WO2021132589A1 - ポリヌクレオチド及び医薬組成物 - Google Patents
ポリヌクレオチド及び医薬組成物 Download PDFInfo
- Publication number
- WO2021132589A1 WO2021132589A1 PCT/JP2020/048799 JP2020048799W WO2021132589A1 WO 2021132589 A1 WO2021132589 A1 WO 2021132589A1 JP 2020048799 W JP2020048799 W JP 2020048799W WO 2021132589 A1 WO2021132589 A1 WO 2021132589A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- nucleotides
- sugar
- nucleotide
- modified
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)C(NC(NC1=O)=Nc2c1nc[n]2[C@@](C1*)O[C@@](C*)C1O)=O Chemical compound CC(C)C(NC(NC1=O)=Nc2c1nc[n]2[C@@](C1*)O[C@@](C*)C1O)=O 0.000 description 4
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/67—General methods for enhancing the expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/33—Chemical structure of the base
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/50—Vector systems having a special element relevant for transcription regulating RNA stability, not being an intron, e.g. poly A signal
Definitions
- the present invention relates to a polynucleotide and a pharmaceutical composition containing the polynucleotide.
- mRNA messenger RNA
- RNA synthase RNA synthase
- ribosomes bind to the transcribed single-stranded mRNA to synthesize proteins by translation. It is transmitted by being done. This form of transmission is called the "central dogma” in molecular biology and is a basic principle common to both prokaryotes and eukaryotes.
- MRNA which is an intermediate substance for genetic signal transduction, has base sequence information and structure for being directly recognized by the ribosome and translated into a protein.
- a polynucleotide as mRNA (hereinafter, also referred to as "artificial mRNA”) can be used as a protein replacement therapy type nucleic acid drug by enhancing or enhancing expression or a vaccine therapy type nucleic acid drug by peptide expression.
- artificial mRNA composed of only natural bases is introduced into cells from the outside, an immune response is rapidly evoked by binding to intracellular Toll-like receptors (TLR3, TLR7, TLR8, RIG-I, etc.).
- TLR3, TLR7, TLR8, RIG-I, etc. intracellular Toll-like receptors
- Non-Patent Document 2 polynucleotides containing sugar-modified nucleotides such as 2'-O-methylated RNA, 2'-F-modified RNA and cross-linked nucleic acids reduce the immunoreactivity of nucleic acid drugs and resist nucleic acid-degrading enzymes. It has been shown that it is effective for all of the granting of (Non-Patent Document 3).
- Non-Patent Document 4 In recent years, there has been an active movement to use mRNA artificially synthesized in vitro by an in vitro transcription method (hereinafter referred to as "IVT") as a pharmaceutical product (Non-Patent Document 4).
- IVT in vitro transcription method
- Non-Patent Document 5 reports, in a clinical trial of an artificial mRNA cancer vaccine for melanoma patients, the incidence of metastasis is significantly reduced after the start of administration of the cancer vaccine. , Certain results are being reported.
- these clinically applied artificial mRNAs are produced by IVT. Artificial mRNA produced by IVT has the following two problems. First, the introduction position of the modified nucleotide introduced for the purpose of reducing the immunoreactivity and imparting stability to the nucleolytic enzyme cannot be controlled.
- Patent Document 1 discloses a case in which peptide translation ability is attenuated or eliminated in artificial mRNA into which 2'-F modified RNA is introduced by IVT. Second, only modified nucleotides recognized as substrates in the RNA synthase used in IVT can be introduced. Similarly, Patent Document 1 discloses that artificial mRNA containing 2'-O-methylated RNA is difficult to prepare by an IVT reaction using a general RNA polymerase. Therefore, it cannot be said that the position and type of the modified nucleotide have been thoroughly investigated in the artificial mRNA produced by introducing the modified nucleotide by IVT.
- Non-Patent Documents 6 and 7 A method of artificially synthesizing mRNA using a technique of chemically linking a plurality of RNAs has been reported (Non-Patent Documents 6 and 7). Using this method, it is possible to introduce a modified nucleotide containing a sugar moiety modification at an arbitrary position of an artificial mRNA containing a coding sequence (hereinafter, may be referred to as “CDS”). In fact, Non-Patent Documents 6 and 7 disclose that artificial mRNA in which 2'-O-methylated RNA was introduced into one place in the CDS of mRNA was prepared and its peptide translating ability was confirmed. ing.
- the modified nucleotide is required to achieve sufficiently low immunoreactivity and high stability as a nucleic acid drug. Further knowledge is needed regarding the modification rate, position and type of.
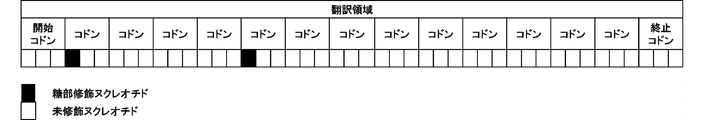
- the translational activity is maintained even if the sugar portion of the first nucleotide among the first, second and third nucleotides contained in each of the plurality of codons constituting the translation region is modified. Found to be done.
- the present invention includes the following embodiments.
- [1] Contains the translation region from the start codon to the stop codon
- the translation region contains n codons, where n is a positive integer greater than or equal to 2.
- the n codons contain the first, second and third nucleotides, respectively.
- the first nucleotide in at least two of the n codons is a sugar-modified nucleotide.
- Polynucleotide [2] The polynucleotide according to [1], wherein the sugar moiety-modified nucleotide contains a sugar moiety modified at least at the 2'position. [3]
- the sugar moiety modified at least in the 2'position is selected from the following.
- the sugar-modified nucleotide contains a base corresponding to a base selected from the group consisting of adenine, guanine, cytosine and uracil.
- [6-1] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 5% or more.
- [6-2] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 10% or more.
- [6-3] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 15% or more.
- [6-13] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 65% or more.
- [6-14] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 70% or more.
- [6-15] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 75% or more.
- [6-16] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 80% or more.
- [6-17] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 90% or more.
- [6-18] The polynucleotide according to any one of [1] to [5], wherein the ratio of the first nucleotide in the n codons being a sugar-modified nucleotide is 95% or more.
- [10-7] The polynucleotide according to any one of [1] to [10], wherein the proportion of the third nucleotide in the n codons being a sugar-modified nucleotide is 45% or less.
- [10-8] The polynucleotide according to any one of [1] to [10], wherein the proportion of the third nucleotide in the n codons being a sugar-modified nucleotide is 40% or less.
- [10-9] The polynucleotide according to any one of [1] to [10], wherein the proportion of the third nucleotide in the n codons being a sugar-modified nucleotide is 35% or less.
- [10-16] The above-mentioned one of [1] to [6-18] and [9] to [9-11], wherein the ratio of the third nucleotide in the n codons being a sugar-modified nucleotide is 0%.
- [11] The polynucleotide according to any one of [1] to [10-16], wherein n is an integer of 2 to 2000.
- [11-1] The polynucleotide according to any one of [1] to [10-16], wherein n is an integer of 2 to 1500.
- [11-2] The polynucleotide according to any one of [1] to [10-16], wherein n is an integer of 2 to 1000.
- n is an integer of 200 to 500.
- the 5'side untranslated region contains a base-modified nucleotide containing the following base: [In the formula, R is an alkyl group having 1 to 6 carbon atoms] [12] The polynucleotide according to.
- X 3 represents OH, SH or salts thereof, However, X 1 and X 2 are not O at the same time]
- the present invention further includes the following embodiments.
- Method. [1C] Use of the polynucleotide according to any one of [1] to [24] or the pharmaceutical composition according to [25] for treating a disease.
- [1D] Use of the polynucleotide according to any one of [1] to [24] in the manufacture of a medicament for treating a disease.
- [1E] The polynucleotide according to any one of [1] to [24], for use in the manufacture of a medicament for treating a disease.
- [1F] A kit for use in the treatment of a disease, which comprises the polynucleotide according to any one of [1] to [24] or the pharmaceutical composition and instruction manual according to [25].
- the present invention further includes the following embodiments.
- [2A] Includes translated region and 5'untranslated region A polynucleotide in which the first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- [2B] Includes translated region and 3'untranslated region A polynucleotide in which the first, second, and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- [2C] Includes translated region, 5'side untranslated region and 3'side untranslated region The first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- [2D] Includes translated region and 5'untranslated region A polynucleotide in which the 1st to 3rd nucleotides from the 5'end of the 5'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by a phosphorothioate.
- [2E] Includes translated region and 3'untranslated region A polynucleotide in which the 1st to 3rd nucleotides from the 3'end of the 3'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by a phosphorothioate.
- [2F] Includes translated region, 5'side untranslated region and 3'side untranslated region The 1st to 3rd nucleotides from the 5'end of the 5'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by phosphorothioates.
- [2G] Includes translated region and 5'untranslated region The first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- [2H] Includes translated region and 3'untranslated region The first, second and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- [2I] Includes translated region, 5'side untranslated region and 3'side untranslated region
- the first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- the 1st to 3rd nucleotides from the 5'end of the 5'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by phosphorothioates.
- the first, second and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- a polynucleotide in which the 1st to 3rd nucleotides from the 3'end of the 3'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by a phosphorothioate.
- Each lane is as follows (1-3: Compound R17) (concentrations in the reaction solution are 1, 3, 5 ⁇ M, respectively), 4-6: Compound R1 (concentrations in the reaction solution are 1, 3, 3, respectively). 5 ⁇ M), 7: No RNA, M: Protein size marker (Precision Plus Protein Dual Extra Standards (BIORAD))).
- the numbers on the left of the figure represent the molecular weight of the protein, and the arrows indicate the translation products produced.
- the results of Western blot analysis of the translation reaction performed using RRL using compound R2 and compound R18 as substrates are shown.
- Each lane is as follows (1: No RNA, 2: Compound R18 (5 ⁇ g), 3: Compound R2 (5 ⁇ g), M: Protein size marker (Precision Plus Protein Dual Extra Standards (BIORAD))) ..
- the numbers on the left of the figure represent the molecular weight of the protein, and the arrows indicate the translation products produced.
- the translation region from the start codon to the termination codon is included, the translation region contains n codons, the n is a positive integer of 2 or more, and the n codons are:
- the present invention relates to a polynucleotide containing the first, second and third nucleotides, respectively, in which the first nucleotide in at least two codons of the n codons is a sugar-modified nucleotide.
- FIG. 1 shows a schematic diagram of a translation region in which the first nucleotide in any two codons is a sugar-modified nucleotide.
- the polynucleotide of the present embodiment Since the translation activity is maintained even if the sugar portion of the first nucleotide in the plurality of codons constituting the translation region is modified, the polynucleotide of the present embodiment has the translation activity while having the modification site in the translation region. Maintained.
- the term "translational activity” means an activity in which mRNA is translated to synthesize a polypeptide ("polypeptide" in the present specification includes a protein).
- the polynucleotide of the present embodiment also has excellent stability to an enzyme (eg, a nucleolytic enzyme (nuclease)).
- maintaining translational activity means that translational activity is 60% or more as compared with a polynucleotide in which a polynucleotide in which the sugar portion of the first nucleotide in a plurality of codons is modified is not modified. Indicates that there is.
- the translational activity of the modified polynucleotide is preferably 70% or more, 80% or more, 90% or more, or 100% or more as compared with the unmodified polynucleotide.
- the polynucleotide of the present embodiment is characterized in that the translation region is translated into a polypeptide, for example, mRNA, small open reading frame (smORF), non-canonical open reading frame, long noncoding RNA (lncRNA), ply-microRNA. It is understood as a polynucleotide having a function equivalent to (pri-miRNA).
- the polynucleotide of this embodiment includes a translation region.
- the translated region is also referred to as a code sequence (CDS).
- One polynucleotide may contain a plurality of translation regions.
- a translation region is a region composed of a plurality of codons from a start codon to a stop codon (or a stop codon), and is translated to synthesize a polypeptide.
- a codon is a unit that encodes each amino acid that constitutes a polypeptide, and the unit is composed of three nucleotides.
- examples of the start codon include AUG encoding methionine.
- Non-normal start codons other than AUG can also include CUG, GUG, UUG, ACG, AUC, AUU, AAG, AUA, and AGG.
- Stop codons include, for example, UAA, UAG and UGA.
- the type of codons constituting the translation region is not particularly limited, and can be appropriately selected depending on the target polypeptide.
- the number of codons (n) constituting the translation region is preferably an integer of 2 to 2000, more preferably an integer of 2 to 1500, still more preferably an integer of 2 to 1000, and most preferably 2 to 2. It is an integer of 500. Further, the lower limit of the numerical range may be changed to 5, 10, 50, 100, 200 and the like.
- the number of codons (n) constituting the translation region when the lower limit is changed is preferably an integer of 5 to 2000, 10 to 2000, 50 to 2000, 100 to 2000 or 200 to 2000, and more preferably 5.
- Each codon contains the 1st, 2nd and 3rd nucleotides.
- the start codon (AUG)
- the first nucleotide is A
- the second nucleotide is U
- the third nucleotide is G.
- Nucleotides usually include a sugar part, a base part and a phosphoric acid part.
- the sugar part is the part contained in the nucleotide corresponding to the sugar
- the base part is the part contained in the nucleotide corresponding to the base
- the phosphoric acid part is the part contained in the nucleotide corresponding to the phosphoric acid. Is.
- nucleotide modified with a sugar moiety is referred to as a "sugar moiety-modified nucleotide”
- a nucleotide with a modified base moiety is referred to as a “base-modified nucleotide”
- a nucleotide with a modified phosphate moiety is referred to as "phosphate moiety-modified nucleotide”.
- phosphate moiety-modified nucleotide phosphate moiety-modified nucleotide.
- modification means changing the structure of a sugar portion, a base portion or a phosphoric acid portion. The change in structure due to modification is not particularly limited. Modifications include, for example, substitution of any site with any substituent.
- OH bonded to the 2'-carbon of the sugar moiety is replaced with H (that is, the ribose moiety is replaced with the 2'-deoxyribose moiety), and the 2'-carbon of the sugar moiety is substituted.
- Substituting H bound to OH with OH ie, substituting the 2'-deoxyribose moiety with the ribose moiety) is not included in the modification of the sugar moiety. Therefore, when ribonucleotides are used as a reference, the 2'-deoxyribonucleotide corresponding to the ribonucleotide is not a "sugar-modified nucleotide".
- the ribonucleotide corresponding to the 2'-deoxyribonucleotide is not a "sugar-modified nucleotide".
- the unmodified sugar moiety is preferably a sugar moiety corresponding to ribose or 2'-deoxyribose, and more preferably a sugar moiety corresponding to ribose. That is, in the polynucleotide of the present embodiment, the nucleotide other than the sugar moiety-modified nucleotide preferably contains a sugar moiety corresponding to ribose or 2'-deoxyribose, and more preferably contains a sugar moiety corresponding to ribose.
- At least two of the first nucleotides contained in the plurality of codons constituting the translation region are sugar-modified nucleotides.
- the position of the codon containing the sugar-modified nucleotide is not particularly limited.
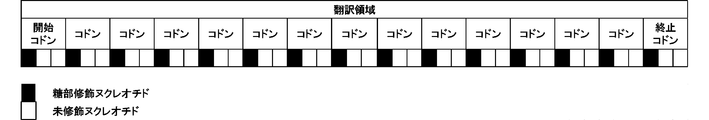
- the proportion of the first nucleotide being a sugar-modified nucleotide is 5% or more, 10% or more, 15% or more, 20% or more, 25% or more, 30% or more, 35% or more, 40% or more, 45% or more, It is preferably 50% or more, 55% or more, 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more or 100%.
- the ratio is 100%, it means that all of the first nucleotides are sugar-modified nucleotides. The larger the ratio, the better the stability to the enzyme.
- FIG. 2 shows a schematic diagram of a translation region in which all of the first nucleotides are sugar-modified nucleotides.
- the substituent at the 2'position of the sugar moiety of the first nucleotide is preferably fluorine.
- At least one of the second nucleotides contained in the plurality of codons constituting the translation region may be a sugar moiety-modified nucleotide, but the sugar moiety of the second nucleotide is modified. It does not have to be.
- the proportion of the second nucleotide being a sugar-modified nucleotide is 50% or less, 45% or less, 40% or less, 35% or less, 30% or less, 25% or less, 20% or less, 15% or less, 10% or less, It may be 5% or less, or 0%.
- the ratio is 0%, it means that not all of the second nucleotides are sugar-modified nucleotides.
- the substituent at the 2'position of the sugar moiety of the second nucleotide is preferably fluorine.
- At least one of the third nucleotides contained in the plurality of codons constituting the translation region may be a sugar-modified nucleotide.
- the proportion of the third nucleotide being a sugar-modified nucleotide is 100%, 90% or less, 80% or less, 70% or less, 60% or less, 50% or less, 45% or less, 40% or less, 35% or less, 30. % Or less, 25% or less, 20% or less, 15% or less, 10% or less, 5% or less, or 0%.
- the first, second, and third nucleotides of the stop codon may be sugar-modified nucleotides from the viewpoint of improving translational activity.
- FIG. 3 shows a schematic diagram of a translation region in which all of the first nucleotides and all of the nucleotides of the stop codon are sugar-modified nucleotides.
- the first, second, and third nucleotides of the start codon may be sugar-modified nucleotides from the viewpoint of improving the stability to the nucleolytic enzyme.
- the substituent at the 2'position of the sugar portion of the first, second and third nucleotides of the start codon is fluorine.
- the sugar moiety-modified nucleotide is not particularly limited as long as the sugar moiety of the nucleotide is modified, but preferably contains a sugar moiety modified at least at the 2'position.
- the sugar moiety modified at least at the 2'position may be a sugar moiety in which the 2'position and the 4'position are crosslinked.
- modified sugar moiety examples include the following.
- M is R 1, OR 1, R 2 OR 1, SH, SR 1, NH 2, NHR 1, NR 1 2, N 3, CN, F, Cl, Br or I
- R 1 is independently alkyl or aryl, preferably alkyl having 1 to 6 carbon atoms, and more preferably alkyl having 1 to 3 carbon atoms.
- R 2 is an alkylene, preferably an alkylene having 1 to 6 carbon atoms]
- examples of the alkyl having 1 to 6 carbon atoms include linear or branched alkyl having 1 to 6 carbon atoms.
- linear alkyl having 1 to 6 carbon atoms include methyl, ethyl, propyl, butyl, pentyl and hexyl.
- alkyl chain having 1 to 6 carbon atoms include isopropyl, isobutyl, sec-butyl, tert-butyl, and methyl-substituted pentyl.
- alkyl having 1 to 3 carbon atoms include methyl, ethyl, propyl and isopropyl.
- aryl examples include phenyl which may be substituted and naphthyl which may be substituted.
- the alkylene having 1 to 6 carbon atoms is a group excluding one hydrogen atom bonded to the carbon atom of the alkyl having 1 to 6 carbon atoms.
- the modified sugar moiety indicates a modified sugar structure contained in the sugar moiety-modified nucleotide.
- M of the modified sugar moiety 2- (methoxy) ethoxy, 3-aminopropoxy, 2-[(N, N-dimethylamino) oxy] ethoxy, 3- (N, N-dimethylamino) propoxy, 2- [2- (N, N-dimethylamino) ethoxy] ethoxy, 2- (methylamino) -2-oxoethoxy, 2- (N-methylcarbamoyl) ethoxy], and 2-cyanoethoxy can also be mentioned.
- the modified sugar moiety further includes the sugar moiety of the following nucleic acids: -Locked Nucleic Acid (LNA) [Tetrahedron Letters, 38, 8735 (1997) and Tetrahedron, 54, 3607 (1998)]; ⁇ Ethylene bridged nucleic acid (ENA) [Nucleic Acids Research, 32, e175 (2004)]; ⁇ Constrained Ethyl (cEt) [The Journal of Organic Chemistry 75, 1569 (2010)]; ⁇ Amido-Bridged Nucleic Acid (AmNA) [Chem Bio Chem 13, 2513 (2012)]; ⁇ 2'-O, 4'-c-Spirocyclopropylene bridged nucleic acid (scpBNA) [Chem.
- LNA Locked Nucleic Acid
- EDA Ethylene bridged nucleic acid
- AmNA AmNA
- the modified sugar moiety is not particularly limited, but is preferably selected from the following.
- the sugar moiety-modified nucleotide preferably contains a base moiety corresponding to a base selected from the group consisting of adenine (A), guanine (G), cytosine (C) and uracil (U), and the type of the base is at least. Two types are preferable. Here, “there are at least two types of bases” means that, for example, one sugar-modified nucleotide contains a base portion corresponding to adenine, and another sugar-modified nucleotide contains a base portion corresponding to guanine. Means.
- the sugar-modified nucleotide may be a base-modified nucleotide and / or a phosphate-modified nucleotide (in other words, the sugar-modified nucleotide may further contain a modified base and / or a modified phosphate moiety. ). At least one sugar-modified nucleotide may contain a modified base.
- the translation region may contain base-modified nucleotides.
- the position where the base-modified nucleotide is present in the translation region is not particularly limited.
- the base-modified nucleotide may be a sugar-modified nucleotide and / or a phosphate-modified nucleotide (in other words, the base-modified nucleotide may further contain a modified sugar and / or a modified phosphate. ).
- the base-modified nucleotide is not particularly limited as long as the base of the nucleotide is modified.
- the unmodified base portion include base portions corresponding to adenine, guanine, cytosine and uracil.
- the modified base portion include a base portion in which oxygen in the unmodified base portion is replaced with sulfur, a base portion in which hydrogen in the unmodified base portion is substituted with alkyl or halogen having 1 to 6 carbon atoms, and an unmodified base.
- base-modified nucleotide examples include 5-methylcytosine (5-me-C), 5-hydroxymethylcytosine, xanthin, hypoxanthin, 2-aminoadenine, 6-methyladenine, 6-methylguanine, 2 -Propyladenine, 2-propylguanine, 2-thiouracil, 2-thiotimine, 2-thiocitosine, 5-propynyluracil, 5-propynylcitosine, 6-azouracil, 6-azocitosine, 6-azotimine, 5-pseudouracil, 4- Thiouracil, 8-haloadenine, 8-haloguanine, 8-aminoadenine, 8-aminoguanine, 8-mercaptoadenine, 8-mercaptoguanine, 8-alkylthioadenine, 8-alkylthioguanine, 8-hydroxyadenine, 8-hydroxyguanine, 5-bromouracil, 5-bromo
- the translation region may contain phosphate modified nucleotides.
- the position where the phosphate-modified nucleotide is present in the translation region is not particularly limited.
- the phosphate-modified nucleotide may be a sugar-modified nucleotide and / or a base-modified nucleotide (in other words, the phosphoric acid-modified nucleotide may further contain a modified sugar and / or a modified base. ).
- the phosphoric acid portion-modified nucleotide is not particularly limited as long as the phosphoric acid portion (phosphodiester bond) of the nucleotide is modified.
- the modified phosphoric acid moiety include a phosphorothioate bond, a phosphorodithioate bond, an alkylphosphonate bond, and a phosphoromidate bond.
- the translated region may contain a phosphoric acid moiety-modified nucleotide whose modified phosphoric acid moiety is an optical isomer (Rp, Sp).
- Rp, Sp an optical isomer
- Methods for selectively synthesizing optical isomers of phosphorothioate bonds include, for example, J. Am. Chem. Soc., 124, 4962 (2002), Nucleic Acids Research, 42, 13546 (2014), and Science 361, 1234 ( It is disclosed in 2018).
- the polynucleotide of this embodiment may further include a 5'side untranslated region (5'UTR).
- the 5'-side untranslated region is a region that exists upstream of the translation region (5'-terminal side) and is not translated for polypeptide synthesis.
- the number of nucleotides constituting the 5'-side untranslated region is preferably an integer of 1 to 1000, more preferably an integer of 1 to 500, still more preferably an integer of 1 to 250, and particularly preferably 1. It is an integer of ⁇ 100.
- the 5'side untranslated region may contain sugar-modified nucleotides.
- the position of the sugar-modified nucleotide is not particularly limited, but from the viewpoint of improving translational activity, the first, second, and third nucleotides from the 5'end may be sugar-modified nucleotides.
- nucleotides in the 5'side untranslated region may be sugar-modified nucleotides.
- modified sugar moiety of the sugar moiety-modified nucleotide include those described in the item of [sugar moiety-modified nucleotide] in the above (translation region).
- the 5'side untranslated region may contain a base-modified nucleotide.
- the position where the base-modified nucleotide is present in the 5'-side untranslated region is not particularly limited.
- the base-modified nucleotide may be a sugar-modified nucleotide and / or a phosphate-modified nucleotide (in other words, the base-modified nucleotide may further contain a modified sugar and / or a modified phosphate. ).
- the modified base portion of the base-modified nucleotide include those described in the item of [Base-modified nucleotide] in the above (translated region).
- the 5'-side untranslated region preferably contains the following modified base portion from the viewpoint of improving translation activity.
- R is an alkyl group having 1 to 6 carbon atoms
- the alkyl group R of the modified base portion is preferably methyl or ethyl.
- alkyl having 1 to 6 carbon atoms include those described in the item of [sugar-modified nucleotide] in the above (translated region).
- the 3'side untranslated region may contain a poly A chain.
- the 3'side untranslated region may contain both a polynucleotide other than the poly A chain and the poly A chain, or may contain only one of them. The inclusion of the poly A chain tends to improve the translational activity.
- modified base portion of the base-modified nucleotide include those described in the item of [Base-modified nucleotide] in the above (translation region).
- the nucleotides on the left and right sides of the connecting portion are the two nucleotides constituting the polynucleotide of the present embodiment.
- the translational activity can be maintained even if the connecting portion is included.
- Nucleotide A on the right side (5'end side) and nucleotide B on the left side (3'end side) in the connecting portion nucleotide C on the 3'end side adjacent to the nucleotide B, and 3'adjacent to the nucleotide C.
- Nucleotide D on the terminal side does not have to be modified.
- the salt of OH and SH of X 3 of the connecting portion for example, pharmaceutically acceptable salts thereof.
- Pharmaceutically acceptable salts include, for example, alkali metal salts, alkaline earth metal salts, ammonium salts, organic amine salts, and amino acid salts.
- alkali metal salt include sodium salt, lithium salt, and potassium salt.
- alkaline earth metal salt include a calcium salt and a magnesium salt.
- connecting portion includes the following. [In the formula, R 1 , R 2 , B 1 , B 2 , and X 3 are as described above]
- the position where the connecting portion exists is not particularly limited.
- the connecting portion may exist in any of the translation region, the 5'side untranslated region, and the 3'side untranslated region, but when the connecting portion exists, the connecting portion exists at least in the translation region. Is preferable.
- the number of the connecting portions is not particularly limited, and can be appropriately selected depending on the length of the polynucleotide.
- the number of connecting portions is, for example, 1 to 200, 1 to 100, 1 to 50, 1 to 20, 1 to 10, 1 to 8, 1 to 6, 1 to 4, 1 ⁇ 3, or 1 or 2 may be mentioned.
- the first nucleotide and the second nucleotide in at least one codon among the plurality of codons constituting the translation region may be linked by a phosphorothioate.
- the number of phosphorothioate bonds is not particularly limited and can be appropriately selected depending on the length of the polynucleotide.
- the number of phosphorothioate bonds is, for example, 1 to 200, 1 to 100, 1 to 50, 1 to 20, 1 to 10, 1 to 8, 1 to 6, 1 to 4, 1 ⁇ 3, or 1 or 2 may be mentioned.
- the 1st to 2nd nucleotides from the 5'end of the 5'side untranslated region, the 1st to 3rd nucleotides, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are phosphorothioates. May be concatenated by. For example, when the 1st to 3rd nucleotides are linked by a phosphorothioate, the 1st and 2nd nucleotides are linked by a phosphorothioate, and the 2nd and 3rd nucleotides are linked by a phosphorothioate. It means that they are connected.
- the 1st to 2nd nucleotides from the 3'end of the 3'side untranslated region, the 1st to 3rd nucleotides, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are phosphorothioates. May be concatenated by.
- Another embodiment of the invention includes a translated region and a 5'side untranslated region. It relates to a polynucleotide in which the first, second and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- Another embodiment of the invention includes a translated region and a 3'side untranslated region. It relates to a polynucleotide in which the first, second and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- Another embodiment of the present invention includes a translation region, a 5'side untranslated region and a 3'side untranslated region.
- the first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides. It relates to a polynucleotide in which the first, second and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- Another embodiment of the present invention includes a translation region, a 5'side untranslated region and a 3'side untranslated region.
- the first, second, and third nucleotides from the 5'end of the 5'side untranslated region are sugar-modified nucleotides.
- the 1st to 3rd nucleotides from the 5'end of the 5'side untranslated region, the 1st to 4th nucleotides, or the 1st to 5th nucleotides are linked by phosphorothioates.
- the first, second and third nucleotides from the 3'end of the 3'side untranslated region are sugar-modified nucleotides.
- the polynucleotide of this embodiment can be produced, for example, by chemical synthesis. Specifically, the polynucleotide of the present embodiment can be produced by introducing a predetermined sugar-modified nucleotide at a predetermined position while extending the polynucleotide chain using a known chemical synthesis method. .. Known chemical synthesis methods include, for example, the phosphoramidite method, the phosphorothioate method, the phosphotriester method, and the CEM method (see Nucleic Acids Research, 35, 3287 (2007)). Further, an ABI3900 high-throughput nucleic acid synthesizer (manufactured by Applied Biosystems) can also be used.
- Step A Compound (b) is prepared by reacting compound (a) with compound (a) in a solvent in the presence of a base, for example, at a temperature between 0 ° C. and 80 ° C. for 10 minutes to 3 days.
- a base for example, at a temperature between 0 ° C. and 80 ° C. for 10 minutes to 3 days.
- the solvent include DMF, DMA, NMP and the like, and these are used alone or in combination.
- Examples of the base include imidazole, triethylamine, diisopropylethylamine and the like.
- the silylating agent include bis (trifluoromethanesulfonic acid) di-tert-butylsilyl and the like.
- Step B Compound (c) can be produced by reacting compound (b) with a corresponding alkylating agent in a solvent in the presence of a base at a temperature between 0 ° C. and 150 ° C. for 10 minutes to 3 days. it can.
- the reaction can also be accelerated with suitable additives.
- the solvent include DMF, pyridine, dichloromethane, THF, ethyl acetate, 1,4-dioxane, NMP and the like, and these are used alone or in combination.
- Examples of the base include aqueous sodium hydroxide solution, potassium carbonate, pyridine, triethylamine, N-ethyl-N, N-diisopropylamine and the like.
- the alkylating agent include methyl iodide, ethyl iodide, methyl bromide and the like.
- the additive include tetrabutylammonium bromide.
- Step E Compound (f) can be produced by reacting compound (e) and compound (g) in a solvent at a temperature between 0 ° C. and 100 ° C. for 10 seconds to 24 hours in the presence of a base.
- a solvent include dichloromethane, acetonitrile, toluene, ethyl acetate, THF, 1,4-dioxane, DMF, NMP and the like, and these are used alone or in combination.
- Examples of the base include triethylamine, N, N-diisopropylethylamine, pyridine and the like, and these are used alone or in combination.
- the 5'cap structure can be introduced using known methods (eg, enzymatic and chemical synthesis methods).
- known methods include, for example, the methods described in Top. Curr. Chem. (Z) (2017) 375: 16 and Beilstein J. Org. Chem. 2017, 13, 2819-2832.
- the linking method is not particularly limited, and examples thereof include an enzymatic method and a chemical synthesis method.
- Examples of the ligation by the enzymatic method include ligase-based ligation.
- Examples of the ligase include T4DNA Ligase, T4RNA Ligase1, T4RNALigase2, T4RNALigase2, truncated T4RNALigase2, truncated KQ, E.ColiDNALigase, Taq.DNALigase, etc. It can be mixed and used.
- polydisperse polyethylene glycol may be used in order to promote the ligation reaction by the molecular crowding effect.
- the polydisperse PEG include PEG4000, PEG6000, PEG8000, PEG10000 and the like, and these can be used alone or in combination.
- the condensation reaction is carried out in the presence of a template DNA containing a nucleotide chain complementary to the 3'terminal nucleotide chain of the 5'terminal polynucleotide unit and the 5'terminal nucleotide chain of the 3'terminal polynucleotide unit. It is preferable to carry out.
- the template DNA is from the 3'end of the 5'terminal polynucleotide unit, preferably from the 2 to 50 base length, more preferably 5 to 40 base length nucleotide strands, and from the 5'end of the 3'end side polynucleotide unit.
- Additives may be added in the condensation reaction.
- Additives include, for example, 1-hydroxybenzotriazole (HOBt) and 4-dimethylaminopyridine (DMAP).
- HOBt 1-hydroxybenzotriazole
- DMAP 4-dimethylaminopyridine
- the condensation reaction may be carried out in the presence of a buffer solution.
- a buffer solution examples include acetate buffer solution, Tris buffer solution, citrate buffer solution, phosphate buffer solution, and water.
- the temperature of the condensation reaction is not particularly limited, but may be, for example, room temperature to 200 ° C.
- the time of the condensation reaction is not particularly limited, but may be, for example, 5 minutes to 100 hours.
- B p represents a base that may be protected by a protecting group
- B represents a base
- Polymer represents a solid phase support.
- R 4 is a protecting group that can be selectively deprotected, for example, tert-butyldimethylsilyl group, triethylsilyl group
- R 3 is a protecting group used in solid phase synthesis of nucleic acid, for example, p, p.
- X a represents a nucleic acid sequence
- Y a and Y b are each independently leaving groups, for example, halogen, preferably chlorine atom or bromine atom.
- a nucleic acid sequence is a partial structure in a nucleic acid that forms a nucleic acid together with a compound that binds to each other.
- each B may be the same or different.
- Step 1 Compound (B) can be produced by reacting compound (A) in a solvent at a temperature between 60 ° C. and the boiling point of the solvent used for 10 seconds to 3 days.
- the solvent include toluene, xylene, 1,2-dichloroethane, 1,4-dioxane, N, N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), 1,2-dichlorobenzene, water and the like. These can be used alone or in combination.
- Compound (A) is, for example, J.A. Am. Chem. Soc. (1999), 121,5661-5665. It can be manufactured by the method described in 1.
- the B p in the compound (A) is not particularly limited, but any of the following structures is preferable.
- R 6 is a group that constitutes a part of the protecting group of the base, and represents, for example, a methyl group, an isopropyl group, a phenyl group that may have a substituent, and the like.
- the substituent in the phenyl group which may have a substituent represents, for example, a methyl group, an isopropyl group or a tert-butyl group.
- Step 2 Compound (C) contains compound (B) in a solvent in the presence of 1 to 100 equivalents of an oxidizing agent at a temperature between 0 ° C. and the boiling point of the solvent used, preferably 1 to 3 days, preferably 1 to 3 days. It can be produced by reacting with 100 equivalents of an additive.
- the solvent include an aprotic solvent such as chloroform and dichloromethane, and these can be used alone or in combination.
- the oxidizing agent include organic oxidizing agents such as Jones reagent, chromic acid, pyridinium dichromate, ruthenium tetroxide, sodium chlorite, and Dess-Martin reagent, and inorganic substances such as pyridinium chlorochromate.
- Examples thereof include system oxidizing agents, and these can be used alone or in combination.
- Examples of the additive include pyridine, triethylamine, N, N-diisopropylethylamine and the like, and these can be used alone or in combination.
- Step 3 Compound (D) is prepared by reacting compound (C) in a solvent such as pyridine in the presence of hydroxylamine hydrochloride at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days. Can be manufactured.
- a solvent such as pyridine
- Step 4 Compound (E) is prepared by reacting compound (D) in a solvent in the presence of 1 to 100,000 equivalents of a deprotecting agent at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days.
- a deprotecting agent at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days.
- the solvent include toluene, xylene, water and the like, and these can be used alone or in combination.
- Examples of the deprotecting agent include trifluoroacetic acid, trichloroacetic acid, acetic acid, hydrochloric acid and the like, and these can be used alone or in combination.
- Compound (F) can be produced by reacting compound (E) in a solvent in the presence of a reducing agent at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days. ..
- a reducing agent examples include trifluoroacetic acid, trichloroacetic acid, acetic acid, hydrochloric acid, toluene, xylene, toluene, xylene, tetrahydrofuran, methanol, ethanol, 1,4-dioxane, water and the like, and these may be used alone or in combination.
- the reducing agent include sodium borohydride, sodium cyanoborohydride, lithium borohydride, sodium triacetoxyborohydride and the like.
- Step 6 Compound (G) is produced by reacting compound (F) in a solvent, in the presence of a catalyst, in a hydrogen atmosphere, at a temperature between 0 ° C. and the boiling point of the solvent used, for 10 seconds to 3 days.
- a catalyst in a hydrogen atmosphere, at a temperature between 0 ° C. and the boiling point of the solvent used, for 10 seconds to 3 days.
- the solvent include trifluoroacetic acid, acetic acid, dilute hydrochloric acid, methanol, ethanol, isopropanol, water and the like, and these can be used alone or in combination.
- the catalyst include palladium carbon, ruthenium carbon and the like.
- Compound (G) can also be produced, for example, by the method described in International Publication No. 2017/1236669.
- Step 7 Compound (H) contains compound (G) in a solvent in the presence of 1 to 100 equivalents of compound (G') and a base at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days. It can be produced, preferably by reacting with 1 to 1000 equivalents of base.
- the solvent include methanol, ethanol, isopropanol, dichloromethane, acetonitrile, toluene, ethyl acetate, tetrahydrofuran (THF), 1,4-dioxane, N, N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), and the like.
- Examples thereof include water, which can be used alone or in combination.
- Examples of the base include pyridine, triethylamine, N-ethyl-N, N-diisopropylamine, 2,6-lutidine and the like, and these can be used alone or in combination.
- Step 8 Compound (I) comprises compound (H) and p, p'-dimethoxytrityl chloride in a solvent such as pyridine, optionally in the presence of a co-solvent, at a temperature between 0 ° C and 100 ° C for 5 minutes. It can be produced by reacting for ⁇ 100 hours.
- a co-solvent include methanol, ethanol, dichloromethane, chloroform, 1,2-dichloroethane, toluene, ethyl acetate, acetonitrile, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, dioxane, N, N-dimethylformamide (DMF). ), N, N-dimethylacetamide, N-methylpyrrolidone, triethylamine, N, N-diisopropylethylamine, water and the like, and these can be used alone or in combination.
- Step 9 Compound (J) is produced by reacting compound (I) with an additive of 1 to 10 equivalents in a solvent at a temperature between 0 ° C. and the boiling point of the solvent used for 10 minutes to 10 days.
- a solvent include dichloromethane, acetonitrile, toluene, ethyl acetate, THF, 1,4-dioxane, DMF, DMA, NMP and the like, and these can be used alone or in combination.
- the additive include tetrabutylammonium fluoride, triethylamine hydrofluoric acid salt and the like, and these can be used alone or in combination.
- Step 10 Compound (K) is produced by reacting compound (J) with succinic anhydride in a solvent in the presence of 1 to 30 equivalents of a base at a temperature between room temperature and 200 ° C. for 5 minutes to 100 hours. can do.
- the solvent include methanol, ethanol, dichloromethane, chloroform, 1,2-dichloroethane, toluene, ethyl acetate, acetonitrile, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, dioxane, N, N-dimethylformamide (DMF).
- N, N-dimethylacetamide, N-methylpyrrolidone, pyridine, water and the like examples include cesium carbonate, potassium carbonate, potassium hydroxide, sodium hydroxide, sodium methoxydo, potassium tert-butoxide, triethylamine, diisopropylethylamine, N-methylmorpholin, pyridine, 1,8-diazabicyclo [5.4]. .0] -7-Undecene (DBU), N, N-dimethyl-4-aminopyridine (DMAP) and the like can be mentioned, and these can be used alone or in combination.
- DBU cesium carbonate
- potassium carbonate potassium hydroxide
- sodium hydroxide sodium methoxydo
- potassium tert-butoxide triethylamine, diisopropylethylamine, N-methylmorpholin
- pyridine 1,8-diazabicyclo [5.4]. .0] -7-Undecene (DBU), N, N
- Step 11 Compound (L) comprises 1 to 30 equivalents of base, condensing agent and optionally 0.01 to the solvent-free or solvent-free or solvent-free or solvent-free or solvent-free or solvent-free or solvent-free or solvent-free compound (K) compound (K).
- the reaction was carried out in an acetic anhydride / pyridine solution at a temperature between room temperature and 200 ° C. for 5 minutes to 100 hours. It can be manufactured by allowing it to be produced.
- the solvent include those exemplified in step 4.
- Examples of the base include cesium carbonate, potassium carbonate, potassium hydroxide, sodium hydroxide, sodium methoxydo, potassium tert-butoxide, triethylamine, diisopropylethylamine, N-methylmorpholin, pyridine, 1,8-diazabicyclo [5.4]. .0] -7-Undecene (DBU), N, N-dimethyl-4-aminopyridine (DMAP) and the like can be mentioned, and these can be used alone or in combination.
- DBU cesium carbonate
- potassium carbonate potassium hydroxide
- sodium hydroxide sodium methoxydo
- potassium tert-butoxide triethylamine
- diisopropylethylamine N-methylmorpholin
- pyridine 1,8-diazabicyclo [5.4]. .0] -7-Undecene (DBU), N, N-dimethyl-4-aminopyridine (DMAP) and the like can be mentioned, and these can
- Step 13 Compound (N) is prepared by reacting compound (M) with compound (M) in a buffer solution in the presence of 1 to 1000 equivalents of compound (O) at a temperature between room temperature and 100 ° C. for 5 minutes to 100 hours.
- a buffer solution examples include acetic acid buffer solution, Tris buffer solution, citric acid buffer solution, phosphate buffer solution, water and the like, and these can be used alone or in combination.
- the compound (O) a commercially available product can be used.
- B p represents a base that may be protected by a protecting group
- B represents a base
- R 7 is a protecting group, for example, a tert-butyldimethylsilyl group or a triethylsilyl group
- Yc represents a base.
- it represents a chlorine atom, a bromine atom, and a tosylate group
- X b represents a nucleic acid sequence.
- each B may be the same or different.
- Step 14 Compound (Q) is produced by reacting compound (P) in a solvent in the presence of additives and bases at a temperature between 0 ° C. and the boiling point of the solvent used for 10 seconds to 3 days.
- the solvent include dichloromethane, acetonitrile, toluene, ethyl acetate, THF, 1,4-dioxane, DMF, DMA, NMP and the like, and these can be used alone or in combination.
- the additive include tosyl anhydride, tosyl chloride, thionyl chloride, oxalyl chloride and the like, and these can be used alone or in combination.
- Examples of the base include pyridine, triethylamine, N-ethyl-N, N-diisopropylamine, potassium carbonate, potassium hydroxide, sodium hydroxide, sodium methoxydo, potassium tert-butoxide, triethylamine, diisopropylethylamine, N-methylmorpholin. , Pyridine, 1,8-diazabicyclo [5.4.0] -7-undecene (DBU), N, N-dimethyl-4-aminopyridine (DMAP) and the like, and these are used alone or in combination. be able to.
- DBU 1,8-diazabicyclo [5.4.0] -7-undecene
- DMAP N-dimethyl-4-aminopyridine
- Step 17 Compound (T) can be produced by adding a reducing agent to compound (S) in a solvent and reacting the compound (S) at a temperature between room temperature and the boiling point of the solvent used for 10 seconds to 3 days.
- a reducing agent examples include methanol, ethanol, dichloromethane, chloroform, 1,2-dichloroethane, toluene, ethyl acetate, acetonitrile, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, dioxane, N, N-dimethylformamide (DMF).
- the reducing agent include sodium borohydride, sodium cyanoborohydride, lithium borohydride, sodium triacetoxyborohydride, palladium carbon in a hydrogen atmosphere, and the like.
- Compound (V) can be produced by reacting compound (U) and compound (AA) in a solvent at a temperature between 0 ° C. and 100 ° C. for 10 seconds to 24 hours in the presence of a base. ..
- a solvent include dichloromethane, acetonitrile, toluene, ethyl acetate, THF, 1,4-dioxane, DMF, NMP and the like, and these can be used alone or in combination.
- Examples of the base include triethylamine, N, N-diisopropylethylamine, pyridine and the like, and these can be used alone or in combination.
- As the compound (AA), a commercially available product can be used.
- One embodiment of the present invention relates to a pharmaceutical composition containing the polynucleotide.
- the pharmaceutical composition of the present embodiment By administering the pharmaceutical composition of the present embodiment to a patient having a disease, the polynucleotide is translated, the polypeptide encoded by the polynucleotide is synthesized, and the disease is treated.
- Diseases are not particularly limited, but are, for example, cancer and proliferative diseases, infectious and parasitic diseases, blood and hematopoietic diseases, autoimmune diseases, endocrine, nutritional and metabolic diseases (congenital metabolic disorders). ), Mental, nervous system diseases, skin and subcutaneous tissue diseases, eye diseases, ear diseases, respiratory diseases, digestive system diseases, renal urinary tract genital diseases, cardiovascular diseases, cerebrovascular diseases, musculoskeletal diseases Diseases of the gut system and connective tissues, abortion, perinatal diseases, congenital malformations, acquired injuries, and addiction.
- the pharmaceutical composition may be administered in the form of a predetermined preparation.
- Formulations include, for example, liquid dosage forms for oral or parenteral administration, and liquid dosage forms include, for example, pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups, etc. And an elixir agent.
- the liquid dosage form in addition to the active ingredient, is an inert diluent commonly used in the art (eg, water or other solvent), solubilizers and emulsifiers (eg, ethyl alcohol, isopropyl alcohol, ethyl carbonate).
- the pharmaceutical product for oral administration may contain at least one of an adjuvant (for example, a wetting agent, an emulsifying agent and a suspending agent), a sweetening agent, a flavoring agent, and a flavoring agent.
- an adjuvant for example, a wetting agent, an emulsifying agent and a suspending agent
- a sweetening agent for example, a sweetening agent, a flavoring agent, and a flavoring agent.
- Formulations for parenteral administration may contain solubilizers (eg, Cremophor®, alcohols, oils, denaturing oils, glycols, polysorbates, cyclodextrins, polymers, and combinations thereof).
- the pharmaceutical composition may contain an optional component in addition to the polynucleotide.
- Optional components include, for example, solvents, aqueous solvents, non-aqueous solvents, dispersion media, diluents, dispersions, suspension aids, surfactants, isotonic agents, thickeners, emulsifiers, preservatives, lipids, liposomes.
- One or more pharmaceutically acceptable additives selected from liposomes, lipid nanoparticles, core-shell nanoparticles, polymers, lipoplexes, peptides, proteins, cells, hyaluronidases, and mixtures thereof. Can be mentioned.
- the reagents used in the synthesis of the compounds were purchased from Sigma-Aldrich, Tokyo Chemical Industry Co., Ltd., Wako Pure Chemical Industries, Ltd., and Kanto Chemical Co., Inc. without purification.
- the anhydrous solvent was prepared by drying on activated molecular sieve 4A for 12 hours, or a commercially available anhydrous grade solvent was used.
- the reaction was followed by thin layer silica gel chromatography (silica gel 70F254 TLC plate-Wako, Wako Pure Chemical Industries, Ltd.). Silica gel 60N (spherical, neutral, particle size 40 to 50 ⁇ m) for flash chromatography (Kanto Chemical Co., Inc.) was used for purification of the compound.
- NMR NMR was measured using a deuterated solvent (CDCl 3 , CD 3 OD, DMSO-d 6 ) (Kanto Kagaku Co., Ltd.) and JEOL ECS 400 MHz (JEOL Ltd.).
- the acquired NMR data was analyzed using JEOL Delta (JEOL Ltd.) as software, and the chemical shift value was the residual signal in the deuterated solvent (CDCl 3 : 7.26, CD 3 OD: 3.31, DMSO-d 6). : 2.50) (Organometallics 2010, 29, 2176-2179) was used for correction.
- Step 1 Synthesis of compound 4 N-(9-((2R, 3S, 4S, 5R) -3- (tert-butyldimethylsilyloxy) -5-((tert-butyldimethylsilyloxy) methyl) -4-hydroxy-tetrahydrofuran-2-yl) -6-oxo- 6,9-dihydro-1H-purin-2-yl) isobutyramide
- Compound 3 obtained by the method described in the literature (J. Am. Chem. Soc., 1999, 121, 5661-5665) was used, dissolved in 1,2-dichlorobenzene (2.0 mL), and placed on an oil bath (160). The mixture was stirred at (° C.) for 4 hours.
- Step 3 Synthesis of compound 6 N-(9-((2R, 3S, 5S) -3- (tert-butyldimethylsilyloxy) -4- (hydroxyimino) -5- (hydroxymethyl) -tetrahydrofuran-2-yl) -6-oxo-6,9-dihydro -1H-purin-2-yl) isobutyramide
- An ice-cooled 90% aqueous trifluoroacetic acid solution (1.0 mL) was added to compound 5 (129 mg, 0.22 mmol), and the mixture was stirred on an ice bath for 30 minutes.
- the reaction mixture was concentrated under reduced pressure, and the obtained residue was azeotroped with toluene and water (1: 1, v / v) under reduced pressure three times.
- Step 4 Synthesis of compound 7 N-(9-((2R, 3S, 4S, 5S) -4-amino-3- (tert-butyldimethylsilyloxy) -5- (hydroxymethyl) -tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro -1H-purin-2-yl) isobutyramide
- Sodium borohydride (15 mg, 0.38 mmol) was added to a solution of compound 6 (93 mg, 0.19 mmol) in acetic acid (1.9 mL), and the mixture was stirred at room temperature for 1 hr.
- Step 5 Synthesis of Compound 8 N-(9-((2R, 3S, 4S, 5S) -4-amino-3- (tert-butyldimethylsilyloxy) -5- (hydroxymethyl) -tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro -1H-purin-2-yl) isobutyramide 10% Palladium on carbon (20 mg) was added to a 90% aqueous acetic acid solution (1.5 mL) of compound 7 (50 mg, 0.10 mmol), and the mixture was stirred at room temperature for 18 hours under a hydrogen atmosphere.
- Step 6 Synthesis of compound 9 N-(9-((2R, 3S, 4R, 5S) -3- (tert-butyldimethylsilyloxy) -5- (hydroxymethyl) -4- (2,2,2-trifluoroacetamido) -tetrahydrofuran-2-yl) -6 -oxo-6,9-dihydro-1H-purin-2-yl) isobutyramide
- Ethyl trifluoroacetate (0.76 mL) was added to a solution of compound 8 (40 mg, 0.076 mmol) and triethylamine (45 L, 0.38 mmol) known in the literature (WO2017 / 123669) in methanol (0.76 mL), and the mixture was added at room temperature 24 Stirred for hours.
- Step 7 Synthesis of compound 10 N-(9-((2R, 3S, 4R, 5S) -5-((bis (4-methoxyphenyl) (phenyl) methoxy) methyl) -3- (tert-butyldimethylsilyloxy) -4- (2,2,2) -trifluoroacetamido) -tetrahydrofuran-2-yl) -6-oxo-6,9-dihydro-1H-purin-2-yl) isobutyramide Dimethoxytrityl chloride (18 mg, 0.053 mmol) was added to a solution of compound 9 (10 mg, 0.017 mmol) in anhydrous pyridine (1 mL), and the mixture was stirred at room temperature for 1.5 hours.
- Step 8 Synthesis of compound 11 N-(9-((2R, 3S, 4S, 5S) -5-((bis (4-methoxyphenyl) (phenyl) methoxy) methyl) -3-hydroxy-4- (2,2,2-trifluoroacetamido)- tetrahydrofuran-2-yl) -6-oxo-6,9-dihydro-1H-purin-2-yl) isobutyramide Tetrahydrofuran ammonium fluoride (1M tetrahydrofuran solution, 19 ⁇ L, 0.019 mmol) was added to a solution of compound 10 (14 mg, 0.016 mmol) in tetrahydrofuran (1 mL), and the mixture was stirred at room temperature for 1 hr.
- Step 9 Synthesis of Compound 12 Compound 11 (0.90 g, 1.20 mmol), triethylamine (0.42 mL, 3.0 mmol) in acetonitrile (12 mL) solution with succinic anhydride (0.24 g, 2.40 mmol), dimethylaminopyridine (29 mg) , 0.24 mmol) was added, and the mixture was stirred at room temperature for 1 hour. After confirming the disappearance of the raw material by thin layer chromatography, the reaction solution was concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed twice with saturated aqueous sodium hydrogen carbonate solution and then with saturated brine.
- Compound 12 can also be synthesized by obtaining Intermediate 6 from the following starting material 13.
- Step 10 Synthesis of compound 14 N-(9-((2R, 3R, 5S) -5-((bis (4-methoxyphenyl) (phenyl) methoxy) methyl) -3-((tert-butyldimethylsilyl) oxy) -4- (hydroxyimino) tetrahydrofuran- 2-yl) -6-oxo-6,9-dihydro-1H-purin-2-yl) isobutyramide Under an argon atmosphere, compound 13 (ChemGenes, 5.0 g, 6.5 mmol) was dissolved in dehydrated dichloromethane (50 mL) and stirred while cooling in an ice bath.
- reaction solution was transferred to an eggplant flask while being washed with chloroform (containing 1% triethylamine) and concentrated. The residue was added to saturated aqueous sodium hydrogen carbonate, stirred for 15 minutes, and then extracted twice with chloroform. The organic layers were united, washed once with saturated brine, and dehydrated with anhydrous sodium sulfate. After the desiccant was filtered off, the filtrate was concentrated to give compound 14 (4.13 g, diastereomer mixture, 2-step yield 81%) as an orange foam.
- Step 11 Synthesis of Compound 6 from Compound 14 Using Compound 14 (3.80 g) obtained in Step 10, Compound 6 (2.12 g, 4.41 mmol, yield 91%) was obtained in the same manner as in Step 3. The detailed data of compound 6 is as described in step 3.
- the obtained solid phase carrier (2.0 mg) was weighed in a 2 mL volumetric flask, and a deblocking reagent was added to make the total volume 2 mL and mixed by inversion, which was used as a measurement sample. After performing a blank measurement using a 3 w / v% trichloroacetic acid / dichloromethane solution, the measurement was performed using a measurement sample. Absorbance at 504 nm: From 0.377, supported amount: 24.8 ⁇ mol / g)
- Step 12 Synthesis of compound 17 N-(9-((3aR, 4R, 6R, 6aR) -6- (hydroxymethyl) -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) -9H-purin- 6-yl) benzamide
- N6-Benzyladenosine Compound 16 (100 g, 269 mmol, 1.0 eq.), Acetone (2.70 L), Dimethoxypropane (166 mL, 1.35 mol, 5.0 eq.) In a 10 L 4-neck flask under an argon atmosphere. Was added in sequence.
- Step 13 Synthesis of compound 18 ((3aR, 4R, 6R, 6aR) -6- (6-benzamido-9H-purin-9-yl) -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) methyl methanesulfonate Under an argon atmosphere, compound 17 (222 g) and pyridine (520 mL) obtained in step 12 were added to a 2 L 4-neck flask, the reaction solution was cooled in an ice bath, and the internal temperature was 4 ° C to 9 ° C.
- Methanesulfonyl chloride (25.0 mL, 321 mmol, 1.2 eq.) was added dropwise over 15 minutes, and the mixture was stirred for 2 hours. After confirming the progress of the reaction by LC / MS, add distilled water (500 mL) to the reaction solution, extract the solution with ethyl acetate (1.0 L) three times, and then extract the organic layer with 1N hydrochloric acid (1.0 L ⁇ 1,500). The mixture was washed successively with mL ⁇ 2), saturated aqueous sodium hydrogen carbonate solution (500 mL ⁇ 2) and saturated brine (500 mL ⁇ 2), and dehydrated with anhydrous sodium sulfate.
- Step 14 Synthesis of compound 19 N-(9-((3aR, 4R, 6R, 6aR) -6- (azidomethyl) -2,2-dimethyltetrahydrofuro [3,4-d] [1,3] dioxol-4-yl) -9H-purin- 6-yl) benzamide
- compound 18 150 g
- dehydrated DMF 1.26 L
- Sodium azide 82.8 g, 1.26 mol, 5.0 eq.
- the reaction solution was slowly cooled to room temperature, and distilled water (1.0 L) and ethyl acetate (600 mL) were added. Distilled water (3.0 L) was added to the obtained solution, and the aqueous layer was extracted 6 times with ethyl acetate (500 mL). The organic layer was washed twice with distilled water (800 mL) and twice with saturated brine (800 mL) and dehydrated with anhydrous sodium sulfate.
- Step 15 Synthesis of compound 20 N-(9-((3aR, 4R, 6R, 6aR) -2,2-dimethyl-6-((2,2,2-trifluoroacetamido) methyl) tetrahydrofuro [3,4-d] [1,3] dioxol -4-yl) -9H-purin-6-yl) benzamide Under an argon atmosphere, compound 19 (55.7 g, 128 mmol, 1.0 eq.) And methanol (1.28 L) obtained in step 14 were added to a 3 L 4-neck flask.
- Step 16 Synthesis of compound 21 N-(9-((2R, 3R, 4S, 5R) -3,4-dihydroxy-5-((2,2,2-trifluoroacetamido) methyl) tetrahydrofuran-2-yl) -9H-purin-6-yl ) benzamide
- Compound 20 (10.0 g, 19.8 mmol, 10 eq.) Obtained in step 15 and distilled water (50.0 mL) were added to a 1 L eggplant flask, and the solution was cooled in an ice bath.
- Step 17 Synthesis of compound 22 N-(9-((2R, 3R, 4R, 5R) -3-((tert-butyldimethylsilyl) oxy) -4-hydroxy-5-((2,2,2-trifluoroacetamido) methyl) tetrahydrofuran-2-yl )-9H-purin-6-yl) benzamide
- Compound 21 (15.6 g, 33.6 mmol, 1.0 eq.) Obtained in step 16 and dehydrated DMF (111 mL) were added to a 500 mL eggplant flask under an argon atmosphere, and the solution was cooled in an ice bath.
- the compound 6a which is a raw material for the polynucleotide, was synthesized by the following scheme.
- Step 1 Synthesis of compound 2a N-(9-((3aR, 5R, 6R, 6aS) -2,2-di-tert-butyl-6-methoxytetrahydrofuro [2,3-d] [1,3,2] dioxasilol-5-yl)- 9H-purin-6-yl) benzamide
- Di-t-butylsilylbis (trifluoromethanesulfonate) (68.6 g, 156 mmol) was slowly added to a solution of commercially available compound 1a (30.0 g, 78.0 mmol) in DMF (300 mL) under ice cooling.
- Step 2 Synthesis of Compound 3a N-(9-((3aR, 5R, 6R, 6aS) -2,2-di-tert-butyl-6-methoxytetrahydrofuro [2,3-d] [1,3,2] dioxasilol -5-yl) -9H-purin-6-yl) -N-methylbenzamide
- Compound 2a (10.0 g, 19.0 mmol) was dissolved in dichloromethane (50 mL), and tetrabutylammonium bromide (9.20 g, 28.5 mmol) and 1 M aqueous sodium hydroxide solution (50 ml) were added.
- Step 3 Synthesis of compound 4a N-(9-((2R, 3R, 4S, 5S) -4,5-dihydroxy-3-methoxytetrahydrofuran-2-yl) -9H-purin-6-yl)-N-methylbenzamide
- Compound 3a (6.25 g, 11.6 mmol) was dissolved in tetrahydrofuran (63 mL) and cooled in an ice bath.
- Triethylamine (8.07 ml, 57.9 mmol) and triethylamine hydrofluorate (1.89 ml, 11.6 mmol) were added, and the mixture was stirred for 1 hour and 5 minutes while cooling in an ice bath.
- Step 4 Synthesis of compound 5a N-(9-((2R, 3R, 4S, 5S) -5- (bis (4-methoxyphenyl) (phenyl) methoxy) -4-hydroxy-3-methoxytetrahydrofuran-2-yl) -9H-purin-6- yl)-N-methylbenzamide
- Compound 4a (4.25 g, 10.6 mmol) was dissolved in pyridine (43 mL) and stirred in an ice bath.
- 4,4'-Dimethoxytrityl chloride (5.41 g, 20.0 mmol) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hours and 25 minutes.
- reaction solution was added to ice-cooled aqueous sodium hydrogen carbonate, quenched, and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated.
- the compound 6b which is a raw material for the polynucleotide, was synthesized by the following scheme.
- Step 1 Synthesis of compound 2b N-(9-((4aR, 6R, 7R, 7aS) -2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo [3,2-d] [1,3,2] dioxasilin-6 -yl) -9H-purin-6-yl) benzamide
- Di-t-butylsilylbis (trifluoromethanesulfonate) (70.8 g, 161 mmol) was slowly added to a solution of commercially available compound 1b (30.0 g, 80.4 mmol) in DMF (300 mL) under ice cooling.
- Step 2 Synthesis of compound 3b N-(9-((4aR, 6R, 7R, 7aS) -2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo [3,2-d] [1,3,2] dioxasilin-6 -yl) -9H-purin-6-yl) -N-methylbenzamide
- Compound 2b (10.0 g, 19.5 mmol) was dissolved in dichloromethane (50 mL), and tetrabutylammonium bromide (9,41 g, 29.2 mmol) and 1 M aqueous sodium hydroxide solution (50 ml) were added.
- Step 4 Synthesis of compound 5b N-(9-((2R, 3R, 4R, 5R) -5-((bis (4-methoxyphenyl) (phenyl) methoxy) methyl) -3-fluoro-4-hydroxytetrahydrofuran-2-yl) -9H-purin -6-yl)-N-methylbenzamide
- Compound 4b (4.93 g, 12.7 mmol) was dissolved in pyridine (49 mL) and stirred in an ice bath.
- 4,4'-Dimethoxytrityl chloride (6.47 g, 29.2 mmol) was added to the reaction mixture, and the mixture was stirred at room temperature for 1 hour and 20 minutes.
- reaction solution was added to ice-cooled aqueous sodium hydrogen carbonate, quenched, and extracted with ethyl acetate.
- the organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated.
- the compound 6c which is a raw material for the polynucleotide, was synthesized by the following scheme.
