WO2021131181A1 - 生分解性樹脂用分解促進剤、生分解性樹脂組成物、生分解性樹脂成形体、及び生分解性樹脂用分解促進剤の製造方法 - Google Patents
生分解性樹脂用分解促進剤、生分解性樹脂組成物、生分解性樹脂成形体、及び生分解性樹脂用分解促進剤の製造方法 Download PDFInfo
- Publication number
- WO2021131181A1 WO2021131181A1 PCT/JP2020/035289 JP2020035289W WO2021131181A1 WO 2021131181 A1 WO2021131181 A1 WO 2021131181A1 JP 2020035289 W JP2020035289 W JP 2020035289W WO 2021131181 A1 WO2021131181 A1 WO 2021131181A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- biodegradable resin
- aliphatic
- decomposition accelerator
- mass
- biodegradable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/36—Means for plasticising or homogenising the moulding material or forcing it through the nozzle or die
- B29C48/395—Means for plasticising or homogenising the moulding material or forcing it through the nozzle or die using screws surrounded by a cooperating barrel, e.g. single screw extruders
- B29C48/40—Means for plasticising or homogenising the moulding material or forcing it through the nozzle or die using screws surrounded by a cooperating barrel, e.g. single screw extruders using two or more parallel screws or at least two parallel non-intermeshing screws, e.g. twin screw extruders
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/16—Compositions of unspecified macromolecular compounds the macromolecular compounds being biodegradable
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/0001—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor characterised by the choice of material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/022—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the choice of material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/92—Measuring, controlling or regulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
- C08G63/18—Dicarboxylic acids and dihydroxy compounds the acids or hydroxy compounds containing carbocyclic rings
- C08G63/181—Acids containing aromatic rings
- C08G63/183—Terephthalic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L5/00—Compositions of polysaccharides or of their derivatives not provided for in groups C08L1/00 or C08L3/00
- C08L5/14—Hemicellulose; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L97/00—Compositions of lignin-containing materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L97/00—Compositions of lignin-containing materials
- C08L97/005—Lignin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L97/00—Compositions of lignin-containing materials
- C08L97/02—Lignocellulosic material, e.g. wood, straw or bagasse
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/05—Filamentary, e.g. strands
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/07—Flat, e.g. panels
- B29C48/08—Flat, e.g. panels flexible, e.g. films
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/09—Articles with cross-sections having partially or fully enclosed cavities, e.g. pipes or channels
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/12—Articles with an irregular circumference when viewed in cross-section, e.g. window profiles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/15—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor incorporating preformed parts or layers, e.g. extrusion moulding around inserts
- B29C48/154—Coating solid articles, i.e. non-hollow articles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/16—Articles comprising two or more components, e.g. co-extruded layers
- B29C48/18—Articles comprising two or more components, e.g. co-extruded layers the components being layers
- B29C48/21—Articles comprising two or more components, e.g. co-extruded layers the components being layers the layers being joined at their surfaces
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/30—Extrusion nozzles or dies
- B29C48/305—Extrusion nozzles or dies having a wide opening, e.g. for forming sheets
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2067/00—Use of polyesters or derivatives thereof, as moulding material
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/06—Biodegradable
Definitions
- the present invention relates to a decomposition accelerator capable of promoting biodegradation of a biodegradable resin and a method for producing the same.
- the present invention also relates to a biodegradable resin composition containing the decomposition accelerator for biodegradable resin, and a biodegradable resin molded product obtained by extrusion molding or injection molding of the biodegradable resin composition.
- biodegradable plastics for example, polybutylene terephthalate / adipate (hereinafter abbreviated as PBAT), polylactic acid (hereinafter abbreviated as PLA), polybutylene succinate (hereinafter abbreviated as PBS).
- PBAT polybutylene terephthalate / adipate
- PLA polylactic acid
- PBS polybutylene succinate
- PBSA Polybutylene succinate / adipate
- PHA polyhydroxyalkanol
- PHA includes poly (3-hydroxybutyrate) (hereinafter abbreviated as PHB), poly (3-hydroxybutyrate / 3-hydroxyvalerate) (hereinafter abbreviated as PHBV), and poly (3-hydroxybutyrate). Rate / 3-hydroxyhexanoate) (hereinafter abbreviated as PHBH), poly (3-hydroxybutyrate / 4-hydroxybutyrate) and the like.
- each resin differs not only in the biodegradation rate and decomposition rate of the resin alone, but also in the mechanical properties such as tensile elongation at break and flexural modulus. For this reason, in general, each resin is combined or mixed with other auxiliary materials and additives to improve physical properties such as mechanical strength according to the intended use and place of use.
- Patent Document 1 also takes into consideration the final disposal treatment after the product has been used, and is a biodegradable resin that decomposes at the time of landfill and does not remain in the environment, and does not emit harmful substances such as dioxin at the time of incineration.
- a molding material composed of a biomass material and an unsaturated carboxylic acid or an unsaturated carboxylic acid with respect to 5 to 95% by weight of an aliphatic aromatic polyester (A component) and 95 to 5% by weight of cellulose, lignocellulose or starch-based substance (B component).
- a method for producing a composite resin composition having excellent molding processability and strength characteristics by heating and kneading a composition containing the derivative and an organic peroxide (C component) is disclosed.
- Patent Document 2 describes a plant material containing cellulose in a high-molecular-weight aliphatic polyester, for example, a material consisting of the epidermis or skin layer of a plant, a seed coat material, a material for fruit skin, bagasse, milling waste, or sugar cane.
- a resin composition and a molded product containing bagasse of the stem of sugar cane are disclosed.
- biodegradation rate and decomposition rate are more severe than the conventional conditions generally considered to be biodegradable.
- conventional biodegradable resins such as PBAT, PBS, PBSA, PLA, and PHBH, and to decompose more in the same time. It is said that.
- An object of the present invention is to increase and arbitrarily control the biodegradable rate and decomposition rate of biodegradable resins such as aliphatic polyester resins, aliphatic-aromatic polyester resins, and aliphatic oxycarboxylic acid resins. It is an object of the present invention to provide a decomposition accelerator suitable for a biodegradable resin and a method for producing the same, a biodegradable resin composition containing the decomposition accelerator, and a molded product thereof.
- biodegradable resins such as aliphatic polyester resins, aliphatic-aromatic polyester resins, and aliphatic oxycarboxylic acid resins.
- the present inventor has focused on nitrogen and carbon, three components of cellulose, hemicellulose and lignin, or three components of soluble nitrogen-free substance, cellulose and lignin, and these components decompose biodegradable resins. I thought that it would contribute to or be involved in the promotion of. Based on this idea, by adjusting these blending ratios, etc., when blended with biodegradable resin, the biodegradation rate and decomposition rate are made higher than the original biodegradation rate and decomposition rate of biodegradable resin. I found that I could do it.
- the gist of the present invention lies in the following [1] to [12].
- a decomposition accelerator for biodegradable resins containing 20% by mass or more of a soluble nitrogen-free substance and having a total content of cellulose and lignin of 50% by mass or less.
- the biodegradable resin is one or more selected from the group consisting of an aliphatic polyester resin (A), an aliphatic-aromatic polyester resin (B) and an aliphatic oxycarboxylic acid resin (C).
- the main constituent units of the aliphatic-aromatic polyester resin (B) are a repeating unit derived from an aliphatic diol, a repeating unit derived from an aliphatic dicarboxylic acid, and a repeating unit derived from an aromatic dicarboxylic acid.
- a biodegradable resin molded product which is an extrusion molded product or an injection molded product of the biodegradable resin composition according to any one of [4] to [7].
- a method for producing a decomposition accelerator for a biodegradable resin which comprises crushing a raw material and then selecting a powder having a predetermined particle size from the obtained powder to obtain a decomposition accelerator for a biodegradable resin.
- the decomposition accelerator for biodegradable resin contains cellulose, hemicellulose and lignin, and the mass ratio of nitrogen to carbon in the decomposition accelerator for biodegradable resin is 0.04 or more, and cellulose.
- the biodegradable resin decomposition accelerator contains 20% by mass or more of a soluble nitrogen-free substance, and the total content of cellulose and lignin is 50% by mass or less, [9] to [ 11]
- the method for producing a decomposition accelerator for a biodegradable resin according to any one of.
- the biodegradability rate and decomposition rate of biodegradable resins such as aliphatic polyester resins, aliphatic-aromatic polyester resins, and aliphatic oxycarboxylic acid resins. Can be higher than the original biodegradable rate and decomposition rate of the biodegradable resin.
- biodegradable resin composition of the present invention containing such a decomposition accelerator for a biodegradable resin of the present invention, a biodegradable resin molded product having excellent biodegradability is provided.
- the present invention is not limited to the following description, and can be arbitrarily modified and carried out without departing from the gist of the present invention.
- a numerical value or a physical property value is sandwiched before and after using "-"
- it is used as including the values before and after that.
- the decomposition accelerator for biodegradable resin according to the first aspect of the present invention (hereinafter, may be referred to as “decomposition accelerator I of the present invention”) contains cellulose, hemicellulose and lignin as essential components. Therefore, the mass ratio of nitrogen (nitrogen element) to carbon (carbon element) in the decomposition accelerator for biodegradable resin (hereinafter, may be referred to as “nitrogen / carbon ratio”) is 0.04 or more. In addition, the mass ratio of the content of hemicellulose to the total content of cellulose and lignin (hereinafter, may be referred to as “hemicellulose / (cellulose + lignin) ratio”) is 0.2 or more. And.
- the decomposition accelerator for biodegradable resin according to the second aspect of the present invention (hereinafter, may be referred to as “decomposition accelerator II of the present invention”) contains 20% by mass or more of a soluble nitrogen-free substance. Moreover, the total content of cellulose and lignin is 50% by mass or less.
- the decomposition accelerator I of the present invention and the decomposition accelerator II of the present invention are collectively referred to as the "decomposition accelerator of the present invention".
- the decomposition accelerator I of the present invention has a nitrogen / carbon ratio of 0.04 or more.
- the reason why the decomposition of the biodegradable resin is promoted by the decomposition accelerator I whose nitrogen / carbon ratio satisfies this range is presumed to be as follows. Microorganisms such as bacteria and fungi that decompose biodegradable resins require nitrogen as well as carbon as a nutrient source. On the other hand, biodegradable resins are mainly composed of carbon, oxygen and hydrogen, and have a low nitrogen content.
- the decomposition accelerator I of the present invention contains a certain amount or more of nitrogen with respect to carbon, it is presumed that it promotes the growth of microorganisms and the secretion of degrading enzymes, and promotes the decomposition of biodegradable resins.
- the nitrogen / carbon ratio of the decomposition accelerator I of the present invention is preferably 0.05 or more, more preferably 0.06 or more.
- the upper limit of the nitrogen / carbon ratio is not particularly limited, but is preferably 5 or less. If the nitrogen / carbon ratio exceeds 5, the carbon content may be insufficient and sufficient effects may not be exhibited.
- the decomposition accelerator I of the present invention has a mass ratio (hemicellulose / (cellulose + lignin) ratio) of the content of hemicellulose to the total content of cellulose and lignin of 0.2 or more.
- the reason why the decomposition accelerator I whose hemicellulose / (cellulose + lignin) ratio satisfies this range promotes the decomposition of the biodegradable resin is presumed to be as follows. Hemicellulose tends to be decomposed and absorbed by microorganisms faster than cellulose and lignin.
- the decomposition accelerator I of the present invention having a hemicellulose / (cellulose + lignin) ratio of 0.2 or more, when the resin composition is released into the natural world, the growth of microorganisms is promoted particularly at an early stage and the surface area is increased. It is presumed that increasing the amount promotes the decomposition of the biodegradable resin.
- the hemicellulose / (cellulose + lignin) ratio of the decomposition accelerator I of the present invention is preferably 0.6 or more, more preferably 1.0 or more.
- the upper limit of the hemicellulose / (cellulose + lignin) ratio is not particularly limited, but is usually 10 or less. This is because in order to make this ratio larger than 10, it is necessary to remove cellulose and / or lignin contained in the raw material, which increases the production cost.
- the decomposition accelerator I of the present invention preferably contains cellulose, hemicellulose, and lignin in a total amount of 5 to 50% by mass, particularly about 10 to 45% by mass, in 100% by mass of the decomposition accelerator I.
- the decomposition accelerator I of the present invention contains water, it is preferably dried to reduce the water content to less than 5% by mass. If the water content is 5% by mass or more, the biodegradable resin may be hydrolyzed when mixed with the biodegradable resin, the molecular weight may be lowered, and the mechanical strength may not be sufficient.
- the contents of nitrogen, carbon, cellulose, hemicellulose and lignin in the decomposition accelerator I of the present invention can be measured and calculated by the methods described in Examples described later.
- the contents of nitrogen, carbon, cellulose, hemicellulose and lignin of each raw material depend on the mixing ratio of the raw materials, and the nitrogen, carbon, cellulose, hemicellulose and lignin of each raw material are used. Can be obtained as the sum of the contents of the product multiplied by the ratio.
- the decomposition accelerator II of the present invention contains 20% by mass or more of a soluble nitrogen-free substance. It is presumed that the reason why the decomposition of the biodegradable resin is promoted by the decomposition accelerator whose soluble nitrogen-free content is within this range is as follows.
- the soluble nitrogen-free substance is an analytical value generally used for displaying components of feed and the like, and the component containing the soluble nitrogen-free substance has a property of being easily decomposed and absorbed by microorganisms.
- natural soil and mature compost usually have a low number of microorganisms and activity of degrading enzymes because there are few easily degradable organic substances available to microorganisms such as bacteria and fungi.
- the decomposition accelerator II of the present invention promotes the growth of microorganisms and the activation of degrading enzymes and promotes the decomposition of biodegradable resins by containing a certain amount or more of soluble nitrogen-free substances.
- the content of the soluble nitrogen-free substance of the decomposition accelerator II of the present invention is preferably 30% by mass or more, more preferably 40% by mass or more.
- the upper limit of the content of the soluble nitrogen-free substance is not particularly limited, but it is usually 99% by mass or less because the cost for purification is high in order to make it 100% by mass.
- the decomposition accelerator II of the present invention has a total content of cellulose and lignin of 50% by mass or less.
- Cellulose and lignin have the property of being decomposed and absorbed by microorganisms later than soluble nitrogen-free substances. If the total content of cellulose and lignin in the decomposition accelerator II of the present invention exceeds 50% by mass, the rate of biodegradation may decrease.
- the total content of cellulose and lignin in the decomposition accelerator II of the present invention is preferably 30% by mass or less, more preferably 20% by mass or less.
- the lower limit of the total content of cellulose and lignin is not particularly limited, but is usually 5% by mass or more.
- the decomposition accelerator II of the present invention contains water, it is preferable to dry it to reduce the water content to less than 5% by mass. If the water content is 5% by mass or more, the biodegradable resin may be hydrolyzed when mixed with the biodegradable resin, the molecular weight may be lowered, and the mechanical strength may not be sufficient.
- the contents of the soluble nitrogen-free substance, cellulose and lignin in the decomposition accelerator II of the present invention can be measured and calculated by the methods described in Examples described later. Further, in the case of the decomposition accelerator II using two or more kinds of raw materials, the contents of the soluble nitrogen-free substance, cellulose and lignin of each raw material are determined according to the mixing ratio of the raw materials. And the content of lignin can be calculated as the sum of the products multiplied by the ratio.
- the decomposition accelerator II of the present invention contains 20% by mass or more of a soluble nitrogen-free substance, and the total content of cellulose and lignin is 50% by mass or less, but the mass ratio of nitrogen to carbon is 0. It is preferable that the content is 04 or more and the mass ratio of the hemicellulose content to the total content of cellulose and lignin is 0.2 or more.
- the decomposition accelerator I of the present invention can be obtained by using a substance containing cellulose, hemicellulose and lignin as a raw material and adjusting the concentration and ratio thereof.
- raw materials residue of squeezed plant seeds and fruits, removed shells and bran, bran, and lees generated in the process of oil refining and brewing can be used.
- Specific examples include peanuts, rice, wheat, barley, oil palm, cotton, soybeans, rapeseed, and corn.
- An organic substance or an inorganic substance containing nitrogen may be added to the decomposition accelerator I of the present invention in order to adjust the nitrogen / carbon ratio within a predetermined range.
- the decomposition accelerator II of the present invention can be obtained by using a material containing a soluble nitrogen-free substance as a raw material and adjusting its concentration and ratio.
- raw materials residue of squeezed plant seeds and fruits, removed shells and bran, bran, and lees generated in the process of oil refining and brewing can be used.
- Specific examples include pistachios, walnuts, chestnuts, peanuts, rice, wheat, barley, oil palm, bagasse, cotton, soybeans, rapeseed, and corn.
- the decomposition accelerator of the present invention can be obtained by pulverizing the above raw materials to a certain particle size.
- the raw materials may be crushed separately or a mixture of the raw materials mixed in advance may be crushed.
- each raw material is crushed in advance and mixed. Is preferable.
- each raw material is crushed in advance and mixed. It is preferable to do so.
- the raw material can be crushed using, for example, a crusher or the like.
- a crusher for example, various crushers such as a cutter mill, a millstone, a crusher, a jet mill, a ball mill, and a pin mill can be used.
- a jet mill, a ball mill, and a pin mill are preferable in order to obtain a decomposition accelerator having a predetermined particle size, and a jet mill is particularly preferable because it can be continuously and efficiently pulverized.
- the crushing method is roughly divided into wet crushing using a medium such as water and dry crushing using no medium. Dry pulverization is preferable because it is necessary to dry to a predetermined moisture content.
- the particle size after pulverization is usually 500 ⁇ m or less, preferably 300 ⁇ m or less, from the viewpoint that when used with a biodegradable resin, it is more easily mixed with the biodegradable resin and is more effective as a decomposition accelerator. , More preferably 100 ⁇ m or less.
- the decomposition accelerator of the present invention preferably has a 50% cumulative particle size of 60 ⁇ m or less, more preferably 40 ⁇ m or less, and particularly preferably 20 ⁇ m or less. The lower the value of the 50% mass cumulative particle size of this decomposition accelerator, the better the appearance of the biodegradable resin composition obtained by mixing with the biodegradable resin and the biodegradable resin molded product obtained by molding the biodegradable resin composition.
- the lower limit of the 50% cumulative mass particle size is not particularly limited, but is preferably 1 ⁇ m or more. When the 50% mass cumulative particle size is less than 1 ⁇ m, it is easy to scatter, and when mixed with a biodegradable resin, problems such as blocking and classification may occur, which may make handling difficult.
- the 50% mass cumulative particle size of the decomposition accelerator of the present invention is measured by the method described in the section of Examples below.
- a mesh sieve or screen having a particle size of that particle size, a centrifugal classifier, or the like is used to sort out those having a predetermined particle size. Is preferable.
- crushing and sorting may be performed at the same time using a crusher provided with a sorting mechanism.
- the powder obtained by pulverizing each raw material is sorted in this way, and the powder having a predetermined particle size is blended in a specific ratio as needed to obtain the decomposition accelerator of the present invention.
- the powder preferably used at this time is not particularly limited as long as it is the powder of the above-mentioned raw material, and examples thereof include rice bran, wheat bran, soybean meal, and rapeseed meal powder.
- the decomposition accelerator of the present invention When mixing the decomposition accelerator of the present invention with the biodegradable resin, it is preferable to dry it in advance. The reason is that moisture in the atmosphere (air) in the storage atmosphere is adsorbed during storage, and it becomes difficult for the moisture to mix with the biodegradable resin. Further, as described above, when the decomposition accelerator contains water, the biodegradable resin is hydrolyzed to reduce the molecular weight, and the decomposition accelerator can be dried from the viewpoint of impairing the mechanical strength. preferable.
- the decomposition accelerator of the present invention after the above-mentioned pulverization and particle size adjustment, further drying is preferably performed.
- it may be heated at a temperature of about 40 to 200 ° C. using a general dryer.
- the water content of the decomposition accelerator is less than 5% by mass, particularly 0 to 3% by mass.
- the water content of the decomposition accelerator is the mass W x of the decomposition accelerator and the mass after the decomposition accelerator is dried to an absolute dry state according to the method for measuring the water content described in the section of Examples below. from W y, it is calculated by the following formula.
- Moisture content (mass%) ⁇ (W x- W y ) / W x ⁇ x 100
- the biodegradable resin composition of the present invention contains 2 parts by mass or more and 250 parts by mass or less of the decomposition accelerator of the present invention and 100 parts by mass of the biodegradable resin.
- the content of the decomposition accelerator of the present invention when the content of the decomposition accelerator of the present invention is less than 2 parts by mass with respect to 100 parts by mass of the biodegradable resin, the biodegradability due to the inclusion of the decomposition accelerator of the present invention The improvement effect of is not sufficiently obtained. If the content of the decomposition accelerator of the present invention exceeds 250 parts by mass, the mechanical properties of the obtained molded product are impaired.
- the biodegradable resin composition of the present invention preferably contains 10 parts by mass or more and 100 parts by mass or less of the decomposition accelerator of the present invention with respect to 100 parts by mass of the biodegradable resin, and contains 15 parts by mass or more and 80 parts by mass or less. Is more preferable.
- the biodegradable resin contained in the biodegradable resin composition of the present invention may be any biodegradable resin, and is not particularly limited, but is an aliphatic polyester resin (A) or an aliphatic-aromatic resin. Examples thereof include polyester-based resin (B) and aliphatic oxycarboxylic acid-based resin (C). Two or more of the aliphatic polyester resin (A), the aliphatic-aromatic polyester resin (B), and the aliphatic oxycarboxylic acid resin (C) may be mixed and used.
- the aliphatic polyester-based resin (A), the aliphatic-aromatic polyester-based resin (B), and the aliphatic oxycarboxylic acid-based resin (C) are polymers each having a repeating unit. It is also called a compound unit for the compound from which each repeating unit is derived.
- a repeating unit derived from an aliphatic diol is an "aliphatic diol unit”
- a repeating unit derived from an aliphatic dicarboxylic acid is a "aliphatic dicarboxylic acid unit”
- a repeating unit derived from an aromatic dicarboxylic acid is a "aromatic dicarboxylic acid”.
- acid unit also called "acid unit”.
- the "main constituent unit" in the aliphatic polyester-based resin (A), the aliphatic-aromatic polyester-based resin (B), and the aliphatic oxycarboxylic acid-based resin (C) is usually the polyester-based constituent unit. It is a structural unit contained in the resin in an amount of 80% by mass or more, and may not contain any structural unit other than the main structural unit.
- aliphatic polyester resin (A) As the aliphatic polyester resin (A), an aliphatic polyester resin containing an aliphatic diol unit and an aliphatic dicarboxylic acid unit as main constituent units is preferable.
- the ratio of the succinic acid unit to the total dicarboxylic acid unit is preferably 5 mol% or more and 100 mol% or less.
- the aliphatic polyester resin (A) may be a mixture of aliphatic polyester resins having different amounts of succinic acid units.
- an aliphatic polyester-based resin that does not contain an aliphatic dicarboxylic acid unit other than succinic acid containing only a succinic acid unit as an aliphatic dicarboxylic acid unit
- an aliphatic polyester-based resin that contains an aliphatic dicarboxylic acid unit other than succinic acid It is also possible to blend with the resin and adjust the amount of succinic acid unit in the aliphatic polyester resin (A) within the above preferable range before use.
- the aliphatic polyester resin (A) is a polyester resin containing an aliphatic diol unit represented by the following formula (1) and an aliphatic dicarboxylic acid unit represented by the following formula (2). Is. -OR 1 -O- (1) -OC-R 2- CO- (2)
- R 1 represents a divalent aliphatic hydrocarbon group.
