WO2021100738A1 - 樹脂組成物及び成形体 - Google Patents
樹脂組成物及び成形体 Download PDFInfo
- Publication number
- WO2021100738A1 WO2021100738A1 PCT/JP2020/042905 JP2020042905W WO2021100738A1 WO 2021100738 A1 WO2021100738 A1 WO 2021100738A1 JP 2020042905 W JP2020042905 W JP 2020042905W WO 2021100738 A1 WO2021100738 A1 WO 2021100738A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- block copolymer
- resin composition
- rubber
- mass
- polymer block
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L21/00—Compositions of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
- C08L9/06—Copolymers with styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/02—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type
- C08F297/04—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type polymerising vinyl aromatic monomers and conjugated dienes
- C08F297/044—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the anionic type polymerising vinyl aromatic monomers and conjugated dienes using a coupling agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/04—Reduction, e.g. hydrogenation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L53/00—Compositions of block copolymers containing at least one sequence of a polymer obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L7/00—Compositions of natural rubber
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
Definitions
- the present invention relates to a resin composition containing at least a rubber, a block copolymer having a structural unit derived from farnesene, and a cross-linking agent, and a molded product of the resin composition.
- a rubber composition obtained by mixing a thermoplastic elastomer such as a styrene elastomer or an olefin elastomer with various rubbers has excellent mechanical properties and flexibility, so that it is used for footwear soles, various tires, building materials such as packing, mechanical parts, etc. It is used in a wide range of fields.
- the rubber composition containing the thermoplastic elastomer becomes slippery due to the adhesion of moisture, which poses a problem because it poses a high risk especially in applications such as soles for footwear and various tires.
- Patent Documents 1 to 8 cannot produce a resin composition having an excellent balance of flexibility, tear strength, tensile properties, dry grip properties and wet grip properties, and further improvement is required. ing. Further, Patent Documents 1 to 8 describe block copolymers containing a polymer block containing a structural unit derived from an aromatic vinyl compound and a polymer block containing a structural unit derived from farnesene, and hydrogenated products thereof. There is no. Further, although the resin composition disclosed in Patent Document 9 has an excellent balance of flexibility, tear strength, tensile property, dry grip property and wet grip property, it is difficult for the molded product to slip, such as grip property under various environments. Further improvement is required for the performance of. In addition, it is also required to have excellent wear resistance for applications such as soles for footwear and various tires.
- an object of the present invention is to provide a resin composition that is not slippery and can be a molded product having excellent wear resistance, and a molded product of the resin composition.
- the present invention is as follows.
- [1] Contains rubber (I), block copolymer (II) and cross-linking agent (III), The mass ratio [(I) / (II)] of the rubber (I) and the block copolymer (II) is 99/1 to 55/45.
- the block copolymer (II) contains a polymer block (A) containing a structural unit derived from an aromatic vinyl compound and a polymer block (B) containing a structural unit derived from farnesene (b1).
- [2] A molded product of the resin composition according to the above [1].
- [3] A shoe sole using at least a part of the resin composition according to the above [1].
- the resin composition of this embodiment is Contains rubber (I), block copolymer (II) and cross-linking agent (III), The mass ratio [(I) / (II)] of the rubber (I) and the block copolymer (II) is 99/1 to 55/45.
- the block copolymer (II) contains a polymer block (A) containing a structural unit derived from an aromatic vinyl compound and a polymer block (B) containing a structural unit derived from farnesene (b1). It is characterized by being a copolymer.
- the block copolymer (II) containing the structural unit (b1) derived from farnesene By using the block copolymer (II) containing the structural unit (b1) derived from farnesene, flexibility is imparted to the resin composition as described later, and the resin composition has road surface followability, so that it can be expelled (water repellent performance). Is high. Furthermore, since the block copolymer (II) has excellent water-repellent performance, it is particularly subjected to any of dry conditions (dry surface), wet conditions (wet surface), and ice conditions (snow top surface to ice top surface). However, it can be expected that the molded product is excellent in slip resistance (hereinafter, "slip resistance” may be referred to as "grip property").
- the resin composition of the present embodiment has an excellent balance of tear strength and tensile properties in addition to the above performance, and further contains a large amount of rubber (I) as compared with the block copolymer (II), resulting in cost reduction. Can be expected to be connected.
- Rubber (I) examples of the rubber (I) contained in the resin composition of the present embodiment include natural rubber and various synthetic rubbers used as a rubber raw material for rubber soles such as footwear soles.
- the rubber (I) does not include the block copolymer (II).
- Natural rubber is for tire industry or footwear such as TSR (Technically Specified Rubber) such as SMR (Malaysia TSR), SIR (Indonesia TSR), STR (Thai TSR) and RSS (Ribbed Smoked Sheet).
- TSR Technically Specified Rubber
- SMR Magnetic Reliable TSR
- SIR Indonesia TSR
- STR Thai TSR
- RSS Rabbed Smoked Sheet
- natural rubber generally used in various fields of application such as shoe soles, high-purity natural rubber, epoxidized natural rubber, hydroxylated natural rubber, hydrogenated natural rubber, and modified natural rubber such as grafted natural rubber. ..
- Examples of synthetic rubber include styrene-butadiene copolymer rubber (SBR), polyisoprene rubber (IR), polybutadiene rubber (BR), acrylic nitrile butadiene copolymer rubber (NBR), and ethylene-propylene-diene copolymer (EP, EPM). , Ethylene-propylene-non-conjugated diene copolymer rubber (EPDM), butyl rubber (isobutylene-isoprene rubber (IIR)), halogenated butyl rubber modified from butyl rubber and the like.
- SBR styrene-butadiene copolymer rubber
- IR polyisoprene rubber
- BR polybutadiene rubber
- NBR acrylic nitrile butadiene copolymer rubber
- EP ethylene-propylene-diene copolymer
- EPDM Ethylene-propylene-non-conjugated diene copolymer
- styrene-butadiene copolymerization from the viewpoint of physical properties such as strength, abrasion resistance, elasticity, and flexibility required for various uses, and compatibility with various components such as block copolymer (II).
- block copolymer (II) At least one selected from rubber, natural rubber, polyisoprene rubber, polybutadiene rubber, acrylic nitrile butadiene copolymer rubber, ethylene-propylene-diene copolymer, butyl rubber, and butyl gum halide modified butyl rubber is preferable.
- the rubber (I) may be used alone or in combination of two or more.
- the rubber (I) is used in combination include, for example, two or more kinds selected from NR, SBR, and BR. It is preferable to use these two or more kinds of rubbers as the rubber (I) from the viewpoint of the balance between grip property, wear property and mechanical physical properties.
- NR is used as the rubber (I)
- a softener described later in combination it is preferable to use a softener described later in combination.
- liquid polydiene and its hydrogenated product or a modified product thereof (sometimes simply referred to as "liquid polydiene") are suitable. It is considered that the combined use of liquid polydiene lowers the viscosity of NR and enhances the dispersibility in the composition, so that it becomes easier to obtain a molded product having excellent grip.
- the liquid polydiene may be used alone or in combination of two or more.
- the weight average molecular weight (Mw) of the liquid polydiene is preferably 100 to 200,000, more preferably 1,000 to 130,000, further preferably 2,000 to 60,000, and preferably 5,000 to 50,000. Even more preferable. When the weight average molecular weight is within the above range, the workability is improved and the bleed-out of the resin composition can be suppressed.
- the number average molecular weight (Mn) of the liquid polydiene is preferably 2,000 to 180,000, more preferably 3,000 to 130,000, further preferably 4,000 to 50,000, and 5,000 to 35, 000 is even more preferable. When the quantity average molecular weight is within the above range, the processability is improved and the bleed-out of the resin composition can be suppressed.
- the molecular weight distribution (Mw / Mn) of the liquid polydiene is preferably 1 to 6, more preferably 1 to 4, further preferably 1 to 3, and even more preferably 1 to 2.
- Mw and Mn of the liquid polydiene are values obtained from the measurement of gel permeation chromatography (GPC), and mean the values measured by the method described in Examples described later.
- the combination of NR, SBR, and BR can further contain liquid polydiene.
- the content ratio of NR is 50% by mass or more in the above combination, the advantage of using the liquid polydiene in combination is more satisfactorily exhibited.
- the content of the liquid polydiene as the softening agent is the total amount of the rubber (I), the block copolymer (II), and the liquid polybutadiene.
- 100 parts by mass 0.1 to 10 parts by mass is preferable, 0.5 to 9 parts by mass is more preferable, 1 to 8 parts by mass is further preferable, and 1.5 to 7 parts by mass is further preferable.
- the rubber (I) does not contain the above liquid polydiene.
- SBR for example, any of emulsion-polymerized styrene-butadiene rubber (E-SBR), solution-polymerized styrene-butadiene rubber (S-SBR), modified styrene-butadiene rubber (modified SBR) and the like can be used.
- E-SBR emulsion-polymerized styrene-butadiene rubber
- S-SBR solution-polymerized styrene-butadiene rubber
- modified SBR modified styrene-butadiene rubber
- a commercially available SBR can be used, in which the styrene content is preferably 0.1 to 70% by mass, more preferably 5 to 50% by mass, further preferably 15 to 35% by mass, and 15 to 35% by mass. 25% by mass is even more preferable.
- SBR has a good balance of physical properties such as elasticity and strength, is also good for kneading and molding
- IR a commercially available IR can be used, but it must be an IR polymerized by a Ziegler-based catalyst having a high cis-form content or an IR having an ultra-high cis-form content obtained by using a lanthanoid-based rare earth metal catalyst. Is preferable.
- the weight average molecular weight (Mw) of IR is preferably 90,000 to 2 million, more preferably 150,000 to 1.5 million. When Mw is in the above range, moldability, tear strength, and tensile properties are good.
- the weight average molecular weight of IR is a polystyrene-equivalent weight average molecular weight obtained from the measurement of gel permeation chromatography (GPC).
- the vinyl content of IR is preferably 50% by mass or less, more preferably 40% by mass or less, still more preferably 30% by mass or less. IR has a good balance of physical properties such as mechanical strength, and is also suitable from the viewpoint of easy availability.
- BR a commercially available BR can be used, but it must be a BR polymerized by a Ziegler-based catalyst having a high cis-form content or an ultra-high cis-form content BR obtained by using a lanthanoid-based rare earth metal catalyst. Is preferable.
- the weight average molecular weight (Mw) of BR is preferably 90,000 to 2 million, more preferably 150,000 to 1,500,000, further preferably 250,000 to 1,000,000, and 350,000 to 700,000. Is even more preferable. When Mw is in the above range, moldability, tear strength, and tensile properties are good.
- the vinyl content of BR is preferably 50% by mass or less, more preferably 40% by mass or less, and further preferably 30% by mass or less. BR is particularly suitable from the viewpoint that it can be expected to impart excellent wear resistance to the molded product of the resin composition.
- the acrylonitrile content in the NBR is preferably 20 to 41% by mass, more preferably 25 to 41% by mass.
- the acrylonitrile content is in the above range, it can be expected to impart better grip and abrasion resistance to the molded product of the resin composition under various environments.
- EP and EPDM can be used as the ethylene-propylene-diene copolymer (EP, EPM) and the ethylene-propylene-non-conjugated diene copolymer rubber (EPDM).
- Examples of the non-conjugated diene constituting EPDM include 5-ethylidene-2-norbornene (ENB), 1,4-hexadiene, 5-methylene-2-norbornene (MNB), 1,6-octadene, 5-methyl-1, 4-Hexadiene, 3,7-dimethyl-1,6-octadiene, 1,3-cyclopentadiene, 1,4-cyclohexadiene, tetrahydroinden, methyltetrahydroinden, dicyclopentadiene, 5-isopropylidene-2-norbornene, 5-Vinyl-norbornene, dicyclooctadiene, methylenenorbornene and the like can be mentioned
- 5-ethylidene-2-norbornene is preferable in terms of vulcanization rate.
- EMB 5-ethylidene-2-norbornene
- EP, EPM ethylene-propylene-diene copolymer
- EPDM ethylene-propylene-non-conjugated diene copolymer rubber
- EPDM Diene copolymer rubber
- the IIR may be non-halogenated butyl rubber, regenerated butyl rubber, or the like, and examples thereof include those used in the field of tire industry or soles for footwear.
- the halogenated butyl rubber is modified by introducing halogen into the molecule of IIR, and examples thereof include brominated butyl rubber (Br-IIR) and chlorinated butyl rubber (Cl-IIR).
- a brominated isobutylene-p-methylstyrene copolymer or the like can also be used.
- the block copolymer (II) may be an unhydrogenated block copolymer (hereinafter, may be referred to as “block copolymer (P)”), and may be a block copolymer as described later.
- (P) may be a hydrogenated block copolymer (hereinafter, may be referred to as “hydrogenated block copolymer (Q)”) hydrogenated at a specific hydrogenation rate.
- the polymer block (A) contains a structural unit derived from an aromatic vinyl compound.
- aromatic vinyl compounds include styrene, ⁇ -methylstyrene, 2-methylstyrene, 3-methylstyrene, 4-methylstyrene, 4-propylstyrene, 4-t-butylstyrene, 4-cyclohexylstyrene, and 4-.
- the polymer block (A) contains a structural unit derived from a monomer other than the aromatic vinyl compound, for example, another monomer such as a monomer constituting the polymer block (B) described later. May be good.
- the content of the structural unit derived from the aromatic vinyl compound in the polymer block (A) is preferably 60% by mass or more, more preferably 70% by mass or more, further preferably 80% by mass or more, and 90% by mass or more. Is even more preferable, and 100% by mass is particularly preferable.
- the content of the polymer block (A) in the block copolymer (II) is preferably 10 to 45% by mass, more preferably 10 to 40% by mass, and further preferably 10 to 30% by mass. %, More preferably 15 to 25% by mass.
- the content is 10% by mass or more, the molding processability of the resin composition is good, more excellent wear resistance is likely to be exhibited, and the tear strength and tensile properties can be made suitable.
- the content is 45% by mass or less, sufficient flexibility can be obtained, slip resistance is likely to be exhibited, and good molding processability, tear strength, tensile properties and the like are easily exhibited.
- the polymer block (B) contains a structural unit (b1) derived from farnesene (hereinafter, also simply referred to as “structural unit (b1)”).
- the farnesene may be either ⁇ -farnesene or ⁇ -farnesene represented by the following formula (1), but ⁇ -farnesene is preferable from the viewpoint of ease of production of the block copolymer (II).
- ⁇ -farnesene and ⁇ -farnesene may be used in combination.
- the content of the farnesene-derived structural unit (b1) in the polymer block (B) is preferably 1 to 100% by mass. Since the polymer block (B) contains the structural unit (b1) derived from farnesene, the flexibility is improved and the grip property is excellent. From this point of view, the content of the structural unit (b1) derived from farnesene in the polymer block (B) is more preferably 10 to 100% by mass, further preferably 20 to 100% by mass, and more preferably 30 to 100% by mass. More preferably, it is particularly preferably 50 to 100% by mass, and most preferably 100% by mass, that is, the polymer block (B) is composed of the structural unit (b1).
- the content of the farnesene-derived structural unit (b1) in the polymer block (B) is preferably 50 to 100% by mass, more preferably 60 to 100% by mass, and further 70 to 100% by mass. Preferably, 80 to 100% by mass is even more preferable.
- the content of the farnesene-derived structural unit (b1) in the polymer block (B) is preferably 1% by mass or more, 10 By mass% or more is more preferable, 20% by mass or more is further preferable, 30% by mass or more is further preferable, and 50% by mass or more is particularly preferable.
- the polymer block (B) may contain a structural unit (b2) derived from a conjugated diene other than farnesene (hereinafter, also simply referred to as “structural unit (b2)”), and the structure in the polymer block (B).
- the content of the unit (b2) is preferably 0 to 99% by mass.
- conjugated diene include isoprene, butadiene, 2,3-dimethyl-butadiene, 2-phenyl-butadiene, 1,3-pentadiene, 2-methyl-1,3-pentadiene, 1,3-hexadiene, and 1,3.
- the structural unit (b2) is contained in the polymer block (B), the content of the structural unit (b2) is more preferably 90% by mass or less, further preferably 80% by mass or less, and more preferably 70% by mass or less. More preferably, 50% by mass or less is particularly preferable.
- the polymer block (B) may contain other structural units other than the structural unit (b1) and the structural unit (b2).
- the total content of the structural unit (b1) and the structural unit (b2) in the polymer block (B) is preferably 60% by mass or more, more preferably 80% by mass or more, still more preferably 100% by mass.
- the block copolymer (II) is a polymer block (C) containing a structural unit (c2) derived from a conjugated diene other than farnesene in addition to the above-mentioned polymer block (A) and polymer block (B). Can be included.
- the polymer block (C) has a content of the structural unit (c1) derived from farnesene of 0% by mass or more and less than 1% by mass, and the content of the structural unit (c2) derived from a conjugated diene other than farnesene is 1 to 1 to It is preferably a polymer block of 100% by mass.
- Farnesene which constitutes a structural unit (c1) derived from farnesene (hereinafter, also simply referred to as “structural unit (c1)”), and a structural unit (c2) derived from a conjugated diene other than farnesene (hereinafter, simply “structural unit (c2)).
- the conjugated diene constituting includes farnesene constituting the structural unit (b1) derived from farnesene described above and the same conjugated diene constituting the structural unit (b2) derived from the conjugated diene other than farnesene. ..
- conjugated diene constituting the structural unit (c2) derived from the conjugated diene other than farnesene isoprene, butadiene and myrcene are preferable, and isoprene and butadiene are more preferable. These may be used alone or in combination of two or more.
- the content of the farnesene-derived structural unit (c1) in the polymer block (C) is preferably 0% by mass.
- the content of the structural unit (c2) derived from the conjugated diene other than farnesene in the polymer block (C) is more preferably 60 to 100% by mass, further preferably 80 to 100% by mass, and more preferably 90 to 100% by mass. It is more preferably 100% by mass, and particularly preferably 100% by mass.
- the hydrogenated block copolymer (Q) of the block copolymer (P) is crosslinked with a cross-linking agent (III) described later, it has excellent wear resistance and can exhibit tear strength while maintaining tensile properties. It is possible to achieve both tensile properties and tear strength.
- the polymer block (C) may contain a structural unit (c1) derived from farnesene and a structural unit other than the structural unit derived from a conjugated diene other than farnesene (c2).
- the total content of the structural unit (c1) and the structural unit (c2) in the polymer block (C) is preferably 60% by mass or more, more preferably 80% by mass or more, still more preferably 100% by mass.
- the block copolymer (II) is a block copolymer (P) or a hydrogenated block copolymer (Q) containing at least one polymer block (A) and one polymer block (B).
- the bonding form of the polymer block (A) and the polymer block (B) is not particularly limited, and may be linear, branched, radial, or a combination of two or more thereof. Among these, a form in which each block is linearly connected is preferable.
- the binding form represented by n can be exemplified.
- the l, m, and n independently represent an integer of 1 or more.
- the block copolymer (II) contains at least one polymer block (A) and one polymer block (B), the polymer block (A), the polymer block (B), and the polymer block (A)
- the bond form has blocks in the order of ABA and is a triblock copolymer represented by ABA. That is, the block copolymer (II) is preferably a triblock copolymer represented by ABA, and the triblock copolymer is a hydrogenated additive even if it is an unwatered additive. There may be.
- the block copolymer (II) contains the polymer block (A), the polymer block (B) and the polymer block (C), the block copolymer (II) is at least one polymer block ( It is preferably a block copolymer having B) at the end, and the block copolymer may be an unwatered additive or a hydrogenated additive. Moldability is improved by having at least one polymer block (B) at the end of the polymer chain. From this point of view, when the block copolymer (II) is linear, it is more preferable to have the polymer blocks (B) at both ends thereof. When the block copolymer (II) is branched or radial, the number of polymer blocks (B) present at the ends is preferably 2 or more, and more preferably 3 or more.
- the block copolymer (II) is a block copolymer containing at least two polymer blocks (A), at least one polymer block (B), and at least one polymer block (C). It contains at least two polymer blocks (A), at least one polymer block (B), and at least one polymer block (C), and at least one polymer. It is more preferable to have one or more of the blocks (B) at the end.
- the bonding form of the plurality of polymer blocks is not particularly limited and is linear. It may be branched, radial or a combination of two or more thereof. Among these, a form in which each block is linearly connected is preferable.
- the block copolymer (II) has a structure having blocks in the order of the polymer block (B), the polymer block (A), and the polymer block (C) (that is, the structure of BAC). Is preferable.
- the block copolymer (II) is a tetrablock copolymer represented by BACA, a pentablock copolymer represented by BACAB, and the like.
