WO2022149617A1 - 化合物ライブラリ - Google Patents
化合物ライブラリ Download PDFInfo
- Publication number
- WO2022149617A1 WO2022149617A1 PCT/JP2022/001007 JP2022001007W WO2022149617A1 WO 2022149617 A1 WO2022149617 A1 WO 2022149617A1 JP 2022001007 W JP2022001007 W JP 2022001007W WO 2022149617 A1 WO2022149617 A1 WO 2022149617A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- equivalents
- carrier
- library
- compound
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images














Classifications
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B50/00—Methods of creating libraries, e.g. combinatorial synthesis
- C40B50/14—Solid phase synthesis, i.e. wherein one or more library building blocks are bound to a solid support during library creation; Particular methods of cleavage from the solid support
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
- C40B30/04—Methods of screening libraries by measuring the ability to specifically bind a target molecule, e.g. antibody-antigen binding, receptor-ligand binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/38—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/66—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings being part of condensed ring systems and singly-bound oxygen atoms, bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/21—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/44—Radicals substituted by doubly-bound oxygen, sulfur, or nitrogen atoms, or by two such atoms singly-bound to the same carbon atom
- C07D213/46—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/65—One oxygen atom attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
- C07D217/06—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines with the ring nitrogen atom acylated by carboxylic or carbonic acids, or with sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/192—Radicals derived from carboxylic acids from aromatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/26—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/20—Radicals substituted by singly bound hetero atoms other than halogen by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/10—Libraries containing peptides or polypeptides, or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/14—Libraries containing macromolecular compounds and not covered by groups C40B40/06 - C40B40/12
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B50/00—Methods of creating libraries, e.g. combinatorial synthesis
- C40B50/14—Solid phase synthesis, i.e. wherein one or more library building blocks are bound to a solid support during library creation; Particular methods of cleavage from the solid support
- C40B50/18—Solid phase synthesis, i.e. wherein one or more library building blocks are bound to a solid support during library creation; Particular methods of cleavage from the solid support using a particular method of attachment to the solid support
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
- G01N33/54366—Apparatus specially adapted for solid-phase testing
Definitions
- the present disclosure relates to a compound library having structural diversity, a method for producing the same, a screening method, and the like.
- Non-Patent Documents 1 and 2 Expansion of compound libraries with structural diversity and screening methods are key techniques for acquiring hit compounds (hit generation) (Non-Patent Documents 1 and 2).
- hit generation hit compounds
- Non-Patent Documents 30 As a method for searching for a new compound from a known compound, the concept of scaffold hopping (identifying a compound having both "isotactical molecular structures" and "significantly differential molecular backbones” for the parent compound) was proposed in 1999.
- Non-Patent Document 30 widely accepted and used in the field of medical chemistry (Non-Patent Document 31).
- Non-Patent Document 27 Toug target (a general term for drug efficacy targets characterized by not having deep holes to which small molecules can bind, which was difficult with conventional small molecule drug discovery.
- Non-Patent Document 28 For example, inhibition of protein-protein interaction, inhibition of RNA-protein interaction, In contrast to (including inhibition of nucleic acid-nucleic acid interaction), it has been pointed out that compounds within the range of rule of 5 may not have sufficient affinity for proteins (Non-Patent Document 28). In addition, the relationship between the three-dimensionality of molecules and the probability of success in clinical trials (Non-Patent Document 29) has attracted attention, and a method for supplying a compound library rich in three-dimensionality has also been emphasized.
- Non-Patent Document 5 a method of acquiring a hit compound by FBDD (fragment-based drug discovery) is also used (Non-Patent Document 5), and acquisition of a hit compound leading to drug discovery has been reported for a plurality of target molecules (non-patent).
- Document 6 due to the small molecular weight of the fragment compound used, the affinity of the obtained hit compound is weak, and a growing or linking approach is required to improve the activity (Non-Patent Document 7).
- Structural information of the target molecule is indispensable for greatly increasing the activity, and it is not suitable for the target molecule without structural information (Non-Patent Document 2).
- Non-Patent Document 8 For target molecules with structural information, an approach that utilizes a computational method is used to search for the optimal linker that links fragments to each other (Non-Patent Document 8).
- a peptide library using amino acids as a building block is known (Non-Patent Document 9).
- the connection mode of each building block is limited to the amide bond by the amidation reaction, and the building block used is also limited to amino acids.
- each library was a library (Structurally Homogeneus Library) in which building blocks were connected around a common structure (Scaffold) (Non-Patent Document 10).
- the Patentularly Heterogeneus Library is a library in which the compounds constituting the library do not have a specific common structure (Structure) and are constructed by sequentially connecting building blocks, so that the structural diversity of the compounds can be enhanced. It has been reported (Non-Patent Document 11, Non-Patent Document 12).
- Non-Patent Document 14 a building block that does not utilize a protecting group is used.
- Non-Patent Document 16 a DNA-encoded library technique (DELT; DNA encoded-library technology) using a building block not limited to amino acids is known (Non-Patent Document 16).
- DELT has a feature of utilizing a DNA tag for decoding a compound having binding activity with a target molecule. Decoding by a DNA tag is to correctly show the structural information of each library compound in a one-to-one relationship by the sequence information. Therefore, the DNA tag needs to be bound in the same molecule as the library compound (for example, Non-Patent Document 20 and Non-Patent Document 21).
- Non-Patent Document 17 the DNA tag is damaged by the chemical reaction used. It has been reported that damage to this DNA tag makes it impossible to correctly decode compounds that bind to target molecules, and in fact, DELT libraries increase false negatives on a scale of 10 to the 8th power or higher.
- Non-Patent Document 22 a method using AS-MS (Affinity Selection Mass Spectrometry) when screening a molecule having binding activity with a target molecule is also known (Non-Patent Document 22).
- AS-MS Affinity Selection Mass Spectrometry
- This technique is recognized as an unbiased assay for screening compounds using affinity with target molecules without labeling or immobilization, and can also identify allosteric inhibitors.
- AS-MS an example of screening by mixing existing compounds existing as a single compound (Non-Patent Documents 23 and 24), and an example of utilization for derivative evaluation rather than use for hit generation (Non-Patent Documents). 25 and 26) have also been reported, and their usefulness as a method for screening compounds has been recognized.
- Non-Patent Document 25 There is also a report that a library synthesized by reagent mixture synthesis (one of the methods of Real mixture synthesis, a method of using a mixture of a plurality of building blocks to be combined) was screened by AS-MS (Non-Patent Document 25). , The use of the reagent library centering on the scaffold structure is limited. There is a need for a new compound library with structural diversity that can be used for the development of pharmaceuticals, but there is room for improvement in various respects in establishing such a compound library.
- JP2019-527672 (A) JP2020-524701 (A)
- the viewpoint of diversity of library compounds will be described.
- the majority of conventional compound libraries have been composed of a group of compounds with a defined scaffold (Structurally Homogeneous Library).
- Structurally Homogeneous Library By binding a group of building blocks having a common reactive functional group to a certain scaffold, the diversity of the pharmacophore derived from the structural diversity of the building blocks is generated.
- structural diversity is generated by combining multiple pharmacophores.
- attention has been focused only on the structural diversity of the pharmacophore, which is derived from the structure of the building block.
- the diversity of connecting parts (linkers) that connect building blocks has not been noticed.
- the compound library that includes diversity in the bonding mode that connects them has diversity in the configuration of the pharmacophore (that is, the distance and angle between the functional groups that form the pharmacophore).
- the linker will acquire affinity with the target molecule. Therefore, a library in which a core block containing a functional group forming a pharmacophore can be freely selected and a linker in between can be freely selected is extremely valuable because it can significantly expand the structural diversity.
- a library that considers the diversity of linkers using a building block having two or more reactive functional groups is not fully utilized.
- Structurally Heterogeneous Library is constructed by solid-phase synthesis
- the use of multifactional manufacturing block is limited, and a plurality of binding modes are used at one bonding point, but one specific type. It is limited to application to capping of reactive functional groups (introduction of building blocks to molecular ends) (Non-Patent Document 12). DELT using a plurality of linkers when introducing a chanin block inside a molecule has been reported (Non-Patent Document 21), but a quality problem described later remains.
- Non-Patent Document 8 is limited to searching for linkers and bonds while fixing the above.
- Non-Patent Document 15 discloses a peptide library formed only by an amide bond, 100,000 or more compounds are included in one reaction space for synthesis.
- Non-Patent Document 15 since there are many sequences that cannot be synthesized even though only the amide bond is used, false negatives occur, and it is difficult to obtain information on the affinity of the designed compound for the target.
- impurities due to the progress of a reaction other than the intended one and impurities due to the remaining unreacted synthetic intermediates are mixed, and as a result, false positives due to unintentionally produced compounds cannot be excluded.
- the present inventors limit the number of compounds contained in one reaction space and limit the number of flasks in order to rapidly synthesize a large-scale (for example, 1 million or more) compounds as a quality-guaranteed mixture.
- a large-scale (for example, 1 million or more) compounds as a quality-guaranteed mixture.
- a molecule is constructed using a building block (multifunctional binding-block) having two or more reactive functional groups in the molecule.
- the core block of the building block having a reactive functional group is not limited to a specific scaffold unlike many conventional libraries.
- various protecting groups can also be utilized for the preparation of a library that simultaneously achieves the above-mentioned junction diversity and quality assurance. That is, by utilizing a protecting group, avoidance of side reactions in the reaction at the time of bond formation, realization of junction diversity by exposing the functional group to be used in an arbitrary step, and arbitrary hydrogen bond donating property to the target compound. It can be possible to include a group.
- this method can actually significantly improve the molecular diversity of the combination of the core block and the linker, and the library can be obtained.
- a library that guarantees the quality of the constituent compounds (presence / absence of each compound, abundance ratio, and presence / absence of impurities) can be constructed.
- the library provides a hit compound that can specifically bind to the target molecule.
- the compound has the following structure in which the first core block (first CB), the first linker (first L), and the second core block (second CB) are covalently linked: (1st CB)-(1st L)-(2nd CB) Including
- the library comprises two or more first CBs, two or more first Ls, and two or more second CBs.
- the library is composed of one or more mixtures containing the compound of 1 ⁇ 10 2 to 1 ⁇ 10 5 . The library.
- [A2] The following structure in which the compound is further covalently linked to a second linker (second L) and a third core block (third CB): (1st CB)-(1st L)-(2nd CB)-(2nd L)-(3rd CB) Including The library according to [A1], wherein the library includes two or more types of second L and two or more types of third CB.
- [A3] The following structure in which the compound is further covalently linked to a third linker (third L) and a fourth core block (fourth CB): (1st CB) — (1st L) — (2nd CB) — (2nd L) — (3rd CB) — (3rd L) — (4th CB); (1st CB) — (1st L) — (2nd CB) (— (3rd L) — (4th CB)) — (2nd L) — (3rd CB); Including The library according to [A1] or [A2], wherein the library comprises two or more types of 3L and two or more types of 4CB.
- [A4] The library according to any one of [A1] to [A3], wherein the compound does not contain an oligonucleotide tag.
- the 1st L, 2nd L, and 3rd L are amide, sulfonamide, acylsulfonamide, amino, ether, thioether, ester, ketone, sulfonamide, single bond, ethine-1,2-diyl (-C ⁇ C). -),
- the library according to any one of [A1] to [A4] which comprises a structure independently selected from the group consisting of a nitrogen-containing 5-membered heteroarylene.
- [A7] The library according to [A5] or [A6], wherein the single bond is a carbon-nitrogen single bond or a carbon-carbon single bond.
- the first L, the second L, and the third L are amidation, sulfon amidation, acyl sulfon amidation, reductive amination, etherification, thioetherification, esterification, O-alkylation, N-alkylation, It is formed by a reaction independently selected from the group consisting of a ketone bond forming reaction, a carbon-nitrogen bond forming reaction, a carbon-carbon bond forming reaction, and a nitrogen-containing 5-membered heteroarylene bond forming reaction, [A1] to [ A7] The library described in any one of.
- the first L, the second L, and the third L are independent of the group consisting of amidation, sulfonamide, acyl sulfonamide, reductive amination, O-alkylation, and carbon-carbon bond formation reaction.
- the library according to any one of [A1] to [A8] formed by the selected reaction.
- the carbon-carbon bond forming reaction is an aromatic nucleophilic substitution reaction and / or a Buchwald-Hartwig coupling, and the carbon-carbon bond forming reaction is a Suzuki coupling, a Negishi coupling, or a still cup.
- the library according to any one of [A1] to [A9] which comprises 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more compounds.
- [A12] 1 ⁇ 10 7 or less, 5 ⁇ 10 6 or less, 1 ⁇ 10 6 or less, 5 ⁇ 10 5 or less, 1 ⁇ 10 5 or less, 50,000 or less, 20000 or less, 10000 or less, 8000 or less, 7000 or less, 6000 or less.
- the compounds constituting the compound library are 1) The standard deviation of the clogp value calculated for each compound is 1.2 or more, 1.3 or more, 1.4 or more, or 1.5 or more; 2) The variance of the clogp value calculated for each compound is 1.5 or more, 1.75 or more, 2.0 or more, or 2.5 or more; 3) The median Fsp3 of [(number of sp3 carbon atoms contained in the molecule) / number of atoms other than hydrogen contained in the molecule] calculated for each compound is 0.10 or more, 0.15 or more, and 0.
- the standard deviation of the molecular weight calculated for each compound is 25 or more, 50 or more, or 75 or more; 5)
- the median number of hydrogen bond-accepting groups in each compound is 2 to 30, 3 to 20, or 3 to 18; 6)
- the difference between the largest and smallest compounds for the number of hydrogen bond-donating groups in each compound is 0-10, 0-8, or 1-5; and 7) tPSA / Tropical Polar Surface calculated for each compound.
- Area / Topological Polar Surface Area Dispersion is 20 or greater, 25 or greater, 30 or greater,; and 8) Extended-concentivity hydrogens up to 6 bonds distinguished at the atomic level with Tanimoto Similarity (ECFP_6: atomic level distinction) calculated for each compound.
- the value calculated by ECFP_6 using Pipeline Topology 2017HF2 of Dasso Systems has a median value of 0.98 or less, 0.95 or less, 0.90 or less, 0.85 or less, or 0.80 or less ,; 9) Tanimoto Simality calculated for each compound (ECFP_6: Extended-concentrivity fingers up to 6 bonds distinguished at the atomic level, eg, ECFP_6 value calculated using a central value of Pipeline Pilot 2017HF2 of Dassault Systèmes). Is 0.50 or more, 0.55 or more, 0.60 or more, 0.65 or more, 0.70 or more, or 0.75 or more; The library according to any one of [A1] to [A12], which satisfies the condition of 1 or more.
- [A14] The library according to any one of [A1] to [A13], wherein the library is produced as a mixture of compounds of 1 ⁇ 10 2 to 1 ⁇ 10 5 .
- [A15] For one, two, or all selected from the group consisting of the first CB, the second CB, and the third CB. 1) The total number of carbon atoms, nitrogen atoms, and oxygen atoms contained in the constituent atoms is 1 or more and 18 or less, 2 or more and 17 or less, 3 or more and 16 or less, 4 or more and 15 or less, 5
- [A16] The description in any one of [A1] to [A15] containing at least one cyclic structure for one, two, or all selected from the group consisting of the first CB, the second CB, and the third CB. Library.
- [A17] For one, two, three, or all selected from the group consisting of the first CB, the second CB, the third CB, and the fourth CB. 1) The total number of carbon atoms, nitrogen atoms, and oxygen atoms contained in the constituent atoms is 1 or more and 18 or less, 2 or more and 17 or less, 3 or more and 16 or less, 4 or more and 15 or less, 5
- [A18] [A1] to [A17] containing at least one cyclic structure for one, two, three, or all selected from the group consisting of the first CB, the second CB, the third CB, and the fourth CB.
- the ratio of the compound (compound A) contained in the maximum molar amount to the compound (compound B) contained in the minimum molar amount is the ratio (compound A / compound B).
- [B1] A method for screening a compound that binds to a target molecule, and (1) providing the library according to any one of [A1] to [A23]; (2) Contacting the one or more mixtures constituting the library with the target molecule, respectively; (3) The method comprising identifying a compound that binds to a target molecule.
- [B2] The method according to [B1], wherein the target molecule is a protein, nucleic acid, polypeptide, or sugar chain.
- [B3] The method according to [B1] or [B2], wherein the identification of the compound that binds to the target molecule is carried out by AS-MS.
- [B4] The method according to any one of [B1] to [B3], wherein the mixture comprises 100 to 50,000, 100 to 10000, 200 to 5000, 400 to 3000, or 600 to 1500 compounds.
- [B5] Any of [B1] to [B4], in which a mixture of 2 to 100,000, 2 to 50,000, 2 to 20000, 2 to 10000, 2 to 5000, or 2 to 2500 contained in the library is screened. The method described.
- [C1] A method for manufacturing a library.
- a carrier carrier (first BB carrier carrier) to which the first building block (first BB) is covalently linked is prepared, wherein the first BB carrier is a na type of first building block (first BB 1 ⁇ . It is a solid phase carrier (first BB 1-na supported carrier) containing the first BB na ), and each of the first BB 1-na portions of the first BB 1-na supported carrier has a bond with the second building block (second BB). It has a reactive functional group for forming, and the reactive functional group contained in the first BB 1-na supporting carrier is one kind or two or more kinds.
- a mixture of supporting carriers having a common reactive functional group is prepared, and each mixture is used as one kind of second building block introduction reagent (second BB introduction reagent) or two or more kinds.
- second BB-1 BB carrier carrier in which the second BB is covalently linked to the first BB by reacting with a mixture of the second BB introduction reagents of the above, the carrier carrier obtained here is of the nb type.
- na is an integer of 2 or more
- nb is an integer of 2 or more
- the obtained compound library contains a compound of 100 or more as a mixture.
- a mixture of supported carriers having a common reactive functional group among the first BB 1-na supported carriers is prepared, and each mixture is reacted with one of the nb types of second BB introduction reagents.
- the 2nd BB 1-nb-1st BB 1-na supporting carrier each has a reactive functional group capable of forming a linker with the 3rd building block (3rd BB), and each 1st BB 1-na portion
- the reactive functional group contained at one in the second BB 1-nb portion is one type or two or more types, and further (c) among the second B 1-nb- 1st BB 1-na supporting carriers, the reactive functional group.
- a mixture of supported carriers having a common group is prepared, and each mixture is reacted with one kind of third building block introduction reagent (third BB introduction reagent) or a mixture of two or more kinds of third BB introduction reagents to be shared by the third BB.
- a carrier carrier (third BB-second BB-first BB carrier carrier or second BB- (third BB-) first BB carrier carrier) linked to the first BB or the second BB by binding is prepared, and the carrier carrier obtained here is A carrier carrier (3rd BB 1 -nc ⁇ 2nd BB 1-nb ⁇ 1st BB 1 -na carrying carrier, or 2nd BB 1-nb ⁇ (No.
- any of the 3rd BB 1 introduction reagent to the 3rd BB nc introduction reagent is the reactivity of each of the 2nd BB 1-nb-1st BB 1-na supporting carrier. Reacts with a functional group to form a second linker (2L),
- [C4] The method according to [C3], wherein the supported carrier obtained in (c) is a third BB 1-nc- 2nd BB 1 -nb- 1st BB 1 -na carrier.
- the 3rd BB 1-nc ⁇ 2nd BB 1-nb ⁇ 1st BB 1 -na supporting carrier or the 2nd BB 1-nb ⁇ (3rd BB 1-nc ⁇ ) 1st BB 1 -na supporting carrier is the 4th building, respectively.
- the above which has a reactive functional group for forming a bond with a block (4th BB) and is contained in each of the 1st BB 1 -na portion, the 2nd BB 1-nb portion, or the 3rd BB 1-nc portion.
- the number of reactive functional groups is one or more, and (d) 3rd BB 1-nc ⁇ 2nd BB 1-na ⁇ 1st BB 1 -na supporting carrier or 2nd BB 1-nb ⁇ (3rd BB 1-nc ).
- a mixture of supporting carriers having a common reactive functional group is prepared, and each mixture is used as one kind of 4th building block introduction reagent (4th BB introduction reagent) or 2 or more kinds. Reacting with a mixture of 4th BB-introducing reagents to prepare a carrier on which the 4th BB is covalently linked to the 3rd BB, 2nd BB, or 1st BB, where the carrier obtained is the nd type 4th building block.
- any one of the 4th BB 1 -introducing reagent to the 4th BB nd -introducing reagent is a 3rd BB 1-nc- 2nd BB 1 -nb- 1st BB 1 -na supporting carrier.
- it reacts with the respective reactive functional groups of the 2nd BB 1 -nb ⁇ (3rd BB 1-nc ⁇ ) 1st BB 1 -na supporting carrier to form a third linker.
- the method according to any one of [C1] to [C4], wherein nd is an integer of 2 or more, and the first linker, the second linker, or the third linker of the compound contained in the library is two or more kinds. ..
- the carrier obtained in (d) is the carrier. 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ 1st BB 1 -na supporting carrier; 4th BB 1 -nd ⁇ (3rd BB 1-nc ⁇ ) 2nd BB 1-nb ⁇ 1st BB 1 -na supporting carrier; 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ (4th BB 1 -nd ⁇ ) 1st BB 1-na supporting carrier; 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ (2nd BB 1-nc ⁇ ) 1st BB 1 -na supporting carrier; or 4th BB 1 -nd ⁇ 2nd BB 1-nb ⁇ (3rd BB 1-nc ⁇ )
- the method according to [C5] which is a 1BB 1-na supported carrier.
- the carrier obtained in (d) is the carrier. 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ 1st BB 1 -na supporting carrier; The method according to [C5] or [C6].
- the first linker and the second linker are 1-1) There are two or more types of first linkers; 1-2) Two or more types of second linker; and 1-3) Three or more types of first or second linker; Satisfy one or more of the above, or.
- the first linker, the second linker and the third linker are 2-1) There are two or more types of first linkers; 2-2) There are two or more types of second linkers; 2-3) There are two or more types of third linkers; 2-4) There are three or more types of first linker, second linker or third linker; The method according to any one of [C1] to [C7], which satisfies the condition of 1 or more.
- na is an integer of 2 to 1000
- nb is an integer of 2 to 1000
- either na or nb is an integer of 3 or more
- na is an integer of 2 to 1000
- nb is.
- the first BB-supporting carrier obtained by reacting one of the first BB 1 -introducing reagents to the 1st BB na -introducing reagent with a solid-phase carrier to introduce the first BB into the solid-phase carrier is used.
- [C11] A mixture of 2 to na types of the 1st BB introduction reagent of the 1st BB 1 introduction reagent to the 1st BB na introduction reagent is reacted with the solid phase carrier to introduce the 1st BB into the solid phase carrier, and the 1st BB 1-
- Each of the 1st BB 1-na moieties of the 1st BB 1-na carrying carrier has one reactive functional group to form a bond with the 2nd building block (2nd BB) and / or.
- the second BB 1-nb portion of the second BB 1- nb -1st BB 1-na carrying carrier each has one reactive functional group to form a bond with the third building block (3rd BB) [C1]. ] ⁇ [C11].
- Each of the 1st BB 1-na moieties of the 1st BB 1-na carrying carrier has one reactive functional group to form a bond with the 2nd building block (2nd BB) and / or.
- Each of the 1st BB 1-na moieties of the 2nd BB 1-nb-1st BB 1-na carrying carrier has one reactive functional group for forming a bond with the 3rd building block (3rd BB) [C1]. ] ⁇ [C11].
- [C14] The method according to any one of [C1] to [C13], further comprising a step of deprotecting, protecting, or inducing the reactive functional group.
- [C15] The method according to [C14], which comprises a step of washing the solid phase-supported carrier with a solution mixed with an acidic component after the step of deprotecting the reactive functional group.
- [C16] The method according to [C15], wherein the acidic component is an acidic compound having a pKa of 0 to 10 in water.
- [C17] The group consisting of 1-hydroxy-7-azabenzotriazole (HOAt), 1-hydroxybenzotriazole (HOBt), ethyl (hydroxyimino) ethyl cyanoacetate (Oxyma) and 4-nitrophenol.
- [C18] The method according to any one of [C15] to [C17], wherein the functional group produced by the step of deprotecting the reactive functional group is either carboxyl or hydroxy substituted with an aromatic ring.
- [C19] The method according to any one of [C1] to [C18], further comprising the step of linking two crosslinkable groups present in the molecule to obtain a cyclic compound.
- [C20] The method according to any one of [C1] to [C19], further comprising modifying the functional group contained in the compound cut out from the solid phase carrier in the step (e).
- [C21] The method according to [C20], wherein the functional group is a functional group generated by excision of a compound from a solid phase carrier.
- [C22] The method according to either [C20] or [C21], wherein the modification is a C 1-6 alkylation of hydroxy or carboxy.
- a different second BB introduction reagent having higher reactivity is used in the first BB-supporting carrier in which the conversion rate of the reaction between the second BB introduction reagent and the reactive functional group of the first BB is less than 100%. Further comprising reacting with the remaining reactive functional group of the first BB and / or in (c) above, the conversion rate of the reaction between the third BB introduction reagent and the reactive functional group is less than 100% of the second BB-.
- the first BB-carrying carrier further comprises reacting a different, more reactive third BB-introducing reagent with the remaining reactive functional groups, and / or in (d) above, the fourth BB-introducing reagent and the reactive functional group.
- the UV tag has the formula 1: [In the formula, ring A is selected from benzene, naphthalene, anthracene and biphenyl; The benzene, the naphthalene, the anthracene and the biphenyl are halogen atoms, nitro, cyano, carboxy, sulfoxy, -COR 1 , -OR 1 , -NR 1 R 2 , -CONR 1 R 2 , or -SO 2 NR 1 .
- the UV tag is the formula 2 or the formula 3: (In the formula, X is a divalent group selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O-, the group is bound to the building block, and Y and Z is selected from the group consisting of hydrogen atom, halogen, nitro, cyano, carboxy, sulfoxyl, -COR 1 , -OR 1 , -NR 1 R 2 , -CONR 1 R 2 , or -SO 2 NR 1 R 2 . It may be substituted with the same or different 1 to 3 groups, and R 1 and R 2 may independently form a C 1-6 alkyl group or two together to form a heterocycle).
- the method according to [C24] which is a structure represented by.
- the UV tag has the formula 3: (In the formula, X is one selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O-, and represents that it is bound to a building block, and Y and Z are hydrogen. From the group consisting of atom, hydrogen atom, nitro, cyano, carboxy, sulfoxyl, -COR 1 , -O-R 1 , -NR 1 R 2 , -CO-NR 1 R 2 , or -SO 2 -NR 1 R 2 . It may be substituted with one to three selected identical or different groups, even if R 1 and R 2 are independently C 1-6 alkyl groups or two together to form a heterocycle. good) The method according to [C24], which is a structure represented by.
- the UV tag is the formula 4: (In the formula, X represents one selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O- and is bound to the building block, where R 1 and R 2 are. Each independently may form a heterocycle with a C 1-6 alkyl or a nitrogen atom to which they are linked).
- [C29] The method according to any one of [C1] to [C28], wherein the compound contained in the library does not contain an oligonucleotide tag.
- [C30] The method according to any one of [C1] to [C23] and [C29], wherein the compound contained in the library does not contain a UV tag.
- [C31] Further, the following steps: 1) A step of obtaining a solid phase-supported carrier obtained in any of the steps; 2) A step of cutting out the intermediate compound from the solid phase-supported carrier obtained in step 1); and 3) a step of analyzing the intermediate compound; In addition, optionally, a step of analyzing the compound cut out in the step (e).
- the method according to any one of [C1] to [C30] which comprises.
- [C32] The method according to [C31], wherein the analysis is a step of analyzing using a liquid chromatogram.
- the first L, the second L, the third L and the fourth L are amidation, sulfon amidation, acyl sulfon amidation, reductive amination, etherification, thioetherification, esterification, O-alkylation, N-alkyl. It is formed by a reaction independently selected from the group consisting of chemical formation, ketone bond formation reaction, carbon-nitrogen bond formation reaction, carbon-carbon bond formation reaction, and nitrogen-containing 5-membered heteroarylene formation reaction, [C1] ⁇ . The method according to any one of [C32].
- the 1st L, 2nd L, 3rd L and 4th L are amide, sulfonamide, acyl sulfonamide, amino, ether, thioether, ester, ketone, sulfon, single bond, etin-1,2-diyl (-C). ⁇ C-), and the method according to any of [C1] to [C33], which comprises a structure independently selected from the group consisting of a nitrogen-containing 5-membered heteroarylene.
- the carbon-nitrogen bond forming reaction is an aromatic nucleophilic substitution reaction and / or a buckwald-heartwig coupling, and the carbon-carbon bond forming reaction is a Suzuki coupling, a Negishi coupling, a still cup.
- [C36] The method according to any one of [C1] to [C35], wherein the obtained library contains 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more compounds.
- the obtained compound library is 1 ⁇ 10 8 or less, 1 ⁇ 10 7 or less, 5 ⁇ 10 6 or less, 1 ⁇ 10 6 or less, 500000 or less, 100000 or less, 50,000 or less, 20000 or less, 10000 or less, 8000 or less,
- the method according to any one of [C1] to [C36] which comprises a compound of 7000 or less, 6000 or less, 5000 or less, 4000 or less, 3000 or less, 2000 or less, 1500 or less, or 1200 or less.
- [C38] The library according to any one of [C1] to [C37], wherein the production of 90% or more of the target compounds for which synthesis is set as the compound constituting the library is confirmed.
- [C39] Of the target compounds whose synthesis was set as the compounds constituting the library, production was confirmed for 91% or more, 92% or more, 93% or more, 94% or more, 95% or more, or 96% or more of the compounds.
- [C40] The library according to [C38] or [C39], wherein the confirmation of synthesis is based on the retention time and mass-to-charge ratio (m / z) in a liquid chromatograph mass spectrometer (LC-MS).
- [C41] The method according to any one of [C1] to [C37], wherein the obtained library is the library according to any one of [A1] to [A23].
- [C42] 1 ⁇ 10 2 to 1 ⁇ 10 8 , 3 ⁇ 10 2 to 1 ⁇ 10 8 , 1 including the combination of compound libraries prepared by the method according to any one of [C1] to [C40].
- [D1] A method for forming a carbon-carbon bond by a Suzuki coupling reaction, in which a compound V having a desorbing group X8 on a carbon atom of an aromatic ring is substituted with the desorbing group to form a carbon-carbon bond.
- 1,3,5,7-Tetramethyl-6-phenyl-2,4,6-trioxa-6-phosphaadamantan is a boron atom-containing compound W in which a carbon atom is linked to a carbon atom, which is capable of forming a reaction.
- X 8 is a halogen atom or -O-SO 2 -R 5 ; R 5 is C 1-6 alkyl optionally substituted with one or more fluorine atoms, or substituted with one or more fluorine atoms or fluorine atoms.
- [D2] The method according to [D1], wherein the organic base is selected from the group consisting of organic bases having a conjugate acid pKa of 23 or more in acetonitrile, and two or more different bases may be used.
- [D3] The method according to any one of [D1] or [D2], wherein the organic base is selected from the group consisting of amidines, guanidines, and phosphazenes.
- the organic base is 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), 7-methyl-1,5,7-triazabicyclo [4.4.0] deca. -5-ene (MTBD), 1-ethyl-2,2,4,4,4-pentakis (dimethylamino) -2 ⁇ 5 , 4 ⁇ 5 -catenadi (phosphazen) (P2Et), 1-tert-butyl-2, 2,4,4,4-pentakis (dimethylamino) -2 ⁇ 5 , 4 ⁇ 5 -catenadi (phosphazen) (P2tBu), tert-butylimino-tris (dimethylamino) phosphorane (P1tBu), 2-tert-butylimino-2- Select from the group consisting of diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorin (BEMP), tert-butylimino-
- [D5] The method according to any one of [D1] to [D4], wherein the organic base is 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG).
- BTMG 2-tert-butyl-1,1,3,3-tetramethylguanidine
- the resin for solid-phase synthesis has compound Va in the side chain via a degradable linker.
- the compound Va comprises a compound represented by X8 - Ar8 as a part of a chemical structure.
- the Ar 8 contains phenyl, naphthyl, pyrrolyl, thienyl, frill, pyridyl, thiazolyl, isothiazolyl, pyrazolyl, oxazolyl, isoxazolyl, imidazolyl, triallyl, pyrimidyl, pyrazinyl, pyridadinyl, quinolinyl, isoquinolinyl, 4H-quinolinyl, phthalazinyl.
- the solid-phase synthesis resin has a boron atom-containing compound Wa in the side chain via a decomposable linker.
- the boron atom-containing compound Wa is a compound represented by X 9 -Ar 9 or X 9 -CR 6 R 7 R 8 having a boron atom-containing group linked to a carbon atom by a boron atom.
- X 9 is -B (-OR 10 ) (-OR 11 ) , -BF 3K, or (a), (b), (c), or (d) below.
- M is lithium, sodium, or potassium
- R 10 and R 11 are independently hydrogen atoms, or C 1-6 alkyl which may be substituted with C 1-6 alkoxy or C 6-10 aryl, respectively, or R 10 and R 11 intervene. Together with the oxygen and boron atoms form a 5- to 8-membered saturated or unsaturated ring, the ring being selected from C 1-6 alkyl groups, C 1-6 alkoxy and C 6-10 aryl. It may be substituted with one or more substituents.
- Ar 9 is a C 6-10 aryl, or a 5- to 10-membered heteroaryl containing one or more ring heteroatoms independently selected from O, N and S, each of which is a fluorine atom, cyano, C. 1-6 alkyl, C 1-6 alkoxy, (C 1-6 alkoxy) carbonyl, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, (C 6-10 aryl) carbonyl amino, A 5- to 10-membered heteroarylcarbonylamino, a di (C 1-6 alkyl) amino, a 4- to 8-membered cyclic amino, an aminocarbonyl, containing one or more ring heteroatoms independently selected from O, N and S.
- R 6 , R 7 and R 8 are independent hydrogen atoms, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3-8 cycloalkyl, C 7-14 aralkyl, (C 1 ).
- the boron atom-containing compound Wa is X9 - Ar9 .
- Ar 9 is a C 6-10 aryl, or a 5- to 10-membered heteroaryl containing one or more ring hetero atoms independently selected from O, N and S, each of which is a fluorine atom, cyano, C.
- [D13] The method according to any one of [D1] to [D12], wherein the reaction is carried out in the presence of a solvent.
- the solvent is selected from the group consisting of halogen-based solvents, nitrile-based solvents, amide-based solvents, ether-based solvents, and aromatic hydrocarbon-based solvents, and two or more of them may be used.
- [D15] The method according to [D13], wherein the solvent is selected from the group consisting of ether solvents, and two or more kinds may be used.
- the ether solvent is tetrahydrofuran, diethyl ether, 2-methyltetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, 1,3-dioxolane, diisopropyl ether, cyclopentylmethyl ether, t-butylmethyl ether.
- the method according to [D15] which is selected from the group consisting of 4-methyltetrahydropyran and two or more thereof may be used.
- [D17] The method according to [D15], wherein the ether solvent is tetrahydrofuran.
- [D18] The method according to any one of [D1] to [D17], wherein the reaction is carried out at a reaction temperature of 0 ° C to 100 ° C.
- [D19] The method according to [D18], wherein the reaction temperature is 20 ° C to 90 ° C, 40 ° C to 80 ° C, 50 ° C to 70 ° C, or 55 ° C to 65 ° C.
- [D22] The method according to any one of [D1] to [D21], wherein the reaction is carried out in a mixture containing a resin for solid phase synthesis containing two or more different compounds Va in the side chain as a substrate.
- [D23] The method according to any one of [D1] to [D22], wherein the reaction is carried out in a mixture containing a resin for solid phase synthesis containing two or more different boron atom-containing compounds Wa in the side chain as a substrate. ..
- the mixture contains, as a substrate, a resin for solid-phase synthesis to which two or more different compounds Va are bonded, or a resin for solid-phase synthesis to which two or more different boron atom-containing compounds Wa are bonded, and is bonded to individual resins.
- the method for producing a compound constituting a compound library which comprises a method for forming a carbon-carbon bond by the method according to any one of [D1] to [D24].
- the present specification provides a library composed of compounds having various chemical structures. Further provided, an assay method using the library and a method for producing the library.
- FIG. 1 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 3.
- FIG. 2 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 4.
- FIG. 3 is a diagram showing an X-ray crystal structure (6QGG) of a complex of Bcl-2 protein registered in PDB (Protein Data Bank) and ABT-737 analog, and a chemical structural formula of ABT-737 analog. Is.
- FIG. 4 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 8.
- FIG. 1 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 3.
- FIG. 2 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 4.
- FIG. 3 is
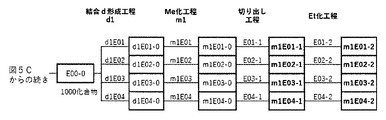
- FIG. 5A is a diagram showing a process name, a mixture name, and a library number for library synthesis in Example 8.
- the process name is written in the upper part of the horizontal line in FIGS. 5A, 5B, and 5C, and the mixture name is written in the square.
- the process name and the mixture name in FIGS. 5A, 5B, and 5C are described by omitting the library number "2-1".
- “h01” represents the process "2-2-h01”
- “h01A01-0” represents the mixture "2-1-h01A01-0”.
- FIG. 5B is a part of FIG. 5 showing the process name, mixture name, and library number of the library synthesis in Example 8.
- FIG. 5C is a part of FIG.
- FIG. 5 showing the process name, mixture name, and library number of the library synthesis in Example 8.
- FIG. 6 is a diagram showing "diversity of core blocks" and "diversity of linkers" included in the library synthesized in Example 9.
- FIG. 7A is a diagram showing a process name, a mixture name, and a library number for library synthesis in Example 9. The process name is written in the upper part of the horizontal line in FIGS. 7A, 7B, and 7C, and the mixture name is written in the square. The process name and mixture name in FIGS. 7A, 7B, and 7C are described by omitting the library number "2-2-". For example, “h01” represents the process “2-2-h01", and "a1B01-0" represents the mixture "2-2-a1B01-0".
- FIG. 7B is a part of FIG. 7 showing the process name, mixture name, and library number of the library synthesis in Example 9.
- FIG. 7C is a part of FIG. 7 showing the process name, mixture name, and library number of the library synthesis in Example 9.
- FIG. 8 is a diagram showing "diversity of core blocks” and “diversity of linkers” included in the library synthesized in Example 10 (case 1).
- FIG. 9 is a diagram showing "diversity of core blocks” and "diversity of linkers” included in the library synthesized in Example 10 (case 2).
- FIG. 10 is a diagram showing a process name, a mixture name, and a library number for library synthesis in Example 10 (case 1). The process name is written in the upper part of the horizontal line in FIG.
- FIG. 11 is a diagram showing a process name, a mixture name, and a library number for library synthesis in Example 10 (case 2). The process name is written in the upper part of the horizontal line in FIG. 11, and the mixture name is written in the square.
- the process name and mixture name in FIG. 11 are described by omitting the library number “4-2-2”. For example, “a1B05” represents the process “4-2-a1B05”, and “a1B05-0” represents the mixture "4-1-1B05-0”.
- C 1-6 alkyl is a monovalent derivative derived by removing one arbitrary hydrogen atom from linear and branched saturated aliphatic hydrocarbons having 1 to 6 carbon atoms. It is the basis. Specifically, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, t-butyl, 1-methylpropyl, n-pentyl, isopentyl, 2-methylbutyl, 1,1-dimethylpropyl, Examples include 1-ethylpropyl, hexyl, 4-methylpentyl, and 2-ethylbutyl.
- C 3-7 cycloalkyl means a cyclic saturated aliphatic hydrocarbon group having 3 to 7 carbon atoms. Specific examples thereof include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and the like.
- C 6-10 aryl means a monovalent aromatic hydrocarbon ring group.
- Examples of the C 6-10 aryl include phenyl, 1-naphthyl, 2-naphthyl and the like.
- halogen atom means a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like.
- preferred halogen atoms include a fluorine atom, a chlorine atom and a bromine atom.
- halogen atom is an alkyl or a substituent of a group containing an alkyl as a part thereof (alkoxy, alkenyl, alkylthio, etc.)
- a fluorine atom is mentioned as a preferable halogen atom.
- Specific examples of the group having a halogen atom as a substituent include trifluoromethyl, pentafluoroethyl, trifluoromethoxy, pentafluoroethoxy, trifluoromethylthio, and pentafluoroethylthio.
- a library composed of compounds having a given chemical structure is provided.
- the number of compounds constituting the library is not particularly limited as long as it can be used for screening, and the number of compounds may be, for example, 1 ⁇ 10 2 to 1 ⁇ 10 8 , 3 ⁇ 10 2 to 1 ⁇ 10 7 . It may be 5 ⁇ 10 2 to 1 ⁇ 10 6 and the like.
- the number of compounds means the number of compounds that are different in chemical structure, and tautomers and the like are the same compound.
- compounds having different chemical structures such as positional isomers and stereoisomers are understood as different compounds.
- a compound containing isotopes in a ratio significantly different from the natural abundance can be understood as a compound.
- the lower limit of the number of compounds constituting the library 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more are exemplified.
- the upper limit of the number of the compounds 1 ⁇ 10 8 or less, 1 ⁇ 10 7 or less, 5 ⁇ 10 6 or less, 1 ⁇ 10 6 or less, 500000 or less, 100000 or less, 50,000 or less, 20000 or less, 10000 or less, 8000 or less. , 7000 or less, 6000 or less, 5000 or less, 4000 or less, 3000 or less, 2000 or less, 1500 or less, or 1200 or less are exemplified.
- the compounds constituting the library are not particularly limited as long as they are compounds that can stably exist under screening conditions.
- the compounds may be prepared synthetically or may be partially obtained by purchase.
- the compound comprises an atom selected from a hydrogen atom, a carbon atom, an oxygen atom, a nitrogen atom, a sulfur atom, a halogen atom, and a phosphorus atom.
- the compound is only an atom selected from hydrogen atom, carbon atom, oxygen atom, nitrogen atom, sulfur atom, halogen atom (eg, fluorine atom, chlorine atom, bromine atom, iodine atom), and phosphorus atom. Consists of.
- the compound constituting the library is not particularly limited, but has, for example, a molecular weight in the range of 200 to 3000, specifically 300 to 2000, more specifically 300 to 1800, and more specifically 400 to 1800. ..
- the standard deviation of the molecular weights of the compounds constituting the library is not particularly limited, but is, for example, 25 or more, specifically 50 or more, and more specifically 75 or more.
- the compounds constituting the library are not particularly limited, but are, for example, 7 to 200, specifically 11 to 130, more specifically 11 to 120 carbon atoms; for example, 0 to 40, specifically. 2 to 27 nitrogen atoms, more specifically 2 to 24 nitrogen atoms; for example, 0 to 30, specifically 2 to 20, more specifically 2 to 18 oxygen atoms. include.
- each compound constituting the library is not particularly limited as long as it can be used for screening, and each compound is, for example, 0.001 to 50,000 ⁇ g, specifically 0.1 to 40,000 ⁇ g, and particularly specifically 1 to 30,000 ⁇ g. It may be in the range of.
- the ratio (Compound A / Compound B) of the compound (Compound A) contained in the maximum molar amount to the compound (Compound B) contained in the minimum molar amount in the mixture is 500 or less.
- the number of compounds contained in one mixture is not particularly limited as long as it is applicable to screening, and for example, 100 to 100,000, 100 to 10000, or 100 to 5000 different compounds are contained in the mixture. included.
- the compounds constituting the library are included in the mixture.
- the mixture is not particularly limited as long as it can be used for screening using a mixture of compounds as a sample, and may be a mixture of concentrated compounds or a solution diluted with a solvent or the like.
- the mixture may be prepared by mixing individually synthesized or obtained compounds, or a mixture obtained as a product by a chemical reaction using the mixture as a substrate may be used.
- the library is produced, for example, as a mixture of 100-100,000, 100-10000, or 100-5000 different compounds.
- the compounds that make up the library can be characterized by the number of sp3 carbon atoms contained in their chemical structure.
- the sp3 carbon atom means a carbon atom in which all bonds are only single bonds.
- the median value of [(the number of sp3 carbon atoms contained in the molecule) / the number of atoms other than hydrogen contained in the molecule] calculated for each compound is, for example, 0.1 or more, specifically. More specifically, it is 0.15 or more, more specifically, 0.2 or more, more specifically, 0.25 or more, and most specifically, 0.3 or more.
- the compounds that make up the library can be characterized by the number of oxygen atoms contained in their chemical structure.
- the oxygen atom means all the oxygen atoms contained in the chemical structure of the compound, and includes the oxygen atoms contained in the hydroxy group, the ether structure, the carbonyl group and the like.
- the number of oxygen atoms contained in the chemical structure of each compound is, for example, in the range of 0 to 30, specifically 2 to 20, and more specifically 2 to 18.
- the value of [(the number of oxygen atoms contained in the molecule) / the number of atoms other than hydrogen contained in the molecule] calculated for each compound is, for example, 0 to 0.20, specifically. Is in the range of 0.01 to 0.20, more specifically 0.03 to 0.17, and more specifically 0.06 to 0.14.
- the compounds that make up the library can be characterized by the number of nitrogen atoms contained in their chemical structure.
- the nitrogen atom means all nitrogen atoms contained in the chemical structure of the compound, and includes an intra-ring nitrogen atom such as an amino group and a pyridine ring, and a nitrogen atom contained in an amide group, an imino group and the like.
- the number of nitrogen atoms contained in the chemical structure of each compound is, for example, in the range of 0 to 40, specifically 2 to 27, and more specifically 2 to 24.
- the value of [(the number of nitrogen atoms contained in the molecule) / the number of atoms other than hydrogen contained in the molecule] calculated for each compound is, for example, 0 to 0.2, specifically. Is in the range of 0 to 0.18, more specifically 0.05 to 0.18.
- the total number of oxygen atoms and nitrogen atoms contained in the chemical structure of each compound is, for example, in the range of 1 to 70, specifically 2 to 60, and more specifically 2 to 40.
- the value of [(the number of oxygen atoms and nitrogen atoms contained in the molecule) / the number of atoms other than hydrogen contained in the molecule] calculated for each compound is, for example, 0.01 to 0. 35, specifically in the range of 0.02 to 0.3, more specifically in the range of 0.01 to 0.27.
- the total number of atoms other than carbon and hydrogen atoms contained in the chemical structure of each compound ranges from, for example, 1 to 85, specifically 2 to 56, and more specifically 3 to 50. It becomes the range of.
- the value of [(the number of carbon atoms and atoms other than hydrogen atoms contained in the molecule) / the number of atoms other than hydrogen contained in the molecule] calculated for each compound is, for example, 0.05. It is in the range of ⁇ 0.45, specifically 0.05 to 0.4, and more specifically 0.1 to 0.4.
- the compounds that make up the library can be characterized by the number of basic nitrogen atoms contained in their chemical structure.
- the basic nitrogen atom is a nitrogen atom contained in the basic compound
- the pKb of the compound in water is, for example, 14 or less, specifically 13.5 or less, more specifically 13 or less.
- the number of basic nitrogen atoms of the compounds constituting the library is, for example, in the range of 0 to 30, specifically 0 to 20, and more specifically 0 to 18.
- the compounds that make up the library can be characterized by the number of hydrogen bond-donating groups contained in their chemical structure.
- the hydrogen bond-donating group has a hydrogen atom bonded by a covalent bond, and is a non-covalent attractive mutual bond with an isolated electron pair such as nitrogen, oxygen, sulfur, fluorine, or a ⁇ -electron system located in the vicinity. It is a group that can cause action.
- the hydrogen atom possessed by the group is not particularly limited as long as the electron negative degree of the bonded atom is higher than that of the hydrogen atom. Can be mentioned.
- the number of hydrogen bond-donating groups of the compounds constituting the library is, for example, in the range of 0 to 20, specifically 0 to 15, and more specifically 0 to 10.
- the difference between the largest compound and the smallest compound in terms of the number of hydrogen bond-donating groups of each compound is, for example, 1 or more, specifically 2 or more, and more specifically 3 or more.
- the compounds that make up the library can be characterized by the number of hydrogen bond accepting groups contained in their chemical structure.
- the hydrogen bond-accepting group is a group having an isolated electron pair such as nitrogen, oxygen, sulfur, fluorine, and a ⁇ -electron system, and has a non-covalent bond attraction with a hydrogen atom bonded by a covalent bond located in the vicinity. It is a group that can cause a target interaction.
- the group having an isolated electron pair of the group is not particularly limited as long as it can exist stably, and for example, a carbonyl group such as a ketone group, an ester group, or an amide group, an ether group, an amino group, an imino group, and the like.
- the number of hydrogen bond accepting groups of the compounds constituting the library is, for example, in the range of 0 to 40, specifically 0 to 30, and more specifically 0 to 20.
- the difference between the largest compound and the smallest compound in terms of the number of hydrogen bond-donating groups of each compound is, for example, 1 or more, specifically 2 or more, and more specifically 3 or more.
- the compounds constituting the library have a chemical structure in which the first core block (first CB), the first linker (first L), and the second core block (second CB) are covalently linked.
- the following formula: (1st CB)-(1st L)-(2nd CB) It is represented by and is characterized by sharing the above chemical structure.
- the first CB and the second CB are arbitrary chemical structures containing at least one atom, and are not particularly limited as long as they are partial structures of the compound.
- the first CB and the second CB may have any chemical structure as long as they form a compound different from the other compounds constituting the library.
- the first CB and the second CB have the same chemical structure. May be good.
- first CB and the second CB may contain a chemical structure specified as a third core block (third CB) and / or a fourth core block (fourth CB) as a substitution as described later.
- first L is not particularly limited as long as it is an arbitrary chemical structure that connects the first CB and the second CB, and may be, for example, a direct bond (single bond, double bond, triple bond).
- the compounds constituting the library have the following structure in which the first CB, the first L, the second CB, the second linker (second L), and the third core block (third CB) are covalently linked. : (1st CB)-(1st L)-(2nd CB)-(2nd L)-(3rd CB) It is characterized by including.
- the first CB, the first L, and the second CB are as described above.
- the third CB is an arbitrary chemical structure containing at least one atom, and is not particularly limited as long as it is a partial structure of the compound.
- the third CB may have any chemical structure as long as it forms a compound different from the other compounds constituting the library, and may be combined with, for example, the same chemical structure as the first CB and / or the second CB. .. Further, the first CB, the second CB, and the third CB may contain a chemical structure specified as a fourth core block (fourth CB) as a substitution as described later.
- the second L is not particularly limited as long as it is an arbitrary chemical structure that connects the second CB and the third CB, and may be, for example, a direct bond (single bond, double bond, triple bond).
- the compounds constituting the library are covalently bound to the first CB, the first L, the second CB, the second L, the third CB, the third linker (third L), and the fourth core block (fourth CB).
- the following structure connected by: (1st CB)-(1st L)-(2nd CB)-(2nd L)-(3rd CB)-(3rd L)-(4th CB); (1st CB) — (1st L) — (2nd CB) (— (3rd L) — (4th CB)) — (2nd L) — (3rd CB); It is characterized by including.
- the second of the above equations is — (1L) — (1st CB), — (2nd L) — (3rd CB), and — (3L) — (4th CB). It means that it is connected to 2CB.
- the compound represented by the above two formulas is included in the range of the compounds represented by the above (first CB)-(first L)-(second CB).
- the fourth CB is an arbitrary chemical structure containing at least one atom, and is not particularly limited as long as it is a partial structure of the compound.
- the third CB may have any chemical structure as long as it forms a compound different from the other compounds constituting the library, and may be combined with, for example, the same chemical structure as the first CB and / or the second CB. ..
- the first CB, the second CB, and the third CB may contain a chemical structure specified as a fourth core block (fourth CB) as a substitution as described later.
- the second L is not particularly limited as long as it is an arbitrary chemical structure that connects the second CB and the third CB, and may be, for example, a direct bond (single bond, double bond, triple bond).
- (1st CB) — (1st L) — (2nd CB) — (2nd L) — (3rd CB) — (3rd L) — (4th CB); and (1st CB) — (1st L) — (2nd CB) (-(3L)-(4th CB))-(2L)-(3rd CB); Includes compounds represented by.
- the first CB, the second CB, the third CB, and the fourth CB may further have a fifth core block (— (4L) — (5th CB)) linked via a fourth linker.
- a compound having a group — (4L) — (5th CB) the 4th L and the 5th CB are herein as the 1st L, the 2nd L, and the 3rd L, and the 1st CB, the 2nd CB, the 3rd CB, and the 4th CB, respectively. It may have the chemical structure specified in the book.
- the compound may further have a sixth core block (— (5L) — (6th CB)) linked via a fifth linker.
- Compounds having the groups — (4L) — (5th CB) and — (5L) — (6th CB) are synthesized by a method of sequentially introducing each of the groups based on the methods described herein. be able to.
- One aspect of the present invention is characterized in that the compounds constituting the library do not contain tags for decoding.
- the tag for decoding is not particularly limited as long as it can be used to specify the structure of the compound, and examples thereof include an oligonucleotide tag.
- DNA-encoded library (DEL) is known as a library having a tag for decoding.
- the library of the invention does not include a library encoded by an oligonucleotide represented by DEL.
- the linkers contained in the compound are amides, sulfone amides, acyl sulfone amides, aminos, ethers, thioethers, esters, ketones, sulfones, single bonds, It contains a structure independently selected from the group consisting of ethins-1,2-diyl (-C ⁇ C-), and nitrogen-containing 5-membered heteroarylene.
- R 1 and R 2 are independently hydrogen atoms or C 1-6 alkyl]. It is represented by an expression selected from.
- the single bond is a carbon-nitrogen single bond or a carbon-carbon single bond.
- the above linker covalently binds each core block, eg, two core blocks selected from a first CB, a second CB, a third CB, and a fourth CB, by reaction with a reactive functional group. It is derived from the chemical structure generated by linking.
- the first L, second L, and third L are amidation, sulfon amidation, acyl sulfon amidation, reductive amination, etherification, thioetherification, esterification, O-alkylation, N-alkyl.
- the carbon-nitrogen bond forming reaction is, for example, an aromatic nucleophilic substitution reaction and / or a buckwald-heartwig coupling
- the carbon-carbon bond forming reaction is a Suzuki coupling or a Negishi cup. Examples include rings, stille couplings and / or sonogashira couplings.
- the compound constituting the library is a compound containing the first CB to the third CB and the first L and the second L, and at least one selected from the group consisting of the first CB, the second CB, and the third CB.
- At least two, or all have a total number of carbon atoms, nitrogen atoms, and oxygen atoms of 2 or more and 17 or less, 3 or more and 16 or less, 4 or more and 15 or less, and 5 or more and 14 or less, respectively.
- Or 6 or more and 13 or less One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- the compound constituting the library is a compound containing the first CB to the third CB and the first L and the second L, and at least one selected from the group consisting of the first CB, the second CB, and the third CB. , At least two or all of them are characterized in that the total number of carbon atoms, nitrogen atoms and oxygen atoms is 1 or more and 18 or less, respectively.
- One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- the compound constituting the library is a compound containing the first CB to the third CB and the first L and the second L, and at least one selected from the group consisting of the first CB, the second CB, and the third CB.
- At least two, or all, are characterized by containing at least one or two annular structures.
- One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- the compound constituting the library is a compound containing the first CB to the fourth CB and the first L to the third L, and at least one of the first CB, the second CB, the third CB, and the fourth CB, at least two. At least three or all of them have a total number of carbon atoms, nitrogen atoms, and oxygen atoms of 2 or more and 17 or less, 3 or more and 16 or less, 4 or more and 15 or less, and 5 or more and 14 or less, respectively. , Or 6 or more and 13 or less.
- One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- the compound constituting the library is a compound containing the first CB to the fourth CB and the first L to the third L, and at least one of the first CB, the second CB, the third CB, and the fourth CB, at least two.
- One, at least three or all of them are characterized in that the total number of carbon atoms, nitrogen atoms and oxygen atoms is 1 or more and 18 or less, respectively.
- One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- the compound constituting the library is a compound containing the first CB to the fourth CB and the first L to the third L, and at least one of the first CB, the second CB, the third CB, and the fourth CB, at least two.
- One, at least three or all are characterized by containing at least one or two annular structures.
- One aspect of the present invention is characterized in that all the compounds constituting the library satisfy the above conditions.
- a library comprising 1 ⁇ 10 2 to 1 ⁇ 10 8 compounds, wherein the library has two or more first CBs, two or more first Ls, and two or more second CBs. Includes 2 or more 1st CBs, 2 or more 1Ls, 2 or more 2nd CBs, 2 or more 2nd Ls, and 2 or more 3rd CBs; or 2 or more 1st CBs, 2 Includes one or more types of 1L, two or more types of second CB, two or more types of second L, two or more types of third CB, two or more types of third L, and two or more types of fourth CB.
- the total number of types of the first L, the second L, and the third L is 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, or 8 or more. In another aspect of the invention, the total number of first L, second L, and third L types is 3-30, 4-30, 5-30, 6-30, 7-20, or 8-15. be.
- the present invention provides the following in one aspect: A library containing 1 ⁇ 10 2 to 1 ⁇ 10 8 compounds, wherein the compounds constituting the library have the following structures in which the first CB, the first L, the second CB, the second L, and the third CB are covalently linked: (1st CB)-(1st L)-(2nd CB)-(2nd L)-(3rd CB)
- the total number of first L and second L types is 3 or more, and the total number of first CB, second CB, and third CB types is 6 or more, and 1 ⁇ 10 2 to 1 ⁇ 10 5
- It is composed of one or more mixtures containing the above-mentioned compounds, and 90% or more of the compounds constituting the library have a retention time and a mass-to-charge ratio (m /) in a liquid chromatograph mass spectrometer (LC-MS).
- the library detected by z).
- the present invention provides the following in one aspect: A library containing 1 ⁇ 10 2 to 1 ⁇ 10 4 compounds, wherein the first CB, the first L, the second CB, the second L, the third CB, the third L, and the fourth CB are covalently bonded.
- the library is 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 20 or more, 30 or more, 40 or more, 50 or more, or 100 or more of the above libraries.
- the library is 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 or more, 20 or more, 30 or more, 40 or more, 50 or more.
- library aggregates prepared in combination of 100 or more are provided.
- the library aggregate is, for example, 200 or more, 300 or more, 400 or more, 500 or more, 600 or more, 700 or more, 800 or more, 900 or more, 1000 or more, 2000 or more, 4000 or more, 10000 or more, 50,000 or more, 100,000.
- the above compounds can be included.
- the individual libraries constituting the library assembly described above can be a mixture of compounds.
- Screening can be performed using the libraries described herein. That is, in one embodiment of the invention, a method of screening for a compound that binds to a target molecule is provided, which method (1) provides the library described herein; (2) Contacting the one or more mixtures constituting the library with the target molecule, respectively; (3) It is characterized by comprising identifying a compound that binds to a target molecule.
- the target molecule is not particularly limited as long as it is a molecule that is a target in the search for a drug, and for example, a protein, nucleic acid, polypeptide, sugar chain, etc., specifically, an enzyme, a G-protein coupled receptor, an ion channel, etc. , Transporters, nuclear receptors, etc.
- Specific examples of the target molecule include Bcl-2, IL-6, A2aR, NMDA-type glutamate receptor, estrogen receptor and the like.
- the method for identifying a compound that binds to a target molecule is not particularly limited as long as it is a method known to those skilled in the art in the technical field to which the present invention belongs, but the method can specify the chemical structure of the compound that binds to the target molecule. Method is preferable.
- a method using AS-MS Affinity Selection Mass Spectrometry
- SPR Surface Plasma Resonance, surface plasmon resonance
- X-ray crystal structure analysis a cryo-electron microscope were used.
- a method, and the like can be mentioned, and a preferred example is a method using AS-MS.
- the method using AS-MS can be carried out by the method described in, for example, Current Opinion in Chemical Biology 2007, 11: 518-526, for example, 96-well Macro Spin column, P-2 Material (Harvard, 74-5655). ), Panquish UHPLC (Thermo Fisher Scientific) and the like.
- Examples of the number of compounds contained in the mixture include 100 to 50,000, 100 to 10000, 200 to 5000, 400 to 3000, 600 to 1500 and the like.
- the number of mixtures to be screened is not particularly limited and may be one mixture, and the lower limit is 1 or more, 2 or more, 3 or more, 4 or more, 5 or more, 6 or more, 7 or more, 8 or more, 9 or more, 10 20 or more, 30 or more, 40 or more, 50 or more, or 100 or more are exemplified, and the upper limit is 100,000 or less, 50,000 or less, 20000 or less, 10000 or less, 5000 or less, 3000 or less, 2000 or less, or 1000 or less. Then, for example, screening may be performed for each of 1 to 5000, 1 to 3000, 2 to 2000, or 2 to 1500 mixtures.
- a carrying carrier in which the first BB is covalently linked by using (a) a first building block introduction reagent (first BB introduction reagent) of the na type. 1BB 1-na carrying carrier) is prepared, (b) a mixture of carrying carriers having a common reactive functional group among the first BB 1-na carrying carriers is prepared, and each mixture is used as one kind of second building block.
- a carrier carrier in which the second BB of nb type is covalently linked to the first BB by reacting with the introduction reagent (second BB introduction reagent) or a mixture of two or more kinds of second BB introduction reagents.
- the introduction reagent second BB introduction reagent
- the compound represented by the second BB 1-nb-1st BB 1-na can be obtained as a mixture thereof in part or in whole.
- a library can be constructed from the compound.
- the compounds represented by the second BB 1-nb-1st BB 1 -na are na type 1st BB carrier introduction reagent ( 1st BB 1-na introduction reagent) and nb type 2nd BB introduction reagent (2nd BB 1-nb ). It includes a group of compounds of up to na ⁇ nb or a part thereof, which are produced from a carrier-introducing reagent), and a group of compounds obtained by further modifying the group of compounds.
- na is an integer of 2 or more, 3 or more, 4 or more, or 5 or more
- nb is an integer of 2 or more, 3 or more, 4 or more, or 5 or more.
- the resulting compound library can contain 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more compounds as a mixture.
- the first BB-supporting carrier is a solid-phase carrier (first BB 1-na- supporting carrier) that supports the first building block (first BB 1-na ) of the na type, and the first BB 1- na -supporting carrier.
- first BB 1-na- supporting carrier supports the first building block (first BB 1-na ) of the na type, and the first BB 1- na -supporting carrier.
- Each of the 1st BB 1-na moieties of the carrier has a reactive functional group for forming a bond with the 2nd building block (2nd BB).
- the reactive functional group contained in the first BB 1-na supporting carrier is one type, two or more types, or three or more types.
- the solid-phase carrier and one kind of the first BB - introduced reagent are reacted using each of the first BB 1 -supported carrier to the first BB na -supported carrier, and the first BB 1-supported carrier to the first BB na -supported carrier are supported.
- Each of the carriers can be obtained as one type of first BB-supported carrier and used to obtain a mixture of first BB-supported carriers (first BB 1-na- supported carrier).
- the mixture of the first BB 1-na- supported carrier obtained by reacting the mixture of the first BB introduction reagent (for example, the mixture of the first BB 1-na introduction reagent) with the solid phase carrier can be used in (b). ..
- a mixture of a part of the first BB introduction reagent may be used as a mixture of a part of the first BB 1-na carrying carrier, and the reaction of the first BB introduction reagent and the first BB introduction reagent to be included in the mixture. It can be selected based on the type of sex functional group.
- the carrier obtained here is a carrier carrier (second BB 1-nb-1st BB 1-na carrier carrier) containing nb types of second building blocks (second BB 1 to 2nd BB nb ).
- 2nd BB 1 -introduced reagent to 2nd BB nb -introduced reagent reacts with each reactive functional group of the 1st BB 1-na- supported carrier to form a first linker (1L).
- the solid phase carrier that can be used in the method for producing the library described in the present specification is not particularly limited as long as it is usually used.
- Examples thereof include CTC resin, Trt resin, SASRIN resin, Linkamido resin, Merrifield resin, Wang resin, 2- (4-bromomethylphenoxy) ethyl polystyrene, and carboxyl group, amino group, aminomethyl group on the polystyrene.
- Examples thereof include a solid phase carrier having an arbitrary functional group such as a hydroxy group and a hydroxymethyl group.
- an arbitrary linker that covalently connects the carrier and the first BB may be used, and the design may be such that cutting can be performed between the linker and the first BB.
- the carrier is not particularly limited, and examples thereof include PEG (polyethylene glycol) in addition to polystyrene.
- the conditions for the reaction between the first BB and the solid phase carrier can be appropriately set by those skilled in the art based on the methods described in publicly known documents and the like.
- the reaction conditions for cutting out the compound from the solid phase carrier can be appropriately set by those skilled in the art based on the chemical structure of the solid phase carrier used.
- the reagent used for cutting include hydrochloric acid, carboxylic acids such as trifluoroacetic acid (TFA), 2,2,2-trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoro.
- fluoroalcohols such as isopropyl alcohol (HFIP), blended acids having a pKa of 10 or less in water, or any Lewis acid can be used.
- the compound excised from the solid phase carrier is obtained as a mixture.
- a method for producing a library comprising (a), (b), and (e), wherein the second BB 1-nb-1st BB 1-na supporting carrier, respectively, is the first. It has a reactive functional group capable of forming a linker with 3 building blocks (3rd BB), and one kind of the reactive functional group is contained in each 1st BB 1-na part or 2nd BB 1-nb part.
- a method comprising preparing a second BB-first BB-supported carrier, or a second BB- (third BB-) first BB-supported carrier) is provided.
- the obtained carrier is a carrier (3rd BB 1-nc ⁇ 2nd BB 1-nb ⁇ 1st BB 1 -na ) containing a third building block (3rd BB 1 to 3rd BB nc ) of nc type. It is a carrier carrier or a second BB 1-nb ⁇ (3rd BB 1-nc ⁇ ) 1st BB 1 -na carrier carrier), and any one of the 3rd BB 1 introduction reagent to the 3rd BB nc introduction reagent is the 2nd BB 1-nb ⁇ . It reacts with each reactive functional group of the first BB 1-na supporting carrier to form a second linker (second L).
- nc is an integer of 2 or more, 3 or more, 4 or more, or 5 or more
- the first linker or the second linker of the compound contained in the library is two or more kinds.
- the third BB 1-nc moiety is linked to the first BB 1 -na moiety via the second linker.
- a reactive functional group having a structure and protected by a protecting group is present in the first BB 1-na moiety, and after introducing the second BB 1-nb moiety, the group is deprotected to deprotect the third BB 1- . It can be introduced by forming a second linker with the reactive functional group of the nc introduction reagent.
- the carrier obtained in (c) is a third BB 1-nc- 2nd BB 1 -nb- 1st BB 1 -na carrier and a second BB 1-nb-1st BB 1-na carrier.
- Each of the second BB 1-nb moieties has one reactive functional group for forming a bond with the third building block (third BB).
- One aspect of the present invention is a method for producing a library, which comprises (a), (b), (c), and (e), wherein the third BB 1-nc ⁇ the second BB 1-nb ⁇ first.
- the reactive functional group having a group and contained one in each of the first BB 1 -na part, the second BB 1-nb part, or the third BB 1-nc part is one kind, two kinds or more, or three kinds or more.
- a mixture of supported carriers having a common reactive functional group is prepared, and each mixture is reacted with one kind of 4th building block introduction reagent (4th BB introduction reagent) or a mixture of 2 or more kinds of 4th BB introduction reagents, and the first
- a method is provided comprising preparing a carrier in which 4BB is covalently linked to a third BB, a second BB, or a first BB.
- the supported carrier obtained in (e) is a supported carrier containing an nd type of fourth building block (4th BB 1 to 4th BB nd ), and any one of the 4th BB 1 introduction reagent to the 4th BB nd introduction reagent is the first. Reacts with the respective reactive functional groups of 3BB 1- nc ⁇ 2nd BB 1- nb ⁇ 1st BB 1 -na supporting carrier or 2nd BB 1-nb ⁇ (3rd BB 1-nc ⁇ ) 1st BB 1 -na supporting carrier.
- the fourth BB is introduced by forming the third linker.
- the reactive functional group forming the 4th BB introduction reagent and the 3rd linker may have a protecting group, in which case deprotection is performed before the reaction with the 4th BB introduction reagent.
- nd is an integer of 2 or more, 3 or more, 4 or more, or 5 or more, and the first linker, the second linker, or the third linker of the compound contained in the library is two or more kinds.
- the carrier carrier obtained in (d) is 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ 1st BB 1 -na supporting carrier; 4th BB 1 -nd ⁇ (3rd BB 1-nc ⁇ ) 2nd BB 1-nb ⁇ 1st BB 1 -na supporting carrier; 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ (4th BB 1 -nb ⁇ ) 1st BB 1-na supporting carrier; 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ (2nd BB 1-nc ⁇ ) 1st BB 1 -na supporting carrier; or 4th BB 1 -nd ⁇ 2nd BB 1-nb ⁇ (3rd BB 1-nc ⁇ ) It is a 1BB 1-na supporting carrier.
- the carrier obtained in (d) may be a mixture of two or more supported carriers represented by the above formulas.
- the carrier obtained in (d) is the 4th BB 1 -nd ⁇ 3rd BB 1-nc ⁇ 2nd BB 1 -nb ⁇ 1st BB 1 -na carrier.
- the first linker and the second linker are 1-1) There are two or more types of first linkers, three or more types, or four or more types; 1-2) 2 or more types of 2nd linker, 3 or more types, or 4 or more types; and 1-3) 1st or 2nd linker is 3 or more types, 4 or more types, or 5 or more types. ; It is characterized by satisfying one or more conditions of.
- first linker, the second linker and the third linker are 2-1) There are two or more types of first linkers, three or more types, or four or more types; 2-2) There are two or more types of second linkers, three or more types, or four or more types; 2-3) 2 or more types of 3rd linker, 3 or more types, or 4 or more types; and 2-4) 3 or more types of 1st, 2nd or 3rd linker, 4 or more types, or 5 More than a kind; It is characterized by satisfying one or more conditions of.
- na is an integer of 2 to 1000
- nb is an integer of 2 to 1000
- either na or nb is an integer of 3 or more.
- na is an integer of 2 to 1000
- nb is an integer of 2 to 1000
- nc is an integer of 2 to 1000
- any of na, nb or nc is 3. It is the above integer.
- the method according to any one of [C1] to [C33], wherein the obtained library contains 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more compounds.
- the number of compounds contained in the library obtained by the library synthesis method is 800 or less, 1000 or less, 1200 or less, 1500 or less, 1700 or less, 2000 or less, 2500 or less, 3000 or less, 4000 or less, 5000 or less.
- 6000 or less, 7000 or less, 8000 or less, 10000 or less, 20000 or less, 50,000 or less, or 100,000 or less can be exemplified with an upper limit of 100 or more, 200 or more, 300 or more, 400 or more, 500 or more, or 1000 or more. Can be exemplified as the lower limit.
- the library synthesis method may further comprise the steps of deprotecting and / or protecting the reactive functional groups.
- the supporting carrier can be washed with a solution mixed with an acidic component to remove impurities.
- an acidic component an acidic compound having a pKa of 0 to 10 in water can be used.
- the acidic component include 1-hydroxy-7-azabenzotriazole (HOAt), 1-hydroxybenzotriazole (HOBt), ethyl (hydroxyimino) ethyl cyanoacetate (Oxyma) and 4-nitrophenol. ..
- the functional group produced by the deprotection step of the reactive functional group is not particularly limited as long as it is a group having appropriate reactivity, and may be, for example, carboxyl or hydroxy substituted with an aromatic ring.
- the library synthesis method may further include the step of linking two crosslinkable groups present in the molecule to obtain a cyclic compound.
- the cyclization step can be performed, for example, after cutting out the compound from the solid phase carrier. Alternatively, it can be performed on a solid phase carrier before cutting (also referred to as cyclization on a resin or cyclization on a resin).
- the two crosslinkable groups present in the molecule include, for example, a combination of a carboxyl group and an amino group, a combination of a carboxyl group and a hydroxy group, a combination of an acetylene group and an azido group, and an arbitrary substituent substituted at an arbitrary position. Examples include combinations of alkens and combinations of thiol groups.
- the cyclization step can be carried out using a mixture of the obtained compounds as a substrate after cutting out the compound from the solid phase carrier.
- the method of synthesizing a library may further comprise modifying (or sometimes referred to as inducing) a functional group contained in a compound contained in the resulting library.
- the functional group to be modified is not particularly limited, and examples thereof include a hydroxy group, a carboxyl group, and an amino group.
- the functional group obtained by modification include ether, amide, ester, sulfonamide, and acyl sulfonamide.
- the step required for modifying the functional group may be a single step or a plurality of steps.
- the functional group is a functional group produced by excision of a compound from a solid phase carrier, and examples of the modification include C 1-6 alkylation of hydroxy or carboxy.
- the reaction between the building block introduction reagent and the reactive functional group preferably has a conversion rate of 100%, but a reaction that does not necessarily reach 100% is also assumed.
- the reactivity is higher in the first BB-supporting carrier in which the conversion rate of the reaction between the second BB introduction reagent and the reactive functional group of the first BB is less than 100%. It further comprises reacting the second BB introduction reagent with the remaining reactive functional groups of the first BB.
- the reactivity is higher in the second BB-1st BB carrying carrier in which the conversion rate of the reaction between the third BB introduction reagent and the reactive functional group is less than 100%.
- the conversion rate of the reaction between the 4th BB introduction reagent and the reactive functional group is less than 100%, the third BB-2nd BB-1st BB carrying carrier or the 2nd BB-.
- the first BB carrying carrier further comprising reacting a different, more reactive fourth BB-introducing reagent with the remaining reactive functional groups.
- one or more compounds in the library may or may not contain UV tags. If one or more compounds in the library, eg, all compounds, contain UV tags, then as UV tags, eg, Formula 1: [In the formula, ring A is selected from benzene, naphthalene, anthracene and biphenyl; The benzene, the naphthalene, the anthracene and the biphenyl are halogen atoms, nitro, cyano, carboxy, sulfoxy, -COR 1 , -OR 1 , -NR 1 R 2 , -CONR 1 R 2 , or -SO 2 NR 1 .
- R 2 It may be substituted with the same or different 1 to 3 groups selected from the group consisting of R 2 , where R 1 and R 2 are independently combined with a C 1-6 alkyl or linked nitrogen atom, respectively. Therefore, a heterocycle may be formed, and X is one selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O-, and is bound to the building block. show) A group having the structure represented by can be introduced into the compound.
- Equation 2 or Equation 3 (In the formula, X is a divalent group selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O-, the group is bound to the building block, and Y and Z is selected from the group consisting of hydrogen atom, halogen, nitro, cyano, carboxy, sulfoxyl, -COR 1 , -OR 1 , -NR 1 R 2 , -CONR 1 R 2 , or -SO 2 NR 1 R 2 . It may be substituted with the same or different 1 to 3 groups, and R 1 and R 2 may independently form a C 1-6 alkyl group or two together to form a heterocycle). A group having the structure represented by may be introduced into the compound.
- formula 3 (In the formula, X is one selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O-, and represents that it is bound to a building block, and Y and Z are hydrogen. From the group consisting of atom, hydrogen atom, nitro, cyano, carboxy, sulfoxyl, -COR 1 , -O-R 1 , -NR 1 R 2 , -CO-NR 1 R 2 , or -SO 2 -NR 1 R 2 . It may be substituted with one to three selected identical or different groups, even if R 1 and R 2 are independently C 1-6 alkyl groups or two together to form a heterocycle. good) A group having the structure represented by may be introduced into the compound.
- the formula 4 (In the formula, X represents one selected from the group consisting of -CONH-, -NHCO-, -CH 2 O- and -O- and is bound to the building block, where R 1 and R 2 are. Each independently may form a heterocycle with a C 1-6 alkyl or a nitrogen atom to which they are linked). A group having the structure represented by may be introduced into the compound.
- the compound contained in the library is characterized by containing no UV tag and / or an oligonucleotide tag.
- the compounds constituting the library contain UV tags and can be used for the analysis of intermediate compounds in solid phase synthesis of the compounds.
- the method of making the library further 1) A step of obtaining a supported carrier obtained in any of the steps; 2) A step of cutting out the intermediate compound from the carrier obtained in step 1); and 3) a step of analyzing the intermediate compound;
- a step of analyzing the compound cut out in the step (e) including.
- the results obtained from the analysis of intermediate compounds can be used for optimizing the reaction conditions of solid-phase reactions, which enables the production of high-quality libraries.
- the analysis can be performed using a liquid chromatogram.
- the first linker, second linker, third linker and fourth linker formed in the method for producing a library are amidated, sulfonated, acylsulfonamided, reductive amidation, and the like.
- the chemical structure formed by the reaction to be carried out is exemplified.
- the first linker, second linker, third linker and fourth linker are amides, sulfonamides, acylsulfonamides, aminos, ethers, thioethers, esters, ketones, sulfones, single bonds, ethins-1. , 2-Zyl (-C ⁇ C-), and contains structures independently selected from the group consisting of nitrogen-containing 5-membered heteroarylenes.
- the method for producing a library can be performed for producing the library according to any one of [A1] to [A23].
- the first linker, the second linker, the third linker and the fourth linker formed in the method for producing a library are the first linker, the second linker formed in the method for producing a library defined with respect to the above library.
- reagents base, condensing agent, catalyst, ligand, etc.
- base condensing agent, catalyst, ligand, etc.
- reagents having the same function may be used in combination at the same time (for example, DIPEA as a base). Potassium phosphate, etc.).
- the equivalent of the reagents shown below indicates how many times the number of moles of the reagent is used with respect to the total number of moles of the functional groups used for forming the linker in each compound of the mixture on the solid phase side.
- the equivalent of the substrate on the liquid phase side can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to the substrate on the solid phase side. It can be equivalent, or 1 to 10 equivalents.
- reaction time can be appropriately set by those skilled in the art, and for example, it is necessary to reach a sufficient conversion rate in a study experiment (also referred to as a model experiment) using the corresponding substrate or a similar substrate. It is possible to determine or confirm the reaction completion time by cutting out the product from a part of the solid phase carrier, or the time expected by some method from the examination result.
- a and B shown in the following formulas each indicate a core block to which a reactive functional group is linked, and each core block may be further linked to another core block via a linker. Further, either A or B includes a solid phase carrier.
- R1 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom and B to be bonded].
- the amide bond as a linker can be formed by the reaction of an amine with a carboxylic acid or a derivative thereof.
- X of the reactive functional group linked to A is a hydroxy or halogen atom.
- a condensing agent can be used to form an amide bond as a linker by subjecting it to a reaction with an amine containing an amino group linked to B.
- the condensing agent is not particularly limited as long as it can be usually used, but for example, N, N'-dicyclohexylcarbodiimide, N, N'-diisopropylcarbodiimide, 1-ethyl-3- (3-dimethylaminopropyl).
- the number of equivalents of the condensing agent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to the carboxylic acid containing A. It can be ⁇ 10 equivalents.
- the equivalent number of amines to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to carboxylic acid containing A. It can be 10 equivalents.
- R1 in the above equation is as already defined.
- Preferred examples of R1 are hydrogen atoms, C 1-6 alkyls which may have substituents, such as methyl and ethyl, and aromatic rings which may have substituents, such as benzene and ethyl. Examples include pyridine.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 200 ° C., 0 to 150 ° C., 0 to 100 ° C., 0 to 80 ° C., or 10 to 80 ° C.
- Bases may be used if desired, eg, pyridine, 2,6-lutidine, 2,4,6-colysine, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, potassium hydride, etc.
- bases are used as bases. be able to.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to carboxylic acid containing A. It can be 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and the reaction time required in the model experiment or the reaction end time can be determined or confirmed by cutting out the product from a part of the solid phase carrier. After completion of the reaction, the desired compound can be obtained by a usual post-treatment method.
- X is a halogen atom
- specific examples thereof include a chlorine atom or a fluorine atom.
- the reaction between the acid halide and the amine may be carried out in the presence of a base, or may be carried out using 2 equivalents or more of the amine.
- the bases that can be used here include pyridine, 2,6-lutidine, 2,4,6-coridine, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, potassium hydride, sodium hydride, and the like.
- Lithium hydride sodium amide, lithium amide, KOH, NaOH, LiOH, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, sodium phosphate, potassium phosphate, K 2 HPO 4, KHCO 3 , Na 2 HPO 4, NaHCO 3 , Sodium tetraborate, DBU, DBN, MTBD, BTMG, TMG, 1,8-bis (dimethylamino) naphthalene, LiOtBu, NaOtBu, KOtBu, potassium t-pentoxide, sodium t-pentoxide, lithium t-pentoxide, LiHMDS, NaHMDS, KHMDS, HMDS, Lithium Diisopropylamide, Lithium Dicyclohexylamide, 2,2,6,6-Tetramethylpiperidinyl Lithium, 2,2,6,6-Tetramethylpiperidinyl Magnesium Chloride Lithium Chloride Complex, Examples thereof include N, N-d
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, relative to acid halides containing A. It can be 10 equivalents.
- the equivalent number of amines to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, relative to acid halides containing A. It can be 10 equivalents.
- R 1 in the above equation is as already defined. Preferred examples of R 1 include hydrogen atoms, C 1-6 alkyls such as methyl and ethyl.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, -80 to 150 ° C, -80 to 100 ° C, -80 to 80 ° C, -40 to 80 ° C, or 0 to 80 ° C.
- the acid halide used in the reaction may be prepared from the corresponding carboxylic acid in the reaction system.
- the reagent is not particularly limited as long as it can be usually used, but for example, thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride, phosphoryl chloride, sulfryl chloride, (chloromethylene) dimethyliminium chloride (chloromethylene) dimethyliminium chloride ( Vilsmeier reagent), 1-chloro-N, N, 2-trimethyl-1-propenylamine (Ghosez reagent), chloro-N, N, N', N'-bis (tetramethylene) formamidinium tetrafluoroboic acid, PyClU, PipClU, Chloro-N, N, N', N'-Tetramethylformamidinium Hexafluorophosphate, Fluoro-N, N, N', N'-T
- an amide solvent such as DMF may coexist.
- Bases may be used if desired, eg, pyridine, 2,6-lutidine, 2,4,6-colysine, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, potassium hydride, etc.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imithazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.),
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 40 to 150 ° C., ⁇ 40 to 100 ° C., ⁇ 40 to 80 ° C., ⁇ 20 to 80 ° C., or 0 to 80 ° C.
- the equivalent number of the reagents and bases to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, 1 to carboxylic acid containing A. It can be ⁇ 20 equivalents, 1 ⁇ 15 equivalents, 1 ⁇ 10 equivalents, or 1 ⁇ 5 equivalents.
- the equivalent number of amines to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, 1 to 20 with respect to the carboxylic acid containing A.
- the equivalent can be 1 to 15 equivalents, 1 to 10 equivalents, or 1 to 5 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- X of the reactive functional group linked to A is a hydroxy or halogen atom.
- a condensing agent can be used to form an amide bond as a linker by subjecting it to a reaction with an amine containing an amino group linked to B.
- the reagents and other reaction conditions used here are the same as when A contains a solid phase carrier.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to the amine containing B. It can be equivalent, or 1 to 10 equivalents.
- the equivalent number of the carboxylic acid to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to an amine containing B. It can be 30 equivalents, or 1 to 10 equivalents.
- A is an acid halide
- the equivalent number of bases to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to an amine containing B. It can be 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of the acid halide used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing B. It can be 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- R 1 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom and A to be bonded].
- the sulfonamide bond as a linker can be formed by the reaction of the amine with a sulfonyl halide or a derivative thereof.
- X of the reactive functional group linked to B is a halogen atom, for example, a chlorine atom or a fluorine atom, and an amine containing an amino group linked to A using a base is used.
- a sulfonamide bond as a linker can be formed.
- the base is not particularly limited as long as it can be usually used, but for example, pyridine, 2,6-lutidine, 2,4,6-colysium, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, and the like.
- NMI NMI, DMAP, potassium hydride, sodium hydride, lithium hydride, sodium amide, lithium amide, KOH, NaOH, LiOH, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, potassium phosphate, sodium phosphate, K 2 HPO 4, KHCO 3 , Na 2 HPO 4, Sodium tetraborate, DBU, DBN, MTBD, BTMG, TMG, 1,8-bis (dimethylamino) naphthalene, LiOtBu, NaOtBu, KOtBu, potassium t-pentoxide, sodium t-pentoxide, LiHMDS, NaHMDS, KHMDS, HMDS, lithium diisopropylamide, lithium dicyclohexylamide, 2,2,6,6-tetramethylpiperidinyllithium, 2,2,6,6-tetramethylpiperidinylmagnesium chloride Lithium chloride complex, N, N-dimethylaniline, P1-t
- the equivalent number of sulfonyl halides to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing A. It can be 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to an amine containing A. Can be equivalent.
- a solvent can be used in the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF (tetratetra), 2-methyltetrachloride, 4 -Ether-based solvents such as methyltetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethylether, diglyme, triglym, tetraglyme, acetone, MEK, MIBK, 4-methyl-2-pentanone, cyclo Ketone solvents such as pentanone, cyclohexanone and diethyl ketone, urea solvents such as TMU, DMI and DMPU, amide solvents such as DMF, DMA, NFM, NMP, NEP and N
- aromatic solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate, prop
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 100 ° C., 0 to 60 ° C., or 0 to 30 ° C.
- an additive may be used, and for example, NMI, DMAP, HOAt, oxyma, pentafluorophenol, pyridine N-oxide, NaI and the like can be used as the additive.
- the number of equivalents of the additive to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing A. It can be 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- X of the reactive functional group linked to B is a halogen atom.
- X may be prepared from hydroxy.
- the reagent is not particularly limited as long as it can be usually used, and for example, phosphorus oxychloride, phosphorus trichloride, thionyl chloride, phosgene and the like can be used.
- An amide-based solvent such as DMF may coexist.
- the reagents and other reaction conditions used here are the same as when A contains a solid phase carrier.
- the equivalent number of bases to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, and 1 to 50 equivalents with respect to the sulfonic acid halide containing B. It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of amines to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to sulfonic acid halide containing B. It can be ⁇ 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- R 2 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom, a sulfur atom and B to be bonded].
- the acyl sulfonamide bond as a linker can be formed by the reaction of a carboxylic acid or a derivative thereof with a sulfonamide.
- the reactive functional group X linked to A is hydroxy, and by subjecting it to a reaction with an amine containing an amino group linked to B using a condensing agent. It is possible to form an acyl sulfonamide as a linker.
- the condensing agent is not particularly limited as long as it can be usually used, but for example, DCC (N, N'-dicyclohexylcarbodiimide), DIC (N, N'-diisopropylcarbodiimide), EDCI (1-ethyl-).
- Carbodiimide-based condensing agents such as 3- (3-dimethylaminopropyl) carbodiimide hydrochloride) and HOAt (1-hydroxy-7-azabenzotriazole), HOBt (1-hydroxybenzotriazole), HOOBt (3,4-dihydro-
- activators such as 3-hydroxy-4-oxo-1,2,3-benzotriazole) and oxyma (cyano (hydroxyimino) ethyl acetate), HATU (O- (7-aza-1H-benzotriazole-).
- CFH chloro-N, N, N', N'-tetramethylformamidinium hexafluorophosphate
- PyCIU N, N, N', N'-bis (tetramethylene) chloroformamidinium hexafluorophosphate) Acid
- BTFFH fluoro-N, N, N', N'-bis (tetramethylene) formamidinium hexafluorophosphate
- TFFH fluoro-N, N, N', N'-tetramethylami Dinium hexafluorophosphate
- TCFH chloro-N, N, N', N'-tetramethylformamidinium hexafluorophosphate
- PyClU (1- (chloro-1-pyrrolidinylmethylene) pyrrolidi Nium hexafluorophosphate
- formamidinium salt-based condensing agents such as PipClU (chlorodipiperidino
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- reaction temperature can be appropriately set by those skilled in the art, and may be, for example, 0 to 25 ° C, 0 to 50 ° C, 0 to 80 ° C, -20 to 80 ° C, -20 to 140 ° C, or 0 to 140 ° C. can.
- Bases may be used if desired, eg, pyridine, 2,6-lutidine, 2,4,6-colysine, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, potassium hydride, etc.
- the number of equivalents of the condensing agent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to the carboxylic acid containing A. It can be ⁇ 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to carboxylic acid containing A. It can be 10 equivalents.
- the equivalent number of the sulfonamide containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, and 1 to 30 equivalents with respect to the carboxylic acid containing A. , Or 1 to 10 equivalents.
- R 1 in the above equation is as already defined.
- Preferred examples of R 1 include hydrogen atoms, C 1-6 alkyls such as methyl and ethyl.
- the reagent is not particularly limited as long as it can be usually used, but for example, thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus pentachloride, phosphoryl chloride, sulfryl chloride, (chloromethylene) dimethyliminium chloride (chloromethylene) dimethyliminium chloride ( Vilsmeier reagent), 1-chloro-N, N, 2-trimethyl-1-propenylamine (Ghosez reagent), chloro-N, N, N', N'-bis (tetramethylene) formamidinium tetrafluoroboic acid, PyClU, PipClU, Chloro-N, N, N', N'-Tetramethylformamidinium Hexafluorophosphate, Fluoro-N,
- an amide solvent such as DMF may coexist.
- Bases may be used if desired, eg, pyridine, 2,6-lutidine, 2,4,6-colysine, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, potassium hydride, etc.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 40 to 150 ° C., ⁇ 40 to 100 ° C., ⁇ 40 to 80 ° C., ⁇ 20 to 80 ° C., or 0 to 80 ° C.
- the equivalent number of the reagents and bases to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, 1 to carboxylic acid containing A. It can be ⁇ 20 equivalents, 1 ⁇ 15 equivalents, 1 ⁇ 10 equivalents, or 1 ⁇ 5 equivalents.
- the equivalent number of the sulfonamide containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, and 1 to 30 equivalents with respect to the carboxylic acid containing A. It can be 1 to 20 equivalents, 1 to 15 equivalents, 1 to 10 equivalents, or 1 to 5 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- X of the reactive functional group linked to A is halide (specifically, a chlorine atom or a fluorine atom) or hydroxy, and in the case of hydroxy, a condensing agent is used. It can be used to form an acylsulfonamide as a linker by subjecting it to a reaction with a sulfonamide containing an amino group linked to B.
- the acyl sulfonamide formation reaction can be carried out using the reagents and reaction conditions already described.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to a sulfonamide containing B. It can be equivalent, or 1 to 10 equivalents.
- the number of equivalents of the condensing agent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to sulfonamide containing B. It can be ⁇ 10 equivalents.
- the equivalent number of the carboxylic acid or acid halide containing A to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to the sulfone amide containing B. It can be up to 30 equivalents, or 1 to 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- R 3 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom and B to be bonded].
- the amine bond as a linker uses an aromatic nucleophilic substitution reaction using an amine with an aryl halide or a derivative thereof, a catalytic coupling reaction using a metal catalyst, or an alkyl halide, a hydroxyalkyl or a derivative thereof and an amine. It can be formed by the nucleophilic substitution reaction.
- the reaction described in the above formula can form a linker represented by ⁇ NR 3 ⁇ .
- a reaction between a primary amide or a secondary amide and an aryl halide or a derivative thereof, a nucleophilic substitution reaction using a primary amide or a secondary amide and an alkyl halide or a hydroxyalkyl derivative, and a primary sulfonamide or 2 It can also be formed by a reaction between a primary sulfonamide and an aryl halide or a derivative thereof, or a nucleophilic substitution reaction using a primary sulfonamide or a secondary sulfonamide and an alkyl halide or a hydroxyalkyl derivative.
- X of the reactive functional group linked to A is a group derived from a halogen or a hydroxy group.
- the group derived from the hydroxy group include methanesulfonyl group, trifluoromethanesulfonyl group, nonafluorobutanesulfonyl group, benzenesulfonyl group, paratoluenesulfonyl group, 2-nitrobenzenesulfonyl group, 4-nitrobenzenesulfonyl group, 2,4. -Dinitrobenzenesulfonyl groups and the like can be exemplified.
- AX can be subjected to a reaction with an amine linked to B in the presence of a base.
- the base is not particularly limited as long as it can be normally used, but for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium phosphate, potassium phosphate, and hydrogen phosphate di.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX.
- the equivalent number of the amine containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- R 3 in the above equation is as already defined.
- Preferred examples of R 3 are a hydrogen atom, a C 1-6 alkyl optionally having a substituent, such as methyl and ethyl, and an aromatic ring optionally having a substituent, such as benzene, pyridine. And so on.
- examples of the ring structure formed together with the nitrogen atom and B to be bonded include pyrrolidine, piperidine, piperazine, morpholine and the like, and examples of the heterocyclic aromatic amine include pyrrol, imidazole, pyrazole, indole, benzimidazole and the like. ..
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 150 ° C., 10 to 120 ° C., 20 to 100 ° C., 20 to 80 ° C., or 20 to 60 ° C.
- a metal catalyst may be used, for example, for palladium, for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allyl palladium chloride dimer, palladium ( ⁇ ).
- copper oxide (I) (Cu 2 O), copper iodide (I) (CuI), copper bromide ( I) (CuBr), copper (I) chloride (CuCl), copper acetate (Cu (OAc) 2 ), bis (acetylacetonato) copper (II) (Cu (acac) 2), copper 2-thiophenecarboxylate ( I) (CuTc) or the like can be used as a metal catalyst.
- a ligand may be used, and as the ligand of palladium, for example, trialkylphosphine (Cy 3 (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A ( Di (1-adamantyl) -n-butylphosphine), (tBu) 2 PMe (di-tert-butylmethylphosphine), P (Ad) 3 (tri (1-adamantyl) phosphine), neopentyl (tBu) 2 P ( Di-tert-butyl neopentylphosphine), triarylphosphine (PPh 3 (triphenylphosphine), etc.), dialkylbiarylphosphine (XPhos (2-dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl), etc.
- ligand can be used as a ligand.
- ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- diamine ligands include ditertbutylbipyridine (dtBBPy (4,4'-di-tert-butyl-2,2'-dipyridyl)) and dimethylethylenediamine (DMEDA (N, N'). -Diamineethylenediamine)), cyclohexyldimethylethylenediamine (CyDMEDA (trans-N, N'-dimethylcyclohexane-1,2-diamine)), 1,10-phenanthroline (Phen), etc.
- dtBBPy ditertbutylbipyridine
- DMEDA dimethylethylenediamine
- CyDMEDA cyclohexyldimethylethylenediamine
- Phen 1,10-phenanthroline
- 1,1 , 1-Trifluoro-3-phenyl-2- (1H-pyrrole-2-yl) propan-2-ol, 2-methyl-8-hydroxyquinoline and the like can be used as the ligand.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0 with respect to AX. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX.
- the equivalent number of the catalyst precursor to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the aromatic nucleophilic substitution reaction with an amine and the catalytic coupling reaction using a metal catalyst are the reagents already described. It can be carried out depending on the reaction conditions.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to the amine containing B. It can be equivalent, or 1 to 10 equivalents.
- the equivalent number of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to an amine containing B. It can be ⁇ 10 equivalents.
- the equivalent number of the metal catalyst can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 with respect to the amine containing B. It can be equivalent, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the catalyst precursor to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 with respect to the amine containing B. It can be equivalent, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the X of the reactive functional group linked to A is a group derived from a desorbing group, for example, a halogen or a hydroxy group, and is hydroxy.
- the groups derived from the groups include methanesulfonyl group, trifluoromethanesulfonyl group, nonafluorobutanesulfonyl group, benzenesulfonyl group, paratoluenesulfonyl group, 2-nitrobenzenesulfonyl group, 4-nitrobenzenesulfonyl group and 2,4-di. Groups such as the nitrobenzenesulfonyl group are exemplified.
- the linker represented by ⁇ NR 3 ⁇ can be formed by subjecting it to a reaction with an amine linked to B in the presence of a base.
- the base is not particularly limited as long as it can be normally used, but for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, lithium acetate, sodium acetate, potassium acetate, cesium acetate, etc.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX.
- the equivalent number of the amine containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- R 3 in the above equation is as already defined.
- Preferred examples of R 3 are a hydrogen atom, a C 1-6 alkyl optionally having a substituent, such as methyl and ethyl, and an aromatic ring optionally having a substituent, such as benzene, pyridine. And so on.
- examples of the ring structure formed together with the nitrogen atom and B to be bonded include pyrrolidine, piperidine, piperazine, morpholine and the like, and examples of the heterocyclic aromatic amine include pyrrol, imidazole, pyrazole, indole, benzimidazole and the like. ..
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrahydrogen, 4-methyltetrahydro.
- Ether-based solvents such as pyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethyl ether, diglyme, triglyme, tetraglyme, urea-based solvents such as TMU, DMI and DMPU, DMF, DMA, NFM and NMP.
- NEP, NBP and other amide solvents DMSO and other sulfoxide solvents, diphenyl sulfone, dimethyl sulfone, diethyl sulfone, sulfolane, 3-methyl sulfolane, ethyl methyl sulfone, ethyl isopropyl sulfone and other sulfonate solvents, anisole, toluene, etc.
- aromatic solvents such as xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene and pyridine
- solvents such as acetonitrile and limonene can be used.
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 120 ° C., ⁇ 20 to 80 ° C., 0 to 80 ° C., or 0 to 60 ° C.
- Additives may be used if required, eg 12-crown-4, 15-crown-5, 18-crown-6, HMPA, TMEDA, tetrabutylammonium chloride, tetrabutylammonium bromide, tetrabutylammonium iodide.
- Tetrabutylammonium hydrogensulfate can be used as an additive.
- the desired compound can be obtained by a usual post-treatment method.
- the nucleophilic substitution reaction with an amine can be carried out by the reagents and reaction conditions already described.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to the amine containing B. It can be equivalent, or 1 to 10 equivalents.
- the equivalent number of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to an amine containing B. It can be ⁇ 10 equivalents.
- R 1 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom to be bonded, a carbonyl carbon atom and A].
- the amide bond as a linker can also be formed by reacting a primary amide or a secondary amide with an aryl halide or a derivative thereof.
- A contains a solid phase carrier
- a base is used and X of the reactive functional group linked to B is a halogen or hydroxy group.
- the base is not particularly limited as long as it can be usually used, but for example, NaOtBu, KOtBu, K2 CO 3 , Cs 2 CO 3 , K 3 PO 4 , NaOH, NaOPh , KOPh, NaBHT, DBU, etc.
- TEA TMG, BTMG, MTBD, BTPP, P1tBu, P2Et, P2tBu, LiHMDS, NaHMDS, KHMDS, potassium hydride, sodium hydride, lithium hydride, sodium amide, lithium amide, KOH, NaOH, LiOH, sodium carbonate, carbonic acid.
- the number of equivalents of BX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to an amide containing A. It can be ⁇ 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to the amide containing A. Can be equivalent.
- a solvent can be used in the above reaction, and the solvent is not particularly limited.
- an ether solvent tetratetra, 2-methyltetrachloride, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, cyclopentylmethyl ether, t-Butylmethyl ether, 4-methyltetrahydropyran, diglyme, triglym, tetraglym, etc.), amide solvent (N, N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), N, N-dimethylacetamide (N, N-dimethylacetamide) DMA), N-ethylpyrrolidone (NEP), N-butylpyrrolidone (NBP), etc.), Sulfoxide solvent (dimethylsulfoxide (DMSO), methylphenylsulfoxide), urea solvent (1,3-dimethyl-2-imidazolidi) Non (DM
- a solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, for example, 0 to 200 ° C, 0 to 150 ° C, 20 to 150 ° C. , 40 to 120 ° C, or 60 to 100 ° C.
- a metal solvent may be used, for example, PdCl 2 , Pd (OAc) 2 , Pd 2 (dba) 3 -CHCl 3 and the like.
- NiCl 2 nickel chloride (II)
- Ni (COD) 2 bis (1,5-cyclooctadiene) nickel (0)
- nickel salts such as CuI, CuBr, CuCl, Cu (OAc) 2
- copper salts such as copper (II) oxide and copper (I)
- iron (III) chloride can be used as the metal catalyst.
- a ligand may be used.
- trialkylphosphine tBu 3P ( tri -tert-butylphosphine), Cy 3P (tricyclohexylphosphine), tBu2 MeP (di-tert-butylmethylphosphine)
- biaryldialkylphosphin XPhos ( 2 -dicyclohexylphos)
- ligand ′ -Dimethylcyclohexane-1,2-diamine), Phen (1,10-phenanthroline), etc.
- a metal ligand complex may be used as a catalyst, for example, Buchwald catalyst precursors such as XPhos Pd G1 to 4, Xantphos Pd G3, 4, tBuBrettPhos Pd G4, AlPhos Pd G6 Br (CAS: 2097600-15-0).
- NHC-Pd such as body and PEPPSI-IPr, [(NHC) Pd (allyl) Cl] (allyl [1,3-bis (2,6-diisopropylphenyl) imidazole-2-iriden] chloropalladium (II)).
- the complex can be used as a catalyst.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, with respect to the amide containing A. It can be 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 with respect to the amide containing A. It can be equivalent, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the metal ligand complex to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0, with respect to the amide containing A. It can be 0.01 to 7.5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the desired compound can be obtained by a usual post-treatment method.
- B contains a solid phase carrier in the above formula
- the reaction between the primary amide or secondary amide containing A and the aryl halide or its derivative (BX) can be carried out by the reagents and reaction conditions already described. ..
- the equivalent number of the primary amide or the secondary amide including A to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, and 1 to 50 with respect to BX.
- the equivalent can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to BX. Can be.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0 with respect to BX. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to BX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the metal ligand complex to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents with respect to BX, 0. It can be 001 to 7.5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- R 1 is a C 1-6 alkyl which may have a hydrogen atom and a substituent, and may form a ring together with a nitrogen atom, a sulfur atom and B to be bonded].
- Sulfonamide bonds as linkers can also be formed by reaction of primary or secondary sulfonamides with aryl halides or derivatives thereof.
- a sulfonamide bond as a linker can be formed by subjecting it to a reaction with a compound containing a sulfonamide group linked to B.
- the base is not particularly limited as long as it can be usually used, but for example, NaOtBu, KOtBu, K2 CO 3 , Cs 2 CO 3 , K 3 PO 4 , NaOH, NaOPh , KOPh, NaBHT, DBU, etc.
- TEA TMG, BTMG, MTBD, BTPP, P1tBu, P2Et, P2tBu, LiHMDS, NaHMDS, KHMDS, potassium hydride, sodium hydride, lithium hydride, sodium amide, lithium amide, KOH, NaOH, LiOH, sodium carbonate, carbonic acid.
- a solvent can be used in the above reaction, and the solvent is not particularly limited.
- an ether solvent tetratetra, 2-methyltetrachloride, 1,4-dioxane, 1,2-dimethoxyethane, diisopropyl ether, cyclopentylmethyl ether, t-Butylmethyl ether, 4-methyltetrahydropyran, diglyme, triglym, tetraglym, etc.), amide solvent (N, N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), N, N-dimethylacetamide (N, N-dimethylacetamide) DMA), N-ethylpyrrolidone (NEP), N-butylpyrrolidone (NBP), etc.), Sulfoxide solvent (dimethylsulfoxide (DMSO), methylphenylsulfoxide), urea solvent (1,3-dimethyl-2-imidazolidi) Non (DM
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 150 ° C, 20 to 150 ° C, 40 to 120 ° C, or 60 to 100 ° C.
- a metal catalyst may be used, for example, palladium salts such as PdCl 2 , Pd (OAc) 2 , Pd 2 (dba) 3 -CHCl 3 and NiCl 2 (nickel chloride (II)), Ni ( COD) 2 (Nis (1,5-cyclooctadiene) nickel (0)), nickel salts, CuI, CuBr, CuCl, Cu (OAc) 2 , copper oxide (II), copper oxide (I), etc. Copper salt can be used as a metal catalyst.
- a ligand may be used, for example, trialkylphosphin (tBu 3 P (tri-tert-butyl phosphine), Cy 3 P (tricyclohexylphosphine), tBu 2 MeP (di-tert-butylmethylphosphine).
- tBu 3 P tri-tert-butyl phosphine
- Cy 3 P tricyclohexylphosphine
- tBu 2 MeP di-tert-butylmethylphosphine
- Biaryldialkylphosphin (XPhos (2-dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl), tBuXPhos (2-di-tert-butylphosphino-2', 4', 6'- Triisopropylbiphenyl), Me 4 tBuXPhos (2-di-tert-butylphosphino-3,4,5,6-tetramethyl-2', 4', 6'-triisopropyl-1,1'-biphenyl), RuPhos (2-dicyclohexylphosphino-2', 6'-diisopropoxybiphenyl), BrettPhos (2- (dicyclohexylphosphino) -3,6-dimethoxy-2', 4', 6'-triisopropyl-1, 1'-biphenyl), tBuBrettPhos (2- (di-tert)
- Butyl-2,2'-dipyridyl DMEDA (N, N'-dimethylethylenediamine), CyDMEDA (trans-N, N'-dimethylcyclohexane-1,2-diamine), Phen (1,10-phenanthroline), etc.) Can be used as a ligand.
- a metal ligand complex may be used as a catalyst, for example, Buchwald catalyst precursors such as XPhos Pd G1 to 4, Xantphos Pd G3, 4, tBuBrettPhos Pd G4, AlPhos Pd G6 Br (CAS: 2097600-15-0).
- NHC-Pd such as body and PEPPSI-IPr, [(NHC) Pd (allyl) Cl] (allyl [1,3-bis (2,6-diisopropylphenyl) imidazole-2-iriden] chloropalladium (II)).
- a complex or the like can be used as a catalyst.
- the equivalent number of the sulfone amide containing B to be used can be appropriately set by those skilled in the art, and for example, 1 to 100 equivalents, 1 to 1 to 100 equivalents with respect to the aryl halide containing A or a derivative thereof (AX). It can be 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, and 1 to 50 equivalents with respect to an aryl halide containing A or a derivative thereof (AX). It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0 with respect to AX. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the metal ligand complex to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents with respect to AX, 0. It can be 001 to 7.5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- B contains a solid phase carrier in the above formula
- the reaction between an aryl halide containing A or a derivative thereof (AX) and a primary sulfonamide or a secondary sulfonamide containing B is described in the reagents and reaction conditions already described. Can be done by.
- the equivalent number of the aryl halide or its derivative (AX) containing A to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents with respect to the sulfone amide containing B, 1 to 1 to. It can be 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to 1 to 100 equivalents with respect to a sulfonamide containing B. It can be 10 equivalents.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to the sulfone amide containing B. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7. It can be 5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the metal ligand complex to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents with respect to the sulfone amide containing B. It can be 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- R 4 is a hydrogen atom, a C 1-6 alkyl which may have a substituent, and may form a ring together with a carbon atom and A to be bonded
- R 3 is a hydrogen atom. It is a C 1-6 alkyl which may have a substituent and may form a ring with a hydrogen atom and B to be bonded.
- Amine bonds as linkers can also be formed by reductive amination reactions using aldehydes and amines, or ketones and amines.
- R 4 is C 1-6 alkyl, C 6-10 aryl, 5 ⁇ . It is selected from 10-membered heteroaryl, C 3-7 cycloalkyl.
- a reducing agent can be used to form a carbon-nitrogen single bond by subjecting it to a reaction with an amine containing an amino group linked to B.
- the reducing agent is not particularly limited as long as it can be usually used, but is not particularly limited, but is pyridineborane, 2-picolinborane, tert-butylamineborane, dimethylsulfideborane, sodiumtriacetoxyborohydride, sodium hydride, cyano.
- the number of equivalents of the reducing agent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to aldehydes containing A. It can be 10 equivalents.
- the equivalent number of the amine containing B to be used can be appropriately set by those skilled in the art, and for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, or 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, or 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, with respect to an aldehyde containing A. It can be 1 to 10 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone)) , DMPU (N, N'-dimethylpropylene urea), etc.), amide solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N) -Methyl-2-pyrrolidone), NEP (N
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 200 ° C, 20 to 200 ° C, 20 to 150 ° C, 40 to 150 ° C, or 60 to 150 ° C.
- an acid may be used, and for example, acetic acid, formic acid, trifluoroacetic acid, hydrochloric acid, sulfuric acid and the like can be used as the acid.
- Lewis acid may be used, for example, zinc chloride, titanium tetrachloride, titanium trichloride, boron trifluoride-ethyl ether complex, tetraethyl orthotitanium, tetraisopropyl orthotitanium, nickel chloride, lithium chloride, etc. It can be used as Lewis acid.
- the equivalent number of the acid to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0.01 to 0.01 to aldehyde containing A. It can be 50 equivalents, 0.01-30 equivalents, 0.1-30 equivalents, or 0.1-10 equivalents.
- the equivalent number of Lewis acid to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0. It can be 01 to 50 equivalents, 0.01 to 30 equivalents, 0.1 to 30 equivalents, or 0.1 to 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- B contains a solid phase carrier in the above formula
- the equivalent number of the aldehyde or ketone containing A to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 with respect to the amine containing B. It can be up to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of the reducing agent can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing B. It can be 10 equivalents.
- the equivalent number of the acid to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0.01 to 0.01 to the amine containing B. It can be 50 equivalents, 0.01-30 equivalents, 0.1-30 equivalents, or 0.1-10 equivalents.
- Lewis acid the equivalent number of Lewis acid to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0. It can be 01 to 50 equivalents, 0.01 to 30 equivalents, 0.1 to 30 equivalents, or 0.1 to 10 equivalents.
- the ether bond as a linker can be formed by a reaction between a hydroxyaryl and an alcohol, a reaction between an aryl halide or a derivative thereof and a hydroxyaryl or an alcohol, or a reaction between a hydroxyaryl or an alcohol and an alkyl halide.
- A contains a solid phase carrier and A—X is hydroxyaryl
- X of the reactive functional group linked to A is a hydroxy group
- an azodicarboxylic acid derivative and a phosphin or a cyanomethylenephosphorane derivative are used. It can be used to form an ether bond as a linker by subjecting the reactive functional group Y linked to B to a reaction with an alcohol which is a hydroxy group.
- the azodicarboxylate derivative is not particularly limited as long as it can be usually used, but for example, diethyl azodicarboxylate (DEAD), bis azodicarboxylate (2,2,2-trichloroethyl), N, N.
- N', N'-tetramethylazodicarboxyxamide TMAD
- di-tert-butyl azodicarboxylate dimethyl azodicarboxylate, dimethyl azodicarboxylate, dibenzyl azodicarboxylate, dibenzyl azodicarboxylate, diisopropyl azodicarboxylate ( DIAD), 1,1'-(azodicarbonyl) dipiperidin (ADDP), N, N, N', N'-tetraisopropylazodicarboxyxamide (TIPA), 1,6-dimethyl-1,5,7-hexahydro -1,4,6,7-tetrazosin-2,5-dione (DHTD) bis (2-methoxyethyl) azodicarboxylate can be used.
- DIAD 1,1'-(azodicarbonyl) dipiperidin
- TIPA 1,6-dimethyl-1,5,7-hexahydro -1,4,
- the phosphine is not particularly limited as long as it can be usually used, and for example, triphenylphosphine (TPP), tributylphosphine (TBP), 4- (dimethylamino) phenyldiphenylphosphine, isopropyldiphenylphosphine, phenoxydiphenylphosphine, and tri.
- TPP triphenylphosphine
- TBP tributylphosphine
- 4- (dimethylamino) phenyldiphenylphosphine isopropyldiphenylphosphine
- phenoxydiphenylphosphine and tri.
- -N-octylphosphine diphenyl-2-pyridylphosphine, tri-tert-butylphosphine, dicyclohexylphenylphosphine, tricyclohexylphosphine, trihexylphos
- the cyanomethylenephosphorane derivative is not particularly limited as long as it can be usually used, and for example, cyanomethylenetributylphosphorane (CMBP), cyanomethylenetrimethylphosphorane (CMMP) and the like can be used.
- CMBP cyanomethylenetributylphosphorane