- R 2 represents a divalent aliphatic hydrocarbon group.
- the aliphatic diol unit and the aliphatic dicarboxylic acid unit represented by the formulas (1) and (2) may be derived from a compound derived from petroleum or a compound derived from a plant material. However, it is desirable that it is derived from a compound derived from a plant material.
- the aliphatic polyester resin (A) is a copolymer, even if the aliphatic polyester resin (A) contains two or more kinds of aliphatic diol units represented by the formula (1). Often, the aliphatic polyester resin (A) may contain two or more kinds of aliphatic dicarboxylic acid units represented by the formula (2).
- the aliphatic dicarboxylic acid unit represented by the formula (2) preferably contains 5 mol% or more and 100 mol% or less of the succinic acid unit with respect to the total dicarboxylic acid unit.
- the amount of the succinic acid constituent unit in the aliphatic polyester resin (A) is preferably 10 mol% or more, more preferably 50 mol% or more, still more preferably 64 mol% or more, based on the total dicarboxylic acid unit. , Particularly preferably 68 mol% or more.
- the ratio of the succinic acid unit to the total dicarboxylic acid unit in the aliphatic polyester resin (A) may be referred to as "succinic acid unit amount".
- the aliphatic dicarboxylic acid unit represented by the formula (2) contains 5 mol% or more and 50 mol% or less of one or more kinds of aliphatic dicarboxylic acid units in addition to succinic acid with respect to the total dicarboxylic acid units. Is more preferable.
- the amount of the aliphatic dicarboxylic acid unit other than succinic acid in the aliphatic polyester resin (A) is preferably 10 mol% or more and 45 mol% or less, more preferably 15 mol, based on the total dicarboxylic acid unit. % Or more and 40 mol% or less.
- the aliphatic diol giving the diol unit represented by the formula (1) is not particularly limited, but from the viewpoint of moldability and mechanical strength, an aliphatic diol having 2 or more and 10 or less carbon atoms is preferable, and 4 or more carbon atoms are preferable. Aliphatic diols of 6 or less are particularly preferred. For example, ethylene glycol, 1,3-propanediol, 1,4-butanediol, 1,4-cyclohexanedimethanol and the like can be mentioned, and 1,4-butanediol is particularly preferable. Two or more kinds of the above aliphatic diols can be used.
- the aliphatic dicarboxylic acid component that gives the aliphatic dicarboxylic acid unit represented by the formula (2) is not particularly limited, but an aliphatic dicarboxylic acid having 2 or more and 40 or less carbon atoms and a derivative such as an alkyl ester thereof are preferable. Derivatives such as an aliphatic dicarboxylic acid having 4 or more and 10 or less carbon atoms and an alkyl ester thereof are particularly preferable.
- Derivatives of aliphatic dicarboxylic acids having 4 or more and 10 or less carbon atoms other than succinic acid and alkyl esters thereof include, for example, adipic acid, suberic acid, sebacic acid, dodecanedioic acid, dimer acid and the like and alkyl esters thereof. Derivatives can be mentioned. Of these, adipic acid and sebacic acid are preferable, and adipic acid is particularly preferable. Two or more kinds of the above aliphatic dicarboxylic acid components can be used, and in this case, a combination of succinic acid and adipic acid is preferable.
- the aliphatic polyester resin (A) may have a repeating unit (aliphatic oxycarboxylic acid unit) derived from the aliphatic oxycarboxylic acid.
- aliphatic oxycarboxylic acid unit derived from the aliphatic oxycarboxylic acid.
- Specific examples of the aliphatic oxycarboxylic acid component giving the aliphatic oxycarboxylic acid unit include lactic acid, glycolic acid, 2-hydroxy-n-butyric acid, 2-hydroxycaproic acid, 6-hydroxycaproic acid, and 2-hydroxy. Examples thereof include -3,3-dimethylbutyric acid, 2-hydroxy-3-methylbutyric acid, 2-hydroxyisocaproic acid and the like, or derivatives such as lower alkyl esters or intramolecular esters thereof.
- any of D-form, L-form, and racemic form may be used, and the form may be any of solid, liquid, and aqueous solution.
- particularly preferred are lactic acid or glycolic acid or derivatives thereof.
- These aliphatic oxycarboxylic acids can be used alone or as a mixture of two or more kinds.
- the content thereof is 100 mol% of all the constituent units constituting the aliphatic polyester resin (A) from the viewpoint of moldability. It is preferably 20 mol% or less, more preferably 10 mol% or less, further preferably 5 mol% or less, and most preferably 0 mol% (not included).
- the aliphatic polyester resin (A) is a trifunctional or higher aliphatic polyhydric alcohol, a trifunctional or higher functional aliphatic polyvalent carboxylic acid or an acid anhydride thereof, or a trifunctional or higher functional aliphatic polyvalent oxycarboxylic acid component.
- the melt viscosity may be increased by polymerization.
- trifunctional aliphatic polyhydric alcohol examples include trimethylolpropane, glycerin and the like.
- tetrafunctional aliphatic polyhydric alcohol examples include pentaerythritol and the like. These can be used alone or in combination of two or more.
- trifunctional aliphatic polyvalent carboxylic acid or its acid anhydride examples include propanetricarboxylic acid or its acid anhydride.
- tetrafunctional polyvalent carboxylic acid or its acid anhydride examples include cyclopentanetetracarboxylic acid or its acid anhydride. These can be used alone or in combination of two or more.
- the trifunctional aliphatic oxycarboxylic acid includes (i) a type having two carboxyl groups and one hydroxyl group in the same molecule, and (ii) a type having one carboxyl group and two hydroxyl groups. It is divided into two types, and any type can be used. From the viewpoint of moldability, mechanical strength and appearance of the molded product, (i) a type having two carboxyl groups and one hydroxyl group in the same molecule, such as malic acid, is preferable, and more specifically, malic acid is preferable. Used.
- the tetrafunctional aliphatic oxycarboxylic acid component is (i) a type in which three carboxyl groups and one hydroxyl group are shared in the same molecule, and (ii) two carboxyl groups and two hydroxyl groups. (Iii) is divided into a type that shares three hydroxyl groups and one carboxyl group in the same molecule, and any type can be used.
- the tetrafunctional aliphatic oxycarboxylic acid component preferably has a plurality of carboxyl groups, and more specific examples thereof include citric acid and tartaric acid. These can be used alone or in combination of two or more.
- the aliphatic polyester resin (A) contains a structural unit derived from such a trifunctional or higher functional component, the content thereof is 100 mol% with all the structural units constituting the aliphatic polyester resin (A) as 100 mol%.
- the lower limit is usually 0 mol% or more, preferably 0.01 mol% or more, and the upper limit is usually 5 mol% or less, preferably 2.5 mol% or less.
- the method for producing the aliphatic polyester resin (A) a known method for producing polyester can be adopted.
- the polycondensation reaction at this time is not particularly limited as appropriate conditions that have been conventionally adopted can be set.
- a method of further increasing the degree of polymerization by performing a reduced pressure operation after advancing the esterification reaction is adopted.
- the object of the produced aliphatic polyester resin (A) is The amount of the diol component and the dicarboxylic acid component used is set so as to have the composition of. Normally, the diol component and the dicarboxylic acid component react in substantially equal molar amounts, but since the diol component is distilled off during the esterification reaction, it is usually 1 to 20 mol% more than the dicarboxylic acid component. Used.
- the aliphatic polyester resin (A) contains a component (arbitrary component) other than essential components such as an aliphatic oxycarboxylic acid unit and a polyfunctional component unit
- the aliphatic oxycarboxylic acid unit and the polyfunctional component unit are also included.
- the corresponding compounds (monomers and oligomers) are subjected to the reaction so as to obtain the desired composition.
- there is no limitation on the timing and method of introducing the above-mentioned arbitrary component into the reaction system and it is arbitrary as long as the aliphatic polyester resin (A) suitable for the present invention can be produced.
- the timing and method for introducing the aliphatic oxycarboxylic acid into the reaction system are not particularly limited as long as it is before the polycondensation reaction between the diol component and the dicarboxylic acid component, and examples thereof include the following (1) and (2). .. (1) A method in which the catalyst is previously dissolved in an aliphatic oxycarboxylic acid solution and mixed (2) A method in which the catalyst is introduced into the reaction system at the time of raw material preparation and simultaneously mixed.
- the compound forming the polyfunctional component unit may be introduced at the same time as other monomers or oligomers in the initial stage of polymerization, or may be charged after the transesterification reaction and before the start of reduced pressure. It is preferable that the compound forming the polyfunctional component unit is charged at the same time as other monomers or oligomers in terms of simplification of the process.
- the aliphatic polyester resin (A) is usually produced in the presence of a catalyst.
- a catalyst that can be used in the production of a known polyester-based resin can be arbitrarily selected as long as the effects of the present invention are not significantly impaired.
- metal compounds such as germanium, titanium, zirconium, hafnium, antimony, tin, magnesium, calcium and zinc are suitable. Of these, germanium compounds and titanium compounds are preferable.
- Examples of the germanium compound that can be used as a catalyst include an organic germanium compound such as tetraalkoxygermanium and an inorganic germanium compound such as germanium oxide and germanium chloride. Among them, germanium oxide, tetraethoxygermanium, tetrabutoxygermanium and the like are preferable, and germanium oxide is particularly preferable, from the viewpoint of price and availability.
- titanium compound that can be used as a catalyst examples include organic titanium compounds such as tetraalkoxytitanium such as tetrapropyl titanate, tetrabutyl titanate, and tetraphenyl titanate. Among them, tetrapropyl titanate, tetrabutyl titanate and the like are preferable from the viewpoint of price and availability.
- One type of catalyst may be used alone, or two or more types may be used in combination in any combination and ratio.
- the amount of the catalyst used is arbitrary as long as the effect of the present invention is not significantly impaired, but is usually 0.0005% by mass or more, more preferably 0.001% by mass or more, and usually 3% by mass with respect to the amount of the monomer used. % Or less, preferably 1.5% by mass or less. If it falls below the lower limit of this range, the effect of the catalyst may not appear. Exceeding the upper limit of this range may result in high manufacturing costs, significant coloration of the resulting polymer, and reduced hydrolysis resistance.
- the introduction time of the catalyst is not particularly limited as long as it is before the polycondensation reaction, and it may be introduced at the time of raw material preparation or at the start of decompression.
- an aliphatic oxycarboxylic acid unit it is introduced at the same time as a monomer or oligomer that forms an aliphatic oxycarboxylic acid unit such as lactic acid or glycolic acid at the time of raw material preparation, or the catalyst is dissolved in an aqueous aliphatic oxycarboxylic acid solution.
- the method of introducing the above is preferable.
- a method of dissolving and introducing a catalyst in an aqueous solution of an aliphatic oxycarboxylic acid is preferable in terms of increasing the polymerization rate.
- the reaction conditions such as temperature, polymerization time, and pressure for producing the aliphatic polyester resin (A) are arbitrary as long as the effects of the present invention are not significantly impaired.
- the reaction temperature of the esterification reaction and / or transesterification reaction between the dicarboxylic acid component and the diol component is usually 150 ° C. or higher, preferably 180 ° C. or higher, and usually 260 ° C. or lower, preferably 250 ° C. or lower.
- the reaction atmosphere is usually an inert atmosphere such as nitrogen or argon.
- the reaction pressure is usually normal pressure to 10 kPa, but normal pressure is particularly preferable.
- the reaction time is usually 1 hour or more, usually 10 hours or less, preferably 6 hours or less, and more preferably 4 hours or less. If the reaction temperature is too high, overproduction of unsaturated bonds may occur, gelation due to unsaturated bonds may occur, and it may be difficult to control the polymerization.
- the polycondensation reaction conditions after the ester reaction and / or transesterification reaction between the dicarboxylic acid component and the diol component are as follows.
- the pressure is usually 0.01 ⁇ 10 3 Pa or more, preferably 0.03 ⁇ 10 3 Pa or more, usually 1.4 ⁇ 10 3 Pa or less, preferably 0.4 ⁇ 10 3 Pa or less under a vacuum degree. It is desirable to do it.
- the reaction temperature is usually 150 ° C. or higher, preferably 180 ° C. or higher, and usually 260 ° C. or lower, preferably 250 ° C. or lower.
- the reaction time is usually 2 hours or more, usually 15 hours or less, preferably 10 hours or less. If the reaction temperature is too high, overproduction of unsaturated bonds may cause gelation due to unsaturated bonds, making it difficult to control polymerization.
- a chain extender such as a carbonate compound or a diisocyanate compound can also be used.
- the amount of the chain extender is usually 10 as the ratio of carbonate bonds and urethane bonds in the aliphatic polyester resin (A) when all the constituent units constituting the aliphatic polyester resin are 100 mol%. It is mol% or less, preferably 5 mol% or less, and more preferably 3 mol% or less.
- the presence of urethane bonds or carbonate bonds in the aliphatic polyester resin (A) may inhibit biodegradability. Therefore, in the present invention, all the constituent units constituting the aliphatic polyester resin (A) are used.
- the carbonate bond is less than 1 mol%, preferably 0.5 mol% or less, more preferably 0.1 mol% or less, and the urethane bond is 0.55 mol% or less, preferably 0.3 mol% or less. It is more preferably 0.12 mol% or less, still more preferably 0.05 mol% or less.
- this amount is 0.9 parts by mass or less, preferably 0.5 parts by mass or less, more preferably 0.2 parts by mass or less, and further preferably 0. . 1 part by mass or less.
- smoke or odor from the molten film from the die outlet may become a problem due to decomposition of the urethane bond in the film forming process or the like, and in the molten film.
- the film may break due to foaming and stable molding may not be possible.
- the amount of carbonate bond and the amount of urethane bond in the aliphatic polyester resin (A) can be calculated and obtained from the NMR measurement results such as 1 H-NMR and 13 C-NMR.
- the carbonate compound as a chain extender examples include diphenyl carbonate, ditril carbonate, bis (chlorophenyl) carbonate, m-cresyl carbonate, dinaphthyl carbonate, dimethyl carbonate, diethyl carbonate, dibutyl carbonate, ethylene carbonate, and the like. Examples thereof include diamyl carbonate and dicyclohexyl carbonate.
- carbonate compounds composed of the same or different types of hydroxy compounds derived from hydroxy compounds such as phenols and alcohols can also be used.
- diisocyanate compound examples include 2,4-tolylene diisocyanate, a mixture of 2,4-tolylene diisocyanate and 2,6-tolylene diisocyanate, 1,5-naphthylene diisocyanate, and xylylene diisocyanate.
- Xylylene diisocyanate hydride hexamethylene diisocyanate, isophorone diisocyanate, 4,4'-dicyclohexylmethane diisocyanate, tetramethylxylylene diisocyanate, 2,4,6-triisopropylphenyldiisocyanate, 4,4'-diphenylmethane diisocyanate, trizine diisocyanate, etc.
- dioxazoline dioxazoline
- silicic acid ester examples include tetramethoxysilane, dimethoxydiphenylsilane, dimethoxydimethylsilane, and diphenyldihydroxysilane.
- High molecular weight polyester resins using these chain extenders can also be manufactured using conventional techniques. After the completion of polycondensation, the chain extender is added to the reaction system in a uniform molten state without a solvent, and is reacted with the polyester obtained by polycondensation.
- the terminal group obtained by catalytically reacting the diol component and the dicarboxylic acid component has substantially a hydroxyl group and has a weight average molecular weight (Mw) of 20,000 or more, preferably 40,000.
- Mw weight average molecular weight
- a polyester resin having a higher molecular weight can be obtained.
- Prepolymers with a weight average molecular weight of 20,000 or more are not affected by the remaining catalyst even under harsh conditions such as a molten state by using a small amount of chain extender, so that they are high without forming a gel during the reaction.
- a polyester resin having a molecular weight can be produced.
- the weight average molecular weight (Mw) of the polyester resin is obtained as a conversion value using monodisperse polystyrene from a value measured by gel permeation chromatography (GPC) at a measurement temperature of 40 ° C. using chloroform as a solvent.
- a prepolymer having a weight average molecular weight of 20,000 or more, preferably 40,000 or more is used.
- the weight average molecular weight is less than 20,000, the amount of the diisocyanate compound used for increasing the molecular weight may increase and the heat resistance may decrease.
- a polyester resin having a urethane bond having a linear structure linked via a urethane bond derived from a diisocyanate compound is produced.
- the pressure at the time of chain extension is usually 0.01 MPa or more and 1 MPa or less, preferably 0.05 MPa or more and 0.5 MPa or less, more preferably 0.07 MPa or more and 0.3 MPa or less, but normal pressure is the most preferable.
- the reaction temperature during chain extension is usually 100 ° C. or higher, preferably 150 ° C. or higher, more preferably 190 ° C. or higher, most preferably 200 ° C. or higher, usually 250 ° C. or lower, preferably 240 ° C. or lower, more preferably 230 ° C. or higher. It is as follows. If the reaction temperature is too low, the viscosity is high and uniform reaction is difficult, and high stirring power tends to be required. If the reaction temperature is too high, gelation and decomposition of the polyester resin tend to occur at the same time.
- the reaction time at the time of chain extension is usually 0.1 minutes or more, preferably 1 minute or more, more preferably 5 minutes or more, usually 5 hours or less, preferably 1 hour or less, more preferably 30 minutes or less, most preferably. It is less than 15 minutes. If the reaction time is too short, the effect of adding the chain extender tends not to be exhibited. If the reaction time is too long, gelation and decomposition of the polyester resin tend to occur at the same time.
- the molecular weight of the aliphatic polyester resin (A) is a weight average molecular weight (Mw) of monodisperse polystyrene as a standard substance measured by gel permeation chromatography (GPC), and is usually 10,000 or more and 1,000,000 or more. It is as follows. This Mw is preferably 20,000 or more and 500,000 or less, and more preferably 50,000 or more and 400,000 or less because it is advantageous in terms of moldability and mechanical strength.
- Mw weight average molecular weight of monodisperse polystyrene as a standard substance measured by gel permeation chromatography (GPC), and is usually 10,000 or more and 1,000,000 or more. It is as follows. This Mw is preferably 20,000 or more and 500,000 or less, and more preferably 50,000 or more and 400,000 or less because it is advantageous in terms of moldability and mechanical strength.
- the melt flow rate (MFR) of the aliphatic polyester resin (A) is a value measured at 190 ° C. and a load of 2.16 kg based on JIS K7210 (1999), and is usually 0.1 g / 10 minutes or more and 100 g / 10. Less than a minute. From the viewpoint of moldability and mechanical strength, this MFR is preferably 50 g / 10 minutes or less, and particularly preferably 40 g / 10 minutes or less.
- the MFR of the aliphatic polyester resin (A) can be adjusted by the molecular weight.
- the melting point of the aliphatic polyester resin (A) is preferably 70 ° C. or higher, more preferably 75 ° C. or higher, preferably 170 ° C. or lower, more preferably 150 ° C. or lower, and particularly preferably lower than 130 ° C. When there are a plurality of melting points, it is preferable that at least one melting point is within the above range. If the melting point is out of the above range, the moldability is poor.
- the elastic modulus of the aliphatic polyester resin (A) is preferably 180 to 1000 MPa. If the elastic modulus is less than 180 MPa, a problem is likely to occur in molding workability. If the elastic modulus exceeds 1000 MPa, the impact resistance tends to deteriorate.
- the method for adjusting the melting point and elastic modulus of the aliphatic polyester resin (A) is not particularly limited.
- the melting point and elastic modulus are adjusted by selecting the type of copolymerization component of an aliphatic dicarboxylic acid component other than succinic acid, adjusting the copolymerization ratio of each, or combining them. It is possible.
- the aliphatic polyester resin (A) is not limited to one type, and two or more types of aliphatic polyester resins (A) having different types of constituent units, constituent unit ratios, manufacturing methods, physical characteristics, etc. can be blended and used.
- aliphatic-aromatic polyester resin (B) As the aliphatic-aromatic polyester resin (B), at least a part of the repeating unit of the above-mentioned aliphatic polyester resin (A) is replaced with an aromatic compound unit, preferably the above-mentioned aliphatic compound.
- a system resin is exemplified.
- the aromatic compound unit examples include an aromatic diol unit having an aromatic hydrocarbon group which may have a substituent and an aromatic dicarboxylic acid having an aromatic hydrocarbon group which may have a substituent.
- aromatic dicarboxylic acid unit having an aromatic heterocyclic group which may have a substituent examples include an aromatic dicarboxylic acid unit having an aromatic heterocyclic group which may have a substituent, an aromatic oxycarboxylic acid unit having an aromatic hydrocarbon group which may have a substituent, and the like. ..
- the aromatic hydrocarbon group and the aromatic heterocyclic group may be a single ring, or may be one in which a plurality of rings are bonded or condensed with each other.
- aromatic hydrocarbon group examples include 1,2-phenylene group, 1,3-phenylene group, 1,4-phenylene group, dinaphthylene group, diphenylene group and the like.
- aromatic heterocyclic group examples include a 2,5-furandyl group.
- aromatic dicarboxylic acid component that gives an aromatic dicarboxylic acid unit
- aromatic dicarboxylic acid component that gives an aromatic dicarboxylic acid unit
- isophthalic acid naphthalenedicarboxylic acid
- diphenyldicarboxylic acid 2,5-frangicarboxylic acid.
- terephthalic acid is preferable.
- the aromatic dicarboxylic acid component may be a derivative of an aromatic dicarboxylic acid compound.
- the derivative of the aromatic dicarboxylic acid component exemplified above is preferable, and among them, a lower alkyl ester having 1 or more and 4 or less carbon atoms, an acid anhydride and the like can be mentioned.
- Specific examples of the derivative of the aromatic dicarboxylic acid compound include lower alkyl esters such as methyl ester, ethyl ester, propyl ester and butyl ester of the above-exemplified aromatic dicarboxylic acid component; and the above-exemplified aromatic dicarboxylic acid such as succinic anhydride. Cyclic acid anhydride as an acid component; and the like. Of these, dimethyl terephthalate is preferable.
- the aromatic diol component may be a derivative of an aromatic diol compound. Further, it may be a compound having a structure in which a plurality of aliphatic diol compounds and / or aromatic diol compounds are dehydrated and condensed with each other.
- aromatic oxycarboxylic acid component that gives the aromatic oxycarboxylic acid unit include p-hydroxybenzoic acid and p- ⁇ -hydroxyethoxybenzoic acid.
- the aromatic oxycarboxylic acid component may be a derivative of an aromatic oxycarboxylic acid compound. Further, it may be a compound (oligomer) having a structure in which a plurality of aliphatic oxycarboxylic acid compounds and / or aromatic oxycarboxylic acid compounds are dehydrated and condensed with each other. That is, an oligomer may be used as a raw material.
- any of the D-form, the L-form, and the racemic form may be used.
- the aromatic compound component is not limited to the above example as long as it can provide an aromatic compound unit.
- One type of aromatic compound component may be used alone, or two or more types may be used in any combination and in a ratio.
- an aromatic dicarboxylic acid component As the aliphatic-aromatic polyester resin (B), it is preferable to use an aromatic dicarboxylic acid component as a component that gives an aromatic compound unit, and the content of the aromatic dicarboxylic acid unit in this case is the aliphatic dicarboxylic acid. Based on the total amount of the unit and the aromatic dicarboxylic acid unit (100 mol%), it is preferably 10 mol% or more and 80 mol% or less. It is preferable to use terephthalic acid or 2,5-furandicarboxylic acid as the aromatic dicarboxylic acid component.
