WO2022003710A1 - A process for the preparation of vanillin and other substituted phenylaldehydes - Google Patents
A process for the preparation of vanillin and other substituted phenylaldehydes Download PDFInfo
- Publication number
- WO2022003710A1 WO2022003710A1 PCT/IN2021/050495 IN2021050495W WO2022003710A1 WO 2022003710 A1 WO2022003710 A1 WO 2022003710A1 IN 2021050495 W IN2021050495 W IN 2021050495W WO 2022003710 A1 WO2022003710 A1 WO 2022003710A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- och
- formula
- compound
- substituted
- vanillin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/32—Preparation of ethers by isomerisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
- C07C45/28—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation of CHx-moieties
Definitions
- the present invention relates to a chemical process for the production of substituted phenylaldehydes such as vanillin from substituted phenylpropenes or substituted phenylpropenes enriched essential oils such as eugenol or eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylprop-2-enes such as isoeugenol, an intermediate compound.
- the invention relates to the conversion of substituted phenylpropenes to other substituted phenylaldehydes, particularly vanillin without the protection of the phenolic group, therefore, it offers a step economy.
- the present chemical process involves the use of environment-friendly solvents and devoid of the use of any chlorinated solvent.
- This novel process developed for the production of vanillin and also provides ease of preparation for other substituted phenylaldehydes derivatives.
- Vanillin (4-hydroxy-3-methoxybenzaldehyde, la) is the highest volume aroma chemical produced worldwide ( sue et al., 2016, ACS Sustainable Chem. Eng., 4(1), 35-46).
- Natural vanilla is extracted from the cured pods of different species of Vanilla (Orchidaceae), V. Planifolia Jacks, ex Andrews (syn. V.fragrans (Salisbury) Ames), V. tahitensis J.W. Moore, and V. pompona Scheide ( Sinha et al., 2008, J. Food Sci. Nutr., 59(4), 299-326 ).
- vanillin is widely used as a flavoring agent in confectionery, chocolates, baked goods, non-alcoholic and alcoholic beverages, and many other foods. Non-food applications of vanillin are found in cosmetics, personal care products, detergents, and perfumery.
- vanillin is a starting material in the manufacture of several drugs (for example L-3,4- DihydroxyPhenylAlanine, L-DOPA) (Kaiser et al. 1976, US 3969397 and Knowles, W. S., 2002, Angew. Chem., Int. Ed., 41, 1998-2007; Knowles, W.
- drugs for example L-3,4- DihydroxyPhenylAlanine, L-DOPA
- synthesis of vanillin involves the treatment of eugenol with potassium hydroxide at around 150°C for isomerization of eugenol to isoeugenol which is subsequently oxidized to vanillin using nitro-benzol or mono nitro-toluol as an oxidizing agent.
- the drawback of this process is the use of a large amount of aniline (toxic vapors) and the formation of azo-benzol as a major by-product.
- isoeugenol is treated with hydrogen peroxide in presence of anhydrous tertiary amyl alcohol and vanadium pentoxide ⁇ Milas et al. 1947, US 2414385 and Milas, N. A., 1937, J. Am. Chem. Soc., 59, 2342).
- this process is also associated with prolonging reaction time (12-15 h) and tedious isolation of vanillin through the p- nitrophenylhydrazone formation.
- the preparation of vanillin from isoeugenol in only 23% yield by the use of a two-step methodology is also reported ⁇ Pappo, R., 1956, et al. J. Org.
- Lampman et al. reported the preparation of vanillin from eugenol via a two-step chemical process using KOH, dimethylsulfoxide, and nitrobenzene ⁇ Lampman et al., 1963, J. Chem. Educ., 60, 503-504).
- the caustic effluent and azobenzene as major wastes in this process area significant drawbacks of the process.
- CN 1289836A developed by Shanghai Tianxiang Fine Chemical Co., Ltd., disclosed a chemical method of preparing vanillin (in 50% yield) from clove oil using sodium hydroxide, potassium permanganate, p-aminobenzene sulfonic acid, nitrobenzene, and carbonyl-based mixed catalyst at 100-300°C.
- the waste generated by the reaction is difficult to deal with, resulting in serious environmental pollution.
- Natural-grade vanillin prepared from eugenol has an enticing aroma, food safety, and high practical value, thus attracts wide attention and in-depth research.
- the preparation of vanillin from eugenol traditionally involves mainly two steps (1) isomerization of eugenol to isoeugenol and (2) oxidation of isoeugenol to vanillin.
- transition metal compounds such as compounds of palladium, cobalt, ruthenium, copper, etc ( Parreira, L. et al., Adv. Synth.
- the present invention relates to the development of an efficient, convenient, and safe chemical process for vanillin production from eugenol and eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of isoeugenol, an intermediate compound.
- This novel process also provides ease of preparation for other substituted phenylaldehydes from either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylpropenes and substituted phenylpropenes rich essential oils.
- the main objective of the present invention is to provide a novel, convenient, efficient, and safe chemical process for preparing vanillin and other substituted phenylaldehydes from substituted phenylpropenes or substituted phenylpropenes enriched essential oils (Scheme 1).
- Another objective of the present invention is to provide a convenient, efficient, and safe chemical process for preparing vanillin from eugenol and eugenol rich essential oils.
- Yet another objective of the invention is to provide an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safrole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
- Another objective of the invention is to develop a chemical process for the production of vanillin and other substituted phenylaldehydes with high purity and minimum side products.
- Yet another objective of the invention is to provide an effective chemical process that uses environmentally safe solvents e.g. class 3 and 4 solvents.
- Yet another objective of the invention is to provide an effective, convenient, and safe chemical process that is devoid of the use of any chlorinated solvent (starting from reaction medium to work up process).
- Yet another objective of the invention is to provide an effective, convenient, and safe chemical process which does not require any specific reaction conditions e.g. anhydrous or inert reaction conditions.
- methyl-substituted pyridines as additives such as 2,6-dimethylpyridine (2,6-lutidine), 2-methylpyridine, 4-methylpyridine, 2,4,6- trimethylpyridine and tertiary amine N-oxides such as pyridine-N-oxide, 2-picoline-N-oxide, 3- picoline-N
- Yet another objective of the invention is to provide an effective, convenient, and safe chemical process for value addition of essential oils rich in substituted phenylpropene(s) through the preparation of important substituted phenylaldehydes.
- Yet another objective of the invention is to provide an effective, convenient, and safe chemical process for large scale production of vanillin to the industrial level.
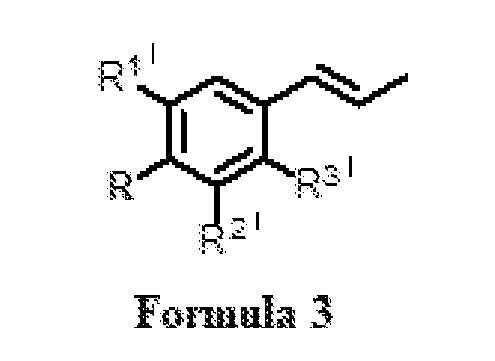
- R 1 and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula
- R is selected from OH and OCH 3 ;
- R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , b. oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); c. oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
- essential oil herein refers to the concentrated volatile oil extracted from plants which retains the natural odour and flavor of the plant from which it was extracted.
- essential oil includes but not limited to essential oils obtained from eugenol rich plants such as Syzygium aromaticum, Ocimum tenuiflorum, and Ocimum gratissimum.
- oxidizing agent herein refers to the substance used to oxidize a reactant in a chemical reaction.
- the term “oxidizing agent” includes but not limited to OsO 4 /NaIO 4 , K 2 [OsO 2 (OH) 4 ]/NaIO 4 , OsCl 3 /NaIO 4 , Polyurea-encapsulated OsO 4 /NaIO 4 , Poly 4- vinylpyridine crosslinked supported OsO 4 /NaIO 4 and Styrene-divinylbenzene crosslinked supported OsO 4 /NaIO 4 .
- biphasic medium herein refers to a medium comprising two different phases for a reaction mixture.
- the term “biphasic medium” includes but not limited to a mixture of water and non-prodc solvent in a ratio 1:4.
- non-protic solvent refers to the solvent which does not release any proton (H + ion) upon dissociation.
- non-protic solvent includes but not limited to tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether and tert-butanol.
- Ratios, concentrations, amounts, and other numerical data may be presented herein in a range format. It is to be understood that such range format is used merely for convenience and brevity and should be interpreted flexibly to include not only the numerical values explicitly recited as the limits of the range, but also to include all the individual numerical values or sub-ranges encompassed within that range as if each numerical value and sub-range is explicitly recited.
- a temperature in the range of 60 °C to 220 °C should be interpreted to include not only the explicitly recited limits of 60 °C to 220 °C but also to include sub-ranges, such as 75 °C to 208 °C, and so forth, as well as individual amounts, within the specified ranges, such as 109.6 °C, and
- the present invention provides an effective, convenient, and safe chemical process for the preparation of vanillin from eugenol and eugenol rich essential oils as such from any eugenol rich plant such as Syzygium aromaticum (L) Merr. and L.M. Penny (Clove leaf/bud oil), Ocimum tenuiflorum cv. CIM-ayu, Ocimum gratissimum (Clocimum) with high purity and minimum side products.
- This process is also efficient for the preparation of other important substituted phenylaldehydes from substituted phenylpropenes and substituted phenylpropenes rich essential oils.
- This process is useful for value addition of essential oils rich in substituted phenylpropenes(s)/undesirable essential oils rich in phenylpropenes(s) from sassafras and banned plant varieties such as tetraploid or hexaploid varieties of Acorus calamus.
- the proven working efficiency of this process offers conversion of eugenol to vanillin up to lOg scale which can be up- scaled up to industrial level, even under non-specific reaction conditions such as anhydrous or inert reaction conditions.
- the present invention also provides an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, saffole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, Ocimum gratissimum, rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a
- step (a) there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the solvent used in step (a) is selected from the group consisting of choline hydroxide and choline chloride.
- the solvent used in step (a) is choline hydroxide.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent, wherein the solvent is selected from the group consisting of choline hydroxide and choline chloride, and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and O
- step (a) there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the metal hydroxide used in step (a) is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide.
