WO2021244557A1 - 分析来自细胞的目标核酸的方法 - Google Patents
分析来自细胞的目标核酸的方法 Download PDFInfo
- Publication number
- WO2021244557A1 WO2021244557A1 PCT/CN2021/097800 CN2021097800W WO2021244557A1 WO 2021244557 A1 WO2021244557 A1 WO 2021244557A1 CN 2021097800 W CN2021097800 W CN 2021097800W WO 2021244557 A1 WO2021244557 A1 WO 2021244557A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nucleic acid
- target nucleic
- sequence
- strand
- attached
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6816—Hybridisation assays characterised by the detection means
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
Definitions
- This application relates to the field of biomedicine, in particular to a method for analyzing target nucleic acids from cells and related preparations.
- nucleic acid sequencing technology has undergone rapid and tremendous progress. Sequencing technology generates a large amount of sequence data, which can be used for research and interpretation of genomes and genomic regions, and provides information that is widely used in conventional biological research and diagnosis. Genome sequencing can be used to obtain information on a variety of biomedical backgrounds, including diagnostics, prognosis, biotechnology, and forensic biology. Sequencing includes Maxam-Gilbert sequencing and chain termination or de novo sequencing (including shotgun sequencing and bridge PCR), or next-generation methods, including polymerase clone sequencing, 454 pyrosequencing, Illumina sequencing, SOLiD sequencing, Ion Torrent Semiconductor sequencing, HeliScope single molecule sequencing, [image] sequencing, etc. For most sequencing applications, samples such as nucleic acid samples are processed before being introduced into the sequencer.
- the final signal value is the average of multiple cells, and the information of cell heterogeneity is lost.
- the current analysis of the mRNA content of cells by direct sequencing relies on the analysis of a large amount of mRNA obtained from a tissue sample containing millions of cells, which means that when the gene expression is analyzed in a large amount of mRNA, the expression in a single cell A lot of functional information will be lost or blurred; in addition, dynamic processes such as the cell cycle cannot be observed based on the overall average.
- certain cell types in complex tissues for example, the brain can only be studied by analyzing cells individually.
- This application provides a method for analyzing target nucleic acid from a cell, the method comprising:
- a target nucleic acid derived from a single cell wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid;
- a solid support attached with at least one oligonucleotide tag wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and is located in the barcode sequence 3'end of the hybridization sequence, the second strand includes a first portion complementary to the hybridization sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid Part, and the first strand and the second strand form a partially double-stranded structure or the second strand and the attached target nucleic acid form a partially double-stranded structure;
- the oligonucleotide tag is connected to the attached target nucleic acid, thereby generating a barcoded target nucleic acid.
- the oligonucleotide tag is releasably attached to the solid support.
- it includes releasing the at least one oligonucleotide tag from the solid support, and in b) making the released oligonucleotide tag and the attached target Nucleic acids are linked to produce barcoded target nucleic acids.
- the oligonucleotide tag is directly or indirectly attached to the solid support through the 5' end of its first strand.
- a ligase is further included in the discrete partition, and the ligase connects the oligonucleotide tag to the attached target nucleic acid.
- the ligase includes T4 ligase.
- the target nucleic acid sequence is located at the 3'end of the barcode sequence.
- the solid support is a bead.
- the beads are magnetic beads.
- the discrete partitions are holes or droplets.
- the barcode sequence includes a cell barcode sequence, and each oligonucleotide tag attached to the same solid support contains the same cell barcode sequence.
- the cell barcode sequence comprises at least 2 cell barcode segments separated by a linker sequence.
- a) includes co-distributing the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag into the discrete partitions.
- b) includes connecting the hybridizing sequence of the first strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the Barcoded target nucleic acid.
- b) includes hybridizing the second portion of the second strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, and allowing the The hybridization sequence of the first strand of the oligonucleotide tag is connected to the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the barcoded target nucleic acid.
- the attached target nucleic acid includes a unique molecular identification region.
- the unique molecular identification region is located between the oligonucleotide adaptor sequence and the target nucleic acid sequence.
- the oligonucleotide tag further includes an amplification primer recognition region.
- the amplification primer recognition region is a universal amplification primer recognition region.
- the method further includes:
- the method further includes, after b) and before c), releasing the barcoded target nucleic acid from the discrete partition.
- c) includes sequencing the barcoded target nucleic acid to obtain the characterization result.
- the method further comprises assembling contiguous nucleic acid sequences of at least a portion of the genome of the single cell from the sequence of the barcoded target nucleic acid.
- the single cell is characterized based on the nucleic acid sequence of at least a portion of the genome of the single cell.
- each of the discrete partitions includes at most the target nucleic acid derived from a single cell.
- the method further includes identifying a single nucleic acid sequence in the barcoded target nucleic acid as derived from a given nucleic acid in the target nucleic acid based at least in part on the existence of the unique molecular identification region.
- the target nucleic acid includes an exogenous nucleic acid
- the exogenous nucleic acid includes an exogenous nucleic acid linked to a protein, lipid, and/or small molecule compound, and the protein, lipid, and/or small molecule The compound can bind to the target molecule in the cell.
- the method further comprises determining the amount of a given nucleic acid in the target nucleic acid based on the presence of the unique molecular identification region.
- it includes pre-treating the cells before a).
- the pretreatment includes fixing the cells.
- the cells are fixed using a fixative, and the fixative is selected from one or more of the following group: formaldehyde, paraformaldehyde, methanol, ethanol, acetone, glutaraldehyde, osmium Acid and potassium dichromate.
- the fixative is selected from one or more of the following group: formaldehyde, paraformaldehyde, methanol, ethanol, acetone, glutaraldehyde, osmium Acid and potassium dichromate.
- the pretreatment includes exposing the nucleus of the cell.
- the pretreatment includes treating the cells with a detergent, the detergent including Triton, Tween, SDS, NP-40, and/or digitonin.
- the target nucleic acid includes one or more selected from the group consisting of DNA, RNA and cDNA.
- it further includes, after b) and before c), amplifying the barcoded target nucleic acid.
- the barcoded target nucleic acid is released from the discrete partition, and the amplification is performed after the barcoded target nucleic acid is released from the discrete After the partition is released.
- amplification primers are used in the amplification, and random guide sequences are included in the amplification primers.
- the random leader sequence is a random hexamer.
- the amplifying includes at least partially hybridizing the random leader sequence with the barcoded target nucleic acid and extending the random leader sequence in a template-directed manner.
- it includes releasing at least a portion of the target nucleic acid from the single cell in the discrete partition to the outside of the cell, and in b) the released target nucleic acid and the oligonucleotide
- the nucleotide tag is connected to produce a barcoded target nucleic acid.
- it includes allowing at least a portion of the oligonucleotide tag released from the solid support to enter the single cell, and to link with the target nucleic acid in b), thereby generating a barcoded Target nucleic acid.
- it includes using a microfluidic device to co-distribute the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag into the discrete partitions.
- the discrete partitions are droplets
- the microfluidic device is a droplet generator.
- the microfluidic device includes a first input channel and a second input channel that meet at a junction fluidly connected to the output channel.
- the method further includes introducing a sample containing the target nucleic acid into the first input channel, and introducing the solid support to which at least one oligonucleotide tag is attached into the first input channel. Two input channels, thereby generating a mixture of the sample and the solid support in the output channel.
- the output channel and the third input channel are fluidly connected at the junction.
- it further includes introducing oil into the third input channel, so that aqueous droplets in the water-in-oil emulsion are formed as the discrete partitions.
- each of the discrete partitions contains at most the target nucleic acid from a single cell.
- the first input channel and the second input channel form a substantially perpendicular angle to each other.
- the target nucleic acid includes cDNA derived from RNA in the single cell.
- the RNA includes mRNA.
- it includes reverse transcription of the RNA before a) and the production of the attached target nucleic acid.
- a reverse transcription primer is used in the reverse transcription, and the reverse transcription primer includes the oligonucleotide adaptor sequence and the polyT sequence in a 5'to 3'direction.
- the reverse transcription includes hybridizing the polyT sequence with the RNA and extending the polyT sequence in a template-directed manner.
- the target nucleic acid includes DNA derived from the single cell.
- the DNA includes genomic DNA, open chromatin DNA, protein-bound DNA regions, and/or exogenous nucleic acids linked to proteins, lipids, and/or small molecule compounds. And/or small molecule compounds can bind to target molecules in cells.
- it includes fragmenting the DNA derived from a single cell before a).
- the attached target nucleic acid is produced after or during the fragmentation.
- the fragmentation includes the use of ultrasonic fragmentation, and then adding a sequence containing the oligonucleotide adaptor to the fragmented DNA, thereby obtaining the attached target nucleic acid.
- the fragmentation includes using DNA endonuclease or DNA exonuclease to break, and then adding a sequence containing the oligonucleotide adaptor to the fragmented DNA to obtain the Attached target nucleic acid
- the fragmentation includes using a transposase-nucleic acid complex to integrate the sequence comprising the oligonucleotide adaptor into the DNA, and releasing the transposase to obtain the Attached target nucleic acid.
- the transposase-nucleic acid complex includes a transposase and a transposon end nucleic acid molecule, wherein the transposon end nucleic acid molecule includes the oligonucleotide adaptor sequence.
- the transposase includes Tn5.
- the DNA includes a DNA region that binds to a protein
- the transposase-nucleic acid complex also includes a portion that directly or indirectly recognizes the protein.
- the part that directly or indirectly recognizes the protein includes one or more of the following group: an antibody that specifically binds to the protein and protein A or protein G.
- the present application also provides a composition
- a composition comprising: a plurality of solid supports, each of which is attached with at least one oligonucleotide tag, wherein each of the oligonucleotides
- the acid tag includes a first strand and a second strand
- the first strand includes a barcode sequence and a hybridization sequence located at the 3'end of the barcode sequence
- the second strand includes a hybrid sequence complementary to the hybrid sequence of the first strand.
- the barcode sequence of the oligonucleotide tag includes a common barcode domain and a variable domain, and the common barcode domain is in the oligonucleotide tag attached to the same solid support The same, and the common barcode domain is different between two or more solid supports in the plurality of solid supports.
- the present application also provides a kit for analyzing target nucleic acids from cells, which includes the composition described in the present application.
- the kit includes a transposase.
- the kit further includes at least one of a nucleic acid amplification agent, a reverse transcription agent, a fixative, a permeabilizing agent, a linking agent, and a lysis agent.
- a method for amplifying a target nucleic acid from a cell comprising:
- a) Provide discrete partitions comprising: i. a target nucleic acid derived from a single cell, wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid; and ii. A solid support attached with at least one oligonucleotide tag, wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and a 3'end of the barcode sequence.
- a hybridizing sequence comprising a first portion complementary to the hybridizing sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and
- the first strand and the second strand form a partially double-stranded structure or the second strand and the attached target nucleic acid form a partially double-stranded structure;
- the oligonucleotide tag is releasably attached to the solid support.
- it includes releasing the at least one oligonucleotide tag from the solid support, and in b) making the released oligonucleotide tag and the attached target Nucleic acids are linked to produce barcoded target nucleic acids.
- the oligonucleotide tag is directly or indirectly attached to the solid support through the 5' end of its first strand.
- a ligase is further included in the discrete partition, and the ligase connects the oligonucleotide tag to the attached target nucleic acid.
- the ligase includes T4 ligase.
- the target nucleic acid sequence is located at the 3'end of the barcode sequence.
- the solid support is a bead.
- the discrete partitions are holes or droplets.
- the barcode sequence includes a cell barcode sequence, and each oligonucleotide tag attached to the same solid support contains the same cell barcode sequence.
- the cell barcode sequence comprises at least 2 cell barcode segments separated by a linker sequence.
- a) includes co-distributing the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag into the discrete partitions.
- b) includes connecting the hybridizing sequence of the first strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the Barcoded target nucleic acid.
- b) includes hybridizing the second portion of the second strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, and allowing the The hybridization sequence of the first strand of the oligonucleotide tag is connected to the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the barcoded target nucleic acid.
- the attached target nucleic acid includes a unique molecular identification region.
- the unique molecular identification region is located between the oligonucleotide adaptor sequence and the target nucleic acid sequence.
- the oligonucleotide tag further includes an amplification primer recognition region.
- the amplification primer recognition region is a universal amplification primer recognition region.
- the barcoded target nucleic acid is released from the discrete partition, and the amplification is performed after the barcoded target nucleic acid is released from the discrete After the partition is released.
- amplification primers are used in the amplification, and random guide sequences are included in the amplification primers.
- the random leader sequence is a random hexamer.
- the amplifying includes at least partially hybridizing the random leader sequence with the barcoded target nucleic acid and extending the random leader sequence in a template-directed manner.
- this application also provides a method for sequencing a target nucleic acid from a cell, the method comprising:
- a) Provide discrete partitions comprising: i. a target nucleic acid derived from a single cell, wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid; and ii. A solid support attached with at least one oligonucleotide tag, wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and a 3'end of the barcode sequence.
- a hybridizing sequence comprising a first portion complementary to the hybridizing sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and
- the first strand and the second strand form a partially double-stranded structure or the second strand and the attached target nucleic acid form a partially double-stranded structure;
- the oligonucleotide tag is releasably attached to the solid support.
- it includes releasing the at least one oligonucleotide tag from the solid support, and in b) making the released oligonucleotide tag and the attached target Nucleic acids are linked to produce barcoded target nucleic acids.
- the oligonucleotide tag is directly or indirectly attached to the solid support through the 5' end of its first strand.
- a ligase is further included in the discrete partition, and the ligase connects the oligonucleotide tag to the attached target nucleic acid.
- the ligase includes T4 ligase or T7 ligase.
- the target nucleic acid sequence is located at the 3'end of the barcode sequence.
- the solid support is a bead.
- the discrete partitions are holes or droplets.
- the barcode sequence includes a cell barcode sequence, and each oligonucleotide tag attached to the same solid support contains the same cell barcode sequence.
- the cell barcode sequence comprises at least 2 cell barcode segments separated by a linker sequence.
- a) includes co-distributing the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag into the discrete partitions.
- b) includes connecting the hybridizing sequence of the first strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the Barcoded target nucleic acid.
- b) includes hybridizing the second portion of the second strand of the oligonucleotide tag with the oligonucleotide adaptor attached to the target nucleic acid, and allowing the The hybridization sequence of the first strand of the oligonucleotide tag is connected to the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the barcoded target nucleic acid.
- the attached target nucleic acid includes a unique molecular identification region.
- the unique molecular identification region is located between the oligonucleotide adaptor sequence and the target nucleic acid sequence.
- the oligonucleotide tag further includes an amplification primer recognition region.
- the amplification primer recognition region is a universal amplification primer recognition region.
- it further comprises a continuous nucleic acid sequence that assembles at least a part of the genome of the single cell from the sequence of the barcoded target nucleic acid.
- the single cell is characterized based on the nucleic acid sequence of at least a portion of the genome of the single cell.
- each of the discrete partitions includes at most the target nucleic acid derived from a single cell.
- it further includes identifying a single nucleic acid sequence in the barcoded target nucleic acid as derived from a given nucleic acid in the target nucleic acid based at least in part on the existence of the unique molecular identification region.
- it further includes determining the amount of a given nucleic acid in the target nucleic acid based on the existence of the unique molecular identification region.
- Figure 1 shows a schematic diagram of the PCR method in this application for generating nucleotide tags suitable for non-transcriptome analysis.
- Figure 2 shows a schematic diagram of the T4 ligase method in this application for generating nucleotide tags suitable for non-transcriptome analysis.
- Figure 3 shows a schematic diagram of the PCR method in this application for generating nucleotide tags suitable for transcriptome analysis.
- Figure 4 shows a schematic diagram of the T4 ligase method in this application for generating nucleotide tags suitable for non-transcriptome analysis.
- Figure 5 shows the fragment length distribution diagram of the ATAC sequencing results of human 293T cells mediated by the Tn5 transposition reaction in the present application.
- FIGS 6A and 6B show the signal-enriched transcription start site (TSS) map of the human 293T cell ATAC sequencing result mediated by the Tn5 transposition reaction in the present application.
- TSS signal-enriched transcription start site
- FIG. 7 shows the ratio diagram of different types of sequences of the ATAC sequencing results of human 293T cells mediated by the Tn5 transposition reaction in the present application.
- Figure 8 shows a schematic diagram of the microfluidic chip in this application.
- Figure 9 shows the stacking curve of the ATAC sequencing results in this application based on the number of reads in each barcode.
- Figure 10 shows the distribution of the number of unique mapped reads in a single cell as a result of ATAC sequencing in this application.
- Figure 11 shows the distribution map of the ATAC data of the cells in this application in the gene region.
- Figure 12 shows the result of the ATAC signal correlation analysis of single cells in the present application.
- Figure 13 shows the results of the Cuttag library fragment distribution in this application.
- Fig. 14 shows the result of the position of the cut tag fragments in the transcription initiation site in this application.
- Figure 15 shows the result of the proportion of Cuttag fragments distributed in the genome in this application.
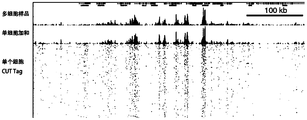
- Figure 16 shows the results of the single-cell Cut tag distribution results in this application.
- Figure 17 shows the result of clearly distinguishing single cells of mixed cells according to the single-cell transcriptome in this application.
- Figure 18 shows the distribution results of the number of transcripts and genes detected in each cell in this application.
- Figure 19 shows the result of clearly distinguishing single cells of mixed cells according to the single-cell genome in the present application.
- Figure 20 shows the results of single-cell sequencing in this application with different degrees of coverage for each cell and each genomic site.
- Fig. 21 shows the result of clearly distinguishing single cells of mixed cells based on single-cell DNA modification in the present application.
- Figure 22 shows the results of the methylation modification distribution detected in each cell in this application.
- Figure 23 shows the 5hmC modification distribution results detected in each cell in this application.
- FIG. 24 shows the result of the single cell in the mixed cell can be well distinguished according to the transcriptome and ATAC in this application.
- FIG. 25 shows the result of the single cell in the mixed cell can be well distinguished according to the transcriptome and the cut tag in this application.
- Figure 26 shows the result of the transcriptome and methylome of the same cell in this application that can be well matched with the gene model and the known methylation sites.
- Figure 27 shows a schematic diagram of a spatial lattice chip in this application.
- Figure 28 shows the result of the number of genes after the HE staining of the slices and the space lattice chip are superimposed in this application.
- sequencing generally refers to a technology for obtaining sequence information of nucleic acid molecules.
- analysis of the base sequence of a specific DNA fragment for example, the arrangement of adenine (A), thymine (T), cytosine (C) and guanine (G), etc.
- sequencing methods can include Sanger dideoxy chain termination Method (Chain Termination Method), Pyrosequencing method, and "Synthetic Parallel Sequencing” or "Connected Sequencing” platforms used by Illumina, Life Technologies, and Roche for next-generation sequencing, sequencers from MGI/Complete Genomics; usually It may also include nanopore sequencing methods, such as the method developed by Oxford Nanopore Technology, PacBio's third-generation sequencer, or electronic detection-based methods, such as Ion Torrent technology launched by Life Technologies.
- characterization result generally refers to the information description of nucleic acids and other related molecules obtained by sequencing or other biological analysis methods such as genomics and/or proteomics.
- it can include sequence information of whole genome sequencing, accessible chromatin sequence and distribution information, nucleic acid sequence and its binding factor binding information, pathogenic gene mutation information, single nucleotide polymorphism (SNP), nucleotide methyl Chemistry, transcriptome information (such as temporal or spatial changes in gene expression levels), etc.
- protein A generally refers to a cell-derived protein that can bind to the conserved region of the antibody heavy chain derived from different species (ie, the recognition protein of the antibody). For example, it can bind to the Fc fragment in human and various mammalian serum IgG molecules.