- CMMP cyanomethylenetrimethylphosphorane
- the number of equivalents of alcohol containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- the equivalent number of the azodicarboxylic acid derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 with respect to AX.
- the equivalent can be 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of phosphine to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents with respect to AX. It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of the cyanomethylenephosphorane derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents and 1 to 100 equivalents with respect to AX. It can be 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 200 ° C., 0 to 150 ° C., 0 to 120 ° C., or 0 to 100 ° C.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the desired compound can be obtained by a usual post-treatment method.
- B contains a solid phase carrier and A—X is hydroxyaryl
- X of the reactive functional group linked to A is a hydroxy group
- an azodicarboxylic acid derivative and a phosphin or a cyanomethylenephosphorane derivative are used.
- Using the reactive functional group Y linked to B to form an ether bond as a linker by reacting with an alcohol which is a hydroxy group can be carried out by the above-mentioned reagents and reaction conditions. ..
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents with respect to the alcohol containing B. It can be 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to alcohol containing B. It can be up to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 75 equivalents, relative to the alcohol containing B, for example. It can be 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to alcohol containing B. It can be ⁇ 10 equivalents.
- A contains a solid phase carrier and A—X is an aryl halide or a derivative thereof
- X of the reactive functional group linked to A is a halogen or a hydroxy group derivative
- B is used using a base.
- An ether bond as a linker can be formed by subjecting Y of the reactive functional group linked to to the reaction with hydroxyaryl or alcohol which is a hydroxy group.
- the base is not particularly limited as long as it can be normally used, but for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium phosphate, potassium phosphate, dihydrogen phosphate.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, and for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 140 ° C, 20 to 110 ° C, or 40 to 80 ° C.
- a metal catalyst may be used, for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allylpalladium chloridedimer, palladium ( ⁇ -thinnamyl).
- Chloride dimer (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadien) bis (trimethylsilylmethyl) palladium (II) and the like can be used.
- a ligand may be used, for example, trialkylphosphin (PCy 3 (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A (di (1-adamantyl)).
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- the equivalent number of hydroxyaryl or alcohol containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, and 1 to 30 equivalents with respect to AX. , Or 1 to 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the number of equivalents of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0 with respect to AX. It can be 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the precursor of the palladium catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 with respect to AX. It can be equivalent, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the desired compound can be obtained by a usual post-treatment method.
- B contains a solid phase carrier and A—X is an aryl halide or an analog thereof
- X of the reactive functional group linked to A is a halogen or hydroxy group derivative
- a base is used.
- the formation of an ether bond as a linker by subjecting Y of the reactive functional group linked to B to a reaction with hydroxyaryl or an alcohol which is a hydroxy group can be carried out by the above-mentioned reagents and reaction conditions.
- the equivalent number can be appropriately set by those skilled in the art. It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents with respect to hydroxyaryl or alcohol containing B. , Or 1 to 10 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0 with respect to hydroxyaryl or alcohol containing B. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a ligand When a ligand is used, its equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, with respect to hydroxyaryl or alcohol containing B. It can be 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, with respect to hydroxyaryl or alcohol containing B. It can be 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- A contains a solid phase carrier and A—X is a hydroxyaryl or an alcohol
- X of the reactive functional group linked to A is a hydroxy group
- the reactive group linked to B using a base is reactive.
- An ether bond as a linker can be formed by subjecting the functional group to a reaction with an alkyl halide in which Y is a halogen or an alkylating agent in which Y is a hydroxy group derivative.
- the base is not particularly limited as long as it can be usually used, but for example, in addition to pyridine derivatives such as pyridine, 2,6-lutidine, 2,4,6-colysium, DTBP, and DTBMP, triethylamine and DIPEA.
- Tertiary amines such as 4-methylmorpholin, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, lithium hydroxide, sodium hydroxide, potassium hydroxide, tetraalkylammonium hydroxide (tetraalkyl hydroxide) (Methylammonium, tetrabutylammonium hydroxide, etc.), sodium hydride, potassium hydride, DBU, DBN, MTBD, BTMG, 1,8-bis (dimethylamino) naphthalene, BEMP, BTPP, P2Et, P2tBu, potassium phosphate, LiOtBu, NaOtBu, KOtBu, LiHMDS, NaHMDS, KHMDS, HMDS, Triethylamine, DIPEA, 4-Methylmorpholine, Lithium hydride, Sodium amide, Lithiumamide, Sodium phosphate, Potassium phosphate, KHCO 3
- a solvent can be used in the above reaction, and the reaction is not particularly limited.
- a solvent can be used in the reaction, and the reaction is not particularly limited.
- Solvents diethyl ethers, diisopropyl ethers, THF (tetrahydrogen), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ethers, isosorbidodimethyl ethers, digrims, triglycrims, tetragrims, etc.
- Ether solvents acetone, MEK, MIBK, 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethyl ketone and other ketone solvents, TMU, DMI, DMPU and other urea solvents, DMF, DMA, NFM, NMP. , NEP, NBP and other amide solvents, DMSO and other sulfoxide solvents, methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate, GVL ( ⁇ -valerolactone), pentyl acetate, methyl propionate, etc.
- Ester-based solvents diphenylsulfone, dimethylsulfone, diethylsulfone, sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone and other sulfonate-based solvents, anisole, toluene, xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene, pyridine and other aromatic solvents, as well as carbonate solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate and propylene carbonate, acetonitrile, A solvent such as limonene can be used.
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 120 ° C, 20 to 100 ° C, or 20 to 60 ° C.
- Additives may be used if required, eg Ag 2O , 12-Crown-4, 15-Crown-5, 18-Crown-6, HMPA, TMEDA, Tetrabutylammonium chloride, Tetrabutylammonium bromide, Tetra.
- Butylammonium iodide and tetrabutylammonium hydrogensulfate can be used as additives.
- the equivalent number of the alkyl halide or hydroxy group derivative containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 1 to AX. It can be 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the number of equivalents of the additive to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to AX. Can be equivalent.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 1 to an alkyl halide or an alkylating agent containing B. It can be 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to an alkyl halide or an alkylating agent containing B. It can be 30 equivalents, or 1 to 10 equivalents.
- Carbon-carbon single bond formation reaction A carbon-carbon single bond as a linker can be formed, for example, by reacting an aryl halide with a boron derivative or a zinc derivative.
- the reactive functional group X linked to A is halogen or —O — SO2 - R5; R5 may be substituted with one or more fluorine atoms.
- a carbon-carbon single bond as a linker can be formed by subjecting the functional group to a reaction with a boronic acid analog which is a boron derivative or an alkylboran reagent derived from a carbon-carbon terminal double bond.
- the equivalents of the boronic acid derivative and the alkylborane reagent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, and 1 to 30 equivalents with respect to AX. , Or 1 to 10 equivalents.
- the metal catalyst is not particularly limited as long as it can be usually used, but for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allylpalladium chloride.
- Dimer palladium ( ⁇ -cinnamyl) chloride dimer, (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadien) bis (trimethylsilylmethyl) palladium (II) and the like can be used.
- a ligand may be used, for example, trialkylphosphine (PCy (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A (di (1-adamantyl)-.
- n-butylphosphine n-butylphosphine
- (tBu) 2 PMe di-tert - butylmethylphosphine
- P (Ad) 3 neopentyl (tBu) 2P (di-tert-butylneopentylphosphine), etc.
- triarylphosphine PPh 3 (triphenylphosphin), etc.
- Dialkylbiarylphosphine XPhos (2-dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl
- SPhos (2-dicyclohexylphosphino-2', 6'- Dimethoxybiphenyl
- RuPhos (2-dicyclohexylphosphino-2', 6'-diisopropoxybiphenyl
- BrettPhos (2- (dicyclohexylphosphino) -3,6-dime
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- Phosphin) Palladium (I) Dimer, etc. Phosphin ⁇ -allyl palladium catalyst (XPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 4', 6'-triisopropyl-1,1) '-Biphenyl) palladium (II)), SPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-dimethoxy-1,1'-biphenyl) palladium (II)), RuPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-diisopropoxy-1,1'-biphenyl) palladium (II)), [BretPhos Pd (crotyl)] OTf (crotyl) (2-D
- the number of equivalents of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0 with respect to AX. It can be 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the precursor of the catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents with respect to AX. , 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 200 ° C, 0 to 140 ° C, 10 to 100 ° C, or 20 to 60 ° C.
- a base may be used, for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium phosphate, potassium phosphate, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide, tetraalkyl hydroxide.
- Ammonium tetramethylammonium hydroxide, tetrabutylammonium hydroxide, etc.
- NaOtBu, KOtBu sodium fluoride, potassium fluoride, cesium fluoride, triethylamine
- DIPEA dimethylamino
- DBU dimethylammonium hydroxide
- DBN diBN
- MTBD triethylamine
- TMG BTMG
- P1tBu MTBD
- BEMP Based on BTPP, P2Et, P2tBu, sodium tetraborate, 1,8-bis (dimethylamino) naphthalene, potassium t-pentoxide, sodium t-pentoxide, lithium t-pentoxide, tetrabutylammonium fluoride, cesium fluoride, etc.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the reactive functional group X linked to A is halogen or —O — SO2 ⁇ R5 ;
- R5 may be substituted with one or more fluorine atoms, C 1-6 alkyl, or 1 It is a phenyl that may be substituted with the above-mentioned fluorine atom or C 1-6 alkyl which may be substituted with a fluorine atom, and
- Y of the reactive functional group linked to B using a metal catalyst is a zinc derivative.
- the number of equivalents of the zinc derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to AX. Can be equivalent.
- the metal catalyst is not particularly limited as long as it can be usually used, but for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allyl.
- Palladium chloride dimer palladium ( ⁇ -cinnamyl) chloride dimer, (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadien) bis (trimethylsilylmethyl) palladium (II) and the like can be used. Etc. can be used. If necessary, a ligand may be used, for example, trialkylphosphin (PCy 3 (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A (di (1-adamantyl)).
- PCy 3 tricyclohexylphosphine
- P (tBu) 3 tri-tert-butylphosphine
- cataCXium A di (1-adamantyl)
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- the number of equivalents of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0 with respect to AX. It can be 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the precursor of the catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents with respect to AX. , 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 40 to 100 ° C., ⁇ 20 to 70 ° C., or 20 to 60 ° C.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- Etin-1,2-diyl group formation reaction The ethine-1,2-diyl bond as a linker can be formed by the reaction of an aryl halide with an acetylene derivative.
- the reactive functional group X linked to A is a halogen
- the reactive functional group Y linked to B using a metal catalyst is an acetylene derivative.
- an ethine-1,2-diyl group as a linker can be formed.
- the equivalent number of the acetylene derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to AX. Can be equivalent.
- the metal catalyst is not particularly limited as long as it can be usually used, but for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allylpalladium chloride.
- Dimer palladium ( ⁇ -cinnamyl) chloride dimer, (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadiene) bis (trimethylsilylmethyl) palladium (II) copper, for example, copper oxide (I) (Cu 2 O) ), Copper oxide (II) copper iodide (I) (CuI), copper bromide (I) (CuBr), copper chloride (I) (CuCl), copper acetate (Cu (OAc) 2 ), copper sulfate, Ullmann ⁇ -type conditions (eg, in an aprotonic solvent such as DMF, metal catalysts such as copper (II) acetate, Buchwald-type conditions (eg, carbonates of alkali metals such as cesium carbonate; BINAP; Pd2 (dba) 3, etc.
- aprotonic solvent such as DMF
- metal catalysts such as copper (II) acetate
- Buchwald-type conditions
- a palladium catalyst such as Pd (OAc) 2 can be used.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents with respect to AX. It can be 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, and for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- Acetonitrile, water, silene and limonene can be used.
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 140 ° C., 5 to 120 ° C., 10 to 100 ° C. or 20 to 80 ° C.
- a ligand may be used, and as the ligand of palladium, for example, trialkylphosphine (PCy 3 (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A ( Di (1-adamantyl) -n-butylphosphine), (tBu) 2 PMe (di-tert-butylmethylphosphine), P (Ad) 3 (tri (1-adamantyl) phosphine), neopentyl tBu) 2 P, etc.) , Triarylphosphine (PPh 3 (triphenylphosphine), etc.), Dialkylbiarylphosphine (XPhos, SPhos (2-dicyclohexylphosphino-2', 6'-dimethoxybiphenyl), RuPhos (2-dicyclohexylphosphino-2', 6'-d
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- copper ligands examples include ditertbutylbipyridine (dtBBPy (4,4'-di-tert-butyl-2,2'-dipyridyl)) and dimethylethylenediamine (DMEDA (N, N'). -Dimethylethylenediamine)), cyclohexyldimethylethylenediamine (CyDMEDA (trans-N, N'-dimethylcyclohexane-1,2-diamine)), 1,10-phenanthlorin (Phen), etc.
- dtBBPy ditertbutylbipyridine
- DMEDA dimethylethylenediamine
- CyDMEDA cyclohexyldimethylethylenediamine
- Phen 1,10-phenanthlorin
- diamide-based ligands such as N, N'-dimethylglycine, sarcosin (N-methylglycine), L-proline, 4-hydroxy-L-proline, 6-methylpicolinic acid, for example, N1- ( 2 -methyl) -1-naphthalenyl) -N 2- (phenylmethyl) ethanediamide, N 1 , N 2 -bis (phenylmethyl) ethanediamine, N 1 , N 2 -bis (2,6-dimethylphenyl) ethanediamine, N 1 , N 2 -bis (2-furanylmethyl) ethanediamine, N1, N2 - bis (2,4,6-trimethoxyphenyl) ethanediamine, N1, N2 - bis (5-methyl [1,1'-) Biphenyl] -2-yl) -etanjiamide, [(2,6-dimethylphenyl) amino] (oxo) acetic acid (DMPAO), N
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the catalyst precursor can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a base may be used, for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, sodium phosphate, potassium phosphate, disodium hydrogen phosphate, dipotassium hydrogen phosphate, Sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide, tetraalkylammonium hydroxide (tetramethylammonium hydroxide, tetrabutylammonium hydroxide, etc.), cesium fluoride, potassium fluoride, LiOtBu, NaOtBu, KOtBu, Sodium hydride, potassium hydride, lithium bis (trimethylsilyl) amide, sodium bis (trimethylsilyl) amide, potassium bis (trimethylsilyl) amide, lithium diisopropylamide, methyllithium, butyllithium, pyridine, alkyl substituted pyridine (2,6-lutidin) , DTBP, DTBMP, DMAP, etc.
- Additives may be used if necessary, such as zinc, zinc chloride, zinc bromide, zinc iodide, zinc acetate, zinc trifluoromethanesulfonate, tetrabutylammonium bromide, tetrabutylammonium chloride, tetrabutylammonium iodide, etc. Can be used as an additive.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the number of equivalents of the additive to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to AX. Can be equivalent.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- ester bond formation reaction The ester bond as a linker can be formed by the reaction of a carboxylic acid or a derivative thereof with an alcohol.
- the reactive functional group X linked to A is hydroxy, and by subjecting it to a reaction with an alcohol containing a hydroxy group linked to B using a condensing agent. It is possible to form an ester bond as a linker.
- the number of equivalents of alcohol containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- the condensing agent is not particularly limited as long as it can be usually used, but for example, carbodiimide-based condensing agents such as DCC, DIC, N, N'-di-tert-butylcarbodiimide, EDCI, and EDCI / HCl.
- carbodiimide-based condensing agents such as DCC, DIC, N, N'-di-tert-butylcarbodiimide, EDCI, and EDCI / HCl.
- HOAt HOBt
- activators such as oxyma, HATU, TATU (1- [bis (dimethylamino) methylene] -1H-1,2,3-triazolo [4,5-b] pyridinium 3-oxide tetra Fluorobolate), uronium salt-based condensing agents such as COMU, PyBOP (1H-benzotriazole-1-yloxytripyrrolidinophosphonium hexafluorophosphate), PyOxim ([ethylcyano (hydroxyimino) acetato-O 2 ] tri- 1-Pyrrolidinylphosphonium Hexafluorophosphate and other phosphonium salt-based condensing agents, TCFH (chloro-N, N, N', N'-tetramethylformamidinium hexafluorophosphate), PyClU (1- (chloro) -1-pyrrolidinyl methylene)
- Acid anhydrides such as benzoic acid anhydrides, isobutyl chloroate, 4-nitrophenyl chloroate, 4-nitrobenzyl chloroate, acidifieds such as 2,4,6-trichlorobenzoylchloride, triphosgen and the like.
- a phosgen derivative, a carbodiimidazole derivative such as 1,1'-carbonyldiimidazole, or the like can be used.
- the equivalent number of the condensing agent to be used can be appropriately set by those skilled in the art, for example, with respect to AX. It can be 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- a solvent can be used in the above reaction, and the reaction is not particularly limited, and for example, a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrachloride, 4-methyl Ethereal solvents such as tetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethyl ether, diglyme, triglym, tetraglyme, acetone, MEK, MIBK, 4-methyl-2-pentanone, cyclopentanone.
- a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrachloride, 4-methyl Ethereal solvents such as te
- Cyclohexanone ketone solvents such as diethylketone, urea solvents such as TMU, DMI, DMPU, amide solvents such as DMF, DMA, NFM, NMP, NEP, NBP, sulfoxide solvents such as DMSO, diphenylsulfones, dimethyl.
- ketone solvents such as diethylketone
- urea solvents such as TMU, DMI, DMPU
- amide solvents such as DMF, DMA, NFM, NMP, NEP, NBP
- sulfoxide solvents such as DMSO, diphenylsulfones, dimethyl.
- Sulfonate solvents such as sulfone, diethylsulfone, sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate, GVL ( ⁇ -valerolactone) , Estel solvents such as pentyl acetate, methyl propionate, anisole, toluene, xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene, pyridine, pyridine In addition to aromatic solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate,
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 100 ° C, 20 to 80 ° C, or 20 to 50 ° C.
- a base may be used, for example, pyridine, 2,6-lutidine, 2,4,6-colysium, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, sodium carbonate, carbonic acid.
- Bases such as potassium, cesium carbonate, sodium hydrogen carbonate, potassium phosphate, K 2 HPO 4, potassium hydrogen carbonate, Na 2 HPO 4 , DBU, DBN, MTBD, BTMG, TMG, 1,8-bis (dimethylamino) naphthalene, etc. Can be used as.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the desired compound can be obtained by a usual post-treatment method.
- X of the reactive functional group linked to A is hydroxy
- an azodicarboxylic acid derivative and a phosphine or a cyanomethylenephosphorane derivative are used to react with an alcohol containing a hydroxy group linked to B.
- an ether bond as a linker can be formed.
- the number of equivalents of alcohol containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to AX. It can be 30 equivalents, or 1 to 10 equivalents.
- the azodicarboxylate derivative is not particularly limited as long as it can be usually used, but for example, diethyl azodicarboxylate (DEAD), bis azodicarboxylate (2,2,2-trichloroethyl), N, N.
- DEAD diethyl azodicarboxylate
- bis azodicarboxylate (2,2,2-trichloroethyl) N, N.
- N', N'-tetramethylazodicarboxyxamide (TMAD), di-tert-butyl azodicarboxylate, dimethyl azodicarboxylate, dibenzyl azodicarboxylate, diisopropyl azodicarboxylate (DIAD), 1,1'-(azo Dicarbonyl) Dipiperidin (ADDP), N, N, N', N'-tetraisopropylazodicarboxyxamide (TIPA), 1,6-dimethyl-1,5,7-hexahydro-1,4,6,7-tetrazocin ⁇ 2,5-dione (DHTD), bis azodicarboxylate (2-methoxyethyl) and the like can be used.
- TMAD N', N'-tetramethylazodicarboxyxamide
- TMAD di-tert-butyl azodicarboxylate, dimethyl azodicarboxylate, dibenzyl azodicarboxylate
- the phosphine is not particularly limited as long as it can be usually used, and for example, triphenylphosphine (TPP), tributylphosphine (TBP), 4- (dimethylamino) phenyldiphenylphosphine, isopropyldiphenylphosphine, phenoxydiphenylphosphine, and tri.
- TPP triphenylphosphine
- TBP tributylphosphine
- 4- (dimethylamino) phenyldiphenylphosphine isopropyldiphenylphosphine
- phenoxydiphenylphosphine and tri.
- -N-octylphosphine diphenyl-2-pyridylphosphine, tri-tert-butylphosphine, dicyclohexylphenylphosphine, tricyclohexylphosphine, trihexylphos
- the cyanomethylenephosphorane derivative is not particularly limited as long as it can be usually used, and for example, cyanomethylenetributylphosphorane (CMBP), cyanomethylenetrimethylphosphorane (CMMP) and the like can be used.
- CMBP cyanomethylenetributylphosphorane
- CMMP cyanomethylenetrimethylphosphorane
- the equivalent number of the azodicarboxylic acid derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to AX. It can be 30 equivalents, or 1 to 10 equivalents.
- the number of equivalents of phosphine to be used can be appropriately set by those skilled in the art, and for example, 1 to 150 equivalents, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, etc. Alternatively, it can be 1 to 10 equivalents.
- the equivalent number of the cyanomethylenephosphorane derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 150 equivalents and 1 to 100 equivalents with respect to AX. It can be 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents.
- a solvent can be used in the above reaction, and the reaction is not particularly limited, and for example, a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrachloride, 4-methyl Ethereal solvents such as tetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethyl ether, diglyme, triglym, tetraglyme, acetone, MEK, MIBK, 4-methyl-2-pentanone, cyclopentanone.
- a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrachloride, 4-methyl Ethereal solvents such as te
- Ketone solvent such as diethyl ketone
- Urea solvent such as TMU, DMI, DMPU
- Amid solvent such as DMF, DMA, NFM, NMP, NEP, NBP
- Sulfoxide solvent such as DMSO, Methyl acetate, Acetic acid.
- Estel-based solvents such as ethyl, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate, GVL ( ⁇ -valerolactone), pentyl acetate, methyl propionate, diphenylsulfone, dimethylsulfone, diethylsulfone, sulfolane, 3-methylsulfolane, Cyclic solvents such as ethylmethylsulfone and ethylisopropylsulfone, anisole, toluene, xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene, pyridine, etc.
- GVL ⁇ -valerolactone
- Cyclic solvents such as ethyl
- hydrocarbon solvents such as hexane, pentane, heptane, and cyclohexane
- carbonate solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate, and propylene carbonate
- solvents such as acetonitrile and limonene
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 150 ° C., 0 to 120 ° C., or 20 to 100 ° C.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- ester bond formation reaction The ester bond as a linker can be formed by reacting a carboxylic acid or a derivative thereof with an alkyl halide in addition to the above-mentioned method.
- the reactive functional group X linked to A is hydroxy
- the reactive functional group Y linked to B using a base is an alkyl halide or a halogen.
- An ester bond as a linker can be formed by subjecting Y to a reaction with an alkylating agent which is a hydroxy group derivative.
- the number of equivalents of the alkylating agent containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, 1 to 30 equivalents, relative to AX. Alternatively, it can be 1 to 10 equivalents.
- the base is not particularly limited as long as it can be usually used, but for example, in addition to alkylpyridine derivatives such as 2,6-lutidin and 2,4,6-colysium, triethylamine, DIPEA, 4-methylmorpholin and the like.
- alkylpyridine derivatives such as 2,6-lutidin and 2,4,6-colysium, triethylamine, DIPEA, 4-methylmorpholin and the like.
- Tertiary amines sodium carbonate, potassium carbonate, cesium carbonate, lithium hydroxide, sodium hydroxide, potassium hydroxide, barium hydroxide, tetraalkylammonium hydroxide (tetramethylammonium hydroxide, tetrabutylammonium hydroxide, etc.) , Lithium hydride, sodium hydride, potassium hydride, DBU, DBN, MTBD, BTMG, TMG, 1,8-bis (dimethylamino) naphthalene, BEMP, BTPP, P2Et, P2tBu, potassium phosphate, sodium phosphate, LiOtBu, NaOtBu, KOtBu, LiHMDS, NaHMDS, KHMDS, HMDS and the like can be used.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to carboxylic acid containing A. It can be 10 equivalents.
- a solvent can be used in the above reaction, and the reaction is not particularly limited.
- a solvent can be used in the reaction, and the reaction is not particularly limited.
- Solvents diethyl ethers, diisopropyl ethers, THF, 2-methyltetrahexyl, 4-methyltetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ethers, isosorbidodimethyl ethers, digrims, trigrims, tetraglycims and other ether solvents , Acetone, MEK, MIBK, 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethyl ketone and other ketone solvents, TMU, DMI, DMPU and other urea solvents, DMF, DMA, NFM, NMP, NEP, Amido-based solvents such as NBP, sulfoxide-based solvents such as DMSO, ester-based solvents such as methyl acetate, ethyl acetate, propyl acetate, isoprop
- Solvents such as diphenylsulfone, dimethylsulfone, diethylsulfone, sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, anisole, toluene, xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , In addition to aromatic solvents such as ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene and pyridine, carbonate solvents such as dimethyl carbonate, diethyl carbonate, ethylene carbonate and propylene carbonate, acetonitrile and limonene, etc.
- sulfonic solvents such as diphenylsulfone, dimethylsulfone, diethylsulfone, sulfolane, 3-methylsulfolane,
- Solvents can be used. A single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used. Additives may be used if required, eg Ag 2O , 12-Crown-4, 15-Crown-5, 18-Crown-6, HMPA, TMEDA, Tetrabutylammonium chloride, Tetrabutylammonium bromide, Tetra. Butylammonium iodide can be used as an additive. When an additive is used, the equivalent number of the additive to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0.01 with respect to AX.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 150 ° C., 0 to 120 ° C., 20 to 100 ° C., or 20 to 60 ° C.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the sulfone bond as a linker can be formed, for example, by reacting an aryl halide or a derivative thereof with an alkyl halide or a derivative thereof and sulfur dioxide or a sulfur dioxide equivalent.
- A comprises a solid phase carrier in the above formula
- the reactive functional group X linked to A is halogen or —O — SO2 - R5; R5 may be substituted with one or more fluorine atoms.
- a sulfone bond as a linker can be formed by subjecting a hydroxyalkyl derivative having a functional group Y of halogen or —O — SO2 ⁇ R5 to a reaction with sulfur dioxide or a sulfur dioxide equivalent.
- the sulfur dioxide equivalent is not particularly limited as long as it can be usually used, and for example, bis (sulfur dioxide) -1,4-diazabicyclo [2.2.2] octane adduct or sulfur dioxide 1-methyl. Pyrrolidine adducts, potassium pyrosulfite, etc. can be used.
- the equivalent number of the alkyl halide or its derivative containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to AX. It can be 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of sulfur dioxide or sulfur dioxide equivalent to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents with respect to AX. It can be 1 to 10 equivalents, or 1 to 5 equivalents.
- the metal catalyst is not particularly limited as long as it can be usually used, but for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allylpalladium chloridedimer, palladium.
- ( ⁇ -Cinnamyl) chloride dimer, (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadien) bis (trimethylsilylmethyl) palladium (II) and the like can be used.
- a ligand may be used, for example, trialkylphosphine (PCy (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A (di (1-adamantyl)-.
- PCy tricyclohexylphosphine
- P (tBu) 3 tri-tert-butylphosphine
- cataCXium A di (1-adamantyl)-.
- n-butylphosphine n-butylphosphine
- (tBu) 2 PMe di-tert - butylmethylphosphine
- P (Ad) 3 neopentyl (tBu) 2P (di-tert-butylneopentylphosphine), etc.
- triarylphosphine PPh 3 (triphenylphosphin), etc.
- Dialkylbiarylphosphine XPhos (2-dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl
- SPhos (2-dicyclohexylphosphino-2', 6'- Dimethoxybiphenyl
- RuPhos (2-dicyclohexylphosphino-2', 6'-diisopropoxybiphenyl
- BrettPhos (2- (dicyclohexylphosphino) -3,6-dime
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- Phosphin) Palladium (I) Dimer, etc. Phosphin ⁇ -allyl palladium catalyst (XPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 4', 6'-triisopropyl-1,1) '-Biphenyl) palladium (II)), SPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-dimethoxy-1,1'-biphenyl) palladium (II)), RuPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-diisopropoxy-1,1'-biphenyl) palladium (II)), [BretPhos Pd (crotyl)] OTf (crotyl) (2-D
- the number of equivalents of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0 with respect to AX. It can be 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the precursor of the catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents with respect to AX. , 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 200 ° C, 0 to 140 ° C, 10 to 100 ° C, or 20 to 60 ° C.
- a base may be used, for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium phosphate, potassium phosphate, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide, tetraalkyl hydroxide.
- Ammonium tetramethylammonium hydroxide, tetrabutylammonium hydroxide, etc.
- NaOtBu, KOtBu sodium fluoride, potassium fluoride, cesium fluoride, triethylamine
- DIPEA dimethylamino
- DBU dimethylammonium hydroxide
- DBN diBN
- MTBD triethylamine
- DIPEA dimethylamino
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX. Can be.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the equivalent number of the aryl halide containing A to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, and 1 to 50 equivalents with respect to the alkyl halide containing B. It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the equivalent number of sulfur dioxide or sulfur dioxide equivalent can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to an alkyl halide containing B. It can be ⁇ 30 equivalents, 1 ⁇ 10 equivalents, or 1 ⁇ 5 equivalents.
- the equivalent number of the metal catalyst used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 with respect to alkyl halides containing B. It can be equivalent, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 0.001 to alkyl halides containing B. It can be 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of precursors of the catalyst used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 with respect to alkyl halides containing B. It can be ⁇ 5 equivalents, 0.01 ⁇ 5 equivalents, or 0.01 ⁇ 3 equivalents.
- the thioether bond as a linker uses an aromatic nucleophilic substitution reaction using an aryl halide or a derivative thereof and a thiol, a catalytic coupling reaction using a metal catalyst, or an alkyl halide, a hydroxyalkyl or a derivative thereof and a thiol. It can be formed by the nucleophilic substitution reaction.
- A contains a solid phase carrier and A—X is an aryl halide or a derivative thereof, X of the reactive functional group linked to A is a group derived from a halogen or a hydroxy group.
- Examples of the group derived from the hydroxy group include methanesulfonyl group, trifluoromethanesulfonyl group, nonafluorobutanesulfonyl group, benzenesulfonyl group, paratoluenesulfonyl group, 2-nitrobenzenesulfonyl group, 4-nitrobenzenesulfonyl group, 2,4. -Dinitrobenzenesulfonyl groups and the like can be exemplified.
- AX can be subjected to a reaction with a thiol linked to B in the presence of a base.
- the base is not particularly limited as long as it can be normally used, but for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, sodium phosphate, potassium phosphate, and hydrogen phosphate di.
- DMAP N, N-dimethylaniline, N-methylmorpholine, triethylamine
- DIPEA DBU
- DBN DBN
- MTBD TMG
- BTMG P1tBu
- BEMP BTPP
- P2Et P2tBu
- NMI sodium tetraborate
- 1,8 -Bis (dimethylamino) naphthalene N, N-dimethylaniline, tetrabutylammonium fluoride, cesium fluoride and the like can be used.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX.
- the equivalent number of thiols containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 150 ° C., 10 to 120 ° C., 20 to 100 ° C., 20 to 80 ° C., or 20 to 60 ° C.
- a metal catalyst may be used, for example, for palladium, for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allyl palladium chloride dimer, palladium ( ⁇ ).
- -Cinnamyl) chloride dimer 1,3-methylallyl) palladium chloride dimer, (1,5-cyclooctadiene) bis (trimethylsilylmethyl) palladium (II) for nickel, for example, NiCl 2 , (nickel chloride (II)), Ni ( COD) 2 (bis (1,5-cyclooctadiene) nickel (0)), etc.
- nickel for example, NiCl 2 , (nickel chloride (II)), Ni ( COD) 2 (bis (1,5-cyclooctadiene) nickel (0)), etc.
- copper for example, copper oxide (I) (Cu 2 O), copper iodide (I) (CuI), copper bromide ( I) (CuBr), copper (I) chloride (CuCl), copper acetate (Cu (OAc) 2 ) and the like can be used as the metal catalyst.
- a ligand may be used, and as the ligand of palladium, for example, trialkylphosphine (Cy 3 (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A ( Di (1-adamantyl) -n-butylphosphine), (tBu) 2 PMe (di-tert-butylmethylphosphine), P (Ad) 3 (tri (1-adamantyl) phosphine), neopentyl (tBu) 2 P ( Di-tert-butyl neopentylphosphine), triarylphosphine (PPh 3 (triphenylphosphine), etc.), dialkylbiarylphosphine (XPhos (2-dicyclohexylphosphino-2', 4', 6'-triisopropylbiphenyl), etc.
- ligand can be used as a ligand.
- ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- copper ligands examples include ditertbutylbipyridine (dtBBPy (4,4'-di-tert-butyl-2,2'-dipyridyl)) and dimethylethylenediamine (DMEDA (N, N') for diamine ligands. -Diamineethylenediamine)), cyclohexyldimethylethylenediamine (CyDMEDA (trans-N, N'-dimethylcyclohexane-1,2-diamine)), 1,10-phenanthroline (Phen), etc.
- diamine-based ligands such as N, N'-dimethylglycine, sarcosin (N-methylglycine), L-proline, 4-hydroxy-L-proline, for example, N1- (2-methyl- 1 -naphthalenyl)-.
- DMPAO 2- (phenylmethyl) ethanediamide
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0 with respect to AX. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the catalyst precursor to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to AX. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- the aromatic nucleophilic substitution reaction with thiol and the catalytic coupling reaction using a metal catalyst are the reagents already described. It can be carried out depending on the reaction conditions.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to thiol containing B. It can be equivalent, or 1 to 10 equivalents.
- the number of equivalents of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to thiols containing B. It can be ⁇ 10 equivalents.
- the equivalent number of the metal catalyst can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0. It can be 001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 with respect to the thiol containing B. It can be equivalent, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the catalyst precursor to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 with respect to thiol containing B. It can be equivalent, 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the X of the reactive functional group linked to A is a group derived from a desorbing group, for example, a halogen or a hydroxy group, and is hydroxy.
- the groups derived from the groups include methanesulfonyl group, trifluoromethanesulfonyl group, nonafluorobutanesulfonyl group, benzenesulfonyl group, paratoluenesulfonyl group, 2-nitrobenzenesulfonyl group, 4-nitrobenzenesulfonyl group and 2,4-di.
- a thioether bond as a linker can be formed by subjecting it to a reaction with a thiol linked to B in the presence of a base.
- the base is not particularly limited as long as it can be normally used, but for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogencarbonate, potassium hydrogencarbonate, lithium hydroxide, sodium hydroxide, potassium hydroxide, and the like.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 equivalents with respect to AX.
- the equivalent number of thiols containing B to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to AX. It can be ⁇ 10 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent such as DCM, chloroform, DCE, carbon tetrachloride, diethyl ether, diisopropyl ether, THF, 2-methyltetrahydrogen, 4-methyltetrahydro.
- Ether-based solvents such as pyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethyl ether, diglyme, triglyme, tetraglyme, urea-based solvents such as TMU, DMI and DMPU, DMF, DMA, NFM and NMP.
- NEP, NBP and other amide solvents DMSO and other sulfoxide solvents, diphenyl sulfone, dimethyl sulfone, diethyl sulfone, sulfolane, 3-methyl sulfolane, ethyl methyl sulfone, ethyl isopropyl sulfone and other sulfonate solvents, anisole, toluene, etc.
- aromatic solvents such as xylene, chlorobenzene, bromobenzene, ethylbenzene, nitrobenzene, ⁇ , ⁇ , ⁇ -trifluorotoluene, 1,2-dichlorobenzene, benzene and pyridine
- solvents such as acetonitrile and limonene can be used.
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 120 ° C., ⁇ 20 to 80 ° C., 0 to 80 ° C., or 0 to 60 ° C.
- Additives may be used if required, eg 12-crown-4, 15-crown-5, 18-crown-6, HMPA, TMEDA, tetrabutylammonium chloride, tetrabutylammonium bromide, tetrabutylammonium iodide.
- Tetrabutylammonium hydrogensulfate can be used as an additive.
- the desired compound can be obtained by a usual post-treatment method.
- the nucleophilic substitution reaction with thiol can be carried out by the reagents and reaction conditions already described.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 with respect to thiol containing B. It can be equivalent, or 1 to 10 equivalents.
- the number of equivalents of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to thiols containing B. It can be ⁇ 10 equivalents.
- the ketone bond as a linker can be formed by reacting a carboxylic acid or a derivative thereof with a boron derivative or a zinc derivative.
- A contains a solid phase carrier in the above formula
- the reactive functional group X linked to A is a hydroxy, an alkoxy group, an aryloxy group, an alkylthio group, or an arylthio group.
- X is hydroxy, it can be subjected to a ketone bond formation reaction by adding an acid anhydride or by forming a mixed acid anhydride in advance or in a reaction system.
- the acid anhydride is not particularly limited as long as it can be usually used, and for example, benzoic acid anhydride, anhydrous acetic acid, isobutyric acid anhydride, isovaleric acid anhydride, pivalic acid anhydride and the like are used. be able to.
- the number of equivalents of the acid anhydride to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 75 equivalents, 1 to 50 equivalents, or 1 to 30 equivalents, with respect to the carboxylic acid containing A. It can be 1 to 10 equivalents.
- the equivalent number of the boron derivative or the zinc derivative can be appropriately set by those skilled in the art, and for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 30 equivalents, or 1 to 50 equivalents, or 1 to 30 equivalents, or 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, or 1 to 100 equivalents, 1 to 75 equivalents, or 1 to 30 equivalents, with respect to the carboxylic acid containing A. It can be 1 to 10 equivalents.
- a boronic acid analog in which Y of the reactive functional group linked to B is a boron derivative using a metal catalyst, an alkylboran reagent derived from a carbon-carbon terminal double bond, or a compound in which Y is a zinc derivative.
- a ketone bond as a linker can be formed.
- the equivalent number of the boronic acid derivative, the alkylborane reagent, and the zinc derivative to be used can be appropriately set by those skilled in the art, and for example, 1 to 100 equivalents, 1 to 75 equivalents, and 1 to 50 equivalents with respect to A-CO-X. It can be 1 to 30 equivalents, or 1 to 10 equivalents.
- the metal catalyst is not particularly limited as long as it can be usually used, but for example, palladium acetate, Pd (dba) 2 , Pd 2 (dba) 3 , Pd 2 (dba) 3 CHCl 3 , allylpalladium chloride.
- Dimer, palladium ( ⁇ -cinnamyl) chloride dimer, (1-methylallyl) palladium chloride dimer, (1,5-cyclooctadien) bis (trimethylsilylmethyl) palladium (II) and the like can be used.
- a ligand may be used, for example, phosphite esters (P (OEt) 3 (triethylphosphite), P (OPh) 3 (triphenylphosphite), etc.), trialkylphosphine ( PCy (tricyclohexylphosphine), P (tBu) 3 (tri-tert-butylphosphine), cataCXium A (di (1-adamantyl) -n-butylphosphine), (tBu) 2 PMe (di-tert-butylmethylphosphine) ), P (Ad) 3 , neopentyl (tBu) 2 P (di-tert-butyl neopentylphosphine, etc.), triarylphosphine (PPh 3 (triphenylphosphine), tri (2-furyl) phosphine, etc.), dialkyl Biarylphosphin (XPhos), tri
- ligands can be used as ligands. can.
- the above-mentioned ligand and a precursor of a palladium catalyst may be used, for example, Buchwald 1st generation catalyst precursor (G1) (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- G1 Buchwald 1st generation catalyst precursor (XPhos Pd G1, SPhos Pd G1, RuPhos Pd G1, BrettPhos Pd G1, tBuXPhos Pd).
- Phosphin) Palladium (I) Dimer, etc. Phosphin ⁇ -allyl palladium catalyst (XPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 4', 6'-triisopropyl-1,1) '-Biphenyl) palladium (II)), SPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-dimethoxy-1,1'-biphenyl) palladium (II)), RuPhos Pd (crotyl) Cl (chloro (crotyl) (2-dicyclohexylphosphino-2', 6'-diisopropoxy-1,1'-biphenyl) palladium (II)), [BretPhos Pd (crotyl)] OTf (crotyl) (2-D
- the number of equivalents of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, 0 with respect to AX. It can be 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number of the ligand to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents, and 0.001 to 5 equivalents with respect to AX. It can be 0.01 to 5 equivalents or 0.01 to 3 equivalents.
- the equivalent number of the precursor of the catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 equivalents with respect to AX. , 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- a solvent can be used for the above reaction, and the reaction is not particularly limited, but for example, a halogen-based solvent (DCM (dioxide), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl).
- DCM dioxide
- chloroform chloroform
- DCE 1,2-dichloroethane
- carbon tetrachloride etc.
- ether-based solvent diethyl
- Ether diisopropyl ether, THF (tetrahexyl), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl) Ether), isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethylketone, etc.) ), Urea solvent (TMU (N, N, N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.
- Amid solvent (DMF (N, N-dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl) -2-pyrrolidone), NBP (N-butyl-2-pyrrolidone), formamide, etc.), Sulfoxide-based solvent (DMSO (dimethylsulfoxide), methylphenylsulfoxide, etc.), Sulfon-based solvent (diphenylsulfone, dimethylsulfone, diethylsulfone, etc.) Sulfolane, 3-methylsulfolane, ethylmethylsulfone, ethylisopropylsulfone, etc.), ester solvents (methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobuty
- reaction temperature can be appropriately set by those skilled in the art, and can be, for example, 0 to 200 ° C, 0 to 140 ° C, 10 to 100 ° C, or 20 to 90 ° C.
- a base may be used, for example, sodium carbonate, potassium carbonate, cesium carbonate, sodium phosphate, potassium phosphate, sodium hydroxide, potassium hydroxide, cesium hydroxide, barium hydroxide, tetraalkyl hydroxide.
- Ammonium tetramethylammonium hydroxide, tetrabutylammonium hydroxide, etc.
- NaOtBu, KOtBu sodium fluoride, potassium fluoride, cesium fluoride, triethylamine
- DIPEA dimethylamino
- DBU dimethylammonium hydroxide
- DBN diBN
- MTBD triethylamine
- TMG BTMG
- P1tBu MTBD
- BEMP Based on BTPP, P2Et, P2tBu, sodium tetraborate, 1,8-bis (dimethylamino) naphthalene, potassium t-pentoxide, sodium t-pentoxide, lithium t-pentoxide, tetrabutylammonium fluoride, cesium fluoride, etc.
- the number of equivalents of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to A-CO-X. It can be 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- X of the reactive functional group linked to A is a hydroxy, an alkoxy group, an aryloxy group, an alkylthio group, an arylthio group, and Y of the reactive functional group linked to B.
- Y of the reactive functional group linked to B is a boron derivative or a zinc derivative.
- the formation of a ketone bond as a linker can be performed by the reagents and reaction conditions already described.
- the equivalent number can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, and 1 to 30 equivalents with respect to BY. , Or 1 to 10 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 5 with respect to BY. It can be equivalent, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 0.001 to BY. It can be 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the equivalent number can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents, 0.001 to 7.5 equivalents, 0.001 to 0.001 to BY. It can be 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the number of equivalents of AX to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to BY. It can be 10 equivalents.
- a bond using a triazole ring which is an example of a nitrogen-containing 5-membered heteroarylene as a linker, can be formed by a reaction between an azide derivative and an acetylene derivative.
- a contains a solid phase carrier in the above formula a metal catalyst can be used to form a bond using a triazole ring as a linker by subjecting it to a reaction with an acetylene derivative containing a propargyl group linked to B. can.
- the number of equivalents of the acetylene derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 with respect to the azide derivative containing A.
- the metal catalyst is not particularly limited as long as it can be normally used, but for example, metallic copper, copper chloride (I), copper chloride (II), copper bromide (I), copper bromide (II). , Copper iodide, Copper (I) acetate, Copper (II) acetate, Copper (I) oxide, Copper (II) oxide, Copper sulfate, Zinc acetate, Chloro (H5-pentamethylcyclopentadiene) [Bis (triphenylphosphine) )] Luthenium (II), carbonyl (dihydride) tris (triphenylphosphine) ruthenium (II) and the like can be used.
- the equivalent number of the metal catalyst to be used can be appropriately set by those skilled in the art, for example, 0.001 to 10 equivalents and 0.001 to 7.5 equivalents with respect to the azide derivative containing A. , 0.001 to 5 equivalents, 0.01 to 5 equivalents, or 0.01 to 3 equivalents.
- the additive to be used for example, sodium L-ascorbic acid, cellulose, amberlist, tetrabutylammonium bromide and the like can be used.
- the equivalent number of the additive to be used can be appropriately set by those skilled in the art, for example, 0.01 to 100 equivalents, 0.01 to 75 equivalents, 0 with respect to the azide derivative containing A. It can be 0.01 to 50 equivalents, 0.01 to 30 equivalents, or 0.01 to 10 equivalents.
- a solvent can be used for the above reaction, and the present invention is not particularly limited, but a halogen-based solvent (DCM (dichloromethane), chloroform, DCE (1,2-dichloroethane), carbon tetrachloride, etc.), an ether-based solvent (diethyl ether, etc.) Diisopropyl ether, THF (tetratetra), 2-methyltetrachloride, 4-methyltetrahydropyran, 1,4-dioxane, DME (1,2-dimethoxyethane), TBME (t-butylmethyl ether), CPME (cyclopentylmethyl ether) , Isosorbidodimethylether, diglyme, triglyme, tetraglym, etc.), ketone solvents (acetone, MEK (methylethylketone), MIBK (methylisobutylketone), 4-methyl-2-pentanone, cyclopentanone,
- Acids, silenes and limonene can be used.
- a single solvent may be used, or a mixed medium in which a plurality of solvents are combined may be used.
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, ⁇ 20 to 110 ° C., 0 to 100 ° C., 10 to 90 ° C., or 20 to 80 ° C.
- the reaction can be carried out even under high temperature conditions of 100 ° C. or higher by using an excessive amount of the reagent without using a solvent.
- a base may be used, for example, pyridine, 2,6-lutidine, 2,4,6-colysium, quinoline, DTBP, DTBMP, triethylamine, DIPEA, 4-methylmorpholin, NMI, DMAP, 1, 4-Diazabicyclo [2.2.2] Octane (DABCO) Potassium Hydrochloride, Sodium Hydrochloride, Sodium Amid, Lithiumamide, KOH, NaOH, LiOH, Sodium Carbonate, Potassium Carbonate, Cesium Carbonate, Sodium Hydrogen Carbonate, Potassium Phosphate , K 2 HPO 4, KHCO 3 , NaHCO 3 , Na 2 HPO 4, Sodium tetraborate, DBU, DBN, MTBD, BTMG, TMG, 1,8-bis (dimethylamino) naphthalene, LiOtBu, NaOtBu, KOtBu, potassium Use t-pentoxide, sodium t-pentoxide, N
- the equivalent number of the base to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 1 to the azide derivative containing A. It can be 30 equivalents, or 1 to 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the two or more core blocks are linked via a linker to form the chemical structure of the compounds contained in the library.
- the compounds constituting the library can be prepared by linking core blocks by a linker formation reaction.
- the core block is not particularly limited as long as it has a chemical structure linked via a linker in the compound molecule.
- the molecular structure of a compound contained in a library is specified with each of two or more chemical structures separated by a linker as a core block, where the linker means a linking moiety formed in compound synthesis.
- the compound contained in the library is prepared by a linker forming reaction with a reactive functional group of a building block and means two or more chemical structures other than the linker of the branched structure of the compound.
- the library comprises one or more compounds prepared by linking core blocks by a linker forming reaction, further comprising additional compounds comprising a linker corresponding to the linker contained in the compound. It may be included.
- the additional compound may be a compound prepared without undergoing a linker formation reaction, or may be a compound obtained by purchase, for example.
- the linker when the single bond is a linker, the linker is selected from C 5-10 aryl, 5-10 membered heteroaryl, 5-8 membered heterocyclyl, and C 3-10 cycloalkyl 2
- the two chemical structures are linked together and the chemical structures constitute all or part of the core block linked by the linker.
- the core blocks are 1 to trivalent core blocks independently represented by the following equations: 1) Divalent core block: 1-1) -A 1- ; 1-2) -X 1 -Q 1 -X 2- ; or 1-3) -X 3 -Q 2 -X 4 -Q 3 -X 5- ;
- a 1 is a C 1-10 alkylene, which may have one or more substituents independently selected from Ra
- Q 1 is selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, each of which is independently selected from Rb 1 It may have the above substituents
- X 1 and X 2 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra
- Q 2 and Q 3 are independently selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocycl
- Each may have one or more substituents independently selected from Rb;
- X 3 , X 4 and X 5 are independently single-bonded or C 1-10 alkylenes, which may have one or more substituents independently selected from Ra;
- a 1 , X 1 , X 2 , X 3 , X 4 and X 5 are C 2-10 alkylenes, then the alkylene is selected from -O-, -S-, or -NR 11- .
- R 11 is a hydrogen atom or C 1-6 alkyl
- Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- 4-12 member non-aromatic heterocyclyls which may be substituted with substituents of C 6-10 aryloxy, which may be substituted with one or more substituents selected from Rd, selected from Rd.
- Rd Independently selected from the group consisting of C 6-10 arylsulfanyl, which may be substituted with one or more substituents, and adamantyl;
- Rb is substituted with one or more substituents selected from halogen atoms, hydroxys, Ra and C 1-6 alkyl, which may be substituted with one or more substituents selected from Ra.
- Rc may be C 1-6 alkyl substituted with nitro, hydroxy, 1 or 2 or more halogen atoms, C 1-6 alkoxy optionally substituted with 1 or 2 or more halogen atoms, halogen atom, Amino, cyano, C 1-6 alkyl amino optionally substituted with 1 or 2 or more hydroxy groups, di (C 1-6 alkyl) amino, C optionally substituted with 1 or 2 or more hydroxy groups One or more selected from 3-10 cycloalkylamino, (C 1-6 alkyl) carbonyl, (C 1-6 alkoxy) carbonyl, C 1-6 alkyl sulfonyl, C 3-10 cycloalkyl sulfonyl, Re C 7-14 Aralkyl, which may be substituted with a substituent, C 6-10 aryl, which may be substituted with 1 or 2 or more substituents selected from Re, 1 or 2 or more selected from Re.
- Rd may be C 1-6 alkyl substituted with nitro, hydroxy, 1 or 2 or more halogen atoms, C 1-6 alkoxy optionally substituted with 1 or 2 or more halogen atoms, halogen atom, Amino, cyano, C 1-6 alkyl amino, di (C 1-6 alkyl) amino, C 1-6 alkyl carbonyl, (C 1-6 alkoxy) carbonyl, (C 1-6 alkoxy) carbonyl amino, N- ( C 1-6 alkoxy) substituted with one or more substituents selected from carbonyl-N- (C 1-6 alkyl) amino, C 1-6 alkyl sulf
- Re may be substituted with one or more substituents selected from nitro, hydroxy, halogen atom and hydroxy C 1-6 alkyl, halogen atom, amino, cyano, C 1-6 alkylamino, di.
- (C 1-6 alkyl) C 1-6 alkyl optionally substituted with 1 or 2 or more substituents selected from amino, phenyl and hydroxy 1 or 2 or more substitutions selected from C 1-6 alkyl carbonyl, phenyl and hydroxy May be substituted with a group (C 1-6 alkoxy) carbonyl, C 1-6 alkyl sulfonyl, C 3-8 cycloalkyl sulfonyl, C 1-6 alkyl sulfonyl amino, C 3-8 cycloalkyl sulfonyl amino, as well.
- R 21 may be substituted with 1 or 2 or more substituents selected from the hydrogen atom, Ra, C 1-6 alkyl, even if substituted with 1 or 2 or more substituents selected from Rd.
- Good C 6-10 aryl, 4-12 member heterocyclyls optionally substituted with 1 or 2 or more substituents selected from Rc, substituted with 1 or 2 or more substituents selected from Rc May be 5-10 membered heteroaryl, optionally substituted with 1 or 2 or more substituents selected from Ra (C 1-6 alkoxy) carbonyl, 1 or 2 or more substituents selected from Ra.
- Di (C 1-6 alkyl) aminocarbonyl substituted with 1 or 2 or more substituents of choice may be substituted with 1 or 2 or more halogen atoms C 1-6 alkylsulfonyl, C 7-14 Aralkyl sulfonyl, C 3-10 cycloalkyl sulfonyl, amino sulfonyl, C 1-6 alkyl amino sulfonyl, di (C 1-6 alkyl) amino sulfonyl, and di (C 1-6 alkyl) phosphono Selected;
- R 22 is selected from the group consisting of a hydrogen atom and a C 1-6 alkyl optionally substituted with one or more substituents selected from Ra;
- R 23 may be substituted with a hydrogen atom, 1 or 2 or more substituents selected from Ra, C 1-6 alkyl, even if substituted with 1 or 2 or more substituents selected from Ra.
- Good [(C 1-6 alkyl) amino] C 1-6 alkyl, optionally substituted with one or more substituents selected from Ra [di (C 1-6 alkyl) amino] C 1- 6 Alkyl, may be substituted with 1 or 2 or more substituents selected from Rc C 3-10 Cycloalkyl, may be substituted with 1 or 2 or more substituents selected from Rd C 6 -10 aryl, 5-10 membered heteroaryl which may be substituted with 1 or 2 or more substituents selected from Rc, and 1 or 2 or more substituents selected from Rc.
- R 24 is selected from the group consisting of a hydrogen atom and a C 1-6 alkyl optionally substituted with one or more substituents selected from Ra].
- a 2 is a C 1-10 alkyl, which may have one or more substituents independently selected from Ra;
- Q 1 is selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, each of which is independently selected from Rb 1 It may have the above substituents;
- X 6 and X 7 are independently hydrogen atoms or C 1-10 alkyl, and the alkylene may have one or more substituents independently selected from Ra;
- Q 2 and Q 3 are independently selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, the group of which is independent.
- Each may have one or more substituents independently selected from Rb;
- X 1 , X 3 and X 4 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra;
- a 1 , X 3 , and X 4 are C 2-10 alkylenes, 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted into the alkylene.
- R 11 may be a hydrogen atom or a C 1-6 alkyl;
- X 2 and X 5 are C 2-10 alkyl, even if 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted in the alkylene.
- R 11 is a hydrogen atom or C 1-6 alkyl
- Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- 4-12 member non-aromatic heterocyclyls which may be substituted with substituents of C 6-10 aryloxy, which may be substituted with one or more substituents selected from Rd, selected from Rd.
- Rd Independently selected from the group consisting of C 6-10 arylsulfanyl, which may be substituted with one or more substituents, and adamantyl;
- Rb is substituted with one or more substituents selected from halogen atoms, hydroxys, Ra and C 1-6 alkyl, which may be substituted with one or more substituents selected from Ra.
- Rc may be C 1-6 alkyl substituted with nitro, hydroxy, 1 or 2 or more halogen atoms, C 1-6 alkoxy optionally substituted with 1 or 2 or more halogen atoms, halogen atom, Amino, cyano, C 1-6 alkyl amino optionally substituted with 1 or 2 or more hydroxy groups, di (C 1-6 alkyl) amino, C optionally substituted with 1 or 2 or more hydroxy groups One or more selected from 3-10 cycloalkylamino, (C 1-6 alkyl) carbonyl, (C 1-6 alkoxy) carbonyl, C 1-6 alkyl sulfonyl, C 3-10 cycloalkyl sulfonyl, Re C 7-14 Aralkyl, which may be substituted with a substituent, C 6-10 aryl, which may be substituted with 1 or 2 or more substituents selected from Re, 1 or 2 or more selected from Re.
- Rd may be C 1-6 alkyl substituted with nitro, hydroxy, 1 or 2 or more halogen atoms, C 1-6 alkoxy optionally substituted with 1 or 2 or more halogen atoms, halogen atom, Amino, cyano, C 1-6 alkyl amino, di (C 1-6 alkyl) amino, C 1-6 alkyl carbonyl, (C 1-6 alkoxy) carbonyl, (C 1-6 alkoxy) carbonyl amino, N- ( C 1-6 alkoxy) substituted with one or more substituents selected from carbonyl-N- (C 1-6 alkyl) amino, C 1-6 alkyl sulf
- Re may be substituted with one or more substituents selected from nitro, hydroxy, halogen atom and hydroxy C 1-6 alkyl, halogen atom, amino, cyano, C 1-6 alkylamino, di.
- (C 1-6 alkyl) C 1-6 alkyl optionally substituted with 1 or 2 or more substituents selected from amino, phenyl and hydroxy 1 or 2 or more substitutions selected from C 1-6 alkyl carbonyl, phenyl and hydroxy May be substituted with a group (C 1-6 alkoxy) carbonyl, C 1-6 alkyl sulfonyl, C 3-8 cycloalkyl sulfonyl, C 1-6 alkyl sulfonyl amino, C 3-8 cycloalkyl sulfonyl amino, as well.
- R 21 may be substituted with 1 or 2 or more substituents selected from the hydrogen atom, Ra, C 1-6 alkyl, even if substituted with 1 or 2 or more substituents selected from Rd.
- Good C 6-10 aryl, 4-12 member heterocyclyls optionally substituted with 1 or 2 or more substituents selected from Rc, substituted with 1 or 2 or more substituents selected from Rc May be 5-10 membered heteroaryl, optionally substituted with 1 or 2 or more substituents selected from Ra (C 1-6 alkoxy) carbonyl, 1 or 2 or more substituents selected from Ra.
- Di (C 1-6 alkyl) aminocarbonyl substituted with 1 or 2 or more substituents of choice may be substituted with 1 or 2 or more halogen atoms C 1-6 alkylsulfonyl, C 7-14 Aralkyl sulfonyl, C 3-10 cycloalkyl sulfonyl, amino sulfonyl, C 1-6 alkyl amino sulfonyl, di (C 1-6 alkyl) amino sulfonyl, and di (C 1-6 alkyl) phosphono Selected;
- R 22 is selected from the group consisting of a hydrogen atom and a C 1-6 alkyl optionally substituted with one or more substituents selected from Ra;
- R 23 may be substituted with a hydrogen atom, 1 or 2 or more substituents selected from Ra, C 1-6 alkyl, even if substituted with 1 or 2 or more substituents selected from Ra.
- Good [(C 1-6 alkyl) amino] C 1-6 alkyl, optionally substituted with one or more substituents selected from Ra [di (C 1-6 alkyl) amino] C 1- 6 Alkyl, may be substituted with 1 or 2 or more substituents selected from Rc C 3-10 Cycloalkyl, may be substituted with 1 or 2 or more substituents selected from Rd C 6 -10 aryl, 5-10 membered heteroaryl which may be substituted with 1 or 2 or more substituents selected from Rc, and 1 or 2 or more substituents selected from Rc.
- R 24 is selected from the group consisting of a hydrogen atom and a C 1-6 alkyl optionally substituted with one or more substituents selected from Ra].
- Trivalent core block In the divalent core block, a trivalent core block having one further linker binding position at an arbitrary position.
- the core blocks are mono- or divalent core blocks independently represented by the following equations: 1) Divalent core block: 1-1) -A 1- ; 1-2) -X 1 -Q 1 -X 2- ; or 1-3) -X 3 -Q 2 -X 4 -Q 3 -X 5- ;
- a 1 is a C 1-10 alkylene, which may have one or more substituents independently selected from Ra
- Q 1 is selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, each of which is independently selected from Rb 1 It may have the above substituents
- X 1 and X 2 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra
- Q2 and Q3 are independently selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocycl
- Each may have one or more substituents independently selected from Rb;
- X 3 , X 4 and X 5 are independently single-bonded or C 1-10 alkylenes, which may have one or more substituents independently selected from Ra;
- a 1 , X 1 , X 2 , X 3 , X 4 and X 5 are C 2-10 alkylenes, then the alkylene is selected from -O-, -S-, or -NR 11- .
- R 11 is a hydrogen atom or C 1-6 alkyl
- Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- Rc substituents selected from Rc, 1 or 2 or more.
- Rb is substituted with one or more substituents selected from halogen atom, hydroxy, Ra and C 1-6 alkyl which may be substituted with one or more substituents selected from Ra.
- a 2 is a C 1-10 alkyl, which may have one or more substituents independently selected from Ra;
- Q 1 is selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, each of which is independently selected from Rb 1 It may have the above substituents;
- X 6 and X 7 are independently hydrogen atoms or C 1-10 alkyl, and the alkylene may have one or more substituents independently selected from Ra;
- Q 2 and Q 3 are independently selected from C 5-10 arylene, 5-10 membered heteroarylene, 5-8 membered non-aromatic heterocyclylene, and C 3-10 cycloalkylene, the group of which is independent.
- Each may have one or more substituents independently selected from Rb;
- X 1 , X 3 and X 4 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra;
- a 1 , X 3 , and X 4 are C 2-10 alkylenes, 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted into the alkylene.
- R 11 may be a hydrogen atom or a C 1-6 alkyl;
- X 2 and X 5 are C 2-10 alkyl, even if 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted in the alkylene.
- R 11 is a hydrogen atom or C 1-6 alkyl
- Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- Rc substituents selected from Rc, 1 or 2 or more.
- Rb is substituted with one or more substituents selected from halogen atom, hydroxy, Ra and C 1-6 alkyl which may be substituted with one or more substituents selected from Ra.
- the core blocks are mono- or divalent core blocks independently represented by the following equations: 1) Divalent core block: 1-1) -A 1- ; 1-2) -X 1 -Q 1 -X 2- ; or 1-3) -X 3 -Q 2 -X 4 -Q 3 -X 5- ; [In the formula, A 1 is a C 1-10 alkylene, which may have one or more substituents independently selected from Ra; Q1 , Q2 and Q3 consist of a group consisting of a benzene ring, a pyridine ring, a piperidine ring, a piperazine ring, a pyrimidine ring, a pyrazine ring, a pyridazine ring, a pyrazole ring, an imidazole ring, a thiophene ring, a furan proposal, and a naphthalene ring.
- X 1 and X 2 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra;
- X 3 , X 4 and X 5 are independently single-bonded or C 1-10 alkylenes, which may have one or more substituents independently selected from Ra;
- a 1 , X 1 , X 2 , X 3 , X 4 and X 5 are C 2-10 alkylenes, then the alkylene is selected from -O-, -S-, or -NR 11- .
- R 11 is a hydrogen atom or C 1-6 alkyl
- Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- Rc substituents selected from Rc, 1 or 2 or more.
- Rb is substituted with one or more substituents selected from halogen atom, hydroxy, Ra and C 1-6 alkyl which may be substituted with one or more substituents selected from Ra.
- a 2 is a C 1-10 alkyl, which may have one or more substituents independently selected from Ra;
- Q1 , Q2 and Q3 consist of a group consisting of a benzene ring, a pyridine ring, a piperidine ring, a piperazine ring, a pyrimidine ring, a pyrazine ring, a pyridazine ring, a pyrazole ring, an imidazole ring, a thiophene ring, a furan proposal, and a naphthalene ring.
- X 6 and X 7 are independently hydrogen atoms or C 1-10 alkyl, and the alkylene may have one or more substituents independently selected from Ra;
- X 1 , X 3 and X 4 are independently single-bonded or C 1-10 alkylene, and the alkylene may have one or more substituents independently selected from Ra;
- a 1 , X 3 , and X 4 are C 2-10 alkylenes, 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted into the alkylene.
- R 11 may be a hydrogen atom or a C 1-6 alkyl; In the above definition, when X 2 and X 5 are C 2-10 alkyl, even if 1 to 3 groups selected from -O-, -S-, or -NR 11- are inserted in the alkylene. Often, R 11 is a hydrogen atom or C 1-6 alkyl; Ra is a halogen atom, hydroxy, C 1-6 alkoxy, cyano, (C 1-6 alkoxy) carbonyl, carboxy, (C 1-6 alkoxy) carbonyl amino, (C 1-6 alkyl) carbonyl amino, amino, C.
- Rc substituents selected from Rc, 1 or 2 or more.
- Rb is substituted with one or more substituents selected from halogen atom, hydroxy, Ra and C 1-6 alkyl which may be substituted with one or more substituents selected from Ra.
- the reactive functional group derivatization step means a step of converting a reactive functional group into another group, for example, a step of introducing a protective group into a reactive functional group (protection step), a reactive functional group.
- a reactive functional group examples thereof include a step of deprotecting a protective group contained in a group, a step of converting a reactive functional group into a group for forming a linker, and a step of converting a reactive functional group into another functional group.
- the reactive functional group derivatization step is not particularly limited, but for example, a step of converting a primary or secondary amino group into a sulfonamide group, a step of converting a primary or secondary amino group into an amide group, and the like.
- a step of converting a primary or secondary amino group into a secondary or tertiary amino group a step of converting a carboxyl group into an amide group, a step of converting a carboxyl group into an acylsulfonamide group, a step of converting a carboxyl group into an ester group.
- Conversion step carboxyl group to hydroxymethyl group, formyl group to amino group, ketone group to amino group, halogen atom to hydroxy group, hydroxy group to ether group Conversion of hydroxy group to ester group, conversion of hydroxy group to sulfonic acid ester group, conversion of hydroxy group to halogen atom, conversion of hydroxymethyl group to formyl group, hydroxymethyl
- a step of converting a group into a carboxyl group a step of converting an olefin group into a boron atom-containing group by reacting it with a boron reagent, a step of converting a halogen atom into a boron atom-containing group by reacting it with a boron reagent, and a halogen.
- Examples thereof include a step of converting an atom into a carboxy group, a step of converting a carboxy group into a thioester group, a step of converting a thioester group into a boron atom-containing group, a step of converting an amino group into a halogen atom, and the like.
- the step of converting the halogen atom to the hydroxy group is not particularly limited, but in a solvent such as NMP, DMF, THF, DMPr (N, N-dimethylpropionamide), DEAc (N, N-diethylacetamide) and the like. It can be converted into a hydroxy group by reacting a halogen atom with water at 20 to 80 ° C.
- the step of converting the hydroxy group to the sulfonic acid ester group is not particularly limited, and is, for example, MsCl, TsCl, NsCl, DNsCl, chloromethanesulfonyl chloride, nonafluorobutanesulfonyl chloride, methanesulfonic acid anhydride, trifluoromethanesulfonic acid.
- An anhydride, nonafluorobutane sulfonic acid anhydride, or the like can be reacted with a hydroxy group at ⁇ 78 to 80 ° C. in a solvent such as THF or DCM. If necessary, it can also be carried out in the presence of a tertiary amine such as triethylamine and an aromatic base such as pyridine or 2,6-lutidine.
- the step of converting an olefin group into a boron atom-containing group by reacting it with a boron reagent or the like is not particularly limited, but for example, 9-BBN, pinacol borane, catechol borane, bis (pinacolat) diborone or the like is converted into THF or the like. It can be converted into a boron atom-containing group by reacting with an olefin group at 20 to 80 ° C. in a solvent. If necessary, it can be carried out in the presence of a metal catalyst such as a palladium catalyst, an iridium catalyst, a rhodium catalyst, a ruthenium catalyst, or a copper catalyst.
- a metal catalyst such as a palladium catalyst, an iridium catalyst, a rhodium catalyst, a ruthenium catalyst, or a copper catalyst.
- the step of converting a halogen atom into a boron atom-containing group by reacting it with a boron reagent is not particularly limited, but for example, in the presence of a metal catalyst such as a palladium catalyst or a copper catalyst, bis (pinacholate) diboron or the like is THF. It can be converted into a boron atom-containing group by reacting with a halogen atom at 20 to 100 ° C. in a solvent such as.
- the step of converting the halogen atom to the carboxy group is not particularly limited, but is limited to carbon monoxide or a reagent that produces carbon monoxide in the reaction system in the presence of a metal catalyst such as a palladium catalyst (such as oxalic acid and N-formyl saccharin). ) Can be converted into a carboxy group by reacting with a halogen atom at 20 to 100 ° C. in a solvent such as DMF.
- a metal catalyst such as a palladium catalyst (such as oxalic acid and N-formyl saccharin).
- the step of converting the carboxy group to the thioester group is not particularly limited, but the carboxy group and the thiol are reacted at 20 to 80 ° C. in a solvent such as DCM or NMP in the presence of a condensing agent such as DIC, PyClU or PipClU. This can be converted to a thioester group.
- the step of converting the thioester group to the boron atom-containing group is not particularly limited, but for example, in the presence of a metal catalyst such as a rhodium catalyst, bis (pinacholate) diboron or the like is thioestered at 20 to 100 ° C. in a solvent such as THF. By reacting the groups, it can be converted into a boron atom-containing group.
- a metal catalyst such as a rhodium catalyst, bis (pinacholate) diboron or the like is thioestered at 20 to 100 ° C. in a solvent such as THF.
- the product A—X is an aryl halide
- the reactive functional group X linked to A is a halogen atom such as a fluorine atom, a chlorine atom, a bromine atom, or iodine. It is possible to carry out the step of converting to a halogen atom by subjecting it to a reaction with an amino group of a reactive functional group linked to A of the raw material using an atom, a nitrite derivative, an acid, and a halogen source. can.
- the nitrite derivative is not particularly limited as long as it can be usually used, but for example, sodium nitrite, potassium nitrite, methyl nitrite, ethyl nitrite, propyl nitrite, isopropyl nitrite, tert-nitrite. Butyl, isobutyl nitrite, butyl nitrite, amyl nitrite, isoamyl nitrite, hexyl nitrite and the like can be used.
- the acid is not particularly limited as long as it can be usually used, but for example, hydrochloric acid, hydrobromic acid, sulfuric acid, methanesulfonic acid, p-toluenesulfonic acid, camphor-sulfonic acid, acetic acid, tetrafluoroboric acid and the like are used. be able to.
- the halogen source is not particularly limited as long as it can be usually used, but for example, hydrochloric acid, hydrobromic acid, bromine, copper (I) chloride (CuCl), copper (I) bromide (CuBr), copper iodide ( I) (CuI), Copper Bromide (II) (CuBr 2 ), Potassium Bromide (KBr), Sodium Bromide (NaBr), Potassium Iodine (KI), Lithium Chloride (LiCl), Bromoform (CHBr 3 ), Dibromomethane (CH 2 Br 2 ), diiodomethane (CH 2 I 2 ), bromotrichloromethane (BrCCl 3 ), bromotrimethylsilane (TMSBr), tetrabutylammonium bromide (TBAB), benzylammonium chloride, N-bromosuccinimide (NBS). ), N-chlorosuccinimide (NCS), N-
- the equivalent number of the nitrite derivative to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to an amine containing A. It can be ⁇ 20 equivalents.
- the number of equivalents of the acid used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 10 with respect to an amine containing A. Can be equivalent.
- the number of equivalents of the halogen source to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing A. It can be 20 equivalents.
- a solvent can be used in the above reaction, and the reaction is not particularly limited, but for example, halogen-based solvents such as DCM, dibromomethane (DBM), chloroform, bromoform, DCE, dibromoethane, carbon tetrachloride, diethyl ether, diisopropyl ether, etc.
- halogen-based solvents such as DCM, dibromomethane (DBM), chloroform, bromoform, DCE, dibromoethane, carbon tetrachloride, diethyl ether, diisopropyl ether, etc.
- Ether-based solvents such as THF (tetrahydrogen), 2-methyltetrahydrogen, 4-methyltetrahydropyran, 1,4-dioxane, DME, TBME, CPME, diisopropyl ether, isosorbidodimethylether, diglyme, triglyme, tetraglyme, acetone, MEK, Ketone-based solvents such as MIBK, 4-methyl-2-pentanone, cyclopentanone, cyclohexanone, diethyl ketone, urea-based solvents such as TMU, DMI, and DMPU, and amide-based solvents such as DMF, DMA, NFM, NMP, NEP, and NBP.
- THF tetrahydrogen
- 2-methyltetrahydrogen 4-methyltetrahydropyran
- 1,4-dioxane 1,4-dioxane
- DME TBME
- CPME diis
- Ester solvents such as methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate, GVL ( ⁇ -valerolactone), pentyl acetate, methyl propionate, urea solvents (TMU (N, N,) N', N'-tetramethylurea), DMI (1,3-dimethyl-2-imidazolidinone), DMPU (N, N'-dimethylpropylene urea), etc.), amide solvent (DMF (N, N-) Dimethylformamide), DMA (N, N-dimethylacetamide), NFM (N-formylmorpholin), NMP (N-methyl-2-pyrrolidone), NEP (N-ethyl-2-pyrrolidone), NBP (N-butyl- 2-pyrrolidone), formamide, etc.), sulfoxide-based solvent (DMF (N
- the reaction temperature can be appropriately set by those skilled in the art, and can be, for example, -20 to 100 ° C, -100 to 80 ° C, -5 to 60 ° C, 0 to 45 ° C, or 0 to 30 ° C.
- an additive may be used, and for example, copper (II) oxide (CuO), copper sulfate (II) (CuSO4), iron (II) sulfate (FeSO4) and the like can be used as the additive. ..
- the number of equivalents of the additive to be used can be appropriately set by those skilled in the art, for example, 1 to 100 equivalents, 1 to 75 equivalents, 1 to 50 equivalents, 1 to 30 equivalents, or 1 to 1 to an amine containing A. It can be 10 equivalents.
- the reaction time can be appropriately set by those skilled in the art, and for example, the time required to reach a sufficient conversion rate in a study experiment (also called a model experiment) using the corresponding substrate or a similar substrate, or something from the study result
- the time expected by the technique, or the product excision from some solid phase carriers, can be used to determine or confirm the end of the reaction.
- the desired compound can be obtained by a usual post-treatment method.
- Any protective group, introduction condition, or deprotection condition known in the present art can be used for the step of introducing the protecting group into the reactive functional group and the step of deprotecting the protecting group, and the process is not particularly limited.
- it can be appropriately set and used with reference to "Greene's Protective Groups in Organic Synthesis, Fifth edition” and the like.
- the step of introducing a protective group into a reactive functional group is not particularly limited, but for example, as a group for introducing a protective group into a primary or secondary amino group, for example, a Boc group, an Fmoc group, a Cbz group, or a Teoc group. , Troc group, Alloc group, Trt group, Ns group, DNs group, Ts group, PMB group, DMB group, trifluoroacetyl group, THP group, SEM group and the like.
- a group for introducing a protective group into a primary or secondary amino group for example, a Boc group, an Fmoc group, a Cbz group, or a Teoc group.
- Troc group Alloc group, Trt group, Ns group, DNs group, Ts group, PMB group, DMB group, trifluoroacetyl group, THP group, SEM group and the like.
- the form in which the protective group of the carboxyl group is introduced is not particularly limited, and for example, methyl ester, ethyl ester, propyl ester, isopropyl ester, isobutyl ester, t-butyl ester, allyl ester, trityl ester, 2-chlorotrityl ester, and the like.
- Examples thereof include benzyl ester and OBO ester.
- the protective group to be introduced into the hydroxy group is not particularly limited, but for example, an acetyl group, a pivaloyl group, a t-butyl group, a trityl group, a 2-chlorotrityl group, a THP group, an MOM group, a TMS group, a TES group and a triisopropylsilyl.
- Examples thereof include a group, a TBDMS group, a TBDPS group, an SEM group, a tert-butyldiphenylcyrylylethyl group, an allyl group, a benzyl group, a phenylcarbamate group, a methylacetal group and the like.
- the protecting group to be introduced into the formyl group is not particularly limited, and examples thereof include a dimethyl acetal group, a diisopropyl acetal group, and an ethylidene acetal group.
- the protecting group to be introduced into the sulfonamide group is not particularly limited, and examples thereof include a PMB group, a DMB group, a Trt group, an SEM group, an allyl group, and a benzyl group.
- the protecting group to be introduced into a nitrogen atom such as a pyrazole ring, an imidazole ring, an indole ring, and an indazole ring is not particularly limited, but for example, a PMB group, a DMB group, a Trt group, an SEM group, an allyl group, a benzyl group, a THP group and the like can be used. Can be mentioned.
- a step of deprotecting the protecting group introduced into the reactive functional group for example, an amino group, a carboxy group, a hydroxy group, a formyl group, a ketone group, a thiol group and the like can be mentioned as a group generated by the deprotection.
- the protective group removed from the amino group for example, Boc group, Fmoc group, Cbz group, Teoc group, Troc group, Alloc group, Trt group, Ns group, DNs group, Ts group, PMB group, DMB group and trifluoroacetyl group. , THP group, SEM group and the like.
- Pre-stage forms of exposing carboxyl groups by deprotection include, for example, methyl esters, ethyl esters, propyl esters, isopropyl esters, isobutyl esters, t-butyl esters, allyl esters, trityl esters, 2-chlorotrityl esters, benzyl esters. , OBO ester, and the like.
- Protective groups removed from the hydroxy group include, for example, acetyl group, pivaloyl group, t-butyl group, trityl group, 2-chlorotrityl group, THP group, MOM group, TMS group, TES group, triisopropylsilyl group, TBDMS group. , TBDPS group, SEM group, tert-butyldiphenylcyrylylethyl group, allyl group, benzyl group and the like.
- the protecting group removed from the formyl group is not particularly limited, and examples thereof include a dimethyl acetal group, a diisopropyl acetal group, and an ethylidene acetal group.
- the protecting group removed from the sulfonamide group is not particularly limited, and examples thereof include a PMB group, a DMB group, a Trt group, an SEM group, an allyl group, and a benzyl group.
- the protecting group removed from the nitrogen atom such as the pyrazole ring, the imidazole ring, the indole ring, and the indazole ring is not particularly limited, and examples thereof include a PMB group, a DMB group, a Trt group, an SEM group, an allyl group, a benzyl group, and a THP group. Can be mentioned.
- a method for producing a carbon-carbon bond forming reaction by Suzuki coupling which has a high yield in solid phase synthesis. Specifically, when meCgPPhPdG4 and (dtbpf) PdCl 2 are used as the palladium catalyst, cutting out from the solid phase is reduced, and the Suzuki coupling reaction can be carried out in high yield.
- any linker that covalently connects the resin for solid phase synthesis and the first BB has a benzyl ether structure
- the benzyl ether structure is cleaved in the carbon-carbon bond formation reaction catalyzed by palladium, and the progress of cleavage of the compound is confirmed.
- the substrate contained on the solid phase side has low reactivity and / or the reactivity of the reagent is low, it may take a long reaction time to complete the reaction, and the progress of excision from the solid phase progresses. It can occur that the yield decrease in the solid phase reaction is greatly affected.
- ASA indicates an accessible surface area and is used as an index indicating the contactable area of another molecule around a specific atom.
- Science. 1983 Aug19; 221 (4612): 709-13., J. Org. Chem. 2011, 76, 2, 435-440 ASA generated conformations in Schrödinger's Maestro MacroModel (searching from the most stable conformation to the high energy conformation up to 30 kJ / mol in CHCl 3 ), and for each of the obtained conformations.
- the ASA was calculated by Schrodinger's Maestro Advanced Surfaces (Type: Extended Surface), and the largest ASA among the multiple conformations was selected.
- the 0.04 M P (Cy) 3Pd G3 1,4-dioxane solution used in the examples was prepared as follows. It was prepared by adding 1,4-dioxane (1 mL) to P (Cy) 3Pd G3 (26 mg, 0.04 mmol) in a 2 mL glass vial under a nitrogen atmosphere and shaking to dissolve.
- the 1.5 M aqueous solution of K3 PO4 used in the examples was prepared as follows. Using a 10 mL volumetric flask under a nitrogen atmosphere, K3 PO 4 ( 3.18 g, 15 mmol) was prepared by measuring up with distilled water.
- the 0.02M P (Cy) 3Pd G3 NMP solution used in the examples was prepared as follows. Dissolve by adding NMP (2.98 mL) to P (Cy) 3Pd G3 (38.7 mg, 60 ⁇ mol) and 9-methylcarbazole (5.7 mg, 31 ⁇ mol) in a 4 mL glass vial under a nitrogen atmosphere and shaking. And prepared.
- reaction tracking and purity measurement are carried out using SQD (Waters) or 2020 (Shimadzu) and mass spectrometer under the analytical conditions shown below to measure retention time and mass spectrometry. I went there.
- the descriptions m / z [M + H] + and (M + H) + described in the analysis results of LCMS in the examples are all the values detected in the positive mode. Further, the UV area% in LCMS showed a value in PDA (210-400 nm) unless otherwise specified. When a specific wavelength (for example, 319 nm, 290 nm, 280 nm, etc.) is described, the UV area% at a wavelength up to +/- 4 nm centered on the described wavelength is described.
- Conversion rate and conversion rate are synonymous with each other and indicate the degree of progress of the reaction, and are calculated by the following formulas unless otherwise specified.
- Silica gel for column chromatography is SHOKO Scientific Purif-Pack (registered trademark) SI (60 ⁇ m), SHOKO Purif-Pack (registered trademark) -EX SI (50 ⁇ m), Biotage (registered trademark) SNAP Cartridge KP-Si, registered trademark. ), SNAP ⁇ Ltra, Biotage (registered trademark) Subar D (Duo) (60 ⁇ m), Biotage (registered trademark) Subar HC D (Duo) (20 ⁇ m) and the like were appropriately used.
- Phase Separator refers to an operation of removing water contained in an organic phase using, for example, Biotage (registered trademark) ISOLUTE Phase Separators.
- concentration concentration under reduced pressure or “concentration under reduced pressure” refer to the evaporation and removal of solvent under reduced pressure by a rotary evaporator, mechanical oil vacuum pump or mechanical oil-free vacuum pump.
- the expressions “overnight” and “all night” refer to about 8 to 14 hours, but this is not the case.
- the solid phase reaction can be carried out in any suitable container, eg, a glass vial that can be sealed with a cap with Teflon packing, a frit filter and a column with a suitable stopper.
- the container size allows the resin to be effectively agitated, taking into account that there is sufficient space for the solvent and that certain resins can swell significantly when treated with an organic solvent. Select a size that has ample room.
- “Shaking” in solid phase reactions is a shaker suitable for ensuring sufficient mixing, which is a commonly accepted factor for successful reaction on resin (eg, Tokyo Rika Kikai, EYELA, MMS). -320, MMS-220H, or AS ONE, MyBL-100CS, or TAITEC, M.BR-104) was used at 50-200 rpm or 600-1000 rpm.
- the equivalent of the reagents is a multiple (equivalent) number of moles with respect to the total number of moles of functional groups used in the reaction of each compound of the mixture on the solid phase side. Indicates whether to use the reagent of. For example, when describing 10 equivalents, it is recommended to use a reagent having a molar number 10 times (10 equivalents) the total number of moles of functional groups used in the reaction of each compound of the mixture on the solid phase side. show.
- the equivalent of the reagents when the substrate is a mixture is a number of moles (equivalents) of the total number of moles of the functional groups used in the reaction of each compound of the mixture. Indicates whether to use. For example, when it is described as 10 equivalents, it means that a reagent having 10 times (10 equivalents) the number of moles is used with respect to the total number of moles of functional groups used in the reaction of each compound of the mixture.
- the number of moles of the mixture for example, the following cases can be considered. Unless otherwise specified, any value may be adopted. When individual compounds whose number of moles is known in advance are mixed, the total may be considered as the number of moles of the mixture.
- the total sum of the prepared solid-phase-supported substrate-supported amount (mmol / g) multiplied by the amount of the solid-phase-supported substrate used is added. It can be thought of as the number of moles of the mixture.
- the conversion rate of each step may be considered as 100% (that is, the theoretical amount), and when cut out from the solid phase, it is cut out.
- the reaction may be carried out in the subsequent liquid phase, assuming that the efficiency is 100% (that is, the theoretical amount has been cut out).
- the solid-phase supporting compound used for solid-phase synthesis shows the loading rate (mmol / g), which indicates the loading rate (initial loading rate) calculated when the starting material is supported on the solid phase. There is.
- the loading rate after the reaction may differ from the initial loading rate because the molecular weight changes due to the ligation reaction of the building blocks, but the initial loading rate may be used throughout the synthesis process.
- the loading rate may differ depending on the lot, but the same compound number may be used for the compound number.
- filtering with a filtered vial refers to the operation of removing insoluble matter contained in the reaction solution or diluted solution, and refers to an LC vial with a filter (for example, Thomason SINGLE StEP Filter Vials, TH-). It is shown that the LC solution is prepared by adding the solution into 35541) and pushing the inner vial.
- the notation of indicates polystyrene resin, and indicates a state in which the compound is supported on the solid phase.
- the number in “-01R” indicating that the compound was supported on the solid phase used in the examples indicates the type of resin used.
- "01" of “-01R” is supported on brominated (brominatedo) Wang resin, 4- (bromomethyl) phenoxymethylpolystyrene (for example, Merck, 4- (benzyloxy) benzyl bromide, polymer-supported).
- the compound in the state of benzene is shown.
- "02” in “-02R” is 4- (bromomethyl) phenoxyethyl polystyrene (for example, Merck & Co., 2- (4-bromomethylphenoxy) ethyl polystyrene, sometimes referred to as modified Wang resin in the examples).
- the solid-phase-supported compounds used in Examples 8, 9 and 10 are all polystyrene carboxylic acids (for example, CHEM-IMPEX, polystyrene carboxylate, sometimes referred to as carboxylic acid resin in Examples).
- CHEM-IMPEX polystyrene carboxylate, sometimes referred to as carboxylic acid resin in Examples.
- a compound in a state of being carried via a linker portion is used.
- Mixture numbers may also be used to indicate the mixtures synthesized in Examples 8, 9 and 10. For example, “2-1" in “2-1-a1B01-0” indicates the type of library, and "a1B01" indicates the type of mixture. "-0" of "-0” indicates a state supported on the solid phase, and "-1" indicates a state cut out from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID may be used to indicate each compound contained in the mixture.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID may be used to indicate each compound contained in the mixture.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID may be used to indicate each compound contained in the mixture.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is performed after excision from the solid phase.
- ID indicates a state in which functional group conversion is
- the obtained crude product is dissolved in ethyl acetate (100 mL) and hexane (200 mL), washed 3 times with water (200 mL) and once with saturated aqueous sodium chloride solution (100 mL), and concentrated under reduced pressure to form a white solid.
- the title compound D066 (1.44 g, 2.71 mmol, 83%) was obtained.
- tert-butyl 4- [4- (hydroxymethyl) phenoxy] piperidine-1-carboxylate (D062) and methyl 3 Methyl 3- [4-[(4-piperidin-4-yloxyphenyl) methoxy] phenyl] benzoato (D068) can be synthesized from ⁇ (4-hydroxyphenyl) benzoato (D070).
- D068, which illustrates the synthetic method in Examples 1-1-5, may be referred to as compound C101 in subsequent examples.
- Carboxylic acid resin (C001-03R) (carrying amount 2.19 mmol / g, 4.50 g, 9.86 mmol) and NMP (67.5 mL) were added to a 100 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour. Finally. Methyl 3- [4-[(4-piperidin-4-yloxyphenyl) methoxy] phenyl] benzoat (C101) (1.315 g, 3.15 mmol) and HOAt (1.34 g, 9.86 mmol), DIC (1) (5.44 mL, 9.86 mmol) was added, and the mixture was shaken at room temperature for 22 hours. Piperidine (5.85 mL, 59.1 mmol), HOAt (8.05 g, 59.1 mmol) and DIC (9.21 mL, 59.1 mmol) were added and shaken overnight at room temperature.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.010 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.2 M pentamethylbenzene for 2 minutes. After filtration, NMP (0.25 mL) was added to the filtrate. Acetonitrile (0.25 mL) was added to 0.050 mL of the obtained solution to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, 100% of the target object C002 was observed.
- Examples 1-1-7 Synthesis of compound C003-03R
- compound C002-03R (supported amount 0.507 mmol / g, 6.79 g, 3.44 mmol), methanol (4.07 mL) and THF (36.6 mL) were added to a 50 mL glass vial at room temperature. Shake for 1 hour. Aqueous sodium hydroxide solution (5M, 3.55 mL, 17.8 mmol) was added and shaken at 60 ° C. for 3 hours.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.010 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.2 M pentamethylbenzene for 2 minutes. After filtration, NMP (0.25 mL) was added to the filtrate. Acetonitrile (0.25 mL) was added to 0.050 mL of the obtained solution to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, 100% of the target object C003 was observed.
- Carboxylic acid resin (C001-03R) (supported amount 2.19 mmol / g, 1.00 g, 2.19 mmol) and NMP (15 mL) were added to a 20 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour. ..
- reaction solution and the solid phase suspension (10 ⁇ L) were transferred onto a filter and washed 3 times with NMP (0.1 mL), 3 times with methanol (0.1 mL) and 3 times with DCM (0.1 mL). Then, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.1 M pentamethylbenzene for 2 minutes. After filtration, the solid phase was washed with NMP (0.05 mL), acetonitrile (0.25 mL) was added to the filtrate, LC samples were prepared, and the reaction progress was measured by LCMS measurement. As a result, 100% of the target object C004 was observed.
- reaction solution and the solid phase suspension (10 ⁇ L) were transferred onto a filter and washed 3 times with NMP (0.1 mL), 3 times with methanol (0.1 mL) and 3 times with DCM (0.1 mL). Then, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.1 M pentamethylbenzene for 2 minutes. After filtration, the solid phase was washed with NMP (0.05 mL), acetonitrile (0.25 mL) was added to the filtrate, LC samples were prepared, and the reaction progress was measured by LCMS measurement. As a result, 100% of the target object C005 was observed.
- Carboxylic acid resin (C001-03R) (supported amount 2.19 mmol / g, 1.00 g, 2.19 mmol) and NMP (15 mL) were added to a 20 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour. ..
- compound C007-03R (carrying amount 0.050 mmol / g, 0.030 g, 1.5 ⁇ mol) and NMP (0.300 mL) were added to a 0.6 mL glass vial, and the mixture was shaken at room temperature for 1 hour.
- Add piperazine 0.5 MnmP solution, 30.0 ⁇ L, 0.015 mmol
- HOAt 0.5 MnmP solution, 30.0 ⁇ L, 0.015 mmol
- DIC (2.34 ⁇ L, 0.015 mmol
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.020 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.02 M pentamethylbenzene for 5 minutes. After filtration, the resin on the filter was washed with NMP (0.05 mL). Acetonitrile (0.25 mL) was added to 50 ⁇ L of the obtained filtrate to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, the target product AA001 was observed with a purity of 88.9%.
- AA001 LRMS m / z 283 [M + H] + .
- Retention time 0.471 minutes (analytical conditions SMD-FA05-1, 280 nm).
- AA001-03R which illustrates the synthetic method in Examples 1-1-12, may be referred to as compound D021-03R in subsequent examples.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.020 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.02 M pentamethylbenzene for 5 minutes. After filtration, the resin on the filter was washed with NMP (0.05 mL). Acetonitrile (0.25 mL) was added to 50 ⁇ L of the obtained filtrate to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, the target product AA003 was observed with a purity of 95.6%.
- Examples 1-1-15 Synthesis of AA015-03R Compound AA001-03R (carrying amount 0.195 mmol / g, 10.0 mg, 1.95 ⁇ mol) and NMP (150 mL) were added to a 0.6 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour. 3-Fluoro-4-nitrophenol (AA014, 1.53 mg, 9.75 ⁇ mol) and DIPEA (3.41 ⁇ L, 0.020 mmol) were added, and the mixture was shaken at 100 ° C. for 24 hours.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.020 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.02 M pentamethylbenzene for 5 minutes. After filtration, the resin on the filter was washed with NMP (0.05 mL). Acetonitrile (0.25 mL) was added to 50 ⁇ L of the obtained filtrate to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, the target product AA015 was observed with a purity of 96.9%.
- Examples 1-1-16 Synthesis of AA017-03R Examples using the compound AA001-03R (supported amount 0.195 mmol / g, 37.0 mg, 7.4 ⁇ mol) and 4-hydroxy-3,5-dimethylbenzoic acid (AA016, 3.60 mg, 0.0022 mmol). Synthesis was carried out in the same manner as in 1-1-13 to obtain compound AA017-03R (supported amount 0.189 mmol / g, 32.1 mg, 6.1 ⁇ mol).
- Example 1-2 Synthesis of substrate for reaction study mainly used in Example 2-2, building block used in a mixture library, etc.
- Example 1-21 4- (4-bromophenyl) -1-hydroxy- Synthesis method of N, N-dimethyl-2-naphthamide (Compound E002)
- the naphthol reagents (Compound E002, Compound E003) used in Examples 1-2-7 and 1-2-9 were synthesized by the methods shown below. did.
- Example 1-2-2 Synthesis of 4-bromo-1-((4-methoxybenzyl) oxy) -N, N-dimethyl-2-naphthamide (Compound E005) 4-bromo-1-hydroxy-N, N -Dimethyl-2-naphthamide (Compound E004) (60 g, 204 mmol) in DMA (510 mL) solution with 1- (chloromethyl) -4-methoxybenzene (33.3 mL, 245 mmol) and tBuOK (25.2 g, 224 mmol). Added at room temperature. After stirring at 90 ° C.
- Examples 1-2-3 1-((4-methoxybenzyl) oxy) -N, N-dimethyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-) Il) Synthetic of -2-naphthamide (Compound E006) Under a nitrogen atmosphere, 4-bromo-1-((4-methoxybenzyl) oxy) -N, N-dimethyl-2-naphthamide (Compound E005) (85 g, 205 mmol) , Bis (pinacholate) diboron, Pd (dpppf) Cl 2.
- Example 1-2-4 Synthesis of 4- (4-bromophenyl) -1-((4-methoxybenzyl) oxy) -N, N-dimethyl-2-naphthamide (Compound E007) Under a nitrogen atmosphere, the previous step.
- Example 1-2-5 Synthesis of 4- (4-bromophenyl) -1-hydroxy-N, N-dimethyl-2-naphthamide (Compound E002) (4- (4-bromophenyl) -1-((( Add TFA (48.8 mL, 638 mmol) to a solution of 4-methoxybenzyl) oxy) -N, N-dimethyl-2-naphthamide) (62.6 g, 128 mmol) (Compound E007) in DCM (638 mL) and add 1 at room temperature. Stir for hours. The reaction solution was concentrated under reduced pressure, ethyl acetate (200 mL) was added, and the reaction solution was concentrated under reduced pressure again.
- Example 1-2-6 Synthesis of 4- (5-bromopyridin-2-yl) -1-hydroxy-N, N-dimethyl-2-naphthamide (Compound E003) using 5-bromo-2-iodopyridine Then, it was synthesized by the same method as in Example 1-2-4 (Compound E007) and Example 1-2-5 (Compound E002).
- the reaction suspension was transferred to a column with a filter, nitrogen pressure was applied to remove the reaction solution, and then the resin was washed three times with NMP (200 mL). Further, the resin was washed repeatedly with water (200 mL) three times, methanol (200 mL) three times, and DCM (200 mL) three times, and the obtained resin was dried under reduced pressure overnight to give the title compound (Compound EA01-). 01-01R) was obtained (10.2 g, loading amount: 0.4 mmol / g).
- Example 1-2-8 Determination of carrier amount of compound EA01-01-01R
- the carrier amount (mmol / g) carried on the resin was determined by the following method. The following is an example of an experimental operation, and does not uniformly specify the loading amount for all lots.
- the concentration in the solution was calculated from the obtained UV area% of E002 and the calibration curve, and the amount of E002 supported on the resin was determined. As a result, the loading amount was calculated to be 0.40 mmol / g.
- Analytical conditions were analyzed under SMD-FA05-1, and the UV area% was confirmed at a UV wavelength of 319 nm.
- LCMS m / z 370.0, 372.0 [M + H] + Holding time: 1.45 minutes.
- Example 1-2-10 The amount of compound EA01-02-01R carried was determined using the same method as in Example 1-2-8. LCMS: m / z 371.0, 373.0 [M + H] + . Holding time: 1.21 minutes.
- Example 1-2-11 Method for synthesizing ⁇ 1- [3-ethoxycarbonyl-5- (trifluoromethyl) phenyl] pyrazole-4-yl ⁇ boronic acid (BB06)
- Examples 1-2-12 Synthesis of 3- (4-chloro-1H-pyrazole-1-yl) -5- (trifluoromethyl) ethyl benzoate (Compound E010) 3-bromo-5- (trifluoromethyl) ) Ethyl benzoate (Compound E009) (2.08 g, 7 mmol), 4-chloro-1H-pyrazole (1.08 g, 10.5 mmol), copper iodide (0.133 g, 0.700 mmol), N, N' -A solution of dimethylethylenediamine (0.30 mL, 2.8 mmol) and potassium carbonate (2.90 g, 21.00 mmol) in dioxane (17.5 mL) was nitrogen substituted three times.
- Examples 1-2-13 3- (4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole-1-yl) -5-( Synthesis of Ethyl Trifluoromethyl (Compound E011) (3- (4-Chloro-1H-pyrazole-1-yl) -5- (Trifluoromethyl) Ethyl Benzoate (Compound E010) (1.93 g, 6) .06 mmol), bis (pinacholate) diboron (1.92 g, 7.57 mmol), RuPhos (424 mg, 0.908 mmol), allylpalladium (II) chloride dimer (111 mg, 0.303 mmol) and potassium acetate (1.49 g, A solution of 15.1 mmol) of dioxane (30 mL) was replaced with nitrogen and stirred at 90 ° C.
- Example 1-2-14 Pre-synthesis step of (1- (3- (ethoxycarbonyl) -5- (trifluoromethyl) phenyl) -1H-pyrazole-4-yl) boronic acid (BB06) (Example 1) 3- (4- (4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole-1-yl) -5- obtained in -2-13) Ethyl (trifluoromethyl) benzoate (Compound E011) (equivalent to 6.06 mmol), sodium periodate (6.48 g, 30.3 mmol), ammonium acetate (2.34 g, 30.3 mmol), acetone (40 mL) , Water (20 mL) was stirred at room temperature for 19 hours.
- Example 1-3 Synthesis of substrate for reaction study mainly used in Example 2-3, and building block used in a mixture library
- Example 1-3-1 3-ethynyl-5- (trifluoromethyl) benzoic acid Synthesis of ethyl acid (BB25)
- Examples 1-3-3 Synthesis of 3- (trifluoromethyl) ethyl benzoate (BB25) 3- (trifluoromethyl) -5-((trimethylsilyl) ethynyl) ethyl benzoate (Compound E012, A solution of 1.64 g (5.22 mmol) and potassium carbonate (0.721 g, 5.22 mmol) in ethanol (20.8 mL) was stirred at room temperature for one and a half hours. The mixture was filtered through cerite and washed 3 times with methanol (20 mL) and 3 times with ethyl acetate (20 mL). The filtrate was concentrated.
- Examples 1-4 Synthesis of reaction study substrates mainly used in Examples 2-4 and 2-5, and building blocks used in a mixture library
- Examples 1-4-1 4- (piperazine-1-yl) . ) Synthetic method of allyl benzoate (BB27)
- Example 1-4-2 6- [4-[(2-Methylpropan-2-yl) oxycarbonyl] piperazine-1-yl] pyridazine-3-carboxylic acid (AC012) synthesis method
- 6-chloropyridazine-3-carboxylic acid AC010, CAS: 5096-73-1, 1.00 g, 6.31 mmol
- tert-butylpiperazin-1-carboxylate AC011, CAS: 57260-.
- DIPEA 3.30 mL, 18.9 mmol, 3.0 eq
- acetonitrile 15.00 mL
- Example 1-4-3 Method for synthesizing prop-2-enyl 6- [4-[(2-methylpropan-2-yl) oxycarbonyl] piperazine-1-yl] pyridazine-3-carboxylate (AC013)
- Example 1-4-4 Synthesis method of 6- (piperazine-1-yl) pyridazine-3-carboxylate allyl (BB28)
- Example 1-4-5 Method for synthesizing tert-butyl 6-piperazine-1-ylpyridazine-3-carboxylate (BB29)
- Propan-2-yl 6-chloropyridazine-3-carboxylate (AC018, CAS: 321946-09-2, 100 mg, 0.498 mmol) and piperazine (AC016, 129 mg, 1.50 mmol, 3.0) in a 20 mL eggplant flask. Equivalent), DMAP (6.1 mg, 0.05 mmol, 0.1 equivalent), acetonitrile (2.49 mL), DIPEA (131 ⁇ L, 0.748 mmol, 1.5 equivalent) were added and at 70 ° C. under a nitrogen atmosphere. The mixture was heated and stirred for 1.5 hours.
- reaction solution was diluted with AcOEt (30 mL) and washed with saturated aqueous ammonium chloride solution (10 mL), water (10 mL) and saturated brine (10 mL).
- the organic layer was filtered through a phase separator, and the filtrate was concentrated under reduced pressure.
- the obtained residue was purified by silica gel column chromatography (NH-silica gel) (ethyl acetate / hexane, 0-20%, then DCM / methanol, 0-10%), and the title compound (AC019, 52%, 65 mg) was purified. , 0.260 mmol) was obtained.
- reaction tracking At any time, 10 ⁇ L of the reaction solution and resin suspension was sampled in a nitrogen atmosphere and transferred to a pipette tip with a filter, 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), DCM (100 ⁇ L). ), Then immersed in a 10% TFA / DCM solution (50 ⁇ L) containing 0.1 Mtrimethylbenzene for 5 minutes. After filtration, the filtrate was dissolved in TFE (1000 ⁇ L), an LC sample was prepared, and the reaction progress was measured by performing LCMS measurement. The compounds shown below were targeted for detection and analyzed under the analysis conditions SMD-FA05-1, and the UV area% was confirmed at a UV wavelength of 319 nm.
- Compound EA01-02-01R (20 mg, carrying amount 0.37 mmol / g) and 1,4-dioxane (200 ⁇ L) were added to a 0.6 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour.
- reaction tracking At any time, 10 ⁇ L of the reaction solution and resin suspension was sampled in a nitrogen atmosphere and transferred to a pipette tip with a filter, 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), DCM (100 ⁇ L). ), Then immersed in a 10% TFA / DCM solution (50 ⁇ L) containing 0.1 Mtrimethylbenzene for 5 minutes. After filtration, the filtrate was dissolved in DMI (250 ⁇ L). The solution (50 ⁇ L) was diluted with acetonitrile (250 ⁇ L) to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. The compounds shown below were targeted for detection and analyzed under the analysis conditions SMD-FA05-1, and the UV area% was confirmed at a UV wavelength of 319 nm.
- Compound EA01-02-01R (20 mg, carrying amount 0.37 mmol / g) and 1,4-dioxane (200 ⁇ L) were added to a 0.6 mL glass vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour.
- reaction tracking At any time, 10 ⁇ L of the reaction solution and resin suspension was sampled in a nitrogen atmosphere and transferred to a pipette tip with a filter, 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), DCM (100 ⁇ L). ), Then immersed in a 10% TFA / DCM solution (50 ⁇ L) containing 0.05 Mtrimethylbenzene for 5 minutes. After filtration, the filtrate was dissolved in DMI (250 ⁇ L), the solution (50 ⁇ L) was diluted with acetonitrile (250 ⁇ L) to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. The compounds shown below were targeted for detection and analyzed under the analysis conditions SMD-FA05-1, and the UV area% was confirmed at a UV wavelength of 319 nm.
- compound EA01-02-01R (338 mg, loading amount 0.37 mmol / g, 0.125 mmol) and NMP (5.68 mL) were added to a 10 mL glass vial and shaken at room temperature for 1 hour.
- (4-Acetylphenyl) Boronic acid (BB18) (205 mg, 1.250 mmol, 10 eq)
- K 3 PO 4 (159 mg, 0.750 mmol, 6 eq)
- P (Cy) 3 Pd G3 (16 mg, 0.025 mmol) , 0.2 eq) and degassed water (568 ⁇ L) were added and shaken at 90 ° C. for 20 hours.
- Compound B013-01R was sampled, transferred to a pipette tip with a filter, washed 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), 3 times with DCM (100 ⁇ L), and then containing 0.05 Mtrimethylbenzene 10 Soaked in% TFA / DCM solution (50 ⁇ L) for 5 minutes. After filtration, the filtrate was dissolved in TFE-acetonitrile (200-800 ⁇ L) to prepare an LC sample, and the purity was measured by performing LCMS measurement.
- Compound B021-01R was sampled, transferred to a pipette tip with a filter, washed 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), 3 times with DCM (100 ⁇ L), and then containing 0.05 Mtrimethylbenzene 10 Soaked in% TFA / DCM solution (50 ⁇ L) for 5 minutes. After filtration, the filtrate was dissolved in TFE-acetonitrile (200-800 ⁇ L) to prepare an LC sample, and the purity was measured by performing LCMS measurement.
- Compound B025-01R was sampled, transferred to a pipette tip with a filter, washed 3 times with NMP (100 ⁇ L), 3 times with methanol (100 ⁇ L), 3 times with DCM (100 ⁇ L), and then containing 0.05 Mtrimethylbenzene 10 Soaked in% TFA / DCM solution (100 ⁇ L) for 5 minutes. After filtration, the filtrate was dissolved in TFE (200 ⁇ L), an LC sample was prepared, and the purity was measured by performing LCMS measurement.
- Example 1-5 Synthesis of substrate for reaction study mainly used in Examples 2-5 and 2-7, and building block used in a mixture library
- Example 1-5-1 Synthesis of compound AE025-01R
- Example 1-5-2 Synthesis of compound AE026-01R Compound AE025-01R (1.102 g), NMP (16 mL) and methanol (4 mL) were added to a 30 mL glass vial, and the mixture was shaken at room temperature for 1 hour. A 5 M aqueous sodium hydroxide solution (0.6 mL, 3.00 mmol) was added, and the mixture was shaken at room temperature for 4 hours.
- AE029 was observed by performing LC / MS measurements on a portion of the solution.
- AE029-01R which illustrates the synthetic method in Examples 1-5-4, may be referred to as compound BAD006-01R in subsequent examples.
- Example 1-6 Synthesis of reaction study substrate mainly used in Example 2-8, and building blocks used in a mixture library
- Example 1-6-1 2-bromo-4- (3,3,4) , 4,5,5,6,6,7,7,8,8,9,9,10,10,10-Synthesis of benzaldehyde (compound AE003)
- AE011 (202 mg, 0.907 mmol) and acetic acid (109 mg, 1.81 mmol) were added to a DCM (7.56 mL) solution of AE010 (500 mg, 0.756 mmol), and the mixture was stirred at room temperature for 10 minutes.
- Sodium triacetoxyborohydride (320 mg, 1.51 mmol) was added, and the mixture was stirred at room temperature for 6 hours.
- the mixture was diluted with dichloromethane, TFA was added, and then purified by reverse phase column chromatography (C18, 40-85% 0.1% TFA acetonitrile solution / 0.1% formic acid aqueous solution), and the title compound AE012 (554 mg, 0) was used. .639 mmol, 84%) was obtained.
- LRMS m / z 868.1 [M + H] + .
- Retention time 0.39 minutes (analytical conditions SMD-TFA50-1).
- EB20-02-01R used in this example was synthesized in Example 2-2-4.
- Aqueous NaOH solution (5M, 228 ⁇ L) was added and the mixture was shaken at room temperature for 2 hours.
- AE016-01R (0.37 mmol / g, 350 mg) and NMP (5.95 mL, 17v / w) were added to an 8.0 mL screw cap vial under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour.
- BB28 (96 mg, 3.0 eq), HOAt (106 mg, 6.0 eq), DIC (121 ⁇ L, 6.0 eq) were added and the mixture was shaken at room temperature for 14 hours.
- the aromatic aldehyde AC009-01R used in this example was synthesized in Examples 1-4-11.
- solid-phase-supported aromatic aldehydes AC009-01R (0.37 mmol / g, 20 mg) and DCE (400 ⁇ L, 20 v / w) were added, and the temperature was 1 hour at room temperature. I shook it.
- BB29 (11.7 mg, 6.0 eq) and Ti (OtBu) 4 (28.6 ⁇ L, 10 eq) were added and the mixture was shaken at 60 ° C. for 1 hour.
- Aqueous LiOH solution (2M, 22 ⁇ L) was added and the mixture was shaken at room temperature for 22 hours.
- the suspension of resin was transferred to a column with a filter using methanol, 3 times with NMP (432 ⁇ L), 3 times with tetrabutylammonium hydrogen sulfate (0.05 MnmP solution, 432 ⁇ L), 3 times with NMP (432 ⁇ L). , MeOH (432 ⁇ L) 3 times, DCM (432 ⁇ L) 3 times and then dried under reduced pressure to give the title compound AE022-01R.
- Example 1-7 Synthesis of substrate for reaction study mainly used in Examples 2-9 and 2-10, and building block used in a mixture library
- Example 1-7-1 Synthesis of compound C008-03R Under a nitrogen atmosphere, compound C003-03R (carrying amount 0.511 mmol / g, containing dichloromethane (0.050 g), 0.950 g, 0.460 mmol) and NMP (13.5 mL) were added to a 20 mL glass vial at room temperature. I shook it for an hour.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.010 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.2 M pentamethylbenzene for 2 minutes. After filtration, NMP (0.25 mL) was added to the filtrate. Acetonitrile (0.25 mL) was added to 0.050 mL of the obtained solution to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, 100% of the target object C008 was observed. Compound C008 LRMS: m / z 382, 384 [M + H] + . Retention time: 1.232 minutes (analytical conditions SMD-FA05-1, 280 nm).
- Example 1-7-2 Synthesis of compound C009-03R Compound C005-03R (carrying amount 0.201 mmol / g, 250 mg, 0.0502 mmol), NMP (3.75 mL), 3-bromoaniline (C104) (20 ⁇ L, 0.19 mmol) in a 5 mL glass vial under a nitrogen atmosphere. ), HOAt (26 mg, 0.19 mmol) and DIC (29 ⁇ L, 0.19 mmol) were added and shaken at 60 ° C. for 15 hours.
- Examples 1-7-3 Synthesis of compound C010-03R Compound C010-03R (supported amount 0.194 mmol / g) using compound C005-03R (supported amount 0.201 mmol / g) and 3-bromo-4-methylaniline (C103) by the same method as compound C009-03R. ) Can be synthesized.
- Example 1-8 Synthesis of substrate for reaction study mainly used in Example 2-11, and building block used in a mixture library
- Example 1-8-1 Solid phase-supported Ar-NH2 (Compound D001-03R) Synthetic solid-phase-supported Ar-NH2 (compound D001-03R) was synthesized as follows by condensation of solid-phase-supported carboxylic acid (C007-03R) and 3- (aminomethyl) aniline (D038).
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (0.020 mL) onto a filter, NMP (0.1 mL) 3 times, methanol (0.1 mL) 3 times, DCM (0.1 mL) 3 times. After washing once, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.02 M pentamethylbenzene for 5 minutes. After filtration, the resin on the filter was washed with NMP (0.05 mL). Acetonitrile (0.50 mL) was added to the obtained filtrate to prepare an LC sample, and the reaction progress was measured by performing LCMS measurement. As a result, the target product D001 was observed with a purity of 98.2%.
- Example 1-8-2 Synthesis of solid phase-supported Ar-NHMe (Compound D020-03R)
- the solid-phase-supported Ar-NHMe (D020-03R) is solid-phase-supported by the same method as in Example 1-8-1. It was synthesized by condensation of a supported carboxylic acid (C007-03R) and 3- (aminomethyl) -N-methylaniline (D039). Solid phase purification and reaction tracking (compound to be tracked; D020) were also carried out in the same manner as in Example 1-8-1.
- Example 1-9 Synthesis of substrate for reaction study mainly used in Example 3 and building block used in a mixture library
- Example 1-9-1 Synthesis of compound E015-02R
- Brominated modified Wang resin (Compound E014-02R, Sigma-Aldrich, 534471, 100-200 mesh, carrying amount: 1.23 mmol / g, copolymer (styrene-1% DVB), 1.50 g, 1.845 mmol under nitrogen atmosphere. ) was added with NMP (15 mL) and shaken at room temperature for 1 hour. 3-bromophenol (Compound E015,638 mg, 3.690 mmol) and tBuOK (393 mg, 3.51 mmol) were added, and the mixture was shaken at room temperature for 20 hours.
- Example 1-9-2 Calculation of supported amount of compound E015-02R The supported amount (mmol / g) carried on the resin was determined by the following method. The following is an example of an experimental operation, and does not uniformly specify the loading amount for all lots.
- Examples 1-9-4 Calculation of the supported amount of compound E016-02R
- the supported amount (mmol / g) carried on the resin was determined by the following method. The following is an example of an experimental operation, and does not uniformly specify the loading amount for all lots.
- the concentration in the solution was calculated from the obtained UV area% of E016 and the calibration curve, and the amount of E016 supported on the resin was determined. As a result, the loading amount was calculated to be 0.733 mmol / g.
- Example 1-10 Synthesis of the compound mainly used in Example 7
- Example 1-10-1 Synthesis of 4- (2-phenylsulfanilethylamino) -3- (trifluoromethyl) benzenesulfonamide (AG010)
- Examples 1-10-2 4- [4- [(2-bromophenyl) methyl] piperazine-1-yl] -N- [3-nitro-4- (2-phenylsulfanylethylamino) phenyl] sulfonylbenzamide Synthesis of (AG003)
- Examples 1-10-6 4- [4- [[2- (3-ethoxyphenyl) phenyl] methyl] piperazine-1-yl] -N- [4- (2-phenylsulfanylethylamino) -3- Synthesis of (trifluoromethyl) phenyl] sulfonylbenzamide [AG012 (B2R045-01)]
- Examples 1-10-7 4- [4- [[2- (5-ethoxypyridin-3-yl) phenyl] methyl] piperazine-1-yl] -N- [4- (2-phenylsulfanylethylamino) ) -3- (Trifluoromethyl) phenyl] sulfonylbenzamide [AG013 (B2R046-01)] synthesis
- Example 1-11 Synthesis of other compounds
- Example 1-11-1 Synthesis of compound A102-3R
- Carboxylic Resin (A101-03R) (Rapp Polymer, supported amount 1.70 mmol / g, 40.0 g) and NMP (600 mL) were added to an empty column with an 800 mL filter under a nitrogen atmosphere, and the mixture was shaken at room temperature for 1 hour. ..
- HOAt (9.26 g, 68.0 mmol) and DIC (10.6 mL, 68.0 mmol), ethyl 4- [4-[(4-piperidin-4-yloxyphenyl) methoxy] phenyl] benzoat (C102) (4) .13 g, 9.58 mmol) was added and shaken at room temperature for 3.5 hours.
- HOAt (18.5 g, 136 mmol), DIC (21.2 mL, 136 mmol) and piperidine (16.8 mL, 170 mmol) were added and shaken overnight at room temperature.
- Reaction follow -up Transfer the reaction solution and the solid phase suspension (10 ⁇ L) onto a filter and wash with NMP (0.1 mL) 3 times, MeOH (0.1 mL) 3 times, and DCM (0.1 mL) 3 times. Then, it was immersed in a 10% TFA / DCM solution (0.1 mL) containing 0.05 M pentamethylbenzene for 5 minutes. After filtration, the solid phase was washed with DMF (0.05 mL), MeCN (0.25 mL) was added to the filtrate, an LC sample was prepared, and the reaction progress was measured by performing LCMS measurement. As a result, 100% of the target object A102 was observed.
- Compound A 102LRMS m / z 243 [M + H] + Retention time: 1.081 minutes (analytical conditions FA05-1, 299 nm).
- compound A102-03R (carrying amount 0.200 mmol / g, 47.9 g), NMP (503 mL) and 2-methyl-2-butanol (144 mL) were added to a 2 L separable flask at room temperature. Was stirred for 1 hour. After cooling the suspension to 5 ° C., an aqueous solution of normal tetrabutylammonium hydroxide (1M, 14.4 mL, 14.4 mmol) was added dropwise, and the mixture was stirred at room temperature for 3.5 hours. An aqueous solution of normal tetrabutylammonium hydroxide (1M, 2.39 mL, 2.39 mmol) was added dropwise, and the operation of stirring at room temperature for 1 hour was repeated twice.
- the solid phase purification reaction solution and the solid phase suspension were prepared 3 times with HOAt (0.1 M in NMP, 800 mL), 3 times with NMP (800 mL), 3 times with MeOH (800 mL), and 3 times with DCM (800 mL).
- the mixture was washed 3 times with heptane (800 mL) and the obtained solid phase was dried under reduced pressure for 6 days to obtain compound A103-3R (carrying amount 0.201 mmol / g, 49.8 g, 10.0 mmol). ..
- reaction solution and the solid phase suspension (12 ⁇ L) were transferred onto a filter and washed 3 times with NMP (0.1 mL), 3 times with MeOH (0.1 mL), and 3 times with DCM (0.1 mL). Then, it was immersed in a 10% TFA / DCM solution (0.05 mL) containing 0.02 M pentamethylbenzene for 5 minutes. After filtration, the solid phase was washed with DMF (0.05 mL), MeCN (0.25 mL) was added to the filtrate, an LC sample was prepared, and the reaction progress was measured by performing LCMS measurement. As a result, 100% of the target object A103 was observed.
- Compound A101-03R (supported amount 1.70 mmol / g) and 3- [4-[(4-piperidin-4-yloxyphenyl) methoxy] phenyl] benzoato (C101) were prepared by the same method as for compound A102-03R.
- Compound A130-03R (supported amount 0.200 mmol / g) can be synthesized by using the compound A130-03R.
- Compound A131-03R (supported amount 0.201 mmol / g) can be synthesized using compound A130-03R (supported amount 0.200 mmol / g) by the same method as compound A103-03R.
- Methyl 2- [3- [tert-butyl (dimethyl) silyl] oxyphenyl] acetate Synthesis of compound BP114 Methyl 2- (3-hydroxyphenyl) acetate (compound BP113, CAS) in a 500 mL flask under a nitrogen atmosphere. : 42058-59-3) (15.0 g, 90.3 mmol, 1.0 eq), imidazole (18.4 g, 270.8 mmol, 3.0 eq) and DMF (150 mL) were added and stirred at room temperature for 10 minutes. TBDMSCl (20.4 g, 135.4 mmol, 1.5 eq) was added dropwise to this reaction mixture.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Structural Engineering (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- General Physics & Mathematics (AREA)
- Biotechnology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Cell Biology (AREA)
- Pathology (AREA)
- Microbiology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Pyridine Compounds (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Description
構造多様性を有する化合物ライブラリの拡充とスクリーニング手法は、ヒット化合物を取得(ヒットジェネレーション:hit generation)する上で鍵となる技術である(非特許文献1および2)。新規化合物の探索を既知化合物から行う手法として、スキャホールドホッピング(親化合物に対して「isofunctional molecular structures」と「significantly different molecular backbones」を併せ持つ化合物を同定すること)の考え方が1999年に提唱され(非特許文献30)、メディシナルケミストリーの分野で広く受け入れられ利用されている(非特許文献31)。
(a)としては、HTS(ハイスループットスクリーニング)ライブラリが挙げられ、長らくヒット化合物を取得するための手法として中心的な役割を担っていた(非特許文献3)。HTSライブラリの拡充は、Lipinskiが提唱したrule of 5の影響を強く受け、その多くは一定の範囲の中(例えば分子量500を超えない)で実施されてきた(非特許文献4)。また、FBDD(フラグメントベースドドラッグディスカバリー)によりヒット化合物を取得する手法も利用され(非特許文献5)、複数の標的分子に対して、創薬に繋がるヒット化合物の取得が報告されている(非特許文献6)。しかし、用いるフラグメント化合物の分子量の小ささが理由となり、得られるヒット化合物のアフィニティが弱く、活性の向上に向けてgrowingやlinkingのアプローチが必要となる(非特許文献7)。大きく活性を上昇させるためには標的分子の構造情報が不可欠であり、構造情報のない標的分子に対しては適していない(非特許文献2)。構造情報のある標的分子に対しては、フラグメント同士を連結する最適なリンカーの探索に対して計算手法を活用するアプローチが用いられている(非特許文献8)。
(b)の新たに化合物ライブラリを拡充する手法の一つとして、アミノ酸類をビルディングブロックとしたペプチドライブラリが知られている(非特許文献9)。各ビルディングブロックの連結様式は、アミド化反応によるアミド結合に限定され、利用するビルディングブロックもアミノ酸に限定されている。
混合物でライブラリ合成を行う一つの例として、アミノ酸に限定しないビルディングブロックを利用したDNA−encodedライブラリ技術(DELT;DNA encoded−library technology)が知られている(非特許文献16)。DELTにおいては、標的分子と結合活性を有する化合物のデコーディングのためにDNAタグを利用する特徴を有する。DNAタグによるデコーディングとは、その配列情報によりライブラリ化合物一つ一つの構造情報を正しく1対1の関係で示すことである。そのため、DNAタグはライブラリ化合物と同一分子内に結合させる必要がある(例えば、非特許文献20、非特許文献21)。DNAタグはデコーディングの役割を担うものの、DNAタグがライブラリ化合物と同一の分子内に存在するが故にライブラリ合成に使用できる化学反応の反応条件には制約が生じる。結果として、合成可能な化合物の範囲が限定されることになる(非特許文献17)。また、利用する化学反応によってDNAタグの損傷が発生していることも報告されている(非特許文献18)。このDNAタグの損傷により、標的分子と結合する化合物のデコーディングを正しく行うことが出来なくなり、実際にDELTのライブラリは10の8乗以上の規模では偽陰性(false negative)が増加すると報告されている(非特許文献19)。
医薬品の開発に利用できる、構造多様性を有した新たな化合物ライブラリが求められるが、そのような化合物ライブラリの確立には様々な点で改善の余地がある。
一方、混合物でライブラリ化合物を合成する際には試薬の除去を目的とした固相抽出ならびに液液抽出、減圧留去または固相試薬を用いる除去以外の精製操作は行わないため、ビルディングブロック同士を結合させる際に目的以外の反応が進行すると、意図しない化合物の存在率が増加し、除去されないままアッセイを実施することになる。多くの化合物及び官能基が共存した状態で反応を行うことでこの問題はより増大し、目的化合物の収量低下は顕著となる。
また、混合物を利用する合成手法においては、反応系に加えるビルディングブロックを混ぜて基質に加えるため、ビルディングブロックの反応性差に起因して生成する化合物の収量に差が生じ、混合物をアッセイする際には各化合物間に濃度差が容易に生じてしまうと考えられる。加えるビルディングブロックのモル数を調整する手法も考えられるが、用いるビルディングブロックについて反応性を事前に比較することが現実的ではないため、反応コントロールが困難であり、得られる混合物の品質が担保されないことが問題となる。
上記のように混合物としてライブラリ化合物を合成する従来のライブラリは、(ア)1フラスコ当たりの化合物数が多い、(イ)DNAタグの存在による利用可能な反応の制限とDNAタグの損傷、(ウ)混合物であることに起因する反応コントロールの困難性、(エ)保護基活用の不足、が理由となり混合物に含まれる化合物の品質の担保ができていない。従来技術においては上記の技術的課題が未解決であり、混合物を利用して調製する化合物ライブラリにおいて、コアブロックを連結するリンカーに多様性を有する化合物ライブラリを調製できる状況に至っていない。
すなわち、構造の多様性を追求すると同時に、品質の担保がなされたライブラリ構築の方法論の例はこれまでにない。
本発明はこのような状況を踏まえてなされたものであり、本発明は多様な結合様式を含む化合物ライブラリ、およびその製造方法を提供することを課題とする。
分子内に2つ以上の反応性官能基を有するビルディングブロック(multifunctional building−block)を用い、分子を構築していく。このとき、反応性官能基を有するビルディングブロックのコアブロックは、従来の多くのライブラリと異なり、特定のスキャホールドに限定されない。第1ビルディングブロック(第1BB)の一方の反応性官能基を介して固相試薬に連結すると、もう一方の反応性官能基を有する固相担持された試薬を調製できる。この固相試薬群のうち、次工程で反応に用いる第1ビルディングブロック上の反応性官能基が共通する群を均一に混合する。これを複数の容器に再度分割し、それぞれの容器に異なる第2ビルディングブロックを反応させて、第1BBと第2BBが第1リンカー(第1L)を介して連結して担持された固相試薬が調製できる。再び、次工程で反応に用いる第2ビルディングブロック上の反応性官能基が共通する群を均一に混合後、複数の容器に再度分割する。これらの工程を必要な回数繰り返した後に、固相から切り出すことにより、同じフラスコ内にて指数関数的な多様性を有すると同時にコアブロックとリンカーに多様性を有する化合物群を提供できる。
また、上記のような結合多様性と品質の担保を同時に達成したライブラリの調製には、種々保護基を活用することもできる。すなわち、保護基を活用することにより、結合形成時の反応における副反応の回避、使用する官能基を任意の工程で露出させることによる結合多様性の実現、および目的化合物に任意の水素結合供与性基を含ませることを可能とすることができる。
前記化合物は、第1コアブロック(第1CB)、第1リンカー(第1L)、および第2コアブロック(第2CB)が共有結合で連結した以下の構造:
(第1CB)—(第1L)—(第2CB)
を含み、
前記ライブラリは、2種類以上の第1CB、2種類以上の第1L、および2種類以上の第2CBを含み、
前記ライブラリは、1×102~1×105の前記化合物を含む1または2以上の混合物により構成される、
前記ライブラリ。
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)
を含み、
前記ライブラリが、2種類以上の第2L、および2種類以上の第3CBを含む、[A1]に記載のライブラリ。
[A3]前記化合物が、さらに第3リンカー(第3L)、および第4コアブロック(第4CB)が共有結合で連結した以下の構造:
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB);または、
(第1CB)—(第1L)—(第2CB)(—(第3L)—(第4CB))—(第2L)—(第3CB);
を含み、
前記ライブラリが、2種類以上の第3L、および2種類以上の第4CBを含む、[A1]または[A2]に記載のライブラリ。
[A5]第1L、第2L、および第3Lが、アミド、スルホンアミド、アシルスルホンアミド、アミノ、エーテル、チオエーテル、エステル、ケトン、スルホン、単結合、エチン−1,2−ジイル(−C≡C−)、および含窒素5員ヘテロアリーレンからなる群から独立して選択される構造を含む、[A1]~[A4]のいずれかに記載のライブラリ。
−C(=O)−NR1−
−NR1−C(=O)−
−SO2−NR1−
−NR1−SO2−
−SO2−NR1−C(=O)−
−C(=O)−NR1−SO2−
−NR1−
−O−
−S−
−C(=O)−O−
−O−C(=O)−
−C(=O)−
−SO2−
−C≡C−
−CHR2−
単結合