- the metal hydroxide used in step (a) is potassium hydroxide.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide, wherein the metal hydroxide is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide, to obtain a compound of Formula (3); wherein R
- a process for the preparation of a compound Formula (1) as disclosed herein wherein the oxidizing agent used in step (b) is selected from the group consisting of OsO 4 /NaIO 4 , K 2 [OsO 2 (OH) 4 ]/NaIO 4 , OsCl 3 /NaIO 4 , Polyurea-encapsulated OsO 4 /NaIO 4 , Poly 4-vinylpyridine crosslinked supported OsO 4 /NaIO 4 , Styrene-divinylbenzene crosslinked supported OsO 4 /NaIO 4 .
- the oxidizing agent used in step (b) is OsO 4 /NaIO 4 .
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, Ocimum gratissimum, rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent, wherein
- step (b) is a mixture of water and non-protic solvent in a ratio of 1:4.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- a process for the preparation of a compound Formula (1) as disclosed herein wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol.
- the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol.
- a process for the preparation of a compound Formula (1) as disclosed herein wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2- methyltetrahydrofuran, 1,4-dioxane, and t-butylmethyl ether.
- the non-protic solvent is tetrahydrofuran.
- step (b) is a mixture of water and non-protic solvent
- the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol, in a ratio of 1:4.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- step (b) a process for the preparation of a compound Formula (1) as disclosed herein, wherein the additive used in step (b) is selected from the group consisting of (a) tertiary amine N -oxides selected from pyridine-N-oxide, 2,6- dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N-oxide, N,N- dimethylaniline-N-oxide, N,N-diethylaniline-N-oxide and triethylamine-N-oxide, and (b) methyl- substituted pyridines selected from 2-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine), 4- methylpyridine, 2,4,6-trimethylpyridine.
- the additive used in step (b) is selected from the group consisting of (a) tertiary amine N -oxides selected from pyridine-N-oxide, 2,6- dimethyl pyr
- step (b) there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the additive used in step (b) is selected from the group consisting of (a) tertiary amine N-oxides selected from pyridine-N- oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N- oxide, and (b) methyl-substituted pyridines selected from 2-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine) and 2,4,6-trimethylpyridine.
- the additive used in step (b) is 2,6-dimethyl pyridine.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- a process for the preparation of a compound Formula (1) as disclosed herein wherein the oxidizing agent used in step (b) is in the range of between 0.05 to 2.0 mole % and metaperiodate used in step (c) is the range of between 1 to 5 moles %.
- the oxidizing agent used in step (b) is 0.2 mole % and metaperiodate used in step (c) is 2.5 moles %.
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- a process for the preparation of a compound Formula (1) wherein R 1 is CHO; R 2 , R 3 , R 4 , R 5 , and R 6 are independently selected from H, OH, and OCH 3 ; or R 4 and R 5 together form -OCH 2 O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R 1 ’ together form -OCH 2 O-; and R 3 ’ is selected from H and OCH 3 , in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH 3 ; R 1 ’ and R 2 ’ are independently selected from H and OCH 3 or R and R and R
- the present invention reports a chemical process for the preparation of substituted phenylaldehydes such as vanillin from substituted phenylpropenes and substituted phenylpropenes rich essential oils such as eugenol (2a) and eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of isoeugenol, an intermediate compound (Scheme 1 ).
- the invention particularly relates to the conversion of phenylpropenes and substituted phenylpropenes rich essential oils to substituted phenylaldehydes such as vanillin without the protection of the phenolic group, therefore, offers step economy.
- the present chemical process involves the use of environment-friendly solvents and devoid of the use of any chlorinated solvent. This process also provides ease of preparation for substituted phenylaldehydes of formula 1 (wherein R 1 is -CHO, and R 2 , R 3 , R 4 , R 5 , R 6 , may be (a) a hydrogen atom; (b) an alkoxy group having one or more carbon atom with at least two of R 2 , R 3 , R 4 , R 5 , R 6 , being a hydrogen atom; (c) an alkoxy group having one or more carbon atom with at least one of R 2 , R 3 , R 4 , R 5 , R 6 , being a hydroxyl group; (d) an alkoxy group having one or more carbon atom with one of R 2 , R 3 , R 4 , R 5 , R 6 , being a methylenedioxy group in combination with either a hydrogen atom or a hydroxyl group or an alkoxy group
- Scheme 1 The process for the preparation of substituted phenylaldehydes from substituted phenylpropenes through either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylprop-2-enes, an intermediate compounds.
- the present invention also provides an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safrole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
- Synthesis The process for the preparation of vanillin from eugenol comprises two synthetic steps; Step 1: Conversion of eugenol (2a) to isoeugenol (3a,); Step 2: Conversion of isoeugenol to vanillin (la).
- Step-1 Conversion of eugenol (2a) to isoeugenol (3a)
- eugenol (2a) into isoeugenol (3a)
- alkali-metal hydroxide s
- s alkali-metal hydroxide
- choline based eutectic solvents such as choline hydroxide, choline chloride, etc.
- Step-2 Conversion of isoeugenol (3a) to vanillin (la)
- isoeugenol (3a) was treated with an aqueous solution of osmium tetroxide (OsO 4 ) ranging from 0.05 to 2.0-mole % and substituted pyridines such as 2-methylpyridine, 3-methylpyridine, 4-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine), 2,4,6-trimethylpyridine, 4-dimethylaminopyridine or tertiary amine N-oxides such as pyridine-N-oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4- picoline-N-oxide, ranging from 0.5 to 5 moles in biphasic reaction medium having a mixture of water and non-protic solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dio
- Table 1 Physical data of vanillin (la), veratraldehyde (3,4-dimethoxybenzaldehyde, lb), 4-anisaldehyde (4- methoxybenzaldehyde, lc), 2,3-dimethxoy-4,5-methylenedixybenzaldehyde (Id), and 2,4,5 -trimethoxybenzaldehyde
- Step 1 Preparation of isoeugenol (3a) form eugenol (2a) [0067]
- eugenol 0.1 g, 0.61 mmol
- choline hydroxide 46% assay w/win aqueous solution, 0.5 g, 4.27 mmol
- potassium hydroxide 0.2 g, 3.65 mmol
- the organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material.
- the crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil.
- the crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil.
- the crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil.
- the progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, the reaction was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin(la) as a white solid in 49% yield.
- sodium metaperiodate (NalCU, 32.49 g, 152.43 mmol) was added to the reaction mixture portion- wise (in about 20 min) and stirring was continued for 3h at 25°C.
- the progress of the reaction was monitored by TLC (20% ethyl acetate- hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- the crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 70% yield.
- sodium metaperiodate NaIO 4 , 1.63 g, 7.62 mmol
- reaction mixture portion-wise in about 7 min
- stirring was continued for 3h at 25°C.
- the progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- sodium metaperiodate NaIO 4 , 1.63 g, 7.62 mmol
- reaction mixture portion-wise in about 7 min
- stirring was continued for 3h at 25 °C.
- the progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- the crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 65.2% yield.
- the crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 58.6% yield.
- the crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 59% yield.
- the crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil.
- Step-2 Preparation of vanillin (la) from isoeugenol (3a) (obtained from modified clove oil): [0083] On ice bath, isoeugenol (3a, 10.00g, 60.97 mmol) was taken in around bottom flask and dissolved in 300 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO 4 , 0.2% aqueous solution, 100mL) was dropwise added to the solution (in about 20- 30 min). After 10 min, 2,6-dimethyl pyridine (6.80g, 60.97 mmol) was added to the reaction mixture which was stirred further for 30 min.
- THF tetrahydrofuran
- OsO 4 osmium tetroxide
- sodium metaperiodate (NalO 4 , 32.49g, 152.43 mmol) was added to the reaction mixture portion- wise (in about 20 min) and stirring was continued for 3h at 25°C.
- the progress of the reaction was monitored by TLC (20% ethyl acetate- hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 , and concentrated.
- the crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 70% yield.
- Step 1 Preparation of methyl isoeugenol (3b) from methyl eugenol (2b):
- Step 2 Preparation of veratraldehyde (3, 4-dimethoxybenzaldehyde, lb) from methyl isoeugenol
- Step 1 Preparation of anethole (3c) from methyl chavicol (2c):
- the crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure anethole (3c, a mixture of cis and trans) as oil.
- Step 2 Preparation of 4-anisaldehyde (4-methoxy benzaldehyde, lc) from anethole (3c):
- anethole (3c, 0.20 g, 1.35 mmol) was taken in around bottom flask and dissolved in a 6 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO 4 , 0.2% aqueous solution, 2 mL) was dropwise added to the solution (in about 5 min). After 10 min, 2,6-dimethyl pyridine (0.14g, 1.35 mmol) was added to the reaction mixture which was stirred further for 30 min.
- THF tetrahydrofuran
- OsO 4 osmium tetroxide
- sodium metaperiodate NaIO 4 ,0.72g, 3.38 mmol
- reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25 °C.
- the progress of the reaction was monitored by TLC (15% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- Step 1 Preparation of isodillapiole (3d) from dillapiole (2d):
- sodium metaperiodate NaIO 4 ,0.07g, 0.34 mmol
- reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25°C.
- the progress of the reaction was monitored by TLC (5% ethyl acetate-hexane, 2 runs). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- the crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (1:99) as eluent which yielded pure 2,3-dimethoxy-4,5- methylenedioxybenzaldehyde (Id) as a white solid in 70% yield.
- sodium metaperiodate NaIO 4 ,0.15 g, 0.72 mmol
- reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25°C.
- the progress of the reaction was monitored by TLC (10% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na 2 SO 4 ), and concentrated.
- the crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate- hexane (1.5:98.5) as eluent which yielded pure 2,4,5-trimethoxybenzaldehyde (le) as a white solid in 75% yield.
- Example 7 Preparation of 2,4,5-trimethoxybenzaldehyde (le) from ⁇ -asarone (3f): [0092] On ice bath, ⁇ -asarone (3f, 0.04 g, 0.19 mmol) was taken in around bottom flask and dissolved in 3 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO 4 , 0.2% aqueous solution, 0.04 mL) was dropwise added to the solution (in about 1- 2 min). After 10 min, 2,6-dimethyl pyridine (0.02 g, 0.19 mmol) was added to the reaction mixture which was stirred further for 30 min.
- THF tetrahydrofuran
- OsO 4 osmium tetroxide
- the crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate- hexane (1.5:98.5) as eluent which yielded pure 2,4,5-trimethoxybenzaldehyde (le) as a white solid in 73% yield.
- a chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safirole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
- the process does not require the protection of the phenolic group of eugenol/isoeugenol or purification/enrichment of essential oils and therefore offers a step economy.
