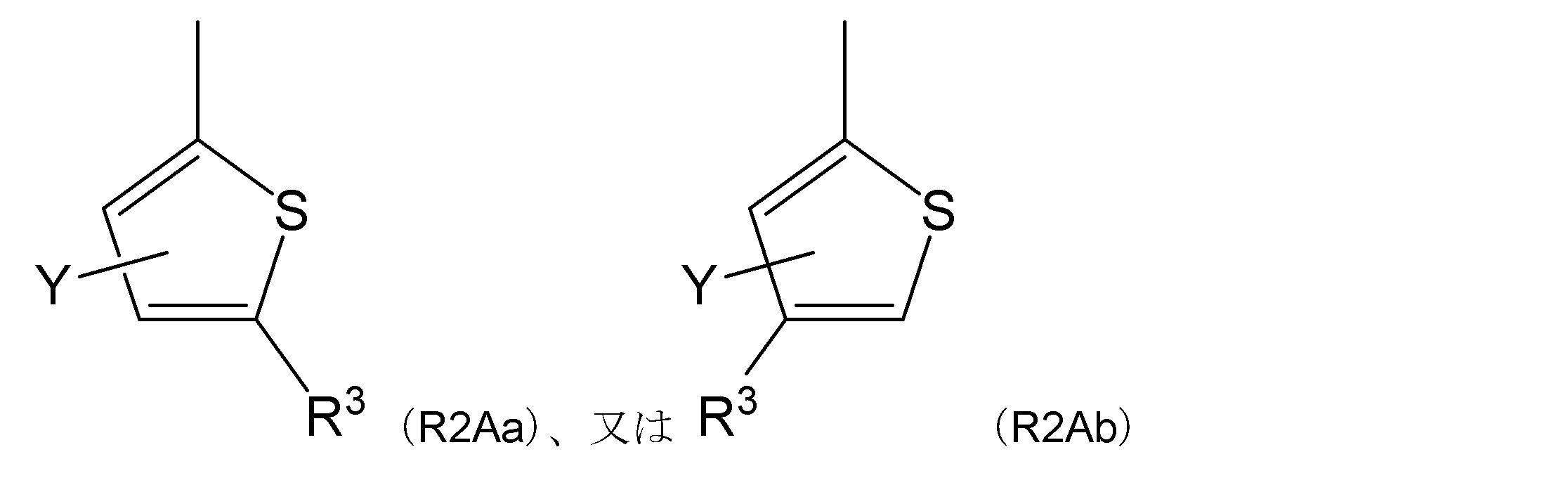
で表される基である、項1~5のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
前記R3がアセチル基であり、
前記R4及び前記R5が同一又は異なって、水素原子又はアルキル基であり、
前記R6が置換されていてもよいエチル基であり、
前記A1、前記A2、及び前記A4が全てCHであり、前記A3がCH又はNであり、
前記Yが水素原子、アルキル基、ハロゲン原子又はアルコキシ基であり、且つ
前記Zが水素原子である、
項5~9のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
前記R2が一般式(R2Aa):
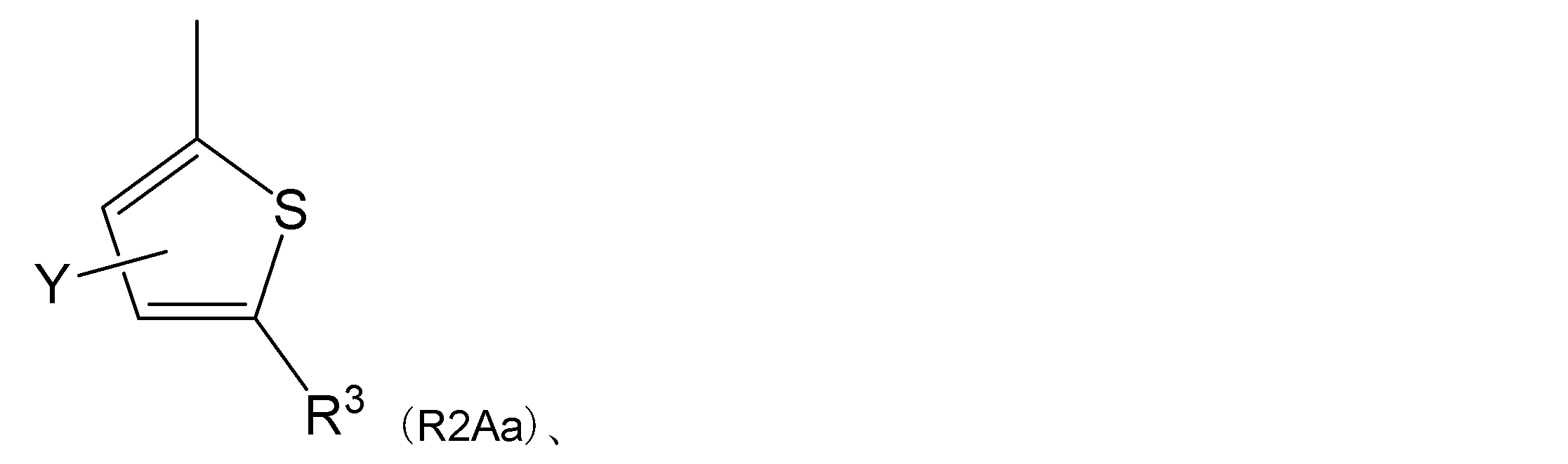
であり、
前記R4は水素原子であり、
前記R5はアルキル基であり、好ましくは該アルキル基の炭素数は1~3であり、
前記R6が置換されていてもよいエチル基であり、好ましくは該エチル基が有していてもよい置換基としてはアリールアルコキシ基であり、特に好ましくはベンジルオキシ基であり、
前記A1、前記A2、及び前記A4が全てCHであり、前記A3がCH又はNであり、且つ
前記Zが水素原子である、
項5~10のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物1)、
3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物2)、
3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物3)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物4)
3-(5-アセチルチオフェン-2-イル)-3-[3-({4-[2-(ベンジロキシ)エチル]-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル}メチル)-4-メチルフェニル]-2-メチルプロパン酸(化合物13)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物16)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物48)、
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物49)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物50)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物51)。
本発明は、その一態様において、一般式(I):

R1a及びR1bは同一又は異なって、水素原子、アルキル基又はハロゲン原子を示す。
R2は置換されていてもよいヘテロ環由来の基を示し、且つ前記ヘテロ環はチオフェン、フラン、ピロール、若しくはチアゾール、又はこれらのいずれかのヘテロ環を含む縮合環を示す。

で表される基である。

で表される基が挙げられ、さらに好ましくは一般式(R2Aa)又は(R2Ab):

で表される基が挙げられる。
R4及びR5は同一又は異なって、水素原子又は置換されていてもよいアルキル基を示す、或いはR4及びR5は互いに結合して-NH-CH=N-を形成する。

R6は置換されていてもよいアルキル基を示す。
A1、A2、A3及びA4は同一又は異なって、CH又はNを示す(但し、Nは1つ以下である。)。
Zは水素原子又はハロゲン原子を示す。
本発明の一態様において、一般式(I)の中でも、好ましくは一般式(IA):

が挙げられ、より好ましくは一般式(IAA):
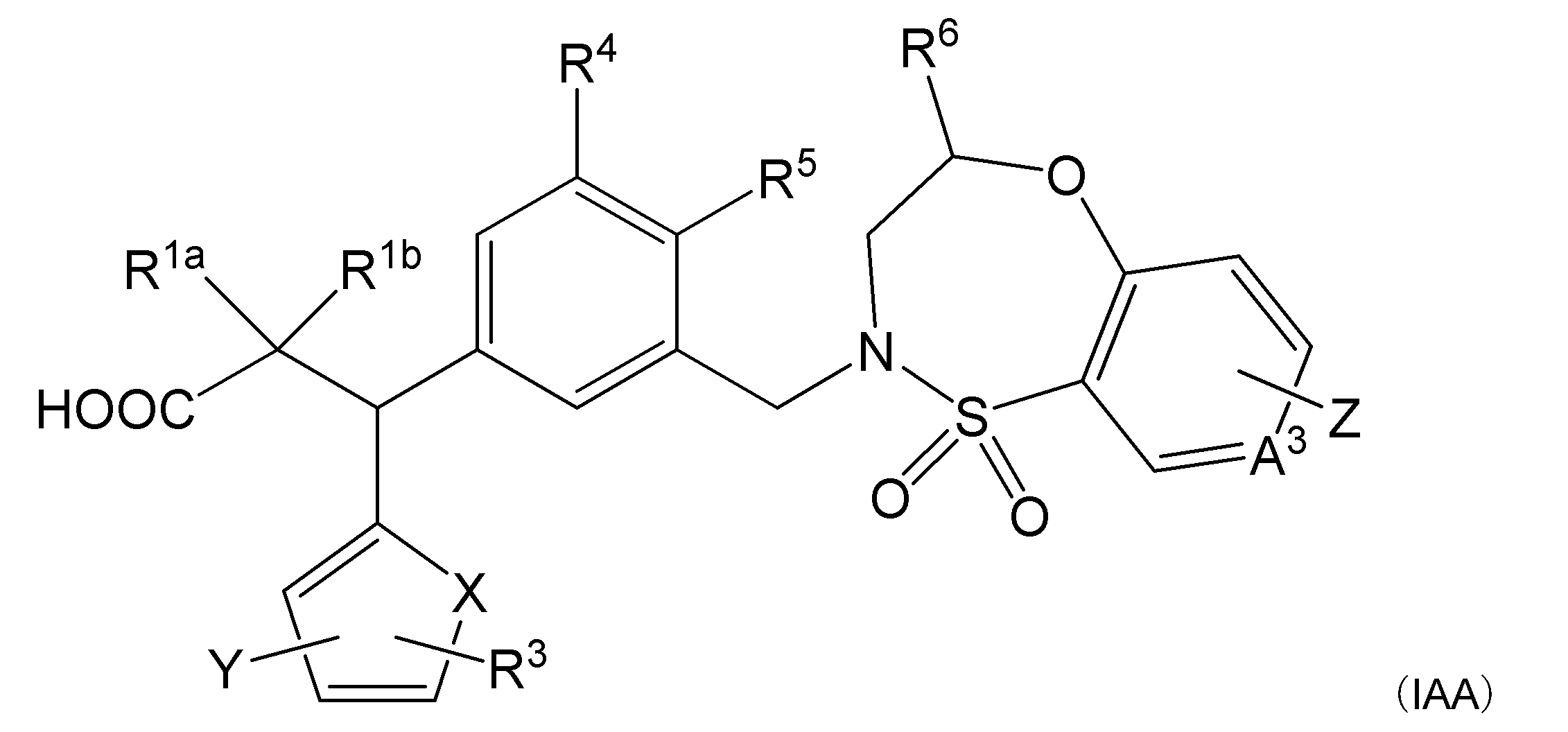
が挙げられ、さらに好ましくは一般式(IAAA):

が挙げられ、よりさらに好ましくは一般式(IAAAA1):

、又は一般式(IAAAA2):
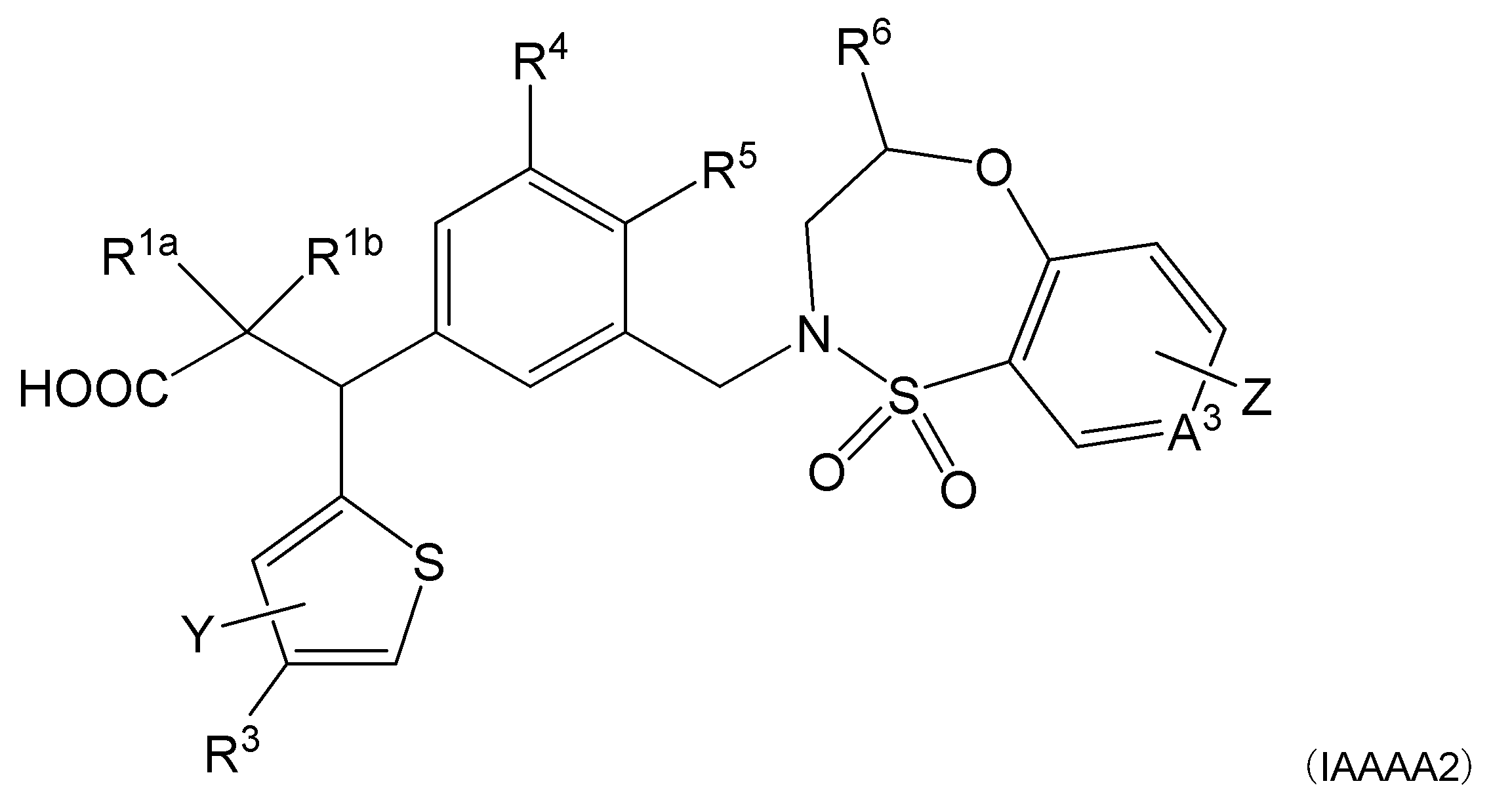
が挙げられる。
R1a及びR1bが同一又は異なって、水素原子又はアルキル基であり、
R3がアセチル基であり、
R4及びR5が同一又は異なって、水素原子又はアルキル基であり、
R6が置換されていてもよいエチル基であり、
A1、A2、及びA4が全てCHであり、A3がCH又はNであり、
Yが水素原子、アルキル基、ハロゲン原子又はアルコキシ基であり、且つ
Zが水素原子である、
ことが好ましい。
前記R2が一般式(R2Aa):

であり、
前記R4は水素原子であり、
前記R5はアルキル基であり、好ましくは該アルキル基の炭素数は1~3であり、
前記R6が置換されていてもよいエチル基であり、好ましくは該エチル基が有していてもよい置換基としてはアリールアルコキシ基であり、特に好ましくはベンジルオキシ基であり、
前記A1、前記A2、及び前記A4が全てCHであり、前記A3がCH又はNであり、且つ
前記Zが水素原子である、
ことが好ましい。
3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物2)、
3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物3)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物4)
3-(5-アセチルチオフェン-2-イル)-3-[3-({4-[2-(ベンジロキシ)エチル]-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル}メチル)-4-メチルフェニル]-2-メチルプロパン酸(化合物13)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物16)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物48)、
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物49)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物50)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物51)。
一般式(I)で表される化合物の塩は、薬学的に許容される塩である限り、特に制限されるものではない。該塩としては、酸性塩、塩基性塩のいずれも採用することができる。酸性塩の例としては、塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、リン酸塩等の無機酸塩; 酢酸塩、プロピオン酸塩、酒石酸塩、フマル酸塩、マレイン酸塩、リンゴ酸塩、クエン酸塩、メタンスルホン酸塩、パラトルエンスルホン酸塩等の有機酸塩が挙げられ、塩基性塩の例としては、ナトリウム塩、及びカリウム塩等のアルカリ金属塩; 並びにカルシウム塩、マグネシウム塩等のアルカリ土類金属塩; アンモニアとの塩; モルホリン、ピペリジン、ピロリジン、モノアルキルアミン、ジアルキルアミン、トリアルキルアミン、モノ(ヒドロキシアルキル)アミン、ジ(ヒドロキシアルキル)アミン、トリ(ヒドロキシアルキル)アミン等の有機アミンとの塩等が挙げられる。

本発明の化合物(I)は様々な方法で製造することができる。

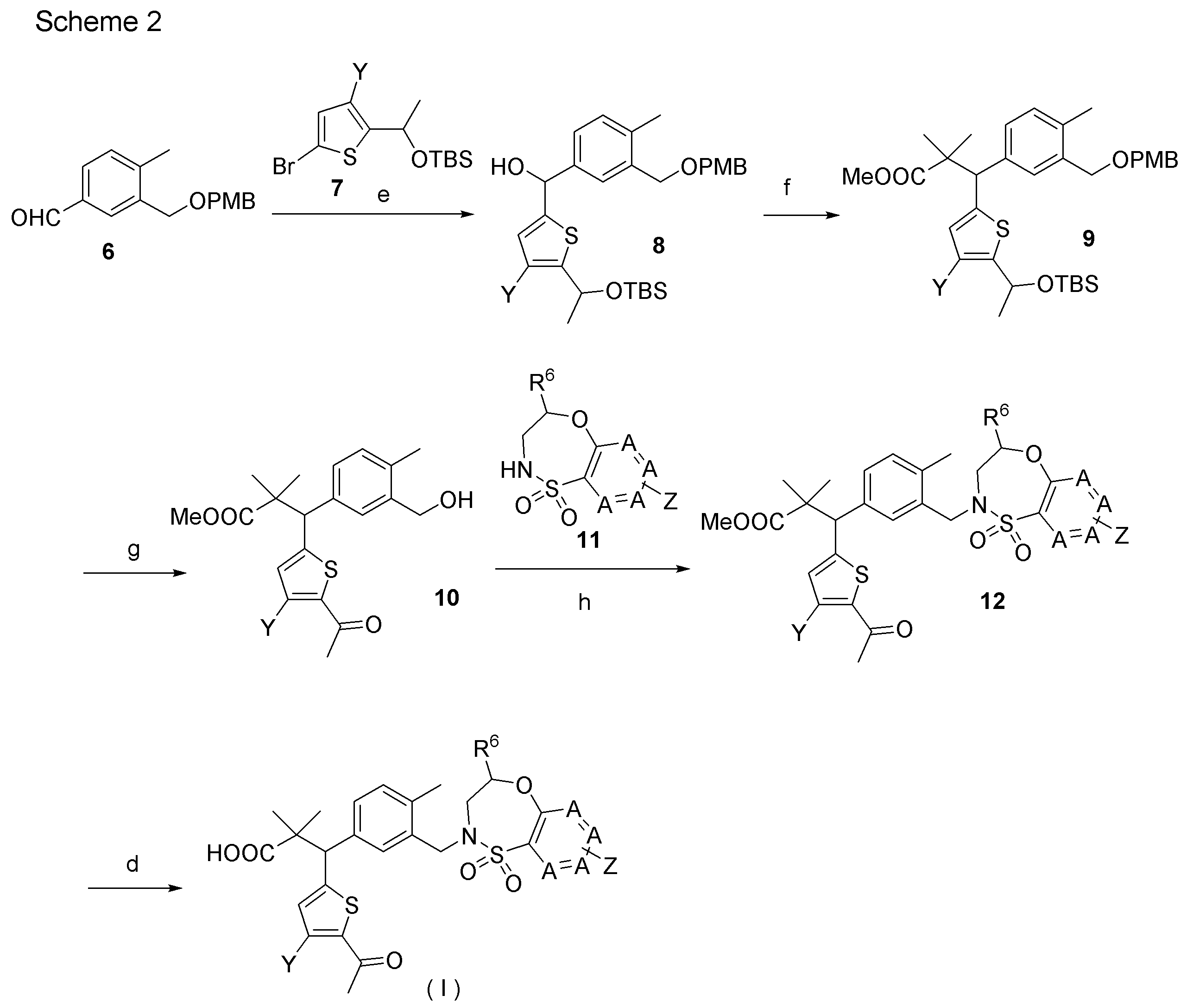


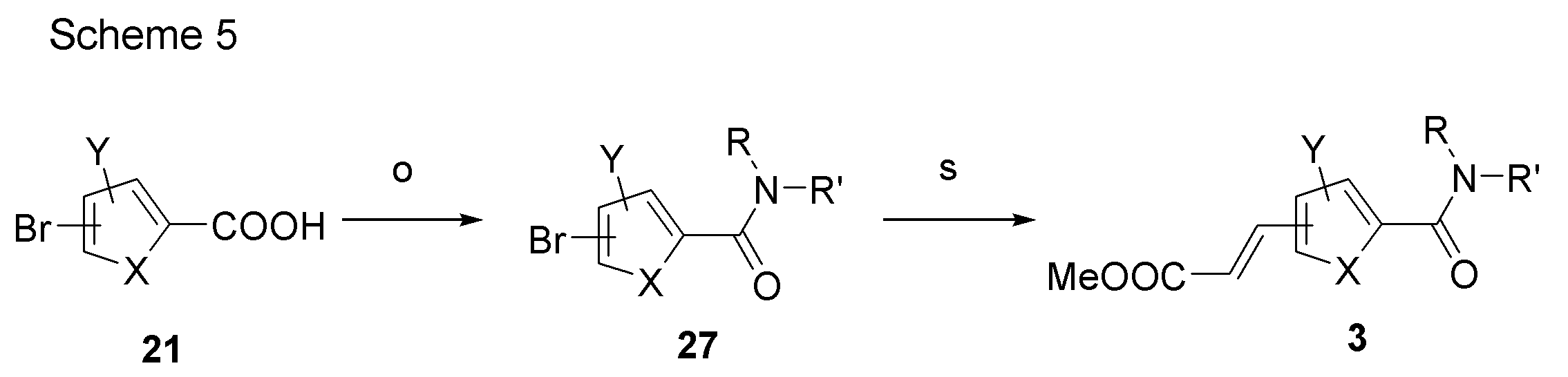

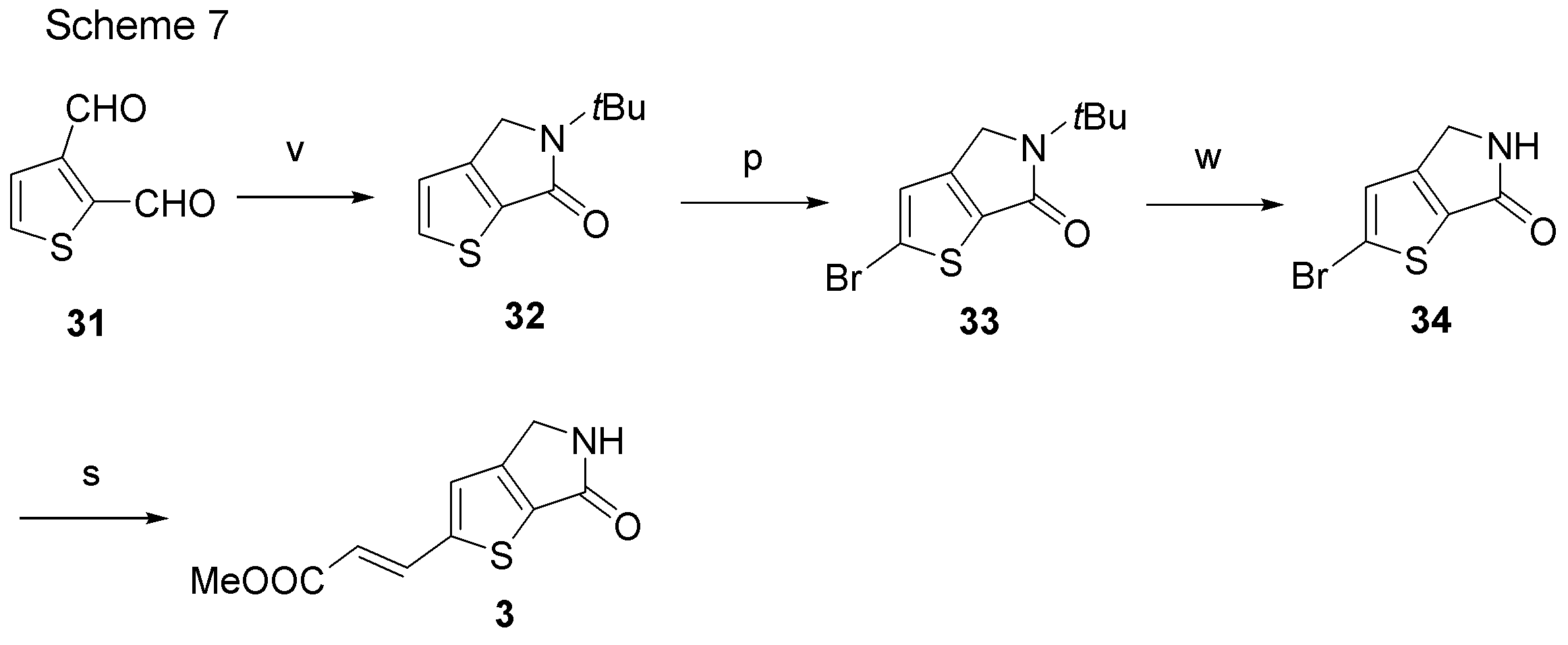


工程a-1では、合成中間体42から合成中間体43を製造する。合成中間体42に対して、トリチルクロリドを例えば1~3当量、好ましくは1.1~1.3当量使用することができ、塩基を例えば1~3当量、好ましくは1.1~1.7当量使用することができる。塩基としては、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジン、N-メチルモルホリン等が挙げられ、好ましくはN-メチルモルホリン又はトリエチルアミンを挙げることができる。反応は、通常、溶媒中で行われる。溶媒としては反応に悪影響を及ぼさない限り特に限定されず、例えば、ジクロロメタン、クロロホルム、テトラヒドロフラン、1,4-ジオキサン、1,2-ジメトキシエタン、ジエチルエーテル、アセトニトリル、N,N-ジメチルホルムアミド等が挙げられ、好ましくはジクロロメタンを挙げることができる。反応時間は、用いる試薬又は溶媒により異なるが、通常1時間~12時間、好ましくは1~2時間である。反応温度は、用いる試薬又は溶媒により異なるが、通常0 ℃~100 ℃、好ましくは40 ℃~60 ℃である。

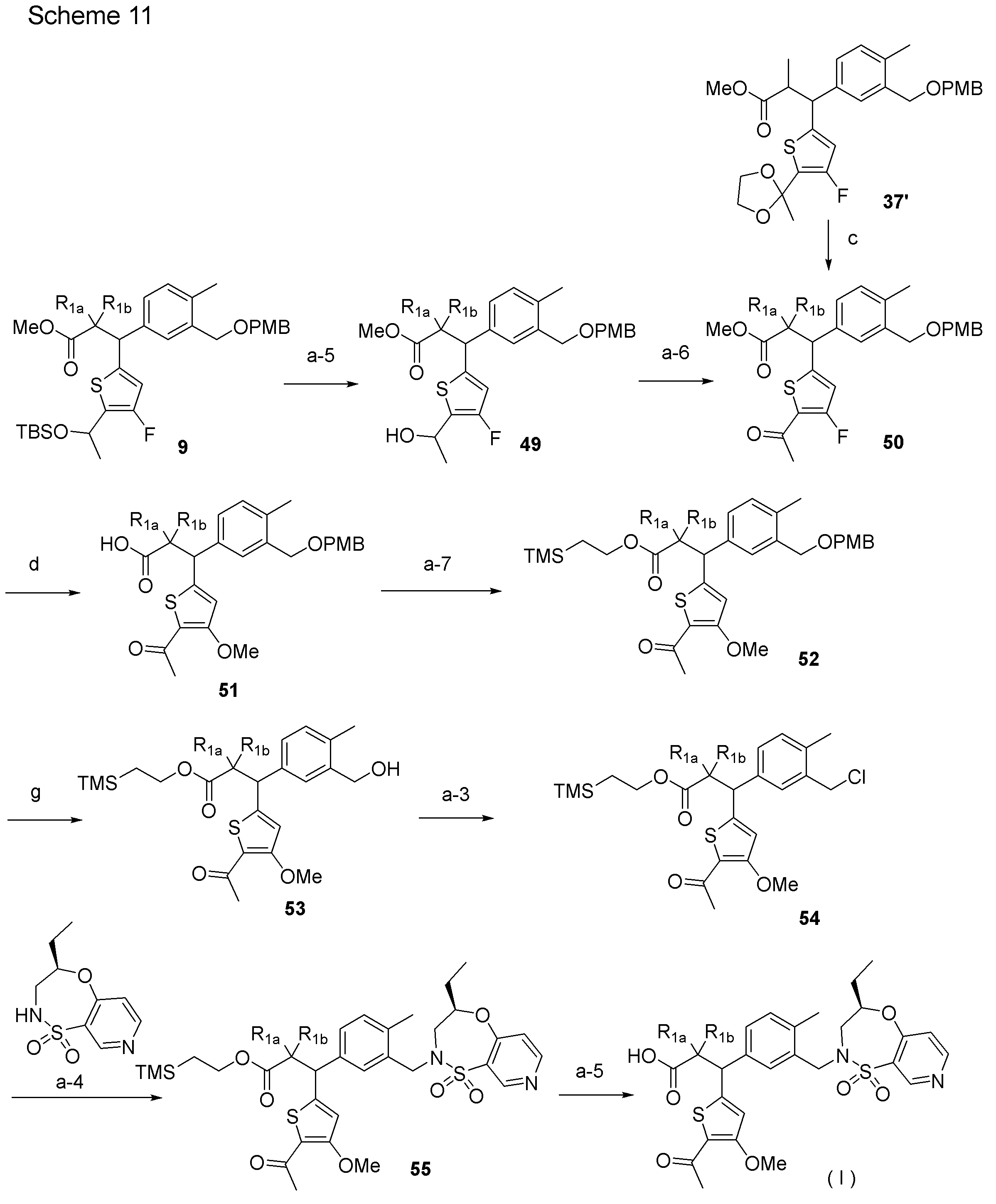
工程a-5では、化合物9から化合物49を製造する。化合物9に対して、テトラブチルアンモニウムフルオリドを2~5当量、好ましくは2~3当量用いる。溶媒は反応に悪影響を及ぼさない限り特に限定されず、テトラヒドロフラン、N,N-ジメチルホルムアミド等が挙げられ、好ましくはテトラヒドロフランを用いる。反応時間は、用いる試薬又は溶媒により異なるが、通常1~24時間、好ましくは1~3時間である。反応温度は用いる試薬又は溶媒により異なるが、通常0℃~100℃、好ましくは25℃~40℃である。
工程a-6では、化合物49から化合物50を製造する。化合物49に対して、二酸化マンガンを10~50当量、好ましくは25~50当量用いる。溶媒は反応に悪影響を及ぼさない限り特に限定されず、ジクロロメタン、アセトン、ベンゼン、トルエン、テトラヒドロフラン、1,4-ジオキサン等が挙げられ、好ましくはジクロロメタンを用いる。反応時間は、用いる試薬又は溶媒により異なるが、通常1~24時間、好ましくは2~24時間である。反応温度は用いる試薬又は溶媒により異なるが、通常0℃~100℃、好ましくは25℃~40℃である。
工程a-7では、化合物51から化合物52を製造する。化合物51に対して、TMSエタノールを1~5当量、好ましくは3~5当量用いる。縮合剤を通常1~3当量、好ましくは1.5~2当量、塩基を通常1~5当量、好ましくは2~5当量用いる。縮合剤としては、例えば、N,N’-ジシクロヘキシルカルボジイミド、1,1’-カルボニルジイミダゾール、1-エチル-3-[3-(ジメチルアミノ)プロピル]カルボジイミド塩酸塩、ベンゾトリアゾール-1-イルオキシトリピロリジノホスホニウム ヘキサフルオロリン酸塩、O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩、シアノリン酸ジエチル、アジ化ジフェニルホスホリル、トリフルオロ酢酸ペンタフルオロフェニルエステル、イソプロピルクロロホルメート等が挙げられ、好ましくはO-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩又はO-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩を挙げることができる。塩基としては、例えば、トリエチルアミン、ピリジン、N,N-ジイソプロピルエチルアミン、4-ジメチルアミノピリジン等が挙げられ、好ましくはトリエチルアミン、N,N-ジイソプロピルエチルアミン、又は4-ジメチルアミノピリジンを挙げることができる。溶媒としては反応に悪影響を及ぼさない限り特に限定されず、例えば、ジクロロメタン、クロロホルム、テトラヒドロフラン、1,4-ジオキサン、1,2-ジメトキシエタン、ジエチルエーテル、アセトニトリル、N,N-ジメチルホルムアミド等が挙げられ、好ましくはジクロロメタン又はN,N-ジメチルホルムアミドを挙げることができる。反応時間は、用いる試薬又は溶媒により異なるが、通常1時間~24時間、好ましくは3時間~24時間である。反応温度は、用いる試薬又は溶媒により異なるが、通常0℃~100℃、好ましくは25℃~40℃である。
本発明の化合物(I)は、Nrf2活性化作用を有する。このため、一般式(I)で表される化合物、その塩、及びそれらの溶媒和物からなる群より選択される少なくとも1種(本明細書において、「本発明の有効成分」と示すこともある。)は、例えば、医薬、試薬等として利用することができる。この観点から、本発明は、その一態様において、本発明の有効成分を含有する、医薬(本明細書において、「本発明の医薬」と示すこともある。)、に関する。
mCPBA: 三クロロ過安息香酸
M: モル濃度
N: 規定度
CDCl3:重クロロホルム
CD3OD:重メタノール
DMSO-d6: 重ジメチルスルホキシド
1H NMR: プロトン核磁気共鳴
DBU: 1,8-ジアザビシクロ [5.4.0] -7-ウンデセン
DDQ: 2,3-ジクロロ-5,6-ジシアノ-p-ベンゾキノン
DEAD: アゾジカルボン酸ジエチル
DIPEA: N,Nージイソプロピルエチルアミン
DMF: N,Nージメチルホルムアミド
DMA: N,Nージメチルアセトアミド
DMSO: ジメチルスルホキシド
EDCI: 1ーエチルー3-(3ージメチルアミノプロピル)カルボジイミド塩酸塩
HOBt: 1ーヒドロキシベンゾトリアゾール
LDA: リチウム ジイソプロピルアミド
PdCl2(dppf)2-CH2Cl2: [1,1'-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド ジクロロメタン付加物
TEA: トリエチルアミン
TFA: トリフルオロ酢酸
THF: テトラヒドロフラン
p-TsOH-H2O: p-トルエンスルホン酸一水和物
[RhCl(cod)]2: クロロ(1,5-シクロオクタジエン)ロジウム(I) (ダイマー)
HATU: O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩
HBTU: O-(ベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロリン酸塩。
(R)-4-エチル-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 7.83 (1H, dd, J = 7.6, 1.6 Hz), 7.47 (1H, td, J = 7.6, 1.6 Hz), 7.19 (1H, td, J = 7.6, 1.2 Hz), 7.17 (1H, dd, J = 7.6, 1.2 Hz), 4.66 (1H, br s), 3.89-3.82 (1H, m), 3.70-3.62 (1H, m), 3.39 (1H, ddd, J =14.8, 5.2, 2.0 Hz), 1.83-1.71 (1H, m), 1.67-1.56 (1H, m), 1.13 (3H, t, J = 7.2 Hz)。
(R)-4-メチル-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 7.81 (1H, dd, J = 8.0, 1.6 Hz), 7.46 (1H, td, J = 8.0, 1.6 Hz), 7.19 (1H, td, J = 8.0, 1.2 Hz), 7.15 (1H, dd, J = 8.0, 1.2 Hz), 4.75 (1H, br s), 4.19-4.12 (1H, m), 3.67-3.59 (1H, m), 3.41 (1H, ddd, J = 14.8, 5.2, 2.0 Hz), 1.40 (3H, d, J = 6.4 Hz)。
(R)-4-エチル-3,4-ジヒドロ-2H-ピリド[2,3-b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 8.40 (1H, dd, J = 4.8, 2.0 Hz), 8.15 (1H, dd, J = 7.6, 2.0 Hz), 7.19 (1H, dd, J = 7.6, 4.8 Hz), 6.34 (1H, br s), 4.26-4.21 (1H, m), 3.66-3.58 (1H, m), 3.50-3.44 (1H, m), 1.84-1.75 (1H, m), 1.71-1.65 (1H, m), 1.12 (3H, t, J = 7.6 Hz)。
(R)-4-エチル-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 8.82 (1H, s), 8.53 (1H, d, J = 5.6 Hz), 7.04 (1H, d, J = 5.6, Hz), 5.98 (1H, br s), 4.29-4.23 (1H, m), 3.56-3.46 (2H, m), 1.81-1.74 (1H, m), 1.72-1.63 (1H, m), 1.10 (3H, t, J = 7.6 Hz)。
(R)-2-フルオロ-N-(2-ヒドロキシブチル)ベンゼンスルホンアミドの製造

1H NMR (400 MHz, CDCl3) δ 7.51 (1H, m), 7.04 (2H, t, J = 8.4 Hz), 3.73-3.67 (1H, m), 3.28 (1H, dd, J = 12.8, 3.2 Hz), 2.96 (1H, d, J = 12.8, 8.0 Hz), 1.55-1.41 (2H, m), 0.94 (3H, t, J =7.6 Hz)。
(R)-4-エチル-9-フルオロ-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR (400 MHz, CDCl3) δ 7.38-7.33 (1H, m), 6.93-6.88 (2H, m), 5.26 (1H, br t, J = 6.0 Hz), 4.34-4.27 (1H, m), 3.54 (1H, ddd, J = 14.0, 6.4, 4.0 Hz), 3.23 (1H, ddd, J= 14.0, 11.2, 6.8 Hz), 1.78-1.58 (2H, m), 1.07 (3H, t, J = 7.6 Hz)。
[(ブタ-3-エン-1-イルオキシ)メチル]ベンゼンの製造
1H NMR (400 MHz, CDCl3) δ 7.38-7.26 (5H, m), 5.91-5.79 (1H, m), 5.15-5.07 (1H, m), 5.08-5.02 (1H, m), 4.53 (2H, s), 3.54 (2H, t, J = 6.8 Hz), 2.43-2.35 (2H, m)。
2-[2-(ベンジロキシ)エチル]オキシランの製造
1H NMR (400 MHz, CDCl3) δ 7.38-7.26 (5H, m), 4.54 (2H, s), 3.68-3.58 (2H, m), 3.12-3.06 (1H, m), 2.79 (1H, dd, J = 5.0, 4.2 Hz), 2.54 (1H, dd, J = 5.0, 3.7 Hz), 1.97-1.86 (1H, m), 1.84-1.73 (1H, m)。
1-アミノ-4-(ベンジルオキシ)ブタン-2-オールの製造
1H NMR (400 MHz, CDCl3) δ 7.37-7.26 (5H, m), 4.52 (2H, s), 3.77-3.63 (3H, m), 2.79 (1H, dd, J = 12.8, 3.6 Hz), 2.61 (1H, dd, J = 12.8, 7.6 Hz), 1.77-1.71 (2H, m)。
4-[2-(ベンジルオキシ)エチル]-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 7.83 (1H, d,J = 7.6 Hz), 7.42 (1H, td, J = 7.6, 1.6 Hz), 7.33-7.26 (5H, m), 7.20 (1H, t, J = 7.6 Hz), 6.96 (1H, d, J= 7.6 Hz), 4.63 (1H, br s), 4.55 (1H, d,J = 12.0 Hz), 4.49 (1H, d, J = 12.0 Hz), 4.11 (1H, br t, J = 9.6 Hz), 3.85-3.63 (3H, m), 3.39-3.35 (1H, m), 2.00-1.86 (2H, m)。
(5-ブロモ-2-メチルフェニル)メタノールの製造

1H NMR(400 MHz, CDCl3)δ 7.53 (1H, d, J= 2.0 Hz), 7.32 (1H, dd, J = 8.0, 2.0 Hz), 7.03 (1H, d, J = 8.0 Hz), 4.66 (2H, d, J = 5.6 Hz), 2.27 (3H, s), 1.64 (1H, t, J = 5.6 Hz)。
(R)-2-(5-ブロモ-2-メチルベンジル)-4-エチル-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J= 7.6, 1.6 Hz), 7.52 (1H, td, J = 7.6, 1.6 Hz), 7.39 (1H, d, J = 2.0 Hz), 7.35 (1H, dd, J = 8.0, 2.0 Hz), 7.26 (1H, td, J = 7.6, 1.2 Hz), 7.21 (1H, dd, J = 7.6, 1.2 Hz), 7.06 (1H, d, J = 8.0 Hz), 4.51 (1H, d, J = 14.4 Hz), 4.08-4.02 (1H, m), 3.85 (1H, d, J =14.4 Hz), 3.76 (1H, dd, J= 15.2, 10.8 Hz), 3.06 (1H, dd, J = 15.2, 2.0 Hz), 2.29 (3H, s), 1.79-1.70 (1H, m), 1.56-1.50 (1H, m), 1.11 (3H, t, J = 7.2 Hz)。
(R)-4-エチル-2-[2-メチル-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ベンジル]-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン1,1-ジオキシドの製造

1H NMR(400 MHz, CDCl3)δ 7.90 (1H, d, J= 7.6 Hz), 7.68 (1H, d, J = 7.6 Hz), 7.54 (1H, s), 7.52 (1H, t, J = 7.6 Hz), 7.25 (1H, t, J = 7.6 Hz), 7.22-7.20 (2H, m), 4.64 (1H, d, J = 13.6 Hz), 4.09-4.04 (1H, m), 3.83 (1H, d, J =13.6 Hz), 3.74 (1H, dd, J= 15.2, 10.8 Hz), 2.97 (1H, dd, J = 15.2, 1.2 Hz), 2.43 (3H, s), 1.77-1.70 (1H, m), 1.51-1.42 (1H, m), 1.32 (12H, s), 1.05 (3H, t, J = 7.2 Hz)。
4-ブロモ-2-{[(4-メトキシベンジル)オキシ]メチル}-1-メチルベンゼンの製造

1H NMR(400 MHz, CDCl3)δ 7.50 (1H, d, J= 2.0 Hz), 7.32-7.28 (3H, m), 7.02 (1H, d, J = 8.0 Hz), 6.90 (2H, d, J = 8.4 Hz), 4.51 (2H, s), 4.46 (2H, s), 3.81 (3H, s), 2.24 (3H, s)。
3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルベンズアルデヒドの製造

1H NMR(400 MHz, CDCl3)δ 9.97 (1H, s), 7.87 (1H, d, J = 1.6 Hz), 7.72 (1H, dd, J= 8.0, 1.6 Hz), 7.33-7.26 (3H, m), 6.90 (2H, br d, J = 8.8 Hz), 4.56 (2H, s), 4.54 (2H, s), 3.82 (3H, s), 2.39 (3H, s)。
2-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-4,4,5,5-テトラメチル-1,3,2-ジオキサボロランの製造

1H NMR(400 MHz, CDCl3)δ 7.73 (1H, s), 7.65 (1H, dd, J = 7.2, 1.2 Hz), 7.27 (2H, d, J= 8.8 Hz), 7.18 (1H, d, J = 7.2 Hz), 6.88 (2H, br d, J = 8.8 Hz), 4.52 (2H, s), 4.47 (2H, s), 3.81 (3H, s), 2.36 (3H, s), 1.34 (12H, s)。
メチル 3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.28-7.24 (3H, m), 7.11-7.10 (2H, m), 6.88 (2H, br d, J= 8.4 Hz), 6.81 (1H, d, J = 3.6 Hz), 6.65 (1H, dd, J = 3.6, 0.8 Hz), 4.67 (1H, t, J = 7.6 Hz), 4.48 (2H, s), 4.46 (2H, s), 4.00-3.95 (2H, m), 3.94-3.89 (2H, m), 3.81 (3H, s), 3.59 (3H, s), 3.08 (1H, dd, J = 15.6, 7.6 Hz), 2.99 (1H, dd, J = 15.6, 7.6 Hz), 2.28 (3H, s), 1.71 (3H, s)。
メチル 3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチル-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