- B-A- (C-A) p -B, B-A- (C-A-B) q, B- (A-C-A-B) r (p, q, r are independently 2 It is preferably a copolymer represented by (representing the above integer), and more preferably a pentablock copolymer represented by BACAB. That is, the block copolymer (II) is preferably a pentablock copolymer represented by BACAB, and even if the pentablock copolymer is an unhydrogenated additive. It may be a hydrogenated additive.
- the entire bonded polymer blocks are treated as one polymer block.
- the polymer block which should be strictly described as AXA (X represents the coupling agent residue) is displayed as A as a whole.
- this type of polymer block containing a coupling agent residue is treated as described above, for example, it contains a coupling agent residue and is strictly BACXC-.
- the block copolymer to be described as AB is described as BACAB and is treated as an example of the pentablock copolymer.
- the two or more polymer blocks (A) in the above-mentioned block copolymer (II) may be a polymer block composed of the same structural unit or a polymer block composed of different structural units. Good.
- the block copolymer (II) has two or more polymer blocks (B) or two or more polymer blocks (C)
- each polymer block is composed of the same structural unit. It may be a polymer block or a polymer block composed of different structural units.
- the respective aromatic vinyl compounds may be the same type or different.
- the block copolymer (II) contains the polymer block (A) and the polymer block (B) and does not contain the polymer block (C)
- the polymer block (A) and the polymer block (B) The mass ratio [(A) / (B)] of is 1/99 to 70/30, preferably 5/95 to 60/40, more preferably 10/90 to 50/50, and 15/85 to 40. / 60 is even more preferred, and 15/85 to 35/65 is even more preferred. Within this range, a resin composition having excellent flexibility and excellent grip can be obtained.
- [(A) / (B)] is 1/99 to 70/30, preferably 5/95 to 60/40, more preferably 10/90 to 50/50, and 20/80 to 40/60. More preferably, 25/75 to 35/65 is even more preferable. Within this range, a resin composition having excellent flexibility and excellent grip can be obtained.
- the mass ratio of the polymer block (A) and the total amount of the polymer block (B) and the polymer block (C) [(A) / ((B) + (C) ))] Is preferably 1/99 to 70/30. Within this range, a resin composition having excellent tensile properties, molding processability and grip properties can be obtained. From this point of view, the mass ratio [(A) / ((B) + (C))] is more preferably 1/99 to 60/40, further preferably 10/90 to 40/60, and 10/90 to. 30/70 is even more preferred, and 15/85 to 25/75 is even more preferred.
- the block copolymer (II) contains the polymer block (A), the polymer block (B) and the polymer block (C), the polymer block (B) and the polymer block (B) in the block copolymer (
- the total content of the structural unit (b1) and the structural unit (c1) relative to the total amount of C) [((b1) + (c1)) / ((B) + (C))] is the flexibility, tensile properties and From the viewpoint of excellent grip, 40 to 90% by mass is preferable, 50 to 80% by mass is more preferable, and 60 to 70% by mass is further preferable.
- the block copolymer (II) contains the polymer block (A) and the polymer block (B) and does not contain the polymer block (C), the polymer block (A) and the polymer block (A) in the block copolymer
- the total content of the polymer block (B) is preferably 80% by mass or more, more preferably 90% by mass or more, further preferably 95% by mass or more, still more preferably 100% by mass.
- the block copolymer (II) contains the polymer block (A), the polymer block (B) and the polymer block (C)
- the total content of (C) is preferably 80% by mass or more, more preferably 90% by mass or more, further preferably 95% by mass or more, still more preferably 100% by mass.
- -A hydrogenated block copolymer (Q) in which a block copolymer (P) containing a polymer block (A), a polymer block (B) and a polymer block (C) is hydrogenated.
- the polymer block (C) has a content of the structural unit (c1) derived from farnesene of 0% by mass or more and less than 1% by mass, and the content of the structural unit (c2) derived from a conjugated diene other than farnesene is 1.
- the block copolymer (II) is composed of the polymer block (A), the polymer block (B), the polymer block (C), and other monomers as long as the effects of the present invention are not impaired. It may contain a polymer block.
- Examples of such other monomers include propylene, 1-butene, 1-pentene, 4-methyl-1-pentene, 1-hexene, 1-octene, 1-decene, 1-undecene, 1-dodecene, 1-.
- Unsaturated hydrocarbon compounds such as tridecene, 1-tetradecene, 1-pentadecene, 1-hexadecene, 1-heptadecene, 1-octadecene, 1-nonadecene, 1-eicosene; acrylic acid, methacrylate, methyl acrylate, methyl methacrylate , Acrylonitrile, methacrylonitrile, maleic acid, fumaric acid, crotonic acid, itaconic acid, 2-acryloylethanesulfonic acid, 2-methacryloylethanesulfonic acid, 2-acrylamide-2-methylpropanesulfonic acid, 2-methacrylamide-2 -Functional group-containing unsaturated compounds such as methylpropane sulfonic acid, vinyl sulfonic acid, vinyl acetate and methyl vinyl ether; and the like. These may be used alone or in combination of two or more. When the block copolymer (II)
- the block copolymer (II) contains, for example, a block copolymer (P) containing a polymer block (A) and a polymer block (B), or a polymer block (C), the polymer block ( In the case of the block copolymer (P) containing A), the polymer block (B) and the polymer block (C), it can be suitably produced by the polymerization step obtained by anion polymerization. Further, when the block copolymer (II) is a hydrogenated block copolymer (Q), the carbon-carbon double bond in the structural unit derived from the conjugated diene in the block copolymer (P) is hydrogenated. It can be suitably produced by the process.
- the block copolymer (P) can be produced by a solution polymerization method or the method described in JP-A-2012-502135 and JP-A-2012-502136.
- the solution polymerization method is preferable, and for example, a known method such as an ionic polymerization method such as anionic polymerization or cationic polymerization, or a radical polymerization method can be applied.
- the anion polymerization method is preferable.
- a block copolymer (P) is obtained by sequentially adding an aromatic vinyl compound, farnesene and / or a conjugated diene other than farnesene in the presence of a solvent, an anionic polymerization initiator, and if necessary, a Lewis base.
- the anion polymerization initiator include alkali metals such as lithium, sodium and potassium; alkaline earth metals such as beryllium, magnesium, calcium, strontium and barium; lanthanoid rare earth metals such as lanthanum and neodymium; the alkali metals and alkalis. Examples thereof include earth metals and compounds containing lanthanoid-based rare earth metals. Of these, compounds containing alkali metals and alkaline earth metals are preferable, and organic alkali metal compounds are more preferable.
- organic alkali metal compound examples include methyllithium, ethyllithium, n-butyllithium, sec-butyllithium, t-butyllithium, hexyllithium, phenyllithium, stillbenlithium, dilithiomethane, dilythionaphthalene, and 1,4-dilithiobtan. , 1,4-Dilithio-2-ethylcyclohexane, 1,3,5-trilithiobenzene and other organolithium compounds; sodium naphthalene, potassium naphthalene and the like.
- the organic alkali metal compound may be used as an organic alkali metal amide by reacting with a secondary amine such as diisopropylamine, dibutylamine, dihexylamine, or dibenzylamine.
- the amount of the organic alkali metal compound used for the polymerization varies depending on the molecular weight of the block copolymer (P), but is usually 0.01 to 3 with respect to the total amount of the aromatic vinyl compound, farnesene and conjugated diene other than farnesene. It is in the range of mass%.
- the solvent is not particularly limited as long as it does not adversely affect the anion polymerization reaction.
- saturated aliphatic hydrocarbons such as n-pentane, isopentane, n-hexane, n-heptane, and isooctane; cyclopentane, cyclohexane, and methylcyclopentane.
- Saturated alicyclic hydrocarbons such as; aromatic hydrocarbons such as benzene, toluene, xylene and the like. These may be used alone or in combination of two or more. There is no particular limitation on the amount of solvent used.
- the Lewis base has a role of controlling the microstructure in the structural unit derived from farnesene and the structural unit derived from conjugated diene other than farnesene.
- Lewis bases include ether compounds such as dibutyl ether, diethyl ether, tetrahydrofuran, dioxane, and ethylene glycol diethyl ether; pyridine; tertiary amines such as N, N, N', N'-tetramethylethylenediamine, and trimethylamine; potassium.
- Alkali metal alkoxides such as t-butoxide; phosphine compounds and the like can be mentioned.
- the amount is usually preferably in the range of 0.01-1000 mol equivalents per 1 mol of anionic polymerization initiator.
- the temperature of the polymerization reaction is usually in the range of about ⁇ 80 to 150 ° C., preferably 0 to 100 ° C., and more preferably 10 to 90 ° C.
- the type of the polymerization reaction may be a batch type or a continuous type. Each monomer is continuously or intermittently supplied into the polymerization reaction solution so that the abundance of the aromatic vinyl compound, farnesene and / or conjugated diene other than farnesene in the polymerization reaction system is within a specific range.
- the block copolymer (P) can be produced by sequentially polymerizing each monomer in the polymerization reaction solution so as to have a specific ratio.
- the polymerization reaction can be stopped by adding an alcohol such as methanol or isopropanol as a polymerization terminator.
- the obtained polymerization reaction solution is poured into a poor solvent such as methanol to precipitate the block copolymer (P), or the polymerization reaction solution is washed with water, separated, and dried to obtain the block copolymer (P). Can be isolated.
- the block copolymer (II) preferably contains a structure having a polymer block (B), a polymer block (A), and a polymer block (C) in this order. Therefore, a step of obtaining a block copolymer (P) by producing a polymer block (B), a polymer block (A), and a polymer block (C) in this order, and in the case of a hydrogenated product, further It is more preferable to produce the hydrogenated block copolymer (Q) by a method including a step of hydrogenating the obtained block copolymer (P).
- block copolymer (II) has a polymer block (B) only at one end of the polymer chain, after polymerizing each of the other polymer blocks so as to bond linearly. Finally, it may be obtained by a method for producing a polymer block (B).
- the block copolymer (II) comprises at least two polymer blocks (A), at least one polymer block (B), and at least one polymer block (C), and at least one.
- the method for producing the block copolymer (P) is as follows. , [I] A method of polymerizing a polymer block (B), a polymer block (A), a polymer block (C), and a polymer block (A) in this order, and [ii] a polymer block (B), a weight.
- Examples thereof include a method of polymerizing a coalesced block (A) and a polymer block (C) in this order, and coupling the ends of the polymer blocks (C) with each other using a coupling agent.
- the latter method [ii] using a coupling agent is preferable from the viewpoint of efficient production.
- the coupling agent examples include divinylbenzene; polyvalent epoxy compounds such as epoxidized 1,2-polybutadiene, epoxidized soybean oil, and tetraglycidyl-1,3-bisaminomethylcyclohexane; tin tetrachloride, tetrachlorosilane, and the like.
- Halogens such as trichlorosilane, trichloromethylsilane, dichlorodimethylsilane, dibromodimethylsilane; methyl benzoate, ethyl benzoate, phenyl benzoate, diethyl oxalate, diethyl malonate, diethyl adipate, dimethyl phthalate, dimethyl terephthalate Etc.; Carbonated ester compounds such as dimethyl carbonate, diethyl carbonate, diphenyl carbonate, etc .; Diethoxydimethylsilane, trimethoxymethylsilane, triethoxymethylsilane, tetramethoxysilane, tetraethoxysilane, tetrabutoxysilane, tetrakis (2- Epoxide silane compounds such as ethylhexyloxy) silane, bis (triethoxysilyl) ethane, and 3-aminopropyltri
- an unmodified block copolymer may be obtained as described above, but a modified block copolymer as described below may be obtained.
- the block copolymer (P) may be modified before the hydrogenation step described later.
- the functional group that can be introduced include an amino group, an alkoxysilyl group, a hydroxyl group, an epoxy group, a carboxyl group, a carbonyl group, a mercapto group, an isocyanate group, an acid anhydride group and the like.
- Examples of the method for modifying the block copolymer (P) include tin tetrachloride, tetrachlorosilane, dichlorodimethylsilane, dimethyldiethoxysilane, and tetramethoxysilane that can react with the polymerization active terminal before adding the polymerization terminator.
- Tetraethoxysilane, 3-aminopropyltriethoxysilane, tetraglycidyl-1,3-bisaminomethylcyclohexane, 2,4-tolylene diisocyanate and other coupling agents, and 4,4'-bis (diethylamino) benzophenone examples thereof include a method of adding a polymerization terminal modifier such as N-vinylpyrrolidone or another modifier described in JP-A-2011-132298. Further, maleic anhydride or the like can be grafted onto the isolated copolymer and used. The position where the functional group is introduced may be the polymerization terminal of the block copolymer (P) or the side chain. Further, the functional group may be used alone or in combination of two or more.
- the modifier is preferably in the range of 0.01 to 10 molar equivalents with respect to 1 mol of the anionic polymerization initiator.
- the block copolymer (II) can be used as a hydrogenated block copolymer (Q) by subjecting it to a step of hydrogenating the block copolymer (P) obtained by the above method or a modified block copolymer. Good.
- a known method can be used as the method of hydrogenation. For example, in a solution prepared by dissolving the block copolymer (P) in a solvent that does not affect the hydrogenation reaction, a Cheegler-based catalyst; nickel, platinum, palladium, ruthenium or rhodium supported on carbon, silica, rhodium, etc.
- Rhodium metal catalyst an organic metal complex having cobalt, nickel, palladium, rhodium or ruthenium metal is allowed to exist as a hydrogenation catalyst to carry out a hydrogenation reaction.
- a hydrogenation catalyst may be added to a polymerization reaction solution containing the block copolymer obtained by the above-mentioned method for producing block copolymer (P) to carry out a hydrogenation reaction.
- the hydrogenation catalyst is preferably palladium carbon in which palladium is supported on carbon.
- the hydrogen pressure is preferably 0.1 to 20 MPa
- the reaction temperature is preferably 100 to 200 ° C.
- the reaction time is preferably 1 to 20 hours.
- One of the preferred embodiments of the block copolymer (II) is a hydrogenated block copolymer (Q) in which 70 mol% or more of carbon-carbon double bonds in the structural unit derived from the conjugated diene are hydrogenated.
- the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene is preferably 70 to 98 mol%, and is preferably 70 to 98 mol%. 97 mol% is more preferable, 80 to 96 mol% is further preferable, 85 to 96 mol% is further preferable, and 87 to 96 mol% is particularly preferable.
- the hydrogenation rate is the hydrogenation rate of carbon-carbon double bonds in all the conjugated diene-derived structural units present in the block copolymer (P).
- Examples of the carbon-carbon double bond in the structural unit derived from the conjugated diene present in the block copolymer (P) include the carbon-carbon double bond in the structural unit derived from the conjugated diene in the polymer block (B). And when the block copolymer (P) further contains the polymer block (C), carbon in the polymer block (B) and in the structural unit derived from the conjugated diene in the polymer block (C). -Carbon double bonds can be mentioned.
- the hydrogenated block copolymer (Q) contains the polymer block (B) and the polymer block (C)
- the structural unit derived from the conjugated diene of the polymer block (B) and the polymer block (C) It is preferable that the hydrogenation rate of the carbon-carbon double bond is different.
- the difference in the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene between the polymer block (B) and the polymer block (C) may be, for example, 1 to 20 mol% and 3 to 15 mol. It may be%, and it may be 5 to 10 mol%.
- the degree of cross-linking by the cross-linking agent (III) described later differs for each of the polymerization blocks (B) and (C), whereby the hydrogenated block copolymer (Q) is hard and soft. It is considered that different portions of the above can be present to exhibit excellent effects such as tear strength and tensile properties. Further, from the viewpoint of exhibiting remarkably excellent tensile properties, it is more preferable that the hydrogenation rate of the polymer block (C) is higher than that of the polymer block (B).
- the polymer block (B) and the polymer block (C) in the hydrogenated block copolymer (Q) are hydrogenated, but they are referred to as "polymer block” as before hydrogenation. (B) ”and“ polymer block (C) ”.
- the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene of the polymer block (B) in the hydrogenated block copolymer (Q) is preferably 70 to 98 mol%, more preferably 70 to 98 mol%. It is 70 to 95 mol%, more preferably 70 to 95 mol%, even more preferably 70 to 95 mol%, and particularly preferably 80 to 95 mol%.
- the block copolymer (P) contains the polymer block (C)
- the hydrogenation rate of the above is preferably 95 to 99.9 mol%, more preferably 96 to 99.5 mol%, and further preferably 97 to 99.5 mol%.
- the hydrogenation rate can be calculated by measuring 1 H-NMR of the block copolymer (P) and the hydrogenated block copolymer (Q) after hydrogenation.
- the peak top molecular weight (Mp) of the block copolymer (P) is preferably 4,000 to 1,500,000, more preferably 9,000 to 1,000,000, from the viewpoint of molding processability, and is 30,000. -800,000 is even more preferred, and 50,000-500,000 is even more preferred.
- the molecular weight distribution (Mw / Mn) of the block copolymer (P) is preferably 1 to 6, more preferably 1 to 4, further preferably 1 to 3, and even more preferably 1 to 2. When the molecular weight distribution is within the above range, the variation in the viscosity of the block copolymer (P) is small, and it is easy to handle.
- the peak top molecular weight (Mp) of the hydrogenated block copolymer (Q) is preferably 4,000 to 1,500,000, more preferably 9,000 to 1,000,000, and 30 More preferably, 000 to 800,000, even more preferably 50,000 to 500,000.
- the molecular weight distribution (Mw / Mn) of the hydrogenated block copolymer (Q) is preferably 1 to 6, more preferably 1 to 4, further preferably 1 to 3, and even more preferably 1 to 2. When the molecular weight distribution is within the above range, the variation in viscosity of the hydrogenated block copolymer (Q) is small, and it is easy to handle.
- the peak top molecular weight (Mp) and the molecular weight distribution (Mw / Mn) in the present specification mean values measured by the method described in Examples described later.
- the peak top molecular weight of the polymer block (A) is preferably 2,000 to 100,000, more preferably 4,000 to 80,000, and even more preferably 5,000 to 50,000 from the viewpoint of molding processability. , 6,000 to 30,000 are even more preferred.
- the peak top molecular weight of the polymer block (B) is preferably 2,000 to 200,000, more preferably 3,000 to 150,000, and even more preferably 4,000 to 100,000 from the viewpoint of molding processability. ..
- the peak top molecular weight of the polymer block (C) is preferably 4,000 to 200,000, more preferably 4,500 to 150,000, and 5,000 to 100,000 from the viewpoint of molding processability. More preferred.
- block copolymer (II) has the characteristic of easily repelling water.
- the above-mentioned "easiness of repelling water” is that water is dripped on a quartz plate, a molded product made of block copolymer (II) is placed on the quartz plate, and pressure is applied to the molded product from above. It can be evaluated by observing the state of water at that time from the quartz plate side.
- a molded body made of block copolymer (II) can repel most of the water between the molded body and the quartz plate. More specifically, the ease of repelling water can be evaluated by the method described in Examples described later.
- the resin composition of this embodiment contains a cross-linking agent (III).
- the cross-linking agent (III) is preferably one that undergoes a cross-linking reaction only with a carbon-carbon double bond or ⁇ -methylene thereof in a structural unit derived from a conjugated diene.
- ⁇ -methylene indicates methylene adjacent to a carbon-carbon double bond.
- cross-linking agent (III) examples include radical generators, sulfur and sulfur-containing compounds, and more preferably sulfur and / or sulfur-containing compounds from the viewpoint of tear strength and tensile properties. It is sulfur.
- radical generator examples include dicumyl peroxide, dit-butyl peroxide, 2,5-dimethyl-2,5-di (t-butylperoxy) hexane, and 2,5-dimethyl-2,5-di (t).
- the sulfur can be used without particular limitation, and may be, for example, fine powdered sulfur, precipitated sulfur, colloidal sulfur, insoluble sulfur, or the like.
- the sulfur-containing compound include sulfur monochloride, sulfur dichloride, disulfide compounds, triazine thiols and the like.
- Other cross-linking agents (III) include phenolic resins such as alkylphenol resins and brominated alkylphenol resins; combinations of p-quinonedioxime and lead dioxide, p, p'-dibenzoylquinonedioxime and trilead tetroxide, etc. Can also be used.
- the mass ratio [(I) / (II)] of the rubber (I) and the block copolymer (II) is 99/1 to 55/45. If the mass ratio [(I) / (II)] is out of the above range, excellent wear resistance cannot be obtained. Further, when the mass ratio is within the above range, the appearance of slip resistance becomes more remarkable. That is, with the above mass ratio, the resin composition can be made into a molded product that is less slippery and has excellent wear resistance. Further, in order to dramatically improve the slip resistance such as dry grip property and wet grip property and further improve the wear resistance, the mass ratio is preferably 99/1 to 60/40, and 99/1 to 65.
- the resin composition is imparted with flexibility by containing the block copolymer (II), the grip property tends to be improved as the content ratio of the block copolymer (II) is higher.
- the grip property is unexpectedly improved. It turned out to improve.