- the process uses environmentally safe solvents e.g. class 3 and 4 solvents, accepted in the pharmaceutical industry. [0100] The process does not require any specific reaction conditions e.g. anhydrous or inert reaction conditions.
- the process offers significantly improved product yield(s) (up to 29%) over the existing process.
- the process is useful for value addition of essential oils rich in phenylpropenes(s) or undesirable essential oils rich in phenylpropenes(s) from banned plant varieties such as tetraploid or hexaploid varieties of Acorus calamus, through the preparation of important phenylaldehydes useful in different industries.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
The present invention relates to a chemical process for the production of substituted phenylaldehydes such as vanillin (1a) from substituted phenylpropenes or substituted phenylpropenes enriched essential oils such as eugenol (2a) or eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylprop-2-enes such as isoeugenol, an intermediate compound. The invention relates to the conversion of substituted phenylpropenes to other substituted phenylaldehydes, particularly vanillin without the protection of the phenolic group, therefore, it offers a step economy. The present chemical process involves the use of class 3 and 4 solvents thereby devoid of the use of any chlorinated solvent.
Description
A PROCESS FOR THE PREPARATION OF VANILLIN AND OTHER SUBSTITUTED
PHENYLALDEHYDES
FIELD OF THE INVENTION
[0001] The present invention relates to a chemical process for the production of substituted phenylaldehydes such as vanillin from substituted phenylpropenes or substituted phenylpropenes enriched essential oils such as eugenol or eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylprop-2-enes such as isoeugenol, an intermediate compound. The invention relates to the conversion of substituted phenylpropenes to other substituted phenylaldehydes, particularly vanillin without the protection of the phenolic group, therefore, it offers a step economy. The present chemical process involves the use of environment-friendly solvents and devoid of the use of any chlorinated solvent. This novel process developed for the production of vanillin and also provides ease of preparation for other substituted phenylaldehydes derivatives. BACKGROUND OF THE INVENTION
[0002] Vanillin (4-hydroxy-3-methoxybenzaldehyde, la) is the highest volume aroma chemical produced worldwide ( Fache et al., 2016, ACS Sustainable Chem. Eng., 4(1), 35-46). Natural vanilla is extracted from the cured pods of different species of Vanilla (Orchidaceae), V. Planifolia Jacks, ex Andrews (syn. V.fragrans (Salisbury) Ames), V. tahitensis J.W. Moore, and V. pompona Scheide ( Sinha et al., 2008, J. Food Sci. Nutr., 59(4), 299-326 ). It is used widely in both food and non-food applications, in fragrances, and as a flavoring in pharmaceutical preparations. In the food industry, vanillin is widely used as a flavoring agent in confectionery, chocolates, baked goods, non-alcoholic and alcoholic beverages, and many other foods. Non-food applications of vanillin are found in cosmetics, personal care products, detergents, and perfumery. In the pharmaceutical industry, vanillin is a starting material in the manufacture of several drugs (for example L-3,4- DihydroxyPhenylAlanine, L-DOPA) (Kaiser et al. 1976, US 3969397 and Knowles, W. S., 2002, Angew. Chem., Int. Ed., 41, 1998-2007; Knowles, W. S., 1986, Chem. Educ, 63, 222-225; Inukai, et al., 1998, JP 10136994 A). Approximately 60% of industrial vanillin is used in the food industry; 33% as a fragrance in perfumes and cosmetics; and 7% in pharmaceuticals ( Priefert , et. al., 2001, Appl. Microbiol. Biot., 56(3-4), 296-314). Reports suggest that less than 1% of the global production of vanillin is derived from vanilla pods; the majority is produced synthetically using,
e.g. petroleum products, lignin, and eugenol as starting materials ( Walton et al., 2000, Curr. Opin. Biotechnol., 11, 490-496 ).
[0003] In 1858, for the first time, Gobley isolated and identified the vanillin constituent of the vanilla bean and confirmed that vanillin was the chief flavor component of vanilla. The vanilla aroma was so popular that in 1875, less than 20 years from its initial isolation, synthetic vanillin prepared from eugenol by its isomerization followed by oxidation became available in France and the United States (Bots R. H., 1927, US 1643,804). In this process, synthesis of vanillin involves the treatment of eugenol with potassium hydroxide at around 150°C for isomerization of eugenol to isoeugenol which is subsequently oxidized to vanillin using nitro-benzol or mono nitro-toluol as an oxidizing agent. The drawback of this process is the use of a large amount of aniline (toxic vapors) and the formation of azo-benzol as a major by-product.
[0004] In another process for vanillin production, isoeugenol is treated with hydrogen peroxide in presence of anhydrous tertiary amyl alcohol and vanadium pentoxide {Milas et al. 1947, US 2414385 and Milas, N. A., 1937, J. Am. Chem. Soc., 59, 2342). However, this process is also associated with prolonging reaction time (12-15 h) and tedious isolation of vanillin through the p- nitrophenylhydrazone formation. The preparation of vanillin from isoeugenol in only 23% yield by the use of a two-step methodology is also reported {Pappo, R., 1956, et al. J. Org. Chem., 21, 478-479). This methodology involves the protection of the phenolic group of isoeugenol followed by oxidation using osmium tetroxide/sodium periodate. The disadvantages of this process for the preparation of vanillin from eugenol are three chemical steps and low product yield. Further, in 1957, Frainacci, N. T., reported the preparation of acetyl vanillin from acetyl isoeugenol in 90% yield using chromium trioxide (CrO3), Cone, sulfuric acid, and acetic acid (US 2794813). However, this method does not disclose preparation vanillin as such and the use of excess sulfuric acid is the main disadvantage of this method. In 1963, Lampman et al. reported the preparation of vanillin from eugenol via a two-step chemical process using KOH, dimethylsulfoxide, and nitrobenzene {Lampman et al., 1963, J. Chem. Educ., 60, 503-504). The caustic effluent and azobenzene as major wastes in this process area significant drawbacks of the process. In 1970, Fiechi et al. disclosed another method for the preparation of vanillin by isomerization of eugenol to isoeugenol using aqueous alkali metal hydroxide and subsequent oxidation of isoeugenol to vanillin by sodium meta-nitrobenzenesulfonate (US 3544621). The use of azobenzene or aniline as a solvent and excess mineral acids such as nitric acid, sulfuric acid is associated with the
disadvantages of this process. Further, in 2001, CN 1289836A developed by Shanghai Tianxiang Fine Chemical Co., Ltd., disclosed a chemical method of preparing vanillin (in 50% yield) from clove oil using sodium hydroxide, potassium permanganate, p-aminobenzene sulfonic acid, nitrobenzene, and carbonyl-based mixed catalyst at 100-300°C. The waste generated by the reaction is difficult to deal with, resulting in serious environmental pollution. In 2002, Sinha et. al. derived a microwave-assisted process for the preparation of phenylaldehydes from substituted phenylpropenes involving osmium tetroxide and sodium meta-periodate in presence of cocatalysts namely amberlite IRA-410 and quaternary ammonium salt (WO 02/079132 and EP 1373178A1). However, despite the author’s claim, there is no actual experimental data for the application of this process to vanillin production or any other phenylpropene having a free phenolic group. Additionally, the use of a chlorinated solvent(s), and microwave assistance are disadvantages of this method.
[0005] While efforts are on for the preparation of vanillin from eugenol, the production of vanillin from the lignin, another natural source for vanillin production, began in around 1936 (Sandbom et al., 1936, US 2057117 and Schulz L., 1940, US 2187366). This method rapidly became the main process for vanillin production. In this process, the preparation of vanillin involves the use of nitrobenzene or nitrobenzene sulfonic acid, NaOH, or KOH and was associated with the disadvantages such as prolong reaction time and low yield of vanillin. However, almost all the lignin-to- vanillin plants were closed at the end of the 1990s due to increased environmental concerns towards the caustic effluents of the process, decreased lignin availability due to the increasing use of the Kraft process for pulping - in which lignin is burnt to produce energy, and also due to the arising of cheap chemical intermediates from petroleum such guaiacol for vanillin synthesis which started in 1936 (Boedecker and Volk, US 2,062,205). Since then, the dominant feedstock for vanillin is petroleum, a non-renewable source ( Hocking , M. B., 1997, J. Chem. Educ., 74, 1055·, Fache et al., 2016, ACS Sustainable Chem. Eng., 4(1), 35-46; Corbet, M. et al., 2013,
WO 2013/166642 Al).
[0006] Natural-grade vanillin prepared from eugenol has an enticing aroma, food safety, and high practical value, thus attracts wide attention and in-depth research. In general, the preparation of vanillin from eugenol traditionally involves mainly two steps (1) isomerization of eugenol to isoeugenol and (2) oxidation of isoeugenol to vanillin. There have been continuous investigations for new methods for either isomerization of eugenol by the use of transition metal compounds such
as compounds of palladium, cobalt, ruthenium, copper, etc ( Parreira, L. et al., Adv. Synth. Catal., 352, 1533-1538·, Mao, H., et al., 2016, J. Chin. Chem. Soc., 63, 261-266·, Sanchez-Gonalez, E., et al., 2017, Cryst. Eng. Comm., 19, 4142-4146). However, except for a few reports of academic interest, oxidation of isoeugenol to vanillin is mainly realized by the conventional methods described above ( Liu, B., et al., 2019, Chem. Comm., 55, 4817-4820·, Adilina, I. B., et al., 2012, J.
Mol. Catal. A-Chem., 361-362, 72-79', Franco, A., et al., 2017, Beilstein J. Org. Chem., 13, 1439- 1445·, Gusevskaya, E. V., et al., 2012, J. Mol. Catal. A-Chem., 363-364, 140-147).
[0007] Despite the above protocols for vanillin production from eugenol, there has been a continuous surge for the exploration of more efficient and safe procedures for vanillin production. The present invention relates to the development of an efficient, convenient, and safe chemical process for vanillin production from eugenol and eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of isoeugenol, an intermediate compound. This novel process also provides ease of preparation for other substituted phenylaldehydes from either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylpropenes and substituted phenylpropenes rich essential oils.
OBJECTIVE OF THE INVENTION
[0008] The main objective of the present invention is to provide a novel, convenient, efficient, and safe chemical process for preparing vanillin and other substituted phenylaldehydes from substituted phenylpropenes or substituted phenylpropenes enriched essential oils (Scheme 1).
[0009] Another objective of the present invention is to provide a convenient, efficient, and safe chemical process for preparing vanillin from eugenol and eugenol rich essential oils.