- the mammals can include pigs, dogs, rabbits, humans, monkeys, mice, mice, and cattle, etc.; protein A binds to IgG.
- Classes can mainly include IgG1, IgG2 and IgG4; besides binding to IgG, protein A can also bind to IgM and IgA in the serum.
- protein A may include protein A (SPA) from Staphylococcus aureus.
- SPA is the main component of cell wall antigens. Almost 90% of Staphylococcus aureus strains contain this component, but the content of different strains varies greatly.
- the ability of protein A to bind to antibodies can be used to locate and/or analyze the target protein by forming a target protein-antibody-protein A complex.
- solid support generally refers to any material that is suitable or can be modified to be suitable for attaching the oligonucleotide tags, barcode sequences, primers, etc. described herein.
- a solid support includes an array of holes or recesses located in the surface. These can be manufactured using a variety of technologies, such as photolithography, stamping technology, molding technology, and microetching technology; the composition and geometry of the solid support can be based on Its use varies.
- the solid support can be a planar structure (such as a slide, chip, microchip, and/or array, etc.); for example, the solid support or its surface can also be non-planar, such as that of a tube or container.
- the inner or outer surface; for example, the solid support may also include microspheres or beads.
- beads or “microspheres” or “parcitiles” generally refer to small discrete particles.
- Suitable bead compositions include, but are not limited to: plastics, ceramics, glass, polystyrene, methyl styrene, acrylic polymers, paramagnetic materials, thorium oxide sol, carbon graphite, titanium dioxide, latex or cross-linked dextran (Such as agarose), cellulose, nylon, cross-linked micelles and Teflon, and any other materials for solid supports outlined in this article can all be used. Fishers Ind.) Microsphere Detection Guide; in some embodiments, the microspheres may be magnetic microspheres or beads.
- unique molecular identification area can also be referred to as “molecular barcode”, “molecular marker”, “unique identifier (UID)”, “unique molecular identifier (UMI)”, etc., usually referring to A unique sequence code attached to each original nucleotide fragment of the same sample.
- the subsequent amplification bias can be corrected by directly counting the unique molecular identifiers (UMI) sequenced after amplification.
- UMI unique molecular identifiers
- UMI can be designed, incorporated, and applied according to methods known in the art, for example, by WO2012/142213, Islam et al. (Nat. Methods) (2014) 11:163-166, and Kivioja, T. et al. The publication of (Nat. Methods) (2012) 9:72-74 is exemplified, and the document is incorporated herein by reference in its entirety.
- the term "amplification primer recognition region” generally refers to a nucleotide sequence capable of complementary hybridization with the primer sequence for amplifying the target nucleic acid.
- the combination of the primer and the primer can trigger nucleotide extension, ligation and/or synthesis, for example, to increase the copy number of the target nucleic acid (ie amplification) under the action of polymerase chain reaction, and in some embodiments, it also includes an oligonucleotide tag. , Amplification of sequences such as molecular unique identifiers.
- the term "discrete partition” generally refers to independent spatial units that contain the target substance to be analyzed.
- the discrete partitions may also contain other Other substances, such as dyes, emulsifiers, surfactants, stabilizers, polymers, aptamers, reducing agents, initiators, biotin markers, fluorophores, buffers, acidic solutions, alkaline solutions, Light-sensitive enzymes, pH-sensitive enzymes, aqueous buffers, detergents, ionic detergents, non-ionic detergents, etc.
- the term "releasably attached” generally means that the connection between the oligonucleotide tag and the solid support is releasable, cleavable or reversible or destructible and destructible.
- the connection between the oligonucleotide tag and the solid support contains unstable bonds, such as chemical, thermal or light-sensitive bonds, such as disulfide bonds, UV-sensitive bonds, etc., which are destroyed by corresponding treatments.
- connection between the oligonucleotide tag and the solid support includes a specific base that can be recognized by a nuclease, such as dU, which can be cleaved by the action of the UNG enzyme; for example,
- the connection between the oligonucleotide tag and the solid support contains an endonuclease recognition sequence, which can be cleaved by the action of nuclease; for example, the solid support is degradable and passes through the solid Degradation of the support releases the oligonucleotide tag, enabling releasable attachment and the like.
- linker generally refers to a nucleotide sequence that connects various functional sequences together, and can also include a molecular sequence (nucleic acid, polypeptide or other Chemical connection structure, etc.) wherein the functional sequence may include cell barcode segment, barcode sequence, amplification primer recognition region, sequencing primer recognition region, unique molecular identifier, etc.
- the nucleotide It can be a fixed nucleotide sequence.
- the linker can also include chemical modifications.
- random leader sequence generally refers to a random primer that can present a fourfold degeneracy at each position.
- the random guide sequence recognizes and binds to the corresponding region of the target nucleic acid (including the target nucleic acid sequence and other nucleotide sequences attached thereto) to realize the synthesis and/or amplification of the nucleotide sequence.
- barcode sequence generally refers to a nucleotide sequence capable of identifying a target nucleic acid or its derivative or modified form.
- cell barcode sequence generally refers to a nucleotide sequence that can be used to identify the source of a target nucleic acid sample.
- the source can be, for example, from the same cell or different cells.
- different cell barcode sequences can be used to label the nucleic acid in each source, so that the source of the sample can be identified.
- Bar codes also commonly referred to as indexes, labels, etc.
- any suitable bar code or bar code group can be used, such as the cell bar code sequence described in the publication of US2013/0274117.
- cell barcode segment generally refers to the barcode nucleotide units constituting the cell barcode sequence
- N of the cell barcode segments can form a cell barcode segment through the action of PCR or DNA ligase.
- N can be greater than or equal to 1, so that the cell barcode sequence formed is sufficient to identify the cell source of each nucleic acid sample derived from multiple sources.
- oligonucleotide adaptor generally refers to a nucleotide sequence that is attached to a target nucleic acid and includes a sequence that is capable of complementary hybridization to the oligonucleotide tag.
- the nucleotide sequence may be a partially double-stranded structure, for example, it may have a protruding sequence that hybridizes with the oligonucleotide tag; in some embodiments, the oligonucleotide adaptor may also include a transposase (such as Tn5 transposition Enzyme) binding sequence; in some embodiments, the oligonucleotide adaptor may also include an amplification primer recognition sequence; in some embodiments, the oligonucleotide adaptor may also include a reverse transcription primer sequence.
- a transposase such as Tn5 transposition Enzyme
- barcoded target nucleic acid generally refers to a target nucleic acid to which at least a cell barcode sequence is attached.
- the term "common barcode domain” generally refers to a barcode sequence used to identify the source of the target nucleic acid.
- the common barcode domains contained in oligonucleotide tags attached to the same solid support are the same, and the common barcode domains contained in oligonucleotide tags attached to different solid supports are mutually exclusive.
- the oligonucleotide tag released from the same solid support is connected to the target nucleic acid derived from one cell, and its cellular origin can be identified through the common barcode domain.
- variable domain generally refers to a nucleotide sequence set according to different needs outside the common barcode domain.
- linker sequence for example, linker sequence, amplification primer recognition sequence, sequencing primer recognition sequence, etc.
- transposase-nucleic acid complex generally refers to a complex formed by a transposase and a sequence containing the oligonucleotide adaptor.
- Transposase usually refers to an enzyme that can bind to the end of a transposon and catalyze its movement to other parts of the genome through a cut, paste mechanism or a replicative transposition mechanism.
- a transposon usually refers to a segment of nucleotides that can freely jump in the genome. It was proposed by Barbara McClintock in the late 1940s when he was studying the genetic mechanism of maize. Later other research groups described the transposable molecule.
- chromosome fragments can change position, jumping from one chromosome to another.
- the relocation of these transposons can change the expression of other genes.
- transposition in corn can cause color changes, and in other organisms such as bacteria, it can cause antibiotic resistance in the process of human evolution.
- the transposase-nucleic acid complex can include two dimers formed by the transposases respectively combined with oligonucleotide adaptors, and the two transposases can be the same transposase or different ,
- the oligonucleotide adaptors that they bind respectively can be the same or different.
- Tn5 generally refers to the Tn5 transposase, which is a member of the ribonuclease (RNase) superfamily.
- RNase ribonuclease
- Tn5 can be found in Shewanella and Escherichia coli.
- Tn5 can include the naturally occurring Tn5 transposase and various active mutant forms;
- Tn5 like most other transposases, contains the DDE motif, which is the active site that catalyzes the transfer of the transposon.
- DDE motifs can coordinate with divalent metal ions (such as magnesium and manganese) and play an important role in catalyzing reactions.
- the transposase Tn5 may increase the transposition activity through mutations in the DDE region and catalyze the movement of the transposon.
- the glutamic acid at position 326 is converted to aspartic acid
- the two aspartic acids at position 97 and 188 are converted to glutamic acid (amino acid numbering based on the amino acid sequence of GenBank Accession No. YP_001446289) and so on.
- microfluidic device generally refers to a device or system capable of implementing microfluidic control.
- microfluidics usually refers to a technology for precise control and manipulation of micro-scale fluids, especially those with sub-micron structures.
- Micro usually refers to tiny volumes or volumes (such as nanoliters, picoliters, and other types of microfluidics). ).
- Microfluidic technology has been widely used in many fields, such as the field of biomedicine, for example, enzyme analysis in molecular biology methods (such as glucose and lactate analysis), DNA analysis (such as polymerase chain reaction and high-throughput sequencing), Proteomics analysis, etc.
- the main structure of the microfluidic device may include a simple reservoir connected to it, a fluid pipe that delivers fluid from external sources, manifolds, fluid flow units (for example, actuators, pumps, compressors), etc., and distributes microfluidics Delivery to subsequent processing operations, fluid conduits of instruments or components, etc.
- a fluid pipe that delivers fluid from external sources, manifolds, fluid flow units (for example, actuators, pumps, compressors), etc., and distributes microfluidics Delivery to subsequent processing operations, fluid conduits of instruments or components, etc.
- hybridization generally refers to the nucleus contained in nucleic acid (such as RNA, DNA) under in vitro and/or in vivo conditions at a suitable temperature and ionic strength of the solution.
- nucleic acid such as RNA, DNA
- the nucleotide sequence enables it to specifically non-covalently bind (ie form Watson-Crick base pairs and/or G/U base pairs) to another nucleic acid sequence.
- Watson-Crick base pairing includes: adenine/adenosine (A) paired with thymidine/thymine (T), A paired with uracil/uridine
- the hybridization between two RNA molecules for example, dsRNA
- G guanine/guanosine
- C cytosine/cytidine
- the hybridization between two RNA molecules for example, dsRNA
- G can also be U base pairing.
- Hybridization requires that the two nucleic acids contain complementary sequences, but possible mismatches between bases cannot be ruled out.
- the conditions suitable for hybridization between two nucleic acids depend on the length and degree of complementarity of the nucleic acids, which are well known in the art. The greater the degree of complementarity between two nucleotide sequences, the greater the value of melting temperature (Tm) of hybrids of nucleic acids having these complementary sequences.
- read length refers to reads, which usually refers to a sequence obtained by a reaction in nucleotide sequencing. Reads can be a short sequencing fragment, which is the base sequence data obtained by a single sequencing by a sequencer. The length of reads can be different for different sequencing instruments.
- the present application provides a method for analyzing target nucleic acid from a cell, the method comprising:
- a target nucleic acid derived from a single cell wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid;
- a solid support attached with at least one oligonucleotide tag wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and is located in the barcode sequence 3'end of the hybridization sequence, the second strand includes a first portion complementary to the hybridization sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid Part, and the first chain and the second chain form a partially double-stranded structure;
- Two parts, and the second strand and the attached target nucleic acid form a partially double-stranded structure
- the oligonucleotide tag is connected to the attached target nucleic acid, thereby generating a barcoded target nucleic acid.
- it further includes:
- this application also provides a method for amplifying a target nucleic acid from a cell, the method comprising:
- a) Provide discrete partitions comprising: i. a target nucleic acid derived from a single cell, wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid; and ii. A solid support attached with at least one oligonucleotide tag, wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and a 3'end of the barcode sequence.
- a hybridizing sequence, the second strand comprising a first portion complementary to the hybridizing sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and
- the first strand and the second strand form a partially double-stranded structure; or the step ii.
- each of the oligonucleotide tags Contains a first strand and a second strand, the first strand includes a barcode sequence and a hybridization sequence located at the 3'end of the barcode sequence, and the second strand includes a first portion complementary to the hybrid sequence of the first strand And a second part complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and the second strand and the attached target nucleic acid form a partially double-stranded structure;
- this application also provides a method for sequencing a target nucleic acid from a cell, the method comprising:
- a) Provide discrete partitions comprising: i. a target nucleic acid derived from a single cell, wherein at least part of the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid; and ii. A solid support attached with at least one oligonucleotide tag, wherein each of the oligonucleotide tags includes a first strand and a second strand, and the first strand includes a barcode sequence and a 3'end of the barcode sequence.
- a hybridizing sequence, the second strand comprising a first portion complementary to the hybridizing sequence of the first strand and a second portion complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and
- the first strand and the second strand form a partially double-stranded structure; or the step ii.
- each of the oligonucleotide tags Contains a first strand and a second strand, the first strand includes a barcode sequence and a hybridization sequence located at the 3'end of the barcode sequence, and the second strand includes a first portion complementary to the hybrid sequence of the first strand And a second part complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and the second strand and the attached target nucleic acid form a partially double-stranded structure;
- the oligonucleotide tag in the present application may include a first strand and a second strand, and the first strand and the second strand may be provided at the same time or separately.
- the first chain and the second chain when the first chain and the second chain are provided at the same time, the first chain and the second chain may form a partially double-stranded structure; when the first chain and the second chain are When the two strands are provided separately, the second strand may form a partially double-stranded structure with the attached target nucleic acid.
- the barcoded target nucleic acid is generated by linking the oligonucleotide tag with the attached target nucleic acid.
- the hybridization sequence of the first strand of the oligonucleotide tag is connected to the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the barcoded target nucleic acid.
- the second portion of the second strand of the oligonucleotide tag is hybridized with the oligonucleotide adaptor attached to the target nucleic acid, and the second portion of the oligonucleotide tag is hybridized
- One strand of the hybridization sequence is connected to the oligonucleotide adaptor attached to the target nucleic acid, thereby generating the barcoded target nucleic acid.
- the conditions suitable for hybridization between two nucleic acids depend on the length and degree of complementarity of the nucleic acids, which are well known in the art. The greater the degree of complementarity between two nucleotide sequences, the greater the value of melting temperature (Tm) of hybrids of nucleic acids having these complementary sequences.
- the length of the second part of the second strand of the oligonucleotide tag is sufficient for its complementary sequence (the oligonucleotide adaptor sequence attached to the target nucleic acid or a partial sequence thereof) Form a double-stranded structure.
- the length of the second part of the second strand may be 1 nucleotide or more, 2 nucleotides or more, 3 nucleotides or more, 5 nucleotides or More, 8 nucleotides or more, 10 nucleotides or more, 12 nucleotides or more, 15 nucleotides or more, 20 nucleotides or more, 22 nucleotides Nucleotides or more, 25 nucleotides or more or 30 nucleotides or more.
- the hybridization does not exclude possible mismatches between bases.
- the sequence of the first part of the second strand or the second part of the second strand need not be 100% complementary to the sequence of the hybridizing sequence.
- it can be 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more More, 98% or more, 99% or more, 99.5% or more complementary.
- the remaining non-complementary nucleotides can be clustered or interspersed with complementary nucleotides and need not be adjacent to each other or complementary nucleotides.
- polynucleotides can hybridize on one or more segments so that no intermediate or adjacent segments are involved in the hybridization event (e.g., forming a hairpin structure, "bumps", etc.).
- a ligation reaction is used to ligate the oligonucleotide tag with the attached target nucleic acid.
- the linking may include joining two nucleic acid segments together by catalyzing the formation of a phosphodiester bond, such as the hybridizing sequence of the first strand of the oligonucleotide tag and the attachment to the target nucleic acid.
- the ligation reaction can include DNA ligase, such as E. coli DNA ligase, T4 DNA ligase, T7 DNA ligase, mammalian ligase (for example, DNA ligase I, DNA ligase III, DNA ligase IV), thermostable Ligase etc.
- T4 DNA ligase can join segments containing DNA, oligonucleotides, RNA and RNA-DNA hybrids.
- the ligation reaction may not include DNA ligase, but instead use alternatives such as topoisomerase.
- Using high concentration of DNA ligase and including PEG can achieve rapid ligation.
- the optimum temperature of the DNA ligase for example, 37°C
- the melting temperature of the DNA to be ligated can be considered.
- the target nucleic acid and the barcoded solid support can be suspended in a suitable buffer to minimize the effects of ions that may affect the connection.
- the releasing at least a portion of the target nucleic acid from the single cell in the discrete partition to the outside of the cell includes releasing at least a portion of the target nucleic acid from the single cell in the discrete partition to the outside of the cell, and linking the released target nucleic acid to the oligonucleotide tag in b) , So as to produce barcoded target nucleic acid.
- the releasing at least a portion of the target nucleic acid from the single cell in the discrete partition to the outside of the cell may include contacting the cell with a lysis reagent to release the contents of the cell in the discrete partition.
- the lytic agent may include a biologically active agent, for example, a lytic enzyme used to lyse different cell types (such as gram positive or negative bacteria, plants, yeast, mammals, etc.), such as lysozyme, leuco peptide Enzymes, lysostaphin, thioglucosidase kitalase, lyticase, and other commercially available lytic enzymes.
- a surfactant-based dissolving solution may also be used to dissolve the cells.
- the dissolving solution may include nonionic surfactants such as Triton X-100 and Tween 20.
- the dissolving solution may include ionic surfactants such as sodium lauryl sarcosinate and sodium dodecyl sulfate (SDS).
- ionic surfactants such as sodium lauryl sarcosinate and sodium dodecyl sulfate (SDS).
- SDS sodium dodecyl sulfate
- other methods that can be used such as electroporation, heat, sound, or mechanical cell destruction can also be used for lysis.
- the releasing at least a portion of the target nucleic acid from the single cell in the discrete partition to the outside of the cell may include at least 5%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45% of the target nucleic acid is released outside the cell from the single cell in the discrete partition.
- it includes allowing at least a portion of the oligonucleotide tag released from the solid support to enter the single cell, and to connect with the target nucleic acid in b), thereby generating a barcoded target nucleic acid.
- the release of at least a portion of the oligonucleotide tag from the solid support into the single cell may include at least 25%, at least 30%, at least 35%, at least 40%, at least 50%, At least 55%, at least 60%, at least 70%, at least 75%, at least 75% of the oligonucleotide tag enters the single cell.
- the oligonucleotide tag is releasably attached to the solid support.
- an oligonucleotide tag that is releasably, cleavably or reversibly attached to the solid support includes being released by the cleavage/disruption of the linkage between the oligonucleotide tag molecule and the solid support Or a releasable oligonucleotide tag, or an oligonucleotide tag released by the degradation of the solid support itself, so that the oligonucleotide tag can be accessed or accessible by other reagents, or both.
- the acrydite moiety connected to the solid support precursor, another substance connected to the solid support precursor, or the precursor itself contains an unstable bond, for example, a chemical, heat, or light sensitive bond, for example, a disulfide bond , UV sensitive keys, etc.
- the unstable bond can be used to reversibly link (covalently link) a substance (such as an oligonucleotide tag) to a solid support.
- a thermally labile bond may include attachment based on nucleic acid hybridization (e.g., when an oligonucleotide hybridizes to a complementary sequence attached to a solid support) such that the thermal melting of the hybrid is removed from the solid support (or Beads) release oligonucleotides, for example, sequences containing oligonucleotide tags.
- nucleic acid hybridization e.g., when an oligonucleotide hybridizes to a complementary sequence attached to a solid support
- Beads Beads
- adding multiple types of unstable bonds to a gel solid support can lead to the production of a solid support that can respond to different stimuli.