- the resin composition of the present embodiment can be a molded product having excellent slip resistance and wear resistance.
- the content of the cross-linking agent (III) is more likely to be improved in slip resistance and abrasion resistance with respect to 100 parts by mass of the total content of the rubber (I) and the block copolymer (II), and the flexibility, tear strength, etc. From the viewpoint that the balance of tensile properties can also be exhibited, 0.1 to 10 parts by mass is preferable, 0.5 to 6.0 parts by mass is more preferable, and 1.0 to 4.0 parts by mass is further preferable. , 1.5 to 2.5 parts by mass is even more preferable.
- the total content of the rubber (I), the block copolymer (II) and the cross-linking agent (III) in the resin composition of the present embodiment is likely to be more excellent in slip resistance and abrasion resistance, and is flexible and tearable. From the viewpoint of being able to exhibit a balance between strength and tensile properties, 60 to 80% by mass is preferable, and 65 to 80% by mass is more preferable.
- the resin composition of this embodiment preferably contains silica.
- silica include wet silica (hydrous silicic acid), dry silica (silicic anhydride), calcium silicate, aluminum silicate and the like.
- wet silica is preferable from the viewpoint of further improving molding processability, mechanical strength and wear resistance of the obtained molded product, and further improving slip resistance such as dry grip property and wet grip property.
- These silicas may be used alone or in combination of two or more.
- the specific surface area of silica is preferably 20 m 2 / g or more, more preferably 50 m 2 / g or more, further preferably 100 m 2 / g or more, still more preferably 150 m 2 / g or more.
- the upper limit of the specific surface area of silica may be a range that does not impair the effects of the present invention.
- Silicas having different specific surface areas may be used in combination.
- a part of the silica contained in the resin composition (for example, 50% by mass or more of the total amount of silica) is silica having a specific surface area of 150 m 2 / g or more, the above-mentioned remarkably excellent grip property has a specific surface area of less than 150 m 2 / g. It can be expected to be expressed even if the above silica is used in combination.
- the specific surface area of the silica can be measured by, for example, the BET method.
- the silica content is preferably 1 to 50 parts by mass, more preferably 10 to 45 parts by mass, and 20 to 40 parts by mass with respect to 100 parts by mass of the total of rubber (I) and block copolymer (II). More preferred. When the silica content is within the above range, mechanical strength such as excellent tear strength is exhibited, and grip and wear resistance are further improved.
- the resin composition of this embodiment preferably contains a silane coupling agent.
- the silane coupling agent include sulfide compounds, mercapto compounds, vinyl compounds, amino compounds, glycidoxy compounds, nitro compounds, chloro compounds and the like.
- the silane coupling agent preferably has an alkoxysilyl group from the viewpoint of reactivity with silica, and among them, methoxysilane and ethoxysilane are preferable. These silane coupling agents may be used alone or in combination of two or more.
- sulfide compound examples include bis (3-triethoxysilylpropyl) tetrasulfide, bis (2-triethoxysilylethyl) tetrasulfide, bis (3-trimethoxysilylpropyl) tetrasulfide, and bis (2-trimethoxy).
- Cyrilethyl) tetrasulfide bis (3-triethoxysilylpropyl) trisulfide, bis (3-trimethoxysilylpropyl) trisulfide, bis (3-triethoxysilylpropyl) disulfide, bis (3-trimethoxysilylpropyl) Disulfide, 3-trimethoxysilylpropyl-N, N-dimethylthiocarbamoyltetrasulfide, 3-triethoxysilylpropyl-N, N-dimethylthiocarbamoyltetrasulfide, 2-trimethoxysilylethyl-N, N-dimethylthiocarbamoyl Examples thereof include tetrasulfide, 3-trimethoxysilylpropylbenzothiazole tetrasulfide, 3-triethoxysilylpropylbenzothiazole tetrasulfide, 3-trie
- Examples of the mercapto compound include 3-mercaptopropyltrimethoxysilane, 3-mercaptopropyltriethoxysilane, 3-mercaptopropyl-di (tridecane-1-oxy-13-penta (ethyleneoxide)) ethoxysilane, and 2-mercapto. Examples thereof include ethyltrimethoxysilane and 2-mercaptoethyltriethoxysilane.
- Examples of the vinyl compound include vinyltriethoxysilane and vinyltrimethoxysilane.
- Examples of amino compounds include 3-aminopropyltriethoxysilane, 3-aminopropyltrimethoxysilane, 3- (2-aminoethyl) aminopropyltriethoxysilane, and 3- (2-aminoethyl) aminopropyltri. Examples thereof include methoxysilane.
- Examples of the glycidoxy compound include ⁇ -glycidoxypropyltriethoxysilane, ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -glycidoxypropylmethyldiethoxysilane, and ⁇ -glycidoxypropylmethyldimethoxysilane.
- Examples of the nitro compound include 3-nitropropyltrimethoxysilane and 3-nitropropyltriethoxysilane.
- Examples of the chloro compound include 3-chloropropyltrimethoxysilane, 3-chloropropyltriethoxysilane, 2-chloroethyltrimethoxysilane, and 2-chloroethyltriethoxysilane.
- silane coupling agents from the viewpoint of wear resistance, bis (3-triethoxysilylpropyl) disulfide, bis (3-triethoxysilylpropyl) tetrasulfide, 3-mercaptopropyltrimethoxysilane, and 3-mercapto Propyltriethoxysilane is preferred.
- the content of the silane coupling agent is preferably 0.01 to 30 parts by mass, more preferably 0.05 to 25 parts by mass, further preferably 1 to 20 parts by mass, and 3 to 15 parts by mass with respect to 100 parts by mass of silica.
- the unit is even more preferable.
- the content of the silane coupling agent is within the above range, the dispersibility and abrasion resistance in the composition are further improved.
- the resin composition of the present embodiment preferably does not contain a polyolefin-based resin, or the content of the polyolefin-based resin with respect to 100 parts by mass of the block copolymer (II) is less than 10 parts by mass. If the content is less than 10 parts by mass, melt-kneading can be preferably performed. It is more preferable that the resin composition of the present embodiment does not contain a polyolefin resin.
- the polyolefin-based resin that can be contained include polyethylene, polypropylene, polybutene-1, polyhexene-1, poly-3-methyl-butene-1, poly-4-methyl-pentene-1, and ethylene-vinyl acetate. Examples thereof include polymers and ethylene-acrylic acid copolymers.
- the resin composition of the present embodiment may contain a cross-linking accelerator.
- the cross-linking accelerator include N, N-diisopropyl-2-benzothiazole-sulfenamide, 2-mercaptobenzothiazole, di-2-benzothiazolyl disulfide, 2- (4-morpholinodithio) benzothiazole and the like.
- Thiazols such as diphenylguanidine and triphenylguanidine; aldehyde-amine-based or aldehyde-ammonia-based reactants such as butylaldehyde-aniline reactants and hexamethylenetetramine-acetaldehyde reactants; 2-mercaptoimidazoline and the like.
- Imidazolines such as thiocarbanilide, diethylurea, dibutylthiourea, trimethylthiourea, diorsotrilthiourea; thiurammono or polysulfides such as tetramethylthium monosulfide, tetramethylthium disulfide, pentamethylene thiuram tetrasulfide; dimethyldithiocarbamine Thiocarbamates such as zinc acid, zinc ethylphenyldithiocarbamate, sodium dimethyldithiocarbamate, selenium dimethyldithiocarbamate, telluryl diethyldithiocarbamate; xanthogenates such as zinc dibutylxanthogenate; saltylic acid and the like can be mentioned.
- thioureas such as thiocarbanilide, diethylurea, dibutylthiourea, trimethylthiourea, diorso
- cross-linking accelerators may be used alone or in combination of two or more.
- the content of the cross-linking accelerator is preferably 0.05 to 15.0 parts by mass, preferably 0.1 to 5.0 parts by mass, based on 100 parts by mass of the total of rubber (I) and block copolymer (II). Is more preferable, and 0.5 to 4.0 parts by mass is further preferable.
- the resin composition of the present embodiment may contain a filler other than the above-mentioned silica.
- a filler an inorganic filler and / or an organic filler can be used.
- Preferred inorganic fillers are preferably silicate compounds, metal oxides and the like.
- the silicate compound is preferably one or more selected from the group consisting of kaolin, talc, clay, pyrophyllite, mica, montmorillonite, bentonite, wallastonite, sepiolite, zonotrite, zeolite, diatomaceous earth, and halloysite. It is a silicate compound.
- the metal oxide is selected from the group consisting of titanium oxide, iron oxide, magnesium oxide, aluminum oxide, cerium oxide, antimony oxide, tin oxide, lead oxide, chromium oxide, cobalt oxide, tungsten oxide, and copper oxide.
- One or more metal oxides are selected from the group consisting of titanium oxide, iron oxide, magnesium oxide, aluminum oxide, cerium oxide, antimony oxide, tin oxide, lead oxide, chromium oxide, cobalt oxide, tungsten oxide, and copper oxide.
- One or more metal oxides are selected from the group consisting of titanium oxide, iron oxide, magnesium oxide, aluminum oxide, cerium oxide, antimony oxide, tin oxide, lead oxide, chromium oxide, cobalt oxide, tungsten oxide, and copper oxide.
- a fine particle type organic filler and / or a fiber type organic filler is exemplified.
- a preferred example of the fine particle type organic filler is at least one selected from the group consisting of urethane fine particles, acrylic fine particles, styrene fine particles, acrylic / styrene fine particles, styrene olefin fine particles, fluorine fine particles, polyethylene fine particles and silicone fine particles.
- Organic fillers can be mentioned.
- the preferable average particle size of the fine particle type organic filler is 2 to 50 ⁇ m. The average particle size can be measured by a laser diffraction method based on JIS Z 8825-1.
- various organic fibers can be used, for example, 6-nylon fiber, 6,6-nylon fiber, 4,6-nylon fiber, aromatic polyamide fiber, para-aramid fiber, meta.
- Aramid fiber polyethylene terephthalate (PET) fiber, polyethylene naphthalate (PEN) fiber, polyparaphenylene benzoxazole (PBO) fiber, PVA fiber (viniron), polyarylene sulfide fiber, 2,6-hydroxynaphthoic acid Parahydroxybenzoic acid fiber (Vectran), polyethylene (PE) fiber, polypropylene (PP) fiber, polylactic acid (PLA) fiber, polybutylene succinate (PBS) fiber, polyethylene succinate fiber, syndiotactic-1,2- Synthetic fibers such as polybutadiene (SPB) fibers and polyvinyl chloride (PVC) fibers, and natural (regenerated) fibers such as cotton, hemp, rayon, and cellulose fibers are preferably exemplified.
- the organic fibers can be roughly classified into organic microfibers and organic nanofibers according to their average fiber diameter.
- the organic microfibers are organic fibers having an average fiber diameter on the order of micrometers, and the average fiber diameter is preferably 1 to 200 ⁇ m, more preferably 10 to 100 ⁇ m, and even more preferably 15 to 90 ⁇ m.
- the average fiber length of the organic microfibers is not particularly limited, but is preferably in the range of 0.1 to 20 mm, more preferably in the range of 0.5 to 15 mm, and even more preferably in the range of 1 to 10 mm. .. By having such an average particle size and an average length, the balance between the processability of the resin composition and the effect of improving the obtained wet grip property is excellent.
- the organic nanofibers are organic fibers having an average fiber diameter on the order of nanometers, and the average fiber diameter is preferably in the range of 1 to 900 nm, more preferably in the range of 1 to 700 nm. More preferably, it is in the range of 1 to 500 nm.
- the average fiber length of the organic nanofibers is preferably in the range of 0.1 to 1000 ⁇ m, more preferably in the range of 1 to 750 ⁇ m, and even more preferably in the range of 5 to 600 ⁇ m.
- the types of the organic microfibers and the organic nanofibers are not particularly limited, but are polyethylene terephthalate (PET) fibers, PVA-based fibers (vinylon), 2,6-hydroxynaphthoic acid / parahydroxybenzoic acid fibers (Vectran), and polylactic acid.
- PET polyethylene terephthalate
- PVA-based fibers vinylylon
- Vectran parahydroxybenzoic acid fibers
- PVA fiber and cellulose fiber are preferable
- PVA-based fiber (vinylon) and cellulose fiber are more preferable
- cellulose fiber is further preferable from the viewpoint of improving grip property.
- As the organic microfiber made of a cellulosic fiber for example, a cellulosic microfiber sold under the product name "ARBOCEL" manufactured by Rettenmeier is preferably exemplified.
- Examples of the organic nanofibers made of cellulosic fibers include cellulosic nanofibers such as Celish KY-100G (average fiber length: 500 ⁇ m, average fiber diameter: 20 nm, solid content: 10% by mass) manufactured by Daicel Finechem Co., Ltd. Fibers are preferably exemplified.
- the content of the filler is preferably 0 to 120 parts by mass, and 0.5 to 80 parts by mass with respect to 100 parts by mass of the total content of the rubber (I) and the block copolymer (II). It may be 1 to 50 parts by mass, or 1 to 30 parts by mass.
- the resin composition of the present embodiment may contain a softening agent as long as the effects of the present invention are not impaired.
- the resin composition does not have to contain a softening agent because it has sufficient fluidity for molding due to the above-mentioned structure.
- the softeners paraffin-based, naphthen-based and aromatic process oils, mineral oils, white oils and other oil-based softeners may exude oil over time when formed into a molded product. Therefore, it is preferable not to include it in the resin composition.
- softening agent examples include phthalic acid derivatives such as dioctyl phthalate and dibutyl phthalate; liquid copolymers of ethylene and ⁇ -olefin; liquid paraffin; polybutene; low molecular weight polyisobutylene; liquid polybutadiene, liquid polyisoprene, and the like.
- Liquid polydiene such as liquid ⁇ -polyfarnesene, liquid polyisoprene / butadiene copolymer, liquid isoprene / ⁇ -farnesene, liquid butadiene / ⁇ -farnesene, liquid styrene / butadiene copolymer, liquid styrene / isoprene copolymer and its water. Examples thereof include an adjunct or a modified product thereof.
- the content of the resin composition is preferably 0.1 to 30% by mass, more preferably 0.5 to 20% by mass, still more preferably 1.0 to 15% by mass.
- additives Other additives other than those described above can be added to the resin composition of the present embodiment as long as the effects of the present invention are not impaired.
- additives include heat aging agents, antioxidants, light stabilizers, antistatic agents, mold release agents, flame retardants, foaming agents, pigments, dyes, whitening agents and the like. These additives may be used alone or in combination of two or more.
- the method for producing the resin composition of the present embodiment is, for example, by melt-kneading each component excluding the cross-linking agent (III) and the cross-linking accelerator added as necessary, and then cross-linking by the cross-linking method described later. Can be manufactured. There is no particular limitation on the method of melt-kneading each component except the cross-linking agent (III) and the cross-linking accelerator added as necessary, and rubber (I), block copolymer (II) and other optional components can be used.
- a kneading device for example, a uniaxial extruder, a multi-screw extruder, a Banbury mixer, a brabender mixer, an open roll, a heating roll, various kneaders, and the like for melt kneading.
- a method in which rubber (I), block copolymer (II) and other optional components are supplied from separate charging ports and melt-kneaded, or block copolymer (II) and silica, a silane coupling agent, etc.
- melt-kneaded product and other optional components may be used.
- the temperature at the time of melt-kneading can be arbitrarily selected in the range of usually 20 to 270 ° C.
- a cross-linking agent (III) and a cross-linking accelerator added as necessary are added, and a vulcanization temperature is usually about 120 to 200 ° C., preferably about 120 to 200 ° C. Is 140 to 200 ° C., the vulcanization pressure is usually about 0.5 to 10 MPa, and a method of cross-linking is usually performed by maintaining the vulcanization pressure for about 1 minute to 2 hours.
- the resin composition of the present embodiment can be evaluated for slip resistance by the coefficient of static friction measured according to ASTM D-1894.
- the preferable numerical range of the coefficient of static friction varies depending on the type of rubber (I) used and cannot be unconditionally specified, but the larger the value of the coefficient of static friction, the less slippery it is.
- the coefficient of static friction is preferably 4.5 or more, more preferably 5.0 or more under dry conditions. Further, in the case of wet matter, it is preferably 1.5 or more, more preferably 2.0 or more.
- the dry condition and the wet condition mean the measurement conditions described in Examples described later.
- the wear resistance of the resin composition of the present embodiment can be evaluated by the amount of wear measured by a DIN wear test (rotary cylindrical wear tester) according to JIS K 6264-2: 2005.
- the amount of wear is preferably 200 mm 3 or less, more preferably 190 mm 3 or less, and further, 180 mm 3 or less and 170 mm 3 or less.
- the resin composition of the present embodiment has an excellent vulcanization rate, that is, the time required for vulcanization (90% vulcanization time) is short. Can be expected.
- the vulcanization rate is high, the viscosity of the resin composition increases rapidly during vulcanization, and the vulcanizing agent, which is sulfur and / or a sulfur-containing compound in the resin composition, a cross-linking accelerator, a cross-linking aid, and antioxidant. Since it becomes difficult for agents, anti-aging agents, plasticizing agents such as softeners, etc.
- the vulcanization rate of the resin composition is 90, which is obtained by measuring the torque of the resin composition at 160 ° C. using a vibration type vulcanization tester (curast meter) according to JIS K 6300-2: 2001. % The time required to reach the vulcanization amount (90% vulcanization time).
- the vulcanization rate Tc (90) of the resin composition is preferably 13 minutes or less, more preferably 12 minutes or less. The lower limit is not particularly limited, but is usually 1 minute or more.
- the resin composition of this embodiment has a specific gravity measured according to ISO 1183: 1987, preferably 0.88 to 1.30, and more preferably 0.90 to 1.20.
- a molded product made of a resin composition does not lack practicality. For example, when the resin composition is used for the sole of footwear, it does not become too heavy and the weight of footwear can be expected to be reduced.
- the resin composition of the present embodiment has a hardness (hereinafter, also referred to as “A hardness”) according to the type A durometer method of JIS K 6253-: 2012, preferably 90 or less, more preferably 85 or less. Further, the A hardness is preferably 25 or more, more preferably 30 or more, and further preferably 35 or more. When the A hardness is in the above range, the molding processability is good, the frictional force is high due to the good flexibility, and the grip property and the wear resistance are also excellent.
- the resin composition of the present embodiment has a tensile strength at cutting (tensile breaking strength) measured according to JIS K 6251: 2010, preferably 5.0 MPa or more, more preferably 10.0 MPa or more, still more preferably 10.0 MPa or more. It is 13.0 MPa or more. When the tensile breaking strength is 5.0 MPa or more, the tensile characteristics are good.
- the elongation at cutting (tensile elongation at break) measured according to JIS K 6251: 2010 is preferably 100% or more, more preferably 150% or more, still more preferably. It is 200% or more, and even more preferably 370% or more. When the tensile elongation at break is 100% or more, the tensile characteristics are good.
- the resin composition of the present embodiment has a tear strength (tear strength) measured according to JIS K 6252-1: 2015, preferably 2.0 kN / m or more, more preferably 2.5 kN / m or more. More preferably, it is 3.0 kN / m or more.
- the molded product in this embodiment is obtained by using the above-mentioned resin composition.
- the shape of the molded body may be any molded body that can be produced using the above-mentioned resin composition, and is molded into various shapes such as pellets, films, sheets, plates, pipes, tubes, rods, and granules. can do.
- the method for producing this molded product is not particularly limited, and it can be molded by various conventional molding methods such as injection molding, blow molding, press molding, extrusion molding, and calendar molding. From the above-mentioned resin composition, a press-molded product can be particularly preferably obtained.
- the resin composition of the present embodiment can be suitably used as a molded product such as a shoe sole using at least a part thereof.
- shoe soles for footwear such as hiking boots, sandals, safety shoes, mountain climbing shoes, marathon shoes, underground socks and boots; sports equipment such as underwater glasses, snorkels, ski boots and skis / snowboards; pens and scissors, etc.
- Equipment to be used, and various grips such as knives;
- Parts of home appliances such as refrigerators, vacuum cleaners and waterproof bodies (mobile phones, etc.);
- Tread members such as tires, especially winter tires, studless tires, all-season tires, fuel-efficient tires and heavy-duty tires
- Office equipment such as copy machine feed rollers and take-up rollers
- Parts used for furniture such as sofas and chair seats
- Rubber parts such as switch covers, stoppers, casters and foot rubbers
- Building materials such as coated plywood and coated steel plates
- Industrial use It can be suitably used for industrial members such as belts and industrial rubber hoses.
- ⁇ -Farnesene (purity 97.6% by mass, manufactured by Amiris, Incorporated) is purified by a 3 ⁇ molecular sieve and distilled in a nitrogen atmosphere to produce gingiberene, bisabolen, farnesene epoxide, farnesol. Hydrocarbon impurities such as isomers, E, E-farnesol, squalene, ergosterol and several dimerses of farnesene were removed and used for the following polymerization.