から選択される式により表される、[A1]~[A5]のいずれかにのいずれかに記載のライブラリ。
[A8]第1L、第2L、および第3Lが、アミド化、スルホンアミド化、アシルスルホンアミド化、還元的アミノ化、エーテル化、チオエーテル化、エステル化、O−アルキル化、N−アルキル化、ケトン結合形成反応、炭素—窒素結合形成反応、炭素—炭素結合形成反応、および含窒素5員ヘテロアリーレン結合形成反応からなる群から独立して選択される反応により形成される、[A1]~[A7]のいずれかに記載のライブラリ。
[A9]第1L、第2L、および第3Lが、アミド化、スルホンアミド化、アシルスルホンアミド化、還元的アミノ化、O−アルキル化、および炭素—炭素結合形成反応からなる群から独立して選択される反応により形成される、[A1]~[A8]のいずれかに記載のライブラリ。
[A11]100以上、200以上、300以上、400以上、500以上、または1000以上の化合物を含む、[A1]~[A9]のいずれかに記載のライブラリ。
[A12]1×107以下、5×106以下、1×106以下、5×105以下、1×105以下、50000以下、20000以下、10000以下、8000以下、7000以下、6000以下、5000以下、4000以下、3000以下、2000以下、1500以下、または1200以下の化合物を含む、[A1]~[A11]のいずれかに記載のライブラリ。
1)各化合物について算出されるclogp値の標準偏差が1.2以上、1.3以上、1.4以上、または1.5以上;
2)各化合物について算出されるclogp値の分散が1.5以上、1.75以上、2.0以上、または2.5以上;
3)各化合物について算出される[(分子に含まれるsp3炭素原子の数)/分子に含まれる水素以外の原子の数]のFsp3の中央値が、0.10以上、0.15以上、0.20以上、0.25以上、または0.30以上;
4)各化合物について算出される分子量の標準偏差が25以上、50以上、または75以上;
5)各化合物の水素結合受容性基の数の中央値が2~30、3~20、または3~18;
6)各化合物の水素結合供与性基の数について最大の化合物と最小の化合物の差が0~10、0~8、または1~5;および
7)各化合物について算出されるtPSA/Topological Polar Surface Area/位相幾何学的極性表面積の分散が20以上、25以上、30以上、;および
8)各化合物について算出されるTanimoto Similarity(ECFP_6:原子レベルで区別する6結合までのExtended−connectivity fingerprintsであり、例えばダッソーシステムズ社のPipeline Pilot 2017HF2を用いてECFP_6によって算出される値)の中央値が0.98以下、0.95以下、0.90以下、0.85以下、または0.80以下、;
9)各化合物について算出されるTanimoto Similarity(ECFP_6:原子レベルで区別する6結合までのExtended−connectivity fingerprintsであり、例えばダッソーシステムズ社のPipeline Pilot 2017HF2を用いてECFP_6によって算出される値)の中央値が0.50以上、0.55以上、0.60以上、0.65以上、0.70以上、または0.75以上;
の1以上の条件を満たす、[A1]~[A12]のいずれかに記載のライブラリ。
[A15]第1CB、第2CB、および第3CBからなる群より選択される1つ、2つ、または全てについて、
1)構成する原子に含まれる炭素原子、窒素原子、酸素原子の合計数が、1以上18以下である、2以上17以下である、3以上16以下である、4以上15以下である、5以上14以下である、または6以上13以下である、[A1]~[A14]のいずれかに記載のライブラリ。
[A1]~[A15]のいずれかに記載のライブラリ。
[A17]第1CB、第2CB、第3CB、および第4CBからなる群より選択される1つ、2つ、3つ、または全てについて、
1)構成する原子に含まれる炭素原子、窒素原子、酸素原子の合計数が、1以上18以下である、2以上17以下である、3以上16以下である、4以上15以下である、5以上14以下である、または6以上13以下である、[A1]~[A16]のいずれか1項に記載のライブラリ。
[A20]前記ライブラリを構成する化合物のうち、80%以上の化合物が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)により検出されている、[A1]~[A19]のいずれかに記載のライブラリ。
[A21]前記ライブラリを構成する化合物のうち、90%以上の化合物が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)により検出されている、[A1]~[A20]のいずれかに記載のライブラリ。
[A22]前記ライブラリを構成する化合物のうち、91%以上、92%以上、93%以上、94%以上、95%以上、または96%以上の化合物が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)により検出されている、[A1]~[A21]のいずれかに記載のライブラリ。
[A23][A1]~[A22]のいずれかに記載のライブラリを2以上組み合わせて調製される、200以上の化合物を含むライブラリ集合体。
(1)[A1]~[A23]のいずれかに記載のライブラリを提供すること;
(2)前記ライブラリを構成する前記1または2以上の混合物をそれぞれ標的分子と接触させること;
(3)標的分子に結合する化合物を同定すること
を含む、前記方法。
[B3]標的分子に結合する化合物の同定がAS−MSにより実施される、[B1]または[B2]に記載の方法。
[B5]前記ライブラリに含まれる2~100000、2~50000、2~20000、2~10000、2~5000、または2~2500の混合物についてスクリーニングを行う、[B1]~[B4]のいずれかに記載の方法。
(a)第1ビルディングブロック(第1BB)が共有結合で連結する担持担体(第1BB担持担体)を調製すること、ここで第1BB担持担体は、na種類の第1ビルディングブロック(第1BB1~第1BBna)を含む固相担体(第1BB1−na担持担体)であり、第1BB1−na担持担体の第1BB1−na部分はそれぞれ、第2ビルディングブロック(第2BB)との結合を形成するための反応性官能基を有し、第1BB1−na担持担体に含まれる前記反応性官能基は1種類または2種類以上である、
(b)第1BB1−na担持担体のうち、反応性官能基が共通する担持担体の混合物を調製し、各混合物を1種類の第2ビルディングブロック導入試薬(第2BB導入試薬)または2種類以上の第2BB導入試薬の混合物と反応させて、第2BBが共有結合で第1BBに連結する担持担体(第2BB−第1BB担持担体)を調製すること、ここで得られる担持担体はnb種類の第2ビルディングブロック(第2BB1~第2BBnb)を含む担持担体(第2BB1−nb−第1BB1−na担持担体)であり、第2BB1導入試薬~第2BBnb導入試薬のいずれかが第1BB1−na担持担体のそれぞれの反応性官能基と反応して第1リンカー(第1L)を形成する、
(e)固相担体から化合物を切り出すこと
を含み、naは2以上の整数であり、nbは2以上の整数であり、得られる化合物ライブラリが100以上の化合物を混合物として含む、前記方法。
[C3]第2BB1−nb−第1BB1−na担持担体がそれぞれ、第3ビルディングブロック(第3BB)とのリンカー形成が可能な反応性官能基を有し、それぞれの第1BB1−na部分または第2BB1−nb部分に1つ含まれる該反応性官能基は1種類または2種類以上であり、さらに
(c)第2B1−nb−第1BB1−na担持担体のうち、反応性官能基が共通する担持担体の混合物を調製し、各混合物を1種類の第3ビルディングブロック導入試薬(第3BB導入試薬)または2種類以上の第3BB導入試薬の混合物と反応させて、第3BBが共有結合で第1BBまたは第2BBに連結する担持担体(第3BB−第2BB−第1BB担持担体、または第2BB−(第3BB−)第1BB担持担体)を調製すること、ここで得られる担持担体はnc種類の第3ビルディングブロック(第3BB1~第3BBnc)を含む担持担体(第3BB1−nc−第2BB1−nb−第1BB1−na担持担体、または第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体)であり、第3BB1導入試薬~第3BBnc導入試薬のいずれかが第2BB1−nb−第1BB1−na担持担体のそれぞれの反応性官能基と反応して第2リンカー(第2L)を形成する、
を含み、ncは2以上の整数であり、ライブラリに含まれる化合物の第1リンカーまたは第2リンカーが2種類以上である、[C1]または[C2]に記載の方法。
[C5]第3BB1−nc−第2BB1−nb−第1BB1−na担持担体または第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体がそれぞれ、第4ビルディングブロック(第4BB)との結合を形成するための反応性官能基を有し、それぞれの第1BB1−na部分、第2BB1−nb部分、または第3BB1−nc部分に1つ含まれる前記反応性官能基は1種類または2種類以上であり、さらに
(d)第3BB1−nc−第2BB1−na−第1BB1−na担持担体または第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体のうち、反応性官能基が共通する担持担体の混合物を調製し、各混合物を1種類の第4ビルディングブロック導入試薬(第4BB導入試薬)または2種類以上の第4BB導入試薬の混合物と反応させて、第4BBが共有結合で第3BB、第2BB、または第1BBに連結する担持担体を調製すること、ここで得られる担持担体はnd種類の第4ビルディングブロック(第4BB1~第4BBnd)を含む担持担体であり、第4BB1導入試薬~第4BBnd導入試薬のいずれかが第3BB1−nc−第2BB1−nb−第1BB1−na担持担体または第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体のそれぞれの反応性官能基と反応して第3リンカーを形成する、
を含み、ndは2以上の整数であり、ライブラリに含まれる化合物の第1リンカー、第2リンカーまたは第3リンカーが2種類以上である、[C1]~[C4]のいずれかに記載の方法。
第4BB1−nd−第3BB1−nc−第2BB1−nb−第1BB1−na担持担体;
第4BB1−nd−(第3BB1−nc−)第2BB1−nb−第1BB1−na担持担体;
第3BB1−nc−第2BB1−nb−(第4BB1−nd−)第1BB1−na担持担体;
第4BB1−nd−第3BB1−nc−(第2BB1−nb−)第1BB1−na担持担体;または
第4BB1−nd−第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体
である、[C5]に記載の方法。
第4BB1−nd−第3BB1−nc−第2BB1−nb−第1BB1−na担持担体;
である、[C5]または[C6]に記載の方法。
[C8]前記第1リンカーおよび前記第2リンカーが、
1−1)第1リンカーが2種類以上である;
1−2)第2リンカーが2種類以上である;および
1−3)第1リンカーまたは第2リンカーが3種類以上である;
の1以上の条件を満たす、または。
前記第1リンカー、前記第2リンカーおよび前記第3リンカーが、
2−1)第1リンカーが2種類以上である;
2−2)第2リンカーが2種類以上である;
2−3)第3リンカーが2種類以上である;
2−4)第1リンカー、第2リンカーまたは第3リンカーが3種類以上である;
の1以上の条件を満たす、[C1]~[C7]のいずれかに記載の方法。
naが2~1000の整数であり、nbが2~1000の整数であり、ncが2~1000の整数であり、かつ、na、nbまたはncのいずれかが3以上の整数である、[C1]~[C8]のいずれかに記載の方法。
[C11]第1BB1導入試薬~第1BBna導入試薬のうちの2~na種類の第1BB導入試薬の混合物を固相担体と反応させて第1BBを固相担体に導入して第1BB1−na担持担体を調製する、[C1]~[C10]のいずれかに記載の方法。
[C13]第1BB1−na担持担体の第1BB1−na部分はそれぞれ、第2ビルディングブロック(第2BB)との結合を形成するための1つの反応性官能基を有し、および/または、第2BB1−nb−第1BB1−na担持担体の第1BB1−na部分はそれぞれ、第3ビルディングブロック(第3BB)との結合を形成するための1つの反応性官能基を有する、[C1]~[C11]のいずれかに記載の方法。
[C15]前記反応性官能基の脱保護工程の後、上記固相担持担体を、酸性成分を混合した溶液を用いて洗浄する工程を含む、[C14]に記載の方法。
[C16]上記酸性成分が、水中でのpKaが0~10の酸性化合物である、[C15]に記載の方法。
[C18]前記反応性官能基の脱保護工程によって生成される官能基が、カルボキシルまたは芳香環に置換したヒドロキシのいずれかである、[C15]~[C17]のいずれかに記載の方法。
[C20](e)の工程で、固相担体から切り出された化合物に含まれる官能基を修飾することをさらに含む、[C1]~[C19]のいずれかに記載の方法。
[C21]前記官能基が、固相担体から化合物の切り出しにより生じる官能基である[C20]に記載の方法。
[C22]前記修飾がヒドロキシまたはカルボキシのC1−6アルキル化である、[C20]または[C21]のいずれかに記載の方法。
前記(c)において、第3BB導入試薬と反応性官能基との反応の変換率が100%に満たない第2BB−第1BB担持担体において、反応性がより高い異なる第3BB導入試薬を残存する反応性官能基と反応させることをさらに含む、および/または
前記(d)において、第4BB導入試薬と反応性官能基との反応の変換率が100%に満たない第3BB−第2BB−第1BB担持担体または第2BB−(第3BB−)第1BB担持担体において、反応性がより高い異なる第4BB導入試薬を残存する反応性官能基と反応させることをさらに含む、[C1]~[C22]のいずれかに記載の方法。
[C25]前記UVタグが、式1:

該ベンゼン、該ナフタレン、該アントラセンおよび該ビフェニルは、ハロゲン原子、ニトロ、シアノ、カルボキシ、スルホキシ、−COR1、−OR1、−NR1R2、−CONR1R2、又は−SO2NR1R2からなる群より選ばれる同一または異なる1~3個の基で置換されていてもよく、R1およびR2はそれぞれ独立してC1−6アルキル、または連結する窒素原子と一緒になって、ヘテロ環を形成していてもよく、Xは、−CONH−、−NHCO−、−CH2O−及び−O−からなる群より選ばれる1種でビルディングブロックと結合していることを表す)
で表される構造である、[C24]に記載の方法。

で表される構造である、[C24]に記載の方法。
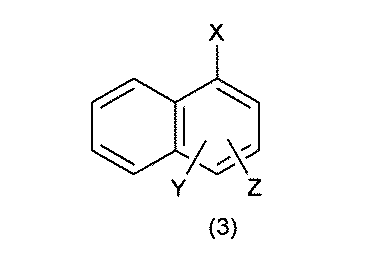
で表される構造である、[C24]に記載の方法。
で表される構造である、[C24]に記載の方法。
[C30]前記ライブラリに含まれる前記化合物が、UVタグを含まない、[C1]~[C23]および[C29]のいずれかに記載の方法。
[C31]さらに以下の工程:
1)いずれかの工程で得られた固相担持担体を得る工程;
2)工程1)で得られた固相担持担体から中間体化合物を切り出す工程;および
3)前記中間体化合物を分析する工程;
並びに、任意で、工程(e)で切り出された化合物を分析する工程。
を含む、[C1]~[C30]のいずれかに記載の方法。
[C33]第1L、第2L、第3Lおよび第4Lが、アミド化、スルホンアミド化、アシルスルホンアミド化、還元的アミノ化、エーテル化、チオエーテル化、エステル化、O−アルキル化、N−アルキル化、ケトン結合形成反応、炭素—窒素結合形成反応、炭素—炭素結合形成反応、および含窒素5員ヘテロアリーレン形成反応からなる群から独立して選択される反応により形成される、[C1]~[C32]のいずれかに記載の方法。
[C35]前記炭素−窒素結合形成反応が、芳香族求核置換反応および/またはバックワルド・ハートウィッグカップリングであり、前記炭素—炭素結合形成反応が、鈴木カップリング、根岸カップリング、スティレカップリングおよび/または薗頭カップリングである、[C1]~[C34]のいずれかに記載の方法。
[C37]得られる化合物ライブラリが1×108以下、1×107以下、5×106以下、1×106以下、500000以下、100000以下、50000以下、20000以下、10000以下、8000以下、7000以下、6000以下、5000以下、4000以下、3000以下、2000以下、1500以下、または1200以下化合物を含む、[C1]~[C36]のいずれかに記載の方法。
[C39]前記ライブラリを構成する化合物として合成を設定した目的化合物のうち、91%以上、92%以上、93%以上、94%以上、95%以上、または96%以上の化合物について製造が確認される、[A1]~[C38]のいずれかに記載のライブラリ。
[C40]合成の確認が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)による、[C38]または[C39]に記載のライブラリ。
[C42][C1]~[C40]のいずれかに記載の方法により調製される化合物ライブラリを組み合わせることを含む、1×102~1×108、3×102~1×108、1×102~1×107、または5×102~1×106の化合物を含む化合物ライブラリ集合体の製造方法。
X8は、ハロゲン原子または−O−SO2−R5であり;R5は1以上のフッ素原子で置換されてもよいC1−6アルキル、または1以上のフッ素原子もしくはフッ素原子で置換されていてもよいC1−6アルキルで置換されてもよいフェニルであり;
該化合物Vが固相合成用樹脂であるか、または該ホウ素原子含有化合物Wが固相合成用樹脂である、前記方法。
[D3]前記有機塩基が、アミジン類、グアニジン類、およびホスファゼン類からなる群より選択される、[D1]または[D2]のいずれかに記載の方法。
[D6]化合物Vが、固相合成用樹脂が、分解可能なリンカーを介して化合物Vaを側鎖に有し、
化合物Vaが、X8−Ar8で表される化合物を化学構造の一部に含む、[D1]~[D5]のいずれかに記載の方法。
[D8]前記X8が、臭素原子である、[D1]~[D7]のいずれかに記載の方法。
[D9]前記Ar8が、フェニル、ナフチル、ピロリル、チエニル、フリル、ピリジル、チアゾリル、イソチアゾリル、ピラゾリル、オキサゾリル、イソキサゾリル、イミダゾリル、トリアリル、ピリミジル、ピラジニル、ピリダジニル、キノリニル、イソキノリニル、4H−キノリジニル、フタラジニル、ナフチリジニル、キノキサリニル、キナゾリニル、シンノリニル、プテリジニル、インドリル、インドリニル、ベンゾチオフェニル、ベンゾフラニル、ベンゾイソチアゾリル、ベンゾイソオキサゾリル、インダゾリル、ベンゾイミダゾリル、ベンゾトリアゾリル、アザインドリル、およびイミダゾピリジルからなる群より独立して選択され、各々置換されていてもよい、[D1]~[D8]のいずれかに記載の方法。
[D11]前記固相合成用樹脂が、分解可能なリンカーを介してホウ素原子含有化合物Waを側鎖に有し、
ホウ素原子含有化合物Waが、X9−Ar9またはX9−CR6R7R8で表される、ホウ素原子により炭素原子に連結するホウ素原子含有基を有する化合物であり、
X9は、−B(−OR10)(−OR11)、−BF3K、または下記の(a)、(b)、(c)、もしくは(d)

R10およびR11はそれぞれ独立して水素原子、またはC1−6アルコキシまたはC6−10アリールで置換されていてもよいC1−6アルキルであり、またはR10およびR11は、介在する酸素原子及びホウ素原子と一緒になって5~8員の飽和または不飽和環を形成し、当該環は、C1−6アルキル基、C1−6アルコキシおよびC6−10アリールから選択される1以上の置換基により置換されていてもよく、
Ar9は、C6−10アリール、またはO、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールであり、その各々は、フッ素原子、シアノ、C1−6アルキル、C1−6アルコキシ、(C1−6アルコキシ)カルボニル、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、(C6−10アリール)カルボニルアミノ、O、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールカルボニルアミノ、ジ(C1−6アルキル)アミノ、4~8員環状アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、および4~8員環状アミノカルボニルからなる群より独立して選択される1つまたは複数の基によって置換されていてもよく、
R6、R7およびR8はそれぞれ独立して水素原子、C1−6アルキル、C2−6アルケニル、C2−6アルキニル、C3−8シクロアルキル、C7−14アラルキル、(C1−6アルキル)カルボニル、(C6−10アリール)カルボニル、O、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールカルボニル、C6−10アリール、またはO、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールであり、その各々は、フッ素原子、シアノ、C1−6アルキル、C1−6アルコキシ、(C1−6アルコキシ)カルボニル、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、(C6−10アリール)カルボニルアミノ、O、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールカルボニルアミノ、ジ(C1−6アルキル)アミノ、4~8員環状アミノ、およびアミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、および4~8員環状アミノカルボニルからなる群より独立して選択される1以上の基によって置換されていてもよい、[D1]~[D10]のいずれかに記載の方法。
Ar9は、C6−10アリール、またはO、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールであり、その各々は、フッ素原子、シアノ、C1−6アルキル、C1−6アルコキシ、(C1−6アルコキシ)カルボニル、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、(C6−10アリール)カルボニルアミノ、O、NおよびSから独立して選択される1以上の環ヘテロ原子を含む5~10員ヘテロアリールカルボニルアミノ、ジ(C1−6アルキル)アミノ、4~8員環状アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、および4~8員環状アミノカルボニルからなる群より独立して選択される1つまたは複数の基によって置換されていてもよい、[D1]~[D11]のいずれかに記載の方法。
[D14]前記溶媒が、ハロゲン系溶媒、ニトリル系溶媒、アミド系溶媒、エーテル系溶媒、および芳香族炭化水素系溶媒からなる群より選択され、2種以上を使用してもよい、[D13]に記載の方法。
[D16]前記エーテル系溶媒が、テトラヒドロフラン、ジエチルエーテル、2−メチルテトラヒドロフラン、1,4−ジオキサン、1,2−ジメトキシエタン、1,3−ジオキソラン、ジイソプロピルエーテル、シクロペンチルメチルエーテル、t−ブチルメチルエーテル、および4−メチルテトラヒドロピランからなる群より選択され、2種以上を使用してもよい、[D15]に記載の方法。
[D18]前記反応が、0℃~100℃の反応温度で行われる、[D1]~[D17]のいずれかに記載の方法。
[D19]前記反応温度が、20℃~90℃、40℃~80℃、50℃~70℃、または55℃~65℃である、[D18]に記載の方法。
[D21]前記ベンジルエーテル構造が以下である、[D20]に記載の方法。

[D23]前記反応が、2以上の異なるホウ素原子含有化合物Waを側鎖に含む固相合成用樹脂を基質として含む混合物中で行われる、[D1]~[D22]のいずれかに記載の方法。
[D25]化合物ライブラリを構成する化合物の製造方法であって、[D1]~[D24]のいずれか1項に記載の方法により炭素—炭素結合を形成する方法を含む、前記方法。
本明細書において「C3−7シクロアルキル」とは、炭素数が3~7の環状の飽和脂肪族炭化水素基を意味する。具体的には、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、およびシクロヘプチルなどが挙げられる。
本明細書において「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子、ヨウ素原子などを意味する。本発明においてハロゲン原子が、アリール、ヘテロアリールなどの置換基となる場合、好ましいハロゲン原子として、フッ素原子、塩素原子および臭素原子が挙げられる。本発明においてハロゲン原子がアルキル、またはアルキルをその一部に含む基(アルコキシ、アルケニル、アルキルチオなど)の置換基となる場合、好ましいハロゲン原子として、フッ素原子が挙げられる。ハロゲン原子を置換基として有する基の具体例としては、トリフルオロメチル、ペンタフルオロエチル、トリフルオロメトキシ、ペンタフルオロエトキシ、トリフルオロメチルチオ、およびペンタフルオロエチルチオなどが挙げられる。
ここでライブラリを構成する化合物の数の下限として、100以上、200以上、300以上、400以上、500以上、または1000以上が例示される。さらに該化合物の数の上限として、1×108以下、1×107以下、5×106以下、1×106以下、500000以下、100000以下、50000以下、20000以下、10000以下、8000以下、7000以下、6000以下、5000以下、4000以下、3000以下、2000以下、1500以下、または1200以下が例示される。
本発明の一つの側面において、ライブラリを構成する化合物は混合物中に含まれる。試料となる化合物の混合物を用いるスクリーニングに使用できる限りにおいて、該混合物は特に限定されず、濃縮した化合物の混合物であっても、溶媒などで希釈した溶液であってもよい。該混合物は、個々に合成または入手した化合物を混合して調製してもよく、または混合物を基質とする化学反応により生成物として得られる混合物を使用してもよい。本発明の一つの態様において、ライブラリは、例えば100~100000、100~10000、または100~5000の異なる化合物の混合物として製造される。
(第1CB)—(第1L)—(第2CB)
により表され、上記化学構造を共有することを特徴とする。ここで、第1CB、および第2CBは、少なくとも1原子を含む任意の化学構造であり、化合物の部分構造であれば特に限定されない。第1CB、および第2CBは、ライブラリを構成する他の化合物と異なる化合物を形成する限りにおいては、いかなる化学構造であってもよく、例えば、第1CB、および第2CBは同一の化学構造であってもよい。また第1CB、および第2CBは、後述するように第3コアブロック(第3CB)、および/または第4コアブロック(第4CB)として特定される化学構造を置換として含んでいてもよい。ここで、第1Lは、第1CBおよび第2CBを連結する任意の化学構造であれば特に限定されず、例えば、直接結合(単結合、二重結合、三重結合)であってもよい。
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)
を含むことを特徴とする。ここで、第1CB、第1L、および第2CBについては既に述べたとおりである。第3CBは、少なくとも1原子を含む任意の化学構造であり、化合物の部分構造であれば特に限定されない。第3CBは、ライブラリを構成する他の化合物と異なる化合物を形成する限りにおいては、いかなる化学構造であってもよく、例えば、第1CB、および/または第2CBと同一の化学構造で合ってもよい。また第1CB、第2CB、および第3CBは、後述するように第4コアブロック(第4CB)として特定される化学構造を置換として含んでいてもよい。ここで、第2Lは、第2CBおよび第3CBを連結する任意の化学構造であれば特に限定されず、例えば、直接結合(単結合、二重結合、三重結合)であってもよい。
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB);または、
(第1CB)—(第1L)—(第2CB)(—(第3L)—(第4CB))—(第2L)—(第3CB);
を含むことを特徴とする。なお上記式のうちの2番目の式は、—(第1L)—(第1CB)、—(第2L)—(第3CB)、および—(第3L)—(第4CB)は、いずれも第2CBに連結していることを意味する。一態様において、上記2式で表される化合物は、上記の(第1CB)—(第1L)—(第2CB)により表される化合物の範囲に包含される。
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB);および
(第1CB)—(第1L)—(第2CB)(—(第3L)—(第4CB))—(第2L)—(第3CB);
により表される化合物が含まれる。別の態様においては一つの混合物中に下式:
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB);または
(第1CB)—(第1L)—(第2CB)(—(第3L)—(第4CB))—(第2L)—(第3CB);
のいずれかにより表される化合物が含まれる。別の態様においては一つの混合物中に下式:
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB);
により表される化合物が含まれる。
−C(=O)−NR1−
−NR1−C(=O)−
−SO2−NR1−
−NR1−SO2−
−SO2−NR1−C(=O)−
−C(=O)−NR1−SO2−
−NR1−
−O−
−S−
−C(=O)−O−
−O−C(=O)−
−C(=O)−
−SO2−
−C≡C−
−CHR2−
単結合

から選択される式により表される。ここで単結合は、炭素—窒素間単結合または炭素—炭素間単結合である。
1×102~1×108の化合物を含むライブラリであって、ライブラリを構成する化合物は、第1CB、第1L、第2CB、第2L、および第3CBが共有結合で連結した以下の構造:
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)
を含み、第1Lおよび第2Lの種類の数の合計が3以上であり、第1CB、第2CB、および第3CBの種類の数の合計が6以上であり、1×102~1×105の前記化合物を含む1または2以上の混合物により構成され、該ライブラリを構成する化合物のうち90%以上の化合物が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)により検出されている、前記ライブラリ。
1×102~1×104の化合物を含むライブラリであって、ライブラリを構成する化合物は、第1CB、第1L、第2CB、第2L、第3CB、第3L、および第4CBが共有結合で連結した以下の構造:
(第1CB)—(第1L)—(第2CB)—(第2L)—(第3CB)—(第3L)—(第4CB)
を含み、第1L、第2L、および第3Lの種類の数の合計が5以上であり、第1CB、第2CB、第3CB、および第4CBの種類の数の合計が8以上であり、1×102~5×103の前記化合物を含む1または2以上の混合物により構成され、該ライブラリを構成する化合物のうち95%以上の化合物が液体クロマトグラフ質量分析計(LC−MS)における保持時間および質量電荷比(m/z)により検出されている、前記ライブラリ。
(1)本明細書に記載のライブラリを提供すること;
(2)前記ライブラリを構成する前記1または2以上の混合物をそれぞれ標的分子と接触させること;
(3)標的分子に結合する化合物を同定すること
を含むことを特徴とする。
上記(e)について、固相担体からの化合物を切り出しの反応条件は、使用した固相担体の化学構造に基づいて当業者であれば適宜設定することができる。切り出しに使用する試薬としては、例えば、塩酸、トリフルオロ酢酸(TFA)などのカルボン酸、2,2,2−トリフルオロエタノール(TFE)や1,1,1,3,3,3−ヘキサフルオロイソプロピルアルコール(HFIP)などのフルオロアルコールの他、水中でのpKaが10以下のブレンステッド酸、もしくは任意のルイス酸を用いることができる。一つの態様において、固相担体から切り出しされた化合物は混合物として得られる。
(d)において得られる担持担体が、
第4BB1−nd−第3BB1−nc−第2BB1−nb−第1BB1−na担持担体;
第4BB1−nd−(第3BB1−nc−)第2BB1−nb−第1BB1−na担持担体;
第3BB1−nc−第2BB1−nb−(第4BB1−nb−)第1BB1−na担持担体;
第4BB1−nd−第3BB1−nc−(第2BB1−nb−)第1BB1−na担持担体;または
第4BB1−nd−第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体
である。一つの態様において、(d)で得られる担持担体は2以上の上記式で表される担持担体の混合物であってもよい。一つの態様において、(d)において得られる担持担体は第4BB1−nd−第3BB1−nc−第2BB1−nb−第1BB1−na担持担体である。
1−1)第1リンカーが2種類以上、3種類以上、または4種類以上である;
1−2)第2リンカーが2種類以上、3種類以上、または4種類以上である;および
1−3)第1リンカーまたは第2リンカーが3種類以上、4種類以上、または5種類以上である;
の1以上の条件を満たすことを特徴とする。さらに別の側面において、第1リンカー、第2リンカーおよび前記第3リンカーが、
2−1)第1リンカーが2種類以上、3種類以上、または4種類以上である;
2−2)第2リンカーが2種類以上、3種類以上、または4種類以上である;
2−3)第3リンカーが2種類以上、3種類以上、または4種類以上である;および
2−4)第1リンカー、第2リンカーまたは第3リンカーが3種類以上、4種類以上、または5種類以上である;
の1以上の条件を満たすことを特徴とする。
得られるライブラリが100以上、200以上、300以上、400以上、500以上、または1000以上の化合物を含む、[C1]~[C33]のいずれかに記載の方法。
本発明の一つの側面において、ライブラリ合成方法により得られるライブラリが含む化合物の数について、800以下、1000以下、1200以下、1500以下、1700以下、2000以下、2500以下、3000以下、4000以下、5000以下、6000以下、7000以下、8000以下、10000以下、20000以下、50000以下、または100000以下を上限として例示することができ、100以上、200以上、300以上、400以上、500以上、または1000以上を下限として例示することができる。

該ベンゼン、該ナフタレン、該アントラセンおよび該ビフェニルは、ハロゲン原子、ニトロ、シアノ、カルボキシ、スルホキシ、−COR1、−OR1、−NR1R2、−CONR1R2、又は−SO2NR1R2からなる群より選ばれる同一または異なる1~3個の基で置換されていてもよく、R1およびR2はそれぞれ独立してC1−6アルキル、または連結する窒素原子と一緒になって、ヘテロ環を形成していてもよく、Xは、−CONH−、−NHCO−、−CH2O−及び−O−からなる群より選ばれる1種でビルディングブロックと結合していることを表す)
で表される構造の基を化合物に導入することができる。

で表される構造の基を化合物に導入してもよい。
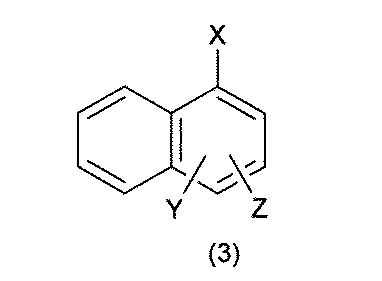
で表される構造の基を化合物に導入してもよい。

で表される構造の基を化合物に導入してもよい。
本発明の一つの側面において、ライブラリを構成する化合物はUVタグを含み、該化合物の固相合成において、中間体化合物の分析に用いることができる。一つの態様において、ライブラリの製造方法はさらに、
1)いずれかの工程で得られた担持担体を得る工程;
2)工程1)で得られた担持担体から中間体化合物を切り出す工程;および
3)前記中間体化合物を分析する工程;
並びに、任意で、工程(e)で切り出された化合物を分析する工程、
を含む。中間体化合物の分析で得られる結果は、固相反応の反応条件の最適化などに利用することができ、それにより質の高いライブラリの製造が可能となる。一つの態様において、前記分析は液体クロマトグラムを用いて行うことができる。

リンカーとしてのアミド結合は、カルボン酸またはその誘導体とアミンの反応により形成することができる。
上記式のR1は既に定義した通りである。R1の好ましい例としては、水素原子、C1−6アルキル、例えば、メチル、およびエチルなどが挙げられる。

リンカーとしてのスルホンアミド結合は、アミンとスルホニルハライドまたはその誘導体との反応により形成することができる。

リンカーとしてのアシルスルホンアミド結合は、カルボン酸またはその誘導体とスルホンアミドの反応により形成することができる。


リンカーとしてのアミド結合は、1級アミドまたは2級アミドとハロゲン化アリールまたはその誘導体との反応により形成することもできる。
上記式においてBが固相担体を含む場合、Aを含む1級アミドまたは2級アミドとハロゲン化アリールまたはその誘導体(B−X)との反応は既に述べた試薬や反応条件により行うことができる。ここで、使用するAを含む1級アミドまたは2級アミドの当量数は当業者が適宜設定可能であり、例えば、B−Xに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用する塩基の当量数は当業者が適宜設定可能であり、例えば、B−Xに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。金属触媒を使用する場合、使用する金属触媒の当量数は当業者が適宜設定可能であり、例えば、B−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。配位子を使用する場合、使用する配位子の当量数は当業者が適宜設定可能であり、例えば、B−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。金属配位子複合体を使用する場合、使用する金属配位子複合体の当量数は当業者が適宜設定可能であり、例えば、B−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。
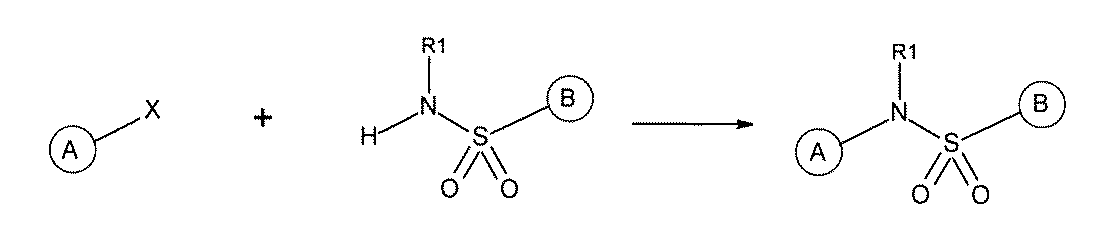
リンカーとしてのスルホンアミド結合はまた、1級スルホンアミドまたは2級スルホンアミドとハロゲン化アリールまたはその誘導体との反応により形成することもできる。
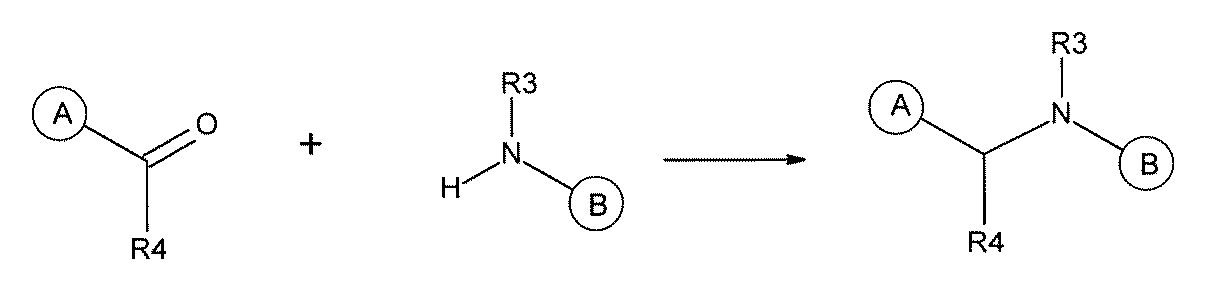
リンカーとしてのアミン結合は、アルデヒドとアミン、またはケトンとアミンを用いる還元的アミノ化反応によっても形成することができる。

