WO2021137299A1 - 植物の改変方法 - Google Patents
植物の改変方法 Download PDFInfo
- Publication number
- WO2021137299A1 WO2021137299A1 PCT/JP2020/049058 JP2020049058W WO2021137299A1 WO 2021137299 A1 WO2021137299 A1 WO 2021137299A1 JP 2020049058 W JP2020049058 W JP 2020049058W WO 2021137299 A1 WO2021137299 A1 WO 2021137299A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- shoot
- apex
- plant
- shoot apex
- fine particles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8201—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation
- C12N15/8206—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation by physical or chemical, i.e. non-biological, means, e.g. electroporation, PEG mediated
- C12N15/8207—Methods for introducing genetic material into plant cells, e.g. DNA, RNA, stable or transient incorporation, tissue culture methods adapted for transformation by physical or chemical, i.e. non-biological, means, e.g. electroporation, PEG mediated by mechanical means, e.g. microinjection, particle bombardment, silicon whiskers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/89—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microinjection
- C12N15/895—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation using microinjection using biolistic methods
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
Definitions
- the present invention relates to a method for modifying a plant.
- the general transformation methods for plants that are currently in widespread use are the electroporation method, the Agrobacterium tumefaciens bacterium, or the particle gun method, etc. It is a method to introduce by.
- these methods have a problem that even if a gene is introduced into a cell or a tissue, it is difficult to culture the tissue depending on the plant variety, and it is difficult to regenerate the plant to prepare a transformant.
- the transformation efficiency is not sufficiently high, and therefore there is a problem that a selectable marker gene must be introduced to perform marker selection.
- somatic mutations somatic mutations (somaclonal mutations) frequently occur with the need for long-term tissue culture. Therefore, from the viewpoint of reducing the labor for producing transformed plants and the safety of transformed plants, it has been required to develop a method for modifying plants without tissue culture.
- Non-Patent Document 1 a method of directly introducing a gene into the shoot apex of an exposed immature embryo or a ripe embryo by using a particle gun method is known.
- Patent Document 1 and Non-Patent Document 2 describe a method of infecting a ripe embryo immediately after germination with Agrobacterium and introducing a gene.
- Non-Patent Document 1 has not yet demonstrated that the transgene is transmitted to the next generation. Due to these factors, the above method has not been generalized until now.
- the present invention provides a method for modifying a plant without using callus or tissue fragments in an in vitro culture system.
- the present invention also provides a method for modifying a plant without introducing a selectable marker gene.
- the present invention further provides a method for editing the genome of a plant and a method for producing a plant having excellent reproducibility.
- the present inventors have completed the present invention as a result of diligent studies for solving the above-mentioned problems.
- the present invention includes the following.
- a method for modifying a plant wherein the fine particles are coated with at least one nucleic acid and / or at least one protein, and the coated fine particles are shot into the stem apex of the plant.
- the stem apex includes a step of growing a stem apex shot with fine particles to obtain a plant body and a step of selecting a modified plant body from the plant body, and the stem apex is (1) a stem apex of a ripe seed embryo.
- ⁇ 4> The method according to ⁇ 1> above, wherein the growth of the shoot apex by shooting the coated fine particles is carried out in a medium containing no antibiotics and plant hormones.
- ⁇ 5> The method according to ⁇ 1>, wherein the shooting of the plant into the stem apex is performed by shooting fine particles coated on L2 layer cells of the stem apex.
- ⁇ 6> The method according to ⁇ 1> above, wherein the plant is shot into the shoot apex by shooting fine particles coated on the stem apex stem cells that migrate to the germline of the shoot apex.
- ⁇ 7> The method according to ⁇ 1>, wherein the fine particles have a diameter of 0.3 ⁇ m or more and 1.5 ⁇ m or less.
- the stem apex of the plant is the stem apex of the embryo of the (1) ripe seed, and the stem apex of the embryo of the (1) ripe seed is the endosperm, sheath leaf, leaf primordia, and leaf primordia from the ripe seed.
- the shoot apex of the plant is the shoot apex of the (2) shoot, and the shoot apex of the (2) shoot is exposed by removing tubers and leaf primordia from the shoot. The method according to ⁇ 1> above.
- the stem apex of the plant is the stem apex of the (3) apical bud or lateral bud, and the stem apex of the (3) apical bud or lateral bud removes the leaf primordia from the apical bud or lateral bud and exposes it.
- the stem apex of the plant is (4) the stem apex of the immature embryo, and the stem apex of the (4) immature embryo is from immature seeds 8 to 35 days after pollination to endosperm, sheath leaf, leaf primordia.
- the fine particles are coated with linear DNA containing a nucleic acid cassette for which the fine particles are to be introduced.
- the linear DNA is a linear plasmid and further contains a nucleic acid of 0.8 kb or more and 1.2 kb or less located at each end of the nucleic acid cassette.
- the coating of the fine particles is performed by coating the fine particles with a nucleic acid encoding a modifying enzyme.
- ⁇ 15> The method according to ⁇ 1> above, wherein the modifying enzyme is a nuclease or deaminase.
- the fine particle coating is performed by coating the fine particles with nucleic acids linked to promoters and terminators, respectively, and the nucleic acids encode nucleic acids capable of expressing at least one guide RNA and Casnuclease protein.
- ⁇ 17> The method according to ⁇ 1>, wherein the coating of the fine particles is performed by coating the fine particles with a nuclease.
- the nuclease is a Cas nuclease.
- the coating of the fine particles is performed by coating the fine particles with a protein, and the shooting into the stem apex of the plant is performed using a hydrophilic macrocarrier film having the fine particles coated with the protein.
- ⁇ 20> The method according to ⁇ 1>, wherein the ripe seed is a ripe seed containing roots having a length of 1 mm or less.
- ⁇ 21> The method according to ⁇ 1>, wherein the plant is selected from the group consisting of wheat, barley, rice, corn, soybean, potato and apple.
- ⁇ 22> The step of bringing the shoot apex of the ripe seed embryo, the shoot apex of the young shoot, the shoot apex of the apical bud or the shoot apex, or the shoot apex of the immature embryo into contact with the hyperosmotic solution is further included in ⁇ 1>.
- the method described. ⁇ 23> The method according to ⁇ 22>, wherein the step of contacting with the high osmotic solution is before or after the step of shooting the coated fine particles.
- ⁇ 24> The method according to ⁇ 22>, wherein the step of contacting with the high osmotic solution is before and after the step of shooting the coated fine particles.
- a method for editing a plant genome which comprises a step of coating fine particles with at least one nucleic acid and / or at least one protein, a step of shooting the coated fine particles into the stem apex of a plant, and the coating.
- the stem apex includes a step of growing a stem apex in which the fine particles are shot to obtain a plant body and a step of selecting a modified plant body from the plant body, and the stem apex is (1) a shoot apex of a ripe seed embryo.
- the stem apex includes a step of growing a stem apex shot with fine particles to obtain a plant body and a step of selecting a modified plant body from the plant body, and the stem apex is (1) a stem apex of a ripe seed embryo.
- the target gene can be efficiently introduced into shoot apical stem cells that migrate to the germline in the shoot apex.
- FIG. 1 is a diagram showing the state of expression of the GFP protein at the shoot apex according to Example 1 of the present invention.
- Figure 2 is a graph showing the results of transgene confirmation by genomic PCR in T 1 generation transformant according to Example 2 of the present invention.
- FIG. 3 is a photograph showing the results of the Southern analysis of the transgenic individual (Example 3).
- FIG. 4A is an overall photograph of T 1st generation seeds observed by GFP fluorescence according to Example 4 of the present invention.
- 4B is a diagram of GFP fluorescence observation photograph of T 1 generation of half seeds according to Example 4 of the present invention.
- Figure 4C is a diagram of a GFP fluorescence photograph of the T 1 generation of young leaves according to a fourth embodiment of the present invention.
- FIG. 4D is a diagram of a Western blot photograph of detecting the GFP protein T 1 generation of mature leaf according to a fourth embodiment of the present invention.
- Figure 5A is a photograph showing the results of Southern analysis of the resulting genomic DNA from T 1 seeds young leaves obtained from the main stem (Example 5).
- Figure 5B is a photograph showing the results of Southern analysis of the resulting genomic DNA from T 1 seeds young leaves obtained from the main stem and tiller (Example 5).
- FIG. 6 is a photograph showing the transient expression of GFP in maize (Example 6).
- FIG. 7 is a photograph showing the transient expression of GFP in soybean (Example 7).
- FIG. 8 is a photograph showing the transient expression of GFP in barley (Example 8).
- FIG. 9 is a photograph showing the transient expression of GFP in potatoes (Example 9).
- FIG. 10 is a diagram showing a target gene-introduced individual (Example 13).
- FIG. 11 is a diagram showing the mutagenesis of the target gene in the shoot apex one week after the gene transfer (Example 14).
- Figure 12 is a diagram showing a target gene introduction individuals in T 0 generation adult leaves (Example 14).
- Figure 13 is a diagram showing a target gene mutation in T 1 generation leaves (Example 14).
- FIG. 14 is a diagram showing a target gene-introduced individual by direct introduction of Cas9 protein (Example 15).
- FIG. 15A is a photograph showing immature embryos of isolated maize (17 days after pollination).
- FIG. 15A is a photograph showing immature embryos of isolated maize (17 days after pollination).
- FIG. 15B is a photograph showing immature embryos after culturing.
- FIG. 15C is a photograph showing an immature embryo having a germ length of about 7 to 15 mm (Example 23).
- FIG. 16A is a photograph showing immature maize embryos before gene transfer.
- FIG. 16B is a photograph showing immature maize embryos undergoing gene transfer.
- FIG. 16C is a fluorescence observation photograph showing the transient expression of tdTomato in maize 20 hours after gene transfer (Example 23).
- FIG. 17A is a photograph showing immature maize embryos that have been osmotic-treated before gene transfer.
- FIG. 17B is a photograph showing immature maize embryos undergoing gene transfer.
- FIG. 17A is a photograph showing immature maize embryos that have been osmotic-treated before gene transfer.
- FIG. 17B is a photograph showing immature maize embryos undergoing gene transfer.
- FIG. 17C is a fluorescence observation photograph showing the transient expression of tdTomato in maize 20 hours after gene transfer (Example 24).
- FIG. 18A is a photograph showing immature maize embryos that have been osmotic-treated before gene transfer.
- FIG. 18B is a photograph showing immature maize embryos undergoing gene transfer.
- FIG. 18C is a photograph showing immature maize embryos that have been osmotic-treated after gene transfer.
- FIG. 18D is a fluorescence observation photograph showing the transient expression of tdTomato in maize 20 hours after gene transfer (Example 25).
- FIG. 19A is a fluorescence observation photograph showing the transient expression of tdTomato in maize 20 hours after gene transfer.
- FIG. 19B is a fluorescence observation photograph showing an immature embryo having a strong fluorescence signal. Arrows indicate shoot apical meristems.
- FIG. 19C is a photograph showing selected embryos 10 days after gene transfer.
- FIG. 19D is a photograph showing selected embryos 40 days after gene transfer (Example 26).
- FIG. 20A is a photograph showing the shoot apex of the embryo 7 days after the gene transfer. The sampled shoot apex area is shown by a black line.
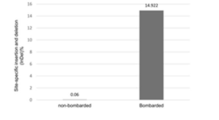
- FIG. 21A is a graph showing the rate of site-specific insertions and deletions (Indel%) in shoot apical meristems of immature maize embryos (Example 26).
- FIG. 21B is a diagram showing site-specific deletions in shoot apical meristems of immature maize embryos.
- the PAM site (TTTA) and the expected LbCpf1 cleavage site are shown (Example 26).
- FIG. 22A is a fluorescence observation photograph showing the transient expression of mNeongreen in maize 20 hours after gene transfer (Example 27).
- FIG. 22B shows a close-up photograph of the selected embryo. Arrows indicate shoot apical meristems (Example 27).
- FIG. 23 is a fluorescence observation photograph showing transient expression of GFP in wheat 20 hours after gene transfer (Example 29).
- a step of coating the fine particles with at least one nucleic acid and / or at least one protein a step of shooting the coated fine particles into the stem apex of the plant, and the coating.
- the stem apex includes a step of growing a stem apex shot with fine particles to obtain a plant body and a step of selecting a modified plant body from the plant body, and the stem apex is (1) a stem apex of a ripe seed embryo. (2) shoot apex of young shoot, (3) shoot apex of apical or lateral shoot, or (4) shoot apex of immature embryo.
- the "seed” includes not only natural seeds obtained by cultivating (or culturing) a plant under natural or similar conditions, but also artificial seeds, but is preferably natural seeds. However, if an artificial seed that can obtain a transformable shoot apex is developed, it can be used. Natural seeds include not only seeds obtained in outdoor fields, but also seeds obtained by greenhouse cultivation and seeds obtained from tissue cultures such as in vitro seedlings. Seeds obtained by tissue culture (direct reprogramming) or the like can also be used as the seeds of the present invention as long as the shoot apex can be obtained.
- Ripe seeds are those that have completed the ripening process after pollination and are fully ripe as seeds. Whether or not the ripening process is completed can be determined by whether or not the water content of the seed is 35% or less.
- Rhizome is a general term for tubers located underground, and refers to tubers with many buds, bulbs with spherical and large apical buds, and bulbs with enlarged scaly leaves attached to shortened stems. Immature seeds are seeds that are not fully ripe.
- the stem apex means a tissue consisting of a growth point (stem apical meristem) at the tip of the stem and several leaf primordia generated from the growth point and the growth point.
- a growth point stem apical meristem
- leaf primordia generated from the growth point and the growth point.
- only the hemispherical (dome-shaped) growth point from which the leaf primordia is removed may be used as the stem apex, or the stem apex containing the growth point and the leaf primordia and the plant tissue including the stem apex may be used.
- a virus-free tissue can be obtained by using only the growth points from which the leaf primordia have been removed.
- the "germ cell lineage” is a general term for germ cells from the primordial germ cell which is the source of the germ cell to the egg cell and the sperm cell which are the final products, and the "L2 layer” is used.
- the second cell layer from the outside of the shoot apical meristem.
- the "young shoot” is a shoot at the apex of the embryo of a higher plant, and can be identified by growing after germination and becoming a rhizome.
- the "immature embryo” means an embryo of an immature seed, which is a seed. It can be determined by the degree of ripening.
- the method for modifying a plant according to the present invention can be widely applied to plants that generally produce seeds, plants that produce rhizomes, and plants that can be cultivated at the shoot apex. Therefore, the target plants of the plant modification method according to the present invention are seed plants including angiosperms and gymnosperms. Angiosperms include monocotyledonous plants and dicotyledonous plants.
- the implanter transformation method is generally a transformation method that does not include the operation of tissue culture, and is a method of transforming cells at a growth point site while the plant is growing. However, in the present specification, the implanter transformation method is used in the sense that it includes only the tissue culture method using shoot apex culture and does not include other methods including tissue culture operations. The implanter method has the same meaning.
- the monocotyledonous plant may be of any kind, and examples thereof include grasses, liliaceae, bananas, bromeliaceae, orchids, and the like.
- Examples of gramineous plants include rice, wheat, barley, corn, embaku, shiva, sorghum, rye, millet, and sugar cane.
- Examples of Liliaceae plants include leeks and asparagus.
- Examples of Musaceae plants include bananas.
- Examples of plants of the Bromeliaceae family include pineapples.
- Examples of orchidaceous plants include orchids.
- the dicotyledonous plants include, for example, Abranaceae plants, legumes, eggplants, melons, hirugaos, roses, mulberry plants, aois, sardines, sardines, and tades. Examples include family plants.
- Examples of cruciferous plants include Arabidopsis thaliana, Chinese cabbage, rapeseed, cabbage, cauliflower, and radish.
- Examples of legumes include soybeans, adzuki beans, common beans, peas, cowpeas, alfalfa and the like.
- Examples of Solanaceae plants include tomatoes, eggplants, potatoes, tobacco, and capsicum.
- Examples of Cucurbitaceae plants include oriental melon, cucumber, melon, and watermelon.
- Examples of Convolvulaceae plants include morning glory, sweet potato (convolvulaceae), bindweed and the like.
- Examples of Rosaceae plants include roses, strawberries, and apples.
- Examples of Morus alba plants include morus alba, figs, and rubber trees.
- Examples of mallow plants include cotton and kenaf.
- Examples of Asteraceae plants include lettuce.
- Examples of Amaranthaceae plants include sugar beet (Amaranthus tricolor).
- gymnosperms include pine, sugi, ginkgo and cycad.
- the ripe seeds or immature seeds of the plant are made to absorb water. If necessary, vernalization may be performed before water absorption. Water absorption is performed by soaking the seeds in water and incubating them. Alternatively, the embryo excluding the endosperm and the like may be incubated on an agar medium.
- the water absorption temperature is preferably 15 to 25 ° C. for wheat, barley or rice, and 25 to 35 ° C. for corn or soybean, for example. At this time, the water may be changed at least once.
- the water absorption period is preferably before the radicles start to grow (roots of 1 mm or less) or before new leaf primordia are formed.
- water absorption time it is less than 16 hours after water absorption, preferably 12 hours, although it depends on the dormant state of the seed.
- the shoot apex of the embryo in the seed absorbed above is exposed.
- the shoot apex is exposed by removing the pods and leaf primordia.
- the seed coat and cotyledons are removed to expose the shoot apex.
- the shoots are sprouted from the seed potatoes, and the tubers and leaf primordia are removed from the cut sprouts to expose the shoot apex.
- the shoot apex is exposed by removing the leaf primordia from the seed embryo or the extracted apical or lateral bud.
- the exposure means may be any one as long as it can remove the sheath leaves and leaf primordia or seed coats and cotyledons under a stereomicroscope, and for example, a needle having a diameter of about 0.2 mm is pierced. Instruments for installation, tweezers, pipettes, syringes, and cutting instruments such as scalpels and cutters. Next, the endosperm and excess scutellum are excised using a cutting instrument such as a scalpel, and the embryo and scutellum containing the exposed shoot apex are placed on an agar medium with the shoot apex facing upward.
- a cutting instrument such as a scalpel
- the scalpel may be replaced with a new sterile scalpel at the final stage of cutting the shoot apex to obtain a virus-free shoot apex.
- a virus-free transformant can be obtained.
- immature seeds it is preferable to use embryos 8 to 35 days after pollination, more preferably 10 to 20 days after pollination, and 15 to 20 days after pollination. Is more preferred.
- immature seeds 1/4 of the grains are removed from the upper end of the immature ears having embryos from which the shell and silk have been removed, and the immature embryos extracted from the grains can be used.
- nucleic acid and / or protein into cells at shoot apex
- a recombinant vector containing the target gene is prepared to target the shoot apex of a ripe embryo or an immature embryo, and the Agrobacterium method, electroporation method, particle gun method, PEG-calcium phosphate method, and liposomes are used.
- Nucleic acid (recombinant vector, etc.), protein, etc. can be introduced by the method, microinjection method, whisker method, plasma method, laser injection method, etc. Alternatively, a method of shooting into an immature seed embryo is preferable.
- the means for shooting the fine particles is not particularly limited as long as the fine particles can be shot into the plant cells, and examples thereof include a particle gun (gene gun) in the particle gun method.
- a particle gun gene gun
- a method of introducing into a ripe seed embryo or an immature seed embryo by using a particle gun method is preferable from the viewpoint of introduction efficiency into a plant.
- the particle gun method is also effective in introducing genes into the shoot apex of potatoes.
- the particle gun method is a method of coating metal fine particles with nucleic acid and / or protein and driving them into a cell tissue, and is effective when the infection efficiency of Agrobacterium is low as in monocotyledonous plants.
- the vector used in the present invention is not particularly limited, and for example, pAL system (pAL51, pAL156, etc.), pUC system (pUC18, pUC19, pUC9, etc.), pBI system (pBI121, pBI101, pBI221, pBI2113, pBI101.2, etc.), A pPZP system, a pSMA system, an intermediate vector system (pLGV23Neo, pNCAT, etc.), cauliflower mosaic virus (CaMV), green beans mosaic virus (BGMV), tobacco mosaic virus (TMV), and the like can be used.
- pAL system pAL51, pAL156, etc.
- pUC system pUC18, pUC19, pUC9, etc.
- pBI system pBI121, pBI101, pBI221, pBI2113, pBI101.2, etc.
- a pPZP system a pSMA system
- an intermediate vector system p
- a vector containing the target gene can be prepared, for example, as follows.
- a method is used in which the purified DNA is cleaved with an appropriate restriction enzyme, inserted into a restriction enzyme site or a multicloning site of an appropriate vector DNA, and ligated to the vector. Can be done.
- the target gene may be inserted into the intermediate vector by double cross-over, and TA cloning, In-Fusion cloning, or the like may be used.
- the target gene or the target gene for genome editing is not particularly limited as long as the expression or suppression of the expression of the gene is desired, and it may be an endogenous gene or a foreign gene of the target plant.
- a different species may be used, and for example, genes such as animals, plants, microorganisms, and viruses can be used.
- genes include, for example, sugar metabolism-related genes, lipid metabolism-related genes, useful substance (pharmaceuticals, enzymes, pigments, aromatic components, etc.) production-related genes, plant growth control (promotion / suppression) -related genes, and flowering regulation-related genes.
- Disease resistance insect feeding resistance, nematodes, mold (fungi) and bacterial disease resistance, virus (disease) resistance, etc.
- Related genes environmental stress (low temperature, high temperature, dryness, salt, photodamage, ultraviolet rays) Examples include related resistance genes, transporter-related genes, flour-making characteristics / bread-making characteristics / noodle-making characteristics-related genes, and site-specific nuclease genes.
- the target gene may be introduced so as to express antisense, ribozyme, RNAi, etc., depending on the purpose of gene transfer.
- nuclease gene to be introduced may be of a different species from which it is derived, and for example, genes such as animals, plants, microorganisms, and viruses, or artificially synthesized genes can be used. Examples of such genes include meganuclease genes, TALEN, CRISPR-CAS system, TARGET AID and the like.
- the desired mutation may be introduced by inserting or substituting foreign DNA between the cleaved sites by recombination.
- gene editing is a part of a technique called new bleeding technique (NBT), in which a specific gene on the genome is cleaved and a mutation is introduced by using a meganuclease, CRISPR-CAS, or the like.
- NBT new bleeding technique
- Gene disruption, site-specific DNA fragment insertion or substitution, or an artificial enzyme complex in which the nuclease activity is removed from the CRISPR system and the deaminase deaminase is added it is not limited to these, and any technique that can edit the genome may be used, although it includes, but is not limited to, the introduction of a targeted point mutation with high efficiency by expressing it in a cell. By using genome editing technology, the targeted gene can be destroyed with high efficiency.
- Genome editing can be said to be a technique different from the conventional plant transformation methods in which foreign genes are integrated almost randomly, such as the direct introduction method and the Agrobacterium method, and the genome is defined from the definition of the transformation method.
- Genome editing technology is characterized by including the step of cleaving genomic DNA using a nuclease or a guide RNA and a nuclease capable of targeting the cleavage site, and is a conventional transformation method using such a targetable nuclease or a guide RNA and a nuclease. Can be distinguished from.
- nuclease protein may be introduced into the cell, and the DNA and / or RNA encoding the nuclease gene is introduced into the cell to express the nuclease protein. It means that it may be.
- guide RNA RNA may be introduced into cells, or DNA capable of expressing guide RNA may be introduced to express guide RNA.
- Examples of the protein encoded by the site-specific nuclease gene include zinc finger nuclease, protein expressing zinc finger nuclease activity, and TAL effector nucleoase (TALEN).
- Zinc finger nucleases are fusion proteins of several zinc finger motifs that recognize specific bases and FokI nucleases.
- TALLEN is a fusion protein of Transcriction Activator Like (TAL) effector and FokI nuclease.
- Site-specific nucleases are composed of other additional targeting techniques, such as meganucleases, RNA-induced CRISPR-Cas9, or leucine zippers.
- the expression of one or more gene products can be modified or modified by introducing a site-specific nuclease into the shoot apex, incorporating it into the genome, expressing it, and editing the genome. That is, a CRISPR-Cas system containing a DNA molecule encoding one or more gene products and capable of containing one or more guide RNAs targeting the Cas protein and the DNA molecule into cells that can express it is introduced.
- One or more guide RNAs target the genomic locus of a DNA molecule encoding one or more gene products
- the Cas protein cleaves the genomic locus of a DNA molecule encoding one or more gene products.
- nuclease gene or protein When a nuclease gene or protein is introduced into a cell for genome editing, it is not always necessary to integrate it into the genome to obtain a stable transformant.
- the nuclease gene can be transiently expressed, thereby allowing genome editing.
- Genome editing can also be performed by introducing a protein (and guide RNA) such as Cas protein into cells. According to these methods, the obtained genome-edited individual may not be a genetically modified product.
- the Cas protein and guide RNA may be naturally occurring (combinations) or may be non-naturally occurring combinations. In the present invention, the expression of two or more gene products may be modified or modified.
- the guide RNA may contain a guide sequence fused to the tracr sequence.
- the length of the guide RNA is at least 15, 16, 17, 18, 19, 20 nucleotides, and the preferred upper limit number of nucleotides is 30 or less, more preferably 25 or less, still more preferably 22 or less, and most preferably 20 or less. Is.
- the cell to be transformed, or the cell subject to genome editing is a plant cell, more preferably a shoot apical meristem cell, and even more preferably an L2 cell in the shoot apical meristem. Most preferably, the cells migrate to the germline in the shoot apical meristem.
- a protein such as Cas protein may contain one or more nuclear localization signals (NLS).
- the Cas protein is a type II CRISPR-based enzyme.
- the Cas protein is a Cas9 protein.
- the Cas9 protein is Streptococcus pneumoniae (S. pneumoniae), Streptococcus pyogenes (S. pyogenes), or S. pneumoniae. It is S. thermophilus Cas9 and may contain mutant Cas9 derived from those organisms.
- the protein can be a Cas9 homolog or ortholog.
- Proteins such as Cas protein may be codon-optimized for expression in eukaryotic cells.
- the Cas protein can direct the cleavage of one or two strands in the localization of the target sequence.
- the expression of the gene product is reduced and the gene product is a protein.
- a addition signal, selectable marker gene, etc. can be linked to the vector.
- the target gene to be inserted into the vector may be a plurality of types per vector, and the recombinant vector to be coated on the fine particles may be a plurality of types per fine particle.
- a recombinant vector containing a target gene or a nuclease gene and a recombinant vector containing a drug resistance gene may be prepared separately, mixed, coated with fine particles, and shot into a plant tissue.
- the target gene for genome editing may be two or more, or a vector may be constructed so that two or more target genes can be cleaved.
- two or more kinds of vectors may be prepared, or a plurality of guide RNAs may be inserted so as to be expressed on one plasmid. It may also contain a guide sequence in which the guide RNA is fused to the tracr sequence.
- the double strand may be cleaved site-specifically to promote homologous recombination and site-specific genome editing may be performed.
- DNA having homologous sequences on both sides of the cleavage region may be introduced to cause double crossover recombination.
- Homologous recombination can also be performed using a D10A mutant of Cas9, which is known to function as a nickase (a DNA cleaving enzyme that inserts a nick into only one DNA strand).
- a protein can be translated from a plurality of coding regions by inserting an IRES (internal ribosome entry site) as a translation start region on the 3'-downstream side of the promoter and on the 5'-upstream side of the translation start codon.
- IRES internal ribosome entry site
- the terminator may be any sequence that can terminate the transcription of the gene transcribed by the promoter and has a poly A addition signal.
- a terminator for a nopaline synthase (NOS) gene and a terminator for an octopine synthase (OCS) gene for example, a terminator for a nopaline synthase (NOS) gene and a terminator for an octopine synthase (OCS) gene.
- NOS nopaline synthase
- OCS octopine synthase
- the selection marker gene examples include a herbicide resistance gene (biaraphos resistance gene, glyphosate resistance gene (EPSPS), sulfonylurea resistance gene (ALS)), drug resistance gene (tetracycline resistance gene, ampicillin resistance gene, canamycin resistance gene). , Hyglomycin resistance gene, Spectinomycin resistance gene, Chloramphenicol resistance gene, Neomycin resistance gene, etc.), Fluorescent or luminescent reporter genes (luciferase, ⁇ -galactosidase, ⁇ -glucuronidase (GUS), green fluorescence protein (GFP) ) Etc.), neomycin phosphotransferase II (NPT II), dihydrofolate reductase and other enzyme genes.
- EPSPS glyphosate resistance gene
- ALS sulfonylurea resistance gene
- drug resistance gene tetracycline resistance gene, ampicillin resistance gene, canamycin resistance gene.
- Hyglomycin resistance gene Spectinomycin
- the vector containing the target gene to be shot into a plant may be a circular plasmid, and may be a linear DNA obtained by cutting the plasmid with a restriction enzyme, a linear plasmid, a nucleic acid cassette fragment obtained by cutting out only the DNA fragment to be introduced, or a cassette fragment. It may be a DNA fragment in which a nucleic acid of 0.8 kb or more and 1.2 kb or less is added to one or both ends. These DNA fragments may be DNA fragments amplified by PCR.
- the nucleic acid to be added is not particularly limited and may be a sequence derived from a vector, but the sequence of the target site for introduction is more preferably used.
- the lower limit of the length of the nucleic acid added to the end of the cassette fragment is 0.5 kb or more, more preferably 0.8 kb or more, further preferably 1.0 kb or more, and the upper limit is 3.0 kb or less. It is preferably 2.0 kb or less, more preferably 1.5 kb or less.
- the target gene or a vector containing a nuclease targeting the target gene is shot into a ripe embryo, larva, apical bud, lateral bud, or immature embryo of a plant.
- Nucleic acids and / or proteins (such as nucleases) encoding the gene of interest or the nuclease gene that targets the gene of interest can be coated on the surface of microparticles (microcarriers) and shot into plant cells.
- the fine particles are not particularly limited as long as they have a high specific density, are chemically inactive and do not easily harm the living body in order to enhance the penetrating power into the cells, and examples thereof include metal fine particles and ceramic fine particles. Be done.
- the metal fine particles may be single metal fine particles or alloy fine particles, and among the single metal fine particles, gold particles, tungsten particles, and the like are particularly preferably used.
- the target gene can be introduced into plant cells as follows. First, fine particles such as gold particles or tungsten particles are washed and sterilized, and the fine particles, nucleic acids (recombinant vector, linear DNA, RNA, etc.) and / or proteins, CaCl 2 , and spermidine are added while stirring with a vortex mixer or the like. Then, DNA / RNA and / or protein is coated on gold particles or tungsten particles, and washed with ethanol or phosphate buffered physiological saline (PBS, etc.). The coating may be the entire surface or a part of the metal fine particles.
- PBS phosphate buffered physiological saline
- the particle size (diameter) of the fine particles is preferably 0.3 ⁇ m or more and 1.5 ⁇ m or less, more preferably 0.4 ⁇ m or more, still more preferably 0.5 ⁇ m or more, and particularly preferably 0.6 ⁇ m.
- the upper limit of the particle size is 1.4 ⁇ m or less, 1.3 ⁇ m or less, 1.2 ⁇ m or less, 1.1 ⁇ m or less, 1.0 ⁇ m or less, 0.9 ⁇ m or less, 0.8 ⁇ m or less, 0.7 ⁇ m or less, 0.6 ⁇ m. It is preferable in the following order.
- Gold particles or tungsten particles are applied to the macrocarrier film as uniformly as possible using a pipetman or the like, and then dried in a sterile environment such as a clean bench.
- a sterile environment such as a clean bench.
- protein-coated fine particles it is preferable to use a hydrophilic macrocarrier film.
- hydrophilic macrocarrier film a hydrophilic film may be attached to the macrocarrier film, or a hydrophilic coating may be applied.
- a hydrophilic coating may be applied.
- the method for making the film hydrophilic include a method using a surfactant, a photocatalyst, and a hydrophilic polymer.
- hydrophilic polymer used in the above method examples include polyethylene glycol, hydroxyethyl methacrylate, hydroxypropyl methacrylate, dihydroxyethyl methacrylate, diethylene glycol methacrylate, triethylene glycol methacrylate, polyethylene glycol methacrylate, vinylpyrrolidone, acrylic acid, acrylamide, dimethylacrylamide, and gluco.
- Examples thereof include polymers of hydrophilic monomers such as xyoxyethyl methacrylate, 3-sulfopropylmethacryloxyethyldimethylammonium betaine, 2-methacryloyloxyethyl phosphorylcholine, and 1-carboxydimethylmethacryloyloxyethylmethaneammonium.
- hydrophilic monomers such as xyoxyethyl methacrylate, 3-sulfopropylmethacryloxyethyldimethylammonium betaine, 2-methacryloyloxyethyl phosphorylcholine, and 1-carboxydimethylmethacryloyloxyethylmethaneammonium.
- a plate on which the macrocarrier film, the target ripe embryo, the bud, the apical bud, the lateral bud, or the shoot apex of the immature embryo was placed was installed in the particle gun device, and high-pressure helium gas was directed from the gas acceleration tube toward the macrocarrier film. And fire.
- the macrocarrier film stops at the stopping plate, but the gold particles pass through the stopping plate and penetrate the target placed under the stopping plate, and the target gene is introduced.
- the distance between the stopping plate and the target shoot apex depends on the particle size of the fine particles, but is, for example, preferably 9 cm or less, more preferably 8 cm or less, still more preferably 7 cm or less, and particularly preferably 6 cm or less.
- the lower limit of the distance is, for example, preferably 2 cm or more, more preferably 3 cm or more, still more preferably 4 cm or more.
- the optimum value of the distance between the stopping plate and the target can be appropriately determined by a transient expression experiment or the like according to the type, particle size, gas pressure, etc. of the fine particles.
- the gas pressure depends on the type of fine particles and the distance from the target, but is preferably 1,100 to 1,600 psi, more preferably 1,200 to 1,500 psi.
- the optimum pressure can be appropriately determined by a transient expression experiment or the like according to the type of fine particles, the type of target, the distance between the target and the stopping plate, and the like.
- the number of times the fine particles are shot into the stem apex is preferably 2 times or more, more preferably 3 times or more, and more preferably 4 times or more.
- the upper limit of the number of times the fine particles are shot into the shoot apex is preferably 20 times or less, more preferably 15 times or less, and further preferably 10 times or less.
- the optimum number can be appropriately determined by a transient expression experiment or the like.
- nucleic acids and / or proteins are released from the fine particles, and the nucleic acids are integrated into genomic DNA to obtain transformed cells.
- a nucleic acid that proliferates in the form of a plasmid such as geminivirus or an artificial chromosome is introduced, it may be transformed without integration.
- nucleic acids and / or proteins are released from the fine particles in the cells into which the fine particles are shot, and the nucleic acids migrate to the nucleus to express nuclease to cleave the genomic DNA.
- mutations occur and genome-edited cells are obtained.
- genome editing occurs by translocating to the nucleus (organelle) by a nuclear localization signal (which may be an organelle translocation signal) and cleaving DNA.
- genome editing can be applied not only to nuclear genome genes but also to organelles (intracellular organelles such as chloroplasts and mitochondria).
- the transfer signal to each organelle may be introduced by ligating it to a nuclease, or a promoter ligating to a nuclease or the like may be used only in each organelle.
- the modified ripe embryos, buds, apical buds, lateral buds, or shoot apex of immature embryos are grown on an agar medium for about 1 month and then transplanted into soil.
- transformants can also be obtained by growing in a normal medium (without antibiotics, plant hormones, etc.) without applying selective pressure with drugs, but drug resistance genes May be further introduced.
- a genome editing individual can be selected by simultaneously introducing a nuclease and a drug resistance gene. When a drug resistance gene is introduced, transformed cells can be selectively cultured by the drug.
- a selective drug suitable for shoot apex culture for example, a sulfonyl urea-based herbicide chlorosulfuron (resistance can be acquired by introducing a mutant ALS gene (acetobutyric acid synthase gene)) is known.
- the drug resistance gene When introducing a drug resistance gene, the drug resistance gene may be on the same vector as the target gene or the nuclease gene, or may be on a different vector. When inserted on separate vectors, when they are integrated into different chromosomes, the plant individual into which the gene of interest has been introduced by self-pollination or backcrossing to progeny, or the genome-edited plant individual It has the advantage that it can be separated into individual plants having a drug resistance gene.
- high osmotic pressure is applied to the shoot apex of a ripe seed embryo, the shoot apex of a young shoot, the shoot apex of an apical bud or a lateral shoot, or the shoot apex of an immature embryo from the viewpoint of increasing the modification efficiency.
- the step of contacting with the solution can be further included.
- a solution having a high osmotic pressure as compared with the plant cytoplasm can be used, and examples thereof include a solution containing sugar or a sugar alcohol.
- the sugar or sugar alcohol include sorbitol, mannitol, and sucrose, but sorbitol, mannitol, and sucrose are preferably contained from the viewpoint of efficiently performing high osmotic treatment.
- the osmotic potential of the high osmotic solution include those equal to the osmotic potential of at least an 11% sucrose aqueous solution.
- Examples of the contact method include a method of immersing the shoot apex of an embryo of the ripe seed, the shoot apex of a young shoot, the shoot apex of an apical bud or a lateral shoot, or the shoot apex of an immature embryo in the hyperosmotic solution, and the ripe seed.
- the contact environment may be a bright place or a dark place, but from the viewpoint of germination efficiency, a dark place is preferable in the case of dark germinated seeds, and a bright place is used in the case of light germinated seeds. Is preferable.
- the contact time may be before or after the step of shooting the coated fine particles. From the viewpoint of increasing the modification efficiency, it is preferable to include a step of contacting the coated fine particles before and after the step of shooting them.
- the contact time before the step of shooting the coated fine particles is preferably 1 hour or more and 20 hours or less, more preferably 2 hours or more and 10 hours or less, and further preferably 3 hours or more and 5 hours or less.
- the contact time after the step of shooting the coated fine particles is preferably 1 hour or more and 60 hours or less, preferably 10 hours or more and 30 hours or less, and further preferably 15 hours or more and 25 hours or less.
- a plant into which a target gene has been introduced or a modified plant whose genome has been edited can be created, and the plant can be further grown.
- the plant thus produced stably expresses the trait of the target gene or the genome-edited gene, or the expression of the target gene is suppressed, and the plant is normally inherited (transmitted) to progeny. ).
- the gene transfer efficiency, genome editing efficiency, and target gene expression efficiency (transformation efficiency) into plants can be examined as follows.
- the efficiency of gene transfer is determined by whether or not the target gene or target gene has been genome-edited by PCR method and / or electrophoresis or Southern blot method by extracting DNA from a growing individual after modification treatment or gene transfer treatment.
- the gene transfer efficiency is calculated from the number of explants used for transfer and the number of growing individuals having foreign genes.
- RNA expressed from the target gene is examined in the growing individual in which the gene transfer has been confirmed.
- the presence or absence of RNA can be confirmed by, for example, the RT-PCR method. It may be detected by Northern blotting.
- a protein expressed from a target gene or a genome-edited target gene It is also possible to examine the presence or absence of a protein expressed from a target gene or a genome-edited target gene.
- the presence or absence of protein can be confirmed, for example, by staining plant pieces, electrophoresis, ELISA, RIA, dot immunobiding assay, and / or Western blotting.
- the number of explants used for introduction and the number of growing individuals in which the presence of the protein expressed from the target gene was confirmed, or the number of explants used for introduction and the presence of a protein whose genome was edited for the target gene (or non-existence) From the number of growing individuals whose existence) has been confirmed, the target gene expression efficiency (transformation efficiency) or the genome editing efficiency of the target gene is calculated.
- Example 1 To find the study appropriate gold particle size of optimal gold particle size gene transfer, the presence of the transgene in the presence or absence or a contemporary GFP fluorescence in shoot apex after gene introduction treatment (T 0 generation) investigated.
- a plasmid DNA solution (1 ⁇ g / ⁇ L) purified by Qiagen Plasmamid Midi Kit (Qiagen) was placed so as to be 5 ⁇ g per 750 ⁇ g of gold particles.
- the sterilized gold particle-containing solution was carefully suspended using an ultrasonic generator (Ultrasonic Cleaner UW-25 manufactured by Taga Electric Co., Ltd.), placed in an appropriate amount in the above tube, and stirred by pipetting.
- the vortex was vigorously suspended for 5 minutes. After allowing to stand at room temperature for 10 minutes, it was centrifuged at 9,100 ⁇ g for 2 seconds. The supernatant was removed and washed with 70% ethanol and 99.5% ethanol. Finally, the supernatant was removed and 24 ⁇ L of 99.5% ethanol was added and suspended well. In a clean bench, 6 ⁇ L was poured into the center of the macro carrier and air-dried.
- the particle gun uses Biolithic (registered trademark) PDS-1000 / He Party Delivery System (BIO-RAD), and the pressure at the time of shooting is about 94.9 kgf / cm 2 (1,350 psi), and the target is The distance to the tissue was 5 cm (particle diameter of 0.6 ⁇ m or more) or 3.5 cm (particle diameter of less than 0.6 ⁇ m). I shot four times per petri dish. After shooting, it was allowed to stand overnight at 22 ° C. in a dark place.
- the gene transfer efficiency at the shoot apex was higher when the gold particle diameters were 0.8 ⁇ m, 0.6 ⁇ m, and 0.3 ⁇ m as compared with the gold particle diameters of 1.6 ⁇ m and 1.0 ⁇ m (Table 1).
- the gene transfer efficiency at the shoot apex was the highest at 73.3% as compared with other gold particle sizes (Table 1).
- the gene transfer efficiency was also higher in the gold particles of 0.6 ⁇ m than in the gold particles of 1.0 ⁇ m.
- PCR is performed using TakaRa PCR Thermal Cycler Dice (registered trademark) at 95 ° C. for 3 minutes, followed by a reaction at 95 ° C. for 30 seconds, 60 ° C. for 30 seconds, and 72 ° C. for 1 minute as one cycle. I went through a cycle. After the PCR reaction, 1.0% agarose gel electrophoresis was performed, and the PCR product was detected by staining with ethidium bromide. Those in which the expected 601 bp GFP gene fragment was detected were judged to be transgenic individuals.
- the gene transfer efficiency per number of mature embryos subjected to gene transfer treatment from the obtained number of gene transfer individuals (number of gene transfer individuals / treatment). The number of ripe embryos ⁇ 100) was calculated.
- Example 2 to find the study appropriate gas pressure optimum gas pressure in gene transfer was investigated whether the transgene and the presence or absence of GFP fluorescence in the shoot apex after gene introduction treatment in the contemporary (T 0 generation) .
- 1. Investigation using the transient expression system of GFP fluorescent protein as an index (1) Preparation of ripe wheat seeds The method described in Example 1-1- (1) was followed. (2) Exposure of shoot apex in ripe seed embryo The method described in Example 1-1- (2) was followed. (3) Gene transfer The gold particle size was 0.6 ⁇ m, the gas pressure was 1,100 psi or 1,350 psi, and the method described in Example 1-1- (3) was followed.
- Example 1-1- (4) Investigation of transient expression efficiency of GFP protein The method described in Example 1-1- (4) was followed. As a result, the gene transfer efficiency described in Example 1-1- (4) was 74.2% at a gas pressure of 1,350 psi, which was higher than 13.3% at a gas pressure of 1,100 psi. (Table 3). Further, as described later, for the T 0 generation gene transfer efficiency was higher gas pressure 1,350psi than the gas pressure 1,100Psi.
- Example 1-2- (5) was followed.
- the gene transfer efficiency described in Example 1-2- (5) was 4.2% at a gas pressure of 1,350 psi, which was higher than 0.8% at a gas pressure of 1,100 psi. (Table 3). From this, the gene transfer efficiency was improved by using the gas pressure of 1,350 psi, which had a higher transient expression efficiency of the GFP protein in Example 2-1 (4) than the gas pressure of 1,100 psi. I found out that I would do it.
- Example 3 Examination of optimum water absorption time for gene transfer In order to find an appropriate water absorption time, the presence or absence of GFP fluorescence on the shoot apex after gene transfer treatment and its current generation (T 0 generation) and next generation (T 1 generation) The presence or absence of the transgene was investigated.
- T 0 generation and T 1 survey a genomic PCR of indicators in generation plants (1) an individual transient GFP fluorescence in the shoot apex in growth examples of transformation processing individual 3-1- (3) it was observed , Grow by the method described in Example 1-2- (4).
- T 1 plant leaves Presence or absence of transgene in T 1 plant leaves
- the individuals in which the transgene was confirmed in (2) above were bred to obtain T 1 seeds.
- Each T 1 seeds were sown and develop about 10 to 20 grains were first leaf a (50 mg) was sampled.
- Genomic PCR and electrophoresis were based on Example 1-2- (5).
- each resulting transgene confirmed individuals (T 1 generation) from per number mature embryos were transgenic process gene transfer efficiency (the transgenic population / processing The number of ripe embryos ⁇ 100) was calculated.
- the gene transfer efficiencies when ripe seeds absorbed in water for 6 to 12 hours and 12 to 18 hours were used in the experiment were 3.1% (Fig.
- transgene confirmation individuals T 1 generation 5 lines shown in Table 4, by Southern blot analysis in which the GFP gene region as a probe, it was confirmed that the GFP gene is inserted into the genomic DNA (Fig. 3).
- T 1 introduced in transgenic wheat made by confirming the gene introduction method of gene expression in generation gene (GFP gene) expression.
- T 1 and T 1 seeds obtained in GFP fluorescence observation Example 3-2- (3) in seeds placed in a sterile Petri dish, LAS 3000 (FujiFilm) below, result of observation of GFP fluorescence of T 1 seeds (filter 510DF10), GFP fluorescence was observed in the seeds (Fig. 4A). Next, the seeds in which GFP fluorescence was observed were cut in half, and GFP fluorescence observation of endosperm (excitation: 470/40, absorption: 525/50) was performed under a stereoscopic fluorescence microscope (MZFLIII manufactured by Leica). As a result, GFP fluorescence was observed in the endosperm (E), and GFP fluorescence was strongly observed in the aleurone layer (Al) (FIG. 4B).
- MZFLIII stereoscopic fluorescence microscope
- total protein was extracted from each adult leaf of the wild-type strain and the transformant (gene transfer individual), and an attempt was made to detect GFP fluorescent protein by Western blotting.
- a protein extraction buffer (0.25 M sorbitol, 50 mM Tris / acetate, pH 7.5, 1 mM EDTA, 2 mM DTT, 1% PVP, 10 ⁇ M PMSF).
- the extract was centrifuged (1,100 xg, 15 minutes, 4 ° C.) and the supernatant was further centrifuged (12,800 xg, 15 minutes, 4 ° C.).
- the supernatant obtained after this centrifugation was used as the total protein fraction.
- 20 ⁇ g of the total protein fraction was electrophoresed on a 12.5% SDS polyacrylamide gel and transferred to a nitrocellulose membrane by a semi-dry method.
- the membrane was shaken with blocking buffer (5% [w / v] Skim Milk Powerer in TTBS (10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20, pH 7.5)) for 1 hour. After washing the membrane with TTBS for 5 minutes ⁇ 3 times, the primary antibody was reacted for 1 hour. After washing the membrane with TTBS for 5 minutes ⁇ 3 times, the secondary antibody was reacted for 1 hour.
- blocking buffer 5% [w / v] Skim Milk Powerer in TTBS (10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20, pH 7.5)
- transformants can be obtained by essentially the same method as wheat.
- T 0 generation 1 fabricated transgenic individuals in genotype confirmed the method in the T 1 generation each individual acquired from the system (T 0 generation), cells with different genetic information in the same individual becomes a mixed chimera. In order to investigate whether it has a different genetic information more T 1 seeds obtained from transgenic individuals of a certain system (T 0 generation), Southern blot analysis was performed.
- T 0 generation FG1 system of individuals obtained in Example 3-2- (3) From T 0 generation FG1 system of individuals obtained in Example 3-2- (3), and harvested T 1 seed per panicle from the main stem and five tillers were seeded multiple individuals from there ..
- the seeded T 1 generation individuals were grown, and the presence or absence of a transgene in each T 1 generation individual was investigated according to the method described in Example 3-2- (3).
- the transgene Table 5
- T 1 seeds obtained from transgenic individuals of a certain system were found to have the same genetic information. This suggests that the number of shoot apical stem cells that migrate to the germline is very small, and it was found that this method enables gene transfer into this very small number of shoot apical stem cells.
- the gene transfer efficiency at the shoot apex was higher when the gold particle diameters were 0.8 ⁇ m, 0.6 ⁇ m, and 0.3 ⁇ m as compared with the gold particle diameters of 1.6 ⁇ m and 1.0 ⁇ m (Table 6).
- the gene transfer efficiency at the shoot apex was the highest at 85% as compared with other gold particle sizes (Table 6).
- Example 7 Acquisition of transformant in soybean In order to confirm whether a transformant of soybean can be obtained by the same method as wheat, the presence or absence of GFP fluorescence on the shoot apex after gene transfer treatment and the subsequent acquisition of the transformant The efficiency was investigated.
- BM medium (4.3 g / L MS salt, MS vitamin, 3% sucrose, 0.50 g / L MES, 3% PPM (plant preservative mixture, Nacalai Tesque), 6. The medium was placed at 0 g / L phytagel, pH 5.7).
- Example 1-1- (3) A 35S promoter of cauliflower mosaic virus (CaMV) was used as a gene transfer promoter, and the method described in Example 1-1- (3) was followed.
- CaMV cauliflower mosaic virus
- T 0 survey and genomic PCR indicators in generation plants (1) stand overnight growth transformants treated individuals transformants treated individuals RM medium (4.3g / L MS salt, MS vitamin, 3% sucrose, 0 Transplanted into a disposable plant cell culture vessel (Sigma) containing .50 g / L MES, 3% PPM (transformant transformative mixture, Nacalai Tesque), 0.3% gellite, pH 5.7) and incubator (23 ° C., 16 hours). Raised in (Hicho). When root growth and leaf differentiation from the shoot apex were observed, the plants were transplanted to a cell tray containing horticultural seedling cultivation soil. After that, it was grown up to the second leaf in the same incubator.
- PCR was performed using TakaRa PCR Thermal Cycler Dice at 95 ° C. for 3 minutes, followed by 32 cycles of 95 ° C. for 30 seconds, 60 ° C. for 30 seconds, and 72 ° C. for 1 minute as one cycle. After the PCR reaction, 1.0% agarose gel electrophoresis was performed, stained with ethidium bromide, and the PCR product was detected by ultraviolet irradiation. The predicted 601 bp GFP gene fragment was detected as a gene-introduced individual. It was judged.
- the gene transfer efficiency per number of mature embryos subjected to gene transfer treatment from the obtained number of gene transfer individuals (number of gene transfer individuals / treatment). The number of ripe embryos ⁇ 100) was calculated.
- transgene confirmation individuals (T 0 generation) of the processing mature embryos 470 individuals was 63 individuals (gene transfer efficiency 13.4%). From this, it is possible to obtain a soybean gene-introduced individual with high efficiency by using a gold particle size of 0.6 ⁇ m, which had a high transient expression efficiency in Example 7-1- (4). I understood it.
- Example 8 Acquisition of transformants in barley In order to confirm whether a transformant of barley can be obtained by the same method as that of wheat, the presence or absence of GFP fluorescence in the shoot apex after gene transfer treatment was investigated.
- Example 1-1- (4) Investigation of transient expression efficiency of GFP protein The method described in Example 1-1- (4) was followed. As a result, the gene transfer efficiency at the shoot apex was higher when the gold particle diameters were 0.8 ⁇ m, 0.6 ⁇ m, and 0.3 ⁇ m as compared with the gold particle diameters of 1.6 ⁇ m and 1.0 ⁇ m (Table 8). In particular, when 0.3 ⁇ m was used, the gene transfer efficiency at the shoot apex was the highest at 80.0% as compared with other gold particle diameters (FIGS. 8 and 8).
- Example 9 Acquisition of transformant in potato In order to confirm whether a transformant of potato can be obtained by the same method as wheat, the presence or absence of GFP fluorescence in the shoot apex after gene transfer treatment was investigated.
- Primer sequence CGACGGCCAGTGCCAAAGCTT (SEQ ID NO: 4) Primer sequence: ATGACCATGATTACGAATTC (SEQ ID NO: 5) Primer sequence: AAGCTAGAGTAAGTAGTAGTCGCCA (SEQ ID NO: 6) Primer sequence: ATACTGTCCTTCTAGTGTAGCCG (SEQ ID NO: 7)
- PCR was carried out using TakaRa PCR Thermal Cycler Dice for 40 cycles, with 98 ° C. for 10 seconds, 60 ° C. for 15 seconds, and 68 ° C. for 2 minutes and 30 seconds as one cycle.
- the PCR product was purified by ethanol precipitation and dissolved in sterile water to a concentration of 1 ⁇ g / ⁇ L, which was used in the following experiments.
- a linear vector (nucleic acid cassette to be introduced) is preferable for gene transfer, and more preferably, a linear vector in which 0.8 kb or more of nucleic acid is added to both ends of the nucleic acid cassette to be introduced. I found that it was to use.
- a transformant can be obtained by a method essentially the same as that for wheat. 1.
- Ripe seeds of threshed rice (variety: Nihonbare) are sterilized and washed by the method described in Example 1-1- (1), and then placed on a Kim towel or filter paper moistened with sterilized water. Incubated at ° C for 24 to 48 hours is used in the following experiments.
- Example 12 Acquisition of transformant in apple A transformant can be obtained for apple by a method essentially the same as that for wheat. 1. 1. Preparation of apple shoot apex The shoot apex of an apple (variety: Jonagold) is prepared according to the induction conditions for shoot apex culture described in Plant Tissue Culture, 9 (2), 69-73 (1992).
- Example 1-1- (3) As the gene transfer promoter, the 35S promoter of cauliflower mosaic virus (CaMV) is used, and the method described in Example 1-1- (3) is followed. 4. Selection using the transient expression of GFP protein as an index Five spots in shoot apical tissue in an individual transformed with a gold particle size of 0.6 ⁇ m according to the method described in Example 1-1- (4). Those in which the above GFP fluorescence is observed are selected. 5. Growth of transformed individual The transformed individual is grown according to the method described in Example 1-2- (4). 6. Confirmation of transgene in T 0 plant leaves Acquire a gene transfer-confirmed individual according to the method described in Example 1-2- (5). A stable transformant can be obtained by raising the obtained individual and obtaining next-generation seeds.
- CaMV cauliflower mosaic virus
- Example 13 Target mutation introduction using the gene transfer method
- mutation introduction of a target sequence by RNA-induced CRISPR / Cas9 is performed. I tried. From Example 1-1- (4), in order to prepare a stable transformant using the gene transfer method, a gold particle size of 0.3 ⁇ m or more and 0.9 ⁇ m or less is preferable, but a genome-edited individual is prepared. In order to do so, the nuclease and the like may be transiently expressed in the nucleus of the plant cell or in the organelle, and thus the particle size is not limited to the above. Therefore, the range of gold particle size in which the genome of wheat can be edited using the gene transfer method was examined.
- a target plasmid DNA in which a 20 bp partial sequence (CCGTCACGCAGGACCAATCTCC: SEQ ID NO: 8) inside the TaMLO gene was inserted as a target sequence immediately after the start codon of the fluorescent reporter gene (GFP) was used.
- the gene is designed to be expressed under the control of the maize ubiquitin promoter and first intron.
- a terminator of the nopaline synthase (NOS) gene is added.
- Cas9 cleaves the TaMLO gene target sequence, and two bases are deleted or one base is inserted in the repair process.
- a translation frameshift occurs, the codon reading frame of the GFP gene is restored, and the active GFP protein is expressed.
- the gRNA in the integrated plasmid was designed to be expressed under the control of the wheat-derived U6 promoter.
- the Cas9 gene is designed to be expressed under the control of the maize ubiquitin promoter and the first intron.
- a terminator of the nopaline synthase (NOS) gene is added as the terminator.
- the gold particles were prepared and introduced according to the method of Example 2-1 (3). However, the target plasmid DNA solution and the gRNA / Cas9 integrated plasmid were added so as to be 2.5 ⁇ g and 7.5 ⁇ g per 750 ⁇ g of gold particles, respectively. The amount of gold particles was 375 ⁇ g for a gold particle diameter of 0.3 ⁇ m and 187.5 ⁇ g for other gold particle diameters per shot.
- the experiment was performed twice, and the average value of the target sequence cleavage introduction efficiency is shown in Table 10.
- Table 10 As a result, when the target plasmid DNA and the gRNA / Cas9 integrated plasmid were co-introduced, GFP fluorescence indicating target sequence cleavage was observed at gold particle diameters of 0.3 ⁇ m to 1.0 ⁇ m, but at gold particle diameter of 1.6 ⁇ m. Not observed (Table 10).
- the efficiency was the highest at 23.5% when a gold particle diameter of 0.6 ⁇ m was used (Tables 10 and 10). From these results, it was found that the gold particle size is preferably less than 1.6 ⁇ m and preferably the gold particle size of 0.6 ⁇ m in order to prepare a genome-edited individual using the gene transfer method.
- Example 14 targeted mutation introduced by using the gene transfer method (T 0 generation, T 1 generation)
- TaQsd1 gene alanne aminotransphase gene
- TaLOX2 gene lipoxygenase gene
- TaLOX2 gene lipoxygenase gene
- TaQsd1 target sequence ACGGATCCACCTCCCCTGCAGCGG (SEQ ID NO: 9)
- TaLOX2 target sequence GTGCCGCGCGCGACGAGCTCTTCGG (SEQ ID NO: 10)
- TaGASR7 target sequence CCGCCGGGGCACCTACCGCAAC (SEQ ID NO: 11)
- the above-mentioned gRNA expression plasmid, Cas9 gene expression plasmid, and GFP expression plasmid were added in an amount of 5 ⁇ g, 2.5 ⁇ g, and 2.5 ⁇ g, respectively, per 750 ⁇ g of gold particles.
- the diameter of the gold particles was 0.6 ⁇ m, and the amount of gold particles was 187.5 ⁇ g per shot.
- MS-maltose medium was used for individuals in which GFP fluorescence (excitation: 470/40 absorption: 525/50) at the shoot apex was observed under a solid fluorescence microscope (Leica, MZFLIII). It was transplanted to a disposable plant cell culture vessel (Sigma) containing the above, and grown for a long day (22 ° C., 16 hours day length). After about 1 week, the shoot apex in each individual was cut out and placed in an 8-unit PCR tube supplemented with 2 ⁇ L of DNAzol Direct (registered trademark, Cosmo Bio) to extract genomic DNA.
- DNAzol Direct registered trademark, Cosmo Bio
- TaQsd1-F CAGCCTGGGAGGGAATGACC (SEQ ID NO: 12)
- TaQsd1-R ACCTGGTGGAATCCAGAGC (SEQ ID NO: 13)
- TaLOX2-F CGTCTACCGCTACCGACGTTACAACG (SEQ ID NO: 14)
- TaLOX2-R GGTCGCCGTACTTGCTCGATCAAGT (SEQ ID NO: 15)
- TaGASR7-F CCTTCATCCTTCAGCCATGCAT (SEQ ID NO: 16)
- TaGASR7-R CCACTAAATGCCTATCACATACG (SEQ ID NO: 17)
- PCR reaction solution 0.5 ⁇ L of genomic DNA, 0.4 U of KODFX Neo (TOYOBO), 2 ⁇ 10 ⁇ L of attached buffer, 0.4 mM dNTPs, and 0.2 ⁇ M of each primer were added, and the mixture was filled up to a total of 20 ⁇ L with sterile distilled water.
- PCR was carried out using TakaRa PCR Thermal Cycler Dice for 35 cycles with 98 ° C. for 10 seconds and 68 ° C. for 1 minute as one cycle. After the PCR reaction, 1 ⁇ L of each PCR product, 10 ⁇ 1 ⁇ L of attached buffer, and 5 U of an appropriate restriction enzyme were added, and the mixture was filled up with sterile distilled water to a total of 10 ⁇ L. After reacting overnight at 37 ° C., 2.0% agarose gel electrophoresis was performed and stained with ethidium bromide.
- T according to the method described in 0 generation mature leaves and T 1 generation target gene mutation confirmed in young leaves
- Preparation Example of wheat mature seed 13-1- (1).
- Exposure of shoot apex in ripe seed embryo The method described in Example 13-1- (2) was followed.
- Gene transfer The method described in Example 13-1- (3) was followed.
- transgenic individuals that had been allowed to stand overnight were transplanted into a disposable plant cell culture vessel (Sigma) containing MS-maltose medium and kept for a long day (22 ° C., 16 hours). Raised. After growing for 2 weeks, when the 2-3 leaves were observed, they were transplanted to a pot containing horticultural seedling cultivation soil. After that, it was cultivated in a long-day artificial weather room (24 ° C., 16-hour day length, humidity 50-70%).
- Genomic DNA was extracted from the 5th leaf or flag leaf (50 mg) using the benzyl chloride method, and a PCR reaction was carried out using this as a template and primers prepared from sequences specific to each gene.
- PCR reaction solution 0.7 ⁇ L of genomic DNA, 0.4 U of KODFX Neo (TOYOBO), 2 ⁇ 5 ⁇ L of attached buffer, 0.4 mM dNTPs, and 0.2 ⁇ M of each primer were added, and the mixture was filled up to a total of 10 ⁇ L with sterile distilled water.
- PCR was carried out using TakaRa PCR Thermal Cycler Dice for 32 cycles with 98 ° C. for 10 seconds and 68 ° C. for 1 minute as one cycle. PCR / restriction enzyme analysis was performed according to the method described in Example 13-1- (4).
- the PCR product was completely cleaved by the restriction enzyme, whereas in the mutant-introduced individual, the PCR product was left uncut. DNA was extracted and purified from the uncut band and subjected to sequence analysis, and those having a mutation introduced into the target gene sequence were judged to be individuals with the target gene mutation introduced. As a result, for all the target genes was confirmed target gene mutation in T 0 generation adult leaves (12, Table 11).
- T 1 seeds were collected from TaQsd1 mutation-introduced individuals confirmed in (5) above, and benzyl chloride method was used from young leaves (50 mg) of T 1 generation plants. The genomic DNA was extracted using. Subsequent confirmation of the introduction of the target gene mutation was carried out according to the method described in (5) above. As a result, the T 1 generation 2 lines were confirmed target gene mutation in T 1 generation young leaves. 13, showing an analysis result in the T 1 generation 1 system. It was confirmed that mutations were introduced into the target gene in 9 out of 16 individuals. By using this gene transfer method, it was found that it is possible to edit the genome of shoot apical stem cells that migrate to the germline in the shoot apex.
- Example 15 Introducing a target mutation by introducing a Cas9 protein using the gene transfer method It was verified whether or not genome editing of a target gene inherent in wheat is possible by introducing a gRNA / Cas9 protein using the gene transfer method. .. For verification, a complex of gRNA and Cas9 protein targeting the TaLOX2 gene was introduced into the wheat shoot apex.
- gRNA / Cas9 protein The gene transfer of ripe wheat embryos into the shoot apex was carried out as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) was used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes was used.
- NEB EnGen Cas9 NLS
- crRNA GUGCCGGCGCGACGAGGCUCUUguuuugagcuauugcuguuguug (SEQ ID NO: 18)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19)
- the gold particle diameter was 0.6 ⁇ m, the amount of gold particles was set to 187.5 or 375 ⁇ g per shot, and four shots were performed per plate.
- a hydrophilic film (3M, SH2CLHF) cut into 1.0 to 1.5 cm squares was attached to the center of the macro carrier in a clean bench. 8 ⁇ L of the above gold particle / Cas9 complex was poured into this and air-dried. The shooting conditions were in accordance with Example 1-1- (3).
- Target mutation-introduced soybeans using the gene transfer method As for soybeans, target gene mutation-introduced individuals can be obtained by essentially the same method as wheat.
- each of the above gRNA expression plasmids, Cas9 gene expression plasmids, and GFP expression plasmids per 750 ⁇ g of gold particles, respectively.
- the gold particle diameter is 0.6 ⁇ m, and the amount of gold particles is 187.5 ⁇ g per shot.
- a target gene mutation-introduced individual can be obtained by essentially the same method as wheat.
- Glyma A complex of gRNA and Cas9 protein targeting the 10G2444.01 gene is introduced into the shoot apex. 1. 1. Preparation of ripe soybean seeds is carried out according to the method described in Example 15-1. 2. Exposure of shoot apex in ripe seed embryos follow the method described in Example 15-2.
- gRNA / Cas9 protein transfer Gene transfer into the shoot apex of ripe soybean embryos is performed as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) is used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes is used.
- NEB EnGen Cas9 NLS
- crRNA GGUGGGCGGAGCCUUGGGGCGGguuuuuuagagcuauugcuguuuuuuug (SEQ ID NO: 21)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19) The shooting conditions follow the method described in Example 14-1- (3).
- Example 15-4 Selection using the transient expression of GFP protein as an index follow the method described in Example 15-4. 5. Growth of transgenic individuals follow the method described in Example 15-5. 6. T 0 according to the method described in confirmation Example 15-6 of the target gene mutation in plant leaves, can be obtained stably mutation in the target gene has been introduced individual.
- a target gene mutation-introduced individual can be obtained by essentially the same method as wheat.
- a complex of gRNA and Cas9 protein targeting the OsPDS (rice phytoene desaturase) gene is introduced into the rice shoot apex.
- gRNA / Cas9 protein transfer Gene transfer into the shoot apex of ripe rice embryos is performed as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) is used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes is used.
- NEB EnGen Cas9 NLS
- crRNA GUUGGUCUUGCUCCUGCAGguuuuuaggcuauugcuguuuuug (SEQ ID NO: 22)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19) The shooting conditions follow the method described in Example 14-1- (3).
- Example 10-4 Selection using the transient expression of GFP protein as an index follow the method described in Example 10-4. 5. Growth of transgenic individuals follow the method described in Example 10-5. 6. T 0 according to the method described in confirmation Example 15-6 of the target gene mutation in plant leaves, can be obtained stably mutation in the target gene has been introduced individual.
- Example 19 Introducing a target mutation into an apple by introducing a Cas9 protein using the gene transfer method
- an individual having a target gene mutation introduced can be obtained by a method essentially similar to that of wheat.
- a complex of gRNA and Cas9 protein targeting the apple PDS (phytoene desaturase) gene is introduced into the apple shoot apex.
- crRNA ACCUGAUCGAGUAACUACAGguuuuuaggcuauugcuguuguug (SEQ ID NO: 23)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19)
- the shooting conditions follow the method described in Example 14-1- (3). However, the GFP vector described in Example 7-1- (3) is used.
- Example 11-4 Selection using the transient expression of GFP protein as an index The method described in Example 11-4 is followed. 5. Growth of transgenic individuals follow the method described in Example 11-5. 6. T 0 according to the method described in confirmation Example 15-6 of the target gene mutation in plant leaves, can be obtained stably mutation in the target gene has been introduced individual.
- Example 20 Introduction of target mutation into potato by introduction of Cas9 protein using the gene transfer method
- an individual having a target gene mutation introduced can be obtained by essentially the same method as wheat.
- a complex of gRNA and Cas9 protein targeting the StIAA2 (an Auxin / Indole-3-Acetic Family member) gene is introduced into the potato shoot apex.
- gRNA / Cas9 protein transfer The gene transfer into the shoot apex of potato is performed as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) is used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes is used.
- NEB EnGen Cas9 NLS
- crRNA GAUGUUUAGCUCCUUUACUAguuuuuuagagcuauugcuguuguug (SEQ ID NO: 24)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19)
- the shooting conditions follow the method described in Example 14-1- (3). However, the GFP vector described in Example 7-1- (3) is used.
- Example 21 Introduction of target mutation into corn by introducing Cas9 protein using the gene transfer method
- an individual having a target gene mutation introduced can be obtained by a method essentially similar to that of wheat.
- a complex of gRNA and Cas9 protein targeting the ZmALS2 (maize acetolactate synthase) gene is introduced into the maize shoot apex.
- gRNA / Cas9 protein transfer The gene transfer into the shoot apex of maize is carried out as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) is used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes is used.
- NEB EnGen Cas9 NLS
- crRNA GCUGCUCGAUCCGUCCCCAguuuugagcuauugcuguuguug (SEQ ID NO: 25)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19) The shooting conditions follow the method described in Example 14-1- (3).
- Example 22 Introducing a target mutation into barley by introducing Cas9 protein using the gene transfer method
- an individual having a target gene mutation introduced can be obtained by essentially the same method as wheat.
- a complex of gRNA and Cas9 protein targeting the HvPM19 (barley ABA-inducible plasma membrane protein) gene is introduced into the barley shoot apex.
- gRNA / Cas9 protein transfer The gene transfer into the barley shoot apex is performed as follows using the particle gun method. In the examples, a mixture (0.5 ⁇ g / ⁇ L) of crRNA and tracrRNA (Fasmac) is used as the gRNA. As the Cas9 protein, EnGen Cas9 NLS (NEB) derived from Streptococcus pyogenes is used.
- NEB EnGen Cas9 NLS
- crRNA GCUCUCCACUCUGGGCUCUUguuuuuagagcuaugcuguuuuuug (SEQ ID NO: 26)
- tracrRNA AAACAGCAUAAGCAAGUUAAAAAUAAGGCUAGUCCUCUUAUCACUUGAAAAAGUGGCACCGAGUCGGUGCU (SEQ ID NO: 19) The shooting conditions follow the method described in Example 14-1- (3).
- MSO medium MS salt, MS vitamin, 2 g / L myo-inositol, 20 g / L sucrose, 8 g / L Bacto-agar, pH
- the medium was placed at 5.8 (Fig. 15A). It was cultured at 25 ° C. for 2 days in a bright place (about 50 ⁇ molm -2 s -1 ), and the one in which the sucrose leaves began to appear was used in the following experiment (Fig.). 15B and C).
- HMG13 target sequence CTCGTCACGATTCCCCCTTCCC (SEQ ID NO: 27) 10 mg of 0.4 ⁇ m or 0.6 ⁇ m gold particles were weighed, 1 mL of 70% ethanol was added, and the mixture was well suspended in a vortex. Then, the gold particles were precipitated by centrifugation to remove ethanol. Then, 1 mL of 50% glycerol was added to prepare a sterilized gold particle solution.
- the particle gun uses Biolithic (registered trademark) PDS-1000 / He Party Delivery System (BIO-RAD), and the pressure at the time of shooting is about 94.9 kgf / cm 2 (1,350 psi), and the target is The distance to the tissue was 5 cm (particle diameter of 0.6 ⁇ m or more) or 3.5 cm (particle diameter of less than 0.6 ⁇ m). I shot 4 times per petri dish (Figs. 16A and 16B).
- Example 24 Effect of osmotic treatment before firing on the acquisition of transformants in maize using immature maize embryos 1. Investigation using the transient expression system of fluorescent protein as an index (1) Preparation of immature maize embryos The method described in Example 23-1- (1) was followed.
- Example 23-1- (4) Investigation of transient expression efficiency of tdTomato protein The method described in Example 23-1- (4) was followed. Compared with the case where the osmotic pressure treatment was not performed (FIG. 16C), the fluorescence intensity derived from the tdTomato protein was improved when the osmotic pressure treatment was performed for 4 hours before the shooting (FIG. 17C). From this, it was found that the osmotic treatment improves the transient expression efficiency of the fluorescent reporter gene.
- Example 25 Effect of osmotic treatment before and after shooting on the acquisition of transformants in maize using immature maize embryos 1. Investigation using the transient expression system of fluorescent protein as an index (1) Preparation of immature maize embryos The method described in Example 23-1- (1) was followed.
- Example 23-1- (4) Investigation of transient expression efficiency of tdTomato protein The method described in Example 23-1- (4) was followed. Compared to the case where only the osmotic pressure treatment for 4 hours before the shot was performed (Fig. 17C), the case where the osmotic pressure treatment for 4 hours before the shot and the osmotic pressure treatment for 20 hours after the shot was performed were derived from the tdTomato protein. The fluorescence intensity was improved (Fig. 18D). From this, it was found that the transient expression efficiency of the fluorescent reporter gene is dramatically improved by the osmotic pressure treatment before and after the shot.
- Example 26 Editing the inplanta genome in immature maize shoot apical meristem (1) Preparation of immature maize embryos The method described in Example 23-1- (1) was followed.
- Example 25-1- (3) Gene transfer The method described in Example 25-1- (3) was followed. (4) Selection and culture based on transient expression of tdTomato protein TdTomato fluorescence at shoot apex (maximum excitation 554 nm and maximum emission) under a stereoscopic fluorescence microscope (Nikon SMZ 18 with a DS-Ri 2 camera) 20 hours after implantation. 581 nm) was observed (FIG. 19A) and embryos with a strong fluorescence signal in the shoot apical meristem were selected (FIG. 19B). The selected embryos were transferred onto MSO medium in a disposable plant cell culture vessel (Sigma) and cultured at 25 ° C.
- FIG. 19D A photograph 40 days after the shooting is shown in FIG. 19D.
- the library was prepared by two-step PCR in which the target region was amplified and a sequence adapter was added. Barcodes were designed using primers and added in the first PCR step to distinguish the samples. The adapter was added in the second PCR step for sequencing.
- NGS Next Generation Sequencing
- the NGS results show that transient expression of Cpf1 (pGEP359) and guide RNA (pGEP324) delivered by violistic bombardment can result in significant genome-editing events in shoot apical meristems of immature corn embryos.
- pGEP359 and guide RNA (pGEP324) delivered by violistic bombardment can result in significant genome-editing events in shoot apical meristems of immature corn embryos.
- Fig. 21 Compared to the non-impact control, the impacted shoot apex contained a 14.9% insertion deletion rate (Indel%) at the target site (FIG. 21A).
- NGS results also revealed the chimera of genome-editing modifications in shoot apical meristems, where multiple deletions of various sizes (eg, 5 bp, 6 bp, and 8 bp, etc.) were detected (FIG. 21B). ..
- Example 27 in planta genome editing gene transfer method of corn T 0 leaf in order to assess whether you can use the genome editing of target genes present in corn, tried to mutagenesis of hmg13 gene by MAD7. Maize genotype A188 was used for mutagenesis of the hmg13 gene. 1. 1. Confirmation of marker gene (mNeongreen) in shoot apical meristem after gene transfer (1) Preparation of immature maize embryo The method described in Example 23-1- (1) was followed.
- the introduced genes are plasmid DNA (pGEP837) containing MAD7 nuclease expressed using a promoter derived from the corn ubiquitin 1 gene (ZmUbi1) and the fluorescent reporter gene mNeongreen expressed using the d35S promoter, and corn HMG13, which targets ZmUbi1.
- a plasmid DNA (pGEP842) containing a guide RNA to be expressed using was used.
- the particle gun uses Biolithic (registered trademark) PDS-1000 / He Party Delivery System (BIO-RAD), and the pressure at the time of shooting is about 94.9 kgf / cm 2 (1,350 psi), and the target is The distance to the tissue was 5 cm. I shot four times per petri dish. After bombardment, the sample was left overnight at 25 ° C. in the dark.
- Genomic DNA was extracted from leaves of 3 or later leaves using the following method, and whether or not mutation was introduced into the target gene of the obtained plant. I examined.
- Digital droplet PCR was performed using genomic DNA as a template and primers prepared based on sequences specific to each gene.
- efficiency of targeted gene mutagenesis in T 0 plants obtained (19.6%, OS1-8) in the case of MS-maltose medium (2. It was significantly higher than 1%, NT1-9) (Table 13).
- osmotic treatment increased the proportion of gene-edited plants with high Indel% (70% ⁇ ) compared to untreated osmotic (Table 14).
- Example 28 Editing the in-plata genome of maize T 1 leaf A T 0 plant derived from OS1 to 8 obtained in Example 27-1- (6) was grown to grow a next-generation T 1 plant. It was confirmed whether the mutation of the hmg13 gene was inherited. T 1 seeds harvested from 8 lines of T 0 plants were sown and young leaves were sampled. DNA was extracted according to the method described in Example 27-1- (6), and site-specific sequence modification was evaluated using a next-generation sequencer (NGS) (Table 15). As a result, mutation of the target gene was confirmed in 6 of the 8 strains, and 1 strain was a homozygous mutant. From this, it was found that by applying osmotic pressure treatment to immature maize embryos, next-generation genome-edited individuals can be obtained with high efficiency even when the inplanta method is used.
- NGS next-generation sequencer
- Example 29 Effect of osmotic treatment on wheat in planet genome editing It was investigated whether the effect of osmotic treatment on wheat in planet genome editing confirmed in Example 27 was also observed on wheat. 1. 1. Confirmation of introduction of target gene mutation in the first week after gene transfer (1) Preparation of ripe wheat seeds Using ripe seeds of wheat (Triticum aestivum cv. Haruyokoi), the method of Example 1-1- (1) was followed.
- the PCR product In wild-type strains, the PCR product is completely cleaved by restriction enzymes, whereas in individuals into which the mutation has been introduced, the PCR product is left uncut. DNA was extracted and purified from the uncut band and subjected to sequence analysis, and those having a mutation introduced into the target gene sequence were judged to be individuals with the target gene mutation introduced. As a result, compared with the case of MSOSM medium (with osmotic pressure treatment before and after shooting) (16.3%) and the case of MS-maltose medium (without osmotic pressure treatment) (2.4%), T 0 leaves were used. It was found that the efficiency of target gene mutation introduction was improved (Table 16). Similar to the results in the maize of Example 27, it was confirmed that the genome editing efficiency was improved by the osmotic treatment in the wheat.
- the present invention can be used in agriculture, pharmaceutical industry, enzyme industry, etc.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Medicinal Chemistry (AREA)
- Cell Biology (AREA)
- Breeding Of Plants And Reproduction By Means Of Culturing (AREA)
Abstract
植物の改変方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、 該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
Description
本発明は、植物の改変方法に関する。
現在普及している植物の一般的な形質転換法は、in vitro培養系のプロトプラスト、カルス又は組織片などに外来遺伝子を、電気穿孔法、アグロバクテリウム・ツメファシエンス菌を介して又はパーティクルガン法などにより導入する方法である。しかしこれらの方法では、遺伝子が細胞または組織に導入されたとしても、植物品種によっては組織培養が困難で、植物体を再生して形質転換体を作製することが難しいという問題があった。また、形質転換効率が十分に高くなく、そのため選択マーカー遺伝子を導入してマーカー選抜を行わなければならないという問題もあった。さらに、長期の組織培養を必要とすることに伴い、体細胞変異(ソマクローナル変異)が頻繁に生じる点も問題であった。そのため、形質転換植物作製の労力軽減や形質転換植物の安全性の観点から、組織培養を伴わない植物の改変方法の開発が求められていた。
一方、コムギやイネなどにおいて、in vitro培養系のカルス又は組織片を用いない形質転換法(インプランタ形質転換法)も知られている。インプランタ形質転換方法としては、露出させた未熟胚又は完熟胚の茎頂に、パーティクルガン法を用いて直接遺伝子導入する方法が知られている(非特許文献1)。また、特許文献1及び非特許文献2には、発芽直後の完熟胚にアグロバクテリウムを感染させ、遺伝子導入する方法が記載されている。
しかし、これらの文献に記載の方法は、共通して、実験者の手技に大きく依存するため遺伝子導入効率が低調であり、再現性の点において改善の余地がある。また、非特許文献1は、導入遺伝子が次世代に伝達されることの実証に至っていない。このような要因から、上記の手法は、現在に至るまで汎用化されていない。
Bilang et al. (1993) Transient gene expression in vegetative shot apical meristems of wheat after ballistic microtargeting. Plant Journal (1993) 4, 735-744
Supartana et al. (2005) Development of simple and efficient in planta transformation for rice (Oryza sativa L.) using Agrobacterium tumefaciencs. Journal of Bioscience AND Bioengineering (2005) 4, 391-397
本発明は、in vitro培養系のカルス又は組織片を用いない植物の改変方法を提供する。本発明はまた、選択マーカー遺伝子を導入しない植物の改変方法を提供する。本発明はさらに、植物のゲノム編集方法、及び再現性に優れる植物の作製方法を提供する。
本発明者らは、前述の課題解決のために鋭意検討を行なった結果、本発明を完成するに至った。
すなわち、本発明は以下を包含する。
<1> 植物の改変方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
<2> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質を含まない培地で行われる、前記<1>に記載の方法。
<3> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、植物ホルモンを含まない培地で行われる、前記<1>に記載の方法。
<4> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質、及び植物ホルモンを含まない培地で行われる、前記<1>に記載の方法。
<5> 前記植物の茎頂への撃ち込みは、茎頂のL2層細胞に被覆した微粒子を撃ち込むことによって行われる、前記<1>に記載の方法。
<6> 前記植物の茎頂への撃ち込みは、茎頂の生殖細胞系列へ移行する茎頂幹細胞に被覆した微粒子を撃ち込むことによって行われる、前記<1>に記載の方法。
<7> 前記微粒子が、直径0.3μm以上1.5μm以下である、前記<1>に記載の方法。
<8> 前記植物の茎頂は、前記(1)完熟種子の胚の茎頂であり、前記(1)完熟種子の胚の茎頂が、該完熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、前記<1>に記載の方法。
<9> 前記植物の茎頂は、前記(2)幼芽の茎頂であり、前記(2)幼芽の茎頂が、該幼芽から塊茎、葉原基を除去し、露出させた茎頂である、前記<1>に記載の方法。
<10> 前記植物の茎頂は、前記(3)頂芽又は側芽の茎頂であり、前記(3)頂芽又は側芽の茎頂が、頂芽又は側芽から葉原基を除去し、露出させた茎頂である、前記<1>に記載の方法。
<11> 前記植物の茎頂は、(4)未熟胚の茎頂であり、前記(4)未熟胚の茎頂が、受粉後8日から35日の未熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、前記<1>に記載の方法。
<12> 前記微粒子の被覆は、微粒子を導入目的とする核酸カセットを含む線状DNAで被覆することにより行われる、前記<1>に記載の方法。
<13> 前記線状DNAは線状プラスミドであり、それぞれ核酸カセットの各末端に位置する0.8kb以上1.2kb以下の核酸をさらに含む、前記<12>に記載の方法。
<14> 前記微粒子の被覆は、微粒子を修飾酵素をコードする核酸で被覆することにより行われる、前記<1>に記載の方法。
<15> 前記修飾酵素が、ヌクレアーゼまたはデアミナーゼである、前記<1>に記載の方法。
<16> 前記微粒子被覆は、微粒子をプロモーターおよびターミネーターにそれぞれ連結された核酸で被覆することにより行われ、前記核酸は、少なくとも1つのガイドRNAを発現することができる核酸およびCasヌクレアーゼタンパク質をコードする核酸を含む、前記<1>に記載の方法。
<17> 前記微粒子の被覆は、微粒子をヌクレアーゼで被覆することにより行われる、前記<1>に記載の方法。
<18> 前記ヌクレアーゼがCasヌクレアーゼである、前記<17>に記載の方法。
<19> 前記微粒子の被覆は、微粒子をタンパク質で被覆することにより行われ、前記植物の茎頂への撃ち込みは、タンパク質で被覆された微粒子を有する親水性マクロキャリアフィルムを用いて行われる、前記<1>に記載の方法。
<20> 前記完熟種子は、長さが1mm以下の根を含む完熟種子である、前記<1>に記載の方法。
<21> 前記植物が、コムギ、オオムギ、イネ、トウモロコシ、ダイズ、ジャガイモおよびリンゴからなる群から選択される、前記<1>に記載の方法。
<22> 前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を高浸透圧溶液に接触させる工程をさらに含む、前記<1>に記載の方法。
<23> 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前または後である、前記<22>に記載の方法。
<24> 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前および後である、前記<22>に記載の方法。
<25> 改変された植物体を成長させる工程をさらに含む、前記<1>に記載の方法。
<26> 植物のゲノム編集方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であり、前記ゲノムは、茎頂分裂組織中の生殖細胞系列またはそれに移行し得る幹細胞内に存在し、前記方法はインプランタ法であることを特徴とする方法。
<27> 改変された植物体を成長させる工程をさらに含む、前記<26>に記載の方法。
<28> 植物の作製方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
<1> 植物の改変方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
<2> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質を含まない培地で行われる、前記<1>に記載の方法。
<3> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、植物ホルモンを含まない培地で行われる、前記<1>に記載の方法。
<4> 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質、及び植物ホルモンを含まない培地で行われる、前記<1>に記載の方法。
<5> 前記植物の茎頂への撃ち込みは、茎頂のL2層細胞に被覆した微粒子を撃ち込むことによって行われる、前記<1>に記載の方法。
<6> 前記植物の茎頂への撃ち込みは、茎頂の生殖細胞系列へ移行する茎頂幹細胞に被覆した微粒子を撃ち込むことによって行われる、前記<1>に記載の方法。
<7> 前記微粒子が、直径0.3μm以上1.5μm以下である、前記<1>に記載の方法。
<8> 前記植物の茎頂は、前記(1)完熟種子の胚の茎頂であり、前記(1)完熟種子の胚の茎頂が、該完熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、前記<1>に記載の方法。
<9> 前記植物の茎頂は、前記(2)幼芽の茎頂であり、前記(2)幼芽の茎頂が、該幼芽から塊茎、葉原基を除去し、露出させた茎頂である、前記<1>に記載の方法。
<10> 前記植物の茎頂は、前記(3)頂芽又は側芽の茎頂であり、前記(3)頂芽又は側芽の茎頂が、頂芽又は側芽から葉原基を除去し、露出させた茎頂である、前記<1>に記載の方法。
<11> 前記植物の茎頂は、(4)未熟胚の茎頂であり、前記(4)未熟胚の茎頂が、受粉後8日から35日の未熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、前記<1>に記載の方法。
<12> 前記微粒子の被覆は、微粒子を導入目的とする核酸カセットを含む線状DNAで被覆することにより行われる、前記<1>に記載の方法。
<13> 前記線状DNAは線状プラスミドであり、それぞれ核酸カセットの各末端に位置する0.8kb以上1.2kb以下の核酸をさらに含む、前記<12>に記載の方法。
<14> 前記微粒子の被覆は、微粒子を修飾酵素をコードする核酸で被覆することにより行われる、前記<1>に記載の方法。
<15> 前記修飾酵素が、ヌクレアーゼまたはデアミナーゼである、前記<1>に記載の方法。
<16> 前記微粒子被覆は、微粒子をプロモーターおよびターミネーターにそれぞれ連結された核酸で被覆することにより行われ、前記核酸は、少なくとも1つのガイドRNAを発現することができる核酸およびCasヌクレアーゼタンパク質をコードする核酸を含む、前記<1>に記載の方法。
<17> 前記微粒子の被覆は、微粒子をヌクレアーゼで被覆することにより行われる、前記<1>に記載の方法。
<18> 前記ヌクレアーゼがCasヌクレアーゼである、前記<17>に記載の方法。
<19> 前記微粒子の被覆は、微粒子をタンパク質で被覆することにより行われ、前記植物の茎頂への撃ち込みは、タンパク質で被覆された微粒子を有する親水性マクロキャリアフィルムを用いて行われる、前記<1>に記載の方法。
<20> 前記完熟種子は、長さが1mm以下の根を含む完熟種子である、前記<1>に記載の方法。
<21> 前記植物が、コムギ、オオムギ、イネ、トウモロコシ、ダイズ、ジャガイモおよびリンゴからなる群から選択される、前記<1>に記載の方法。
<22> 前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を高浸透圧溶液に接触させる工程をさらに含む、前記<1>に記載の方法。
<23> 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前または後である、前記<22>に記載の方法。
<24> 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前および後である、前記<22>に記載の方法。
<25> 改変された植物体を成長させる工程をさらに含む、前記<1>に記載の方法。
<26> 植物のゲノム編集方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であり、前記ゲノムは、茎頂分裂組織中の生殖細胞系列またはそれに移行し得る幹細胞内に存在し、前記方法はインプランタ法であることを特徴とする方法。
<27> 改変された植物体を成長させる工程をさらに含む、前記<26>に記載の方法。
<28> 植物の作製方法であって、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
本発明の方法によれば、微粒子を撃ち込む標的として完熟種子胚、幼芽、頂芽、側芽、又は未熟胚中の茎頂を用いることで、カルス化及び選択マーカー遺伝子を必要とせずに、再現性良く植物の改変体を取得することができる。本発明によれば、茎頂中の生殖細胞系列へ移行する茎頂幹細胞に目的遺伝子を効率よく導入することができる。
以下、本発明を詳細に説明する。
本発明に係る植物の改変法においては、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂である。
本発明に係る植物の改変法においては、微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、該被覆した微粒子を植物の茎頂に撃ち込む工程と、該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、該植物体から改変された植物体を選択する工程とを含み、前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂である。
本明細書において、「種子」とは、植物を天然またはそれに近い条件下で栽培(または培養)して得られる天然の種子のみでなく、人工種子も含むが、好ましくは天然の種子である。ただし、形質転換可能な茎頂が取得できる人工種子が開発されればそれを用いることも可能である。天然の種子には、野外の畑などで得られる種子のみでなく、温室栽培により得られる種子や、インビトロ苗などの組織培養物から得られる種子も含まれる。組織培養(ダイレクト・リプログラミング)などにより得られる種子も茎頂が取得できる限り本発明の種子として使用し得る。
なお、本発明においては、茎頂を取得する材料として完熟種子、地下茎、または未熟種子を用いるのが好ましい。完熟種子とは、受粉後の登熟過程が終了して種子として完熟しているものをいう。なお、前記登熟過程が終了したか否かは、種子の水分含量が35%以下であるか否かで判別することができる。地下茎とは、地下にある茎の総称で、塊状で多くの芽をもつ塊茎,球状で大きな頂芽をもつ球茎,短縮した茎に肥大した鱗片葉がついて球状となった鱗茎などをいう。未熟種子とは、種子として完熟していないものをいう。
本明細書において、茎頂とは、茎の先端の成長点(茎頂分裂組織)ならびに成長点および成長点から生じた数枚の葉原基からなる組織を含む意味である。本発明においては、葉原基を除去した半球状(ドーム状)の成長点のみを茎頂として用いても良く、成長点および葉原基を含む茎頂や該茎頂を含む植物組織を用いてもよい。葉原基を除去した成長点のみを用いることでウイルスフリーの組織が得られる。また、本明細書において、「生殖細胞系列」とは、生殖細胞の源である始原生殖細胞から最終産物である卵細胞や精細胞に至るまでの生殖細胞の総称であり、「L2層」とは、茎頂分裂組織の外側から2番目の細胞層をいう。「幼芽」とは、高等植物の胚の頂端にある芽であり、発芽後に生長し地上茎になることにより判別することができ、「未熟胚」とは、未熟種子の胚をいい、種子の登熟度により判別することができる。
1.形質転換の前処理工程
本発明に係る植物の改変法は、広く一般に種子を生じる植物、地下茎を生じる植物および茎頂培養可能な植物に適用することができる。従って、本発明に係る植物の改変法の対象植物は、被子植物及び裸子植物を含む種子植物である。被子植物には、単子葉植物及び双子葉植物が含まれる。インプランタ形質転換法とは、一般には組織培養の操作を含まない形質転換法であり,植物が成長する状態のまま成長点部位の細胞を形質転換させる方法である。ただし、本明細書においては、インプランタ形質転換法は、茎頂培養を用いる組織培養方法のみは含み、その他の組織培養の操作を含む方法は含まれないという意味で使用する。インプランタ法も同様の意味である。
本発明に係る植物の改変法は、広く一般に種子を生じる植物、地下茎を生じる植物および茎頂培養可能な植物に適用することができる。従って、本発明に係る植物の改変法の対象植物は、被子植物及び裸子植物を含む種子植物である。被子植物には、単子葉植物及び双子葉植物が含まれる。インプランタ形質転換法とは、一般には組織培養の操作を含まない形質転換法であり,植物が成長する状態のまま成長点部位の細胞を形質転換させる方法である。ただし、本明細書においては、インプランタ形質転換法は、茎頂培養を用いる組織培養方法のみは含み、その他の組織培養の操作を含む方法は含まれないという意味で使用する。インプランタ法も同様の意味である。
単子葉植物としては、いずれの種類であってもよいが、例えば、イネ科植物、ユリ科植物、バショウ科植物、パイナップル科植物、ラン科植物などが挙げられる。
イネ科植物としては、イネ、コムギ、オオムギ、トウモロコシ、エンバク、シバ、ソルガム、ライムギ、アワ、サトウキビなどが挙げられる。ユリ科植物としては、ネギ、アスパラガスなどが挙げられる。バショウ科植物としては、バナナなどが挙げられる。パイナップル科植物としては、パイナップルなどが挙げられる。ラン科植物としては、ランなどが挙げられる。
双子葉植物としては、例えば、アブラナ科植物、マメ科植物、ナス科植物、ウリ科植物、ヒルガオ科植物、バラ科植物、クワ科植物、アオイ科植物、キク科植物、ヒユ科植物、およびタデ科植物などが挙げられる。
アブラナ科植物としては、シロイヌナズナ、ハクサイ、ナタネ、キャベツ、カリフラワー、ダイコンなどが挙げられる。マメ科植物としては、ダイズ、アズキ、インゲンマメ、エンドウ、ササゲ、アルファルファなどが挙げられる。ナス科植物としては、トマト、ナス、ジャガイモ、タバコ、トウガラシなどが挙げられる。ウリ科植物としては、マクワウリ、キュウリ、メロン、スイカなどが挙げられる。ヒルガオ科植物としては、アサガオ、サツマイモ(カンショ)、ヒルガオなどが挙げられる。バラ科植物としては、バラ、イチゴ、リンゴなどが挙げられる。クワ科植物としては、クワ、イチジク、ゴムノキなどが挙げられる。アオイ科植物としては、ワタ、ケナフなどが挙げられる。キク科植物としては、レタスなどが挙げられる。ヒユ科植物としては、テンサイ(サトウダイコン)などが挙げられる。タデ科植物としては、ソバなどが挙げられる。
一方、裸子植物としては、マツ、スギ、イチョウ及びソテツなどが挙げられる。
本発明に係る植物の改変法の1実施態様としては、まず植物の完熟種子、又は未熟種子を吸水させる。必要により吸水前に春化処理をしてもよい。吸水は、種子を水に浸種し、インキュベートすることにより行われる。また、胚乳等を除いた胚を寒天培地上でインキュベートして行ってもよい。吸水温度は、例えば、コムギ、オオムギまたはイネの場合には、15~25℃が好ましく、トウモロコシまたはダイズでは25~35℃が好ましい。この際、一度以上、水を交換してもよい。吸水期間は、例えば、コムギの場合には、幼根が伸長を開始する前まで(1mm以下の根)、又は新しい葉原基が形成される前までが好ましい。吸水時間で表すと、種子の休眠状態にもよるが、吸水後16時間未満であり、好ましくは12時間である。この吸水工程により、種子を柔らくすることで茎頂を露出させ易くすることができる。
2.種子における胚の茎頂を露出させる工程
次いで、上記で吸水させた種子における胚の茎頂を露出させる。コムギ、オオムギ、イネ又はトウモロコシの場合には、鞘葉及び葉原基を除去することで、茎頂を露出させる。ダイズの場合には、種皮及び子葉を除去することで、茎頂を露出させる。ジャガイモの場合には、種芋から芽出しを行い、切り出した幼芽より、塊茎、葉原基を除去することで、茎頂を露出させる。リンゴの場合には、種子胚または摘出した頂芽もしくは側芽から葉原基を除去することで、茎頂を露出させる。露出手段としては、実体顕微鏡下において、鞘葉及び葉原基または種皮及び子葉を除去することができるものであればいずれのものであってよいが、例えば、直径0.2mm程度の針などの穿設するための器具、ピンセット、ピペット、注射器、並びにメスおよびカッターなどの切断器具が挙げられる。次いで、メスなどの切断器具を用いて胚乳及び余分な胚盤部分を切除し、露出した茎頂を含む胚及び胚盤を寒天培地上に、茎頂を上向きに置床する。ウイルスフリーの茎頂を得るために、茎頂を切り出す最終段階でメスを新規な滅菌メスに取り替えてもよい。この場合はウイルスフリーの形質転換体を得ることができる。
未熟種子を用いる場合には、受粉後8日~35日の胚を使用することが好ましく、受粉後10日~20日の胚を使用することがより好ましく、受粉後15日~20日の胚を使用することがさらに好ましい。
また、未熟種子を用いる場合には、殻とシルクを取り除いた、胚を有する未熟穂の上端から1/4の穀粒を削除し、穀粒から引き抜いた未熟胚を使用することができる。
次いで、上記で吸水させた種子における胚の茎頂を露出させる。コムギ、オオムギ、イネ又はトウモロコシの場合には、鞘葉及び葉原基を除去することで、茎頂を露出させる。ダイズの場合には、種皮及び子葉を除去することで、茎頂を露出させる。ジャガイモの場合には、種芋から芽出しを行い、切り出した幼芽より、塊茎、葉原基を除去することで、茎頂を露出させる。リンゴの場合には、種子胚または摘出した頂芽もしくは側芽から葉原基を除去することで、茎頂を露出させる。露出手段としては、実体顕微鏡下において、鞘葉及び葉原基または種皮及び子葉を除去することができるものであればいずれのものであってよいが、例えば、直径0.2mm程度の針などの穿設するための器具、ピンセット、ピペット、注射器、並びにメスおよびカッターなどの切断器具が挙げられる。次いで、メスなどの切断器具を用いて胚乳及び余分な胚盤部分を切除し、露出した茎頂を含む胚及び胚盤を寒天培地上に、茎頂を上向きに置床する。ウイルスフリーの茎頂を得るために、茎頂を切り出す最終段階でメスを新規な滅菌メスに取り替えてもよい。この場合はウイルスフリーの形質転換体を得ることができる。
未熟種子を用いる場合には、受粉後8日~35日の胚を使用することが好ましく、受粉後10日~20日の胚を使用することがより好ましく、受粉後15日~20日の胚を使用することがさらに好ましい。
また、未熟種子を用いる場合には、殻とシルクを取り除いた、胚を有する未熟穂の上端から1/4の穀粒を削除し、穀粒から引き抜いた未熟胚を使用することができる。
3.茎頂の細胞に核酸および/またはタンパク質を導入する工程
核酸および/またはタンパク質の導入には、公知の遺伝子工学的手法を用いることができ、特に限定されない。一般的には、目的遺伝子を含む組換えベクターを作製して、完熟胚、または未熟胚の茎頂を標的に、アグロバクテリウム法、エレクトロポレーション法、パーティクルガン法、PEG-リン酸カルシウム法、リポソーム法、マイクロインジェクション法、ウィスカー法、プラズマ法、レーザーインジェクション法などにより、核酸(組換えベクターなど)やタンパク質などを導入することができるが、核酸および/またはタンパク質を被覆した微粒子を完熟種子胚、または未熟種子胚に撃ち込む方法が好ましい。微粒子を撃ち込む手段としては、微粒子を植物細胞に撃ち込むことができるものであれば特に制限はなく、パーティクルガン法におけるパーティクルガン(遺伝子銃)などが挙げられる。コムギ、イネ、トウモロコシまたはダイズでは、植物体への導入効率から、パーティクルガン法を用いて完熟種子胚、または未熟種子胚に導入する方法が好ましい。また、パーティクルガン法はジャガイモの茎頂に遺伝子を導入するのにも有効である。パーティクルガン法は、金属微粒子に核酸および/またはタンパク質をコーティングして細胞組織に打ち込む方法であり、単子葉植物のようにアグロバクテリウムの感染効率が低い場合に有効である。
核酸および/またはタンパク質の導入には、公知の遺伝子工学的手法を用いることができ、特に限定されない。一般的には、目的遺伝子を含む組換えベクターを作製して、完熟胚、または未熟胚の茎頂を標的に、アグロバクテリウム法、エレクトロポレーション法、パーティクルガン法、PEG-リン酸カルシウム法、リポソーム法、マイクロインジェクション法、ウィスカー法、プラズマ法、レーザーインジェクション法などにより、核酸(組換えベクターなど)やタンパク質などを導入することができるが、核酸および/またはタンパク質を被覆した微粒子を完熟種子胚、または未熟種子胚に撃ち込む方法が好ましい。微粒子を撃ち込む手段としては、微粒子を植物細胞に撃ち込むことができるものであれば特に制限はなく、パーティクルガン法におけるパーティクルガン(遺伝子銃)などが挙げられる。コムギ、イネ、トウモロコシまたはダイズでは、植物体への導入効率から、パーティクルガン法を用いて完熟種子胚、または未熟種子胚に導入する方法が好ましい。また、パーティクルガン法はジャガイモの茎頂に遺伝子を導入するのにも有効である。パーティクルガン法は、金属微粒子に核酸および/またはタンパク質をコーティングして細胞組織に打ち込む方法であり、単子葉植物のようにアグロバクテリウムの感染効率が低い場合に有効である。
本発明に用いるベクターは特に限定されず、例えば、pAL系(pAL51、pAL156など)、pUC系(pUC18、pUC19、pUC9など)、pBI系(pBI121、pBI101、pBI221、pBI2113、pBI101.2など)、pPZP系、pSMA系、中間ベクター系(pLGV23Neo、pNCATなど)、カリフラワーモザイクウイルス(CaMV)、インゲンマメモザイクウイルス(BGMV)、タバコモザイクウイルス(TMV)などを用いることができる。
目的遺伝子を含むベクターは、例えば以下のようにして作製することができる。ベクターに目的遺伝子を挿入するには、例えば、精製されたDNAを適当な制限酵素で切断し、適当なベクターDNAの制限酵素部位又はマルチクローニングサイトなどに挿入してベクターに連結する方法を用いることができる。また、二重交叉組換え(double cross-over)により中間ベクターに目的遺伝子を挿入してもよく、TAクローニング、In-Fusionクローニングなどを用いてもよい。
目的遺伝子、またはゲノム編集のターゲットとなる遺伝子は特に限定されず、その遺伝子の発現または発現の抑制が望まれるものであればよく、対象となる植物の内因性遺伝子または外来遺伝子でもよい。外来遺伝子としては、生物種が異なるものであってもよく、例えば、動物、植物、微生物、ウイルスなどの遺伝子を用いることができる。そのような遺伝子として、例えば、糖代謝関連遺伝子、脂質代謝関連遺伝子、有用物質(医薬、酵素、色素、芳香成分など)生産関連遺伝子、植物生長制御(促進/抑制)関連遺伝子、開花調節関連遺伝子、耐病虫害性(昆虫食害抵抗性、線虫、カビ(菌類)及び細菌病抵抗性、ウイルス(病)抵抗性など)関連遺伝子、環境ストレス(低温、高温、乾燥、塩、光障害、紫外線)関連耐性遺伝子、トランスポーター関連遺伝子、製粉特性・製パン特性・製麺特性関連遺伝子、部位特異的ヌクレアーゼ遺伝子などが挙げられる。また、目的遺伝子は、センス鎖以外に、遺伝子導入の目的に応じてアンチセンス、リボザイム、RNAiなどを発現するように導入してもよい。また、導入するヌクレアーゼ遺伝子は、由来する生物種が異なるものであってもよく、例えば、動物、植物、微生物、ウイルスなどの遺伝子や、人工合成遺伝子を用いることができる。そのような遺伝子としては、例えば、メガヌクレアーゼ遺伝子、TALEN、CRISPR-CASシステム、TARGET AIDなどがあげられる。
ゲノム編集の標的となる遺伝子については、切断した部位の間に組換えにより外来DNA挿入または置換することにより目的の変異を導入してもよい。
本明細書において、「ゲノム編集」とは、ニューブリーディングテクニック(NBT)といわれる技術の一部で、メガヌクレアーゼ、CRISPR-CASなどを用いて、ゲノム上の特定の遺伝子を切断して変異を導入することで遺伝子を破壊すること、部位特異的にDNA断片を挿入または置換すること、または、CRISPRシステムから、ヌクレアーゼ活性を除去したものに、脱アミノ化酵素であるデアミナーゼを付加した人工酵素複合体を構築し、細胞中で発現させることで、狙った点変異を高効率に導入して遺伝子機能を改変することも含まれるがこれらに限られず、ゲノムを編集できる技術であればよい。ゲノム編集技術を用いることで、狙った遺伝子を高い効率で破壊できる。遺伝子破壊の場合、遺伝子組換えの痕跡を残さずに狙った遺伝子のみを破壊できることから組換え植物と取り扱わない国もある。また、ゲノム編集によれば、切断配列の両側の配列に相同な断片を、その部位に導入したいDNA断片の両側に連結しておくことで、効率よく部位特異的なDNA断片の挿入または置換が可能である。
上記意味で、ゲノム編集は、外来遺伝子がほぼランダムに組み込まれる従来型の植物の形質転換法、例えば、直接導入法やアグロバクテリウム法などとは異なる技術とも言え、形質転換法の定義からゲノム編集技術を除く考え方もあり得る。ゲノム編集技術では、切断部位をターゲティングできるヌクレアーゼまたはガイドRNAとヌクレアーゼを用いてゲノムDNAを切断する工程を含むことが特徴で、かかるターゲティングできるヌクレアーゼまたはガイドRNAとヌクレアーゼを用いない従来型の形質転換法とは区別できる。ここで、「ヌクレアーゼまたはガイドRNAとヌクレアーゼを用いて」という意味は、ヌクレアーゼタンパク質を細胞に導入してもよく、ヌクレアーゼ遺伝子をコードするDNAおよび/またはRNAを細胞に導入してヌクレアーゼタンパク質を発現させてもよいという意味である。また、ガイドRNAについてもRNAを細胞に導入してもよく、ガイドRNAを発現し得るDNAを導入してガイドRNAを発現させてもよい趣旨である。
部位特異的ヌクレアーゼ遺伝子がコードするタンパク質としては、例えば、ジンクフィンガーヌクレアーゼ、ジンクフィンガーヌクレアーゼ活性を発現するタンパク質またはTAL effector nuclease(TALEN)などが挙げられる。ジンクフィンガーヌクレアーゼは、特定の塩基を認識するジンクフィンガーモチーフ数個とFokIヌクレアーゼの融合タンパク質である。TALLENはTranscription Activator Like (TAL) effectorとFokIヌクレアーゼの融合タンパク質である。部位特異的ヌクレアーゼは、他の追加の標的化技術、例えば、メガヌクレアーゼ、RNA誘発性CRISPR-Cas9、またはロイシンジッパーなどで構成される。
部位特異的ヌクレアーゼを茎頂に導入して、ゲノム中に組み込み、発現させてゲノム編集することにより、1つ以上の遺伝子産物の発現を変更または改変することができる。すなわち、1つ以上の遺伝子産物をコードするDNA分子を含有し、発現し得る細胞中に、Casタンパク質およびDNA分子をターゲティングする1つ以上のガイドRNAを含み得るCRISPR-Casシステムを導入し、それにより1つ以上のガイドRNAが、1つ以上の遺伝子産物をコードするDNA分子のゲノム遺伝子座をターゲティングし、Casタンパク質が、1つ以上の遺伝子産物をコードするDNA分子のゲノム遺伝子座を開裂し、それにより1つ以上の遺伝子産物の発現を変更または改変することができる。
ヌクレアーゼ遺伝子またはタンパク質を細胞に導入してゲノム編集を行う場合、必ずしもゲノムにインテグレートさせて安定な形質転換体を取る必要は無い。ヌクレアーゼ遺伝子を一過的に発現させ、それによりゲノム編集を行うことができる。また、Casタンパク質等のタンパク質(およびガイドRNA)を細胞に導入することによってもゲノム編集を行うことができる。これらの方法によれば、得られたゲノム編集個体は遺伝子組換え体とはならない場合がある。
Casタンパク質およびガイドRNAは、天然に存在するもの(組み合わせ)であってもよく、天然には存在しない組み合わせであってもよい。本発明においては、2つ以上の遺伝子産物の発現を変更または改変してもよい。ガイドRNAが、tracr配列に融合しているガイド配列を含んでいてもよい。
ガイドRNAの長さは、少なくとも15、16、17、18、19、20ヌクレオチドであり、好ましい上限のヌクレオチド数は、30以下、より好ましくは25以下、さらに好ましくは22以下、最も好ましくは20以下である。
好ましい実施形態において、形質転換される細胞、またはゲノム編集の対象となる細胞は、植物細胞であり、より好ましくは、茎頂分裂組織の細胞であり、さらに好ましくは茎頂分裂組織中のL2細胞であり、もっとも好ましくは茎頂分裂組織中の生殖細胞系列に移行する細胞である。
本発明においては、Casタンパク質等のタンパク質が1つ以上の核局在化シグナル(NLS)を含んでいてもよい。一部の実施形態において、Casタンパク質は、II型CRISPR系酵素である。一部の実施形態において、Casタンパク質は、Cas9タンパク質である。一部の実施形態において、Cas9タンパク質は、肺炎連鎖球菌(S.pneumoniae)、化膿性連鎖球菌(S.pyogenes)、またはS.サーモフィラス(S.thermophilus)Cas9であり、それらの生物に由来する突然変異Cas9を含み得る。タンパク質は、Cas9ホモログまたはオルソログであり得る。
Casタンパク質等のタンパク質は、真核細胞中の発現のためにコドン最適化されていてもよい。Casタンパク質は、標的配列の局在における1または2つの鎖の開裂を指向し得る。本発明の他の側面において、遺伝子産物の発現を減少させ、遺伝子産物はタンパク質である。
ベクターには、目的遺伝子のほかに、例えばプロモーター、エンハンサー、インシュレーター、イントロン、ターミネーター、ポリA付加シグナル、選抜マーカー遺伝子などを連結することができる。
ベクターに挿入する目的遺伝子は1ベクターあたり複数種であってもよく、微粒子に被覆する組換えベクターも1微粒子あたり複数種であってもよい。例えば、目的遺伝子、またはヌクレアーゼ遺伝子を含む組換えベクターと、薬剤耐性遺伝子を含む組換えベクターを別々に作製し、混合して微粒子を被覆して植物組織に撃ち込んでもよい。
ゲノム編集の目的遺伝子は2以上であってもよく、2以上の目的遺伝子を切断できるようにベクターを構築してもよい。その場合、2種類以上のベクターを作製してもよく、1つのプラスミド上に複数のガイドRNAを発現するように挿入してもよい。また、ガイドRNAがtracr配列に融合しているガイド配列を含んでいてもよい。
ゲノム編集により、部位特異的に二本鎖を切断し、相同組換えを促進して部位特異的ゲノム編集を行ってもよい。この場合は切断領域の両側に相同な配列を有するDNAを導入して二重交叉組換えを起こせばよい。また、ニッカーゼ(一方のDNA鎖のみにnickを入れるDNA切断酵素)として機能することが知られているCas9のD10A変異体を用いても同様に相同組換えを行うことができる。
プロモーターとしては、植物体内または植物細胞において機能し、構成的に発現するか、植物の特定の組織内あるいは特定の発育段階において発現を導くことのできるDNAであれば、植物由来のものでなくてもよい。具体例としては、例えば、カリフラワーモザイクウイルス(CaMV)35Sプロモーター、El2-35Sオメガプロモーター、ノパリン合成酵素遺伝子のプロモーター(Pnos)、トウモロコシ由来ユビキチンプロモーター、イネ由来のアクチンプロモーター、タバコ由来PRタンパク質プロモーター、ADHプロモーター、RuBiscoプロモーターなどが挙げられる。翻訳活性を高める配列、例えば、タバコモザイクウイルスのオメガ配列を用いて翻訳効率を上げることができる。また、翻訳開始領域としてIRES(internal ribosomal entry site)をプロモーターの3’-下流側で、翻訳開始コドンの5’-上流側に挿入することで複数のコーディング領域からタンパク質を翻訳させることもできる。
ターミネーターとしては、前記プロモーターにより転写された遺伝子の転写を終結でき、ポリA付加シグナルを有する配列であればよく、例えば、ノパリン合成酵素(NOS)遺伝子のターミネーター、オクトピン合成酵素(OCS)遺伝子のターミネーター、CaMV 35S ターミネーターなどが挙げられる。
選抜マーカー遺伝子としては、例えば、除草剤耐性遺伝子(ビアラホス耐性遺伝子、グリフォセート耐性遺伝子(EPSPS)、スルホニル尿素系耐性遺伝子(ALS))、薬剤耐性遺伝子(テトラサイクリン耐性遺伝子、アンピシリン耐性遺伝子、カナマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、スペクチノマイシン耐性遺伝子、クロラムフェニコール耐性遺伝子、ネオマイシン耐性遺伝子など)、蛍光または発光レポーター遺伝子(ルシフェラーゼ、β-ガラクトシダーゼ、β-グルクロニターゼ(GUS)、グリーンフルオレッセンスプロテイン(GFP)など)、ネオマイシンホスホトランスフェラーゼII(NPT II)、ジヒドロ葉酸レダクターゼなどの酵素遺伝子が挙げられる。ただし、本発明によれば選抜マーカー遺伝子を導入しなくても形質転換体、またはゲノム編集された植物体の作製は可能である。
植物に撃ち込む目的遺伝子を含むベクターとしては、環状のプラスミドでも良く、プラスミドを制限酵素等で切断した線状DNA、線状プラスミド、導入するDNA断片のみを切り出した核酸カセット断片、または、カセット断片の一方もしくは両方の端に0.8kb以上、1.2kb以下の核酸が付加されたDNA断片であってもよい。これらのDNA断片は、PCRによって増幅されたDNA断片であってもよい。この場合付加される核酸としては、特に制限されず、ベクター由来の配列であってもよいが、導入の標的部位の配列がより好ましく用いられる。カセット断片の端に付加される核酸の長さの下限としては、0.5kb以上、より好ましくは0.8kb以上、さらに好ましくは1.0kb以上であり、上限としては、3.0kb以下、より好ましくは2.0kb以下、さらに好ましくは1.5kb以下である。
上記目的遺伝子、又は上記目的遺伝子を標的とするヌクレアーゼを含むベクターを、植物の完熟胚、幼芽、頂芽、側芽、または未熟胚に撃ち込む。目的遺伝子、又は目的遺伝子を標的とするヌクレアーゼ遺伝子をコードする核酸および/またはタンパク質(ヌクレアーゼなど)は微粒子(マイクロキャリア)の表面に被覆して植物細胞に撃ち込むことができる。微粒子としては、細胞内への貫通力を高めるため、高比重で、かつ、化学的に不活性であり生体に害を及ぼしにくいものであれば特に制限はなく、金属微粒子、セラミック微粒子などが挙げられる。金属微粒子としては、金属単体微粒子であってもよく、合金微粒子であってもよく、前記金属単体微粒子の中でも、金粒子、タングステン粒子などが特に好ましく用いられる。
以下のようにして目的遺伝子を植物細胞に導入することができる。まず、金粒子またはタングステン粒子などの微粒子を洗浄滅菌し、該微粒子、核酸(組換えベクター、直鎖状DNA、RNAなど)および/またはタンパク質、CaCl2、スペルミジンをボルテックスミキサーなどで攪拌しながら加えて、DNA・RNAおよび/またはタンパク質を金粒子またはタングステン粒子にコーティング(被覆)し、エタノールまたはリン酸緩衝生理食塩水(PBSなど)で洗浄する。なお、前記被覆は、前記金属微粒子の全表面であってもよいし、一部であってもよい。
前記微粒子の粒径(直径)は0.3μm以上1.5μm以下が好ましく、より好ましい粒径は0.4μm以上、さらに好ましくは0.5μm以上、特に好ましくは0.6μmを用いる。粒径の上限としては、1.4μm以下、1.3μm以下、1.2μm以下、1.1μm以下、1.0μm以下、0.9μm以下、0.8μm以下、0.7μm以下、0.6μm以下の順に好ましい。
金粒子またはタングステン粒子は、ピペットマンなどを用いてマクロキャリアフィルムに可能な限り均一に塗布した後、クリーンベンチなどの無菌環境中で乾燥させる。タンパク質をコートした微粒子の場合は、親水性のマクロキャリアフィルムを用いるのが好ましい。
親水性のマクロキャリアフィルムは、マクロキャリアフィルムに親水性フィルムを貼付しても良いし、親水性コーティングを施しても良い。フィルムを親水化する手法としては、界面活性剤や光触媒、親水性ポリマーを利用する手法等が挙げられる。
上記手法に用いられる親水性ポリマーとしては、ポリエチレングリコール、ヒドロキシエチルメタクリレート、ヒドロキシプロピルメタクリレート、ジヒドロキシエチルメタクリレート、ジエチレングリコールメタクリレート、トリエチレングリコールメタクリレート、ポリエチレングリコールメタクリレート、ビニルピロリドン、アクリル酸、アクリルアミド、ジメチルアクリルアミド、グルコキシオキシエチルメタクリレート、3-スルホプロピルメタクリルオキシエチルジメチルアンモニウムベタイン、2-メタクリロイルオキシエチルホスホリルコリン、1-カルボキシジメチルメタクリロイルオキシエチルメタンアンモニウム等の親水性モノマーの重合体が挙げられる。
そして、マクロキャリアフィルム、ターゲットの完熟胚、幼芽、頂芽、側芽、または未熟胚の茎頂を置床したプレートをパーティクルガン装置に設置し、ガス加速管から高圧ヘリウムガスをマクロキャリアフィルムに向かって発射する。マクロキャリヤアフィルムはストッピングプレートで止まるが、金粒子はストッピングプレートを通過して、ストッピングプレートの下に設置したターゲットに貫入し、目的遺伝子が導入される。
ストッピングプレートとターゲットとなる茎頂との距離は、微粒子の粒径にもよるが、例えば、9cm以下が好ましく、より好ましくは8cm以下、さらに好ましくは7cm以下、特に好ましくは6cm以下であり、距離の下限としては、例えば、2cm以上が好ましく、より好ましくは3cm以上、さらに好ましくは4cm以上である。ストッピングプレートとターゲットの距離は、微粒子の種類、粒径、ガス圧などに応じて、一過的発現実験などにより、適宜最適な値を決定することができる。
ガス圧としては、微粒子の種類、ターゲットとの距離にもよるが、好ましくは、例えば、1,100~1,600psiが好ましく,より好ましくは1,200~1,500psiである。ガス圧については、微粒子の種類、ターゲットの種類、ターゲットとストッピングプレートとの距離などに応じて、一過的発現実験などにより、適宜最適な圧力を決定することができる。
本発明の植物の改変方法においては、茎頂に微粒子を撃ち込む回数としては、2回以上が好ましく、さらに好ましくは3回以上、より好ましくは4回以上である。茎頂に微粒子を撃ち込む回数の上限としては、20回以下が好ましく、より好ましくは15回以下、さらに好ましくは10回以下である。撃ち込む回数については、一過的発現実験などにより、適宜最適な回数を決定することができる。
微粒子を撃ち込まれた細胞中では、核酸および/またはタンパク質が微粒子から遊離し、核酸がゲノムDNAにインテグレートすることで形質転換細胞が得られる。ただし、ジェミニウイルスのようなプラスミド状で増殖する核酸や人工染色体を導入した場合はインテグレートせずに形質転換する場合もあり得る。また、本発明の形質転換法によれば、オルガネラへの外来遺伝子の導入も可能である。その場合にはオルガネラで特異的に発現するプロモーターを機能的に連結された遺伝子を用いるのが好ましい。
本発明の植物のゲノム編集方法では、微粒子を撃ち込まれた細胞中では、核酸および/またはタンパク質が微粒子から遊離し、核酸が核に移行してヌクレアーゼを発現することでゲノムDNAを切断し、その修復の過程で変異が起こりゲノム編集された細胞が得られる。タンパク質の場合は、核移行シグナル(オルガネラ移行シグナルであってもよい)により核(オルガネラ)に移行し、DNAを切断することにより、ゲノム編集が起こる。ゲノム編集は核のゲノム遺伝子のみでなく、オルガネラ(葉緑体、ミトコンドリアなどの細胞内小器官)の遺伝子についても適用可能である。この場合は、各オルガネラへの移行シグナルをヌクレアーゼに連結させて導入してもよく、ヌクレアーゼなどに連結するプロモーターを各オルガネラでのみ発現するプロモーターを用いてもよい。
本発明の植物のゲノム編集方法では、微粒子を撃ち込まれた細胞中では、核酸および/またはタンパク質が微粒子から遊離し、核酸が核に移行してヌクレアーゼを発現することでゲノムDNAを切断し、その修復の過程で変異が起こりゲノム編集された細胞が得られる。タンパク質の場合は、核移行シグナル(オルガネラ移行シグナルであってもよい)により核(オルガネラ)に移行し、DNAを切断することにより、ゲノム編集が起こる。ゲノム編集は核のゲノム遺伝子のみでなく、オルガネラ(葉緑体、ミトコンドリアなどの細胞内小器官)の遺伝子についても適用可能である。この場合は、各オルガネラへの移行シグナルをヌクレアーゼに連結させて導入してもよく、ヌクレアーゼなどに連結するプロモーターを各オルガネラでのみ発現するプロモーターを用いてもよい。
改変処理した完熟胚、幼芽、頂芽、側芽、または未熟胚の茎頂を寒天培地上で1ヶ月程度生育させた後、土へ移植する。茎頂への撃ち込みの場合、薬剤などによる選択圧をかけずに(抗生物質、植物ホルモンなどを含まない)通常の培地で生育させることによっても形質転換体を得ることができるが、薬剤耐性遺伝子をさらに導入してもよい。本発明の植物のゲノム編集方法では、ヌクレアーゼと薬剤耐性遺伝子を同時に導入することでゲノム編集個体を選択することもできる。薬剤耐性遺伝子を導入した場合は、薬剤により形質転換細胞を選択的に培養することができる。茎頂培養に適した選択薬剤としては、例えば、スルフォニル尿素系除草剤クロロスルフロン(変異型ALS遺伝子(アセト酪酸合成酵素遺伝子)導入により耐性獲得可能)などが知られている。
薬剤耐性遺伝子を導入する場合、該薬剤耐性遺伝子は目的遺伝子、又はヌクレアーゼ遺伝子と同じベクター上にあってもよく別のベクター上にあってもよい。別々のベクター上に挿入した場合は、それらが別々の染色体に組み込まれた場合、自家受粉や戻し交配を行って後代を取ることにより目的遺伝子が導入された植物個体、又はゲノム編集された植物個体と薬剤耐性遺伝子を有する植物個体とに分離できるというメリットがある。
本発明の植物の改変方法においては、改変効率を増加させる点から、完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を高浸透圧溶液に接触させる工程をさらに含むことができる。
前記高浸透圧溶液としては、植物細胞質と比較して、浸透圧が高い溶液を用いることができ、糖、または糖アルコールを含む溶液などが挙げられる。
前記糖、または糖アルコールとしては、ソルビトール、マンニトール、スクロースなどが挙げられるが、効率よく高浸透圧処理を行う点から、ソルビトール、マンニトール、及びスクロースを含むことが好ましい。
前記高浸透圧溶液の浸透電位としては、少なくとも11%スクロース水溶液の浸透電位に等しいものなどが挙げられる。
前記糖、または糖アルコールとしては、ソルビトール、マンニトール、スクロースなどが挙げられるが、効率よく高浸透圧処理を行う点から、ソルビトール、マンニトール、及びスクロースを含むことが好ましい。
前記高浸透圧溶液の浸透電位としては、少なくとも11%スクロース水溶液の浸透電位に等しいものなどが挙げられる。
前記接触させる方法としては、前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を前記高浸透圧溶液に浸漬させる方法、前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を前記高浸透圧溶液の培地に置床させる方法、前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂に前記高浸透圧溶液を噴霧する方法などが挙げられる。
前記接触させる環境としては、明所であっても暗所であってもよいが、発芽効率の点から暗発芽種子の場合は、暗所であることが好ましく、光発芽種子の場合は明所が好ましい。
前記接触させる時期としては、前記被覆した微粒子を撃ち込む工程の前または後が挙げられる。改変効率を増加させる点から、前記被覆した微粒子を撃ち込む工程の前及び後に接触させる工程を含むことが好ましい。
前記被覆した微粒子を撃ち込む工程の前における、前記接触させる時間としては、1時間以上20時間以下が好ましく、2時間以上10時間以下がより好ましく、3時間以上5時間以下がさらに好ましい。
前記被覆した微粒子を撃ち込む工程の後における、前記接触させる時間としては、1時間以上60時間以下が好ましく、10時間以上30時間以下が好ましく、15時間以上25時間以下がさらに好ましい。
前記被覆した微粒子を撃ち込む工程の後における、前記接触させる時間としては、1時間以上60時間以下が好ましく、10時間以上30時間以下が好ましく、15時間以上25時間以下がさらに好ましい。
以上の手法により、目的遺伝子を導入した植物体やゲノム編集した改変された植物体を作出することができ、さらに前記植物体を成長させることができる。また、このようにして作出された植物は、安定的に目的遺伝子、又はゲノム編集された遺伝子の形質が発現し、または、目的遺伝子の発現が抑制され、後代に正常に遺伝する(伝達される)。
植物への遺伝子導入効率、又はゲノム編集効率および目的遺伝子発現効率(形質転換効率)は、以下のようにして調べることができる。
遺伝子の導入効率は、改変処理後、又は遺伝子導入処理後の生育個体からDNAを抽出して、PCR法および/または電気泳動又はサザンブロット法により、目的遺伝子、又は目的遺伝子がゲノム編集されたか否かを検出することができ、導入に用いた外植片数と外来遺伝子を有していた生育個体数から、遺伝子導入効率を算出する。
目的遺伝子の発現効率、又は目的遺伝子のゲノム編集効率は、遺伝子の導入が確認された生育個体について、目的遺伝子から発現されたRNAの有無を調べる。RNAの有無は、例えばRT-PCR法などで確認することができる。ノザンブロッティングにより検出してもよい。
また、目的遺伝子、又はゲノム編集された目的遺伝子から発現されたタンパク質の有無を調べることもできる。タンパク質の有無は、例えば植物片の染色、電気泳動、ELISA法、RIA、ドットイミュノバイディングアッセイ、および/またはウエスタンブロット法などで確認することができる。そして導入に用いた外植片数と目的遺伝子から発現されたタンパク質の存在が確認された生育個体数、又は導入に用いた外植片数と目的遺伝子がゲノム編集されたタンパク質の存在(または非存在)が確認された生育個体数から、目的遺伝子発現効率(形質転換効率)、又は目的遺伝子のゲノム編集効率を算出する。
以下、実施例により本発明を具体的に説明するが、本発明はこれらの実施例により何ら限定されるものではない。
[実施例1]遺伝子導入に最適な金粒子径の検討
適切な金粒子径を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無又はその当代(T0世代)における導入遺伝子の有無を調査した。
適切な金粒子径を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無又はその当代(T0世代)における導入遺伝子の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%、花王株式会社)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、休眠打破のため4℃にて2日間インキュベートした。その後、22℃で約12時間インキュベートし、以下の実験に用いた。
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%、花王株式会社)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、休眠打破のため4℃にて2日間インキュベートした。その後、22℃で約12時間インキュベートし、以下の実験に用いた。
(2)完熟種子胚中の茎頂の露出
実体顕微鏡下において、上記発芽種子の胚部分の鞘葉及び第1葉原基から第3葉原基を針(直径0.20mm)の先端を用いて除去した。その後、滅菌済みナイフ(または滅菌済みメス)を用いて、胚乳及び余分な胚盤部分を除去し、茎頂部分を完全に露出させた。これを30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt,MS vitamin,30g/L マルトース,0.98g/L MES,3%PPM(plant preservative mixture,ナカライテスク),7.0g/L phytagel(登録商標、シグマアルドリッチ),pH5.8)に置床した。
実体顕微鏡下において、上記発芽種子の胚部分の鞘葉及び第1葉原基から第3葉原基を針(直径0.20mm)の先端を用いて除去した。その後、滅菌済みナイフ(または滅菌済みメス)を用いて、胚乳及び余分な胚盤部分を除去し、茎頂部分を完全に露出させた。これを30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt,MS vitamin,30g/L マルトース,0.98g/L MES,3%PPM(plant preservative mixture,ナカライテスク),7.0g/L phytagel(登録商標、シグマアルドリッチ),pH5.8)に置床した。
(3)遺伝子導入
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、導入遺伝子は蛍光レポーター遺伝子GFP(S65T)を含むプラスミドDNA(pUC系プラスミド)を用いた。当該遺伝子は、トウモロコシのユビキチンプロモーターと第一イントロンの制御下で発現するよう設計されたものである。ターミネーターとしては、ノパリン合成酵素(NOS)遺伝子のターミネーターが付加されている。
0.3μmから1.6μmの各金粒子30mgをはかり取り、70%エタノール500μLを加え、ボルテックスにてよく懸濁した。その後、遠心により金粒子を沈殿させ、エタノールを除去した。その後、50%グリセロール500μLを加え滅菌金粒溶液とした。
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、導入遺伝子は蛍光レポーター遺伝子GFP(S65T)を含むプラスミドDNA(pUC系プラスミド)を用いた。当該遺伝子は、トウモロコシのユビキチンプロモーターと第一イントロンの制御下で発現するよう設計されたものである。ターミネーターとしては、ノパリン合成酵素(NOS)遺伝子のターミネーターが付加されている。
0.3μmから1.6μmの各金粒子30mgをはかり取り、70%エタノール500μLを加え、ボルテックスにてよく懸濁した。その後、遠心により金粒子を沈殿させ、エタノールを除去した。その後、50%グリセロール500μLを加え滅菌金粒溶液とした。
1.5mLチューブに、Qiagen Plasmid Midi Kit(Qiagen)により精製したプラスミドDNA溶液(1μg/μL)を、金粒子750μgあたり5μgになるように入れた。滅菌金粒子含有溶液は使用前に、超音波発生器(多賀電機製超音波洗浄機UW-25)を使って念入りに懸濁し、上記チューブに適当量入れピペッティングにより攪拌した。次に上記チューブに、金粒子750μgあたり2.5M CaCl2(nacalai tesque)25μLと0.1M Spermidine(nacalai tesque)10μLを加えた。混合後すぐに、ボルテックスにより5分間激しく懸濁した。10分間室温にて静置後、9,100×gで2秒間遠心した。上清を除去し、70%エタノール及び99.5%エタノールで洗浄した。最後に、上清を除去して99.5%エタノールを24μL添加してよく懸濁した。クリーンベンチ内でマクロキャリアの中央に6μLずつ注ぎ、風乾させた。
パーティクルガン(遺伝子銃)は、Biolistic(登録商標) PDS-1000/He Particle Delivery System(BIO-RAD)を使用し、撃ち込み時の圧力は約94.9kgf/cm2(1,350psi)とし、標的組織までの距離は5cm(粒子の直径0.6μm以上)または3.5cm(粒子の直径0.6μm未満)とした。1シャーレにつき4回ずつ撃ち込んだ。撃ち込み後、22℃、暗所において一晩静置した。
(4)GFPタンパク質の一過的な発現効率の調査
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図1)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表1)。特に、0.6μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が73.3%と最も高かった(表1)。また、後述するように、遺伝子導入効率についても、金粒子1.0μmよりも0.6μmの方が高かった。
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図1)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表1)。特に、0.6μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が73.3%と最も高かった(表1)。また、後述するように、遺伝子導入効率についても、金粒子1.0μmよりも0.6μmの方が高かった。

2.T0世代植物におけるゲノミックPCRを指標とした調査
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に準拠した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
0.6μm、1.0μmの2種の金粒子径を用い、実施例1-1-(3)に記載の方法に準拠した。
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に準拠した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
0.6μm、1.0μmの2種の金粒子径を用い、実施例1-1-(3)に記載の方法に準拠した。
(4)形質転換処理個体の生育
一晩静置した形質転換処理個体をMS-maltose培地を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。3-4週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて第4-6葉が出るまで育成した。
一晩静置した形質転換処理個体をMS-maltose培地を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。3-4週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて第4-6葉が出るまで育成した。
(5)T0植物葉における導入遺伝子の有無
得られた植物体において、蛍光レポーター遺伝子であるGFP遺伝子の有無をPCR法により調査した。第4-6葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、GFP遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
プライマーの配列:ACGGCCACAAGTTCAGCGT(配列番号1)
プライマーの配列:ACCATGTGATCGCGCTTCT(配列番号2)
得られた植物体において、蛍光レポーター遺伝子であるGFP遺伝子の有無をPCR法により調査した。第4-6葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、GFP遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
プライマーの配列:ACGGCCACAAGTTCAGCGT(配列番号1)
プライマーの配列:ACCATGTGATCGCGCTTCT(配列番号2)
PCR反応液には、ゲノミックDNA20ng、ExTaqHS(登録商標、TaKaRa)0.25U、10×添付バッファー1.5μL、2mM dNTPs、プライマー対各2.5pmolを加え、滅菌蒸留水で合計15μLにフィルアップした。
PCRは、TaKaRa PCR Thermal Cycler Dice(登録商標)を用いて、95℃で3分間処理後、95℃を30秒間、60℃を30秒間及び72℃を1分間の反応を1サイクルとしてこれを33サイクル行った。PCR反応後、1.0%のアガロースゲル電気泳動を行い、エチジウムブロマイドで染色してPCR産物を検出し、予想される601bpのGFP遺伝子断片が検出されたものを、遺伝子導入個体と判断した。
遺伝子導入に用いた金粒子径1.0μm、0.6μmの2通りにおいて、それぞれ得られた遺伝子導入個体数から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。
その結果、1.0μm径の金粒子を用いた場合において、導入遺伝子は検出されなかった(表2)。一方、0.6μm径の金粒子を用いた場合において、導入遺伝子確認個体(T0世代)は3個体(遺伝子導入効率1.4%)検出された(表2)。このことから、金粒子径1.0μmと比較し、実施例1-1-(4)において一過的な発現効率が高かった金粒子径0.6μmを用いることで、遺伝子導入効率が向上することがわかった。

[実施例2]遺伝子導入に最適なガス圧の検討
適切なガス圧を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無とその当代(T0世代)における導入遺伝子の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に準拠した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
金粒子径を0.6μm、ガス圧を1,100psiまたは1,350psiとし、実施例1-1-(3)に記載の方法に準拠した。
適切なガス圧を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無とその当代(T0世代)における導入遺伝子の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に準拠した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
金粒子径を0.6μm、ガス圧を1,100psiまたは1,350psiとし、実施例1-1-(3)に記載の方法に準拠した。
(4)GFPタンパク質の一過的な発現効率の調査
実施例1-1-(4)に記載の方法に準拠した。その結果、実施例1-1-(4)に記載の遺伝子導入効率は、ガス圧1,350psiの場合では74.2%となり、ガス圧1,100psiの場合の13.3%に比べ高かった(表3)。また、後述するように、T0世代の遺伝子導入効率についても、ガス圧1,100psiよりもガス圧1,350psiの方が高かった。
実施例1-1-(4)に記載の方法に準拠した。その結果、実施例1-1-(4)に記載の遺伝子導入効率は、ガス圧1,350psiの場合では74.2%となり、ガス圧1,100psiの場合の13.3%に比べ高かった(表3)。また、後述するように、T0世代の遺伝子導入効率についても、ガス圧1,100psiよりもガス圧1,350psiの方が高かった。
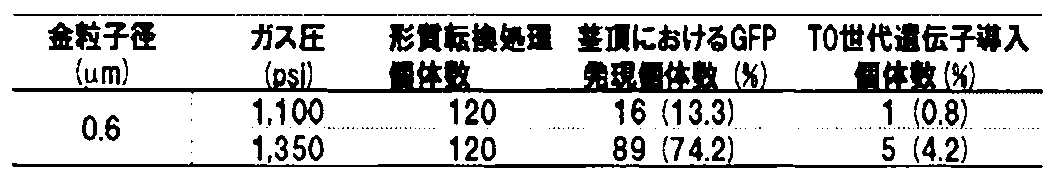
2.T0世代植物におけるゲノミックPCRを指標とした調査
(1)形質転換処理個体の生育
実施例1-2-(4)に記載の方法に準拠した。
(2)T0植物葉における導入遺伝子の有無
実施例1-2-(5)に記載の方法に準拠した。その結果、実施例1-2-(5)に記載の遺伝子導入効率は、ガス圧1,350psiの場合では4.2%となり、ガス圧1,100psiの場合の0.8%に比べ高かった(表3)。このことから、ガス圧1,100psiと比較し、実施例2-1-(4)においてGFPタンパク質の一過的な発現効率が高かったガス圧1,350psiを用いることで、遺伝子導入効率が向上することがわかった。
(1)形質転換処理個体の生育
実施例1-2-(4)に記載の方法に準拠した。
(2)T0植物葉における導入遺伝子の有無
実施例1-2-(5)に記載の方法に準拠した。その結果、実施例1-2-(5)に記載の遺伝子導入効率は、ガス圧1,350psiの場合では4.2%となり、ガス圧1,100psiの場合の0.8%に比べ高かった(表3)。このことから、ガス圧1,100psiと比較し、実施例2-1-(4)においてGFPタンパク質の一過的な発現効率が高かったガス圧1,350psiを用いることで、遺伝子導入効率が向上することがわかった。
[実施例3]遺伝子導入に最適な吸水時間の検討
適切な吸水時間を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無とその当代(T0世代)及び次世代(T1世代)における導入遺伝子の有無を調査した。
適切な吸水時間を見出すため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無とその当代(T0世代)及び次世代(T1世代)における導入遺伝子の有無を調査した。
1.完熟胚の調製及び遺伝子導入操作
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、4℃にて2日間インキュベートした。その後、22℃で6~12時間、12~18時間の各時間インキュベートし、以下の実験に用いた。種子根長は、根鞘を切り開いた後に幼根の長さを測定することで調査した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
0.6μmの金粒子径を用い、実施例1-1-(3)に記載の方法に準拠した。
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、4℃にて2日間インキュベートした。その後、22℃で6~12時間、12~18時間の各時間インキュベートし、以下の実験に用いた。種子根長は、根鞘を切り開いた後に幼根の長さを測定することで調査した。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠した。
(3)遺伝子導入
0.6μmの金粒子径を用い、実施例1-1-(3)に記載の方法に準拠した。
2.T0世代及びT1世代植物におけるゲノミックPCRを指標とした調査
(1)形質転換処理個体の生育
実施例3-1-(3)において茎頂における一過的なGFP蛍光が観察された個体について、実施例1-2-(4)に記載の方法により育成した。
(1)形質転換処理個体の生育
実施例3-1-(3)において茎頂における一過的なGFP蛍光が観察された個体について、実施例1-2-(4)に記載の方法により育成した。
(2)T0植物葉における導入遺伝子の有無
実施例1-2-(5)に記載の方法に準拠した。各吸水時間後の完熟胚を形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T0世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、6~12時間、12~18時間吸水させた完熟種子を実験に用いた場合の遺伝子導入効率は、それぞれ3.1%、1.3%となり(表4)、吸水6~12時間後の完熟胚で特に高かった(表4)。
実施例1-2-(5)に記載の方法に準拠した。各吸水時間後の完熟胚を形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T0世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、6~12時間、12~18時間吸水させた完熟種子を実験に用いた場合の遺伝子導入効率は、それぞれ3.1%、1.3%となり(表4)、吸水6~12時間後の完熟胚で特に高かった(表4)。

(3)T1植物葉における導入遺伝子の有無
上記(2)で導入遺伝子が確認された個体を育成し、T1種子を取得した。各T1種子を10~20粒程度播種・育成し、第1葉(50mg)をサンプリングした。ゲノミックPCR及び電気泳動は実施例1-2-(5)に準拠した。各吸水時間後の完熟胚を形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T1世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、6~12時間、12~18時間吸水させた完熟種子を実験に用いた場合の遺伝子導入効率はそれぞれ3.1%(図2系統1及び2、表4)、0.7%(図2系統3~5、表4)となり、吸水6~12時間後の完熟胚で特に高かった。これらの結果から、遺伝子導入には完熟種子の吸水初期(6時間後から18時間程度であり、種子根長1.0mm以下)が適切であり、特に吸水6~12時間後が好ましいと判断した。
上記(2)で導入遺伝子が確認された個体を育成し、T1種子を取得した。各T1種子を10~20粒程度播種・育成し、第1葉(50mg)をサンプリングした。ゲノミックPCR及び電気泳動は実施例1-2-(5)に準拠した。各吸水時間後の完熟胚を形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T1世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、6~12時間、12~18時間吸水させた完熟種子を実験に用いた場合の遺伝子導入効率はそれぞれ3.1%(図2系統1及び2、表4)、0.7%(図2系統3~5、表4)となり、吸水6~12時間後の完熟胚で特に高かった。これらの結果から、遺伝子導入には完熟種子の吸水初期(6時間後から18時間程度であり、種子根長1.0mm以下)が適切であり、特に吸水6~12時間後が好ましいと判断した。
なお、表4に示した導入遺伝子確認個体(T1世代)5系統については、GFP遺伝子領域をプローブとしたサザンブロット解析によっても、GFP遺伝子がゲノミックDNAへ挿入されているのを確認した(図3)。
[実施例4]T1世代における遺伝子発現の確認
当該遺伝子導入法により作製された形質転換コムギにおける導入遺伝子(GFP遺伝子)発現の有無を調査した。
当該遺伝子導入法により作製された形質転換コムギにおける導入遺伝子(GFP遺伝子)発現の有無を調査した。
1.T1種子におけるGFP蛍光観察
実施例3-2-(3)で取得したT1種子を滅菌シャーレに置き、LAS3000(FujiFilm)下で、T1種子のGFP蛍光(フィルター510DF10)を観察した結果、種子においてGFP蛍光が観察された(図4A)。次に、GFP蛍光が観察された種子を半切し、実体蛍光顕微鏡(Leica社製MZFLIII)下で、胚乳のGFP蛍光観察(励起:470/40、吸収:525/50)を行った。その結果、胚乳(E)においてGFP蛍光が観察された他、アリューロン層(Al)においてGFP蛍光が強く観察された(図4B)。
実施例3-2-(3)で取得したT1種子を滅菌シャーレに置き、LAS3000(FujiFilm)下で、T1種子のGFP蛍光(フィルター510DF10)を観察した結果、種子においてGFP蛍光が観察された(図4A)。次に、GFP蛍光が観察された種子を半切し、実体蛍光顕微鏡(Leica社製MZFLIII)下で、胚乳のGFP蛍光観察(励起:470/40、吸収:525/50)を行った。その結果、胚乳(E)においてGFP蛍光が観察された他、アリューロン層(Al)においてGFP蛍光が強く観察された(図4B)。
2.T1世代幼葉におけるGFP蛍光タンパク質の発現解析
実施例3-2-(3)で育成させたT1世代植物の幼葉をサンプリングし、実体蛍光顕微鏡(Leica社製MZFLIII)下で、葉におけるGFP蛍光(励起:470/40、吸収:525/50)を観察した。その結果、図4Cに示すように、ゲノミックPCR陽性個体(形質転換体)では葉の気孔細胞においてGFP蛍光が観察された。
実施例3-2-(3)で育成させたT1世代植物の幼葉をサンプリングし、実体蛍光顕微鏡(Leica社製MZFLIII)下で、葉におけるGFP蛍光(励起:470/40、吸収:525/50)を観察した。その結果、図4Cに示すように、ゲノミックPCR陽性個体(形質転換体)では葉の気孔細胞においてGFP蛍光が観察された。
次に、野生型株及び形質転換体(遺伝子導入個体)のそれぞれの成葉から全タンパク質を抽出し、ウェスタンブロット法によるGFP蛍光タンパク質の検出を試みた。成葉1gを液体窒素で凍結・粉砕した後、タンパク抽出バッファー(0.25M sorbitol、50mM Tris/acetate、pH7.5、1mM EDTA、2mM DTT、1%PVP、10μM PMSF)に懸濁した。この抽出液を遠心分離(1,100×g、15分間、4℃)し、その上清をさらに遠心分離(12,800×g、15分間、4℃)した。この遠心分離の後得られた上清を全タンパク質画分とした。全タンパク質画分20μgを12.5%のSDSポリアクリルアミドゲルを用いて電気泳動し、セミドライ法によってニトロセルロースメンブレンに転写した。メンブレンをブロッキングバッファー(5%[w/v] Skim Milk Powder in TTBS(10mM Tris-HCl、150mM NaCl、0.05% Tween-20、pH 7.5))で1時間振盪した。メンブレンをTTBSで5分間×3回洗浄した後、一次抗体を1時間反応させた。メンブレンをTTBSで5分間×3回洗浄した後、二次抗体を1時間反応させた。メンブレンをTTBSで5分間×3回洗浄した後、Amersham ECL Western blotting analysis system(GE Healthcare)のinstruction manualに記載の方法に従って検出を行った。なお、一次抗体として、マウスIgG1κ由来GFP抗体(ROCHE)(2000倍希釈)を用い、二次抗体として上記キットに添付されたペルオキシダーゼ標識抗マウスIgG抗体(5000倍希釈)を使用した。
ウェスタンブロット解析の結果、形質転換体ではGFPタンパク質の推定分子量27kDa付近にシグナルが見られたが、野生型株ではシグナルは検出されなかった(図4D)。これらの結果から、当該遺伝子導入法により作製された形質転換植物では、正常に導入遺伝子が発現していることが確認された。
オオムギ、トウモロコシ、イネ、ダイズ、ジャガイモおよびリンゴについても、本質的にはコムギと同様の手法により形質転換体を得ることができる。
[実施例5]T0世代1系統から取得したT1世代各個体における遺伝型の確認
本法において作製された遺伝子導入個体(T0世代)は、同一個体内において異なった遺伝情報を持つ細胞が混在したキメラになる。そこで、ある1系統の遺伝子導入個体(T0世代)から得られた複数のT1種子がそれぞれ異なる遺伝情報を有するか否かを調査するため、サザンブロット解析を行った。
本法において作製された遺伝子導入個体(T0世代)は、同一個体内において異なった遺伝情報を持つ細胞が混在したキメラになる。そこで、ある1系統の遺伝子導入個体(T0世代)から得られた複数のT1種子がそれぞれ異なる遺伝情報を有するか否かを調査するため、サザンブロット解析を行った。
実施例3-2-(3)で得られたT0世代のFG1系統の個体から、主茎および5つの分げつ由来の穂ごとにT1種子を収穫し、そこから複数個体を播種した。播種したT1世代個体を生育し、実施例3-2-(3)に記載の方法に従い、各T1世代個体における導入遺伝子の有無を調査した。その結果、分げつ2以外の主茎および分げつ由来の穂から収穫したT1世代において、導入遺伝子を確認した(表5)。

次に、主茎から得られたT1種子14個体(FG1-主茎-1~14)の幼葉から得られたゲノミックDNAに対しHindIII処理を行い、GFP遺伝子領域をプローブとしてサザンブロット解析を行った。その結果、全ての個体について同様のシグナルパターンが得られた(図5A)。さらに、主茎および分げつ1、3、4、5から得られたT1種子(FG1-主茎-1、FG1-分げつ1-1、FG1-分げつ3-1、FG1-分げつ4-1、FG1-分げつ5-1)の幼葉から得られたゲノミックDNAに対しHindIII処理を行い、上記と同様にサザンブロット解析を行った。その結果、全ての個体について同様のシグナルパターンが得られた(図5B)。これらの結果から、ある1系統の遺伝子導入個体(T0世代)から得られた複数のT1種子は同じ遺伝情報を有することがわかった。これは、生殖細胞系列へ移行する茎頂幹細胞は極少数であることを示唆しており、当該方法によりこの極少数の茎頂幹細胞へ遺伝子導入が可能であることがわかった。
[実施例6]トウモロコシにおける形質転換体の取得
コムギと同様の手法によりトウモロコシの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
コムギと同様の手法によりトウモロコシの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ完熟種子の調製
トウモロコシ(スノーデント王夏)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、30℃で約36時間インキュベートし、以下の実験に用いた。
(1)トウモロコシ完熟種子の調製
トウモロコシ(スノーデント王夏)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、30℃で約36時間インキュベートし、以下の実験に用いた。
(2)完熟種子胚中の茎頂の露出
実体顕微鏡下において、滅菌済みナイフ(または滅菌済みメス)を用いて、胚乳及び余分な胚盤部分を除去した。その後、上記発芽種子の胚部分の子葉鞘及び葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを10個/プレートとなるようにMS-maltose培地(4.3g/L MS salt ,MS vitamin,30g/L マルトース,0.98g/L MES ,3%PPM(plant preservative mixture,登録商標、ナカライテスク),7.0g/L phytagel,pH5.8)に置床した。一処理区につき上記プレートを2枚用意した。
実体顕微鏡下において、滅菌済みナイフ(または滅菌済みメス)を用いて、胚乳及び余分な胚盤部分を除去した。その後、上記発芽種子の胚部分の子葉鞘及び葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを10個/プレートとなるようにMS-maltose培地(4.3g/L MS salt ,MS vitamin,30g/L マルトース,0.98g/L MES ,3%PPM(plant preservative mixture,登録商標、ナカライテスク),7.0g/L phytagel,pH5.8)に置床した。一処理区につき上記プレートを2枚用意した。
(3)遺伝子導入
実施例1-1-(3)に記載の方法に従った。
実施例1-1-(3)に記載の方法に従った。
(4)GFPタンパク質の一過的な発現効率の調査
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図6)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表6)。特に、0.6μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が85%と最も高かった(表6)。
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図6)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表6)。特に、0.6μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が85%と最も高かった(表6)。

[実施例7]ダイズにおける形質転換体の取得
コムギと同様の手法によりダイズの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無及びその後の形質転換体取得効率を調査した。
コムギと同様の手法によりダイズの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無及びその後の形質転換体取得効率を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)ダイズ完熟種子の調製
ダイズ(ユキホマレ)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で3分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、23℃で約40時間インキュベートし、以下の実験に用いた。
(1)ダイズ完熟種子の調製
ダイズ(ユキホマレ)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で3分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、23℃で約40時間インキュベートし、以下の実験に用いた。
(2)完熟種子胚中の茎頂の露出
実体顕微鏡下において、滅菌済みナイフ(または滅菌済みメス)を用いて、子葉を全てカットし、胚軸のみにした。その後、上記胚軸の初生葉と基部を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを約15個/プレートとなるようにBM培地(4.3g/L MS salt 、MS vitamin、3% スクロース、0.50g/L MES 、3%PPM(plant preservative mixture、ナカライテスク)、6.0g/L phytagel、pH5.7)に置床した。
実体顕微鏡下において、滅菌済みナイフ(または滅菌済みメス)を用いて、子葉を全てカットし、胚軸のみにした。その後、上記胚軸の初生葉と基部を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを約15個/プレートとなるようにBM培地(4.3g/L MS salt 、MS vitamin、3% スクロース、0.50g/L MES 、3%PPM(plant preservative mixture、ナカライテスク)、6.0g/L phytagel、pH5.7)に置床した。
(3)遺伝子導入
プロモーターとして、カリフラワーモザイクウイルス(CaMV)の35Sプロモーターを用い、実施例1-1-(3)に記載の方法に従った。
プロモーターとして、カリフラワーモザイクウイルス(CaMV)の35Sプロモーターを用い、実施例1-1-(3)に記載の方法に従った。
(4)GFPタンパク質の一過的な発現効率の調査
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図7)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μmおよび1.0μmに比べ、金粒子径0.6μmの場合では茎頂における遺伝子導入効率が85.0%と高かった(表7)。
実体蛍光顕微鏡(Leica社製MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで(図7)、茎頂におけるGFP遺伝子の導入効率を算出した。形質転換処理した完熟胚中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを遺伝子導入個体として、遺伝子導入効率(遺伝子導入個体/処理完熟胚数×100)を算出した。その結果、金粒子径1.6μmおよび1.0μmに比べ、金粒子径0.6μmの場合では茎頂における遺伝子導入効率が85.0%と高かった(表7)。

2.T0世代植物におけるゲノミックPCRを指標とした調査
(1)形質転換処理個体の生育
一晩静置した形質転換処理個体をRM培地(4.3g/L MS salt 、MS vitamin、3% スクロース、0.50g/L MES 、3%PPM(plant preservative mixture、ナカライテスク)、0.3% ゲルライト、pH5.7)を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、インキュベーター(23℃、16時間日長)で育成した。根の生育及び茎頂からの葉の分化が観察された時点で、園芸用育苗培土を入れたセルトレイへ移植した。その後、同インキュベーターにて第2葉目まで育成した。
(1)形質転換処理個体の生育
一晩静置した形質転換処理個体をRM培地(4.3g/L MS salt 、MS vitamin、3% スクロース、0.50g/L MES 、3%PPM(plant preservative mixture、ナカライテスク)、0.3% ゲルライト、pH5.7)を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、インキュベーター(23℃、16時間日長)で育成した。根の生育及び茎頂からの葉の分化が観察された時点で、園芸用育苗培土を入れたセルトレイへ移植した。その後、同インキュベーターにて第2葉目まで育成した。
(2)T0植物葉における導入遺伝子の有無
得られた植物体において、蛍光レポーター遺伝子であるGFP遺伝子の有無をPCR法により調査した。第2葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、GFP遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
プライマーの配列:ACGGCCACAAGTTCAGCGT(配列番号1)
プライマーの配列:ACCATGTGATCGCGCTTCT(配列番号2)
得られた植物体において、蛍光レポーター遺伝子であるGFP遺伝子の有無をPCR法により調査した。第2葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、GFP遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
プライマーの配列:ACGGCCACAAGTTCAGCGT(配列番号1)
プライマーの配列:ACCATGTGATCGCGCTTCT(配列番号2)
PCR反応液には、ゲノミックDNA20ng、ExTaqHS(TaKaRa)0.25U、10×添付バッファー1.5μL、2mM dNTPs、プライマー対各2.5pmolを加え、滅菌蒸留水で合計15μLにフィルアップした。
PCRは、TaKaRa PCR Thermal Cycler Diceを用いて、95℃で3分間処理後、95℃を30秒間、60℃を30秒間及び72℃を1分間の反応を1サイクルとしてこれを32サイクル行った。PCR反応後、1.0%のアガロースゲル電気泳動を行い、エチジウムブロマイドで染色して紫外線照射によりPCR産物を検出し、予想される601bpのGFP遺伝子断片が検出されたものを、遺伝子導入個体と判断した。
遺伝子導入に用いた金粒子径1.0μm、0.6μmの2通りにおいて、それぞれ得られた遺伝子導入個体数から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。
その結果、0.6μm径の金粒子を用いた場合において、処理完熟胚470個体のうち導入遺伝子確認個体(T0世代)は63個体(遺伝子導入効率13.4%)であった。このことから、実施例7-1-(4)において一過的な発現効率が高かった金粒子径0.6μmを用いることで、高効率でダイズの遺伝子導入個体を取得することが可能であることがわかった。
[実施例8]オオムギにおける形質転換体の取得
コムギと同様の手法によりオオムギの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
コムギと同様の手法によりオオムギの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)オオムギ完熟種子の調製
オオムギ(ワセドリ二条)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、4℃にて2日間インキュベートした後、22℃で6~12時間インキュベートし、以下の実験に用いた。
(1)オオムギ完熟種子の調製
オオムギ(ワセドリ二条)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、4℃にて2日間インキュベートした後、22℃で6~12時間インキュベートし、以下の実験に用いた。
(2)完熟種子胚中の茎頂の露出
実体顕微鏡下において、滅菌済みメスを用いて、胚乳及び余分な胚盤部分を除去した。その後、上記発芽種子の胚部分の子葉鞘及び葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt 、MS vitamin、30g/L マルトース、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク),7.0g/L phytagel,pH5.8)に置床した。
実体顕微鏡下において、滅菌済みメスを用いて、胚乳及び余分な胚盤部分を除去した。その後、上記発芽種子の胚部分の子葉鞘及び葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt 、MS vitamin、30g/L マルトース、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク),7.0g/L phytagel,pH5.8)に置床した。
(3)遺伝子導入
実施例1-1-(3)に記載の方法に従った。
実施例1-1-(3)に記載の方法に従った。
(4)GFPタンパク質の一過的な発現効率の調査
実施例1-1-(4)に記載の方法に従った。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表8)。特に、0.3μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が80.0%と最も高かった(図8、表8)。
実施例1-1-(4)に記載の方法に従った。その結果、金粒子径1.6μm及び1.0μmに比べ、金粒子径0.8μm、0.6μm、及び0.3μmの場合では茎頂における遺伝子導入効率が高かった(表8)。特に、0.3μmを用いたときは、他の金粒子径と比較して、茎頂における遺伝子導入効率が80.0%と最も高かった(図8、表8)。

[実施例9]ジャガイモにおける形質転換体の取得
コムギと同様の手法によりジャガイモの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
コムギと同様の手法によりジャガイモの形質転換体が得られるか確認するため、遺伝子導入処理後の茎頂におけるGFP蛍光の有無を調査した。
1.GFP蛍光タンパク質の一過的発現系を指標とした調査
(1)ジャガイモの調製
種芋(男爵)をハイター(次亜塩素酸濃度1%)で消毒し、水洗い、乾燥後、22℃で1-2週間蛍光灯下でインキュベートし、芽出しを行った。
(1)ジャガイモの調製
種芋(男爵)をハイター(次亜塩素酸濃度1%)で消毒し、水洗い、乾燥後、22℃で1-2週間蛍光灯下でインキュベートし、芽出しを行った。
(2)茎頂の露出
実体顕微鏡下において、萌芽より葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを40個/プレートとなるようにMS培地(4.3g/L MS salt 、MS vitamin、30g/L ショ糖、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク)、7.0g/L phytagel、pH5.8)に置床した。
実体顕微鏡下において、萌芽より葉原基を針(直径0.20mm)の先端を用いて除去し、茎頂部分を完全に露出させた。これを40個/プレートとなるようにMS培地(4.3g/L MS salt 、MS vitamin、30g/L ショ糖、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク)、7.0g/L phytagel、pH5.8)に置床した。
(3)遺伝子導入
導入ベクターをpUC18-35S-GFPとし、実施例1-1-(3)に記載の方法に従った。
導入ベクターをpUC18-35S-GFPとし、実施例1-1-(3)に記載の方法に従った。
(4)GFPタンパク質の一過的な発現効率の調査
実施例1-1-(4)に記載の方法に従った。その結果、金粒子径0.6μmを用いることで、茎頂におけるGFP蛍光を観察することができた。(図9)。
実施例1-1-(4)に記載の方法に従った。その結果、金粒子径0.6μmを用いることで、茎頂におけるGFP蛍光を観察することができた。(図9)。
[実施例10]線状プラスミドを用いた形質転換
形質転換効率改善のため、線状プラスミドを用いた遺伝子導入処理後の茎頂におけるGFP蛍光の有無及びその後の形質転換体取得効率を調査した。
形質転換効率改善のため、線状プラスミドを用いた遺伝子導入処理後の茎頂におけるGFP蛍光の有無及びその後の形質転換体取得効率を調査した。
1.T0世代植物におけるゲノミックPCRを指標とした調査
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に従った。
(1)コムギ完熟種子の調製
実施例1-1-(1)に記載の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に従った。
実施例1-1-(2)に記載の方法に従った。
(3)線状ベクターの調整
実施例1-1-(3)で用いたGFPベクター(配列番号3)を鋳型として、ユビキチンプロモーター及びNosターミネーターに特異的な配列(配列番号4及び5)又はベクターに特異的な配列(配列番号6及び7)から作製したプライマーを用いてPCR反応を行った。その後、PCR産物をエタノール沈殿により精製することで、線状ベクター1(Pubi-GFP-Tnos断片)及び線状ベクター2(Pubi-GFP-Tnos断片-1kb付加)を作製した。なお線状ベクター2においては、線状ベクター1の5’末端に約1.2kbp、3’末端に0.8kbpのGFPベクター由来配列が付加されている。
プライマーの配列:CGACGGCCAGTGCCAAGCTT(配列番号4)
プライマーの配列:ATGACCATGATTACGAATTC(配列番号5)
プライマーの配列:AAGCTAGAGTAAGTAGTTCGCCA(配列番号6)
プライマーの配列:ATACTGTCCTTCTAGTGTAGCCG(配列番号7)
実施例1-1-(3)で用いたGFPベクター(配列番号3)を鋳型として、ユビキチンプロモーター及びNosターミネーターに特異的な配列(配列番号4及び5)又はベクターに特異的な配列(配列番号6及び7)から作製したプライマーを用いてPCR反応を行った。その後、PCR産物をエタノール沈殿により精製することで、線状ベクター1(Pubi-GFP-Tnos断片)及び線状ベクター2(Pubi-GFP-Tnos断片-1kb付加)を作製した。なお線状ベクター2においては、線状ベクター1の5’末端に約1.2kbp、3’末端に0.8kbpのGFPベクター由来配列が付加されている。
プライマーの配列:CGACGGCCAGTGCCAAGCTT(配列番号4)
プライマーの配列:ATGACCATGATTACGAATTC(配列番号5)
プライマーの配列:AAGCTAGAGTAAGTAGTTCGCCA(配列番号6)
プライマーの配列:ATACTGTCCTTCTAGTGTAGCCG(配列番号7)
PCR反応液には、ベクターDNA10ng、PrimeSTAR(登録商標) GXL DNA Polymerase(TaKaRa)1.0U、5×添付バッファー4.0μL、0.2mM dNTPs、プライマー対各2.5pmolを加え、滅菌蒸留水で合計20μLにフィルアップした。
PCRは、TaKaRa PCR Thermal Cycler Diceを用いて、98℃を10秒間、60℃を15秒間及び68℃を2分30秒間の反応を1サイクルとしてこれを40サイクル行った。PCR産物をエタノール沈殿により精製し、1μg/μLになるように滅菌水に溶解し、これを以下の実験に用いた。
(4)遺伝子導入
0.6μmの金粒子径を用い、GFPベクター又は線状ベクター溶液(1μg/μL)を、金粒子750μgあたり10μgになるように入れ、実施例1-1-(3)に記載の方法に準拠した。
0.6μmの金粒子径を用い、GFPベクター又は線状ベクター溶液(1μg/μL)を、金粒子750μgあたり10μgになるように入れ、実施例1-1-(3)に記載の方法に準拠した。
(5)形質転換処理個体の生育
一晩静置した形質転換処理個体をMS-maltose培地を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。3-4週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて第4-6葉が出るまで育成した。
一晩静置した形質転換処理個体をMS-maltose培地を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。3-4週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて第4-6葉が出るまで育成した。
(6)T0植物葉における導入遺伝子の有無
実施例1-2-(5)に記載の方法に準拠した。各種ベクターを形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T0世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、従来のGFPベクターを導入した場合(4.2%)に比べ、線状ベクター1及び線状ベクター2を実験に用いた場合の遺伝子導入効率は、それぞれ7.1%、14.2%となり(表9)、線状ベクター2で特に高かった。これらの結果から、遺伝子導入には線状ベクター(導入目的とする核酸カセット)が好ましく、より好ましくは導入目的とする核酸カセットの両末端に0.8 kb以上の核酸が付加された線状ベクターを用いることだとわかった。
実施例1-2-(5)に記載の方法に準拠した。各種ベクターを形質転換処理した場合において、それぞれ得られた導入遺伝子確認個体(T0世代)から遺伝子導入処理を行った完熟胚数当たりの遺伝子導入効率(遺伝子導入個体数/処理完熟胚数×100)を算出した。その結果、従来のGFPベクターを導入した場合(4.2%)に比べ、線状ベクター1及び線状ベクター2を実験に用いた場合の遺伝子導入効率は、それぞれ7.1%、14.2%となり(表9)、線状ベクター2で特に高かった。これらの結果から、遺伝子導入には線状ベクター(導入目的とする核酸カセット)が好ましく、より好ましくは導入目的とする核酸カセットの両末端に0.8 kb以上の核酸が付加された線状ベクターを用いることだとわかった。

[実施例11]イネにおける形質転換体の取得
イネについても、本質的にはコムギと同様の手法により形質転換体を得ることができる。
1.イネ完熟種子の調製
脱穀したイネ(品種:日本晴)の完熟種子を実施例1-1-(1)に記載の方法で殺菌、洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、25℃で24時間~48時間インキュベートしたものを以下の実験に用いる。
イネについても、本質的にはコムギと同様の手法により形質転換体を得ることができる。
1.イネ完熟種子の調製
脱穀したイネ(品種:日本晴)の完熟種子を実施例1-1-(1)に記載の方法で殺菌、洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、25℃で24時間~48時間インキュベートしたものを以下の実験に用いる。
2.完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に従い、完熟種子胚中の茎頂を完全露出させる。茎頂を露出させた完熟胚を約30個/プレートとなるようにMS-maltose培地に置床する。
実施例1-1-(2)に記載の方法に従い、完熟種子胚中の茎頂を完全露出させる。茎頂を露出させた完熟胚を約30個/プレートとなるようにMS-maltose培地に置床する。
3.遺伝子導入
イネ完熟胚の茎頂への遺伝子導入は、実施例1-1-(3)に記載の方法に従って行う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを選抜する。
5.形質転換処理個体の生育
実施例1-2-(4)に記載の方法に従い、形質転換処理個体を生育する。
6.T0植物葉における導入遺伝子の確認
実施例1-2-(5)に記載の方法に従い、遺伝子導入確認個体を取得する。得られた個体を育成し、次世代種子を取得することで、安定的な形質転換体を取得することができる。
イネ完熟胚の茎頂への遺伝子導入は、実施例1-1-(3)に記載の方法に従って行う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを選抜する。
5.形質転換処理個体の生育
実施例1-2-(4)に記載の方法に従い、形質転換処理個体を生育する。
6.T0植物葉における導入遺伝子の確認
実施例1-2-(5)に記載の方法に従い、遺伝子導入確認個体を取得する。得られた個体を育成し、次世代種子を取得することで、安定的な形質転換体を取得することができる。
[実施例12]リンゴにおける形質転換体の取得
リンゴについても、本質的にはコムギと同様の手法により形質転換体を得ることができる。
1.リンゴ茎頂の調製
リンゴ(品種:ジョナゴールド)の茎頂の調製は、植物組織培養, 9(2), 69-73 (1992)に記載の茎頂培養の誘導条件に沿って実施する。
リンゴについても、本質的にはコムギと同様の手法により形質転換体を得ることができる。
1.リンゴ茎頂の調製
リンゴ(品種:ジョナゴールド)の茎頂の調製は、植物組織培養, 9(2), 69-73 (1992)に記載の茎頂培養の誘導条件に沿って実施する。
2.茎頂の露出
1.の手法にて調製した茎頂を、約30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt 、MS vitamin、30g/L マルトース、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク)、7.0g/L phytagel、pH5.8)に置床する。
1.の手法にて調製した茎頂を、約30個/プレートとなるようにMS-maltose培地(4.3g/L MS salt 、MS vitamin、30g/L マルトース、0.98g/L MES 、3%PPM(plant preservative mixture、登録商標、ナカライテスク)、7.0g/L phytagel、pH5.8)に置床する。
3.遺伝子導入
プロモーターとして、カリフラワーモザイクウイルス(CaMV)の35Sプロモーターを用い、実施例1-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを選抜する。
5.形質転換処理個体の生育
実施例1-2-(4)に記載の方法に従い、形質転換処理個体を生育する。
6.T0植物葉における導入遺伝子の確認
実施例1-2-(5)に記載の方法に従い、遺伝子導入確認個体を取得する。得られた個体を育成し、次世代種子を取得することで、安定的な形質転換体を取得することができる。
プロモーターとして、カリフラワーモザイクウイルス(CaMV)の35Sプロモーターを用い、実施例1-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織において5スポット以上のGFP蛍光が観察されたものを選抜する。
5.形質転換処理個体の生育
実施例1-2-(4)に記載の方法に従い、形質転換処理個体を生育する。
6.T0植物葉における導入遺伝子の確認
実施例1-2-(5)に記載の方法に従い、遺伝子導入確認個体を取得する。得られた個体を育成し、次世代種子を取得することで、安定的な形質転換体を取得することができる。
[実施例13]当該遺伝子導入法を利用した標的変異導入
当該遺伝子導入法を利用してコムギのゲノム編集が可能か否かを検証するため、RNA誘発性CRISPR/Cas9による標的配列の変異導入を試みた。なお、実施例1-1-(4)より当該遺伝子導入法を用いて安定な形質転換体を作製する上では、金粒子径0.3μm以上0.9μm以下が良いが、ゲノム編集個体を作製する上では、植物細胞核内又はオルガネラ内で一過的にヌクレアーゼ等が発現すれば良いため、上記粒子径に限定されない。そこで、当該遺伝子導入法を用いてコムギのゲノム編集が可能な金粒子径の範囲を検討した。
当該遺伝子導入法を利用してコムギのゲノム編集が可能か否かを検証するため、RNA誘発性CRISPR/Cas9による標的配列の変異導入を試みた。なお、実施例1-1-(4)より当該遺伝子導入法を用いて安定な形質転換体を作製する上では、金粒子径0.3μm以上0.9μm以下が良いが、ゲノム編集個体を作製する上では、植物細胞核内又はオルガネラ内で一過的にヌクレアーゼ等が発現すれば良いため、上記粒子径に限定されない。そこで、当該遺伝子導入法を用いてコムギのゲノム編集が可能な金粒子径の範囲を検討した。
1.GFP蛍光を指標とした標的変異導入確認
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、休眠打破のため4℃にて2日間インキュベートした。その後、22℃で約12時間インキュベートし、以下の実験に用いた。
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Fielder)の完熟種子をハイター(次亜塩素酸濃度6%)に浸漬し、室温で20分間振とう後、クリーンベンチ内で滅菌水で洗浄した。洗浄後、滅菌水で湿らせたキムタオル又はろ紙上に載せ、休眠打破のため4℃にて2日間インキュベートした。その後、22℃で約12時間インキュベートし、以下の実験に用いた。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例1-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、蛍光レポーター遺伝子(GFP)の開始コドン直後に標的配列としてTaMLO遺伝子内部の20bp部分配列(CCGTCACGCAGGACCCAATCTCC:配列番号8)が挿入された標的プラスミドDNA(pUC系プラスミド)を用いた。当該遺伝子は、トウモロコシのユビキチンプロモーターと第一イントロンの制御下で発現するよう設計されたものである。ターミネーターとしては、ノパリン合成酵素(NOS)遺伝子のターミネーターが付加されている。当該ベクターと、gRNA/Cas9一体型プラスミドを共導入することで、TaMLO遺伝子標的配列をCas9が切断し、修復過程で2塩基が欠失または1塩基が挿入される。その結果、翻訳フレームシフトが起こり、GFP遺伝子のコドンの読み枠が回復し、活性型のGFPタンパク質が発現する。
当該一体型プラスミド(pUC系プラスミド)中のgRNAはコムギ由来U6プロモーターの制御下で発現するよう設計されたものである。当該Cas9遺伝子はトウモロコシのユビキチンプロモーターと第一イントロンの制御下で発現するよう設計されたものである。ターミネーターとしては、ノパリン合成酵素(NOS)遺伝子のターミネーターが付加されている。
金粒子の調製及び導入操作は、実施例2-1-(3)の方法に準拠して行った。但し、標的プラスミドDNA溶液及びgRNA/Cas9一体型プラスミドを、金粒子750μgあたりそれぞれ2.5μg及び7.5μgになるように入れた。金粒子量は1回の撃ち込みあたり、金粒子径0.3μmでは375μg、それ以外の金粒子径では187.5μgになるように入れた。
(4)TaMLO遺伝子配列切断によるGFP蛍光の確認
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで、TaMLO遺伝子標的配列の切断の有無を調査した。遺伝子導入処理した完熟胚中、茎頂組織において1スポット以上のGFP蛍光が観察されたものを標的配列切断個体として、標的配列切断導入効率(標的配列切断個体/処理完熟胚数×100)を算出した。実験は2度行い、標的配列切断導入効率の平均値を表10に示した。その結果、標的プラスミドDNA及びgRNA/Cas9一体型プラスミドを共導入した場合、金粒子径0.3μmから1.0μmでは標的配列切断を示すGFP蛍光が観察されたが、金粒子径1.6μmでは観察されなかった(表10)。また、その効率は金粒子径0.6μmを用いた場合、23.5%と最も高かった(表10,図10)。これらの結果から、当該遺伝子導入法を用いてゲノム編集個体を作製するためには、金粒子径が1.6μm未満が良く、金粒子径0.6μmが好ましいことがわかった。
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)を観察することで、TaMLO遺伝子標的配列の切断の有無を調査した。遺伝子導入処理した完熟胚中、茎頂組織において1スポット以上のGFP蛍光が観察されたものを標的配列切断個体として、標的配列切断導入効率(標的配列切断個体/処理完熟胚数×100)を算出した。実験は2度行い、標的配列切断導入効率の平均値を表10に示した。その結果、標的プラスミドDNA及びgRNA/Cas9一体型プラスミドを共導入した場合、金粒子径0.3μmから1.0μmでは標的配列切断を示すGFP蛍光が観察されたが、金粒子径1.6μmでは観察されなかった(表10)。また、その効率は金粒子径0.6μmを用いた場合、23.5%と最も高かった(表10,図10)。これらの結果から、当該遺伝子導入法を用いてゲノム編集個体を作製するためには、金粒子径が1.6μm未満が良く、金粒子径0.6μmが好ましいことがわかった。

[実施例14]当該遺伝子導入法を利用した標的変異導入(T0世代、T1世代)
当該遺伝子導入法を利用してコムギに内在する標的遺伝子のゲノム編集が可能か否かを検証するため、RNA誘発性CRISPR/Cas9によるTaQsd1遺伝子(alanine aminotransferase遺伝子)、TaLOX2遺伝子(lipoxygenase遺伝子)、及びTaGASR7遺伝子(Snakin/GASA遺伝子)の変異導入を試みた。なお、TaQsd1遺伝子の変異導入には実用品種「春よ恋」を、その他には「Bobwhite」を用いた。
当該遺伝子導入法を利用してコムギに内在する標的遺伝子のゲノム編集が可能か否かを検証するため、RNA誘発性CRISPR/Cas9によるTaQsd1遺伝子(alanine aminotransferase遺伝子)、TaLOX2遺伝子(lipoxygenase遺伝子)、及びTaGASR7遺伝子(Snakin/GASA遺伝子)の変異導入を試みた。なお、TaQsd1遺伝子の変異導入には実用品種「春よ恋」を、その他には「Bobwhite」を用いた。
1.遺伝子導入後一週間目における標的遺伝子変異導入確認
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Bobwhite、Triticum aestivum cv. Haruyokoi)の完熟種子を用い、実施例1-1-(1)の方法に従った。
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Bobwhite、Triticum aestivum cv. Haruyokoi)の完熟種子を用い、実施例1-1-(1)の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠して行った。
実施例1-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、gRNA発現用プラスミドとして、pTAKN-2ベクターのマルチクローニングサイトへコムギU6プロモーター及びgRNA scaffoldをクローニングした。その後、内部のBbsIサイトへ各標的遺伝子の標的配列を導入し、各gRNA発現用プラスミドとした。
TaQsd1標的配列:ACGGATCCACCTCCCTGCAGCGG (配列番号9)
TaLOX2標的配列:GTGCCGCGCGACGAGCTCTTCGG(配列番号10)
TaGASR7標的配列:CCGCCGGGCACCTACGGCAAC(配列番号11)
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、gRNA発現用プラスミドとして、pTAKN-2ベクターのマルチクローニングサイトへコムギU6プロモーター及びgRNA scaffoldをクローニングした。その後、内部のBbsIサイトへ各標的遺伝子の標的配列を導入し、各gRNA発現用プラスミドとした。
TaQsd1標的配列:ACGGATCCACCTCCCTGCAGCGG (配列番号9)
TaLOX2標的配列:GTGCCGCGCGACGAGCTCTTCGG(配列番号10)
TaGASR7標的配列:CCGCCGGGCACCTACGGCAAC(配列番号11)
上記各gRNA発現用プラスミド、Cas9遺伝子発現用プラスミド及びGFP発現用プラスミド(実施例1-1-(3)参照)を金粒子750μgあたり各々5μg、2.5μg、2.5μg入れた。金粒子径は0.6μmとし、金粒子量は1回の撃ち込みあたり、187.5μgになるように入れた。
(4)各標的遺伝子配列切断による変異導入の確認
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)が見られた個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。約1週間後に各個体中の茎頂を切り出し、2μLのDNAzol Direct(登録商標、コスモバイオ)が添加された8連PCRチューブへ入れ、ゲノミックDNAを抽出した。
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)が見られた個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。約1週間後に各個体中の茎頂を切り出し、2μLのDNAzol Direct(登録商標、コスモバイオ)が添加された8連PCRチューブへ入れ、ゲノミックDNAを抽出した。
これを鋳型として、各遺伝子に特異的な配列から作製されたプライマーを用いてPCR反応を行った。
TaQsd1-F:CAGCCTGGAGGGAATGACC (配列番号12)
TaQsd1-R:ACCTGGTGGAATCCAGAGC (配列番号13)
TaLOX2-F:CGTCTACCGCTACGACGTCTACAACG(配列番号14)
TaLOX2-R:GGTCGCCGTACTTGCTCGGATCAAGT(配列番号15)
TaGASR7-F:CCTTCATCCTTCAGCCATGCAT(配列番号16)
TaGASR7-R:CCACTAAATGCCTATCACATACG(配列番号17)
TaQsd1-F:CAGCCTGGAGGGAATGACC (配列番号12)
TaQsd1-R:ACCTGGTGGAATCCAGAGC (配列番号13)
TaLOX2-F:CGTCTACCGCTACGACGTCTACAACG(配列番号14)
TaLOX2-R:GGTCGCCGTACTTGCTCGGATCAAGT(配列番号15)
TaGASR7-F:CCTTCATCCTTCAGCCATGCAT(配列番号16)
TaGASR7-R:CCACTAAATGCCTATCACATACG(配列番号17)
PCR反応液には、ゲノミックDNA0.5μL、KODFXNeo(TOYOBO)0.4U、2×添付バッファー10μL、0.4mMdNTPs、プライマー対各0.2μMを加え、滅菌蒸留水で合計20μLにフィルアップした。
PCRは、TaKaRa PCR Thermal Cycler Diceを用いて、98℃を10秒間、68℃を1分間の反応を1サイクルとしてこれを35サイクル行った。PCR反応後、各PCR産物を1μL、10×添付バッファー1μL,適当な制限酵素5Uを加え、滅菌蒸留水で合計10μLにフィルアップした。37℃、オーバーナイトで反応させた後、2.0%のアガロースゲル電気泳動を行い、エチジウムブロマイドで染色した。
野生型株ではPCR産物が制限酵素により完全に切断されるのに対し、変異が導入された個体ではPCR産物に切れ残りが生じた。切れ残ったバンドからDNAを抽出・精製し、シークエンス解析にかけ、標的遺伝子配列に変異が導入されているものについて、標的遺伝子変異導入個体と判断した(図11)。その結果、全ての標的遺伝子について、ゲノム編集用ベクター導入後1週間目の茎頂中の細胞群における変異導入が確認された。
2.T0世代成葉及びT1世代幼葉における標的遺伝子変異導入確認
(1)コムギ完熟種子の調製
実施例13-1-(1)に記載の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例13-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
実施例13-1-(3)に記載の方法に従った。
(1)コムギ完熟種子の調製
実施例13-1-(1)に記載の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例13-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
実施例13-1-(3)に記載の方法に従った。
(4)遺伝子導入個体の生育
一晩静置した遺伝子導入個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。2週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて育成した。
一晩静置した遺伝子導入個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。2週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて育成した。
(5)T0植物葉における標的遺伝子変異導入の確認
得られた植物体において、標的遺伝子に変異が導入されているかを調査した。第5葉又は止葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、各遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
得られた植物体において、標的遺伝子に変異が導入されているかを調査した。第5葉又は止葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、各遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行った。
PCR反応液には、ゲノミックDNA0.7μL、KODFXNeo(TOYOBO)0.4U、2×添付バッファー5μL、0.4mMdNTPs、プライマー対各0.2μMを加え、滅菌蒸留水で合計10μLにフィルアップした。
PCRは、TaKaRa PCR Thermal Cycler Diceを用いて、98℃を10秒、68℃を1分の反応を1サイクルとしてこれを32サイクル行った。PCR/制限酵素解析は実施例13-1-(4)に記載の方法に従った。
野生型株ではPCR産物が制限酵素により完全に切断されるのに対し、変異が導入された個体ではPCR産物に切れ残りが生じた。切れ残ったバンドからDNAを抽出・精製し、シークエンス解析にかけ、標的遺伝子配列に変異が導入されているものについて、標的遺伝子変異導入個体と判断した。その結果、全ての標的遺伝子について、T0世代成葉における標的遺伝子変異導入を確認した(図12、表11)。

(6)T1植物葉における標的遺伝子変異導入の確認
上記(5)で変異が確認されたTaQsd1変異導入個体からT1種子を回収し、T1世代植物の幼葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出した。その後の標的遺伝子変異導入の確認は、上記(5)に記載の方法に従った。その結果、T1世代2系統について、T1世代幼葉における標的遺伝子変異導入を確認した。図13に、T1世代1系統における解析結果を示した。16個体中9個体について標的遺伝子に変異が導入されていることを確認した。当該遺伝子導入法を用いることで、茎頂中の生殖細胞系列へ移行する茎頂幹細胞をゲノム編集することが可能だとわかった。
上記(5)で変異が確認されたTaQsd1変異導入個体からT1種子を回収し、T1世代植物の幼葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出した。その後の標的遺伝子変異導入の確認は、上記(5)に記載の方法に従った。その結果、T1世代2系統について、T1世代幼葉における標的遺伝子変異導入を確認した。図13に、T1世代1系統における解析結果を示した。16個体中9個体について標的遺伝子に変異が導入されていることを確認した。当該遺伝子導入法を用いることで、茎頂中の生殖細胞系列へ移行する茎頂幹細胞をゲノム編集することが可能だとわかった。
[実施例15]当該遺伝子導入法を利用したCas9タンパク質導入による標的変異導入
当該遺伝子導入法を利用したgRNA/Cas9タンパク質導入によって、コムギに内在する標的遺伝子のゲノム編集が可能か否かを検証した。検証するにあたり、TaLOX2遺伝子を標的としたgRNA及びCas9タンパク質の複合体をコムギ茎頂へ導入した。
当該遺伝子導入法を利用したgRNA/Cas9タンパク質導入によって、コムギに内在する標的遺伝子のゲノム編集が可能か否かを検証した。検証するにあたり、TaLOX2遺伝子を標的としたgRNA及びCas9タンパク質の複合体をコムギ茎頂へ導入した。
1.遺伝子導入後一週間目における標的遺伝子変異導入確認
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv.Bobwhite)の完熟種子を用い、実施例1-1-(1)の方法に従った。
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv.Bobwhite)の完熟種子を用い、実施例1-1-(1)の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠して行った。
実施例1-1-(2)に記載の方法に準拠して行った。
(3)gRNA/Cas9タンパク質導入
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いた。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いた。
コムギ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いた。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いた。
crRNA:
GUGCCGCGCGACGAGCUCUUguuuuagagcuaugcuguuuug(配列番号18)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
GUGCCGCGCGACGAGCUCUUguuuuagagcuaugcuguuuug(配列番号18)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
上記各gRNA 4μg、Cas9タンパク質 7μg、10×Cas9バッファー2μL、Ribolock RNase Inhibitor 0.5μLを加え、滅菌蒸留水で合計20μLにフィルアップした。室温で15分間静置した後、金粒子750μgまたは1500μgを入れ、氷上で10分間静置した。上清を捨てた後、GFP発現用プラスミド(実施例1-1-(3)参照)2.5μgおよび滅菌蒸留水を32μL加え、これを金粒子/Cas9複合体と使用した。なお、金粒子径は0.6μmとし、金粒子量は1回の撃ち込みあたり、187.5または375μgになるように入れ、1プレートあたり4回の撃ち込みを行った。クリーンベンチ内でマクロキャリアの中央に、1.0~1.5cm角に切った親水性フィルム(3M、SH2CLHF)を貼りつけた。これに上記金粒子/Cas9複合体を8μLずつ注ぎ、風乾させた。撃ち込み条件は実施例1-1-(3)に従った。
(4)TaLOX2標的遺伝子配列切断による変異導入の確認
解析は実施例13-1-(4)に記載の方法に従った。
親水性フィルムを未使用時には、茎頂におけるGFP蛍光が観察されなかったのに対し、使用時にはGFP蛍光が観察された(表12)。さらに、1撃ち込み当たりの金粒子量を187.5μgから375μgに増やすことで、GFP蛍光の発現効率が23.3%から66.7%に向上した(表12)。親水性フィルムを使用することで、水系溶媒に分散させた金粒子が効率的に茎頂に導入されることがわかった。GFP蛍光が観察された個体について実施例13-1-(4)に従ってPCR/制限酵素解析を行った結果、野生型株ではPCR産物が制限酵素により完全に切断されるのに対し、変異が導入された個体ではPCR産物に切れ残りが生じた。切れ残ったバンドからDNAを抽出・精製し、シークエンス解析にかけた結果、標的遺伝子配列に変異が導入されていることが確認された(図14)。その結果、TaLOX2標的遺伝子について、gRNA/Cas9タンパク質導入後1週間目の茎頂中の細胞群における変異導入が確認された。
解析は実施例13-1-(4)に記載の方法に従った。
親水性フィルムを未使用時には、茎頂におけるGFP蛍光が観察されなかったのに対し、使用時にはGFP蛍光が観察された(表12)。さらに、1撃ち込み当たりの金粒子量を187.5μgから375μgに増やすことで、GFP蛍光の発現効率が23.3%から66.7%に向上した(表12)。親水性フィルムを使用することで、水系溶媒に分散させた金粒子が効率的に茎頂に導入されることがわかった。GFP蛍光が観察された個体について実施例13-1-(4)に従ってPCR/制限酵素解析を行った結果、野生型株ではPCR産物が制限酵素により完全に切断されるのに対し、変異が導入された個体ではPCR産物に切れ残りが生じた。切れ残ったバンドからDNAを抽出・精製し、シークエンス解析にかけた結果、標的遺伝子配列に変異が導入されていることが確認された(図14)。その結果、TaLOX2標的遺伝子について、gRNA/Cas9タンパク質導入後1週間目の茎頂中の細胞群における変異導入が確認された。

[実施例16]当該遺伝子導入法を利用したダイズにおける標的変異導入
ダイズについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。
ダイズについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。
1.ダイズ完熟種子の調製
ダイズ(フクユタカ、エンレイ)の完熟種子を用い、実施例7-1-(1)に記載の方法に従い調製する。
ダイズ(フクユタカ、エンレイ)の完熟種子を用い、実施例7-1-(1)に記載の方法に従い調製する。
2.完熟種子胚中の茎頂の露出
実施例7-1-(2)に記載の方法に従う。
実施例7-1-(2)に記載の方法に従う。
3.遺伝子導入
完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNA発現用プラスミドとして、pTAKN-2ベクターのマルチクローニングサイトへダイズU6プロモーター及びgRNA scaffoldをクローニングする。その後、内部のBbsIサイトへ標的遺伝子の標的配列を導入し、各gRNA発現用プラスミドとする。
Glyma.10G244400.1標的配列:CCTCCGCCCAAGGCTCCGCCACC (配列番号20)
Glyma.20G150000.1標的配列:CCTCCGCCCAAGGCTCCGCCACC(配列番号20)
完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNA発現用プラスミドとして、pTAKN-2ベクターのマルチクローニングサイトへダイズU6プロモーター及びgRNA scaffoldをクローニングする。その後、内部のBbsIサイトへ標的遺伝子の標的配列を導入し、各gRNA発現用プラスミドとする。
Glyma.10G244400.1標的配列:CCTCCGCCCAAGGCTCCGCCACC (配列番号20)
Glyma.20G150000.1標的配列:CCTCCGCCCAAGGCTCCGCCACC(配列番号20)
上記各gRNA発現用プラスミド、Cas9遺伝子発現用プラスミド及びGFP発現用プラスミド(実施例7-1-(3)参照)を金粒子750μgあたり各々5μg、2.5μg、2.5μg入れる。金粒子径は0.6μmとし、金粒子量は1回の撃ち込みあたり、187.5μgになるように入れる。
4.GFPタンパク質の一過的な発現を指標とした選抜
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)が見られた個体をRM培地(4.3g/L MS salt ,MS vitamin,3% スクロース,0.50g/L MES ,3%PPM(plant preservative mixture,ナカライテスク),0.3% ゲルライト,pH5.7)を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、インキュベーター(23℃、16時間日長)で育成する。
実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40 吸収:525/50)が見られた個体をRM培地(4.3g/L MS salt ,MS vitamin,3% スクロース,0.50g/L MES ,3%PPM(plant preservative mixture,ナカライテスク),0.3% ゲルライト,pH5.7)を入れたディスポーザブル植物細胞培養容器(Sigma)に移植し、インキュベーター(23℃、16時間日長)で育成する。
5.遺伝子導入個体の生育
実施例7-2-(1)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
得られた植物体において、標的遺伝子に変異が導入されているかを調査する。新たに生育した葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、各遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行う。PCR/制限酵素解析は実施例13-1-(4)に記載の方法に従って行う。変異が導入された個体をそのまま育成し、次世代の種子を取得することで、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例7-2-(1)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
得られた植物体において、標的遺伝子に変異が導入されているかを調査する。新たに生育した葉(50mg)から塩化ベンジル法を用いてゲノミックDNAを抽出し、これを鋳型として、各遺伝子に特異的な配列から作製したプライマーを用いてPCR反応を行う。PCR/制限酵素解析は実施例13-1-(4)に記載の方法に従って行う。変異が導入された個体をそのまま育成し、次世代の種子を取得することで、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例17]当該遺伝子導入法を利用したCas9タンパク質導入によるダイズへの標的変異導入
ダイズについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばGlyma.10G244400.1遺伝子を標的としたgRNA及びCas9タンパク質の複合体を茎頂へ導入する。
1.ダイズ完熟種子の調製
実施例15-1に記載の方法に従って行う。
2.完熟種子胚中の茎頂の露出
実施例15-2に記載の方法に従って行う。
ダイズについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばGlyma.10G244400.1遺伝子を標的としたgRNA及びCas9タンパク質の複合体を茎頂へ導入する。
1.ダイズ完熟種子の調製
実施例15-1に記載の方法に従って行う。
2.完熟種子胚中の茎頂の露出
実施例15-2に記載の方法に従って行う。
3.gRNA/Cas9タンパク質導入
ダイズ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GGUGGCGGAGCCUUGGGCGGguuuuagagcuaugcuguuuug(配列番号21)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
ダイズ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GGUGGCGGAGCCUUGGGCGGguuuuagagcuaugcuguuuug(配列番号21)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例15-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例15-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例15-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例15-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例18]当該遺伝子導入法を利用したCas9タンパク質導入によるイネへの標的変異導入
イネについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばOsPDS(イネphytoene desaturase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をイネ茎頂へ導入する。
イネについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばOsPDS(イネphytoene desaturase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をイネ茎頂へ導入する。
1.イネ完熟種子の調製
実施例10-1に記載の方法に従って行う。
2.完熟種子胚中の茎頂の露出
実施例10-2に記載の方法に従って行う。
実施例10-1に記載の方法に従って行う。
2.完熟種子胚中の茎頂の露出
実施例10-2に記載の方法に従って行う。
3.gRNA/Cas9タンパク質導入
イネ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GUUGGUCUUUGCUCCUGCAGguuuuagagcuaugcuguuuug(配列番号22)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
イネ完熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GUUGGUCUUUGCUCCUGCAGguuuuagagcuaugcuguuuug(配列番号22)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例10-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例10-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例10-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例10-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例19]当該遺伝子導入法を利用したCas9タンパク質導入によるリンゴへの標的変異導入
リンゴについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばリンゴPDS(phytoene desaturase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をリンゴ茎頂へ導入する。
リンゴについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばリンゴPDS(phytoene desaturase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をリンゴ茎頂へ導入する。
1.リンゴ茎頂の調製
実施例11-1に記載の方法に従う。
2.茎頂の露出
実施例11-2に記載の方法に従う。
3.gRNA/Cas9タンパク質導入
リンゴの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
ACCUGAUCGAGUAACUACAGguuuuagagcuaugcuguuuug(配列番号23)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。但し、実施例7-1-(3)に記載のGFPベクターを用いる。
実施例11-1に記載の方法に従う。
2.茎頂の露出
実施例11-2に記載の方法に従う。
3.gRNA/Cas9タンパク質導入
リンゴの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
ACCUGAUCGAGUAACUACAGguuuuagagcuaugcuguuuug(配列番号23)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。但し、実施例7-1-(3)に記載のGFPベクターを用いる。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例11-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例11-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例11-4の記載の方法に従う。
5.遺伝子導入個体の生育
実施例11-5に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例20]当該遺伝子導入法を利用したCas9タンパク質導入によるジャガイモへの標的変異導入
ジャガイモについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばStIAA2(an Auxin/Indole-3-Acetic Acid family member)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をジャガイモ茎頂へ導入する。
ジャガイモについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばStIAA2(an Auxin/Indole-3-Acetic Acid family member)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をジャガイモ茎頂へ導入する。
1.ジャガイモ茎頂の調製
実施例9-1に記載の方法に従う。
2.茎頂の露出
実施例9-2に記載の方法に従う。
実施例9-1に記載の方法に従う。
2.茎頂の露出
実施例9-2に記載の方法に従う。
3.gRNA/Cas9タンパク質導入
ジャガイモの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GAUGUUUAGCUCCUUUACUAguuuuagagcuaugcuguuuug(配列番号24)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。但し、実施例7-1-(3)に記載のGFPベクターを用いる。
ジャガイモの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GAUGUUUAGCUCCUUUACUAguuuuagagcuaugcuguuuug(配列番号24)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。但し、実施例7-1-(3)に記載のGFPベクターを用いる。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例9-(4)の記載の方法に従う。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例9-(4)の記載の方法に従う。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例21]当該遺伝子導入法を利用したCas9タンパク質導入によるトウモロコシへの標的変異導入
トウモロコシについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばZmALS2(トウモロコシacetolactate synthase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をトウモロコシ茎頂へ導入する。
トウモロコシについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばZmALS2(トウモロコシacetolactate synthase)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をトウモロコシ茎頂へ導入する。
1.トウモロコシ茎頂の調製
実施例6-1-(1)に記載の方法に従う。
2.茎頂の露出
実施例6-1-(2)に記載の方法に従う。
実施例6-1-(1)に記載の方法に従う。
2.茎頂の露出
実施例6-1-(2)に記載の方法に従う。
3.gRNA/Cas9タンパク質導入
トウモロコシの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GCUGCUCGAUUCCGUCCCCAguuuuagagcuaugcuguuuug(配列番号25)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
トウモロコシの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GCUGCUCGAUUCCGUCCCCAguuuuagagcuaugcuguuuug(配列番号25)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織においてGFP蛍光が観察されたものを選抜する。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織においてGFP蛍光が観察されたものを選抜する。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例22]当該遺伝子導入法を利用したCas9タンパク質導入によるオオムギへの標的変異導入
オオムギについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばHvPM19(オオムギABA-inducible plasma membrane protein)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をオオムギ茎頂へ導入する。
オオムギについても、本質的にはコムギと同様の手法により標的遺伝子変異導入個体を得ることができる。例えばHvPM19(オオムギABA-inducible plasma membrane protein)遺伝子を標的としたgRNA及びCas9タンパク質の複合体をオオムギ茎頂へ導入する。
1.オオムギ茎頂の調製
実施例8-1-(1)に記載の方法に従う。
2.茎頂の露出
実施例8-1-(2)に記載の方法に従う。
実施例8-1-(1)に記載の方法に従う。
2.茎頂の露出
実施例8-1-(2)に記載の方法に従う。
3.gRNA/Cas9タンパク質導入
オオムギの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GCUCUCCACUCUGGGCUCUUguuuuagagcuaugcuguuuug(配列番号26)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
オオムギの茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行う。
実施例においては、gRNAとしてcrRNA及びtracrRNA(ファスマック)の混合物(0.5μg/μL)を用いる。Cas9タンパク質として、Streptococcus pyogenes 由来EnGen Cas9 NLS(NEB)を用いる。
crRNA:
GCUCUCCACUCUGGGCUCUUguuuuagagcuaugcuguuuug(配列番号26)
tracrRNA:
AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU(配列番号19)
撃ち込み条件は、実施例14-1-(3)に記載の方法に従う。
4.GFPタンパク質の一過的な発現を指標とした選抜
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織においてGFP蛍光が観察されたものを選抜する。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
実施例1-1-(4)に記載の方法に従い、金粒子径0.6μmを用いて形質転換処理した個体中、茎頂組織においてGFP蛍光が観察されたものを選抜する。
5.遺伝子導入個体の生育
実施例1-2-(4)に記載の方法に従う。
6.T0植物葉における標的遺伝子変異導入の確認
実施例15-6に記載の方法に従って、安定的に標的遺伝子に変異が導入された個体を取得することができる。
[実施例23]トウモロコシ未熟胚を用いたトウモロコシにおける形質転換体の取得
1.蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ未熟胚の調製
全ての殻とシルクを取り除いた、2~5mmの胚を有する受粉10~20日後のトウモロコシA188の未熟穂を、70%エタノールに5分間浸漬した後、10%の漂白剤(8.25%の次亜塩素酸ナトリウム)と0.1%のTween 20を含む溶液に20分間浸漬し、滅菌水で洗浄した。洗浄後、滅菌フード内で5~10分間乾燥した。その後、滅菌済みメスを用いて、穂の上端から1/4の穀粒を削除し、スパチュラを用いて、穀粒から未熟胚を慎重に引き抜いた。新鮮な単離した未熟胚を、胚盤側を下にしてMSOプレート(MSO培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、8g/LのBacto-agar、pH 5.8に置床した(図15A)。25℃で2日間、明所(約50μmolm-2s-1)で培養し、子鞘葉が出現し始めたものを以下の実験に用いた(図15BおよびC)。
(1)トウモロコシ未熟胚の調製
全ての殻とシルクを取り除いた、2~5mmの胚を有する受粉10~20日後のトウモロコシA188の未熟穂を、70%エタノールに5分間浸漬した後、10%の漂白剤(8.25%の次亜塩素酸ナトリウム)と0.1%のTween 20を含む溶液に20分間浸漬し、滅菌水で洗浄した。洗浄後、滅菌フード内で5~10分間乾燥した。その後、滅菌済みメスを用いて、穂の上端から1/4の穀粒を削除し、スパチュラを用いて、穀粒から未熟胚を慎重に引き抜いた。新鮮な単離した未熟胚を、胚盤側を下にしてMSOプレート(MSO培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、8g/LのBacto-agar、pH 5.8に置床した(図15A)。25℃で2日間、明所(約50μmolm-2s-1)で培養し、子鞘葉が出現し始めたものを以下の実験に用いた(図15BおよびC)。
(2)未熟胚中の茎頂の露出
実体顕微鏡下において、微小ピンセット、及び微小針(33 G NanoPass針)を用いて、子葉鞘、第1葉原基、及び第2葉原基を除去し、茎頂部分を完全に露出させた。MSOボンバードメントプレートに簡単に配置できるよう、左右の胚盤をトリミングした。
実体顕微鏡下において、微小ピンセット、及び微小針(33 G NanoPass針)を用いて、子葉鞘、第1葉原基、及び第2葉原基を除去し、茎頂部分を完全に露出させた。MSOボンバードメントプレートに簡単に配置できるよう、左右の胚盤をトリミングした。
(3)遺伝子導入
トウモロコシ未熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
導入遺伝子としては、トウモロコシユビキチン1遺伝子由来のプロモーター(ZmUbi1)を用いて発現させるLpCpf1ヌクレアーゼ、及びd35Sプロモーター用いて発現させる蛍光レポーター遺伝子tdTomatoを含むプラスミドDNA(pGEP359)、およびトウモロコシHMG13を標的とし、ZmUbi1を用いて発現させるガイドRNAを含むプラスミドDNA(pGEP324)を用いた。
HMG13標的配列:CTCGTCACGATTCCCCTCTCC (配列番号27)
0.4μmまたは0.6μmの金粒子10mgをはかり取り、70%エタノール1mLを加え、ボルテックスにてよく懸濁した。その後、遠心により金粒子を沈殿させ、エタノールを除去した。その後、50%グリセロール1mLを加え滅菌金粒子溶液とした。
トウモロコシ未熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
導入遺伝子としては、トウモロコシユビキチン1遺伝子由来のプロモーター(ZmUbi1)を用いて発現させるLpCpf1ヌクレアーゼ、及びd35Sプロモーター用いて発現させる蛍光レポーター遺伝子tdTomatoを含むプラスミドDNA(pGEP359)、およびトウモロコシHMG13を標的とし、ZmUbi1を用いて発現させるガイドRNAを含むプラスミドDNA(pGEP324)を用いた。
HMG13標的配列:CTCGTCACGATTCCCCTCTCC (配列番号27)
0.4μmまたは0.6μmの金粒子10mgをはかり取り、70%エタノール1mLを加え、ボルテックスにてよく懸濁した。その後、遠心により金粒子を沈殿させ、エタノールを除去した。その後、50%グリセロール1mLを加え滅菌金粒子溶液とした。
50%グリセロール中の金粒子各100μLに、10μLのプラスミドDNA溶液(1.0μgのpGEP359および1.5μgのpGEP324)を加えた。滅菌金粒子含有溶液は使用前に、超音波発生器を使って念入りに懸濁し、上記チューブに適当量入れピペッティングにより攪拌した。次に、100μLの2.5M CaCl2、および40μLの0.1M Spermidineを加えた。混合後、室温で5~10分間、または4℃で30分間ボルテックスを続けた。DNA被覆金粒子を1分間沈降させ、最高速度で5秒間スピンして上清を除去し、500μLの100%エタノールで洗浄した。最後に、上清を除去して120μLの100%エタノールを添加して懸濁した(10回のボンバードメント用)。クリーンベンチ内でマクロキャリアの中央に10μLずつ注ぎ、風乾させた。
パーティクルガン(遺伝子銃)は、Biolistic(登録商標)PDS-1000/He Particle Delivery System(BIO-RAD)を使用し、撃ち込み時の圧力は約94.9kgf/cm2(1,350psi)とし、標的組織までの距離は5cm(粒子の直径0.6μm以上)または3.5cm(粒子の直径0.6μm未満)とした。1シャーレにつき4回ずつ撃ち込んだ(図16AおよびB)。
(4)tdTomatoタンパク質の一過的な発現効率の調査
撃ち込み20時間後に、実体蛍光顕微鏡(Nikon SMZ 18 with a DS-Ri 2 camera)下で茎頂におけるtdTomato蛍光(最大励起554nmおよび最大発光581nm)を観察した(図16C)。
撃ち込み20時間後に、実体蛍光顕微鏡(Nikon SMZ 18 with a DS-Ri 2 camera)下で茎頂におけるtdTomato蛍光(最大励起554nmおよび最大発光581nm)を観察した(図16C)。
[実施例24]トウモロコシ未熟胚を用いたトウモロコシにおける形質転換体の取得における撃ち込み前の浸透圧処理の影響
1.蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
1.蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
(2)未熟胚中の茎頂の露出
実施例23-1-(2)に記載の方法に従った。
実施例23-1-(2)に記載の方法に従った。
(3)遺伝子導入
撃ち込み前に、未熟胚を、浸透性ボンバードメントプレート(MSOSM培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、0.2Mのマンニトール(36.4g/L)+0.2Mのソルビトール(36.4g/L)、15g/LのBacto-agar、pH5.8)で、25℃で4時間暗所でインキュベートした(図17A)以外は、実施例23-1-(3)に記載の方法に従った。
撃ち込み前に、未熟胚を、浸透性ボンバードメントプレート(MSOSM培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、0.2Mのマンニトール(36.4g/L)+0.2Mのソルビトール(36.4g/L)、15g/LのBacto-agar、pH5.8)で、25℃で4時間暗所でインキュベートした(図17A)以外は、実施例23-1-(3)に記載の方法に従った。
(4)tdTomatoタンパク質の一過的な発現効率の調査
実施例23-1-(4)に記載の方法に従った。
浸透圧処理を行わない場合(図16C)と比較し、撃ち込み前の4時間の浸透圧処理を施した場合には、tdTomatoタンパク質由来の蛍光強度が向上した(図17C)。これより、浸透圧処理により、蛍光レポーター遺伝子の一過的な発現効率が向上することがわかった。
実施例23-1-(4)に記載の方法に従った。
浸透圧処理を行わない場合(図16C)と比較し、撃ち込み前の4時間の浸透圧処理を施した場合には、tdTomatoタンパク質由来の蛍光強度が向上した(図17C)。これより、浸透圧処理により、蛍光レポーター遺伝子の一過的な発現効率が向上することがわかった。
[実施例25]
トウモロコシ未熟胚を用いたトウモロコシにおける形質転換体の取得における撃ち込み前後の浸透圧処理の影響
1.蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
トウモロコシ未熟胚を用いたトウモロコシにおける形質転換体の取得における撃ち込み前後の浸透圧処理の影響
1.蛍光タンパク質の一過的発現系を指標とした調査
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
(2)未熟胚中の茎頂の露出
実施例23-1-(2)に記載の方法に従った。
実施例23-1-(2)に記載の方法に従った。
(3)遺伝子導入
撃ち込み前に、未熟胚を、浸透性ボンバードメントプレート(MSOSM培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、0.2Mのマンニトール(36.4g/L)+0.2Mのソルビトール(36.4g/L)、15g/LのBacto-agar、pH5.8)で、25℃で4時間暗所でインキュベートし、撃ち込み後に、未熟胚を、浸透性ボンバードメントプレートで、20時間暗所でインキュベートした(図18C)以外は、実施例23-1-(3)に記載の方法に従った。
撃ち込み前に、未熟胚を、浸透性ボンバードメントプレート(MSOSM培地:MS塩、MSビタミン、2g/Lのミオイノシトール、20g/Lのスクロース、0.2Mのマンニトール(36.4g/L)+0.2Mのソルビトール(36.4g/L)、15g/LのBacto-agar、pH5.8)で、25℃で4時間暗所でインキュベートし、撃ち込み後に、未熟胚を、浸透性ボンバードメントプレートで、20時間暗所でインキュベートした(図18C)以外は、実施例23-1-(3)に記載の方法に従った。
(4)tdTomatoタンパク質の一過的な発現効率の調査
実施例23-1-(4)に記載の方法に従った。
撃ち込み前の4時間の浸透圧処理のみを行った場合(図17C)に比べ、撃ち込み前の4時間の浸透圧処理、及び撃ち込み後の20時間の浸透圧処理を行った場合、tdTomatoタンパク質由来の蛍光強度が向上した(図18D)。これより、撃ち込み前後の浸透圧処理により、蛍光レポーター遺伝子の一過的な発現効率が飛躍的に向上することがわかった。
実施例23-1-(4)に記載の方法に従った。
撃ち込み前の4時間の浸透圧処理のみを行った場合(図17C)に比べ、撃ち込み前の4時間の浸透圧処理、及び撃ち込み後の20時間の浸透圧処理を行った場合、tdTomatoタンパク質由来の蛍光強度が向上した(図18D)。これより、撃ち込み前後の浸透圧処理により、蛍光レポーター遺伝子の一過的な発現効率が飛躍的に向上することがわかった。
[実施例26]トウモロコシ未熟の茎頂分裂組織におけるin plantaゲノム編集
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
(2)未熟胚中の茎頂の露出
実施例23-1-(2)に記載の方法に従った。
実施例23-1-(2)に記載の方法に従った。
(3)遺伝子導入
実施例25-1-(3)に記載の方法に従った。
(4)tdTomatoタンパク質の一過的な発現に基づく選択及び培養
撃ち込み20時間後に、実体蛍光顕微鏡(Nikon SMZ 18 with a DS-Ri 2 camera)下で茎頂におけるtdTomato蛍光(最大励起554nmおよび最大発光581nm)を観察し(図19A)、茎頂分裂組織において強い蛍光シグナルを有する胚を選択した(図19B)。
選択した胚を、ディスポーザブル植物細胞培養容器(Sigma)中のMSO培地上に移し、胚の発芽および成長のために25℃明所(50-100μmolm-2s-1)で培養した(図19C)。2-4枚の葉を有する生存種子を土壌に移し、25℃明所(200μmolm-2s-1)のグロースチャンバー内で成長させた。撃ち込みの40日後の写真を図19Dに示した。
実施例25-1-(3)に記載の方法に従った。
(4)tdTomatoタンパク質の一過的な発現に基づく選択及び培養
撃ち込み20時間後に、実体蛍光顕微鏡(Nikon SMZ 18 with a DS-Ri 2 camera)下で茎頂におけるtdTomato蛍光(最大励起554nmおよび最大発光581nm)を観察し(図19A)、茎頂分裂組織において強い蛍光シグナルを有する胚を選択した(図19B)。
選択した胚を、ディスポーザブル植物細胞培養容器(Sigma)中のMSO培地上に移し、胚の発芽および成長のために25℃明所(50-100μmolm-2s-1)で培養した(図19C)。2-4枚の葉を有する生存種子を土壌に移し、25℃明所(200μmolm-2s-1)のグロースチャンバー内で成長させた。撃ち込みの40日後の写真を図19Dに示した。
(5)標的ゲノム編集の評価
衝撃を受けた胚の茎頂を、撃ち込みの7日後に微小NanoPass針でサンプリングした(図20A)。サンプリングした茎頂を、DNA抽出のためにクリーンな1.7ml遠心管(低保持)に慎重に入れた。DNA単離のために、DNAzolダイレクト(Molecular Research Cent、Inc.、Chincinnati、OH、USA)を製造説明書に従い、用いた。以下のとおり、次世代シーケンサー(NGS、Illumina Miseq 150 PEplatform)を用いて部位特異的配列修飾を評価した(図20BおよびC)。
衝撃を受けた胚の茎頂を、撃ち込みの7日後に微小NanoPass針でサンプリングした(図20A)。サンプリングした茎頂を、DNA抽出のためにクリーンな1.7ml遠心管(低保持)に慎重に入れた。DNA単離のために、DNAzolダイレクト(Molecular Research Cent、Inc.、Chincinnati、OH、USA)を製造説明書に従い、用いた。以下のとおり、次世代シーケンサー(NGS、Illumina Miseq 150 PEplatform)を用いて部位特異的配列修飾を評価した(図20BおよびC)。
(ライブラリープレパレーション)
ライブラリーは、ターゲット領域を増幅し、シーケンスアダプターを追加する2工程PCRにより準備した。バーコードはプライマーを使用して設計され、サンプルの区別のために最初のPCR工程で追加した。アダプターは、シーケンスのために2回目のPCR工程で追加した。
ライブラリーは、ターゲット領域を増幅し、シーケンスアダプターを追加する2工程PCRにより準備した。バーコードはプライマーを使用して設計され、サンプルの区別のために最初のPCR工程で追加した。アダプターは、シーケンスのために2回目のPCR工程で追加した。
(次世代シーケンシング(NGS))
単位複製配列(アプリコン)は、Illumina Miseq 150 PEplatformを用いて決定した。ボンバードメントしたコムギ葉サンプルは50,000×カバレッジで配列決定した。
単位複製配列(アプリコン)は、Illumina Miseq 150 PEplatformを用いて決定した。ボンバードメントしたコムギ葉サンプルは50,000×カバレッジで配列決定した。
(データ解析)
QCおよび逆多重化の読取りには、FastQC + Jemultiplexer + Trimmomaticを使用し、標的におけるバリエーション識別にはCRISPRessoを使用し、イベント呼び出しの編集には、インハウスbash customer scriptを使用した。インハウスbash customer scriptを使用して、分析パイプライン全体を自動的に実行した。
QCおよび逆多重化の読取りには、FastQC + Jemultiplexer + Trimmomaticを使用し、標的におけるバリエーション識別にはCRISPRessoを使用し、イベント呼び出しの編集には、インハウスbash customer scriptを使用した。インハウスbash customer scriptを使用して、分析パイプライン全体を自動的に実行した。
NGSの結果は、バイオリスティックボンバードメントによって送達されたCpf1(pGEP359)およびガイドRNA(pGEP324)の一過性発現が、トウモロコシ未熟胚の茎頂分裂組織において有意なゲノム編集事象を生じさせることができることを示した(図21)。非衝撃対照と比較して、衝撃を受けた茎頂は標的部位に14.9%の挿入欠失率(InDel%)を含んでいた(図21A)。NGSの結果はまた、茎頂分裂組織におけるゲノム編集修飾のキメラ性を明らかにし、そこでは様々なサイズ(例えば5bp、6bp、および8bpなど)での複数の欠失が検出された(図21B)。
[実施例27]トウモロコシT0葉のin plantaゲノム編集
遺伝子導入方法がトウモロコシに存在する標的遺伝子のゲノム編集に使用できるかどうかを評価するために、MAD7によるhmg13遺伝子の突然変異誘発を試みた。トウモロコシ遺伝子型A188をhmg13遺伝子の突然変異誘発に使用した。
1.遺伝子導入後の茎頂分裂組織におけるマーカー遺伝子(mNeongreen)の確認
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
遺伝子導入方法がトウモロコシに存在する標的遺伝子のゲノム編集に使用できるかどうかを評価するために、MAD7によるhmg13遺伝子の突然変異誘発を試みた。トウモロコシ遺伝子型A188をhmg13遺伝子の突然変異誘発に使用した。
1.遺伝子導入後の茎頂分裂組織におけるマーカー遺伝子(mNeongreen)の確認
(1)トウモロコシ未熟胚の調製
実施例23-1-(1)に記載の方法に従った。
(2)未熟胚中の茎頂の露出
実施例23-1-(2)に記載の方法に従った。
実施例23-1-(2)に記載の方法に従った。
(3)遺伝子導入
解剖後、露出された茎頂分裂組織を有する胚をMS-maltoseボンバードメントプレート(MS-maltose培地)または浸透性ボンバードメントプレート(MSOSM培地)に移した。プレートをパラフィルムで覆い、ボンバードメント前にそれらを4時間25℃で暗所でインキュベートした。
トウモロコシ未熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
導入遺伝子は、トウモロコシユビキチン1遺伝子由来のプロモーター(ZmUbi1)を用いて発現させるMAD7ヌクレアーゼ、及びd35Sプロモーター用いて発現させる蛍光レポーター遺伝子mNeongreenを含むプラスミドDNA(pGEP837)、およびトウモロコシHMG13を標的とし、ZmUbi1を用いて発現させるガイドRNAを含むプラスミドDNA(pGEP842)を用いた。
HMG13標的配列:CTCGTCACGATTCCCCTCTCC (配列番号27)
解剖後、露出された茎頂分裂組織を有する胚をMS-maltoseボンバードメントプレート(MS-maltose培地)または浸透性ボンバードメントプレート(MSOSM培地)に移した。プレートをパラフィルムで覆い、ボンバードメント前にそれらを4時間25℃で暗所でインキュベートした。
トウモロコシ未熟胚の茎頂への遺伝子導入は、パーティクルガン法を用いて以下のようにして行った。
導入遺伝子は、トウモロコシユビキチン1遺伝子由来のプロモーター(ZmUbi1)を用いて発現させるMAD7ヌクレアーゼ、及びd35Sプロモーター用いて発現させる蛍光レポーター遺伝子mNeongreenを含むプラスミドDNA(pGEP837)、およびトウモロコシHMG13を標的とし、ZmUbi1を用いて発現させるガイドRNAを含むプラスミドDNA(pGEP842)を用いた。
HMG13標的配列:CTCGTCACGATTCCCCTCTCC (配列番号27)
0.6μmの金粒子30mgをはかり取り、70%エタノール500μLを加え、ボルテックスにてよく懸濁した。その後、遠心により金粒子を沈殿させ、エタノールを除去した。その後、500μLの蒸留水を加え滅菌金粒含有溶液とした。
プラスミドDNA溶液(7μgのpGEP837および3μgのpGEP842)を1.5mLの試験管に加えた。使用前に超音波発生装置(超音波洗浄機UW-25、多賀電気株式会社製)を用いて十分に懸濁した、滅菌金粒子含有溶液を、上記チューブに適量入れ、ピペッティングしながら攪拌した。次に、750μgの金粒子あたり25μLの2.5M CaCl2および10μLの0.1MSpermidineを上記チューブに加えた。混合直後に、得られた混合物をボルテックスミキサーを用いて5分間激しく懸濁した。混合物を室温で10分間放置し、次いで9,100×gで2秒間遠心分離した。上澄み液を除去し、沈殿物を70%エタノール、次いで99.5%エタノールで洗浄した。最後に上清を取り除き、99.5%エタノール24μLを加えてよく懸濁した。クリーンベンチ内で、6μLの懸濁液をマクロキャリアの中心に注ぎ、そして次にマクロキャリアを風乾させた。
プラスミドDNA溶液(7μgのpGEP837および3μgのpGEP842)を1.5mLの試験管に加えた。使用前に超音波発生装置(超音波洗浄機UW-25、多賀電気株式会社製)を用いて十分に懸濁した、滅菌金粒子含有溶液を、上記チューブに適量入れ、ピペッティングしながら攪拌した。次に、750μgの金粒子あたり25μLの2.5M CaCl2および10μLの0.1MSpermidineを上記チューブに加えた。混合直後に、得られた混合物をボルテックスミキサーを用いて5分間激しく懸濁した。混合物を室温で10分間放置し、次いで9,100×gで2秒間遠心分離した。上澄み液を除去し、沈殿物を70%エタノール、次いで99.5%エタノールで洗浄した。最後に上清を取り除き、99.5%エタノール24μLを加えてよく懸濁した。クリーンベンチ内で、6μLの懸濁液をマクロキャリアの中心に注ぎ、そして次にマクロキャリアを風乾させた。
パーティクルガン(遺伝子銃)は、Biolistic(登録商標)PDS-1000/He Particle Delivery System(BIO-RAD)を使用し、撃ち込み時の圧力は約94.9kgf/cm2(1,350psi)とし、標的組織までの距離は5cmとした。1シャーレにつき4回ずつ撃ち込んだ。ボンバードメント後、試料を25℃暗所で一晩放置した。
(4)mNeongreenタンパク質の一過的な発現効率の調査
撃ち込み20時間後に、実体蛍光顕微鏡下(Nikon SMZ 18 with a DS-Ri 2 camera)で茎頂におけるGFP蛍光(励起:470/40、吸収:525/50)を観察し(図22)、茎頂におけるmNeongreen遺伝子の導入効率を計算した。
形質転換プロセスを受けた未熟胚のうち、茎頂組織に観察される1つ以上の蛍光スポットを有するものをトランスジェニック個体とした。そして遺伝子導入効率を計算した(トランスジェニック個体数/処理された未熟胚の数×100)。その結果、MSOSM培地(撃ち込み前及び後の浸透圧処理あり)の場合(OS1~8)の茎頂における遺伝子導入効率は、MS-maltose培地(浸透圧処理なし)の場合(NT1~9)よりも高いことがわかった(表13)。
撃ち込み20時間後に、実体蛍光顕微鏡下(Nikon SMZ 18 with a DS-Ri 2 camera)で茎頂におけるGFP蛍光(励起:470/40、吸収:525/50)を観察し(図22)、茎頂におけるmNeongreen遺伝子の導入効率を計算した。
形質転換プロセスを受けた未熟胚のうち、茎頂組織に観察される1つ以上の蛍光スポットを有するものをトランスジェニック個体とした。そして遺伝子導入効率を計算した(トランスジェニック個体数/処理された未熟胚の数×100)。その結果、MSOSM培地(撃ち込み前及び後の浸透圧処理あり)の場合(OS1~8)の茎頂における遺伝子導入効率は、MS-maltose培地(浸透圧処理なし)の場合(NT1~9)よりも高いことがわかった(表13)。

(5)衝撃を受けた未熟胚の成長
一晩放置した衝撃を受けた全ての胚を、MSO培地を入れた植物細胞培養用の使い捨て容器(Sigma)に移し、長日条件(25℃、日照16時間)で1-2週間成長させた。
一晩放置した衝撃を受けた全ての胚を、MSO培地を入れた植物細胞培養用の使い捨て容器(Sigma)に移し、長日条件(25℃、日照16時間)で1-2週間成長させた。
(6)T0植物の葉における標的遺伝子変異導入の確認
以下の方法を用いて3葉以降の葉からゲノムDNAを抽出し、得られた植物体の目的遺伝子に変異が導入されているか否かを調べた。
以下の方法を用いて3葉以降の葉からゲノムDNAを抽出し、得られた植物体の目的遺伝子に変異が導入されているか否かを調べた。
-ゲノムDNAの抽出方法-
2mLチューブに葉組織(2-5mm2)を集め、300μLの抽出バッファー(20mM Tris-HCl pH7.5、25mM NaCl、2.5mM EDTA、0.05%SDS)をチューブに添加した。組織ホモジナイザー(Geno/Grinder)で組織を粉砕し、1,700×gで10分間遠心した。DNAテンプレートとして上清50μLを集めた。
2mLチューブに葉組織(2-5mm2)を集め、300μLの抽出バッファー(20mM Tris-HCl pH7.5、25mM NaCl、2.5mM EDTA、0.05%SDS)をチューブに添加した。組織ホモジナイザー(Geno/Grinder)で組織を粉砕し、1,700×gで10分間遠心した。DNAテンプレートとして上清50μLを集めた。
ゲノムDNAを鋳型として用い、各遺伝子に特異的な配列に基づいて作製したプライマーを用いてデジタルドロップレットPCRを行った。
その結果、MSOSM培地により浸透圧処理を行った場合において、得られたT0植物における標的遺伝子突然変異誘発の効率(19.6%、OS1-8)は、MS-maltose培地の場合(2.1%、NT1-9)より顕著に高かった(表13)。
さらに、浸透圧処理は、浸透圧未処理と比較して、高いInDel%(70%<)の遺伝子編集植物の比率を増加させた(表14)。
その結果、MSOSM培地により浸透圧処理を行った場合において、得られたT0植物における標的遺伝子突然変異誘発の効率(19.6%、OS1-8)は、MS-maltose培地の場合(2.1%、NT1-9)より顕著に高かった(表13)。
さらに、浸透圧処理は、浸透圧未処理と比較して、高いInDel%(70%<)の遺伝子編集植物の比率を増加させた(表14)。

[実施例28]トウモロコシT1葉のin plantaゲノム編集
実施例27-1-(6)で得られたOS1~8に由来するT0植物体を生育させ、次世代であるT1植物に、hmg13遺伝子の変異が遺伝しているかを確認した。
8系統のT0植物体から収穫したT1種子を播種し、幼葉をサンプリングした。実施例27-1-(6)に記載の方法にDNAを抽出し、次世代シーケンサー(NGS)を用いて部位特異的配列修飾を評価した(表15)。その結果、8系統中6系統において、標的遺伝子の変異を確認し、1系統においてはホモ変異体であった。このことから、トウモロコシの未熟胚を材料に、浸透圧処理を施すことにより、in planta法を用いた場合においても高効率に次世代のゲノム編集個体を取得できることがわかった。
実施例27-1-(6)で得られたOS1~8に由来するT0植物体を生育させ、次世代であるT1植物に、hmg13遺伝子の変異が遺伝しているかを確認した。
8系統のT0植物体から収穫したT1種子を播種し、幼葉をサンプリングした。実施例27-1-(6)に記載の方法にDNAを抽出し、次世代シーケンサー(NGS)を用いて部位特異的配列修飾を評価した(表15)。その結果、8系統中6系統において、標的遺伝子の変異を確認し、1系統においてはホモ変異体であった。このことから、トウモロコシの未熟胚を材料に、浸透圧処理を施すことにより、in planta法を用いた場合においても高効率に次世代のゲノム編集個体を取得できることがわかった。

[実施例29]コムギin plantaゲノム編集における浸透圧処理の効果
実施例27で確認されたin plantaゲノム編集における浸透圧処理の効果が、コムギにおいても見られるか調査した。
1.遺伝子導入後一週間目における標的遺伝子変異導入確認
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Haruyokoi)の完熟種子を用い、実施例1-1-(1)の方法に従った。
実施例27で確認されたin plantaゲノム編集における浸透圧処理の効果が、コムギにおいても見られるか調査した。
1.遺伝子導入後一週間目における標的遺伝子変異導入確認
(1)コムギ完熟種子の調製
コムギ(Triticum aestivum cv. Haruyokoi)の完熟種子を用い、実施例1-1-(1)の方法に従った。
(2)完熟種子胚中の茎頂の露出
実施例1-1-(2)に記載の方法に準拠して行った。
実施例1-1-(2)に記載の方法に準拠して行った。
(3)遺伝子導入
コムギ完熟胚の茎頂への遺伝子導入は、実施例14-1-(3)に準拠して行った。ただし、標的遺伝子としてTaGASR7を用い、浸透圧処理としては実施例27-1-(3)の方法に準拠して行った。
(4)GFPタンパク質の一過的発現の観察
撃ち込み20時間後に、実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40、吸収:525/50)を観察した。その結果、MSOSM培地(撃ち込み前及び後の浸透圧処理あり)の場合、MS-maltose培地(浸透圧処理なし)の場合に比べ、完熟胚におけるGFPタンパク質の一過的発現の強度が増加していた(図23)。
コムギ完熟胚の茎頂への遺伝子導入は、実施例14-1-(3)に準拠して行った。ただし、標的遺伝子としてTaGASR7を用い、浸透圧処理としては実施例27-1-(3)の方法に準拠して行った。
(4)GFPタンパク質の一過的発現の観察
撃ち込み20時間後に、実体蛍光顕微鏡(Leica、MZFLIII)下で茎頂におけるGFP蛍光(励起:470/40、吸収:525/50)を観察した。その結果、MSOSM培地(撃ち込み前及び後の浸透圧処理あり)の場合、MS-maltose培地(浸透圧処理なし)の場合に比べ、完熟胚におけるGFPタンパク質の一過的発現の強度が増加していた(図23)。
(5)遺伝子導入個体の生育
一晩静置した遺伝子導入個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。2週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて育成した。
(6)T0植物葉における標的遺伝子変異導入の確認
得られた植物体において、標的遺伝子に変異が導入されているかを調査した。第5葉をサンプリングし、実施例27-1-(6)に記載の方法にDNAを抽出した。DNA抽出以降、実施例14-2-(5)の記載の方法に従い、TaGASR7遺伝子における変異導入の有無を調査した。
一晩静置した遺伝子導入個体をMS-maltose培地を入れたディスポ-ザブル植物細胞培養容器(Sigma)に移植し、長日(22℃、16時間日長)で育成した。2週間生育させた後、第2-3葉が観察された時点で、園芸用育苗培土を入れたポットへ移植した。その後、長日の人工気象室(24℃、16時間日長、湿度50-70%)にて育成した。
(6)T0植物葉における標的遺伝子変異導入の確認
得られた植物体において、標的遺伝子に変異が導入されているかを調査した。第5葉をサンプリングし、実施例27-1-(6)に記載の方法にDNAを抽出した。DNA抽出以降、実施例14-2-(5)の記載の方法に従い、TaGASR7遺伝子における変異導入の有無を調査した。
野生型株ではPCR産物が制限酵素により完全に切断されるのに対し、変異が導入された個体ではPCR産物に切れ残りが生じる。切れ残ったバンドからDNAを抽出・精製し、シークエンス解析にかけ、標的遺伝子配列に変異が導入されているものについて、標的遺伝子変異導入個体と判断した。その結果、MSOSM培地(撃ち込み前及び後の浸透圧処理あり)の場合(16.3%)、MS-maltose培地(浸透圧処理なし)の場合(2.4%)に比べ、T0葉における標的遺伝子変異導入の効率が向上することがわかった(表16)。実施例27のトウモロコシにおける結果と同様に、コムギにおいても浸透圧処理によるゲノム編集効率の向上が確認された。

本発明は、農業、医薬産業、酵素産業などに利用できる。
Al アリューロン層
E 胚乳
E 胚乳
Claims (28)
- 植物の改変方法であって、
微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、
該被覆した微粒子を植物の茎頂に撃ち込む工程と、
該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、
該植物体から改変された植物体を選択する工程とを含み、
前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。 - 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質を含まない培地で行われる、請求項1に記載の方法。
- 前記被覆した微粒子を撃ち込んだ茎頂の生育は、植物ホルモンを含まない培地で行われる、請求項1に記載の方法。
- 前記被覆した微粒子を撃ち込んだ茎頂の生育は、抗生物質、及び植物ホルモンを含まない培地で行われる、請求項1に記載の方法。
- 前記植物の茎頂への撃ち込みは、茎頂のL2層細胞に被覆した微粒子を撃ち込むことによって行われる、請求項1から4のいずれかに記載の方法。
- 前記植物の茎頂への撃ち込みは、茎頂の生殖細胞系列へ移行する茎頂幹細胞に被覆した微粒子を撃ち込むことによって行われる、請求項1から4のいずれかに記載の方法。
- 前記微粒子が、直径0.3μm以上1.5μm以下である、請求項1から6のいずれかに記載の方法。
- 前記植物の茎頂は、前記(1)完熟種子の胚の茎頂であり、
前記(1)完熟種子の胚の茎頂が、該完熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、請求項1から7のいずれかに記載の方法。 - 前記植物の茎頂は、前記(2)幼芽の茎頂であり、
前記(2)幼芽の茎頂が、該幼芽から塊茎、葉原基を除去し、露出させた茎頂である、請求項1から7のいずれかに記載の方法。 - 前記植物の茎頂は、前記(3)頂芽又は側芽の茎頂であり、
前記(3)頂芽又は側芽の茎頂が、頂芽または側芽から葉原基を除去し、露出させた茎頂である、請求項1から7のいずれかに記載の方法。 - 前記植物の茎頂は、(4)未熟胚の茎頂であり、
前記(4)未熟胚の茎頂が、受粉後8日から35日の未熟種子から胚乳、鞘葉、葉原基、および余分な胚盤を除去し、露出させた茎頂である、請求項1から7のいずれかに記載の方法。 - 前記微粒子の被覆は、微粒子を導入目的とする核酸カセットを含む線状DNAで被覆することにより行われる、請求項1から11のいずれかに記載の方法。
- 前記線状DNAは線状プラスミドであり、それぞれ核酸カセットの各末端に位置する0.8kb以上1.2kb以下の核酸をさらに含む、請求項12に記載の方法。
- 前記微粒子の被覆は、微粒子を修飾酵素をコードする核酸で被覆することにより行われる、請求項1から11のいずれかに記載の方法。
- 前記修飾酵素が、ヌクレアーゼまたはデアミナーゼである、請求項14に記載の方法。
- 前記微粒子被覆は、微粒子をプロモーターおよびターミネーターにそれぞれ連結された核酸で被覆することにより行われ、
前記核酸は、少なくとも1つのガイドRNAを発現することができる核酸およびCasヌクレアーゼタンパク質をコードする核酸を含む、請求項1から11のいずれかに記載の方法。 - 前記微粒子の被覆は、微粒子をヌクレアーゼで被覆することにより行われる、請求項1から11のいずれかに記載の方法。
- 前記ヌクレアーゼがCasヌクレアーゼである、請求項17に記載の方法。
- 前記微粒子の被覆は、微粒子をタンパク質で被覆することにより行われ、
前記植物の茎頂への撃ち込みは、タンパク質で被覆された微粒子を有する親水性マクロキャリアフィルムを用いて行われる、請求項1から11のいずれかに記載の方法。 - 前記完熟種子は、長さが1mm以下の根を含む完熟種子である、請求項1から19のいずれかに記載の方法。
- 前記植物が、コムギ、オオムギ、イネ、トウモロコシ、ダイズ、ジャガイモおよびリンゴからなる群から選択される、請求項1から20のいずれかに記載の方法。
- 前記完熟種子の胚の茎頂、幼芽の茎頂、頂芽又は側芽の茎頂、または未熟胚の茎頂を高浸透圧溶液に接触させる工程をさらに含む、請求項1から21のいずれかに記載の方法。
- 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前または後である、請求項22に記載の方法。
- 前記高浸透圧溶液に接触させる工程が、前記被覆した微粒子を撃ち込む工程の前および後である、請求項22に記載の方法。
- 改変された植物体を成長させる工程をさらに含む、請求項1から24のいずれかに記載の方法。
- 植物のゲノム編集方法であって、
微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、
該被覆した微粒子を植物の茎頂に撃ち込む工程と、
該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、
該植物体から改変された植物体を選択する工程とを含み、
前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であり、
前記ゲノムは、茎頂分裂組織中の生殖細胞系列またはそれに移行し得る幹細胞内に存在し、
前記方法はインプランタ法であることを特徴とする方法。 - 改変された植物体を成長させる工程をさらに含む、請求項26に記載の方法。
- 植物の作製方法であって、
微粒子を、少なくとも1種の核酸および/または少なくとも1種のタンパク質により被覆する工程と、
該被覆した微粒子を植物の茎頂に撃ち込む工程と、
該被覆した微粒子を撃ち込んだ茎頂を生育し、植物体を得る工程と、
該植物体から改変された植物体を選択する工程とを含み、
前記茎頂は、(1)完熟種子の胚の茎頂、(2)幼芽の茎頂、(3)頂芽又は側芽の茎頂、または(4)未熟胚の茎頂であることを特徴とする方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20910123.7A EP4086353A4 (en) | 2020-01-03 | 2020-12-28 | METHOD FOR PLANT MODIFICATION |
| CN202080091555.5A CN115087739A (zh) | 2020-01-03 | 2020-12-28 | 植物的修饰方法 |
| JP2021568473A JP7665906B2 (ja) | 2020-01-03 | 2020-12-28 | 植物の改変方法、植物のゲノム編集方法、及び植物の作製方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/733,993 US11499158B2 (en) | 2016-05-13 | 2020-01-03 | Method for modifying plant |
| US16/733,993 | 2020-01-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021137299A1 true WO2021137299A1 (ja) | 2021-07-08 |
Family
ID=76685922
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/049058 Ceased WO2021137299A1 (ja) | 2020-01-03 | 2020-12-28 | 植物の改変方法 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4086353A4 (ja) |
| JP (1) | JP7665906B2 (ja) |
| CN (1) | CN115087739A (ja) |
| WO (1) | WO2021137299A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI826265B (zh) * | 2023-02-17 | 2023-12-11 | 國立成功大學 | 植物表面修飾物及修飾方法 |
| GB2628406A (en) * | 2023-03-23 | 2024-09-25 | Green Bioactives Ltd | Methods of using vascular plant stem cells |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004506427A (ja) * | 2000-08-11 | 2004-03-04 | シンジェンタ パーティシペーションズ アクチェンゲゼルシャフト | 植物の安定した形質転換の方法 |
| WO2005024034A1 (ja) | 2003-09-08 | 2005-03-17 | Kumiai Chemical Industry Co., Ltd. | 植物のインプランタ形質転換法 |
| JP2008212048A (ja) * | 2007-03-02 | 2008-09-18 | National Agriculture & Food Research Organization | コムギにおける導入された目的遺伝子の発現効率を向上させる方法 |
| WO2017090761A1 (ja) * | 2015-11-27 | 2017-06-01 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換する、単子葉植物のゲノム配列の変換方法、及びそれに用いる分子複合体 |
| JP2017205104A (ja) * | 2016-05-13 | 2017-11-24 | 株式会社カネカ | 植物のゲノム編集方法 |
| JP2017205103A (ja) * | 2016-05-13 | 2017-11-24 | 株式会社カネカ | 形質転換植物の作製方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3095870A1 (en) * | 2015-05-19 | 2016-11-23 | Kws Saat Se | Methods for the in planta transformation of plants and manufacturing processes and products based and obtainable therefrom |
| CN112585269B (zh) * | 2018-06-15 | 2025-01-07 | 科沃施种子欧洲股份两合公司 | 改善植物中基因组工程化和再生的方法ii |
-
2020
- 2020-12-28 WO PCT/JP2020/049058 patent/WO2021137299A1/ja not_active Ceased
- 2020-12-28 JP JP2021568473A patent/JP7665906B2/ja active Active
- 2020-12-28 EP EP20910123.7A patent/EP4086353A4/en active Pending
- 2020-12-28 CN CN202080091555.5A patent/CN115087739A/zh active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004506427A (ja) * | 2000-08-11 | 2004-03-04 | シンジェンタ パーティシペーションズ アクチェンゲゼルシャフト | 植物の安定した形質転換の方法 |
| WO2005024034A1 (ja) | 2003-09-08 | 2005-03-17 | Kumiai Chemical Industry Co., Ltd. | 植物のインプランタ形質転換法 |
| JP2008212048A (ja) * | 2007-03-02 | 2008-09-18 | National Agriculture & Food Research Organization | コムギにおける導入された目的遺伝子の発現効率を向上させる方法 |
| WO2017090761A1 (ja) * | 2015-11-27 | 2017-06-01 | 国立大学法人神戸大学 | 標的化したdna配列の核酸塩基を特異的に変換する、単子葉植物のゲノム配列の変換方法、及びそれに用いる分子複合体 |
| JP2017205104A (ja) * | 2016-05-13 | 2017-11-24 | 株式会社カネカ | 植物のゲノム編集方法 |
| JP2017205103A (ja) * | 2016-05-13 | 2017-11-24 | 株式会社カネカ | 形質転換植物の作製方法 |
Non-Patent Citations (5)
| Title |
|---|
| BILANG ET AL.: "Transient gene expression in vegetative shot apical meristems of wheat after ballistic microtargeting", PLANT JOURNAL, vol. 4, 1993, pages 735 - 744, XP055555119, DOI: 10.1046/j.1365-313X.1993.04040735.x |
| HAMADA HARUYASU, LINGHU QIANYAN, NAGIRA YOZO, MIKI RYUJI, TAOKA NAOAKI, IMAI RYOZO: "An in planta biolistic method for stable wheat transformation", SCIENTIFIC REPORTS, vol. 7, no. 11443, 2017, pages 1 - 8, XP055838382 * |
| PLANT TISSUE CULTURE LETTERS, vol. 9, no. 2, 1992, pages 69 - 73 |
| See also references of EP4086353A4 |
| SUPARTANA ET AL.: "Development of simple and efficient in planta transformation for rice (Oryza sativa L.) using Agrobacterium tumefaciencs", JOURNAL OF BIOSCIENCE AND, vol. 4, 2005, pages 391 - 397, XP005667212, DOI: 10.1263/jbb.100.391 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4086353A1 (en) | 2022-11-09 |
| CN115087739A (zh) | 2022-09-20 |
| EP4086353A4 (en) | 2024-03-06 |
| JPWO2021137299A1 (ja) | 2021-07-08 |
| JP7665906B2 (ja) | 2025-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7321477B2 (ja) | 植物のゲノム編集方法 | |
| JP6967217B2 (ja) | 形質転換植物の作製方法 | |
| EP3095870A1 (en) | Methods for the in planta transformation of plants and manufacturing processes and products based and obtainable therefrom | |
| CN103975067B (zh) | 幼体和成熟柑橘植物的转化 | |
| JP2018503392A (ja) | 遺伝子一過性発現により完全な植物において部位特異的改変を行うための方法 | |
| JP2018504133A (ja) | 色素体形質転換方法 | |
| JP7665906B2 (ja) | 植物の改変方法、植物のゲノム編集方法、及び植物の作製方法 | |
| WO2021108337A1 (en) | Methods of transformation | |
| CN112522283B (zh) | 一种花粉发育相关基因及其应用 | |
| US11499158B2 (en) | Method for modifying plant | |
| JP7616514B2 (ja) | 腋芽メリステムを用いた植物の改変方法 | |
| JP7605390B2 (ja) | コムギのゲノム編集方法、及びその利用 | |
| US20240052002A1 (en) | Tomato-derived sijul gene regulating phloem development and use thereof | |
| WO2022154115A1 (ja) | 形質転換またはゲノム編集された次世代植物体の製造方法 | |
| CN116286963B (zh) | 耐盐相关蛋白GhIPUT1及其编码基因的应用 | |
| WO2025079531A1 (ja) | ゲノム編集方法及びゲノム編集された植物の製造方法 | |
| Wani et al. | Transgenesis: An efficient tool in mulberry breeding | |
| AU2024361001A1 (en) | Genome editing method and method for producing genome-edited plant | |
| CN121825996A (zh) | 辣椒细胞周期基因CaTAM及其应用 | |
| WO2022186295A1 (ja) | 受精を介さず種子植物の胚発生を誘導する方法、それに用いられるタンパク質、核酸、及びベクター、並びに受精を介さず胚を発生しうる組換え種子植物 | |
| DEEPAK | MASTER OF SCIENCE (Agriculture) | |
| Desai et al. | I nternational Journal of Scientific Research and Reviews |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20910123 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021568473 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020910123 Country of ref document: EP Effective date: 20220803 |