- Each type of unstable bond can be sensitive to related stimuli (eg, chemical stimulation, light, temperature, etc.), so that the release of substances attached to the solid support through each type of unstable bond can be controlled by applying appropriate stimuli .
- agents can be provided that are releasably attached to a solid support or otherwise arranged in discrete partitions, such that once delivered to a desired set of agents (for example, by co-dispensing)
- the activatable group can react with the desired reagent.
- activatable groups include caged groups, removable blocking or protecting groups, for example, photolabile groups, thermally labile groups, or chemically removable groups.
- ester linkages e.g., acids, bases, or Hydroxylamine cleavable
- adjacent diol linkage e.g., cleavable by sodium periodate
- Diels-Alder linkage e.g., thermally cleavable
- sulfone linkage e.g., cleavable by alkali
- monosilane Base ether linkage e.g., cleavable by acid
- glycoside linkage e.g., cleavable by amylase
- peptide linkage e.g., cleavable by protease
- phosphodiester linkage e.g., cleavable by nuclease ( DNA enzyme) cleaved.
- the oligonucleotide tag is directly or indirectly attached to the solid support through the 5' end of its first strand. For example, including releasing the at least one oligonucleotide tag from the solid support, and linking the released oligonucleotide tag with the attached target nucleic acid in b), thereby producing Barcoded target nucleic acid.
- the target nucleic acid sequence is located at the 3'end of the barcode sequence.
- the target nucleic acid can be directly connected to the 3'end of the barcode sequence; for example, the target nucleic acid is not directly connected to the 3'end of the barcode sequence, and the target nucleic acid can be directly connected to the barcode sequence.
- the target nucleic acid can be directly connected to the barcode sequence.
- the barcoded target nucleic acid is amplified.
- the barcoded target nucleic acid is released from the discrete partition, and the amplification is performed after the barcoded target nucleic acid is released from the discrete partition.
- further chemical or enzymatic modification may be performed, for example, the modification may include bisulfite conversion, 5hmc conversion, etc., before amplification.
- amplification primers are used in the amplification.
- the amplification may also include further modification of the barcoded target nucleic acid so that it also has a fixed sequence on the other side that can be used for PCR amplification.
- the modification may include reverse transcription. Chain switching, second-strand synthesis, terminal transferase (terminal transferase) reaction, and connection of a second adaptor (adaptor) can be used.
- the amplification primers may also include universal primers.
- an amplification primer is used in the amplification, and the amplification primer may include a random guide sequence.
- the random leader sequence includes random primers that can exhibit four-fold degenerate at each position.
- random primers include any nucleic acid primers having various random sequence lengths known in the art.
- random primers can include lengths of 3, 4, 5, 6, 7, 8, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 or more nucleotides Random sequence.
- the plurality of random primers may include random primers having different lengths.
- the plurality of random primers may include random primers having an equal length.
- the plurality of random objects may include random sequences of about 5 to about 18 nucleotides in length.
- the plurality of random objects includes random hexamers.
- the random hexamer is commercially available and widely used in amplification reactions, such as multiple displacement amplification (MDA), for example, REPLI-g Whole Genome Amplification Kit (QIAGEN, Valencia, CA) as an example .
- MDA multiple displacement amplification
- REPLI-g Whole Genome Amplification Kit QIAGEN, Valencia, CA
- Random primers of any suitable length can be used in the methods and compositions described in this application.
- the amplifying includes at least partially hybridizing the random leader sequence with the barcoded target nucleic acid and extending the random leader sequence in a template-directed manner.
- the oligonucleotide tag includes a first strand and a second strand
- the first strand includes a barcode sequence and a hybridization sequence located at the 3'end of the barcode sequence
- the second strand includes A first part complementary to the hybridizing sequence of the first strand and a second part complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and the first strand and the second strand form Partially double-stranded structure.
- the oligonucleotide tag includes a first strand and a second strand
- the first strand includes a barcode sequence and a hybridization sequence located at the 3'end of the barcode sequence
- the second strand includes The first portion of the first strand that is complementary to the hybridization sequence and the second portion that is complementary to the oligonucleotide adaptor sequence attached to the target nucleic acid, and the second strand and the attached
- the connected target nucleic acid forms a partially double-stranded structure.
- the conditions suitable for hybridization between two nucleic acids depend on the length and degree of complementarity of the nucleic acids, which are well known in the art. The greater the degree of complementarity between two nucleotide sequences, the greater the value of melting temperature (Tm) of hybrids of nucleic acids having these complementary sequences.
- Tm melting temperature
- the length of the first part of the second strand or the second part of the second strand is sufficient for its complementary sequence (for example, the hybridization of the first strand at the 3'end of the barcode sequence)
- the sequence for example, the oligonucleotide adaptor sequence attached to the target nucleic acid or a partial sequence thereof) forms a double-stranded structure.
- the length of the first part of the second strand or the second part of the second strand may be 1 nucleotide or more, 2 nucleotides or more, and 3 nucleosides. Acid or more, 5 nucleotides or more, 8 nucleotides or more, 10 nucleotides or more, 12 nucleotides or more, 15 nucleotides or more, 20 nucleotides or more, 22 nucleotides or more, 25 nucleotides or more, or 30 nucleotides or more.
- the length of the sequence of the first part of the second strand and the second part of the second strand may be the same or different.
- the double-stranded structure does not exclude possible mismatches between bases.
- the sequence of the first part of the second strand or the second part of the second strand need not be 100% complementary to the sequence of the hybridizing sequence.
- it can be 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more More, 98% or more, 99% or more, 99.5% or more complementary.
- the remaining non-complementary nucleotides can be clustered or interspersed with complementary nucleotides and need not be adjacent to each other or complementary nucleotides.
- polynucleotides can hybridize on one or more segments so that no intermediate or adjacent segments are involved in the hybridization event (e.g., forming a hairpin structure, "bumps", etc.).
- the second part of the oligonucleotide tag attached to the same solid support may be the same.
- the second part of the oligonucleotide tag attached to the same solid support may be different.
- the second part of each oligonucleotide tag attached to the same solid support may include one or more nucleotide sequences, for example, the sequence of the second part may be 2.
- the number of the oligonucleotide tags attached to the same solid support and containing the same second part may be 1 or more, for example, 50 or more, 100 or more, 500 One or more, 1,000 or more, 1,500 or more, 2,000 or more, 3,000 or more, 5,000 or more, 8,000 or more, 10,000 or more, 12,000 or more, 15,000 or more, 18,000 One or more, 20,000 or more, 22,000 or more, 25,000 or more, 28,000 or more, 30,000 or more, 35,000 or more, 40,000 or more, 45,000 or more, 50,000 or more.
- the number of the oligonucleotide tags containing different second parts attached to the same solid support can be set to different ratios as needed, so as to be connected to the corresponding attached target nucleic acid.
- the barcode sequence includes a cell barcode sequence, and each oligonucleotide tag attached to the same solid support contains the same cell barcode sequence.
- the oligonucleotide tags attached to the same solid support may include 1 or more oligonucleotide tags, for example, 50 or more, 100 or more, 500 or more , 1000 or more, 1500 or more, 2000 or more, 3000 or more, 5000 or more, 8000 or more, 10000 or more, 12000 or more, 15000 One or more, 18,000 or more, 20,000 or more, 22,000 or more, 25,000 or more, 28,000 or more, 30,000 or more, 35,000 or more, 40,000 or More, 45,000 or more, 50,000 or more, 55,000 or more, 60,000 or more, 65,000 or more, 70,000 or more, 75,000 or more, 80,000 or more , 85,000 or more, 90,000 or more, 95,000 or more, 100,000 or more, 110,000 or more, 120,000 or more, the cell barcode sequences of these oligonucleotide tags are the same
- the sequence of the second part of the second strand may be one or more, for example, the sequence of the second part is 2 or more
- the cell barcode sequences contained in the oligonucleotide tag sets attached to different solid supports are different from each other, and the oligonucleotide tag sets may be all the barcodes attached to the same solid support.
- the cell barcode sequence includes at least 2 cell barcode segments.
- the cell barcode segment is 4 or more nucleotides (nt), for example, 5 or more, for example, 10 or more, 12 or more, 15 or more, 18 or more, 20 or more, 21 or more, 22 or more, 23 or more, 24 or more, 25 or more, 26 or more, 27 or more, 28 or more, 29 or more, 30 or more, 31 or more, 32 or more, 33 or more, 34 or more, or 35 or more.
- the cell barcode sequence includes at least 2 cell barcode segments, at least 3 cell barcode segments, at least 4 cell barcode segments, at least 5 cell barcode segments, and at least 6 cell barcode segments. Segment, at least 7 cell barcode segments, at least 8 cell barcode segments, the cell barcode segment is encoded as a cell barcode segment 1 in the sequence from the 5'end to the 3'end in the oligonucleotide tag , Cell barcode section 2, Cell barcode section 3, Cell barcode section 4, Cell barcode section 5...Cell barcode section n.
- the at least two cell barcode segments can form the cell barcode sequence by PCR or DNA ligase.
- the cell barcode sequence can be generated by the following method:
- At least one solid support into at least 2 primary aliquots, for example, at least 8 aliquots, at least 16 aliquots, at least 24 aliquots, at least 32 aliquots, at least 40 equals, at least 48 equals, at least 56 equals, at least 64 equals, at least 72 equals, at least 80 equals, at least 88 equals, at least 96 equals;
- each of the primary aliquots with at least 1 cell barcode segment 1, for example, at least 1000 cell barcode segment 1, for example, at least 10,000 cell barcode segment 1, for example, at least 100,000 cells
- Barcode section for example, at least 1,000,000 cells.
- Barcode section for example, at least 10,000,000 cells. Barcode section 1.
- the cell barcode section 1 in each aliquot and the cells in any other aliquot The sequence and/or length of barcode segment 1 are different from each other;
- At least 1 cell barcode segment 2 or its complementary sequence to each of the secondary aliquots for example, at least 1000 cell barcode segment 2 or its complementary sequence, for example, at least 10,000 cell barcode regions Segment 2 or its complementary sequence, for example, at least 100,000 cell barcode segment 2 or its complementary sequence, for example, at least 1,000,000 cell barcode segment 2 or its complement, for example, at least 10 million cell barcode segment 2 or its complement Sequence, the cell barcode segment 2 or its complementary sequence in each aliquot is different from the cell barcode segment 2 or its complementary sequence in any other aliquot in sequence and/or length;
- steps 4)-6) can be repeated, the number of repetitions can be n, n can be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more to connect cell barcode segment 3.
- Cell barcode section 4, cell barcode section 5...Cell barcode section n to generate a cell barcode with a unique sequence for each cell, so that the target nucleic acid in the first cell can have a first cell barcode with a unique sequence .
- the target nucleic acid in the second cell may have a second cell barcode with a unique sequence, the target nucleic acid in the second cell may have a second cell barcode with a unique sequence, and so on.
- the barcoded target nucleic acid is released from the discrete partition.
- c) is further performed: sequencing the barcoded target nucleic acid to obtain the characterization result.
- the characterization result may include the nucleotide sequence information of the barcoded target nucleic acid, for example, including the cell barcode nucleotide sequence information, the nucleotide sequence information of the target nucleic acid, and UMI sequence information.
- a continuous nucleic acid sequence of at least a part of the genome of the single cell is assembled from the sequence of the barcoded target nucleic acid.
- the single cell is characterized based on the nucleic acid sequence of at least a portion of the genome of the single cell.
- the oligonucleotide tag further includes a linker sequence 1, and the 5'end of the cell barcode segment 1 can be connected to a solid support through the linker sequence 1.
- the linker sequence 1 may include acrydite modification, photocleavage modification, S-S modification, dU base modification and other sequences, which can be disconnected by various methods to release the oligonucleotide tag.
- the oligonucleotide tag also includes other functional sequences, and the other functional sequences may be located between the cell barcode segment 1 and the linker sequence 1, for example, a complete or partial functional sequence (e.g., Primer sequence (for example, universal primer sequence, targeting primer sequence, random primer sequence) recognition region, primer annealing sequence, attachment sequence, sequencing primer recognition region, amplification primer recognition region (for example, universal amplification primer recognition region), etc. , For subsequent processing.
- a complete or partial functional sequence e.g., Primer sequence (for example, universal primer sequence, targeting primer sequence, random primer sequence) recognition region, primer annealing sequence, attachment sequence, sequencing primer recognition region, amplification primer recognition region (for example, universal amplification primer recognition region), etc.
- the subsequent processing includes amplification.
- the amplification may include PCR amplification (for example, Taq DNA polymerase amplification, Super Taq DNA polymerase amplification, LA Taq DNA polymerase amplification, Pfu DNA polymerase amplification, Phusion DNA polymerase amplification , KOD DNA polymerase amplification, etc.), isothermal amplification (for example, loop-mediated isothermal amplification (LAMP), helicase-dependent amplification (HDA), recombinase polymerase amplification (RPA), Strand displacement amplification (SDA), nucleic acid sequence-based amplification (NASBA), transcription-mediated amplification (TMA), etc.), T7 promoter linear amplification, degenerate oligonucleotide primer PCR amplification (DOP-PCR) ), Multiple Displacement Amplification (MDA), Multiple Annealing Circular Cycle Amplification (MALBAC), etc.
- PCR amplification for example, Taq DNA
- the cell barcode may not contain a linker, and the cell barcode may be a separate nucleic acid sequence synthesized by other methods.
- the universal primer sequence may include P5 or other suitable primers.
- Universal primers (for example, P5) are also compatible with the sequencing device, for example, can be attached to the flow cell in the sequencing device.
- such universal primer sequences can provide complementary sequences of oligonucleotides constrained on the surface of the flow cell in the sequencing device, so that the barcoded target nucleic acid sequence can be immobilized on the surface for sequencing.
- an amplification primer sequence is a primer sequence used for an amplification or replication process (for example, extending the primer along the target nucleic acid sequence), so as to generate an amplified barcoded target nucleic acid sequence.
- the resulting amplified target sequence will contain such primers and be easily transferred to the sequencing system.
- the sequencing primer sequence may include the R1 primer sequence and the R2 primer sequence.
- the oligonucleotide tag may comprise a T7 promoter sequence.
- the T7 promoter sequence includes the nucleotide sequence shown in SEQ ID NO:1 (TAATACGACTCACTATAG).
- the oligonucleotide tag may contain at least 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, and any one of SEQ ID NO: 6-9. 78%, 79%, 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94% , 95%, 96%, 97%, 98%, 99%, or 100% identity regions.
- the nucleotide adaptor sequence may include a P5 sequence.
- the nucleotide adaptor sequence includes the P7 sequence.
- the cell barcode segment 1 and the linker sequence 1 may include any sequence or a combination of the above-mentioned multiple functional sequences.
- these oligonucleotides may include any one or more of the following: P5, R1 and R2 sequences, non-cleavable 5'acrydite-P5, cleavable 5'acrydite-SS-P5, R1c, sequencing primers, reading Take primers, universal primers, P5_U, universal read primers and/or binding sites of any of these primers.
- the cell barcode sequence includes at least two cell barcode segments separated by a linker sequence.
- the 3'end of the cell barcode segment 1 has a linker sequence 2
- the 5'end and the 3'end of the cell barcode segment 2 have linker sequences 3 and 4, respectively
- the 5'end and 3 of the cell barcode segment 3 The'end has linker sequences 5 and 6, respectively
- the 5'end and 3'end of cell barcode segment 4 have linker sequences 7 and 8, respectively, and so on, the 5'end and 3'end of cell barcode segment n
- linker sequences 2n-1 and 2n respectively; linker sequence 2 and linker sequence 3 can be at least partially complementary paired to form a double-stranded structure, and linker sequence 4 and linker sequence 5 can be at least partially complementary paired to form a double-stranded structure.
- the subsequence 6 and the linker sequence 7 can be at least partially complementary paired to form a double-stranded structure, and so on, to activate the cell barcode section 1, the cell barcode section 2, the cell barcode section 3, the cell barcode section 4, and so on. Connection of bar code segment n.
- a ligation reaction is used to ligate barcode segments of each cell to form an oligonucleotide tag.
- the linking may include joining two nucleic acid segments together by catalyzing the formation of a phosphodiester bond, such as cell barcode segment 1 and the aforementioned functional sequence, for example, linker sequence 2 and cell barcode segment 2, linking Subsequence 3 and cell barcode segment 3, linker sequence 4 and cell barcode segment 4, linker sequence 5 and cell barcode segment 5, linker sequence 6 and cell barcode segment 6, and so on.
- the ligation reaction may include DNA ligase, such as E.
- T4 DNA ligase can join segments containing DNA, oligonucleotides, RNA and RNA-DNA hybrids.
- the ligation reaction may not include DNA ligase, but instead use alternatives such as topoisomerase.
- Using high concentration of DNA ligase and including PEG can achieve rapid ligation.
- the optimum temperature of the DNA ligase for example, 37° C.
- the melting temperature of the DNA to be ligated can be considered.
- the sample and barcoded solid support can be suspended in a buffer to minimize the effects of ions that may affect the connection.
- the cell barcode segment provided in each round may contain the following structure: the cell barcode segment and the linker sequence located at the 3'end of the cell barcode segment are double-stranded structures located in the cell
- the linker sequence at the 5'end of the barcode segment is a protruding single-stranded structure, and a double-stranded structure is formed by at least partially complementary pairing with the linker sequence at the 5'end of the previous cell barcode segment.
- an example of using a ligation reaction to ligate barcode segments of each cell to form an oligonucleotide tag can be as shown in FIG. 2 or FIG. 4.
- each cell barcode segment is connected to form an oligonucleotide tag.
- the polymerase chain reaction can be performed by any one or more of the following polymerases: Taq DNA polymerase, Super Taq DNA polymerase, LA Taq DNA polymerase, UltraPF DNA polymerase, Tth DNA polymerase, Pfu DNA Polymerase, VentR DNA polymerase, Phusion DNA polymerase, KOD DNA polymerase, Iproof DNA polymerase.
- the polymerase chain reaction may further include a buffer solution and metal ions that enable the polymerase to maintain activity; for example, the polymerase chain reaction may also include dNTP and its modified derivatives.
- each round provides the complementary sequence of the cell barcode segment, the complementary sequence is a single-stranded structure, and the 5'end and the 3'end each have A linker sequence of a single-stranded structure, wherein the linker sequence at the 5'end can be at least partially complementary to the linker sequence at the 3'end of the cell barcode segment connected in the previous round to form a double-stranded structure, and the linker sequence at the 3'end can be paired with The linker sequence at the 5'end of the cell barcode segment connected in the latter round is at least partially complementary paired to form a double-stranded structure.
- PCR polymerase chain reaction
- PCR polymerase chain reaction
- the target nucleic acid includes one or more selected from the group consisting of DNA, RNA and cDNA.
- the target nucleic acid includes cDNA derived from RNA in the single cell.
- the RNA includes mRNA.
- the target nucleic acid is added with an oligonucleotide adaptor sequence to become an attached target nucleic acid.
- the oligonucleotide adaptor sequence is located at the 5'end of the target nucleic acid.
- the oligonucleotide adaptor sequence may include a nucleotide sequence L that is complementary to the second part of the second strand in the oligonucleotide tag, and the nucleotide sequence L is The length may be the same as or different from the length of the second part of the second strand in the oligonucleotide tag; for example, the length of the nucleotide sequence L may be 1 nucleotide or more.
- nucleotides or more More, 2 nucleotides or more, 3 nucleotides or more, 5 nucleotides or more, 8 nucleotides or more, 10 nucleotides or more, 12 nuclei Nucleotides or more, 15 nucleotides or more, 20 nucleotides or more, 22 nucleotides or more, 25 nucleotides or more or 30 nucleotides or more .
