WO2022083679A1 - Prodrugs for sustained releasing therapeutic agents and uses thereof - Google Patents
Prodrugs for sustained releasing therapeutic agents and uses thereof Download PDFInfo
- Publication number
- WO2022083679A1 WO2022083679A1 PCT/CN2021/125237 CN2021125237W WO2022083679A1 WO 2022083679 A1 WO2022083679 A1 WO 2022083679A1 CN 2021125237 W CN2021125237 W CN 2021125237W WO 2022083679 A1 WO2022083679 A1 WO 2022083679A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pharmaceutically acceptable
- acceptable salt
- prodrug compound
- alkyl
- direct bond
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
- C07D239/545—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/553—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals with other hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms with halogen atoms or nitro radicals directly attached to ring carbon atoms, e.g. fluorouracil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/645—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having two nitrogen atoms as the only ring hetero atoms
- C07F9/6509—Six-membered rings
- C07F9/6512—Six-membered rings having the nitrogen atoms in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/24—Condensed ring systems having three or more rings
- C07H15/252—Naphthacene radicals, e.g. daunomycins, adriamycins
Definitions
- the present disclosure relates to prodrugs for sustained releasing therapeutic agents, and methods for using such prodrugs for the treatment of diseases.
- the therapeutic agents are delivered to the body systemically via oral/GI absorption or systemic injection. Then the therapeutic agents are delivered to the action site by the blood circulation, thus, the therapeutic agent are exposed to the entire body. In some cases, the unintended exposure of the therapeutic agents in the other parts of the body may cause side effects, sometimes serious side effects.
- many locally delivered dosage forms are developed.
- One good example is the inhalation dosage form to treat respiratory diseases, such as COPD and asthma.
- These therapeutic agents target receptors on the airway and make the airway open for more efficient oxygen exchange.
- local delivered drugs such as topical corticosteroids for skin rash, intravitreal injection of anti-VEGF for wet AMD (age-related macular degeneration) , intra-articular injection of corticosteroids to treat osteoarthritis.
- the purpose of these local delivered dosage form is to have high local drug concentration to have the therapeutic effective, yet to reduce systemic exposure of the drug to have minimum side effects.
- Local delivery dosage form are difficult to use in most cases, especially the locally injected dosage forms. Therefore, it is desirable to have the delivery system, e.g., sustained or controlled release dosage form, that can release the drug slowly to have prolonged duration to reduce the frequency of dosing.
- the prodrug approach is one of the sustained release mechanism often used by people known in this field.
- Drugs in the systemic circulation are metabolized and eliminated from the system by enzymes or excretion.
- a peak concentration much higher than the effective concentration is needed so that the trough between each dosing would be above the effective concentrations.
- the peak concentration can cause undesirable side effect.
- the most popular approach is to use polymers to slow down the drug release from the dosing system. Many orally administrated sustained release/controlled release systems using this approach. There are pros and cons with this approach in some incidents, especially for locally injected delivery system targeting release duration in weeks or months. Using polymer system to control drug release may also have challenges in manufacturing process and quality control of the products.
- Another approach to have more precise control of the drug release is to have a prodrug that can release the drug via chemical bond breakage.
- the release mechanism is essentially a chemical reaction, more specifically, it’s a first order chemical reaction.
- the rate of the drug release is much more predictable due to consistency of the environment in the biology system.
- by controlling the solubility of the prodrug it is possible to create a system that can release the drug in a zero order or pseudo-zero manner using solubility as rate limiting step.
- the present disclosure is related to a prodrug system that controls drug release rate via chemical structure of the prodrug moiety for the intended location and indication.
- the present disclosure provides a prodrug compound comprising a parent drug moiety and a tail moiety, wherein
- the parent drug moiety is derived from a parent drug comprising a reactive group selected from the group consisting of amine, amino, hydroxyl, carboxylate, ketone, and amide,
- the tail moiety is covalently linked to the parent drug moiety and has a Formula (I) :
- L 1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage
- L is a direct bond or alkyl
- U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
- V is a direct bond or alkyl
- W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
- Z is selected from the group consisting of a direct bond, alkyl, aryl, NR 1 R 2 , and OR 3 , wherein said alkyl and aryl are optionally substituted with one or more R 4 ;
- R 1 , R 2 and R 3 are independently hydrogen, alkyl or cycloalkyl
- R 4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,
- the present disclosure provides a prodrug compound having a formula selected from the group consisting of:
- the present disclosure provides a prodrug compound having a formula selected from the group consisting of:
- the present disclosure provides a prodrug compound having a formula of:
- the present disclosure provides a prodrug compound having a formula of:
- the present disclosure provides a pharmaceutical composition
- a pharmaceutical composition comprising the prodrug compound provided herein or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable excipient.
- the present disclosure provides a method of treating diseases in a subject in need thereof, comprising administering to the subject a therapeutic effective amount of the prodrug compound provided herein or pharmaceutically acceptable salts thereof, or the pharmaceutical composition provided herein.
- Figures 1 and 2 depict the plasma and gastric concentration of fluorouracil following the continuous intragastric administration of the exemplary compound of the present disclosure and the continuous intravenous infusion or single oral administration of free fluorouracil to male Sprague Dawley rats, respectively
- linking substituents are described. It is specifically intended that each linking substituent includes both the forward and backward forms of the linking substituent.
- -NR (CR’R”) -includes both -NR (CR’R”) -and - (CR’R”) NR-.
- the Markush variables listed for that group are understood to be linking groups. For example, if the structure requires a linking group and the Markush group definition for that variable lists “alkyl” , then it is understood that the “alkyl” represents a linking alkylene group.
- any variable e.g., R i
- its definition at each occurrence is independent of its definition at every other occurrence.
- R i the definition at each occurrence is independent of its definition at every other occurrence.
- the group may optionally be substituted with up to two R i moieties and R i at each occurrence is selected independently from the definition of R i .
- combinations of substituents and/or variables are permissible, but only if such combinations result in stable compounds.
- C i-j indicates a range of the carbon atoms numbers, wherein i and j are integers and the range of the carbon atoms numbers includes the endpoints (i.e. i and j) and each integer point in between, and wherein j is greater than i.
- C 1-6 indicates a range of one to six carbon atoms, including one carbon atom, two carbon atoms, three carbon atoms, four carbon atoms, five carbon atoms and six carbon atoms.
- the term “C 1-12 ” indicates 1 to 12, particularly 1 to 10, particularly 1 to 8, particularly 1 to 6, particularly 1 to 5, particularly 1 to 4, particularly 1 to 3 or particularly 1 to 2 carbon atoms.
- alkyl refers to a saturated linear or branched-chain hydrocarbon radical, which may be optionally substituted independently with one or more substituents described below.
- C i-j alkyl refers to an alkyl having i to j carbon atoms.
- alkyl groups contain 1 to 10 carbon atoms.
- alkyl groups contain 1 to 9 carbon atoms.
- alkyl groups contain 1 to 8 carbon atoms, 1 to 7 carbon atoms, 1 to 6 carbon atoms, 1 to 5 carbon atoms, 1 to 4 carbon atoms, 1 to 3 carbon atoms, or 1 to 2 carbon atoms.
- C 1-10 alkyl examples include, but are not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, and decyl.
- C 1-6 alkyl are methyl, ethyl, propyl, isopropyl, n-butyl, i-butyl, s-butyl, t-butyl, n-pentyl, 2-pentyl, 3-pentyl, 2-methyl-2-butyl, 3-methyl-2-butyl, 3-methyl-1-butyl, 2-methyl-1-butyl, 1-hexyl, 2-hexyl, 3-hexyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 3-methyl-3-pentyl, 2-methyl-3-pentyl, 2, 3-dimethyl-2-butyl, 3, 3-dimethyl-2-butyl, and the like.
- alkenyl refers to linear or branched-chain hydrocarbon radical having at least one carbon-carbon double bond, which may be optionally substituted independently with one or more substituents described herein, and includes radicals having “cis” and “trans” orientations, or alternatively, “E” and “Z” orientations.
- alkenyl groups contain 2 to 12 carbon atoms. In some embodiments, alkenyl groups contain 2 to 11 carbon atoms.
- alkenyl groups contain 2 to 11 carbon atoms, 2 to 10 carbon atoms, 2 to 9 carbon atoms, 2 to 8 carbon atoms, 2 to 7 carbon atoms, 2 to 6 carbon atoms, 2 to 5 carbon atoms, 2 to 4 carbon atoms, 2 to 3 carbon atoms, and in some embodiments, alkenyl groups contain 2 carbon atoms.
- alkenyl group include, but are not limited to, ethylenyl (or vinyl) , propenyl (allyl) , butenyl, pentenyl, 1-methyl-2 buten-1-yl, 5-hexenyl, and the like.
- alkynyl refers to a linear or branched hydrocarbon radical having at least one carbon-carbon triple bond, which may be optionally substituted independently with one or more substituents described herein.
- alkenyl groups contain 2 to 12 carbon atoms. In some embodiments, alkynyl groups contain 2 to 11 carbon atoms.
- alkynyl groups contain 2 to 11 carbon atoms, 2 to 10 carbon atoms, 2 to 9 carbon atoms, 2 to 8 carbon atoms, 2 to 7 carbon atoms, 2 to 6 carbon atoms, 2 to 5 carbon atoms, 2 to 4 carbon atoms, 2 to 3 carbon atoms, and in some embodiments, alkynyl groups contain 2 carbon atoms.
- alkynyl group include, but are not limited to, ethynyl, 1-propynyl, 2-propynyl, and the like.
- amine refers to derivatives of ammonia, wherein one or more hydrogen atoms are replaced by a substituent, and can be represented by N (H) n (R’) 3-n wherein n is 0, 1, or 2, and each R’ is independently hydroxyl, nitro, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups, or two R’ together with the nitrogen atom to which they are attached form an optionally substituted heterocyclyl or heteroaryl.
- amino refers to —NH 2 .
- acetal refers to –O-CH (R’) -O-, wherein R’ represents alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
- aryl refers to monocyclic and polycyclic ring systems having a total of 5 to 20 ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 12 ring members.
- aryl include, but are not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Also included within the scope of the term “aryl” , as it is used herein, is a group in which an aromatic ring is fused to one or more additional rings.
- polycyclic ring system In the case of polycyclic ring system, only one of the rings needs to be aromatic (e.g., 2, 3-dihydroindole) , although all of the rings may be aromatic (e.g., quinoline) .
- the second ring can also be fused or bridged.
- polycyclic aryl include, but are not limited to, benzofuranyl, indanyl, phthalimidyl, naphthimidyl, phenanthridinyl, or tetrahydronaphthyl, and the like.
- Aryl groups can be substituted at one or more ring positions with substituents as described above.
- cycloalkyl refers to a monovalent non-aromatic, saturated or partially unsaturated monocyclic and polycyclic ring system, in which all the ring atoms are carbon and which contains at least three ring forming carbon atoms.
- the cycloalkyl may contain 3 to 12 ring forming carbon atoms, 3 to 10 ring forming carbon atoms, 3 to 9 ring forming carbon atoms, 3 to 8 ring forming carbon atoms, 3 to 7 ring forming carbon atoms, 3 to 6 ring forming carbon atoms, 3 to 5 ring forming carbon atoms, 4 to 12 ring forming carbon atoms, 4 to 10 ring forming carbon atoms, 4 to 9 ring forming carbon atoms, 4 to 8 ring forming carbon atoms, 4 to 7 ring forming carbon atoms, 4 to 6 ring forming carbon atoms, 4 to 5 ring forming carbon atoms.
- Cycloalkyl groups may be saturated or partially unsaturated. Cycloalkyl groups may be substituted. In some embodiments, the cycloalkyl group may be a saturated cyclic alkyl group. In some embodiments, the cycloalkyl group may be a partially unsaturated cyclic alkyl group that contains at least one double bond or triple bond in its ring system. In some embodiments, the cycloalkyl group may be monocyclic or polycyclic.
- Examples of monocyclic cycloalkyl group include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl and cyclododecyl.
- polycyclic cycloalkyl group examples include, but are not limited to, adamantyl, norbornyl, fluorenyl, spiro-pentadienyl, spiro [3.6] -decanyl, bicyclo [1, 1, 1] pentenyl, bicyclo [2, 2, 1] heptenyl, and the like.
- heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen (including N-oxides) .
- heteroaryl refers to an aryl group having, in addition to carbon atoms, one or more heteroatoms.
- the heteroaryl group can be monocyclic. Examples of monocyclic heteroaryl include, but are not limited to, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, benzofuranyl and pteridinyl.
- the heteroaryl group also includes polycyclic groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring.
- polycyclic heteroaryl include, but are not limited to, indolyl, isoindolyl, benzothienyl, benzofuranyl, benzo [1, 3] dioxolyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzthiazolyl, quinolyl, isoquinolyl, dihydroquinolinyl, dihydroisoquinolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H-quinolizinyl, carbazolyl, acridinyl
- heterocyclyl refers to a saturated or partially unsaturated carbocyclyl group in which one or more ring atoms are heteroatoms independently selected from oxygen, sulfur, nitrogen, phosphorus, and the like, the remaining ring atoms being carbon, wherein one or more ring atoms may be optionally substituted independently with one or more substituents.
- the heterocyclyl is a saturated heterocyclyl.
- the heterocyclyl is a partially unsaturated heterocyclyl having one or more double bonds in its ring system.
- the heterocyclyl may contains any oxidized form of carbon, nitrogen or sulfur, and any quaternized form of a basic nitrogen.
- Heterocyclyl also includes radicals wherein the heterocyclyl radicals are fused with a saturated, partially unsaturated, or fully unsaturated (i.e., aromatic) carbocyclic or heterocyclic ring.
- the heterocyclyl radical may be carbon linked or nitrogen linked where such is possible.
- the heterocycle is carbon linked.
- the heterocycle is nitrogen linked.
- a group derived from pyrrole may be pyrrol-1-yl (nitrogen linked) or pyrrol-3-yl (carbon linked) .
- a group derived from imidazole may be imidazol-1-yl (nitrogen linked) or imidazol-3-yl (carbon linked) .
- 3-to 12-membered heterocyclyl refers to a 3-to 12-membered saturated or partially unsaturated monocyclic or polycyclic heterocyclic ring system having 1 to 3 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- the fused, spiro and bridged ring systems are also included within the scope of this definition.
- monocyclic heterocyclyl examples include, but are not limited to oxetanyl, 1, 1-dioxothietanylpyrrolidyl, tetrahydrofuryl, tetrahydrothienyl, pyrrolyl, furanyl, thienyl, pyrazolyl, imidazolyl, triazolyl, oxazolyl, thiazolyl, piperidyl, piperazinyl, piperidinyl, morpholinyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, pyridonyl, pyrimidonyl, pyrazinonyl, pyrimidonyl, pyridazonyl, pyrrolidinyl, triazinonyl, and the like.
- fused heterocyclyl examples include, but are not limited to, phenyl fused ring or pyridinyl fused ring, such as quinolinyl, isoquinolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, quinoxalinyl, quinolizinyl, quinazolinyl, azaindolizinyl, pteridinyl, chromenyl, isochromenyl, indolyl, isoindolyl, indolizinyl, indazolyl, purinyl, benzofuranyl, isobenzofuranyl, benzimidazolyl, benzothienyl, benzothiazolyl, carbazolyl, phenazinyl, phenothiazinyl, phenanthridinyl, imidazo [1, 2-a] pyridinyl, [1, 2, 4] triazolo [4, 3-a
- spiro heterocyclyl examples include, but are not limited to, spiropyranyl, spirooxazinyl, and the like.
- bridged heterocyclyl examples include, but are not limited to, morphanyl, hexamethylenetetraminyl, 3-aza-bicyclo [3.1.0] hexane, 8-aza-bicyclo [3.2.1] octane, 1-aza-bicyclo [2.2.2] octane, 1, 4-diazabicyclo [2.2.2] octane (DABCO) , and the like.
- hydroxyl refers to —OH.
- linkage refers to bonds or chemical moiety formed from a chemical reaction between the functional groups of at least two entities to be linked, thereby forming one molecule or maintaining association of the entities in sufficiently close proximity.
- a linkage can be integrated in the resulting linked molecule or structure, with or without its reacted functional groups.
- Such linkages may be covalent or non-covalent.
- Hydrolytically unstable or degradable linkages mean that the linkages are degradable in water or in aqueous solutions, including for example, body fluid such as blood.
- Enzymatically unstable or degradable linkages mean that the linkage can be degraded by one or more enzymes.
- Such degradable linkages include, but are not limited to ester linkages formed by the carboxylic acid in one entity with alcohol groups on a biologically active agent, wherein such ester groups generally hydrolyze under physiological conditions to release the biologically active agent.
- Other hydrolytically degradable linkages include but are not limited to carbonate linkages, imine linkages resulted from reaction of an amine and an aldehyde, phosphate ester linkages resulted from reaction of a phosphate group and an alcohol, hydrazone linkages resulted from reaction of a hydrazide and an aldehyde, acetal linkages resulted from reaction of an aldehyde and an alcohol, amide linkages resulted from reaction of an amine group and a carboxyl group.
- partially unsaturated refers to a radical that includes at least one double or triple bond.
- partially unsaturated is intended to encompass rings having multiple sites of unsaturation, but is not intended to include aromatic (i.e., fully unsaturated) moieties.
- the term “pharmaceutically acceptable” indicates that the substance or composition is compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the subjects being treated therewith.
- substitution or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and that the substitution results in a stable or chemically feasible compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc.
- an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. It will be understood by those skilled in the art that substituents can themselves be substituted, if appropriate. Unless specifically stated as “unsubstituted” , references to chemical moieties herein are understood to include substituted variants. For example, reference to an “aryl” group or moiety implicitly includes both substituted and unsubstituted variants.
- therapeutic agent refers to any substance which can affect any physical or biochemical properties of a biological organism, including but not limited to viruses, bacteria, fungi, plants, animals, and human.
- therapeutic agents include any substance intended for diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, or to otherwise enhance physical or mental well-being of humans or animals.
- Drug delivery of therapeutic agents to specific tissues or sites within a body presents a variety of challenges, particularly where local delivery of a therapeutic agent to a specific tissue and sustained release of the therapeutic agent are desired, and where avoidance of high systemic concentration of the therapeutic agent leading to toxic side effects is desired.
- the present disclosure provides a prodrug compound capable of locally delivering a therapeutic agent and releasing the therapeutic agent in a controlled and sustained manner with reduced systemic side effect. This is achieved by deliberately designing a prodrug compound which achieves a balance of solubility of the prodrug compound and the rate of the prodrug compound for releasing the parent drug.
- the present disclosure provides a prodrug compound comprising a parent drug moiety and a tail moiety, wherein
- the parent drug moiety is derived from a parent drug comprising a reactive group selected from the group consisting of amine, amino, hydroxyl, and amide,
- the tail moiety is covalently linked to the parent drug moiety and has a Formula (I) :
- L 1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage
- L is a direct bond or alkyl
- U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
- V is a direct bond or alkyl
- W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
- Z is selected from the group consisting of a direct bond, alkyl, aryl, NR 1 R 2 , and OR 3 , wherein said alkyl and aryl are optionally substituted with one or more R 4 ;
- R 1 , R 2 and R 3 are independently hydrogen, alkyl or cycloalkyl
- R 4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,
- the parent drug comprises at least one reactive group, which is capable of reacting with a reactive functional group of a second entity (e.g., the tail moiety provided herein) and an optional co-reactant to form a cleavable linkage, thereby linking the parent drug moiety to the second entity (e.g., the tail moiety) .
- a reactive functional group of a second entity e.g., the tail moiety provided herein
- an optional co-reactant to form a cleavable linkage, thereby linking the parent drug moiety to the second entity (e.g., the tail moiety) .
- a "cleavable linkage” is a relatively labile bond that cleaves under physiological conditions.
- An exemplary releasable linkage is a hydrolyzable bond that cleaves upon reaction with water (i.e., is hydrolyzed) .
- the tendency of a bond to hydrolyze in water may depend not only on the general type of linkage connecting two atoms but also on the substituents attached to these atoms.
- Appropriate hydrolytically unstable or weak linkages include but are not limited to carboxylate ester, phosphate ester, anhydrides, acetals, ketals, amide, acyloxyalkyl ether, imines, hydrazone, orthoesters, peptides, oligonucleotides, thioesters, urea, thiourea, carbamate, thiocabamate, phosphoramidate, phosphonamidate, carbonates and thiocarbonate.
- Certain functional groups have atoms that may be chemically degraded by a process other than hydrolysis. Exemplary releasable linkages in this category include certain carbamates and Fmoc derivatives.
- cleavable linkage is an enzymatically cleavable linkage.
- An "enzymatically cleavable linkage” means a linkage that is subject to cleavage by one or more enzymes.
- the parent drug is selected from the group consisting of anti-cancer agent, anti-inflammatory drugs, antibiotics, anti-fugus agent, JAK inhibitors, and VEGF inhibitors.
- the parent drug is an anti-cancer agent.
- the parent drug is an anti-inflammatory drug.
- the parent drug is an antibiotic.
- the parent drug is an anti-fugus agent.
- the parent drug is selected from the group consisting of Fluorouracil, Temozolomide, Daunorubicin, 10-hydroxyl-camptothecine, and 7-ethyl-10-hydroxyl-camptothecine.
- the reactive group of the parent drug reacts with the reactive functional group of the tail moiety and an optional co-reactant to form a cleavable linkage selected from the group consisting of carbonate, thiocarbonate, carbamate, thiocarbamate, carboxylate ester, phosphate ester, amide, imine, hydrazone, phosphonamidate and acetal.
- L is a direct bond
- L is alkyl. In certain embodiments, L is C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl, or C 1-2 alkyl.
- L is methyl, ethyl or hexyl.
- U is a direct bond
- U is heterocyclyl. In certain embodiments, U is saturated heterocyclyl. In certain embodiments, U is partially unsaturated heterocyclyl.
- U is 5-to 12-membered heterocyclyl, 5-to 11-membered heterocyclyl, 5-to 10-membered heterocyclyl, 5-to 9-membered heterocyclyl, 5-to 8-membered heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered heterocyclyl.
- U is 5-to 12-membered saturated heterocyclyl, 5-to 11-membered saturated heterocyclyl, 5-to 10-membered saturated heterocyclyl, 5-to 9-membered saturated heterocyclyl, 5-to 8-membered saturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered saturated heterocyclyl.
- U is piperidinyl
- U is 5-to 12-membered partially unsaturated heterocyclyl, 5-to 11-membered partially unsaturated heterocyclyl, 5-to 10-membered partially unsaturated heterocyclyl, 5-to 9-membered partially unsaturated heterocyclyl, 5-to 8-membered partially unsaturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered partially unsaturated heterocyclyl.
- U is 1, 2, 3, 4-tetrahydro-isoquinolinyl.
- U is aryl. In certain embodiments, U is 5 to 12 membered aryl, 5 to 10 membered aryl, 5 to 8 membered aryl, or 5 to 6 membered aryl.
- U is phenyl
- L is a direct bond
- U is a direct bond, heterocyclyl or aryl.
- L is a direct bond
- U is a direct bond, 5-to 12-membered saturated or partially unsaturated heterocyclyl, or 5 to 12 membered aryl.
- L is a direct bond
- U is selected from the group consisting of a direct bond, 1, 2, 3, 4-tetrahydro-isoquinolinyl and phenyl.
- L is alkyl
- U is a direct bond, heterocyclyl or aryl.
- L is C 1-6 alkyl
- U is a direct bond, 5-to 12-membered saturated or partially unsaturated heterocyclyl, or 5 to 12 membered aryl.
- L is C 1-6 alkyl
- U is selected from the group consisting of a direct bond, piperidinyl and phenyl.
- V is a direct bond
- V is alkyl. In certain embodiments, V is C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl, or C 1-2 alkyl.
- V is methyl
- W is a direct bond
- W is aryl. In certain embodiments, W is 5 to 12 membered aryl, 5 to 10 membered aryl, 5 to 8 membered aryl, or 5 to 6 membered aryl.
- W is phenyl
- W is heterocyclyl. In certain embodiments, W is saturated heterocyclyl. In certain embodiments, W is partially unsaturated heterocyclyl.
- W is 5-to 12-membered heterocyclyl, 5-to 11-membered heterocyclyl, 5-to 10-membered heterocyclyl, 5-to 9-membered heterocyclyl, 5-to 8-membered heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered heterocyclyl.
- W is 5-to 12-membered saturated heterocyclyl, 5-to 11-membered saturated heterocyclyl, 5-to 10-membered saturated heterocyclyl, 5-to 9-membered saturated heterocyclyl, 5-to 8-membered saturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered saturated heterocyclyl.
- W is pyrrolidinyl, piperidinyl or piperazinyl.
- Z is a direct bond
- Z is alkyl optionally substituted with one or more R 4 . In certain embodiments, Z is C 1-8 alkyl optionally substituted with one or more R 4 .
- R 4 is cycloalkyl or aryl.
- Z is C 1-8 alkyl.
- Z is C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl or C 1- 2 alkyl, each of which is substituted with one or two R 4 , and R 4 is independently cycloalkyl or aryl.
- Z is methyl or ethyl, which is substituted with one or two R 4 , and R 4 is independently adamantyl or phenyl.
- Z is aryl optionally substituted with one or more R 4 . In certain embodiments, Z is 5-to 12-membered aryl optionally substituted with one or more R 4 .
- R 4 is alkyl
- Z is phenyl optionally substituted with one or more R 4 , wherein R 4 is C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl, or C 1-2 alkyl.
- Z is phenyl substituted with one or more R 4 , wherein R 4 is methyl or ethyl.
- Z is NR 1 R 2 .
- R 1 and R 2 are independently alkyl or cycloalkyl.
- R 1 and R 2 are independently C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl, or C 1-2 alkyl.
- R 1 and R 2 are independently C 3-6 cycloalkyl.
- Z is OR 3 .
- R 3 is alkyl. In certain embodiments, R 3 is C 1-6 alkyl, C 1-5 alkyl, C 1-4 alkyl, C 1-3 alkyl, or C 1-2 alkyl. In certain embodiments, R 3 is methyl.
- the present disclosure provides a prodrug compound having a formula selected from the group consisting of:
- prodrug compound having a Formula (II) , (III) , (IV) or (V) in prodrug compound having a Formula (II) , (III) , (IV) or (V) ,
- U is heterocyclyl, aryl or heteroaryl
- V is a direct bond or alkyl
- W is a direct bond, heterocyclyl or aryl
- Z is alkyl, aryl, NR 1 R 2 , or OR 3 , wherein said alkyl and aryl are optionally substituted with one or more R 4 ,
- R 1 , R 2 , R 3 and R 4 are defined as supra.
- the present disclosure provides a prodrug compound having a formula selected from the group consisting of:
- prodrug compound having a Formula (VI) , (VII) , (VIII) or (IX) in prodrug compound having a Formula (VI) , (VII) , (VIII) or (IX) ,
- L is a direct bond or alkyl
- U is a direct or aryl
- V is a direct bond
- W is a direct bond or heterocyclyl
- Z is NR 1 R 2 or alkyl optionally substituted with one or more R 4 .
- R 1 , R 2 and R 4 are defined as supra.
- the present disclosure provides a prodrug compound having a formula of:
- the present disclosure provides a prodrug compound having a formula of:
- the prodrug compound provided herein has a lower solubility than the parent drug at biological pH.
- the present disclosure provides a prodrug compound selected from the group consisting of:
- prodrug compounds provided herein are described with reference to both generic formulae and specific compounds.
- the prodrug compounds of the present disclosure may exist in a number of different forms or derivatives, all within the scope of the present disclosure. These include, for example, tautomers, stereoisomers, racemic mixtures, regioisomers, salts, solvated forms, amorphous forms, different crystal forms or polymorphs.
- the prodrug compounds of present disclosure can comprise one or more asymmetric centers depending on substituent selection, and thus can exist in various stereoisomeric forms, e.g., enantiomers and/or diastereomers.
- the prodrug compounds provided herein may have an asymmetric carbon center, and thus compounds provided herein may have either the (R) or (S) stereo-configuration at a carbon asymmetric center. Therefore, the prodrug compounds of the present disclosure may be in the form of an individual enantiomer, diastereomer or geometric isomer, or may be in the form of a mixture of stereoisomers.
- the term “enantiomer” refers to two stereoisomers of a compound which are non-superimposable mirror images of one another.
- the term “diastereomer” refers to a pair of optical isomers which are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities.
- a particular enantiomer may, in some embodiments be provided substantially free of the opposite enantiomer, and may also be referred to as “optically enriched” .
- “Optically enriched” means that the compound is made up of a significantly greater proportion of one enantiomer. In certain embodiments, the compound is made up of at least about 90%by weight of a preferred enantiomer. In other embodiments, the compound is made up of at least about 95%, 98%, or 99%by weight of a preferred enantiomer.
- Preferred enantiomers may be isolated from racemic mixtures by any method known to those skilled in the art, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization can be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound provided herein or it can be done on a final racemic product.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration.
- absolute stereochemistry may be determined by Vibrational Circular Dichroism (VCD) spectroscopy analysis.
- VCD Vibrational Circular Dichroism
- mixtures of diastereomers for example mixtures of diastereomers enriched with 51%or more of one of the diastereomers, including for example 60%or more, 70%or more, 80%or more, or 90%or more of one of the diastereomers are provided.
- the present disclosure additionally encompasses the compounds as individual isomers substantially free of other isomers and alternatively, as mixtures of various isomers, e.g., racemic mixtures of enantiomers.