上記式においてBが固相担体を含み、A—Xがヒドロキシアリールの場合、Aに連結する反応性官能基のXはヒドロキシ基であり、アゾジカルボン酸誘導体とホスフィン、またはシアノメチレンホスホラン誘導体を使用して、Bに連結する反応性官能基のYがヒドロキシ基であるアルコールとの反応に付すことによりリンカーとしてのエーテル結合を形成することは、既に述べた試薬や反応条件により行うことができる。ここで、アゾジカルボン酸誘導体を使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むアルコールに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。ホスフィンを使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むアルコールに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。シアノメチレンホスホラン誘導体を使用する場合には、その当量数は当業者が適宜設定可能であり、例えば、Bを含むアルコールに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用するA−Xの当量数は当業者が適宜設定可能であり、例えば、Bを含むアルコールに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。
上記式においてAが固相担体を含み、A—Xがハロゲン化アリールまたはその誘導体の場合、Aに連結する反応性官能基のXはハロゲンもしくはヒドロキシ基誘導体であり、塩基を使用して、Bに連結する反応性官能基のYがヒドロキシ基であるヒドロキシアリールもしくはアルコールとの反応に付すことによりリンカーとしてのエーテル結合を形成することができる。ここで、塩基としては、通常使用できるものであれば特に限定されないが、例えば、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、炭酸水素カリウム、リン酸ナトリウム、リン酸カリウム、リン酸水素二ナトリウム、リン酸水素二カリウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化バリウム、水酸化テトラアルキルアンモニウム(水酸化テトラメチルアンモニウム、水酸化テトラブチルアンモニウムなど)、LiOtBu、NaOtBu、KOtBu、水素化ナトリウム、水素化カリウム、リチウムビス(トリメチルシリル)アミド、ナトリウムビス(トリメチルシリル)アミド、カリウムビス(トリメチルシリル)アミド、リチウムジイソプロピルアミド、メチルリチウム、ブチルリチウム、ピリジン、アルキル置換ピリジン(2、6−ルチジン、DTBP、DTBMPなど)、DMAP、N,N−ジメチルアニリン、N−メチルモルホリン、トリエチルアミン、DIPEA、DBU、DBN、MTBD、TMG、BTMG、P1tBu、BEMP、BTPP、P2Et、P2tBu、NMI、水素化リチウム、ナトリウムアミド、リチウムアミド、四ホウ酸ナトリウム、1,8−ビス(ジメチルアミノ)ナフタレン、カリウムt−ペントキシド、ナトリウムt−ペントキシド、リチウムt−ペントキシド、リチウムジシクロヘキシルアミド、2,2,6,6−テトラメチルピペリジニルリチウム、2,2,6,6−テトラメチルピペリジニルマグネシウム クロリド リチウム クロリド錯体、テトラブチルアンモニウムフルオリド、セシウムフルオリド、酸化銀、炭酸銀などを使用することができる。
使用する金属触媒の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。
使用するパラジウム触媒の前駆体の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。
上記式においてBが固相担体を含み、A—Xがハロゲン化アリールまたはその類縁体の場合、Aに連結する反応性官能基のXはハロゲンもしくはヒドロキシ基誘導体であり、塩基を使用して、Bに連結する反応性官能基のYがヒドロキシ基であるヒドロキシアリールもしくはアルコールとの反応に付すことによりリンカーとしてのエーテル結合を形成することは既に述べた試薬や反応条件により行うことができる。ここで、塩基を使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むヒドロキシアリールもしくはアルコールに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用するA−Xの当量数は当業者が適宜設定可能であり、例えば、Bを含むヒドロキシアリールもしくはアルコールに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。金属触媒を使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むヒドロキシアリールもしくはアルコールに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。配位子を使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むヒドロキシアリールもしくはアルコールに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。触媒前駆体を使用する場合、その当量数は当業者が適宜設定可能であり、例えば、Bを含むヒドロキシアリールもしくはアルコールに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。
上記式においてAが固相担体を含み、A—Xがヒドロキシアリールもしくはアルコールの場合、Aに連結する反応性官能基のXはヒドロキシ基であり、塩基を使用して、Bに連結する反応性官能基のYがハロゲンであるアルキルハライドもしくはYがヒドロキシ基誘導体であるアルキル化剤との反応に付すことによりリンカーとしてのエーテル結合を形成することができる。ここで、塩基としては、通常使用できるものであれば特に限定されないが、例えば、ピリジン、2,6−ルチジン、2,4,6−コリジン、DTBP、DTBMPなどのピリジン誘導体の他、トリエチルアミン、DIPEA、4−メチルモルホリンなどの第三級アミン、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、炭酸水素カリウム、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化テトラアルキルアンモニウム(水酸化テトラメチルアンモニウム、水酸化テトラブチルアンモニウムなど)、水素化ナトリウム、水素化カリウム、DBU、DBN、MTBD,BTMG、1,8−ビス(ジメチルアミノ)ナフタレン、BEMP、BTPP、P2Et、P2tBu、リン酸カリウム、LiOtBu、NaOtBu、KOtBu、LiHMDS,NaHMDS,KHMDS、HMDS、トリエチルアミン、DIPEA、4−メチルモルホリン、水素化リチウム、ナトリウムアミド、リチウムアミド、リン酸ナトリウム、リン酸カリウム、KHCO3、Na2HPO4、四ホウ酸ナトリウム、カリウムt−ペントキシド、ナトリウムt−ペントキシド、リチウムt−ペントキシド、リチウムジイソプロピルアミド、リチウムジシクロヘキシルアミド、2,2,6,6−テトラメチルピペリジニルリチウム、2,2,6,6−テトラメチルピペリジニルマグネシウム クロリド リチウム クロリド錯体、N,N−ジメチルアニリン、P1−tBu、P2tBu、ブチルリチウム、メチルリチウム、テトラブチルアンモニウムフルオリド、セシウムフルオリド、酸化銀、炭酸銀などを使用することができる。
上記式においてBが固相担体を含み、A—Xがヒドロキシアリールもしくはアルコールの場合、Aに連結する反応性官能基のXはヒドロキシ基であり、塩基を使用して、Bに連結する反応性官能基のYがハロゲンであるアルキルハライドもしくはYがヒドロキシ基誘導体であるアルキル化剤との反応に付すことによりリンカーとしてのエーテル結合を形成することは、既に述べた試薬や反応条件により行うことができる。ここで、塩基を使用する場合、その等量数は当業者が適宜設定可能であり、例えば、Bを含むアルキルハライドもしくはアルキル化剤に対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用するA−Xの当量数は当業者が適宜設定可能であり、例えば、Bを含むアルキルハライドもしくはアルキル化剤に対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。
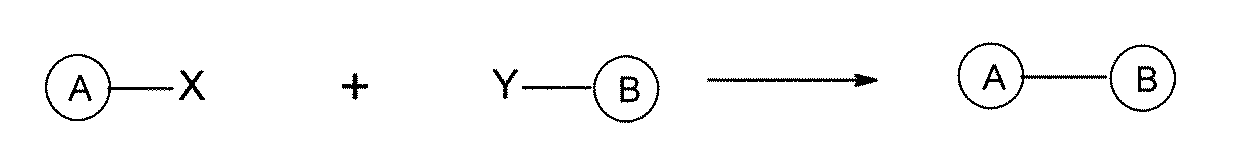


上記式において、Aに連結する反応性官能基のXはヒドロキシであり、アゾジカルボン酸誘導体とホスフィン、またはシアノメチレンホスホラン誘導体を使用して、Bに連結するヒドロキシ基を含むアルコールとの反応に付すことによりリンカーとしてのエーテル結合を形成することができる。使用するBを含むアルコールの当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。ここで、アゾジカルボン酸誘導体としては、通常使用できるものであれば特に限定されないが、例えば、アゾジカルボン酸ジエチル(DEAD)、アゾジカルボン酸ビス(2,2,2−トリクロロエチル)、N,N,N′,N′−テトラメチルアゾジカルボキサミド(TMAD)、アゾジカルボン酸ジ−tert−ブチル、アゾジカルボン酸ジメチル、アゾジカルボン酸ジベンジル、アゾジカルボン酸ジイソプロピル(DIAD)、1,1′−(アゾジカルボニル)ジピペリジン(ADDP)、N,N,N′,N′−テトライソプロピルアゾジカルボキサミド(TIPA)、1,6−ジメチル−1,5,7−ヘキサヒドロ−1,4,6,7−テトラゾシン−2,5−ジオン(DHTD)、アゾジカルボン酸ビス(2−メトキシエチル)などを使用することができる。ホスフィンとしては、通常使用できるものであれば特に限定されないが、例えば、トリフェニルホスフィン(TPP)、トリブチルホスフィン(TBP)、4−(ジメチルアミノ)フェニルジフェニルホスフィン、イソプロピルジフェニルホスフィン、フェノキシジフェニルホスフィン、トリ−n−オクチルホスフィン、ジフェニル−2−ピリジルホスフィン、トリ−tert−ブチルホスフィン、ジシクロヘキシルフェニルホスフィン、トリシクロヘキシルホスフィン、トリヘキシルホスフィンなどを使用することができる。シアノメチレンホスホラン誘導体としては、通常使用できるものであれば特に限定されないが、例えば、シアノメチレントリブチルホスホラン(CMBP)、シアノメチレントリメチルホスホラン(CMMP)などを使用することができる。使用するアゾジカルボン酸誘導体の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用するホスフィンの当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。シアノメチレンホスホラン誘導体を使用する場合には、使用するシアノメチレンホスホラン誘導体の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~150当量、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。

上記反応には溶媒を用いることができ、特に限定はされないが、例えば、上記反応には溶媒を用いることができ、特に限定はされないが、例えばDCM、クロロホルム、DCE、四塩化炭素などのハロゲン系溶媒、ジエチルエーテル、ジイソプロピルエーテル、THF、2−メチルテトラヒドロフラン、4−メチルテトラヒドロピラン、1,4−ジオキサン、DME、TBME、CPME、ジイソプロピルエーテル、イソソルビドジメチルエーテル、ジグリム、トリグリム、テトラグリムなどのエーテル系溶媒、アセトン、MEK、MIBK、4−メチル−2−ペンタノン、シクロペンタノン、シクロヘキサノン、ジエチルケトンなどのケトン系溶媒、TMU、DMI、DMPUなどのウレア系溶媒、DMF、DMA、NFM、NMP、NEP,NBPなどのアミド系溶媒、DMSOなどのスルホキシド系溶媒、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸イソブチル、GVL(γ−バレロラクトン)、酢酸ペンチル、プロピオン酸メチルなどのエステル系溶媒、ジフェニルスルホン、ジメチルスルホン、ジエチルスルホン、スルホラン、3−メチルスルホラン、エチルメチルスルホン、エチルイソプロピルスルホンなどのスルホン系溶媒、アニソール、トルエン、キシレン、クロロベンゼン、ブロモベンゼン、エチルベンゼン、ニトロベンゼン、α,α,α—トリフルオロトルエン、1,2−ジクロロベンゼン、ベンゼン、ピリジンなどの芳香族系溶媒の他、炭酸ジメチル、炭酸ジエチル、炭酸エチレン、炭酸プロピレンなどのカーボネート系溶媒などの他、アセトニトリル、リモネンなどの溶媒を用いることができる。単一の溶媒を用いてもよいし、複数の溶媒を組み合わせた混媒を用いてもよい。必要な場合は添加剤を用いてもよく、例えば、Ag2O、12−クラウン−4、15−クラウンー5、18−クラウン−6、HMPA、TMEDA、テトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、テトラブチルアンモニウムヨージドを添加剤として使用することができる。添加剤を使用する場合、使用する添加剤の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.01~100当量、0.01~75当量、0.01~50当量、0.01~30当量、0.1~30当量、または0.1~10当量とすることができる。反応温度は当業者が適宜設定可能であり、例えば、−20~150℃、0~120℃、20~100℃、または20~60℃とすることができる。反応時間は当業者が適宜設定可能であり、例えば、該当の基質、もしくは類似基質を用いた検討実験(モデル実験とも呼ぶ)において充分な変換率に達するのに要した時間、もしくは検討結果から何らかの手法によって予想される時間、または一部の固相担体から生成物の切り出しを行って反応終了時点を決定または確認することができる。反応終了後、通常の後処理方法にて目的の化合物を得ることができる。


上記式においてAが固相担体を含み、A—Xがハロゲン化アリールまたはその誘導体の場合、Aに連結する反応性官能基のXはハロゲンもしくはヒドロキシ基から誘導される基である。ヒドロキシ基から誘導される基として、例えば、メタンスルホニル基、トリフルオロメタンスルホニル基、ノナフルオロブタンスルホニル基、ベンゼンスルホニル基、パラトルエンスルホニル基、2−ニトロベンゼンスルホニル基、4−ニトロベンゼンスルホニル基、2,4−ジニトロベンゼンスルホニル基などを例示することができる。A−Xは、塩基の存在下、Bに連結するチオールとの反応に付すことができる。ここで、塩基としては、通常使用できるものであれば特に限定されないが、例えば、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、炭酸水素カリウム、リン酸ナトリウム、リン酸カリウム、リン酸水素二ナトリウム、リン酸水素二カリウム、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化バリウム、テトラアルキルアンモニウムヒドロキシド(テトラメチルアンモニウムヒドロキシド、テトラブチルアンモニウムヒドロキシド、ベンジルトリエチルアンモニウムヒドロキシド、など)、フッ化セシウム、フッ化カリウム、LiOtBu、NaOtBu、KOtBu、カリウムt−ペントキシド、ナトリウムt−ペントキシド、リチウムt−ペントキシド、水素化ナトリウム、水素化カリウム、水素化リチウム、ナトリウムアミド、リチウムアミド、リチウムビス(トリメチルシリル)アミド、ナトリウムビス(トリメチルシリル)アミド、カリウムビス(トリメチルシリル)アミド、ヘキサメチルジシラザン、リチウムジイソプロピルアミド、リチウムジシクロヘキシルアミド、2,2,6,6−テトラメチルピペリジニルリチウム、2,2,6,6−テトラメチルピペリジニルマグネシウムクロリド リチウムクロリド錯体、メチルリチウム、ブチルリチウム、ピリジン、アルキル置換ピリジン(2、6−ルチジン、2,4,6−コリジン、DTBP、DTBMPなど)、DMAP、N,N−ジメチルアニリン、N−メチルモルホリン、トリエチルアミン、DIPEA、DBU、DBN、MTBD、TMG、BTMG、P1tBu、BEMP、BTPP、P2Et、P2tBu、NMI、四ホウ酸ナトリウム、1,8−ビス(ジメチルアミノ)ナフタレン、N,N−ジメチルアニリン、テトラブチルアンモニウムフルオリド、セシウムフルオリドなどを使用することができる。使用する塩基の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。使用するBを含むチオールの当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。上記反応には溶媒を用いることができ、特に限定はされないが、例えば、ハロゲン系溶媒(DCM(ジクロロメタン)、クロロホルム、DCE(1,2−ジクロロエタン)、四塩化炭素など)、エーテル系溶媒(ジエチルエーテル、ジイソプロピルエーテル、THF(テトラヒドロフラン)、2−メチルテトラヒドロフラン、4−メチルテトラヒドロピラン、1,4−ジオキサン、DME(1,2−ジメトキシエタン)、TBME(t−ブチルメチルエーテル)、CPME(シクロペンチルメチルエーテル)、イソソルビドジメチルエーテル、ジグリム、トリグリム、テトラグリムなど)、ケトン系溶媒(アセトン、MEK(メチルエチルケトン)、MIBK(メチルイソブチルケトン)、4−メチル−2−ペンタノン、シクロペンタノン、シクロヘキサノン、ジエチルケトンなど)、ウレア系溶媒(TMU(N,N,N′,N′−テトラメチル尿素)、DMI(1、3−ジメチル−2−イミダゾリジノン)、DMPU(N,N’−ジメチルプロピレン尿素)など)、アミド系溶媒(DMF(N,N−ジメチルホルムアミド)、DMA(N,N−ジメチルアセトアミド)、NFM(N−ホルミルモルホリン)、NMP(N−メチル−2−ピロリドン)、NEP(N−エチル−2−ピロリドン)、NBP(N−ブチル−2−ピロリドン)、ホルムアミドなど)、スルホキシド系溶媒(DMSO(ジメチルスルホキシド)、メチルフェニルスルホキシドなど)、スルホン系溶媒(ジフェニルスルホン、ジメチルスルホン、ジエチルスルホン、スルホラン、3−メチルスルホラン、エチルメチルスルホン、エチルイソプロピルスルホンなど)、エステル系溶媒(酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸イソブチル、GVL(γ−バレロラクトン)、酢酸ペンチル、プロピオン酸メチルなど)、芳香族炭化水素系溶媒(アニソール、トルエン、キシレン、クロロベンゼン、ブロモベンゼン、エチルベンゼン、ニトロベンゼン、α,α,α—トリフルオロトルエン、1,2−ジクロロベンゼン、ベンゼン、テトラリン、ピリジン、キノリンなど)、アルコール系溶媒(エタノール、イソプロパノール、tertブタノール、tertアミルアルコールなど)、炭化水素系溶媒(ヘキサン、ペンタン、ヘプタン、シクロヘキサン、メチルシクロヘキサンなど)などの他、アセトニトリル、水、ジメチルアセタール、ジメチル硫酸、シレン、リモネンなどを使用することができる。単一の溶媒を用いてもよいし、複数の溶媒を組み合わせた混媒を用いてもよい。反応温度は当業者が適宜設定可能であり、例えば、0~150℃、10~120℃、20~100℃、20~80℃、または20~60℃とすることができる。必要な場合は金属触媒を用いてもよく、例えば、パラジウムでは例えば酢酸パラジウム、Pd(dba)2、Pd2(dba)3、Pd2(dba)3CHCl3、アリルパラジウムクロリドダイマー、パラジウム(π−シンナミル)クロリドダイマー、(1−メチルアリル)パラジウムクロリドダイマー、(1,5−シクロオクタジエン)ビス(トリメチルシリルメチル)パラジウム(II)ニッケルでは例えば、NiCl2、(塩化ニッケル(II))、Ni(COD)2(ビス(1,5−シクロオクタジエン)ニッケル(0))、など、銅では例えば酸化銅(I)(Cu2O)、ヨウ化銅(I)(CuI)、臭化銅(I)(CuBr)、塩化銅(I)(CuCl)、酢酸銅(Cu(OAc)2)などを金属触媒として使用することができる。

上記式においてAが固相担体を含む場合、Aに連結する反応性官能基のXはヒドロキシ、アルコキシ基、アリールオキシ基、アルキルチオ基、アリールチオ基である。Xがヒドロキシの場合、酸無水物を添加することで、事前に、もしくは反応系中で混合酸無水物を形成することにより、ケトン結合形成反応に付すことができる。ここで、酸無水物としては、通常使用できるものであれば特に限定されないが、例えば、安息香酸無水物、無水酢酸、イソ酪酸無水物、イソ吉草酸無水物、ピバル酸無水物などを使用することができる。使用する酸無水物の当量数は当業者が適宜設定可能であり、例えば、Aを含むカルボン酸に対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。ホウ素誘導体または亜鉛誘導体の当量数は当業者が適宜設定可能であり、例えば、Aを含むカルボン酸に対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。金属触媒を使用して、Bに連結する反応性官能基のYがホウ素誘導体であるボロン酸類縁体、もしくは炭素−炭素末端二重結合から誘導したアルキルボラン試薬、もしくはYが亜鉛誘導体である化合物との反応に付すことによりリンカーとしてのケトン結合を形成することができる。使用するボロン酸誘導体およびアルキルボラン試薬、亜鉛誘導体の当量数は当業者が適宜設定可能であり、例えば、A−CO−Xに対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。ここで、金属触媒としては、通常使用できるものであれば特に限定されないが、例えば、酢酸パラジウム、Pd(dba)2、Pd2(dba)3、Pd2(dba)3CHCl3、アリルパラジウムクロリドダイマー、パラジウム(π−シンナミル)クロリドダイマー、(1−メチルアリル)パラジウムクロリドダイマー、(1,5−シクロオクタジエン)ビス(トリメチルシリルメチル)パラジウム(II)などを使用することができる。必要な場合は配位子を用いてもよく、例えば、亜リン酸エステル類(P(OEt)3(トリエチルホスファイト)、P(OPh)3(トリフェニルホスファイト)など)、トリアルキルホスフィン(PCy(トリシクロヘキシルホスフィン)、P(tBu)3(トリ−tert−ブチルホスフィン)、cataCXium A(ジ(1−アダマンチル)−n−ブチルホスフィン)、(tBu)2PMe(ジ−tert−ブチルメチルホスフィン)、P(Ad)3、ネオペンチル(tBu)2P(ジ−tert−ブチルネオペンチルホスフィン)など)、トリアリールホスフィン(PPh3(トリフェニルホスフィン)、トリ(2−フリル)ホスフィンなど)、ジアルキルビアリールホスフィン(XPhos(2−ジシクロヘキシルホスフィノ−2′,4′,6′−トリイソプロピルビフェニル)、SPhos(2−ジシクロヘキシルホスフィノ−2’,6’−ジメトキシビフェニル)、RuPhos(2−ジシクロヘキシルホスフィノ−2′,6′−ジイソプロポキシビフェニル)、BrettPhos(2−(ジシクロヘキシルホスフィノ)−3,6−ジメトキシ−2’,4’,6’−トリイソプロピル−1,1’−ビフェニル)、tBuXPhos(2−ジ−tert−ブチルホスフィノ−2′,4′,6′−トリイソプロピルビフェニル)、CPhos(2−ジシクロヘキシルホスフィノ−2’,6’−bis(N,N−ジメチルアミノ)ビフェニル)、DavePhos(2−ジシクロヘキシルホスフィノ−2′−(N,N−ジメチルアミノ)ビフェニル)、RockPhos(2−ジ(ターシャル−ブチル)ホスフィノ−2’,4’,6’−トリイソプロピル−3−メトキシ−6−メチルビフェニル,ジ‐ターシャル−ブチル(2’,4’,6’−トリイソプロピル−3−メトキシ−6−メチル−[1,1’−ビフェニル]−2−イル)ホスフィン)、Me4tBuXPhos(2−ジ−tert−ブチルホスフィノ−3,4,5,6−テトラメチル−2′,4′,6′−トリイソプロピル−1,1′−ビフェニル)、tBuBrettPhos(2−(ジ−tert−ブチルホスフィノ)−2’,4’,6’−トリイソプロピル−3,6−ジメトキシ−1,1’−ビフェニル)、AdBrettPhos(2−(ジ−1−アダマンチルホスフィノ)−2’,4’,6’−トリイソプロピル−3,6−ジメトキシ−1,1’−ビフェニル)など)、ジアルキルモノアリールホスフィン(APhos((4−(N,N−ジメチルアミノ)フェニル)ジ−tert−ブチルホスフィン)、tBu)2PPh、MorDalPhos(2−モルホリノフェニルジ(1−アダマンチル)ホスフィン)など)、二座ホスフィン配位子(dppf(1,1′−ビス(ジフェニルホスフィノ)フェロセン)、dtbpf(1,1’−ビス(ジ−tert−ブチルホスフィノ)フェロセン)、BINAP(2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル)、XantPhos(4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン)、Josiphos((R)−1−[(Sp)−2−(ジシクロヘキシルホスフィノ)フェロセニル]エチルジ−tert−ブチルホスフィン)など)、NHC配位子(IPr(1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾール−2−イリデン)、SIPr(1,3−ビス(2,6−ジ−i−プロピルフェニル)イミダゾリジン−2−イリデン)、IMes(1,3−ビス(2,4,6−トリメチルフェニル)イミダゾール−2−イリデン)、IPent(1,3−ビス(2,6−ジ−3−ペンチルフェニル)イミダゾール−2−イリデン))などを配位子として使用することができる。また、上記の配位子とパラジウム触媒の前駆体を用いてもよく、例えばBuchwald第1世代触媒前駆体(G1)(XPhos Pd G1、SPhos Pd G1、RuPhos Pd G1、BrettPhos Pd G1、tBuXPhos Pd G1など)、Buchwald第2世代触媒前駆体(G2)(XPhos Pd G2、SPhos Pd G2、RuPhos Pd G2、BrettPhos Pd G2、tBuXPhos Pd G2、CPhos Pd G2、DavePhos Pd G2、APhos Pd G2、tBu)2PPh Pd G2、MorDalPhos Pd G2、PCy3 Pd G2、P(tBu)3 Pd G2、cataCXium A Pd G2、XantPhos Pd G2など)、Buchwald第3世代触媒前駆体(G3)(XPhos Pd G3、SPhos Pd G3、RuPhos Pd G3、BrettPhos Pd G3、tBuXPhos Pd G3、CPhos Pd G3、DavePhos Pd G3、RockPhos Pd G3、Me4tBuXPhos Pd G3、tBuBrettPhos Pd G3、AdBrettPhos Pd G3、APhos Pd G3、(tBu)2PPh Pd G3、MorDalPhos Pd G3、PCy3 Pd G3、P(tBu)3 Pd G3、cataCXium A Pd G3、tBu)2PMe Pd G3、ネオペンチルtBu)2P Pd G3、dppf Pd G3、dtbpf Pd G3、BINAP Pd G3、XantPhos Pd G3、Josiphos Pd G3など)、Buchwald第4世代触媒前駆体(G4)(XPhos Pd G4、SPhos Pd G4、RuPhos Pd G4、BrettPhos Pd G4、tBuXPhos Pd G4、CPhos Pd G4、tBuBrettPhos Pd G4、APhos Pd G4、PCy3 Pd G4、P(tBu)3 Pd G4、cataCXium A Pd G4、dppf Pd G4、BINAP Pd G4、XantPhos Pd G4など)、ジクロロパラジウム(II)錯体(Pd(PPh3)2Cl2、Pd(PCy3)2Cl2、Pd(dppf)Cl2、Pd(dtbpf)Cl2、(APhos)2PdCl2など)、パラジウム(0)錯体(テトラキストリフェニルホスフィンパラジウム(0)、ビス(トリシクロヘキシルホスフィン)パラジウム(0)、ビス(トリ−tert−ブチルホスフィン)パラジウム(0)など)、ハロゲン化パラジウム(I)錯体(ブロモ(トリ−tert−ブチルホスフィン)パラジウム(I)ダイマー、ヨード(トリ−tert−ブチルホスフィン)パラジウム(I)ダイマーなど)、ホスフィンπ−アリルパラジウム触媒(XPhos Pd(クロチル)Cl(クロロ(クロチル)(2−ジシクロヘキシルホスフィノ−2’,4’,6’−トリイソプロピル−1,1’−ビフェニル)パラジウム(II))、SPhos Pd(クロチル)Cl(クロロ(クロチル)(2−ジシクロヘキシルホスフィノ−2’,6’−ジメトキシ−1,1’−ビフェニル)パラジウム(II))、RuPhos Pd(クロチル)Cl(クロロ(クロチル)(2−ジシクロヘキシルホスフィノ−2’,6’−ジイソプロポキシ−1,1’−ビフェニル)パラジウム(II))、[BrettPhos Pd(クロチル)]OTf(クロチル(2−ジシクロヘキシルホスフィノ−2’,4’,6’−トリイソプロピル−3,6−ジメトキシ−1,1’−ビフェニル)パラジウム(II)トリフレート)、[tBuXPhos Pd(アリル)]OTf(アリル(2−ジ−tert−ブチルホスフィノ−2’,4’,6’−トリイソプロピル−1,1’−ビフェニル)パラジウム(II)トリフレート)、[tBuBrettPhos Pd(アリル)]OTf(アリル(2−ジ−tert−ブチルホスフィノ−3,6−ジメトキシ−2’,4’,6’−トリイソプロピル−1,1’−ビフェニル)パラジウム(II)トリフレート)、AmPhos Pd(クロチル)Cl(クロロ(クロチル)[(p−ジメチルアミノフェニル)(ジ−tert−ブチルホスフィン)]パラジウム(II))、P(Cy)3 Pd(クロチル)Cl(クロロ(クロチル)(トリシクロヘキシルホスフィン)パラジウム(II))、[P(tBu)3] Pd(クロチル)Cl(トリ−tert−ブチルホスフィン(クロロ)(クロチル)パラジウム(II))、[BINAP Pd(アリル)]Cl((R)−BINAP Pd(アリル)]Cl;アリル[(R)−2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフタレン]パラジウム(II)クロリド)、[XantPhos Pd(アリル)]Cl(アリル[4,5−ビス(ジフェニルホスフィノ)−9,9−ジメチルキサンテン]パラジウム(II)クロリド)など)、PEPPSI触媒(Pd−PEPPSI IPr、Pd−PEPPSI SIPr((1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾリデン)(3−クロロピリジル)パラジウム(II)ジクロリド)、Pd−PEPPSI IPent(1,3−ビス(2,6−ジ−3−ペンチルフェニル)イミダゾール−2−イリデン)など)、(NHC)Pd(アリル)Cl触媒(アリル[1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾール−2−イリデン]クロロパラジウム(II)、アリル[1,3−ビス(2,6−ジイソプロピルフェニル)−2−イミダゾリジニリデン]クロロパラジウム(II)、[1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾール−2−イリデン]クロロ[3−フェニルアリル]パラジウム(II)など)、NHC−Pdナフトキノン触媒(1,3−ビス(2,6−ジイソプロピルフェニル)イミダゾール−2−イリデン(1,4−ナフトキノン)パラジウム(0)ダイマーなど)などをパラジウム触媒の前駆体として用いることができる。使用する金属触媒の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。使用する配位子の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。使用する触媒の前駆体の当量数は当業者が適宜設定可能であり、例えば、A−Xに対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。

上記式においてAが固相担体を含む場合、金属触媒を使用して、Bに連結するプロパルギル基を含むアセチレン誘導体との反応に付すことによりリンカーとしてのトリアゾール環を用いた結合を形成することができる。使用するアセチレン誘導体の当量数は当業者が適宜設定可能であり、例えば、Aを含むアジド誘導体に対して、1~100当量、1~75当量、1~50当量、1~30当量、または1~10当量とすることができる。ここで、金属触媒としては、通常使用できるものであれば特に限定されないが、例えば、金属銅、塩化銅(I),塩化銅(II)、臭化銅(I)、臭化銅(II)、ヨウ化銅、酢酸銅(I)、酢酸銅(II)、酸化銅(I)、酸化銅(II)、硫酸銅、酢酸亜鉛、クロロ(H5−ペンタメチルシクロペンタジエン)[ビス(トリフェニルホスフィン)]ルテニウム(II)、カルボニル(ジヒドリド)トリス(トリフェニルホスフィン)ルテニウム(II)などを使用することができる。金属触媒を使用する場合、使用する金属触媒の当量数は当業者が適宜設定可能であり、例えば、Aを含むアジド誘導体に対して、0.001~10当量、0.001~7.5当量、0.001~5当量、0.01~5当量、または0.01~3当量とすることができる。使用する添加剤として例えば、L−アスコルビン酸ナトリウム、セルロース、アンバーリスト、テトラブチルアンモニウムブロミドなどを使用することができる。添加剤を使用する場合、使用する添加剤の当量数は当業者が適宜設定可能であり、例えば、Aを含むアジド誘導体に対して、0.01~100当量、0.01~75当量、0.01~50当量、0.01~30当量、または0.01~10当量とすることができる。
本発明の一つの側面において、2以上のコアブロックは、リンカーを介して連結し、ライブラリに含まれる化合物の化学構造を構成する。一つの態様において、ライブラリを構成する化合物はリンカー形成反応によりコアブロックを連結させることにより調製することができる。ここでコアブロックは、化合物分子においてリンカーを介して連結する化学構造であれば特に限定されない。一つの態様において、ライブラリに含まれる化合物の分子構造は、リンカーにより隔てられる2以上の化学構造のそれぞれをコアブロックとして特定され、ここでリンカーは化合物合成において形成される連結部分を意味する。別の態様において、ライブラリに含まれる化合物は、ビルディングブロックの反応性官能基によるリンカー形成反応により調製し、該化合物の分枝構造のリンカー以外の2以上の化学構造を意味する。
本発明の一つの側面において、単結合がリンカーの場合、該リンカーは、C5−10アリール、5~10員ヘテロアリール、5~8員ヘテロシクリル、およびC3−10シクロアルキルから選択される2つの化学構造を連結し、該化学構造が該リンカーにより連結されるコアブロックの全部または一部を構成する。
1)2価のコアブロック:
1−1)−A1−;
1−2)−X1−Q1−X2−;または
1−3)−X3−Q2−X4−Q3−X5−;
[式中、A1はC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q1はC5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X1およびX2は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q2およびQ3は、独立に、C5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから独立に選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X3、X4およびX5は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X1、X2、X3、X4、およびX5がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリールオキシ、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1− 6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、−NR21R22、−CONR23R24、ジ(C1−6アルキル)ホスホノ、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびRcから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリルからなる群から独立に選択され;
Rcは、ニトロ、ヒドロキシ、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルコキシ、ハロゲン原子、アミノ、シアノ、1または2以上のヒドロキシ基で置換されていてもよいC1−6アルキルアミノ、1または2以上のヒドロキシ基で置換されていてもよいジ(C1−6アルキル)アミノ、C3−10シクロアルキルアミノ、(C1−6アルキル)カルボニル、(C1−6アルコキシ)カルボニル、C1−6アルキルスルホニル、C3−10シクロアルキルスルホニル、Reから選択される1または2以上の置換基で置換されていてもよいC7−14アラルキル、Reから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Reから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Reから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリル、Reから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびオキソからなる群から独立に選択され;
Rdは、ニトロ、ヒドロキシ、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルコキシ、ハロゲン原子、アミノ、シアノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、C1−6アルキルカルボニル、(C1−6アルコキシ)カルボニル、(C1−6アルコキシ)カルボニルアミノ、N−(C1−6アルコキシ)カルボニル−N−(C1−6アルキル)アミノ、C1−6アルキルスルホニル、C3−8シクロアルキルスルホニル、Reから選択される1または2以上の置換基で置換されていてもよいC7−14アラルキル、Reから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Reから選択される1または2以上の置換基で置換されていてもよいC3−8シクロアルキル、Reから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリル、およびReから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリールからなる群から独立に選択され;
Reは、ニトロ、ヒドロキシ、ハロゲン原子およびヒドロキシから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、ハロゲン原子、アミノ、シアノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、フェニルおよびヒドロキシから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルカルボニル、フェニルおよびヒドロキシから選択される1または2以上の置換基で置換されていてもよい(C1−6アルコキシ)カルボニル、C1−6アルキルスルホニル、C3−8シクロアルキルスルホニル、C1−6アルキルスルホニルアミノ、C3−8シクロアルキルスルホニルアミノ、ならびにオキソからなる群から独立に選択され;
R21は、水素原子、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員のヘテロシクリル、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルコキシ)カルボニル、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルキル)カルボニル、(C3−10シクロアルキル)カルボニル、Rdから選択される1または2以上の置換基で置換されていてもよい(C6−10アリール)カルボニル、Rcから選択される1または2以上の置換基で置換されていてもよい(3~12員の非芳香族ヘテロシクリル)カルボニル、Rgから選択される1または2以上の置換基で置換されていてもよい(5~10員のヘテロアリール)カルボニル、アミノカルボニル、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルキル)アミノカルボニル、Raから選択される1または2以上の置換基で置換されていてもよいジ(C1−6アルキル)アミノカルボニル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキルスルホニル、C7−14アラルキルスルホニル、C3−10シクロアルキルスルホニル、アミノスルホニル、C1−6アルキルアミノスルホニル、ジ(C1−6アルキル)アミノスルホニル、およびジ(C1−6アルキル)ホスホノからなる群から選択され;
R22は、水素原子、およびRaから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルからなる群から選択され;
R23は、水素原子、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよい[(C1−6アルキル)アミノ]C1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよい[ジ(C1−6アルキル)アミノ]C1−6アルキル、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびRcから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリルからなる群から選択され;
R24は、水素原子、およびRaから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルからなる群から選択される]
により表される2価のコアブロック。
2)1価のコアブロック:
2−1)−A2;
2−2)−X1−Q1−X6;または
2−3)−X3−Q2−X4−Q3−X7;
[式中、A2はC1−10アルキルであり、該アルキルはRaから独立に選択される1以上の置換基を有していてもよく;
Q1はC5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X6およびX7は、独立に、水素原子またはC1−10アルキルであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q2およびQ3は、独立に、C5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから独立に選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X1、X3およびX4は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X3、およびX4がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
上記定義において、X2、およびX5がC2−10アルキルの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリールオキシ、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、−NR21R22、−CONR23R24、ジ(C1−6アルキル)ホスホノ、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびRcから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリルからなる群から独立に選択され;
Rcは、ニトロ、ヒドロキシ、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルコキシ、ハロゲン原子、アミノ、シアノ、1または2以上のヒドロキシ基で置換されていてもよいC1−6アルキルアミノ、1または2以上のヒドロキシ基で置換されていてもよいジ(C1−6アルキル)アミノ、C3−10シクロアルキルアミノ、(C1−6アルキル)カルボニル、(C1−6アルコキシ)カルボニル、C1−6アルキルスルホニル、C3−10シクロアルキルスルホニル、Reから選択される1または2以上の置換基で置換されていてもよいC7−14アラルキル、Reから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Reから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Reから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリル、Reから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびオキソからなる群から独立に選択され;
Rdは、ニトロ、ヒドロキシ、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルコキシ、ハロゲン原子、アミノ、シアノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、C1−6アルキルカルボニル、(C1−6アルコキシ)カルボニル、(C1−6アルコキシ)カルボニルアミノ、N−(C1−6アルコキシ)カルボニル−N−(C1−6アルキル)アミノ、C1−6アルキルスルホニル、C3−8シクロアルキルスルホニル、Reから選択される1または2以上の置換基で置換されていてもよいC7−14アラルキル、Reから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Reから選択される1または2以上の置換基で置換されていてもよいC3−8シクロアルキル、Reから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリル、およびReから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリールからなる群から独立に選択され;
Reは、ニトロ、ヒドロキシ、ハロゲン原子およびヒドロキシから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、ハロゲン原子、アミノ、シアノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、フェニルおよびヒドロキシから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルカルボニル、フェニルおよびヒドロキシから選択される1または2以上の置換基で置換されていてもよい(C1−6アルコキシ)カルボニル、C1−6アルキルスルホニル、C3−8シクロアルキルスルホニル、C1−6アルキルスルホニルアミノ、C3−8シクロアルキルスルホニルアミノ、ならびにオキソからなる群から独立に選択され;
R21は、水素原子、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員のヘテロシクリル、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルコキシ)カルボニル、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルキル)カルボニル、(C3−10シクロアルキル)カルボニル、Rdから選択される1または2以上の置換基で置換されていてもよい(C6−10アリール)カルボニル、Rcから選択される1または2以上の置換基で置換されていてもよい(3~12員の非芳香族ヘテロシクリル)カルボニル、Rgから選択される1または2以上の置換基で置換されていてもよい(5~10員のヘテロアリール)カルボニル、アミノカルボニル、Raから選択される1または2以上の置換基で置換されていてもよい(C1−6アルキル)アミノカルボニル、Raから選択される1または2以上の置換基で置換されていてもよいジ(C1−6アルキル)アミノカルボニル、1または2以上のハロゲン原子で置換されていてもよいC1−6アルキルスルホニル、C7−14アラルキルスルホニル、C3−10シクロアルキルスルホニル、アミノスルホニル、C1−6アルキルアミノスルホニル、ジ(C1−6アルキル)アミノスルホニル、およびジ(C1−6アルキル)ホスホノからなる群から選択され;
R22は、水素原子、およびRaから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルからなる群から選択され;
R23は、水素原子、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよい[(C1−6アルキル)アミノ]C1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよい[ジ(C1−6アルキル)アミノ]C1−6アルキル、Rcから選択される1または2以上の置換基で置換されていてもよいC3−10シクロアルキル、Rdから選択される1または2以上の置換基で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、およびRcから選択される1または2以上の置換基で置換されていてもよい3~12員の非芳香族ヘテロシクリルからなる群から選択され;
R24は、水素原子、およびRaから選択される1または2以上の置換基で置換されていてもよいC1−6アルキルからなる群から選択される]
により表される1価のコアブロック。
前記2価のコアブロックにおいて、任意の位置にさらに1つのリンカー結合位置を有する3価のコアブロック。
1)2価のコアブロック:
1−1)−A1−;
1−2)−X1−Q1−X2−;または
1−3)−X3−Q2−X4−Q3−X5−;
[式中、A1はC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q1はC5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X1およびX2は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q2およびQ3は、独立に、C5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから独立に選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X3、X4およびX5は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X1、X2、X3、X4、およびX5がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールオキシ、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、1または2以上のハロゲン原子で置換されていてもよい5~10員のヘテロアリール、および3~12員の非芳香族ヘテロシクリルからなる群から独立に選択される]
により表される2価のコアブロック。
2−1)−A2;
2−2)−X1−Q1−X6;または
2−3)−X3−Q2−X4−Q3−X7;
[式中、A2はC1−10アルキルであり、該アルキルはRaから独立に選択される1以上の置換基を有していてもよく;
Q1はC5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X6およびX7は、独立に、水素原子またはC1−10アルキルであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q2およびQ3は、独立に、C5−10アリーレン、5~10員ヘテロアリーレン、5~8員非芳香族ヘテロシクリレン、およびC3−10シクロアルキレンから独立に選択され、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X1、X3およびX4は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X3、およびX4がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
上記定義において、X2、およびX5がC2−10アルキルの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールオキシ、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、1または2以上のハロゲン原子で置換されていてもよい5~10員のヘテロアリール、および3~12員の非芳香族ヘテロシクリルからなる群から独立に選択される]
により表される1価のコアブロック。
1)2価のコアブロック:
1−1)−A1−;
1−2)−X1−Q1−X2−;または
1−3)−X3−Q2−X4−Q3−X5−;
[式中、A1はC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
Q1、Q2およびQ3は、ベンゼン環、ピリジン環、ピペリジン環、ピペラジン環、ピリミジン環、ピラジン環、ピリダジン環、ピラゾール環、イミダゾール環、チオフェン環、フラン案、およびナフタレン環からなる群から選択される環からなる2価の基であり、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X1およびX2は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
X3、X4およびX5は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X1、X2、X3、X4、およびX5がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールオキシ、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、1または2以上のハロゲン原子で置換されていてもよい5~10員のヘテロアリール、および3~12員の非芳香族ヘテロシクリルからなる群から独立に選択される]
により表される2価のコアブロック。
2−1)−A2;
2−2)−X1−Q1−X6;または
2−3)−X3−Q2−X4−Q3−X7;
[式中、A2はC1−10アルキルであり、該アルキルはRaから独立に選択される1以上の置換基を有していてもよく;
Q1、Q2およびQ3は、ベンゼン環、ピリジン環、ピペリジン環、ピペラジン環、ピリミジン環、ピラジン環、ピリダジン環、ピラゾール環、イミダゾール環、チオフェン環、フラン案、およびナフタレン環からなる群から選択される環からなる2価の基であり、該基はそれぞれRbから独立に選択される1以上の置換基を有していてもよく;
X6およびX7は、独立に、水素原子またはC1−10アルキルであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
X1、X3およびX4は、独立に、単結合またはC1−10アルキレンであり、該アルキレンはRaから独立に選択される1以上の置換基を有していてもよく;
上記定義において、A1、X3、およびX4がC2−10アルキレンの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
上記定義において、X2、およびX5がC2−10アルキルの場合、該アルキレンに−O−、−S−、または−NR11−から選択される1~3の基が挿入されていてもよく、R11は水素原子またはC1−6アルキルであり;
Raは、ハロゲン原子、ヒドロキシ、C1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、(C1−6アルコキシ)カルボニルアミノ、(C1−6アルキル)カルボニルアミノ、アミノ、C1−6アルキルアミノ、ジ(C1−6アルキル)アミノ、アミノカルボニル、(C1−6アルキル)アミノカルボニル、ジ(C1−6アルキル)アミノカルボニル、C1−6アルキルスルホニルアミノ、C3−10シクロアルキルスルホニルアミノ、C7−14アラルキル、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、Rcから選択される1または2以上の置換基で置換されていてもよい5~10員のヘテロアリール、Rcから選択される1または2以上の置換基で置換されていてもよい4~12員の非芳香族ヘテロシクリル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールオキシ、1または2以上のハロゲン原子で置換されていてもよいC6−10アリールスルファニル、およびアダマンチルからなる群から独立に選択され;
Rbは、ハロゲン原子、ヒドロキシ、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルキル、Raから選択される1または2以上の置換基で置換されていてもよいC1−6アルコキシ、シアノ、(C1−6アルコキシ)カルボニル、カルボキシ、C3−10シクロアルキル、1または2以上のハロゲン原子で置換されていてもよいC6−10アリール、1または2以上のハロゲン原子で置換されていてもよい5~10員のヘテロアリール、および3~12員の非芳香族ヘテロシクリルからなる群から独立に選択される]
により表される1価のコアブロック。
酸としては通常使用できるものであれば特に限定されないが、例えば、塩酸、臭化水素酸、硫酸、メタンスルホン酸、p−トルエンスルホン酸、カンファ—スルホン酸、酢酸、テトラフルオロほう酸などを使用することができる。
反応時間は当業者が適宜設定可能であり、例えば、該当の基質、もしくは類似基質を用いた検討実験(モデル実験とも呼ぶ)において充分な変換率に達するのに要した時間、もしくは検討結果から何らかの手法によって予想される時間、または一部の固相担体から生成物の切り出しを行って反応終了時点を決定または確認することができる。反応終了後、通常の後処理方法にて目的の化合物を得ることができる。
スルホンアミド基に導入する保護基として特に限定されないが、例えば、PMB基、DMB基、Trt基、SEM基、アリル基、ベンジル基、などが挙げられる。
反応性官能基に導入した保護基を脱保護する工程について、例えば、脱保護により生じる基としてアミノ基、カルボキシ基、ヒドロキシ基、ホルミル基、ケトン基、チオール基などが挙げられる。アミノ基から取り除かれる保護基として例えば、Boc基、Fmoc基、Cbz基、Teoc基、Troc基、Alloc基、Trt基、Ns基、DNs基、Ts基、PMB基、DMB基、トリフルオロアセチル基、THP基、SEM基などが挙げられる。
ヒドロキシ基から取り除かれる保護基として、例えば、アセチル基、ピバロイル基、t−ブチル基、トリチル基、2−クロロトリチル基、THP基、MOM基、TMS基、TES基、トリイソプロピルシリル基、TBDMS基、TBDPS基、SEM基、tert−ブチルジフェニルシリリルエチル基、アリル基、ベンジル基、などが挙げられる。
スルホンアミド基から取り除かれる保護基として特に限定されないが、例えば、PMB基、DMB基、Trt基、SEM基、アリル基、ベンジル基、などが挙げられる。
ピラゾール環、イミダゾール環、インドール環、インダゾール環などの窒素原子から取り除かれる保護基として特に限定されないが、例えば、PMB基、DMB基、Trt基、SEM基、アリル基、ベンジル基、THP基などが挙げられる。






明細書中もしくは実施例中で使用している「cpKa」はpKaの計算値を示し、ADMET predictorのバージョン9.5を用いて計算した値を利用した。
本発明の内容を以下の実施例及び参考例でさらに説明するが、本発明はその内容に限定されるものではない。
実施例中にて使用した0.04MのP(Cy)3Pd G3の1,4−ジオキサン溶液は以下のように調製した。窒素雰囲気下、2mLのガラスバイアルにP(Cy)3Pd G3(26mg、0.04mmol)に1,4−ジオキサン(1mL)を加えて振とうすることで溶解し調製した。


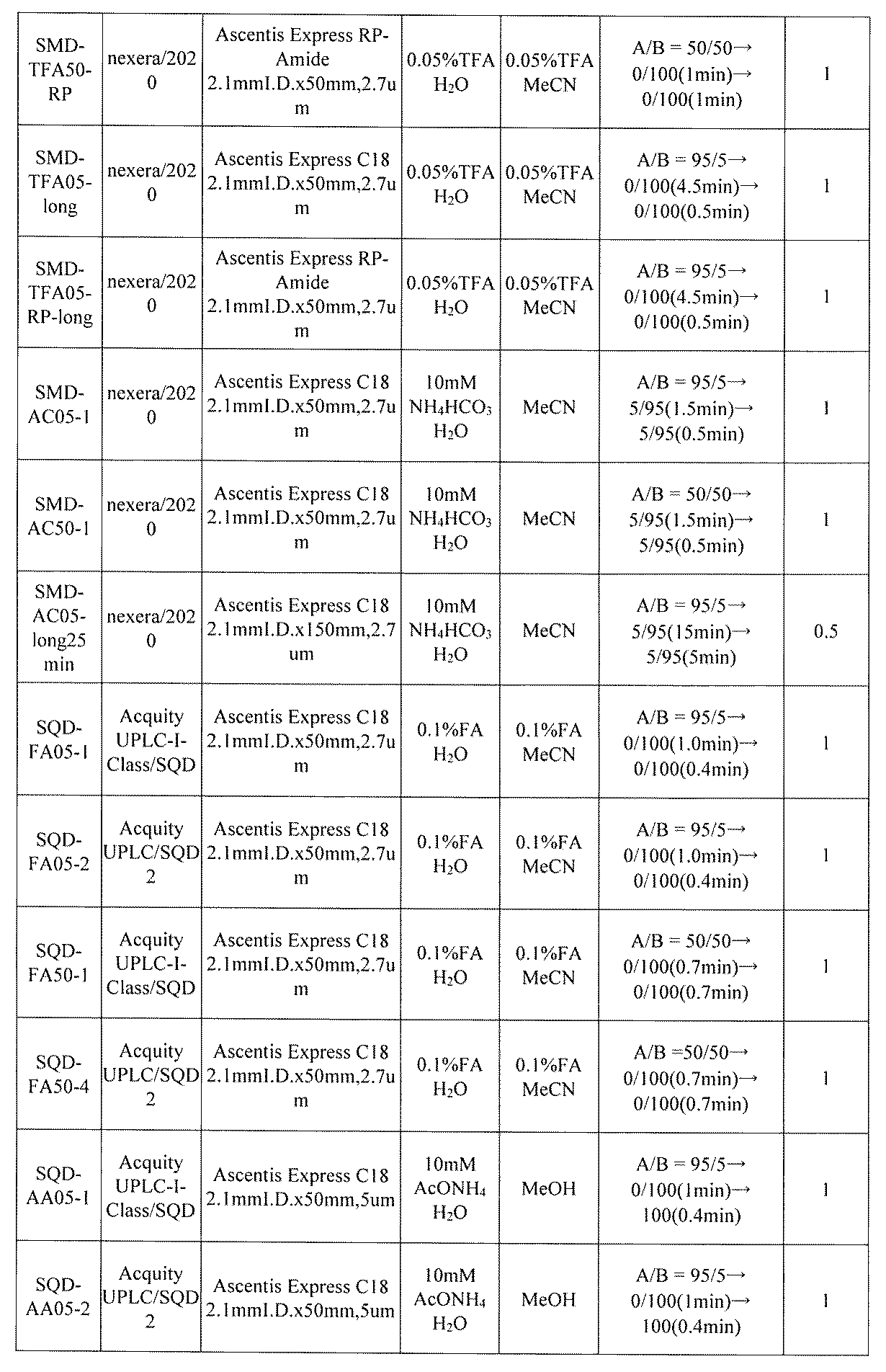


「変換率」および「転換率」(%)=(目的物のUV面積%)/((出発物質のUV面積%)+(目的物のUV面積%)+(副生成物のUV面積%))×100
「濃縮」、「減圧濃縮」または「減圧下濃縮」という表現は、ロータリー・エバポレーター、機械式オイル真空ポンプもしくは機械式オイルフリー真空ポンプによる減圧下での溶媒の蒸発除去を示す。
「減圧下で一晩乾燥」という表現は、ロータリー・エバポレーター、機械式オイル真空ポンプもしくは機械式オイルフリー真空ポンプによる減圧下での溶媒の蒸発除去を示す。
固相反応は、任意の適切な容器、例えばテフロンパッキンを備えたキャップにより密栓が可能なガラスバイアル、フリットフィルタおよび適切な栓を有するカラム中で実施することができる。容器サイズは、溶媒にとって十分な空間が存在し、かつ、ある一定の樹脂は、有機溶媒で処理すると、著しく膨潤する可能性があることを考慮して、樹脂が効果的に撹拌されるのに十分な余地があるようなサイズを適宜選択する。
固相反応における溶媒の使用量は、反応に使用したレジンの重量に対して任意の倍量の容量(v/w)を利用した。たとえば、20mgのレジンを使用し、20v/wの溶媒を使用した場合には、20mg×20v/w=400μLの溶媒を使用した。




実施例8、9および10にて合成している混合物を示す場合には、混合物番号を利用することもある。例えば、「2−1−a1B01−0」の「2−1」はライブラリの種類を示し、「a1B01」は混合物の種類を示す。「−0」の「−0」は固相に担持された状態を示し、「−1」の場合には固相から切り出した状態を示す。また、「−2」の場合には固相からの切り出し後に官能基変換を行った状態を示す。混合物に含まれる各化合物を示す際は、IDを利用することもある。例えば、ID「2−1−a1B01−0−0001」の場合、「0001」は混合物「2−1−a1B01−0」中に含まれる任意の1化合物を特定する番号であり、IDにより混合物および含まれる化合物を特定する。
「2−1−a1B01−0−0001」の表記例
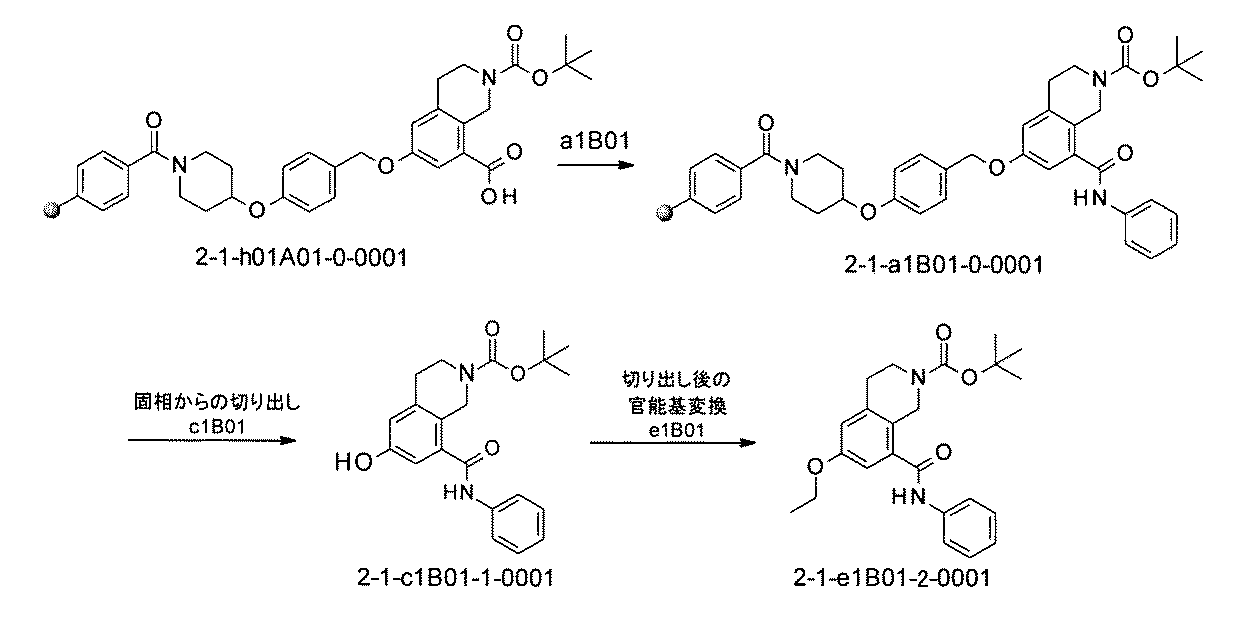
実施例1−1:主に実施例2−1で用いる反応検討用基質、ならびに混合物ライブラリで用いるビルディングブロックなどの合成

LCMS(ESI) m/z=279.1[M+H]+。
保持時間:0.75分(分析条件SQD−FA05−1)。

LCMS(ESI) m/z=293.2[M+H]+。
保持時間:0.78分(分析条件SQD−FA05−1)。

LCMS:m/z 554[M+Na]+。
保持時間:1.684分(分析条件 SMD−FA05−1、290nm)。

LCMS:m/z 432[M+H]+。
保持時間:0.904分(分析条件 SMD−FA05−1、290nm)。
実施例1−1−4で合成法を例示したD067は、これ以降の実施例において、化合物C102と表記することもある。

実施例1−1−5で合成法を例示したD068は、これ以降の実施例において、化合物C101と表記することもある。

反応液と固相の懸濁液を全てフィルター上に移し、NMP(90mL)で3回、メタノール(90mL)で3回、DCM(90mL)で3回、ヘプタン(90mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C002−03R(担持量0.507mmol/g、6.87g、3.48mmol)を得た。
反応液と固相の懸濁液(0.010mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.2Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、ろ液にNMP(0.25mL)を加えた。得られた溶液のうち0.050mLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C002が100%観測された。
LRMS:m/z 229[M+H]+。
保持時間:1.006分(分析条件SMD−FA05−1、280nm)。
反応液と固相の懸濁液を全てフィルター上に移し、NMP(130mL)で3回、水(130mL)で3回、HOAt(0.2M、NMP溶液 130mL)で3回、NMP(130mL)で3回、メタノール(130mL)で3回、DCM(130mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C003−03R(担持量0.511mmol/g、5.88g、3.00mmol)を得た。
反応液と固相の懸濁液(0.010mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.2Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、ろ液にNMP(0.25mL)を加えた。得られた溶液のうち0.050mLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C003が100%観測された。
極大波長:267nm。
保持時間:0.806分(分析条件SMD−FA05−1、280nm)。
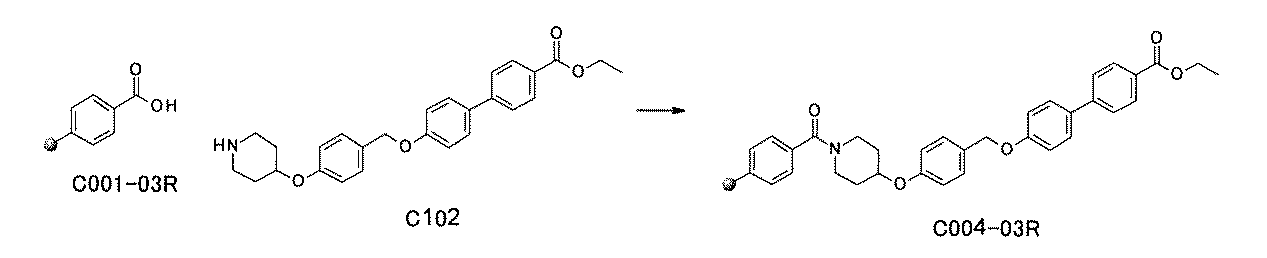
反応液と固相の懸濁液を全てフィルター上に移し、NMP(20mL)で3回、メタノール(20mL)で3回、DCM(20mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C004−03R(担持量0.200mmol/g、1.25g、0.250mmol)を得た。
反応液と固相の懸濁液(10μL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をNMP(0.05mL)で洗浄し、ろ液にアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C004が100%観測された。

LRMS:m/z 243[M+H]+。
保持時間:1.085分(分析条件SMD−FA05−1、299nm)。

反応液と固相の懸濁液を全てフィルター上に移し、NMP(20mL)で3回、水(20mL)で3回、HOAt(0.2M、NMP溶液、20mL)で3回、NMP(20mL)で3回、メタノール(20mL)で3回、DCM(20mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C005−03R(担持量0.201mmol/g、1.16g、0.232mmol)を得た。
反応液と固相の懸濁液(10μL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をNMP(0.05mL)で洗浄し、ろ液にアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C005が100%観測された。
極大波長:293nm。
保持時間:0.775分(分析条件SMD−FA05−1、299nm)。
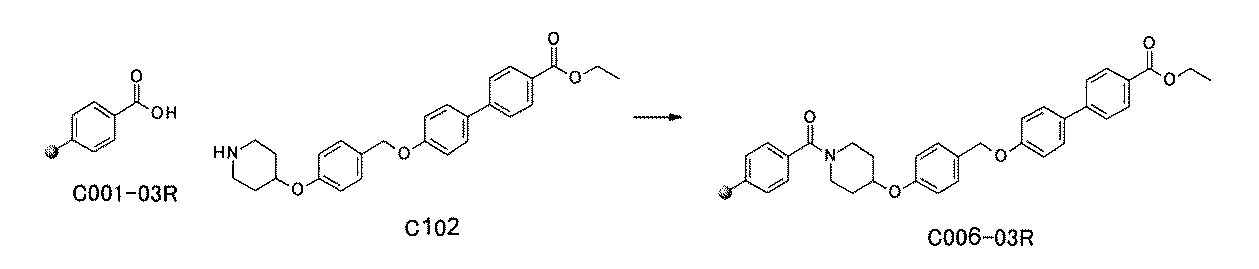
反応液と固相の懸濁液を全てフィルター上に移し、NMP(20mL)で3回、メタノール(20mL)で3回、DCM(20mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C006−03R(担持量0.050mmol/g、1.18g、0.059mmol)を得た。

反応液と固相の懸濁液を全てフィルター上に移し、NMP(20mL)で3回、水(20mL)で3回、HOAt(0.2M、NMP溶液、20mL)で3回、NMP(20mL)で3回、メタノール(20mL)で3回、DCM(20mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C007−03R(担持量0.050mmol/g、1.10g、0.055mmol)を得た。

反応液と固相の懸濁液をNMP(0.6mL)を用いてフィルター上に移し、NMP(0.6mL)で2回、メタノール(0.6mL)で3回、DCM(0.6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物AA001−03R(担持量0.050mmol/g、25.6mg、1.28mmol)を得た。
反応液と固相の懸濁液(0.020mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液50μLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物AA001が88.9%の純度で観測された。

LRMS:m/z 283[M+H]+。
保持時間:0.471分(分析条件SMD−FA05−1、280nm)。
実施例1−1−12で合成法を例示したAA001−03Rは、これ以降の実施例において、化合物D021−03Rと表記することもある。

反応液と固相の懸濁液をNMP(5.96mL)を用いてフィルター上に移し、NMP(5.96mL)で2回、水で(5.96mL)で3回、HOAt(0.2M、NMP溶液、5.96mL)で3回、NMP(5.96mL)で3回、メタノール(5.96mL)で3回、DCM(5.96mL)で3回、ヘプタン(5.96mL)で3回、洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物AA003−03R(担持量0.189mmol/g、275.2mg、52.0μmol)を得た。
反応液と固相の懸濁液(0.020mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液50μLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物AA003が95.6%の純度で観測された。

LCMS:m/z 448[M+H]+。
保持時間:0.817分(分析条件SMD−FA05−1、280nm)。
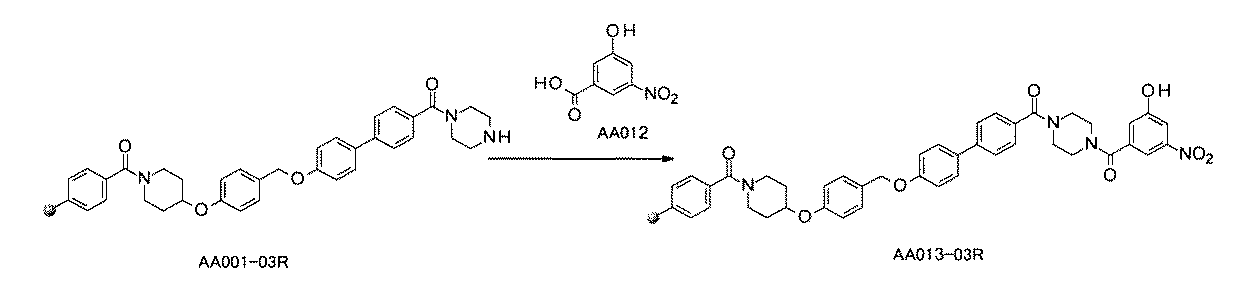
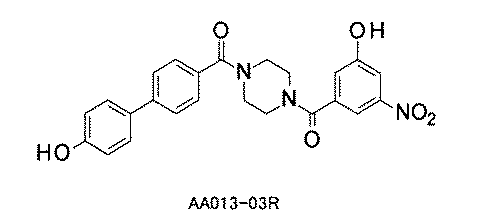
LCMS:m/z 448[M+H]+。
保持時間:0.852分(分析条件SMD−FA05−1、280nm)。
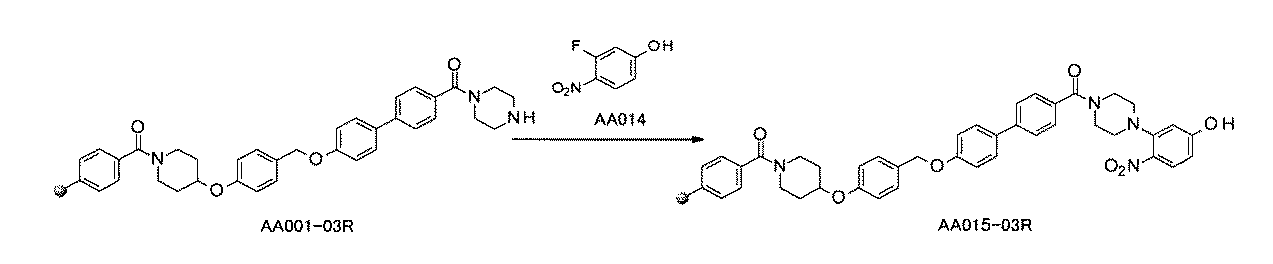
反応液と固相の懸濁液をNMP(0.2mL)を用いてフィルター上に移し、NMP(0.2mL)で2回、水で(0.2mL)で3回、NMP(0.2mL)で3回、HOAt(0.2M、NMP溶液、0.2mL)で3回、NMP(0.2mL)で3回、メタノール(0.2mL)で3回、DCM(0.2mL)で3回、ヘプタン(0.2mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物AA015−03R(担持量0.189mmol/g、10.0mg、1.89μmol)を得た。
反応液と固相の懸濁液(0.020mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液50μLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物AA015が96.9%の純度で観測された。

LCMS:m/z 420[M+H]+。
保持時間:0.937分(分析条件SMD−FA05−1、280nm)。
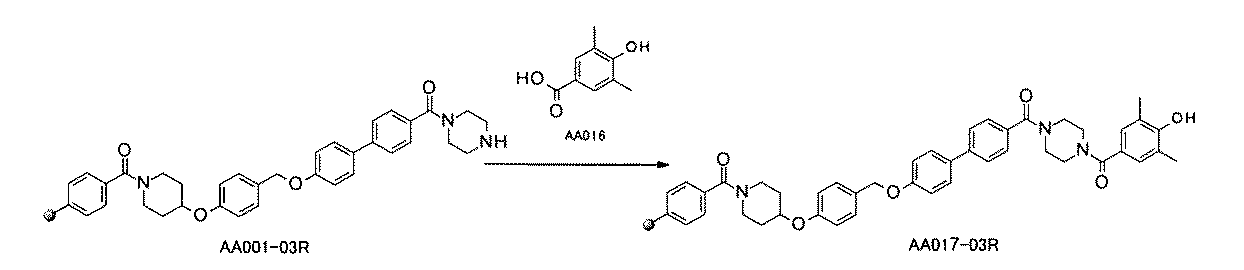

LCMS:m/z 431[M+H]+。
保持時間:0.843分(分析条件SMD−FA05−1、280nm)。
実施例1−2−1:4−(4−ブロモフェニル)−1−ヒドロキシ−N,N−ジメチル−2−ナフトアミド(化合物E002)の合成法
実施例1−2−7および実施例1−2−9で使用したナフトール試薬(化合物E002、化合物E003)は以下に示す方法で合成した。

4−ブロモ−1−ヒドロキシ−N,N−ジメチル−2−ナフトアミド(化合物E004)(60g、204mmol)のDMA(510mL)溶液に1−(クロロメチル)−4−メトキシベンゼン(33.3mL,245mmol)とtBuOK(25.2g,224mmol)を室温で加えた。90℃で1時間30分攪拌した後、反応溶液に水(1L),EtOAc(300mL)とヘキサン(200mL)を加え、有機層を抽出した。有機層を食塩水で洗浄した後、減圧濃縮し、標題化合物(化合物E005)を粗生成物として得た。これ以上の精製は実施せずに次の反応を行った。
LCMS:m/z 412[M—H]−。
保持時間:1.31分(分析条件 SMD−FA05−1)。
窒素雰囲気下、4−ブロモ−1−((4−メトキシベンジル)オキシ)−N,N−ジメチル−2−ナフトアミド(化合物E005)(85g,205mmol)、ビス(ピナコレート)ジボロン,Pd(dppf)Cl2・DCM(8.38g,10.3mmol)と酢酸カリウム(60.4g,615mmol)にジオキサン(1026mL)を室温で加え、90℃で2時間加熱し、標題化合物(化合物E006)を粗生成物の溶液として得た。これ以上の精製は実施せずに次の反応を行った。
LCMS:m/z 462[M+H]+。
保持時間:1.45分(分析条件 SMD−FA05−1)。
窒素雰囲気下、前工程(実施例1−2−2)で得た、1−((4−メトキシベンジル)オキシ)−N,N−ジメチル−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−2−ナフトアミド(化合物E006)(95g,205mmol)を含む反応溶液に1−ブロモ−4−ヨードベンゼン(69.6g、246mmol)とPd(dppf)Cl2・DCM(8.37g,10.3mmol)を室温で加え攪拌した。反応溶液にリン酸カリウム(131g,615mmol)と水(137mL)を室温で加えた後、90℃で2時間攪拌した。反応溶液の有機層をシリカゲル(300mL)でろ過し、酢酸エチル(800mL)で溶出した。
ろ液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製し(酢酸エチル/ヘキサン,0−35%)、茶色アモルファスの粗生成物を得た。得られた粗生成物を酢酸エチル(20mL)とヘキサン(180mL)の混合溶媒で粉砕洗浄し、得られた個体をろ過して溶媒を除去し、白色固体の標題化合物(化合物E007、51%、50.7g、103mmol)を得た。
LCMS:m/z 488[M—H]−。
保持時間:1.50分(分析条件 SMD−FA05−1)。
(4−(4−ブロモフェニル)−1−((4−メトキシベンジル)オキシ)−N,N−ジメチル−2−ナフトアミド)(62.6g,128mmol)(化合物E007)のDCM(638mL)溶液にTFA(48.8mL,638mmol)を加え、室温で1時間攪拌した。反応溶液を減圧下濃縮した後に、酢酸エチル(200mL)を加え再び減圧下濃縮した。ヘキサン(500mL)を加えて粉砕洗浄し、得られた個体を酢酸エチル(30mL)とヘキサン(270mL)の混合溶媒で洗浄し、白色固体を得た。得られた個体を酢酸エチル(500mL)に溶解し、炭酸水素ナトリウム水溶液(300mL)を加えた。有機層を抽出し、減圧下濃縮した。残渣を酢酸エチル(50mL)とヘキサン(500mL)で粉砕洗浄し、白色固体の標題化合物(化合物E002、85%、40.1g、103mmol)を得た。
LCMS:m/z 370[M+H]+。
保持時間:1.41分(分析条件 SMD−FA05−1)。
5−ブロモ−2−ヨードピリジンを用いて、実施例1−2−4(化合物E007)、実施例1−2−5(化合物E002)と同様の方法で合成した。

LCMS:m/z 491[M+H]+。
保持時間:1.32分(分析条件 SMD−FA05−1)。
LCMS:m/z 371[M+H]+。
保持時間:1.17分(分析条件 SMD−FA05−1)。

レジンに担持された担持量(mmol/g)は、以下の方法で決定した。
以下は実験操作の一例であり、全てのロットに関する担持量を一律で規定するものではない。

LCMS:m/z 370.0,372.0[M+H]+
保持時間:1.45分。

(担持量:0.37mmol/g)
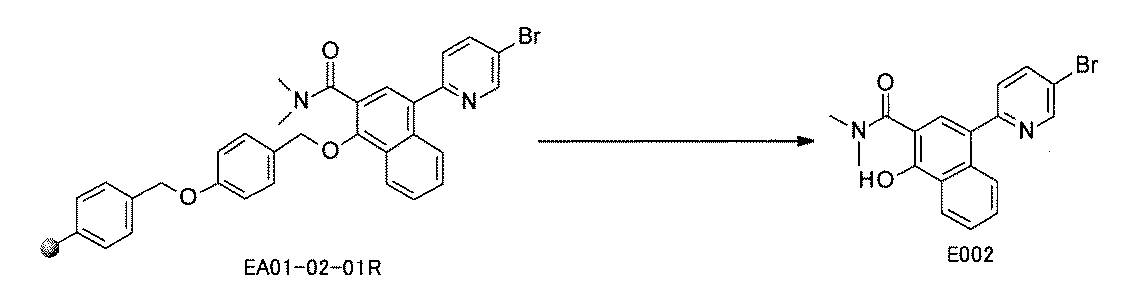
保持時間:1.21分。
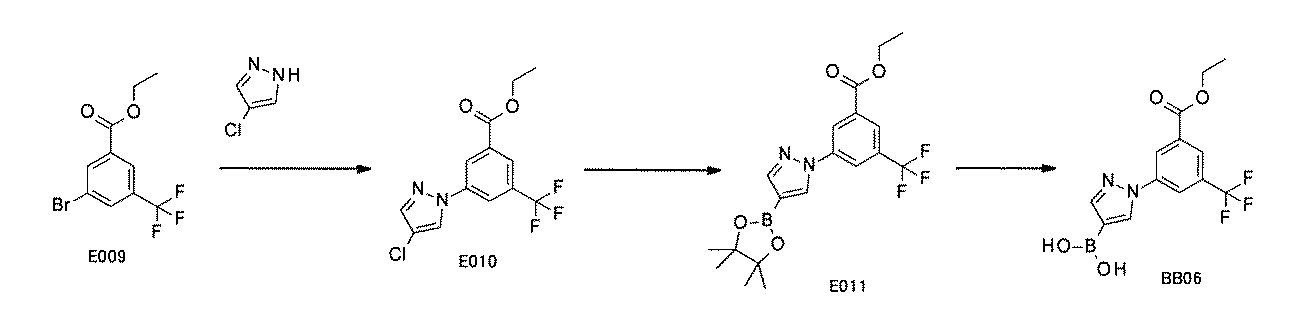
3−ブロモ−5−(トリフルオロメチル)安息香酸エチル(化合物E009)(2.08g,7mmol),4−クロロ−1H−ピラゾール(1.08g,10.5mmol),ヨウ化銅(0.133g,0.700mmol),N,N’−ジメチルエチレンジアミン(0.30mL,2.8mmol)と炭酸カリウム(2.90g,21.00mmol)のジオキサン(17.5mL)溶液を、3回窒素置換した。110℃で16時間攪拌した。混合物をセライトでろ過し、酢酸エチル(25mL)で5回洗浄した。ろ液を塩化アンモニウム水溶液(40mL)で2回、水(40mL)で1回、飽和食塩水(40mL)で1回洗浄した後、硫酸ナトリウムで乾燥し、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製し(酢酸エチル/ヘキサン,20%)、白色固体の標題化合物(化合物E010、87%、1.93g、6.07mmol)を得た。
LCMS:m/z 319[M+H]+。
保持時間:1.33分(分析条件 SMD−TFA05−1)。
(3−(4−クロロ−1H−ピラゾール−1−イル)−5−(トリフルオロメチル)安息香酸エチル(化合物E010)(1.93g,6.06mmol),ビス(ピナコラート)ジボロン(1.92g,7.57mmol)、RuPhos(424mg,0.908mmol),アリルパラジウム(II)クロリド ダイマー(111mg,0.303mmol)と酢酸カリウム(1.49g,15.1mmol)のジオキサン(30mL)溶液を、窒素置換した。90℃で16.5時間攪拌した。混合物をセライトでろ過し、酢酸エチル(20mL)で5回洗浄した。ろ液を飽和食塩水(40mL)で洗浄した後、硫酸ナトリウムで乾燥し、減圧下濃縮した。得られた淡黄色油状の標題化合物(化合物E011)は精製せず、混合物のまま次の工程に用いた。
LCMS:m/z 411[M+H]+。
保持時間:1.53分(分析条件 SMD−FA05−1)。
前工程(実施例1−2−13)で得られた3−(4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1H−ピラゾール−1−イル)−5−(トリフルオロメチル)安息香酸エチル(化合物E011)(6.06mmol相当)と、過ヨウ素酸ナトリウム(6.48g,30.3mmol)、酢酸アンモニウム(2.34g,30.3mmol)、アセトン(40mL)、水(20mL)を室温で19時間攪拌した。混合物に酢酸エチル(100mL)と水(50mL)を加えた。有機層を抽出後、さらに酢酸エチル(100mL)で2回抽出した。有機層を硫酸ナトリウムで乾燥し、減圧下濃縮した。残渣を逆相カラムクロマトグラフィー(0.1%ギ酸アセトニトリル溶液/0.1%ギ酸水溶液、50−100%)にて精製した。得られた固体を桐山ロートを使い、水(15mL)で5回洗浄した。得られた固体を減圧下乾燥し、標題化合物(BB06、55%、1.09g、3.32mmol)を得た。
LCMS:m/z 329[M+H]+。
保持時間:1.02分(分析条件 SMD−FA05−1)。
実施例1−3−1:3−エチニル−5−(トリフルオロメチル)安息香酸エチル(BB25)の合成
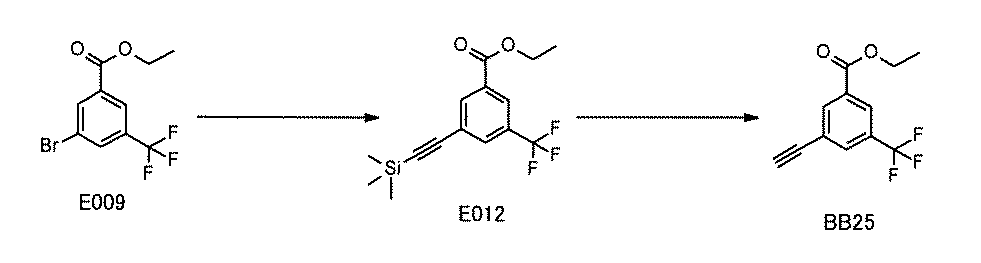
窒素雰囲気下、3−ブロモ−5−(トリフルオロメチル)安息香酸エチル(化合物E009、1.49g,5mmol),トリメチルシリルアセチレン(1.39mL,10.0mmol),PdCl2(PPh3)2(0.175g,0.25mmol),ヨウ化銅(48mg,0.25mmol)のトリエチルアミン(14.9mL,107mmol)溶液を、90℃で2.5時間攪拌した。混合物をセライトでろ過し、酢酸エチル(20mL)で4回洗浄した。ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(NH−シリカゲル)で精製(酢酸エチル/ヘキサン,10%)し、淡茶色オイルの標題化合物(化合物E012、104%、1.64g、5.22mmol)を得た。
保持時間:1.70分(分析条件 SMD−FA05−1)。
1H−NMR(400MHz,DMSO−d6)δ:8.18(s,1H),8.15(s,1H),8.10(s,1H),4.36(q,J=7.0Hz,2H),1.35(t,J=7.0Hz,3H),0.26(s,9H)。
3−(トリフルオロメチル)−5−((トリメチルシリル)エチニル)安息香酸エチル(化合物E012、1.64g、5.22mmol)と炭酸カリウム(0.721g,5.22mmol)のエタノール(20.8mL)溶液を室温で1時間半攪拌した。混合物をセライトでろ過し、メタノール(20mL)で3回、酢酸エチル(20mL)で3回洗浄した。ろ液を濃縮した。残渣をDMSOと0.1%ギ酸水溶液に溶解し、逆相カラムクロマトグラフィーで精製(0.1%ギ酸アセトニトリル溶液/0.1%ギ酸水溶液、50−100%)した。濃縮後、得られた残渣に酢酸エチルと水を加えた。有機層を抽出し、炭酸水素ナトリウム水溶液と飽和食塩水で洗浄し、硫酸ナトリウムで乾燥した。濃縮し、淡茶色オイルの標題化合物(BB25、89%、1.12 g、4.62mmol)を得た。
保持時間:1.32分(分析条件 SMD−FA05−1)
極大波長:213nm
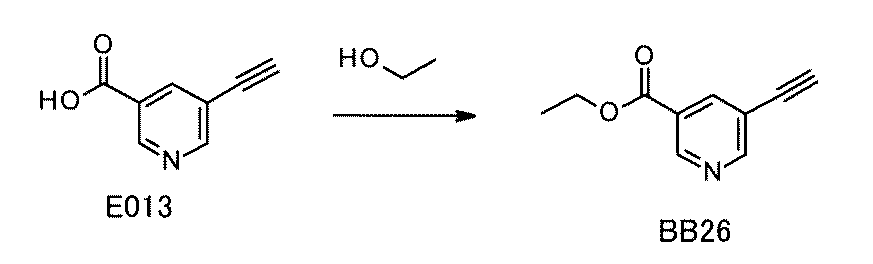
LCMS:m/z 176[M+H]+。
保持時間:0.91分(分析条件 SMD−FA05−1)。
実施例1−4−1:4−(ピペラジン−1−イル)安息香酸アリル(BB27)の合成法
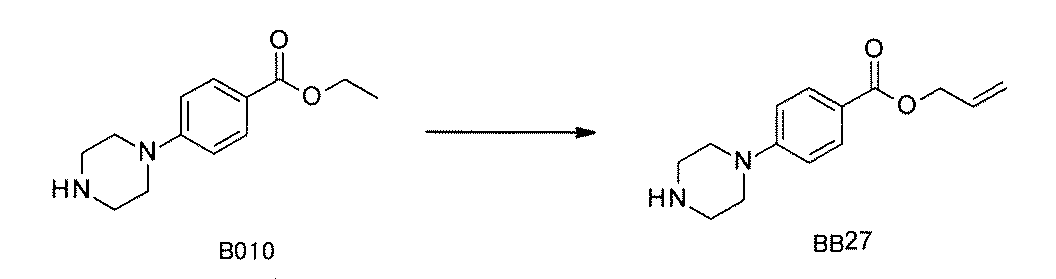
LCMS(ESI,m/z):247.1[M+H]+。
保持時間:0.595分(分析条件 SMD−FA05−1)。
実施例1−4−1で合成法を例示したBB27は、これ以降の実施例において、化合物D049と表記することもある。
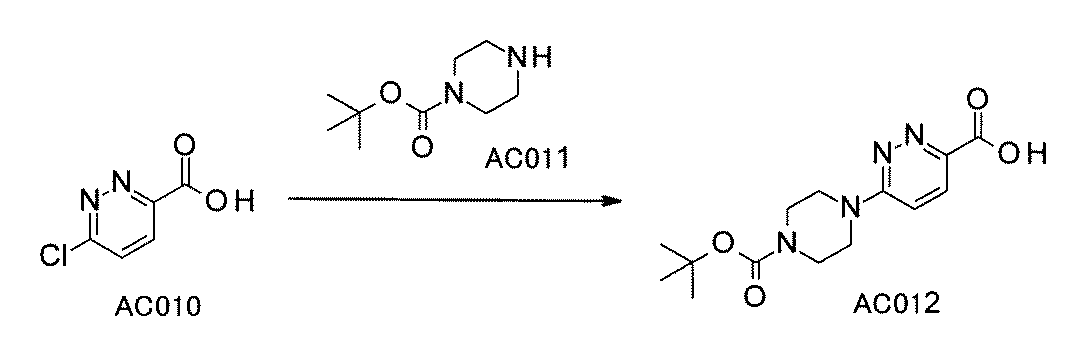
LCMS:m/z 309.2[M+H]+。
保持時間:0.70分(分析条件 SMD−FA05−1)。
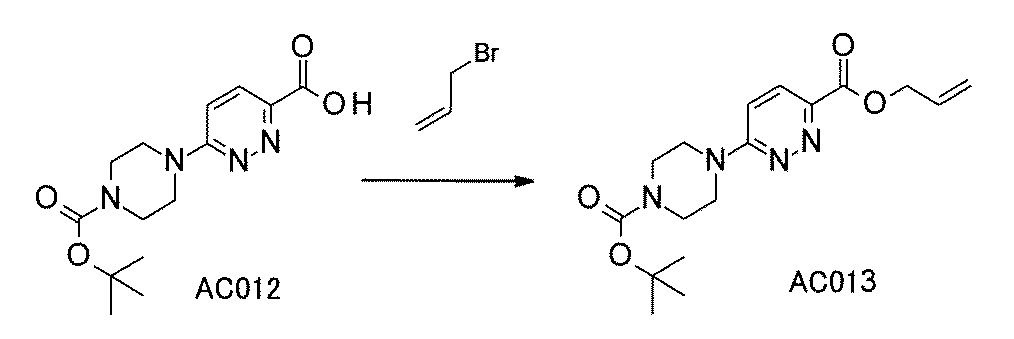
LCMS:m/z 349.2[M+H]+。
保持時間:1.06分(分析条件 SMD−FA05−1)。
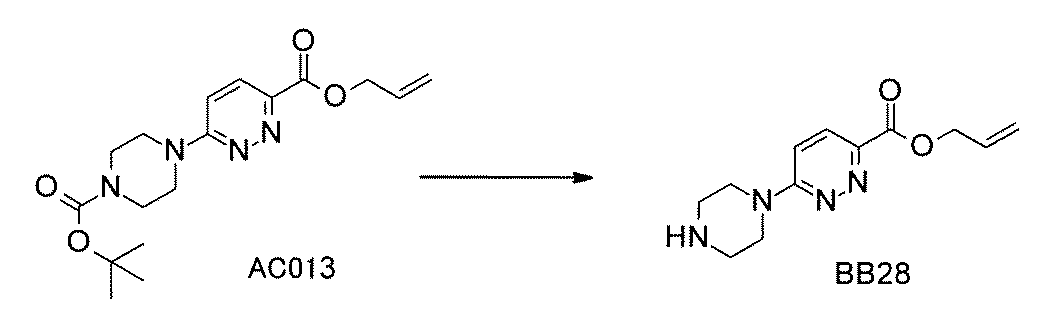
LCMS:m/z 249.2[M+H]+。
保持時間:0.39、0.46分(二峰性のピーク形状のため)(分析条件 SMD−FA05−1)。

保持時間:0.53分(分析条件 SMD−FA05−1)。
1H−NMR(400MHz,CDCl3)δ:7.83(d,J=9.7Hz,1H),6.83(d,J=9.7Hz,1H),3.77−3.75(m,4H),3.01−2.99(m,4H)。

保持時間:0.46、0.48分(二峰性のピーク形状のため)(分析条件 SMD−FA05−1)。
1H−NMR(400MHz,CDCl3)δ:7.87(d,J=9.7Hz,1H),6.84(d,J=9.7Hz,1H),5.37−5.28(m,1H),3.78−3.75(m,4H),3.01−2.99(m,4H),1.41(d,J=6.4Hz,6H)。
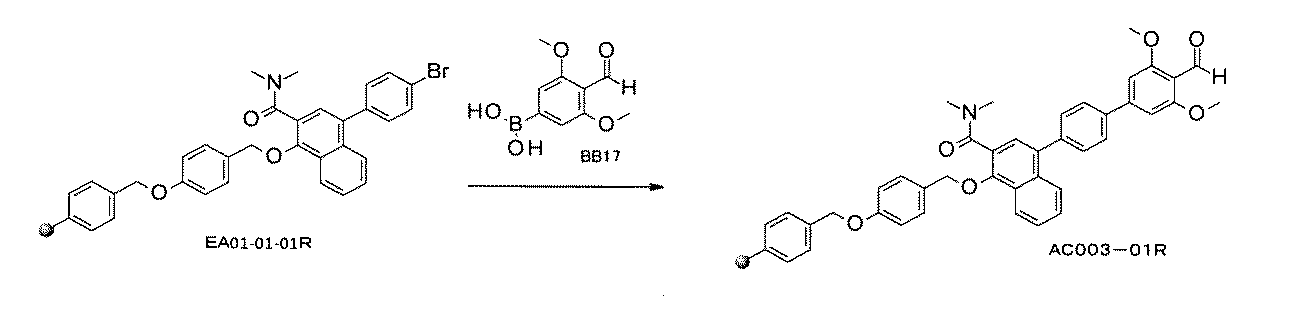
任意の時間経過時に、窒素雰囲気下で反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、メタノール(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(1000μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.30分。

任意の時間経過時に、窒素雰囲気下で反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、メタノール(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させた。その溶液(50μL)をアセトニトリル(250μL)にて希釈してLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.14分。

任意の時間経過時に、窒素雰囲気下で反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、メタノール(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.10分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50 μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.50分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.30分。


LCMS:m/z[M+H]+411.1。
保持時間:1.164分(分析条件 SMD−FA05−1,319nm)。
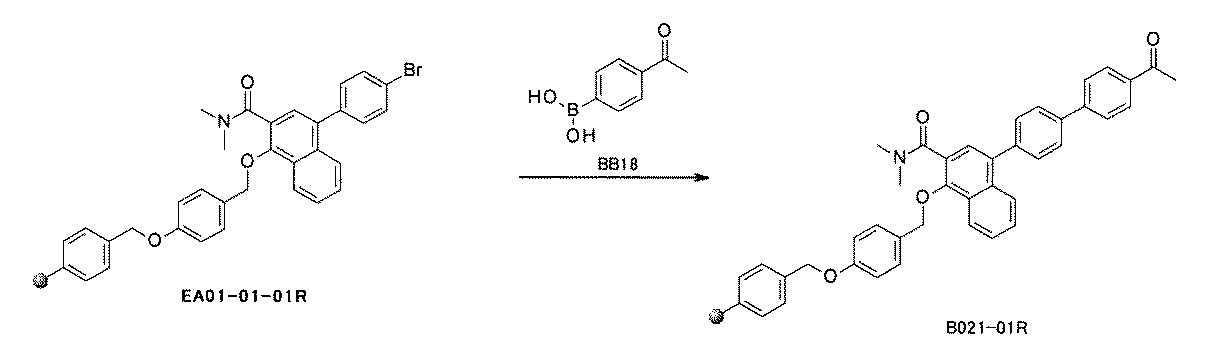

LCMS:m/z 410.2[M+H]+。
保持時間:1.368分(分析条件 SMD−FA05−1,319nm)。


LCMS:m/z 428.1[M+H]+。
保持時間:1.461分(分析条件 SMD−FA05−1,319nm)。
実施例1−5−1:化合物AE025−01Rの合成

得られた固相の一部を0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液に浸した。溶液の一部のLC/MS測定を行うことで、AE025とその加水分解体AE026が観測された。
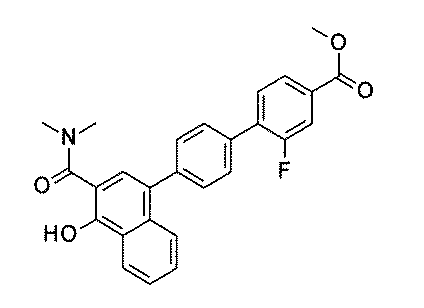
LRMS:m/z 444[M+H]+。
保持時間:1.493分(分析条件SMD−FA05−1、319nm)。


LRMS:m/z 430[M+H]+
保持時間:1.224分(分析条件SMD−FA05−1、319nm)

反応液と固相の懸濁液を全てフィルター上に移し、NMP(20mL)で3回、メタノール(20mL)で3回、DCM(20mL)で3回、ヘプタン(20mL)で3回洗浄し、得られた固相を減圧下で1時間乾燥させ、化合物AE028−01R(1.124g)を得た。

LRMS:m/z 658[M+H]+。
保持時間:1.492分(分析条件SMD−FA05−1、319nm)。
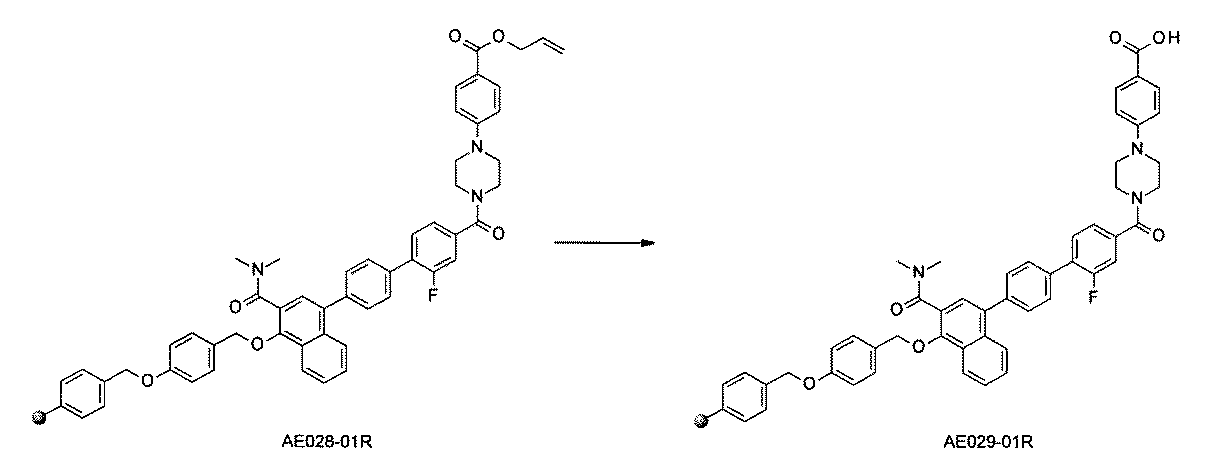
反応液と固相の懸濁液を全てフィルター上に移し、NAC(0.05M、NMP/水=1/1溶液、22mL)で3回、マロン酸(0.05M、NMP溶液、22mL)で3回、HOAt(0.2M、NMP溶液、22mL)で3回、水(22mL)で3回、メタノール(22mL)で3回、DCM(22mL)で3回、ヘプタン(22mL)で3回洗浄し、得られた固相を減圧下で1時間乾燥させ、化合物AE029−01R(1.054g)を得た。

LRMS:m/z 618[M+H]+。
保持時間:1.216分(分析条件SMD−FA05−1、319nm)。
実施例1−5−4で合成法を例示したAE029−01Rは、これ以降の実施例において、化合物BAD006−01Rと表記することもある。
実施例1−6—1:2−ブロモ−4−(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10−ヘプタデカフルオロデシルスルファニル)ベンズアルデヒド(化合物AE003)の合成

極大波長:309nm。
保持時間:1.156分(分析条件SMD−FA50−2)。
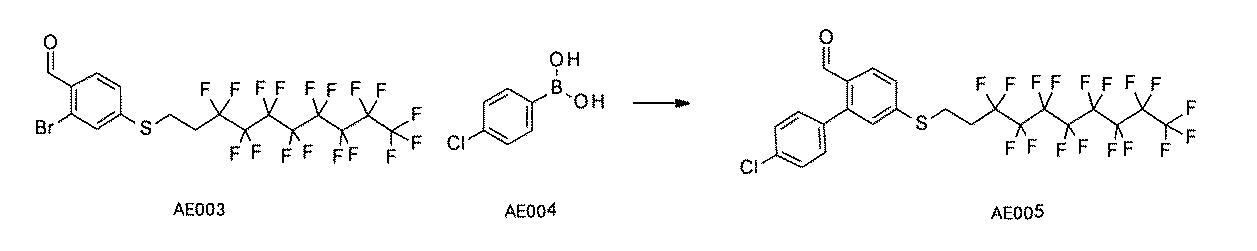
極大波長:308nm。
保持時間:1.229分(分析条件SMD−FA50−2)。

LRMS:m/z 913[M+H]+。
保持時間:1.065分(分析条件SMD−FA50−2)。

LRMS:m/z 885[M+H]+。
保持時間:0.788分(分析条件SMD−TFA50−1)。
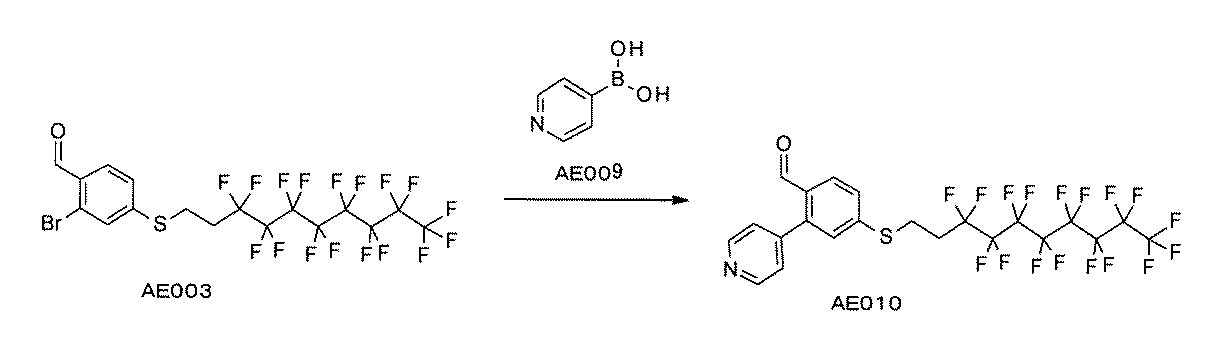
MS:m/z 661.9[M+H]+。
保持時間:0.92分(分析条件SMD−FA50−2)。

LRMS:m/z 868.1[M+H]+。
保持時間:0.39分(分析条件SMD−TFA50−1)。

保持時間:1.05分(分析条件SMD−TFA05−1)。
1H−NMR(400MHz,MeOD)δ:8.96(d,J=6.4Hz,2H),8.24−8.10(m,3H),7.97(d,J=8.1Hz,1H),7.74(t,J=8.3Hz,2H),7.51(d,J=1.6Hz,1H),4.47(s,2H),3.66(s,2H),3.37(m,2H),2.61(m,2H)。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.01分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:1.07分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
分析対象化合物;6−[4−[4−[6−[3−(ジメチルカルバモイル)−4−ヒドロキシナフタレン−1−イル]ピリジン−3−イル]−3−フルオロベンゾイル]ピペラジン−1−イル]ピリダジン−3−カルボン酸(AE019)

保持時間:0.86分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:0.92分。

乾燥後の一部のレジンをフィルター付きピペットチップに移し、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をアセトニトリル(250μL)にて希釈しLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

保持時間:0.78分。
実施例1−7−1:化合物C008−03Rの合成

反応液と固相の懸濁液(0.010mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.2Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、ろ液にNMP(0.25mL)を加えた。得られた溶液のうち0.050mLにアセトニトリル(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C008が100%観測された。

LRMS:m/z 382、384[M+H]+。
保持時間:1.232分(分析条件SMD−FA05−1、280nm)。
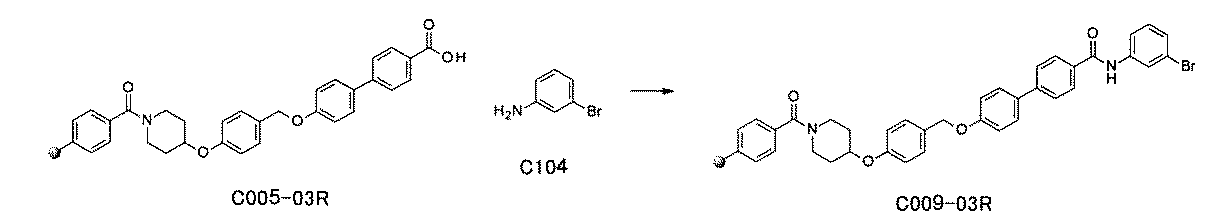
反応液と固相の懸濁液を全てフィルター上に移し、NMP(5mL)で3回、NMP/メタノール=1:1(5mL)で1回、メタノール(5mL)で3回、DCM(5mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C009−03R(担持量0.195mmol/g、248mg、0.0484mmol)を得た。

LRMS:m/z 368、370[M+H]+。
保持時間:1.166分(分析条件SMD−FA05−1、299nm)。

C010−03Rを0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸し、NMP(0.05mL)、アセトニトリル(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物C010が観測された。

LRMS:m/z 382、384[M+H]+。
保持時間:1.221分(分析条件SMD−FA05−1、299nm)。

固相を、NMP(15mL)で2回、MeOH(15mL)で3回、DCM(1mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物C011−03R(担持量0.195 mmol/g、1.04g、0.203mmol)を得た。

LRMS:m/z369、371[M+H]+。
保持時間:1.014分(分析条件SMD−FA05−1、299nm)。
実施例1−8−1:固相担持Ar−NH2(化合物D001−03R)の合成
固相担持Ar−NH2(化合物D001−03R)は、固相担持カルボン酸(C007−03R)と3−(アミノメチル)アニリン(D038)の縮合により、以下のように合成した。
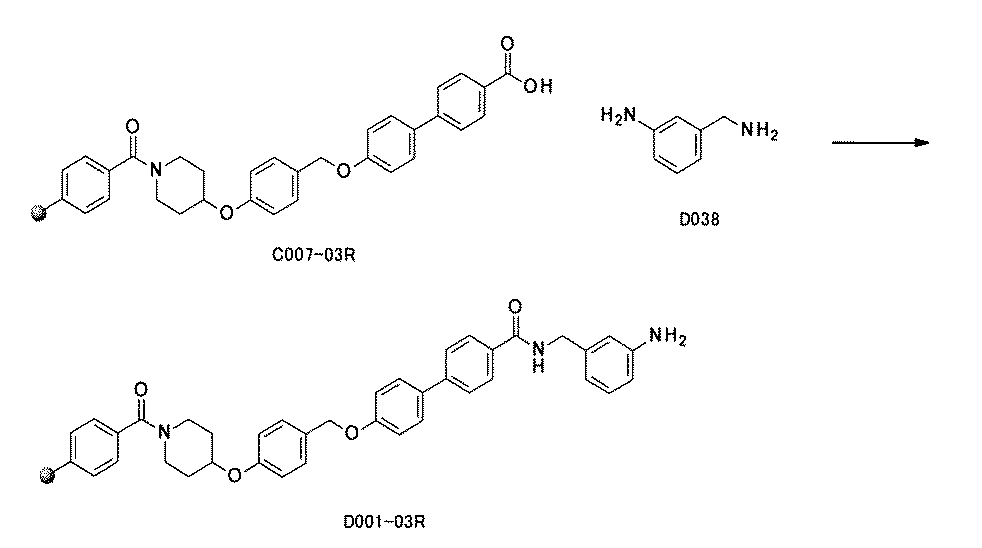
反応液と固相の懸濁液をNMP(6mL)を用いてフィルター上に移し、NMP(6mL)で2回、メタノール(6mL)で3回、DCM(6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D001−03R(担持量0.050mmol/g、450mg、0.0225mmol)を得た。
反応液と固相の懸濁液(0.020mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D001が98.2%の純度で観測された。

LRMS:m/z 319[M+H]+。
保持時間:0.707分(分析条件SMD−TFA05−RP、290nm)。
固相担持Ar−NHMe(D020−03R)は、実施例1−8−1と同様の手法で、固相担持カルボン酸(C007−03R)と3−(アミノメチル)−N−メチルアニリン(D039)の縮合により合成した。固相精製、反応追跡(追跡対象化合物;D020)も実施例1−8−1と同様に行った。

LRMS:m/z 333[M+H]+。
保持時間:0.679分(分析条件SMD−FA05−1)。
実施例1−9−1:化合物E015−02Rの合成

レジンに担持された担持量(mmol/g)は、以下の方法で決定した。以下は実験操作の一例であり、全てのロットに関する担持量を一律で規定するものではない。

得られたE015のUV面積%と検量線から溶液中の濃度を算出し、レジンに担持されているE015の量を求めた。その結果担持量を0.556mmol/gと算出した。

レジンに担持された担持量(mmol/g)は、以下の方法で決定した。以下は実験操作の一例であり、全てのロットに関する担持量を一律で規定するものではない。

実施例1−10−1:4−(2−フェニルスルファニルエチルアミノ)−3−(トリフルオロメチル)ベンゼンスルホンアミド(AG010)の合成
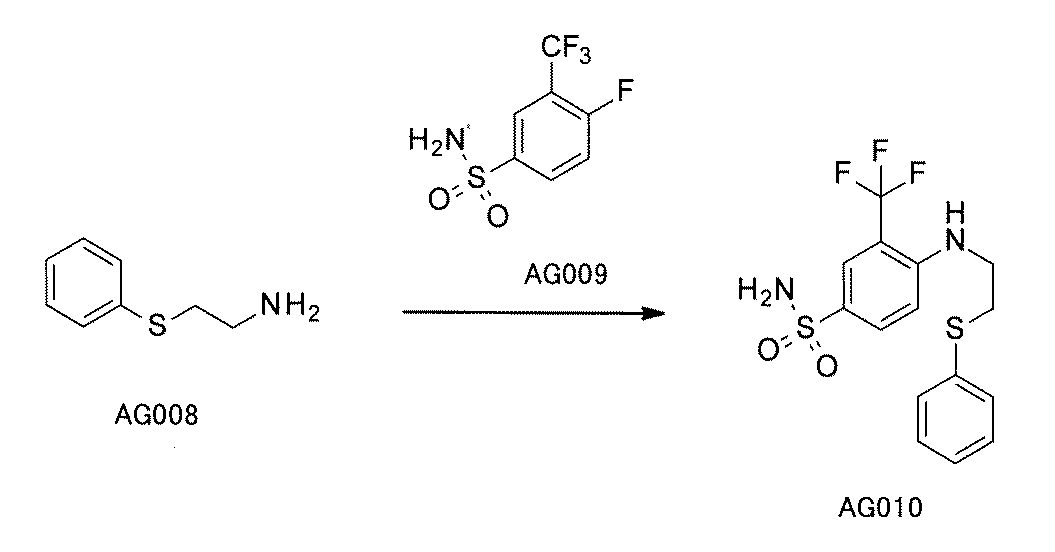
LRMS(ESI):m/z 377[M+H]+。

LRMS(ESI):m/z 710[M+H]+。
保持時間:1.166分(分析条件PH−TFA05−1)。

LRMS(ESI):m/z 752[M+H]+。
実施例1−10−3で合成法を例示したAG005は、これ以降の実施例において、化合物B2Q045−01と表記することもある。

LRMS(ESI):m/z 753[M+H]+。
実施例1−10−4で合成法を例示したAG007は、これ以降の実施例において、化合物B2Q046−01と表記することもある。

LRMS(ESI):m/z 733[M+H]+。
保持時間:1.319分(分析条件PH−FA05−2)。

LRMS(ESI):m/z 775[M+H]+。
実施例1−10−6で合成法を例示したAG012は、これ以降の実施例において、化合物B2R045−01と表記することもある。

LRMS(ESI):m/z 776[M+H]+。
実施例1−10−7で合成法を例示したAG013は、これ以降の実施例において、化合物B2R046−01と表記することもある。
実施例1−11−1:化合物A102−3Rの合成

反応液と固相の懸濁液を全て2Lのセパラブルフラスコに移し、NMP(800mL)で3回、MeOH(800mL)で3回、DCM(800mL)で3回、ヘプタン(800mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A102−03R(担持量0.200mmol/g、47.9g)を得た。
反応液と固相の懸濁液(10μL)をフィルター上に移し、NMP(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.1mL)に5分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物A102が100%観測された。

102LRMS:m/z 243[M+H]+
保持時間:1.081分(分析条件FA05−1、299nm)。

反応液と固相の懸濁液を、HOAt(0.1M in NMP、800mL)で3回、NMP(800mL)で3回、MeOH(800mL)で3回、DCM(800mL)で3回、ヘプタン(800mL)で3回洗浄し、得られた固相を減圧下で6日乾燥させ、化合物A103−3R(担持量0.201mmol/g、49.8g、10.0mmol)を得た。
反応液と固相の懸濁液(12μL)をフィルター上に移し、NMP(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物A103が100%観測された。
極大波長:294nm
保持時間:0.761分(分析条件FA05−1、299nm)。

極大波長:266nm
保持時間:0.797分(分析条件FA05−1、299nm)。

窒素雰囲気下500mLフラスコに、メチル 2−(3−ヒドロキシフェニル)アセテート(化合物BP113,CAS:42058−59−3)(15.0g,90.3mmol,1.0eq)、イミダゾール(18.4g,270.8mmol,3.0eq)とDMF(150mL)を加え、室温で10分撹拌した。この反応混合物にTBDMSCl(20.4g,135.4mmol,1.5eq)を滴下して加えた。この反応混合物を室温で16時間撹拌し、酢酸エチル(450mL)で希釈した。得られた有機層を水(150mL)で3回洗浄し、Na2SO4で乾燥した。固体をろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(0~20%酢酸エチル/石油エーテル)で精製し、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシフェニル]アセテート(化合物BP114)を無色オイルとして得た(24.4g,93.5%)。
LCMS(ESI,m/z):281[M+H]+。
保持時間:0.825分。
分析条件:PH−FA−15。
窒素雰囲気下500mLフラスコに、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシフェニル]アセテート(化合物BP114)(16.4g,58.5mmol,1.0eq)とTHF(550mL)を加え、−78℃に冷却した。この反応液にLDA(2M THF溶液,38mL,76.0mmol,1.3eq)を滴下して加え、−78℃で1時間撹拌した。得られた反応液にMeI(10.0g,70.2mmol,1.2eq)を−78℃で加え、反応混合物を室温下16時間撹拌した。反応を水(200mL)で停止させ酢酸エチル(400mL)で3回抽出した。有機層を混合しNa2SO4で乾燥させた。固体をろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(0~12%酢酸エチル/石油エーテル)で精製し、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシフェニル]プロパノエート(化合物BP115)を黄色オイルとして得た(16.2g,89.2%)。
LCMS(ESI,m/z):295[M+H]+。
保持時間:0.851分。
分析条件:PH−FA−15。
窒素雰囲気下500mLフラスコに、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシフェニル]プロパノエート(化合物BP115)(10.0g,34.0mmol,1.0eq)、B2Pin2(8.6g,34.0mmol,1.0eq)、[Ir(cod)OMe]2(2.3g,3.4mmol,0.1eq)、Dtbpy(1.8g,6.8mmol,0.2eq)とCPME(シクロペンチルメチルエーテル)(200mL)を加えた。この反応混合物を30℃で16時間撹拌した。反応混合物を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(0~30%酢酸エチル/石油エーテル)と逆相シリカゲルカラムクロマトグラフィー(C18)により精製(30~80%CH3CN/水)で精製し、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシ−5−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)フェニル]プロパノエート(化合物BP116)を淡黄色オイルとして得た(6.6g,41.4%)。
LCMS(ESI,m/z):421[M+H]+。
保持時間:0.883分。
分析条件:PH−FA−15。
窒素雰囲気下300mLフラスコに、メチル 2−[3−[tert−ブチル(ジメチル)シリル]オキシ−5−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)フェニル]プロパノエート(化合物BP116)(3.9g,9.3mmol,1.0eq)、CuBr2(6.2g,27.8mmol,3.0eq)、MeOH(60mL)と水(60mL)を加えた。この反応混合物を80℃で5時間撹拌した。反応混合物を酢酸エチル(250mL)で希釈し、有機層を分離した。有機層を飽和NaCl水溶液(100mL)で洗浄し、Na2SO4で乾燥させた。固体をろ過後、ろ液を減圧下濃縮した。残差を逆相シリカゲルカラムクロマトグラフィー(C18)により精製(5~55%CH3CN/水)で精製し、メチル 2−[3−ブロモ−5−ヒドロキシフェニル]プロパノエート(化合物D084)を無色オイルとして得た(0.9g,36.6%)。
LCMS(ESI,m/z):257,259[M−H]+。
保持時間:0.926分。
分析条件:PH−FA−16。

LCMS:m/z570,572[M+Na]+。
保持時間:1.589分(分析条件SMD−FA05−1)

LCMS:m/z448,450[M+H]+。
保持時間:0.863分(分析条件SMD−FA05−1)。
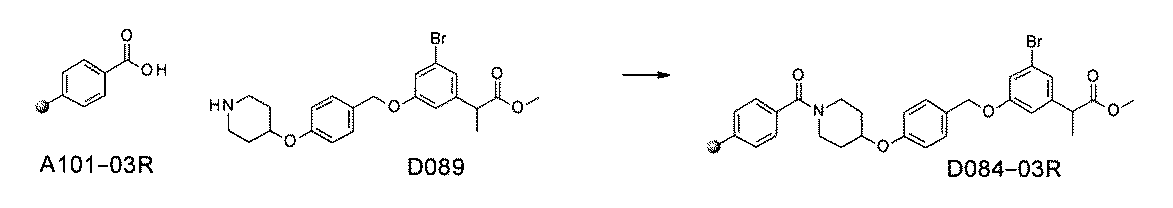
D084−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.10mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物D084が99.2%観測された。
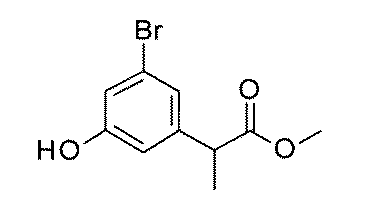
極大波長:278nm
保持時間:0.983分(分析条件FA05−1、277nm)。

窒素雰囲気下、250mLのフィルター付きカラムに固相担持エステル(化合物D084−03R)(11.58g、0.200mmol/g)、NMP(162mL)、tAmylOH(46.3mL)を加え、室温で1時間振盪した。1M TBAOH水溶液(4.63mL、4.63mmol)を加え、室温で7.5時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(208mL)で3回、NMP(208mL)で3回、0.05M TBAHSO4+DTBP in NMP(208mL)で3回、NMP/水=1/1(208mL)で3回、NMP(208mL)で3回、MeOH(208mL)で3回、DCM(208mL)で3回、ヘプタン(208mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、標題化合物D127−03R(13.8g、0.201mmol/g)を得た。
反応液と固相の懸濁液(0.015mL)をフィルター上に移し、DMA(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.10mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D127が98.4%の純度で観測された。
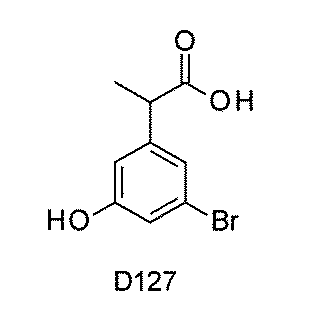
極大波長:278nm。
保持時間:0.807(分析条件 SMD−FA05−1、280nm)。

LCMS(ESI,m/z):321[M+H]+。
保持時間:1.250分。
分析条件:PH−TFA−23。
実施例1−11−10:メチル 5−[tert−ブチル(ジメチル)シリル]オキシ−7−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1,2,3,4−テトラヒドロナフタレン−1−カルボキシラート(BP125)の合成

LCMS(ESI,m/z):447[M+H]+。
保持時間:1.337分。
分析条件:PH−TFA−23。

LCMS(ESI,m/z):283[M−H]+。
保持時間:0.995分。
分析条件:PH−FA−16。

LRMS(ESI):m/z572、574[M+H]+。
保持時間:1.651分(分析条件SMD−FA05−1)。
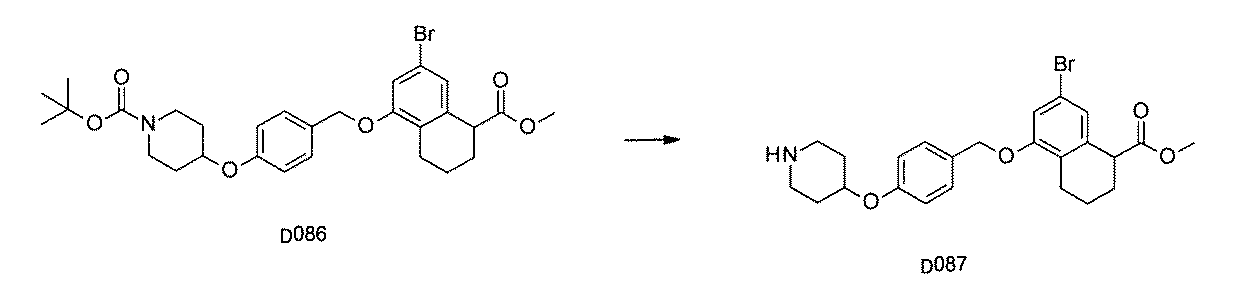
LCMS:m/z472,474[M+H]+。
保持時間:0.906分(分析条件SMD−FA05−1)。


LRMS:m/z 285、287[M+H]+
保持時間:1.051分(分析条件FA05−1、277nm)。


極大波長:285nm。
保持時間:0.831(分析条件SMD−FA05−1、280nm)。

窒素雰囲気下1Lフラスコに、4−フルオロベンズアルデヒド(化合物BP106,CAS:459−57ー4)(53.1g,427.4mmol,1.0eq)、プロプ−2−エニル 4−ピペリジン−1−カルボキシラート(化合物BP105,CAS:187265−40−3)(95.0g,512.9mmol,1.2eq)、Cs2CO3(334.2g,1025.8mmol,2.4eq)とDMF(480mL)を加えた。この反応混合物を100℃で16時間撹拌した。反応混合物を室温に低下させ、水(400mL)を加え酢酸エチル(500mL)で3回抽出した。有機層を混合し、飽和NaCl溶液(500mL)で洗浄し、Na2SO4で乾燥した。固体をろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィーで精製(0~50%酢酸エチル/石油エーテル)し、プロプ−2−エニル 4−(4−ホルミルフェノキシ)ピペリジン−1−カルボキシラート(化合物BP107)を淡黄色オイルとして得た(89.4g,61.5%)。
LCMS(ESI,m/z):290[M+H]+。
保持時間:0.704分。
分析条件:PH−TFA−22。
窒素雰囲気下3Lフラスコに、プロプ−2−エニル 4−(4−ホルミルフェノキシ)ピペリジン−1−カルボキシラート(化合物BP107)(89.4g,309.0mmol,1.0eq)とEtOH(900mL)を加えた。この反応混合物に氷冷下、NaBH4(23.4g,618.0mmol,2.0eq)を少しずつ添加し、得られた反応混合物を室温で2時間撹拌した。水(200mL)を加えて反応を停止し、反応混合物をろ過した。濾別した固体を水(200mL)で洗浄し、さらにジオキサン(300mL)とヘキサン(300mL)で通液洗浄し、プロプ−2−エニル 4−[4−(ヒドロキシメチル)フェノキシ]ピペリジン−1−カルボキシラート(化合物D081)を白色固体として得た(70.7g,76.2%)。
LCMS(ESI,m/z):292[M+H]+。
保持時間:0.797分。
分析条件:PH−TFA−22。

窒素雰囲気下500mLフラスコに、tert−ブチル 8−ブロモ−3,4−ジヒドロ−1H−イソキノリン−2−カルボキシラート(化合物BP109,CAS:893566−75−1)(24.0g,76.9mmol,1.0eq)、B2Pin2(19.5g,76.9mmol,1.0eq)、[Ir(cod)OMe]2(5.1g,7.7mmol,0.1eq)、Dtbpy(4.1g,15.4mmol,0.2eq)とTHF(240mL)を加えた。この反応混合物を30℃で16時間撹拌した。反応混合物を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(0~15%酢酸エチル/石油エーテル)と逆相シリカゲルカラムクロマトグラフィー(C18)により精製(30~80%CH3CN/水)で精製し、tert−ブチル 8−ブロモ−6−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−3,4−ジヒドロ−1H−イソキノリン−2−カルボキシラート(化合物BP110)を褐色オイルとして得た(24.0g,60.6%)。
LCMS(ESI,m/z):423,425[M+H]+。
保持時間:1.070分。
分析条件:PH−FA−12。
窒素雰囲気下5L反応容器に、tert−ブチル 8−ブロモ−6−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−3,4−ジヒドロ−1H−イソキノリン−2−カルボキシラート(化合物BP110)(24.0g,54.8mmol,1.0eq)、PdCl2(dppf)‐DCM(6.7g,8.2mmol,0.15eq)、TEA(16.6g,164.3mmol,3.0eq)、とEtOH(3.7L)を加えた。この反応混合物を15atmのCO雰囲気下、100℃で3日間撹拌した。COガスを除去後、反応混合物を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(0~30%酢酸エチル/石油エーテル)で精製し、tert−ブチル 8−ブロモ−6−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−3,4−ジヒドロ−1H−イソキノリン−2−カルボキシラート(化合物BP111)を褐色オイルとして得た(21.0g,38.2%)。
LCMS(ESI,m/z):432[M+H]+。
保持時間:0.991分。
分析条件:PH−FA−12。
窒素雰囲気下500mLフラスコに、2−O−tert−ブチル 8−O−エチル 6−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−3,4−ジヒドロ−1H−イソキノリン−2,8−ジカルボキシラート(化合物BP111)(22.0g,51.0mmol,1.0eq)、AcOH(50mL)と水(50mL)を加えた。この反応混合物に室温下、過酸化水素水(33mL,970.2mmol,19.0eq)を滴下しながら加えた。得られた反応混合物を室温で16時間撹拌し、水(100mL)で希釈した。反応混合物を飽和炭酸ナトリウム水溶液(200mL)で塩基性に調整(pH~8)し、固体をろ過した。濾別した固体を逆相シリカゲルカラムクロマトグラフィー(C18)により精製(30~50%CH3CN/水)で精製し、さらにヘキサン(300mL)、次いで水(200mL)で懸濁精製し、2−O−tert−ブチル 8−O−エチル 6−ヒドロキシ−3,4−ジヒドロ−1H−イソキノリン−2,8−ジカルボキシラート(化合物D082)を黄色固体として得た(7.3g,42.4%)。
LRMS(ESI):m/z 320[M−H]+。
保持時間:1.039分。
分析条件:PH−FA−13。

LRMS(ESI):m/z 617[M+Na]+。
保持時間:1.563分(分析条件 SMD−FA05−1)

LRMS(ESI):m/z511[M+H]+。
保持時間:2.572分(分析条件SMD−AC05−long)。
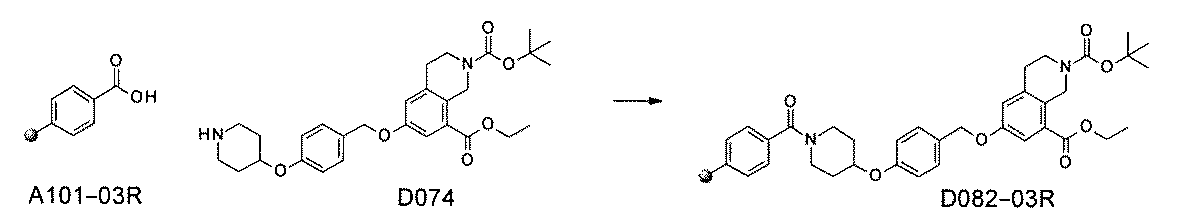
D082−03Rを0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.10mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物D082が81.1%、D082−Bocが18.9%観測された。

LRMS:m/z 266[M−tBu+H]+
保持時間:1.099分(分析条件FA05−1、299nm)。

LRMS:m/z 222[M+H]+
保持時間:0.460分(分析条件FA05−1、299nm)。
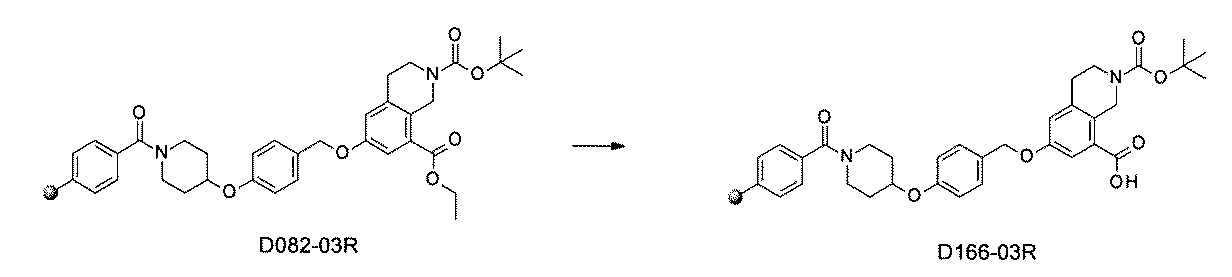
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(172mL)で3回、0.05M TBAHSO4+DTBP in NMP(172mL)で3回、NMP/水=1/1(172mL)で3回、NMP(172mL)で3回、MeOH(172mL)で3回、DCM(172mL)で3回、ヘプタン(172mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、化合物D166−03R(8.59 g、0.201mmol/g)を得た。実施例1−11−8と同様の切り出し操作を行い、目的物D166が96.9面積%観測された。

極大波長:304nm。
保持時間:0.871(分析条件 SMD−FA05−1、280nm)。

窒素雰囲気下500mLフラスコに、(4−ピペリジン−4−イルオキシフェニル)メタノール(化合物BP103,CAS:1251336−69−2)(8.7g,41.8mmol,1.0eq)とDMF(85mL)を加えた。
この溶液に(4−ニトロフェニル)2−トリメチルシリルエチルカルボナテ(化合物BP112)(11.6g,41.0mmol,1.0eq)のDCM(85mL)溶液を室温下添加した。得られた反応混合物を16時間撹拌し、反応液を減圧下濃縮した。残差を逆相シリカゲルカラムクロマトグラフィー(C18)により精製(5~70%CH3CN/水)で精製し、2−トリメチルシリルエチル4−[4−(ヒドロキシメチル)フェノキシ]ピペリジン−1−カルボキシラート(化合物D081)を白色固体として得た(5.4g,36.7%)。
LCMS(ESI,m/z):374[M+Na]+。
保持時間:1.180分。
分析条件:PH−TFA−24。
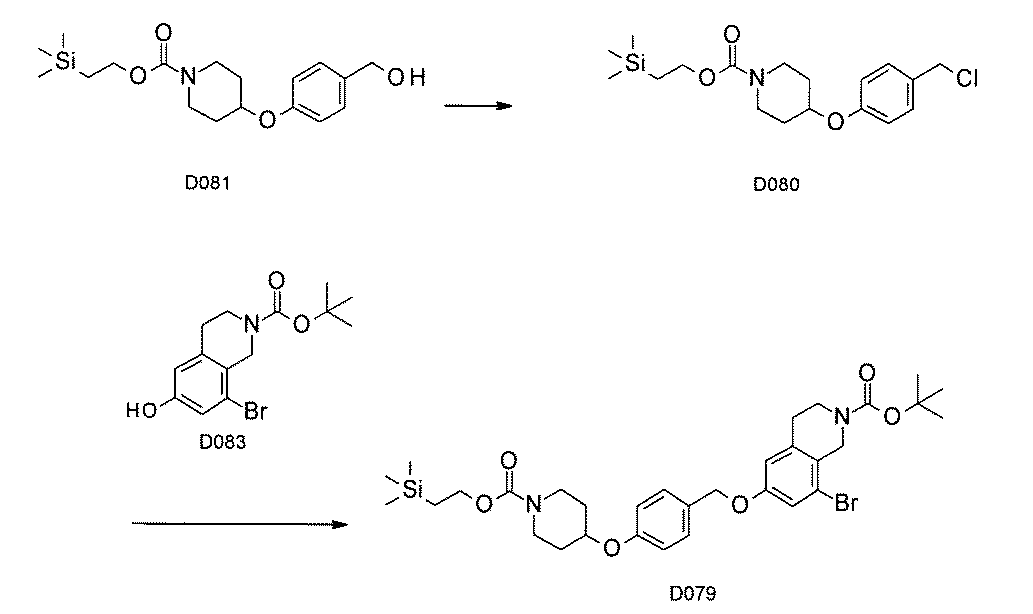
LRMS(ESI):m/z683、685[M+Na]+。
保持時間:1.826分(分析条件SMD−FA05−1)。

1H−NMR(400MHz,CDCl3)δ:7.31(2H,d,J=9.1Hz),7.08(1H,d,J=2.7Hz),6.92(2H,d,J=8.6Hz),6.71(1H,s),4.93(2H,s),4.47(2H,s),4.41−4.35(1H,m),3.60(2H,t,J=5.9Hz),3.17−3.12(2H,m),2.78−2.70(4H,m),2.02−1.99(2H,m),1.71−1.66(2H,m),1.50(9H,s)。
LRMS(ESI):m/z517、519[M+H]+。
保持時間:0.994分(分析条件SMD−FA05−1)。


LRMS:m/z 272、274[M−tBu+H]+
保持時間:1.159分(分析条件FA05−1、277nm)。

反応液と固相の懸濁液を全てフィルター上に移し、NMP:H2O(1:1)(v/v、5mL)で3回、NMP(5mL)で3回、MeOH(5.2mL)で3回、DCM(5.2mL)で、ヘプタン(5.2mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物B101−03R(担持量0.194mmol/g、1.00g)を得た。

LRMS:m/z 398、400[M+H]+
保持時間:1.228(分析条件FA05−1、299nm)。

B133−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A133が99.6%観測された。

LRMS:m/z 368、370[M+H]+
保持時間:1.161(分析条件FA05−1、299nm)。

B107−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物B107が100%観測された。

LRMS:m/z 314[M+H]+
保持時間:1.083(分析条件FA05−1、299nm)。

B102−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物B102が99.1%観測された。

LRMS:m/z 388、390[M+H]+
保持時間:1.165(分析条件FA05−1、299nm)。

D090−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物D090が100%観測された。

LRMS:m/z 416[M+H]+
保持時間:1.192(分析条件FA05−1、299nm)。

A109−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A109が100%観測された。

LRMS:m/z 348[M+H]+
保持時間:1.036(分析条件FA05−1、299nm)。


LRMS:m/z 416[M+H]+
保持時間:1.236(分析条件FA05−1、299nm)。

LRMS:m/z 334[M+H]+
保持時間:0.821(分析条件FA05−1、299nm)。


LRMS:m/z 367[M+H]+
保持時間:0.917(分析条件FA05−1、299nm)。

A122−03Rを0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A122が87.0%、A122−Bocが12.2%観測された。

LRMS:m/z 399[M—tBu+H]+
保持時間:1.060(分析条件FA05−1、299nm)。

LRMS:m/z 355[M+H]+
保持時間:0.548(分析条件FA05−1、299nm)。

反応液と固相の懸濁液を全てフィルター上に移し、NMP:H2O(1:1)(v/v、5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物B108−03R(担持量0.196mmol/g、528mg)を得た。

LRMS:m/z 331[M+H]+
保持時間:1.125(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP(45mL)を用いてフィルター上に移し、NMP(45mL)で4回、メタノール(45mL)で4回、DCM(45mL)で4回、ヘプタン(45mL)で4回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物B105−03R(担持量0.197mmol/g、3.13g)を得た。
B105−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物B105が98.4%観測された。
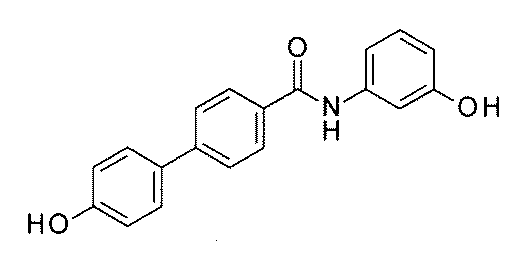
LRMS:m/z 306[M+H]+
保持時間:0.868(分析条件FA05−1、299nm)。
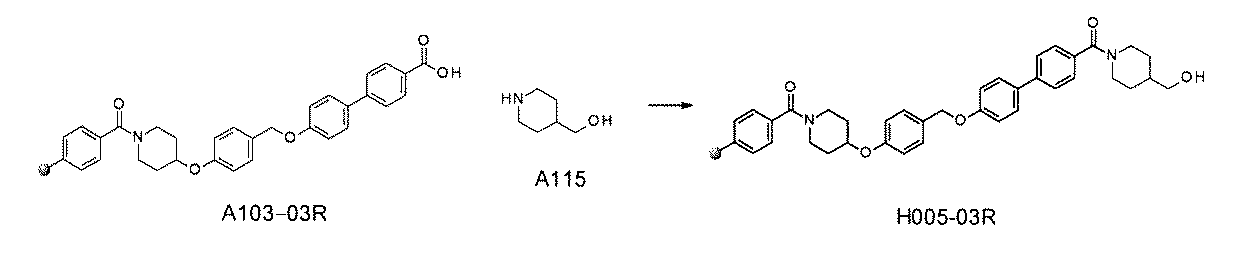
H005−03Rを0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.10mL)に5分間浸し、DMF(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物H005が96.7%観測された。

LRMS:m/z 312[M+H]+
保持時間:0.712(分析条件FA05−1、299nm)。

H002−03Rを0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物H002が97.2%観測された。H002−03RはD020−03Rと同一化合物だが、固相担持量が異なる
LRMS:m/z 333[M+H]+
保持時間:0.675(分析条件FA05−1、299nm)。

A118−03Rを0.2Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMA(0.20mL)、MeCN(0.10mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A118が98.1%観測された。A118−03RはD001−03Rと同一化合物だが、固相担持量が異なる
LRMS:m/z 319[M+H]+
保持時間:0.625(分析条件FA05−1、299nm)。
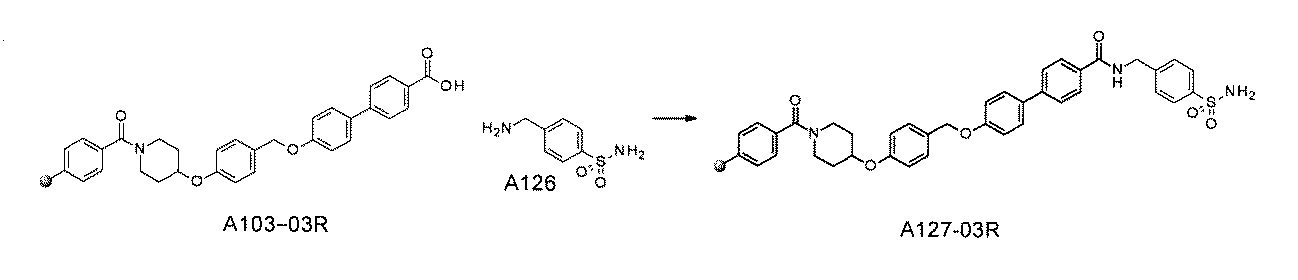
A127−03Rを0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A127が100%観測された。
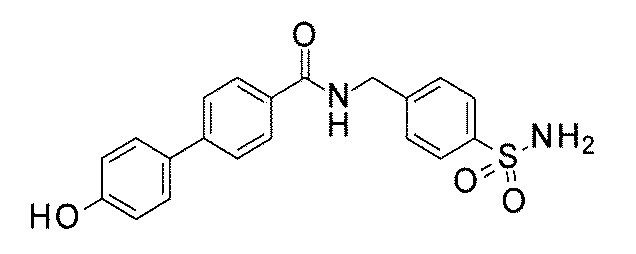
LRMS:m/z 383[M+H]+
保持時間:0.771(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP:H2O(1:1)(v/v、10mL)を用いて2個のフィルター上に移し、それぞれをNMP:H2O(1:1)(v/v、5mL)で3回、NMP(5mL)で3回、0.05M TBAHSO4・DTBPのNMP溶液(5mL)で3回、0.05M NMMのNMP溶液(5mL)で3回、NMP:H2O(1:1)(v/v、5mL)で3回、NMP(5mL)で3回、メタノール(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物H004−03R(担持量0.192mmol/g、523mg)を得た。

LRMS:m/z 442[M+H]+
保持時間:0.901(分析条件FA05−1、299nm)。
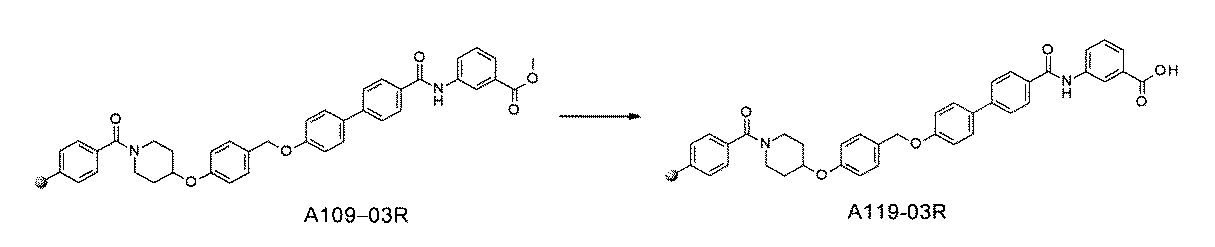
反応液と固相の懸濁液をNMP:H2O(1:1)(v/v、5mL)を用いて8個のフィルター上に移し、それぞれをNMP:H2O(1:1)(v/v、5mL)で3回、0.05M TBAHSO4・DTBPのNMP溶液(5mL)で3回、NMP:H2O(1:1)(v/v、5mL)で3回、、NMP(5mL)で3回、メタノール(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A119−03R(担持量0.196mmol/g、2.05g)を得た。
A119−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、DMF(0.05mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A119が98.8%観測された。

LRMS:m/z 334[M+H]+
保持時間:0.875分(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP:H2O(1:1)(v/v、10mL)を用いて2個のフィルター上に移し、それぞれをNMP:H2O(1:1)(v/v、5mL)で3回、NMP(5mL)で3回、メタノール(5.5mL)で3回、DCM(5.5mL)で3回、ヘプタン(5.5mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A120−03R(担持量0.190mmol/g、507mg)を得た。

LRMS:m/z 518[M+H]+
保持時間:1.868(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP(100mL)で3回、0.3M N−アセチルシステインのNMP:H2O(5:1、v/v)溶液(100mL)で3回、0.2M DIPEAのNMP溶液(100mL)で3回、NMP(100mL)で3回、メタノール(100mL)で3回、DCM(100mL)で3回、ヘプタン(100mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物H003−03R(担持量0.197mmol/g、4.71g)を得た。
H003−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物H003が96.8%観測された。H003−03Rは、D021−03Rと同一化合物であるが、固相担持量が異なる。
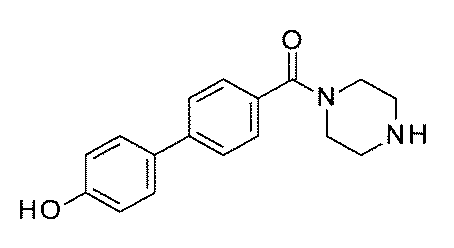
LRMS:m/z 283[M+H]+
保持時間:0.472(2峰性)(分析条件FA05−1、299nm)。
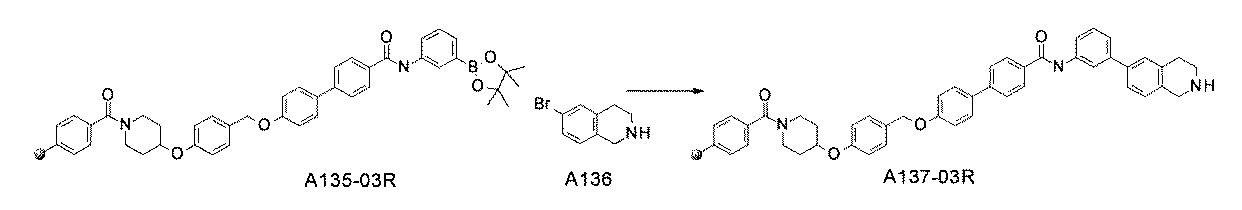
反応液と固相の懸濁液をNMP(5mL)を用いて3個のフィルター上に移し、それぞれをNMP(5.0mL)で3回、0.2M N−アセチルシステインのNMP:H2O(5:1、v/v)溶液(5.0mL)で3回、NMP(5.0mL)で3回、メタノール(5.0mL)で3回、DCM(5.0mL)で3回、ヘプタン(5.0mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A137−03R(担持量0.193mmol/g、753mg)を得た。
A137−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A137が94.0%観測された。

LRMS:m/z 421[M+H]+
保持時間:0.748(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP:H2O(1:1)(v/v、60mL)を用いて8個のフィルター上に移し、それぞれをNMP:H2O(1:1)(v/v、60mL)で3回、0.05M TBAHSO4・DTBPのNMP溶液(60mL)で3回、、NMP:H2O(1:1)(v/v、60mL)で3回、NMP(60mL)で3回、メタノール(60mL)で3回、DCM(60mL)で3回、ヘプタン(60mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A123−03R(担持量0.193mmol/g、3.0g)を得た。
A123−03Rを0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸し、NMP(0.10mL)、MeCN(0.25mL)を加えてろ過したろ液のLCMS測定を行うことで目的物A123が96.3%、A123—Bocが3.7%観測された。
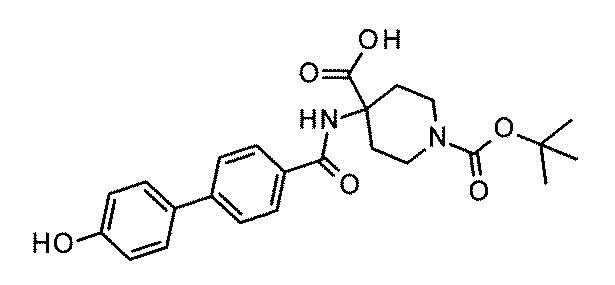
LRMS:m/z 385[M—tBu+H]+
保持時間:0.945(分析条件FA05−1、299nm)。

LRMS:m/z 341[M+H]+
保持時間:0.464(分析条件FA05−1、299nm)。

反応液と固相の懸濁液をNMP(60mL)で3回、NMP:H2O(1:1)(v/v、60mL)で3回、NMP(60mL)で3回、メタノール(60mL)で3回、DCM(60mL)で3回、ヘプタン(60mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物A125−03R(担持量0.185mmol/g、3.35g)を得た。

LRMS:m/z 620[M+H]+
保持時間:1.119(分析条件FA05−1、299nm)。

LRMS:m/z 520[M+H]+
保持時間:0.713(分析条件FA05−1、299nm)。

それぞれの反応液と固相の懸濁液を0.2M DMEDAのNMP溶液(3mL)で3個のフィルター上に移し、0.2M DMEDAのNMP溶液(5.0mL)で3回、NMP(5.0mL)で3回、0.05M TBAHSO4・DTBPのNMP溶液(5.0mL)で3回、0.05M BTMGのNMP溶液(5.0mL)で3回、0.05M DTBPのNMP溶液(5.0mL)で3回、NMP:H2O(1:1)(v/v、5.0mL)で3回、NMP(5.0mL)で3回、メタノール(5.0mL)で3回、DCM(5.0mL)で3回、ヘプタン(5.0mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物H001−03R(担持量0.188mmol/g、895mg)を得た。

LRMS:m/z 520 [M+H]+。
保持時間:0.708(分析条件FA05−1、299nm)。

LRMS:m/z 340 [M+H]+。
保持時間:0.448(分析条件FA05−1、299nm)。

窒素雰囲気下500mLフラスコに、tert−ブチル 4−[4−(ヒドロキシメチル)フェノキシ]ピペリジン−1−カルボキシラート(化合物BP102,CAS:321337−38−6)(20.0g,65.1mmol,1.0eq)、TEA(19.8g,195.8mmol,3.0eq)とDCM(100mL)を加えた。この反応混合物を0℃に冷却し、MsCl(14.9g,130.1mmol,2.0eq)を滴下して加えた。この反応混合物を室温で2時間撹拌し、DCM(250mL)で希釈した。得られた有機層を水(100mL)で洗浄し、Na2SO4で乾燥した。固体をろ過後、ろ液を減圧下濃縮し、粗生成物として、tert−ブチル 4−[4−(クロロメチル)フェノキシ]ピペリジン−1−カルボキシラート(化合物BP126)を褐色オイルとして得た(20.0g)。
LCMS(ESI,m/z):226[M−Boc+H]+。
保持時間:1.171分。
分析条件:PH−FA−17。
窒素雰囲気下300mLフラスコに、tert−ブチル 4−[4−(クロロメチル)フェノキシ]ピペリジン−1−カルボキシラート(化合物BP126)(粗生成物20.0g)、K2CO3(7.2g,51.9mmol,1.0eq)、4−ブロモフェノール(9.0g,51.9mmol,1.0eq)とDMF(100mL)を加えた。この反応混合物を70℃で16時間撹拌した。反応混合物をろ過し、濾別した固体を酢酸エチル(300mL)で洗浄した。ろ液を分離し、分離した有機層を水(100mL)で洗浄し、Na2SO4で乾燥させた。固体をろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(0~15%酢酸エチル/石油エーテル)で精製し、tert−ブチル 4−[4−[(4−ブロモフェノキシ)メチル]フェノキシ]ピペリジン−1−カルボキシラート(化合物D120)を白色固体として得た(11.0g,45.4%)。
LCMS(ESI,m/z):362,364[M+H]+:脱Boc体として観測した。
保持時間:2.187分。
分析条件:PH−AC−12。

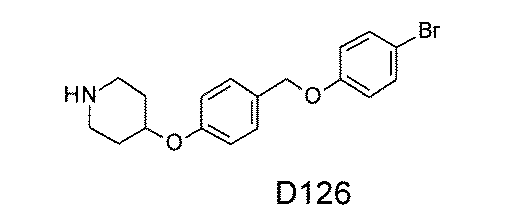
LCMS:m/z 362,364[M+H]+。
保持時間:0.780分(分析条件 SMD−FA05−1、305±95nm)。


極大波長:226,281nm。
保持時間:0.880分(分析条件SMD−FA05−1、280nm)。
固相担持固相担持ArBr(D121−03R)と4,4,5,5−テトラメチル−2−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1,3,2−ジオキサボロラン(D125)を用いてArボロン酸エステル(D122−03R)を合成した固相実験(反応操作の例示)
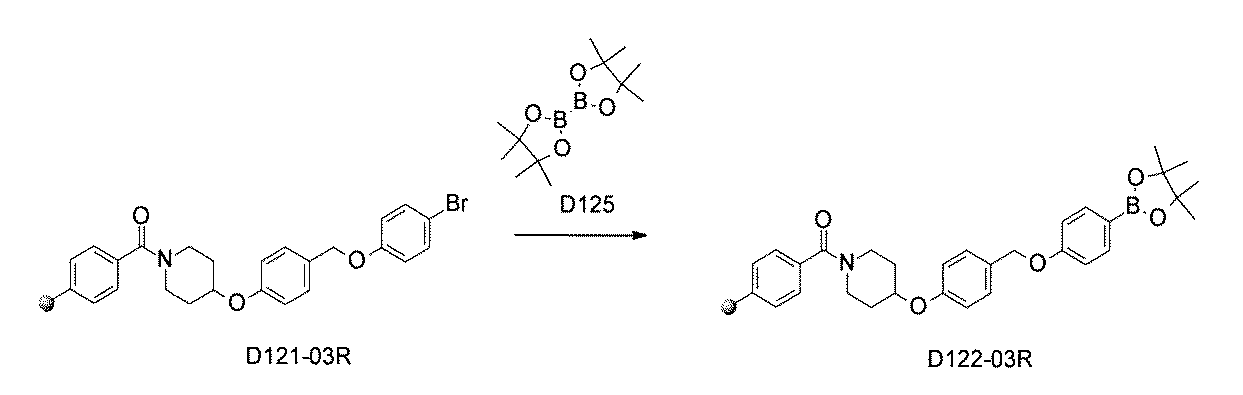
反応液と固相の懸濁液をNMP(0.4mL)を用いてフィルター上に移し、NMP(0.4mL)で3回、水(0.4mL)で3回、NMP/水=/1(0.4mL)で3回、NMP(0.4mL)で3回、メタノール(0.4mL)で3回、DCM(0.4mL)で3回で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D122−03R(担持量0.198mmol/g、16.0mg)を得た。
窒素雰囲気下、レジンの懸濁液20μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をNMP(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D122が100面積%観測された。

極大波長:238nm。
保持時間:1.007分(分析条件SMD−FA05−1、250nm)。

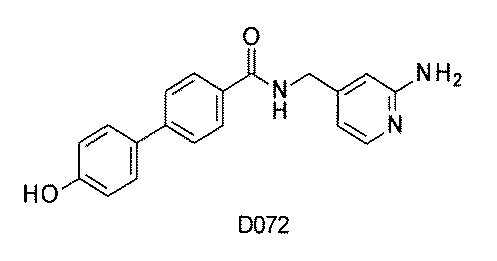
LRMS:m/z 320[M+H]+。
保持時間:0.681分(分析条件SMD−TFA05−RP、299nm)。
固相担持ヘテロAr−NH2(化合物D106−03R)は、固相担持アミン(H003−03R)と5−アミノピリジン−2−カルボン酸(D108)の縮合により、以下のように合成した。

反応液と固相の懸濁液をNMP(2mL)を用いてフィルター上に移し、NMP(4mL)で3回、NMP/水=1/1(4mL)で3回、NMP(4mL)で3回、メタノール(4mL)で3回、DCM(4mL)で3回、ヘプタン(4mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D106−03R(担持量0.192mmol/g、434mg)を得た。
反応液と固相の懸濁液(0.012mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D106が93.2%の純度で観測された。

LRMS:m/z 403[M+H]+。
保持時間:0.667分(分析条件SMD−FA05−1、299nm)

反応液と固相の懸濁液をNMP(1mL)を用いてフィルター上に移し、NMP(2mL)で3回、0.05M NAC in NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(3mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させた。得られた固相を用いて再度同様の反応および固相精製を行うことで、化合物D116−03R(担持量0.187mmol/g、109mg)を得た。
反応液と固相の懸濁液(0.012mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D116が86.0%の純度で観測された。
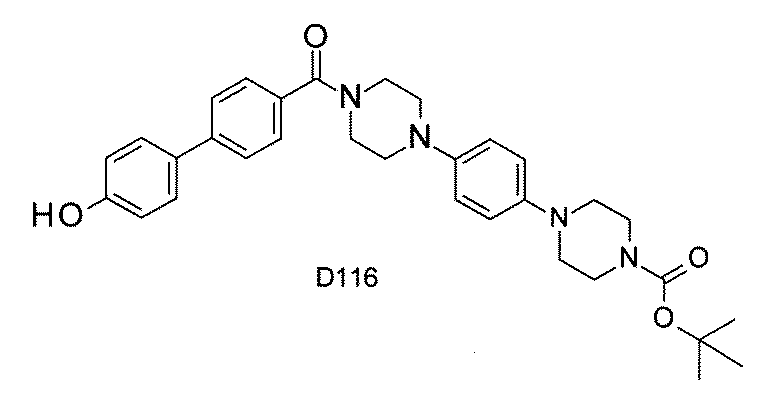
LRMS:m/z 543[M+H]+。
保持時間:1.055分(分析条件SMD−FA05−1、290nm)
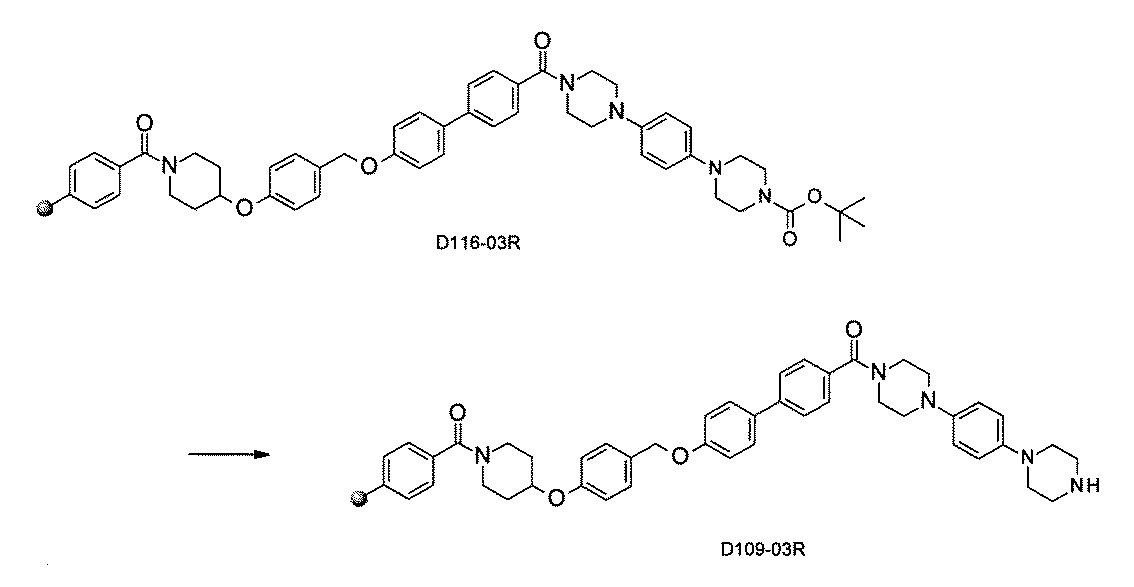
反応液に1MのDMEDAinNMP(0.935mL、0.935mmol)を加え、室温で2時間振盪した。反応液と固相の懸濁液をNMP(1mL)を用いてフィルター上に移し、0.2M DMEDA in NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、0.2M TBAHSO4+DTBP in NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(2mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D109−03R(担持量0.191mmol/g、107.3mg)を得た。
反応液と固相の懸濁液(0.012mL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをNMP(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D109が92.5%の純度で観測された。
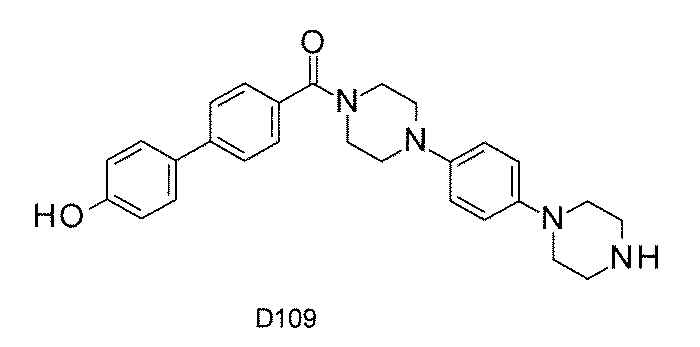
LRMS:m/z 443[M+H]+。
保持時間:0.605分(分析条件SMD−FA05−1、290nm)
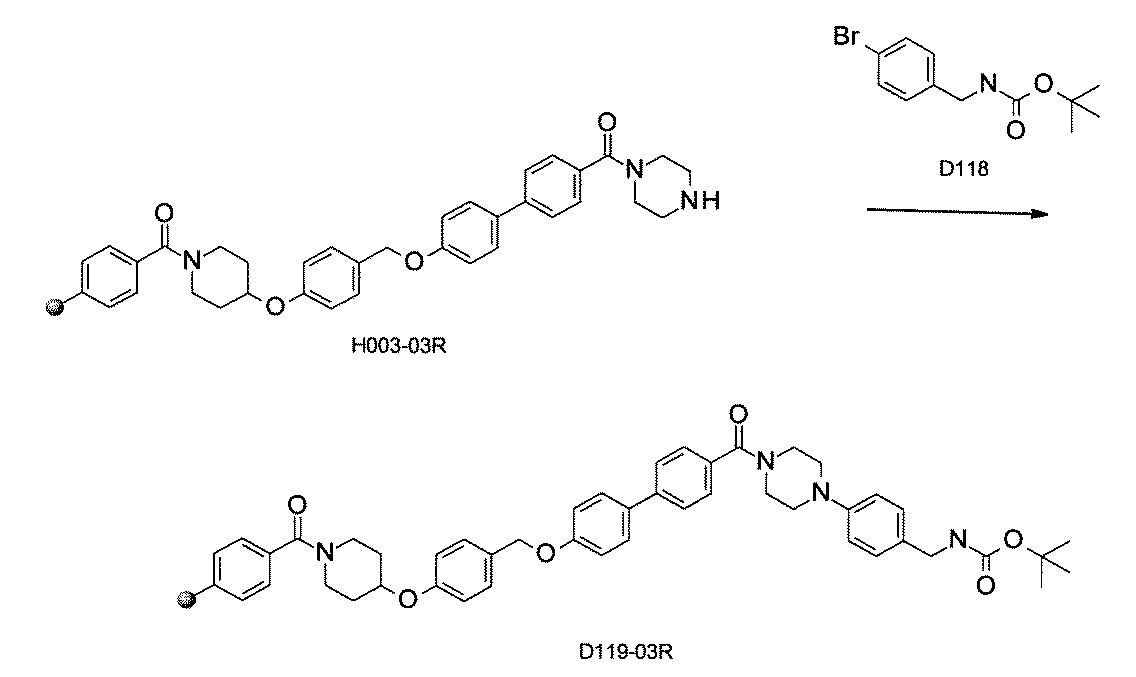

LRMS:m/z 488[M+H]+。
保持時間:1.087分(分析条件SMD−FA05−1、290nm)


極大波長:267nm。
保持時間:0.611分(分析条件SMD−FA05−1、290nm)
固相担持スルフィン酸(化合物D093−03R)は、固相担持ArI(D090−03R)から以下のように合成した。

反応液と固相の懸濁液をNMP(0.4mL)を用いてフィルター上に移し、NMP/水(0.4mL)で3回、NMP(0.4mL)で3回、0.2M NAC in NMP/水=5/1(0.4mL)で3回、0.05M TBAHSO4+DTBP in NMP(0.4mL)で3回、0.05M NMM in NMP(0.4mL)で3回、NMP/水=1/1(0.4mL)で3回、NMP(0.4mL)で3回、MeOH(0.4mL)で3回、DCM(0.4mL)で3回、ヘプタン(0.4mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D093−03R(担持量0.196mmol/g、18.9mg)を得た。
反応液と固相の懸濁液(12μL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D093が96.5%の純度で観測された。
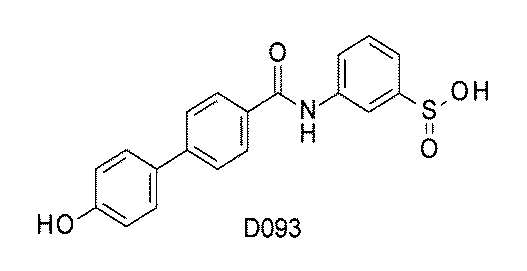
LRMS:m/z 354[M+H]+。
保持時間:0.736分(分析条件SMD−FA05−1、299nm)。
固相担持スルホニルクロリド(化合物D095−03R)は、固相担持N−Meアニリン(H002−03R)とベンゼン−1,3−ジスルホニルクロリド(D096)のスルホンアミド化により、以下のように合成した。

反応液と固相の懸濁液をDCM(0.4mL)を用いてフィルター上に移し、DCM(0.4mL)で3回、ヘプタン(0.4mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、化合物D095−03R(19.9mg、0.187mmol/g)を得た。
反応液と固相の懸濁液(12μL)をフィルター上に移し、DMA(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D095が91.0%の純度で観測された。

LRMS:m/z 571[M+H]+。
保持時間:1.156分(分析条件SMD−FA05−1、299nm)

反応液と固相の懸濁液(15μL)をフィルター上に移し、NMP(100μL)で3回、メタノール(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に2分間浸した。ろ過後、ろ液にDMA(200μL)を加えた。得られた溶液のうち50μLにアセトニトリル(100μL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物B103が99%観測された。

LRMS:m/z 320[M+H]+。
保持時間:0.867分(分析条件SMD−FA05−1、299nm)。

反応液と固相の懸濁液(15μL)をフィルター上に移し、NMP(100μL)で3回、メタノール(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に2分間浸した。ろ過後、ろ液にDMA(200μL)を加えた。得られた溶液のうち50μLにアセトニトリル(100μL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物B104が97%観測された。

LRMS:m/z 324[M+H]+。
保持時間:0.857分(分析条件SMD−FA05−1、299nm)。
ビルディングブロック(BB)同士を反応させて、コアブロックをリンカーで連結させることによる多様性を持たせたライブラリを供給するためには、ライブラリ合成時の反応条件における基質適用範囲を明確にし、高収率かつ高純度での反応実施が不可欠である。または、より広範囲の基質を高収率かつ高純度で反応実施できる反応条件を見いだし、その反応条件をライブラリ合成に適用していくことが不可欠である。
さらに、ある任意の反応において、一般的に知られている純度を低減し得る課題がある場合には、その課題を解決可能な反応条件をライブラリ合成に適用していくことが好ましい。実施例2においては、各結合形成反応において、ライブラリ合成に適用可能な反応条件の探索について例示する。
以下では、光延反応(Ar−O−Rの結合形成)において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
Ar−OH(アレノール)としてフェノールに種々の置換基を導入した基質にて、反応性の違いを確認した。なお、光延反応に影響を与える因子として求核種(Ar−OH(アレノール))の電子的要因が挙げられる。
窒素雰囲気下、3mLのスクリューキャップバイアルに、フェノール(10.00mg,0.106mmol,1.0当量)とTHF(1.06mL,0.10M)を加えた。続いて、1−プロパノール(31.9μL,0.425mmol,4.0当量)、TMAD(36.6mg,0.213mmol,2.0当量)、トリブチルホスフィン(52.4μL,0.213mmol,2.0当量)を室温にて加え、得られた混合物を室温にて撹拌した。
途中、反応開始から30分の段階で、窒素雰囲気下、反応混合物を1μL採取し、アセトニトリル200μLに溶解させ、LCサンプルを調製し、LCMS測定(分析条件SQD−FA05−1)を行うことで反応追跡を行った。
保持時間:0.93分(分析条件SQD−FA05−1)
(原料のフェノールの保持時間は、0.50分(分析条件SQD−FA05−1))
Ar−OH(1.0当量)、THF(0.10M)、1−プロパノール(4.0当量)、TMAD(2.0当量)、トリブチルホスフィン(2.0当量)、室温撹拌。
その結果は、以下の表X1の通りである。
また、反応変換率(conv.)は、特に記載しない限り、以下の通り計算した。
反応変換率(conv.)=生成物(Ar−O−nPr)の280nmでのUV面積%/(生成物(Ar−O−nPr)の280nmでのUV面積%+原料(Ar−OH)の280nmでのUV面積%)

光延反応において、R−OH(アルコール)を1−プロパノールに固定した場合に、求核種(Ar−OH(アレノール))の立体的要因が反応に及ぼす影響について、以下の通り確認した。
なお、特に記載がない限り、反応追跡は30分、1時間、2時間、3時間を基本として、LCMS測定(測定条件SQD−FA05−1)にて行い、原料Ar−OH(アレノール)の消失を確認した時間を記載した。
また、反応変換率(conv.)は、特に記載しない限り、以下の通り計算した。
反応変換率(conv.)= 生成物(Ar−O−nPr)の280nmでのUV面積%/(生成物(Ar−O−nPr)の280nmでのUV面積%+ 原料(Ar−OH)の280nmでのUV面積%)
表X−2のrun7およびrun8については、280nmの替わりに270nmでのUV面積にて確認を行った。

実施例2−1−1および実施例2−1−2の結果をあわせると、確認を行った範囲において、反応の完結の可否に影響を及ぼす要因として電子的要因の影響が大きいことが確認された。
以上の結果から、実施例2−1−3以降では、反応性を確認する基質として反応完結までに時間を要した4−ニトロフェノールをAr−OH(アレノール)として固定し、種々のR−OH(アルコール)との反応性の確認を行うこととした。
Ar−OH(アレノール)として4−ニトロフェノールに固定し、実施例2−1−1と同様の条件および操作によって反応実施をした。
4−ニトロフェノール(1.0当量)、THF(0.10M)、R−OH(4.0当量)、TMAD(2.0当量)、トリブチルホスフィン(2.0当量)、室温撹拌。
反応変換率(conv.)は、特に記載しない限り、以下の通り計算した。
反応変換率(conv.)= 生成物(Ar−O−nPr)の280nmでのUV面積%/(生成物(Ar−O−nPr)の280nmでのUV面積%+ 原料(Ar−OH)の280nmでのUV面積%)

続いて、2級アルコールを基質とした場合について反応確認した。3−ペンタノールを用いた場合には、20時間で完全に反応が完結することが確認できた(run7)。これにより、例えば、イソプロピルアルコールや2−ブタノールといった3−ペンタノールよりも嵩の低い範囲については同様に高反応変換率を達成できる可能性が極めて高い。
環状2級アルコールを基質とした場合には、5員環であるシクロペンチルアルコールでは反応を完結させられた一方で、6員環であるシクロヘキシルアルコールの反応性は極端に低下することが確認された(run11およびrun12)。
なお、基質内にアミノ基を含む場合にも、特に問題なく反応を完結させられることが確認できた(run15)。
もしくは固相側にAr−OH(アレノール)がある場合には、反応に用いた液相を排出し、試薬を入れ替えて、同じ反応を繰り返す操作(ダブルカップリングとも呼ばれる)においても、高反応変換率を達成できることを示している。
以上の液相での反応の確認結果を踏まえ、実施例2−1−5では実際にライブラリ合成をおこなう固相での反応確認をおこなった。
光延反応は、至適な酸性度の範囲の求核種であれば、Ar−OH(アレノール)に限らず、スルホンアミドも適用可能であることが知られている。スルホンアミドを基質とした場合の反応の成否と、反応速度の確認をおこなった。


LCMS(ESI)M/z=321.1(M+H)+。
保持時間:0.90分(分析条件SQD−FA05−1)。


LCMS(ESI)M/z=335.3(M+H)+。
保持時間:0.93分(分析条件SQD−FA05−1)。
実施例2−1−1~2−1−3の液相実験にて確認した反応条件を適用して、固相担持されたAr−OHと表AA−01に示すAA004~AA007のアルコールとの光延反応を実施し、ライブラリ合成に適用可能なビルディングブロックの範囲を確認した。

任意の時間経過時に、窒素雰囲気下でレジンの懸濁液20μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、表AA−02に示す化合物を目的物(TM)の追跡対象とし、原料(SM)については下に示すAA003を追跡対象とした。分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長280nmにて確認した。

保持時間:0.82分(分析条件SQDFA05−1)。

なお、ここで示す基質範囲の場合においても、固相担持されたAr−OHは変換されずに残存していることから、反応時間の延長、反応温度、または試薬量の調整によって、反応を高変換率化できる余地はある。換言すれば、一態様において、当該炭素原子の嵩高さをひとつの指標としてライブラリ合成の事前の難易度判定に活用可能であるが、これよりも嵩高い場合においても必ずしも本反応が適用できないとは限らない。
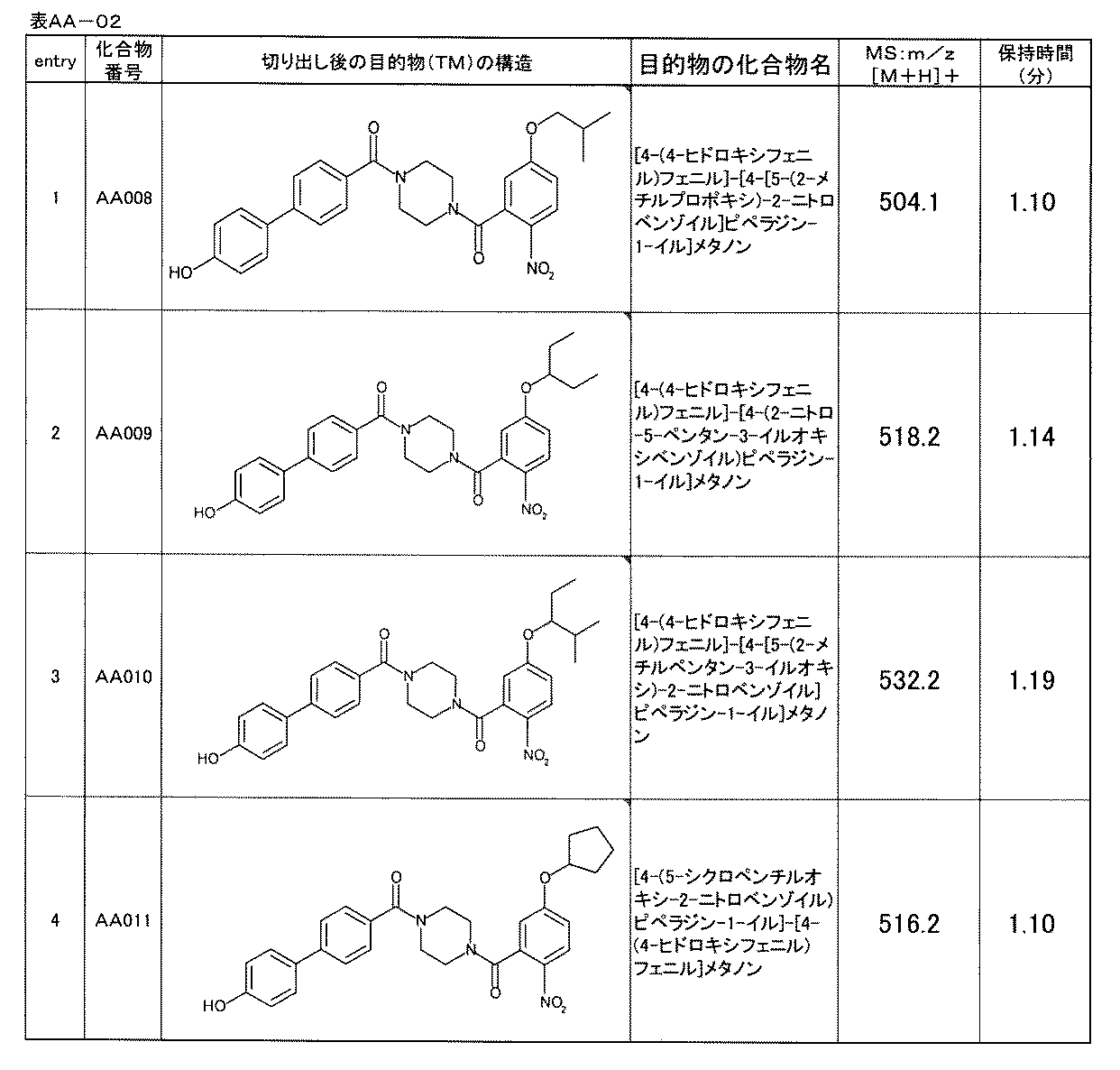

保持時間:0.82分。

任意の時間経過時に、窒素雰囲気下でレジンの懸濁液20μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、表AA−04に示す化合物を追跡対象として、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長280nmにて確認した。
22時間経過時におけるLCMSの結果について表AA−03に示す。Ar−OHであるAA013−03R、AA015−03R、AA017−03Rを用いた場合、目的物のエーテル体が75.0面積%以上で確認された。
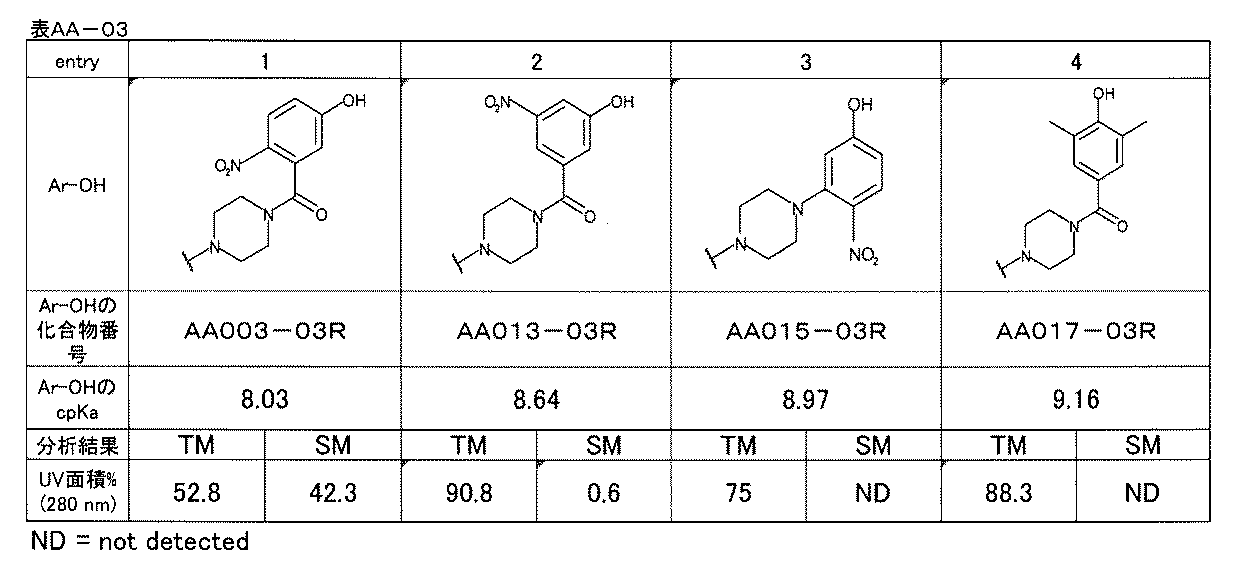
追跡対象の化合物について表AA−04に示す。
[表AA−04]追跡対象化合物の構造

本発明の手法による光延反応が固相反応においてもライブラリ合成に適用可能な実用的なレベルにあることが示された。また、本実施例はライブラリ合成実施前に適用可能な範囲を特定する方法の一例を例示したものである。結果としては、光延反応におけるAr−O−R結合形成が固相においてもライブラリ合成を行う上で実用的なレベルにあることが示されているが、反応条件の改善、および追加の基質検討や実際のライブラリ合成の知見を適用することで、Ar−OHならびにアルコールの基質範囲は、本実施例で示した範囲よりも拡大することもある。
実施例2−1−5の固相実験にて確認した反応条件を適用し、ライブラリ合成に適用可能な固相担持ArOH、並びにビルディングブロックであるアルコールの範囲を確認した。まず固相担持されたortho置換Ar−OHを使用して各種のArBrを有するアルコールとの光延反応を実施し、反応条件の確認を行った。
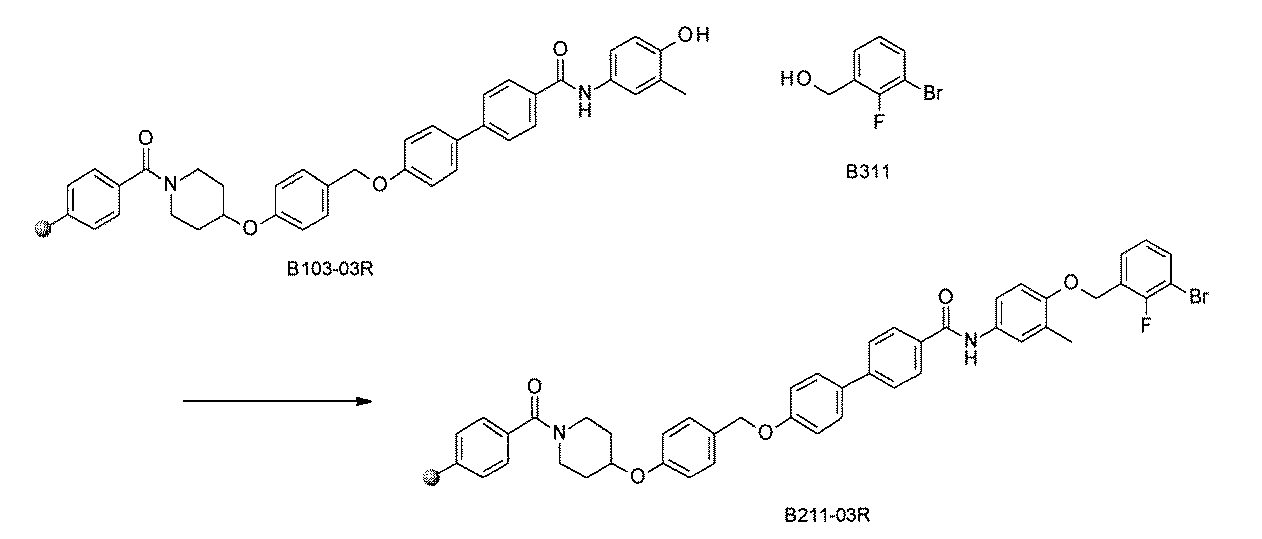
反応液と固相の懸濁液をフィルター上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物B211が95%観測された。

LRMS:m/z 506,508[M+H]+
保持時間:1.355分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 510,512[M+H]+
保持時間:1.301分(分析条件SMD−FA05−1、299nm)

LRMS:m/z 502,504[M+H]+
保持時間:1.376分(分析条件SMD−FA05−1、299nm)
実施例2−1−6−1~3では光延試薬としてnBu3P/TMADを使用しており、2級アルコールである(1−(3−ブロモフェニル)エタン−1−オール)との反応では生成物のエーテルを59%観測した。光延試薬を変更した場合に、エーテルの生成比を向上させることができるか確認する目的で、光延試薬をCMMPに変更して光延反応を実施した。



LRMS:m/z 502,504[M+H]+
保持時間:1.419分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 516,518[M+H]+
保持時間:1.449分(分析条件SMD−FA05−1、299nm)



LRMS:m/z 506,508[M+H]+
保持時間:1.349分(分析条件SMD−FA05−1、299nm)

LRMS:m/z 506,508[M+H]+
保持時間:1.381分(分析条件SMD−FA05−1、299nm)
ライブラリ合成に向けてより温和な反応条件を見出すことを目的として、反応温度を低下させて室温(25℃)下での光延反応を、固相担持フェノール誘導体(B105−03R)を基質として反応の確認を行った。


LRMS:m/z 492,494[M+H]+
保持時間:1.323分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 488,490[M+H]+
保持時間:1.385分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 488,490[M+H]+
保持時間:1.355分(分析条件SMD−FA05−1、299nm)
これらの結果から、CMMPを光延試薬として固相担持ArOHとアルコール、例えば置換ベンジルアルコール、置換フェネチルアルコールなどとの光延反応が、不特定多数の化合物を扱うライブラリ合成においてより好ましい温和な条件である室温(25℃)で反応を行える場合があることを確認した。
以下では、鈴木カップリングの適切な反応条件を選択することによって、再現性を高める条件を一例として例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
実施例2−2−0:鈴木カップリング反応の固相でのライブラリ合成時の課題抽出

表ER1−1A


LCMS:m/z 468[M+H]+。
保持時間:1.53分(分析条件 SMD−FA05−1)。
実施例2−2−0で特定した課題に対して解決できた実験は以下で示される。

振とう開始から1,2,27時間経過時にサンプリングを行った。固相上の反応変換率を確認する目的でEB02−05を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(200μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
窒素雰囲気下、4mLのガラスバイアルにP(Cy)3Pd G3(78mg、60μmol)にNMP3mLを加えて振とうすることで溶解し調製した。
結果は[表ER1−1B]、runBB04−2,3,4に示すとおりだった。runBB04−3に示すように、試薬であるBB04を78%残した状態で、原料が消失し反応が完結する条件を見出した。


LCMS:m/z 468[M+H]+。
保持時間:1.53分(分析条件 SMD−FA05−1)。
原料として化合物EA01−02−01Rを用いた場合においても、原料としてEB02−05−01Rを用いた場合と同様の条件で反応進行率を追跡し、鈴木カップリングが完結した実験は以下で示される。

振とう開始から1,2,27時間経過時にサンプリングを行った。
固相上の反応変換率を確認する目的でEB02−06を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(200μL)に溶解させ、その溶液にをTFE(800μL)を加えて希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
BB04を検出対象に、上記操作で行った、NMP(100μL)で3回洗浄したろ液をフィルター付きバイアルでろ過して、LCサンプルを調製した。BB04標準溶液のピーク面積と濃度を計算して比較することで試薬の消失速度進行度を測定した。なお、UV面積%は、PDA(210−400nm)にて確認した。


振とう開始から1,2時間経過時にサンプリングを行った。
固相上の反応変換率を確認する目的でEB06−02を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(200μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
表ER1−2


LCMS:m/z 575[M+H]+。
保持時間:1.47分(分析条件 SMD−FA05−1)。
不安定で消失速度が速いボロン酸を試薬として使用した場合、ピナコールを添加し、ボロン酸を安定なピナコールエステル体として存在させ、水を添加せず塩基をK3PO4−H2Oに変更することで、ボロン酸の消失を抑えながら反応を完結させた例を以下に示す。

窒素雰囲気下、2mLのガラスバイアルにP(Cy)3Pd G3(26mg、0.04mmol)に1,4−ジオキサン 1mLを加えて振とうすることで溶解し調製した。
反応追跡は以下のように実施した。振とう開始から1,2,14時間経過時にサンプリングを行った。
固相上の反応変換率を確認する目的で化合物EB20−02を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
[表ER1−3]


LCMS:m/z 445[M+H]+。
保持時間:1.25分(分析条件 SMD−FA05−1)。
以下の実施例では、特定のボロン酸における問題点の特定と、それらの解決策を特定した例について示す。
本実施例では、ボロン酸BB19を使用した際の反応を例示し、分子内にホルミル基を有する化合物を目的物とする鈴木カップリング反応の問題点を示す。
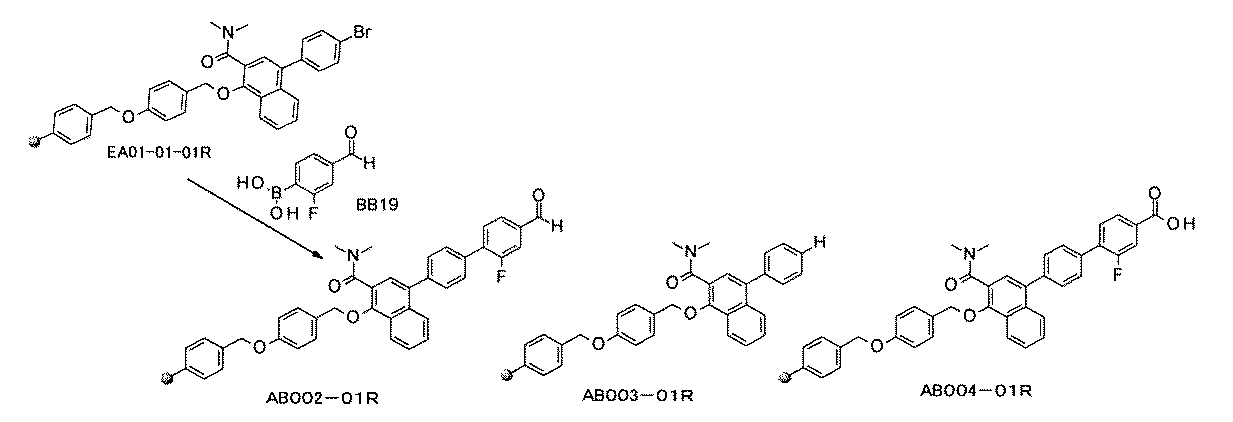
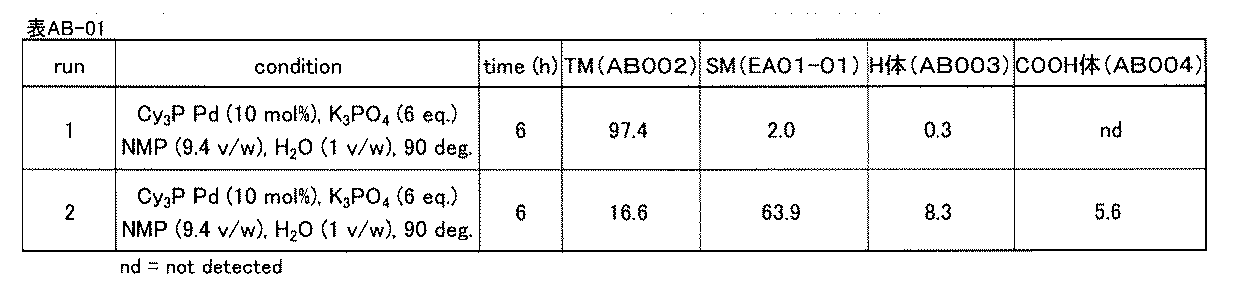
特定の理論に限定されないが、以上の結果から分子内にホルミル基を有する化合物を目的物とする鈴木カップリング反応、特にボロン酸が液相において消費してしまう副反応が進行する場合においては、固相上の原料の変換率の再現性を担保することに課題があることが特定された。
以下に代表的な実験操作として、表AB−01のrun1の実験について示す。
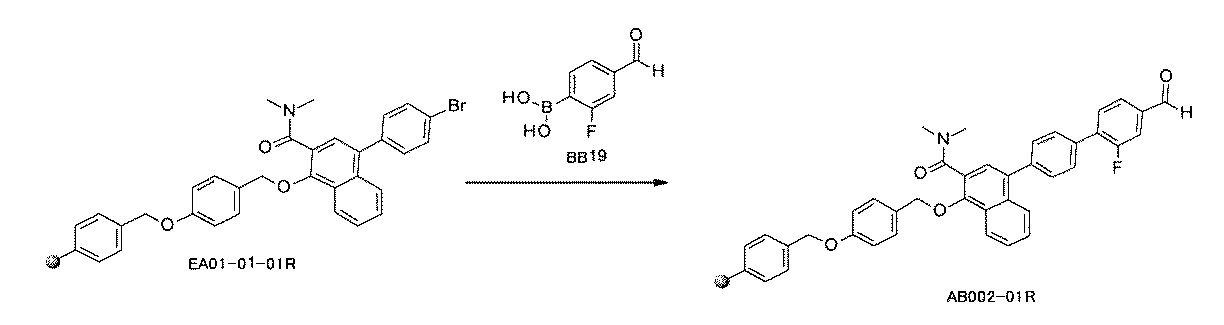
振とう開始から任意の時間経過時に、固相側、液相側共にサンプリングを行った。
固相上の反応変換率を確認する目的で、表AB−02に示した化合物を追跡対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(100μL)に5分間浸した。ろ過後、ろ液をTFE(250μL)に溶解させることでLCサンプルを調製し、分析条件SMD−FA05−1にてLCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。

(2−フルオロ−4−ホルミルフェニル)ボロン酸(BB19)

保持時間:0.61分(分析条件 SMD−FA05−1)。
液相中での消失が早いボロン酸に対して、非特許参考文献(ACS Catal.2018,8,4,2989−2994)を参考に、ピナコールを添加することでボロン酸を安定なピナコールエステル体として存在させつつ、水の当量を制限してボロン酸の消失を抑制する反応条件の探索を行った。その結果、水を添加せず塩基をK3PO4−H2Oに変更することで、ボロン酸の消失を抑えながら反応を完結させた。操作の一例を示す。

表ER1−4


保持時間:1.14分(分析条件 SMD−FA05−1)
実施例2−2−6で合成法を例示したEB19−02−01Rは、これ以降の実施例において、化合物AC005−01Rと表記することもある。
ホルミル基を有する目的物の場合に、実施例2−2−0~2−2−4の条件ではCannizzaro反応と思われる副反応が進行したが、その問題が解決した条件で反応が完結した例を示す。レジンを膨潤させる工程で水を添加することにより、塩基のNMP−水の混合溶媒に対する溶解度の再現性を高め、反応進行率が遅い条件の場合でも、反応の進行率の再現性を高めバッチ間でのブレを少なくした一例を示す。

0.04MのP(Cy)3Pd G3のNMP溶液はEB02−05−01Rと同様に調製した。
振とう開始から1,3,19,25,91時間経過時にサンプリングを行った。
固相上の反応変換率を確認する目的でEB08−15を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(200μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
結果は表ER1−5、runBB15−1,2,3,4、5に示すとおりだった。


LCMS:m/z 440[M+H]+。
保持時間:1.47分(分析条件 SMD−FA05−1)。
溶媒をNMPから4−MeTHPに変更し、ボロン酸の消費を抑え、Cannizzaro反応の問題を解決し、反応が完結した一例を示す。
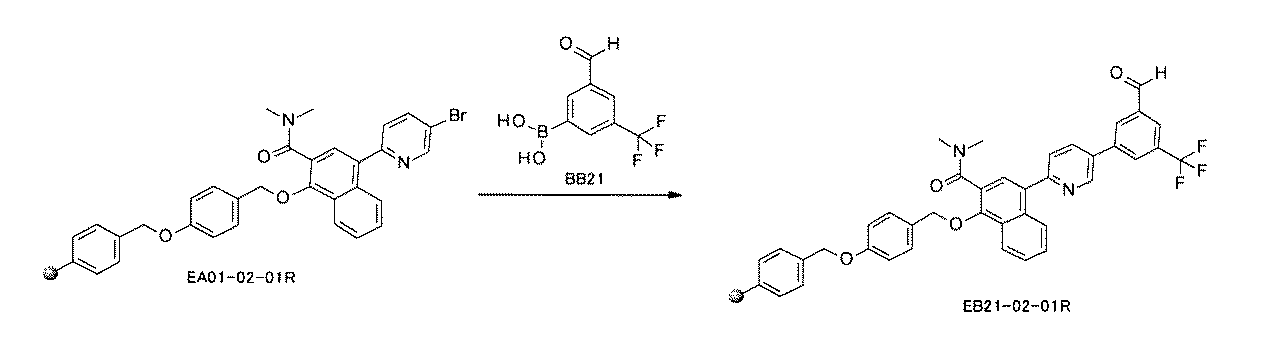
窒素雰囲気下、2mLのガラスバイアルにP(Cy)3Pd G3(26mg、0.04mmol)に4−MeTHP溶液1mLを加えて振とうすることで溶解し調製した。
振とう開始から1,3,21時間経過時にサンプリングを行った。
固相上の反応変換率を確認する目的でEB21−02を検出対象に、窒素雰囲気下にて反応液とレジンの懸濁液10μLを必ずレジンが含まれるようサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(200μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、UV面積%は、UV波長319nmにて確認した。
表ER1−6

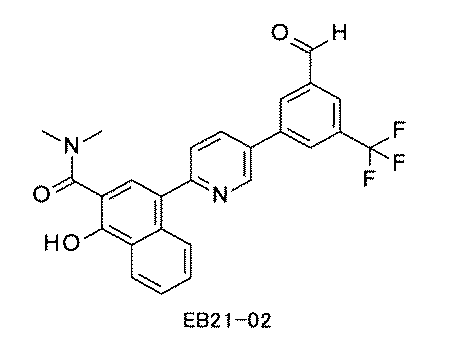
LCMS:m/z 411[M+H]+。
保持時間:1.27分(分析条件 SMD−FA05−1)。
実施例2−2−8で合成法を例示したEB21−02−01Rは、これ以降の実施例において、化合物AC007−01Rと表記することもある。
有機塩基を用いることで、幅広い基質に対して官能基許容性高く、鈴木カップリングが適用可能となった条件を示す。
実施例2−2−9−1:固相担持ArBr(C011−03R)と2,6−ジメチルフェニルボロン酸(C305)の鈴木カップリングによるC207−03Rの合成
有機塩基としてBTMGを用いた鈴木カップリング反応において、Pd触媒によっては目的物の固相からの切り出しが進行してしまうのに対し、Pd触媒としてmeCgPPhPdG4や(dtbpf)PdCl2を用いた場合には固相からの切り出しなく高収率にて鈴木カップリング反応が行える一例を示す。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
実施例2−2−9−1−1:Pd触媒としてmeCgPPhPdG4を使用した鈴木カップリング反応

反応液の上清のうち3μLをMeCN(0.5mL)に溶解した溶液のLCMS測定を行った結果、固相から切り出された目的物C207は観測されなかった。
反応液と固相の懸濁液のうち12μLをフィルター付きチップ上に移し、NMP(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した。ペンタメチルベンゼンの10%TFA/DCM溶液(0.1M、0.05mL)に2分間浸してろ過し、NMP(0.05mL)で洗浄し、合わせたろ液をMeCN(0.25mL)で薄めた溶液のLCMS測定を行うことで、目的物C207が99.8%観測された。ただし、波長299nm(±4nm)で切り出して解析している。

LRMS:m/z395[M+H]+。
保持時間:1.040分(分析条件SMD−FA05−1)。
実施例2−2−9−1−2:Pd触媒として(dtbpf)PdCl 2 を使用した鈴木カップリング反応
meCgPPhPdG4の代わりに(dtbpf)PdCl2(2.5mg、0.0039mmol)を用いて、実施例2−2−9−1−1と同様の操作を行い、60℃で24時間振盪した。
反応液の上清のうち3μLをMeCN(0.5mL)に溶解した溶液のLCMS測定を行った結果、固相から切り出された目的物C207は観測されなかった。
反応液と固相の懸濁液のうち12μLをフィルター付きチップ上に移し、NMP(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した。ペンタメチルベンゼンの10%TFA/DCM溶液(0.1M、0.05mL)に2分間浸してろ過し、NMP(0.05mL)で洗浄し、合わせたろ液をMeCN(0.25mL)で薄めた溶液のLCMS測定を行うことで、目的物C207が99.2%観測された。ただし、波長299nm(±4nm)で切り出して解析している。
実施例2−2−9−1−3:Pd触媒としてXPhosPdG4やRuPhosPdG4、APhosPdG4、cataCXiumPdG4を使用した鈴木カップリング反応
meCgPPhPdG4の代わりにXPhosPdG4(3.4mg、0.0039mmol)もしくはRuPhosPdG4(3.3mg、0.0039mmol)、APhosPdG4(2.5mg、0.0039mmol)、CataCXiumPdG4(2.9mg、0.0039mmol)を用いて、いずれにおいても実施例2−2−9−1−1と同様の操作を行い、60℃で24時間振盪した。
反応液の上清のうち3μLをMeCN(0.5mL)に溶解した溶液のLCMS測定を行った結果、いずれの条件においても固相から切り出された目的物C207が観測された。
実施例2−2−9−1−1~3の結果から、Wangレジンに代表されるp−アルコキシベンジルリンカーを介してアレノールがエーテル結合にて固相担持された基質に対して、Pd触媒としてXPhosPdG4やRuPhosPdG4、APhosPdG4、cataCXiumPdG4を用いた場合には鈴木カップリング反応時に目的物の固相からの切り出しが進行し収率低下の原因となるのに対して、Pd触媒としてmeCgPPhPdG4や(dtbpf)PdCl2を用いた場合には固相からの切り出しなく高収率にて鈴木カップリング反応が行えることが示された。
実施例2−2−9−2:固相担持ArBr(C009−03R)と2,6−ジメチルフェニルボロン酸(C305)の鈴木カップリングによるC204−03Rの合成

LRMS:m/z 394[M+H]+
保持時間:1.335分(分析条件 SMD−FA05−1)。
実施例2−2−9−1~2の結果より、固相担持ArBrとボロン酸との鈴木カップリング反応が、有機塩基BTMGを用いることで、60℃という温和な条件下、実施可能であることが示された。
固相担持Arボロン酸エステル(D122−03R)とエチル 4−(4−ヒドロキシフェニル)ベンゾアト(D124)を用いた鈴木カプリングの固相実験(反応操作の例示)

窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(100μL)に5分間浸した。切り出し液をろ過し、固相をNMP(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D123が98.4面積%観測された。
LRMS:m/z 243[M+H]+。
保持時間:1.085分(分析条件SMD−FA05−1、295nm)。
以下では、アセチレンカップリング反応の適切な反応条件を選択することによって、堅牢性を高める条件を一例として例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
広範囲の基質に適用可能と考えられる反応条件として、公知文献(J.Org.Chem.2013,78,568−581,J.Orgnomet.Chem.1975,93,253,並びにJ.Organomet.Chem.1975,93,259)を参考にしてCuIを用いない条件を選択した。塩基は固相反応に適応容易な有機塩基からDIAを選択し、Pd触媒としてPd(PPh3)4を選択してアセチレン誘導体とのカップリング反応を実施した。

途中、反応開始から3時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(1000μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるArBrが2.7%残存し、生成物のアセチレン体(B003)は95.9%観測された。
また、反応終了時の22時間経過時には原料のArBrが消失し、反応が完結していることを確認した。

LCMS:m/z 450.1[M+H]+。
保持時間:1.514分(分析条件SMD−FA05−1,319nm)。
(原料ArBrの保持時間は、1.421分(分析条件SMD−FA05−1,319nm)。
電子求引性基であるトリフルオロメチル基とエステルを有するアセチレン誘導体とのカップリング反応において、Pd(PPh3)4を触媒とした際にカップリング反応が完結しない反応例を以下に示す。

反応開始から15時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(100μL)に5分間浸した。ろ過後、ろ液をTFE(100μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるArBrが35.1%残存し、生成物のアセチレン体(B005)は54.1%観測された。

LCMS:m/z 532.1[M+H]+。
保持時間:1.204分(分析条件SMD−FA50−1,319nm)。
(原料のArBrの保持時間は、0.786分(分析条件SMD−FA50−1,319nm)。
実施例2−3−4で合成法を例示したB034−01Rは、これ以降の実施例において、化合物AF001−01Rと表記することもある。
実施例2−3−2にて、反応の変換率が35%にとどまってしまった基質について、反応条件を検討した結果、触媒をXphosPd G4に変更することで反応を完結させることが可能であることを確認した。以下に、具体的な操作を示す。

途中、反応開始から1時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(1000μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるArBrが36.5%残存し、生成物のアセチレン体(B005)が61.9%観測された。
その後経時的に反応を追跡し(3,5,7,22時間)、22時間経過時に原料が消失しアセチレン体(B005)を98.8%観測した。
この結果から、Pd(PPh3)4では十分に反応が進行しないことが確認された電子不足アセチレンとの反応の場合においても、触媒をXphosPd G4とすることで、反応を完結させることができる反応条件を見出した。
実施例2−3−3にて見出した反応条件において、固相側の基質としてピリジン環を有するArBrと電子不足アセチレン誘導体との反応を確認した。

途中、反応開始から1時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(1000μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるArBrが25.8%残存し、生成物のアセチレン体(B008)を71.6%観測された。
この結果から、実施例2−3−3にて見いだした条件は、固相側の幅広いArBrに対して、電子不足アセチレン誘導体とのカップリング反応を高い変換率にて達成できることを確認した。
LCMS:m/z 533.1[M+H]+。
保持時間:1.034分(分析条件SMD−FA50−1,319nm)。
(原料のArBrの保持時間は、0.501分(分析条件SMD−FA50−1,319nm)。
実施例2−3−4で合成法を例示したB034−01Rは、これ以降の実施例において、化合物AF001−01Rと表記することもある。
以下では還元的アミノ化反応のライブラリ合成に適用可能な実施方法を一例として示し、適切な安定性の保護基の選択・活用例についても例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
還元的アミノ化反応の条件設定の過程において、問題点を発見し解決策を特定する例を示す。
還元的アミノ化反応に用いられる一般的な還元剤、酸、溶媒を用いて、固相における反応進行を確認した。還元剤としてテトラメチルアンモニウムトリアセトキシボロヒドリド、酸としてAcOH、溶媒としてDCEを選択し、アルデヒドAC005−01RとアミンBB28を基質とした還元的アミノ化反応を実施した。アルデヒドAC005−01Rは実施例2−2−6にて、アミンBB28は実施例1−4−4にて合成した。以下に操作の一例を示す。
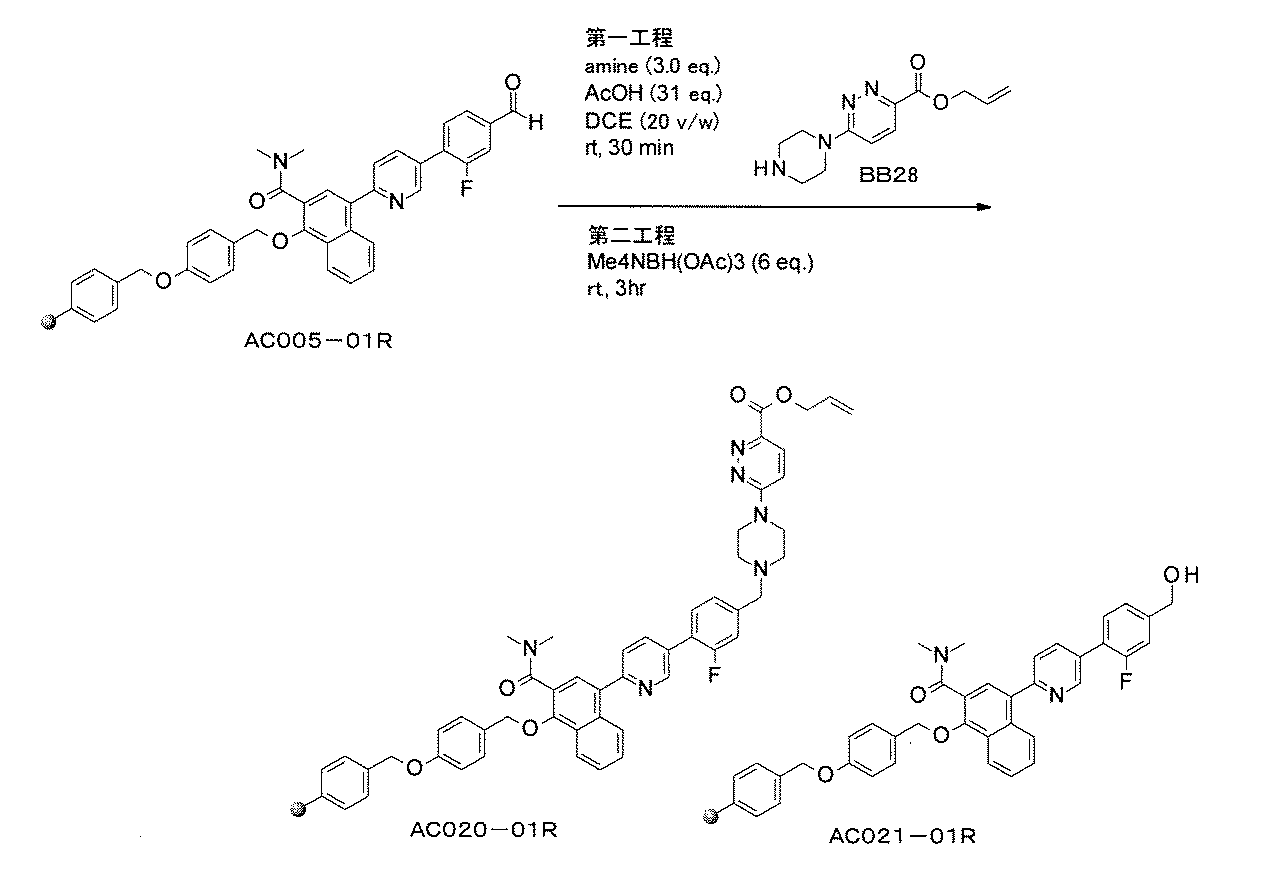
窒素雰囲気下、任意の時間において、レジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.2Mトリメチルベンゼンを含む10%TFA/DCM溶液(100μL)に5分間浸した。ろ過後、ろ液をTFE:トルエン(9:1、500μL)に溶解させることでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を追跡対象とし、分析条件SMD−AC05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
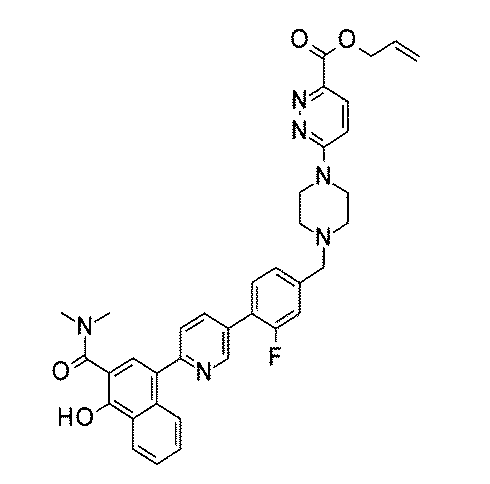
MS:m/z 647.2[M+H]+
保持時間:1.28分
(原料のArCHOの保持時間は、1.11分)

MS:m/z 417.0[M+H]+。
保持時間:0.96分。
(原料のArCHOの保持時間は、1.11分)。

窒素雰囲気下、任意の時間において、レジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、表AC−02に示す化合物を追跡対象とし、分析条件SMD−AC05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
[表AC−01]
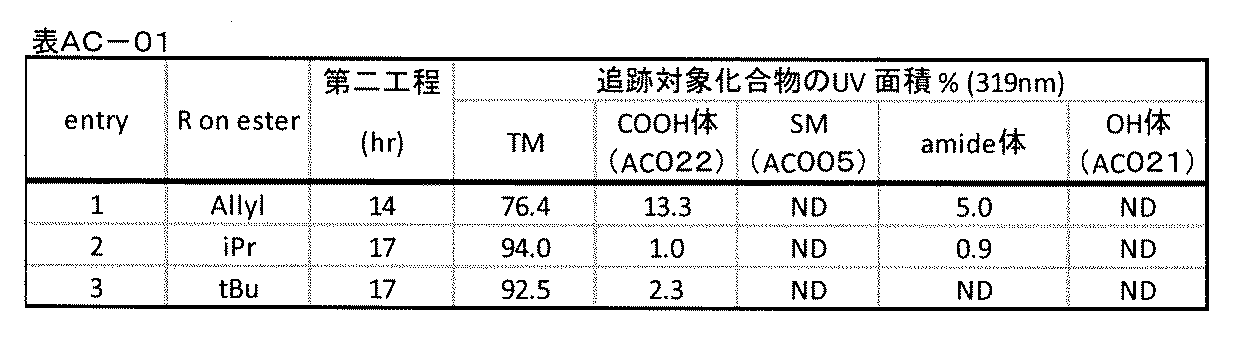
本実験の結果より、本発明の手法によって、アルデヒドに対して還元剤としてトリアセトキシ水素化ホウ素ナトリウム、ルイス酸としてTi(OtBu)4、添加剤としてAcOH、溶媒としてDCEを用いることで、固相においてもOH体の副生を回避できることが明らかとなった。さらには、適切な保護基を選択することでアミド体の副生を回避し、アミノカルボン酸エステルを高収率で還元的アミノ化に用いることが可能であることが明らかになった。


様々な官能基を有し反応性の異なるアルデヒドとBB29との還元的アミノ化反応を行い、基質適用範囲を明らかにした。実験は、実施例2−4−1にて見出した反応条件を参考に、表AC−03に示した化合物とBB29を用いた。アルデヒドAC005−01Rは実施例2−2−6にて、AC004−01Rは実施例1−4−8にて、AC006−01Rは実施例1−4−9にて、AC007−01Rは実施例2−2−8、AC003−01Rは実施例1−4−7にて合成した。

窒素雰囲気下にてレジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に表AC−04に示す化合物を追跡対象とし、分析条件SMD−AC05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
[表AC−03]




窒素雰囲気下にてレジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をDMI(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に表AC−06に示す化合物を追跡対象とし、分析条件SMD−AC05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
[表AC−05]
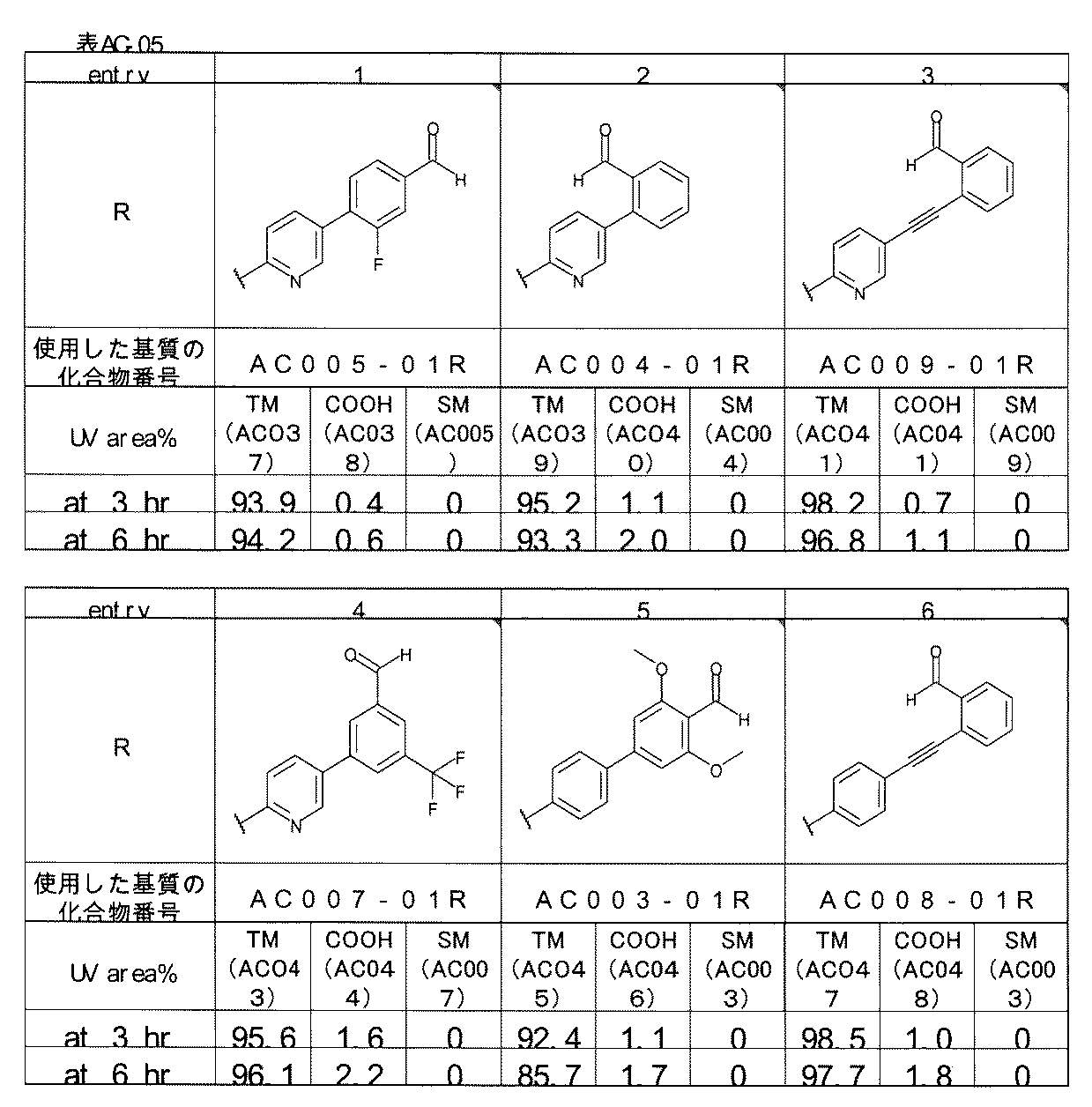



保護基を選択する際には、当然のことながら脱保護法の確認も重要である。特にライブラリ合成においては、複数の基質中に複数の官能基の存在下、高い官能基耐性を維持した上での脱保護が望ましい。今回の例においては、嵩高いエステルであるtBuエステルの脱保護が他の官能基に影響を与えることなく脱保護反応が高収率で進行することは、ライブラリの純度担保の上で必須である。

反応追跡は以下のように実施した。
[表AC−07]

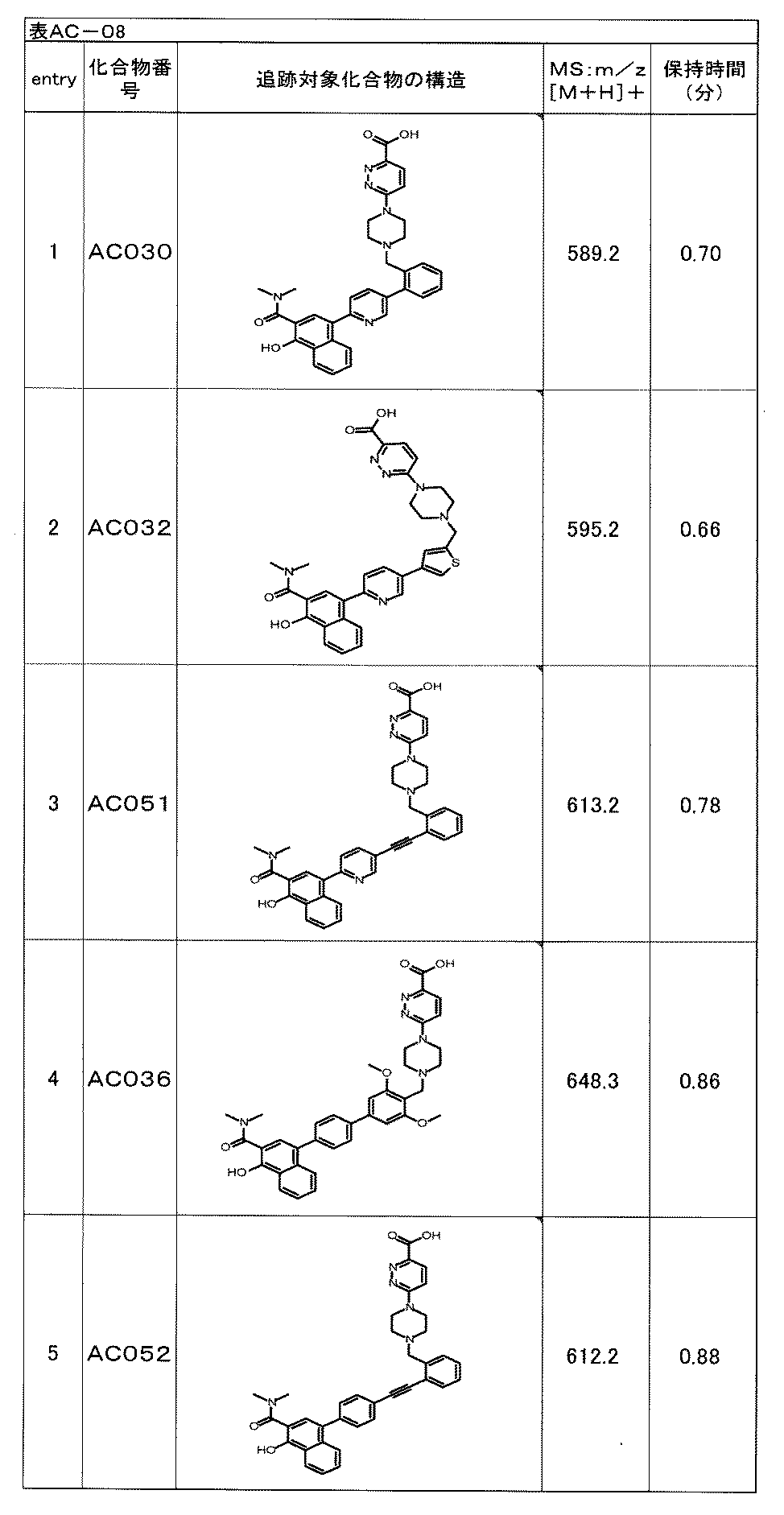
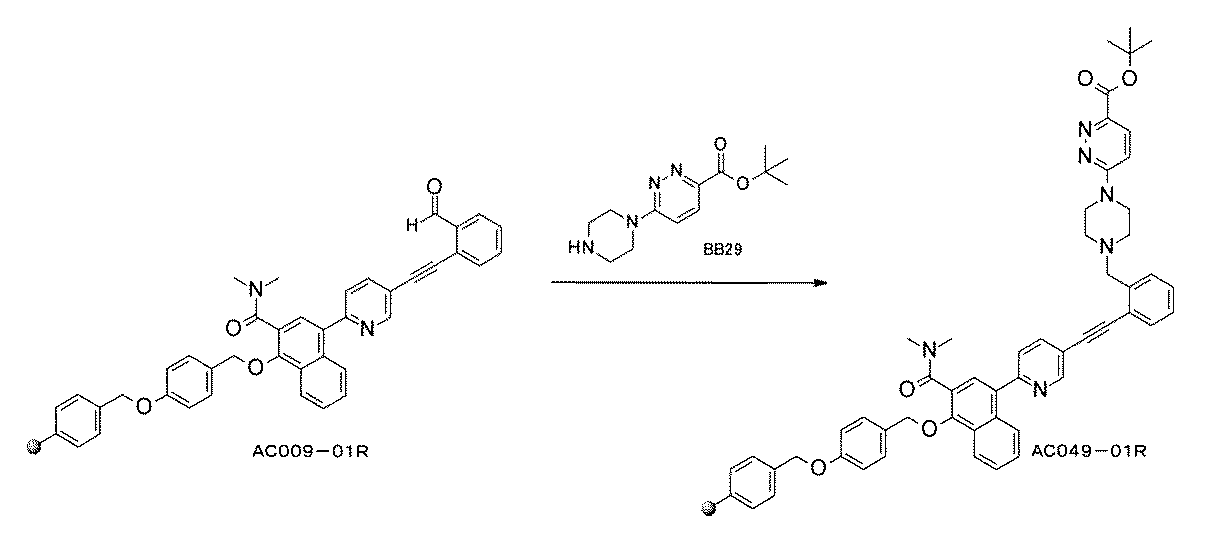
窒素置換されたグローブバッグ中にてレジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、MeOH(100μL)で3回、DCM(100μL)で3回繰り返して洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA−DCM(切り出し溶液)(50μL)に5分間浸した。切り出し液をろ過し、濾液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を追跡対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

MS:m/z 669.3[M+H]+。
保持時間:0.92分。
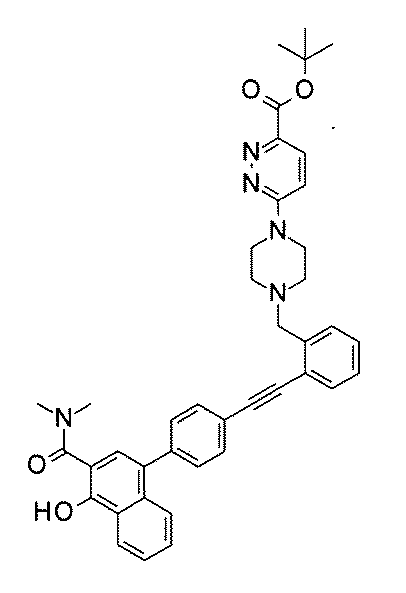
MS:m/z 668.4[M+H]+。
保持時間:1.02分。
固相担持アミンとアルデヒドに対する還元的アミノ化反応を、還元剤としてNaBH(OAc)3を選択しNMP中酢酸を添加する条件で実施した。

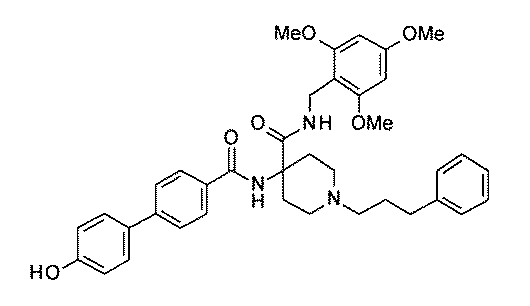
LRMS:m/z 638[M+H]+
保持時間:0.839分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 451[M+H]+
保持時間:1.056分(分析条件SMD−FA05−1、299nm)

反応液と固相の懸濁液をフィルター上に移し、DMA(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMA(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物H103が89%観測された。

LRMS:m/z 660[M+H]+
保持時間:0.848分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 624[M+H]+
保持時間:0.817分(分析条件SMD−FA05−1、299nm)
以下ではケトンに対する還元的アミノ化反応のライブラリ合成に適用可能な実施方法を一例として示し、適切な安定性の保護基の選択・活用例についても例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
公知文献(J.Med.Chem.2007,50,3528)を参考にしてTi(OiPr)4をケトンの活性化剤として選択した。また、高温での加熱反応を実施するためにエーテル系溶媒としてMe−THPを反応溶媒として選択し、固相アリールケトンとピペリジンとの還元的アミノ化反応を実施した。

反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(17.80mg、0.084mmol,12当量)を加え、100℃で15時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから15時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、10%TFA/DCM溶液(100μL)に5分間浸した。ろ過後、ろ液をTFE(100μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるケトン(B006)が23%残存し、生成物のエチルエステル体(B014)は11%,イソプロピルエステル体(B015)が48%であった。
高純度で混合物ライブラリを調製するためには、原料のケトンの残存を低減させ(高い反応変換率の達成)、かつエステル交換が進行しにくい(望まない副反応の低減)反応条件を検討する必要性が確認できた。

LCMS:m/z 629.2[M+H]+。
保持時間:1.594分(分析条件SMD−FA05−1,319nm)。
(原料ケトンの保持時間は、1.106分(分析条件SMD−FA05−1,319nm)。

LCMS:m/z 643.2[M+H]+。
保持時間:1.663分(分析条件SMD−FA05−1,319nm)。
エステル交換を低減させる目的で活性化剤をTi(OtBu)4に変更して、固相担持ケトン(B013−01R)と4−(ピペラジン−1−イル)安息香酸エチル(B010)との還元的アミノ化反応を実施した。

反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(17.80mg、0.084mmol,12当量)を加え、100℃で15時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから15時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、10%TFA/DCM溶液(100μL)に5分間浸した。ろ過後、ろ液をTFE(100μL)に溶解させ、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
その結果、原料であるケトン(B013)の残存量が7%に低減し、生成物のエチルエステル体(B014)が74%に増加した。また、Ti(OiPr)4の替わりにTi(OtBu)4を用いることで、実施例2−5−1にて見られたイソプロピルエステル体などのエステル交換体の生成は回避できた。Ti(OtBu)4を活性化剤として使用することが変換率向上に有効であることを確認した。一方、ケトンの還元体であるアルコール(B016)が11%観測された。
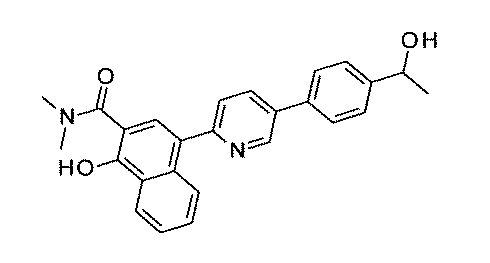
LCMS:m/z 413.1[M+H]+。
保持時間:0.810分(分析条件SMD−TFA05−1,319nm)。
変換率をさらに向上する目的で、反応温度を高くするためにアニソールを反応溶媒として選択し、反応温度を120℃に設定して還元的アミノ化反応を実施した。
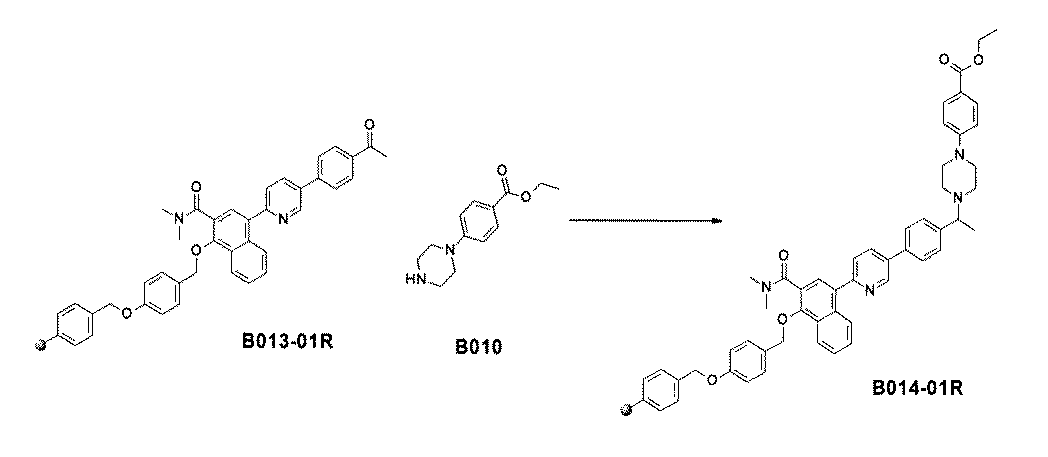
反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(12.2mg,0.058mmol,12当量)を加え、120℃で15時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから15時間経過時に、実施例2−5−2と同様に測定を行った。
その結果、原料であるケトン(B013)の残存量が3%に低減、生成物のエチルエステル体(B014)が84%に増加した。また、ケトンの還元体であるアルコール(B016)が12%観測されたため、特定の理論に限定されないが、一段階目のイミニウム形成が不十分であることが明らかとなった。
次に、4−(ピペラジン−1−イル)安息香酸アリル(BB27)を用いた還元的アミノ化を実施した。

反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(12.21mg、0.058mmol,12当量)を加え、120℃で18時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから18時間経過時に、実施例2−5−2と同様に測定を行った。
その結果、原料であるケトン(B013)の残存量が25%、生成物のアリルエステル体(B018)が40%、また、ケトンの還元体であるアルコール(B016)が15%観測された。

LCMS:m/z 641.1[M+H]+。
保持時間:0.910分(分析条件SMD−FA05−1,319nm)。
変換率を向上する目的で、反応温度を140℃に設定し、イミニウム形成を促進するためにアミン、Ti(OtBu)4の当量を増加して反応を実施した。
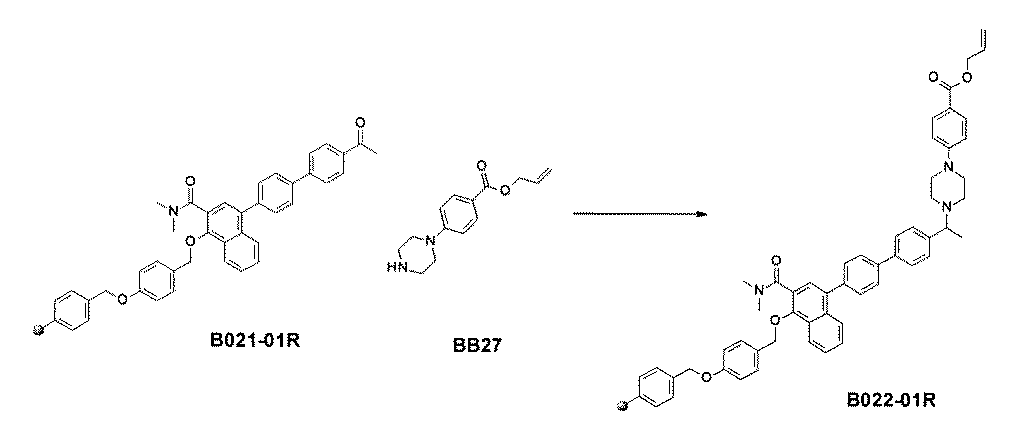
反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(16.00mg、0.077mmol,12当量)を加え、140℃で5時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから5時間経過時に、実施例2−5−2と同様に測定を行った。
結果を表2−5−5,Run1に示す。
窒素雰囲気下、1.5mLのスクリューキャップバイアルに、固相担持アリールケトンB021−01R(0.32mmol/g,20.0mg,6.4μmol,1.0当量)とアニソール(500μL)を加え、室温下1.5時間振とうした。窒素雰囲気下で4−(ピペラジン−1−イル)安息香酸アリル(BB27)(19.00mg,0.077mmol,12当量)、Ti(OtBu)4(49.0μL,0.128mmol,20当量)を加え、得られた混合物を140℃で21時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから5時間経過時に、実施例2−5−2と同様に測定を行った。結果を表2−5−5,Run2に示す。

LCMS:m/z 640.2[M+H]+。
保持時間:1.067分(分析条件SMD−FA05−1,319nm)。

LCMS:m/z 656[M+H]+。
保持時間:1.127分(分析条件SMD−FA05−1,319nm)。

LCMS:m/z 600[M+H]+。
保持時間:0.941分(分析条件SMD−FA05−1,319nm)。
[表2−5−5]

さらに変換率を向上させる目的で、トリアセトキシ水素化ホウ素ナトリウムの当量を増加した反応とスケールアップ反応による反応条件の確認を実施した。

窒素雰囲気下、1.5mLのスクリューキャップバイアルに、固相担持アリールケトンB021−01R(0.35mmol/g,20.0mg,7.0μmol,1.0当量)とアニソール(500μL)を加え、室温下1.5時間振とうした。窒素雰囲気下で4−(ピペラジン−1−イル)安息香酸アリル(BB27)(21.00mg,0.084mmol,12当量)、Ti(OtBu)4(54.0μL,0.140mmol,20当量)を加え、得られた混合物を140℃で22時間振とうした。
反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(45.00mg,0.210mmol,30当量)を加え、140℃で20時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから20時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(200μL)とアセトニトリル(800μL)で希釈してLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。結果を表2−5−6,Run1に示す。
窒素雰囲気下、4.0mLのスクリューキャップバイアルに、固相担持アリールケトンB021−01R(0.35mmol/g,100.0mg,35μmol,1.0当量)とアニソール(2.5mL)を加え、室温下1.5時間振とうした。窒素雰囲気下で4−(ピペラジン−1−イル)安息香酸アリル(BB27)(103.0mg,0.42mmol,12当量)、Ti(OtBu)4(270.0μL,0.700mmol,20当量)を加え、得られた混合物を140℃で21時間振とうした。
反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(223.0mg、1.05mmol,30当量)を加え、140℃で18時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから18時間経過時に、実施例2−5−6−1と同様に測定を行った。結果を表2−5−6,Run2に示す。
[表2−5−6]

固相担持ケトンB021−01Rで設定した還元的アミノ化反応条件下、適用基質を拡大する目的でピリジンを有する基質B013−01Rで反応進行確認を実施した。

反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(235.0mg、1.10mmol,30当量)を加え、140℃で20時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから20時間経過時に、実施例2−5−6−1と同様に測定を行った。結果を表2−5−7に示す。
[表2−5−7]

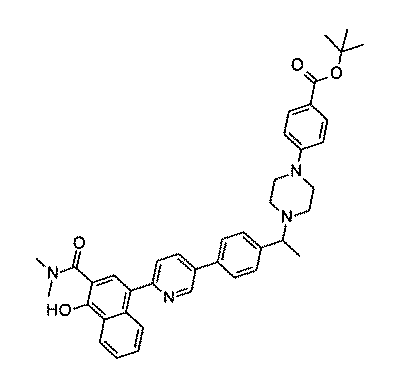
LCMS:m/z 657[M+H]+。
保持時間:0.957分(分析条件SMD−FA05−1,319nm)。
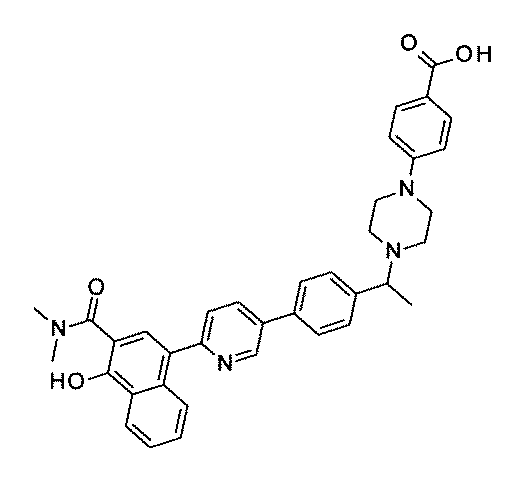
LCMS:m/z 601.3[M+H]+。
保持時間:0.760分(分析条件SMD−FA05−1,319nm)。
これまでに設定した還元的アミノ化反応条件を用い、適用基質を拡大する目的でフッ素を有する基質(B025−01R)で反応が進行することを確認した。

反応液を室温に冷却後、窒素雰囲気下でトリアセトキシ水素化ホウ素ナトリウム(45.00mg、0.210mmol,30当量)を加え、140℃で20時間振とうした。
トリアセトキシ水素化ホウ素ナトリウムを加えてから20時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLサンプリングしてフィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をTFE(200μL)とアセトニトリル(800μL)で希釈してLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。結果を表2−5−8に示す。


LCMS:m/z 658.3[M+H]+。
保持時間:1.112分(分析条件SMD−FA05−1,319nm)。

LCMS:m/z 674.3[M+H]+。
保持時間:1.181分(分析条件SMD−FA05−1,319nm)。

LCMS:m/z 618.3[M+H]+。
保持時間:0.941分(分析条件SMD−FA05−1,319nm)。
混合物ライブラリの合成において高い反応変換率を達成していくためには、個々の反応条件を適切に設定するだけでなく、反応実施後の洗浄工程についても工夫が必要である。以下では、エステル部分の加水分解反応後の洗浄を適切な方法を選択することによって、続くアミドカップリング反応の堅牢性を高める方法を一例として例示する。
塩基を用いてエステル部位を加水分解し、次の結合形成反応に用いるカルボン酸を露出させる場合、該当カルボン酸がフリー体ではなく、用いた塩基のカウンターカチオンとの塩を形成した状態で存在する可能性がある(式AF−1)。例えば、次の結合形成反応が縮合剤を用いたアミド化である場合、縮合剤の種類によっては、カルボン酸がフリー体でなく塩形成をしていることに由来する反応変換率の低下が懸念される。以下では、エステル部位の加水分解後の洗浄方法の違いが次工程のアミドカップリング反応に与える影響について検証した。
[式AF−1]

加水分解後のレジン洗浄においては、種々カルボン酸のpKaを考慮し、カルボン酸塩をプロトン化し得る試薬溶液として、ここでは硫酸水素テトラブチルアンモニウム塩/NMP溶液、もしくはHOAt/NMP溶液を選択した。アミドカップリング反応においては、アミンと共存可能な縮合剤、すなわち、アミンとの意図しない反応にて消費されない縮合剤としてDICを、また、カルボン酸とDICによって生じる一次活性エステルからのウレア転位抑制の添加剤としてHOAtを選択した。検討の結果について表AF−01に示し、実験の一例として、run4に示した反応の操作について示す。
[式AF−2]

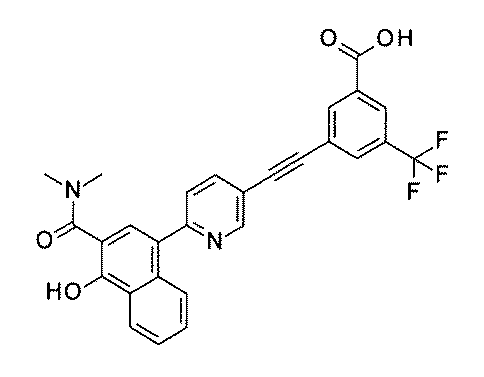
LRMS:m/z 505.1[M+H]+
保持時間:1.30分(分析条件 SMD−FA05−1)
窒素雰囲気下、0.6mLのスクリューキャップバイアルに、AF002−01R(0.37mmol/g,15mg,1.0当量)、NMP(255μL,17v/w)を加え、室温にて1時間振とうした。HOAt(5.3mg,6.0当量)DIC(6.0mg,6.0当量)を加え、混合物を40℃で22時間振とうした。
表AF−01に表記した時間経過時に、窒素雰囲気下にてレジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−FA05−1にて分析し、UV面積%は、UV波長319nmにて確認した。

LRMS:m/z 733.2[M+H]+。
保持時間:1.51分(分析条件 SMD−FA05−1)。

以下では、アミドカップリング反応において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
なお、アミドカップリングに限定されないが、大規模な混合物ライブラリ合成を高反応変換率および高純度にて実施するに当たっては、個々の基質での反応の成否をひとつずつ実験にて確認していくことは一局面において現実的ではない。そのため、検討未実施の不特定多数の基質での反応の成否を予測できると非常に価値が高い。実施例2−7においては、アミドカップリング反応の適用基質範囲確認と同時に、適用範囲を実験にて確認していない基質の反応の成否を予測する手法の一例についても示す。
ここで、ASAとはaccessible surface areaの略であり、ある特定の原子周辺への他分子の接触可能面積を示す指標である。ここでは、求電子種の反応点に対してどれだけ求電子種の反応点が接近しうる面積を有するかを表す指標として用いる。ASAの値が小さいほど、他分子が該当原子に対して接近しにくく、その原子の周辺の立体障害が大きいことを意味する。(公知文献;Science.1983 Aug 19;221(4612):709−13.,J.Org.Chem.2011,76,2,435−440)。
Schrodinger社のMaestroのMacroModelで配座を発生させた(CHCl3中、最安定配座から30kJ/molまでエネルギーの高い配座まで探索)。得られた複数の配座各々に対して、同じくSchrodinger社のMaestroのAdvanced SurfacesでASAを計算し(Type:Extended Surface)、複数の配座のASAの中で最大のASAを選択した。
本実施例2−7においては、求核種であるアミン化合物(分子)について配座を発生させ、得られた複数の配座各々に対して、求核部位となるアミノ基の窒素原子のASAを計算し、複数の配座のASAの中で最大のASAを選択した。
アミドカップリング反応に一般的に利用される試薬の中から、液相に存在するアミンとの反応が遅く共存可能な縮合剤、すなわち、アミンとの意図しない反応にて消費されない縮合剤としてDICを、また、カルボン酸とDICによって生じる一次活性エステルからのウレア転位抑制の添加剤としてHOAtを選択した。[表AD−01]に示すBB30~BB33、AD013~AD021のアミンに対してアミドカップリング反応を実施し、適用可能なビルディングブロックの範囲を確認した。

[表AD−01]



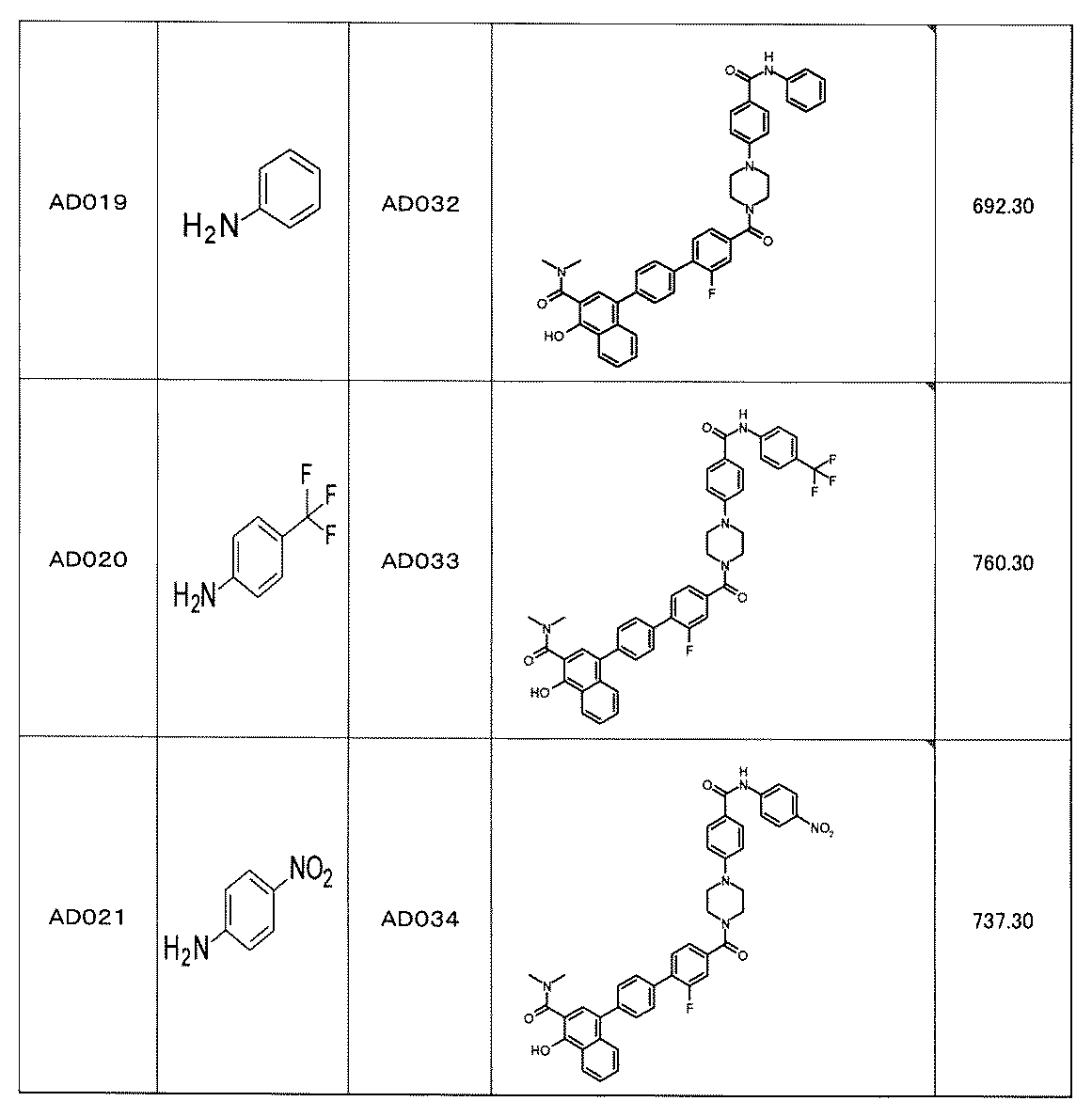
任意の反応時間経過時に、窒素雰囲気下でレジン懸濁液10μLをサンプリング後、1.5mLガラスバイアル中のn−PrNH2(20μL)に吐出した。数回ピペッティングした後に、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(50μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。
LCMSの結果について表AD−03に示す。AD007~AD010ならびにAD019をアミンとして用いた場合、22時間経過時に目的物のアミド体が94.3面積%以上で確認された。

[表AD−04]第一級アミンの嵩高さの影響

この傾向から、第一級アミンの窒素原子のASAが73.8を下回る場合には、反応完結が難しくなり、混合物ライブラリの純度低下を引き起こすことが懸念される。逆に73.8を上回る場合には、純度低下を引き起こすことなく高反応変換率かつ高純度の混合物ライブラリを提供できることが期待される。
[表AD−05]第二級アミンの嵩高さの影響

このことから、第二級アミンの窒素原子のASAが20を下回る場合には、目的化合物を全く得られなくなる懸念がある一方で、30を超える場合には反応完結が可能であり、純度低下を引き起こすことなく高反応変換率かつ高純度の混合物ライブラリを提供できることが期待される。
[表AD−06]芳香族アミンの嵩高さの影響

芳香族アミンにおいては、窒素原子のASAが>78.2のアミンを用いた場合、本実施例における反応条件において、88.4面積%以上で反応が進行した。
なお、本実施例は、混合物ライブラリに適用できる基質範囲の特定方法の一例の例示であり、実際のライブラリ合成への適用において制限されるものではない。特に、ASAが上記で示した範囲外の場合においても、固相担持されたAr−COOHもしくはその活性エステルは変換されずに残存していることから、反応時間の延長、反応温度、または試薬量の調整によって、反応を高変換率化できる余地はある。換言すれば、一態様において、ASAをひとつの指標としてライブラリ合成の事前の難易度判定に活用可能であるが、ASAが上記で示した範囲外の場合に、必ずしも本反応が適用できないとは限らない。また、追加の実験検討や実際の混合物ライブラリ合成時の結果から、適用できる基質の範囲は拡大することもある。
以下ではアシルスルホンアミド化反応のに適用可能な実施方法を一例として例示する。また、反応変換率が十分でない場合に、混合物ライブラリの純度低下を抑制する手法についても例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
アシルスルホンアミド化反応の条件設定の過程において、問題点を発見し解決策を特定する例を下記に示す。
公知文献(J.Med.Chem.2006,49,1165−1181)を参考にして、縮合剤としてEDCI−HCl、活性化剤としてDMAP、溶媒としてDCMを選択し、Ar−COOHであるAE008とスルホンアミドBB40を用いてアシルスルホンアミド化反応を実施した。AE008は、実施例1−6—4に示した方法で合成した。


LRMS:m/z 1096.2[M+H]+。
保持時間:1.01分(分析条件 SMD−TFA05−1)。

LRMS:m/z 1040.2[M+H]+。
保持時間:0.59(分析条件 SMD−TFA05−1)。

反応追跡は以下のように実施した。

[表AE−01]
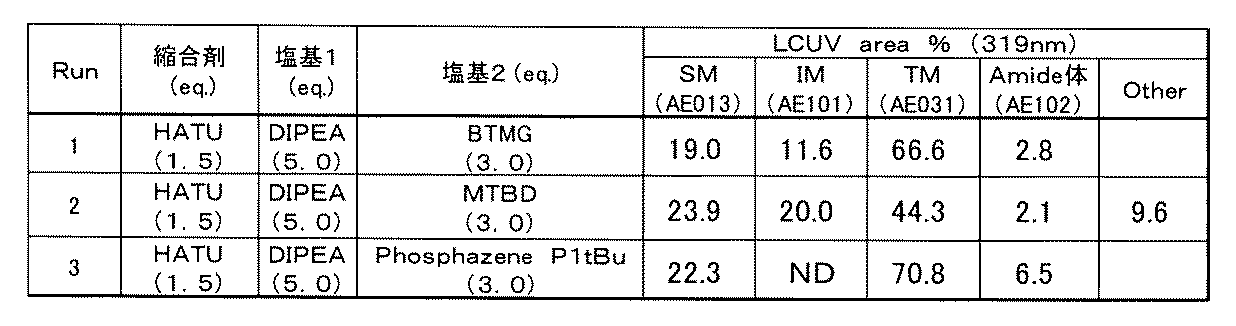
本実験の結果より、Ar−COOHに対してHATUを縮合剤として利用する事で、カルボジイミド系縮合剤を使用した際に観測された副生成物を回避できることが明らかとなった。さらには、カルボン酸の活性化を行った後に加える塩基2としてホスファゼンP1tBuを用いることで、スルホンアミドを高収率でカップリングさせることが可能であることが明らかになった。
前述した条件を最適化する過程において発見した問題と解決策の一例を示す。Ar−COOHであるAE032と(4−クロロフェニル)メタンスルホンアミド:AE033を用いて、以下に示す操作で実施した。以下に一例として表AE−02中のrun4の例を示す。
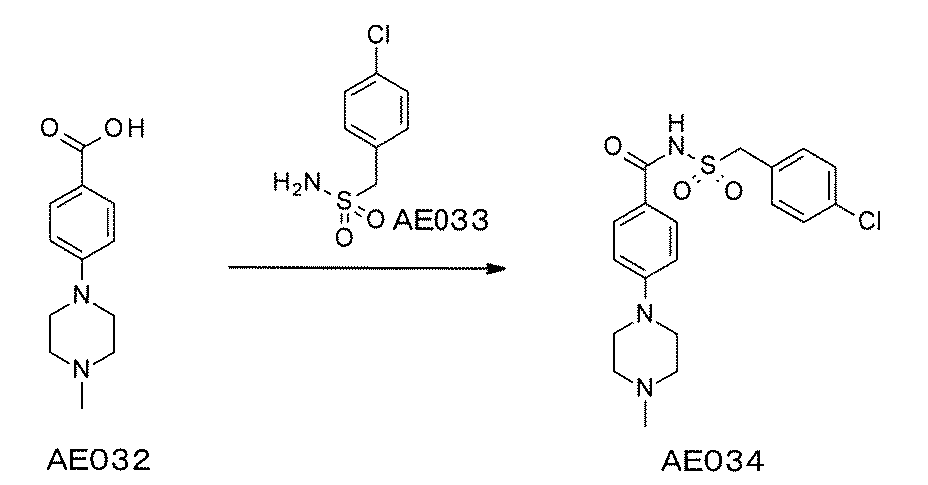
その他のrunにおいて使用した溶媒、試薬ならびにそれらの使用量を表AE−02に示す。反応追跡は以下のように実施した。

LRMS:m/z 408.0[M+H]+。
保持時間:0.79分(分析条件 SMD−TFA05−1)。

LRMS:m/z 262.1[M+H]+。
保持時間:0.54分(分析条件 SMD−TFA05−1)。

LRMS:m/z 276.1[M+H]+。
保持時間:0.60分(分析条件 SMD−TFA05−1)。

[表AE−02]
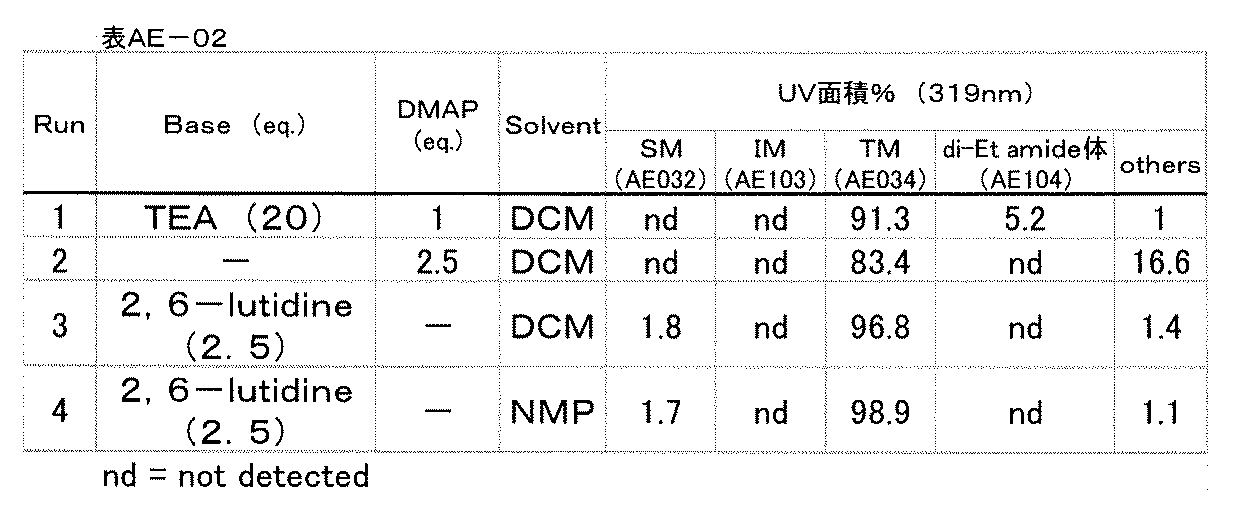
本実験の結果より、本発明の手法によって、Ar−COOHに対してHATUを縮合剤、2,6−ルチジンを塩基として利用し、カルボン酸の活性化を行った後にホスファゼンP1tBuのような強塩基と共にスルホンアミドを添加することで、アシルスルホンアミド化反応を高収率で進行させることが可能であることが示された。
実施例2−8−2にて液相実験で見出した条件を固相反応へと適用し、固相反応において更なる改善を行った。固相の実験は、固相担持されたAr−COOHであるAE019−01RとスルホンアミドAE033を用いて以下に示す操作で実施した。以下に一例として[表AE−03]中のrun4の例を示す。

途中、反応開始から1、18時間経過時に、窒素雰囲気下にて反応液とレジンの懸濁液10μLをサンプリング後、1.5mLガラスバイアル中のn−PrNH2(30μL)に吐出した。数回ピペッティングした後に、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。なお、以下に示す化合物を検出対象とし、分析条件SMD−AC05−1にて分析し、UV面積%は、UV波長319nmにて確認した。
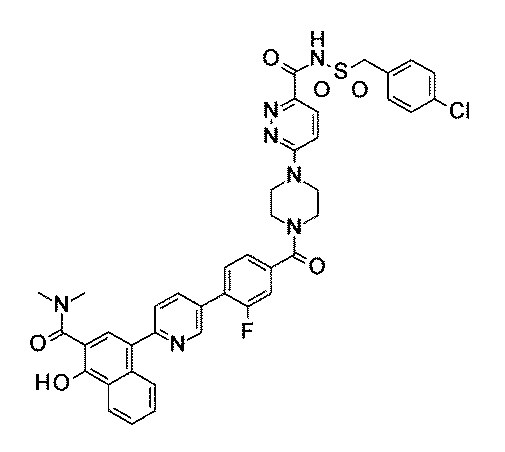
LRMS:m/z 808.1[M+H]+。
保持時間:0.89分(分析条件 SMD−AC05−1)。

LRMS:m/z 662.2[M+H]+。
保持時間:1.05分(分析条件 SMD−AC05−1)。

[表AE−03]

本実験の結果より、本発明の手法によって、固相担持されたAr−COOHに対してHATUを縮合剤、2,6−ルチジンを塩基として利用し、カルボン酸の活性化を行った後にホスファゼンP1tBuのような強塩基と共にスルホンアミドを添加することで、固相反応においてもアシルスルホンアミド化反応が高収率させることが可能であることが示された。
反応変換率が十分でない場合に、原料もしくは活性中間体を混合物ライブラリに含まれる別の化合物へと変換できれば、意図しない化合物の混入を軽減した「質の高い」ライブラリ供給が可能となるため、極めて重要な手法となり得る。
以下では、アシルスルホンアミド化反応の際に残存する活性中間体に対して適切な処理を施すことで原料へと戻ることを回避し、結果としてライブラリ成分以外の不純物が増加することを防ぐ手法の一例を示す。具体的には、反応性が低いビルディングブロックとの反応を行った後、混合物ライブラリに含める予定であり、かつ、より反応性の高いビルディングブロックを反応系内に添加することで、活性中間体をライブラリ成分へと変換する手法の例示である。これにより、混合物ライブラリへの意図しない未反応体や活性中間体などの不純物の混入を低減することができる。以下には、カルボン酸としてAE022−01R、スルホンアミドのビルディングブロックとしてAE049、より反応性の高いアミンビルディングブロックとしてAE023を用いた実験について、一例として示す。ここではAE049、AE023共に、split & pool法による混合物ライブラリ作成時に利用するビルディングブロックを想定している。また、AE022−01Rは、実施例1−6−12に示す方法にて合成した。


LRMS:m/z 948.3[M+H]+。
保持時間:1.13分(分析条件 SMD−FA05−1)。
保持時間:1.17分(分析条件 SMD−AC05−1)。

LRMS:m/z 654.3[M+H]+。
保持時間:0.88分(分析条件 SMD−FA05−1)。

LRMS:m/z 720.1[M+H]+。
保持時間:1.47分(分析条件 SMD−AC05−1)。

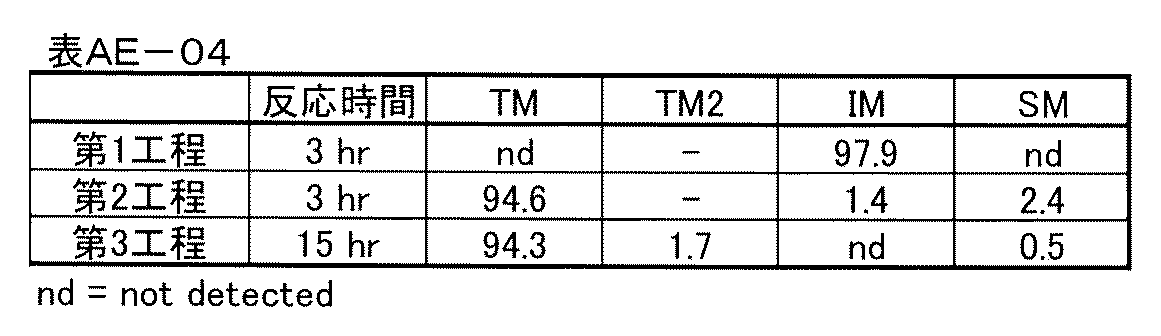
本発明の本結果より、残存する反応中間体に対して適切な処理を施すことで原料へと戻ることを回避し、ライブラリ成分以外の不純物が増加することを防ぐことが可能であることが明らかになった。本発明の本手法は、本実施例にて例示したアシルスルホンアミド化の場合に限定されず、「質の高い」ライブラリを供給するために、デザインされた化合物以外の含有量を抑制する、あらゆる反応の際に広く適用可能な手法である。
実施例A−E2、3にて見出した条件を表AE−05に示すAE037、AE038、AE040~AE049のスルホンアミドを用いてアシルスルホンアミド化反応を実施し、ライブラリ合成に適用可能なビルディングブロックの範囲を確認した。
実験の一例として、AE029−01RとAE037(BB36)の反応の操作について下記に示す。

任意の時間において、窒素雰囲気下、レジンの懸濁液10μLをサンプリング後、1.5mLガラスバイアル中のn−PrNH2(20μL)に吐出した。数回ピペッティングした後に、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.1Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。

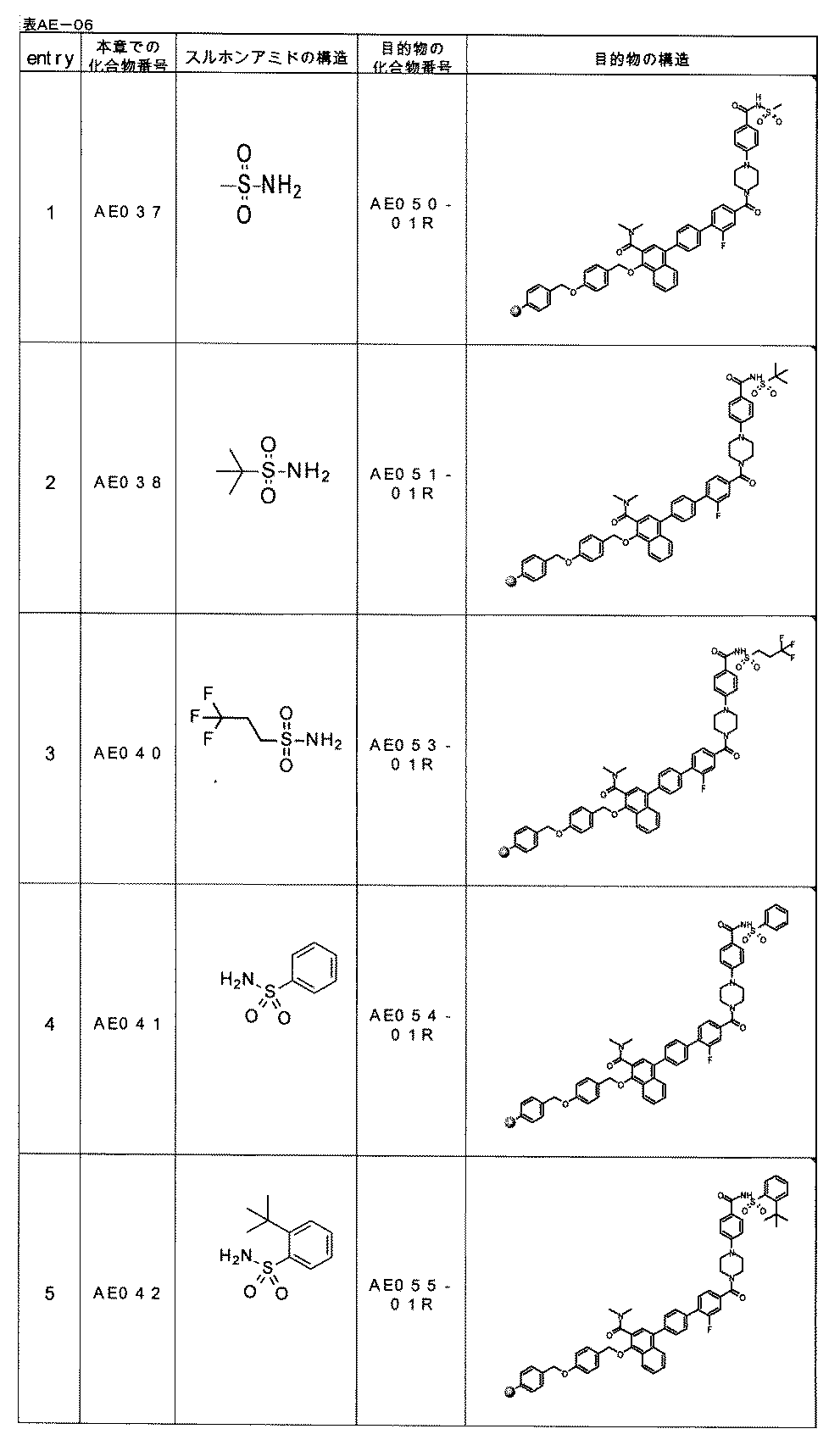


[表AE−07]

実施例2−8−3にて見出した反応条件を利用して、固相に担持されたスルホンアミドに対するアシル化反応を実施し、アシルスルホンアミドリンカーの結合方向性を拡大した。
実験の一例として、A127−03Rに対するA128を用いたアシル化反応の操作について下記に示す。

窒素雰囲気下、0.6mLのガラスバイアルに化合物A127−03R(担持量0.194mmol/g、20mg)とNMP(160μL)を加え、室温で1時間振とうした後、上記で調製した反応液とPhoaphazene P1tBu(9.9μL、39μmmol)を加え、室温で24時間振とうした。
レジンの懸濁液15μLをサンプリング後、フィルター付きピペットチップに移し、DMF(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。ろ過後、NMP(100μL)でフィルターを洗浄し、合わせた溶液(100μL)をMeCN(250μL)で希釈し、LCMS測定を行うことで目的物A129が98.2%観測された。

LRMS:m/z 633、635[M+H]+
保持時間:1.179(分析条件FA05−1、299nm)。
以下では、sp2−sp3カップリング反応において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
公知文献(Org.Lett.2015,17,3370−3383.)を参考にして塩基として有機塩基のP2tBuを選択し、Pd触媒としてRuPhos Pd G4を選択してアルキルボロン酸との鈴木カップリング反応を実施した。

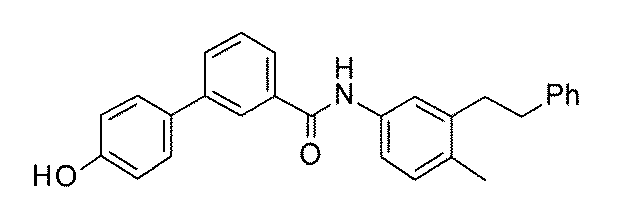
LRMS:m/z 408[M+H]+。
保持時間:1.351分(分析条件SMD−FA05−1、280nm)。


LRMS:m/z 472[M+H]+。
保持時間:1.852分(分析条件SMD−FA05−1、280nm)。

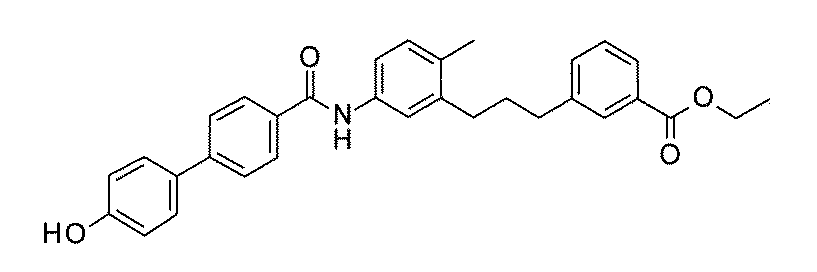
LRMS:m/z 494[M+H]+
保持時間:1.417分(分析条件 SMD−FA05−1)
公知文献(Chem.Commun.2011,47,5181−5183.)を参考にして、Pd触媒としてPd−PEPPSI−IPentを選択してアルキル亜鉛化合物との鈴木カップリング反応を実施した。


LRMS:m/z 346[M+H]+。
保持時間:1.260分(分析条件SMD−FA05−1、280nm)。
実施例2−9−1および実施例2−9−2の結果から、アリール基とアルキル基を結合させる際、つまりsp2−sp3カップリングを行う際に、基質の反応性や入手性に応じて少なくとも本実施例にて示した反応条件を自在に選択可能であることを示している。
以下では、C−Nカップリング反応において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
公知文献(Org.Lett.2015,17,3370−3383.)を参考にして塩基として有機塩基のP2tBuもしくはNaOtBuを選択し、Pd触媒としてRuPhos Pd G4を選択して脂肪族アミンとのBuchwald−Hartwigアミノ化反応を実施した。
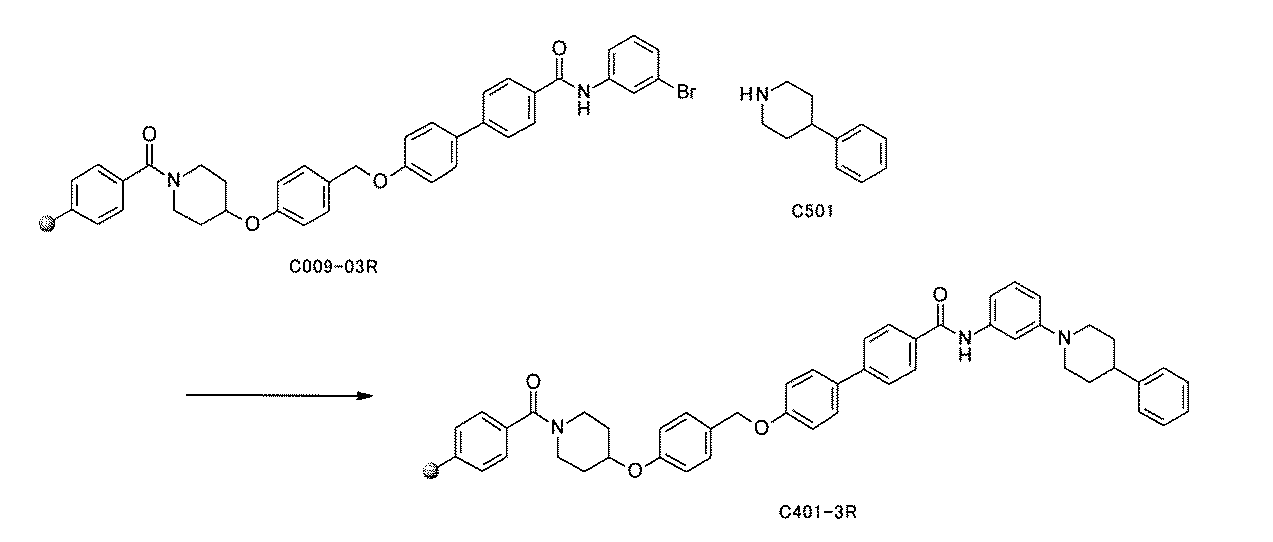
反応液と固相の懸濁液をフィルター上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.2Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物C401が94%観測された。
LRMS:m/z 449[M+H]+。
保持時間:1.115分(分析条件SMD−FA05−1.299nm)。


LRMS:m/z 437[M+H]+。
保持時間:1.213分(分析条件SMD−FA05−1、299nm)。

LRMS:m/z 463[M+H]+。
保持時間:1.344分(分析条件SMD−FA05−1、299nm)。


LRMS:m/z 451[M+H]+。
保持時間:1.021分(分析条件SMD−FA05−1、299nm)。
以上の結果から、種々の基質の組み合わせに対して、固相においてもBuchwald−Hartwigアミノ化反応による結合形成が可能であり、混合物ライブラリの合成にも適用可能な反応条件を確認した。
以下では、固相担持ArBrと芳香族アミン、ヘテロ芳香族アミンとのC−Nカップリング反応において、広い基質適用範囲が期待される反応条件での結合形成反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
実施例2−10−2−1:固相担持ArBr(C010−03R)と4−(メチルアミノ)ベンゾニトリル(C503)のカップリングによるC405−03Rの合成
反応液と固相の懸濁液のうち12μLをフィルター付きチップ上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した。ペンタメチルベンゼンの10%TFA/DCM溶液(0.1M、0.05mL)に2分間浸してろ過し、DMF(0.05mL)で洗浄し、合わせたろ液をMeCN(0.25mL)で薄めた溶液のLCMS測定を行うことで、目的物C405が89.5%観測された。ただし、波長299nm(±4nm)で切り出して解析している。

LRMS:m/z 434[M+H]+
保持時間:2.708分(分析条件 SMD−FA05−long)

LRMS:m/z 474[M+H]+
保持時間:1.129分(分析条件 SMD−FA05−1)
実施例2−10−3:固相担持アミンとアリールブロミドのC−Nカップリング反応
以下では、固相担持アミンとアリールブロミドとのC−Nカップリング反応において、広い基質適用範囲が期待される反応条件での結合形成反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
実施例2−10−3−1:固相担持アミン(A137−03R)と5−ブロモチオフェン−2−カルボン酸エチル(C801)のカップリングによるC407−03Rの合成

反応液と固相の懸濁液のうち12μLをフィルター付きチップ上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した。ペンタメチルベンゼンの10%TFA/DCM溶液(0.1M、0.05mL)に2分間浸してろ過し、DMF(0.05mL)で洗浄し、合わせたろ液をMeCN(0.25mL)で薄めた溶液のLCMS測定を行うことで、目的物C407が69.8%観測された。ただし、波長299nm(±4nm)で切り出して解析している。

LRMS:m/z 575[M+H]+
保持時間:1.427分(分析条件 SMD−FA05−1)
実施例2−10−3の結果から、固相担持アミンに対してアリールブロミドとのC−Nカップリング反応が適用可能であることが示された。
実施例2−10−4:固相担持ArBrと5員環HetArとのCu試薬を用いたC−N結合形成反応の固相実験
非特許文献(J.Am.Chem.Soc.2001,123,7727.)を参考にして塩基として有機塩基のMTBD(7−メチル−1,5,7−トリアザビシクロ[4.4.0]デカ−5−エン)を選択し、Cu触媒としてCuIを、リガンドとしてDMEDA(N,N′−ジメチルエチレンジアミン)を選択してピラゾール−3−カルボン酸エチルとのC−N結合形成反応を実施した。
実施例2−10−4−1:固相担持ArBr(C009−03R)とピラゾール−3−カルボン酸エチル(B321)とのカップリング反応

反応液と固相の懸濁液をフィルター上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物B226が77%観測された。

LRMS:m/z428[M+H]+。
保持時間:1.096分(分析条件SMD−FA05−1,299nm)。
この結果からCuIを使用したHeteroArのN−Hと固相ArBrとのC−N結合形成反応を実施可能であることを確認した。
以下では、スルホンアミド化反応において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
また、本実施例中における固相担持Ar−NH2とは、Ar−NH2を部分構造として有する化合物が担持されたレジン(例としてD001−03R)のことを表す。同様に、固相担持Ar−NHMeとはAr−NHMeを部分構造として有する化合物が担持されたレジン(例としてD020−03R)のことを表す。固相担持アルキルアミンとは、アルキルアミンを部分構造として有する化合物が担持されたレジン(例としてD021−03R)のことを表す。
公知文献(Eur.J.Org.Chem.2012,764−771 並びにEur.J.Med.Chem.2018,143、48−65)を参考に、溶媒としてTHF、塩基としてピリジンもしくはトリエチルアミンを用いて固相担持Ar−NH2のスルホンアミド化を実施した。その際、公知文献(Chem.Pharm.Bull.1999、47、809−812)で示されるようなアニリンに対するスルホニルクロリドの過剰反応が懸念された。そこで、幅広い固相担持Ar−NH2に対して過剰反応を抑制できる反応条件を見出すために、反応性が高いと予想されるスルホニルクロリドを用いて反応条件の検討を行った。具体的には、芳香環上に電子求引性基であるニトロ基を有する4−ニトロベンゼンスルホニルクロリド(D002)を用いて、固相担持Ar−NH2(D001−03R)のスルホンアミド化を実施した。溶媒としてTHF、塩基としてピリジンもしくはトリエチルアミン用いて固相担持Ar−NH2(D001−03R)と4−ニトロベンゼンスルホニルクロリド(D002)のスルホンアミド化を行った反応結果を[表2−11−1]に示す。また塩基としてピリジンを用いた場合の実験操作(表2−11−1中、Run2)を例として以下に示す。

反応追跡は以下のように実施した。
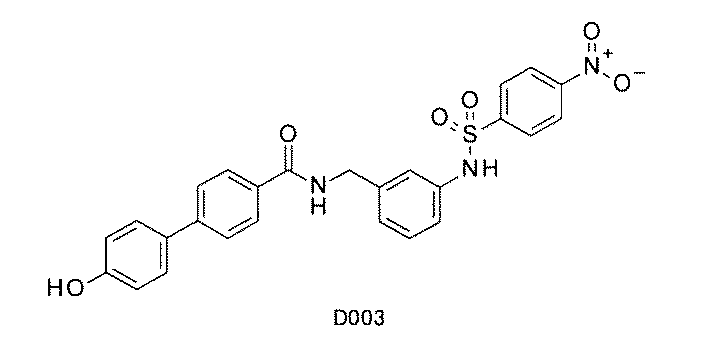
LCMS:m/z 504[M+H]+。
保持時間:1.018分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 689[M+H]+。
保持時間:1.222分(分析条件 SMD−FA05−1、290nm)。
混合物ライブラリを構築するためには反応性が異なる様々な基質の混合物に対して、高収率で望みの反応が進行する条件を設定する必要がある。例えば、反応性が高い基質の生成物は、反応性が低い基質が十分な反応変換率に達するまで反応条件下にさらされることになる。そのため、その間に過剰反応等が起こらないことを確認した上で、混合物ライブラリ合成の反応条件として設定することが好ましい。実施例2−11−1におけるTHFとピリジンを用いた条件は、反応時間の延長に伴う過剰反応が進行しなかったことから、混合物ライブラリ構築に適したスルホンアミド化条件であると判断した。
実施例2−11−1で見出したスルホンアミド化条件を用いて、固相担持Ar−NH2(D001‐03R)のスルホンアミド化におけるスルホニルクロリドの適用範囲を確認した。実施例2−11−1と同様の実験操作を行い、スルホニルクロリド(D005~D011)を用いてスルホンアミド化を実施した。
使用したスルホニルクロリド(D005~D011)の構造を表2−11−2−1に、得られた結果を表2−11−2−2に示す。

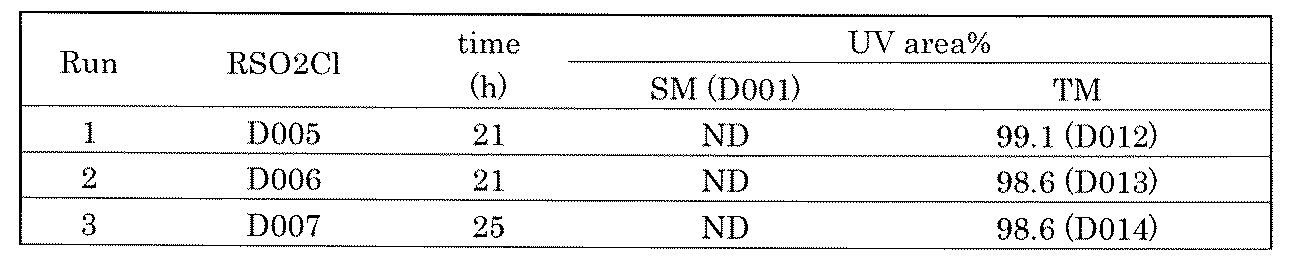


実施例2−11−2において、チオフェン−2−スルホニルクロリド(D011)を用いた場合に室温21時間の時点で40.9面積%の原料(D001)が未反応であった。そこで、D011のように反応性が低いスルホニルクロリドを用いた場合でも高純度で目的生成物が得られる反応条件を設定することとした。公知文献(Tetrahedron Lett.2001、42、979−982)を参考に、塩基としてNMI(45当量)を用いる条件およびピリジン(45当量)にNMI(5当量)を添加する条件を実施し、NMIに反応を加速させる効果があるか調査した。また、反応性が高い4−ニトロベンゼンスルホニルクロリド(D002)を用いた場合に過剰反応が進行しないかどうかも同時に確かめた。検討結果を表2−11−3に示す。また、表2−11−3中、ピリジン(45当量)にNMI(5当量)を添加し、チオフェン−2−スルホニルクロリド(D011)を用いてスルホンアミド化を行った実施例(表2−11−3中、Run3)を以下に示す。
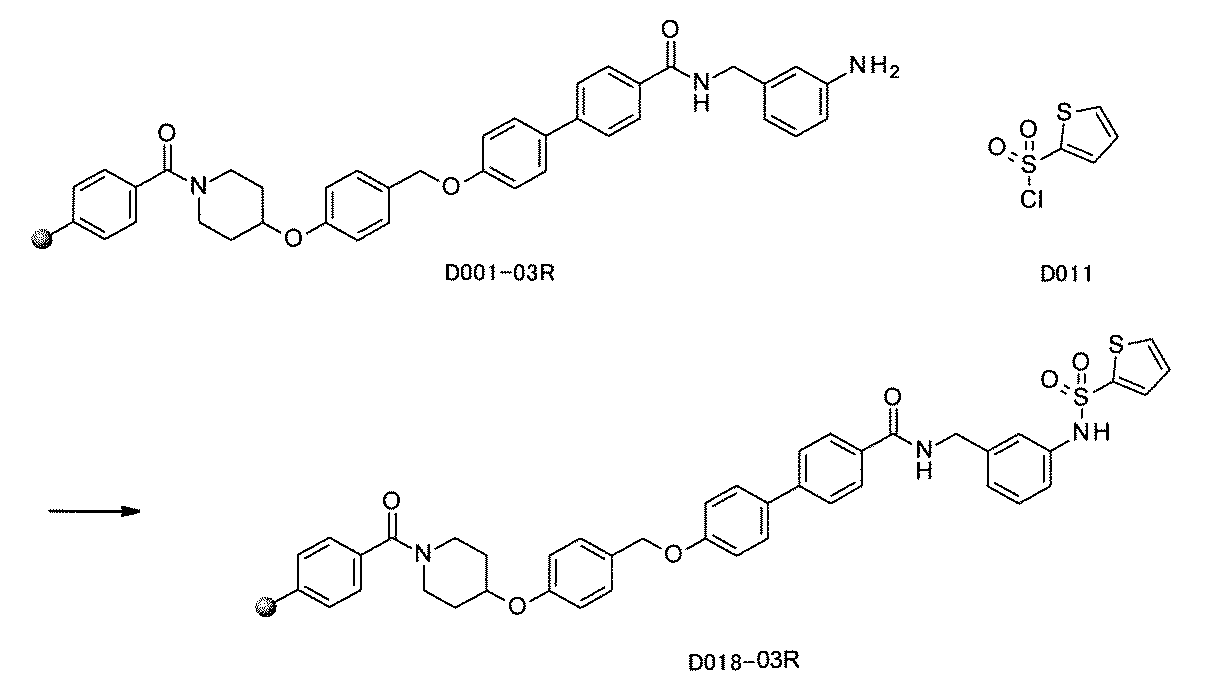
窒素雰囲気下、ピリジン(3.6μL、0.045mmol)、NMI(10v/v、THF溶液、4.0μL、5.0μmol)とチオフェン−2−スルホニルクロリド(D011)(5.48mg、0.0300mmol)を加え、得られた混合物を室温で21時間振とうした。
反応追跡は以下のように実施した。
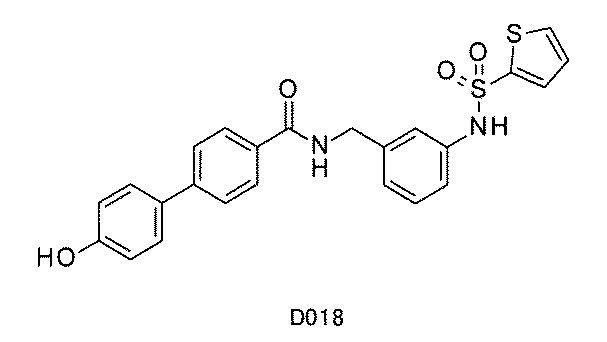
LCMS:m/z 465[M+H]+。
保持時間:1.046分(分析条件 SMD−TFA05−RP、290nm)。

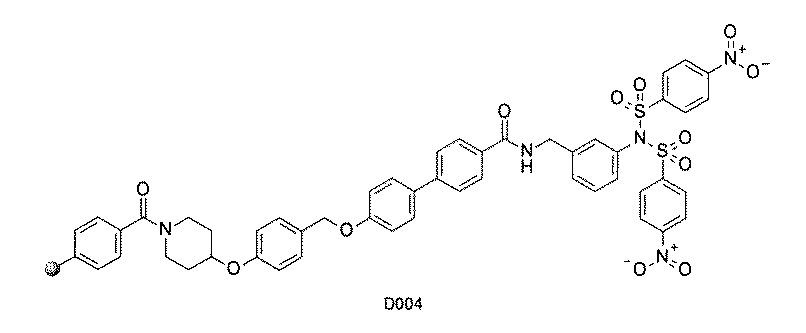
LCMS:m/z 689[M+H]+。
保持時間:1.244分(分析条件 SMD−TFA05−RP、290nm)。

LCMS:m/z 504[M+H]+。
保持時間:1.244分(分析条件 SMD−TFA05−RP、290nm)。
実施例2−11−3で見出したTHF中でピリジン(45当量)とNMI(5当量)用いる反応条件の適用範囲を確認した。実施例2−11−3と同様の実験操作により、固相担持Ar−NH2(D001−03R)、Ar−NHMe(D020−03R)もしくはアルキルアミン(D021−03R)とスルホニルクロリド(D002、D005、D007、D011、D008、D019)を用いたスルホンアミド化反応を実施した。

化合物番号:D019
得られた結果を表2−11−4−1に示す。また表2−11−4−1中、固相担持Ar−NHMe(D020−03R)もしくはアルキルアミン(D021−03R)とスルホニルクロリド(D011)を用いたスルホンアミド化における反応操作を代表例として示す。

窒素雰囲気下、ピリジン(3.6μL、0.045mmol)、NMI(10v/v、THF溶液,4.0μL、5.0μmol)とチオフェン−2−スルホニルクロリド(D011)(5.48mg、0.0300mmol)を加え、得られた混合物を室温で21時間振とうした。
反応追跡は以下のように実施した。
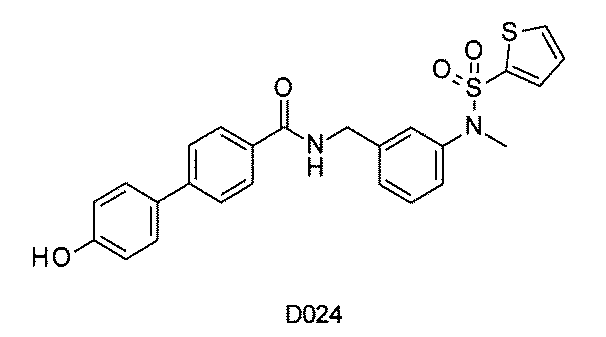
LCMS:m/z 479[M+H]+。
保持時間:1.103分(分析条件 SMD−TFA05−RP、290nm)。


LCMS:m/z 429[M+H]+。
保持時間:1.032分(分析条件 SMD−TFA05−RP、280nm)。

[表2−11−4−2]
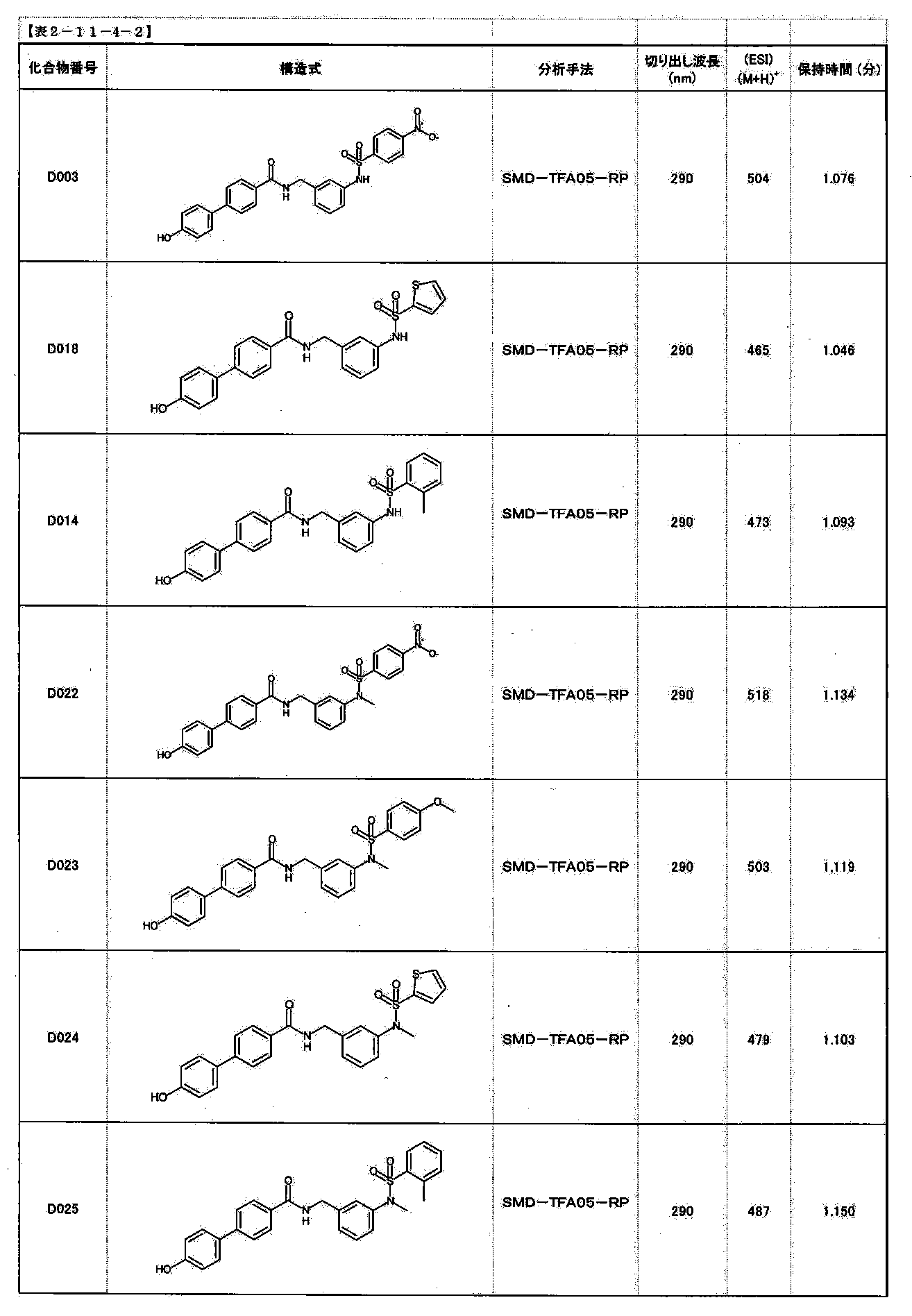

実施例2−11−3で見出した塩基としてピリジンとNMI用いる反応条件を用いて、固相担持ヘテロAr−NH2のスルホンアミド化を行った。実施例2−11−3と同様の実験操作により、固相担持ヘテロAr−NH2(D072−03R)と4−メトキシベンゼンスルホニルクロリド(D006)を用いたスルホンアミド化反応を実施した。得られた結果を表2−11−4−3中、Run1に示す。また、Run1から溶媒をDCMに変更した場合の結果をRun2に示す。さらに、DCM溶媒中塩基としてNMI(15当量)もしくは3、4−ルチジン(15当量)を使用した場合の結果をRun3、 4にそれぞれ示す。表2−11−4−3中、DCM中で3、4−ルチジンを用いる反応条件(Run4)を用いたスルホンアミド化における反応操作を代表例として示す。Run1~3も同様の実験操作を行った。

窒素雰囲気下、3,4−ルチジン(6.6μL、59μmol)と4−メトキシベンゼンスルホニルクロリド(D005)(8.1mg、0.039mmol)を加え、得られた混合物を室温で24時間振とうした。
窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D073が90.8面積%観測された。

LCMS:m/z 490[M+H]+。
保持時間:0.800分(分析条件 SMD−FA05−1、299nm)。
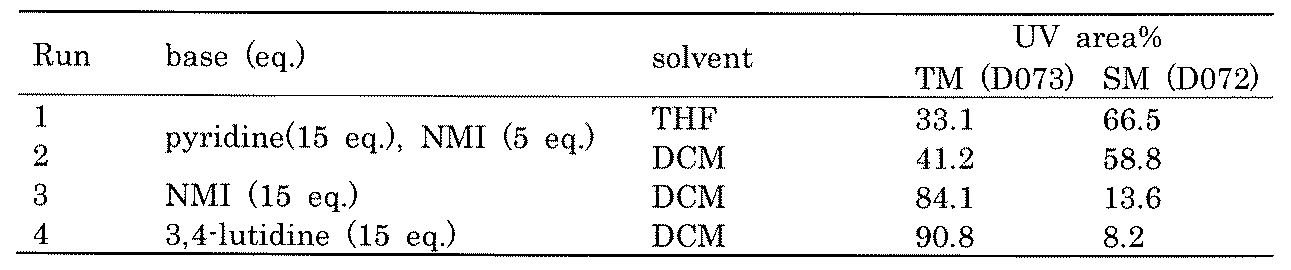
本条件は混合物ライブラリ合成に適用することができ、その基質適用範囲も制限されない。
実施例2−11−1で見出した、THF中ピリジンを用いる条件を用いて芳香環上にフッ素を有する4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)と、固相担持Ar−NHMe(D020)のスルホンアミド化を実施した(表2−11−5、Run1)。その結果、スルホニルクロリドに含まれるフッ素原子が塩基として用いたピリジンによって置換された副生成物4−(4−ヒドロキシフェニル)−N−[[3−[メチル−(3−ニトロ−4−ピリジン−1−イウム−1−イルフェニル)スルホニルアミノ]フェニル]メチル]ベンズアミド(D036)が7.0面積%確認された。そのため、固相担持Ar−NH2(D001−03R)もしくはAr−NHMe(D020−03R)と4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)のスルホンアミド化反応は別条件にて実施することとした。固相担持Ar−NHMe(D020−03R)に対してはTHF溶媒中、塩基としてピリジン、2,6−ルチジンもしくは2,6−ジ−tert−ブチルピリジン(DTBP)を用いた検討を行った。固相担持Ar−NH2(D001−03R)に対してはTHF溶媒中2,6−ルチジンを用いた条件、もしくはDCE溶媒中2,6−ルチジンもしくは2,6−ジ−tert−ブチルピリジンを用いた条件を検討した。得られた結果を表2−11−5に示す。また、表2−11−5、Run2に示したTHF溶媒中、塩基として2,6−ルチジンを用いて固相担持Ar−NHMe(D020−03R)と4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)のスルホンアミド化を行った場合の反応操作を代表例として示す。Run1、3、4、5についてもRun2と同様の実験操作を行った。
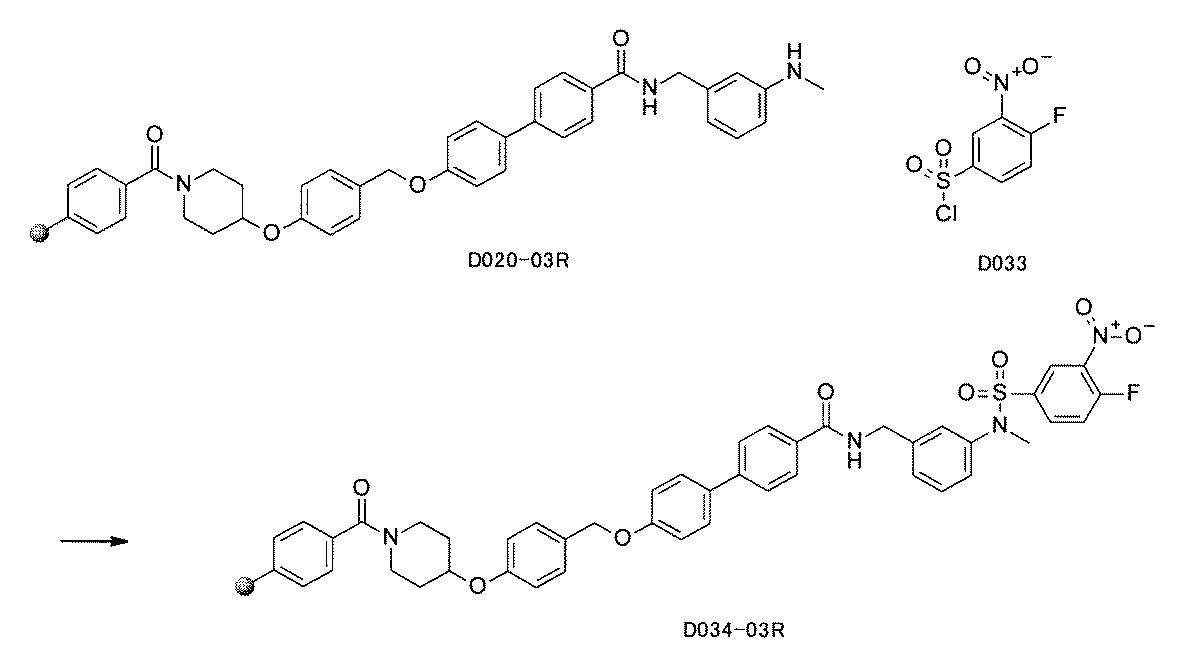
窒素雰囲気下、2,6−ルチジン(5.2μL、0.045mmol)と4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)(7.2mg、0.0300mmol)を加え、得られた混合物を室温で35時間振とうした。
反応追跡は以下のように実施した。窒素雰囲気下、レジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.02Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をNMP(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D034が97.1面積%観測された。

LCMS:m/z 536[M+H]+。
保持時間:1.142分(分析条件 SMD−TFA05−RP、290nm)。
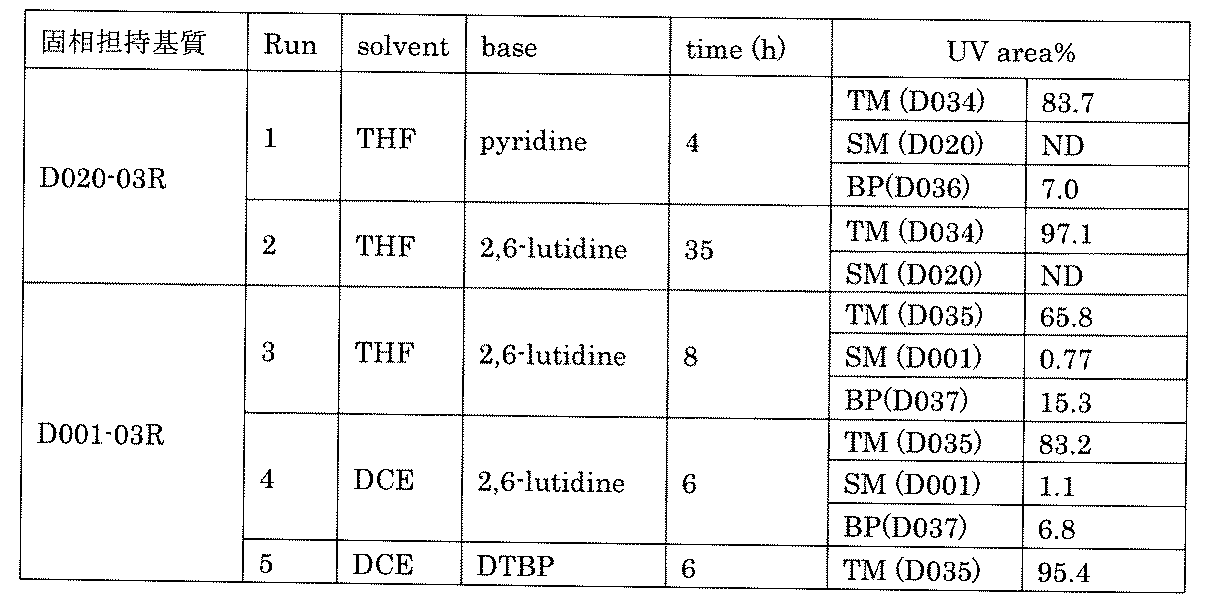


LCMS:m/z 595[M+H]+。
保持時間:0.870分(分析条件 SMD−TFA05−RP、290nm)。
表2−11−5中、Run3における主生成物(D035)と副生成物(D037、構造は推定)の分析結果を以下に示す。
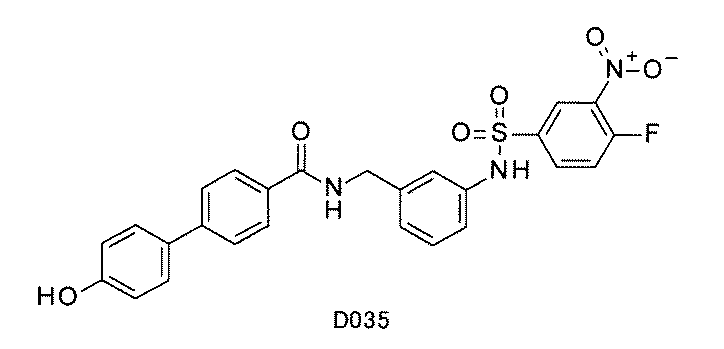
LCMS:m/z 522[M+H]+。
保持時間:1.104分(分析条件 SMD−TFA05−RP、290nm)。

LCMS:m/z 538[M+H]+。
保持時間:1.256分(分析条件 SMD−TFA05−RP、290nm)。
実施例2−11−5で見出した、DCE中DTBPを用いる条件を用いて、芳香環上にフッ素を有する4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)と、固相担持ヘテロAr−NH2(D106−03R)のスルホンアミド化を実施した。その結果、目的物であるD107が89.4面積%観測された。このことから、DCE中DTBPを使用する反応条件は、ヘテロAr−NH2のスルホンアミド化にも適用可能であることを確認した。

窒素雰囲気下、DTBP(52.8μL、0.240mmol)と、4−フルオロ−3−ニトロベンゼンスルホニルクロリド(D033)(9.20mg、0.038mmol)のDCE(50μL)溶液を加え、得られた混合物を60℃で23時間振とうした。
反応追跡は以下のように実施した。窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をNMP(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D107が89.4面積%観測された。

LCMS:m/z 606[M+H]+。
保持時間:0.928分(分析条件 SMD−FA05−1、299nm)。
固相担持アルキルアミン(D109−03R)のスルホンアミド化反応を行い、化合物(D110−03R)は以下のように合成した。(反応操作の例示)


LCMS:m/z 646[M+H]+。
保持時間:1.113分(分析条件 SMD−FA05−1、290nm)。
実施例2−11−7−1と同様の反応条件を用いて、固相担持アルキルアミン(D111−03R)とスルホニルクロリド(D033)のスルホンアミド化を行った(表2−11−6、Run1)。その結果、目的物(D114)が31.0面積%で観測された。同時に、固相担持アルキルアミン(D111−03R)がスルホニルクロリド(D033)に含まれるフッ素原子と芳香族求核置換反応を起こすことによって生じたと推定される副生成物(D115)が50.1面積%生じた。Run2では、望まぬ芳香族求核置換反応を防ぐ目的で、芳香環状に塩素原子を有する4−クロロ−3−ニトロベンゼンスルホニルクロリド(D113)を2,6−ルチジンと共に使用したところ、目的物D112が87.0面積%観測された。表2−11−6中、Run2の反応操作を代表例として示す。Run1についてもRun2と同様の実験操作を行った。

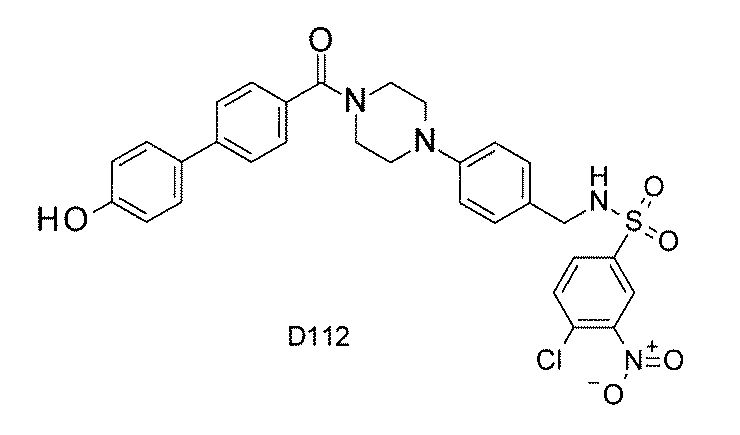
LCMS:m/z 607[M+H]+。
保持時間:1.099分(分析条件 SMD−FA05−1、299nm)。

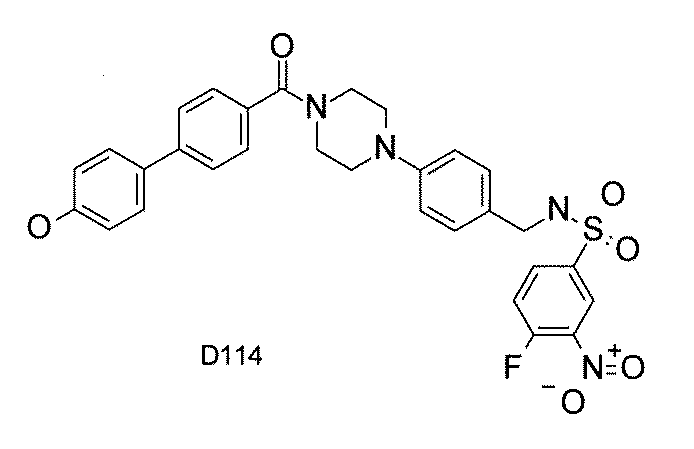
LCMS:m/z 591[M+H]+。
保持時間:1.061分(分析条件 SMD−FA05−1、299nm)。

LCMS:m/z 607[M+H]+。
保持時間:1.255分(分析条件 SMD−FA05−1、299nm)。
実施例2−11−3で見出した、THF中で塩基としてピリジンとNMI用いる反応条件を用いて、固相担持Ar−NHMe(D020−03R)とフェニルメタンスルホニルクロリド(D100)を用いてスルホンアミド化を実施した。
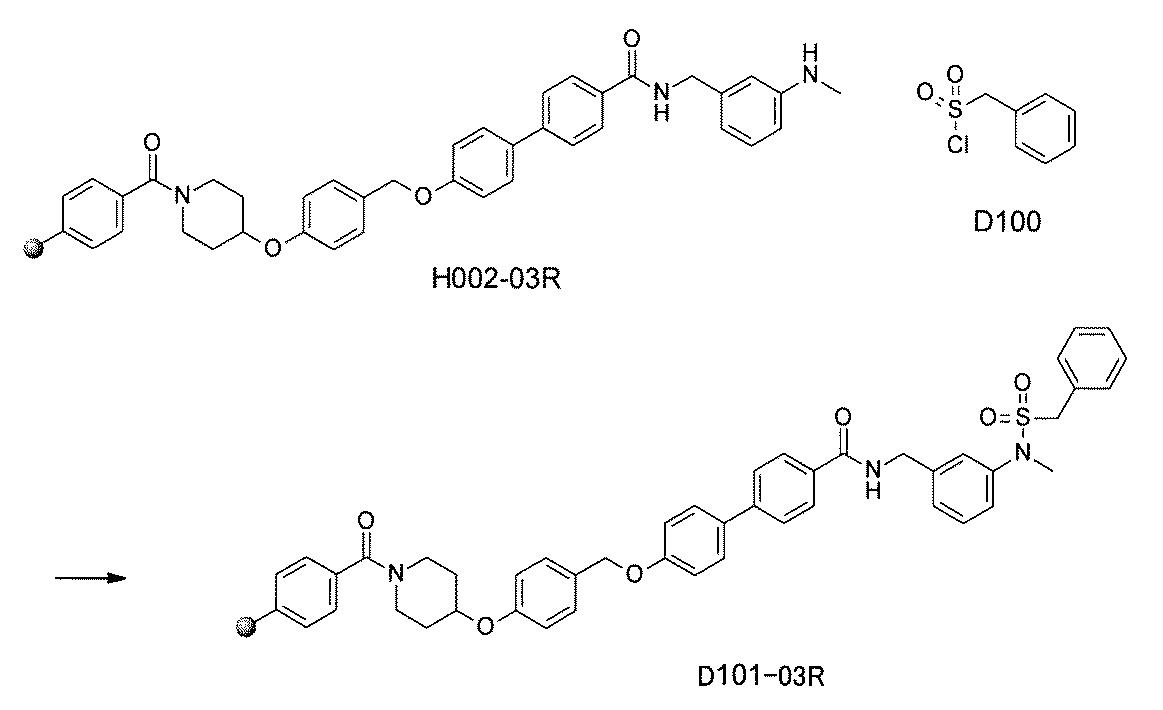
窒素雰囲気下、ピリジン(4.8μL、0.059mmol)、NMI(1.6μL、2.0μmol)とフェニルメタンスルホニルクロリド(D100)(7.5mg、0.039mmol)を加え、室温で24時間振とうした。
反応追跡は以下のように実施した。
窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物(D101)が95.9面積%観測された。
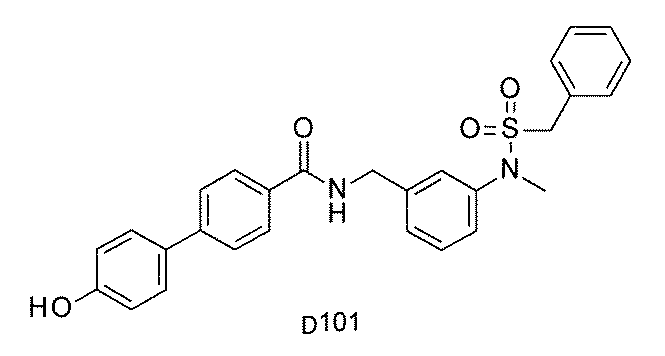
LCMS:m/z 487[M+H]+。
保持時間:1.056分(分析条件 SMD−FA05−1、299nm)。
実施例2−11−6−1−1と同様の手法で、固相担持Ar−NHMe(D020−03R)と2−ナフタレン−1−イルエタンスルホニルクロリド(D102)を用いてスルホンアミド化を実施した。得られた結果を表2−11−7中Run1に示す。まずTHF中で塩基としてピリジン(15当量)とNMI(5当量)を用いて室温で24時間反応を行った場合、目的物D103が82.5面積%で得られた一方、原料H002が14.4%残存した(Run1)。次に溶媒をDCMに変更したところ、反応の効率が向上し、目的物が91.8面積%で得られた(Run2)。Run3で塩基を4−tBu−pryridine(15当量)に変更したところ、目的物(D103)の純度が92.2%に向上した。更にTHF中3、4−ルチジン(15当量)を用いる条件を試したところ、目的物D103が93.8%面積%生成することを観測した。
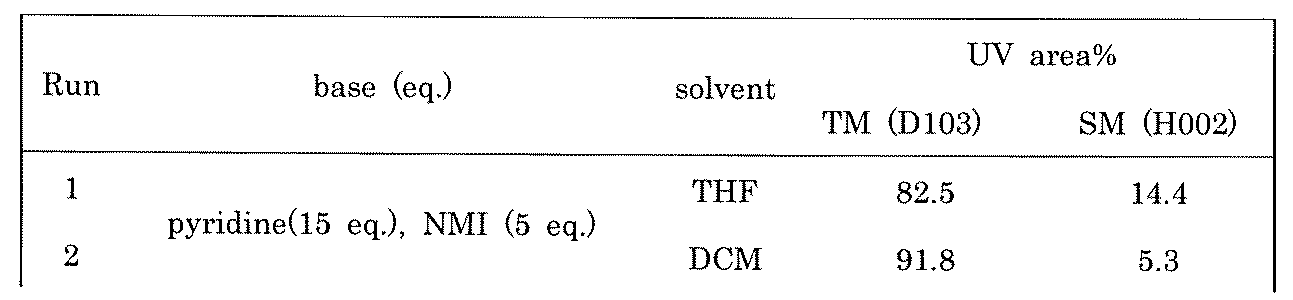

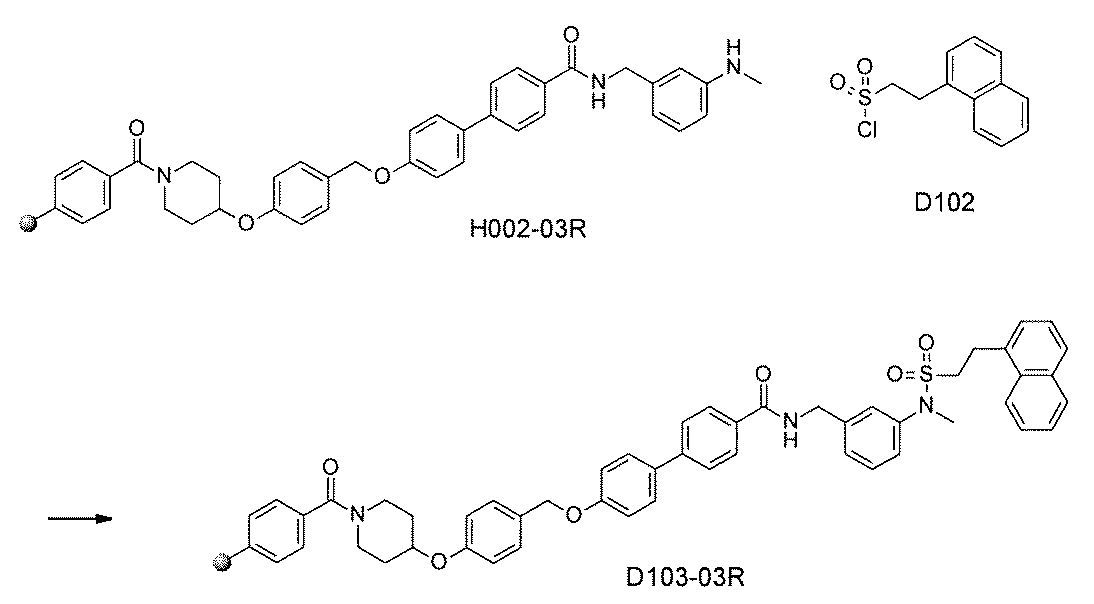
窒素雰囲気下、3、4−ルチジン(6.56μL、0.059mmol)と2−ナフタレン−1−イルエタンスルホニルクロリド(D102)(9.4mg、0.039mmol)を加え、得られた混合物を室温で24時間振とうした。
窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D103が93.8面積%観測された。
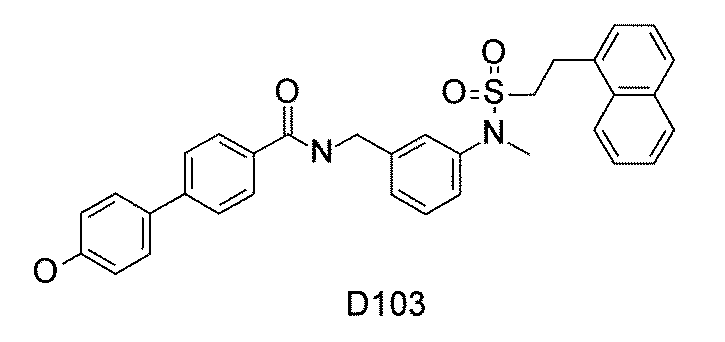
LCMS:m/z 551[M+H]+。
保持時間:1.199分(分析条件 SMD−FA05−1、299nm)。
実施例2−11−3で見出した、THF中で塩基としてピリジンとNMI用いる反応条件を用いて、固相担持アミン(D021−03R)とフェニルメタンスルホニルクロリド(D100)のスルホンアミド化を実施した。実験操作を以下に示す。

窒素雰囲気下、ピリジン(4.8μL、0.059mmol)、NMI(1.6μL、2.0μmol)とフェニルメタンスルホニルクロリド(D100)(7.5mg、0.039mmol)を加え、得られた混合物を室温で7時間振とうした。
窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D104が97.2面積%観測された。

LCMS:m/z 437[M+H]+。
保持時間:0.975分(分析条件 SMD−FA05−1、299nm)。
実施例2−11−6−1−2で見出した、DCM中で塩基としてピリジンとNMI用いる反応条件を用いて、固相担持アミン(H003−03R)と2−ナフタレン−1−イルエタンスルホニルクロリド(D102)を用いてスルホンアミド化を実施した。実験操作を以下に示す。

窒素雰囲気下、ピリジン(4.8μL、0.059mmol)、NMI(1.6μL、2.0μmol)と2−ナフタレン−1−イルエタンスルホニルクロリド(D102)(10.4mg、0.039mmol)を加え、得られた混合物を室温で7時間振とうした。
窒素雰囲気下、レジンの懸濁液12μLをサンプリング後、フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(500μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D105が94.8面積%観測された。

LCMS:m/z 501[M+H]+。
保持時間:1.144分(分析条件 SMD−FA05−1、299nm)。
固相担持スルホニルクロリド(D095−03R)のスルホンアミド化により、化合物D097−03Rは以下のように合成した。

反応液と固相の懸濁液(12μL)をフィルター上に移し、DMA(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.1mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D097が85.2%の純度で観測された。

LRMS:m/z 620[M+H]+。
保持時間:2.483分(分析条件SMD−TFA05−RP−long、299nm)。
固相担持スルフィン酸(化合物D093−03R)とNCS、およびピペリジンを用いて、スルホンアミド(D094−03R)は以下のように合成した。

反応液と固相の懸濁液(12μL)をフィルター上に移し、NMP(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.50mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D094が86.7%の純度で観測された。

LRMS:m/z 437[M+H]+。
保持時間:1.132分(分析条件SMD−FA05−1、299nm)。
非特許文献(Angew.Chem.Int.Ed.2014,53,10204−10208)を参考に、固相担持ArI(D090−03R)とDABSOを用いてスルホン(D099−03R)を合成した。
固相担持ArI(D090−03R)とDABSOを用いてスルホン(D099−03R)を合成した固相実験(反応操作の例示)
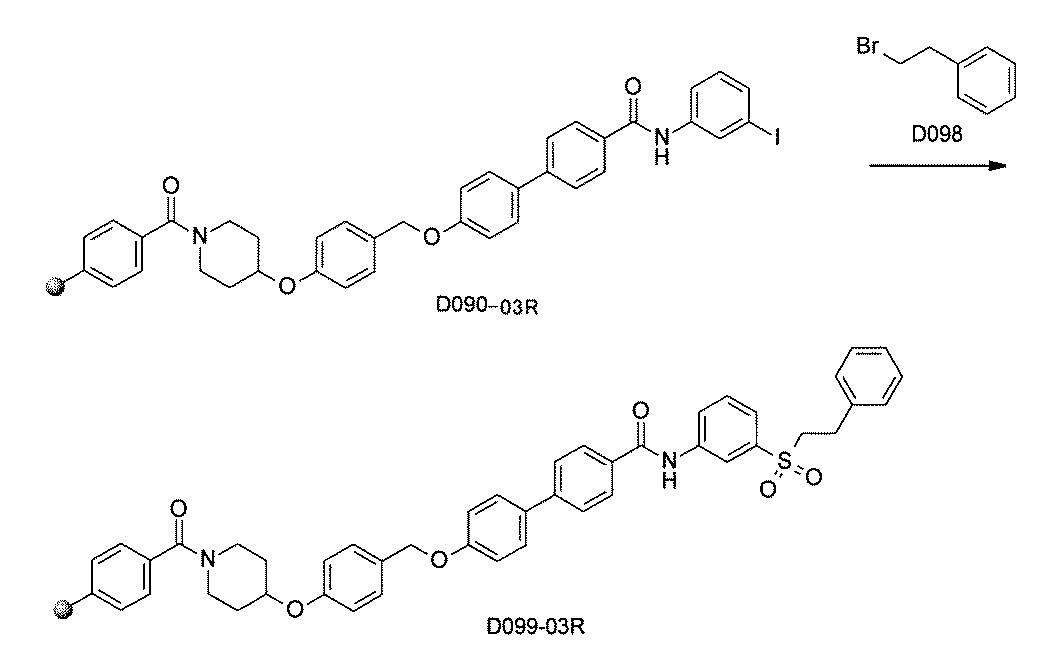

LCMS:m/z 458[M+H]+。
保持時間:1.135分(分析条件SMD−FA05−1、299nm)。
以下では、C−N結合形成反応において広い基質適用範囲が期待される反応条件での、適用可能な基質範囲の特定方法の一例を例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
2級アミンに対するアルキル化の方法論として還元的アミノ化反応以外にもハロゲン化アルキルもしくはアルコール誘導体との求核置換反応が広く受け入れられている。以下では、固相アミンに対するハロゲン化アルキルもしくはアルコール誘導体によるアルキル化反応において広い基質適用範囲が期待される反応条件でのC−N結合形成の可否を確認した。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
固相担持アミンのハロゲン化アルキルに対する求核置換反応を、塩基としてNa2CO3を選択しNMP中で実施した。より具体的には、Na2CO3を塩基として使用し、固相担持アミン(B012−03R)と(2−ブロモエチル)ベンゼン(H204)との求核置換反応(H105−03Rの合成)を例示する。
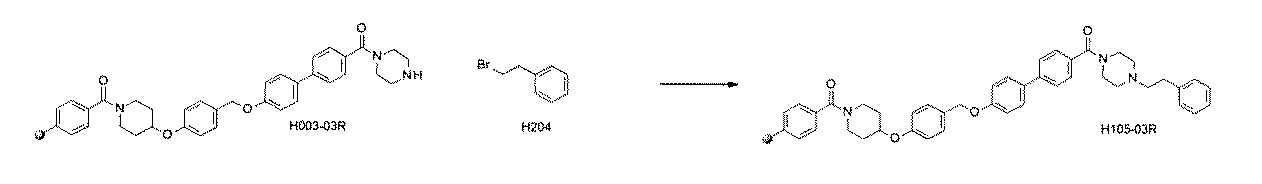

LRMS:m/z 387[M+H]+
保持時間:0.677分(分析条件SMD−FA05−1、299nm)
固相担持アミンのアルコール誘導体に対する求核置換反応を、活性化剤としてテトラキストリフェニルホスフィンパラジウムを選択しTHF中で実施した。より具体的には、テトラキストリフェニルホスフィンパラジウムを活性化剤として使用した、固相担持アミン(H003−03R)とアリルメチルカーボネート(H205)との求核置換反応(H106−03Rの合成)を例示する。


LRMS:m/z 323[M+H]+
保持時間:0.537分(分析条件SMD−FA05−1、299nm)
以上、C−N結合を構築する際には先に示した還元的アミノ化反応に加えて塩基もしくは遷移金属を利用した、アミンのアルコール誘導体もしくはハロゲン化アルキルへの求核置換反応も適用可能であることを示した。
2級アミンに対するアルキル化反応を上記実施例に示したが、一般的に一級アミンに対しては過剰反応が懸念される。そこでオルトニトロベンゼンスルホンアミド誘導体に対するアルキル化に続くスルホニル基の脱保護による、一級アミンの形式的なモノアルキル化が広く用いられている。以下では、固相スルホンアミドに対するハロゲン化アルキルもしくはアルコールによるアルキル化反応の一例を例示する。なお、以下で示す基質は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
固相担持アミンのアルコール誘導体に対する求核置換反応を、活性化剤としてCMMPを選択しTHF中で実施した。より具体的には、CMMPを活性化剤として使用し、固相担持スルホンアミド(H004−03R)と2−(3−ブロモフェニル)エタノール(H206)の求核置換反応(H107−03Rの合成)を例示する
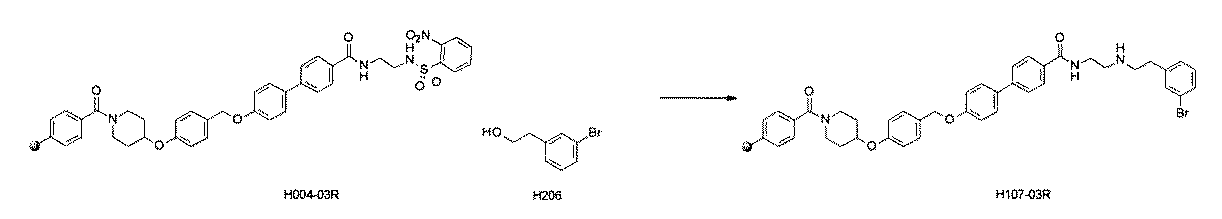
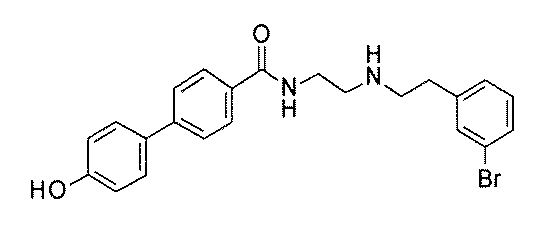
LRMS:m/z 439,441[M+H]+
保持時間:0.768分(分析条件SMD−FA05−1、299nm)
固相担持スルホンアミドのハロゲン化アルキルに対する求核置換反応を、ホスファゼン塩基P1tBuを塩基として選択しNMP中で実施した。より具体的には、ホスファゼン塩基P1tBuを塩基として使用した、固相担持スルホンアミド(H004−03R)と(2−ブロモエチル)ベンゼン(H204)の求核置換反応(H108−03Rの合成)を例示する。


LRMS:m/z 361[M+H]+
保持時間:0.697分(分析条件SMD−FA05−1、299nm)
非特許文献(Nature,2018,557,228−232.)を参考にして塩基として有機塩基のMTBD(7−メチル−1,5,7−トリアザビシクロ[4.4.0]デカ−5−エン)を選択し、Pd触媒としてtBuBrettphos Pd G4を選択してアミドとのPd試薬を用いたカップリング反応を実施した。

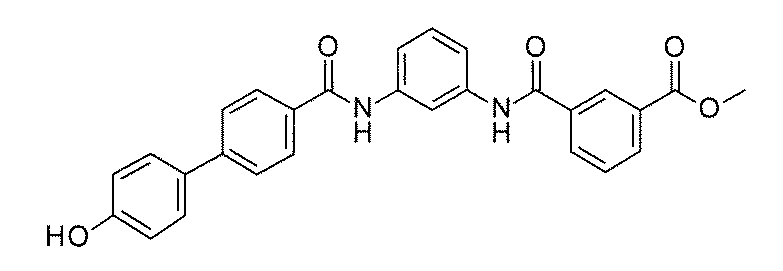
LRMS:m/z 467[M+H]+
保持時間:1.060分(分析条件SMD−FA05−1、299nm)
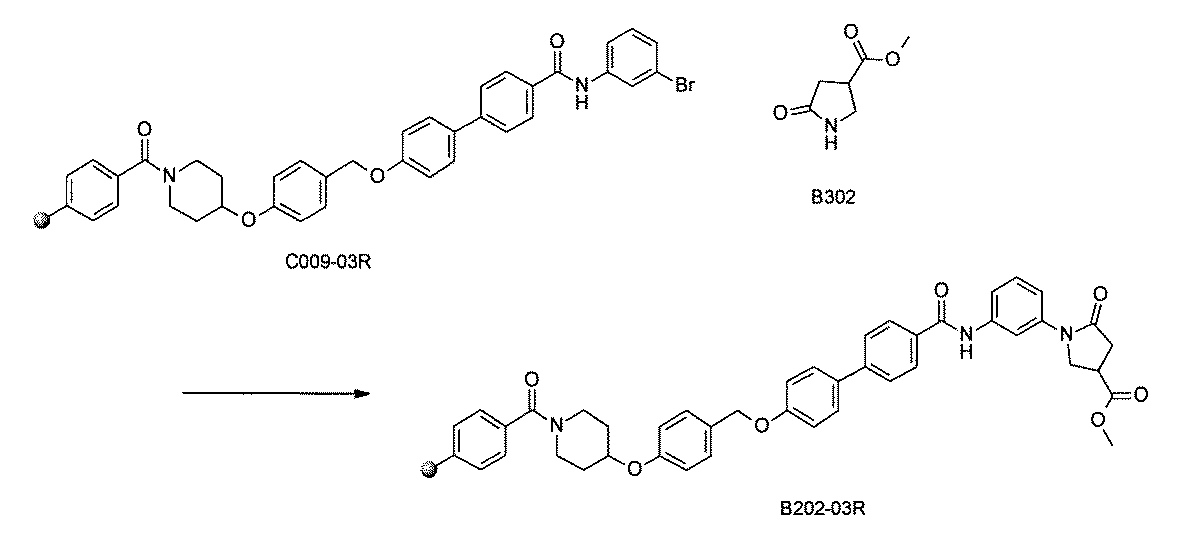
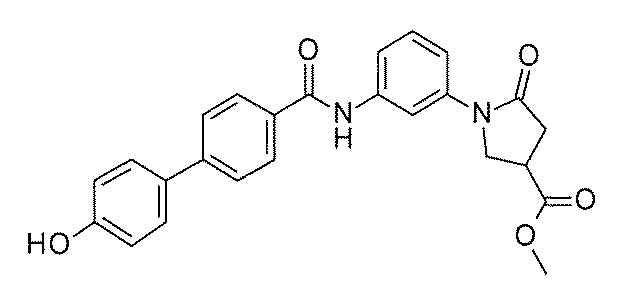
LRMS:m/z 431[M+H]+
保持時間:0.923分(分析条件SMD−FA05−1、299nm)
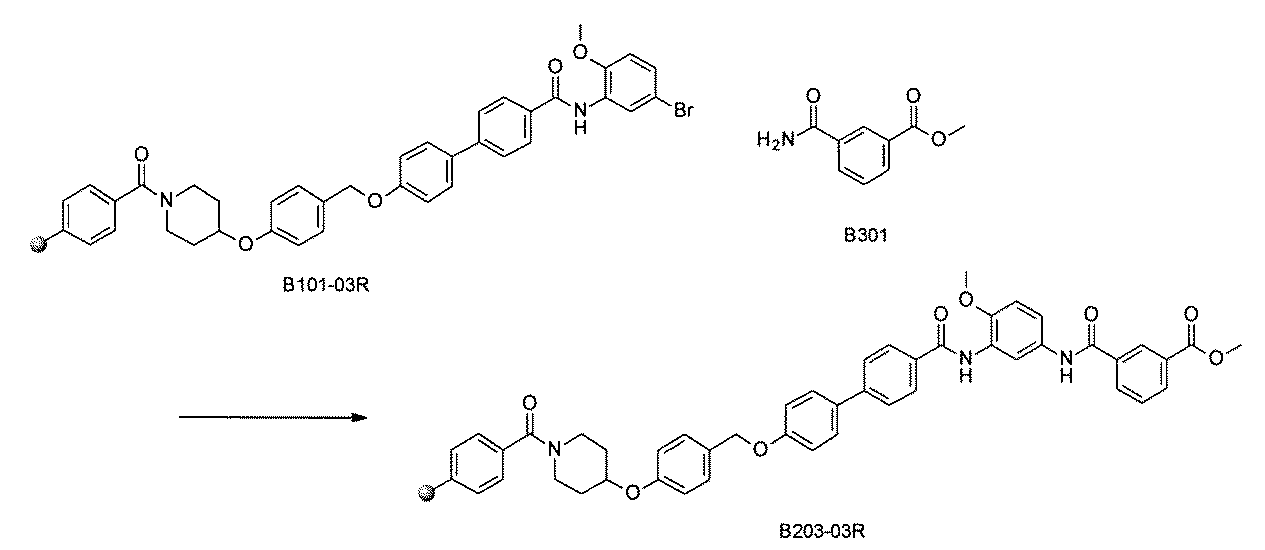
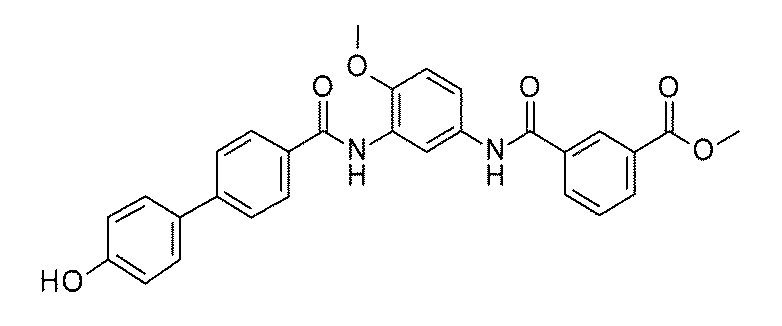
LRMS:m/z 497[M+H]+
保持時間:1.089分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 461[M+H]+
保持時間:0.943分(分析条件SMD−FA05−1、299nm)
これらの結果から、MTBDを塩基、tBuBrettphos Pd G4をPd触媒とした、ArBrとアミドとのカップリング反応が実施可能であることを確認した。
非特許文献(Nature,2018,557,228−232.)を参考にして塩基として有機塩基のP2Etを選択し、Pd触媒としてtBuBrettphos Pd G4を選択してスルホンアミドとのカップリング反応を実施した。


LRMS:m/z 517[M+H]+
保持時間:1.060分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 465[M+H]+
保持時間:1.040分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 527[M+H]+
保持時間:1.091分(分析条件SMD−FA05−1、299nm)

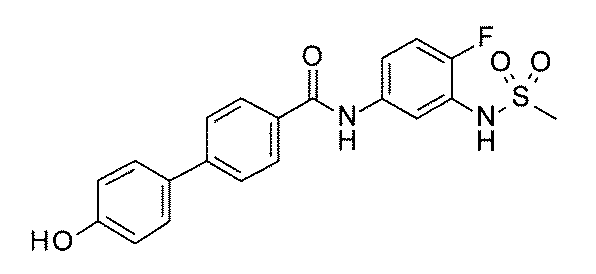
LRMS:m/z 401[M+H]+
保持時間:0.884分(分析条件SMD−FA05−1、299nm)
これらの結果から、P2Etを塩基、tBuBrettphos Pd G4をPd触媒とした、ArBrとスルホンアミドとのカップリング反応が実施可能であることを確認した。
実施例2−14−1:ROHとハロゲン化アルキルとの求核置換反応に関する固相実験
以下では、固相アルコールのハロゲン化アルキルに対する求核置換反応に関しての一例を例示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
固相担持アルコールのハロゲン化アルキルに対する求核置換反応を、塩基としてKOtBuを選択しTHF中で実施した。より具体的には、KOtBuを塩基として使用した、固相担持アルコール(H005−03R)とブロモメチルベンゼン(H207)の求核置換反応(H109−03Rの合成)を例示する。
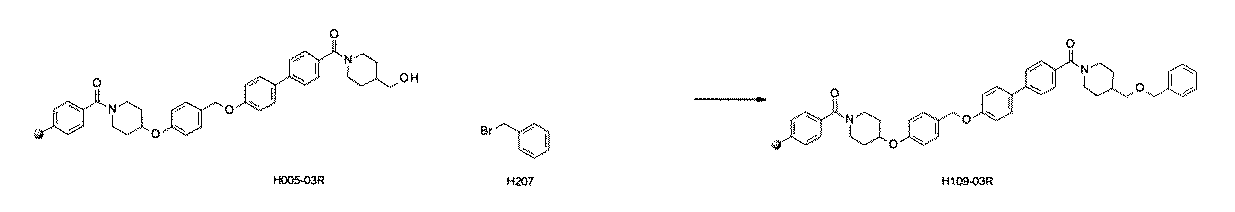
反応液と固相の懸濁液をフィルター上に移し、DMA(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.1Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMA(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物H109が92%観測された。

LRMS:m/z 402[M+H]+
保持時間:1.155分(分析条件SMD−FA05−1、299nm)
以下では、固相担持ArBrとアレノールとのC−Oカップリング反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。


LRMS:m/z 410[M+H]+
保持時間:1.352分(分析条件 SMD−FA05−1)
この結果から、固相ArBrに対してアレノールとのC−Oカップリング反応が適用可能であることが示された。
以下では、固相担持ArOHとアリールブロミドとのC−Oカップリング反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
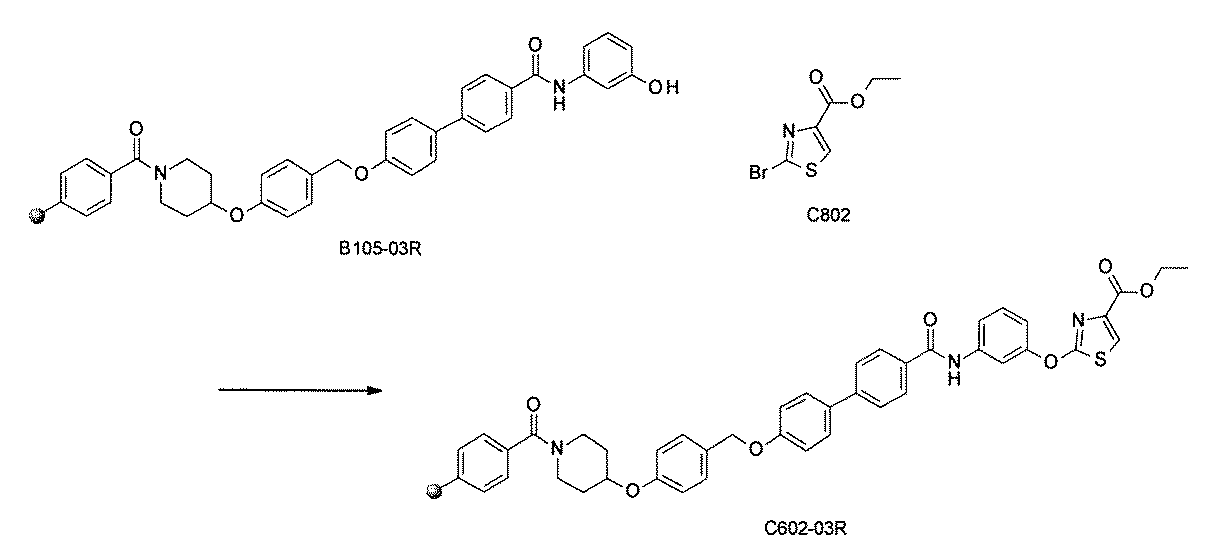
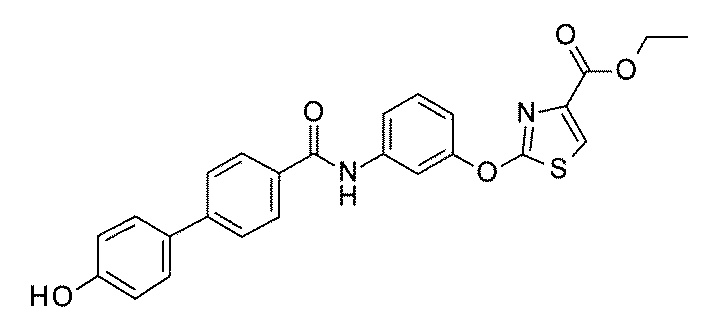
LRMS:m/z 461[M+H]+
保持時間:1.140分(分析条件 SMD−FA05−1)
この結果から、固相ArOHに対してアリールブロミドとのC−Oカップリング反応が適用可能であることが示された。
以下では、固相担持ArBrとチオールとのC−Sカップリング反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。

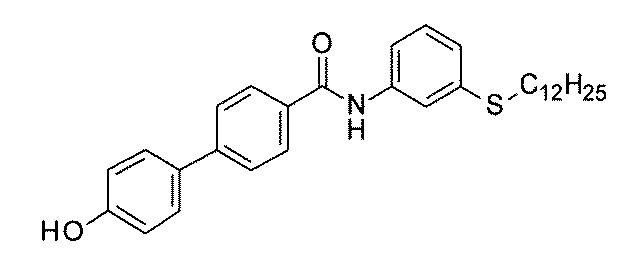
LRMS:m/z 490[M+H]+
保持時間:1.829分(分析条件 SMD−FA05−1)
この結果から、固相ArBrに対してチオールとのC−Sカップリング反応が適用可能であることが示された。
以下では、固相担持カルボン酸とチオールとの縮合反応によって得られる固相チオエステルと、ボロン酸とのカップリングによるケトン合成反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。

LRMS:m/z 394[M+H]+
保持時間:1.149分(分析条件 SMD−FA05−1)
この結果から、固相担持カルボン酸から固相チオエステルを経由して、ボロン酸とのカップリング反応によるケトンの合成が可能であることが示された。
以下では、エステル結合形成反応(R1CO2H+R2OH)の適切な反応条件を選択することによって、再現性を高める条件を一例として例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
非特許文献(RSC Advances,2018,8,25168.)を参考にして縮合剤としてWSC・HClを選択し、塩基としてDMAPを使用して固相担持されたカルボン酸とアルコールとのエステル化反応を実施した。

極大波長:300.4nm
保持時間:1.144分(分析条件SMD−FA05−1、299nm)


LRMS:m/z 271[M+H]+
保持時間:1.256分(分析条件SMD−FA05−1、299nm)


極大波長:294.4nm
保持時間:1.344分(分析条件SMD−FA05−1、299nm)
これらの結果から、WSC・HClを縮合剤としDMAPを塩基とした縮合条件において、固相担持ArCO2Hと1級、並びに2級アルコールとのエステル化反応が実施できることを確認した。
非特許文献(Tetrahedron Lett.1995,36,2529.)と、固相エーテル化反応(実施例2−1−6−4~13)を参考に、CMMPを光延試薬として選択し固相担持ArCO2H(B106−03R)とアルコールとのエステル化反応を実施した。

反応液と固相の懸濁液をフィルター上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に2分間浸した。ろ過後、固相をDMF(0.05mL)で洗浄し、ろ液にMeCN(0.25mL)を加え、LCサンプルを調整し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物B222が98%観測された。

これらの結果から、CMMPを光延試薬としたエステル化条件において、固相担持ArCO2Hと1級、並びに2級アルコールとのエステル化反応が実施できることを確認した。
以下では、固相担持カルボン酸とアレノールとの縮合反応によるエステル合成反応の一例を提示する。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。

反応液と固相の懸濁液のうち12μLをフィルター付きチップ上に移し、DMF(0.1mL)で3回、MeOH(0.1mL)で3回、DCM(0.1mL)で3回洗浄した。ペンタメチルベンゼンの10%TFA/DCM溶液(0.02M、0.05mL)に1分間浸してろ過し、DMF(0.05mL)で洗浄し、合わせたろ液をMeCN(0.25mL)で薄めた溶液のLCMS測定を行うことで、目的物であるC604が98.4%観測された。ただし、波長299nm(±4nm)で切り出して解析している。

LRMS:m/z424[M+H]+。
保持時間:1.283分(分析条件SMD−FA05−1)。
この結果から、固相担持カルボン酸とアレノールの縮合反応が実施できることが示された。
実施例2−18:含窒素5員環HetAr形成の適用基質範囲確認のための実験
以下では、含窒素5員環HetAr形成の適切な合成反応条件を選択することによって、リンカーとして含窒素5員環HetArを合成する条件を一例として例示する。なお、以下で示す範囲は特定方法の一例の例示であり、実際のライブラリ合成への適用において、以下に示す範囲に制限されるものではない。
非特許文献(J.Org.Chem.2011,76,6832.)を参考にしてCuI,DIPEA,AcOHを反応促進剤として選択し、固相担持アセチレン化合物ArCCH(B107−03R)とエチルアジドアセテート(B319)とのHuisgen環化反応を実施した。


LRMS:m/z 443[M+H]+
保持時間:0.984分(分析条件SMD−FA05−1、299nm)
非特許文献(J.Org.Chem.2011,76,6832.)を参考にしてCuI,DIPEA,AcOHを反応促進剤として選択し、固相担持アジド化合物ArN3(B108−03R)と4−エチニルトルエン(B320)とのHuisgen環化反応を実施した。

LRMS:m/z 447[M+H]+
保持時間:1.219分(分析条件SMD−FA05−1、299nm)
これらの結果からHuisgen環化反応を実施することで、リンカーとして含窒素5員環HetArを、固相担持アセチレンとアジド化合物、または、固相担持アジド化合物とアセチレン化合物との反応で形成可能であることを確認した。
非特許文献(J.Am.Chem.Soc.2001,123,7727.)を参考にして塩基として有機塩基のMTBD(7−メチル−1,5,7−トリアザビシクロ[4.4.0]デカ−5−エン)を選択し、Cu触媒としてCuIを、リガンドとしてDMEDA(N,N′−ジメチルエチレンジアミン)を選択してピラゾール−3−カルボン酸エチルとのC−N結合形成反応を実施した。


LRMS:m/z 428[M+H]+
保持時間:1.096分(分析条件SMD−FA05−1、299nm)
この結果からCuIを使用したHeteroArのN−Hと固相ArBrとのC−N結合形成反応を実施可能であることを確認した。
実施例2−19−1:固相担持ArBr(C009−03R)から固相担持カルボン酸(C119−03R)への官能基変換反応
以下では、固相担持ArBrから固相担持カルボン酸への官能基変換反応の一例を提示する。固相担持カルボン酸は実施例2−7、2−8、2−16、2−17に示すように、アミド、アシルスルホンアミド、ケトン、エステルを得る際に重要な基質である。固相担持ArBrから固相担持カルボン酸への官能基変換が適用可能となることで、固相担持ArBrからアミド、アシルスルホンアミド、ケトン、エステルが合成可能となる。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。
この結果から、固相においてアリールブロミドからカルボン酸への変換を行えることが示された。
以下では、固相担持カルボン酸とチオールとの縮合反応によって得られる固相チオエステルから、固相担持ボロン酸エステルまたはボロン酸への変換反応の一例を提示した。固相担持ボロン酸エステルは実施例2−2−10に示すように、鈴木カップリング反応に適用可能な重要な基質である。なお、実際のライブラリ合成への適用において、以下に示す基質に制限されるものではない。


LRMS:m/z 416[M+H]+
保持時間:1.224分(分析条件 SMD−FA05−1)
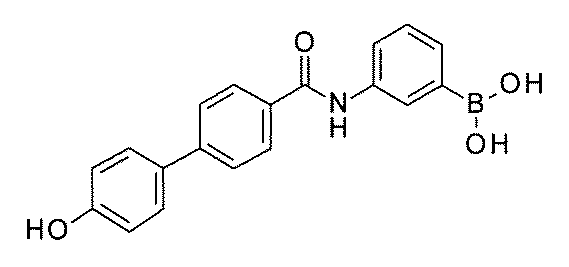
LRMS:m/z 334[M+H]+
保持時間:0.809分(分析条件 SMD−FA05−1)
この結果から、固相においてカルボン酸からチオエステルの形成を経て、ボロン酸への変換を行えることが示された。
ArNH2を出発物質とした、結合形成反応の利用可能な範囲を拡充させる一つの方法として、ArBrへ変換し結合形成反応を実施する方法が挙げられる。ArNH2からArBrへ変換させる方法としてSandmeyer反応を選択した。有機酸を使用した温和な反応条件下での反応を実施している非特許文献(Org.Chem.2017,19,2518.)を参考にしてジアゾ化試薬としてNaNO2(亜硝酸ナトリウム)、Br化剤としてBrCCl3(ブロモトリクロロメタン)を、酸としてAcOHを、反応溶媒としてTHF−H2Oを選択し、固相ArNH2へのSandmeyerブロモ化反応を実施した。

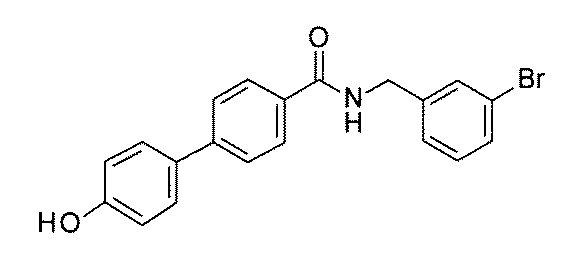
LRMS:m/z 382、284[M+H]+
保持時間:1.081分(分析条件SMD−FA05−1、299nm)

窒素雰囲気下、0.6mLのガラスバイアルに、固相担持ArNH2(A118−03R)(担持量0.197mmol/g、20mg、0.0039mmol)と1,4−ジオキサン(0.3mL)を加え、室温で1時間振とうした。NaNO2(亜硝酸ナトリウム、13.6mg、0.197mmol)、BrCCl3(ブロモトリクロロメタン、19.4μL、0.197mmol)、AcOH(22.6μL、0.394mmol)とH2O(0.1mL)を加え、室温で3時間振とうした。
亜硝酸ナトリウムを使用する反応条件では、亜硝酸ナトリウムを溶解するために水を使用する。固相反応、特にライブラリ合成時には、レジンがより膨潤しやすい有機溶媒を選択することが好ましい。そこで有機溶媒中でジアゾ化試薬として亜硝酸エステルを使用し、広範囲の基質に適用可能と考えられる反応条件として、非特許文献(Org.Biomol.Chem.2019,17,88.)、または非特許文献(Tetrahedron,2013,69,3511.)を参考に、ジアゾ化試薬として亜硝酸ヘキシル、Br化剤としてTMSBr(ブロモトリメチルシラン)を、酸としてPPTS(p−トルエンスルホン酸ピリジニウム)を選択し、反応溶媒としてDCE(ジクロロエタン)を反応溶媒として使用してSandmeyer反応を実施した。
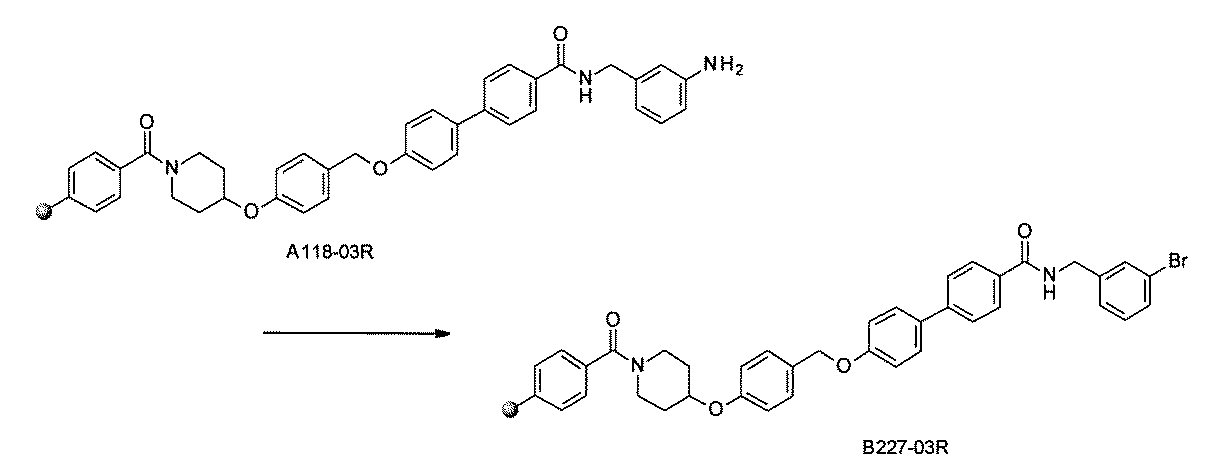

LRMS:m/z 338[M+H]+
保持時間:1.047分(分析条件SMD−FA05−1、299nm)
特定の理論に限定されないが、実施例2−19−3−3において観測されたArCl(B228)は、反応溶媒として使用したDCE(ジクロロエタン)がクロロ源として作用することで生成したと考えられるので、特許文献(WO2020178282)中で使用実績があるDBM(ジブロモメタン)を反応溶媒と選択して反応を実施した。

反応のスケールを増加しても、固相でのSandmeyer反応が問題なく進行することを確認するために、5倍のスケールで反応を実施した。

これらの結果から、亜硝酸ナトリウム、もしくは亜硝酸ヘキシルをジアゾ化試薬として使用するSandmeyer反応により、ArNH2をArBrへ変換可能であることを確認した。
ライブラリ化合物の多様性の確保とライブラリの品質の担保は、共にライブラリの使用目的である化合物スクリーニングにおける成否に関わる重要な要素であり、化合物ライブラリを供給する際の重要な課題である。
多様性の課題に対する解決策として、一般的なライブラリ合成で考慮されているビルディングブロックの多様性に加えて、本発明の着想であるビルディングブロック同士をつなぐ連結部分(リンカー)に多様性を持たせ、それらの多様性を掛け合わせた化合物の混合物ライブラリの合成について一例を示す。また、品質の担保に関しての課題の解決策の一例として、「意図したライブラリ化合物が合成されて混合物ライブラリ中に存在していること」に加え、「各ライブラリ化合物間の含量の差が小さいこと」を確認する工程の実践例を示す。
[表LBB01]
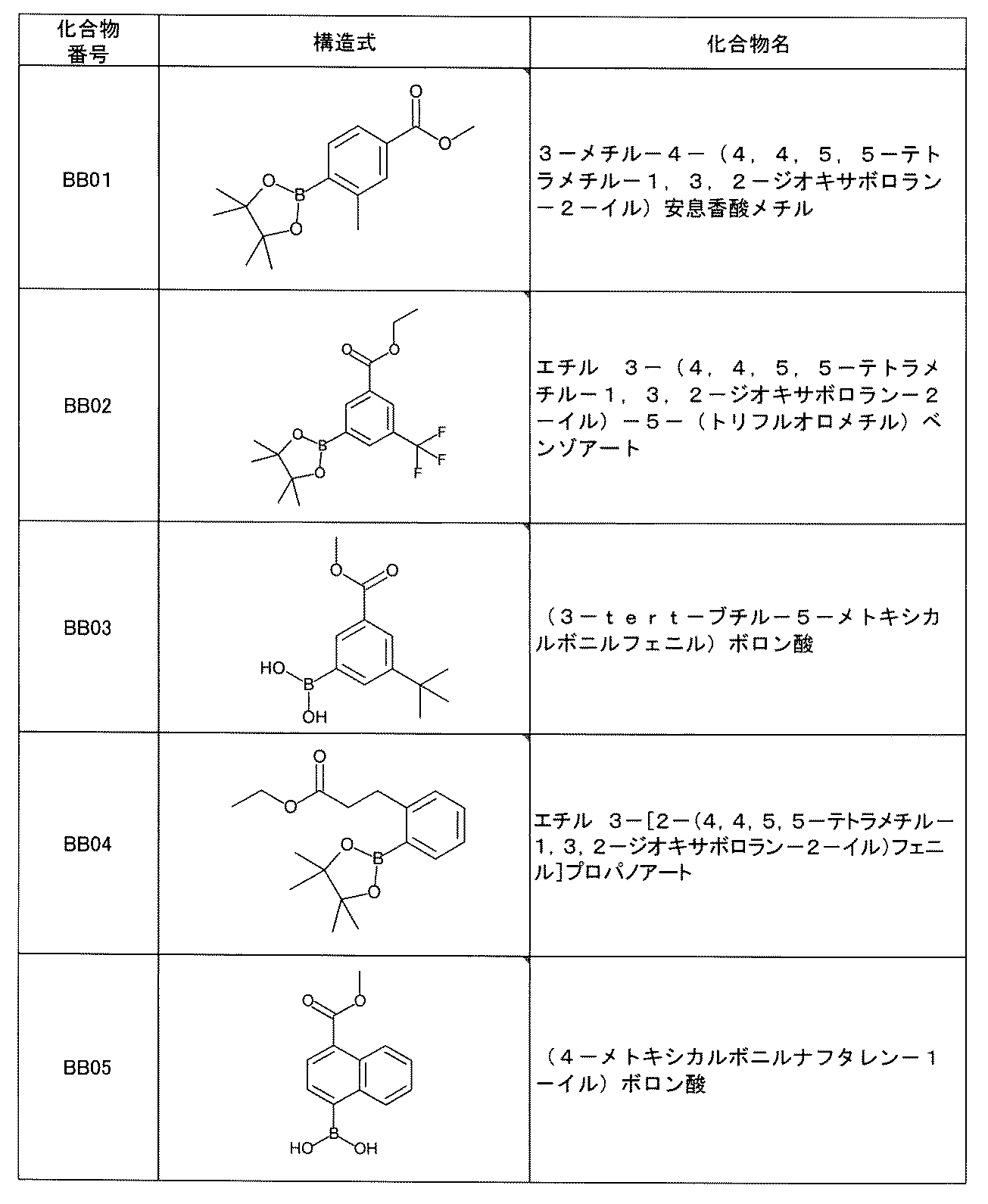



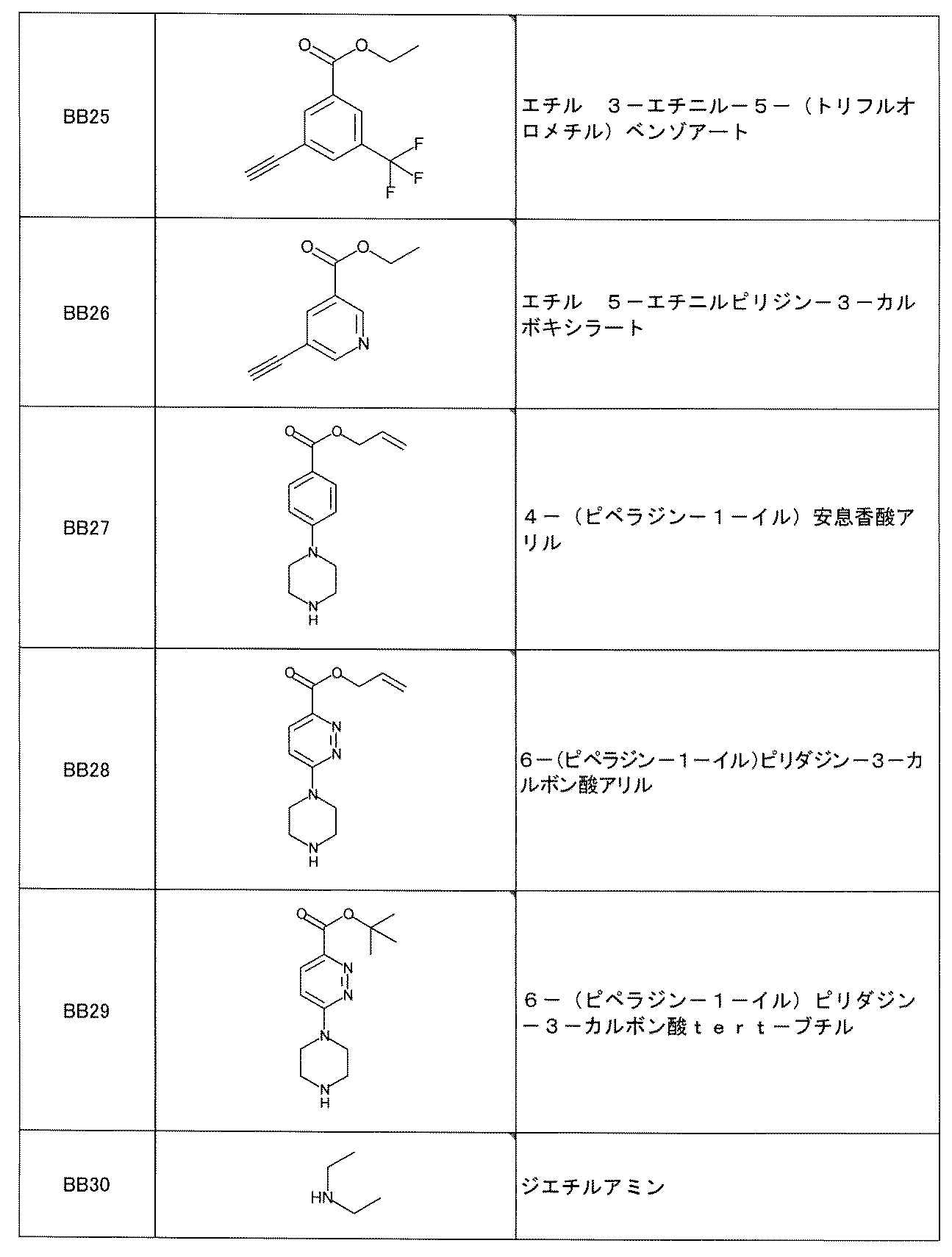


実施例3−1−1:mixEB01−01Rの合成
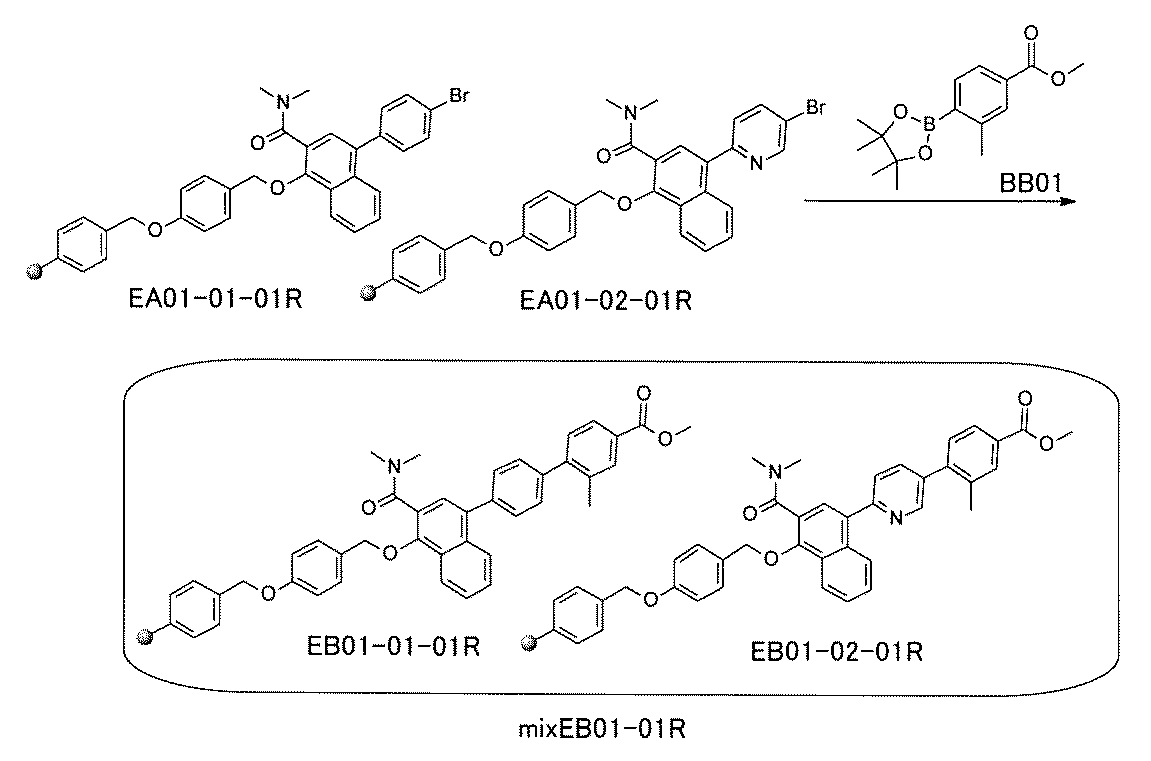

化合物EA01−01−01R(48mg、担持量0.4mmol/g,0.019mmol)、EA01−02−01R(52mg、担持量0.37mmol/g,0.019mmol)と表LBB01に示したボロン酸またはボロン酸エステル試薬(BB02~04)を用いて、実施例3−1−1と同様の方法で[表EB02]に示した標題化合物を合成した。用いたビルディングブロックと分析対象の化合物の構造、反応時間、分析データについても[表EB02]に示す。



化合物EA01−01−01R(48mg、担持量0.4mmol/g,0.019mmol)、EA01−02−01R(52mg、担持量0.37mmol/g,0.019mmol)と表LBB01に示した[1−[3−エトキシカルボニル−5−(トリフルオロメチル)フェニル]ピラゾル−4−イル]ボロン酸(BB06)を用いて、実施例3−1−3と同様の方法で[表EB06]に示した標題化合物を合成した。用いたビルディングブロックと分析対象の化合物の構造、反応時間、分析データについても[表EB06]に示す。



化合物EA01−01−01R(48mg、担持量0.4mmol/g,0.019mmol)、化合物EA01−02−01R(52mg、担持量0.37mmol/g,0.019mmol)と表LBB01に示したボロン酸試薬(BB08~18)を用いて、実施例3−1−5と同様の方法で[表EB08]に示した標題化合物を合成した。用いたビルディングブロックと分析対象の化合物の構造、反応時間、分析データについても[表EB08]に示す。


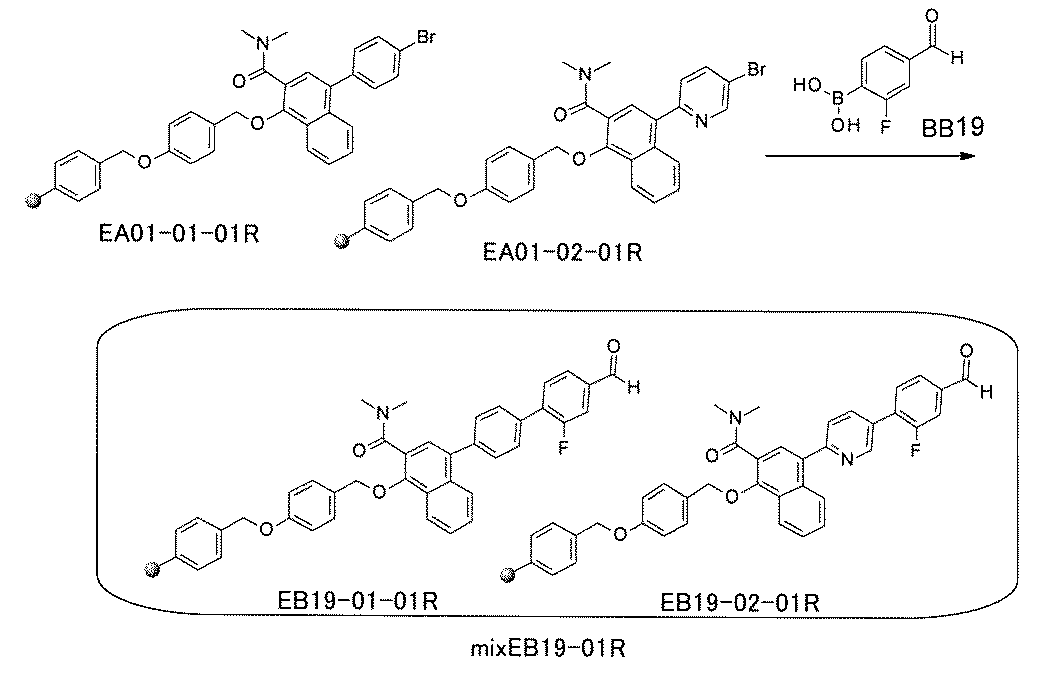
[表EB19]

化合物EA01−01−01R(48mg、担持量0.4mmol/g,0.019mmol)、EA01−02−01R(52mg、担持量0.37mmol/g,0.019mmol)と表LBB01に示した3−フルオロ−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)安息香酸メチル(BB20)を用いて、実施例3−1−7と同様の方法で[表EB20]に示した標題化合物を合成した。用いたビルディングブロックと分析対象の化合物の構造、反応時間、分析データについても[表EB20]に示す。
[表EB20]



化合物EA01−01−01R(48mg、担持量0.4mmol/g,0.019mmol)、EA01−02−01R(52mg、担持量0.37mmol/g,0.019mmol)と表LBB01に示した[3−ホルミル−5−(トリフルオロメトキシ)フェニル]ボロン酸(BB22)を用いて、実施例3−1−9と同様の方法で[表EB22]に示した標題化合物を合成した。用いたビルディングブロックと分析対象の化合物の構造、反応時間、分析データについても[表EB22]に示す。