- ⁇ Rubber (I)> -Rubber (I): SBR Product name JSR 1502, manufactured by JSR Corporation, styrene-butadiene copolymer rubber (E-SBR), manufactured by emulsification polymerization method, styrene content 23.5% by mass
- ⁇ Crosslinking agent (III)> -Sulfur Fine sulfur 200 mesh, manufactured by Tsurumi Chemical Industry Co., Ltd.
- Zinc oxide Zinc oxide 1 type, manufactured by Sakai Chemical Industry Co., Ltd.
- Stearic acid Product name Lunac S-20, manufactured by Kao Corporation
- the details of each measuring method for the polymer obtained in the production example are as follows. (1) Measurement of peak top molecular weight and molecular weight distribution The peak top molecular weight (Mp) and molecular weight distribution (Mw / Mn) of the styrene block and the polymer are determined by GPC (gel permeation chromatography) using the standard polystyrene-equivalent molecular weight. The peak top molecular weight (Mp) was determined from the position of the peak of the molecular weight distribution.
- the measuring device and conditions are as follows.
- Block copolymer (P) and block copolymer after hydrogenation (II) (referred to as “hydrogenated block copolymer (II)") was dissolved in a deuterated chloroform solvent, and 1 H-NMR was measured at 50 ° C. using "Lambda-500” manufactured by Nippon Denshi Co., Ltd.
- the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene in the hydrogenated block copolymer (II) is the carbon-carbon double bond appearing at 4.5 to 6.0 ppm in the obtained spectrum. It was calculated from the peak of the proton possessed by the following formula.
- Hydrogenation rate (mol%) ⁇ 1- (number of moles of carbon-carbon double bond contained per mole of hydrogenated block copolymer (II)) / (per mole of block copolymer (P)) Number of moles of carbon-carbon double bond contained) ⁇ ⁇ 100 (2-2) Further, the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene in the polymer block (B), and the carbon in the structural unit derived from the conjugated diene in the polymer block (C). -The hydrogenation rate of the carbon double bond was calculated in the same manner as above from the proton peaks appearing in the respective spectra obtained above.
- a molded product (25 mm in diameter and 2 mm in thickness) made of a polymer produced in the production example is formed by dropping water (1 mL) containing food coloring on a colorless and transparent quartz plate (5 cm ⁇ 5 cm) having a smooth surface. A flat sheet) was installed, and a load of 2 kg was applied to the molded product with a tensile tester.
- the food coloring is added to water from the viewpoint of easy observation, and does not affect the ease of repelling water.
- the state of water on the ground contact surface between the molded body and the quartz plate was visually observed from the quartz plate side, and evaluated according to the following evaluation criteria A to C.
- Coverage% [(Area of red area on the ground plane between the molded body and the quartz plate) / (Area of the ground plane between the molded body and the quartz plate)] ⁇ 100
- Hydrogenated block copolymer (II-1) In a pressure vessel substituted with nitrogen and dried, 50.0 kg of cyclohexane was charged as a solvent, 0.1905 kg of sec-butyllithium (10.5 mass% cyclohexane solution) as an anionic polymerization initiator, and 0.40 kg of tetrahydrofuran as a Lewis base were charged. After the temperature is raised to 50 ° C., 6.34 kg of ⁇ -farnesene is added and polymerized for 2 hours, then 2.50 kg of styrene (1) is added and polymerized for 1 hour, and 3.66 kg of butadiene is further added for 1 hour. Polymerization was performed.
- a reaction solution containing a polystyrene-poly ( ⁇ -farnesene) -polystyrene triblock copolymer (P2) was obtained.
- Palladium carbon (palladium-supported amount: 5% by mass) was added to this reaction solution as a hydrogenation catalyst in an amount of 5% by mass based on the block copolymer (P2), and the reaction was carried out at a hydrogen pressure of 2 MPa and 150 ° C. for 10 hours.
- a hydrogen pressure of 2 MPa and 150 ° C. for 10 hours was carried out.
- Hydrogenated block copolymer (II-2) was obtained.
- the above physical characteristics of the hydrogenated block copolymer (II-2) were measured. The results are shown in Table 1.
- Unhydrogenated block copolymer (II'-1) After charging 50.0 kg of cyclohexane as a solvent and 0.1010 kg of sec-butyl styrene (10.5 mass% cyclohexane solution) as an anionic polymerization initiator in a pressure-resistant container that has been replaced with nitrogen and dried, the temperature is raised to 50 ° C. , Styrene (1) 1.70 kg was added and polymerized for 1 hour, 0.065 kg of N, N, N', N'-tetramethylethylenediamine (TMEDA) was added as a Lewis base, and then 13.30 kg of isoprene was added.
- TMEDA N, N, N', N'-tetramethylethylenediamine
- the polymerization was carried out for 2 hours, and 1.70 kg of styrene (2) was further added and polymerized for 1 hour to obtain a reaction solution containing a styrene-isoprene-styrene triblock copolymer (P'1).
- the polymerization reaction was stopped by adding 200 mL of methanol to this reaction solution. By washing the obtained mixed solution with water and then re-precipitating it in a large amount of methanol, it is also referred to as a styrene-isoprene-styrene triblock copolymer (hereinafter, "unhydrogenated block copolymer (II'-1)"). I got).
- the above physical characteristics of the unhydrogenated block copolymer (II'-1) were measured. The results are shown in Table 1.
- Table 1 Each notation in Table 1 is as follows.
- * 1: (A) / (B) indicates the mass ratio of the content of the polymer block (A) to the content of the polymer block (B).
- * 2: (A) / ((B) + (C)) is the sum of the content of the polymer block (A) and the contents of each of the polymer block (B) and the polymer block (C). Shows the mass ratio.
- * 3: (b1) / (B) indicates the content (mass%) of the structural unit (b1) derived from farnesene in the polymer block (B).
- F-St-Bd-St-F represents a poly ( ⁇ -farnesene) -polystyrene-polybutadiene-polystyrene-poly ( ⁇ -farnesene) pentablock copolymer.
- St-F-St represents a polystyrene-poly ( ⁇ -farnesene) -polystyrene triblock copolymer.
- St-Ip-St represents a polystyrene-polyisoprene-polystyrene triblock copolymer.
- the hydrogenation rate of (II) indicates the hydrogenation rate of carbon-carbon double bonds in the structural unit derived from the conjugated diene in the block copolymer (P).
- the hydrogenation rate of the polymer block (B) indicates the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene in the polymer block (B). However, it is shown only when it has a polymer block (B).
- the hydrogenation rate of the polymer block (C) indicates the hydrogenation rate of the carbon-carbon double bond in the structural unit derived from the conjugated diene in the polymer block (C). However, it is shown only when it has a polymer block (C).
- the melt-kneaded product is placed in a mixing roll (step 4), the cross-linking agent (III) and the cross-linking accelerator are added (step 5), re-kneaded and taken out (step 6) to obtain a resin composition. It was.
- the coefficient of static friction was measured by the same method as in (1) Dry above, except that 1 cc of distilled water was dropped on the wet friction table. The larger the coefficient of static friction, the greater the frictional force, the less slippery, and the better the wet grip.
- the amount of DIN wear was measured by a DIN wear test according to JIS K 6264-2: 2005. The smaller the value, the better the wear resistance.
- the vulcanization rate The vulcanization rate (Tc90) was measured according to JIS K 6300-2 using a vibration vulcanization tester (curast meter) at 160 ° C. of the above resin composition obtained in Examples and Comparative Examples. The vulcanization time up to 90% vulcanization amount was defined as the vulcanization rate (t90). The smaller the value, the better the vulcanization rate.
- Examples 10 to 13 [Comparative Examples 6 and 7] According to the blending ratio (parts by mass) shown in Table 4, the same procedure as in Example 1 was carried out to obtain a resin composition and a rubber sheet, and the physical characteristics were evaluated.
- the rubber (I) and the cross-linking accelerators (3) to (5) in Table 4 are as follows, and the other materials are as shown above.
- Example 14 [Comparative Examples 8 and 9] According to the blending ratio (parts by mass) shown in Table 5, the same procedure as in Example 1 was carried out to obtain a resin composition and a rubber sheet, and the physical characteristics were evaluated.
- the rubber (I) in Table 5 is as follows, and the other materials are as shown above. ⁇ Rubber (I)> -Rubber (I): NR Natural rubber, from Indonesia, RSS No. 1
- Example 14 From the comparison between Example 14 and Comparative Example 8, it can be seen that the inclusion of the block copolymer (II) increases the coefficient of static friction under both dry and wet conditions and is excellent in slip resistance. Further, from the comparison between Example 14 and Comparative Example 9, by setting the mass ratio of the rubber (I) and the block copolymer (II) to a specific numerical range, the resistance to slipping is maintained while maintaining excellent slip resistance. It can be seen that it is also excellent in abrasion resistance. Further, from Example 14, it can be seen that the resin composition of the present embodiment is not slippery, has excellent wear resistance, and can obtain a molded product having an excellent balance of tear strength, tensile properties, and the like.
- Example 15 to 20 [Comparative Example 10] According to the blending ratio (parts by mass) shown in Table 6, the same procedure as in Example 1 was carried out to obtain a resin composition and a rubber sheet, and the physical characteristics were evaluated. The materials in Table 6 are as shown above.
- Examples 21 to 33 [Comparative Examples 11 and 12] According to the blending ratios (parts by mass) shown in Tables 8 and 9, the same procedure as in Example 1 was carried out to obtain a resin composition and a rubber sheet, and the physical characteristics were evaluated. As the softeners in Tables 8 and 9, the softeners produced in the following Production Examples 4 to 8 were used. The fillers (cellulose fibers) in Table 9 are as follows, and the other materials are as shown above.