- prodrug compounds of the present disclosure may also exist in different tautomeric forms, and all such forms are embraced within the scope of the present disclosure.
- tautomer or “tautomeric form” refers to structural isomers of different energies which are interconvertible via a low energy barrier.
- proton tautomers include interconversions via migration of a proton, such as keto-enol, amide-imidic acid, lactam-lactim, imine-enamine isomerizations and annular forms where a proton can occupy two or more positions of a heterocyclic system (for example, 1H-and 3H-imidazole, 1H-, 2H-and 4H-1, 2, 4-triazole, 1H-and 2H-isoindole, and 1H-and 2H-pyrazole) .
- Valence tautomers include interconversions by reorganization of some of the bonding electrons. Tautomers can be in equilibrium or sterically locked into one form by appropriate substitution.
- Compounds of the present disclosure identified by name or structure as one particular tautomeric form are intended to include other tautomeric forms unless otherwise specified.
- the present disclosure is also intended to include all isotopes of atoms in the compounds.
- Isotopes of an atom include atoms having the same atomic number but different mass numbers.
- hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine, bromide or iodine in the compounds of present disclosure are meant to also include their isotopes, such as but not limited to 1 H, 2 H, 3 H, 11 C, 12 C, 13 C, 14 C, 14 N, 15 N, 16 O, 17 O, 18 O, 31 P, 32 P, 32 S, 33 S, 34 S, 36 S, 17 F, 18 F, 19 F, 35 Cl, 37 Cl, 79 Br, 81 Br, 124 I, 127 I and 131 I.
- hydrogen includes protium, deuterium and tritium.
- carbon includes 12 C and 13 C.
- Isotopically-enriched compounds of Formula (I) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- prodrug compounds of the present disclosure can be formulated as or be in the form of pharmaceutically acceptable salts. Unless specified to the contrary, a prodrug compound provided herein includes pharmaceutically acceptable salts of such compound.
- the term “pharmaceutically acceptable” indicates that the substance or composition is compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the subjects being treated therewith.
- the term “pharmaceutically acceptable salt” includes salts that retain the biological effectiveness of the free acids and bases of the specified compound and that are not biologically or otherwise undesirable.
- Contemplated pharmaceutically acceptable salt forms include, but are not limited to, mono, bis, tris, tetrakis, and so on.
- Pharmaceutically acceptable salts are non-toxic in the amounts and concentrations at which they are administered. The preparation of such salts can facilitate the pharmacological use by altering the physical characteristics of a compound without preventing it from exerting its physiological effect. Useful alterations in physical properties include lowering the melting point to facilitate transmucosal administration and increasing the solubility to facilitate administering higher concentrations of the drug.
- Pharmaceutically acceptable salts include acid addition salts such as those containing sulfate, chloride, hydrochloride, fumarate, maleate, phosphate, sulfamate, acetate, citrate, lactate, tartrate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, cyclohexylsulfamate and quinate.
- acid addition salts such as those containing sulfate, chloride, hydrochloride, fumarate, maleate, phosphate, sulfamate, acetate, citrate, lactate, tartrate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, cyclohexylsulfamate and quinate.
- Pharmaceutically acceptable salts can be obtained from acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
- acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
- Pharmaceutically acceptable salts also include basic addition salts such as those containing benzathine, chloroprocaine, choline, diethanolamine, ethanolamine, t-butylamine, ethylenediamine, meglumine, procaine, aluminum, calcium, lithium, magnesium, potassium, sodium, ammonium, alkylamine, and zinc, when acidic functional groups, such as carboxylic acid or phenol are present.
- acidic functional groups such as carboxylic acid or phenol are present.
- salts can be prepared by standard techniques.
- the free-base form of a compound can be dissolved in a suitable solvent, such as an aqueous or aqueous-alcohol solution containing the appropriate acid and then isolated by evaporating the solution.
- the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
- an inorganic acid such as hydrochloric acid
- the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary) , an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- an inorganic or organic base such as an amine (primary, secondary or tertiary) , an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- suitable salts include organic salts derived from amino acids, such as L-glycine, L-lysine, and L-arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as hydroxyethylpyrrolidine, piperidine, morpholine or piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- amino acids such as L-glycine, L-lysine, and L-arginine
- ammonia primary, secondary, and tertiary amines
- cyclic amines such as hydroxyethylpyrrolidine, piperidine, morpholine or piperazine
- inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- the compounds of present disclosure can exist in unsolvated forms, solvated forms (e.g., hydrated forms) , and solid forms (e.g., crystal or polymorphic forms) , and the present disclosure is intended to encompass all such forms.
- solvate or “solvated form” refers to solvent addition forms that contain either stoichiometric or non-stoichiometric amounts of solvent. Some compounds have a tendency to trap a fixed molar ratio of solvent molecules in the crystalline solid state, thus forming a solvate. If the solvent is water the solvate formed is a hydrate; and if the solvent is alcohol, the solvate formed is an alcoholate. Hydrates are formed by the combination of one or more molecules of water with one molecule of the substance in which the water retains its molecular state as H 2 O. Examples of solvents that form solvates include, but are not limited to, water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, and ethanolamine.
- crystal form As used herein, the terms “crystal form” , “crystalline form” , “polymorphic forms” and “polymorphs” can be used interchangeably, and mean crystal structures in which a compound (or a salt or solvate thereof) can crystallize in different crystal packing arrangements, all of which have the same elemental composition. Different crystal forms usually have different X-ray diffraction patterns, infrared spectral, melting points, density hardness, crystal shape, optical and electrical properties, stability and solubility. Recrystallization solvent, rate of crystallization, storage temperature, and other factors may cause one crystal form to dominate. Crystal polymorphs of the compounds can be prepared by crystallization under different conditions.
- the present disclosure is also intended to include all isotopes of atoms in the compounds.
- Isotopes of an atom include atoms having the same atomic number but different mass numbers.
- hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine, bromide or iodine in the compounds of present disclosure are meant to also include their isotopes, such as but not limited to 1 H, 2 H, 3 H, 11 C, 12 C, 13 C, 14 C, 14 N, 15 N, 16 O, 17 O, 18 O, 31 P, 32 P, 32 S, 33 S, 34 S, 36 S, 17 F, 18 F, 19 F, 35 Cl, 37 Cl, 79 Br, 81 Br, 124 I, 127 I and 131 I.
- hydrogen includes protium, deuterium and tritium.
- carbon includes 12 C and 13 C.
- prodrug compounds provided herein Synthesis of the prodrug compounds provided herein is illustrated in the synthetic schemes in the examples.
- the prodrug compounds provided herein can be prepared using any known organic synthesis techniques and can be synthesized according to any of numerous possible synthetic routes, and thus these schemes are illustrative only and are not meant to limit other possible methods that can be used to prepare the compounds provided herein. Additionally, the steps in the schemes are for better illustration and can be changed as appropriate.
- the embodiments of the prodrug compounds in examples were synthesized for the purposes of research and potentially submission to regulatory agencies.
- the reactions for preparing the prodrug compounds of the present disclosure can be carried out in suitable solvents, which can be readily selected by one skilled in the art of organic synthesis.
- suitable solvents can be substantially non-reactive with the starting materials (reactants) , the intermediates, or products at the temperatures at which the reactions are carried out, e.g. temperatures that can range from the solvent’s freezing temperature to the solvent's boiling temperature.
- a given reaction can be carried out in one solvent or a mixture of more than one solvent.
- suitable solvents for a particular reaction step can be selected by one skilled in the art.
- Preparation of the prodrug compounds of the present disclosure can involve the protection and deprotection of various chemical groups.
- the need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art.
- the chemistry of protecting groups can be found, for example, in T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999) , in P. Kocienski, Protecting Groups, Georg Thieme Verlag, 2003, and in Peter G.M. Wuts, Greene's Protective Groups in Organic Synthesis, 5 th Edition, Wiley, 2014, all of which are incorporated herein by reference in its entirety.
- Reactions can be monitored according to any suitable method known in the art.
- product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g. 1 H or 13 C) , infrared spectroscopy, spectrophotometry (e.g. UV-visible) , mass spectrometry, or by chromatographic methods such as high performance liquid chromatography (HPLC) , liquid chromatography-mass spectroscopy (LCMS) , or thin layer chromatography (TLC) .
- HPLC high performance liquid chromatography
- LCMS liquid chromatography-mass spectroscopy
- TLC thin layer chromatography
- Compounds can be purified by one skilled in the art by a variety of methods, including high performance liquid chromatography (HPLC) ( “Preparative LC-MS Purification: Improved Compound Specific Method Optimization” Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6 (6) ,
- the known starting materials of the present disclosure can be synthesized by using or according to the known methods in the art, or can be purchased from commercial suppliers. Unless otherwise noted, analytical grade solvents and commercially available reagents were used without further purification.
- the reactions of the present disclosure were all done under a positive pressure of nitrogen or argon or with a drying tube in anhydrous solvents, and the reaction flasks were typically fitted with rubber septa for the introduction of substrates and reagents via syringe. Glassware was oven dried and/or heat dried.
- the present disclosure provides a prodrug compound capable of locally delivering a therapeutic agent and releasing the therapeutic agent in a controlled and sustained manner with reduced systemic exposure and potential side effect due to systemic exposure.
- the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof are useful as therapeutic or prophylactic agent for various diseases, depending on the parent drug selected to be released.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total) , whether detectable or undetectable. “Therapy” can also mean prolonging survival as compared to expected survival if not receiving it.
- Those in need of therapy include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
- the term “therapy” also encompasses prophylaxis unless there are specific indications to the contrary.
- the terms “therapeutic” and “therapeutically” should be interpreted in a corresponding manner.
- treatment is used synonymously with “therapy” .
- treat can be regarded as “applying therapy” where “therapy” is as defined herein.
- prophylaxis is intended to have its normal meaning and includes primary prophylaxis to prevent the development of the disease and secondary prophylaxis whereby the disease has already developed and the patient is temporarily or permanently protected against exacerbation or worsening of the disease or the development of new symptoms associated with the disease.
- the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof shows a desired overall release rate of parent drug by controlling its solubility at a biological pH and the release of parent drug at different pHs.
- the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof has a lower solubility than the parent drug at a biological pH.
- the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof shows a solubility at acidic pH that is higher than that at biological pH.
- the ratio of the solubility of the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof at acidic pH to that at biological pH is greater than 5, greater than 10, greater than 20, greater than 30, greater than 40, greater than 50, greater than 60, greater than 70, greater than 80, greater than 90, greater than 100, greater than 200, greater than 300, greater than 400, greater than 500, greater than 600, greater than 700, greater than 800, greater than 900, greater than 1000, greater than 1100, greater than 1200, greater than 1300, greater than 1400, greater than 1500 or even higher.
- the reduced solubility of the prodrug compounds provided herein may avoid the high local concentration after administration, thereby providing a solubility controlled zero order sustained release mechanism.
- the parent drug may be released from the prodrug compound provided herein through the cleavage of the linkage between the parent drug moiety and the tail moiety.
- the release of the parent drug may involve enzymatic or non-enzymatic processes.
- the parent drug is released from the prodrug compound provided herein via hydrolysis process.
- the release of the parent drug may be affected by a variety of factors, for example, the selection of specific parent drug, the linkage between the parent drug moiety and tail moiety, the tail moiety, and the administration of the prodrug compound (for example, administration site, administration route) .
- the present disclosure contemplates parent drugs with varying reactive groups and linkages with the tail moiety.
- the present disclosure also contemplates varying administration of the prodrug compounds provided herein.
- the prodrug compounds provided herein is locally administered to a subject in need thereof.
- the prodrug compounds provided herein is locally administered to a subject in need thereof via injection.
- the prodrug compounds provided herein is locally administered to a subject in need thereof via oral dosage form.
- the prodrug compounds provided herein is locally administered to a subject in need thereof via inhalation.
- the prodrug compounds provided herein is locally administered to a subject in need thereof via implant.
- the prodrug compounds provided herein is locally administered to a subject in need thereof via topical application.
- release of parent drug may occur in a variety of locations upon administration to a subject.
- the prodrug compounds provided herein or a pharmaceutically acceptable salt thereof may release the parent drugs via hydrolysis at different rates.
- the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the percent of the parent drug released from the prodrug compound at pH 7.4 in 6 hours after administration.
- the release rate of the parent drug from the prodrug compound may vary in a range of about 50%to about 100%in 6 hours after administration, for example, about 55%to about 100%, about 60%to about 100%, about 65%to about 100%, about 70%to about 100%, about 75%to about 100%, about 80%to about 100%, about 85%to about 100%, about 90%to about 100%, about 95%to about 100%, about 96%to about 100%, about 97%to about 100%, about 98%to about 100%, about 99%to about 100%, or even about 100%.
- the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the percent of the parent drug released from the prodrug compound at pH 7.4 in 0.5 hours after administration.
- the release rate of the parent drug from the prodrug compound may vary in a range of about 5%to about 100%in 0.5 hours after administration, for example, about 10%to about 100%, about 20%to about 100%, about 30%to about 100%, about 40%to about 100%, about 50%to about 100%, about 60%to about 100%, about 70%to about 100%, about 80%to about 100%, about 90%to about 100%, about 95%to about 100%, about 96%to about 100%, about 97%to about 100%, about 98%to about 100%, about 99%to about 100%, or even about 100%.
- the release rate of the parent drug from the prodrug compound is not greater than 70%in 6 hours after administration, for example, not greater than 65%, not greater than 60%, not greater than 55%, not greater than 50%, not greater than 45%, not greater than 40%, not greater than 35%, not greater than 30%, not greater than 25%, not greater than 20%, not greater than 15%, not greater than 10%, not greater than 5%, not greater than 4%, not greater than 3%, not greater than 2%, or not greater than 1%.
- the prodrug compound provided herein remains stable without releasing the parent drug via hydrolysis.
- the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the hydrolysis constant (K h ) of the prodrug compound in 6 hours after administration.
- the prodrug compound provided herein may have a K h value in a range of about 0.1 to about 15, for example, about 0.1 to about 14, about 0.1 to about 13, about 0.1 to about 12, about 0.1 to about 11, about 0.1 to about 10, about 0.1 to about 9, about 0.1 to about 8, about 0.1 to about 7, about 0.1 to about 6, about 0.1 to about 5, about 0.1 to about 4, about 0.1 to about 3, about 0.1 to about 2, about 0.1 to about 1, about 0.1 to about 0.5, about 0.1 to about 0.4, about 0.1 to about 0.3, or about 0.1 to about 0.2.
- the prodrug compound provided herein may have a K h value not greater than 0.2, for example, not greater than not greater than 0.15, not greater than 0.1, not greater than 0.09, not greater than 0.08, not greater than 0.07, not greater than 0.06, not greater than 0.05, not greater than 0.04, not greater than 0.03, not greater than 0.02, or not greater than 0.01.
- the prodrug compound provided herein may have a K h value of 0.
- the prodrug compound provided herein release more parent drug in 6 hours after administration at pH 7.4 than at pH 2.0.
- the ratio of the percent of the parent drug released from the prodrug compound provided herein in 6 hours after administration at pH 7.4 to that at pH 2.0 is greater than 1, for example, greater than 1.1, greater than 1.2, greater than 1.3, greater than 1.4, greater than 1.5, greater than 2, great than 2.5, greater than 3, greater than 3.5, greater than 4, greater than 4.5, greater than 5, greater than 5.5, greater than 6, greater than 7, greater than 8, greater than 9, greater than 10, and the like.
- the ratio of K h value of the prodrug compound provided herein at pH 7.4 to that at pH 2.0 is greater than 1, for example, greater than 1.5, greater than 2, greater than 3, greater than 4, greater than 5, greater than 10, greater than 20, greater than 30, greater than 40, greater than 50, greater than 60, greater than 70, greater than 80, greater than 90, greater than 100, greater than 150, greater than 200, greater than 250, greater than 300, greater than 350, greater than 400, and the like.
- the prodrug compound provided herein may provide sustained release of parent drug over a period of 1-12 hours.
- compositions comprising the prodrug compounds of the present disclosure.
- compositions comprising the prodrug compounds of the present disclosure, and at least one pharmaceutical acceptable excipient.
- the term “pharmaceutical composition” refers to a formulation containing the drug delivery system of the present disclosure in a form suitable for administration to a subject.
- the term “pharmaceutically acceptable excipient” means an excipient that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes excipient that is acceptable for veterinary use as well as human pharmaceutical use.
- a “pharmaceutically acceptable excipient” as used herein includes both one and more than one such excipient.
- pharmaceutically acceptable excipient also encompasses “pharmaceutically acceptable carrier” and “pharmaceutically acceptable diluent” .
- compositions provided herein can be in any form that allows for the composition to be administered to a subject, including, but not limited to a human, and formulated to be compatible with an intended route of administration.
- compositions provided herein may be supplied in bulk or in unit dosage form depending on the intended administration route.
- powders, granules, tablets, pills, capsules, gelcaps, and caplets may be acceptable as solid dosage forms
- emulsions, syrups, elixirs, suspensions, and solutions may be acceptable as liquid dosage forms.
- gel, solutions, emulsions and suspensions may be acceptable as liquid dosage forms
- a powder suitable for reconstitution with an appropriate solution as solid dosage forms for inhalation administration, solutions, sprays, dry powders, and aerosols may be acceptable dosage form.
- powders, sprays, ointments, pastes, creams, lotions, gels, solutions, and patches may be acceptable dosage form.
- pessaries, tampons, creams, gels, pastes, foams and spray may be acceptable dosage form.
- solid, semi-solid, gel may be acceptable dosage form.
- compositions of the present disclosure may be in a form of formulation for oral administration.
- compositions of the present disclosure may be in a form of formulation for injection administration.
- compositions of the present disclosure may be in a form of formulation for inhalation administration.
- compositions of the present disclosure may be in a form of formulation for topical administration.
- compositions provided herein may be formulated in the form of skin patches that are well known to those of ordinary skill in the art.
- excipients and carriers are generally known to those skilled in the art and are thus included in the present disclosure.
- excipients and carriers are described, for example, in “Remingtons Pharmaceutical Sciences” Mack Pub. Co., New Jersey (1991) , in “Remington: The Science and Practice of Pharmacy” , Ed. University of the Sciences in Philadelphia, 21 st Edition, LWW (2005) , which are incorporated herein by reference.
- the pharmaceutical compositions of the present disclosure can be formulated as a single dose.
- the amount of the prodrug compounds provided herein in the single dose will vary depending on the subject treated and particular mode of administration.
- the pharmaceutical compositions of the present disclosure can be formulated to be administered to a subject at a time interval of a few days, a few weeks, a few months or even longer.
- compositions comprise the drug delivery system of the present disclosure, as two or more combination therapy.
- a method of treating diseases in a subject in need thereof comprising administering to the subject a therapeutic effective amount of the prodrug compound or the pharmaceutical composition provided herein.
- the diseases to be treated depends on the selected parent drug in the prodrug or the pharmaceutical composition provided herein.
- the disease can be selected from the group consisting of anal cancer, breast cancer, colorectal cancer, esophageal cancer, pancreatic cancer, head and neck cancer, brain cancer, liver cancer, gastric cancer, bladder cancer, oral mucosal cancer, esophageal cancer, anaplastic astrocytoma, glioblastoma multiforme, acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, Kaposi’s sarcoma, and neuroblastoma.
- the parent drug is selected from an anti-cancer agent, an anti-inflammatory drug, an antibiotic, an anti-fugus agent, a JAK inhibitor, or a VEGF inhibitor.
- the parent drug selected is Fluorouracil
- the prodrug compound provided herein is useful in the treatment of cancers, including colon, esophageal, gastric, rectum, breast, biliary tract, stomach, head and neck, cervical, pancreas, renal cell, and carcinoid.
- the parent drug selected is Temozolomide, and thus the prodrug compound provided herein is useful in the treatment of anaplastic astrocytoma and glioblastoma multiforme.
- the parent drug selected is Daunorubicin, and thus the prodrug compound provided herein is useful in the treatment of acute nonlymphocytic leukemia (myelogenous, monocytic, erythroid) and acute lymphocytic leukemia.
- the parent drug selected is 10-hydroxyl-camptothecine or 7-ethyl-10-hydroxyl-camptothecine, and thus the prodrug compound provided herein is useful in the treatment of cancers, including gastric, esophagus, cardiac, colon, liver, lung, bladder, acute leukemia, chronic myelogenous leukemia, and chorionic epithelioma.
- terapéuticaally effective amount refers to an amount of a therapeutic agent selected in the drug delivery system provided herein or pharmaceutically acceptable salts thereof which is effective to provide “therapy” in a subject, or to “treat” disorders, diseases or conditions in a subject.
- non-exemplified compounds according to the present disclosure may be successfully performed by modifications apparent to those skilled in the art, e.g., by appropriately protecting interfering groups, by utilizing other suitable reagents and building blocks known in the art other than those described, and/or by making routine modifications of reaction conditions.
- other reactions disclosed herein or known in the art will be recognized as having applicability for preparing other compounds of the present disclosure.
- MS mass spectra
- ESI electrospray ionization
- reaction mixture was filtered and purified by preparative chromatography eluting with MeCN and 0.1%TFA-H 2 O to give 4'- (dipropylamino) - [1, 1'-biphenyl] -4-yl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (2) (39mg) .
- MS-ESI (m/z) 456 [M+1] + .
- a suspension of daunorubicin hydrochloride (656mg, 1.24mmol) in trimethyl orthoformate (10mL) was heated at 110°C for 3 h.
- the reaction was concentrated under reduced pressure.
- the residue was diluted with acetone (40mL) followed by addition of 1N HCl (4mL) and conc. HCl (0.1mL) .
- the pH2.0 buffer solution was prepared by adding 50 mL of 0.1M phosphoric acid solution in a 200 mL volumetric flask, adjusting the pH value to 2.0 with a 0.1M sodium dihydrogen phosphate solution, then diluting to 200mL with water.
- the pH7.4 buffer solution was prepared by adding 50mL of 0.1M disodium hydrogen phosphate solution in a 200 mL volumetric flask, adjusting the pH value to 7.4 with a 0.1M phosphoric acid solution, then diluting to 200mL with water.
- the compounds were dissolved in pH7.4/pH2.0 buffer solution, placed in a constant temperature shaker at 37°C and 200rpm, and sampled at 0h, 0.5h and 6h respectively. The residual contents of the compounds relative to 0h were tested at each time point.
- the hydrolysis constant (K h ) of the present disclosed compounds at pH 7.4 are greater than that at pH 2.
- the objective of this study was to assess the pharmacokinetics of free fluorouracil and an exemplary compound of the present disclosure in stomach and plasma following the continuous intragastric administration of the exemplary compound of the present disclosure and the continuous intravenous infusion or single oral administration of free fluorouracil to male Sprague Dawley rats.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Provided are prodrugs for sustained releasing therapeutic agents, and methods for using such prodrugs for the treatment of diseases.
Description
FIELD OF THE DISCLOSURE
The present disclosure relates to prodrugs for sustained releasing therapeutic agents, and methods for using such prodrugs for the treatment of diseases.
BACKGROUND OF THE DISCLOSURE
Most of the therapeutic agents are delivered to the body systemically via oral/GI absorption or systemic injection. Then the therapeutic agents are delivered to the action site by the blood circulation, thus, the therapeutic agent are exposed to the entire body. In some cases, the unintended exposure of the therapeutic agents in the other parts of the body may cause side effects, sometimes serious side effects. To reduce systemic exposure caused side effects, many locally delivered dosage forms are developed. One good example is the inhalation dosage form to treat respiratory diseases, such as COPD and asthma. These therapeutic agents target receptors on the airway and make the airway open for more efficient oxygen exchange. There are many other examples of local delivered drugs, such as topical corticosteroids for skin rash, intravitreal injection of anti-VEGF for wet AMD (age-related macular degeneration) , intra-articular injection of corticosteroids to treat osteoarthritis. The purpose of these local delivered dosage form is to have high local drug concentration to have the therapeutic effective, yet to reduce systemic exposure of the drug to have minimum side effects. Local delivery dosage form, however, are difficult to use in most cases, especially the locally injected dosage forms. Therefore, it is desirable to have the delivery system, e.g., sustained or controlled release dosage form, that can release the drug slowly to have prolonged duration to reduce the frequency of dosing. The prodrug approach is one of the sustained release mechanism often used by people known in this field.
Drugs in the systemic circulation are metabolized and eliminated from the system by enzymes or excretion. In order to maintain a concentration above the effective concentration at all time, a peak concentration much higher than the effective concentration is needed so that the trough between each dosing would be above the effective concentrations. The peak concentration, however, in some cases, can cause undesirable side effect. There are many ways to reduce peak concentration without proportionally reduce trough concentration. The most popular approach is to use polymers to slow down the drug release from the dosing system. Many orally administrated sustained release/controlled release systems using this approach. There are pros and cons with this approach in some incidents, especially for locally injected delivery system targeting release duration in weeks or months. Using polymer system to control drug release may also have challenges in manufacturing process and quality control of the products. Another approach to have more precise control of the drug release is to have a prodrug that can release the drug via chemical bond breakage. In this case, the release mechanism is essentially a chemical reaction, more specifically, it’s a first order chemical reaction. The rate of the drug release is much more predictable due to consistency of the environment in the biology system. Furthermore, by controlling the solubility of the prodrug, it is possible to create a system that can release the drug in a zero order or pseudo-zero manner using solubility as rate limiting step.
The present disclosure is related to a prodrug system that controls drug release rate via chemical structure of the prodrug moiety for the intended location and indication.
SUMMARY OF THE DISCLOSURE
In one aspect, the present disclosure provides a prodrug compound comprising a parent drug moiety and a tail moiety, wherein
the parent drug moiety is derived from a parent drug comprising a reactive group selected from the group consisting of amine, amino, hydroxyl, carboxylate, ketone, and amide,
the tail moiety is covalently linked to the parent drug moiety and has a Formula (I) :
wherein:
L
1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage;
L is a direct bond or alkyl;
U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
V is a direct bond or alkyl;
W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
Z is selected from the group consisting of a direct bond, alkyl, aryl, NR
1R
2, and OR
3, wherein said alkyl and aryl are optionally substituted with one or more R
4;
R
1, R
2 and R
3 are independently hydrogen, alkyl or cycloalkyl; and
R
4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,
provided that when U is not a direct bond, V, W and Z are not direct bonds at the same time,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the present disclosure provides a prodrug compound having a formula selected from the group consisting of:

or a pharmaceutically acceptable salt thereof.
In some embodiments, the present disclosure provides a prodrug compound having a formula selected from the group consisting of:


or a pharmaceutically acceptable salt thereof.
In some embodiments, the present disclosure provides a prodrug compound having a formula of:

or a pharmaceutically acceptable salt thereof.
In some embodiments, the present disclosure provides a prodrug compound having a formula of:

or a pharmaceutically acceptable salt thereof.
In a further aspect, the present disclosure provides a pharmaceutical composition comprising the prodrug compound provided herein or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable excipient.
In another aspect, the present disclosure provides a method of treating diseases in a subject in need thereof, comprising administering to the subject a therapeutic effective amount of the prodrug compound provided herein or pharmaceutically acceptable salts thereof, or the pharmaceutical composition provided herein.
DESCRIPTION OF THE DRAWINGS
Figures 1 and 2 depict the plasma and gastric concentration of fluorouracil following the continuous intragastric administration of the exemplary compound of the present disclosure and the continuous intravenous infusion or single oral administration of free fluorouracil to male Sprague Dawley rats, respectively
DETAILED DESCRIPTION OF THE DISCLOSURE
Reference will now be made in detail to certain embodiments of the present disclosure, examples of which are illustrated in the accompanying structures and formulas. While the present disclosure will be described in conjunction with the enumerated embodiments, it will be understood that they are not intended to limit the present disclosure to those embodiments. On the contrary, the present disclosure is intended to cover all alternatives, modifications, and equivalents, which may be included within the scope of the present disclosure as defined by the claims. One skilled in the art will recognize many methods and materials similar or equivalent to those described herein, which could be used in the practice of the present disclosure. The present disclosure is in no way limited to the methods and materials described. In the event that one or more of the incorporated references and similar materials differs from or contradicts this application, including but not limited to defined terms, term usage, described techniques, or the like, the present disclosure controls. All references, patents, patent applications cited in the present disclosure are hereby incorporated by reference in their entireties.
It is appreciated that certain features of the present disclosure, which are, for clarity, described in the context of separate embodiments, can also be provided in combination in a single embodiment. Conversely, various features of the present disclosure, which are, for brevity, described in the context of a single embodiment, can also be provided separately or in any suitable sub-combination. It must be noted that, as used in the specification and the appended claims, the singular forms “a, ” “an, ” and “the” include plural forms of the same unless the context clearly dictates otherwise.
Definitions
Definitions of specific functional groups and chemical terms are described in more detail below. For purposes of this disclosure, the chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75
th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Organic Chemistry, Thomas Sorrell, 2
nd Edition, University Science Books, Sausalito, 2006; Smith and March March’s Advanced Organic Chemistry, 6
th Edition, John Wiley & Sons, Inc., New York, 2007; Larock, Comprehensive Organic Transformations, 3
rd Edition, VCH Publishers, Inc., New York, 2018; Carruthers, Some Modern Methods of Organic Synthesis, 4
th Edition, Cambridge University Press, Cambridge, 2004; the entire contents of each of which are incorporated herein by reference.
At various places in the present disclosure, linking substituents are described. It is specifically intended that each linking substituent includes both the forward and backward forms of the linking substituent. For example, -NR (CR’R”) -includes both -NR (CR’R”) -and - (CR’R”) NR-. Where the structure clearly requires a linking group, the Markush variables listed for that group are understood to be linking groups. For example, if the structure requires a linking group and the Markush group definition for that variable lists “alkyl” , then it is understood that the “alkyl” represents a linking alkylene group.
When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom in the ring. When a substituent is listed without indicating the atom via which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such formula. Combinations of substituents and/or variables are permissible, but only if such combinations result in stable compounds.
When any variable (e.g., R
i) occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at every other occurrence. Thus, for example, if a group is shown to be substituted with 0-2 R
i moieties, then the group may optionally be substituted with up to two R
i moieties and R
i at each occurrence is selected independently from the definition of R
i. Also, combinations of substituents and/or variables are permissible, but only if such combinations result in stable compounds.
As used herein, the term “C
i-j” indicates a range of the carbon atoms numbers, wherein i and j are integers and the range of the carbon atoms numbers includes the endpoints (i.e. i and j) and each integer point in between, and wherein j is greater than i. For examples, C
1-6 indicates a range of one to six carbon atoms, including one carbon atom, two carbon atoms, three carbon atoms, four carbon atoms, five carbon atoms and six carbon atoms. In some embodiments, the term “C
1-12” indicates 1 to 12, particularly 1 to 10, particularly 1 to 8, particularly 1 to 6, particularly 1 to 5, particularly 1 to 4, particularly 1 to 3 or particularly 1 to 2 carbon atoms.
As used herein, the term “alkyl” , whether as part of another term or used independently, refers to a saturated linear or branched-chain hydrocarbon radical, which may be optionally substituted independently with one or more substituents described below. The term “C
i-j alkyl” refers to an alkyl having i to j carbon atoms. In some embodiments, alkyl groups contain 1 to 10 carbon atoms. In some embodiments, alkyl groups contain 1 to 9 carbon atoms. In some embodiments, alkyl groups contain 1 to 8 carbon atoms, 1 to 7 carbon atoms, 1 to 6 carbon atoms, 1 to 5 carbon atoms, 1 to 4 carbon atoms, 1 to 3 carbon atoms, or 1 to 2 carbon atoms. Examples of “C
1-10 alkyl” include, but are not limited to, methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, and decyl. Examples of “C
1-6 alkyl” are methyl, ethyl, propyl, isopropyl, n-butyl, i-butyl, s-butyl, t-butyl, n-pentyl, 2-pentyl, 3-pentyl, 2-methyl-2-butyl, 3-methyl-2-butyl, 3-methyl-1-butyl, 2-methyl-1-butyl, 1-hexyl, 2-hexyl, 3-hexyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 3-methyl-3-pentyl, 2-methyl-3-pentyl, 2, 3-dimethyl-2-butyl, 3, 3-dimethyl-2-butyl, and the like.
As used herein, the term “alkenyl” , whether as part of another term or used independently, refers to linear or branched-chain hydrocarbon radical having at least one carbon-carbon double bond, which may be optionally substituted independently with one or more substituents described herein, and includes radicals having “cis” and “trans” orientations, or alternatively, “E” and “Z” orientations. In some embodiments, alkenyl groups contain 2 to 12 carbon atoms. In some embodiments, alkenyl groups contain 2 to 11 carbon atoms. In some embodiments, alkenyl groups contain 2 to 11 carbon atoms, 2 to 10 carbon atoms, 2 to 9 carbon atoms, 2 to 8 carbon atoms, 2 to 7 carbon atoms, 2 to 6 carbon atoms, 2 to 5 carbon atoms, 2 to 4 carbon atoms, 2 to 3 carbon atoms, and in some embodiments, alkenyl groups contain 2 carbon atoms. Examples of alkenyl group include, but are not limited to, ethylenyl (or vinyl) , propenyl (allyl) , butenyl, pentenyl, 1-methyl-2 buten-1-yl, 5-hexenyl, and the like.
As used herein, the term “alkynyl” , whether as part of another term or used independently, refers to a linear or branched hydrocarbon radical having at least one carbon-carbon triple bond, which may be optionally substituted independently with one or more substituents described herein. In some embodiments, alkenyl groups contain 2 to 12 carbon atoms. In some embodiments, alkynyl groups contain 2 to 11 carbon atoms. In some embodiments, alkynyl groups contain 2 to 11 carbon atoms, 2 to 10 carbon atoms, 2 to 9 carbon atoms, 2 to 8 carbon atoms, 2 to 7 carbon atoms, 2 to 6 carbon atoms, 2 to 5 carbon atoms, 2 to 4 carbon atoms, 2 to 3 carbon atoms, and in some embodiments, alkynyl groups contain 2 carbon atoms. Examples of alkynyl group include, but are not limited to, ethynyl, 1-propynyl, 2-propynyl, and the like.
As used herein, the term “amide” refers to -C (=O) NR’-, wherein R’ represents hydrogen, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “amine” refers to derivatives of ammonia, wherein one or more hydrogen atoms are replaced by a substituent, and can be represented by N (H)
n (R’)
3-n wherein n is 0, 1, or 2, and each R’ is independently hydroxyl, nitro, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups, or two R’ together with the nitrogen atom to which they are attached form an optionally substituted heterocyclyl or heteroaryl.
As used herein, the term “amino” refers to –NH
2.
As used herein, the term “acetal” refers to –O-CH (R’) -O-, wherein R’ represents alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “aryl” , whether as part of another term or used independently, refers to monocyclic and polycyclic ring systems having a total of 5 to 20 ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains 3 to 12 ring members. Examples of “aryl” include, but are not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Also included within the scope of the term “aryl” , as it is used herein, is a group in which an aromatic ring is fused to one or more additional rings. In the case of polycyclic ring system, only one of the rings needs to be aromatic (e.g., 2, 3-dihydroindole) , although all of the rings may be aromatic (e.g., quinoline) . The second ring can also be fused or bridged. Examples of polycyclic aryl include, but are not limited to, benzofuranyl, indanyl, phthalimidyl, naphthimidyl, phenanthridinyl, or tetrahydronaphthyl, and the like. Aryl groups can be substituted at one or more ring positions with substituents as described above.
As used herein, the term “cycloalkyl” , whether as part of another term or used independently, refer to a monovalent non-aromatic, saturated or partially unsaturated monocyclic and polycyclic ring system, in which all the ring atoms are carbon and which contains at least three ring forming carbon atoms. In some embodiments, the cycloalkyl may contain 3 to 12 ring forming carbon atoms, 3 to 10 ring forming carbon atoms, 3 to 9 ring forming carbon atoms, 3 to 8 ring forming carbon atoms, 3 to 7 ring forming carbon atoms, 3 to 6 ring forming carbon atoms, 3 to 5 ring forming carbon atoms, 4 to 12 ring forming carbon atoms, 4 to 10 ring forming carbon atoms, 4 to 9 ring forming carbon atoms, 4 to 8 ring forming carbon atoms, 4 to 7 ring forming carbon atoms, 4 to 6 ring forming carbon atoms, 4 to 5 ring forming carbon atoms. Cycloalkyl groups may be saturated or partially unsaturated. Cycloalkyl groups may be substituted. In some embodiments, the cycloalkyl group may be a saturated cyclic alkyl group. In some embodiments, the cycloalkyl group may be a partially unsaturated cyclic alkyl group that contains at least one double bond or triple bond in its ring system. In some embodiments, the cycloalkyl group may be monocyclic or polycyclic. Examples of monocyclic cycloalkyl group include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-1-enyl, 1-cyclopent-2-enyl, 1-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-cyclohex-2-enyl, 1-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl and cyclododecyl. Examples of polycyclic cycloalkyl group include, but are not limited to, adamantyl, norbornyl, fluorenyl, spiro-pentadienyl, spiro [3.6] -decanyl, bicyclo [1, 1, 1] pentenyl, bicyclo [2, 2, 1] heptenyl, and the like.
As used herein, the term “carboxylate” or “carboxylate ester” refers to -C (=O) O-.
As used herein, the term “phosphate ester” refers to –OP (=O) (OR’) O-, wherein R’ represents hydrogen, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups
As used herein, the term “carbamate” refers to –NR’ (C=O) O–, wherein R’ represents hydrogen, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “thiocarbamate” refers to –NR’ (C=S) O–, wherein R’ represents hydrogen, an N-protecting group, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “carbonate” refers to –OC (=O) O-.
As used herein, the term “thiocarbonate” refers to –OC (=S) O-.
As used herein, the term “heteroatom” refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen (including N-oxides) .
As used herein, the term “heteroaryl” , whether as part of another term or used independently, refers to an aryl group having, in addition to carbon atoms, one or more heteroatoms. The heteroaryl group can be monocyclic. Examples of monocyclic heteroaryl include, but are not limited to, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, benzofuranyl and pteridinyl. The heteroaryl group also includes polycyclic groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring. Examples of polycyclic heteroaryl include, but are not limited to, indolyl, isoindolyl, benzothienyl, benzofuranyl, benzo [1, 3] dioxolyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzthiazolyl, quinolyl, isoquinolyl, dihydroquinolinyl, dihydroisoquinolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H-quinolizinyl, carbazolyl, acridinyl, phenazinyl, phenothiazinyl, phenoxazinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, and the like.
As used herein, the term “heterocyclyl” refers to a saturated or partially unsaturated carbocyclyl group in which one or more ring atoms are heteroatoms independently selected from oxygen, sulfur, nitrogen, phosphorus, and the like, the remaining ring atoms being carbon, wherein one or more ring atoms may be optionally substituted independently with one or more substituents. In some embodiments, the heterocyclyl is a saturated heterocyclyl. In some embodiments, the heterocyclyl is a partially unsaturated heterocyclyl having one or more double bonds in its ring system. In some embodiments, the heterocyclyl may contains any oxidized form of carbon, nitrogen or sulfur, and any quaternized form of a basic nitrogen. “Heterocyclyl” also includes radicals wherein the heterocyclyl radicals are fused with a saturated, partially unsaturated, or fully unsaturated (i.e., aromatic) carbocyclic or heterocyclic ring. The heterocyclyl radical may be carbon linked or nitrogen linked where such is possible. In some embodiments, the heterocycle is carbon linked. In some embodiments, the heterocycle is nitrogen linked. For example, a group derived from pyrrole may be pyrrol-1-yl (nitrogen linked) or pyrrol-3-yl (carbon linked) . Further, a group derived from imidazole may be imidazol-1-yl (nitrogen linked) or imidazol-3-yl (carbon linked) .
In some embodiments, the term “3-to 12-membered heterocyclyl” refers to a 3-to 12-membered saturated or partially unsaturated monocyclic or polycyclic heterocyclic ring system having 1 to 3 heteroatoms independently selected from nitrogen, oxygen, or sulfur. The fused, spiro and bridged ring systems are also included within the scope of this definition. Examples of monocyclic heterocyclyl include, but are not limited to oxetanyl, 1, 1-dioxothietanylpyrrolidyl, tetrahydrofuryl, tetrahydrothienyl, pyrrolyl, furanyl, thienyl, pyrazolyl, imidazolyl, triazolyl, oxazolyl, thiazolyl, piperidyl, piperazinyl, piperidinyl, morpholinyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, pyridonyl, pyrimidonyl, pyrazinonyl, pyrimidonyl, pyridazonyl, pyrrolidinyl, triazinonyl, and the like. Examples of fused heterocyclyl include, but are not limited to, phenyl fused ring or pyridinyl fused ring, such as quinolinyl, isoquinolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, quinoxalinyl, quinolizinyl, quinazolinyl, azaindolizinyl, pteridinyl, chromenyl, isochromenyl, indolyl, isoindolyl, indolizinyl, indazolyl, purinyl, benzofuranyl, isobenzofuranyl, benzimidazolyl, benzothienyl, benzothiazolyl, carbazolyl, phenazinyl, phenothiazinyl, phenanthridinyl, imidazo [1, 2-a] pyridinyl, [1, 2, 4] triazolo [4, 3-a] pyridinyl, [1, 2, 3] triazolo [4, 3-a] pyridinyl groups, and the like. Examples of spiro heterocyclyl include, but are not limited to, spiropyranyl, spirooxazinyl, and the like. Examples of bridged heterocyclyl include, but are not limited to, morphanyl, hexamethylenetetraminyl, 3-aza-bicyclo [3.1.0] hexane, 8-aza-bicyclo [3.2.1] octane, 1-aza-bicyclo [2.2.2] octane, 1, 4-diazabicyclo [2.2.2] octane (DABCO) , and the like.
As used herein, the term “hydrazone” refers to -C (R’) =N-NH-, wherein R’ represents hydrogen, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “hydroxyl” refers to –OH.
As used herein, the term “ketone” refers to -C (=O) -.
As used herein, the term “phosphonamidate” refers to –OP (=O) (R’) (NR”) -, wherein R’ and R” are independently hydrogen, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “imine” refers to -C (R’) =N-, wherein R’ represents hydrogen, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl and other suitable organic groups.
As used herein, the term “linkage” or “linker” refers to bonds or chemical moiety formed from a chemical reaction between the functional groups of at least two entities to be linked, thereby forming one molecule or maintaining association of the entities in sufficiently close proximity. A linkage can be integrated in the resulting linked molecule or structure, with or without its reacted functional groups. Such linkages may be covalent or non-covalent. Hydrolytically unstable or degradable linkages mean that the linkages are degradable in water or in aqueous solutions, including for example, body fluid such as blood. Enzymatically unstable or degradable linkages mean that the linkage can be degraded by one or more enzymes. Such degradable linkages include, but are not limited to ester linkages formed by the carboxylic acid in one entity with alcohol groups on a biologically active agent, wherein such ester groups generally hydrolyze under physiological conditions to release the biologically active agent. Other hydrolytically degradable linkages include but are not limited to carbonate linkages, imine linkages resulted from reaction of an amine and an aldehyde, phosphate ester linkages resulted from reaction of a phosphate group and an alcohol, hydrazone linkages resulted from reaction of a hydrazide and an aldehyde, acetal linkages resulted from reaction of an aldehyde and an alcohol, amide linkages resulted from reaction of an amine group and a carboxyl group.
As used herein, the term “partially unsaturated” refers to a radical that includes at least one double or triple bond. The term “partially unsaturated” is intended to encompass rings having multiple sites of unsaturation, but is not intended to include aromatic (i.e., fully unsaturated) moieties.
As used herein, the term “pharmaceutically acceptable” indicates that the substance or composition is compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the subjects being treated therewith.
As used herein, the term “substituted” , whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent. It will be understood that “substitution” or “substituted with” includes the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and that the substitution results in a stable or chemically feasible compound, e.g., which does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. Unless otherwise indicated, an “optionally substituted” group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position. It will be understood by those skilled in the art that substituents can themselves be substituted, if appropriate. Unless specifically stated as “unsubstituted” , references to chemical moieties herein are understood to include substituted variants. For example, reference to an “aryl” group or moiety implicitly includes both substituted and unsubstituted variants.
As used herein, the terms “therapeutic agent” , “drug” , “biologically active molecule” , “biologically active agent” , “active agent” and the like refer to any substance which can affect any physical or biochemical properties of a biological organism, including but not limited to viruses, bacteria, fungi, plants, animals, and human. In particular, as used herein, therapeutic agents include any substance intended for diagnosis, cure, mitigation, treatment, or prevention of disease in humans or other animals, or to otherwise enhance physical or mental well-being of humans or animals.
Drug delivery of therapeutic agents to specific tissues or sites within a body presents a variety of challenges, particularly where local delivery of a therapeutic agent to a specific tissue and sustained release of the therapeutic agent are desired, and where avoidance of high systemic concentration of the therapeutic agent leading to toxic side effects is desired.
Compound
Therefore, the present disclosure provides a prodrug compound capable of locally delivering a therapeutic agent and releasing the therapeutic agent in a controlled and sustained manner with reduced systemic side effect. This is achieved by deliberately designing a prodrug compound which achieves a balance of solubility of the prodrug compound and the rate of the prodrug compound for releasing the parent drug.
In one aspect, the present disclosure provides a prodrug compound comprising a parent drug moiety and a tail moiety, wherein
the parent drug moiety is derived from a parent drug comprising a reactive group selected from the group consisting of amine, amino, hydroxyl, and amide,
the tail moiety is covalently linked to the parent drug moiety and has a Formula (I) :
wherein:
L
1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage;
L is a direct bond or alkyl;
U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
V is a direct bond or alkyl;
W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;
Z is selected from the group consisting of a direct bond, alkyl, aryl, NR
1R
2, and OR
3, wherein said alkyl and aryl are optionally substituted with one or more R
4;
R
1, R
2 and R
3 are independently hydrogen, alkyl or cycloalkyl; and
R
4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,
provided that when U is not a direct bond, V, W and Z are not direct bonds at the same time,
or a pharmaceutically acceptable salt thereof.
In some embodiments, the parent drug comprises at least one reactive group, which is capable of reacting with a reactive functional group of a second entity (e.g., the tail moiety provided herein) and an optional co-reactant to form a cleavable linkage, thereby linking the parent drug moiety to the second entity (e.g., the tail moiety) .
A "cleavable linkage" is a relatively labile bond that cleaves under physiological conditions. An exemplary releasable linkage is a hydrolyzable bond that cleaves upon reaction with water (i.e., is hydrolyzed) . The tendency of a bond to hydrolyze in water may depend not only on the general type of linkage connecting two atoms but also on the substituents attached to these atoms. Appropriate hydrolytically unstable or weak linkages include but are not limited to carboxylate ester, phosphate ester, anhydrides, acetals, ketals, amide, acyloxyalkyl ether, imines, hydrazone, orthoesters, peptides, oligonucleotides, thioesters, urea, thiourea, carbamate, thiocabamate, phosphoramidate, phosphonamidate, carbonates and thiocarbonate. Certain functional groups have atoms that may be chemically degraded by a process other than hydrolysis. Exemplary releasable linkages in this category include certain carbamates and Fmoc derivatives. Certain molecules containing these kinds of functionalities appropriately bonded may undergo chemical degradation (release) upon action of a base. In such cases “cleave” may occur at higher values of pH or through the action of biological molecules that contain basic moieties (e.g. histidines) . Another exemplary cleavable linkage is an enzymatically cleavable linkage. An "enzymatically cleavable linkage" means a linkage that is subject to cleavage by one or more enzymes.
In certain embodiments, the parent drug is selected from the group consisting of anti-cancer agent, anti-inflammatory drugs, antibiotics, anti-fugus agent, JAK inhibitors, and VEGF inhibitors.
In some embodiments, the parent drug is an anti-cancer agent.
In some embodiments, the parent drug is an anti-inflammatory drug.
In some embodiments, the parent drug is an antibiotic.
In some embodiments, the parent drug is an anti-fugus agent.
In some embodiments, the parent drug is selected from the group consisting of Fluorouracil, Temozolomide, Daunorubicin, 10-hydroxyl-camptothecine, and 7-ethyl-10-hydroxyl-camptothecine.
In some embodiments, the reactive group of the parent drug reacts with the reactive functional group of the tail moiety and an optional co-reactant to form a cleavable linkage selected from the group consisting of carbonate, thiocarbonate, carbamate, thiocarbamate, carboxylate ester, phosphate ester, amide, imine, hydrazone, phosphonamidate and acetal.
In some embodiments, L
1 is selected from a direct bond, *-CH
2OC (=O) O-, *-CH
2OC (=S) O-, *-C (=O) O-, *-OC (=S) -, *-C (=O) -, *-C (=O) N (R
a) -, *-C (=S) N (R
a) -, *-CH
2OP (=O) (R
a) O-, and *-P (=O) (R
a) N (R
a) -, wherein R
a is hydrogen, alkyl, alkenyl or alkynyl, and the*end of L
1 is connected to the parent drug moiety.
In some embodiments, L
1 is selected from the group consisting of a direct bond, *-CH
2OC (=O) O-, *-C (=O) O-, *-C (=O) -, *-C (=O) N (R
a) -, *-CH
2OP (=O) (R
a) O-, and *-P (=O) (R
a) N (R
a) -.
In some embodiments, the parent drug comprises amine group which reacts with a reactive functional group of the tail moiety and an optional co-reactant, such that L
1 selected from *-CH
2OC (=O) O-, *-C (=O) O-, *-C (=O) -, and *-CH
2OP (=O) (R
a) O-is formed.
In some embodiments, the parent drug comprises amino group which reacts with a reactive functional group of the tail moiety and an optional co-reactant, such that L
1 selected from a direct bond and *-C (=O) O-is formed.
In some embodiments, the parent drug comprises hydroxyl group which reacts with a reactive functional group of the tail moiety and an optional co-reactant, such that L
1 selected from *-C (=O) -, *-C (=O) O-, *-C (=O) N (R
a) -and *-P (=O) (R
a) N (R
a) -is formed.
In some embodiments, L is a direct bond.
In some embodiments, L is alkyl. In certain embodiments, L is C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl, or C
1-2 alkyl.
In certain embodiments, L is methyl, ethyl or hexyl.
In some embodiments, U is a direct bond.
In some embodiments, U is heterocyclyl. In certain embodiments, U is saturated heterocyclyl. In certain embodiments, U is partially unsaturated heterocyclyl.
In certain embodiments, U is 5-to 12-membered heterocyclyl, 5-to 11-membered heterocyclyl, 5-to 10-membered heterocyclyl, 5-to 9-membered heterocyclyl, 5-to 8-membered heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered heterocyclyl.
In certain embodiments, U is 5-to 12-membered saturated heterocyclyl, 5-to 11-membered saturated heterocyclyl, 5-to 10-membered saturated heterocyclyl, 5-to 9-membered saturated heterocyclyl, 5-to 8-membered saturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered saturated heterocyclyl.
In certain embodiments, U is piperidinyl.
In certain embodiments, U is 5-to 12-membered partially unsaturated heterocyclyl, 5-to 11-membered partially unsaturated heterocyclyl, 5-to 10-membered partially unsaturated heterocyclyl, 5-to 9-membered partially unsaturated heterocyclyl, 5-to 8-membered partially unsaturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered partially unsaturated heterocyclyl.
In certain embodiments, U is 1, 2, 3, 4-tetrahydro-isoquinolinyl.
In some embodiments, U is aryl. In certain embodiments, U is 5 to 12 membered aryl, 5 to 10 membered aryl, 5 to 8 membered aryl, or 5 to 6 membered aryl.
In certain embodiments, U is phenyl.
In some embodiments, L is a direct bond, and U is a direct bond, heterocyclyl or aryl.
In certain embodiments, L is a direct bond, and U is a direct bond, 5-to 12-membered saturated or partially unsaturated heterocyclyl, or 5 to 12 membered aryl.
In certain embodiments, L is a direct bond, and U is selected from the group consisting of a direct bond, 1, 2, 3, 4-tetrahydro-isoquinolinyl and phenyl.
In some embodiments, L is alkyl, and U is a direct bond, heterocyclyl or aryl.
In certain embodiments, L is C
1-6 alkyl, and U is a direct bond, 5-to 12-membered saturated or partially unsaturated heterocyclyl, or 5 to 12 membered aryl.
In certain embodiments, L is C
1-6 alkyl, and U is selected from the group consisting of a direct bond, piperidinyl and phenyl.
In some embodiments, V is a direct bond.
In some embodiments, V is alkyl. In certain embodiments, V is C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl, or C
1-2 alkyl.
In certain embodiments, V is methyl.
In some embodiments, W is a direct bond.
In some embodiments, W is aryl. In certain embodiments, W is 5 to 12 membered aryl, 5 to 10 membered aryl, 5 to 8 membered aryl, or 5 to 6 membered aryl.
In certain embodiments, W is phenyl.
In some embodiments, W is heterocyclyl. In certain embodiments, W is saturated heterocyclyl. In certain embodiments, W is partially unsaturated heterocyclyl.
In certain embodiments, W is 5-to 12-membered heterocyclyl, 5-to 11-membered heterocyclyl, 5-to 10-membered heterocyclyl, 5-to 9-membered heterocyclyl, 5-to 8-membered heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered heterocyclyl.
In certain embodiments, W is 5-to 12-membered saturated heterocyclyl, 5-to 11-membered saturated heterocyclyl, 5-to 10-membered saturated heterocyclyl, 5-to 9-membered saturated heterocyclyl, 5-to 8-membered saturated heterocyclyl, 5-to 7-membered heterocyclyl or 5-to 6-membered saturated heterocyclyl.
In certain embodiments, W is pyrrolidinyl, piperidinyl or piperazinyl.
In some embodiments, Z is a direct bond.
In some embodiments, Z is alkyl optionally substituted with one or more R
4. In certain embodiments, Z is C
1-8 alkyl optionally substituted with one or more R
4.
In certain embodiments, R
4 is cycloalkyl or aryl.
In certain embodiments, Z is C
1-8 alkyl.
In certain embodiments, Z is C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl or C
1-
2 alkyl, each of which is substituted with one or two R
4, and R
4 is independently cycloalkyl or aryl.
In certain embodiments, Z is methyl or ethyl, which is substituted with one or two R
4, and R
4 is independently adamantyl or phenyl.
In some embodiments, Z is aryl optionally substituted with one or more R
4. In certain embodiments, Z is 5-to 12-membered aryl optionally substituted with one or more R
4.
In certain embodiments, R
4 is alkyl.
In certain embodiments, Z is phenyl optionally substituted with one or more R
4, wherein R
4 is C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl, or C
1-2 alkyl.
In certain embodiments, Z is phenyl substituted with one or more R
4, wherein R
4 is methyl or ethyl.
In some embodiments, Z is NR
1R
2.
In certain embodiments, R
1 and R
2 are independently alkyl or cycloalkyl.
In certain embodiments, R
1 and R
2 are independently C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl, or C
1-2 alkyl.
In certain embodiments, R
1 and R
2 are independently C
3-6 cycloalkyl.
In some embodiments, Z is OR
3.
In certain embodiments, R
3 is alkyl. In certain embodiments, R
3 is C
1-6 alkyl, C
1-5 alkyl, C
1-4 alkyl, C
1-3 alkyl, or C
1-2 alkyl. In certain embodiments, R
3 is methyl.
In some embodiments, the present disclosure provides a prodrug compound having a formula selected from the group consisting of:


or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, Z and R
a are defined as supra.
In certain embodiments, in prodrug compound having a Formula (II) , (III) , (IV) or (V) ,
L is a direct bond,
U is heterocyclyl, aryl or heteroaryl,
V is a direct bond or alkyl,
W is a direct bond, heterocyclyl or aryl;
Z is alkyl, aryl, NR
1R
2, or OR
3, wherein said alkyl and aryl are optionally substituted with one or more R
4,
wherein R
1, R
2, R
3 and R
4 are defined as supra.
In some embodiments, the present disclosure provides a prodrug compound having a formula selected from the group consisting of:

or a pharmaceutically acceptable salt thereof, wherein Q is hydrogen or ethyl, and L, U, V, W, Z and R
a are as defined as supra.
In certain embodiments, in prodrug compound having a Formula (VI) , (VII) , (VIII) or (IX) ,
L is a direct bond or alkyl,
U is a direct or aryl,
V is a direct bond,
W is a direct bond or heterocyclyl;
Z is NR
1R
2 or alkyl optionally substituted with one or more R
4.
wherein R
1, R
2 and R
4 are defined as supra.
In some embodiments, the present disclosure provides a prodrug compound having a formula of:

or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined as supra.
In some embodiments, the present disclosure provides a prodrug compound having a formula of:

or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined as supra.
In some embodiments, the prodrug compound provided herein has a lower solubility than the parent drug at biological pH.
In some embodiments, the present disclosure provides a prodrug compound selected from the group consisting of:



The prodrug compounds provided herein are described with reference to both generic formulae and specific compounds. In addition, the prodrug compounds of the present disclosure may exist in a number of different forms or derivatives, all within the scope of the present disclosure. These include, for example, tautomers, stereoisomers, racemic mixtures, regioisomers, salts, solvated forms, amorphous forms, different crystal forms or polymorphs.
The prodrug compounds of present disclosure can comprise one or more asymmetric centers depending on substituent selection, and thus can exist in various stereoisomeric forms, e.g., enantiomers and/or diastereomers. For example, the prodrug compounds provided herein may have an asymmetric carbon center, and thus compounds provided herein may have either the (R) or (S) stereo-configuration at a carbon asymmetric center. Therefore, the prodrug compounds of the present disclosure may be in the form of an individual enantiomer, diastereomer or geometric isomer, or may be in the form of a mixture of stereoisomers.
As used herein, the term “enantiomer” refers to two stereoisomers of a compound which are non-superimposable mirror images of one another. The term “diastereomer” refers to a pair of optical isomers which are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities.
Where a particular enantiomer is preferred, it may, in some embodiments be provided substantially free of the opposite enantiomer, and may also be referred to as “optically enriched” . “Optically enriched” , as used herein, means that the compound is made up of a significantly greater proportion of one enantiomer. In certain embodiments, the compound is made up of at least about 90%by weight of a preferred enantiomer. In other embodiments, the compound is made up of at least about 95%, 98%, or 99%by weight of a preferred enantiomer. Preferred enantiomers may be isolated from racemic mixtures by any method known to those skilled in the art, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis. Optionally a derivatization can be carried out before a separation of stereoisomers. The separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound provided herein or it can be done on a final racemic product. Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration. Alternatively, absolute stereochemistry may be determined by Vibrational Circular Dichroism (VCD) spectroscopy analysis. See, for example, Jacques, et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981) ; Wilen, S.H., et al., Tetrahedron 33: 2725 (1977) ; Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962) ; Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972) .
In some embodiments, mixtures of diastereomers, for example mixtures of diastereomers enriched with 51%or more of one of the diastereomers, including for example 60%or more, 70%or more, 80%or more, or 90%or more of one of the diastereomers are provided.
The present disclosure additionally encompasses the compounds as individual isomers substantially free of other isomers and alternatively, as mixtures of various isomers, e.g., racemic mixtures of enantiomers.
The prodrug compounds of the present disclosure may also exist in different tautomeric forms, and all such forms are embraced within the scope of the present disclosure. The term “tautomer” or “tautomeric form” refers to structural isomers of different energies which are interconvertible via a low energy barrier. For example, proton tautomers (also known as prototropic tautomers) include interconversions via migration of a proton, such as keto-enol, amide-imidic acid, lactam-lactim, imine-enamine isomerizations and annular forms where a proton can occupy two or more positions of a heterocyclic system (for example, 1H-and 3H-imidazole, 1H-, 2H-and 4H-1, 2, 4-triazole, 1H-and 2H-isoindole, and 1H-and 2H-pyrazole) . Valence tautomers include interconversions by reorganization of some of the bonding electrons. Tautomers can be in equilibrium or sterically locked into one form by appropriate substitution. Compounds of the present disclosure identified by name or structure as one particular tautomeric form are intended to include other tautomeric forms unless otherwise specified.
The present disclosure is also intended to include all isotopes of atoms in the compounds. Isotopes of an atom include atoms having the same atomic number but different mass numbers. For example, unless otherwise specified, hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine, bromide or iodine in the compounds of present disclosure are meant to also include their isotopes, such as but not limited to
1H,
2H,
3H,
11C,
12C,
13C,
14C,
14N,
15N,
16O,
17O,
18O,
31P,
32P,
32S,
33S,
34S,
36S,
17F,
18F,
19F,
35Cl,
37Cl,
79Br,
81Br,
124I,
127I and
131I. In some embodiments, hydrogen includes protium, deuterium and tritium. In some embodiments, carbon includes
12C and
13C. Isotopically-enriched compounds of Formula (I) can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
The prodrug compounds of the present disclosure can be formulated as or be in the form of pharmaceutically acceptable salts. Unless specified to the contrary, a prodrug compound provided herein includes pharmaceutically acceptable salts of such compound.
As used herein, the term “pharmaceutically acceptable” indicates that the substance or composition is compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the subjects being treated therewith.
As used herein, the term “pharmaceutically acceptable salt” , unless otherwise indicated, includes salts that retain the biological effectiveness of the free acids and bases of the specified compound and that are not biologically or otherwise undesirable. Contemplated pharmaceutically acceptable salt forms include, but are not limited to, mono, bis, tris, tetrakis, and so on. Pharmaceutically acceptable salts are non-toxic in the amounts and concentrations at which they are administered. The preparation of such salts can facilitate the pharmacological use by altering the physical characteristics of a compound without preventing it from exerting its physiological effect. Useful alterations in physical properties include lowering the melting point to facilitate transmucosal administration and increasing the solubility to facilitate administering higher concentrations of the drug.
Pharmaceutically acceptable salts include acid addition salts such as those containing sulfate, chloride, hydrochloride, fumarate, maleate, phosphate, sulfamate, acetate, citrate, lactate, tartrate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, cyclohexylsulfamate and quinate. Pharmaceutically acceptable salts can be obtained from acids such as hydrochloric acid, maleic acid, sulfuric acid, phosphoric acid, sulfamic acid, acetic acid, citric acid, lactic acid, tartaric acid, malonic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclohexylsulfamic acid, fumaric acid, and quinic acid.
Pharmaceutically acceptable salts also include basic addition salts such as those containing benzathine, chloroprocaine, choline, diethanolamine, ethanolamine, t-butylamine, ethylenediamine, meglumine, procaine, aluminum, calcium, lithium, magnesium, potassium, sodium, ammonium, alkylamine, and zinc, when acidic functional groups, such as carboxylic acid or phenol are present. For example, see Remington's Pharmaceutical Sciences, 19
thed., Mack Publishing Co., Easton, PA, Vol. 2, p. 1457, 1995; “Handbook of Pharmaceutical Salts: Properties, Selection, and Use” by Stahl and Wermuth, Wiley-VCH, Weinheim, Germany, 2002. Such salts can be prepared using the appropriate corresponding bases.
Pharmaceutically acceptable salts can be prepared by standard techniques. For example, the free-base form of a compound can be dissolved in a suitable solvent, such as an aqueous or aqueous-alcohol solution containing the appropriate acid and then isolated by evaporating the solution. Thus, if the particular compound is a base, the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
Similarly, if the particular compound is an acid, the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary) , an alkali metal hydroxide or alkaline earth metal hydroxide, or the like. Illustrative examples of suitable salts include organic salts derived from amino acids, such as L-glycine, L-lysine, and L-arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as hydroxyethylpyrrolidine, piperidine, morpholine or piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
It is also to be understood that the compounds of present disclosure can exist in unsolvated forms, solvated forms (e.g., hydrated forms) , and solid forms (e.g., crystal or polymorphic forms) , and the present disclosure is intended to encompass all such forms.
As used herein, the term “solvate” or “solvated form” refers to solvent addition forms that contain either stoichiometric or non-stoichiometric amounts of solvent. Some compounds have a tendency to trap a fixed molar ratio of solvent molecules in the crystalline solid state, thus forming a solvate. If the solvent is water the solvate formed is a hydrate; and if the solvent is alcohol, the solvate formed is an alcoholate. Hydrates are formed by the combination of one or more molecules of water with one molecule of the substance in which the water retains its molecular state as H
2O. Examples of solvents that form solvates include, but are not limited to, water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid, and ethanolamine.
As used herein, the terms “crystal form” , “crystalline form” , “polymorphic forms” and “polymorphs” can be used interchangeably, and mean crystal structures in which a compound (or a salt or solvate thereof) can crystallize in different crystal packing arrangements, all of which have the same elemental composition. Different crystal forms usually have different X-ray diffraction patterns, infrared spectral, melting points, density hardness, crystal shape, optical and electrical properties, stability and solubility. Recrystallization solvent, rate of crystallization, storage temperature, and other factors may cause one crystal form to dominate. Crystal polymorphs of the compounds can be prepared by crystallization under different conditions.
The present disclosure is also intended to include all isotopes of atoms in the compounds. Isotopes of an atom include atoms having the same atomic number but different mass numbers. For example, unless otherwise specified, hydrogen, carbon, nitrogen, oxygen, phosphorous, sulfur, fluorine, chlorine, bromide or iodine in the compounds of present disclosure are meant to also include their isotopes, such as but not limited to
1H,
2H,
3H,
11C,
12C,
13C,
14C,
14N,
15N,
16O,
17O,
18O,
31P,
32P,
32S,
33S,
34S,
36S,
17F,
18F,
19F,
35Cl,
37Cl,
79Br,
81Br,
124I,
127I and
131I. In some embodiments, hydrogen includes protium, deuterium and tritium. In some embodiments, carbon includes
12C and
13C.
Synthesis of Compound
Synthesis of the prodrug compounds provided herein is illustrated in the synthetic schemes in the examples. The prodrug compounds provided herein can be prepared using any known organic synthesis techniques and can be synthesized according to any of numerous possible synthetic routes, and thus these schemes are illustrative only and are not meant to limit other possible methods that can be used to prepare the compounds provided herein. Additionally, the steps in the schemes are for better illustration and can be changed as appropriate. The embodiments of the prodrug compounds in examples were synthesized for the purposes of research and potentially submission to regulatory agencies.
The reactions for preparing the prodrug compounds of the present disclosure can be carried out in suitable solvents, which can be readily selected by one skilled in the art of organic synthesis. Suitable solvents can be substantially non-reactive with the starting materials (reactants) , the intermediates, or products at the temperatures at which the reactions are carried out, e.g. temperatures that can range from the solvent’s freezing temperature to the solvent's boiling temperature. A given reaction can be carried out in one solvent or a mixture of more than one solvent. Depending on the particular reaction step, suitable solvents for a particular reaction step can be selected by one skilled in the art.
Preparation of the prodrug compounds of the present disclosure can involve the protection and deprotection of various chemical groups. The need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art. The chemistry of protecting groups can be found, for example, in T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999) , in P. Kocienski, Protecting Groups, Georg Thieme Verlag, 2003, and in Peter G.M. Wuts, Greene's Protective Groups in Organic Synthesis, 5
th Edition, Wiley, 2014, all of which are incorporated herein by reference in its entirety.
Reactions can be monitored according to any suitable method known in the art. For example, product formation can be monitored by spectroscopic means, such as nuclear magnetic resonance spectroscopy (e.g.
1H or
13C) , infrared spectroscopy, spectrophotometry (e.g. UV-visible) , mass spectrometry, or by chromatographic methods such as high performance liquid chromatography (HPLC) , liquid chromatography-mass spectroscopy (LCMS) , or thin layer chromatography (TLC) . Compounds can be purified by one skilled in the art by a variety of methods, including high performance liquid chromatography (HPLC) ( “Preparative LC-MS Purification: Improved Compound Specific Method Optimization” Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6 (6) , 874-883, which is incorporated herein by reference in its entirety) , and normal phase silica chromatography.
The known starting materials of the present disclosure can be synthesized by using or according to the known methods in the art, or can be purchased from commercial suppliers. Unless otherwise noted, analytical grade solvents and commercially available reagents were used without further purification.
Unless otherwise specified, the reactions of the present disclosure were all done under a positive pressure of nitrogen or argon or with a drying tube in anhydrous solvents, and the reaction flasks were typically fitted with rubber septa for the introduction of substrates and reagents via syringe. Glassware was oven dried and/or heat dried.
Use of Compounds
In an aspect, the present disclosure provides a prodrug compound capable of locally delivering a therapeutic agent and releasing the therapeutic agent in a controlled and sustained manner with reduced systemic exposure and potential side effect due to systemic exposure. Thus, the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof are useful as therapeutic or prophylactic agent for various diseases, depending on the parent drug selected to be released.
As used herein, the term “therapy” is intended to have its normal meaning of dealing with a disease in order to entirely or partially relieve one, some or all of its symptoms, or to correct or compensate for the underlying pathology, thereby achieving beneficial or desired clinical results. For purposes of this disclosure, beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total) , whether detectable or undetectable. “Therapy” can also mean prolonging survival as compared to expected survival if not receiving it. Those in need of therapy include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented. The term “therapy” also encompasses prophylaxis unless there are specific indications to the contrary. The terms “therapeutic” and “therapeutically” should be interpreted in a corresponding manner.
The term “treatment” is used synonymously with “therapy” . Similarly the term “treat” can be regarded as “applying therapy” where “therapy” is as defined herein.
As used herein, the term “prophylaxis” is intended to have its normal meaning and includes primary prophylaxis to prevent the development of the disease and secondary prophylaxis whereby the disease has already developed and the patient is temporarily or permanently protected against exacerbation or worsening of the disease or the development of new symptoms associated with the disease.
The prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof shows a desired overall release rate of parent drug by controlling its solubility at a biological pH and the release of parent drug at different pHs.
In some embodiments, the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof has a lower solubility than the parent drug at a biological pH.
In certain embodiments, the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof shows a solubility at acidic pH that is higher than that at biological pH. In some embodiments, the ratio of the solubility of the prodrug compound of the present disclosure or a pharmaceutically acceptable salt thereof at acidic pH to that at biological pH is greater than 5, greater than 10, greater than 20, greater than 30, greater than 40, greater than 50, greater than 60, greater than 70, greater than 80, greater than 90, greater than 100, greater than 200, greater than 300, greater than 400, greater than 500, greater than 600, greater than 700, greater than 800, greater than 900, greater than 1000, greater than 1100, greater than 1200, greater than 1300, greater than 1400, greater than 1500 or even higher.
The reduced solubility of the prodrug compounds provided herein may avoid the high local concentration after administration, thereby providing a solubility controlled zero order sustained release mechanism.
The parent drug may be released from the prodrug compound provided herein through the cleavage of the linkage between the parent drug moiety and the tail moiety. The release of the parent drug may involve enzymatic or non-enzymatic processes. In some embodiments, the parent drug is released from the prodrug compound provided herein via hydrolysis process.
The release of the parent drug may be affected by a variety of factors, for example, the selection of specific parent drug, the linkage between the parent drug moiety and tail moiety, the tail moiety, and the administration of the prodrug compound (for example, administration site, administration route) . The present disclosure contemplates parent drugs with varying reactive groups and linkages with the tail moiety.
The present disclosure also contemplates varying administration of the prodrug compounds provided herein. In some embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof. In certain embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof via injection. In certain embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof via oral dosage form. In certain embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof via inhalation. In certain embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof via implant. In certain embodiments, the prodrug compounds provided herein is locally administered to a subject in need thereof via topical application. Depending on the specific parent drug, cleavable linkage and tail moiety combination, release of parent drug may occur in a variety of locations upon administration to a subject.
In some embodiments, the prodrug compounds provided herein or a pharmaceutically acceptable salt thereof may release the parent drugs via hydrolysis at different rates.
In some embodiments, the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the percent of the parent drug released from the prodrug compound at pH 7.4 in 6 hours after administration. In some embodiments, at pH 7.4, the release rate of the parent drug from the prodrug compound may vary in a range of about 50%to about 100%in 6 hours after administration, for example, about 55%to about 100%, about 60%to about 100%, about 65%to about 100%, about 70%to about 100%, about 75%to about 100%, about 80%to about 100%, about 85%to about 100%, about 90%to about 100%, about 95%to about 100%, about 96%to about 100%, about 97%to about 100%, about 98%to about 100%, about 99%to about 100%, or even about 100%.
In some embodiments, the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the percent of the parent drug released from the prodrug compound at pH 7.4 in 0.5 hours after administration. In some embodiments, at pH 7.4, the release rate of the parent drug from the prodrug compound may vary in a range of about 5%to about 100%in 0.5 hours after administration, for example, about 10%to about 100%, about 20%to about 100%, about 30%to about 100%, about 40%to about 100%, about 50%to about 100%, about 60%to about 100%, about 70%to about 100%, about 80%to about 100%, about 90%to about 100%, about 95%to about 100%, about 96%to about 100%, about 97%to about 100%, about 98%to about 100%, about 99%to about 100%, or even about 100%.
In some embodiments, at pH 2.0, the release rate of the parent drug from the prodrug compound is not greater than 70%in 6 hours after administration, for example, not greater than 65%, not greater than 60%, not greater than 55%, not greater than 50%, not greater than 45%, not greater than 40%, not greater than 35%, not greater than 30%, not greater than 25%, not greater than 20%, not greater than 15%, not greater than 10%, not greater than 5%, not greater than 4%, not greater than 3%, not greater than 2%, or not greater than 1%. In certain embodiments, at pH 2.0, the prodrug compound provided herein remains stable without releasing the parent drug via hydrolysis.
In some embodiments, the release rate of the parent drug from the prodrug compound provided herein or a pharmaceutically acceptable salt thereof may be characterized by the hydrolysis constant (K
h) of the prodrug compound in 6 hours after administration. In some embodiments, at pH 7.4, the prodrug compound provided herein may have a K
h value in a range of about 0.1 to about 15, for example, about 0.1 to about 14, about 0.1 to about 13, about 0.1 to about 12, about 0.1 to about 11, about 0.1 to about 10, about 0.1 to about 9, about 0.1 to about 8, about 0.1 to about 7, about 0.1 to about 6, about 0.1 to about 5, about 0.1 to about 4, about 0.1 to about 3, about 0.1 to about 2, about 0.1 to about 1, about 0.1 to about 0.5, about 0.1 to about 0.4, about 0.1 to about 0.3, or about 0.1 to about 0.2.
In some embodiments, at pH 2.0, the prodrug compound provided herein may have a K
h value not greater than 0.2, for example, not greater than not greater than 0.15, not greater than 0.1, not greater than 0.09, not greater than 0.08, not greater than 0.07, not greater than 0.06, not greater than 0.05, not greater than 0.04, not greater than 0.03, not greater than 0.02, or not greater than 0.01. In certain embodiments, the prodrug compound provided herein may have a K
h value of 0.
In some embodiments, the prodrug compound provided herein release more parent drug in 6 hours after administration at pH 7.4 than at pH 2.0.
In certain embodiments, the ratio of the percent of the parent drug released from the prodrug compound provided herein in 6 hours after administration at pH 7.4 to that at pH 2.0 is greater than 1, for example, greater than 1.1, greater than 1.2, greater than 1.3, greater than 1.4, greater than 1.5, greater than 2, great than 2.5, greater than 3, greater than 3.5, greater than 4, greater than 4.5, greater than 5, greater than 5.5, greater than 6, greater than 7, greater than 8, greater than 9, greater than 10, and the like.
In certain embodiments, the ratio of K
h value of the prodrug compound provided herein at pH 7.4 to that at pH 2.0 is greater than 1, for example, greater than 1.5, greater than 2, greater than 3, greater than 4, greater than 5, greater than 10, greater than 20, greater than 30, greater than 40, greater than 50, greater than 60, greater than 70, greater than 80, greater than 90, greater than 100, greater than 150, greater than 200, greater than 250, greater than 300, greater than 350, greater than 400, and the like.
By deliberately selecting suitable parent drug and the tail moiety to achieve the desired combination of solubility and release profile, the prodrug compound provided herein may provide sustained release of parent drug over a period of 1-12 hours.
Pharmaceutical Compositions
In a further aspect, there is provided pharmaceutical compositions comprising the prodrug compounds of the present disclosure.
In another aspect, there is provided pharmaceutical compositions comprising the prodrug compounds of the present disclosure, and at least one pharmaceutical acceptable excipient.
As used herein, the term “pharmaceutical composition” refers to a formulation containing the drug delivery system of the present disclosure in a form suitable for administration to a subject.
As used herein, the term “pharmaceutically acceptable excipient” means an excipient that is useful in preparing a pharmaceutical composition that is generally safe, non-toxic and neither biologically nor otherwise undesirable, and includes excipient that is acceptable for veterinary use as well as human pharmaceutical use. A “pharmaceutically acceptable excipient” as used herein includes both one and more than one such excipient. The term “pharmaceutically acceptable excipient” also encompasses “pharmaceutically acceptable carrier” and “pharmaceutically acceptable diluent” .
The pharmaceutical compositions provided herein can be in any form that allows for the composition to be administered to a subject, including, but not limited to a human, and formulated to be compatible with an intended route of administration.
A variety of routes are contemplated for the pharmaceutical compositions provided herein, and accordingly the pharmaceutical composition provided herein may be supplied in bulk or in unit dosage form depending on the intended administration route. For example, for oral, buccal, and sublingual administration, powders, granules, tablets, pills, capsules, gelcaps, and caplets may be acceptable as solid dosage forms, and emulsions, syrups, elixirs, suspensions, and solutions may be acceptable as liquid dosage forms. For injection administration, gel, solutions, emulsions and suspensions may be acceptable as liquid dosage forms, and a powder suitable for reconstitution with an appropriate solution as solid dosage forms. For inhalation administration, solutions, sprays, dry powders, and aerosols may be acceptable dosage form. For topical (including buccal and sublingual) or transdermal administration, powders, sprays, ointments, pastes, creams, lotions, gels, solutions, and patches may be acceptable dosage form. For vaginal administration, pessaries, tampons, creams, gels, pastes, foams and spray may be acceptable dosage form. For implant administration, solid, semi-solid, gel may be acceptable dosage form.
In some embodiments, the pharmaceutical compositions of the present disclosure may be in a form of formulation for oral administration.
In some embodiments, the pharmaceutical compositions of the present disclosure may be in a form of formulation for injection administration.
In some embodiments, the pharmaceutical compositions of the present disclosure may be in a form of formulation for inhalation administration.
In some embodiments, the pharmaceutical compositions of the present disclosure may be in a form of formulation for topical administration.
In certain embodiments, the pharmaceutical compositions provided herein may be formulated in the form of skin patches that are well known to those of ordinary skill in the art.
Besides those representative dosage forms described above, pharmaceutically acceptable excipients and carriers are generally known to those skilled in the art and are thus included in the present disclosure. Such excipients and carriers are described, for example, in “Remingtons Pharmaceutical Sciences” Mack Pub. Co., New Jersey (1991) , in “Remington: The Science and Practice of Pharmacy” , Ed. University of the Sciences in Philadelphia, 21
st Edition, LWW (2005) , which are incorporated herein by reference.
In some embodiments, the pharmaceutical compositions of the present disclosure can be formulated as a single dose. The amount of the prodrug compounds provided herein in the single dose will vary depending on the subject treated and particular mode of administration.
In some embodiments, the pharmaceutical compositions of the present disclosure can be formulated to be administered to a subject at a time interval of a few days, a few weeks, a few months or even longer.
In a further aspect, there is also provided pharmaceutical compositions comprise the drug delivery system of the present disclosure, as two or more combination therapy.
Method of treatment of disease
In a further aspect, there is provided a method of treating diseases in a subject in need thereof, comprising administering to the subject a therapeutic effective amount of the prodrug compound or the pharmaceutical composition provided herein.
The diseases to be treated depends on the selected parent drug in the prodrug or the pharmaceutical composition provided herein. In some embodiments, the disease can be selected from the group consisting of anal cancer, breast cancer, colorectal cancer, esophageal cancer, pancreatic cancer, head and neck cancer, brain cancer, liver cancer, gastric cancer, bladder cancer, oral mucosal cancer, esophageal cancer, anaplastic astrocytoma, glioblastoma multiforme, acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, Kaposi’s sarcoma, and neuroblastoma.
In some embodiments, the parent drug is selected from an anti-cancer agent, an anti-inflammatory drug, an antibiotic, an anti-fugus agent, a JAK inhibitor, or a VEGF inhibitor.
In some embodiments, the parent drug selected is Fluorouracil, and thus the prodrug compound provided herein is useful in the treatment of cancers, including colon, esophageal, gastric, rectum, breast, biliary tract, stomach, head and neck, cervical, pancreas, renal cell, and carcinoid.
In some embodiments, the parent drug selected is Temozolomide, and thus the prodrug compound provided herein is useful in the treatment of anaplastic astrocytoma and glioblastoma multiforme.
In some embodiment, the parent drug selected is Daunorubicin, and thus the prodrug compound provided herein is useful in the treatment of acute nonlymphocytic leukemia (myelogenous, monocytic, erythroid) and acute lymphocytic leukemia.
In some embodiment, the parent drug selected is 10-hydroxyl-camptothecine or 7-ethyl-10-hydroxyl-camptothecine, and thus the prodrug compound provided herein is useful in the treatment of cancers, including gastric, esophagus, cardiac, colon, liver, lung, bladder, acute leukemia, chronic myelogenous leukemia, and chorionic epithelioma.
In this context, the term “therapeutically effective amount” refers to an amount of a therapeutic agent selected in the drug delivery system provided herein or pharmaceutically acceptable salts thereof which is effective to provide “therapy” in a subject, or to “treat” disorders, diseases or conditions in a subject.
EXAMPLES
For the purpose of illustration, the following examples are included. However, it is to be understood that these examples do not limit the present disclosure and are only meant to suggest a method of practicing the present disclosure. Persons skilled in the art will recognize that the chemical reactions described may be readily adapted to prepare a number of other compounds of the present disclosure or a pharmaceutically acceptable salt thereof, and alternative methods for preparing the compounds of the present disclosure or a pharmaceutically acceptable salt thereof are deemed to be within the scope of the present disclosure. For example, the synthesis of non-exemplified compounds according to the present disclosure may be successfully performed by modifications apparent to those skilled in the art, e.g., by appropriately protecting interfering groups, by utilizing other suitable reagents and building blocks known in the art other than those described, and/or by making routine modifications of reaction conditions. Alternatively, other reactions disclosed herein or known in the art will be recognized as having applicability for preparing other compounds of the present disclosure.
As used herein the symbols and conventions used in these processes, schemes and examples are consistent with those used in the contemporary scientific literature, for example, the Journal of the American Chemical Society or the Journal of Biological Chemistry. Unless otherwise noted, all starting materials were obtained from commercial suppliers and used without further purification. For example, the following abbreviations may be used in the examples and throughout the specification: g (grams) ; mg (milligrams) ; L (liters) ; mL (milliliters) ; μL (microliters) ; psi (pounds per square inch) ; M (molar) ; mM (millimolar) ; i.v. (intravenous) ; Hz (Hertz) ; MHz (megahertz) ; aq. (aqueous solution) ; mol (moles) ; mmol (millimoles) ; r.t. (room temperature) ; min (minutes) ; h (hours) ; mp (melting point) ; TLC (thin layer chromatography) ; Rt (retention time) ; RP (reverse phase) ; AcOH (acetic acid) ; MeOH (methanol) ; i-PrOH (isopropanol) ; TEA (triethylamine) ; TFA (trifluoroacetic acid) ; THF (tetrahydrofuran) ; DMSO (dimethyl sulfoxide) ; EtOAc (ethyl acetate) ; DCM (dichloromethane) ; HCHO (formaldehyde) ; MeCN (acetonitrile) ; DIPEA (N, N-diisopropylethylamine) ; PE (petroleum ether) ; DMF (N, Ν-dimethylformamide) ; Pd
2dba
3 (tris (dibenzylideneacetone) dipalladium) ; NMP (1-methyl-2-pyrrolidinone) ; HATU (2- (7-azabenzotriazol-1-yl) -N, N, N', N'-tetramethyluronium hexafluorophosphate) ; BOP ( (benzotriazol-1-yloxy) tris (dimethylamino) phosphonium hexafluorophosphate) .
Unless otherwise indicated, all temperatures are expressed in ℃ (degrees Centigrade) . All reactions were conducted under an inert atmosphere at r.t. unless otherwise noted.
1H NMR spectra were recorded on an Agilent 400MR NMR Spectrometer. Chemical shifts are expressed in parts per million (ppm) . Coupling constants are in units of hertz (Hz) . Splitting patterns describe apparent multiplicities and are designated as s (singlet) , d (doublet) , t (triplet) , q (quartet) , m (multiplet) and br (broad) .
Low-resolution mass spectra (MS) and compound purity data were acquired on an Agilent LC/MS single quadrapole system equipped with electrospray ionization (ESI) source, and UV detector (215 and 254 nm) .
Example 1
2- (1- (4- (Dibutylamino) phenyl) piperidin-4-yl) ethyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (1)

Synthetic Route of 1

5-Fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a)

A reaction of 5-Fluorouracil (10g, 76.9mmol) and 37%HCHO (aq. ) (100mL) in sealed tube was stirred at 60℃ for 5 hours, and the mixture was concentrated to give the crude product of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) , which was used for next step directly. MS-ESI (m/z) : 161 [M+1]
+.
Ethyl 2- (1- (4-nitrophenyl) piperidin-4-yl) acetate (1b)

A reaction of 1-fluoro-4-nitrobenzene (248mg, 1.76mmol) , ethyl 2- (piperidin-4-yl) acetate hydrochloride (443mg, 2, 13mmol) and K
2CO
3 (738mg, 5, 34mmol) in MeCN (10mL) was stirred at 110℃ in sealed tube for 5 hours, and the mixture was filtered and concentrated to give the crude product of ethyl 2- (1- (4-nitrophenyl) piperidin-4-yl) acetate (1b) , which was used for next step directly. MS-ESI (m/z) : 293 [M+1]
+.
Ethyl 2- (1- (4-aminophenyl) piperidin-4-yl) acetate (1c)

A mixture of ethyl 2- (1- (4-nitrophenyl) piperidin-4-yl) acetate (1b) (415mg, 1.42mmol) , Fe powder (1.5g, 26.8mmol) , (NH
4)
2SO
4 (108mg, 0.81mmol) and 1M HCl (2.5mL) in EtOH (20mL) was refluxed for 3 hours. The mixture was filtered and concentrated to give the crude product of ethyl 2- (1- (4- aminophenyl) piperidin-4-yl) acetate (1c) , which was used for next step directly. MS-ESI (m/z) : 263 [M+1]
+.
Ethyl 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) acetate (1d)

To a solution of ethyl 2- (1- (4-aminophenyl) piperidin-4-yl) acetate (1c) (370mg, 1.41mmol) in MeOH (20mL) , AcOH (1mL) , n-butyraldehyde (415μL, 4.7mmol) and NaBH
3CN (820mg, 13.0mmol) was added. The mixture was stirred for 1.5 h , then concentrated to remove MeOH, dissolved in EtOAc and washed with NaHCO
3 (aq) , dried and concentrated to give the crude product of ethyl 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) acetate (1d) , which was used for next step directly. MS-ESI (m/z) : 375 [M+1]
+.
2- (1- (4- (Dibutylamino) phenyl) piperidin-4-yl) ethan-1-ol (1e)

To a solution of ethyl 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) acetate (1d) (2.1g, 5.6mmol) in THF (24mL) , was added 1M LiAlH
4-THF (35mL, 35mmol) . The mixture was stirred overnight, and quenched with water, extracted with EtOAc, dried and concentrated. The residue was purified by column chromatography on silica gel eluting with petroleum ether and EtOAc to give 304 mg of 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) ethan-1-ol (1e) . MS-ESI (m/z) : 333 [M+1]
+.
2- (1- (4- (Dibutylamino) phenyl) piperidin-4-yl) ethyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (1)

To a solution of 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) ethan-1-ol (1e) (160mg, 0.48mmol) in DCM (5mL) cooled below -50℃, was added the solution of DIPEA (850μL, 4.8mmol) and triphosgene (142mg, 0.48mmol) in DCM (1.4mL) . The mixture was stirred at ambient temperature for 2 h, and added to the solution of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (310mg, 1.9mmol) in MeCN (5mL) . The reaction was stirred overnight, and purified by preparative chromatography eluting with MeCN and 0.1%TFA-H
2O to give 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) ethyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (1) (39 mg) . MS-ESI (m/z) : 519 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.03 (d, J = 5.0 Hz, 1H) , 8.14 (d, J = 6.5 Hz, 1H) , 7.38 (brs, 2H) , 6.67 (brs, 2H) , 5.58 (s, 2H) , 4.18 (t, J = 6.2 Hz, 2H) , 3.61 –2.93 (m, 8H) , 2.03 –0.58 (m, 21H) .
Example 2
4'- (Dipropylamino) - [1, 1'-biphenyl] -4-yl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (2)

Synthetic Route of 2

4'-Amino- [1, 1'-biphenyl] -4-ol (2a)

A reaction of 4'-nitro- [1, 1'-biphenyl] -4-ol (500mg, 2.32mmol) , Fe powder (2.63g, 47.0mmol) , NH
4Cl (125mg, 2.32mmol) , AcOH (500μL) and 1M HCl (250μL) in EtOH (40mL) was refluxed for 3.5h. The mixture was filtered and concentrated to give the crude product of 4'-amino- [1, 1'-biphenyl] -4-ol (2a) , which was used for next step directly. MS-ESI (m/z) : 186 [M+1]
+.
4'- (Dipropylamino) - [1, 1'-biphenyl] -4-ol (2b)

4'-Amino- [1, 1'-biphenyl] -4-ol (2a) (400mg, 2.16mmol) was dissolved in MeOH (40mL) and treated with propionaldehyde (760μL, 10.4mmol) , AcOH (500μL) and NaBH
3CN (1.75g, 27.9mmol) . The mixture was stirred overnight. The reaction mixture was filtered and concentrated, the residue was diluted with EtOAc and washed with NaHCO
3 (aq. ) . The aqueous layer was extracted with EtOAc twice. The combined layers were dried over Na
2SO
4, and concentrated to dryness. The residue was purified by column chromatography on silica gel (PE: EtOAc = 10: 1 to 3: 1) to give the product of 4'- (dipropylamino) - [1, 1'-biphenyl] -4-ol (2b) (510mg) . MS-ESI (m/z) : 270 [M+1]
+.
4'- (Dipropylamino) - [1, 1'-biphenyl] -4-yl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (2)

To a solution of 4'- (dipropylamino) - [1, 1'-biphenyl] -4-ol (2b) (270mg, 1mmol) in DCM (5mL) cooled below -50℃ was added DIPEA (1.8mL, 10mmol) and triphosgene (300mg, 1mmol) in DCM (3mL) , the mixture was stirred below -50℃ for 2h. A suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (640mg) in MeCN (5mL) was added into the mixture and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography eluting with MeCN and 0.1%TFA-H
2O to give 4'- (dipropylamino) - [1, 1'-biphenyl] -4-yl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (2) (39mg) . MS-ESI (m/z) : 456 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.06 (d, J = 5.0 Hz, 1H) , 8.16 (d, J = 6.5 Hz, 1H) , 7.67 –7.39 (m, 4H) , 7.27 (d, J = 8.3 Hz, 2H) , 6.72 (brs, 2H) , 5.70 (s, 2H) , 3.28 (brs, 4H) , 1.50 (brs, 4H) , 0.87 (t, J = 7.4 Hz, 6H) .
Example 3
4- (Dipentylamino) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (3)

Synthetic Route of 3

4- (Dipentylamino) phenol (3a)

To a mixture of 4-aminophenol (4.0g, 36.6mmol) , 1-iodopentane (17.1g, 86.5mmol) in DMF (72mL) was added K
2CO
3 (5.2g, 37.4mmol) . The reaction was stirred at 75℃ for 1.5h. The reaction mixture was poured into water (300mL) , and extracted with EtOAc 3 times. The combined organic layers were washed with Na
2S
2O
3 (aq. ) , dried over Na
2SO
4, and evaporated to dryness. The residue was purified by silica gel chromatography eluted with PE and EA to give 4- (dipentylamino) phenol (3a) . MS-ESI (m/z) : 250 [M+1]
+.
4- (Dipentylamino) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (3)

To a solution of 4- (dipentylamino) phenol (3a) (250mg, 1mmol) in DCM (5mL) cooled below -50℃ was added DIPEA (1.8mL, 10mmol) and triphosgene (300mg, 1mmol) , the mixture was stirred for 1h. The mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (800mg) in MeCN (5mL) dried over
 molecular sieves and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give 4- (dipentylamino) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (3) (95mg) . MS-ESI (m/z) : 436 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.05 (d, J = 5.0 Hz, 1H) , 8.14 (d, J = 6.5 Hz, 1H) , 7.28 –6.40 (m, 4H) , 5.67 (s, 2H) , 3.47 –3.11 (m, 4H) , 1.42 (s, 4H) , 1.31 –1.13 (m, 8H) , 0.83 (t, J = 6.8 Hz, 6H) .
molecular sieves and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give 4- (dipentylamino) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (3) (95mg) . MS-ESI (m/z) : 436 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.05 (d, J = 5.0 Hz, 1H) , 8.14 (d, J = 6.5 Hz, 1H) , 7.28 –6.40 (m, 4H) , 5.67 (s, 2H) , 3.47 –3.11 (m, 4H) , 1.42 (s, 4H) , 1.31 –1.13 (m, 8H) , 0.83 (t, J = 6.8 Hz, 6H) .
Example 4
4- (1- (3, 5-Dimethylphenyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (4)

Synthetic Route of 4

4- (1- (3, 5-Dimethylphenyl) piperidin-4-yl) phenol (4a)

A mixture of 1-bromo-3, 5-dimethylbenzene (0.74g, 4mmol) , 4- (piperidin-4-yl) phenol hydrobromide (1.04g, 4mmol) , 2- (di-t-butylphosphino) ) -1, 1'-biphenyl (300mg, 1mmol) , Pd
2dba
3 (370mg, 0.40mmol) and t-BuONa (0.76g, 7.9mmol) in dioxone (40mL) under nitrogen gas was stirred at 55℃ for 3h. And 2- (di-t-butylphosphino) ) -1, 1'-biphenyl (300mg, 1mmol) , Pd
2dba
3 (370mg, 0.40mmol) and t-BuONa (0.76g, 7.9mmol) was added into the reaction additionally. The reaction was stirred at 55℃ for 5h. The mixture was filtered and washed with EtOAc and the filtrate was concentrated. The residue was purified by column chromatography on silica gel to give 4- (1- (3, 5-dimethylphenyl) piperidin-4-yl) phenol (4a) (638mg) . MS-ESI (m/z) : 282 [M+1]
+.
4- (1- (3, 5-Dimethylphenyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (4)

To a solution of 4- (1- (3, 5-dimethylphenyl) piperidin-4-yl) phenol (4a) (158mg, 0.56mmol) in DCM (4mL) cooled below -50℃ was added DIPEA (1.0mL, 5.6mmol) and triphosgene (167mg, 0.56mmol) , the mixture was stirred for 1h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (450mg) in MeCN (5mL) and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give 4- (1- (3, 5-dimethylphenyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (4) (40mg) . MS-ESI (m/z) : 468 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.05 (d, J = 5.0 Hz, 1H) , 8.14 (d, J = 6.6 Hz, 1H) , 7.33 (d, J = 8.4 Hz, 2H) , 7.21 (d, J = 8.3 Hz, 2H) , 7.07 –6.60 (m, 3H) , 5.69 (s, 2H) , 4.57 –3.59 (m, 4H) , 2.83 (brs, 1H) , 2.25 (s, 6H) , 2.04 –1.76 (m, 4H) .
Example 5
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-yl) carbonate (5)

Synthetic Route of 5

2- (4-Propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-ol (5a)

A suspension of 1, 2, 3, 4-tetrahydroisoquinolin-6-ol (0.50g, 3.36mmol) , 1-bromo-4-propylbenzene (0.75g, 3.75mmol) , Pd
2dba
3 (200mg, 0.22mmol) , dicyclohexyl (2', 4', 6'-triisopropyl- [1, 1'-biphenyl] -2-yl) phosphane (300mg, 0.63mmol) and Cs
2CO
3 (3.0g, 9.2mmol) in PhMe (15mL) was stirred in sealed tube at 110℃ for 7h. The mixture was diluted with EtOAc and filtered. The filtrate was concentrated and purified by column chromatography on silica gel to give 2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-ol (5a) (109mg) . MS-ESI (m/z) : 268 [M+1]
+.
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-yl) carbonate (5)

To a solution of 2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-ol (5a) (26.7mg, 0.1mmol) in DCM (1mL) cooled below -50℃ was added DIPEA (200μL, 1.1mmol) and triphosgene (30mg, 0.1mmol) , the mixture was stirred for 1h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (80mg) in MeCN (1mL) dried over
 molecular sieves and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-yl) carbonate (5) (2.0mg) . MS-ESI (m/z) : 454 [M+1]
+.
molecular sieves and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (2- (4-propylphenyl) -1, 2, 3, 4-tetrahydroisoquinolin-6-yl) carbonate (5) (2.0mg) . MS-ESI (m/z) : 454 [M+1]
+.
Example 6
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (4- ( (1- (4-propylphenyl) piperidin-4-yl) methyl) phenyl) carbonate (6)

Synthetic Route of 6

(4-Methoxyphenyl) (1- (4-propylphenyl) piperidin-4-yl) methanone (6a)

A suspension of (4-methoxyphenyl) (piperidin-4-yl) methanone hydrochloride (1.0g, 3.9mmol) , 1-bromo-4-propylbenzene (0.8g, 4.0mmol) , Pd
2dba
3 (732mg, 0.8mmol) , dicyclohexyl (2', 4', 6'-triisopropyl- [1, 1'-biphenyl] -2-yl) phosphane (1.15g, 2.4mmol) and t-BuONa (1.1g, 11.4mmol) in PhMe (20mL) was stirred in sealed tube under nitrogen gas at 110℃ for 4h. The mixture was filtered. The filtrate was concentrated and purified by column chromatography on silica gel to give (4-methoxyphenyl) (1- (4-propylphenyl) piperidin-4-yl) methanone (6a) (1.09g) . MS-ESI (m/z) : 338 [M+1]
+.
4- (4-Methoxybenzyl) -1- (4-propylphenyl) piperidine (6b)

To a solution of (4-methoxyphenyl) (1- (4-propylphenyl) piperidin-4-yl) methanone (6a) (600mg, 1.78mmol) in MeOH (20mL) was added NaBH
4 (200mg, 5.33mmol) . After being stirred for 40min, NaBH
4 (200mg, 5.33mmol) was added into the mixture additionally. The reaction was stirred for 1.5h and quenched with NaHCO
3 aqueous solution. The mixture was extracted with EtOAc, dried over Na
2SO
4, and evaporated to dryness to afford as a black oily residue.
The residue was dissolved in TFA (20mL) followed by addition of triethylsilane (3mL) . The reaction was stirred at 80℃ for 1h and quenched with NaHCO
3 aqueous solution. The mixture was extracted with EtOAc to give the crude product of 4- (4-methoxybenzyl) -1- (4-propylphenyl) piperidine (6b) (1.87g) , was used for next step directly. MS-ESI (m/z) : 324 [M+1]
+.
4- ( (1- (4-Propylphenyl) piperidin-4-yl) methyl) phenol (6c)

To a solution of 4- (4-methoxybenzyl) -1- (4-propylphenyl) piperidine (6b) (1.87g) in DCM (10mL) was added 1M BBr
3-DCM solution (6mL) . The reaction was stirred for 1h. After quenching with NaHCO
3 aqueous solution, the mixture was extracted with DCM. The crude product was purified by silica gel chromatography to give 4- ( (1- (4-propylphenyl) piperidin-4-yl) methyl) phenol (6c) (400mg) . MS-ESI (m/z) : 310 [M+1]
+.
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (4- ( (1- (4-propylphenyl) piperidin-4-yl) methyl) phenyl) carbonate (6)

To a solution of 4- ( (1- (4-propylphenyl) piperidin-4-yl) methyl) phenol (6c) (320mg, 1.0mmol) in DCM (5mL) cooled below -50℃ was added DIPEA (1.7mL, 10.3mmol) and triphosgene (307mg, 1.0mmol) , the mixture was stirred for 1h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (830mg) in MeCN (5mL) and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (4- ( (1- (4-propylphenyl) piperidin-4-yl) methyl) phenyl) carbonate (6) (49mg) . MS-ESI (m/z) : 496 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.05 (d, J = 5.0 Hz, 1H) , 8.14 (d, J = 6.5 Hz, 1H) , 7.43 –7.09 (m, 8H) , 5.69 (s, 2H) , 3.85 –2.96 (m, 4H) , 2.60 (d, J = 6.7 Hz, 2H) , 2.52 (d, J = 7.6 Hz, 2H) , 1.78 (d, J = 14.3 Hz, 3H) , 1.53 (dt, J = 16.6, 8.4 Hz, 4H) , 0.85 (t, J = 7.3 Hz, 3H) .
Example 7
4- (1- (2, 2-Diphenylethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (7)

Synthetic Route of 7

4- (1- (2, 2-Diphenylethyl) piperidin-4-yl) phenol (7a)

To a solution of 4- (piperidin-4-yl) phenol hydrobromide (500mg, 1.9mmol) , 2, 2-diphenylacetaldehyde (400mg, 2.0mmol) and AcOH (0.5mL) in MeOH (20mL) was added NaBH
3CN (1.0g, 15.9mmol) . The reaction was stirred at ambient temperature overnight. After removal of solvent by evaporation, the residue was partitioned between EtOAc and NaHCO
3 (aq. ) . The separated organic layer was dried over Na
2SO
4 and evaporated to dryness. The crude product was purified by silica gel chromatography to give 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) phenol (7a) (400mg) . MS-ESI (m/z) : 358 [M+1]
+.
4- (1- (2, 2-Diphenylethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (7)

To a solution of 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) phenol (7a) (380mg, 1.06mmol) in DCM (5mL) cooled below -50℃ was added DIPEA (1.8mL, 10.3mmol) and triphosgene (315mg, 1.06mmol) , the mixture was stirred for 0.5h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (855mg) in MeCN (5mL) and stirred for 2h. The reaction mixture was filtered and purified by preparative chromatography to give 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (7) (39mg) . MS-ESI (m/z) : 544 [M+1]
+.
Example 8
4- (1- (Adamantan-1-ylmethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (8)

Synthetic Route of 8

Adamantan-1-yl (4- (4-hydroxyphenyl) piperidin-1-yl) methanone (8a)

To a mixture of 4- (piperidin-4-yl) phenol hydrobromide (520mg, 2mmol) and adamantane-1-carbonyl chloride (400mg, 2mmol) in DCM (20mL) was added DIPEA (1mL, 5.7mmol) , the reaction was stirred at ambient temperature for 4 h followed by evaporation to give the crude product of adamantan-1-yl (4- (4-hydroxyphenyl) piperidin-1-yl) methanone (8a) , which was used for next step directly. MS-ESI (m/z) : 340 [M+1]
+.
4- (1- (Adamantan-1-ylmethyl) piperidin-4-yl) phenol (8b)

Adamantan-1-yl (4- (4-hydroxyphenyl) piperidin-1-yl) methanone (8a) (650mg, 1.9mmol) was dissolved in THF (20mL) followed by addition of 1M LiAlH
4-THF (6mL, 6mmol) at ambient temperature. The reaction was stirred for 0.5h and quenched by addition of NaOH solution. The mixture was filtered and the filtrate was concentrated. The crude product was purified by column chromatography to give the product of 4- (1- (adamantan-1-ylmethyl) piperidin-4-yl) phenol (8b) (595mg) . MS-ESI (m/z) : 326 [M+1]
+.
4- (1- (Adamantan-1-ylmethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (8)

To a solution of 4- (1- (adamantan-1-ylmethyl) piperidin-4-yl) phenol (8b) (575mg, 1.77mmol) in DCM (5mL) cooled below -50℃ was added DIPEA (2.9mL, 16.6mmol) and triphosgene (524mg, 1.77mmol) , the mixture was stirred for 0.5h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (1.13g) in MeCN (6mL) and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give 4- (1- (adamantan-1-ylmethyl) piperidin-4-yl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (8) (167mg) . MS-ESI (m/z) : 512 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.05 (d, J = 5.0 Hz, 1H) , 8.13 (d, J = 6.6 Hz, 1H) , 7.41 –7.19 (m, 4H) , 5.69 (s, 2H) , 3.76 –2.77 (m, 7H) , 2.18 –1.55 (m, 19H) .
Example 9
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (9)

Synthetic Route of 9

(1-Benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanone (9a)

A suspension of (4-methoxyphenyl) (piperidin-4-yl) methanone hydrochloride (520mg, 2.0mmol) , (bromomethylene) dibenzene (520mg, 2.1mmol) , KI (340mg, 2.0mmol) and K
2CO
3 (820mg, 5.9mmol) in MeCN (20mL) was stirred at 90℃overnight. The mixture was filtered and the filtrate was evaporated to dryness to give the crude product of (1-benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanone (9a) . MS-ESI (m/z) : 386 [M+1]
+.
(1-Benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanol (9b)

(1-Benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanone (9a) (750mg, 1.94mmol) was dissolved in THF (20mL) followed by addition of 1M LiAlH
4-THF solution (4mL, 4mmol) . The reaction was stirred for 0.5h and quenched with NaOH (aq. ) solution. The mixture was filtered and the filtrate was evaporated to dryness to give the crude product of (1-benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanol (9b) . MS-ESI (m/z) : 388 [M+1]
+.
1-Benzhydryl-4- (4-methoxybenzyl) piperidine (9c)

(1-Benzhydrylpiperidin-4-yl) (4-methoxyphenyl) methanol (9b) (939mg, 2.43mmol) was dissolved in TFA (5mL) followed by addition HSiEt
3 (2.5mL) . The reaction was stirred at for 1h and concentrated. The residue was dissolved in DCM, washed with NaHCO
3 solution. The organic layer was dried over Na
2SO
4 and concentrated to give the crude product of 1-benzhydryl-4- (4-methoxybenzyl) piperidine (9c) . MS-ESI (m/z) : 372 [M+1]
+.
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenol (9d)

1-Benzhydryl-4- (4-methoxybenzyl) piperidine (9c) (1.07g, 2.88mmol) was dissolved in DCM (8mL) followed by addition of 1M BBr
3-DCM solution (6mL, 6mmol) . The mixture was stirred at room temperature for 5h. The reaction was diluted with DCM and quenched with NaHCO
3 (aq. ) solution. The aqueous layer was extracted with DCM. The combined DCM layers were dried over Na
2SO
4, and concentrated to dryness. The residue was purified by column chromatography on silica gel (PE: EtOAc = 10: 1 to 3: 1) to give the product of 4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenol (9d) (504mg) . MS-ESI (m/z) : 358 [M+1]
+.
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (9)

To a solution of 4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenol (9d) (100mg, 0.28mmol) in DCM (3mL) cooled below -50℃ was added DIPEA (700μL, 4.2mmol) and triphosgene (100mg, 0.34mmol) and the mixture was stirred for 0.5h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (180mg) in MeCN (3mL) and stirred for 1h. The reaction mixture was filtered and purified by preparative chromatography to give 4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) carbonate (9) (27mg) . MS-ESI (m/z) : 544 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.61 (d, J = 5.2 Hz, 1H) , 7.37 (d, J = 7.5 Hz, 4H) , 7.29 –7.10 (m, 8H) , 7.05 (d, J = 8.5 Hz, 2H) , 5.74 (s, 2H) , 4.21 (s, 1H) , 2.84 (d, J = 11.4 Hz, 2H) , 2.53 (d, J = 6.6 Hz, 2H) , 2.35 (s, 1H) , 1.76 (t, J = 11.5 Hz, 2H) , 1.61 –1.42 (m, 3H) , 1.38 –1.24 (m, 2H) .
Example 10
4- (4-Butylpiperidin-1-yl) phenethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (10)

Synthetic Route of 10

Methyl 2- (4- (4-butylpiperidin-1-yl) phenyl) acetate (10a)

A suspension of methyl 2- (4-bromophenyl) acetate (241.2mg, 1.05mmol) , 4-butylpiperidine hydrochloride (207.6g, 1.17mmol) , Pd
2dba
3 (33mg, 0.035mmol) , dicyclohexyl (2', 4', 6'-triisopropyl- [1, 1'-biphenyl] -2-yl) phosphane (67mg, 0.14mmol) and Cs
2CO
3 (1.14g, 3.51mmol) in PhMe (10mL) was stirred in sealed tube at 110℃for 18h. The mixture was diluted with MeCN and filtered. The filtrate was concentrated to give the crude product of methyl 2- (4- (4-butylpiperidin-1-yl) phenyl) acetate (10a) , which was used for next step directly. MS-ESI (m/z) : 290 [M+1]
+.
2- (4- (4-Butylpiperidin-1-yl) phenyl) ethan-1-ol (10b)

Methyl 2- (4- (4-butylpiperidin-1-yl) phenyl) acetate (10a) (300mg, 1.03mmol) was dissolved in THF (11mL) followed by addition of 1M LiAlH
4-THF (3mL) . The mixture was stirred at room temperature overnight. The reaction was quenched with water. The aqueous layer was extracted with EtOAc 3 times. The combined EtOAc layers were dried over Na
2SO
4 and evaporated to dryness. The residue was purified by preparative chromatography to give 2- (4- (4-butylpiperidin-1-yl) phenyl) ethan-1-ol (10b) (281mg) . MS-ESI (m/z) : 262 [M+1]
+.
4- (4-Butylpiperidin-1-yl) phenethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (10)

A mixture of 2- (4- (4-butylpiperidin-1-yl) phenyl) ethan-1-ol (10b) (94mg, 0.36mmol) , triphosgene (108mg, 0.36mmol) and DIPEA (650μL) in DCM (3.5mL) cooled below -50℃ was stirred for 2h, followed by addition of the suspension of fluorouracil (50mg, 0.38mmol) in MeCN (3.6mL) . The reaction was stirred at room temperature for 2h followed by concentration to dryness. The crude product was purified by preparative chromatography to give 4- (4-butylpiperidin-1-yl) phenethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (10) (35mg) . MS-ESI (m/z) : 418 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.00 (d, J = 4.9 Hz, 1H) , 8.12 (d, J = 7.2 Hz, 1H) , 7.44 –7.16 (m, 4H) , 4.45 (t, J = 6.6 Hz, 2H) , 3.62 –2.94 (m, 4H) , 1.83 (d, J = 13.2 Hz, 2H) , 1.58 –0.96 (m, 11H) , 0.90 –0.77 (m, 3H) .
Example 11
2- (1- (4- (Dibutylamino) phenyl) piperidin-4-yl) ethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (11)

Synthetic Route of 11

2- (1- (4- (Dibutylamino) phenyl) piperidin-4-yl) ethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (11)

To the solution of 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) ethan-1-ol (1e) (278mg, 0.84mmol) in DCM (7mL) cooled below -50℃, was added the solution of DIPEA (1.4mL, 8.4mmol) and triphosgene (248mg, 0.84mmol) in DCM (2.5mL) . The mixture was stirred at ambient temperature for 1.5 h, and added to the suspension of fluorouracil (109mg, 0.84mmol) in MeCN (10mL) . The reaction was stirred overnight, and purified by preparative chromatography eluting with MeCN and 0.1%TFA-H
2O to give 2- (1- (4- (dibutylamino) phenyl) piperidin-4-yl) ethyl 5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidine-1 (2H) -carboxylate (11) (99mg) . MS-ESI (m/z) : 489 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.02 (s, 1H) , 8.21 (d, J = 7.2 Hz, 1H) , 7.39 (brs, 2H) , 6.67 (brs, 2H) , 4.37 (t, J = 6.1 Hz, 2H) , 3.90 –3.15 (m, 8H) , 2.12 – 1.05 (m, 15H) , 0.92 –0.68 (m, 6H) .
Example 12
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) methylphosphonate (12)

Synthetic Route of 12

4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) methylphosphonate (12)

To a solution of 4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenol (9d) (200mg, 0.56mmol) in DCM (2mL) at ambient temperature was added NEt
3 (310μL, 2.24mmol) and methyl phosphonic dichloride (90mg, 0.67mmol) , the mixture was stirred for 0.5h. The reaction mixture was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (360mg) in NMP (2mL) and stirred for 3h. The reaction mixture was filtered and purified by preparative chromatography to give 4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenyl ( (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl) methylphosphonate (12) (90mg) . MS-ESI (m/z) : 578 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.41 –7.29 (m, 5H) , 7.24 –7.18 (m, 4H) , 7.18 –7.09 (m, 2H) , 7.07 –6.89 (m, 4H) , 5.72 –5.31 (m, 2H) , 4.22 (s, 1H) , 2.95 –2.78 (m, 2H) , 2.55 –2.42 (m, 2H) , 1.83 –1.65 (m, 4H) , 1.55 –1.20 (m, 6H) .
Example 13
(S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl 2- (1- (2, 2-diphenylethyl) piperidin-4-yl) acetate (13)

Synthetic Route of 13

2- (1- (2, 2-Diphenylethyl) piperidin-4-yl) acetic acid (13a)

To a suspension of 2- (piperidin-4-yl) acetic acid hydrochloride (2.3g, 12.8mmol) , 2, 2-diphenylacetaldehyde (2.5g, 12.7mmol) and AcONa (3.1g, 37.8mmol) in MeOH (25mL) was added NaBH
3CN (2.0g, 31.8mmol) . The reaction was stirred at ambient temperature for 4.5 h. After removal of solvent by evaporation, the residue was partitioned between EtOAc and H
2O. The separated organic layer was dried over Na
2SO
4, and evaporated to dryness. The crude product was purified by silica gel chromatography to give 2- (1- (2, 2-diphenylethyl) piperidin-4-yl) acetic acid (13a) (517mg) . MS-ESI (m/z) : 324 [M+1]
+.
(S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl 2- (1- (2, 2-diphenylethyl) piperidin-4-yl) acetate (13)

A mixture of 10-hydroxycamptothecin (36mg, 0.1mmol) , 2- (1- (2, 2-diphenylethyl) piperidin-4-yl) acetic acid (13a) (32mg, 0.1mmol) , HATU (114mg, 0.3mmol) , DIPEA (105μL, 0.6mmol) in NMP (2 mL) was stirred at room temperature for 23h. The reaction mixture was purified by preparative chromatography to give (S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl 2- (1- (2, 2-diphenylethyl) piperidin-4-yl) acetate (13) as a trifluoroacetate salt. MS-ESI (m/z) : 670 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.99 (s, 1H) , 8.65 (s, 1H) , 8.20 (d, J = 9.2 Hz, 1H) , 7.89 (d, J = 2.8 Hz, 1H) , 7.65 (dd, J = 9.3, 2.6 Hz, 1H) , 7.47 –7.18 (m, 11H) , 6.52 (brs, 1H) , 5.41 (s, 2H) , 5.28 (s, 2H) , 4.60 –4.53 (m, 1H) , 3.95–3.85 (m, 2H) , 3.51 (d, J = 12.0 Hz, 2H) , 3.31 –2.85 (m, 2H) , 2.63 (d, J = 7.0 Hz, 2H) , 2.15 –1.72 (m, 5H) , 1.60 –1.45 (m, 1H) , 1.12 –0.96 (m, 1H) , 0.86 (t, J = 7.3 Hz, 3H) .
Example 14
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl benzhydrylprolinate (14)

Synthetic Route of 14

Benzhydrylproline (14a)

A reaction mixture of methyl prolinate hydrochloride (6.0g, 36.2mmol) , chlorodiphenylmethane (8.1g, 40.0mmol) , K
2CO
3 (15.0g, 108.5mmol) and KI (6.0g, 36.1mmol) in MeCN (120mL) was stirred at 60℃ for 24 hours. The mixture was filtered and concentrated, the residue was dissolved in EtOAc and washed with water. The aqueous layer was extracted with EtOAc. The combined layers were washed with Na
2S
2O
3 aqueous solution, dried over Na
2SO
4, and concentrated to give the crude product of methyl benzhydrylprolinate (12.0g) , which was used for next step directly. MS-ESI (m/z) : 296 [M+1]
+.
To a solution of the above crude product (12.0g) in MeOH (50mL) , NaOH (4.9g, 122.5mmol) in water (50mL) was added. The reaction was stirred at 50℃ for 17 hours. The mixture was concentrated at 45℃ under reduced pressure to remove MeOH and the leftover aqueous solution washed with methyl t-butyl ether. The aqueous phase was acidified with 1M HCl till pH = 5-6 and extracted with DCM six times. The combined organic layers were dried over Na
2SO
4 and concentrated to give benzhydrylproline (14a) (7.8g) . MS-ESI (m/z) : 282 [M+1]
+.
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl benzhydrylprolinate (14)

A mixture of (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (50mg, 0.13mmol) , benzhydrylproline (14a) (72mg, 0.26mmol) , HATU (145mg, 0.38mmol) , and DIPEA (135μL, 0.76mmol) in NMP (2 mL) was stirred at room temperature for 3.5h. The reaction mixture was purified by preparative chromatography to give (S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl benzhydrylprolinate (14) (31mg) . MS-ESI (m/z) : 656 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.16 (d, J = 9.1 Hz, 1H) , 7.56 (dd, J = 5.3, 2.8 Hz, 5H) , 7.44 –7.21 (m, 8H) , 5.42 (s, 2H) , 5.33 (s, 2H) , 5.04 (brs, 1H) , 3.92 (brs, 1H) , 3.16 (q, J = 7.6 Hz, 2H) , 3.00 (brs., 1H) , 2.80 –2.60 (m, 1H) , 2.44 –2.30 (m, 1H) , 2.25 –2.12 (m, 1H) , 2.05 –1.78 (m, 4H) , 1.31 (t, J = 7.5 Hz, 3H) , 0.86 (t, J = 7.3 Hz, 3H) .
Example 15
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl N-benzhydryl-P-methylphosphonamidate (15)

Synthetic Route of 15

(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl N-benzhydryl-P-methylphosphonamidate (15)

To a suspension of (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (120mg, 0.3mmol) and DIPEA (280μL, 1.6mmol) in DCM (4mL) was added methyl phosphonic dichloride (60mg, 0.45mmol) . The reaction was stirred at room temperature for 45 minutes, followed by treatment with a solution of aminodiphenylmethane (80 μL, 0.46mmol) and DIPEA (80μL, 0.46 mmol) in DCM (1mL) . The mixture was stirred for 35 minutes and concentrated to dryness. The residue was purified by preparative chromatography to give (S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl N-benzhydryl-P- methylphosphonamidate (15) (34mg) . MS-ESI (m/z) : 636 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.10 (d, J = 9.2 Hz, 1H) , 7.81 (s, 1H) , 7.66 (s, 1H) , 7.50 –7.43 (m, 1H) , 7.35 –6.98 (m, 10H) , 5.80 (brs, 1H) , 5.73 (d, J = 16.3 Hz, 1H) , 5.50 (t, J = 9.75 Hz, 1H) , 5.35 –5.25 (m, 2H) , 5.21 (s, 2H) , 3.04 –2.90 (m, 2H) , 1.95 –1.82 (m, 2H) , 1.66 (d, J = 16.7 Hz, 3H) , 1.18 (t, J = 7.0 Hz, 3H) , 1.03 (t, J = 7.3 Hz, 3H) .
Example 16
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-benzhydrylpyrrolidin-2-yl) methyl) carbamate (16)

Synthetic Route of 16

1-Benzhydrylpyrrolidine-2-carboxamide (16a)

To a mixture of benzhydrylproline (14a) (1.0g, 3.6mmol) and DIPEA (2mL, 11.5mmol) in MeCN (20mL) cooled in ice water was added benzyl chloroformate (0.8mL, 5.6mmol) . The reaction was stirred for 3h followed by treatment with ammonium hydroxide (4mL) and then kept at room temperature for overnight. After removal of solvent, the crude product of 1-benzhydrylpyrrolidine-2-carboxamide (16a) was obtained as a brown oil, which was used for next step directly. MS-ESI (m/z) : 281 [M+1]
+.
(1-Benzhydrylpyrrolidin-2-yl) methanamine (16b)

1-Benzhydrylpyrrolidine-2-carboxamide (16a) (3.6mmol) was dissolved in THF (25mL) followed by addition of 1M LiAlH
4-THF solution (22mL, 22mmol) . The reaction was stirred for overnight and quenched with NaOH (aq. ) solution. The mixture was filtered and the filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel (PE: EtOAc = 10: 1 to 5: 1) to give the product of (1-benzhydrylpyrrolidin-2-yl) methanamine (16b) (600mg) . MS-ESI (m/z) : 267 [M+1]
+.
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-benzhydrylpyrrolidin-2-yl) methyl) carbamate (16)

A solution of 4-nitrophenyl chloroformate (58mg, 0.29mmol) in NMP (0.25mL) was added into a solution of (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (50mg, 0.13mmol) and DIPEA (0.2mL, 1.15mmol) in NMP (1mL) at -10℃. The mixture was stirred for 1.5h followed by addition of (1-benzhydrylpyrrolidin-2-yl) methanamine (16b) (77mg, 0.29mmol) in NMP (0.25mL) . The reaction mixture was stirred for 2.5h and purified by preparative chromatography to give (S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-benzhydrylpyrrolidin-2-yl) methyl) carbamate (16) (36mg) . MS-ESI (m/z) : 685 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.17 (d, J = 9.2 Hz, 1H) , 8.08 (s, 1H) , 7.79 –7.63 (m, 5H) , 7.50 –7.34 (m, 5H) , 7.33 –7.22 (m, 2H) , 5.74 (d, J = 16.3 Hz, 1H) , 5.29 (d, J = 26.2 Hz, 3H) , 4.88 (s, 1H) , 4.02 (s, 1H) , 3.88 –3.76 (m, 1H) , 3.54 (dt, J = 15.1, 7.2 Hz, 1H) , 3.43 –3.32 (m, 1H) , 3.26 –3.08 (m, 3H) , 2.43 –2.31 (m, 1H) , 2.27 –2.04 (m, 3H) , 1.97 –1.82 (m, 2H) , 1.39 (t, J = 7.6 Hz, 3H) , 1.02 (t, J = 7.3 Hz, 3H) .
Example 17
(S) -4- (1-butylpiperidin-4-yl) phenyl (4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (17)

Synthetic Route of 17

4- (1-Butylpiperidin-4-yl) phenol (17a)

To a mixture of 4- (piperidin-4-yl) phenol hydrobromide (500mg, 1.94mmol) , n-butyraldehyde (200μL, 2.17mmol) and AcOH (2mL) in MeOH (20mL) was added sodium cyanoborohydride (250mg, 3.98mmol) , and the reaction was stirred at room temperature for overnight. The mixture was concentrated to remove MeOH, diluted with EtOAc (30mL) and washed with NaHCO
3 (aq. ) (30mL) . The aqueous layer was extracted with EtOAc (30mL) and the combined organic layers were dried over Na
2SO
4, and evaporated to dryness to afford the crude product of 4- (1-butylpiperidin-4-yl) phenol (17a) , which was used for next step directly (380mg) . MS-ESI (m/z) : 234 [M+1]
+.
(S) -4- (1-butylpiperidin-4-yl) phenyl (4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (17)

To a solution of 4- (1-butylpiperidin-4-yl) phenol (17a) (240mg, 1.0mmol) and DIPEA (1.9mL, 10.9mmol) in DCM (10mL) cooled below -50℃ was added a solution of triphosgene (360mg, 1.2mmol) in DCM (3.6mL) . the mixture was stirred for 1h followed by addition of (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (300mg, 0.76mmol) . The reaction was allowed to stirred at room temperature for 1h. The reaction mixture was diluted with MeCN and purified by preparative chromatography to give (S) -4- (1-butylpiperidin-4-yl) phenyl (4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (17) (5mg) . MS-ESI (m/z) : 652 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.27 (d, J = 9.2 Hz, 1H) , 8.04 (d, J = 2.6 Hz, 1H) , 7.72 (dd, J = 9.4, 2.5 Hz, 1H) , 7.65 (s, 1H) , 7.36 –7.09 (m, 4H) , 5.75 (d, J = 16.4 Hz, 1H) , 5.35 –5.24 (m, 3H) , 3.80 –3.69 (m, 2H) , 3.17 (q, J = 7.8 Hz, 2H) , 3.05 –2.65 (m, 4H) , 2.43 –2.22 (m, 1H) , 2.08 –1.65 (m, 8H) , 1.48 –1.16 (m, 5H) , 1.08 –0.86 (m, 6H) .
Example 18
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-heptylpiperidin-4-yl) methyl) carbonate (18)

Synthetic Route of 18

(1-Heptylpiperidin-4-yl) methanol (18a)

To a mixture of piperidin-4-ylmethanol (2.0g, 17.4mmol) , n-heptanal (2.0g, 17.4mmol) and AcOH (6mL) in MeOH (60mL) was added sodium cyanoborohydride (2.2g, 34.8mmol) , and the reaction was stirred at room temperature for overnight. The mixture was distilled to remove MeOH and dissolved with EtOAc (100mL) and washed with NaHCO
3 (aq. ) (100mL) . The aqueous layer was extracted with EtOAc (100mL) and the combined organic layers were dried over Na
2SO
4, and evaporated to dryness to afford the crude product of (1-heptylpiperidin-4-yl) methanol (18a) , which was used for next step directly (3.8g) . MS-ESI (m/z) : 214 [M+1]
+.
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-heptylpiperidin-4-yl) methyl) carbonate (18)
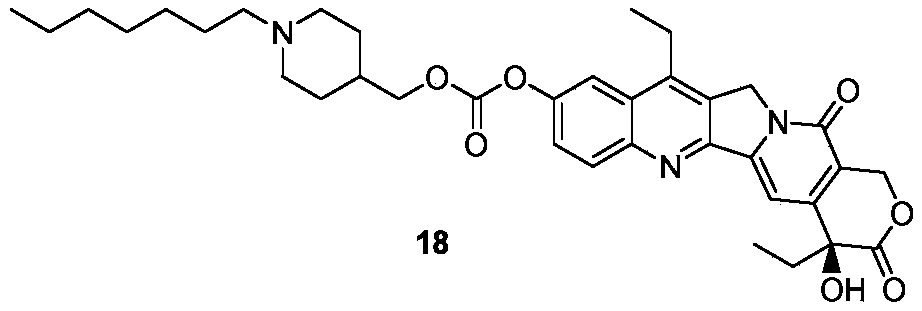
A solution of 4-nitrophenyl chloroformate (82mg, 0.41mmol) in NMP (0.3mL) was added into a solution of (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (55mg, 0.14mmol) and DIPEA (0.2mL, 1.2mmol) in NMP (1mL) at -10℃. The mixture was stirred for 1 h followed by addition of (1-heptylpiperidin-4-yl) methanol (18a) (94mg, 0.43 mmol) in NMP (0.2mL) . The reaction mixture was stirred for 4.5 h and purified by preparative chromatography to give (S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl ( (1-heptylpiperidin-4-yl) methyl) carbonate (18) (4mg) . MS-ESI (m/z) : 632 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.23 (d, J = 9.2 Hz, 1H) , 7.91 (s, 1H) , 7.65 (s, 1H) , 7.62 (d, J = 9.2 Hz, 1H) , 5.74 (d, J = 16.3 Hz, 1H) , 5.29 (d, J = 24.0 Hz, 3H) , 4.22 (d, J = 5.6 Hz, 2H) , 3.77 (d, J = 11.9 Hz, 2H) , 3.15 (q, J = 7.7 Hz, 2H) , 2.99 (s, 2H) , 2.74 –2.59 (m, 2H) , 2.14 –1.62 (m, 3H) , 1.44 –1.14 (m, 14H) , 1.03 (t, J = 7.4 Hz, 3H) , 0.95 –0.70 (m, 6H) .
Example 19
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl dicyclohexylglycinate (19)

Synthetic Route of 19

Ethyl dicyclohexylglycinate (19a)
A solution of dicyclohexylamine (3.66g, 20.2mmol) and ethyl 2-bromoacetate (1.67g, 10mmol) in EtOAc (20mL) was heated at 70℃ for overnight. The mixture was filtered and the filtrate was concentrated to dryness. The residue was purified by column chromatography on silica gel (PE: EtOAc = 10: 1 to 3: 1) to give the product of ethyl dicyclohexylglycinate (19a) (2.42g) . MS-ESI (m/z) : 268 [M+1]
+.
Dicyclohexylglycine (19b)
To a solution of ethyl dicyclohexylglycinate (19a) (2.42g, 9.1mmol) in MeOH (5mL) was added a solution of LiOH (650mg, 27.1mmol) in water (5mL) . The reaction mixture was stirred at 50℃ for overnight and concentrated under reduced pressure. The residue was diluted with water (30mL) and EtOAc (60mL) , and acidified with formic acid (2mL) . The aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na
2SO
4 and concentrated to give dicyclohexylglycine (19b) (870mg) . MS-ESI (m/z) : 240 [M+1]
+.
(S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl dicyclohexylglycinate (19)

A mixture of dicyclohexylglycine (19b) (37mg, 0.15mmol) , (S) -4, 11-diethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (42mg, 0.11mmol) , HATU (113mg, 0.30mmol) and DIPEA (105μL, 0.60mmol) in NMP (1mL) was stirred at room temperature for overnight. The reaction mixture was purified by preparative chromatography to give (S) -4, 11-diethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl dicyclohexylglycinate (19) (6.4mg) . MS-ESI (m/z) : 614 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 8.95 (s, 1H) , 8.28 (d, J = 9.1 Hz, 1H) , 8.07 (d, J = 2.5 Hz, 1H) , 7.74 (dd, J = 9.2, 2.5 Hz, 1H) , 7.32 (s, 1H) , 5.42 (s, 2H) , 5.35 (s, 2H) , 4.69 (s, 2H) , 3.18 (q, J = 7.7 Hz, 2H) , 2.09 –1.94 (m, 4H) , 1.94 –1.75 (m, 6H) , 1.75 –1.04 (m, 17H) , 0.86 (t, J = 7.3 Hz, 3H) .
Example 20
(1-Benzhydrylpyrrolidin-2-yl) methyl ( (S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (20)

Synthetic Route of 20

(1-Benzhydrylpyrrolidin-2-yl) methanol (20a)
1M LiAlH
4-THF (10mL, 10mmol) was added dropwise into a solution of benzhydrylproline (14a) (1.0g, 3.6mmol) in THF (20mL) . The reaction mixture was quenched with NaHCO
3 (aq. ) and extracted with EtOAc twice. The organic layers were combined, dried over Na
2SO
4 and concentrated under reduced pressure to provide (1-benzhydrylpyrrolidin-2-yl) methanol (20a) (900mg) . MS-ESI (m/z) : 268 [M+1]
+.
(1-Benzhydrylpyrrolidin-2-yl) methyl ( (S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (20)

A solution of 4-nitrophenyl chloroformate (150mg, 0.74mmol) in NMP (0.5mL) was added into a solution of (S) -4-ethyl-4, 9-dihydroxy-1, 12-dihydro-14H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinoline-3, 14 (4H) -dione (100mg, 0.27mmol) and DIPEA (0.4mL, 2.3mmol) in NMP (1mL) at -10℃. The mixture was stirred for 2 h followed by addition of (1-benzhydrylpyrrolidin-2-yl) methanol (20a) (98mg, 0.37mmol) and DMAP (54mg, 0.46mmol) in NMP (0.5mL) . The reaction mixture was stirred for 1 h and purified by preparative chromatography to give (1-benzhydrylpyrrolidin-2-yl) methyl ( (S) -4-ethyl-4-hydroxy-3, 14-dioxo-3, 4, 12, 14-tetrahydro-1H-pyrano [3', 4': 6, 7] indolizino [1, 2-b] quinolin-9-yl) carbonate (20) (30mg) . MS-ESI (m/z) : 658 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.36 (s, 1H) , 8.22 (d, J = 9.2 Hz, 1H) , 7.78 (d, J = 2.6 Hz, 1H) , 7.76 –7.66 (m, 5H) , 7.59 (dd, J = 9.2, 2.5 Hz, 1H) , 7.50 –7.33 (m, 6H) , 5.74 (d, J = 16.4 Hz, 1H) , 5.35 –5.26 (m, 3H) , 5.05 (s, 1H) , 4.46 –4.36 (m, 2H) , 4.10 (brs, 1H) , 4.12 –3.80 (m, 2H) , 3.24 (s, 1H) , 2.48 –2.36 (m, 1H) , 2.36 –2.26 (m, 1H) , 2.23 –2.08 (m, 2H) , 1.98 –1.82 (m, 2H) , 1.03 (t, J = 7.4 Hz, 3H) .
Example 21
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl (3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl) carbamate (21)

Synthetic Route of 21

3-Methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl isocyanate (21a)
A mixture of temozolomide (200mg, 1.0mmol) and oxalyl chloride (175μL, 2.1mmol) in 1, 2-dichloroethane (25mL) was stirred at 85℃ for 4 h. The reaction mixture was concentrated under reduced pressure to give 3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl isocyanate (21a) , which was used for next step directly. MS-ESI (m/z) : 275 [M+MeOH+Na]
+ (Sample quenched with MeOH) .
4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl (3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl) carbamate (21)
A mixture of 3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl isocyanate (21a) (1mmol) in DCM was added into a solution of 4- ( (1- benzhydrylpiperidin-4-yl) methyl) phenol (9d) (1mmol) and DIPEA (900μL, 5.2mmol) in DCM (10mL) . The reaction mixture was stirred at room temperature for 1.5 h followed by evaporation. The residue was purified by preparative chromatography to give 4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl (3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl) carbamate (21) (109mg) . MS-ESI (m/z) : 578 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 9.63 (s, 1H) , 8.46 (s, 1H) , 7.69 –7.56 (m, 4H) , 7.43 –7.31 (m, 6H) , 7.19 –7.04 (m, 4H) , 4.89 (s, 1H) , 4.08 (s, 3H) , 3.47 (d, J = 12.0 Hz, 2H) , 2.63 –2.46 (m, 4H) , 1.92 –1.64 (m, 5H) .
Example 22
4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenyl (3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carbonyl) carbamate (22)

Synthetic Route of 22

1-Benzhydrylpiperidin-4-one (22a)

A suspension of 4-piperidinonehydrochloride (1.0g, 7.4mmol) , (chloromethylene) dibenzene (1.57g, 7.7mmol) , K
2CO
3 (22.1mmol) and KI (7.4mmol) in MeCN (30mL) was stirred at 60℃ for overnight. The mixture was filtered and the filtrate was concentrated to dryness. The residue was dissolved with EtOAc (50mL) and washed with water (50mL) and the organic layer was dried over Na
2SO
4 evaporated to dryness to give the crude product of 1-benzhydrylpiperidin-4-one (22a) (2.2g) , which was used for next step directly. MS-ESI (m/z) : 266 [M+1]
+.
4- ( (1-Benzhydrylpiperidin-4-ylidene) methyl) benzonitrile (22b)

To a solution of dimethyl (4-cyanobenzyl) phosphonate (2.2g, 9.8mmol) in THF (20mL) cooled at -50℃ was added 60%NaH (2.3g) in portions and then followed by addition of 1-benzhydrylpiperidin-4-one (22a) (7.7mmol) in THF (20mL) . The reaction was stirred for 4 h and quenched with water (100mL) . The mixture was extracted with EtOAc twice and the combined organic layers was dried over Na
2SO
4 and evaporated to dryness to give 4- ( (1-Benzhydrylpiperidin-4-ylidene) methyl) benzonitrile (22b) . MS-ESI (m/z) : 365 [M+1]
+.
(4- ( (1-Benzhydrylpiperidin-4-yl) methyl) phenyl) methanamine (22c)

A suspension of 4- ( (1-benzhydrylpiperidin-4-ylidene) methyl) benzonitrile (22b) (1.4g, 3.8mmol) and 10%Pd/C (200mg) in THF (20mL) was degassed under vacuum and purged with H
2 several times. The mixture was stirred with a H
2 balloon at room temperature for overnight. The suspension was filtered and to the filtrate was added LiAlH
4 (50mmol) in THF (35mL) . The reaction was stirred for 3.5 h followed by addition of 2mL of NaOH (aq. ) , the suspension was filtered and the filter cake was washed with EtOAc. The combined filtrates were concentrated to dryness to give (4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenyl) methanamine (22c) (1.26g) , which was used for next step directly. MS-ESI (m/z) : 371 [M+1]
+.
N- (4- ( (1-benzhydrylpiperidin-4-yl) methyl) benzyl) -3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carboxamide (22)

To a solution of 3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1-d] [1, 2, 3, 5] tetrazine-8-carboxylic acid (200mg, 1.0mmol) and DIPEA (1mL, 5.7mmoL) in DCM (20mL) was added HATU (1.15g, 3.0mmol) and (4- ( (1-benzhydrylpiperidin-4-yl) methyl) phenyl) methanamine (22c) (400mg, 1.1mmol) . The reaction was stirred at room temperature for 3 h followed by addition of water and DCM. The organic layer was dried over Na
2SO
4 and evaporated to dryness. The crude product was purified by silica gel chromatography and preparative chromatography to give N- (4- ( (1-benzhydrylpiperidin-4-yl) methyl) benzyl) -3-methyl-4-oxo-3, 4-dihydroimidazo [5, 1- d] [1, 2, 3, 5] tetrazine-8-carboxamide (22) (43mg) . MS-ESI (m/z) : 548 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 8.37 (s, 1H) , 7.68 –7.57 (m, 4H) , 7.43 –7.31 (m, 6H) , 7.22 (d, J = 7.7 Hz, 2H) , 7.02 (d, J = 7.6 Hz, 2H) , 4.69 –4.60 (m, 2H) , 4.02 (s, 3H) , 3.55 –3.42 (m, 4H) , 2.56 –2.46 (m, 3H) , 1.96 –1.55 (m, 5H) .
Example 23
(2S, 3S, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -2-methyl-4- ( ( (4- ( (4-methylpiperazin-1-yl) methyl) phenoxy) carbonyl) amino) tetrahydro-2H-pyran-3-yl acetate (23)

Synthetic Route of 23

4- ( (4-Methylpiperazin-1-yl) methyl) phenol (23a)

A mixture of 4-hydroxybenzaldehyde (2.0g, 20mmol) , 1-methylpiperazine (2.44g, 20mmol) , AcOH (8mL) and NaBH
3CN (2.51g, 40mmol) in MeOH (80mL) was stirred at room temperature for overnight. The reaction mixture was evaporated to dryness and dissolved with EtOAc (50mL) and NaHCO
3 (aq. ) (50mL) . The organic layer was dried over Na
2SO
4 and evaporated to dryness to give 4- ( (4-methylpiperazin-1-yl) methyl) phenol (23a) (2.37g) , which was used for next step directly. MS-ESI (m/z) : 207 [M+1]
+.
(2S, 3R, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -4- (l2-chloraneyl) -2-methyltetrahydro-2H-pyran-3-yl acetate hydrochloride (23b)

A suspension of daunorubicin hydrochloride (656mg, 1.24mmol) in trimethyl orthoformate (10mL) was heated at 110℃ for 3 h. The reaction was concentrated under reduced pressure. The residue was diluted with acetone (40mL) followed by addition of 1N HCl (4mL) and conc. HCl (0.1mL) . The mixture was evaporated to dryness and re-crystallization from MeOH (4mL) , EtOAc (40mL) and PE (40mL) to give (2S, 3R, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -4- (l2-chloraneyl) -2-methyltetrahydro-2H-pyran-3-yl acetate hydrochloride (23b) (622mg) . MS-ESI (m/z) : 570 [M+1]
+.
(2S, 3S, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -2-methyl-4- ( ( (4- ( (4-methylpiperazin-1-yl) methyl) phenoxy) carbonyl) amino) tetrahydro-2H-pyran-3-yl acetate (23)

A solution of 4-nitrophenyl chloroformate (80mg, 0.2mmol) in NMP (0.5mL) was added into a solution of 4- ( (4-methylpiperazin-1-yl) methyl) phenol (23a) (40mg, 0.1mmol) and DIPEA (90μL, 0.5mmol) in NMP (1mL) at -10℃. The mixture was stirred for 4 h followed by addition of (2S, 3R, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -4- (l2-chloraneyl) -2-methyltetrahydro-2H-pyran-3-yl acetate hydrochloride (23b) (50mg, 0.083mmol) in NMP (0.5mL) . The reaction mixture was stirred for 1 h and purified by preparative chromatography to give (2S, 3S, 4S, 6R) -6- ( ( (1S, 3S) -3-acetyl-3, 5, 12-trihydroxy-10-methoxy-6, 11-dioxo-1, 2, 3, 4, 6, 11-hexahydrotetracen-1-yl) oxy) -2-methyl-4- ( ( (4- ( (4-methylpiperazin-1-yl) methyl) phenoxy) carbonyl) amino) tetrahydro-2H-pyran-3-yl acetate (23) (25mg) . MS-ESI (m/z) : 802 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 13.26 (s, 1H) , 7.95 –7.84 (m, 3H) , 7.69 –7.62 (m, 1H) , 7.29 (d, J = 8.2 Hz, 2H) , 7.02 (d, J = 8.2 Hz, 2H) , 5.54 (brs, 1H) , 5.33 (s, 1H) , 5.02 –4.92 (m, 2H) , 4.39 (q, J = 5.6 Hz, 1H) , 3.97 (s, 3H) , 4.15 –3.55 (m, 10H) , 3.35 (brs, 1H) , 2.98 (brs, 1H) , 2.93 (s, 2H) , 2.73 (s, 2H) , 2.33 –2.05 (m, 8H) , 1.95 –1.83 (m, 1H) , 1.68 –1.56 (m, 1H) , 1.03 (d, J = 6.4 Hz, 3H) .
Example 24
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl hexyl carbonate (24)

Synthetic Route of 24

(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl hexyl carbonate (24)

To a solution of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (640mg) and hexyl carbonochloridate (165μL, 1.0mmol) in MeCN (5mL) was added DIPEA (1mL, 5.6mmol) . The reaction was stirred at room temperature for 2h followed by prep. HPLC purification to afford (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl hexyl carbonate (24) . MS-ESI (m/z) : 289 [M+1]
+.
1H NMR (400 MHz, DMSO-d
6) δ 12.01 (s, 1H) , 8.12 (d, J = 6.6 Hz, 1H) , 5.56 (s, 2H) , 4.09 (t, J = 6.6 Hz, 2H) , 1.61 –1.52 (m, 2H) , 1.31 –1.18 (m, 6H) , 0.87 –0.79 (m, 3H) .
Example 25
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (4-methoxyphenyl) carbonate (25)