[0010] Yet another objective of the invention is to provide an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safrole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
[0011] It is also an objective of the invention to provide a process for the preparation of other substituted phenylaldehydes from either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylpropenes.
[0012] Another objective of the invention is to develop a chemical process for the production of vanillin and other substituted phenylaldehydes with high purity and minimum side products. [0013] Yet another objective of the invention is to provide an effective chemical process that uses environmentally safe solvents e.g. class 3 and 4 solvents. [0014] Yet another objective of the invention is to provide an effective, convenient, and safe chemical process that is devoid of the use of any chlorinated solvent (starting from reaction medium to work up process).
[0015] Yet another objective of the invention is to provide an effective, convenient, and safe chemical process which does not require any specific reaction conditions e.g. anhydrous or inert reaction conditions.
[0016] It is also an objective of the invention to provide an effective, convenient, and safe chemical process for vanillin production from isoeugenol, with improved product yield(s) significantly (up to 29%) as compared with the existing process by the use of methyl-substituted pyridines as additives such as 2,6-dimethylpyridine (2,6-lutidine), 2-methylpyridine, 4-methylpyridine, 2,4,6- trimethylpyridine and tertiary amine N-oxides such as pyridine-N-oxide, 2-picoline-N-oxide, 3- picoline-N-oxide, 4-picoline-N-oxide.
[0017] Yet another objective of the invention is to provide an effective, convenient, and safe chemical process for value addition of essential oils rich in substituted phenylpropene(s) through the preparation of important substituted phenylaldehydes. [0018] Yet another objective of the invention is to provide an effective, convenient, and safe chemical process for large scale production of vanillin to the industrial level.
SUMMARY OF THE INVENTION
[0019] In an aspect of the present disclosure, there is provided a process for the preparation of a compound Formula ( 1 ),
 wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3;
or R4 and R5 together form -OCH2O-, comprising the steps of: a. isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3;
or R4 and R5 together form -OCH2O-, comprising the steps of: a. isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
 wherein R is selected from OH and OCH3;
wherein R is selected from OH and OCH3;
R1 and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula
(3);
 wherein R is selected from OH and OCH3;
wherein R is selected from OH and OCH3;
R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, b. oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); c. oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0020] These and other features, aspects, and advantages of the present subject matter will be better understood with reference to the following description and appended claims. This summary is provided to introduce a selection of concepts in a simplified form. This summary is not intended
to identify key features or essential features of the claimed subject matter, nor is it intended to be used to limit the scope of the claimed subject matter.
DETAILED DESCRIPTION OF THE INVENTION [0021] The invention will now be described in detail in connection with certain preferred and optional embodiments, so that various aspects thereof may be more fully understood and appreciated.
Definitions:
[0022] For convenience, before further description of the present disclosure, certain terms employed in the specification, and examples are delineated here. These definitions should be read in the light of the remainder of the disclosure and understood as by a person of skill in the art. The terms used herein have the meanings recognized and known to those of skill in the art, however, for convenience and completeness, particular terms and their meanings are set forth below.
[0023] The articles "a", "an" and "the" are used to refer to one or to more than one (i.e., to at least one) of the grammatical object of the article.
[0024] The terms "comprise" and "comprising" are used in the inclusive, open sense, meaning that additional elements may be included. It is not intended to be construed as "consists of only". [0025] Throughout this specification, unless the context requires otherwise the word "comprise", and variations such as "comprises" and "comprising", will be understood to imply the inclusion of a stated element or step or group of element or steps but not the exclusion of any other element or step or group of element or steps.
[0026] The term “essential oil” herein refers to the concentrated volatile oil extracted from plants which retains the natural odour and flavor of the plant from which it was extracted. In the present disclosure, the term “essential oil” includes but not limited to essential oils obtained from eugenol rich plants such as Syzygium aromaticum, Ocimum tenuiflorum, and Ocimum gratissimum.
[0027] The term “oxidizing agent” herein refers to the substance used to oxidize a reactant in a chemical reaction. In the present disclosure, the term “oxidizing agent” includes but not limited to OsO4/NaIO4, K2[OsO2(OH)4]/NaIO4, OsCl3/NaIO4, Polyurea-encapsulated OsO4/NaIO4, Poly 4- vinylpyridine crosslinked supported OsO4/NaIO4 and Styrene-divinylbenzene crosslinked supported OsO4/NaIO4.
[0028] The term “biphasic medium” herein refers to a medium comprising two different phases for a reaction mixture. In the present disclosure, the term “biphasic medium” includes but not limited to a mixture of water and non-prodc solvent in a ratio 1:4.
[0029] The term “non-protic solvent” herein refers to the solvent which does not release any proton (H+ ion) upon dissociation. In the present disclosure, the term “non-protic solvent” includes but not limited to tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether and tert-butanol.
[0030] Ratios, concentrations, amounts, and other numerical data may be presented herein in a range format. It is to be understood that such range format is used merely for convenience and brevity and should be interpreted flexibly to include not only the numerical values explicitly recited as the limits of the range, but also to include all the individual numerical values or sub-ranges encompassed within that range as if each numerical value and sub-range is explicitly recited. For example, a temperature in the range of 60 °C to 220 °C should be interpreted to include not only the explicitly recited limits of 60 °C to 220 °C but also to include sub-ranges, such as 75 °C to 208 °C, and so forth, as well as individual amounts, within the specified ranges, such as 109.6 °C, and
187.3 °C.
[0031] The present disclosure is not to be limited in scope by the specific embodiments described herein, which are intended for the purposes of exemplification only. Funcdonally-equivalent products, compositions, and methods are clearly within the scope of the disclosure, as described herein.
[0032] The present invention provides an effective, convenient, and safe chemical process for the preparation of vanillin from eugenol and eugenol rich essential oils as such from any eugenol rich plant such as Syzygium aromaticum (L) Merr. and L.M. Penny (Clove leaf/bud oil), Ocimum tenuiflorum cv. CIM-ayu, Ocimum gratissimum (Clocimum) with high purity and minimum side products. This process is also efficient for the preparation of other important substituted phenylaldehydes from substituted phenylpropenes and substituted phenylpropenes rich essential oils. This process is useful for value addition of essential oils rich in substituted phenylpropenes(s)/undesirable essential oils rich in phenylpropenes(s) from sassafras and banned plant varieties such as tetraploid or hexaploid varieties of Acorus calamus. The proven working efficiency of this process offers conversion of eugenol to vanillin up to lOg scale which can be up-
scaled up to industrial level, even under non-specific reaction conditions such as anhydrous or inert reaction conditions.
[0033] Further, the present invention also provides an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, saffole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents.
[0034] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1),
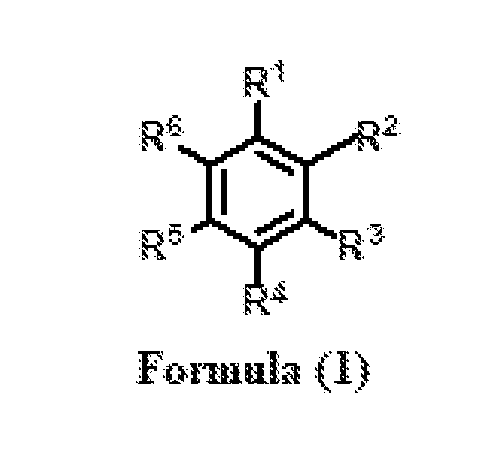 wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
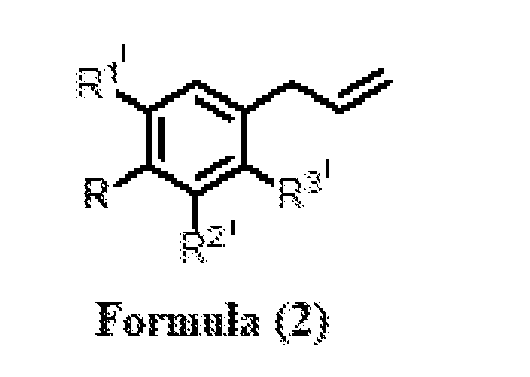 wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3);
wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3);
 wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1 together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a
biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30°C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1 together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a
biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30°C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0035] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with osmium tetroxide and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0036] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, Ocimum gratissimum. [0037] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, Ocimum gratissimum, rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected
from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1). [0038] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the solvent used in step (a) is selected from the group consisting of choline hydroxide and choline chloride. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the solvent used in step (a) is choline hydroxide. [0039] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent, wherein the solvent is selected from the group consisting of choline hydroxide and choline chloride, and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0040] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the metal hydroxide used in step (a) is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the metal hydroxide used in step (a) is potassium hydroxide.
[0041] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide, wherein the metal hydroxide is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide, to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0042] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the oxidizing agent used in step (b) is selected from the group consisting of OsO4/NaIO4, K2[OsO2(OH)4]/NaIO4, OsCl3/NaIO4, Polyurea-encapsulated OsO4/NaIO4, Poly 4-vinylpyridine crosslinked supported OsO4/NaIO4, Styrene-divinylbenzene crosslinked supported OsO4/NaIO4. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the oxidizing agent used in step (b) is OsO4/NaIO4. [0043] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected
from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent, wherein the oxidizing agent is selected from the group consisting of OsO4/NaIO4, K2[OsO2(OH)4]/NaIO4, OsCl3/NaIO4, Polyurea-encapsulated OsO4/NaIO4, Poly 4-vinylpyridine crosslinked supported OsO4/NaIO4, Styrene-divinylbenzene crosslinked supported OsO4/NaIO4, and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0044] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, Ocimum gratissimum, rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent, wherein the solvent is selected from the group consisting of choline hydroxide and choline chloride, and a metal hydroxide, wherein the metal hydroxide is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide, to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent, wherein the oxidizing agent is selected from the group consisting of OsO4/NaIO4,
K2[OsO2(OH)4]/NaIO4, OsCl3/NaIO4, Polyurea-encapsulated OsO4/NaIO4, Poly 4-vinylpyridine crosslinked supported OsO4/NaIO4, Styrene-divinylbenzene crosslinked supported OsO4/NaIO4, and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0045] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the biphasic reaction medium of step (b) is a mixture of water and non-protic solvent in a ratio of 1:4.