- the nucleotide sequence L may be complementary to the second part of the second strand in the oligonucleotide tag to form a double-stranded structure.
- the double-stranded structure cannot exclude possible mismatches between bases.
- the sequence of the nucleotide sequence L need not be 100% complementary to the sequence of the second part of the second strand in the oligonucleotide tag.
- it can be 60% or more, 65% or more, 70% or more, 75% or more, 80% or more, 85% or more, 90% or more, 95% or more More, 98% or more, 99% or more, 99.5% or more complementary.
- the remaining non-complementary nucleotides can be clustered or interspersed with complementary nucleotides and need not be adjacent to each other or complementary nucleotides.
- a polynucleotide can hybridize on one or more segments so that no intermediate or adjacent segments are involved in the hybridization event (e.g., forming a hairpin structure, "bulge", etc.).
- the nucleotide adaptor sequence includes a transposon end sequence.
- the transposon end sequence is Tn5 or a modified Tn5 transposon end sequence.
- the transposon end sequence is the Mu transposon end sequence.
- the Tn5 or modified Tn5 transposon end sequence or Mu transposon end sequence may comprise 15 to 25 nucleotides, for example, 16 nucleotides, 17 nucleotides, 18 nucleotides , 19 nucleotides, 20 nucleotides, 21 nucleotides, 22 nucleotides, 23 nucleotides, 24 nucleotides.
- Tn5 chimeric end sequence A14 Tn5MEA
- Tn5 chimeric end sequence B15 Tn5MEB
- NTS complementary non-transferred sequence
- Tn5MEA 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3’; (SEQ ID NO: 2)
- Tn5MEB 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3’; (SEQ ID NO: 3)
- Tn5NTS 5’-CTGTCTCTTATACACATCT-3’.
- the RNA is reverse transcribed before step a) in the method of the present application, and the attached target nucleic acid is produced.
- the first strand synthesis primer is used to synthesize the first strand of cDNA from mRNA in each mRNA sample.
- the first-strand synthetic primer includes an oligo dT primer.
- the first-strand synthetic primer used in the reverse transcription may be a reverse transcription primer that contains the oligonucleotide adaptor sequence and the polyT sequence in a 5'to 3'direction.
- the reverse transcription includes hybridizing the polyT sequence with the RNA and extending the polyT sequence in a template-directed manner.
- the first-strand synthetic primer is random.
- the first-strand synthetic primer is a mixture of an oligo dT primer and a random substance.
- the method further includes incorporating template-switching oligonucleotide primers (TSO primers) together with a mixture of oligo dT primers and random substances.
- TSO primers template-switching oligonucleotide primers
- the second strand of cDNA is synthesized using TSO primers.
- the second strand of cDNA is synthesized using a second amplification primer that is complementary to the first strand of cDNA, and the first strand extends beyond the mRNA template to include the complementary TSO strand.
- the target nucleic acid includes DNA derived from the single cell.
- the DNA includes genomic DNA
- the DNA includes genomic DNA, open chromatin DNA, protein-bound DNA regions, and/or exogenous nucleic acids linked to proteins, lipids and/or small molecule compounds, the proteins, lipids and/or small molecules
- the compound can bind to the target molecule in the cell.
- the protein may include antibodies and antigens.
- the target molecule may include a target nucleic acid sequence to be analyzed in a cell.
- the DNA derived from a single cell is fragmented before step a) in the method described in this application.
- DNA fragmentation can include separating or breaking DNA strands into small pieces or segments.
- a variety of methods can be used to fragment DNA, for example, the sequence of the oligonucleotide adaptor is attached after the DNA is fragmented (the sequence of the attached oligonucleotide adaptor under this condition is not Including the transposon end sequence), including restriction digestion or various methods of generating shear force.
- restriction digestion can use restriction enzymes to make nicks in the DNA sequence by cutting the blunt ends of the two strands or by uneven cutting to create sticky ends.
- shear force-mediated DNA strand destruction can include sonication, acoustic shearing, needle shearing, pipetting, or atomization.
- Sonication is a type of hydrodynamic shear that exposes DNA sequences to short-term shear forces, which can produce fragment sizes of about 700 bp.
- Acoustic shearing applies high-frequency acoustic energy to the DNA sample in the bowl-shaped transducer.
- Needle shearing generates shearing force by passing DNA through a small diameter needle to physically tear the DNA into smaller segments.
- the atomizing force can be generated by passing DNA through the small holes of the nebulizer unit, where the resulting DNA fragments are collected from the fine mist leaving the unit.
- these fragments can be any length between about 200 to about 100,000 bases.
- the fragment will be about 200 bp to about 500 bp, about 500 bp to about 1 kb, about 1 kb to about 10 kb, or about 5 kb to about 50 kb, or about 10 kb to about 30 kb, for example, about 15 kb to about 25 kb.
- the fragmentation of larger genetic components can be performed by any convenient method, for example, including commercially available shear-based fragmentation systems (for example, Covaris fragmentation system), size-targeted fragmentation systems (For example, Blue Pippin (Sage Science)), enzyme fragmentation methods (for example, DNA endonuclease, DNA exonuclease) and so on.
- the fragmentation includes using ultrasonic fragmentation, and then adding a sequence containing the oligonucleotide adaptor to the fragmented DNA, thereby obtaining the attached target nucleic acid.
- the attached target nucleic acid is produced after the fragmentation or during the fragmentation.
- the fragmentation includes using a transposase-nucleic acid complex to integrate the sequence containing the oligonucleotide adaptor into the DNA, and releasing the transposase to obtain the attached target Nucleic acid.
- the transposase includes Staphylococcus aureus Tn5 (Colegio et al., “Journal of Bacterology” (J. BacterioL), 183: 2384-8, 2001; Kirby C et al., "Molecular Microbiology” (Mol.
- the transposase-nucleic acid complex includes a transposase and a transposon end nucleic acid molecule, wherein the transposon end nucleic acid molecule includes the oligonucleotide adaptor sequence.
- the transposase is Mu transposase.
- the transposase is Tn5 transposase or Tn10 transposase.
- the Tn5 transposase is selected from the group consisting of full-length Tn5 transposase, partial functional domains of Tn5 transposase, mutations of Tn5 transposase.
- the Tn10 transposase is selected from the group consisting of full-length Tn10 transposase, partial functional domains of Tn10 transposase, and Tn10 transposase mutants.
- the Tn5 transposase mutant may be selected from: R30Q, K40Q, Y41H, T47P, E54K/V, M56A, R62Q, D97A, E110K, D188A, Y319A, R322A/K/Q, E326A, K330A/R, K333A, R342A, E344A, E345K, N348A, L372P, S438A, K439A, S445A, G462D, A466D.
- the two transposase molecules can bind to the same or different double-stranded DNA transposons, so that the insertion site is marked by one or two types of DNA.
- the two transposase molecules (such as Tn5 and superactive T years or other types of transposase containing point mutations) can be linked to one of the oligonucleotide adaptor sequence and another standard transposon
- the DNA sequence assembles into a hybrid transposition complex, or only the above-mentioned double-stranded structure 2 is used to form a single Tn5 transcomplex.
- the standard transposon DNA sequence may include an amplification primer sequence and/or a sequencing primer sequence.
- the DNA may include a DNA region that binds to a protein
- the transposase-nucleic acid complex may also include a portion that directly or indirectly recognizes the protein.
- the part that directly or indirectly recognizes the protein may include Staphylococcus aureus protein A (ProteinA), streptococcal protein G (ProteinG), streptococcal protein L (ProteinL) or other protein analogs that have the function of binding antibodies.
- the portion that directly or indirectly recognizes the protein may also include an antibody that specifically binds to the protein.
- Staphylococcus aureus protein A (ProteinA), streptococcal protein G (ProteinG), streptococcal protein L (ProteinL) or other protein analogs with the function of binding antibodies can each bind to the specific binding to the protein.
- Staphylococcus aureus protein A (ProteinA)
- streptococcal protein G (ProteinG)
- streptococcal protein L ProteinL
- other protein analogs with the function of binding antibodies can each bind to the specific binding to the protein.
- the transposase forms a fusion protein with the Staphylococcus aureus protein A (ProteinA), streptococcal protein G (ProteinG), streptococcal protein L (ProteinL) or other protein analogs that have the function of binding antibodies.
- Staphylococcus aureus protein A ProteinA
- streptococcal protein G ProteinG
- streptococcal protein L ProteinL
- other protein analogs that have the function of binding antibodies.
- the fusion protein binds to the antibody that specifically binds to the protein to form a complex, and then targets the protein.
- the antibody that specifically binds to the protein binds to the protein, and then the fusion protein binds to the antibody to target the protein.
- the oligonucleotide adaptor sequence may also include an antibody recognition sequence, which is used to recognize/track different antibodies.
- the antibody recognition sequence can be generated in a manner similar to random primers.
- the attached target nucleic acid includes a unique molecular identification region.
- the unique molecular identification region refers to a unique nucleic acid sequence attached to each of a plurality of nucleic acid molecules.
- UMI can be used to correct subsequent amplification bias by directly counting the unique molecular identification regions (UMI) sequenced after amplification.
- UMI includes identifying a single nucleic acid sequence in the barcoded target nucleic acid as derived from a given nucleic acid in the target nucleic acid based at least in part on the existence of the unique molecular identification region.
- UMI can be designed, incorporated and applied in a manner known in the art, for example, through WO 2012/142213, Islam “Nat.Methods” (2014) 11:163-166, and Kivioja, T. et al. "Nat.Methods” (2012) 9:72-74 As shown in the publication, each of the documents is incorporated herein by reference in its entirety.
- the unique molecular identification region is located between the oligonucleotide adaptor sequence and the target nucleic acid sequence.
- the target nucleic acid may also include an exogenous nucleic acid, which includes an exogenous nucleic acid linked to a protein, lipid, and/or small molecule compound, and the protein, lipid, and/or small molecule compound can interact with The target molecule within the cell binds.
- the protein may include antibodies and antigens.
- the target molecule may include a target nucleic acid sequence to be analyzed in a cell.
- the transposition reactions and methods described herein are performed in batches, and then the biological particles (e.g., nuclei/cells/chromatin from a single cell) are distributed so that multiple discrete partitions are covered by the biological particles (e.g., cells, nuclei). , Chromatin or cell beads) alone occupy.
- the biological particles e.g., cells, nuclei). , Chromatin or cell beads
- a plurality of biological particles may be allocated to a plurality of discrete partitions such that the discrete partitions of the plurality of discrete partitions include a single biological particle.
- the solid support may include beads.
- the beads may be porous, non-porous, and/or a combination thereof.
- the beads may be solid, semi-solid, semi-fluid, fluid, and/or combinations thereof.
- the beads may be soluble, destructible, and/or degradable.
- the beads may be non-degradable.
- the beads may be gel beads.
- the gel beads may be hydrogel beads. Gel beads can be formed from molecular precursors, such as polymers or monomer substances.
- the semi-solid beads may be liposomal beads.
- the solid beads may contain metals, including iron oxide, gold, and silver.
- the beads may be silica beads.
- the beads are magnetic beads.
- the beads can be rigid.
- the beads may be flexible and/or compressible.
- the beads can have any suitable shape.
- the shape of the beads may include, but is not limited to, spherical, non-spherical, elliptical, oblong, amorphous, circular, cylindrical, and deformed forms thereof.
- the beads may have a uniform size or a non-uniform size.
- the diameter of the beads may be at least about 10 nm, 100 nm, 500 nm, 1 ⁇ m, 5 ⁇ m, 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 70 ⁇ m, 80 ⁇ m, 90 ⁇ m, 100 ⁇ m, 250 ⁇ m, 500 ⁇ m, 1 mm or more.
- the diameter of the beads may be less than about 10 nm, 100 nm, 500 nm, 1 ⁇ m, 5 ⁇ m, 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 70 ⁇ m, 80 ⁇ m, 90 ⁇ m, 100 ⁇ m, 250 ⁇ m, 500 ⁇ m, 1 mm or less.
- the diameter of the beads can be in the range of about 40-75 ⁇ m, 30-75 ⁇ m, 20-75 ⁇ m, 40-85 ⁇ m, 40-95 ⁇ m, 20-100 ⁇ m, 10-100 ⁇ m, 1-100 ⁇ m, 20-250 ⁇ m, or 20-500 ⁇ m Inside.
- the beads may be provided in a bead population or multiple beads having a relatively monodisperse size distribution.
- maintaining relatively consistent bead characteristics can contribute to overall consistency.
- the beads described herein may have a coefficient of variation of their cross-sectional dimensions of less than 50%, less than 40%, less than 30%, less than 20%, and for example less than 15%, less than 10%, less than 5% or less The size distribution.
- the beads may comprise natural and/or synthetic materials.
- the beads may comprise natural polymers, synthetic polymers, or natural and synthetic polymers.
- Natural polymers may include proteins and sugars, such as deoxyribonucleic acid, rubber, cellulose, starch (e.g., amylose, pullulan), protein, enzyme, polysaccharide, silk, polyhydroxyalkanoate, chitosan , Dextran, collagen, carrageenan, plantago ovata, gum arabic, agar, gelatin, shellac, karaya, xanthan gum, corn syrup, guar gum, karaya, agarose, Alginic acid, alginate or its natural polymer.
- proteins and sugars such as deoxyribonucleic acid, rubber, cellulose, starch (e.g., amylose, pullulan), protein, enzyme, polysaccharide, silk, polyhydroxyalkanoate, chitosan , Dextran, collagen, carrageenan,
- Synthetic polymers can include acrylic, nylon, siloxane, spandex, viscose rayon, polycarboxylic acid, polyvinyl acetate, polyacrylamide, polyacrylate, polyethylene glycol, polyurethane, polylactic acid, dioxide Silicon, polystyrene, polyacrylonitrile, polybutadiene, polycarbonate, polyethylene, polyethylene terephthalate, polychlorotrifluoroethylene, polyethylene oxide, polyethylene terephthalate Glycol ester, polyisobutylene, polymethyl methacrylate, polyoxymethylene, polypropylene, polystyrene, polytetrafluoroethylene, polyvinyl alcohol, polyvinyl chloride, polyvinylidene chloride, polyvinylidene fluoride, poly Vinyl fluoride and/or combinations thereof (e.g., copolymers).
- the beads can also be formed of materials other than polymers, such as lipids, micelles, ceramics, glass ceramics, material composites,
- the beads may contain molecular precursors (e.g., monomers or polymers), which can form a polymer network through the polymerization of the molecular precursors.
- the precursor may be an already polymerized substance, which can be further polymerized by, for example, chemical crosslinking.
- the precursor may include one or more of acrylamide or methacrylamide monomers, oligomers, or polymers.
- the beads may contain a prepolymer, which is an oligomer that can be further polymerized.
- prepolymers can be used to prepare polyurethane beads.
- the beads may contain separate polymers that can be further polymerized together.
- beads can be produced by the polymerization of different precursors so that they comprise mixed polymers, copolymers and/or block copolymers.
- the beads can include covalent or ionic bonds between polymer precursors (e.g., monomers, oligomers, linear polymers), nucleic acid molecules (e.g., oligonucleotides), primers, and other entities.
- the covalent bond may be a carbon-carbon bond, a thioether bond, or a carbon-heteroatom bond.
- crosslinking can be permanent or reversible, depending on the specific crosslinking agent used.
- Reversible crosslinking can allow the polymer to be linearized or dissociated under appropriate conditions.
- reversible crosslinking can also allow the binding substance to be reversibly attached to the surface of the bead.
- crosslinking agents can form disulfide bonds.
- the chemical crosslinking agent that forms disulfide bonds may be cystamine or modified cystamine.
- disulfide bonds can be formed between molecular precursor units (e.g., monomers, oligomers, or linear polymers) or precursors incorporated into the beads and nucleic acid molecules (e.g., oligonucleotides).
- cystamine including modified cystamine
- cystamine is an organic reagent containing disulfide bonds, which can be used as a crosslinking agent between individual monomers of beads or polymer precursors.
- Polyacrylamide can be polymerized in the presence of cystamine or a substance containing cystamine (e.g., modified cystamine) to produce polyacrylamide gel beads containing disulfide bonds (e.g., containing chemically reducible cross-linked Chemically degradable beads). Disulfide bonds can allow the beads to degrade or dissolve when they are exposed to a reducing agent.
- chitosan a linear polysaccharide polymer
- glutaraldehyde a hydrophilic chain
- the cross-linking of chitosan polymers can be achieved by chemical reactions triggered by heat, pressure, pH changes and/or radiation.
- the beads can be macromolecules of single or mixed monomers polymerized by various monomers such as agarose, polyenamide, PEG, or macromolecular gels such as chitin, hyaluronic acid, and dextran.
- monomers such as agarose, polyenamide, PEG, or macromolecular gels such as chitin, hyaluronic acid, and dextran.
- macromolecular gels such as chitin, hyaluronic acid, and dextran.
- the microfluidic droplet platform in which the droplets aggregate into gel beads of uniform size.
- the beads may comprise an acrydite portion, which in certain aspects can be used to attach one or more nucleic acid molecules (e.g., barcode sequence, barcoded nucleic acid molecule, barcoded oligonucleotide, primer or other oligonucleotide) Receive beads.
- the acrydite moiety can refer to acrydite analogs produced by the reaction of acrydite with one or more substances, such as the reaction of acrydite with other monomers and crosslinkers during the polymerization reaction.
- the acrydite moiety can be modified to form a chemical bond with the substance to be attached, such as a nucleic acid molecule (e.g., barcode sequence, barcoded nucleic acid molecule, barcoded oligonucleotide, primer or other oligonucleotide).
- the acrydite moiety can be modified with a thiol group capable of forming a disulfide bond, or it can be modified with a group that already contains a disulfide bond. Thiols or disulfides (via disulfide exchange) can be used as anchor points for the substance to be attached, or another part of the acrydite part can be used for attachment.
- the attachment may be reversible, such that when the disulfide bond is broken (e.g., in the presence of a reducing agent), the attached substance is released from the beads.
- the acrydite moiety can contain reactive hydroxyl groups that can be used for attachment.
- it can also include other release methods, such as UV photo-induced release, or it can be released by enzymes.
- the present application provides a device for co-dispensing a solid support (such as beads) with a sample, for example, for co-dispensing sample components and beads to the same discrete partition.
- a solid support such as beads
- sample components and beads for example, for co-dispensing sample components and beads to the same discrete partition.
- the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag are co-distributed into the discrete partitions.
- the device can be formed of any suitable material.
- the device may be formed of a material selected from the group consisting of fused silica, soda lime glass, borosilicate glass, poly(methyl methacrylate) PMMA, PDMS, sapphire, silicon, germanium, cycloolefin copolymer, Polyethylene, polypropylene, polyacrylate, polycarbonate, plastics, thermosetting plastics, hydrogels, thermoplastics, paper, elastomers, and combinations thereof.
- the discrete partitions may include holes or droplets.
- the target nucleic acid derived from a single cell and the solid support to which at least one oligonucleotide tag is attached are co-dispensed into the wells or droplets.
- the wells may include sample loading holes of a cell culture plate or any other container wells that can cooperate with the device and are suitable for co-dispensing.
- the discrete partitions are droplets.
- each of the discrete partitions includes at most the target nucleic acid derived from a single cell.
- the target nucleic acid is located in a single cell or cell nucleus.
- a microfluidic device is used to co-distribute the target nucleic acid derived from a single cell and the solid support attached with at least one oligonucleotide tag into the discrete partitions.
- discrete partitions e.g., droplets or wells
- discrete partitions contain single cells and are processed according to the methods described in this application.
- discrete partitions contain single cells and/or single cell nuclei.
- Single cells and/or single cell nuclei can be allocated and processed according to the methods described in this application.
- a single cell nucleus can be an integral part of a cell.
- discrete partitions contain chromatin from a single cell or single cell nucleus (e.g., a single chromosome or other part of the genome), and are distributed and processed according to the methods described in this application.