- Poly ⁇ -farnesene (IV-1) In a pressure-resistant container that has been replaced with nitrogen and dried, 40 kg of hexane as a solvent and 0.55 kg of n-butyllithium (17 mass% hexane solution) as an initiator are charged, the temperature is raised to 50 ° C., and then 40 kg of ⁇ -farnesene is added. Polymerized for 1 hour. The obtained polymerization reaction solution was treated with methanol, and the polymerization reaction solution was washed with water. The polymerization reaction solution after washing and water were separated and dried at 70 ° C. for 12 hours to obtain poly ⁇ -farnesene (IV-1) having the physical characteristics shown in Table 7. The number average molecular weight (Mn) and the molecular weight distribution (Mw / Mn) were measured in the same manner as in the above [Measurement method].
- Poly ⁇ -farnesene (IV-2) 40 kg of hexane as a solvent and 1.84 kg of n-butyllithium (17 mass% hexane solution) as an initiator are charged in a pressure-resistant container that has been replaced with nitrogen and dried, and the temperature is raised to 50 ° C., and then 40 kg of ⁇ -farnesene is added. Polymerized for 1 hour. The obtained polymerization reaction solution was treated with methanol, and the polymerization reaction solution was washed with water. The polymerization reaction solution after washing and water were separated and dried at 70 ° C. for 12 hours to obtain poly ⁇ -farnesene (IV-2) having the physical characteristics shown in Table 7. The number average molecular weight (Mn) and the molecular weight distribution (Mw / Mn) were measured in the same manner as in the above [Measurement method].
- Butadiene- ⁇ -farnesene copolymer (IV-3) 40 kg of hexane as a solvent and 0.72 kg of n-butyllithium (17 mass% hexane solution) as an initiator were charged in a pressure-resistant container that had been replaced with nitrogen and dried, and the temperature was raised to 50 ° C., and then 24.0 kg prepared in advance. A mixture of ⁇ -farnesene and 16.0 kg of butadiene was added and polymerized for 1 hour. The obtained polymerization reaction solution was treated with methanol, and the polymerization reaction solution was washed with water. The polymerization reaction solution after washing and water were separated and dried at 70 ° C.
- Hydrogenated polyisoprene (IV-4) In a pressure-resistant container that has been replaced with nitrogen and dried, 40 kg of hexane as a solvent and 0.77 kg of n-butyllithium (17 mass% hexane solution) as an initiator are charged, heated to 50 ° C., and then 40 kg of isoprene is added for 1 hour. Polymerized. The obtained polymerization reaction solution was treated with methanol, and the polymerization reaction solution was washed with water. The polymerization reaction solution after washing and water were separated and dried at 70 ° C. for 12 hours to obtain polyisoprene.
- Modified liquid polyisoprene (IV-5) 40 kg of hexane and 0.90 kg of n-butyllithium (17 mass% hexane solution) were charged in a pressure-resistant container substituted with nitrogen and dried, the temperature was raised to 70 ° C., 41 kg of isoprene was added, and the mixture was polymerized for 1 hour. After adding methanol to the obtained polymerization reaction solution, the polymerization reaction solution was washed with water. Water was separated and the polymerization reaction solution was vacuum dried at 70 ° C. for 12 hours to obtain unmodified polyisoprene (IV-5).
- Example 31 containing the block copolymer (II-1) has a better coefficient of static friction.
- Example 32 in which a part of the silica (1) of Example 31 is replaced with a cellulose fiber, the grip property is further improved.
- Example 33 in which a part of the softening agent of Example 32 was replaced with modified liquid polyisoprene (IV-5), the numerical value of the DIN wear amount was reduced while maintaining the coefficient of static friction of Example 32.
- the resin composition of the present invention can be a molded product that is not slippery and has excellent wear resistance, and is expected to have an excellent balance of tear strength and tensile properties. Therefore, the resin composition of the present invention is used for footwear soles; sports equipment; various grips such as writing tools, tools and electric tools; household appliances parts such as refrigerators; automobile accessories such as side moldings; tread members such as tires; Office equipment such as take-up rollers; parts used for furniture such as sofas; rubber parts such as foot rubber; building materials such as coated plywood and coated steel plates; industrial members such as industrial belts can be suitably used.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
例えば、熱可塑性エラストマーの数平均分子量やゴム成分のガラス転移温度を特定し、あるいは粘着付与樹脂や水添ブロック共重合体を含有させた樹脂組成物が提案されている(例えば、特許文献1~8)。また、ファルネセン由来の構造単位を含有する重合体ブロックを含む水添ブロック共重合体を含有させた樹脂組成物が提案されている(例えば、特許文献9)。
また、特許文献9に開示された樹脂組成物は、柔軟性、引裂強度、引張特性、ドライグリップ性及びウェットグリップ性のバランスに優れるものの、様々な環境下におけるグリップ性といった成形体の滑りにくさの性能に更なる向上が求められている。加えて、履物用靴底や各種タイヤ等の用途には優れた耐摩耗性を有することも要求される。
すなわち、本発明は下記のとおりである。
前記ゴム(I)と前記ブロック共重合体(II)との質量比[(I)/(II)]が99/1~55/45であり、
前記ブロック共重合体(II)が、芳香族ビニル化合物由来の構造単位を含有する重合体ブロック(A)と、ファルネセン由来の構造単位(b1)を含有する重合体ブロック(B)とを含むブロック共重合体である、樹脂組成物。
[2]前記[1]に記載の樹脂組成物の成形体。
[3]前記[1]に記載の樹脂組成物を少なくとも一部に用いた、靴底。
また本明細書において、実施態様の好ましい形態を示すが、個々の好ましい形態を2つ以上組み合わせたものもまた、好ましい形態である。数値範囲で示した事項について、いくつかの数値範囲がある場合、それらの下限値と上限値とを選択的に組み合わせて好ましい形態とすることができる。
なお、本明細書において、「XX~YY」との数値範囲の記載がある場合、「XX以上YY以下」を意味する。
本実施態様の樹脂組成物は、
ゴム(I)、ブロック共重合体(II)及び架橋剤(III)を含有し、
前記ゴム(I)と前記ブロック共重合体(II)との質量比[(I)/(II)]が99/1~55/45であり、
前記ブロック共重合体(II)が、芳香族ビニル化合物由来の構造単位を含有する重合体ブロック(A)と、ファルネセン由来の構造単位(b1)を含有する重合体ブロック(B)とを含むブロック共重合体であることを特徴とする。
加えて、上記質量比[(I)/(II)]を上記特定の数値範囲とすることにより、滑りにくく耐摩耗性の両方に優れさせることができる。
また、本実施態様の樹脂組成物は、上記性能に加え、引裂強度及び引張特性のバランスに優れ、更にブロック共重合体(II)に比べゴム(I)の含有量が多いことからコスト削減にも繋がることが期待できる。
本実施態様の樹脂組成物に含有されるゴム(I)としては、履物用靴底等のゴム底のゴム原料として用いられている天然ゴムや各種合成ゴム等が挙げられる。
なお、ゴム(I)には、前記ブロック共重合体(II)は含まれない。
上記ゴムの中でも、各種用途において求められる強度、耐摩耗性、弾性、及び柔軟性等の物性、並びにブロック共重合体(II)等の各種成分との相容性の観点から、スチレンブタジエン共重合ゴム、天然ゴム、ポリイソプレンゴム、ポリブタジエンゴム、アクリルニトリルブタジエン共重合ゴム、エチレン-プロピレン-ジエン共重合体、ブチルゴム、及びブチルゴムを変性したハロゲン化ブチルゴムから選ばれる少なくとも1種が好ましい。
ゴム(I)は1種を単独で又は2種以上を併用してもよい。
また、ゴム(I)として少なくともNRを用いる場合、後述する軟化剤を併用することが好ましい。軟化剤の中でも、液状ポリジエン及びその水添物又はその変性物(単に「液状ポリジエン」ということがある。)が好適である。液状ポリジエンを併用することにより、NRの粘度が下がり組成物への分散性が高まると考えられるため、優れたグリップ性を有する成形体をより得やすくなる。液状ポリジエンは、1種を単独で又は2種以上を併用してもよい。
上記液状ポリジエンの数平均分子量(Mn)は、2,000~180,000が好ましく、3,000~130,000がより好ましく、4,000~50,000が更に好ましく、5,000~35,000がより更に好ましい。数量平均分子量が上記範囲内であると加工性が向上し、また樹脂組成物のブリードアウトが抑制できる。
上記液状ポリジエンの分子量分布(Mw/Mn)は、1~6が好ましく、1~4がより好ましく、1~3が更に好ましく、1~2がより更に好ましい。分子量分布が上記範囲内であると、液状ポリジエンの粘度のばらつきが小さく、取り扱いが容易である。
液状ポリジエンのMw及びMnは、ゲルパーミエーションクロマトグラフィー(GPC)の測定から求めた値であり、後述する実施例に記載した方法で測定した値を意味する。
また、ゴム(I)として少なくともNRを用いる場合の好ましい実施態様の一例として、軟化剤としての上記液状ポリジエンの含有量は、ゴム(I)、ブロック共重合体(II)、及び液状ポリブタジエンの総量100質量部に対して、0.1~10質量部が好ましく、0.5~9質量部がより好ましく、1~8質量部が更に好ましく、1.5~7質量部がより更に好ましい。
なお、ゴム(I)には、上記液状ポリジエンは含まれない。
また、市販のSBRを用いることができ、スチレン含量が0.1~70質量%のものが好ましく、5~50質量%のものがより好ましく、15~35質量%のものが更に好ましく、15~25質量%のものがより更に好ましい。
SBRは、弾性や強度等の物性バランスがよく、混練りや成型加工等にも良好で、入手容易の観点からも好適である。
IRの重量平均分子量(Mw)は、9万~200万であることが好ましく、15万~150万であることがより好ましい。Mwが上記範囲にある場合、成形加工性、引裂強度、及び引張特性が良好となる。なお、IRの重量平均分子量とは、ゲルパーミエーションクロマトグラフィー(GPC)の測定から求めたポリスチレン換算の重量平均分子量である。
IRのビニル含量は好ましくは50質量%以下、より好ましくは40質量%以下、更に好ましくは30質量%以下である。
IRは、機械的強度等の物性バランスにも良好で、入手容易の観点からも好適である。
BRの重量平均分子量(Mw)は、9万~200万であることが好ましく、15万~150万であることがより好ましく、25万~100万であることが更に好ましく、35万~70万であることがより更に好ましい。Mwが上記範囲にある場合、成形加工性、引裂強度、及び引張特性が良好となる。
BRのビニル含量は、好ましくは50質量%以下、より好ましくは40質量%以下、更に好ましくは30質量%以下である。
BRは、特に、優れた耐摩耗性を樹脂組成物の成形体に付与することが期待できる観点からも好適である。
本実施態様の樹脂組成物は、ブロック共重合体(II)を含有することにより、柔軟性が良好となり、滑りにくく、耐摩耗性を発現することができ、更に引張特性及び引裂強度の発現も期待することができる。
ブロック共重合体(II)は、未水添のブロック共重合体(以下、「ブロック共重合体(P)」と称すことがある。)であってもよく、後述するようにブロック共重合体(P)が特定の水素添加率で水素添加された水添ブロック共重合体(以下、「水添ブロック共重合体(Q)」と称すことがある。)であってもよい。
重合体ブロック(A)は、芳香族ビニル化合物由来の構造単位を含有する。かかる芳香族ビニル化合物としては、例えばスチレン、α-メチルスチレン、2-メチルスチレン、3-メチルスチレン、4-メチルスチレン、4-プロピルスチレン、4-t-ブチルスチレン、4-シクロヘキシルスチレン、4-ドデシルスチレン、2,4-ジメチルスチレン、2,4-ジイソプロピルスチレン、2,4,6-トリメチルスチレン、2-エチル-4-ベンジルスチレン、4-(フェニルブチル)スチレン、1-ビニルナフタレン、2-ビニルナフタレン、ビニルアントラセン、N,N-ジエチル-4-アミノエチルスチレン、ビニルピリジン、4-メトキシスチレン、モノクロロスチレン、ジクロロスチレン及びジビニルベンゼン等が挙げられる。これらの芳香族ビニル化合物は、1種を単独で又は2種以上を併用してもよい。これらの中でも、スチレン、α-メチルスチレン、4-メチルスチレンが好ましく、スチレンがより好ましい。
重合体ブロック(B)は、ファルネセン由来の構造単位(b1)(以下、単に「構造単位(b1)」ともいう)を含有する。
上記ファルネセンとしては、α-ファルネセン、又は下記式(1)で表されるβ-ファルネセンのいずれでもよいが、ブロック共重合体(II)の製造容易性の観点から、β-ファルネセンが好ましい。なお、α-ファルネセンとβ-ファルネセンとは組み合わせて用いてもよい。
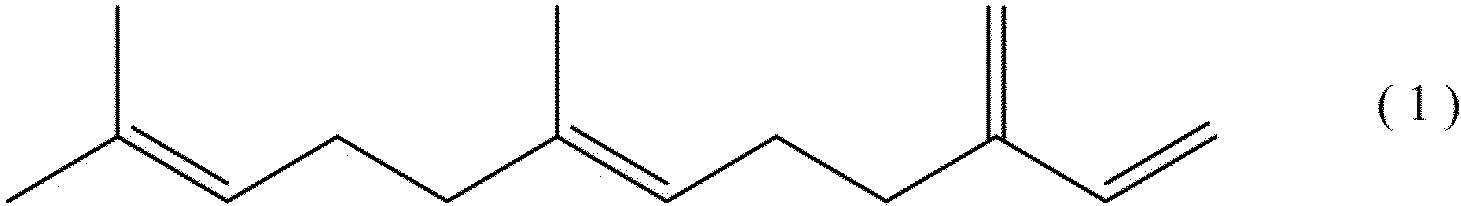
また、ファルネセンがバイオ由来である場合には、石油由来のブタジエン、イソプレンといったファルネセン以外の共役ジエンの使用量を抑制し、石油依存度を低減することができる。当該観点からは、重合体ブロック(B)中のファルネセン由来の構造単位(b1)の含有量は、50~100質量%が好ましく、60~100質量%がより好ましく、70~100質量%が更に好ましく、80~100質量%がより更に好ましい。
また、重合体ブロック(B)中に後述する構造単位(b2)を含有する場合、重合体ブロック(B)中のファルネセン由来の構造単位(b1)の含有量は1質量%以上が好ましく、10質量%以上がより好ましく、20質量%以上が更に好ましく、30質量%以上がより更に好ましく、50質量%以上が特に好ましい。
かかる共役ジエンとしては、例えばイソプレン、ブタジエン、2,3-ジメチル-ブタジエン、2-フェニル-ブタジエン、1,3-ペンタジエン、2-メチル-1,3-ペンタジエン、1,3-ヘキサジエン、1,3-オクタジエン、1,3-シクロヘキサジエン、2-メチル-1,3-オクタジエン、1,3,7-オクタトリエン、ミルセン及びクロロプレン等が挙げられる。これらは1種を単独で又は2種以上を併用してもよい。これらの中でも、イソプレン、ブタジエン及びミルセンが好ましく、イソプレン、ブタジエンがより好ましい。
重合体ブロック(B)中に構造単位(b2)を含有する場合、構造単位(b2)の含有量は、90質量%以下がより好ましく、80質量%以下が更に好ましく、70質量%以下がより更に好ましく、50質量%以下が特に好ましい。
ブロック共重合体(II)は、前述の重合体ブロック(A)及び重合体ブロック(B)に加えて、更にファルネセン以外の共役ジエン由来の構造単位(c2)を含有する重合体ブロック(C)を含むことができる。
重合体ブロック(C)は、ファルネセン由来の構造単位(c1)の含有量が0質量%以上かつ1質量%未満であり、ファルネセン以外の共役ジエン由来の構造単位(c2)の含有量が1~100質量%である重合体ブロックであることが好ましい。
ファルネセン以外の共役ジエン由来の構造単位(c2)を構成する共役ジエンとしては、それらの中でも、イソプレン、ブタジエン及びミルセンが好ましく、イソプレン及びブタジエンがより好ましい。これらは1種を単独で又は2種以上を併用してもよい。
重合体ブロック(C)中におけるファルネセン以外の共役ジエン由来の構造単位(c2)の含有量は、60~100質量%がより好ましく、80~100質量%が更に好ましく、90~100質量%がより更に好ましく、100質量%であることが特に好ましい。構造単位(c2)を含む重合体ブロック(C)を用いることにより、ブロック共重合体(P)は前述の構造単位(b1)及び構造単位(c2)の異なる共役ジエンを有することになり、当該ブロック共重合体(P)の水添ブロック共重合体(Q)を後述する架橋剤(III)により架橋しても、耐摩耗性に優れ、引張特性を維持しつつ引裂強度を発現することができ、引張特性と引裂強度の両立を図ることができる。
重合体ブロック(C)中における構造単位(c1)及び構造単位(c2)の合計含有量は、60質量%以上が好ましく、80質量%以上がより好ましく、100質量%が更に好ましい。
ブロック共重合体(II)は、重合体ブロック(A)及び重合体ブロック(B)をそれぞれ少なくとも1個含むブロック共重合体(P)又は水添ブロック共重合体(Q)である。
重合体ブロック(A)及び重合体ブロック(B)の結合形態は特に制限されず、直線状、分岐状、放射状又はそれらの2つ以上の組み合わせであってもよい。これらの中でも、各ブロックが直線状に結合した形態が好ましい。