[0046] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium, wherein the biphasic reaction medium is a mixture of water and non-protic solvent in a ratio of 1 :4, to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0047] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein,
wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2- methyltetrahydrofuran, 1,4-dioxane, and t-butylmethyl ether. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the non-protic solvent is tetrahydrofuran. [0048] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the biphasic reaction medium of step (b) is a mixture of water and non-protic solvent, wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol, in a ratio of 1:4. [0049] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium, wherein the biphasic reaction medium is a mixture of water and non-protic solvent in a ratio of 1:4, wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol, to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0050] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the additive used in step (b) is selected from the group consisting of (a) tertiary amine N -oxides selected from pyridine-N-oxide, 2,6- dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N-oxide, N,N-
dimethylaniline-N-oxide, N,N-diethylaniline-N-oxide and triethylamine-N-oxide, and (b) methyl- substituted pyridines selected from 2-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine), 4- methylpyridine, 2,4,6-trimethylpyridine.
[0051] In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the additive used in step (b) is selected from the group consisting of (a) tertiary amine N-oxides selected from pyridine-N- oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N- oxide, and (b) methyl-substituted pyridines selected from 2-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine) and 2,4,6-trimethylpyridine. In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the additive used in step (b) is 2,6-dimethyl pyridine.
[0052] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive, wherein the additive is selected from the group consisting of (a) tertiary amine N-oxides selected from pyridine-N-oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline- N-oxide, 4-picoline-N-oxide, N,N-dimethylaniline-N-oxide, N,N-diethylaniline-N-oxide and triethylamine-N-oxide, and (b) methyl-substituted pyridines selected from 2-methylpyridine, 2,6- dimethylpyridine (2,6-lutidine), 4-methylpyridine, 2,4,6-trimethylpyridine, in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0053] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the oxidizing agent used in step (b) is in the range of between 0.05 to 2.0 mole % and metaperiodate used in step (c) is the range of between 1 to 5 moles %. [0054] In another embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the oxidizing agent used in step (b) is 0.2 mole % and metaperiodate used in step (c) is 2.5 moles %.
[0055] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent, wherein the oxidizing agent is in the range of between 0.05 to 2.0 mole %, and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate, wherein the metaperiodate is in the range of between 1 to 5 moles %, in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
[0056] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1) as disclosed herein, wherein the yield of the compound of Formula (1) is in the range of 58 to 70%.
[0057] In an embodiment of the present disclosure, there is provided a process for the preparation of a compound Formula (1), wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: (a) isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2), wherein R is selected from OH and OCH3; R1’ and R2’ are
independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula (3); wherein R is selected from OH and OCH3; R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, (b) oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3); (c) oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1), wherein the yield of the compound of Formula (1) is in the range of 58 to 70%. EXAMPLES
[0058] The following examples are given by way of illustration and should not construe to limit the scope of the invention. The synthesis of representative compounds has been given. Essential oil of Syzygium aromaticum(L) Merr. and L.M.Penny are obtained from Avra synthesis, Cat. No. ASC-2642. The Essential oil of Ocimum gratissimum (L) and Ocimum tenuiflorum cv. CIM-ayu are obtained from CSLRCLMAP, Farm, Lucknow.
[0059] The present invention reports a chemical process for the preparation of substituted phenylaldehydes such as vanillin from substituted phenylpropenes and substituted phenylpropenes rich essential oils such as eugenol (2a) and eugenol rich essential oils indiscriminately through either cis or trans or a mixture of cis and trans isomer(s) of isoeugenol, an intermediate compound (Scheme 1 ). The invention particularly relates to the conversion of phenylpropenes and substituted phenylpropenes rich essential oils to substituted phenylaldehydes such as vanillin without the protection of the phenolic group, therefore, offers step economy. The present chemical process involves the use of environment-friendly solvents and devoid of the use of any chlorinated solvent. This process also provides ease of preparation for substituted phenylaldehydes of formula 1 (wherein R1 is -CHO, and R2, R3, R4, R5, R6, may be (a) a hydrogen atom; (b) an alkoxy group having one or more carbon atom with at least two of R2, R3, R4, R5, R6, being a hydrogen atom; (c)
an alkoxy group having one or more carbon atom with at least one of R2, R3, R4, R5, R6, being a hydroxyl group; (d) an alkoxy group having one or more carbon atom with one of R2, R3, R4, R5, R6, being a methylenedioxy group in combination with either a hydrogen atom or a hydroxyl group or an alkoxy group having at least one carbon atoms; (f) a protected hydroxyl group with at least one of R2, R3, R4, R5, R6, being a hydrogen atom in combination with either an alkoxy group having one or more carbon atom, a hydroxyl group, a methylenedioxy group) from substituted phenylpropenes or substituted phenylpropenes enriched oils indiscriminately from either cis or trans or a mixture of cis and trans isomer(s).

Scheme 1: The process for the preparation of substituted phenylaldehydes from substituted phenylpropenes through either cis or trans or a mixture of cis and trans isomer(s) of substituted phenylprop-2-enes, an intermediate compounds.
[0060] Further, the present invention also provides an efficient, convenient, and safe chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safrole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents. [0061] Synthesis: The process for the preparation of vanillin from eugenol comprises two synthetic steps; Step 1: Conversion of eugenol (2a) to isoeugenol (3a,); Step 2: Conversion of isoeugenol to vanillin (la).
Step-1: Conversion of eugenol (2a) to isoeugenol (3a) [0062] Different attempts were made to transform eugenol (2a) into isoeugenol (3a) which involved the use of (i) alkali-metal hydroxide (s) such as sodium hydroxide, potassium hydroxide, barium hydroxide, etc., in a variety of choline based eutectic solvents such as choline hydroxide, choline chloride, etc., at a temperature ranging between 60-220 °C, however, in particular, for vanillin production from eugenol, we have used potassium hydroxide in Choline hydroxide at 140°C and 220°C which transformed eugenol (2a) to isoeugenol (3a) with 51 and 64 % conversion, respectively (GC analysis, cis:trans = 19:39 and 21 :43 respectively) Scheme-2. The transformation of eugenol (2a) into isoeugenol (3a) was also performed in neat Choline hydroxide which yielded isoeugenol (3a) in relatively low yield.
[0063] Accordingly, isomerization of other substituted phenylpropenes such as methyl eugenol (2b), methyl chavicol (2c), and dillapiole (2d) to methyl isoeugenol (3b), anethole (3c), and isodillapiole (3d) respectively, were prepared using methods.
Step-2: Conversion of isoeugenol (3a) to vanillin (la)
[0064] For conversion of isoeugenol (3a) to vanillin (la), isoeugenol (3a) was treated with an aqueous solution of osmium tetroxide (OsO4) ranging from 0.05 to 2.0-mole % and substituted pyridines such as 2-methylpyridine, 3-methylpyridine, 4-methylpyridine, 2,6-dimethylpyridine (2,6-lutidine), 2,4,6-trimethylpyridine, 4-dimethylaminopyridine or tertiary amine N-oxides such as pyridine-N-oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4- picoline-N-oxide, ranging from 0.5 to 5 moles in biphasic reaction medium having a mixture of water and non-protic solvents such as tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, isopropyl ether and t-butylmethyl ether in an appropriate ratio ranging from 0.025:4.75 to 2:3. Under these conditions, an osmate ester of isoeugenol (3a) was formed which was oxidized to
vanillin by the addition of sodium metaperiodate ranging from 1 to 5 moles in the same reaction at 20-30°C. However, in particular, we used 0.2 mole% of aqueous osmium tetroxide (OSO4) and 1.0 moles of 2,6-dimethylpyridine (2,6-luddine), 2.5 moles of sodium metaperiodate in water and tetrahydrofuran mixture (1:4), scheme-2. The same reaction was performed using 2- methyltetrahydrofuran, 1 ,4-dioxane, isopropyl ether, and t-butyl methyl ether respectively, in place of tetrahydrofuran. The efficiency of 2-methyltetrahydrofuran as solvent appeared to be marginally lower than tetrahydrofuran, however, other solvents such as 1 ,4-dioxane, isopropyl ether and t- butyl methyl ether gave vanillin in low yields. Also, the use of 2-methylpyridine, 3- methylpyridine,4-methylpyridine, 2,4,6-trimethylpyridine, 4-dimethylaminopyridine, and 2- aminopyridine ranging from 0.5 to 5 moles gave vanillin in relatively low yields in comparison to product yield obtained without any additive. Under similar reaction conditions, other additives such as pyridine-N-oxide, 2,6-dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N-oxide were also used for this conversion which yielded vanillin in improved yields. Accordingly, other substituted phenylaldehydes such as veratraldehyde (3,4- dimethoxybenzaldehyde, lb), 4-anisaldehyde (4-methoxybenzaldehyde, lc), 2,3-dimethxoy-4,5- methylenedixybenzaldehyde (Id), and 2,4,5-trimethoxybenzaldehyde (le) from either cis or trans or a mixture of cis and trans isomer(s) of methyl isoeugenol (3b), anethole (3c), either cis or trans or a mixture of cis and trans isomer(s) of isodillapiole (3d) and a-and β-asarone (a-and β-, 3e&3f) respectively, were prepared using developed process. [0065] The schematic diagram for the process of preparation is represented hereunder;

Scheme-2; Reaction conditions and reagents: (a) KOH, choline hydroxide (46% assay in aqueous solution), 220°C, 2.5h; (b) OsO4/NaIO4, tetrahydrofuran-water, 25°C, about 5.30h; (c) OsO4/NaIO4, tetrahydrofuran-water, 2,6-dimethylpyridine, 25°C, about 4.30h.
[0066] The details of vanillin (la), veratraldehyde (3,4-dimethoxybenzaldehyde, lb), 4- anisaldehyde (4-methoxybenzaldehyde, lc), 2,3-dimethxoy-4,5-methylenedixybenzaldehyde (Id), and 2,4,5-trimethoxybenzaldehyde (le) prepared using additives for the oxidation of isoeugenol, are given in Table- 1.
Table 1: Physical data of vanillin (la), veratraldehyde (3,4-dimethoxybenzaldehyde, lb), 4-anisaldehyde (4- methoxybenzaldehyde, lc), 2,3-dimethxoy-4,5-methylenedixybenzaldehyde (Id), and 2,4,5 -trimethoxybenzaldehyde
(le).