- a ligase is also included in the discrete partition, and the ligase connects the oligonucleotide tag to the attached target nucleic acid.
- the discrete partition includes but is not limited to ligase, and may also include other required enzymes.
- DNA polymerases, DNA endonucleases, DNA exonucleases, terminal transferases, and light-sensitive enzymes capable of releasing the oligonucleotide tag from the solid support are pH-sensitive enzymes.
- the ligase includes T4 ligase, but is not limited to T4 ligase. For example, it may also include E.
- DNA ligase for example, DNA ligase I, DNA ligase Enzyme III, DNA ligase IV), thermostable ligase, etc.
- the device is formed in a manner that includes fluid flow channels. Any suitable channel can be used.
- the device includes one or more fluid input channels (e.g., inlet channels) and one or more fluid outlet channels.
- the inner diameter of the fluid channel may be about 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 85 ⁇ m, 90 ⁇ m, 100 ⁇ m, 125 ⁇ m, or 150 ⁇ m.
- the inner diameter of the fluid channel may be greater than 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 85 ⁇ m, 90 ⁇ m, 100 ⁇ m, 125 ⁇ m, 150 ⁇ m or more.
- the inner diameter of the fluid channel may be less than about 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 85 ⁇ m, 90 ⁇ m, 100 ⁇ m, 125 ⁇ m, or 150 ⁇ m.
- the volumetric flow rate in the fluid channel can be any flow rate known in the art.
- the microfluidic device is a droplet generator.
- a microfluidic device can be used to make the solid support attached with at least one oligonucleotide tag and the solid support attached with at least one oligonucleotide tag and the sample simultaneously formed A combination of samples (e.g., samples containing target nucleic acid).
- the small aqueous droplets serve as discrete partitions.
- the aqueous droplet may be an aqueous core surrounded by an oil phase, for example, an aqueous droplet in a water-in-oil emulsion.
- the aqueous droplet may contain one or more solid supports to which at least one oligonucleotide tag is attached, a sample, an amplification reagent, and a reducing agent.
- the aqueous droplet may contain one or more of the following: water, nuclease-free water, solid support attached with at least one oligonucleotide tag, acetonitrile, solid support, gel solid support Compounds, polymer precursors, polymer monomers, polyacrylamide monomers, acrylamide monomers, degradable crosslinkers, non-degradable crosslinkers, disulfide bonds, acrydite parts, PCR reagents, cells, nuclei , Chloroplasts, mitochondria, ribosomes, primers, polymerases, barcodes, polynucleotides, oligonucleotides, DNA, RNA, peptide polynucleotides, complementary DNA (cDNA), double-stranded DNA (dsDNA),
- the aqueous droplets can have a uniform size or an uneven size.
- the diameter of the aqueous droplet may be about 1 ⁇ m, 5 ⁇ m, 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 45 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 90 ⁇ m, 100 ⁇ m, 250 ⁇ m, 500 ⁇ m, or 1 mm.
- the fluid droplet may have a diameter of at least about 1 ⁇ m, 5 ⁇ m, 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 45 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 90 ⁇ m, 100 ⁇ m, 250 ⁇ m, 500 ⁇ m, 1 mm or more. .
- the fluid droplet may have a diameter of less than about 1 ⁇ m, 5 ⁇ m, 10 ⁇ m, 20 ⁇ m, 30 ⁇ m, 40 ⁇ m, 45 ⁇ m, 50 ⁇ m, 60 ⁇ m, 65 ⁇ m, 70 ⁇ m, 75 ⁇ m, 80 ⁇ m, 90 ⁇ m, 100 ⁇ m, 250 ⁇ m, 500 ⁇ m, or 1 mm.
- the fluid droplet may have a diameter of about 40-75 ⁇ m, 30-75 ⁇ m, 20-75 ⁇ m, 40-85 ⁇ m, 40-95 ⁇ m, 20-100 ⁇ m, 10-100 ⁇ m, 1-100 ⁇ m, 20-250 ⁇ m, or 20-500 ⁇ m. The diameter within the range.
- the microfluidic device e.g., a small droplet generator
- a sample e.g., nucleic acid sample
- a first fluid input channel fluidly connected to a first fluid intersection (e.g., a first fluid junction).
- a pre-formed solid support for example, a solid support to which at least one oligonucleotide tag is attached, such as a degradable solid support
- the second fluid input channel that is also fluidly connected to the first fluid intersection point , wherein the first fluid input channel and the second fluid input channel meet at the intersection of the first fluid.
- the sample and the solid support to which at least one oligonucleotide tag is attached can be mixed at the first fluid intersection to form a mixture (e.g., an aqueous mixture).
- the fourth fluid input channel can be provided with a reducing agent (or other required reagents, such as surfactants, stabilizers, polymers, aptamers, initiators, biotin markers, fluorophores, buffers, acidic solutions, Alkaline solution, light-sensitive enzyme, pH-sensitive enzyme, aqueous buffer, etc.), the fourth fluid input channel is also fluidly connected to the first fluid intersection, and is connected to the first and second fluid input channels in the first fluid Intersection points meet.
- the reducing agent can then be mixed with the solid support to which at least one oligonucleotide tag is attached and the sample at the first fluid intersection.
- the reducing agent (or other required reagents, such as surfactants, stabilizers, polymers, aptamers, initiators, biotin markers, fluorophores, buffers, acidic Solutions, alkaline solutions, light-sensitive enzymes, pH-sensitive enzymes, aqueous buffers, etc.) are premixed with the sample and/or the solid support to which at least one oligonucleotide tag is attached so as to pass through the first fluid input channel
- a sample is provided to the microfluidic device and/or a solid support to which at least one oligonucleotide tag is attached is provided to the microfluidic device through the second fluid input channel.
- the sample containing the target nucleic acid and the solid support mixture to which at least one oligonucleotide tag is attached can pass through a first fluid connected to the first fluid intersection (and to any fluid channel that constitutes the first fluid intersection).
- the outlet channel leaves the first fluid intersection.
- the mixture may be provided to a second fluid intersection (e.g., a second fluid junction) fluidly connected to the first outlet channel.
- a second fluid intersection e.g., a second fluid junction
- an oil (or other suitable immiscible) fluid can be fluidly connected from the point of intersection with the second fluid (and to any fluid channel that constitutes that point of intersection) and meets the first outlet channel at the second fluid intersection point.
- One or more separate fluid input channels enter the second fluid intersection.
- oil can be provided in one or two separate fluid input channels that are fluidly connected to the second fluid intersection (and to the first outlet channel) and that intersect with the first outlet channel and each other at the second fluid intersection point (or Other suitable immiscible fluids).
- the oil and the mixture of the sample and the solid support to which at least one oligonucleotide tag is attached can be mixed at the second fluid intersection.
- the formed aqueous droplets can be transported within the oil through the second fluid outlet channel exiting from the second fluid intersection.
- the formed aqueous droplets may also exit the second outlet channel from the first fluid intersection point and the fluid droplets may be dispensed into the holes for further processing.
- the sample containing the target nucleic acid is formed into droplets such that at least 50%, 60%, 70%, 80%, 90% or more of the droplets contain no more than one label with at least one oligonucleotide attached.
- Solid support allowing at least 50%, 60%, 70%, 80%, 90%, or more of the sample containing the target nucleic acid to form a droplet includes exactly one solid support to which at least one oligonucleotide tag is attached.
- the sample before the mixture enters the microfluidic device, the sample can be combined with a solid support that contains any other reagents (for example, an amplifying agent, a reducing agent, etc., required for sample amplification) to which at least one oligonucleotide tag is attached.
- the substance e.g., a degradable solid support
- the mixture can flow from the first fluid input channel and enter the fluid intersection.
- the oil phase may enter the fluid intersection from a second fluid input channel (for example, a fluid channel perpendicular or substantially perpendicular to the first fluid input channel) that is also fluidly connected to the fluid intersection.
- the aqueous mixture and oil can be mixed at the point of fluid intersection, so that a water-in-oil emulsion (e.g., a solid support-water-oil emulsion) is formed.
- the emulsion may contain a plurality of small aqueous droplets (e.g., small droplets containing an aqueous reaction mixture) in a continuous oil phase.
- each aqueous droplet may contain a single solid support (e.g., a gel solid support attached to the same set of barcodes), an aliquot of a sample (e.g., target nucleic acid from one cell), and any other reagents (E.g., reducing agent, reagents required for sample amplification, etc.).
- the fluid droplet may comprise a plurality of solid supports to which at least one oligonucleotide tag is attached.
- the droplets can be transported by the continuous oil phase through the fluid outlet channel away from the fluid intersection.
- the fluid droplets leaving the outlet channel can be dispensed into the holes for further processing.
- the fluid droplets formed at the second fluid intersection may contain the reducing agent.
- the reducing agent can degrade or dissolve the solid support contained in the fluid droplet when the droplet travels through the exit channel leaving the intersection of the second fluid.
- a microfluidic device may contain three discrete fluid intersection points in parallel. Liquid droplets can be formed at any of the three fluid intersection points.
- the sample and the solid support to which at least one oligonucleotide tag is attached can be mixed in any of the three fluid intersections.
- a reducing agent or other arbitrary and required reagents, such as a permeabilizing agent, an amplifying agent, a cutting agent that releases the oligonucleotide tag from the solid support
- Oil can be added at any of the intersections of these three fluids.
- the microfluidic device includes a first input channel and a second input channel, which merge at a junction fluidly connected to the output channel.
- the outlet channel may be fluidly connected with the third input channel at the junction.
- the method further includes introducing a sample containing the target nucleic acid into the first input channel, and introducing the solid support attached with at least one oligonucleotide tag into the second input channel, thereby A mixture of the sample and the solid support to which at least one oligonucleotide tag is attached is generated in the output channel.
- a fourth input channel may also be included and it may intersect the third input channel and the outlet channel at the junction.
- the microfluidic device may include first, second, and third input channels, where the third input channel intersects the first input channel, the second input channel, or the junction of the first input channel and the second input channel.
- the output channel and the third input channel are fluidly connected at the junction.
- the first input channel and the second input channel form a substantially perpendicular angle to each other.
- each of the discrete partitions contains at most the target nucleic acid from a single cell.
- oil can be used to produce droplets.
- the oil may include fluorinated oil, silicone oil, mineral oil, vegetable oil, and combinations thereof.
- the aqueous fluid in the microfluidic device may also contain alcohol.
- the alcohol can be glycerol, ethanol, methanol, isopropanol, pentanol, ethane, propane, butane, pentane, hexane, and combinations thereof.
- the alcohol can be at about 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19% or 20% (v/v) exists in the aqueous fluid.
- the alcohol may be at least about 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, Concentrations of 19%, 20% or higher (v/v) are present in the aqueous fluid.
- the alcohol may be less than about 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19% or 20% (v/v) is present in the aqueous fluid.
- the oil may also contain surfactants to stabilize the emulsion.
- the surfactant may be a fluorosurfactant, Krytox lubricant, Krytox FSH, engineered fluid, HFE-7500, silicone compound, PEG-containing silicon compound, such as bis krytoxpeg (BKP).
- the surfactant can be used at about 0.1%, 0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 5% or 10% %(W/w) exists.
- the surfactant can be at least about 0.1%, 0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 5 %, 10% (w/w) or higher concentration exists.
- the surfactant may be less than about 0.1%, 0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 5 % Or 10% (w/w) is present.
- accelerators and/or initiators can be added to the oil.
- the accelerator may be tetramethylethylenediamine (TMEDA or TEMED).
- the initiator may be ammonium persulfate or calcium ion.
- the accelerator can be at a rate of about 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5% , 1.6%, 1.7%, 1.8%, 1.9% or 2% (v/v) are present.
- the accelerator may be at least about 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%, 1.4% , 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, or 2% (v/v) or higher concentration.
- the accelerator may be less than about 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%, 1.4% , 1.5%, 1.6%, 1.7%, 1.8%, 1.9% or 2% (v/v) are present.
- the cell is a cell of any organism.
- the cells of the organism may be in vitro cells (for example, an established cultured cell line), or may be isolated cells (cultured cells from an individual, primary cells).
- the cell may be a cell in the body (a cell in a biological individual), for example, a cell from various tissues.
- the biological cells may include animal cells, plant cells, and microbial cells.
- the plant cell may include Arabidopsis thaliana cells, and may also include cells of agricultural crops, such as plant somatic cells such as wheat, corn, rice, sorghum, millet, soybean, etc.; the plant cells may also include cells of fruit and nut plants , For example, produce apricots, oranges, lemons, apples, plums, pears, almonds, walnuts and other plants.
- the plant cell may be a cell derived from any part of the plant body, for example, root cells, leaf cells, xylem cells, phloem cells, cambium cells, apical meristem cells, parenchyma cells.
- the microbial cells may include bacteria (e.g. Escherichia coli, archaea), fungi (e.g. yeast), actinomycetes, rickettsiae, mycoplasma, chlamydia, spirochetes, and the like.
- the animal cell may include invertebrate (e.g., Drosophila, nematode, planarian, etc.) cells, vertebrate (e.g., zebrafish, chicken, mammalian) cells.
- invertebrate e.g., Drosophila, nematode, planarian, etc.
- vertebrate e.g., zebrafish, chicken, mammalian cells.
- the mammalian cells may include mice, rats, rabbits, pigs, dogs, cats, monkeys, humans, and the like.
- the animal cells may include cells from any tissue of the organism, such as stem cells, induced pluripotent stem (iPS) cells, germ cells (eg oocytes, egg cells, sperm cells, etc.), adult stem cells, somatic cells ( For example, fibroblasts, hematopoietic cells, cardiomyocytes, neurons, muscle cells, bone cells, liver cells, pancreatic cells, epithelial cells, immune cells and those derived from lung, spleen, kidney, stomach, large intestine, small intestine and other organs or tissues Any cell) and embryos at any stage in vitro or in vivo.
- stem cells eg oocytes, egg cells, sperm cells, etc.
- germ cells eg oocytes, egg cells, sperm cells, etc.
- adult stem cells e.g., etc.
- somatic cells fibroblasts, hematopoietic cells, cardiomyocytes, neurons, muscle cells, bone cells, liver cells, pancreatic cells, epit
- the cell may be a cell derived from a biological fluid.
- the body fluid of the organism may include cerebrospinal fluid, aqueous humor, lymph, digestive juice (e.g., saliva, gastric juice, small intestinal fluid, bile, etc.), breast milk, blood, urine, sweat, tears, feces, respiratory secretions, reproduction Organ secretions (such as semen, cervical mucus), etc.
- the sample includes the cell and/or the nucleus obtained therefrom.
- the sample may include nucleic acid molecules of the organism.
- the nucleic acid molecule can be isolated and extracted from any organism by the technical means known to those skilled in the art to separate nucleic acid molecules, including DNA and RNA.
- the nucleic acid molecule is extracted from the aforementioned biological cells or body fluids of the biological body.
- the target nucleic acid may include nucleic acid derived from any of the aforementioned cells.
- nucleic acid in a single cell may be included in the target nucleic acid.
- the target nucleic acid may be derived from a single cell polynucleotide, for example, double-stranded DNA.
- the double-stranded DNA may include genomic DNA, for example, coding DNA and non-coding DNA; for example, open chromatin region DNA, protein binding site DNA, mitochondrial DNA and chloroplast DNA, for example, the polynucleotide may include RNA, for example Ribosomal RNA, mRNA.
- the target nucleic acid can also be a sample containing cells from a formalin-fixed and paraffin-embedded (Formalin-Fixed and Parrffin-Embedded, FFPE).
- FFPE Formin-Fixed and Parrffin-Embedded
- the target nucleic acid may also include a sequence containing a SNP site in the genome of an organism, and a nucleotide sequence modified by methylation or hydroxymethylation.
- the cells can also be pretreated.
- the pretreatment also includes exposing the nucleus of the cell.
- cell nuclei can be exposed by treatment with lysis buffer and concentrated sucrose solution.
- the cell and/or the cell nucleus exposed (obtained) therefrom may be encapsulated in a suitable matrix to form microspheres, and the microspheres are used as a sample for reaction.
- the pretreatment includes fixing the cell and/or the cell nucleus exposed (obtained) therefrom.
- a fixative is used to fix the cells, and the fixative is selected from one or more of the following group: formaldehyde, paraformaldehyde, methanol, ethanol, acetone, glutaraldehyde, osmic acid and dichromic acid Potassium.
- the pretreatment includes treating the cells or cell nucleus with a detergent, and the detergent includes Triton, NP-40 and/or digitonin.
- the pretreatment may also include the removal of organelles such as mitochondria, chloroplasts, and ribosomes.
- organelles such as mitochondria, chloroplasts, and ribosomes.
- the cells can be dispensed with the lysis reagent to release the contents of the cells in the discrete subregions.
- the lytic agent is brought into contact with the cell suspension at the same time that the cells are introduced into the droplet generation area through the additional channel, or when the cells are about to be introduced into the droplet generation area.
- the lysing agent may include biologically active agents, such as lysing enzymes for lysing different cell types (e.g., gram positive or negative bacteria, plants, yeast, mammals, etc.), such as lysozyme, leuco peptidase, Lysostaphin, thioglucosidase kitalase, lyticase, and other commercially available lytic enzymes.
- lytic agents can also be co-partitioned with the cells so that the contents of the cells are released into discrete partitions.
- a surfactant-based dissolving solution may be used to dissolve the cells, for example, the dissolving solution may include a nonionic surfactant such as Triton X-100 and Tween 20.
- the dissolving solution may include ionic surfactants such as sodium lauryl sarcosinate and sodium dodecyl sulfate (SDS).
- SDS sodium dodecyl sulfate
- other methods that can be used such as electroporation, heat, sound, or mechanical cell destruction
- electroporation, heat, sound, or mechanical cell destruction can also be used for lysis.
- the present application also provides a composition
- a composition comprising: a plurality of solid supports, each of said solid supports is attached with at least one oligonucleotide tag, wherein each of said oligonucleotide tags comprises a first A strand and a second strand, the first strand includes a barcode sequence and a hybridization sequence at the 3'end of the barcode sequence, and the second strand includes a first portion complementary to the hybrid sequence of the first strand and The second part of the sequence of the nucleic acid to be tested is complementary, and the first strand and the second strand form a partially double-stranded structure or the second strand and the attached target nucleic acid form a partially double-stranded structure Structure; the barcode sequence of the oligonucleotide tag includes a common barcode domain and a variable domain, and the common barcode domain is the same in the oligonucleotide tag attached to the same solid support, and The common barcode domain is different between two or more solid supports in
- the application also provides a kit for analyzing target nucleic acid from cells, which includes the composition described in the application.
- the kit may also include a transposase.
- the kit further includes at least one of a nucleic acid amplification agent, a reverse transcription agent, a fixative, a permeabilizing agent, a linking agent, and a lysing agent.
- the nucleotide tag has two strands, forming a partial double-stranded structure 1, as shown below:
- Chain I solid support ⁇ attachment sequence-barcode sequence (barcode)-hybridization sequence (fixed sequence, hybridizing with the complementary part of chain II), wherein the barcode sequence (barcode) is (barcode-linker) n greater than or equal to 1.
- Bead-acrydite-S-S-ACACTCTTTCCCTACACGACGCTCTTCCGATCT read1, SEQ ID NO: 6
- barcode-ATCCACGTGCTTGAG SEQ ID NO: 12
- Strand II Hybrid sequence (fixed sequence, hybridized with fixed DNA sequence in strand I)-a sequence complementary to the 5'end of strand I of the transposon complex
- the solid support is polyacrylamide microspheres, which are prepared by a microfluidic device.
- Acrylamide Bis mixture, acrydite-DNA primer and APS inducer are mixed in a microfluidic device to form droplets, which contain TEMED catalyst, The droplets will spontaneously polymerize into gel microspheres, and then the microspheres will be labeled according to the barcode synthesis method.