直線状の結合形態としては、重合体ブロック(A)をA、重合体ブロック(B)をBで表したときに、(A-B)l、A-(B-A)m、又はB-(A-B)nで表される結合形態等を例示することができる。なお、前記l、m及びnはそれぞれ独立して1以上の整数を表す。
ブロック共重合体(II)が、重合体ブロック(A)及び重合体ブロック(B)をそれぞれ少なくとも1個含む場合、重合体ブロック(A)、重合体ブロック(B)、重合体ブロック(A)の順にブロックを有する結合形態であって、A-B-Aで表されるトリブロック共重合体であることが好ましい。
すなわち、ブロック共重合体(II)は、A-B-Aで表されるトリブロック共重合体であることが好ましく、当該トリブロック共重合体は未水添加物であっても水素添加物であってもよい。
ブロック共重合体(II)は、重合体ブロック(B)、重合体ブロック(A)、及び重合体ブロック(C)の順にブロックを有する構造(すなわち、B-A-Cの構造)であることが好ましい。
具体的には、ブロック共重合体(II)は、B-A-C-Aで表されるテトラブロック共重合体、B-A-C-A-Bで表されるペンタブロック共重合体、B-A-(C-A)p-B、B-A-(C-A-B)q、B-(A-C-A-B)r(p、q、rはそれぞれ独立して2以上の整数を表す)で表される共重合体であることが好ましく、中でもB-A-C-A-Bで表されるペンタブロック共重合体であることがより好ましい。
すなわち、ブロック共重合体(II)は、B-A-C-A-Bで表されるペンタブロック共重合体であることが好ましく、当該ペンタブロック共重合体は未水添加物であっても水素添加物であってもよい。
ブロック共重合体(II)が、重合体ブロック(A)、重合体ブロック(B)及び重合体ブロック(C)を含む場合、重合体ブロック(A)と重合体ブロック(B)との質量比[(A)/(B)]は、1/99~70/30であり、5/95~60/40が好ましく、10/90~50/50がより好ましく、20/80~40/60が更に好ましく、25/75~35/65がより更に好ましい。当該範囲内であると、柔軟性に優れ、優れたグリップ性を有する樹脂組成物を得ることができる。
また、ブロック共重合体(II)が、重合体ブロック(A)、重合体ブロック(B)及び重合体ブロック(C)を含む場合、ブロック共重合体中における、これら重合体ブロック(A)~(C)の合計含有量は、80質量%以上が好ましく、90質量%以上がより好ましく、95質量%以上が更に好ましく、100質量%がより更に好ましい。
・重合体ブロック(A)、重合体ブロック(B)及び重合体ブロック(C)を含むブロック共重合体(P)が水素添加された水添ブロック共重合体(Q)であり、
・重合体ブロック(C)が、ファルネセン由来の構造単位(c1)の含有量が0質量%以上かつ1質量%未満であり、ファルネセン以外の共役ジエン由来の構造単位(c2)の含有量が1~100質量%である重合体ブロックであり、
・重合体ブロック(A)と、重合体ブロック(B)と重合体ブロック(C)の合計量との質量比[(A)/((B)+(C))]が10/90~30/70であり、
・少なくとも2個の前記重合体ブロック(A)、少なくとも1個の前記重合体ブロック(B)、及び少なくとも1個の重合体ブロック(C)を含み、かつ少なくとも1個の重合体ブロック(B)を末端に有し、
・水添ブロック共重合体(Q)中の共役ジエン由来の構成単位における炭素-炭素二重結合の水素添加率が70モル%以上である。
ブロック共重合体(II)は、重合体ブロック(A)、重合体ブロック(B)及び重合体ブロック(C)のほか、本発明の効果を阻害しない限り、他の単量体で構成される重合体ブロックを含有していてもよい。
ブロック共重合体(II)が他の重合体ブロックを有する場合、その含有量は10質量%以下が好ましく、5質量%以下がより好ましい。
ブロック共重合体(II)が、例えば重合体ブロック(A)及び重合体ブロック(B)を含有するブロック共重合体(P)、あるいは重合体ブロック(C)を含む場合には重合体ブロック(A)、重合体ブロック(B)及び重合体ブロック(C)を含有するブロック共重合体(P)の場合、アニオン重合により得る重合工程により好適に製造できる。更に、ブロック共重合体(II)が水添ブロック共重合体(Q)である場合、前記ブロック共重合体(P)中の共役ジエン由来の構造単位における炭素-炭素二重結合を水素添加する工程により好適に製造できる。
ブロック共重合体(P)は、溶液重合法又は特表2012-502135号公報、特表2012-502136号公報に記載の方法等により製造することができる。これらの中でも溶液重合法が好ましく、例えば、アニオン重合やカチオン重合等のイオン重合法、ラジカル重合法等の公知の方法を適用できる。これらの中でもアニオン重合法が好ましい。アニオン重合法としては、溶媒、アニオン重合開始剤、及び必要に応じてルイス塩基の存在下、芳香族ビニル化合物、ファルネセン及び/又はファルネセン以外の共役ジエンを逐次添加して、ブロック共重合体(P)を得る。
アニオン重合開始剤としては、例えば、リチウム、ナトリウム、カリウム等のアルカリ金属;ベリリウム、マグネシウム、カルシウム、ストロンチウム、バリウム等のアルカリ土類金属;ランタン、ネオジム等のランタノイド系希土類金属;前記アルカリ金属、アルカリ土類金属、ランタノイド系希土類金属を含有する化合物等が挙げられる。中でもアルカリ金属及びアルカリ土類金属を含有する化合物が好ましく、有機アルカリ金属化合物がより好ましい。
重合に用いる有機アルカリ金属化合物の使用量は、ブロック共重合体(P)の分子量によっても異なるが、通常、芳香族ビニル化合物、ファルネセン及びファルネセン以外の共役ジエンの総量に対して0.01~3質量%の範囲である。
重合反応は、メタノール、イソプロパノール等のアルコールを重合停止剤として添加して停止できる。得られた重合反応液をメタノール等の貧溶媒に注いでブロック共重合体(P)を析出させるか、重合反応液を水で洗浄し、分離後、乾燥することによりブロック共重合体(P)を単離できる。
なお、前記ブロック共重合体(II)が、ポリマー鎖の片末端にのみ重合体ブロック(B)を有する場合には、直線状に結合するように他の各重合体ブロックを重合させた後で、最後に重合体ブロック(B)を製造する方法により得てもよい。
〔i〕重合体ブロック(B)、重合体ブロック(A)、重合体ブロック(C)、及び重合体ブロック(A)をこの順に重合する方法、及び
〔ii〕重合体ブロック(B)、重合体ブロック(A)、及び重合体ブロック(C)をこの順に重合し、重合体ブロック(C)の末端同士を、カップリング剤を用いてカップリングすることにより製造する方法、等が挙げられる。
本実施態様においては、効率的に製造する観点から、カップリング剤を用いる後者の方法〔ii〕が好ましい。
変性したブロック共重合体の場合、後述の水素添加工程の前に、前記ブロック共重合体(P)を変性してもよい。導入可能な官能基としては、例えばアミノ基、アルコキシシリル基、水酸基、エポキシ基、カルボキシル基、カルボニル基、メルカプト基、イソシアネート基、酸無水物基等が挙げられる。
ブロック共重合体(P)の変性方法としては、例えば、重合停止剤を添加する前に、重合活性末端と反応し得る四塩化錫、テトラクロロシラン、ジクロロジメチルシラン、ジメチルジエトキシシラン、テトラメトキシシラン、テトラエトキシシラン、3-アミノプロピルトリエトキシシラン、テトラグリシジル-1,3-ビスアミノメチルシクロヘキサン、2,4-トリレンジイソシアネート等のカップリング剤や、4,4’-ビス(ジエチルアミノ)ベンゾフェノン、N-ビニルピロリドン等の重合末端変性剤、又は特開2011-132298号公報に記載のその他の変性剤を添加する方法が挙げられる。また、単離後の共重合体に無水マレイン酸等をグラフト化して用いることもできる。
官能基が導入される位置はブロック共重合体(P)の重合末端でも、側鎖でもよい。また上記官能基は1種又は2種以上を組み合わせてもよい。上記変性剤は、アニオン重合開始剤1モルに対して、0.01~10モル当量の範囲であることが好ましい。
ブロック共重合体(II)は、前記方法により得られたブロック共重合体(P)又は変性されたブロック共重合体を水素添加する工程に付すことにより、水添ブロック共重合体(Q)としてよもよい。
水素添加する方法は公知の方法を用いることができる。例えば、水素添加反応に影響を及ぼさない溶媒にブロック共重合体(P)を溶解させた溶液に、チーグラー系触媒;カーボン、シリカ、けいそう土等に担持されたニッケル、白金、パラジウム、ルテニウム又はロジウム金属触媒;コバルト、ニッケル、パラジウム、ロジウム又はルテニウム金属を有する有機金属錯体等を、水素添加触媒として存在させて水素化反応を行う。
水素添加工程においては、前記したブロック共重合体(P)の製造方法によって得られたブロック共重合体を含む重合反応液に水素添加触媒を添加して水素添加反応を行ってもよい。本発明において水素添加触媒は、パラジウムをカーボンに担持させたパラジウムカーボンが好ましい。
水素添加反応において、水素圧力は0.1~20MPaが好ましく、反応温度は100~200℃が好ましく、反応時間は1~20時間が好ましい。
なお、上記水素添加率はブロック共重合体(P)中に存在する全ての共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率である。
水添ブロック共重合体(Q)が重合体ブロック(B)及び重合体ブロック(C)を含有する場合、重合体ブロック(B)と重合体ブロック(C)との共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率は異なることが好ましい。重合体ブロック(B)と重合体ブロック(C)との共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率の差は、例えば、1~20モル%でもよく、3~15モル%でもよく、5~10モル%であってもよい。上記水素添加率が異なることによって、後述する架橋剤(III)による架橋度合いが重合ブロック(B)及び(C)ごとに異なるものとなり、これによって水添ブロック共重合体(Q)中に硬柔の異なる部分を存在させ、引裂強度や引張特性等の優れた効果を発現させることができると考えられる。また、顕著に優れた引張特性を発現させる観点から、重合体ブロック(B)よりも重合体ブロック(C)の上記水素添加率が高い方がより好ましい。
なお本明細書において、水添ブロック共重合体(Q)中の重合体ブロック(B)及び重合体ブロック(C)は水素添加されているが、水素添加前と同様にこれらを「重合体ブロック(B)」及び「重合体ブロック(C)」と表記する。
ブロック共重合体(P)が重合体ブロック(C)を含有する場合、水添ブロック共重合体(Q)中の重合体ブロック(C)の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率は、好ましくは95~99.9モル%であり、より好ましくは96~99.5モル%であり、更に好ましくは97~99.5モル%である。
なお、水素添加率は、ブロック共重合体(P)及び水素添加後の水添ブロック共重合体(Q)の1H-NMRを測定することにより算出できる。
ブロック共重合体(P)の分子量分布(Mw/Mn)は1~6が好ましく、1~4がより好ましく、1~3が更に好ましく、1~2がより更に好ましい。分子量分布が前記範囲内であると、ブロック共重合体(P)の粘度のばらつきが小さく、取り扱いが容易である。
水添ブロック共重合体(Q)の分子量分布(Mw/Mn)は1~6が好ましく、1~4がより好ましく、1~3が更に好ましく、1~2がより更に好ましい。分子量分布が前記範囲内であると、水添ブロック共重合体(Q)の粘度のばらつきが小さく、取り扱いが容易である。
なお、本明細書におけるピークトップ分子量(Mp)及び分子量分布(Mw/Mn)は後述する実施例に記載した方法で測定した値を意味する。
水のはじきやすさは、より具体的には後述する実施例に記載の方法で評価することができる。
本実施態様の樹脂組成物は、架橋剤(III)を含有する。
架橋剤(III)としては、共役ジエン由来の構造単位における炭素-炭素二重結合若しくはそのα-メチレンのみで架橋反応するものであることが好ましい。ここで、α-メチレンは炭素-炭素二重結合に隣接するメチレンを示す。この様な架橋剤(III)を含有することで、ブロック共重合体(II)中の(水添ブロック共重合体の場合は、該共重合体中に残存する)上記炭素-炭素二重結合若しくは上記炭素-炭素二重結合のα-メチレンのみが架橋され、優れた柔軟性を維持しつつ、滑りにくく耐摩耗性に優れるものとすることができ、良好な引張特性及び引裂強度も期待できる。
ラジカル発生剤としては、例えば、ジクミルペルオキシド、ジt-ブチルペルオキシド、2,5-ジメチル-2,5-ジ(t-ブチルペルオキシ)ヘキサン、2,5-ジメチル-2,5-ジ(t-ブチルペルオキシ)ヘキシン-3、1,3-ビス(t-ブチルペルオキシイソプロピル)ベンゼン、α,α’-ビス(t-ブチルペルオキシ)ジイソプロピルベンゼン、3,3,5-トリメチルシクロヘキサン、n-ブチル-4,4-ビス(t-ブチルペルオキシ)バレレート、ベンゾイルペルオキシド、p-クロロベンゾイルペルオキシド、2,4-ジクロロベンゾイルペルオキシド、t-ブチルペルオキシベンゾエート、t-ブチルペルオキシイソプロピルカーボネート、ジアセチルペルオキシド、ラウロイルペルオキシド、t-ブチルクミルペルオキシド等の有機過酸化物が挙げられる。これらは、1種を単独で又は2種以上を併用してもよい。
硫黄含有化合物としては、例えば、一塩化硫黄、二塩化硫黄、ジスルフィド化合物、トリアジンチオール類等が挙げられる。
架橋剤(III)としては、その他にアルキルフェノール樹脂、臭素化アルキルフェノール樹脂等のフェノール系樹脂;p-キノンジオキシムと二酸化鉛、p,p’-ジベンゾイルキノンジオキシムと四酸化三鉛の組み合わせ等も使用することができる。
前記ゴム(I)と前記ブロック共重合体(II)との質量比[(I)/(II)]が、99/1~55/45である。質量比[(I)/(II)]が上記範囲外であると、優れた耐摩耗性を得ることができない。また、質量比が上記範囲内であれば、滑りにくさの発現がより顕著なものとなる。すなわち、上記質量比であれば、より滑りにくく、耐摩耗性に優れる成形体とすることができる樹脂組成物となる。更に、ドライグリップ性及びウェットグリップ性といった滑りにくさを飛躍的に向上させ、かつ耐摩耗性を更に優れさせるには、上記質量比は99/1~60/40が好ましく、99/1~65/35が好ましく、99/1~70/30が好ましく、95/5~70/30が好ましい。
ブロック共重合体(II)を含有することにより、樹脂組成物に柔軟性が付与されるため、ブロック共重合体(II)の含有割合が高いほどグリップ性が向上する傾向にある。しかし、上記質量比[(I)/(II)]が示すような特定の範囲において、ブロック共重合体(II)よりもゴム(I)の含有割合が高くなるに従い、意外にもグリップ性が向上することがわかった。加えて、優れた耐摩耗性も発現されるので、本実施態様の樹脂組成物は、滑りにくさと耐摩耗性の両方に優れた成形体とすることができる。
(シリカ)
本実施態様の樹脂組成物は、シリカを含有することが好ましい。
シリカとしては、例えば、湿式シリカ(含水ケイ酸)、乾式シリカ(無水ケイ酸)、ケイ酸カルシウム、ケイ酸アルミニウム等が挙げられる。これらシリカの中でも、成形加工性、得られる成形体の機械強度及び耐摩耗性を一層向上させ、ドライグリップ性及びウェットグリップ性といった滑りにくさを一層向上させる観点から、湿式シリカが好ましい。これらシリカは、1種を単独で又は2種以上を併用してもよい。
上記シリカの比表面積は、例えばBET法により測定することができる。
本実施態様の樹脂組成物は、シランカップリング剤を含有することが好ましい。
シランカップリング剤としては、例えば、スルフィド系化合物、メルカプト系化合物、ビニル系化合物、アミノ系化合物、グリシドキシ系化合物、ニトロ系化合物、クロロ系化合物等が挙げられる。
また、シランカップリング剤は、シリカとの反応性の観点から、アルコキシシリル基を有ることが好ましく、中でもメトキシシラン、エトキシシランであることが好ましい。これらシランカップリング剤は、1種を単独で又は2種以上を併用してもよい。
メルカプト系化合物としては、例えば3-メルカプトプロピルトリメトキシシラン、3-メルカプトプロピルトリエトキシシラン、3-メルカプトプロピル-ジ(トリデカン-1-オキシ-13-ペンタ(エチレンオキサイド))エトキシシラン、2-メルカプトエチルトリメトキシシラン及び2-メルカプトエチルトリエトキシシラン等が挙げられる。
アミノ系化合物としては、例えば、3-アミノプロピルトリエトキシシラン、3-アミノプロピルトリメトキシシラン、3-(2-アミノエチル)アミノプロピルトリエトキシシラン、及び3-(2-アミノエチル)アミノプロピルトリメトキシシラン等が挙げられる。
グリシドキシ系化合物としては、例えば、γ-グリシドキシプロピルトリエトキシシラン、γ-グリシドキシプロピルトリメトキシシラン、γ-グリシドキシプロピルメチルジエトキシシラン、及びγ-グリシドキシプロピルメチルジメトキシシラン等が挙げられる。
ニトロ系化合物としては、例えば、3-ニトロプロピルトリメトキシシラン、及び3-ニトロプロピルトリエトキシシラン等が挙げられる。
クロロ系化合物としては、例えば、3-クロロプロピルトリメトキシシラン、3-クロロプロピルトリエトキシシラン、2-クロロエチルトリメトキシシラン、及び2-クロロエチルトリエトキシシラン等が挙げられる。
これらシランカップリング剤の中でも、耐摩耗性の観点から、ビス(3-トリエトキシシリルプロピル)ジスルフィド、ビス(3-トリエトキシシリルプロピル)テトラスルフィド、3-メルカプトプロピルトリメトキシシラン、及び3-メルカプトプロピルトリエトキシシランが好ましい。
本実施態様の樹脂組成物は、ポリオレフィン系樹脂を含有しないか又はブロック共重合体(II)100質量部に対するポリオレフィン系樹脂の含有量が10質量部未満であることが好ましい。当該含有量が10質量部未満であれば、好適に溶融混練することができる。本実施態様の樹脂組成物は、ポリオレフィン系樹脂を含有しないことがより好ましい。
含有させることができるポリオレフィン系樹脂としては、例えば、ポリエチレン、ポリプロピレン、ポリブテン-1、ポリヘキセン-1、ポリ-3-メチル-ブテン-1、ポリ-4-メチル-ペンテン-1、エチレン-酢酸ビニル共重合体、エチレン-アクリル酸共重合体等が挙げられる。
本実施態様の樹脂組成物は、架橋促進剤を含有していてもよい。
架橋促進剤としては、例えば、N,N-ジイソプロピル-2-ベンゾチアゾール-スルフェンアミド、2-メルカプトベンゾチアゾール、ジ-2-ベンゾチアゾリルジスルフィド、2-(4-モルホリノジチオ)ベンゾチアゾール等のチアゾール類;ジフェニルグアニジン、トリフェニルグアニジン等のグアニジン類;ブチルアルデヒド-アニリン反応物、ヘキサメチレンテトラミン-アセトアルデヒド反応物等のアルデヒド-アミン系反応物ないしはアルデヒド-アンモニア系反応物;2-メルカプトイミダゾリン等のイミダゾリン類;チオカルバニリド、ジエチルウレア、ジブチルチオウレア、トリメチルチオウレア、ジオルソトリルチオウレア等のチオウレア類;テトラメチルチウラムモノスルフィド、テトラメチルチウラムジスルフィド、ペンタメチレンチウラムテトラスルフィド等のチウラムモノないしポリスルフィド類;ジメチルジチオカルバミン酸亜鉛、エチルフェニルジチオカルバミン酸亜鉛、ジメチルジチオカルバミン酸ナトリウム、ジメチルジチオカルバミン酸セレン、ジエチルジチオカルバミン酸テルル等のチオカルバミン酸塩類;ジブチルキサントゲン酸亜鉛等のキサントゲン酸塩類;サルチル酸等が挙げられる。これらの架橋促進剤は、1種を単独で又は2種以上を併用してもよい。
架橋促進剤の含有量は、ゴム(I)及びブロック共重合体(II)の合計100質量部に対して、0.05~15.0質量部が好ましく、0.1~5.0質量部がより好ましく、0.5~4.0質量部が更に好ましい。
本実施態様の樹脂組成物は、上述のシリカ以外のフィラーを含有してもよい。前記フィラーとしては、無機フィラー及び/又は有機フィラーが利用可能である。好ましい無機フィラーとしては、ケイ酸塩化合物及び金属酸化物等が好ましく例示される。ケイ酸塩化合物は、好ましくはカオリン、タルク、クレー、パイロフィライト、マイカ、モンモリロナイト、ベントナイト、ワラストナイト、セピオライト、ゾノトライト、ゼオライト、ケイソウ土、及びハロイサイトからなる群から選択される1種以上のケイ酸塩化合物である。また、金属酸化物は、酸化チタン、酸化鉄、酸化マグネシウム、酸化アルミニウム、酸化セリウム、酸化アンチモン、酸化スズ、酸化鉛、酸化クロム、酸化コバルト、酸化タングステン、及び酸化銅からなる群から選択される1種以上の金属酸化物である。
上記フィラーの含有量は、ゴム(I)及びブロック共重合体(II)の合計含有量100質量部に対して、好ましくは0~120質量部であり、0.5~80質量部であってもよく、1~50質量部であってもよく、1~30質量部であってもよい。
本実施態様の樹脂組成物は、発明の効果を損なわない範囲で、軟化剤を含有してもよい。しかし、上記樹脂組成物は、前述の構成によって成形加工に十分な流動性を有していることから軟化剤を含有しなくともよい。特に、軟化剤の中でもパラフィン系、ナフテン系及び芳香族系等のプロセスオイル、ミネラルオイル、ホワイトオイル等のオイル系軟化剤は、成形体とした際に経時的にオイルが染み出てくるおそれがあるため樹脂組成物に含有させないことが好ましい。
含有させることができる軟化剤としては、例えばジオクチルフタレート、ジブチルフタレート等のフタル酸誘導体;エチレンとα-オレフィンとの液状コオリゴマー;流動パラフィン;ポリブテン;低分子量ポリイソブチレン;液状ポリブタジエン、液状ポリイソプレン、液状β-ポリファルネセン、液状ポリイソプレン/ブタジエン共重合体、液状イソプレン/β-ファルネセン、液状ブタジエン/β-ファルネセン、液状スチレン/ブタジエン共重合体、液状スチレン/イソプレン共重合体等の液状ポリジエン及びその水添物又はその変性物等が挙げられる。
軟化剤を含有する場合の含有量は、上記樹脂組成物中、0.1~30質量%が好ましく、0.5~20質量%がより好ましく、1.0~15質量%が更に好ましい。
本実施態様の樹脂組成物には、本発明の効果を損なわない範囲において、上述したもの以外のその他の添加剤を添加することができる。その他の添加剤としては、例えば、熱老化防止剤、酸化防止剤、光安定剤、帯電防止剤、離型剤、難燃剤、発泡剤、顔料、染料、増白剤等が挙げられる。これらの添加剤は、1種を単独で又は2種以上を併用してもよい。
本実施態様の樹脂組成物の製造方法は、例えば、前記架橋剤(III)及び必要に応じて添加される架橋促進剤を除く各成分を溶融混練した後、後述の架橋方法で架橋することにより製造することができる。
架橋剤(III)及び必要に応じて添加される架橋促進剤を除く各成分を溶融混練する方法に特に制限はなく、ゴム(I)、ブロック共重合体(II)及びその他の任意成分を、同時に混練装置、例えば、一軸押出機、多軸押出機、バンバリーミキサー、ブラベンダーミキサー、オープンロール、加熱ロール、各種ニーダー等に供給して溶融混練する方法が挙げられる。また、ゴム(I)、ブロック共重合体(II)及びその他の任意成分を別々の仕込み口から供給して溶融混練する方法、あるいはブロック共重合体(II)とシリカ及びシランカップリング剤等の一部の任意成分とを予め溶融混練し、当該溶融混練物とその他の任意成分と溶融混練する方法等であってもよい。