 Example-1
Example-1
Step 1: Preparation of isoeugenol (3a) form eugenol (2a)
[0067] (a) In a round bottom flask, eugenol (0.1 g, 0.61 mmol), choline hydroxide (46% assay w/win aqueous solution, 0.5 g, 4.27 mmol), and potassium hydroxide (0.2 g, 3.65 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was neutralized with con. HC1 (0.5 mL) followed by its extraction with ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 64% (GC analysis; cis: trans = 21:43). [0068] (b) In a round bottom flask, eugenol (0.2 g, 1.21 mmol), choline hydroxide (46% assay w/w in aqueous solution, 1.0 g, 8.53mmol), and sodium hydroxide (0.30 g, 7.31 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was neutralized with con. HQ (0.5 mL) followed by its extraction with ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 41% (GC analysis; cis: trans = 14:27).
[0069] (c) In a round bottom flask, eugenol (0.2 g, 1.21 mmol), choline hydroxide (46% assay w/w in aqueous solution, 1.0 g, 8.53mmol),and barium hydroxide (1.25 g, 7.31 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was neutralized with con. HQ (0.5 mL) followed by its extraction with ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 20% (GC analysis; cis: trans = 7:13).
[0070] (d) In a round bottom flask, eugenol (0.1 g, 0.61 mmol), choline hydroxide (46% assay w/w in aqueous solution, 0.5 g, 4.27 mmol) was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture extracted with ethyl acetate and water. The organic layer was dried over
anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 30% (GC analysis, cis: trans = 7:23). [0071](e) In a round bottom flask, eugenol (0.1 g, 0.61 mmol), choline chloride (0.5 g, 3.66 mmol), and potassium hydroxide (0.2 g, 3.65 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was neutralized with con. HC1 (0.5 mL) followed by its extraction with ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol(3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 28% (GC analysis; cis: trans = 9:19). Step 2: Preparation of vanillin(la) from isoeugenol(3a)
[0072](a) On ice bath, isoeugenol (3a, l.OOg, 6.09mmol) was taken in around bottom flask and dissolved in 40mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 10 mL) was dropwise added to the solution (in about 5-8 min). The reaction mixture was stirred for 2 h. After 2h, sodium metaperiodate (NaKU, 3.24g, 15.24 mmol) was added portion-wise to the reaction mixture (in about lOmin) and the reaction mixture was further stirred for 3 h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, the reaction was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin(la) as a white solid in 49% yield.
[0073](b) On ice bath, isoeugenol (3a, 10.00g, 60.97 mmol) was taken in around bottom flask and dissolved in 300 mL mixture of tetrahydrofuran (THF) and water (4: 1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 100mL) was dropwise added to the solution (in about 20- 30 min). After 10 min, 2,6-dimethylpyridine (6.80g, 60.97 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NalCU, 32.49
g, 152.43 mmol) was added to the reaction mixture portion- wise (in about 20 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate- hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 70% yield.
[0074] (c) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in a round bottom flask and dissolved in 15 mL mixture of 2-methyltetrahydrofuran (2-MeTHF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, 2,6-dimethylpyridine (0.32 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4, 1.63 g, 7.62 mmol) was added to reaction mixture portion-wise (in about 7 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin(la) as a white solid in 64% yield. [0075](d) On ice bath, isoeugenol(3a, 0.2 g, 1.21 mmol) was taken in a round bottom flask and dissolved in 10 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 2 mL) was dropwise added to the solution (in about 2 min). After 10 min, 2-methylpyridine (0.11 g, 1.21 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4, 0.65 g, 3.04 mmol) was added to reaction mixture portion-wise (in about 3 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 66 % yield. [0076](e) On ice bath, isoeugenol(3a, 0.22 g, 1.34 mmol) was taken in a round bottom flask and dissolved in 10 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium
tetroxide (OsO4, 0.2% aqueous solution, 2.2 mL) was dropwise added to the solution (in about 2 min). After 10 min, 2,4,6-trimethylpyridine (0.16 g, 1.34 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4, 0.71 g, 3.35 mmol) was added to reaction mixture portion-wise (in about 3 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 60% yield. [0077] (f) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in around bottom flask and dissolved in 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, pyridine N-oxide(0.28 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaKX 1.63 g, 7.62 mmol) was added to reaction mixture portion- wise (in about 7 min) and stirring was continued for 3h at
25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 56.5% yield.
[0078] (g) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in around bottom flask and dissolved in 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, 2,6-dimethyl pyridine N-oxide (0.37 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4, 1.63 g,
7.62 mmol) was added to reaction mixture portion-wise (in about 7 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 70% yield.
[0079] (h) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in around bottom flask and dissolved in 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, 2-picoline N-oxide (0.33 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4, 1.63 g, 7.62 mmol) was added to reaction mixture portion-wise (in about 7 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 65.2% yield.
[0080] (i) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in around bottom flask and dissolved in 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, 3-picoline N-oxide (0.33 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NalCU, 1.63 g, 7.62 mmol) was added to reaction mixture portion-wise (in about 7 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 58.6% yield.
[0081] (j) On ice bath, isoeugenol (3a, 0.5 g, 3.04 mmol) was taken in around bottom flask and dissolved in 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in about 2-3 min). After 10 min, 4-picoline N-oxide (0.33 g, 3.04 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NalCU, 1.63 g, 7.62 mmol) was added to reaction mixture portion-wise (in about 7 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (20% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material
was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate- hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 59% yield.
Example 2 Step-1: Preparation of isoeugenol (3a) from eugenol (2a) (Isomerisation of clove oil, 89% eugenol):
[0082] In a round bottom flask, clove oil (89% eugenol, 0.1 g, 0.61 mmol), choline hydroxide (46% assay w/w in aqueous solution, 0.5 g, 4.27 mmol), and potassium hydroxide (0.2 g, 3.65 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was neutralized with con. HQ (0.5 mL) followed by its extraction with ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure isoeugenol (3a, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 53% (GC analysis; cis: trans = 18:35). Step-2: Preparation of vanillin (la) from isoeugenol (3a) (obtained from modified clove oil): [0083] On ice bath, isoeugenol (3a, 10.00g, 60.97 mmol) was taken in around bottom flask and dissolved in 300 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 100mL) was dropwise added to the solution (in about 20- 30 min). After 10 min, 2,6-dimethyl pyridine (6.80g, 60.97 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NalO4, 32.49g, 152.43 mmol) was added to the reaction mixture portion- wise (in about 20 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (20% ethyl acetate- hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4 , and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate-hexane (10:90) as eluent which yielded pure vanillin (la) as a white solid in 70% yield.
[0084] The preparation of vanillin as obtained from Syzygium aromaticum (L) Merr. and L.M. Penny (Qove leaf/bud oil) may also be done from Ocimum tenuiflorum cv. CIM-Ayu, Ocimum gratissimum (Clocimum) as these plants are a rich source of eugenol.
Example 3
Step 1: Preparation of methyl isoeugenol (3b) from methyl eugenol (2b):
[0085] In a round bottom flask, methyl eugenol (O.lg, 0.56 mmol), choline hydroxide (46% assay w/w in aqueous solution, 0.5g, 4.48 mmol) and potassium hydroxide (0.1 g, 3.37 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by GC analysis. After completion of the reaction, the reaction mixture was worked up using ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure methyl isoeugenol
(3b, a mixture of cis and trans) as oil. The conversion from eugenol to methyl isoeugenol was 82% (GC analysis; cis: trans = 16:66).
Step 2: Preparation of veratraldehyde (3, 4-dimethoxybenzaldehyde, lb) from methyl isoeugenol
(3b): [0086] On ice bath, methyl isoeugenol (3b, 0.50g, 2.81 mmol) was taken in around bottom flask and dissolved in a 15 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 5 mL) was dropwise added to the solution (in 5min). After 10 min, 2,6-dimethyl pyridine (0.30g, 2.81 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4,1.50g, 7.02 mmol) was added to reaction mixture portion-wise (in about 15 min) and stirring was continued for 3h at 25°C.
The progress of the reaction was monitored by TLC (15% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate-hexane (5:95) as eluent which yielded pure veratraldehyde (3, 4-dimethoxybenzaldehyde, lb) as oil in 82% yield.
Example 4
Step 1: Preparation of anethole (3c) from methyl chavicol (2c):
[0087] In a round bottom flask, methyl chavicol (0. lg, 0.68 mmol), choline hydroxide (46% assay w/w in aqueous solution, 0.5g, 4.48 mmol) and potassium hydroxide (0.1 g, 4.08 mmol) was taken and was allowed to reflux for 2.5 h at temp 220°C. The progress of the reaction was monitored by
GC analysis. After completion of the reaction, the reaction mixture was worked up using ethyl acetate and water. The organic layer was dried over anhydrous sodium sulfate and concentrated to an oily crude material. The crude material was purified by column chromatography on silica gel (100-200 mesh size) using ethyl acetate-hexane as eluent which yielded pure anethole (3c, a mixture of cis and trans) as oil. The conversion from eugenol to isoeugenol was 84% (GC analysis; cis: trans = 14:70).
Step 2: Preparation of 4-anisaldehyde (4-methoxy benzaldehyde, lc) from anethole (3c):
[0088] On ice bath, anethole (3c, 0.20 g, 1.35 mmol) was taken in around bottom flask and dissolved in a 6 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 2 mL) was dropwise added to the solution (in about 5 min). After 10 min, 2,6-dimethyl pyridine (0.14g, 1.35 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4,0.72g, 3.38 mmol) was added to reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (15% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate- hexane (2:98) as eluent which yielded pure 4-anisaldehyde (lc) as a white solid in 59% yield. Example 5
Step 1: Preparation of isodillapiole (3d) from dillapiole (2d):
[0089] Under an inert atmosphere, dillapiole (2d, 0.05 g, 0.23 mmol) was taken in around bottom flask and dissolved in 3 mL of tetrahydrofuran. To this mixture, PdCl2 (0.002g) was added. The reaction mixture was allowed to stir at 30°C for 6 h. The completion of the reaction was confirmed by GC analysis. After completion of the reaction, the reaction mixture was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude was purified by column chromatography on silica gel (100-200 mesh size) using hexane as eluent which yielded pure isodillapiole (3d, a mixture of cis and trans) as oil. The conversion from dillapiole (2d) to isodillapiole(3d) was 76% (GC analysis, cis: trans = 6:70). Step 2: Preparation of 2,3-dimethoxy-4,5-methylenedioxybenzaldehyde (Id) from isodillapiole
(3d):
[0090] On ice bath, isodillapiole (3d, 0.03 g, 0.14 mmol) was taken in around bottom flask and dissolved in 3 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 0.03 mL) was dropwise added to the solution (in about 2 min). After 10 min, 2,6-dimethyl pyridine (0.02 g, 0.14 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4,0.07g, 0.34 mmol) was added to reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (5% ethyl acetate-hexane, 2 runs). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel 100-200 mesh size using ethyl acetate-hexane (1:99) as eluent which yielded pure 2,3-dimethoxy-4,5- methylenedioxybenzaldehyde (Id) as a white solid in 70% yield.