- the solution contains 10mM DTT, and the S-S bond can be reduced to release the primer.
- Chain A Phosphate group-a sequence that is at least partially complementary to the fixed DNA sequence in chain I or chain II of the nucleic acid molecule in chain II-(UMI)-the sequence bound by Tn5 transposase
- AGGCCAGAGCATTCGNNNNNNNAGATGTGTATAAGAGACAG (SEQ ID NO: 5)
- Chain B Tn5 transposase binding sequence (sequence complementary to the sequence binding to the transposon protein (Tn5) in chain A)-phosphate group
- the UMI in the A chain is not necessary; the sequences in (1) and (2) can contain modified bases, such as 5mC.
- the Tn5 transposable complex is a dimer.
- Two Tn5 proteins can bind to the same or different partial double-stranded DNA transposons, so that the insertion site is marked by one or two types of DNA;
- Tn5 protein (which can contain point mutation super Active or other types of transposase) can be combined with the above double-stranded structure 2 and another standard transposon DNA to assemble into a hybrid transposable complex, or only the above-mentioned double-stranded structure 2 can be used to form a single Tn5 transposition.
- Complex can be used to form a single Tn5 transposition.
- samples can be non-fixed cells or nuclei, formaldehyde (or other fixatives) fixed cells or nuclei, non-fixed or fixed tissue sections, etc.
- the fixed or non-fixed sample is treated with a buffer containing detergents (Triton, NP-40 or Digitonin, etc.), and it can also include an intermediate step of lysing cells (non-fixed samples) to obtain cell nuclei.
- Typical penetrant solutions can include Tris, sucrose, sodium chloride, detergents.
- Tn5 enzyme buffer containing divalent metal ion for example, magnesium ion
- the reaction system includes: cell or cell nucleus or tissue; Tn5 transposition complex; buffer. After the reaction, the sample was washed with buffer to remove unreacted Tn5 enzyme.
- the reaction system includes: cell or nucleus or tissue (after the transposition reaction); T4 DNA ligase, nucleotide tag, after the reaction, add excess free and nucleotide tag complementary sequences to the ligation reaction system to block excess unreacted Nucleotide tag.
- Tn5 enzyme purchased from Epicenter
- 10uM Tn5 enzyme purchased from Epicenter
- the transposon formed by the Top1/Bottom double strand and Tn5 is p-Tn5
- the Top2/Bottom double strand is formed by Tn5.
- the transposon is Tn5-B.
- the PCR adaptor sequence is ACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 6)
- the connection sequence 1 (Linker1) is CGACTCACTACAGGG (SEQ ID NO: 7)
- the connection sequence 2 (Linker 2) sequence is TCGGTGACACGATCG (SEQ ID NO: 8)
- the synthesized microspheres were evenly divided into 96-well plates, and PCR handle-96xbarcode1-linker1 was added respectively, and the first round of barcoding reaction was performed.
- the reaction system and process are as follows: 10ul microspheres+2ul BstI buffer+1ul 10uM dNTP+1ul 100uM PCR handle-96xbarcode1-linker1, then keep at 95°C for 5min, 60°C for 20min; then add 1ul BstI+5ul H 2 O, hold at 60°C for 60 min.
- microspheres After the microspheres are washed, they are annealed with the complementary sequence CGAATGCTCTGGCCTCAAGCACGTGGAT (SEQ ID NO: 9) to form a partial double-stranded structure, and finally the following micro-beads with a partial double-stranded structure attached are obtained.
- ball the complementary sequence CGAATGCTCTGGCCTCAAGCACGTGGAT (SEQ ID NO: 9) to form a partial double-stranded structure, and finally the following micro-beads with a partial double-stranded structure attached are obtained.
- the human 293T cell line was resuspended in lysis buffer (10mM Tris-Cl, pH 7.4; 10mM NaCl; 3mM MgCl2; 0.01% NP-40) to lyse the cells to obtain cell nuclei.
- reaction system Take 100,000 cell nuclei and react with the p-Tn5 and Tn5-B obtained in step (1).
- the reaction system is as follows:
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- step D Add an equal volume of perfluorooctanol breaker droplets to the droplets in step D, centrifuge, extract the water phase, use Qiagen DNA purification kit to purify the DNA in the water phase, and use the following reaction system to amplify the DNA to obtain the final sequencing library: 36ul DNA Template, 10ul 5xPCR Buffer, 1ul 10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul Taq, 94°C 2min, 94°C 30sec, 55°C 30sec, 72c 30sec, 18 cycles.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- the above library invested about 500 cells, each cell was sequenced 100,000 PE150reads, and the total data volume was 15G.
- the sequenced fragment size presents a typical ATAC nucleosome gradient (see Figure 5), and the signal-enriched transcription start site (TSS) presents a typical ATAC signal (see Figures 6A and 6B).
- TSS signal-enriched transcription start site
- the peaks overlap with the known open area (see Figure 7), where the total peak value is 11898, and the peaks overlapped with union DHS ratio (Peaks overlapped with union DHS ratio) is 74.0%.
- the peak ratio of the blacklist Peaks overlaped with blacklist ratio
- FRiP Fraction of reads in peaks, the read length score falling into the peak domain
- CUT&Tag is the latest method to study the interaction between DNA and protein, instead of the traditional ChIP-seq method. Its principle is to use a protein A (a cell-derived protein that can bind to the conserved region of antibody heavy chains from different species). ) A protein fused with Tn5, through the binding of protein A and antibody, the Tn5 enzyme is targeted to the target protein bound by the antibody, and the DNA fragment is directly inserted into the DNA region bound by the target protein through the transposition activity of the Tn5 enzyme. This product is amplified and sequenced to directly obtain the binding position information of the protein.
- the molecular product of CUT&Tag is the same as ATAC, the difference is that the Tn5 enzyme insertion site in ATAC is in the open chromatin region, and the Tn5 insertion site in CUT&Tag is around the target protein, so it can be used similar to ATAC in Example 1.
- Method to label this product the DNA transposon used is similar to ATAC, and it can also assemble one or hybrid Tn5 transposition complex. The different steps are: use protein A or G-Tn5 fusion protein to assemble the Tn5 transposition complex; in order to distinguish between multiple antibodies, in addition to the ATAC Tn5 sequence, the DNA transposon can also contain antibody identification codes at different positions. Used to distinguish multiple antibodies.
- Sample preparation it can be non-fixed cells or nuclei, cells or nuclei fixed with formaldehyde (or other fixatives), non-fixed or fixed tissue sections, etc.
- the fixed or non-fixed sample is treated with a buffer containing detergents (Triton, NP-40 or Digitonin, etc.), and it can also include an intermediate step of lysing cells (non-fixed samples) to obtain cell nuclei. Permeate the cell and nucleus so that the Tn5 enzyme can enter the nucleus for action.
- a buffer containing detergents Triton, NP-40 or Digitonin, etc.
- Transposition reaction Use protein A-Tn5 fusion protein (primary antibody-protein A-Tn5 fusion protein complex) to bind the sample, wash the excess enzyme, and then add the Tn5 reaction solution containing divalent ions to the sample to carry out the transposition reaction, (37°C, 30 minutes to 2 hours).
- protein A-Tn5 fusion protein primary antibody-protein A-Tn5 fusion protein complex
- the nucleotide tag has two strands, forming a partial double-stranded structure 1, as shown below:
- Chain I solid support ⁇ attachment sequence-barcode sequence (barcode)-hybridization sequence (fixed sequence, hybridizing with the complementary part of chain II), wherein the barcode sequence (barcode) is (barcode-linker) n greater than or equal to 1.
- Bead-acrydite-S-S-ACACTCTTTCCCTACACGACGCTCTTCCGATCT read1, SEQ ID NO: 6
- barcode-ATCCACGTGCTTGAG SEQ ID NO: 12
- Strand II Hybrid sequence (fixed sequence, hybridized with fixed DNA sequence in strand I)-a sequence complementary to the 5'end of strand I of the transposon complex
- the solid support is polyacrylamide microspheres, which are prepared by a microfluidic device.
- Acrylamide Bis mixture, acrydite-DNA primer and APS inducer are mixed in a microfluidic device to form droplets, which contain TEMED catalyst, The droplets will spontaneously polymerize into gel microspheres, and then the microspheres will be labeled according to the barcode synthesis method.
- the solution contains 10mM DTT, and the S-S bond can be reduced to release the primer.
- Chain A Phosphate group-a sequence that is at least partially complementary to the fixed DNA sequence in chain I or chain II of the nucleic acid molecule in chain II-(UMI)-the sequence bound by Tn5 transposase
- AGGCCAGAGCATTCGNNNNNNNAGATGTGTATAAGAGACAG (SEQ ID NO: 5)
- Chain B Tn5 transposase binding sequence (sequence complementary to the sequence binding to the transposon protein (Tn5) in chain A)-phosphate group
- the UMI in the A chain is not necessary; the sequences in (1) and (2) can contain modified bases, such as 5mC.
- the Tn5 transposition complex is a dimer.
- Two pA-Tn5 proteins can bind to the same or different partial double-stranded DNA transposons, so that the insertion site is marked by one or two types of DNA;
- pA-Tn5 protein can be Containing point mutation hyperactivity or other types of transposase
- the equimolar concentration of pA-Tn5 protein and the annealed double-stranded primer are mixed and placed at room temperature for more than 1 hour to form a functional transposon complex.
- samples can be non-fixed cells or nuclei, formaldehyde (or other fixatives) fixed cells or nuclei, non-fixed or fixed tissue sections, etc.
- the fixed or non-fixed sample is treated with a buffer containing detergents (Triton, NP-40 or Digitonin, etc.), and it can also include an intermediate step of lysing cells (non-fixed samples) to obtain cell nuclei. Permeate the cell and the nucleus, so that the antibody and pA-Tn5 enzyme can enter the nucleus for action.
- Typical penetrant solutions can include Tris, sucrose, sodium chloride, detergents.
- the antibody against the target protein is incubated with the sample so that the antibody specifically binds to the target protein, and the unbound antibody is removed by washing. Then incubate the pA-Tn5 transposon with the sample, so that the pA-Tn5 protein binds to the antibody, thereby positioning it near the target protein.
- Tn5 enzyme buffer containing divalent metal ion for example, magnesium ion
- the transposition reaction 37°C, 30 minutes to 2 hours. That is, the reaction system includes: cell or cell nucleus or tissue; buffer. After the reaction, the sample is washed with buffer to remove unreacted reagents.
- the reaction system includes: cell or nucleus or tissue (after the transposition reaction); T4 DNA ligase, nucleotide tag, after the reaction, add excess free and nucleotide tag complementary sequences to the ligation reaction system to block excess unreacted Nucleotide tag.
- transposome A.pA-Tn5 transposome (transposome)
- the pA-Tn5 transposon was assembled into a 10uM concentration of pA-Tn5 transposon.
- the transposon formed by the Top1/Bottom double strand and Tn5 is p-pA-Tn5
- Top2/Bottom The transposon formed by the double strand and Tn5 is pA-Tn5-B.
- the PCR adaptor sequence is ACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 6)
- the connection sequence 1 (Linker1) is CGACTCACTACAGGG (SEQ ID NO: 7)
- the connection sequence 2 (Linker 2) sequence is TCGGTGACACGATCG (SEQ ID NO: 8)
- the synthesized microspheres were evenly divided into 96-well plates, and PCR handle-96xbarcode1-linker1 was added respectively, and the first round of barcoding reaction was performed.
- the reaction system and process are as follows: 10ul microspheres+2ul BstI buffer+1ul 10uM dNTP+1ul 100uM PCR handle-96xbarcode1-linker1, then keep at 95°C for 5min, 60°C for 20min; then add 1ul BstI+5ul H 2 O, hold at 60°C for 60 min.
- microspheres After the microspheres are washed, they are annealed with the complementary sequence CGAATGCTCTGGCCTCAAGCACGTGGAT (SEQ ID NO: 9) to form a partial double-stranded structure, and finally the following micro-beads with a partial double-stranded structure attached are obtained.
- ball the complementary sequence CGAATGCTCTGGCCTCAAGCACGTGGAT (SEQ ID NO: 9) to form a partial double-stranded structure, and finally the following micro-beads with a partial double-stranded structure attached are obtained.
- the human cell line 293T were resuspended in lysis buffer (10mM Tris-Cl, pH 7.4 ; 10mM NaCl; 3mM MgCl 2; 0.01% NP-40) cells were lysed to obtain a cell nucleus.
- the binding conditions are as follows: 0.05% Digitonin, 20mM HEPES, pH 7.5, 300mM NaCl, 0.5mM Spermidine, 1X Protease inhibitor (Roche) buffer In, the antibody concentration is 1ug/100ul, bind at room temperature for 1hr or 4 degrees Celsius overnight.
- the target protein antibody such as anti-histone H3K4me3 antibody (Abcam)
- the binding conditions are as follows: 0.05% Digitonin, 20mM HEPES, pH 7.5, 300mM NaCl, 0.5mM Spermidine, 1X Protease inhibitor (Roche) buffer
- the antibody concentration is 1ug/100ul, bind at room temperature for 1hr or 4 degrees Celsius overnight.
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- cell nucleus solution 100 cell nucleus/ul concentration
- cell nucleus solution 100 cell nucleus/ul concentration
- concentration 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- step D Add an equal volume of perfluorooctanol breaker droplets to the droplets in step D, centrifuge, extract the water phase, use Qiagen DNA purification kit to purify the DNA in the water phase, and use the following reaction system to amplify the DNA to obtain the final sequencing library: 36ul DNA Template, 10ul 5xPCR Buffer, 1ul 10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul Taq, 94°C2min, 94°C30sec, 55°C30sec, 72°C30sec, 18 cycles.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- the above library invested about 500 cells, each cell was sequenced 100,000 PE150reads, and the total data volume was 15G.
- FIG. 13 shows the results of the Cuttag library fragment distribution.
- Figure 14 shows the results of the distribution of Cuttag fragments at the transcription start site.
- Figure 15 shows the proportion of Cuttag fragments distributed in the genome.
- Figure 16 shows the distribution results of single-cell Cuttag results.
- the superimposed single-cell data shows typical H3K4me3 histone modification distribution characteristics, which are highly similar to the experimental results of multi-cell samples, indicating that the single-cell data obtained by this method is true and accurate sex.
- a buffer containing detergents Triton, NP-40, Digitonin, etc.
- Triton, NP-40, Digitonin, etc. may include intermediate steps of lysing cells (non-fixed samples) to obtain nuclei, detergent lysis or permeabilization Cells and nuclei allow molecular biology reagents such as enzymes to enter the cells or nucleus.
- step (3) Use the reverse transcription primer of step (1) to provide a reverse transcriptase reaction system, add a chain conversion template, and perform an intracellular reverse transcription reaction on the sample. After the reaction, the cell/nucleus is still in an independent and complete form.
- the reaction system and conditions are as follows: cells/tissues, reverse transcriptase buffer, RNase inhibitor, dNTP, TSO chain conversion primer, reverse transcription primer; 50-55°C, 5 minutes, 4°C + reverse transcriptase, 42°C. Wash and remove primers and enzyme system, and carry out nucleotide tag ligation reaction on cells or tissues. After the end, add primers to neutralize excess primers.
- Purify mRNA/cDNA directly purify mRNA/cDNA from non-fixed tissue, and purify mRNA/cDNA after uncrosslinking of fixed tissue; perform PCR amplification of cDNA on mRNA/cDNA to obtain cDNA library, and use Tn5 or other DNA interruption methods to cDNA library Construct a sequencing library.
- Cell nucleus homogenize the tissue in 10mM Tris-Cl, pH 7.4; 10mM NaCl; 3mM MgCl 2 ; 0.01% NP-40 buffer, lyse the cells, centrifuge at 500g for 5min, resuspend once with buffer, centrifuge at 500g for 5min, and resuspend in the above buffer.
- each component is as follows: 1000/ul cell nucleus, 1x RT Buffer, 1uM dNTP, 1uM above reverse transcription primer, 1u/ul RNase enzyme inhibitor, 1uM TSO primer primer sequence (5′-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG(SEQ ID) NO:14)-3', where the G at the 3 end can be rG, rG represents ribose guanine, 1 unit/ul RT enzyme (Superscript II reverse transcriptase); reaction conditions: 50°C 5min, 4°C 5min, 42°C 60min, Wash the nucleus with PBS, centrifuge at 500g 5min and wash twice to remove unreacted enzymes and primers.
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme. 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles. Purify the library with AMPure XP magnetic beads in a 1:1 volume.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- the above library invested about 500 cells, each cell was sequenced 100,000 PE150reads, and the total data volume was 15G.
- Figure 17 shows that the single cell result clearly distinguishes a single cell of the two types of cells.
- Figure 18 shows the distribution of the number of transcripts and genes detected in each cell. The method of this application can be used for single cell transcriptome detection.
- a single-cell genome experiment can be performed on the two mixed cells 293T (human) and 3T3 (mouse) using the method of this application. According to the ratio of the measured sequence alignment to the human or mouse genome, Figure 19 shows that the single cell result clearly distinguishes a single cell of the two types of cells.
- FIG. 20 shows that the coverage of the genome of a single human cell, according to the arrangement of the chromosomes, shows that single-cell sequencing has a different degree of coverage in each cell and each genome site.
- the method of this application can be used for single cell genome detection.
- the method of the present application can also be used to distinguish single cells from various cells in mixed cells through genome and transcriptome detection.
- nucleotide tag is the same as in Example 1. The only difference is that in the 5'amine-S-S-ACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 6) sequence coupled to the microsphere, all C bases are replaced with 5mC modified bases.
- Sample Fixed single cell or cell nucleus.
- Sample processing method Cells or cell nuclei are treated with a certain concentration of SDS and/or other detergents and heated for a certain period of time to remove the protein bound to the DNA, but does not untie the cross-links, so the DNA is still fixed In the cell structure.
- the reaction system includes: cell or cell nucleus or tissue (after transposition reaction); T4 DNA ligase, nucleotide tag, T4 DNA ligase. After the reaction, excess free and nucleotide tag complementary sequences are added to the ligation reaction system to block the excess unreacted nucleotide tag.
- CNV copy number information
- SNV point mutation information
- the connecting primer is designed to resist the transformed base or the modified base to ensure amplification.
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- a transformation kit such as EpiTect Fast Bisulfite Conversion Kit or NEB Enzymatic Methylation conversion kit to transform the genomic DNA obtained above, for example, Qiagen kit as an example, refer to the instructions to configure the bisulfite conversion reagent.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Recover DNA 12.5 ⁇ L 4X Enzyme Reaction Buffer, 10ul 5-hmC Modifying Enzyme, add water to 50ul, and react at 30°C for 1hr. Purify DNA with a 1:1 volume of magnetic beads.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Figure 23 shows the results of the distribution of 5hmC modification sites in a single cell.
- the single-cell 5hmC modification data obtained in this application is true and accurate.
- the dT primer and Tn5 enzyme containing the same 5'end linking sequence were used. Prepare the cell nucleus, and then perform RT (reverse transcription) reaction on the cells, wash and remove the RT reaction system, and then perform the Tn5 ATAC reaction, and then the mRNA and ATAC in the cell will be labeled at the same time. Then connect to the primer released on the microsphere. Recover ATAC DNA and RT mRNA/cDNA mixture.
- the human cell line 293T were resuspended in lysis buffer (10mM Tris-Cl, pH 7.4 ; 10mM NaCl; 3mM MgCl 2; 0.01% NP-40) cells were lysed to obtain a cell nucleus.
- reaction system Take 100,000 cell nuclei and react with p-Tn5 and Tn5-B obtained in the examples of this application.