- polybutylene terephthalate adipate and / or polybutylene terephthalate succinate resin is preferable.
- a polybutylene-2,5-flange carboxylate resin is also preferable.
- the aliphatic-aromatic polyester resin (B) can be produced in the same manner as the above-mentioned aliphatic polyester resin (A) by using at least an aromatic compound component as a raw material.
- the molecular weight of the aliphatic-aromatic polyester resin (B) is a weight average molecular weight (Mw) of monodisperse polystyrene as a standard substance measured by gel permeation chromatography (GPC), and is usually 10,000 or more. It is less than one million. This Mw is preferably 30,000 or more and 800,000 or less, and more preferably 50,000 or more and 600,000 or less because it is advantageous in terms of moldability and mechanical strength.
- Mw weight average molecular weight of monodisperse polystyrene as a standard substance measured by gel permeation chromatography (GPC), and is usually 10,000 or more. It is less than one million. This Mw is preferably 30,000 or more and 800,000 or less, and more preferably 50,000 or more and 600,000 or less because it is advantageous in terms of moldability and mechanical strength.
- the melt flow rate (MFR) of the aliphatic-aromatic polyester resin (B) is a value measured at 190 ° C. and a load of 2.16 kg based on JIS K7210 (1999), and is usually 0.1 g / 10 minutes or more. It is 100 g / 10 minutes or less. From the viewpoint of moldability and mechanical strength, this MFR is preferably 50 g / 10 minutes or less, and particularly preferably 30 g / 10 minutes or less.
- the MFR of the aliphatic-aromatic polyester resin (B) can be adjusted by the molecular weight.
- the melting point of the aliphatic-aromatic polyester resin (B) is usually 60 ° C. or higher, preferably 70 ° C. or higher, more preferably 80 ° C. or higher, preferably 150 ° C. or lower, more preferably 140 ° C. or lower, particularly preferably. It is 120 ° C. or lower. When there are a plurality of melting points, it is preferable that at least one melting point is within the above range. If the melting point is out of the above range, the moldability is poor.
- the elastic modulus of the aliphatic-aromatic polyester resin (B) is preferably 180 to 1000 MPa. If the elastic modulus is less than 180 MPa, a problem is likely to occur in molding workability. If the elastic modulus exceeds 1000 MPa, the impact resistance tends to deteriorate.
- the method for adjusting the melting point and elastic modulus of the aliphatic-aromatic polyester resin (B) is not particularly limited.
- the type of copolymerization component of the aliphatic dicarboxylic acid component other than the aromatic dicarboxylic acid component adjusting the copolymerization ratio of each, or combining them, the melting point and elastic modulus can be obtained. It is possible to adjust.
- the aliphatic-aromatic polyester resin (B) is not limited to one type, and two or more types of aliphatic-aromatic polyester resin (B) having different types of constituent units, composition unit ratios, manufacturing methods, physical characteristics, etc. are used. It can be blended and used.
- the aliphatic oxycarboxylic acid-based resin (C) contains an aliphatic oxycarboxylic acid unit as a main constituent unit.
- the aliphatic oxycarboxylic acid unit is preferably represented by the following formula (3). -OR 3- CO- (3)
- R 3 represents a divalent aliphatic hydrocarbon group or a divalent alicyclic hydrocarbon group.
- aliphatic oxycarboxylic acid component giving the aliphatic oxycarboxylic acid unit of the formula (3) include lactic acid, glycolic acid, 2-hydroxy-n-butyric acid, 2-hydroxycaproic acid, 2-hydroxy-3, Examples thereof include 3-dimethylbutyric acid, 2-hydroxy-3-methylbutyric acid, 2-hydroxyisocaproic acid, and mixtures thereof.
- optical isomers are present in these, either D-form or L-form may be used.
- lactic acid or glycolic acid is preferred. Two or more kinds of these aliphatic oxycarboxylic acid components can be mixed and used.
- polylactic acid (PLA) is particularly preferable.
- Urethane bonds, amide bonds, carbonate bonds, ether bonds, etc. can be introduced into the aliphatic oxycarboxylic acid resin (C) as long as it does not affect the biodegradability.
- the method for producing the aliphatic oxycarboxylic acid resin (C) is not particularly limited, and it can be produced by a known method such as a direct polymerization method of oxycarboxylic acid or a ring-opening polymerization method of a cyclic body.
- the molecular weight of the aliphatic oxycarboxylic acid resin (C) is a weight average molecular weight (Mw) of monodisperse polystyrene as a standard substance measured by gel permeation chromatography (GPC), and is usually 10,000 or more and 1,000 or more. It is less than 000. This Mw is preferably 20,000 or more and 500,000 or less, and more preferably 50,000 or more and 400,000 or less because it is advantageous in terms of moldability and mechanical strength.
- the melt flow rate (MFR) of the aliphatic oxycarboxylic acid resin (C) is a value measured at 190 ° C. and a load of 2.16 kg based on JIS K7210 (1999), and is usually 0.1 g / 10 minutes or more and 100 g. / 10 minutes or less. From the viewpoint of moldability and mechanical strength, this MFR is preferably 50 g / 10 minutes or less, and particularly preferably 40 g / 10 minutes or less.
- the MFR of the aliphatic oxycarboxylic acid resin (C) can be adjusted by the molecular weight.
- the melting point of the aliphatic oxycarboxylic acid resin (C) is preferably 70 ° C. or higher, more preferably 75 ° C. or higher, preferably 170 ° C. or lower, more preferably 150 ° C. or lower, and particularly preferably lower than 130 ° C. When there are a plurality of melting points, it is preferable that at least one melting point is within the above range. If the melting point is out of the above range, the moldability is poor.
- the elastic modulus of the aliphatic oxycarboxylic acid resin (C) is preferably 180 to 1000 MPa. If the elastic modulus is less than 180 MPa, a problem is likely to occur in molding workability. If the elastic modulus exceeds 1000 MPa, the impact resistance tends to deteriorate.
- the method for adjusting the melting point and elastic modulus of the aliphatic oxycarboxylic acid resin (C) is not particularly limited.
- the polyhydroxyalkanoate (D) described below can also be preferably used.
- the polyhydroxyalkanoate (hereinafter, may be referred to as PHA) (D) preferably used in the present invention has a general formula: [-CHR-CH2-CO-O-] (in the formula, R has 1 carbon atom). It is an aliphatic polyester containing a repeating unit represented by (15) alkyl groups), and is a copolymer containing a 3-hydroxybutyrate unit and a 3-hydroxyhexanoate unit as main constituent units.
- the polyhydroxyalkanoate (D) preferably contains 80 mol% or more of 3-hydroxybutyrate units as a constituent component, and more preferably 85 mol% or more. Further, those produced by microorganisms are preferable.
- polyhydroxyalkanoate (D) examples include poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) copolymer resin and poly (3-hydroxybutyrate-co-3-hydroxyvalerate-). Co-3-hydroxyhexanoate) copolymer resin and the like can be mentioned.
- a poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) copolymer resin, that is, PHBH is preferable from the viewpoint of molding processability and physical properties of the obtained molded product.
- polyhydroxyalkanoate (D) a comonomer such as 3-hydroxybutyrate (hereinafter, may be referred to as 3HB) and 3-hydroxyhexanoate copolymerized (hereinafter, may be referred to as 3HH).
- 95/5 to 85/15 (mol% / mol%) is more preferable.
- the comonomer ratio is less than 3 mol%, it may be difficult to perform molding because the molding processing temperature and the thermal decomposition temperature are close to each other. If the comonomer ratio exceeds 20 mol%, the crystallization of polyhydroxyalkanoate (D) may be delayed and the productivity may be deteriorated.
- the ratio of each monomer in polyhydroxyalkanoate (D) can be measured by gas chromatography as follows.
- the molecular weight of polyhydroxyalkanoate (D) is a weight average molecular weight (Mw) of monodisperse polystyrene as a standard substance measured by the above-mentioned gel permeation chromatography (GPC), and is usually 200,000 or more and 2,500. It is 000 or less. This Mw is preferably 250,000 or more and 2,000,000 or less, and more preferably 300,000 or more and 1,000,000 or less because it is advantageous in terms of moldability and mechanical strength. If Mw is less than 200,000, the mechanical properties and the like may be inferior. If Mw exceeds 2.5 million, molding may become difficult.
- Mw weight average molecular weight
- the melt flow rate (MFR) of polyhydroxyalkanoate (D) is a value measured at 190 ° C. and a load of 2.16 kg based on JIS K7210 (1999), and is preferably 1 g / 10 minutes or more and 100 g / 10 minutes or less. Is. From the viewpoint of moldability and mechanical strength, this MFR is more preferably 80 g / 10 minutes or less, and particularly preferably 50 g / 10 minutes or less.
- the MFR of polyhydroxy alkanoate (D) can be adjusted by molecular weight.
- the melting point of polyhydroxyalkanoate (D) is preferably 100 ° C. or higher, more preferably 120 ° C. or higher, 180 ° C. or lower, more preferably 170 ° C. or lower, and particularly preferably less than 160 ° C. When there are a plurality of melting points, it is preferable that at least one melting point is within the above range.
- Polyhydroxyalkanoate (D) is, for example, Alcaligenes europhos AC32 strain in which a PHA synthase gene derived from Aeromonas caviae is introduced into Alcaligenes europhos (international deposit based on the Budapest Treaty, international deposit authority: Patent of the Institute of Industrial Technology, Incorporated Administrative Agency) Biological Deposit Center (1-1-1, Central 6th, Higashi, Tsukuba-shi, Ibaraki, Japan), Hara Deposit date: Transferred on August 12, 1996, August 7, 1997, Deposit number FERM BP-6038 (Hara) It is produced by microorganisms such as Deposited FERM P-15786)) (J. Bacteriol., 179, 4821 (1997)).
- polyhydroxy alkanoate (D) As the polyhydroxy alkanoate (D), a commercially available product can also be used. Commercially available products of polyhydroxyalkanoate (D) containing 3-hydroxybutyrate unit and 3-hydroxyhexanoate unit as main constituent units include "PHBH X331N”, “PHBH X131A”, and "PHBH X151A” manufactured by Kaneka Corporation. Etc. can be used.
- the aliphatic oxycarboxylic acid resin (C) including the polyhydroxy alkanoate (D) is not limited to one type, and two or more types having different types of constituent units, constituent unit ratios, production methods, physical properties, etc.
- the aliphatic oxycarboxylic acid resin (C) of the above can be blended and used.
- the biodegradable resin composition of the present invention includes fillers (fillers), plasticizers, antistatic agents, antioxidants, light stabilizers, ultraviolet absorbers, dyes, pigments, etc. Antistatic agents, crystal nucleating agents, anti-blocking agents, weather resistant agents, heat stabilizers, flame retardants, mold release agents, antifogging agents, surface wetting improvers, incineration aids, dispersion aids, various surfactants, slips One or more of various additives such as agents may be contained as "other components".
- the biodegradable resin composition of the present invention may also contain a freshness-preserving agent, an antibacterial agent, etc. as a functional additive.
- the total amount of the other components is the raw material of the present invention. It is preferably 0.01% by mass or more and 40% by mass or less with respect to the total amount of the degradable resin composition.
- the biodegradable resin composition of the present invention is produced by kneading the above-mentioned degradable resin and the decomposition accelerator of the present invention in a kneader and dispersing the decomposition accelerator in the biodegradable resin.
- other resins and other components used as needed may be mixed in the kneader together with the decomposition accelerator and the biodegradable resin.
- the decomposition accelerator and biodegradable resin of the present invention are mixed with other resins and other components used as necessary in a predetermined ratio at the same time or in any order, in a tumbler and a V-type blender. , Nauter mixer, tumbler mixer, kneading roll, extruder and other mixers, and preferably further melt kneading.
- the kneader used in the mixing step may be a melt kneader.
- the extruder may be either a twin-screw extruder or a single-screw extruder, but a twin-screw extruder is more preferable.
- the temperature during melt-kneading is preferably 140 to 220 ° C. Within this temperature range, the time required for the melting reaction can be shortened, deterioration of the color tone due to deterioration of the resin and carbonization of the decomposition accelerator can be prevented, and impact resistance, moisture heat resistance, etc. The physical characteristics in terms of practical use can be further improved. From the same viewpoint, the melt-kneading temperature is more preferably 150 to 210 ° C.
- the melt-kneading time should avoid unnecessary lengthening from the viewpoint of more reliably avoiding resin deterioration and the like, and is preferably 20 seconds or more and 20 minutes or less, more preferably 30 seconds or more and 15 seconds. Less than a minute. Therefore, it is preferable to set the conditions of the melt-kneading temperature and time so as to satisfy the melt-kneading condition.
- the biodegradable resin composition of the present invention can be molded by various molding methods applied to general-purpose plastics.
- the molding method include compression molding (compression molding, laminate molding, stampable molding), injection molding, extrusion molding and co-extrusion molding (film molding by inflation method and T-die method, laminate molding, pipe molding, electric wire / cable molding). , Deformed material molding), hot press molding, hollow molding (various blow molding), calendar molding, solid molding (uniaxial stretching molding, biaxial stretching molding, roll rolling molding, stretch orientation non-woven molding, thermal molding (vacuum molding, pressure emptying) Molding), plastic processing, powder molding (rotary molding), various non-woven molding (dry method, bonding method, entanglement method, spunbond method, etc.), etc.
- injection molding, extrusion molding, compression molding, or hot pressing. Molding is preferably applied.
- As a specific shape of the molded product application to a sheet, a film, or a container is preferable.
- the biodegradable resin molded product of the present invention formed by molding the decomposition accelerator of the present invention has a chemical function, an electrical function, a magnetic function, a mechanical function, a friction / wear / lubrication function, and an optical function. It is also possible to perform various secondary processes for the purpose of imparting thermal functions, surface functions such as biocompatibility, and the like. Examples of secondary processing include embossing, painting, adhesion, printing, metallizing (plating, etc.), machining, surface treatment (antistatic treatment, corona discharge treatment, plasma treatment, photochromism treatment, physical vapor deposition, chemical vapor deposition, etc.) Coating, etc.) and the like.
- the biodegradable resin molded product of the present invention comprising the biodegradable resin composition of the present invention is used as a packaging material for packaging liquids, powders and solids such as various foods, chemicals and miscellaneous goods, and for agriculture. It is suitably used in a wide range of applications such as materials and building materials. Specific uses include injection molded products (eg, fresh food trays, fast food containers, coffee capsule containers, cutlery, outdoor leisure products, etc.), extruded products (eg, films, sheets, fishing threads, fishing nets, etc.). Examples include vegetation nets, secondary processing sheets, water retention sheets, etc.), hollow molded products (bottles, etc.), and the like.
- injection molded products eg, fresh food trays, fast food containers, coffee capsule containers, cutlery, outdoor leisure products, etc.
- extruded products eg, films, sheets, fishing threads, fishing nets, etc.
- Examples include vegetation nets, secondary processing sheets, water retention sheets, etc.), hollow molded products (bot
- the biodegradable resin molded product of the present invention is suitable as a container for food such as a film for food packaging, a tray for fresh food, a container for fast food, and a lunch box.
- the nitrogen and carbon contents were measured using a commercially available automatic combustion method analyzer capable of simultaneously analyzing nitrogen and carbon.
- the nitrogen content is based on the total nitrogen measuring method or nitrogen measuring device conforming to JIS K0102, JIS K6451-1, JIS M8819, JIS Z7302-8, etc., and the carbon content is JIS K0102, JIS M8819, JIS Z7302-8, etc. It can also be measured individually by a carbon measuring device suitable for.
- ⁇ Measurement and calculation of cellulose, lignin, and hemicellulose contents The contents of cellulose, lignin, and hemicellulose in the decomposition accelerator I are as follows from the values of heat-resistant ⁇ -amylase-treated neutral detergent fiber (aNDFom), acidic detergent fiber (ADFom), and acidic detergent lignin (ADL). It was calculated by the formula of.
- aNDFom heat-resistant ⁇ -amylase-treated neutral detergent fiber
- ADFom acidic detergent fiber
- ADL acidic detergent lignin
- Cellulose (%) ADFom (%) -ADL (%)
- Lignin (%) ADL (%)
- the cellulose and lignin contents of the decomposition accelerator II were also measured and calculated in the same manner as above from the values of the acidic detergent fiber (ADFom) and the acidic detergent lignin (ADL).
- moisture As a method for quantifying water content, a heat weight loss method was used. In this method, an automatic moisture measuring device was used to dry the sample at 135 ° C. until the rate of weight change became constant or less, and the moisture content was determined from the weight change before and after that, and the weighed sample was measured at 135 ° C. for 2 hours. It may be dried, allowed to cool in a desiccator, weighed, and the weight loss may be calculated as the amount of water. In addition, the Karl Fischer method or the like can also be used as a method for quantifying water content.
- the Kjeldahl method was used as a method for quantifying crude protein.
- sulfuric acid and a decomposition accelerator are first added to the sample and heated to convert nitrogen in the sample into an ammonium salt.
- sodium hydroxide is added and heated, and the generated ammonia is collected with sulfuric acid having a known concentration.
- the amount of nitrogen is measured by titrating this solution with an aqueous solution of sodium hydroxide having a known concentration.
- the value of the obtained amount of nitrogen (mass%) is multiplied by the nitrogen-protein conversion coefficient (6.25) to obtain the crude protein mass.
- an automatic analyzer by the combustion method Dumas method
- ⁇ Crude fat As a method for quantifying crude fat, a diethyl ether extraction method was used. In this method, the fat in the sample is extracted with diethyl ether using a Soxhlet extractor, and the extract obtained by volatilizing diethyl ether is weighed as crude fat. As a method for quantifying crude fat, an acid decomposition diethyl ether extraction method or the like can also be used.
- ⁇ Crude fiber ⁇ A filtration method was used as a method for quantifying crude fibers.
- sulfuric acid is first added to the sample, boiled and filtered.
- an aqueous sodium hydroxide solution is added to the insoluble material, the mixture is boiled and filtered, and the obtained residue is dried and weighed.
- the residue is heated and incinerated at 550 to 600 ° C. and weighed. The amount of crude fiber is calculated from the weight difference before and after heat ashing.
- the direct ashing method was used as the method for quantifying the crude ash content.
- the amount of crude ash is determined by heating and ashing the sample at 550 to 600 ° C. and measuring the weight.
- the particle size distribution of the decomposition accelerator was measured using a laser diffraction type particle size distribution measuring device (“SALD-2300” manufactured by Shimadzu Corporation).
- SALD-2300 laser diffraction type particle size distribution measuring device
- the particles of the decomposition accelerator dispersed in a dispersion medium such as water are irradiated with laser light, and the observed pattern of diffracted scattered light and the decomposition accelerator are arranged in a large number of spherical particles having different diameters.
- the distribution of particle size and frequency so that they match was calculated.
- the particle size (referred to as 50% particle size or medium diameter) which becomes 50% of the total mass when the mass is integrated in order from the particles having the smallest diameter is defined as the 50% cumulative mass particle size.
- the weight change before and after standing was measured, and the rate of change (ratio of mass loss to mass before standing) was calculated.
- the calculated rate of change was calculated as a ratio to the rate of change in the case of only the aliphatic polyester resin (A) of Comparative Example I-1 or II-1, and used as the biodegradability improvement rate.
- the Elmendorf tear strength was measured according to ISO6383-2 (1983). The larger this value is, the more preferable it is.
- Biodegradable resin In the following Examples and Comparative Examples, the following aliphatic polyester resin (A) was used as the biodegradable resin (A): Polybutylene succinate adipate (PBSA). Product name BioPBS TM FD92PB manufactured by PTTMCC Biochem Melting point: 89 ° C
- Table 1 shows the nitrogen, carbon, cellulose, lignin, and hemicellulose contents (% by mass), nitrogen / carbon ratio, and hemicellulose / (cellulose + lignin) ratio obtained for each raw material.
- Examples I-1 to I-3, Comparative Examples I-1 to I-5 The aliphatic polyester resin (A) and the decomposition accelerator are blended in the ratios shown in Table 2, and using a small twin-screw kneader (“Xplore Micro 15cc Twin Screw Compounder” manufactured by DSM), 170 under a nitrogen atmosphere. Melt kneading was performed at ° C. for 4 minutes.
- the biodegradability of the biodegradable resin can be greatly improved by using the decomposition accelerator I of the present invention in which the nitrogen / carbon ratio and the hemicellulose / (cellulose + lignin) ratio are within the predetermined ranges. I know I can do it.
- Table 3 shows the soluble nitrogen-free substances obtained for each raw material, the contents of cellulose and lignin, and the total (mass%) thereof.
- Example II-1 to II-3 Comparative Examples II-1 to II-4
- the aliphatic polyester resin (A) and the decomposition accelerator are blended at the ratios shown in Table 4, and using a small twin-screw kneader (“Xplore Micro 15cc Twin Screw Composer” manufactured by DSM), 170 under a nitrogen atmosphere. Melt kneading was performed at ° C. for 4 minutes.
- Uncrushed wheat bran (manufactured by Showa Sangyo Co., Ltd.) is pre-crushed with a cutter mill until it passes through a sieve with a mesh size of 500 ⁇ m, and then a jet mill crusher (“Nanojet Mizer NJ-50” manufactured by Aisin Nano Technologies) is used.
- the wheat bran was finely pulverized under the conditions of an air pressure of 1.4 MPa and a treatment rate of 60 g / h, and then dried at 80 ° C. for 8 hours to produce wheat bran having a 50% cumulative particle size of 9 ⁇ m.
- the moisture content of each of the above 50% cumulative particle sizes 56 ⁇ m, 35 ⁇ m, and 9 ⁇ m was 8.2 mass% before drying and 2.3 mass% after drying.
- Examples III-1 to III-3 As a decomposition accelerator, wheat bran having a cumulative particle size of 56 ⁇ m, 35 ⁇ m, or 9 ⁇ m obtained as described above was used in an amount of 11.1 parts by mass with respect to 100 parts by mass of the aliphatic polyester resin (A). The mixture was blended at a ratio and melt-kneaded at 170 ° C. for 4 minutes in a nitrogen atmosphere using a small twin-screw kneader (“Xplore Micro 15cc Twin Screen Compounder” manufactured by DSM).
- the obtained resin composition was molded into a film having a thickness of about 120 ⁇ m with a hot press molding machine at 170 ° C., and cut out into a shape specified in each test standard.
- the stress at break, the Ermendorf tear strength, and the puncture impact strength were evaluated. The results are shown in Table 5.