Example 6 Preparation of 2,4,5-trimethoxybenzaldehyde (le) from a-asarone (3e):
[0091] On ice bath, a-asarone (3e, 0.06 g, 0.29 mmol)was taken in around bottom flask and dissolved in 3 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 0.06 mL) was dropwise added to the solution (in about 2- 3 min). After 10 min, 2,6-dimethyl pyridine (0.03 g, 0.29 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIO4,0.15 g, 0.72 mmol) was added to reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25°C. The progress of the reaction was monitored by TLC (10% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate- hexane (1.5:98.5) as eluent which yielded pure 2,4,5-trimethoxybenzaldehyde (le) as a white solid in 75% yield.
Example 7 Preparation of 2,4,5-trimethoxybenzaldehyde (le) from β-asarone (3f):
[0092] On ice bath, β-asarone (3f, 0.04 g, 0.19 mmol) was taken in around bottom flask and dissolved in 3 mL mixture of tetrahydrofuran (THF) and water (4:1). After 15 min, osmium tetroxide (OsO4, 0.2% aqueous solution, 0.04 mL) was dropwise added to the solution (in about 1- 2 min). After 10 min, 2,6-dimethyl pyridine (0.02 g, 0.19 mmol) was added to the reaction mixture which was stirred further for 30 min. Subsequently, sodium metaperiodate (NaIC>4,0.10g, 0.48 mmol) was added to reaction mixture portion-wise (in about 5 min) and stirring was continued for 3h at 25 °C. The progress of the reaction was monitored by TLC (10% ethyl acetate-hexane). After completion of the reaction, it was worked up using ethyl acetate and water. The organic layer was separated, dried over anhydrous sodium sulfate (Na2SO4), and concentrated. The crude material was purified by column chromatography over silica gel (100-200 mesh size) using ethyl acetate- hexane (1.5:98.5) as eluent which yielded pure 2,4,5-trimethoxybenzaldehyde (le) as a white solid in 73% yield.
ADVANTAGES OF THE PRESENT INVENTION [0093] An efficient and safe chemical process for vanillin production from eugenol and eugenol rich essential oils.
[0094] A chemical process for the isomerization of eugenol and other substituted phenylpropenes such as phenylpropene, safirole, methyl chavicol, methyl eugenol, chavibetol, apiole, dillapiole, etc. which uses environmentally safe solvents. [0095] The process does not require the protection of the phenolic group of eugenol/isoeugenol or purification/enrichment of essential oils and therefore offers a step economy.
[0096] The process gives the same product yield from either cis or trans or a mixture of cis and trans isomer(s) of isoeugenol and therefore does not require any chromatographic separation of cis- and trans-isomers of isoeugenol or selective synthesis of cis- or trans-isomer of isoeugenol. [0097] This chemical process is also efficient for the preparation of other important substituted phenylaldehydes from either cis or trans or a mixture of cis and trans isomer(s) of phenylpropenes. [0098] The process offers production of vanillin and other substituted phenylaldehydes with high purity and minimum side products.
[0099] The process uses environmentally safe solvents e.g. class 3 and 4 solvents, accepted in the pharmaceutical industry.
[0100] The process does not require any specific reaction conditions e.g. anhydrous or inert reaction conditions.
[0101] The process offers significantly improved product yield(s) (up to 29%) over the existing process. [0102] The process is useful for value addition of essential oils rich in phenylpropenes(s) or undesirable essential oils rich in phenylpropenes(s) from banned plant varieties such as tetraploid or hexaploid varieties of Acorus calamus, through the preparation of important phenylaldehydes useful in different industries.
[0103] This effective chemical process is applicable for large scale production of vanillin.
Claims
1. A process for the preparation of a compound Formula (1),
 wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: a. isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
wherein R1 is CHO; R2, R3, R4, R5, and R6 are independently selected from H, OH, and OCH3; or R4 and R5 together form -OCH2O-, comprising the steps of: a. isomerization of substituted phenylpropenes of Formula (2) or essential oil rich in substituted phenylpropenes of Formula (2),
 wherein R is selected from OH and OCH3;
wherein R is selected from OH and OCH3;
R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3, in the presence of a solvent and a metal hydroxide to obtain a compound of Formula
(3);
 wherein R is selected from OH and OCH3;
wherein R is selected from OH and OCH3;
R1’ and R2’ are independently selected from H and OCH3 or R and R1’ together form -OCH2O-; and R3’ is selected from H and OCH3,
b. oxidizing the compound of Formula (3) as obtained in step (a) with an oxidizing agent and an additive in a biphasic reaction medium to obtain a reaction mixture comprising osmate ester of the compound of Formula (3) and c. oxidizing the osmate ester of the compound of Formula (3) by adding sodium metaperiodate in the reaction mixture as obtained in step (b) with stirring at a temperature ranging between 20 to 30 °C for a period of ranging between 1 to 5 hours to obtain the compound of Formula (1).
2. The process as claimed in claim 1, wherein the essential oil is obtained from eugenol rich plants selected from the group consisting of Syzygium aromaticum, Ocimum tenuiflorum, and Ocimum gratissimum.
3. The process as claimed in claim 1 , wherein the solvent used in step (a) is selected from the group consisting of choline hydroxide and choline chloride;
4. The process as claimed in claim 1, wherein the metal hydroxide used in step (a) is selected from the group consisting of potassium hydroxide, barium hydroxide, and sodium hydroxide.
5. The process as claimed in claim 1, wherein the oxidizing agent used in step (b) is selected from the group consisting of OsO4/NaIO4, K2[OsO2(OH)4]/NaIO4, OsCl3/NaIO4, Polyurea- encapsulated OsO4/NaIO4, Poly 4-vinylpyridine crosslinked supported OsO4/NaIO4, Styrene-divinylbenzene crosslinked supported OsO4/NaIO4.
6. The process as claimed in claim 1, wherein the biphasic reaction medium of step (b) is a mixture of water and non-protic solvent in a ratio 1:4.
7. The process as claimed in claim 6, wherein the non-protic solvent is selected from the group consisting of tetrahydrofuran, 2-methyltetrahydrofuran, 1,4-dioxane, acetone, acetonitrile, methyl tert-butylether, tert-butanol.
8. The process as claimed in claim 1, wherein the additive used in step (b) is selected from the group consisting of (a) tertiary amine N-oxides selected from pyridine-N-oxide, 2,6- dimethyl pyridine N-oxide, 2-picoline-N-oxide, 3-picoline-N-oxide, 4-picoline-N- oxide,N,N-dimethylaniline-N-oxide, N,N-diethylaniline-N-oxide and triethylamine-N- oxide, and (b) methyl-substituted pyridines selected from 2-methylpyridine, 2,6- dimethylpyridine (2,6-lutidine), 4-methylpyridine, 2,4,6-trimethylpyridine.
9. The process as claimed in claim 1, wherein the oxidizing agent used in step (b) is in the range of between 0.05 to 2.0 mole % and sodium metaperiodate used in step (c) is the range of between 1 to 5 moles %.
10. The process as claimed in claim 1, wherein the yield of the compound of Formula (1) is in the range of 58 to 70%.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21832872.2A EP4175937A4 (en) | 2020-07-03 | 2021-05-21 | PROCESS FOR THE PRODUCTION OF VANILLIN AND OTHER SUBSTITUTED PHENYLALDEHYDES |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN202011028345 | 2020-07-03 | ||
| IN202011028345 | 2020-07-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022003710A1 true WO2022003710A1 (en) | 2022-01-06 |
Family
ID=79315686
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2021/050495 Ceased WO2022003710A1 (en) | 2020-07-03 | 2021-05-21 | A process for the preparation of vanillin and other substituted phenylaldehydes |
Country Status (2)
| Country | Link |
|---|---|
| EP (1) | EP4175937A4 (en) |
| WO (1) | WO2022003710A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116510539A (en) * | 2023-05-04 | 2023-08-01 | 安徽普朗膜技术有限公司 | Method for preparing polyketone separation membrane by low-temperature thermal-induced phase separation method |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1643804A (en) | 1926-01-25 | 1927-09-27 | Richard H Bots | Process of manufacturing vanillin |
| US2057117A (en) | 1933-03-28 | 1936-10-13 | Marathon Paper Mills Co | Process of making vanillin |
| US2062205A (en) | 1932-03-21 | 1936-11-24 | Boedecker Friedrich | Process of producing the m-alkylethers of protocatechuic aldehyde |
| US2187366A (en) | 1937-02-03 | 1940-01-16 | Schimmel & Co A G | Manufacture of vanillin |
| US2414385A (en) | 1942-03-16 | 1947-01-14 | Research Corp | Hydroxylation of unsaturated organic compounds containing an alcohol or ether group |
| US2794813A (en) | 1953-09-15 | 1957-06-04 | Farinacci Nicholas Thomas | Oxidation of organic compounds with a chromic oxidizing agent |
| US3544621A (en) | 1967-12-20 | 1970-12-01 | Collins Chem Co Inc | Method of preparing vanillin from eugenol |
| US3969397A (en) | 1968-12-27 | 1976-07-13 | Hoffmann-La Roche Inc. | Process for the preparation of l-dopa |