溶融混練時の温度は、通常20~270℃の範囲で任意に選択することができる。
(滑りにくさ)
本実施態様の樹脂組成物は、滑りにくさを、ASTM D-1894に準じて測定される静摩擦係数により評価することができる。静摩擦係数の好ましい数値範囲は、用いるゴム(I)の種類によって異なり一概には特定できないが、静摩擦係数の数値が大きいほど滑りにくいことを示す。例えば、ゴム(I)としてSBRを用いた場合、静摩擦係数が、ドライ条件で、好ましくは4.5以上、より好ましくは5.0以上である。また、ウェットの件で、好ましくは1.5以上、より好ましくは2.0以上である。
ドライ条件及びウェット条件については、後述する実施例に記載した測定条件を意味する。
本実施態様の樹脂組成物は、耐摩耗性を、JIS K 6264-2:2005に準じてDIN摩耗試験(回転円筒型摩耗試験機)により測定される摩耗量により評価することができる。摩耗量は、好ましくは200mm3以下、より好ましくは190mm3以下であり、更には180mm3以下、170mm3以下とすることもできる。
本実施態様の樹脂組成物は、架橋剤(III)として硫黄及び/又は硫黄含有化合物を用いた場合、加硫速度に優れる、すなわち、加硫に要する時間(90%加硫時間)が短いことが期待できる。
加硫速度が速いと、加硫する時の樹脂組成物の粘度上昇が速く、樹脂組成物中の硫黄及び/又は硫黄含有化合物である加硫剤や、架橋促進剤、架橋助剤、酸化防止剤、老化防止剤、軟化剤等の可塑剤等が、ゴム(I)やブロック共重合体(II)の表面へブリードしにくくなるため、加工用の金型の汚染を抑制することができる。
本明細書において樹脂組成物の加硫速度は、JIS K 6300-2:2001にしたがって、振動式加硫試験機(キュラストメーター)を用いて樹脂組成物の160℃におけるトルクを測定し、90%加硫量に至るまでに要する時間(90%加硫時間)とする。樹脂組成物の加硫速度Tc(90)としては、13分以下が好ましく、12分以下がより好ましい。下限は特に限定されないが、通常1分以上である。
本実施態様の樹脂組成物は、ISO 1183:1987に準じて測定される比重が、好ましくは0.88~1.30、より好ましくは0.90~1.20である。上記範囲であれば樹脂組成物からなる成形体は、実用性に欠けることはない。例えば樹脂組成物を履物用靴底に用いる場合、重くなり過ぎず、履物の軽量化も期待できる。
本実施態様の樹脂組成物は、JIS K 6253-2:2012のタイプAデュロメータ法による硬度(以下、「A硬度」ともいう)が、好ましくは90以下、より好ましくは85以下である。また、A硬度は、好ましくは25以上であり、より好ましくは30以上であり、更に好ましくは35以上である。A硬度が上記範囲であれば成形加工性が良好となり、また良好な柔軟性から摩擦力が高くなり、グリップ性及び耐摩耗性にも優れたものとなる。
本実施態様の樹脂組成物は、JIS K 6251:2010に準じて測定される切断時引張強さ(引張破断強度)が、好ましくは5.0MPa以上、より好ましくは10.0MPa以上、更に好ましくは13.0MPa以上である。引張破断強度が5.0MPa以上であれば、引張特性が良好なものとなる。
本実施態様の樹脂組成物は、JIS K 6251:2010に準じて測定される切断時伸び(引張破断伸び)が、好ましくは100%以上であり、より好ましくは150%以上であり、更に好ましくは200%以上であり、より更に好ましくは370%以上である。引張破断伸びが100%以上であれば、引張特性が良好なものとなる。
本実施態様の樹脂組成物は、JIS K 6252-1:2015に準じて測定される引裂き強さ(引裂強度)が、好ましくは2.0kN/m以上、より好ましくは2.5kN/m以上、更に好ましくは3.0kN/m以上である。
本実施態様における成形体は、上述の樹脂組成物を用いて得られるものである。
成形体の形状は、上述の樹脂組成物を用いて製造できる成形体であればいずれでもよく、例えばペレット、フィルム、シート、プレート、パイプ、チューブ、棒状体、粒状体等、種々の形状に成形することができる。この成形体の製造方法は特に制限はなく、従来からの各種成形法、例えば、射出成形、ブロー成形、プレス成形、押出成形、カレンダー成形等により成形することができる。上述の樹脂組成物は、特にプレス成形体を好適に得ることができる。
上記成形体は、滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性のバランスに優れることも期待できるため、これら特性が要求される成形品に用いることができる。特に、本実施態様の樹脂組成物を少なくとも一部に用いた靴底等の成形品として好適に用いることができる。
具体的には、ハイキングブーツ、サンダル、安全靴、登山靴、マラソンシューズ、地下足袋及び長靴等の履物用靴底;水中眼鏡、シュノーケル、スキーブーツ及びスキー・スノーボード等のスポーツ用品;ペン及びはさみ等の筆記用具、ドライバー、ペンチ及びレンチ等の工具及び電気工具、歯ブラシ及びキッチン用品(包丁及びヘラ等)等の水周り用品、ゴルフクラブ、スキーのストック、自転車、バイク等のスポーツ及びフィットネス等に用いられる器具、並びにナイフ等の各種グリップ;冷蔵庫、掃除機及び防水ボディー(携帯電話等)等の家電の部品;サイドモール、ラック・オピニオンブーツ、サスペンションブーツ、等速ジョイントブーツ、ウェザーストリップ、マットガード、フロアーマット、アームレスト、ベルトラインモール、フラッシュマウント及び自動車内外装(ギア及びノブ等)の自動車用品;タイヤ、特にウィンタータイヤ、スタッドレスタイヤ、オールシーズンタイヤ、低燃費タイヤ及び重荷重用タイヤ等のトレッド部材;コピー機送りローラー及び巻き取りローラー等の事務機器;ソファー及びチェアーシート等の家具に用いられる部品;スイッチカバー、ストッパー、キャスター及び足ゴム等のゴム部品;被覆合板及び被覆鋼板等の建材;工業用ベルト、工業用ゴムホース等の工業用部材等に好適に用いることができる。
・ゴム(I):SBR
製品名JSR 1502、JSR株式会社製、スチレンブタジエン共重合ゴム(E-SBR)、乳化重合法で製造、スチレン含有量=23.5質量%
・水添ブロック共重合体(II-1)
後述の製造例1の水添ブロック共重合体(II-1)
・水添ブロック共重合体(II-2)
後述の製造例2の水添ブロック共重合体(II-2)
・未水添ブロック共重合体(II’-1)
後述の製造例3の未水添ブロック共重合体(II’-1)
・硫黄:微粉硫黄200メッシュ、鶴見化学工業株式会社製
<架橋促進剤>
・架橋促進剤(1):製品名ノクセラーCZ、大内新興化学工業株式会社製、N-シクロヘキシル-2-ベンゾチアゾリルフェンアミド
・架橋促進剤(2):製品名ノクセラーTT、大内新興化学工業株式会社製、テトラメチルチウラムジスルフィド)
・シリカ(1):製品名ULTRASIL7000GR、エボニック デグサ ジャパン製、湿式シリカ、BET法比表面積175m2/g
・シリカ(2):製品名Nipsil VN3、東ソー・シリカ株式会社製、湿式シリカ、BET法比表面積200m2/g
・シリカ(3):製品名Nipsil ER、東ソー・シリカ株式会社製、湿式シリカ、BET法比表面積70-100m2/g
・シランカップリング剤(1):製品名Si75、エボニック デグサ ジャパン製、ビス(3-トリエトキシシリルプロピル)ジスルフィド
・シランカップリング剤(2):製品名Si69、エボニック デグサ ジャパン製、ビス(3-トリエトキシシリルプロピル)テトラスルフィド
・シランカップリング剤(3):製品名Silquest A189、モメンティブ製、3-メルカプトプロピルトリメトキシシラン
・酸化亜鉛:酸化亜鉛1種、堺化学工業株式会社製
・ステアリン酸:製品名ルナックS-20、花王株式会社製
製造例で得られた重合体についての各測定方法の詳細は次のとおりである。
(1)ピークトップ分子量及び分子量分布の測定
スチレンブロック及び重合体のピークトップ分子量(Mp)、及び分子量分布(Mw/Mn)は、GPC(ゲルパーミエーションクロマトグラフィー)により標準ポリスチレン換算分子量で求め、分子量分布のピークの頂点の位置からピークトップ分子量(Mp)を求めた。測定装置及び条件は、以下のとおりである。
・装置 :東ソー株式会社製 GPC装置「HLC-8320GPC」
・分離カラム :東ソー株式会社製 カラム「TSKgelSuperHZ4000」
・溶離液 :テトラヒドロフラン
・溶離液流量 :0.7mL/min
・サンプル濃度:5mg/10mL
・カラム温度 :40℃
(2-1)ブロック共重合体(P)、及び水素添加後のブロック共重合体(II)(「水添ブロック共重合体(II)」と表記する)をそれぞれ重クロロホルム溶媒に溶解し、日本電子株式会社製「Lambda-500」を用いて50℃で1H-NMRを測定した。
水添ブロック共重合体(II)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率は、得られたスペクトルの4.5~6.0ppmに現れる炭素-炭素二重結合が有するプロトンのピークから、下記式により算出した。
水素添加率(モル%)={1-(水添ブロック共重合体(II)1モルあたりに含まれる炭素-炭素二重結合のモル数)/(ブロック共重合体(P)1モルあたりに含まれる炭素-炭素二重結合のモル数)}×100
(2-2)また、重合体ブロック(B)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率、及び重合体ブロック(C)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率について、上記で得られたそれぞれのスペクトルに現れるプロトンピークから、上記と同様に算出した。
無色透明かつ表面が平滑である石英板(5cm×5cm)に食紅を添加した水(1mL)を垂らし、その上に製造例で製造した重合体からなる成形体(直径25mm、厚み2mmの丸型平面状シート)を設置し、該成形体に引張試験機で2kgの荷重をかけた。なお、食紅は観察容易の観点から水に添加したものであり、水のはじきやすさに影響を与えるものではない。
成形体と石英板との接地面における水の様子を石英板側から目視で観察し、下記評価基準A~Cにより評価した。
A:上記接地面に食紅が見えない
B:上記接地面に食紅が少し見える
C:上記接地面に食紅が多く見える
また、成形体と石英板との接地面における水で覆われている割合(被覆率%)を、石英板側から撮影した観察画像を画像解析ソフトウェア(三谷商事株式会社製、製品名「WinROOF ver.5.7.2」)により画像解析し、下記式から定量した。
被覆率%=[(成形体と石英板との接地面における赤い領域の面積)/(成形体と石英板との接地面の面積)]×100
水添ブロック共重合体(II-1):
窒素置換し、乾燥させた耐圧容器に、溶媒としてシクロヘキサン50.0kg、アニオン重合開始剤としてsec-ブチルリチウム(10.5質量%シクロヘキサン溶液)0.1905kg、ルイス塩基としてテトラヒドロフラン0.40kgを仕込み、50℃に昇温した後、β-ファルネセン6.34kgを加えて2時間重合を行い、引き続いてスチレン(1)2.50kgを加えて1時間重合させ、更にブタジエン3.66kgを加えて1時間重合を行った。続いてこの重合反応液にカップリング剤としてジクロロジメチルシラン0.02kgを加え1時間反応させることで、ポリ(β-ファルネセン)-ポリスチレン-ポリブタジエン-ポリスチレン-ポリ(β-ファルネセン)ペンタブロック共重合体(P1)を含む反応液を得た。
この反応液に、水素添加触媒としてパラジウムカーボン(パラジウム担持量:5質量%)を前記ブロック共重合体(P1)に対して5質量%添加し、水素圧力2MPa、150℃の条件で10時間反応を行った。放冷、放圧後、濾過によりパラジウムカーボンを除去し、濾液を濃縮し、更に真空乾燥することにより、ポリ(β-ファルネセン)-ポリスチレン-ポリブタジエン-ポリスチレン-ポリ(β-ファルネセン)ペンタブロック共重合体の水素添加物(II-1)(以下、「水添ブロック共重合体(II-1)」という。)を得た。
得られた水添ブロック共重合体(II-1)について、上記の物性を測定した。結果を表1に示す。
水添ブロック共重合体(II-2):
窒素置換し、乾燥させた耐圧容器に、溶媒としてシクロヘキサン50.0kg、アニオン重合開始剤としてsec-ブチルリチウム(10.5質量%シクロヘキサン溶液)0.0413kgを仕込み、50℃に昇温した後、スチレン(1)1.12kgを加えて1時間重合を行い、続いてβ-ファルネセン10.25kgを加えて2時間重合を行い、更にスチレン(2)1.12kgを加えて1時間重合を行い、ポリスチレン-ポリ(β-ファルネセン)-ポリスチレントリブロック共重合体(P2)を含む反応液を得た。
この反応液に、水素添加触媒としてパラジウムカーボン(パラジウム担持量:5質量%)を前記ブロック共重合体(P2)に対して5質量%添加し、水素圧力2MPa、150℃の条件で10時間反応を行った。放冷、放圧後、濾過によりパラジウムカーボンを除去し、濾液を濃縮し、更に真空乾燥することにより、ポリスチレン-ポリ(β-ファルネセン)-ポリスチレントリブロック共重合体の水素添加物(以下、「水添ブロック共重合体(II-2)」ともいう)を得た。
水添ブロック共重合体(II-2)について、上記の物性を測定した。結果を表1に示す。
未水添ブロック共重合体(II’-1):
窒素置換し、乾燥させた耐圧容器に、溶媒としてシクロヘキサン50.0kg、アニオン重合開始剤としてsec-ブチルリチウム(10.5質量%のシクロヘキサン溶液)0.1010kgを仕込み、50℃に昇温した後、スチレン(1)1.70kgを加えて1時間重合を行い、ルイス塩基としてN,N,N’,N’-テトラメチルエチレンジアミン(TMEDA)0.065kgを加え、続いてイソプレン13.30kgを加えて2時間重合を行い、更にスチレン(2)1.70kgを加えて1時間重合を行い、スチレン-イソプレン-スチレントリブロック共重合体(P’1)を含む反応液を得た。
この反応液に、メタノール200mLを加えることによって重合反応を停止させた。得られた混合液を水洗し、次いで大量のメタノール中に再沈させることによって、スチレン-イソプレン-スチレントリブロック共重合体(以下、「未水添ブロック共重合体(II’-1)」ともいう)を得た。
未水添ブロック共重合体(II’-1)について、上記の物性を測定した。結果を表1に示す。

*1:(A)/(B)は、重合体ブロック(A)の含有量と重合体ブロック(B)の含有量との質量比を示す。
*2:(A)/((B)+(C))は、重合体ブロック(A)の含有量と、重合体ブロック(B)及び重合体ブロック(C)それぞれの含有量の合計との質量比を示す。
*3:(b1)/(B)は、重合体ブロック(B)中のファルネセン由来の構造単位(b1)の含有量(質量%)を示す。
*4:((b1)+(c1))/((B)+(C))は、重合体ブロック(B)及び重合体ブロック(C)の合計量に対する構造単位(b1)及び構造単位(c1)の合計含有量(質量%)を示す。
*5: F-St-Bd-St-Fは、ポリ(β-ファルネセン)-ポリスチレン-ポリブタジエン-ポリスチレン-ポリ(β-ファルネセン)ペンタブロック共重合体を示す。
St-F-Stは、ポリスチレン-ポリ(β-ファルネセン)-ポリスチレントリブロック共重合体を示す。
St-Ip-Stは、ポリスチレン-ポリイソプレン-ポリスチレントリブロック共重合体を示す。
*6:(II)の水素添加率は、ブロック共重合体(P)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率を示す。
*7:重合体ブロック(B)の水素添加率は、重合体ブロック(B)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率を示す。ただし、重合体ブロック(B)を有する場合のみ示す。
*8:重合体ブロック(C)の水素添加率は、重合体ブロック(C)中の共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率を示す。ただし、重合体ブロック(C)を有する場合のみ示す。
(1)溶融混練(樹脂組成物の製造)
表3に記載した配合割合(質量部)に従って、架橋剤(III)及び架橋促進剤を除く各成分を、表2に示す条件にてミキシングロール(14インチロール 関西ロール株式会社製)に投入して溶融混練した(工程1及び2)。その後、溶融混練物をオープンロールから取り出した(工程3)。
次いで、この溶融混練物をミキシングロールに入れ(工程4)、架橋剤(III)及び架橋促進剤を投入し(工程5)、再混練して、取り出す(工程6)ことで樹脂組成物を得た。
(2)成形(成形体の製造)
また、上記で得られた樹脂組成物をプレス成形(160℃、7~40分)して架橋させつつゴムシート(厚み0.5mm)を得た。
得られた樹脂組成物又はゴムシートを用い、下記の評価方法に基づき物性を評価した。結果を表3に示す。

(静摩擦係数)
(1)ドライ
ASTM D-1894に準じて、実施例及び比較例で得られた樹脂組成物のゴムシート表面の静摩擦係数を測定した。
上記ゴムシートから縦110mm、横63.5mm、厚さ0.5mmの試験片を切り出してセル(重さ200g、63.5mm×63.5mm)に巻き付け、摩擦係数測定装置の試験片台が水平になるようにオートグラフヘッドに固定した。摩擦台の材質をアルミとし、引張速度150mm/分で静摩擦係数を測定した。静摩擦係数の数値が大きいほど摩擦力が大きくて滑りにくく、ドライグリップ性に優れる。
(2)ウェット
摩擦台に蒸留水を1cc垂らした以外は、上記(1)ドライと同様の方法で静摩擦係数を測定した。静摩擦係数の数値が大きいほど摩擦力が大きくて滑りにくく、ウェットグリップ性に優れる。
JIS K 6264-2:2005に準じてDIN摩耗試験によりDIN摩耗量を測定した。数値が小さいほど耐摩耗性に優れる。
(加硫速度)
加硫速度(Tc90)の測定は、JIS K 6300-2に準じ、振動式加硫試験機(キュラストメーター)を使用して、実施例及び比較例において得られた上記樹脂組成物の160℃におけるトルクを測定し、90%加硫量にいたる加硫時間を加硫速度(t90)とした。数値が小さいほど加硫速度に優れる。
ISO 1183:1987に準じて測定した。全ての実施例及び比較例において比重は1.1g/cm3であった。
(硬度)
実施例及び比較例で得られた樹脂組成物のゴムシートからJIS K 6251:2010に準拠した打ち抜き刃を用い、ダンベル3号形試験片(厚さ0.5mm)を得た。
得られた試験片を12枚重ねて厚み6mmとし、硬度をタイプAデュロメータの圧子を用い、JIS K 6253-3:2012に準拠して測定した。なお、硬度の数値が小さいほど柔軟性に優れる。
上記(硬度)測定と同様の方法で作製したダンベル3号形試験片(0.5mm)を用い、JIS K 6251:2010に準じて、引張破断強度及び引張破断伸びを測定した。引張破断強度及び引張破断伸びの数値が大きいほど引張特性に優れる。
(引裂強度)
JIS K 6252-1:2015に準じて、実施例及び比較例で得られた樹脂組成物のゴムシートから、打ち抜き刃を用い切り込みなしアングル形試験片(厚さ0.5mm)を得た。得られた試験片を用い、引張速度500mm/min、温度23℃で引裂強さを測定した。数値が大きいほど引裂強度に優れる。
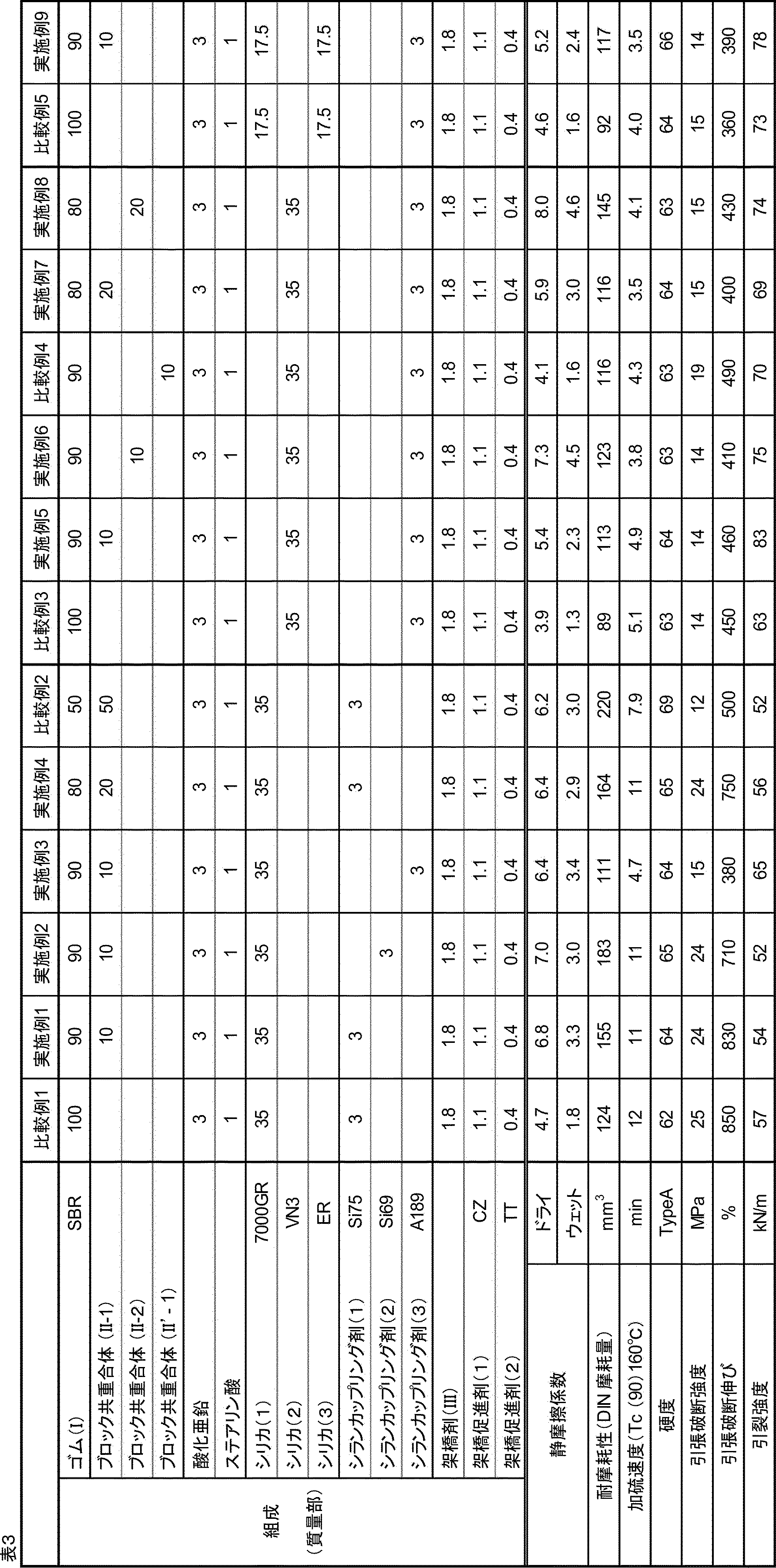
また、実施例1及び4と比較例2との対比から、ゴム(I)とブロック共重合体(II)との質量比を特定の数値範囲とすることにより、優れた滑りにくさを保持しつつ耐摩耗性にも優れることがわかる。
更に、表3に示す全ての実施例から、本実施形態の樹脂組成物であれば滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性等のバランスに優れる成形体が得られることがわかる。
表4に記載した配合割合(質量部)に従って、実施例1と同様に行い樹脂組成物及びゴムシートを得て、物性を評価した。
また、表4におけるゴム(I)及び架橋促進剤(3)~(5)は次のとおりであり、また他の材料は、前記に示したとおりである。
<ゴム(I)>
・ゴム(I):BR
製品名BR-01、JSR株式会社製、ポリブタジエンゴム(BR)、重量平均分子量(Mw)=55万、シス体含量=95質量%、ビニル含量=2.5質量%
<架橋促進剤>
・架橋促進剤(3):製品名ノクセラーDM、大内新興化学工業株式会社製、ジ-2-ベンゾチアゾリルジスルフィド
・架橋促進剤(4):製品名ノクセラーTBT、大内新興化学工業株式会社製、テトラブチルチウラムジスルフィド)
・架橋促進剤(5):サルチル酸

また、実施例10及び11と比較例7との対比から、ゴム(I)とブロック共重合体(II)との質量比を特定の数値範囲とすることにより、優れた滑りにくさを保持しつつ耐摩耗性にも優れることがわかる。
更に、表4に示す全ての実施例から、本実施形態の樹脂組成物であれば滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性等のバランスに優れる成形体が得られることがわかる。
表5に記載した配合割合(質量部)に従って、実施例1と同様に行い樹脂組成物及びゴムシートを得て、物性を評価した。
また、表5におけるゴム(I)は次のとおりであり、また他の材料は、前記に示したとおりである。
<ゴム(I)>
・ゴム(I):NR
天然ゴム、インドネシア産、RSS No.1

また、実施例14と比較例9との対比から、ゴム(I)とブロック共重合体(II)との質量比を特定の数値範囲とすることにより、優れた滑りにくさを保持しつつ耐摩耗性にも優れることがわかる。
更に、実施例14から、本実施形態の樹脂組成物であれば滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性等のバランスに優れる成形体が得られることがわかる。
表6に記載した配合割合(質量部)に従って、実施例1と同様に行い樹脂組成物及びゴムシートを得て、物性を評価した。
また、表6における材料は、前記に示したとおりである。

更に、表6に示す全ての実施例から、本実施形態の樹脂組成物であれば滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性等のバランスに優れる成形体が得られることがわかる。
表8,9に記載した配合割合(質量部)に従って、実施例1と同様に行い樹脂組成物及びゴムシートを得て、物性を評価した。
また、表8,9における軟化剤は次の製造例4~8で製造した軟化剤を用いた。表9におけるフィラー(セルロース繊維)は次のとおりであり、他の材料は前記に示したとおりである。
[製造例4]
ポリβ-ファルネセン(IV-1):
窒素置換し乾燥させた耐圧容器に、溶媒としてヘキサン40kg、開始剤としてn-ブチルリチウム(17質量%ヘキサン溶液)0.55kgを仕込み、50℃に昇温した後、β-ファルネセン40kgを加えて1時間重合した。得られた重合反応液をメタノールで処理し、水を用いて重合反応液を洗浄した。洗浄後の重合反応液と水とを分離して、70℃で12時間乾燥することにより、表7に示す物性を有するポリβ-ファルネセン(IV-1)を得た。