Landscapes
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Mechanical Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Manufacturing & Machinery (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Biological Depolymerization Polymers (AREA)
Abstract
脂肪族ポリエステル系樹脂、脂肪族-芳香族ポリエステル系樹脂、脂肪族オキシカルボン酸系樹脂などの生分解性樹脂の生分解速度や分解率を高め、かつ任意に制御することができる、生分解性樹脂に適した分解促進剤と、この分解促進剤を含む生分解性樹脂組成物を提供する。セルロース、ヘミセルロース及びリグニンを含む生分解性樹脂用分解促進剤であって、該生分解性樹脂用分解促進剤中の炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、生分解性樹脂用分解促進剤。
Description
本発明は、生分解性樹脂の生分解を促進することが可能な分解促進剤及びその製造方法に関する。本発明はまた、この生分解性樹脂用分解促進剤を含む生分解性樹脂組成物、及びこの生分解性樹脂組成物を押出成形又は射出成形してなる生分解性樹脂成形体に関する。
近年、プラスチック製品の海洋廃棄等による、生態系や環境汚染の懸念が顕在化している。世界各国において、環境汚染防止の観点などから様々な規制が制定されつつある。例えば、欧州では、小売業における使い捨てプラスチック製レジ袋や使い捨てプラスチック容器のカップや皿の使用を禁じる規制や法律が制定されつつある。この法律の使用禁止措置の対象外とするためには、ここで定められたバイオマス必要最低含有量以上のバイオマスを原料とし、かつ一般家庭でも堆肥にすることが可能な製品(ホームコンポスト可能な製品)であることが必要となる。また、最近では海洋に流出したプラスチック製品が海中でも分解する、海洋生分解可能なプラスチック製品も希求されてきている。
従来から、生分解性を有するプラスチック(樹脂)として、例えばポリブチレンテレフタレート/アジペート(以下、PBATと略する)、ポリ乳酸(以下、PLAと略記する)、ポリブチレンサクシネート(以下、PBSと略記する)、ポリブチレンサクシネート/アジペート(以下、PBSAと略記する)等の脂肪族ポリエステル、ポリヒドロキシアルカノール(以下、PHAと略記する)などが知られている。PHAには、ポリ(3-ヒドロキシブチレート)(以下、PHBと略する)、ポリ(3-ヒドロキシブチレート/3-ヒドロキシバレレート)(以下、PHBVと略する)、ポリ(3-ヒドロキシブチレート/3-ヒドロキシヘキサノエート)(以下、PHBHと略する)、ポリ(3-ヒドロキシブチレート/4-ヒドロキシブチレート)などがある。
これらの樹脂は、樹脂単独での生分解の速度や分解率が異なるだけでなく、引張破断伸びや曲げ弾性率などの機械的特性なども異なる。このため、一般的には、使用する用途や場所などに合わせて、それぞれの樹脂を組み合わせたり、他の副資材や添加剤と混合して機械強度などの物性を向上させて使用されている。
特許文献1には、製品の使用を終えた後の最終的廃棄処理をも考慮に入れ、埋め立て時には分解して環境に残存せず、焼却時にはダイオキシン等の有害物質を排出しない、生分解性樹脂とバイオマス材料から成る成形材料として、脂肪族芳香族ポリエステル(A成分)5~95重量%、セルロース、リグノセルロース又はデンプン系物質(B成分)95~5重量%に対して、不飽和カルボン酸又はその誘導体及び有機過酸化物(C成分)を含有する組成物を加熱混練するなどして、成形加工性に優れ、強度特性にも優れた複合樹脂組成物を製造する方法が開示されている。
特許文献2には、高分子量の脂肪族ポリエステルにセルロースを含有する植物材料、例えば、植物の表皮又は皮層からなるもの、又は、種皮材料、堅果の果皮の材料、おがくず、製粉くず、又は、サトウキビの茎のバガスなどを配合した樹脂組成物及び成形体が開示されている。
ホームコンポスト可能なプラスチック製品や海洋生分解可能なプラスチック製品にあっては、その生分解速度や分解率は、一般的に生分解可能であると考えられてきた従来の条件よりもより厳しい条件下(たとえば、低温かつ分解菌が少ない環境下)で、従来のPBATやPBS、PBSA、PLA、PHBHなどの生分解性樹脂の生分解速度よりも速いこと、そして同じ時間でも多く分解することが必要とされる。
特許文献1~2に記載の樹脂組成物やそれからなる成形体は、機械強度はある程度向上されるが、ここでは生分解速度や分解率には着目されておらず、実際に生分解速度や分解率が、従来の生分解性樹脂よりもどの程度高くなったのかは不明である。また、使用する添加剤や副資材によっては、分解率が悪化し、機械強度が向上しないものもあった。
本発明の目的は、脂肪族ポリエステル系樹脂、脂肪族-芳香族ポリエステル系樹脂、脂肪族オキシカルボン酸系樹脂などの生分解性樹脂の生分解速度や分解率を高め、かつ任意に制御することができる、生分解性樹脂に適した分解促進剤及びその製造方法と、この分解促進剤を含む生分解性樹脂組成物及びその成形体を提供することにある。
本発明者は、窒素及び炭素と、セルロース、ヘミセルロース及びリグニンの3つの成分、或いは、可溶無窒素物、セルロース及びリグニンの3つの成分に着目し、これらの成分が、生分解性樹脂の分解の促進に寄与ないし関与しているであろうと考えた。この考えのもと、これらの配合比率等を調整することにより、生分解性樹脂に配合した場合に、その生分解速度や分解率を生分解性樹脂本来の生分解速度や分解率よりも高めることができることを見出した。
本発明の要旨は、下記の[1]~[12]に存する。
[1] セルロース、ヘミセルロース及びリグニンを含む生分解性樹脂用分解促進剤であって、該生分解性樹脂用分解促進剤中の炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、生分解性樹脂用分解促進剤。
[2] 可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下である、生分解性樹脂用分解促進剤。
[3] 水分率が5質量%未満である、[1]又は[2]に記載の生分解性樹脂用分解促進剤。
[4] [1]~[3]のいずれかに記載の生分解性樹脂用分解促進剤2質量部以上250質量部以下と生分解性樹脂100質量部を含む生分解性樹脂組成物。
[5] 前記生分解性樹脂が脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)及び脂肪族オキシカルボン酸系樹脂(C)からなる群より選ばれる1種以上である、[4]に記載の生分解性樹脂組成物。
[6] 前記脂肪族ポリエステル系樹脂(A)が脂肪族ジオールに由来する繰返し単位と脂肪族ジカルボン酸に由来する繰返し単位とを主構成単位として含む、[5]に記載の生分解性樹脂組成物。
[7] 前記脂肪族-芳香族ポリエステル系樹脂(B)が脂肪族ジオールに由来する繰返し単位と脂肪族ジカルボン酸に由来する繰返し単位と芳香族ジカルボン酸に由来する繰り返し単位とを主構成単位として含む、[5]又は[6]に記載の生分解性樹脂組成物。
[8] [4]~[7]のいずれかに記載の生分解性樹脂組成物の押出成形体又は射出成形体である生分解性樹脂成形体。
[9] 原料を粉砕した後、得られた粉体から所定の粒度のものを選別して、生分解性樹脂用分解促進剤を得る、生分解性樹脂用分解促進剤の製造方法。
[10] 更に乾燥する工程を有する、[9]に記載の生分解性樹脂用分解促進剤の製造方法。
[11] 前記生分解性樹脂用分解促進剤が、セルロース、ヘミセルロース及びリグニンを含み、該生分解性樹脂用分解促進剤中の炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、[9]又は[10]に記載の生分解性樹脂用分解促進剤の製造方法。
[12] 前記生分解性樹脂用分解促進剤が、可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下である、[9]~[11]のいずれかに記載の生分解性樹脂用分解促進剤の製造方法。
本発明の生分解性樹脂用分解促進剤によれば、脂肪族ポリエステル系樹脂、脂肪族-芳香族ポリエステル系樹脂、脂肪族オキシカルボン酸系樹脂などの生分解性樹脂の生分解速度や分解率を当該生分解性樹脂本来の生分解速度や分解率よりも高めることができる。
このような本発明の生分解性樹脂用分解促進剤を含む本発明の生分解性樹脂組成物によれば、生分解性に優れた生分解性樹脂成形体が提供される。
以下に本発明の実施の形態を詳細に説明する。
本発明は以下の説明に限定されるものではなく、本発明の要旨を逸脱しない範囲において、任意に変形して実施することができる。
本明細書において、「~」を用いてその前後に数値又は物性値を挟んで表現する場合、その前後の値を含むものとして用いることとする。
本明細書において、「~」を用いてその前後に数値又は物性値を挟んで表現する場合、その前後の値を含むものとして用いることとする。
[生分解性樹脂用分解促進剤]
本発明の第1の態様に係る生分解性樹脂用分解促進剤(以下、「本発明の分解促進剤I」と称す場合がある。)は、セルロース、ヘミセルロース及びリグニンを必須成分として含むものであって、該生分解性樹脂用分解促進剤中の炭素(炭素元素)に対する窒素(窒素元素)の質量比(以下、「窒素/炭素比」と称す場合がある。)が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合(以下、「ヘミセルロース/(セルロース+リグニン)比」と称す場合がある。)が0.2以上であることを特徴とする。
本発明の第1の態様に係る生分解性樹脂用分解促進剤(以下、「本発明の分解促進剤I」と称す場合がある。)は、セルロース、ヘミセルロース及びリグニンを必須成分として含むものであって、該生分解性樹脂用分解促進剤中の炭素(炭素元素)に対する窒素(窒素元素)の質量比(以下、「窒素/炭素比」と称す場合がある。)が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合(以下、「ヘミセルロース/(セルロース+リグニン)比」と称す場合がある。)が0.2以上であることを特徴とする。
本発明の第2の態様に係る生分解性樹脂用分解促進剤(以下、「本発明の分解促進剤II」と称す場合がある。)は、可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下であることを特徴とする。
以下において、本発明の分解促進剤Iと本発明の分解促進剤IIをまとめて「本発明の分解促進剤」と総称する。
<分解促進剤I>
本発明の分解促進剤Iは、窒素/炭素比が0.04以上である。窒素/炭素比がこの範囲を満足する分解促進剤Iによって生分解性樹脂の分解が促進される理由として、次のようなことが推測される。
生分解性樹脂を分解する細菌や真菌等の微生物は、栄養源として炭素とともに窒素を必要とする。一方、生分解性樹脂は主に炭素、酸素、水素から構成され、窒素の含有率は低い。そのため、微生物が生分解性樹脂を分解する際に、窒素が不足して増殖や分解酵素の分泌が抑えられ、十分な生分解速度が得られない傾向がある。
本発明の分解促進剤Iは、炭素に対して一定量以上の窒素を含むため、微生物の増殖と分解酵素の分泌を促し、生分解性樹脂の分解を促進すると推測される。
本発明の分解促進剤Iは、窒素/炭素比が0.04以上である。窒素/炭素比がこの範囲を満足する分解促進剤Iによって生分解性樹脂の分解が促進される理由として、次のようなことが推測される。
生分解性樹脂を分解する細菌や真菌等の微生物は、栄養源として炭素とともに窒素を必要とする。一方、生分解性樹脂は主に炭素、酸素、水素から構成され、窒素の含有率は低い。そのため、微生物が生分解性樹脂を分解する際に、窒素が不足して増殖や分解酵素の分泌が抑えられ、十分な生分解速度が得られない傾向がある。
本発明の分解促進剤Iは、炭素に対して一定量以上の窒素を含むため、微生物の増殖と分解酵素の分泌を促し、生分解性樹脂の分解を促進すると推測される。
本発明の分解促進剤Iの窒素/炭素比は、好ましくは0.05以上、より好ましくは0.06以上である。窒素/炭素比の上限は、特に限定されないが5以下が好ましい。窒素/炭素比が5を超えると炭素分が不足し、十分な効果を発揮できないことがある。
本発明の分解促進剤Iは、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合(ヘミセルロース/(セルロース+リグニン)比)が0.2以上である。ヘミセルロース/(セルロース+リグニン)比がこの範囲を満足する分解促進剤Iによって生分解性樹脂の分解が促進される理由として、次のようなことが推測される。
ヘミセルロースは、セルロースやリグニンと比べて早く微生物により分解・吸収される傾向がある。ヘミセルロース/(セルロース+リグニン)比が0.2以上である本発明の分解促進剤Iを用いることにより、樹脂組成物が自然界に放出された際に、特に初期に微生物の増殖を促すとともに表面積を増加させることで、生分解性樹脂の分解を促進すると推測される。
ヘミセルロースは、セルロースやリグニンと比べて早く微生物により分解・吸収される傾向がある。ヘミセルロース/(セルロース+リグニン)比が0.2以上である本発明の分解促進剤Iを用いることにより、樹脂組成物が自然界に放出された際に、特に初期に微生物の増殖を促すとともに表面積を増加させることで、生分解性樹脂の分解を促進すると推測される。
本発明の分解促進剤Iのヘミセルロース/(セルロース+リグニン)比は、好ましくは0.6以上、より好ましくは1.0以上である。ヘミセルロース/(セルロース+リグニン)比の上限は、特に限定されないが、通常は10以下である。この比を10よりも大きくするためには、原料に含まれるセルロース及び/又はリグニンを取り除く必要があり、製造コストが高くなるためである。
本発明の分解促進剤Iは、セルロース、ヘミセルロース及びリグニンをその合計量が分解促進剤Iの100質量%中に5~50質量%、特に10~45質量%程度含まれていることが好ましい。
本発明の分解促進剤Iは、水分を含んでいる場合は、乾燥して水分率を5質量%未満にすることが好ましい。水分率が5質量%以上であると、生分解性樹脂と混ぜる際、生分解性樹脂が加水分解を起こして、分子量が低下して、機械的強度が十分でなくなることがある。
本発明の分解促進剤I中の窒素、炭素、セルロース、ヘミセルロース及びリグニンの含有量は、後述の実施例に記載の方法で測定並びに算出することができる。
原料を2種類以上用いた分解促進剤Iの場合の、窒素、炭素、セルロース、ヘミセルロース及びリグニンの含有量は、原料の配合割合に応じて、それぞれの原料の窒素、炭素、セルロース、ヘミセルロース及びリグニンの含有量を割合比に乗じたものの総和として求めることができる。
原料を2種類以上用いた分解促進剤Iの場合の、窒素、炭素、セルロース、ヘミセルロース及びリグニンの含有量は、原料の配合割合に応じて、それぞれの原料の窒素、炭素、セルロース、ヘミセルロース及びリグニンの含有量を割合比に乗じたものの総和として求めることができる。
<分解促進剤II>
本発明の分解促進剤IIは、可溶無窒素物を20質量%以上含有する。可溶無窒素物含有量がこの範囲を満たす分解促進剤によって、生分解性樹脂の分解が促進される理由として、次のようなことが推測される。
可溶無窒素物は飼料等の成分表示で一般的に使われる分析値であり、可溶無窒素物を含む成分は、微生物によって分解、吸収されやすい性質を持つ。一方、自然界の土壌や熟成の進んだコンポストは通常、細菌や真菌等の微生物が利用できる易分解性の有機物が少ないため、微生物の数や分解酵素の活性が低い状態にある。
本発明の分解促進剤IIは一定量以上の可溶無窒素物を含むことにより、微生物の増殖と分解酵素の活性化を促し、生分解性樹脂の分解を促進すると推測される。
本発明の分解促進剤IIは、可溶無窒素物を20質量%以上含有する。可溶無窒素物含有量がこの範囲を満たす分解促進剤によって、生分解性樹脂の分解が促進される理由として、次のようなことが推測される。
可溶無窒素物は飼料等の成分表示で一般的に使われる分析値であり、可溶無窒素物を含む成分は、微生物によって分解、吸収されやすい性質を持つ。一方、自然界の土壌や熟成の進んだコンポストは通常、細菌や真菌等の微生物が利用できる易分解性の有機物が少ないため、微生物の数や分解酵素の活性が低い状態にある。
本発明の分解促進剤IIは一定量以上の可溶無窒素物を含むことにより、微生物の増殖と分解酵素の活性化を促し、生分解性樹脂の分解を促進すると推測される。
本発明の分解促進剤IIの可溶無窒素物の含有量は、好ましくは30質量%以上、より好ましくは40質量%以上である。可溶無窒素物の含有量の上限は特に限定されないが、100質量%にするためには精製のためのコストが高くなるため、通常は99質量%以下である。
本発明の分解促進剤IIは、セルロースとリグニンの含有量の合計が50質量%以下である。セルロースやリグニンは、可溶無窒素物より遅れて微生物により分解、吸収される性質がある。本発明の分解促進剤II中のセルロースとリグニンの含有量の合計が50質量%を超えると、生分解の速度が低下することがある。本発明の分解促進剤II中のセルロースとリグニンの含有量の合計は好ましくは30質量%以下、より好ましくは20質量%以下である。セルロースとリグニンの含有量の合計の下限には特に制限はないが、通常5質量%以上である。
本発明の分解促進剤IIは、水分を含んでいる場合は、乾燥して水分率を5質量%未満にすることが好ましい。水分率が5質量%以上であると、生分解性樹脂と混ぜる際、生分解性樹脂が加水分解を起こして、分子量が低下して、機械的強度が十分でなくなることがある。
本発明の分解促進剤II中の可溶無窒素物、セルロース及びリグニンの含有量は、後述の実施例に記載の方法で測定並びに算出することができる。
また、原料を2種類以上用いた分解促進剤IIの場合の、可溶無窒素物、セルロース及びリグニンの含有量は、原料の配合割合に応じて、それぞれの原料の可溶無窒素物、セルロース及びリグニンの含有量を割合比に乗じたものの総和として求めることができる。
また、原料を2種類以上用いた分解促進剤IIの場合の、可溶無窒素物、セルロース及びリグニンの含有量は、原料の配合割合に応じて、それぞれの原料の可溶無窒素物、セルロース及びリグニンの含有量を割合比に乗じたものの総和として求めることができる。
本発明の分解促進剤IIは、可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下であるが、炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、ことが好ましい。
<分解促進剤の製造方法>
本発明の分解促進剤Iは、セルロース、ヘミセルロース及びリグニンを含むものを原料として、その濃度や割合を調整しながら、得ることができる。原料としては、植物の種子や実を搾った残渣、取り除いた殻や糠、ふすま、製油や醸造の過程で発生する粕類を使用することができる。具体的には落花生、米、小麦、大麦、アブラヤシ、綿、大豆、菜種、トウモロコシ等が挙げられる。
本発明の分解促進剤Iは、セルロース、ヘミセルロース及びリグニンを含むものを原料として、その濃度や割合を調整しながら、得ることができる。原料としては、植物の種子や実を搾った残渣、取り除いた殻や糠、ふすま、製油や醸造の過程で発生する粕類を使用することができる。具体的には落花生、米、小麦、大麦、アブラヤシ、綿、大豆、菜種、トウモロコシ等が挙げられる。
本発明の分解促進剤Iには、窒素/炭素比を所定の範囲に調整するために、窒素を含む有機物や無機物を添加してもよい。
本発明の分解促進剤IIは、可溶無窒素物を含むものを原料として、その濃度や割合を調整しながら、得ることができる。原料としては、植物の種子や実を搾った残渣、取り除いた殻や糠、ふすま、製油や醸造の過程で発生する粕類を使用することができる。具体的にはピスタチオ、クルミ、栗、落花生、米、小麦、大麦、アブラヤシ、バガス、綿、大豆、菜種、トウモロコシ等が挙げられる。
本発明の分解促進剤は、上記の原料を粉砕して、ある一定の粒度にすることで得ることができる。2種以上の原料を用いる場合、原料別に粉砕してもあらかじめ原料を混合した混合物を粉砕してもよい。
本発明の分解促進剤Iの製造において、窒素/炭素比、及びヘミセルロース/(セルロース+リグニン)比を上記の範囲に制御しやすいという観点から、予め原料毎に粉砕しておき、それらを混合する方が好ましい。
本発明の分解促進剤IIの製造において、上記の可溶無窒素物、セルロース及びリグニンの割合や比率を上記の範囲に制御しやすいという観点から、予め原料毎に粉砕しておき、それらを混合する方が好ましい。
原料の粉砕は、例えば、粉砕機などを用いて行うことができる。
粉砕機としては、例えばカッターミル、石臼、擂潰機、ジェットミル、ボールミル、ピンミル等の種々の粉砕機を用いることができる。中でも、所定の粒子径の分解促進剤を得るには、ジェットミル、ボールミル、ピンミルが好ましく、連続的かつ効率的に粉砕できる点から、ジェットミルが特に好ましい。
粉砕機としては、例えばカッターミル、石臼、擂潰機、ジェットミル、ボールミル、ピンミル等の種々の粉砕機を用いることができる。中でも、所定の粒子径の分解促進剤を得るには、ジェットミル、ボールミル、ピンミルが好ましく、連続的かつ効率的に粉砕できる点から、ジェットミルが特に好ましい。
粉砕方法は、水等の媒体を用いる湿式粉砕と、媒体を用いない乾式粉砕とに大別される。所定の水分率まで乾燥させる必要があることから、乾式粉砕が好ましい。
粉砕後の粒度は、生分解性樹脂に対して使用する際、より生分解性樹脂と混ざりやすく、分解促進剤として効果を得やすいという観点から、通常、粒径が500μm以下、好ましくは300μm以下、より好ましくは100μm以下である。
本発明の分解促進剤は、50%質量累積粒度が60μm以下が好ましく、より好ましくは40μm以下であり、特に好ましくは20μm以下である。この分解促進剤の50%質量累積粒度の値が低くなるほど、生分解性樹脂と混合して得られる生分解性樹脂組成物及びそれを成形して得られる生分解性樹脂成形体の外見が良好(例えば、シート表面上の凹凸が無くなり、透明性が向上する)なものとなり、且つ、生分解性樹脂成形体としての、機械物性、例えば、降伏応力、破断時応力、エルメンドルフ引裂強度、パンクチャー衝撃強度、破断時応力などが向上するため、好ましい。
50%質量累積粒度の下限については特に限定されないが、1μm以上が好ましい。50%質量累積粒度が1μm未満の場合、飛散しやすく、また、生分解性樹脂と混合する際にブロッキングや分級などの問題が起こるなど、取り扱いが難しくなることがある。
本発明の分解促進剤は、50%質量累積粒度が60μm以下が好ましく、より好ましくは40μm以下であり、特に好ましくは20μm以下である。この分解促進剤の50%質量累積粒度の値が低くなるほど、生分解性樹脂と混合して得られる生分解性樹脂組成物及びそれを成形して得られる生分解性樹脂成形体の外見が良好(例えば、シート表面上の凹凸が無くなり、透明性が向上する)なものとなり、且つ、生分解性樹脂成形体としての、機械物性、例えば、降伏応力、破断時応力、エルメンドルフ引裂強度、パンクチャー衝撃強度、破断時応力などが向上するため、好ましい。
50%質量累積粒度の下限については特に限定されないが、1μm以上が好ましい。50%質量累積粒度が1μm未満の場合、飛散しやすく、また、生分解性樹脂と混合する際にブロッキングや分級などの問題が起こるなど、取り扱いが難しくなることがある。
本発明の分解促進剤の50%質量累積粒度は後掲の実施例の項に記載の方法で測定される。
上述の好適な粒径及び50%質量累積粒度とするには、その粒径の目の大きさを持つメッシュ篩やスクリーン、又は遠心分級機等を使って、所定の粒度のものを選別することが好ましい。あるいは、選別する機構を備えた粉砕機を用いて粉砕と選別を同時に行っても良い。
上述の好適な粒径及び50%質量累積粒度とするには、その粒径の目の大きさを持つメッシュ篩やスクリーン、又は遠心分級機等を使って、所定の粒度のものを選別することが好ましい。あるいは、選別する機構を備えた粉砕機を用いて粉砕と選別を同時に行っても良い。
