WO2022102731A1 - Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 - Google Patents
Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 Download PDFInfo
- Publication number
- WO2022102731A1 WO2022102731A1 PCT/JP2021/041650 JP2021041650W WO2022102731A1 WO 2022102731 A1 WO2022102731 A1 WO 2022102731A1 JP 2021041650 W JP2021041650 W JP 2021041650W WO 2022102731 A1 WO2022102731 A1 WO 2022102731A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- therapy
- administered
- administration
- alkyl
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/555—Heterocyclic compounds containing heavy metals, e.g. hemin, hematin, melarsoprol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/28—Compounds containing heavy metals
- A61K31/282—Platinum compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against enzymes
Definitions
- the present disclosure relates to a cancer treatment method using a combination of standard treatment and an EP4 antagonist and an immune checkpoint inhibitor.
- Prostaglandin E 2 is known as a metabolite in the arachidonic acid cascade, and has cytoprotective action, uterine contraction action, pain threshold lowering action, gastrointestinal peristaltic motility promoting action, wakefulness action, and gastric acid. It is known to have a secretory inhibitory effect, a blood pressure lowering effect, a diuretic effect, and the like.
- Non-Patent Document 1 PGE2 receptors have subtypes with different roles. There are roughly four subtypes known at present, and they are called EP 1 , EP 2 , EP 3 , and EP 4 , respectively.
- the EP4 receptor suppresses MCP-1 production from macrophages, suppresses TNF- ⁇ , IL-2, and IFN- ⁇ production from lymphocytes, and anti-inflammatory by enhancing IL-10 production. , Vascular dilation, angiogenesis, suppression of elastic fiber formation, and regulation of MMP-9 expression.
- EP4 receptors are thought to be involved in cancer immunoregulation via bone marrow-derived immunosuppressive cells (Myeloid Derived Suppressor Cells), regulatory T cells and natural killer cells.
- Non-Patent Document 2-7 compounds that strongly bind to the EP4 receptor and have an antagonistic effect are diseases caused by activation of the EP4 receptor, such as bone disease, cancer, systemic granuloma, immune disease, allergy, and atopy.
- diseases caused by activation of the EP4 receptor such as bone disease, cancer, systemic granuloma, immune disease, allergy, and atopy.
- Stress, endometriosis, uterine adenomyosis, neonatal arterial duct patency, cholelithiasis, etc. are considered to be useful for
- Patent Document 1 discloses that the compound represented by the general formula (I) has an EP4 antagonism and is useful as a therapeutic agent for cancer (see Patent Document 1).
- the immune checkpoint inhibitor is a new therapeutic method that cancels the immunosuppressive mechanism and activates the immune response to cancer.
- anti-CTLA-4 cytotoxic T lymphocyte-associated antibody
- anti-PD-1 programmed cell death-1 antibody nivolumab and pembrolizumab have been approved in Japan and overseas and are used in cancer treatment. ..
- Patent Document 2 discloses that a combination of a compound represented by the general formula (I) and an immune checkpoint inhibitor is useful for cancer treatment (see Patent Document 2).
- the main treatment for unresectable advanced / recurrent colorectal cancer is drug therapy, and standard treatment with fluorinated pyrimidine antineoplastic agents, oxaliplatin, irinotecan, etc. is one of them.
- XELOX and bevacizumab (hereinafter, may be abbreviated as "XELOX + bevacizumab therapy").
- FOLFIRINOX therapy using a regimen containing fluorouracil (5-FU) in combination with oxaliplatin and irinotecan (hereinafter, “. It may be abbreviated as "FFX therapy"), and in the hope of reducing toxicity from FFX therapy, rapid administration of fluorouracil may be omitted and modified FOLFIRINOX therapy (hereinafter, abbreviated as "mFFX therapy”) in which the dose of irinotecan is reduced. ) Or a combination of gemcitabine and nabpacritaxel (hereinafter, may be abbreviated as "GnP therapy”).
- FFX therapy modified FOLFIRINOX therapy
- GnP therapy a combination of gemcitabine and nabpacritaxel
- drug therapy is the main treatment for stage IV or relapsed non-small cell lung cancer, and combination therapy with docetaxel and ramucirumab (hereinafter, may be abbreviated as "DTX + RAM therapy”) or docetaxel therapy as standard therapy. (Hereinafter, it may be abbreviated as "DTX therapy”).
- An object of the present invention is to provide a new treatment method for cancer (for example, colorectal cancer, pancreatic cancer, lung cancer).
- cancer for example, colorectal cancer, pancreatic cancer, lung cancer.
- EP4 receptor antagonist and immune checkpoint inhibitor in standard treatment can be an effective cancer treatment method.
- further administration of EP4 receptor antagonists or EP4 receptor antagonists and immune checkpoint inhibitors to patients who have undergone prechemoradiation therapy may be an effective cancer treatment (both).
- the treatment method of the present invention may be abbreviated as the treatment method of the present invention.
- [1] Standard therapies ((i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vi) docetaxel therapy, or (vi) dosetaxel and ramsilmab therapy), and A cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an EP4 antagonist as an active ingredient, characterized by administration in combination with administration of an immune checkpoint inhibitor, or [2] standard therapy (((2] standard therapy.
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an immune checkpoint inhibitor as an active ingredient, characterized by being administered in combination.
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an immune checkpoint inhibitor as an active ingredient, which is characterized by being administered.
- the present invention provides a new cancer treatment method.
- FIG. 1 evaluates the tolerability, safety, and efficacy of compound A, nivolumab, described below, in combination with XELOX + bevacizumab therapy in patients with unresectable advanced or recurrent colorectal cancer.
- FIG. 2 shows the safety, efficacy and pharmacokinetics of compound A and nivolumab described below as neoadjuvant therapy after preoperative chemoradiotherapy for locally advanced rectal cancer that can be curatively resected.
- FIG. 1 evaluates the tolerability, safety, and efficacy of compound A, nivolumab, described below, in combination with XELOX + bevacizumab therapy in patients with unresectable advanced or recurrent colorectal cancer.
- FIG. 2 shows the safety, efficacy and pharmacokinetics of compound A and nivolumab described below as neoadjuvant therapy after preoperative chemoradiotherapy for locally advanced rectal cancer
- FIG. 3 is a multicenter non-cooperative evaluation of the tolerability, safety and efficacy of compound A, nivolumab described below in combination with mFFX therapy or GnP therapy in patients with pancreatic cancer with distant metastasis. The outline of the blinded uncontrolled trial is shown.
- FIG. 4 shows the compounds A, nivolumab and docetaxel described below in patients with advanced or recurrent non-small cell lung cancer refractory to combination therapy including anti-PD-1 antibody or anti-PD-L1 antibody and platinum preparations.
- the EP4 antagonist is not particularly limited as long as it is a compound having an EP4 antagonist action, but in one embodiment, the general formula (I):
- R 1 represents COOR 8 , tetrazole, SO 3 H, SO 2 NH 2 , SO 2 NHR 8-1 , CONHSO 2 R 8-1 , SO 2 NHCOR 8-1 , or hydroxamic acid.
- R 8 represents a hydrogen atom, C1-4 alkyl, or benzyl
- R 8-1 represents C1-4 alkyl, C1-4 haloalkyl, C3-10 carbocycle, or 3-10 membered heterocycle, and the C3-10 carbocycle and 3-10 membered heterocycle, respectively, is C1.
- L 1 represents C1-5 alkylene, C2-5 alkenylene, or C2-5 alkinylene, R2 is halogen, C1-4alkyl, C1-4alkoxy, C1-4alkylthio, C2-4alkenyl, C2-4alkynyl, -O (C1-4haloalkyl), -S (C1-4haloalkyl),- C (O) (C1-4 alkyl), -SO 2 (C1-4 alkyl), -CONH (C1-4 alkyl), -CON (C1-4 alkyl) 2 , -NHC (O) (C1-4 alkyl) ), -N (C1-4 alkyl) C (O) (C1-4 alkyl) (C1-4 alkyl) (C1-4 alkyl) (C1-4 alkyl)
- the C1-4 alkyl may be substituted with halogen and may be substituted.
- (C1-4alkyl) 2 in the R2 represents two independent C1-4 alkyls, each of which may be the same or different.
- X 1 represents CR 6 or nitrogen atom
- R 6 represents hydrogen atom or
- X 2 represents CR 7 or nitrogen atom
- R 7 represents hydrogen atom
- R 2 or -L 3 -R 9 and L 3 represents methylene, oxygen atom or sulfur atom which may be oxidized.
- R 9 represents a 4-10 membered heterocycle which may be substituted with a substituent selected from the group consisting of halogen, C1-4alkyl, and C1-4haloalkyl.
- each said C1-4 alkyl may be substituted with a halogen.
- ring represents a benzene ring or a 5-6 member monocyclic aromatic heterocycle.
- R5 is ( 1 ) halogen, (2) C1-4alkyl, (3) carboxyl, (4) nitrile, (5) -CONHR 11 , (6) -C (O) R 12 , (7) -OR.
- R 14 (8) -S (O) t R 15 , (9) -CH 2 R 16 , (10) -NR 17 R 17 , (11) -NHCOR 11 , (12) C4-10 carbocycle, or ( 13) Represents a 4-10 membered heterocycle, the C4-10 membered ring or the 4-10 membered heterocycle may be substituted with 1 to 3 R18s , and if there are multiple R18s , then R Each of the 18 may be the same or different independently.
- R 11 represents a C1-6 alkyl, C3-6 cycloalkyl, phenyl, or 4-6 membered heterocycle, where R 11 may be substituted with 1 to 3 R 13s , wherein the R 13s are plural.
- R 13 may be independently the same or different, respectively.
- R 13 represents a halogen, C1-6 alkyl, C3-6 cycloalkyl, C1-4 alkoxy, hydroxyl group, -NR 20 R 21 , benzene, or 4-6 membered heterocycle.
- R 20 and R 21 independently represent a hydrogen atom or C1-4 alkyl, respectively.
- R 12 represents a C1-6 alkyl, C3-6 cycloalkyl, benzene, or 4-6 membered heterocycle, and the C3-6 cycloalkyl, benzene, or 4-6 membered heterocycle is independent of each other. It may be substituted with halogen, C1-4alkyl, or C1-4alkoxy.
- R 14 represents a hydrogen atom, C1-6 alkyl, C3-6 cycloalkyl, benzene, or benzyl, wherein the C1-6 alkyl may be substituted with 1 to 3 R 19s , where the R 19 is. When there are a plurality of R 19 , each of them may be independently the same or different. R 19 is substituted with a substituent selected from the group consisting of C1-4 alkoxy, -CONH (C1-4 alkyl), -CON (C1-4 alkyl) 2 , or C1-4 alkyl and C1-4 haloalkyl. Represents a 5-6 member monocyclic aromatic heterocycle which may be present.
- the (C1-4 alkyl) 2 in the R 19 represents two independent C1-4 alkyls, each of which may be the same or different.
- R 15 represents C1-6 alkyl, C3-6 cycloalkyl, benzene, or benzyl.
- R 16 represents a hydroxyl group or C1-4 alkoxy and represents R 17 independently represent a hydrogen atom, C1-6 alkyl, or C3-6 cycloalkyl, respectively.
- R18 is halogen, C1-6alkyl, C3-6cycloalkyl , C1-4alkoxy, oxo, nitrile, hydroxyl group, hydroxymethyl, 1-methyl-1-hydroxyethyl, (C1-4alkyl) SO 2- , Represents a 4-6 membered heterocycle, (C1-4 alkyl) NH-, or (C1-4 alkyl) 2 N-.
- the (C1-4 alkyl) 2 in the R 18 represents two independent C1-4 alkyls, each of which may be the same or different.
- m represents an integer of 1 to 4
- n represents an integer of 0 to 4
- p represents an integer of 0 to 2
- q represents an integer of 0 to 6
- r represents an integer of 0 to 6.
- R 2 , R 3 , R 4 , and R 5 may be independent and may be the same or different. ), Or a salt thereof.
- EP4 antagonist AN0025, E7046, IK-007, RMX-1002, grapprant, AAT-007, CR6086, INV-1120, BYD-001, TT-038, DT095895, P-001, ER-819762, MK-2894, MF498, animal page, CJ-042794, EP4A, BGC201531, CJ-023423, GW627368, AH23848, DT-9081, or WO2001 / 062708, WO2002 / 020462, WO2002 / 0232900, WO2002.
- C1-4 alkyl is, for example, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, and isobutyl.
- C1-3 alkyl is, for example, methyl, ethyl, n-propyl, and isopropyl.
- C1-5 alkylene is, for example, methylene, ethylene, propylene, butylene, and pentylene.
- C2-5 alkenylene means, for example, ethenylene, 1-propenylene, 2-propenylene, 1-butenylene, 2-butenylene, 3-butenylene, 1-pentenylene, 2-pentenylene, 3 -Pentenylene, 4-pentenylen, etc.
- C2-5 alkynylene means, for example, ethynylene, 1-propinilen, 2-propinilen, 1-butinilen, 2-butinilen, 3-butinilen, 1-pentinylene, 2-pentinylene, 3-pentinylene, and 4-Pentinylene, etc.
- halogen is fluorine, chlorine, bromine, and iodine.
- C1-4 alkoxy is, for example, methoxy, ethoxy, propoxy, isopropoxy, butoxy, 1-methylpropoxy, tert-butoxy, isobutoxy and the like.
- C1-4 alkylthio is, for example, methylthio, ethylthio, propylthio, isopropylthio, butylthio, 1-methylpropylthio, tert-butylthio, isobutylthio and the like.
- the "C2-4 alkenyl” is, for example, ethenyl, 1-propenyl, 2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl and the like.
- C2-4 alkynyl is, for example, ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl and the like.
- C1-4 haloalkyl means C1-4 alkyl substituted with halogen, for example, monofluoromethyl, difluoromethyl, trifluoromethyl, 2-fluoroethyl, 1-fluoroethyl, 2, 2-Difluoroethyl, 1,2-difluoroethyl, 1,1-difluoroethyl, 2,2,2-trifluoroethyl, 1,2,2-trifluoroethyl, 1,1,2-trifluoroethyl, 1 , 2,2,2-Tetrafluoroethyl, 1,1,2,2-Tetrafluoroethyl, Pentafluoroethyl, 1,2-Dibromo-1,2,2-Trifluoroethyl, 1-Chloro-1,2 , 2,2-Tetrafluoroethyl, 3-fluoropropyl, 3-chloropropyl, 2-fluoropropyl, 2-chloropropyl, 2-chloro
- the "sulfur atom which may be oxidized" refers to sulfur (S), sulfoxide (S (O)), and sulfone (SO 2 ).
- the "4-10-membered heterocycle” is a 4-10-membered monocyclic or bicyclic heterocycle containing 1 to 5 heteroatoms selected from an oxygen atom, a nitrogen atom and a sulfur atom.
- Means for example, oxetane, azetidine, pyrrolidine, pyrrol, imidazole, triazole, tetrazole, pyrazole, pyridine, piperidine, piperazin, pyrazine, pyrimidine, pyridazine, azepine, diazepine, furan, pyran, oxepine, thiophene, Thiopirane, Thiepin, Oxazole, Isoxazole, Thiazol, Isothiazole, Frazan, Oxazole, Oxazole, Oxazole, Oxazolepine, Oxazolezepine, Thiasiazol, Thiazin, Thiasiazin, Thiazepine, Thiasiazepin
- the "3-10 membered heterocycle” is a 3-10 membered monocyclic or bicyclic heterocycle containing 1 to 5 heteroatoms selected from an oxygen atom, a nitrogen atom and a sulfur atom.
- the "5-10-membered aromatic heterocycle” is a 5-10-membered monocyclic or bicyclic ring containing 1 to 4 heteroatoms selected from an oxygen atom, a nitrogen atom and a sulfur atom. It means an aromatic heterocycle and means, for example, pyrrole, imidazole, triazole, tetrazole, pyrazole, furan, thiophene, oxazole, isooxazole, thiazole, isothiazole, frazan, oxadiazole, thiadiazole, pyridine, pyrazine, pyrimidine, pyridazine, Indole, isoindole, benzofuran, isobenzofuran, benzothiophene, isobenzothiophene, indazole, purine, benzoxazole, benzothiazole, benzoimidazole, benzoflazan, benzothiazole, benzotriazole,
- the "5-6-membered monocyclic aromatic heterocycle” means, for example, pyrrole, imidazole, triazole, tetrazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, furan, thiophene, oxazole, isoxazole, thiazole. , Isothiazole, Frazan, Oxadiazole, and Thiadiazole rings.
- C4-10 carbocycle means a monocyclic or bicyclic carbocycle of C4 to 10, for example, cyclobutane, cyclopentane, cyclohexane, cycloheptane, cyclooctane, cyclononane, cyclodecane, cyclopentene.
- Cyclohexene Cycloheptene, Cyclooctene, Cyclopentadiene, Cyclohexadiene, Cycloheptadiene, Cyclooctane
- Benzene Pentalene, Perhydropentalene, Azulene, Perhydroazulene, Inden, Perhydroinden, Indan, Naphthalene, Dihydronaphthalene, Tetrahydronaphthalene, perhydronaphthalene ring and the like.
- C3-10 carbocycle means a monocyclic or bicyclic carbocycle of C3-10, for example, cyclopropane, the carbocycle described in the above-mentioned "C4-10 carbocycle", and the like. Is.
- C1-6 alkyl means, for example, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, isobutyl, pentyl, 1-methylbutyl, 2-methylbutyl, 3 -Methylbutyl, 1,1-dimethylpropyl, 1,2-dimethylpropyl, 2,2-dimethylpropyl, hexyl, 1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 1,1- Dimethylbutyl, 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 1-methyl-1-ethylpropyl, 2-methyl-2-ethylpropyl, 1 -Ethylbutyl, 2-ethylbutyl and the like.
- C3-6 cycloalkyl is cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- the "4-6 membered heterocycle” means a 4-6 membered monocyclic heterocycle containing 1 to 4 heteroatoms selected from an oxygen atom, a nitrogen atom and a sulfur atom.
- R 1 is preferably COOR 8 .
- R8 is preferably a hydrogen atom or a C1-4 alkyl, and more preferably a hydrogen atom.
- R8-1 is preferably C1-4alkyl, benzene or pyridine, the benzene and pyridine being C1-4alkyl, C1-4haloalkyl, C1-4alkoxy, -O (C1-4haloalkyl). ), C1-4 alkylthio, -S (C1-4 haloalkyl), halogen, or nitrile may be substituted.
- L 1 is preferably C1-5 alkylene or C2-5 alkenylene, more preferably C1-5 alkylene, and particularly preferably propylene.
- R 2 is preferably fluorine.
- X 1 is preferably CR 6 .
- R6 is preferably a hydrogen atom or fluorine, and more preferably a hydrogen atom.
- X 2 is preferably CR 7 .
- R 7 is preferably fluorine, nitrile, -CH 2 R 9 , or -OR 9 , and more preferably nitrile.
- R9 is preferably a 4-10-membered heterocycle optionally substituted with methyl or trifluoromethyl
- the 4-10-membered heterocycle is preferably a 5-10-membered aromatic heterocycle.
- More preferably 5-10-membered nitrogen-containing aromatic heterocycles eg, pyrazole, imidazole, triazole, pyrrolopyridine, pyrrolopyrimidine, pyropyridazine, imidazolipyridazine, imidazolepyridine, imidazolipyrimidine, imidazolepyrazine, pyrazolopyridine, pyrazolo). (Pyrimidine, etc.).
- R3 is preferably fluorine.
- R4 is preferably methyl, ethyl, or trifluoromethyl, more preferably methyl.
- X3 is preferably methylene or an oxygen atom, and more preferably an oxygen atom.
- R10 is preferably methyl, ethyl, methylcarbonyl, ethylcarbonyl, methylsulfonyl, ethylsulfonyl, or tert-butoxycarbonyl.
- the ring is preferably a benzene, thiophene, or pyrazole ring, and more preferably a benzene ring.
- R5 is preferably ⁇ CONHR 11 , fluorine, methoxy, benzene ring, or 4-10 membered heterocycle
- the 4-10 membered heterocycle is preferably azetidine, pyrridine, piperidine, oxazolidine, oxadi.
- R11 is preferably C1-6alkyl, C3-6cycloalkyl , pyran, pyrrolidine, piperidine, pyrazole, thiazole, oxazole, isoxazole, pyridine, pyridazine, or pyrimidine ring, and more preferably C1-. It is 6-alkyl.
- R 13 is preferably halogen, C1-6 alkyl, C3-6 cycloalkyl, C1-4 alkoxy, hydroxyl group, ⁇ NR 20 R 21 , benzene, oxetane, pyridine, pyrazole, or oxazole ring. More preferably, fluorine, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, isobutyl, cyclopentyl, cyclobutyl, oxetane, hydroxyl group, methoxy, ethoxy, propoxy, isopropoxy, dimethylamino, benzene. , Pyridine, pyrazole, or oxazole ring.
- R20 is preferably a hydrogen atom or methyl.
- R 21 is preferably a hydrogen atom or methyl.
- R12 is preferably C1-3 alkyl, C3-6 cycloalkyl, benzene, or a 4-6 membered heterocycle.
- the 4-6-membered heterocycle is preferably oxetane, azetidine, pyrrolidine, piperidine, pyrazine, pyrazine, thiopyran, oxazine, oxadiazine, thiazine, thiadiazine, pyrrol, imidazole, triazole, tetrazole, pyrazole, pyridine, pyrazine, pyrimidine, and the like.
- the 4-6 membered heterocycle may be substituted with C1-4 alkoxy.
- R14 is preferably a hydrogen atom, methyl, ethyl, benzene, or benzyl.
- R 19 is preferably methoxy, -CONHCH 3 , -CON (CH 3 ) 2 , oxazole, thiazole, pyrazole, or a pyridine ring.
- R15 is preferably methyl, cyclopropyl, or benzene.
- R 16 is preferably a hydroxyl group.
- R 17 is preferably methyl, ethyl, or cyclopropyl, and more preferably methyl.
- R18 is preferably fluorine, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, isobutyl, cyclopropyl, methoxy, ethoxy, n-propoxy, isopropoxy, oxo. , Nitrile, hydroxyl group, hydroxymethyl, 1-methyl-1-hydroxyethyl, methylsulfonyl, pyridine, dimethylamino.
- m is preferably an integer of 1 to 2, and more preferably 1.
- n is preferably an integer of 0 to 1, and more preferably 1.
- p is preferably 0.
- q is preferably 0.
- r is preferably an integer of 0 to 4, and more preferably an integer of 0 to 2.
- s is preferably an integer of 0 to 2, and more preferably 1 or 2.
- t is preferably an integer of 0 to 2.
- X 3a is preferably an oxygen atom.
- na is preferably an integer of 0 to 1, and more preferably 1.
- qa is preferably 0.
- ra is preferably an integer of 0 to 2.
- the general formula (I) is preferably ring, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 8-1 , R 9 , R. 10 , R 11 , R 12 , R 13 , R 14 , R 15 , R 16 , R 17 , R 18 , R 19 , R 20 , R 21, R 21 , L 1 , L 2 , L 3 , X 1 , X 2 , X 3 , a combination of preferred definitions of each of m, n, p, q, r, s, and t.
- the compound represented by the general formula (I) is preferably the general formula (Ia).
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above.
- a salt thereof more preferably the general formula (I-1).
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4
- X 3a represents a methylene or oxygen atom, and other symbols represent the above. It has the same meaning as), or a salt thereof.
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above.
- a salt thereof more preferably the general formula (Ic).
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above.
- a salt thereof still more preferably the general formula (Id).
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above.
- na represents an integer of 0 to 1
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above.
- a salt thereof particularly preferably the general formula (I-2).
- R 2a represents a halogen
- R 6a represents a hydrogen atom or a halogen
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4
- other symbols have the same meanings as above. Represented
- a salt thereof most preferably the general formula (I-4).
- R 2a represents a halogen
- R 6a represents a hydrogen atom or a halogen
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4, and other symbols have the same meanings as above. Represented
- a salt thereof or a salt thereof.
- R 5a is a C4-10 carbocycle or a 4-10-membered heterocycle that may be substituted with 1 to 3 R 18 , and the R 18 is plural, then the R 18 is They may be the same or different independently, na represents an integer of 0 to 1, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and other symbols have the same meanings as above. It is a compound represented by (1) or a salt thereof, and more preferably the general formula (Ig).
- R 5a is a C4-10 carbocycle or a 4-10-membered heterocycle that may be substituted with 1 to 3 R 18 , and the R 18 is plural, then the R 18 is They may be the same or different independently, na represents an integer of 0 to 1, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and other symbols have the same meanings as above. It is a compound represented by (1) or a salt thereof, and more preferably the general formula (Ih).
- R 5a is a C4-10 carbocycle or a 4-10-membered heterocycle that may be substituted with 1 to 3 R 18 , and the R 18 is plural, then the R 18 is They may be the same or different independently, na represents an integer of 0 to 1, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and other symbols have the same meanings as above. It is a compound represented by (1) or a salt thereof, and more preferably the general formula (Ii).
- R 5a is a C4-10 carbocycle or a 4-10-membered heterocycle that may be substituted with 1 to 3 R 18 , and the R 18 is plural, then the R 18 is They may be the same or different independently, na represents an integer of 0 to 1, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and other symbols have the same meanings as above. It is a compound represented by (1) or a salt thereof, and is particularly preferably the general formula (I-3).
- R 2a represents a halogen
- R 6a represents a hydrogen atom or a halogen
- R 5a may be substituted with 1 to 3 R 18s , a C4-10 carbocycle or a 4-10 member complex.
- R 18s when the R 18s are plural, the R 18s may be independently the same or different, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and others.
- the symbol has the same meaning as above.
- Is a compound represented by the above, or a salt thereof, and most preferably the general formula (I-5).
- R 2a represents a halogen
- R 6a represents a hydrogen atom or a halogen
- R 5a may be substituted with 1 to 3 R 18s , a C4-10 carbocycle or a 4-10 member complex.
- R 18s when the R 18s are plural, the R 18s may be independently the same or different, qa represents an integer of 0 to 3, ra represents an integer of 0 to 4, and others.
- the symbol has the same meaning as above.
- It is a compound represented by) or a salt thereof.
- L 1 is preferably propylene.
- the compound described in the examples of WO2016 / 111347, or a salt thereof is more preferable as an EP4 antagonist. More preferably (1) 4- [4-Cyano-2-( ⁇ [(2'R, 4S) -6- (methylcarbamoyl) -2,3-dihydrospiro [chromen-4,1'-cyclopropane] -2' -Il] carbonyl ⁇ amino) phenyl] butanoic acid, (2) 4- ⁇ 4-Cyano-2-[( ⁇ (2'R, 4S) -6-[(cyclopropylmethyl) carbamoyl] -2,3-dihydrospiro [chromen-4,1'-cyclopropane] ] -2'-yl ⁇ carbonyl) amino] phenyl ⁇ butanoic acid, (3) 4- ⁇ 4-Cyano-2-[( ⁇ (2'R, 4S) -6-[(2-methoxyethyl) carb
- the EP4 antagonist is preferably (1) 4- [ 4 -cyano-2-( ⁇ [(2'R, 4S) -6- (5-methyl-1,3,4-). Oxadiazole-2-yl) -2,3-dihydrospiro [chromen-4,1'-cyclopropane] -2'-yl] carbonyl ⁇ amino) phenyl] butanoic acid, (2) 4- [4-Cyano-2- ( ⁇ [(2'R, 4S) -6- (5-cyclopropyl-1,3,4-oxadiazole-2-yl) -2,3- Dihydrospiro [chromen-4,1'-cyclopropane] -2'-yl] carbonyl ⁇ amino) phenyl] butanoic acid, (3) 4- [4-Cyano-2-( ⁇ [(2'R, 4S) -6- (3-methyl-1,2,4-oxadiazole-5-yl) -2,3-dihydro
- 4- [4-cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyran-4,1'- Cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butane acid is 4- [4-cyano-2-( ⁇ [(2'R, 4S) -6- (isopropylcarbamoyl) -2,3-dihydrospiroyl). It may also be named [chromen-4,1'-cyclopropane] -2'-yl] carbonyl ⁇ amino) phenyl] butanoic acid.
- Compound A can be produced according to a known method, for example, the method described in Example 2-13 of WO2016 / 111347.
- compound B 4- ⁇ 4-cyano-2-[( ⁇ (2'R, 4S) -6-[(2-methoxyethyl) carbamoyl] -2,3-dihydrospiro [chromen-4,1'-" Cyclopropane] -2'-yl ⁇ carbonyl) amino] phenyl ⁇ butanoic acid (hereinafter, may be abbreviated as compound B), or a salt.
- Compound B can be produced according to a known method, for example, the method described in Example 2-2 of WO2016 / 111347.
- isomers include all of them unless otherwise specified.
- alkyl groups, alkoxy groups, alkylene groups and the like include linear and branched chains.
- R S isomers, ⁇ , ⁇ isomers, enantiomers, diastereomers.
- D D, L, d, l bodies
- chromatographically separated polar bodies high polar and low polar
- equilibrium compounds rotational isomers, mixtures of any proportions thereof
- All chiral mixtures are included in the present invention.
- the present invention also includes all isomers due to tautomerism.
- the compound represented by the general formula (I) is converted into a salt by a known method.
- the salt is preferably a pharmaceutically acceptable salt.
- the salt is preferably water-soluble.
- Pharmaceutically acceptable salts include, for example, acid addition salts, alkali metal salts, alkaline earth metal salts, ammonium salts, amine salts and the like.
- Examples of the acid addition salt include hydrochloride, hydrobromide, hydroiodide, sulfate, phosphate, inorganic acid salt such as nitrate, or acetate, lactate, tartrate, and benzoic acid.
- Organic acid salts such as salts, citrates, methanesulfonates, ethanesulfonates, trifluoroacetates, benzenesulfonates, toluenesulfonates, isethionates, glucronates, or gluconates
- alkali metal salt include salts with potassium and sodium.
- alkaline earth metal salt include salts with calcium, magnesium and the like.
- Examples of the ammonium salt include salts with tetramethylammonium and the like.
- Amine salts include, for example, triethylamine, methylamine, dimethylamine, cyclopentylamine, benzylamine, phenethylamine, piperidine, monoethanolamine, diethanolamine, tris (hydroxymethyl) aminomethane, lysine, arginine, and N-methyl-D-. Examples include salt with glucamine.
- the compound used in the present invention can be made into an N-oxide compound by any method.
- the N-oxide compound represents a compound in which the nitrogen atom represented by the general formula (I) is oxidized.
- the compound represented by the general formula (I) and a salt thereof may exist in a non-solvate form, or may exist in a form solvated with a pharmaceutically acceptable solvent such as water or ethanol. ..
- the solvate is preferably a hydrate.
- the compound represented by the general formula (I) can form a co-crystal with a suitable co-crystal forming agent.
- a pharmaceutically acceptable one formed with a pharmaceutically acceptable co-crystal forming agent is preferable.
- a co-crystal is typically defined as a crystal in which two or more different molecules are formed by an intramolecular interaction different from that of an ionic bond.
- the co-crystal may be a complex of a neutral molecule and a salt.
- Cocrystals can be prepared by known methods, eg, by melt crystallization, by recrystallization from a solvent, or by physically grinding the components together. Suitable co-crystal forming agents include those described in WO2006 / 007448.
- all references to the compound represented by the general formula (I) are the compound represented by the general formula (I), a salt thereof, an N-oxide form thereof, a solvate thereof (for example, a hydrate), or a hydrate thereof.
- the co-crystal or N-oxide of a salt of the compound represented by the general formula (I), a solvate thereof (for example, a hydrate), or a co-crystal thereof is included. That is, in the present invention, the compound represented by the general formula (I) or a salt thereof is a solvent product (for example, a hydrate) of the compound represented by the general formula (I), an N-oxide form thereof, or a salt thereof. It includes a co-crystal or an N-oxide compound of a salt of a compound represented by the general formula (I), a solvate thereof (for example, a hydrate), or a co-crystal thereof.
- the compound represented by the general formula (I) can be administered as a prodrug.
- the prodrug of the compound represented by the general formula (I) means a compound converted into the compound represented by the general formula (I) by a reaction with an enzyme, gastric acid or the like in a living body.
- Examples of the prodrug of the compound represented by the general formula (I) include, for example, when the compound represented by the general formula (I) has an amino group, the compound in which the amino group is acylated, alkylated or phosphorylated (for example,).
- the amino group of the compound represented by the general formula (I) is eicosanoylation, alanylation, pentylaminocarbonylation, (5-methyl-2-oxo-1,3-dioxolen-4-yl) methoxycarbonylation, tetrahydrofuranyl. (Pyrrolidylmethylation, pivaloyloxymethylation, acetoxymethylation, tert-butylated compound, etc.); When the compound represented by the general formula (I) has a hydroxyl group, the hydroxyl group is acylated or alkylated.
- the carboxy group of the compound is ethyl esterification, phenyl esterification, carboxymethyl esterification, dimethylaminomethyl esterification, pivaloyloxymethyl esterification, 1- ⁇ (ethoxycarbonyl) oxy ⁇ ethyl esterification, phthalidyl esterification, ( 5-Methyl-2-oxo-1,3-dioxolen-4-yl) methyl esterification, 1- ⁇ [(cyclohexyloxy) carbonyl] oxy ⁇ ethyl esterification, methylamidated compounds, etc.) and the like.
- These compounds can be produced by methods known per se.
- the prodrug of the compound represented by the general formula (I) may be either a hydrated substance or a non-hydrated substance.
- the prodrug of the compound represented by the general formula (I) is a general formula under physiological conditions as described in Hirokawa Shoten 1990, "Development of Pharmaceuticals", Vol. 7, “Molecular Design", pp. 163 to 198. It may be changed to the compound represented by (I).
- each atom constituting the compound represented by the general formula (I) is an isotope thereof (for example, 2H , 3H , 13C , 14C , 15N , 16N , 17O , 18O , 18F . , 35 S, 36 Cl, 77 Br, 125 I, etc.) and the like.
- the EP4 antagonist can be produced by a known method, for example, the compound represented by the general formula (I) can be produced according to the method described in WO2016 / 111347.
- the EP4 antagonist is usually formulated with a pharmaceutically acceptable carrier such as various additives or solvents and then administered systemically or topically, orally or parenterally.
- a pharmaceutically acceptable carrier means a substance other than the active ingredient, which is generally used for pharmaceutical preparations.
- the pharmaceutically acceptable carrier preferably has no pharmacological action at the dose of the preparation, is harmless, and does not interfere with the therapeutic effect of the active ingredient.
- the pharmaceutically acceptable carrier can also be used for the purpose of enhancing the usefulness of the active ingredient and the preparation, facilitating the formulation, stabilizing the quality, or improving the usability.
- a substance as described in Yakuji Nippo Co., Ltd. 2016 "Pharmaceutical Additives Dictionary 2016" (edited by Japan Pharmaceutical Additives Association) may be appropriately selected according to the purpose.
- Examples of the dosage form used for administration include oral preparations (eg, tablets, capsules, granules, powders, oral solutions, syrups, oral jelly, etc.), oral preparations (eg, oral tablets, etc.). Oral sprays, semi-solids for the oral cavity, gargling agents, etc.), injection formulations (eg, injections, etc.), dialysis formulations (eg, dialysis agents, etc.), inhalation formulations (eg, inhalants, etc.), Ophthalmic preparations (eg, eye drops, ointment, etc.), otologic preparations (eg, ear drops, etc.), nasal preparations (eg, nasal drops, etc.), rectal preparations (eg, suppositories, etc.) Semi-solid preparations for the rectal, intestinal injections, etc.), vaginal preparations (eg, vaginal tablets, suppositories for vagina, etc.), and skin preparations (eg, solids for external use, liquids for external
- the dose of the EP4 antagonist used in the present invention varies depending on age, body weight, symptoms, therapeutic effect, administration method, treatment time, etc., but is usually in the range of 1 ng to 1000 mg per adult. Orally administered once to several times daily, or parenterally administered once to several times daily in the range of 0.1 ng to 100 mg per adult, or 1 hour to 24 times daily. It is continuously administered intravenously over a period of time.
- a dose smaller than the above dose may be sufficient, or administration beyond the range may be necessary.
- one aspect of the dose is about 1-100 mg orally administered 1 to 3 times daily, preferably 1 mg, 5 mg, 10 mg, 15 mg, 20 mg at a time. , 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg, 55 mg, 60 mg, 65 mg, 70 mg, 75 mg, 80 mg, 85 mg, 90 mg, 95 mg or 100 mg orally administered 1 to 3 times a day, more preferably 1 mg once.
- 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 30 mg, 35 mg, 40 mg, 45 mg, 50 mg are orally administered 1 to 3 times a day, and more preferably 5 mg, 10 mg, 20 mg or 40 mg is orally administered once a day. It is administered, more preferably 20 mg or 40 mg orally once daily.
- an immune checkpoint molecule means a molecule that exerts an immunosuppressive function by transmitting an inhibitory co-signal.
- the immune checkpoint molecule include CTLA-4, PD-1, PD-L1 (programmed cell date-ligand 1), PD-L2 (programmed cell death-ligand 2), LAG-3 (Lymphometry activation 3), and T.
- T cell immunoglobulin and mucin-3 T cell immunoglobulin and mucin-3
- BTLA Band T lymphocyte attenuator
- B7H3, B7H4 CD160, CD39, CD73
- A2aR adenosine A2areceptor
- IDO1 Indoreamine 2,3-dioxygenase
- ArginaseI T cell immunoglobulin and ITIM doman
- CD115 etc. 2012, Cancer Cell, 27, 450-461, 2015
- the molecule is not particularly limited as long as it has a function consistent with the definition.
- the immune checkpoint inhibitor is a substance that inhibits the function of the immune checkpoint molecule.
- the immune checkpoint inhibitor is not particularly limited as long as it is a substance that can suppress the function (signal) of the immune checkpoint molecule.
- immune checkpoint inhibitor examples include anti-PD-1 antibodies (eg, Nivolumab, Cemiplimab (REGN-2810), Pembrolizumab (MK-3475), Spartanlizumab (PDR-001), Tremelimumab (BGB-A317)).
- anti-PD-1 antibodies eg, Nivolumab, Cemiplimab (REGN-2810), Pembrolizumab (MK-3475), Spartanlizumab (PDR-001), Tremelimumab (BGB-A317).
- anti-PD-L2 antibody PD-L1 fusion protein, PD-L2 fusion protein (eg, AMP- 224), anti-Tim-3 antibody (eg, MBG453), anti-LAG-3 antibody (eg, BMS-986016, LAG525), anti-KIR antibody (eg, Lilylumab) and the like.
- Antibodies comprising the heavy and light chain complementarity determining regions (CDRs) or variable regions (VR) of the known antibodies are also aspects of immune checkpoint inhibitors.
- further embodiments of anti-PD-1 antibodies include antibodies comprising, for example, Nivolumab heavy and light chain complementarity determining regions (CDRs) or variable regions (VR).
- the immune checkpoint inhibitor is preferably an anti-PD-1 antibody, an anti-PD-L1 antibody and an anti-CTLA-4 antibody, and more preferably an anti-PD-1 antibody or an anti-PD-L1 antibody.
- the anti-PD-1 antibody is preferably Nivolumab, Cemiplimab, Pembrolizumab, Spartanismab, Tisslerizumab, Tremelimumab, Sintilimab and Camrelizumab
- the anti-PD-L1 antibody is preferably AlibaB
- Durvalumab -4 Antibodies are preferably Ipilimumab and Tremelimumab.
- the anti-PD-1 antibody is more preferably Nivolumab, Cemiplimab and Pembrolizumab, and even more preferably Nivolumab.
- the immune checkpoint inhibitor is preferably an anti-PD-1 antibody, more preferably Nivolumab.
- the immune checkpoint inhibitor can be produced by a known method.
- Nivolumab can be produced according to the method described in WO2006 / 121168
- Pembrolizumab can be produced according to the method described in WO2008 / 156712
- BMS-936559 can be produced according to WO2007 / 005874.
- Ipilimumab can be produced according to the method described in WO2001 / 014424.
- any one or any of these immune checkpoint inhibitors can be used in combination with administration of EP4 antagonists and standard therapy.
- the dose of the immune checkpoint inhibitor used in the combination of the present invention varies depending on age, body weight, symptoms, therapeutic effect, administration method, treatment time, etc., but is adjusted to bring about the optimum desired effect.
- about 1 to 10 mg / kg (body weight) or about 200 to 1200 mg once with an immune checkpoint inhibitor as an active ingredient is applied at intervals of 2 to 4 weeks for about 30 minutes to about 60 minutes or about 60 minutes or more.
- the dose in terms of body weight per administration for example, 1 mg / kg, 2 mg / kg, 3 mg / kg, 4 mg / kg, 5 mg / kg, 6 mg / kg, 7 mg / kg, 8 mg / kg, 9 mg.
- the dose per dose includes, for example, 200 mg, 240 mg, 250 mg, 280 mg, 300 mg, 320 mg, 350 mg, 360 mg, 400 mg, 420 mg, 450 mg, 480 mg, 500 mg, etc. Included are 540 mg, 560 mg, 600 mg, 640 mg, 700 mg, 720 mg, 750 mg, 800 mg, 840 mg, 900 mg, 1000 mg, 1080 mg, 1100 mg, 1120 mg or 1200 mg.
- the administration interval may be, for example, 2 weeks, 3 weeks or 4 weeks, and the single administration time may be, for example, about 30 minutes, about 60 minutes or about 60 minutes or more.
- Nivolumab which is an anti-PD-1 antibody
- the immune checkpoint inhibitor is Nivolumab
- Nivolumab which is an anti-PD-1 antibody
- it is administered at the following dosage and administration. That is, for patients with malignant melanoma, Nivolumab was administered by intravenous drip infusion at 3 mg / kg (body weight) once at 2 week intervals or 2 mg / kg (body weight) at 3 week intervals, or 240 mg once for 2 weeks. Intravenous infusion of 480 mg at intervals or once every 4 weeks.
- nivolumab For patients with non-small cell lung cancer, renal cell cancer, classical Hodgkin lymphoma, head and neck cancer, gastric cancer and malignant pleural mesothelioma, 3 mg / kg (body weight) of nivolumab was instilled at 2-week intervals. Injection is administered.
- malignant melanoma, non-small cell lung cancer, renal cell cancer, urinary tract epithelial cancer, MSI-H or dMMR-positive colorectal cancer including patients of children aged 12 years or older.
- Gastric cancer, hepatocellular carcinoma, small cell lung cancer and malignant thoracic mesothelioma are administered by intravenous drip infusion of 240 mg of nivolumab at 2-week intervals or 480 mg at 4-week intervals. ..
- 1 mg / kg (body weight) of Nivolumab is intravenously infused four times at 3 week intervals to patients with malignant melanoma, and then Nivolumab.
- 3 mg / kg (body weight) is intravenously infused at 2-week intervals, or 80 mg of Nivolumab is infused 4 times at 3-week intervals, and then 240 mg of Nivolumab is infused at 2-week intervals. Or 480 mg once may be infused intravenously at 4-week intervals.
- 240 mg of nivolumab is intravenously infused four times at 3 week intervals to patients with renal cell carcinoma or colorectal cancer, and then 240 mg of nivolumab is administered once for 2 weeks. Intravenous infusion of 480 mg at intervals or once every 4 weeks may be performed.
- Pembrolizumab which is the same anti-PD-1 antibody
- it is administered at the following dosage and administration. That is, malignant melanoma, non-small cell lung cancer, classical Hodgkin lymphoma, head and neck cancer, MSI-H or dMMR-positive solid or colon cancer, urinary epithelial cancer, cervical cancer, primary mediastinct.
- Patients with B-cell lymphoma, hepatocellular carcinoma, gastric cancer, and Merkel cell carcinoma receive 200 mg of pembrolizumab at 3-week intervals or 400 mg at 6-week intervals by intravenous drip infusion.
- Pembrolizumab As another dosage and administration, for example, for patients with classical Hodgkin lymphoma, MSI-H or dMMR-positive solid tumor or colorectal cancer and primary mediastinal B-cell lymphoma in children over 2 years old, Pembrolizumab As a result, 2 mg / kg (body weight) (up to 200 mg once) is administered by intravenous drip infusion at intervals of 3 weeks.
- Avelumab which is an anti-PD-L1 antibody
- 10 mg / kg (body weight) of Avelumab is administered once for 2 weeks to each patient of Merkel cell cancer and urothelial cancer. It is administered by intravenous drip infusion at intervals.
- Atezolizumab the same PD-L1 antibody, is administered to patients with non-small cell lung cancer, urinary tract epithelial cancer, and hepatocellular carcinoma by intravenous drip infusion of 1200 mg of atezolizumab at 3-week intervals, and triple-negative breast cancer.
- Patients receive 840 mg of atezolizumab in combination with paclitaxel at 2-week intervals by intravenous drip infusion.
- Durvalumab which is the same PD-L1 antibody, is administered to patients with non-small cell lung cancer and urinary tract epithelial cancer by intravenous drip infusion of 10 mg / kg (body weight) once as Durvalumab at 2-week intervals.
- 1500 mg of Durvalumab is administered by intravenous drip infusion at 4-week intervals.
- Ipilimumab which is an anti-CTLA-4 antibody
- Intravenous drip infusion was given to patients with renal cell carcinoma and MSI-H or dMMR-positive colon cancer at 1 mg / kg (body weight) once daily as ipilimumab four times at 3-week intervals in combination with nivolumab.
- Intravenous drip infusion is given to patients with non-small cell lung cancer at 1 mg / kg (body weight) of Ipilimumab at 6-week intervals.
- the administration form of the immune checkpoint inhibitor in the present invention is preferably parenteral administration, and examples of parenteral administration include subcutaneous administration, intradermal administration, intraperitoneal administration, intramuscular administration, intravenous administration and the like. However, subcutaneous administration or intravenous administration is preferable. More preferably, it is administered intravenously. As the administration form of intravenous administration, intravenous drip infusion is preferable.
- the above dosage can also be used in the therapeutic method of the present invention.
- the "XELOX therapy” in the present invention is a cancer treatment method using a combination of capecitabine and oxaliplatin (L-OHP).
- the "XELOX + bevacizumab therapy" in the present invention is a cancer treatment method using a combination of capesitabin, oxaliplatin and bevacizumab, and in one embodiment, bevacizumab 7.5 mg / kg intravenously over about 30 to 90 minutes.
- oxaliplatin 130 mg / m 2 body surface area
- bevacizumab 1000 mg / m 2 / time body surface area 1.36 m 2 or less.
- Withdrawal, dose reduction, and resumption of XELOX + bevacizumab therapy will be carried out at the discretion of the doctor with reference to the latest package insert.
- Compound A an EP4 antagonist in combination with XELOX + bevacizumab therapy, is, in one embodiment, 5 mg orally once daily, 10 mg orally once daily, 20 mg orally once daily or It is orally administered at 40 mg once a day. Preferably, 20 mg is orally administered once a day or 40 mg is orally administered once a day.
- the dose of Compound A may be reduced or the administration itself may be discontinued. Further, if the criteria for resumption are satisfied, the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg
- the two-step dose reduction is 10 mg
- the administration of compound A as an EP4 antagonist is started.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- Nivolumab as an immune checkpoint inhibitor in combination with XELOX + bevacizumab therapy in one embodiment, is 240 mg once at 2-week intervals, 360 mg at 3-week intervals, or 480 mg at 4-week intervals as Nivolumab. It is administered by intravenous drip infusion. Preferably, 360 mg of nivolumab is administered by intravenous drip infusion at intervals of 3 weeks. The administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient. In addition, administration of nivolumab can be resumed if the criteria for resumption are met.
- the cancer to which this combination -1 can be applied is not particularly limited as long as it is a cancer to which this combination -1 can exert an effect, but in one embodiment, it is colorectal cancer, preferably colon / rectal cancer. More preferably, it is unresectable advanced or recurrent colorectal cancer.
- This combination-1 is administered to a patient who has not been treated for colorectal cancer in one embodiment.
- the EP4 antagonist preferably compound A
- the immune checkpoint inhibitor preferably anti-PD-1 antibody (preferably nivolumab)
- XELOX + bevasizumab therapy when administered on the same day, one embodiment.
- EP4 antagonist and immune checkpoint inhibitor are administered first.
- the EP4 antagonist and immune checkpoint inhibitor are administered followed by bevacizumab, oxaliplatin, and capecitabine in that order.
- EP4 antagonist preferably compound A
- an immune checkpoint inhibitor preferably anti-PD-1 antibody (preferably nivolumab)
- XELOX + Bevasizumab therapy may be given first or at the same time as administration of EP4 antagonists and immune checkpoint inhibitors.
- the order of administration of EP4 antagonist, immune checkpoint inhibitor, bevacizumab, oxaliplatin and capecitabine may be initiated from any agent unless otherwise defined, and two or more agents. It may be administered at the same time.
- the "preoperative chemoradiotherapy” in the present invention is a combination of irradiation and chemotherapy such as fluorouracil (5-FU) or capecitabine. In the present invention, a combination of irradiation and capecitabine administration is preferred.
- the preoperative chemoradiotherapy in the present invention in one embodiment, is 45 Gy / 25 doses of pelvic cavity irradiation and 5.4 Gy / 3 doses of boost to the primary lesion, and 825 mg / m 2 capecitabin (starting dose).
- capecitabin is taken orally for a period equivalent to 75% of the 28 irradiations.
- the dose can be appropriately reduced for 21 days or 42 times or more as oral administration in the morning and evening), depending on the degree of expression of side effects of the patient.
- Compound A as an EP4 antagonist used as neoadjuvant therapy after preoperative chemoradiation therapy is, in one embodiment, 5 mg orally once daily, 10 mg orally once daily, 20 mg. It is orally administered once a day or 40 mg orally once a day. Preferably, 20 mg is orally administered once a day or 40 mg is orally administered once a day. More preferably, compound A is orally administered at 40 mg once daily. Depending on the degree of side effects of the patient, the dose of Compound A may be reduced or the administration itself may be discontinued.
- the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg and the two-step dose reduction is 10 mg.
- Nivolumab as an immune checkpoint inhibitor used as neoadjuvant therapy after preoperative chemoradiation therapy in one embodiment, is 240 mg once every 2 weeks, once 360 mg every 3 weeks, or 1 as Nivolumab.
- a dose of 480 mg is administered by intravenous drip infusion at 4-week intervals.
- 240 mg of nivolumab is administered by intravenous drip infusion at 2-week intervals.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- One embodiment of this combination-2 is administration of an EP4 antagonist, and one embodiment is a combination administration of an EP4 antagonist and an immune checkpoint inhibitor.
- the cancer to which this combination-2 is applied is not particularly limited as long as it is a cancer for which this neoadjuvant therapy can be effective, but in one embodiment, it is colorectal cancer, preferably colon / rectal cancer. .. More preferably, it is locally advanced rectal cancer that can be curatively resected.
- it is administered to a patient who can undergo curative resection without distant metastasis on diagnostic imaging after preoperative chemoradiotherapy.
- it is administered to a patient who is capable of (curative) resection in neoadjuvant therapy.
- FFX therapy is oxaliplatin (L-OHP), irinotecan hydrochloride hydrate (irinotecan, CPT-11), levofolinate calcium (levofolinate, l-LV). It is a cancer treatment method using a combination of 4 drugs of fluorouracil (5-FU), and as a recommended dosage, for example, oxaliplatin 85 mg / m 2 (body surface area) is intravenously administered over 2 hours, and then levofolinate 200 mg.
- L-OHP oxaliplatin
- irinotecan hydrochloride hydrate irinotecan, CPT-11
- levofolinate calcium levofolinate calcium
- / M 2 is intravenously administered over 2 hours
- irinotecan 180 mg / m 2 is intravenously administered over 1.5 hours from 30 minutes after the start of levofolinate administration
- fluorouracil 400 mg / m 2 is further administered after the end of levofolinate administration. Is a rapid intravenous administration, and then fluorouracil 2400 mg / m 2 is intravenously administered over 46 hours, and the series of administrations is carried out at 2-week intervals.
- the "weight loss regimen" of the FFX therapy is to reduce the dose of any of the four drugs administered in the FFX therapy from the first administration or to discontinue the administration itself, or to administer any of the first cycle and thereafter. Depending on the degree of occurrence of side effects observed in the above, the dose is reduced in any of the subsequent administrations after the second cycle, or the administration of any of the four agents is discontinued. In that embodiment, for example, it is not necessary to perform rapid intravenous administration of fluorouracil from the first administration, and the dose of oxaliplatin is between 65 mg / m 2 , 50 mg / m 2 or 85 to 50 mg / m 2 .
- the dose of irinotecan may be any dose between 150 mg / m 2 , 120 mg / m 2 , 90 mg / m 2 or 180-90 mg / m 2 .
- the dose of fluorouracil administered intravenously may be any dose between 1800 mg / m 2 , 1200 mg / m 2 and 2400 to 1200 mg / m 2 .
- rapid intravenous administration of fluorouracil may be discontinued depending on the degree of side effects of the patient in any administration after the second cycle in the FFX therapy.
- the dose of oxaliplatin may be reduced to any dose between 65 mg / m 2 , 50 mg / m 2 or 85-50 mg / m 2 or discontinuation of oxaliplatin.
- the dose of irinotecan may be reduced to any dose between 150 mg / m 2 , 120 mg / m 2 , 90 mg / m 2 or 180-90 mg / m 2 , depending on the extent of the patient's onset of side effects.
- administration of irinotecan may be discontinued, and the dose of fluorouracil administered intravenously may be 1800 mg / m 2 , 1200 mg / m 2 or 2400 to 1200 mg / m 2 , depending on the degree of side effects of the patient.
- the dose may be reduced to any dose during the period or administration of the fluorouracil may be discontinued.
- mFFX therapy modified FOLFIRINOX therapy
- the recommended dosage is, for example, oxaliplatin 85 mg / m 2 (body surface area) intravenously over 2 hours.
- levoleucovorin 200 mg / m 2 was intravenously administered over 2 hours
- irinotecan 150 mg / m 2 was intravenously administered over 1.5 hours from 30 minutes after the start of levoleucovorin administration
- fluorouracil 2400 mg was further administered after the end of levoleucovorin administration.
- This is a treatment method in which / m 2 is intravenously administered over 46 hours, and the series of administrations is performed at 2-week intervals.
- the dose of oxaliplatin may be any dose between 65 mg / m 2 , 50 mg / m 2 or 85-50 mg / m 2 .
- the dose of irinotecan may be any dose between 140 mg / m 2 , 120 mg / m 2 or 140-120 mg / m 2
- the dose of fluorouracil administered intravenously continuously is 2400 mg. It may be any dose between / m 2 , 1800 mg / m 2 , 1200 mg / m 2 or 2400 to 1200 mg / m 2 .
- the dose of oxaliplatin may be 65 mg / m 2 , 50 mg / m 2 or 85 to 50 mg / m 2 depending on the degree of the occurrence of side effects of the patient.
- the dose may be reduced to any dose during the period or the administration of oxaliplatin may be discontinued, and the dose of irinotecan may be adjusted to 120 mg / m 2 , 90 mg / m 2 or 150 to 90 mg, depending on the degree of side effects of the patient.
- the dose may be reduced to any dose between / m 2 or the administration of irinotecan may be discontinued, and the dose of fluorouracil administered intravenously may be 1800 mg / m 2 , depending on the degree of side effects of the patient.
- the dose may be reduced to any dose between 1200 mg / m 2 or 2400 to 1200 mg / m 2 or administration of the fluorouracil may be discontinued.
- the interval between a series of administrations of the FFX therapy or its weight loss regimen is set to a 3-week interval or a 4-week interval temporarily or continuously depending on the degree of side effect occurrence of the patient. You may. Withdrawal / reduction / resumption of FFX therapy or its weight loss regimen (eg, mFFX therapy) will be carried out at the discretion of the physician with reference to the latest package insert.
- FFX therapy or its combination with a weight loss regimen (hereinafter, may be abbreviated as this combination-3).
- the EP4 antagonist in combination with FFX therapy or a weight loss regimen thereof (eg, mFFX therapy) is administered in one embodiment at the dosages described in (1) EP4 antagonist.
- compound A which is an EP4 antagonist, is orally administered at 5 mg once a day, at 10 mg once a day, at 20 mg once a day, or at 40 mg once a day. Will be done.
- 20 mg is orally administered once a day or 40 mg is orally administered once a day. More preferably, it is orally administered at 20 mg once a day.
- the dose of Compound A may be reduced or the administration itself may be discontinued. Further, if the criteria for resumption are satisfied, the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg
- the two-step dose reduction is 10 mg
- the administration of compound A as an EP4 antagonist is started.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- the immune checkpoint inhibitor in this combination-3 is administered in one embodiment at the dosage as described in (2) Immune checkpoint inhibitor.
- nivolumab as an immune checkpoint inhibitor is administered by intravenous drip infusion of 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals.
- 480 mg of nivolumab is administered by intravenous drip infusion at 4-week intervals.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- EP4 antagonist preferably compound A
- immune checkpoint inhibitor preferably anti-PD-1 antibody (preferably Nivolumab)
- FFX therapy or a weight loss regimen thereof eg, mFFX therapy
- the EP4 antagonist and the immune checkpoint inhibitor are administered first.
- FFX therapy or a weight loss regimen thereof eg, mFFX therapy
- an EP4 antagonist preferably compound A
- an immune checkpoint inhibitor preferably an anti-PD-1 antibody (preferably Nivolumab)
- FFX therapy or a weight loss regimen thereof eg, mFFX therapy
- FFX therapy or a weight loss regimen thereof may be given first, or may be co-administered with an EP4 antagonist and an immune checkpoint inhibitor.
- the order of administration of the EP4 antagonist, immune checkpoint inhibitor, FFX therapy or a weight loss regimen thereof may be initiated from any drug unless otherwise defined. Further, two or more drugs may be administered at the same time.
- GnP therapy in the present invention is a therapy in which gemcitabine and nabupaclitaxel are combined, and in one embodiment, the GnP therapy is, for example, 1000 mg / m 2 of gemcitabine as a recommended dosage.
- Body surface area was intravenously administered over 30 minutes
- nabupaclitaxel was intravenously administered at 125 mg / m 2 (body surface area) over 30 minutes
- the series of administrations was performed at 1-week intervals and continued for 3 weeks. It is a therapy that takes a week off.
- gemcitabine may be reduced to 800 mg / m 2 or 600 mg / m 2 depending on the degree of side effect of the patient in any administration after the second cycle in the GnP therapy.
- the dose may be reduced to 100 mg / m 2 or 75 mg / m 2 .
- Withdrawal, dose reduction, and resumption of GnP therapy will be carried out at the discretion of the doctor with reference to the latest package insert.
- Combination with GnP therapy (hereinafter, may be abbreviated as this combination-4).
- the EP4 antagonist in this combination- 4 is administered in one embodiment at the dosage as described in (1) EP4 antagonist.
- compound A which is an EP4 antagonist, is orally administered at 5 mg once a day, at 10 mg once a day, at 20 mg once a day, or at 40 mg once a day. Will be done.
- 20 mg is orally administered once a day or 40 mg is orally administered once a day. More preferably, compound A is orally administered at 40 mg once daily.
- the dose of Compound A may be reduced or the administration itself may be discontinued.
- the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg and the two-step dose reduction is 10 mg.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- the immune checkpoint inhibitor in this combination-4 is administered in one embodiment at the dosage as described in (2) Immune checkpoint inhibitor.
- nivolumab as an immune checkpoint inhibitor is administered by intravenous drip infusion of 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals.
- 480 mg of nivolumab is administered by intravenous drip infusion at 4-week intervals.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- the "docetaxel and ramucirumab therapy" in the present invention is a cancer treatment method using a combination of docetaxel and ramucirumab, and the recommended dosage is, for example, docetaxel 60 mg / m 2 (body surface area) for 60 minutes. It is a treatment method in which ramucirumab 10 mg / kg is intravenously administered over 60 minutes, and the series of administrations is carried out at 3-week intervals. As one embodiment, in any administration after the second cycle in the DTX + RAM therapy, docetaxel is increased or decreased to 75 mg / m 2 or 50 mg / m 2 or administration of docetaxel is discontinued depending on the degree of side effect of the patient.
- the dose of ramucirumab may be reduced to 8 mg / kg or 6 mg / kg, or the administration of ramucirumab may be discontinued, or the administration time of ramucirumab may be shortened to 30 to 60 minutes. Withdrawal, dose reduction, and resumption of DTX + RAM therapy will be carried out at the discretion of the doctor with reference to the latest package insert. In one embodiment, administration of docetaxel and ramucirumab is started on the same day.
- the EP4 antagonist in this combination-5 is administered in one embodiment at the dosages described in (1) EP4 antagonist.
- compound A which is an EP4 antagonist, is orally administered at 5 mg once a day, at 10 mg once a day, at 20 mg once a day, or at 40 mg once a day. Will be done.
- 20 mg is orally administered once a day or 40 mg is orally administered once a day.
- the dose of Compound A may be reduced or the administration itself may be discontinued.
- the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg
- the two-step dose reduction is 10 mg
- the administration of compound A as an EP4 antagonist is started.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- the immune checkpoint inhibitor in this combination-5 is administered in one embodiment at the dosage as described in (2) Immune checkpoint inhibitor.
- nivolumab as an immune checkpoint inhibitor is administered by intravenous drip infusion of 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals.
- 360 mg of nivolumab is administered by intravenous drip infusion at intervals of 3 weeks.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- administration of DTX + RAM therapy, immune checkpoint inhibitors and EP4 antagonists is initiated on the same day.
- EP4 antagonist preferably compound A
- immune checkpoint inhibitor preferably anti-PD-1 antibody (preferably Nivolumab)
- DTX + RAM therapy is performed after administration of EP4 antagonists and immune checkpoint inhibitors.
- EP4 antagonist preferably compound A
- an immune checkpoint inhibitor preferably anti-PD-1 antibody (preferably Nivolumab)
- DTX + RAM therapy May be performed first or at the same time as the administration of the EP4 antagonist and the immune checkpoint inhibitor.
- the order of administration of EP4 antagonist, immune checkpoint inhibitor, docetaxel and ramucirumab may be started from any drug unless otherwise defined, and two or more drugs may be administered simultaneously. You may.
- the "docetaxel therapy" in the present invention is a cancer treatment method using docetaxel, and in one embodiment, the recommended dosage is, for example, 60 mg / m 2 (body surface area) of docetaxel for 60 minutes. It is a therapy in which intravenous administration is performed over the above and the administration is performed at 3-week intervals. As one embodiment, in any administration after the second cycle in the DTX therapy, docetaxel may be increased or decreased to 75 mg / m 2 or 50 mg / m 2 or administration of docetaxel may be discontinued depending on the patient's condition. Withdrawal, dose reduction, and resumption of DTX therapy will be carried out at the discretion of the doctor with reference to the latest package insert.
- the EP4 antagonist in this combination-6 is administered in one embodiment at the dosages described in (1) EP4 antagonist.
- compound A which is an EP4 antagonist, is orally administered at 5 mg once a day, at 10 mg once a day, at 20 mg once a day, or at 40 mg once a day. Will be done.
- 20 mg is orally administered once a day or 40 mg is orally administered once a day.
- the dose of Compound A may be reduced or the administration itself may be discontinued.
- the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg and the two-step dose reduction is 10 mg.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- nivolumab as an immune checkpoint inhibitor is administered by intravenous drip infusion of 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals.
- 360 mg of nivolumab is administered by intravenous drip infusion at intervals of 3 weeks.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- administration of DTX therapy, immune checkpoint inhibitors and EP4 antagonists is initiated on the same day.
- EP4 antagonists and immune checkpoint inhibitors are administered first.
- DTX therapy is performed after administration of EP4 antagonists and immune checkpoint inhibitors.
- EP4 antagonists and immune checkpoint inhibitors are administered on the same day, DTX therapy. May be performed first or at the same time as the administration of the EP4 antagonist and the immune checkpoint inhibitor.
- the order of administration of the EP4 antagonist, immune checkpoint inhibitor, and docetaxel may be started from any drug unless otherwise defined, and two or more drugs may be administered simultaneously. May be good.
- the "ramucirumab therapy (hereinafter, may be abbreviated as” RAM therapy ”)" in the present invention is a cancer treatment method using ramucirumab, and in one embodiment, as a recommended dosage.
- it is a treatment method in which ramucirumab 10 mg / kg is intravenously administered over 60 minutes, and the series of administrations is carried out at 3-week intervals.
- the dose of ramucirumab may be reduced to 8 mg / kg or 6 mg / kg or the administration of ramucirumab may be discontinued depending on the patient's condition in any of the administrations after the second cycle in the RAM therapy.
- the administration time of ramucirumab may be shortened to 30 to 60 minutes.
- the EP4 antagonist in this combination-7 is administered in one embodiment at the dosages described in (1) EP4 antagonist.
- compound A which is an EP4 antagonist, is orally administered at 5 mg once a day, at 10 mg once a day, at 20 mg once a day, or at 40 mg once a day. Will be done.
- 20 mg is orally administered once a day or 40 mg is orally administered once a day.
- the dose of Compound A may be reduced or the administration itself may be discontinued.
- the administration of compound A can be resumed, and when resuming, the dose of compound A can be reduced step by step and resumed at the discretion of the doctor.
- the one-step dose reduction is 20 mg and the two-step dose reduction is 10 mg.
- the one-step dose reduction is 10 mg and the two-step dose reduction is 5 mg.
- the immune checkpoint inhibitor in this combination-7 is administered in one embodiment at the dosage as described in (2) Immune checkpoint inhibitor.
- nivolumab as an immune checkpoint inhibitor is administered by intravenous drip infusion of 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals.
- 360 mg of nivolumab is administered by intravenous drip infusion at intervals of 3 weeks.
- the administration of nivolumab itself may be discontinued depending on the degree of side effects of the patient.
- administration of nivolumab can be resumed if the criteria for resumption are met.
- administration of RAM therapy, immune checkpoint inhibitors and EP4 antagonists is initiated on the same day.
- EP4 antagonists and immune checkpoint inhibitors are administered first.
- RAM therapy is performed after administration of EP4 antagonists and immune checkpoint inhibitors.
- EP4 antagonists and immune checkpoint inhibitors are administered on the same day, RAM therapy. May be performed first or at the same time as the administration of the EP4 antagonist and the immune checkpoint inhibitor.
- the order of administration of the EP4 antagonist, immune checkpoint inhibitor, and ramucirumab may be started from any drug unless otherwise defined, and two or more drugs may be administered simultaneously. May be good.
- the disease to which the therapeutic method of the present invention is applied is cancer.
- the cancers include, for example, leukemia (eg, acute myeloid leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia), malignant lymphoma (hodgkin lymphoma, non-hodgkin lymphoma (eg, adult)).
- leukemia eg, acute myeloid leukemia, chronic myeloid leukemia, acute lymphocytic leukemia, chronic lymphocytic leukemia
- malignant lymphoma hodgkin lymphoma, non-hodgkin lymphoma (eg, adult)
- T-cell leukemia follicular lymphoma, diffuse large B-cell lymphoma)
- multiple myeloma myelodystrophy syndrome, head and neck cancer
- esophageal cancer esophageal adenocarcinoma
- gastric cancer esophagogastric junction Cancer
- duodenal cancer colon cancer, colon cancer, rectal cancer
- liver cancer eg, hepatocellular carcinoma
- bile sac / bile duct cancer biliary tract cancer
- pancreatic cancer eg, pancreatic duct cancer
- Insulinoma intraductal papillary mucinous tumor
- thyroid cancer lung cancer
- non-small cell lung cancer eg, flat epithelial non-small cell lung cancer, non-flat epithelial non-small cell lung cancer
- small cell lung cancer breast cancer
- ovary Cancer eg, serous ovarian cancer
- cervical cancer uterine body cancer
- endometrial cancer vagina
- Diseases to which the therapeutic method of the present invention is applied are preferably colon cancer, pancreatic cancer, and lung cancer, and more preferably unresectable advanced or recurrent colon / rectal cancer, and curatively resectable locally advanced cancer.
- Lung cancer that has undergone combination therapy including rectal cancer, pancreatic cancer with distant metastasis, anti-PD-1 antibody or anti-PD-L1 antibody, and platinum preparation, and has progressed or recurred refractory.
- unresectable advanced or recurrent colorectal cancer curatively resectable locally advanced cancer, rectal cancer, pancreatic duct cancer with distant metastasis, and anti-PD-1 antibody or anti-PD- Non-small cell lung cancer with refractory progression or recurrence after receiving combination therapy including L1 antibody and platinum preparation.
- treatment refers to, for example, (i) reducing the growth of tumor cells, (ii) reducing the symptoms caused by the cancer, and (iii) the quality of life of a cancer patient. Includes treatments performed to improve, (iv) reduce the dose of other anti-cancer drugs or cancer treatment aids already administered, and / or (v) prolong the survival of cancer patients.
- “suppressing the progression” of a cancer means delaying the progression of the cancer, stabilizing the symptoms caused by the cancer, and slowing the progression of the symptoms.
- “suppression of recurrence” of cancer means to prevent recurrence of cancer in patients whose cancer lesions have completely or substantially disappeared or have been completely or substantially eliminated by cancer treatment or excision surgery. do.
- the treatment method of the present invention is described in the following cancer patients, that is, (a) patients whose therapeutic effect with an anticancer drug is insufficient or insufficient, or patients whose exacerbation after treatment with an anticancer drug, (b) curative or curative. Patients with unresectable, metastatic, recurrent, refractory and / or distant metastatic cancer, (c) Tumor Proportion Score (hereinafter abbreviated as "TPS”) is 50% or more, 25% or more, 10 % Or more, 5% or more or 1% or more of cancer patients, (d) Combined Positive Score (hereinafter abbreviated as "CPS”) is 20% or more, 10% or more, 5% or more or 1% or more.
- TPS Tumor Proportion Score
- CPS Combined Positive Score
- TMB Tumor mutation load
- the treatment method of the present invention comprises the following cancer patients, that is, (g) patients who have not been treated with an anticancer drug, (h) TPS is less than 50%, less than 25%, and less than 10%.
- cancer patients with cancer less than 5% or less than 1%, (i) Patients with cancer having CPS less than 20%, less than 10%, less than 5% or less than 1%, (j) dMMR and / or MSI -Prescription for patients with cancer who do not have H or who have low-frequency microsatellite instability (hereinafter abbreviated as "MSI-L"), or (k) patients with cancer who have a low frequency of TMB. May be more required.
- cancer patients for whom the treatment method of the present invention is required include (i) unresectable advanced or recurrent cancer patients, particularly colon / rectal cancer patients, and (ii) curative resectable topical cancer patients.
- cancer patients with advanced cancer especially patients with rectal cancer, (iii) cancer patients who have not been treated with anticancer drugs and / or have distant metastases, especially patients with pancreatic cancer, or (iv). Cancer patients who have undergone combination therapy including anti-PD-1 antibody or anti-PD-L1 antibody and platinum preparations and have progressed or relapsed refractory, especially patients with lung cancer.
- a patient with unresectable advanced or recurrent colorectal cancer may be mentioned.
- a patient who has not been treated for colorectal cancer may be mentioned.
- a patient with rectal cancer which is locally advanced cancer that can be curatively resected, is mentioned.
- One embodiment includes a patient who has not been treated for rectal cancer.
- a patient with pancreatic cancer who has not been treated with an anticancer drug and / or has distant metastasis can be mentioned.
- a patient who has not been treated for pancreatic cancer can be mentioned.
- a patient who has not been treated with a systemic malignant tumor agent for pancreatic cancer is mentioned.
- a systemic antineoplastic agent for pancreatic cancer having distant metastasis.
- a lung cancer patient who has received combination therapy including an anti-PD-1 antibody or an anti-PD-L1 antibody and a platinum preparation and has shown refractory progression or recurrence.
- a patient confirmed to have non-small cell lung cancer can be mentioned.
- Patients with stage IV or recurrent non-small cell lung cancer are preferred. More preferably, there is a patient who has received a combination therapy including an anti-PD-1 antibody or an anti-PD-L1 antibody as a first-line treatment and a platinum preparation, and has shown refractory progression or recurrence.
- the therapeutic method of the present invention maximizes its antitumor effect. It can be expected to be demonstrated. Further, according to the therapeutic method of the present invention, it is possible to reduce the dose of each drug and administer it, which is expected to reduce side effects.
- the therapeutic method of the present invention exerts a synergistic effect.
- the therapeutic effect of immune checkpoint inhibitors or EP4 receptor antagonists, or standard therapy alone, or immune checkpoint inhibitors and standard therapies, EP4 receptor antagonists and standard therapies, or immunity exerts a synergistic effect compared to the therapeutic effect of the combined use of checkpoint inhibitors and EP4 receptor antagonists.
- the treatment method of the present invention reduces side effects. Also, in one embodiment, the therapeutic effect of immune checkpoint inhibitors or EP4 receptor antagonists, or standard therapy alone, or immune checkpoint inhibitors and standard therapies, EP4 receptor antagonists and standard therapies, or immunity. It reduces side effects compared to the therapeutic effect of combination with checkpoint inhibitors and EP4 receptor antagonists.
- timing of administration of each drug used for concomitant administration in the combination of the present invention may be simultaneous or separate.
- the therapeutic method of the present invention can be applied to the treatment of metastatic cancer and the suppression of metastasis.
- the therapeutic method of the present invention in one aspect, suppresses recurrence.
- treatment is associated with reduction in tumor size, suppression of tumor growth (delay or arrest), suppression of tumor metastasis (delay or arrest), suppression of recurrence (prevention or delay), and cancer. It means producing at least one of alleviations of one or more symptoms.
- the therapeutic method of the present invention is used for (1) complementing and / or enhancing the therapeutic effect, (2) improving kinetics / absorption, reducing the dose, and / or (3) reducing side effects. May be used in combination with the following drugs (eg, known anti-cancer treatments).
- standard therapy is a standard treatment method that is based on scientific evidence and is recommended for general patients in a certain condition, and examples thereof include chemotherapy.
- One embodiment includes (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpaclitaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsylmab therapy.
- the present invention provides, for example, the following embodiments.
- Standard therapies preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsylumab therapy, more preferably.
- bevasizumab and XELOX therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) dosetaxel and ramsilmab therapy, more preferably (i).
- Bevasizumab and XELOX therapy (iii) FOLFIRINOX therapy or its weight loss regimen, (iv) gemcitabine and nabpacritaxel therapy, or (vi) docetaxel and ramsilmab therapy), and administration in combination with immunocheckpoint inhibitor.
- a characteristic therapeutic agent for suppressing the progression, suppressing recurrence, and / or suppressing the progression of cancer which comprises an EP4 antagonist as an active ingredient.
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsilmab therapy, more preferably.
- a therapeutic agent for suppressing the progression, suppressing recurrence, and / or suppressing the progression of cancer which comprises an immune checkpoint inhibitor as an active ingredient.
- EP4 antagonist is a general formula (I):
- R 1 represents COOR 8 , tetrazole, SO 3 H, SO 2 NH 2 , SO 2 NHR 8-1 , CONHSO 2 R 8-1 , SO 2 NHCOR 8-1 , or hydroxamic acid.
- R 8 represents a hydrogen atom, C1-4 alkyl, or benzyl
- R 8-1 represents C1-4 alkyl, C1-4 haloalkyl, C3-10 carbocycle, or 3-10 membered heterocycle, and the C3-10 carbocycle and 3-10 membered heterocycle, respectively, is C1.
- L 1 represents C1-5 alkylene, C2-5 alkenylene, or C2-5 alkinylene, R2 is halogen, C1-4alkyl, C1-4alkoxy, C1-4alkylthio, C2-4alkenyl, C2-4alkynyl, -O (C1-4haloalkyl), -S (C1-4haloalkyl),- C (O) (C1-4 alkyl), -SO 2 (C1-4 alkyl), -CONH (C1-4 alkyl), -CON (C1-4 alkyl) 2 , -NHC (O) (C1-4 alkyl) ), -N (C1-4 alkyl) C (O) (C1-4 alkyl) (C1-4 alkyl) (C1-4 alkyl) (C1-4 alkyl)
- the C1-4 alkyl may be substituted with halogen and may be substituted.
- (C1-4alkyl) 2 in the R2 represents two independent C1-4 alkyls, each of which may be the same or different.
- X 1 represents CR 6 or nitrogen atom
- R 6 represents hydrogen atom or
- X 2 represents CR 7 or nitrogen atom
- R 7 represents hydrogen atom
- R 2 or -L 3 -R 9 and L 3 represents methylene, oxygen atom or sulfur atom which may be oxidized.
- R 9 represents a 4-10 membered heterocycle which may be substituted with a substituent selected from the group consisting of halogen, C1-4alkyl, and C1-4haloalkyl.
- each said C1-4 alkyl may be substituted with a halogen.
- ring represents a benzene ring or a 5-6 member monocyclic aromatic heterocycle.
- R5 is ( 1 ) halogen, (2) C1-4alkyl, (3) carboxyl, (4) nitrile, (5) -CONHR 11 , (6) -C (O) R 12 , (7) -OR.
- R 14 (8) -S (O) t R 15 , (9) -CH 2 R 16 , (10) -NR 17 R 17 , (11) -NHCOR 11 , (12) C4-10 carbocycle, or ( 13) Represents a 4-10 membered heterocycle, the C4-10 membered ring or the 4-10 membered heterocycle may be substituted with 1 to 3 R18s , and if there are multiple R18s , then R Each of the 18 may be the same or different independently.
- R 11 represents a C1-6 alkyl, C3-6 cycloalkyl, phenyl, or 4-6 membered heterocycle, where R 11 may be substituted with 1 to 3 R 13s , wherein the R 13s are plural.
- R 13 may be independently the same or different, respectively.
- R 13 represents a halogen, C1-6 alkyl, C3-6 cycloalkyl, C1-4 alkoxy, hydroxyl group, -NR 20 R 21 , benzene, or 4-6 membered heterocycle.
- R 20 and R 21 independently represent a hydrogen atom or C1-4 alkyl, respectively.
- R 12 represents a C1-6 alkyl, C3-6 cycloalkyl, benzene, or 4-6 membered heterocycle, and the C3-6 cycloalkyl, benzene, or 4-6 membered heterocycle is independent of each other. It may be substituted with halogen, C1-4alkyl, or C1-4alkoxy.
- R 14 represents a hydrogen atom, C1-6 alkyl, C3-6 cycloalkyl, benzene, or benzyl, wherein the C1-6 alkyl may be substituted with 1 to 3 R 19s , where the R 19 is. When there are a plurality of R 19 , each of them may be independently the same or different. R 19 is substituted with a substituent selected from the group consisting of C1-4 alkoxy, -CONH (C1-4 alkyl), -CON (C1-4 alkyl) 2 , or C1-4 alkyl and C1-4 haloalkyl. Represents a 5-6 member monocyclic aromatic heterocycle which may be present.
- the (C1-4 alkyl) 2 in the R 19 represents two independent C1-4 alkyls, each of which may be the same or different.
- R 15 represents C1-6 alkyl, C3-6 cycloalkyl, benzene, or benzyl.
- R 16 represents a hydroxyl group or C1-4 alkoxy and represents R 17 independently represent a hydrogen atom, C1-6 alkyl, or C3-6 cycloalkyl, respectively.
- R18 is halogen, C1-6alkyl, C3-6cycloalkyl , C1-4alkoxy, oxo, nitrile, hydroxyl group, hydroxymethyl, 1-methyl-1-hydroxyethyl, (C1-4alkyl) SO 2- , Represents a 4-6 membered heterocycle, (C1-4 alkyl) NH-, or (C1-4 alkyl) 2 N-.
- the (C1-4 alkyl) 2 in the R 18 represents two independent C1-4 alkyls, each of which may be the same or different.
- m represents an integer of 1 to 4
- n represents an integer of 0 to 4
- p represents an integer of 0 to 2
- q represents an integer of 0 to 6
- r represents an integer of 0 to 6.
- the EP4 antagonist is the general formula (I-2).
- R 2a represents a halogen
- R 6a represents a hydrogen atom or a halogen
- qa represents an integer of 0 to 3
- ra represents an integer of 0 to 4
- other symbols are the same as in claim 1.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-].
- the immune checkpoint inhibitor is an anti-PD-1 antibody, an anti-PD-L1 antibody, or an anti-CTLA-4 antibody.
- the immune checkpoint inhibitor is an anti-PD-1 antibody.
- the immune checkpoint inhibitor is an anti-PD-L1 antibody
- the anti-PD-L1 antibody is Atezolizumab, Avelumab, Durvalumab, BMS-936559, STI-1014, KN035 (Envafolimab), LY3300054 (Lodapolyma).
- Nivolumab 3 mg / kg (body weight) or 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 240 mg once for 2 weeks).
- the agent according to the above [17] wherein 360 mg once at intervals is administered at intervals of 3 weeks, or 480 mg once at intervals of 4 weeks).
- the bevacizumab and CAPOX therapy (I) Bevacizumab is administered once at 7.5 mg / kg at 3-week intervals. (Ii) Oxaliplatin 130 mg / m 2 is administered at 3-week intervals, and (iii) capecitabine 1000 mg / m 2 is orally administered twice daily for 14 days, followed by a 7-day rest period.
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an EP4 antagonist as an active ingredient, characterized by administration in combination with bevasizumab and XELOX therapy, as well as immune checkpoint inhibitors.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane]. -4,1'-cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butane acid, or a salt thereof, 5 mg to 40 mg (preferably 20 mg or 40 mg once) as an EP4 antagonist daily.
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an immune checkpoint inhibitor as an active ingredient, characterized by administration in combination with bevasizumab and XELOX therapy, and EP4 antagonists.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- the colorectal cancer is unresectable advanced or recurrent colorectal cancer (preferably unresectable advanced or recurrent colorectal cancer without treatment history).
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiroyl].
- Agent. [2-13] The agent according to the above [2-12], wherein the immune checkpoint inhibitor is an anti-PD-1 antibody.
- Anti-PD-1 antibody is Nivolumab, Cemiplimab, Pembrolizumab, Spartanismab, Tissellizumab, AMP-514, Dostarlimab, Toriparimab, Toripalimab, Camrelizumab, Gen Retifanlimab), AGEN2034 (Balstilimab), CS1003, HLX10 (Serplelimab), BAT-1306, AK105, AK103, BI 754991, LZM009, CMAB819, Sym021, GB226 (Geptanolimab) ), BCD-100 (Prolgolimab), PF-06801591 (Sasanlimab), CX-188, JNJ-63722383 (Cemiplimab) or AB122 (Zimberelimab), the agent according to the above [2-13].
- the immune checkpoint inhibitor is an anti-PD-L1 antibody
- the anti-PD-L1 antibody is Atezolizumab, Avelumab, Durvalumab, BMS-936559, STI-1014, KN035 (Envafolimab), LY3300054 (Lodapolima).
- Nivolumab 3 mg / kg (body weight) or 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals (preferably 240 mg once).
- the agent according to the above [2-17] wherein 360 mg is administered once at 2-week intervals, or 480 mg once at 4-week intervals).
- 240 mg of Nivolumab is intravenously administered at intervals of 2 weeks for about 30 minutes.
- Preoperative chemoradiotherapy includes 45 Gy / 25 doses of pelvic cavity and 5.4 Gy / 3 doses of boost to the primary lesion, and capecitabine at 825 mg / m 2 twice daily.
- Cancer containing an EP4 antagonist as an active ingredient which is characterized by being administered to a cancer patient who has undergone preoperative chemoradiotherapy (preferably colorectal cancer, more preferably colon / rectum).
- preoperative chemoradiotherapy preferably colorectal cancer, more preferably colon / rectum.
- a suppressive, recurrence-suppressing and / or therapeutic agent for cancer preferably curatively resectable locally advanced rectal cancer.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- Preoperative chemoradiotherapy is a combination of radiation therapy and capecitabin (preferably 45 Gy / 25 doses to the pelvic cavity and 5.4 Gy / 3 doses to the primary lesion, and capecitabin. 825 mg / m 2 is administered twice a day for 21 days or 42 times or more), Agent.
- the agent according to the above [2-39], which is further administered in combination with an immune checkpoint inhibitor, wherein the immune checkpoint inhibitor is nivolumab, and 240 mg at a time as anti-Nivolumab is 2
- Colorectal cancer containing an immune checkpoint inhibitor as an active ingredient which is characterized by being administered in combination with an EP4 antagonist to cancer patients who have undergone preoperative chemoradiotherapy. Progression suppression, recurrence suppression and / or therapeutic agent,
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is Nivolumab, 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals (preferably 240 mg once at 2-week intervals).
- Intervals) and (iii) preoperative chemoradiotherapy is a combination of radiation therapy and capecitabin (preferably 45 Gy / 25 doses of pelvic cavity and 5.4 Gy / 3 doses to the primary lesion). Bost irradiation, and 825 mg / m 2 of capecitabin twice daily for 21 days or 42 times or more). Agent.
- [3-1] The agent according to the above [1] or [2], wherein the standard therapy is FOLFIRINOX therapy or a weight loss regimen thereof.
- [3-2] The agent according to the above [3-1], wherein the EP4 antagonist is the compound represented by the general formula (I) described in the above [3] or a salt thereof.
- [3-3] The agent according to the above [3-1] or [3-2], wherein the cancer is pancreatic cancer (preferably pancreatic duct cancer, more preferably invasive pancreatic duct cancer).
- [3-4] The agent according to the above [3-3], wherein the pancreatic cancer is pancreatic cancer having distant metastasis.
- Anti-PD-1 antibodies include Nivolumab, Cemiplimab, Pembrolizumab, Spartanismab, Tissellizumab, AMP-514, Dostarlimab, Toriparimab, Toripalimab, Camrelizumab, Gen Retifanlimab), AGEN2034 (Balstilimab), CS1003, HLX10 (Serplelimab), BAT-1306, AK105, AK103, BI 754091, LZM009, CMAB819, Sym021, GB226 (Geptanolimab) ), BCD-100 (Prolgolimab), PF-06801591 (Sasanlimab), CX-188, JNJ-63722383 (Cemiplimab) or AB122 (Zimberelimab), the agent according to the above [3-11].
- the immune checkpoint inhibitor is an anti-PD-L1 antibody, and the anti-PD-L1 antibody is Atezolizumab, Avelumab, Durvalumab, BMS-936559, STI-1014, KN035 (Envafolimab), LY3300054 (Lodapolima). , HLX20, SHR-1316, CS1001, MSB2311, BGB-A333, KL-A167, CK-301, AK106, AK104, ZKAB001, FAZ053, CBT-502, JS003 and CX-072, in the above [3-10]. The agent described.
- Nivolumab 3 mg / kg (body weight) or 240 mg once at 2-week intervals, 360 mg once at 3-week intervals, or 480 mg once at 4-week intervals (preferably 240 mg once).
- the agent according to the above [3-15] wherein 360 mg is administered once at 2-week intervals, or 480 mg once at 4-week intervals).
- 480 mg of Nivolumab is intravenously administered at intervals of 4 weeks for about 30 minutes.
- FOLFIRINOX therapy or a weight loss regimen thereof is a therapy in which (i) oxaliplatin, (ii) levoleucovorinate calcium, (iii) irinotecan hydrochloride hydrate, and (iv) fluorouracil are administered in combination.
- FOLFIRINOX therapy Intravenously administer oxaliplatin 50-85 mg / m 2 (preferably 50 mg / m 2 , 65 mg / m 2 , 85 mg / m 2 , more preferably 85 mg / m 2 ).
- Ii Intravenously administer levoleucovorinate calcium 200 mg / m 2 .
- Irinotecan hydrochloride hydrate 90-180 mg / m 2 preferably 90 mg / m 2 , 120 mg / m 2 , 150 mg / m 2 , 180 mg / m 2 , more preferably 180 mg / m 2 ) intravenously.
- the weight loss regimen for FOLFIRINOX therapy is (I) Intravenously administer oxaliplatin 50-85 mg / m 2 (preferably 50 mg / m 2 , 65 mg / m 2 , 85 mg / m 2 , more preferably 85 mg / m 2 ). (Ii) Intravenously administer levoleucovorinate calcium 200 mg / m 2 . (Iii) Irinotecan hydrochloride hydrate 120-140 mg / m 2 (preferably 140 mg / m 2 ) is administered intravenously.
- Fluorouracil 1200-2400 mg / m 2 (preferably 1200 mg / m 2 , 1800 mg / m 2 , 2400 mg / m 2 , more preferably 2400 mg / m 2 ) is a therapy that is continuously administered intravenously.
- [3-34] FOLFIRINOX therapy or a weight loss regimen thereof administers a series of doses at 2- to 4-week intervals (preferably 2-week intervals, 3-week intervals, 4-week intervals, more preferably 2-week intervals).
- [3-35] FOLFIRINOX therapy Intravenously administer oxaliplatin 85 mg / m 2 over 2 hours.
- levoleucovorinate calcium 200 mg / m 2 is intravenously administered over 2 hours.
- levoleucovorinate calcium is intravenously administered over 1.5 hours.
- fluorouracil 400 mg / m 2 is rapidly intravenously administered.
- fluorouracil 2400 mg / m 2 is continuously administered intravenously over 46 hours, and the series of administrations is carried out at 2-week intervals.
- the agent according to any one of [3-34].
- the weight loss regimen for FOLFIRINOX therapy is (I) Intravenously administer oxaliplatin 85 mg / m 2 over 2 hours. (Ii) Levoleucovorinate calcium 200 mg / m 2 is intravenously administered over 2 hours. (Iii) From 30 minutes after the start of administration of levoleucovorinate calcium, irinotecan hydrochloride hydrate 150 mg / m 2 is intravenously administered over 1.5 hours.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 480 mg once at 4 week intervals). Administered at intervals)
- the FOLFIRINOX therapy is (A) Oxaliplatin 85 mg / m 2 is intravenously administered over 2 hours.
- C Intravenously administer irinotecan hydrochloride hydrate 180 mg / m 2 over 1.5 hours 30 minutes after the start of administration of levoleucovorinate calcium.
- D Further, after the administration of the levoleucovorinate calcium is completed, fluorouracil 400 mg / m 2 is rapidly intravenously administered.
- fluorouracil 2400 mg / m 2 is continuously administered intravenously over 46 hours, and the series of administrations is carried out at 2-week intervals.
- A Oxaliplatin 85 mg / m 2 is intravenously administered over 2 hours.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 480 mg once at 4 week intervals). Administered at intervals)
- the FOLFIRINOX therapy is (A) Oxaliplatin 85 mg / m 2 is intravenously administered over 2 hours.
- C Irinotecan hydrochloride hydrate 180 mg / m 2 is intravenously administered over 1.5 hours from 30 minutes after the start of administration of levoleucovorinate calcium.
- D Further, after the administration of the levoleucovorinate calcium is completed, fluorouracil 400 mg / m 2 is rapidly intravenously administered.
- fluorouracil 2400 mg / m 2 is continuously administered intravenously over 46 hours, and the series of administrations is carried out at 2-week intervals.
- A Oxaliplatin 85 mg / m 2 is intravenously administered over 2 hours.
- pancreatic cancer pancreatic cancer having distant metastasis.
- pancreatic cancer pancreatic cancer having distant metastasis.
- [4-1] The agent according to the above [1] or [2], wherein the standard therapy is gemcitabine and nabupaclitaxel therapy.
- [4-2] The agent according to the above [4-1], wherein the EP4 antagonist is the compound represented by the general formula (I) described in the above [3], or a salt thereof.
- [4-3] The agent according to the above [4-1] or [4-2], wherein the cancer is pancreatic cancer (preferably pancreatic duct cancer, more preferably invasive pancreatic duct cancer).
- [4-4] The agent according to the above [4-3], wherein the pancreatic cancer is pancreatic cancer having distant metastasis.
- Anti-PD-1 antibodies include Nivolumab, Cemiplimab, Pembrolizumab, Spartanismab, Tissellizumab, AMP-514, Dostarlimab, Toriparimab, Toripalimab, Camrelizumab, GenMab, Camrelizumab, Gen Retifanlimab), AGEN2034 (Balstilimab), CS1003, HLX10 (Serplelimab), BAT-1306, AK105, AK103, BI 754091, LZM009, CMAB819, Sym021, GB226 (Geptanolimab) ), BCD-100 (Prolgolimab), PF-06801591 (Sasanlimab), CX-188, JNJ-63722383 (Cemiplimab) or AB122 (Zimberelimab), the agent according to the above [4-11].
- the immune checkpoint inhibitor is an anti-PD-L1 antibody, and the anti-PD-L1 antibody is Atezolizumab, Avelumab, Durvalumab, BMS-936559, STI-1014, KN035 (Envafolimab), LY3300054 (Lodapolima). , HLX20, SHR-1316, CS1001, MSB2311, BGB-A333, KL-A167, CK-301, AK106, AK104, ZKAB001, FAZ053, CBT-502, JS003 and CX-072, in the above [4-10]. The agent described.
- [4-29] The agent according to the above [4-20], wherein 10 mg / kg (body weight) of Durvalumab is administered once at 2-week intervals.
- [4-30] The above-mentioned [4-21], wherein 3 mg / kg (body weight) or 1 mg / kg (body weight) of Ipilimumab is intravenously administered four times at three-week intervals.
- Agent. [4-31] Gemcitabine and nab-paclitaxel therapy, (I) Gemcitabine 600-1000 mg / m 2 (preferably 600 mg / m 2 , 800 mg / m 2 , 1000 mg / m 2 , more preferably 1000 mg / m 2 ) is administered intravenously.
- Nabpaclitaxel 75-125 mg / m 2 (preferably 75 mg / m 2 , 100 mg / m 2 , 125 mg / m 2 , more preferably 125 mg / m 2 ) is administered intravenously.
- Gemcitabine and nab-paclitaxel therapy is a therapy in which a series of administrations are administered at 1-week intervals, continued for 3 weeks, and then withdrawn for 1 week, as described above [4-1] to [4-31].
- [4-33] Gemcitabine and nab-paclitaxel therapy (I) Gemcitabine 1000 mg / m 2 is intravenously administered over 30 minutes. (Ii) Nabpaclitaxel 125 mg / m 2 is intravenously administered over 30 minutes, and the series of administrations is performed at 1-week intervals, continued for 3 weeks, and then withdrawn for 1 week.
- [4-35] The agent according to any one of the above [4-1] to [4-34], which is a therapy in which administration of gemcitabine, nabupaclitaxel, an immune checkpoint inhibitor and an EP4 antagonist is started on the same day. ..
- [4-36] Gemcitabine and nab-paclitaxel therapy, as well as suppression, recurrence suppression and / or treatment of cancer comprising an EP4 antagonist as an active ingredient, characterized by administration in combination with an immune checkpoint inhibitor. It ’s an agent, (I) The EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 480 mg once at 4 week intervals).
- the gemcitabine and nab-paclitaxel therapy is (a) gemcitabine 1000 mg / m 2 intravenously over 30 minutes, (b) nab-paclitaxel 125 mg / m 2 intravenously over 30 minutes, the series. This is a therapy in which the drug is administered at 1-week intervals, continued for 3 weeks, and then withdrawn for 1 week.
- Gemcitabine and nab-paclitaxel therapy, as well as suppression, recurrence suppression and / or treatment of cancer comprising an immune checkpoint inhibitor as an active ingredient, characterized by administration in combination with an EP4 antagonist.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane]. -4,1'-cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butane acid, or a salt thereof, 5 mg to 40 mg (preferably 20 mg or 40 mg once) as an EP4 antagonist daily.
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 480 mg once at 4 week intervals).
- Administered at intervals) (Iii) The gemcitabine and nab-paclitaxel therapy is (a) gemcitabine 1000 mg / m 2 intravenously over 30 minutes, (b) nab-paclitaxel 125 mg / m 2 intravenously over 30 minutes, the series. This is a therapy in which the drug is administered at 1-week intervals, continued for 3 weeks, and then withdrawn for 1 week.
- [4-38] The agent according to the above [4-36] or [4-37], wherein the cancer is pancreatic cancer (preferably pancreatic duct cancer, more preferably invasive pancreatic duct cancer).
- pancreatic cancer preferably pancreatic duct cancer, more preferably invasive pancreatic duct cancer.
- pancreatic cancer is pancreatic cancer having distant metastasis.
- [5-1] The agent according to the above [1] or [2], wherein the standard therapy is docetaxel and / or ramucirumab therapy.
- [5-2] The agent according to the above [5-1], wherein the standard therapy is docetaxel therapy.
- [5-3] The agent according to the above [5-1], wherein the standard therapy is ramucirumab therapy.
- [5-4] The agent according to the above [5-1], wherein the standard therapy is docetaxel and ramucirumab therapy.
- [5-5] The agent according to the above [5-1] to [5-4], wherein the EP4 antagonist is the compound represented by the general formula (I) described in the above [3] or a salt thereof.
- [5-6] The agent according to any one of the above [5-1] to [5-5], wherein the cancer is lung cancer (preferably non-small cell lung cancer).
- lung cancer is non-small cell lung cancer with advanced or recurrent stage IV or recurrence (preferably non-small cell lung cancer with advanced or recurrent refractory to combination therapy including anti-PD-1 antibody or anti-PD-L1 antibody, and platinum preparations.
- the agent according to the above [5-6] which is (cell lung cancer).
- [5-8] The above-mentioned [5-8], which is administered to a patient who has received a combination therapy containing an anti-PD-1 antibody or an anti-PD-L1 antibody and a platinum preparation and has progressed or relapsed refractory.
- the EP4 antagonist is any of the above-mentioned [5-1] to [5-8], wherein the EP4 antagonist is the compound represented by the general formula (I-2) described in the above [8] or a salt thereof.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiroyl].
- Anti-PD-1 antibodies include Nivolumab, Cemiplimab, Pembrolizumab, Spartanismab, Tissellizumab, AMP-514, Dostarlimab, Toriparimab, Toripalimab, Camrelizumab, Gen Retifanlimab), AGEN2034 (Balstilimab), CS1003, HLX10 (Serplelimab), BAT-1306, AK105, AK103, BI 754091, LZM009, CMAB819, Sym021, GB226 (Geptanolimab) ), BCD-100 (Prolgolimab), PF-06801591 (Sasanlimab), CX-188, JNJ-63722383 (Cemiplimab) or AB122 (Zimberelimab), the agent according to the above [5-14].
- the immune checkpoint inhibitor is an anti-PD-L1 antibody
- the anti-PD-L1 antibody is Atezolizumab, Avelumab, Durvalumab, BMS-936559, STI-1014, KN035 (Envafolimab), LY3300054 (Lodapolima).
- Ipilimumab is administered intravenously at a dose of 3 mg / kg (body weight) or 1 mg / kg (body weight) four times at 3-week intervals or at a dose of 1 mg / kg (body weight) at 6-week intervals.
- Docetaxel therapy is a therapy in which docetaxel 50 to 75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) is administered intravenously.
- Docetaxel therapy is intravenous docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) over 60 minutes.
- Ramucirumab therapy is a therapy in which ramucirumab 6 to 10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) is intravenously administered.
- Ramucirumab therapy is 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) of ramucirumab for 30-60 minutes (preferably 60 minutes). ), And the series of administrations is performed at 3-week intervals.
- Docetaxel and ramucirumab therapy (I) Docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) is administered intravenously. (Ii) Ramucirumab 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) is administered intravenously.
- the agent according to any one of the above-mentioned [5-1], [5-4] to [5-33], which is a therapy.
- Docetaxel and ramucirumab therapy (I) Docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) is administered intravenously over 60 minutes. (Ii) Ramucirumab 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) administered intravenously over 30-60 minutes (preferably 60 minutes). The series of doses will be given at 3-week intervals.
- Agent. [5-41] A drug containing an EP4 antagonist as an active ingredient, which is characterized by being administered in combination with docetaxel therapy and an immune checkpoint inhibitor, for suppressing the progression, suppressing recurrence, and / or treating the cancer.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane]. -4,1'-cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butane acid, or a salt thereof, 5 mg to 40 mg (preferably 20 mg or 40 mg once) as an EP4 antagonist daily.
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Docetaxel therapy is intravenous administration of docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) over 60 minutes.
- a drug which is a therapy in which the administration is performed at 3-week intervals.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane]. -4,1'-cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butane acid, or a salt thereof, 5 mg to 40 mg (preferably 20 mg or 40 mg once) as an EP4 antagonist daily.
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Administered at intervals) (Iii) Docetaxel therapy is intravenous administration of docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) over 60 minutes.
- a drug which is a therapy in which the administration is performed at 3-week intervals.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Ramucirumab therapy takes 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) over 30-60 minutes (preferably 60 minutes).
- a drug that is administered intravenously and the series of administrations is performed at 3-week intervals.
- a therapeutic agent for suppressing the progression, suppressing recurrence, and / or suppressing the progression of cancer containing an immune checkpoint inhibitor as an active ingredient which is characterized by being administered in combination with ramucirumab therapy and an EP4 antagonist.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Ramucirumab therapy takes 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) over 30-60 minutes (preferably 60 minutes).
- Ramsylumab 4- [4-cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyran-] 4,1'-Cyclopropane] -2'-carbonyl ⁇ amino) phenyl] butanoic acid, or a salt thereof, and Nivolumab are therapies that begin on the same day (preferably 4-when administered on the same day).
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an EP4 antagonist as an active ingredient, characterized by administration in combination with docetaxel and ramucirumab therapy and immune checkpoint inhibitors.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Docetaxel and ramucirumab therapy (A) Docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) is intravenously administered over 60 minutes. (B) Ramucirumab 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) administered intravenously over 30-60 minutes (preferably 60 minutes).
- the agent which is a therapy in which the series of administrations is performed at 3-week intervals.
- a cancer progression-suppressing, recurrence-suppressing and / or therapeutic agent comprising an immune checkpoint inhibitor as an active ingredient, characterized by administration in combination with docetaxel and ramucirumab therapy, and EP4 antagonists.
- the EP4 antagonist is 4- [ 4 -cyano-2-( ⁇ (2'R, 4S) -6-[(propane-2-yl) carbamoyl] -2,3-dihydrospiro [1-benzopyrane].
- the immune checkpoint inhibitor is nivolumab, 240 mg once at 2 week intervals, 360 mg once at 3 week intervals, or 480 mg once at 4 week intervals (preferably 360 mg once for 3 weeks).
- Docetaxel and ramucirumab therapy (A) Docetaxel 50-75 mg / m 2 (preferably 50 mg / m 2 , 60 mg / m 2 , 75 mg / m 2 , more preferably 60 mg / m 2 ) is intravenously administered over 60 minutes. (B) Ramucirumab 6-10 mg / kg (preferably 6 mg / kg, 8 mg / kg, 10 mg / kg, more preferably 10 mg / kg) administered intravenously over 30-60 minutes (preferably 60 minutes).
- the agent which is a therapy in which the series of administrations is performed at 3-week intervals.
- Non-small advanced or recurrent lung cancer refractory to stage IV or recurrent non-small cell lung cancer preferably anti-PD-1 or anti-PD-L1 antibody, and combination therapy including platinum preparations
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsilmab therapy.
- bevasizumab and XELOX therapy preferably, (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsilmab therapy, more preferably (vi).
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or its weight loss regimen, (iv) gemcitabine and nabpacritaxel therapy, or (vi) docetaxel and ramsilmab therapy, and administered in combination with immunocheckpoint inhibitor.
- An EP4 antagonist used to control the progression, control and / or treatment of cancer.
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsilmab therapy.
- bevasizumab and XELOX therapy Preferably, (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsilmab therapy, more preferably (vi).
- bevasizumab and XELOX therapy iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (vi) docetaxel and ramsilmab therapy
- An immune checkpoint inhibitor used to control the progression, control and / or treatment of cancer.
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsylumab therapy.
- bevasizumab and XELOX therapy preferably, (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsilmab therapy, more preferably (vi).
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsylumab therapy.
- bevasizumab and XELOX therapy preferably, (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsilmab therapy, more preferably (vi).
- Standard therapy preferably (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, or (v) docetaxel and / or ramsilmab therapy.
- bevasizumab and XELOX therapy preferably, (i) bevasizumab and XELOX therapy, (iii) FOLFIRINOX therapy or a weight loss regimen thereof, (iv) gemcitabine and nabpacritaxel therapy, (vii) docetaxel therapy, or (vi) docetaxel and ramsilmab therapy, more preferably (vi).
- Example 1 An open-label, uncontrolled study in patients with unresectable advanced or recurrent colorectal cancer in combination with Compound A, Nivolumab as first-line treatment and XELOX + bevasizumab therapy as standard treatment.
- the purpose of this study is to examine the tolerability, safety and efficacy of compound A, Nivolumab and XELOX + bevasizumab therapy as first-line treatment in patients with unresectable advanced or recurrent colorectal cancer. do.
- the clinical trial can evaluate the combined effect of compound A, nivolumab and XELOX + bevacizumab therapy.
- Target patients Patients with unresectable advanced or recurrent colon / rectal cancer (2) Patient selection criteria Treatment with systemic antineoplastic agents for unresectable advanced or recurrent colon / rectal cancer at the time of enrollment Patients with no history and who met all of the prescribed patient selection criteria determined in light of the patient selection criteria in each of the trials conducted so far for Compound A, Nivolumab and XELOX + bevasizumab therapy were selected. If it becomes clear that the criteria are not met from registration to before the initial administration of the investigational drug, administration of the investigational drug will not be started and the study will be discontinued.
- Compound A was orally administered at 20 mg or 40 mg once daily, and continued to be administered until the prescribed discontinuation criteria for compound A were met.
- compound A, nivolumab and XELOX + bevacizumab therapy were administered on the same day, compound A and nivolumab were administered first, compound A and nivolumab were administered, and then XELOX + bevacizumab therapy was started.
- Nivolumab Nivolumab 360 mg was administered intravenously over 30 minutes at 3-week intervals and continued until the prescribed discontinuation criteria for nivolumab were met.
- nivolumab The administration of nivolumab was carried out at least 14 days after the previous administration and after the 15th day.
- Oxaliplatin was administered intravenously at 130 mg / m 2 (body surface area) over 2 hours.
- Capecitabine was orally administered at 1000 mg / m 2 twice daily after breakfast and after dinner, and after oral administration for 14 days, the drug was withdrawn for 7 days.
- Commercially available products were used for bevacizumab, oxaliplatin, and capecitabine.
- Clinical trial schedule The study consists of a screening phase, a treatment phase and a follow-up phase.
- the outline of the clinical trial schedule is shown in FIG.
- the screening period was within 28 days before the administration of the study drug, and the investigator or investigator met the above selection criteria and did not violate the above exclusion criteria, and included patients who were judged to be eligible for this study.
- the treatment period was 21 days per cycle, and the date of administration of the first investigational drug was the first day of cycle 1.
- the first day of each cycle after the second cycle was set to [21X (number of cycles-1) + 1] days.
- Administration of Compound A, Nivolumab and XELOX + bevacizumab therapy was started according to the above dosage and administration, respectively, and administration was continued according to the administration criteria, dose reduction criteria and dose at the time of dose reduction for Compound A, nivolumab and XELOX + bevacizumab therapy.
- the end of the treatment period is the time when the evaluation at the end (discontinuation) of the administration of Compound A, nivolumab and XELOX + bevacizumab therapy is completed.
- the subjects who discontinued or discontinued the administration of compound A, nivolumab and XELOX + bevacizumab therapy will be evaluated at the end of administration (discontinuation) and will shift to the follow-up period.
- a follow-up survey will be conducted after the follow-up period.
- CT / MRI imaging in the head fluorodeoxyglucose-positron emission tomography (FDG-PET) (or bone) Scinchography) etc. were performed to confirm the presence or absence of brain metastasis or bone metastasis.
- FDG-PET fluorodeoxyglucose-positron emission tomography
- the investigator or investigator measured the tumor diameter of the target lesion according to RECIST Guidelines 1.1, and determined the antitumor effect. Baseline evaluation was performed using the latest imaging tests within 28 days prior to study drug administration and confirmed to have one or more measurable lesions as defined in RECIST Guidelines version 1.1. The diagnostic imaging during the treatment period was performed at 6-week intervals from the 1st day of the first cycle to 6 weeks (+7 days) to 54 weeks (+7 days), and at 12-week intervals ( ⁇ 7 days) thereafter.
- Progression-free survival (day) "the day when the overall effect was determined to be PD, or the earliest day of death due to any cause"-"the date when the investigational drug was started” + 1
- PD progressing disease
- the date on which the last evaluable diagnostic imaging was performed is the discontinuation date.
- Subjects who have not undergone evaluable diagnostic imaging and have not died will have the study drug administration start date as the discontinuation date.
- Subjects who receive post-treatment for cancer before the overall effect is determined to be PD or die will have the date of the last evaluable diagnostic imaging prior to the start of post-treatment for cancer as the termination date. ..
- Response period The response period is calculated from the following formula.
- Response period (day) "The day when the overall effect is judged to be PD for the first time after the response is confirmed or the earliest day of death due to any cause"-"The first judgment date of the confirmed CR or PR" + 1
- the evaluation target is a subject who showed CR or PR confirmed through the clinical trial.
- Period until response The period until response is calculated from the following formula.
- Duration to response (day) "First determination date of confirmed CR or PR"-"Start date of study drug administration" + 1 (7)
- Rate of change in the sum of tumor diameters of the target lesion The rate of change in the sum of tumor diameters of the target lesion was calculated using the following formula for subjects having the target lesion. However, the rate of change in the sum of tumor diameters of the target lesion after post-treatment is not calculated.
- the maximum rate of change in the sum of tumor diameters of the target lesion is the rate of change at the time when the sum of tumor diameters of the target lesion becomes the smallest.
- the sum of tumor diameters of the target lesion after the overall effect is determined to be PD or after post-treatment is not used to calculate the maximum rate of change.
- Example 2 An open-label, uncontrolled study in which compound A or compound A and Nivolumab are used in combination as neoadjuvant therapy after preoperative chemoradiotherapy in curatively resectable locally advanced rectal cancer.
- Effective as neoadjuvant therapy after preoperative chemoradiation therapy (CRT) for curatively resectable locally advanced rectal cancer when compound A is administered alone and / or compound A and Nivolumab are administered in combination.
- CRT preoperative chemoradiation therapy
- the purpose is to examine sex, pharmacokinetics and safety.
- the study can evaluate the effect of compound A and / or the combination of compound A and nivolumab as neoadjuvant therapy.
- Target curative resectable locally advanced rectal cancer (2) Patient selection criteria At the time of enrollment, the prescribed criteria for Nivolumab and Compound A were determined in consideration of the patient selection criteria in each clinical trial conducted so far. Patients who met all of the patient selection criteria were selected. If it becomes clear that the criteria are not met between registration and the initial administration of the investigational drug, administration of the investigational drug will not be started and the study will be discontinued.
- preoperative CRT As preoperative CRT, it was administered to patients who were treated to meet the following conditions. -45 Gy / 25 times of irradiation to the pelvic cavity and 5.4 Gy / 3 times of bomb irradiation to the primary lesion were performed. It was started as capecitabine at a daily dose of 1,650 mg / m 2 (825 mg / m 2 twice daily). The starting dose is determined based on the usage / dose D method of Xeloda® Tablets 300.
- capecitabine was taken orally for a period corresponding to 75% of the 28 times of radiation irradiation (eg, in the case of 50.4 Gy / 28 times irradiation, it was taken for 21 days or 42 times in the morning and evening. Oral administration more than once). It doesn't matter if you lose weight or not.
- Compound A Compound A was orally administered at 40 mg once daily, and administration was continued until the prescribed discontinuation criteria for compound A were met.
- Nivolumab Nivolumab was administered intravenously at 240 mg every 2 weeks for 30 minutes and continued until the prescribed discontinuation criteria for nivolumab were met. Nivolumab administration was performed at least 10 days after the day following the previous administration, and after the 11th day.
- Clinical trial schedule The study consists of a screening phase, a therapeutic phase, a perioperative phase and a follow-up phase. The outline of the clinical trial design is shown in FIG.
- perioperative period in principle, surgery is performed 7 days after the final administration date of compound A, 14 days after the final administration date of nivolumab, and within 14 weeks after the end of preoperative CRT.
- the condition of the subject should be given top priority, and if surgery is required immediately, such as exacerbation of the underlying disease, or if surgery should be postponed, such as treatment for adverse events. Not limited.
- various final examinations are performed starting from 30 days after the end of the surgical treatment or the time when the investigator or the investigator determines that the complications due to the surgery have been alleviated, whichever is later.
- Imaging and endoscopy determined the presence or absence of tumor exacerbations from colonoscopy, CT or MRI images. The implementation period is not affected by the suspension of the investigational drug. In addition, if brain metastasis is suspected due to clinical symptoms during the screening period (within 14 days before administration of the investigational drug), CT / MRI imaging in the head, FDG-PET (or bone scintigraphy), etc. are performed to perform brain metastasis. Or, the presence or absence of bone metastasis was confirmed.
- pCR rate The percentage of subjects judged to be AJCC Tumor regression grade 0 by the pathologist of each implementing medical institution is defined as the pCR rate. Calculated. The pCR was one in which no viable tumor cells were found not only in the primary lesion but also in the regional lymph nodes (ypT0N0).
- MPR rate The percentage of subjects judged to be AJCC Tumor regression grade 0 or 1 by the pathologist of each implementing medical institution is calculated as the MPR rate.
- Example 3 An open-label, uncontrolled study in patients with pancreatic cancer with distant metastasis using compound A, Nivolumab and standard therapies mFFX or GnP therapy as first-line therapy.
- the purpose of this study is to examine the tolerability, safety and efficacy of compound A, Nivolumab and mFFX therapy or GnP therapy in combination as first-line treatment in patients with pancreatic cancer.
- the clinical trial can evaluate the combined effect of compound A, nivolumab and mFFX therapy or GnP therapy.
- Compound A was orally administered at 20 mg or 40 mg once daily, and continued to be administered until the prescribed discontinuation criteria for compound A were met.
- compound A, nivolumab and mFFX therapy or GnP therapy were administered on the same day, compound A and nivolumab were administered first, compound A and nivolumab were administered, and then mFFX therapy or GnP therapy was started.
- Nivolumab Nivolumab 480 mg was administered intravenously over 30 minutes at 4-week intervals and continued until the prescribed discontinuation criteria for nivolumab were met.
- nivolumab The administration of nivolumab was carried out at least 24 days after the previous administration and after the 25th day.
- Oxaliplatin was administered intravenously at 85 mg / m 2 (body surface area) over 2 hours.
- Irinotecan was administered intravenously at 150 mg / m 2 (body surface area) over 90 minutes.
- Levoleucovorinate was administered intravenously at 200 mg / m 2 (body surface area) over 2 hours.
- Fluorouracil was administered intravenously at 2400 mg / m 2 (body surface area) over 46 hours. The series of administrations was performed at 2-week intervals.
- Commercially available products were used for oxaliplatin, irinotecan, levofolinate, and fluorouracil.
- [GnP therapy] Gemcitabine was administered intravenously at 1000 mg / m 2 (body surface area) over 30 minutes and was withdrawn for at least 6 days.
- Nabpaclitaxel was administered intravenously at 125 mg / m 2 (body surface area) over 30 minutes and was withdrawn for at least 6 days. The series of administrations was performed at 1-week intervals, continued for 3 weeks, and then withdrawn for 1 week.
- the treatment period was 28 days per cycle, and the date of administration of the first investigational drug was the first day of cycle 1.
- the first day of each cycle after the second cycle was set to [28 ⁇ (number of cycles-1) + 1] days.
- Start administration of compound A, nivolumab and mFFX therapy or GnP therapy according to the above dosage and administration and continue administration according to the administration criteria, dose reduction criteria and dose at the time of dose reduction for compound A, nivolumab and mFFX therapy or GnP therapy. bottom.
- the end of the treatment period was defined as the time when the evaluation at the end (discontinuation) of the administration of compound A, nivolumab and mFFX therapy or GnP therapy was completed.
- the administration should be postponed until the clinical laboratory test values on the scheduled administration date recover to the condition satisfying the criteria, and the administration should be confirmed after confirming that the contraindications for each drug do not apply.
- Weight loss and weight loss criteria for mFFX therapy Weight loss will be implemented according to the latest package insert.
- [Criteria for discontinuing mFFX therapy] During the treatment phase, subjects who meet any of the prescribed discontinuation criteria determined in consideration of the discontinuation criteria in each clinical trial conducted so far for mFFX therapy will discontinue administration of mFFX therapy.
- the administration should be postponed until the clinical laboratory test values on the scheduled administration date recover to the condition satisfying the criteria, and the administration should be confirmed after confirming that the contraindications for each drug do not apply.
- Weight loss and weight loss criteria for GnP therapy Weight loss will be implemented according to the latest package insert.
- [Criteria for discontinuing GnP therapy] During the treatment period, subjects who meet any of the prescribed discontinuation criteria determined in consideration of the discontinuation criteria in each clinical trial conducted so far for GnP therapy discontinue administration of GnP therapy.
- Effectiveeness evaluation criteria (Diagnostic imaging) CT / magnetic resonance imaging (MRI) imaging of the chest, abdomen and pelvis was performed.
- CT / MRI imaging in the head fluorodeoxyglucose-positron emission tomography (FDG-PET) (or bone) Scinchography) etc. were performed to confirm the presence or absence of brain metastasis or bone metastasis.
- FDG-PET fluorodeoxyglucose-positron emission tomography
- the investigator or investigator measured the tumor diameter of the target lesion according to RECIST Guidelines 1.1, and determined the antitumor effect. Baseline evaluation was performed using the latest imaging tests within 28 days prior to study drug administration and confirmed to have one or more measurable lesions as defined in RECIST Guidelines version 1.1. The diagnostic imaging during the treatment period was performed 8 weeks after the 1st day of the first cycle (+7 days), and the tumor diameter was measured.
- the response rate indicates the proportion of subjects whose best overall effect was determined to be CR or PR.
- Disease control rate indicates the proportion of subjects whose best overall effect was determined to be CR, PR or SD.
- Progression-free survival The progression-free survival is calculated from the following formula.
- Progression-free survival (day) "the day when the overall effect was determined to be PD, or the earliest day of death due to any cause"-"the date when the investigational drug was started” + 1
- the day on which the last evaluable diagnostic imaging was performed is set as the discontinuation date.
- Subjects who have not undergone evaluable diagnostic imaging and have not died will have the study drug administration start date as the discontinuation date.
- Subjects who receive post-treatment for cancer before the overall effect is determined to be PD or die will have the date of the last evaluable diagnostic imaging prior to the start of post-treatment for cancer as the termination date. .. (5) Response period
- the response period is calculated from the following formula.
- Response period (day) "The day when the overall effect is judged to be PD for the first time after the response is confirmed or the earliest day of death due to any cause"-"The first judgment date of the confirmed CR or PR" + 1
- the evaluation target is a subject who showed CR or PR confirmed through the clinical trial.
- Duration to response (day) "First determination date of confirmed CR or PR"-"Start date of study drug administration" + 1 (7)
- Rate of change in the sum of tumor diameters of the target lesion The rate of change in the sum of tumor diameters of the target lesion was calculated using the following formula for subjects having the target lesion. However, the rate of change in the sum of tumor diameters of the target lesion after post-treatment is not calculated.
- the maximum rate of change in the sum of tumor diameters of the target lesion is the rate of change at the time when the sum of tumor diameters of the target lesion becomes the smallest.
- the sum of tumor diameters of the target lesion after the overall effect is determined to be PD or after post-treatment is not used to calculate the maximum rate of change.
- Example 4 Compound A, nivolumab and standard as second-line treatment in patients with advanced or recurrent non-small cell lung cancer refractory to combination therapy with anti-PD-1 or anti-PD-L1 antibodies and platinum preparations.
- An open-label, uncontrolled study with the therapy docetaxel and ramucirumab This study is an advanced or recurrent non-small cell lung cancer refractory to combination therapy with anti-PD-1 or anti-PD-L1 antibodies and platinum. The purpose of this study is to examine the tolerability, safety and efficacy of compound A, nivolumab, docetaxel and ramucirumab in combination as second-line treatment in patients.
- the clinical trial can evaluate the combined effect of compound A, nivolumab, docetaxel and ramucirumab.
- Target patients Patients with stage IV or recurrent non-small cell lung cancer
- Patient selection criteria At the time of enrollment, received combination therapy including anti-PD-1 antibody or anti-PD-L1 antibody as first-line treatment, and platinum preparations.
- Prescribed patient selection criteria determined in consideration of the patient selection criteria in each of the trials conducted so far for patients with refractory progression or recurrence and for Compound A, Nivolumab, docetaxel and ramucirumab. Patients who meet all were selected. If it became clear that the study drug did not meet the criteria from registration to before the first administration, administration of the study drug was not started and the study was discontinued.
- Patient exclusion criteria At the time of enrollment, patient exclusion criteria in each clinical trial for compound A, Nivolumab, docetaxel and ramucirumab, or patient selection in each proper use guide or proper use information for compound A, Nivolumab and docetaxel and ramucirumab. Patients who were considered to meet any of the prescribed patient exclusion criteria determined in consideration of the precautions were excluded. If it became clear that the study drug did not meet the criteria from registration to before the first administration, administration of the study drug was not started and the study was discontinued.
- Compound A was orally administered at 20 mg or 40 mg once daily, and continued to be administered until the prescribed discontinuation criteria for compound A were met.
- compound A, nivolumab, docetaxel and ramucirumab were administered on the same day, administration of compound A and nivolumab was first followed by administration of docetaxel and ramucirumab.
- Nivolumab Nivolumab 360 mg was administered intravenously over about 30 minutes at 3-week intervals and continued until the prescribed discontinuation criteria for nivolumab were met. The administration of nivolumab was carried out at least 14 days after the previous administration and after the 15th day.
- Docetaxel and ramucirumab The dosage of docetaxel and ramucirumab follows the procedure of the implementing medical institution, but the recommended dosage of docetaxel is 60 mg / m 2 (body surface area) intravenously over 60 minutes at 3-week intervals.
- Ramucirumab was administered intravenously at 10 mg / kg over 60 minutes at 3-week intervals.
- the 3-week interval means that the drug is administered between the 22nd and 29th days, with the previous administration day of docetaxel or ramucirumab as the 1st day.
- Commercially available products were used for docetaxel and ramucirumab.
- the treatment period was 21 days per cycle, and the date of administration of the first investigational drug was the first day of cycle 1.
- the first day of each cycle after the second cycle is [21 ⁇ (number of cycles-1) + 1] days.
- Administration of compound A, nivolumab, docetaxel and ramucirumab was started according to the above dosage and administration, respectively, and administration was continued according to the administration criteria, dose reduction criteria and dose at the time of dose reduction for compound A, nivolumab, docetaxel and ramucirumab.
- the end of the treatment period was defined as the time when the evaluation at the end (discontinuation) of the administration of compound A, nivolumab, docetaxel and ramucirumab was completed.
- the investigator or investigator measured the tumor diameter of the target lesion according to RECIST Guidelines 1.1, and determined the antitumor effect. Baseline evaluation was performed using the latest imaging tests within 28 days prior to study drug administration and confirmed to have one or more measurable lesions as defined in RECIST Guidelines Version 1.1. The diagnostic imaging during the treatment period was performed at 6-week intervals ( ⁇ 7 days) from the first day to 54 weeks after the first cycle, and at 12-week intervals ( ⁇ 7 days) thereafter, and the tumor diameter was measured.
- the overall effect and the best overall effect were the results of diagnostic imaging evaluated based on RECIST Guidelines Version 1.1.
- Evaluation item (1) Response rate (ORR), (2) Disease control rate (DCR), (3) Overall survival (OS), (4) Progression-free survival (PFS), (5) Duration of response (DOR), ( 6) Duration to response (TTR), (7) Best overall effect (BOR), (8) Rate of change in tumor diameter of target lesion, (9) Maximum rate of change in tumor diameter of target lesion, (9) 10) Changes in tumor markers
- ORR Response rate
- DCR Disease control rate
- DCR Disease control rate
- OS Overall survival
- PFS Progression-free survival
- DOR Duration of response
- TTR Duration to response
- BOR Best overall effect
- Rate of change in tumor diameter of target lesion (9) Maximum rate of change in tumor diameter of target lesion, (9) 10) Changes in tumor markers
- Response rate Response rate indicates the proportion of subjects whose best overall effect was determined to be CR or PR.
- (2) Disease control rate The disease control rate indicates the proportion
- Subjects who have not undergone evaluable diagnostic imaging and have not died will have the study drug administration start date as the discontinuation date.
- Subjects who receive post-treatment for cancer before the overall effect is determined to be PD or die will have the date of the last evaluable diagnostic imaging prior to the start of post-treatment for cancer as the termination date. .. (5)
- Response period The response period is calculated from the following formula.
- Response period (day) "The day when the overall effect is judged to be PD for the first time after the response is confirmed or the earliest day of death due to any cause"-"The first judgment date of the confirmed CR or PR" + 1
- the evaluation target is a subject who showed CR or PR confirmed through the clinical trial.
- (6) Period until response The period until response is calculated from the following formula.
- Duration to response (day) "First determination date of confirmed CR or PR"-"Start date of study drug administration" + 1 (7)
- Rate of change in the sum of tumor diameters of the target lesion was calculated using the following formula for subjects having the target lesion. However, the rate of change in the sum of tumor diameters of the target lesion after post-treatment is not calculated.
- the maximum rate of change in the sum of tumor diameters of the target lesion is the rate of change at the time when the sum of tumor diameters of the target lesion becomes the smallest.
- the sum of tumor diameters of the target lesion after the overall effect is determined to be PD or after post-treatment is not used to calculate the maximum rate of change.
- the present invention provides a new cancer treatment method and is useful.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本発明の課題は、有効ながん治療法を提供することにある。標準治療とEP4拮抗薬および免疫チェックポイント阻害物質(たとえば、抗PD-1抗体など)とを組み合わせたがん治療法を提供する。本発明の治療法は、がん治療に有用である。
Description
本開示は、標準治療とEP4拮抗薬および免疫チェックポイント阻害物質との併用によるがん治療法等に関する。
プロスタグランジンE2(PGE2)は、アラキドン酸カスケード中の代謝産物として知られており、細胞保護作用、子宮収縮作用、疼痛閾値の低下作用、消化管の蠕動運動促進作用、覚醒作用、胃酸分泌抑制作用、血圧降下作用、利尿作用等を有していることが知られている。
近年の研究の中で、PGE2受容体には、それぞれ役割の異なったサブタイプが存在することがわかってきた。現時点で知られているサブタイプは、大別して4つあり、それぞれEP1、EP2、EP3、EP4と呼ばれている(非特許文献1)。
これらのサブタイプのうち、EP4受容体は、マクロファージからのMCP-1産生抑制、リンパ球からのTNF-α、IL-2、およびIFN-γ産生抑制、並びにIL-10産生増強による抗炎症、血管拡張、血管新生、弾性線維の形成抑制、MMP-9発現制御に関与していると考えられている。さらに、EP4受容体は、骨髄(ミエロイド)由来免疫抑制細胞(Myeloid Derived Suppressor Cells)、制御性T細胞およびナチュラルキラー細胞を介したがん免疫制御にも関与していると考えられている。
これらのことより、EP4受容体に強く結合し、拮抗作用を有する化合物は、EP4受容体の活性化による疾患、例えば、骨疾患、がん、全身性肉芽腫、免疫疾患、アレルギー、アトピー、喘息、歯槽膿漏、歯肉炎、歯周病、アルツハイマー、川崎病、熱傷、多臓器不全、慢性頭痛、疼痛、血管炎、静脈不全、静脈瘤、動脈瘤、大動脈瘤、痔瘻、尿崩症、ストレス、子宮内膜症、子宮腺筋症、新生児動脈管開存症、胆石症等の疾患治療に有用であると考えられる(非特許文献2-7)。
特許文献1には、一般式(I)で示される化合物が、EP4拮抗作用を有し、がん治療薬として有用であることが開示されている(特許文献1参照)。
一方、がん細胞やがんの微小環境には、がんに対する免疫応答を妨げる種々の免疫チェックポイント分子が存在する。免疫チェックポイント阻害物質は、免疫抑制機構を解除し、がんに対する免疫反応を活性化する新たな治療法であり、既に、免疫チェックポイント阻害物質として、抗CTLA-4(cytotoxic T lymphocyte-associated antigen-4)抗体のイピリムマブ(ipilimumab)や抗PD-1(programmed cell death-1)抗体のニボルマブ(nivolumab)およびペンブロリズマブ(pembrolizumab)等が国内外で承認を得て、がん治療で使用されている。
特許文献2には、一般式(I)で示される化合物と免疫チェックポイント阻害物質とを組み合わせることががん治療に有用であることが開示されている(特許文献2参照)。
治癒切除不能な進行・再発の結腸・直腸がんの、主な治療としては薬物療法であり、フッ化ピリミジン系抗悪性腫瘍剤、オキサリプラチンおよびイリノテカンなどを用いた標準治療があり、その一つに、XELOXおよびベバシズマブの併用(以下、「XELOX+ベバシズマブ療法」と略記することがある。)がある。
また、遠隔転移を有する切除不能な膵臓がんの主な治療としては薬物療法であり、標準療法として、フルオロウラシル(5-FU)を含むレジメンをオキサリプラチンおよびイリノテカンと併用するFOLFIRINOX療法(以下、「FFX療法」と略記することがある。)、FFX療法から毒性軽減を期待して、フルオロウラシルの急速投与を省略し、イリノテカンを減量したmodified FOLFIRINOX療法(以下、「mFFX療法」と略記することがある。)あるいはゲムシタビンおよびナブパクリタキセルの併用(以下、「GnP療法」と略記することがある。)がある。
さらに、IV期または再発の非小細胞肺がんの主な治療としては薬物療法であり、標準療法として、ドセタキセルおよびラムシルマブの併用療法(以下、「DTX+RAM療法」と略記することがある。)あるいはドセタキセル療法(以下、「DTX療法」と略記することがある。)がある。
それら治療法において一定の効果が認められるものの、更に生存期間の延長をもたらす治療に対するアンメットニーズが存在する。
ジャーナル・オブ・リピッド・メディエーターズ・アンド・セル・シグナリング(Journal of Lipid Mediators and Cell Signalling)、第12巻、379-391ページ、1995年
ファーマコロジカル・レビューズ(Pharmacological Reviews)、第65巻、1010-1052ページ、7月、2013年
第105回 米国がん研究会議(American Association for Cancer Research(AACR))、要旨番号:LB-265、プレゼンテーション標題:ONO-AE3-208 inhibits myeloid derived suppressor cells and glioma growth、プレゼンテーション日時:2014年4月8日
フェブス・レターズ(FEBS Letters)、第364巻、339-341ページ、1995年
キャンサー・サイエンス(Cancer Science)、第105巻、1142-1151ページ、2014年
キャンサー・リサーチ(Cancer Research)、第70巻、1606-1615ページ、2010年
キャンサー・リサーチ(Cancer Research)、第62巻、28-32ページ、2002年
本発明の課題は、がん(例えば、大腸がん、膵臓がん、肺がん)の新たな治療法を提供することである。
本発明者らは、前記課題を解決するため、鋭意研究した結果、標準治療にEP4受容体拮抗薬および免疫チェックポイント阻害物質を併用することが有効ながん治療法となり得ること、また術前化学放射線療法を受けた患者に、さらにEP4受容体拮抗薬またはEP4受容体拮抗薬と免疫チェックポイント阻害物質とを投与することが有効ながん治療法となり得ることを見出した(両方の治療法を合わせて、本発明の治療法と略記することがある。)。
従って、ある態様では、
[1] 標準療法((i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、および免疫チェックポイント阻害物質の投与と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、または
[2] 標準療法((i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、
[3] 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、
[4] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[3]に記載の剤、または
[5] 術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、が提供される。
[1] 標準療法((i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、および免疫チェックポイント阻害物質の投与と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、または
[2] 標準療法((i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、
[3] 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、
[4] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[3]に記載の剤、または
[5] 術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤、が提供される。
本発明は、新たながん治療法を提供する。
(1)EP4拮抗薬
本発明において、EP4拮抗薬は、EP4拮抗作用を有する化合物であれば特に限定されないが、一実施形態において、WO2016/111347に記載された一般式(I):
本発明において、EP4拮抗薬は、EP4拮抗作用を有する化合物であれば特に限定されないが、一実施形態において、WO2016/111347に記載された一般式(I):
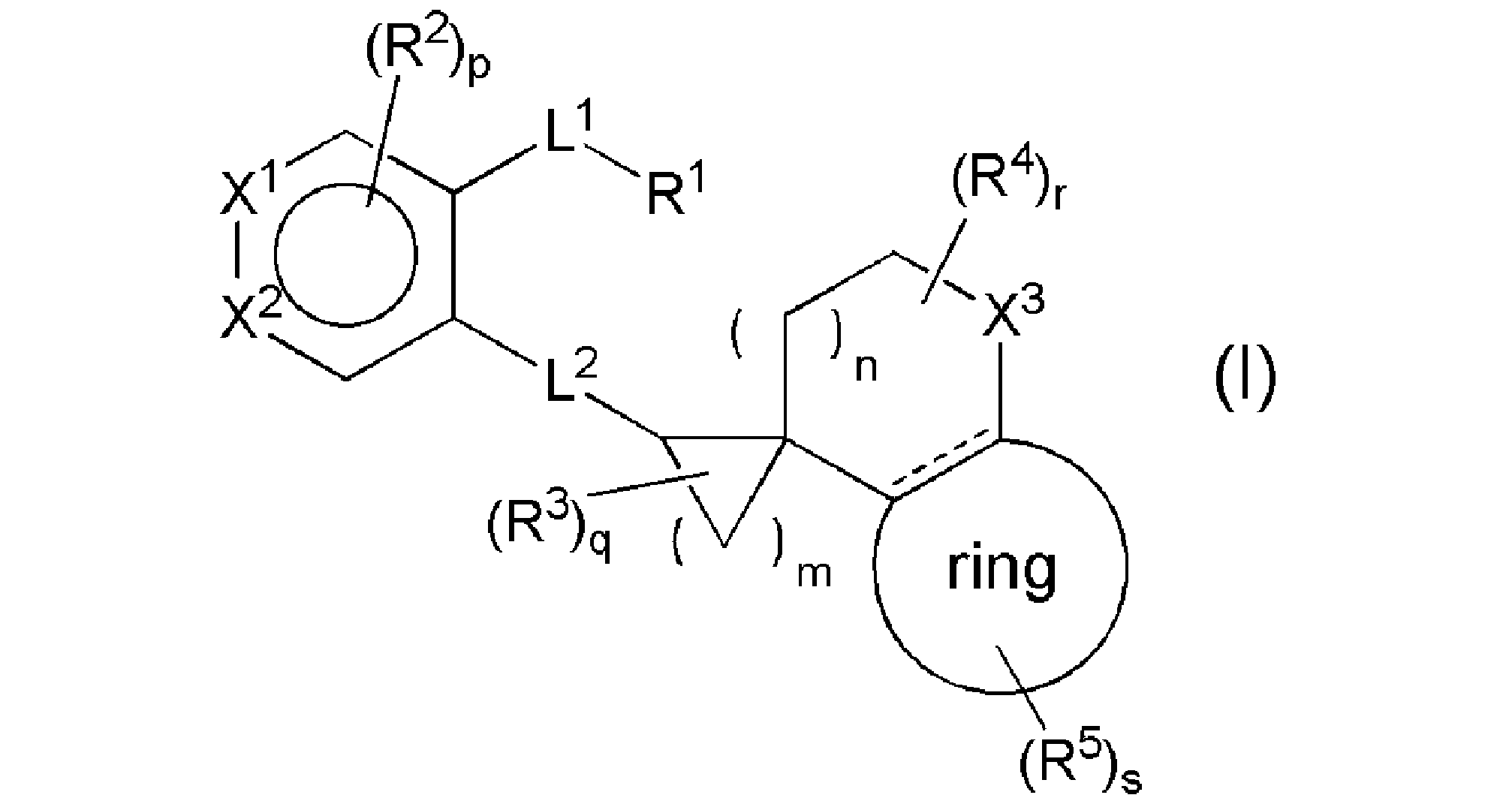
(式中、
R1はCOOR8、テトラゾール、SO3H、SO2NH2、SO2NHR8-1、CONHSO2R8-1、SO2NHCOR8-1、またはヒドロキサム酸を表し、
R8は水素原子、C1-4アルキル、またはベンジルを表し、
R8-1は、C1-4アルキル、C1-4ハロアルキル、C3-10炭素環、または3-10員複素環を表し、各々の該C3-10炭素環および3-10員複素環は、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリル(「-CN」を示す;以下同じ)で置換されていてもよく、
L1は、C1-5アルキレン、C2-5アルケニレン、またはC2-5アルキニレンを表し、
R2は、ハロゲン、C1-4アルキル、C1-4アルコキシ、C1-4アルキルチオ、C2-4アルケニル、C2-4アルキニル、-O(C1-4ハロアルキル)、-S(C1-4ハロアルキル)、-C(O)(C1-4アルキル)、-SO2(C1-4アルキル)、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、-NHC(O)(C1-4アルキル)、-N(C1-4アルキル)C(O)(C1-4アルキル)、-NHSO2(C1-4アルキル)、-N(C1-4アルキル)SO2(C1-4アルキル)、-SO2NH(C1-4アルキル)、-SO2N(C1-4アルキル)2、-NR17R17、ニトロ、ニトリル、水酸基、アルデヒド(ホルミルを示す;以下同じ)、またはカルボキシルを表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
該R2における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
X1はCR6または窒素原子を表し、R6は水素原子またはR2を表し、
X2はCR7または窒素原子を表し、R7は、水素原子、R2、または-L3-R9を表し、L3は、メチレン、酸素原子、または酸化されていてもよい硫黄原子を表し、R9は、ハロゲン、C1-4アルキル、およびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい4-10員複素環を表し、
L2は、-CH2CH2-、-CH=CH-、-CH2O-、-OCH2-、-CH2S-、-SCH2-、-CH2S(O)-、-S(O)CH2-、-CH2SO2-、-SO2CH2-、-CH2NH-、-NHCH2-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-を表し、
R3はC1-4アルキルまたはハロゲンを表し、
R4は、ハロゲン、C1-4アルキル、またはC1-4ハロアルキルを表し、
X3は、メチレン、酸素原子、酸化されていてもよい硫黄原子、またはNR10を表し、R10は、C1-4アルキル、-C(O)(C1-4アルキル)、-C(O)O(C1-4アルキル)、または-SO2(C1-4アルキル)を表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
ringはベンゼン環または5-6員単環式芳香族複素環を表し、
R1はCOOR8、テトラゾール、SO3H、SO2NH2、SO2NHR8-1、CONHSO2R8-1、SO2NHCOR8-1、またはヒドロキサム酸を表し、
R8は水素原子、C1-4アルキル、またはベンジルを表し、
R8-1は、C1-4アルキル、C1-4ハロアルキル、C3-10炭素環、または3-10員複素環を表し、各々の該C3-10炭素環および3-10員複素環は、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリル(「-CN」を示す;以下同じ)で置換されていてもよく、
L1は、C1-5アルキレン、C2-5アルケニレン、またはC2-5アルキニレンを表し、
R2は、ハロゲン、C1-4アルキル、C1-4アルコキシ、C1-4アルキルチオ、C2-4アルケニル、C2-4アルキニル、-O(C1-4ハロアルキル)、-S(C1-4ハロアルキル)、-C(O)(C1-4アルキル)、-SO2(C1-4アルキル)、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、-NHC(O)(C1-4アルキル)、-N(C1-4アルキル)C(O)(C1-4アルキル)、-NHSO2(C1-4アルキル)、-N(C1-4アルキル)SO2(C1-4アルキル)、-SO2NH(C1-4アルキル)、-SO2N(C1-4アルキル)2、-NR17R17、ニトロ、ニトリル、水酸基、アルデヒド(ホルミルを示す;以下同じ)、またはカルボキシルを表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
該R2における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
X1はCR6または窒素原子を表し、R6は水素原子またはR2を表し、
X2はCR7または窒素原子を表し、R7は、水素原子、R2、または-L3-R9を表し、L3は、メチレン、酸素原子、または酸化されていてもよい硫黄原子を表し、R9は、ハロゲン、C1-4アルキル、およびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい4-10員複素環を表し、
L2は、-CH2CH2-、-CH=CH-、-CH2O-、-OCH2-、-CH2S-、-SCH2-、-CH2S(O)-、-S(O)CH2-、-CH2SO2-、-SO2CH2-、-CH2NH-、-NHCH2-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-を表し、
R3はC1-4アルキルまたはハロゲンを表し、
R4は、ハロゲン、C1-4アルキル、またはC1-4ハロアルキルを表し、
X3は、メチレン、酸素原子、酸化されていてもよい硫黄原子、またはNR10を表し、R10は、C1-4アルキル、-C(O)(C1-4アルキル)、-C(O)O(C1-4アルキル)、または-SO2(C1-4アルキル)を表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
ringはベンゼン環または5-6員単環式芳香族複素環を表し、
は単結合または二重結合を表し、
R5は、(1)ハロゲン、(2)C1-4アルキル、(3)カルボキシル、(4)ニトリル、(5)-CONHR11、(6)-C(O)R12、(7)-OR14、(8)-S(O)tR15、(9)-CH2R16、(10)-NR17R17、(11)-NHCOR11、(12)C4-10炭素環、または(13)4-10員複素環を表し、該C4-10炭素環または4-10員複素環は1~3個のR18で置換されていてもよく、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、
R11は、C1-6アルキル、C3-6シクロアルキル、フェニル、または4-6員複素環を表し、R11は1~3個のR13で置換されていてもよく、該R13が複数である場合、R13はそれぞれ独立して同一でも異なっていてもよく、
R13は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、または4-6員複素環を表し、
R20およびR21は、それぞれ独立して、水素原子またはC1-4アルキルを表し、
R12は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環を表し、該C3-6シクロアルキル、ベンゼン、または4-6員複素環は、それぞれ独立して、ハロゲン、C1-4アルキル、またはC1-4アルコキシで置換されていてもよく、
R14は、水素原子、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、該C1-6アルキルは1~3個のR19で置換されていてもよく、該R19が複数である場合、R19はそれぞれ独立して同一でも異なっていてもよく、
R19は、C1-4アルコキシ、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、またはC1-4アルキルおよびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい5-6員単環式芳香族複素環を表し、
該R19における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
R15は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、
R16は水酸基またはC1-4アルコキシを表し、
R17は、それぞれ独立して、水素原子、C1-6アルキル、またはC3-6シクロアルキルを表し、
R18は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、(C1-4アルキル)SO2-、4-6員複素環、(C1-4アルキル)NH-、または(C1-4アルキル)2N-を表し、
該R18における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
mは1~4の整数を表し、nは0~4の整数を表し、pは0~2の整数を表し、qは0~6の整数を表し、rは0~6の整数を表し、sは0~4の整数を表し、tは0~2の整数を表し、
ただし、p、q、r、およびsがそれぞれ2以上の整数を表す場合、R2、R3、R4、およびR5は、それぞれ独立して、同一でも異なっていてもよい。)で示される化合物、またはその塩である。
R5は、(1)ハロゲン、(2)C1-4アルキル、(3)カルボキシル、(4)ニトリル、(5)-CONHR11、(6)-C(O)R12、(7)-OR14、(8)-S(O)tR15、(9)-CH2R16、(10)-NR17R17、(11)-NHCOR11、(12)C4-10炭素環、または(13)4-10員複素環を表し、該C4-10炭素環または4-10員複素環は1~3個のR18で置換されていてもよく、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、
R11は、C1-6アルキル、C3-6シクロアルキル、フェニル、または4-6員複素環を表し、R11は1~3個のR13で置換されていてもよく、該R13が複数である場合、R13はそれぞれ独立して同一でも異なっていてもよく、
R13は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、または4-6員複素環を表し、
R20およびR21は、それぞれ独立して、水素原子またはC1-4アルキルを表し、
R12は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環を表し、該C3-6シクロアルキル、ベンゼン、または4-6員複素環は、それぞれ独立して、ハロゲン、C1-4アルキル、またはC1-4アルコキシで置換されていてもよく、
R14は、水素原子、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、該C1-6アルキルは1~3個のR19で置換されていてもよく、該R19が複数である場合、R19はそれぞれ独立して同一でも異なっていてもよく、
R19は、C1-4アルコキシ、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、またはC1-4アルキルおよびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい5-6員単環式芳香族複素環を表し、
該R19における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
R15は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、
R16は水酸基またはC1-4アルコキシを表し、
R17は、それぞれ独立して、水素原子、C1-6アルキル、またはC3-6シクロアルキルを表し、
R18は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、(C1-4アルキル)SO2-、4-6員複素環、(C1-4アルキル)NH-、または(C1-4アルキル)2N-を表し、
該R18における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
mは1~4の整数を表し、nは0~4の整数を表し、pは0~2の整数を表し、qは0~6の整数を表し、rは0~6の整数を表し、sは0~4の整数を表し、tは0~2の整数を表し、
ただし、p、q、r、およびsがそれぞれ2以上の整数を表す場合、R2、R3、R4、およびR5は、それぞれ独立して、同一でも異なっていてもよい。)で示される化合物、またはその塩である。
また、EP4拮抗薬の別の態様として、AN0025、E7046、IK-007、RMX-1002、grapiprant、AAT-007、CR6086、INV-1120、BYD-001、TT-038、DT095895、P-001、ER-819762、MK-2894、MF498、evatanepag、CJ-042794、EP4A、BGC201531、CJ-023423、GW627368、AH23848、DT-9081、またはWO2001/062708、WO2002/020462、WO2002/032900、WO2002/050031、WO2002/050032、WO2002/050033、WO2002/016311、WO2003/086390、WO2003/087061、WO2003/099857、WO2003/016254、WO2005/021508、WO2004/067524、WO2005/037812、WO2005/061475、WO2005/105732、WO2005/105733、WO2006/122403、WO2007/121578、WO2008/017164、WO2008/104055、WO2008/116304、WO2008/123207、WO2007/143825、WO2009/005076、WO2009/139373、WO2010/019796、WO2010/034110、WO2012/039972、WO2012/043634、WO2012/076063、WO2012/103071、WO2013/004290、WO2013/096496、WO2013/096501、WO2013/101733、WO2013/101598、WO2014/004229、WO2014/004230、WO2014/086739、WO2014/126746、WO2014/186218、WO2014/200075、WO2015/094902、WO2015/094912、WO2015/091475、WO2015/113057、WO2015/147020、WO2016/021742、WO2016/088903、WO2017/014323、WO2017/066633、WO2017/085198、WO2018/195123、WO2018/195123、WO2018/210987、WO2018/210988、WO2018/210992、WO2018/210994、WO2018/210995、WO2018/216640、WO2019/038156、WO2019/245590、WO2019/101171、WO2019/152982、WO2019/166022、CN108929281、WO2020/012305、WO2020/014465、WO2020/151566、WO2020/160075、またはWO2020/161275に記載されている化合物が挙げられる。
本発明において、「C1-4アルキル」とは、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、tert-ブチル、およびイソブチルである。
本発明において、「C1-3アルキル」とは、例えば、メチル、エチル、n-プロピル、およびイソプロピルである。
本発明において、「C1-5アルキレン」とは、例えば、メチレン、エチレン、プロピレン、ブチレン、およびペンチレンである。
本発明において、「C2-5アルケニレン」とは、例えば、エテニレン、1-プロぺニレン、2-プロぺニレン、1-ブテニレン、2-ブテニレン、3-ブテニレン、1-ペンテニレン、2-ペンテニレン、3-ペンテニレン、および4-ペンテニレンなどである。
本発明において、「C2-5アルキニレン」とは、例えば、エチニレン、1-プロピニレン、2-プロピニレン、1-ブチニレン、2-ブチニレン、3-ブチニレン、1-ペンチニレン、2-ペンチニレン、3-ペンチニレン、および4-ペンチニレンなどである。
本発明において、「ハロゲン」とは、フッ素、塩素、臭素、およびヨウ素である。
本発明において、「C1-4アルコキシ」とは、例えば、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、1-メチルプロポキシ、tert-ブトキシ、イソブトキシなどである。
本発明において、「C1-4アルキルチオ」とは、例えば、メチルチオ、エチルチオ、プロピルチオ、イソプロピルチオ、ブチルチオ、1-メチルプロピルチオ、tert-ブチルチオ、イソブチルチオなどである。
本発明において、「C2-4アルケニル」とは、例えば、エテニル、1-プロぺニル、2-プロぺニル、1-ブテニル、2-ブテニル、および3-ブテニルなどである。
本発明において、「C2-4アルキニル」とは、例えば、エチニル、1-プロピニル、2-プロピニル、1-ブチニル、2-ブチニル、および3-ブチニルなどである。
本発明において、「C1-4ハロアルキル」とは、ハロゲンで置換されたC1-4アルキルを示し、例えば、モノフルオロメチル、ジフルオロメチル、トリフルオロメチル、2-フルオロエチル、1-フルオロエチル、2,2-ジフルオロエチル、1,2-ジフルオロエチル、1,1-ジフルオロエチル、2,2,2-トリフルオロエチル、1,2,2-トリフルオロエチル、1,1,2-トリフルオロエチル、1,2,2,2-テトラフルオロエチル、1,1,2,2-テトラフルオロエチル、ペンタフルオロエチル、1,2-ジブロモ-1,2,2-トリフルオロエチル、1-クロロ-1,2,2,2-テトラフルオロエチル、3-フルオロプロピル、3-クロロプロピル、2-フルオロプロピル、2-クロロプロピル、1-フルオロプロピル、1-クロロプロピル、3,3-ジフルオロプロピル、2,3-ジフルオロプロピル、1,3-ジフルオロプロピル、1,2-ジフルオロプロピル、2,2-ジフルオロプロピル、1,1-ジフルオロプロピル、3,3,3-トリフルオロプロピル、2,3,3-トリフルオロプロピル、1,3,3-トリフルオロプロピル、1,2,2-トリフルオロプロピル、1,1,2-トリフルオロプロピル、1,1,3-トリフルオロプロピル、1,1,2,2-テトラフルオロプロピル、2,2,3,3,3-ペンタフルオロプロピル、4-フルオロブチル、4-クロロブチル、3-フルオロブチル、3-クロロブチル、2-フルオロブチル、2-クロロブチル、1-フルオロブチル、1-クロロブチル、3,3-ジフルオロブチル、2,3-ジフルオロブチル、1,3-ジフルオロブチル、1,2-ジフルオロブチル、2,2-ジフルオロブチル、1,1-ジフルオロブチル、3,3,3-トリフルオロブチル、2,3,3-トリフルオロブチル、1,3,3-トリフルオロブチル、1,2,2-トリフルオロブチル、1,1,2-トリフルオロブチル、1,1,3-トリフルオロブチル、1,1,2,2-テトラフルオロブチル、および2,2,3,3,3-ペンタフルオロブチルなどである。
本発明において、「酸化されていてもよい硫黄原子」とは、硫黄(S)、スルホキシド(S(O))、およびスルホン(SO2)を示す。
本発明において、「4-10員複素環」とは、酸素原子、窒素原子および硫黄原子から選択される1~5個のヘテロ原子を含む、4-10員の単環または二環式複素環を意味し、例えば、オキセタン、アゼチジン、ピロリジン、ピロール、イミダゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピぺリジン、ピぺラジン、ピラジン、ピリミジン、ピリダジン、アゼピン、ジアゼピン、フラン、ピラン、オキセピン、チオフェン、チオピラン、チエピン、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、フラザン、オキサジアゾール、オキサジン、オキサジアジン、オキサゼピン、オキサジアゼピン、チアジアゾール、チアジン、チアジアジン、チアゼピン、チアジアゼピン、インドール、イソインドール、インドリジン、ベンゾフラン、イソベンゾフラン、ベンゾチオフェン、イソベンゾチオフェン、インダゾール、キノリン、イソキノリン、キノリジン、プリン、フタラジン、プテリジン、ナフチリジン、キノキサリン、キナゾリン、シンノリン、ベンゾオキサゾール、ベンゾチアゾール、ベンゾイミダゾール、ベンゾジオキソ-ル、ベンゾオキサチオール、クロメン、ベンゾフラザン、ベンゾチアジアゾール、ベンゾトリアゾール、ピロリン、ピロリジン、イミダゾリン、イミダゾリジン、トリアゾリン、トリアゾリジン、テトラゾリン、テトラゾリジン、ピラゾリン、ピラゾリジン、ジヒドロピリジン、テトラヒドロピリジン、ジヒドロピラジン、テトラヒドロピラジン、ジヒドロピリミジン、テトラヒドロピリミジン、パーヒドロピリミジン、ジヒドロピリダジン、テトラヒドロピリダジン、パーヒドロピリダジン、ジヒドロアゼピン、テトラヒドロアゼピン、パーヒドロアゼピン、ジヒドロジアゼピン、テトラヒドロジアゼピン、パーヒドロジアゼピン、ジヒドロフラン、テトラヒドロフラン、ジヒドロピラン、テトラヒドロピラン、ジヒドロオキセピン、テトラヒドロオキセピン、パーヒドロオキセピン、ジヒドロチオフェン、テトラヒドロチオフェン、ジヒドロチオピラン、テトラヒドロチオピラン、ジヒドロチエピン、テトラヒドロチエピン、パーヒドロチエピン、ジヒドロオキサゾール、テトラヒドロオキサゾール(オキサゾリジン)、ジヒドロイソオキサゾール、テトラヒドロイソオキサゾール(イソオキサゾリジン)、ジヒドロチアゾール、テトラヒドロチアゾール(チアゾリジン)、ジヒドロイソチアゾール、テトラヒドロイソチアゾール(イソチアゾリジン)、ジヒドロフラザン、テトラヒドロフラザン、ジヒドロオキサジアゾール、テトラヒドロオキサジアゾール(オキサジアゾリジン)、ジヒドロオキサジン、テトラヒドロオキサジン、ジヒドロオキサジアジン、テトラヒドロオキサジアジン、ジヒドロオキサゼピン、テトラヒドロオキサゼピン、パーヒドロオキサゼピン、ジヒドロオキサジアゼピン、テトラヒドロオキサジアゼピン、パーヒドロオキサジアゼピン、ジヒドロチアジアゾール、テトラヒドロチアジアゾール(チアジアゾリジン)、ジヒドロチアジン、テトラヒドロチアジン、ジヒドロチアジアジン、テトラヒドロチアジアジン、ジヒドロチアゼピン、テトラヒドロチアゼピン、パーヒドロチアゼピン、ジヒドロチアジアゼピン、テトラヒドロチアジアゼピン、パーヒドロチアジアゼピン、テトラヒドロトリアゾロピラジン、モルホリン、チオモルホリン、オキサチアン、インドリン、イソインドリン、ジヒドロベンゾフラン、パーヒドロベンゾフラン、ジヒドロイソベンゾフラン、パーヒドロイソベンゾフラン、ジヒドロベンゾチオフェン、パーヒドロベンゾチオフェン、ジヒドロイソベンゾチオフェン、パーヒドロイソベンゾチオフェン、ジヒドロインダゾール、パーヒドロインダゾール、ジヒドロキノリン、テトラヒドロキノリン、パーヒドロキノリン、ジヒドロイソキノリン、テトラヒドロイソキノリン、パーヒドロイソキノリン、ジヒドロフタラジン、テトラヒドロフタラジン、パーヒドロフタラジン、ジヒドロナフチリジン、テトラヒドロナフチリジン、パーヒドロナフチリジン、ジヒドロキノキサリン、テトラヒドロキノキサリン、パーヒドロキノキサリン、ジヒドロキナゾリン、テトラヒドロキナゾリン、パーヒドロキナゾリン、ジヒドロシンノリン、テトラヒドロシンノリン、パーヒドロシンノリン、ベンゾオキサチアン、ジヒドロベンゾオキサジン、ジヒドロベンゾチアジン、ピラジノモルホリン、ジヒドロベンゾオキサゾール、パーヒドロベンゾオキサゾール、ジヒドロベンゾチアゾール、パーヒドロベンゾチアゾール、ジヒドロベンゾイミダゾール、パーヒドロベンゾイミダゾール、ジオキソラン、ジオキサン、ジオキサインダン、ベンゾジオキサン、チオクロマン、ジヒドロベンゾジオキシン、ジヒドロベンゾオキサチイン、クロマン、ピラゾロピリミジン、イミダゾピリダジン、イミダゾピリジン、イミダゾピリミジン、ピロロピリジン、ピロロピリミジン、ピロロピリダジン、イミダゾピラジン、ピラゾロピリジン、ピラゾロピリミジン、トリアゾロピリジン、およびジヒドロピリドオキサジン環などである。
本発明において、「3-10員複素環」とは、酸素原子、窒素原子および硫黄原子から選択される1~5個のヘテロ原子を含む、3-10員の単環または二環式複素環を意味し、例えば、アジリジン、オキシラン、チイラン、および前記「4-10員複素環」で記載した複素環などである。
本発明において、「5-10員芳香族複素環」とは、酸素原子、窒素原子および硫黄原子から選択される1~4個のヘテロ原子を含む、5-10員の単環または二環式芳香族複素環を意味し、例えば、ピロール、イミダゾール、トリアゾール、テトラゾール、ピラゾール、フラン、チオフェン、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、フラザン、オキサジアゾール、チアジアゾール、ピリジン、ピラジン、ピリミジン、ピリダジン、インドール、イソインドール、ベンゾフラン、イソベンゾフラン、ベンゾチオフェン、イソベンゾチオフェン、インダゾール、プリン、ベンゾオキサゾール、ベンゾチアゾール、ベンゾイミダゾール、ベンゾフラザン、ベンゾチアジアゾール、ベンゾトリアゾール、キノリン、イソキノリン、フタラジン、プテリジン、ナフチリジン、キノキサリン、キナゾリン、およびシンノリン環などである。
本発明において、「5-6員単環式芳香族複素環」とは、例えば、ピロール、イミダゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピラジン、ピリミジン、ピリダジン、フラン、チオフェン、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、フラザン、オキサジアゾール、およびチアジアゾール環などである。
本発明において、「C4-10炭素環」とは、C4~10の単環または二環式炭素環を意味し、例えば、シクロブタン、シクロペンタン、シクロヘキサン、シクロヘプタン、シクロオクタン、シクロノナン、シクロデカン、シクロペンテン、シクロヘキセン、シクロヘプテン、シクロオクテン、シクロペンタジエン、シクロヘキサジエン、シクロヘプタジエン、シクロオクタジエン、ベンゼン、ペンタレン、パーヒドロペンタレン、アズレン、パーヒドロアズレン、インデン、パーヒドロインデン、インダン、ナフタレン、ジヒドロナフタレン、テトラヒドロナフタレン、およびパーヒドロナフタレン環などである。
本発明において、「C3-10炭素環」とは、C3~10の単環または二環式炭素環を意味し、例えば、シクロプロパン、および前記「C4-10炭素環」で記載した炭素環などである。
本発明において、「C1-6アルキル」とは、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、tert-ブチル、イソブチル、ペンチル、1-メチルブチル、2-メチルブチル、3-メチルブチル、1,1-ジメチルプロピル、1,2-ジメチルプロピル、2,2-ジメチルプロピル、ヘキシル、1-メチルペンチル、2-メチルペンチル、3-メチルペンチル、4-メチルペンチル、1,1-ジメチルブチル、1,2-ジメチルブチル、1,3-ジメチルブチル、2,2-ジメチルブチル、2,3-ジメチルブチル、1-メチル-1-エチルプロピル、2-メチル-2-エチルプロピル、1-エチルブチル、および2-エチルブチルなどである。
本発明において、「C3-6シクロアルキル」とは、シクロプロピル、シクロブチル、シクロペンチル、およびシクロヘキシルである。
本発明において、「4-6員複素環」とは、酸素原子、窒素原子および硫黄原子から選択される1~4個のヘテロ原子を含む、4-6員の単環式複素環を意味し、例えば、オキセタン、アゼチジン、ピロリジン、ピぺリジン、ピラジン、ピラン、チオピラン、オキサジン、オキサジアジン、チアジン、チアジアジン、ピロール、イミダゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピリミジン、ピリダジン、フラン、チオフェン、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、フラザン、オキサジアゾール、およびチアジアゾール環などである。
本発明において、R1として好ましくはCOOR8である。
本発明において、R8として好ましくは水素原子またはC1-4アルキルであり、より好ましくは水素原子である。
本発明において、R8-1として好ましくは、C1-4アルキル、ベンゼンまたはピリジンであり、該ベンゼンおよびピリジンはC1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリルで置換されていてもよい。
本発明において、L1として好ましくはC1-5アルキレンまたはC2-5アルケニレンであり、より好ましくはC1-5アルキレンであり、特に好ましくはプロピレンである。
本発明において、R2として好ましくはフッ素である。
本発明において、X1として好ましくはCR6である。
本発明において、R6として好ましくは水素原子またはフッ素であり、より好ましくは水素原子である。
本発明において、X2として好ましくはCR7である。
本発明において、R7として好ましくはフッ素、ニトリル、-CH2R9、または-OR9であり、より好ましくはニトリルである。
本発明において、R9として好ましくはメチルまたはトリフルオロメチルで置換されていてもよい4-10員複素環であり、該4-10員複素環として好ましくは5-10員芳香族複素環であり、より好ましくは5-10員含窒素芳香族複素環(例えば、ピラゾール、イミダゾール、トリアゾール、ピロロピリジン、ピロロピリミジン、ピロロピリダジン、イミダゾピリダジン、イミダゾピリジン、イミダゾピリミジン、イミダゾピラジン、ピラゾロピリジン、ピラゾロピリミジンなど)である。
本発明において、L2として好ましくは-CH=CH-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-であり、より好ましくは-NHCO-または-CONH-であり、特に好ましくは-NHCO-である。
本発明において、R3として好ましくはフッ素である。
本発明において、R4として好ましくはメチル、エチル、またはトリフルオロメチルであり、より好ましくはメチルである。
本発明において、X3として好ましくはメチレンまたは酸素原子であり、より好ましくは酸素原子である。
本発明において、R10として好ましくはメチル、エチル、メチルカルボニル、エチルカルボニル、メチルスルホニル、エチルスルホニル、またはtert-ブトキシカルボニルである。
本発明において、ringとして好ましくはベンゼン、チオフェン、またはピラゾール環であり、より好ましくはベンゼン環である。
本発明において、R5として好ましくは-CONHR11、フッ素、メトキシ、ベンゼン環、または4-10員複素環であり、該4-10員複素環として好ましくはアゼチジン、ピロリジン、ピペリジン、オキサゾリジン、オキサジアゾール、トリアゾール、チオフェン、フラン、ピラゾール、チアゾール、オキサゾール、イミダゾール、ピリジン、ピラジン、ピリダジン、ピリミジン、ピラゾロピリミジン、ピロロピリミジン、ピラゾロピリジン、ピロロピリジン、またはジヒドロピリドオキサジン環である。
本発明において、R11として好ましくはC1-6アルキル、C3-6シクロアルキル、ピラン、ピロリジン、ピペリジン、ピラゾール、チアゾール、オキサゾール、イソオキサゾール、ピリジン、ピリダジン、またはピリミジン環であり、より好ましくはC1-6アルキルである。
本発明において、R13として好ましくは、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、オキセタン、ピリジン、ピラゾール、またはオキサゾール環であり、より好ましくはフッ素、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、tert-ブチル、イソブチル、シクロペンチル、シクロブチル、オキセタン、水酸基、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ジメチルアミノ、ベンゼン、ピリジン、ピラゾール、またはオキサゾール環である。
本発明において、R20として好ましくは水素原子またはメチルである。
本発明において、R21として好ましくは水素原子またはメチルである。
本発明において、R12として好ましくはC1-3アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環である。該4-6員複素環として好ましくはオキセタン、アゼチジン、ピロリジン、ピぺリジン、ピラジン、ピラン、チオピラン、オキサジン、オキサジアジン、チアジン、チアジアジン、ピロール、イミダゾール、トリアゾール、テトラゾール、ピラゾール、ピリジン、ピラジン、ピリミジン、ピリダジン、フラン、チオフェン、オキサゾール、イソオキサゾール、チアゾール、イソチアゾール、フラザン、オキサジアゾール、またはチアジアゾール環である。該4-6員複素環はC1-4アルコキシで置換されていてもよい。
本発明において、R14として好ましくは水素原子、メチル、エチル、ベンゼン、またはベンジルである。
本発明において、R19として好ましくはメトキシ、-CONHCH3、-CON(CH3)2、オキサゾール、チアゾール、ピラゾール、またはピリジン環である。
本発明において、R15として好ましくはメチル、シクロプロピル、またはベンゼンである。
本発明において、R16として好ましくは水酸基である。
本発明において、R17として好ましくはメチル、エチル、またはシクロプロピルであり、より好ましくはメチルである。
本発明において、R18として好ましくはフッ素、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、tert-ブチル、イソブチル、シクロプロピル、メトキシ、エトキシ、n-プロポキシ、イソプロポキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、メチルスルホニル、ピリジン、ジメチルアミノである。
本発明において、mとして好ましくは1~2の整数であり、より好ましくは1である。
本発明において、nとして好ましくは0~1の整数であり、より好ましくは1である。
本発明において、pとして好ましくは0である。
本発明において、qとして好ましくは0である。
本発明において、rとして好ましくは0~4の整数であり、より好ましくは0~2の整数である。
本発明において、sとして好ましくは0~2の整数であり、より好ましくは1または2である。
本発明において、tとして好ましくは0~2の整数である。
本発明において、X3aとして好ましくは酸素原子である。
本発明において、naとして好ましくは0~1の整数であり、より好ましくは1である。
本発明において、qaとして好ましくは0である。
本発明において、raとして好ましくは0~2の整数である。
本発明において、一般式(I)として好ましくは、前記のring、R1、R2、R3、R4、R5、R6、R7、R8、R8-1、R9、R10、R11、R12、R13、R14、R15、R16、R17、R18、R19、R20、R21、L1、L2、L3、X1、X2、X3、m、n、p、q、r、s、およびtの各々の好ましい定義の組み合わせである。
本発明において、一般式(I)で示される化合物として好ましくは、一般式(I-a)
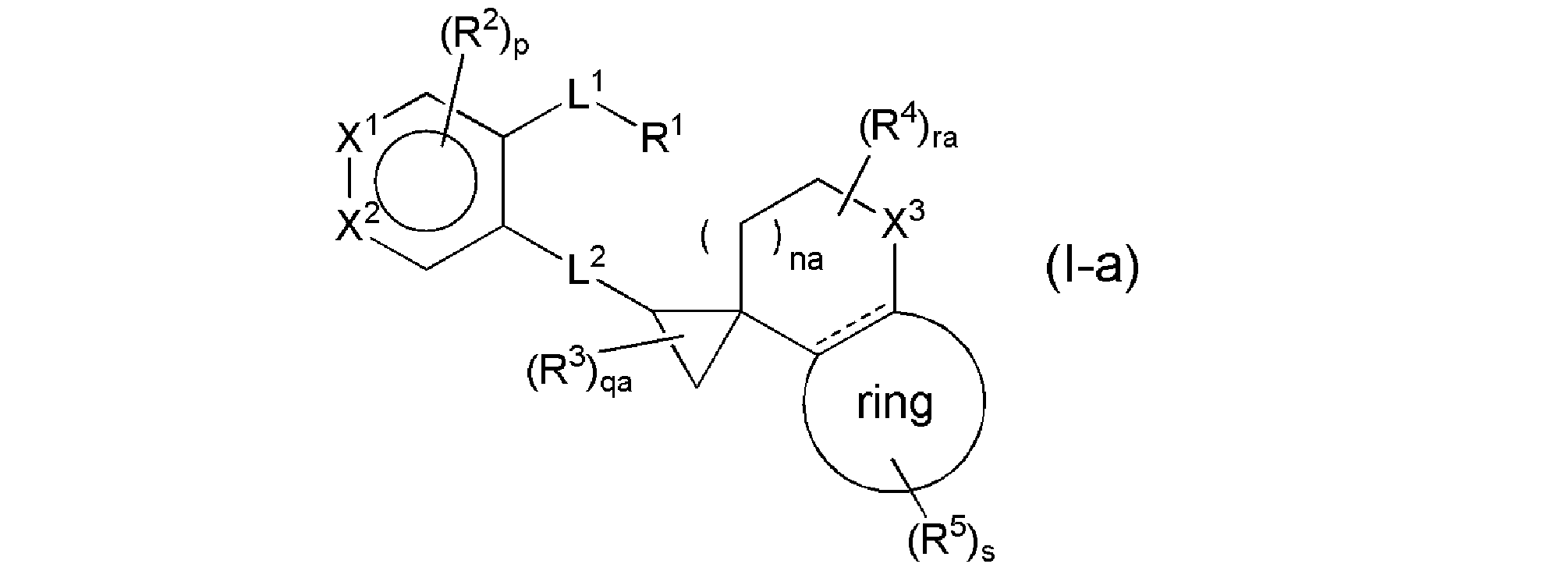
(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、より好ましくは一般式(I-1)
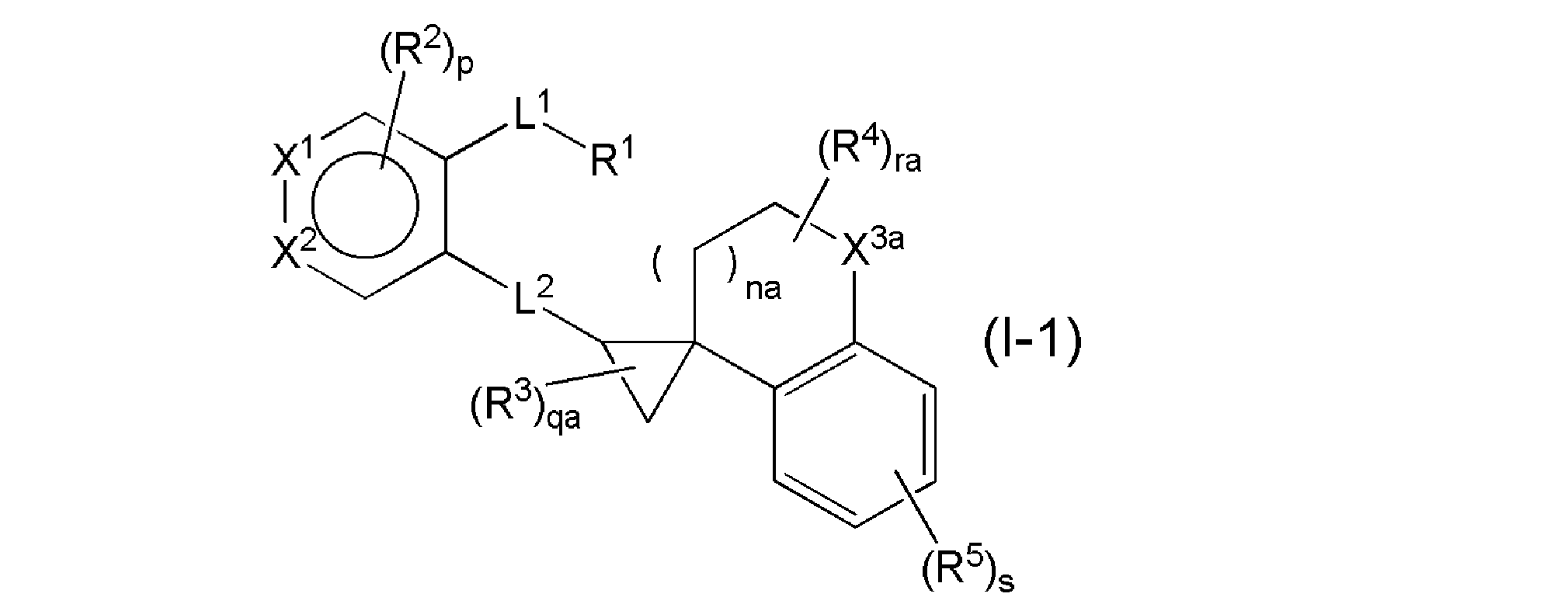
(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、X3aは、メチレンまたは酸素原子を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩である。
本発明において、一般式(I)で示される化合物の別の態様として、より好ましくは、一般式(I-b)
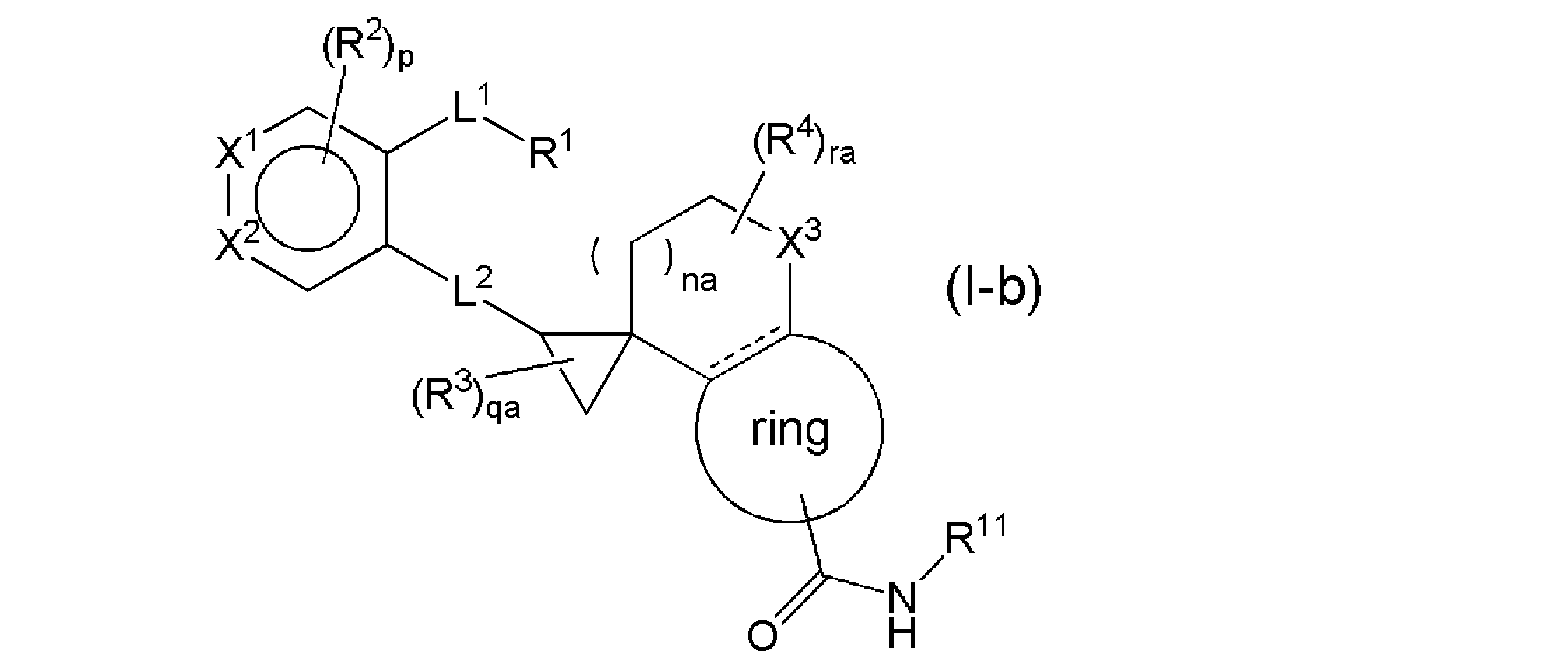
(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、さらに好ましくは一般式(I-c)

(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、なお好ましくは一般式(I-d)

(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、さらになお好ましくは一般式(I-e)

(式中、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、特に好ましくは一般式(I-2)

(式中、R2aはハロゲンを表し、R6aは水素原子またはハロゲンを表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、最も好ましくは一般式(I-4)

(式中、R2aはハロゲンを表し、R6aは水素原子またはハロゲンを表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩である。
本発明において、一般式(I)で示される化合物のさらなる別の態様として、より好ましくは、一般式(I-f)

(式中、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、さらに好ましくは一般式(I-g)

(式中、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、なお好ましくは一般式(I-h)
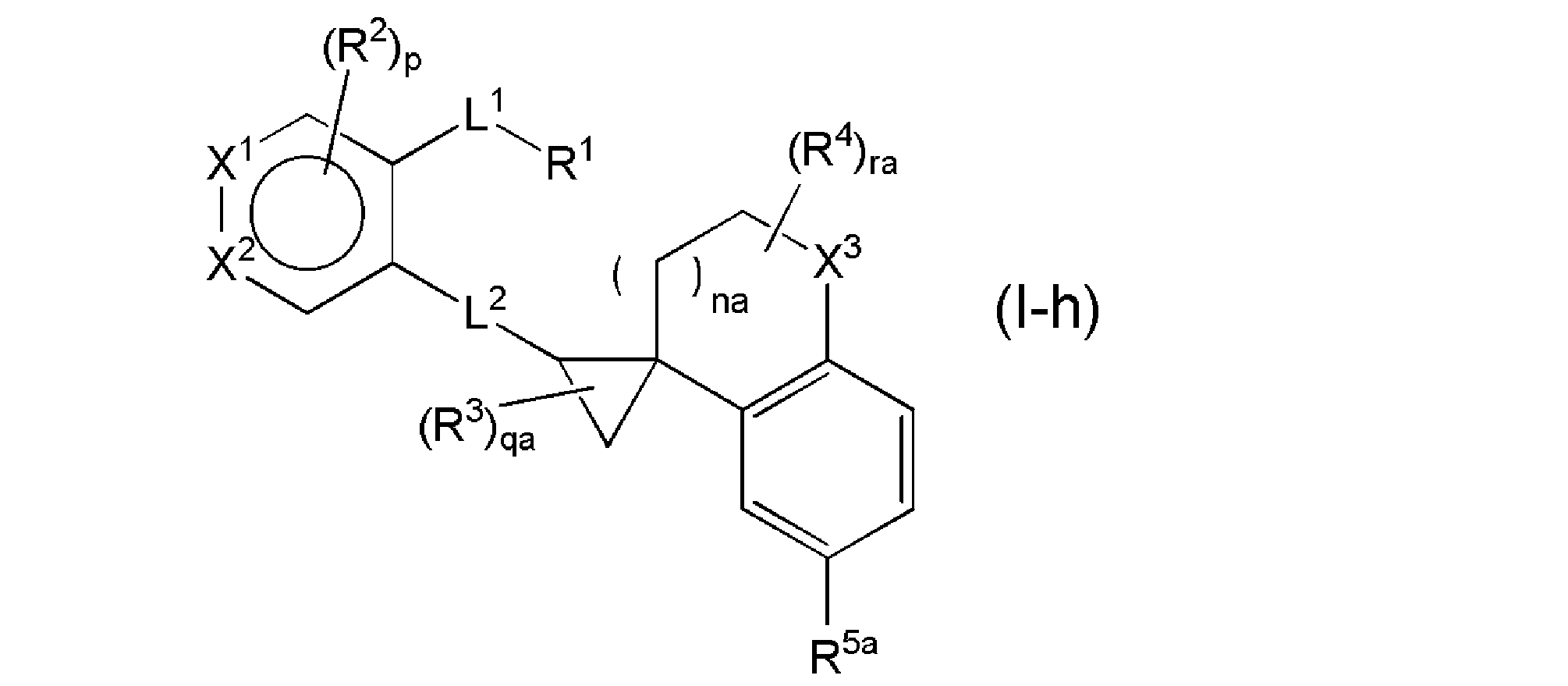
(式中、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、さらになお好ましくは一般式(I-i)

(式中、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、naは0~1の整数を表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、特に好ましくは一般式(I-3)

(式中、R2aはハロゲンを表し、R6aは水素原子またはハロゲンを表し、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩であり、最も好ましくは一般式(I-5)
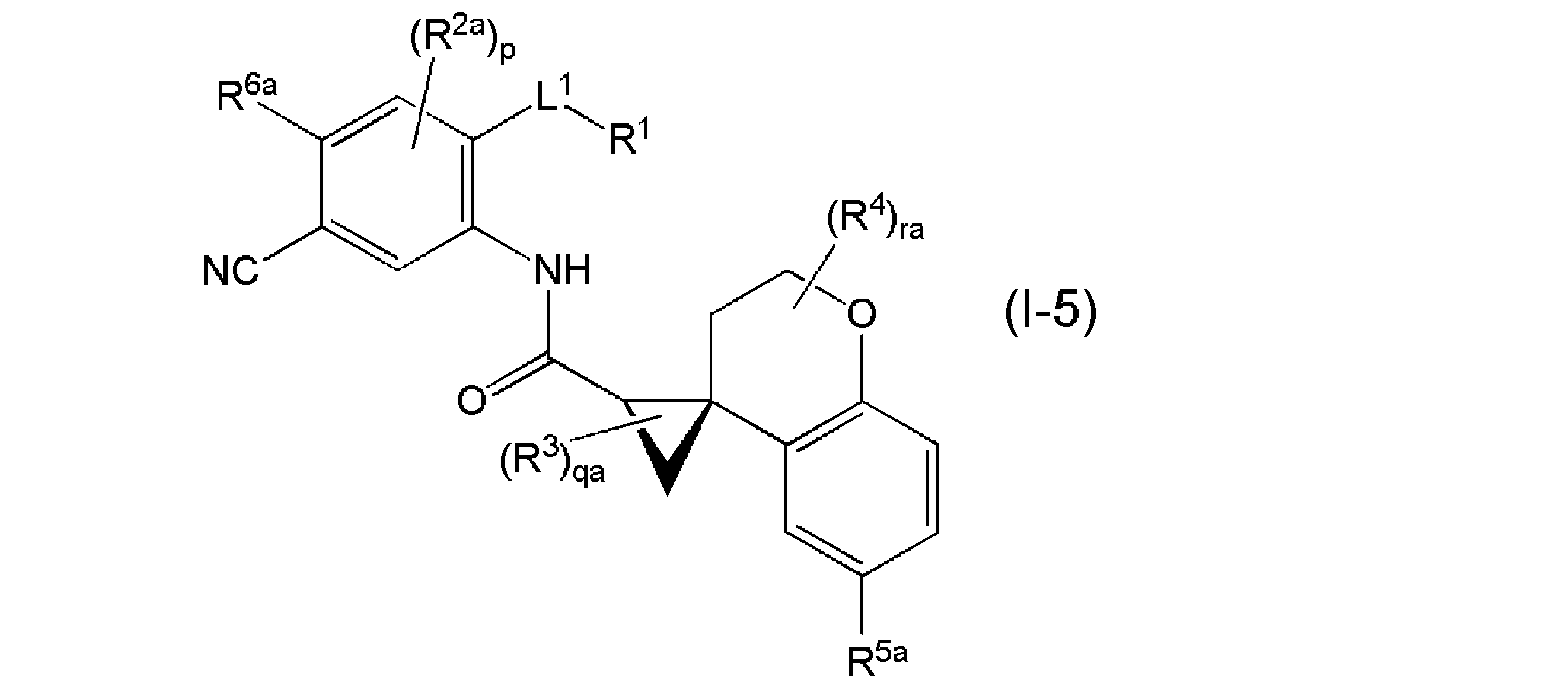
(式中、R2aはハロゲンを表し、R6aは水素原子またはハロゲンを表し、R5aは1~3個のR18で置換されていてもよい、C4-10炭素環または4-10員複素環であって、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は上記と同じ意味を表す。)で示される化合物、またはその塩である。
本発明において、上記の一般式(I-a)、一般式(I-b)、一般式(I-c)、一般式(I-d)、一般式(I-e)、一般式(I-f)、一般式(I-g)、一般式(I-h)、一般式(I-i)、および一般式(I-1)の群から選択される一般式において、それぞれ独立して、好ましくは、L1がプロピレンであり、L2が-CH=CH-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-である。より好ましくは、L1がプロピレンであり、L2が-NHCO-または-CONH-である。さらに好ましくは、L1がプロピレンであり、L2が-NHCO-である。
本発明において、上記の一般式(I-2)、一般式(I-3)、一般式(I-4)、および一般式(I-5)の群から選択される一般式において、それぞれ独立して、好ましくは、L1がプロピレンである。
本発明において、EP4拮抗薬としてより好ましくは、WO2016/111347の実施例に記載された化合物、またはその塩である。さらに好ましくは、
(1) 4-[4-シアノ-2-({[(2’R,4S)-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(2) 4-{4-シアノ-2-[({(2’R,4S)-6-[(シクロプロピルメチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(3) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(4) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-メチル-2-プロパニル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(5) 4-[4-シアノ-2-({[(2’R,4S)-6-{[(2S)-1-メトキシ-2-プロパニル]カルバモイル}-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(6) 4-{4-シアノ-2-[({(2’R,4S)-6-[(1-メチル-1H-ピラゾール-3-イル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(7) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロプロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(8) 4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、
(9) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロペンチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(10) 4-{2-[({(2’R,4S)-6-[(2S)-2-ブタニルカルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]-4-シアノフェニル}ブタン酸、
(11) 4-{4-シアノ-2-[({(2’R,4S)-6-[(トランス-4-ヒドロキシシクロヘキシル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(12) 4-{4-シアノ-2-[({(2’R,4S)-6-[(シス-4-ヒドロキシシクロヘキシル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(13) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-ピリジニルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(14) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリダジニルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(15) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロブチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(16) 4-[4-シアノ-2-({[(2’R,4S)-6-{[1-(2-メチル-2-プロパニル)-1H-ピラゾール-4-イル]カルバモイル}-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(17) 4-[4-シアノ-2-({[(2’R,4S)-6-(テトラヒドロ-2H-ピラン-4-イルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(18) 4-[4-シアノ-2-({[(2’R,4S)-6-(プロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(19) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-エトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、または
(20) 4-[4-シアノ-2-({[(2’R,4S)-6-(エチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(21) 4-[4-シアノ-2-({[(1R,2R)-6’-(メチルカルバモイル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、
(22) 4-{4-シアノ-2-[({(1R,2R)-6’-[(2-メトキシエチル)カルバモイル]-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル}カルボニル)アミノ]フェニル}ブタン酸、
(23) 4-{4-シアノ-2-[({(1R,2R)-6’-[(1-メチル-1H-ピラゾール-4-イル)カルバモイル]-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル}カルボニル)アミノ]フェニル}ブタン酸、
(24) 4-[4-シアノ-2-({[(2’R,4S)-7-フルオロ-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(25) 4-{4-シアノ-2-[({(2’R,4S)-7-フルオロ-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(26) 4-[4-シアノ-2-({[(2’R,4S)-7-フルオロ-6-(イソプロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(27) 4-[4-シアノ-2-({[(2’R,4S)-7-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(28) 4-{4-シアノ-2-[({(2’R,4S)-7-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(29) 4-[4-シアノ-2-({[(2’R,4S)-7-メトキシ-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(30) 4-{4-シアノ-2-[({(2’R,4S)-7-メトキシ-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(31) 4-[4-シアノ-2-({[(2’R,3S)-5-(メチルカルバモイル)-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(32) 4-{4-シアノ-2-[({(2’R,3S)-5-[(2-メトキシエチル)カルバモイル]-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(33) 4-[4-シアノ-2-({[(1S,2R)-6’-[(2-メトキシエチル)カルバモイル]-3’,3’-ジメチル-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、または
(34) 4-[4-シアノ-2-({[(1S,2R)-3’,3’-ジメチル-6’-(メチルカルバモイル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、またはその塩である。
(1) 4-[4-シアノ-2-({[(2’R,4S)-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(2) 4-{4-シアノ-2-[({(2’R,4S)-6-[(シクロプロピルメチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(3) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(4) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-メチル-2-プロパニル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(5) 4-[4-シアノ-2-({[(2’R,4S)-6-{[(2S)-1-メトキシ-2-プロパニル]カルバモイル}-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(6) 4-{4-シアノ-2-[({(2’R,4S)-6-[(1-メチル-1H-ピラゾール-3-イル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(7) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロプロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(8) 4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、
(9) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロペンチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(10) 4-{2-[({(2’R,4S)-6-[(2S)-2-ブタニルカルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]-4-シアノフェニル}ブタン酸、
(11) 4-{4-シアノ-2-[({(2’R,4S)-6-[(トランス-4-ヒドロキシシクロヘキシル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(12) 4-{4-シアノ-2-[({(2’R,4S)-6-[(シス-4-ヒドロキシシクロヘキシル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(13) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-ピリジニルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(14) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリダジニルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(15) 4-[4-シアノ-2-({[(2’R,4S)-6-(シクロブチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(16) 4-[4-シアノ-2-({[(2’R,4S)-6-{[1-(2-メチル-2-プロパニル)-1H-ピラゾール-4-イル]カルバモイル}-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(17) 4-[4-シアノ-2-({[(2’R,4S)-6-(テトラヒドロ-2H-ピラン-4-イルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(18) 4-[4-シアノ-2-({[(2’R,4S)-6-(プロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(19) 4-{4-シアノ-2-[({(2’R,4S)-6-[(2-エトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、または
(20) 4-[4-シアノ-2-({[(2’R,4S)-6-(エチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(21) 4-[4-シアノ-2-({[(1R,2R)-6’-(メチルカルバモイル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、
(22) 4-{4-シアノ-2-[({(1R,2R)-6’-[(2-メトキシエチル)カルバモイル]-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル}カルボニル)アミノ]フェニル}ブタン酸、
(23) 4-{4-シアノ-2-[({(1R,2R)-6’-[(1-メチル-1H-ピラゾール-4-イル)カルバモイル]-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル}カルボニル)アミノ]フェニル}ブタン酸、
(24) 4-[4-シアノ-2-({[(2’R,4S)-7-フルオロ-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(25) 4-{4-シアノ-2-[({(2’R,4S)-7-フルオロ-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(26) 4-[4-シアノ-2-({[(2’R,4S)-7-フルオロ-6-(イソプロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(27) 4-[4-シアノ-2-({[(2’R,4S)-7-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(28) 4-{4-シアノ-2-[({(2’R,4S)-7-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(29) 4-[4-シアノ-2-({[(2’R,4S)-7-メトキシ-6-(メチルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(30) 4-{4-シアノ-2-[({(2’R,4S)-7-メトキシ-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(31) 4-[4-シアノ-2-({[(2’R,3S)-5-(メチルカルバモイル)-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(32) 4-{4-シアノ-2-[({(2’R,3S)-5-[(2-メトキシエチル)カルバモイル]-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(33) 4-[4-シアノ-2-({[(1S,2R)-6’-[(2-メトキシエチル)カルバモイル]-3’,3’-ジメチル-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、または
(34) 4-[4-シアノ-2-({[(1S,2R)-3’,3’-ジメチル-6’-(メチルカルバモイル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、またはその塩である。
または、本発明において、EP4拮抗薬として好ましくは、(1) 4-[4-シアノ-2-({[(2’R,4S)-6-(5-メチル-1,3,4-オキサジアゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(2) 4-[4-シアノ-2-({[(2’R,4S)-6-(5-シクロプロピル-1,3,4-オキサジアゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(3) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-メチル-1,2,4-オキサジアゾール-5-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(4) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(5) 4-[4-シアノ-2-({[(2’R,4S)-6-(1H-ピラゾール-1-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(6) 4-[4-シアノ-2-({[(2’R,4S)-6-(1H-ピラゾール-5-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(7) 4-[4-シアノ-2-({[(2’R,4S)-6-(4-ピリダジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(8) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-オキソ-1-ピロリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(9) 4-[4-シアノ-2-({[(2’R,4S)-6-(6-メトキシ-3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(10) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(1H-ピラゾール-1-イル)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(11) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(ジメチルアミノ)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(12) 4-[4-シアノ-2-({[(2’R,4S)-6-(6-メチル-3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(13) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(メチルアミノ)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(14) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(15) 4-[4-シアノ-2-({[(2’R,4S)-6-(1,3-チアゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(16) 4-[4-シアノ-2-({[(2’R,4S)-6-(1,3-オキサゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(17) 4-[4-シアノ-2-({[(2’R,4S)-6-(1-メチル-1H-1,2,3-トリアゾール-4-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(18) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリダジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(19)4-[4-シアノ-2-({[(2’R,3S)-5-(3-ピリジニル)-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、または
(20)4-[4-シアノ-2-({[(1S,2R)-3’,3’-ジメチル-6’-(3-ピリジニル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、またはその塩である。
(2) 4-[4-シアノ-2-({[(2’R,4S)-6-(5-シクロプロピル-1,3,4-オキサジアゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(3) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-メチル-1,2,4-オキサジアゾール-5-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(4) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(5) 4-[4-シアノ-2-({[(2’R,4S)-6-(1H-ピラゾール-1-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(6) 4-[4-シアノ-2-({[(2’R,4S)-6-(1H-ピラゾール-5-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(7) 4-[4-シアノ-2-({[(2’R,4S)-6-(4-ピリダジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(8) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-オキソ-1-ピロリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(9) 4-[4-シアノ-2-({[(2’R,4S)-6-(6-メトキシ-3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(10) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(1H-ピラゾール-1-イル)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(11) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(ジメチルアミノ)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(12) 4-[4-シアノ-2-({[(2’R,4S)-6-(6-メチル-3-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(13) 4-{4-シアノ-2-[({(2’R,4S)-6-[6-(メチルアミノ)-3-ピリジニル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸、
(14) 4-[4-シアノ-2-({[(2’R,4S)-6-(2-ピリジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(15) 4-[4-シアノ-2-({[(2’R,4S)-6-(1,3-チアゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(16) 4-[4-シアノ-2-({[(2’R,4S)-6-(1,3-オキサゾール-2-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(17) 4-[4-シアノ-2-({[(2’R,4S)-6-(1-メチル-1H-1,2,3-トリアゾール-4-イル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(18) 4-[4-シアノ-2-({[(2’R,4S)-6-(3-ピリダジニル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、
(19)4-[4-シアノ-2-({[(2’R,3S)-5-(3-ピリジニル)-2H-スピロ[1-ベンゾフラン-3,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸、または
(20)4-[4-シアノ-2-({[(1S,2R)-3’,3’-ジメチル-6’-(3-ピリジニル)-2’,3’-ジヒドロスピロ[シクロプロパン-1,1’-インデン]-2-イル]カルボニル}アミノ)フェニル]ブタン酸、またはその塩である。
特に好ましくは、以下の構造式
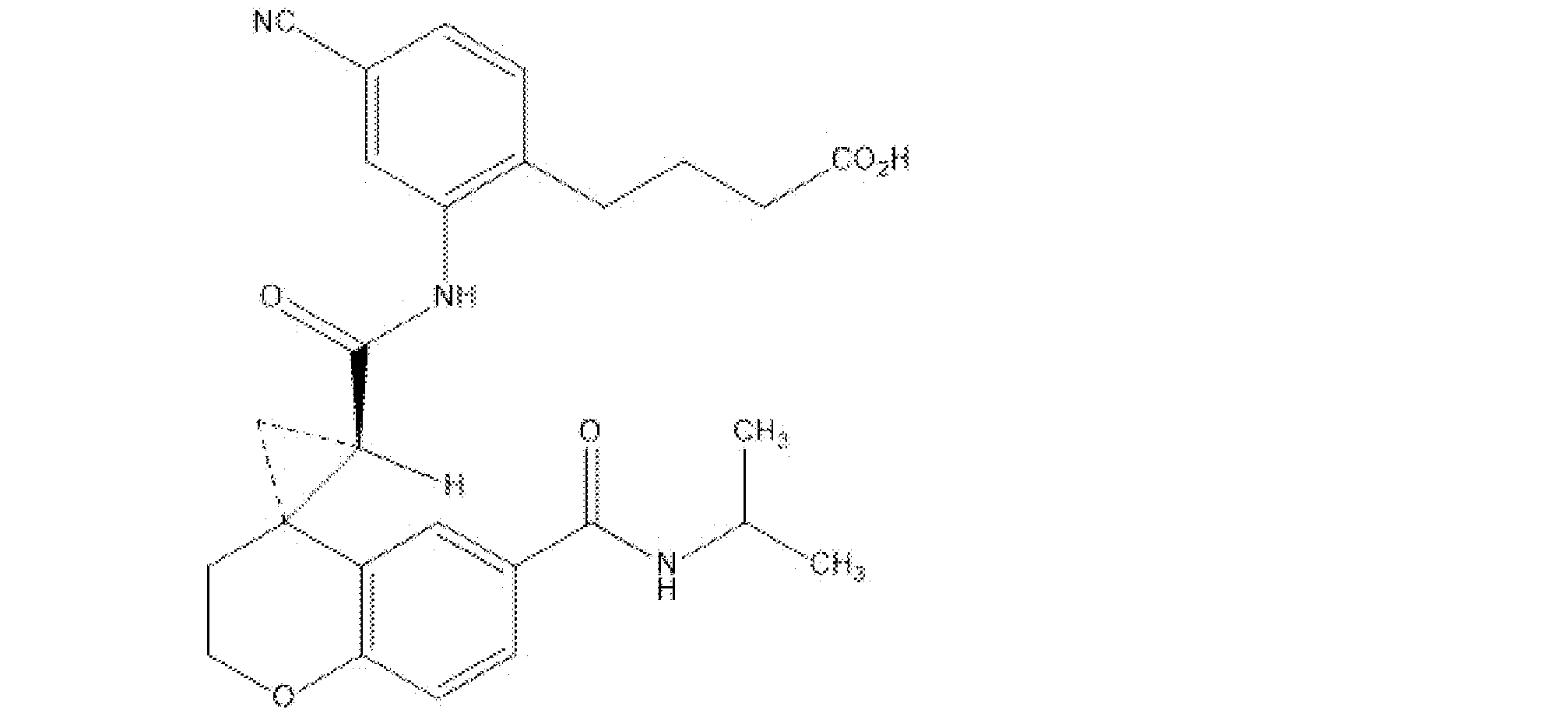
で表される4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸(以下、化合物Aと略記することがある。)、またはその塩である。なお、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸は、4-[4-シアノ-2-({[(2’R,4S)-6-(イソプロピルカルバモイル)-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル]カルボニル}アミノ)フェニル]ブタン酸と命名される場合もある。化合物Aは、公知の方法、例えばWO2016/111347の実施例2-13に記載された方法に従い製造することができる。
特に好ましくは、以下の構造式

で表される4-{4-シアノ-2-[({(2’R,4S)-6-[(2-メトキシエチル)カルバモイル]-2,3-ジヒドロスピロ[クロメン-4,1’-シクロプロパン]-2’-イル}カルボニル)アミノ]フェニル}ブタン酸(以下、化合物Bと略記することがある。)、または塩である。化合物Bは、公知の方法、例えばWO2016/111347の実施例2-2に記載された方法に従い製造することができる。
本発明においては、特に指示しない限り異性体はこれをすべて包含する。例えば、アルキル基、アルコキシ基およびアルキレン基などには直鎖のものおよび分枝鎖のものが含まれる。さらに、二重結合、環、縮合環における異性体(E、Z、シス、トランス体)、不斉炭素の存在などによる異性体(R、S体、α、β体、エナンチオマー、ジアステレオマー)、旋光性を有する光学活性体(D、L、d、l体)、クロマトグラフ分離による極性体(高極性体、低極性体)、平衡化合物、回転異性体、これらの任意の割合の混合物、ラセミ混合物は、すべて本発明に含まれる。また、本発明においては、互変異性による異性体をもすべて包含する。
本発明においては、特に断わらない限り、当業者にとって明らかなように記号
は紙面の向こう側(すなわちα-配置)に結合していることを表し、
は紙面の手前側(すなわちβ-配置)に結合していることを表し、
は、α-配置とβ-配置の任意の混合物であることを表す。
[塩]
一般式(I)で示される化合物は、公知の方法で塩に変換される。
塩としては薬学的に許容される塩が好ましい。
塩は、水溶性のものが好ましい。
薬学的に許容される塩としては、例えば、酸付加塩、アルカリ金属塩、アルカリ土類金属塩、アンモニウム塩、またはアミン塩などが挙げられる。
酸付加塩としては、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、リン酸塩、硝酸塩のような無機酸塩、または酢酸塩、乳酸塩、酒石酸塩、安息香酸塩、クエン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、トリフルオロ酢酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸塩、イセチオン酸塩、グルクロン酸塩、またはグルコン酸塩のような有機酸塩が挙げられる。
アルカリ金属塩としては、例えば、カリウムおよびナトリウムなどとの塩が挙げられる。
アルカリ土類金属塩としては、例えば、カルシウムおよびマグネシウムなどとの塩が挙げられる。
アンモニウム塩としては、例えば、テトラメチルアンモニウムなどとの塩が挙げられる。
アミン塩としては、例えば、トリエチルアミン、メチルアミン、ジメチルアミン、シクロペンチルアミン、ベンジルアミン、フェネチルアミン、ピペリジン、モノエタノールアミン、ジエタノールアミン、トリス(ヒドロキシメチル)アミノメタン、リジン、アルギニン、およびN-メチル-D-グルカミンなどとの塩が挙げられる。
また、本発明に用いられる化合物は、任意の方法でN-オキシド体にすることができる。N-オキシド体とは、一般式(I)で示される化合物の窒素原子が、酸化されたものを表す。
一般式(I)で示される化合物およびその塩は、溶媒和していない形態で存在してもよいし、水、エタノールなどの薬学的に許容できる溶媒と溶媒和した形態で存在してもよい。溶媒和物として好ましくは水和物である。
一般式(I)で示される化合物は、適切な共結晶形成剤と共結晶を形成することができる。共結晶としては、薬学的に許容される共結晶形成剤と形成される、薬学的に許容されるものが好ましい。共結晶は、典型的に、2種以上の異なる分子がイオン結合とは異なる分子間相互作用で形成される結晶として定義される。また、共結晶は中性分子と塩の複合体であってもよい。共結晶は、公知の方法、例えば、融解結晶化により、溶媒からの再結晶により、または成分を一緒に物理的に粉砕することにより、調製することができる。適当な共結晶形成剤としては、WO2006/007448に記載のものを含む。
本発明において、一般式(I)で示される化合物に関する言及はすべて、一般式(I)で示される化合物、その塩、そのN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶、または一般式(I)で示される化合物の塩のN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶を包含する。
すなわち、本発明において、一般式(I)で示される化合物、またはその塩は、一般式(I)で示される化合物の溶媒和物(例えば、水和物)、そのN-オキシド体、もしくはその共結晶、または一般式(I)で示される化合物の塩のN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶を包含する。
一般式(I)で示される化合物は、公知の方法で塩に変換される。
塩としては薬学的に許容される塩が好ましい。
塩は、水溶性のものが好ましい。
薬学的に許容される塩としては、例えば、酸付加塩、アルカリ金属塩、アルカリ土類金属塩、アンモニウム塩、またはアミン塩などが挙げられる。
酸付加塩としては、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、リン酸塩、硝酸塩のような無機酸塩、または酢酸塩、乳酸塩、酒石酸塩、安息香酸塩、クエン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、トリフルオロ酢酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸塩、イセチオン酸塩、グルクロン酸塩、またはグルコン酸塩のような有機酸塩が挙げられる。
アルカリ金属塩としては、例えば、カリウムおよびナトリウムなどとの塩が挙げられる。
アルカリ土類金属塩としては、例えば、カルシウムおよびマグネシウムなどとの塩が挙げられる。
アンモニウム塩としては、例えば、テトラメチルアンモニウムなどとの塩が挙げられる。
アミン塩としては、例えば、トリエチルアミン、メチルアミン、ジメチルアミン、シクロペンチルアミン、ベンジルアミン、フェネチルアミン、ピペリジン、モノエタノールアミン、ジエタノールアミン、トリス(ヒドロキシメチル)アミノメタン、リジン、アルギニン、およびN-メチル-D-グルカミンなどとの塩が挙げられる。
また、本発明に用いられる化合物は、任意の方法でN-オキシド体にすることができる。N-オキシド体とは、一般式(I)で示される化合物の窒素原子が、酸化されたものを表す。
一般式(I)で示される化合物およびその塩は、溶媒和していない形態で存在してもよいし、水、エタノールなどの薬学的に許容できる溶媒と溶媒和した形態で存在してもよい。溶媒和物として好ましくは水和物である。
一般式(I)で示される化合物は、適切な共結晶形成剤と共結晶を形成することができる。共結晶としては、薬学的に許容される共結晶形成剤と形成される、薬学的に許容されるものが好ましい。共結晶は、典型的に、2種以上の異なる分子がイオン結合とは異なる分子間相互作用で形成される結晶として定義される。また、共結晶は中性分子と塩の複合体であってもよい。共結晶は、公知の方法、例えば、融解結晶化により、溶媒からの再結晶により、または成分を一緒に物理的に粉砕することにより、調製することができる。適当な共結晶形成剤としては、WO2006/007448に記載のものを含む。
本発明において、一般式(I)で示される化合物に関する言及はすべて、一般式(I)で示される化合物、その塩、そのN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶、または一般式(I)で示される化合物の塩のN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶を包含する。
すなわち、本発明において、一般式(I)で示される化合物、またはその塩は、一般式(I)で示される化合物の溶媒和物(例えば、水和物)、そのN-オキシド体、もしくはその共結晶、または一般式(I)で示される化合物の塩のN-オキシド体、その溶媒和物(例えば、水和物)、もしくはその共結晶を包含する。
[プロドラッグ]
また、一般式(I)で示される化合物は、プロドラッグとして投与することができる。一般式(I)で示される化合物のプロドラッグとは、生体内において酵素や胃酸などによる反応により一般式(I)で示される化合物に変換される化合物をいう。一般式(I)で示される化合物のプロドラッグとしては、例えば、一般式(I)で示される化合物がアミノ基を有する場合、該アミノ基がアシル化、アルキル化、リン酸化された化合物(例えば、一般式(I)で示される化合物のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化、アセトキシメチル化、tert-ブチル化された化合物など);一般式(I)で示される化合物が水酸基を有する場合、該水酸基がアシル化、アルキル化、リン酸化、ホウ酸化された化合物(例えば、一般式(I)で示される化合物の水酸基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化された化合物など)が挙げられ;一般式(I)で示される化合物がカルボキシ基を有する場合、該カルボキシ基がエステル化、アミド化された化合物(例えば、一般式(I)で示される化合物のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、1-{(エトキシカルボニル)オキシ}エチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、1-{[(シクロヘキシルオキシ)カルボニル]オキシ}エチルエステル化、メチルアミド化された化合物など)などが挙げられる。これらの化合物はそれ自体公知の方法によって製造することができる。また、一般式(I)で示される化合物のプロドラッグは水和物および非水和物のいずれであってもよい。また、一般式(I)で示される化合物のプロドラッグは、廣川書店1990年刊「医薬品の開発」第7巻「分子設計」163~198頁に記載されているような、生理的条件で一般式(I)で示される化合物に変化するものであってもよい。
さらに、一般式(I)で示される化合物を構成する各原子は、その同位元素(例えば、2H、3H、13C、14C、15N、16N、17O、18O、18F、35S、36Cl、77Br、125Iなど)などで置換されていてもよい。
また、一般式(I)で示される化合物は、プロドラッグとして投与することができる。一般式(I)で示される化合物のプロドラッグとは、生体内において酵素や胃酸などによる反応により一般式(I)で示される化合物に変換される化合物をいう。一般式(I)で示される化合物のプロドラッグとしては、例えば、一般式(I)で示される化合物がアミノ基を有する場合、該アミノ基がアシル化、アルキル化、リン酸化された化合物(例えば、一般式(I)で示される化合物のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化、アセトキシメチル化、tert-ブチル化された化合物など);一般式(I)で示される化合物が水酸基を有する場合、該水酸基がアシル化、アルキル化、リン酸化、ホウ酸化された化合物(例えば、一般式(I)で示される化合物の水酸基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化された化合物など)が挙げられ;一般式(I)で示される化合物がカルボキシ基を有する場合、該カルボキシ基がエステル化、アミド化された化合物(例えば、一般式(I)で示される化合物のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、1-{(エトキシカルボニル)オキシ}エチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、1-{[(シクロヘキシルオキシ)カルボニル]オキシ}エチルエステル化、メチルアミド化された化合物など)などが挙げられる。これらの化合物はそれ自体公知の方法によって製造することができる。また、一般式(I)で示される化合物のプロドラッグは水和物および非水和物のいずれであってもよい。また、一般式(I)で示される化合物のプロドラッグは、廣川書店1990年刊「医薬品の開発」第7巻「分子設計」163~198頁に記載されているような、生理的条件で一般式(I)で示される化合物に変化するものであってもよい。
さらに、一般式(I)で示される化合物を構成する各原子は、その同位元素(例えば、2H、3H、13C、14C、15N、16N、17O、18O、18F、35S、36Cl、77Br、125Iなど)などで置換されていてもよい。
[本発明に用いられる化合物の製造方法]
本発明においてEP4拮抗薬は、公知の方法、例えば、一般式(I)で示される化合物は、WO2016/111347に記載された方法に従い製造することができる。
本発明においてEP4拮抗薬は、公知の方法、例えば、一般式(I)で示される化合物は、WO2016/111347に記載された方法に従い製造することができる。
[製剤および投与方法]
本発明においてEP4拮抗薬は、通常、各種の添加剤または溶媒などの薬学的に許容される担体とともに製剤化したうえで、全身的または局所的に、経口または非経口の形で投与される。ここで、薬学的に許容される担体とは、一般的に医薬品の製剤に用いられる、有効成分以外の物質を意味する。薬学的に許容される担体は、その製剤の投与量において薬理作用を示さず、無害で、有効成分の治療効果を妨げないものが好ましい。また、薬学的に許容される担体は、有効成分および製剤の有用性を高める、製剤化を容易にする、品質の安定化を図る、または使用性を向上させるなどの目的で用いることもできる。具体的には、薬事日報社2016年刊「医薬品添加物事典2016」(日本医薬品添加剤協会編集)などに記載されているような物質を、適宜目的に応じて選択すればよい。
本発明においてEP4拮抗薬は、通常、各種の添加剤または溶媒などの薬学的に許容される担体とともに製剤化したうえで、全身的または局所的に、経口または非経口の形で投与される。ここで、薬学的に許容される担体とは、一般的に医薬品の製剤に用いられる、有効成分以外の物質を意味する。薬学的に許容される担体は、その製剤の投与量において薬理作用を示さず、無害で、有効成分の治療効果を妨げないものが好ましい。また、薬学的に許容される担体は、有効成分および製剤の有用性を高める、製剤化を容易にする、品質の安定化を図る、または使用性を向上させるなどの目的で用いることもできる。具体的には、薬事日報社2016年刊「医薬品添加物事典2016」(日本医薬品添加剤協会編集)などに記載されているような物質を、適宜目的に応じて選択すればよい。
投与に用いられる剤型としては、例えば、経口投与用製剤(例:錠剤、カプセル剤、顆粒剤、散剤、経口液剤、シロップ剤、経口ゼリー剤など)、口腔用製剤(例:口腔用錠剤、口腔用スプレー剤、口腔用半固形剤、含嗽剤など)、注射用製剤(例:注射剤など)、透析用製剤(例:透析用剤など)、吸入用製剤(例:吸入剤など)、眼科用製剤(例:点眼剤、眼軟膏剤など)、耳科用製剤(例:点耳剤など)、鼻科用製剤(例:点鼻剤など)、直腸用製剤(例:坐剤、直腸用半固形剤、腸注剤など)、腟用製剤(例:腟錠、腟用坐剤など)、および皮膚用製剤(例:外用固形剤、外用液剤、スプレー剤、軟膏剤、クリーム剤、ゲル剤、貼付剤など)などが挙げられる。
本発明に用いられるEP4拮抗薬の投与量は、年齢、体重、症状、治療効果、投与方法、処理時間等により異なるが、通常、成人一人当たり、一回につき、1ngから1000mgの範囲で一日一回から数回経口投与されるか、または成人一人当たり、一回につき、0.1ngから100mgの範囲で一日一回から数回非経口投与されるか、または一日1時間から24時間の範囲で静脈内に持続投与される。もちろん前記したように、投与量は種々の条件により変動するので、上記投与量より少ない量で十分な場合もあるし、また範囲を越えて投与の必要な場合もある。
ある一態様として、化合物Aを使用する場合、投与量の一態様は、1回につき約1~100mgを1日1~3回経口投与され、好ましくは1回1mg、5mg、10mg、15mg、20mg、25mg、30mg、35mg、40mg、45mg、50mg、55mg、60mg、65mg、70mg、75mg、80mg、85mg、90mg、95mgまたは100mgを1日1~3回経口投与され、さらに好ましくは1回1mg、5mg、10mg、15mg、20mg、25mg、30mg、35mg、40mg、45mg、50mgを1日1~3回経口投与され、さらに好ましくは1日1回5mg、10mg、20mgまたは40mgを1日1回経口投与され、より好ましくは、20mgまたは40mgを1日1回経口投与される。
(2)免疫チェックポイント阻害物質
本発明において、免疫チェックポイント分子は、抑制性共シグナルを伝達することで免疫抑制機能を発揮する分子を意味する。免疫チェックポイント分子としては、CTLA-4、PD-1、PD-L1(programmed cell death-ligand 1)、PD-L2(programmed cell death-ligand 2)、LAG-3(Lymphocyte activation gene 3)、TIM3(T cell immunoglobulin and mucin-3)、BTLA(B and T lymphocyte attenuator)、B7H3、B7H4、CD160、CD39、CD73、A2aR(adenosine A2a receptor)、KIR(killer inhibitory receptor)、VISTA(V-domain Ig-containing suppressor of T cell activation)、IDO1(Indoleamine 2,3-dioxygenase)、ArginaseI、TIGIT(T cell immunoglobulin and ITIM domain)、CD115等が知られているが(Nature Reviews Cancer、12、252-264ページ、2012年、Cancer Cell、27、450-461ページ、2015年を参照)、定義に一致する働きを有する分子であれば特に限定されない。
本発明において、免疫チェックポイント分子は、抑制性共シグナルを伝達することで免疫抑制機能を発揮する分子を意味する。免疫チェックポイント分子としては、CTLA-4、PD-1、PD-L1(programmed cell death-ligand 1)、PD-L2(programmed cell death-ligand 2)、LAG-3(Lymphocyte activation gene 3)、TIM3(T cell immunoglobulin and mucin-3)、BTLA(B and T lymphocyte attenuator)、B7H3、B7H4、CD160、CD39、CD73、A2aR(adenosine A2a receptor)、KIR(killer inhibitory receptor)、VISTA(V-domain Ig-containing suppressor of T cell activation)、IDO1(Indoleamine 2,3-dioxygenase)、ArginaseI、TIGIT(T cell immunoglobulin and ITIM domain)、CD115等が知られているが(Nature Reviews Cancer、12、252-264ページ、2012年、Cancer Cell、27、450-461ページ、2015年を参照)、定義に一致する働きを有する分子であれば特に限定されない。
本発明において免疫チェックポイント阻害物質は、免疫チェックポイント分子の機能を阻害する物質である。免疫チェックポイント阻害物質としては、免疫チェックポイント分子の機能(シグナル)を抑制しうる物質であれば特に限定されない。
免疫チェックポイント阻害物質としては、例えば、抗PD-1抗体(例えば、Nivolumab、Cemiplimab(REGN-2810)、Pembrolizumab(MK-3475)、Spartalizumab(PDR-001)、Tislelizumab(BGB-A317)、AMP-514(MEDI0680)、Dostarlimab(ANB011/TSR-042)、Toripalimab(JS001)、Camrelizumab(SHR-1210)、Genolimzumab(CBT-501)、Sintilimab(IBI308)、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、Retifanlimab(MGA012)、Balstilimab(AGEN2034)、CS1003、Serplulimab(HLX10)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、Geptanolimab(GB226)、SSI-361、JY034、HX008、ISU106、Budigalimab(ABBV181)、Prolgolimab(BCD-100)、Sasanlimab(PF-06801591)、CX-188、Cetrelimab(JNJ-63723283)およびZimberelimab(AB122)等)、抗PD-L1抗体(例えば、Atezolizumab(RG7446/MPDL3280A)、Avelumab(PF-06834635/MSB0010718C)、Durvalumab(MEDI4736)、BMS-936559、STI-1014、Envafolimab(KN035)、Lodapolimab(LY3300054)、HLX20、SHR-1316、CS1001(WBP3155)、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502(TQB2450)、JS003およびCX-072等)または抗CTLA-4抗体(例えば、Ipilimumab(MDX-010)、Zalifrelimab(AGEN1884)およびTremelimumab等)、抗PD-L2抗体、PD-L1融合タンパク質、PD-L2融合タンパク質(例えば、AMP-224)、抗Tim-3抗体(例えば、MBG453)、抗LAG-3抗体(例えば、BMS-986016、LAG525)、抗KIR抗体(例えば、Lirilumab)等である。また、上記既知の抗体の重鎖および軽鎖相補性決定領域(CDRs)または可変領域(VR)を含む抗体も免疫チェックポイント阻害物質の一態様である。例えば、抗PD-1抗体の更なる一態様としては、例えばNivolumabの重鎖および軽鎖相補性決定領域(CDRs)または可変領域(VR)を含む抗体が挙げられる。
本発明において、免疫チェックポイント阻害物質としては好ましくは、抗PD-1抗体、抗PD-L1抗体および抗CTLA-4抗体であり、さらに好ましくは抗PD-1抗体または抗PD-L1抗体である。抗PD-1抗体として好ましくは、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、Toripalimab、SintilimabおよびCamrelizumabであり、抗PD-L1抗体として好ましくは、Atezolizumab、Avelumab、DurvalumabおよびBMS-936559であり、抗CTLA-4抗体として好ましくは、IpilimumabおよびTremelimumabである。さらに、抗PD-1抗体としてより好ましくは、Nivolumab、CemiplimabおよびPembrolizumabであり、さらに好ましくはNivolumabである。本発明において免疫チェックポイント阻害物質として、好ましくは抗PD-1抗体であり、さらに好ましくはNivolumabである。
本発明において、免疫チェックポイント阻害物質は公知の方法で製造することができる。Nivolumabは、WO2006/121168に記載された方法に準じて製造することができ、Pembrolizumabは、WO2008/156712に記載された方法に準じて製造することができ、BMS-936559は、WO2007/005874に記載された方法に準じて製造することができ、Ipilimumabは、WO2001/014424に記載された方法に準じて製造することができる。
本発明では、これら免疫チェックポイント阻害物質のうちのいずれか1種または任意の複数種をEP4拮抗薬の投与および標準療法と組み合わせて用いることができる。
本発明の組み合わせに用いられる免疫チェックポイント阻害物質の投与量は、年齢、体重、症状、治療効果、投与方法、処理時間等により異なるが、最適な所望の効果をもたらすように調整される。
例えば、免疫チェックポイント阻害物質を有効成分として1回約1~10mg/kg(体重)あるいは1回約200~1200mgを2~4週間間隔で、約30分間ないし約60分間あるいは約60分間以上かけて静脈内投与(例えば、点滴静注)することができる。ここで、投与1回当たりの体重換算における投与量として、例えば、1mg/kg、2mg/kg、3mg/kg、4mg/kg、5mg/kg、6mg/kg、7mg/kg、8mg/kg、9mg/kgまたは10mg/kgが挙げられ、一方、投与1回当たりの投与量としては、例えば、200mg、240mg、250mg、280mg、300mg、320mg、350mg、360mg、400mg、420mg、450mg、480mg、500mg、540mg、560mg、600mg、640mg、700mg、720mg、750mg、800mg、840mg、900mg、1000mg、1080mg、1100mg、1120mgまたは1200mgが挙げられる。また、投与間隔としては、例えば、2週間、3週間または4週間が挙げられ、1回の投与時間としては、例えば、約30分間、約60分間または約60分間以上が挙げられる。
免疫チェックポイント阻害物質が抗PD-1抗体であるNivolumabの場合には、次の用法・用量にて投与されている。すなわち、悪性黒色腫患者には、Nivolumabとして1回3mg/kg(体重)を2週間間隔または1回2mg/kg(体重)を3週間間隔で点滴静注投与され、あるいは1回240mgを2週間間隔でまたは1回480mgを4週間間隔で点滴静注される。非小細胞肺がん、腎細胞がん、古典的ホジキンリンパ腫、頭頸部がん、胃がんおよび悪性胸膜中皮腫の各患者には、Nivolumabとして1回3mg/kg(体重)を2週間間隔で点滴静注投与される。また、別の用法・用量として、例えば、悪性黒色腫、非小細胞肺がん、腎細胞がん、尿路上皮がん、MSI-HまたはdMMR陽性大腸癌(12歳以上の小児の患者も含む。)、胃がん、肝細胞がん、小細胞肺がんおよび悪性胸膜中皮腫の各患者には、Nivolumabとして1回240mgを2週間間隔で、あるいは1回480mgを4週間間隔で点滴静注投与される。さらに、別の用法・用量として、例えば、悪性黒色腫患者に対して、Ipilimumabとの併用において、Nivolumabとして1回1mg/kg(体重)を3週間間隔で4回点滴静注され、その後、Nivolumabとして1回3mg/kg(体重)を2週間間隔で点滴静注されるか、あるいはNivolumabとして1回80mgを3週間間隔で4回点滴静注され、その後、Nivolumabとして1回240mgを2週間間隔でまたは1回480mgを4週間間隔で点滴静注される場合がある。また、例えば、腎細胞がん患者または大腸癌患者に対して、Ipilimumabとの併用において、Nivolumabとして1回240mgを3週間間隔で4回点滴静注され、その後、Nivolumabとして1回240mgを2週間間隔または1回480mgを4週間間隔で点滴静注される場合もある。
また、同じ抗PD-1抗体であるPembrolizumabの場合には、次の用法・用量で投与されている。すなわち、悪性黒色腫、非小細胞肺がん、古典的ホジキンリンパ腫、頭頚部がん、MSI-HもしくはdMMR陽性固形がんまたは大腸がん、尿路上皮がん、子宮頸がん、原発性縦隔B細胞リンパ腫、肝細胞がん、胃がんおよびメルケル細胞がんの各患者には、Pembrolizumabとして1回200mgを3週間間隔または1回400mgを6週間間隔で点滴静注投与される。また、別の用法・用量として、例えば、2歳以上の小児の古典的ホジキンリンパ腫、MSI-HもしくはdMMR陽性固形がんまたは大腸がんおよび原発性縦隔B細胞リンパ腫の各患者には、Pembrolizumabとして1回2mg/kg(体重)(1回200mgまで)を3週間間隔で点滴静注投与される。
また、免疫チェックポイント阻害物質が抗PD-L1抗体であるAvelumabの場合には、メルケル細胞がんおよび尿路上皮がんの各患者には、Avelumabとして1回10mg/kg(体重)を2週間間隔で点滴静注投与される。同じPD-L1抗体であるAtezolizumabは、非小細胞肺がん、尿路上皮がんおよび肝細胞がんの各患者には、Atezolizumabとして1回1200mgを3週間間隔で点滴静注投与され、トリプルネガティブ乳癌患者には、パクリタキセルとの併用において、Atezolizumabとして1回840mgを2週間間隔で点滴静注投与される。さらに、同じPD-L1抗体であるDurvalumabは、非小細胞肺がんおよび尿路上皮がんの各患者には、Durvalumabとして1回10mg/kg(体重)を2週間間隔で点滴静注され、進展型小細胞肺がんの患者には、Durvalumabとして1回1500mgを4週間間隔で点滴静注投与される。
また、抗CTLA-4抗体であるIpilimumabの場合には、悪性黒色腫患者に対して、単独もしくはNivolumabとの併用において、Ipilimumabとして1日1回3mg/kg(体重)を3週間間隔で4回点滴静注され、腎細胞がんおよびMSI-HまたはdMMR陽性大腸がんの各患者には、Nivolumabとの併用において、Ipilimumabとして1日1回1mg/kg(体重)を3週間間隔で4回点滴静注され、非小細胞肺がんの患者には、Ipilimumabとして1回1mg/kg(体重)を6週間間隔で点滴静注投与される。
なお、本発明における免疫チェックポイント阻害物質の投与形態としては、非経口投与が好ましく、非経口投与としては、皮下投与、皮内投与、腹腔内投与、筋肉内投与、静脈内投与等が挙げられるが、皮下投与または静脈内投与が好ましい。さらに好ましくは、静脈内投与である。静脈内投与の投与形態としては、点滴静注が好ましい。
本発明において、上記用法用量は本発明の治療法においても使用されうる。
(3)XELOX+ベバシズマブ療法並びにEP4拮抗薬および免疫チェックポイント阻害物質との併用(以下、本併用-1と略記することがある。)
本発明における「XELOX療法」とは、カペシタビンおよびオキサリプラチン(L-OHP)の併用によるがん治療法である。
本発明における「XELOX療法」とは、カペシタビンおよびオキサリプラチン(L-OHP)の併用によるがん治療法である。
本発明における「XELOX+ベバシズマブ療法」とは、カペシタビン、オキサリプラチンおよびベバシズマブの3剤併用によるがん治療法であり、一実施形態において、ベバシズマブ 7.5mg/kgを約30~90分かけて静脈内投与し、オキサリプラチン 130mg/m2(体表面積)を2時間かけて静脈内投与し、当該一連の投与を3週間間隔で実施し、カペシタビン 1000mg/m2/回(体表面積1.36m2未満:1200mg/回、体表面積1.36~1.66m2未満:1500mg/回、体表面積1.66~1.96m2未満:1800mg/回、体表面積1.96m2以上:2100mg/回)を1日2回で14日間経口投与し、7日間休薬する療法である。一態様として、当該XELOX+ベバシズマブ療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、ベバシズマブの投与を中止、カペシタビンの投与量を投与開始用量および体表面積に応じて1800mg/回、1500mg/回、1200mg/回、900mg/回あるいは600mg/回に減量もしくは中止、オキサリプラチンを100mg/m2(体表面積)あるいは85mg/m2(体表面積)に減量もしくは中止してもよい。XELOX+ベバシズマブ療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
XELOX+ベバシズマブ療法との併用における、EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgであり、EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
XELOX+ベバシズマブ療法との併用における、免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回360mgを3週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
本併用-1が適用されうるがんは、本併用-1が効果を発揮しうるがんであれば特に限定されないが、一実施形態において大腸がんであり、好ましくは結腸・直腸がんである。さらに好ましくは、根治切除不能な進行または再発の結腸・直腸がんである。
本併用-1は、一実施形態において、大腸がんに対する治療歴のない患者に投与される。
本併用-1において、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とXELOX+ベバシズマブ療法を同日に投与する場合、一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質を先に投与する。一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質が投与された後にベバシズマブ、オキサリプラチン、カペシタビンの順で投与される。また、別の実施形態として、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とXELOX+ベバシズマブ療法を同日に投与する場合、XELOX+ベバシズマブ療法を先に実施してもよく、EP4拮抗薬および免疫チェックポイント阻害物質の投与と同時でもよい。一実施形態において、EP4拮抗薬、免疫チェックポイント阻害物質、ベバシズマブ、オキサリプラチンおよびカペシタビンの投与の順は他に定義されない限りいずれの薬剤から投与を開始してもよく、また2以上の薬剤を同時に投与してもよい。
(4)術前化学放射線療法後の術前補助療法としてのEP4拮抗薬または同療法としてのEP4拮抗薬と免疫チェックポイント阻害物質との併用(以下、本併用-2と略記することがある。)
本発明における「術前化学放射線療法」とは、放射線照射とフルオロウラシル(5-FU)あるいはカペシタビンなどの化学療法を組み合わせた療法である。本発明において、好ましくは、放射線照射とカペシタビン投与の組み合わせである。
本発明における「術前化学放射線療法」とは、放射線照射とフルオロウラシル(5-FU)あるいはカペシタビンなどの化学療法を組み合わせた療法である。本発明において、好ましくは、放射線照射とカペシタビン投与の組み合わせである。
本発明における術前化学放射線療法は、一実施形態において、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2(開始用量はゼローダ(登録商標)錠300の用法・用量に基づく)の1日2回投与であり、放射線との併用で放射線の照射回数である28回の75%に相当する期間でカペシタビンの内服が行われるが(例:50.4Gy/28回照射の場合、21日間または朝夕の内服として42回以上の内服)、患者の副作用表現の程度に応じて、適宜減量することができる。
術前化学放射線療法後の術前補助療法として使用されるEP4拮抗薬としての化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。より好ましくは、化合物Aは40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。
術前化学放射線療法後の術前補助療法として使用される免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回240mgを2週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
本併用-2の一実施形態は、EP4拮抗薬の投与であり、一実施形態はEP4拮抗薬と免疫チェックポイント阻害物質との併用投与である。
本併用-2が適用されるがんとしては、本術前補助療法が効果を発揮しうるがんであれば特に限定されないが、一実施形態において大腸がんであり、好ましくは結腸・直腸がんである。さらに好ましくは、治癒切除可能な局所進行直腸がんである。
一実施形態において、術前化学放射線療法終了後の画像診断で遠隔転移を認めず、治癒切除が可能な患者に投与される。
一実施形態において、本術前補助療法において(根治)切除が可能となる患者に投与される。
(5)FFX療法、またはその減量レジメン
本発明における「FFX療法」とは、オキサリプラチン(L-OHP)、イリノテカン塩酸塩水和物(イリノテカン、CPT-11)、レボホリナートカルシウム(レボホリナート、l-LV)およびフルオロウラシル(5-FU)の4剤併用によるがん治療法であり、推奨の用法用量として、例えば、オキサリプラチン 85mg/m2(体表面積)を2時間かけて静脈内投与したのち、レボホリナート 200mg/m2を2時間かけて静脈内投与し、レボホリナートの投与開始30分後からイリノテカン 180mg/m2を1.5時間かけて静脈内投与し、さらにレボホリナートの投与終了後に、フルオロウラシル 400mg/m2を急速静脈内投与して、さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内投与し、当該一連の投与を2週間間隔で実施する治療法である。
本発明における「FFX療法」とは、オキサリプラチン(L-OHP)、イリノテカン塩酸塩水和物(イリノテカン、CPT-11)、レボホリナートカルシウム(レボホリナート、l-LV)およびフルオロウラシル(5-FU)の4剤併用によるがん治療法であり、推奨の用法用量として、例えば、オキサリプラチン 85mg/m2(体表面積)を2時間かけて静脈内投与したのち、レボホリナート 200mg/m2を2時間かけて静脈内投与し、レボホリナートの投与開始30分後からイリノテカン 180mg/m2を1.5時間かけて静脈内投与し、さらにレボホリナートの投与終了後に、フルオロウラシル 400mg/m2を急速静脈内投与して、さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内投与し、当該一連の投与を2週間間隔で実施する治療法である。
当該FFX療法の「減量レジメン」とは、当該FFX療法において投与される4剤の何れかの投与量を最初の投与から減量もしくはその投与自体を中止する、または1サイクル目以降の何れかの投与により認められる副作用発現の程度に応じて、その後の2サイクル目以降の何れかの投与において減量する、あるいは4剤の何れかの投与を中止する処方である。その態様として、例えば、最初の投与から、フルオロウラシルの急速静脈内投与を実施しなくてもよく、オキサリプラチンの投与量が、65mg/m2、50mg/m2または85~50mg/m2の間の任意の投与量であってもよく、イリノテカンの投与量が、150mg/m2、120mg/m2、90mg/m2または180~90mg/m2の間の任意の投与量であってもよく、静脈内持続投与されるフルオロウラシルの投与量が、1800mg/m2、1200mg/m2または2400~1200mg/m2の間の任意の投与量であってもよい。
また、当該減量レジメンのもう一つの態様として、当該FFX療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、フルオロウラシルの急速静脈内投与を中止してもよく、患者の副作用発現の程度に応じて、オキサリプラチンの投与量を、65mg/m2、50mg/m2または85~50mg/m2の間の任意の投与量に減量またはオキサリプラチンの投与を中止してもよく、患者の副作用発現の程度に応じて、イリノテカンの投与量を、150mg/m2、120mg/m2、90mg/m2または180~90mg/m2の間の任意の投与量に減量またはイリノテカンの投与を中止してもよく、静脈内持続投与されるフルオロウラシルの投与量を、患者の副作用発現の程度に応じて、1800mg/m2、1200mg/m2または2400~1200mg/m2の間の任意の投与量に減量または当該フルオロウラシルの投与を中止してもよい。
また、modified FOLFIRINOX療法(mFFX療法)として認知されている当該減量レジメンの別の態様は、推奨の用法用量として、例えば、オキサリプラチン 85mg/m2(体表面積)を2時間かけて静脈内投与したのち、レボホリナート 200mg/m2を2時間かけて静脈内投与し、レボホリナート投与開始30分後からイリノテカン 150mg/m2を1.5時間かけて静脈内投与し、さらにレボホリナートの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内投与し、当該一連の投与を2週間間隔で実施する治療法である。
当該mFFX療法のさらなる減量レジメンとして、例えば、最初の投与から、オキサリプラチンの投与量が、65mg/m2、50mg/m2または85~50mg/m2の間の任意の投与量であってもよく、イリノテカンの投与量が、140mg/m2、120mg/m2または140~120mg/m2の間の任意の投与量であってもよく、静脈内持続投与されるフルオロウラシルの投与量が、2400mg/m2、1800mg/m2、1200mg/m2または2400~1200mg/m2の間の任意の投与量であってもよい。
また、当該mFFX療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、オキサリプラチンの投与量を、65mg/m2、50mg/m2または85~50mg/m2の間の任意の投与量に減量またはオキサリプラチンの投与を中止してもよく、患者の副作用発現の程度に応じて、イリノテカンの投与量を、120mg/m2、90mg/m2または150~90mg/m2の間の任意の投与量に減量またはイリノテカンの投与を中止してもよく、静脈内持続投与されるフルオロウラシルの投与量を、患者の副作用発現の程度に応じて、1800mg/m2、1200mg/m2または2400~1200mg/m2の間の任意の投与量に減量または当該フルオロウラシルの投与を中止してもよい。
また、当該FFX療法またはその減量レジメン(例えば、mFFX療法)の一連の投与の間隔を、患者の副作用発現の程度に応じて、一時的にもしくは継続的に、3週間間隔または4週間間隔と設定してもよい。FFX療法またはその減量レジメン(例えば、mFFX療法)の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
(6)FFX療法、またはその減量レジメンとの併用(以下、本併用-3と略記することがある。)
FFX療法またはその減量レジメン(例えば、mFFX療法)との併用におけるEP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。さらに好ましくは、20mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgであり、EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
FFX療法またはその減量レジメン(例えば、mFFX療法)との併用におけるEP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。さらに好ましくは、20mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgであり、EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-3における、免疫チェックポイント阻害物質は、一実施形態において、(2)免疫チェックポイント阻害物質に記載の用法用量で投与される。免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回480mgを4週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
本併用-3において、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とFFX療法またはその減量レジメン(例えば、mFFX療法)を同日に投与する場合、一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質を先に投与する。一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質が投与された後にFFX療法またはその減量レジメン(例えば、mFFX療法)が実施される。また、別の実施形態として、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とFFX療法またはその減量レジメン(例えば、mFFX療法)を同日に投与する場合、FFX療法またはその減量レジメン(例えば、mFFX療法)を先に実施してもよく、EP4拮抗薬および免疫チェックポイント阻害物質の投与と同時でもよい。一実施形態において、EP4拮抗薬、免疫チェックポイント阻害物質、FFX療法またはその減量レジメン(例えば、mFFX療法)の投与の順は他に定義されない限りいずれの薬剤から投与を開始してもよく、また2以上の薬剤を同時に投与してもよい。
(7)GnP療法
本発明における「GnP療法」とは、ゲムシタビンとナブパクリタキセルを組み合わせた療法であり、一実施形態において、GnP療法は、推奨の用法用量として、例えば、ゲムシタビンを1000mg/m2(体表面積)を30分かけて静脈内投与し、ナブパクリタキセルを125mg/m2(体表面積)を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する療法である。一態様として、当該GnP療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、ゲムシタビンを800mg/m2もしくは600mg/m2に減量してもよく、ナブパクリタキセルを100mg/m2もしくは75mg/m2に減量してもよい。GnP療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
本発明における「GnP療法」とは、ゲムシタビンとナブパクリタキセルを組み合わせた療法であり、一実施形態において、GnP療法は、推奨の用法用量として、例えば、ゲムシタビンを1000mg/m2(体表面積)を30分かけて静脈内投与し、ナブパクリタキセルを125mg/m2(体表面積)を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する療法である。一態様として、当該GnP療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、ゲムシタビンを800mg/m2もしくは600mg/m2に減量してもよく、ナブパクリタキセルを100mg/m2もしくは75mg/m2に減量してもよい。GnP療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
(8)GnP療法との併用(以下、本併用-4と略記することがある。)
本併用-4における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。より好ましくは、化合物Aは40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-4における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。より好ましくは、化合物Aは40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用―4における、免疫チェックポイント阻害物質は、一実施形態において、(2)免疫チェックポイント阻害物質に記載の用法用量で投与される。免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回480mgを4週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
(9)DTX+RAM療法
本発明における「ドセタキセルおよびラムシルマブ療法」とは、ドセタキセルとラムシルマブの併用によるがん治療法であり、推奨の用法用量として、例えば、ドセタキセル 60mg/m2(体表面積)を60分以上かけて静脈内投与し、ラムシルマブ 10mg/kgを60分かけて静脈内投与し、当該一連の投与を3週間間隔で実施する治療法である。一態様として、当該DTX+RAM療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、ドセタキセルを75mg/m2もしくは50mg/m2に増減またはドセタキセルの投与を中止してもよく、ラムシルマブを8mg/kgもしくは6mg/kgに減量またはラムシルマブの投与を中止してもよく、ラムシルマブの投与時間を30~60分に短縮してもよい。DTX+RAM療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。一実施形態において、ドセタキセルとラムシルマブの投与は同日に開始される。
本発明における「ドセタキセルおよびラムシルマブ療法」とは、ドセタキセルとラムシルマブの併用によるがん治療法であり、推奨の用法用量として、例えば、ドセタキセル 60mg/m2(体表面積)を60分以上かけて静脈内投与し、ラムシルマブ 10mg/kgを60分かけて静脈内投与し、当該一連の投与を3週間間隔で実施する治療法である。一態様として、当該DTX+RAM療法における2サイクル目以降の何れかの投与において、患者の副作用発現の程度に応じて、ドセタキセルを75mg/m2もしくは50mg/m2に増減またはドセタキセルの投与を中止してもよく、ラムシルマブを8mg/kgもしくは6mg/kgに減量またはラムシルマブの投与を中止してもよく、ラムシルマブの投与時間を30~60分に短縮してもよい。DTX+RAM療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。一実施形態において、ドセタキセルとラムシルマブの投与は同日に開始される。
(10)DTX+RAM療法との併用(以下、本併用-5と略記することがある。)
本併用-5におけるEP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgであり、EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-5におけるEP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgであり、EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-5における、免疫チェックポイント阻害物質は、一実施形態において、(2)免疫チェックポイント阻害物質に記載の用法用量で投与される。免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回360mgを3週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
一実施形態において、DTX+RAM療法、免疫チェックポイント阻害物質およびEP4拮抗薬の投与は同日に開始される。
本併用-5において、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とDTX+RAM療法を同日に投与する場合、一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質を先に投与する。一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質が投与された後にDTX+RAM療法が実施される。また、別の実施形態として、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とDTX+RAM療法を同日に投与する場合、DTX+RAM療法を先に実施してもよく、EP4拮抗薬および免疫チェックポイント阻害物質の投与と同時でもよい。一実施形態において、EP4拮抗薬、免疫チェックポイント阻害物質、ドセタキセルおよびラムシルマブの投与の順は他に定義されない限りいずれの薬剤から投与を開始してもよく、また2以上の薬剤を同時に投与してもよい。
(11)DTX療法
本発明における「ドセタキセル療法」とは、ドセタキセルによるがん治療法であり、一実施形態において、推奨の用法用量として、例えば、ドセタキセルを60mg/m2(体表面積)を60分以上かけて静脈内投与し、当該投与を3週間間隔で行う療法である。一態様として、当該DTX療法における2サイクル目以降の何れかの投与において、患者の状態に応じて、ドセタキセルを75mg/m2もしくは50mg/m2に増減またはドセタキセルの投与を中止してもよい。DTX療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
本発明における「ドセタキセル療法」とは、ドセタキセルによるがん治療法であり、一実施形態において、推奨の用法用量として、例えば、ドセタキセルを60mg/m2(体表面積)を60分以上かけて静脈内投与し、当該投与を3週間間隔で行う療法である。一態様として、当該DTX療法における2サイクル目以降の何れかの投与において、患者の状態に応じて、ドセタキセルを75mg/m2もしくは50mg/m2に増減またはドセタキセルの投与を中止してもよい。DTX療法の休薬・減量・再開は、最新の添付文書を参考に医師の判断で実施される。
(12)DTX療法との併用(以下、本併用-6と略記することがある。)
本併用-6における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-6における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-6における、免疫チェックポイント阻害物質は、一実施形態において、(2)免疫チェックポイント阻害物質に記載の用法用量で投与される。免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回360mgを3週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
一実施形態において、DTX療法、免疫チェックポイント阻害物質およびEP4拮抗薬の投与は同日に開始される。
本併用-6において、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とDTX療法を同日に投与する場合、一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質を先に投与する。一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質が投与された後にDTX療法が実施される。また、別の実施形態として、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とDTX療法を同日に投与する場合、DTX療法を先に実施してもよく、EP4拮抗薬および免疫チェックポイント阻害物質の投与と同時でもよい。一実施形態において、EP4拮抗薬、免疫チェックポイント阻害物質、およびドセタキセルの投与の順は他に定義されない限りいずれの薬剤から投与を開始してもよく、また2以上の薬剤を同時に投与してもよい。
(13)RAM療法
本発明における「ラムシルマブ療法(以下、「RAM療法」と略記することがある。)」とは、ラムシルマブによるがん治療法であり、一実施形態において、推奨の用法用量として、例えば、ラムシルマブ 10mg/kgを60分かけて静脈内投与し、当該一連の投与を3週間間隔で実施する治療法である。一態様として、当該RAM療法における2サイクル目以降の何れかの投与において、患者の状態に応じて、ラムシルマブを8mg/kgもしくは6mg/kgに減量またはラムシルマブの投与を中止してもよく、また患者の副作用の程度に応じて、ラムシルマブの投与時間を30~60分に短縮してもよい。
本発明における「ラムシルマブ療法(以下、「RAM療法」と略記することがある。)」とは、ラムシルマブによるがん治療法であり、一実施形態において、推奨の用法用量として、例えば、ラムシルマブ 10mg/kgを60分かけて静脈内投与し、当該一連の投与を3週間間隔で実施する治療法である。一態様として、当該RAM療法における2サイクル目以降の何れかの投与において、患者の状態に応じて、ラムシルマブを8mg/kgもしくは6mg/kgに減量またはラムシルマブの投与を中止してもよく、また患者の副作用の程度に応じて、ラムシルマブの投与時間を30~60分に短縮してもよい。
(14)RAM療法との併用(以下、本併用-7と略記することがある。)
本併用-7における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-7における、EP4拮抗薬は、一実施形態において、(1)EP4拮抗薬に記載の用法用量で投与される。EP4拮抗薬である化合物Aは、一実施形態において、5mgで1日一回経口投与、10mgで1日一回経口投与、20mgで1日一回経口投与または40mgで1日一回経口投与される。好ましくは、20mgで1日一回経口投与または40mgで1日一回経口投与される。患者の副作用発現の程度に応じて、化合物Aの投与量を減量するか、または投与自体を中止してもよい。また、再開の基準を満たせば、化合物Aの投与を再開することができ、再開する場合、医師の判断により、化合物Aの用量を1段階ずつ減量して再開することができる。一実施形態において、EP4拮抗薬としての化合物Aを投与開始用量40mgで投与する場合、1段階減量は20mgであり、2段階減量は10mgである。EP4拮抗薬としての化合物Aを投与開始用量20mgで投与する場合、1段階減量は10mgであり、2段階減量は5mgである。
本併用-7における、免疫チェックポイント阻害物質は、一実施形態において、(2)免疫チェックポイント阻害物質に記載の用法用量で投与される。免疫チェックポイント阻害物質としてのNivolumabは、一実施形態において、Nivolumabとして1回240mgを2週間間隔、1回360mgを3週間間隔、または1回480mgを4週間間隔で点滴静注投与される。好ましくは、Nivolumabとして1回360mgを3週間間隔で点滴静注投与される。患者の副作用発現の程度に応じて、Nivolumabの投与自体を中止してもよい。また、再開の基準を満たせば、Nivolumabの投与を再開することができる。
一実施形態において、RAM療法、免疫チェックポイント阻害物質およびEP4拮抗薬の投与は同日に開始される。
本併用-7において、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とRAM療法を同日に投与する場合、一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質を先に投与する。一実施形態において、EP4拮抗薬および免疫チェックポイント阻害物質が投与された後にRAM療法が実施される。また、別の実施形態として、EP4拮抗薬(好ましくは化合物A)および免疫チェックポイント阻害物質(好ましくは抗PD-1抗体(好ましくはNivolumab))とRAM療法を同日に投与する場合、RAM療法を先に実施してもよく、EP4拮抗薬および免疫チェックポイント阻害物質の投与と同時でもよい。一実施形態において、EP4拮抗薬、免疫チェックポイント阻害物質、およびラムシルマブの投与の順は他に定義されない限りいずれの薬剤から投与を開始してもよく、また2以上の薬剤を同時に投与してもよい。
[適用疾患および患者]
本発明の治療法の適用疾患は、がんである。
本発明の治療法の適用疾患は、がんである。
より具体的には、がんとして、例えば、白血病(例えば、急性骨髄性白血病、慢性骨髄性白血病、急性リンパ性白血病、慢性リンパ性白血病)、悪性リンパ腫(ホジキンリンパ腫、非ホジキンリンパ腫(例えば、成人T細胞白血病、濾胞性リンパ腫、びまん性大細胞型B細胞性リンパ腫))、多発性骨髄腫、骨髄異形成症候群、頭頸部がん、食道がん、食道腺がん、胃がん、食道胃接合部がん、十二指腸がん、大腸がん、結腸がん、直腸がん、肝臓がん(例えば、肝細胞がん)、胆嚢・胆管がん、胆道がん、膵臓がん(例えば、膵管がん、インスリノーマ、膵管内乳頭粘液性腫瘍)、甲状腺がん、肺がん(例えば、非小細胞肺がん(例えば、扁平上皮非小細胞肺がん、非扁平上皮非小細胞肺がん)、小細胞肺がん)、乳がん、卵巣がん(例えば、漿液性卵巣がん)、子宮頚がん、子宮体がん、子宮内膜がん、膣がん、外陰部がん、腎がん(例えば、腎細胞がん)、腎盂・尿管がん、尿路上皮がん(例えば、膀胱がん、上部尿路がん)、陰茎がん、前立腺がん、精巣腫瘍(例えば、胚細胞腫瘍)、骨・軟部肉腫、悪性骨腫瘍、皮膚がん(例えば、ブドウ膜悪性黒色腫、悪性黒色腫、メルケル細胞がん)、胸腺腫、中皮腫、悪性胸膜中皮腫、膠芽腫、血液がん、および原発不明がんなどが挙げられる。
本発明の治療法の適用疾患として、好ましくは大腸がん、膵臓がん、肺がんであり、より好ましくは根治切除不能な進行または再発の結腸・直腸がん、治癒切除可能な局所進行がんである直腸がん、遠隔転移を有する膵臓がん、抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた肺がんである。また、さらに好ましくは根治切除不能な進行または再発の結腸・直腸がん、治癒切除可能な局所進行がんである直腸がん、遠隔転移を有する膵管がん、および抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた非小細胞肺がんである。本明細書においてがんの「治療」とは、例えば、(i)腫瘍細胞の増殖を減少させる、(ii)がんに起因する症状を低減させる、(iii)がん患者の生活の質を向上させる、(iv)既に投与されている他の抗がん薬またはがん治療補助薬の用量を低減させる、および/または(v)がん患者の生存を延長させるために行われる治療を含み、がんの「進行抑制」とは、がんの進行を遅延、がんに起因する症状を安定化および症状の進行を後退させることを意味する。また、がんの「再発抑制」とは、がん治療あるいは切除手術によってそのがん病変が完全にあるいは実質的に消滅あるいは取り除かれた患者におけるがんの再発を予防的に抑止することを意味する。
本発明の治療法は、次のがん患者、すなわち、(a)抗がん薬による治療効果が不十分あるいは十分ではない患者もしくは抗がん薬による治療後に増悪した患者、(b)根治もしくは切除不能、転移性、再発性、難治性および/または遠隔転移性のがんの患者、(c)Tumor Proportion Score(以下、「TPS」と略記する。)が50%以上、25%以上、10%以上、5%以上もしくは1%以上であるがんの患者、(d)Combined Positive Score(以下、「CPS」と略記する。)が20%以上、10%以上、5%以上もしくは1%以上であるがんの患者、(e)ミスマッチ修復欠損(以下、「dMMR」と略記する。)および/または高頻度マイクロサテライト不安定性(以下、「MSI-H」と略記する。)を有するがんの患者、(f)腫瘍変異負荷(以下、「TMB」と略記する。)が高頻度であるがんの患者に処方することがある。また、一方で、本発明の治療法は、次のがん患者、すなわち、(g)抗がん薬による治療歴のない患者、(h)TPSが50%未満、25%未満、10%未満、5%未満もしくは1%未満であるがんの患者、(i)CPSが20%未満、10%未満、5%未満もしくは1%未満であるがんの患者、(j)dMMRおよび/またはMSI-Hを有しない、もしくは低頻度マイクロサテライト不安定性(以下、「MSI-L」と略記する。)を有するがんの患者、または(k)TMBが低頻度であるがんの患者への処方がより求められる場合もある。特に、本発明の治療法の処方が求められるがん患者としては、(i)根治切除不能な進行または再発のがん患者、特に結腸・直腸がんの患者、(ii)治癒切除可能な局所進行がんであるがん患者、特に直腸がんの患者、(iii)抗がん薬による治療歴のないおよび/または遠隔転移を有するがん患者、特に、膵臓がんの患者、または(iv)抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められたがん患者、特に、肺がんの患者が挙げられる。
一実施形態において、根治切除不能な進行または再発の結腸・直腸がんの患者が挙げられる。一実施形態において、結腸・直腸がんに対する治療歴のない患者が挙げられる。好ましくは、結腸・直腸がんに対する全身性抗悪性腫瘍剤による治療歴のない患者が挙げられる。さらに好ましくは、根治切除不能な進行性または再発の結腸・直腸がんに対する全身性抗悪性腫瘍剤による治療歴のない患者が挙げられる。
一実施形態において、治癒切除可能な局所進行がんである直腸がんの患者が挙げられる。一実施形態として、直腸がんに対して治療歴のない患者が挙げられる。
一実施形態において、抗がん薬による治療歴のないおよび/または遠隔転移を有する膵臓がんの患者が挙げられる。
一実施形態において、膵臓がんに対する治療歴のない患者が挙げられる。好ましくは、膵臓がんに対する全身性悪性腫瘍剤による治療歴のない患者が挙げられる。さらに好ましくは、遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者が挙げられる。
一実施形態において、抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた肺がんの患者が挙げられる。
一実施形態において、非小細胞肺がんであると確認された患者が挙げられる。好ましくは、IV期または再発の非小細胞肺がんの患者が挙げられる。さらに好ましくは、一次治療としての抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者が挙げられる。
このうち、例えば、免疫チェックポイント阻害物質またはEP4受容体拮抗薬単独での治療効果が十分ではないがん患者に対して、本発明の治療法は、特に、その抗腫瘍効果を最大限に発揮することが期待できる。また、本発明の治療法により、ぞれぞれの薬剤の用量を下げて投与することも可能となり、副作用の軽減が期待できる。
本発明の治療法は、一態様として、相乗効果を発揮する。また、一態様として、免疫チェックポイント阻害物質またはEP4受容体拮抗薬、もしくは標準療法単独での治療効果、または免疫チェックポイント阻害物質および標準療法、EP4受容体拮抗薬および標準療法、または免疫チェックポイント阻害物質およびEP4受容体拮抗薬との併用による治療効果に比べて、相乗効果を発揮する。
本発明の治療法は、一態様として、副作用を軽減する。また、一態様として、免疫チェックポイント阻害物質またはEP4受容体拮抗薬、もしくは標準療法単独での治療効果、または免疫チェックポイント阻害物質および標準療法、EP4受容体拮抗薬および標準療法、または免疫チェックポイント阻害物質およびEP4受容体拮抗薬との併用による治療効果に比べて、副作用を軽減する。
本発明の組み合わせにおける併用投与に用いる各薬剤の投与タイミングは、他に定義されない限り、同時でも別々でもよい。
本発明の治療法は、一態様として、転移性がんの治療や転移の抑制にも適用可能である。
本発明の治療法は、一態様として、再発を抑制する。
本発明において、治療は、腫瘍サイズの低下、腫瘍の成長の抑制(遅延または停止)、腫瘍の転移の抑制(遅延または停止)、再発の抑制(防止または遅延)、およびがんと関連する一つまたは複数の症状の緩和のうち少なくとも1つを生じさせることを意味する。
本発明において、本発明の治療法は、(1)治療効果の補完および/または増強、(2)動態・吸収改善、投与量の低減、および/または(3)副作用の軽減のために、他の薬物(例えば、公知の抗がん治療)と組み合わせて使用されてもよい。
本明細書において、「約」とは、表示される数値を10%以内の範囲内で下回って、または上回って変化してよいことを意味する。または、約30分は30分±5分、約60分は60分±5分を意味する。
本明細書において、「標準療法」とは、科学的根拠に基づく、ある状態の一般的な患者に行われることが推奨される標準治療法であり、例えば、化学療法等が挙げられる。一態様として、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法が挙げられる。
本発明は、例えば、下記の実施態様を提供する。
[1] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、および免疫チェックポイント阻害物質の投与と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤。
[2] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤。
[3] EP4拮抗薬が一般式(I):
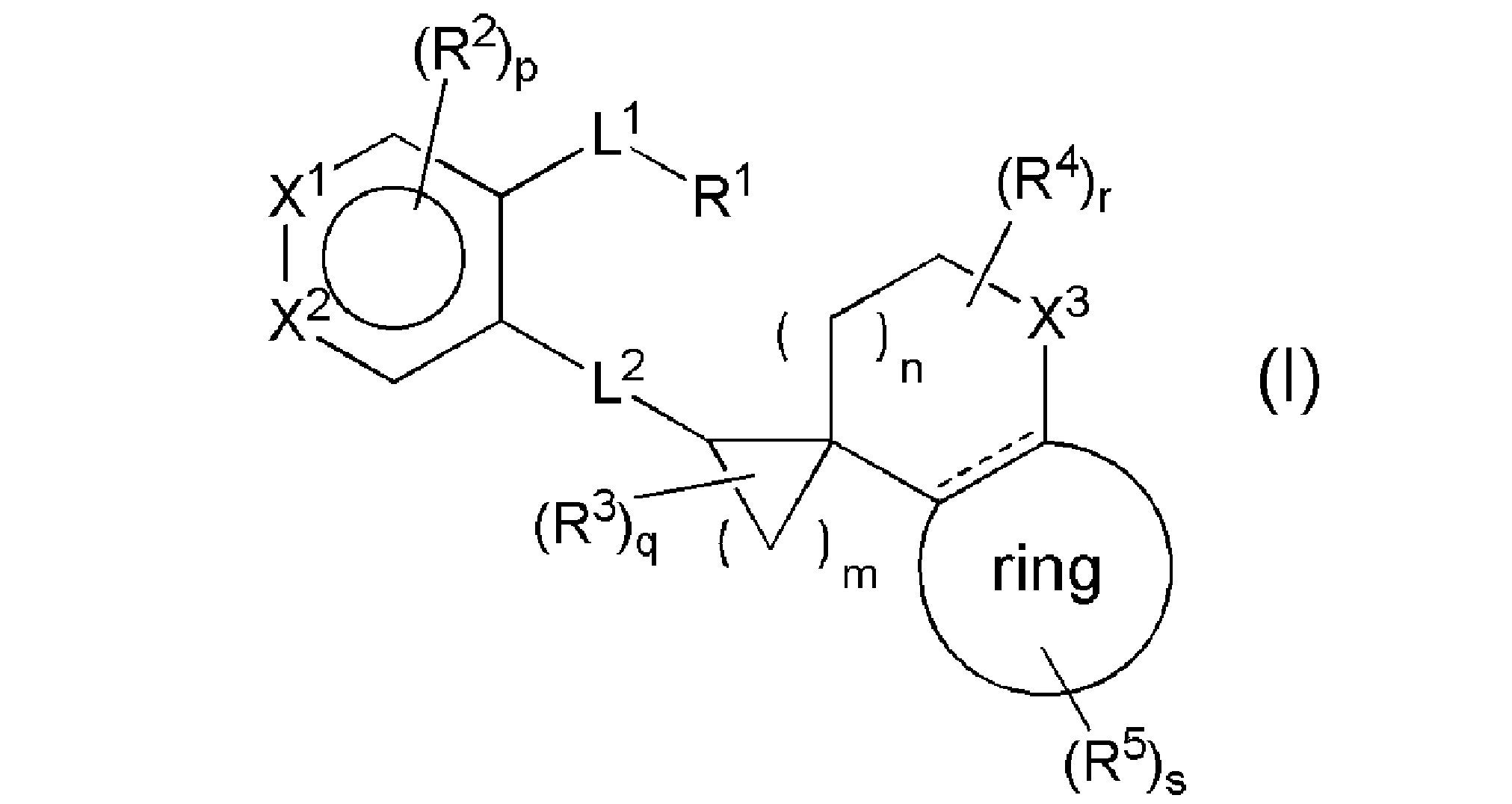
(式中、
R1はCOOR8、テトラゾール、SO3H、SO2NH2、SO2NHR8-1、CONHSO2R8-1、SO2NHCOR8-1、またはヒドロキサム酸を表し、
R8は水素原子、C1-4アルキル、またはベンジルを表し、
R8-1は、C1-4アルキル、C1-4ハロアルキル、C3-10炭素環、または3-10員複素環を表し、各々の該C3-10炭素環および3-10員複素環は、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリル(「-CN」を示す;以下同じ)で置換されていてもよく、
L1は、C1-5アルキレン、C2-5アルケニレン、またはC2-5アルキニレンを表し、
R2は、ハロゲン、C1-4アルキル、C1-4アルコキシ、C1-4アルキルチオ、C2-4アルケニル、C2-4アルキニル、-O(C1-4ハロアルキル)、-S(C1-4ハロアルキル)、-C(O)(C1-4アルキル)、-SO2(C1-4アルキル)、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、-NHC(O)(C1-4アルキル)、-N(C1-4アルキル)C(O)(C1-4アルキル)、-NHSO2(C1-4アルキル)、-N(C1-4アルキル)SO2(C1-4アルキル)、-SO2NH(C1-4アルキル)、-SO2N(C1-4アルキル)2、-NR17R17、ニトロ、ニトリル、水酸基、アルデヒド(ホルミルを示す;以下同じ)、またはカルボキシルを表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
該R2における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
X1はCR6または窒素原子を表し、R6は水素原子またはR2を表し、
X2はCR7または窒素原子を表し、R7は、水素原子、R2、または-L3-R9を表し、L3は、メチレン、酸素原子、または酸化されていてもよい硫黄原子を表し、R9は、ハロゲン、C1-4アルキル、およびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい4-10員複素環を表し、
L2は、-CH2CH2-、-CH=CH-、-CH2O-、-OCH2-、-CH2S-、-SCH2-、-CH2S(O)-、-S(O)CH2-、-CH2SO2-、-SO2CH2-、-CH2NH-、-NHCH2-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-を表し、
R3はC1-4アルキルまたはハロゲンを表し、
R4は、ハロゲン、C1-4アルキル、またはC1-4ハロアルキルを表し、
X3は、メチレン、酸素原子、酸化されていてもよい硫黄原子、またはNR10を表し、R10は、C1-4アルキル、-C(O)(C1-4アルキル)、-C(O)O(C1-4アルキル)、または-SO2(C1-4アルキル)を表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
ringはベンゼン環または5-6員単環式芳香族複素環を表し、
R1はCOOR8、テトラゾール、SO3H、SO2NH2、SO2NHR8-1、CONHSO2R8-1、SO2NHCOR8-1、またはヒドロキサム酸を表し、
R8は水素原子、C1-4アルキル、またはベンジルを表し、
R8-1は、C1-4アルキル、C1-4ハロアルキル、C3-10炭素環、または3-10員複素環を表し、各々の該C3-10炭素環および3-10員複素環は、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリル(「-CN」を示す;以下同じ)で置換されていてもよく、
L1は、C1-5アルキレン、C2-5アルケニレン、またはC2-5アルキニレンを表し、
R2は、ハロゲン、C1-4アルキル、C1-4アルコキシ、C1-4アルキルチオ、C2-4アルケニル、C2-4アルキニル、-O(C1-4ハロアルキル)、-S(C1-4ハロアルキル)、-C(O)(C1-4アルキル)、-SO2(C1-4アルキル)、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、-NHC(O)(C1-4アルキル)、-N(C1-4アルキル)C(O)(C1-4アルキル)、-NHSO2(C1-4アルキル)、-N(C1-4アルキル)SO2(C1-4アルキル)、-SO2NH(C1-4アルキル)、-SO2N(C1-4アルキル)2、-NR17R17、ニトロ、ニトリル、水酸基、アルデヒド(ホルミルを示す;以下同じ)、またはカルボキシルを表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
該R2における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
X1はCR6または窒素原子を表し、R6は水素原子またはR2を表し、
X2はCR7または窒素原子を表し、R7は、水素原子、R2、または-L3-R9を表し、L3は、メチレン、酸素原子、または酸化されていてもよい硫黄原子を表し、R9は、ハロゲン、C1-4アルキル、およびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい4-10員複素環を表し、
L2は、-CH2CH2-、-CH=CH-、-CH2O-、-OCH2-、-CH2S-、-SCH2-、-CH2S(O)-、-S(O)CH2-、-CH2SO2-、-SO2CH2-、-CH2NH-、-NHCH2-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-を表し、
R3はC1-4アルキルまたはハロゲンを表し、
R4は、ハロゲン、C1-4アルキル、またはC1-4ハロアルキルを表し、
X3は、メチレン、酸素原子、酸化されていてもよい硫黄原子、またはNR10を表し、R10は、C1-4アルキル、-C(O)(C1-4アルキル)、-C(O)O(C1-4アルキル)、または-SO2(C1-4アルキル)を表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
ringはベンゼン環または5-6員単環式芳香族複素環を表し、
は単結合または二重結合を表し、
R5は、(1)ハロゲン、(2)C1-4アルキル、(3)カルボキシル、(4)ニトリル、(5)-CONHR11、(6)-C(O)R12、(7)-OR14、(8)-S(O)tR15、(9)-CH2R16、(10)-NR17R17、(11)-NHCOR11、(12)C4-10炭素環、または(13)4-10員複素環を表し、該C4-10炭素環または4-10員複素環は1~3個のR18で置換されていてもよく、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、
R11は、C1-6アルキル、C3-6シクロアルキル、フェニル、または4-6員複素環を表し、R11は1~3個のR13で置換されていてもよく、該R13が複数である場合、R13はそれぞれ独立して同一でも異なっていてもよく、
R13は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、または4-6員複素環を表し、
R20およびR21は、それぞれ独立して、水素原子またはC1-4アルキルを表し、
R12は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環を表し、該C3-6シクロアルキル、ベンゼン、または4-6員複素環は、それぞれ独立して、ハロゲン、C1-4アルキル、またはC1-4アルコキシで置換されていてもよく、
R14は、水素原子、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、該C1-6アルキルは1~3個のR19で置換されていてもよく、該R19が複数である場合、R19はそれぞれ独立して同一でも異なっていてもよく、
R19は、C1-4アルコキシ、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、またはC1-4アルキルおよびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい5-6員単環式芳香族複素環を表し、
該R19における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
R15は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、
R16は水酸基またはC1-4アルコキシを表し、
R17は、それぞれ独立して、水素原子、C1-6アルキル、またはC3-6シクロアルキルを表し、
R18は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、(C1-4アルキル)SO2-、4-6員複素環、(C1-4アルキル)NH-、または(C1-4アルキル)2N-を表し、
該R18における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
mは1~4の整数を表し、nは0~4の整数を表し、pは0~2の整数を表し、qは0~6の整数を表し、rは0~6の整数を表し、sは0~4の整数を表し、tは0~2の整数を表し、
ただし、p、q、r、およびsがそれぞれ2以上の整数を表す場合、R2、R3、R4、およびR5は、それぞれ独立して、同一でも異なっていてもよい。)で示される化合物、またはその塩である、前記[1]または[2]に記載の剤。
[4] 標準療法がベバシズマブおよびXELOX療法である、前記[1]~[3]のいずれかに記載の剤。
[5] がんが大腸がんである、前記[1]~[4]のいずれかに記載の剤。
[6] 大腸がんが、結腸・直腸がんである、前記[5]記載の剤。
[7] 結腸・直腸がんが、根治切除不能な進行または再発の結腸・直腸がん(好ましくは、治療歴のない根治切除不能な進行または再発の結腸・直腸がん)である、前記[6]記載の剤。
R5は、(1)ハロゲン、(2)C1-4アルキル、(3)カルボキシル、(4)ニトリル、(5)-CONHR11、(6)-C(O)R12、(7)-OR14、(8)-S(O)tR15、(9)-CH2R16、(10)-NR17R17、(11)-NHCOR11、(12)C4-10炭素環、または(13)4-10員複素環を表し、該C4-10炭素環または4-10員複素環は1~3個のR18で置換されていてもよく、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、
R11は、C1-6アルキル、C3-6シクロアルキル、フェニル、または4-6員複素環を表し、R11は1~3個のR13で置換されていてもよく、該R13が複数である場合、R13はそれぞれ独立して同一でも異なっていてもよく、
R13は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、または4-6員複素環を表し、
R20およびR21は、それぞれ独立して、水素原子またはC1-4アルキルを表し、
R12は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環を表し、該C3-6シクロアルキル、ベンゼン、または4-6員複素環は、それぞれ独立して、ハロゲン、C1-4アルキル、またはC1-4アルコキシで置換されていてもよく、
R14は、水素原子、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、該C1-6アルキルは1~3個のR19で置換されていてもよく、該R19が複数である場合、R19はそれぞれ独立して同一でも異なっていてもよく、
R19は、C1-4アルコキシ、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、またはC1-4アルキルおよびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい5-6員単環式芳香族複素環を表し、
該R19における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
R15は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、
R16は水酸基またはC1-4アルコキシを表し、
R17は、それぞれ独立して、水素原子、C1-6アルキル、またはC3-6シクロアルキルを表し、
R18は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、(C1-4アルキル)SO2-、4-6員複素環、(C1-4アルキル)NH-、または(C1-4アルキル)2N-を表し、
該R18における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
mは1~4の整数を表し、nは0~4の整数を表し、pは0~2の整数を表し、qは0~6の整数を表し、rは0~6の整数を表し、sは0~4の整数を表し、tは0~2の整数を表し、
ただし、p、q、r、およびsがそれぞれ2以上の整数を表す場合、R2、R3、R4、およびR5は、それぞれ独立して、同一でも異なっていてもよい。)で示される化合物、またはその塩である、前記[1]または[2]に記載の剤。
[4] 標準療法がベバシズマブおよびXELOX療法である、前記[1]~[3]のいずれかに記載の剤。
[5] がんが大腸がんである、前記[1]~[4]のいずれかに記載の剤。
[6] 大腸がんが、結腸・直腸がんである、前記[5]記載の剤。
[7] 結腸・直腸がんが、根治切除不能な進行または再発の結腸・直腸がん(好ましくは、治療歴のない根治切除不能な進行または再発の結腸・直腸がん)である、前記[6]記載の剤。
[8] EP4拮抗薬が、一般式(I-2)

(式中、R2aはハロゲンを表し、R6aは水素原子またはハロゲンを表し、qaは0~3の整数を表し、raは0~4の整数を表し、その他の記号は請求項1と同じ意味を表す。)で示される化合物またはその塩である、前記[1]~[7]のいずれかに記載の剤。
[9] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[1]~[8]のいずれかに記載の剤。
[10] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[9]に記載の剤。
[11] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[9]に記載の剤。
[12] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[1]~[11]のいずれかに記載の剤。
[13] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[12]に記載の剤。
[9] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[1]~[8]のいずれかに記載の剤。
[10] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[9]に記載の剤。
[11] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[9]に記載の剤。
[12] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[1]~[11]のいずれかに記載の剤。
[13] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[12]に記載の剤。
[14] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[13]に記載の剤。
[15] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003またはCX-072である、前記[12]に記載の剤。
[16] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[12]に記載の剤。
[17] 抗PD-1抗体が、Nivolumabである前記[13]に記載の剤。
[15] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003またはCX-072である、前記[12]に記載の剤。
[16] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[12]に記載の剤。
[17] 抗PD-1抗体が、Nivolumabである前記[13]に記載の剤。
[18] 抗PD-1抗体が、Pembrolizumabである前記[13]に記載の剤。
[19] 抗PD-1抗体が、Cemiplimabである前記[13]に記載の剤。
[20] 抗PD-L1抗体が、Avelumabである前記[15]に記載の剤。
[21] 抗PD-L1抗体が、Atezolizumabである前記[15]に記載の剤。
[22] 抗PD-L1抗体が、Durvalumabである前記[15]に記載の剤。
[23] 抗CTLA-4抗体が、Ipilimumabである前記[16]に記載の剤。
[24] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[17]に記載の剤。
[25] Nivolumabとして1回360mgを3週間間隔で静脈内投与されることを特徴とする、前記[17]に記載の剤。
[26] Nivolumabとして1回360mgを3週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[17]に記載の剤。
[27] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔または1回400mgを6週間間隔で投与されることを特徴とする、前記[18]に記載の剤。
[28] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[19]に記載の剤。
[29] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[20]に記載の剤。
[30] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[21]に記載の剤。
[31] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[22]に記載の剤。
[32] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[23]に記載の剤。
[19] 抗PD-1抗体が、Cemiplimabである前記[13]に記載の剤。
[20] 抗PD-L1抗体が、Avelumabである前記[15]に記載の剤。
[21] 抗PD-L1抗体が、Atezolizumabである前記[15]に記載の剤。
[22] 抗PD-L1抗体が、Durvalumabである前記[15]に記載の剤。
[23] 抗CTLA-4抗体が、Ipilimumabである前記[16]に記載の剤。
[24] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[17]に記載の剤。
[25] Nivolumabとして1回360mgを3週間間隔で静脈内投与されることを特徴とする、前記[17]に記載の剤。
[26] Nivolumabとして1回360mgを3週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[17]に記載の剤。
[27] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔または1回400mgを6週間間隔で投与されることを特徴とする、前記[18]に記載の剤。
[28] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[19]に記載の剤。
[29] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[20]に記載の剤。
[30] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[21]に記載の剤。
[31] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[22]に記載の剤。
[32] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[23]に記載の剤。
[33] 当該ベバシズマブおよびXELOX療法が、
(i)ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、
(ii)オキサリプラチン 130mg/m2を3週間間隔で投与する、および
(iii)カペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、前記[1]~[32]のいずれかに記載の剤。
[34] ベバシズマブ、オキサリプラチンおよびカペシタビンの投与を同日に開始する療法である、前記[1]~[33]のいずれかに記載の剤。
[35] ベバシズマブ、オキサリプラチン、カペシタビン、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[1]~[34]のいずれかに記載の剤。
[36] ベバシズマブおよびXELOX療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)当該ベバシズマブおよびXELOX療法が、ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、オキサリプラチン 130mg/m2を3週間間隔で投与する、およびカペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、剤。
(i)ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、
(ii)オキサリプラチン 130mg/m2を3週間間隔で投与する、および
(iii)カペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、前記[1]~[32]のいずれかに記載の剤。
[34] ベバシズマブ、オキサリプラチンおよびカペシタビンの投与を同日に開始する療法である、前記[1]~[33]のいずれかに記載の剤。
[35] ベバシズマブ、オキサリプラチン、カペシタビン、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[1]~[34]のいずれかに記載の剤。
[36] ベバシズマブおよびXELOX療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)当該ベバシズマブおよびXELOX療法が、ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、オキサリプラチン 130mg/m2を3週間間隔で投与する、およびカペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、剤。
[37] ベバシズマブおよびXELOX療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)当該ベバシズマブおよびXELOX療法が、ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、オキサリプラチン 130mg/m2を3週間間隔で投与する、およびカペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、剤。
[38] ベバシズマブ、オキサリプラチン、カペシタビン、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である、前記[36]または[37]記載の剤。
[39] がんが大腸がんである、前記[36]~[38]のいずれかに記載の剤。
[40] 大腸がんが、結腸・直腸がんである、前記[39]記載の剤。
[41] 結腸・直腸がんが、根治切除不能な進行または再発の結腸・直腸がん(好ましくは、治療歴のない根治切除不能な進行または再発の結腸・直腸がん)である、前記[40]記載の剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)当該ベバシズマブおよびXELOX療法が、ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、オキサリプラチン 130mg/m2を3週間間隔で投与する、およびカペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、剤。
[38] ベバシズマブ、オキサリプラチン、カペシタビン、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である、前記[36]または[37]記載の剤。
[39] がんが大腸がんである、前記[36]~[38]のいずれかに記載の剤。
[40] 大腸がんが、結腸・直腸がんである、前記[39]記載の剤。
[41] 結腸・直腸がんが、根治切除不能な進行または再発の結腸・直腸がん(好ましくは、治療歴のない根治切除不能な進行または再発の結腸・直腸がん)である、前記[40]記載の剤。
[2-1] 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤。
[2-2] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[2-1]に記載の剤。
[2-3] 術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤。
[2-4] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[2-1]~[2-3]のいずれかに記載の剤。
[2-5] がんが大腸がん(好ましくは結腸・直腸がん)である、前記[2-1]~[2-4]のいずれかに記載の剤。
[2-6] 大腸がんが、治癒切除可能な局所進行直腸がんである、前記[2-5]に記載の剤。
[2-7] 免疫チェックポイント阻害物質および/またはEP4拮抗薬が術前化学放射線療法(CRT)後の術前補助療法として使用される、前記[2-1]~[2-6]の何れかに記載の剤。
[2-8] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[2-1]~[2-7]のいずれかに記載の剤。
[2-9] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[2-1]~[2-8]のいずれかに記載の剤。
[2-10] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[2-9]に記載の剤。
[2-11] EP4拮抗薬として1回20mgまたは40mg(好ましくは、1回40mg)を1日1回経口投与されることを特徴とする、前記[2-9]に記載の剤。
[2-12] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[2-2]~[2-11]のいずれかに記載の剤。
[2-13] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[2-12]に記載の剤。
[2-2] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[2-1]に記載の剤。
[2-3] 術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤。
[2-4] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[2-1]~[2-3]のいずれかに記載の剤。
[2-5] がんが大腸がん(好ましくは結腸・直腸がん)である、前記[2-1]~[2-4]のいずれかに記載の剤。
[2-6] 大腸がんが、治癒切除可能な局所進行直腸がんである、前記[2-5]に記載の剤。
[2-7] 免疫チェックポイント阻害物質および/またはEP4拮抗薬が術前化学放射線療法(CRT)後の術前補助療法として使用される、前記[2-1]~[2-6]の何れかに記載の剤。
[2-8] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[2-1]~[2-7]のいずれかに記載の剤。
[2-9] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[2-1]~[2-8]のいずれかに記載の剤。
[2-10] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[2-9]に記載の剤。
[2-11] EP4拮抗薬として1回20mgまたは40mg(好ましくは、1回40mg)を1日1回経口投与されることを特徴とする、前記[2-9]に記載の剤。
[2-12] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[2-2]~[2-11]のいずれかに記載の剤。
[2-13] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[2-12]に記載の剤。
[2-14] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[2-13]に記載の剤。
[2-15] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003またはCX-072である、前記[2-12]に記載の剤。
[2-16] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[2-12]に記載の剤。
[2-17] 抗PD-1抗体が、Nivolumabである前記[2-13]に記載の剤。
[2-18] 抗PD-1抗体が、Pembrolizumabである前記[2-13]に記載の剤。
[2-19] 抗PD-1抗体が、Cemiplimabである前記[2-13]に記載の剤。
[2-20] 抗PD-L1抗体が、Avelumabである前記[2-15]に記載の剤。
[2-21] 抗PD-L1抗体が、Atezolizumabである前記[2-15]に記載の剤。
[2-22] 抗PD-L1抗体が、Durvalumabである前記[2-15]に記載の剤。
[2-23] 抗CTLA-4抗体が、Ipilimumabである前記[2-16]に記載の剤。
[2-16] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[2-12]に記載の剤。
[2-17] 抗PD-1抗体が、Nivolumabである前記[2-13]に記載の剤。
[2-18] 抗PD-1抗体が、Pembrolizumabである前記[2-13]に記載の剤。
[2-19] 抗PD-1抗体が、Cemiplimabである前記[2-13]に記載の剤。
[2-20] 抗PD-L1抗体が、Avelumabである前記[2-15]に記載の剤。
[2-21] 抗PD-L1抗体が、Atezolizumabである前記[2-15]に記載の剤。
[2-22] 抗PD-L1抗体が、Durvalumabである前記[2-15]に記載の剤。
[2-23] 抗CTLA-4抗体が、Ipilimumabである前記[2-16]に記載の剤。
[2-24] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[2-17]に記載の剤。
[2-25] Nivolumabとして1回240mgを2週間間隔で静脈内投与されることを特徴とする、前記[2-17]に記載の剤。
[2-26] Nivolumabとして1回240mgを2週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[2-17]に記載の剤。
[2-27] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔で投与されることを特徴とする、前記[2-18]に記載の剤。
[2-28] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[2-19]に記載の剤。
[2-29] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[2-20]に記載の剤。
[2-30] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[2-21]に記載の剤。
[2-31] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[2-22]に記載の剤。
[2-32] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[2-23]に記載の剤。
[2-33] 術前化学放射線療法終了後14日以内に投与を開始する、前記[2-1]~[2-32]のいずれかに記載の剤。
[2-25] Nivolumabとして1回240mgを2週間間隔で静脈内投与されることを特徴とする、前記[2-17]に記載の剤。
[2-26] Nivolumabとして1回240mgを2週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[2-17]に記載の剤。
[2-27] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔で投与されることを特徴とする、前記[2-18]に記載の剤。
[2-28] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[2-19]に記載の剤。
[2-29] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[2-20]に記載の剤。
[2-30] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[2-21]に記載の剤。
[2-31] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[2-22]に記載の剤。
[2-32] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[2-23]に記載の剤。
[2-33] 術前化学放射線療法終了後14日以内に投与を開始する、前記[2-1]~[2-32]のいずれかに記載の剤。
[2-34] 手術を、EP4拮抗薬の最終投与日の7日以降および免疫チェックポイント阻害物質の最終投与日の14日後以降、かつ術前化学放射線療法終了後14週間以内に行う、前記[2-2]~[2-33]のいずれかに記載の剤。
[2-35] 術前化学放射線療法終了後の画像診断で遠隔転移を認めなかった患者に投与される、前記[2-1]~[2-34]のいずれかに記載の剤。
[2-36] 術前化学放射線療法終了後の画像診断で遠隔転移を認めず、治癒切除が可能な患者に投与される、前記[2-1]~[2-35]のいずれかに記載の剤。
[2-37] 術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである、前記[2-1]~[2-36]のいずれかに記載の剤。
[2-38] 術前化学放射線療法が、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である、前記[2-1]~[2-37]のいずれかに記載の剤。
[2-39] 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含むがん(好ましくは大腸がん、さらに好ましくは結腸・直腸がん(好ましくは、治癒切除可能な局所進行直腸がん))の進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg、さらに好ましくは40mg)を1日1回経口投与され、
(ii)術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである(好ましくは、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である)、
剤。
[2-40] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[2-39]に記載の剤であって、免疫チェックポイント阻害物質がNivolumabであり、抗Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回240mgを2週間間隔)で投与される、剤。
[2-35] 術前化学放射線療法終了後の画像診断で遠隔転移を認めなかった患者に投与される、前記[2-1]~[2-34]のいずれかに記載の剤。
[2-36] 術前化学放射線療法終了後の画像診断で遠隔転移を認めず、治癒切除が可能な患者に投与される、前記[2-1]~[2-35]のいずれかに記載の剤。
[2-37] 術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである、前記[2-1]~[2-36]のいずれかに記載の剤。
[2-38] 術前化学放射線療法が、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である、前記[2-1]~[2-37]のいずれかに記載の剤。
[2-39] 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含むがん(好ましくは大腸がん、さらに好ましくは結腸・直腸がん(好ましくは、治癒切除可能な局所進行直腸がん))の進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg、さらに好ましくは40mg)を1日1回経口投与され、
(ii)術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである(好ましくは、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である)、
剤。
[2-40] さらに免疫チェックポイント阻害物質と組み合わせて投与される、前記[2-39]に記載の剤であって、免疫チェックポイント阻害物質がNivolumabであり、抗Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回240mgを2週間間隔)で投与される、剤。
[2-41]術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含む大腸がんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg、さらに好ましくは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回240mgを2週間間隔)で投与され、および(iii)術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである(好ましくは、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である)、
剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg、さらに好ましくは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回240mgを2週間間隔)で投与され、および(iii)術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである(好ましくは、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である)、
剤。
[3-1] 標準療法がFOLFIRINOX療法またはその減量レジメンである、前記[1]または[2]に記載の剤。
[3-2] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[3-1]に記載の剤。
[3-3] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[3-1]または[3-2]に記載の剤。
[3-4] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[3-3]に記載の剤。
[3-5] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[3-1]~[3-4]のいずれかに記載の剤。
[3-6] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[3-1]~[3-5]のいずれかに記載の剤。
[3-7] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[3-1]~[3-6]のいずれかに記載の剤。
[3-8] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[3-7]に記載の剤。
[3-9] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[3-7]に記載の剤。
[3-10] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[3-1]~[3-9]のいずれかに記載の剤。
[3-11] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[3-10]に記載の剤。
[3-2] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[3-1]に記載の剤。
[3-3] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[3-1]または[3-2]に記載の剤。
[3-4] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[3-3]に記載の剤。
[3-5] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[3-1]~[3-4]のいずれかに記載の剤。
[3-6] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[3-1]~[3-5]のいずれかに記載の剤。
[3-7] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[3-1]~[3-6]のいずれかに記載の剤。
[3-8] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[3-7]に記載の剤。
[3-9] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[3-7]に記載の剤。
[3-10] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[3-1]~[3-9]のいずれかに記載の剤。
[3-11] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[3-10]に記載の剤。
[3-12] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[3-11]に記載の剤。
[3-13] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[3-10]に記載の剤。
[3-14] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[3-10]に記載の剤。
[3-15] 抗PD-1抗体が、Nivolumabである前記[3-11]に記載の剤。
[3-16] 抗PD-1抗体が、Pembrolizumabである前記[3-11]に記載の剤。
[3-17] 抗PD-1抗体が、Cemiplimabである前記[3-11]に記載の剤。
[3-18] 抗PD-L1抗体が、Avelumabである前記[3-13]に記載の剤。
[3-19] 抗PD-L1抗体が、Atezolizumabである前記[3-13]に記載の剤。
[3-20] 抗PD-L1抗体が、Durvalumabである前記[3-13]に記載の剤。
[3-21] 抗CTLA-4抗体が、Ipilimumabである前記[3-14]に記載の剤。
[3-13] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[3-10]に記載の剤。
[3-14] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[3-10]に記載の剤。
[3-15] 抗PD-1抗体が、Nivolumabである前記[3-11]に記載の剤。
[3-16] 抗PD-1抗体が、Pembrolizumabである前記[3-11]に記載の剤。
[3-17] 抗PD-1抗体が、Cemiplimabである前記[3-11]に記載の剤。
[3-18] 抗PD-L1抗体が、Avelumabである前記[3-13]に記載の剤。
[3-19] 抗PD-L1抗体が、Atezolizumabである前記[3-13]に記載の剤。
[3-20] 抗PD-L1抗体が、Durvalumabである前記[3-13]に記載の剤。
[3-21] 抗CTLA-4抗体が、Ipilimumabである前記[3-14]に記載の剤。
[3-22] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[3-15]に記載の剤。
[3-23] Nivolumabとして1回480mgを4週間間隔で静脈内投与されることを特徴とする、前記[3-15]に記載の剤。
[3-24] Nivolumabとして1回480mgを4週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[3-15]に記載の剤。
[3-25] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔または1回400mgを6週間間隔で投与されることを特徴とする、前記[3-16]に記載の剤。
[3-26] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[3-17]に記載の剤。
[3-27] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[3-18]に記載の剤。
[3-28] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[3-19]に記載の剤。
[3-29] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与または1回1500mgを4週間間隔で4回静脈内投与されることを特徴とする、前記[3-20]に記載の剤。
[3-23] Nivolumabとして1回480mgを4週間間隔で静脈内投与されることを特徴とする、前記[3-15]に記載の剤。
[3-24] Nivolumabとして1回480mgを4週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[3-15]に記載の剤。
[3-25] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔または1回400mgを6週間間隔で投与されることを特徴とする、前記[3-16]に記載の剤。
[3-26] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[3-17]に記載の剤。
[3-27] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[3-18]に記載の剤。
[3-28] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[3-19]に記載の剤。
[3-29] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与または1回1500mgを4週間間隔で4回静脈内投与されることを特徴とする、前記[3-20]に記載の剤。
[3-30] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与または1回1mg/kg(体重)を6週間間隔で投与されることを特徴とする、前記[3-21]に記載の剤。
[3-31] FOLFIRINOX療法またはその減量レジメンが、(i)オキサリプラチン、(ii)レボホリナートカルシウム、(iii)イリノテカン塩酸塩水和物、(iv)フルオロウラシルを組み合わせて投与する、療法である、前記[3-1]~[3-30]のいずれかに記載の剤。
[3-32] FOLFIRINOX療法が、
(i)オキサリプラチン 50~85mg/m2(好ましくは、50mg/m2、65mg/m2、85mg/m2、より好ましくは、85mg/m2)を静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を静脈内投与する、
(iii)イリノテカン塩酸塩水和物 90~180mg/m2(好ましくは、90mg/m2、120mg/m2、150mg/m2、180mg/m2、より好ましくは、180mg/m2)を静脈内投与する、
(iv)フルオロウラシル 400mg/m2を急速静脈内投与する、
(v)さらにフルオロウラシル 1200~2400mg/m2(好ましくは、1200mg/m2、1800mg/m2、2400mg/m2、より好ましくは、2400mg/m2)を静脈内持続投与する、療法である、前記[3-1]~[3-31]のいずれかに記載の剤。
[3-31] FOLFIRINOX療法またはその減量レジメンが、(i)オキサリプラチン、(ii)レボホリナートカルシウム、(iii)イリノテカン塩酸塩水和物、(iv)フルオロウラシルを組み合わせて投与する、療法である、前記[3-1]~[3-30]のいずれかに記載の剤。
[3-32] FOLFIRINOX療法が、
(i)オキサリプラチン 50~85mg/m2(好ましくは、50mg/m2、65mg/m2、85mg/m2、より好ましくは、85mg/m2)を静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を静脈内投与する、
(iii)イリノテカン塩酸塩水和物 90~180mg/m2(好ましくは、90mg/m2、120mg/m2、150mg/m2、180mg/m2、より好ましくは、180mg/m2)を静脈内投与する、
(iv)フルオロウラシル 400mg/m2を急速静脈内投与する、
(v)さらにフルオロウラシル 1200~2400mg/m2(好ましくは、1200mg/m2、1800mg/m2、2400mg/m2、より好ましくは、2400mg/m2)を静脈内持続投与する、療法である、前記[3-1]~[3-31]のいずれかに記載の剤。
[3-33] FOLFIRINOX療法の減量レジメンが、
(i)オキサリプラチン 50~85mg/m2(好ましくは、50mg/m2、65mg/m2、85mg/m2、より好ましくは、85mg/m2)を静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を静脈内投与する、
(iii)イリノテカン塩酸塩水和物 120~140mg/m2(好ましくは、140mg/m2)を静脈内投与する、
(iv)フルオロウラシル 1200~2400mg/m2(好ましくは、1200mg/m2、1800mg/m2、2400mg/m2、より好ましくは、2400mg/m2)を静脈内持続投与する、療法である、前記[3-1]~[3-31]のいずれかに記載の剤。
[3-34] FOLFIRINOX療法またはその減量レジメンが、一連の投与を2~4週間間隔(好ましくは、2週間間隔、3週間隔、4週間間隔、より好ましくは2週間間隔)で実施する、療法である、前記[3-1]~[3-33]のいずれかに記載の剤。
[3-35] FOLFIRINOX療法が、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)当該オキサリプラチンの投与終了後、レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(v)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、前記[3-1]~[3-32]、[3-34]のいずれかに記載の剤。
[3-36] FOLFIRINOX療法の減量レジメンが、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、前記[3-1]~[3-31]または[3-33]、[3-34]のいずれかに記載の剤。
[3-37] FOLFIRINOX療法もしくはその減量レジメン、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[3-1]~[3-36]のいずれかに記載の剤。
(i)オキサリプラチン 50~85mg/m2(好ましくは、50mg/m2、65mg/m2、85mg/m2、より好ましくは、85mg/m2)を静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を静脈内投与する、
(iii)イリノテカン塩酸塩水和物 120~140mg/m2(好ましくは、140mg/m2)を静脈内投与する、
(iv)フルオロウラシル 1200~2400mg/m2(好ましくは、1200mg/m2、1800mg/m2、2400mg/m2、より好ましくは、2400mg/m2)を静脈内持続投与する、療法である、前記[3-1]~[3-31]のいずれかに記載の剤。
[3-34] FOLFIRINOX療法またはその減量レジメンが、一連の投与を2~4週間間隔(好ましくは、2週間間隔、3週間隔、4週間間隔、より好ましくは2週間間隔)で実施する、療法である、前記[3-1]~[3-33]のいずれかに記載の剤。
[3-35] FOLFIRINOX療法が、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)当該オキサリプラチンの投与終了後、レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(v)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、前記[3-1]~[3-32]、[3-34]のいずれかに記載の剤。
[3-36] FOLFIRINOX療法の減量レジメンが、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、前記[3-1]~[3-31]または[3-33]、[3-34]のいずれかに記載の剤。
[3-37] FOLFIRINOX療法もしくはその減量レジメン、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[3-1]~[3-36]のいずれかに記載の剤。
[3-38] FOLFIRINOX療法またはその減量レジメン、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該FOLFIRINOX療法が、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(e)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法であり、当該FOLFIRINOX療法の減量レジメンが、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該FOLFIRINOX療法が、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(e)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法であり、当該FOLFIRINOX療法の減量レジメンが、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、剤。
[3-39] FOLFIRINOX療法またはその減量レジメン、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該FOLFIRINOX療法が、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(e)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法であり、当該FOLFIRINOX療法の減量レジメンが、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該FOLFIRINOX療法が、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(e)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法であり、当該FOLFIRINOX療法の減量レジメンが、
(a)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(b)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(c)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5かけて静脈内投与する、
(d)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、療法である、剤。
[3-40] FOLFIRINOX療法もしくはその減量レジメン、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である、[3-38]または[3-39]に記載の剤。
[3-41] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[3-38]~[3-40]に記載の剤。
[3-42] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[3-41]に記載の剤。
[3-43] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[3-38]~[3-42]のいずれかに記載の剤。
[3-41] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[3-38]~[3-40]に記載の剤。
[3-42] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[3-41]に記載の剤。
[3-43] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[3-38]~[3-42]のいずれかに記載の剤。
[4-1] 標準療法がゲムシタビンおよびナブパクリタキセル療法である、前記[1]または[2]に記載の剤。
[4-2] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[4-1]に記載の剤。
[4-3] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[4-1]または[4-2]に記載の剤。
[4-4] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[4-3]に記載の剤。
[4-5] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[4-1]~[4-4]のいずれかに記載の剤。
[4-6] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[4-1]~[4-5]のいずれかに記載の剤。
[4-7] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[4-1]~[4-6]のいずれかに記載の剤。
[4-8] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[4-7]に記載の剤。
[4-9] EP4拮抗薬として1回20mgまたは40mg(好ましくは、1回40mg)を1日1回経口投与されることを特徴とする、前記[4-7]に記載の剤。
[4-10] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[4-1]~[4-9]のいずれかに記載の剤。
[4-11] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[4-10]に記載の剤。
[4-2] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[4-1]に記載の剤。
[4-3] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[4-1]または[4-2]に記載の剤。
[4-4] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[4-3]に記載の剤。
[4-5] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[4-1]~[4-4]のいずれかに記載の剤。
[4-6] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[4-1]~[4-5]のいずれかに記載の剤。
[4-7] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[4-1]~[4-6]のいずれかに記載の剤。
[4-8] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[4-7]に記載の剤。
[4-9] EP4拮抗薬として1回20mgまたは40mg(好ましくは、1回40mg)を1日1回経口投与されることを特徴とする、前記[4-7]に記載の剤。
[4-10] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[4-1]~[4-9]のいずれかに記載の剤。
[4-11] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[4-10]に記載の剤。
[4-12] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[4-11]に記載の剤。
[4-13] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[4-10]に記載の剤。
[4-13] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[4-10]に記載の剤。
[4-14] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[4-10]に記載の剤。
[4-15] 抗PD-1抗体が、Nivolumabである前記[4-11]に記載の剤。
[4-16] 抗PD-1抗体が、Pembrolizumabである前記[4-11]に記載の剤。
[4-17] 抗PD-1抗体が、Cemiplimabである前記[4-11]に記載の剤。
[4-18] 抗PD-L1抗体が、Avelumabである前記[4-13]に記載の剤。
[4-19] 抗PD-L1抗体が、Atezolizumabである前記[4-13]に記載の剤。
[4-20] 抗PD-L1抗体が、Durvalumabである前記[4-13]に記載の剤。
[4-21] 抗CTLA-4抗体が、Ipilimumabである前記[4-14]に記載の剤。
[4-22] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[4-15]に記載の剤。
[4-23] Nivolumabとして1回480mgを4週間間隔で静脈内投与されることを特徴とする、前記[4-15]に記載の剤。
[4-24] Nivolumabとして1回480mgを4週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[4-15]に記載の剤。
[4-15] 抗PD-1抗体が、Nivolumabである前記[4-11]に記載の剤。
[4-16] 抗PD-1抗体が、Pembrolizumabである前記[4-11]に記載の剤。
[4-17] 抗PD-1抗体が、Cemiplimabである前記[4-11]に記載の剤。
[4-18] 抗PD-L1抗体が、Avelumabである前記[4-13]に記載の剤。
[4-19] 抗PD-L1抗体が、Atezolizumabである前記[4-13]に記載の剤。
[4-20] 抗PD-L1抗体が、Durvalumabである前記[4-13]に記載の剤。
[4-21] 抗CTLA-4抗体が、Ipilimumabである前記[4-14]に記載の剤。
[4-22] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[4-15]に記載の剤。
[4-23] Nivolumabとして1回480mgを4週間間隔で静脈内投与されることを特徴とする、前記[4-15]に記載の剤。
[4-24] Nivolumabとして1回480mgを4週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[4-15]に記載の剤。
[4-25] Pembrolizumabとして1回2mg/kg(体重)または1回200mgを3週間間隔で投与されることを特徴とする、前記[4-16]に記載の剤。
[4-26] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[4-17]に記載の剤。
[4-27] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[4-18]に記載の剤。
[4-28] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[4-19]に記載の剤。
[4-29] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[4-20]に記載の剤。
[4-30] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[4-21]に記載の剤。
[4-31] ゲムシタビンおよびナブパクリタキセル療法が、
(i)ゲムシタビン 600~1000mg/m2(好ましくは、600mg/m2、800mg/m2,1000mg/m2、より好ましくは、1000mg/m2)を静脈内投与する、
(ii)ナブパクリタキセル 75~125mg/m2(好ましくは、75mg/m2、100mg/m2、125mg/m2、より好ましくは125mg/m2)を静脈内投与する、
療法である、前記[4-1]~[4-30]のいずれかに記載の剤。
[4-32] ゲムシタビンおよびナブパクリタキセル療法が、一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、前記[4-1]~[4-31]のいずれかに記載の剤。
[4-33] ゲムシタビンおよびナブパクリタキセル療法が、
(i)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、
(ii)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、
療法である、前記[4-1]~[4-32]のいずれかに記載の剤。
[4-34] ゲムシタビン、ナブパクリタキセルの投与を同日に開始する療法である、前記[4-1]~[4-33]のいずれかに記載の剤。
[4-35] ゲムシタビン、ナブパクリタキセル、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[4-1]~[4-34]のいずれかに記載の剤。
[4-26] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[4-17]に記載の剤。
[4-27] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[4-18]に記載の剤。
[4-28] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[4-19]に記載の剤。
[4-29] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[4-20]に記載の剤。
[4-30] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与されることを特徴とする、前記[4-21]に記載の剤。
[4-31] ゲムシタビンおよびナブパクリタキセル療法が、
(i)ゲムシタビン 600~1000mg/m2(好ましくは、600mg/m2、800mg/m2,1000mg/m2、より好ましくは、1000mg/m2)を静脈内投与する、
(ii)ナブパクリタキセル 75~125mg/m2(好ましくは、75mg/m2、100mg/m2、125mg/m2、より好ましくは125mg/m2)を静脈内投与する、
療法である、前記[4-1]~[4-30]のいずれかに記載の剤。
[4-32] ゲムシタビンおよびナブパクリタキセル療法が、一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、前記[4-1]~[4-31]のいずれかに記載の剤。
[4-33] ゲムシタビンおよびナブパクリタキセル療法が、
(i)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、
(ii)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、
療法である、前記[4-1]~[4-32]のいずれかに記載の剤。
[4-34] ゲムシタビン、ナブパクリタキセルの投与を同日に開始する療法である、前記[4-1]~[4-33]のいずれかに記載の剤。
[4-35] ゲムシタビン、ナブパクリタキセル、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[4-1]~[4-34]のいずれかに記載の剤。
[4-36] ゲムシタビンおよびナブパクリタキセル療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該ゲムシタビンおよびナブパクリタキセル療法が、(a)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、(b)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、剤。
[4-37] ゲムシタビンおよびナブパクリタキセル療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該ゲムシタビンおよびナブパクリタキセル療法が、(a)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、(b)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、剤。
[4-38] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[4-36]または[4-37]に記載の剤。
[4-39] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[4-38]に記載の剤。
[4-40] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[4-36]~[4-39]のいずれかに記載の剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該ゲムシタビンおよびナブパクリタキセル療法が、(a)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、(b)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、剤。
[4-37] ゲムシタビンおよびナブパクリタキセル療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回480mgを4週間間隔)で投与され、
(iii)当該ゲムシタビンおよびナブパクリタキセル療法が、(a)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、(b)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、療法である、剤。
[4-38] がんが膵臓がん(好ましくは、膵管がん、より好ましくは浸潤性膵管がん)である、前記[4-36]または[4-37]に記載の剤。
[4-39] 膵臓がんが、遠隔転移を有する膵臓がんである、前記[4-38]に記載の剤。
[4-40] 遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者に投与されることを特徴とする、前記[4-36]~[4-39]のいずれかに記載の剤。
[5-1] 標準療法がドセタキセルおよび/またはラムシルマブ療法である、前記[1]または[2]に記載の剤。
[5-2] 標準療法がドセタキセル療法である、前記[5-1]に記載の剤。
[5-3] 標準療法がラムシルマブ療法である、前記[5-1]に記載の剤。
[5-4] 標準療法がドセタキセルおよびラムシルマブ療法である、前記[5-1]に記載の剤。
[5-5] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[5-1]~[5-4]に記載の剤。
[5-6] がんが肺がん(好ましくは、非小細胞肺がん)である、前記[5-1]~[5-5]のいずれかに記載の剤。
[5-7] 肺がんが、IV期または再発の非小細胞肺がん(好ましくは、抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん)である、前記[5-6]記載の剤。
[5-8]抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者に投与されることを特徴とする、前記[5-1]~[5-7]のいずれかに記載の剤。
[5-9] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[5-1]~[5-8]のいずれか一項に記載の剤。