- Step 1 Synthesis of compound 3c N-(9-((4aR, 6R, 7R, 7aS) -2,2-di-tert-butyl-7-methoxytetrahydro-4H-furo [3,2-d] [1,3,2] dioxasilin-6 -yl) -9H-purin-6-yl) -N-ethylbenzamide
- Compound 2a (11.7 g, 22.3 mmol) was dissolved in dichloromethane (58.5 mL), and tetrabutylammonium bromide (10.8 g, 33.4 mmol) and 1 M aqueous sodium hydroxide solution (58.5 ml) were added.
- Step 2 Synthesis of compound 4c N-ethyl-N-(9-((2R, 3R, 4R, 5R) -4-hydroxy-5- (hydroxymethyl) -3-methoxytetrahydrofuran-2-yl) -9H-purin-6-yl) benzamide
- Compound 3c (6.14 g, 11.1 mmol) was dissolved in tetrahydrofuran (61.4 mL) and cooled in an ice bath.
- Triethylamine (7.73 ml, 55.4 mmol) and triethylamine hydrofluorate (1.81 ml, 11.1 mmol) were added, and the mixture was stirred for 2 hours while cooling in an ice bath.
- the compound 6d which is a raw material for the polynucleotide, was synthesized by the following scheme.
- Step 1 Synthesis of compound 3d N-(9-((4aR, 6R, 7R, 7aS) -2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo [3,2-d] [1,3,2] dioxasilin-6 -yl) -9H-purin-6-yl) -N-ethylbenzamide
- Compound 2b (1.00 g, 1.95 mmol) was dissolved in dichloromethane (5.0 mL), and tetrabutylammonium bromide (0.942 g, 2.92 mmol) and 1 M aqueous sodium hydroxide solution (5.0 ml) were added.
- Step 3 Synthesis of compound 5d N-(9-((2R, 3R, 4R, 5R) -5-((bis (4-methoxyphenyl) (phenyl) methoxy) methyl) -3-fluoro-4-hydroxytetrahydrofuran-2-yl) -9H-purin -6-yl)-N-ethylbenzamide
- Compound 5d was obtained in the same manner as in the process of obtaining compound 5c.
- RNA oligonucleotides are 2'-TOM (triisopropylsilyloxymethyl) protected ⁇ -cyanoethyl phosphoramidite (DMT-2'-O-TOM-rA (Ac), DMT-2'-O-TOM-rG (Ac)) , DMT-2'-O-TOM-rC (Ac), DMT-2'-O-TOM-rU) (Glen Research or ChemGenes, respectively), and the DNA oligonucleotide is ⁇ -cyanoethyl phosphoramidite (DMT).
- -dA (Bz), DMT-dG (iBu), DMT-dC (Ac), DMT-T) were used.
- Each phosphoromidite monomer is prepared so as to be a 0.05 mol / L acetonitrile solution, and a DNA / RNA solid phase synthesizer (NTS M-2-MX, Nippon Techno Service Co., Ltd.) is used using a solid phase carrier of 0.2 ⁇ mol or 0.8 ⁇ mol. ).
- NTS M-2-MX Nippon Techno Service Co., Ltd.
- CPG 1000A dA-CPG, dG-CPG, Ac-dC-CPG, dT-CPG
- the condensation time was 2 minutes.
- RNA having a phosphate group at the 5'end 5'-monophosphate RNA
- Universal UnyLinker Support 2000A (ChemGenes) was used as a solid phase carrier, and the condensation time of the first base was 15 minutes, and after that, 3 minutes each. And said. Phosphorylation of the hydroxyl group at the 5'end was performed using a chemical phosphorylation reagent (0.05 mol / L acetonitrile solution) (Glen Research or ChemGenes). Solid-phase synthesis of an RNA oligonucleotide in which a 3'-aminoguanosine monomer was introduced at the 3'end was performed using Compound 15. The condensation time for the first base was 15 minutes, and thereafter it was 3 minutes each.
- the following reagents were used for the solid phase synthesizer.
- a commercially available deblocking reagent (Deblocking Solution-1, 3 w / v% trichloroacetic acid / dichloromethane solution) (Wako Pure Chemical Industries, Ltd.) and react for 10 seconds. went.
- a commercially available activator solution (activator solution 3) (Wako Pure Chemical Industries, Ltd.) was used as an activator for phosphoramidite.
- Capping of the unreacted 5'-terminal hydroxyl group was performed by reacting for 10 seconds using commercially available capping solutions (Cap A solution-2 and Cap B solution-2) (Wako Pure Chemical Industries, Ltd.).
- Cap A solution-2 and Cap B solution-2 As an oxidizing agent for producing a phosphoric acid ester, a solution containing pyridine, THF, water and iodine (Oxidizer, 0.01 M iodine, 29.2% water, 6.3% pyridine, 64.5% acetonitrile), Honeywell) was used. Reacted for 10 seconds. After solid-phase synthesis, the dimethoxytrityl group of the hydroxyl group at the 5'end of the RNA oligonucleotide was deprotected on the solid-phase carrier.
- RNA oligonucleotides were deresined and deprotected according to a conventional method (concentrated ammonia water, 55 ° C, 12 hours). DNA oligonucleotides were purified using a cartridge column (MicroPure II Column, LGC Biosearch Technology) according to the product protocol. RNA oligonucleotides are completely dried by concentration with a centrifugal evaporator, and then tetrabutylammonium fluoride (1M tetrahydrofuran solution) (1 mL) is used to remove the TOM protecting group of the 2'hydroxyl group.
- tetrabutylammonium fluoride (1M tetrahydrofuran solution)
- Tris-HCl buffer (hereinafter, Tris-HCl) (1M, pH 7.4) (1 mL) was added to the solution and mixed, and then tetrahydrofuran was removed by concentration with a centrifugal evaporator.
- the obtained solution was treated by a product protocol using a gel filtration column (NAP-25, GE Healthcare) equilibrated with ultrapure water.
- the fraction containing the obtained RNA oligonucleotide was concentrated by a centrifugal evaporator and then purified using a modified polyacrylamide gel (hereinafter, dPAGE).
- RNA fragments using dPAGE An aqueous solution of ammonium persulfate (hereinafter, APS) and N, N, N', N'-tetramethylethylenediamine (hereinafter, TEMED) are added as a polymerization agent to an acrylamide gel solution (including 7M urea as a denaturant) to solidify.
- APS ammonium persulfate
- TEMED N, N, N', N'-tetramethylethylenediamine
- a gel was prepared by allowing (room temperature, 6 to 12 hours).
- RNA samples were mixed with gel loading buffer (80% formamide, TBE), heated at 90 ° C. for 3 minutes and then loaded onto a gel. After electrophoresis, RNA bands were detected by UV irradiation (254 nm) and excised from the gel using a razor blade.
- RNA pellets were rinsed with 80% ethanol and then air dried at room temperature for 1 hour. The obtained RNA pellets are dissolved in ultrapure water, diluted to an appropriate concentration, and then the absorbance at 260 nm is measured by ultraviolet-visible absorptiometry (NanoDrop, Thermo scientific), and the concentration is determined from the molar extinction coefficient of each RNA sequence.
- the structure of the purified oligonucleotide was determined by mass spectrometry using MALDI-TOF MS (Ultraflex III, Bruker Daltonics) (matrix: 3-hydroxypicolinic acid) or analysis using modified polyacrylamide gel electrophoresis.
- RNA bands were detected by gel staining (room temperature, 15 minutes) using SYBR® Green II Nucleic Acid Stain (Lonza) diluted 10,000 times with ultrapure water (equipment used). : ChemiDoc, BIORAD). The yield of the chemical ligation reaction was calculated by comparing the band strength of the RNA conjugate product with the conjugate product isolated and purified by dPAGE as a reference substance.
- RNA linkage product obtained by the chemical ligation reaction was recovered as RNA pellets from the reaction solution by ethanol precipitation (0.3M sodium acetate (pH 5.2) / 70% ethanol), and then purified by dPAGE.
- each nucleotide N represents RNA
- N (M) represents 2'-O-methyl modified RNA
- N (F) represents 2'-F modified RNA
- dN represents DNA.
- p indicates that the 3'or 5'end is phosphorylated.
- the underlined "AUG” represents the start codon and the underlined "UGA” represents the stop codon.
- the diagonal line (/) in the sequence indicates that the polynucleotides are linked at that portion.
- Example 1 An ultra-pure aqueous solution of RNA fragment E1-1 (10 nmol) obtained as sequence 2 by chemical synthesis, 5'phosphate RNA fragment E1-2 (10 nmol) obtained as sequence 3, and template DNA1 (10 nmol). 200 ⁇ L, final nucleic acid concentration; 50 ⁇ M) were prepared in 3 batches. 100 ⁇ L of T4 RNA Ligase 2 Reaction Buffer (10X) (manufactured by New England BioLabs) and 440 ⁇ L of ultrapure water were added to each prepared solution, heated at 90 ° C for 5 minutes, and then returned to room temperature over 30 minutes. .. A 60% PEG6000 aqueous solution was added to each of these solutions to a final concentration of 15%.
- T4 RNA Ligase 2 Reaction Buffer (10X) manufactured by New England BioLabs
- 440 ⁇ L of ultrapure water were added to each prepared solution, heated at 90 ° C for 5 minutes, and then returned to room temperature over 30 minutes. ..
- RNA Ligase 2 (manufactured by New England BioLabs) (10 units / ⁇ L) (10 ⁇ L) was added to this solution, mixed, and then allowed to stand on a temperature-controlled heat block (37 ° C., 16 hours). Chloroform of the same volume was added to the reaction solution, mixed by vortex, centrifuged, the upper layer was taken, and RNA pellets were obtained by alcohol precipitation (0.3 M aqueous sodium acetate solution (pH 5.2) / 70% ethanol). The RNAs obtained in each batch were combined and purified on a 7.5% denatured polyacrylamide gel to give RNA conjugate E1 (9.7 nmol, 32% yield).
- Example 2 Using the RNA fragment E2-1 obtained as sequence 6 by chemical synthesis, the 5'phosphate RNA fragment E2-2 obtained as sequence 7, and the template DNA1, the RNA conjugate product E2 (in the same manner as in Example 1) 10.9 nmol, yield 36%) was obtained.
- Example 3 Using the RNA fragment E3-1 obtained as sequence 9 by chemical synthesis, the 5'phosphate RNA fragment E3-2 obtained as sequence 10, and the template DNA1, RNA conjugate product E3 (in the same manner as in Example 1). 13.4 nmol, yield 45%) was obtained.
- Example 4 Using the RNA fragment E4-1 obtained as the sequence 12 by chemical synthesis, the 5'phosphate RNA fragment E4-2 obtained as the sequence 13, and the template DNA1, the RNA conjugate product E4 (in the same manner as in Example 1) 6.8 nmol, yield 34%) was obtained.
- Each nucleotide N in Tables 3 to 8 is RNA, N (M) is 2'-O-methyl modified RNA, N (F) is 2'-F modified RNA, and N (L) is LNA.
- N (MOE) represents 2'-O-methoxyethyl modified RNA and dN represents DNA.
- a (m6) indicates that the base part is N6-methyladenine.
- p indicates that the 3'or 5'end is phosphorylated
- p (S) indicates that the 3'or 5'end is phosphorylated.
- NH2 indicates that the hydroxyl group at the 3'or 5'end is replaced with an amino group.
- -N / P- and -P / N- indicate that the phosphate bond of the nucleotide is replaced with a phosphate amide bond
- -NHAc / S- indicates that the phosphate bond of the nucleotide is -NHC (O) -CH2.
- the underlined "AUG” represents the start codon and the underlined "UGA” represents the stop codon.
- a diagonal line (/) in the sequence indicates that the polynucleotide was linked at that portion.
- Reference example 1 3'-aminoRNA fragment R1-1 (200 ⁇ M) obtained as sequence 15 by chemical synthesis, 5'phosphate RNA fragment R1-2 (200 ⁇ M) obtained as sequence 16, and template DNA2 (200 ⁇ M).
- the ultrapure aqueous solution was heated at 90 ° C. for 3 minutes and then returned to room temperature over 30 minutes or longer.
- To this solution add the same volume of a separately prepared buffer solution of condensing agent (500 mM HEPES-NaOH (pH 8.5), 200 mM NaCl, 200 mM MgCl 2 of 1M EDC-HCl / 1M HOBt) and mix. The ligation reaction was started.
- the reaction solution (nucleic acid concentration: 100 ⁇ M, 0.5M EDC-HCl / 0.5M HOBt, 250 mM HEPES-NaOH (pH 8.5), 100 mM NaCl, 100 mM MgCl 2 ) is a temperature-controlled heat block (25 ° C). Let it sit on top all night. After the reaction, RNA pellets were obtained by alcohol precipitation (0.3 M aqueous sodium acetate solution (pH 5.2) / 70% ethanol). This RNA was analyzed on a 7.5% modified polyacrylamide gel, and the reaction yield (yield 52%) was calculated.
- Reference example 5 Phosphate buffer (50 mM) and MiliQ water were added to R5-1 (100 ⁇ M) obtained by chemical synthesis, a DMF solution of iodoacetic acid NHS ester (5 mM) was added, and the mixture was incubated at 30 ° C. for 2 hours. Subsequently, pellets of R5-1 iodine-acetylated by alcohol precipitation (0.3 M aqueous sodium acetate solution (pH 5.2) / 70% ethanol) were obtained.
- R5-2 (50 ⁇ M) obtained by chemical synthesis, template DNA1 (50 ⁇ M), aqueous sodium chloride solution (100 mM), and phosphate buffer (20 mM, pH 7.5) are mixed and heated at 90 ° C for 3 minutes. After that, it was returned to room temperature for 30 minutes or more.
- the above iodine-acetylated R5-1 (50 ⁇ M) was added to this solution, and the mixture was incubated at 30 ° C. for 6 hours.
- RNA pellets were obtained by alcohol precipitation (0.3 M aqueous sodium acetate solution (pH 5.2) / 70% ethanol). This RNA was purified by 7.5% denatured polyacrylamide gel to obtain RNA linkage product R5 (2.32 nmol, yield 7.3%).
- RNA conjugate product R6 (11.1 nmol, yield 37%) was obtained in the same manner as in Example 1.
- RNA conjugate product R7 (3.1 nmol, yield 10%) was obtained in the same manner as in Example 1.
- RNA conjugate product R8 (2.6 nmol, yield 9%) was obtained in the same manner as in Example 1.
- RNA conjugate product R9 (5.9 nmol, yield 20%) was obtained in the same manner as in Example 1.
- RNA conjugate product R10 (2.1 nmol, yield 9%) was obtained in the same manner as in Example 1.
- Reference example 11 An ultrapure aqueous solution of RNA fragment R11-1 (200 ⁇ M) obtained as sequence 49 by chemical synthesis, 5'-aminoRNA fragment R11-2 (200 ⁇ M) obtained as sequence 50, and template DNA1 (200 ⁇ M). After heating at 90 ° C. for 3 minutes, the temperature was returned to room temperature over 30 minutes. To this solution, add the same volume of a separately prepared buffer solution of condensing agent (500 mM HEPES-NaOH (pH 8.5), 200 mM NaCl, 200 mM MgCl 2 of 1M EDC-HCl / 1M HOBt) and mix. The ligation reaction was started.
- condensing agent 500 mM HEPES-NaOH (pH 8.5)
- 200 mM NaCl 200 mM MgCl 2 of 1M EDC-HCl / 1M HOBt
- the reaction solution (nucleic acid concentration: 100 ⁇ M, 0.5M EDC-HCl / 0.5M HOBt, 250 mM HEPES-NaOH (pH 8.5), 100 mM NaCl, 100 mM MgCl 2 ) is a temperature-controlled heat block (25 ° C). Allowed to stand on (7 hours).
- RNA pellets were obtained by alcohol precipitation (0.3 M aqueous sodium acetate solution (pH 5.2) / 70% ethanol). This RNA was purified by 7.5% denatured polyacrylamide gel to obtain RNA linkage product R11 (1.2 nmol, yield 15%).
- Reference example 12 1x T4 DNA ligase buffer solution (66 mM Tris-HCl (pH 7.6), 6.6 mM MgCl 2 , 10) of R12-1 (10 ⁇ M), R12-2 (10 ⁇ M) and template DNA 1 (10 ⁇ M) obtained by chemical synthesis.
- mM DTT, 0.1 mM ATP) (Takarabio) (2.1 mL in total) was heated at 90 ° C. for 5 minutes and then slowly cooled to room temperature. 60% PEG6000 was added to this solution to a final concentration of 15%.
- RNA linkage product R12 (1.4 nmol, yield 27%).
- RNA conjugate product R13 (1.3 nmol, yield 26%) was obtained in the same manner as in Reference Example 12.
- RNA conjugate product R14 (9.4 nmol, yield 31%) was obtained in the same manner as in Example 1.
- RNA conjugate product R15 8.6 nmol, yield 29%) was obtained in the same manner as in Example 1.
- RNA conjugate product R16 (1.1 nmol, yield) was used in the same manner as in Reference Example 12. 37%) was obtained.
- RNA linkage product L84 (PO) (30 nmol, yield 22%).
- RNA linkage product R17 (360 pmol, yield 36%).
- RNA conjugate product R18 (11.9 nmol, yield 40%) was obtained in the same manner as in Example 1.
- Test Example 1 Translation reaction of mRNA sample
- the translation reaction was performed according to the product protocol using a commercially available translation kit.
- the translation reaction in the prokaryotic cell system was carried out using compound R1 and compound R17 as substrate RNA and PURExpress (registered trademark) (New England BioLabs) as a reagent.
- the translation reaction in the eukaryotic cell system was carried out using compound R2 and compound R18 as substrate RNA and Rabbit Reticulocyte Lysate System, Nuclease Treated (hereinafter, RRL) (Promega) as a reagent.
- RRL Rabbit Reticulocyte Lysate System, Nuclease Treated
- Rabbit Reticulocyte Lysate System Kit included Rabbit Reticulocyte Lysate (14.0 ⁇ L), Amino Acid Mixture --Met (0.20 ⁇ L), Amino Acid Mixture --Leu (0.20 ⁇ L), Murine RNase Inhibitor (New England BioLabs) (0.4 ⁇ L), Ultrapure Water A mixture of water (5.2 ⁇ L) was used as the translation reaction solution. A translation reaction was carried out by adding and mixing this solution to a tube containing an RNA sample (30 ° C, 2 hours).
- Loading buffer for 2x SDS-PAGE (125 mM Tris-HCl (pH 6.8), 30 (v / v)% glycerol, 4% sodium dodecyl sulfate (SDS), 0.03% bromophenol blue) (Hereinafter, BPB)) was mixed and then heated at 90 ° C. for 3 minutes to prepare a sample for SDS-PAGE analysis. Samples for SDS-PAGE analysis were immediately electrophoresed on an SDS-PAGE gel (using 25 mM Tris, 192 mM Glycine, 0.1% SDS as a buffer for electrophoresis).
- the translation product on the gel is a membrane for Western blotting by a semi-dry method (Immobilon (registered trademark) -P) (IPVH00010, Millipore) (The membrane is hydrophilized with methanol as a pretreatment, and then the blotting buffer Transferred to (using 25 mM Tris, 192 mM Glycine, 20% MeOH). In the transfer, a constant current condition (energization time: 1 hour) was used, and the current value to be used was determined from the size of the membrane. That is, the current value (mA) was set so that the membrane area (cm 2) x 2 was obtained.
- Immobilon (registered trademark) -P) IPVH00010, Millipore
- the TBS-T solution of 5% ECL Prime used in the subsequent operations is Amersham ECL Prime (GE Healthcare) and TBS-T (0.05M Tris-HCl (pH 7.4), 150 mM NaCl, 0.05% tween 20). Prepared by mixing. The membrane to which the translation product was transferred was blocked (5% ECL Prime TBS-T solution, room temperature, shake for 1 hour) and then treated with primary antibody (4,000-fold dilution, 0.5% ECL Prime TBS-T solution, 4 ° C).
- Test Example 2 Translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate
- a rabbit erythrocyte lysate For each compound obtained in Examples 1 to 3 and Reference Examples 6 to 10, 14, 15, and 18, using the Rabbit-Reticulocyte-Lysate-System-Nuclease-Treated kit (manufactured by Promega, catalog number L4960). The translational activity in the nuclear cell line was evaluated. First, 2 ⁇ L of each mRNA sample diluted with THE RNA storage solution (Thermofisher Scientific, Catalog No. AM7001) so that the final concentration of each compound becomes 0.3 ⁇ M is placed on a 96-well PCR plate (manufactured by AS ONE). Dispensed.
- THE RNA storage solution Thermofisher Scientific, Catalog No. AM7001
- the translation product in the reaction solution after the translation reaction was detected by the sandwich ELISA method described below.
- 6 * His, His-Tag antibody (Protein Tech, Catalog No. 66005-1-Ig) was diluted to 3 ⁇ g / mL with 0.1 M Carbonate buffer (pH 9.4), and 96 Well ELISA plate (Nunk). 50 ⁇ L per well was dispensed into (manufactured by the company) and allowed to stand overnight at 4 ° C. to prepare a plate on which the antibody was immobilized. Subsequently, the plate was washed with Tris Buffered Saline with Tween 20 (Santa Kurtz, Catalog No.
- washing solution sc-24953 (hereinafter referred to as a washing solution) diluted 1-fold with purified water, and then bovine serum albumin (Wako Jun). Yakusha, Catalog No. 017-22231) was diluted to a final concentration of 3%, and 200 ⁇ L of a washing solution (hereinafter referred to as blocking solution) was dispensed per well and allowed to stand at room temperature for 1 hour. After washing the plate with the washing solution, 50 ⁇ L of the translation reaction solution diluted 100-fold with the blocking solution was dispensed per well and allowed to stand at room temperature for 1 hour.
- the translation product polypeptide preparation (manufactured by Scrum Co., Ltd.) was similarly diluted with a blocking solution to each concentration and dispensed into a plate.
- HRP Monoclonal ANTI-FLAG M2-Peroxidase
- each compound produced a polypeptide encoded in the gene sequence by the translation system of eukaryotic cells after being added to rabbit erythrocyte lysate.
- the translation product in the reaction solution after the translation reaction was prepared by using the sandwich ELISA method described in Test Example 2 (Translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate), and the translation reaction solution was mixed with a blocking solution 20. The procedure was exactly the same except that it was double diluted and added to the plate. As a result of the measurement, the translation product concentration ( ⁇ M) in each translation reaction solution and the compound R18 having no sugar modification, which were quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation, were set to 1. The relative translation product amounts at the time are shown in Tables 12-14.
- each compound produced a polypeptide encoded in the gene sequence by the translation system of human cells after being added to Hela cell lysate.
- the culture supernatant is removed from the cells, washed once with ice-cooled D-PBS (-) (manufactured by Nacalai Tesque), and then 2% protease inhibitor cocktail (for animal cell extract). 20 ⁇ L of NP-40 (Invitrogen, FNN0021) containing the above was added per well and vigorously infiltrated for 5 minutes to lyse the cells.
- the translation product in the obtained cytolytic solution was prepared by using the sandwich ELISA method described in Test Example 2 (translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate) in a blocking solution of the cytolytic solution.
- each compound produced a polypeptide encoded in the gene sequence after being added to Hela cells.
- Test Example 3 Stability test of each compound by phosphodiesterase I
- RNA solution prepared in Example 1 and Reference Examples 10 and 18, the stability to phosphodiesterase I (SVPD) was evaluated by the following method.
- the compound having a sugar moiety modification has improved resistance to phosphodiesterase I as compared with the compound R18 having no sugar moiety modification.
- Test Example 4 Stability test of mRNA sample by phosphodiesterase I
- the enzyme stability of each of the compounds obtained in Examples 1 to 4 and Reference Examples 6, 14, 15 and 18 was evaluated using phosphodiesterase I (warthington, Catalog No. 3926).
- phosphodiesterase I was added to SVPD stock solution (110 mM Tris-HCl (Nippon Gene, Catalog No. 314-90401), 110 mM NaCl (Ambion, Catalog No. AM9759), 15 mM MgCl 2 (Nakalitesk, Catalog No. 20942-34)).
- the final concentration was adjusted to 55 U / mL, and this was further diluted 10-fold with 5 ⁇ SVPD reaction buffer (100 mM Tris-HCl (Ambion, catalog number AM9855G), 500 mM NaCl, 75 mM MgCl 2). Then, 5.5 U / mL SVPD enzyme solution was prepared. Each compound was diluted with THE RNA storage solution (Thermo Fisher Scientific, Catalog No. AM7001) to a final concentration of 5 ⁇ M. Distilled water 30 ⁇ L, 5 ⁇ M mRNA 12 ⁇ L, and 5 ⁇ SVPD reaction buffer 12 ⁇ L were added in this order to a 96-well PCR plate to prepare an enzyme reaction premix solution.
- 40.5 ⁇ L of the enzyme reaction premix solution was dispensed into another 96-well PCR plate, and 4.5 ⁇ L of 5.5 U / mL SVPD enzyme solution was added and mixed well.
- RT primer 5'-TCAGTGGTGGTGGTGGTGGTGTTTG-3' (SEQ ID NO: 75)
- Fw primer 5'-ATCTTGTCGTCGTCGTCCTT-3' (SEQ ID NO: 76)
- Rv primer 5'-GAATACAAGCTACTTGTTCTTTT-3'(SEQ ID NO: 77)
- Taqman MGB Probe 5'-CAGCCACCATG-3' (SEQ ID NO: 78)
- Test Example 5 Translation reaction in eukaryotic cell lineage
- RNA RNA obtained in Reference Examples 3 to 5, 11 to 13, 16 and 18, the translation reaction in the eukaryotic cell line was evaluated by the following method.
- the solution contained in Rabbit-Reticulocyte-Lysate-System_-Nuclease-Treated (Promega L4960), RNase inhibitor Murine (New England Biolab, M0314S) and RNA were mixed in the following composition.
- the solution was incubated at 37 ° C. for 1 hour. After incubation, 5 ⁇ L of the solution was dispensed and diluted 100-fold with 495 ⁇ L of Blocking buffer (3% BSA / TBST). 100 ⁇ L of diluted translation solution was used for ELISA.
- the translation reaction solution was added at 100 ⁇ L / well and incubated at room temperature for 1 hour.
- the translation reaction solution in the plate was discarded and washed 3 times with 200 ⁇ L / well TBST.
- a 100 ⁇ L / well Anti-Flag solution (Cell Signaling Tecnorogy, Catalog No. 2368) (1: 1000 in bloking buffer) was added. It was covered with parafilm to prevent dry concentration and incubated overnight at 4 ° C.
- the solution in the plate was discarded and washed 3 times with 200 ⁇ L / well TBST.
- 100 ⁇ L / well Anti-rabbit IgG HRP solution (Sigma-Aldrich) (1: 10000 in bloking buffer) was added and incubated at room temperature for 1 hour.
- the solution in the plate was discarded and washed 4 times with 200 ⁇ L / well TBST.
- 100 ⁇ L / well TMB substrate solution was added and incubated for 5 minutes at room temperature.
- a 100 ⁇ L / well 2M H 2 SO 4 solution was added to the well to which the TBM subustrate solution had been added.
- a plate reader (Mithras LB940 (Berthold)) was used to measure the absorbance at 450 nm to evaluate translation efficiency.
- the translation product concentration ( ⁇ M) in each translation reaction solution and R18 without sugar modification were set to 1.
- the relative translation product amounts of are shown in Tables 18 to 20.
- each compound produced a polypeptide encoded in the gene sequence by the translation system of eukaryotic cells after being added to rabbit erythrocyte lysate.
- each nucleotide N is RNA
- each nucleotide n lower is DNA
- N (M) is 2'-O-methyl modified RNA
- N (F) is 2'-F modified.
- RNA N (L) stands for LNA
- N (MOE) stands for 2'-O-methoxyethyl modified RNA.
- Am6 indicates that the base portion is N6-methyladenine
- Ae6 indicates that the base portion is N6-ethyladenine.
- p indicates that the 3'or 5'end is phosphorylated.
- N (B) represents 2', 4'-BNA NC (Me) containing the sugar part shown below.
- BDBD represents an artificial dangling end having the following structure.
- Test Example 6 Translation reaction test using Hela cell line lysate of mRNA sample
- the translational activity in human cell lines was evaluated using the 1-Step Human Coupled IVT Kit (Cat. No. 88882, manufactured by Thermo Fisher Scientific).
- the translation reaction was carried out under the same conditions as in Test Example 2 (translation reaction test using Hela cell line lysate of mRNA sample).
- the translation product in the reaction solution after the translation reaction was detected by the sandwich ELISA method described below. First, 6 * His, His-Tag antibody (Protein Tech, Catalog No.
- 66005-1-Ig was diluted to 3 ⁇ g / mL with 0.1 M Carbonate buffer (pH 9.4), and 96 Well ELISA plate (Nunk). 50 ⁇ L per well was dispensed into (manufactured by the company) and allowed to stand overnight at 4 ° C. to prepare a plate on which the antibody was immobilized. Subsequently, the plate was washed with Tris Buffered Saline with Tween 20 (Santa Kurtz, Catalog No. sc-24953) (hereinafter referred to as a washing solution) diluted 1-fold with purified water, and then bovine serum albumin (Wako Jun). Yakusha, Catalog No.
- a washing solution hereinafter referred to as blocking solution
- 50 ⁇ L of the translation reaction solution diluted with the blocking solution was dispensed per well and allowed to stand at room temperature for 1 hour.
- the translation product polypeptide preparation shown in SEQ ID NO: 539 was similarly diluted with a blocking solution to each concentration and dispensed into a plate.
- HRP Monoclonal ANTI-FLAG M2-Peroxidase Ab produced in mouse (SIGMA, catalog antibody A8592-1MG) diluted 10,000-fold with blocking solution after washing the plate with a washing solution. It was dispensed and allowed to stand at room temperature for 1 hour. After washing the plate with a washing solution, 50 ⁇ L of 1-Step Ultra TMB-ELISA (Thermo Fisher Scientific, Catalog No. 34028) was dispensed per well and allowed to stand at room temperature for several minutes.
- HRP Monoclonal ANTI-FLAG M2-Peroxidase
- each compound having a sugar moiety modification was added to Hela cell lysate, and then a polypeptide encoded in the gene sequence was produced by the translation system of eukaryotic cells.
- Test Example 7 In vitro translation reaction test using Hela cell line of mRNA sample, for each of the compounds listed in Tables 21-56, in vitro translation activity was evaluated using Hela cell lines. First, Hela cells suspended in RPMI medium (manufactured by Nacalai Tesque) containing 10% bovine fetal serum were seeded on a culture plate for 96-well adherent cells so that the number of cells per well was 10,000 cells / 100 ⁇ L. The cells were cultured overnight at 37 ° C. under 5% CO2 conditions. After removing the culture supernatant from the cells after culturing overnight and adding RPMI medium containing 40 ⁇ L of 10% bovine fetal serum per well, each compound and final concentration were adjusted to 0.1 ⁇ M.
- RPMI medium manufactured by Nacalai Tesque
- Lipofectamin MessengerMAX Transfection Reagent (Cermo Fisher Scientific, Catalog No .: LMRNA008) with a concentration of 0.3% is diluted with Optimem (Thermo Fisher Scientific, Catalog No .: 31985-070) and mixed.
- Optimem Thermo Fisher Scientific, Catalog No .: 31985-070
- Table 103 shows a 2% protease inhibitor cocktail (animal cells).
- each mRNA having a sugar moiety modification produces a polypeptide encoded by a gene sequence after being added to Hela cells, and its activity is equal to or higher than that of an mRNA having no sugar moiety modification. there were.
- Test Example 8 In vitro translation reaction test using mouse primary hepatocytes of mRNA sample
- in vitro translational activity was evaluated using primary mouse hepatocytes (Cat. No. MSCP10, manufactured by Thermo Fisher Scientific).
- primary mouse hepatocytes Cat. No. MSCP10, manufactured by Thermo Fisher Scientific.
- Hepatocytes were seeded on a 96-well collagen I-coated culture plate (Corning, catalog number: 356407) so that the number of cells per well was 10,000 cells / 100 ⁇ L, and the cells were seeded under 37 ° C. and 5% CO2 conditions for 5 hours. It was cultured. The culture supernatant was removed from the cells after culturing, and William's E Medium, no phenol red containing Primary Hepatocyte Maintenance Supplements (Cat. No. CM4000, manufactured by Thermo Fisher Scientific) was added to 100 ⁇ L per well. In addition, the cells were cultured at 37 ° C. under 5% CO2 conditions for 5 hours.
- each compound was adjusted to a final concentration of 0.1 ⁇ M.
- the compound and Lipofectamin MessengerMAX Transfection Reagent with a final concentration of 0.3% were diluted with Optimem and mixed, and the mixture was added to each culture plate to a concentration of 10 ⁇ L per well, and 5 under 37 ° C. and 5% CO2 conditions. Incubated for hours.
- the culture supernatant was removed from the cells, 100 ⁇ L of William's E Medium, no phenol red containing Primary Hepatocyte Maintenance Supplements was added per well, and then the cells were further cultured for 1 hour under 37 ° C. and 5% CO2 conditions. did. After culturing for 1 hour, the culture supernatant is removed from the cells, washed once with ice-cooled D-PBS (-) (manufactured by Nacalai Tesque), and then a 2% protease inhibitor cocktail (for animal cell extracts). 20 ⁇ L of NP-40 (Invitrogen, FNN0021) containing the above was added per well and vigorously infiltrated for 30 seconds to lyse the cells.
- the translation product in the obtained cytolyte was carried out in the same manner as the sandwich ELISA method described in Test Example 6 (translation reaction test using Hela cell line lysate of mRNA sample).
- the translation product concentration (nM) in each translation reaction solution which was quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation, is shown in the table below.
- each mRNA having a sugar moiety modification produces a polypeptide encoded by a gene sequence after being added to mouse hepatocytes, and its activity is equal to or higher than that of an mRNA having no sugar moiety modification. Met.
- Test Example 9 In vitro translation reaction test using Hela cell line of mRNA sample
- each compound was diluted with THE RNA Storage Solution (Cat. No. AM7000, manufactured by Thermo Fisher Scientific) to 20 ⁇ M.
- Hela cell lines were suspended in a mixture of SE Cell Line Nucleofector Solution and Supplement 1 attached to SE Cell Line 96-well Nucleofector Kit (Lonza, Catalog No. V4SC-1096) to 200,000 cells / 19 ⁇ L.
- the prepared nucleic acid solution and Hela cell suspension were mixed at a volume ratio of 1:19, and then electroporation was performed under pulse conditions FF-137 using a Nucleofector (tm) 96-well Shuttl system (manufactured by Lonza). .. Cells 10 minutes after electroporation were suspended in RPMI medium containing 10% bovine fetal serum (manufactured by Nacalai Tesque), and the number of cells per well was 50,000 cells / 145 ⁇ L. The cells were seeded and cultured for 3 hours under the conditions of 37 ° C. and 5% CO2.
- an mRNA having a sugar moiety modification produces a polypeptide encoded by a gene sequence after electroporation into Hela cells, and its activity is an mRNA having no sugar moiety modification in the translation region. It was superior to.
- Test Example 10 Translation reaction test using Hela cell line lysate of mRNA sample
- the translational activity in human cell lines was evaluated using the 1-Step Human Coupled IVT Kit (Cat. No. 88882, manufactured by Thermo Fisher Scientific Co., Ltd.).
- the translation reaction was carried out in the same manner as in Test Example 2 (translation reaction test using Hela cell line lysate of mRNA sample) under the condition that the nucleic acid concentration was 1 ⁇ M.
- the translation product in the reaction solution after the translation reaction was subjected to SEQ ID NO: 540 as a translation product polypeptide preparation according to the method of the sandwich ELISA method described in Test Example 6 (translation reaction test using Hela cell line lysate of mRNA sample). The same method was used except that the peptide shown in (1) (manufactured by Cosmobio) was used.
- the translation product concentration (nM) in each translation reaction solution and each compound in the table without sugar modification in the translation region, which was quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation, were 1
- the relative translation product amount at the time of is shown in the table below.
- Translation product polypeptide preparation NH2-MDYKDDDDKGGHHHHHH-COOH (SEQ ID NO: 540)
- Test Example 11 In vitro translation reaction test using Hela cell line of mRNA sample
- in vitro translation activity was evaluated using Hela cell lines. Transfection of each compound into the cell line and preparation of the cell lysate were carried out in the same manner as in Test Example 7 (in vitro translation reaction test using Hela cell line of mRNA sample), and cell lysis was performed at 2%. It was performed using an iScript RT-qPCR Sample Preparation Reagent containing a protease inhibitor cocktail (for animal cell extracts). The translation product in the obtained cytolyte was carried out in the same manner as the sandwich ELISA method described in Test Example 10 (translation reaction test using Hela cell line lysate of mRNA sample). As a result of the measurement, the translation product concentration (nM) in each translation reaction solution, which was quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation, is shown in the table below.
- each mRNA having a sugar moiety modification produces a polypeptide encoded by the gene sequence after being added to Hela cells, and its activity is superior to that of an mRNA having no sugar moiety modification in the translation region.
- Test Example 12 Translation reaction test using Hela cell line lysate of mRNA sample
- the translational activity in human cell lines was evaluated using the 1-Step Human Coupled IVT Kit (Cat. No. 88882, manufactured by Thermo Fisher Scientific).
- the translation reaction was carried out under the same conditions as in Test Example 2 (translation reaction test using Hela cell line lysate of mRNA sample) under the condition that the nucleic acid concentration was 1 ⁇ M.
- the translation product in the reaction solution after the translation reaction was subjected to SEQ ID NO: 541 as a translation product polypeptide preparation according to the method of the sandwich ELISA method described in Test Example 6 (translation reaction test using Hela cell line lysate of mRNA sample). The same method was used except that the peptide shown in (1) (manufactured by Cosmobio) was used.
- the translation product concentration (nM) in each translation reaction solution and E82, which has no sugar modification in the translation region were set to 1 as quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation.
- the relative translation product amounts are listed in Tables 124-133.
- Translation product polypeptide preparation NH2-MDYKDDDDKGGDYKDDDDKHHHHHH-COOH (SEQ ID NO: 541)
- each mRNA compound having a sugar moiety modification was added to Hela cell lysate, and then a polypeptide encoded in the gene sequence was produced by the translation system of eukaryotic cells.
- Test Example 13 In vitro translation reaction test using Hela cell line of mRNA sample
- in vitro translational activity was evaluated using Hela cell lines. Transfection of each compound into the cell line and preparation of the cell lysate were carried out in the same manner as in Test Example 7 (in vitro translation reaction test using Hela cell line of mRNA sample), and cell lysis was 2%. It was performed using an iScript RT-qPCR Sample Preparation Reagent containing a protease inhibitor cocktail (for animal cell extracts). The translation product in the obtained cytolyte was carried out in the same manner as the sandwich ELISA method described in Test Example 12 (translation reaction test using Hela cell line lysate of mRNA sample). As a result of the measurement, the translation product concentration (nM) in each translation reaction solution, which was quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation, is shown in the table below.
- each mRNA having a sugar moiety modification produces a polypeptide encoded by a gene sequence after being added to Hela cells, and its activity is superior to that of an mRNA having no sugar moiety modification. It was.
- Test Example 14 Translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate
- the translational activity in the eukaryotic cell line was evaluated using the Rabbit-Reticulocyte-Lysate-System-Nuclease-Treated kit.
- the translation reaction was carried out in the same manner as in Test Example 2 (translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate).
- the translation product in the reaction solution after the translation reaction was detected by the sandwich ELISA method described below.
- Anti-Human EGF antibody (Peprotech, catalog number 500-P45) was diluted to 3 ⁇ g / mL with 0.1 M Carbonate buffer (pH 9.4) and placed on a 96 Well ELISA plate (Nunk). An antibody-immobilized plate was prepared by dispensing 50 ⁇ L per well and allowing it to stand overnight at 4 ° C. Subsequently, the plate was washed with Tris Buffered Saline with Tween 20 (hereinafter referred to as a washing solution) diluted 1-fold with purified water, and then bovine serum albumin was diluted to a final concentration of 3%.
- Tris Buffered Saline with Tween 20 hereinafter referred to as a washing solution
- blocking solution (Hereinafter referred to as blocking solution) was dispensed at a rate of 200 ⁇ L per well and allowed to stand at room temperature for 1 hour. After washing the plate with the washing solution, 50 ⁇ L of the translation reaction solution diluted 100-fold with the blocking solution was dispensed per well and allowed to stand at room temperature for 1 hour. 50 ⁇ L of Monoclonal ANTI-FLAG M2-Peroxidase (HRP) Ab produced in mouse (SIGMA, catalog antibody A8592-1MG) diluted 10,000-fold with blocking solution after washing the plate with a washing solution. It was dispensed and allowed to stand at room temperature for 1 hour.
- HRP Monoclonal ANTI-FLAG M2-Peroxidase
- each mRNA compound having a sugar moiety modification was added to rabbit erythrocyte lysate, and then produced a FLAG-EGF peptide encoded in the gene sequence by a translation system of eukaryotic cells.
- Test Example 15 Translation reaction test using Hela cell line lysate of mRNA sample
- the translational activity in human cell lines was evaluated using the 1-Step Human Coupled IVT Kit (Cat. No. 88882, manufactured by Thermo Fisher Scientific).
- the translation reaction was carried out under the same conditions as in Test Example 2 (translation reaction test using Hela cell line lysate of mRNA sample) under the condition that the nucleic acid concentration was 1 ⁇ M.
- the translation product in the reaction solution after the translation reaction is prepared by blocking the translation reaction solution according to the method of the sandwich ELISA method described in Test Example 14 (Translation reaction in eukaryotic cell line; Translation reaction test using rabbit erythrocyte lysate). It was carried out in the same manner except that it was diluted 20 times with.
- the absorbance at the time of measuring each translation reaction solution (the absorbance of the measurement wavelength minus the absorbance of the reference wavelength) is shown in the table below.
- each mRNA compound having a sugar moiety modification was added to Hela cell lysate, and then produced a FLAG-EGF peptide encoded in the gene sequence by a eukaryotic translation system.
- Test Example 16 In vitro translation reaction test using Hela cell line of mRNA sample
- in vitro translational activity was evaluated using Hela cell lines. Transfection of each compound into the cell line and preparation of the cell lysate were carried out in the same manner as in Test Example 7 (in vitro translation reaction test using Hela cell line of mRNA sample), and cell lysis was performed at 2%. It was performed using an iScript RT-qPCR Sample Preparation Reagent containing a protease inhibitor cocktail (for animal cell extracts).
- the translation product in the obtained cytolytic solution blocks the cytolytic solution according to the method of the sandwich ELISA method described in Test Example 14 (translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate). The procedure was the same except that it was diluted 30-fold with the solution.
- the absorbance at the time of measuring each cytolyte (the absorbance of the measurement wavelength minus the absorbance of the reference wavelength) is shown in the table below.
- each mRNA compound having a sugar moiety modification produces a FLAG-EGF peptide encoded in the gene sequence by the eukaryotic cell translation system after being added to Hela cells, and its activity is the sugar moiety. It was superior to unmodified mRNA.
- Test Example 17 Translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate
- the translational activity in the eukaryotic cell line was evaluated using the Rabbit-Reticulocyte-Lysate-System-Nuclease-Treated kit.
- the translation reaction was carried out in the same manner as in Test Example 2 (translation reaction in eukaryotic cell line; translation reaction test using rabbit erythrocyte lysate).
- the translation product in the reaction solution after the translation reaction was subjected to SEQ ID NO: 542 as a translation product polypeptide preparation according to the method of the sandwich ELISA method described in Test Example 6 (translation reaction test using Hela cell line lysate of mRNA sample). The same method was used except that the peptide shown in (1) (manufactured by Cosmobio) was used.
- the translation product concentration (nM) in each translation reaction solution and E6 without sugar modification in the translation region were set to 1 as quantified using a calibration curve prepared based on the absorbance of the polypeptide preparation.
- the relative translation product amounts are listed in the table below.
- Translation product polypeptide preparation NH2-MDYKDDDDKIIDYKDDDDKGGDYKDDDDKSIINFEKLHHHHHH-COOH (SEQ ID NO: 542)
- each mRNA compound having a sugar moiety modification was added to rabbit erythrocyte lysate, and then a peptide encoded in the gene sequence was produced by a translation system of eukaryotic cells.
- Test Example 18 Translation reaction test using Hela cell line lysate of mRNA sample
- the translational activity in human cell lines was evaluated using the 1-Step Human Coupled IVT Kit (Cat. No. 88882, manufactured by Thermo Fisher Scientific).
- the translation reaction was carried out under the same conditions as in Test Example 2 (translation reaction test using Hela cell line lysate of mRNA sample) under the condition that the nucleic acid concentration was 1 ⁇ M.
- the translation product in the reaction solution after the translation reaction is a blocking solution of the translation reaction solution according to the method of the sandwich ELISA method described in Test Example 17 (Translation reaction in eukaryotic cell line; Translation reaction test using rabbit erythrocyte lysate).
- each mRNA compound having a sugar moiety modification was added to Hela cell lysate, and then a peptide encoded in the gene sequence was produced by the translation system of eukaryotic cells.
- Test Example 19 serum stability test of mRNA sample
- serum nucleic acid stability was evaluated using commercially available mouse sera (Takara Bio, Catalog No. 2311B, lot No. AJ10759A).
- mouse serum was diluted 15-fold with UltraPure (tm) DNase / RNase-Free Distilled Water (DW) (invitrogen, Catalog No. 10977-015) to prepare a diluted serum solution.
- DW DNase / RNase-Free Distilled Water
- Each compound was diluted with THE RNA storage solution (Thermo Fisher Scientific, Catalog No. AM7001) to a final concentration of 5 ⁇ M.
- 28 ⁇ L of diluted serum solution and 7 ⁇ L of 5 ⁇ M mRNA were added to another 96-well PCR plate and mixed well.
- Reverse transcriptase cDNA was prepared using this diluted sample 5 ⁇ L and 2 ⁇ M RT primer (Sigma Aldrich) 1 ⁇ L using the TaqMan MicroRNA RT kit (Thermo Fisher Scientific, Catalog No. 4366597). The reaction temperature was 16 ° C 30 min ⁇ 42 ° C 30 min ⁇ 85 ° C 5 min.
- RT primer 5'-TCAGTGGTGGTGGTGGTGGTGTTTG-3' (SEQ ID NO: 75)
- Fw primer 5'-ATCTTGTCGTCGTCGTCCTT-3' (SEQ ID NO: 76)
- Rv primer 5'-GAATACAAGCTACTTGTTCTTTT-3'(SEQ ID NO: 77)
- Taqman MGB Probe 5'-CAGCCACCATG-3' (SEQ ID NO: 78)
- mRNA having a sugar moiety modification has improved degradation resistance in serum as compared with mRNA having no sugar moiety modification.
- Test Example 20 Translation reaction of VEGF protein
- RNA Ribonucleic acid
- M 2'-O-methyl modified RNA
- N (F) 2'-F modified RNA
- N (MOE) 2'-O-methoxyethyl.
- Modified RNA, dN represents DNA.
- Comparative compound RN was obtained by the following series of operations.
- the plasmid DNA used was a commercially available pUC19 vector Sma I site in which the artificially synthesized gene sequence GN shown in SEQ ID NO: 546 was inserted (manufactured by Fasmac Co., Ltd.).
- PCR was performed using plasmid DNA, FW and RV primers.
- the final concentration of the PCR solution is as follows; template DNA 0.2 ng / ⁇ L, dNTP 250 ⁇ M, primer 0.3 ⁇ M, PrimeSTAR HS DNA Polymerase (Takara) 0.025 U / ⁇ L, PrimeSTAR Buffer (Mg 2 + plus) (Takara) 1x. Temperature The cycles are as follows; 94 oC 5 min, [98 oC 10 sec, 55 oC 30 sec, 72 oC 60 sec] x 30 cycles. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation. Subsequently, the template plasmid was decomposed and removed by DpnI treatment.
- the final concentration of the reaction solution was PCR product 3.43 ⁇ g / ⁇ L, Dpn I (Takara) 1 U / ⁇ L, T buffer (Takara) 1x, and incubation was performed at 37 ° C. for 1 hour. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation.
- a transcription reaction with T7 RNA polymerase was performed.
- the final concentration of the reaction solution is as follows; DNA 10 ng / ⁇ L, DTT 5 mM, ATP, CTP, UTP 2 mM each, GMP 2 mM, GTP 0.5 mM, T7 RNA polymerase (Takara) 2.5 U / ⁇ L, T7 RNA Polymerese buffer (Takara) 1x, RNase Inhibitor, Murine (NEB) 0.2 U / ⁇ L. Incubation was performed at 37 degrees for 2 hours.
- DNase recombinant, RNase-free, Takara
- Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation to obtain a 3'-terminal polynucleotide fragment RN-2.
- RNA linkage product by RNA ligase 2 The ligation reaction by RNA ligase 2 was carried out using the 3'terminal polynucleotide fragment RN-1 obtained by chemical synthesis according to a conventional method, the RNA fragment RN-2 obtained by in vitro transcription, and the template DNA-A. Final concentrations are RN-1 2 ⁇ M, RN-2 2 ⁇ M, DNAN 4 ⁇ M, PEG8000 10% (v / v), T4 RNA ligase 2 (NEB) 1 U / ⁇ L, T4 RNA ligase 2 buffer (NEB) 1x , RNase inhibitor Murine (NEB) 1 U / ⁇ L.
- Substrates and reagents other than PEG8000, T4 RNA ligase 2, and RNase inhibitor were added to the tube, and then heated at 90 ° C for 3 minutes to naturally dissipate heat to room temperature. The remaining reagents and enzymes were added and incubated at 45 ° C for 1 hour. DNase was added at a final concentration of 0.33 U / ⁇ L and incubated at 37 ° C for 15 minutes. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation to obtain ligated RNA. Reaction samples were analyzed by dPAGE (5% acrylamide, 7M urea). Gel purification was performed as follows.
- RNA conjugate product (Linkage sequence analysis of RNA conjugate product)
- the sequence of the linking part of the RNA conjugate product RN was analyzed using the Smarter RACE 5'3'Kit (manufactured by Takara Bio, Catalog No. 634859) by the following series of operations.
- the RN prepared to 100 ng / ⁇ L was analyzed. Take 1 ⁇ L, mix with 1 ⁇ L of 5'CDS Primer A and 9 ⁇ L of Nuclease-free water, heat at 72 ° C for 4 minutes and cool at 4 ° C for 2 minutes. 1 ⁇ L SMARTER II A Oligo, 4 in reaction.
- RNA was synthesized by adding 240 ⁇ L of TE Buffer and mixing. Subsequently, a PCR reaction was carried out using the obtained template RNA as follows.
- RV Primer 2 5'-GATTACGCCAAGCTTTGG CTTGAAGATGTA CTCGATCTCATCAGG-3' ⁇ ⁇ ⁇ SEQ ID NO: 548
- Sequence primer M13FW 5'-CTCGATCTCATCAGG-3' ⁇ ⁇ ⁇ SEQ ID NO: 549
- Sequence primer M13RV 5'-CAGGAAACAGCTATGAC -3' ⁇ ⁇ ⁇ SEQ ID NO: 550
- RNA Ribonucleic acid
- M 2'-O-methyl modified RNA
- N (F) 2'-F modified RNA
- N (MOE) 2'-O-methoxyethyl.
- Modified RNA, dN represents DNA.
- Comparative compound RN' was obtained by the following series of operations.
- the plasmid DNA used was a commercially available pUC19 vector EcoRV site and XbaI site in which the artificially synthesized gene sequence GN'shown in SEQ ID NO: 552 was inserted (manufactured by Genewith Co., Ltd.).
- the plasmid DNA was linearized with the restriction enzyme XbaI.
- the final concentration of the reaction solution was plasmid DNA 20 ng / ⁇ L, XbaI (Takara) 0.75 U / ⁇ L, M buffer (Takara) 1x, BSA 0.01%, and the mixture was incubated at 37 ° C. for 1 hour. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation. A transcription reaction was carried out using the obtained linearized DNA.
- the final concentration of the reaction solution is as follows; DNA 10 ng / ⁇ L, DTT 5 mM, ATP, CTP, UTP 2 mM each, GMP 2 mM, GTP 0.5 mM, T7 RNA polymerase (Takara) 2.5 U / ⁇ L, T7 RNA Polymerese buffer (Takara) 1x, RNase Inhibitor, Murine (NEB) 0.2 U / ⁇ L. Incubation was performed at 37 degrees for 2 hours. Subsequently, DNase (recombinant, RNase-free, Takara) was added at a final concentration of 0.1 U / ⁇ L and incubated at 37 ° C. for 30 minutes. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation to obtain a 3'terminal polynucleotide fragment RN'-2.
- RNA ligase 2 using 5'terminal polynucleotide fragment RN'-1 obtained by chemical synthesis according to a conventional method, 3'terminal polynucleotide fragment RN'-2 obtained by in vitro transcription, and template DNA-B.
- the ligation reaction was carried out with.
- Final concentrations are RN-1 2 ⁇ M, RN-2 2 ⁇ M, DNAN 4 ⁇ M, PEG8000 10% (v / v), T4 RNA ligase 2 (NEB) 1 U / ⁇ L, T4 RNA ligase 2 buffer (NEB) 1x , RNase inhibitor Murine (NEB) 1 U / ⁇ L.
- Substrates and reagents other than PEG8000, T4 RNA ligase 2, and RNase inhibitor were added to the tube, and then heated at 90 ° C for 3 minutes to naturally dissipate heat to room temperature. The remaining reagents and enzymes were added and incubated at 45 ° C for 1 hour. DNase was added at a final concentration of 0.33 U / ⁇ L and incubated at 37 ° C for 15 minutes. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation to obtain ligated RNA. Reaction samples were analyzed by dPAGE (5% acrylamide, 7M urea). Gel purification was performed as follows.
- each nucleotide N in the table is RNA
- N (M) is 2'-O-methyl modified RNA
- N (F) is 2'-F modified RNA
- N (MOE) is 2'-O-methoxyethyl.
- Modified RNA, dN represents DNA.
- Compound EN with sugar modification was obtained by the following series of operations. (Preparation of linearized plasmid DNA and preparation of RNA fragment by in vitro transcriotion) The fragment on the 3'side was carried out in the same manner as the above RN'-2 preparation method.
- RNA linkage product by RNA ligase Using RNA fragment EN-1 obtained by chemical synthesis, RNA fragment RN'-2, and template DNA-C, a ligation reaction was carried out by RNA ligase 2. Final concentrations are EN-1 2 ⁇ M, RN'-2 2 ⁇ M, template DNA 4 ⁇ M, PEG8000 10% (v / v), T4 RNA ligase 2 (NEB) 1 U / ⁇ L, T4 RNA ligase 2 buffer (NEB) ) 1x, RNase inhibitor Murine (NEB) 1 U / ⁇ L.
- Substrates and reagents other than PEG8000, T4 RNA ligase 2, and RNase inhibitor were added to the tube, and then heated at 90 ° C for 3 minutes to naturally dissipate heat to room temperature. The remaining reagents and enzymes were added and incubated at 45 ° C for 1 hour. DNase (recombinant, RNase-free, Takara) was added at a final concentration of 0.33 U / ⁇ L and incubated at 37 ° C. for 15 minutes. Rough purification was performed by phenol-chloroform extraction and isopropanol precipitation to obtain ligated RNA. Reaction samples were analyzed by dPAGE (5% acrylamide, 7M urea). Gel purification was performed as follows.
- each nucleotide N (uppercase) represents RNA
- each nucleotide N (F) represents 2'-F modified RNA
- N (MOE) represents 2'-O-methoxyethyl modified RNA.
- RN For RN, RN', and EN, in vitro translation activity was evaluated using Hela cell lines.
- RPMI medium manufactured by Nacalai Tesque
- 10% bovine fetal serum were seeded on a culture plate for 96-well adherent cells so that the number of cells per well was 10,000 cells / 100 ⁇ L.
- the cells were cultured overnight under 37 ° C. and 5% CO2 conditions.
- RPMI medium containing 40 ⁇ L of 10% bovine fetal serum per well, the final concentrations of each compound were 0.3, 1, 3, and 10 nM.
- the amount of VEGF protein in the obtained culture supernatant was measured using Human VEGE Quantikine ELISA (manufactured by R & D, catalog number DVE00) according to the manual attached to the kit.
- the VEGF protein concentration (ng / mL) in each culture supernatant quantified as a result of the measurement is shown in the table below.
- SEQ ID NO: 1 Compound E1 SEQ ID NO: 2: Compound E1-1 SEQ ID NO: 3: Compound E1-2 SEQ ID NO: 4: Template DNA1 SEQ ID NO: 5: Compound E2 SEQ ID NO: 6: Compound E2-1 SEQ ID NO: 7: Compound E2-2 SEQ ID NO: 8: Compound E3 SEQ ID NO: 9: Compound E3-1 SEQ ID NO: 10: Compound E3-2 SEQ ID NO: 11: Compound E4 SEQ ID NO: 12: Compound E4-1 SEQ ID NO: 13: Compound E4-2 SEQ ID NO: 14: Compound R1 SEQ ID NO: 15: Compound R1-1 SEQ ID NO: 16: Compound R1-2 SEQ ID NO: 17: Template DNA2 SEQ ID NO: 18: Compound R2 SEQ ID NO: 19: Compound R2-1 SEQ ID NO: 20: Compound R2-2 SEQ ID NO: 21: Compound R3 SEQ ID NO: 22: Compound R3-1 SEQ ID NO: 23: Compound
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Crystallography & Structural Chemistry (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
遺伝情報伝達の中間物質であるmRNAは、リボソームに直接認識されタンパク質に翻訳されるための塩基配列情報と構造を有している。
しかしながら、天然塩基のみから構成される人工mRNAを外から細胞内に導入すると、細胞内のToll様レセプター(TLR3、TLR7、TLR8、RIG-I等)に結合することで速やかに免疫応答が惹起され、炎症反応及びタンパク質の翻訳量の低下が引き起こされることが知られている(非特許文献1)。細胞内でタンパク質を発現させるためには、何らかの形で人工mRNA自体の免疫反応性を低減し、かつ翻訳量を低下させない工夫が必要となる。また、天然塩基のみから構成されるRNAは核酸分解酵素に対して脆弱であることから、安定性付与の観点からも修飾ヌクレオチドの導入が必要とされている(非特許文献2)。修飾ヌクレオチドの中でも、2'-O-メチル化RNA、2'-F化RNAや架橋型核酸などの糖部修飾ヌクレオチドを含むポリヌクレオチドは、核酸医薬の免疫反応性の低減や核酸分解酵素に対する耐性の付与のいずれにも有効であることが示されている(非特許文献3)。
例えば、非特許文献5に、メラノーマ患者を対象とした人工mRNAがんワクチンの臨床試験では、がんワクチンの投与が開始されてから転移発症率が大幅に減少することが報告されているように、一定の成果も報告されつつある。
しかしながら、これらの臨床応用されている人工mRNAはIVTによって作製されたものである。IVTで製造される人工mRNAは以下の2つの課題を持つ。第1に、免疫反応性の低減や核酸分解酵素に対する安定性付与を目的として導入される修飾ヌクレオチドの導入位置は、制御ができない。2'-F化修飾RNAをIVTにより導入した人工mRNAでは、ペプチド翻訳能が減弱・消失する事例が特許文献1に開示されている。第2に、IVTで用いられるRNA合成酵素に基質として認識される修飾ヌクレオチド以外は導入不可能である。同じく、特許文献1には、2'-O-メチル化修飾RNAを含む人工mRNAは、一般的なRNAポリメラーゼを用いたIVT反応では調製が困難であることが開示されている。
したがって、IVTにより修飾ヌクレオチドを導入して作製された人工mRNAでは、修飾ヌクレオチドの位置及び種類に関して、十全な検討がなされているとは言い難い。
[1]
開始コドンから終止コドンまでの翻訳領域を含み、
前記翻訳領域がn個のコドンを含み、前記nが2以上の正の整数であり、
前記n個のコドンが、それぞれ、1番目、2番目及び3番目のヌクレオチドを含み、
前記n個のコドンのうちの少なくとも2個のコドンにおける1番目のヌクレオチドが、糖部修飾ヌクレオチドである、
ポリヌクレオチド。
[2]
前記糖部修飾ヌクレオチドが、少なくとも2'位が修飾された糖部を含む、[1]に記載のポリヌクレオチド。
[3]
前記少なくとも2'位が修飾された糖部が、以下から選択される、
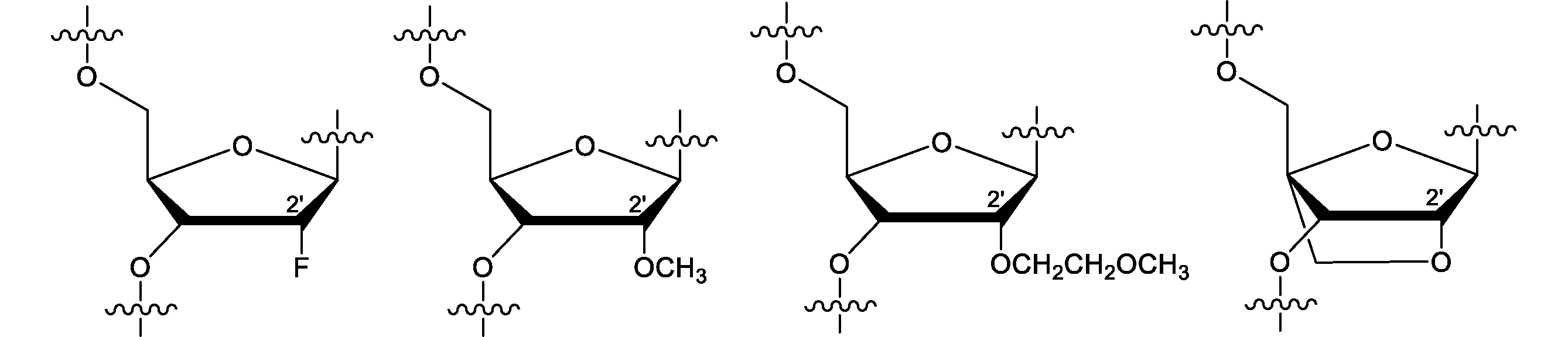
[4]
前記糖部修飾ヌクレオチドが、アデニン、グアニン、シトシン及びウラシルからなる群から選択される塩基に対応する塩基部を含み、
前記塩基の種類が少なくとも2種類である、[1]~[3]のいずれかに記載のポリヌクレオチド。
[5]
前記糖部修飾ヌクレオチドの少なくとも1個が、修飾塩基部を含む、[1]~[4]のいずれかに記載のポリヌクレオチド。
[6]
前記n個のコドンの全てにおける1番目のヌクレオチドが、糖部修飾ヌクレオチドである、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-1]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、5%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-2]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、10%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-3]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、15%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-4]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、20%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-5]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、25%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-6]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、30%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-7]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、35%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-8]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、40%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-9]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、45%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-10]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、50%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-11]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、55%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-12]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、60%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-13]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、65%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-14]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、70%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-15]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、75%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-16]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、80%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-17]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、90%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[6-18]
前記n個のコドンにおける1番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、95%以上である、[1]~[5]のいずれかに記載のポリヌクレオチド。
[7]
前記終止コドンの1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、[1]~[6-18]のいずれかに記載のポリヌクレオチド。
[8]
前記開始コドンの1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、[1]~[7]のいずれかに記載のポリヌクレオチド。
[9]
前記n個のコドンのうちの少なくとも1個のコドンにおける2番目のヌクレオチドが、糖部修飾ヌクレオチドである、[1]~[8]のいずれかに記載のポリヌクレオチド。
[9-1]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、50%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-2]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、45%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-3]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、40%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-4]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、35%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-5]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、30%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-6]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、25%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-7]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、20%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-8]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、15%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-9]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、10%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-10]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、5%以下である、[1]~[9]のいずれかに記載のポリヌクレオチド。
[9-11]
前記n個のコドンにおける2番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、0%である、[1]~[6-18]のいずれかに記載のポリヌクレオチド。
[10]
前記n個のコドンのうちの少なくとも1個のコドンにおける3番目のヌクレオチドが、糖部修飾ヌクレオチドである、[1]~[9-11]のいずれかに記載のポリヌクレオチド。
[10-1]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、100%である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-2]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、90%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-3]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、80%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-4]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、70%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-5]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、60%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-6]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、50%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-7]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、45%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-8]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、40%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-9]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、35%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-10]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、30%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-11]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、25%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-12]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、20%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-13]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、15%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-14]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、10%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-15]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、5%以下である、[1]~[10]のいずれかに記載のポリヌクレオチド。
[10-16]
前記n個のコドンにおける3番目のヌクレオチドが糖部修飾ヌクレオチドである割合が、0%である、[1]~[6-18]及び[9]~[9-11]のいずれかに記載のポリヌクレオチド。
[11]
前記nが2~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-1]
前記nが2~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-2]
前記nが2~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-3]
前記nが2~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-4]
前記nが5~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-5]
前記nが5~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-6]
前記nが5~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-7]
前記nが5~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-8]
前記nが10~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-9]
前記nが10~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-10]
前記nが10~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-11]
前記nが10~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-12]
前記nが50~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-13]
前記nが50~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-14]
前記nが50~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-15]
前記nが50~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-16]
前記nが100~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-17]
前記nが100~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-18]
前記nが100~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-19]
前記nが100~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-20]
前記nが200~2000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-21]
前記nが200~1500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-22]
前記nが200~1000の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[11-23]
前記nが200~500の整数である、[1]~[10-16]のいずれかに記載のポリヌクレオチド。
[12]
5'側非翻訳領域を更に含む、[1]~[11-23]のいずれかに記載のポリヌクレオチド。
[13]
前記5'側非翻訳領域が、以下の塩基部を含む塩基部修飾ヌクレオチドを含む、