原料毎に粉砕して得られた粉体をこのように選別し、所定の粒度としたものを、必要に応じて特定の割合で配合して、本発明の分解促進剤を得ることができる。この際に好ましく用いられる粉体としては、上記原料の粉体であれば特に限定されないが、米糠、小麦ふすま、大豆粕、菜種粕の粉などが挙げられる。
本発明の分解促進剤を生分解性樹脂と混ぜる際には、予め乾燥させておくことが好ましい。その理由としては、保管中に保管雰囲気における大気(空気)中の水分が吸着してしまい、湿気により生分解性樹脂と混ざりにくくなるためである。また、前述の通り、分解促進剤が水分を含んでいると、生分解性樹脂が加水分解を起こして分子量が低下し、機械的強度が損なわれる観点からも、分解促進剤を乾燥することが好ましい。
このため、本発明の分解促進剤の製造時及び/又は使用時には、上記の粉砕及び粒度調整後、好ましくは更に乾燥を行う。
乾燥には、一般的な乾燥機を用いて、40~200℃程度の温度で加熱すればよい。
このような乾燥処理を行うことで、分解促進剤の水分量を5質量%未満、特に0~3質量%とすることが好ましい。
なお、分解促進剤の水分率は、分解促進剤の質量Wxと、この分解促進剤を後掲の実施例の項に記載の水分量の測定方法に従って絶乾状態まで乾燥させた後の質量Wyから、下記式で算出される。
水分率(質量%)={(Wx-Wy)/Wx}×100
乾燥には、一般的な乾燥機を用いて、40~200℃程度の温度で加熱すればよい。
このような乾燥処理を行うことで、分解促進剤の水分量を5質量%未満、特に0~3質量%とすることが好ましい。
なお、分解促進剤の水分率は、分解促進剤の質量Wxと、この分解促進剤を後掲の実施例の項に記載の水分量の測定方法に従って絶乾状態まで乾燥させた後の質量Wyから、下記式で算出される。
水分率(質量%)={(Wx-Wy)/Wx}×100
[生分解性樹脂組成物]
本発明の生分解性樹脂組成物は、本発明の分解促進剤2質量部以上250質量部以下と生分解性樹脂100質量部を含むものである。本発明の生分解性樹脂組成物において、生分解性樹脂100質量部に対する本発明の分解促進剤の含有量が2質量部未満であると、本発明の分解促進剤を含むことによる生分解性の向上効果を十分に得ることができない。本発明の分解促進剤の含有量が250質量部を超えると、得られる成形体の機械特性が損なわれる。
本発明の生分解性樹脂組成物は生分解性樹脂100質量部に対して本発明の分解促進剤を10質量部以上100質量部以下含むことが好ましく、15質量部以上80質量部以下含むことがより好ましい。
本発明の生分解性樹脂組成物は、本発明の分解促進剤2質量部以上250質量部以下と生分解性樹脂100質量部を含むものである。本発明の生分解性樹脂組成物において、生分解性樹脂100質量部に対する本発明の分解促進剤の含有量が2質量部未満であると、本発明の分解促進剤を含むことによる生分解性の向上効果を十分に得ることができない。本発明の分解促進剤の含有量が250質量部を超えると、得られる成形体の機械特性が損なわれる。
本発明の生分解性樹脂組成物は生分解性樹脂100質量部に対して本発明の分解促進剤を10質量部以上100質量部以下含むことが好ましく、15質量部以上80質量部以下含むことがより好ましい。
本発明の生分解性樹脂組成物に含まれる生分解性樹脂としては、生分解性を有するものであればよく、特に制限はないが、脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)及び脂肪族オキシカルボン酸系樹脂(C)等が挙げられる。
脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)、脂肪族オキシカルボン酸系樹脂(C)のうちの2種以上を混合して用いてもよい。
脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)、脂肪族オキシカルボン酸系樹脂(C)のうちの2種以上を混合して用いてもよい。
以下に各生分解性樹脂について説明する。
脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)、脂肪族オキシカルボン酸系樹脂(C)は、それぞれ繰返し単位を有する重合体であるが、それぞれの繰返し単位は、それぞれの繰返し単位の由来となる化合物に対する化合物単位とも呼ぶ。例えば、脂肪族ジオールに由来する繰返し単位を「脂肪族ジオール単位」、脂肪族ジカルボン酸に由来する繰返し単位を「脂肪族ジカルボン酸単位」、芳香族ジカルボン酸に由来する繰返し単位を「芳香族ジカルボン酸単位」とも呼ぶ。
脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)、脂肪族オキシカルボン酸系樹脂(C)中の「主構成単位」とは、通常、その構成単位が当該ポリエステル系樹脂中に80質量%以上含まれる構成単位のことであり、主構成単位以外の構成単位が全く含まれない場合もある。
<脂肪族ポリエステル系樹脂(A)>
脂肪族ポリエステル系樹脂(A)としては、脂肪族ジオール単位及び脂肪族ジカルボン酸単位を主構成単位として含む脂肪族ポリエステル系樹脂が好ましい。
脂肪族ポリエステル系樹脂(A)としては、脂肪族ジオール単位及び脂肪族ジカルボン酸単位を主構成単位として含む脂肪族ポリエステル系樹脂が好ましい。
脂肪族ポリエステル系樹脂(A)は、全ジカルボン酸単位中のコハク酸単位の割合が5モル%以上100モル%以下であることが好ましい。脂肪族ポリエステル系樹脂(A)は、コハク酸単位の量が異なる脂肪族ポリエステル系樹脂の混合物であってもよい。例えば、コハク酸以外の脂肪族ジカルボン酸単位を含まない(脂肪族ジカルボン酸単位としてコハク酸単位のみを含む)脂肪族ポリエステル系樹脂と、コハク酸以外の脂肪族ジカルボン酸単位を含む脂肪族ポリエステル系樹脂とをブレンドして、脂肪族ポリエステル系樹脂(A)におけるコハク酸単位量を上記好適範囲内に調整して使用することも可能である。
より具体的には、脂肪族ポリエステル系樹脂(A)は、下記式(1)で表される脂肪族ジオール単位、および下記式(2)で表される脂肪族ジカルボン酸単位を含むポリエステル系樹脂である。
-O-R1-O- (1)
-OC-R2-CO- (2)
-O-R1-O- (1)
-OC-R2-CO- (2)
式(1)中、R1は、2価の脂肪族炭化水素基を表す。式(2)中、R2は、2価の脂肪族炭化水素基を表す。
式(1)、(2)で表される脂肪族ジオール単位、脂肪族ジカルボン酸単位は、石油から誘導された化合物由来であっても、植物原料から誘導された化合物由来であってもかまわないが、植物原料から誘導された化合物由来であることが望ましい。
式(1)、(2)で表される脂肪族ジオール単位、脂肪族ジカルボン酸単位は、石油から誘導された化合物由来であっても、植物原料から誘導された化合物由来であってもかまわないが、植物原料から誘導された化合物由来であることが望ましい。
脂肪族ポリエステル系樹脂(A)が共重合体である場合には、脂肪族ポリエステル系樹脂(A)中に2種以上の式(1)で表される脂肪族ジオール単位が含まれていてもよく、脂肪族ポリエステル系樹脂(A)中に2種以上の式(2)で表される脂肪族ジカルボン酸単位が含まれていてもよい。
前述の通り、式(2)で表される脂肪族ジカルボン酸単位には、コハク酸単位が、全ジカルボン酸単位に対して5モル%以上100モル%以下含まれることが好ましい。脂肪族ポリエステル系樹脂(A)におけるコハク酸構成単位量を上記所定範囲内とすることで、成形性が向上するとともに耐熱性、分解性にも優れた生分解性樹脂組成物を得ることが可能となる。同様の理由から、脂肪族ポリエステル系樹脂(A)中のコハク酸単位量は、全ジカルボン酸単位に対して好ましくは10モル%以上、より好ましくは50モル%以上、更に好ましくは64モル%以上、特に好ましくは68モル%以上である。
以下、脂肪族ポリエステル系樹脂(A)中の全ジカルボン酸単位に対するコハク酸単位の割合を「コハク酸単位量」と称す場合がある。
以下、脂肪族ポリエステル系樹脂(A)中の全ジカルボン酸単位に対するコハク酸単位の割合を「コハク酸単位量」と称す場合がある。
式(2)で表される脂肪族ジカルボン酸単位には、コハク酸の他に1種類以上の脂肪族ジカルボン酸単位が全ジカルボン酸単位に対して5モル%以上50モル%以下含まれていることがより好ましい。コハク酸以外の脂肪族ジカルボン酸単位を上記所定範囲内共重合することで、脂肪族ポリエステル系樹脂(A)の結晶化度を下げることができ、生分解速度を速くすることが可能である。同様の理由から、脂肪族ポリエステル系樹脂(A)中のコハク酸以外の脂肪族ジカルボン酸単位量は、全ジカルボン酸単位に対して好ましくは10モル%以上45モル%以下、より好ましくは15モル%以上40モル%以下である。
式(1)で表されるジオール単位を与える脂肪族ジオールとしては、特に限定されないが、成形性や機械強度の観点から、炭素数が2以上10以下の脂肪族ジオールが好ましく、炭素数4以上6以下の脂肪族ジオールが特に好ましい。例えば、エチレングリコール、1,3-プロパンジオール、1,4-ブタンジオール、1,4-シクロヘキサンジメタノール等が挙げられ、中でも1,4-ブタンジオールが特に好ましい。上記脂肪族ジオールは、2種類以上を用いることもできる。
式(2)で表される脂肪族ジカルボン酸単位を与える脂肪族ジカルボン酸成分としては、特に限定されないが、炭素数が2以上40以下の脂肪族ジカルボン酸及びそのアルキルエステル等の誘導体が好ましく、炭素数が4以上10以下の脂肪族ジカルボン酸及びそのアルキルエステル等の誘導体が特に好ましい。コハク酸以外の炭素数が4以上10以下の脂肪族ジカルボン酸及びそのアルキルエステル等の誘導体としては、例えば、アジピン酸、スベリン酸、セバシン酸、ドデカン二酸、ダイマー酸等及びそのアルキルエステル等の誘導体が挙げられる。中でもアジピン酸、セバシン酸が好ましく、アジピン酸が特に好ましい。上記脂肪族ジカルボン酸成分は、2種類以上を用いることもでき、この場合、コハク酸とアジピン酸との組み合わせが好ましい。
脂肪族ポリエステル系樹脂(A)は、脂肪族オキシカルボン酸に由来する繰返し単位(脂肪族オキシカルボン酸単位)を有していてもよい。脂肪族オキシカルボン酸単位を与える脂肪族オキシカルボン酸成分の具体例としては、例えば、乳酸、グリコール酸、2-ヒドロキシ-n-酪酸、2-ヒドロキシカプロン酸、6-ヒドロキシカプロン酸、2-ヒドロキシ-3,3-ジメチル酪酸、2-ヒドロキシ-3-メチル酪酸、2-ヒドロキシイソカプロン酸等、又はこれらの低級アルキルエステル若しくは分子内エステル等の誘導体が挙げられる。これらに光学異性体が存在する場合には、D体、L体又はラセミ体の何れでもよく、形態としては固体、液体又は水溶液のいずれであってもよい。これらの中で特に好ましいものは、乳酸又はグリコール酸或いはその誘導体である。これら脂肪族オキシカルボン酸は単独でも、2種以上の混合物としても使用することもできる。
脂肪族ポリエステル系樹脂(A)がこれらの脂肪族オキシカルボン酸単位を含む場合、その含有量は、成形性の観点から、脂肪族ポリエステル系樹脂(A)を構成する全構成単位を100モル%として20モル%以下であることが好ましく、より好ましくは10モル%以下、更に好ましくは5モル%以下、最も好ましくは0モル%(含まない)である。
脂肪族ポリエステル系樹脂(A)は3官能以上の脂肪族多価アルコール、3官能以上の脂肪族多価カルボン酸又はその酸無水物、或いは3官能以上の脂肪族多価オキシカルボン酸成分を共重合することによって、溶融粘度が高められたものであってもよい。
3官能の脂肪族多価アルコールの具体例としては、トリメチロールプロパン、グリセリン等が挙げられる。4官能の脂肪族多価アルコールの具体例としては、ペンタエリスリトール等が挙げられる。これらは単独でも2種以上混合して使用することもできる。
3官能の脂肪族多価カルボン酸又はその酸無水物の具体例としては、プロパントリカルボン酸又はその酸無水物が挙げられる。
4官能の多価カルボン酸又はその酸無水物の具体例としては、シクロペンタンテトラカルボン酸又はその酸無水物等が挙げられる。
これらは単独でも2種以上混合して使用することもできる。
4官能の多価カルボン酸又はその酸無水物の具体例としては、シクロペンタンテトラカルボン酸又はその酸無水物等が挙げられる。
これらは単独でも2種以上混合して使用することもできる。
3官能の脂肪族オキシカルボン酸は、(i)カルボキシル基が2個とヒドロキシル基が1個を同一分子中に有するタイプと、(ii)カルボキシル基が1個とヒドロキシル基が2個のタイプとに分かれ、何れのタイプも使用可能である。成形性、機械強度や成形品外観の観点からリンゴ酸等の(i)カルボキシル基が2個とヒドロキシル基が1個を同一分子中に有するタイプが好ましく、より具体的には、リンゴ酸が好ましく用いられる。
4官能の脂肪族オキシカルボン酸成分は、(i)3個のカルボキシル基と1個のヒドロキシル基とを同一分子中に共有するタイプ、(ii)2個のカルボキシル基と2個のヒドロキシル基とを同一分子中に共有するタイプ、(iii)3個のヒドロキシル基と1個のカルボキシル基とを同一分子中に共有するタイプに分かれ、何れのタイプも使用可能である。4官能の脂肪族オキシカルボン酸成分は、カルボキシル基を複数有するものが好ましく、より具体的には、クエン酸、酒石酸等が挙げられる。
これらは単独でも2種以上混合して使用することもできる。
4官能の脂肪族オキシカルボン酸成分は、(i)3個のカルボキシル基と1個のヒドロキシル基とを同一分子中に共有するタイプ、(ii)2個のカルボキシル基と2個のヒドロキシル基とを同一分子中に共有するタイプ、(iii)3個のヒドロキシル基と1個のカルボキシル基とを同一分子中に共有するタイプに分かれ、何れのタイプも使用可能である。4官能の脂肪族オキシカルボン酸成分は、カルボキシル基を複数有するものが好ましく、より具体的には、クエン酸、酒石酸等が挙げられる。
これらは単独でも2種以上混合して使用することもできる。
脂肪族ポリエステル系樹脂(A)がこのような3官能以上の成分由来の構成単位を含む場合、その含有量は、脂肪族ポリエステル系樹脂(A)を構成する全構成単位を100モル%として、下限が通常0モル%以上、好ましくは0.01モル%以上であり、上限が通常5モル%以下、好ましくは2.5モル%以下である。
脂肪族ポリエステル系樹脂(A)の製造方法は、ポリエステルの製造に関する公知の方法が採用できる。この際の重縮合反応は、従来から採用されている適切な条件を設定することができ、特に制限されない。通常、エステル化反応を進行させた後、減圧操作を行うことによって更に重合度を高める方法が採用される。
脂肪族ポリエステル系樹脂(A)の製造時に、ジオール単位を形成するジオール成分とジカルボン酸単位を形成するジカルボン酸成分とを反応させる場合には、製造される脂肪族ポリエステル系樹脂(A)が目的とする組成を有するようにジオール成分およびジカルボン酸成分の使用量を設定する。通常、ジオール成分とジカルボン酸成分とは実質的に等モル量で反応するが、ジオール成分は、エステル化反応中に留出することから、通常はジカルボン酸成分よりも1~20モル%過剰に用いられる。
脂肪族ポリエステル系樹脂(A)に脂肪族オキシカルボン酸単位や多官能成分単位等の必須成分以外の成分(任意成分)を含有させる場合、その脂肪族オキシカルボン酸単位や多官能成分単位もそれぞれ目的とする組成となるように、それぞれに対応する化合物(モノマーやオリゴマー)を反応に供するようにする。このとき、上記の任意成分を反応系に導入する時期および方法に制限はなく、本発明に好適な脂肪族ポリエステル系樹脂(A)を製造できる限り任意である。
例えば脂肪族オキシカルボン酸を反応系に導入する時期および方法は、ジオール成分とジカルボン酸成分との重縮合反応以前であれば特に限定されず、以下の(1)、(2)などが挙げられる。
(1)予め触媒を脂肪族オキシカルボン酸溶液に溶解させた状態で混合する方法
(2)原料仕込み時に触媒を反応系に導入すると同時に混合する方法
(1)予め触媒を脂肪族オキシカルボン酸溶液に溶解させた状態で混合する方法
(2)原料仕込み時に触媒を反応系に導入すると同時に混合する方法
多官能成分単位を形成する化合物の導入時期は、重合初期の他のモノマーやオリゴマーと同時に仕込むようにしてもよく、エステル交換反応後、減圧を開始する前に仕込むようにしてもよい。多官能成分単位を形成する化合物は、他のモノマーやオリゴマーと同時に仕込む方が工程の簡略化の点で好ましい。
脂肪族ポリエステル系樹脂(A)は、通常、触媒の存在下で製造される。触媒としては、公知のポリエステル系樹脂の製造に用いることのできる触媒を、本発明の効果を著しく損なわない限り任意に選択することができる。その例を挙げると、ゲルマニウム、チタン、ジルコニウム、ハフニウム、アンチモン、スズ、マグネシウム、カルシウム、亜鉛等の金属化合物が好適である。中でもゲルマニウム化合物、チタン化合物が好適である。
触媒として使用できるゲルマニウム化合物としては、例えば、テトラアルコキシゲルマニウム等の有機ゲルマニウム化合物、酸化ゲルマニウム、塩化ゲルマニウム等の無機ゲルマニウム化合物などが挙げられる。中でも、価格や入手の容易さなどから、酸化ゲルマニウム、テトラエトキシゲルマニウムおよびテトラブトキシゲルマニウムなどが好ましく、特に酸化ゲルマニウムが好適である。
触媒として使用できるチタン化合物としては、例えば、テトラプロピルチタネート、テトラブチルチタネート、テトラフェニルチタネート等のテトラアルコキシチタンなどの有機チタン化合物が挙げられる。中でも、価格や入手の容易さなどから、テトラプロピルチタネート、テトラブチルチタネートなどが好ましい。
本発明の目的を損なわない限り、他の触媒の併用を妨げない。
触媒は1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で併用してもよい。
触媒は1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で併用してもよい。
触媒の使用量は、本発明の効果を著しく損なわない限り任意であるが、使用するモノマー量に対して、通常0.0005質量%以上、より好ましくは0.001質量%以上で、通常3質量%以下、好ましくは1.5質量%以下である。この範囲の下限を下回ると触媒の効果が現れないおそれがある。この範囲の上限を上回ると製造費が高くなったり得られるポリマーに著しい着色を生じたり、耐加水分解性が低下したりするおそれがある。
触媒の導入時期は、重縮合反応以前であれば特に限定されず、原料仕込み時に導入しておいてもよく、減圧開始時に導入してもよい。脂肪族オキシカルボン酸単位を導入する場合は、原料仕込み時に乳酸やグリコール酸等の脂肪族オキシカルボン酸単位を形成するモノマーやオリゴマーと同時に導入するか、又は脂肪族オキシカルボン酸水溶液に触媒を溶解して導入する方法が好ましい。特に、重合速度が大きくなるという点で脂肪族オキシカルボン酸水溶液に触媒を溶解して導入する方法が好ましい。
脂肪族ポリエステル系樹脂(A)を製造する際の温度、重合時間、圧力などの反応条件は、本発明の効果を著しく損なわない限り任意である。
ジカルボン酸成分とジオール成分とのエステル化反応および/又はエステル交換反応の反応温度は、通常150℃以上、好ましくは180℃以上で、通常260℃以下、好ましくは250℃以下である。
反応雰囲気は、通常、窒素、アルゴン等の不活性雰囲気下である。
反応圧力は、通常、常圧~10kPaであるが、中でも常圧が好ましい。
反応時間は、通常1時間以上で、通常10時間以下、好ましくは6時間以下、より好ましくは4時間以下である。
反応温度が高すぎると、不飽和結合の過剰生成が起こり、不飽和結合が要因となるゲル化が起こり、重合の制御が困難になることがある。
ジカルボン酸成分とジオール成分とのエステル化反応および/又はエステル交換反応の反応温度は、通常150℃以上、好ましくは180℃以上で、通常260℃以下、好ましくは250℃以下である。
反応雰囲気は、通常、窒素、アルゴン等の不活性雰囲気下である。
反応圧力は、通常、常圧~10kPaであるが、中でも常圧が好ましい。
反応時間は、通常1時間以上で、通常10時間以下、好ましくは6時間以下、より好ましくは4時間以下である。
反応温度が高すぎると、不飽和結合の過剰生成が起こり、不飽和結合が要因となるゲル化が起こり、重合の制御が困難になることがある。
ジカルボン酸成分とジオール成分とのエステル反応および/又はエステル交換反応後の重縮合反応条件は以下の通りである。
圧力は、通常0.01×103Pa以上、好ましくは0.03×103Pa以上で、通常1.4×103Pa以下、好ましくは0.4×103Pa以下の真空度下で行うことが望ましい。
反応温度は、通常150℃以上、好ましくは180℃以上で、通常260℃以下、好ましくは250℃以下である。
反応時間は、通常2時間以上で、通常15時間以下、好ましくは10時間以下である。
反応温度が高すぎると、不飽和結合の過剰生成で不飽和結合が要因となるゲル化が起こり、重合の制御が困難になることがある。
圧力は、通常0.01×103Pa以上、好ましくは0.03×103Pa以上で、通常1.4×103Pa以下、好ましくは0.4×103Pa以下の真空度下で行うことが望ましい。
反応温度は、通常150℃以上、好ましくは180℃以上で、通常260℃以下、好ましくは250℃以下である。
反応時間は、通常2時間以上で、通常15時間以下、好ましくは10時間以下である。
反応温度が高すぎると、不飽和結合の過剰生成で不飽和結合が要因となるゲル化が起こり、重合の制御が困難になることがある。
脂肪族ポリエステル系樹脂(A)の製造時には、カーボネート化合物やジイソシアネート化合物等の鎖延長剤を使用することもできる。この場合、鎖延長剤の量は、脂肪族ポリエステル系樹脂を構成する全構成単位を100モル%とした場合の脂肪族ポリエステル系樹脂(A)中のカーボネート結合やウレタン結合の割合として、通常10モル%以下、好ましくは5モル%以下、より好ましくは3モル%以下である。
脂肪族ポリエステル系樹脂(A)中にウレタン結合やカーボネート結合が存在すると、生分解性を阻害する可能性があるため、本発明では、脂肪族ポリエステル系樹脂(A)を構成する全構成単位に対し、カーボネート結合は1モル%未満、好ましくは0.5モル%以下、より好ましくは0.1モル%以下であり、ウレタン結合は0.55モル%以下、好ましくは0.3モル%以下、より好ましくは0.12モル%以下、更に好ましくは0.05モル%以下とするのがよい。この量は、脂肪族ポリエステル系樹脂(A)100質量部あたりに換算すると、0.9質量部以下、好ましくは0.5質量部以下、より好ましくは0.2質量部以下、さらに好ましくは0.1質量部以下である。特に、ウレタン結合量が上記上限値を上回ると、成膜工程等において、ウレタン結合の分解のため、ダイス出口からの溶融膜からの発煙や臭気が問題となる場合があり、また、溶融膜中に発泡による膜切れが起こって安定的に成形できないことがある。
脂肪族ポリエステル系樹脂(A)中のカーボネート結合量やウレタン結合量は、1H-NMRや13C-NMR等のNMR測定結果から算出して求めることができる。
鎖延長剤としてのカーボネート化合物としては、具体的には、ジフェニルカーボネート、ジトリールカーボネート、ビス(クロロフェニル)カーボネート、m-クレジルカーボネート、ジナフチルカーボネート、ジメチルカーボネート、ジエチルカーボネート、ジブチルカーボネート、エチレンカーボネート、ジアミルカーボネート、ジシクロヘキシルカーボネートなどが例示される。その他、フェノール類、アルコール類のようなヒドロキシ化合物から誘導される、同種、又は異種のヒドロキシ化合物からなるカーボネート化合物も使用可能である。
ジイソシアネート化合物としては、具体的には、2,4-トリレンジイソシアネート、2,4-トリレンジイソシアネートと2,6-トリレンジイソシアネートとの混合体、1,5-ナフチレンジイソシアネート、キシリレンジイソシアネート、水素化キシリレンジイソシアネート、ヘキサメチレンジイソシアネート、イソホロンジイソシアネート、4,4’-ジシクロヘキシルメタンジイソシアネート、テトラメチルキシリレンジイソシアネート、2,4,6-トリイソプロピルフェニルジイソシアネート、4,4’-ジフェニルメタンジイソシアネート、トリジンジイソシアネート等の公知のジイソシアネートなどが例示される。
その他の鎖延長剤として、ジオキサゾリン、珪酸エステルなどを使用してもよい。
珪酸エステルとしては、具体的には、テトラメトキシシラン、ジメトキシジフェニルシラン、ジメトキシジメチルシラン、ジフェニルジヒドロキシシラン等が例示される。