[5-10] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[5-1]~[5-9]のいずれか一項に記載の剤。
[5-11] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[5-10]に記載の剤。
[5-12] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[5-10]に記載の剤。
[5-2] 標準療法がドセタキセル療法である、前記[5-1]に記載の剤。
[5-3] 標準療法がラムシルマブ療法である、前記[5-1]に記載の剤。
[5-4] 標準療法がドセタキセルおよびラムシルマブ療法である、前記[5-1]に記載の剤。
[5-5] EP4拮抗薬が前記[3]に記載の一般式(I)で示される化合物、またはその塩である、前記[5-1]~[5-4]に記載の剤。
[5-6] がんが肺がん(好ましくは、非小細胞肺がん)である、前記[5-1]~[5-5]のいずれかに記載の剤。
[5-7] 肺がんが、IV期または再発の非小細胞肺がん(好ましくは、抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん)である、前記[5-6]記載の剤。
[5-8]抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者に投与されることを特徴とする、前記[5-1]~[5-7]のいずれかに記載の剤。
[5-9] EP4拮抗薬が、前記[8]に記載の一般式(I-2)で示される化合物、またはその塩である、前記[5-1]~[5-8]のいずれか一項に記載の剤。
[5-10] EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、前記[5-1]~[5-9]のいずれか一項に記載の剤。
[5-11] EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、前記[5-10]に記載の剤。
[5-12] EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、前記[5-10]に記載の剤。
[5-13] 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、前記[5-1]~[5-12]のいずれかに記載の剤。
[5-14] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[5-13]に記載の剤。
[5-15] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[5-14]に記載の剤。
[5-16] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[5-13]に記載の剤。
[5-14] 免疫チェックポイント阻害物質が、抗PD-1抗体である、前記[5-13]に記載の剤。
[5-15] 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012(Retifanlimab)、AGEN2034(Balstilimab)、CS1003、HLX10(Serplulimab)、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226(Geptanolimab)、SSI-361、JY034、HX008、ISU106、ABBV181(Budigalimab)、BCD-100(Prolgolimab)、PF-06801591(Sasanlimab)、CX-188、JNJ-63723283(Cetrelimab)またはAB122(Zimberelimab)である、前記[5-14]に記載の剤。
[5-16] 免疫チェックポイント阻害物質が抗PD-L1抗体であって、抗PD-L1抗体が、Atezolizumab、Avelumab、Durvalumab、BMS-936559、STI-1014、KN035(Envafolimab)、LY3300054(Lodapolimab)、HLX20、SHR-1316、CS1001、MSB2311、BGB-A333、KL-A167、CK-301、AK106、AK104、ZKAB001、FAZ053、CBT-502、JS003およびCX-072である、前記[5-13]に記載の剤。
[5-17] 免疫チェックポイント阻害物質が抗CTLA-4抗体であって、抗CTLA-4抗体が、Ipilimumab、AGEN1884またはTremelimumabである、前記[5-13]に記載の剤。
[5-18] 抗PD-1抗体が、Nivolumabである前記[5-14]に記載の剤。
[5-19] 抗PD-1抗体が、Pembrolizumabである前記[5-14]に記載の剤。
[5-20] 抗PD-1抗体が、Cemiplimabである前記[5-14]に記載の剤。
[5-21] 抗PD-L1抗体が、Avelumabである前記[5-16]に記載の剤。
[5-22] 抗PD-L1抗体が、Atezolizumabである前記[5-16]に記載の剤。
[5-23] 抗PD-L1抗体が、Durvalumabである前記[5-16]に記載の剤。
[5-24] 抗CTLA-4抗体が、Ipilimumabである前記[5-17]に記載の剤。
[5-25] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[5-18]記載の剤。
[5-26] Nivolumabとして1回360mgを3週間間隔で静脈内投与されることを特徴とする、前記[5-18]記載の剤。
[5-27] Nivolumabとして1回360mgを3週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[5-18]記載の剤。
[5-28] Pembrolizumabとして1回2mg/kg(体重)、または1回200mgを3週間間隔、または1回400mgを6週間間隔で投与されることを特徴とする、前記[5-19]記載の剤。
[5-18] 抗PD-1抗体が、Nivolumabである前記[5-14]に記載の剤。
[5-19] 抗PD-1抗体が、Pembrolizumabである前記[5-14]に記載の剤。
[5-20] 抗PD-1抗体が、Cemiplimabである前記[5-14]に記載の剤。
[5-21] 抗PD-L1抗体が、Avelumabである前記[5-16]に記載の剤。
[5-22] 抗PD-L1抗体が、Atezolizumabである前記[5-16]に記載の剤。
[5-23] 抗PD-L1抗体が、Durvalumabである前記[5-16]に記載の剤。
[5-24] 抗CTLA-4抗体が、Ipilimumabである前記[5-17]に記載の剤。
[5-25] Nivolumabとして1回3mg/kg(体重)もしくは1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは、1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔)で投与されることを特徴とする、前記[5-18]記載の剤。
[5-26] Nivolumabとして1回360mgを3週間間隔で静脈内投与されることを特徴とする、前記[5-18]記載の剤。
[5-27] Nivolumabとして1回360mgを3週間間隔で約30分間かけて静脈内投与されることを特徴とする、前記[5-18]記載の剤。
[5-28] Pembrolizumabとして1回2mg/kg(体重)、または1回200mgを3週間間隔、または1回400mgを6週間間隔で投与されることを特徴とする、前記[5-19]記載の剤。
[5-29] Cemiplimabとして1回350mgを3週間間隔で投与されることを特徴とする、前記[5-20]記載の剤。
[5-30] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[5-21]記載の剤。
[5-31] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[5-22]記載の剤。
[5-32] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与、または1回1500mgを4週間間隔で4回静脈内投与されることを特徴とする、前記[5-23]に記載の剤。
[5-33] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与または1回1mg/kg(体重)を6週間間隔で投与されることを特徴とする、前記[5-24]に記載の剤。
[5-34] ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を静脈内投与する療法である、前記[5-1]、[5-2]、[5-4]~[5-33]のいずれかに記載の剤。
[5-35] ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、前記[5-1]、[5-2]、[5-4]~[5-34]のいずれかに記載の剤。
[5-36] ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を静脈内投与する療法である、前記[5-1]、[5-3]、[5-4]~[5-33]のいずれかに記載の剤。
[5-37] ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、前記[5-1]、[5-3]、[5-4]~[5-33]、[5-36]のいずれかに記載の剤。
[5-30] Avelumabとして1回10mg/kg(体重)を2週間間隔で投与されることを特徴とする、前記[5-21]記載の剤。
[5-31] Atezolizumabとして1回1200mgを3週間間隔で投与されることを特徴とする、前記[5-22]記載の剤。
[5-32] Durvalumabとして1回10mg/kg(体重)を2週間間隔で投与、または1回1500mgを4週間間隔で4回静脈内投与されることを特徴とする、前記[5-23]に記載の剤。
[5-33] Ipilimumabとして1回3mg/kg(体重)または1回1mg/kg(体重)を3週間間隔で4回静脈内投与または1回1mg/kg(体重)を6週間間隔で投与されることを特徴とする、前記[5-24]に記載の剤。
[5-34] ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を静脈内投与する療法である、前記[5-1]、[5-2]、[5-4]~[5-33]のいずれかに記載の剤。
[5-35] ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、前記[5-1]、[5-2]、[5-4]~[5-34]のいずれかに記載の剤。
[5-36] ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を静脈内投与する療法である、前記[5-1]、[5-3]、[5-4]~[5-33]のいずれかに記載の剤。
[5-37] ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、前記[5-1]、[5-3]、[5-4]~[5-33]、[5-36]のいずれかに記載の剤。
[5-38] ドセタキセルおよびラムシルマブ療法が、
(i)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を静脈内投与する、
(ii)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を静脈内投与する、
療法である、前記[5-1]、[5-4]~[5-33]のいずれかに記載の剤。
[5-39] ドセタキセルおよびラムシルマブ療法が、
(i)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(ii)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される、
療法である、前記[5-1]、[5-4]~[5-33]、[5-38]のいずれかに記載の剤。
[5-40] ドセタキセルおよび/またはラムシルマブ療法、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[5-1]~[5-39]のいずれかに記載の剤。
[5-41] ドセタキセル療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、剤。
(i)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を静脈内投与する、
(ii)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を静脈内投与する、
療法である、前記[5-1]、[5-4]~[5-33]のいずれかに記載の剤。
[5-39] ドセタキセルおよびラムシルマブ療法が、
(i)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(ii)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される、
療法である、前記[5-1]、[5-4]~[5-33]、[5-38]のいずれかに記載の剤。
[5-40] ドセタキセルおよび/またはラムシルマブ療法、免疫チェックポイント阻害物質およびEP4拮抗薬の投与を同日に開始する療法である、前記[5-1]~[5-39]のいずれかに記載の剤。
[5-41] ドセタキセル療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、剤。
[5-42] ドセタキセル療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセル療法が、ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは60mg/m2)を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、剤。
[5-43] ドセタキセル、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である(同日に投与される場合、好ましくは、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与後にドセタキセルが投与される)、前記[5-41]または[5-42]に記載の剤。
[5-44] ラムシルマブ療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-44] ラムシルマブ療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-45] ラムシルマブ療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-46] ラムシルマブ、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である(同日に投与される場合、好ましくは、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与後にラムシルマブが投与される)、前記[5-44]または[5-45]に記載の剤。
[5-47] ドセタキセルおよびラムシルマブ療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセルおよびラムシルマブ療法が、
(a)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(b)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ラムシルマブ療法が、ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-46] ラムシルマブ、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である(同日に投与される場合、好ましくは、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与後にラムシルマブが投与される)、前記[5-44]または[5-45]に記載の剤。
[5-47] ドセタキセルおよびラムシルマブ療法、並びに免疫チェックポイント阻害物質と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセルおよびラムシルマブ療法が、
(a)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(b)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-48] ドセタキセルおよびラムシルマブ療法、並びにEP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセルおよびラムシルマブ療法が、
(a)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(b)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
(i)EP4拮抗薬が4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩であり、EP4拮抗薬として1回5mg~40mg(好ましくは1回20mgまたは40mg)を1日1回経口投与され、
(ii)免疫チェックポイント阻害物質がNivolumabであり、Nivolumabとして1回240mgを2週間間隔で、1回360mgを3週間間隔で、または1回480mgを4週間間隔(好ましくは1回360mgを3週間間隔)で投与され、
(iii)ドセタキセルおよびラムシルマブ療法が、
(a)ドセタキセル 50~75mg/m2(好ましくは、50mg/m2、60mg/m2、75mg/m2、より好ましくは、60mg/m2)を60分以上かけて静脈内投与する、
(b)ラムシルマブ 6~10mg/kg(好ましくは、6mg/kg、8mg/kg、10mg/kg、より好ましくは、10mg/kg)を30~60分(好ましくは、60分)かけて静脈内投与し、当該一連の投与が3週間間隔で実施される療法である、剤。
[5-49] ドセタキセル、ラムシルマブ、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与を同日に開始する療法である(同日に投与される場合、好ましくは、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩、およびNivolumabの投与後にドセタキセルおよびラムシルマブが投与される)、前記[5-47]または[5-48]に記載の剤。
[5-50] がんが肺がん(好ましくは、非小細胞肺がん)である、前記[5-41]~[5-49]のいずれかに記載の剤。
[5-51] 肺がんが、IV期または再発の非小細胞肺がん(好ましくは、抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん)である、前記[5-50]記載の剤。
[5-52]抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者に投与されることを特徴とする、前記[5-41]~[5-51]のいずれかに記載の剤。
[5-50] がんが肺がん(好ましくは、非小細胞肺がん)である、前記[5-41]~[5-49]のいずれかに記載の剤。
[5-51] 肺がんが、IV期または再発の非小細胞肺がん(好ましくは、抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん)である、前記[5-50]記載の剤。
[5-52]抗PD-1抗体または抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者に投与されることを特徴とする、前記[5-41]~[5-51]のいずれかに記載の剤。
[6-1] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、および免疫チェックポイント阻害物質の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療に使用される、EP4拮抗薬。
[6-2] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療に使用される、免疫チェックポイント阻害物質。
[6-2] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療に使用される、免疫チェックポイント阻害物質。
[6-3] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、および免疫チェックポイント阻害物質の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療剤の製造における、EP4拮抗薬の使用。
[6-4] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療剤の製造における、免疫チェックポイント阻害物質の使用。
[6-5] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)の有効量と免疫チェックポイント阻害物質の有効量およびEP4拮抗薬の有効量とをがん治療を必要とする患者に、別々または同時に投与することを含むがんの進行抑制、再発抑制および/または治療方法。
[6-4] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療剤の製造における、免疫チェックポイント阻害物質の使用。
[6-5] 標準療法(好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(v)ドセタキセルおよび/またはラムシルマブ療法、より好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、(vii)ドセタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法、さらに好ましくは、(i)ベバシズマブおよびXELOX療法、(iii)FOLFIRINOX療法またはその減量レジメン、(iv)ゲムシタビンおよびナブパクリタキセル療法、または(vi)ドセタキセルおよびラムシルマブ療法)の有効量と免疫チェックポイント阻害物質の有効量およびEP4拮抗薬の有効量とをがん治療を必要とする患者に、別々または同時に投与することを含むがんの進行抑制、再発抑制および/または治療方法。
他に定義されない限り、本明細書中で使用される全ての技術的、科学的用語、および略語は、本発明の分野に属する当業者によって普通に理解されるものと同様の意味を有する。
また、本明細書において、明示的に引用される全ての特許文献および非特許文献もしくは参考文献の内容は、全て本明細書の一部としてここに引用し得る。
以下、実施例によって本発明を詳述するが、本発明はこれらに限定されるものではない。
一般式(I)に示されるEP4受容体拮抗薬として、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸(化合物A)を使用した。化合物Aは、公知の方法、例えばWO2016/111347の実施例2-13に記載された方法に従い製造することができる。
一般式(I)に示されるEP4受容体拮抗薬として、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸(化合物A)を使用した。化合物Aは、公知の方法、例えばWO2016/111347の実施例2-13に記載された方法に従い製造することができる。
実施例1:根治切除不能な進行または再発の結腸・直腸がん患者を対象に、一次治療として化合物A、Nivolumabおよび標準治療であるXELOX+ベバシズマブ療法を併用する非盲検非対照試験
本試験は、根治切除不能な進行または再発の結腸・直腸がん患者を対象に、一次治療として化合物A、NivolumabおよびXELOX+ベバシズマブ療法を併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、NivolumabおよびXELOX+ベバシズマブ療法との併用効果が評価できる。
(1)対象患者
根治切除不能な進行または再発の結腸・直腸がん患者
(2)患者選択基準
登録時に、根治切除不能な進行または再発の結腸・直腸がんに対する全身性抗悪性腫瘍剤による治療歴のない患者で、かつ化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
(3)患者除外基準
登録時に、化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関する各々の治験における患者除外基準、あるいは化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
本試験は、根治切除不能な進行または再発の結腸・直腸がん患者を対象に、一次治療として化合物A、NivolumabおよびXELOX+ベバシズマブ療法を併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、NivolumabおよびXELOX+ベバシズマブ療法との併用効果が評価できる。
(1)対象患者
根治切除不能な進行または再発の結腸・直腸がん患者
(2)患者選択基準
登録時に、根治切除不能な進行または再発の結腸・直腸がんに対する全身性抗悪性腫瘍剤による治療歴のない患者で、かつ化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
(3)患者除外基準
登録時に、化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関する各々の治験における患者除外基準、あるいは化合物A、NivolumabおよびXELOX+ベバシズマブ療法に関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
(4)用法・用量および投与期間
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、NivolumabおよびXELOX+ベバシズマブ療法を同日に投与する場合、化合物AおよびNivolumabを先に投与し、化合物AおよびNivolumabを投与した後、XELOX+ベバシズマブ療法を開始した。
[Nivolumab]
Nivolumab 360mgを3週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも14日以上空け、15日目以降に実施した。
[XELOX+ベバシズマブ療法]
ベバシズマブは7.5mg/kgを90分間かけて静脈内投与し、忍容性が良好であれば2回目は60分間、それ以降のサイクルでは30分間かけて静脈内投与した。オキサリプラチンは130mg/m2(体表面積)を2時間かけて静脈内投与した。カペシタビンは1000mg/m2を朝食後および夕食後の1日2回経口投与し、14日間経口投与後、7日間休薬した。なお、ベバシズマブ、オキサリプラチンおよびカペシタビンはそれぞれ市販品を用いた。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図1に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、NivolumabおよびXELOX+ベバシズマブ療法を同日に投与する場合、化合物AおよびNivolumabを先に投与し、化合物AおよびNivolumabを投与した後、XELOX+ベバシズマブ療法を開始した。
[Nivolumab]
Nivolumab 360mgを3週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも14日以上空け、15日目以降に実施した。
[XELOX+ベバシズマブ療法]
ベバシズマブは7.5mg/kgを90分間かけて静脈内投与し、忍容性が良好であれば2回目は60分間、それ以降のサイクルでは30分間かけて静脈内投与した。オキサリプラチンは130mg/m2(体表面積)を2時間かけて静脈内投与した。カペシタビンは1000mg/m2を朝食後および夕食後の1日2回経口投与し、14日間経口投与後、7日間休薬した。なお、ベバシズマブ、オキサリプラチンおよびカペシタビンはそれぞれ市販品を用いた。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図1に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
治療期は1サイクル21日間とし、初回の治験薬投与日をサイクル1の1日目とした。第2サイクル以降の各サイクルの1日目は、[21X(サイクル数-1)+1]日とした。化合物A、NivolumabおよびXELOX+ベバシズマブ療法についてそれぞれ上記用法・用量に従って、投与を開始し、化合物A、NivolumabおよびXELOX+ベバシズマブ療法についての投与基準、減量基準および減量時の投与量に従って投与を継続した。化合物A、NivolumabおよびXELOX+ベバシズマブ療法の投与終了(中止)時の評価が終了した時点を治療期終了とする。治験薬を投与したすべての被験者のうち、化合物A、NivolumabおよびXELOX+ベバシズマブ療法の投与を中止または終了した被験者は、投与終了(中止)時の評価を実施し、後観察期に移行する。後観察期終了後は、追跡調査を実施する。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準のすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[XELOX+ベバシズマブ療法の投与基準]
被験者は、毎回の投与開始時において、XELOX+ベバシズマブ療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準にすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[XELOX+ベバシズマブ療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[XELOX+ベバシズマブ療法の中止基準]
治療期において、XELOX+ベバシズマブ療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はXELOX+ベバシズマブ療法の投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準のすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[XELOX+ベバシズマブ療法の投与基準]
被験者は、毎回の投与開始時において、XELOX+ベバシズマブ療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準にすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[XELOX+ベバシズマブ療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[XELOX+ベバシズマブ療法の中止基準]
治療期において、XELOX+ベバシズマブ療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はXELOX+ベバシズマブ療法の投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
治験責任医師または治験分担医師は、RECISTガイドライン1.1版に準じて標的病変の腫瘍径を計測し、抗腫瘍効果を判定した。ベースライン評価は、治験薬投与前28日以内の最新の画像検査を用いて行い、RECISTガイドライン1.1版に定義される測定可能病変を一つ以上有することを確認した。治療期の画像診断は、第1サイクル1日目から6週後(+7日)から54週後(+7日)までは6週間間隔、それ以降は12週間間隔(±7日)で計測した。
なお、総合効果および最良総合効果とは、RECISTガイドライン1.1版に基づき評価された画像診断結果とした。
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏功期間(DOR)、(6)奏功に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移(CEAおよびCA19-9)
(1)奏効率
奏効率は、最良総合効果が完全奏功(complete responese、以下、「CR」と略記する。)または部分奏効(partial response、以下、「PR」と略記する。)と判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたは安定(stable disease、以下、「SD」と略記する。)と判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果が進行(progressive disease、以下、「PD」と略記する。)と判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏功期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏功に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏功期間(DOR)、(6)奏功に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移(CEAおよびCA19-9)
(1)奏効率
奏効率は、最良総合効果が完全奏功(complete responese、以下、「CR」と略記する。)または部分奏効(partial response、以下、「PR」と略記する。)と判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたは安定(stable disease、以下、「SD」と略記する。)と判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果が進行(progressive disease、以下、「PD」と略記する。)と判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏功期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏功に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
(8)標的病変の腫瘍径和の最大変化率
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
[安全性の評価項目]
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから10カ月が経過した時点において、PRと判断された症例は2例であった。
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから10カ月が経過した時点において、PRと判断された症例は2例であった。
実施例2:治癒切除可能な局所進行の直腸がんを対象に、術前化学放射線療法後の術前補助療法として、化合物Aまたは化合物AおよびNivolumabを併用する非盲検非対照試験
本試験は、治癒切除可能な局所進行直腸がんを対象とした術前化学放射線療法(CRT)後の術前補助療法として、化合物Aを単剤投与および/または化合物AとNivolumabを併用投与したときの有効性、薬物動態および安全性を検討することを目的とする。当該治験により、術前補助療法としての化合物Aおよび/または化合物AとNivolumabの併用の効果が評価できる。
(1)対象
治癒切除可能な局所進行直腸がん
(2)患者選択基準
登録時に、Nivolumabおよび化合物Aに関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準のすべてを満たす患者を選択した。なお、登録から治験薬の初回投与前までに基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず治験中止とする。
本試験は、治癒切除可能な局所進行直腸がんを対象とした術前化学放射線療法(CRT)後の術前補助療法として、化合物Aを単剤投与および/または化合物AとNivolumabを併用投与したときの有効性、薬物動態および安全性を検討することを目的とする。当該治験により、術前補助療法としての化合物Aおよび/または化合物AとNivolumabの併用の効果が評価できる。
(1)対象
治癒切除可能な局所進行直腸がん
(2)患者選択基準
登録時に、Nivolumabおよび化合物Aに関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準のすべてを満たす患者を選択した。なお、登録から治験薬の初回投与前までに基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず治験中止とする。
術前CRTとして、下記の条件を満たした治療が行われた患者に投与した。
・45Gy/25回の骨盤腔への照射と5.4Gy/3回の原発巣へのboost照射が行われた。
・カペシタビンとして1日1,650mg/m2(825mg/m21日2回)の用量で開始された。開始用量はゼローダ(登録商標)錠300の用法・用量のD法に基づいて決定。
・放射線との併用で放射線の照射回数である28回の75%に相当する期間でカペシタビンの内服が行われた(例:50.4Gy/28回照射の場合、21日間または朝夕の内服として42回以上の内服)。減量の有無は問わない。
・45Gy/25回の骨盤腔への照射と5.4Gy/3回の原発巣へのboost照射が行われた。
・カペシタビンとして1日1,650mg/m2(825mg/m21日2回)の用量で開始された。開始用量はゼローダ(登録商標)錠300の用法・用量のD法に基づいて決定。
・放射線との併用で放射線の照射回数である28回の75%に相当する期間でカペシタビンの内服が行われた(例:50.4Gy/28回照射の場合、21日間または朝夕の内服として42回以上の内服)。減量の有無は問わない。
術前CRTとしてカペシタビンおよび放射線照射による治療が終了後、14日間以内に治験薬投与を開始できる患者に投与される。
術前CRT終了後の画像診断で遠隔転移を認めず、治癒切除が可能な患者に投与される。なお、登録前の画像診断は、術前CRT終了14日前から登録日の範囲で撮影した画像に基づき診断することとする。
(3)患者除外基準
登録時に、化合物AおよびNivolumabに関する各々の治験における患者除外基準、あるいは化合物AおよびNivolumabに関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
(4)用法・用量および投与期間
[化合物A]
化合物Aを40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで投与を継続した。
[Nivolumab]
Nivolumabは 240mgを2週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。Nivolumab投与は、前回の投与の次の日から少なくとも10日以上空け、11日目以降に実施した。
[治験スケジュール]
本治験は、スクリーニング期、治療期、周術期および後観察期で構成される。治験デザインの概要を図2に示す。
(3)患者除外基準
登録時に、化合物AおよびNivolumabに関する各々の治験における患者除外基準、あるいは化合物AおよびNivolumabに関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とする。
(4)用法・用量および投与期間
[化合物A]
化合物Aを40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで投与を継続した。
[Nivolumab]
Nivolumabは 240mgを2週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。Nivolumab投与は、前回の投与の次の日から少なくとも10日以上空け、11日目以降に実施した。
[治験スケジュール]
本治験は、スクリーニング期、治療期、周術期および後観察期で構成される。治験デザインの概要を図2に示す。
スクリーニング期では、術前CRTを施された治癒切除可能な局所進行直腸がんで、本治験の対象として適格と判断した患者を登録し、治療期に移行する。
治療期では、術前CRT終了後14日以内に治験薬の投与を開始した。化合物A(40mg、1日1回(QD)、経口投与)を単剤で一定期間投与後、化合物A(40mg、QD、経口投与)とNivolumab(240mg、2週間間隔(Q2W)、静脈内投与)との併用で一定期間投与する群(コホート1)、および化合物A(40mg、QD、経口投与)とNivolumab(240mg、Q2W、静脈内投与)との併用で一定期間投与する群(コホート2)を設定した。なお、コホート1への組み入れ終了後、コホート2への組み入れを開始する。治験薬投与を完了後、すべての被験者は投与終了(中止)時検査を実施し、治療期から周術期に移行する。
周術期では、原則化合物Aの最終投与日の7日後以降およびNivolumabの最終投与日の14日後以降、かつ術前CRT終了後14週間以内に手術を実施する。なお、手術の時期については被験者の状態を最優先に考慮し、原疾患の増悪など、早急に手術が必要となった場合、有害事象に対する処置など、手術の延期が必要となった場合はこの限りでない。後観察期では、手術治療終了後30日あるいは治験責任医師または治験分担医師が手術による合併症が軽快したと判断した時点のいずれか遅い時点を起点日として、各種最終検査を行う。ただし、起点日までに臨床上の必要性から原疾患(直腸がん)に対する後治療を開始する場合は、後治療を開始する前に最終検査を行う。また、その後に追跡調査を実施する。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、ベネフィット・リスクのバランスを考慮し、投与継続による有用性が期待されると治験責任医師または治験分担医師が判断する場合、治験薬の投与を継続できる場合がある。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、ベネフィット・リスクのバランスを考慮し、投与継続による有用性が期待されると治験責任医師または治験分担医師が判断する場合、治験薬の投与を継続できる場合がある。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[有効性の評価項目]
(画像診断・内視鏡検査)
画像診断および内視鏡検査は、大腸内視鏡、CTまたはMRIの撮影像から腫瘍の増悪の有無を判定した。実施時期は、治験薬の休薬の影響を受けない。また、スクリーニング期(治験薬投与前14日以内)に、臨床症状などにより脳転移が疑われる場合、頭部におけるCT/MRI撮影、FDG-PET(または骨シンチグラフィ)などを実施し、脳転移または骨転移の有無を確認した。
(評価項目)
(1)病理学的完全奏効(pCR)率、(2)Major pathological response(MPR)率、(3)全生存期間(OS)、(4)無再発生存期間(RFS)、(5)無イベント生存期間(EFS)、(6)腫瘍マーカー(CEA およびCA19-9)の推移
(1)pCR率
各実施医療機関の病理医により、AJCC Tumor regression grade 0と判定された被験者の割合をpCR率として算出した。なお、原発巣のみならず、所属リンパ節にもviableな腫瘍細胞を認めないもの(ypT0N0)をpCRとした。
(2)MPR率
各実施医療機関の病理医により、AJCC Tumor regression grade 0または1と判定された被験者の割合をMPR率として算出する。
[手術の評価項目]
(1)根治切除術実施率、(2)根治切除術までの治療完遂率、(3)治験実施計画書に規定された期間における根治切除術実施率、(4)切除標本の肉眼的評価
[安全性の評価項目]
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)有害事象、(2)周術期合併症、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから10カ月が経過した時点において、pCRと判断された症例は3例であった。
(画像診断・内視鏡検査)
画像診断および内視鏡検査は、大腸内視鏡、CTまたはMRIの撮影像から腫瘍の増悪の有無を判定した。実施時期は、治験薬の休薬の影響を受けない。また、スクリーニング期(治験薬投与前14日以内)に、臨床症状などにより脳転移が疑われる場合、頭部におけるCT/MRI撮影、FDG-PET(または骨シンチグラフィ)などを実施し、脳転移または骨転移の有無を確認した。
(評価項目)
(1)病理学的完全奏効(pCR)率、(2)Major pathological response(MPR)率、(3)全生存期間(OS)、(4)無再発生存期間(RFS)、(5)無イベント生存期間(EFS)、(6)腫瘍マーカー(CEA およびCA19-9)の推移
(1)pCR率
各実施医療機関の病理医により、AJCC Tumor regression grade 0と判定された被験者の割合をpCR率として算出した。なお、原発巣のみならず、所属リンパ節にもviableな腫瘍細胞を認めないもの(ypT0N0)をpCRとした。
(2)MPR率
各実施医療機関の病理医により、AJCC Tumor regression grade 0または1と判定された被験者の割合をMPR率として算出する。
[手術の評価項目]
(1)根治切除術実施率、(2)根治切除術までの治療完遂率、(3)治験実施計画書に規定された期間における根治切除術実施率、(4)切除標本の肉眼的評価
[安全性の評価項目]
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)有害事象、(2)周術期合併症、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから10カ月が経過した時点において、pCRと判断された症例は3例であった。
実施例3:遠隔転移を有する膵臓がん患者を対象に、一次治療として化合物A、Nivolumabおよび標準療法であるmFFX療法またはGnP療法を併用する非盲検非対照試験
本試験は、遠隔転移を有する膵臓がん患者を対象に、一次治療として化合物A、NivolumabおよびmFFX療法またはGnP療法を併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、NivolumabおよびmFFX療法またはGnP療法との併用効果が評価できる。
(1)対象患者
遠隔転移を有する膵臓がん患者
(2)患者選択基準
登録時に、遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者で、かつ化合物A、NivolumabおよびmFFX療法またはGnP療法に関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(3)患者除外基準
登録時に、化合物A、NivolumabおよびmFFX療法またはGnP療法に関する各々の治験における患者除外基準、あるいは化合物A、NivolumabおよびmFFX療法またはGnP療法に関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
本試験は、遠隔転移を有する膵臓がん患者を対象に、一次治療として化合物A、NivolumabおよびmFFX療法またはGnP療法を併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、NivolumabおよびmFFX療法またはGnP療法との併用効果が評価できる。
(1)対象患者
遠隔転移を有する膵臓がん患者
(2)患者選択基準
登録時に、遠隔転移を有する膵臓がんに対する全身性抗悪性腫瘍剤による治療歴のない患者で、かつ化合物A、NivolumabおよびmFFX療法またはGnP療法に関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(3)患者除外基準
登録時に、化合物A、NivolumabおよびmFFX療法またはGnP療法に関する各々の治験における患者除外基準、あるいは化合物A、NivolumabおよびmFFX療法またはGnP療法に関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(4)用法・用量および投与期間
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、NivolumabおよびmFFX療法またはGnP療法を同日に投与する場合、化合物AおよびNivolumabを先に投与し、化合物AおよびNivolumabを投与した後、mFFX療法またはGnP療法を開始した。
[Nivolumab]
Nivolumab 480mgを4週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも24日以上空け、25日目以降に実施した。
[mFFX療法]
オキサリプラチンは85mg/m2(体表面積)を2時間かけて静脈内投与した。イリノテカンは150mg/m2(体表面積)を90分かけて静脈内投与した。レボホリナートは200mg/m2(体表面積)を2時間かけて静脈内投与した。フルオロウラシルは2400mg/m2(体表面積)を46時間かけて静脈内投与した。当該一連の投与は2週間間隔で実施した。なお、オキサリプラチン、イリノテカン、レボホリナートおよびフルオロウラシルはそれぞれ市販品を用いた。
[GnP療法]
ゲムシタビンは1000mg/m2(体表面積)を30分かけて静脈内投与し、少なくとも6日間休薬した。ナブパクリタキセルは125mg/m2(体表面積)を30分かけて静脈内投与し、少なくとも6日間休薬した。当該一連の投与は、1週間間隔で行い、3週間続けた後、1週間休薬した。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図3に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、NivolumabおよびmFFX療法またはGnP療法を同日に投与する場合、化合物AおよびNivolumabを先に投与し、化合物AおよびNivolumabを投与した後、mFFX療法またはGnP療法を開始した。
[Nivolumab]
Nivolumab 480mgを4週間間隔で30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも24日以上空け、25日目以降に実施した。
[mFFX療法]
オキサリプラチンは85mg/m2(体表面積)を2時間かけて静脈内投与した。イリノテカンは150mg/m2(体表面積)を90分かけて静脈内投与した。レボホリナートは200mg/m2(体表面積)を2時間かけて静脈内投与した。フルオロウラシルは2400mg/m2(体表面積)を46時間かけて静脈内投与した。当該一連の投与は2週間間隔で実施した。なお、オキサリプラチン、イリノテカン、レボホリナートおよびフルオロウラシルはそれぞれ市販品を用いた。
[GnP療法]
ゲムシタビンは1000mg/m2(体表面積)を30分かけて静脈内投与し、少なくとも6日間休薬した。ナブパクリタキセルは125mg/m2(体表面積)を30分かけて静脈内投与し、少なくとも6日間休薬した。当該一連の投与は、1週間間隔で行い、3週間続けた後、1週間休薬した。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図3に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
治療期は1サイクル28日間とし、初回の治験薬投与日をサイクル1の1日目とした。第2サイクル以降の各サイクルの1日目は、[28×(サイクル数-1)+1]日とした。化合物A、NivolumabおよびmFFX療法またはGnP療法についてそれぞれ上記用法・用量に従って、投与を開始し、化合物A、NivolumabおよびmFFX療法またはGnP療法についての投与基準、減量基準および減量時の投与量に従って投与を継続した。化合物A、NivolumabおよびmFFX療法またはGnP療法の投与終了(中止)時の評価が終了した時点を治療期終了とした。治験薬を投与したすべての被験者のうち、化合物A、NivolumabおよびmFFX療法またはGnP療法の投与を中止または終了した被験者は、投与終了(中止)時の評価を実施し、後観察期に移行する。後観察期終了後は、追跡調査を実施する。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[mFFX療法の投与基準]
被験者は、毎回の投与開始時において、mFFX療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[mFFX療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[mFFX療法の中止基準]
治療期において、mFFX療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はmFFX療法の投与を中止する。
[GnP療法の投与基準]
被験者は、毎回の投与開始時において、GnP療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[GnP療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[GnP療法の中止基準]
治療期において、GnP療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はGnP療法の投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[mFFX療法の投与基準]
被験者は、毎回の投与開始時において、mFFX療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[mFFX療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[mFFX療法の中止基準]
治療期において、mFFX療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はmFFX療法の投与を中止する。
[GnP療法の投与基準]
被験者は、毎回の投与開始時において、GnP療法に関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべて合致しなければならない。投与予定日の臨床検査値が当該基準の条件を満たす状態へ回復するまで投与を延期し、各薬剤の禁忌に該当しないことを確認した上で投与する。
[GnP療法の減量および減量基準]
最新の添付文書に準じて減量を実施する。
[GnP療法の中止基準]
治療期において、GnP療法に関してこれまでに実施された各治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はGnP療法の投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
治験責任医師または治験分担医師は、RECISTガイドライン1.1版に準じて標的病変の腫瘍径を計測し、抗腫瘍効果を判定した。ベースライン評価は、治験薬投与前28日以内の最新の画像検査を用いて行い、RECISTガイドライン1.1版に定義される測定可能病変を一つ以上有することを確認した。治療期の画像診断は、第1サイクル1日目から8週後(+7日)に行い、腫瘍径を計測した。
なお、総合効果および最良総合効果とは、RECISTガイドライン1.1版に基づき評価された画像診断結果とした。
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏効期間(DOR)、(6)奏効に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移(CEAおよびCA19-9)
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏効期間(DOR)、(6)奏効に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移(CEAおよびCA19-9)
(1)奏効率
奏効率は、最良総合効果がCRまたはPRと判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたはSDと判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果がPDと判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏効期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏効に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
奏効率は、最良総合効果がCRまたはPRと判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたはSDと判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果がPDと判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏効期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏効に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
(8)標的病変の腫瘍径和の最大変化率
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
(9)腫瘍マーカーの変化率の推移(CEAおよびCA19-9)
腫瘍マーカーの変化率を以下の計算式を用いて算出する。ただし、後治療が行われた後の腫瘍マーカーの変化率は算出しない。
腫瘍マーカーの変化率を以下の計算式を用いて算出する。ただし、後治療が行われた後の腫瘍マーカーの変化率は算出しない。
[安全性の評価項目]
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性(DLT)、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
化合物A、NivolumabおよびmFFX療法との併用について、1例目の被験者が登録されてから9ヶ月が経過した時点において、PRと判断された症例が1例であった。
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性(DLT)、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
化合物A、NivolumabおよびmFFX療法との併用について、1例目の被験者が登録されてから9ヶ月が経過した時点において、PRと判断された症例が1例であった。
また、化合物A、NivolumabおよびGnP療法との併用について、1例目の被験者が登録されてから9カ月が経過した時点において、PRと判断された症例は2例であった。
実施例4:抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん患者を対象に、二次治療として化合物A、Nivolumabおよび標準療法であるドセタキセルおよびラムシルマブを併用する非盲検非対照試験
本試験は、抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん患者を対象に、二次治療として化合物A、Nivolumab、ドセタキセルおよびラムシルマブを併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、Nivolumab、ドセタキセルおよびラムシルマブとの併用効果が評価できる。
(1)対象患者
IV期または再発の非小細胞肺がん患者
(2)患者選択基準
登録時に、一次治療としての抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者で、かつ化合物A、Nivolumab、ドセタキセルおよびラムシルマブに関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(3)患者除外基準
登録時に、化合物A、Nivolumab、ドセタキセルおよびラムシルマブに関する各々の治験における患者除外基準、あるいは化合物A、Nivolumabおよびドセタキセルおよびラムシルマブに関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
本試験は、抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法に不応の進行または再発の非小細胞肺がん患者を対象に、二次治療として化合物A、Nivolumab、ドセタキセルおよびラムシルマブを併用したときの忍容性、安全性および有効性を検討することを目的とする。当該治験により、化合物A、Nivolumab、ドセタキセルおよびラムシルマブとの併用効果が評価できる。
(1)対象患者
IV期または再発の非小細胞肺がん患者
(2)患者選択基準
登録時に、一次治療としての抗PD-1抗体もしくは抗PD-L1抗体、およびプラチナ製剤を含む併用療法を受け、不応の進行または再発が認められた患者で、かつ化合物A、Nivolumab、ドセタキセルおよびラムシルマブに関して、これまでに実施された各々の治験における患者選択基準を考慮して決定された所定の患者選択基準をすべて満たす患者を選択した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(3)患者除外基準
登録時に、化合物A、Nivolumab、ドセタキセルおよびラムシルマブに関する各々の治験における患者除外基準、あるいは化合物A、Nivolumabおよびドセタキセルおよびラムシルマブに関して各々の適正使用ガイドあるいは適正使用情報における患者選択時の注意事項を考慮して決定された所定の患者除外基準のいずれかに該当すると考えられる患者は除外した。なお、登録から治験薬を初回投与前までに当該基準を満たさないことが明らかとなった場合は、治験薬の投与を開始せず、治験中止とした。
(4)用法・用量および投与期間
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、Nivolumab、ドセタキセルおよびラムシルマブを同日に投与する場合、化合物AおよびNivolumabを先に投与し後、ドセタキセルおよびラムシルマブの投与を開始した。
[Nivolumab]
Nivolumab 360mgを3週間間隔で約30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも14日以上空け、15日目以降に実施した。
[ドセタキセルおよびラムシルマブ]
ドセタキセルおよびラムシルマブの用法用量は、実施医療機関の手順に従うが、推奨される用法用量としては、ドセタキセルは60mg/m2(体表面積)を60分以上かけて3週間間隔で静脈内投与した。ラムシルマブは10mg/kgを60分かけて3週間間隔で静脈内投与した。なお、3週間間隔とは、前回のドセタキセルもしくはラムシルマブの投与日を1日目として22日目から29日目の間に投与することである。なお、ドセタキセルおよびラムシルマブはそれぞれ市販品を用いた。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図4に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
[化合物A]
化合物Aを20mgもしくは40mg1日1回経口投与し、化合物Aに関する所定の投与中止基準に該当するまで継続投与した。なお、化合物A、Nivolumab、ドセタキセルおよびラムシルマブを同日に投与する場合、化合物AおよびNivolumabを先に投与し後、ドセタキセルおよびラムシルマブの投与を開始した。
[Nivolumab]
Nivolumab 360mgを3週間間隔で約30分間かけて静脈内投与し、Nivolumabに関する所定の投与中止基準に該当するまで継続投与した。なお、Nivolumabの投与は、前回の投与から少なくとも14日以上空け、15日目以降に実施した。
[ドセタキセルおよびラムシルマブ]
ドセタキセルおよびラムシルマブの用法用量は、実施医療機関の手順に従うが、推奨される用法用量としては、ドセタキセルは60mg/m2(体表面積)を60分以上かけて3週間間隔で静脈内投与した。ラムシルマブは10mg/kgを60分かけて3週間間隔で静脈内投与した。なお、3週間間隔とは、前回のドセタキセルもしくはラムシルマブの投与日を1日目として22日目から29日目の間に投与することである。なお、ドセタキセルおよびラムシルマブはそれぞれ市販品を用いた。
[治験スケジュール]
本試験は、スクリーニング期、治療期および後観察期からなる。治験スケジュールの概要を図4に示す。
スクリーニング期は治験薬投与前28日以内とし、治験責任医師または治験分担医師は上記選択基準を満たし、かつ上記除外基準に抵触せず、本治験の対象として適格と判断した患者を組み入れた。
治療期は1サイクル21日間とし、初回の治験薬投与日をサイクル1の1日目とした。第2サイクル以降の各サイクルの1日目は、[21×(サイクル数-1)+1]日とする。化合物A、Nivolumab、ドセタキセルおよびラムシルマブについてそれぞれ上記用法・用量に従って、投与を開始し、化合物A、Nivolumab、ドセタキセルおよびラムシルマブについての投与基準、減量基準および減量時の投与量に従って投与を継続した。化合物A、Nivolumab、ドセタキセルおよびラムシルマブの投与終了(中止)時の評価が終了した時点を治療期終了とした。治験薬を投与したすべての被験者のうち、化合物A、Nivolumab、ドセタキセルおよびラムシルマブの投与を中止または終了した被験者は、投与終了(中止)時の評価を実施し、後観察期に移行する。後観察期終了後は、追跡調査を実施する。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[ドセタキセルおよびラムシルマブの投与基準]
最新の添付文書に準じて投与した。
[ドセタキセルおよびラムシルマブの減量および減量基準]
最新の添付文書に準じて減量を実施する。
[ドセタキセルおよびラムシルマブの中止基準]
最旬の添付文書に準じて投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
[化合物AおよびNivolumabの投与基準]
被験者は、毎回の投与開始時において、化合物AおよびNivolumabに関してこれまでに実施された治験における投与基準を考慮して決定された所定の投与基準をすべてに合致しなければならない。当該基準のいずれかに合致しない場合、予定された化合物AおよびNivolumabの投与を休薬する。ただし、当該基準のいずれかに合致しない場合でも、病勢進行によると判断される臨床症状の悪化を認めず、投与継続による臨床的ベネフィットが期待され、被験者のベースラインおよび併用薬の影響を考慮して安全に化合物AおよびNivolumabの投与を継続可能と判断される場合に限り、化合物AおよびNivolumabの投与継続を可能とする。
[化合物Aの投与中止基準]
治療期において、化合物Aに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者は化合物Aの投与を中止する。
[Nivolumabの投与中止基準]
治療期において、Nivolumabに関してこれまでに実施された治験における投与中止基準を考慮して決定された所定の投与中止基準のいずれかに該当した被験者はNivolumabの投与を中止する。
[ドセタキセルおよびラムシルマブの投与基準]
最新の添付文書に準じて投与した。
[ドセタキセルおよびラムシルマブの減量および減量基準]
最新の添付文書に準じて減量を実施する。
[ドセタキセルおよびラムシルマブの中止基準]
最旬の添付文書に準じて投与を中止する。
[有効性の評価基準]
(画像診断)
胸部、腹部および骨盤のCT/核磁気共鳴画像(MRI)撮影等を実施した。また、スクリーニング期(治験薬投与前28日以内)に、臨床症状等により脳転移が疑われる場合、頭部におけるCT/MRI撮影、フルオロデオキシグルコース―ポジトロン断層撮影法(FDG-PET)(または骨シンチグラフィ)等を実施し、脳転移または骨転移の有無を確認した。
治験責任医師または治験分担医師は、RECISTガイドライン1.1版に準じて標的病変の腫瘍径を計測し、抗腫瘍効果を判定した。ベースライン評価は、治験薬投与前28日以内の最新の画像検査を用いて行い、RECISTガイドライン1.1版に定義される測定可能病変を一つ以上有することを確認した。治療期の画像診断は、第1サイクル1日目から54週後まで6週間隔(±7日)、それ以降は12週間隔(±7日)で行い、腫瘍径を計測した。
なお、総合効果および最良総合効果とは、RECISTガイドライン1.1版に基づき評価された画像診断結果とした。
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏効期間(DOR)、(6)奏効に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移
(1)奏効率
奏効率は、最良総合効果がCRまたはPRと判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたはSDと判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果がPDと判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏効期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏効に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
(評価項目)
(1)奏効率(ORR)、(2)病勢制御率(DCR)、(3)全生存期間(OS)、(4)無増悪生存期間(PFS)、(5)奏効期間(DOR)、(6)奏効に至るまでの期間(TTR)、(7)最良総合効果(BOR)、(8)標的病変の腫瘍径和の変化率、(9)標的病変の腫瘍径和の最大変化率、(10)腫瘍マーカーの推移
(1)奏効率
奏効率は、最良総合効果がCRまたはPRと判定された被験者の割合を示す。
(2)病勢制御率
病勢制御率は、最良総合効果がCR、PRまたはSDと判定された被験者の割合を示す。
(3)全生存期間
全生存期間は、以下の式から算出する。
全生存期間(日)=「あらゆる原因による死亡日」-「治験薬投与開始日」+1
なお、追跡不能となった、またはデータカットオフ日までに死亡していない被験者は、最終生存確認日を打切り日とする。
(4)無増悪生存期間
無増悪生存期間は、以下の式から算出する。
無増悪生存期間(日)=「総合効果がPDと判定された日、またはあらゆる原因による死亡日のうち早い日」-「治験薬投与開始日」+1
なお、総合効果がPDと判定されておらず、かつ死亡もしていない被験者は、最後の評価可能な画像診断が行われた日を打切り日とする。評価可能な画像診断が行われておらず、かつ死亡もしていない被験者は、治験薬投与開始日を打切り日とする。総合効果がPDと判定されるまたは死亡する前に、がんに対する後治療を受けた被験者は、がんに対する後治療開始前における最後の評価可能な画像診断が行われた日を打切り日とする。
(5)奏効期間
奏効期間は、以下の式から算出する。
奏効期間(日)=「奏効確定後初めて総合効果がPDと判定された日またはあらゆる原因による死亡日のうち早い日」-「確定されたCRまたはPRの最初の判定日」+1
なお、評価対象は治験を通じて確定されたCRまたはPRを示した被験者とする。
(6)奏効に至るまでの期間
奏効に至るまでの期間は、以下の式から算出する。
奏効に至るまでの期間(日)=「確定されたCRまたはPRの最初の判定日」-「治験薬投与開始日」+1
(7)標的病変の腫瘍径和の変化率
標的病変を有する被験者を対象に、標的病変の腫瘍径和の変化率を以下の計算式を用いて算出した。ただし、後治療が行われた後の標的病変の腫瘍径和の変化率は算出しない。
(8)標的病変の腫瘍径和の最大変化率
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
標的病変の腫瘍径和の最大変化率は、標的病変の腫瘍径和が最も小さくなった時点の変化率とする。ただし、総合効果がPDと判定された後または後治療が行われた後の標的病変の腫瘍径和は最大変化率の算出に使用しない。
(9)腫瘍マーカーの変化率の推移
腫瘍マーカーの変化率を以下の計算式を用いて算出する。ただし、後治療が行われた後の腫瘍マーカーの変化率は算出しない。
腫瘍マーカーの変化率を以下の計算式を用いて算出する。ただし、後治療が行われた後の腫瘍マーカーの変化率は算出しない。
[安全性の評価項目]
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性(DLT)、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから8ヶ月が経過した時点において、PRと判断された症例は1例であった。
以下の項目について治験責任医師などにより、既定の時期に測定、検査および調査を実施した。
(1)用量制限毒性(DLT)、(2)有害事象、(3)臨床検査(血液学的検査、血液生化学的検査、ホルモン調査、血液凝固系検査、尿定性検査、免疫学的検査)、(4)バイタルサイン(収縮期血圧/拡張期血圧、脈拍数、体温)、経皮的酸素飽和度(SpO2)、体重、(5)12誘導心電図、(6)ECOG Performance Status、(7)胸部X線、(8)眼科検査(視力検査、眼圧検査、細隙灯顕微鏡検査、および(9)CT検査
[有効性評価結果]
1例目の被験者が登録されてから8ヶ月が経過した時点において、PRと判断された症例は1例であった。
本発明は、新たながん治療法を提供するものであり、有用である。
Claims (36)
- 標準療法、および免疫チェックポイント阻害物質の投与と組み合わせて投与されることを特徴とする、EP4拮抗薬を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、前記EP4拮抗薬が一般式(I):

R1はCOOR8、テトラゾール、SO3H、SO2NH2、SO2NHR8-1、CONHSO2R8-1、SO2NHCOR8-1、またはヒドロキサム酸を表し、
R8は水素原子、C1-4アルキル、またはベンジルを表し、
R8-1は、C1-4アルキル、C1-4ハロアルキル、C3-10炭素環、または3-10員複素環を表し、各々の該C3-10炭素環および3-10員複素環は、C1-4アルキル、C1-4ハロアルキル、C1-4アルコキシ、-O(C1-4ハロアルキル)、C1-4アルキルチオ、-S(C1-4ハロアルキル)、ハロゲン、またはニトリル(「-CN」を示す;以下同じ)で置換されていてもよく、
L1は、C1-5アルキレン、C2-5アルケニレン、またはC2-5アルキニレンを表し、
R2は、ハロゲン、C1-4アルキル、C1-4アルコキシ、C1-4アルキルチオ、C2-4アルケニル、C2-4アルキニル、-O(C1-4ハロアルキル)、-S(C1-4ハロアルキル)、-C(O)(C1-4アルキル)、-SO2(C1-4アルキル)、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、-NHC(O)(C1-4アルキル)、-N(C1-4アルキル)C(O)(C1-4アルキル)、-NHSO2(C1-4アルキル)、-N(C1-4アルキル)SO2(C1-4アルキル)、-SO2NH(C1-4アルキル)、-SO2N(C1-4アルキル)2、-NR17R17、ニトロ、ニトリル、水酸基、アルデヒド(ホルミルを示す;以下同じ)、またはカルボキシルを表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
該R2における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
X1はCR6または窒素原子を表し、R6は水素原子またはR2を表し、
X2はCR7または窒素原子を表し、R7は、水素原子、R2、または-L3-R9を表し、L3は、メチレン、酸素原子、または酸化されていてもよい硫黄原子を表し、R9は、ハロゲン、C1-4アルキル、およびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい4-10員複素環を表し、
L2は、-CH2CH2-、-CH=CH-、-CH2O-、-OCH2-、-CH2S-、-SCH2-、-CH2S(O)-、-S(O)CH2-、-CH2SO2-、-SO2CH2-、-CH2NH-、-NHCH2-、-NHCO-、-CONH-、-NHSO2-、または-SO2NH-を表し、
R3はC1-4アルキルまたはハロゲンを表し、
R4は、ハロゲン、C1-4アルキル、またはC1-4ハロアルキルを表し、
X3は、メチレン、酸素原子、酸化されていてもよい硫黄原子、またはNR10を表し、R10は、C1-4アルキル、-C(O)(C1-4アルキル)、-C(O)O(C1-4アルキル)、または-SO2(C1-4アルキル)を表し、各々の該C1-4アルキルはハロゲンで置換されていてもよく、
ringはベンゼン環または5-6員単環式芳香族複素環を表し、

R5は、(1)ハロゲン、(2)C1-4アルキル、(3)カルボキシル、(4)ニトリル、(5)-CONHR11、(6)-C(O)R12、(7)-OR14、(8)-S(O)tR15、(9)-CH2R16、(10)-NR17R17、(11)-NHCOR11、(12)C4-10炭素環、または(13)4-10員複素環を表し、該C4-10炭素環または4-10員複素環は1~3個のR18で置換されていてもよく、該R18が複数である場合、R18はそれぞれ独立して同一でも異なっていてもよく、
R11は、C1-6アルキル、C3-6シクロアルキル、フェニル、または4-6員複素環を表し、R11は1~3個のR13で置換されていてもよく、該R13が複数である場合、R13はそれぞれ独立して同一でも異なっていてもよく、
R13は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、水酸基、-NR20R21、ベンゼン、または4-6員複素環を表し、
R20およびR21は、それぞれ独立して、水素原子またはC1-4アルキルを表し、
R12は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、または4-6員複素環を表し、該C3-6シクロアルキル、ベンゼン、または4-6員複素環は、それぞれ独立して、ハロゲン、C1-4アルキル、またはC1-4アルコキシで置換されていてもよく、
R14は、水素原子、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、該C1-6アルキルは1~3個のR19で置換されていてもよく、該R19が複数である場合、R19はそれぞれ独立して同一でも異なっていてもよく、
R19は、C1-4アルコキシ、-CONH(C1-4アルキル)、-CON(C1-4アルキル)2、またはC1-4アルキルおよびC1-4ハロアルキルからなる群より選択される置換基で置換されていてもよい5-6員単環式芳香族複素環を表し、
該R19における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
R15は、C1-6アルキル、C3-6シクロアルキル、ベンゼン、またはベンジルを表し、
R16は水酸基またはC1-4アルコキシを表し、
R17は、それぞれ独立して、水素原子、C1-6アルキル、またはC3-6シクロアルキルを表し、
R18は、ハロゲン、C1-6アルキル、C3-6シクロアルキル、C1-4アルコキシ、オキソ、ニトリル、水酸基、ヒドロキシメチル、1-メチル-1-ヒドロキシエチル、(C1-4アルキル)SO2-、4-6員複素環、(C1-4アルキル)NH-、または(C1-4アルキル)2N-を表し、
該R18における(C1-4アルキル)2とは、独立した2個のC1-4アルキルを表し、それぞれのC1-4アルキルは同一でも異なっていてもよく、
mは1~4の整数を表し、nは0~4の整数を表し、pは0~2の整数を表し、qは0~6の整数を表し、rは0~6の整数を表し、sは0~4の整数を表し、tは0~2の整数を表し、
ただし、p、q、r、およびsがそれぞれ2以上の整数を表す場合、R2、R3、R4、およびR5は、それぞれ独立して、同一でも異なっていてもよい。)で示される化合物、またはその塩である剤。 - 標準療法、およびEP4拮抗薬の投与と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含むがんの進行抑制、再発抑制および/または治療剤であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩である剤。
- がんが大腸がんである、請求項1もしくは請求項2に記載の剤。
- 標準療法がベバシズマブおよびXELOX療法である、請求項1~3のいずれかに記載の剤。
- ベバシズマブおよびXELOX療法が、
(i)ベバシズマブ 1回7.5mg/kgを3週間間隔で投与する、
(ii)オキサリプラチン 130mg/m2を3週間間隔で投与する、および
(iii)カペシタビン 1000mg/m2を1日2回で14日間経口投与し、7日間休薬する、
療法である、請求項1~4のいずれかに記載の剤。 - がんが膵臓がんである、請求項1もしくは請求項2に記載の剤。
- 標準療法がFOLFIRINOX療法またはその減量レジメンである、請求項1、請求項2、または請求項6のいずれかに記載の剤。
- FOLFIRINOX療法が、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウムの投与開始30分後からイリノテカン塩酸塩水和物 180mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後、フルオロウラシル 400mg/m2を急速静脈内投与する、
(v)さらにフルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、
療法である、請求項7に記載の剤。 - FOLFIRINOX療法の減量レジメンが、
(i)オキサリプラチン 85mg/m2を2時間かけて静脈内投与する、
(ii)レボホリナートカルシウム 200mg/m2を2時間かけて静脈内投与する、
(iii)レボホリナートカルシウム投与開始30分後からイリノテカン塩酸塩水和物 150mg/m2を1.5時間かけて静脈内投与する、
(iv)さらに当該レボホリナートカルシウムの投与終了後に、フルオロウラシル 2400mg/m2を46時間かけて静脈内持続投与し、当該一連の投与が2週間間隔で実施される、
療法である、請求項7に記載の剤。 - 標準療法がゲムシタビンおよびナブパクリタキセル療法である、請求項1、請求項2または請求項6のいずれかに記載の剤。
- ゲムシタビンおよびナブパクリタキセル療法が、
(i)ゲムシタビン 1000mg/m2を30分かけて静脈内投与する、
(ii)ナブパクリタキセル 125mg/m2を30分かけて静脈内投与し、当該一連の投与を1週間間隔で行い、3週間続けた後、1週間休薬する、
療法である、請求項10に記載の剤。 - がんが肺がんである、請求項1もしくは請求項2に記載の剤。
- 標準療法がドセタキセルおよび/またはラムシルマブ療法である、請求項1または請求項2または請求項12のいずれかに記載の剤。
- 標準療法がドセタキセル療法である、請求項13に記載の剤。
- ドセタキセル療法が、ドセタキセル 60mg/m2を60分以上かけて静脈内投与し、当該投与を3週間間隔で実施される療法である、請求項14に記載の剤。
- 標準療法がドセタキセルおよびラムシルマブ療法である、請求項1、請求項2、請求項12または請求項13のいずれかに記載の剤。
- ドセタキセルおよびラムシルマブ療法が、
(i)ドセタキセル 60mg/m2を60分以上かけて静脈内投与する、および
(ii)ラムシルマブ 10mg/kgを60分かけて静脈内投与し、当該一連の投与が3週間間隔で実施される、
療法である、請求項16に記載の剤。 - 術前化学放射線療法を受けたがん患者に投与されることを特徴とする、EP4拮抗薬を有効成分として含む大腸がんの進行抑制、再発抑制および/または治療剤であって、前記EP4拮抗薬が請求項1記載の一般式(I)で示される化合物、またはその塩である剤。
- さらに免疫チェックポイント阻害物質と組み合わせて投与される、請求項18に記載の剤。
- 術前化学放射線療法を受けたがん患者を対象に、EP4拮抗薬と組み合わせて投与されることを特徴とする、免疫チェックポイント阻害物質を有効成分として含む大腸がんの進行抑制、再発抑制および/または治療剤であって、前記EP4拮抗薬が請求項1記載の一般式(I)で示される化合物、またはその塩である剤。
- 免疫チェックポイント阻害物質および/またはEP4拮抗薬が術前化学放射線療法(CRT)後の術前補助療法として使用される、請求項18~20のいずれかに記載の剤。
- 術前化学放射線療法が、放射線治療とカペシタビンの組み合わせである、請求項18~21のいずれか一項に記載の剤。
- 術前化学放射線療法が、45Gy/25回の骨盤腔への照射および5.4Gy/3回の原発巣へのboost照射、およびカペシタビンを825mg/m2を1日2回で21日間もしくは42回以上投与する療法である、請求項18~22のいずれかに記載の剤。
- EP4拮抗薬が、4-[4-シアノ-2-({(2’R,4S)-6-[(プロパン-2-イル)カルバモイル]-2,3-ジヒドロスピロ[1-ベンゾピラン-4,1’-シクロプロパン]-2’-カルボニル}アミノ)フェニル]ブタン酸、またはその塩である、請求項1~23のいずれかに記載の剤。
- EP4拮抗薬として1回5mg~40mgを1日1回経口投与されることを特徴とする、請求項24に記載の剤。
- EP4拮抗薬として1回20mgまたは40mgを1日1回経口投与されることを特徴とする、請求項25に記載の剤。
- 免疫チェックポイント阻害物質が、抗PD-1抗体、抗PD-L1抗体または抗CTLA-4抗体である、請求項1~17、または19~26のいずれかに記載の剤。
- 免疫チェックポイント阻害物質が、抗PD-1抗体である、請求項1~17、または19~27のいずれかに記載の剤。
- 抗PD-1抗体が、Nivolumab、Cemiplimab、Pembrolizumab、Spartalizumab、Tislelizumab、AMP-514、Dostarlimab、Toripalimab、Camrelizumab、Genolimzumab、Sintilimab、STI-A1110、ENUM 388D4、ENUM 244C8、GLS010、MGA012、AGEN2034、CS1003、HLX10、BAT-1306、AK105、AK103、BI 754091、LZM009、CMAB819、Sym021、GB226、SSI-361、JY034、HX008、ISU106、ABBV181、BCD-100、PF-06801591、CX-188、JNJ-63723283またはAB122である、請求項28に記載の剤。
- 抗PD-1抗体がNivolumabである、請求項29に記載の剤。
- Nivolumabとして
(i)1回240mgを2週間間隔で投与される、
(ii)1回360mgを3週間間隔で投与される、もしくは
(iii)1回480mgを4週間間隔で投与されることを特徴とする、
請求項30記載の剤。 - 標準療法、および免疫チェックポイント阻害物質の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療に使用される、EP4拮抗薬であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩であるEP4拮抗薬。
- 標準療法、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療に使用される、免疫チェックポイント阻害物質であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩である免疫チェックポイント阻害物質。
- 標準療法、および免疫チェックポイント阻害物質の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療剤の製造における、EP4拮抗薬の使用であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩である使用。
- 標準療法、およびEP4拮抗薬の投与と組み合わせて投与される、がんの進行抑制、再発抑制および/または治療剤の製造における、免疫チェックポイント阻害物質の使用であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩である使用。
- 標準療法の有効量と免疫チェックポイント阻害物質の有効量およびEP4拮抗薬の有効量とをがん治療を必要とする患者に、別々または同時に投与することを含むがんの進行抑制、再発抑制および/または治療方法であって、前記EP4拮抗薬が請求項1に記載の一般式(I)で示される化合物、またはその塩である方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18/035,608 US20230390303A1 (en) | 2020-11-13 | 2021-11-12 | Cancer treatment by combination of ep4 antagonist and immune checkpoint inhibitor |
| KR1020237015423A KR20230107228A (ko) | 2020-11-13 | 2021-11-12 | Ep4 길항약과 면역 체크포인트 저해 물질의 병용에 의한 암 치료 |
| EP21891976.9A EP4245301A4 (en) | 2020-11-13 | 2021-11-12 | TREATMENT OF CANCER BY COMBINED USE OF AN EP4 ANTAGONIST AND AN IMMUNE CHECKPOINT INHIBITOR |
| JP2022562190A JP7816164B2 (ja) | 2020-11-13 | 2021-11-12 | Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020189152 | 2020-11-13 | ||
| JP2020-189152 | 2020-11-13 | ||
| JP2020-211006 | 2020-12-21 | ||
| JP2020211006 | 2020-12-21 | ||
| JP2021-006828 | 2021-01-20 | ||
| JP2021006828 | 2021-01-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022102731A1 true WO2022102731A1 (ja) | 2022-05-19 |
Family
ID=81602339
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/041650 Ceased WO2022102731A1 (ja) | 2020-11-13 | 2021-11-12 | Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20230390303A1 (ja) |
| EP (1) | EP4245301A4 (ja) |
| JP (1) | JP7816164B2 (ja) |
| KR (1) | KR20230107228A (ja) |
| TW (1) | TW202228674A (ja) |
| WO (1) | WO2022102731A1 (ja) |
Citations (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| WO2001062708A1 (fr) | 2000-02-22 | 2001-08-30 | Ono Pharmaceutical Co., Ltd. | Derives d'acide benzoique, procede de production desdits derives, et medicament contenant ces derives comme principe actif |
| WO2002016311A1 (en) | 2000-08-22 | 2002-02-28 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives, process for producing the same and drugs containing the same as the active ingredient |
| WO2002020462A1 (en) | 2000-09-01 | 2002-03-14 | Ono Pharmaceutical Co., Ltd. | Benzoic acid derivatives and drugs containing the same as the active ingredient |
| WO2002032900A2 (en) | 2000-10-19 | 2002-04-25 | Pfizer Pharmaceuticals Inc. | Aryl or heteroaryl fused imidazole compounds as anti-inflammatory and analgesic agents |
| WO2002050031A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Indole derivatives |
| WO2002050033A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Benzo(f) isoindol derivatives and their use as ep4 receptor ligands |
| WO2002050032A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Naphthalene derivatives which bind to the ep4 receptor |
| WO2003016254A1 (en) | 2001-08-09 | 2003-02-27 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivative compounds and drugs comprising these compounds as the active ingredient |
| WO2003087061A1 (en) | 2002-04-12 | 2003-10-23 | Pfizer Japan Inc. | Pyrazole compounds as anti-inflammatory and analgesic agents |
| WO2003086390A1 (en) | 2002-04-12 | 2003-10-23 | Pfizer Japan Inc. | Imidazole compounds as anti-inflammatory and analgesic agents |
| WO2003099857A1 (en) | 2002-05-23 | 2003-12-04 | Theratechnologies Inc. | Antagonistic peptides of prostaglandin e2 receptor subtype ep4 |
| WO2004067524A1 (en) | 2003-01-29 | 2004-08-12 | Pharmagene Laboratories Limited | Ep4 receptor antagonists |
| WO2005021508A1 (en) | 2003-09-03 | 2005-03-10 | Pfizer Inc. | Phenyl or pyridyl amide compounds as prostaglandin e2 antagonists |
| WO2005037812A1 (en) | 2003-10-16 | 2005-04-28 | Asterand Uk Limited | Furan derivatives as ep4 receptor antagonists |
| WO2005061475A2 (en) | 2003-12-22 | 2005-07-07 | Astellas Pharma Inc. | Ornithine derivatives as prostaglandin e2 agonists or antagonists |
| WO2005105732A1 (en) | 2004-05-04 | 2005-11-10 | Pfizer Japan Inc. | Substituted methyl aryl or heteroaryl amide compounds |
| WO2005105733A1 (en) | 2004-05-04 | 2005-11-10 | Pfizer Japan Inc. | Ortho substituted aryl or heteroaryl amide compounds |
| WO2006007448A2 (en) | 2004-06-17 | 2006-01-19 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions and related methods of use |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| WO2006122403A1 (en) | 2005-05-19 | 2006-11-23 | Merck Frosst Canada Ltd. | Quinoline derivatives as ep4 antagonists |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007121578A1 (en) | 2006-04-24 | 2007-11-01 | Merck Frosst Canada Ltd. | Indole amide derivatives as ep4 receptor antagonists |
| WO2007143825A1 (en) | 2006-06-12 | 2007-12-21 | Merck Frosst Canada Ltd. | Indoline amide derivatives as ep4 receptor ligands |
| WO2008017164A1 (en) | 2006-08-11 | 2008-02-14 | Merck Frosst Canada Ltd. | Thiophenecarboxamide derivatives as ep4 receptor ligands |
| WO2008104055A1 (en) | 2007-02-26 | 2008-09-04 | Merck Frosst Canada Ltd. | Indole and indoline cyclopropyl amide derivatives as ep4 receptor antagonists |
| WO2008116304A1 (en) | 2007-03-26 | 2008-10-02 | Merck Frosst Canada Ltd. | Naphthalene and quinoline sulfonylurea derivatives as ep4 receptor antagonists |
| WO2008123207A1 (ja) | 2007-03-26 | 2008-10-16 | Astellas Pharma Inc. | オルニチン誘導体 |
| WO2008156712A1 (en) | 2007-06-18 | 2008-12-24 | N. V. Organon | Antibodies to human programmed death receptor pd-1 |
| WO2009005076A1 (ja) | 2007-07-03 | 2009-01-08 | Astellas Pharma Inc. | アミド化合物 |
| WO2009139373A1 (ja) | 2008-05-14 | 2009-11-19 | アステラス製薬株式会社 | アミド化合物 |
| WO2010019796A1 (en) | 2008-08-14 | 2010-02-18 | Chemietek, Llc | Heterocyclic amide derivatives as ep4 receptor antagonists |
| WO2010034110A1 (en) | 2008-09-25 | 2010-04-01 | Merck Frosst Canada Ltd. | Beta-carboline sulphonylurea derivatives as ep4 receptor antagonists |
| WO2012039972A1 (en) | 2010-09-21 | 2012-03-29 | Eisai R&D Management Co., Ltd. | Pharmaceutical composition |
| WO2012043634A1 (ja) | 2010-09-29 | 2012-04-05 | 株式会社エヌビィー健康研究所 | ヒトプロスタグランジンe2受容体ep4に対する抗体 |
| WO2012076063A1 (en) | 2010-12-10 | 2012-06-14 | Rottapharm S.P.A. | Pyridine amide derivatives as ep4 receptor antagonists |
| WO2012103071A2 (en) | 2011-01-25 | 2012-08-02 | Eisai R&D Management Co., Ltd. | Compounds and compositions |
| WO2013004290A1 (en) | 2011-07-04 | 2013-01-10 | Rottapharm S.P.A. | Cyclic amine derivatives as ep4 receptor antagonists |
| WO2013096501A1 (en) | 2011-12-21 | 2013-06-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013096496A2 (en) | 2011-12-21 | 2013-06-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013101598A1 (en) | 2011-12-27 | 2013-07-04 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013101733A1 (en) | 2011-12-27 | 2013-07-04 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2014004230A1 (en) | 2012-06-29 | 2014-01-03 | Eli Lilly And Company | Dimethyl-benzoic acid compounds |
| WO2014004229A1 (en) | 2012-06-29 | 2014-01-03 | Eli Lilly And Company | Phenoxyethyl piperidine compounds |
| WO2014086739A1 (de) | 2012-12-06 | 2014-06-12 | Bayer Pharma Aktiengesellschaft | Neuartige benzimidazolderivate als ep4-antagonisten |
| WO2014126746A1 (en) | 2013-02-15 | 2014-08-21 | Eli Lilly And Company | Phenoxyethoxy compounds |
| WO2014186218A1 (en) | 2013-05-17 | 2014-11-20 | Eli Lilly And Company | Phenoxyethyl dihydro-1h-isoquinoline compounds |
| WO2014200075A1 (ja) | 2013-06-12 | 2014-12-18 | 科研製薬株式会社 | 4-アルキニルイミダゾール誘導体およびそれを有効成分として含有する医薬 |
| WO2015094902A1 (en) | 2013-12-17 | 2015-06-25 | Eli Lilly And Company | Phenoxyethyl cyclic amine derivatives and their activity as ep4 receptor modulators |
| WO2015094912A1 (en) | 2013-12-17 | 2015-06-25 | Eli Lilly And Company | Dimethylbenzoic acid compounds |
| WO2015091475A1 (de) | 2013-12-19 | 2015-06-25 | Bayer Pharma Aktiengesellschaft | Benzimidazolderivate als ep4-liganden |
| WO2015113057A1 (en) | 2014-01-27 | 2015-07-30 | Allergan, Inc. | Antagonists acting at multiple prostaglandin receptors for the treatment of inflammation |
| WO2015147020A1 (ja) | 2014-03-26 | 2015-10-01 | アステラス製薬株式会社 | アミド化合物 |
| WO2016021742A1 (en) | 2014-08-07 | 2016-02-11 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds as ep4 receptor antagonists |
| WO2016088903A1 (en) | 2014-12-05 | 2016-06-09 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds |
| WO2016111347A1 (ja) | 2015-01-09 | 2016-07-14 | 小野薬品工業株式会社 | 三環性スピロ化合物 |
| WO2017014177A1 (ja) * | 2015-07-17 | 2017-01-26 | 凸版印刷株式会社 | 健康状態の評価方法及び抗がん剤に対する長期奏功性の予測方法 |
| WO2017014323A1 (en) | 2015-07-23 | 2017-01-26 | Takeda Pharmaceutical Company Limited | 1-substituted 1,2,3,4-tetrahydro-1,7-naphthyridin-8-amine derivatives and their use as ep4 receptor antagonists |
| WO2017066633A1 (en) | 2015-10-16 | 2017-04-20 | Eisai R&D Management Co., Ltd. | Ep4 antagonists |
| WO2017085198A1 (en) | 2015-11-20 | 2017-05-26 | Actelion Pharmaceuticals Ltd | N-substituted indole derivatives as pge2 receptor modulators |
| WO2018008711A1 (ja) | 2016-07-07 | 2018-01-11 | 小野薬品工業株式会社 | Ep4拮抗薬と免疫チェックポイント阻害薬を含んでなる組み合わせ |
| JP2018021013A (ja) * | 2016-07-07 | 2018-02-08 | 小野薬品工業株式会社 | 医薬用途 |
| WO2018195123A1 (en) | 2017-04-18 | 2018-10-25 | Tempest Therapeutics, Inc. | Bicyclic compounds and their use in the treatment of cancer |
| WO2018210992A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Pyrimidine derivatives |
| WO2018210987A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Benzofurane and benzothiophene derivatives as pge2 receptor modulators |
| WO2018210994A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Phenyl derivatives as pge2 receptor modulators |
| WO2018210988A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Pyrimidine derivatives as pge2 receptor modulators |
| WO2018210995A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | N-substituted indole derivatives |
| WO2018216640A1 (ja) | 2017-05-22 | 2018-11-29 | 小野薬品工業株式会社 | Ep4アンタゴニスト |
| CN108929281A (zh) | 2017-05-27 | 2018-12-04 | 华东师范大学 | 三氮唑类化合物及其合成方法和应用 |
| WO2019038156A1 (en) | 2017-08-22 | 2019-02-28 | Bayer Pharma Aktiengesellschaft | USE OF AN EP4 ANTAGONIST FOR THE TREATMENT OF ARTHRITIS |
| WO2019101171A1 (zh) | 2017-11-27 | 2019-05-31 | 上海邦耀生物科技有限公司 | 噻吩并环类化合物及其合成方法和应用 |
| WO2019117269A1 (ja) * | 2017-12-13 | 2019-06-20 | 国立大学法人広島大学 | 膵がんの検出を補助する方法 |
| WO2019152982A1 (en) | 2018-02-05 | 2019-08-08 | Shenzhen Ionova Life Science Co., Ltd. | Heterobicyclic compounds as ep4 antagonists |
| WO2019166022A1 (en) | 2018-03-02 | 2019-09-06 | Shenzhen Ionova Life Science Co., Ltd. | Heterobicyclic carboxylic acids and salts thereof |
| WO2019245590A1 (en) | 2018-06-18 | 2019-12-26 | Avista Pharma Solutions, Inc. | Chemical compounds |
| WO2020012305A1 (en) | 2018-07-12 | 2020-01-16 | Rottapharm Biotech S.R.L. | (r)-4-(1-(1-(4-(trifluoromethyl)benzyl)pyrrolidine-2-carboxamide)cyclopropyl)-benzoic acid as ep4 receptor antagonist |
| WO2020014465A1 (en) | 2018-07-11 | 2020-01-16 | Arrys Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| WO2020111018A1 (ja) * | 2018-11-27 | 2020-06-04 | 小野薬品工業株式会社 | 免疫チェックポイント阻害薬およびfolfirinox療法との併用によるがん治療 |
| WO2020151566A1 (zh) | 2019-01-22 | 2020-07-30 | 凯复制药有限公司 | 抑制pge2/ep4信号传导的化合物、其制备方法及其在医药上的应用 |
| WO2020160075A1 (en) | 2019-01-30 | 2020-08-06 | Avista Pharma Solutions, Inc. | Chemical compounds |
| WO2020161275A1 (en) | 2019-02-08 | 2020-08-13 | Medibiofarma, S.L. | New n-benzyl-2-phenoxybenzamide derivatives as prostaglandin e2 (pge2) receptors modulators |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7159007B2 (ja) * | 2017-11-01 | 2022-10-24 | 小野薬品工業株式会社 | 脳腫瘍の治療のための医薬 |
-
2021
- 2021-11-12 US US18/035,608 patent/US20230390303A1/en active Pending
- 2021-11-12 KR KR1020237015423A patent/KR20230107228A/ko active Pending
- 2021-11-12 TW TW110142221A patent/TW202228674A/zh unknown
- 2021-11-12 JP JP2022562190A patent/JP7816164B2/ja active Active
- 2021-11-12 WO PCT/JP2021/041650 patent/WO2022102731A1/ja not_active Ceased
- 2021-11-12 EP EP21891976.9A patent/EP4245301A4/en active Pending
Patent Citations (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001014424A2 (en) | 1999-08-24 | 2001-03-01 | Medarex, Inc. | Human ctla-4 antibodies and their uses |
| WO2001062708A1 (fr) | 2000-02-22 | 2001-08-30 | Ono Pharmaceutical Co., Ltd. | Derives d'acide benzoique, procede de production desdits derives, et medicament contenant ces derives comme principe actif |
| WO2002016311A1 (en) | 2000-08-22 | 2002-02-28 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives, process for producing the same and drugs containing the same as the active ingredient |
| WO2002020462A1 (en) | 2000-09-01 | 2002-03-14 | Ono Pharmaceutical Co., Ltd. | Benzoic acid derivatives and drugs containing the same as the active ingredient |
| WO2002032900A2 (en) | 2000-10-19 | 2002-04-25 | Pfizer Pharmaceuticals Inc. | Aryl or heteroaryl fused imidazole compounds as anti-inflammatory and analgesic agents |
| WO2002050031A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Indole derivatives |
| WO2002050033A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Benzo(f) isoindol derivatives and their use as ep4 receptor ligands |
| WO2002050032A1 (en) | 2000-12-21 | 2002-06-27 | Glaxo Group Limited | Naphthalene derivatives which bind to the ep4 receptor |
| WO2003016254A1 (en) | 2001-08-09 | 2003-02-27 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivative compounds and drugs comprising these compounds as the active ingredient |
| WO2003087061A1 (en) | 2002-04-12 | 2003-10-23 | Pfizer Japan Inc. | Pyrazole compounds as anti-inflammatory and analgesic agents |
| WO2003086390A1 (en) | 2002-04-12 | 2003-10-23 | Pfizer Japan Inc. | Imidazole compounds as anti-inflammatory and analgesic agents |
| WO2003099857A1 (en) | 2002-05-23 | 2003-12-04 | Theratechnologies Inc. | Antagonistic peptides of prostaglandin e2 receptor subtype ep4 |
| WO2004067524A1 (en) | 2003-01-29 | 2004-08-12 | Pharmagene Laboratories Limited | Ep4 receptor antagonists |
| WO2005021508A1 (en) | 2003-09-03 | 2005-03-10 | Pfizer Inc. | Phenyl or pyridyl amide compounds as prostaglandin e2 antagonists |
| WO2005037812A1 (en) | 2003-10-16 | 2005-04-28 | Asterand Uk Limited | Furan derivatives as ep4 receptor antagonists |
| WO2005061475A2 (en) | 2003-12-22 | 2005-07-07 | Astellas Pharma Inc. | Ornithine derivatives as prostaglandin e2 agonists or antagonists |
| WO2005105732A1 (en) | 2004-05-04 | 2005-11-10 | Pfizer Japan Inc. | Substituted methyl aryl or heteroaryl amide compounds |
| WO2005105733A1 (en) | 2004-05-04 | 2005-11-10 | Pfizer Japan Inc. | Ortho substituted aryl or heteroaryl amide compounds |
| WO2006007448A2 (en) | 2004-06-17 | 2006-01-19 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions and related methods of use |
| WO2006121168A1 (en) | 2005-05-09 | 2006-11-16 | Ono Pharmaceutical Co., Ltd. | Human monoclonal antibodies to programmed death 1(pd-1) and methods for treating cancer using anti-pd-1 antibodies alone or in combination with other immunotherapeutics |
| WO2006122403A1 (en) | 2005-05-19 | 2006-11-23 | Merck Frosst Canada Ltd. | Quinoline derivatives as ep4 antagonists |
| WO2007005874A2 (en) | 2005-07-01 | 2007-01-11 | Medarex, Inc. | Human monoclonal antibodies to programmed death ligand 1 (pd-l1) |
| WO2007121578A1 (en) | 2006-04-24 | 2007-11-01 | Merck Frosst Canada Ltd. | Indole amide derivatives as ep4 receptor antagonists |
| WO2007143825A1 (en) | 2006-06-12 | 2007-12-21 | Merck Frosst Canada Ltd. | Indoline amide derivatives as ep4 receptor ligands |
| WO2008017164A1 (en) | 2006-08-11 | 2008-02-14 | Merck Frosst Canada Ltd. | Thiophenecarboxamide derivatives as ep4 receptor ligands |
| WO2008104055A1 (en) | 2007-02-26 | 2008-09-04 | Merck Frosst Canada Ltd. | Indole and indoline cyclopropyl amide derivatives as ep4 receptor antagonists |
| WO2008116304A1 (en) | 2007-03-26 | 2008-10-02 | Merck Frosst Canada Ltd. | Naphthalene and quinoline sulfonylurea derivatives as ep4 receptor antagonists |
| WO2008123207A1 (ja) | 2007-03-26 | 2008-10-16 | Astellas Pharma Inc. | オルニチン誘導体 |
| WO2008156712A1 (en) | 2007-06-18 | 2008-12-24 | N. V. Organon | Antibodies to human programmed death receptor pd-1 |
| WO2009005076A1 (ja) | 2007-07-03 | 2009-01-08 | Astellas Pharma Inc. | アミド化合物 |
| WO2009139373A1 (ja) | 2008-05-14 | 2009-11-19 | アステラス製薬株式会社 | アミド化合物 |
| WO2010019796A1 (en) | 2008-08-14 | 2010-02-18 | Chemietek, Llc | Heterocyclic amide derivatives as ep4 receptor antagonists |
| WO2010034110A1 (en) | 2008-09-25 | 2010-04-01 | Merck Frosst Canada Ltd. | Beta-carboline sulphonylurea derivatives as ep4 receptor antagonists |
| WO2012039972A1 (en) | 2010-09-21 | 2012-03-29 | Eisai R&D Management Co., Ltd. | Pharmaceutical composition |
| WO2012043634A1 (ja) | 2010-09-29 | 2012-04-05 | 株式会社エヌビィー健康研究所 | ヒトプロスタグランジンe2受容体ep4に対する抗体 |
| WO2012076063A1 (en) | 2010-12-10 | 2012-06-14 | Rottapharm S.P.A. | Pyridine amide derivatives as ep4 receptor antagonists |
| WO2012103071A2 (en) | 2011-01-25 | 2012-08-02 | Eisai R&D Management Co., Ltd. | Compounds and compositions |
| WO2013004290A1 (en) | 2011-07-04 | 2013-01-10 | Rottapharm S.P.A. | Cyclic amine derivatives as ep4 receptor antagonists |
| WO2013096501A1 (en) | 2011-12-21 | 2013-06-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013096496A2 (en) | 2011-12-21 | 2013-06-27 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013101598A1 (en) | 2011-12-27 | 2013-07-04 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2013101733A1 (en) | 2011-12-27 | 2013-07-04 | Allergan, Inc. | Compounds acting at multiple prostaglandin receptors giving a general anti-inflammatory response |
| WO2014004230A1 (en) | 2012-06-29 | 2014-01-03 | Eli Lilly And Company | Dimethyl-benzoic acid compounds |
| WO2014004229A1 (en) | 2012-06-29 | 2014-01-03 | Eli Lilly And Company | Phenoxyethyl piperidine compounds |
| WO2014086739A1 (de) | 2012-12-06 | 2014-06-12 | Bayer Pharma Aktiengesellschaft | Neuartige benzimidazolderivate als ep4-antagonisten |
| WO2014126746A1 (en) | 2013-02-15 | 2014-08-21 | Eli Lilly And Company | Phenoxyethoxy compounds |
| WO2014186218A1 (en) | 2013-05-17 | 2014-11-20 | Eli Lilly And Company | Phenoxyethyl dihydro-1h-isoquinoline compounds |
| WO2014200075A1 (ja) | 2013-06-12 | 2014-12-18 | 科研製薬株式会社 | 4-アルキニルイミダゾール誘導体およびそれを有効成分として含有する医薬 |
| WO2015094902A1 (en) | 2013-12-17 | 2015-06-25 | Eli Lilly And Company | Phenoxyethyl cyclic amine derivatives and their activity as ep4 receptor modulators |
| WO2015094912A1 (en) | 2013-12-17 | 2015-06-25 | Eli Lilly And Company | Dimethylbenzoic acid compounds |
| WO2015091475A1 (de) | 2013-12-19 | 2015-06-25 | Bayer Pharma Aktiengesellschaft | Benzimidazolderivate als ep4-liganden |
| WO2015113057A1 (en) | 2014-01-27 | 2015-07-30 | Allergan, Inc. | Antagonists acting at multiple prostaglandin receptors for the treatment of inflammation |
| WO2015147020A1 (ja) | 2014-03-26 | 2015-10-01 | アステラス製薬株式会社 | アミド化合物 |
| WO2016021742A1 (en) | 2014-08-07 | 2016-02-11 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds as ep4 receptor antagonists |
| WO2016088903A1 (en) | 2014-12-05 | 2016-06-09 | Takeda Pharmaceutical Company Limited | Heterocyclic compounds |
| WO2016111347A1 (ja) | 2015-01-09 | 2016-07-14 | 小野薬品工業株式会社 | 三環性スピロ化合物 |
| WO2017014177A1 (ja) * | 2015-07-17 | 2017-01-26 | 凸版印刷株式会社 | 健康状態の評価方法及び抗がん剤に対する長期奏功性の予測方法 |
| WO2017014323A1 (en) | 2015-07-23 | 2017-01-26 | Takeda Pharmaceutical Company Limited | 1-substituted 1,2,3,4-tetrahydro-1,7-naphthyridin-8-amine derivatives and their use as ep4 receptor antagonists |
| WO2017066633A1 (en) | 2015-10-16 | 2017-04-20 | Eisai R&D Management Co., Ltd. | Ep4 antagonists |
| WO2017085198A1 (en) | 2015-11-20 | 2017-05-26 | Actelion Pharmaceuticals Ltd | N-substituted indole derivatives as pge2 receptor modulators |
| WO2018008711A1 (ja) | 2016-07-07 | 2018-01-11 | 小野薬品工業株式会社 | Ep4拮抗薬と免疫チェックポイント阻害薬を含んでなる組み合わせ |
| JP2018021013A (ja) * | 2016-07-07 | 2018-02-08 | 小野薬品工業株式会社 | 医薬用途 |
| WO2018195123A1 (en) | 2017-04-18 | 2018-10-25 | Tempest Therapeutics, Inc. | Bicyclic compounds and their use in the treatment of cancer |
| WO2018210992A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Pyrimidine derivatives |
| WO2018210987A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Benzofurane and benzothiophene derivatives as pge2 receptor modulators |
| WO2018210994A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Phenyl derivatives as pge2 receptor modulators |
| WO2018210988A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | Pyrimidine derivatives as pge2 receptor modulators |
| WO2018210995A1 (en) | 2017-05-18 | 2018-11-22 | Idorsia Pharmaceuticals Ltd | N-substituted indole derivatives |
| WO2018216640A1 (ja) | 2017-05-22 | 2018-11-29 | 小野薬品工業株式会社 | Ep4アンタゴニスト |
| CN108929281A (zh) | 2017-05-27 | 2018-12-04 | 华东师范大学 | 三氮唑类化合物及其合成方法和应用 |
| WO2019038156A1 (en) | 2017-08-22 | 2019-02-28 | Bayer Pharma Aktiengesellschaft | USE OF AN EP4 ANTAGONIST FOR THE TREATMENT OF ARTHRITIS |
| WO2019101171A1 (zh) | 2017-11-27 | 2019-05-31 | 上海邦耀生物科技有限公司 | 噻吩并环类化合物及其合成方法和应用 |
| WO2019117269A1 (ja) * | 2017-12-13 | 2019-06-20 | 国立大学法人広島大学 | 膵がんの検出を補助する方法 |
| WO2019152982A1 (en) | 2018-02-05 | 2019-08-08 | Shenzhen Ionova Life Science Co., Ltd. | Heterobicyclic compounds as ep4 antagonists |
| WO2019166022A1 (en) | 2018-03-02 | 2019-09-06 | Shenzhen Ionova Life Science Co., Ltd. | Heterobicyclic carboxylic acids and salts thereof |
| WO2019245590A1 (en) | 2018-06-18 | 2019-12-26 | Avista Pharma Solutions, Inc. | Chemical compounds |
| WO2020014465A1 (en) | 2018-07-11 | 2020-01-16 | Arrys Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| WO2020012305A1 (en) | 2018-07-12 | 2020-01-16 | Rottapharm Biotech S.R.L. | (r)-4-(1-(1-(4-(trifluoromethyl)benzyl)pyrrolidine-2-carboxamide)cyclopropyl)-benzoic acid as ep4 receptor antagonist |
| WO2020111018A1 (ja) * | 2018-11-27 | 2020-06-04 | 小野薬品工業株式会社 | 免疫チェックポイント阻害薬およびfolfirinox療法との併用によるがん治療 |
| WO2020151566A1 (zh) | 2019-01-22 | 2020-07-30 | 凯复制药有限公司 | 抑制pge2/ep4信号传导的化合物、其制备方法及其在医药上的应用 |
| WO2020160075A1 (en) | 2019-01-30 | 2020-08-06 | Avista Pharma Solutions, Inc. | Chemical compounds |
| WO2020161275A1 (en) | 2019-02-08 | 2020-08-13 | Medibiofarma, S.L. | New n-benzyl-2-phenoxybenzamide derivatives as prostaglandin e2 (pge2) receptors modulators |
Non-Patent Citations (14)
| Title |
|---|
| "105th Annual Meeting of American Association for Cancer Research (AACR), Abstract: LB-265", TITLE OF PRESENTATION: ONO-AE3-208 INHIBITS MYELOID DERIVED SUPPRESSOR CELLS AND GLIOMA GROWTH, DATE OF PRESENTATION, 8 April 2014 (2014-04-08) |
| "Molecular Design", vol. 7, 1990, HIROKAWA PUBLISHING COMPANY, article "Development of Drugs", pages: 163 - 198 |
| CANCER CELL, vol. 27, 2015, pages 450 - 461 |
| CANCER RESEARCH, vol. 62, 2002, pages 28 - 32 |
| CANCER RESEARCH, vol. 70, 2010, pages 1606 - 1615 |
| CANCER SCIENCE, vol. 105, 2014, pages 1142 - 1151 |
| CHAN KELVIN, SHAH KEYA, LIEN KELLY, COYLE DOUG, LAM HENRY, KO YOO-JOUNG: "A Bayesian Meta-Analysis of Multiple Treatment Comparisons of Systemic Regimens for Advanced Pancreatic Cancer", PLOS ONE, vol. 9, no. 10, 6 October 2014 (2014-10-06), pages e108749, XP055929395, DOI: 10.1371/journal.pone.0108749 * |
| EDWARD B GARON, TUDOR-ELIADE CIULEANU, OSCAR ARRIETA, KUMAR PRABHASH, KONSTANTINOS N SYRIGOS, TUNCAY GOKSEL, KEUNCHIL PARK, VERA G: "Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial", THE LANCET, ¬J.B. FLINT|, vol. 384, no. 9944, 1 August 2014 (2014-08-01), pages 665 - 673, XP055184786, ISSN: 01406736, DOI: 10.1016/S0140-6736(14)60845-X * |
| FEBS LETTERS, vol. 364, 1995, pages 339 - 341 |
| JOURNAL OF LIPID MEDIATORS AND CELL SIGNALLING, vol. 12, 1995, pages 379 - 391 |
| NATURE REVIEWS CANCER, vol. 12, 2012, pages 252 - 264 |
| PHARMACOLOGICAL REVIEWS, vol. 65, July 2013 (2013-07-01), pages 1010 - 1052 |
| See also references of EP4245301A4 |
| YOH, TOMOAKI: "A Case of Effective Neoadjuvant Chemoradiotherapy with Capecitabine for Locally Advanced Sigmoid Colon Cancer", JAPANESE JOURNAL OF CANCER AND CHEMOTHERAPY, vol. 38, no. 6, 1 January 2011 (2011-01-01), JP , pages 1021 - 1024, XP009536660, ISSN: 0385-0684 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20230390303A1 (en) | 2023-12-07 |
| TW202228674A (zh) | 2022-08-01 |
| JP7816164B2 (ja) | 2026-02-18 |
| EP4245301A4 (en) | 2024-08-21 |
| JPWO2022102731A1 (ja) | 2022-05-19 |
| EP4245301A1 (en) | 2023-09-20 |
| KR20230107228A (ko) | 2023-07-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6885390B2 (ja) | Axl阻害剤と免疫チェックポイント阻害剤とを組み合わせて投与することを特徴とする癌治療のための医薬 | |
| KR102800132B1 (ko) | Sting 작동 화합물 | |
| JP7020218B2 (ja) | Ep4拮抗薬と免疫チェックポイント阻害薬を含んでなる組み合わせ | |
| JP7156287B2 (ja) | Axl阻害剤を有効成分として含むがん治療剤 | |
| JP6698648B2 (ja) | ユビキチン活性化酵素阻害物質及び化学療法剤の投与 | |
| US10208034B2 (en) | Quinoline derivative | |
| JP6697453B2 (ja) | ユビキチン活性化酵素阻害物質及び放射線の投与 | |
| WO2019074116A1 (ja) | Axl阻害剤を有効成分として含む固形がん治療剤 | |
| EP3560926B1 (en) | 6-amino-7,9-dihydro-8h-purin-8-one compounds as brk inhibitors | |
| JP6022691B2 (ja) | ジオキシノ−及びオキサジン−[2,3−d]ピリミジンpi3k阻害剤化合物及び使用方法 | |
| JPWO2018216640A1 (ja) | Ep4アンタゴニスト | |
| TWI781405B (zh) | 使用磺醯胺化合物與免疫調節劑之癌併用療法 | |
| JP7816164B2 (ja) | Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 | |
| JP2026067904A (ja) | Ep4拮抗薬と免疫チェックポイント阻害物質との併用によるがん治療 | |
| RU2820817C2 (ru) | Комбинированное лечение злокачественного новообразования с использованием сульфонамидного соединения и иммунорегулятора | |
| RU2798265C2 (ru) | Соединение, обладающее агонистической активностью в отношении sting |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21891976 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022562190 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021891976 Country of ref document: EP Effective date: 20230613 |