- the reaction system is as follows:
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution bead solution, oil (FC40 fluorocarbon oil, containing 1% surfactant FluoroSurfactant, Ran Biotech) form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- oil FC40 fluorocarbon oil, containing 1% surfactant FluoroSurfactant, Ran Biotech
- reaction system to amplify DNA to obtain the final sequencing library: 36ul DNA template, 10ul 5xPCR Buffer, 1ul 10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul 10mM ISPCR primer, 1ul Taq, 72°C 5min, 94°C 2min, 94°C30sec, 55°C30sec, 72°C3min, 12 cycles.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme, 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- This application is used to detect the transcriptome and ATAC of the same cell at the same time.
- Figure 24 shows that a single cell of the two types of cells can be well distinguished based on the transcriptome and ATAC genome.
- the method of this application is used to simultaneously detect the transcriptome and ATAC of the same cell with accuracy.
- the human cell line 293T were resuspended in lysis buffer (10mM Tris-Cl, pH 7.4 ; 10mM NaCl; 3mM MgCl 2; 0.01% NP-40) cells were lysed to obtain a cell nucleus.
- the binding conditions are as follows: 0.05% Digitonin, 20mM HEPES, pH 7.5, 300mM NaCl, 0.5mM Spermidine, 1X Protease inhibitor (Roche) buffer In, the antibody concentration is 1ug/100ul, bind at room temperature for 1hr or 4°C overnight.
- the target protein antibody such as anti-histone H3K4me3 antibody (Abcam)
- the binding conditions are as follows: 0.05% Digitonin, 20mM HEPES, pH 7.5, 300mM NaCl, 0.5mM Spermidine, 1X Protease inhibitor (Roche) buffer
- the antibody concentration is 1ug/100ul, bind at room temperature for 1hr or 4°C overnight.
- the cell nucleus obtained above is subjected to the following RT reaction and the final concentration of each component is as follows: 1000/ul cell nucleus, 1x RT Buffer, 1uM dNTP, 1uM above reverse transcription primer, 1u/ul RNase enzyme inhibitor, 1uM TSO primer primer sequence (5′- AAGCAGTGGTATCAACGCAGAGTACATrGrGrG(SEQ ID NO: 14)-3', where the G at the 3 end can be rG, rG represents ribose guanine, 1 unit/ul RT enzyme (Superscript II reverse transcriptase); reaction conditions: 50°C for 5 minutes, 4°C for 5 minutes, At 42°C for 60 minutes, wash the nucleus with PBS, centrifuge at 500g for 5 minutes and wash twice to remove unreacted enzymes and primers.
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- reaction system uses the following reaction system to amplify DNA to obtain the final sequencing library: 36ul DNA template, 10ul 5xPCR Buffer, 1ul10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul 10mM ISPCR primer, 1ul Taq, 72°C 5min, 94°C 2min, 94 °C30sec, 55°C30sec, 72°C3min, 12 cycles.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- DNA template 10ul 5xPCR Buffer, 1ul 10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul Taq 94°C2min, 94°C30sec, 55°C30sec, 72°C30sec, 18 cycles
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme, 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Figure 25 shows that, according to both the transcriptome and the cut tag group, a single cell of the two types of cells can be well distinguished.
- the method of this application is used to simultaneously detect the transcriptome and cut tag of the same cell with accuracy.
- the sample is processed in the same way as simply detecting genomic DNA.
- the nucleus is first stripped, then the Tn5 transposition reaction is performed, and then the RT (reverse transcription) reaction is performed, and then the processing is performed in the same manner as in Example 5.
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- reaction system to amplify DNA to obtain the final sequencing library: 36ul DNA template, 10ul 5xPCR Buffer, 1ul 10mM dNTP, 1ul 10uM primer TrueseqD501, 1ul 10uM primer N701, 1ul 10mM ISPCR primer, 1ul Taq, 72°C 5min, 94°C 2min, 94°C30sec, 55°C30sec, 72°C3min, 12 cycles.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme, 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- This application is used to detect the transcriptome and genome of the same cell at the same time, which can well distinguish a single cell of two types of cells. It is accurate in detecting the transcriptome and cut tag of the same cell at the same time.
- the cell nucleus obtained above undergoes the following RT reaction
- microfluidic chip as shown in Figure 8 is used for cell labeling, the bead channel: 100um, and the nuclei channel: 50um.
- Cell nucleus solution 1ml (100 cell nucleus/ul concentration), including: 200ul 10xT4 DNA ligase Buffer, 10ul T4 DNA ligase, 10ul 1M DTT, 780ul nucleus/water.
- Bead solution (100 bead/ul concentration): Bead in PBS.
- Cell nucleus solution, bead solution, and oil form a 120um diameter drop collection on the microfluidic chip, and connect for 1 hour at 37°C.
- a transformation kit such as EpiTect Fast Bisulfite Conversion Kit or NEB Enzymatic Methylation conversion kit to transform the genomic DNA obtained above, for example, Qiagen kit as an example, refer to the instructions to configure the bisulfite conversion reagent.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Recover DNA 12.5 ⁇ L 4X Enzyme Reaction Buffer, 10ul 5-hmC Modifying Enzyme, add water to 50ul, and react at 30°C for 1hr. Purify DNA with a 1:1 volume of magnetic beads.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme, 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- This application is used to detect the transcriptome and methylation of the same cell at the same time.
- Figure 26 shows that the transcriptome and methylation of the same cell can perform well with gene models and known methylation sites. match.
- the method of this application is used to simultaneously detect the transcriptome and methylation of the same cell with accuracy.
- Spatial lattice chip There are fixed-space DNA oligo clusters on the chip, the structure is as follows:
- the space lattice is synthesized by microarray in-situ synthesis method (Affymetrix, NimbleGene) or other methods, including transfer from the existing array by PCR method, and extension by sequential labeling method.
- microarray in-situ synthesis method Affymetrix, NimbleGene
- transfer from the existing array by PCR method and extension by sequential labeling method.
- tissue sections stick frozen sections of non-fixed tissues on a cover glass, add 1% formaldehyde, fix the tissues, and wash.
- NNNNNNNN in the lattice DNA sequence is an 8bp specific primer sequence, and each point on the lattice corresponds to a specific 8bp sequence.
- the OCT-embedded tissue was sliced with a cryostat and attached to a glass slide treated with polylysine.
- the tissue on the slide was treated with lysis solution (10mM Tris-Cl, pH 7.4; 10mM NaCl; 3mM MgCl 2 ; 0.01% NP-40) at room temperature for 5 minutes.
- the p-Tn5 and Tn5-B obtained in the examples of this application are used to react on the glass slide, and the reaction system is as follows:
- the G can be rG, rG represents ribose guanine, 1 unit/ul RT enzyme (Superscript II reverse transcriptase); reaction conditions: 50°C for 5 min, 4°C for 5 min, 42°C for 60 min, and wash the section with PBS.
- the tissue is contacted with the synthetic primer matrix glass slide, and 1xT4 ligase buffer, 1 unit/ul T4 DNA ligase is added, so that part of the double-stranded adaptor on the slide is connected with the RT product and the AATC product on the tissue section.
- Recover cDNA and ATACDNA Add proteinase K reaction buffer and proteinase K at the top of the slice, de-crosslink at 55-65°C, and purify the DNA, then use Qiagen kit to obtain genomic DNA and reverse transcribed mRNA/cDNA
- Primer TrueseqD501 sequence AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO: 10)
- Primer N701 sequence CAAGCAGAAGACGGCATACGAGATATCGGCTAGTCTCGTGGGCTCGG (SEQ ID NO: 11)
- reaction system 25ul or more reaction system, 1ul 10uM primer TrueseqD501, 1ul 10uM primer Nextera N701 primer, 1ul Taq enzyme, 72°C5min, 94°C2min, 94°C30sec, 60°C30sec, 72°C3sec, 18 cycles.
- Illumina Novaseq measures 100,000 PE150 reads per cell.
- Figure 28 shows that the slice HE staining is superimposed on the spatial lattice chip, and the color intensity of each dot represents the number of genes obtained by measurement.
- the method of this application can be used for the research of spatial multi-omics technology platform.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
Claims (114)
- 一种分析来自细胞的目标核酸的方法,所述方法包括:a)提供包含下述的离散分区:ⅰ.源于单个细胞的目标核酸,其中至少部分所述目标核酸被添加了寡核苷酸衔接子序列而成为经附接的目标核酸;以及ⅱ.附接有至少一个寡核苷酸标签的固体支持物,其中每个所述寡核苷酸标签包含第一链以及第二链,所述第一链包含条码序列以及位于所述条码序列3’端的杂交序列,所述第二链包含与所述第一链的所述杂交序列互补的第一部分以及与附接至所述目标核酸的所述寡核苷酸衔接子序列互补的第二部分,且所述第一链与所述第二链形成部分双链的结构或者所述第二链与所述经附接的目标核酸形成部分双链的结构;b)在所述离散分区中,使所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的目标核酸。
- 根据权利要求1所述的方法,其中所述寡核苷酸标签可释放地附接至所述固体支持物。
- 根据权利要求1-2中任一项所述的方法,其包括从所述固体支持物上释放所述至少一个寡核苷酸标签,并在b)中使经释放的所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的目标核酸。
- 根据权利要求1-3中任一项所述的方法,其中所述寡核苷酸标签通过其第一链的5’端直接或间接附接至所述固体支持物。
- 根据权利要求1-4中任一项所述的方法,其中所述离散分区中还包含连接酶,且所述连接酶使所述寡核苷酸标签与所述经附接的目标核酸连接。
- 根据权利要求5所述的方法,其中所述连接酶包括T4连接酶。
- 根据权利要求1-6中任一项所述的方法,其中在所述条码化的目标核酸中,所述目标核酸序列位于所述条码序列的3’端。
- 根据权利要求1-7中任一项所述的方法,其中所述固体支持物为珠粒。
- 根据权利要求8所述的方法,其中所述珠粒为磁性珠粒。
- 根据权利要求1-9中任一项所述的方法,其中所述离散分区为孔或微滴。
- 根据权利要求1-10中任一项所述的方法,其中所述条码序列包含细胞条码序列,且附接至同一个固体支持物上的各寡核苷酸标签所包含的细胞条码序列相同。
- 根据权利要求11所述的方法,其中所述细胞条码序列包含由连接子序列间隔开的至少2个细胞条码区段。
- 根据权利要求1-12中任一项所述的方法,其中a)包括将所述源于单个细胞的目标核酸与所 述附接有至少一个寡核苷酸标签的固体支持物共分配至所述离散分区中。
- 根据权利要求1-13中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求1-14中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第二链的所述第二部分与附接至所述目标核酸的所述寡核苷酸衔接子杂交,以及使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求1-15中任一项所述的方法,其中所述经附接的目标核酸中包含独特分子鉴别区。
- 根据权利要求16所述的方法,其中所述独特分子鉴别区位于所述寡核苷酸衔接子序列与所述目标核酸序列之间。
- 根据权利要求1-17中任一项所述的方法,其中所述寡核苷酸标签还包含扩增引物识别区。
- 根据权利要求18所述的方法,其中所述扩增引物识别区为通用扩增引物识别区。
- 根据权利要求1-19中任一项所述的方法,其进一步包括:c)获得所述条码化的目标核酸的表征结果;以及d)至少部分基于c)中获得的所述表征结果中存在相同的所述细胞条码序列而将所述目标核酸的序列识别为源于所述单个细胞。
- 根据权利要求20所述的方法,其进一步包括,在b)之后并且在c)之前,从所述离散分区中释放所述条码化的目标核酸。
- 根据权利要求20-21中任一项所述的方法,其中c)包括对所述条码化的目标核酸进行测序,从而获得所述表征结果。
- 根据权利要求20-22中任一项所述的方法,其进一步包括由所述条码化的目标核酸的序列组装所述单个细胞的基因组的至少一部分的连续核酸序列。
- 根据权利要求23所述的方法,其中基于所述单个细胞的所述基因组的至少一部分的所述核酸序列来表征所述单个细胞。
- 根据权利要求1-24中任一项所述的方法,其中每个所述离散分区至多包括源自单个细胞的所述目标核酸。
- 根据权利要求20-25中任一项所述的方法,其进一步包括至少部分基于所述独特分子鉴别区的存在将所述条码化的目标核酸中的单个核酸序列鉴别为源于所述目标核酸中的给定核酸。
- 根据权利要求20-26中任一项所述的方法,所述目标核酸包括外源核酸,所述外源核酸包括 与蛋白、脂类和/或小分子化合物连接的外源核酸,所述蛋白、脂类和/或小分子化合物能够与细胞内的靶分子结合。
- 根据权利要求27所述的方法,其进一步包括基于所述独特分子鉴别区的存在确定所述目标核酸中给定核酸的量。
- 根据权利要求1-28中任一项所述的方法,其包括在a)之前对所述细胞进行预处理。
- 根据权利要求29所述的方法,其中所述预处理包括固定所述细胞。
- 根据权利要求30所述的方法,其中使用固定剂对所述细胞进行固定,所述固定剂选自下组中的一种或多种:甲醛、多聚甲醛、甲醇、乙醇、丙酮、戊二醛、锇酸和重铬酸钾。
- 根据权利要求29-31中任一项所述的方法,其中所述预处理包括使所述细胞的细胞核被暴露。
- 根据权利要求29-32中任一项所述的方法,其中所述预处理包括使用去垢剂处理所述细胞,所述去垢剂包括Triton、NP-40和/或digitonin。
- 根据权利要求1-33中任一项所述的方法,其中所述目标核酸包括选自下组的一种或多种:DNA、RNA和cDNA。
- 根据权利要求20-34中任一项所述的方法,其进一步包括,在b)之后并且在c)之前,对所述条码化的目标核酸进行扩增。
- 根据权利要求35所述的方法,其包括在b)之后并且在c)之前,从所述离散分区中释放所述条码化的目标核酸,且所述扩增在所述条码化的目标核酸从所述离散分区中释放后进行。
- 根据权利要求35-36中任一项所述的方法,其中所述扩增中使用扩增引物,且所述扩增引物中包含随机引导序列。
- 根据权利要求37所述的方法,其中所述随机引导序列为随机六聚体。
- 根据权利要求35-38中任一项所述的方法,其中所述扩增包括使所述随机引导序列与所述条码化的目标核酸至少部分杂交并且以模板定向的方式延伸所述随机引导序列。
- 根据权利要求1-39中任一项所述的方法,其包括使至少一部分所述目标核酸从所述离散分区中的所述单个细胞中释放到细胞外,并在b)中使经释放的所述目标核酸与所述寡核苷酸标签连接,从而产生条码化的目标核酸。
- 根据权利要求1-40中任一项所述的方法,其包括使至少一部分从所述固体支持物释放的所述寡核苷酸标签进入所述单个细胞中,并在b)中与所述目标核酸连接,从而产生条码化的目标核酸。
- 根据权利要求1-41中任一项所述的方法,其包括使用微流控装置将所述源于单个细胞的目标核酸与所述附接有至少一个寡核苷酸标签的固体支持物共分配至所述离散分区中。
- 根据权利要求42所述的方法,其中所述离散分区为微滴,且所述微流控装置为微滴发生器。
- 根据权利要求42-43中任一项所述的方法,其中所述微流控装置包括第一输入通道和第二输入通道,它们在与输出通道流体连接的接合处汇合。
- 根据权利要求44所述的方法,其中所述方法还包括将包含所述目标核酸的样品引入所述第一输入通道,且将附接有至少一个寡核苷酸标签的所述固体支持物引入所述第二输入通道,从而在所述输出通道中生成所述样品与所述固体支持物的混合物。
- 根据权利要求45所述的方法,其中所述输出通道与第三输入通道在接合处流体连接。
- 根据权利要求46所述的方法,其还包括将油引入所述第三输入通道,使得形成油包水乳液内的水性小滴作为所述离散分区。
- 根据权利要求47所述的方法,其中每个所述离散分区中至多包含来自单个细胞的所述目标核酸。
- 根据权利要求44-48中任一项所述的方法,其中所述第一输入通道和所述第二输入通道彼此之间形成基本上垂直的角度。
- 根据权利要求1-49中任一项所述的方法,其中所述目标核酸包括源自所述单个细胞中RNA的cDNA。
- 根据权利要求50所述的方法,其中所述RNA包括mRNA。
- 根据权利要求34-51中任一项所述的方法,其包括在a)之前对所述RNA进行反转录,并产生所述经附接的目标核酸。
- 根据权利要求52所述的方法,其中所述反转录中使用反转录引物,所述反转录引物以5‘至3’的方向包含所述寡核苷酸衔接子序列以及polyT序列。
- 根据权利要求53所述的方法,其中所述反转录包括使所述polyT序列与所述RNA杂交并且以模板定向的方式延伸所述polyT序列。
- 根据权利要求1-54中任一项所述的方法,其中所述目标核酸包括源自所述单个细胞的DNA。
- 根据权利要求55所述的方法,其中所述DNA包括基因组DNA、开放染色质DNA、蛋白质结合的DNA区域和/或与蛋白、脂类和/或小分子化合物连接的外源核酸,所述蛋白、脂类和/或小分子化合物能够与细胞内的靶分子结合。
- 根据权利要求56所述的方法,其包括在a)之前对源自单个细胞的所述DNA进行片段化。
- 根据权利要求57所述的方法,其中在所述片段化之后或者在所述片段化的过程中产生所述经附接的目标核酸。
- 根据权利要求57-58中任一项所述的方法,其中所述片段化包括使用超声断裂,而后在经断 裂的所述DNA上添加包含所述寡核苷酸衔接子的序列,从而获得所述经附接的目标核酸。
- 根据权利要求57-59中任一项所述的方法,其中所述片段化包括使用DNA内切酶、外切酶打断,而后在经断裂的所述DNA上添加包含所述寡核苷酸衔接子的序列,从而获得所述经附接的目标核酸。
- 根据权利要求57-60中任一项所述的方法,其中所述片段化包括使用转座酶-核酸复合物将包含所述寡核苷酸衔接子的序列整合到所述DNA中,并释放所述转座酶以获得所述经附接的目标核酸。
- 根据权利要求61所述的方法,其中所述转座酶-核酸复合物包含转座酶以及转座子末端核酸分子,其中所述转座子末端核酸分子包含所述寡核苷酸衔接子序列。
- 根据权利要求61-62中任一项所述的方法,其中所述转座酶包括Tn5。