Synthetic Route of 25

(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl (4-methoxyphenyl) carbonate (25)

The title compound (25) was prepared according to the method of Example 24 by using 4-methoxyphenyl carbonochloridate instead of hexyl carbonochloridate. MS-ESI (m/z) : 311 [M+1]
+.
Example 26
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) benzoate (26)

Synthetic Route of 26

4- (1- (2, 2-Diphenylethyl) piperidin-4-yl) benzoic acid (26a)

To a solution of 4- (piperidin-4-yl) benzoic acid hydrochloride (317mg, 1.3mmol) , 2, 2-diphenylacetaldehyde (510mg, 2.6mmol) and AcOH (1.0mL) in MeOH (13mL) was added NaBH
3CN (250mg, 3.9mmol) . The mixture was stirred at ambient temperature for 2 h. After removal of the solvent by evaporation, the residue was dissolved in EtOAc and washed NaHCO
3 (aq. ) solution. The organic layer was dried over Na
2SO
4, and evaporated to dryness. The crude product was purified by silica gel chromatography to give 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) benzoic acid (26a) (340mg) . MS-ESI (m/z) : 386 [M+1]
+.
(5-Fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) benzoate (26)

To a solution of 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) benzoic acid (26a) (300mg, 0.78mmol) and 1H-benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate (BOP) (520mg, 1.2mmol) in MeCN (8mL) was added DIPEA (690ul, 3.9mmol) . The mixture was stirred at room temperature for 1h and then was added into the suspension of 5-fluoro-1- (hydroxymethyl) pyrimidine-2, 4 (1H, 3H) -dione (1a) (435mg) in MeCN (7mL) and stirred for 1h. The suspension was filtered and the filtrate was distilled to a residue. The residue was purified by column chromatography on silica gel followed by prep. HPLC to afford (5-fluoro-2, 4-dioxo-3, 4-dihydropyrimidin-1 (2H) -yl) methyl 4- (1- (2, 2-diphenylethyl) piperidin-4-yl) benzoate (26) (192mg) . MS-ESI (m/z) : 528 [M+1]
+.
1H NMR (400 MHz, Chloroform-d) δ 7.61 (d, J = 5.2 Hz, 1H) , 7.37 (d, J = 7.4 Hz, 4H) , 7.29 –7.20 (m, 4H) , 7.20 –7.10 (m, 4H) , 7.06 (d, J = 8.6 Hz, 2H) , 5.74 (s, 2H) , 4.22 (s, 1H) , 2.85 (d, J = 11.5 Hz, 2H) , 2.53 (d, J = 6.7 Hz, 2H) , 1.77 (t, J = 11.3 Hz, 2H) , 1.63 –1.43 (m, 3H) , 1.39 –1.25 (m, 2H) .
Example 27
Drug release assay
Method
Preparation of pH2.0 buffer solution
The pH2.0 buffer solution was prepared by adding 50 mL of 0.1M phosphoric acid solution in a 200 mL volumetric flask, adjusting the pH value to 2.0 with a 0.1M sodium dihydrogen phosphate solution, then diluting to 200mL with water.
Preparation of pH7.4 buffer solution
The pH7.4 buffer solution was prepared by adding 50mL of 0.1M disodium hydrogen phosphate solution in a 200 mL volumetric flask, adjusting the pH value to 7.4 with a 0.1M phosphoric acid solution, then diluting to 200mL with water.
HPLC condition

Method of hydrolysis rate test
The compounds were dissolved in pH7.4/pH2.0 buffer solution, placed in a constant temperature shaker at 37℃ and 200rpm, and sampled at 0h, 0.5h and 6h respectively. The residual contents of the compounds relative to 0h were tested at each time point.
The release rates (%) were calculated using the following equation: Release rate (%) = [A (0h) -A (xh) ] /A (0h) ×100%, (x=0, 0.5, 6) , A (xh) is the peak area of compounds tested by HPLC at the indicated time point.
The hydrolysis constants (K
h) were calculated using the following equation: Hydrolysis constant K
h = -ln [100%-Release rate (%) ] /t
Results
Results of the parent drug release for exemplary prodrug compounds of the present disclosure are shown in Table 1.
Table 1


As shown in Table 1, the hydrolysis constant (K
h) of the present disclosed compounds at pH 7.4 are greater than that at pH 2.
Example 28
Rat Pharmacokinetics Assay
The objective of this study was to assess the pharmacokinetics of free fluorouracil and an exemplary compound of the present disclosure in stomach and plasma following the continuous intragastric administration of the exemplary compound of the present disclosure and the continuous intravenous infusion or single oral administration of free fluorouracil to male Sprague Dawley rats.
Following the administration of the exemplary compound and free fluorouracil at the same molar amount, the plasma and gastric concentration of fluorouracil were tested and are shown in Figure 1 and Figure 2, respectively.
From Figure 1, it can be seen that the concentration of fluorouracil released from the exemplary compound in plasma is lower compared with that achieved by continuous intravenous infusion or single oral administration of free fluorouracil. This indicated the prodrug of the present disclosure has lower systematic exposure than parent drug and reduced systematic side effects of fluorouracil. From Figure 2, it can be seen that the concentration of fluorouracil released from the exemplary compound in stomach tissue is higher compared with that achieved by continuous intravenous infusion or single oral administration of free fluorouracil. This indicated the prodrug of the present disclosure achieved sustained release of fluorouracil at stomach tissue.
The foregoing description is considered as illustrative only of the principles of the present disclosure. Further, since numerous modifications and changes will be readily apparent to those skilled in the art, it is not desired to limit the invention to the exact construction and process shown as described above. Accordingly, all suitable modifications and equivalents may be considered to fall within the scope of the invention as defined by the claims that follow.
Claims (55)
- A prodrug compound comprising a parent drug moiety and a tail moiety, whereinthe parent drug moiety is derived from a parent drug comprising a reactive group selected from the group consisting of amine, amino, hydroxyl, and amide,the tail moiety is covalently linked to the parent drug moiety and has a Formula (I) :
 wherein:L 1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage;L is a direct bond or alkyl;U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;V is a direct bond or alkyl;W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;Z is selected from the group consisting of a direct bond, alkyl, aryl, NR 1R 2, and OR 3, wherein said alkyl and aryl are optionally substituted with one or more R 4;R 1, R 2 and R 3 are independently hydrogen, alkyl or cycloalkyl; andR 4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,provided that when U is not a direct bond, V, W and Z are not direct bonds at the same time,or a pharmaceutically acceptable salt thereof.
wherein:L 1 is connected to the parent drug moiety through the reactive group of the parent drug to form a cleavable linkage;L is a direct bond or alkyl;U is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;V is a direct bond or alkyl;W is selected from the group consisting of a direct bond, cycloalkyl, heterocyclyl, aryl and heteroaryl;Z is selected from the group consisting of a direct bond, alkyl, aryl, NR 1R 2, and OR 3, wherein said alkyl and aryl are optionally substituted with one or more R 4;R 1, R 2 and R 3 are independently hydrogen, alkyl or cycloalkyl; andR 4 is selected from the group consisting of alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl,provided that when U is not a direct bond, V, W and Z are not direct bonds at the same time,or a pharmaceutically acceptable salt thereof. - The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the parent drug is selected from the group consisting of anti-cancer agent, anti-inflammatory drugs, antibiotics, anti-fugus agent, JAK inhibitors, and VEGF inhibitors.
- The prodrug compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein the parent drug is anti-cancer agent.
- The prodrug compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein the parent drug is anti-inflammatory drugs.
- The prodrug compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein the parent drug is antibiotics.
- The prodrug compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein the parent drug is anti-fugus agent.
- The prodrug compound of claim 2, or a pharmaceutically acceptable salt thereof, wherein the parent drug is selected from the group consisting of Fluorouracil, Temozolomide, Daunorubicin, 10-hydroxyl-camptothecine, and 7-ethyl-10-hydroxyl-camptothecine.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L 1 is connected to the parent drug moiety through the reactive group of the parent drug moiety to form a cleavable linkage selected from the group consisting of carbonate, thiocarbonate, carbamate, thiocarbamate, carboxylate ester, phosphate ester, amide, imine, hydrazone, phosphonamidate and acetal.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L 1 is selected from a direct bond, *-CH 2OC (=O) O-, *-CH 2OC (=S) O-, *-C (=O) O-, *-OC (=S) -, *-C (=O) -, *-C (=O) N (R a) -, *-C (=S) N (R a) -, *-CH 2OP (=O) (R a) O-, and *-P (=O) (R a) N (R a) -, wherein R a is hydrogen, alkyl, alkenyl or alkynyl, and the*end of L 1 is connected to the parent drug moiety.
- The prodrug compound of claim 9, or a pharmaceutically acceptable salt thereof, wherein L 1 is selected from the group consisting of a direct bond, *-CH 2OC (=O) O-, *-C (=O) O-, *-C (=O) -, *-C (=O) N (R a) -, *-CH 2OP (=O) (R a) O-, and *-P (=O) (R a) N (R a) -.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L is a direct bond.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L is alkyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L is C 1-6 alkyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein U is a direct bond.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein U is heterocyclyl.
- The prodrug compound of claim 15, or a pharmaceutically acceptable salt thereof, wherein U is 5-to 12-membered heterocyclyl.
- The prodrug compound of claim 16, or a pharmaceutically acceptable salt thereof, wherein U is piperidinyl or 1, 2, 3, 4-tetrahydro-isoquinolinyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein U is aryl.
- The prodrug compound of claim 18, or a pharmaceutically acceptable salt thereof, wherein U is 5-to 12-membered aryl.
- The prodrug compound of claim 19, or a pharmaceutically acceptable salt thereof, wherein U is phenyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L is a direct bond, and U is a direct bond, heterocyclyl or aryl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein L is alkyl, and U is a direct bond, heterocyclyl or aryl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein V is a direct bond.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein V is alkyl.
- The prodrug compound of claim 24, or a pharmaceutically acceptable salt thereof, wherein V is C 1-6 alkyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein W is a direct bond.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein W is aryl.
- The prodrug compound of claim 27, or a pharmaceutically acceptable salt thereof, wherein W is 5-to 12-membered aryl.
- The prodrug compound of claim 28, or a pharmaceutically acceptable salt thereof, wherein W is phenyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein W is heterocyclyl.
- The prodrug compound of claim 30, or a pharmaceutically acceptable salt thereof, wherein W is 5-to 12-membered heterocyclyl.
- The prodrug compound of claim 31, or a pharmaceutically acceptable salt thereof, wherein W is pyrrolidinyl, piperidinyl or piperazinyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is a direct bond.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is alkyl optionally substituted with one or more R 4.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is C 1-8 alkyl optionally substituted with one or more R 4.
- The prodrug compound of claim 34 or 35, or a pharmaceutically acceptable salt thereof, wherein R 4 is cycloalkyl or aryl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is aryl optionally substituted with one or more R 4.
- The prodrug compound of claim 37, or a pharmaceutically acceptable salt thereof, wherein Z is 5-to 12-membered aryl optionally substituted with one or more R 4.
- The prodrug compound of claim 38, or a pharmaceutically acceptable salt thereof, wherein Z is phenyl optionally substituted with one or more R 4.
- The prodrug compound of any one of claims 37 to 39, or a pharmaceutically acceptable salt thereof, wherein R 4 is alkyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is NR 1R 2.
- The prodrug compound of claim 41, or a pharmaceutically acceptable salt thereof, wherein R 1 and R 2 are independently alkyl or cycloalkyl.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein Z is OR 3.
- The prodrug compound of claim 43, or a pharmaceutically acceptable salt thereof, wherein R 3 is alkyl.
- The prodrug compound of claim 1 having a formula selected from the group consisting of:
 or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, Z and R a are as defined in claim 1.
or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, Z and R a are as defined in claim 1. - The prodrug compound of claim 45, or a pharmaceutically acceptable salt thereof, whereinL is a direct bond,U is heterocyclyl, aryl or heteroaryl,V is a direct bond or alkyl,W is a direct bond, heterocyclyl or aryl;Z is alkyl, aryl, NR 1R 2, or OR 3, wherein said alkyl and aryl are optionally substituted with one or more R 4.
- The prodrug compound of claim 1 having a formula selected from the group consisting of:
 or a pharmaceutically acceptable salt thereof, wherein Q is hydrogen or ethyl, and L, U, V, W, Z and R a are as defined in claim 1.
or a pharmaceutically acceptable salt thereof, wherein Q is hydrogen or ethyl, and L, U, V, W, Z and R a are as defined in claim 1. - The prodrug compound of claim 47, or a pharmaceutically acceptable salt thereof, whereinL is a direct bond or alkyl,U is a direct bond or aryl,V is a direct bond,W is a direct bond or heterocyclyl;Z is NR 1R 2 or alkyl optionally substituted with one or more R 4.
- The prodrug compound of claim 1 having a formula of:
 or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined in claim 1.
or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined in claim 1. - The prodrug compound of claim 1 having a formula of:
 or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined in claim 1.
or a pharmaceutically acceptable salt thereof, wherein L, U, V, W, and Z are as defined in claim 1. - The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the prodrug compound has a lower solubility than the parent drug at biological pH.
- The prodrug compound of claim 1, or a pharmaceutically acceptable salt thereof, wherein the compound is selected from the group consisting of:



- A pharmaceutical composition comprising the prodrug compound according to any one of claims 1-52 or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable excipient.
- A method of treating diseases in a subject in need thereof, comprising administering to the subject a therapeutic effective amount of the prodrug compound according to any one of claims 1-52 or pharmaceutically acceptable salts thereof, or the pharmaceutical composition according to claim 53.
- The method of claim 54, wherein the disease is selected from the group consisting of anal cancer, breast cancer, colorectal cancer, esophageal cancer, pancreatic cancer, head and neck cancer, brain cancer, liver cancer, gastric cancer, bladder cancer, oral mucosal cancer, esophageal cancer, anaplastic astrocytoma, glioblastoma multiforme, acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, Kaposi’s sarcoma, and neuroblastoma.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21882094.2A EP4232437A4 (en) | 2020-10-23 | 2021-10-21 | PROMEDICAMENTS FOR DELAYED-RELEASE THERAPEUTIC AGENTS AND THEIR USES |
| CN202180072494.2A CN116529252B (en) | 2020-10-23 | 2021-10-21 | Prodrugs for sustained-release therapeutic agents and their uses |
| US18/033,210 US20230391731A1 (en) | 2020-10-23 | 2021-10-21 | Prodrugs for sustained releasing therapeutic agents and uses thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNPCT/CN2020/123167 | 2020-10-23 | ||
| CN2020123167 | 2020-10-23 | ||
| CN2021117394 | 2021-09-09 | ||
| CNPCT/CN2021/117394 | 2021-09-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022083679A1 true WO2022083679A1 (en) | 2022-04-28 |
Family
ID=81289697
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2021/125237 Ceased WO2022083679A1 (en) | 2020-10-23 | 2021-10-21 | Prodrugs for sustained releasing therapeutic agents and uses thereof |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20230391731A1 (en) |
| EP (1) | EP4232437A4 (en) |
| CN (1) | CN116529252B (en) |
| TW (1) | TW202227401A (en) |
| WO (1) | WO2022083679A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024124092A3 (en) * | 2022-12-09 | 2024-07-18 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Prodrugs of temozolomide to facilitate delivery into brain tumors |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5610160A (en) * | 1991-04-05 | 1997-03-11 | Sloan; Kenneth B. | Topical 5-fluorouracil prodrug composition and method |
| US20070161668A1 (en) * | 2003-12-23 | 2007-07-12 | American Bioscience, Inc. | Di-ester prodrugs of camptothecin, process for their preparation and their therapeutical applications |
| US20130331422A1 (en) * | 2010-12-17 | 2013-12-12 | Neonc Technologies Inc. | Methods and devices for using isoperilly alcohol |
| CN105315294A (en) * | 2014-06-26 | 2016-02-10 | 王杭祥 | 7-ethyl-10-hydroxycamptothecine drug precursor, preparation method and application thereof |
| CN112618550A (en) * | 2021-01-15 | 2021-04-09 | 中国医学科学院医药生物技术研究所 | Antineoplastic uracil compound and lipid composition thereof |
| CN112724088A (en) * | 2021-01-15 | 2021-04-30 | 中国医学科学院医药生物技术研究所 | Uracil compound and anti-tumor application thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992019233A2 (en) * | 1991-04-29 | 1992-11-12 | Control Delivery Systems, Inc. | Sustained release composition and methods of use thereof |
| WO1999058543A1 (en) * | 1998-05-13 | 1999-11-18 | Instytut Farmaceutyczny | New n-alkyloxycarbonyl derivatives of monosaccharides l-acosamine and l-daunosamine |
| UY29827A1 (en) * | 2005-10-03 | 2007-05-31 | Astrazeneca Ab | 2-AMINA-PYRIMIDINE-4- (2-METHYL-1- (TETRAHIDRO-2H-PIRAN-4-IL) -1-IMIDAZOL-5-Y1) SUBSTITUTED AND ITS DERIVATIVES, PHARMACEUTICAL COMPOSITIONS CONTAINING THEM, PROCESSES FOR PREPARATION AND APPLICATIONS |
| WO2007095389A2 (en) * | 2006-02-17 | 2007-08-23 | Novacea, Inc. | Treatment of hyperproliferative diseases with camptothecine n-oxide and analogs |
| CN105175285B (en) * | 2015-10-10 | 2017-05-03 | 山东大学 | Multiple target point type Tamibarotene derivative as well as preparation method and application thereof |
-
2021
- 2021-10-20 TW TW110138814A patent/TW202227401A/en unknown
- 2021-10-21 US US18/033,210 patent/US20230391731A1/en active Pending
- 2021-10-21 WO PCT/CN2021/125237 patent/WO2022083679A1/en not_active Ceased
- 2021-10-21 CN CN202180072494.2A patent/CN116529252B/en active Active
- 2021-10-21 EP EP21882094.2A patent/EP4232437A4/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5610160A (en) * | 1991-04-05 | 1997-03-11 | Sloan; Kenneth B. | Topical 5-fluorouracil prodrug composition and method |
| US20070161668A1 (en) * | 2003-12-23 | 2007-07-12 | American Bioscience, Inc. | Di-ester prodrugs of camptothecin, process for their preparation and their therapeutical applications |
| US20130331422A1 (en) * | 2010-12-17 | 2013-12-12 | Neonc Technologies Inc. | Methods and devices for using isoperilly alcohol |
| CN105315294A (en) * | 2014-06-26 | 2016-02-10 | 王杭祥 | 7-ethyl-10-hydroxycamptothecine drug precursor, preparation method and application thereof |
| CN112618550A (en) * | 2021-01-15 | 2021-04-09 | 中国医学科学院医药生物技术研究所 | Antineoplastic uracil compound and lipid composition thereof |
| CN112724088A (en) * | 2021-01-15 | 2021-04-30 | 中国医学科学院医药生物技术研究所 | Uracil compound and anti-tumor application thereof |
Non-Patent Citations (7)
| Title |
|---|
| ABRAHAM SUNNY ET AL: "Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES, NATIONAL ACADEMY OF SCIENCES, vol. 104, no. 13, 1 March 2007 (2007-03-01), pages 5584 - 5589, XP002542194, ISSN: 0027-8424, DOI: 10.1073/pnas.0700223104 * |
| BUUR, A. ; BUNDGAARD, H.: "Prodrugs of 5-fluorouracil. VIII. Improved rectal and oral delivery of 5-fluorouracil via various prodrugs. Structure-rectal absorption relationships", INTERNATIONAL JOURNAL OF PHARMACEUTICS, ELSEVIER, NL, vol. 36, no. 1, 1 April 1987 (1987-04-01), NL , pages 41 - 49, XP025516422, ISSN: 0378-5173, DOI: 10.1016/0378-5173(87)90234-1 * |
| See also references of EP4232437A4 * |
| SUDA YASUO ET AL: "The synthesis and in vitro and in vivo stability of 5-fluorouracil prodrugs which possess serum albumin binding potency", vol. 16, no. 9, 1 January 1993 (1993-01-01), pages 876 - 878, XP002337007 * |
| T. NAGASE, K. SHIRAISHI, Y. YAMADA: "5-Fluorouracil derivatives. XIV. Selective synthesis of 1-[[(alkoxycarbonyl)oxy]methyl]-5-fluorouracils via 1,3-bis(hydroxymethyl)-5-fluorouracil", HETEROCYCLES, vol. 27, no. 5, 31 December 1988 (1988-12-31), JP , pages 1155 - 1158, XP009536010, ISSN: 1881-0942 * |
| ZHANG LINA : "Behavior of short-chain chemotherapeutic polyelectrolytes in solutions", CHEMICAL JOURNAL OF CHINESE UNIVERSITIES,, vol. 7, no. 5, 31 December 1986 (1986-12-31), CN , pages 465 - 469, XP009536009, ISSN: 0251-0790 * |
| ZHU QIWEN; YU XUMENG; SHEN QIANQIAN; ZHANG QIUMENG; SU MINGBO; ZHOU YUBO; LI JIA; CHEN YI; LU WEI: "A series of camptothecin prodrugs exhibit HDAC inhibition activity", BIOORGANIC, ELSEVIER, AMSTERDAM, NL, vol. 26, no. 16, 4 August 2018 (2018-08-04), AMSTERDAM, NL, pages 4706 - 4715, XP085456960, ISSN: 0968-0896, DOI: 10.1016/j.bmc.2018.08.008 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024124092A3 (en) * | 2022-12-09 | 2024-07-18 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Prodrugs of temozolomide to facilitate delivery into brain tumors |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116529252A (en) | 2023-08-01 |
| EP4232437A1 (en) | 2023-08-30 |
| EP4232437A4 (en) | 2025-11-05 |
| CN116529252B (en) | 2025-11-14 |
| TW202227401A (en) | 2022-07-16 |
| US20230391731A1 (en) | 2023-12-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021027911A1 (en) | Novel spirocyclic k-ras g12c inhibitor | |
| US20240376123A1 (en) | Kras g12d inhibitors and uses thereof | |
| US20230174526A1 (en) | Benzothiazolyl biaryl compound, and preparation method and use | |
| CN114163457A (en) | Pyrimido five-membered nitrogen heterocyclic compound and use thereof | |
| CN106749233B (en) | A class of sulfonamide derivatives and their applications | |
| BR112019014688A2 (en) | 1,1,1-TRIFLUORO-3-HYDROXYPROPAN-2-IL CARBAMATE DERIVATIVES AS MAGL INHIBITORS | |
| ES2899694T3 (en) | Novel compounds, their synthesis and their uses | |
| CN108727378A (en) | Novel isoquinoline compound and its medical usage | |
| KR20090040923A (en) | Compounds and Methods for Inhibiting the Interaction of BCL Proteins with Binding Partners | |
| CN107001378A (en) | Pyrrolopyrimidine compounds | |
| TW202345836A (en) | Pyrido-[3,4-d]pyridazine amine derivatives useful as nlrp3 derivatives | |
| WO2022121981A1 (en) | Novel camptothecin derivative, composition containing same, and use thereof | |
| CN114746414A (en) | Aza-quinoline compounds and uses thereof | |
| US20230295166A1 (en) | Atr inhibitors and uses thereof | |
| TW200835495A (en) | Substituted 8-piperidinyl-2-pyridinyl-pyrimido[1,2-a] pyrimidin-6-one and 8-piperidinyl-2-pyrimidinyl-pyrimido[1,2-a] pyrimidin-6-one derivatives | |
| CN112142747A (en) | A kind of pyrazolone pyrimidine compound, its preparation method and application | |
| EP4497752A1 (en) | Pyrimido-pyridazinone compound as toll-like receptor agonist | |
| CN105541693A (en) | Aromatic heterocyclic derivative and application thereof in medicines | |
| KR20200078610A (en) | Amino-substituted nitrogen-containing condensed-ring compounds and preparation methods and uses thereof | |
| WO2022199676A1 (en) | Fused tetracyclic compound, preparation method therefor and application thereof in medicine | |
| AU2017266878A1 (en) | Pyridinethiones, pharmaceutical compositions thereof, and their therapeutic use for treating a proliferative, inflammatory, neurodegenerative, or immune-mediated disease | |
| CN116964061A (en) | Tricyclic pyrimidines as cyclin-dependent kinase 7 (CDK7) inhibitors | |
| WO2022063297A1 (en) | Quinazoline derivative, preparation method therefor and use thereof | |
| WO2022083679A1 (en) | Prodrugs for sustained releasing therapeutic agents and uses thereof | |
| WO2022222911A1 (en) | Pyrimidone compound and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21882094 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202180072494.2 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021882094 Country of ref document: EP Effective date: 20230523 |