珪酸エステルとしては、具体的には、テトラメトキシシラン、ジメトキシジフェニルシラン、ジメトキシジメチルシラン、ジフェニルジヒドロキシシラン等が例示される。
これらの鎖延長剤(カップリング剤)を用いた高分子量ポリエステル系樹脂についても従来の技術を用いて製造することが可能である。鎖延長剤は、重縮合終了後、均一な溶融状態で無溶媒にて反応系に添加し、重縮合により得られたポリエステルと反応させる。
より具体的には、ジオール成分とジカルボン酸成分とを触媒反応させて得られる、末端基が実質的にヒドロキシル基を有し、重量平均分子量(Mw)が20,000以上、好ましくは40,000以上のポリエステルに上記鎖延長剤を反応させることにより、より高分子量化したポリエステル系樹脂を得ることができる。重量平均分子量が20,000以上のプレポリマーは、少量の鎖延長剤の使用で、溶融状態といった苛酷な条件下でも、残存する触媒の影響を受けないので反応中にゲルを生ずることなく、高分子量のポリエステル系樹脂を製造することができる。
ポリエステル系樹脂の重量平均分子量(Mw)は、溶媒をクロロホルムとし、測定温度40℃でゲルパーミエーションクロマトグラフィー(GPC)による測定値から単分散ポリスチレンによる換算値として求められる。
したがって、例えば鎖延長剤として上記のジイソシアネート化合物を用いて、ポリエステル系樹脂を更に高分子量化する場合には、重量平均分子量が20,000以上、好ましくは40,000以上のプレポリマーを用いることが好ましい。重量平均分子量が20,000未満であると、高分子量化するためのジイソシアネート化合物の使用量が多くなり耐熱性が低下する場合がある。このようなプレポリマーを用いてジイソシアネート化合物に由来するウレタン結合を介して連鎖した線状構造を有するウレタン結合を有するポリエステル系樹脂が製造される。
鎖延長時の圧力は、通常0.01MPa以上1MPa以下、好ましくは0.05MPa以上0.5MPa以下、より好ましくは0.07MPa以上0.3MPa以下であるが、常圧が最も好ましい。
鎖延長時の反応温度は、通常100℃以上、好ましくは150℃以上、より好ましくは190℃以上、最も好ましくは200℃以上で、通常250℃以下、好ましくは240℃以下、より好ましくは230℃以下である。反応温度が低すぎると粘度が高く均一な反応が難しく、高い攪拌動力も要する傾向がある。反応温度が高すぎると、ポリエステル系樹脂のゲル化や分解が併発する傾向がある。
鎖延長時の反応時間は、通常0.1分以上、好ましくは1分以上、より好ましくは5分以上で、通常5時間以下、好ましくは1時間以下、より好ましくは30分以下、最も好ましくは15分以下である。反応時間が短すぎる場合には、鎖延長剤の添加効果が発現しない傾向がある。反応時間が長すぎる場合には、ポリエステル系樹脂のゲル化や分解が併発する傾向がある。
脂肪族ポリエステル系樹脂(A)の分子量は、ゲルパーミエーションクロマトグラフィー(GPC)により測定した、単分散ポリスチレンを標準物質とした重量平均分子量(Mw)で、通常10,000以上1,000,000以下である。このMwは、成形性と機械強度の点において有利なため、好ましくは20,000以上500,000以下、より好ましくは50,000以上400,000以下である。
脂肪族ポリエステル系樹脂(A)のメルトフローレート(MFR)は、JIS K7210(1999年)に基づいて190℃、荷重2.16kgで測定した値で、通常0.1g/10分以上100g/10分以下である。成形性と機械強度の観点から、このMFRは好ましくは50g/10分以下、特に好ましくは40g/10分以下である。脂肪族ポリエステル系樹脂(A)のMFRは、分子量により調節することが可能である。
脂肪族ポリエステル系樹脂(A)の融点は70℃以上が好ましく、より好ましくは75℃以上で、170℃以下が好ましく、より好ましくは150℃以下、特に好ましくは130℃未満である。融点が複数存在する場合には、少なくとも1つの融点が上記範囲内にあることが好ましい。融点が上記範囲外では成形性に劣る。
脂肪族ポリエステル系樹脂(A)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族ポリエステル系樹脂(A)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族ポリエステル系樹脂(A)の融点や弾性率の調整法は特に限定されない。例えば、コハク酸以外の脂肪族ジカルボン酸成分の共重合成分の種類を選択したり、ぞれぞれの共重合比率を調節したり、それらを組み合わせたりすることにより、融点や弾性率を調節することが可能である。
脂肪族ポリエステル樹脂(A)は1種に限らず、構成単位の種類や構成単位比、製造方法、物性等の異なる2種以上の脂肪族ポリエステル樹脂(A)をブレンドして用いることができる。
<脂肪族-芳香族ポリエステル系樹脂(B)>
脂肪族-芳香族ポリエステル系樹脂(B)としては、上述の脂肪族ポリエステル系樹脂(A)の繰り返し単位の少なくとも一部が、芳香族化合物単位に置き換えられたもの、好ましくは、上述の脂肪族ポリエステル系樹脂(A)の脂肪族ジカルボン酸単位の一部が芳香族ジカルボン酸単位に置き換えられた、脂肪族ジオール単位と脂肪族ジカルボン酸単位と芳香族ジカルボン酸単位とを主構成単位として含むポリエステル系樹脂が例示される。
脂肪族-芳香族ポリエステル系樹脂(B)としては、上述の脂肪族ポリエステル系樹脂(A)の繰り返し単位の少なくとも一部が、芳香族化合物単位に置き換えられたもの、好ましくは、上述の脂肪族ポリエステル系樹脂(A)の脂肪族ジカルボン酸単位の一部が芳香族ジカルボン酸単位に置き換えられた、脂肪族ジオール単位と脂肪族ジカルボン酸単位と芳香族ジカルボン酸単位とを主構成単位として含むポリエステル系樹脂が例示される。
芳香族化合物単位としては、例えば、置換基を有していてもよい芳香族炭化水素基を有する芳香族ジオール単位、置換基を有していてもよい芳香族炭化水素基を有する芳香族ジカルボン酸単位、置換基を有していてもよい芳香族複素環基を有する芳香族ジカルボン酸単位、置換基を有していてもよい芳香族炭化水素基を有する芳香族オキシカルボン酸単位等が挙げられる。芳香族炭化水素基、芳香族複素環基は、単環でもよいし、複数の環が互いに結合、又は縮合したものでもよい。芳香族炭化水素基の具体例としては、1,2-フェニレン基、1,3-フェニレン基、1,4-フェニレン基、ジナフチレン基、ジフェニレン基等が挙げられる。芳香族複素環基の具体例としては2,5-フランジイル基等が挙げられる。
芳香族ジカルボン酸単位を与える芳香族ジカルボン酸成分の具体例としては、例えば、テレフタル酸、イソフタル酸、ナフタレンジカルボン酸、ジフェニルジカルボン酸、2,5-フランジカルボン酸等が挙げられる。中でも、テレフタル酸が好ましい。
芳香族ジカルボン酸成分としては、芳香族ジカルボン酸化合物の誘導体でもよい。例えば、上記に例示した芳香族ジカルボン酸成分の誘導体が好ましく、中でも、炭素数1以上4以下である低級アルキルエステルや、酸無水物等が挙げられる。芳香族ジカルボン酸化合物の誘導体の具体例としては、上記例示した芳香族ジカルボン酸成分のメチルエステル、エチルエステル、プロピルエステル、ブチルエステル等の低級アルキルエステル;無水コハク酸等の上記例示した芳香族ジカルボン酸成分の環状酸無水物;等が挙げられる。中でも、ジメチルテレフタレートが好ましい。
芳香族ジオール単位を与える芳香族ジオール成分の具体例としては、例えば、キシリレングリコール、4,4’-ジヒドロキシビフェニル、2,2-ビス(4’-ヒドロキシフェニル)プロパン、2,2-ビス(4’-β-ヒドロキシエトキシフェニル)プロパン、ビス(4-ヒドロキシフェニル)スルホン、ビス(4-β-ヒドロキシエトキシフェニル)スルホン酸等が挙げられる。芳香族ジオール成分としては、芳香族ジオール化合物の誘導体でもよい。また、複数の脂肪族ジオール化合物及び/又は芳香族ジオール化合物が互いに脱水縮合した構造を有する化合物であってもよい。
芳香族オキシカルボン酸単位を与える芳香族オキシカルボン酸成分の具体例としては、例えば、p-ヒドロキシ安息香酸、p-β-ヒドロキシエトキシ安息香酸等が挙げられる。芳香族オキシカルボン酸成分としては、芳香族オキシカルボン酸化合物の誘導体でもよい。また、複数の脂肪族オキシカルボン酸化合物及び/又は芳香族オキシカルボン酸化合物が互いに脱水縮合した構造を有する化合物(オリゴマー)であってもよい。即ち、原料物質としてオリゴマーを用いてもよい。
これら芳香族化合物単位を与える芳香族化合物成分に光学異性体が存在する場合には、D体、L体、及びラセミ体のいずれを用いてもよい。
芳香族化合物成分は、芳香族化合物単位を与えることができれば、上記の例に限定されるものではない。
芳香族化合物成分は1種を単独で用いてもよく、2種以上を任意の組み合わせ、及び比率で併用してもよい。
芳香族化合物成分は、芳香族化合物単位を与えることができれば、上記の例に限定されるものではない。
芳香族化合物成分は1種を単独で用いてもよく、2種以上を任意の組み合わせ、及び比率で併用してもよい。
脂肪族-芳香族ポリエステル系樹脂(B)としては、芳香族化合物単位を与える成分として芳香族ジカルボン酸成分を用いることが好ましく、この場合の芳香族ジカルボン酸単位の含有量は、脂肪族ジカルボン酸単位と芳香族ジカルボン酸単位の全量を基準(100モル%)として、10モル%以上80モル%以下であることが好ましい。
芳香族ジカルボン酸成分としてはテレフタル酸又は2,5-フランジカルボン酸を用いることが好ましい。その場合、脂肪族-芳香族ポリエステル系樹脂(B)としては、ポリブチレンテレフタレートアジペート及び/又はポリブチレンテレフタレートサクシネート系樹脂が好ましい。脂肪族-芳香族ポリエステル系樹脂(B)としては、ポリブチレン-2,5-フランジカルボキシレート系樹脂も好ましい。
芳香族ジカルボン酸成分としてはテレフタル酸又は2,5-フランジカルボン酸を用いることが好ましい。その場合、脂肪族-芳香族ポリエステル系樹脂(B)としては、ポリブチレンテレフタレートアジペート及び/又はポリブチレンテレフタレートサクシネート系樹脂が好ましい。脂肪族-芳香族ポリエステル系樹脂(B)としては、ポリブチレン-2,5-フランジカルボキシレート系樹脂も好ましい。
脂肪族-芳香族ポリエステル系樹脂(B)は、原料に少なくとも芳香族化合物成分を用いて、前述の脂肪族ポリエステル系樹脂(A)と同様に製造することができる。
脂肪族-芳香族ポリエステル系樹脂(B)の分子量は、ゲルパーミエーションクロマトグラフィー(GPC)により測定した、単分散ポリスチレンを標準物質とした重量平均分子量(Mw)で、通常10,000以上1,000,000以下である。このMwは、成形性と機械強度の点において有利なため、好ましくは30,000以上800,000以下、より好ましくは50,000以上600,000以下である。
脂肪族-芳香族ポリエステル系樹脂(B)のメルトフローレート(MFR)は、JIS K7210(1999年)に基づいて190℃、荷重2.16kgで測定した値で、通常0.1g/10分以上100g/10分以下である。成形性と機械強度の観点から、このMFRは、好ましくは50g/10分以下、特に好ましくは30g/10分以下である。脂肪族-芳香族ポリエステル系樹脂(B)のMFRは、分子量により調節することが可能である。
脂肪族-芳香族ポリエステル系樹脂(B)の融点は通常60℃以上で、70℃以上が好ましく、より好ましくは80℃以上で、150℃以下が好ましく、より好ましくは140℃以下、特に好ましくは120℃以下である。融点が複数存在する場合には、少なくとも1つの融点が上記範囲内にあることが好ましい。融点が上記範囲外では成形性に劣る。
脂肪族-芳香族ポリエステル系樹脂(B)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族-芳香族ポリエステル系樹脂(B)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族-芳香族ポリエステル系樹脂(B)の融点や弾性率の調整法は特に限定されない。例えば、芳香族ジカルボン酸成分以外の脂肪族ジカルボン酸成分の共重合成分の種類を選択したり、ぞれぞれの共重合比率を調節したり、それらを組み合わせたりすることにより、融点や弾性率を調節することが可能である。
脂肪族-芳香族ポリエステル系樹脂(B)は1種に限らず、構成単位の種類や構成単位比、製造方法、物性等の異なる2種以上の脂肪族-芳香族ポリエステル系樹脂(B)をブレンドして用いることができる。
<脂肪族オキシカルボン酸系樹脂(C)>
脂肪族オキシカルボン酸系樹脂(C)は、脂肪族オキシカルボン酸単位を主構成単位とするものである。その脂肪族オキシカルボン酸単位は、下記式(3)で表されることが好ましい。
-O-R3-CO- (3)
式(3)中、R3は2価の脂肪族炭化水素基又は2価の脂環式炭化水素基を表す。
脂肪族オキシカルボン酸系樹脂(C)は、脂肪族オキシカルボン酸単位を主構成単位とするものである。その脂肪族オキシカルボン酸単位は、下記式(3)で表されることが好ましい。
-O-R3-CO- (3)
式(3)中、R3は2価の脂肪族炭化水素基又は2価の脂環式炭化水素基を表す。
式(3)の脂肪族オキシカルボン酸単位を与える脂肪族オキシカルボン酸成分の具体例としては、乳酸、グリコール酸、2-ヒドロキシ-n-酪酸、2-ヒドロキシカプロン酸、2-ヒドロキシ-3,3-ジメチル酪酸、2-ヒドロキシ-3-メチル酪酸、2-ヒドロキシイソカプロン酸、あるいはこれらの混合物等が挙げられる。これらに光学異性体が存在する場合には、D体、L体のいずれでもよい。これらの中で好ましいものは、乳酸又はグリコール酸である。これら脂肪族オキシカルボン酸成分は、2種類以上を混合して用いることもできる。
脂肪族オキシカルボン酸系樹脂(C)としては、特にポリ乳酸(PLA)が好ましい。
生分解性に影響を与えない範囲で、脂肪族オキシカルボン酸系樹脂(C)にはウレタン結合、アミド結合、カーボネート結合、エーテル結合等を導入することができる。
脂肪族オキシカルボン酸系樹脂(C)の製造方法は、特に限定されるものではなく、オキシカルボン酸の直接重合法、あるいは環状体の開環重合法等公知の方法で製造することができる。
脂肪族オキシカルボン酸系樹脂(C)の分子量は、ゲルパーミエーションクロマトグラフィー(GPC)により測定した、単分散ポリスチレンを標準物質とした重量平均分子量(Mw)で、通常10,000以上1,000,000以下である。このMwは、成形性と機械強度の点において有利なため、好ましくは20,000以上500,000以下、より好ましくは50,000以上400,000以下である。
脂肪族オキシカルボン酸系樹脂(C)のメルトフローレート(MFR)は、JIS K7210(1999年)に基づいて190℃、荷重2.16kgで測定した値で、通常0.1g/10分以上100g/10分以下である。成形性と機械強度の観点から、このMFRは、好ましくは50g/10分以下、特に好ましくは40g/10分以下である。脂肪族オキシカルボン酸系樹脂(C)のMFRは、分子量により調節することが可能である。
脂肪族オキシカルボン酸系樹脂(C)の融点は70℃以上が好ましく、より好ましくは75℃以上で、170℃以下が好ましく、より好ましくは150℃以下、特に好ましくは130℃未満である。融点が複数存在する場合には、少なくとも1つの融点が上記範囲内にあることが好ましい。融点が上記範囲外では成形性に劣る。
脂肪族オキシカルボン酸系樹脂(C)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族オキシカルボン酸系樹脂(C)の弾性率は180~1000MPaであることが好ましい。
弾性率が180MPa未満では成形加工性に問題が起こり易い。弾性率が1000MPaを超えると耐衝撃強度が悪くなる傾向にある。
脂肪族オキシカルボン酸系樹脂(C)の融点や弾性率の調整法は特に限定されない。例えば、脂肪族オキシカルボン酸以外の共重合成分の種類を選択したり、ぞれぞれの共重合比率を調節したり、それらを組み合わせたりすることにより、融点や弾性率を調節することが可能である。
脂肪族オキシカルボン酸系樹脂(C)としては、以下に説明するポリヒドロキシアルカノエート(D)も好ましく用いることができる。
本発明において好適に用いられる用いるポリヒドロキシアルカノエート(以下、PHAと称することがある)(D)は、一般式:[-CHR-CH2-CO-O-](式中、Rは炭素数1~15のアルキル基である。)で示される繰り返し単位を含む脂肪族ポリエステルであり、3-ヒドロキシブチレート単位と3-ヒドロキシヘキサノエート単位を主たる構成単位として含む共重合体である。
ポリヒドロキシアルカノエート(D)は、成形性、熱安定性の観点から、構成成分として3-ヒドロキシブチレート単位を80モル%以上含むことが好ましく、85モル%以上含むことがより好ましい。また、微生物によって生産されたものが好ましい。
ポリヒドロキシアルカノエート(D)の具体例としては、ポリ(3-ヒドロキシブチレート-コ-3-ヒドロキシヘキサノエート)共重合樹脂、ポリ(3-ヒドロキシブチレート-コ-3-ヒドロキシバレレート-コ-3-ヒドロキシヘキサノエート)共重合樹脂等が挙げられる。
特に、成形加工性および得られる成形体の物性の観点から、ポリ(3-ヒドロキシブチレート-コ-3-ヒドロキシヘキサノエート)共重合樹脂、即ちPHBHが好ましい。
特に、成形加工性および得られる成形体の物性の観点から、ポリ(3-ヒドロキシブチレート-コ-3-ヒドロキシヘキサノエート)共重合樹脂、即ちPHBHが好ましい。
ポリヒドロキシアルカノエート(D)において、3-ヒドロキシブチレート(以下、3HBと称する場合がある)と、共重合している3-ヒドロキシヘキサノエート(以下、3HHと称する場合がある)等のコモノマーとの構成比、即ち共重合樹脂中のモノマー比率は、成形加工性および成形体品質等の観点から、3-ヒドロキシブチレート/コモノマー=97/3~80/20(モル%/モル%)が好ましく、95/5~85/15(モル%/モル%)がより好ましい。このコモノマー比率が3モル%未満であると、成形加工温度と熱分解温度が近接するため成形加工し難い場合がある。コモノマー比率が20モル%を超えると、ポリヒドロキシアルカノエート(D)の結晶化が遅くなるため生産性が悪化する場合がある。
ポリヒドロキシアルカノエート(D)中の各モノマー比率は、以下のようにガスクロマトグラフィーによって測定できる。
乾燥PHA約20mgに、2mlの硫酸/メタノール混液(15/85(質量比))と2mlのクロロホルムを添加して密栓し、100℃で140分間加熱して、PHA分解物のメチルエステルを得る。冷却後、これに1.5gの炭酸水素ナトリウムを少しずつ加えて中和し、炭酸ガスの発生が止まるまで放置する。4mlのジイソプロピルエーテルを添加してよく混合した後、上清中のPHA分解物のモノマーユニット組成をキャピラリーガスクロマトグラフィーにより分析することにより、共重合樹脂中の各モノマー比率を求められる。
ポリヒドロキシアルカノエート(D)の分子量は、前記のゲルパーミエーションクロマトグラフィー(GPC)により測定した、単分散ポリスチレンを標準物質とした重量平均分子量(Mw)で、通常200,000以上2,500,000以下である。このMwは、成形性と機械強度の点において有利なため、好ましくは250,000以上2,000,000以下、より好ましくは300,000以上1,000,000以下である。Mwが200,000未満では、機械物性等が劣る場合がある。Mwが2,500,000超えると、成形加工が困難となる場合がある。
ポリヒドロキシアルカノエート(D)のメルトフローレート(MFR)は、JIS K7210(1999年)に基づいて190℃、荷重2.16kgで測定した値で、好ましくは1g/10分以上100g/10分以下である。成形性と機械強度の観点から、このMFRはより好ましくは80g/10分以下、特に好ましくは50g/10分以下である。ポリヒドロキシアルカノエート(D)のMFRは、分子量により調節することが可能である。
ポリヒドロキシアルカノエート(D)の融点は100℃以上が好ましく、より好ましくは120℃以上で、180℃以下が好ましく、より好ましくは170℃以下、特に好ましくは160℃未満である。融点が複数存在する場合には、少なくとも1つの融点が上記範囲内にあることが好ましい。
ポリヒドロキシアルカノエート(D)は、例えば、Alcaligenes eutrophusにAeromonas caviae由来のPHA合成酵素遺伝子を導入したAlcaligenes eutrophus AC32株(ブダペスト条約に基づく国際寄託、国際寄託当局:独立行政法人産業技術総合研究所特許生物寄託センター(日本国茨城県つくば市東1丁目1番地1中央第6)、原寄託日:平成8年8月12日、平成9年8月7日に移管、寄託番号FERM BP-6038(原寄託FERM P-15786より移管))(J.Bacteriol.,179,4821(1997))等の微生物によって産生される。
ポリヒドロキシアルカノエート(D)は、市販品を用いることもできる。3-ヒドロキシブチレート単位及び3-ヒドロキシヘキサノエート単位を主構成単位として含むポリヒドロキシアルカノエート(D)の市販品としては、カネカ社製「PHBH X331N」、「PHBH X131A」、「PHBH X151A」等を用いることができる。
本発明では、ポリヒドロキシアルカノエート(D)を含め、脂肪族オキシカルボン酸系樹脂(C)は1種に限らず、構成単位の種類や構成単位比、製造方法、物性等の異なる2種以上の脂肪族オキシカルボン酸系樹脂(C)をブレンドして用いることができる。
<その他の成分>
本発明の生分解性樹脂組成物には、本発明の分解促進剤以外に、フィラー(充填剤)、可塑剤、帯電防止剤、酸化防止剤、光安定剤、紫外線吸収剤、染料、顔料、加水分解防止剤、結晶核剤、アンチブロッキング剤、耐候剤、熱安定剤、難燃剤、離型剤、防曇剤、表面ぬれ改善剤、焼却補助剤、分散助剤、各種界面活性剤、スリップ剤等の各種添加剤の1種又は2種以上が「その他の成分」として含まれていてもよい。
本発明の生分解性樹脂組成物にはまた、機能性添加剤として、鮮度保持剤、抗菌剤等が含有されていてもよい。
本発明の生分解性樹脂組成物には、本発明の分解促進剤以外に、フィラー(充填剤)、可塑剤、帯電防止剤、酸化防止剤、光安定剤、紫外線吸収剤、染料、顔料、加水分解防止剤、結晶核剤、アンチブロッキング剤、耐候剤、熱安定剤、難燃剤、離型剤、防曇剤、表面ぬれ改善剤、焼却補助剤、分散助剤、各種界面活性剤、スリップ剤等の各種添加剤の1種又は2種以上が「その他の成分」として含まれていてもよい。
本発明の生分解性樹脂組成物にはまた、機能性添加剤として、鮮度保持剤、抗菌剤等が含有されていてもよい。
これらのその他の成分は、本発明の効果を損なわない範囲で任意に配合することができ、1種を単独で用いてもよく、2種以上を混合して使用してもよい。
本発明の生分解性樹脂組成物におけるこれらのその他の成分の含有量は、通常、本発明の生分解性樹脂組成物の物性を損なわないために、その他の成分の総量は、本発明の生分解性樹脂組成物の総量に対して0.01質量%以上40質量%以下であることが好ましい。
<生分解性樹脂組成物の製造方法>
本発明の生分解性樹脂組成物は、上述の分解性樹脂と本発明の分解促進剤とを混練機内で混練し、該分解促進剤を生分解性樹脂中に分散させることにより製造される。また、分解促進剤及び生分解性樹脂と共に、必要に応じて用いられるその他の樹脂やその他の成分を混練機内で混合してもよい。
本発明の生分解性樹脂組成物は、上述の分解性樹脂と本発明の分解促進剤とを混練機内で混練し、該分解促進剤を生分解性樹脂中に分散させることにより製造される。また、分解促進剤及び生分解性樹脂と共に、必要に応じて用いられるその他の樹脂やその他の成分を混練機内で混合してもよい。
この混合工程は、本発明の分解促進剤及び生分解性樹脂と、必要に応じて用いられるその他の樹脂やその他の成分を、所定の割合で同時に、又は任意の順序で、タンブラー、V型ブレンダー、ナウターミキサー、バンバリーミキサー、混練ロール、押出機等の混合機により混合し、好ましくはさらに溶融混錬することにより行われる。
混合工程で使用される混練機は溶融混練機であってもよい。押出機は二軸押出機、単軸押出機のいずれでもよいが、二軸押出機がより好ましい。