[12]に記載のポリヌクレオチド。
[14]
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、[12]又は[13]に記載のポリヌクレオチド。
[15]
5'キャップ構造を更に含む、[12]~[14]のいずれかに記載のポリヌクレオチド。
[16]
3'側非翻訳領域を更に含む、[1]~[15]のいずれかに記載のポリヌクレオチド。
[17]
前記3'側非翻訳領域が、ポリA鎖を含む、[16]に記載のポリヌクレオチド。
[18]
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、[16]又は[17]に記載のポリヌクレオチド。
[19]
5'側非翻訳領域及び/又は3'側非翻訳領域が、糖部修飾ヌクレオチドを含む、[12]~[18]のいずれかに記載のポリヌクレオチド。
[20]
以下の構造を含む、

R1及びR2は、それぞれ独立して、H、OH、F又はOCH3を表し、
B1及びB2は、それぞれ独立して、塩基部を表し、
X1は、O、S又はNHを表し、
X2は、O、S、NH又は以下の構造を表し、

ただし、X1及びX2は、同時にOではない]
請求項[1]~[19]のいずれかに記載のポリヌクレオチド。
[21]
ホスホロチオエート構造を含む、請求項[1]~[20]のいずれかに記載のポリヌクレオチド。
[22]
前記n個のコドンのうちの少なくとも1個のコドンにおける1番目のヌクレオチドと2番目のヌクレオチドとが、ホスホロチオエートによって連結されている、[1]~[21]のいずれかに記載のポリヌクレオチド。
[23]
前記5'側非翻訳領域の5'末端から1~2番目のヌクレオチド、1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、[1]~[22]のいずれかに記載のポリヌクレオチド。
[24]
前記3'側非翻訳領域の3'末端から1~2番目のヌクレオチド、1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、[1]~[23]のいずれかに記載のポリヌクレオチド。
[25]
請求項[1]~[24]のいずれかに記載のポリヌクレオチドを含む、医薬組成物。
[1A]
疾患の治療に使用するための、[1]~[24]のいずれかに記載のポリヌクレオチド又は[25]に記載の医薬組成物。
[1B]
疾患を治療する方法であって、その必要のある患者に治療有効量の[1]~[24]のいずれかに記載のポリヌクレオチド又は[25]に記載の医薬組成物を投与することを含む方法。
[1C]
疾患を治療するための、[1]~[24]のいずれかに記載のポリヌクレオチド又は[25]に記載の医薬組成物の使用。
[1D]
疾患を治療するための医薬の製造における、[1]~[24]のいずれかに記載のポリヌクレオチドの使用。
[1E]
疾患を治療するための医薬の製造に使用するための、[1]~[24]のいずれかに記載のポリヌクレオチド。
[1F]
[1]~[24]のいずれかに記載のポリヌクレオチド又は[25]に記載の医薬組成物及び使用説明書を含む、疾患の治療に使用するためのキット。
[2A]
翻訳領域及び5'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチド。
[2B]
翻訳領域及び3'側非翻訳領域を含み、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチド。
[2C]
翻訳領域、5'側非翻訳領域及び3'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチド。
[2D]
翻訳領域及び5'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
[2E]
翻訳領域及び3'側非翻訳領域を含み、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
[2F]
翻訳領域、5'側非翻訳領域及び3'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されており、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
[2G]
翻訳領域及び5'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
[2H]
翻訳領域及び3'側非翻訳領域を含み、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
[2I]
翻訳領域、5'側非翻訳領域及び3'側非翻訳領域を含み、
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されており、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチド。
本発明の一実施形態は、開始コドンから終止コドンまでの翻訳領域を含み、前記翻訳領域がn個のコドンを含み、前記nが2以上の正の整数であり、前記n個のコドンが、それぞれ、1番目、2番目及び3番目のヌクレオチドを含み、前記n個のコドンのうちの少なくとも2個のコドンにおける1番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチドに関する。
本実施形態のポリヌクレオチドは、翻訳領域を含む。翻訳領域はコード配列(CDS)とも称される。一つのポリヌクレオチドが複数の翻訳領域を含んでいてもよい。翻訳領域は、開始コドンから終止コドン(又は終結コドンという)までの複数のコドンで構成され、かつ翻訳されてポリペプチドが合成される領域である。コドンは、ポリぺプチドを構成する各アミノ酸をコードする単位であり、前記単位は3個のヌクレオチドから構成される。
本実施形態のポリヌクレオチドでは、翻訳領域を構成する複数のコドンに含まれる1番目のヌクレオチドの少なくとも2個が、糖部修飾ヌクレオチドである。糖部修飾ヌクレオチドを含むコドンの位置は特に限定されない。1番目のヌクレオチドが糖部修飾ヌクレオチドである割合は、5%以上、10%以上、15%以上、20%以上、25%以上、30%以上、35%以上、40%以上、45%以上、50%以上、55%以上、60%以上、65%以上、70%以上、75%以上、80%以上、85%以上、90%以上、95%以上又は100%であることが好ましい。前記割合が100%とは、1番目のヌクレオチドの全てが糖部修飾ヌクレオチドであることを意味する。前記割合が大きいほど、酵素に対する安定性に優れる傾向にある。図2に、1番目のヌクレオチドの全てが糖部修飾ヌクレオチドである翻訳領域の概略図を示す。特に限定するものではないが、1番目のヌクレオチドが糖部修飾ヌクレオチドである場合、1番目のヌクレオチドの糖部の2'位の置換基はフッ素であることが好ましい。

Mは、R1、OR1、R2OR1、SH、SR1、NH2、NHR1、NR1 2、N3、CN、F、Cl、Br又はIであり、
R1は、それぞれ独立して、アルキル又はアリールであり、好ましくは炭素数1~6のアルキルであり、より好ましくは炭素数1~3のアルキルであり、
R2は、アルキレンであり、好ましくは炭素数1~6のアルキレンである]
炭素数1~3のアルキルとしては、例えば、メチル、エチル、プロピル及びイソプロピルが挙げられる。
・Locked Nucleic Acid (LNA) [Tetrahedron Letters, 38, 8735 (1997)及びTetrahedron, 54, 3607 (1998)];
・Ethylene bridged nucleic acid (ENA) [Nucleic Acids Research, 32, e175 (2004)];
・Constrained Ethyl (cEt) [The Journal of Organic Chemistry 75, 1569 (2010)];
・Amido-Bridged Nucleic Acid (AmNA) [Chem Bio Chem 13, 2513 (2012)];
・2'-O,4'-c-Spirocyclopropylene bridged nucleic acid (scpBNA) [Chem. Commun., 51, 9737 (2015)];
・tricycloDNA (tcDNA) [Nat. Biotechnol., 35, 238 (2017)];
・Unlocked Nucleic Acid (UNA) [Mol. Ther. Nucleic Acids 2, e103 (2013)];
・3'-フルオロヘキシトール核酸 (FHNA) [Nat. Biotechnol., 35, 238 (2017)];
・ペプチド核酸 (PNA) [Acc. Chem. Res., 32, 624 (1999)];
・オキシペプチド核酸 (OPNA) [J. Am. Chem. Soc., 123, 4653 (2001)];
・ペプチドリボ核酸 (PRNA) [J. Am. Chem. Soc., 122, 6900 (2000)]。
翻訳領域は塩基部修飾ヌクレオチドを含んでいてもよい。翻訳領域において塩基部修飾ヌクレオチドが存在する位置は、特に限定されない。塩基部修飾ヌクレオチドは、糖部修飾ヌクレオチド及び/又はリン酸部修飾ヌクレオチドであってもよい(言い換えると、塩基部修飾ヌクレオチドは修飾糖部及び/又は修飾リン酸部を更に含んでいてもよい。)。
翻訳領域はリン酸部修飾ヌクレオチドを含んでいてもよい。翻訳領域においてリン酸部修飾ヌクレオチドが存在する位置は、特に限定されない。リン酸部修飾ヌクレオチドは、糖部修飾ヌクレオチド及び/又塩基部修飾ヌクレオチドであってもよい(言い換えると、リン酸部修飾ヌクレオチドは修飾糖部及び/又は修飾塩基部を更に含んでいてもよい。)。
本実施形態のポリヌクレオチドは、5'側非翻訳領域(5'UTR)を更に含んでいてもよい。5'側非翻訳領域は、翻訳領域の上流(5'末端側)に存在し、ポリペプチド合成のための翻訳が行われない領域である。5'側非翻訳領域を構成するヌクレオチドの数は、好ましくは1~1000の整数であり、より好ましくは1~500の整数であり、更に好ましくは1~250の整数であり、特に好ましくは1~100の整数である。

修飾塩基部のアルキル基Rは、メチル又はエチルであることが好ましい。
本実施形態のポリヌクレオチドは、5'キャップ構造を更に含んでいてもよい。5'キャップ構造は、5'側非翻訳領域の上流に存在する。5'キャップ構造を含むことによって、翻訳活性が向上する傾向にある。
本実施形態のポリヌクレオチドは、3'側非翻訳領域(3'UTR)を更に含んでいてもよい。3'側非翻訳領域は、翻訳領域の下流に存在し、ポリペプチド合成のための翻訳が行われない領域である。
本実施形態のポリヌクレオチドは、以下の連結部を含んでいてもよい。

R1及びR2は、それぞれ独立して、H、OH、F又はOCH3を表し、
B1及びB2は、それぞれ独立して、塩基部を表し、
X1は、O、S又はNHを表し、
X2は、O、S、NH又は以下の構造を表し、

ただし、X1及びX2は、同時にOではない]

前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチドに関する。
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチドに関する。
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、ポリヌクレオチドに関する。
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されており、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記5'側非翻訳領域の5'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されており、
前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドであり、
前記3'側非翻訳領域の3'末端から1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、ポリヌクレオチドに関する。
本実施形態のポリヌクレオチドは、Kozak配列及び/又はRibosome Binding sequence (RBS)を更に含んでいてもよい。
本実施形態のポリヌクレオチドは、例えば、化学合成によって製造することができる。具体的には、公知の化学合成法を用いて、ポリヌクレオチド鎖を伸長させながら、所定の位置で所定の糖部修飾ヌクレオチドを導入することによって、本実施形態のポリヌクレオチドを製造することができる。公知の化学合成法としては、例えば、ホスホロアミダイト法、ホスホロチオエート法、ホスホトリエステル法、及びCEM法(Nucleic Acids Research, 35, 3287 (2007)を参照)が挙げられる。また、ABI3900ハイスループット核酸合成機(アプライドバイオシステムズ社製)を利用することもできる。
・テトラへドロン(Tetrahedron)、第48巻、第12号、2223-2311頁(1992年);
・カレント・プロトコールズ・イン・ヌクレイック・アシッズ・ケミストリー(Current Protocols in Nucleic Acids Chemistry)、John Wiley & Sons(2000年);
・プロトコールズ・フォー・オリゴヌクレオチズ・アンド・アナログズ(Protocols for Oligonucleotides and Analogs)、Human Press(1993年);
・ケミストリー・アンド・バイオロジー・オブ・アーティフィシャル・ヌクレイック・アシッズ(Chemistry and Biology of Artificial Nucleic Acids)、Wiley-VCH(2012年);
・ゲノムケミストリー 人工核酸を活用する科学的アプローチ、講談社(2003年);
・核酸化学のニュートレンド、化学同人(2011年)。
塩基部修飾ヌクレオチドの原材料となるホスホロアミダイト(f)の合成方法について以下に示す。

化合物(b)は、溶媒中、塩基存在下、化合物(a)と、例えば相当するシリル化剤を、0 ℃と80 ℃の間の温度で、10分間から3日間反応させることにより製造することができる。
溶媒としては、例えばDMF、DMA、NMP等が挙げられ、これらは単独でまたは混合して用いられる。
塩基としては、例えばイミダゾール、トリエチルアミン、ジイソプロピルエチルアミン等が挙げられる。
シリル化剤としては、例えば、ビス(トリフルオロメタンスルホン酸)ジ-tert-ブチルシリル等が挙げられる。
化合物(c)は、溶媒中、塩基存在下、化合物(b)と、相当するアルキル化剤を、0 ℃と150 ℃の間の温度で、10分間から3日間反応させることにより製造することができる。適当な添加剤により、反応を促進させることもできる。
溶媒としては、例えばDMF、ピリジン、ジクロロメタン、THF、酢酸エチル、1,4-ジオキサン、NMP等が挙げられ、これらを単独でまたは混合して用いられる。
塩基としては、例えば、水酸化ナトリウム水溶液、炭酸カリウム、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン等が挙げられる。
アルキル化剤としては、例えば、ヨウ化メチル、ヨウ化エチル、臭化メチル等が挙げられる。
添加化剤としては、例えば、テトラブチルアンモニウムブロミドが挙げられる。
化合物(d)は、溶媒中、化合物(c)とフッ素試薬を-80 ℃と200 ℃の間の温度で、10秒間から72時間反応させることにより製造することができる。このとき、塩基を加えることもできる。
フッ素試薬としては、例えばフッ化水素、フッ化水素酸トリエチルアミン塩、フッ化テトラブチルアンモニウム(TBAF)等が挙げられる。
塩基としては、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン等が挙げられる。
溶媒としては、例えばジクロロメタン、クロロホルム、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、N,N-ジメチルアセトアミド(DMA)、NMP、ジメチルスルホキシド(DMSO)等が挙げられる。
化合物(e)は、溶媒中、塩基存在下、化合物(d)と、相当するアルキル化剤を、0 ℃と150 ℃の間の温度で、10分間から3日間反応させることにより製造することができる。適当な活性化剤により、反応を促進させることもできる。
溶媒としては、例えばDMF、ピリジン、ジクロロメタン、THF、酢酸エチル、1,4-ジオキサン、NMP等が挙げられ、これらを単独でまたは混合して用いられる。
塩基としては、例えば、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン、2,6-ルチジン等が挙げられる。
アルキル化剤としては、例えば、トリチルクロリド、p,p'-ジメトキシトリチルクロリド等が挙げられる。
活性化剤としては、例えば、4-ジメチルアミノピリジン等が挙げられる。
化合物(f)は、溶媒中、化合物(e)と化合物(g)を、塩基存在下、0 ℃と100 ℃の間の温度で、10秒間から24時間反応させることにより製造することができる。
溶媒としては、例えばジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、NMP等が挙げられ、これらを単独でまたは混合して用いられる。
塩基としては、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン等が挙げられ、これらを単独でまたは混合して用いられる。

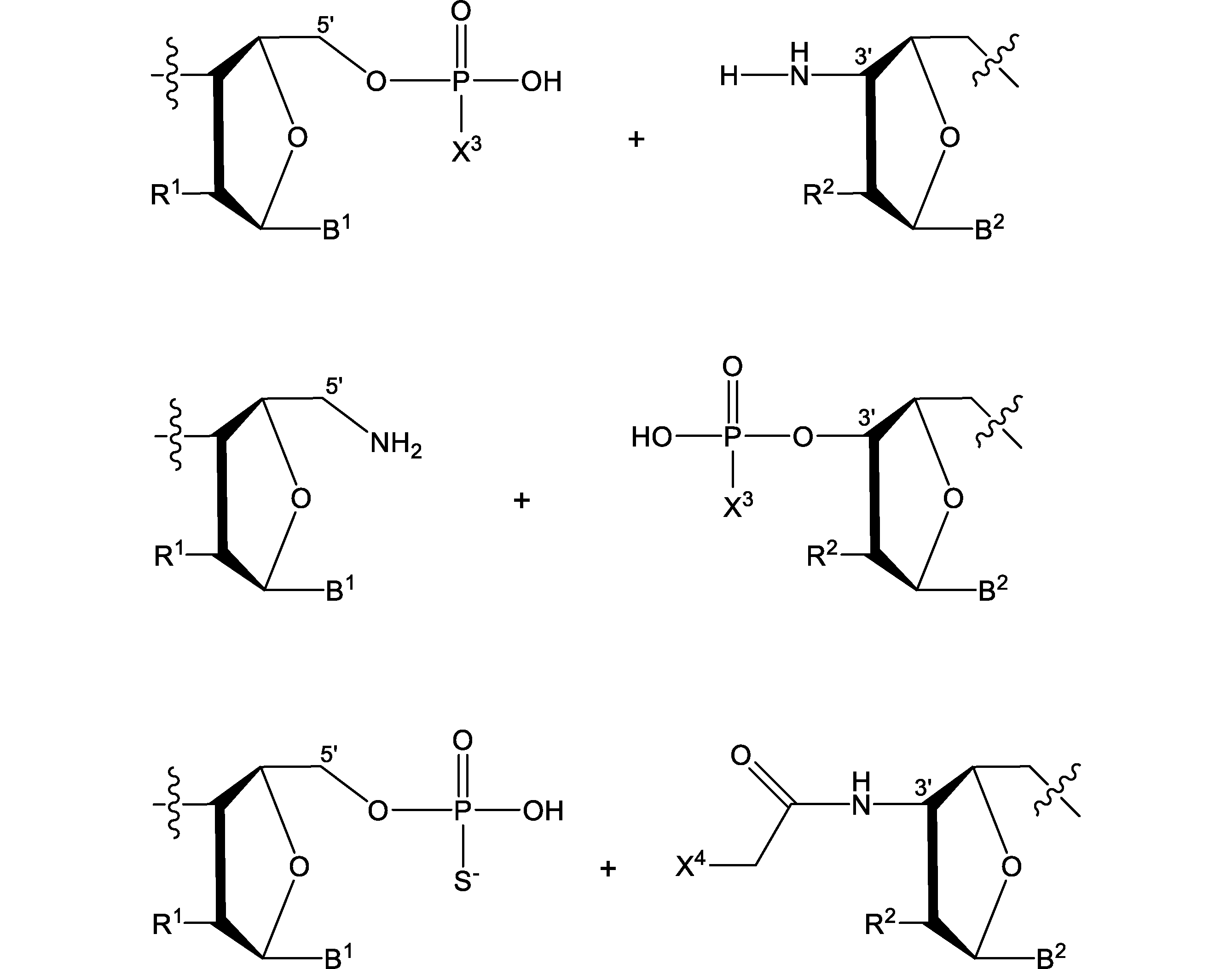
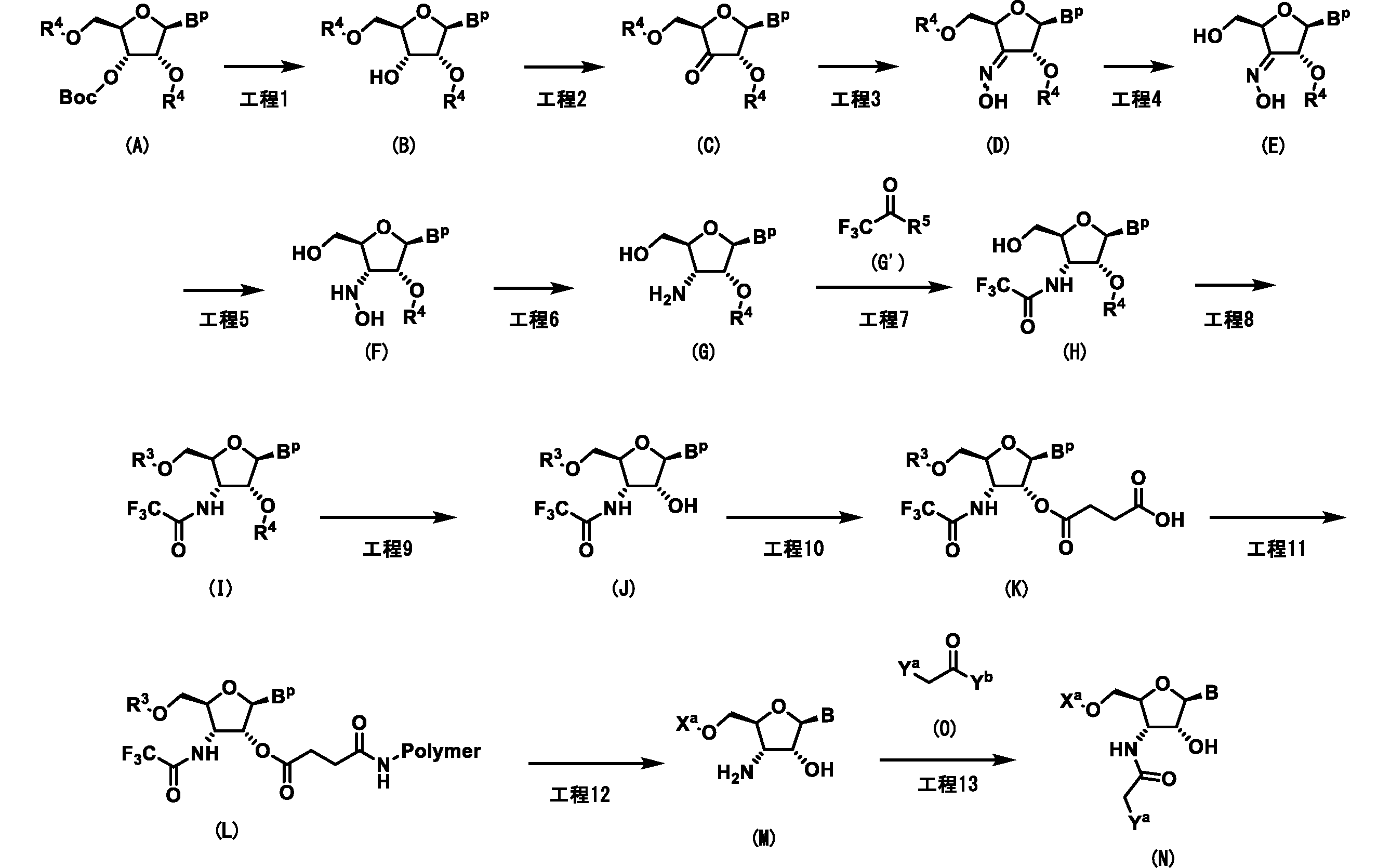
化合物(B)は、化合物(A)を溶媒中、60℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、トルエン、キシレン、1,2-ジクロロエタン、1,4-ジオキサン、N,N-ジメチルホルムアミド(DMF)、N-メチルピロリドン(NMP)、1,2-ジクロロベンゼン、水等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(A)は、例えば、J.Am.Chem.Soc.(1999),121,5661-5665.に記載の方法等により製造することができる。
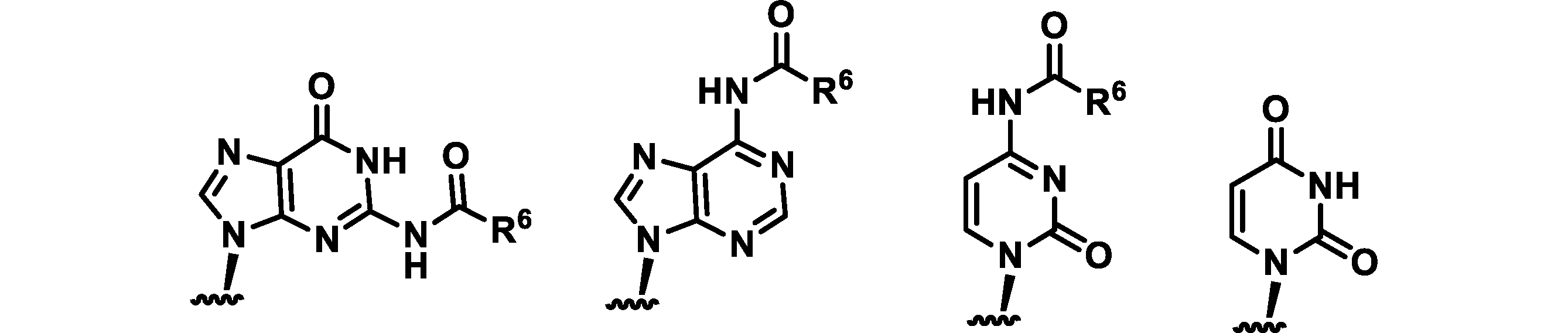
化合物(C)は、化合物(B)を溶媒中、1~100当量の酸化剤の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、好ましくは1~100当量の添加剤と反応させることで製造することができる。
溶媒としては、例えば、クロロホルムやジクロロメタン等の非プロトン性溶媒等が挙げられ、これらを単独で又は混合して用いることができる。
酸化剤としては、例えば、ジョーンズ試薬、クロム酸、二クロム酸ピリジニウム、四酸化ルテニウム、亜塩素酸ナトリウム、デス-マーチン(Dess-Martin)試薬等の有機系酸化剤又はクロロクロム酸ピリジニウム等の無機系酸化剤等が挙げられ、これらを単独で又は混合して用いることができる。
添加剤として、例えば、ピリジン、トリエチルアミン、N,N-ジイソプロピルエチルアミン等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(D)は、化合物(C)をピリジン等の溶媒中、ヒドロキシルアミン塩酸塩の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
化合物(E)は、化合物(D)を溶媒中、1~100000当量の脱保護剤の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、トルエン、キシレン、水等が挙げられ、これらを単独で又は混合して用いることができる。
脱保護剤としては、例えば、トリフルオロ酢酸、トリクロロ酢酸、酢酸、塩酸等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(F)は、化合物(E)を溶媒中、還元剤の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒は、例えば、トリフルオロ酢酸、トリクロロ酢酸、酢酸、塩酸、トルエン、キシレン、トルエン、キシレン、テトラヒドロフラン、メタノール、エタノール、1,4-ジオキサン、水等が挙げられ、これらを単独で又は混合して用いることができる。
還元剤は、例えば、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化ホウ素リチウム、トリアセトキシ水素化ホウ素ナトリウム等が挙げられる。
化合物(G)は、化合物(F)を溶媒中、触媒の存在下、水素雰囲気下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒は、例えば、トリフルオロ酢酸、酢酸、希塩酸、メタノール、エタノール、イソプロパノール、水等が挙げられ、これらを単独で又は混合して用いることができる。
触媒は、例えば、パラジウム炭素、ルテニウム炭素等が挙げられる。
化合物(G)は、例えば、国際公開第2017/123669号に記載の方法等でも製造することができる。
化合物(H)は、化合物(G)を溶媒中、1~100当量の化合物(G')及び塩基の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、好ましくは1~1000当量の塩基と反応させることにより製造することができる。
溶媒としては、例えば、メタノール、エタノール、イソプロパノール、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、テトラヒドロフラン(THF)、1,4-ジオキサン、N,N-ジメチルホルムアミド(DMF)、N-メチルピロリドン(NMP)、水等が挙げられ、これらを単独で又は混合して用いることができる。
塩基としては、例えば、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン、2,6-ルチジン等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(G')は、市販品を用いることができる。
化合物(I)は、化合物(H)とp,p'-ジメトキシトリチルクロリドを、ピリジン等の溶媒中、必要に応じて共溶媒の存在下、0℃と100℃の間の温度で、5分間~100時間、反応させることで製造することができる。
共溶媒としては、例えば、メタノール、エタノール、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トルエン、酢酸エチル、アセトニトリル、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン、ジオキサン、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド、N-メチルピロリドン、トリエチルアミン、N,N-ジイソプロピルエチルアミン、水等が挙げられ、これらは単独で又は混合して用いることができる。
化合物(J)は、化合物(I)を溶媒中、0℃と、用いる溶媒の沸点との間の温度で、10分間から10日間、1~10当量の添加剤と反応させることにより製造することができる。
溶媒としては、例えば、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、DMA、NMP等が挙げられ、これらを単独で又は混合して用いることができる。
添加剤としては、例えば、テトラブチルアンモニウムフルオリド、トリエチルアミン三フッ化水素酸塩等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(K)は、化合物(J)とコハク酸無水物を、溶媒中、1~30当量の塩基存在下、室温と200℃の間の温度で、5分間~100時間、反応させることで製造することができる。
溶媒としては、例えば、メタノール、エタノール、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トルエン、酢酸エチル、アセトニトリル、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン、ジオキサン、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド、N-メチルピロリドン、ピリジン、水等があげられ、これらは単独で又は混合して用いることができる。
塩基としては、例えば、炭酸セシウム、炭酸カリウム、水酸化カリウム、水酸化ナトリウム、ナトリウムメトキシド、カリウム tert-ブトキシド、トリエチルアミン、ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)、N,N-ジメチル-4-アミノピリジン(DMAP)等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(L)は、化合物(K)と、末端がアミノ化された固相支持体とを、無溶媒で又は溶媒中、1~30当量の塩基、縮合剤及び必要に応じて0.01~30当量の添加剤の存在下、室温と200℃の間の温度で、5分間~100時間反応した後、無水酢酸/ピリジン溶液中、室温と200℃の間の温度で5分間~100時間反応させることにより製造することができる。
溶媒としては、工程4で例示したものが挙げられる。
塩基としては、例えば、炭酸セシウム、炭酸カリウム、水酸化カリウム、水酸化ナトリウム、ナトリウムメトキシド、カリウム tert-ブトキシド、トリエチルアミン、ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)、N,N-ジメチル-4-アミノピリジン(DMAP)等が挙げられ、これらを単独で又は混合して用いることができる。
縮合剤としては、例えば、1,3-ジシクロヘキサンカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド・塩酸塩(EDC)、カルボニルジイミダゾール、ベンゾトリアゾール-1-イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルホロホスファート、(ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウム ヘキサフルオロホスファート、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウム ヘキサフルオロホスファート(HATU)、O-(ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウム ヘキサフルオロホスファート(HBTU)、ヨウ化 2-クロロ-1-メチルピリジニウム等が挙げられる。
添加剤としては、例えば、1-ヒドロキシベンゾトリアゾール(HOBt)、4-ジメチルアミノピリジン(DMAP)等が挙げられ、これらを単独で又は混合して用いることができる。
固相支持体としては、固相合成を行う上で公知のアミノ化された固相支持体が用いられる限り特に限定されるものではないが、例えば、長鎖アルキルアミノ基で修飾されたCPG(controlled pore glass)やPS(polystyrene resign)等の固相支持体が挙げられる。
例えば、長鎖アルキルアミン細孔性ガラス(LCAA-CPG)は、市販品を用いることができる。
化合物(M)は、化合物(L)を用い、公知のオリゴヌクレオチド化学合成法で、対応するヌクレオチド鎖を伸長した後に、固相からの脱離、保護基の脱保護及び精製を行うことで製造することができる。
固相からの脱離、脱保護は、オリゴヌクレオチド化学合成後、溶媒又は無溶媒中、-80℃から200℃の間の温度で、10秒間から72時間塩基で処理することにより製造することができる。
塩基としては、例えば、アンモニア、メチルアミン、ジメチルアミン、エチルアミン、ジエチルアミン、イソプロピルアミン、ジイソプロピルアミン、ピペリジン、トリエチルアミン、エチレンジアミン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)、炭酸カリウム等が挙げられ、これらを単独で又は混合して用いることができる。
溶媒としては、例えば、水、メタノール、エタノール、THF等が挙げられ、これらを単独で又は混合して用いることができる。
オリゴヌクレオチドの精製は、C18逆相カラムあるいは陰イオン交換カラム、好ましくは前述2つの手法を組み合わせにより可能である。
精製後の核酸複合体純度は、好ましくは90%以上、より好ましくは95%以上である。
化合物(N)は、化合物(M)を用い、緩衝液中、1~1000当量の化合物(O)の存在下、室温と100℃の間の温度で、5分間~100時間、反応させることで製造することができる。
緩衝液としては、例えば、酢酸緩衝液、トリス緩衝液、クエン酸緩衝液、リン酸緩衝液、水等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(O)は、市販品を用いることができる。
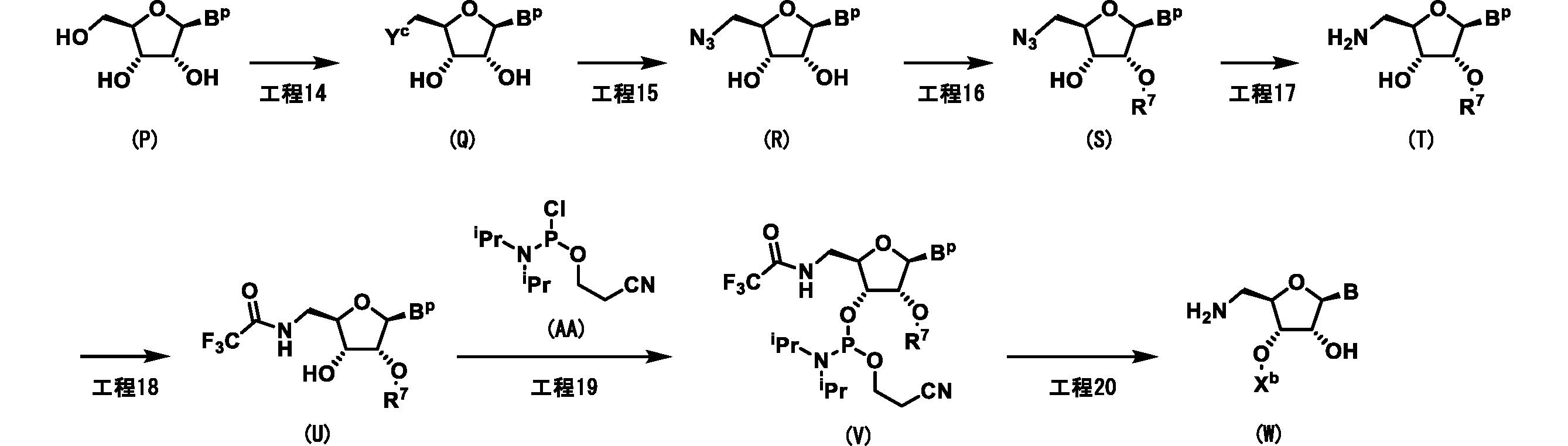
化合物(Q)は、化合物(P)を溶媒中、添加剤及び塩基の存在下、0℃と、用いる溶媒の沸点との間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、DMA、NMP等が挙げられ、これらを単独で又は混合して用いることができる。
添加剤としては、例えば、トシル酸無水物、トシルクロリド、チオニルクロリド、オキザリルクロリド等が挙げられ、これらを単独で又は混合して用いることができる。
塩基としては、例えば、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン、炭酸カリウム等が挙げられ、これらを単独又は混合して用いることができる。
化合物(P)は市販品を用いることができる。
化合物(R)は、化合物(Q)を溶媒中、アジド化剤と必要に応じて塩基の存在下、室温と、用いる溶媒の沸点の間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、DMA、NMP等が挙げられ、これらを単独で又は混合して用いることができる。
アジド化剤としては、例えば、アジ化ナトリウム等が挙げられる。
塩基としては、例えば、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン、炭酸カリウム等が挙げられ、これらを単独又は混合して用いることができる。
化合物(S)は、化合物(R)を溶媒中、シリル化剤と塩基の存在下、室温と、用いる溶媒の沸点の間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、DMA、NMP等が挙げられ、これらを単独で又は混合して用いることができる。
シリル化剤としては、例えば、tert-ブチルジメチルシリルクロリド、tert-ブチルジメチルシリルトリフラート、トリエチルシリルクロリド等が挙げられる。
塩基としては、例えば、ピリジン、トリエチルアミン、N-エチル-N,N-ジイソプロピルアミン、炭酸カリウム、水酸化カリウム、水酸化ナトリウム、ナトリウムメトキシド、カリウム tert-ブトキシド、トリエチルアミン、ジイソプロピルエチルアミン、N-メチルモルホリン、ピリジン、1,8-ジアザビシクロ[5.4.0]-7-ウンデセン(DBU)、N,N-ジメチル-4-アミノピリジン(DMAP)等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(T)は、化合物(S)を溶媒中、還元剤を加え、室温と、用いる溶媒の沸点の間の温度で、10秒間から3日間、反応させることで製造することができる。
溶媒としては、例えば、メタノール、エタノール、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、トルエン、酢酸エチル、アセトニトリル、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン、ジオキサン、N,N-ジメチルホルムアミド(DMF)、N,N-ジメチルアセトアミド、N-メチルピロリドン、トリエチルアミン、N,N-ジイソプロピルエチルアミン、酢酸、水等が挙げられ、これらは単独で又は混合して用いることができる。
還元剤としては、例えば、水素化ホウ素ナトリウム、水素化シアノホウ素ナトリウム、水素化ホウ素リチウム、トリアセトキシ水素化ホウ素ナトリウム、水素雰囲気下におけるパラジウム炭素等が挙げられる。
化合物(U)は、化合物(T)を用いて、工程7と同様にして製造することができる。
化合物(V)は、溶媒中、化合物(U)と化合物(AA)を、塩基存在下、0℃と100℃の間の温度で、10秒間から24時間、反応させることで製造することができる。
溶媒としては、例えば、ジクロロメタン、アセトニトリル、トルエン、酢酸エチル、THF、1,4-ジオキサン、DMF、NMP等が挙げられ、これらを単独で又は混合して用いることができる。
塩基としては、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン、ピリジン等が挙げられ、これらを単独で又は混合して用いることができる。
化合物(AA)は、市販品を用いることができる。
化合物(W)は、化合物(V)を用いて、工程12と同様にして製造することができる。
・アールエヌエー・メソッズ・イン・モレキュラーバイオロジー・メソッズ・アンド・プロトコルズ(RNA, Methods in Moleculer Biology (Methods and Protocols))、第703巻、第3章(2011年);
・カルジアック・ジーン・セラピー・メソッズ・イン・モレキュラーバイオロジー・メソッズ・アンド・プロトコルズ(Cardiac Gene Therapy: Methods in Moleculer Biology (Methods and Protocols))、第1521巻、第8章(2016年);
・ジャーナル・オブ・モレキュラー・バイオロジー(Journal of Molecular Biology)、第249巻、398ページ~408ページ(1995)
本発明の一実施形態は、前記ポリヌクレオチドを含む医薬組成物に関する。本実施形態の医薬組成物を、疾患を有する患者に投与することにより、ポリヌクレオチドが翻訳され、前記ポリヌクレオチドがコードするポリペプチドが合成され、前記疾患が治療される。