なお、数平均分子量(Mn)及び分子量分布(Mw/Mn)は、上記[測定方法]と同様に測定した。
ポリβ-ファルネセン(IV-2):
窒素置換し乾燥させた耐圧容器に、溶媒としてヘキサン40kg、開始剤としてn-ブチルリチウム(17質量%ヘキサン溶液)1.84kgを仕込み、50℃に昇温した後、β-ファルネセン40kgを加えて1時間重合した。得られた重合反応液をメタノールで処理し、水を用いて重合反応液を洗浄した。洗浄後の重合反応液と水とを分離して、70℃で12時間乾燥することにより、表7に示す物性を有するポリβ-ファルネセン(IV-2)を得た。
なお、数平均分子量(Mn)及び分子量分布(Mw/Mn)は、上記[測定方法]と同様に測定した。
ブタジエン-β-ファルネセン共重合体(IV-3):
窒素置換し乾燥させた耐圧容器に、溶媒としてヘキサン40kg、開始剤としてn-ブチルリチウム(17質量%ヘキサン溶液)0.72kgを仕込み、50℃に昇温した後、予め調製した24.0kgのβ-ファルネセンと16.0kgのブタジエンの混合液を加えて1時間重合した。得られた重合反応液をメタノールで処理し、水を用いて重合反応液を洗浄した。洗浄後の重合反応液と水とを分離して、70℃で12時間乾燥することにより、表7に示す物性を有するブタジエン-β-ファルネセン共重合体(IV-3)を得た。
なお、数平均分子量(Mn)及び分子量分布(Mw/Mn)は、上記[測定方法]と同様に測定した。
水添ポリイソプレン(IV-4):
窒素置換し乾燥させた耐圧容器に、溶媒としてヘキサン40kg、開始剤としてn-ブチルリチウム(17質量%ヘキサン溶液)0.77kgを仕込み、50℃に昇温した後、イソプレン40kgを加えて1時間重合した。得られた重合反応液をメタノールで処理し、水を用いて重合反応液を洗浄した。洗浄後の重合反応液と水とを分離して、70℃で12時間乾燥することにより、ポリイソプレンを得た。
上記ポリイソプレン40kgを、溶媒であるシクロヘキサン50kgとオートクレーブ中で混合し、50℃まで昇温した。続いて、トリイソブチルアルミニウムと2-エチルヘキサン酸ニッケルとを3:1のモル比で混合したものを水素添加触媒として、上記ポリイソプレンが含有する全不飽和結合のモル数に対し、水素添加触媒を構成しているニッケル金属として1.5×10-3倍モル加え、80℃まで昇温した。水素圧力を1.0MPaに保つように随時水素を供給しながら攪拌を続け、10時間反応を行った。放冷、放圧後、濾過により、トリイソブチルアルミニウムと2-エチルヘキサン酸ニッケルを除去し、濾液を濃縮し、真空乾燥することにより、表7に示す物性を有する水添ポリイソプレン(IV-4)を得た。
なお、数平均分子量(Mn)、分子量分布(Mw/Mn)、及び水素添加率(モル%)は、上記[測定方法]と同様に測定した。
変性液状ポリイソプレン(IV-5):
窒素置換し、乾燥させた耐圧容器に、ヘキサン40kg、n-ブチルリチウム(17質量%ヘキサン溶液)0.90kgを仕込み、70℃に昇温した後、イソプレン41kgを加えて1時間重合した。得られた重合反応液にメタノールを添加後、重合反応液を水で洗浄した。水を分離して、重合反応液を70℃で12時間真空乾燥することにより、未変性ポリイソプレン(IV-5)を得た。続いて、未変性ポリイソプレン(IV-5)30kgと無水マレイン酸0.45kgを仕込み、160℃で20時間反応させて、表7に示す物性を有する無水マレイン酸変性液状ポリイソプレン(IV-5)を得た。
なお、数平均分子量(Mn)及び分子量分布(Mw/Mn)は、上記[測定方法]と同様に測定した。
変性剤の反応率は99%であり、変性液状ポリイソプレン(IV-5)中に付加された官能基は、未変性重合体100質量部に対し1.5質量部であった。
・セルロース繊維:製品名ARBOCEL-BWW40、レッテンマイヤー社製、繊維長200μm、繊維径20μm、繊維密度110-145
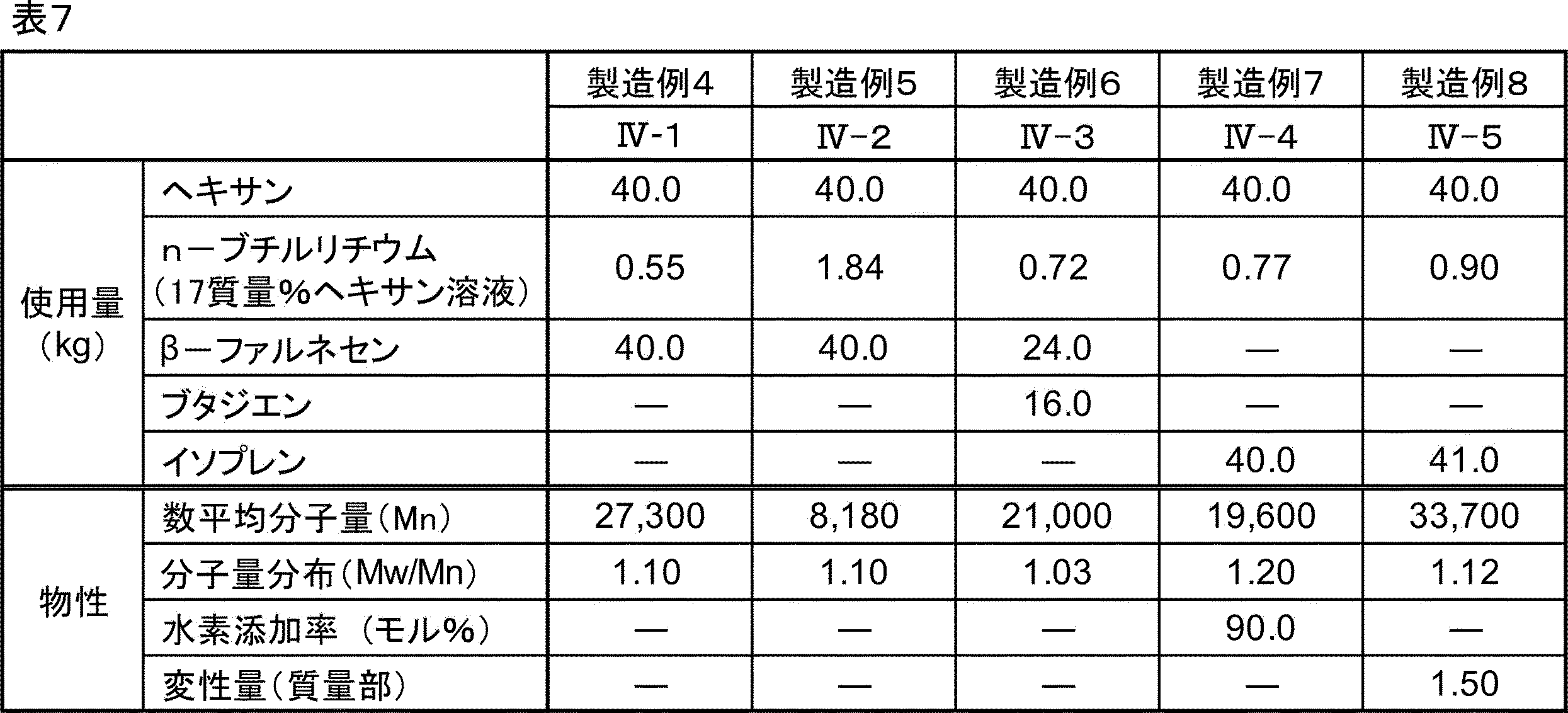

更に、表8に示す全ての実施例から、本実施形態の樹脂組成物であれば滑りにくく、耐摩耗性に優れ、加えて引裂強度及び引張特性等のバランスに優れる成形体が得られることがわかる。
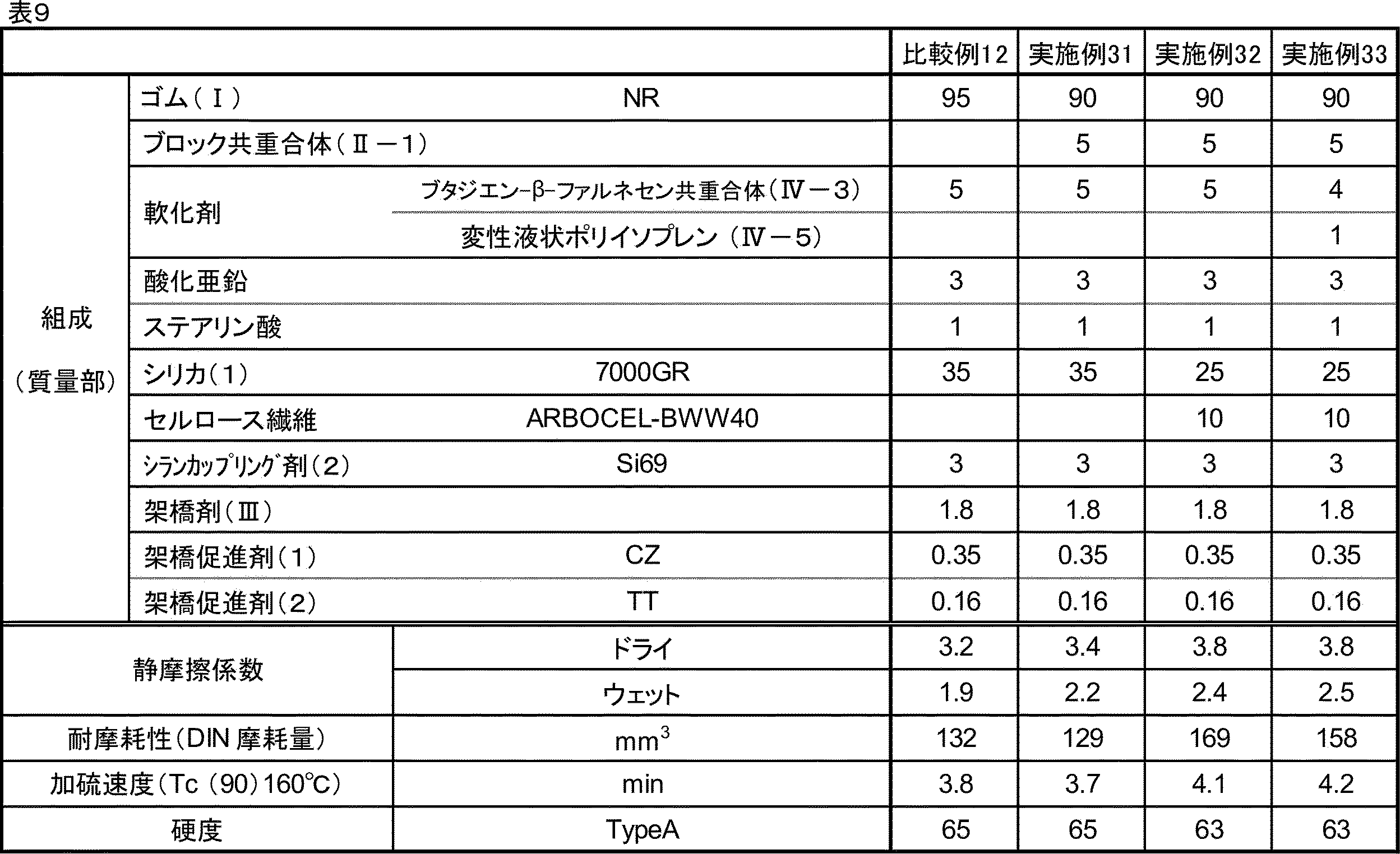
Claims (15)
- ゴム(I)、ブロック共重合体(II)及び架橋剤(III)を含有し、
前記ゴム(I)と前記ブロック共重合体(II)との質量比[(I)/(II)]が99/1~55/45であり、
前記ブロック共重合体(II)が、芳香族ビニル化合物由来の構造単位を含有する重合体ブロック(A)と、ファルネセン由来の構造単位(b1)を含有する重合体ブロック(B)とを含むブロック共重合体である、樹脂組成物。 - 前記ゴム(I)と前記ブロック共重合体(II)との質量比[(I)/(II)]が99/1~70/30である、請求項1に記載の樹脂組成物。
- 前記ブロック共重合体(II)が、更にファルネセン以外の共役ジエン由来の構造単位(c2)を含有する重合体ブロック(C)を含む、請求項1又は2に記載の樹脂組成物。
- 前記ブロック共重合体(II)が、少なくとも2個の前記重合体ブロック(A)、少なくとも1個の前記重合体ブロック(B)、及び少なくとも1個の前記重合体ブロック(C)を含有し、かつ少なくとも1個の前記重合体ブロック(B)を末端に有する、請求項3に記載の樹脂組成物。
- 前記ブロック共重合体(II)が、共役ジエン由来の構造単位における炭素-炭素二重結合が70モル%以上水素添加された水添ブロック共重合体(Q)である、請求項1~4のいずれかに記載の樹脂組成物。
- 前記ブロック共重合体(II)が、共役ジエン由来の構造単位における炭素-炭素二重結合が70モル%以上水素添加された水添ブロック共重合体(Q)であって、
前記重合体ブロック(B)と前記重合体ブロック(C)との共役ジエン由来の構造単位における炭素-炭素二重結合の水素添加率が異なる、請求項3又は4に記載の樹脂組成物。 - 前記ゴム(I)が、スチレンブタジエン共重合ゴム、天然ゴム、ポリイソプレンゴム、ポリブタジエンゴム、アクリルニトリルブタジエン共重合ゴム、エチレン-プロピレン-ジエン共重合体、ブチルゴム、及びブチルゴムを変性したハロゲン化ブチルゴムから選ばれる少なくとも1種である、請求項1~6のいずれかに記載の樹脂組成物。
- 前記架橋剤(III)が、硫黄及び/又は硫黄含有化合物である、請求項1~7のいずれかに記載の樹脂組成物。
- 前記架橋剤(III)の含有量が、前記ゴム(I)及び前記ブロック共重合体(II)の合計含有量100質量部に対し、0.1~10質量部である、請求項1~8のいずれかに記載の樹脂組成物。
- 更にシリカを含有する、請求項1~9のいずれかに記載の樹脂組成物。
- 更にシランカップリング剤を含有する、請求項1~10のいずれかに記載の樹脂組成物。
- 前記ゴム(I)が少なくとも天然ゴムを含み、更に軟化剤を含有する、請求項1~11のいずれかに記載の樹脂組成物。
- JIS K 6264-2:2005に準じてDIN摩耗試験により測定される摩耗量が、200mm3以下である、請求項1~12のいずれかに記載の樹脂組成物。
- 請求項1~13のいずれかに記載の樹脂組成物の成形体。
- 請求項1~13のいずれかに記載の樹脂組成物を少なくとも一部に用いた、靴底。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/777,134 US20220403145A1 (en) | 2019-11-18 | 2020-11-18 | Resin composition and molded article |
| JP2021558411A JP7492973B2 (ja) | 2019-11-18 | 2020-11-18 | 樹脂組成物及び成形体 |
| EP20889212.5A EP4063417B1 (en) | 2019-11-18 | 2020-11-18 | Resin composition and molded article |
| CN202080079033.3A CN114641525B (zh) | 2019-11-18 | 2020-11-18 | 树脂组合物和成形体 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-207920 | 2019-11-18 | ||
| JP2019207920 | 2019-11-18 | ||
| JP2020123089 | 2020-07-17 | ||
| JP2020-123089 | 2020-07-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021100738A1 true WO2021100738A1 (ja) | 2021-05-27 |
Family
ID=75980719
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/042905 Ceased WO2021100738A1 (ja) | 2019-11-18 | 2020-11-18 | 樹脂組成物及び成形体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20220403145A1 (ja) |
| EP (1) | EP4063417B1 (ja) |
| JP (1) | JP7492973B2 (ja) |
| CN (1) | CN114641525B (ja) |
| TW (1) | TWI854063B (ja) |
| WO (1) | WO2021100738A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023127895A1 (ja) * | 2021-12-28 | 2023-07-06 | 株式会社クラレ | 熱可塑性エラストマー組成物、熱可塑性エラストマー組成物のペレット、樹脂組成物、熱可塑性エラストマー組成物の製造方法、及び樹脂組成物の製造方法 |
| JPWO2023195167A1 (ja) * | 2022-04-08 | 2023-10-12 | ||
| JP2024021539A (ja) * | 2022-08-04 | 2024-02-16 | 住友ゴム工業株式会社 | シート搬送ローラ用ゴム組成物およびシート搬送ローラ |
| WO2024262423A1 (ja) * | 2023-06-20 | 2024-12-26 | 株式会社クラレ | メタクリル系樹脂組成物とその製造方法、及び成形体 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011132298A (ja) | 2009-12-22 | 2011-07-07 | Sumitomo Rubber Ind Ltd | 変性共重合体、それを用いたゴム組成物および空気入りタイヤ |
| JP2012502136A (ja) | 2008-09-04 | 2012-01-26 | アムイリス ビオテクフノロジエス,インコーポレイテッド | ファルネセン共重合体 |
| JP2012502135A (ja) | 2008-09-04 | 2012-01-26 | アムイリス ビオテクフノロジエス,インコーポレイテッド | ポリファルネセンを含む接着剤組成物 |
| JP2014208796A (ja) * | 2013-03-29 | 2014-11-06 | 株式会社クラレ | ゴム組成物、加硫ゴム及びタイヤ |
| JP2016113614A (ja) * | 2014-12-10 | 2016-06-23 | 株式会社クラレ | 熱可塑性エラストマー組成物、架橋物、成形体、部材、ウェザーシール、及びウェザーシール用コーナー部材 |
| JP2018024776A (ja) * | 2016-08-10 | 2018-02-15 | 株式会社クラレ | 熱可塑性エラストマー組成物、積層構造体及び該積層構造体の製造方法 |
| JP2019026826A (ja) * | 2017-07-31 | 2019-02-21 | 株式会社クラレ | 樹脂組成物及び成形体 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103958600B (zh) * | 2011-12-01 | 2016-08-24 | Jsr株式会社 | 橡胶组合物、橡胶弹性体、轮胎以及嵌段共聚物 |
| RU2629197C2 (ru) * | 2012-04-04 | 2017-08-25 | Курарей Ко., Лтд. | Сополимер, резиновая композиция с его использованием и шина |
| CA2869393C (en) * | 2012-04-04 | 2020-03-24 | Kuraray Co., Ltd. | Rubber composition comprising isoprene-farnesene copolymer and tire produced therefrom |
| KR102028232B1 (ko) * | 2012-06-08 | 2019-10-02 | 주식회사 쿠라레 | 수소 첨가 블록 공중합체 및 그 제조 방법 |
| US9834666B2 (en) * | 2013-09-30 | 2017-12-05 | Kuraray Co., Ltd. | Polyolefin-based resin composition and molded body |
| JP5763865B1 (ja) * | 2013-09-30 | 2015-08-12 | 株式会社クラレ | 熱可塑性エラストマー組成物及び成形体 |
| EP3081613A4 (en) * | 2013-12-11 | 2017-08-09 | Kuraray Co., Ltd. | Sealant |
| TW201529691A (zh) * | 2013-12-11 | 2015-08-01 | Kuraray Co | 樹脂組成物、成形體、及樹脂改質劑 |
| WO2016125899A1 (ja) * | 2015-02-06 | 2016-08-11 | 株式会社クラレ | 水添ブロック共重合体 |
| TWI612067B (zh) * | 2015-08-24 | 2018-01-21 | Asahi Chemical Ind | 氫化嵌段共聚物與使用其之聚丙烯系樹脂組合物及其成型體 |
| JP6993188B2 (ja) * | 2017-11-16 | 2022-01-13 | Toyo Tire株式会社 | タイヤ用ゴム組成物、及びそれを用いた空気入りタイヤ |
-
2020
- 2020-11-18 TW TW109140331A patent/TWI854063B/zh active
- 2020-11-18 CN CN202080079033.3A patent/CN114641525B/zh active Active
- 2020-11-18 US US17/777,134 patent/US20220403145A1/en active Pending
- 2020-11-18 JP JP2021558411A patent/JP7492973B2/ja active Active
- 2020-11-18 EP EP20889212.5A patent/EP4063417B1/en active Active
- 2020-11-18 WO PCT/JP2020/042905 patent/WO2021100738A1/ja not_active Ceased
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012502136A (ja) | 2008-09-04 | 2012-01-26 | アムイリス ビオテクフノロジエス,インコーポレイテッド | ファルネセン共重合体 |
| JP2012502135A (ja) | 2008-09-04 | 2012-01-26 | アムイリス ビオテクフノロジエス,インコーポレイテッド | ポリファルネセンを含む接着剤組成物 |
| JP2011132298A (ja) | 2009-12-22 | 2011-07-07 | Sumitomo Rubber Ind Ltd | 変性共重合体、それを用いたゴム組成物および空気入りタイヤ |
| JP2014208796A (ja) * | 2013-03-29 | 2014-11-06 | 株式会社クラレ | ゴム組成物、加硫ゴム及びタイヤ |
| JP2016113614A (ja) * | 2014-12-10 | 2016-06-23 | 株式会社クラレ | 熱可塑性エラストマー組成物、架橋物、成形体、部材、ウェザーシール、及びウェザーシール用コーナー部材 |
| JP2018024776A (ja) * | 2016-08-10 | 2018-02-15 | 株式会社クラレ | 熱可塑性エラストマー組成物、積層構造体及び該積層構造体の製造方法 |
| JP2019026826A (ja) * | 2017-07-31 | 2019-02-21 | 株式会社クラレ | 樹脂組成物及び成形体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4063417A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023127895A1 (ja) * | 2021-12-28 | 2023-07-06 | 株式会社クラレ | 熱可塑性エラストマー組成物、熱可塑性エラストマー組成物のペレット、樹脂組成物、熱可塑性エラストマー組成物の製造方法、及び樹脂組成物の製造方法 |
| JPWO2023195167A1 (ja) * | 2022-04-08 | 2023-10-12 | ||
| WO2023195167A1 (ja) * | 2022-04-08 | 2023-10-12 | 株式会社アシックス | 靴底用部材及び靴 |
| JP2024021539A (ja) * | 2022-08-04 | 2024-02-16 | 住友ゴム工業株式会社 | シート搬送ローラ用ゴム組成物およびシート搬送ローラ |
| WO2024262423A1 (ja) * | 2023-06-20 | 2024-12-26 | 株式会社クラレ | メタクリル系樹脂組成物とその製造方法、及び成形体 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2021100738A1 (ja) | 2021-05-27 |
| TWI854063B (zh) | 2024-09-01 |
| CN114641525B (zh) | 2025-07-29 |
| EP4063417A1 (en) | 2022-09-28 |
| US20220403145A1 (en) | 2022-12-22 |
| CN114641525A (zh) | 2022-06-17 |
| JP7492973B2 (ja) | 2024-05-30 |
| TW202124557A (zh) | 2021-07-01 |
| EP4063417B1 (en) | 2026-03-11 |
| EP4063417A4 (en) | 2023-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7492973B2 (ja) | 樹脂組成物及び成形体 | |
| US7371805B2 (en) | Hydrogenated copolymer and composition thereof | |
| JP4367338B2 (ja) | 共役ジエン系ゴム、その製造方法、ゴム組成物およびゴム架橋物 | |
| JP6699853B2 (ja) | 熱可塑性エラストマー組成物、架橋物、成形体、部材、ウェザーシール、及びウェザーシール用コーナー部材 | |
| JP2010043257A (ja) | 炭化水素ポリマー添加物を添加した、優れた不透過性を有するエラストマ組成物 | |
| JP7073857B2 (ja) | 樹脂組成物及び成形体 | |
| JP2009052057A (ja) | 熱可塑性エラストマー組成物の製造方法 | |
| JPWO2019172185A1 (ja) | 変性液状ジエン系重合体およびゴム組成物 | |
| CN111465645B (zh) | 橡胶组合物和充气轮胎 | |
| CN113474413A (zh) | 交联物及轮胎 | |
| JP7453877B2 (ja) | 共役ジエン系重合体組成物 | |
| CN115135714B (zh) | 树脂组合物和成型体 | |
| WO2023112364A1 (ja) | タイヤ用ゴム組成物、トレッドゴム及びタイヤ | |
| WO2026083987A1 (ja) | 樹脂組成物及び成形体 | |
| JP2023144632A (ja) | 変性共役ジエン系重合体、それを含む靴用ゴム組成物、及びアウトソール | |
| WO2023127895A1 (ja) | 熱可塑性エラストマー組成物、熱可塑性エラストマー組成物のペレット、樹脂組成物、熱可塑性エラストマー組成物の製造方法、及び樹脂組成物の製造方法 | |
| JP2025178621A (ja) | ゴム組成物 | |
| JP2025117871A (ja) | 変性共役ジエン系重合体、靴用ゴム組成物、及びアウトソール | |
| JP2025117902A (ja) | 変性共役ジエン系重合体、靴用ゴム組成物、及びアウトソール | |
| WO2025109830A1 (ja) | タイヤ用ゴム組成物、タイヤ用トレッドゴム、及びタイヤ | |
| JP2004307565A (ja) | スチレン系熱可塑性エラストマー組成物の製造方法 | |
| JP2019218489A (ja) | ゴム組成物、タイヤ、コンベヤベルト、ゴムクローラ、防振装置、免震装置及びホース | |
| HK1077310B (en) | Hydrogenated copolymer and composition thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20889212 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021558411 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020889212 Country of ref document: EP Effective date: 20220620 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 202080079033.3 Country of ref document: CN |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2020889212 Country of ref document: EP |