| JPH10136994A (en) | 1996-11-14 | 1998-05-26 | Sankyo Co Ltd | Production of l-veratrylglycine |
| CN1289836A (en) | 2000-10-16 | 2001-04-04 | 上海天香精细化工有限公司 | Process for preparing vanillin from clove oil |
| WO2002079132A1 (en) | 2001-03-29 | 2002-10-10 | Council Of Scientific And Industrial Research | Microwave assisted process for the preparation of substituted phenylaldehydes from trans and cis-phenylpropenes |
| EP1373178A1 (en) | 2001-03-29 | 2004-01-02 | Council Of Scientific And Industrial Research | Microwave assisted process for the preparation of substituted phenylaldehydes from trans and cis-phenylpropenes |
| WO2013166642A1 (en) | 2012-05-07 | 2013-11-14 | Rhodia Operations | Process for production of vanillin and vanillin derivatives |
| CN103626643A (en) * | 2013-12-03 | 2014-03-12 | 南昌航空大学 | Preparation method for synthesizing vanillin by using natural eugenol as raw material |
| CN104402691A (en) | 2014-11-06 | 2015-03-11 | 江西理工大学 | Preparation method of vanillin |
-
2021
- 2021-05-21 WO PCT/IN2021/050495 patent/WO2022003710A1/en not_active Ceased
- 2021-05-21 EP EP21832872.2A patent/EP4175937A4/en active Pending
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1643804A (en) | 1926-01-25 | 1927-09-27 | Richard H Bots | Process of manufacturing vanillin |
| US2062205A (en) | 1932-03-21 | 1936-11-24 | Boedecker Friedrich | Process of producing the m-alkylethers of protocatechuic aldehyde |
| US2057117A (en) | 1933-03-28 | 1936-10-13 | Marathon Paper Mills Co | Process of making vanillin |
| US2187366A (en) | 1937-02-03 | 1940-01-16 | Schimmel & Co A G | Manufacture of vanillin |
| US2414385A (en) | 1942-03-16 | 1947-01-14 | Research Corp | Hydroxylation of unsaturated organic compounds containing an alcohol or ether group |
| US2794813A (en) | 1953-09-15 | 1957-06-04 | Farinacci Nicholas Thomas | Oxidation of organic compounds with a chromic oxidizing agent |
| US3544621A (en) | 1967-12-20 | 1970-12-01 | Collins Chem Co Inc | Method of preparing vanillin from eugenol |
| US3969397A (en) | 1968-12-27 | 1976-07-13 | Hoffmann-La Roche Inc. | Process for the preparation of l-dopa |
| JPH10136994A (en) | 1996-11-14 | 1998-05-26 | Sankyo Co Ltd | Production of l-veratrylglycine |
| CN1289836A (en) | 2000-10-16 | 2001-04-04 | 上海天香精细化工有限公司 | Process for preparing vanillin from clove oil |
| WO2002079132A1 (en) | 2001-03-29 | 2002-10-10 | Council Of Scientific And Industrial Research | Microwave assisted process for the preparation of substituted phenylaldehydes from trans and cis-phenylpropenes |
| EP1373178A1 (en) | 2001-03-29 | 2004-01-02 | Council Of Scientific And Industrial Research | Microwave assisted process for the preparation of substituted phenylaldehydes from trans and cis-phenylpropenes |
| WO2013166642A1 (en) | 2012-05-07 | 2013-11-14 | Rhodia Operations | Process for production of vanillin and vanillin derivatives |
| CN103626643A (en) * | 2013-12-03 | 2014-03-12 | 南昌航空大学 | Preparation method for synthesizing vanillin by using natural eugenol as raw material |
| CN104402691A (en) | 2014-11-06 | 2015-03-11 | 江西理工大学 | Preparation method of vanillin |
Non-Patent Citations (25)
| Title |
|---|
| "Comprehensive Organic Synthesis", 30 November 1990, PERGAMON PRESS, Oxford, ISBN: 0-08-040598-3, article DONALD G. LEE; TAO CHEN: "3.8 - Cleavage Reactions", pages: 541 - 591, XP009534326, DOI: 10.1016/B978-0-08-052349-1.00202-X * |
| FACHE ET AL., ACS SUSTAINABLE CHEM. ENG., vol. 4, no. 1, 2016, pages 35 - 46 |
| FRANCO, A. ET AL., BEILSTEIN J. ORG. CHEM., vol. 13, 2017, pages 1439 - 1445 |
| GADILOHAR BALU L.; SHANKARLING GANAPATI S.: "Choline based ionic liquids and their applications in organic transformation", JOURNAL OF MOLECULAR LIQUIDS, vol. 227, 1 January 1900 (1900-01-01), NL , pages 234 - 261, XP029882534, ISSN: 0167-7322, DOI: 10.1016/j.molliq.2016.11.136 * |
| GUSEVSKAYA, E. V. ET AL., J. MOL. CATAL. A-CHEM., vol. 363-364, 2012, pages 140 - 147 |
| HOCKING, M. B., J. CHEM. EDUC., vol. 74, 1997, pages 1055 |
| JOSHI R K: "Chemical Composition, In Vitro Antimicrobial and Antioxidant Activities of the Essential Oils of Ocimum Gratissimum, O. Sanctum and their Major Constituents", INDIAN JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 75, no. 4, 1 January 2013 (2013-01-01), pages 457 - 462, XP055897648, DOI: 10.4103/0250-474X.119834 * |
| KNOWLES, W. S., ANGEW. CHEM., INT. ED., vol. 41, 2002, pages 1998 - 2007 |
| KNOWLES, W. S., CHEM. EDUC, vol. 63, 1986, pages 222 - 225 |
| KOTHARI S K , BHATTACHARYA A K , SINGH C P , SINGH K , SYAMASUNDAR K V , RAMESH S , KUMAR S: "A new methyl eugenol-rich and short duration strain of Ocimum tenuiflorum", JOURNAL OF MEDICINAL AND AROMATIC PLANT SCIENCES, vol. 22, no. 4a, 2000, pages 385 - 388, XP009534000, ISSN: 0253-7125 * |
| LAMPMAN ET AL., J. CHEM. EDUC., vol. 60, 1963, pages 503 - 504 |
| LAMPMAN GARY M, ANDREWS JENNIFER, BRATZ WAYNE, HANSSEN OTTO, KELLEY KENNETH, PERRY DANA, RIDGEWAY ANTHONY: "The preparation of vanillin from eugenol and sawdust ", JOURNAL OF CHEMICAL EDUCATION, vol. 54, no. 12, 1 January 1977 (1977-01-01), pages 776 - 778, XP055897644 * |
| LIU, B. ET AL., CHEM. COMM., vol. 55, 2019, pages 4817 - 4820 |
| MAO, H. ET AL., J. CHIN. CHEM. SOC., vol. 63, 2016, pages 261 - 266 |
| MILAS, N. A., J. AM. CHEM. SOC., vol. 59, 1937, pages 2342 |
| PAPPO, R. ET AL., J. ORG. CHEM., vol. 21, 1956, pages 478 - 479 |
| PARREIRA, L. ET AL., ADV. SYNTH. CATAL., vol. 352, pages 1533 - 1538 |
| PRAMOD KANNISSERY, ANSARI SHAHID H., ALI JAVED: "Eugenol: A Natural Compound with Versatile Pharmacological Actions", NATURAL PRODUCT COMMUNICATIONS, vol. 5, no. 12, 1 December 2010 (2010-12-01), US , pages 1 - 8, XP055897646, ISSN: 1934-578X, DOI: 10.1177/1934578X1000501236 * |
| PRIEFERT, APPL. MICROBIOL. BIOT., vol. 56, no. 3-4, 2001, pages 296 - 314 |
| SANCHEZ-GONALEZ, E. ET AL., CRYST. ENG. COMM., vol. 19, 2017, pages 4142 - 4146 |
| See also references of EP4175937A4 |
| SHARMA UK ET AL., MED CHEM RES, vol. 22, no. 11, 2013, pages 5129 - 5140 |
| SHINHA AK ET AL., CHEM LETT, vol. 32, no. 8, 2003, pages 780 - 781 |
| SINHA ET AL., J. FOOD SCI. NUTR., vol. 59, no. 4, 2008, pages 299 - 326 |
| WALTON ET AL., CURR. OPIN. BIOTECHNOL., vol. 11, 2000, pages 490 - 496 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116510539A (en) * | 2023-05-04 | 2023-08-01 | 安徽普朗膜技术有限公司 | Method for preparing polyketone separation membrane by low-temperature thermal-induced phase separation method |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4175937A1 (en) | 2023-05-10 |
| EP4175937A4 (en) | 2024-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3045444B1 (en) | Method of preparing vanillin | |
| Shi et al. | Catalyst-and additive-free sunlight-induced autoxidation of aldehydes to carboxylic acids | |
| CN108752178A (en) | A kind of preparation method of 2,7- dimethyl -2,4,6- sarohornene -1,8- dialdehyde | |
| US6015905A (en) | Process for the preparation of 2-(6-substituted pyrid-2-yloxymethyl) phenylacetate | |
| WO2022003710A1 (en) | A process for the preparation of vanillin and other substituted phenylaldehydes | |
| CN114351173B (en) | Electrochemical synthesis method of 4-methoxybenzaldehyde | |
| JPH05502232A (en) | Method for producing sclareolide | |
| GB2038823A (en) | Phenoxypyridine derivatives | |
| CH626357A5 (en) | Process for the preparation of 4-halodihydropyranones | |
| Das et al. | A Rapid and Efficient Stereoselective Synthesis of (Z)‐and (E)‐Allyl Bromides from Baylis–Hillman Adducts Using Bromo (dimethyl) sulfonium Bromide | |
| Singh et al. | An environmentally benign process to synthesize vanillin and other substituted phenyl aldehydes using natural phenylpropenes | |
| WO2000055135A1 (en) | Process for producing pyridine derivative through reaction of azametal lacyclo pentadiene with alkyne | |
| CN115417756B (en) | Process for preparing vanillin from eugenol by one-pot method | |
| Shi et al. | β-Cyclodextrin promoted oxidation of aldehydes to carboxylic acids in water | |
| CN110483261B (en) | A kind of method for catalytic dehydrogenation of aryl secondary alcohol into ketone | |
| Steves et al. | Copper‐Catalyzed Aerobic Alcohol Oxidation | |
| CN110483264A (en) | A kind of synthetic method of veratraldehyde | |
| Prakash et al. | Oxidation of α-benzoyl-o-hydroxyacetophenones using iodobenzene diacetate in methanolic potassium hydroxide: A new synthesis of 2-benzoylcoumaran-3-ones | |
| Shono et al. | Synthesis of 2-methyl-3-hydroxy-4H-pyran-4-one and 4-hydroxy-5-methyl-2H-furan-3-one from carbohydrates | |
| Saidi et al. | A new protocol for O-methylation of phenolic compounds with trimethyl phosphite or trimethyl phosphate under solvent-free condition and microwave irradiation | |
| Jia et al. | O-Alkylation of oxime with N-vinyl lactams induced by radical cation | |
| CN113372209B (en) | A kind of synthetic method of gigantotrienone | |
| CN109651270A (en) | A method of synthesis 2- acetyl group -3- methylpyrazine | |
| Rieck et al. | A new synthesis of anthraquinones | |
| CN115925524B (en) | Method for preparing vanillin from 4-methyl guaiacol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21832872 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2021832872 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |