WO2022077034A1 - Heteroaryl amide inhibitors of cd38 - Google Patents
Heteroaryl amide inhibitors of cd38 Download PDFInfo
- Publication number
- WO2022077034A1 WO2022077034A1 PCT/US2021/071805 US2021071805W WO2022077034A1 WO 2022077034 A1 WO2022077034 A1 WO 2022077034A1 US 2021071805 W US2021071805 W US 2021071805W WO 2022077034 A1 WO2022077034 A1 WO 2022077034A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- formula
- group
- och
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present disclosure relates to biochemistry, and medicine. More specifically, this disclosure relates to novel compounds, processes for their preparation, and pharmaceutical formulations and methods of treating diseases by modulating the level of cellular NAD+ and related metabolites thereof through the inhibition of the CD38 enzyme.
- Nicotinamide Adenine Dinucleotide is a biochemical that is found in all cells performing its critical role in oxidoreductase reactions.
- NAD+ and its related pyridine nucleotides NADH, nicotinamide adenine dinucleotide phosphate (NADP+), and NADPH are recognized as major redox carriers in all organisms.
- These pyridine dinucleotides regulate the cytosolic and mitochondrial redox state and are key participants monitoring the metabolic status of the cell (Houtkooper el al. (2010) Endo. Rev. 31(2): 194-223); Koch-Nolte et al. (2009) Sei. Signal. 2(57); Houtkooper et al. (2012) J. Cell Biol.) 199(2):205-209).
- NAD+ is also a substrate for various enzymes, where it is consumed in the process of donating its adenosine diphosphate (ADP) ribose to acceptor molecules or in the process of hydrolysis or cyclization.
- ADP adenosine diphosphate
- the enzymes that are the major consumers of NAD+ are the ADP ribosyl transferases (i.e., poly( ADP-ribose) polymerase (PARP) and ADP-ribosyltransferase (ART) family of enzymes), the sirtuins (Sirtl- 7), and the ADP ribosyl cyclases/hydrolases (CD38/CD157). These enzymes are involved in pathways that regulate Ca2+ signalling, gene transcription, DNA repair, cell survival, energy metabolism, and oxidative stress.
- ADP ribosyl transferases i.e., poly( ADP-ribose) polymerase (PARP) and ADP-ribosyltransferase (ART) family of enzymes
- sirtuins Sirtl- 7
- CD38/CD157 ADP ribosyl cyclases/hydrolases
- NAD+ is also a component of the circadian cycle with daily oscillations that, tie cellular metabolism to chromatin remodelling and gene transcription. It is known that exercise and caloric restriction elevate NAD+ levels while aging and obesity decrease cellular NAD+ levels.
- Cellular NAD+ is produced by either the de novo synthesis pathway from tryptophan or by the Preiss-Handler and/or the salvage synthesis pathways from precursors such as nicotinic acid (niacin), nicotinamide (NAM), nicotinamide riboside (NR), and nicotinamide mononuscleotide (NMN), which are imported into the cells.
- the modulation of cellular NAD+ levels can be achieved by blocking the consumption of NAD + by inhibiting enzymes that consume NAD+.
- CD38 is one of such NAD+ consuming enzymes and reported to be the main cellular NAD+ consumer.
- CD38 is a type II membrane- anchored enzyme. It efficiently catalyzes the breakdown of NAD+ to nicotinamide (NAM) and ADP ribose (ADPR) and hydrolyzes NAADP to ADPR phosphate (ADPRP). CD38 acts as a cyclase converting NAD+ to cyclic ADPR (cADPR). Finally, ADPR is also a breakdown product of cADPR hydrolysis mediated by CD38.
- ADP ribose (ADPR) and cyclic ADPR (cADPR) are metabolites of NAD+ generated by CD38 ⁇ mediated hydrolysis or cyclization and they play a key role as intracellular Ca2+ mobilizing second messengers.
- cADPR is mainly involved stimulating Ca2+ release from the endoplasmic reticulum via ryanodine receptors, whereas ADPR activates the plasma membrane cation channel TRPM2 (Transient receptor potential melastatin 2) facilitating calcium entry into the cells.
- TRPM2 Transient receptor potential melastatin 2
- Aberrant TRPM2 activation has been shown to induce abnormal intracellular Ca2+ accumulation and cell death in a variety of cell types, including neurons, and is implicated in several neurological disorders.
- Nicotinamide is a precursor for NAD + and is a key molecule involved in energy metabolism. NAM is converted into nicotinamide mononucleotide (NMN) by the enzyme nicotinamide phosphoribosyltransferase (NAMPT). Alternatively, NAM can be irreversibly methylated by Nicotinamide N ⁇ methyltransferase (NNMT) enzyme and excreted from the body.
- NAM nicotinamide mononucleotide
- NAMPT nicotinamide phosphoribosyltransferase
- NAM can be irreversibly methylated by Nicotinamide N ⁇ methyltransferase (NNMT) enzyme and excreted from the body.
- NAM N1 -methylnicotinamide
- NAM supplementation has shown positive effects, high levels of NAM can exert negative effects through multiple routes, including inhibition of PARPs and sirtuins and alteration of methyl metabolism.
- NAM supplementation has shown to cause a significant decrease of insulin sensitivity in human subjects, neurotoxicity and hepatotoxicity.
- Certain heteroaryl amides are known such as N-(3-chloro-2-methylpyridin-4-yl)- 6-imidazol-l-ylpyridine-2-carboxamide, pubchem.ncbi.nlm.nih.gov/compound/ 99607495, however, no biological data for the compound is reported.
- Int. Patent Pub. WO 2015/187499 refers to certain unrelated reverse amides as ASK1 inhibitors.
- Int. Patent Pub. WO 2009/014637 refers to certain benzimidazolylpyridines as protein kinase inhibitors.
- U.S. 7,919,487 refers to certain heteroaryl hydrazones.
- the present disclosure provides novel compounds, processes for their preparation, and pharmaceutical formulations and methods of treating diseases by modulating the level of cellular NAD+ and related metabolites thereof through the inhibition of the CD38 enzyme
- This disclosure pertains to a compound of Formula I or a pharmaceutically acceptable salt, ester, or prodrug thereof
- R l is selected from the group consisting of H, halo, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 /Jalkoxy, and perfluoro(C 1 -C 6 )alkoxy-; wherein (C 1 -C 6 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ( (C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 2 is H, halo, -CN, (C 1 -C 6 )alkyl, (O.-CcOalkoxy, perfluoro(C]-C6)alkyl, perfluoro(C 1 - C 6 )alkoxy-, cycloalkyl, cycloalkyl-O-, heterocycloalkyl, heterocycloalkyl-O-, aryl, aryl-O-, R 5 -(C(R 4 ) 2 )n-O- wherein (C 1 -C 6 dalkyl, cycloalkyl, heterocycloalkyl, and aryl are each optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NIh, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N ⁇ , -CF 3
- R 3 is H, halo, (C 1 -C 3 )alkyl, -CF 3 , (C 1 -C 3 )alkoxy, -OCF 3 or (R 7 ) 2 N-; wherein R 7 is H or (C 1 -C 3 )alkyl; each R 4 is independently H or (C 3 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ; R 5 is selected from the group consisting of (C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkyl,
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1 -3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ; n is an integer from one to three;
- R 8 is H, -CH 3 or ⁇ CF 3 ;
- Het is a heterocycle of the formula each R y is independently selected from H, halo, (C 1 -C 6 )alkyl,-CF 3 , (C 1 -C 6 )alkoxy, -OCF 3 ,
- R 11 is independently H or (C 1 -C 3 )alkyl
- R 12 is H or (Cj-C3)alkyl
- R 13 is (C 1 -C 3 )alkyl.
- Het is a ring of the
- R 8 is -CH 3 or -CF 3 ; and W is a compound of
- R f is selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -OCH 3 , and -OCF 3 .
- R 2 is selected from the group consisting of H, (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy-, perfluoro(C 1 - C 6 )alkyl, perfluoro(C 1 -C 6 )a1koxy-, cycloalkyl, cycloalkyl-O-, heterocycloalkyl, aryl, R 3 - (C(R 4 ) 2 )n-O- or (R 6 )2N-; wherein (C 1 -C 6 )alkyl, cycloalkyl, heterocycloalkyl and aryl is optionally substituted with 1 -3 substituents independently selected from the group consisting of II, halo, - CN, (C 1 -C 3 )alkyl, -NH2, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 ,
- R 5 is selected from the group consisting of (C 1 -C 3 )alkyl, cycloalky], heterocycloalkyl, and aryl; wherein (C 1 -C 6 )alkyl, cycloalkyl, heterocycloalkyl and aryl is optionally substituted with 1- 3 substituents independently selected from the group consisting of H, halo, -CN, (C’.-Cslalkyl, - NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 , and -OCF 3 ; and R 6 is independently H or (C 1 -C 3 )alkyl optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )al
- R 3 is selected from the group consisting of H, halo, (C 1 -C 3 )alkyl, -CF 3 , --OCH 3 , -OCF 3 , and (R 7 ) 2 N ⁇ ; and wherein R' is H or (C 1 -C 3 )alkyl.
- R 1 is selected from the group consisting of H, F, -CH 3 , and -OCH 3
- R 1 is H.
- R 2 is selected from the group consisting of FI, (C 1 -C 3 )alkyl, (C 1 - C3)alkoxy-, perfluorofC (C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkoxy-, 3- to 10-membered cycloalkyl, 3- to 10-membered cycloalkyl-O-, 5- to 10-membered heterocycloalkyl, 6- to 10-membered aryl, R 5 - (C(R 4 ) 2 )n-O- or (iV'y-X-i wherein (C 1 -C 3 )alkyl, 3- to 10-membered cycloalkyl, 3- to 10- membered cycloalkyl-O-, 5- to 10-membered heterocycloalkyl, 6- to 10-membered and is optionally substituted with 1 -3
- R 2 is selected from the group consisting of methoxy-, cyclopropoxy- or R 5 -(C(R 4 ) 2 .)-O ⁇ ; and each R 4 is H; wherein R 5 is selected from the group consisting of C 1 -alkyl and tetrahydropyran, and wherein said C 1 -alkyl is substituted with -OCH 3 .
- R 3 is selected from the group consisting of H, F, -CH3, -OCH 3 , or H2N-. In certain embodiments, R 3 is H.
- R 9 is selected from the group consisting of H, halo, (C 1 -C 3 )alkyi, -CF 3 , -OCH3, -OCF 3 , -CN, R 12 O((C 1 -C 3 )alkyl) each R 11 is independently selected from H, (C 1 -Csjalkyl; R 12 is H or (C 1 -Csjalkyl.
- At least one R y is selected from the group consisting of F, (C 1 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 , -CN, and in a specific embodiment, at least one R 9 is -CF.
- R 10 is H, and in certain embodiments, R 10 is H.
- Het is a ring of the Formula iii: wherein: one R 9 is H, and the other R 9 is -CF 3 ,, and R 10 is H.
- R 8 is H.
- the compound is a compound of Formula 1A or a pharmaceutially acceptable salt, ester, or prodrug thereof:
- the compound of Formula I* is a compound of Formula I* A or a pharmaceutically acceptable salt, ester, or prodrug thereof
- R 1 is selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -OCH 3 , and -OCF 3 ;
- R 2 is H, (C 1 -C 6 )alkyl, (C 1 -C 6 )alkyl , perfluoro(C 1 -C 6 )alkyl, perfluoro(C 1 -C 6 )alkyl -, cycloalkyl, cycloalkyl-O, heterocycloalkyl, aryl, wherein (C 1 -C 6 )alkyl, cycloalkyl, cycloalkyl-O, heterocycloalkyl and aryl are optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 , and - OCF 3 ; n is an integer from one to three; each R 4
- R 3 is selected from the group consisting of (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and and, wherein (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl and aryl are optionally substituted with 1 -3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, - CF 3, -OCH 3 . and -OCF 3 ,
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting ofH, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R3 is H, halo, (C 1 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 or (R 7 ) 2 N-;
- R 7 is H or (C 1 -C 3 )alkyl
- R 8 is H, -CH 3 or -CF 3 ;
- R 12 is H or (C 1 -C 3 )alkyl.
- the compound is a compound of Formula IB, or a pharmaceutically acceptable salt, ester, or prodrug thereof:
- the compound of Formula I* is a compound of Formula I*B, or a pharmaceutically acceptable salt, ester, or prodrug thereof alkyl, (C 1 -C 3 )alkoxy-, perfluoro(C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkoxy-, cycloalkyl, heterocycloalkyl, aryl, R 5 -(C(R 4 )2)n-O-, or (R 6 ) 2 N-; wherein (C 1 -C 3 )a1ky1 is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2) (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and - OCF 3 ; n is an integer from one to
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -Csjalkyl, - NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 3 is H, halo, (C 1 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 or (R 7 ) 2 N ⁇ , wherein R 7 is H or (C 1 - C 3 )alkyl;
- R 8 is H, -CH 3 or -CF 3 ;
- the compound of is selected from the group consisting of 6-(lH-imidazol-l-yl)-4-((tetrahydro-2H-pyran-4-yl)methoxy)-N-(2-(trifluoromethyl)pyri din-4- yljpicolinamide, 6-(lH-imidazol-l-yl)-4-methoxy-N-(2-(trifluoromethyl)pyridin-4- ylipicolinamide, 2-( lH-imidazol-l-yl)-6-(2-methoxyethoxy)-N-(2-(trifluoromethyl) pyridin-4- yl)pyrimidine-4-carboxamide, 6-(lH-imidazol-l-yl)-4-(2-methoxyethoxy)-N-(2- (trifiuoromethyl)pyridin-4-y])picolinamide, 4-cyclopropoxy-6-(lH-imidazol-l-yl-((tetra
- the disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I or I*, including any compound recited supra, and a pharmaceutically acceptable earner.
- the pharmaceutical formulation comprises therapeutically effective amount of at least one additional medicinal or pharmaceutical agent.
- this additional agent is a therapeutically effect amount of an anti-aging agent.
- this additional agent is a therapeutically effect amount of an anti- rheumatoid arthritis agent.
- the disclosure provides a method of treating a disease or medical disorder in a subject suffering therefrom and which benefits from modulation of NA.D+ level or related metabolites thereof level , comprising administering to the subject a pharmaceutical formulation in an amount effective to modulate the level of NAD+ or related metabolites thereof, the formulation comprising a therapeutically effective amount of a compound of Formula I or I*, including any compound recited supra, and a pharmaceutically acceptable carrier, and optionally comprising another agent which modulate the level of N AD or related metabolites thereof.
- the disease or disorder is or is related to nonalcoholic stetohepatits, aging, aging chronical condition, senescence, immunometabolism, sepsis, inflammation, infection, arthritis, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, lupus, lupus eiythematosus, Crohn disease, ulcerative colitis, plaque psoriasis, ankylosing spondylitis, juvenile idiopathic arthritis, hidradenitis suppurativa, fibrosis, hepatic fibrosis, renal fibrosis, pulmonary fibrosis, cardiac fibrosis, cancer, multiple myeloma, cardiovascular disorder, neurological disorder, infertility, loss of ovarian follicles, decreased oocyte quality and quantity, ovarian senescence, transient receptor potential melastatin 2 (TRPM2) regulation, calcium flux regulation, ischemia-reperfusion-
- TRPM2 transient receptor
- the disease or disorder is or is related to nonalcoholic steatohepatitis or the like.
- the disease or disorder is or is related to an age-related disease or disorder or the like.
- the disease or disorder is or is related to a fibrotic disease of the digestive system, lung, heart, kidney, liver, or lung or the like.
- the disease or disorder is or is related to Multiple Myeloma or the like, and the method further comprises administering an immuno-oncology drag to the subject in need thereof.
- the disclosure provides the use of a pharmaceutical formulation comprising a compound of Formula 1 , including any compound recited above, to treat a disease or disorder in a subject that benefits from modultation of the level of NAD+ or related metabolites thereof.
- the pharmaceutical formulation comprising a compound of Formula 1, including any compound recited above is used to treat a disease or disorder in a subject that benefits from the subject benefiting from inhibition of CD38.
- the disease or disorder is or is related to agin or the like.
- the pharmaceutical formulation is used to treat a disease or disorder selected from small lung cell carcinoma, renal clear cell carcinoma, chronic lymphocytic leukemiahas, multiple myeloma, hypertension, hypoxic pulmonary vasoconstriction, cardiac hypertrophy, congestive heart failure, stroke, Alzheimer's disease, bipolar disorder, schizophrenia, Huntington's disease, amyotrophic lateral sclerosis, Parkinson's disease, multiple sclerosis, optic neuropathy, epilepsy, idiopathic pulmonary fibrosis, viral-induced fibrosis of the lung, infection-induced fibrosis of the lung, cystic fibrosis, asthma, chronic obstructive pulmonary disease (COPD), metabolic syndrome, obesity, sarcopenic obesity, dyslipidemia, diabetes (such as type I diabetes), diabetic neuropathy, insulin resistance, pancreatitis, acute lung injury' (ALI) acute respiratory distress syndrome (ARDS), hyperphosphatemia, alcohol intolerance, lupus, rheumato
- the pharmaceutical formulation is used to treat a disease or disorder selected from aging, age-related chronic disease, inflammation, cancer, cardiovascular disorder, neurological disorder, pulmonary' disorder, fibrotic diseases, SARS, COVID-19, metabolic disorder, acute lung injury' (ALI), acute respiratory distress syndrome (ARDS), hyperphosphatemia, alcohol intolerance, lupus, arthritis, ataxia-telangiectasia, irritable bowel syndrome, colitis, gout, end stage renal disease, hearing loss, liver disorders, postmenopausal osteoporosis, Hartnup disease, tuberculosis, leishmaniasis, muscular dystrophy, organ reperfusion injury, pellagra, diseases of the skin, damage caused by exposure to radiation, periodontal disease, Leber's hereditary amaurosis, sleep disorder, exercise intolerance, chronic disease associated with cell death.
- a disease or disorder selected from aging, age-related chronic disease, inflammation, cancer, cardiovascular disorder, neurological disorder, pulmonary' disorder, fibrotic diseases, SARS, COVI
- the use is treatment is of Multiple Myeloma, and the treatment further comprises treatment of the subject with an immuno-oncology drug.
- the use is treatment of nonalcoholic steatohepatitis (NASH), and in certain embodiment, the treatment of NASH is with the use of 2-(lH ⁇ imidazol” l-yl)-6-methoxy-N-(2- (trifluoromethyl)pyridin-4-yl)pyrimidine-4-carboxamide.
- Figure 1A depicts the in vitro functional potency for Compound 35 in human CD38+ cells as measured by NAD hydrolase activity assay in primary human activated CD4+ T cells.
- Figure IB depicts the in vitro functional potency for Compound 35 in human CD38+ cells as measured by NAD hydrolase activity assay in primary' human Ml macrophages.
- Figure 2 A depicts the in vitro efficacy of Compound 35 in the Human 3D NASH model as measured by release of inflammatory marker IP-10/CXCL10.
- Figure 213 depicts the in vitro efficacy of Compound 35 in the Human 3D NASH model as measured by release of inflammatory marker IL- 10.
- Figure 2C depicts the in vitro efficacy of Compound 35 in the Human 3D NASH model as measured by release of inflammatory' marker MIP-la/CCL3.
- Figure 2D depicts the in vitro efficacy of Compound 35 in the Human 3 D NASH model as measured by release of inflammatory marker TNFa.
- the response plot represents the average (+/-) standard deviation for the measurement of each cytokine/chemokine released in the supernatant at each condition tested where n :::: 6 biological replicates over one experiment.
- Figures 3 A - 3D depict in vivo efficacy of oral administration of Compound 35 (3 mg/kg) against CD38 in aged mouse model as measured by liver NAD+ (Figure 3 A), NMN ( Figure 3B), NAM ( Figure 3C), and ADPR ( Figure 3D) levels.
- Figures 4A - 4D depict in vivo efficacy of oral administration of Compound 35
- Figure 5 depicts in vitro efficacy of Compound 32 in human CD38+ cells as measured by NAD hydrolase activity assay in primary human Ml macrophages.
- Figures 6A-6D depict in vivo efficacy of acute oral administration of Compound 32 (3 and 10 mg/kg) against CD38 in obese mouse model as measured by liver NAD+ (Figure 6A), NMN ( Figure 6B), NAM ( Figure 6C), and ADPR ( Figure 6D) levels.
- Figures 7A-7D depict in vivo efficacy of chronic oral administration of Compound 32 (10 mg/kg) against CD38 in obese mouse model as measured by liver NAD+ (Figure 7 A), NMN (Figure 7B), NAM ( Figure 7C), and ADPR (Figure 7D) levels.
- Figures 8A-8D depict in vivo efficacy of chronic oral administration of Compound 32 (3 and 10 mg/kg) against CD38 in aged mouse model as measured by liver NAD+ (Figure 8A), NMN ( Figure 8B), NAM ( Figure 8C), and ADPR ( Figure 8D) levels.
- Figure 9 depicts in vitro efficacy of Compound 39 in human CD38+ cells as measured by NAD hydrolase activity assay in primary' human Ml macrophages.
- Figures 10A-10D depict in vivo efficacy of acute oral administration of Compound 39 (3 and 10 mg/kg) against CD38 in obese mouse model as measured by liver NAD+ (Figure 10A), NMN (Figure 10B), NAM (Figure IOC), and ADPR ( Figure 10D) levels.
- the response plot represents the average (+/-) standard deviation for the measurement of NAD metabolites at each condition tested where n ::: 3 over I experiment.
- Figures 11 A-l ID depict in vivo efficacy of chronic oral administration of
- Figures 12A-12D depict in vivo efficacy of chronic oral administration of Compound 39 (3 and 10 mg/kg) against CD38 in aged mouse model as measured by liver NAD+ (Figure 12 A), NMN ( Figure 12B), NAM ( Figure 12C), and .ADPR ( Figure 12D) levels.
- Figures 13 A-13C depict cytokines quantification in plasma for IL-6 (Figure 13A), TNFa ( Figure 13B) and IP-10 ( Figure 13C).
- Figures 14A-14C depict MS analysis of NAD+ (Figure 14A), NAM ( Figure 14B), and ADPR ( Figure 14C) levels in spleen tissue.
- Figures 15A-15C depict MS analysis of NAD+ ( Figure 15 A ), NAM ( Figure 15B), and ADPR ( Figure 15C) levels in live tissue.
- Figure 16 depicts CD38 expression in spleen tissue.
- Figures 17A-17I depict the expression in spleen of MIPla ( Figure 17A), MIP2
- Figure 18 depicts CD38 expression in liver.
- Figures 19A-19I depict the expression in liver of MIPla (Figure 19A), XHP2 (Figure 19B), TNFa (Figure 19C), RANTES ( Figure 19D), MCP1 (Figure 19E), IL-1 p (Figure 19F), IL-6 ( Figure 19G), IP- 10 ( Figure 19H), and IFNy ( Figure 191).
- the response plot represents the average (N-) standard deviation for the measurement of gene expression at each condition tested where n ::: 4 over 1 experiment. *, p ⁇ 0.05; **, p ⁇ 0.01 ; ***, p ⁇ 0.001 ; * ⁇ **, p ⁇ 0.0001 compared with Vehicle sample, one-way ANOVA.
- the term "about” or “approximately” refers to a measurable value such as a parameter, an amount, a temporal duration, and the like, are meant to encompass variations of and from the specified value, such as variations of +/- 10% or less, +/-5% or less, +/- 1% or less, +7-0.5% or less, and +/-0.1 % or less of and from the specified value, insofar such variations are appropriate to perform in the disclosed invention. It is to be understood that the value to which the modifier "about” or “approximately” refers is itself also specifically, and preferably, disclosed.
- alkyl Whenever a numerical range is used in this application, for example when 1 to 6 is used in the definition of “alkyl’'’ means that the alkyl group may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, 4 carbon atoms, 5 carbon atoms, and 6 carbon atoms.
- alkyl encompasses saturated aliphatic hydrocarbons including straight chains and branched chains and 1, 3, 4, 5, and 6 carbon atoms.
- (C 1 -Crdalkyl,” as well as the alkyl moieties of other groups referred to herein refers to linear or branched radicals of 1, 2, 3, 4, 5, and 6 carbon atoms (e.g., methyl, ethyl, n-propyl, isopropyl, n-butyl, iso-butyl, secondary-butyl, tertiary-butyl), optionally substituted by 1, 2, 3, 4, or 5 suitable substituents.
- alkyl also encompasses aliphatic hydrocarbons having at least one carbon-carbon double bond, including straight chains and branched chains having at least one carbon-carbon double bond and 2, 3, 4, 5, and 6 carbon atoms.
- tytytytyjalkyl means straight or branched chain unsaturated radicals of 2 to 6 carbon atoms, including, but not limited to ethenyl, 1 -propenyl, 2-propenyl (allyl), iso-propenyl, 2-methy 1-1 -propenyl, 1-butenyl, 2-butenyl, and the like; optionally substituted by 1 to 5 suitable substituents.
- the alkenyl group may exist as the pure E (ent ought) form, the pure Z (zusammen) form, or any mixture thereof.
- cycloalkyl encompasses saturated or unsaturated (nonaromatic) monocyclic or bicyclic hydrocarbon rings (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl); optionally substituted by 1, 2, 3, 4, and 5 suitable substituents, such as, but not limited to, H, halo, -CN, (C 1 -C 3 )alkyl, -NH2, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 and -OCF 3 .
- suitable substituents such as, but not limited to, H, halo, -CN, (C 1 -C 3 )alkyl, -NH2, (C 1 -C 3 )alkyl-(NH)-
- the cycloalkyl group may have 3 to 12 carbon atoms in the ring(s), such as 3 to 10 carbon atoms, 3 to 8 carbon atoms, 3 to 6 carbon atoms, or 3, 4, 6, 7, 8, 9, 10, 11, or 12 carbon atoms.
- a monocyclic cycloalkyl group may have 3 to 6 carbon atoms, 3 carbon atoms, 4 carbon atoms, 5 carbon atoms, or 6 carbon atoms in the ring, e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl.
- the cycloalkyl may optionally contain one, two or more non-cumulative non-aromatic double or triple bonds.
- heterocycloalkyl includes a monocyclic, bridged, polycyclic or fused polycyclic saturated or unsaturated non-aromatic 3- to 13- membered ring including 1 or more heteroatoms selected from O, S and N, such as a 3 to 10 membered ring, or a 3 to 6 membered ring, a 3-, 4-, 5-, 6-, 7-, 8-, 9-, 1 ()-, 11-, 12-, or 13-membered ring.
- heterocycloalkyl rings include, but are not limited to, azetidinyl, tetrahydrofuranyl, imidazolidinyl, pyrrolidinyl, piperidinyl, piperazinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, thiomorpholinyl, tetrahydrothiazinyl, tetrahydro-thiadiazinyl, morpholinyl, oxetanyl, tetrahydrodiazinyl, oxazinyl, oxathiazinyl, indolinyl, isoindolinyl, quinuclidinyl, chromanyl, isochromanyl, benzoxazinyl, and the like.
- heterocycloalkyl rings are tetrahydrofuran -2-yl, tetrahydrofuran-3-yl, imidazolidin-l-yl, imidazolidin-2-yl, imidazolidin-4-yl, pyrrolidin-l-yl, pyrrolidin-2-yl, pyrrolidin-3-yl, piperidin-l-yl, piperidin-2-yl, piperidin-3-yl, piperazin- 1-yl, piperazin-2-yl, piperazin-3 -yl, l,3-oxazolidin-3-yl, isothi azolidine, l,3-thiazolidin-3-yl, l,2-pyrazolidin-2-yl, 1,3-pyrazolidin-l-yl, 1,2- tetrahydrothiazin-2-yl, 1 , 3 -tetrahydrothi azin-3 -yl, 1 ,
- the heterocycloalkyl ring is optionally substituted by 1 to 5 suitable substituents, or 1 to 3, or 1, 2, 3, 4, or 5 substituents such as, but not limited to, H, halo, -CN, (C 1 AAjalkyl, -NH?, (C 1 -C 1 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 , and -OCF 3
- suitable substituents such as, but not limited to, H, halo, -CN, (C 1 AAjalkyl, -NH?, (C 1 -C 1 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)2N-, -CF 3 , -OCH 3 , and -OCF 3
- aryl is defined to include all-carbon monocyclic or fused-ring polycyclic (/. ⁇ ?., rings which share adjacent pairs of carbon atoms) groups having a completely conjugated pi-electron system.
- the aryl group has 6 to 12, 6 to 10, or 6, 8, 9, 10, or 12 carbon atoms in the ring(s).
- One nonlimiting, exemplary' aryl group is a 6-carbon atom phenyl ring.
- aryl means aromatic radicals containing from 6 to 10 or 6 to 12 carbon atoms such as, but not limited to, phenyl, naphthyl, tetrahydronaphthyl, anthracenyl, indanyl and the like.
- the aryl group is optionally substituted by 1 to 5 suitable substituents, more preferably 1 to 3 substituents such as, but not limited to, H, halo, -CN, (C 1 -Cjjalkyd, -NH?, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -CsjalkyljiN-, -CFj, -OCH 3 and -OCF 3 .
- heteroaryl is defined to include monocyclic or fused- ring polycyclic aromatic heterocyclic groups with one or more heteroatoms selected from O, S and N in the ring.
- the heteroaryl group has 5- to 12-ring atoms including one to 5 heteroatoms selected from O, S, and N, such as 5- to 10-ring atoms, 5- to 8-ring atoms, or 6-, 7 ⁇ , 8-, 9-, 10-, 11-, or 12-ring atoms.
- heteroaryl encompasses aromatic radicals containing at least one ring heteroatom selected from O, S and N and from 1 to 11 carbon atoms, such as from 2 to 9 carbon atoms, from 3 to 8 carbon atoms, or from 3, 4, 5, 6, 7, 8, 9, 10, or 11 carbon atoms, such as, but not limited to, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl, furyl, imidazolyl, pyrrolyl, oxazolyl (e.g, 1 ,3-oxazolyl, 1,2-oxazolyl), thiazolyl (e.g., 1,2-thiazolyl, 1,3-thiazolyl), pyrazolyl, tetrazolyl, triazolyl (e.g., 1,2,3-triazolyl, 1 ,2,4-triazolyl), oxadiazolyl (e.g., 1,
- the heteroaryl group is optionally substituted by 1 to 5 suitable substituents 1 to 3 substituents such as, but not limited to, H, halo, -CN, (C 1 -C 3 )alkyl, -bffih, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and - OCF 3 .
- suitable substituents 1 to 3 substituents such as, but not limited to, H, halo, -CN, (C 1 -C 3 )alkyl, -bffih, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and - OCF 3 .
- alkoxy refers to an alkyl-O- and alkyl is as defined herein.
- alkylaminoalkyl refers to an -alkyl-NR-alkyl group
- amino refers to an -NH2 or an -NRR'group.
- aminoalkyd refers to an -alky-NRR' group.
- aminocarbonyl refers to a -C(O)NRR'.
- arylalkyl refers to -alkylaryl, where alkyl and aryl are defined herein.
- aryloxy refers to both an -O-aryl and an -O-heteroaryl group, as defined herein.
- a “carboxylic acid” group refers to a C-carboxyl group in which R is hydrogen.
- a "cyano" group refers to a -CN group.
- dialkydamino refers to an ⁇ N(alkyl)2 or NR2 group.
- halo or halogen group refers to fluorine, chlorine, bromine or iodine.
- a "hydroxy” group refers to an -OH group.
- N-amido refers to a -R'(C O)XR group.
- a "perfluoroalkyl group” refers to an alkyl group wherein one or more of the hydrogen atoms have been replaced with fluorine atoms.
- a compound of Formula I or “compounds of Formula I”, including compounds of Formula IA- IH, or pharmaceutically acceptable salts, esters, or prodrugs thereof’ encompass all forms of the compound of Formula I, including compounds of Formulae IA- IH, as well as including all hydrates, solvates, isomers, crystalline and noncrystalline forms, isomorphs, polymorphs, metabolites, and prodrugs thereof.
- prodrug means a derivative of a known direct acting drug, which derivative has enhanced delivery characteristics and therapeutic value as compared to the drag, and is transformed into the active drug by an enzymatic or chemical process.
- the phrase “pharmaceutically acceptable” means those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with tissues of humans and animals.
- “pharmaceutically acceptable” means approved by a regulatory' agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
- the present disclosure relates to novel heterocyclic amides of Formula I and I*, to pharmaceutical formulations comprising these heterocyclic amides, and to uses and syntheses thereof.
- the compounds of Formula I have the following structure: or a pharmaceutically acceptable salt, ester, or prodrug thereof, and the compounds of Formula I* have the following structure: or a pharmaceutically acceptable salt, ester, or prodrug thereof, wherein:
- R 1 is selected from the group consisting of H, halo, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy, and perfluoro(C 1 -C 6 )alkoxy-> wherein (C 1 -C 6 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(N1-I) ⁇ , ((C 1 -C 3 )alkyl ) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 2 is H, halo, -CN, (C 1 -C 6 )alkyl, (C 1 -GOalkoxy, pertluoro(C 1 ⁇ C6)alkyl, perfluoro(C 1 - Ce)alkoxy-, cycloalkyl, cycloalkyl-O-, heterocycloalkyl, heterocycloalkyl-O-, and, aryl-O-, R 5 -(C(R 4 ) 2 )n-O- or (R 6 ) 2 N-, wherein (C 1 -C 6 )alkyl, cycloalkyl, heterocycloalkyl, and aryl are each optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl)
- R 3 is H, halo, (C 1 -C 3 )alkyl, -CF 3 , (C 1 -C 3 )alkoxy, -OCF 3 , or (R 7 ) 2 N-, wherein R 7 is H or (C 1 -C 3 )alkyl; n is an integer from one to three; each R 4 is independently H or (C 1 -C 3 )alkyl, wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 5 is selected from the group consisting of (C 1 -C 3 )alkyl, perfluoro(Cj-C 3 )alkyl, HO-(C 2 - C ⁇ alky!, cycloalkyl, heterocycloalkyl, and aryl, wherein (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and aryl are each optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 ,-C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 6 is independently H or (C 1 -C 3 )alkyl, wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N ⁇ , -CF 3, -OCH 3 and -OCR.
- R 8 is H, -CH 3 or -CF 3 ;
- Het is a heterocycle of the formula
- R 12 is H or (C 1 -C 3 )alkyl
- R 13 is (C 1 -C 3 )alkyl.
- the compounds of Formula I-I* may exist in the form of pharmaceutically acceptable salts such as, e.g., acid addition salts and base addition salts of the compounds of Formula I.
- pharmaceutically acceptable salt(s) includes salts of acidic or basic groups which may be present in the compounds of Formula I.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/ dihydrogen phosphate, pyroglutamate, saccharate,
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminum, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts. Hemisalts of acids and bases may also be formed, for example, hemi sulphate and hemicalcium salts.
- suitable salts see Stahl and Wermut (2011) Pharmaceutical Salts: Properties, Selection, and Use, (2nd Revised Edition) pp. 1 -388 (Wiley-VCH), the entire contents of which (and specifically the passages relating to suitable salts) is fully incorporated herein byreference.
- the compounds according to the disclosure may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- amorphous refers to a state in which the material lacks long range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Such materials may not give distinctive X-ray diffraction patterns, and while exhibiting the properties of a solid, are more formally described as a liquid.
- a change from solid to liquid properties occurs which is characterized by a change of state, typically second order (‘glass transition’).
- crystalline refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterized by a phase change, typically first order (‘melting point’).
- the compounds according to the disclosure may also exist in unsolvated and solvated forms.
- solvate is used herein to describe a molecular complex comprising the compound according to the disclosure and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- hydrate is employed when said solvent is water.
- a currently accepted classification system for organic hydrates is one that defines isolated site, channel, or metal-ion coordinated hydrates (see. Polymorphism in Pharmaceutical Solids, (1995) Morris (eel. H. G. Brittain, Marcel Dekker), the entire contents of which (and specifically the passages relating to isolated site, channel, or metal-ion coordinated hydrates) is fully incorporated herein by reference.
- Isolated site hydrates are ones in which the water molecules are isolated from direct contact with each other by intervening organic molecules. In channel hydrates, the water molecules lie in lattice channels where they are next to other water molecules. In metal-ion coordinated hydrates, the water molecules are bonded to the metal ion.
- the complex When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions. In such cases, non-stoichiometry will be the norm.
- multi-component complexes other than salts and solvates
- complexes of this type include clathrates (drug-host inclusion complexes) and co-crystals.
- clathrates drug-host inclusion complexes
- co-crystals The latter are typically defined as crystalline complexes of neutral molecular constituents which are bound together through non-covalent interactions, but could also be a complex of a neutral molecule with a salt.
- Co-crystals may be prepared by melt crystallization, by recry s ⁇ ail isati on from solvents, or by physically grinding the components together (see, Almarsson et. al. (2004) Chem. Commun. 1889-1896), the entire contents of which (and specifically the passages relating to preparation of co-crystals by melt crystallization, recrystallisation from solvents, or grinding) is fully incorporated herein by reference.
- the compounds according to the disclosure may also exist in a mesomorphic state (mesophase or liquid crystal) when subjected to suitable conditions.
- the mesomorphic state is intermediate between the true crystalline state and the true liquid state (either melt or solution).
- Mesomorphism arising as the result of a change in temperature is described as ‘thermotropic’ and that resulting from the addition of a second component, such as, but not limited to, water or another solvent, is described as ‘lyotropic’.
- references to compounds of Formula I-I* include references to salts, solvates, multi-component complexes and liquid crystals thereof and to solvates, multicomponent complexes and liquid crystals of salts thereof.
- the compounds according to the disclosure include compounds of Formula I-I* as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labelled compounds of Formula I.
- the disclosure also relates to prodrugs of the compounds of Formula I.
- prodrugs of the compounds of Formula I.
- certain derivatives of compounds of Formula I-I* which may have little or no pharmacological activity, themselves may, when administered into or onto the body, be converted into compounds of Formula I-I* can have the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as “prodrugs” (see, e.g. Higuchi el al. (1987) “Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series; Bioreversible Carriers in Drug Design, Pergam on Press Ee. E. B. Roche, American Pharmaceutical Association), the entire contents of which documents (and specifically the passages relating to prodrags) are fully incorporated herein by reference.
- Prodrags in accordance with the disclosure be produced, for example, by replacing appropriate functionalities present in the compounds of Formula I-I*with certain moieties known to those skilled in the art as ‘pro-moi eties' (see, Bundgaard (1985) Design of Prodrugs (Elsevier, 1985 ), the entire contents of which (and specifically the passages relating to pro-moieties) is fully incorporated herein by reference.
- prodrugs in accordance with the disclosure include:
- the compounds of Formula I-I* may have asymmetric carbon atoms and may exist as two or more stereoisomers.
- the carbon-carbon bonds of the compounds of Formula I-I* may be depicted herein using a solid line a solid wedge or a dotted wedge.
- the use of a solid line to depict bonds to asymmetric carbon atoms is meant to indicate that all possible stereoisomers (e.g., specific enantiomers, racemic mixtures, etc.) at that carbon atom are included.
- the use of either a solid or dotted wedge to depict bonds to asymmetric carbon atoms is meant to indicate that only the stereoisomer shown is meant to be included. It is possible that compounds of Formula I-I* may contain more than one asymmetric carbon atom.
- a solid line to depict bonds to asymmetric carbon atoms is meant to indicate that all possible stereoisomers are meant to be included.
- the compounds of Formula I-I* may exist as enantiomers and diastereomers or as racemates and mixtures thereof.
- the use of a solid line to depict bonds to one or more asymmetric carbon atoms in a compound of Formula I or I* and the use of a solid or dotted wedge to depict bonds to other asymmetric carbon atoms in the same compound is meant to indicate that a mixture of diastereomers is present.
- Stereoisomers of Formula I-I* include cis and trans isomers, optical isomers such as R and S enantiomers, diastereomers, geometric isomers, rotational isomers, conformational isomers, and tautomers of the compounds of Formula I, including compounds exhibiting more than one type of isomerism; and mixtures thereof (such as racemates and diastereomeric pairs). Also included are acid addition or base addition salts wherein the counterion is optically active, for example, d-lactate or 1-lysine, or racemic, for example, dl -tartrate or di-arginine,
- the first type is the racemic compound (true racemate) referred to above wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts.
- the second type is the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each comprising a single enantiomer.
- the compounds of the Formula I-I* may exhibit the phenomena of tautomerism and structural isomerism.
- the compounds of Formula I-I* may exist in several tautomeric forms, including the enol and imine form, and the keto and enamine form and geometric isomers and mixtures thereof. All such tautomeric forms are included within the scope of compounds of Formula I.
- Tautomers exist as mixtures of a tautomeric set in solution. In solid form, usually one tautomer predominates. Even though one tautomer may be described, the present disclosure includes all tautomers of the compounds of Formula I.
- the present disclosure includes all pharmaceutically acceptable isotopically- labelled compounds of Formula I-I* wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the compounds according to the disclosure include, but are not limited to, isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 3b Cl, fluorine, such as 1S F, iodine, such as 123 I and 125 I, nitrogen, such as l3 N and 13 N, oxygen, such as ! 'O, l7 O and 18 (), phosphorus, such as 32 P, and sulphur, such as 35 S.
- isotopes of hydrogen such as 2 H and 3 H
- carbon such as 11 C, 13 C and 14 C
- chlorine such as 3b Cl
- fluorine such as 1S F
- iodine such as 123 I and 125 I
- nitrogen such as l3 N and 13 N
- oxygen such as ! 'O, l7 O and 18 ()
- phosphorus such as 32 P
- sulphur such as 35 S.
- isotopically-labelled compounds of Formula I for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- Substitution with positron emitting isotopes such as 11 C, l8 F, 15 O and 13 N, are useful in Positron Emission Topography (PEI') studies for examining substrate receptor occupancy.
- substitution with isotopes such as are useful in Single Photon Computed Tomography (SPECT).
- Isotopically -labeled compounds of Formula I-I* may generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically- labeled reagent in place of the non-labeled reagent previously employed.
- metabolites of compounds of Formula I that is, compounds formed in vivo upon administration of the compounds of Formula I
- compounds of the Formula I, and IA-IH may be prepared according to the following reaction schemes and accompanying discussion. Unless otherwise indicated, R 1 through R 13 , W, X, Y, Z, Het, and n, and structural Formula I-I* are as defined above in the reaction schemes and discussion that follow.
- the compounds of this disclosure may be made by processes which include processes analogous to those known in the chemical arts, in light of the description contained herein. Certain processes for the manufacture of the compounds of this disclosure are provided as further features of the disclosure and are illustrated by the following reaction schemes. Other processes may be described in the experimental section.
- certain compounds contain primary amines or carboxylic acid functionalities which may interfere with reactions at other sites of the molecule if left unprotected. Accordingly, such functionalities may be protected by an appropriate protecting group which may be removed in a subsequent step.
- Suitable protecting groups for amine and carboxylic acid protection include those protecting groups commonly used in peptide synthesis (such as, but not limited to, N-t-butoxycarbonyl, benzyloxycarbonyl, and 9- fhiorenylmethylenoxy carbonyl for amines and lower alkyl or benzyl esters for carboxylic acids) which are generally not chemically reactive under the reaction conditions described and can typically be removed without chemically altering other functionality in the Formula I-I* compounds.
- Scheme 1 refers to the preparation of compounds of Formula I from bromo or chloro heteroaryl acids or esters of Formula IV (bromo is depicted but may be replaced with chloro).
- compounds of Formula IV are commercially available or may be made by methods well known to those skilled in the art.
- an activated carboxylate such as, but not limited to, wherein P is an ethyl ester
- a polar solvent such as, but not limited to, DMSO was added copper iodide (0.2 equivalents), L-proline (0.4 equivalents), potassium carbonate (2 equivalents) and W-H, an imidazole, pyrazole, triazole or thiazole (1.5 equivalents).
- the reaction mixture may be heated to between about 80°C to about 110°C, or about 100 °C for about 4 hr to about 24 hr, or for about 16 hr.
- the reaction mixture may then be cooled to RT, diluted with ice-cold water and extracted with a solvent such as, but not limited to, ethyl acetate which may be dried and evaporated under reduced pressure to afford the compound of Formula III.
- the compound of Formula III may be saponified to yield a compound of Formula II by treatment with an excess of lithium hydroxide mono hydrate in a solvent mixture such as, but not limited to, THF, methanol and water.
- a solvent mixture such as, but not limited to, THF, methanol and water.
- the reaction mixture may be allowed to stir at RT for about 8 hr to about 24 hr or for about 16 hr.
- the aqueous layer may be acidified using TV HC1 to adjust the pH to around 2 followed by complete evaporation under reduced pressure to obtain the compound of Formula II.
- the compound of Formula II may be converted to the title compound of Formula I by dissolution in DMF followed by the addition of excess N,N-diisopropylethylamine, i.e. Hunig's base or DIPEA, and excess HATU, i.e. l-[Bis(dimethylamino)methylene]-lH-l,2,3- triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, followed by the addition of the desired amino-Het.
- the reaction mixture may be stirred at RT for about 8 hr to about 24 hr, or for about 16 hr.
- the reaction may be quenched by the addition of water followed by extraction with an organic solvent such as, but not limited to, ethyl acetate to yield the title compound of Formula I.
- a compound of Formula I may be prepared from a compound of Formula V by a so-called Stille reaction with a tributyl stannyl-W, wherein W is an imidazolyl, pyrazolyl, triazolyl or thiazolyl.
- a solution of the bromo or chloro intermediate of Formula V is dissolved in a polar solvent such as, but not limited to, DMF followed by addition of tetrakis(triphenylphosphine)palladium(0) (catalytic).
- the reaction mixture may be purged with Nitrogen gas for 5 minutes then sealed and heated to between about 80°C to about 110 C C or about 100 °C, for about 4 to about 24 hours or for about 16 hours.
- the mixture may be cooled to RT and quenched with water followed by extraction with a solvent such as, but not limited to, ethyl acetate which after diving and evaporation yields the compound of Formula I.
- the compound of Formula V may be prepared from a compound of Formula IV by reaction with Het-NH2 in a solvent such as, but not limited to, toluene and trimethylaluminum solution in toluene.
- the reaction mixture may be stirred in a CEM ® microwave at about 100 °C for about 1 hour.
- the completed reaction mixture may then be cooled to RT, quenched with water then extracted with ethyl acetate to yield the compound of Formula V.
- Compounds of Formula I-I* that, have chiral centers may exist as stereoisomers, such as racemates, enantiomers, or diastereomers.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate using, for example, chiral high pressure liquid chromatography (HPL.C).
- HPL.C high pressure liquid chromatography
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound contains an acidic or basic and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to one skilled in the art.
- Chiral compounds of Formula I-I* may be obtained in enantiomerically- enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% isopropanol, typically from 2% to 20%, and from %0 to 5% of an alkylamine, or 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- Stereoisomeric conglomerates may be moiety, an acid or base such as, but not limited to, tartaric acid or 1- phenylethylamine.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization separated by conventional techniques known to those skilled in the art (see, e.g, Elie (1994) Stereochemistry of Organic Compounds (Wiley, New York), the entire disclosure of which (and specifically the passages relating to the separation of stereoisomeric conglomerates) is incorporated herein by reference.
- a compound of Formula I or I* contains an alkenyl or alkenylene group
- geometric cisltrans (or Z/E) isomers are possible.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallization. Salts of the present disclosure can be prepared according to methods known to those of skill in the art.
- the compounds of Formula I or I* that are basic in nature can form a wide variety of salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is useful to initially isolate the compound of the present disclosure from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent and subsequently convert the latter free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this disclosure can be prepared by treating the base compound with a substantially equivalent amount of the selected mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent, such as, but not limited to, methanol or ethanol. Upon evaporation of the solvent, the desired solid salt is obtained.
- the desired acid salt may also be precipitated from a solution of the free base in an organic solvent by adding an appropriate mineral or organic acid to the solution.
- Those compounds of Formula I-I* that are acidic in nature can form base salts with various pharmacologically acceptable cations.
- examples of such salts include, but are not limited to, the alkali metal or alkaline-earth metal salts and particularly, the sodium and potassium salts. These salts are all prepared by conventional techniques.
- the chemical bases which are used as reagents to prepare the pharmaceutically acceptable base salts of this disclosure are those which form non-toxic base salts with the acidic compounds of Formula I.
- salts may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as, but not limited to, an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- an inorganic or organic base such as, but not limited to, an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- These salts may also be prepared by treating the corresponding acidic compounds with an aqueous solution containing the desired pharmacologically acceptable cations, and then evaporating the resulting solution to dryness, for example, under reduced pressure.
- they may also be prepared by mixing lower alkanolic solutions of the acidic compounds and the desired alkali metal alkoxide together, and then evaporating the resulting solution to dryness in the same manner as before.
- stoichiometric quantities of reagents can
- the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as, but not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as, but not limited to, acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as, but not limited to, glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as, but not limited to, citric acid or tartaric acid, an amino acid, such as, but not limited to, aspartic acid or glutamic acid, an aromatic acid, such as, but not limited to, benzoic acid or cinnamic acid, a
- an inorganic acid such as, but not limited to,
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionization in the resulting salt may vary from completely ionized to almost nonionized.
- Certain compounds of Formula 1 according to the disclosure may exist in more than one crystal form ("polymorphs").
- Polymorphs may be prepared by crystallization under various conditions, for example, using different solvents or different solvent mixtures for recrystallization; crystallization at different temperatures; and/or various modes of cooling, ranging from very fast to very slow cooling during crystallization.
- Polymorphs may also be obtained by heating or melting the compound according to the disclosure followed by gradual or fast cooling. The presence of polymorphs may be determined by solid probe NMR spectroscopy, 1R spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques. Polymorphs may be prepared according to techniques well-known to those skilled in the art.
- Cis! trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallization,
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of Formula I or I* contains an acidic or basic moiety, a base or acid such as, but not limited to, 1 - phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of Formula I or I* contains an acidic or basic moiety, a base or acid such as, but not limited to, 1 - phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds according to the disclosure may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from about 0 to about 50% by volume of isopropanol, from about 2% to about 20%, and from about 0 to about 5% by volume of an alkylamine, or about 0.1% diethylamine.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from about 0 to about 50% by volume of isopropanol, from about 2% to about 20%, and from about 0 to about 5% by volume of an alkylamine, or about 0.1% diethylamine.
- the first type is the racemic compound (true racemate) referred to above wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts.
- the second type is the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each comprising a single enantiomer.
- Racemic mixtures may be separated by conventional techniques known to those skilled in the art (see, for example, Elie et al., (1994) Stereochemistry of Organic Compounds (Wiley)), the entire contents of which, as mentioned previously (and specifically the passages relating to the separation of racemic mixtures) is fully incorporated herein by reference. It will be understood that, the compounds of Formula I-I* are not limited to the particular enantiomer shown, but also include all stereoisomers and mixtures thereof.
- the disclosure also includes isotopically-labeled compounds of Formula I, wherein one or more atoms is replaced by an atom having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- Isotopically-labeled compounds of Formula I-I* may generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed.
- the compounds of Formula I-I* are assessed for their bi opharmaceuti cal properties, such as, but not limited to, solubility and solution stability (across pH), permeability, etc., in order to select the appropriate dosage form and route of administration for treatment of the proposed indication.
- the compounds of Formula I-I* are useful for modulating or inhibiting NAD+ hydrolase activity of CD38 protein. Accordingly, these compounds are useful for the prevention and/or treatment of disease states associated with NAD+ depletion and NAD+ releated metabolites disregulations such as, but not limited to, aging, obesity, diabetes, cancer, heart disease, asthma, and inflammation.
- the disclosure is also directed to pharmaceutical compositions comprising a compound of Formula I or I*, or a pharmaceutically acceptable salt, ester, or prodrug thereof, and a pharmaceutically acceptable carrier.
- Compounds of Formula 1 according to the disclosure intended for pharmaceutical use may be comprised in pharmaceutical formulations. They may be incorporated into these formulations in the form of crystalline or amorphous products.
- the compounds may be, for example, as solid plugs, powders, or films obtained by methods such as, but not limited to, precipitation, ciystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- These compounds may be administered alone, in combination, and/or in cofonnulation with one or more other compounds according to the disclosure, or in combination and/or coformulation with one or more other drugs (or as any combination or coformuation thereof). Generally, they are administered as a combination, formulation, or coformulation in association with one or more pharmaceutically acceptable excipients.
- the compounds disclosed herein can be coformulated with one or more suplements and/or inhibitors, such as NAD supplement and JAK inhibitor.
- excipient is used herein to describe any ingredient other than the compound(s) according to the disclosure.
- the choice of excipient depends to a large extent depend on factors such as, but not limited to, the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- compounds according to the disclosure may be combined with soluble macromolecular entities, such as, but not limited to, cyclodextrin and suitable derivatives thereof or polyethylene glycol -containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as, but not limited to, cyclodextrin and suitable derivatives thereof or polyethylene glycol -containing polymers
- Drug-cyclodextrin complexes may be useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubilizer.
- auxiliary additive i.e. as a carrier, diluent, or solubilizer.
- alpha-, beta- and gamma-cyclodextrins examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148 (specifically page 3, line 25 to page 6, line 8 inclusive).
- the entire contents of WO 91/11172, WO 94/02518 and WO 98/55148 are fully incorporated herein by reference.
- compositions suitable for the delivery of compounds of the present disclosure and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995), the entire contents of which (and specifically the passages relating to pharmaceutical compositions and their methods of preparation) is fully incorporated herein by reference.
- the pharmaceutical formulation according to the disclosure may be administered orally.
- Oral administration may involve swallowing, so that the compound in the formulation enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid, semi-solid and liquid systems such as, but not limited to, tablets; soft or hard capsules containing multi- or nanoparticulates, liquids, or powders, lozenges (including liquid-filled), chews; gels, fast dispersing dosage forms; films; ovules; sprays; and buccal/mucoadhesive patches.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet. [0162] The compounds according to the disclosure may also be used in fast-dissolving, fast-disintegrating dosage forms such as, but not limited to, those described in Liang et al. (2001) Expert Opinion in Therapeutic Patents, 11 (6): 981-986, the entire contents of which (and specifically the passages relating to fast-dissolving, fast-disintegrating dosage forms) is fully incorporated herein by reference.
- the compound of Formula I or I* may make up from about 1 weight % to about 80 weight % of the dosage form, or from about 5 weight % to about 60 weight % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant comprises from about 1 weight % to about 25 weight %, or from about 5 weight. % to about 20 weight % of the dosage form.
- Binders are generally used to impart, cohesive qualities to a tablet formulation. Suitable binders include, but not are not limited to, microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as, but not limited to, lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- Tablets may also optionally comprise surface active agents, such as, but not limited to, sodium lauryl sulfate and polysorbate 80, and glidants such as, but not limited to, silicon dioxide and talc.
- surface active agents such as, but not limited to, sodium lauryl sulfate and polysorbate 80
- glidants such as, but not limited to, silicon dioxide and talc.
- surface active agents may comprise from about 0.2 weight % to about 5 weight % of the tablet, and glidants may comprise from about 0.2 weight % to about 1 weight % of the tablet.
- Tablets also generally contain lubricants such as, but. not limited to, magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from about 0.25 weight % to about 10 w'eight %, or from about 0.5 weight % to about 3 weight % of the tablet.
- compositions include, but are not limited to, anti-oxidants, colorants, flavoring agents, preservatives, taste-masking agents, flavorings and flavor enhancers, salivary stimulating agents, cooling agents, co ⁇ sol vents (including oils), emollients, bulking agents, antifoaming agents, and surfactants.
- Exemplary tablets contain up to about 80% of a compound of Formula I, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % di sintegrant, and from about. 0.25 weight % to about 10 weight % lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tableting.
- the final formulation may comprise one or more layers and may be coated or uncoated, it may be encapsulated.
- Consumable oral films for human or veterinary use are typically pliable water- soluble or water-swell able thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of Formula I, a film-forming polymer, a binder, a solvent, a humectant, a plasticizer, a stabilizer or emulsifier, a viscosity -modifying agent and a solvent. Some components of the formulation may perform more than one function.
- the compound of Formula I or I* may be water-soluble or insoluble.
- a water- soluble compound comprises from about 1 weight % to about 80 weight %, or from about 20 weight % to about 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to about 88 weight % of the solutes, Alternatively, the compound of Formula I or I* may be in the form of multiparticulate beads.
- the film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range of about 30 weight % to about 80 weight %.
- Films in accordance with the disclosure may be prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, for example, by a combined coater dryer, or by freeze-drying or vacuuming,
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Suitable modified release formulations for the purposes according to the disclosure are described in US Patent No. 6,106,864, the entire contents of which (and specifically the modified release formulations from column 2, line 34 to column 4, line 26, in which references to “darifenacin” should be read as referring to the compound of formula I of the disclosure) is fully incorporated herein by reference. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Verma el al. (2001) Pharm. Technol, On-line, 25(2): 1-14, the entire contents of which (and specifically the passages relating to suitable release technologies including high energy dispersions and osmotic and coated particles) is fully incorporated herein by reference.
- the use of chewing gum to achieve controlled release is described in International Patent Publication WO 00/35298, the entire contents of which is fully incorporated herein by reference.
- the pharmaceutical formulations including a compound of Formula I or I* according to the disclosure may also be administered directly into the blood stream, into muscle, or into an internal organ.
- exemplary suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrastemal, intracranial, intramuscular, intrasynovial and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as, but not limited to, salts, carbohydrates and buffering agents (e.g, to a pH of from about 3 to about 9), but, for some applications, they may be formulated as a sterile nonaqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as, but not limited to, sterile, pyrogen-free water.
- excipients such as, but not limited to, salts, carbohydrates and buffering agents (e.g, to a pH of from about 3 to about 9)
- a suitable vehicle such as, but not limited to, sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of Formula I-I* used in the preparation of parenteral pharmaceutical formulations may be increased by the use of appropriate formulation techniques, such as, but not limited to, the incorporation of solubility-enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- compounds according to the disclosure may be formulated as a suspension or as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound.
- Nonlimiting examples of such formulations include drug-coated stents and semi-solids and suspensions comprising drug-loaded poly( ⁇ t7-lactic- coglycolic) acid (PGLA) microspheres.
- the pharmaceutical formulations according to the disclosure may also be administered topically, (intra)dermally, or transdermally to the skin or mucosa.
- Typical formulations for this purpose include, but are not limited to, gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibers, bandages and microemulsions.
- Liposomes may also be used.
- Typical carriers include, but are not limited to, alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol.
- Penetration enhancers may be incorporated (see, Finnin et al. (1999) J. Pharm. Sci. 88 (10):955-958), the entire contents of which (and specifically the passages relating to penetration enhancers) is fully incorporated herein by reference.
- topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g., PowdeijectTM, BiojectTM, etc.) injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Exemplary' modified release formulations include delayed-, sustained-, pulsed- , controlled-, targeted and programmed release.
- the pharmaceutical formulations including a compound of Formula I-l* according to the disclosure may also be administered intranasally or by inhalation, e.g., in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as, but not limited to, phosphatidylcholine) from a dry powder inhaler, as an aerosol spray from a pressurized container, pump, spray, atomizer (or an atomizer using electrohydrodynamics to produce a fine mist), or nebulizer, with or without the use of a suitable propellant, such as, but not limited to, 1,1,1,2-tetrafluoroethane or 1,1,1,2,3,3,3-heptafluoropropane, or as nasal drops.
- the powder may comprise a bioadhesive agent such as, but not limited to, chito
- the pressurized container, pump, spray, atomizer, or nebulizer contains a solution or suspension of the compound according to the disclosure comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilizing, or extending release of the active, a propellant(s) as solvent and an optional surfactant such as, but not limited to, sorbitan trioleate, oleic acid, or an oligolactic acid.
- the pharmaceutical formulation Prior to use in a dry powder or suspension formulation, is micronized to a size suitable for delivery/ by inhalation (e.g., less than about 5 microns). This may be achieved by any appropriate comminuting method, such as, but not limited to, spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenization, or spray diving.
- Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound according to the disclosure, a suitable powder base such as, but not limited to, lactose or starch and a performance modifier such as, but not limited to, /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate.
- Other exemplary excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- An exemplary pharmaceutical formulation for use in an atomizer using electrohydrodynamics to produce a fine mist may contain from about 1 ⁇ g to about 20 mg of the compound according to the disclosure per actuation, and the actuation volume may vary’ from about 1 ⁇ l to about 100 ⁇ l.
- An exemplary formulation comprises a compound of Formula I, propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative exemplary' solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavors such as, but not limited to, menthol and levomenthol, or sweeteners, such as, but not limited to, saccharin or saccharin sodium, may be added to those formulations according to the disclosure intended for inhaled/intranasal administration.
- sweeteners such as, but not limited to, saccharin or saccharin sodium
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the dosage unit may be determined by means of a valve which delivers a metered amount. Units in accordance with the disclosure are provided, e.g., in a metered dose or “puff” containing from about 0.01 pg to about 100 mg of the compound of Formula I.
- the overall daily dose is in the range of about 1 pg to about 200 mg, which may be administered in a single dose or, as divided doses throughout the day and possibly during multiple days.
- compositions according to the disclosure may be administered rectally or vaginally, for example, in the form of a suppository, pessary', or enema.
- Cocoa butter is a traditional suppository' base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed, sustained, pulsed, controlled, targeted, and programmed release.
- the pharmaceutical formulations according to the disclosure may also be administered directly to the eye or ear, for example, in the form of drops of a micronized suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, gels, biodegradable (e.g., absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as, but not limited to, niosomes or liposomes.
- a polymer such as, but not limited to, crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as, but not limited to, benzalkonium chloride. Such formulations may also be delivered by iontophoresis. Formulations for ocular/aural administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
- kits comprises two separate pharmaceutical compositions: a compound of Formula I or I*, or a salt thereof and a second compound as described above.
- the kit comprises means for containing the separate compositions such as, but not limited to, a container, a divided bottle or a divided foil packet.
- the kit comprises directions for the administration of the separate components.
- the kit form is particularly advantageous when the separate components are preferably administered in different dosage forms (e.g., oral and parenteral), are administered at different dosage intervals, or when titration of the individual components of the combination is desired by the prescribing physician.
- Blister packs are well known in the packaging industry and are being widely used for the packaging of pharmaceutical unit dosage forms (tablets, capsules, and the like). Blister packs generally consist of a sheet of relatively stiff material covered with a foil of a transparent plastic material. During the packaging process recesses are formed in the plastic foil. The recesses have the size and shape of the tablets or capsules to be packed. Next, the tablets or capsules are placed in the recesses and the sheet of relatively stiff material is sealed against the plastic foil at the face of the foil which is opposite from the direction in which the recesses were formed. As a result, the tablets or capsules are sealed in the recesses between the plastic foil and the sheet. The strength of the sheet may be such that the tablets or capsules can be removed from the blister pack by manually applying pressure on the recesses whereby an opening is formed in the sheet at the place of the recess. The tablet or capsule can then be removed via said opening.
- a memory aid may be provided on the kit, e.g., in the form of numbers next to the tablets or capsules whereby the numbers correspond with the days of the regimen which the tablets or capsules so specified should be ingested.
- Another example of such a memory aid is a calendar printed on the card, e.g., as follows “First Week, Monday, Tuesday” etc. “Second Week, Monday, Tuesday” etc. Other variations of memory aids will be readily apparent.
- a "daily dose” can be a single tablet or capsule or several pills or capsules to be taken on a given day.
- a daily dose of Formula I or I* compound can consist of one tablet or capsule while a daily dose of the second compound can consist of several tablets or capsules and vice versa.
- the dispenser may be equipped with a memory-aid, so as to further facilitate compliance with the regimen.
- a nonlimiting example of such a memory-aid is a mechanical counter which indicates the number of daily doses that has been dispensed.
- Another nonlimiting example of such a memory -aid is a battery-powered micro-chip memory coupled with a liquid crystal readout, or audible reminder signal which, for example, reads out the date that the last daily dose has been taken and/or reminds one when the next dose is to be taken.
- the disclosure includes a method for treating, retarding, or preventing a disease in a subject, e.g., a mammal such as, but not limited to, a human, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I or I*, or a pharmaceutically acceptable salt, ester, or prodrug thereof.
- the compounds of Formula I or I* are also useful for modulating or inhibiting NAD hydrolase activity of CD38 protein. Accordingly, the disclosure further includes the use of compounds of Formula I-I* for the prevention, retardation, and/or treatment of di sease states associated with NAD+ depletion or NAD+ metabolites dysregulation such as aging (e.g., age- related chronic diseases), cancer, cardiovascular disorders, neurological disorders, pulmonary disorders, fibrotic diseases, metabolic disorders, inflammation, liver disorders, and diseases of the skin., as well as its identification as a cell-surface marker in hematologic cancers such as multiple myeloma (see, Chin et al. (2016) Trends Pharmacol. Sci. 39(4):424-436).
- the methods comprise administering to a subject in need thereof a therapeutically effective amount of a compound of Formula I or I*, or a pharmaceutically acceptable salt, ester, or prodrug thereof.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of cancer, such as small lung cell carcinoma, renal clear cell carcinoma, chronic lymphocytic leukemiahas and multiple myeloma.
- cancer such as small lung cell carcinoma, renal clear cell carcinoma, chronic lymphocytic leukemiahas and multiple myeloma.
- CD38 dysfunction in cancers has been demonstrated such as in small lung cell carcinoma (see, e.g., Blanco et al. (2010) Can. Res.70(10):3896-3904) and in renal clear cell carcinoma (see, Sartini et al. (2006) J. Urol.l76(5):2248-2254).
- the role of CD38 in Chronic lymphocytic leukemia has been described (see, Deaglio et al. (2010) Can. Biol. 20(6) :416-423).
- the role of CD38 in PD-1/PD-L1 resistant cancers has been decribed (see, Verma et ah, (2019) Nature Immunology 20: 1231-1243; Chen et al., (2016) Cancer Discov. 8(9): 1156- 1175).
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of cardiovascular disorders, such as hypertension, hypoxic pulmonary vasoconstriction, cardiac hypertrophy, congestive heart failure and stroke.
- cardiovascular disorders such as hypertension, hypoxic pulmonary vasoconstriction, cardiac hypertrophy, congestive heart failure and stroke.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of neurological disorders, such as Alzheimer's disease, bipolar disorder/ schizophrenia, Huntington's disease, amyotrophic lateral sclerosis, Parkinson's disease, multiple sclerosis, optic neuropathy and epilepsy.
- neurological disorders such as Alzheimer's disease, bipolar disorder/ schizophrenia, Huntington's disease, amyotrophic lateral sclerosis, Parkinson's disease, multiple sclerosis, optic neuropathy and epilepsy.
- the agents described herein may be used as neuroprotective agents.
- the compounds of Formula I-I* may also be administered in the tissue or organ likely to encounter cell death.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of pulmonary disorders, such as idiopathic pulmonary fibrosis, cystic fibrosis, CIVD-19, SARS, asthma, and chronic obstructive pulmonary disease (COPD).
- pulmonary disorders such as idiopathic pulmonary fibrosis, cystic fibrosis, CIVD-19, SARS, asthma, and chronic obstructive pulmonary disease (COPD).
- COPD chronic obstructive pulmonary disease
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of fibrotic diseases, such as idiopathic pulmonary fibrosis and cystic fibrosis.
- Pulmonary disorders have a CD38 dysfunction such as described for idiopathic pulmonary fibrosis, O'Neill et al. ( 1994) Expt, Lung Res. 20(l):41-56.
- the role in cystic fibrosis has been described in Wetmoreet et al. (2010) J. Biolog. Chem. 285(40):30516-30522.
- Asthma is described in Kang et al. (2006) Curr. Res. Med. Rev. 2(2): 143-156.
- COPD is described in Hageman et al. (2003) Free Radical Biol, Med. 35(2): 140-148.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of metabolic disorders, such as metabolic syndrome, obesity, sarcopenic obesity, dyslipidemia, diabetes (such as type I diabetes), diabetic neuropathy, insulin resistance, infection-induced and viral-induced lung fibrosis, and pancreatitis.
- metabolic disorders such as metabolic syndrome, obesity, sarcopenic obesity, dyslipidemia, diabetes (such as type I diabetes), diabetic neuropathy, insulin resistance, infection-induced and viral-induced lung fibrosis, and pancreatitis.
- Use of compounds of Formula I-I* for the prevention and/or treatment of obesity may include wherein the subject has or is likely to develop obesity (e.g. mammals having an elevated risk of developing diet-induced obesity’).
- a mammal may be identified as having or being likely to develop obesity using standard clinical techniques. For example, analysis of a human's family history or eating habits may be used to determine whether the human is likely to develop an obesity condition.
- a mammal identified as having or being susceptible to developing an obesity' condition may be treated by administering a compound of Formula I.
- CD38 dysfunction in metabolic disorders have been described such as in Metabolic Syndrome, Escande Carlos et al. (2013) Diabetes 62(4): 1084-93.
- Obesity/sarcopenic obesity have been described in Maria et al. (2007) FASEB J. 21(13):3629-39.
- Dyslipidemia is described in Surakka Ida et al. (201 1) PLoS Gen. 7(10):e 1002333.
- Dysfunction in Diabetes has been described in Arya et al (2004) Am. J. Hum. Gen. 74(2):272-282 and dysfunction in diabetic neuropathy has been described in Geeta el al. (2010) Neuropharmacol . 58(3):585-92.
- CD38 in Insulin resistance is described in Yoshino et al. (2011) Cell Metab.14(4):528-536.
- CD38 in Type I diabetes is described in Elliott, et al. (1993) Annals NY Acad. Sci. 696: 333-41.
- Pancreatitis has been described in Chan et al. (201 1) Antiox. Redox Sig. 15(10):2743-2755.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of acute lung injury' (ALI) and acute respiratory' distress syndrome (ARDS).
- ALI acute lung injury'
- ARDS acute respiratory' distress syndrome
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of hyperphosphatemia.
- hyperphosphatemia see Takahashi et al. (2004) Kidney Int. 65(3): 1099-1104.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of alcohol intolerance.
- alcohol intolerance see, e.g., Larson et al. (2005) J. Biol. Chem.280 (34 ) : 30550 - 30556.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of lupus.
- lupus see, e.g., Gonzalez -Escribano et al. (2004) Hum. Immunol (2004) 65(6):660-4, Pavon el al. (2013) Cytokine 62(2) :232-243.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of arthritis, such as rheumatoid arthritis.
- arthritis such as rheumatoid arthritis.
- the role of CD38 in rheumatoid arthritis is described in Jorge Postigo et al. (2012) PLoS One 7(3):e33534.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of ataxia- tel angi ectasia.
- the role of CD38 in ataxia-telangiectasia is described in Stern et al. (2012) J. Biol. Chem. 277(l):602-608.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of irritable bowel syndrome and colitis.
- Irritable Bowel Syndrome and colitis involves CD38 dysfunction, for example, Durnin et al. (2012) J, Physiol. (Oxford, UK) 590(8): 1921 -1941.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of gout. See, Nik Cummings et al., European Journal of Human Genetics (2010), 18(11), 1243-7.
- the disclosure includes the use of compounds of Formula I for the prevention and/or treatment of end stage renal disease.
- CD38 The role of CD38 in end stage renal disease is described in Freedman et al. (2005) Nephrol. Dialysis, Transpl. 20(4):712-718.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of hearing loss. Hearing loss is described in Someya el al. (2010) Cell 43(5):802-812.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of liver disorders, such as steatosis and non-alcoholic steatohepatitis (NASH).
- liver disorders such as steatosis and non-alcoholic steatohepatitis (NASH).
- Liver disorders such as steatosis and ⁇ ASH are mediated by CD38, for example Choi et al.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of postmenopausal osteoporosis.
- Postmenopausal osteoporosis disease progression is described in Drummond et al. (2006) J. Bone Mineral Met. 24(l):28-35.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of fertility disorders or disease. Restoration of oocyte quality and enhancement of ovulation rate and fertility is decribed in Bertoldo et al., (2020) Cell Rep 30(6): 1670- 1681.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of Hartnup disease.
- Hartnup disease and CD38 are described in Jepson et al.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of tuberculosis.
- the role of CD38 in tuberculosis is described in Vilcheze et al. (2010) Mol. Microbiol. 76(2):365-377.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of leishmaniasis.
- the role of CD38 in Leishmaniasis is described in Michels et al. (2011) Mol, Microbiol. 82(1 ):4-8.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of muscular dystrophy. Muscular dystrophy and CD38 is described in Goody et al. (2012) PLoS Biology 10(10):el001409.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of organ reperfusion injury.
- Organ reperfusion injury mediated through CD38 is described in Yan Ge et al (2010) Biochem. Biophys. Res. Comm. 399(2): 167-172.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of pellagra.
- Pellagra is also a CD38 mediated disease, see, for example, Williams et a. (2007) Med. Hypoth,69(3):618-628.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of diseases of the skin, such as skin hyperpigmentation, UV skin damage and psoriasis.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of damage caused by exposure to radiation, such as X-ray-induced DNA damage., e.g. by promoting NAD+ modulated DNA repair and/or cell survival.
- the disclosure includes a method of promoting DNA repair in cells. Cells exposed to conditions that may trigger DNA damage, e.g., radiation, may be protected by contacting them before, during and/or after exposure to the DNA damaging agent, with a compound of Formula I or I*.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of periodontal disease.
- the role of CD38 in periodontal disease has also been reported in, e.g., Fujita et al. (2005) J. Periodontal. 76(11): 1960-5.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of Leber's hereditary amaurosis.
- Leber's hereditary amaurosis has also been reported see, for example, in Koenekoop etal. (2012) Nat. Gen.44(9): 1035-1039.
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of sleep disorders. NAD+ involvement in sleep disorders has also been reported (see, Robinson et al. (1977) Biol. Psych.12(1): 139-43).
- the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of exercise intolerance.
- Exercise intolerance has also been reported (see, e.g., Gli ck (1966) Am, J. Physiol. 210(6): 1215-21.
- the compounds of Formula I-I* extend the life span of cells and protects them from stress. Accordingly, the disclosure includes the use of compounds of Formula I-I* for the prevention and/or treatment of diseases, e.g., chronic diseases, associated with cell death, such as, but not limited to, e.g., diseases associated with neural cell death or muscular cell death.
- diseases e.g., chronic diseases
- associated with cell death such as, but not limited to, e.g., diseases associated with neural cell death or muscular cell death.
- the methods may be used to prevent or alleviate neurodegeneration and peripheral neuropathies associated with chemotherapy, such as, but not limited to, cancer chemotherapy (e.g., taxol or cisplatin treatment).
- the compounds of Formula I-I* described herein may be administered to subjects in which caloric restriction or the effects thereof is beneficial.
- Subjects may be subjects suffering from art aging disease, e.g, stroke, heart disease, arthritis, high blood pressure. They may also be administered for treating a metabolic disease, such as, but not limited to, insulin-resistance or other precursor symptom of type II diabetes, type II diabetes or complications thereof. Methods may increase insulin sensitivity or decrease insulin levels in a subject.
- a method may comprise administering to a subject, such as a subject in need thereof, a pharmaceutically effective amount of an agent that increases the activity or protein level of a protein involved in the NAD+ salvage pathway, i.e., in the synthesis of NAD+ and the degradation of nicotinamide.
- a subject in need of such a treatment may be a subject who has insulin resistance or other precursor symptom of type II diabetes, who has type II diabetes, or who is likely to develop any of these conditions.
- the subject may be a subject having insulin resistance, e.g., having high circulating levels of insulin and/or associated conditions, such as impaired glucose tolerance, high blood glucose sugar level and hypertension.
- insulin resistance e.g., having high circulating levels of insulin and/or associated conditions, such as impaired glucose tolerance, high blood glucose sugar level and hypertension.
- Compounds of Formula I-I* may also be used for stimulating fat mobilization, e.g., for treating obesity and any condition resulting therefrom or for reducing weight gain.
- the disclosure provides a method for treating any of the conditions recited above in a mammal, comprises administering a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt, ester, or prodrug thereof to the mammal.
- the mammal may be i a human in need of such treatment or prevention.
- a therapeutically effective amount refers to that amount of the compound being administered which will relieve to some extent one or more of the symptoms of the disorder being treated.
- a therapeutically effective amount refers to that amount which has the effect of reducing or ameliorating the fat and scar tissue present in the liver as well as improving any of the biomarker measures indicative of inflammation.
- treating means reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- treatment refers to the act of treating as “treating” is defined immediately above.
- treating also includes, but not limited to, adjuvant and neo adjuvant treatment of a subject.
- Administration of the compounds of Formula I-I* may be affected by any method that enables delivery of the compounds to the site of action. These methods include oral routes, intraduodenal routes, parenteral injection (including intravenous, transdermal, subcutaneous, intramuscular, intravascular or infusion), intra-articular administration, intravitreal administration, topical, ocular, vaginal rectal administration, and the like.
- Dosage regimens may be adjusted to provide the optimum desired response. For example, a single bolus may be administered, several divided doses may be administered over time or the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation. It is advantageous to formulate parenteral compositions in dosage unit, form for ease of administration and uniformity of dosage.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the mammalian subjects to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic effect in association with the pharmaceutical carrier.
- the dose and dosing regimen is adjusted in accordance with methods well-known in the therapeutic arts. That is, the maximum tolerable dose may be readily established, and the effective amount providing a detectable therapeutic benefit to a patient may also be determined, as may the temporal requirements for administering each agent to provide a detectable therapeutic benefit to the patient. Accordingly, while certain dose and administration regimens are exemplified herein, these examples in no way limit the dose and administration regimen that may be provided to a patient in practicing the present disclosure.
- Dosage values may vary with the type and severity of the condition to be alleviated, and may include single or multiple doses.
- specific dosage regimens may be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions, and that dosage ranges set forth herein are exemplar ⁇ ' only and are not intended to limit the scope or practice of the claimed composition.
- doses may be adjusted based on pharmacokinetic or pharmacodynamic parameters, which may include clinical effects such as toxic effects and/or laboratory values.
- the present disclosure encompasses intra-patient dose-escalation as determined by the skilled artisan. Determining appropriate dosages and regimens for administration of the active agent are well-known in the relevant art and would be understood to be encompassed by the skilled artisan once provided the teachings disclosed herein.
- the amount of the compound of Formula I or I* administered is dependent on the subject being treated, the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician.
- An effective dosage is in the range of about 0.001 mg/kg body weight/day, to about 100 mg/kg body weight/day, or about 1 mg/kg body weight/day to about 35 mg/kg body weight/day in single or divided doses. For a 70 kg human, this amounts to about 0.05 g/day to about 7 g/day or about 0.1 g/day to about 2.5 g/day.
- dosage levels below the lower limit of the aforesaid range may be more than adequate depending on, e.g., the severity of the disorder treated and the age and weight of the subject being treated, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day.
- combination therapy refers to the administration of a compound of Formula I or I* together with an at least one additional pharmaceutical or medicinal agent, either sequentially or simultaneously.
- the present disclosure includes the use of a combination of a compound of Formula I or I* and one or more additional pharmaceutically active agent(s). If a combination of active agents is administered, then they may be administered sequentially or simultaneously, in separate dosage forms or combined in a single dosage form. Accordingly, the present disclosure also includes pharmaceutical compositions comprising an amount of: (a) a first agent comprising a compound of Formula I or I*, or a pharmaceutically acceptable salt, ester, or prodrug thereof of the compound; (b) a second pharmaceutically active agent; and (c) a pharmaceutically acceptable carrier, vehicle or diluent.
- compositions of the present disclosure include, without limitation, the following combination therapies.
- combination therapy includes combinations with agents including, but not limited to, an acetyl-CoA carboxylase (ACC) inhibitor, a ketohex okinase (KHK) inhibitor, a GLP-1 receptor agonist, an FXR agonist, a CB1 antagonist, an ASK1 inhibitor, an inhibitor of CCR2 and/or CCR5, a PNPLA3 inhibitor, a hydroxysteroid 17 ⁇ P dehydrogenase (HSD17B13) inhibitor, a DGATl inhibitor, an FGF21 analog, an FGF19 analog, an SGLT2 inhibitor, a PPAR agonist, an AMPK activator, an SCD1 inhibitor or an MPO inhibitor; Orlistat, TZDs and other insulin-sensitizing agents, FGF21 analogs, Metformin, Omega-3 -acid ethyl esters (e.g., a ketohex okinase (KHK) inhibitor, a GLP-1 receptor agonist, an FXR agonist,
- Lovaza Fibrates, HMG CoA-reductase Inhibitors, Ezetimibe, Probucol, Ursodeoxycholic acid, TGR5 agonists, FXR agonists.
- Vitamin E Betaine, Pentoxifylline, CB1 antagonists.
- Carnitine N- acetyl cysteine, Reduced glutathione, 1 orcaserin, the combination of naltrexone with buproprion, SGLT2 inhibitors (including dapagliflozin, canagliflozin, ernpagliflozin, tofogliflozin, ertugliflozin), Phentermine, Topiramate, GLP-1 receptor agonists, GIP receptor agonists, dual GLP-1 receptor/glucagon receptor agonists, dual GLP-l receptor/GIP receptor agonists (Tirzepatide), Angiotensin-receptor blockers an acetyl-CoA carboxylase (ACC) inhibitor, a BCKDK inhibitor, a ketohexokinase (KHK) inhibitor, ASK1 inhibitors, branched-chain alpha keto acid dehydrogenase kinase inhibitors (BCBK inhibitors), inhibitors of CCR2 and/or CCR5, PNP
- combination therapy also includes combinations with, for example, anti-obesity agents including 1 ip-hydroxy steroid dehydrogenase- 1 (1 ip.- HSD type 1) inhibitors, stearoyl -CoA desaturase- 1 (SCD-l) inhibitor, MCR.-4 agonists, cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (such as, but not limited to, sibutramine), sympathomimetic agents, 3 adrenergic agonists, dopamine agonists (such as, but not limited to, bromocriptine), melanocyte-stimulating hormone analogs, 5HT2c agonists, melanin concentrating hormone antagonists, leptin (the OB protein), leptin analogs, leptin agonists, galanin antagonists, lipase inhibitors (such as, but not limited to, tetrahydrolipstatin,
- anti-obesity agents
- naltrexone available from Regeneron Pharmaceuticals, Inc., Tarrytown, N.Y. and Procter & Gamble Company, Cincinnati, Ohio
- human agouti -related protein (AGRP) inhibitors e.g., ghrelin antagonists, histamine 3 antagonists or inverse agonists, neuromedin U agonists, MTP/ApoB inhibitors (e.g., gut-selective MTP inhibitors, such as, but not limited to, dirlotapide), opioid antagonist, orexin antagonist, the combination of naltrexone with buproprion and the like.
- GPP human agouti -related protein
- ghrelin antagonists e.g., histamine 3 antagonists or inverse agonists
- neuromedin U agonists e.g., MTP/ApoB inhibitors (e.g., gut-selective MTP inhibitors, such as, but not limited to, dirlotapide), opioid antagonist, orexin
- This disclosure also includes combination therapy for the treatment of cancers, such as, but not limited to, Multiple Myeloma.
- treatments include, but are not limited to, combinations of a compound of Formula I or I* with one or more immuno-oncology drugs including, but not limited to, Ipilimumab (Yervoy), Nivolumab (Opdivo), Pembrolizumab (Keytruda), Atezolizumab (Tecentriq), Avelumab (Bavencio), Durvalumab (Imfmzi), and PD- 1ZPD-L1 agonist antibodies.
- immuno-oncology drugs including, but not limited to, Ipilimumab (Yervoy), Nivolumab (Opdivo), Pembrolizumab (Keytruda), Atezolizumab (Tecentriq), Avelumab (Bavencio), Durvalumab (Imfmzi), and PD- 1ZPD-L1 agonist antibodies.
- Combination therapy also includes combination with neurodegenerative disorder therapeutics including, e.g., acetylcholinesterase inhibitors, such as, but not limited to, donepezil hydrochloride, physostigmine salicylate, physostigmine sulfate, metrifonate, neostigmine, ganstigmine, pyridostigmine, ambenonium, demarcarium, rivastigmine, ladostigil, galantamine hydrobromide, tacrine, tolserine, velnacrine maleate, memoquin, huperzine A, phenserine, and edrophonium; amyloid-B or fragments thereof, such as, but not limited to, ABi-i5 conjugated to pan HLA DR-binding epitope; antibodies to amyloid-B, such as, but not limited to, bapineuzumab; amyloid-lowering or -inhibiting agents (including
- Combination therapy also includes combinations with cardiovascular agents including, but not limited to, beta-adrenergic receptor blocking agents (beta blockers), such as, but not limited to, carteolol, esmolol, labetalol, oxprenolol, pindolol, propanolol, sotalol, timolol, acebutolol, nadolol, metoprolol tartrate, metoprolol succinate, atenolol and butoxamine; calcium channel blockers such as, but not limited to, nilvadipine, diperdipine, amlodipine, felodipine, nicardipine, nifedipine, nimodipine, nisoldipine, nitrendipine, lacidipine, lercani dipine, lifarizine, diltiazem, verapamil, and enecadin.
- Combination therapy also includes combinations with anti -rheumatoid arthritis drugs including both symptomatic therapies, including but not limited to NSAIDs and acetaminophen/ paracetamol, and oral and parenterally administered disease-modifying antirheumatic drags (DMARDs), including but not limited to, steroids, methotrexate, anti-IL-6, 11-1 and anti-TNFa antibodies, and JAK inhibitors.
- symptomatic therapies including but not limited to NSAIDs and acetaminophen/ paracetamol
- DMARDs disease-modifying antirheumatic drags
- combination therapy includes combinations with catechol O- methyltransferase (COMT) inhibitors, such as, but not limited to, tolcapone (TASMAR), entacapone (COMTAN), and tropolone.
- catechol O- methyltransferase (COMT) inhibitors such as, but not limited to, tolcapone (TASMAR), entacapone (COMTAN), and tropolone.
- Combination therapy also includes combinations with immunomodulators such as, but not limited to, glatiramer acetate, dimethyl fumarate, fmgolimod, roquinimex, laquinimod, rituximab, alemtuzumab, daclizumab, and natalizumab.
- immunomodulators such as, but not limited to, glatiramer acetate, dimethyl fumarate, fmgolimod, roquinimex, laquinimod, rituximab, alemtuzumab, daclizumab, and natalizumab.
- combination therapy includes combinations with interferons, including, but not limited to, interferon beta-la and interferon beta-lb.
- Combination therapy also includes combinations with neuroprotective drugs such as, but not limited to, 2,3,4,9-tetrahydro-l/f-carbazo1-3-one oxime, desmoteplase, anatibant, astaxanthin, neuropeptide NAP, neurostrol, perampenel, ispronicline, bis(4 ⁇ P-D- glucopyranosyloxybenzyl)-2-p-D-glucopyranosyl-2 ⁇ isobutyltartrate (also known as dactylorhin B or DHB), formobactin, xaliproden, lactacystin, dimeboline hydrochloride, disufenton, arundic acid, citicoline, edaravone, granulocyte-colony stimulating factor, ancrod, 17-B-
- neuroprotective drugs such as
- Combination therapy also includes combinations with trophic factors, such as, but not limited to, nerve growth factor (NGF), basic fibroblast growth factor (bFGF), neurotrophin-3, cardiotrophin-1, brain-derived neurotrophic factor (BDNF), neublastin, meteorin, and glial- derived neurotrophic factor (GDNF), and agents that stimulate production of trophic factors, such as, but not limited to, propentofylline and idebenone.
- trophic factors such as, but not limited to, nerve growth factor (NGF), basic fibroblast growth factor (bFGF), neurotrophin-3, cardiotrophin-1, brain-derived neurotrophic factor (BDNF), neublastin, meteorin, and glial- derived neurotrophic factor (GDNF), and agents that stimulate production of trophic factors, such as, but not limited to, propentofylline and idebenone.
- trophic factors such as, but not limited to, nerve growth factor (NGF), basic fibroblast growth factor (bFGF), neurotroph
- This disclosure also relates to combination therapy for the treatment of aging with a nutraceutical product, i.e., a substance which has physiological benefit or provides protection against chronic disease, including vitamins, e.g., Prenatal Vitamins, Vitamin D3, or Vitamin B 12, Garcinia Cambogia, Raspberry Ketones, Green Tea Supplements, Echinacea, Probiotics, Omega 3 Fatty Acids, Alpha-lipoic Acid, and NAD+ and NAD+ precursors, e.g., NMN, NR, and NA.
- a nutraceutical product i.e., a substance which has physiological benefit or provides protection against chronic disease, including vitamins, e.g., Prenatal Vitamins, Vitamin D3, or Vitamin B 12, Garcinia Cambogia, Raspberry Ketones, Green Tea Supplements, Echinacea, Probiotics, Omega 3 Fatty Acids, Alpha-lipoic Acid, and NAD+ and NAD+ precursors, e.g., NMN, NR, and NA.
- another active agent refers to any therapeutic agent, other than the compound of Formula I, or salt thereof, that is useful for the treatment of a subject suffering from a disease or disorder.
- Active agents include for example, without limitation, anti- rheumatis arthritis drags such as NSAIDs, acetaminophen/paracetamol, disease-modifying antirheumatic drags (DMARDs), steroids, methotrexate, anti -IL-6, II- 1 and anti-TNFa antibodies, JAK inhibitors, and the like.
- a compound of the Formula or a pharmaceutically acceptable salt, ester, or prodrug thereof a compound of Formula l*or a pharmaceutically acceptable salt, ester, or prodrug thereof wherein : if the compound is of Formula I; , , if the compound is of Formula I*;
- R 1 is selected from the group consisting of H, halo, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy, and perfluoro(C 1 -C 6 )alkoxy-; wherein (C 1 -C 6 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH2, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl ) 2 N ⁇ , -CF 3 , -OCH 3 and -OCF 3 ,
- R 2 is H, halo, -CN, (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy, perfluoro(C 1 -C 6 )alkyl, perfluoro(C 1 - C 6 )alkoxy-, cycloalkyl, cycloalkyl-O-, heterocycloalkyl, heterocycloalkyl-O-, aryl, aryl-O-, R 3 -(C(R 4 ) 2 ) n -O- or (R 6 ) 2 N-; wherein (C 1 -C 6 jalkyl, cycloalkyl, heterocycloalkyl, and aryl are each optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -
- R 5 is selected from the group consisting of (C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkyl, HO-(C 2 - C 4 )alkyl-, cycloalkyl, heterocycloalkyl, and aryl; wherein (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and aryl are each optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NHi, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 ,
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1 -3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2, (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N ⁇ , -CF 3 , -OCH 3 and -OCF 3 ;
- R' is H, halo, (C 1 -C 3 )alkyl, -CF 3 , ((C 1 -C 3 )alkoxy, -OCF 3 or (R 7 ) 2 N-; wherein R 7 is H or (C 1 -C 3 )alkyl;
- R 12 is H or (C 1 -C 3 )alkyl
- R 13 is (C 1 -C 3 )alkyl.
- Clause 8 A compound according to Clause 1, wherein Het is a ring of the Formula vii vii
- Clause 1 A compound according to any of Clauses 1-10, wherein W is a group of the compound of Formula (a)
- Clause 14 A compound according to any of Clauses 1-10, wherein W is a group of the compound of Formula (d)
- Clause 16 A compound according to any of Clauses 1-10, wherein W is a group of the compound of Formula (f)
- Clause 17 A compound according to any of Clauses 1 -16, wherein R 1 is selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, (C 1 -CAalkoxy, and perfluoro(C 1 - C3)alkoxy-. [0283] Clause 18. A compound according to any of Clauses 1-17, wherein R 1 is selected from the group consisting of H, F, -CH3, and -OCH 3 .
- Clause 19 A compound according to any of Clauses 1-18, wherein R 1 is H.
- R 2 is selected from the group consisting of H, (O.-Cfijalkyl, (C 1 -C 6 )alkoxy-, perfluoro(C 1 - Ce)alkyl, perfluoro(C 1 -C 6 )alkoxy-, cycloalkyl, cycloalkyl-O-, heterocycloalkyl, aryl, R 5 - (C(R 4 ) 2 )n-O- or (R 6 ) 2 N-; wherein (C 1 -C 6 )alkyl, cycloalkyl, cycloalkyl-O-, heterocycloalkyl and aryl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, OCH 3 and -OCF 3 ; each R 4 is independently FI or (C 1 -C 3 )alkyl optionally substituted with 1 -3 substituents independently selected from the group consisting of H, halo, -CN,
- R 5 is selected from (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and aryl; wherein (C 1 -Csjalkyl, cycloalkyl, heterocycloalkyl and aryl is optionally substituted with 1 -3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ; and
- R 6 is independently H or (C 1 -C 3 )alkyl optionally substituted with 1-3 substituents independently- selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 .
- Clause 21 A compound according to any of Clauses 1-20, wherein R 2 is selected from H, (C 1 -C 3 )alkyl, (C 1 -C 3 )alkoxy-, perfluoro(C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkoxy-, 3- to 10- membered cycloalkyl, 3- to 10-membered cycloalkyl-O-, 5- to 10-membered heterocycloalkyl, 6- to 10-membered aryl, R 5 -(C(R 4 ) 2 )n-O- or (R 6 ) 2 N-; wherein (C 1 -C 3 )alkyl, 3- to 10-membered cycloalkyl, 3- to 10-membered cycloalkyl-O-, 5- to 10-membered heterocycloalkyl, 6- to 10- membered aryl is optionally substituted with 1-3 substituents independently selected from the group consisting
- R 3 is selected from (C 1 -C 3 )alkyl, 3- to 10-membered cycloalkyl, 3- to 10-membered heterocycloalkyl, and 6- to 10-membered aryl; wherein (C 1 -C 3 )alkyl, 3- to 10-membered cycloalkyl, 3- to 10-membered heterocycloalkyl, and 6- to 10-membered aryl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, - CN, (C 1 -C 3 )alkyl, -NH 2 , (C 1 -C 3 )alkyl-(NH)-, ((C 1 -C 3 )alkyl ) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ; and R ,J is independently H or (C 1 -C.3)alkyl optionally substituted with 1 -3 substituents independently selected from the group consisting of H, hal
- Clause 22 A compound according to any of Clauses 1-21, wherein R 2 is selected from the group consisting of methoxy-, cyclopropoxy- or R 5 -(C(R 4 ) 2 )-O-; wherein each R 4 is H; wherein R 3 is selected from C 1 -alkyl and tetrahydropyran; and wherein said C 1 -alkyl is substituted with -OCH 3 .
- Clause 23 A compound according to any of Clauses 1 -22 wherein R 3 is selected from the group consisting of H, halo, (C 3 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 or (R 7 ) 2 N-; wherein R' is H or
- Clause 24 A compound according to any of Clauses 1-23, wherein R 3 is H, F, -CH 3 , - OCH 3 , or H 2 N-.
- Clause 25 A compound according to any Clauses 1-24, wherein EC is H.
- Clause 26 A compound according to any Clauses 1-25, wherein R y is selected from the group consisting of H, halo, (C 3 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 , -CN, is independently selected from H, (C 1 -C 3 )alkyl; and R 12 is H or (C 1 -C 3 )alkyl.
- Clause 27 A compound according to any Clauses 1-26, wherein at least one R 9 is selected from the group consisting of F, (C 1 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 , -CN.
- Clause 28 A compound according to any of Clauses 1-27, wherein at least one R 9 is -
- Clause 29 A compound according to any of Clauses 1, 4 or 11 to 28, wherein at least one R 10 is H.
- Clause 31 A compound according to any of Clauses 1-30, wherein Het is a ring of the formula wherein one R 9 is H and the other R 9 is -CF 3 , and wherein R i0 is H.
- Clause 32 A compound according to any of Clauses 1-31, wherein R s is H.
- Clause 35 A compound according to any of Clauses 1, 4 or 1 1, wherein the compound of Formula I is a compound of Formula IA, or a pharmaceutically acceptable salt, ester, or prodrug thereof the compound of Formula I* is a compound of Formula I*A, or a pharmaceutically acceptable salt, ester, or prodrug thereof wherein :
- R 1 is selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -OCH 3 , and - OCF 3 ;
- R 5 is selected from (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and aryl; wherein (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl and aryl are optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, - NH 2, (C 1 -C 3 )alkyl-(NH)-, ((Ca-C 3 )alkyl) 2 N-, -CF 3 , -OCH 3 and -OCF 3 ;
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, -OCH 3 and -OCF 3 ;
- R 3 is H, halo
- R 7 is H or (C 1 -C 3 )alkyl
- R 8 is H, -CH 3 or -CF 3 ;
- each R u is independently selected from H, (C 1 -C 3 )alkyl
- R 12 is H or (C 1 -C 3 )alkyl.
- R 2 is H, (C 1 -C 3 )alkyl, (C 1 -C 3 )alkoxy-, perfluoro(C 1 -C 3 )alkyl, perfluoro(C 1 -C 3 )alkoxy-, cycloalkyl, heterocycloalkyl, aryl, R 5 -(C(R 4 ) 2 )n-O-, or (R 6 ) 2 N-; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, -OCH 3 and - OCF 3 ; n is an integer from one to three; each R 4 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently- selected from the group consisting of H, halo, -OCH ; and -OCF 3 ;
- R 5 is selected from (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl, and aryl; wherein (C 1 -C 3 )alkyl, cycloalkyl, heterocycloalkyl and aryl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, (C 1 -C 3 )alkyl, -NH 2 , , -OCH 3 and -OCF 3 ;
- R 6 is independently H or (C 1 -C 3 )alkyl; wherein (C 1 -C 3 )alkyl is optionally substituted with 1-3 substituents independently selected from the group consisting of H, halo, -CN, -OCH ; and -OCF 3 ;
- R 3 is H, halo, (C 1 -C 3 )alkyl, -CF 3 , -OCH 3 , -OCF 3 or (R 7 ) 2 N-; wherein R 7 is H or (C 1 - C3)alkyl;
- R 8 is H, -CH 3 or -CF 3 ;
- R 9 is selected from H, halo, (C 1 -C 3 )alkyl, -CF 3 , -OCII3, -OCF 3 , -CN, - and
- R 10 is H or (C 1 -C 3 )alkyl; and R 11 is (C 1 -C 3 )alkyl.
- Clause 37 A compound according to Clause 1, wherein the compound is selected from :
- Clause 38 A pharmaceutical composition comprising a compound, salt, ester, or prodrug of any of Clauses 1-37, and a pharmaceutically acceptable carrier.
- Clause 39 A method of treating a disease or condition in a subject, that benefits from modulation of NAD+ level or related metabolites thereof level, comprising administering to the subject an amount of a compound according to any of Clauses 1 to 37 or a compositon according to Clause 38 effective to modulate NAD+ level or related metabolites thereof level.
- Clause 40 The method of Clause 39, wherein said disease or condition is nonalcoholi c steatohepatitis.
- Clause 41 A compound or composition according to any of Clauses 1 to 38 for use in the treatment of a disease or medical condition in a subject.
- Clause 42 A compound or composition for use according to Clause 41 , wherein the disease or condition benefits from modulation in NAD+level or related metabolites thereof level.
- Clause 43 A compound or composition for use according to Clause 41 or 42, wherein the disease or condition benefits from inhibition of CD38,
- Clause 44 A compound or composition for use according to any of Clauses 41 to 43, wherein the disease or condition is selected from ageing (e.g, age-related chronic disease), inflammation, cancer, such as PD-1/PD-L1 resistant cancers, cardiovascular disorder, neurological disorder, pulmonary disorder, fibrotic diseases, metabolic disorder, acute lung injury (ALT), acute respiratory' distress syndrome (ARDS), hyperphosphatemia, alcohol intolerance, lupus, arthritis, ataxia-telangiectasia, irritable bowel syndrome, colitis, gout, end stage renal disease, hearing loss, liver disorders, postmenopausal osteoporosis, Hartnup disease, tuberculosis, leishmaniasis, muscular dystrophy, organ reperfusion injury, pellagra, diseases of the skin, damage caused by exposure to radiation, periodontal disease, Leber's hereditary amaurosis, sleep disorder, exercise intolerance, chronic disease associated with cell death, and neurodegeneration and peripheral neuropathy associated with chemotherapy.
- ageing e.
- Clause 45 A compound or composition for use according to any of Clauses 41 to 44, wherein the disease or condition is an age-related disease or condition.
- Clause 46 A compound or composition for use according to any of Clauses 41 to 45, wherein the disease or condition is selected from small lung cell carcinoma, renal clear cell carcinoma, chronic lymphocytic leukemiahas, multiple myeloma, hypertension, hypoxic pulmonary vasoconstriction, cardiac hypertrophy, congestive heart failure, stroke, Alzheimer's disease, bipolar disorder, schizophrenia, Huntington's disease, amyotrophic lateral sclerosis, Parkinson's disease, multiple sclerosis, optic neuropathy, epilepsy, idiopathic pulmonary fibrosis, cystic fibrosis, asthma, chronic obstructive pulmonary' disease (COPD), metabolic syndrome, obesity, sarcopenic obesity, dyslipidemia, diabetes (such as type I diabetes), diabetic neuropathy, insulin resistance, pancreatitis, acute lung injury (ALI) acute respiratory' distress syndrome (ARDS), hyperphosphatemia, alcohol intolerance, lupus, rheumatoid arthritis, ataxiatelangiec
- Clause 47 A compound or composition for use according to any of Clauses 41 to 46, wherein the treatment is of multiple myeloma and is a combination treatment with an immunooncology drug.
- Clause 48 A compound or composition for use according to any of Clauses 41 to 46, wherein the disease or condition is nonalcoholic steatohepatitis (NASH).
- NASH nonalcoholic steatohepatitis
- Clause 49 A compound or composition for use according to Clause 48, wherein the compound is 2-(lH-imidazol-l-yl)-6-methoxy-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidine-4- carb oxami de.
- Clause 50 A compound according to any one of claims 1 to 37 for use in a method of treating a disease or disorder in a subject that benefits from modulation the level of NAD+ or related metabolite thereof, comprising administering to the subject a therapeutically effective amount, of the compound.
- Clause 51 The compound for use of claim 50, wherein the disease or disorder is or is related to nonalcoholic steatohepatitis, aging, senescence, immunometabolism, inflammation, infection, sepsis, arthritis, rheumatoid arthritis, psoriatic arthritis, osteoarthritis, lupus erythematosus, Crohn disease, ulcerative colitis, plaque psoriasis, ankylosing spondylitis, juvenile idiopathic arthritis, hidradenitis suppurativa, fibrosis, hepatic fibrosis, renal fibrosis, pulmonary fibrosis, cardiac fibrosis, cancer, multiple myeloma, neurodegeneration, infertility, loss of ovarian follicles, decreased oocyte quality and quantity, ovarian senescence, transient receptor potential melastatin 2 (TRPM2) regulation, calcium flux regulation, ischemiareperfusion-injury,
- TRPM2
- Clause 52 The compound for use of claim 50, wherein the disease or disorder is related to aging.
- Clause 54 The compound for use of claim 52, wherein the disease or disorder is or is related to Senescence, ImmunoMetabolism, fibrotic, neurodegenerative, Multiple Myeloma, or Sepsis.
- Clause 55 The compound for use of claim 54, wherein the disease or disorder is or is related to a fibrotic disease or disorder of the lung, heart, or kidney.
- Clause 56 The compound for use of claim 55, wherein the fibrotic disease is infection-induced fibrosis of the lung or virus-induced infection of the lung.
- Clause 57 The compound for use of claim 54, wherein the disease or disorder is or is related to Multiple Myeloma, and the method further comprising administering an immunooncology drug to the subject.
- NAD+ or related metabolite thereof is selected from the group consisting of NAD+, NMN, ADPR, cADPR, NAM, NAAD, NAADP, NR, MN AM.
- MTBE means methyl t- butyl ether
- NMP means 1 -methyl 2-pyrrolidinone
- TAA means triethyl amine
- TFA means tri fluoroacetic acid
- DCM means dichloromethane
- EtOAc means ethyl acetate
- MgSCh means magnesium sulphate
- NaSOi means sodium sulphate
- MeOH means methanol
- EtOH means ethanol
- H 2 O means water
- HC1 means hydrochloric acid
- POCI 3 means phosphorus oxychloride
- DMSO means dimethyl sulfoxide
- K2CO3 means potassium carbonate
- N means Normal
- M means molar
- mL means milliliter
- 'mmol means millimoles
- pmol means micromoles
- eq.” means equivalent
- °C means degrees Celsius
- Pa means pascal
- Step-1 ethyl 6- imidazol-l-yl)picolinate
- DMSO dimethyl sulfoxide
- a stirred solution of ethyl 6-bromopyridine-2-carboxylate (1 g, 4.35 mmol) in DMSO (10 mL) was added copper iodide (0.276 g, 0.869 mmol), L-proline (0.20 g, 1.74 mmol), potassium carbonate (1.20 g, 8.69 mmol) and imidazole (0.444 g, 6.52 mmol).
- the reaction mixture was heated to 100 °C for 16 h.
- the reaction mixture was cooled to room temperature (RT), and ice-cold water was added and extracted with ethyl acetate.
- Step-3 methyl 5-(6- imidazol-l -yl)picolinamido)picolinate
- Step-2 4-(benzyl oxy)-6-bromopi colinoyl chloride
- Step-4 4-(benz5doxy)-6-(lH-imidazol-l-yl)-N-(2-(trifluoromethyl) pyridin-4-yl) picolinamide
- 4-(benzyloxy)-6-bromO”N-[2-(trifluoromethyl)pyridin-4- yl]pyridine-2-carboxamide (170 mg, 0.38 mmol) in DMF (2 mL) was added IH-i midazole (38.4 mg, 0.57 mmol), copper iodide (15 mg, 0.075 mmol) and cesium carbonate (245 mg, 0.75 mmol).
- the reaction mixture was heated to 100 °C for 16 hr.
- Table 2 shows characterization data for additional compounds of Formula I prepared as described above.
- Table 3 shows characterization data for additional compounds of Formula I prepared by the method shown above.
- Step-3 2-( L ⁇ -imi dazol - 1 -yl)-6-m ethoxy -A%2-(trifluoromethyl)pyri din-4-yl) pyrimidine-4- carboxamide
- Step- 1 6-(l -methyl-1H-imidazol-5-yl)-N-(pyridin-3-yl)picohnannde
- Step- 1 2-chl oro-6-methyl -A'-fpyri din-3 -yl)pyrimidine-4-carboxamide
- Step-1 6-chloro-4-methylpicolinic acid
- Step 2 methyl 5-(6-(lH-imidazol-l-yl)-4-methylpicolinamido)picolinate
- Step-1 To a solution of oxetan-3-one (1.0 g, 13.9 mmol) in Tetrahydrofuran (10 mL) was added bromo(methyl)magnesium (9.25 mL, 27.8 mmol ) at 0°C. The resulting mixture was stirred for 1 hour at room temperature. The progress of reaction was monitored by TLC. The reaction mixture was quenched with saturated ammonium chloride solution (10 mL), extracted with Di chloromethane (50 mL). to afford 3-methyloxetan-3-ol (0.7 g, 57.25% yield) as a light yellow colour liquid.
- reaction mixture was quenched with saturated ammonium chloride solution (10 mL), extracted with ethyl acetate (50 mL).
- the crude residue was purified by gradient column chromatography using 0-30% ethyl acetate in hexane to afford 6-chloro-2-((3-methyloxetan-3-yl)oxy)-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidine-4- carboxamide (0.15 g, 17% yield) and 2-chloro-6-[(3-methyloxetan-3-yl)oxy]-N-[2- (trifluoromethyl)pyridin-4-yl]pyrimidine-4-carboxamide (0.2 g, 22.6% yield) as a offwhite solid.
- reaction mixture was quenched with saturated ammonium chloride solution (10 niL), extracted with ethyl acetate (50 mL).
- the crude residue was purified by gradient column chromatography using 0-30% ethyl acetate in hexane to afford 2-chloro-6-((tetrahydro-2H-pyran-4-yl)methoxy)-N-(2- (t.rifluoromethyl)pyridin-4-yl)pyrimidi ne -4-carboxamide (0.13 g, 52.5% yield) and 6-chloro-2- [(oxan-4-yl)methoxy]-N-[2-(trifluoromethyl)pyridin-4-yl]pyrimidine-4-carboxamide (0.13 g, 52.5% yield) as a off white solids.
- reaction mixture was quenched with saturated ammonium chloride solution (10 mL), extracted with ethyl acetate (50 mL).
- the crude residue was purified by gradient column chromatography using 0-30% ethyl acetate in hexane to afford 2-chloro-6-((tetrahydro-2H-pyran-4-yl )methoxy)-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidi ne -4-carboxamide (0.25 g, 25.27% yield as a off white solid.
- Step-1 2,6-dichloro-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidine-4-carboxamide
- 2-(trifluoromethyl)pyridin-4-amine 783 mg, 4.83 mmol
- Trimethyl aluminum 3.62 mL, 7.25 mmol
- Step-2 2-chloro-6-((tetrahydro-2H-pyran-4-yl)methoxy)-N-(2-(trifluoromethyl)pyridin-4- yl)pyrimidine-4-carboxamide
- Table 16 Provided in Table 16 are characterization data for the compound of Formula I prepared by the method shown in Example 16.
- Table 16 (Compound 74)
- Step-1 2,6-dichloro-N-(2-(trifluoromethyl)pyridm-4-yl)pyrimidine-4-carboxamide
- Step-3 6-(2-methoxyethoxy)-2-(l-methyl-lH-imidazol-5-yl)-N-[2-(trifluoromethyl)pyridin-4- yl]pyrimidine-4-carboxamide
- 2-chloro-6-(2-methoxyethoxy)-N-[2-(trifluoromethyl)pyridin-4- yl]pyrimidine-4-carboxamide 0.1 g, 0,265 mmol
- l-methyl-5- (tributylstannyl)-lH-imidazole 148 mg, 0.398 mmom
- tetrakis(triphenylphosphine)palladium(0) 92 mg, 0,08 mmol
- the resulting mixture was stirred at 100°C for 2 hour in CEM microwave. The progress of reaction was monitored by TEC. The reaction mixture was cooled to room temperature dilute with water (20 mL), extracted with ethyl acetate (20 mL) washed with brain solution.
- Step-1 2,6-dichloro-N-(2-(trifluoromethyl)pyridin-4-yr)pyrimidine-4-carboxamide
- Step-2 2-chloro-6-(2-methoxyethoxy)-N-[2-(trifluoromethyl)pyridm-4-yl]pyrimidine-4- carboxamide
- reaction mixture was quenched with saturated ammonium chloride solution (30 mL), extracted with ethyl acetate (50 mL).
- the crude residue was purified by gradient column chromatography using 0-30% ethyl acetate in hexane to afford 2-chloro-6-(2-methoxyethoxy)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrimidine-4-carboxamide (0.4 g, 34.09% yield) as an off white solid.
- Step-3 6-(2-m ethoxy ethoxy)-2-( 1 -methyl- 1 H-imidazol-2-yl )-N-[2-(trifluoromethyl)pyridin-4- yllpyrimidine-4-carboxamide [0392] To a solution of 2-chloro-6-(2-methoxyethoxy)-N-[2-(trifluoromethyl)pyridin-4- yl]pyrimidine-4-carboxamide (0.13 g, 0.345 mmol) in 1 ,4-dioxane (5 mL) was added 1-methyl- 2-(tributylstannyl)-lH-imidazole (192 mg, 0.518 mmom) and tetrakis(triphenylphosphine)palladium(0) (120 mg, 0, 104 mmol).
- the resulting mixture was stirred at 100°C for 2 hour in CEM microwave. The progress of reaction was monitored by TLC.
- the reaction mixture was cooled to room temperature dilute with water (20 mL), extracted with ethyl acetate (20 mL) washed with brain solution.
- Step-1 2,6-dichloro-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidine-4-carboxamide
- Step-3 6-(2 -methoxy ethoxy)-2-(l -methyl- lH-irnidazol-2-yl)-N-r2-(trifluoromethyl)pyridin-4- yl]pyrimidine-4-carboxamide
- 2-chloro-6-(2-methoxyethoxy)-N-[2-(trifluoromethyl)pyridin-4- yl]pyrimidine-4-carboxamide (0.13 g, 0.345 mmol) in 1 ,4-dioxane (5 mL) was added 1-methyl- 2-(tributylstannyl)-lH-imidazole (192 mg, 0.518 mmom) and tetrakis(triphenylphosphine)palladium(0) (120 mg, 0, 104 mmol).
- the resulting mixture was stirred at 100°C for 2 hour in CEM microwave. The progress of reaction was monitored by TLC.
- the reaction mixture was cooled to room temperature dilute with water (20 mL), extracted with ethyl acetate (20 mL) washed with brain solution.
- Step-1 2,6-dichloro-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidine-4-carboxamide 89879
- the reaction mixture was quenched with saturated ammonium chloride solution (30 niL), extracted with ethyl acetate (20 ml).
- the crude residue was purified by gradient column chromatography using 0-30% ethyl acetate in hexane to afford 2-chl oro-6-(2-hy droxy-2-m ethylpropoxy)-N-( 2-(tri fluorom ethyl)pyridin-4-y l)pyri midine-4- carboxamide (0.25 g, 43.13% yield) as Thick solid.
- Step-3 6-(2-hydroxy-2 -methylpropoxy )-2-( IH-imidazol- 1 -yl)-N-(2-(trifluoromethyl)pyridin-4- yl)pyrimidine-4-carboxamide
- 2-chloro-6-(2-hydroxy-2-methylpropoxy)-N-(2- (trifluoromethyl)pyridin-4-yl)pyrirnidine ⁇ 4-carboxamide 0.2 g, 0.512 mmol
- N,N ⁇ dimethylformamide 5 mL
- caesium carbonate (0.25 g, 0.768 mmol
- diiodocopper 49.0 mg, 0, 154 rmnorn
- IH-imidazole 52.3 mg, 0.768 mmol
- Step-1 6-(methylthio)-N-(pyridin-4-yI)pyrimido[5,4-d]pyrimidin-4-amine
- Step-2 N-]6-methaoespIfinyl-[l,31diazmo[5,4-d]pyrimidin-4-yIlpyridin-4- [0404]
- N-[6-(methylsulfanyl)-[l,3]diazino[5,4-dlpyrimidin-4- yllpyridin-4-amine 200 mg, 740 pmol
- 3 -chlorobenzene- 1 -carboperoxoic acid 255 mg, 2 eq., 1.48 mmol.
- the reaction mixture was stirred at room temperature for 16 hours.
- the reaction mixture was quenched with sat.NaHCO3 solution and extracted in DCM.
- the reaction mixture was cooled to room temperature and extracted in ethyl acetate.
- the organic layer was dried over sodium sulfate and evaporated off to obtain crude which was purified over silica gel flash column chromatography.
- the compound eluted out as a mixture in 5 % MeOH : DCM.
- the fractions were evaporated off to obtain crude which was re-purified by prep TEC.
- the silica gel band was taken and passed through 4g silica gel column. The fractions were evaporated off to obtain the final compound as an off white solid.
- NAD+ hydrolase activity of a human T cell line treated with test compounds was measured using a fluorescence-based assay.
- NH7-dCas9 cells (Jurkat clone) were provided by the Weissman Lab (UCSF 1700 4th St., Byers Hall, Room 403B, San Francisco, CA 94158- 2330). Briefly, cells were centrifuged, resuspended in 44 p ⁇ PBS/10 6 cells and plated in a 96 well black plate (CORNING) at a density of 1 x I0 6 per well.
- Each test compound was received in a powder state and dissolved in DMSO as a 25mM stock solution. Each test compound was serially diluted in order to generate a dose- response curve. The dilution series of each test compound in DMSO was prepared at 50 times the concentrations to be assayed. Each test compound was diluted to generate a 5-fold dilution series of 8 concentrations from 20 ⁇ M to 0.256 nM in order to assay 8 final concentrations ranging from 400 nM to 25.6 ⁇ M. One pl of the 50X concentration compound series was added to each well of the plate containing 1 x 10 6 cells in PBS.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compound 78c at a final concentration of 50 nM and 2 wells containing only PBS (used as a background value).
- the reaction was started by adding nicotinamide l,N 6 -etheno-adenine dinucleotide (Sigma Aldrich) to reach a final concentration of 80 ⁇ M.
- the samples were excited at 321 -15 nm, and the emission of fluorescence was measured at 410-20 nm at 37°C every minute for 1 hr in a Clariostar® microplate reader (BMG LAB TECH).
- NAD+ hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve using the MARS data analysis software (BMG LABTECH).
- the data were uploaded in the Collaborative Drug Design (CDD) Software and for each compound the IC50 was calculated by the software. Each compound was tested in 3 independent experiments on 3 separate days to establish the reproducibility of the results. For each plate, the z-factor was also calculated as a plate quality- using the CDD software.
- NAD+ cyclase activity of the human recombinant CD38 protein treated with test compounds was measured using a fluorescence-based assay. Briefly, recombinant human CD38 Protein (R&D System) was resuspended in 44 ul/well of Sucrose-Tris Buffer (Sucrose 0.25 M, Tris pH 7.4 40 mM) and plated in a 96-well black plate (CORNING) at a density of 125ng/ well.
- R&D System recombinant human CD38 Protein
- Each test compound was received in a powder state and dissolved in DMSO as a 25 mM stock solution. Each test compound was serially diluted in order to generate a dose- response curve. The dilution series of each test compound in DMSO was prepared at 50 times the concentrations to be assayed. Each test compound was diluted to generate a 5-fold dilution series of 8 concentrations from 100 uM to 32 nM in order to assay 8 final concentrations ranging from 2 pM to 0.64 nM. One ul of the SOX concentration compound series was added to each well of the plate containing 125 ng of protein in Sucrose-Tris buffer.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compounds 78c at a final concentration of 500 nM and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- NTD+ nicotinamide guanine dinucleotide
- BMG LABTECH MARS data analysis software
- the performance validation criteria of the assay as a High Throughput Screening (HTS) Z-factor within the plate was greater than 0.9 (optimal HTS z- factor > 0.5) and the Intra-plate, inter-plate and day to day variability (CV) was less than 20%.
- HTS High Throughput Screening
- NAD+ hydrolase activity of the human recombinant CD38 protein treated with test compounds was measured using a fluorescence-based assay. Briefly, recombinant human CD38 Protein (R&D System) was resuspended in 44 pl, -well of Sucrose-Tris Buffer (Sucrose 0.25 M, Tris pH 7.4 40 mM) and plated in a 96-well black plate (CORNING) at a density of 10 ng/ well.
- R&D System recombinant human CD38 Protein
- Each test compound was received in a powder state and dissolved in DMSO as a 25 mM stock solution. Each test compound was serially diluted in order to generate a dose- response curve. The dilution series of each test compound in DMSO was prepared at SOX the concentrations to be assayed. Each test compound was diluted to generate a 5 -fold dilution series of 8 concentrations from 20 pM ⁇ to 0.256 nM in order to assay 8 final concentrations ranging from ranging from 400 nM to 25.6 pM. One pl of the 50X concentration compound series was added to each well of the plate containing 10 ng protein in Sucrose-Tris buffer.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compounds 78c at a final concentration of 50 nM and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- DMSO vehicle only
- 3 wells of cells treated with the reference compounds 78c at a final concentration of 50 nM and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- the reaction was started adding nicotinamide 1 ,N 6 -etheno-adenine dinucleotide (Sigma Aldrich) to reach a final concentration of 80pM..
- the samples were excited at 321-15 nm and the emission of fluorescence was measured at 410-20 nm at 37°C every' minute for
- a Clariostar® microplate reader BMG LABTECH.
- NAD+ hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve using the MARS data analysis software (BMG LABTECH). The data were uploaded in the Collaborative Drug Design (CDD) Software and for each compound the IC50 was calculated by the software. Each compound was tested in 3 independent experiments on 3 separate days to establish the reproducibility of the results. For each plate, the z-factor was also calculated as a plate quality using the CDD software.
- CDD Collaborative Drug Design
- NAD+ cyclase activity of the human recombinant CD38 protein treated with test compounds was measured using a fluorescence-based assay. Briefly, recombinant mouse CD38 Protein (R&.D System) was resuspended in 44 gl/well of Sucrose-Tris Buffer (Sucrose 0.25 M, Tris pH 7.4 40 mM) and plated in a 96-well black plate (CORNING) at a density of 32 ng/ well.
- R&.D System recombinant mouse CD38 Protein
- Each test compound was received in a powder state and dissolved in DMSO as a 25mM stock solution. Each test compound was serially diluted in order to generate a dose- response curve. The dilution series of each test compound in DMSO was prepared at 50 times the concentrations to be assayed. Each test compound was diluted to generate a 5-fold dilution series of 8 concentrations from 100 pM to 32 nM in order to assay 8 final concentrations ranging from 2 p M to 0.64 nM. One pl of the 50X concentration compound series was added to each well of the plate containing 125 ng of protein in Sucrose-Tris buffer.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compounds 78c at a final concentration of 500 nM and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- NAD+ nicotinamide guanine dinucleotide
- BMG LABTECH Clariostar® microplate reader
- NAD+ cyclase activity was calculated as the slope of the linear portion of the fluorescence-time curve using the MARS data analysis software (BMG LABTECH).
- CDD Collaborative Drug Design
- NAD+ hydrolase activity of the mouse recombinant CD38 protein treated with test compounds was measured using a fluorescence-based assay. Briefly, recombinant mouse CD38 Protein (R&D System) was resuspended in 44 pl/well of Sucrose-Tris Buffer (Sucrose
- test compound was received in a powder state and dissolved in DMSO as a
- each test compound was serially diluted in order to generate a dose- response curve.
- the dilution series of each test compound in DMSO was prepared at 50 times the concentrations to be assayed.
- Each test compound was diluted to generate a 5-fold dilution series of 8 concentrations from 20 pM to 0.256 nM in order to assay 8 final concentrations ranging from 400 nM to 25.6 pM.
- One pl of the 50X concentration compound series was added to each well of the plate containing 2.5 ng of protein in Sucrose-Tris buffer.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compounds 78c at a final concentration of 50 nM and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- the reaction was started adding nicotinamide I ,N°-etheno-adenine dinucleotide (Sigma Aldrich) to reach a final concentration of 80uM.
- the samples were excited at 321-15 nm and the emission of fluorescence was measured at 410-20 nm at 37°C every minute for 1 hr in a Clariostar microplate® reader (BMG LABTECH).
- NAD+ hydrolase activity vvas calculated as the slope of the linear portion of the fluorescence-time curve using the MARS data analysis software (BMG LABTECH).
- the data were uploaded in the Collaborative Drug Design (CDD) Software and for each compound the IC50 was calculated by the software. Each compound was tested in 3 independent experiments on 3 separate days to establish the reproducibility of the results. For each plate, the z-factor was also calculated as a plate quality using the CDD software.
- NAD+ hydrolase activity of the rat recombinant CD38 protein treated with test compounds was measured using a fluorescence-based assay. Briefly, recombinant rat CD38 Protein (Sino Biological) was resuspended in 44 pl/well of Sucrose-Tris Buffer (Sucrose 0.25 M, Tris pH 7.4 40 mM) and plated in a 96 well black plate (CORNING) at a density of 2.5 ng/ well.
- Sucrose-Tris Buffer Sucrose 0.25 M, Tris pH 7.4 40 mM
- Each test compound was received in a powder state and dissolved in DMSO as a 25 mM stock solution. Each test compound was serially diluted in order to generate a doseresponse curve. The dilution series of each test compound in DMSO was prepared at 50 times the concentrations to be assayed. Each test compound was diluted to generate a 5-fold dilution series of 8 concentrations from 20 pM to 0.256 nM in order to assay 8 final concentrations ranging from 400 nM to 25.6 pM. One pl of the 50X concentration compound series was added to each well of the plate containing 2.5 ng of protein in Sucrose-Tris buffer.
- Each plate had the following intra plate controls: 3 wells containing cells treated with vehicle only (DMSO), 3 wells of cells treated with the reference compounds 78c at a final concentration of 50 nMI and 2 wells containing only Sucrose-Tris buffer (used as a background value).
- the reaction was started adding nicotinamide l,N°-etheno-adenine dinucleotide (Sigma Aldrich) to reach a final concentration of 80u.M.
- the samples were excited at 321-15 nm and the emission of fluorescence was measured at 410-20 nm at 37°C every minute for 1 hr in a Clariostar microplate® reader (BMG LABTECH).
- NAD+ hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve using the M ARS data analysis software (BMG LABTECH).
- the data were uploaded in the Collaborative Drug Design (CDD) Software and for each compound the IC50 was calculated by the software. Each compound was tested in 3 independent experiments on 3 separate days to establish the reproducibility of the results. For each plate, the z-factor was also calculated as a plate quality using the CDD software.
- the compounds according to the disclosure are potent inhibitors of CD38 and as such possess activity in the treatment of numerous disorders.
- the compounds according to the disclosure that were tested had the following activity in the aforesaid assays (.Assay 1 -6).
- the in vitro potency of Compound 35 against the human CD38 enzyme was determined by a fluorescence-based NAD+ hydrolase activity assay using the NAD+ analog nicotinamide I ,N°-etheno-adenine dinucleotide (ENAD+) as substrate.
- the assay utilized: I) whole primary human CD4+ T cells (from healthy donors) activated with anti-CD3/CD28 antibodies and expressing human CD38, or II) whole primary' human macrophages (from healthy donors) stimulated with LPS to polarize in a pro-inflammatory' Ml state expressing human CD38.
- the cells were incubated in the presence of 8 different concentrations of Compound 35 for 10 minutes and the reaction was started adding the substrate sNAD+.
- the emission of fluorescence was measured at 410-20 nm at 37°C every minute for I hr.
- the NAD+ hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve and used to calculate the IC50 value.
- the IC50 value for Compound 35 was 0. 1.8 nM (0,065 ng/mL) in primary 7 human CD4+ T cells (see Figure 1A) and 2.25 nM (0.817 ng/mL) in primary human Ml macrophages (see Figure IB).
- the efficacy of Compound 35 was also determined in a NASH model (see InSphero Human 3D InSight 1M Brunswick, ME) to investigate the effect of the molecule on the pathophysiological phenotype of NASH such as release of inflammatory markers.
- the microtissue model used is a co-culture of 4 different cell types of human liver cells (hepatocytes, Kupffer cells, endothelial and stellate cells) cultured for 10 days in a NASH induction media containing FFA, LPS and high levels of sugars. In the LEAN group the microtissues were cultured for 10 days in physiological -like medium.
- mice Male C57BL6/J, 22 months old were fasted for 4 hr and compound 35 was administered via oral gavage at a concentration of 3 mg/kg and 10 mg/kg. Mice were humanely euthanized for liver tissue harvesting at 1 3-, and 6-hr post-dosing.
- a vehicle control (DMSO:Solutol: SBE-CD, 2:5:93, v/v) was included to assess baseline NAD+ metabolites levels.
- the samples were vortexed, sonicated briefly in a bath sonicator, and shaken at 55 °C for 3 min at 1,200 rpm. Samples were then centrifuged at 16,000 x g for 10 min at 4 °C, and the supernatant was dried by speedvac. The dried pellet was resuspended in 97% 10 mM ammonium acetate/3% acetonitrile, centrifuged at 16,000 x g for 10 min at 4 °C and 2 pL was injected for LCMS analysis.
- the UPLC-MS/MS analysis to measure NAD+ metabolites was performed using a Vanquish UHPLC system and a Q Exactive Orbitrap mass spectrometer with H-ESI ion source both from Thermo Scientific.
- UPLC separation was performed on a Hypercarb (2.1 x 100 mm, 5 pm particle size, Thermo) column following the method described in Trammell et al. (2013). Acquisition w'as carried out in positive ion, Parallel Reaction Monitoring (PRM) mode and targeted Single Reaction Monitoring (t-SIM).
- PRM Parallel Reaction Monitoring
- t-SIM targeted Single Reaction Monitoring
- Thermo Xcalibur QualBrowser system software (version 4.2.47) and Freestyle (version 1.8.51.0) softwares was used for the data processing.
- peak areas were normalized with respective internal standards, normalized to weight of the tissue sample used, and calculated as fold change relative at 0 h timepoint/vehicle group.
- a single 3 mg/kg dose oral administration of Compound 35 was able to increase NAD+ and NMN levels and to decrease NANI and ADPR levels in liver ( Figures 3 A- 3B).
- a single 10 mg/kg dose oral administration of Compound 35 was able to increase NAD+ and NMN levels and to decrease NAM and ADPR levels in liver ( Figures 4A - 4B).
- the in vitro potency of Compound 32 against the human CD38 enzyme was determined by a fluorescence-based NAD+ hydrolase activity assay using the NAD+ analog nicotinamide 1 ,N6-etheno-adenine dinucleotide (ENAD+) as substrate.
- the assay utilized whole primary human macrophages (from healthy donors) stimulated with LPS to polarize in a pro- inflammatory Ml state expressing human CD38.
- the cells were incubated in the presence of 8 different concentrations of Compound 32 for 10 minutes and the reaction was started adding the substrate eNAD+.
- the emission of fluorescence was measured at 410-20 nm at 37 °C every minute for 1 hr.
- the ⁇ Al.) hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve and used to calculate the IC50 value.
- the IC50 value for Compound 32 was 3.5 nM (1.36 ng/mL) in primary' human Ml macrophages (see Figure 5).
- Obese mice male DIO C57BL6/J, 7,5 months old were fasted for 4 hr and compound 32 was administered via oral gavage at a concentration of 3 mg/kg and 10 mg/kg. Mice were humanely euthanized for liver tissue harvesting at 1-, 3-, and 6-hr post-dosing.
- a vehicle control (DMSO: Solutol HS 15: 80% Captisol (20% in water)(5: 15:80 v/v) was included to assess baseline NAD+ metabolites levels.
- Frozen liver tissue was pulverized and resuspended in the extraction buffer (3 : 1 ethanol : 10 mM aq. HEPES, pH 7.1). The samples were vortexed, sonicated briefly in a bath sonicator, and shaken at 55 °C for 3 min at 1,200 rpm. Samples were then centrifuged at 16,000 x g for 10 min at 4 °C, and the supernatant was dried by speedvac.
- the extraction buffer 3 : 1 ethanol : 10 mM aq. HEPES, pH 7.1
- the dried pellet was resuspended in 97% 10 mM ammonium acetate/3% acetonitrile, centrifuged at 16,000 x g for 10 min at 4 °C and 2 uL was injected for LCMS analysis
- the UPLC-MS/MS analysis to measure NAD+ metabolites was performed using a Vanquish UHPLC system and a Q Exactive Orbitrap mass spectrometer with H-ESI ion source both from Thermo Scientific.
- UPLC separation was performed on a Hypercarb (2.1 x 100 mm, 5 pm particle size, Thermo) column following the method described in Trammell et al. (2013), Acquisition was carried out in positive ion, Parallel Reaction Monitoring (PRM) mode and targeted Single Reaction Monitoring (t-SIM).
- PRM Parallel Reaction Monitoring
- t-SIM targeted Single Reaction Monitoring
- Thermo Xcalibur QualBrowser system version 4.2.47) and Freestyle (version 1.8.51.0) softwares was used for the data processing.
- peak areas were normalized with respective internal standards, normalized to weight of the tissue sample used, and calculated as fold change relative at 0 h timepoint/vehicle group .
- a single 3 mg/kg dose oral administration of Compound 32 was able to increase NAD+ and NMN levels and to decrease NAM and AD PR levels in liver ( Figures 6B-6E).
- a single 10 mg/kg dose oral administration of Compound 32 was able to remarkably increase NAD+ and NMN levels and to decrease NAM and .ADPR levels in liver 1 hour post administration ( Figures 6B-6E).
- Obese mice male DIO C57BL6/J, 65 weeks oid treated for 49 days with compound 32 via oral gavage at a concentration of 10 mg/kg BID. On day 49, mice were humanely euthanized for liver tissue harvesting at 4hr post-dosing. A vehicle control (1% Methylcellulose) was included to assess baseline NAD+ metabolites levels.
- Compound 32 was administered to aged mice (male DIO C57BL6/J, 19-22 months old) via oral gavage at a concentration of 3 mg/kg and 10 mg/kg BID for 5 days. At day 5, mice were humanely euthanized for liver tissue harvesting 3hr post-dosing.
- a vehicle control (DMSO: Solutol HS 15: 80% Captisol (20% in water)(5: 15:80 v/v) was included to assess baseline NAD-f- metabolites levels.
- Frozen liver tissue was pulverized and resuspended in the extraction buffer (3 : 1 ethanol: 10 mM aq. HEPES, pH 7.1). The samples were vortexed, sonicated briefly in a bath sonicator, and shaken at 55 °C for 3 min at 1,200 rpm.
- UPLC separation was performed on a Hypercarb (2.1 x 100 mm, 5 pm particle size, Thermo) column following the method described in Trammell et al. (2013), Acquisition was carried out in positive ion, Parallel Reaction Monitoring (PRM) mode and targeted Single Reaction Monitoring (t-SIM).
- PRM Parallel Reaction Monitoring
- t-SIM Targeted Single Reaction Monitoring
- Thermo Xcalibur QualBrowser system version 4.2.47) and Freestyle (version 1.8.51 .0) softwares was used for the data processing.
- peak areas were normalized with respective internal standards, normalized to weight of the tissue sample used, and calculated as fold change relative at 0 h timepoint/vehicle group .
- the in vitro potency of Compound 39 against the human CD38 enzyme was determined by a fluorescence-based NAD+ hydrolase activity assay using the NAD+ analog nicotinamide I,N6-etheno-adenine dinucleotide (eNAD+) as substrate.
- the assay utilized whole primary human macrophages (from healthy donors) stimulated with LPS to polarize in a pro- inflammatory Ml state expressing human CD38.
- the cells were incubated in the presence of 8 different concentrations of Compound 39 for 10 minutes and the reaction was started adding the substrate sNAD+.
- the emission of fluorescence was measured at 410-20 nm at 37°C every minute for 1 hr.
- the NAD+ hydrolase activity was calculated as the slope of the linear portion of the fluorescence-time curve and used to calculate the IC50 value.
- the IC50 value for Compound 39 was 3.9 nM (1 .6 ng/mL) in primary human Ml macrophages ( Figure 9).
- Obese mice male DIO C57BL6/J, 7.5 months old were fasted for 4 hr and compound 39 was administered via oral gavage at a concentration of 1 mg/kg and 10 mg/kg. Mice were humanely euthanized for liver tissue harvesting at 1-, 3-, and 6-hr post-dosing.
- a vehicle control (DMSO: Solutol HS 15: 80% Captisol (20% in water)(5: 15:80 v/v) was included to assess baseline NAD+ metabolites levels.
- Frozen liver tissue was pulverized and resuspended in the extraction buffer (3 : 1 ethanol: 10 mM aq. HEPES, pH 7.1). The samples were vortexed, sonicated briefly in a bath sonicator, and shaken at 55 °C for 3 min at 1,200 rpm. Samples were then centrifuged at 16,000 x g for 10 min at 4 °C, and the supernatant was dried by speedvac.
- the extraction buffer 3 : 1 ethanol: 10 mM aq. HEPES, pH 7.1
- the dried pellet was resuspended in 97% 10 mM ammonium acetate/3% acetonitrile, centrifuged at 16,000 x g for 10 min at 4 °C and 2 uL was injected for LCMS analysis.
- Obese mice male DIO C57BL6/T 65 weeks old treated for 49 days with compound 39 via oral gavage at a concentration of 10 mg/kg BID. On day 49, mice were humanely euthanized for liver ti ssue harvesting at 4hr post-dosing. A vehicle control (1% Methylcellulose) was included to assess baseline NAD+ metabolites levels.
- Frozen liver tissue was pulverized and resuspended in the extraction buffer (3 : 1 ethanol: 10 mM aq. HEPES, pH 7.1). The samples were vortexed, sonicated briefly in a bath sonicator, and shaken at 55 °C for 3 min at 1,200 rpm. Samples were then centrifuged at 16,000 x g for 10 min at 4 °C, and the supernatant was dried by speedvac.
- the extraction buffer 3 : 1 ethanol: 10 mM aq. HEPES, pH 7.1
- the dried pellet was resuspended in 97% 10 mM ammonium acetate/3% acetonitrile, centrifuged at 16,000 x g for 10 min at 4 C C and 2 pL was injected for LCMS analysis.
- the UPLC-MS/MS analysis to measure NAD+ metabolites was performed using a Vanquish UHPLC system and a Q Exactive Orbitrap mass spectrometer with H-ESI ion source both from Thermo Scientific.
- UPLC separation was performed on a Hypercarb (2.1 x 100 mm, 5 pm particle size, Thermo) column following the method described in Trammell et al. (2013).
- Compound 39 was administered to aged mice (male DIO C57BL6/J, 19-22 months old) via oral gavage at a concentration of 10 mg/kg BID for 5 days. At day 5, mice were humanely euthanized for liver tissue harvesting 3hr post-dosing.
- a vehicle control (DMSO: Solutol HS 15: 80% Captisol (20% in water)(5: 15:80 v/v)) was included to assess baseline NAD+ metabolites levels.
- Cytokines were quantified in plasma: (1) statistically significant decrease of plasma IL-6, TNFa and IP-10 8h post LPS challenge in the two compound 39 treatment arms (lOmg/kg and 30mg/kg); (2) statistically significant decrease of plasma IP-10 4hour post LPS challenge in the trvo compound 39 treatment arms (lOmg/kg and 30mg/kg) and in the control arm (Dexamethasone Img/kg); (3) 43% and 75% decrease of plasma IL-6 4hour post LPS challenge with compound 39 lOmg/kg and 30mg/kg, respectively; (4) 22% and 58% decrease of plasma TNFa 4hour post LPS challenge with compound 39 lOmg/kg and 30mg/kg, respectively; and (5) statistically significant decrease of plasma IL-6, TNFa and IP-10 4hour post LPS challenge in control arm (Dexamethasone Img/kg) ( Figures 13A-13C).
- NAD+ metabolism in spleen was assessed using Mass Spectrometry/ (MS): an increase of NAD+ levels (2.3-fold) in the compound 39 30mg/kg treatment arm at 8 hours post LPS challenge and a statistically significant decrease in ADPR levels (40% decrease) after 8 h treatment with 30 mg/kg compound 39 were observed ( Figures 14A-14C).
- NAD+ metabolism in liver was assessed using Mass Spectrometry’ (MS): a statistically significant increase ofNAD+ levels (2 -fold) with compound 39 lOmg/kg treatment arm at 8 hours post LPS challenge, a significant decrease of NAM levels (38% decrease) in the compound 39 30mg/kg treatment arm at 8 hours post LPS challenge, and a decrease of ADPR levels in the Compound 39 treatment arms at 4 hours post LPS challenge were observed ( Figures 15A-15C).
- the cells were seeded in complete RPMI medium in the presence of absence of 50nM of Compound 32 at a density of 1 X 10 6 cells/ml and then incubated at 37 C C for 24 h. In preparation for flow cytometry, equal numbers of cells were washed and incubated with the membrane-permeable calcium sensor dye eFluor 514 (eBioscience, catalogue no. 65-0859) in PBS for 15 min at room temperature.
- eFluor 514 eBioscience, catalogue no. 65-0859
- FIG. 20A A representative plot of intracellular calcium-flux kinetics in NH7-dCas9 cells in the presence of compound 32 (50 nM) is shown in figure 20A.
- the parameter area under the curve (AUC) relative to the calcium flux was calculated and shown in figure 20B.
- Compound 32 at a final concentration of 50nM was able to significantly decrease the total Ca2+ cellular flux of about 33% ( Figure 20A and 20B).
- the cells were seeded in complete RPMI medium in the presence of absence of 50nM of Compound 39 at a density of 1 X 10 6 cells/ml and then incubated at 37°C for 24 h. In preparation for flow cytometry, equal numbers of cells were washed and incubated with the membrane-permeable calcium sensor dye eFluor 514 (eBioscience, catalogue no. 65-0859) in PBS for 15 min at room temperature.
- eFluor 514 eBioscience, catalogue no. 65-0859
- FIG. 21 A A representative plot of intracellular calcium-flux kinetics in NH7-dCas9 cells in the presence of compound 39 (50 nM) is shown in figure 21 A.
- the parameter area under the curve (AUC) relative to the calcium flux was calculated and shown in figure 21 B.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description


















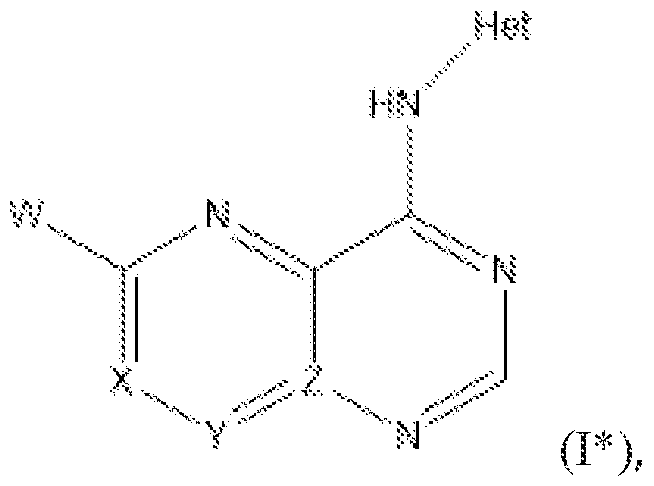










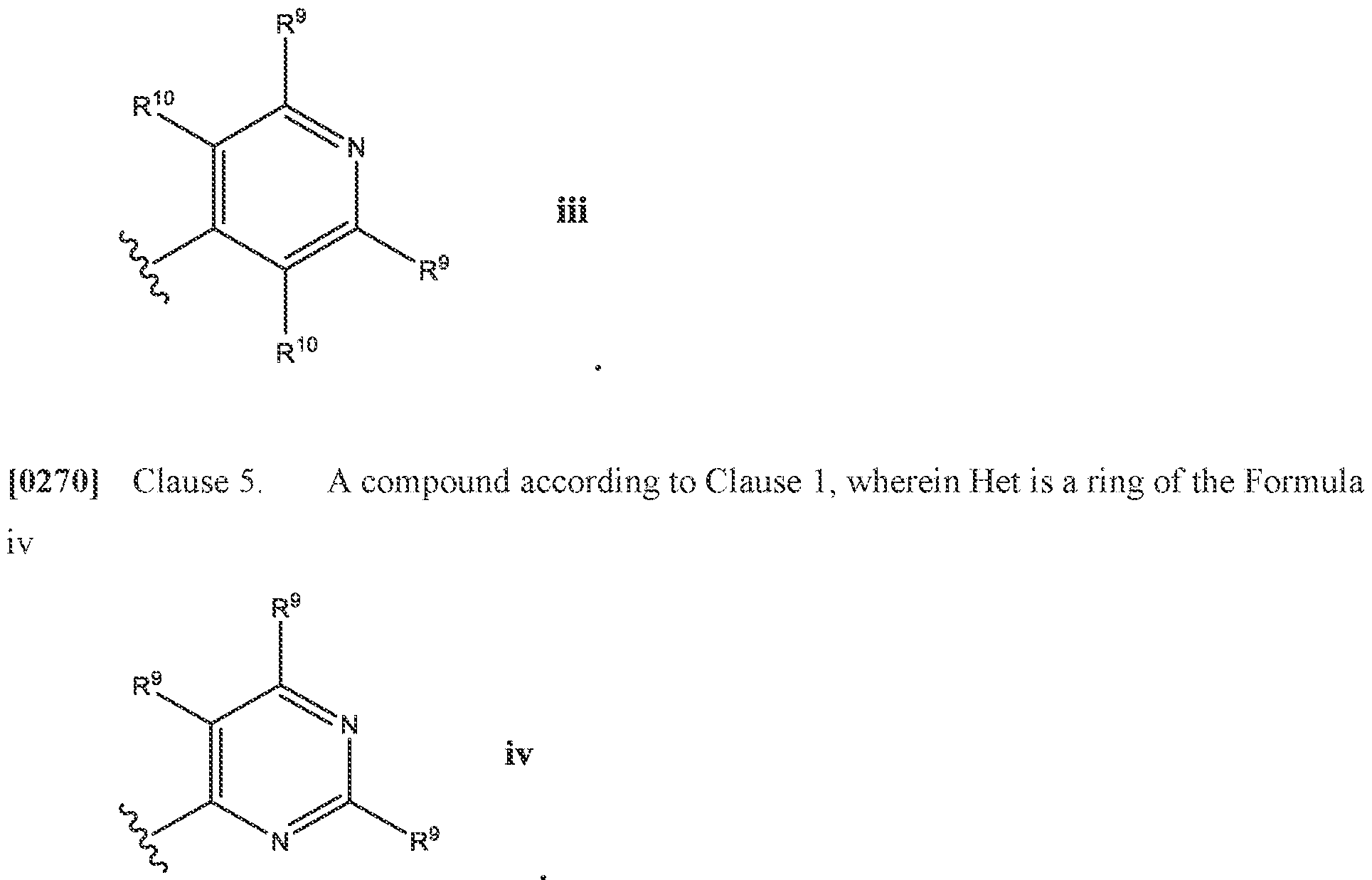

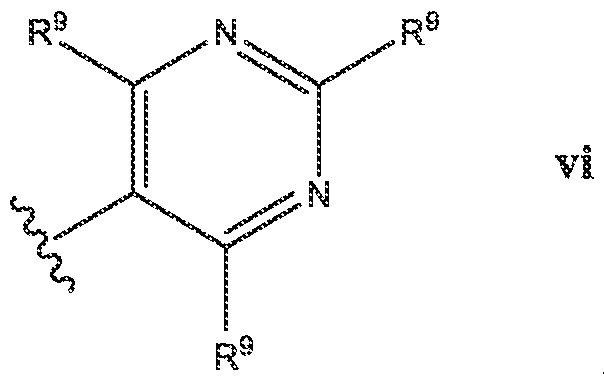
























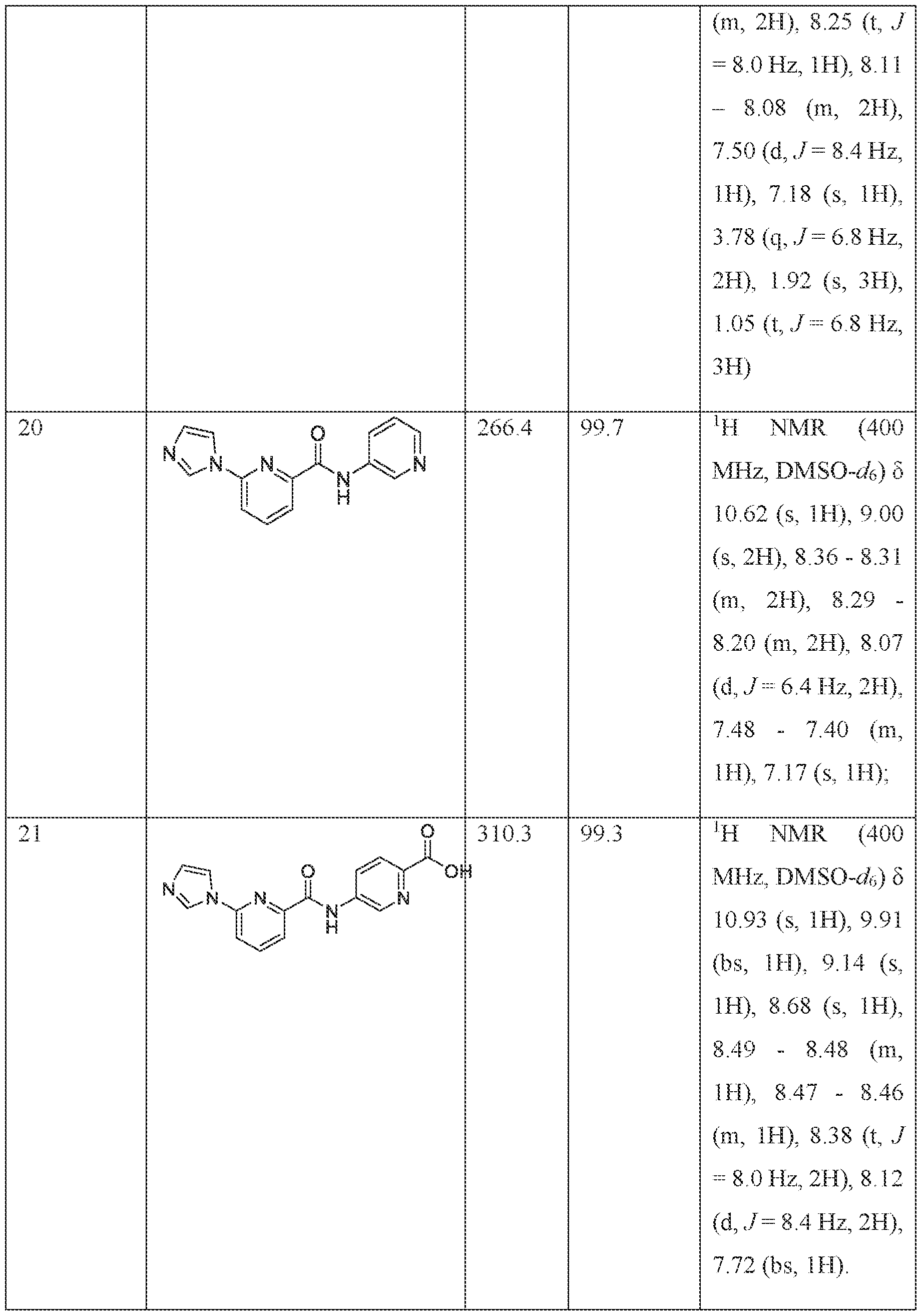


















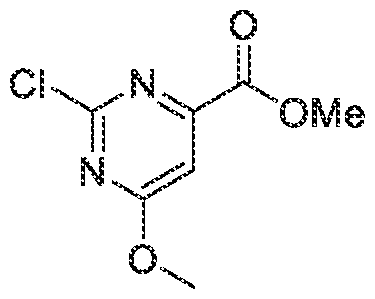





















































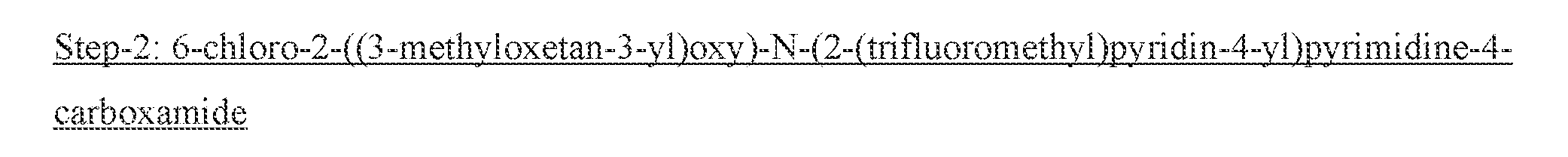





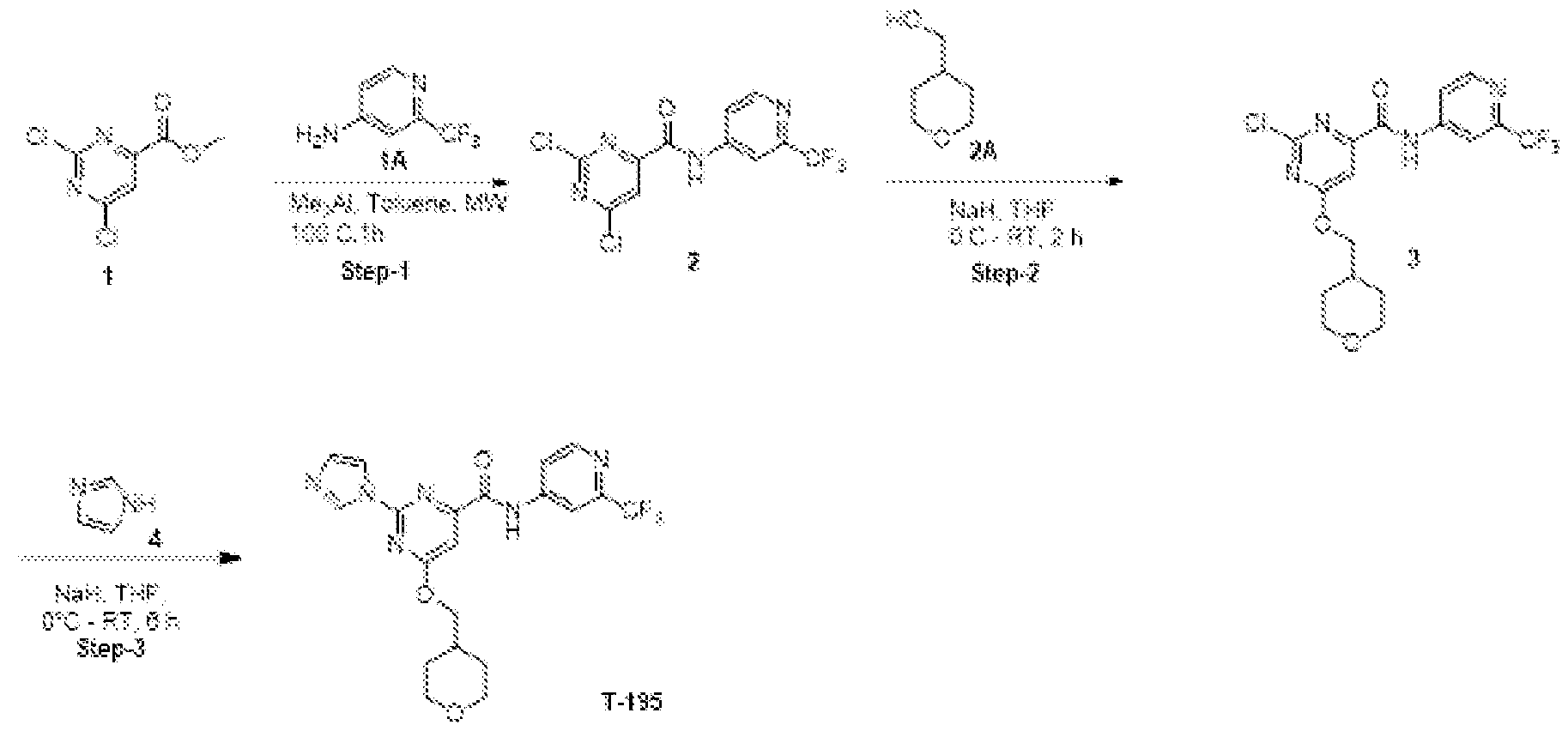





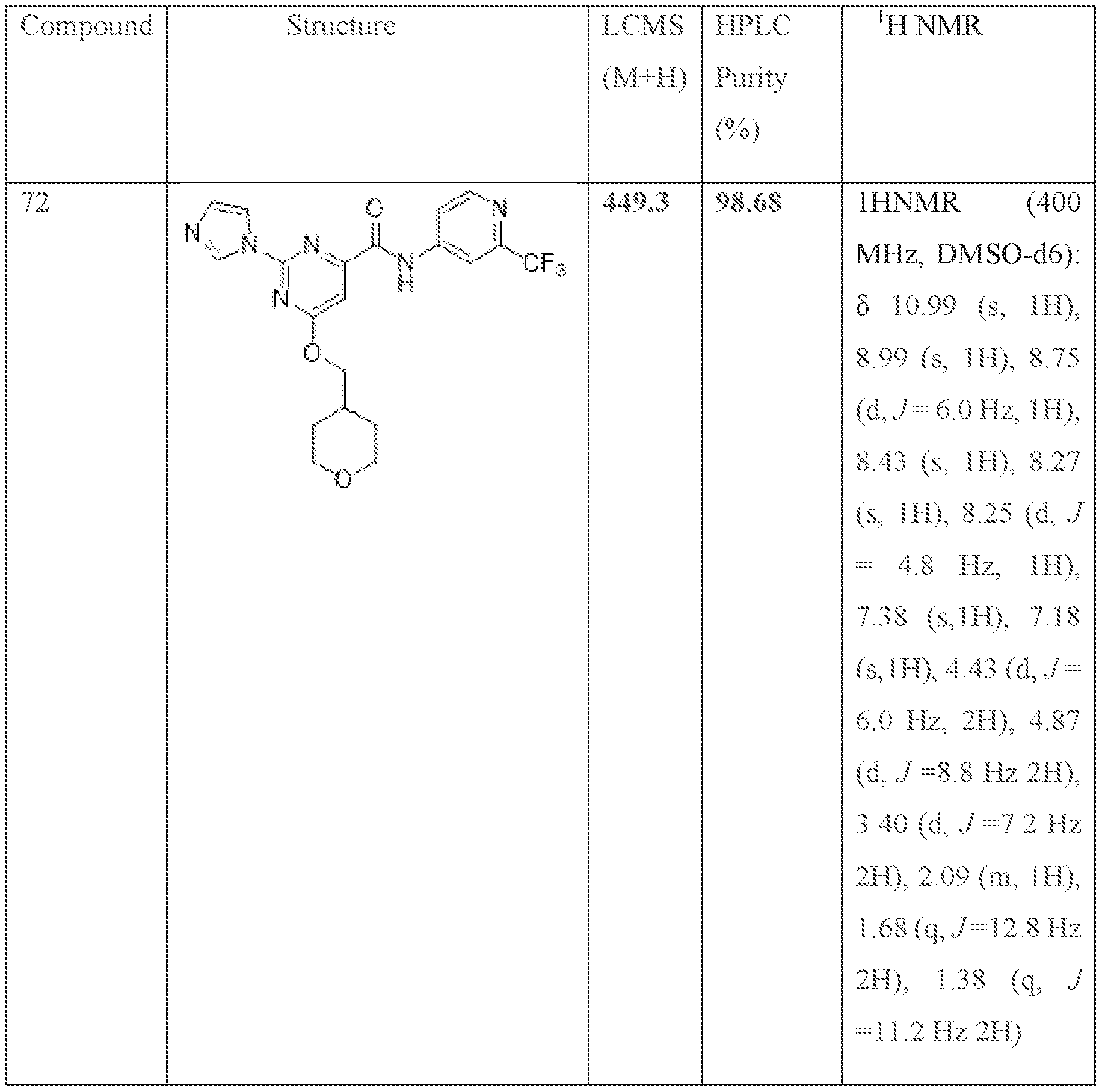












































Claims


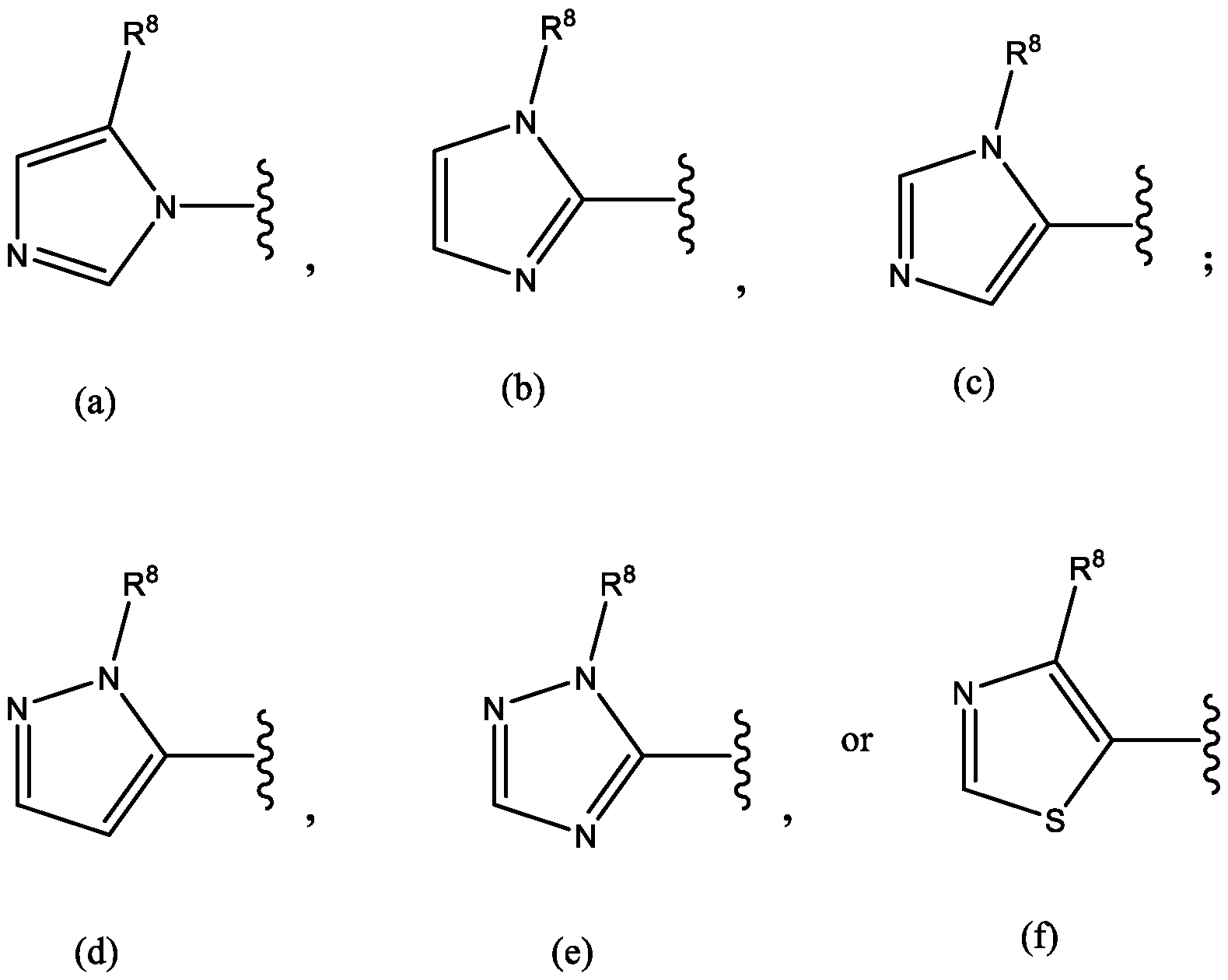





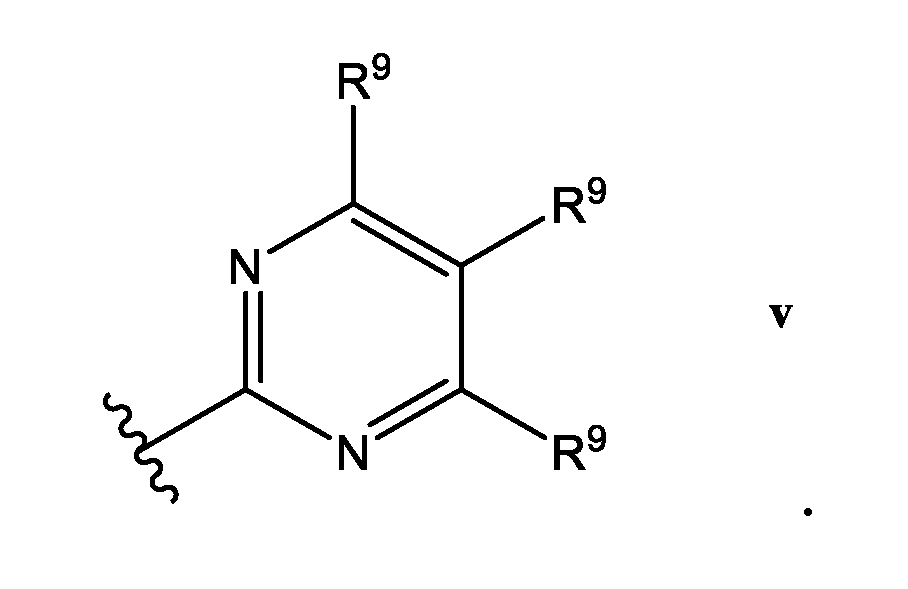


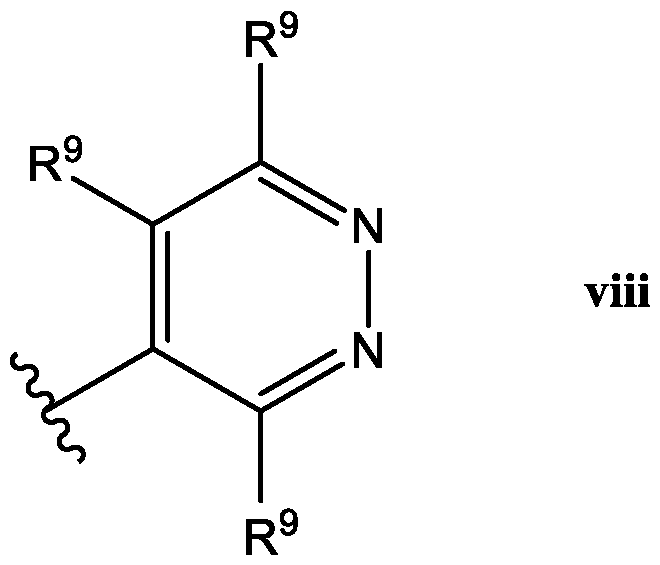







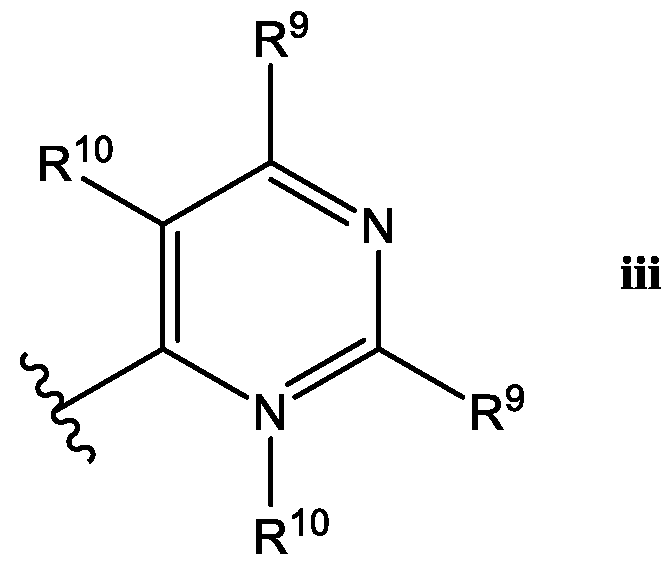




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020237015521A KR20230106605A (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitor of CD38 |
| EP21878737.2A EP4225310A4 (en) | 2020-10-09 | 2021-10-08 | HETEROARYLAMIDE INHIBITORS OF CD38 |
| AU2021356641A AU2021356641A1 (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of cd38 |
| CN202180082975.1A CN116710090A (en) | 2020-10-09 | 2021-10-08 | Heteroarylamide inhibitors of CD38 |
| IL301991A IL301991A (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of cd38 |
| JP2023547331A JP7706558B2 (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of CD38 |
| US18/248,325 US12509451B2 (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of CD38 |
| CA3195121A CA3195121A1 (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of cd38 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202063089818P | 2020-10-09 | 2020-10-09 | |
| US63/089,818 | 2020-10-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022077034A1 true WO2022077034A1 (en) | 2022-04-14 |
Family
ID=81126187
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2021/071805 Ceased WO2022077034A1 (en) | 2020-10-09 | 2021-10-08 | Heteroaryl amide inhibitors of cd38 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US12509451B2 (en) |
| EP (1) | EP4225310A4 (en) |
| JP (1) | JP7706558B2 (en) |
| KR (1) | KR20230106605A (en) |
| CN (1) | CN116710090A (en) |
| AU (1) | AU2021356641A1 (en) |
| CA (1) | CA3195121A1 (en) |
| IL (1) | IL301991A (en) |
| WO (1) | WO2022077034A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023288195A1 (en) * | 2021-07-12 | 2023-01-19 | Cytokinetics, Inc. | Cd38 modulators and methods of use thereof |
| JP2024517750A (en) * | 2021-04-30 | 2024-04-23 | ナンキン イムノファージ バイオテック カンパニー リミテッド | Compounds and their use as CD38 inhibitors - Patents.com |
| WO2024102924A1 (en) * | 2022-11-10 | 2024-05-16 | Flagship Pioneering Innovations, Vi, Llc | Inhibitors of cyclic adp ribose hydrolase methods of use thereof |
| WO2025231370A1 (en) * | 2024-05-02 | 2025-11-06 | Napa Therapeutics Limited | Inhibitors of cd38 |
| WO2026075971A1 (en) | 2024-10-01 | 2026-04-09 | Neolaia Inc. | Combination therapy for treatment of metabolic disorders and inflamation related disorders using incretin compounds and cd38 inhibitors |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011172A1 (en) | 1990-01-23 | 1991-08-08 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1994002518A1 (en) | 1992-07-27 | 1994-02-03 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1998055148A1 (en) | 1997-06-05 | 1998-12-10 | Janssen Pharmaceutica N.V. | Pharmaceutical compositions comprising cyclodextrins |
| WO2000035298A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Chewing gum containing medicament active agents |
| US6106864A (en) | 1995-09-15 | 2000-08-22 | Pfizer Inc. | Pharmaceutical formulations containing darifenacin |
| US20070149547A1 (en) | 2004-02-12 | 2007-06-28 | Celine Bonnefous | Bipyridyl amides as modulators of metabotropic glutamate receptor-5 |
| WO2009014637A2 (en) | 2007-07-19 | 2009-01-29 | Schering Corporation | Heterocyclic amide compounds as protein kinase inhibitors |
| US7919487B2 (en) | 2004-11-10 | 2011-04-05 | Synta Pharmaceuticals Corporation | Heteroaryl compounds |
| US20140249135A1 (en) | 2007-03-01 | 2014-09-04 | Novartis Ag | Pim kinase inhibitors and methods of their use |
| WO2015187499A1 (en) | 2014-06-03 | 2015-12-10 | Gilead Sciences, Inc. | Use of an ask1 inhibitor for the treatment of liver disease, optionally in combination with a loxl2 inhibitor |
| WO2018090869A1 (en) | 2016-11-16 | 2018-05-24 | 广东东阳光药业有限公司 | Amide derivative and use thereof in medicine |
| WO2019014460A1 (en) | 2017-07-12 | 2019-01-17 | The Brigham And Women's Hospital, Inc. | Eaat2 enhancing molecules |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19608653A1 (en) | 1996-03-06 | 1997-09-11 | Thomae Gmbh Dr K | Pyrimido [5,4-d] pyrimidines, medicaments containing these compounds, their use and processes for their preparation |
| CN1627944A (en) * | 2002-01-17 | 2005-06-15 | 神经能质公司 | Substituted quinazolin-4-ylamine analogs as capsaicin modulators |
| JP4116670B2 (en) * | 2004-06-28 | 2008-07-09 | インサイト コーポレイション | 3-Aminocyclopentanecarboxamide as a chemokine receptor modulator |
| MX2009009304A (en) * | 2007-03-01 | 2009-11-18 | Novartis Ag | Pim kinase inhibitors and methods of their use. |
| WO2012161877A1 (en) | 2011-05-23 | 2012-11-29 | Merck Patent Gmbh | Pyridine-and pyrazine derivatives |
| WO2013171641A1 (en) | 2012-05-15 | 2013-11-21 | Novartis Ag | Compounds and compositions for inhibiting the activity of abl1, abl2 and bcr-abl1 |
| RU2685234C1 (en) | 2013-12-09 | 2019-04-17 | Юсб Байофарма Спрл | Condensed bicyclic heteroaromatic derivatives as modulators of tnf activity |
| AU2015356721B2 (en) * | 2014-12-03 | 2018-03-15 | Glaxosmithkline Intellectual Property (No.2) Limited | CD38 inhibitors and methods of treatment |
| WO2018059549A1 (en) * | 2016-09-30 | 2018-04-05 | Novartis Ag | Immune effector cell therapies with enhanced efficacy |
| MX2020012679A (en) | 2018-05-25 | 2021-02-09 | Silverback Therapeutics Inc | Amino-pyrazinecarboxamide compounds, conjugates, and uses thereof. |
| WO2021021986A1 (en) * | 2019-07-31 | 2021-02-04 | Ribon Therapeutics, Inc. | Heterobicyclic amides as inhibitors of cd38 |
-
2021
- 2021-10-08 CN CN202180082975.1A patent/CN116710090A/en active Pending
- 2021-10-08 AU AU2021356641A patent/AU2021356641A1/en active Pending
- 2021-10-08 JP JP2023547331A patent/JP7706558B2/en active Active
- 2021-10-08 WO PCT/US2021/071805 patent/WO2022077034A1/en not_active Ceased
- 2021-10-08 KR KR1020237015521A patent/KR20230106605A/en active Pending
- 2021-10-08 CA CA3195121A patent/CA3195121A1/en active Pending
- 2021-10-08 US US18/248,325 patent/US12509451B2/en active Active
- 2021-10-08 IL IL301991A patent/IL301991A/en unknown
- 2021-10-08 EP EP21878737.2A patent/EP4225310A4/en active Pending
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011172A1 (en) | 1990-01-23 | 1991-08-08 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| WO1994002518A1 (en) | 1992-07-27 | 1994-02-03 | The University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| US6106864A (en) | 1995-09-15 | 2000-08-22 | Pfizer Inc. | Pharmaceutical formulations containing darifenacin |
| WO2000035298A1 (en) | 1996-11-27 | 2000-06-22 | Wm. Wrigley Jr. Company | Chewing gum containing medicament active agents |
| WO1998055148A1 (en) | 1997-06-05 | 1998-12-10 | Janssen Pharmaceutica N.V. | Pharmaceutical compositions comprising cyclodextrins |
| US20070149547A1 (en) | 2004-02-12 | 2007-06-28 | Celine Bonnefous | Bipyridyl amides as modulators of metabotropic glutamate receptor-5 |
| US7919487B2 (en) | 2004-11-10 | 2011-04-05 | Synta Pharmaceuticals Corporation | Heteroaryl compounds |
| US20140249135A1 (en) | 2007-03-01 | 2014-09-04 | Novartis Ag | Pim kinase inhibitors and methods of their use |
| WO2009014637A2 (en) | 2007-07-19 | 2009-01-29 | Schering Corporation | Heterocyclic amide compounds as protein kinase inhibitors |
| WO2015187499A1 (en) | 2014-06-03 | 2015-12-10 | Gilead Sciences, Inc. | Use of an ask1 inhibitor for the treatment of liver disease, optionally in combination with a loxl2 inhibitor |
| WO2018090869A1 (en) | 2016-11-16 | 2018-05-24 | 广东东阳光药业有限公司 | Amide derivative and use thereof in medicine |
| WO2019014460A1 (en) | 2017-07-12 | 2019-01-17 | The Brigham And Women's Hospital, Inc. | Eaat2 enhancing molecules |
Non-Patent Citations (75)
| Title |
|---|
| ALMARSSON, CHEM. COMMUN., 2004, pages 1889 - 1896 |
| AOYAMA ET AL., NEUROSCI. LETT, vol. 298, no. 1, 2001, pages 78 - 80 |
| ARYA ET AL., AM. J. HUM. GEN, vol. 74, no. 2, 2004, pages 272 - 282 |
| BANERJEE ET AL., J. NEUROIM. PHARMACOL, vol. 3, no. 3, 2008, pages 154 - 64 |
| BENAVENTE ET AL., CURR. PHARM. DESIGN, vol. 15, no. 1, 2009, pages 29 - 38 |
| BERTOLDO ET AL., CELL REP, vol. 30, no. 6, 2020, pages 1670 - 1681 |
| BIO. PROTOC, vol. 8, no. 14, 2018 |
| BLANCO ET AL., CAN. RES, vol. 70, no. 10, 2010, pages 3896 - 3904 |
| CAIBIN ET AL., INTERNAT. J. PHYSIOL. PATHOPHYSIOL. PHARMACOL, vol. 4, no. 1, 2012, pages 1 - 9 |
| CHAN ET AL., ANTIOX. REDOX SIG, vol. 15, no. 10, 2011, pages 2743 - 2755 |
| CHEN ET AL., CANCER DISCOV, vol. 8, no. 9, 2018, pages 1156 - 1175 |
| CHIN ET AL., TRENDS PHARMACOL. SCI, vol. 39, no. 4, 2018, pages 424 - 436 |
| CHOE ET AL., PLOS ONE, vol. 6, no. 5, 2011, pages 19046 - 388 |
| CHOI, AGING CELL, vol. 2, no. 6, 2013, pages 1062 - 72 |
| DEAGLIO ET AL., CAN. BIOL, vol. 20, no. 6, 2010, pages 416 - 423 |
| DHOPLE ET AL., MICROBIO. LETTS, vol. 28, no. 109, 1985, pages 17 - 20 |
| DRUMMOND ET AL., J. BONE MINERAL MET, vol. 24, no. 1, 2006, pages 28 - 35 |
| DURNIN ET AL., J. PHYSIOL. (OXFORD, UK, vol. 590, no. 8, 2012, pages 1921 - 1941 |
| ELLIOTT. ET AL., ANNALS NY ACAD. SCI, vol. 696, 1993, pages 333 - 41 |
| ESCANDE CARLOS ET AL., DIABETES, vol. 62, no. 4, 2013, pages 1084 - 93 |
| FINNIN ET AL., J. PHARM. SCI, vol. 88, no. 10, 1999, pages 955 - 958 |
| FREEDMAN ET AL., NEPHROL. DIALYSIS, TRANSPL, vol. 20, no. 4, 2005, pages 712 - 718 |
| FUJITA ET AL., J. PERIODONTOL, vol. 76, no. 11, 2005, pages 1960 - 5 |
| GEETA ET AL., NEUROPHARMACOL, vol. 58, no. 3, 2010, pages 585 - 92 |
| GLICK, AM. J. PHYSIOL., vol. 210, no. 6, 1966, pages 1215 - 21 |
| GONG BING ET AL., NEUROBIOL. AGING, vol. 34, no. 6, 2013, pages 1581 - 8 |
| GONZALEZ-ESCRIBANO ET AL., HUM. IMMUNOL, vol. 65, no. 6, 2004, pages 660 - 4 |
| GOODY ET AL., PLOS BIOLOGY, vol. 10, no. 10, 2012, pages 1001409 |
| GREENE: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
| HAGEMAN ET AL., FREE RADICAL BIOL. MED, vol. 35, no. 2, 2003, pages 140 - 148 |
| HALEBLIA, J. PHARM. SCI, vol. 64, no. 8, 1975, pages 1269 - 1288 |
| HARTSHORNESTUART1970EDWARD ARNOLD: "Crystals and the Polarizing Microscope", 1970 |
| HOUTKOOPER ET AL., ENDO. REV, vol. 31, no. 2, 2010, pages 194 - 223 |
| HOUTKOOPER ET AL., J. CELL BIOL., vol. 199, no. 2, 2012, pages 205 - 209 |
| JEPSON, MET. BASIS INHERITED DIS, 1960, pages 1338 - 64 |
| JORGE POSTIGO ET AL., PLOS ONE, vol. 7, no. 3, 2012, pages 33534 |
| KANG ET AL., CURR. RES. MED. REV, vol. 2, no. 2, 2006, pages 143 - 156 |
| KITAOKA ET AL., J. NEUROPATHOL. EXP. NEUROL, vol. 68, no. 8, 2009, pages 915 - 927 |
| KOCH-NOLTE ET AL., SCI. SIGNAL, vol. 2, no. 57, 2009 |
| KOENEKOOP ET AL., NAT. _GEN, vol. 44, no. 9, 2012, pages 1035 - 1039 |
| LARSON ET AL., J. BIOL. CHEM., vol. 280, no. 34, 2005, pages 30550 - 30556 |
| LAWTON ET AL., AMYOTROPHIC LATERAL SCLEROSIS, vol. 13, no. 1, 2012, pages 110 - 118 |
| LIANG ET AL., EXPERT OPINION IN THERAPEUTIC PATENTS, vol. 11, no. 6, 2001, pages 981 - 986 |
| LIEBERMAN ET AL.: "Pharmaceutical Dosage Forms: Tablets", vol. 1, 1980, MARCEL DEKKER |
| LUCY ET AL., ANN. NEUROL, vol. 51, no. 6, 2002, pages 740 - 9 |
| MARIA ET AL., FASEB J, vol. 21, no. 13, 2007, pages 3629 - 39 |
| MATALONGA ET AL., CELL REP., vol. 18, no. 5, 2017, pages 1241 - 1255 |
| MICHELS ET AL., MOL. MICROBIOL, vol. 82, no. 1, 2011, pages 4 - 8 |
| MORRIS: "Polymorphism in Pharmaceutical Solids", 1995, MACK PUBLISHING COMPANY |
| MULLER ET AL., BIOCHEM J., vol. 212, no. 2, 1983, pages 459 - 464 |
| NIK CUMMINGS ET AL., EUROPEAN JOURNAL OF HUMAN GENETICS, vol. 18, no. 11, 2010, pages 1243 - 7 |
| O'NEILL ET AL., EXPT. LUNG RES, vol. 20, no. 1, 1994, pages 41 - 56 |
| PAVON ET AL., CYTOKINE, vol. 62, no. 2, 2013, pages 232 - 243 |
| ROBINSON ET AL., BIOL. PSYCH, vol. 12, no. 1, 1977, pages 139 - 43 |
| SARTINI ET AL., J. UROL, vol. 176, no. 5, 2006, pages 2248 - 2254 |
| SCHULTZET ET AL., METH. MOL. BIO, vol. 1813, 2018, pages 77 - 90 |
| See also references of EP4225310A4 |
| SOMEYA ET AL., CELL, vol. 43, no. 5, 2010, pages 802 - 812 |
| STERN ET AL., J. BIOL. CHEM., vol. 277, no. 1, 2012, pages 602 - 608 |
| SU ET AL., EUR. RES. J, vol. 30, no. 2, 2007, pages 199 - 204 |
| TAKAHASHI ET AL., KIDNEY INT, vol. 65, no. 3, 2004, pages 1099 - 1104 |
| THAI ET AL., AM. J. RENAL PHYSIOL., vol. 297, no. 1, 2009, pages 169 - 76 |
| TRAMMELL ET AL., COMPU. STRUCT. BIOTECHNOL. J, vol. 20, 2013, pages 4, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3962138/> |
| TRAMMELL ET AL., COMPU. STRUCT. BIOTECHNOL. J., vol. 20, 2013, pages 4, Retrieved from the Internet <URL:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3962138/> |
| URAKKA IDA ET AL., PLOS GEN, vol. 7, no. 10, 2011, pages 1002333 |
| VAN WOERT, LIFE SCI, vol. 6, no. 24, 1967, pages 2605 - 12 |
| VERMA ET AL., NATURE IMMUNOLOGY, vol. 20, 2019, pages 1231 - 1243 |
| VERMA ET AL., PHARM. TECHNOL. ON-LINE, vol. 25, no. 2, 2001, pages 1 - 14 |
| VILCHEZE, MOL. MICROBIOL, vol. 76, no. 2, 2010, pages 365 - 377 |
| WETMOREET ET AL., J. BIOLOG. CHEM, vol. 285, no. 40, 2010, pages 30516 - 30522 |
| WILLIAMS, MED. HYPOTH, vol. 69, no. 3, 2007, pages 618 - 628 |
| WILSON ET AL., J. BIOL. CHEM., vol. 276, no. 14, 2001, pages 11180 - 8 |
| WOZNIACKA ET AL., SKIN PHARMACOL. PHYSIOL, vol. 20, no. 1, 2007, pages 37 - 42 |
| YAN GE ET AL., BIOCHEM. BIOPHYS. RES. COMM., vol. 399, no. 2, 2010, pages 167 - 172 |
| YOSHINO ET AL., CELL METAB, vol. 14, no. 4, 2011, pages 528 - 536 |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2024517750A (en) * | 2021-04-30 | 2024-04-23 | ナンキン イムノファージ バイオテック カンパニー リミテッド | Compounds and their use as CD38 inhibitors - Patents.com |
| EP4330247A4 (en) * | 2021-04-30 | 2025-05-28 | Nanjing Immunophage Biotech Co., Ltd | COMPOUNDS AND THEIR USES AS CD38 INHIBITORS |
| AU2022265301B2 (en) * | 2021-04-30 | 2025-11-20 | Nanjing Immunophage Biotech Co., Ltd | Compounds and their uses as cd38 inhibitors |
| WO2023288195A1 (en) * | 2021-07-12 | 2023-01-19 | Cytokinetics, Inc. | Cd38 modulators and methods of use thereof |
| WO2024102924A1 (en) * | 2022-11-10 | 2024-05-16 | Flagship Pioneering Innovations, Vi, Llc | Inhibitors of cyclic adp ribose hydrolase methods of use thereof |
| WO2025231370A1 (en) * | 2024-05-02 | 2025-11-06 | Napa Therapeutics Limited | Inhibitors of cd38 |
| WO2026075971A1 (en) | 2024-10-01 | 2026-04-09 | Neolaia Inc. | Combination therapy for treatment of metabolic disorders and inflamation related disorders using incretin compounds and cd38 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116710090A (en) | 2023-09-05 |
| IL301991A (en) | 2023-06-01 |
| JP7706558B2 (en) | 2025-07-11 |
| EP4225310A4 (en) | 2024-11-27 |
| KR20230106605A (en) | 2023-07-13 |
| JP2023550537A (en) | 2023-12-01 |
| US20230382901A1 (en) | 2023-11-30 |
| US12509451B2 (en) | 2025-12-30 |
| AU2021356641A1 (en) | 2023-06-15 |
| EP4225310A1 (en) | 2023-08-16 |
| CA3195121A1 (en) | 2022-04-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12509451B2 (en) | Heteroaryl amide inhibitors of CD38 | |
| EP2755652B1 (en) | N-substituted heterocyclyl carboxamides | |
| EP1786785B1 (en) | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors | |
| CN114585614B (en) | Non-peptide somatostatin type 5 receptor agonist and use thereof | |
| JP2024059874A (en) | Octahydrocyclopenta[c]pyrrole allosteric inhibitors of SHP2 | |
| US7183288B2 (en) | Amide derivatives as glycogen synthase kinase 3-β inhibitors | |
| KR100908155B1 (en) | Tetrahydronaphthyridine derivatives useful as histamine H3 receptor ligands | |
| US20060178374A1 (en) | Aminoheteroaryl compounds as protein kinase inhibitors | |
| WO2008025798A1 (en) | Pyridine compounds for treating gpr119 related disorders | |
| US20080207651A1 (en) | Heterocyclic compounds useful in treating diseases and conditions | |
| EP3600312B1 (en) | Piperidinyl- and piperazinyl-substituted heteroaromatic carboxamides as modulators of gpr6 | |
| SK10252003A3 (en) | Pyrimidine compounds | |
| TW200526627A (en) | New compounds | |
| JP2007505888A (en) | Substituted triazole derivatives as oxytocin antagonists | |
| CN116354931A (en) | Substituted oxazole derivatives and their application in medicine | |
| JP2008520644A (en) | Octahydropyrrolo [3,4-c] pyrrole derivative | |
| CA2804924C (en) | Substituted pyridine compound | |
| CN121335897A (en) | 1,3-thiazole and 1,2,4-thiadiazole are CD38 inhibitors used to treat CNS conditions. | |
| WO2025231370A1 (en) | Inhibitors of cd38 | |
| WO2025229624A2 (en) | New cd38 inhibitors | |
| MXPA06000989A (en) | Nicotinamide derivatives useful as pde4 inhibitors. | |
| CN112707873B (en) | Substituted oxazole derivatives and their applications in pharmaceuticals | |
| CN116354862A (en) | Substituted nitrogen heterocyclic compounds and their application in medicine | |
| HK40016591A (en) | Piperidinyl- and piperazinyl-substituted heteroaromatic carboxamides as modulators of gpr6 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21878737 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3195121 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18248325 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023547331 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021878737 Country of ref document: EP Effective date: 20230509 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202180082975.1 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2021356641 Country of ref document: AU Date of ref document: 20211008 Kind code of ref document: A |
|
| WWG | Wipo information: grant in national office |
Ref document number: 18248325 Country of ref document: US |