N-(9-((2R,3S,4S,5R)-3-(tert-butyldimethylsilyloxy)-5-((tert-butyldimethylsilyloxy)methyl)-4-hydroxy-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
文献記載の方法(J. Am. Chem. Soc., 1999, 121, 5661-5665)等により得られる化合物3を用い、1,2-ジクロロベンゼン (2.0 mL) に溶解させ、油浴上 (160℃) にて4時間撹拌した。反応液を室温まで戻した後、濃縮せずにフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 40:1)にて精製し、化合物4を白色固体として得た (0.31 g, 収率53%)。
1H NMR (400 MHz, CDCl3) δ 12.01 (1H, s), 8.50 (1H, s), 8.07 (1H, s), 5.86 (1H, d, J = 6.0 Hz), 4.47 (1H, s), 4.24 - 4.23 (1H, m), 4.22 - 4.21 (1H, m), 3.93 (1H, dd, J = 11.6, 2.0 Hz), 3.82 (1H, dd, J = 11.6, 2.0 Hz), 2.66 (1H, sept., J = 6.8 Hz), 1.27 (3H, d, J = 6.8 Hz), 1.25 (3H, d, J = 6.8 Hz), 0.93 (9H, s), 0.82 (9H, s), 0.13 (3H, s), 0.12 (3H, s), -0.07 (3H, s), -0.20 (3H, s)
13C NMR (100 MHz, CDCl3) δ 179.0, 155.7, 148.5, 148.7, 147.8, 136.8, 121.0, 87.4, 85.4, 77.6, 71.8, 63.6, 36.2, 25.9, 25.4, 19.1, 18.7, 18.3, 17.8, -5.3, -5.4, -5.5, -5.6
ESI-HRMS:calcd for C26H48N5O6Si2 582.31[M+H]+, found : 582.31[M+H]+
N-(9-((2R,3S,5S)-3-(tert-butyldimethylsilyloxy)-5-((tert-butyldimethylsilyloxy) methyl)-4-(hydroxyimino)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl) isobutyramide
クロム酸 (129 mg, 1.29 mmol)の無水ジクロロメタン (2.0 mL)溶液にモレキュラーシーブ3A (粉末状)(258 mg)を加えて氷浴上にて冷却した。この溶液に撹拌下、無水ピリジン (207 μL, 1.29 mmol)を滴下し、氷浴上にて撹拌した。30分後、無水酢酸 (122 μL, 1.29 mmol)を滴下して氷浴上にて撹拌した。30分後、化合物4 (250 mg, 0.43 mmol)のジクロロメタン (1.3 mL)溶液を滴下して室温にて2時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液を酢酸エチルで希釈しシリカパッド (2 cm厚)にてろ過、濾液を減圧濃縮して無色固体が得られた。この粗生成物4'はこのまま次の反応に用いた。
粗生成物4' (0.43 mmolとして)のピリジン (4 mL)溶液にヒドロキシルアミン塩酸塩 (299 mg, 4.30 mmol)を加えて室温にて撹拌した。24時間後、反応液を減圧濃縮し残渣に水を加えて酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウム上にて乾燥させた。有機層を減圧濃縮し、残渣をフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 40 : 1)にて精製し、化合物5を白色固体として得た (255 mg, 収率68% for 2 steps)。
1H NMR (400 MHz, CDCl3) δ 12.14 (1H, s), 9.27 (1H, s), 8.78 (1H, s), 8.11 (1H, s), 5.78 (1H, d, J = 7.6 Hz), 5.09 (1H, s), 4.92 (1H, d, J = 7.2 Hz), 4.14 (1H, d, J = 11.4 Hz), 3.92 (1H, d, J = 11.4 Hz), 2.79 - 2.74 (1H, m), 1.27 - 1.21 (6H, m), 0.91 (9H, s), 0.71 (9H, s), 0.10 (3H, s), 0.07 (3H, s), -0.10 (3H, s), -0.23 (3H, s)
13C NMR (100 MHz, CDCl3) δ 178.9, 157.8, 155.6, 148.7, 147.8, 136.8, 120.8, 87.5, 86.5, 62.2, 36.3, 25.9, 25.5, 25.2, 19.1, 18.8, 18.3, 18.0, -5.0, -5.5, -5.6, -5.7
ESI-HRMS : calcd for C26H47N6O6Si2 595.31[M+H]+, found : 595.31[M+H]+
N-(9-((2R,3S,5S)-3-(tert-butyldimethylsilyloxy)-4-(hydroxyimino)-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
化合物5 (129 mg, 0.22 mmol)に氷冷した90%トリフルオロ酢酸水溶液 (1.0 mL)を加えて氷浴上にて30分間撹拌した。反応液を減圧濃縮し、得られた残渣をトルエンと水 (1 : 1, v/v)にて減圧下にて3回共沸させた。得られた残渣をフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 50 : 1から40 : 1)にて精製し、化合物6を白色固体として得た (96 mg, 収率92%)。
1H NMR (400 MHz, CD3OD) δ 8.36 (1H, s), 5.87 (1H, d, J = 7.6 Hz), 5.18 (1H, dd, J = 7.6, 2.0 Hz), 5.02 (1H, d, J = 2.0 Hz), 4.11 (1H, dd, J = 12.0, 2.0 Hz), 3.92 (1H, d, J = 12.0, 2.0 Hz), 2.71 (1H, sept., J = 7.2 Hz), 1.21 (6H, d, J = 7.2 Hz), 0.72 (9H, s), 0.00 (3H, s), -0.16 (3H, s)
13C NMR (100 MHz, CD3OD) δ 181.8, 157.4, 156.8, 151.0, 150.0, 139.8, 121.3, 88.4, 79.7, 76.5, 61.6, 36.9, 25.9, 19.4, 19.2, -4.5, -5.5
ESI-HRMS : calcd forC20H32N6NaO6Si 503.21[M+Na]+, found : 503.20[M+Na]+
N-(9-((2R,3S,4S,5S)-4-amino-3-(tert-butyldimethylsilyloxy)-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
化合物6 (93 mg, 0.19 mmol)の酢酸 (1.9 mL)溶液に水素化ホウ素ナトリウム(15 mg, 0.38 mmol)を加えて室温にて1時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液を減圧濃縮し、残渣を酢酸エチルに溶解させ、飽和食塩水で洗浄、無水硫酸ナトリウム上にて乾燥させた。有機層を減圧濃縮し、得られた残渣をフラッシュカラムクロマトグラフィー(中性シリカゲル、ジクロロメタン/メタノール = 20 : 1)にて精製し、化合物7を白色固体として得た(51 mg, 収率55%)。
1H NMR (400 MHz, CD3OD) δ 8.34 (1H, s), 6.06 (1H, d, J = 6.0 Hz), 4.75 (1H, t, J = 6.4 Hz), 4.27 (1H, d, J = 2.8 Hz), 3.86 (1H, dd, J = 12.4, 2.0 Hz), 3.73 (1H, d, J = 12.4, 2.0 Hz), 3.62 - 3.60 (1H, m), 2.71 (1H, sept., J = 6.8 Hz), 1.21 (6H, d, J = 6.8 Hz), 0.82 (9H, s), -0.02 (3H, s), -0.23 (3H, s)
13C NMR (100 MHz, CD3OD) δ 181.8, 157.4, 150.8, 149.8, 139.6, 139.4, 121.1, 89.7, 84.5, 77.7, 65.4, 65.2, 36.9, 26.0, -5.2, -5.3
ESI-HRMS : calcd for C20H35N6O6Si 483.24[M+H]+, found : 483.23[M+H]+
N-(9-((2R,3S,4S,5S)-4-amino-3-(tert-butyldimethylsilyloxy)-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
化合物7 (50 mg, 0.10 mmol)の90%酢酸水溶液 (1.5 mL)溶液に10%パラジウム炭素 (20 mg)を加え、水素雰囲気下、室温にて18時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液をメタノールにて希釈しパラジウム炭素をセライト濾過にて除去した。濾液を減圧濃縮し、得られた残渣をフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 15 : 1から10 :1)にて精製し、化合物8を白色固体として得た (酢酸塩として41 mg, 収率75%)。
1H NMR (400 MHz, CD3OD) δ 8.32 (1H, s), 5.99 (1H, s), 4.60 (1H, s), 3.95 - 3.68 (4H, m), 2.73 (1H, br. s), 1.22 (6H, br. s), 0.05 (3H, s), -0.06 (3H, s)
ESI-HRMS : calcd for C20H34N6NaO5Si 489.2258[M+Na]+, found : 489.2231[M+Na]+
N-(9-((2R,3S,4R,5S)-3-(tert-butyldimethylsilyloxy)-5-(hydroxymethyl)-4-(2,2,2-trifluoroacetamido)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
文献既知(WO2017/123669)の化合物8 (40 mg, 0.076 mmol)とトリエチルアミン(45 L, 0.38 mmol)のメタノール溶液 (0.76 mL)溶液にトリフルオロ酢酸エチル (0.76 mL)を加え、室温にて24時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液を減圧濃縮し、得られた残渣をフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 20 : 1から12 :1)にて精製し、化合物9を白色固体として得た (12 mg, 収率28%)。
1H NMR (400 MHz, CDCl3) δ 12.26 (1H, s), 10.11 (1H, s), 7.76 (1H, s), 7.26 (1H, d, J = 3.6 Hz), 5.71 (1H, d, J = 3.6 Hz), 4.98 (1H, dd, J = 6.8 Hz), 4.78 (1H, dd, J = 6.8, 3.6 Hz), 4.21 (1H, d, J = 6.8 Hz), 4.03 (1H, dd, J =11.2 Hz), 3.82 (1H, dd, J =11.2 Hz), 2.79 (1H, sept., J = 6.8 Hz), 1.26 (3H, d, J = 6.8 Hz), 1.24 (3H, d, J = 6.8 Hz), 0.85 (9H, s), -0.01 (3H, s), -0.11 (3H, s)
13C NMR (100 MHz, CDCl3) δ 179.8, 158.0, 157.7, 157.3, 156.9, 155.2, 148.3, 147.3, 138.6, 122.0, 120.0, 117.1, 114.2, 111.3, 91.6, 83.7, 74.5, 61.3, 51.0, 36.1, 25.2, 18.9, 17.7, -5.0, -5.4
ESI-HRMS : calcd for C22H33F3N6NaO6Si 585.21[M+Na]+, found : 585.21[M+Na]+
N-(9-((2R,3S,4R,5S)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-3-(tert-butyldimethylsilyloxy)-4-(2,2,2-trifluoroacetamido)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
化合物9 (10 mg, 0.017 mmol)の無水ピリジン (1 mL)溶液にジメトキシトリチルクロライド (18 mg, 0.053 mmol)を加え、室温にて1.5時間撹拌した。その後ジメトキシトリチルクロライド (18 mg, 0.053 mmol)を追加し、室温にて30分間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液にメタノール (1 mL)を加えた後、減圧濃縮した。残渣を酢酸エチルに溶解させ、水、次いで飽和食塩水で洗浄した。有機層を無水硫酸ナトリウム上にて乾燥させ、減圧濃縮して得られた残渣をフラッシュカラムクロマトグラフィー(中性シリカゲル、ヘキサン/酢酸エチル = 5 : 1から2 :1)にて精製し、化合物10を白色固体として得た (15.2 mg, 収率99%)。
1H NMR (400 MHz, CDCl3) δ 11.99 (1H, s), 10.11 (1H, s), 8.07 (1H, s), 7.81 (1H, s), 7.45 (2H, dd, J = 8.2, 2.0 Hz), 7.32 (4H, dd, J = 9.2, 3.6 Hz), 7.24 - 7.29 (3H, m), 7.01 (1H, d, J = 7.2 Hz), 6.76 (4H, J = 9.2, 3.6 Hz), 5.71 (1H, d, J = 4.2 Hz), 5.16 (1H, dd, J = 6.4, 4.2 Hz), 4.20 - 4.17 (1H, m), 3.76 (3H, s), 3.75 (3H, s), 3.56 (1H, dd, J = 11.2, 2.8 Hz), 3.22 (1H, dd, J = 11.2, 2.8 Hz), 1.82 (1H, d, J = 6.8 Hz), 0.97 (3H, d, J = 6.8 Hz), 0.68 (9H, s), 0.79 (3H, d, J = 6.8 Hz), 0.04 (3H, s), -0.06 (3H, s)
13C NMR (100 MHz, CDCl3) δ 171.2, 158.7, 158.0, 157.6, 157.2, 156.8, 155.4, 147.6, 147.2, 144.8, 139.2, 135.9, 135.4, 130.0, 127.9, 127.1, 122.6, 120.0, 117.0, 114.1, 111.2, 90.1, 86.3, 81.7, 73.4, 62.4, 60.4, 55.2, 51.4, 36.1, 25.4, 18.4, 17.8, -5.0, -5.3
ESI-HRMS : calcd for C43H52F3N6O8Si 865.36[M+H]+, found : 865.35[M+H]+
N-(9-((2R,3S,4S,5S)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-3-hydroxy-4-(2,2,2-trifluoroacetamido)-tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
化合物10 (14 mg, 0.016 mmol)のテトラヒドロフラン (1 mL)溶液にテトラブチルアンモニウムフルオリド (1Mテトラヒドロフラン溶液、19 μL, 0.019 mmol)を加え、室温にて1時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液を減圧濃縮した。得られた残渣をフラッシュカラムクロマトグラフィー (中性シリカゲル、ジクロロメタン/メタノール = 30 : 1から15 :1)にて精製し、化合物11を白色固体として得た (10.8 mg, 収率83%)。
1H NMR (400 MHz, CDCl3) δ 12.12 (1H, br. s), 8.76 (1H, br. s), 7.74 (1H, s), 7.81 (1H, s), 7.68 (1H, d, J = 5.4 Hz), 7.48 (2H, d, J = 7.6 Hz), 7.37 (2H, d, J = 9.2 Hz), 7.34 (2H, d, J = 9.2 Hz), 7.25 - 7.21 (2H, m), 7.17 (1H, t, J = 7.2 Hz), 6.81 (2H, d, J = 9.2 Hz), 6.78 (2H, d, J = 9.2 Hz), 5.80 (1H, d, J = 4.0 Hz), 5.35 (1H, br. s), 5.08 (1H, dd, J = 12.4, 6.4 Hz), 4.30 - 4.29 (1H, m), 3.76 (3H, s), 3.74 (3H, s), 3.57 - 3.53 (1H, m), 3.29 - 3.26 (1H, m), 1.84 - 1.57 (1H, m), 0.94 (3H, d, J = 6.8 Hz), 0.68 (3H, d, J = 6.8 Hz)
13C NMR (100 MHz, CDCl3) δ 179.4, 158.6, 158.4, 158.0, 157.6, 157.3, 147.8, 147.2, 144.8, 139.4, 136.3, 135.7, 130.1, 129.9, 128.1, 128.0, 127.0, 121.0, 120.0, 117.1, 114.2, 111.3, 91.2, 86.1, 82.5, 71.5, 62.7, 55.1, 51.2, 35.9, 18.5, 18.2
ESI-HRMS : calcd for C37H38F3N6O8 751.27[M+H]+, found : 751.27[M+H]+
化合物11 (0.90 g, 1.20 mmol)、トリエチルアミン (0.42 mL, 3.0 mmol)のアセトニトリル (12 mL)溶液にコハク酸無水物 (0.24 g, 2.40 mmol)、ジメチルアミノピリジン (29 mg, 0.24 mmol)を加え、室温にて1時間撹拌した。薄層クロマトグラフィーにて原料の消失を確認後、反応液を減圧濃縮した。残渣を酢酸エチルに溶解させ、飽和炭酸水素ナトリウム水溶液で2回、次いで飽和食塩水で洗浄した。有機層を無水硫酸ナトリウム上にて乾燥させ、減圧濃縮した。残渣に対してジクロロメタン/メタノール溶液 (1 : 1, v/v)にて減圧濃縮による共沸操作を行うことで、白色泡状固体 (トリエチルアミン塩として1.11 g, 97%)を得た。この化合物12はこのまま次の反応に用いた。

N-(9-((2R,3R,5S)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-3-((tert-butyldimethylsilyl)oxy)-4-(hydroxyimino)tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-2-yl)isobutyramide
アルゴン雰囲気下、化合物 13 (ChemGenes社製, 5.0 g, 6.5 mmol) を脱水ジクロロメタン (50 mL) に溶解させ、氷浴で冷却しながら撹拌した。反応液を冷却しつつ炭酸水素ナトリウム (8.2 g, 97.3 mmol)、nor-AZADO (36 mg, 0.260 mmol) を加え、ヨードベンゼンジアセタート (3.14 g, 9.73 mmol) を内温上昇に注意しながら分割して加え、室温に昇温しながら21 時間10 分間撹拌した。原料の消失を確認後、イソプロピルアルコール (7.5 mL) を反応液へ加え4 時間撹拌した (過剰酸化剤のクエンチ)。反応液を氷水に加え、更にクロロホルムを加えて分液し、水層を再度クロロホルムで抽出した。有機層を合一後、水で1 回、飽和食塩水で1 回洗浄した後、無水硫酸ナトリウムで脱水した。乾燥剤をろ別後、ろ液を濃縮することで粗生成物 (9.01 g,一部DMTr 基が脱保護された化合物を含む) を橙色固体として得た。
アルゴン雰囲気下、粗生成物 (9.01 g) を脱水ピリジン (40 mL) に溶解し、氷浴で冷却しながら撹拌した。反応液を冷却しつつヒドロキシルアミン塩酸塩 (4.06 g, 58.7 mmol)を加え、室温に昇温しながら17 時間25 分間撹拌した。原料の消失を確認後、反応液をナスフラスコにクロロホルム (1%トリエチルアミン含有) で洗い込みながら移し、濃縮した。残渣を飽和重曹水に加え15 分間撹拌した後、クロロホルムで2 回抽出した。有機層を合一後、飽和食塩水で1 回洗浄した後、無水硫酸ナトリウムで脱水した。乾燥剤をろ別後、ろ液を濃縮することで化合物 14 (4.13 g, ジアステレオマー混合物、2段階収率81%) を橙色泡状物質として得た。
1H NMR (400MHz, CDCl3) δ: 12.04 (1H, d, J = 23.3 Hz), 9.23 (1H, s), 8.49 (1H, s), 7.89 (1H, s), 7.79 (1H, s), 7.66-7.58 (2H, m), 7.49-7.39 (4H, m), 7.31-7.14 (5H, m), 6.81-6.76 (2H, m), 6.73-6.68 (2H, m), 5.92 (1H, dd, J = 8.0, 1.6 Hz), 5.83 (1H, d, J = 3.7 Hz), 5.64 (1H, d, J = 8.2 Hz), 5.54 (1H, dd, J = 3.9, 1.1 Hz), 5.01 (1H, t, J = 7.3 Hz), 3.80-3.73 (6H, m), 3.54-3.46 (2H, m), 1.28 (1H, m), 1.08 (1H, d, J = 6.9 Hz), 0.99 (1H, d, J = 6.9 Hz), 0.86-0.76 (9H, m), 0.47 (2H, m), 0.11 (1H, s), 0.02--0.02 (3H, m), -0.07 (2H, s)[ジアステレオマー混合物]
ESI-HRMS : calcd for C41H50N6O8Si 781.97[M-H]-, found : 781.84[M-H]-
工程10で得られた化合物14(3.80 g)を用いて、工程3と同様にして、化合物6(2.12 g, 4.41 mmol, 収率91%)を得た。
なお、化合物6の詳細データについては、工程3に記載の通りである。

化合物12の固相上への担持量は以下に示す方法によって算出した。得られた固相担体を規定量とり、デブロッキング試薬 (3 w/v%トリクロロ酢酸/ジクロロメタン溶液)の添加により生じた4,4'-ジメトキシトリチルカチオンの発色を紫外可視吸光光度測定 (石英セル、セル長 : 10 mm)によって測定した。504 nmにおける吸光度と4,4'-ジメトキシトリチルカチオンのモル吸光係数 (波長504 nm : 76,000)から固相上への化合物12の担持量をLambert-Beer式により算出した。すなわち、得られた固相担体 (2.0 mg)を2 mLメスフラスコに量りとり、デブロッキング試薬を加えて全量を2 mLとして転倒混和したものを測定サンプルとした。3 w/v%トリクロロ酢酸/ジクロロメタン溶液を用いてブランク測定を行った後、測定サンプルを用いて測定を行った。504 nmにおける吸光度:0.377より、担持量:24.8 μmol/g)

N-(9-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)benzamide
アルゴン雰囲気下、10Lの4口フラスコに市販のN6-Benzyladenosine(化合物16)(100 g, 269 mmol, 1.0 eq.)、アセトン(2.70 L)、ジメトキシプロパン(166 mL, 1.35 mol, 5.0 eq.)を順次加えた。反応溶液に、濃硫酸(1.44 mL, 26.9 mmol, 0.10 eq.)を加え、室温で15時間攪拌した。原料の残存を確認したので、濃硫酸(1.44 mL, 26.9 mmol, 0.10 eq.)を追加し、24時間攪拌した。原料の残存を確認したので、濃硫酸(1.44 mL, 26.9 mmol, 0.10 eq.)を添加し1時間30分攪拌した後、濃硫酸(2.87 mL, 53.8 mmol, 0.20 eq.)を添加し、4時間攪拌した。
LC/MSで反応の進行を確認後、反応液を氷浴で冷却し、内温が3~5℃となるように飽和炭酸水素ナトリウム水溶液(400 mL)を5分間かけて滴下し、溶液を中性にした。減圧下、反応液を濃縮し、残渣に蒸留水(2.0 L)を加えた。溶液をクロロホルム(1.0 L)で3回抽出し、有機層を無水硫酸ナトリウムで脱水した。ろ過後、溶媒を減圧留去し、化合物17(222 g)を得た。得られた化合物17は更なる精製操作を実施せず、次工程に用いた。
((3aR,4R,6R,6aR)-6-(6-benzamido-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl methanesulfonate
アルゴン雰囲気下、2 Lの4口フラスコに、工程12で得られた化合物17(222 g)、ピリジン(520 mL)を加え、反応液を氷浴で冷却し、内温が4℃~9℃となるようにメタンスルホニルクロリド(25.0 mL, 321 mmol, 1.2 eq.)を15分間かけて滴下し、2時間攪拌した。
LC/MSで反応の進行を確認後、反応液に蒸留水(500 mL)を加え、溶液を酢酸エチル(1.0 L)で3回抽出した後、有機層を1N 塩酸(1.0 L×1, 500 mL×2)、飽和炭酸水素ナトリウム水溶液(500 mL×2)、飽和食塩水(500 mL ×2)で順次洗浄し、無水硫酸ナトリウムで脱水した。ろ過後、溶媒を減圧留去し、得られた残渣をトルエンで共沸することで化合物18(150 g, 17.6 wt %トルエン含有)を得た。得られた化合物18は更なる精製操作を実施せず、次工程に用いた。
N-(9-((3aR,4R,6R,6aR)-6-(azidomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)benzamide
アルゴン雰囲気下、3 Lの4口フラスコに、工程13で得られた化合物18(150 g)と脱水DMF(1.26 L)を加えた。反応液にアジ化ナトリウム(82.8 g, 1.26 mol, 5.0 eq.)を添加し、30分間かけて60℃まで昇温し、60℃で3時間30分間攪拌した。
LC/MSで反応の進行を確認後、反応液を室温まで徐冷し、蒸留水(1.0 L)と酢酸エチル(600 mL)を加えた。得られた溶液に蒸留水(3.0 L)を加え、水層を酢酸エチル(500 mL)で6回抽出した。有機層を蒸留水(800 mL)で2回、飽和食塩水(800 mL)で2回洗浄し、無水硫酸ナトリウムで脱水した。ろ過後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(SiO2 700 g, 酢酸エチル)で精製することで、化合物19(55.7 g, 128 mmol, 収率48% (3 steps from 化合物16))を得た。
N-(9-((3aR,4R,6R,6aR)-2,2-dimethyl-6-((2,2,2-trifluoroacetamido)methyl) tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)benzamide
アルゴン雰囲気下、3 Lの4口フラスコに、工程14で得られた化合物19(55.7 g, 128 mmol, 1.0 eq.)、メタノール(1.28 L)を加えた。反応液に、10% Pd/C(76.8 g, 21.2 mmol, 0.17 eq.)を添加し、反応液中を水素で置換し、室温で16時間攪拌した。
LC/MSで反応の進行を確認後、反応液中をアルゴンガスで置換し、反応液をセライトろ過した。ろ液を減圧濃縮した後、得られた残渣をメタノール(985 mL)に溶解し、3 Lの4口フラスコに移した。溶液を氷浴で冷却し、内温が2~4℃となるように1-(トリフルオロアセチル)イミダゾール(17.0 mL, 149 mmol, 1.2 eq.)を15分間かけて滴下し、4℃で2時間攪拌した。LC/MSで反応の進行を確認後、反応液を減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(SiO2 800 g, ヘプタン/酢酸エチル=1:4)で精製することで、化合物20(21.4 g, 42.2 mmol, 収率33%)を得た。
N-(9-((2R,3R,4S,5R)-3,4-dihydroxy-5-((2,2,2-trifluoroacetamido)methyl) tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide
1Lのナスフラスコに、工程15で得られた化合物20(10.0 g, 19.8 mmol, 10 eq.)、蒸留水(50.0 mL)を加え、溶液を氷浴で冷却した。氷冷下、トリフルオロ酢酸(50.0 mL, 640 mmol, 32.4 eq.)を5分間かけて滴下し、反応液を室温まで昇温し、4時間30分間攪拌した。
LC/MSで反応の進行を確認後、反応液を減圧濃縮し、残渣をトルエン共沸した。得られた残渣にイソプロピルエーテルを加えて、固体を析出させ、ろ取した。得られた固体を減圧下室温にて乾燥し、化合物21(8.86 g, 19.0 mmol, 収率96%)を得た。
N-(9-((2R,3R,4R,5R)-3-((tert-butyldimethylsilyl)oxy)-4-hydroxy-5-((2,2,2-trifluoroacetamido)methyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide
アルゴン雰囲気下、500 mLのナスフラスコに、工程16で得られた化合物21(15.6 g, 33.6 mmol, 1.0 eq.)、脱水DMF(111 mL)を加え、溶液を氷浴で冷却した。氷冷下、内温が6℃未満となるように、イミダゾール(9.16 g, 134 mmol, 4.0eq.)、t-ブチルジメチルシリルクロリド(15.2 g, 101 mmol, 3.0 eq.)を添加し、同温度で30分間攪拌した。
LC/MSで反応の進行を確認後、反応液に氷水を加えた。水層を酢酸エチルで3回抽出し、飽和食塩水で洗浄し、無水硫酸ナトリウムで脱水した。ろ過後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(SiO2 800 g, クロロホルム/2-ブタノン=100:0から85:15)で精製することで、化合物22と化合物23の混合物(10.9 g)を得た。得られた化合物22と化合物23の混合物は更なる精製操作を実施せず、次工程に用いた。
(2R,3R,4R,5R)-5-(6-benzamido-9H-purin-9-yl)-4-((tert-butyldimethylsilyl)oxy)-2-((2,2,2-trifluoroacetamido)methyl)tetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite
アルゴン雰囲気下、200 mLのナスフラスコに、工程17で得られた化合物22と化合物23の混合物(1.19 g, 2.05 mmol, 化合物22:化合物23=9:1)、脱水ジクロロメタン(6.83 mL)を加え、溶液を氷浴で冷却した。氷冷下、ジイソプロピルエチルアミン(0.537 mL, 3.07 mmol, 1.5 eq.)、3-((クロロ(ジイソプロピルアミノ)ホスファニル)オキシ)プロパンニトリル(0.857 mL, 3.07 mmol, 1.5 eq.)のジクロロメタン混合溶液を滴下した後、反応液を室温まで昇温し、室温で2時間攪拌した。
TLCで原料の消失を確認後、反応液に蒸留水を加え、クロロホルム(50 mL)で2回抽出し、有機層を無水硫酸ナトリウムで脱水した。ろ過後、溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(ヘプタン/酢酸エチル=50:50から30:70、0.5%トリエチルアミン含有)で複数回精製することで、目的とする化合物24(608 mg, 0.779 mmol, 収率38%)を淡黄色アモルファス状物質として得た。
1H NMR (400 MHz, CDCl3) δ: 9.82 (1H, d, J = 8.8 Hz), 9.07 (1H, s), 8.83-8.80 (1H, m), 8.06-8.01 (3H, m), 7.66-7.52 (3H, m), 5.86 (1H, d, J = 7.8 Hz), 4.88 (1H, dd, J = 7.8, 5.2 Hz), 4.50 (1H, br s), 4.39-4.29 (1H, m), 4.21-3.88 (3H, m), 3.74-3.63 (1H, m), 3.48-3.35 (1H, m), 2.73-2.66 (2H, m), 1.28-1.23 (12H, m), 1.08-1.04 (1H, m), 0.72-0.68 (9H, m), -0.17 (3H, s), -0.45 (3H, s).
31P NMR(CDCl3) δ: 149.95

N-(9-((3aR,5R,6R,6aS)-2,2-di-tert-butyl-6-methoxytetrahydrofuro[2,3-d][1,3,2]dioxasilol-5-yl)-9H-purin-6-yl)benzamide
市販の化合物1a (30.0 g, 78.0 mmol)のDMF (300 mL)溶液に、氷冷下、ジ-t-ブチルシリルビス(トリフルオロメタンスルホナード) (68.6 g, 156 mmol)をゆっくり加えた。氷冷下、1時間攪拌後、反応液を飽和炭酸水素ナトリウム水溶液に加え、ヘプタン/酢酸エチルの混合溶媒を加え、2回抽出した。有機層を水で2回、飽和食塩水で1回洗浄後、無水硫酸ナトリウムで乾燥した。ろ過後、濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル = 7/3 → 3/7)で精製し、化合物2a (38.7 g, 73.7 mmol)を無色固体として得た(収率95%)。
ESI-MS: 計算値: 524.23 [M-H]-, 実測値: 524.5 [M-H]-
1H-NMR (CDCl3, 400 MHz) δ: 9.32 (s, 1H), 8.76 (s, 1H), 8.05 (s, 1H), 8.03 (t, J = 6.6 Hz, 2H), 7.60 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 6.01 (s, 1H), 4.66 (dd, J = 9.6, 5.0 Hz, 1H), 4.48 (dd, J = 8.9, 4.8 Hz, 1H), 4.31 (d, J = 4.6 Hz, 1H), 4.20 (ddd, J = 10.1, 5.0, 5.0 Hz, 1H), 4.03 (t, J = 9.8 Hz, 1H), 3.70 (s, 3H), 1.10 (s, 9H), 1.06 (s, 9H).
化合物 2a (10.0 g, 19.0 mmol) をジクロロメタン (50 mL) に溶解し、テトラブチルアンモニウムブロミド (9.20 g, 28.5 mmol) 、1 M 水酸化ナトリウム水溶液 (50 ml) を加えた。ヨウ化メチル(4.76 ml, 76.0 mmol) をゆっくりと滴下した。その後、室温で1 時間10 分間攪拌した。原料消失を確認後、反応液を氷冷した水/クロロホルム = 1/1 に加えクエンチした。有機層を水で2 回洗浄後、無水硫酸ナトリウムで脱水し、乾燥剤をろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル = 90/10 → 50/50) にて精製することで化合物 3a (6.25 g, 11.6 mmol) を無色アモスファスとして得た。(収率61%)
ESI-MS: 計算値: 540.26 [M+H]+, 実測値: 540.4 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.56 (s, 1H), 7.94 (s, 1H), 7.49-7.46 (m, 2H), 7.34-7.29 (m, 1H), 7.21 (t, J = 7.6 Hz, 2H), 5.94 (s, 1H), 4.61 (dd, J = 9.6, 5.0 Hz, 1H), 4.46 (dd, J = 9.2, 5.0 Hz, 1H), 4.22 (d, J = 4.6 Hz, 1H), 4.17 (ddd, J = 10.0, 5.2, 5.0 Hz, 1H), 4.00 (dd, J = 10.5, 9.2 Hz, 1H), 3.79 (s, 3H), 3.67 (s, 3H), 1.08 (s, 9H), 1.05 (s, 9H).
N-(9-((2R,3R,4S,5S)-4,5-dihydroxy-3-methoxytetrahydrofuran-2-yl)-9H-purin-6-yl)-N-methylbenzamide
化合物3a (6.25 g, 11.6 mmol) をテトラヒドロフラン(63 mL) に溶解し、氷浴で冷却した。トリエチルアミン (8.07 ml, 57.9 mmol)、トリエチルアミン三フッ化水素酸塩 (1.89 ml, 11.6 mmol) を加え、氷浴で冷却しながら1 時間5 分間攪拌した。原料消失を確認後、トリエチルアミン (10 ml, 76.0 mmol) を加えてクエンチし、クロロホルムで希釈した後に、反応液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (クロロホルム/メタノール = 100/0 → 90/10) にて精製することで化合物4a (4.25 g, 10.6 mmol) を無色アモルファスとして得た。(収率quant.)
ESI-MS: 計算値: 400.16 [M+H]+, 実測値: 400.3 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.56 (s, 1H), 7.97 (s, 1H), 7.50-7.48 (m, 2H), 7.35-7.31 (m, 1H), 7.22 (t, J = 7.8 Hz, 2H), 5.87 (d, J = 7.3 Hz, 1H), 5.86 (dd, J = 11.4, 2.3 Hz, 1H), 4.63 (dd, J = 7.3, 4.6 Hz, 1H), 4.57-4.56 (m, 1H), 4.36-4.34 (m, 1H), 3.99-3.94 (m, 1H), 3.81 (s, 3H), 3.77 (td, J = 12.3, 1.7 Hz, 1H), 3.31 (s, 3H), 2.77 (d, J = 1.4 Hz, 1H).
N-(9-((2R,3R,4S,5S)-5-(bis(4-methoxyphenyl)(phenyl)methoxy)-4-hydroxy-3-methoxytetrahydrofuran-2-yl)-9H-purin-6-yl)-N-methylbenzamide
化合物4a (4.25 g, 10.6 mmol) をピリジン (43 mL) に溶解し氷浴で攪拌した。反応液に 4,4'-ジメトキシトリチルクロリド (5.41 g, 20.0 mmol) を加え、室温で2 時間25 分間攪拌した。原料消失を確認後、反応液を氷冷した重曹水に加えクエンチし、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、ろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル (1%トリエチルアミン含有) = 70/30 → 50/50 ) にて精製することで化合物5a (5.35 g, 7.62 mmol) を無色アモルファスとして得た。(収率71%)
ESI-MS: 計算値: 702.29 [M+H]+, 実測値: 702.6 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.50 (s, 1H), 8.14 (s, 1H), 7.45-7.40 (m, 4H), 7.33-7.22 (m, 8H), 7.16 (t, J = 7.6 Hz, 2H), 6.81 (dd, J = 8.9, 1.1 Hz, 4H), 6.15 (d, J = 3.7 Hz, 1H), 4.48 (dd, J = 11.9, 5.0 Hz, 1H), 4.35 (dd, J = 5.3, 3.9 Hz, 1H), 4.21-4.19 (m, 1H), 3.80 (s, 3H), 3.79 (s, 6H), 3.53 (s, 3H), 3.50 (dd, J = 10.8, 3.0 Hz, 1H), 3.40 (dd, J = 10.8, 4.4 Hz, 1H), 2.66 (d, J = 6.4 Hz, 1H).
(2S,3S,4R,5R)-2-(bis(4-methoxyphenyl)(phenyl)methoxy)-4-methoxy-5-(6-(N-methylbenzamido)-9H-purin-9-yl)tetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite
化合物5a (5.30 g, 7.55 mmol) をジクロロメタン (48 mL) に溶解し、ジイソプロピルエチルアミン (2.64 mL, 15.1 mmol) を加え、氷浴で冷却した。ジクロロメタン (5 mL) に溶解した 2-シアノエチルジイソプロピルクロロホスホロアミジト (2.68 g, 11.3 mmol) を5 分間かけて滴下した。その後、室温まで昇温しながら1 時間10 分間攪拌した。原料消失を確認後、反応液を氷冷した飽和重曹水に加えクエンチした。酢酸エチルを加え抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル (1%トリエチルアミン含有) = 70/30 → 50/50) にて精製することで アミダイト6a (6.22 g, 6.90 mmol) を無色アモルファスとして得た。(収率91%)
ESI-MS: 計算値: 902.40 [M+H]+, 実測値: 902.5 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.47 (s, 0.35H), 8.47 (s, 0.65H), 8.14 (s, 0.35H), 8.09 (s, 0.65H), 7.44-7.39 (m, 4H), 7.33-7.21 (m, 8H), 7.16-7.12 (m, 2H), 6.93-6.78 (m, 4H), 6.12 (d, J = 5.5, 0.65H), 6.10 (d, J = 5.0, 0.35H), 4.66-4.53 (m, 2H), 4.41-4.38 (m, 0.35H), 4.34-4.32 (m, 0.65H), 3.97-3.78 (m, 10H), 3.70-3.44 (m, 7H), 3.36-3.30 (m, 1H), 2.64 (t, J = 6.2 Hz, 1.3H), 2.38 (t, J = 6.4 Hz, 0.70H), 1.22-1.17 (m, 8H), 1.06 (d, J = 6.9 Hz, 4H).
31P-NMR(CDCl3, 162 MHz) δ: 150.70, 150.94.
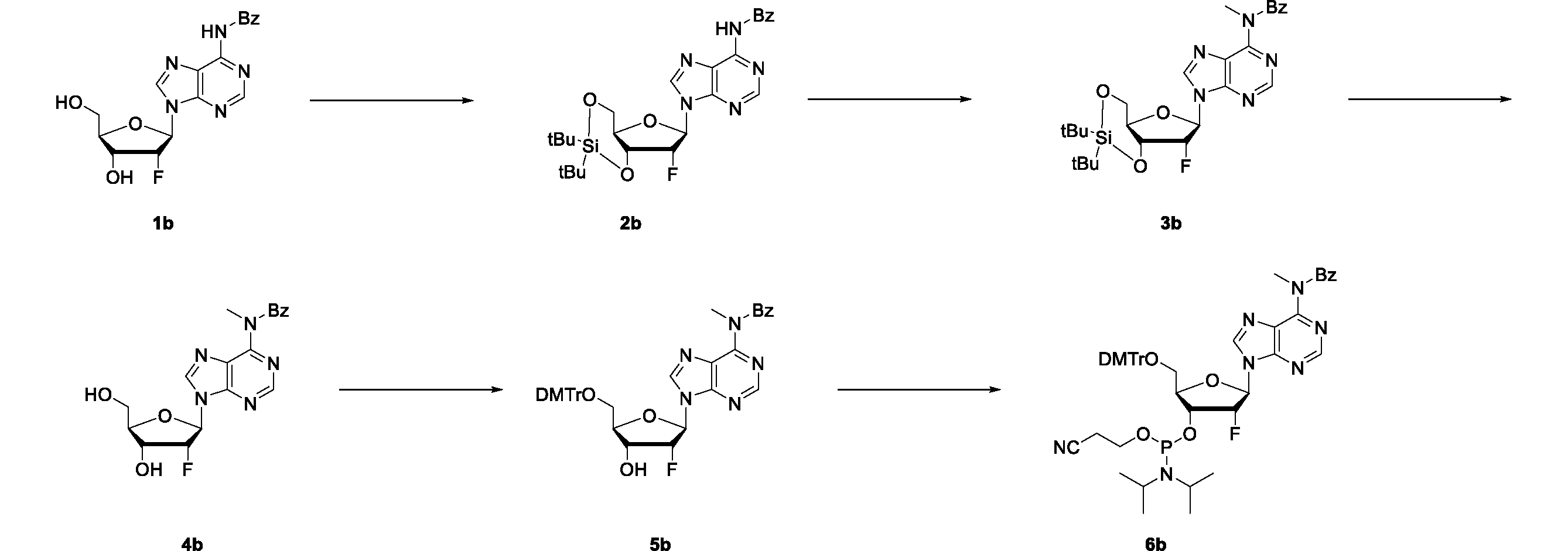
N-(9-((4aR,6R,7R,7aS)-2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo[3,2-d][1,3,2]dioxasilin-6-yl)-9H-purin-6-yl)benzamide
市販の化合物1b (30.0 g, 80.4 mmol)のDMF (300 mL)溶液に、氷冷下、ジ-t-ブチルシリルビス(トリフルオロメタンスルホナード) (70.8 g, 161 mmol)をゆっくり加えた。氷冷下、1時間攪拌後、反応液を飽和炭酸水素ナトリウム水溶液に加え、ヘプタン/酢酸エチルの混合溶媒を加え、2回抽出した。有機層を水で2回、飽和食塩水で1回洗浄後、無水硫酸ナトリウムで乾燥した。ろ過後、濃縮残渣をヘプタン/酢酸エチル = 9/1にてスラリー精製し、化合物2b (38.7 g, 75.4 mmol)を無色固体として得た(収率94%)。
ESI-MS: 計算値: 514.23 [M+H]+, 実測値: 514.5 [M+H]+
1H-NMR (DMSO-d6, 400 MHz) δ: 11.27 (s, 1H), 8.74 (s, 1H), 8.65 (s, 1H), 8.04 (d, J = 8.7 Hz, 2H), 7.65 (t, J = 7.5 Hz, 1H), 7.55 (t, J = 7.5 Hz, 2H), 6.45 (d, J = 23 Hz, 1H), 5.71 (dd, J = 54.5, 4.1 Hz, 1H), 5.03 (m, 1H), 4.44 (q, J = 3.7 Hz, 1H), 4.09 (m, 2H), 1.11 (s, 9H), 1.02 (s, 9H).
N-(9-((4aR,6R,7R,7aS)-2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo[3,2-d][1,3,2]dioxasilin-6-yl)-9H-purin-6-yl)-N-methylbenzamide
化合物2b (10.0 g, 19.5 mmol) をジクロロメタン (50 mL) に溶解し、テトラブチルアンモニウムブロミド (9,41 g, 29.2 mmol) 、1 M 水酸化ナトリウム水溶液 (50 ml) を加えた。ヨウ化メチル(1.83 ml, 29.2 mmol) をゆっくりと滴下した。その後、室温で1 時間攪拌した。原料消失を確認後、反応液を氷冷した水/クロロホルム = 1/1 に加えクエンチした。有機層を水で2 回洗浄後、無水硫酸ナトリウムで脱水し、乾燥剤をろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル = 90/10 → 50/50) にて精製することで化合物3b (6.86 g, 12.8 mmol) を無色アモスファスとして得た。(収率65%)
ESI-MS: 計算値: 528.24 [M+H]+, 実測値: 538.6 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.54 (s, 1H), 7.94 (s, 1H), 7.47 (d, J = 8.1 Hz, 2H), 7.32 (t, J = 7.3 Hz, 2H), 7.21 (t, J = 7.6 Hz, 2H), 6.10 (d, J = 22.0 Hz, 1H), 5.46 (dd, J = 54.5, 4.1 Hz, 1H), 4.86 (ddd, J = 27.2, 9.8, 4.1 Hz, 1H), 4.47 (dd, J = 9.2, 5.0 Hz, 1H), 4.14 (m, 1H), 4.03 (t, J = 9.8 Hz, 1H), 3.78 (s, 3H), 1.11 (s, 9H), 1.05 (s, 9H).
N-(9-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)-N-methylbenzamide
化合物3b (6.67 g, 12.6 mmol) をテトラヒドロフラン(66 mL) に溶解し、氷浴で冷却した。トリエチルアミン (8.81 ml, 63.2 mmol)、トリエチルアミン三フッ化水素酸塩 (2.05 ml, 12.6 mmol) を加え、氷浴で冷却しながら1 時間5 分間攪拌した。原料消失を確認後、トリエチルアミン (10.6 ml, 76.0 mmol) を加えてクエンチし、クロロホルムで希釈した後に、反応液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (クロロホルム/メタノール = 100/0 → 90/10) にて精製することで化合物4b (4.98 g, 12.9 mmol) を無色アモルファスとして得た。(収率quant.)
ESI-MS: 計算値: 388.14 [M+H]+, 実測値: 388.4 [M+H]+
1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.58 (s, 1H), 7.30 (m, 5H), 6.31 (dd, J = 16.9, 2.3 Hz, 1H), 5.75 (d, J = 6.4 Hz, 1H), 5.41 (m, 1H), 5.15 (t, J = 5.3 Hz, 1H), 4.46 (m, 1H), 3.98 (m, 1H), 3.75 (dq, J = 12.4, 2.6 Hz, 1H), 3.67 (s, 3H), 3.61 - 3.56 (m, 1H).
N-(9-((2R,3R,4R,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-3-fluoro-4-hydroxytetrahydrofuran-2-yl)-9H-purin-6-yl)-N-methylbenzamide
化合物4b (4.93 g, 12.7 mmol) をピリジン (49 mL) に溶解し氷浴で攪拌した。反応液に 4,4'-ジメトキシトリチルクロリド (6.47 g, 29.2 mmol) を加え、室温で1時間20 分間攪拌した。原料消失を確認後、反応液を氷冷した重曹水に加えクエンチし、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、ろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル (1%トリエチルアミン含有) = 70/30 → 50/50 ) にて精製することで化合物5b (8.34 g, 12.1 mmol) を無色アモルファスとして得た。(収率95%)
ESI-MS: 計算値: 690.27 [M+H]+, 実測値: 690.7 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.51 (s, 1H), 8.10 (s, 1H), 7.43 (dd, J = 8.2, 1.4 Hz, 2H), 7.37 (dd, 8.2, 1.4 Hz, 2H), 7.28-7.20 (m, 8H), 7.12 (t, J = 7.5 Hz, 2H), 6.79 (d, J = 8.7 Hz), 6.23 (dd, J = 17.1, 2.5 Hz, 1H), 5.58 (dq, J = 52.9, 2.3 Hz, 1H), 4.78 (m, 1H), 4.19 (m, 1H), 3.78 (s, 6H), 3.47 (ddd, J = 57.9, 10.6, 3.5 Hz, 2H), 2.44 (dd, J = 7.5, 2.5 Hz, 1H).
(2R,3R,4R,5R)-2-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-fluoro-5-(6-(N-methylbenzamido)-9H-purin-9-yl)tetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite
化合物5b (10.0 g, 14.6 mmol) をジクロロメタン (80 mL) に溶解し、ジイソプロピルエチルアミン (5.08 mL, 29.1 mmol) を加え、氷浴で冷却した。ジクロロメタン (15 mL) に溶解した 2-シアノエチルジイソプロピルクロロホスホロアミジト (4.18 g, 21.8 mmol) を5 分間かけて滴下した。その後、室温まで昇温しながら1 時間攪拌した。原料消失を確認後、反応液を氷冷した飽和重曹水に加えクエンチした。酢酸エチルを加え抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、乾燥剤をろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル (1%トリエチルアミン含有) = 70/30 → 50/50) にて精製することで アミダイト6b (12.1 g, 13.6 mmol) を無色アモルファスとして得た。(収率93%)
ESI-MS: 計算値: 890.38 [M+H]+, 実測値: 890.8 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.53 (s, 0.49H), 8.50 (s, 0.51H), 8.14 (s, 1H), 7.43-7.39 (m, 2H), 7.37-7.33 (m, 2H), 7.27-7.20 (m, 8H), 7.09-7.04 (m, 2H), 6.77 (t, J = 9.1 Hz, 4H), 6.28-6.19 (m, 1H), 5.74 (dq, J = 18.5, 2.2 Hz, 0.50H), 5.61 (dq, J = 19.2, 2.3 Hz, 0.50H), 5.10-5.00 (m, 0.47H), 4.94-4.85 (m, 0.53H), 4.31 (m, 1H), 3.97-3.82 (m, 1H), 3.79 (s, 3H), 3.79 (s, 3H), 3.63-3.53 (m, 4H), 3.31-3.27 (m, 1H), 2.59 (t, J = 6.2 Hz, 1H), 2.41 (t, J = 6.4 Hz, 1H), 1.20-1.15 (m, 9H), 1.04 (d, J = 6.4 Hz, 3H).
31P-NMR(CDCl3, 162 MHz) δ: 151.97, 151.92, 151.19, 151.11.
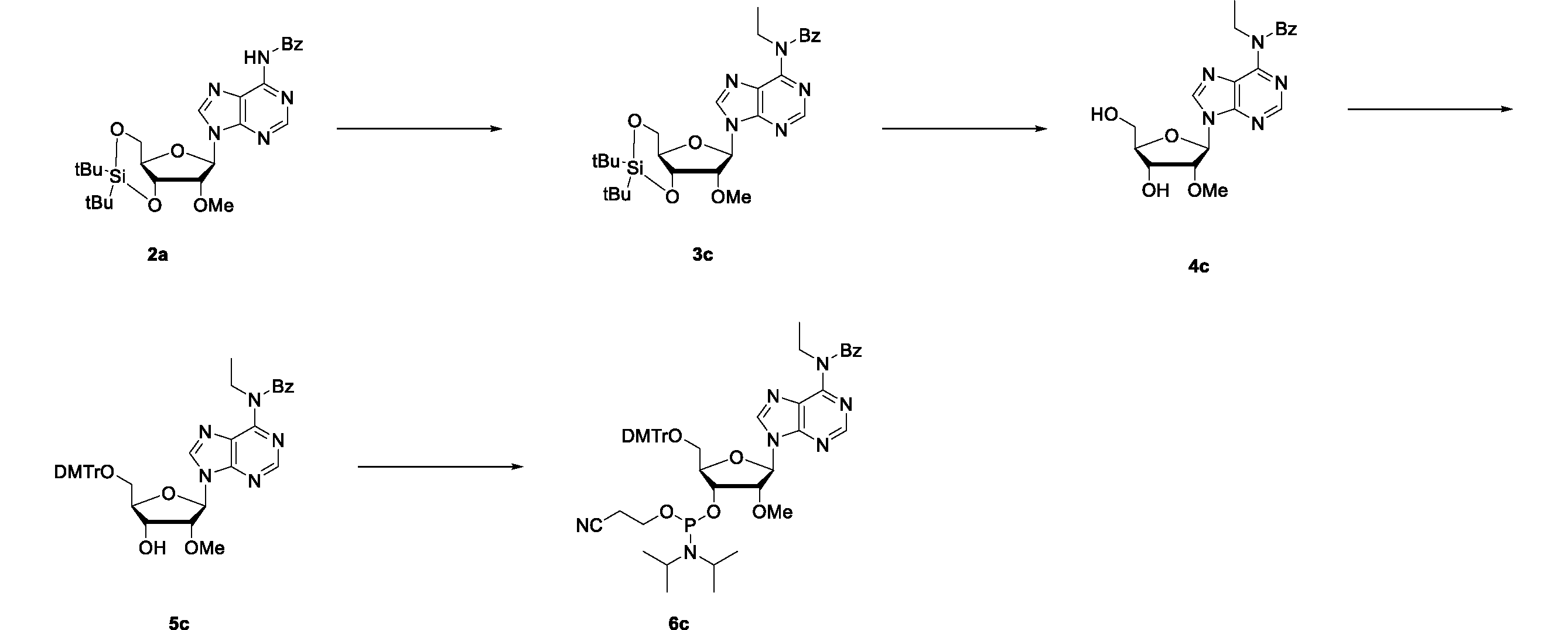
N-(9-((4aR,6R,7R,7aS)-2,2-di-tert-butyl-7-methoxytetrahydro-4H-furo[3,2-d][1,3,2]dioxasilin-6-yl)-9H-purin-6-yl)-N-ethylbenzamide
化合物 2a (11.7 g, 22.3 mmol) をジクロロメタン (58.5 mL) に溶解し、テトラブチルアンモニウムブロミド (10.8 g, 33.4 mmol) 、1 M 水酸化ナトリウム水溶液 (58.5 ml) を加えた。ヨウ化エチル (10.8 ml, 134 mmol) をゆっくりと滴下した。その後、室温で2 時間攪拌した。原料消失を確認後、反応液を氷冷した水/クロロホルム = 1/1 に加えクエンチした。有機層を水で2 回洗浄後、無水硫酸ナトリウムで乾燥させた。ろ過後、ろ液を濃縮した。濃縮残渣をトルエンによりスラリー精製し、再度ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル = 90/10 → 70/30) にて精製することで化合物 3c (6.14 g, 11.1 mmol) を無色アモスファスとして得た。(収率49%)
ESI-MS: 計算値: 554.28 [M+H]+, 実測値: 554.6 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.56 (s, 1H), 7.91 (s, 1H), 7.47-7.45 (m, 2H), 7.31-7.27 (m, 1H), 7.19 (t, J = 7.5 Hz, 2H), 5.94 (s, 1H), 4.61 (dd, J = 9.6, 4.6 Hz, 1H), 4.46 (dd, J = 9.1, 5.0 Hz, 1H), 4.40 (q, J = 7.0 Hz, 2H), 4.22 (d, J = 4.6 Hz, 1H), 4.16 (ddd, J = 10.1, 4.9, 4.8 Hz, 1H), 4.00 (dd, J = 10.5, 9.6 Hz, 1H), 3.67 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H), 1.09 (s, 9H), 1.05 (s, 9H).
N-ethyl-N-(9-((2R,3R,4R,5R)-4-hydroxy-5-(hydroxymethyl)-3-methoxytetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide
化合物3c (6.14 g, 11.1 mmol) をテトラヒドロフラン(61.4 mL) に溶解し、氷浴で冷却した。トリエチルアミン (7.73 ml, 55.4 mmol)、トリエチルアミン三フッ化水素酸塩 (1.81 ml, 11.1 mmol) を加え、氷浴で冷却しながら2 時間攪拌した。原料消失を確認後、トリエチルアミン (10 ml, 76.0 mmol) を加えてクエンチし、クロロホルムで希釈した後に、反応液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (クロロホルム/メタノール= 100/0 → 90/10) にて精製することで化合物4c (4.60 g, 11.1 mmol) を無色アモルファスとして得た。(収率quant.)
ESI-MS: 計算値: 414.18 [M+H]+, 実測値: 414.3 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.56 (s, 1H), 7.92 (s, 1H), 7.50-7.46 (m, 2H), 7.32-7.28 (m, 1H), 7.20 (t, J = 7.5 Hz, 2H), 5.89 (dd, J = 11.6, 2.1 Hz, 1H), 5.85 (d, J = 7.3 Hz, 1H), 4.62 (dd, J = 7.3, 4.6 Hz, 1H), 4.57-4.56 (m, 1H), 4.45-4.39 (m, 2H), 4.36-4.34 (m, 1H), 3.96 (dt, J = 12.9, 1.9, 1H), 3.80-3.73 (m, 1H), 3.29 (s, 3H), 2.70 (d, J = 1.4 Hz, 1H), 1.37 (t, J = 7.1 Hz, 3H).
N-(9-((2R,3R,4R,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-4-hydroxy-3-methoxytetrahydrofuran-2-yl)-9H-purin-6-yl)-N-ethylbenzamide
化合物4c (4.58 g, 11.1 mmol) をピリジン (46 mL) に溶解し氷浴で攪拌した。反応液に 4,4'-ジメトキシトリチルクロリド (5.63 g, 16.6 mmol) を加え、室温で2 時間攪拌した。原料消失を確認後、反応液を氷冷した重曹水に加えクエンチし、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。ろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル (1%トリエチルアミン含有) = 70/30 → 50/50) にて精製することで化合物5c (7.55 g, 10.6 mmol) を無色アモルファスとして得た。(収率95%)
ESI-MS: 計算値: 716.31 [M+H]+, 実測値: 716.2 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.51 (s, 1H), 8.11 (s, 1H), 7.43-7.40 (m, 4H), 7.32-7.22 (m, 8H), 7.13 (t, J = 7.5 Hz, 2H), 6.81 (dd, J = 9.1, 1.4 Hz, 4H), 6.14 (d, J = 3.7 Hz, 1H), 4.47 (dd, J = 5.7, 5.6 Hz, 1H), 4.41 (q, J = 7.0 Hz, 2H), 4.34 (dd, J = 8.4, 4.1 Hz, 1H), 4.21-4.17 (m, 1H), 3.79 (s, 6H), 3.53 (s, 3H), 3.50 (dd, J = 10.5, 3.2 Hz, 1H), 3.39 (dd, J = 10.7, 4.3 Hz, 1H), 2.64 (d, J = 6.4 Hz, 1H), 1.34 (t, J = 7.1 Hz, 3H).
(2R,3R,4R,5R)-2-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-5-(6-(N-ethylbenzamido)-9H-purin-9-yl)-4-methoxytetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite
化合物5c (8.83 g, 12.3 mmol) をジクロロメタン (74 mL) に溶解し、ジイソプロピルエチルアミン (4.31 mL, 24.7 mmol) を加え、氷浴で冷却した。脱水ジクロロメタン (14 mL) に溶解した 2-シアノエチルジイソプロピルクロロホスホロアミジト (4.40 g, 18.6 mmol) を9 分間かけて滴下した。その後、室温まで昇温しながら1 時間攪拌した。原料消失を確認後、反応液を氷冷した飽和重曹水に加えクエンチした。酢酸エチルを加え抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させた。ろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル(1%トリエチルアミン含有) = 70/30 → 50/50) にて精製することでアミダイト6c (10.4 g, 11.3 mmol) を無色アモルファスとして得た。(収率92%)
ESI-MS: 計算値: 916.42 [M+H]+, 実測値: 917.3 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.49 (s, 0.37H), 8.48 (s, 0.63H), 8.11 (s, 0.34H), 8.05 (s, 0.66H), 7.43-7.38 (m, 4H), 7.32-7.21 (m, 8H), 7.11 (t, J = 7.8 Hz, 2H), 6.80 (m, 4H), 6.10 (m, 1H), 4.64-4.52 (m, 2H), 4.43-4.32 (m, 3H), 3.94-3.83 (m, 1H), 3.79-3.78 (m, 6H), 3.67 -3.46 (m, 4H), 3.35-3.29 (m, 1H), 2.64 (t, J = 6.4 Hz, 1.3H), 2.37 (t, J = 6.4 Hz, 0.70H), 1.33 (t, J = 7.1 Hz, 3H), 1.18 (m, 8H), 1.06 (d, J = 6.9 Hz, 4H).
31P-NMR(CDCl3, 162 MHz) δ: 151.67, 150.92.

N-(9-((4aR,6R,7R,7aS)-2,2-di-tert-butyl-7-fluorotetrahydro-4H-furo[3,2-d][1,3,2]dioxasilin-6-yl)-9H-purin-6-yl)-N-ethylbenzamide
化合物2b (1.00 g, 1.95 mmol) をジクロロメタン (5.0 mL) に溶解し、テトラブチルアンモニウムブロミド (0.942 g, 2.92 mmol) 、1 M 水酸化ナトリウム水溶液 (5.0 ml) を加えた。ヨウ化メチル(0.942 ml, 11.7 mmol) をゆっくりと滴下した。その後、室温で2 時間攪拌した。原料消失を確認後、反応液を氷冷した水/クロロホルム = 1/1 に加えクエンチした。有機層を水で2 回洗浄後、無水硫酸ナトリウムで脱水し、乾燥剤をろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル = 80/20 → 70/30) にて精製することで化合物3d (629 mg, 1.16 mmol) を無色アモスファスとして得た。(収率60%)
ESI-MS: 計算値: 542.26 [M+H]+, 実測値: 542.6 [M+H]+
1H-NMR (CDCl3, 400 MHz) δ: 8.55 (s, 1H), 7.91 (s, 1H), 7.46 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.1 Hz, 1H), 7.19 (t, J = 7.8 hz, 2H), 6.09 (d, J = 22.4 Hz, 1H), 5.45 (dd, J = 54.1, 3.9 Hz, 1H), 4.86 (ddd, J = 27.2, 9.8, 4.1 Hz, 1H), 4.48 (dd, J = 9.1, 5.0, 1H), 4.40 (q, J = 7.2 Hz, 2H), 4.04 (t, J = 9.8 Hz, 1H), 1.34 (t, J = 7.1 Hz, 3H), 1.11 (s, 9H), 1.05 (s, 9H).
N-ethyl-N-(9-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-yl)benzamide
化合物4cを得る工程と同様に、化合物4dを得た。
ESI-MS: 計算値: 401.40 [M+H]+, 実測値: 402.1 [M+H]+
N-(9-((2R,3R,4R,5R)-5-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-3-fluoro-4-hydroxytetrahydrofuran-2-yl)-9H-purin-6-yl)-N-ethylbenzamide
化合物5cを得る工程と同様にして化合物5dを得た。
ESI-MS: 計算値: 738.25 [M+Cl]-, 実測値: 738.7 [M+Cl]-
1H-NMR (CDCl3, 400 MHz) δ: 8.52 (s, 1H), 8.07 (s, 1H), 7.43-7.41 (m, 2H), 7.38-7.35 (m, 2H), 7.30-7.20 (m, 8H), 7.10 (t, J = 7.8 Hz, 2H), 6.79 (d, J = 8.2 Hz, 4H), 6.22 (dd, J = 17.4, 2.3 Hz, 1H), 5.58 (ddd, J = 53.0, 2.4, 1.2 Hz, 1H), 4.93-4.74 (m, 1H), 4.40 (q, J = 7.2 Hz, 2H), 4.19-4.16 (m, 1H), 3.78 (s, 6H), 3.54 (dd, J = 11.0, 3.2 Hz, 1H), 3.40 (dd, J = 10.5, 3.1 Hz, 1H), 2.23 (dd, J = 6.9, 2.3 Hz, 1H), 1.33 (t, J = 7.1 Hz, 3H).
(2R,3R,4R,5R)-2-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-5-(6-(N-ethylbenzamido)-9H-purin-9-yl)-4-fluorotetrahydrofuran-3-yl (2-cyanoethyl) diisopropylphosphoramidite
化合物5d(1.93 g, 2.74 mmol) をジクロロメタン (16 mL) に溶解し、ジイソプロピルエチルアミン (0.958 mL, 5.48 mmol) を加え、氷浴で冷却した。脱水ジクロロメタン (3.8 mL) に溶解した 2-シアノエチルジイソプロピルクロロホスホロアミジト (970 mg, 4.11 mmol) を滴下した。その後、室温まで昇温しながら1 時間攪拌した。原料消失を確認後、反応液を氷冷した飽和重曹水に加えクエンチした。酢酸エチルを加え抽出した。有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させた。ろ別後、ろ液を濃縮した。濃縮残渣をシリカゲルカラムクロマトグラフィー (ヘプタン/酢酸エチル(1%トリエチルアミン含有) = 90/10 → 60/40) にて精製することでアミダイト6d (2.29 g, 2.53 mmol) を無色アモルファスとして得た。(収率92%)
ESI-MS: 計算値: 938.36 [M+Cl]-, 実測値: 938.7 [M+Cl]-
1H-NMR (CDCl3, 400 MHz) δ: 8.53 (s, 0.5H), 8.51 (s, 0.5H), 8.11 (s, 1H), 7.41-7.33 (m, 4H), 7.27-7.16 (m, 8H), 7.03 (t, J = 7.8 Hz, 2H), 6.79-6.75 (m, 4H), 6.27-6.18 (m, 1H), 5.78-5.58 (m, 1H), 5.10-4.85 (m, 1H), 4.42-4.38 (m, 2H), 4.31-4.30 (m, 1H), 3.96-3.72 (m, 7H), 3.68-3.51 (m, 4H), 3.29-3.27 (m, 1H), 2.59 (t, J = 6.2 Hz, 1H), 2.40 (t, J = 6.4 Hz, 1H), 1.33-1.31 (m, 3H), 1.19-1.15 (m, 9H), 1.04 (d, J = 6.9 Hz, 3H).
31P-NMR(CDCl3, 162 MHz) δ: 151.94, 151.89, 151.20, 151.11.
DNAオリゴヌクレオチドは固相担体としてCPG 1000A (dA-CPG, dG-CPG, Ac-dC-CPG, dT-CPG)(Glen Research社)を用い、縮合時間は2分間とした。
5'末端にリン酸基を有するRNA (5'-モノリン酸RNA)は固相担体としてUniversal UnyLinker Support 2000A (ChemGenes社)を用い、1塩基目の縮合時間は15分間、それ以降は各3分間とした。5'末端の水酸基のリン酸化は化学リン酸化試薬 (0.05 mol/Lアセトニトリル溶液)(Glen Research社もしくはChemGenes社)を用いて行った。
3'-アミノグアノシンモノマーが3'末端に導入されたRNAオリゴヌクレオチドの固相合成は化合物15を用いて行った。1塩基目の縮合時間は15分間、それ以降は各3分間とした。
アクリルアミドゲル溶液 (変性剤として7M尿素を含む)に過硫酸アンモニウム (以下、APS)の水溶液とN,N,N',N'-テトラメチルエチレンジアミン(以下、TEMED)を重合剤として添加し、固形化 (室温、6~12時間)させることでゲルを作製した。RNAサンプルはゲルローディングバッファー (80%ホルムアミド、TBE)と混合し、90℃にて3分間加熱後、ゲルにロードした。電気泳動後、RNAのバンドはUV光照射 (254 nm)によって検出し、カミソリの刃を用いてゲルから切り出した。切り出したゲル片は細かく破砕した後、超純水にてゲルから抽出した (室温にて6~12時間振とう)。RNAの抽出液はAmicon Ultra 10K (Millipore社)を用いて脱塩・濃縮し、エタノール沈殿 (0.3M酢酸ナトリウム(pH5.2)/70%エタノール)を行うことでRNAのペレットを得た。RNAペレットは80%エタノールでリンス後、室温にて1時間風乾させた。得られたRNAペレットは超純水に溶解させ、適切な濃度に希釈後、紫外可視吸光光度測定 (NanoDrop, Thermo scientific社)によって260 nmの吸光度を測定し、各RNA配列のモル吸光係数から濃度を決定した (各塩基のモル吸光係数として以下の数値を使用した。A = 15300, G = 11800, C = 7400, T = 9300, U = 9900)。
精製したオリゴヌクレオチドの構造決定はMALDI-TOF MS (Ultraflex III, Bruker Daltonics社)(マトリクス:3-ヒドロキシピコリン酸)を用いた質量分析法もしくは変性ポリアクリルアミドゲル電気泳動を用いた分析により行った。
ケミカルライゲーション反応の分析においては、反応液を超純水にて適切に希釈したものをサンプルとして使用した。希釈したサンプルはゲルローディングバッファー (80%ホルムアミド/TBE)と混合し、90℃にて3分間加熱後、ゲルにロードした。電気泳動後、超純水にて10,000倍希釈したSYBR(登録商標) Green II Nucleic Acid Stain (Lonza社)を用いてゲル染色 (室温、15分間)することでRNAのバンドを検出した (使用機器 : ChemiDoc, BIORAD社)。
ケミカルライゲーション反応の収率は、RNA連結産物のバンド強度をdPAGEにて単離精製した連結産物を基準物質として比較することで算出した。
ケミカルライゲーション反応によって得られたRNA連結産物は、反応液からエタノール沈殿 (0.3M酢酸ナトリウム(pH5.2)/70%エタノール)によってRNAペレットとして回収後、dPAGEによって精製を行った。


化学合成により配列2として得られたRNA断片E1-1(10 nmol)ならびに配列3として得られた5'リン酸RNA断片E1-2 (10 nmol)、テンプレートDNA1 (10 nmol )の超純水溶液(200 μL、核酸終濃度;50 μM)を3バッチ調製した。各々の調製溶液にT4 RNA Ligase 2 Reaction Buffer(10X)(New England BioLabs社製)を100 μLならびに超純水440 μLを加え90℃にて5分間加熱後、30分間以上かけて室温に戻した。この各々の溶液に60% PEG6000水溶液を終濃度15%となるように加えた。この溶液にT4 RNA Ligase 2 (New England BioLabs社製) (10 units/μL)(10 μL)を加えて混合した後、温度制御されたヒートブロック上に静置した(37 ℃、16時間)。反応液に同体積のクロロホルムを加えてボルテックスにより混合、遠心分離後、上層をとりアルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。各々のバッチで得られたRNAをまとめ、7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 E1(9.7 nmol, 収率32%)を得た。
化学合成により配列6として得られたRNA断片E2-1ならびに配列7として得られた5'リン酸RNA断片E2-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 E2(10.9 nmol, 収率36%)を得た。
化学合成により配列9として得られたRNA断片E3-1ならびに配列10として得られた5'リン酸RNA断片E3-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 E3 (13.4 nmol, 収率45%)を得た。
化学合成により配列12として得られたRNA断片E4-1ならびに配列13として得られた5'リン酸RNA断片E4-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 E4 (6.8 nmol, 収率34%)を得た。
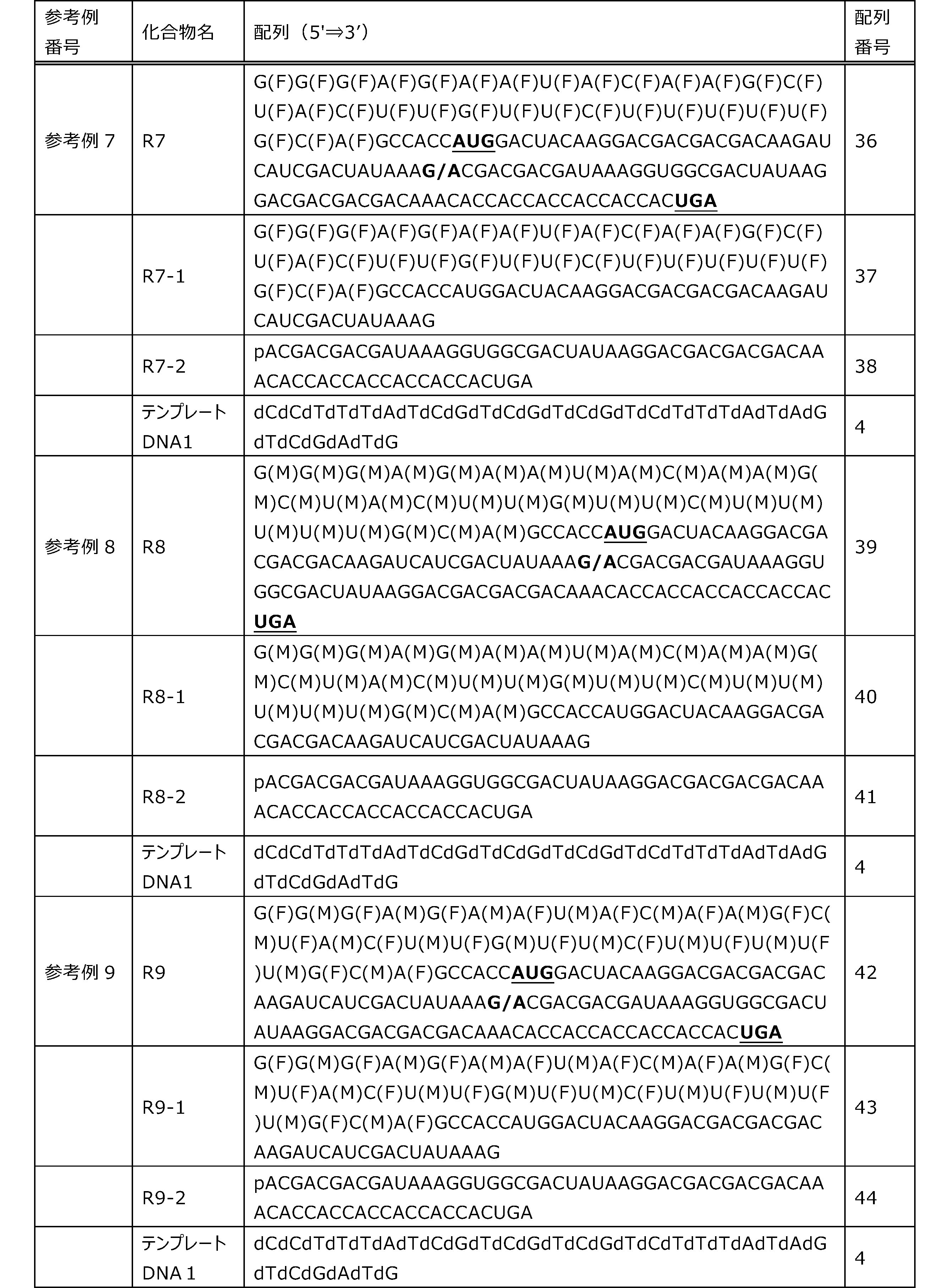
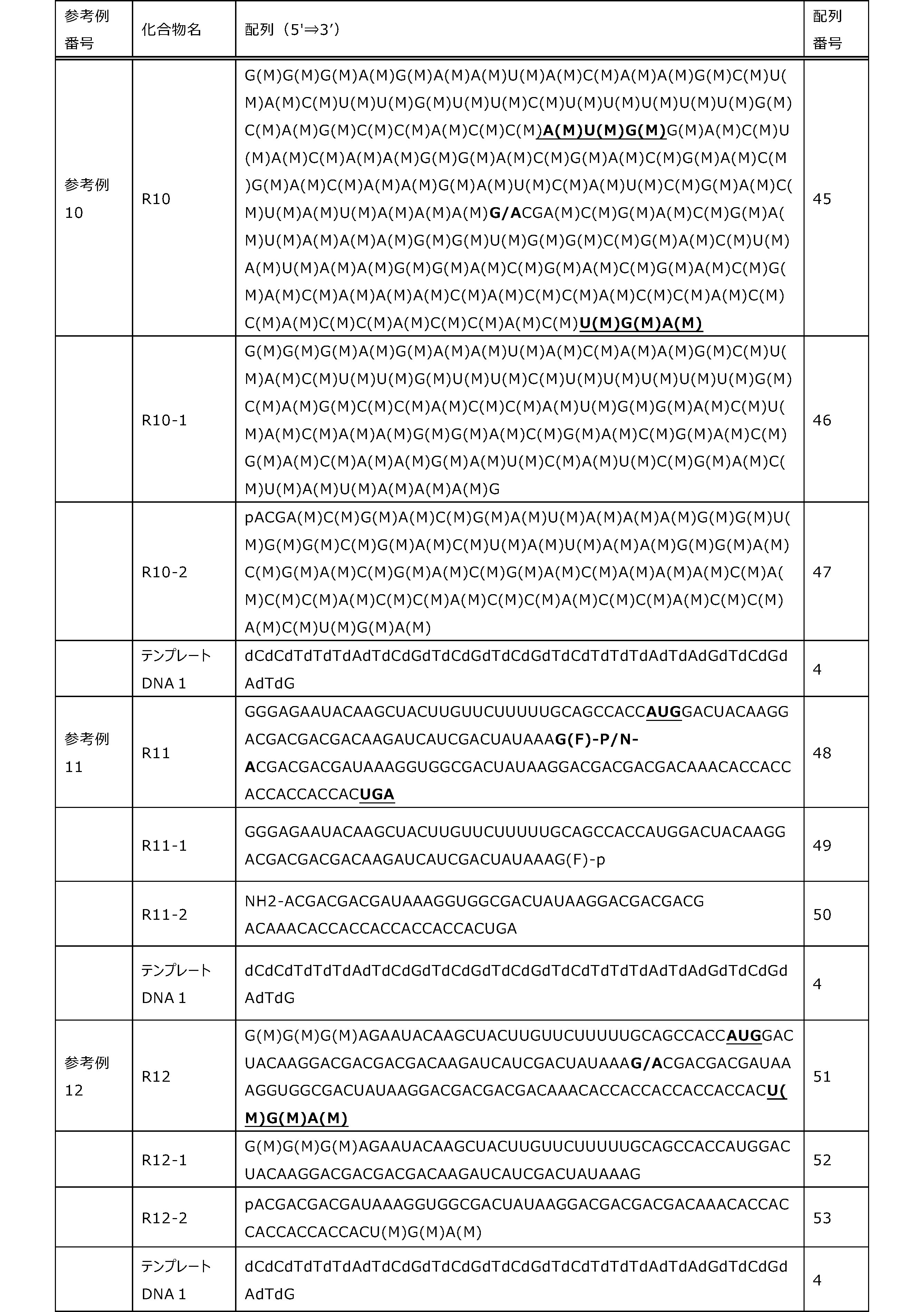
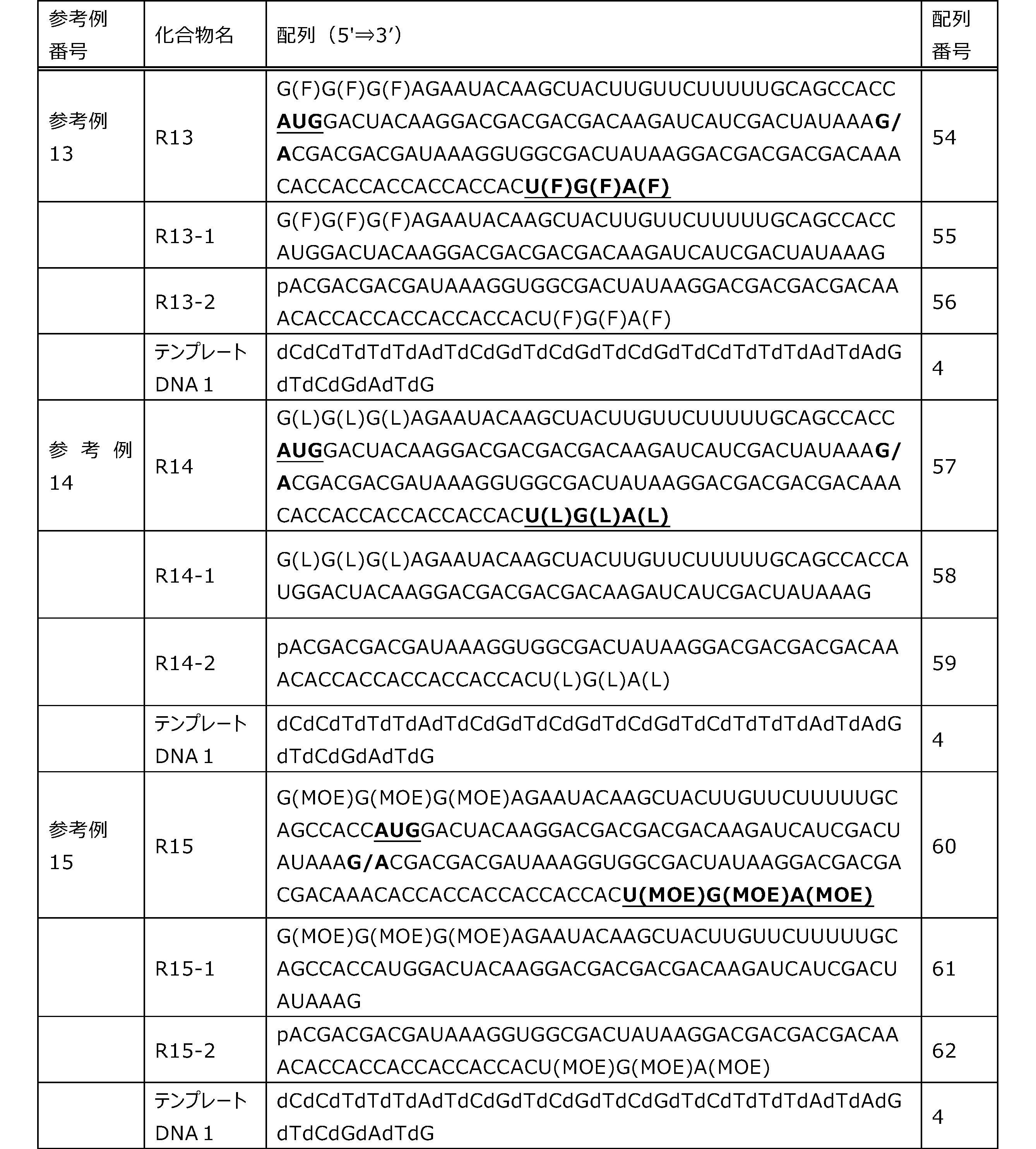

化学合成により配列15として得られた3'-アミノRNA断片R1-1(200 μM)ならびに配列16として得られた5'リン酸RNA断片 R1-2(200 μM)、テンプレートDNA2 (200 μM)の超純水溶液を90℃にて3分間加熱後、30分間以上かけて室温に戻した。この溶液に、別途調製した縮合剤の緩衝溶液 (1M EDC-HCl/1M HOBtの500 mM HEPES-NaOH (pH8.5), 200 mM NaCl, 200 mM MgCl2)を同体積加え、混和することで連結反応を開始した。反応液 (核酸濃度: 100 μM, 0.5M EDC-HCl/0.5M HOBt, 250 mM HEPES-NaOH (pH8.5), 100 mM NaCl, 100 mM MgCl2)は温度制御されたヒートブロック(25℃)上に終夜静置させた。反応後、アルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。
このRNAを7.5%変性ポリアクリルアミドゲルで解析し、反応収率 (収率52%)を算出した。
化学合成により得られたR2-1、R2-2、テンプレートDNA1を用いて参考例1と同様にして、RNA連結産物 R2の生成収率(73%)を算出した。
化学合成により得られたR3-1 (100 μM)、R3-2 (100 μM)、テンプレートDNA3 (100 μM)を用いて参考例1と同様にして、RNA連結産物 R3 (7.7 nmol, 収率26%)を得た。
化学合成により得られたR4-1(100 μM)、R4-2(100 μM)、R4-3(100 μM)、テンプレートDNA1 (100 μM)、テンプレートDNA4 (100 μM)を用いて参考例1と同様にして、RNA連結産物 R4 (2.4 nmol, 収率5.6%)を得た。
化学合成により得られたR5-1 (100 μM)にリン酸バッファー (50 mM), MiliQ水, を加えヨード酢酸 NHSエステル(5 mM)のDMF溶液を加え、30度で2時間インキュベートした。続いてアルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってヨードアセチル化したR5-1のペレットを得た。化学合成により得られたR5-2 (50 μM), テンプレートDNA1 (50 μM), 塩化ナトリウム水溶液 (100 mM)、リン酸バッファー (20 mM, pH 7.5)を混和し、90℃にて3分間加熱後、30分間以上かけて室温に戻した。この溶液に対し、上記ヨードアセチル化したR5-1(50 μM)を添加し、30度で6時間インキュベートした。反応後、アルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。このRNAを7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 R5 (2.32 nmol、収率7.3%)を得た。
化学合成により得られたR6-1、R6-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R6 (11.1 nmol, 収率37%)を得た。
化学合成により得られたR7-1、R7-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R7(3.1 nmol, 収率10%)を得た。
化学合成により得られたR8-1、R8-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R8 (2.6 nmol, 収率9%)を得た。
化学合成により得られたR9-1、R9-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R9 (5.9 nmol, 収率20%)を得た。
化学合成により得られたR10-1、R10-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R10 (2.1 nmol, 収率9%)を得た。
化学合成により配列49として得られたRNA断片R11-1(200 μM)ならびに配列50として得られた5'-アミノRNA断片R11-2(200 μM)、テンプレートDNA1 (200 μM)の超純水溶液を90℃にて3分間加熱後、30分間以上かけて室温に戻した。この溶液に、別途調製した縮合剤の緩衝溶液 (1M EDC-HCl/1M HOBtの500 mM HEPES-NaOH (pH8.5), 200 mM NaCl, 200 mM MgCl2)を同体積加え、混和することで連結反応を開始した。反応液 (核酸濃度: 100 μM, 0.5M EDC-HCl/0.5M HOBt, 250 mM HEPES-NaOH (pH8.5), 100 mM NaCl, 100 mM MgCl2)は温度制御されたヒートブロック(25℃)上に静置させた (7時間)。反応後、アルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。このRNAを7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 R11 (1.2 nmol, 収率15%)を得た。
化学合成により得られたR12-1 (10μM)、R12-2 (10μM)、テンプレートDNA1(10μM)の1x T4 DNA ligaseバッファー溶液 (66 mM Tris-HCl (pH7.6), 6.6 mM MgCl2, 10 mM DTT, 0.1 mM ATP)(タカラバイオ)(計 2.1 mL)を90 ℃で5分間加熱後、室温まで徐冷した。この溶液に60%PEG6000を終濃度15%となるように加えた。この溶液にT4 DNA ligase (タカラバイオ)(350 units/μL)(135 μL)を加えて混合した後、温度制御されたヒートブロック上に静置した(25 ℃、16時間)。 反応液に同体積のクロロホルムを加えてボルテックスにより混合、遠心分離後、上層をとりアルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。このRNAを7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 R12 (1.4 nmol, 収率27%)を得た。
化学合成により得られたR13-1、R13-2、テンプレートDNA1を用いて参考例12と同様にして、RNA連結産物 R13 (1.3 nmol, 収率26%)を得た。
化学合成により得られたR14-1、R14-2、テンプレートDNA1を用いて実施例1と同様にして、RNA連結産物 R14 (9.4 nmol, 収率31%)を得た。
化学合成により得られたR15-1、R15-2、テンプレートDNA1を用いて実施例1と同様にして、RNA連結産物 R15 (8.6 nmol, 収率29%)を得た。
化学合成により得られたR16-1 (10 μM)、R16-2 (10 μM)、テンプレートDNA1(10 μM)を用いて、参考例12と同様にして、RNA連結産物 R16 (1.1 nmol, 収率37%)を得た。
化学合成により得られたR17-1 (50 μM)、R17-2 (50 μM)、テンプレートDNA5 (100 μM)の1x T4 DNA ligaseバッファー溶液 (66 mM Tris-HCl (pH7.6), 6.6 mM MgCl2, 10 mM DTT, 0.1 mM ATP)(タカラバイオ)(計 2.1 mL)を90 ℃で5分間加熱後、室温まで徐冷した。この溶液に50%PEG6000を終濃度10%となるように加えた。この溶液にT4 DNA ligase (タカラバイオ)(350 units/μL)(135 μL)を加えて混合した後、温度制御されたヒートブロック上に静置した(25 ℃、16時間)。 反応液に同体積のクロロホルムを加えてボルテックスにより混合、遠心分離後、上層をとりアルコール沈殿(0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。このRNAを7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 L84(P-O) (30 nmol, 収率22%)を得た。
L84(P-O) (50 μM, 20 μL)、R17-3 (160 μM, 12.5 μL)、テンプレートDNA6 (160 μM, 12.5 μL)の1x T4 DNA ligaseバッファー溶液 (66 mM Tris-HCl (pH7.6), 6.6 mM MgCl2, 10 mM DTT, 0.1 mM ATP)(タカラバイオ)(計 80 μL)を90 ℃で5分間加熱後、室温まで徐冷した。この溶液に50%PEG6000を終濃度10%となるように加えた。この溶液にT4 DNA ligase (タカラバイオ)(350 units/μL)(5 μL)を加えて混合した後、温度制御されたヒートブロック上に静置した (25 ℃、16時間)。 反応液に同体積のクロロホルムを加えてボルテックスにより混合、遠心分離後、上層をとりアルコール沈殿 (0.3M 酢酸ナトリウム水溶液 (pH 5.2)/70%エタノール)によってRNAのペレットを得た。このRNAを7.5%変性ポリアクリルアミドゲルによって精製することでRNA連結産物 R17 (360 pmol, 収率36%)を得た。
化学合成により得られたR18-1、R18-2、テンプレートDNA1を用いて、実施例1と同様にして、RNA連結産物 R18 (11.9 nmol, 収率40%)を得た。
(mRNAサンプルの翻訳反応)
翻訳反応は市販の翻訳キットを用いて製品プロトコールに従って行った。原核細胞系における翻訳反応については、化合物R1及び化合物R17を基質RNAとし、試薬としてPURExpress(登録商標) (New England BioLabs社)を用いて行った。
真核細胞系における翻訳反応については、化合物R2及び化合物R18を基質RNAとし、試薬としてRabbit Reticulocyte Lysate System, Nuclease Treated (以下、RRL) (Promega社)を用いて行った。
それぞれのキットの推奨プロトコールに従って調製した。基質となるRNA以外の材料を含む反応液を翻訳反応液とした。遠心エバポレーターにて乾固したRNAサンプルを含むチューブに対し、翻訳反応液を添加・混合した後、適切な温度のヒートブロック上に置くことで翻訳反応を開始した。翻訳産物は抗FLAG抗体を用いたウエスタンブロッティングによって検出した。一次抗体として抗FLAG抗体 (anti FLAG antibody)(F1804, Sigma社)、二次抗体として抗マウスIgG抗体 (anti-mouse IgG-HRP)(A9044, Sigma社)を使用した。
PURExpress(登録商標)キット付属のSolution A (2 μL), Solution B (1.5 μL), RNase阻害剤 (Murine RNase Inhibitor, New England BioLabs)(0.1 μL), 超純水 (1.4 μL)を混合したものを翻訳反応液とした。この溶液をRNAサンプルを含むチューブに対し、添加・混合することで翻訳反応を行った (37℃, 2時間)。
Rabbit Reticulocyte Lysate Systemキット付属のRabbit Reticulocyte Lysate (14.0 μL), Amino Acid Mixture - Met (0.20 μL), Amino Acid Mixture - Leu (0.20 μL), Murine RNase Inhibitor (New England BioLabs)(0.4 μL), 超純水 (5.2 μL)を混合したものを翻訳反応液とした。この溶液をRNAサンプルを含むチューブに対し、添加・混合することで翻訳反応を行った (30℃, 2時間)。
SDS-PAGEにおいては不連続緩衝液系を用いた。使用したゲルの組成を以下に示す。上層には濃縮ゲル領域 (約1.5 cm)として5%ポリアクリルアミド (アクリルアミド : ビスアクリルアミド= 29 : 1)(0.125M Tris-HCl (pH6.8), 0.1%SDS)、下層には分離ゲル領域として15%ポリアクリルアミド (アクリルアミド : ビスアクリルアミド= 29 : 1)(0.375M Tris-HCl (pH8.8), 0.1%SDS)を使用し、APSの水溶液とTEMEDを重合剤として添加し、固形化 (室温、30分間)させることでゲルを作製した。翻訳反応の反応液を2x SDS-PAGE用ローディングバッファー (125 mM Tris-HCl (pH6.8), 30(v/v)%グリセロール, 4%ドデシル硫酸ナトリウム (以下、SDS), 0.03% ブロモフェノールブルー (以下、BPB))と混合した後、90℃にて3分間加熱したものをSDS-PAGE分析用のサンプルとした。SDS-PAGE分析用のサンプルは直ちにSDS-PAGEゲルにて泳動 (泳動用緩衝液として25mM Tris, 192 mM Glycine, 0.1% SDSを使用)を行った。
電気泳動後、ゲル上の翻訳産物はセミドライ法にてウエスタンブロッティング用のメンブレン (Immobilon(登録商標)-P)(IPVH00010, Millipore社) (メンブレンは前処理としてメタノールにて親水化した後、ブロッティングバッファー (25 mM Tris, 192 mM Glycine, 20% MeOH)に浸したものを使用)に転写した。転写においては定電流条件 (通電時間:1時間)を使用し、メンブレンのサイズから用いる電流値を決定した。すなわちメンブレン面積 (cm2) x 2となるように電流値 (mA)を設定した。
以降の操作において使用している5% ECL Prime のTBS-T溶液はAmersham ECL Prime (GE Healthcare)とTBS-T (0.05M Tris-HCl (pH7.4), 150 mM NaCl, 0.05% tween20)を混合することで調製した。翻訳産物を転写したメンブレンはブロッキング処理 (5% ECL Prime のTBS-T溶液、室温、1時間振とう)した後、一次抗体処理 (4,000倍希釈、0.5% ECL PrimeのTBS-T溶液、4℃、12時間振とう)、洗浄 (TBS-T、5分間x 5回振とう)、二次抗体処理 (50,000倍希釈、0.5% ECL PrimeのTBS-T溶液、室温、1時間振とう)、洗浄 (TBS-T、5分間x 5)を行った。抗体処理後、メンブレン上の翻訳産物は化学発光試薬 (SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific社)を用い、製品推奨のプロトコールに従って検出を行った。化学発光試薬による処理後、ChemiDoc (BIORAD社)を用いて翻訳産物のシグナルを検出した (検出モード:化学発光、露光時間:90 - 300秒)。結果を図4及び 図5に示す。
(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)
実施例1~3および参考例6~10、14、15、18で得られた各化合物について、Rabbit-Reticulocyte-Lysate-System-Nuclease-Treatedキット(プロメガ社製、カタログ番号L4960)を用いて真核細胞系における翻訳活性を評価した。まず、各化合物の終濃度が0.3 μMとなるようTHE RNA storage solution(サーモフィッシャーサイエンティフィック社、カタログ番号AM7001)で希釈した各mRNAサンプルを、96 well PCRプレート(アズワン社製)に2 μLずつ分注した。続いて、1反応あたり7.0 μLのReticulocyte Lysate, Nuclease Treated、1反応あたり0.1 μLのAmino Acid Mixture Minus Leucine、1反応あたり0.1 μLのAmino Acid Mixture Minus Methionine、1反応あたり0.4 μLのRNase Inhibitor, Murine(ニューイングランドバイオラボ社製、カタログ番号M0314)および1反応あたり0.4 μLの精製水を混合してマスターミックスを調製し、mRNAサンプルを添加したPCRプレートに8 μLずつ分注し、添加、混合の後、37℃で1時間静置することにより翻訳反応を行った。
翻訳反応後の反応溶液中の翻訳産物は、以下に記載のサンドイッチELISA法により検出した。まず、6*His, His-Tag 抗体(プロテインテック社、カタログ番号66005-1-Ig)を0.1M Carbonate buffer(pH9.4)にて3 μg/mLに希釈し、96 Well ELISA用プレート(ヌンク社製)に1ウェルあたり50 μLずつ分注し、4℃で一晩静置することにより、抗体の固定化を行ったプレートを作製した。続いて、プレートを精製水で1倍濃度に希釈したTris Buffered Saline with Tween 20 (サンタクルツ社、カタログ番号sc-24953)(以下、洗浄溶液と記載する)で洗浄した後、ウシ血清アルブミン(和光純薬社、カタログ番号017-22231)を終濃度3%となるように希釈した洗浄溶液(以下、ブロッキング溶液と記載する)を1ウェルあたり200 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で100倍希釈した翻訳反応溶液を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。その際、翻訳産物ポリペプチド標品(スクラム社製)を同様にブロッキング溶液で各濃度に希釈してプレートに分注した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で10,000倍に希釈したMonoclonal ANTI-FLAG M2-Peroxidase (HRP) Ab produced in mouse (SIGMA社製、カタログ抗体A8592-1MG)を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、1-Step Ultra TMB-ELISA (サーモフィッシャーサイエンティフィック社製、カタログ番号34028)を、1ウェルあたり50 μLずつ分注し、室温で数分静置した。その後、0.5M硫酸 (和光純薬工業社製)を、1ウェルあたり50 μLずつ分注し反応を停止させた後、吸光光度計(バイオラッド社製)を用いて、測定波長450 nm、参照波長570 nmの吸光度を測定した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(μM)、および糖部修飾を持たないR18を1とした時の相対翻訳産物量を、表9~11に記載した。



実施例1~4および参考例6、10、14、15、18で得られた各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。まず、各化合物の終濃度が0.3 μMとなるようTHE RNA storage solution(サーモフィッシャーサイエンティフィック社、カタログ番号AM7001)で希釈した各化合物を、96 well PCRプレート(アズワン社製)に1 μLずつ分注した。続いて、1反応あたり5.0 μLのHela Lysate、1反応あたり1.0 μLのAccessory Proteins、1反応あたり2.0μLのReaction Mix、1反応あたり0.2 μLのRNase Inhibitor, Murine(ニューイングランドバイオラボ社製、カタログ番号M0314)および1反応あたり0.8 μLの精製水を混合してマスターミックスを調製し、mRNAサンプルを添加したPCRプレートに9 μLずつ分注し、添加、混合の後、37℃で45分間静置することにより翻訳反応を行った。
翻訳反応後の反応溶液中の翻訳産物は、試験例2(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験) に記載のサンドイッチELISA法により、翻訳反応溶液をブロッキング溶液で20倍希釈してプレートに添加すること以外は全く同じ方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(μM)、および糖部修飾を持たない化合物R18を1とした時の相対翻訳産物量を、表12~14に記載した。
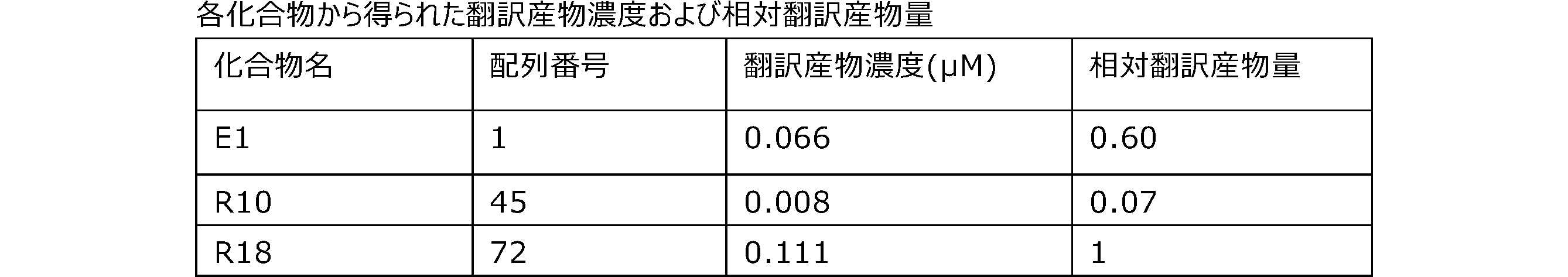


実施例2、3および参考例6、15で得られた各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。まず、10%ウシ胎仔血清を含むRPMI培地(ナカライテスク社製)で懸濁したHela細胞を1ウェルあたり細胞数10,000 cells/100 μLとなるように、96ウェル接着細胞用培養プレートに播種し、37℃、5% CO2条件下で一晩培養した。一晩培養後の細胞から培養上清を除去し、1ウェルあたり40 μLの10%ウシ胎仔血清を含むRPMI培地を添加した後、各化合物の終濃度が0.3 μMとなるよう、各化合物とLipofectamin MessengerMAX Transfection Reagent(サーモフィッシャーサイエンティフィック社製、カタログ番号:LMRNA008)とをオプティメム(サーモフィッシャーサイエンティフィック社製、カタログ番号:31985-070)で希釈して混合し、混合液を1ウェルあたり10 μLとなるよう各々の培養プレートに添加し、37℃、5% CO2条件下で6時間培養した。6時間培養後の細胞から培養上清を除去し、氷冷したD-PBS(-)(ナカライテスク社製)で1度洗浄した後、2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むNP-40(インビトロジェン社、FNN0021)を1ウェルあたり20 μL加え、5分間激しく浸透して細胞を溶解した。
得られた細胞溶解液中の翻訳産物は、試験例2(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験) に記載のサンドイッチELISA法により、細胞溶解液をブロッキング溶液で10倍希釈してプレートに添加すること以外は全く同じ方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および糖部修飾を持たない化合物R6を1とした時の相対翻訳産物量を、表15に記載した。

(各化合物のphosphodiesterase Iによる安定性試験)
実施例1および参考例10、18で作製した各化合物(RNA溶液)を用い、以下の方法で phosphodiesterase I (SVPD)に対する安定性を評価した。MQ水 10 μL, 5 μM RNA溶液 4 μL, 5 x 反応バッファー (Tris-HCl pH 8.0 0.1 M, NaCl 0.5M, MgCl2 0.075 M) 4 μL, 5.5U/μL のSVPD (warthington、カタログ番号3926) 4 μL を添加して37℃でインキュベートした。反応開始後、15, 30, 60分のタイミングで6 μL ずつサンプリングした。
(PAGEによるRNA分解評価)
5%dPAGE(5% acrylamide, 7M urea, small gel, 8 cm x 8 cm)にサンプリングした検体全量をアプライし、300V constant,18 min泳動した。SYBR Green IIで染色し撮影及び定量を行った。各試料中の各化合物の残存量を酵素未反応(0分)に対する相対残存量として算出したものを、表16に記載した。

(mRNAサンプルのphosphodiesterase Iによる安定性試験)
実施例1~4および参考例6、14、15、18で得られた各化合物について、phosphodiesterase I(warthington、カタログ番号3926)を用いて酵素安定性を評価した。まず、phosphodiesterase IをSVPDストック溶液(110 mM Tris-HCl(ニッポンジーン、カタログ番号314-90401)、110 mM NaCl(Ambion、カタログ番号AM9759)、15 mM MgCl2(ナカライテスク、カタログ番号20942-34))で終濃度が55 U/mLとなるように調製し、これをさらに5×SVPD反応buffer(100 mM Tris-HCl(Ambion、カタログ番号AM9855G)、500 mM NaCl、75 mM MgCl2)で10倍希釈して5.5 U/mL SVPD酵素溶液を調製した。各化合物は終濃度が5 μMとなるようTHE RNA storage solution(サーモフィッシャーサイエンティフィック社、カタログ番号AM7001)で希釈した。
96 well PCRプレートにDistilled water 30 μL、5 μM mRNA 12 μL、5×SVPD反応buffer 12 μLを順に加え酵素反応premix溶液を作製した。酵素未反応(0 min)用として別の96 well PCRプレートに酵素反応premix溶液を9 μL分注し、SVPD溶液1 μLと25mM EDTA(Ambion、カタログ番号AM9260G)2.5 μLの混合溶液3.5 μLを添加して-30 ℃で保存した。酵素反応用として、また別の96 well PCRプレートに酵素反応premix溶液を40.5 μL分注し、5.5 U/mL SVPD酵素溶液を4.5 μL添加してよく混合した。これを10 μLずつ新しい96 well PCRプレート 4枚に分注して各プレート所定の時間(15 min、30 min、60 min)37 ℃で反応させた後、25 mM EDTAを2.5 μL添加して測定まで-30 ℃で保存した。
酵素反応後の反応溶液中の残存mRNA量は、以下に記載のRT-qPCR法により検出した。まず、検量線には化合物E4を使用し、THE RNA storage solution によって4 μMから4倍希釈で11点濃度を取り、希釈系列を作製した。検量線及び酵素反応後のサンプル2.5 μLはRibonuclease Inhibitor(タカラバイオ、カタログ番号2311B)を終濃度0.2 U/mLで加えたDistilled waterを用いて1071倍希釈した。この希釈した試料5 μLおよび2μM RT primer(シグマアルドリッチ社) 2 μLを用いてiScript Select cDNA Synthesis Kit(BIO-RAD、カタログ番号1708897)により逆転写産物cDNAを作製した。反応温度は25 ℃ 5 min → 42 ℃ 30 min → 85 ℃ 5 minで実施した。cDNA 2 μL、TaqMan Gene Expression Master Mix 10 μL、Fw primer(シグマアルドリッチ社) 0.28 μL、Rv primer(シグマアルドリッチ社) 0.33 μL、TaqMan MGB Probe(サーモフィッシャーサイエンティフィック社、カタログ番号4316033) 0.38 μLおよびDistilled water 7.01 μLを混合し、qPCR測定を行った。機器はQuantstudio12K Flex(Applied Biosystems)を使用した。また、使用したPrimerおよびTaqman MGB ProbeのDNA配列は以下である。測定の結果、標品のCT値をもとに検量線を用いて、各試料中の各化合物の濃度を定量し、酵素未反応(0分)に対する相対残存量として算出したものを、表17に記載した。
RT primer:5'-TCAGTGGTGGTGGTGGTGGTGTTTG-3' (配列番号75)
Fw primer:5'-ATCTTGTCGTCGTCGTCCTT-3' (配列番号76)
Rv primer:5'-GAATACAAGCTACTTGTTCTTTT-3' (配列番号77)
Taqman MGB Probe:5'-CAGCCACCATG-3' (配列番号78)

同じく、糖部修飾を有するE2およびE3は糖部修飾を有さない化合物R6と比較してphosphodiesteraseIに対する耐性が向上した。
(真核細胞系における翻訳反応)
参考例3~5、11~13、16、18で得られた各化合物(RNA)について、以下の方法で真核細胞系における翻訳反応を評価した。Rabbit-Reticulocyte-Lysate-System_-Nuclease-Treated (Promega L4960)に含まれる溶液、RNase inhibitor Murine(New England Biolab, M0314S)及びRNAを以下の組成で混合した。RRL 7.0 μL, 1 mM AA-Leu 0.1 μL, 1mM AA-Met 0.1 μL, Rnase Inhibitor 0.4 μL, RNA 5μM 2.0 μL, MQ 0.4 μL。この溶液を37℃で1時間インキュベートした。インキュベート後の溶液5 μLを分取しBlocking buffer (3%BSA/TBST) 495μLで100倍希釈した。希釈翻訳液100μLをELISAに使用した。
96wellプレートに100μL/wellの Anti-Histag溶液(プロテインテック社、カタログ番号66005-1-Ig) (3μg/mL in 0.1M Carbonate buffer pH9.4)を添加した。パラフィルムで覆い、4℃で12時間インキュベートした。プレート内の溶液を廃棄し、200μL/wellのTBSTで3回洗浄した。200μL/wellのBlocking buffer(3%BSA/TBST)を添加し室温にて1時間インキュベートした。プレート内の溶液を廃棄し、200μL/wellのTBSTで3回洗浄した。100μL/wellで翻訳反応溶液を添加し室温にて1時間インキュベートした。プレート内の翻訳反応溶液を廃棄し、200μL/wellのTBSTで3回洗浄した。100μL/wellのAnti-Flag溶液(Cell Signaling Tecnorogy社、カタログ番号2368)(1:1000 in bloking buffer)を添加した。乾燥濃縮しないようパラフィルムで覆い、終夜4℃でインキュベートした。プレート内の溶液を廃棄し、200μL/wellのTBSTで3回洗浄した。100μL/wellのAnti-rabbit IgG HRP溶液(シグマアルドリッチ社)(1:10000 in bloking buffer)を添加し、室温で1時間インキュベートした。プレート内の溶液を廃棄し、200μL/wellのTBSTで4回洗浄した。100μL/wellのTMB substrate溶液を添加し、室温にて5分インキュベートした。TBM subustrate溶液を添加してあるwellに100μL/wellの2M H2SO4溶液を添加した。プレートリーダー(Mithras LB940 (Berthold))を使用し、 450nmの吸光度を測定し、翻訳効率を評価した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量し、各翻訳反応溶液中の翻訳産物濃度(μM)、および糖部修飾を持たないR18を1とした時の相対翻訳産物量を表18~20に記載した。

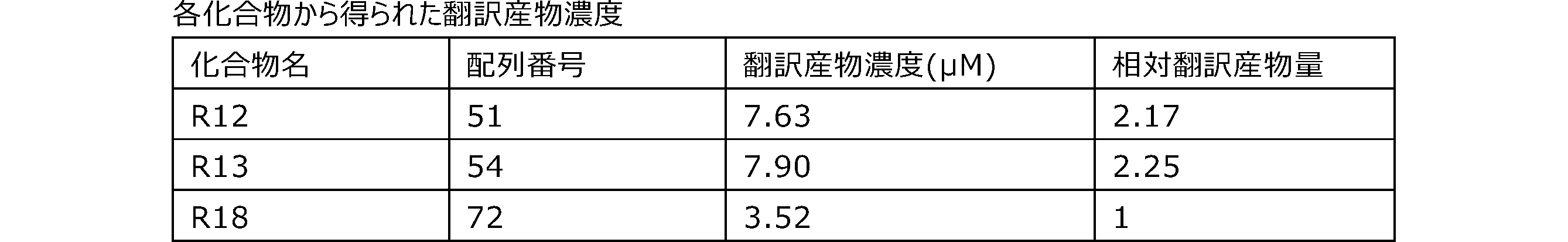
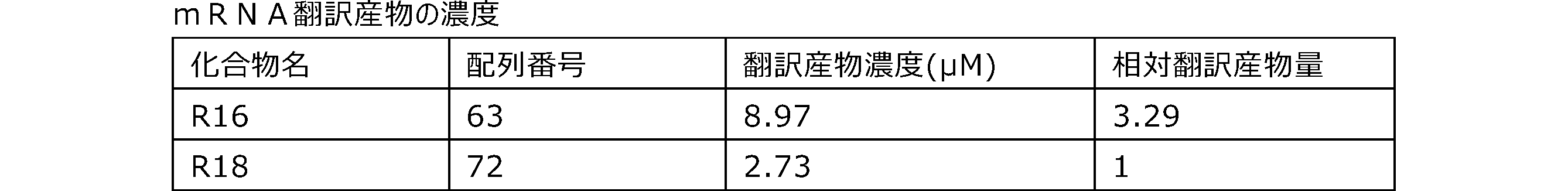
以下表中の各ヌクレオチドN(大文字)はRNAを、各ヌクレオチドn(小文字)はDNAを、 N(M)は2'-O-メチル修飾RNAを、N(F)は2'-F化修飾RNAを、N(L)はLNAを、N(MOE)は2'-O-メトキシエチル修飾RNAを表す。Am6は塩基部がN6-メチルアデニンであること、Ae6は塩基部がN6-エチルアデニンであることを表す。pは3'または5'末端がリン酸化されていることを表す。^は糖部間をつなぐリン酸基がホスホロチオエートであることを表す。N(B)は、下記に示す糖部を含む2',4'-BNANC(Me)を表す。



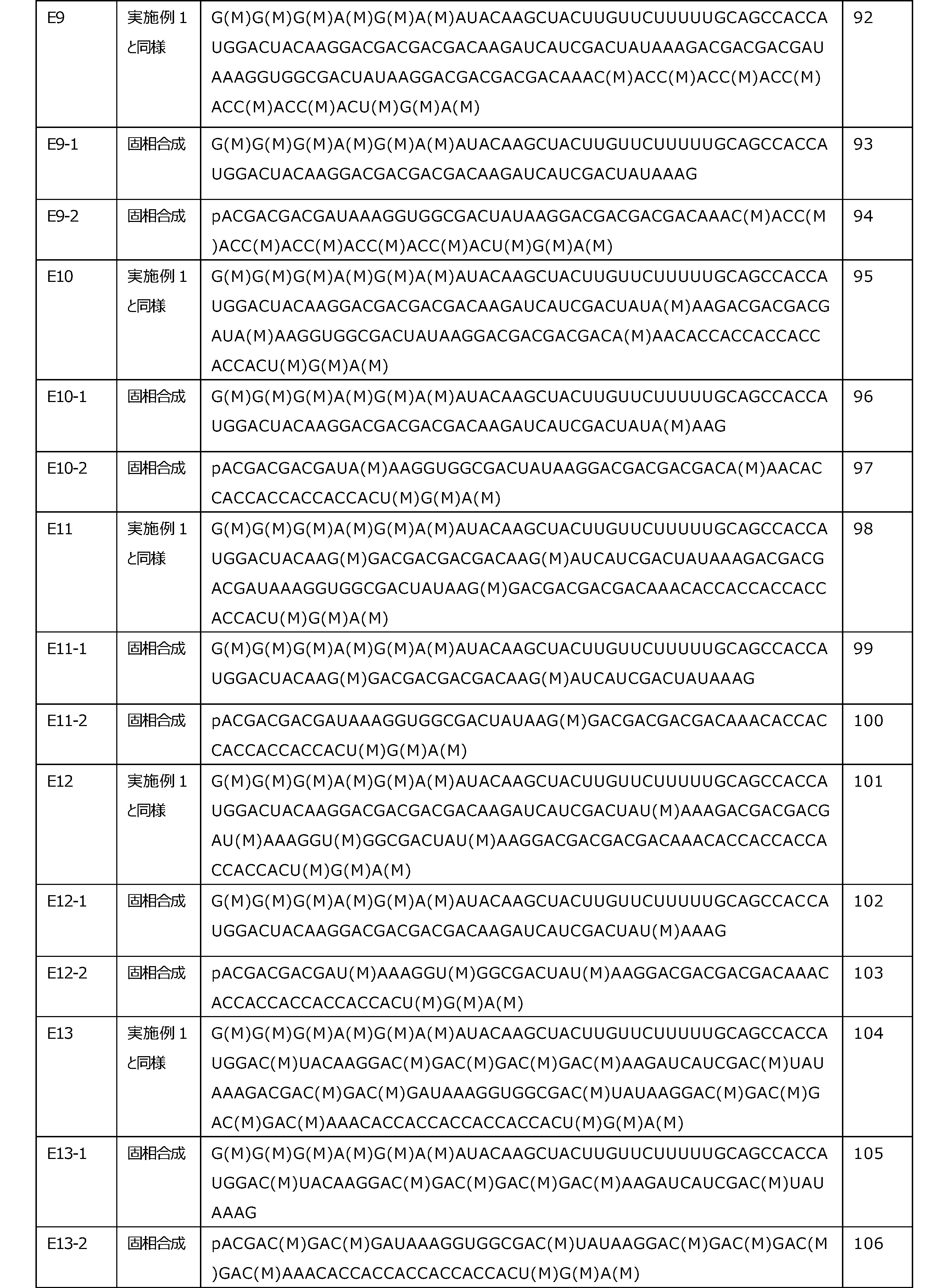
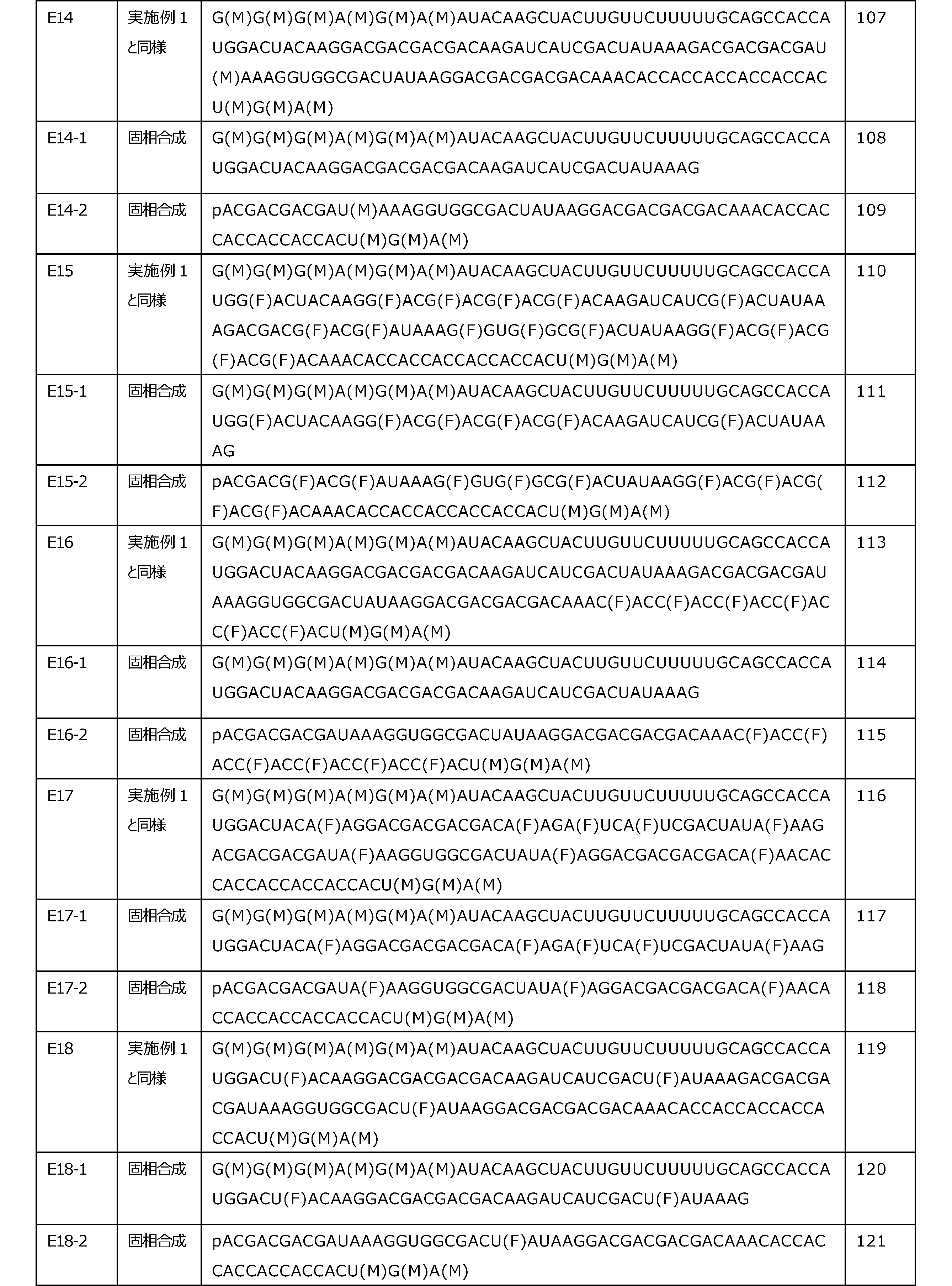
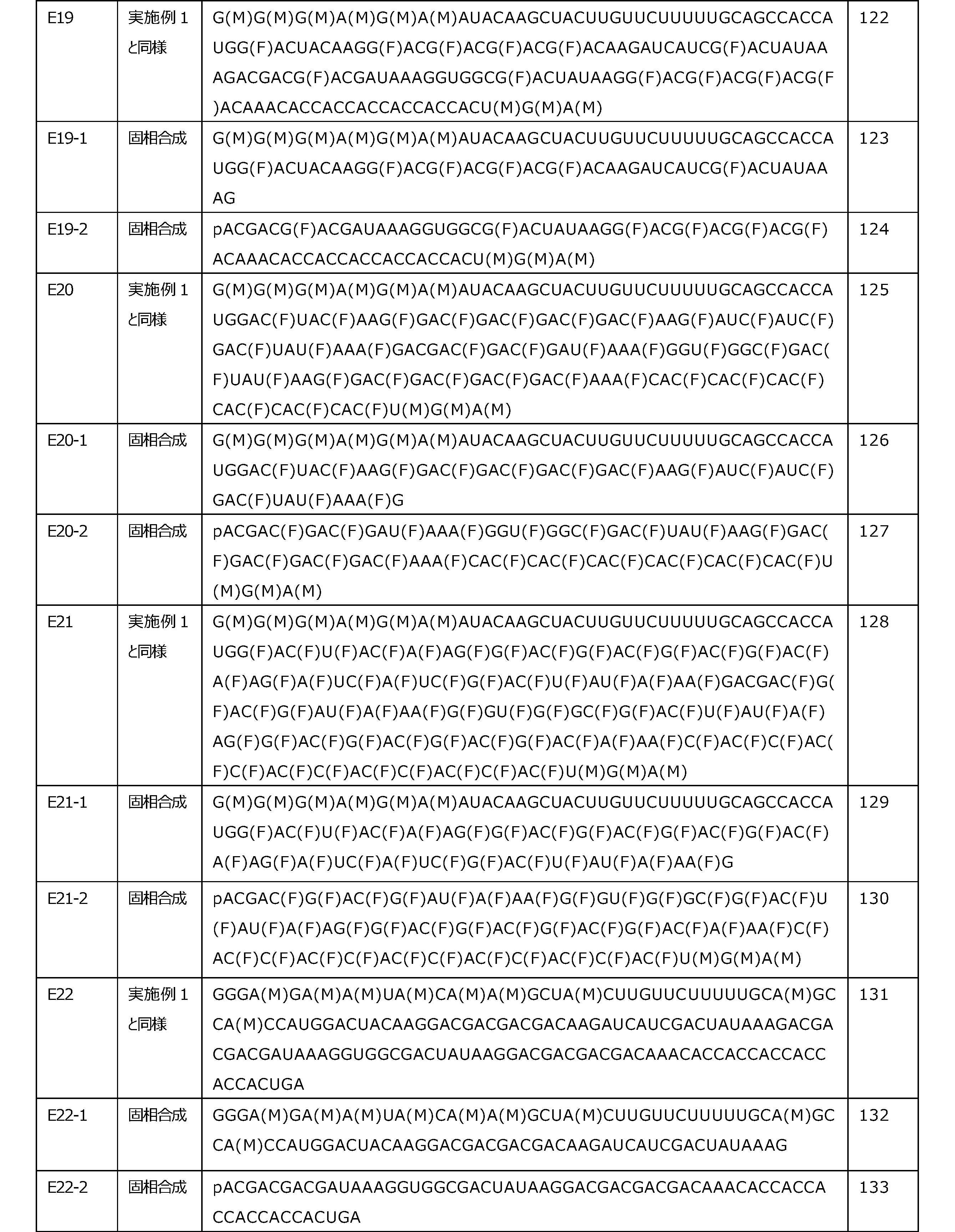



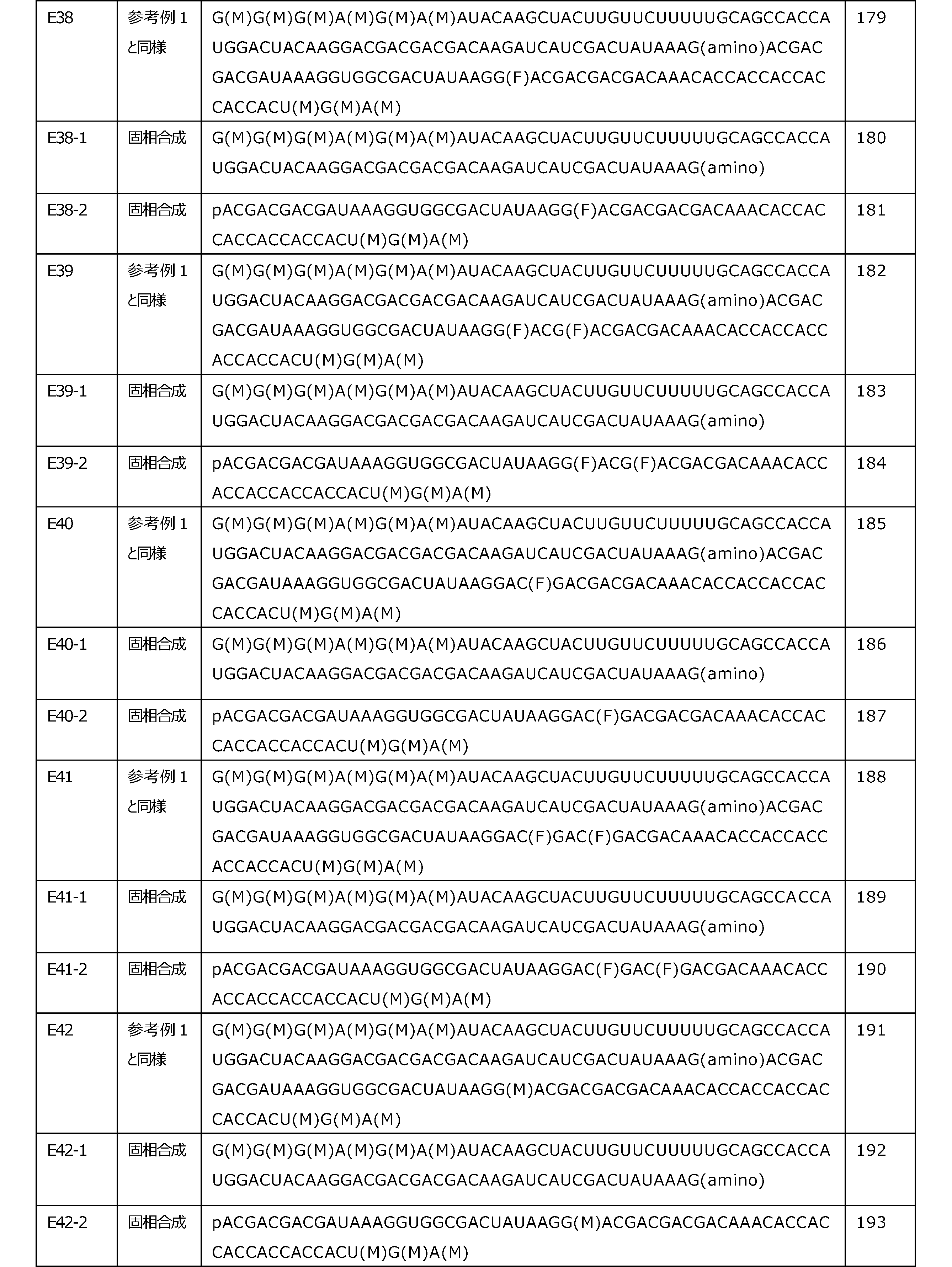






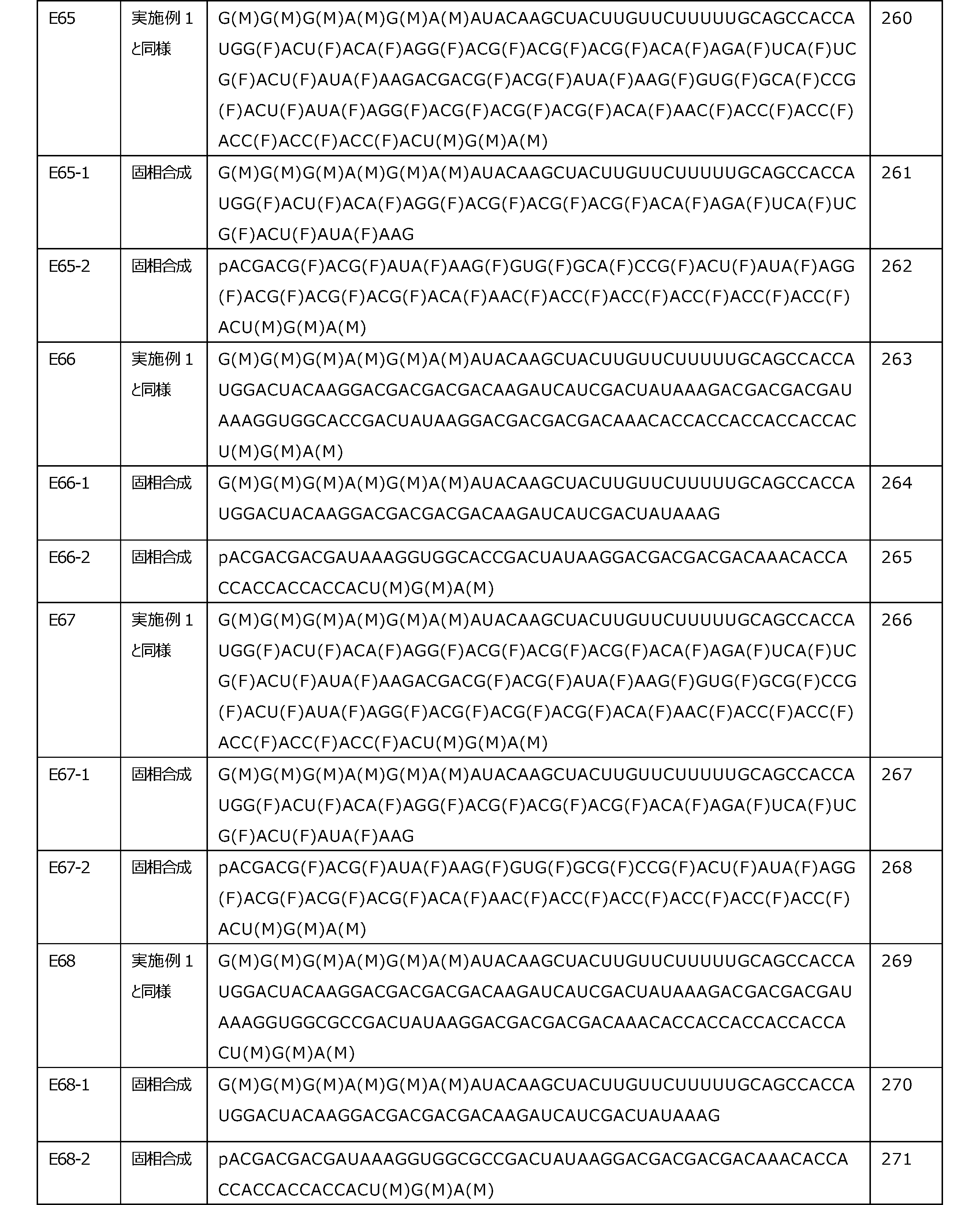
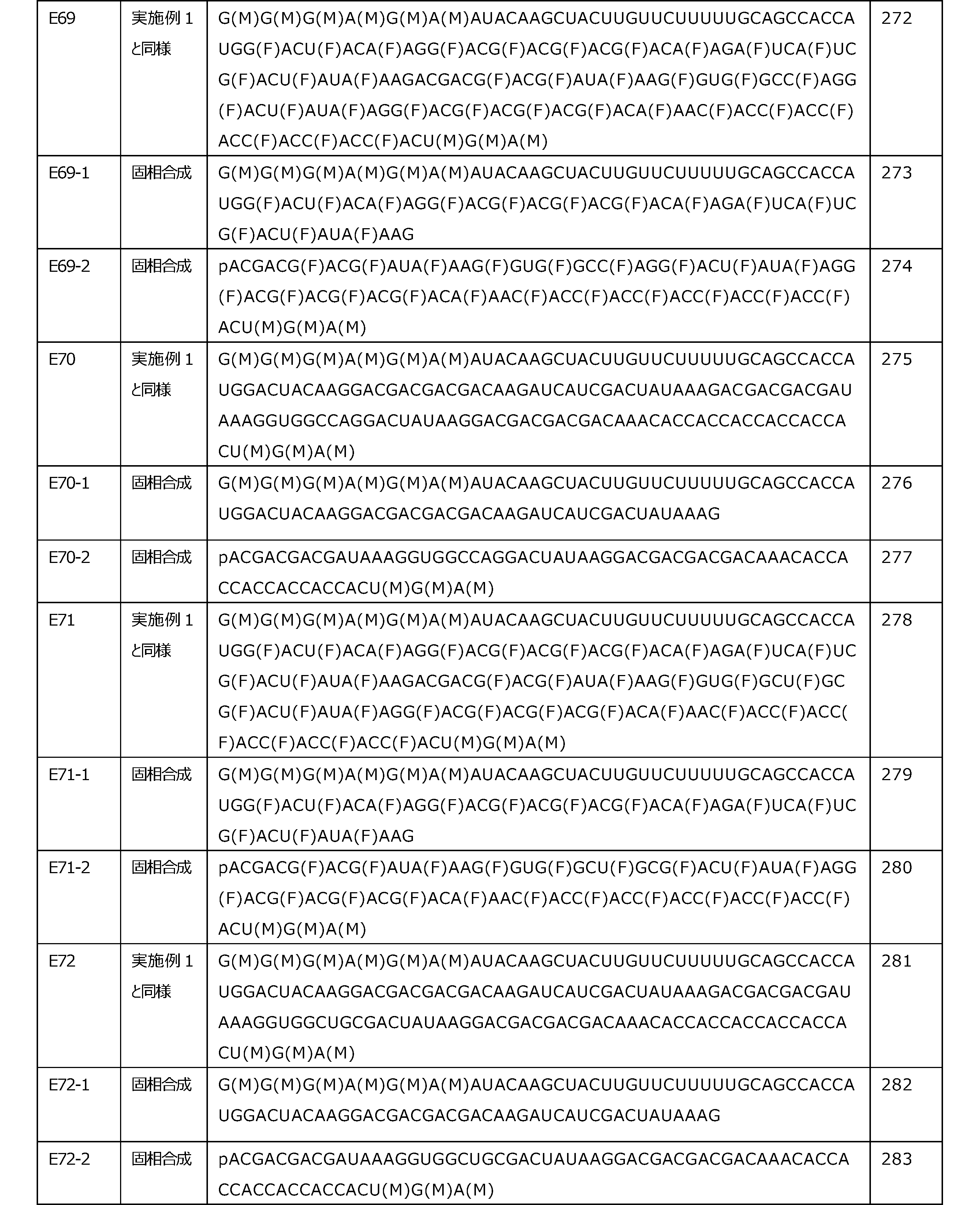




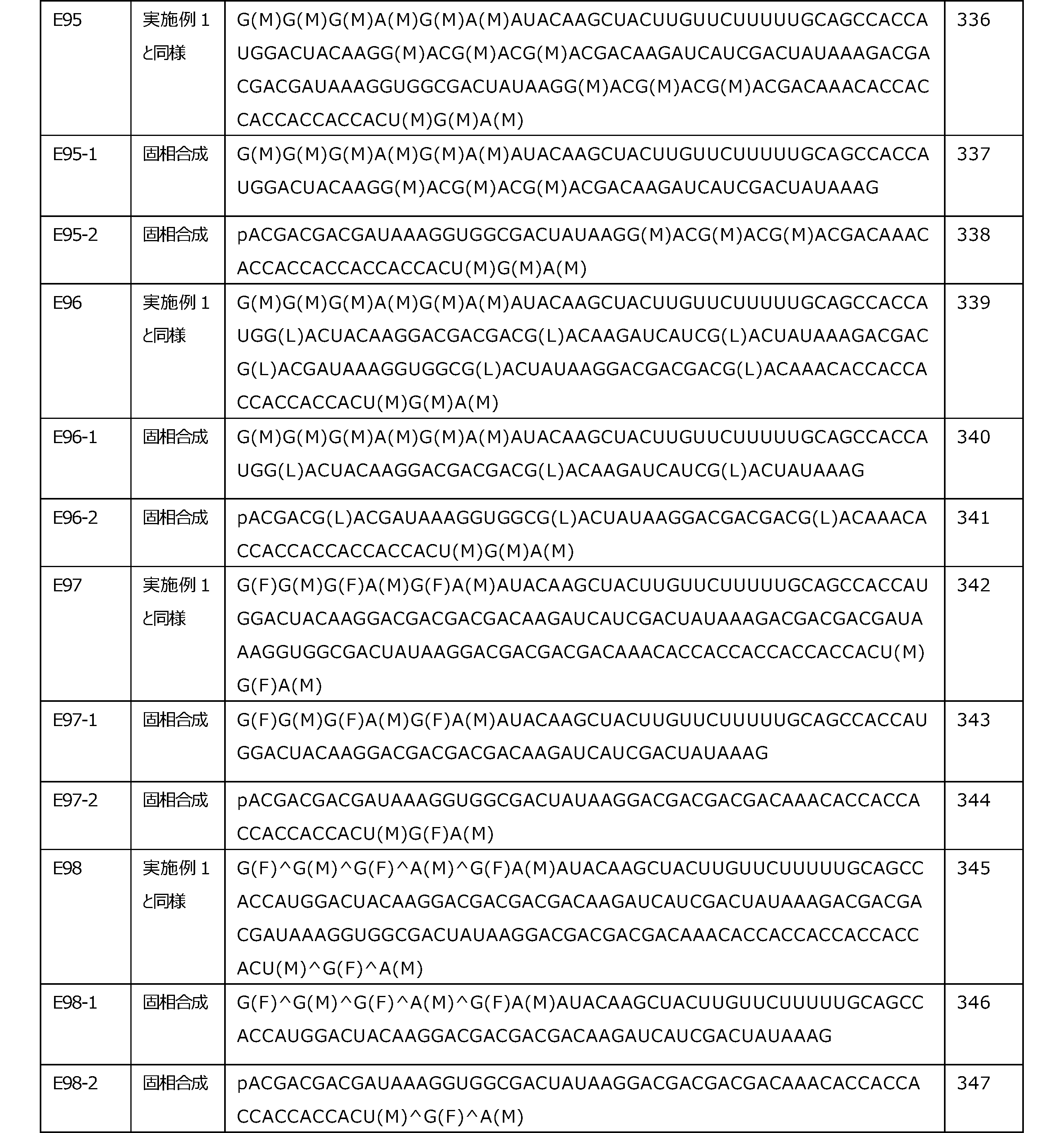


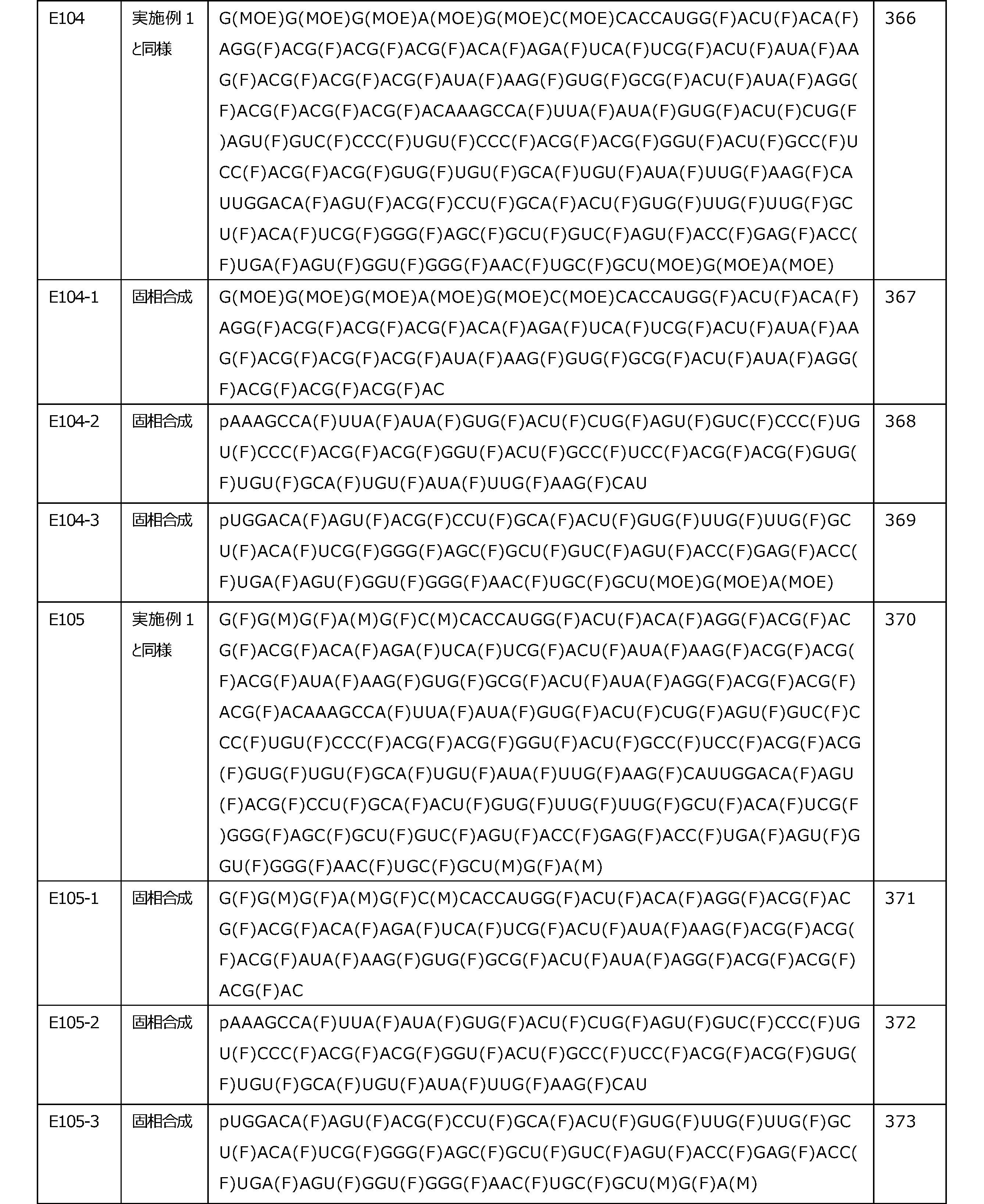


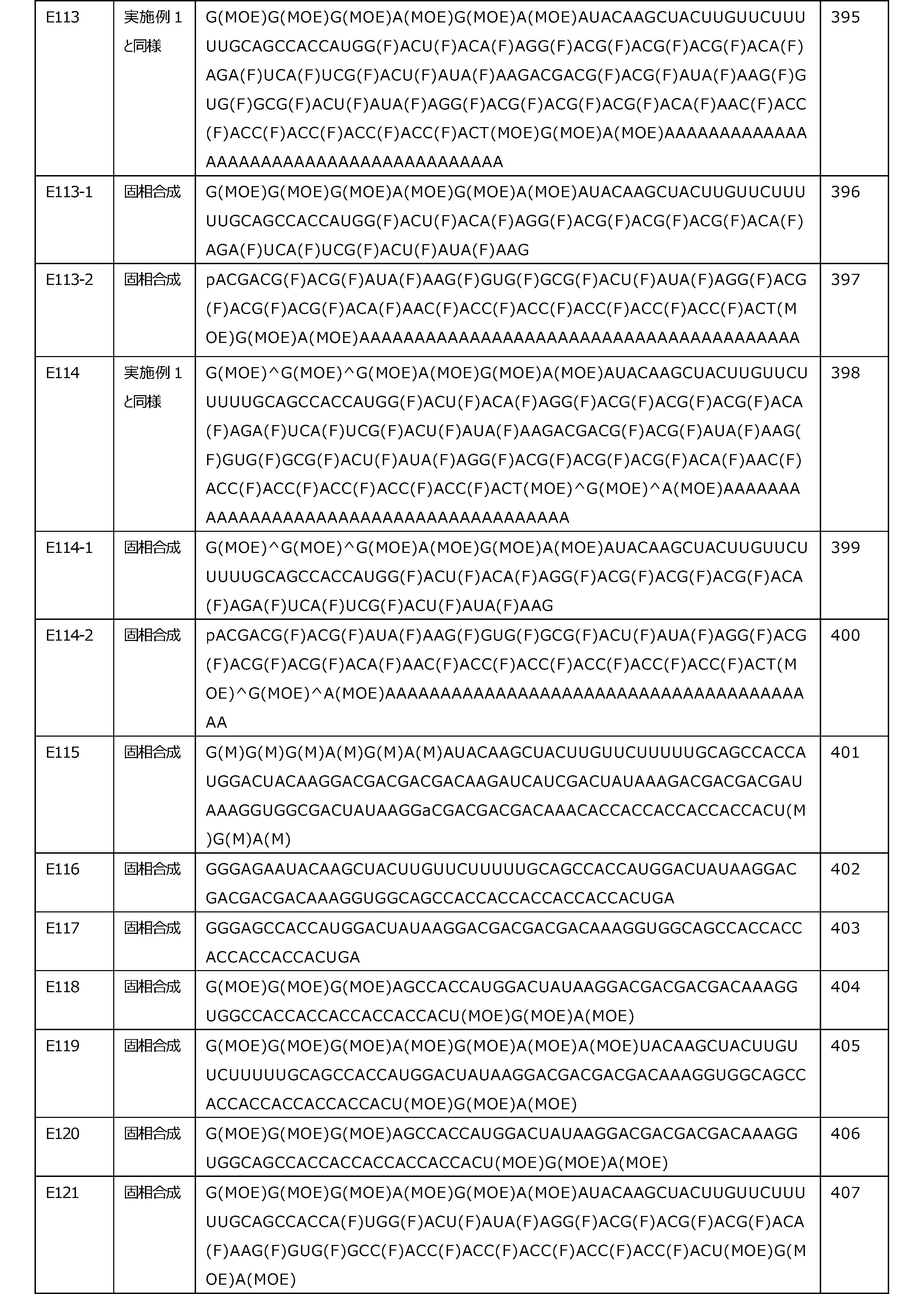


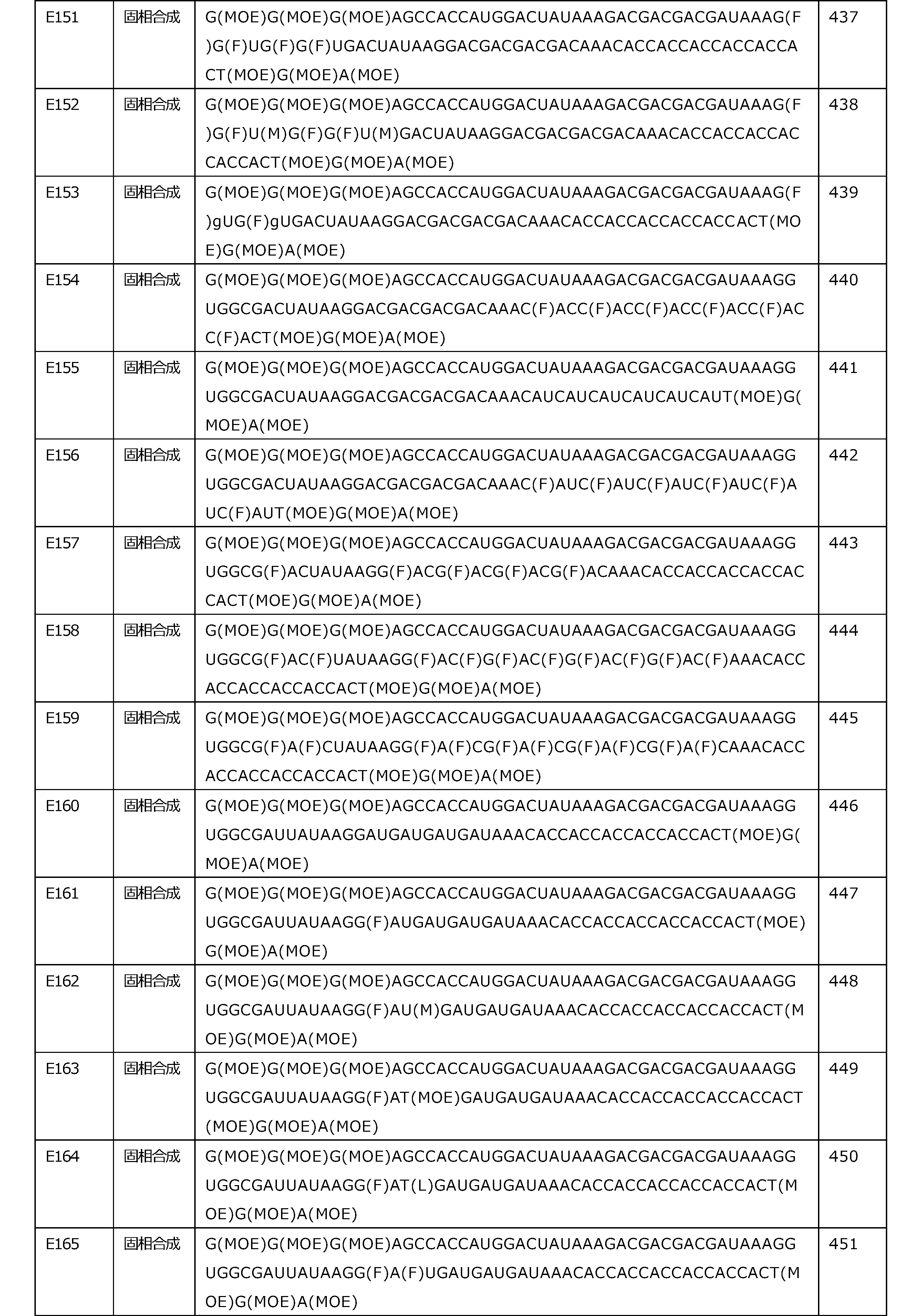
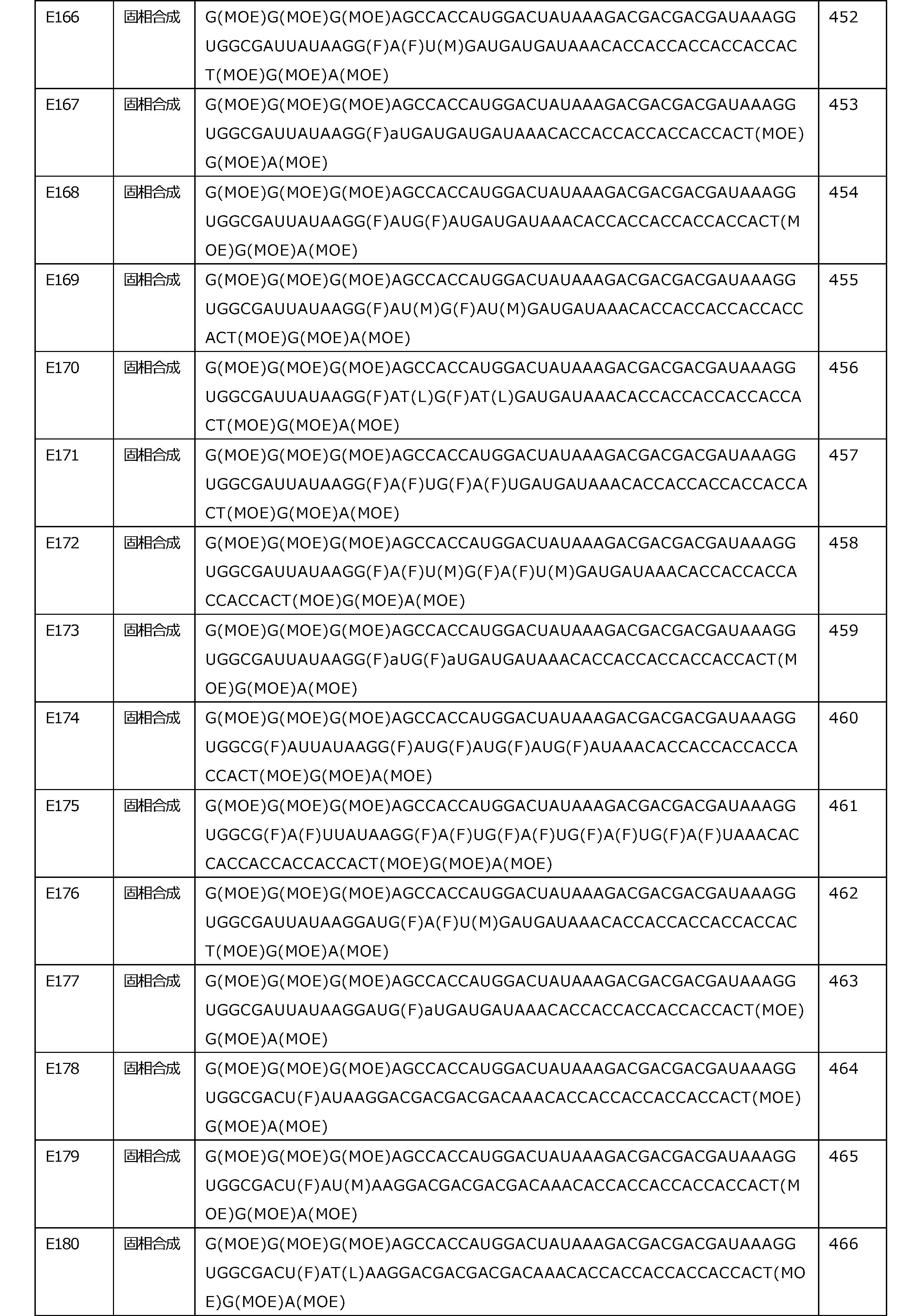


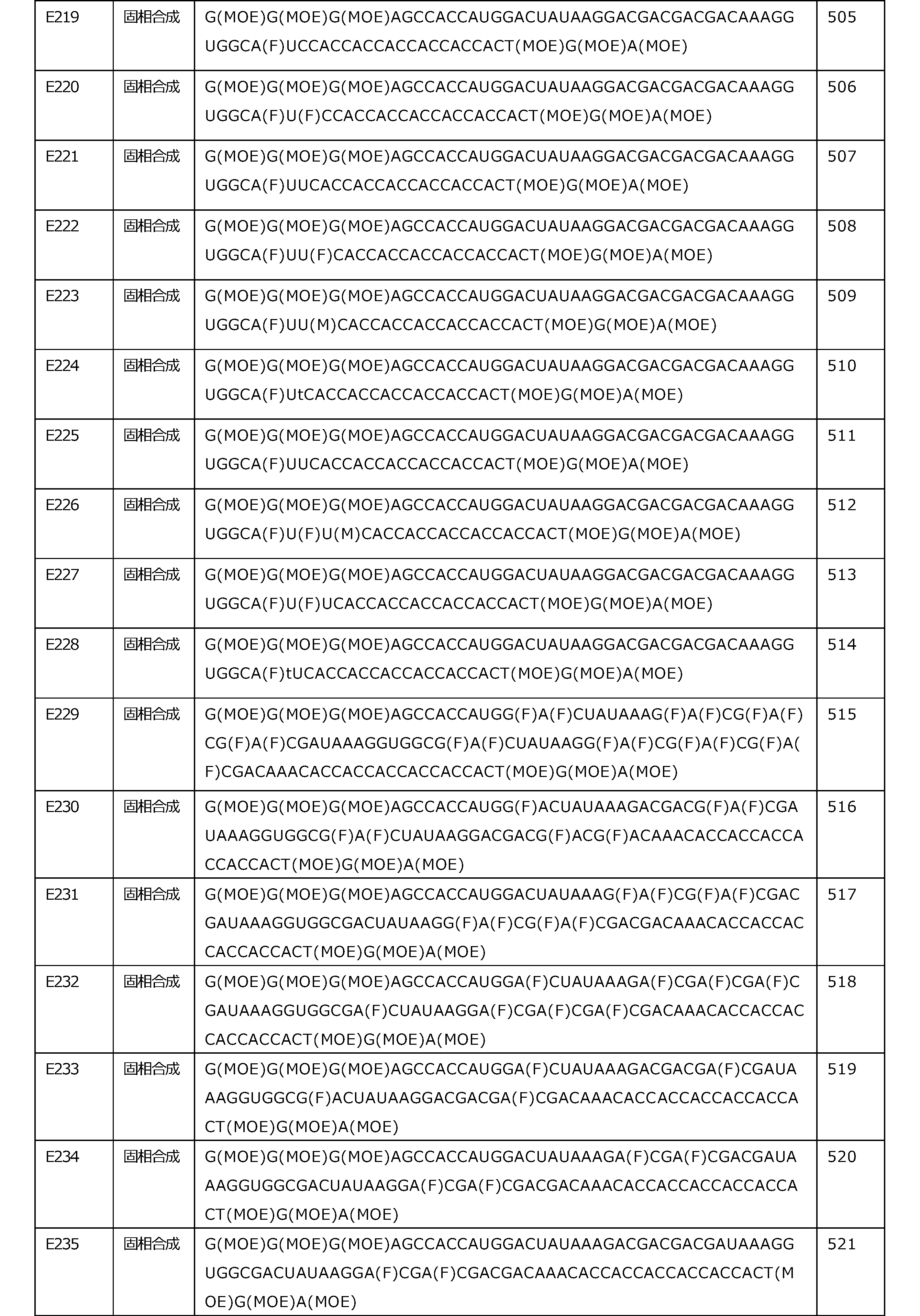


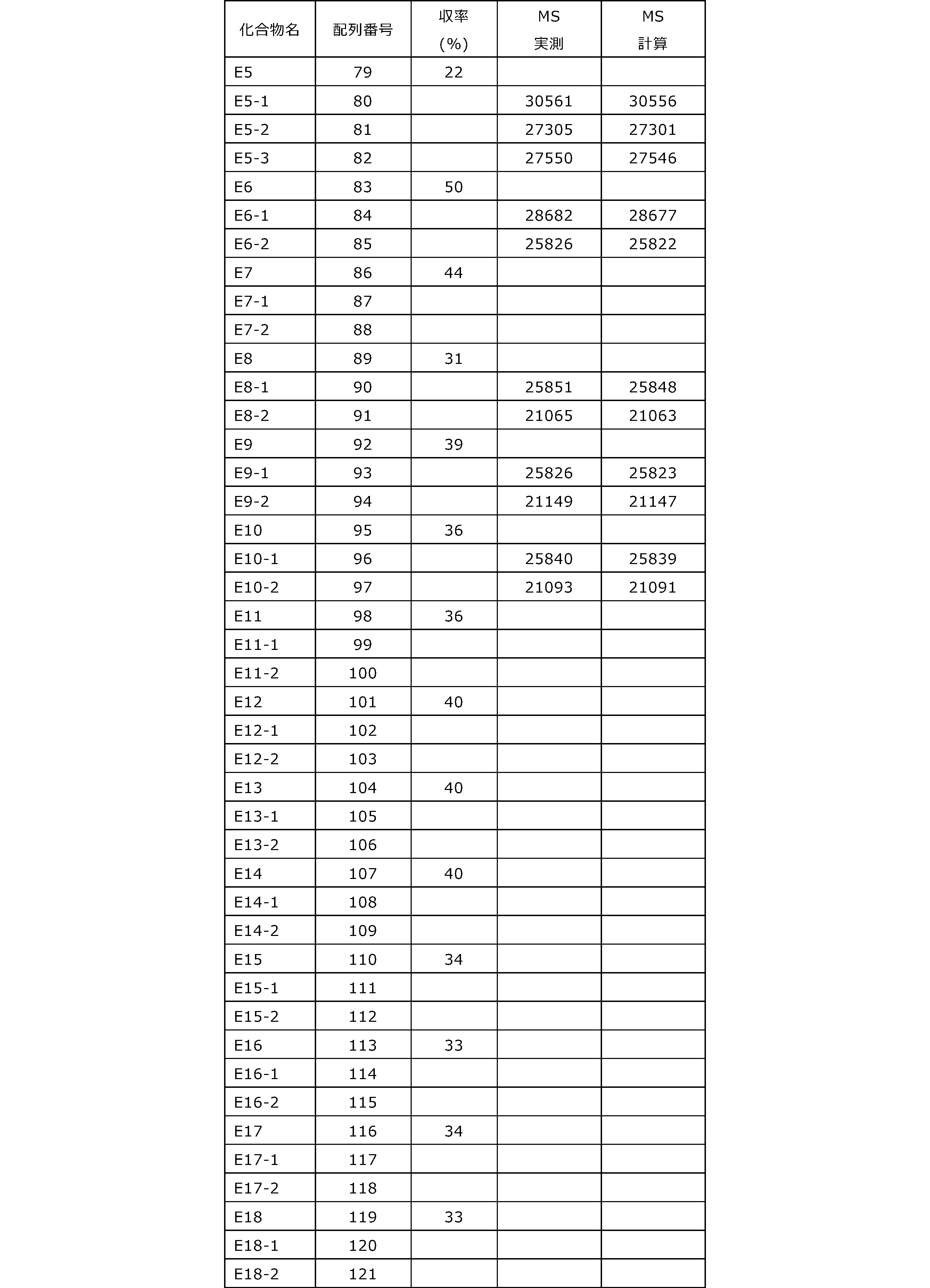










(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)
表21~56に示した各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。翻訳反応は試験例2(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)と同様の条件で行った。
翻訳反応後の反応溶液中の翻訳産物は、以下に記載のサンドイッチELISA法により検出した。まず、6*His, His-Tag 抗体(プロテインテック社、カタログ番号66005-1-Ig)を0.1M Carbonate buffer(pH9.4)にて3 μg/mLに希釈し、96 Well ELISA用プレート(ヌンク社製)に1ウェルあたり50 μLずつ分注し、4℃で一晩静置することにより、抗体の固定化を行ったプレートを作製した。続いて、プレートを精製水で1倍濃度に希釈したTris Buffered Saline with Tween 20 (サンタクルツ社、カタログ番号sc-24953)(以下、洗浄溶液と記載する)で洗浄した後、ウシ血清アルブミン(和光純薬社、カタログ番号017-22231)を終濃度3%となるように希釈した洗浄溶液(以下、ブロッキング溶液と記載する)を1ウェルあたり200 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で希釈した翻訳反応溶液を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。その際、配列番号539に示される翻訳産物ポリペプチド標品(コスモバイオ社製)を同様にブロッキング溶液で各濃度に希釈してプレートに分注した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で10,000倍に希釈したMonoclonal ANTI-FLAG M2-Peroxidase (HRP) Ab produced in mouse (SIGMA社製、カタログ抗体A8592-1MG)を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、1-Step Ultra TMB-ELISA (サーモフィッシャーサイエンティフィック社製、カタログ番号34028)を、1ウェルあたり50 μLずつ分注し、室温で数分静置した。その後、0.5M硫酸 (和光純薬工業社製)を、1ウェルあたり50 μLずつ分注し反応を停止させた後、吸光光度計(バイオラッド社製)を用いて、測定波長450 nm、参照波長570 nmの吸光度を測定した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および糖部修飾を持たないR18を1とした時の相対翻訳産物量を、下記表に記載した。
翻訳産物ポリペプチド標品:NH2-MDYKDDDDKIIDYKDDDDKGGDYKDDDDKHHHHHH-COOH (配列番号539)

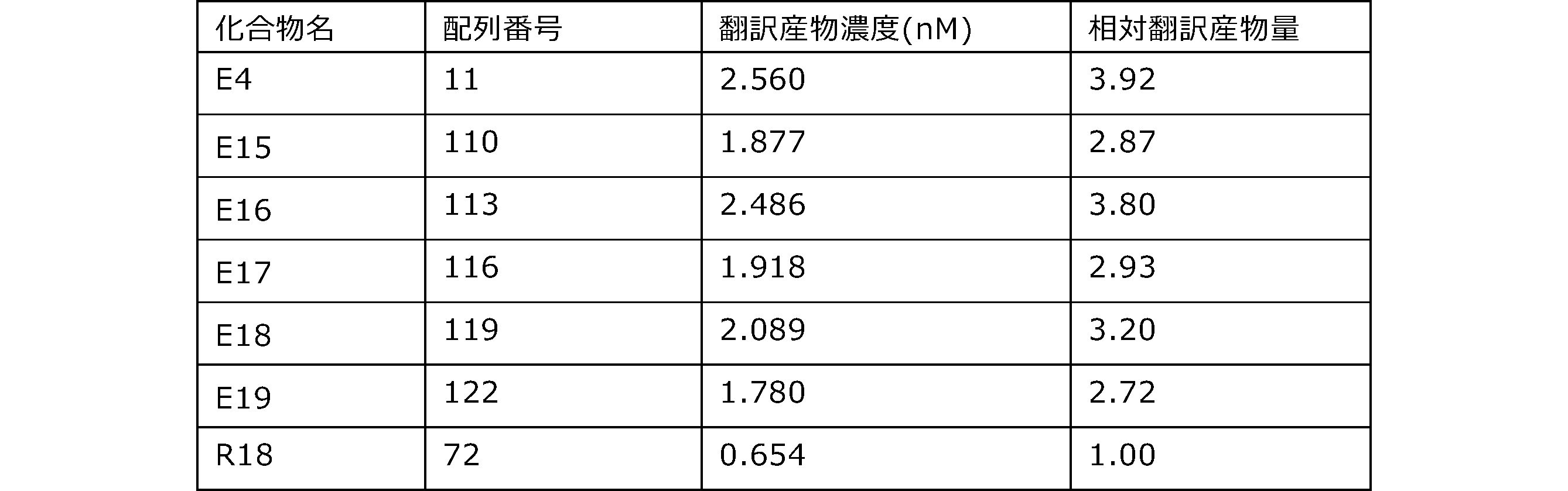




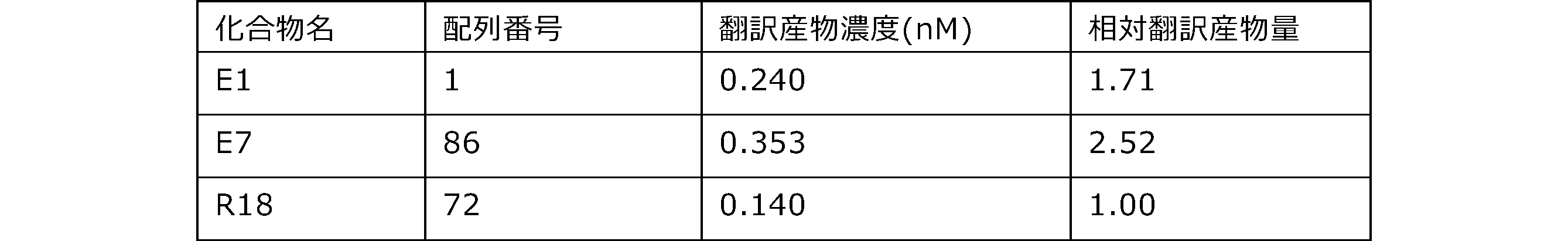




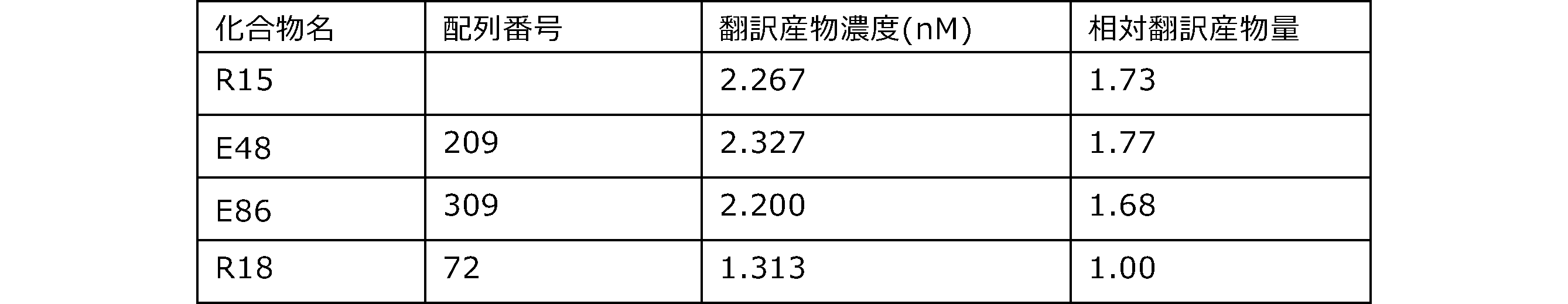





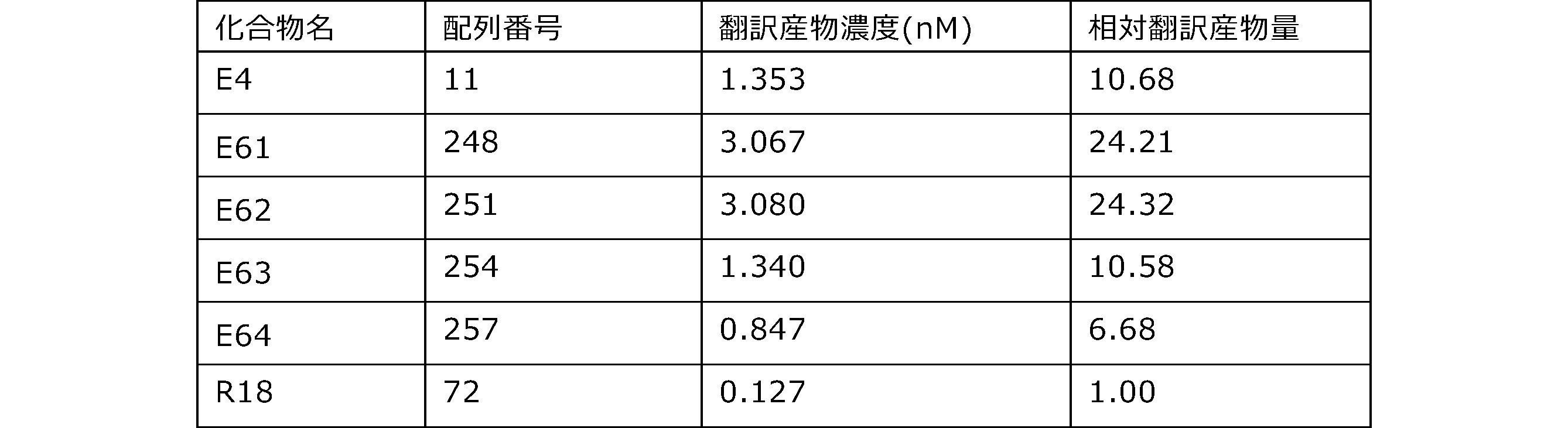


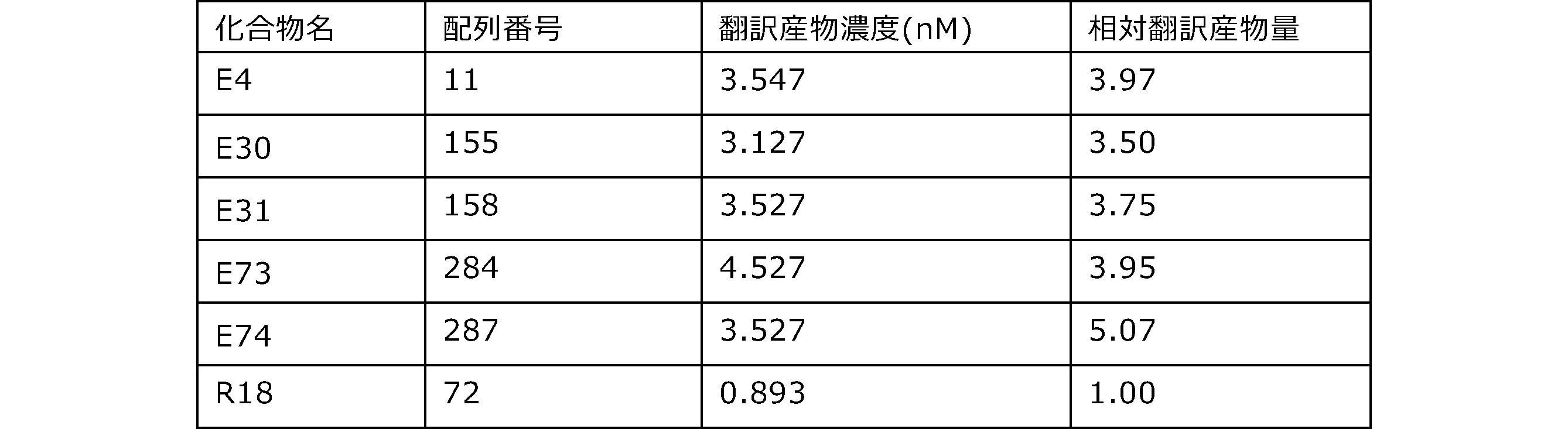




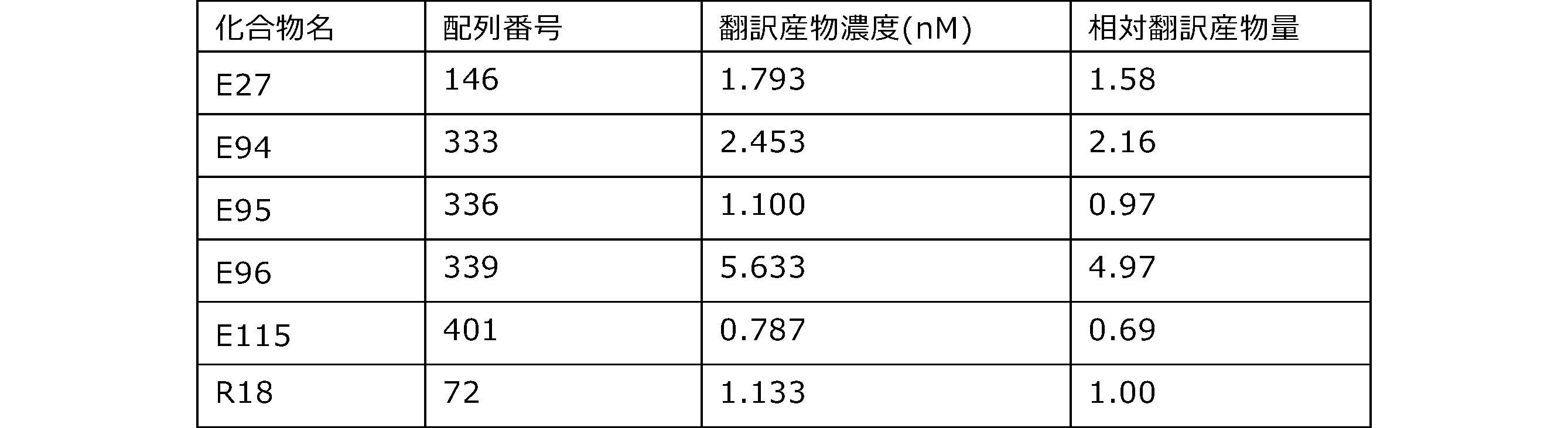
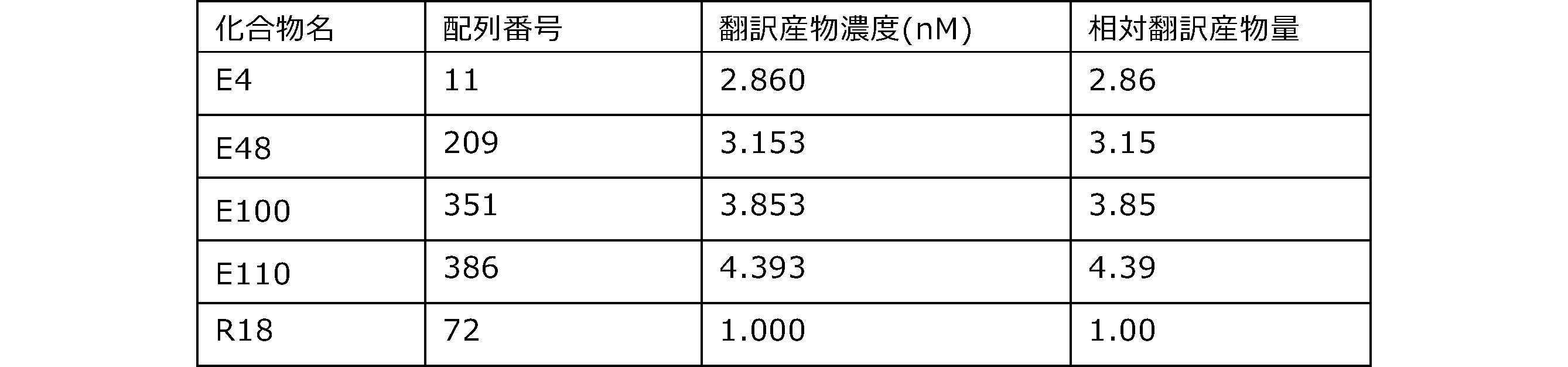





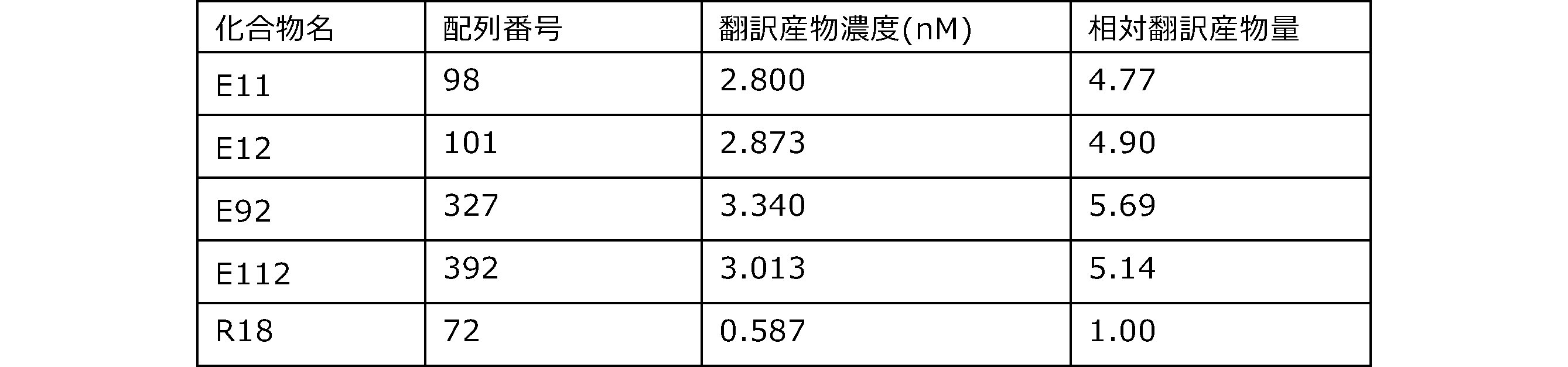


(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)
表21~56に記載の各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。まず、10%ウシ胎仔血清を含むRPMI培地(ナカライテスク社製)で懸濁したHela細胞を1ウェルあたり細胞数10,000 cells/100 μLとなるように、96ウェル接着細胞用培養プレートに播種し、37℃、5% CO2条件下で一晩培養した。一晩培養後の細胞から培養上清を除去し、1ウェルあたり40 μLの10%ウシ胎仔血清を含むRPMI培地を添加した後、各化合物の終濃度が0.1 μMとなるよう、各化合物と終濃度0.3%のLipofectamin MessengerMAX Transfection Reagent(サーモフィッシャーサイエンティフィック社製、カタログ番号:LMRNA008)とをオプティメム(サーモフィッシャーサイエンティフィック社製、カタログ番号:31985-070)で希釈して混合し、混合液を1ウェルあたり10 μLとなるよう各々の培養プレートに添加し、37℃、5% CO2条件下で5時間培養した。5時間培養後の細胞から培養上清を除去し、氷冷したD-PBS(-)(ナカライテスク社製)で1度洗浄した後、表103については2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むNP-40(インビトロジェン社、FNN0021)を1ウェルあたり20 μL加え、30秒間激しく浸透して細胞を溶解した。表104および表105については2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むiScript RT-qPCR Sample Preparation Reagent (バイオラッド社、1708898)を1ウェルあたり20 μL加え、30秒間激しく浸透して細胞を溶解した。
得られた細胞溶解液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法と同様の方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)を、以下表に記載した。




(mRNAサンプルのマウス初代肝細胞を用いたin vitro翻訳反応試験)
表21~56に記載の各化合物について、マウス初代肝細胞(サーモフィッシャーサイエンティフィック社製、カタログ番号MSCP10)を用いてin vitroでの翻訳活性を評価した。まず、Primary Hepatocyte Thawing and Plating Supplements(サーモフィッシャーサイエンティフィック社製、カタログ番号CM3000)を含むWilliam's E Medium, no phenol red(サーモフィッシャーサイエンティフィック社製、カタログ番号A1217601)に懸濁させたマウス初代肝細胞を1ウェルあたり細胞数10,000 cells/100 μLとなるように、96ウェルコラーゲンIコート培養プレート (コーニング社製、カタログ番号:356407)に播種し、37℃、5% CO2条件下で5時間培養した。培養後の細胞から培養上清を除去し、Primary Hepatocyte Maintenance Supplements (サーモフィッシャーサイエンティフィック社製、カタログ番号CM4000)を含むWilliam's E Medium, no phenol redを1ウェルあたり100 μLとなるように添加し、さらに37℃、5% CO2条件下で5時間培養した。一晩培養後の細胞から培養上清を除去し、Primary Hepatocyte Maintenance Supplementsを含むWilliam's E Medium, no phenol redを1ウェルあたり40 μL添加した後、各化合物の終濃度が0.1 μMとなるよう、各化合物と終濃度0.3%のLipofectamin MessengerMAX Transfection Reagentとをオプティメムで希釈して混合し、混合液を1ウェルあたり10 μLとなるよう各々の培養プレートに添加し、37℃、5% CO2条件下で5時間培養した。5時間培養後の細胞から培養上清を除去し、Primary Hepatocyte Maintenance Supplementsを含むWilliam's E Medium, no phenol redを1ウェルあたり100 μL添加した後、さらに37℃、5% CO2条件下で1時間培養した。1時間培養後の細胞から培養上清を除去し、氷冷したD-PBS(-)(ナカライテスク社製)で1度洗浄した後、2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むNP-40(インビトロジェン社、FNN0021)を1ウェルあたり20 μL加え、30秒間激しく浸透して細胞を溶解した。
得られた細胞溶解液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法と同様の方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)を、以下表に記載した。


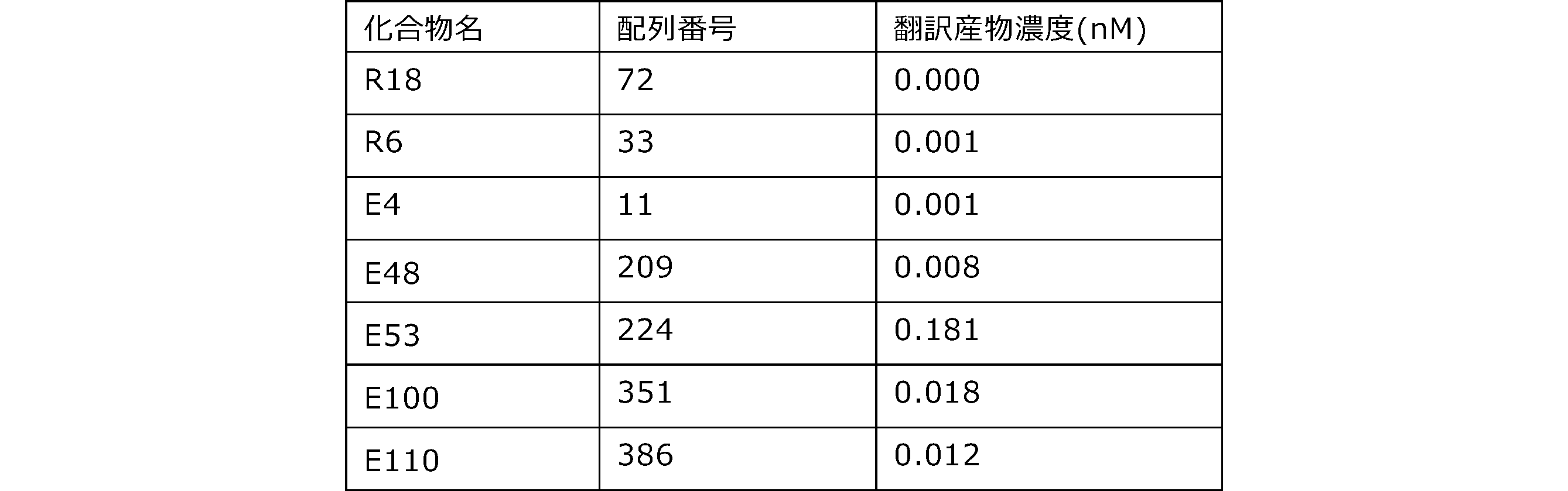
(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)
表21~56に記載した各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。
まず、各化合物を20 μMとなるようにTHE RNA Storage Solution (サーモフィッシャーサイエンティフィック社製、カタログ番号AM7000)で希釈した。Hela細胞株はSE Cell Line 96-well Nucleofector Kit (Lonza社製、カタログ番号V4SC-1096)付属のSE Cell Line Nucleofector SolutionおよびSupplement 1の混合液で200,000 cells/19 μLとなるように懸濁。調製した核酸溶液とHela細胞懸濁液を体積比1:19で混合したのち、Nucleofector(tm) 96-well Shuttl システム(Lonza社製)を用い、パルス条件FF-137でエレクトロポレーションを実施した。エレクトロポレーション10分後の細胞を10%ウシ胎仔血清を含むRPMI培地(ナカライテスク社製)に懸濁し、1ウェルあたり細胞数50,000 cells/145 μLとなるように、96ウェル接着細胞用培養プレートに播種し、37℃、5% CO2条件下で3時間培養した。3時間培養後の細胞から培養上清を除去し、氷冷したD-PBS(-)(ナカライテスク社製)で1度洗浄した後、2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むiScript RT-qPCR Sample Preparation Reagent (バイオラッド社、1708898)を1ウェルあたり20 μL加え、30秒間激しく浸透して細胞を溶解した。
得られた細胞溶解液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法と同様の方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)を、以下表に記載した。

(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)
表21~56に記載した各化合物各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。翻訳反応は試験例2(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)と同様の方法で、核酸濃度が終濃度1 μMとなる条件で行った。
翻訳反応後の反応溶液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、翻訳産物ポリペプチド標品として配列番号540に示されるペプチド(コスモバイオ社製)を用いること以外は同様の方法で実施した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および翻訳領域に糖部修飾を持たない表中の各化合物を1とした時の相対翻訳産物量を、以下表に記載した。
翻訳産物ポリペプチド標品:NH2-MDYKDDDDKGGHHHHHH-COOH (配列番号540)



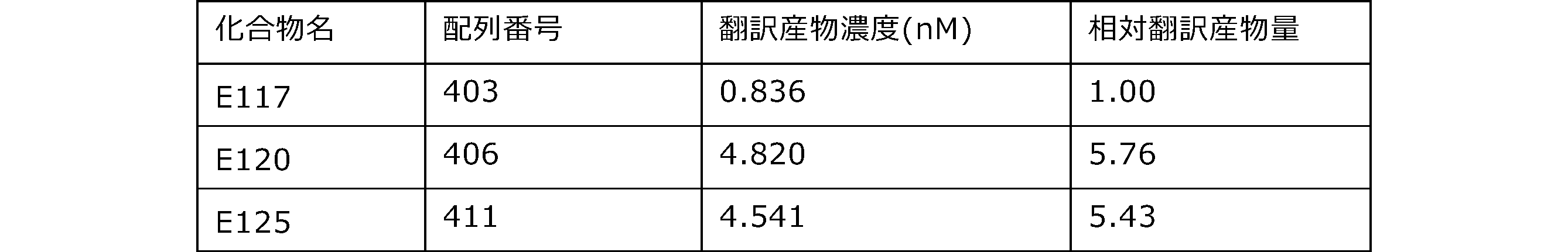

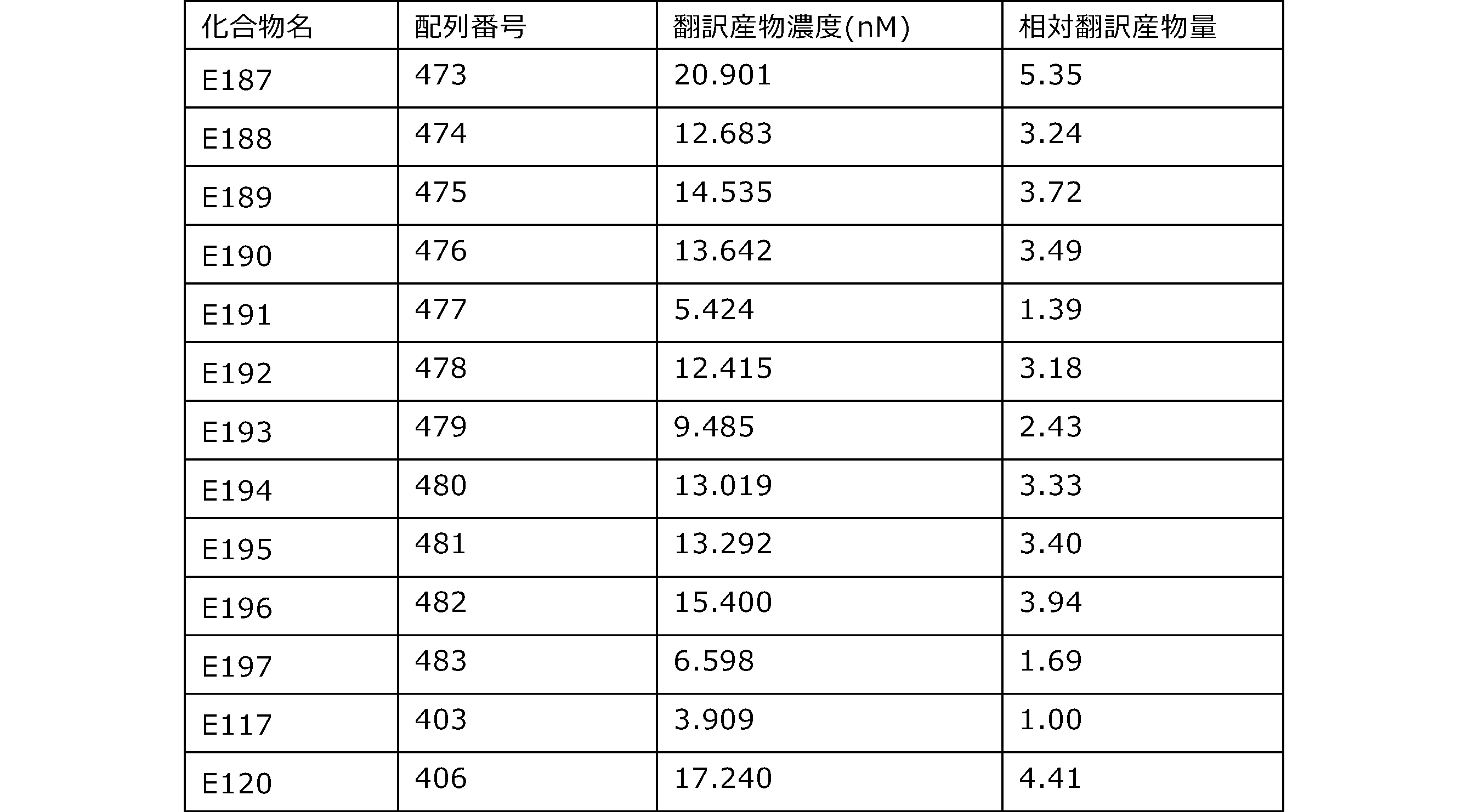
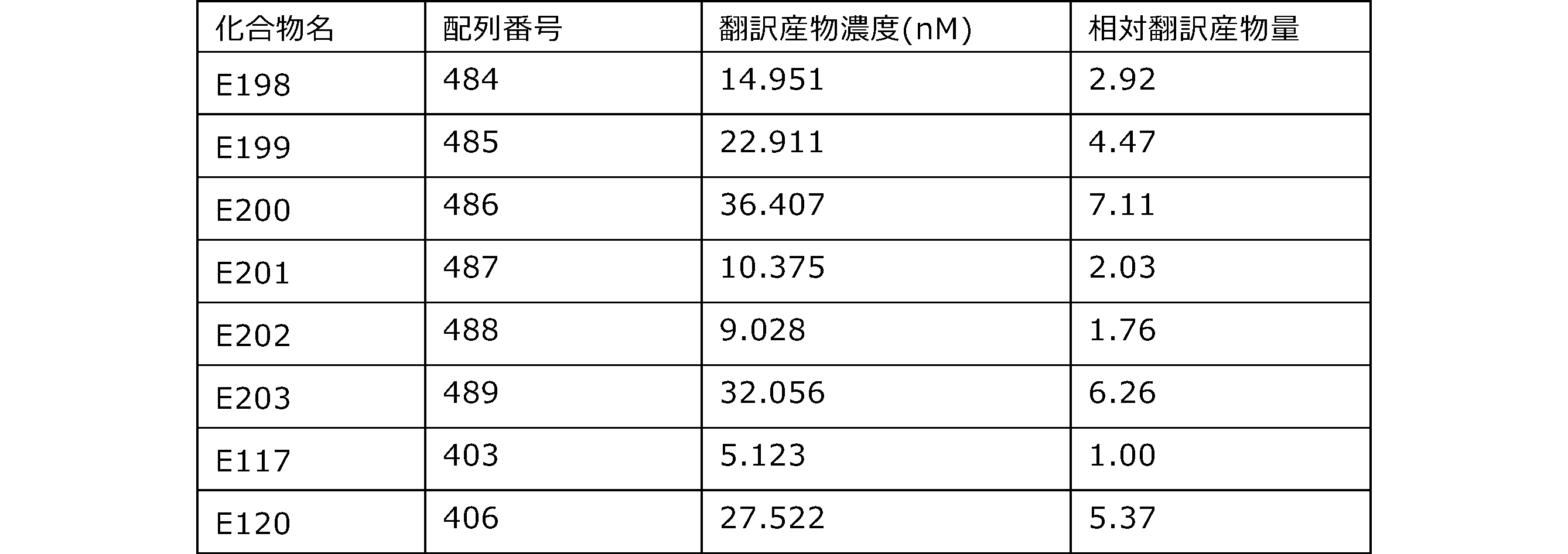
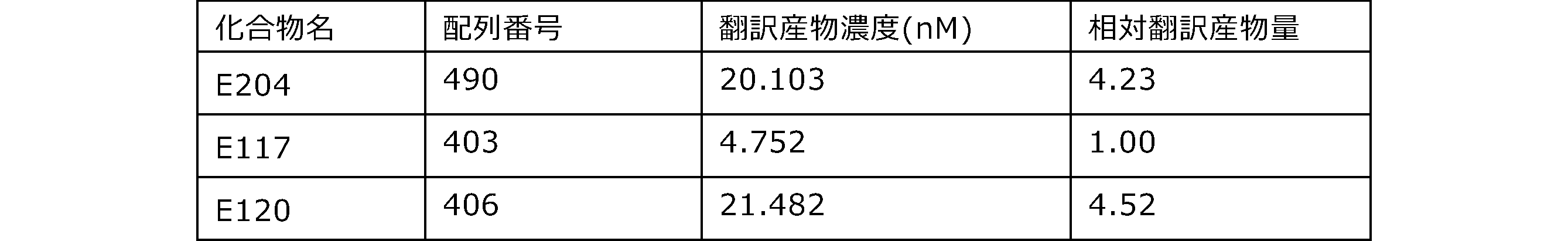
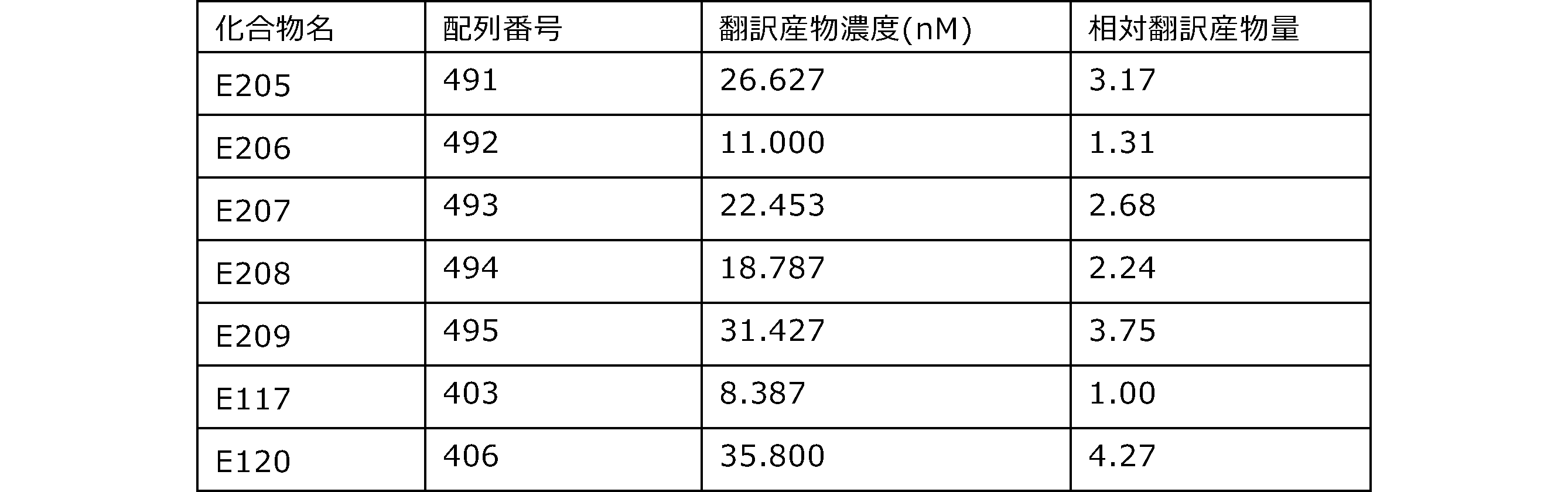


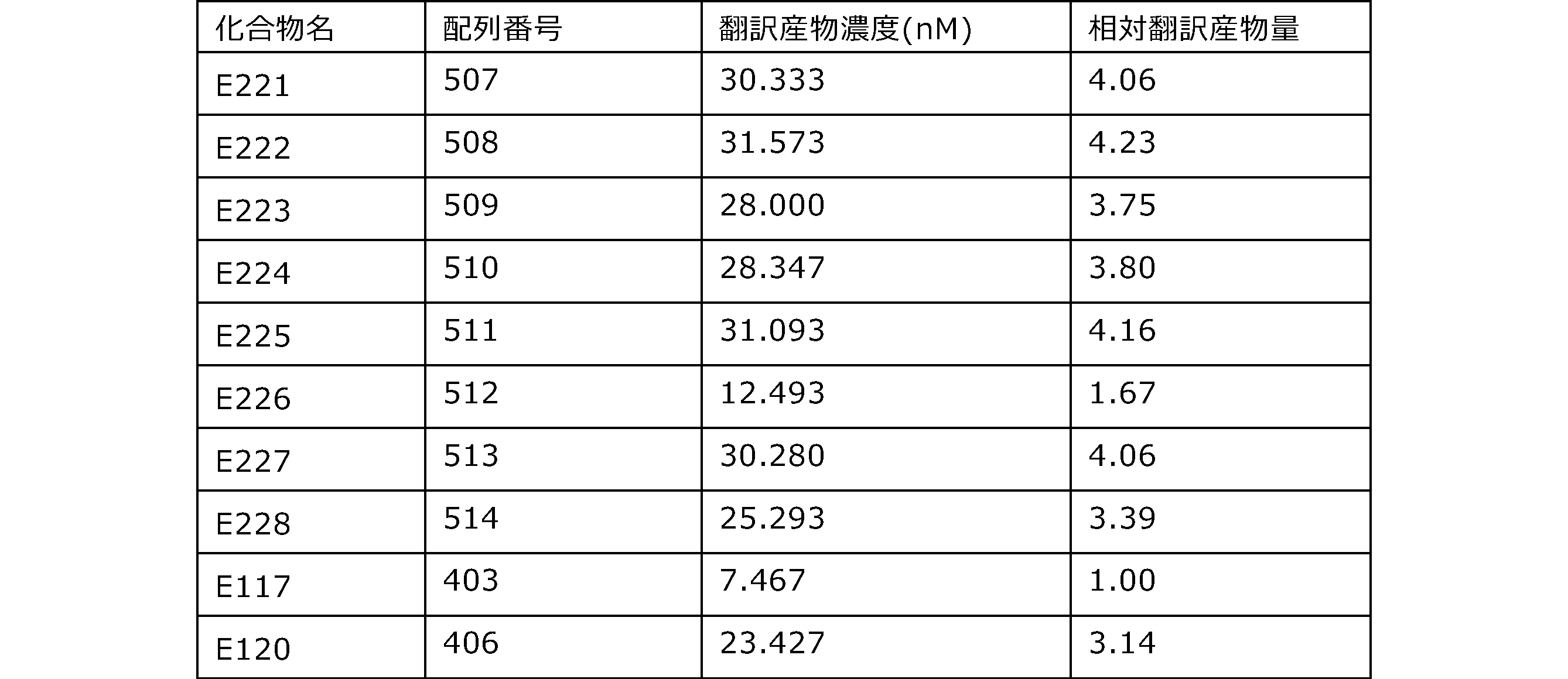
(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)
表21~56に記載の各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。細胞株への各化合物のトランスフェクションと細胞溶解液の調製は試験例7(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)と同様の方法で実施し、細胞の溶解は2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むiScript RT-qPCR Sample Preparation Reagentを用いて実施した。
得られた細胞溶解液中の翻訳産物は、試験例10(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法と同様の方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)を、以下表に記載した。

(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)
表21~56に記載の各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。翻訳反応は試験例2(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)と同様の条件で、核酸濃度が終濃度1 μMとなる条件で行った。
翻訳反応後の反応溶液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、翻訳産物ポリペプチド標品として配列番号541に示されるペプチド(コスモバイオ社製)を用いること以外は同様の方法で実施した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および翻訳領域に糖部修飾を持たないE82を1とした時の相対翻訳産物量を、表124~133に記載した。
翻訳産物ポリペプチド標品:NH2-MDYKDDDDKGGDYKDDDDKHHHHHH-COOH (配列番号541)




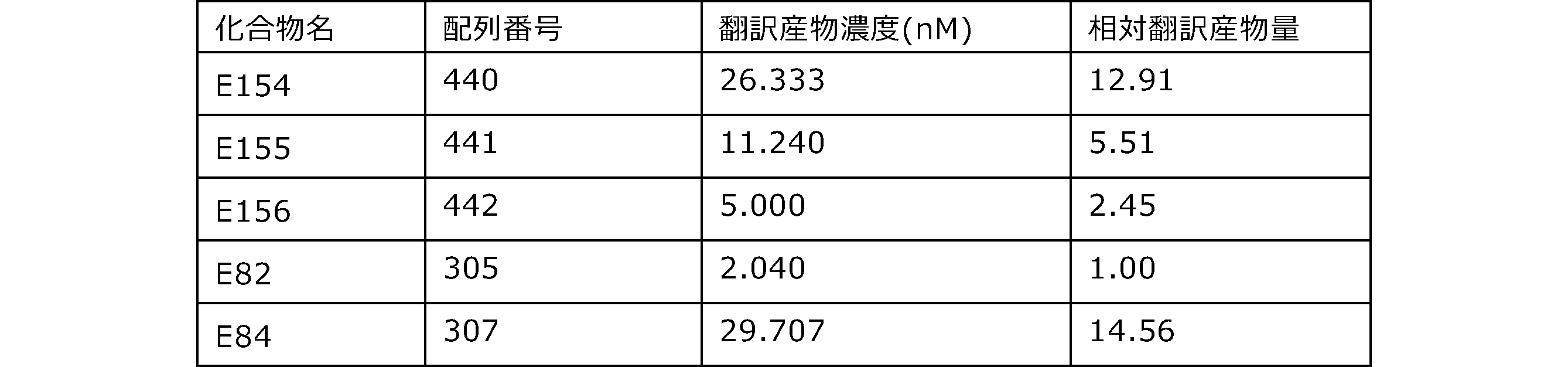




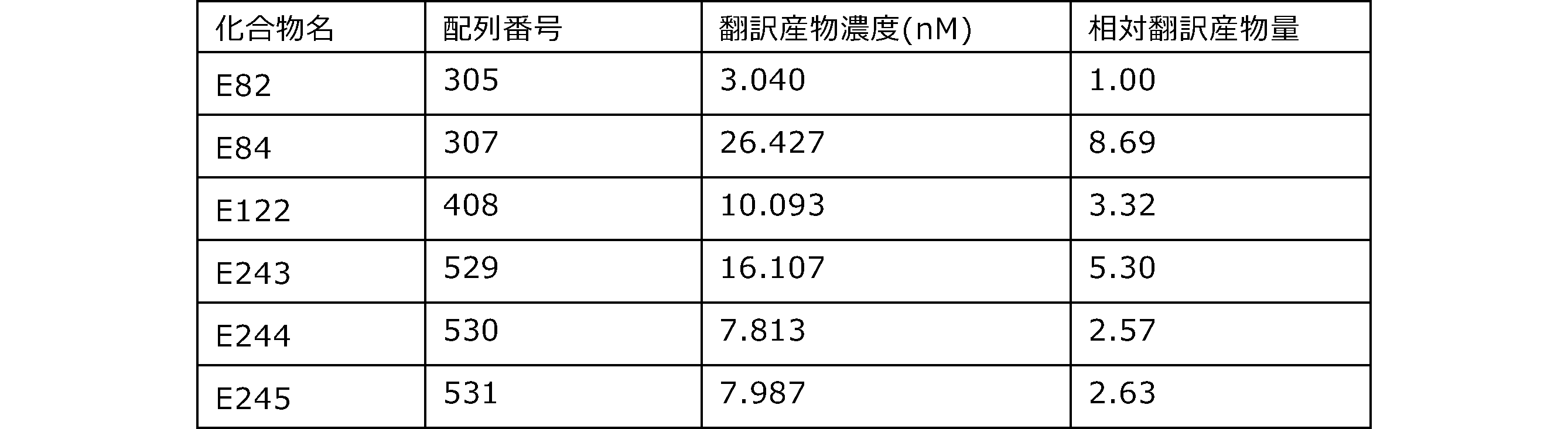
(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)
表21~56に記載された各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。細胞株への各化合物のトランスフェクションと細胞溶解液の調製は試験例7(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)と同様の方法で実施し、細胞の溶解は2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むiScript RT-qPCR Sample Preparation Reagentを用いて実施した。
得られた細胞溶解液中の翻訳産物は、試験例12(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法と同様の方法で実施した。測定の結果、ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)を、以下表に記載した。
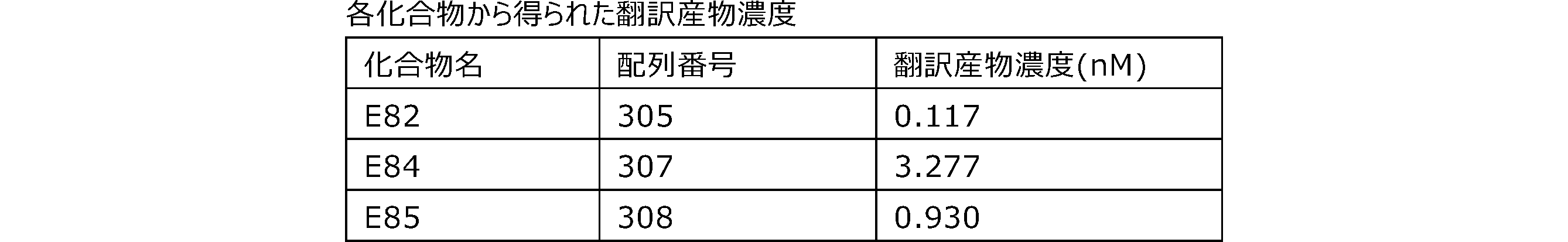
(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)
表21~56に記載される各化合物について、Rabbit-Reticulocyte-Lysate-System-Nuclease-Treatedキットを用いて真核細胞系における翻訳活性を評価した。翻訳反応は試験例2 (真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)と同様の方法で実施した。
翻訳反応後の反応溶液中の翻訳産物は、以下に記載のサンドイッチELISA法により検出した。まず、Anti-Human EGF抗体 (Peprotech社製、カタログ番号500-P45)を0.1M Carbonate buffer(pH9.4)にて3 μg/mLに希釈し、96 Well ELISA用プレート(ヌンク社製)に1ウェルあたり50 μLずつ分注し、4℃で一晩静置することにより、抗体の固定化を行ったプレートを作製した。続いて、プレートを精製水で1倍濃度に希釈したTris Buffered Saline with Tween 20(以下、洗浄溶液と記載する)で洗浄した後、ウシ血清アルブミンを終濃度3%となるように希釈した洗浄溶液(以下、ブロッキング溶液と記載する)を1ウェルあたり200 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で100倍希釈した翻訳反応溶液を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、ブロッキング溶液で10,000倍に希釈したMonoclonal ANTI-FLAG M2-Peroxidase (HRP) Ab produced in mouse (SIGMA社製、カタログ抗体A8592-1MG)を、1ウェルあたり50 μLずつ分注し、室温で1時間静置した。プレートを洗浄溶液で洗浄した後、1-Step Ultra TMB-ELISA (サーモフィッシャーサイエンティフィック社製、カタログ番号34028)を、1ウェルあたり50 μLずつ分注し、室温で数分静置した。その後、0.5M硫酸 (和光純薬工業社製)を、1ウェルあたり50 μLずつ分注し反応を停止させた後、吸光光度計(バイオラッド社製)を用いて、測定波長450 nm、参照波長570 nmの吸光度を測定した。各翻訳反応溶液を測定した際の吸光度(測定波長の吸光度から参照波長の吸光度を差し引いたもの)を以下表に記載した。

(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)
表21~56に記載される各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。翻訳反応は試験例2(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)と同様の条件で、核酸濃度が終濃度1 μMとなる条件で行った。
翻訳反応後の反応溶液中の翻訳産物は、試験例14(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、翻訳反応液をブロッキング溶液で20倍に希釈すること以外は同様の方法で実施した。各翻訳反応溶液を測定した際の吸光度(測定波長の吸光度から参照波長の吸光度を差し引いたもの)を以下表に記載した。

(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)
表21~56に記載された各化合物について、Hela細胞株を用いてin vitroでの翻訳活性を評価した。細胞株への各化合物のトランスフェクションと細胞溶解液の調製は試験例7(mRNAサンプルのHela細胞株を用いたin vitro翻訳反応試験)と同様の方法で実施し、細胞の溶解は2%のプロテアーゼ阻害剤カクテル(動物細胞抽出物用)を含むiScript RT-qPCR Sample Preparation Reagentを用いて実施した。
得られた細胞溶解液中の翻訳産物は、試験例14(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、細胞溶解液液をブロッキング溶液で30倍に希釈すること以外は同様の方法で実施した。各細胞溶解液を測定した際の吸光度(測定波長の吸光度から参照波長の吸光度を差し引いたもの)を以下表に記載した。

(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)
表21~56に記載された各化合物について、Rabbit-Reticulocyte-Lysate-System-Nuclease-Treatedキットを用いて真核細胞系における翻訳活性を評価した。翻訳反応は試験例2 (真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)と同様の方法で実施した。
翻訳反応後の反応溶液中の翻訳産物は、試験例6(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、翻訳産物ポリペプチド標品として配列番号542に示されるペプチド(コスモバイオ社製)を用いること以外は同様の方法で実施した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および翻訳領域に糖部修飾を持たないE6を1とした時の相対翻訳産物量を、以下表に記載した。
翻訳産物ポリペプチド標品:NH2-MDYKDDDDKIIDYKDDDDKGGDYKDDDDKSIINFEKLHHHHHH-COOH (配列番号542)

(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)
表21~56に記載された各化合物について、1-Step Human Coupled IVT Kit(サーモフィッシャーサイエンティフィック社製、カタログ番号88882)を用いてヒト細胞系における翻訳活性を評価した。翻訳反応は試験例2(mRNAサンプルのHela細胞株ライセートを用いた翻訳反応試験)と同様の条件で、核酸濃度が終濃度1 μMとなる条件で行った。
翻訳反応後の反応溶液中の翻訳産物は、試験例17(真核細胞系における翻訳反応;ウサギ赤血球ライセートを用いた翻訳反応試験)に記載のサンドイッチELISA法の方法に従い、翻訳反応液をブロッキング溶液で10倍に希釈すること以外は同様の方法で実施した。ポリペプチド標品の吸光度をもとに作成した検量線を用いて定量した、各翻訳反応溶液中の翻訳産物濃度(nM)、および翻訳領域に糖部修飾を持たないE6を1とした時の相対翻訳産物量を、以下表に記載した。

(mRNAサンプルの血清中安定性試験)
表21~56に記載された各化合物について、市販マウス血清(タカラバイオ、カタログ番号2311B、lot番号AJ10759A)を用いて血清中核酸安定性を評価した。まず、マウス血清をUltraPure(tm) DNase/RNase-Free Distilled Water(DW) (invitrogen、カタログ番号10977-015)で15倍希釈し、希釈血清溶液を調製した。各化合物は終濃度が5 μMとなるようTHE RNA storage solution(サーモフィッシャーサイエンティフィック社、カタログ番号AM7001)で希釈した。
酵素未反応(0 min)用として別の96 well PCRプレートに希釈血清溶液8 μL、6 U/μL Ribonuclease Inhibitor (タカラバイオ、カタログ番号2311B)2.5 μLの混合溶液10.5 μL、および5 μM mRNA 2 μLを添加して-30 ℃で保存した。酵素反応用として、また別の96 well PCRプレートに希釈血清溶液28 μLと5 μM mRNA 7 μLを添加してよく混合した。これを10 μLずつ新しい96 well PCRプレート 4枚に分注して各プレート所定の時間(5min、15 min、30 min、60 min)37 ℃で反応させた後、6 U/μL Rnase inhibitorを2.5 μL添加して測定まで-30 ℃で保存した。
酵素反応後の反応溶液中の残存mRNA量は、以下に記載のRT-qPCR法により検出した。まず、検量線には表140に関しては化合物R18を、表141に関してはそれぞれ対応する化合物を使用し、THE RNA storage solution によって4 μMから4倍希釈で11点濃度を取り、希釈系列を作製した。検量線及び酵素反応後のサンプル2.5 μLはRibonuclease Inhibitorを終濃度0.2 U/mLで加えたDWを用いて1071倍希釈した。この希釈した試料5 μLおよび2μM RT primer(シグマアルドリッチ社) 1 μLを用いてTaqMan Micro RNA RT kit(サーモフィッシャーサイエンティフィック、カタログ番号4366597)により逆転写産物cDNAを作製した。反応温度は16 ℃ 30 min → 42 ℃ 30 min → 85 ℃ 5 minで実施した。cDNA 5 μL、TaqMan Gene Expression Master Mix 10 μL、Fw primer(シグマアルドリッチ社) 0.28 μL、Rv primer(シグマアルドリッチ社) 0.33 μL、TaqMan MGB Probe(サーモフィッシャーサイエンティフィック社、カタログ番号4316033) 0.38 μLおよびDistilled water 4.01 μLを混合し、qPCR測定を行った。機器はQuantstudio12K Flex(Applied Biosystems)を使用した。また、使用したPrimerおよびTaqman MGB ProbeのDNA配列は以下である。測定の結果、標品のCT値をもとに検量線を用いて、各試料中の各化合物の濃度を定量し、酵素未反応(0分)に対する相対残存量として算出したものを、以下表に記載した。
RT primer:5'-TCAGTGGTGGTGGTGGTGGTGTTTG-3' (配列番号75)
Fw primer:5'-ATCTTGTCGTCGTCGTCCTT-3' (配列番号76)
Rv primer:5'-GAATACAAGCTACTTGTTCTTTT-3' (配列番号77)
Taqman MGB Probe:5'-CAGCCACCATG-3' (配列番号78)
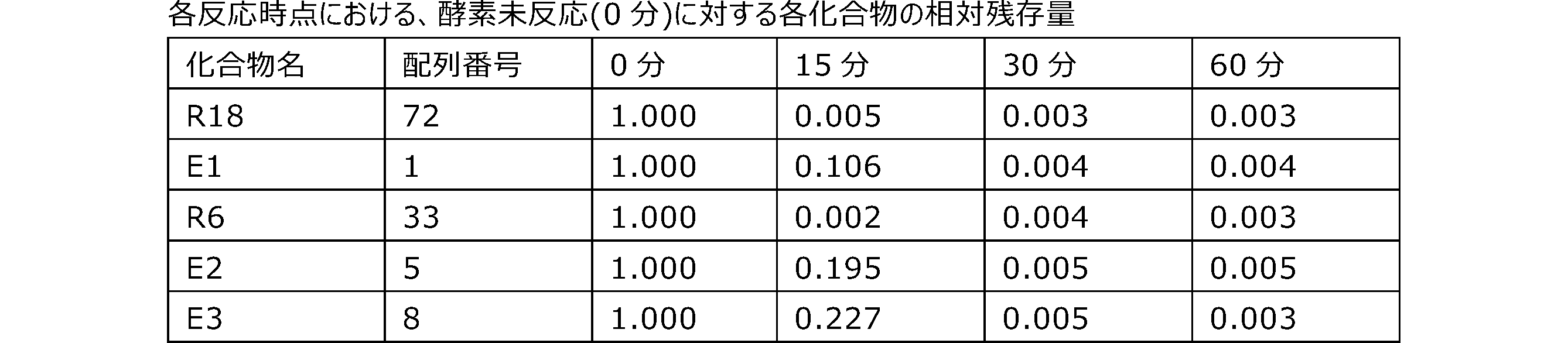

<比較化合物RNの合成>
VEGFタンパク質に翻訳される比較化合物RNの合成に用いた材料(ポリヌクレオチド)の配列情報を示す。表中の各ヌクレオチドNはRNA、N(M)は2'-O-メチル修飾RNAを、N(F)は2'-F化修飾RNAを、N(MOE)は2'-O-メトキシエチル修飾RNAを、dNはDNAを表す。

(PCR反応による二重鎖DNAの調製とIn vitro transcriotionによる3'末端側ポリヌクレオチド断片の調製)
プラスミドDNAは市販のpUC19ベクターのSma Iサイトに、上述の配列番号546に示される人工合成遺伝子配列GNを挿入したものを用いた(株式会社ファスマック社製)。
プラスミドDNA、FWプライマーおよびRVプライマーを用いてPCRを行った。
PCR溶液の終濃度は以下の通り;鋳型DNA 0.2 ng/μL, dNTP 250 μM, プライマー 0.3 μM, PrimeSTAR HS DNA Polymerase (Takara) 0.025 U/μL, PrimeSTAR Buffer(Mg2+ plus)(Takara)1x.温度サイクルは以下の通り;94 oC 5 min, [98 oC 10 sec, 55 oC 30 sec, 72 oC 60 sec] x 30 サイクル。フェノールクロロホルム抽出、イソプロパノール沈殿により、粗精製を行った。続いてDpnI処理により、鋳型plasmidの分解除去を行った。反応液の終濃度 は、PCR産物 3.43 μg/μL, Dpn I (Takara) 1 U/μL, T buffer (Takara) 1xで、37度で1時間インキュベートを行った。フェノールクロロホルム抽出、イソプロパノール沈殿により、粗精製を行った。
定法に従い化学合成により得られた3'末端側ポリヌクレオチド断片RN-1ならびにin vitro transcriptionにより得られたRNA断片RN-2、テンプレートDNA-Aを用いて、RNA ligase 2による連結反応を行った。終濃度は、RN-1 2 μM, RN-2 2 μM, DNAN 4 μM, PEG8000 10% (v/v), T4 RNA ligase 2 (NEB) 1 U/μL, T4 RNA ligase 2 buffer (NEB) 1x, RNase inhibitor Murine (NEB) 1 U/μL。チューブにPEG8000, T4 RNA ligase 2, RNase inhibitor以外の基質・試薬を加えた後、90 度3分加熱し、室温まで自然放熱した。残りの試薬・酵素を添加し、45度で1時間インキュベートした。DNaseを終濃度0.33 U/μL 分添加し、37度で15分インキュベートした。 フェノールクロロホルム抽出、イソプロパノール沈殿により粗精製を行い、連結RNAを得た。反応サンプルは、dPAGE(5% acrylamide, 7M urea)で解析した。ゲル精製を以下の通り行った。dPAGE(6% acrylamide, 7.5 M urea, 25 %フォルムアミド)で泳動後、該当バンドを切り出し、MQ水で12時間抽出を行った。アミコン10K (Merck Millipore)処理, イソプロパノール沈殿により、ゲル抽出産物を得た。
以下の一連の操作により、Smarter RACE 5' 3' Kit(Takara Bio社製、カタログ番号634859を用いてRNA連結産物RNの連結部の配列を分析した。まず、100 ng/μLに調製したRNを1 μL取り、5' CDS Primer A 1 μLおよびNuclease-free water 9 μLと混合し、72℃で4分間加熱し、4℃で2分間冷却した。反応液に1 μLのSMARTER II A Oligo、4 μLの5x First Strand Buffer、0.5 μLの100 mM DTT、1 μLの20 mM dNTPs、0.5 μLの40 U/μL RNase Inhibitor、2 μLのSMARTScribe Reverse Trancriptaseを混合し、42℃で90分間、72℃で10分間加熱し、4℃で冷却したのち、240 μLのTE Bufferを添加し混合することで、鋳型cDNAを合成した。続いて、得られた鋳型cDNAを用いて以下の通りPCR反応を実施した。具体的には、2.5 μLの鋳型cDNA, 5 μLの10x UPM、0.1 μLの100 μM RVプライマー2(配列番号548)、25 μLのSeqAmp Buffer、1 μLのSeqAmp DNA polymerase、16.4 μLのNuclease-free waterを混合し、サーマルサイクラーを用いて94℃で30秒間加熱し、その後68℃30秒間、72℃60秒間の加熱を20回繰り返し、その後4℃で冷却した。得られたPCR産物を1.5%アガロース-TAEゲルで電気泳動し、該当バンドを切り出し、QIAquick Gel Extraction Kit(QIAGEN社製、カタログ番号28704)でPCR産物を精製した。得られたPCR産物1 μLをZero Blunt TOPO PCR Cloning Kit for Sequencing (Invitrogen社製、カタログ番号45-0031)に含まれるTOPO vector 0.5 μL、Salt solution 0.5 μL、Nuclease-free water 1 μLと混合し、室温で5分間反応させた。得られた反応物をCompetent Quick DH5α(東洋紡社製、カタログ番号DNA-913)に形質転換し、100 μg/mLのアンピシリンを含むLB寒天培地プレートに播種し、37℃で一晩培養することによりシングルコロニーを得た。シングルコロニーを100 μg/mLのアンピシリンを含むLB液体培地に植菌して37℃で一晩培養し、得られた大腸菌からPureYield Plasmid Miniprep System(プロメガ社製、カタログ番号A1222)を用いてプラスミドDNAを抽出した。
RVプライマー2:5'- GATTACGCCAAGCTTTGG CTTGAAGATGTA CTCGATCTCATCAGG-3'・・・配列番号548
シーケンスプライマーM13FW:5'-CTCGATCTCATCAGG-3'・・・配列番号549
シーケンスプライマーM13RV:5'-CAGGAAACAGCTATGAC -3'・・・配列番号550
VEGFタンパク質に翻訳される比較化合物RN'の合成に用いた材料(ポリヌクレオチド)の配列情報を示す。表中の各ヌクレオチドNはRNA、N(M)は2'-O-メチル修飾RNAを、N(F)は2'-F化修飾RNAを、N(MOE)は2'-O-メトキシエチル修飾RNAを、dNはDNAを表す。

(線状化プラスミドDNAの調製とIn vitro transcriotionによるRNA断片の調製)
プラスミドDNAは市販のpUC19ベクターのEcoRVサイトおよびXbaIサイトに、配列番号552に示される人工合成遺伝子配列GN'を挿入したものを用いた(株式会社ジーンウィズ製)。プラスミドDNAを制限酵素XbaIを用い直鎖化を行った。反応液の終濃度は、プラスミドDNA 20 ng/μL, XbaI (Takara) 0.75 U/μL, M buffer (Takara) 1x, BSA 0.01%で、37度で1時間インキュベートした。フェノールクロロホルム抽出、イソプロパノール沈殿により粗精製を行った。得られた直鎖化DNAを用いて転写反応を行った。反応液の終濃度は、反応液の終濃度は以下の通り; DNA 10 ng/μL, DTT 5 mM, ATP, CTP, UTP 各2 mM, GMP 2 mM, GTP 0.5 mM, T7 RNA polymerase (Takara) 2.5 U/μL, T7 RNA Polymerese buffer (Takara) 1x, RNase Inhibitor, Murine (NEB) 0.2 U/μL。37度で2時間インキュベートを行った。続いてDNase (recombinant, RNase-free, Takara)を 終濃度0.1 U/μL 分を添加し、 37度で30分インキュベートを行った。フェノールクロロホルム抽出、イソプロパノール沈殿により粗精製を行い、3'末端側ポリヌクレオチド断片RN'-2を得た。
定法に従い化学合成により得られた5'末端側ポリヌクレオチド断片RN'-1ならびにin vitro transcriptionにより得られた3'末端側ポリヌクレオチド断片RN'-2、テンプレートDNA-Bを用いて、RNA ligase 2による連結反応を行った。終濃度は、RN-1 2 μM, RN-2 2 μM, DNAN 4 μM, PEG8000 10% (v/v), T4 RNA ligase 2 (NEB) 1 U/μL, T4 RNA ligase 2 buffer (NEB) 1x, RNase inhibitor Murine (NEB) 1 U/μL。チューブにPEG8000, T4 RNA ligase 2, RNase inhibitor以外の基質・試薬を加えた後、90 度3分加熱し、室温まで自然放熱した。残りの試薬・酵素を添加し、45度で1時間インキュベートした。DNaseを終濃度0.33 U/μL 分添加し、37度で15分インキュベートした。 フェノールクロロホルム抽出、イソプロパノール沈殿により粗精製を行い、連結RNAを得た。反応サンプルは、dPAGE(5% acrylamide, 7M urea)で解析した。ゲル精製を以下の通り行った。dPAGE(6% acrylamide, 7.5 M urea, 25 %フォルムアミド)で泳動後、該当バンドを切り出し、MQ水で12時間抽出を行った。アミコン10K (Merck Millipore)処理, イソプロパノール沈殿により、ゲル抽出産物を得た。
VEGFタンパク質に翻訳される化合物ENの合成に用いた材料(ポリヌクレオチド)の配列情報を示す。表中の各ヌクレオチドNはRNA、N(M)は2'-O-メチル修飾RNAを、N(F)は2'-F化修飾RNAを、N(MOE)は2'-O-メトキシエチル修飾RNAを、dNはDNAを表す。

(線状化プラスミドDNAの調製とIn vitro transcriotionによるRNA断片の調製)
3'側の断片は上記RN'-2調製法と同様の方法で実施した。
化学合成により得られたRNA断片EN-1ならびにRNA断片RN'-2、テンプレートDNA-Cを用いて、RNA ligase 2による連結反応を行った。終濃度は、EN-1 2 μM, RN'-2 2 μM, テンプレートDNA 4 μM, PEG8000 10% (v/v), T4 RNA ligase 2 (NEB) 1 U/μL, T4 RNA ligase 2 buffer (NEB) 1x, RNase inhibitor Murine (NEB) 1 U/μL。チューブにPEG8000, T4 RNA ligase 2, RNase inhibitor以外の基質・試薬を加えた後、90 度3分加熱し、室温まで自然放熱した。残りの試薬・酵素を添加し、45度で1時間インキュベートした。DNase (recombinant, RNase-free, Takara)を終濃度0.33 U/μL 分添加し、37度で15分インキュベートした。 フェノールクロロホルム抽出、イソプロパノール沈殿により粗精製を行い、連結RNAを得た。反応サンプルは、dPAGE(5% acrylamide, 7M urea)で解析した。ゲル精製を以下の通り行った。dPAGE(6% acrylamide, 7.5 M urea, 25 %フォルムアミド)で泳動後、該当バンドを切り出し、MQ水で12時間抽出を行った。アミコン10K (Merck Millipore)処理, イソプロパノール沈殿により、ゲル抽出産物を得た。
上記で得られたRN、RN'およびENの配列情報を表145に示す。表145において、各ヌクレオチドN(大文字)はRNAを、各ヌクレオチドN(F)は2'-F化修飾RNAを、N(MOE)は2'-O-メトキシエチル修飾RNAを表す。


配列番号2:化合物E1-1
配列番号3:化合物E1-2
配列番号4:テンプレートDNA1
配列番号5:化合物E2
配列番号6:化合物E2-1
配列番号7:化合物E2-2
配列番号8:化合物E3
配列番号9:化合物E3-1
配列番号10:化合物E3-2
配列番号11:化合物E4
配列番号12:化合物E4-1
配列番号13:化合物E4-2
配列番号14:化合物R1
配列番号15:化合物R1-1
配列番号16:化合物R1-2
配列番号17:テンプレートDNA2
配列番号18:化合物R2
配列番号19:化合物R2-1
配列番号20:化合物R2-2
配列番号21:化合物R3
配列番号22:化合物R3-1
配列番号23:化合物R3-2
配列番号24:テンプレートDNA3
配列番号25:化合物R4
配列番号26:化合物R4-1
配列番号27:化合物R4-2
配列番号28:化合物R4-3
配列番号29:テンプレートDNA4
配列番号30:化合物R5
配列番号31:化合物R5-1
配列番号32:化合物R5-2
配列番号33:化合物R6
配列番号34:化合物R6-1
配列番号35:化合物R6-2
配列番号36:化合物R7
配列番号37:化合物R7-1
配列番号38:化合物R7-2
配列番号39:化合物R8
配列番号40:化合物R8-1
配列番号41:化合物R8-2
配列番号42:化合物R9
配列番号43:化合物R9-1
配列番号44:化合物R9-2
配列番号45:化合物R10
配列番号46:化合物R10-1
配列番号47:化合物R10-2
配列番号48:化合物R11
配列番号49:化合物R11-1
配列番号50:化合物R11-2
配列番号51:化合物R12
配列番号52:化合物R12-1
配列番号53:化合物R12-2
配列番号54:化合物R13
配列番号55:化合物R13-1
配列番号56:化合物R13-2
配列番号57:化合物R14
配列番号58:化合物R14-1
配列番号59:化合物R14-2
配列番号60:化合物R15
配列番号61:化合物R15-1
配列番号62:化合物R15-2
配列番号63:化合物R16
配列番号64:化合物R16-1
配列番号65:化合物R16-2
配列番号66:化合物R17
配列番号67:化合物R17-1
配列番号68:化合物R17-2
配列番号69:化合物R17-3
配列番号70:テンプレートDNA5
配列番号71:テンプレートDNA6
配列番号72:化合物R18
配列番号73:化合物R18-1
配列番号74:化合物R18-2
配列番号75:RT primer
配列番号76:Fw primer
配列番号77:Rv primer
配列番号78:Taqman MGB Probe
配列番号79~538:化合物E5~E248-1
配列番号539~542:翻訳産物ポリペプチド標品
配列番号543:FWプライマー
配列番号544:RVプライマー1
配列番号545:5'末端ポリヌクレオチド配列RN-1
配列番号546:人工合成遺伝子配列GN
配列番号547:テンプレートDNA-A
配列番号548:RVプライマー2
配列番号549:シーケンスプライマーM13FW
配列番号550:シーケンスプライマーM13RV
配列番号551:5'末端ポリヌクレオチド配列RN'-1
配列番号552:人工合成遺伝子配列GN'
配列番号553:テンプレートDNA-B
配列番号554:EN-1
配列番号555:テンプレートDNA-C
配列番号556:化合物RNおよびRN'
配列番号557:化合物EN
Claims (25)
- 開始コドンから終止コドンまでの翻訳領域を含み、
前記翻訳領域がn個のコドンを含み、前記nが2以上の正の整数であり、
前記n個のコドンが、それぞれ、1番目、2番目及び3番目のヌクレオチドを含み、
前記n個のコドンのうちの少なくとも2個のコドンにおける1番目のヌクレオチドが、糖部修飾ヌクレオチドである、
ポリヌクレオチド。 - 前記糖部修飾ヌクレオチドが、少なくとも2'位が修飾された糖部を含む、請求項1に記載のポリヌクレオチド。
- 前記少なくとも2'位が修飾された糖部が、以下から選択される、

- 前記糖部修飾ヌクレオチドが、アデニン、グアニン、シトシン及びウラシルからなる群から選択される塩基に対応する塩基部を含み、
前記塩基の種類が少なくとも2種類である、請求項1~3のいずれか一項に記載のポリヌクレオチド。 - 前記糖部修飾ヌクレオチドの少なくとも1個が、修飾塩基部を含む、請求項1~4のいずれか一項に記載のポリヌクレオチド。
- 前記n個のコドンの全てにおける1番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項1~5のいずれか一項に記載のポリヌクレオチド。
- 前記終止コドンの1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項1~6のいずれか一項に記載のポリヌクレオチド。
- 前記開始コドンの1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項1~7のいずれか一項に記載のポリヌクレオチド。
- 前記n個のコドンのうちの少なくとも1個のコドンにおける2番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項1~8のいずれか一項に記載のポリヌクレオチド。
- 前記n個のコドンのうちの少なくとも1個のコドンにおける3番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項1~9のいずれか一項に記載のポリヌクレオチド。
- 前記nが2~2000の整数である、請求項1~10のいずれか一項に記載のポリヌクレオチド。
- 5'側非翻訳領域を更に含む、請求項1~11のいずれか一項に記載のポリヌクレオチド。
- 前記5'側非翻訳領域が、以下の塩基部を含む塩基部修飾ヌクレオチドを含む、

請求項12に記載のポリヌクレオチド。 - 前記5'側非翻訳領域の5'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項12又は13に記載のポリヌクレオチド。
- 5'キャップ構造を更に含む、請求項12~14のいずれか一項に記載のポリヌクレオチド。
- 3'側非翻訳領域を更に含む、請求項1~15のいずれか一項に記載のポリヌクレオチド。
- 前記3'側非翻訳領域が、ポリA鎖を含む、請求項16に記載のポリヌクレオチド。
- 前記3'側非翻訳領域の3'末端から1番目、2番目及び3番目のヌクレオチドが、糖部修飾ヌクレオチドである、請求項16又は17に記載のポリヌクレオチド。
- 5'側非翻訳領域及び/又は3'側非翻訳領域が、糖部修飾ヌクレオチドを含む、請求項12~18のいずれか一項に記載のポリヌクレオチド。
- 以下の構造を含む、
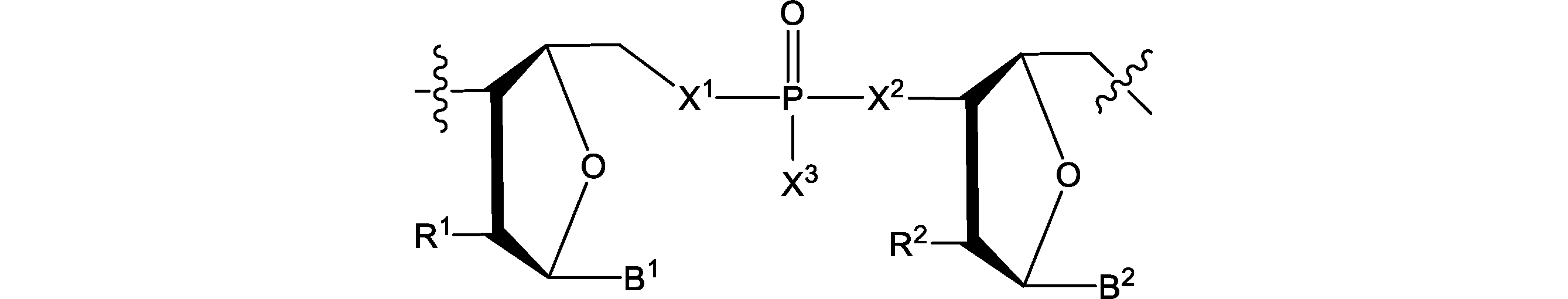
R1及びR2は、それぞれ独立して、H、OH、F又はOCH3を表し、
B1及びB2は、それぞれ独立して、塩基部を表し、
X1は、O、S又はNHを表し、
X2は、O、S、NH又は以下の構造を表し、

ただし、X1及びX2は、同時にOではない]
請求項1~19のいずれか一項に記載のポリヌクレオチド。 - ホスホロチオエート構造を含む、請求項1~20のいずれか一項に記載のポリヌクレオチド。
- 前記n個のコドンのうちの少なくとも1個のコドンにおける1番目のヌクレオチドと2番目のヌクレオチドとが、ホスホロチオエートによって連結されている、請求項1~21のいずれか一項に記載のポリヌクレオチド。
- 前記5'側非翻訳領域の5'末端から1~2番目のヌクレオチド、1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、請求項1~22のいずれか一項に記載のポリヌクレオチド。
- 前記3'側非翻訳領域の3'末端から1~2番目のヌクレオチド、1~3番目のヌクレオチド、1~4番目のヌクレオチド、又は1~5番目のヌクレオチドが、ホスホロチオエートによって連結されている、請求項1~23のいずれか一項に記載のポリヌクレオチド。
- 請求項1~24のいずれか一項に記載のポリヌクレオチドを含む、医薬組成物。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/789,040 US20230073999A1 (en) | 2019-12-26 | 2020-12-25 | Polynucleotide and pharmaceutical composition |
| JP2021567685A JP7787514B2 (ja) | 2019-12-26 | 2020-12-25 | ポリヌクレオチド及び医薬組成物 |
| KR1020227025851A KR20220122701A (ko) | 2019-12-26 | 2020-12-25 | 폴리뉴클레오티드 및 의약 조성물 |
| CA3165956A CA3165956A1 (en) | 2019-12-26 | 2020-12-25 | Polynucleotide and pharmaceutical composition |
| CN202080089509.1A CN115176000A (zh) | 2019-12-26 | 2020-12-25 | 多核苷酸及医药组合物 |
| AU2020414031A AU2020414031A1 (en) | 2019-12-26 | 2020-12-25 | Polynucleotide and pharmaceutical composition |
| EP20906594.5A EP4082579A4 (en) | 2019-12-26 | 2020-12-25 | POLYNUCLEOTIDE AND MEDICINAL COMPOSITION |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019-236399 | 2019-12-26 | ||
| JP2019236399 | 2019-12-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021132589A1 true WO2021132589A1 (ja) | 2021-07-01 |
Family
ID=76575331
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/048799 Ceased WO2021132589A1 (ja) | 2019-12-26 | 2020-12-25 | ポリヌクレオチド及び医薬組成物 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20230073999A1 (ja) |
| EP (1) | EP4082579A4 (ja) |
| JP (1) | JP7787514B2 (ja) |
| KR (1) | KR20220122701A (ja) |
| CN (1) | CN115176000A (ja) |
| AU (1) | AU2020414031A1 (ja) |
| CA (1) | CA3165956A1 (ja) |
| TW (1) | TWI906247B (ja) |
| WO (1) | WO2021132589A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022212442A1 (en) * | 2021-03-31 | 2022-10-06 | Modernatx, Inc. | Synthesis of trinucleotide and tetranucleotide caps for mrna production |
| WO2023277168A1 (ja) * | 2021-06-30 | 2023-01-05 | 協和キリン株式会社 | ポリヌクレオチド及び医薬組成物 |
| US11564893B2 (en) | 2015-08-17 | 2023-01-31 | Modernatx, Inc. | Methods for preparing particles and related compositions |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014093574A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified polynucleotides for altering cell phenotype |
| WO2017123669A1 (en) | 2016-01-11 | 2017-07-20 | Gary Glick | Cyclic dinucleotides for treating conditions associated with sting activity such as cancer |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5678022B2 (ja) | 2012-10-31 | 2015-02-25 | 京セラドキュメントソリューションズ株式会社 | 画像形成装置及び画像形成プログラム |
| US20160022840A1 (en) * | 2013-03-09 | 2016-01-28 | Moderna Therapeutics, Inc. | Heterologous untranslated regions for mrna |
-
2020
- 2020-12-25 JP JP2021567685A patent/JP7787514B2/ja active Active
- 2020-12-25 EP EP20906594.5A patent/EP4082579A4/en active Pending
- 2020-12-25 AU AU2020414031A patent/AU2020414031A1/en active Pending
- 2020-12-25 CN CN202080089509.1A patent/CN115176000A/zh active Pending
- 2020-12-25 WO PCT/JP2020/048799 patent/WO2021132589A1/ja not_active Ceased
- 2020-12-25 TW TW109146310A patent/TWI906247B/zh active
- 2020-12-25 US US17/789,040 patent/US20230073999A1/en active Pending
- 2020-12-25 KR KR1020227025851A patent/KR20220122701A/ko active Pending
- 2020-12-25 CA CA3165956A patent/CA3165956A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014093574A1 (en) | 2012-12-13 | 2014-06-19 | Moderna Therapeutics, Inc. | Modified polynucleotides for altering cell phenotype |
| WO2017123669A1 (en) | 2016-01-11 | 2017-07-20 | Gary Glick | Cyclic dinucleotides for treating conditions associated with sting activity such as cancer |
Non-Patent Citations (38)
| Title |
|---|
| "124", J. AM. CHEM. SOC., 2002, pages 4962 |
| "Chemistry and Biology of Artificial Nucleic Acids", 2012, WILEY-VCH |
| "Current Protocols in Nucleic Acids Chemistry", 2000, JOHN WILEY SONS |
| "Genome Chemistry Jinko Kakusan wo Katsuyo suru Kagakuteki Approach (Scientific approach for utilizing artificial nucleic acids", 2003, KODANSHA LTD. |
| "Protocols for Oligonucleotides and Analogs", 1993, HUMAN PRESS |
| AMIDO-BRIDGED NUCLEIC ACID (AMNA, CHEM BIO CHEM, vol. 13, 2012, pages 2513 |
| BEILSTEIN, J. ORG. CHEM., vol. 13, 2017, pages 2819 - 2832 |
| CARDIAC GENE THERAPY: METHODS IN MOLECULAR BIOLOGY (METHODS AND PROTOCOLS, vol. 1521, 2016 |
| CONSTRAINED ETHYL (CET, THE JOURNAL OF ORGANIC CHEMISTRY, vol. 75, 2010, pages 1569 |
| DRUG DISCOVERY TODAY, vol. 13, no. 19-20, 2008, pages 842 - 855 |
| ETHYLENE BRIDGED NUCLEIC ACID (ENA, NUCLEIC ACIDS RESEARCH, vol. 32, 2004, pages e175 |
| FLUORO HEXITOL NUCLEIC ACID (FHNA: "35", NAT. BIOTECHNOL., vol. 35, 2017, pages 238 |
| GENES, vol. 10, no. 2, 2019, pages 84 |
| HOERNES, THOMAS PHILIPP ET AL.: "Eukaryotic Translation Elongation is Modulated by Single Natural Nucleotide Derivatives in the Coding Sequences of mRNAs", GENES, vol. 10, no. 84, 25 January 2019 (2019-01-25), pages 1 - 12, XP055838407 * |
| HOERNES, THOMAS PHILIPP ET AL.: "Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code", NUCLEIC ACIDS RESEARCH, vol. 44, no. 2, 2016, pages 852 - 862, XP055838402 * |
| J. AM. CHEM. SOC., vol. 121, 1999, pages 5661 - 5665 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 249, 1995, pages 398 - 408 |
| LOCKED NUCLEIC ACID (LNA, TETRAHEDRON LETTERS, vol. 38, 1997, pages 8735 |
| MOLECULAR CELL, vol. 16, 22 October 2004 (2004-10-22), pages 211 - 221 |
| NAKAMOTO, MARGARET A. ET AL.: "mRNA pseudouridylation affects RNA metabolism in the parasite Toxoplasma gondii", RNA, vol. 23, 2017, pages 1834 - 1849, XP055838414 * |
| NATURE BIOTECHNOLOGY, vol. 35, no. 3, 2017, pages 193 - 197 |
| NATURE REVIEWS DRUG DISCOVERY, vol. 13, 2014, pages 759 - 780 |
| NATURE, vol. 547, no. 7662, 2017, pages 222 - 226 |
| NUCLEIC ACIDS RESEARCH, vol. 35, 2007, pages 3287 |
| NUCLEIC ACIDS RESEARCH, vol. 42, 2014, pages 13546 |
| NUCLEIC ACIDS RESEARCH, vol. 44, no. 2, 2015, pages 852 - 862 |
| ORGANOMETALLICS, vol. 29, 2010, pages 2176 - 2179 |
| OXY-PEPTIDE NUCLEIC ACID (OPNA, J. AM. CHEM. SOC., vol. 123, 2001, pages 4653 |
| PEPTIDE NUCLEIC ACID (PNA, ACC. CHEM. RES., vol. 32, 1999, pages 624 |
| PEPTIDE RIBONUCLEIC ACID (PRNA, J. AM. CHEM. SOC., vol. 122, 2000, pages 6900 |
| RNA, METHODS IN MOLECULAR BIOLOGY (METHODS AND PROTOCOLS, vol. 703, 2011 |
| SCIENCE, vol. 361, 2018, pages 1234 |
| See also references of EP4082579A4 |
| SPIROCYCLOPROPYLENE BRIDGED NUCLEIC ACID (SCPBNA, CHEM. COMMUN., vol. 51, 2015, pages 9737 |
| TETRAHEDRON, vol. 48, no. 12, 1992, pages 2223 - 2311 |
| TETRAHEDRON, vol. 54, 1998, pages 3607 |
| TOP. CURR. CHEM, 2017, pages 375 - 16 |
| UNLOCKED NUCLEIC ACID (UNA, MOL. THER. NUCLEIC ACIDS, vol. 2, 2013, pages e103 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11564893B2 (en) | 2015-08-17 | 2023-01-31 | Modernatx, Inc. | Methods for preparing particles and related compositions |
| WO2022212442A1 (en) * | 2021-03-31 | 2022-10-06 | Modernatx, Inc. | Synthesis of trinucleotide and tetranucleotide caps for mrna production |
| WO2023277168A1 (ja) * | 2021-06-30 | 2023-01-05 | 協和キリン株式会社 | ポリヌクレオチド及び医薬組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3165956A1 (en) | 2021-07-01 |
| US20230073999A1 (en) | 2023-03-09 |
| JP7787514B2 (ja) | 2025-12-17 |
| TW202138558A (zh) | 2021-10-16 |
| AU2020414031A1 (en) | 2022-07-14 |
| TWI906247B (zh) | 2025-12-01 |
| EP4082579A1 (en) | 2022-11-02 |
| CN115176000A (zh) | 2022-10-11 |
| JPWO2021132589A1 (ja) | 2021-07-01 |
| EP4082579A4 (en) | 2024-06-26 |
| KR20220122701A (ko) | 2022-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7644229B2 (ja) | 5’-キャッピングされたrna合成用オリゴヌクレオチド | |
| AU2008286771B2 (en) | Tetrahydropyran nucleic acid analogs | |
| KR20220076508A (ko) | 올리고뉴클레오티드 조성물 및 이의 사용 방법 | |
| CN110088113A (zh) | 用于亚磷酰胺和寡核苷酸合成的组合物和方法 | |
| KR20150036642A (ko) | 키랄 제어 | |
| AU2009308217A1 (en) | 5' and 2' bis-substituted nucleosides and oligomeric compounds prepared therefrom | |
| KR20150131365A (ko) | 브리지드 바이사이클릭 뉴클레오시드 | |
| JP7787514B2 (ja) | ポリヌクレオチド及び医薬組成物 | |
| WO2023277168A1 (ja) | ポリヌクレオチド及び医薬組成物 | |
| CA2853609A1 (en) | Inhibition of viral gene expression | |
| TW201718030A (zh) | β2GPI基因表現抑制核酸複合體 | |
| WO2024217535A1 (zh) | 一种寡聚核酸及用于降低脱靶活性的化合物 | |
| CN118813612A (zh) | 一种化合物在RNAi药物中的应用 | |
| JP2022053148A (ja) | 核酸モノマー | |
| CN117769598A (zh) | 多核苷酸及医药组合物 | |
| CN118812614A (zh) | 一类5’-修饰新型核苷酸及其应用 | |
| WO2025188589A1 (en) | NOVEL NUCLEOTIDES AND OLIGONUCLEOTIDES AND RNAi AGENTS COMPRISING THE SAME | |
| WO2010072146A1 (zh) | 一种异核苷化合物及其制备方法和修饰的寡核苷酸 | |
| HK1142909B (en) | Tetrahydropyran nucleic acid analogs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20906594 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021567685 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3165956 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020414031 Country of ref document: AU Date of ref document: 20201225 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20227025851 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020906594 Country of ref document: EP Effective date: 20220726 |