Isomer 1: 1H NMR(400 MHz, CDCl3)δ 7.28-7.26 (3H, m), 7.16 (1H, dd, J = 8.0, 2.0 Hz), 7.06 (1H, d,J = 8.0 Hz), 6.89 (2H, br d, J = 8.8 Hz), 6.82 (1H, d, J = 3.6 Hz), 6.74 (1H, d, J = 3.6 Hz), 4.47 (2H, s), 4.43 (2H, s), 4.25 (1H, d, J = 10.8 Hz), 4.01-3.97 (2H, m), 3.96-3.90 (2H, m), 3.81 (3H, s), 3.43 (3H, s), 3.24-3.16 (1H, m), 2.25 (3H, s), 1.71 (3H, s), 1.20 (3H, d, J = 6.8 Hz)。
Isomer 2: 1H NMR(400 MHz, CDCl3)δ 7.28-7.23 (3H, m), 7.11-7.10 (2H, m), 6.89 (2H, br d, J = 8.8 Hz), 6.77 (1H, d, J = 3.6 Hz), 6.71 (1H, dd, J = 3.6 Hz), 4.48 (2H, s), 4.47 (2H, s), 4.27 (1H, d, J = 11.2 Hz), 3.99-3.95 (2H, m), 3.93-3.82 (2H, m), 3.82 (3H, s), 3.58 (3H, s), 3.21-3.16 (1H, m), 2.28 (3H, s), 1.69 (3H, s), 1.05 (3H, d, J= 6.8 Hz)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造
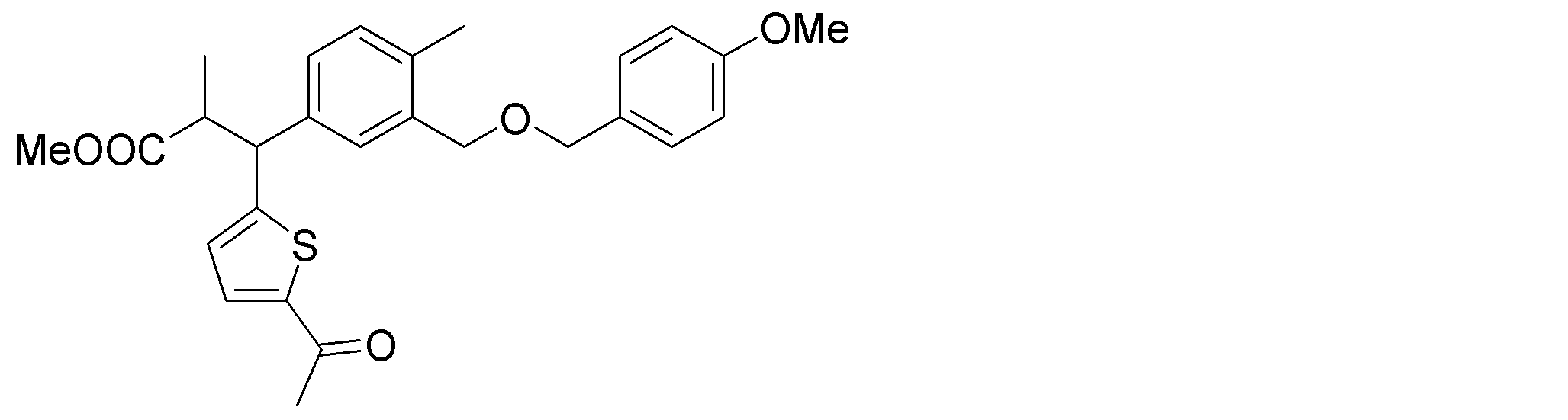
Isomer 1: 1H NMR(400 MHz, CDCl3)δ 7.49 (1H, d, J = 4.0 Hz), 7.28-7.24 (3H, m), 7.17 (1H, dd, J = 8.0, 2.0 Hz), 7.07 (1H, d, J = 8.0 Hz), 6,94-6.88 (3H, m), 4.46 (2H, s), 4.44 (2H, s), 4.36 (1H, d, J = 11.2 Hz), 3.81 (3H, s), 3.45 (3H, s), 3.29-3.21 (1H, m), 2.45 (3H, s), 2.24 (3H, s), 1.21 (3H, d, J = 6.8 Hz)。
Isomer 2: 1H NMR(400 MHz, CDCl3)δ 7.46 (1H, d, J = 4.0 Hz), 7.29-7.24 (3H, m), 7.11 (2H, s), 6,92-6.89 (3H, m), 4.49 (2H, s), 4.48 (2H, s), 4.36 (1H, d, J = 11.2 Hz), 3.82 (3H, s), 3.60 (3H, s), 3.25-3.21 (1H, m), 2.46 (3H, s), 2.26 (3H, s), 1.08 (3H, d, J = 6.8 Hz)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2-メチルプロパノエートの製造

Isomer 1: 1H NMR(400 MHz, CDCl3)δ 7.50 (1H, d, J = 4.0 Hz), 7.30 (1H, br s), 7.16 (1H, dd, J = 7.6, 2.0 Hz), 7.08 (1H, d, J = 7.6 Hz), 6.95 (1H, d, J = 4.0 Hz), 4.63 (2H, d, J = 14.4 Hz), 4.36 (1H, d, J = 10.8 Hz), 3.47 (3H, s), 3.31-3.21 (1H, m), 2.45 (3H, s), 2.26 (3H, s), 1.21 (3H, d, J = 7.2 Hz)。
Isomer 2: 1H NMR(400 MHz, CDCl3)δ 7.46 (1H, d, J = 4.0 Hz), 7.28 (1H, br s), 7.13 (2H, br s), 6.93 (1H, d, J = 4.0 Hz), 4.68 (2H, d, J = 5.6 Hz), 4.37 (1H, d, J = 11.2 Hz), 3.60 (3H, s), 3.27-3.23 (1H, m), 2.46 (3H, s), 2.30 (3H, s), 1.09 (3H, d, J = 6.8 Hz)。
1-(5-ブロモチオフェン-2-イル)エタン-1-オールの製造

1H NMR (400 MHz, CDCl3) δ 6.90 (1H, d, J = 3.6 Hz), 6.72 (1H, d, J= 3.6 Hz), 5.04 (1H, dq, J = 5.6, 6.4 Hz), 1.97 (1H, d, J = 4.8 Hz), 1.57 (3H, d, J = 6.4 Hz)。
[1-(5-ブロモチオフェン-2-イル)エトキシ](tert-ブチル)ジメチルシランの製造

1H NMR (400 MHz, CDCl3), δ 6.86 (1H, d, J = 4.0 Hz), 6.58 (1H, d, J= 4.0 Hz), 5.01 (1H, q, J = 6.4 Hz), 1.47 (3H, d, J = 6.4 Hz), 0.91 (9H, s), 0.09 (3H, s), 0.06 (3H, s)。
(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}チオフェン-2-イル)(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)メタノールの製造
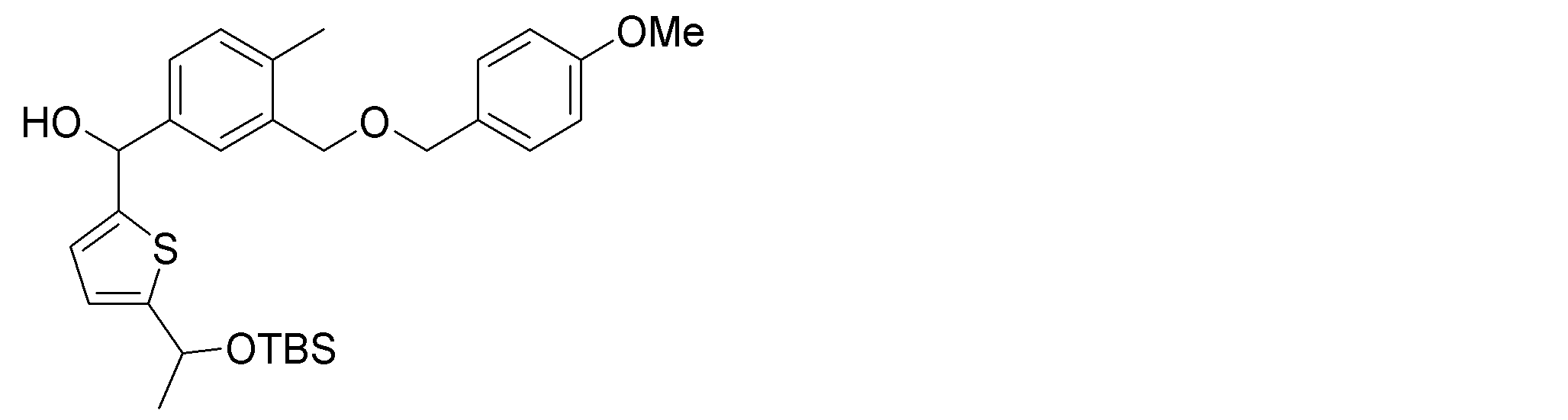
1H NMR (400 MHz, CDCl3) δ 7.41 (1H, s), 7.29-7.26 (3H, m), 7.16 (1H, d, J = 7.6 Hz), 6.88 (1H, d, J = 8.4 Hz), 6.67 (2H, d, J = 4.4 Hz), 5.96 (1H, d, J = 4.0 Hz), 5.17 (1H, q, J = 6.4 Hz), 4.51 (2H, s), 4.48 (2H, s), 3.81 (3H, s), 2.32 (3H, s), 2.29 (1H, d, J = 4.0 Hz), 1.47 (3H, d, J = 6.4 Hz), 0.90 (9H, s), 0.07 (3H, s), 0.03 (3H, br s)。
メチル 3-(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}チオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
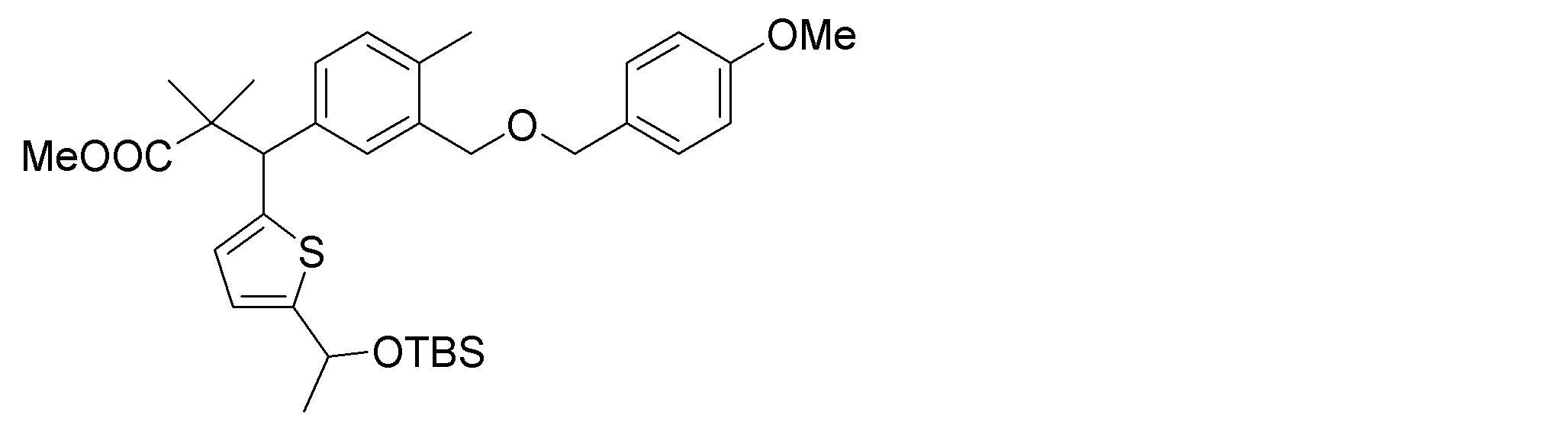
1H NMR (400 MHz, CDCl3) δ 7.32 (1H, dd, J = 4.4, 1.2 Hz), 7.29-7.26 (2H, m), 7.21 (1H, td, J = 7.6, 1.6 Hz), 7.08 (1H, d, J = 7.6 Hz), 6.90 (1H, s), 6.88 (1H, s), 6.76-6.74 (1H, m), 6.64-6.63 (1H, m), 5.00 (1H, q, J = 6.4 Hz), 4.63-4.62 (1H, m), 4.49 (2H, s), 4.47 (2H, br s), 3.81 (3H, s), 3.56 (3H, s), 2.27 (3H, s), 1.45 (3H, d, J = 6.4 Hz), 1.29 (3H, s), 1.22 (3H, s), 0.89 (9H, br s), 0.05 (3H, s), 0.002 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.50 (1H, d, J = 4.0 Hz), 7.34 (1H, d, J = 1.6 Hz), 7.20 (1H, dd, J = 7.6, 1.6 Hz), 7.11 (1H, d, J = 7.6 Hz), 6.97 (1H, d, J = 4.0 Hz), 4.17 (1H, s), 4.68 (2H, d, J = 5.6 Hz), 3.61 (3H, s), 2.49 (3H, s), 2.31 (3H, s), 1.32 (3H, s), 1.24 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.51 (1H, d, J = 4.0 Hz), 7.31-7.24 (2H, m), 7.14 (1H, d, J = 8.0 Hz), 6.87 (1H, d, J = 4.0 Hz), 4.70 (1H, s), 4.57 (2H, s), 3.60 (3H, s), 2.50 (3H, s), 2.38 (3H, s), 1.31 (3H, s), 1.24 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
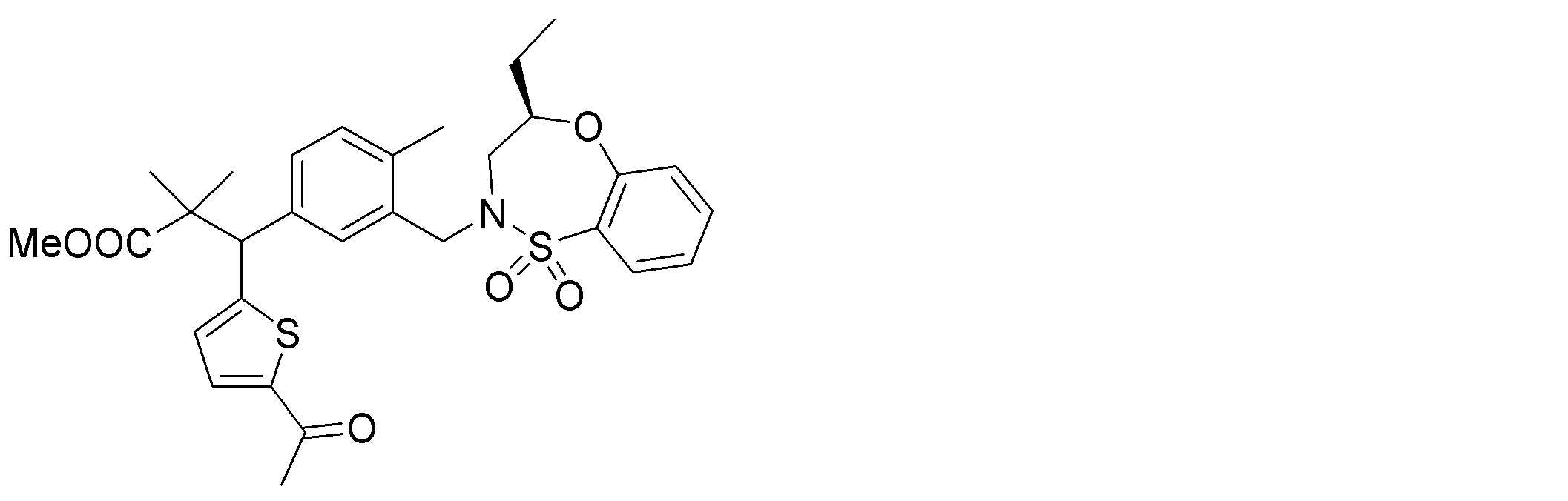
1H NMR (400 Mz, CDCl3) δ 7.87 (1H, dt, J = 7.6, 1.2 Hz), 7.55-7.50 (1H, m), 7.49 (1H, d, J = 4.0 Hz), 7.27-7.11 (5H, m), 6.97-6.95 (1H, m), 4.69 (1H, br s), 4.56-4.55 (1H, m), 3.83-3.80 (1H, m), 4.04-3.97 (1H, m), 3.73-3.71 (1H, m), 3.60 (3H, br s), 2.96-2.93 (1H, m), 2.49-2.48 (3H, m), 2.33 (3H, s), 1.81-1.56 (1H, m), 1.51-1.35 (1H, m), 1.29 (3H, br s), 1.23-1.21 (3H, m), 1.10-1.08 (3H, m)。
メチル 2-アミノ-5-ブロモ-3-ニトロベンゾエートの製造

1H NMR (400 MHz, CDCl3) δ 8.51 (1H, d, J = 2.4 Hz), 8.44 (2H, br s), 8.32 (1H, d, J = 2.4 Hz), 3.93 (3H, s)。
メチル 2,3-ジアミノ-5-ブロモベンゾエートの製造

1H NMR (400 MHz, CDCl3) δ 7.58 (1H, d, J = 1.6 Hz), 6.94 (1H d, J= 1.6 Hz), 5.55 (2H, br s), 3.87 (3H, s), 3.39 (2H, br s)。
メチル 6-ブロモ-1H-ベンゾ[d]イミダゾール-4-カルボキシレートの製造

1H NMR (400 MHz, CDCl3) δ 10.5 (1H, br s), 8.18 (1H, d, J = 0.8 Hz), 8.14 (1H, s), 8.07 (1H, d, J = 0.8 Hz), 4.02 (3H, s)。
(6-ブロモ-1-トリチル-1H-ベンゾ[d]イミダゾール-4-イル)メタノールの製造

1H NMR (400 MHz, CDCl3) δ 7.85 (1H, s), 7.36-7.31 (9H, m), 7.21 (1H, s), 7.17-7.14 (6H, m), 6.50 (1H, s), 5.09 (2H, d, J = 6.4 Hz), 3.67 (1H, t, J = 6.4 Hz),
参考例32
(R)-2-[(6-ブロモ-1-トリチル-1H-ベンゾ[d]イミダゾール-4-イル)メチル]-4-エチル-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR (400 MHz, CDCl3) δ 7.89 (1H, dd, J = 7.6, 1.6 Hz), 7.78 (1H, s), 7.49 (1H, td, J = 7.6, 1.6 Hz), 7.46 (1H, s), 7.34-7.30 (9H, m), 7.23 (1H, t, J = 8.0 Hz), 7.17-7.11 (7H, m), 6.50 (1H, d, J = 1.6 Hz), 4.77 (1H, d, J = 16.0 Hz), 4.55 (1H, d, J = 16.0 Hz), 4.05-3.96 (2H, m), 3.30 (1H, dt, J = 13.6, 6.8 Hz), 1.78-1.67 (1H, m), 1.54-1.44 (1H, m), 1.08 (3H, t, J= 7.2 Hz)。
(R)-4-エチル-2-{[6-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1-トリチル-1H-ベンゾ[d]イミダゾール-4-イル]メチル}-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン 1,1-ジオキシドの製造

1H NMR (400 MHz, CDCl3) δ 7.92 (1H, d, J = 7.6 Hz), 7.84 (1H, s), 7.71 (1H, s), 7.46 (1H, t, J = 7.6 Hz), 7.32-7.28 (9H, m), 7.21 (1H, t, J = 7.6 Hz), 7.16-7.14 (7H, m). 6.77 (1H, s), 4.81 (1H, d, J = 14.8 Hz), 4.58 (1H, d, J = 14.8 Hz), 4.06-3.94 (2H, m), 3.27 (1H, d, J = 14.0 Hz), 1.72-1.62 (1H, m), 1.49-1.40 (1H, m), 1.20 (12H, s), 1.03 (3H, t, J = 7.2 Hz)。
5-ブロモ-3-メチルチオフェン-2-カルバルデヒドの製造

1H NMR (400 MHz, CDCl3) δ 9.90 (1H, s), 6.96 (1H, s), 2.53 (3H, s)。
5-ブロモ-3-メチルチオフェン-2-カルボン酸の製造

1H NMR (400 MHz, DMSO-d6) δ 7.20 (1H, s), 2.43 (3H, s)。
5-ブロモ-N-メチルフラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.04 (1H, d, J= 3.6 Hz), 6.62 (1H, s), 6.42 (1H, d, J= 3.6 Hz), 2.96 (3H, d, J = 4.8 Hz)。
(5-ブロモフラン-2-イル)(ピロリジン-1-イル)メタノンの製造

1H NMR(400 MHz, CDCl3)δ 7.04 (1H, d, J = 3.6 Hz), 6.43 (1H, d, J = 3.6 Hz), 3.82 (2H, t, J = 6.8 Hz), 3.63 (2H, t, J = 6.8 Hz), 2.01 (2H, quin, J = 6.8 Hz), 1.90 (2H, quin, J = 6.8 Hz)。
5-ブロモ-N-イソプロピルフラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.04 (1H, d, J = 3.2 Hz), 6.43 (1H, d, J = 3.2 Hz), 6.09 (1H, br s), 4.24 (1H, septd, J = 6.4, 1.2 Hz), 1.26 (6H, d, J = 6.4 Hz)。
5-ブロモ-N-メチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CD3OD)δ 7.40 (1H, d, J = 4.0 Hz), 7.13 (1H, d, J = 4.0 Hz), 2.86 (3H, s)。
5-ブロモ-N-(2-メトキシエチル)フラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.06 (1H, d, J= 3.6 Hz), 6.63 (1H, br s), 6.43 (1H, d, J= 3.6 Hz), 3.63-3.59 (2H, m), 3.55-3.53 (2H, m), 3.40 (3H, s)。
N,3-ジメチルフラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 6.32 (1H, s), 6.28 (1H, br s), 2.95 (3H, d, J = 5.2 Hz), 2.40 (3H, s)。
4-ブロモ-N-メチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.37 (1H, d,J = 1.2 Hz), 7.35 (1H, d, J = 1.2 Hz), 5.93 (1H, br s), 3.00 (3H, d, J= 4.8 Hz)。
N,4-ジメチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.45 (1H, d, J= 1.2 Hz), 7.29 (1H, br s), 7.00 (1H, br s), 2.96 (3H, d, J = 4.8 Hz), 2.20 (3H, d, J= 0.8 Hz)。
N-メトキシ-N,4-ジメチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.75 (1H, d, J= 1.6 Hz), 7.14 (1H, d, J = 1.6 Hz), 3.76 (3H, s), 3.36 (3H, s), 2.28 (3H, s)。
3-フルオロ-N-メトキシ-N-メチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.38 (1H, dd, J= 5.6, 4.0 Hz), 6.84 (1H, d, J = 5.6 Hz), 3.74 (3H, s), 3.33 (3H, s)。
5-ブロモ-N-(2,4-ジメトキシベンジル)チオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.21 (1H, d, J= 8.0 Hz), 7.19 (1H, d, J = 4.0 Hz), 6.98 (1H, d, J = 4.0 Hz), 6.48 (1H, br s), 6.46 (1H, d, J = 2.4 Hz), 6.43 (1H, dd, J = 8.0, 2.4 Hz), 4.49 (2H, d, J = 5.6 Hz), 3.84 (3H, s), 3.79 (3H, s)。
5-ブロモ-N-メトキシ-N-3-ジメチルチオフェン-2-カルボキサミドの製造

1H NMR (400 MHz, CDCl3) δ 6.87 (1H, s), 3.70 (3H, s), 3.29 (3H, s), 2.51 (3H, s)。
ブロモ-N-メトキシ-N-メチルチアゾール-5-カルボキサミド

1H NMR (400 MHz, CDCl3) δ 8.32 (1H, s), 3.78 (3H, s), 3.36 (3H, s)。
2-ブロモ-N-メトキシ-N-4-ジメチルチアゾール-5-カルボキサミドの製造

1H NMR (400 MHz, CDCl3) δ 3.66 (3H, s), 3.26 (3H, s), 2.68 (3H, s)。
4-ブロモ-N-メトキシ-N-メチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.83 (1H, d, J = 1.6 Hz), 7.44 (1H, d, J = 1.6 Hz), 3.78 (3H, s), 3.37 (3H, s)。
2-ブロモ-N-メトキシ-N-メチルチオフェン-3-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.25 (1H, d, J= 6.0 Hz), 7.04 (1H, d, J = 6.0 Hz), 3.60 (3H, s), 3.34 (3H, s)。
5-ブロモ-N-メトキシ-N-メチルフラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.08 (1H, d,J = 3.6 Hz), 6.44 (1H, d, J = 3.6 Hz), 3.77 (3H, s), 3.33 (3H, s)。
N-[(5-ブロモフラン-2-イル)メチル]プロパン-2-アミンの製造

1H NMR(400 MHz, CDCl3)δ 6.21 (1H, d, J= 3.2 Hz), 6.15 (1H, d, J = 3.2 Hz), 3.75 (2H, s), 2.82 (1H, septd, J = 6.4 Hz), 1.38 (1H, br s), 1.06 (6H, d, J= 6.4 Hz)。
5-(tert-ブチル)-4,5-ジヒドロ-6H-チエノ[2,3-c]ピロール-6-オンの製造

1H NMR (400 MHz, CDCl3) δ 7.56 (1H, d, J = 4.8 Hz), 6.99 (1H, d, J= 4.8 Hz), 4.36 (2H, s), 1.54 (9H, s)
参考例55
2-ブロモ-1,5,6,7-テトラヒドロ-4H-インドール-4-オンの製造

1H NMR(400 MHz, CDCl3)δ 8.49 (1H, br s), 6.51 (1H, d, J = 2.4 Hz), 2.79 (2H, t, J= 6.4 Hz), 2.48 (2H, t, J = 6.4 Hz), 2.15 (2H, quin, J = 6.4 Hz)。
5-ブロモ-N,3-ジメチルフラン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 6.28 (1H, br s), 6.27 (1H, s), 2.95 (3H, d, J = 4.8 Hz), 2.37 (3H, s)。
5-ブロモ-N,4-ジメチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 7.19 (1H, s), 6.23 (1H, br s), 2.95 (3H, d, J = 4.8 Hz), 2.18 (3H, s)。
1-(5-ブロモ-2,4-ジメチル-1H-ピロール-3-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 8.16 (1H, br s), 2.49 (3H, s), 2.42 (3H, s), 2.22 (3H, s)。
1-(5-ブロモ-4-メチルチオフェン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.36 (1H, s), 2.48 (3H, s), 2.22 (3H, s)。
5-ブロモ-3-フルオロ-N-メトキシ-N-メチルチオフェン-2-カルボキサミドの製造

1H NMR(400 MHz, CDCl3)δ 6.89 (1H, s), 3.75 (3H, s), 3.30 (3H, s)。
1-(5-ブロモ-2,4-ジメチル-1H-ピロール-3-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 8.16 (1H, br s), 2.49 (3H, s), 2.42 (3H, s), 2.22 (3H, s)。
1-(5-ブロモ-1-エチル-1H-ピロール-2イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 6.91 (1H, d, J= 1.6 Hz), 6.85 (1H, d, J = 1.6 Hz), 4.33 (2H, q, J = 7.2 Hz), 2.40 (3H, s), 1.35 (3H, t, J = 7.2 Hz)。
(5-ブロモチオフェン-2-イル)(シクロプロピル)メタノンの製造

1H NMR(400 MHz, CDCl3)δ 7.55 (1H, d, J= 4.0 Hz), 7.12 (1H, d, J = 4.0 Hz), 2.47-2.41 (1H, m), 1.25-1.21 (2H, m), 1.05-1.01 (2H, m)。
2-ブロモ-5-(tert-ブチル)-4,5-ジヒドロ-6H-チエノ[2,3-c]ピロール-6-オンの製造

1H NMR (400 MHz, CDCl3) δ 7.01(1H, s), 4.33 (2H, s), 1.53 (9H, s)。
4-ブロモ-1-メチル-1H-ピロール-2-カルボニトリルの製造

1H NMR(400 MHz, CDCl3)δ 6.81 (1H, d, J= 1.6 Hz), 6.75 (1H, d, J = 1.6 Hz), 3.76 (3H, s)。
2-ブロモ-1-メチル-1,5,6,7-テトラヒドロ-4H-インドール-4-オンの製造

1H NMR(400 MHz, CDCl3)δ 6.52 (1H, s), 3.50 (3H, s), 2.73 (2H, t, J = 6.4 Hz), 2.41 (2H, t, J = 6.4 Hz), 2.14 (2H, quin, J = 6.4 Hz)。
1-(5-ブロモ-1,2,4-トリメチル-1H-ピロール-3-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 3.50 (3H, s), 2.50 (3H, s), 2.41 (3H, s), 2.24 (3H, s)。
1-(5-ブロモ-3-フルオロチオフェン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 6.90 (1H, d, J= 0.8 Hz), 2.52 (3H, d, J = 3.2 Hz)。
1-(4-メチルチオフェン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.50 (1H, d, J= 0.8 Hz), 7.22 (1H, br d, J = 0.8 Hz), 2.53 (3H, s), 2.29 (3H, d, J = 0.8 Hz)。
1-(5-ブロモ-3-メチルチオフェン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 6.93 (1H, s), 2.51 (3H, s), 2.47 (3H, s)。
1-(2-ブロモチアゾール-5-イル)エタン-1-オンの製造

1H NMR (400 MHz, CDCl3) δ 8.05, (1H, s), 2.56 (3H, s)。
1-(2-ブロモ-4-メチルチアゾール-5-イル)エタン-1-オンの製造

1H NMR (400 MHz, CDCl3) δ 2.72 (3H, s), 2.51 (3H, s)。
1-(4-ブロモチオフェン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.58 (1H, d, J = 1.2 Hz), 7.53 (1H, d, J = 1.2 Hz), 2.55 (3H, s)。
1-(2-ブロモチオフェン-3-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.35 (1H, d, J = 5.6 Hz), 7.23 (1H, d, J = 5.6 Hz), 2.62 (3H, s)。
1-(5-ブロモフラン-2-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.12 (1H, d,J = 3.6 Hz), 6.49 (1H, d, J = 3.6 Hz), 2.46 (3H, s)。
2-(5-ブロモチオフェン-2-イル)-2-メチル-1,3-ジオキソランの製造

1H NMR(400 MHz, CDCl3)δ 6.90 (1H, d, J= 4.0 Hz), 6.79 (1H, d, J = 4.0 Hz), 4.06-4.00 (m, 2H), 3.99-3.93 (m, 2H), 1.73(s, 3H)。
2-(5-ブロモ-3-メチルチオフェン-2-イル)-2-メチル-1,3-ジオキソロンの製造

1H NMR (400 MHz, CDCl3) δ 6.74 (1H, s), 4.06-3.99 (2H, m), 3.96-3.90 (2H, m), 2.23 (3H, s), 1.71 (3H, s)。
2-ブロモ-5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾールの製造

1H NMR (400 MHz, CDCl3) δ 7.49 (1H, s), 4.13-4.03 (2H, m), 3.99-3.91 (2H, m), 1.75 (3H, s)。
2-ブロモ-4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾールの製造

1H NMR (400 MHz, CDCl3) δ 4.09-4.01 (2H, m), 3.94-3.88 (2H, m), 2.43 (3H, s), 1.71 (3H, s)。
2-(5-ブロモフラン-2-イル)-2-メチル-1,3-ジオキソランの製造

1H NMR(400 MHz, CDCl3)δ 6.29 (1H, d,J = 3.2 Hz), 6.23 (1H, d, J = 3.2 Hz), 4.07-4.03 (2H, m), 4.01-3.97 (2H, m), 1.71 (3H, s)。
2-(5-ブロモチオフェン-3-イル)-2-メチル-1,3-ジオキソランの製造

1H NMR(400 MHz, CDCl3)δ 7.17 (1H, d, J= 1.6 Hz), 7.02 (1H, d, J = 1.6 Hz), 4.07-3.98 (2H, m), 3.91-3.82 (2H, m), 1.64 (3H, s)。
1-(5-ブロモチオフェン-3-イル)エタン-1-オールの製造

1H NMR(400 MHz, CDCl3)δ 6.97 (1H, d, J= 0.8 Hz), 6.94 (1H, br s), 4.76 (1H, q, J= 6.4 Hz), 2.20 (1H, br s), 1.38 (3H, d, J= 6.4 Hz)。
1-(5-ブロモチオフェン-3-イル)エタン-1-オンの製造

1H NMR(400 MHz, CDCl3)δ 7.91 (1H, d, J= 1.6 Hz), 7.49 (1H, d, J = 1.6 Hz), 2.49 (3H, s)。
5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾール-2-カルバルデヒドの製造

1H NMR (400 MHz, CDCl3) δ 9.93 (1H, s), 8.01 (1H, s), 4.12-4.06 (2H, m), 3.99-3.93 (2H, m), 1.80 (3H, s)。
2-ブロモ-4,5-ジヒドロ-6H-チエノ[2,3-c]ピロール-6-オンの製造

1H NMR (400 MHz, CDCl3) δ 7.08 (1H, s), 6.16 (1H, br s), 4.36 (2H, s)。
1-(5-ブロモ-3-フルオロチオフェン-2-イル)エタン-1-オールの製造

1H NMR(400 MHz, CDCl3)δ 6.75 (1H, br s), 5.22-5.17 (1H, m), 2.00 (1H, d, J = 4.0 Hz), 1.54 (1H, d, J = 6.4 Hz)。
[1-(5-ブロモ-3-フルオロチオフェン-2-イル)エトキシ](tert-ブチル)ジメチルシランの製造

1H NMR(400 MHz, CDCl3)δ 6.70 (1H, br s), 5.13 (1H, qd, J = 6.4, 1.2 Hz), 1.45 (3H, d, J = 6.4 Hz), 0.91 (9H, s), 0.082 (3H, s), 0.036 (3H, s)。
メチル (E)-3-[5-(メチルカルバモイル)フラン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.41 (1H, d, J= 16.0 Hz), 7.13 (1H, d, J = 3.6 Hz), 6.67 (1H, d, J = 3.6 Hz), 6.52 (1H, br s), 6.41 (1H, d, J = 16.0 Hz), 3.80 (3H, s), 3.01 (3H, d, J = 4.8 Hz)。
メチル (E)-3-[5-(ピロリジン-1-カルバモイル)フラン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.44 (1H, d, J= 16.0 Hz), 7.14 (1H, d, J = 3.6 Hz), 6.71 (1H, d, J = 3.6 Hz), 6.37 (1H, d, J = 16.0 Hz), 3.90 (2H, t, J = 6.8 Hz), 3.80 (3H, s), 3.65 (2H, t, J = 6.8 Hz), 2.05 (2H, quin, J = 6.8 Hz), 1.92 (2H, quin, J = 6.8 Hz)。
メチル (E)-3-[5-(イソプロピルカルバモイル)フラン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.43 (1H, d, J= 16.0 Hz), 7.13 (1H, d, J = 3.6 Hz), 6.68 (1H, d, J = 3.6 Hz), 6.43 (1H, d, J = 16.0 Hz), 6.15 (1H, br s), 4.27 (1H, septd, J = 6.4, 1.2 Hz), 3.82 (3H, s), 1.28 (6H, d, J = 6.4 Hz)。
参考例91
メチル (E)-3-[5-(メチルカルバモイル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.72 (1H, d,J = 15.6 Hz), 7.39 (1H, d, J = 3.6 Hz), 7.19 (1H, d, J = 3.6 Hz), 6.31 (1H, d, J = 15.6 Hz), 5.96 (1H, br s), 3.80 (3H, s), 3.01 (3H, d, J = 4.8 Hz)。
メチル (E)-3-{5-[(2-メトキシエチル)カルバモイル]フラン-2-イル}アクリレートの製造
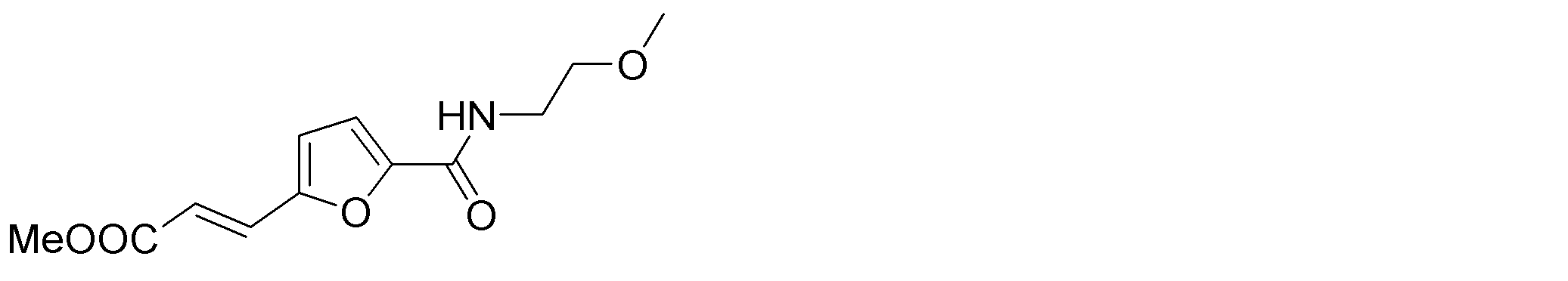
1H NMR(400 MHz, CDCl3)δ 7.42 (1H, d, J = 16.0 Hz), 7.14 (1H, d, J = 3.6 Hz), 6.88 (1H, br s), 6.69 (1H, d, J = 3.6 Hz), 6.46 (1H, d, J = 16.0 Hz), 3.81 (3H, s), 3.66-3.62 (2H, m), 3.58-3.55 (2H, m), 3.41 (3H, s)。
メチル (E)-3-(5-アセチルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.73 (1H, d, J= 16.0 Hz), 7.60 (1H, d, J = 4.0 Hz), 7.24 (1H, d, J = 4.0 Hz), 6.39 (1H, d, J = 16.0 Hz), 3.81 (3H, s), 2.56 (3H, s)。
メチル (E)-3-[4-メチル-5-(メチルカルバモイル)フラン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ7.36 (1H, d, J = 15.6 Hz), 6.53 (1H, s), 6.36 (1H, d, J = 15.6 Hz), 6.34 (1H, br s), 3.80 (3H, s), 2.98 (3H, d, J = 5.2 Hz), 2.39 (3H, s)。
メチル (E)-3-(5-スルファモイルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CD3OD)δ 7.80 (1H, d, J = 16.0 Hz), 7.55 (1H, d, J = 4.0 Hz), 7.37 (1H, d, J = 4.0 Hz), 6.42 (1H, d, J = 16.0 Hz), 3.81 (3H, s)。
メチル (E)-3-(チオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.79 (1H, d, J = 16.0 Hz), 7.37 (1H, d, J = 5.2 Hz), 7.25 (1H, d, J = 3.6 Hz), 7.05 (1H, dd, J= 5.2, 3.6 Hz), 6.24 (1H, d, J = 16.0 Hz), 3.79 (3H, s)。
メチル (E)-3-[5-(メチルカルバモイル)チオフェン-3-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.63 (1H, s), 7.60 (1H, d, J = 16.0 Hz), 7.58 (1H, s), 6.27 (1H, d, J = 16.0 Hz), 6.04 (1H, br s), 3.80 (3H, s), 3.01 (3H, d, J = 4.8 Hz)。
メチル (E)-3{-[5-(イソプロピルアミノ)メチル]フラン-2-イル}アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.38 (1H, d, J = 15.6 Hz), 6.54 (1H, d, J = 3.2 Hz), 6.27 (1H, d, J = 15.6 Hz), 6.26 (1H, d, J = 3.2 Hz), 3.81 (2H, s), 3.78 (3H, s), 2.85 (1H, septd, J = 6.4 Hz), 1.09 (6H, d, J = 6.4 Hz)。
メチル (E)-3-[3-メチル-5-(メチルカルバモイル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.76 (1H, d, J= 16.0 Hz), 7.34 (1H, s), 6.94 (1H, br s), 6.20 (1H, d, J = 16.0 Hz), 3.79 (3H, s), 2.98 (3H, d, J = 4.8 Hz), 2.28 (3H, s)。
メチル (E)-3-(1-メチル-4-オキソ-4,5,6,7-テトラヒドロ-1H-インドール-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.56 (1H, d, J= 15.6 Hz), 7.00 (1H, s), 6.23 (1H, d, J= 15.6 Hz), 3.78 (3H, s), 3.61 (3H, s), 2.79 (2H, t, J = 6.4 Hz), 2.47 (2H, t, J= 6.4 Hz), 2.17 (2H, quin, J = 6.4 Hz)。
メチル (E)-3-(5-アセチル-1-メチル-1H-ピロール-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.64 (1H, d, J= 15.6 Hz), 6.95 (1H, d, J = 4.4 Hz), 6.60 (1H, d, J = 4.4 Hz), 6.37 (1H, d, J = 15.6 Hz), 4.01 (3H, s), 3.81 (3H, s), 2.46 (3H, s)。
メチル (E)-3-(5-アセチル-3-メチルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.76 (1H, d, J = 15.6 Hz), 7.43 (1H, s), 6.29 (1H, d, J = 15.6 Hz), 3.80 (3H, s), 2.52 (3H, s), 2.34 (3H, s)。
メチル (E)-3-(5-アセチル-4-フルオロチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.60 (1H, d, J= 16.0 Hz), 7.00 (1H, s), 6.39 (1H, d, J= 16.0 Hz), 3.81 (3H, s), 2.58 (3H, d, J= 2.8 Hz)。
メチル (E)-3-{5-[(2,4-ジメトキシベンジル)カルバモイル]チオフェン-2-イル}アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.67 (1H, d, J= 16.0 Hz), 7.41 (1H, d, J = 4.0 Hz), 7.19 (1H, d, J = 8.4 Hz), 7.13 (1H, d, J = 4.0 Hz), 6.85 (1H, t, J = 5.6 Hz), 6.43 (1H, d, J = 2.4 Hz), 6.41 (1H, dd, J = 8.0, 2.4 Hz), 6.26 (1H, d, J = 16.0 Hz),4.49 (2H, d, J = 5.6 Hz), 3.81 (3H, s), 3.77 (3H, s), 3.76 (3H, s)。
メチル (E)-3-(5-アセチル-4-メチルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.66 (1H, d, J= 16.0 Hz), 7.07 (1H, s), 6.35 (1H, d, J= 16.0 Hz), 3.81 (3H, s), 2.53 (3H, s), 2.52 (3H, s)。
メチル (E)-3-(4-アセチル-1,3,5-トリメチル-1H-ピロール-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.68 (1H, d, J= 16.0 Hz), 6.03 (1H, d, J = 16.0 Hz), 3.80 (3H, s), 3.59 (3H, s), 2.51 (3H, s), 2.46 (3H, s), 2.40 (3H, s)。
メチル (E)-3-(5-アセチル-1-エチル-1H-ピロール-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.54 (1H, d, J= 16.0 Hz), 7.12 (1H, d, J = 1.6 Hz), 7.08 (1H, d, J = 1.6 Hz), 6.16 (1H, d, J = 16.0 Hz), 4.35 (2H, q, J = 7.2 Hz), 3.77 (3H, s), 2.46 (3H, s), 1.37 (3H, t, J = 7.2 Hz)。
メチル (E)-3-[5-(シクロプロパンカルボニル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.74 (1H, d, J= 16.0 Hz), 7.72 (1H, d, J = 4.0 Hz), 7.27 (1H, d, J = 4.0 Hz), 6.39 (1H, d, J = 16.0 Hz), 3.81 (3H, s), 2.51 (1H, tt, J = 8.0, 4.4 Hz), 1.28-1.25 (2H, m), 1.08-1.04 (2H, m)。
メチル (E)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.71 (1H, d, J= 15.6 Hz), 7.10 (1H, d, J = 3.6 Hz), 6.97 (1H, d, J = 3.6 Hz), 6.18 (1H, d, J = 15.6 Hz), 4.08-4.02 (m, 2H), 3.98-3.95 (m, 2H)、3.78 (3H, s), 1.76 (s, 3H)。
メチル (E)-3-(5-シアノチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.71 (1H, d, J= 16.0 Hz), 7.55 (1H, d, J = 4.0 Hz), 7.23 (1H, d, J = 4.0 Hz), 6.36 (1H, d, J = 16.0 Hz), 3.82 (3H, s)。
メチル (E)-3-[4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]アクリレートの製造

1H NMR (400 MHz, CDCl3) δ 7.61 (1H, d, J = 15.6 Hz), 6.92 (1H, s), 6.11 (1H, d, J = 15.6 Hz), 4.07-3.98 (2H, m), 3.94-3.88 (2H, m), 3.74 (3H, s), 2.23 (3H, s), 1.70 (3H, s)。
メチル (E)-3-(6-オキソ-5,6-ジヒドロ-4H-チエノ[2,3-c]ピロール-2-イル)アクリレートの製造

1H NMR (400 MHz, CDCl3) δ 7.77 (1H, d, J = 15.6 Hz), 7.18 (1H, s), 6.54 (1H, br s), 6.37 (1H, d, J = 15.6 Hz), 4.38 (2H, s), 3.81 (3H, s)。
メチル (E)-3-(5-アセチルフラン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.43 (1H, d, J= 16.0 Hz), 7.17 (1H, d, J = 3.6 Hz), 6.69 (1H, d, J = 3.6 Hz), 6.54 (1H, d, J = 16.0 Hz), 3.79 (1H, s), 2.49 (3H, s)。
メチル (E)-3-(5-アセチルチオフェン-3-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.82 (1H, d, J = 1.2 Hz), 7.74 (1H, d, J = 1.2 Hz), 7.62 (1H, d, J = 16.0 Hz), 6.32 (1H, d, J= 16.0 Hz), 3.81 (3H, s), 2.59 (3H, s)。
メチル (E)-3-(4-アセチルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 8.03 (1H, s), 7.73 (1H, d, J = 16.0 Hz), 7.64 (1H, br s), 6.27 (1H, d, J = 16.0 Hz), 3.80 (3H, s), 2.52 (3H, s)。
メチル (E)-3-(5-シアノ-1-メチル-1H-ピロール-3-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.48 (1H, d,J = 16.0 Hz), 7.01 (1H, d, J = 1.6 Hz), 6.96 (1H, d, J = 1.6 Hz), 6.13 (1H, d, J = 16.0 Hz), 3.80 (3H, s), 3.77 (3H, s)。
メチル (E)-3-(3-アセチルチオフェン-2-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 8.51 (1H, d, J= 16.0 Hz), 7.40 (1H, d, J = 5.6 Hz), 7.28 (1H, d, J = 5.6 Hz), 6.34 (1H, d, J = 16.0 Hz), 3.78 (3H, s), 2.54 (3H, s)。
メチル (E)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)フラン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.39 (1H, d, J= 15.6 Hz), 6.52 (1H, d, J = 3.6 Hz), 6.39 (1H, d, J = 3.6 Hz), 6.32 (1H, d, J = 15.6 Hz), 4.09-4.05 (2H, m), 4.04-4.00 (2H, m), 3.78 (3H, s), 1.75 (3H, s)。
メチル (E)-3-[4-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.72 (1H, d, J= 15.6 Hz), 7.31 (1H, s), 7.21 (1H, s), 6.22 (1H, d, J = 15.6 Hz), 4.06-4.02 (2H, m), 3.89-3.86 (2H, m), 3.79 (3H, s), 1.66 (3H, s)。
エチル (E)-3-(チアゾール-5-イル)アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 8.80 (1H, s), 8.01 (1H, s), 7.82 (1H, d, J = 15.6 Hz), 6.26 (1H, d, J = 15.6 Hz), 4.26 (2H, q, J = 7.2 Hz), 1.33 (3H, t, J = 7.2 Hz)。
メチル (E)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾール-2-イル]アクリレートの製造

1H NMR (400 MHz, CDCl3) δ 7.79 (1H, s), 7.70 (1H, d, J = 15.6 Hz), 6.65 (1H, d, J = 15.6 Hz), 4.14-4.04 (2H, m), 3.99-3.91 (2H, m), 3.82 (3H, s), 1.78 (3H, s)。
メチル (E)-3-[4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾール-2-イル]アクリレートの製造

1H NMR (400 MHz, CDCl3) δ 7.63 (1H, d, J = 15.6 Hz), 6.60 (1H, d, J= 15.6 Hz), 4.08-4.02 (2H, m), 3.94-3.86 (2H, m), 3.80 (3H, s), 2.49 (3H, s), 1.74 (3H, s)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(メチルカルバモイル)フラン-2-イル]プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, d,J = 8.0 Hz), 7.53 (1H, td, J = 8.0, 1.6 Hz), 7.28-7.21 (3H, m), 7.14-7.07 (2H, m), 6.98 (1H, d, J = 3.2 Hz), 6.37 (1H, br s), 6.15 (1H, t, J = 3.2 Hz), 4.55-4.49 (2H, m), 4.04-3.99 (1H, m), 3.87-3.84 (1H, m), 3.80-3.73 (1H, m), 3.62 (3H, br s), 3.12-3.00 (3H, m), 2.95-2.94 (3H, m), 2.30 (3H, br s), 1.76-1.66 (1H, m), 1.53-1.41 (1H, m), 1.09 (3H, t, J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(ピロリジン-1-カルボニル)フラン-2-イル]プロパノエートの製造
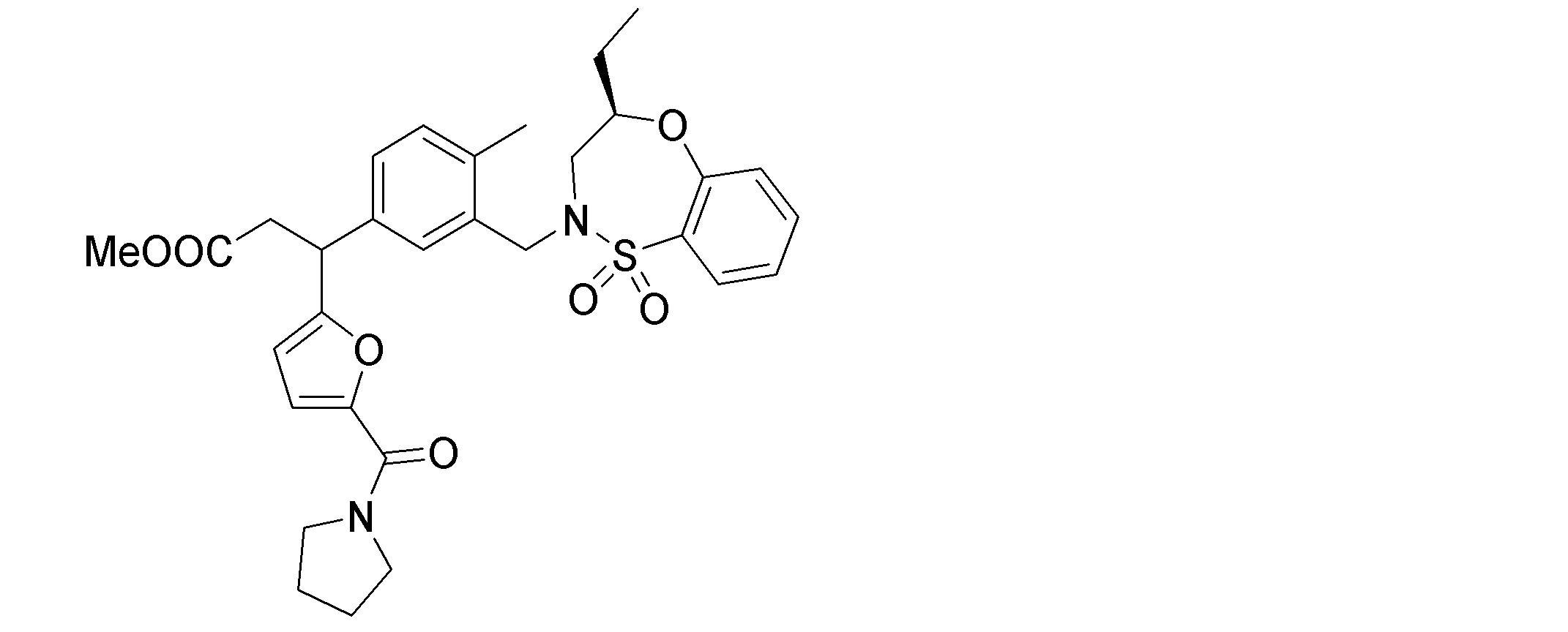
1H NMR(400 MHz, CDCl3)δ 7.88(1H, d,J = 7.6 Hz), 7.52 (1H, t, J = 7.6 Hz), 7.29-7.12 (5H, m), 6.97 (1H, d, J= 3.2 Hz), 6.11 (1H, d, J = 3.2 Hz), 4.57-4.53 (2H, m), 4.03-3.98 (1H, m), 3.81 (1H, d, J = 14.4 Hz), 3.78-3.67 (3H, m), 3.61-3.58 (5H, m), 3.14-3.10 (1H, m), 2.99-2.87 (2H, m), 2.32 (3H, s), 1.99-1.95 (2H, m), 1.88-1.85 (2H, m), 1.74-1.64 (1H, m), 1.50-1.42 (1H, m), 1.08 (3H, t, J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(イソプロピルカルバモイル)フラン-2-イル]プロパノエートの製造
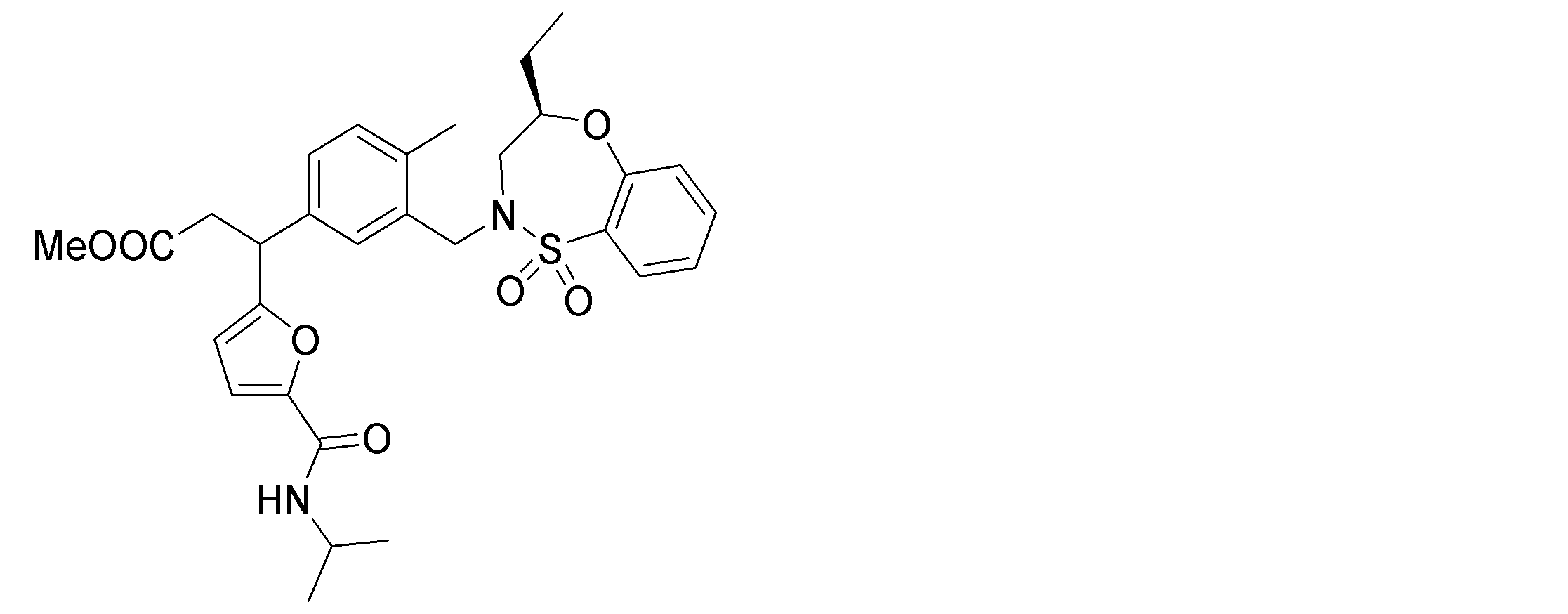
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.53 (1H, td, J= 8.0, 1.6 Hz), 7.26 (1H, t, J = 8.0 Hz), 7.21 (1H, d, J = 8.0 Hz), 7.16-7.08 (3H, m), 6.98 (1H, d, J = 3.6 Hz), 6.14-6.10 (1H, m), 6.05 (1H, br s), 4.56-4.51 (2H, m), 4.26-4.19 (1H, m), 4.03-4.00 (1H, m), 3.87-3.74 (2H, m), 3.64 (3H, s), 3.07-2.99 (2H, m), 2.88-2.85 (1H, m), 2.33 (3H, br s), 1.76-1.65 (1H, m), 1.51-1.42 (1H, m), 1.26-1.22 (6H, m), 1.09-1.08 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(メチルカルバモイル)チオフェン-2-イル]プロパノエートの製造
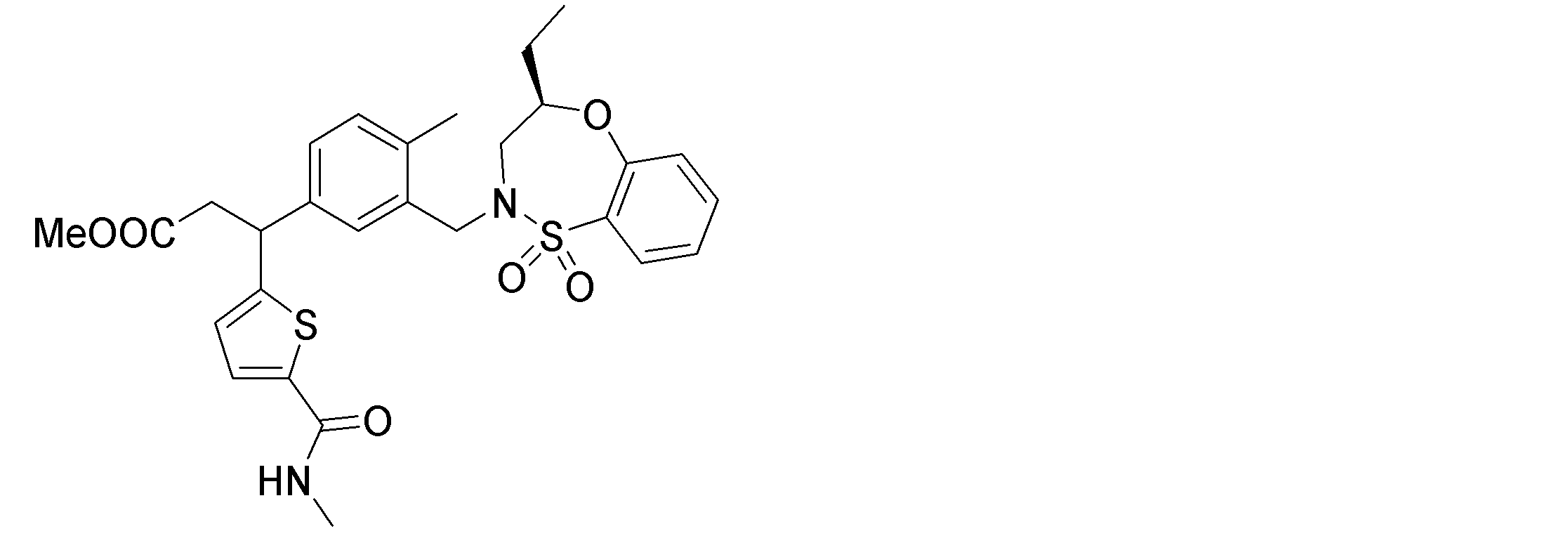
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, br t, J= 8.0 Hz), 7.29-7.19 (3H, m), 7.14 (2H, br s), 7.07 (1H, d, J = 6.8 Hz), 6.78-6.77 (1H, m), 5.99 (1H, br s), 4.69 (1H, t, J = 7.6 Hz), 4.56 (1H, dd, J = 14.0, 5.2 Hz), 4.02-3.95 (1H, m), 3.82-3.69 (2H, m), 3.60 (3H, s), 3.09 (1H, dd, J = 16.0, 7.6 Hz), 3.01-2.90 (5H, m), 2.34 (3H, br s), 1.69-1.62 (1H, m), 1.46-1.38 (1H, m), 1.06 (3H, t,J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-{5-[(2-メトキシエチル)カルバモイル]フラン-2-イル}プロパノエートの製造
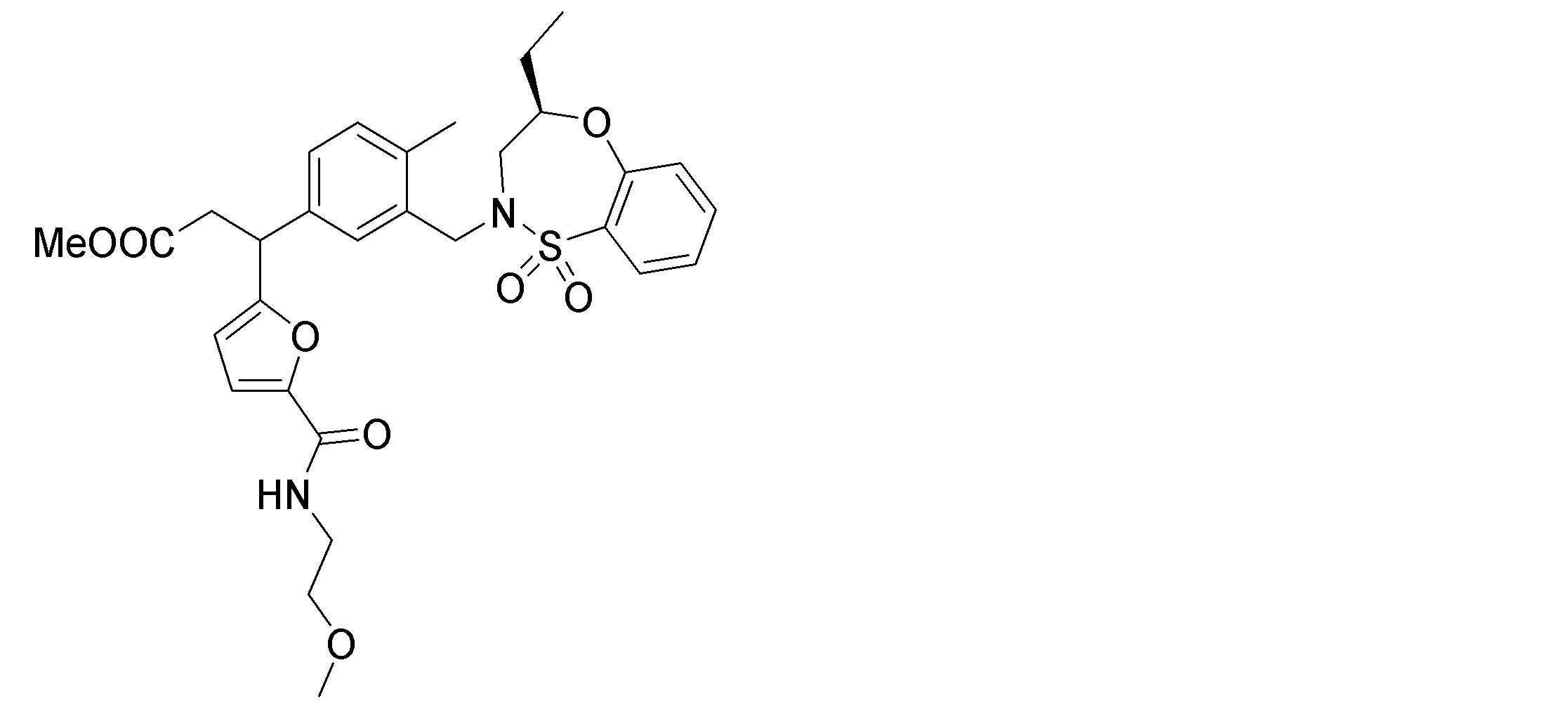
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.26 (1H, t, J = 8.0 Hz), 7.23 (1H, d, J = 8.0 Hz), 7.16-7.09 (3H, m), 6.99 (1H, d, J = 3.6 Hz), 6.55 (1H, br s), 6.12 (1H, t, J= 3.6 Hz), 4.57-4.51 (2H, m), 4.04-4.00 (1H, m), 3.87-3.71 (2H, m), 3.63 (3H, s), 3.60-3.51 (4H, m), 3.37 (3H, s), 3.11-2.85 (3H, m), 2.33 (3H, br s), 1.78-1.67 (1H, m), 1.51-1.41 (1H, m), 1.08 (3H, t, J = 7.6 Hz)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd, J= 8.0, 1.6 Hz), 7.52 (1H, td, J = 8.0, 1.6 Hz), 7.50 (1H, d, J = 4.0 Hz), 7.27-7.20 (2H, m), 7.17-7.12 (2H, m), 7.08 (1H, d, J = 8.4 Hz), 6.87 (1H, br t, J= 2.4 Hz), 4.72 (1H, t, J = 7.6 Hz), 4.58-4.56 (1H, m), 4.03-3.96 (1H, m), 3.83-3.69 (2H, m), 3.61 (3H, s), 3.13-2.91 (3H, m), 2.48 (3H, br s), 2.34 (3H, br s), 1.73-1.63 (1H, m), 1.47-1.39 (1H, m), 1.07 (3H, t, J = 7.6 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-メチル-5-(メチルカルバモイル)フラン-2-イル]プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.6 Hz), 7.53 (1H, td, J= 7.6, 1.6 Hz), 7.27-7.19 (3H, m), 7.13 (1H, d, J = 7.6 Hz), 7.10-7.06 (1H, m), 6.27 (1H, br s), 5.98 (1H, d, J = 3.2 Hz), 4.49-4.44 (2H, m), 4.05-3.98 (1H, m), 3.89-3.72 (2H, m), 3.63 (3H, s), 3.09-2.84 (6H, m), 2.34-2.30 (6H, m), 1.78-1.59 (1H, m), 1.52-1.41 (1H, m), 1.09 (3H, t, J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル)-4-メチルフェニル}-3-(5-スルファモイルチオフェン-2-イル)プロパノエートの製造
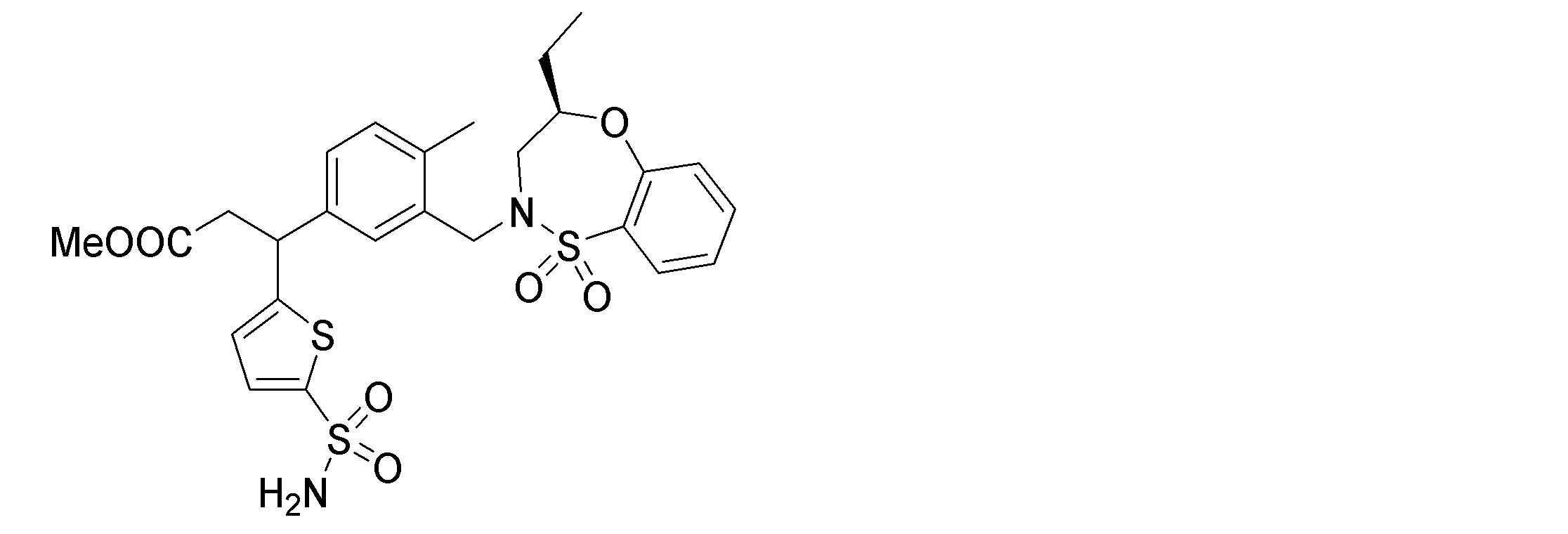
1H NMR(400 MHz, CDCl3)δ 7.84 (1H, br d,J = 8.0 Hz), 7.51 (1H, td, J = 8.0, 1.6 Hz), 7.42 (1H, d, J = 4.0 Hz), 7.27-7.19 (2H, m), 7.15-7.11 (3H, m), 6.78 (1H, d, J = 4.0 Hz), 4.71 (1H, t, J= 8.0 Hz), 4.51 (1H, d, J = 14.4 Hz), 4.03-3.97 (1H, m), 3.83-3.82 (1H, m), 3.77-3.69 (1H, m), 3.61 (3H, s), 3.11-2.95 (3H, m), 2.30 (3H, s), 1.74-1.63 (1H, m), 1.51-1.43 (1H, m), 1.08-1.07 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(チオフェン-2-イル)プロパノエートの製造
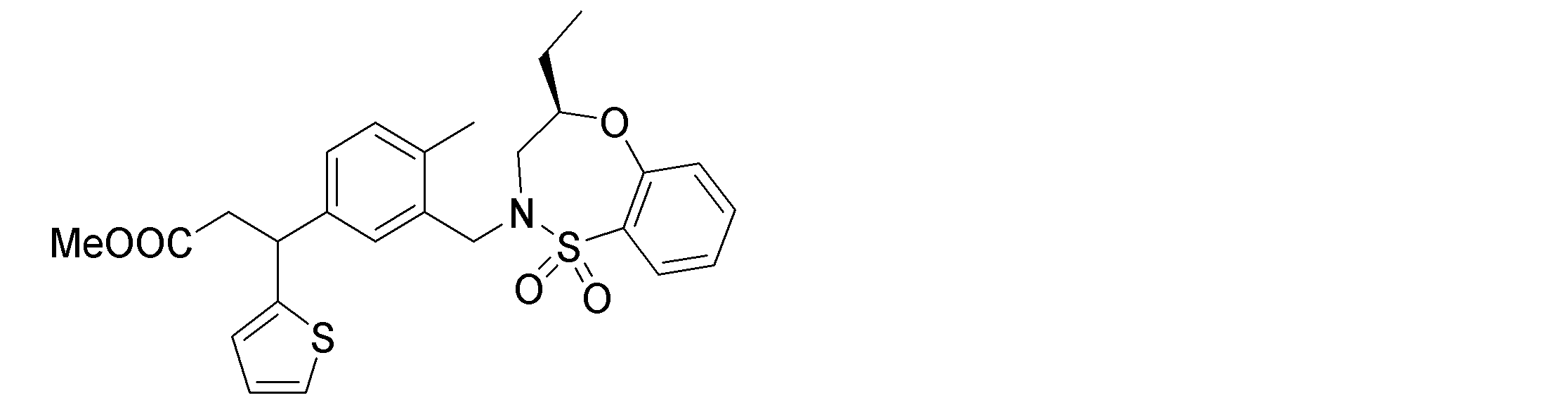
1H NMR(400 MHz, CDCl3)δ7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.51 (1H, td, J= 8.0, 1.6 Hz), 7.26-7.12 (5H, m), 7.03 (1H, d, J = 14.4 Hz), 6.89 (1H, br t,J = 4.0 Hz), 6.81 (1H, br t, J = 4.0 Hz), 4.71 (1H, t, J = 7.6 Hz), 4.60-4.57 (1H, m), 4.01-3.92 (1H, m), 3.80-3.68 (2H, m), 3.59 (3H, br s), 3.13-3.06 (1H, m), 3.02-2.87 (2H, m), 2.36 (3H, br s), 1.71-1.59 (1H, m), 1.44-1.32 (1H, m), 1.07-1.06 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(メチルカルバモイル)チオフェン-3-イル]プロパノエートの製造
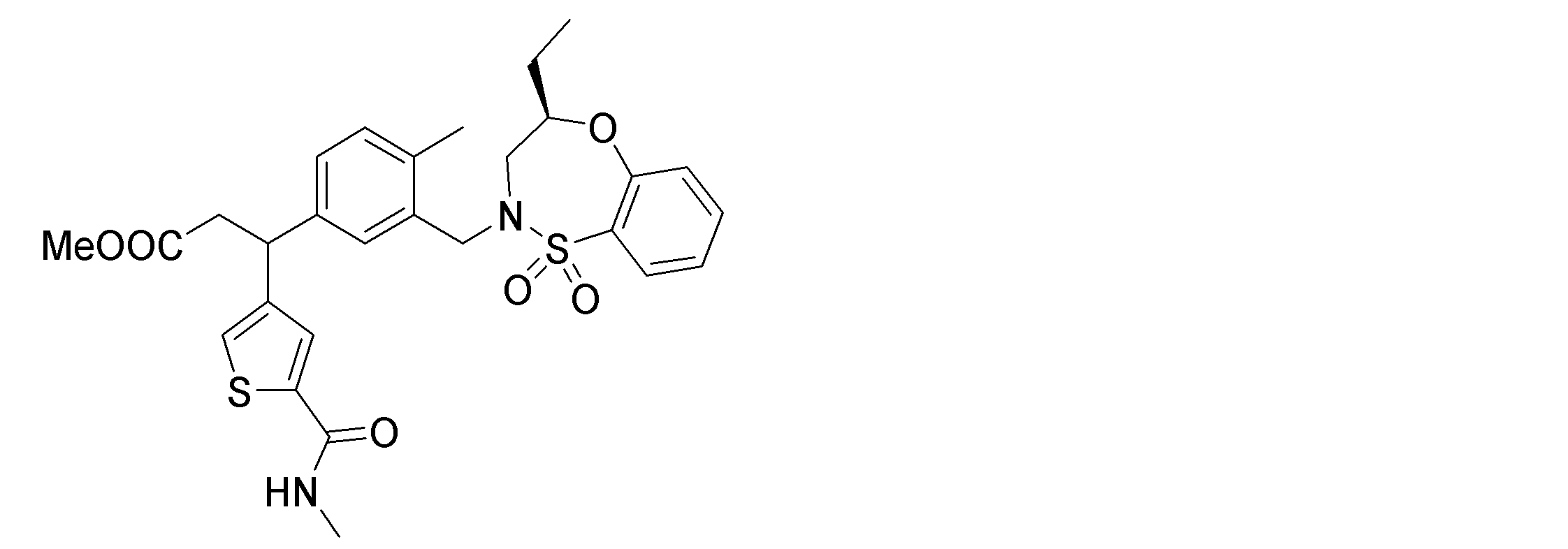
1H NMR(400 MHz, CDCl3)δ 7.85 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.31-7.30 (1H, m), 7.25 (1H, t, J = 8.0 Hz), 7.21 (1H, d, J= 8.0 Hz), 7.11-7.05 (4H, m), 6.37 (1H, br s), 4.51-4.47 (2H, m), 4.02-3.95 (1H, m), 3.84-3.70 (2H, m), 3.58 (3H, s), 3.05-2.92 (3H, m), 2.89 (3H, d, J = 4.8 Hz), 2.28 (3H, s), 1.74-1.61 (1H, m), 1.50-1.39 (1H, m), 1.08-1.07 (3H, m)。
メチル 3-(3-(((R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル)メチル)-4-メチルフェニル)-3-(3-メチル-5-(メチルカルバモイル)チオフェン-2-イル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, br d,J = 8.0 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.26 (1H, d, J = 8.0 Hz), 7.23-7.19 (1H, m), 7.17 (1H, br s), 7.14-7.11 (2H, m), 7.05 (1H, br d, J = 5.6 Hz), 6.08 (1H, br s), 4.72 (1H, t, J = 8.0 Hz), 4.56-4.55 (1H, m), 4.00-3.95 (1H, m), 3.78-3.68 (2H, m), 3.59 (3H, s), 2.99 (2H, dd, J = 7.6, 7.6 Hz), 2.94-2.90 (4H, m), 2.33 (3H, br s), 2.10 (3H, s), 1.73-1.60 (1H, m), 1.45-1.38 (1H, m), 1.07-1.06 (3H, m)。
エチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(チアゾール-5-イル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 8.80 (1H, s), 7.88 (1H, dd, J = 7.6, 1.6 Hz), 7.63 (1H, d, J= 3.6 Hz), 7.52 (1H, td, J = 7.6, 2.0 Hz), 7.25 (1H, td, J = 7.6, 1.2 Hz), 7.20 (1H, dd, J = 8.0, 0.8 Hz), 7.17-7.12 (2H, m), 7.08 (1H, d, J = 7.6 Hz), 4.77 (1H, t, J = 8.0 Hz), 4.57 (1H, dd, J = 14.0, 5.2 Hz), 4.09-3.98 (3H, m), 3.83-3.69 (2H, m), 3.11-2.91 (3H, m), 2.34 (3H, br s), 1.71-1.65 (1H, m), 1.46-1.40 (1H, m), 1.16 (3H, t, J = 7.2 Hz), 1.09-1.05 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(1-メチル-4-オキソ-4,5,6,7-テトラヒドロ-1H-インドール-2-イル)プロパノエートの製造
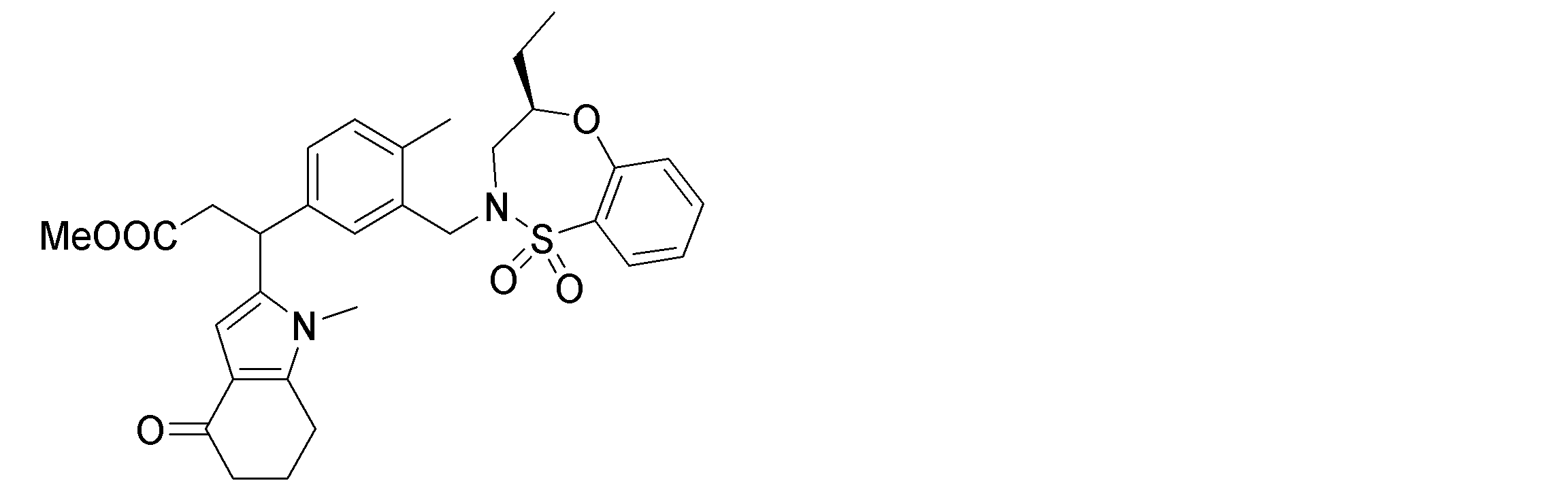
1H NMR(400 MHz, CDCl3)δ 7.87-7.82 (1H, m), 7.55-7.50 (1H, m), 7.28-7.19 (2H, m), 7.13-7.10 (1H, m), 7.06-7.01 (1H, m), 6.96 (1H, s), 6.48 (1H, d, J = 4.0 Hz), 4.52-4.51 (1H, m), 4.41 (1H, t, J = 8.0 Hz), 4.00-3.95 (1H, m), 3.80-3.72 (2H, m), 3.61 (1H, s), 3.23 (3H, br s), 3.04-2.87 (2H, m), 2.80-2.73 (1H, m), 2.67-2.62 (2H, m), 2.46-2.42 (2H, m), 2.34 (3H, br s), 2.14-2.10 (2H, m), 1.75-1.62 (1H, m), 1.50-1.39 (1H, m), 1.11-1.04 (3H, m)。
メチル 3-(5-アセチル-1-メチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
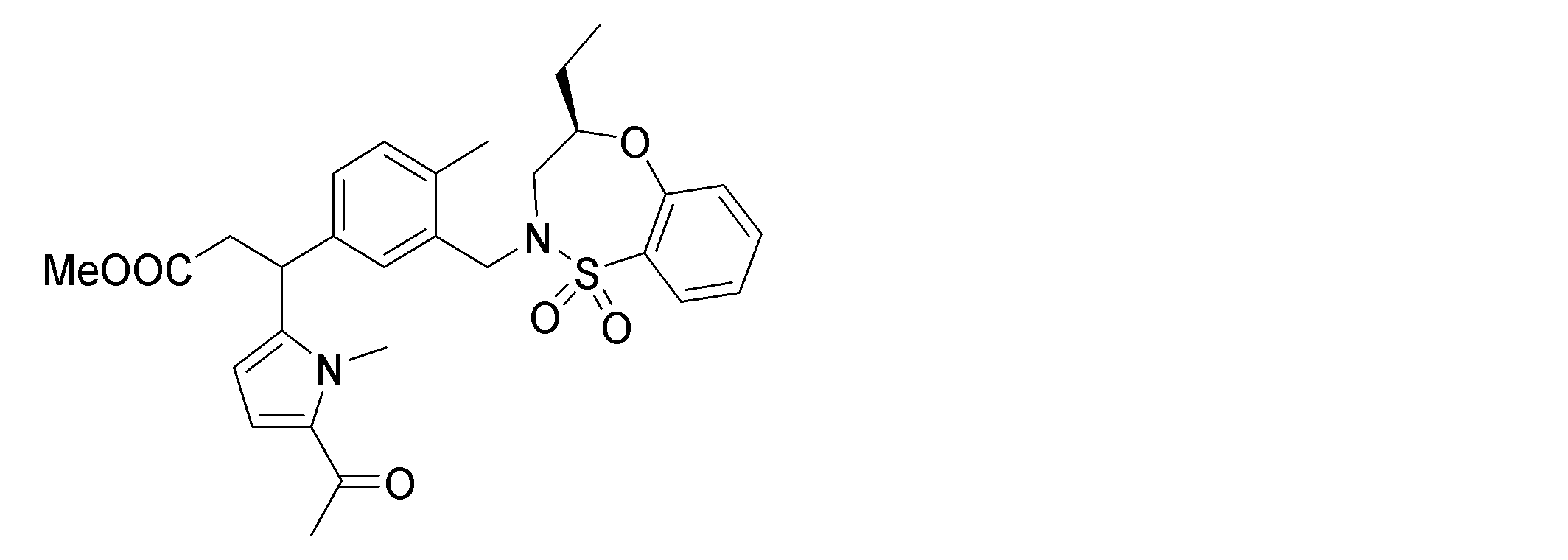
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.25 (1H, t, J = 7.6 Hz), 7.20 (1H, d, J = 7.6 Hz), 7.12-7.10 (1H, m), 7.02-6.92 (3H, m), 6.13 (1H, d,J = 4.4 Hz), 4.57-4.46 (2H, m), 4.00-3.93 (1H, m), 3.80-3.73 (1H, m), 3.68-3.65 (4H, m), 3.63 (3H, br s), 3.05-2.81 (3H, m), 2.39 (3H, s), 2.34-2.31 (3H, m), 1.74-1.61 (1H, m), 1.47-1.40 (1H, m), 1.09-1.05 (3H, m)。
メチル 3-(5-アセチル-3-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ7.87 (1H, dt,J = 8.0, 2.0 Hz), 7.52 (1H, br t, J= 8.0 Hz), 7.38 (1H, s), 7.28-7.20 (2H, m), 7.17-7.12 (2H, m), 7.05 (1H, d, J = 8.0 Hz), 4.74-4.73 (1H, m), 4.58-4.55 (1H, m), 4.03-3.95 (1H, m), 3.80-3.66 (2H, m), 3.60 (3H, br s), 3.07-2.86 (3H, m), 2.46 (3H, br s), 2.35 (3H, br s), 2.16 (3H, s), 1.74-1.60 (1H, m), 1.46-1.36 (1H, m), 1.08-1.07 (3H, m)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.26 (1H, td, J = 8.0, 1.2 Hz), 7.21 (1H, dd, J = 8.0, 1.2 Hz), 7.18-7.07 (3H, m), 6.64 (1H, s), 4.61-4.53 (2H, m), 4.06-3.98 (1H, m), 3.87-3.69 (2H, m), 3.63 (3H, s), 3.09-2.94 (3H, m), 2.50 (3H, br s), 2.35 (3H, br s), 1.74-1.65 (1H, m), 1.49-1.42 (1H, m), 1.08-1.07 (3H, m)。
メチル 3-{5-[(2,4-ジメトキシベンジル)カルバモイル]チオフェン-2-イル}-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, dd,J = 8.0, 1.6 Hz), 7.51 (1H, br t, J= 8.0 Hz), 7.27-7.24 (2H, m), 7.22-7.18 (2H, m), 7.13 (2H, br s), 7.04 (1H, br d, J = 8.0 Hz), 6.76 (1H, br t, J = 3.6 Hz), 6.45-6.40 (3H, m), 4.67 (1H, t, J = 7.6 Hz), 4.56-4.55 (1H, m), 4.47 (2H, d, J = 5.6 Hz), 3.99-3.93 (1H, m), 3.84-3.67 (8H, m), 3.59 (3H, s), 3.09-2.88 (3H, m), 2.33 (3H, s), 1.67-1.58 (1H, m), 1.44-1.36 (1H, m), 1.04-1.03 (3H, m)。
メチル 3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
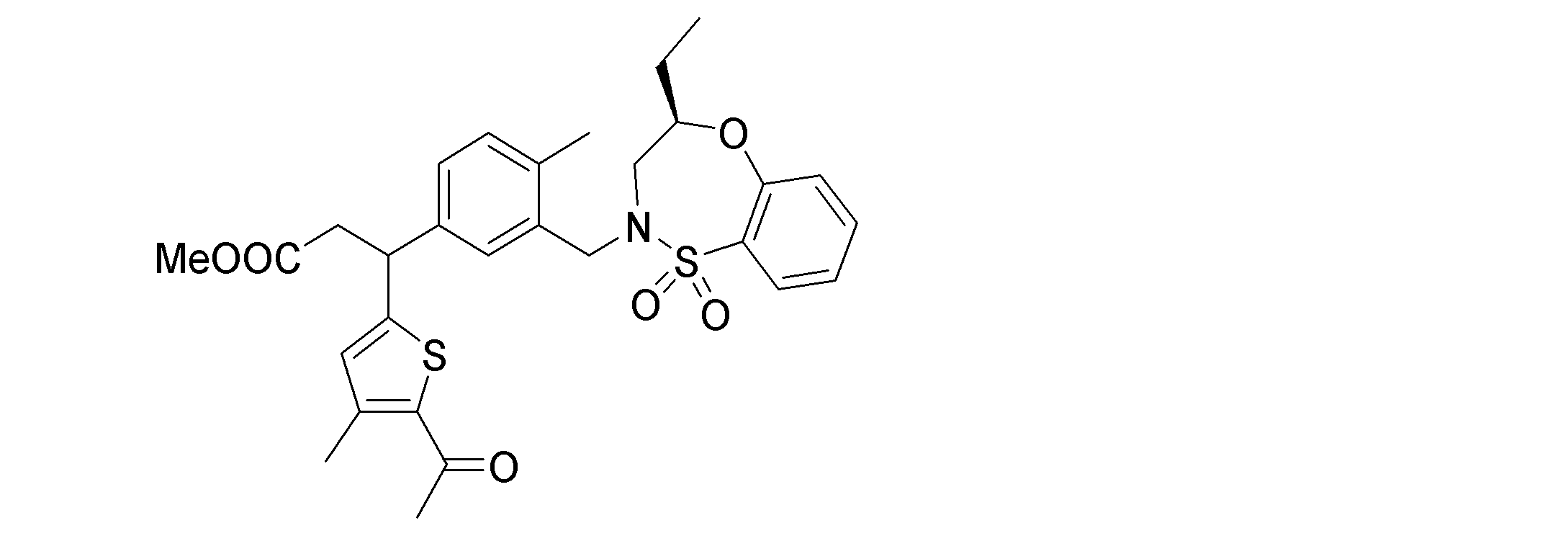
1H NMR(400 MHz, CDCl3)δ 7.89-7.86 (1H, m), 7.52 (1H, br t, J = 8.0 Hz), 7.28-7.08 (5H, m), 6.69 (1H, br s), 4.66 (1H, t, J = 7.6 Hz), 4.60-4.48 (1H, m), 4.04-3.97 (1H, m), 3.85-3.70 (2H, m), 3.62-3.60 (3H, m), 3.12-2.87 (3H, m), 2.45-2.42 (3H, m), 2.35-2.31 (3H, m), 1.73-1.63 (1H, m), 1.47-1.39 (1H, m), 1.08-1.05 (3H, m)。
メチル 3-(4-アセチル-1,3,5-トリメチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.89-7.85 (1H, m), 7.52 (1H, td, J = 8.0, 1.6 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.11 (1H, br d, J = 8.0 Hz), 7.01-6.93 (2H, m), 4.85-4.80 (1H, m), 4.55-4.51 (1H, m), 3.99-3.94 (1H, m), 3.80-3.69 (2H, m), 3.65 (3H, br s), 3.25-3.17 (4H, m), 2.99-2.88 (2H, m), 2.43-2.42 (6H, m), 2.34-2.32 (3H, m), 2.22 (3H, s), 1.75-1.61 (1H, m), 1.48-1.32 (1H, m), 1.07-1.05 (3H, m)。
メチル 3-(5-アセチル-1-エチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.15-7.08 (3H, m), 6.70 (1H, dd, J = 4.8, 2.0 Hz), 6.62 (1H, dd, J = 4.8, 2.0 Hz), 4.55 (1H, d, J = 14.0 Hz), 4.35 (1H, t, J = 8.0 Hz), 4.26 (1H, q, J = 7.2 Hz), 4.03-3.99 (1H, m), 3.84-3.70 (2H, m), 3.60 (3H, s), 3.00-2.93 (2H, m), 2.90-2.82 (1H, m), 2.36 (3H, s), 2.33 (3H, s), 1.73-1.63 (1H, m), 1.47-1.39 (1H, m), 1.30 (3H, t, J = 7.2 Hz), 1.07 (3H, t, J = 7.2 Hz)。
メチル 3-[5-(シクロプロパンカルボニル)チオフェン-2-イル]-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
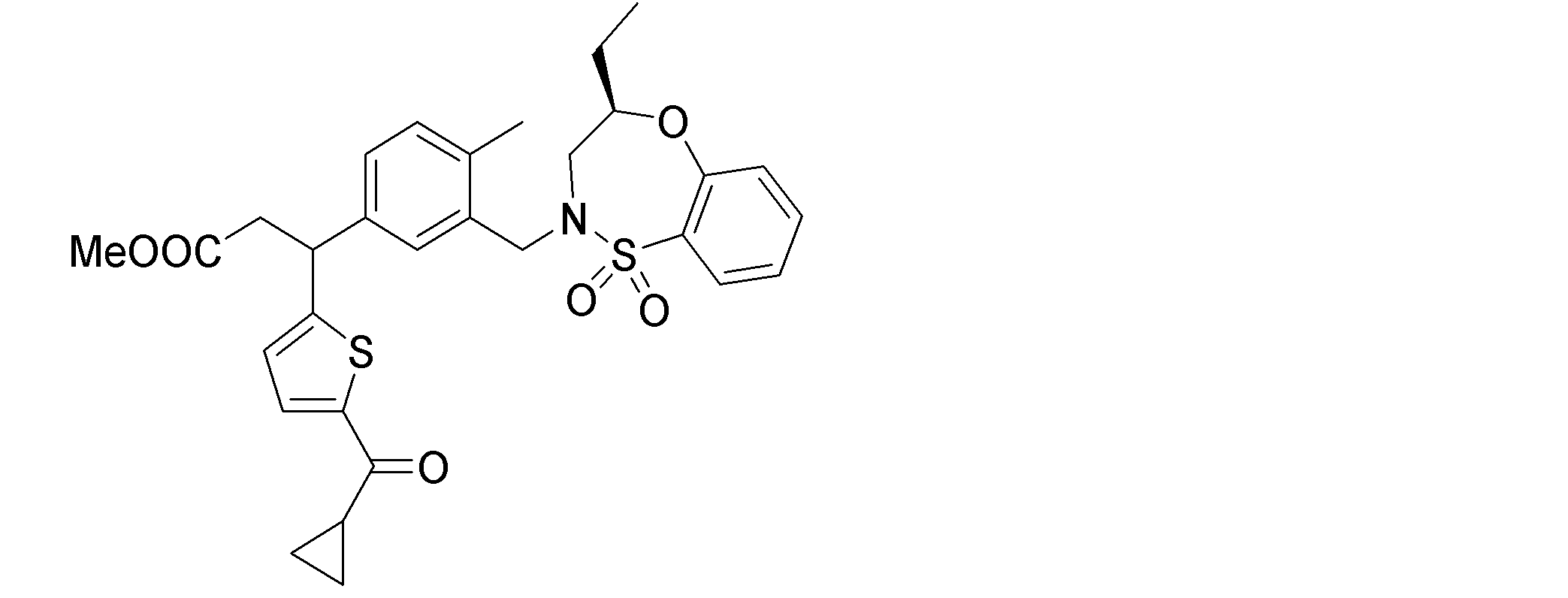
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.6 Hz), 7.63 (1H, d, J= 3.6 Hz), 7.52 (1H, td, J = 7.6, 1.6 Hz), 7.25 (1H, dd, J = 7.6, 0.8 Hz), 7.22 (1H, br d, J = 7.6 Hz), 7.15 (2H, br s), 7.06 (1H, d, J = 10.4 Hz), 6.90-6.88 (1H, m), 4.73 (1H, t, J= 7.6 Hz), 4.60-4.54 (1H, m), 4.00-3.97 (1H, m), 3.81-3.77 (1H, m), 3.76-3.71 (1H, m), 3.61 (3H, s), 3.14-2.89 (3H, m), 2.47-2.43 (1H, m), 2.35 (3H, br s), 1.70-1.61 (1H, m), 1.44-1.39 (1H, m), 1.21-1.17 (2H, m), 1.06 (3H, t, J = 7.2 Hz), 1.00-0.96 (2H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造
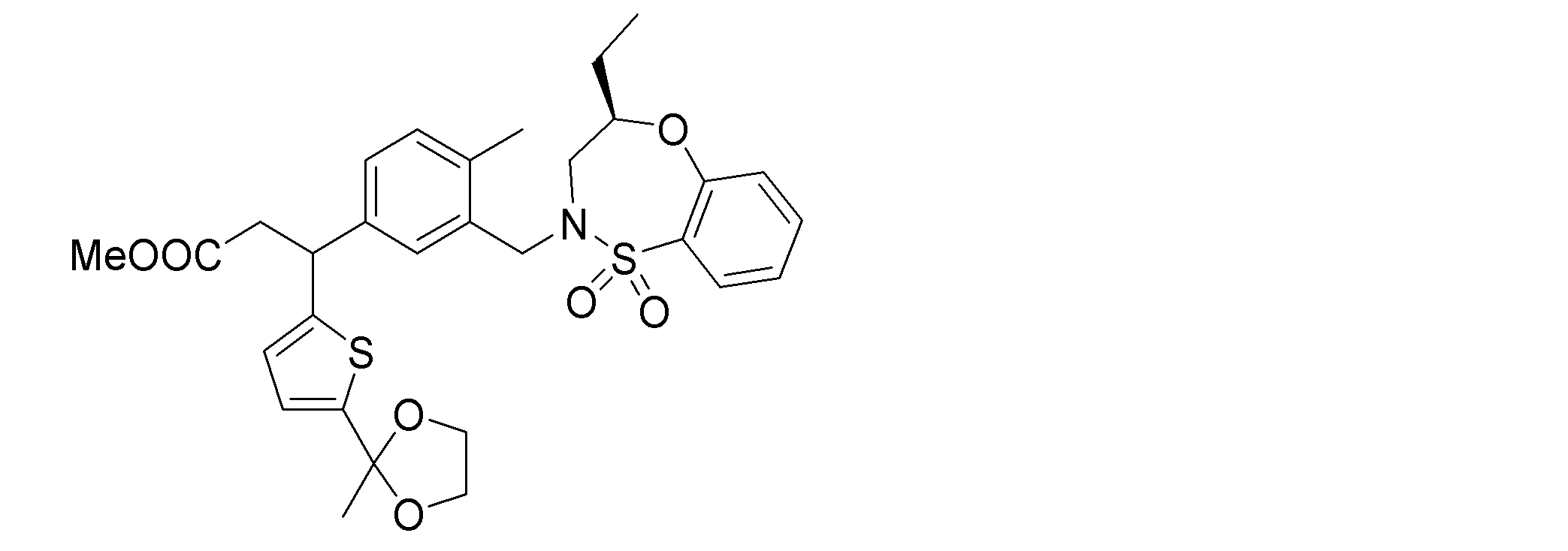
1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J= 8.0, 1.6 Hz), 7.52 (1H, td, J = 8.0, 1.6 Hz), 7.26 (1H, br t, J = 8.0 Hz), 7.21 (1H, dd, J = 8.0, 1.6 Hz), 7.14 (2H, br s), 7.06 (1H, s), 6.81 (1H, d, J= 3.6 Hz), 6.62 (1H, d, J = 3.6 Hz), 4.63 (1H, t, J = 8.0 Hz), 4.58 (1H, d, J = 14.0 Hz), 4.03-3.96 (3H, m), 3.93-3.90 (2H, m), 3.81-3.77 (1H, m), 3.73 (1H, dd, J = 15.2, 10.8 Hz), 3.59 (3H, s), 3.10-3.07 (1H, m), 3.00-2.93 (2H, m), 2.35 (3H, s), 1.76-1.61 (1H, m), 1.70 (3H, s), 1.49-1.41 (1H, m), 1.11.06 (3H, m)。
メチル 3-(5-シアノチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.6 Hz), 7.53 (1H, td, J= 7.6, 1.6 Hz), 7.44 (1H, dd, J = 4.0, 0.8 Hz), 7.28-7.16 (3H, m), 7.13-7.10 (2H, m), 6.85 (1H, br d, J = 4.0 Hz), 4.73 (1H, t, J = 7.6 Hz), 4.58-4.52 (1H, m), 4.06-3.98 (1H, m), 3.88-3.81 (1H, m), 3.76-3.69 (1H, m), 3.63 (3H, s), 3.13-2.93 (3H, m), 2.34 (3H, br s), 1.77-1.64 (1H, m), 1.50-1.39 (1H, m), 1.12-1.06 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.89 (1H, dd, J = 8.0, 1.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.28-7.24 (1H, m), 7.22 (1H, d, J= 8.0 Hz), 7.14 (2H, s), 7.06 (1H, s), 6.45 (1H, s), 4.60-4.53 (2H, m), 4.06-3.95 (3H, m), 3.92-3.86 (2H, m), 3.79 (1H, br d, J = 14.0 Hz), 3.74-3.72 (1H, m), 3.58 (3H, s), 3.05-3.01 (1H, m), 3.00-2.89 (2H, m), 2.35 (3H, s), 2.18 (3H, s), 1.74-1.64 (1H, m), 1.67 (3H, s), 1.52-1.42 (1H, m), 1.09-1.08 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾール-2-イル]プロパノエートの製造
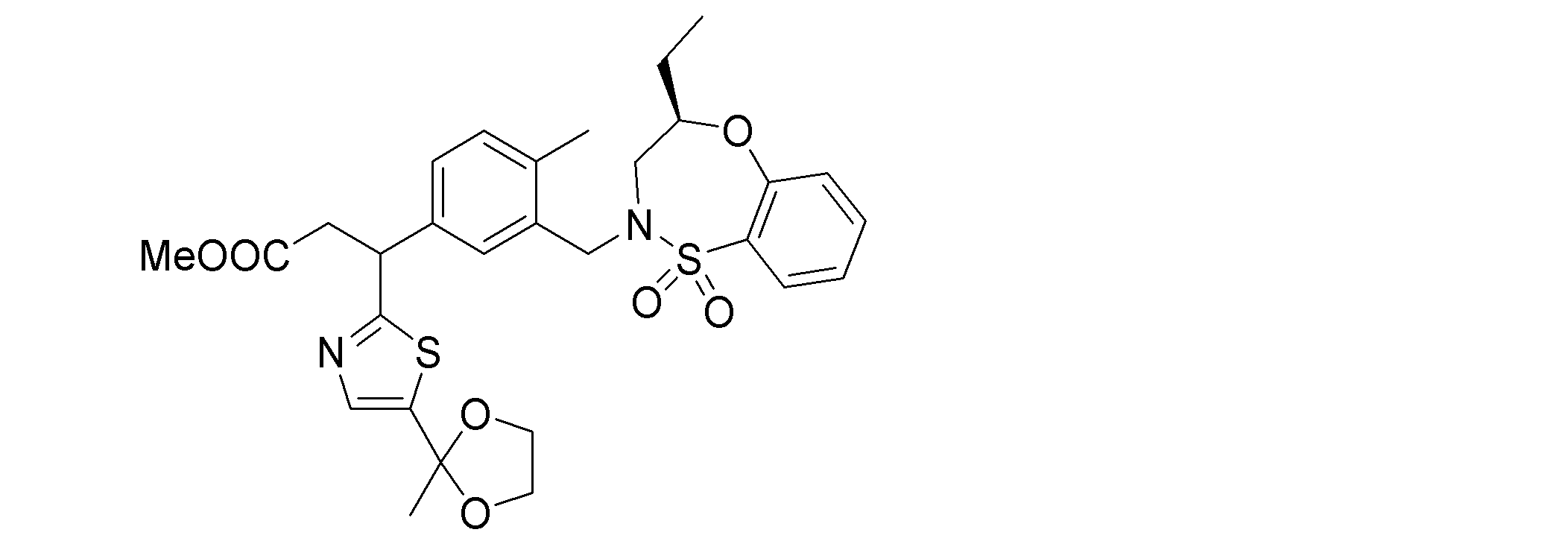
1H NMR (400 MHz, CDCl3) δ 7.89 (1H, dd, J = 7.6, 1.2 Hz), 7.56 (1H, s), 7.52 (1H, td, J = 8.0, 1.2 Hz), 7.24-7.24 (1H, m), 7.22-7.15 (3H, m), 7.13-7.12 (1H, m), 4.72 (1H, t, J = 7.6 Hz), 4.58-4.57 (1H, d, J = 14.0 Hz), 4.05-3.98 (2H, m), 3.94-3.88 (2H, m), 3.81-3.79 (1H, m), 3.74-3.71 (1H, m), 3.61 (3H, s), 3..42-3.35 (1H, ), 3.01-2.92 (2H, m), 2.36 (3H, s), 1.74-1.63 (1H, m), 1.70 (3H, s), 1.53-1.38 (1H, m), 1.09 (3H, t, J = 7.6 Hz)。
参考例148
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チアゾール-2-イル]プロパノエートの製造
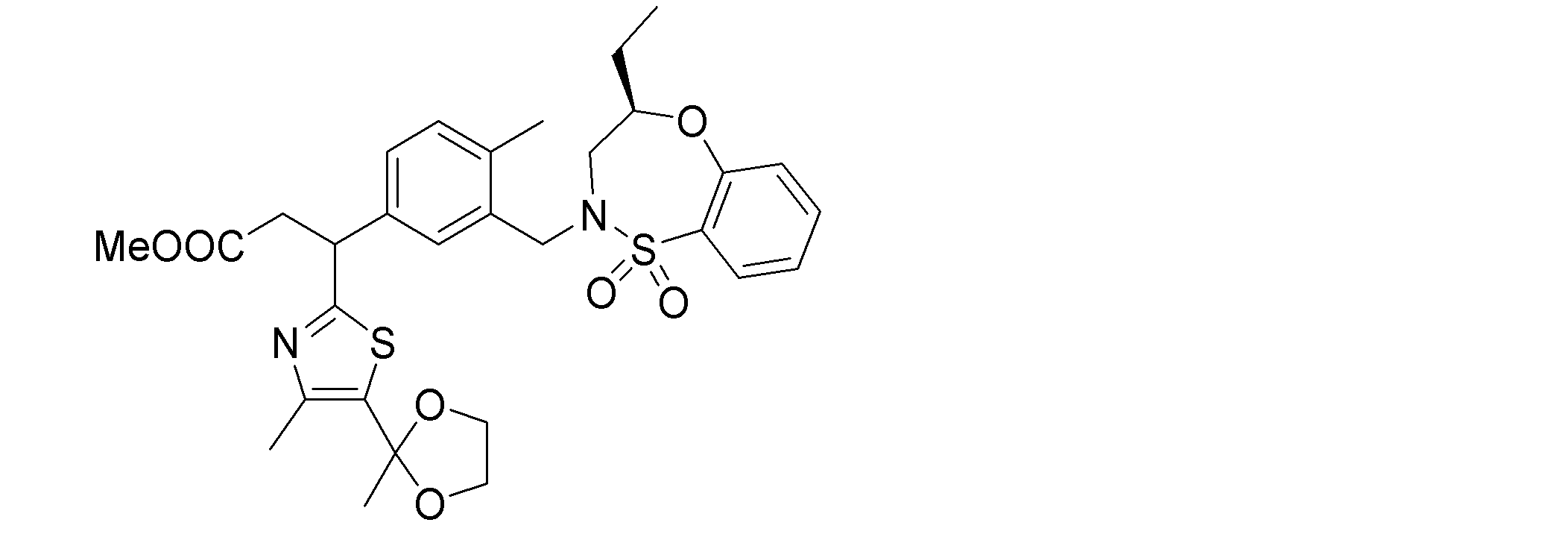
1H NMR (400 MHz, CDCl3) δ 7.90 (1H, d, J = 7.6 Hz), 7.52 (1H t, J= 7.2 Hz), 7.28-7.20 (3H, m), 7.17-7.14 (2H, m), 4.68-4.63 (1H, m), 4.58 (1H, d, J = 14.0 Hz) 4.06-3.94 (3H, m), 3.88-3.79 (3H, m), 3.76-3.70 (1H, m), 3.60 (3H, s), 3.35 (1H, dt, J = 16.0, 6.0 Hz), 3.00-2.93 (2H, m), 2.41 (3H, s), 2.36 (3H, s), 1.74-1.66 (4H, m), 1.51-1.45 (1H, m), 1.09 (3H, t, J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(6-オキソ-5,6-ジヒドロ-4H-チエノ[2,3-c]ピロール-2-イル)プロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.88 (1H, d, J = 7.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.27-7.18 (2H, m), 7.16 (2H, s), 7.11 (1H, d, J = 3.6 Hz), 6.80 (1H, s), 6.47 (1H, s), 4.75 (1H, t, J = 8.0 Hz), 4.56 (1H, d, J =14.4 Hz), 4.28 (2H, s), 4.04-3.97 (1H, m), 3.83-3.81 (1H, m), 3.75-3.68 (1H, m), 3.62 (3H, s), 3.12 (1H, dd, J = 15.6, 8.0 Hz), 3.03 (1H, dd, J = 15.6, 8.0 Hz), 2.96-2.94 (1H, m), 2.33 (3H, s), 1.73-1.64 (1H, m), 1.47-1.39 (1H, m), 1.07-1.06 (3H, m)。
メチル 3-(5-アセチルフラン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.6 Hz), 7.52 (1H, td, J= 7.6, 1.6 Hz), 7.25 (1H, td, J = 7.6, 1.2 Hz), 7.20 (1H, d, J = 8.4 Hz), 7.16-7.11 (3H, m), 7.07 (1H, d, J= 3.6 Hz), 6.16 (1H, t, J = 3.6 Hz), 4.59-4.52 (2H, m), 4.05-3.99 (1H, m), 3.83-3.81 (1H, m), 3.76-3.69 (1H, m), 3.62 (3H, s), 3.17-3.11 (1H, m), 3.00-2.88 (2H, m), 2.40 (3H, s), 2.32 (3H, br s), 1.74-1.66 (1H, m), 1.50-1.41 (1H, m), 1.08-1.07 (3H, m)。
メチル 3-(5-アセチルチオフェン-3-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
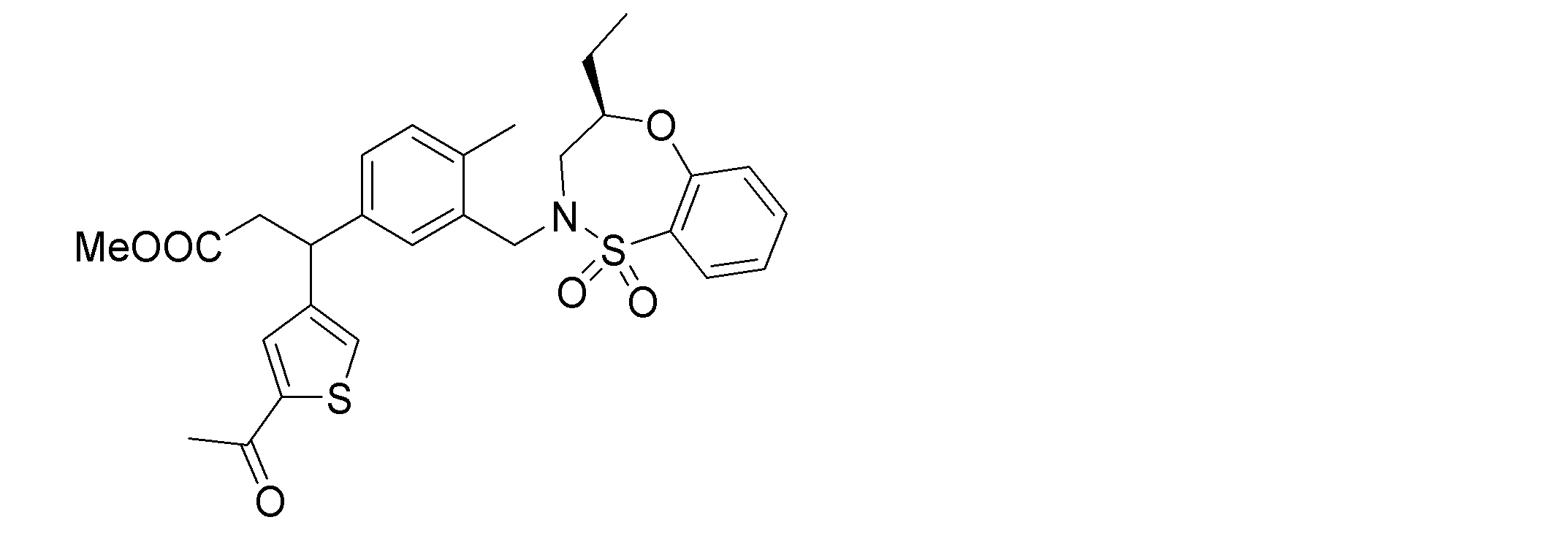
1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dd,J = 8.0, 1.6 Hz), 7.51 (1H, td, J= 8.0, 1.6 Hz), 7.45 (1H, dd, J = 3.2, 1.2 Hz), 7.29 (1H, br s), 7.24 (1H, td,J = 8.0, 1.2 Hz), 7.20 (1H, br d, J= 8.0 Hz), 7.14-7.12 (1H, m), 7.09-7.06 (2H, m), 4.55-4.51 (2H, m), 4.03-3.96 (1H, m), 3.84-3.66 (2H, m), 3.60 (3H, s), 3.06-3.00 (1H, m), 2.98-2.90 (2H, m), 2.48 (3H, s), 2.31 (3H, br s), 1.72-1.62 (1H, m), 1.46-1.37 (1H, m), 1.06 (3H, t, J = 7.2 Hz)。
メチル 3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.84 (1H, d, J= 1.6 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.27-7.19 (3H, m), 7.16-7.11 (2H, m), 7.05 (1H, d, J = 10.4 Hz), 4.66 (1H, t, J= 7.6 Hz), 4.57-4.56 (1H, m), 4.00-3.97 (1H, m), 3.82-3.68 (2H, m), 3.61 (3H, s), 3.10 (1H, dd, J = 15.6, 7.6 Hz), 3.01-2.99 (2H, m), 2.46 (3H, s), 2.35 (3H, br s), 1.72-1.62 (1H, m), 1.48-1.36 (1H, m), 1.06 (3H, t, J = 7.6 Hz)。
メチル 3-(5-シアノ-1-メチル-1H-ピロール-3-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
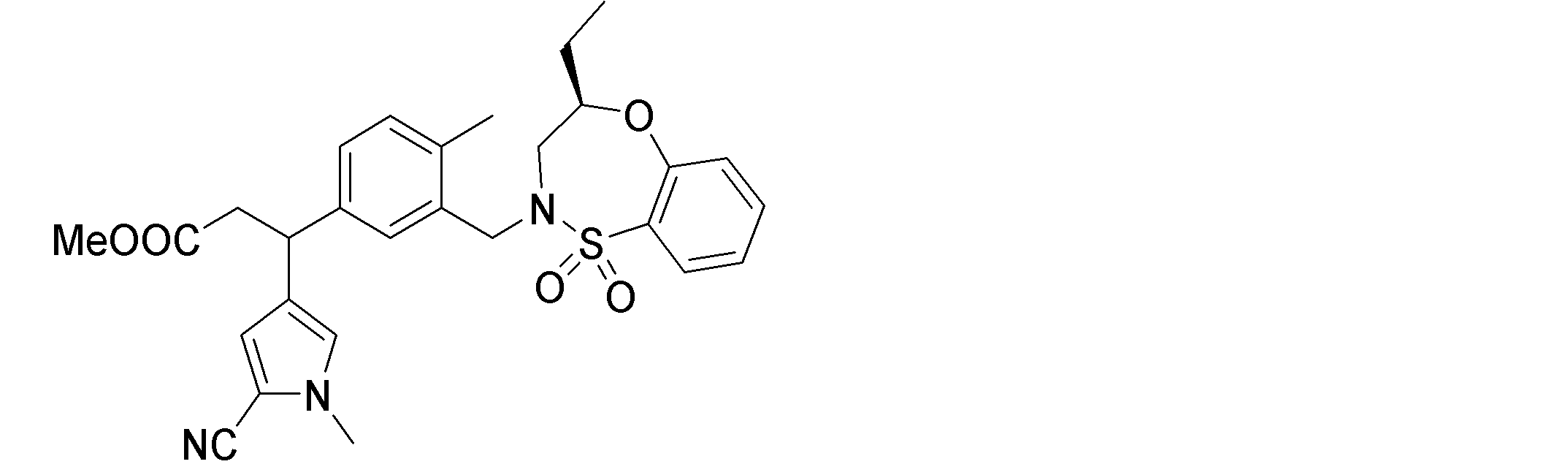
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.25 (1H, td, J = 8.0, 1.2 Hz), 7.21 (1H, d, J = 8.0 Hz), 7.12 (1H, d, J = 8.0 Hz), 7.08-7.04 (2H, m), 6.56-6.53 (2H, m), 4.54-4.53 (1H, m), 4.33 (1H, t, J = 8.0 Hz), 4.05-3.98 (1H, m), 3.86-3.69 (2H, m), 3.68 (3H, s), 3.60 (3H, s), 2.99-2.79 (3H, m), 2.32 (3H, s), 1.75-1.66 (1H, m), 1.51-1.39 (1H, m), 1.09-1.08 (3H, m)。
メチル 3-(3-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, br t, J= 8.0 Hz), 7.34 (1H, d, J = 5.6 Hz), 7.27-7.20 (3H, m), 7.16-7.10 (3H, m), 5.65 (1H, dd, J = 8.0, 8.0 Hz), 4.61-4.59 (1H, m), 4.04-3.99 (1H, m), 3.80-3.72 (2H, m), 3.56 (3H, s), 3.13-3.07 (1H, m), 3.02-2.90 (2H, m), 2.49 (3H, s), 2.35 (3H, br s), 1.71-1.59 (1H, m), 1.51-1.42 (1H, m), 1.13-1.06 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)フラン-2-イル]プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, t, J= 8.0 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.13-7.12 (2H, m), 7.06 (1H, s), 6.17 (1H, d, J= 3.2 Hz), 5.85 (1H, d, J = 3.2 Hz), 4.56 (1H, d, J = 14.0 Hz), 4.48 (1H, t, J = 8.0 Hz), 4.04-3.93 (5H, m), 3.81-3.69 (2H, m), 3.60 (3H, s), 3.07 (1H, d, J = 15.2, 7.2 Hz), 2.97 (1H, d,J = 15.2 Hz), 2.88-2.80 (1H, m), 2.34 (3H, s), 1.75-1.65 (4H, m), 1.50-1.43 (1H, m), 1.08 (3H, t, J = 7.2 Hz)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造
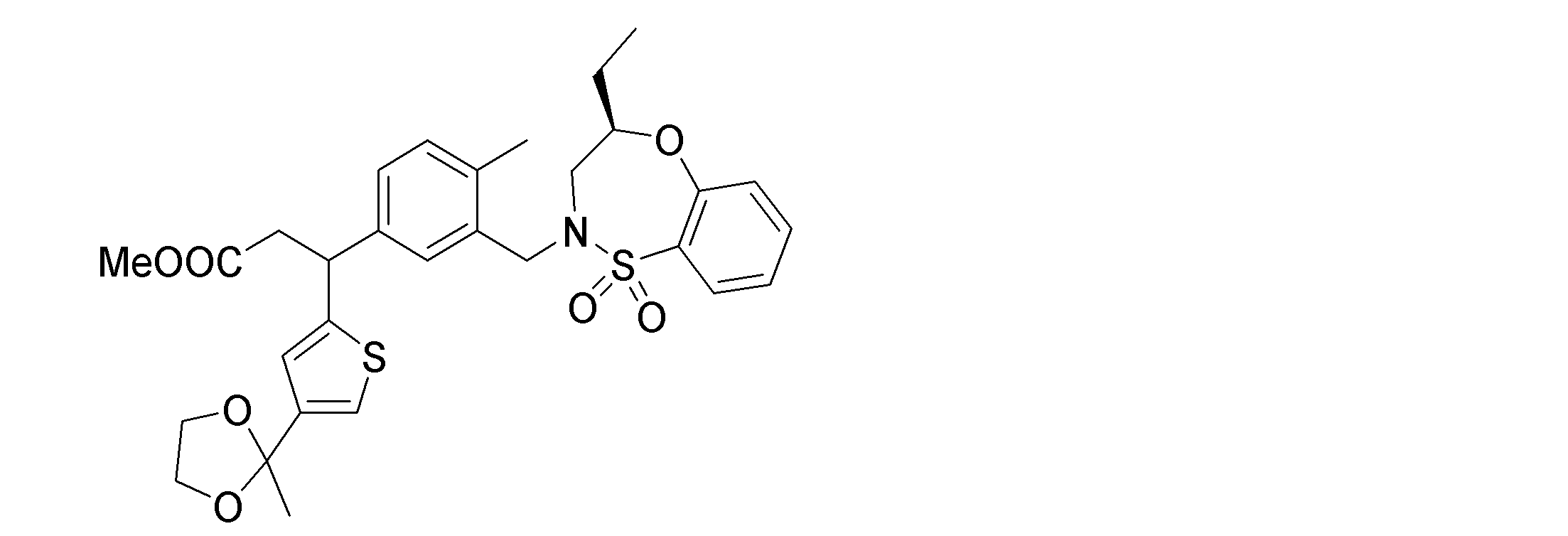
1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J= 8.0, 1.6 Hz), 7.51 (1H, td, J = 8.0, 1.6 Hz), 7.25 (1H, br t, J = 8.0 Hz), 7.20 (1H, br d, J = 8.0 Hz), 7.15-7.14 (2H, m), 7.07 (2H, br s), 6.77 (1H, dt, J = 4.8, 1.2 Hz), 4.64-4.62 (1H, m), 4.58-4.57 (1H, m), 4.03-3.96 (3H, m), 3.89-3.69 (4H, m), 3.59 (3H, s), 3.08 (1H, dd, J = 15.6, 6.8 Hz), 3.00-2.93 (2H, m), 2.35 (3H, br s), 1.73-1.70 (1H, m), 1.62 (3H, s), 1.45-1.41 (1H, m), 1.09-1.07 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチル-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

Isomer 1: 1H NMR(400 MHz, CDCl3)δ 7.90 (1H, dd, J = 8.0, 1.6 Hz), 7.53 (1H, td, J = 8.0, 1.6 Hz), 7.26 (1H, br t, J = 8.0 Hz), 7.22 (1H, dd, J = 8.0, 1.6 Hz), 7.14 (2H, br s), 7.06 (1H, d, J = 11.2 Hz), 6.77 (1H, dd, J = 3.6, 0.8 Hz), 6.69 (1H, br d, J = 3.6 Hz), 4.60-4.57 (1H, m), 4.28-4.25 (1H, m), 4.02-3.88 (5H, m), 3.86-3.69 (2H, m), 3.59 (3H, s), 3.19-3.13 (1H, m), 2.95 (1H, dd, J = 15.2, 2.0 Hz), 2.36-2.34 (3H, m), 1.73-1.62 (4H, m), 1.46-1.38 (1H, m), 1.11-1.04 (6H, m)。
Isomer 2: 1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J = 8.0, 1.6 Hz), 7.53 (1H, td, J = 8.0, 1.6 Hz), 7.26 (1H, br t, J = 8.0 Hz), 7.22 (1H, dd, J = 8.0, 1.6 Hz), 7.20-7.17 (1H, m), 7.11-7.07 (2H, m), 6.82 (1H, d, J = 3.6 Hz), 6.73 (1H, t, J = 3.6 Hz), 4.57 (1H, d, J = 14.0 Hz), 4.21 (1H, d, J = 11.2 Hz), 4.07-3.89 (5H, m), 3.79-3.68 (2H, m), 3.43 (3H, s), 3.21-3.10 (1H, m), 2.94 (1H, dd, J = 15.2, 8.8 Hz), 2.33 (3H, s), 1.73-1.65 (4H, m), 1.51-1.42 (1H, m), 1.20 (3H, d, J = 6.8 Hz), 1.15-1.07 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチル-3-[4-メチル-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造
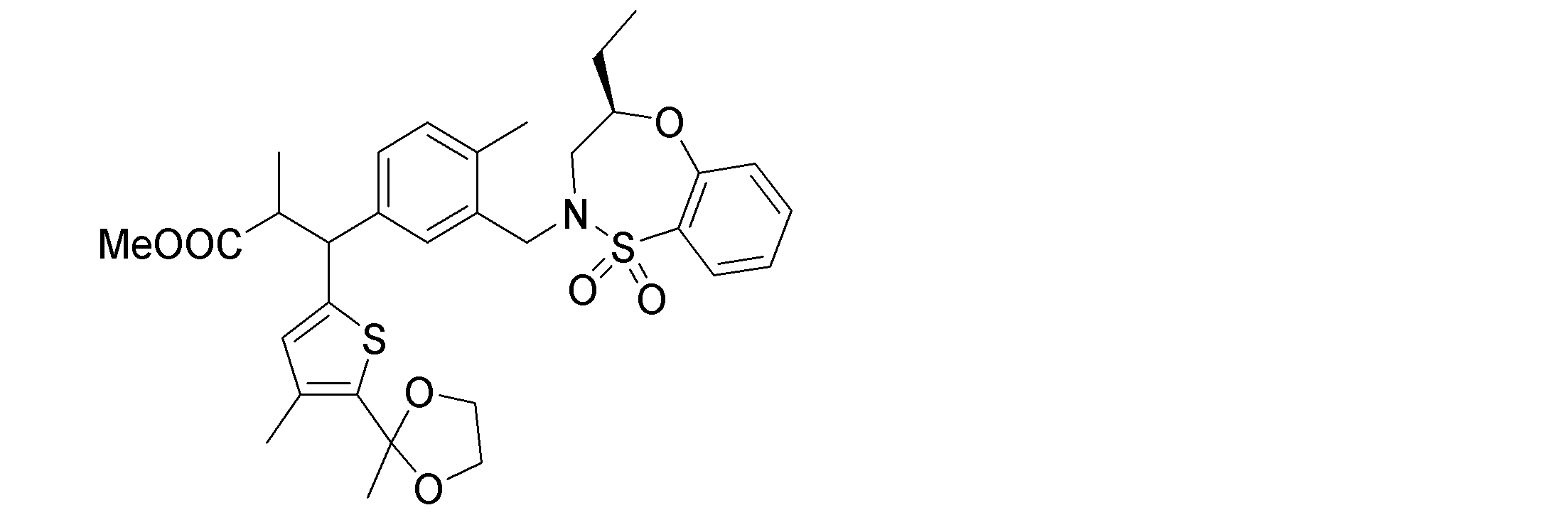
1H NMR (400 MHz, CDCl3) δ 7.89 (1H, d, J = 7.6 Hz), 7.53 (1H, t, J= 7.6 Hz), 7.28-7.21 (2H, m), 7.18-7.04 (3H, m), 6.57-6.51 (1H, m), 4.59-4.57 (1H, m), 4.20-4.19 (1H, m), 4.07-3.94 (3H, m), 3.92-3.82 (3H, m), 3.78-3.69 (1H, m), 3.60-3.42 (3H, m), 3.16-3.08 (1H, m), 3.00-2.92 (1H, m), 2.37-2.33 (3H, m), 2.19-2.16 (3H, m), 1.74-1.62 (1H, m), 1.66-1.65 (3H, m), 1.53-1.37 (1H, m), 1.21-0.99 (6H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチル-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)フラン-2-イル]プロパノエートの製造
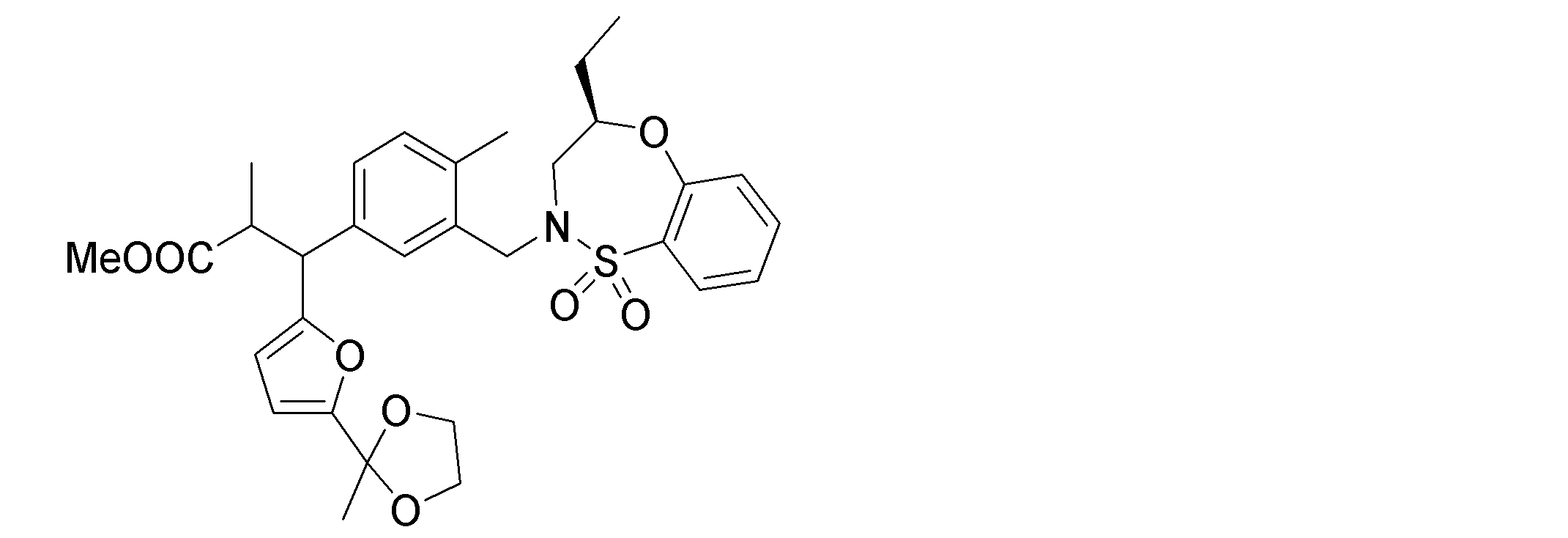
1H NMR(400 MHz, CDCl3)δ 7.89 (1H, br d,J = 8.0 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.28-7.23 (1H, m), 7.22-7.04 (4H, m), 6.18-6.14 (1H, m), 6.01-5.92 (1H, m), 4.58-4.51 (1H, m), 4.15-4.08 (1H, m), 4.04-3.94 (5H, m), 3.85-3.71 (2H, m), 3.63-3.44 (3H, m), 3.23-3.16 (1H, m), 2.98-2.93 (1H, m), 2.35-2.31 (3H, m), 1.69-1.66 (4H, m), 1.52-1.48 (1H, m), 1.16-0.98 (6H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチル-3-[4-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

Isomer 1: 1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J = 7.6, 1.6 Hz), 7.53 (1H, br t, J = 8.0 Hz), 7.28-7.25 (1H, m), 7.24-7.18 (2H, m), 7.11-7.08 (3H, m), 6.86 (1H, dd, J = 6.0, 1.2 Hz), 4.57-4.56 (1H, m), 4.24-4.23 (1H, m), 4.03-3.99 (3H, m), 3.85-3.68 (4H, m), 3.43 (3H, s), 3.19-3.12 (1H, m), 2.96-2.91 (1H, m), 2.32 (3H, s), 1.73-1.69 (1H, m), 1.62 (3H, s), 1.51-1.44 (1H, m), 1.20-1.19 (3H, m), 1.24-1.09 (3H, m)。
Isomer 2: 1H NMR(400 MHz, CDCl3)δ 7.89 (1H, br d, J = 8.0 Hz), 7.53 (1H, td, J = 8.0, 1.6 Hz), 7.28-7.24 (1H, m), 7.22-7.19 (1H, m), 7.16-7.14 (2H, m), 7.09-7.07 (1H, m), 7.03 (1H, br s), 6.83 (1H, br s), 4.60-4.57 (1H, m), 4.28-4.27 (1H, m), 4.04-3.97 (3H, m), 3.87-3.69 (4H, m), 3.58 (3H, br s), 3.19-3.12 (1H, m), 2.97-2.93 (1H, m), 2.36-2.34 (3H, m), 1.71-1.67 (1H, m), 1.62-1.61 (3H, m), 1.46-1.37 (1H, m), 1.11-1.03 (6H, m)。
メチル 3-[5-(ジメチルカルバモイル)フラン-2-イル]-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
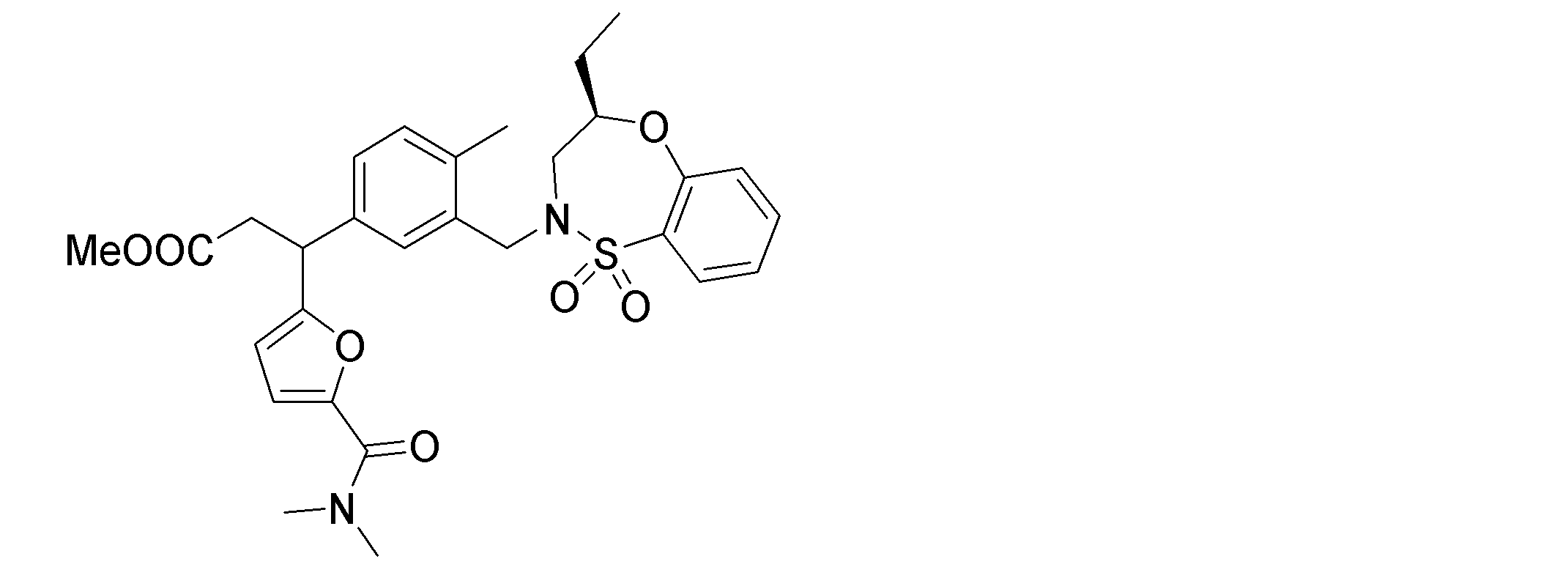
1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd,J = 7.6, 1.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.26 (1H, t, J = 7.6 Hz), 7.21 (1H, d, J = 7.6 Hz), 7.15-7.10 (3H, m), 6.90 (1H, d, J = 3.6 Hz), 6.10 (1H, d, J = 3.6 Hz), 4.56-4.52 (2H, m), 4.04-3.99 (1H, m), 3.82 (1H, d,J = 14 Hz), 3.76-3.69 (1H, m), 3.61 (3H, s), 3.13-2.86 (9H, m), 2.32 (3H, s), 1.75-1.65 (1H, m), 1.52-1.42 (1H, m), 1.08 (3H, t, J = 7.2 Hz)。
メチル 3-(5-カルバモイルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, br t, J= 8.0 Hz), 7.35 (1H, d, J = 3.6 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, br d, J = 8.0 Hz), 7.14 (2H, br s), 7.08 (1H, d, J = 8.4 Hz), 6.81-6.79 (1H, m), 6.01 (2H, br s), 4.70 (1H, t, J = 8.0 Hz), 4.57-4.55 (1H, m), 4.02-3.95 (1H, m), 3.82-3.69 (2H, m), 3.60 (3H, s), 3.09 (1H, dd, J = 15.6, 7.6 Hz), 3.02-2.91 (2H, m), 2.33 (3H, s), 1.72-1.62 (1H, m), 1.46-1.38 (1H, m), 1.07-1.06 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR (400 Mz, CDCl3) δ 7.87 (1H, dt, J = 7.6, 1.2 Hz), 7.55-7.50 (1H, m), 7.49 (1H, d, J = 4.0 Hz), 7.27-7.11 (5H, m), 6.97-6.95 (1H, m), 4.69-4.67 (1H, m), 4.56-4.55 (1H, m), 4.04-3.97 (1H, m), 3.83-3.69 (2H, m), 3.60 (3H, brs), 2.97-2.90 (1H, m), 2.49-2.48 (3H, m), 2.33 (3H, s), 1.81-1.56 (1H, m), 1.51-1.35 (1H, m), 1.29 (3H, br s), 1.23/-1.21 (3H, m), 1.10-1.08 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-9-フルオロ-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.51 (1H, dd, J = 4.0, 1.6 Hz), 7.44-7.39 (1H, m), 7.29/7.28 (1H, m), 7.25-7.22 (1H, m), 7.14 (1H, br d, J = 8.0 Hz), 7.00-6.94 (3H, m), 4.72 (1H, s), 4.58-4.55 (1H, m), 4.50-4.43 (1H, m), 4.31-4.28 (1H, m), 3.62 (3H, s), 3.33 (1H, d, J = 14.4, 4.0 Hz), 3.10 (1H, d, J= 14.4, 1.2 Hz), 2.49 (3H, br s), 2.34 (3H, br s), 1.68-1.56 (1H, m), 1.55-1.45 (1H, m), 1.32 (3H, br s), 1.25 (3H, s), 1.02-1.01 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[2,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 8.51-8.49 (1H, m), 8.26 (1H, br d, J = 8.0 Hz), 7.51 (1H, d, J = 4.0 Hz), 7.30-7.24 (2H, m), 7.21 (1H, s), 7.14 (1H, d, J = 8.0 Hz), 6.98 (1H, t, J = 4.0 Hz), 4.69 (1H, d, J = 5.6 Hz), 4.47-4.42 (2H, m), 4.11-4.05 (1H, m), 3.62-3.59 (4H, m), 3.16 (1H, br d, J = 15.2 Hz), 2.50 (3H, s), 2.33 (3H, br s), 1.81-1.68 (1H, m), 1.60-1.52 (1H, m), 1.30 (3H, s), 1. 24 (3H, s), 1.09 (3H, t, J = 7.2 Hz)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-({4-[2-(ベンジロキシ)エチル]-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル}メチル)-4-メチルフェニル]-2-メチルプロパノエートの製造
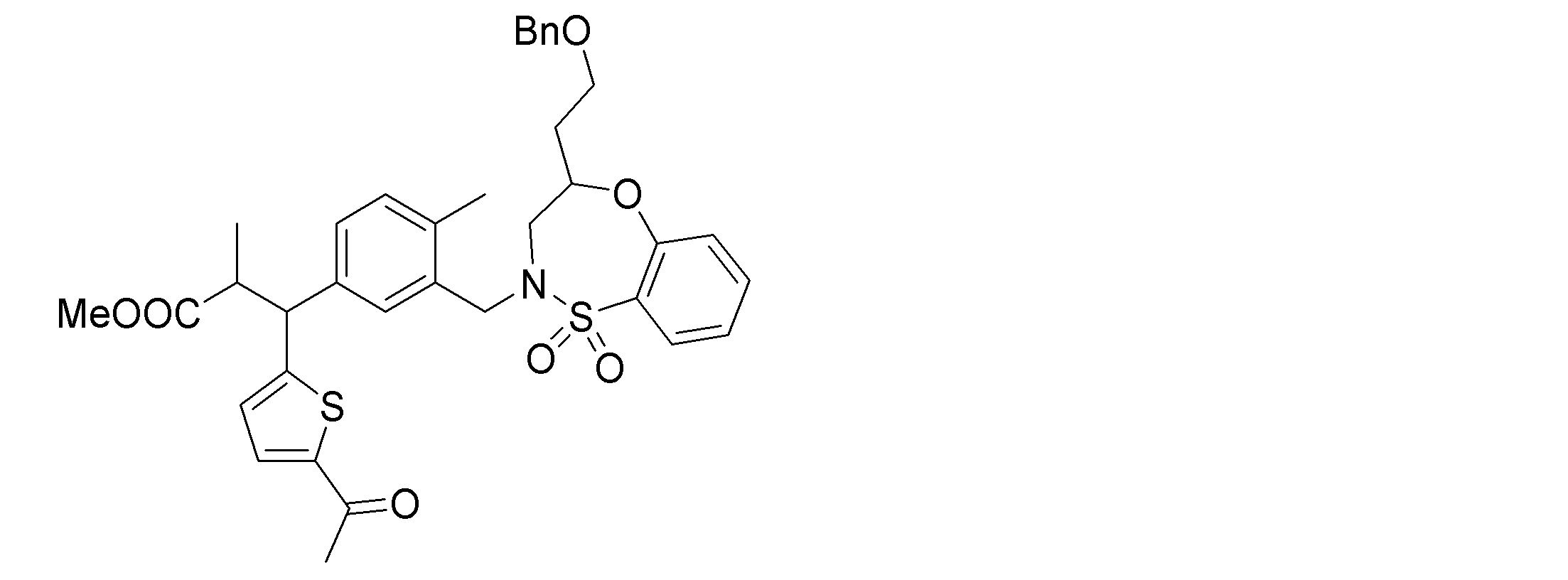
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d,J = 8.0 Hz), 7.49 (1H, br t, J = 8.0 Hz), 7.43 (1H, t, J = 4.0 Hz), 7.35-7.24 (6H, m), 7.14-7.09 (3H, m), 7.00 (1H, br t, J = 8.0 Hz), 6.89 (1H, br t,J = 4.0 Hz), 4.60-4.47 (3H, m), 4.36-4.31 (2H, m), 3.87-3.73 (3H, m), 3.66-3.61 (1H, m), 3.59 (3H, br s), 3.25-3.18 (1H, m), 2.93-2.88 (1H, m), 2.43 (3H, br s), 2.31 (3H, br s), 1.92-1.83 (1H, m), 1.67-1.60 (1H, m), 1.06 (3H, d, J = 7.2 Hz)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 9.03 (1H, s), 8.66 (1H, dd, J = 5.6, 2.0 Hz), 7.47 (1H, dd,J = 4.0, 2.4 Hz), 7.17 (2H, s), 7.12-7.09 (2H, m), 6.93-6.92 (1H, m), 4.47-4.44 (1H, m), 4.38 (2H, d, J = 12.0 Hz), 4.21-4.09 (1H, m), 3.61 (3H, s), 3.51-3.41 (1H, m), 3.23-3.11 (2H, m), 2.47 (3H, br s), 2.33 (3H, s), 1.74-1.63 (1H, m), 1.56-1.47 (1H, m), 1.08-1.02 (6H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[4-(2-ヒドロキシエチル)-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J = 8.0, 1.6 Hz), 7.69-7.44 (3H, m), 7.29-7.22 (1H, m), 7.17-7.10 (3H, m), 6.93 (1H, d, J = 4.0 Hz), 4.77-4.59 (1H, m), 4.44-4.33 (2H, m), 4.02-3.80 (4H, m), 3.60-3.55 (3H, m), 3.31-3.09 (1H, m), 2.92-2.81 (1H, m), 2.49-2.34 (6H, m), 1.96-1.77 (1H, m), 1.62-1.45 (1H, m), 1.07-1.00 (3H, m)。
(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}-4-フルオロチオフェン-2-イル)(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)メタノールの製造

1H NMR(400 MHz, CDCl3)δ 7.31 (1H, br s), 7.22-7.16 (3H, m), 7.10 (1H, d, J = 7.6 Hz), 6.82 (2H, br d, J = 8.8 Hz), 6.40 (1H, d, J = 20.8 Hz), 5.76 (1H, br s), 5.09 (1H, q, J = 6.4 Hz), 4.44-4.43 (4H, m), 3.74 (3H, s), 2.32 (1H, br s), 2.25 (3H, s), 1.39-1.38 (3H, m), 0.82 (9H, br s), 0.01 (3H, br s), -0.05 (3H, br s)。
メチル 3-(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}-4-フルオロチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.25-7.22 (3H, m), 7.13 (1H, br t, J = 8.0 Hz), 7.04 (1H, dd, J = 8.0, 3.2 Hz), 6.84 (2H, br d, J = 8.8 Hz), 6.55 (1H, d, J = 19.6 Hz), 5.07 (1H, q, J = 6.4 Hz), 4.45-4.41 (5H, m), 3.77 (3H, s), 3.54 (3H, s), 2.23 (3H, s), 1.39-1.36 (3H, m), 1.24 (3H, s), 1.15 (3H, s), 0.82 (9H, br s), 0.001 (3H, br s), -0.07 (3H, br s)。
メチル 3-(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}-4-フルオロチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ7.31 (1H, s), 7.18 (1H, br t, J = 8.0 Hz), 7.10 (1H, dd, J= 8.0, 4.0 Hz), 6.60 (1H, d, J = 19.6 Hz), 5.12 (1H, q, J = 6.4 Hz), 4.67 (2H, br s), 4.50-4.47 (1H, m), 3.60 (3H, s), 2.31 (3H, s), 2.32 (3H, s), 1.59 (1H, br s), 1.43-1.42 (3H, m), 1.29 (3H, s), 1.20 (3H, br s), 0.87 (9H, br s), 0.054-0.051 (3H, m), 0.00--0.015 (3H, m)。
メチル 3-(5-{1-[(tert-ブチルジメチルシリル)オキシ]エチル}-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
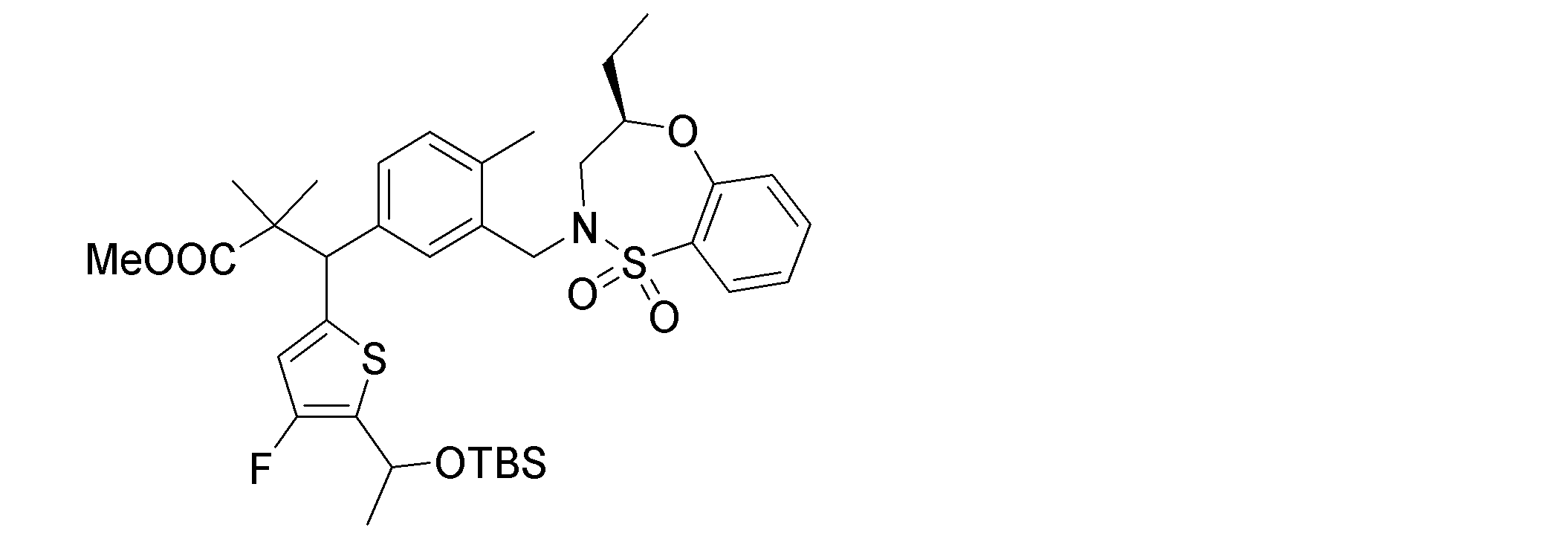
1H NMR(400 MHz, CDCl3)δ 7.84 (1H, d,J = 8.0 Hz), 7.47 (1H, br t, J = 8.0 Hz), 7.23-7.13 (3H, m), 7.11-7.07 (2H, m), 6.57-6.50 (1H, m), 5.09-5.06 (1H, m), 4.52-4.49 (1H, m), 4.42-4.38 (1H, m), 3.99-3.95 (1H, m), 3.80-3.65 (2H, m), 3.55 (3H, br s), 2.95-2.89 (1H, m), 2.29 (3H, br s), 1.69-1.59 (1H, m), 1.39-1.36 (4H, m), 1.22-1.21 (3H, m), 1.14-1.13 (3H, m), 1.06-1.02 (3H, m), 0.82-0.79 (9H, m), 0.004--0.009 (3H, m), -0.052--0.091 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-フルオロ-5-(1-ヒドロキシエチル)チオフェン-2-イル]-2,2-ジメチルプロパン酸の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d,J = 8.0 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.27-7.20 (4H, m), 7.13-7.11 (1H, m), 6.69-6.64 (1H, m), 5.13 (1H, q, J = 6.4 Hz), 4.58-4.48 (2H, m), 4.02-4.00 (1H, m), 3.86-3.73 (2H, m), 3.01-2.96 (1H, m), 2.32 (3H, s), 1.73-1.60 (1H, m), 1.52-1.48 (3H, m), 1.45-1.41 (1H, m), 1.29 (3H, s), 1.18 (3H, s), 1.10-1.06 (3H, m)。
メチル 3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-フルオロ-5-(1-ヒドロキシエチル)チオフェン-2-イル]-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd, J= 8.0, 1.6 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.27-7.17 (4H, m), 7.12 (1H, d, J= 7.6 Hz), 6.63-6.61 (1H, m), 5.15 (1H, q, J= 6.4 Hz), 4.54-4.50 (1H, m), 4.48 (1H, br s), 4.05-4.01 (1H, m), 3.83 (1H, d, J = 14.0 Hz), 3.76-3.70 (1H, m), 3.62 (3H, br s), 3.39-3.35 (1H, m), 3.00-2.95 (1H, m), 2.33 (3H, s), 1.72-1.66 (1H, m), 1.52-1.50 (3H, m), 1.48-1.42 (1H, m), 1.28 (3H, s), 1.19 (3H, br s), 1.09 (3H, t, J = 7.2 Hz)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
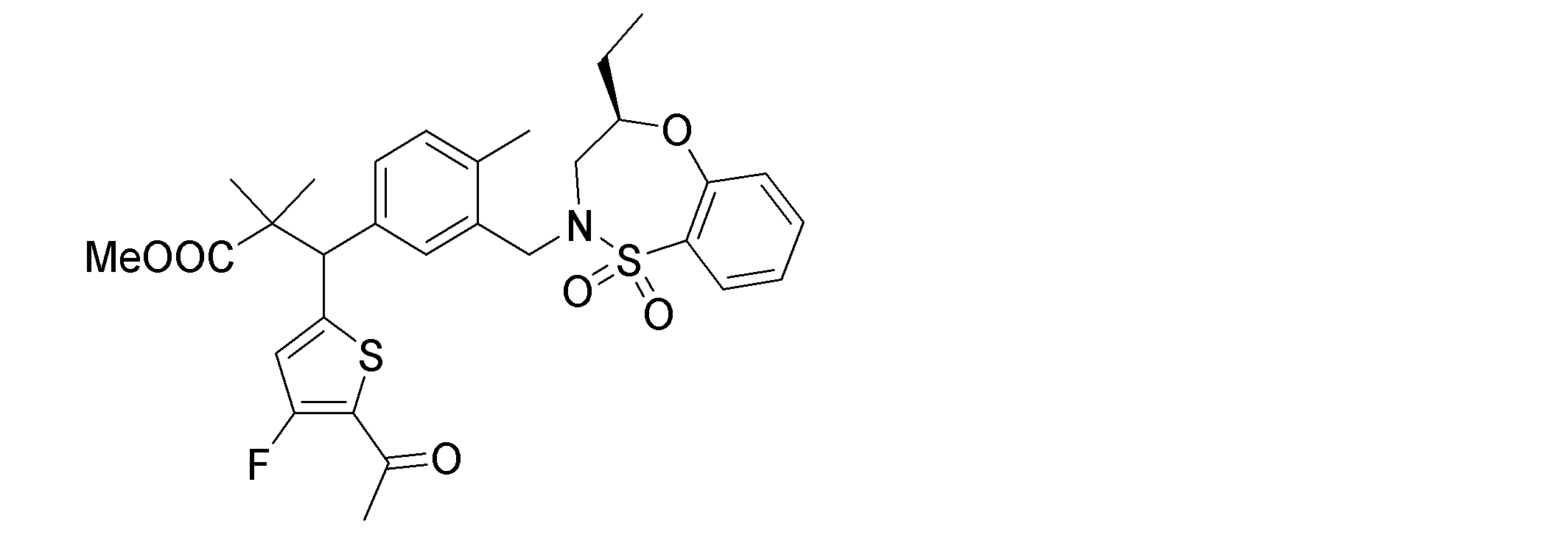
1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dt, J= 8.0, 1.6 Hz), 7.53 (1H, br t, J = 8.0 Hz), 7.29-7.14 (5H, m), 6.75 (1H, br d, J= 6.8 Hz), 4.57-4.54 (2H, m), 4.06-4.00 (1H, m), 3.85-3.82 (1H, m), 3.77-3.71 (1H, m), 3.64 (3H, br s), 3.00-2.93 (1H, m), 2.51 (3H, br s), 2.34 (3H, s), 1.71-1.65 (1H, m), 1.50-1.38 (1H, m), 1.30 (3H, s), 1.20 (3H, s), 1.09-1.08 (3H, m)。
メチル 3-(4-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-1-トリチル-1H-ベンゾ[d]イミダゾール-6-イル)-3-[5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]プロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.88 (1H, d, J = 7.6 Hz), 7.78 (1H, s), 7.47-7.42 (1H, m), 7.31-7.26 (9H, m), 7.22-7.18 (1H, m), 7.15-7.11 (8H, m), 6.70 (1H, d, J = 2.0 Hz), 6.29-6.26 (2H, m), 4.77 (1H, d, J = 15.2 Hz), 4.55-4.54 (1H. m), 4.48 (1H, q, J = 7.2 Hz), 4.03-3.84 (6H, m), 3.53 (3H, s), 3.29-3.23 (1H, m),2.76-2.69 (1H, m), 2.59-2.51 (1H, m), 1.73-1.64 (1H, m), 1.68 (3H, s), 1.45-1.40 (1H, m), 1.03-1.02 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(4-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-1H-ベンゾ[d]イミダゾール-6-イル)プロパノエートの製造
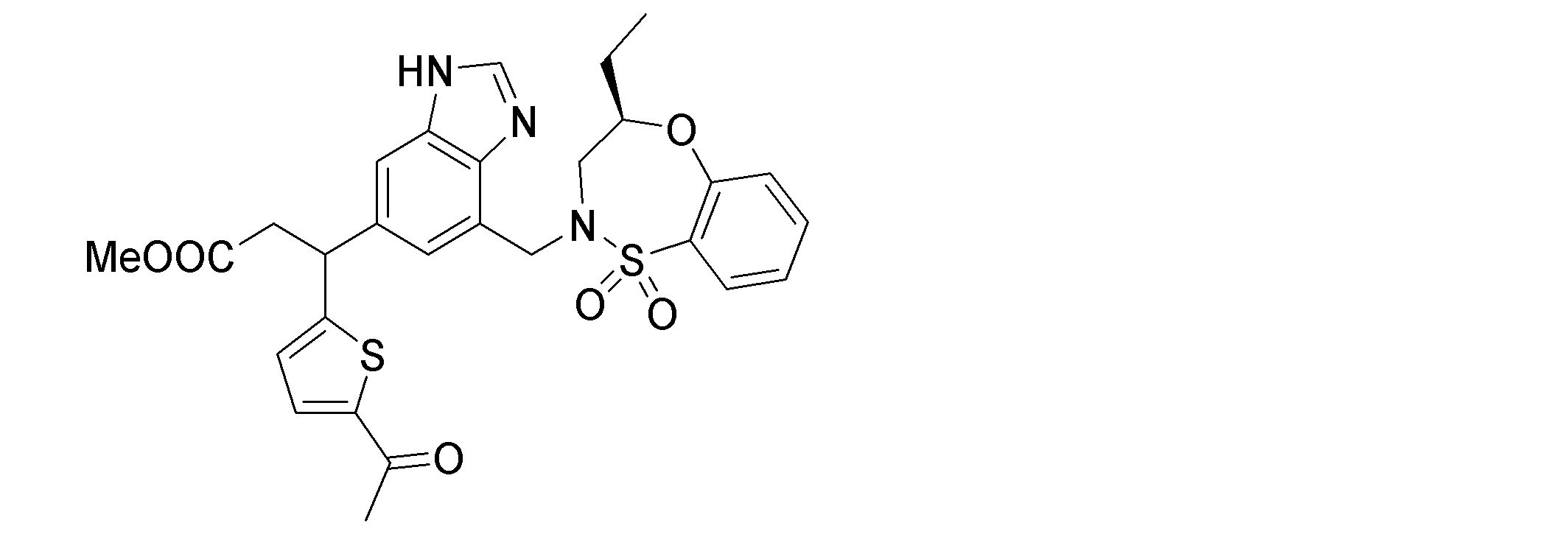
1H NMR (400 MHz, CDCl3) δ 10.6 (1H, s), 8.11 (1H, s), 7.90 (1H, dd, J = 7.6, 1.2 Hz), 7.74 (1H, s), 7.56 (1H, t, J = 7.6 Hz), 7.52-7.48 (1H, m), 7.31-7.24 (2H, m), 6.91 (1H, s), 6.88 (1H, br s), 4.87 (1H, t, J = 7.6 Hz), 4.75-4.72 (1H, m), 4.15-4.07 (1H, m), 3.97-3.93 (1H, m), 3.74-3.71 (1H, m), 3.60 (3H, br s), 3.21-3.07 (3H, m), 2.47 (3H, s), 1.82-1.74 (1H, m), 1.59-1.51 (1H, m), 1.15-1.14 (3H, m)。
メチル 3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造
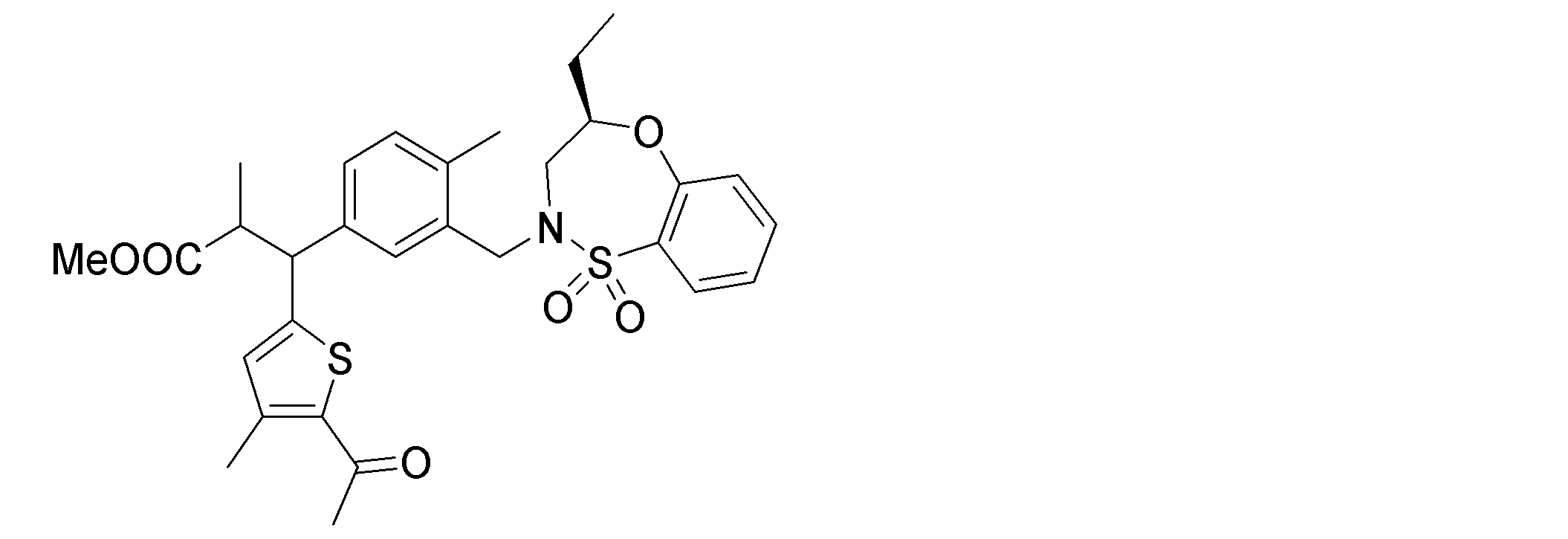
1H NMR (400 MHz, CDCl3) δ 7.89 (1H, d, J = 7.6 Hz), 7.53 (1H, t, J= 7.6 Hz), 7.26 (1H, t, J = 7.6 Hz), 7.22 (1H, d, J = 8.0 Hz), 7.17-7.07 (3H, m), 6.77-6.32 (1H, m), 4.60-4.53 (1H, m), 4.30-4.25 (1H, m), 4.06-3.98 (1H, m), 3.89-3.79 (1H, m), 3.75-3.68 (1H, mz), 3.62-3.46 (3H, m), 3.24-3.14 (1H, m), 2.99-2.92 (1H, m), 2.47-2.42 (6H, m), 2.34-2.31 (3H, m), 1.73-1.62 (1H, m), 1.49-1.33 (1H, m), 1.11-1.04 (6H, m)。
メチル 3-(5-アセチルチアゾール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-プロパノエートの製造
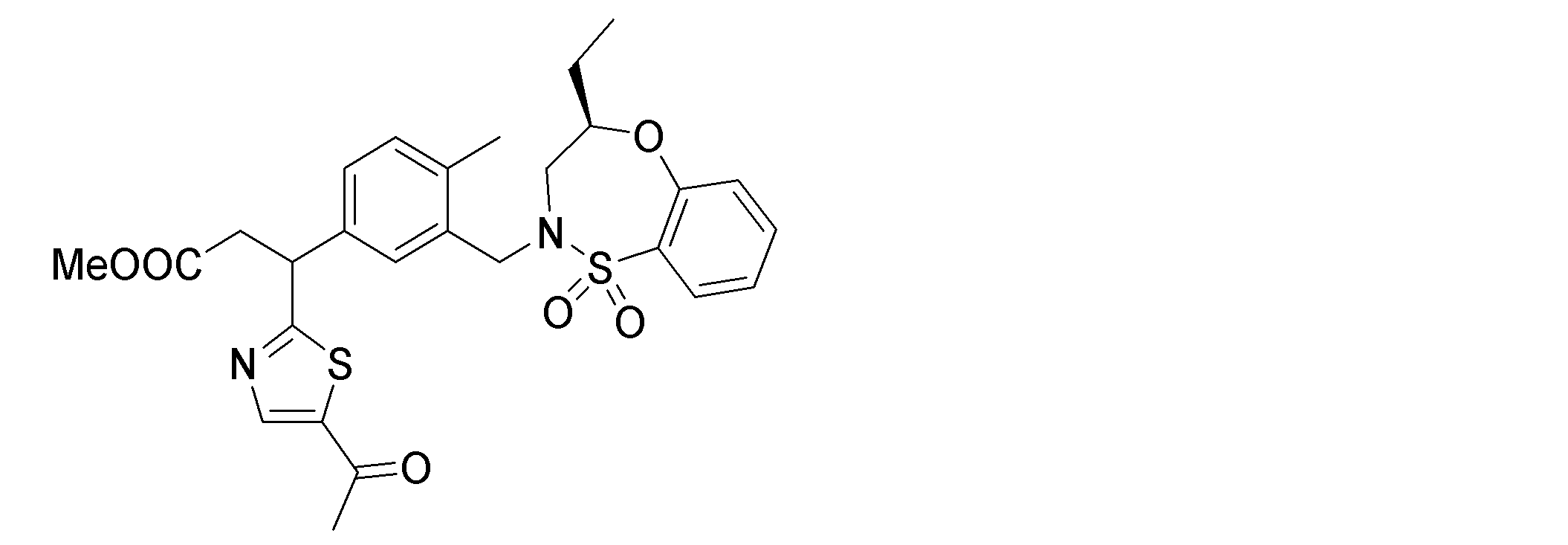
1H NMR (400MHz, CDCl3) δ 8.17 (1H, br s), 7.89 (1H, dd, J = 7.6, 1.6 Hz), 7.52 (1H, td, J = 7.6, 1.6 Hz), 7.28-7.24 (1H, m), 7.22-7.14 (4H, m), 4.79 (1H, t, J = 7.6 Hz), 4.57-4.55 (1H, m), 4.07-3.97 (1H, m), 3.84-3.81 (1H, m), 3.76-3.66 (1H, m), 3.63 (3H, s), 3.43-3.42 (1H, m), 3.02-2.96 (2H, m), 2.52 (3H, s), 2.35 (3H, br s), 1.75-1.64 (1H, m), 1.51-1.39 (1H, m), 1.09-1.07 (3H, m)。
メチル 3-(5-アセチル-4-メチルチアゾール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-プロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.89 (1H, d, J = 7.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.28-7.24 (1H, m), 7.22-7.15 (4H, m), 4.73 (1H, t, J = 7.6 Hz), 4.58-4.56 (1H, m), 4.07-3.98 (1H, m), 3.85-3.82 (1H, m), 3.76-3.70 (1H, m), 3.62 (3H, s), 3.43-3.42 (1H, m), 3.01-2.94 (2H, m,), 2.68 (3H, s), 2.44 (3H, s), 2.36 (3H, br s), 1.77-1.62 (1H, m), 1.52-1.38 (1H, m), 1.11-1.05 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
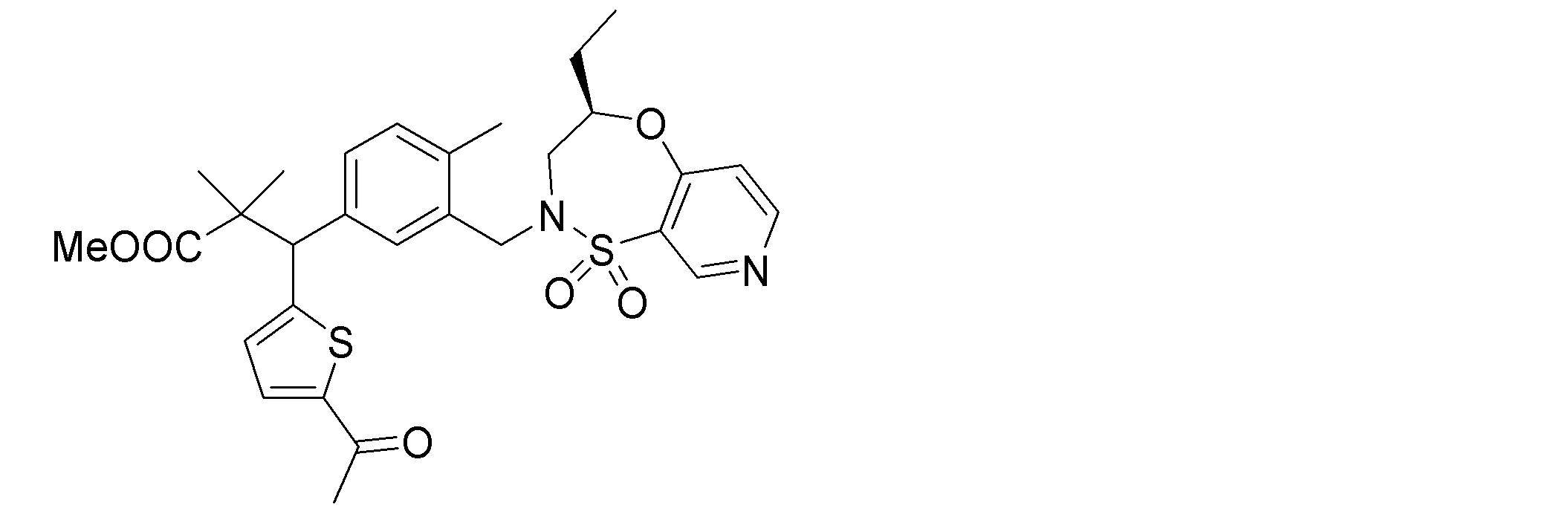
1H NMR (400 Mz, CDCl3) δ 9.03 (1H, s), 8.66 (1H, dd, J = 5.6, 1.6 Hz), 7.51 (1H, dd, J = 4.0, 1.6 Hz), 7.24-7.20 (2H, m), 7.16-7.14 (1H, m), 7.12-7.08 (1H, m), 6.98-6.96 (1H, m), 4.70/4.69 (1H, s), 4.46-4.37 (2H, m), 4.22-4.16 (1H, m), 3.60 (3H, br s), 3.53-3.44 (1H, m), 3.18-3.12 (1H, m), 2.50 (3H, br s), 2.32 (3H, br s), 1.72-1.63 (1H, m), 1.54-1.47 (1H, m), 1.30 (3H, br s), 1.24-1.22 (3H, m), 1.07-1.02 (3H, m)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)プロパノエートの製造
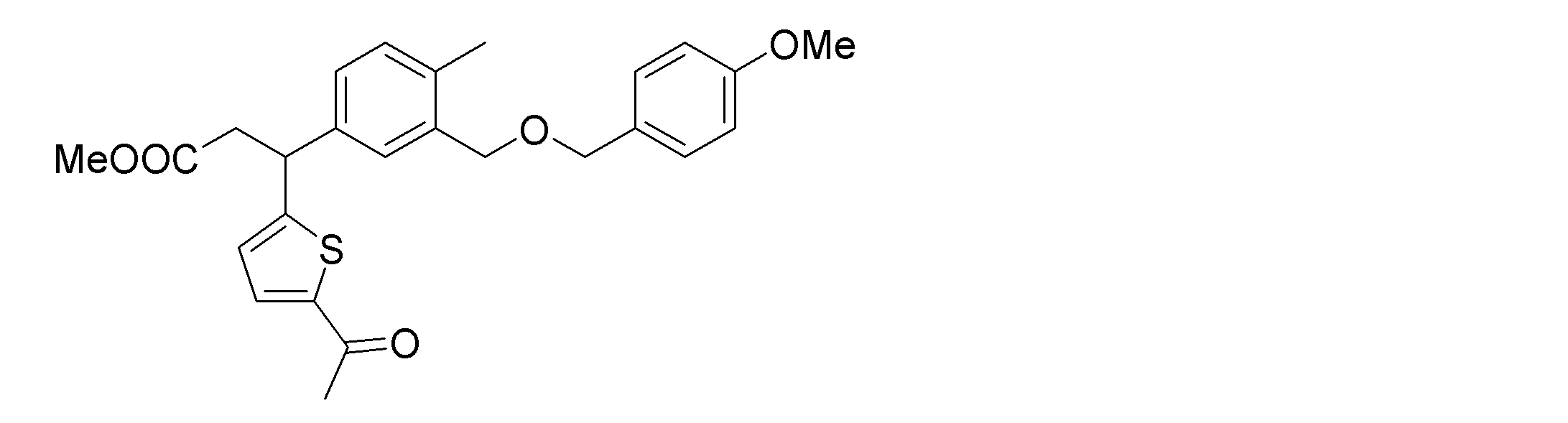
1H NMR(400 MHz, CDCl3)δ 7.49 (1H, d, J = 4.0 Hz), 7.28-7.24 (3H, m), 7.11 (2H, br s), 6.90-6.86 (3H, m), 4.74 (1H, t, J = 8.0 Hz), 4.48 (2H, s), 4.47 (2H, s), 3.82 (3H, s), 3.62 (3H, s), 3.15-3.00 (2H, m), 2.48 (3H, s), 2.27 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.49 (1H, d, J = 4.0 Hz), 7.29 (1H, br s), 7.10 (2H, br s), 6.87 (1H, br d, J = 4.0 Hz), 4.74 (1H, t, J = 8.0 Hz), 4.64 (2H, s), 3.60 (3H, s), 3.15-3.01 (2H, m), 2.46 (3H, s), 2.28 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]プロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.51 (1H, d, J = 4.0 Hz), 7.21 (1H, br s), 7.15 (2H, br s), 6.87 (1H, br d, J = 4.0 Hz), 4.74 (1H, t, J = 8.0 Hz), 4.56 (2H, s), 3.62 (3H, s), 3.15-3.00 (2H, m), 2.48 (3H, s), 2.38 (3H, s)。
メチル 3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパノエートの製造
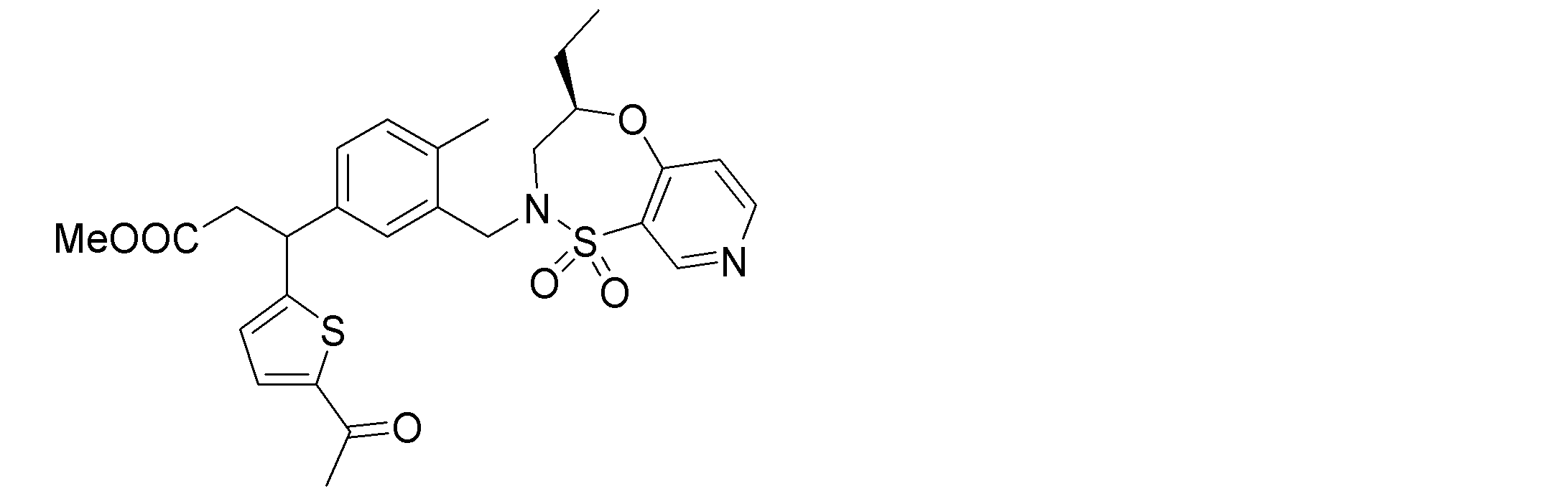
1H NMR (400 Mz, CDCl3) δ 9.02 (1H, s), 8.65 (1H, d, J = 5.6 Hz), 7.50 (1H, br d, J = 4.0 Hz), 7.17 (2H, br s), 7.11-7.08 (2H, m), 6.88 (1H, br d, J= 4.0 Hz), 4.73 (1H, t, J = 7.6 Hz), 4.45-4.37 (2H, m), 4.16-4.09 (1H, m), 3.62 (3H, s), 3.51-3.43 (1H, m), 3.17-3.09 (2H, m), 3.05-3.00 (1H, m), 2.49 (3H, s), 2.33 (3H, s), 1.74-1.65 (1H, m), 1.56-1.50 (1H, m), 1.04 (3H, br t, J = 7.6 Hz)。
メチル 3-[4-フルオロ-5-(1-ヒドロキシエチル)チオフェン-2-イル]-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
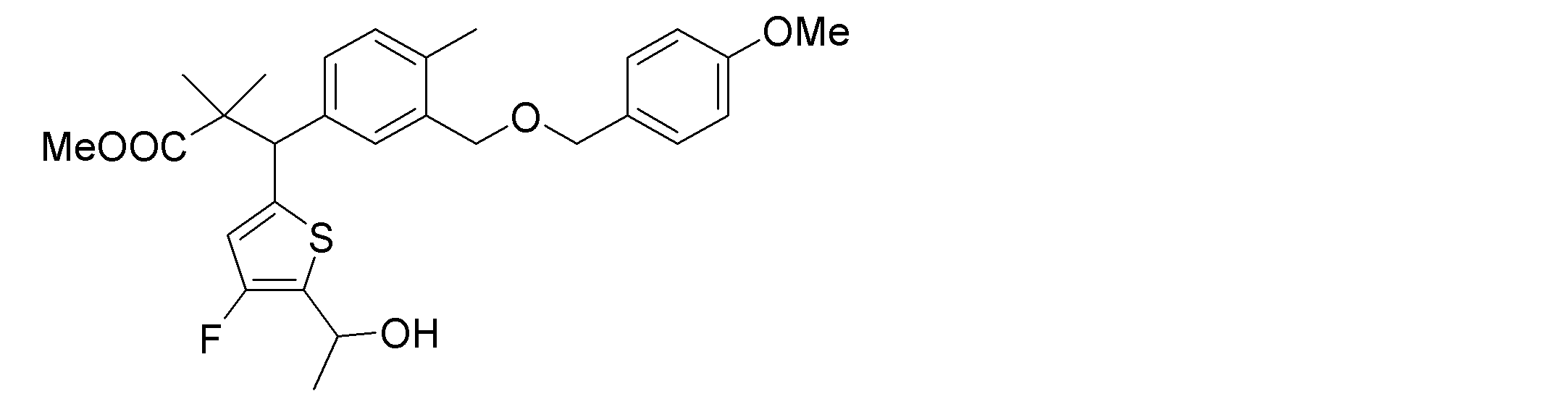
1H NMR(400 MHz, CDCl3)δ 7.29-7.27 (3H, m), 7.16 (1H, d, J = 8.0 Hz), 7.10 (1H, d, J = 8.0 Hz), 6.89 (2H, d, J = 8.4 Hz), 6.64, 6.63 (each 0.5H, s), 5.18-5.12 (1H, m), 4.51-4.48 (5H, m), 3.82 (3H, s), 3.61 (3H, s), 2.28 (3H, s), 1.87 (1H, br s), 1.51, 1.50 (each 1.5H, d, J = 6.4 Hz, thiophene β-CH3 regioisomers), 1.30 (3H, s), 1.20 (3H, s)。
メチル 3-[5-アセチル-4-フルオロチオフェン-2-イル]-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
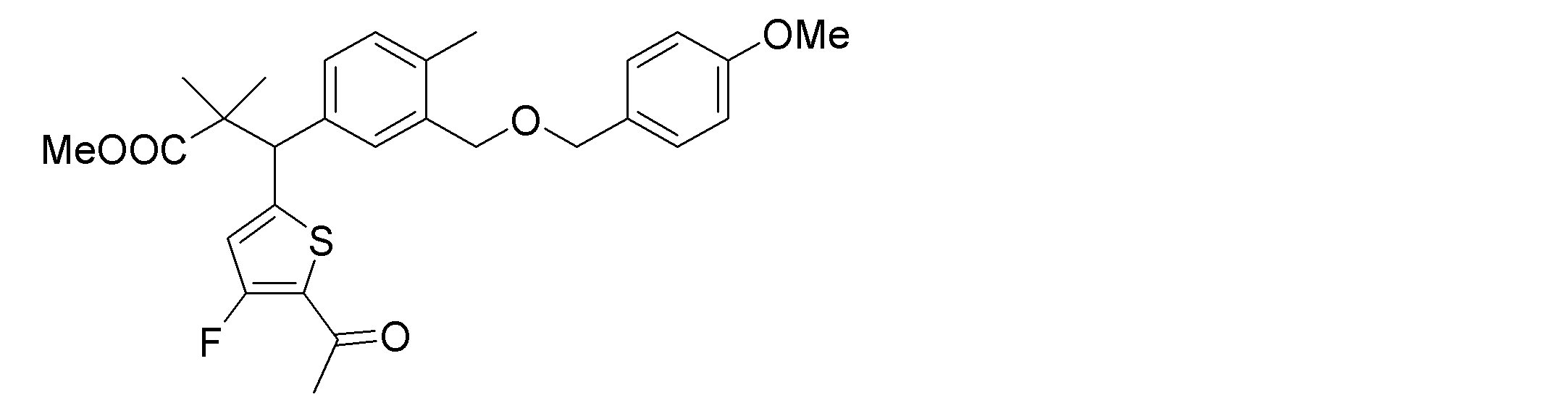
1H NMR(400 MHz, CDCl3)δ 7.29-7.27 (3H, m), 7.15 (1H, dd, J = 7.6, 1.2 Hz), 7.11 (1H, d, J = 7.6 Hz), 6.90 (2H, d, J = 8.4 Hz), 6.75 (1H, s), 4.55 (1H, s), 4.49 (2H, s), 4.48 (2H, s), 3.82 (3H, s), 3.62 (3H, s), 2.51, 2.50 (each 1.5H, s), 2.28 (3H, s), 1.32 (3H, s), 1.21 (3H, s)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.31 (1H, d, J = 1.6 Hz), 7.15 (1H, dd, J= 8.0, 1.6 Hz), 7.11 (1H, d, J = 8.0 Hz), 6.75 (1H, s), 4.67 (2H, s), 4.55 (1H, s), 3.63 (3H, s), 2.50, 2.49 (each 1.5H, s, acetyl regioisomers), 2.29 (3H, s), 1.32 (3H, s), 1.20 (3H, s)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.23 (1H, d, J = 1.6 Hz), 7.21 (1H, dd, J = 8.0, 1.6 Hz), 7.15 (1H, d, J = 8.0 Hz), 6.76 (1H, s), 4.57 (2H, s), 4.44 (1H, s), 3.64 (3H, s), 2.514, 2.507 (each 1.5H, s), 2.39 (3H, s), 1.32 (3H, s), 1.22 (3H, s)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造
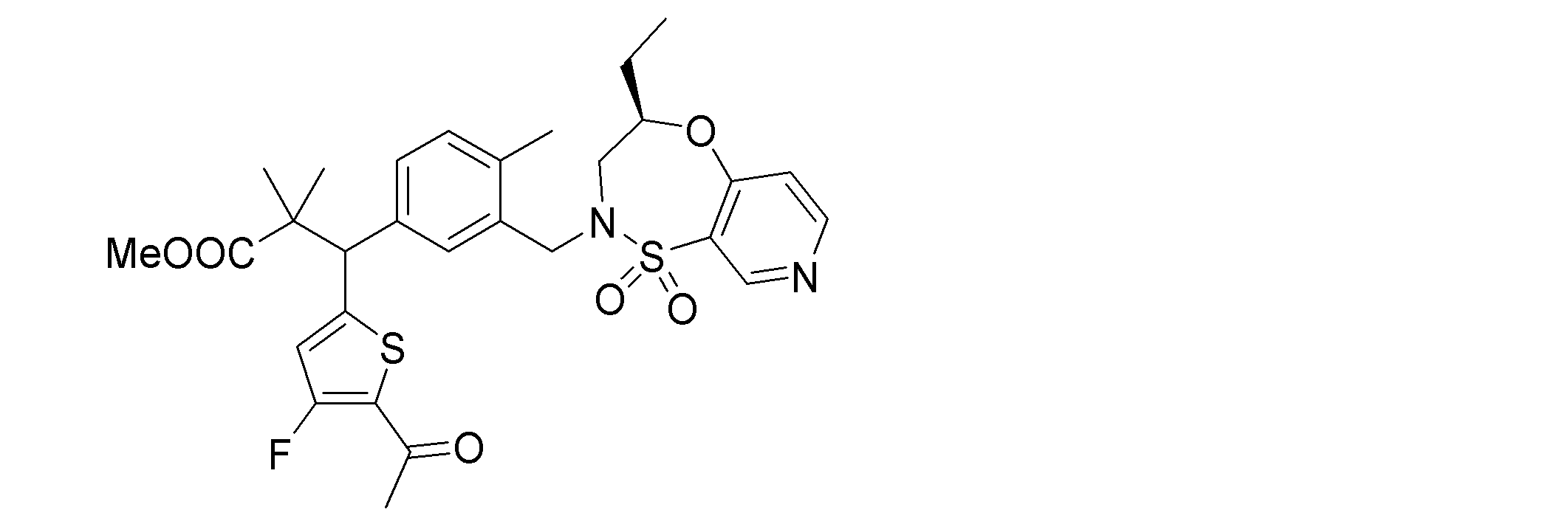
1H NMR (400 Mz, CDCl3) δ 9.02 (1H, s), 8.65 (1H, dd, J = 5.6, 0.4 Hz), 7.21 (1H, dd, J = 7.6, 1.6 Hz), 7.17-7.14 (2H, m), 7.09 (1H, d, J = 5.6, 2.0 Hz), 6.76, 6.75 (each 0.5H, s), 4.55, 4.54 (each 0.5H, s), 4.47-4.36 (2H, m), 4.16, 4.14 (each 0.5H, d, J = 14.4 Hz), 3.640, 3.639 (each 1.5H, s), 3.51-3.43 (1H, m), 3.17 (1H, dt, J= 15.2, 2.8 Hz), 2.51, 2.50 (each 1.5H, s), 2.33, 2.32 (each 1.5H, s), 1.74-1.63 (1H, m), 1.60-1.49 (1H, m), 1.30, 1.29 (each 1.5H, s), 1.20 (3H, s), 1.05 (3H, t, J= 7.2 Hz)。
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸の製造

1H NMR(400 MHz, CDCl3)δ 7.32 (1H, d, J = 2.0 Hz), 7.27 (2H, d, J= 8.8 Hz), 7.21 (1H, dd, J = 8.0, 2.0 Hz), 7.10 (1H, d, J = 8.0 Hz), 6.89 (2H, d, J = 8.8 Hz), 6,73 (1H, s), 4.63 (1H, s), 4.48 (4H, s), 3.87 (3H, s), 3.81 (3H, s), 2.46 (3H, s), 2.27 (3H, s), 1.34 (3H, s), 1.25 (3H s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.31-7.26 (3H, m), 7.18 (1H, dd, J = 7.6, 1.6 H), 7.10 (1H, d, J = 7.6 Hz), 6.89 (2H, d, J = 8.8 Hz), 6.71 (1H, s), 4.61 (1H, s), 4.48 (4H, s), 4.10-4.05 (2H, m), 3.89 (3H, s), 3.81 (3H, s), 2.46, (3H, s), 2.27 (3H, s), 1.32 (3H, s), 1.21 (3H, s), 0.89-0.86 (2H, m), 0.01 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.32 (1H, d, J = 2.0 Hz), 7.21 (1H, dd, J= 8.0, 2.0 Hz), 7.11 (1H, d, J = 8.0 Hz), 6.73 (1H, s), 4.67 (2H, d, J = 5.6 Hz), 4.62 (1H, s), 4.10-4.06 (2H, m), 3.91 (3H ,s), 2.46 (3H,s ), 2.31 (3H, s), 1.56 (1H, t, J = 5.6 Hz), 1.32 (3H, s), 1.22 (3H, s), 0.90-0.86 (2H, m), 0.02 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]-2,2-ジメチルプロパノエートの製造
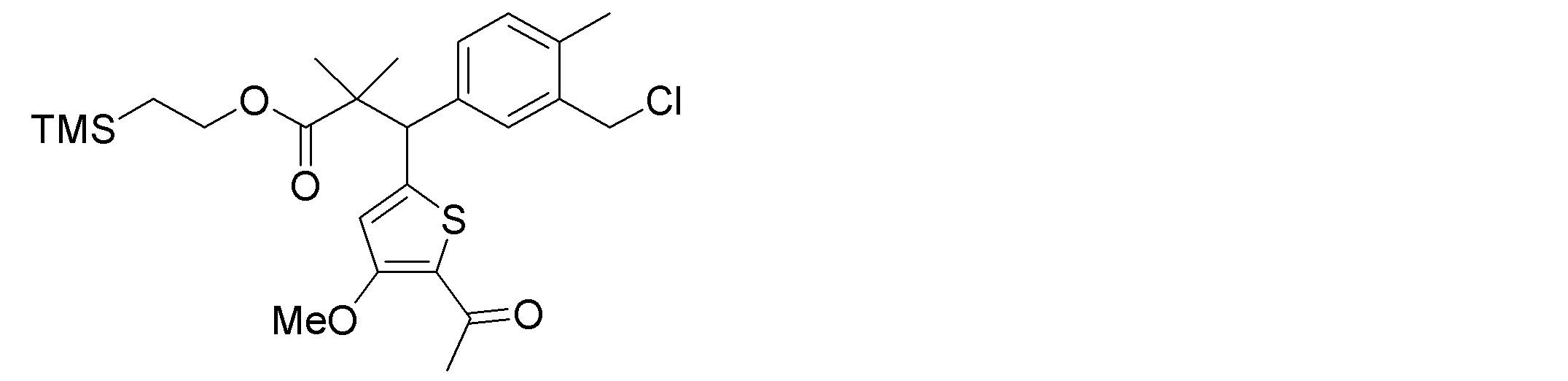
1H NMR (400 MHz, CDCl3) δ 7.26-7.23 (2H, m), 7.12 (1H, d, J = 7.6 Hz), 6.72 (1H, s), 4.60 (1H, s), 4.56 (2H, s), 4.09-4.05 (2H, m), 3.91 (3H, s), 2.73 (3H, s), 2.46 (3H, s), 1.31 (3H, s), 1.22 (3H, s), 0.88-0.84 (2H, m), 0.10 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパノエートの製造

1H NMR (400 Mz, CDCl3) δ 9.01 (1H, s), 8.66, 8.65 (each 0.5H, d, J = 5.6 Hz), 7.24-7.22 (2H, m), 7.14 (1H, d, J = 8.0 Hz), 7.10, 7.09 (each 0.5H, d, J= 5.6 Hz), 6.76, 6.75 (each 0.5H, s), 4.60, 4.59 (each 0.5H, s), 4.45-4.36 (2H, m), 4.16-4.05 (3H, m), 3.933, 3.932 (each 1.5H, s), 3.53-3.44 (1H, m), 3.18-3.11 (1H, m), 2.46 (3H, s), 2.32 (3H, s), 1.73-1.61 (1H, m), 1.58-1.46 (1H, m), 1.30 (3H, s), 1.21, 1.20 (each 1.5H, s), 1.05, 1.03 (each 1.5H, t, J= 7.2 Hz), 0.90-0.86 (2H, m), 0.02, 0.01 (each 4.5H, s)。
2-(5-ブロモ-3-フルオロチオフェン-2-イル)-2-メチル-1,3-ジオキソランの製造

1H NMR(400 MHz, CDCl3)δ 6.78 (1H, br s), 4.07-4.03 (2H, m), 4.02-3.97 (2H, m), 1.77 (3H, s)。
メチル (E)-3-[4-フルオロ-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]アクリレートの製造

1H NMR(400 MHz, CDCl3)δ 7.58 (1H, d, J = 16.0 Hz), 6.93 (1H, s), 6.20 (1H, d, J = 16.0 Hz), 4.10-4.03 (2H, m), 4.02-3.99 (2H, m), 3.79 (3H, s), 1.80 (3H, s)。
メチル 3-[4フルオロ-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)プロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.27 (2H, br d, J = 6.8 Hz), 7.21 (1H, br s), 7.13-7.07 (2H, m), 6.89 (2H, br d, J = 6.8 Hz), 6.52 (1H, br s), 4.55 (1H, t, J = 8.0 Hz), 4.48 (4H, br s), 4.01-3.91 (4H, m), 3.82 (3H, s), 3.61 (3H, s), 3.06-2.92 (2H, m), 2.28 (3H, s), 1.74 (3H, s)。
メチル 3-[4-フルオロ-5-(2-メチル-1,3-ジオキソラン-2-イル)チオフェン-2-イル]-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造
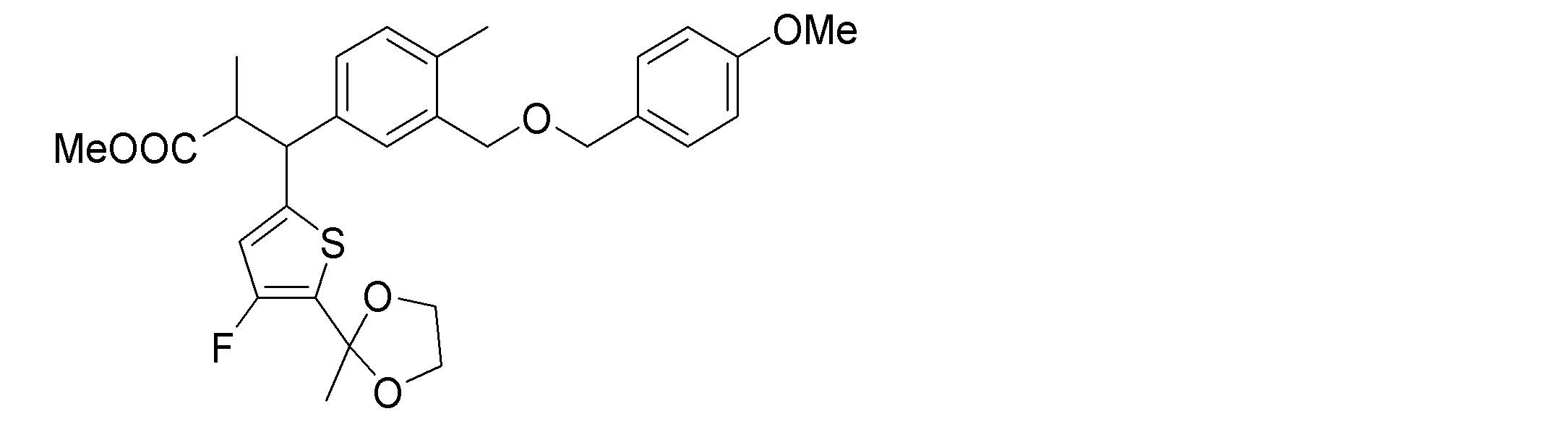
1H NMR(400 MHz, CDCl3)δ 7.27 (2H, br d, J = 8.8 Hz), 7.24-7.20 (1H, m), 7.13-7.06 (2H, m), 6.90 (2H, br d, J= 8.8 Hz), 6.61-6.52 (1H, m), 4.49 (4H, br s), 4.17-4.14 (1H, m), 3.98-3.89 (4H, m), 3.82 (3H, s), 3.63-3.43 (3H, m), 3.19-2.92 (1H, m), 2.28-2.25 (3H, m), 1.74-1.72 (3H, m), 1.23-1.02 (3H, m)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造
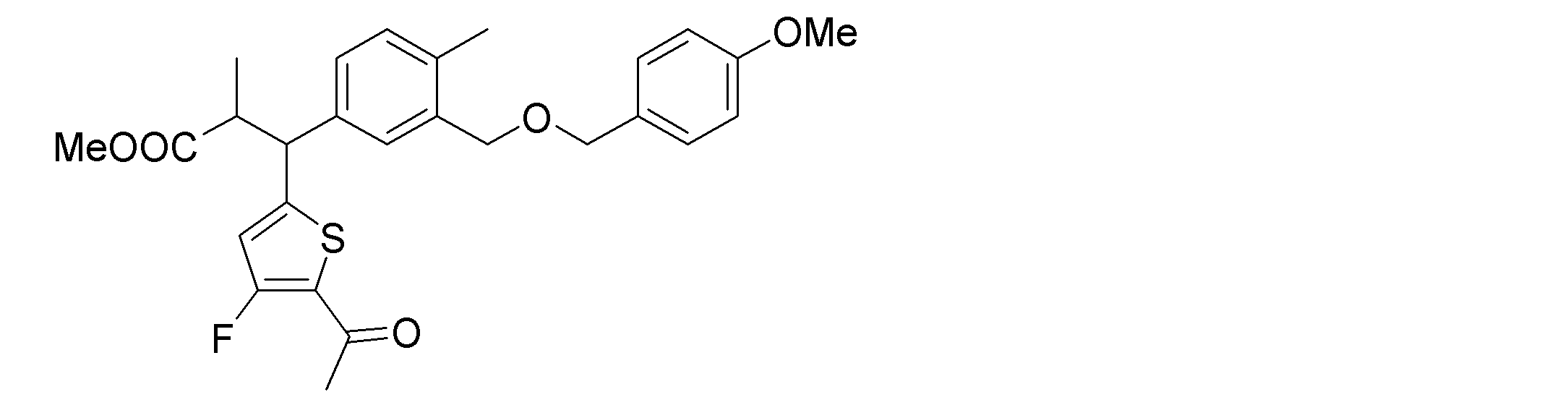
1H NMR(400 MHz, CDCl3)δ 7.28 (2H, d, J = 8.4 Hz), 7.24-7.21 (1H, m), 7.14-7.06 (2H, m), 6.90 (2H, d, J = 8.4 Hz), 6.71-6.63 (1H, m), 4.50-4.46 (4H, m), 4.27-4.22 (1H, m), 3.82 (3H, s), 3.65 (3H, s), 3.23-3.14 (1H, m), 2.50-2.48 (3H, m), 2.27-2.25 (3H, m), 1.24-1.06 (3H, m)。
メチル 3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2-メチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.28-7.26 (1H, m), 7.15-7.04 (2H, m), 6.73-6.65 (1H, m), 4.69-4.64 (2H, m), 4.28-4.23 (1H, m), 3.65-3.48 (3H, m), 3.26-2.98 (1H, m), 2.50-2.47 (3H, m), 2.30-2.28 (3H, m), 1.80 (1H, br s), 1.25-1.07 (3H, m)。
メチル3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]-2-メチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.21-7.11 (3H, m), 6.73-6.70 (1H, m), 4.57-4.55 (2H, m), 4.28-4.21 (1H, m), 3.66-3.48 (3H, m), 3.23-3.14 (1H, m), 2.51-2.48 (3H, m), 2.39-2.36 (3H, m), 1.25-1.07 (3H, m)。
メチル3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造

1H NMR (400 Mz, CDCl3) δ 9.03 (1H, s), 8.66 (1H, br d, J = 5.6 Hz), 7.20-7.08 (4H, m), 6.73-6.70 (1H, m), 4.49-4.40 (2H, m), 4.30-4.13 (2H, m), 3.66-3.40 (4H, m), 3.21-3.13 (2H, m), 2.51-2.48 (3H, m), 2.34-2.32 (3H, m), 1.75-1.65 (1H, m), 1.61-1.50 (1H, m), 1.24-1.03 (6H, m)。
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチルプロパン酸の製造
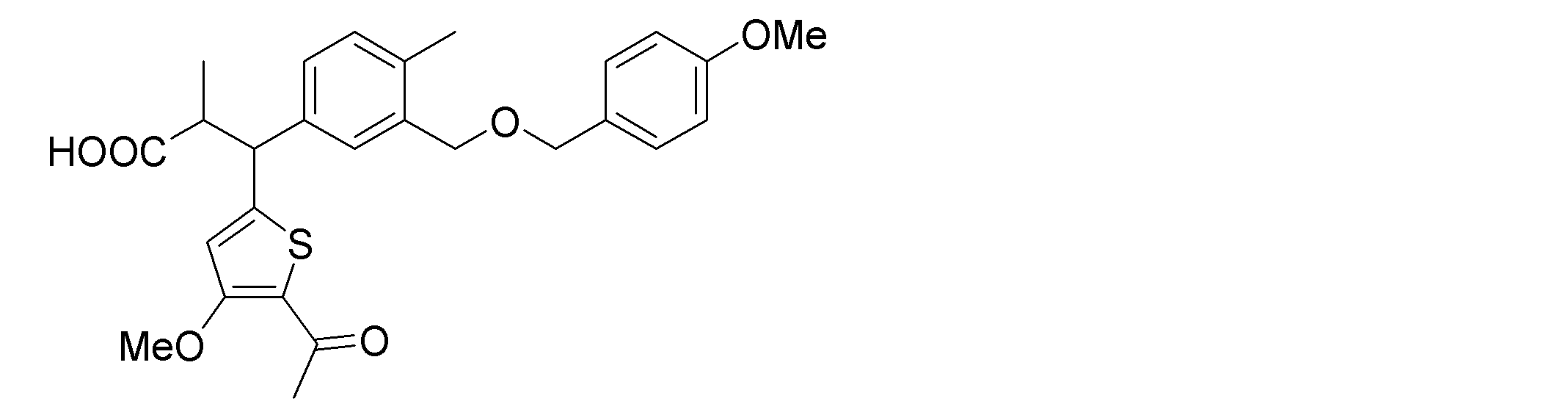
1H NMR(400 MHz, CDCl3)δ 7.28-7.06 (5H, m), 6.90-6.87 (2H, m), 6.70-6.68 (1H, m), 4.49-4.44 (4H, m), 4.25-4.21 (1H, m), 3.91-3.80 (3H, m), 3.81 (3H, br s), 3.25-3.15 (1H, m), 2.44-2.42 (3H, m), 2.26-2.23 (3H, m), 1.25-1.07 (3H, m)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(4-メトキシベンジル)オキシ]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.26 (2H, br d, J = 8.8 Hz), 7.21 (1H, s), 7.09 (2H, s), 6.88 (2H, br d, J = 8.8 Hz), 6.67 (1H, s), 4.47 (2H, s), 4.43 (2H, s), 4.24 (1H, d, J = 11.2 Hz), 4.10-4.05 (2H, m), 3.86 (3H, s), 3.80 (3H, s), 3.17-3.09 (1H, m), 2.41 (3H, s), 2.24 (3H, s), 1.04 (3H, d, J = 6.8 Hz), 0.89-0.85 (2H, m), -0.00 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-[3-(ヒドロキシメチル)-4-メチルフェニル]-2-メチルプロパノエートの製造

1H NMR(400 MHz, CDCl3)δ 7.28 (1H, s), 7.12 (2H, br s), 6.70 (1H, s), 4.67 (2H, br s), 4.27 (1H, d, J = 11.2 Hz), 4.15-4.04 (2H, m), 3.90 (3H, s), 3.19-3.12 (1H, m), 2.43 (3H, s), 2.30 (3H, s), 1.72 (1H, br s), 1.06 (3H, d, J = 7.2 Hz), 0.91-0.87 (2H, m), 0.02 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-[3-(クロロメチル)-4-メチルフェニル]-2-メチルプロパノエートの製造

1H NMR (400 MHz, CDCl3) δ 7.18 (1H, s), 7.13 (2H, br s), 6.68 (1H, s), 4.55 (2H, s), 4.25 (1H, d, J = 11.2 Hz), 4.10-4.05 (2H, m), 3.88 (3H, s), 3.16-3.08 (1H, m), 2.42 (3H, s), 2.36 (3H, s), 1.04 (3H, d, J = 7.2 Hz), 0.89-0.85 (2H, m), -0.00 (9H, s)。
2-(トリメチルシリル)エチル3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパノエートの製造

1H NMR (400 Mz, CDCl3) δ 9.02 (1H, s), 8.66 (1H, br d, J = 5.6 Hz), 7.16-7.09 (4H, m), 6.72 (1H, s), 4.47-4.35 (2H, m), 4.29-4.08 (4H, m), 3.92 (3H, br s), 3.52-3.41 (1H, m), 3.19-3.09 (2H, m), 2.44 (3H, m), 2.33 (3H, s), 1.75-1.64 (1H, m), 1.57-1.49 (1H, m), 1.06-1.02 (6H, m), 0.92-0.87 (2H, m), 0.02 (9H, s)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(メチルカルバモイル)フラン-2-イル]プロパン酸(化合物26)の製造

1H NMR(400 MHz, CDCl3)δ 7.85 (1H, d,J = 8.0 Hz), 7.52 (1H, td, J = 8.0, 1.6 Hz), 7.27-7.20 (3H, m), 7.12-7.06 (2H, m), 6.96 (1H, d, J = 3.2 Hz), 6.59 (1H, br s), 6.14-6.21 (1H, m), 4.53-4.47 (2H, m), 4.01-3.97 (1H, m), 3.87-3.73 (2H, m), 3.13-2.87 (6H, m), 2.28 (3H, br s), 1.73-1.63 (1H, m), 1.50-1.40 (1H, m), 1.07-1.06 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(ピロリジン-1-カルボニル)フラン-2-イル]プロパン酸(化合物28)の製造

1H NMR(400 MHz, CDCl3)δ7.87 (1H, dd,J = 7.6, 1.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.27-7.19 (2H, m), 7.12-7.10 (3H, m), 6.94 (1H, d, J = 3.6 Hz), 6.09 (1H, br s), 4.55-4.51 (2H, m), 4.02-3.96 (1H, m), 3.77 (1H, d,J = 12.4 Hz), 3.76-3.66 (3H, m), 3.58 (1H, t, J = 6.8 Hz), 3.14 (1H, dd, J= 16.0, 8.0 Hz), 2.96 (1H, d, J = 15.2 Hz), 2.99-2.87 (2H, m), 2.31 (3H, s), 1.96-1.91 (2H, m), 1.88-1.71 (2H, m), 1.69-1.61 (1H, m), 1.48-1.42 (1H, m), 1.06-1.05 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(イソプロピルカルバモイル)フラン-2-イル]プロパン酸(化合物29)の製造

1H NMR(400 MHz, CDCl3)δ 7.86(1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, t, J= 8.0 Hz), 7.27-7.06 (5H, m), 6.95 (1H, d,J = 3.6 Hz), 6.21 (1H, d, J = 8.0 Hz), 6.10-6.09 (1H, m), 4.52-4.48 (2H, m), 4.22-4.14 (1H, m), 4.02-3.95 (1H, m), 3.85-3.71 (2H, m), 3.08-2.82 (3H, m), 2.30 (3H, br s), 1.72-1.62 (1H, m), 1.49-1.38 (1H, m), 1.20-1.18 (6H, m), 1.07-1.06 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[5-(メチルカルバモイル)チオフェン-2-イル]プロパン酸(化合物21)の製造
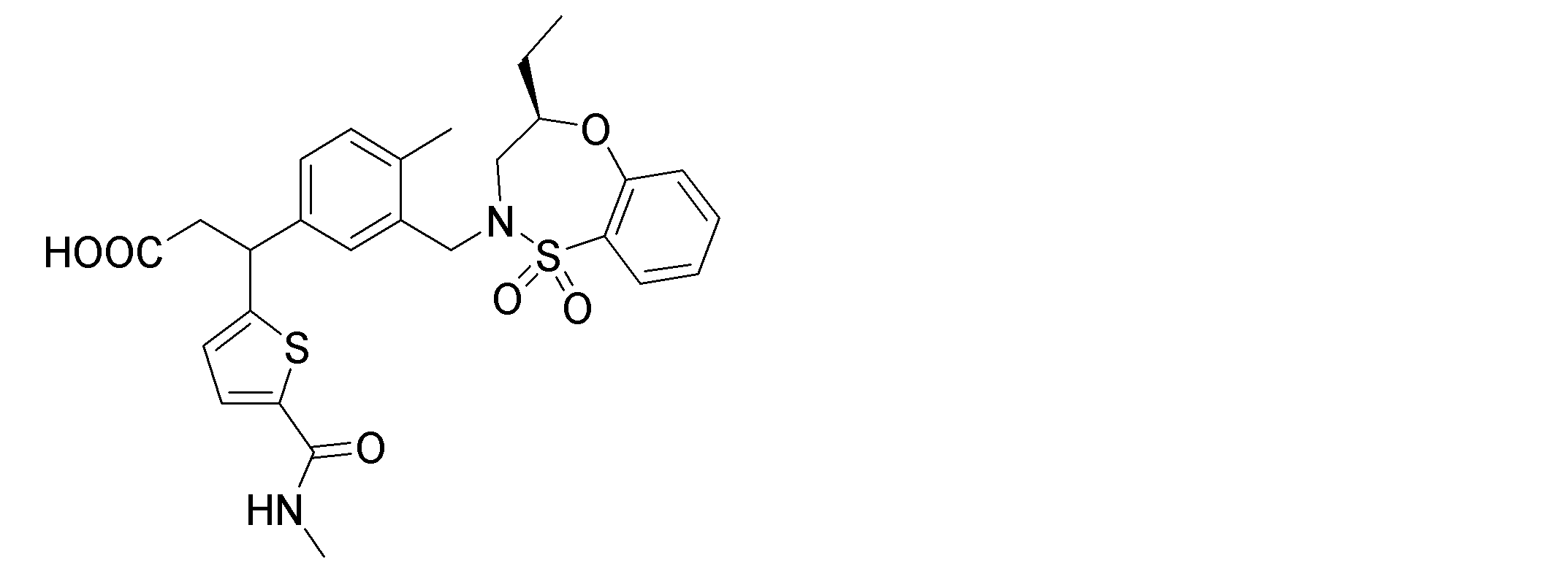
1H NMR(400 MHz, CDCl3)δ7.86 (1H, dd,J = 8.0, 1.6 Hz), 7.51 (1H, td, J= 8.0, 1.6 Hz), 7.29-7.18 (3H, m), 7.12-7.06 (3H, m), 6.76-6.75 (1H, m), 6.32 (1H, br s), 4.65 (1H, t, J = 8.0 Hz), 4.53-4.52 (1H, m), 3.98-3.93 (1H, m), 3.81-3.67 (2H, m), 3.10-2.90 (3H, m), 2.87 (3H, d, J = 4.8 Hz), 2.30 (3H, s), 1.67-1.56 (1H, m), 1.43-1.32 (1H, m), 1.04 (3H, t, J = 7.2 Hz)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-{5-[(2-メトキシエチル)カルバモイル]フラン-2-イル}プロパン酸(化合物30)の製造

1H NMR(400 MHz, CDCl3)δ 7.87(1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.16-7.11 (3H, m), 6.98 (1H, d, J = 3.6 Hz), 6.77 (1H, br s), 6.10-6.09 (1H, m), 4.55-4.51 (2H, m), 4.01-3.98 (1H, m), 3.86-3.70 (2H, m), 3.58-3.53 (4H, m), 3.36 (3H, s), 3.16-3.09 (1H, m), 3.01-2.89 (2H, m), 2.32 (3H, br s), 1.73-1.63 (1H, m), 1.49-1.38 (1H, m), 1.07-1.06 (3H, m)。
3-[5-(ジメチルカルバモイル)フラン-2-イル]-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物27)の製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, t, J= 8.0 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.12 (3H, br s), 6.88 (1H, d, J = 3.6 Hz), 6.08 (1H, d, J = 3.6 Hz), 4.55-4.51 (2H, m), 4.03-3.98 (1H, m), 3.82 (1H, d, J= 14.4 Hz), 3.75-3.68 (1H, m), 3.16-2.86 (9H, m), 2.31 (3H, s), 1.72-1.63 (1H, m), 1.49-1.40 (1H, m), 1.07-1.06 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物5)の製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, br d, J = 8.0 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.50 (1H, d, J = 3.6 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, br d, J = 8.0 Hz), 7.16-7.12 (2H, m), 7.08 (1H, d, J = 9.2 Hz), 6.88-6.86 (1H, m), 4.68 (1H, t, J = 7.6 Hz), 4.55-4.54 (1H, m), 4.02-3.95 (1H, m), 3.82-3.79 (1H, m), 3.74-3.67 (1H, m), 3.12 (1H, dd, J = 16.0, 7.6 Hz), 3.03 (1H, dd, J = 16.0, 8.0 Hz), 2.94-2.92 (1H, m), 2.47 (3H, s), 2.33-2.32 (3H, s), 1.70-1.60 (1H, m), 1.45-1.35 (1H, m), 1.05 (3H, t, J = 7.6 Hz)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[4-メチル-5-(メチルカルバモイル)フラン-2-イル]プロパン酸(化合物31)の製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.27-7.19 (3H, m), 7.12-7.05 (2H, m), 6.38 (1H, br s), 5.96 (1H, d, J = 3.6 Hz), 4.51-4.42 (2H, m), 4.03-3.96 (1H, m), 3.88-3.71 (2H, m), 3.08-2.97 (2H, m), 2.88-2.86 (4H, m), 2.28 (6H, br s), 1.73-1.64 (1H, m), 1.51-1.38 (1H, m), 1.07-1.06 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(5-スルファモイルチオフェン-2-イル)プロパン酸(化合物18)の製造
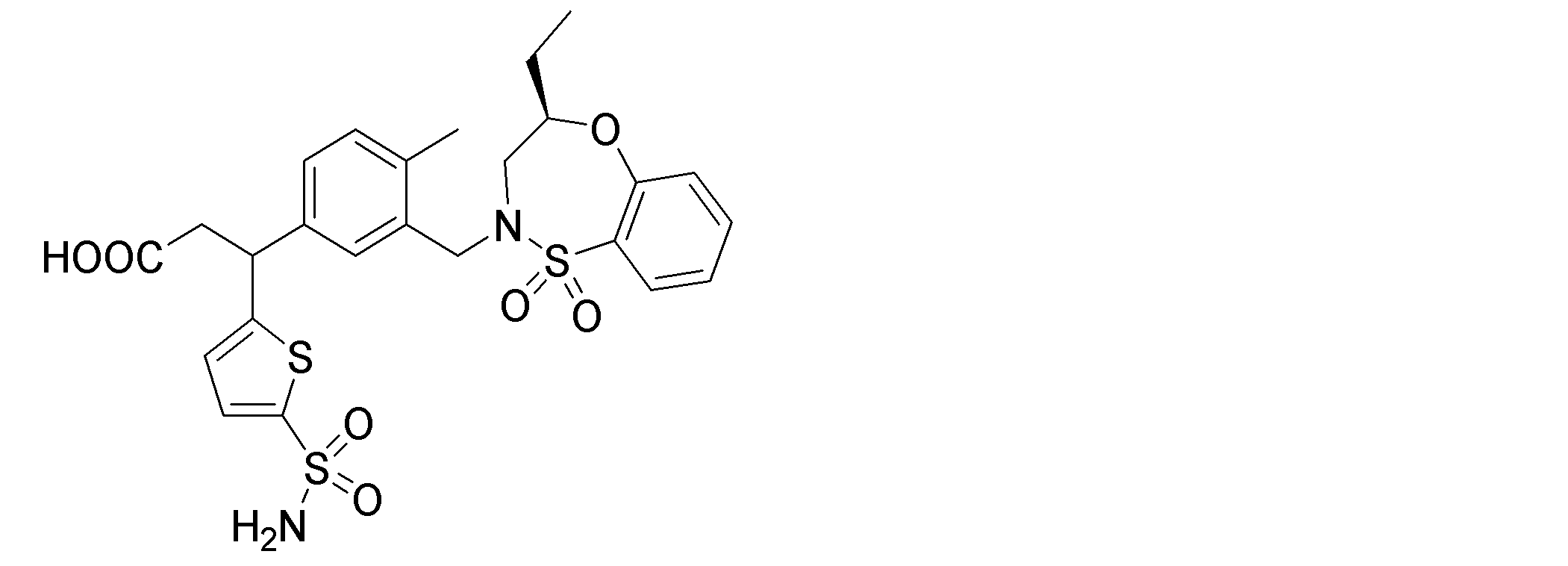
1H NMR(400 MHz, CDCl3)δ 7.82 (1H, dt,J = 8.0, 1.6 Hz), 7.50 (1H, td, J= 8.0, 1.6 Hz), 7.37 (1H, d, J = 4.0 Hz), 7.25-7.09 (5H, m), 6.77 (1H, d, J= 3.6 Hz), 5.40 (2H, br s), 4.67 (1H, t, J= 8.0 Hz), 4.48 (1H, d, J = 14.4 Hz), 4.00-3.95 (1H, m), 3.83-3.82 (1H, m), 3.78-3.68 (1H, m), 3.13-2.94 (3H, m), 2.27 (3H, s), 1.69-1.59 (1H, m), 1.46-1.40 (1H, m), 1.05-1.04 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)- 3-(チオフェン-2-イル)プロパン酸(化合物17)の製造

1H NMR(400 MHz, CDCl3)δ7.86 (1H, dd,J = 8.0, 1.6 Hz), 7.50 (1H, td, J= 8.0, 1.6 Hz), 7.25-7.17 (2H, m), 7.13-7.09 (3H, m), 7.04 (1H, d, J = 9.6 Hz), 6.87-6.85 (1H, m), 6.80-6.78 (1H, m), 4.65 (1H, t, J = 7.6 Hz), 4.54 (1H, d, J = 14.0 Hz), 3.99-3.89 (1H, m), 3.79-3.65 (2H, m), 3.09-2.93 (3H, m), 1.65-1.55 (1H, m), 1.42-1.29 (1H, m), 1.04-1.03 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)- 3-[5-(メチルカルバモイル)チオフェン-3-イル]プロパン酸(化合物22)の製造
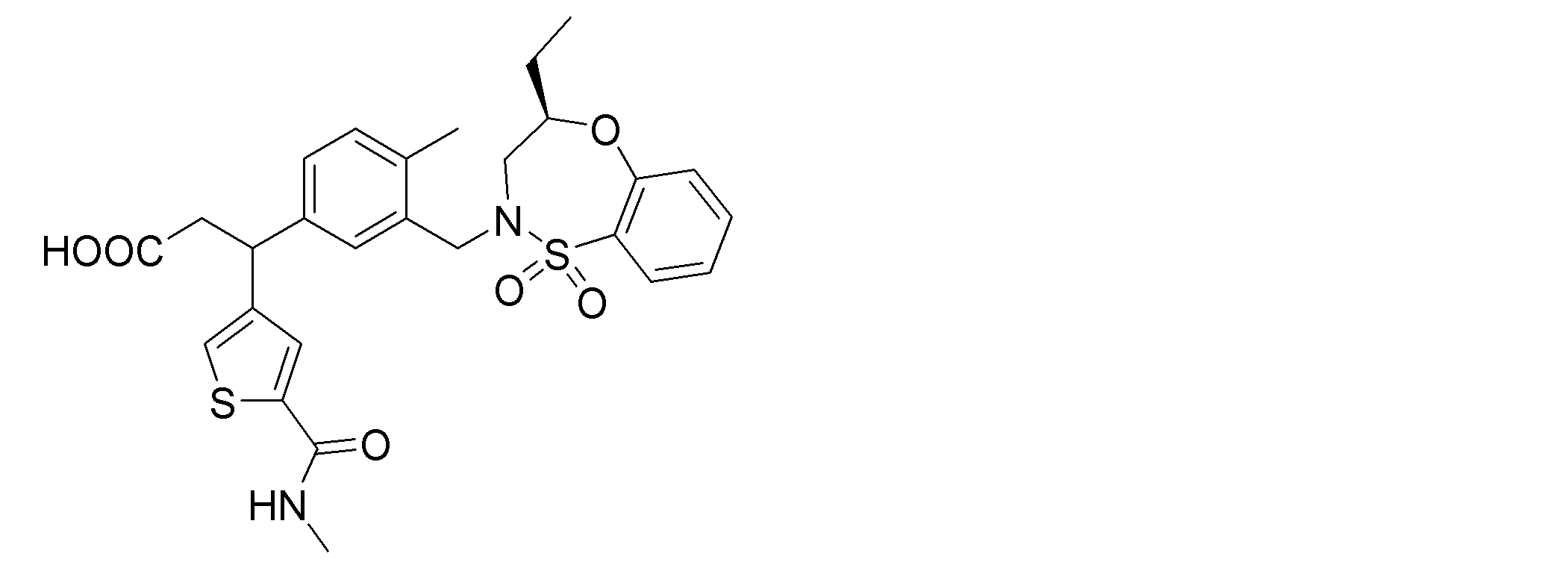
1H NMR(400 MHz, CDCl3)δ7.84 (1H, dt,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.32 (1H, dd, J = 6.0, 1.6 Hz), 7.26-7.19 (2H, m), 7.14-7.03 (4H, m), 6.46 (1H, br s), 4.49-4.43 (2H, m), 3.99-3.94 (1H, m), 3.83-3.82 (1H, m), 3.78-3.70 (1H, m), 3.05-2.90 (3H, m), 2.87 (3H, d, J = 4.8 Hz), 2.27 (3H, s), 1.70-1.61 (1H, m), 1.46-1.37 (1H, m), 1.05 (3H, br t, J = 7.2 Hz)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)- 3-{5-[(イソプロピルアミノ)メチル]フラン-2-イル}プロパン酸(化合物32)の製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, dd,J = 7.6, 1.6 Hz), 7.51 (1H, td, J= 7.6, 1.6 Hz), 7.24 (1H, t, J = 7.6 Hz), 7.20 (1H, d, J = 7.6 Hz), 7.13-7.07 (3H, m), 6.67 (1H, br s), 6.23 (1H, d, J = 3.2 Hz), 5.93 (1H, br s), 4.53-4.46 (2H, m), 4.03-3.94 (1H, m), 3.88 (2H, s), 3.84-3.67 (2H, m), 3.12-3.05 (1H, m), 2.99-2.89 (2H, m), 2.69 (1H, d, J = 14.4, 5.6 Hz), 2.29 (3H, s), 1.72-1.63 (1H, m), 1.48-1.38 (1H, m), 1.24-1.21 (6H, m), 1.07-1.06 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-[3-メチル-5-(メチルカルバモイル)チオフェン-2-イル]プロパン酸(化合物47)の製造

1H NMR(400 MHz, CDCl3)δ 7.84 (1H, br d,J = 7.6 Hz), 7.51 (1H, br t, J = 7.6 Hz), 7.25-7.16 (3H, m), 7.11-7.05 (3H, m), 6.57 (1H, br t, J = 4.8 Hz), 4.67 (1H, t, J = 6.8 Hz), 4.51-4.50 (1H, m), 3.97-3.93 (1H, m), 3.77-3.67 (2H, m), 2.93-2.90 (3H, m), 2.85 (3H, d, J = 4.8 Hz), 2.28 (3H, br s), 2.03 (3H, s), 1.67-1.55 (1H, m), 1.42-1.31 (1H, m), 1.04-1.02 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(チアゾール-5-イル)プロパン酸(化合物39)の製造

1H NMR(400 MHz, CDCl3)δ 8.63 (1H, s), 7.85 (1H, dt, J = 8.0, 1.6 Hz), 7.62 (1H, d, J= 3.6 Hz), 7.50 (1H, td, J = 8.0, 1.6 Hz), 7.23 (1H, br t, J = 8.0 Hz), 7.18 (1H, d, J = 8.0 Hz), 7.12-7.07 (3H, m), 4.73 (1H, t, J = 7.6 Hz), 4.50 (1H, d, J = 14.4 Hz), 4.00-3.92 (1H, m), 3.82-3.67 (2H, m), 3.04-2.91 (3H, m), 2.29 (3H, br s), 1.68-1.60 (1H, m), 1.43-1.27 (1H, m), 1.04-1.03 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(1-メチル-4-オキソ-4,5,6,7-テトラヒドロ-1H-インドール-2-イル)プロパン酸(化合物37)の製造

1H NMR(400 MHz, CDCl3)δ 7.84 (1H, br t,J = 8.0 Hz), 7.54-7.49 (1H, m), 7.27-7.18 (2H, m), 7.11-7.09 (1H, m), 7.06-7.02 (1H, m), 6.98-6.96 (1H, m), 6.56 (1H, s), 4.52-4.50 (1H, m), 4.42 (1H, t, J = 6.4 Hz), 3.99-3.94 (1H, m), 3.80-3.64 (2H, m), 3.22-3.20 (3H, m), 3.06-2.87 (2H, m), 2.77 (1H, dd, J = 16.0, 6.4 Hz), 2.66-2.59 (2H, m), 2.45-2.41 (2H, m), 2.33-2.31 (3H, m), 2.10-2.09 (2H, m), 1.74-1.58 (1H, m), 1.50-1.33 (1H, m), 1.09-1.02 (3H, m)。
3-(5-アセチル-1-メチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物33)の製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, d,J = 8.0 Hz), 7.51 (1H, t, J = 8.0 Hz), 7.26-7.18 (2H, m), 7.12-7.08 (1H, m), 7.02-6.93 (3H, m), 6.15 (1H, d, J = 4.0 Hz), 4.55-4.44 (2H, m), 3.99-3.90 (1H, m), 3.81-3.65 (5H, m), 3.05 (1H, dd, J = 16.0, 8.0 Hz), 2.93-2.82 (2H, m), 2.39 (3H, s), 2.32-2.29 (3H, m), 1.71-1.57 (1H, m), 1.45-1.28 (1H, m), 1.07-1.02 (3H, m)。
3-(5-アセチル-3-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物14)の製造
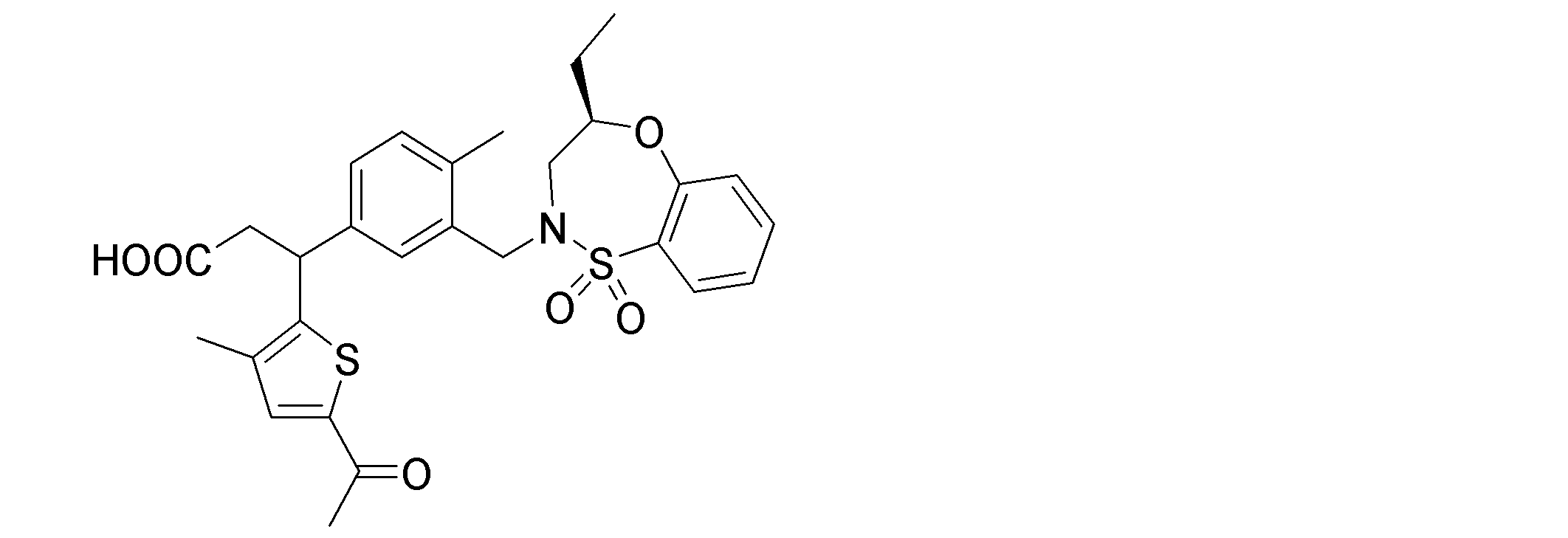
1H NMR(400 MHz, CDCl3)δ7.85 (1H, dd, J= 8.0, 1.6 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.37 (1H, s), 7.27-7.18 (2H, m), 7.14-7.06 (3H, m), 4.70-4.69 (1H, m), 4.54-4.52 (1H, m), 4.00-3.93 (1H, m), 3.80-3.65 (2H, m), 3.05-2.86 (3H, m), 2.45 (3H, br s), 2.31 (3H, br s), 2.14 (3H, s), 1.71-1.56 (1H, m), 1.45-1.31 (1H, m), 1.05-1.04 (3H, m)。
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物8)の製造

1H NMR(400 MHz, CDCl3)δ 7.85 (1H, dd,J = 8.0, 4.0 Hz), 7.51 (1H, td, J= 8.0, 1.6 Hz), 7.25-7.05 (5H, m), 6.62 (1H, br s), 4.56 (1H, t, J = 7.6 Hz), 4.48 (1H, d, J = 14.8 Hz), 4.02-3.94 (1H, m), 3.86-3.69 (2H, m), 3.03-2.88 (3H, m), 2.47 (3H, br s), 2.28 (3H, br s), 1.71-1.62 (1H, m), 1.48-1.40 (1H, m), 1.07-1.06 (3H, m)。
3-(5-カルバモイルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物23)の製造

1H NMR(400 MHz, CDCl3)δ 7.73 (1H, dd,J = 8.0, 1.6 Hz), 7.49 (1H, td, J= 8.0, 1.6 Hz), 7.41-7.40 (1H, m), 7.20 (1H, td, J = 8.0, 1.2 Hz), 7.16-7.06 (3H, m), 7.01 (1H, br d, J = 16.8 Hz), 6.83-6.80 (1H, m),4.59-4.54 (1H, m), 4.49-4.47 (1H, m), 3.92-3.86 (1H, m), 3.71-3.67 (1H, m), 3.60-3.54 (1H, m), 3.02-2.82 (2H, m), 2.78-2.73 (1H, m), 2.25 (3H, br s), 1.53-1.41 (1H, m), 1.34-1.22 (1H, m), 0.95 (3H, t, J = 7.2 Hz)。
3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物7)の製造
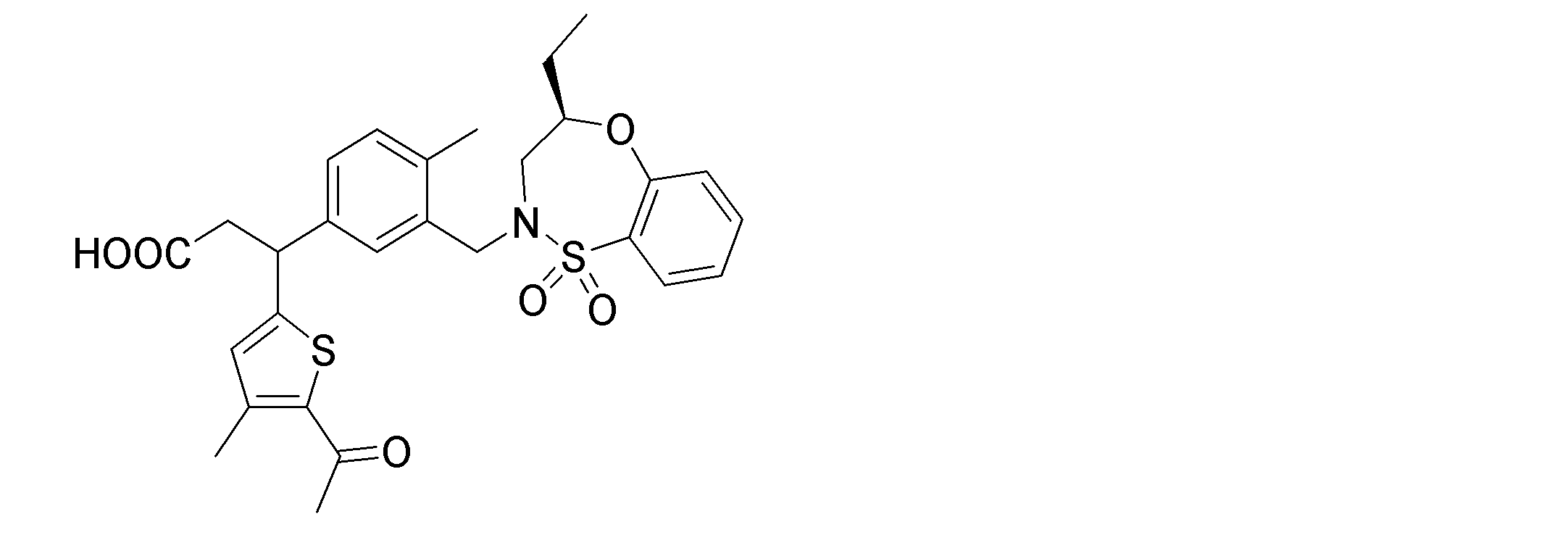
1H NMR(400 MHz, CDCl3)δ 7.89-7.85 (1H, m), 7.54-7.49 (1H, m), 7.27-6.95 (5H, m), 6.69 (1H, br s), 4.62 (1H, t, J= 7.6 Hz), 4.55 (1H, d, J = 14.0 Hz), 4.03-3.98 (1H, m), 3.84-3.68 (2H, m), 3.11 (1H, dd, J = 16.4, 7.2 Hz), 3.04-2.87 (2H, m), 2.45 (3H, s), 2.42 (3H, s), 2.33 (3H, s), 1.70-1.61 (1H, m), 1.45-1.35 (1H, m), 1.07-1.03 (3H, m)。
3-(4-アセチル-1,3,5-トリメチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物35)の製造
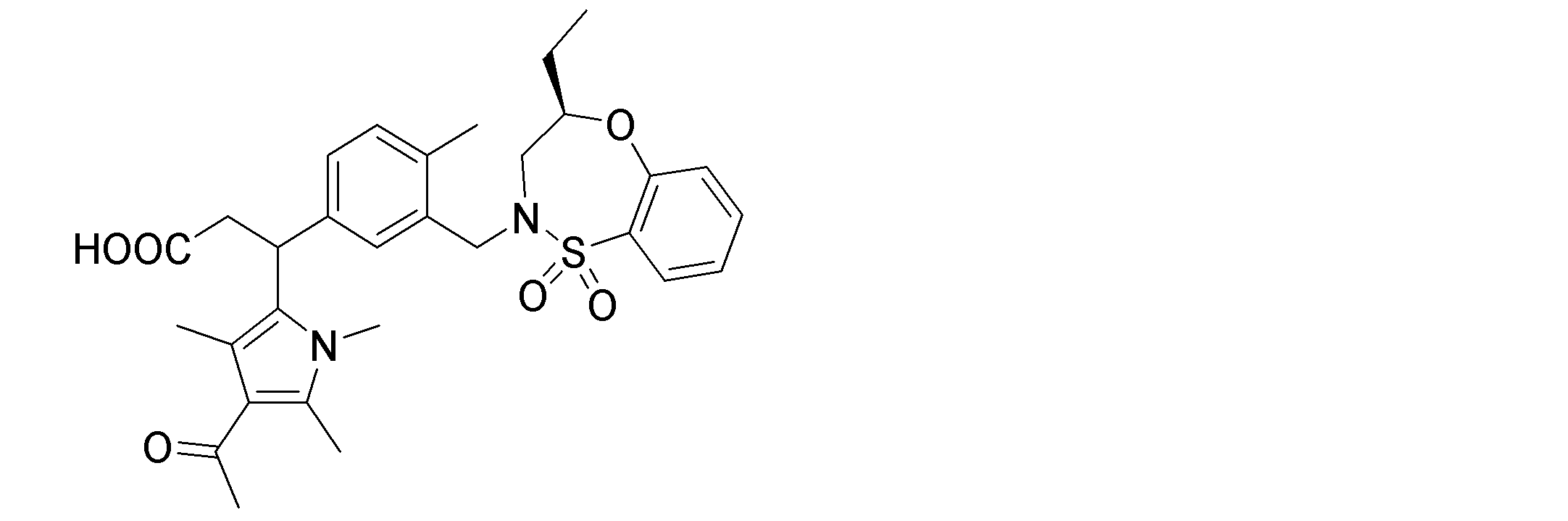
1H NMR(400 MHz, CDCl3)δ 7.88-7.84 (1H, m), 7.52 (1H, br t, J = 8.0 Hz), 7.25 (1H, br t, J = 8.0 Hz), 7.20 (1H, dd, J = 8.0, 3.2 Hz), 7.14-7.11 (1H, m), 7.02-6.99 (1H, m), 6.96-6.95 (1H, m), 4.81 (1H, t, J = 7.6 Hz), 4.54-4.51 (1H, m), 3.99-3.94 (1H, m), 3.80-3.69 (2H, m), 3.28-3.22 (4H, m), 2.99-2.88 (2H, m), 2.42-2.41 (6H, m), 2.34-2.32 (3H, m), 2.21 (3H, s), 1.73-1.60 (1H, m), 1.46-1.32 (1H, m), 1.06-1.04 (3H, m)。
3-(5-アセチル-1-エチル-1H-ピロール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物34)の製造
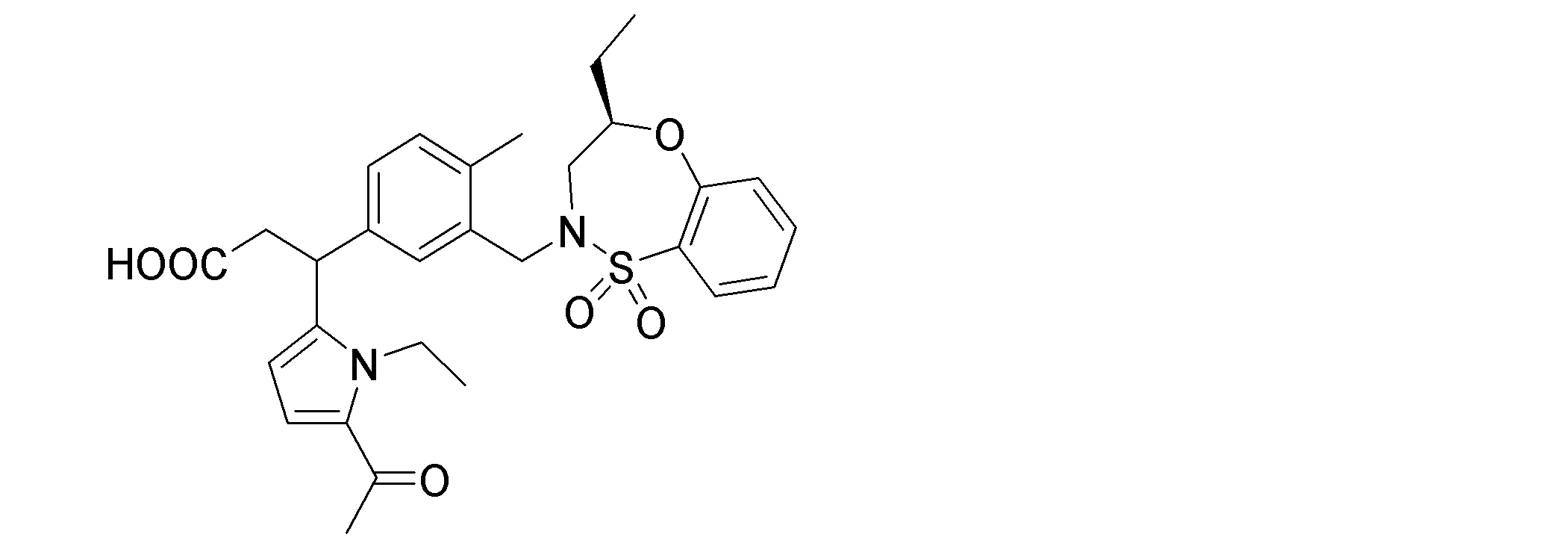
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dt,J = 8.0, 1.6 Hz), 7.51 (1H, t, J= 8.0 Hz), 7.25 (1H, t, J = 8.0 Hz), 7.20 (1H, d, J = 8.0 Hz), 7.14-7.09 (3H, m), 6.71-6.70 (1H, m), 6.63-6.62 (1H, m), 4.54-4.53 (1H, m), 4.35 (1H, t, J = 7.6 Hz), 4.25 (2H, q, J = 7.2 Hz), 4.03-3.97 (1H, m), 3.82-3.81 (1H, m), 3.75-3.68 (1H, m), 3.01-2.85 (3H, m), 2.35 (3H, s), 2.32 (3H, s), 1.70-1.61 (1H, m), 1.45-1.36 (1H, m), 1.29 (3H, t, J = 7.2 Hz), 1.05-1.04 (3H, m)。
3-[5-(シクロプロパンカルボニル)チオフェン-2-イル]-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物20)の製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dt,J = 8.0, 1.6 Hz), 7.62 (1H, d, J= 3.6 Hz), 7.52 (1H, br t, J = 8.0 Hz), 7.25 (1H, br t, J = 8.0 Hz), 7.20 (1H, br d, J = 8.0 Hz), 7.14 (2H, br s), 7.07 (1H, d, J = 10.4 Hz), 6.90-6.88 (1H, m), 4.69 (1H, t, J= 7.6 Hz), 4.57-4.52 (1H, m), 4.02-3.93 (1H, m), 3.83-3.74 (1H, m), 3.74-3.67 (1H, m), 3.13 (1H, dd, J = 16.0, 7.6 Hz), 3.03 (1H, dd, J = 16.0, 7.6 Hz), 2.93 (1H, dd, J = 15.2, 10.0 Hz), 2.47-2.41 (1H, m), 2.33 (3H, br s), 1.69-1.59 (1H, m), 1.43-1.34 (1H, m), 1.20-1.16 (2H, m), 1.04 (3H, t, J = 7.6 Hz), 1.00-0.95 (2H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物1)の製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J = 7.6, 1.6 Hz), 7.53 (1H, td, J = 7.6, 1.6 Hz), 7.47 (1H, dd, J = 4.0, 1.2 Hz), 7.28-7.21 (2H, m), 7.17-7.07 (3H, m), 6.95 (1H, d, J = 4.0 Hz), 4.60-4.52 (1H, m), 4.34 (1H, d, J= 10.8 Hz), 4.06-3.95 (1H, m), 3.89-3.67 (2H, m), 3.23-3.17 (1H, m), 2.98-2.89 (1H, m), 2.46 (3H, s), 2.34-2.32 (3H, m), 1.73-1.61 (1H, m), 1.46-1.33 (1H, m), 1.11-1.05 (6H, m)。
3-(5-シアノチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物19)の製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dd,J = 7.6, 1.6 Hz), 7.53 (1H, td, J= 7.6, 1.6 Hz), 7.43 (1H, dd, J = 3.6, 0.8 Hz), 7.27-7.20 (2H, m), 7.17-7.10 (3H, m), 6.86 (1H, br d, J = 4.0 Hz), 4.70 (1H, t, J = 7.6 Hz), 4.54-4.49 (1H, m), 4.04-3.97 (1H, m), 3.86-3.82 (1H, m), 3.77-3.68 (1H, m), 3.15-3.01 (2H, m), 2.97 (1H, dd, J = 14.0, 5.2 Hz), 2.31 (3H, s), 1.75-1.63 (1H, m), 1.47-1.37 (1H, m), 1.11-1.05 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物6)の製造
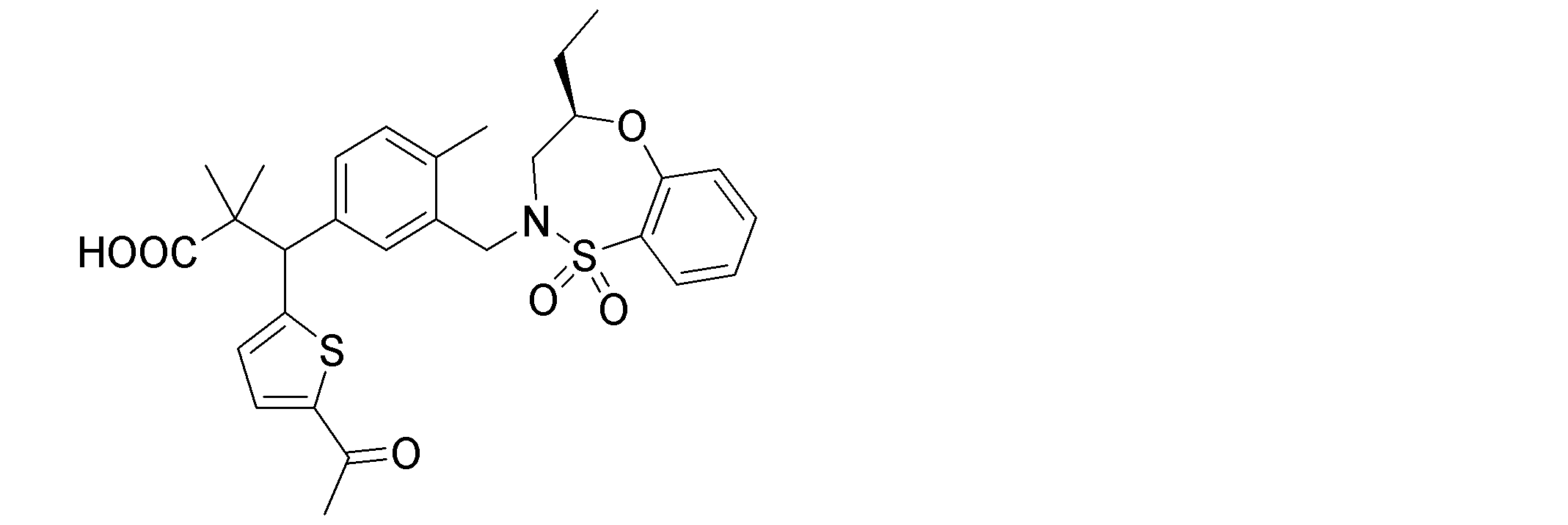
1H NMR (400 MHz, CDCl3) δ 7.88 (1H, dt, J = 7.6, 2.0 Hz), 7.55-7.51 (2H, m), 7.28-7.21 (4H, m), 7.13 (1H, d, J = 7.6 Hz), 7.00 (1H, d, J = 4.0 Hz), 4.72-4.70 (1H, m), 4.55 (1H, d, J = 14.0 Hz), 4.05-3.96 (1H, m), 3.84-3.82 (1H, m), 3.74 (1H, dd, J= 15.2, 10.8 Hz), 2.98-2.97 (1H, m), 2.50 (3H, s), 2.33 (3H, s), 1.75-1.58 (1H, m), 1.50-1.39 (1H, m), 1.32 (3H, s), 1.25 (3H, s), 1.10-1.05 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-9-フルオロ-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物9)の製造
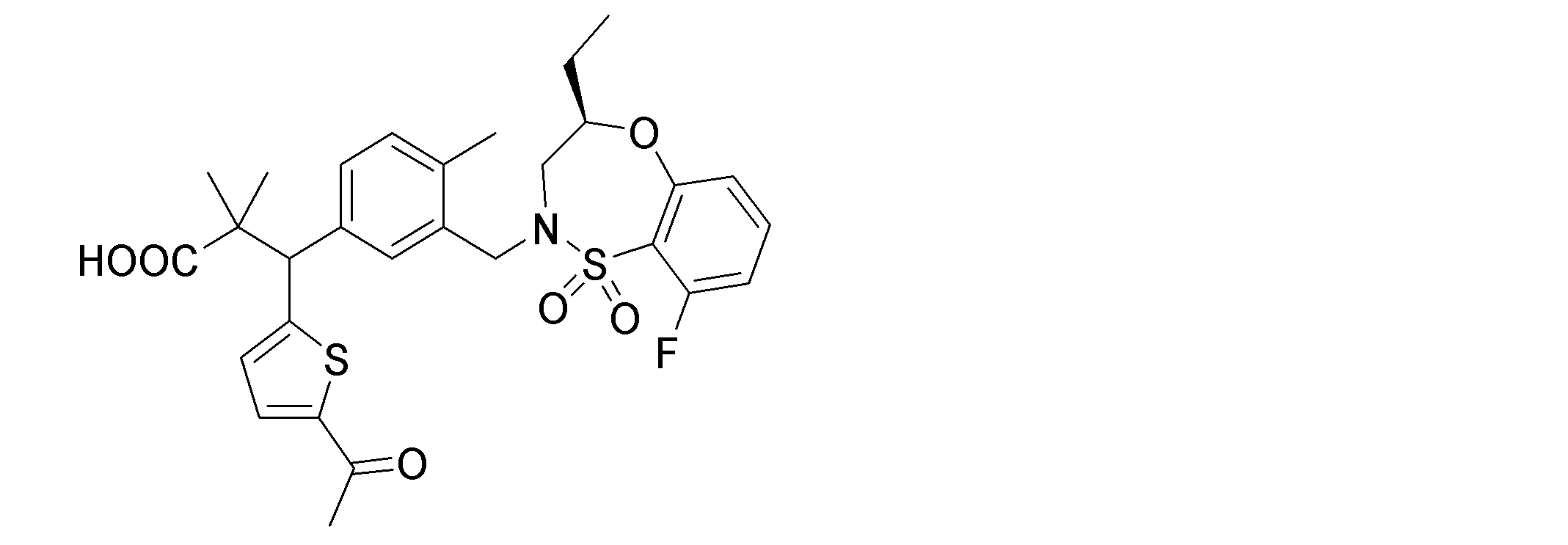
1H NMR (400 MHz, CDCl3) δ 7.52 (1H, br s), 7,44-7.37 (1H, m), 7.37-7.34 (1H, m), 7.27-7.23 (1H, m), 7.14-7.10 (1H, m), 7.04 (1H, br t, 4.0 Hz), 6.99-6.94 (1H, m), 6.77-6.75 (1H, m), 4.78-4.55 (1H, m), 4.75 (1H, s), 4.52-4.42 (1H, m), 4.23-4.18 (1H, m), 3.50-3.32 (1H, m), 3.15-2.84 (1H, m), 2.49 (3H, s), 2.34-2.32 (3H, m), 1.61-1.49 (2H, m), 1.33 (3H, s), 1,26 (3H, s), 1.04-0.96 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(4-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-1H-ベンゾ[d]イミダゾール-6-イル)プロパン酸(化合物45)の製造
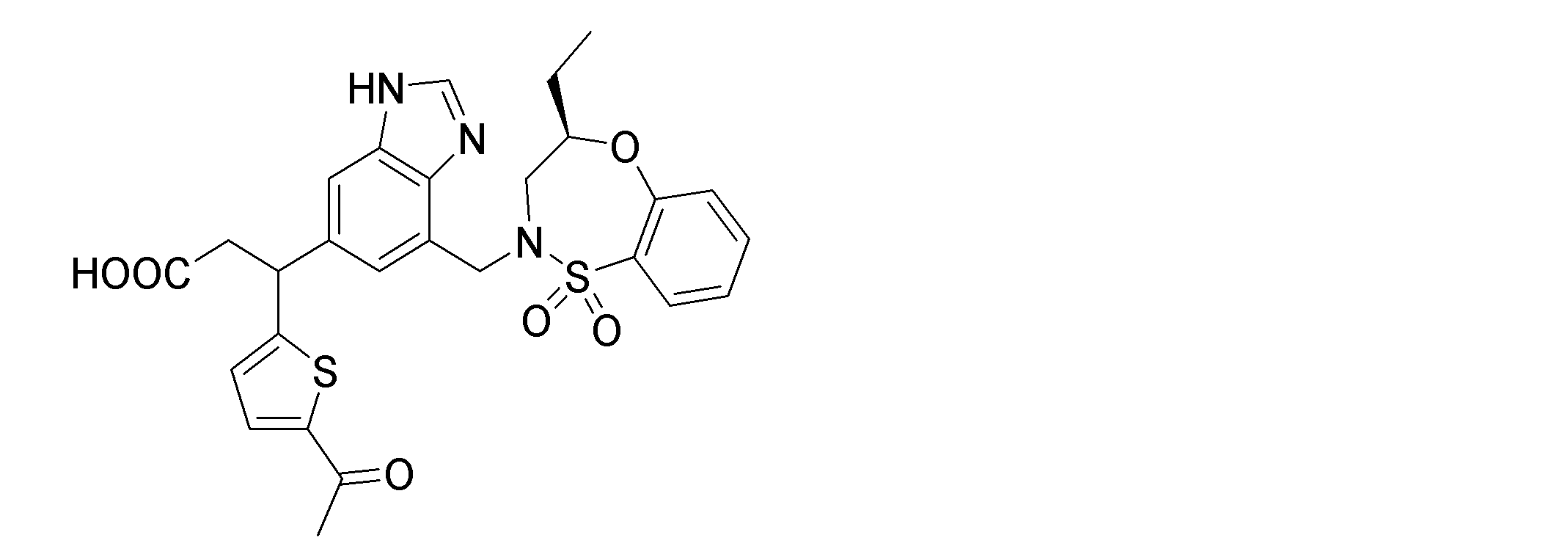
1H NMR (400 MHz, CDCl3) δ 8.11 (1H, s), 7.87 (1H, s), 7.86 (1H, d, J =7.6 Hz), 7.54 (1H, t, J = 7.6 Hz), 7.51 (1H, br s), 7.28-7.24 (1H, m), 7.22 (1H, d, J = 8.0 Hz), 6.95 (1H, s), 6.89 (1H, br s), 4.97-4.95 (1H, m), 4.69-4.66 (1H, m), 4.08-4.00 (1H, m), 3.97-3.92 (1H, m), 3.70-3.62 (1H, m), 3.24-3.22 (1H, m), 3.14-3.11 (1H, m), 3.04-3.01 (1H, m), 2.47 (3H, s), 1.76-1.67 (1H, m), 1.57-1.47 (1H, m), 1.10-1.08 (3H, m)。
3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物2)の製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, d, J =7.6 Hz), 7.53 (1H, d, J = 7.6 Hz), 7.26 (1H, t, J = 7.2 Hz), 7.21 (1H, d, J = 8.0 Hz), 7.18-7.07 (3H, m), 6.78 (1H, s), 4.57-4.54 (1H, d, J = 14.0 Hz), 4.29 (1H, d, J = 10.8 Hz), 4.07-3.98 (1H, m), 3.83-3.82 (1H, m), 3.75-3.67 (1H, m), 3.23-3.17 (1H, m), 2.93-2.47 (1H, m), 2.44 (3H, s), 2.42 (3H, s), 2.34 (3H, br s), 1.75-1.60 (1H, m), 1.44-1.33 (1H, m), 1.11-1.04 (6H, m)。
3-(5-アセチルチアゾール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物40)の製造

1H NMR (400 MHz, CDCl3) δ 8.17 (1H, s), 7.87 (1H, d, J = 7.6 Hz), 7.52 (1H, t, J = 7.6 Hz), 7.25-7.16 (5H, m), 4.78 (1H, br s), 4.53 (1H, d, J = 14.0 Hz), 4.06-3.96 (1H, m), 3.84 (1H, t, J= 13.2 Hz), 3.76-3.61 (1H, m), 3.47-3.40 (1H, m), 3.04-2.93 (2H, m), 2.50 (3H, s), 2.33 (3H, s), 1.73-1.58 (1H, m), 1.50-1.40 (1H, m), 1.08-1.06 (3H, m)。
3-(5-アセチル-4-メチルチアゾール-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物41)の製造
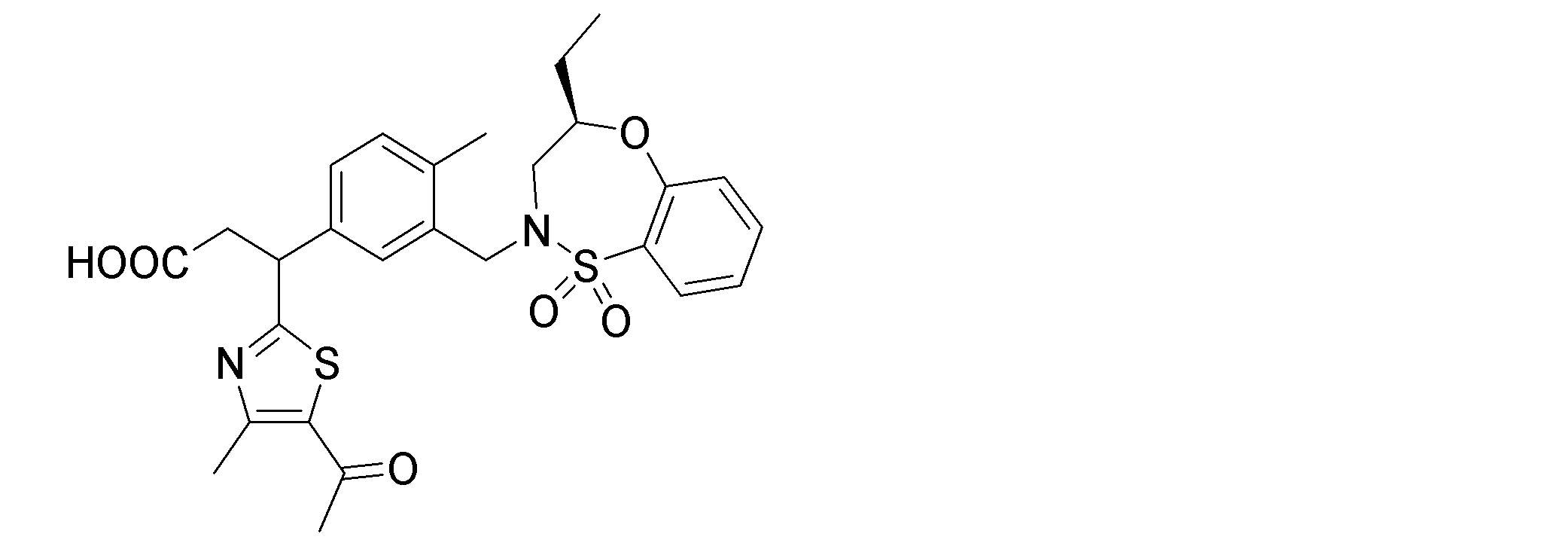
1H NMR (400 MHz, CDCl3) δ 7.87 (1H, d, J = 7.6 Hz), 7.51 (1H, t, J= 7.6 Hz), 7.25 (1H, t, J = 7.6 Hz), 7.20-7.14 (4H, m), 4.69 (1H, t, J = 7.2 Hz), 4.55 (1H, d, J = 14.0 Hz), 4.05-3.96 (1H, m), 3.84-3.82 (1H, m), 3.75-3.70 (1H, m), 3.44 (1H, dd, J = 16.4, 7.6 Hz), 3.04-2.93 (2H, m), 2.67 (3H, s), 2,43 (3H, s), 2.34 (3H, br s), 1.72-1.62 (1H, m), 1.51-1.36 (1H, m), 1,07-1.04 (3H, m)。
3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-3-(6-オキソ-5,6-ジヒドロ-4H-チエノ[2,3-c]ピロール-2-イル)プロパン酸(化合物38)の製造
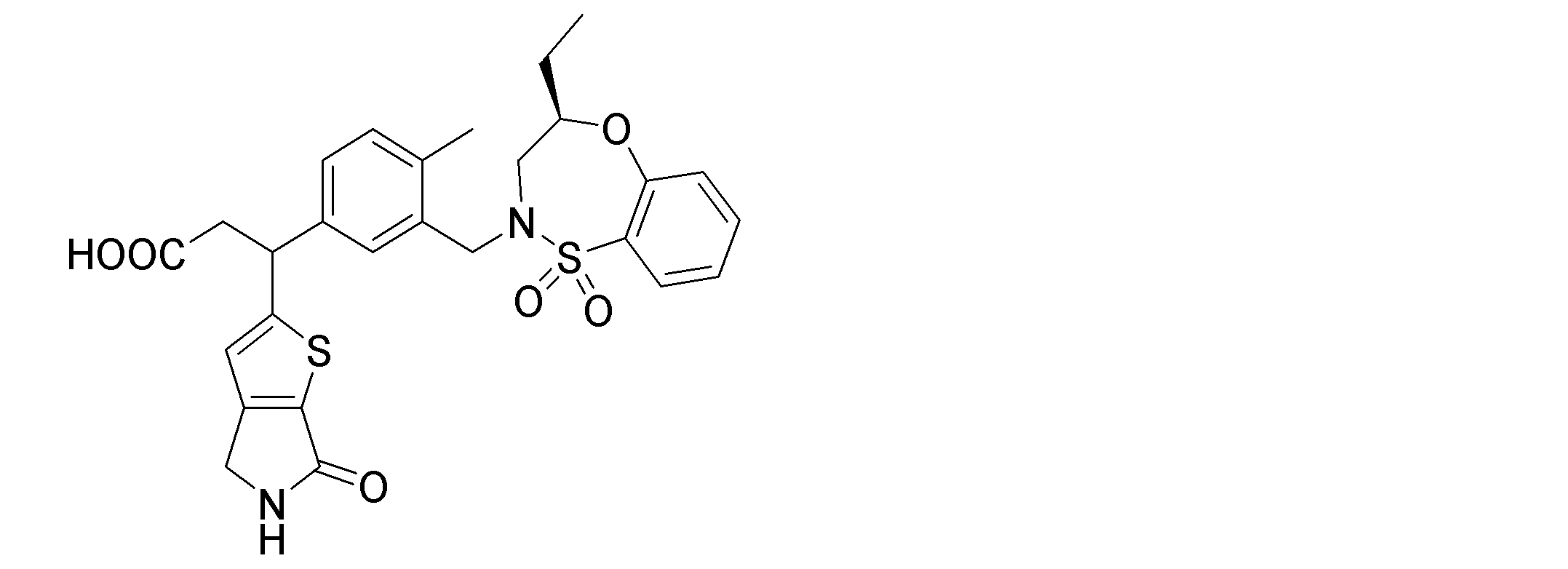
1H NMR (400 MHz, CDCl3) δ 7.87 (1H, d, J = 7.6 Hz), 7.52 (1H, t, J= 7.6 Hz), 7.26-7.23 (1H, m), 7.2 (1H, d, J= 8.0 Hz), 7.15-7.21 (4H, m), 6.86 (1H, s), 4.75 (1H, t, J = 7.2 Hz), 4.54-4.53 (1H, m), 4.25 (2H, s), 4.03-3.96 (1H, m), 3.84-3.82 (1H, m), 3.73-3.65 (1H, m), 3.12 (1H, dd, J = 16.0, 8.0 Hz), 3.03 (1H, d, J= 16.0, 7.2 Hz), 2.96 (1H, d, J = 14.4 Hz), 2.32 (3H, s), 1.71-1.60 (1H, m), 1.46-1.36 (1H, m), 1.05 (3H, t, J = 7.2 Hz)。
3-(5-アセチルフラン-2-イル) -3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物25)の製造

1H NMR(400 MHz, CDCl3)δ 7.87 (1H, dd,J = 8.0, 1.6 Hz), 7.51 (1H, td, J= 8.0, 1.6 Hz), 7.26-7.19 (2H, m), 7.15-7.10 (3H, m), 7.06 (1H, d, J = 3.6 Hz), 6.15-6.13 (1H, m), 4.56-4.50 (2H, m), 4.04-3.96 (1H, m), 3.85-3.80 (1H, m), 3.76-3.68 (1H, m), 3.18-3.12 (1H, m), 3.00-2.89 (2H, m), 2.39 (3H, s), 2.31 (3H, s), 1.70-1.64 (1H, m), 1.49-1.38 (1H, m), 1.08-1.04 (3H, m)。
3-(5-アセチルチオフェン-3-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物15)の製造

1H NMR(400 MHz, CDCl3)δ 7.86 (1H, d,J = 8.0 Hz), 7.51 (1H, br t, J = 8.0 Hz), 7.45 (1H, dd, J = 4.0, 1.2 Hz), 7.29 (1H, br s), 7.26-7.18 (2H, m), 7.13-7.05 (3H, m), 4.53-4.49 (2H, m), 4.01-3.94 (1H, m), 3.84-3.79 (1H, m), 3.74-3.67 (1H, m), 3.07-2.92 (3H, m), 2.47 (3H, s), 2.29 (3H, s), 1.69-1.60 (1H, m), 1.45-1.33 (1H, m), 1.04 (3H, t, J = 7.2 Hz)。
3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物10)の製造
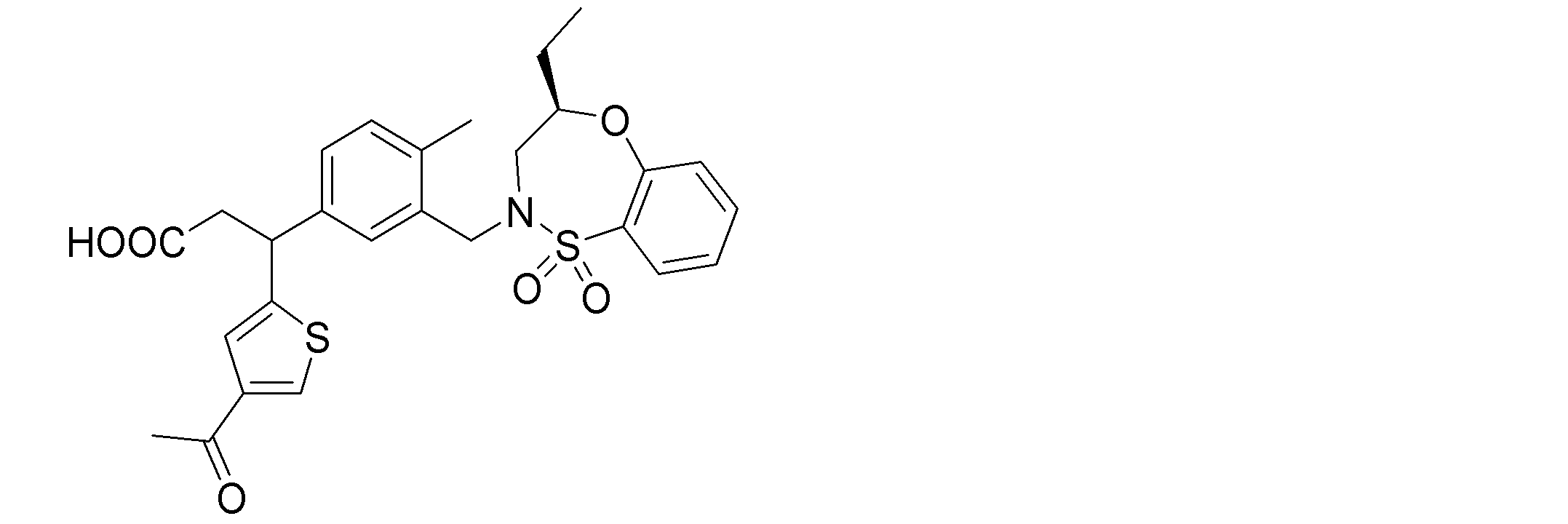
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d,J = 8.0 Hz), 7.84 (1H, br s), 7.52 (1H, br t, J = 8.0 Hz), 7.27-7.23 (2H, m), 7.21-7.18 (1H, m), 7.16-7.11 (2H, m), 7.08-7.05 (1H, m), 4.65-4.61 (1H, m), 4.55 (1H, d, J = 14.0 Hz), 4.02-3.94 (1H, m), 3.83-3.68 (2H, m), 3.15-3.12 (1H, m), 3.04-2.90 (2H, m), 2.47 (3H, s), 2.29 (3H, br s), 1.69-1.60 (1H, m), 1.45-1.33 (1H, m), 1.06-1.02 (3H, m)。
3-(5-シアノ-1-メチル-1H-ピロール-3-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物36)の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 8.0, 1.6 Hz), 7.52 (1H, td, J= 8.0, 1.6 Hz), 7.25 (1H, td, J = 8.0, 1.2 Hz), 7.21 (1H, d, J = 8.0 Hz), 7.13-7.11 (1H, m), 7.08-7.06 (2H, m), 6.57-6.54 (2H, m), 4.53 (1H, d, J = 14.4 Hz), 4.33-4.29 (1H, m), 4.04-3.97 (1H, m), 3.85-3.81 (1H, m), 3.75-3.70 (1H, m), 3.68 (3H, s), 2.99-2.82 (3H, m), 2.31 (3H, s), 1.73-1.64 (1H, m), 1.47-1.38 (1H, m), 1.09-1.06 (3H, m)。
3-(3-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物44)の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d,J = 8.0 Hz), 7.51 (1H, br t, J = 8.0 Hz), 7.33 (1H, d, J = 5.6 Hz), 7.27-7.19 (3H, m), 7.14-7.10 (3H, m), 5.62-5.57 (1H, m), 4.61-4.56 (1H, m), 4.02-3.96 (1H, m), 3.78-3.66 (2H, m), 3.14-3.07 (1H, m), 3.02-2.89 (2H, m), 2.48 (3H, br s), 2.35 (3H, br s), 1.69-1.56 (1H, m), 1.45-1.35 (1H, m), 1.10-1.02 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-2,2-ジメチル-3-(4-メチル-3-{[(R)-4-メチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}フェニル)プロパン酸(化合物12)の製造
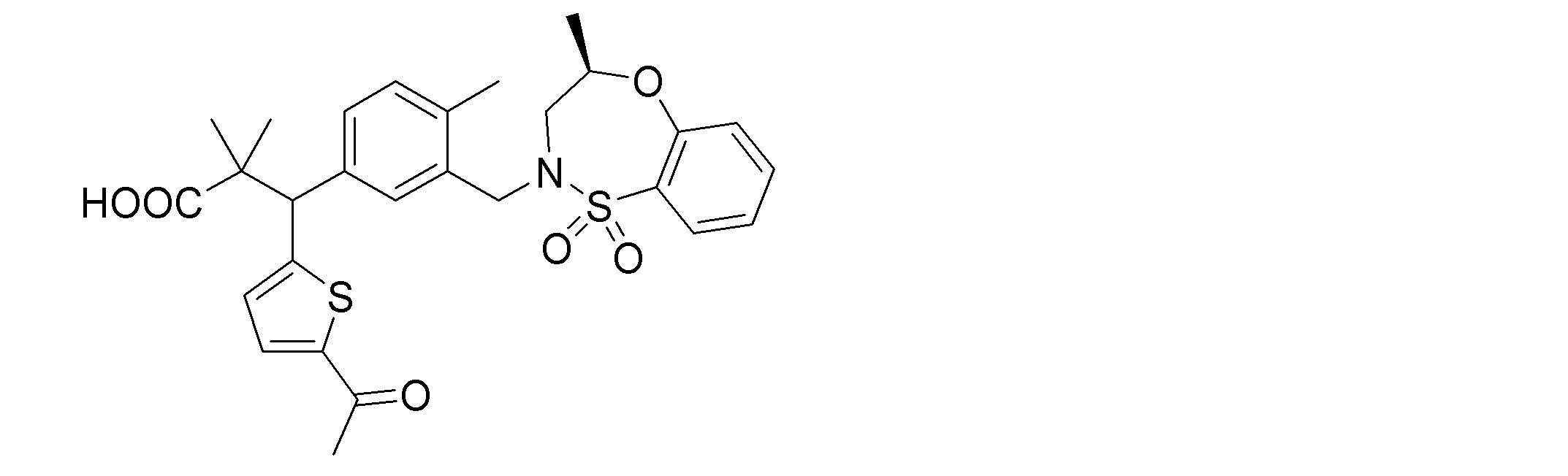
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d, J = 8.0 Hz), 7.55-7.50 (2H, m), 7.28-7.20 (4H, m), 7.15-7.12 (1H, m), 7.01-7.00 (1H, m), 4.71 (1H, d, J= 4.8 Hz), 4.58-4.57 (1H, m), 4.35-4.26 (1H, m), 3.84-3.70 (2H, m), 2.95-2.91 (1H, m), 2.50 (3H, s), 2.34 (3H, br s), 1.34-1.31 (3H, m), 1.28-1.24 (6H, m)。
3-(5-アセチルフラン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物24)の製造

1H NMR(400 MHz, CDCl3)δ 7.89 (1H, dd, J = 8.0, 1.6 Hz), 7.53 (1H, td, J = 8.0, 1.6 Hz), 7.28-7.24 (1H, m), 7.23-7.20 (1H, m), 7.18-7.11 (3H, m), 7.06 (1H, br d, J = 3.6 Hz), 6.25 (1H, d, J= 3.6 Hz), 4.58-4.52 (1H, m), 4.24 (1H, d,J = 10.8 Hz), 4.07-3.97 (1H, m), 3.89-3.81 (1H, m), 3.76-3.68 (1H, m), 3.22-3.16 (1H, m), 3.00-2.93 (1H, m), 2.39 (3H, s), 2.33 (3H, s), 1.74-1.63 (1H, m), 1.49-1.35 (1H, m), 1.10-1.04 (6H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[2,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物46)の製造

1H NMR(400 MHz, CDCl3)δ 8.22 (1H, br t,J = 8.0 Hz), 7.69-7.64 (1H, m), 7.51 (1H, t, J = 3.6 Hz), 7.32-7.19 (2H, m), 7.07-7.03 (2H, m), 6.43-6.38 (1H, m), 4.66-4.38 (3H, m), 3.51-3.25 (3H, m), 2.48-2.46 (3H, m), 2.28 (3H, s), 1.33-1.24 (8H, m), 0.81-0.76 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-[3-({4-[2-(ベンジロキシ)エチル]-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル}メチル)-4-メチルフェニル]-2-メチルプロパン酸(化合物13)の製造
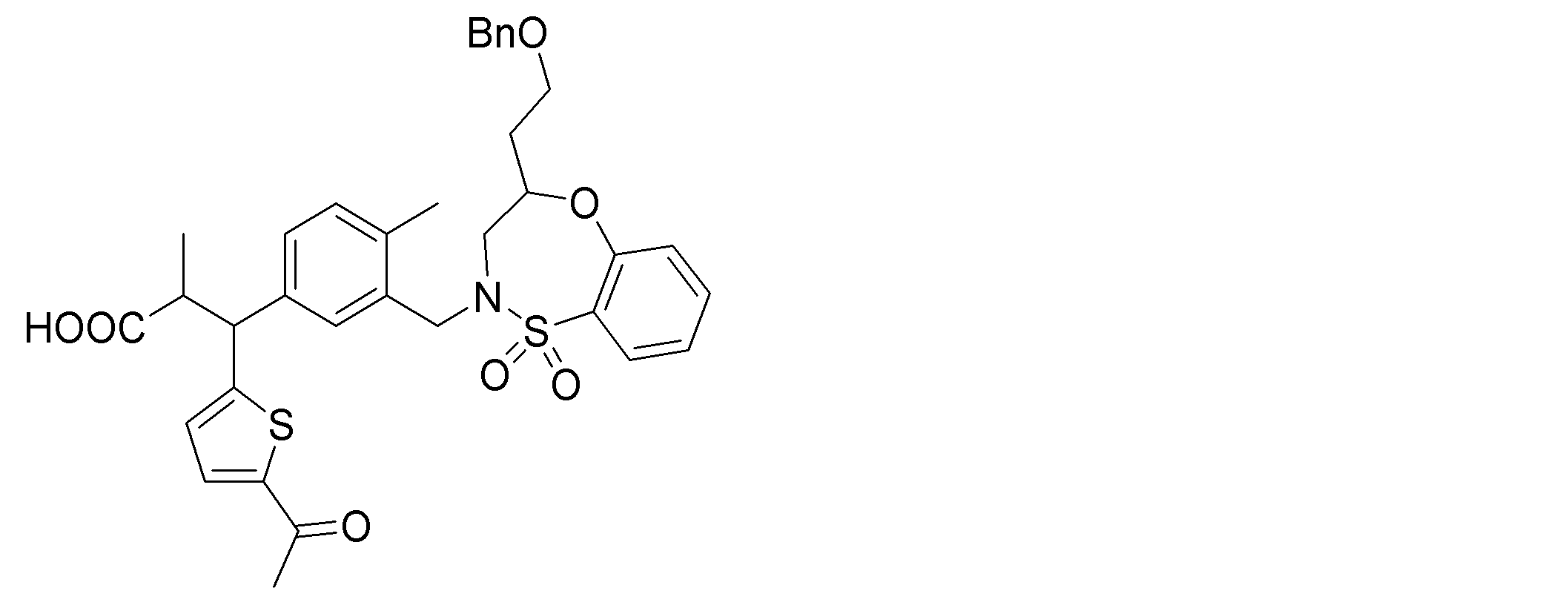
1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dd,J = 7.6, 1.2 Hz), 7.50-7.42 (2H, m), 7.39-7.24 (6H, m), 7.18-7.00 (3H, m), 6.96-6.77 (2H, m), 4.76-4.36 (4H, m), 4.34-4.19 (1H, m), 3.93-3.80 (2H, m), 3.77-3.74 (1H, m), 3.63-3.58 (1H, m), 3.31-3.19 (1H, m), 2.88-2.76 (1H, m), 2.47-2.44 (3H, m), 2.39-2.31 (3H, m), 2.00-1.64 (2H, m), 1.12 (3H, t, J = 6.8 Hz)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[4-(2-ヒドロキシエチル)-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物43a, 43b)の製造
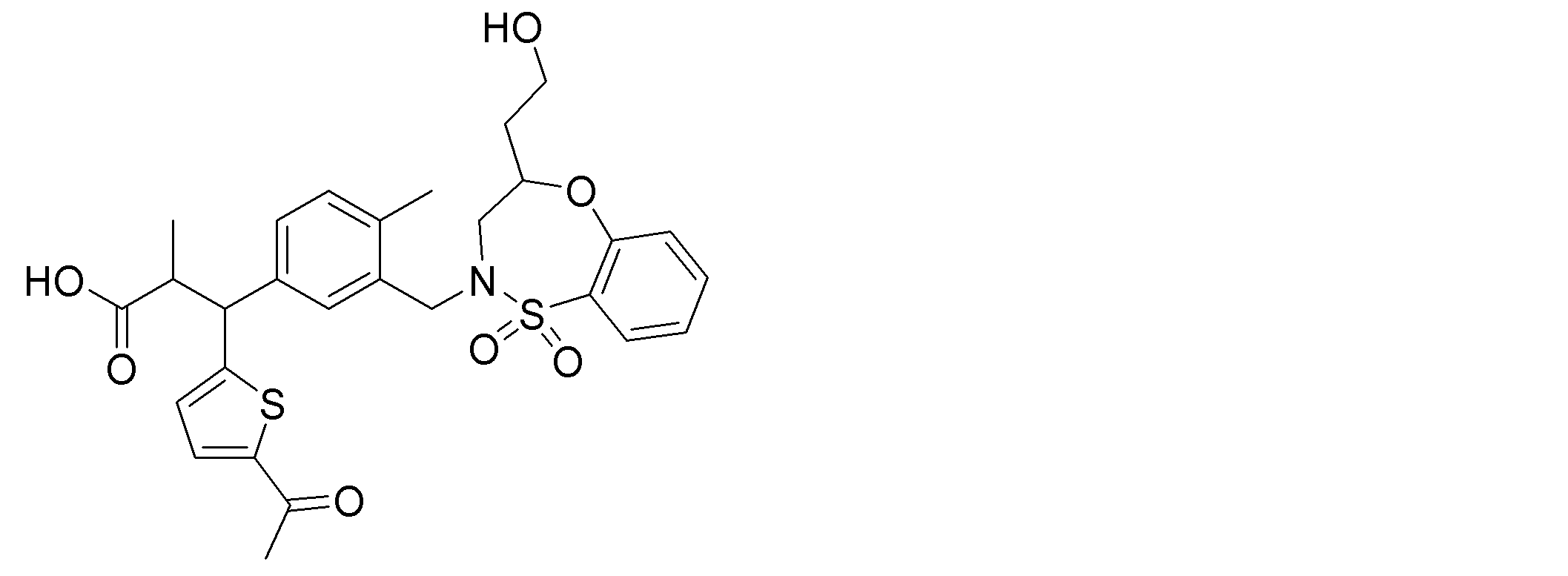
43a: 1H NMR(400 MHz, CDCl3)δ 7.90-7.85 (1H, m), 7.55-7.40 (2H, m), 7.31-7.27 (1H, m), 7.24-7.13 (2H, m), 7.10-7.03 (2H, m), 6.92-6.82 (1H, m), 4.74-4.62 (1H, m), 4.41-4.39 (1H, m), 4.31-4.18 (1H, m), 4.04-3.77 (3H, m), 3.72-3.60 (1H, m), 3.12-3.05 (1H, m), 2.87-2.77 (1H, m), 2.49-2.29 (1H, m), 1.89-1.56 (2H, m), 1.12-0.92 (3H, m)。
43b: 1H NMR(400 MHz, CDCl3)δ 7.90-7.85 (1H, m), 7.55-7.47 (1H, m), 7.42-7.32 (1H, m), 7.29-7.24 (1H, m), 7.22-7.13 (2H, m), 7.11-7.03 (2H, m), 6.93-6.84 (1H, m), 4.75-4.61 (1H, m), 4.44-4.18 (2H, m), 4.05-3.79 (3H, m), 3.73-3.60 (1H, m), 3.15-3.09 (1H, m), 2.88-2.77 (1H, m), 2.49-2.30 (6H, m), 1.91-1.77 (1H, m), 1.63-1.53 (1H, m), 1.14-0.96 (3H, m)。
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物4)の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d, J = 7.6 Hz), 7.53 (1H, br t,J = 7.6 Hz), 7.28-7.20 (5H, m), 7.12 (1H, dd, J = 7.6, 2.4 Hz), 6.82-6.80 (1H, m), 4.60 (1H, d, J = 3.6 Hz), 4.54 (1H, d, J = 14.4 Hz), 4.03-3.99 (1H, m), 3.93 (3H, s), 3.87-3.81 (1H, m), 3.78-3.71 (1H, m), 2.97 (1H, dd, J = 7.6, 1.2 Hz), 2.47 (3H, br s), 2.32 (3H, s), 1.74-1.62 (1H, m), 1.49-1.37 (1H, m), 1.33 (3H, br s), 1.25 (3H, br s), 1.11-1.06 (3H, m)。
3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物3)の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, br d, J = 7.6 Hz), 7.83 (1H, dd, J= 10.4, 1.2 Hz), 7.55-7.50 (1H, m), 7.36-7.35 (1H, m), 7.28-7.25 (1H, m), 7.24-7.19 (1H, m), 7.17-7.04 (3H, m), 4.66-4.53 (1H, m), 4.30 (1H, d, J = 10.8 Hz), 4.03-3.64 (3H, m), 3.31-3.02 (1H, m), 2.96-2.88 (1H, m), 2.46-2.44 (3H, m), 2.35-2.33 (3H, m), 1.70-1.60 (1H, m), 1.44-1.32 (1H, m), 1.09-1.03 (6H, s)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物16)の製造
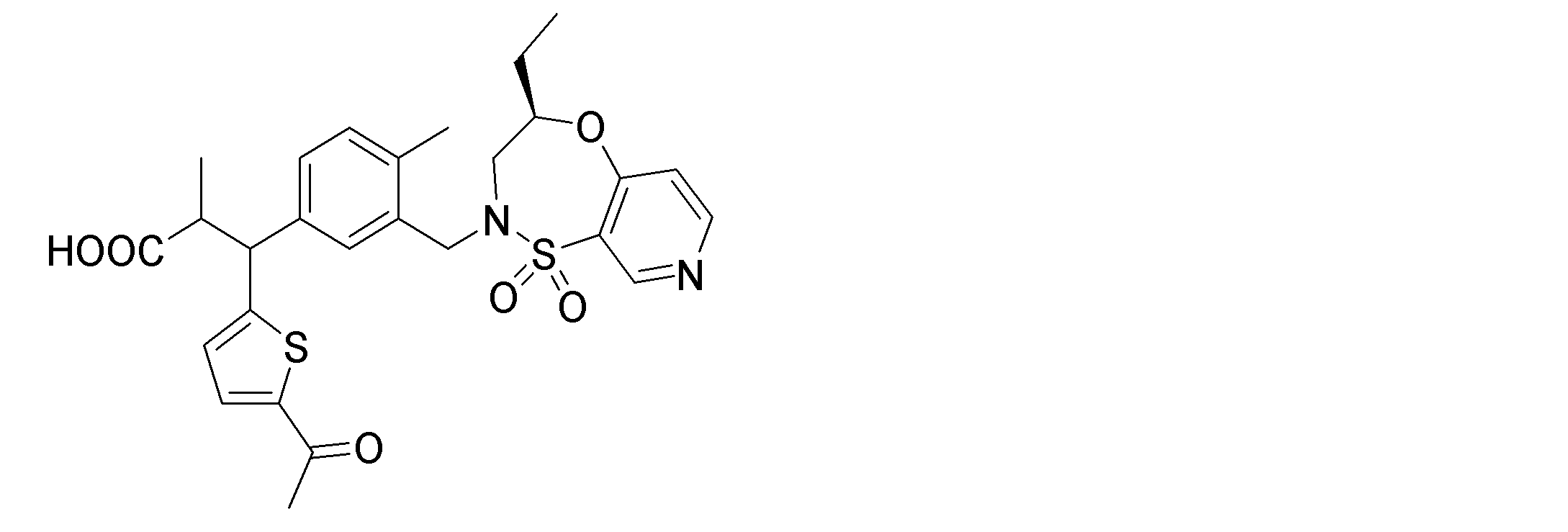
1H NMR(400 MHz, CDCl3)δ 9.02 (1H, s), 8.66 (1H, dd, J = 5.6, 1.6 Hz), 7.49-7.47 (1H, m), 7.17 (2H, s), 7.12-7.10 (2H, m), 6.98-6.97 (1H, m), 4.47-4.36 (3H, m), 4.22-4.10 (1H, m), 3.50-3.40 (1H, m), 3.26-3.12 (2H, m), 2.47 (3H, s), 2.33 (3H, s), 1.74-1.63 (1H, m), 1.56-1.48 (1H, m), 1.12 (3H, d, J = 6.8 Hz), 1.04 (3H, br t, J = 7.2 Hz)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[4-(2-メトキシエチル)-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物42)の製造

1H NMR(400 MHz, CDCl3)δ 7.96-7.89 (1H, m), 7.59-7.54 (1H, m), 7.52-7.46 (1H, m), 7.37-7.27 (2H, m), 7.20-7.10 (3H, m), 6.97-6.88 (1H, m), 4.81-4.73 (1H, m), 4.64-4.43 (1H, m), 4.36-4.21 (1H, m), 3.91-3.72 (3H, m), 3.67-.54 (1H, m), 3.49-3.41 (3H, m), 3.35-3.16 (1H, m), 2.88-2.69 (1H, m), 2.50-2.35 (6H, m), 1.89-1.74 (1H, m), 1.69-1.66 (1H, m), 1.21-1.12 (3H, m)。
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物11)の製造

1H NMR(400 MHz, CDCl3)δ 7.88 (1H, dt,J = 8.0, 1.6 Hz), 7.53 (1H, br t, J= 8.0 Hz), 7.28-7.20 (4H, m), 7.16-7.14 (1H, m), 6.79 (1H, d, J = 5.2 Hz), 4.57-4.54 (2H, m), 4.06-4.01 (1H, m), 3.86-3.84 (1H, m), 3.74 (1H, dd,J = 15.2, 10.4 Hz), 2.97 (1H, dd, J= 15.2, 5.6 Hz), 2.51 (3H, br s), 2.34 (3H, s), 1.74-1.61 (1H, m), 1.49-1.41 (1H, m), 1.33 (3H, s), 1.26 (3H, br s), 1.08 (3H, br t, J = 7.2 Hz)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物48)の製造
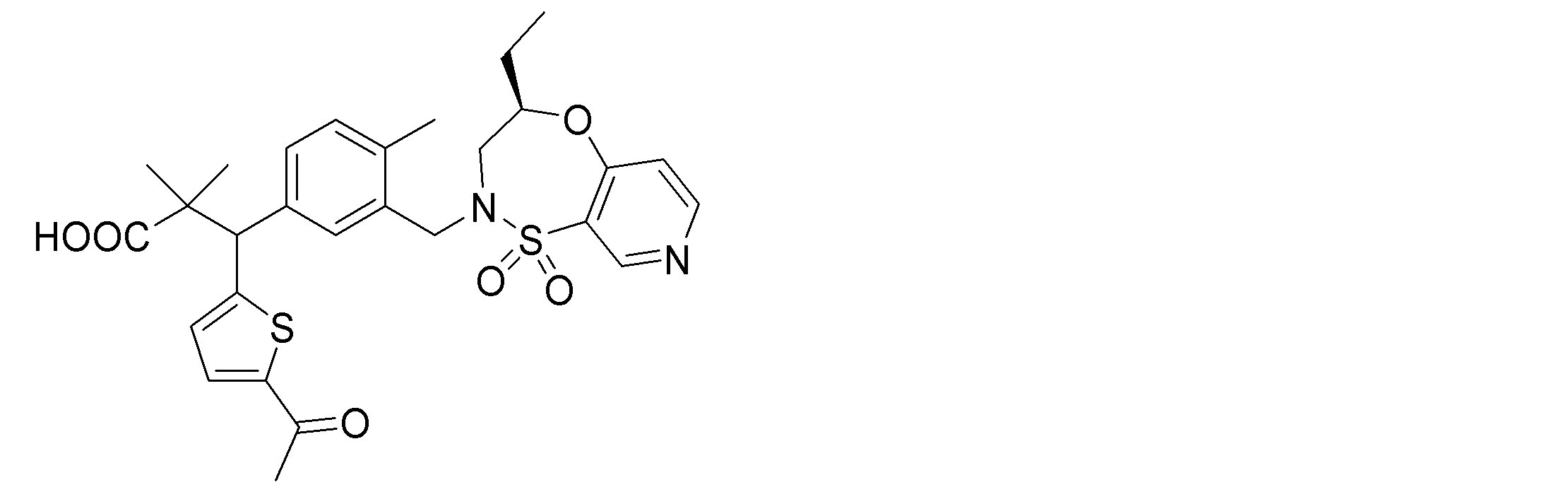
1H NMR (400 MHz, CDCl3) δ 9.00 (1H, br s), 8.66-8.63 (1H, m), 7.54 (1H, d, J = 3.6 Hz), 7.30-7.26 (2H, m), 7.16-7.10 (2H, m), 7.05-7.03 (1H, m), 4.74 (1H, br s), 4.46-4.37 (2H, m), 4.17-4.11 (1H, m), 3.46-3.43 (1H, m), 3.20-3.14 (1H, m), 2.50 (3H, s), 2.32 (3H, s), 1.73-1.63 (1H, m), 1.57-1.47 (1H, m), 1.32 (3H, s), 1.27 (3H, br s), 1.06-1.02 (3H, m)。
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)プロパン酸(化合物53)の製造
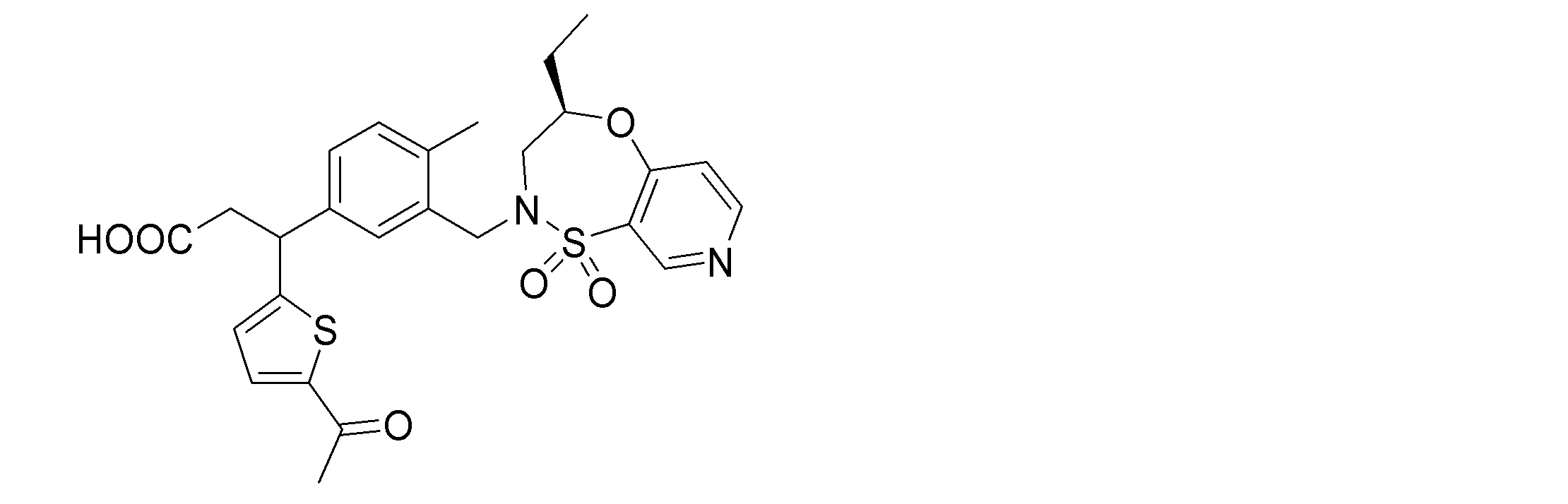
1H NMR(400 MHz, CDCl3)δ 9.00 (1H, s), 8.62 (1H, d, J = 5.6 Hz), 7.49 (1H, br s), 7.16-7.08 (4H, m), 6.88 (1H, br s), 4.71 (1H, t, J = 7.6 Hz) 4.43-4.37 (2H, m), 4.16-4.12 (1H, m), 3.51-3.42 (1H, m), 3.19-3.02 (3H, m), 2.47 (3H, s), 2.32 (3H, s), 1.70-1.61 (1H, m), 1.56-1.48 (1H, m), 1.02 (3H, t, J = 7.6 Hz)。
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物49)の製造
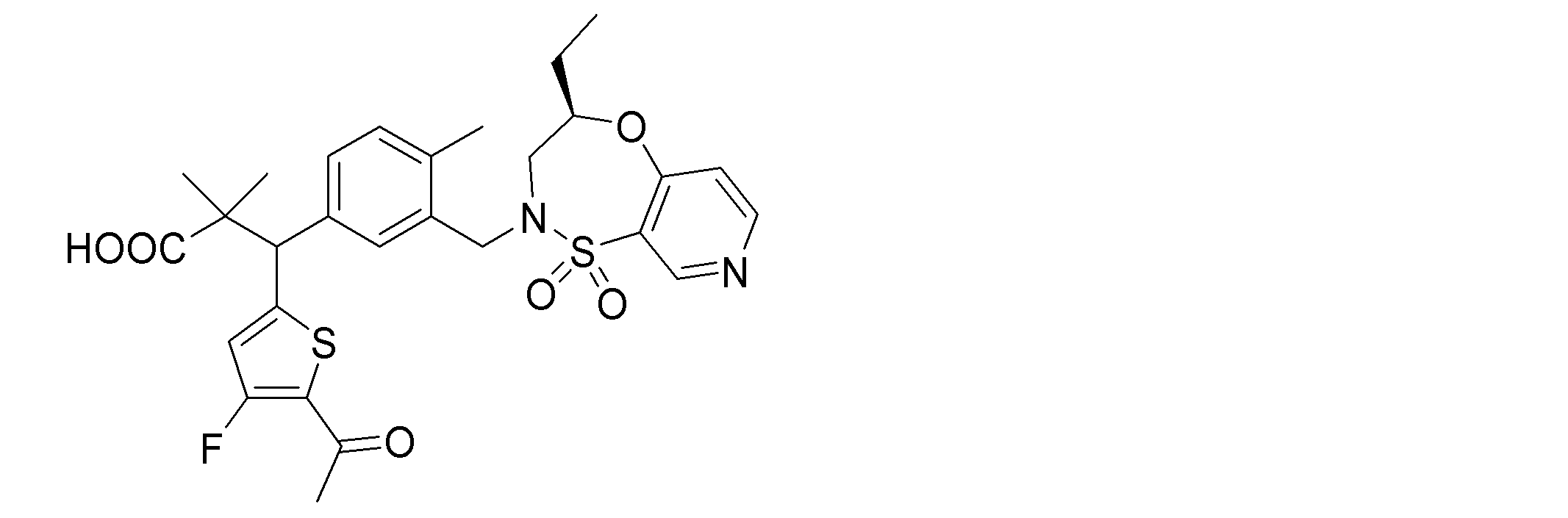
1H NMR (400 MHz, CDCl3) δ 9.00, 8.99 (each 0.5H, s), 8.65, 8.64 (each 0.5H, d, J = 5.6 Hz), 7.26-7.23 (3H, m), 7.16 (1H, d, J = 7.6 Hz), 7.11, 7.10 (each 0.5H, d, J= 5.4 Hz), 6.84, 6.83 (each 0.5H, s), 4.59, 4.58 (each 0.5H, s), 4.51-4.43 (1H, m), 4.39, 4.38 (each 0.5H, d, J = 14.4 Hz), 4.17 (1H, d, J= 14.4 Hz), 3.50-3.42 (1H, m), 3.21, 3.18 (each 0.5H, dd, J = 14.4, 4.8 Hz), 2.52, 2.51 (each 1.5H, s), 2.33, 2.32 (each 1.5H, s), 1.74-1.63 (1H, m), 1.61-1.50 (1H, m), 1.34, 1.33 (each 1.5H, s), 1.25 (3H, s), 1.04 (3H, t, J = 7.2 Hz)。
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物50)の製造

1H NMR (400 MHz, CDCl3) δ 9.01, 9.00 (each 0.5H, s), 8.65, 8.64 (each 0.5H, d, J = 5.6 Hz), 7.29-7.26 (2H, m), 7.14 (1H, d, J = 7.6 Hz), 7.11, 7.10 (each 0,5H, d, J = 5.6 Hz), 6.83, 6.82 (each 0.5H, s), 4.64, 4.61 (each 0.5H, s), 4.48-4.42 (1H, m), 4.37 (1H, d, J = 14.4 Hz), 4.17 (1H, d, J = 14.4 Hz), 3.94 (3H, s), 3.50-3.43 (1H, m), 3.22-3.15 (1H, m), 2.47 (3H, s), 2.312, 2.311 (each 1.5H, s), 1.74-1.64 (1H, m), 1.58-1.50 (1H, m), 1.344, 1.341 (each 1.5H, s), 1.28, 1.27 (each 1.5H, s), 1.05, 1.04 (each 1.5H, t, J = 7.2 Hz)。
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物52)の製造

1H NMR (400 MHz, CDCl3) δ 9.02-9.00 (1H, m), 8.67-8.64 (1H, m), 7.20-7.09 (4H, m), 6.76-6.74 (1H, m), 4.46-4.35 (2H, m), 4.30-4.10 (2H, m), 3.51-3.40 (1H, m), 3.22-3.15 (2H, m), 2.51-2.48 (3H, m), 2.33-2.31 (3H, m), 1.75-1.66 (1H, m), 1.58-1.52 (1H, m), 1.11 (3H, br d, J = 7.2 Hz), 1.05 (3H, br t, J = 7.6 Hz)。
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物51)の製造
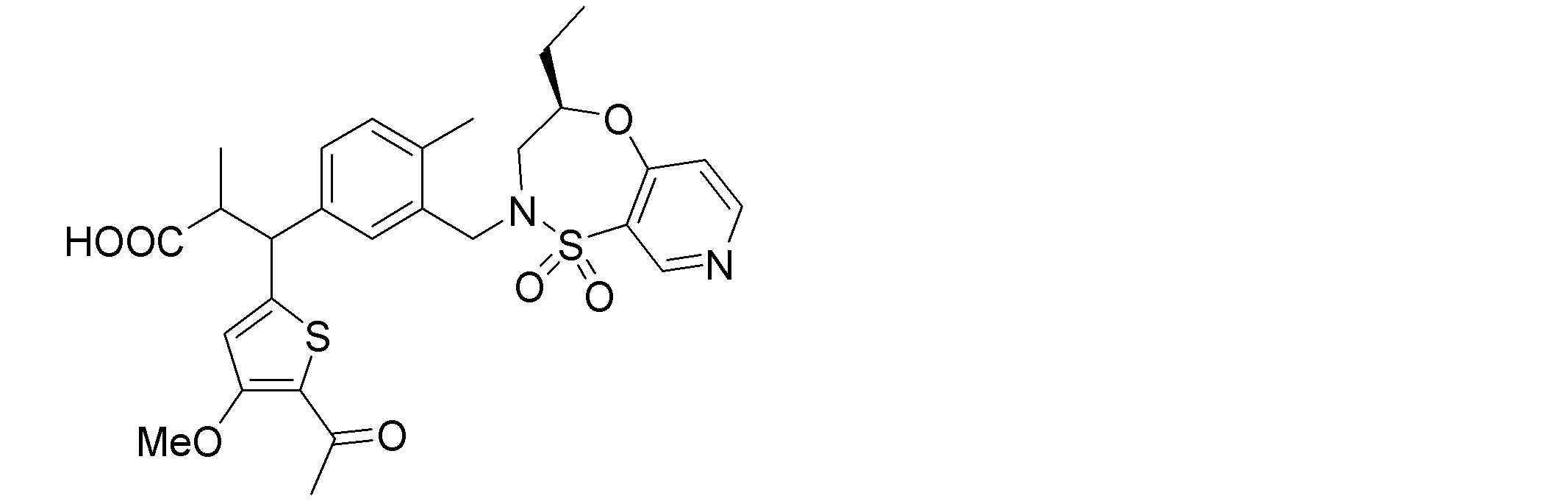
1H NMR (400 MHz, CDCl3) δ 9.02 (1H, s), 8.64 (1H, br d, J = 5.6 Hz), 7.14-708 (4H, m), 6.73 (1H, s), 4.46-4.35 (2H, m), 4.27 (1H, br d, J = 10.8 Hz), 4.22-4.07 (1H, m), 3.87 (3H, br s), 3.55-3.44 (1H, m), 3.22-3.08 (2H, m), 2.38 (3H, br s), 2.31 (3H, s), 1.73-1.65 (1H, m), 1.54-1.49 (1H, m), 1.05-1.01 (6H, m)。
<試験例1-1.Nrf2活性化作用の測定1(Binding assay)>
実施例で製造した化合物1~53を被験物質として用いた。


IC50(M)=10^(LOG(A/B)×(50-C)/(D―C)+LOG(B))
A: 阻害率50%を挟む近傍の2濃度うち、高い濃度
B: 阻害率50%を挟む近傍の2濃度うち、低い濃度
C: Bでの阻害率
D: Aでの阻害率。
化合物の細胞でのNRF2活性可能はレポーター遺伝子アッセイ法を用いて行った。FuGENE HDを用いて、pGL4.37[luc2P/ARE/Hygro](Promega社)をHCE-T細胞に導入し、形質導入の結果、薬剤耐性をもつ細胞を50 μg/mLのハイグロマイシン存在下での培養によって選抜し、クローン株であるHCE-T/ARE-Lucを樹立した。
実施例で製造した化合物の1 mmol/LのDMSO溶液を調製した。この1mmol/L DMSO溶液をアセトニトリルで10 μmol/Lに希釈した後、6.5 mmol/L β-NADPH溶液で200 nmol/Lに希釈し、反応群及び未反応群に50 μLずつ添加した。反応群に0.2 mg protein/mLヒト肝ミクロソーム(Mixed Gender, Pool of 50 livers)(XenoTech, LLC)溶液を50 μLずつ添加し、37 ℃で振とうしながら30分間インキュベーションした。未反応群にメタノール400 μLを添加後、0.2 mg protein/mLヒト肝ミクロソーム溶液を50 μLずつ添加し、組成を合わせた。反応群はインキュベーション後、メタノール400 μLを添加し、反応を停止させた。反応群及び未反応群の試料を-20 ℃で約30分間静置した後、4 ℃、3000 rpmで約10分間遠心分離した。上清をLC/MS/MSにて測定し、被験化合物のピーク面積値から下記式を用いて、被験化合物の残存率を算出した。測定は次に記載の装置、HPLCシステム:高速液体クロマトグラフLC-20Aシリーズ(株式会社島津製作所)及び質量分析装置:API 4000(AB Sciex Pte. Ltd.)、または同等の装置を用いて実施した。
残存率(%)=(反応群における被験化合物のピーク面積値/未反応群における被験化合物のピーク面積値)×100。
結果を表3~11に示す。表3~8及び10はIC50(nM, binding assay)及びMetabolic stabilityの結果を示し、表9及び11はEC50(nM, reporter assay)の結果を示す。

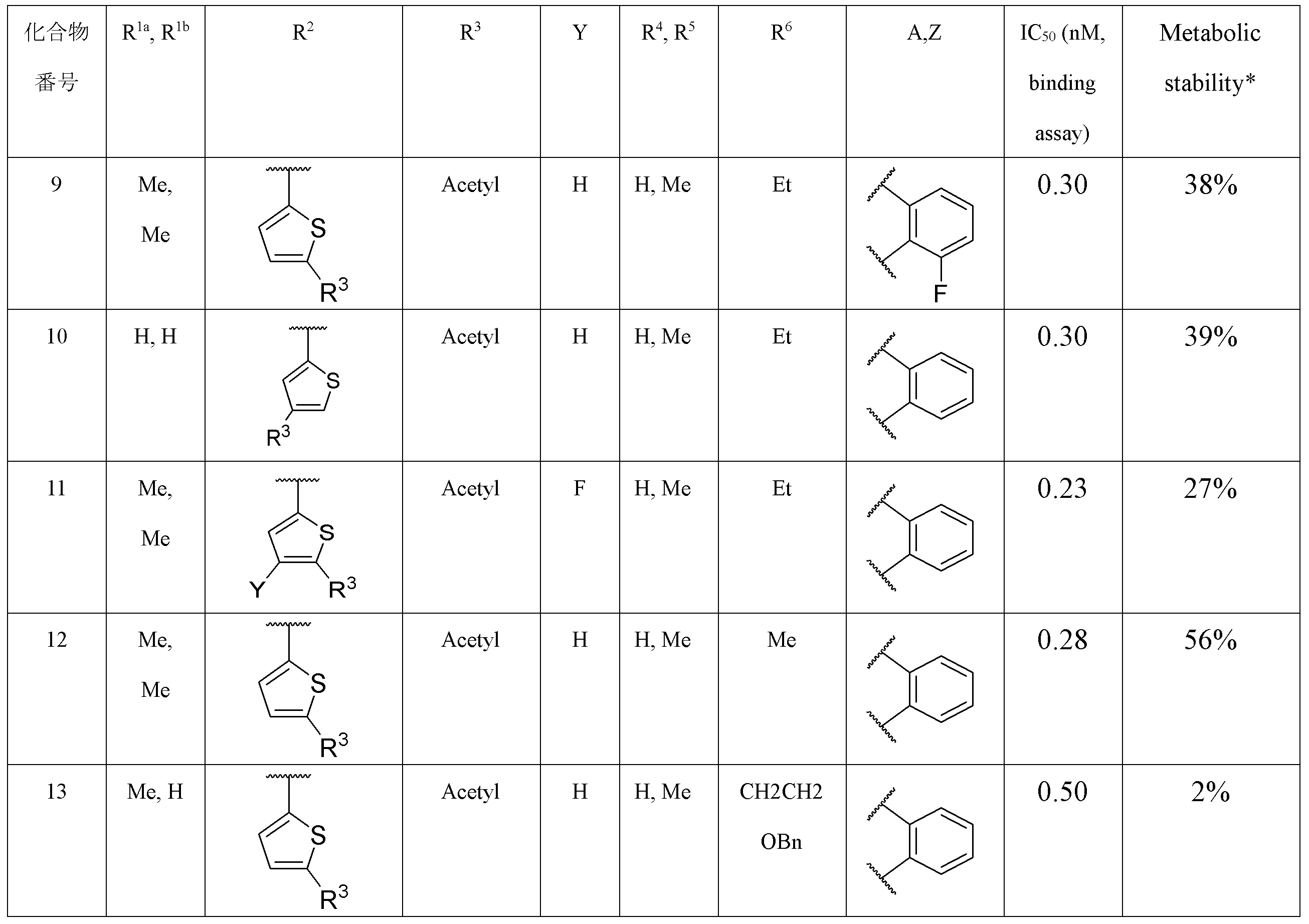



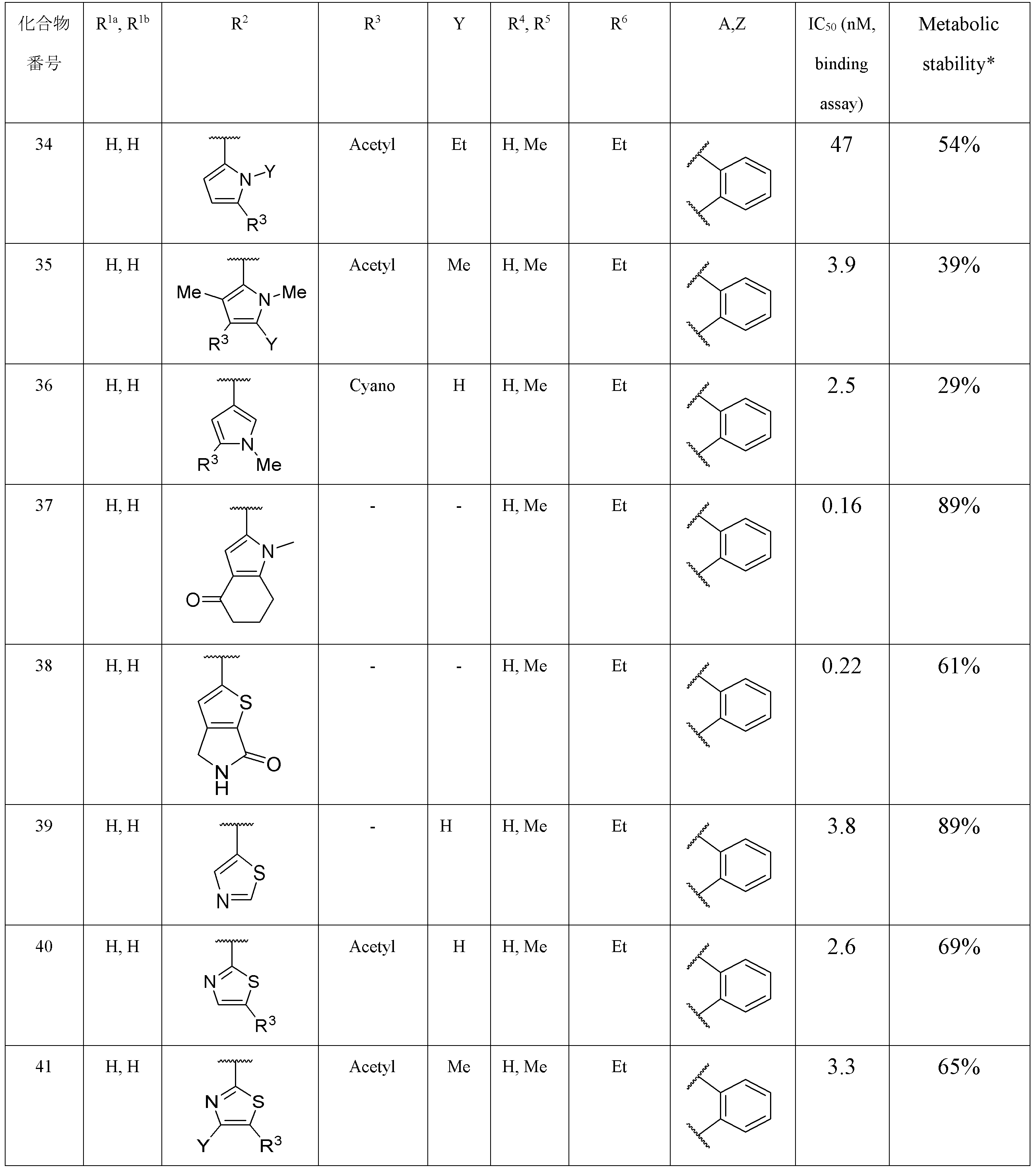
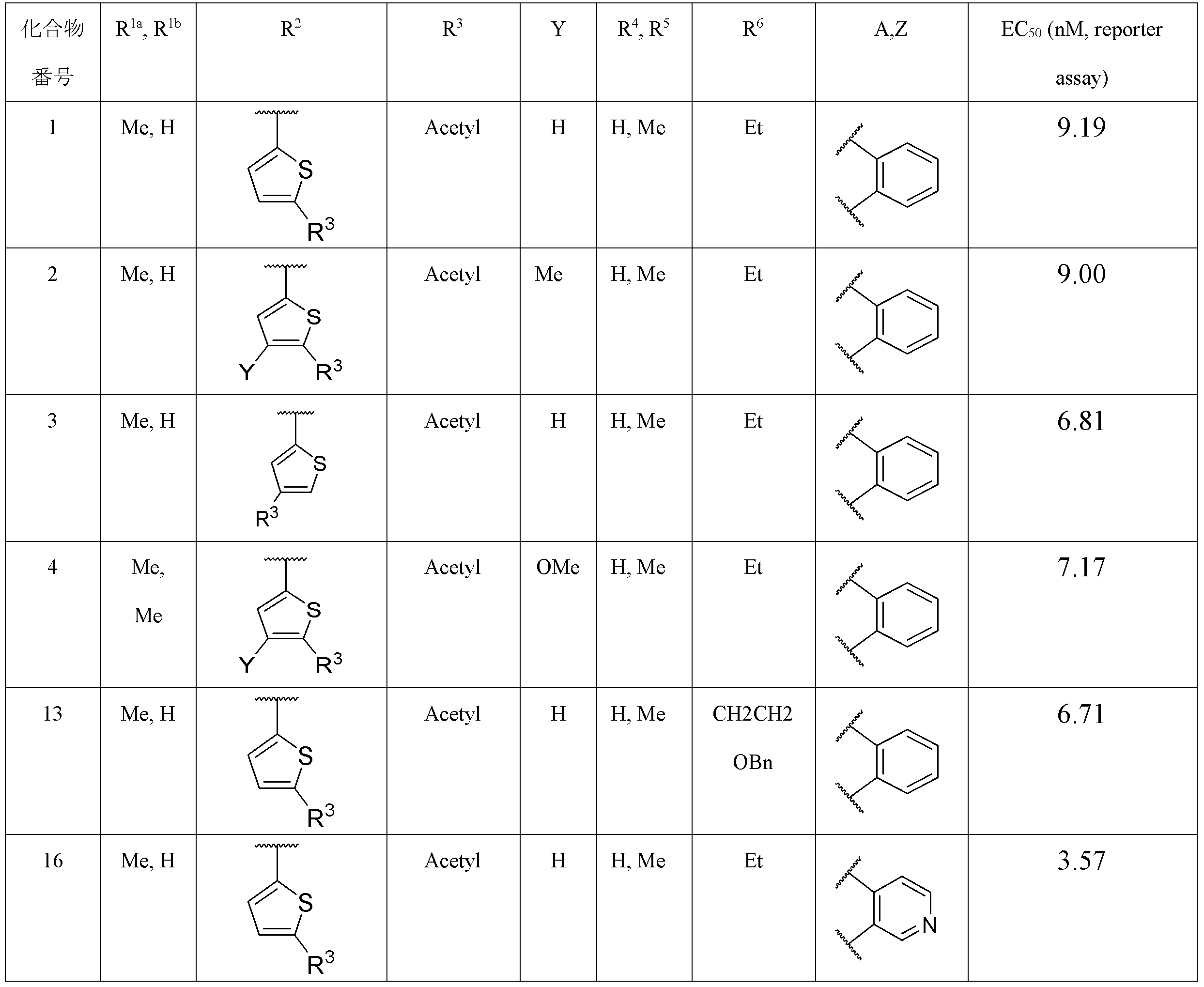
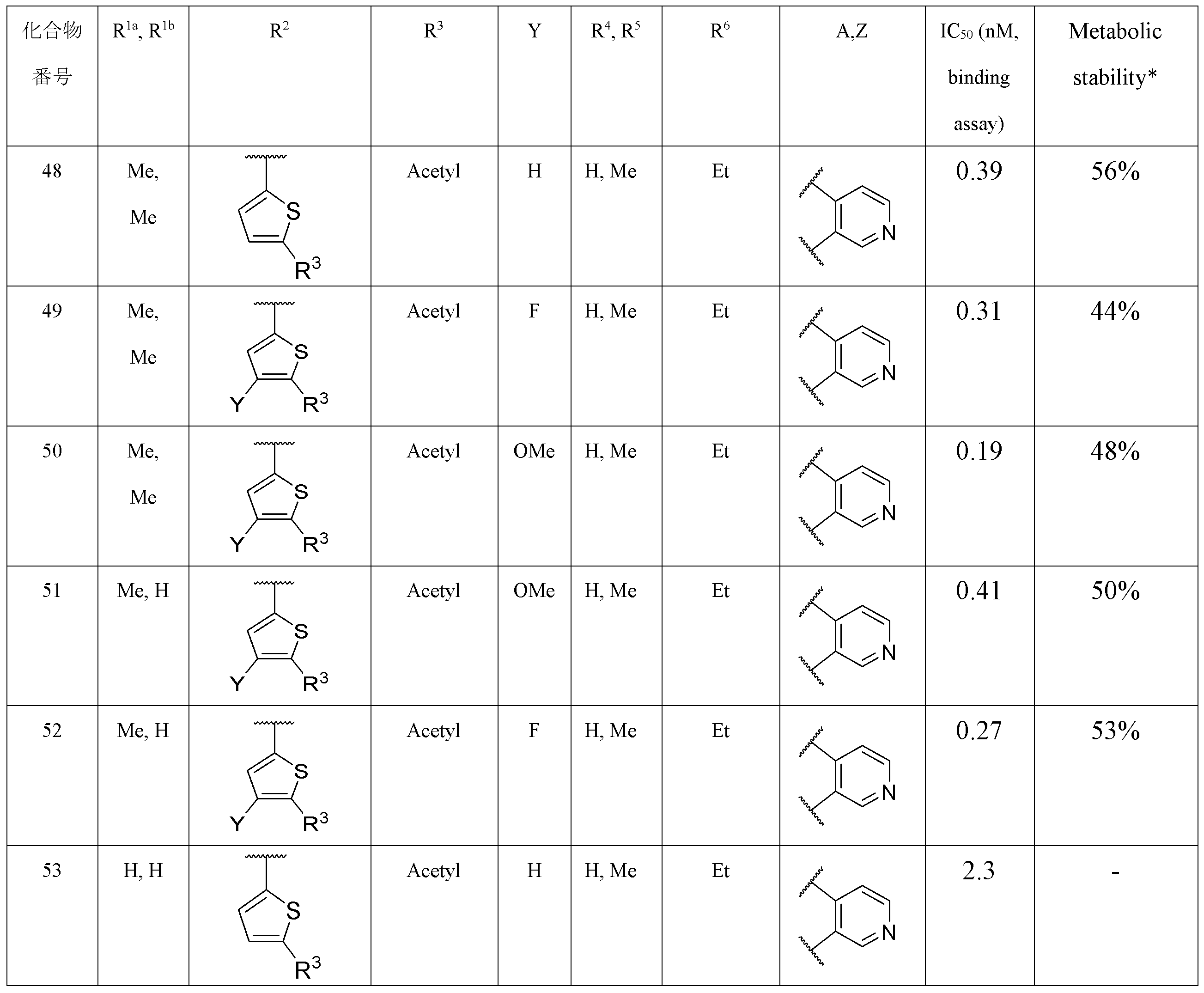

Claims (19)
- 一般式(I):

で表される化合物、その塩、又はそれらの溶媒和物。 - 前記R2が置換されていてもよいチエニル基である、請求項1に記載の化合物、その塩、又はそれらの溶媒和物。
- 前記チエニル基が有していてもよい置換基が電子求引基を含む、請求項2に記載の化合物、その塩、又はそれらの溶媒和物。
- 前記チエニル基が有していてもよい置換基が、アセチル基、アミノスルホニル基、シアノ基又はシクロアルキルカルボニル基を含む、請求項2又は3に記載の化合物、その塩、又はそれらの溶媒和物。
- 前記R2が一般式(R2A):

で表される基である、請求項1~4のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。 - 前記R2が一般式(R2Aa)又は(R2Ab):

で表される基である、請求項1~5のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。 - 前記R3がアセチル基である、請求項5又は6に記載の化合物、その塩、又はそれらの溶媒和物。
- 前記R4及び前記R5が同一又は異なって、水素原子又はアルキル基である、請求項5~7のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
- 前記R1a及び前記R1bが同一又は異なって、水素原子又はアルキル基である、請求項5~8のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。
- 前記R1a及び前記R1bが同一又は異なって、水素原子又はアルキル基であり、
前記R3がアセチル基であり、
前記R4及び前記R5が同一又は異なって、水素原子又はアルキル基であり、
前記R6が置換されていてもよいエチル基であり、
前記A1、前記A2及び前記A4が全てCHであり、前記A3がCH又はNであり、
前記Yが水素原子、アルキル基、ハロゲン原子又はアルコキシ基であり、且つ
前記Zが水素原子である、
請求項5~9のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。 - R1a及びR1bの一方がアルキル基であり、他方が水素原子であるか、R1a及びR1bの両方がアルキル基であり、
前記R2が一般式(R2Aa):

であり、
前記R4は水素原子であり、
前記R5はアルキル基であり、
前記R6が置換されていてもよいエチル基であり、
前記A1、前記A2、及び前記A4が全てCHであり、前記A3がCH又はNであり、且つ
前記Zが水素原子である、
請求項5~10のいずれかに記載の化合物、その塩、又はそれらの溶媒和物。 - 以下のいずれかに記載の化合物、その塩、又はそれらの溶媒和物:
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物1)、
3-(5-アセチル-4-メチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ [b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物2)、
3-(4-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物3)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物4)
3-(5-アセチルチオフェン-2-イル)-3-[3-({4-[2-(ベンジロキシ)エチル]-1,1-ジオキシド-3,4-ジヒドロ-2H-ベンゾ[b][1,4,5]オキサチアゼピン-2-イル}メチル)-4-メチルフェニル]-2-メチルプロパン酸(化合物13)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物16)、
3-(5-アセチルチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物48)、
3-(5-アセチル-4-フルオロチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物49)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2,2-ジメチルプロパン酸(化合物50)、
3-(5-アセチル-4-メトキシチオフェン-2-イル)-3-(3-{[(R)-4-エチル-1,1-ジオキシド-3,4-ジヒドロ-2H-ピリド[4,3-b][1,4,5]オキサチアゼピン-2-イル]メチル}-4-メチルフェニル)-2-メチルプロパン酸(化合物51)。 - 請求項1~12のいずれかに記載の化合物、その塩、及びそれらの溶媒和物からなる群より選択される少なくとも1種を含有する、医薬。
- Nrf2(NF-E2-related factor 2)活性化剤である、請求項13に記載の医薬。
- 局所投与製剤である、請求項13又は14に記載の医薬。
- 眼科用製剤である、請求項13~15のいずれかに記載の医薬。
- 非経口投与製剤である、請求項13又は14に記載の医薬。
- 経静脈投与製剤である、請求項13、14、又は17に記載の医薬。
- 脳疾患、肺疾患、皮膚疾患、耳鼻科疾患、腎疾患又は眼科疾患の予防又は治療用医薬である、請求項11~18のいずれかに記載の医薬。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021529207A JP7583718B2 (ja) | 2019-07-03 | 2020-07-03 | Nrf2活性化化合物 |
| US17/623,983 US12552757B2 (en) | 2019-07-03 | 2020-07-03 | NRF2-activating compound |
| CN202080048742.5A CN114080389B (zh) | 2019-07-03 | 2020-07-03 | Nrf2活性化化合物 |
| EP20835394.6A EP3998262A4 (en) | 2019-07-03 | 2020-07-03 | NRF2 ACTIVATING COMPOUND |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019124336 | 2019-07-03 | ||
| JP2019-124336 | 2019-07-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021002473A1 true WO2021002473A1 (ja) | 2021-01-07 |
Family
ID=74100399
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/026309 Ceased WO2021002473A1 (ja) | 2019-07-03 | 2020-07-03 | Nrf2活性化化合物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US12552757B2 (ja) |
| EP (1) | EP3998262A4 (ja) |
| JP (1) | JP7583718B2 (ja) |
| CN (1) | CN114080389B (ja) |
| WO (1) | WO2021002473A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022145459A1 (ja) * | 2020-12-28 | 2022-07-07 | 千寿製薬株式会社 | Nrf2活性化化合物 |
| USD981851S1 (en) * | 2020-11-09 | 2023-03-28 | Dart Industries Inc. | Manual carry strap |
| JPWO2023210741A1 (ja) * | 2022-04-28 | 2023-11-02 | ||
| US12559504B2 (en) | 2019-05-31 | 2026-02-24 | Ube Corporation | Benzotriazole derivative |
| EP4516792A4 (en) * | 2022-04-28 | 2026-04-01 | Kyoto Pharma Ind | BENZOTHIOPHENE COMPOUND |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008273924A (ja) | 2007-03-30 | 2008-11-13 | Meiji Seika Kaisha Ltd | 2位チエニルカルバペネム誘導体 |
| WO2015092713A1 (en) | 2013-12-18 | 2015-06-25 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| WO2016202253A1 (en) | 2015-06-15 | 2016-12-22 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| WO2018109649A1 (en) * | 2016-12-15 | 2018-06-21 | Glaxosmithkline Intellectual Property Development Limited | Ether linked triazoles as nrf2 regulators |
| WO2018109643A1 (en) * | 2016-12-14 | 2018-06-21 | Glaxosmithkline Intellectual Property Development Limited | Bisaryl heterocycles as nrf2 acti |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6455522B1 (en) * | 1998-02-11 | 2002-09-24 | Bristol-Myers Squibb Pharma Company | Cyclic sulfonamide derivatives as metalloproteinase inhibitors |
| CN108699011B (zh) | 2016-02-03 | 2022-05-06 | 参宿七制药股份有限公司 | Nrf2活化性化合物及其用途 |
| ES2901617T3 (es) * | 2016-12-14 | 2022-03-23 | Glaxosmithkline Ip Dev Ltd | Bisarilamidas como reguladores de NRF2 |
| CN116669744B (zh) * | 2020-12-28 | 2026-03-24 | 千寿制药株式会社 | Nrf2活化化合物 |
-
2020
- 2020-07-03 JP JP2021529207A patent/JP7583718B2/ja active Active
- 2020-07-03 CN CN202080048742.5A patent/CN114080389B/zh active Active
- 2020-07-03 WO PCT/JP2020/026309 patent/WO2021002473A1/ja not_active Ceased
- 2020-07-03 EP EP20835394.6A patent/EP3998262A4/en active Pending
- 2020-07-03 US US17/623,983 patent/US12552757B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008273924A (ja) | 2007-03-30 | 2008-11-13 | Meiji Seika Kaisha Ltd | 2位チエニルカルバペネム誘導体 |
| WO2015092713A1 (en) | 2013-12-18 | 2015-06-25 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| JP2017503786A (ja) * | 2013-12-18 | 2017-02-02 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Nrf2レギュレーター |
| WO2016202253A1 (en) | 2015-06-15 | 2016-12-22 | Glaxosmithkline Intellectual Property Development Limited | Nrf2 regulators |
| WO2018109643A1 (en) * | 2016-12-14 | 2018-06-21 | Glaxosmithkline Intellectual Property Development Limited | Bisaryl heterocycles as nrf2 acti |
| WO2018109649A1 (en) * | 2016-12-15 | 2018-06-21 | Glaxosmithkline Intellectual Property Development Limited | Ether linked triazoles as nrf2 regulators |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3998262A4 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12559504B2 (en) | 2019-05-31 | 2026-02-24 | Ube Corporation | Benzotriazole derivative |
| USD981851S1 (en) * | 2020-11-09 | 2023-03-28 | Dart Industries Inc. | Manual carry strap |
| WO2022145459A1 (ja) * | 2020-12-28 | 2022-07-07 | 千寿製薬株式会社 | Nrf2活性化化合物 |
| JPWO2022145459A1 (ja) * | 2020-12-28 | 2022-07-07 | ||
| CN116669744A (zh) * | 2020-12-28 | 2023-08-29 | 千寿制药株式会社 | Nrf2活化化合物 |
| JP7681044B2 (ja) | 2020-12-28 | 2025-05-21 | 千寿製薬株式会社 | Nrf2活性化化合物 |
| CN116669744B (zh) * | 2020-12-28 | 2026-03-24 | 千寿制药株式会社 | Nrf2活化化合物 |
| JPWO2023210741A1 (ja) * | 2022-04-28 | 2023-11-02 | ||
| WO2023210741A1 (ja) * | 2022-04-28 | 2023-11-02 | 第一三共株式会社 | ベンゾトリアゾール化合物 |
| JP7605446B2 (ja) | 2022-04-28 | 2024-12-24 | 第一三共株式会社 | ベンゾトリアゾール化合物 |
| US12378260B2 (en) | 2022-04-28 | 2025-08-05 | Daiichi Sankyo Company, Limited | Benzotriazole compound |
| EP4516792A4 (en) * | 2022-04-28 | 2026-04-01 | Kyoto Pharma Ind | BENZOTHIOPHENE COMPOUND |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3998262A1 (en) | 2022-05-18 |
| CN114080389A (zh) | 2022-02-22 |
| US12552757B2 (en) | 2026-02-17 |
| CN114080389B (zh) | 2024-08-30 |
| EP3998262A4 (en) | 2023-07-26 |
| JPWO2021002473A1 (ja) | 2021-01-07 |
| US20230024995A1 (en) | 2023-01-26 |
| JP7583718B2 (ja) | 2024-11-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7583718B2 (ja) | Nrf2活性化化合物 | |
| KR102570992B1 (ko) | 타우(Tau)-단백질 표적화 프로탁(PROTAC) 및 관련 사용 방법 | |
| US10442782B2 (en) | Kynurenine-3-monooxygenase inhibitors, pharmaceutical compositions, and methods of use thereof | |
| EP3237411B1 (en) | Compounds, compositions, and methods for increasing cftr activity | |
| US9351954B2 (en) | Multicyclic compounds and methods of use thereof | |
| EP3157917B1 (en) | Compounds, compositions and methods of increasing cftr activity | |
| CN112312904A (zh) | 螺环化合物 | |
| CN114685488A (zh) | 作为sos1抑制剂的化合物及其应用 | |
| KR20220086573A (ko) | 융합된 피리돈 화합물, 및 이의 제조 방법 및 이의 용도 | |
| CN112479996A (zh) | 吡啶氮氧化合物及其制备方法和用途 | |
| CN113557235A (zh) | 用于药物治疗的杂环化合物 | |
| KR20200096261A (ko) | 설폰아마이드 화합물 및 이의 용도 | |
| KR101854879B1 (ko) | 염증성 질환의 치료에 사용하기 위한 n-아실에탄올아민에 특이적인 아미다제의 조절용 조성물 및 조절 방법 | |
| ES2563232T3 (es) | Compuesto de amidina o sal del mismo | |
| US10501433B2 (en) | Kynurenine-3-monooxygenase inhibitors, pharmaceutical compositions, and methods of use thereof | |
| CN107001378A (zh) | 吡咯并嘧啶化合物 | |
| AU2015337607A1 (en) | Substituted dihydropyrrolopyrazole compound | |
| JP2023513373A (ja) | P2x3修飾薬 | |
| EP3441389A1 (en) | Pyrazole-oxazolidinone compound for anti-hepatitis b virus | |
| JP2019112307A (ja) | 置換ジヒドロピロロピラゾール化合物および他の乾癬治療薬が組み合わせて投与される医薬組成物 | |
| JP7681044B2 (ja) | Nrf2活性化化合物 | |
| JP2022538086A (ja) | Rockプロテインキナーゼ阻害薬としてのイソキノリンの誘導体及びその使用 | |
| WO2019037791A1 (zh) | 一种长效雷沙吉兰前药及其制备方法和用途 | |
| CN116194454B (zh) | 含有嘧啶的含氮双环化合物 | |
| HK40061625B (zh) | 吡啶氮氧化合物及其制备方法和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20835394 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021529207 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020835394 Country of ref document: EP Effective date: 20220203 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 17623983 Country of ref document: US |