溶融混練時の温度は140~220℃が好ましい。この温度範囲であれば、溶融反応に要する時間の短縮が可能になり、樹脂の劣化や分解促進剤の炭化に伴う色調の悪化等を防止することができ、また、耐衝撃性や耐湿熱性などの実用面での物理特性をより向上させることができる。同様の観点から、溶融混練温度は150~210℃がより好ましい。
溶融混練時間は、上記と同様に樹脂劣化等をより確実に回避するという観点から無用な長時間化は回避されるべきであり、20秒以上20分以下が好ましく、より好ましくは30秒以上15分以下である。
従って、この溶融混練条件を満たすような溶融混練温度や時間の条件設定を行うことが好ましい。
従って、この溶融混練条件を満たすような溶融混練温度や時間の条件設定を行うことが好ましい。
[成形体]
本発明の生分解性樹脂組成物は、汎用プラスチックに適用される各種成形法により成形することができる。その成形法としては例えば、圧縮成形(圧縮成形、積層成形、スタンパブル成形)、射出成形、押出成形や共押出成形(インフレ法やTダイ法によるフィルム成形、ラミネート成形、パイプ成形、電線/ケーブル成形、異形材の成形)、熱プレス成形、中空成形(各種ブロー成形)、カレンダー成形、固体成形(一軸延伸成形、二軸延伸成形、ロール圧延成形、延伸配向不織布成形、熱成形(真空成形、圧空成形)、塑性加工、粉末成形(回転成形)、各種不織布成形(乾式法、接着法、絡合法、スパンボンド法等)等が挙げられる。中でも、射出成形、押出成形、圧縮成形、又は熱プレス成形が好適に適用される。成形体の具体的な形状としては、シート、フィルム、容器への適用が好ましい。
本発明の生分解性樹脂組成物は、汎用プラスチックに適用される各種成形法により成形することができる。その成形法としては例えば、圧縮成形(圧縮成形、積層成形、スタンパブル成形)、射出成形、押出成形や共押出成形(インフレ法やTダイ法によるフィルム成形、ラミネート成形、パイプ成形、電線/ケーブル成形、異形材の成形)、熱プレス成形、中空成形(各種ブロー成形)、カレンダー成形、固体成形(一軸延伸成形、二軸延伸成形、ロール圧延成形、延伸配向不織布成形、熱成形(真空成形、圧空成形)、塑性加工、粉末成形(回転成形)、各種不織布成形(乾式法、接着法、絡合法、スパンボンド法等)等が挙げられる。中でも、射出成形、押出成形、圧縮成形、又は熱プレス成形が好適に適用される。成形体の具体的な形状としては、シート、フィルム、容器への適用が好ましい。
本発明の分解促進剤を成形してなる本発明の生分解性樹脂成形体には、化学的機能、電気的機能、磁気的機能、力学的機能、摩擦/摩耗/潤滑機能、光学的機能、熱的機能、生体適合性等の表面機能等の付与を目的として、各種の二次加工を施すことも可能である。二次加工の例としては、エンボス加工、塗装、接着、印刷、メタライジング(めっき等)、機械加工、表面処理(帯電防止処理、コロナ放電処理、プラズマ処理、フォトクロミズム処理、物理蒸着、化学蒸着、コーティング等)等が挙げられる。
[用途]
本発明の生分解性樹脂組成物からなる本発明の生分解性樹脂成形体は、各種食品、薬品、雑貨等の液状物や粉粒物、固形物を包装するための包装用資材、農業用資材、建築資材等幅広い用途において好適に用いられる。その具体的用途としては、射出成形品(例えば、生鮮食品のトレー、ファーストフードの容器、コーヒーカプセルの容器、カトラリー、野外レジャー製品等)、押出成形品(例えば、フィルム、シート、釣り糸、漁網、植生ネット、2次加工用シート、保水シート等)、中空成形品(ボトル等)等が挙げられる。更に、その他農業用のフィルム、コーティング資材、肥料用コーティング材、育苗ポット、ラミネートフィルム、板、延伸シート、モノフィラメント、不織布、フラットヤーン、ステープル、捲縮繊維、筋付きテープ、スプリットヤーン、複合繊維、ブローボトル、ショッピングバッグ、ゴミ袋、コンポスト袋、化粧品容器、洗剤容器、漂白剤容器、ロープ、結束材、衛生用カバーストック材、保冷箱、クッション材フィルム、マルチフィラメント、合成紙、医療用として手術糸、縫合糸、人工骨、人工皮膚、マイクロカプセル等のDDS、創傷被覆材等が挙げられる。
本発明の生分解性樹脂組成物からなる本発明の生分解性樹脂成形体は、各種食品、薬品、雑貨等の液状物や粉粒物、固形物を包装するための包装用資材、農業用資材、建築資材等幅広い用途において好適に用いられる。その具体的用途としては、射出成形品(例えば、生鮮食品のトレー、ファーストフードの容器、コーヒーカプセルの容器、カトラリー、野外レジャー製品等)、押出成形品(例えば、フィルム、シート、釣り糸、漁網、植生ネット、2次加工用シート、保水シート等)、中空成形品(ボトル等)等が挙げられる。更に、その他農業用のフィルム、コーティング資材、肥料用コーティング材、育苗ポット、ラミネートフィルム、板、延伸シート、モノフィラメント、不織布、フラットヤーン、ステープル、捲縮繊維、筋付きテープ、スプリットヤーン、複合繊維、ブローボトル、ショッピングバッグ、ゴミ袋、コンポスト袋、化粧品容器、洗剤容器、漂白剤容器、ロープ、結束材、衛生用カバーストック材、保冷箱、クッション材フィルム、マルチフィラメント、合成紙、医療用として手術糸、縫合糸、人工骨、人工皮膚、マイクロカプセル等のDDS、創傷被覆材等が挙げられる。
特に本発明の生分解性樹脂成形体は、食品包装用フィルム、生鮮食品のトレー、ファーストフードの容器、弁当箱等の食品用向けの容器として好適である。
以下、実施例及び比較例を用いて本発明の内容を更に具体的に説明する。本発明はその要旨を超えない限り、以下の実施例によって限定されるものではない。以下の実施例における各種の製造条件や評価結果の値は、本発明の実施態様における上限又は下限の好ましい値としての意味を持つものであり、好ましい範囲は前記した上限又は下限の値と、下記実施例の値又は実施例同士の値との組み合わせで規定される範囲であってもよい。
[分解促進剤の測定]
<窒素、炭素含有量の測定と算出>
窒素と炭素の含有量は、窒素と炭素を同時に分析可能な市販の燃焼法自動分析装置を用いて測定した。
窒素の含有量はJIS K0102、JIS K6451-1、JIS M8819、JIS Z7302-8等に適合する全窒素測定法又は窒素測定装置により、炭素の含有量はJIS K0102、JIS M8819、JIS Z7302-8等に適合する炭素測定装置により、それぞれ個別に測定することもできる。
<窒素、炭素含有量の測定と算出>
窒素と炭素の含有量は、窒素と炭素を同時に分析可能な市販の燃焼法自動分析装置を用いて測定した。
窒素の含有量はJIS K0102、JIS K6451-1、JIS M8819、JIS Z7302-8等に適合する全窒素測定法又は窒素測定装置により、炭素の含有量はJIS K0102、JIS M8819、JIS Z7302-8等に適合する炭素測定装置により、それぞれ個別に測定することもできる。
<セルロース、リグニン、ヘミセルロース含有量の測定と算出>
分解促進剤Iにおけるセルロース、リグニン、ヘミセルロースの含有量は、耐熱性α-アミラーゼ処理中性デタージェント繊維(aNDFom)、酸性デタージェント繊維(ADFom)、酸性デタージェントリグニン(ADL)の値から、以下の式で求めた。
セルロース(%)=ADFom(%)-ADL(%)
リグニン(%)=ADL(%)
ヘミセルロース(%)=aNDFom(%)-ADFom(%)
aNDFom、ADFom、ADLは、常法(例えば日本科学飼料協会発行「飼料分析法・解説 2009」、独立行政法人農林水産消費安全技術センター「飼料分析基準」(http://www.famic.go.jp/ffis/feed/bunseki/bunsekikijun.html)、国立研究開発法人農業・食品産業技術総合研究機構「最近の飼料作物の栄養評価に関する研究の動向」(http://www.naro.affrc.go.jp/nilgs-neo/kenkyukai/files/jikyushiryoriyo2016_koen07.pdf)等を参照)に基づいて測定した。
測定方法の概要は以下の通りである。
分解促進剤Iにおけるセルロース、リグニン、ヘミセルロースの含有量は、耐熱性α-アミラーゼ処理中性デタージェント繊維(aNDFom)、酸性デタージェント繊維(ADFom)、酸性デタージェントリグニン(ADL)の値から、以下の式で求めた。
セルロース(%)=ADFom(%)-ADL(%)
リグニン(%)=ADL(%)
ヘミセルロース(%)=aNDFom(%)-ADFom(%)
aNDFom、ADFom、ADLは、常法(例えば日本科学飼料協会発行「飼料分析法・解説 2009」、独立行政法人農林水産消費安全技術センター「飼料分析基準」(http://www.famic.go.jp/ffis/feed/bunseki/bunsekikijun.html)、国立研究開発法人農業・食品産業技術総合研究機構「最近の飼料作物の栄養評価に関する研究の動向」(http://www.naro.affrc.go.jp/nilgs-neo/kenkyukai/files/jikyushiryoriyo2016_koen07.pdf)等を参照)に基づいて測定した。
測定方法の概要は以下の通りである。
《aNDFom》
試料(質量W1)に亜硫酸ナトリウムと中性デタージェント溶液を加えて煮沸させ、次いで耐熱性α-アミラーゼを加えて煮沸させる。不溶物をガラスフィルター等で濾取し、洗浄・乾燥して秤量する(W2)。次に、この不溶物を加熱灰化して秤量する(W3)。以下の式からaNDFom(%)を求める。
aNDFom(%)=100×(W2-W3)/W1
試料(質量W1)に亜硫酸ナトリウムと中性デタージェント溶液を加えて煮沸させ、次いで耐熱性α-アミラーゼを加えて煮沸させる。不溶物をガラスフィルター等で濾取し、洗浄・乾燥して秤量する(W2)。次に、この不溶物を加熱灰化して秤量する(W3)。以下の式からaNDFom(%)を求める。
aNDFom(%)=100×(W2-W3)/W1
《ADFom及びADL》
試料(質量W4)に酸性デタージェント溶液を加えて煮沸した後、不溶物をガラスフィルター等で濾取し、洗浄・乾燥して秤量する(W5)。次に、この不溶物を72%硫酸で処理した後、不溶物を洗浄・乾燥して秤量する(W6)。最後に、不溶物を加熱灰化して秤量する(W7)。以下の式からADFom(%)及びADL(%)を求める。
ADFom(%)=100×(W5-W7)/W4
ADL(%)=100×(W6-W7)/W4
試料(質量W4)に酸性デタージェント溶液を加えて煮沸した後、不溶物をガラスフィルター等で濾取し、洗浄・乾燥して秤量する(W5)。次に、この不溶物を72%硫酸で処理した後、不溶物を洗浄・乾燥して秤量する(W6)。最後に、不溶物を加熱灰化して秤量する(W7)。以下の式からADFom(%)及びADL(%)を求める。
ADFom(%)=100×(W5-W7)/W4
ADL(%)=100×(W6-W7)/W4
分解促進剤IIにおけるセルロース、リグニン含有量の測定と算出も、酸性デタージェント繊維(ADFom)、酸性デタージェントリグニン(ADL)の値から、上記と同様に求めた。
<可溶無窒素物含有量の測定と算出>
可溶無窒素物は、飼料の公定規格(昭和51年7月24日 農林省告示第756号)に基づき、以下の式で求めた。
可溶無窒素物(%)=100-(水分(%)+粗たん白質(%)+粗脂肪(%)+粗繊維(%)+粗灰分(%))
水分、粗たん白質、粗脂肪、粗繊維、粗灰分の値は、飼料の公定規格又はその他公知の方法を用いて、以下のように定量した。
可溶無窒素物は、飼料の公定規格(昭和51年7月24日 農林省告示第756号)に基づき、以下の式で求めた。
可溶無窒素物(%)=100-(水分(%)+粗たん白質(%)+粗脂肪(%)+粗繊維(%)+粗灰分(%))
水分、粗たん白質、粗脂肪、粗繊維、粗灰分の値は、飼料の公定規格又はその他公知の方法を用いて、以下のように定量した。
《水分》
水分の定量法は、加熱減量法を用いた。
この方法では、自動水分測定装置を用いて、重量変化速度が一定以下になるまで135℃で乾燥させてその前後の重量変化から水分量を求めた、また、秤量した試料を135℃で2時間乾燥し、デシケーター中で放冷後重さを量り、その減量を水分の量として算出してもよい。
このほか、水分の定量法としてはカールフィッシャー法なども用いることができる。
水分の定量法は、加熱減量法を用いた。
この方法では、自動水分測定装置を用いて、重量変化速度が一定以下になるまで135℃で乾燥させてその前後の重量変化から水分量を求めた、また、秤量した試料を135℃で2時間乾燥し、デシケーター中で放冷後重さを量り、その減量を水分の量として算出してもよい。
このほか、水分の定量法としてはカールフィッシャー法なども用いることができる。
《粗たん白質》
粗たん白質の定量法は、ケルダール法を用いた。
この方法では、まず試料に硫酸と分解促進剤を加えて加熱し、試料中の窒素をアンモニウム塩に変換する。次に、水酸化ナトリウムを加えて加熱し、発生するアンモニアを既知濃度の硫酸で捕集する。この溶液を既知濃度の水酸化ナトリウム水溶液で滴定することで、窒素量を測定する。得られた窒素量の値(質量%)に窒素-たん白質換算係数(6.25)を乗じて粗たん白質量を求める。
このほか、窒素量の測定には燃焼法(デュマ法)による自動分析装置を用いることもできる。
粗たん白質の定量法は、ケルダール法を用いた。
この方法では、まず試料に硫酸と分解促進剤を加えて加熱し、試料中の窒素をアンモニウム塩に変換する。次に、水酸化ナトリウムを加えて加熱し、発生するアンモニアを既知濃度の硫酸で捕集する。この溶液を既知濃度の水酸化ナトリウム水溶液で滴定することで、窒素量を測定する。得られた窒素量の値(質量%)に窒素-たん白質換算係数(6.25)を乗じて粗たん白質量を求める。
このほか、窒素量の測定には燃焼法(デュマ法)による自動分析装置を用いることもできる。
《粗脂肪》
粗脂肪の定量法は、ジエチルエーテル抽出法を用いた。
この方法では、ソックスレー抽出装置を用いて試料中の脂肪をジエチルエーテルで抽出し、ジエチルエーテルを揮発させて得られた抽出物を粗脂肪として秤量する。
粗脂肪の定量法としてはその他に酸分解ジエチルエーテル抽出法なども用いることができる。
粗脂肪の定量法は、ジエチルエーテル抽出法を用いた。
この方法では、ソックスレー抽出装置を用いて試料中の脂肪をジエチルエーテルで抽出し、ジエチルエーテルを揮発させて得られた抽出物を粗脂肪として秤量する。
粗脂肪の定量法としてはその他に酸分解ジエチルエーテル抽出法なども用いることができる。
《粗繊維》
粗繊維の定量法は、濾過法を用いた。
この方法では、まず試料に硫酸を加えて煮沸して濾過する。次に、この不溶物に水酸化ナトリウム水溶液を加えて煮沸して濾過し、得られた残留物を乾燥させて重量を測定する。次に、残留物を550~600℃で加熱灰化して重量を測定する。加熱灰化前後の重量差から粗繊維量を求める。
粗繊維の定量法は、濾過法を用いた。
この方法では、まず試料に硫酸を加えて煮沸して濾過する。次に、この不溶物に水酸化ナトリウム水溶液を加えて煮沸して濾過し、得られた残留物を乾燥させて重量を測定する。次に、残留物を550~600℃で加熱灰化して重量を測定する。加熱灰化前後の重量差から粗繊維量を求める。
《粗灰分》
粗灰分の定量法は、直接灰化法を用いた。
この方法では、試料を550~600℃で加熱灰化して重量を測定することで、粗灰分の量を求める。
粗灰分の定量法は、直接灰化法を用いた。
この方法では、試料を550~600℃で加熱灰化して重量を測定することで、粗灰分の量を求める。
<粒径の測定>
分解促進剤の粒度分布は、レーザー回折式粒子径分布測定装置(株式会社島津製作所製「SALD-2300」)を用いて測定した。
この方法では、水等の分散媒中に分散させた分解促進剤の粒子にレーザー光を照射して、観測された回折散乱光のパターンと、分解促進剤を異なる直径を有する多数の球形の粒子の集合体と見なした場合に計算によって推定される回折散乱光のパターンを比較することにより、それらが一致するような粒径と頻度の分布を算出した。粒径の代表値としては、直径が小さい粒子から順に質量を積算したときに全質量の50%になる粒子径(50%粒子径又は中位径と称する)を50%質量累積粒度とする。
分解促進剤の粒度分布は、レーザー回折式粒子径分布測定装置(株式会社島津製作所製「SALD-2300」)を用いて測定した。
この方法では、水等の分散媒中に分散させた分解促進剤の粒子にレーザー光を照射して、観測された回折散乱光のパターンと、分解促進剤を異なる直径を有する多数の球形の粒子の集合体と見なした場合に計算によって推定される回折散乱光のパターンを比較することにより、それらが一致するような粒径と頻度の分布を算出した。粒径の代表値としては、直径が小さい粒子から順に質量を積算したときに全質量の50%になる粒子径(50%粒子径又は中位径と称する)を50%質量累積粒度とする。
[生分解性樹脂組成物及び成形体の評価]
<生分解性試験>
得られた樹脂組成物を170℃の熱プレス成形機で厚さ200μmのフィルムとし、これをISO527-3に準拠したダンベル形状に切り抜き、ポリエチレン製タッパー容器に入れた水分量20%RHの園芸用土(アイリスオーヤマ株式会社製「花と野菜の培養土」)とコンポスト(八幡物産株式会社製、生分解試験用植種源)の質量比1:1混合物中に埋設し、蓋を閉めて28℃の恒温機中に2週間静置した。静置前後の重量変化を測定し、変化率(静置前の質量に対する質量減少量の割合)を算出した。算出された変化率を、比較例I-1又はII-1の脂肪族ポリエステル系樹脂(A)のみの場合の変化率に対する比として算出し、生分解性向上率とした。
<生分解性試験>
得られた樹脂組成物を170℃の熱プレス成形機で厚さ200μmのフィルムとし、これをISO527-3に準拠したダンベル形状に切り抜き、ポリエチレン製タッパー容器に入れた水分量20%RHの園芸用土(アイリスオーヤマ株式会社製「花と野菜の培養土」)とコンポスト(八幡物産株式会社製、生分解試験用植種源)の質量比1:1混合物中に埋設し、蓋を閉めて28℃の恒温機中に2週間静置した。静置前後の重量変化を測定し、変化率(静置前の質量に対する質量減少量の割合)を算出した。算出された変化率を、比較例I-1又はII-1の脂肪族ポリエステル系樹脂(A)のみの場合の変化率に対する比として算出し、生分解性向上率とした。
<引張特性>
フィルム状成形体について、JIS K7127(1999)に準拠して降伏応力及び破断時応力を測定した。これらの値は大きい程好ましい。
フィルム状成形体について、JIS K7127(1999)に準拠して降伏応力及び破断時応力を測定した。これらの値は大きい程好ましい。
<引裂強さ>
フィルム状成形体について、ISO6383-2(1983)に準拠してエルメンドルフ引裂強さを測定した。この値は大きい程好ましい。
フィルム状成形体について、ISO6383-2(1983)に準拠してエルメンドルフ引裂強さを測定した。この値は大きい程好ましい。
<パンクチャー衝撃強度>
幅110mm、長さ1300mmのフィルム状成形体に対して、東洋精機社製打ち抜き衝撃試験機を用いて、先端が直径25mmの半球状の形状をしたアームで直径50mmの穴を12点打ち抜き、パンクチャー衝撃強度を測定した。この値は大きい程好ましい。
幅110mm、長さ1300mmのフィルム状成形体に対して、東洋精機社製打ち抜き衝撃試験機を用いて、先端が直径25mmの半球状の形状をしたアームで直径50mmの穴を12点打ち抜き、パンクチャー衝撃強度を測定した。この値は大きい程好ましい。
[生分解性樹脂]
以下の実施例及び比較例では、生分解性樹脂として、下記脂肪族ポリエステル系樹脂(A)を用いた
脂肪族ポリエステル系樹脂(A):ポリブチレンサクシネートアジペート(PBSA)
PTTMCCBiochem社製 商品名BioPBSTM FD92PB
融点:89℃
以下の実施例及び比較例では、生分解性樹脂として、下記脂肪族ポリエステル系樹脂(A)を用いた
脂肪族ポリエステル系樹脂(A):ポリブチレンサクシネートアジペート(PBSA)
PTTMCCBiochem社製 商品名BioPBSTM FD92PB
融点:89℃
[分解促進剤I及び比較分解促進剤の製造]
各原料は「クラシュミルサー」(岩谷産業社製 IFM-C20G)により粉砕した後、100メッシュ(目開き150μm)の篩を通過したものを乾燥機で80℃にて8時間乾燥を行った。
各原料は「クラシュミルサー」(岩谷産業社製 IFM-C20G)により粉砕した後、100メッシュ(目開き150μm)の篩を通過したものを乾燥機で80℃にて8時間乾燥を行った。
表1に、各原料について求めた窒素、炭素、セルロース、リグニン、ヘミセルロースの含有量(質量%)と、窒素/炭素比、ヘミセルロース/(セルロース+リグニン)比を示す。

[実施例I-1~I-3、比較例I-1~I-5]
脂肪族ポリエステル系樹脂(A)と分解促進剤を表2に示す割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
脂肪族ポリエステル系樹脂(A)と分解促進剤を表2に示す割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
得られた樹脂組成物を用いて前述の生分解性試験を行い、結果を表2に示した。

表2より、窒素/炭素比及びヘミセルロース/(セルロース+リグニン)比が所定の範囲内である本発明の分解促進剤Iを用いることにより、生分解性樹脂の生分解性を大きく向上させることができることが分かる。
[分解促進剤II及び比較分解促進剤の製造]
各原料は「クラシュミルサー」(岩谷産業社製 IFM-C20G)により粉砕した後、100メッシュ(目開き150μm)の篩を通過したものを乾燥機で80℃にて8時間乾燥を行った。
各原料は「クラシュミルサー」(岩谷産業社製 IFM-C20G)により粉砕した後、100メッシュ(目開き150μm)の篩を通過したものを乾燥機で80℃にて8時間乾燥を行った。
表3に、各原料について求めた可溶無窒素物と、セルロース、リグニンの含有量及びその合計(質量%)を示す。
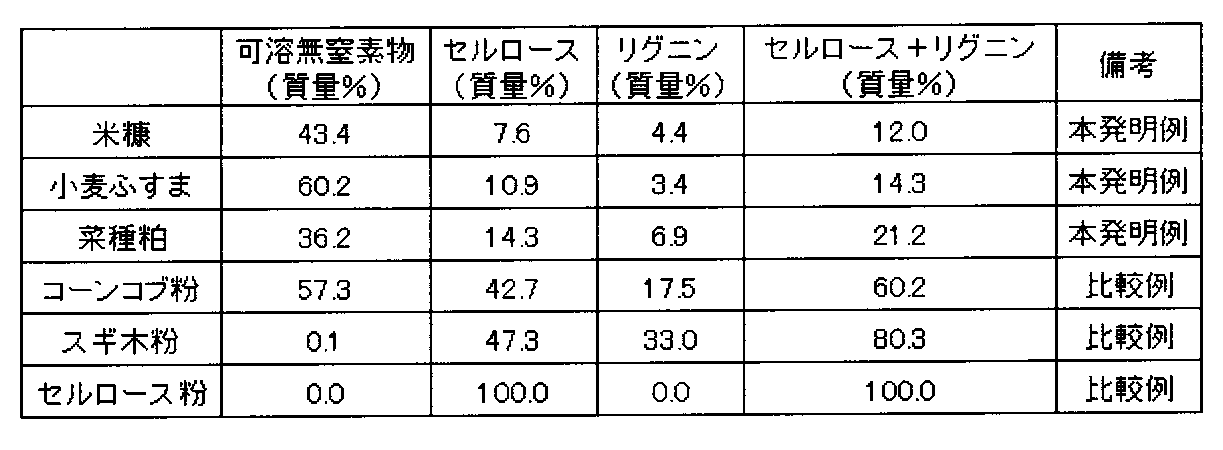
[実施例II-1~II-3、比較例II-1~II-4]
脂肪族ポリエステル系樹脂(A)と分解促進剤を表4に示す割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
脂肪族ポリエステル系樹脂(A)と分解促進剤を表4に示す割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
得られた樹脂組成物を用いて前述の生分解性試験を行い、結果を表4に示した。

表4より、可溶無窒素物及び、セルロースとリグニンの含有量が所定の範囲内である本発明の分解促進剤IIを用いることにより、生分解性樹脂の生分解性を大きく向上させることができることが分かる。
[分解促進剤の製造]
<50%質量累積粒度56μmの小麦ふすま>
粗粉砕した小麦ふすま(昭和産業株式会社製)を、80℃にて8時間乾燥して50%質量累積粒度56μmの小麦ふすまを製造した。
<50%質量累積粒度35μmの小麦ふすま>
粗粉砕した小麦ふすま(昭和産業株式会社製)を、80℃にて8時間乾燥して50%質量累積粒度35μmの小麦ふすまを製造した。
<50%質量累積粒度9μmの小麦ふすま>
未粉砕の小麦ふすま(昭和産業株式会社製)を目開き500μmの篩を通過するまでカッターミルで予備粉砕した後、ジェットミル粉砕機(アイシンナノテクノロジーズ社製「ナノジェットマイザーNJ-50」)を用いて、空気圧1.4MPa、処理速度60g/hの条件で微粉砕し、次いで80℃にて8時間乾燥して50%質量累積粒度9μmの小麦ふすまを製造した。
<50%質量累積粒度56μmの小麦ふすま>
粗粉砕した小麦ふすま(昭和産業株式会社製)を、80℃にて8時間乾燥して50%質量累積粒度56μmの小麦ふすまを製造した。
<50%質量累積粒度35μmの小麦ふすま>
粗粉砕した小麦ふすま(昭和産業株式会社製)を、80℃にて8時間乾燥して50%質量累積粒度35μmの小麦ふすまを製造した。
<50%質量累積粒度9μmの小麦ふすま>
未粉砕の小麦ふすま(昭和産業株式会社製)を目開き500μmの篩を通過するまでカッターミルで予備粉砕した後、ジェットミル粉砕機(アイシンナノテクノロジーズ社製「ナノジェットマイザーNJ-50」)を用いて、空気圧1.4MPa、処理速度60g/hの条件で微粉砕し、次いで80℃にて8時間乾燥して50%質量累積粒度9μmの小麦ふすまを製造した。
上記50%質量累積粒度56μm、35μm、9μmのいずれの小麦ふすまも、乾燥前の水分率は8.2質量%であり、乾燥後の水分率は2.3質量%であった。