- 根据权利要求61-63中任一项所述的方法,其中所述DNA包括与蛋白质结合的DNA区域,且所述转座酶-核酸复合物中还包含直接或间接识别所述蛋白质的部分。
- 根据权利要求64所述的方法,其中所述直接或间接识别所述蛋白质的部分包括下组中的一种或多种:特异性结合所述蛋白质的抗体和蛋白质A或蛋白质G。
- 一种组合物,其包含:多个固体支持物,每个所述固体支持物上附接有至少一个寡核苷酸标签,其中每个所述寡核苷酸标签包含第一链以及第二链,所述第一链包含条码序列以及位于所述条码序列3’端的杂交序列,所述第二链包含与所述第一链的所述杂交序列互补的第一部分以及与待测核酸中的序列互补的第二部分,且所述第一链与所述第二链形成部分双链的结构或者所述第二链与所述经附接的目标核酸形成部分双链的结构;所述寡核苷酸标签的条码序列包含共同条码结构域和可变结构域,所述共同条码结构域在附接于同一个固体支持物的寡核苷酸标签中是相同的,且所述共同条码结构域在所述多个固体支持物中的两个或更多个固体支持物之间是不同的。
- 一种用于分析来自细胞的目标核酸的试剂盒,其包含权利要求66所述的组合物。
- 根据权利要求67所述的试剂盒,其包括转座酶。
- 根据权利要求67-68中任一项所述的试剂盒,其进一步包含核酸扩增剂,逆转录剂,固定剂,通透剂,连接剂和裂解剂中的至少一种。
- 一种扩增来自细胞的目标核酸的方法,所述方法包括:a)提供包含下述的离散分区:i.源于单个细胞的目标核酸,其中至少部分所述目标核酸被添加了寡核苷酸衔接子序列而成为经附接的目标核酸;以及ii.附接有至少一个寡核苷酸标签的固体支持物,其中每个所述寡核苷酸标签包含第一链以及第二链,所述第一链包含条码序列 以及位于所述条码序列3’端的杂交序列,所述第二链包含与所述第一链的所述杂交序列互补的第一部分以及与附接至所述目标核酸的所述寡核苷酸衔接子序列互补的第二部分,且所述第一链与所述第二链形成部分双链的结构或者所述第二链与所述经附接的目标核酸形成部分双链的结构;b)在所述离散分区中,使所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的目标核酸;以及c)对所述条码化的目标核酸进行扩增。
- 根据权利要求70所述的方法,其中所述寡核苷酸标签可释放地附接至所述固体支持物。
- 根据权利要求71所述的方法,其包括从所述固体支持物上释放所述至少一个寡核苷酸标签,并在b)中使经释放的所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的目标核酸。
- 根据权利要求70-72中任一项所述的方法,其中所述寡核苷酸标签通过其第一链的5’端直接或间接附接至所述固体支持物。
- 根据权利要求70-73中任一项所述的方法,其中所述离散分区中还包含连接酶,且所述连接酶使所述寡核苷酸标签与所述经附接的目标核酸连接。
- 根据权利要求74所述的方法,其中所述连接酶包括T4连接酶。
- 根据权利要求70-75中任一项所述的方法,其中在所述条码化的目标核酸中,所述目标核酸序列位于所述条码序列的3’端。
- 根据权利要求70-76中任一项所述的方法,其中所述固体支持物为珠粒。
- 根据权利要求70-77中任一项所述的方法,其中所述离散分区为孔或微滴。
- 根据权利要求70-78中任一项所述的方法,其中所述条码序列包含细胞条码序列,且附接至同一个固体支持物上的各寡核苷酸标签所包含的细胞条码序列相同。
- 根据权利要求79所述的方法,其中所述细胞条码序列包含由连接子序列间隔开的至少2个细胞条码区段。
- 根据权利要求70-80中任一项所述的方法,其中a)包括将所述源于单个细胞的目标核酸与所述附接有至少一个寡核苷酸标签的固体支持物共分配至所述离散分区中。
- 根据权利要求70-81中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求70-82中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第二链的所述 第二部分与附接至所述目标核酸的所述寡核苷酸衔接子杂交,以及使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求70-83中任一项所述的方法,其中所述经附接的目标核酸中包含独特分子鉴别区。
- 根据权利要求84所述的方法,其中所述独特分子鉴别区位于所述寡核苷酸衔接子序列与所述目标核酸序列之间。
- 根据权利要求70-85中任一项所述的方法,其中所述寡核苷酸标签还包含扩增引物识别区。
- 根据权利要求86所述的方法,其中所述扩增引物识别区为通用扩增引物识别区。
- 根据权利要求87所述的方法,其包括在b)之后并且在c)之前,从所述离散分区中释放所述条码化的目标核酸,且所述扩增在所述条码化的目标核酸从所述离散分区中释放后进行。
- 根据权利要求70-88中任一项所述的方法,其中所述扩增中使用扩增引物,且所述扩增引物中包含随机引导序列。
- 根据权利要求89所述的方法,其中所述随机引导序列为随机六聚体。
- 根据权利要求70-90中任一项所述的方法,其中所述扩增包括使所述随机引导序列与所述条码化的目标核酸至少部分杂交并且以模板定向的方式延伸所述随机引导序列。
- 一种对来自细胞的目标核酸进行测序的方法,所述方法包括:a)提供包含下述的离散分区:i.源于单个细胞的目标核酸,其中至少部分所述目标核酸被添加了寡核苷酸衔接子序列而成为经附接的目标核酸;以及ii.附接有至少一个寡核苷酸标签的固体支持物,其中每个所述寡核苷酸标签包含第一链以及第二链,所述第一链包含条码序列以及位于所述条码序列3’端的杂交序列,所述第二链包含与所述第一链的所述杂交序列互补的第一部分以及与附接至所述目标核酸的所述寡核苷酸衔接子序列互补的第二部分,且所述第一链与所述第二链形成部分双链的结构或者所述第二链与所述经附接的目标核酸形成部分双链的结构;b)在所述离散分区中,使所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的目标核酸;以及c)对所述条码化的目标核酸进行测序。
- 根据权利要求92所述的方法,其中所述寡核苷酸标签可释放地附接至所述固体支持物。
- 根据权利要求93所述的方法,其包括从所述固体支持物上释放所述至少一个寡核苷酸标签,并在b)中使经释放的所述寡核苷酸标签与所述经附接的目标核酸连接,从而产生条码化的 目标核酸。
- 根据权利要求92-94中任一项所述的方法,其中所述寡核苷酸标签通过其第一链的5’端直接或间接附接至所述固体支持物。
- 根据权利要求92-95中任一项所述的方法,其中所述离散分区中还包含连接酶,且所述连接酶使所述寡核苷酸标签与所述经附接的目标核酸连接。
- 根据权利要求96所述的方法,其中所述连接酶包括T4连接酶或T7连接酶。
- 根据权利要求92-97中任一项所述的方法,其中在所述条码化的目标核酸中,所述目标核酸序列位于所述条码序列的3’端。
- 根据权利要求92-98中任一项所述的方法,其中所述固体支持物为珠粒。
- 根据权利要求92-99中任一项所述的方法,其中所述离散分区为孔或微滴。
- 根据权利要求92-100中任一项所述的方法,其中所述条码序列包含细胞条码序列,且附接至同一个固体支持物上的各寡核苷酸标签所包含的细胞条码序列相同。
- 根据权利要求101所述的方法,其中所述细胞条码序列包含由连接子序列间隔开的至少2个细胞条码区段。
- 根据权利要求92-102中任一项所述的方法,其中a)包括将所述源于单个细胞的目标核酸与所述附接有至少一个寡核苷酸标签的固体支持物共分配至所述离散分区中。
- 根据权利要求92-103中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求92-104中任一项所述的方法,其中b)包括使所述寡核苷酸标签的第二链的所述第二部分与附接至所述目标核酸的所述寡核苷酸衔接子杂交,以及使所述寡核苷酸标签的第一链的所述杂交序列与附接至所述目标核酸的所述寡核苷酸衔接子连接,从而产生所述条码化的目标核酸。
- 根据权利要求92-105中任一项所述的方法,其中所述经附接的目标核酸中包含独特分子鉴别区。
- 根据权利要求106所述的方法,其中所述独特分子鉴别区位于所述寡核苷酸衔接子序列与所述目标核酸序列之间。
- 根据权利要求92-107中任一项所述的方法,其中所述寡核苷酸标签还包含扩增引物识别区。
- 根据权利要求108所述的方法,其中所述扩增引物识别区为通用扩增引物识别区。
- 根据权利要求92-109中任一项所述的方法,其进一步包括由所述条码化的目标核酸的序列组 装所述单个细胞的基因组的至少一部分的连续核酸序列。
- 根据权利要求110所述的方法,其中基于所述单个细胞的所述基因组的至少一部分的所述核酸序列来表征所述单个细胞。
- 根据权利要求92-111中任一项所述的方法,其中每个所述离散分区至多包括源自单个细胞的所述目标核酸。
- 根据权利要求92-112中任一项所述的方法,其进一步包括至少部分基于所述独特分子鉴别区的存在将所述条码化的目标核酸中的单个核酸序列鉴别为源于所述目标核酸中的给定核酸。
- 根据权利要求113所述的方法,其进一步包括基于所述独特分子鉴别区的存在确定所述目标核酸中给定核酸的量。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21817287.2A EP4163390A4 (en) | 2020-06-03 | 2021-06-02 | METHOD FOR ANALYZING THE TARGET NUCLEIC ACID OF A CELL |
| CN202180039759.9A CN116234926A (zh) | 2020-06-03 | 2021-06-02 | 分析来自细胞的目标核酸的方法 |
| CA3181004A CA3181004A1 (en) | 2020-06-03 | 2021-06-02 | Method for analyzing target nucleic acid from cell |
| JP2022574773A JP7853705B2 (ja) | 2020-06-03 | 2021-06-02 | 細胞に由来する標的核酸を解析する方法 |
| US18/000,665 US20230212648A1 (en) | 2020-06-03 | 2021-06-02 | Method for analyzing target nucleic acid from cell |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010495334.6 | 2020-06-03 | ||
| CN202010495334 | 2020-06-03 | ||
| CN202010506791.0 | 2020-06-05 | ||
| CN202010506791 | 2020-06-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2021244557A1 true WO2021244557A1 (zh) | 2021-12-09 |
Family
ID=78830665
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2021/097800 Ceased WO2021244557A1 (zh) | 2020-06-03 | 2021-06-02 | 分析来自细胞的目标核酸的方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20230212648A1 (zh) |
| EP (1) | EP4163390A4 (zh) |
| CN (1) | CN116234926A (zh) |
| CA (1) | CA3181004A1 (zh) |
| WO (1) | WO2021244557A1 (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114277114A (zh) * | 2021-12-30 | 2022-04-05 | 深圳海普洛斯医学检验实验室 | 一种扩增子测序添加唯一性标识符的方法及应用 |
| CN114574569A (zh) * | 2022-03-28 | 2022-06-03 | 浙江大学 | 一种基于末端转移酶的基因组测序试剂盒和测序方法 |
| CN114574484A (zh) * | 2022-03-17 | 2022-06-03 | 中国科学院北京基因组研究所(国家生物信息中心) | 核酸检测试剂及其应用 |
| CN114807084A (zh) * | 2022-04-26 | 2022-07-29 | 翌圣生物科技(上海)股份有限公司 | 突变型Tn5转座酶及试剂盒 |
| WO2024186877A1 (en) * | 2023-03-07 | 2024-09-12 | Board Of Regents, The University Of Texas System | Methods and compositions for amplification and sequencing of genome and epigenome |
| WO2024244933A1 (zh) * | 2023-05-30 | 2024-12-05 | 深圳赛陆医疗科技有限公司 | 一种snp芯片及其制备方法和应用 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118272509A (zh) * | 2024-03-28 | 2024-07-02 | 浙江大学 | 基于组合索引的单细胞dna-蛋白互作测序试剂盒和方法 |
| CN118638901B (zh) * | 2024-08-15 | 2024-12-03 | 青岛百创智能制造技术有限公司 | 降低微球矩阵重复率的微球合成方法及其产品和应用 |
| CN119876341B (zh) * | 2025-01-21 | 2026-01-06 | 西安交通大学 | 一种条码可重复微球及其制备方法和应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995023875A1 (en) | 1994-03-02 | 1995-09-08 | The Johns Hopkins University | In vitro transposition of artificial transposons |
| WO2012142213A2 (en) | 2011-04-15 | 2012-10-18 | The Johns Hopkins University | Safe sequencing system |
| US20130274117A1 (en) | 2010-10-08 | 2013-10-17 | President And Fellows Of Harvard College | High-Throughput Single Cell Barcoding |
| US20150292988A1 (en) | 2014-04-10 | 2015-10-15 | 10X Genomics, Inc. | Fluidic devices, systems, and methods for encapsulating and partitioning reagents, and applications of same |
| US9834814B2 (en) * | 2013-11-22 | 2017-12-05 | Agilent Technologies, Inc. | Spatial molecular barcoding of in situ nucleic acids |
| CN107735497A (zh) * | 2015-02-18 | 2018-02-23 | 卓异生物公司 | 用于单分子检测的测定及其应用 |
| CN108350497A (zh) * | 2015-08-28 | 2018-07-31 | Illumina公司 | 单细胞核酸序列分析 |
| WO2019165318A9 (en) * | 2018-02-22 | 2019-09-26 | 10X Genomics, Inc. | Ligation mediated analysis of nucleic acids |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114214314A (zh) * | 2014-06-24 | 2022-03-22 | 生物辐射实验室股份有限公司 | 数字式pcr条码化 |
| WO2018118971A1 (en) * | 2016-12-19 | 2018-06-28 | Bio-Rad Laboratories, Inc. | Droplet tagging contiguity preserved tagmented dna |
| WO2019152395A1 (en) * | 2018-01-31 | 2019-08-08 | Bio-Rad Laboratories, Inc. | Methods and compositions for deconvoluting partition barcodes |
| CN120624607A (zh) * | 2018-05-08 | 2025-09-12 | 深圳华大智造科技股份有限公司 | 用于准确且经济高效的测序、单体型分型和组装的基于单管珠粒的dna共条形码化 |
| WO2020041148A1 (en) * | 2018-08-20 | 2020-02-27 | 10X Genomics, Inc. | Methods and systems for detection of protein-dna interactions using proximity ligation |
| EP3844304B1 (en) * | 2018-08-28 | 2024-10-02 | 10X Genomics, Inc. | Methods for generating spatially barcoded arrays |
-
2021
- 2021-06-02 CA CA3181004A patent/CA3181004A1/en active Pending
- 2021-06-02 WO PCT/CN2021/097800 patent/WO2021244557A1/zh not_active Ceased
- 2021-06-02 EP EP21817287.2A patent/EP4163390A4/en active Pending
- 2021-06-02 US US18/000,665 patent/US20230212648A1/en active Pending
- 2021-06-02 CN CN202180039759.9A patent/CN116234926A/zh active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995023875A1 (en) | 1994-03-02 | 1995-09-08 | The Johns Hopkins University | In vitro transposition of artificial transposons |
| US20130274117A1 (en) | 2010-10-08 | 2013-10-17 | President And Fellows Of Harvard College | High-Throughput Single Cell Barcoding |
| WO2012142213A2 (en) | 2011-04-15 | 2012-10-18 | The Johns Hopkins University | Safe sequencing system |
| US9834814B2 (en) * | 2013-11-22 | 2017-12-05 | Agilent Technologies, Inc. | Spatial molecular barcoding of in situ nucleic acids |
| US20150292988A1 (en) | 2014-04-10 | 2015-10-15 | 10X Genomics, Inc. | Fluidic devices, systems, and methods for encapsulating and partitioning reagents, and applications of same |
| CN107735497A (zh) * | 2015-02-18 | 2018-02-23 | 卓异生物公司 | 用于单分子检测的测定及其应用 |
| CN108350497A (zh) * | 2015-08-28 | 2018-07-31 | Illumina公司 | 单细胞核酸序列分析 |
| WO2019165318A9 (en) * | 2018-02-22 | 2019-09-26 | 10X Genomics, Inc. | Ligation mediated analysis of nucleic acids |
Non-Patent Citations (18)
| Title |
|---|
| "GenBank", Database accession no. YP001446289 |
| BOEKECORCES, ANNU REV MICROBIOL., vol. 43, no. 198, pages 403 - 34 |
| BROWN ET AL., PROC NATL ACAD SCI USA, vol. 86, pages 2525 - 9 |
| COLEGIO ET AL., J. BACTERIOL, vol. 183, no. 2384-8, pages 200 |
| CRAIG, N L, SCIENCE, vol. 271, 1996, pages 1512 |
| GLOOR, G B, METHODS MOL. BIOL., vol. 260, no. 200, pages 97 - 114 |
| ICHIKAWAOHTSUBO, J BIOL. CHEM., vol. 265, pages 18829 - 32 |
| ISLAM ET AL., NAT. METHODS, vol. 11, 2014, pages 163 - 166 |
| KIRBY ET AL., MOL. MICROBIOL., vol. 43, pages 173 - 86 |
| KIVIOJA, T ET AL., NAT. METHODS, vol. 9, 2012, pages 72 - 74 |
| KLECKNERN ET AL., CURR TOP MICROBIOL IMMUNOL., vol. 204, no. 199, 1996, pages 49 - 82 |
| LAMPE D J ET AL., EMBO J., vol. 15, 1996, pages 5470 - 9 |
| OHTSUBOSEKINE, CURR. TOP. MICROBIOL. IMMUNOL, vol. 204, no. 199, 1996, pages 1 - 26 |
| PLASTERK R H, CURR. TOPICS MICROBIOL. IMMUNOL., vol. 204, no. 199, 1996, pages 125 - 43 |
| See also references of EP4163390A4 |
| TYLOSIN (TYL, NUCLEIC ACIDS RES., vol. 22, 1994, pages 3765 - 72 |
| WILSON C. ET AL., J. MICROBIOL. METHODS, vol. 71, 2007, pages 332 - 5 |
| ZHANG ET AL., PLOS GENET, vol. 5, 16 October 2009 (2009-10-16), pages 689 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114277114A (zh) * | 2021-12-30 | 2022-04-05 | 深圳海普洛斯医学检验实验室 | 一种扩增子测序添加唯一性标识符的方法及应用 |
| CN114574484A (zh) * | 2022-03-17 | 2022-06-03 | 中国科学院北京基因组研究所(国家生物信息中心) | 核酸检测试剂及其应用 |
| CN114574569A (zh) * | 2022-03-28 | 2022-06-03 | 浙江大学 | 一种基于末端转移酶的基因组测序试剂盒和测序方法 |
| CN114807084A (zh) * | 2022-04-26 | 2022-07-29 | 翌圣生物科技(上海)股份有限公司 | 突变型Tn5转座酶及试剂盒 |
| CN114807084B (zh) * | 2022-04-26 | 2023-05-16 | 翌圣生物科技(上海)股份有限公司 | 突变型Tn5转座酶及试剂盒 |
| WO2024186877A1 (en) * | 2023-03-07 | 2024-09-12 | Board Of Regents, The University Of Texas System | Methods and compositions for amplification and sequencing of genome and epigenome |
| WO2024244933A1 (zh) * | 2023-05-30 | 2024-12-05 | 深圳赛陆医疗科技有限公司 | 一种snp芯片及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023528917A (ja) | 2023-07-06 |
| CA3181004A1 (en) | 2021-12-09 |
| US20230212648A1 (en) | 2023-07-06 |
| EP4163390A1 (en) | 2023-04-12 |
| EP4163390A4 (en) | 2024-08-07 |
| CN116234926A (zh) | 2023-06-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021244557A1 (zh) | 分析来自细胞的目标核酸的方法 | |
| US11035002B2 (en) | Methods and systems for processing polynucleotides | |
| US20220235416A1 (en) | Methods and systems for single cell gene profiling | |
| US10752949B2 (en) | Methods and systems for processing polynucleotides | |
| CN110462060B (zh) | 用于标记细胞的方法和组合物 | |
| EP4240870B1 (en) | SYSTEMS AND METHODS FOR MANUFACTURING SEQUENCE BANK NETWORKS | |
| CN109526228B (zh) | 转座酶可接近性染色质的单细胞分析 | |
| CN113811619A (zh) | 用于处理来自细胞的rna的系统和方法 | |
| US20240254538A1 (en) | Particles associated with oligonucleotides | |
| CN114616341A (zh) | 联接介导的核酸分析 | |
| CN113366117A (zh) | 用于生物样品中转座酶介导的空间标记和分析基因组dna的方法 | |
| CN111051523A (zh) | 功能化凝胶珠 | |
| CN112639985A (zh) | 用于代谢组分析的系统和方法 | |
| CN112272710A (zh) | 高通量多组学样品分析 | |
| CN111699388A (zh) | 用于单细胞处理的系统和方法 | |
| CN106795553A (zh) | 分析来自单个细胞或细胞群体的核酸的方法 | |
| CN110637084A (zh) | Mmlv逆转录酶变体 | |
| JP7853705B2 (ja) | 細胞に由来する標的核酸を解析する方法 | |
| HK40082906A (zh) | 分析来自细胞的目标核酸的方法 | |
| US20240229106A1 (en) | Composition and method for analyzing target molecule from sample |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21817287 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3181004 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2022574773 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021817287 Country of ref document: EP Effective date: 20230103 |