[実施例III-1~III-3]
分解促進剤として、上記のように得られた50%質量累積粒度56μm、35μm、又は9μmの小麦ふすまを、脂肪族ポリエステル系樹脂(A)100質量部に対して、それぞれ11.1質量部の割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
分解促進剤として、上記のように得られた50%質量累積粒度56μm、35μm、又は9μmの小麦ふすまを、脂肪族ポリエステル系樹脂(A)100質量部に対して、それぞれ11.1質量部の割合でブレンドし、小型二軸混練機(DSM社製「Xplore Micro 15cc Twin Screw Compounder」)を使用して、窒素雰囲気下、170℃にて4分間溶融混練を行った。
得られた樹脂組成物を170℃の熱プレス成形機で厚さ約120μmのフィルム状に成形し、それぞれの試験規格に規定された形状に切り抜いて得られた試験片について、前述の降伏応力、破断時応力、エルメンドルフ引裂強度、パンクチャー衝撃強度の評価を行った。結果を表5に示す。

表5から、50%質量累積粒度が小さいほど、生分解性樹脂成形体の機械特性が向上することがわかる。
本発明を特定の態様を用いて詳細に説明したが、本発明の意図と範囲を離れることなく様々な変更が可能であることは当業者に明らかである。
本出願は、2019年12月27日付で出願された日本特許出願2019-238580及び日本特許出願2019-238581に基づいており、その全体が引用により援用される。
本出願は、2019年12月27日付で出願された日本特許出願2019-238580及び日本特許出願2019-238581に基づいており、その全体が引用により援用される。
Claims (12)
- セルロース、ヘミセルロース及びリグニンを含む生分解性樹脂用分解促進剤であって、該生分解性樹脂用分解促進剤中の炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、生分解性樹脂用分解促進剤。
- 可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下である、生分解性樹脂用分解促進剤。
- 水分率が5質量%未満である、請求項1又は2に記載の生分解性樹脂用分解促進剤。
- 請求項1~3のいずれかに記載の生分解性樹脂用分解促進剤2質量部以上250質量部以下と生分解性樹脂100質量部を含む生分解性樹脂組成物。
- 前記生分解性樹脂が脂肪族ポリエステル系樹脂(A)、脂肪族-芳香族ポリエステル系樹脂(B)及び脂肪族オキシカルボン酸系樹脂(C)からなる群より選ばれる1種以上である、請求項4に記載の生分解性樹脂組成物。
- 前記脂肪族ポリエステル系樹脂(A)が脂肪族ジオールに由来する繰返し単位と脂肪族ジカルボン酸に由来する繰返し単位とを主構成単位として含む、請求項5に記載の生分解性樹脂組成物。
- 前記脂肪族-芳香族ポリエステル系樹脂(B)が脂肪族ジオールに由来する繰返し単位と脂肪族ジカルボン酸に由来する繰返し単位と芳香族ジカルボン酸に由来する繰り返し単位とを主構成単位として含む、請求項5又は6に記載の生分解性樹脂組成物。
- 請求項4~7のいずれかに記載の生分解性樹脂組成物の押出成形体又は射出成形体である生分解性樹脂成形体。
- 原料を粉砕した後、得られた粉体から所定の粒度のものを選別して、生分解性樹脂用分解促進剤を得る、生分解性樹脂用分解促進剤の製造方法。
- 更に乾燥する工程を有する、請求項9に記載の生分解性樹脂用分解促進剤の製造方法。
- 前記生分解性樹脂用分解促進剤が、セルロース、ヘミセルロース及びリグニンを含み、該生分解性樹脂用分解促進剤中の炭素に対する窒素の質量比が0.04以上であり、且つ、セルロースとリグニンの合計の含有量に対するヘミセルロースの含有量の質量割合が0.2以上である、請求項9又は10に記載の生分解性樹脂用分解促進剤の製造方法。
- 前記生分解性樹脂用分解促進剤が、可溶無窒素物を20質量%以上含有し、且つ、セルロースとリグニンの含有量の合計が50質量%以下である、請求項9~11のいずれかに記載の生分解性樹脂用分解促進剤の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20906297.5A EP4083130A4 (en) | 2019-12-27 | 2020-09-17 | DEGRADATION ACCELERATOR FOR BIODEGRADABLE RESIN, BIODEGRADABLE RESIN COMPOSITION, BIODEGRADABLE RESIN MOLDED PRODUCT, AND METHOD FOR MANUFACTURING DEGRADATION ACCELERATOR FOR BIODEGRADABLE RESIN |
| JP2021566819A JPWO2021131181A1 (ja) | 2019-12-27 | 2020-09-17 | |
| CN202080066954.6A CN114514289B (zh) | 2019-12-27 | 2020-09-17 | 生物降解性树脂用降解促进剂、生物降解性树脂组合物、生物降解性树脂成型体、以及生物降解性树脂用降解促进剂的制造方法 |
| US17/658,047 US20220282069A1 (en) | 2019-12-27 | 2022-04-05 | Degradation accelerator for biodegradable resin, biodegradable resin composition, biodegradable resin molded product, and method for producing degradation accelerator for biodegradable resin |
| JP2025092663A JP2025116190A (ja) | 2019-12-27 | 2025-06-03 | 生分解性樹脂用分解促進剤、生分解性樹脂組成物、生分解性樹脂成形体、及び生分解性樹脂用分解促進剤の製造方法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-238580 | 2019-12-27 | ||
| JP2019238581 | 2019-12-27 | ||
| JP2019-238581 | 2019-12-27 | ||
| JP2019238580 | 2019-12-27 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/658,047 Continuation US20220282069A1 (en) | 2019-12-27 | 2022-04-05 | Degradation accelerator for biodegradable resin, biodegradable resin composition, biodegradable resin molded product, and method for producing degradation accelerator for biodegradable resin |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021131181A1 true WO2021131181A1 (ja) | 2021-07-01 |
Family
ID=76575884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/035289 Ceased WO2021131181A1 (ja) | 2019-12-27 | 2020-09-17 | 生分解性樹脂用分解促進剤、生分解性樹脂組成物、生分解性樹脂成形体、及び生分解性樹脂用分解促進剤の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220282069A1 (ja) |
| EP (1) | EP4083130A4 (ja) |
| JP (2) | JPWO2021131181A1 (ja) |
| CN (1) | CN114514289B (ja) |
| WO (1) | WO2021131181A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021241712A1 (ja) * | 2020-05-28 | 2021-12-02 | ||
| JP2022142016A (ja) * | 2021-03-16 | 2022-09-30 | 株式会社グリーンテクノプラス | 海洋生分解促進添加剤及びこれを含む海洋生分解性樹脂組成物 |
| CN117089171A (zh) * | 2022-05-21 | 2023-11-21 | 宜科万斯有限公司 | 可生物降解的成型制品和可生物降解的聚酯树脂组合物 |
| EP4279527A1 (en) * | 2022-05-21 | 2023-11-22 | Ecovance Co. Ltd | Biodegradable polyester resin composition |
| US11952459B2 (en) | 2020-12-23 | 2024-04-09 | Kintra Fibers, Inc. | Polyester polymer nanocomposites |
| WO2024202792A1 (ja) * | 2023-03-24 | 2024-10-03 | パナソニックIpマネジメント株式会社 | 複合樹脂組成物及び複合樹脂成形体 |
| US12202967B2 (en) | 2019-06-18 | 2025-01-21 | Kintra Fibers, Inc. | Polyester polymer nanocomposites |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115197405B (zh) * | 2022-07-22 | 2023-05-26 | 中北大学 | 一种基于呋喃环具有水蒸气阻隔性能的可降解PBSeT共聚酯材料及其制备方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11166055A (ja) * | 1997-12-01 | 1999-06-22 | Shokuhin Sangyo Kankyo Hozen Gijutsu Kenkyu Kumiai | 生分解性または生崩壊性素材の製造方法 |
| JP2009019200A (ja) * | 2007-06-11 | 2009-01-29 | Kyoto Univ | リグニン含有ミクロフィブリル化植物繊維及びその製造方法 |
| KR20090065428A (ko) * | 2007-12-17 | 2009-06-22 | 건국대학교 산학협력단 | 버섯부산물(폐배지)을 주원료로 이용한 가축용 발효조사료 제조방법 |
| JP2009538388A (ja) * | 2006-05-26 | 2009-11-05 | エルサム・クラフト・アクティーゼルスカブ | 液化バイオマスから合成ガスを生成する方法 |
| JP2011050359A (ja) * | 2009-09-04 | 2011-03-17 | Musashino Chemical Laboratory Ltd | 新規微生物および該微生物由来の酵素、ならびにこれらを用いた糖化液の製造方法 |
| JP2012158707A (ja) * | 2011-02-02 | 2012-08-23 | Hitachi Chemical Co Ltd | 樹脂組成物及びシート |
| JP2016500558A (ja) * | 2012-10-10 | 2016-01-14 | ザイレコ,インコーポレイテッド | バイオマスの加工処理 |
| JP2017002206A (ja) * | 2015-06-11 | 2017-01-05 | 出光興産株式会社 | 熱可塑性樹脂組成物及び熱可塑性樹脂組成物の製造方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3105451B2 (ja) * | 1996-04-30 | 2000-10-30 | アイセロ化学株式会社 | 生分解性樹脂組成物及びその成形品 |
| JP3646193B2 (ja) * | 2000-05-19 | 2005-05-11 | 宮城県 | 生分解速度が制御された生分解性樹脂組成物およびその製造方法 |
| JP2002088264A (ja) * | 2000-09-12 | 2002-03-27 | Idemitsu Technofine Co Ltd | 生分解性樹脂組成物およびこれを成形してなる成形品、生活用資材、農業用資材 |
| JP4914982B2 (ja) * | 2001-06-01 | 2012-04-11 | 日清製粉株式会社 | 生分解性プラスチック組成物 |
| CN102120870A (zh) * | 2011-02-28 | 2011-07-13 | 殷正福 | 一种可降解塑料及其生产方法 |
| KR101525658B1 (ko) * | 2013-02-21 | 2015-06-03 | 씨제이제일제당 (주) | 식품부산물인 소맥피 또는 대두피를 활용한 바이오 매스 필름용 조성물 및 이를 이용한 바이오매스 필름 |
| KR101383528B1 (ko) * | 2013-08-14 | 2014-04-10 | 플러스폴리 주식회사 | 커피 폐기물을 포함하는 수지 펠릿 및 그 제조방법 |
| US10544301B2 (en) * | 2016-02-09 | 2020-01-28 | Kaneka Corporation | Biodegradable polyester resin composition and molded article formed from said resin composition |
| WO2021241712A1 (ja) * | 2020-05-28 | 2021-12-02 | 三菱ケミカル株式会社 | 生分解性樹脂組成物及び生分解性樹脂成形体 |
-
2020
- 2020-09-17 CN CN202080066954.6A patent/CN114514289B/zh active Active
- 2020-09-17 JP JP2021566819A patent/JPWO2021131181A1/ja active Pending
- 2020-09-17 WO PCT/JP2020/035289 patent/WO2021131181A1/ja not_active Ceased
- 2020-09-17 EP EP20906297.5A patent/EP4083130A4/en active Pending
-
2022
- 2022-04-05 US US17/658,047 patent/US20220282069A1/en active Pending
-
2025
- 2025-06-03 JP JP2025092663A patent/JP2025116190A/ja active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11166055A (ja) * | 1997-12-01 | 1999-06-22 | Shokuhin Sangyo Kankyo Hozen Gijutsu Kenkyu Kumiai | 生分解性または生崩壊性素材の製造方法 |
| JP2009538388A (ja) * | 2006-05-26 | 2009-11-05 | エルサム・クラフト・アクティーゼルスカブ | 液化バイオマスから合成ガスを生成する方法 |
| JP2009019200A (ja) * | 2007-06-11 | 2009-01-29 | Kyoto Univ | リグニン含有ミクロフィブリル化植物繊維及びその製造方法 |
| KR20090065428A (ko) * | 2007-12-17 | 2009-06-22 | 건국대학교 산학협력단 | 버섯부산물(폐배지)을 주원료로 이용한 가축용 발효조사료 제조방법 |
| JP2011050359A (ja) * | 2009-09-04 | 2011-03-17 | Musashino Chemical Laboratory Ltd | 新規微生物および該微生物由来の酵素、ならびにこれらを用いた糖化液の製造方法 |
| JP2012158707A (ja) * | 2011-02-02 | 2012-08-23 | Hitachi Chemical Co Ltd | 樹脂組成物及びシート |
| JP2016500558A (ja) * | 2012-10-10 | 2016-01-14 | ザイレコ,インコーポレイテッド | バイオマスの加工処理 |
| JP2017002206A (ja) * | 2015-06-11 | 2017-01-05 | 出光興産株式会社 | 熱可塑性樹脂組成物及び熱可塑性樹脂組成物の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| J. BACTERIOL., vol. 179, 1997, pages 4821 |
| See also references of EP4083130A4 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12202967B2 (en) | 2019-06-18 | 2025-01-21 | Kintra Fibers, Inc. | Polyester polymer nanocomposites |
| JPWO2021241712A1 (ja) * | 2020-05-28 | 2021-12-02 | ||
| JP7800424B2 (ja) | 2020-05-28 | 2026-01-16 | 三菱ケミカル株式会社 | 生分解性樹脂組成物及び生分解性樹脂成形体 |
| US11952459B2 (en) | 2020-12-23 | 2024-04-09 | Kintra Fibers, Inc. | Polyester polymer nanocomposites |
| US12570796B2 (en) | 2020-12-23 | 2026-03-10 | Kintra Fibers, Inc. | Polyester polymer nanocomposites |
| JP2022142016A (ja) * | 2021-03-16 | 2022-09-30 | 株式会社グリーンテクノプラス | 海洋生分解促進添加剤及びこれを含む海洋生分解性樹脂組成物 |
| CN117089171A (zh) * | 2022-05-21 | 2023-11-21 | 宜科万斯有限公司 | 可生物降解的成型制品和可生物降解的聚酯树脂组合物 |
| EP4279527A1 (en) * | 2022-05-21 | 2023-11-22 | Ecovance Co. Ltd | Biodegradable polyester resin composition |
| WO2024202792A1 (ja) * | 2023-03-24 | 2024-10-03 | パナソニックIpマネジメント株式会社 | 複合樹脂組成物及び複合樹脂成形体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4083130A1 (en) | 2022-11-02 |
| JP2025116190A (ja) | 2025-08-07 |
| CN114514289A (zh) | 2022-05-17 |
| US20220282069A1 (en) | 2022-09-08 |
| JPWO2021131181A1 (ja) | 2021-07-01 |
| EP4083130A4 (en) | 2023-10-11 |
| CN114514289B (zh) | 2024-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114514289B (zh) | 生物降解性树脂用降解促进剂、生物降解性树脂组合物、生物降解性树脂成型体、以及生物降解性树脂用降解促进剂的制造方法 | |
| JP7800424B2 (ja) | 生分解性樹脂組成物及び生分解性樹脂成形体 | |
| TWI599598B (zh) | Biodegradable film material and method of making the same | |
| CN1277882C (zh) | 生物降解聚酯的三元混合物以及由其得到的产品 | |
| KR100620471B1 (ko) | 락트산계 수지 조성물 | |
| CN110678502B (zh) | 用于高度可崩解薄膜的聚合物组合物 | |
| JP7435041B2 (ja) | 生分解性樹脂用分解促進剤、生分解性樹脂組成物及び生分解性樹脂成形体 | |
| JPH10508640A (ja) | 生分解可能なポリマー、その製造及び生分解可能な成形体の製造のためのその使用 | |
| EP2093295A1 (en) | Plant-derived natural biodegradable material | |
| CA2819570C (en) | Polyester composition | |
| JP2022157778A (ja) | 生分解性樹脂組成物及び成形体 | |
| WO2025137759A1 (en) | Biodegradable polymer composites, methods of manufacture and use therefor | |
| CA3149299A1 (en) | Flexible wood composite material | |
| EP4077523B1 (en) | Biodegradable and compostable composition | |
| JP7647408B2 (ja) | 生分解促進剤、生分解性樹脂組成物及び生分解性樹脂成形体 | |
| Bouzidi et al. | Thermoplasticized bread waste and poly (butylene adipate-co-terephthalate) blends: a sustainable alternative to starch in eco-friendly packaging | |
| JP2002088264A (ja) | 生分解性樹脂組成物およびこれを成形してなる成形品、生活用資材、農業用資材 | |
| CN119677805A (zh) | 可生物降解的组合物及制造方法 | |
| JP7613268B2 (ja) | 生分解性樹脂組成物及び生分解性樹脂成形体 | |
| JP2022145493A (ja) | 生分解性樹脂組成物及び生分解性樹脂成形体並びに生分解性樹脂組成物の製造方法 | |
| JP2005060686A (ja) | ポリ乳酸組成物及びそれから得られる成形物 | |
| JP2019172742A (ja) | 樹脂組成物及び樹脂成形品 | |
| Al-Ashor | Sustainable food packaging based on polyhydroxyalkanoate | |
| JP2020164577A (ja) | ポリエステル系樹脂組成物及び成形品 | |
| JP2025069099A (ja) | 樹脂組成物及び成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20906297 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021566819 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020906297 Country of ref document: EP Effective date: 20220727 |