実施例3−2−1:混合物mixEB23−01Rの合成






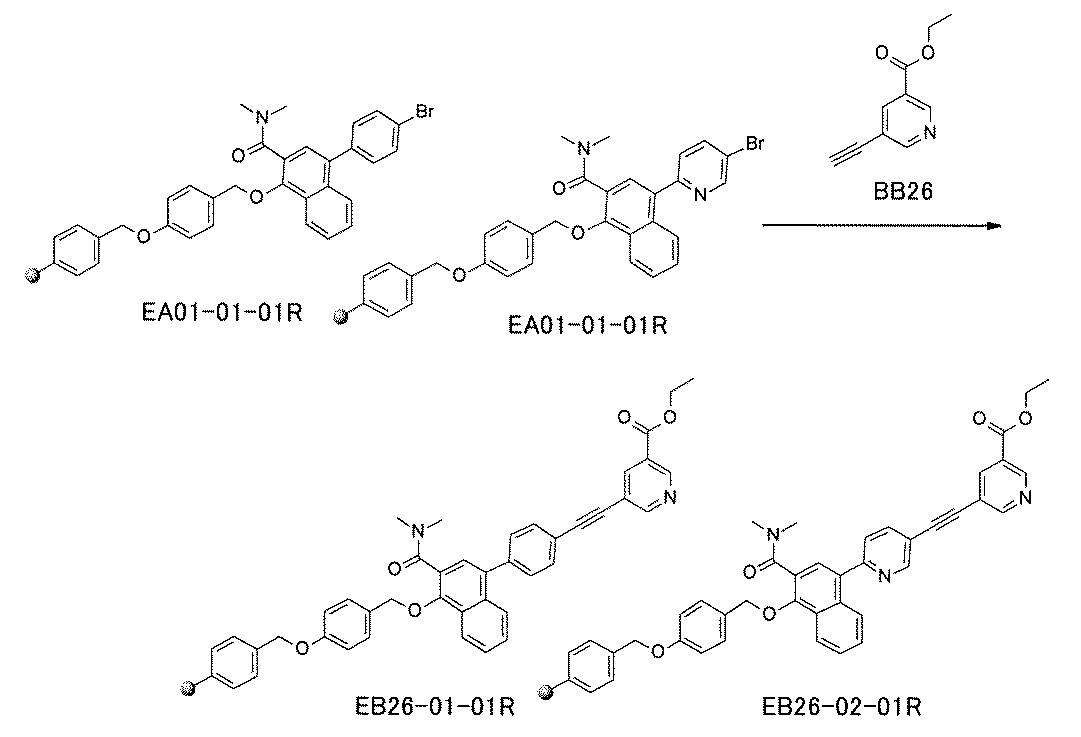

実施例3−3−1:mixAG—C01−01Rの調製
[表AG−C−01]に示す混合物、mixEB07−01R、mixEB08−01R、mixEB09−01R、mixEB10−01R、mixEB13−01R、mixEB14−01R、mixEB15−01R、mixEB16−01R、mixEB17−01R、mixEB19−01R、mixEB21−01R、mixEB22−01R、mixEB23−01Rをガラスバイアル中に移し、実施例3−3−2にて使用する混合物mixAG—C01−01Rとした。各混合物の使用量、含まれる化合物の番号、構造式について[表AG−C−01]に示す。


反応21時間経過時の分析データ(生成物のLCMSと保持時間)について[表AG−C−02]に示す。
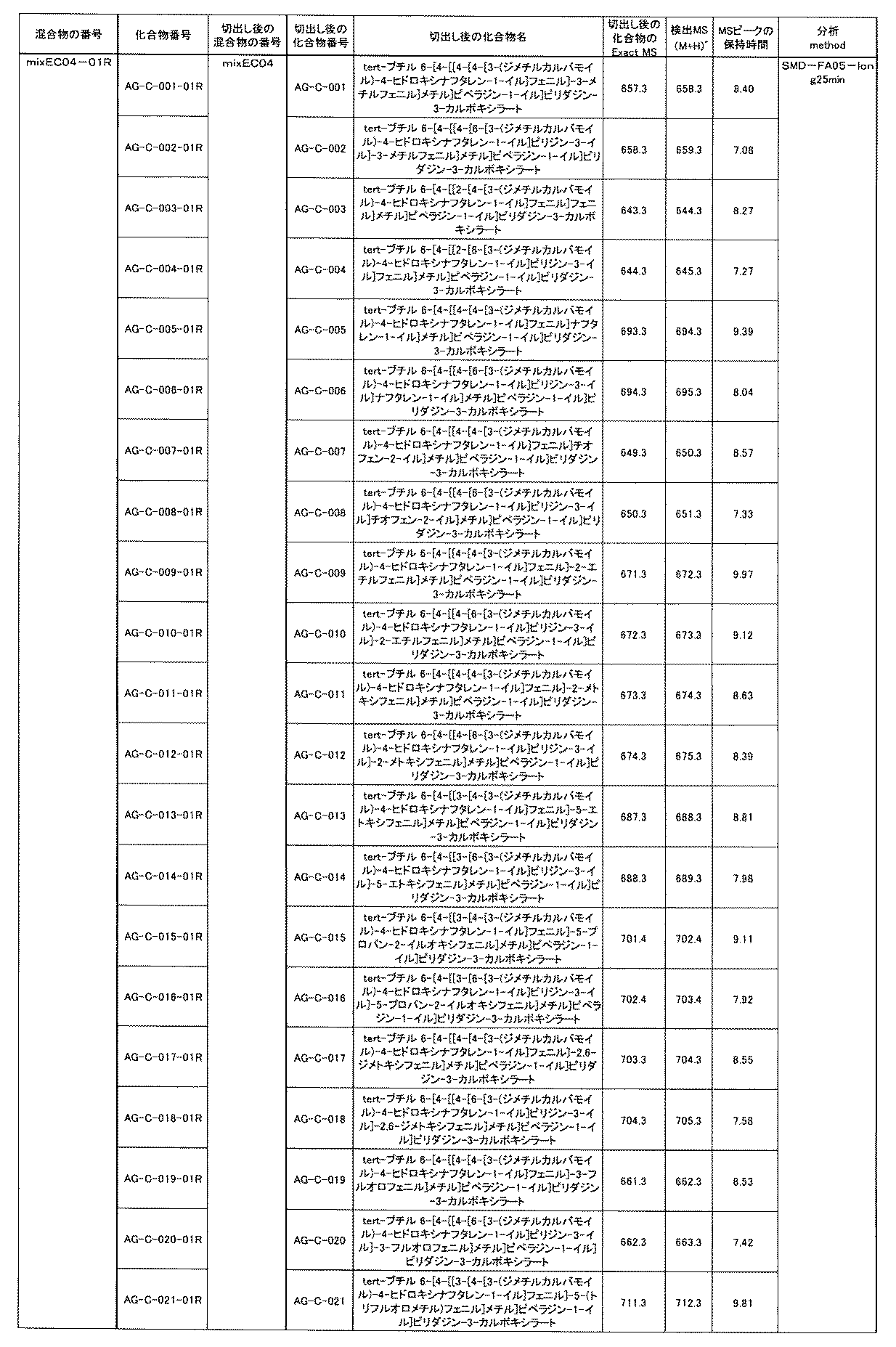

[表AG−C−03]に示す混合物、mixEB07−01R、mixEB08−01R、mixEB09−01R、mixEB10−01R、mixEB13−01R、mixEB14−01R、mixEB15−01R、mixEB16−01R、mixEB17−01R、mixEB19−01R、mixEB21−01R、mixEB22−01R、mixEB23−01Rをガラスバイアル中に移し、実施例3−3−4:にて使用する混合物mixAG—C03−01Rとした。各混合物の使用量、含まれる化合物の番号、構造式について[表AG−C−03]に示す。



[表AG−C−04]


実施例3−4−1:混合物mixEC06−01Rの合成

[表EBC06]


得られたエステル化合物に対して、以下に示した方法でエステルの脱保護を実施し、カルボン酸化合物を合成した。
実施例3−5−1:mixEC01−01Rの合成
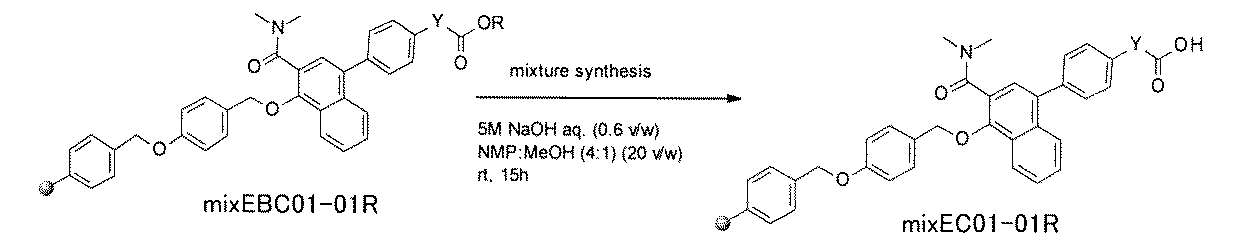




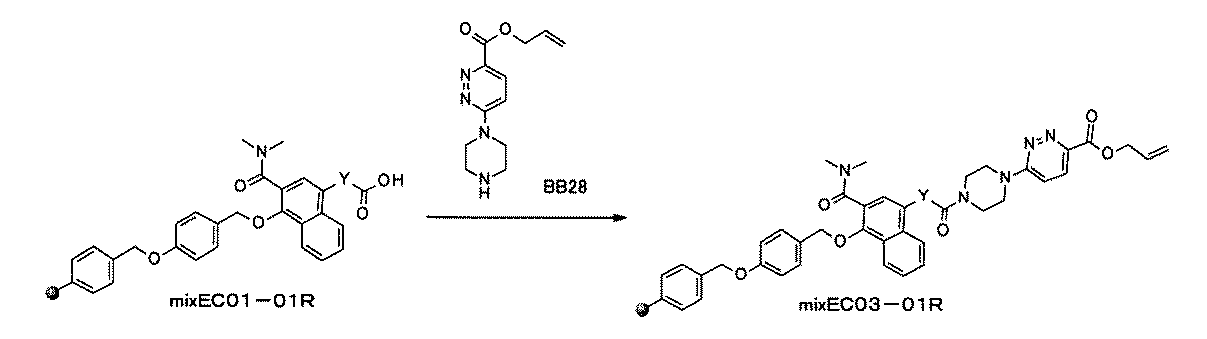
[表EC03]に記載したように、このアミド化反応において、2化合物(EC03−07−01RとEC03−08−01R)が合成されていないことをLC−MSの測定により同定している。目的の化合物が生成しているかどうかを、各工程で分析を実施し確認することで、「質の高い」ライブラリを供給する一例として例示する。

実施例3−4ならびに実施例3−5にて合成したエステル化合物に対して、以下に示した方法でエステルの脱保護を実施し、カルボン酸化合物を合成した。
実施例3−6−1:mixAG—D01−01Rの調製
[表AG−D−01]に示す混合物、mixEC02−01R(200mg)、mixEC03−01R(200mg)、mixEC05−01R(236mg)をガラスバイアル中に移し、実施例3−6−2にて使用する混合物mixAG—D01−01Rを得た。



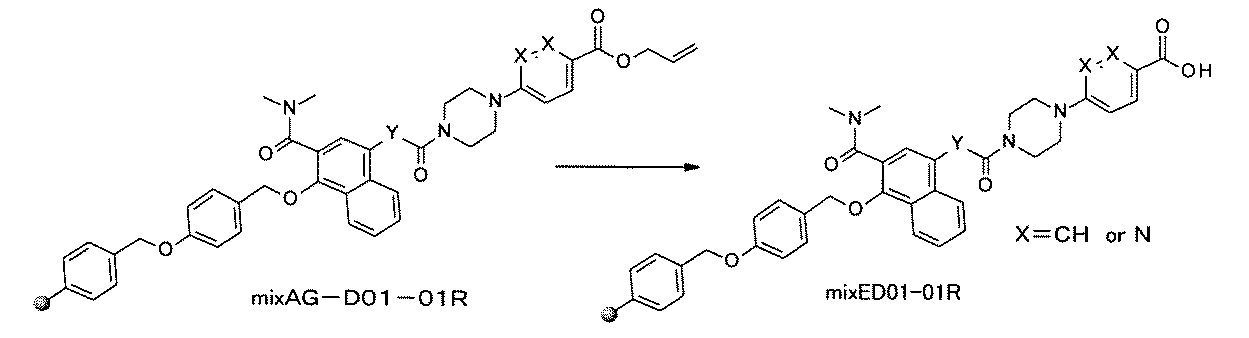
反応3時間経過時の分析データ(生成物のLCMSと保持時間)について[表AG−D−02]に示す。






反応追跡は以下のように実施した。窒素雰囲気下、レジンの懸濁液10μLをサンプリング後、フィルター付きピペットチップに移し、NMP(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回繰り返して洗浄した後、0.05Mトリメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、ろ液をNMP(250μL)に溶解させ、その溶液(50μL)をMeCN(250μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。

[表AG−D−04]に示す混合物、mixED01−01R(421mg)、mixED02−01R(24mg)、mixED03−01R(156mg)、をフィルター付きカラムに移し、NMP(12mL)で3回、MeOH(12mL)で3回、DCM(12mL)で3回、ヘプタン(12mL)で3回で繰り返し洗浄した後に減圧下で乾燥し、実施例3−6−6~実施例3−6−11にて使用する混合物mixED04−01R(601mg)を得た。


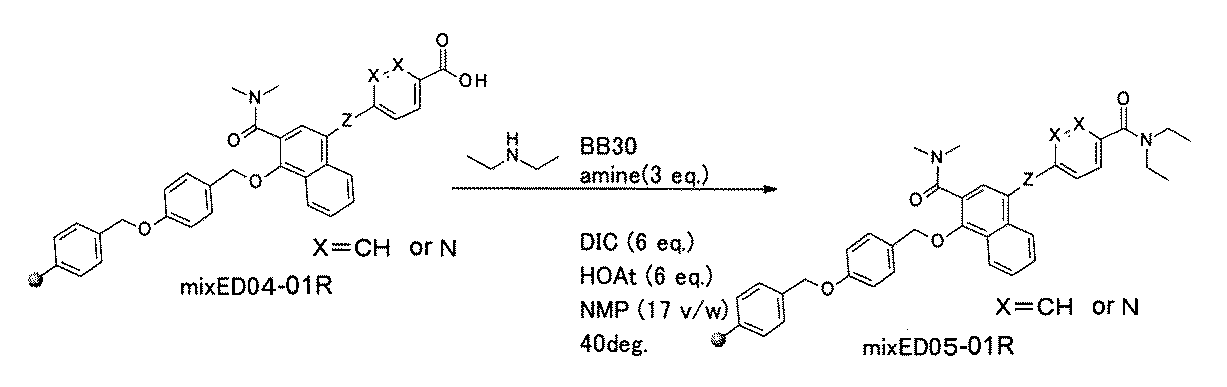




実施例3−6−6と同様の方法で、mixED04−01Rおよび[表LBB01]に示したアミン試薬、BB31を用いて[表ED06]に示した標題混合物(mixED06−01)を、BB32を用いて[表ED07]に示した標題混合物(mixED07−01)を、BB33を用いて[表ED08]に示した標題混合物(mixED08−01)をそれぞれ得た。












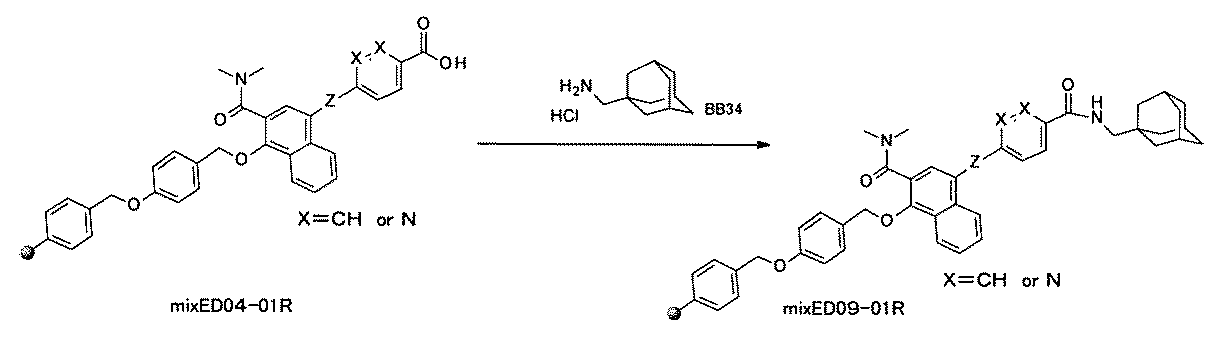




実施例3−6−8と同様の方法で、mixED04−01Rおよび1−(1−アダマンチル)−N−メチルメタンアミン塩酸塩(BB35)を用いて、[表ED10]に示した標題混合物(mixED10−01R)を得た。









実施例3−6−10と同様の方法で、mixED04−01Rおよび[表LBB02]に示したスルホンアミド試薬BB37を用いて[表ED12]に示した標題混合物(mixED12−01)を、BB38を用いて[表ED13]に示した標題混合物(mixED13−01)を、BB39を用いて[表ED14]に示した標題混合物(mixED14−01)を、BB40を用いて[表ED15]に示した標題混合物(mixED15−01)を、BB41を用いて[表ED16]に示した標題混合物(mixED16−01)ををそれぞれ得た。



















固相からの切出し実験を実施し、それぞれの混合物をLC/MSにて分析することで合成したライブラリの評価を行った。




実施例3−7−1と同様の方法で、[表ED06]~[表ED16]に示した混合物を用いて、[表EE02]~[表EE12]に示す混合物を得た。すなわち、mixED06−01Rを用いてmixED06を、mixED07−01Rを用いてmixED07を、mixED08−01Rを用いてmixED08を、mixED09−01Rを用いてmixED09を、mixED10−01Rを用いてmixED10を、mixED11−01Rを用いてmixED11を、mixED12−01Rを用いてmixED12を、mixED13−01Rを用いてmixED13を、mixED14−01Rを用いてmixED14を、mixED15−01Rを用いてmixED15を、mixED16−01Rを用いてmixED16を、それぞれ得た。































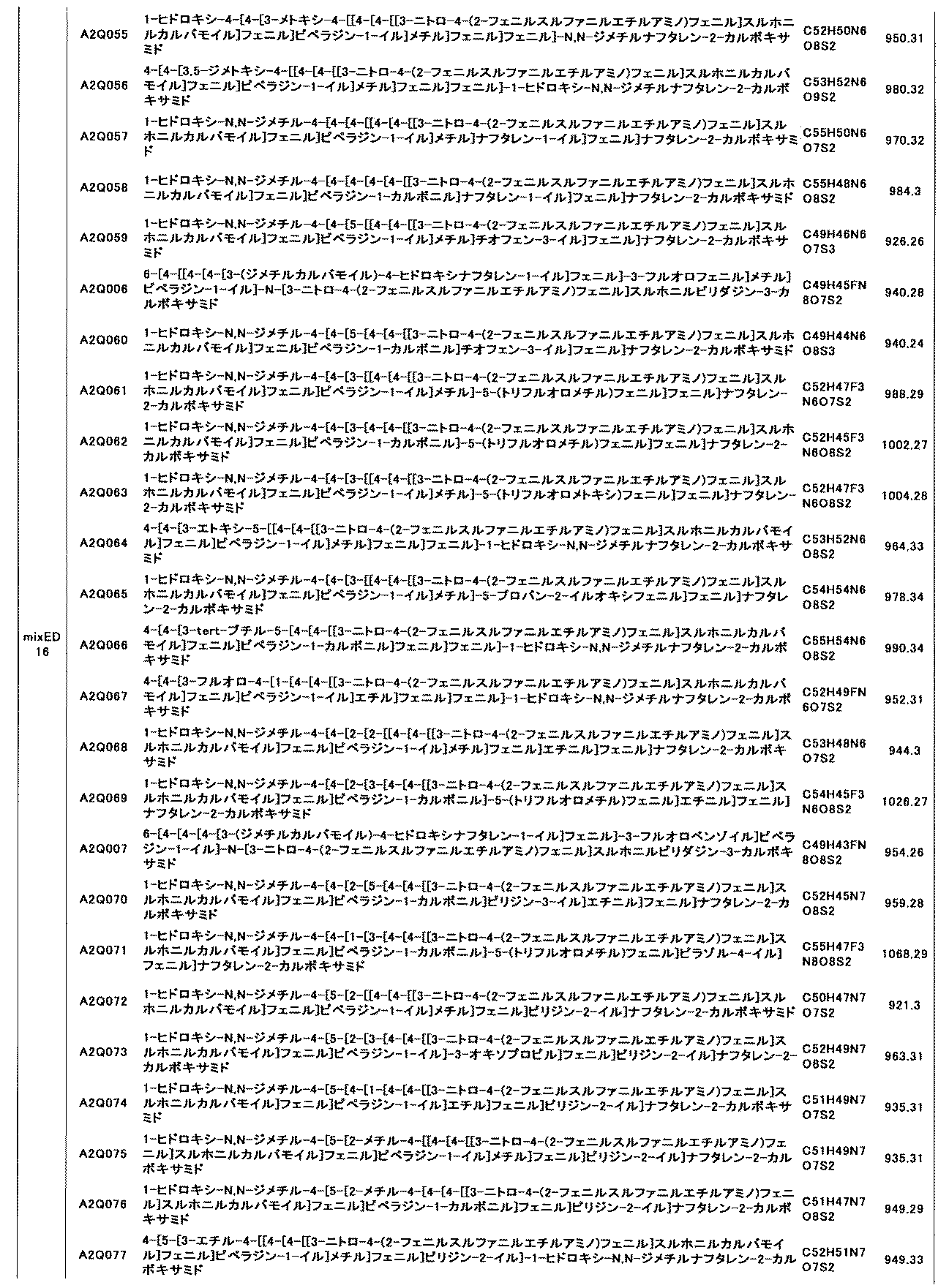

[表AG−E−00]
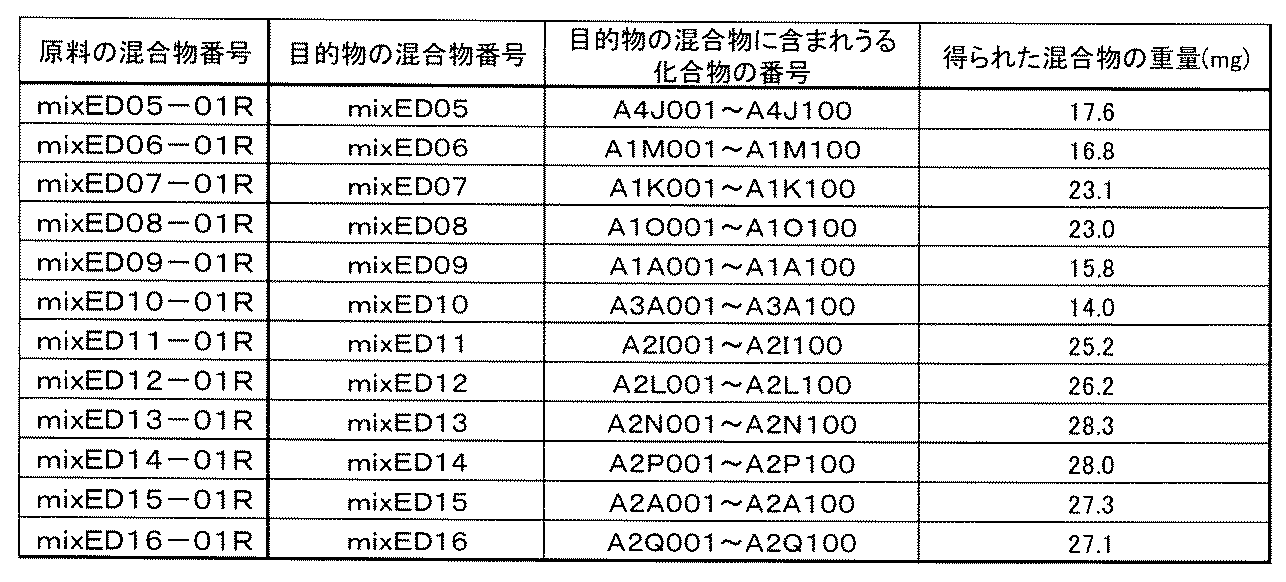
表には、観測されたUVピークの保持時間(min)、計算により算出したUV面積(Cal_UV_area)、そのUVピーク中にて観測された(M+H)+、MSクロマトグラムにおけるMSピークの保持時間、UVピークならびにMSピークに帰属された化合物の番号を記載している。UV面積については、複数の化合物が同一のUVピークに帰属された場合には、帰属された化合物数で均等に除した値を記載している。
100化合物が含まれうる混合物の中には、組成式ならびにExact Massが同一の化合物が複数含まれる場合がある。一例として、[表AG−E—01]中、保持時間3.32分のピーク中に観測されている(M+H)+=674.3を示す化合物は、A4J031、A4J079、A4J092の3つが該当するため、「帰属された化合物の番号」列には3つの化合物番号を記載している。さらに、(M+H)+=674.3は、4.61分ならび8.51分のUVピーク中にも観測されるため、3.32分、4.61分、8.51分のいずれにもA4J31、A4J79、A4J92を記載した。
例えば、[表AG−E—01]中、3.85分のUVピーク中には2つの(M+H)+=704.3と648.2が検出された。そのため、保持時間3.85分のUVピークを2行に渡って記載し、(M+H)+=704.3についてはA4J32を、また(M+H)+=648.2についてはA4J84を帰属した。この場合のUV面積については、3.85分のUVピーク中に2つの化合物が帰属されているため、該当UVピークのUV面積を2で除した数値をCal_UV_areaに記載している。もう一つの例を示すと、5.81分のUVピークの場合には4つの化合物(A4J009、A4J036、A4J047、A4J086)が帰属されているため、該当UVピークのUV面積を4で除した数値をCal_UV_areaに記載している。
また、Cal_UVの数値については、統計的処理により、中央値、平均値、標準偏差、75パーセンタイル、25パーセンタイル、上側境界値、下側境界値、外れ値の数、最大値、最小値を記載した。なお、上側境界値は[式AH01]より、下側境界値は[式AH02]から算出し、外れ値の数は[式AH03]より算出した。
上側境界値=75パーセンタイル+1.5×(75パーセンタイル—25パーセンタイル)
[式AH02]
上側境界値=25パーセンタイル—1.5×(75パーセンタイル—25パーセンタイル)
[式AH03]
外れ値の数=(Cal_UV>上側境界値)のデータ数+(Cal_UV<下側境界値)のデータ数





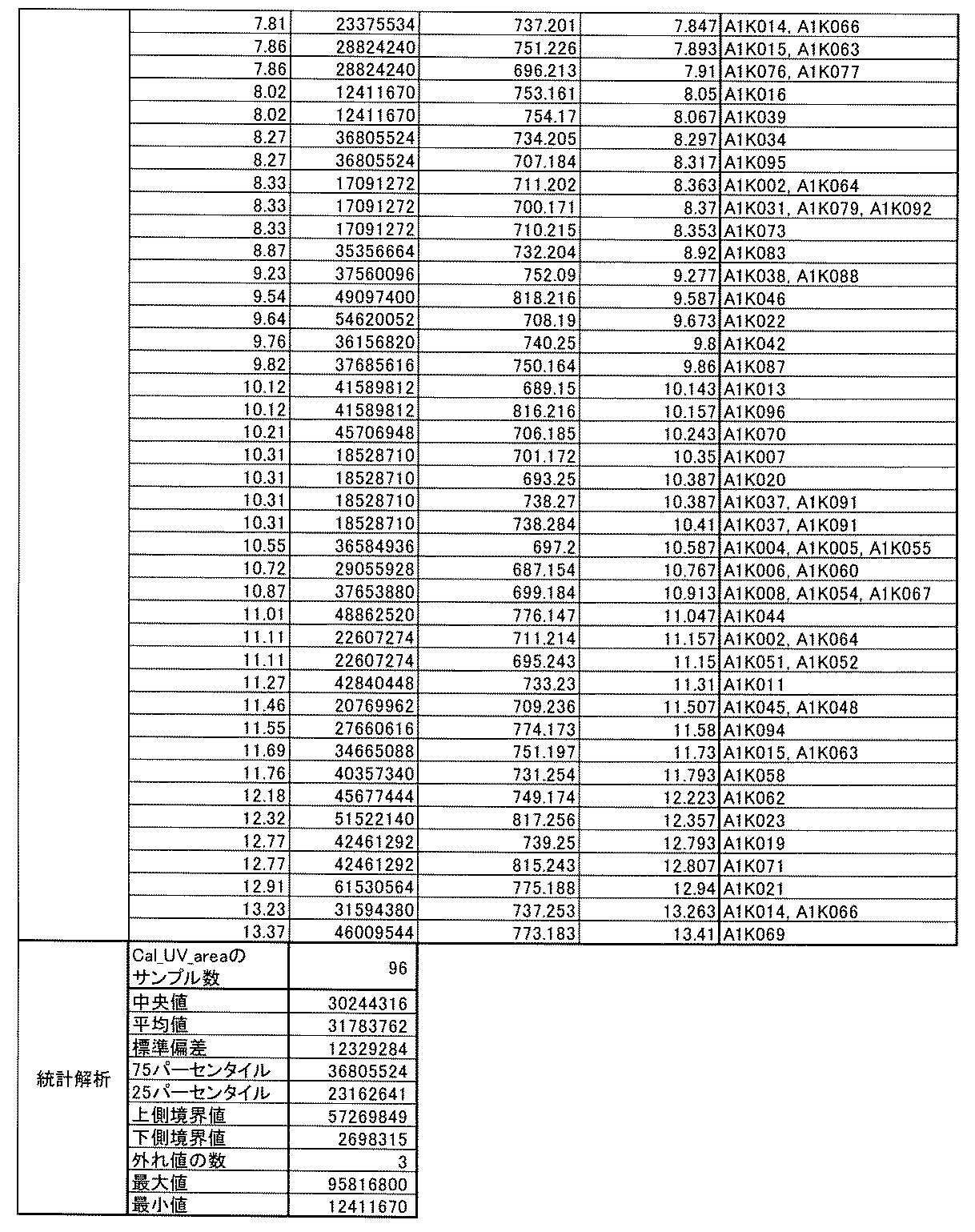





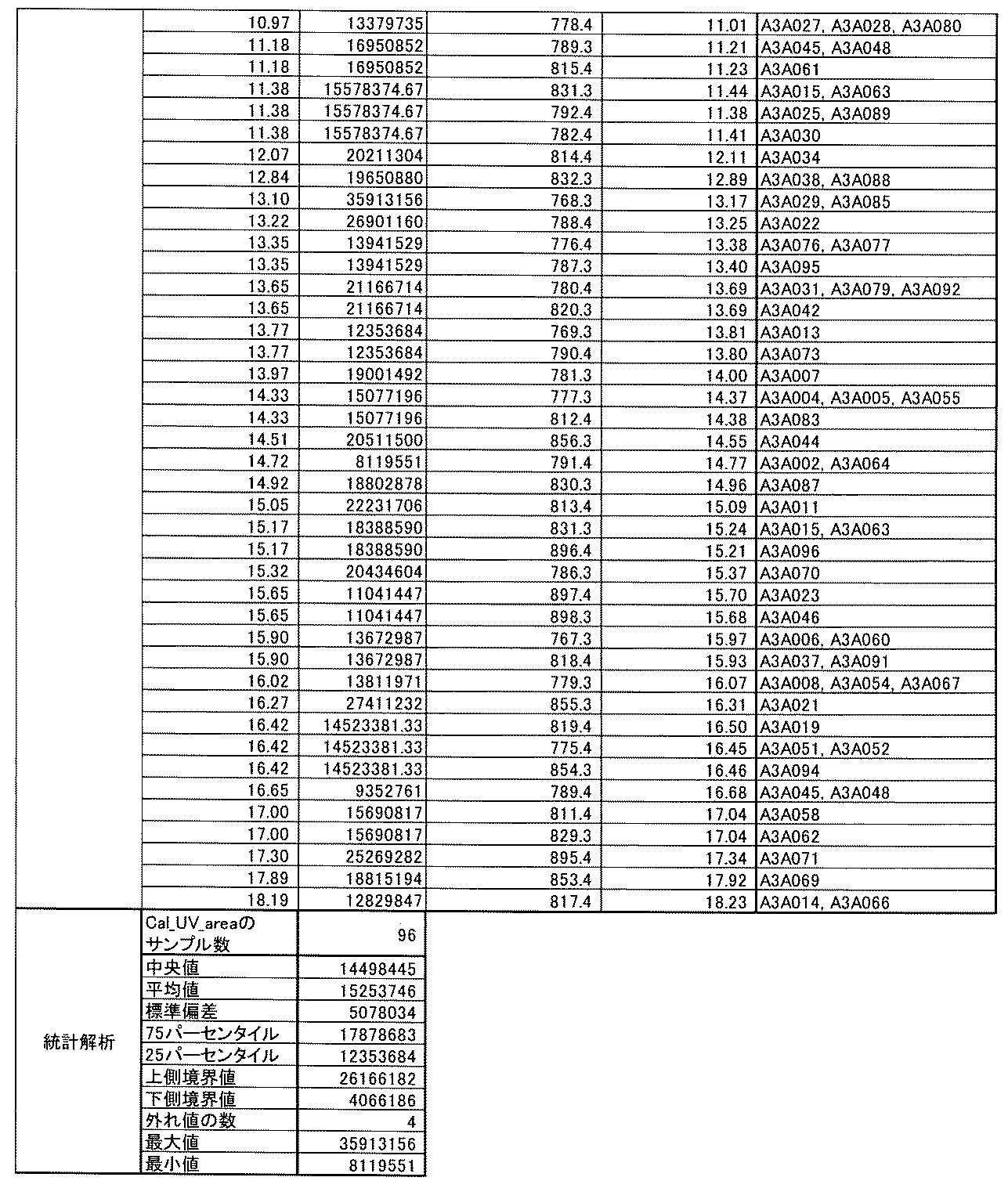



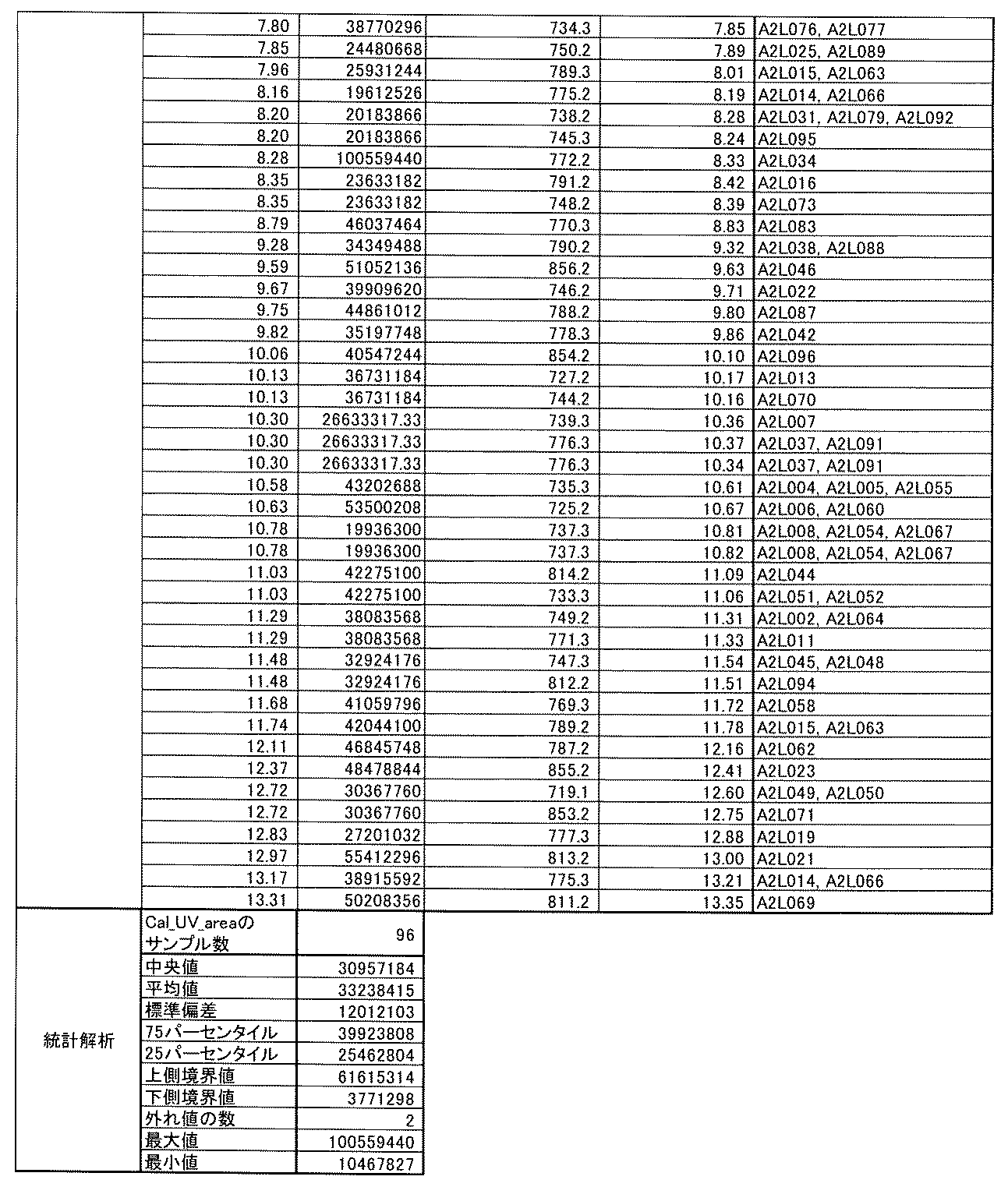



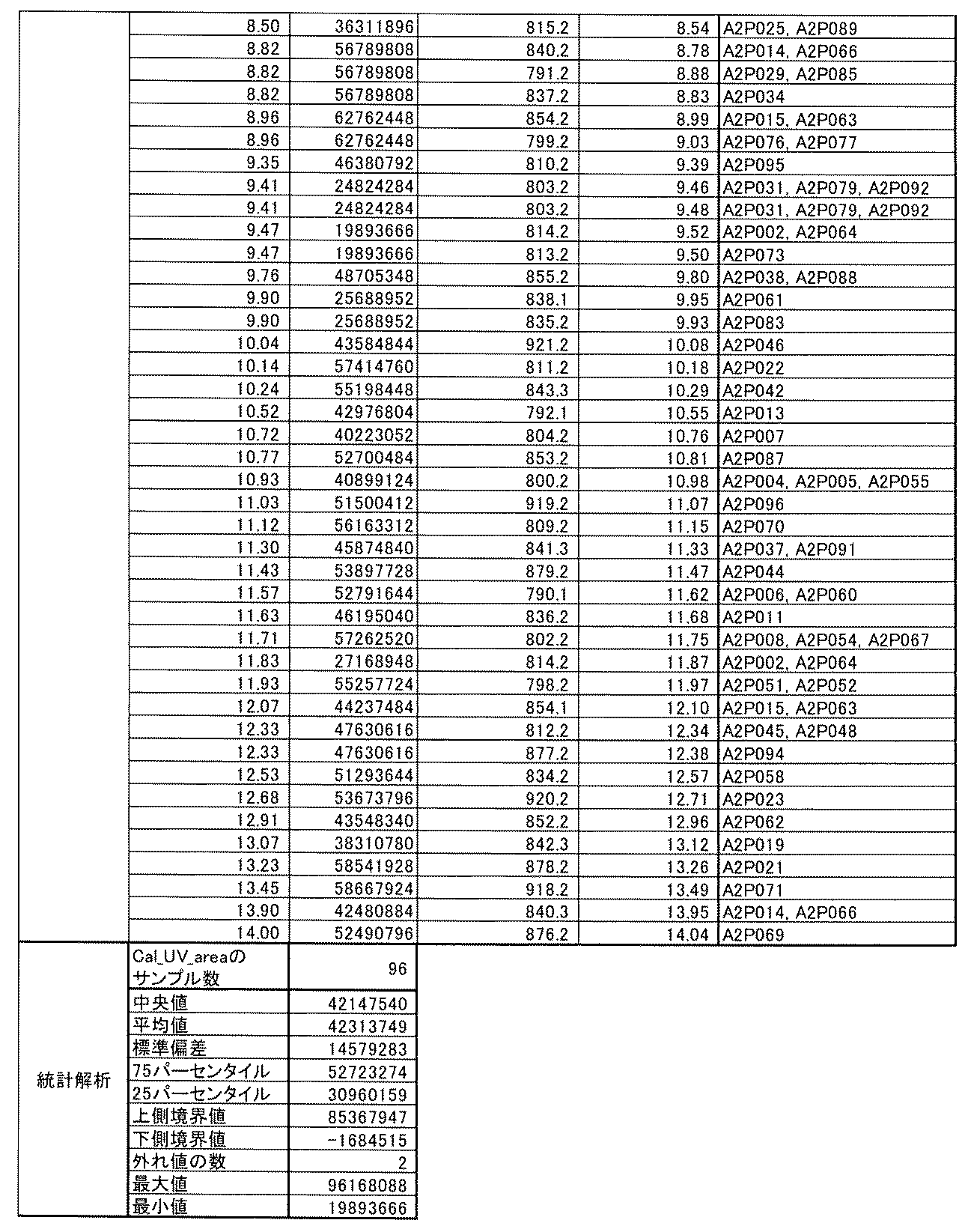




しかしながら、UVタグを含ませて化合物ライブラリ合成を実施することで、「意図したライブラリ化合物が合成されて混合物ライブラリ中に存在していること」ならびに「各ライブラリ化合物間の含量の差が小さいこと」を確認することが可能であることが明らかなり、品質の担保に関しての課題の解決策になることが確認できた。

実施例3では、本発明の手法により、ビルディングブロック同士をつなぐ連結部分(リンカー)にも多様性を有するライブラリを、ライブラリを構成する各化合物の品質の確認を行ったうえで製造することが可能であることを示した。従来のライブラリがビルディングブロックの構造多様性のみに由来したファーマコフォアの多様性を発生させているのに対し、ファーマコフォアを形成するコアブロックのみならず、その間のリンカーも自在に選択できるライブラリとして、構造多様性の著しい拡大がなされたものである。





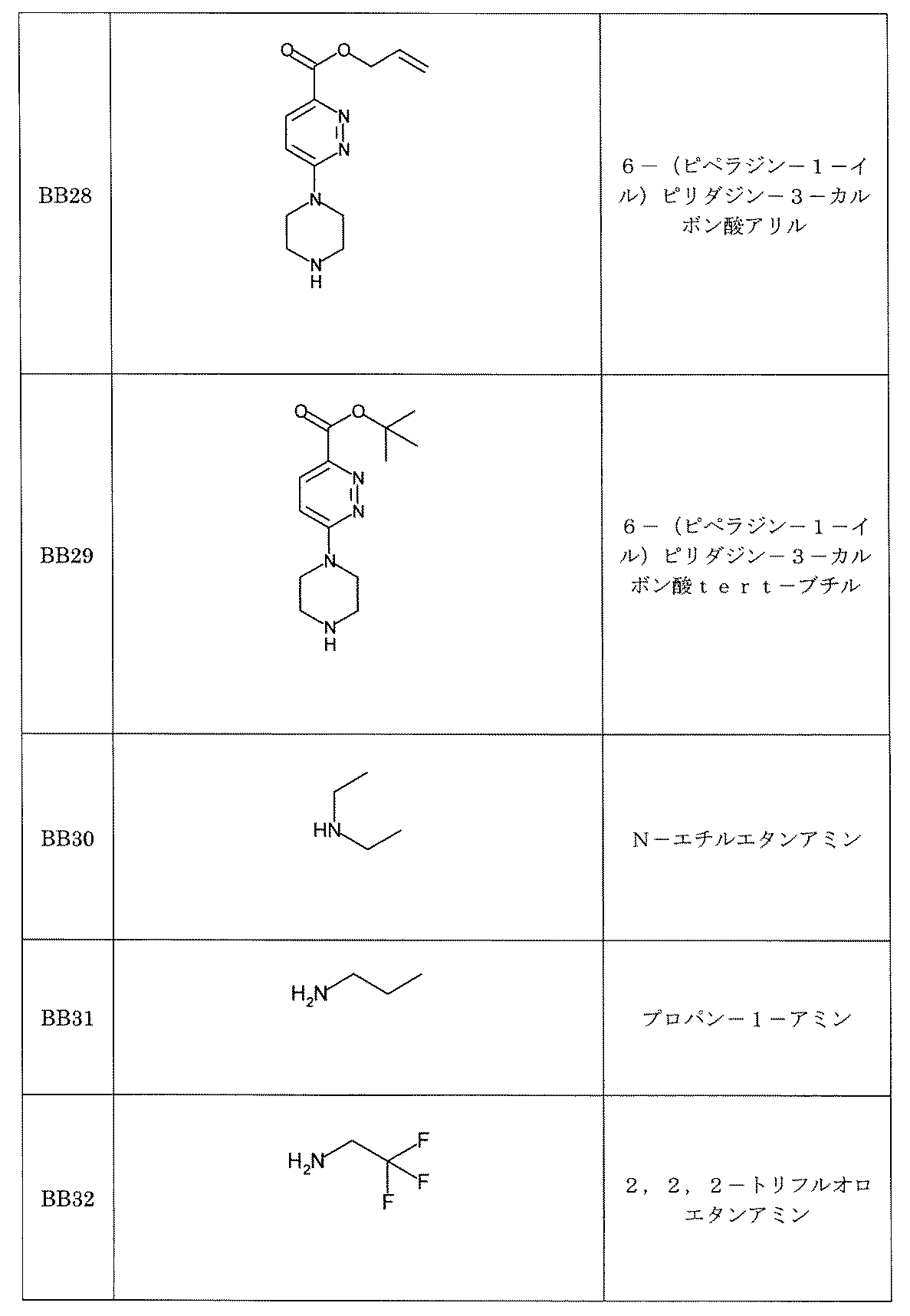
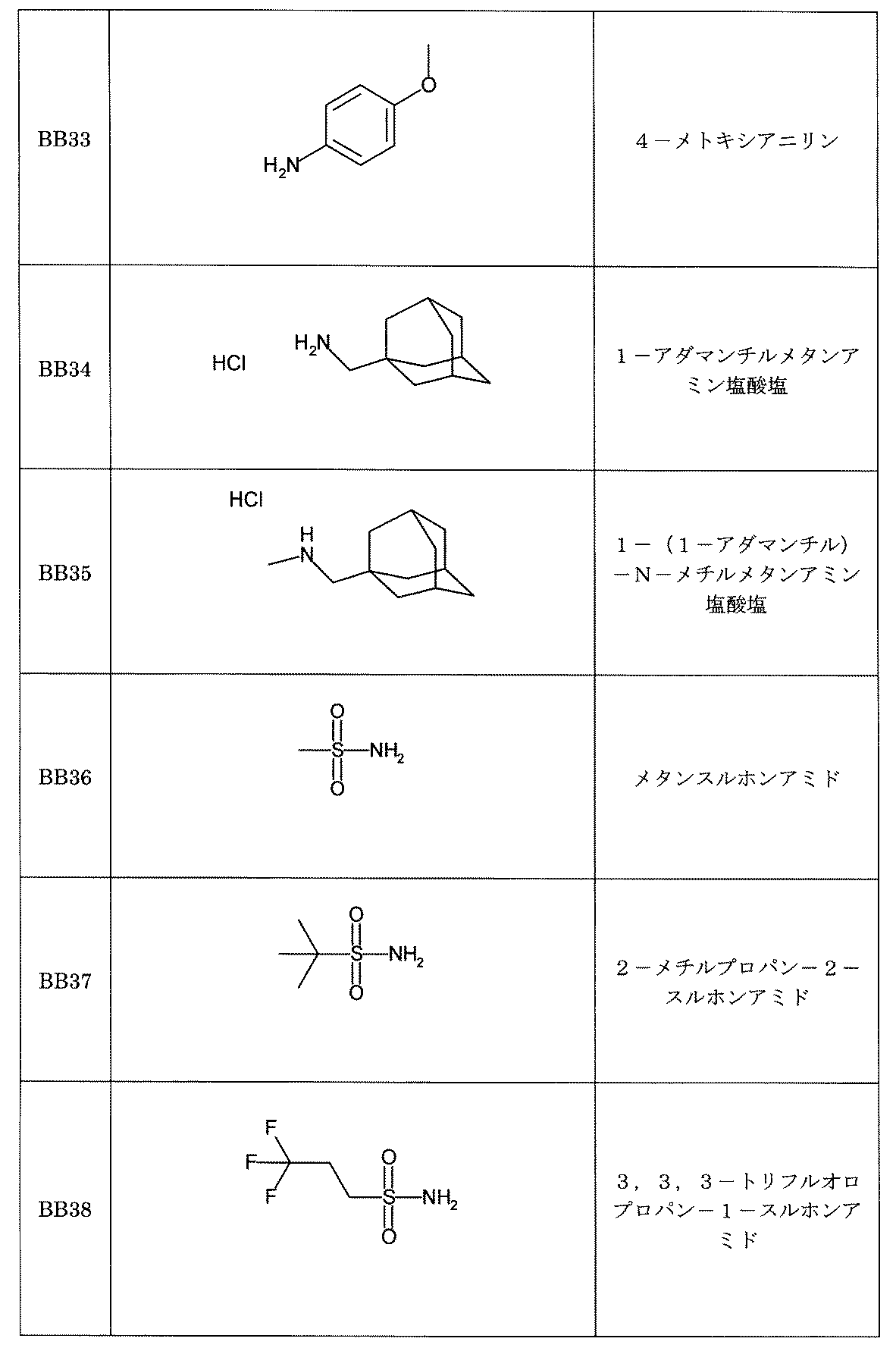

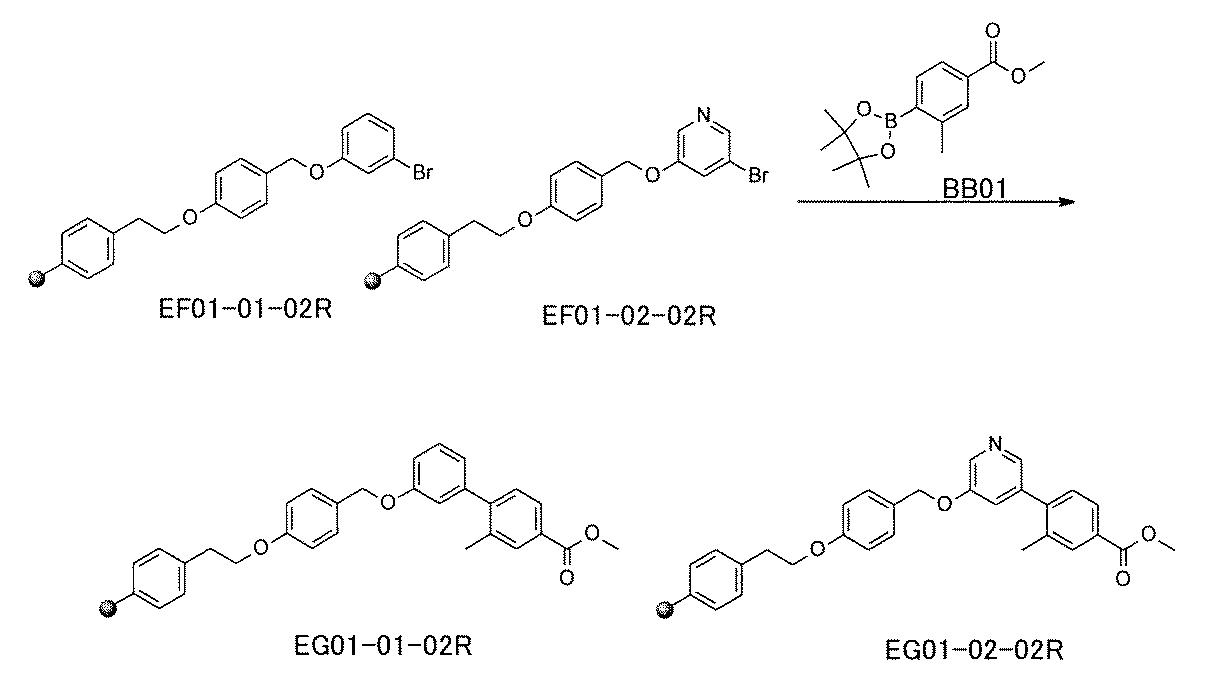
1.7MのK3PO4水溶液は、窒素雰囲気下5mLのメスフラスコを使用し、K3PO4(1.8g、8.48mmol)を蒸留水でメスアップして調製した。
化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)と化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と表LBB02に示したボロン酸またはボロン酸エステル試薬(BB02~04、18)を用いて、実施例4−1−1—Aと同様の方法で[表EG01]に示した標題混合物を合成した。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG01]に示す。


化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)と化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と表LBB02に示した[1−[3−エトキシカルボニル−5−(トリフルオロメチル)フェニル]ピラゾル−4−イル]ボロン酸(BB06)を用いて、実施例4−1−2—Aと同様の方法で[表EG05]に示した標題混合物を合成した。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG05]に示す。


化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)、化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と[表LBB02]に示したボロン酸またはボロン酸エステル試薬(BB08~17)を用いて、実施例4−1−3−Aと同様の方法で[表EG07]に示した標題混合物を合成した。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG07]に示す。。



化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)、化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と表LBB02に示した3−フルオロ−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)安息香酸メチル(BB20)を用いて、実施例4−1−4—Aと同様の方法で[表EG19]に示した標題混合物を合成した。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG19]に示す。

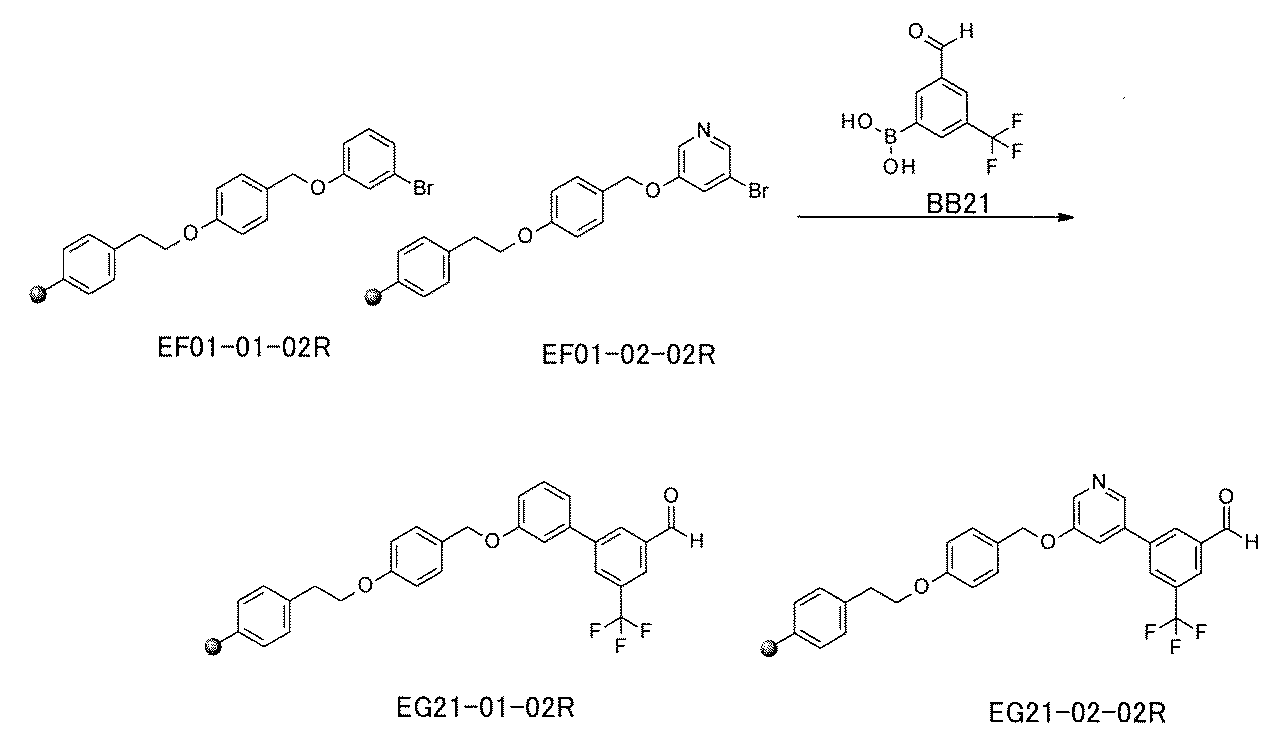

化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)、化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と[表LBB02]に示したボロン酸試薬(BB22)を用いて、実施例4−1−5と同様の方法で合成し、[表EG22]に示した標題混合物mixEG22−02Rを得た。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG22]に示す。


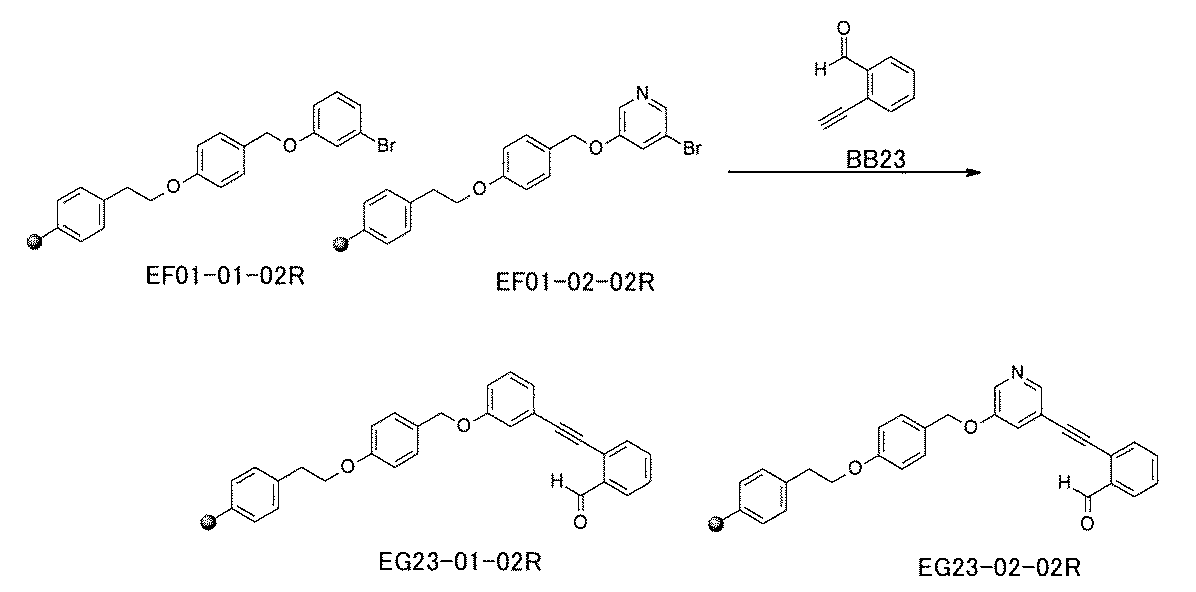
XPhos Pd G4のNMP溶液(0.1M)は、窒素雰囲気下、3mLのガラスバイアルにXPhos Pd G4(86mg、0.1mmol)にNMP(1mL)を加えて振とうすることで溶解し調製した。
化合物EF01−01−02R(57mg、担持量0.556mmol/g,0.032mmol)、化合物EF01−02−02R(43mg、担持量0.733mmol/g,0.032mmol)と[表LBB02]に示したアセチレン試薬(BB23~26)を用いて、実施例4−1−7−Aと同様の方法で[表EG23]に示した標題混合物を合成した。用いたビルディングブロックと得られた混合物に含まれる化合物および反応時間についても[表EG23]に示す。






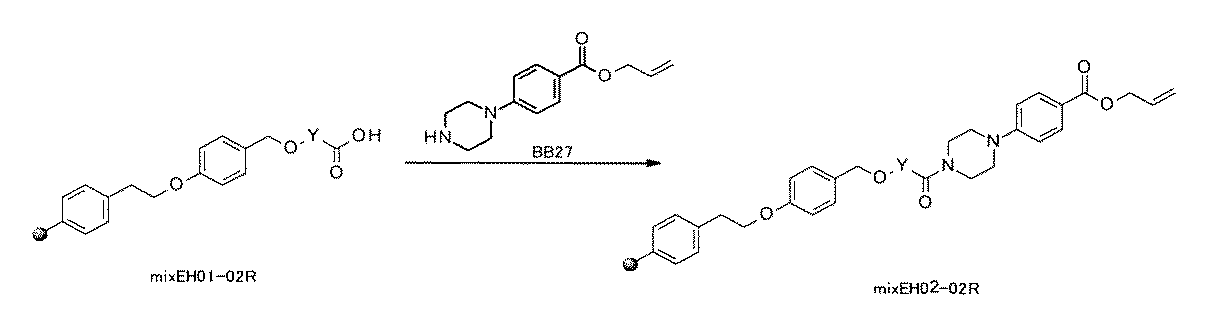
実施例3−5−2の結果から、このアミド化反応において、[表EH02]に記載した化合物のうち2化合物(EH02−07−02RとEH02−08−02R)が欠損している可能性がある。

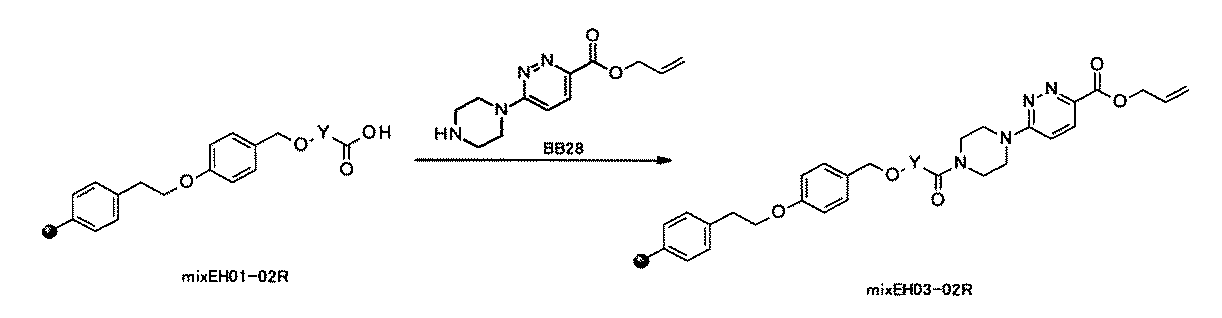
実施例3−5−3の結果から、このアミド化反応において、[表EH03]に記載した化合物のうち2化合物(EH03−07−02RとEH03−08−02R)が欠損している可能性がある。
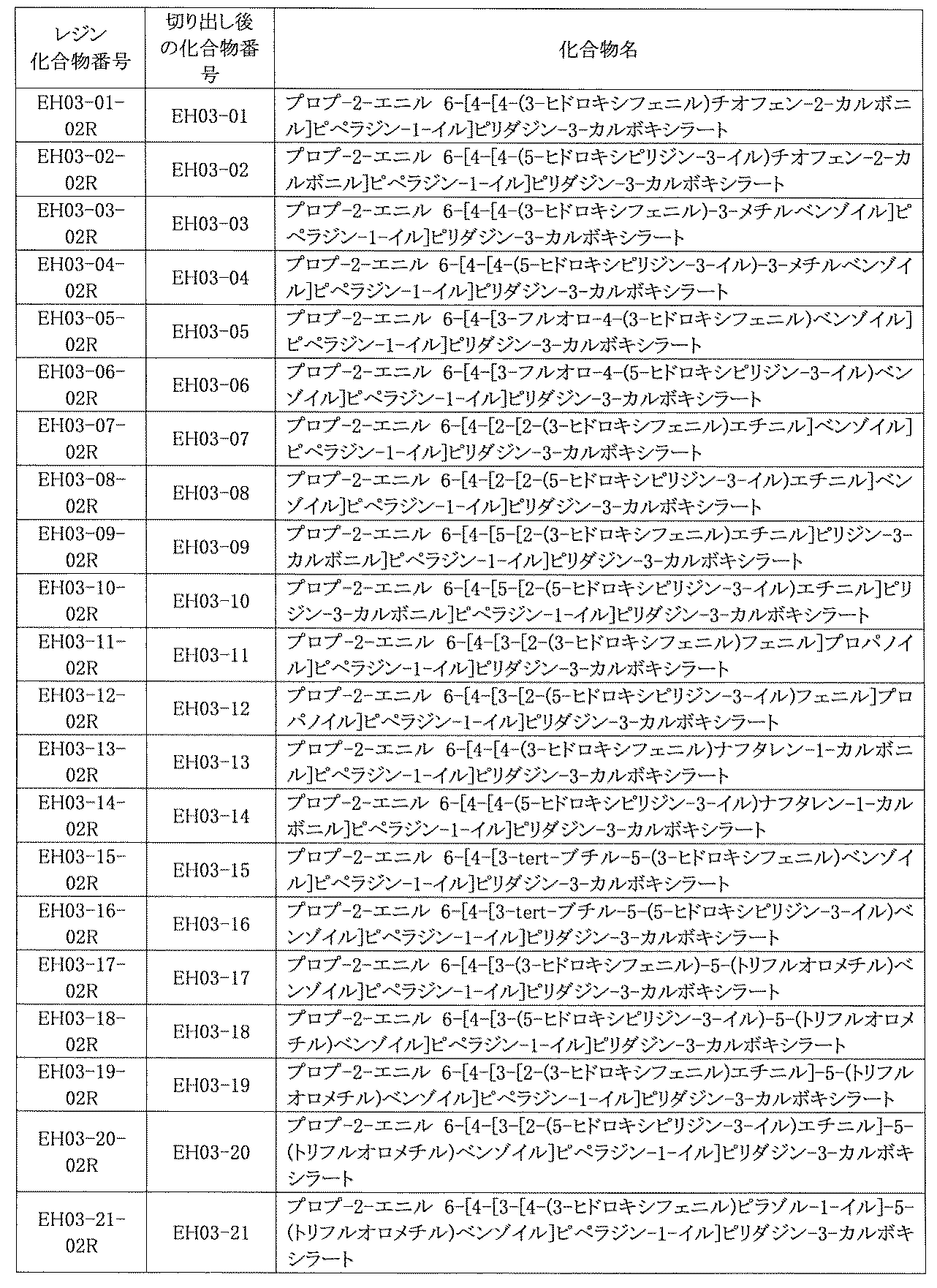









得られた混合物に含まれる化合物について、[表EH06]に示す。


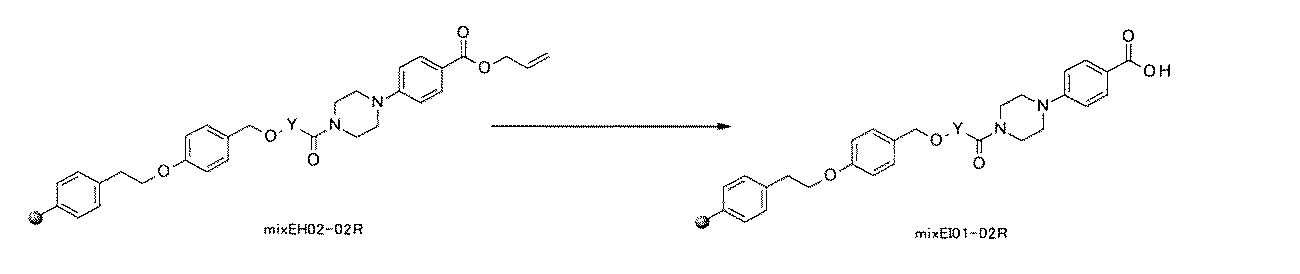





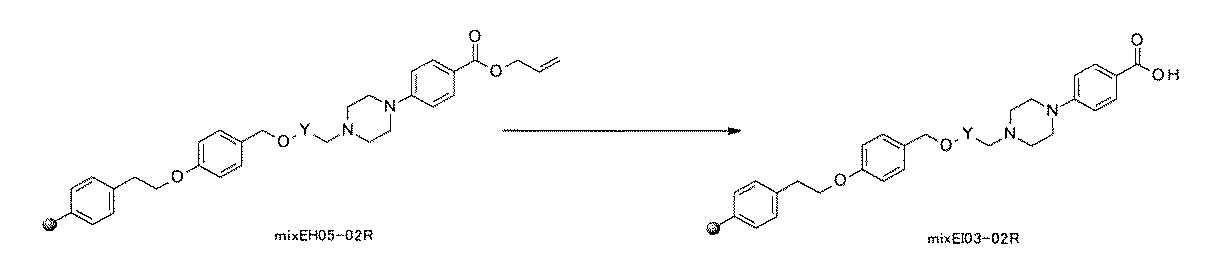

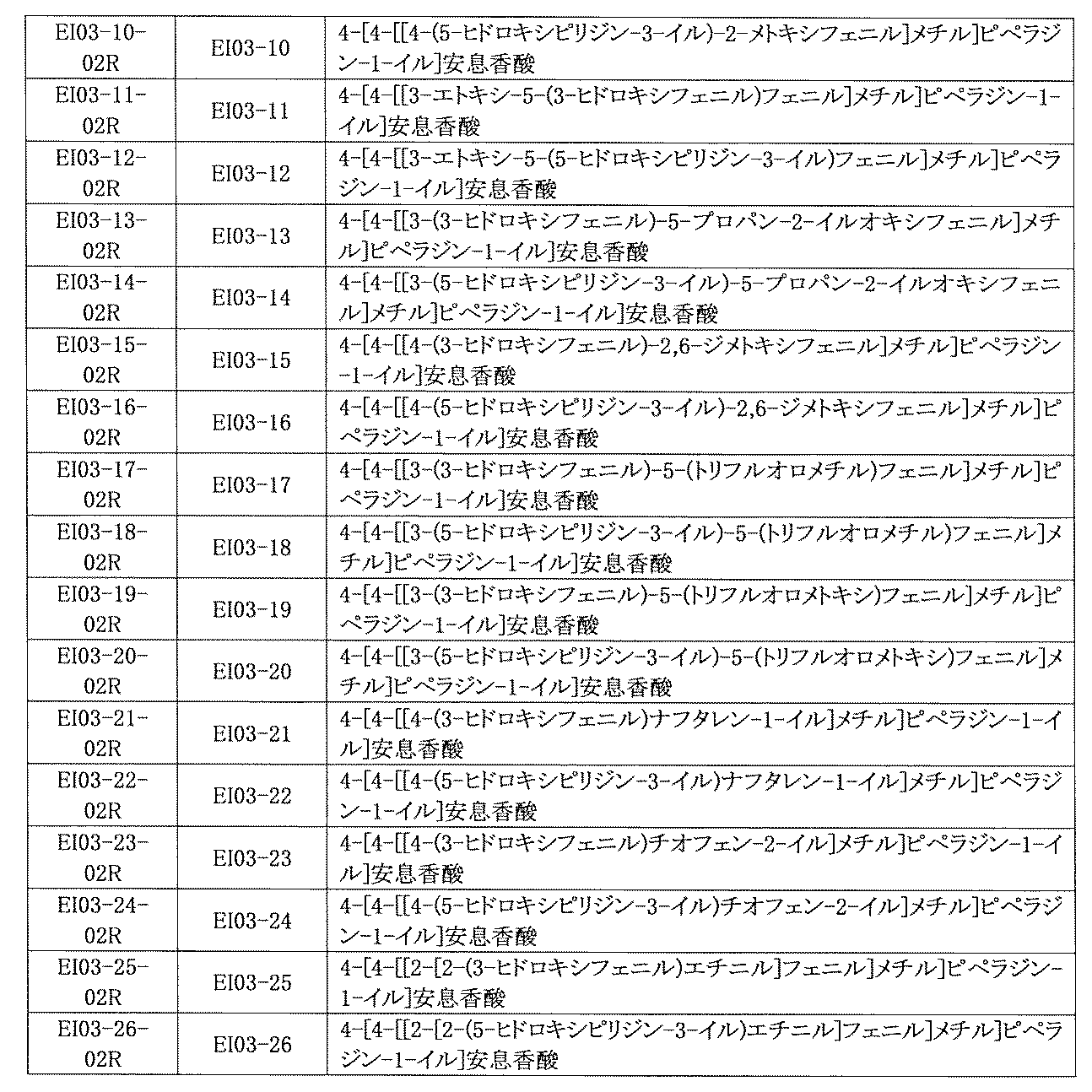



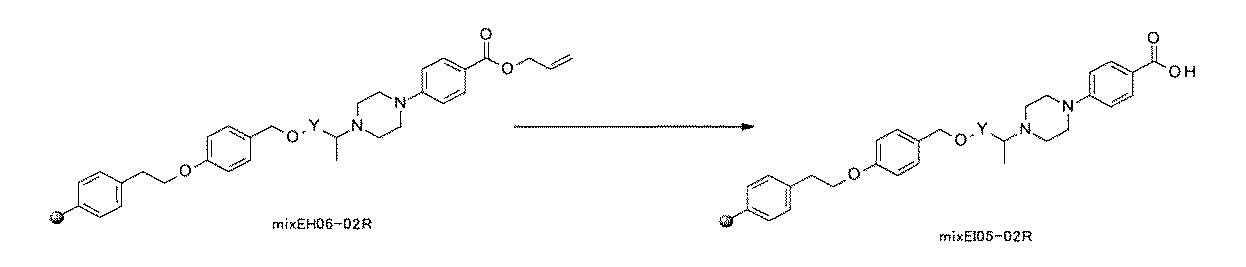





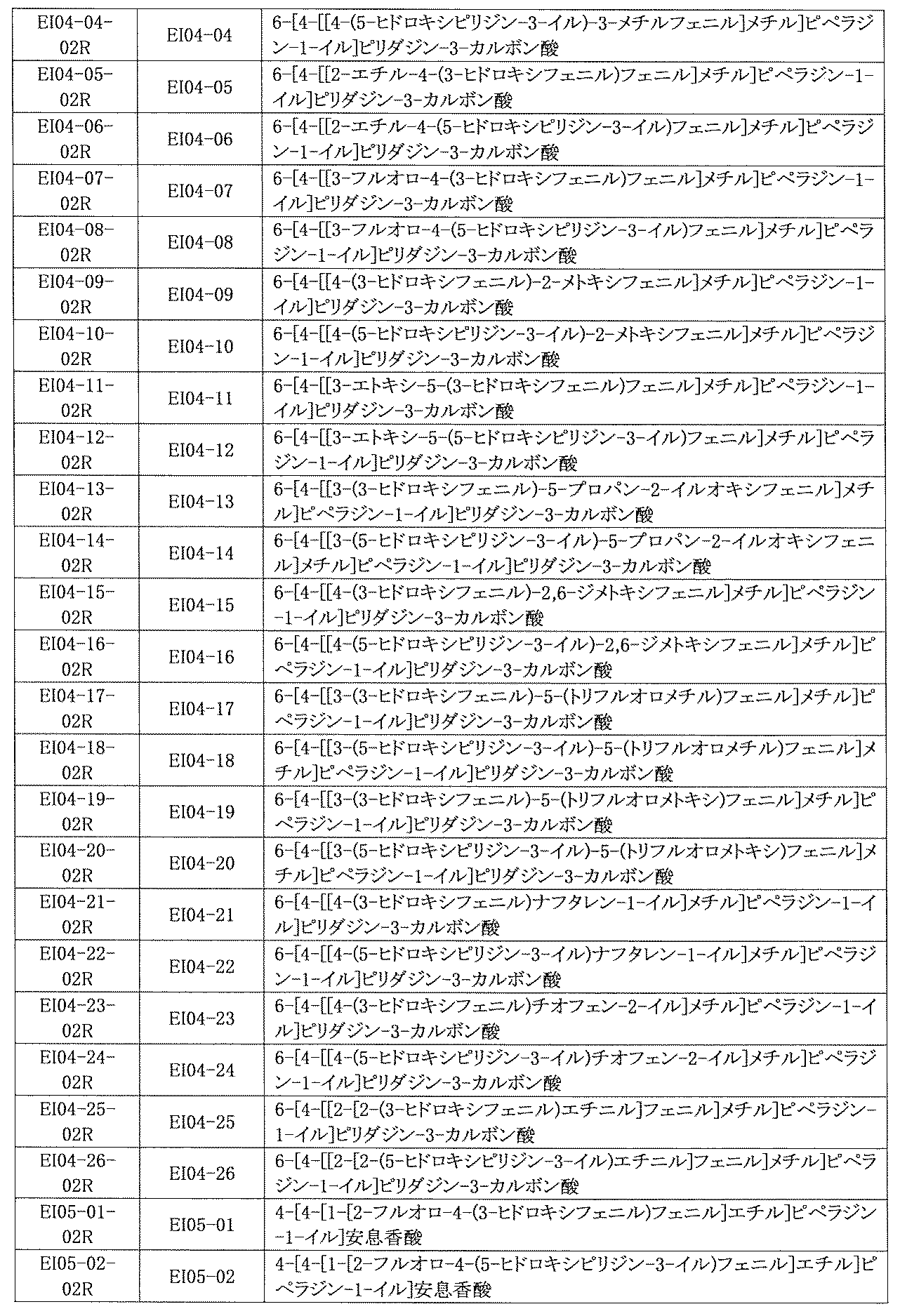





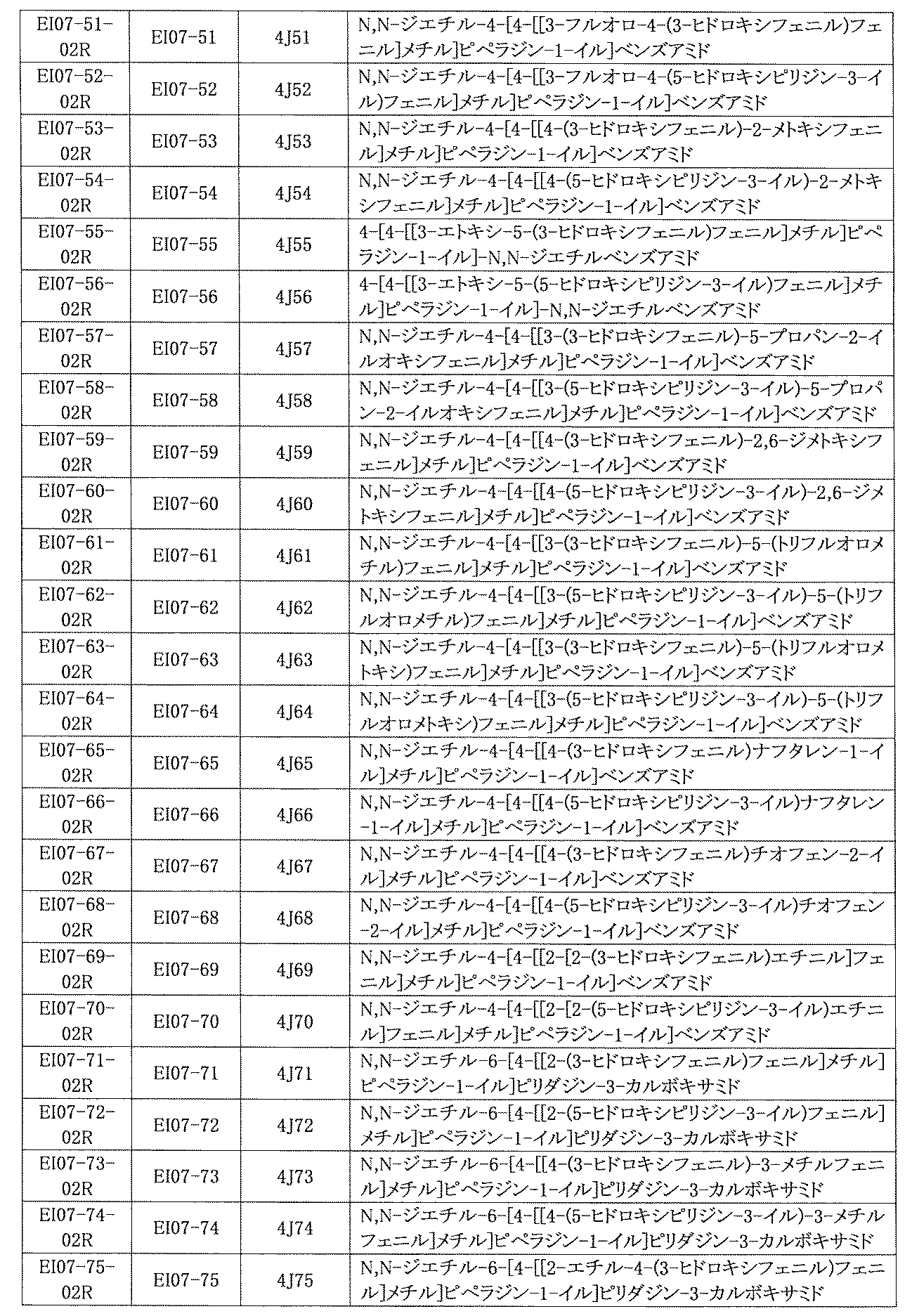


[表EI06]に示した混合物mixEI06−02R(50mg、担持量0.648mmol/g,0.0324mmol)とアミン試薬(BB31~BB33)を用いて、実施例4—3−7と同様の方法で合成し、[表EI08]~[表EI10]に示した標題混合物を得た。得られた混合物に含まれる化合物について、[表EI08]~[表EI10]に示す。


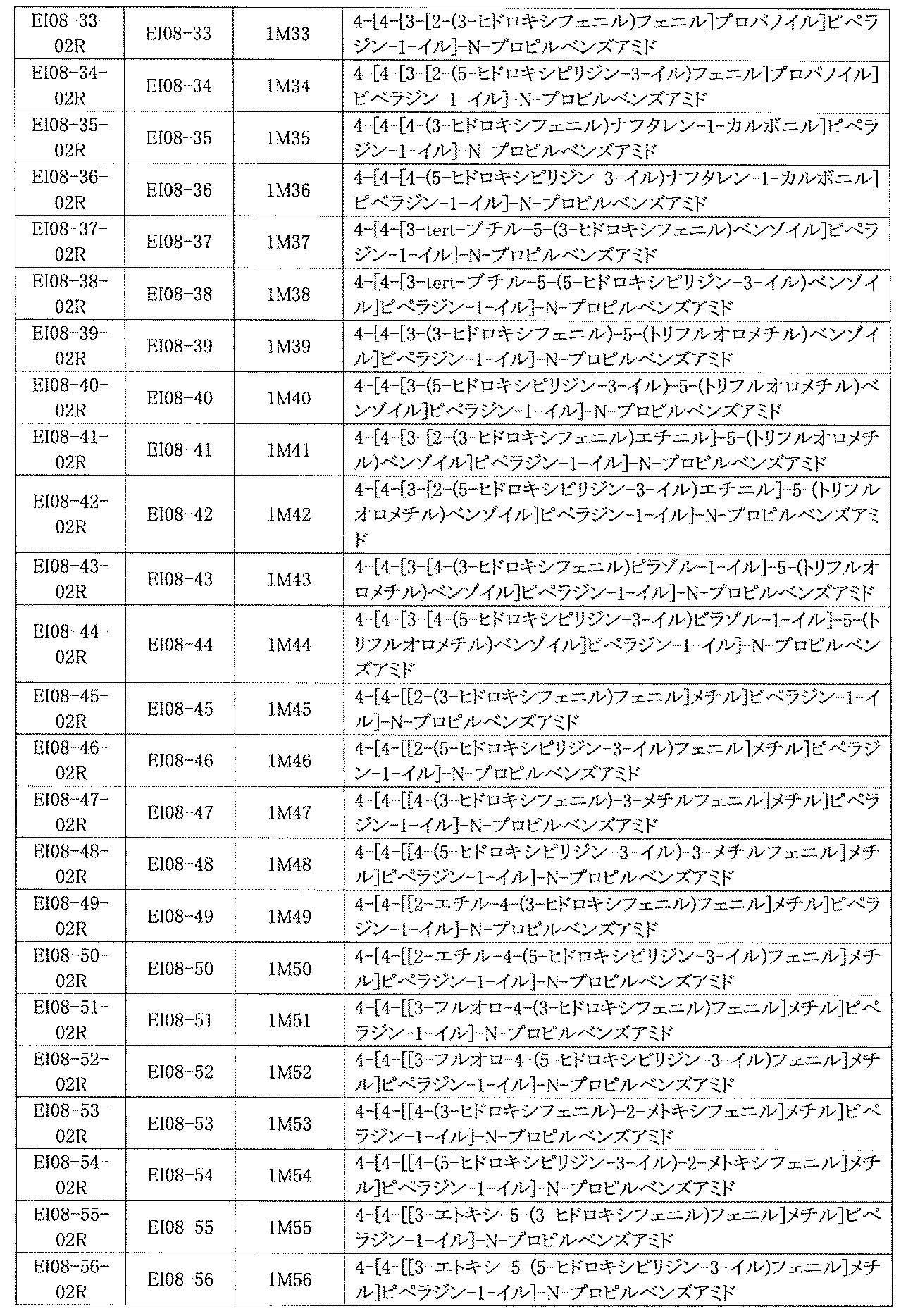
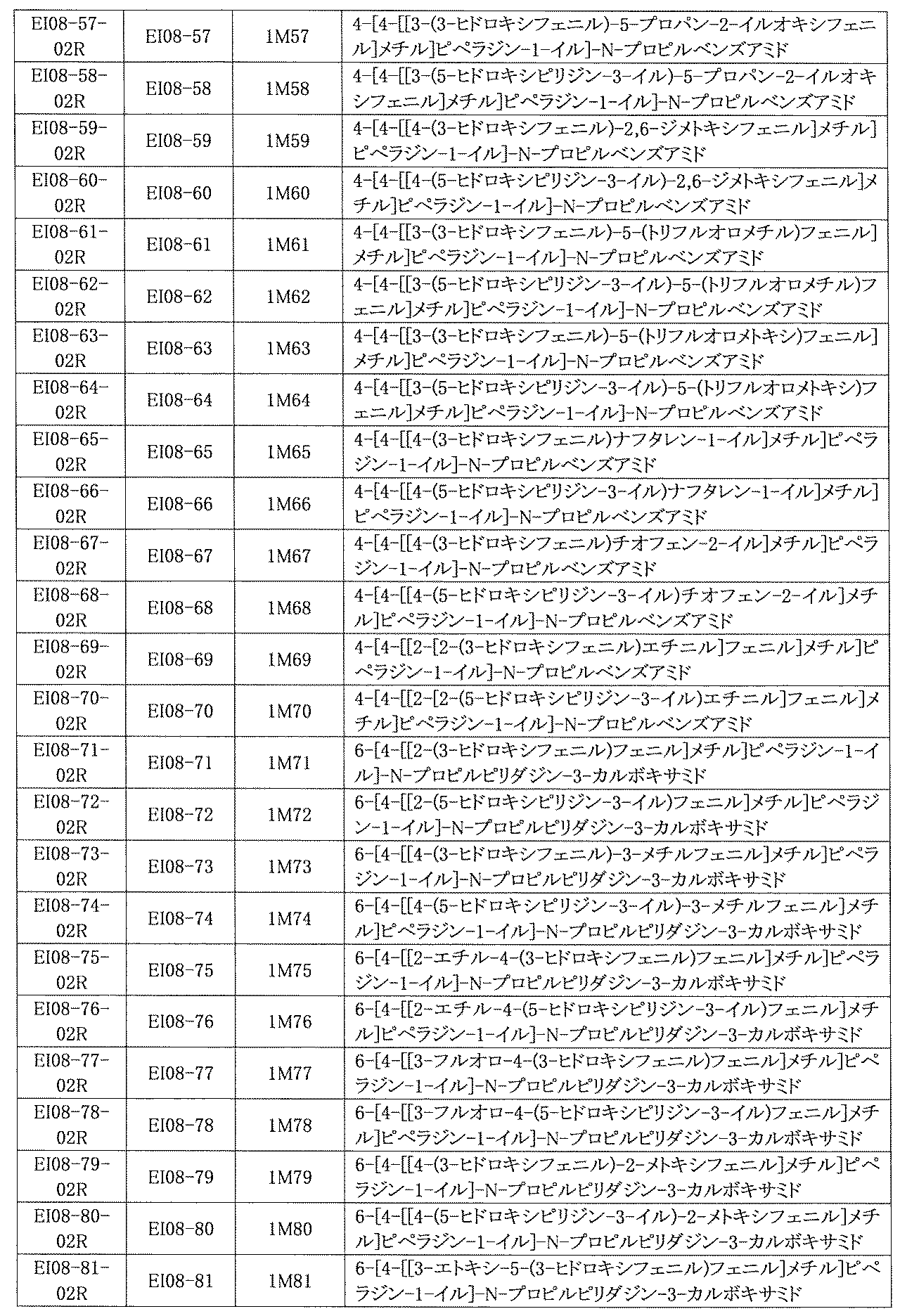








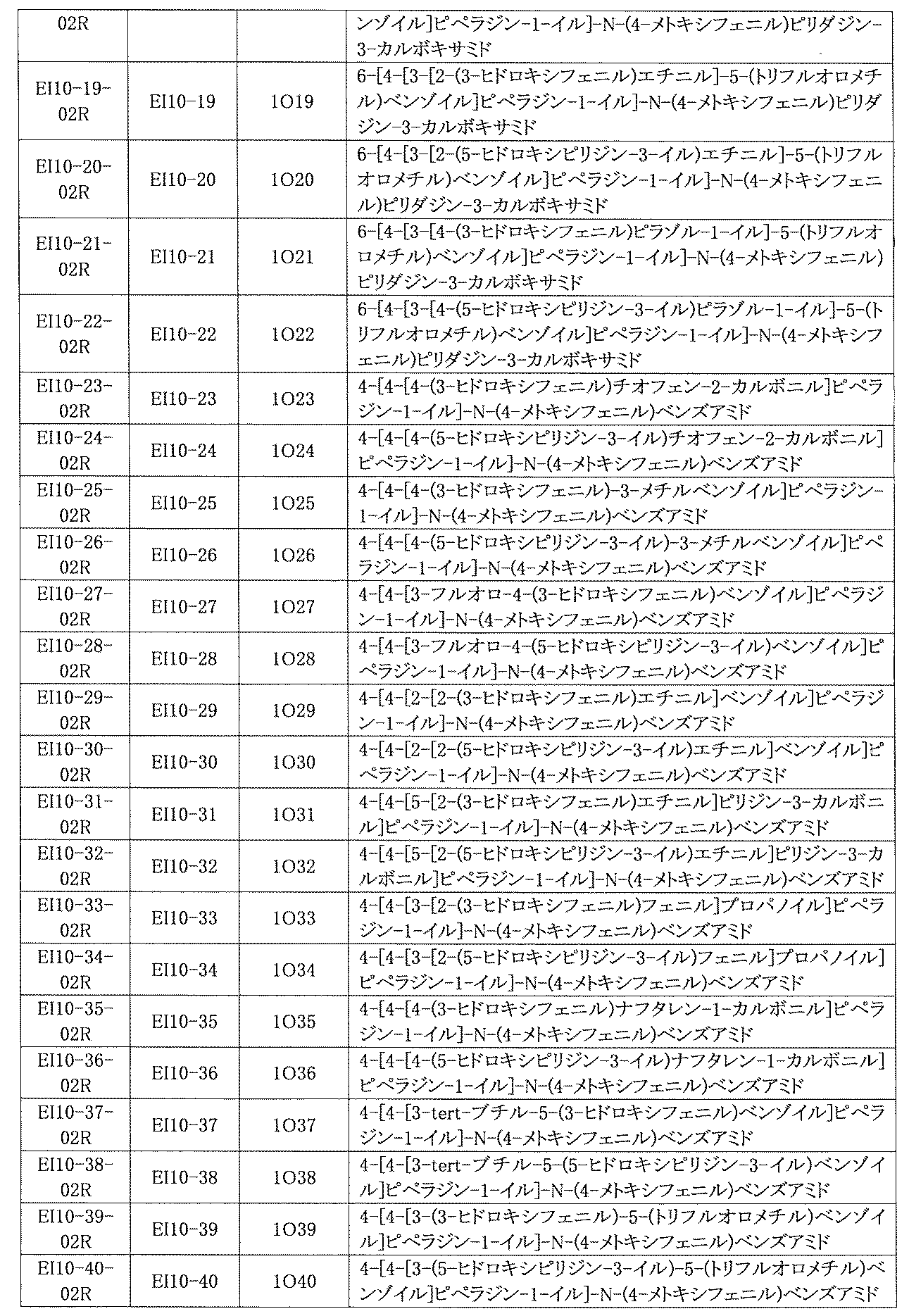







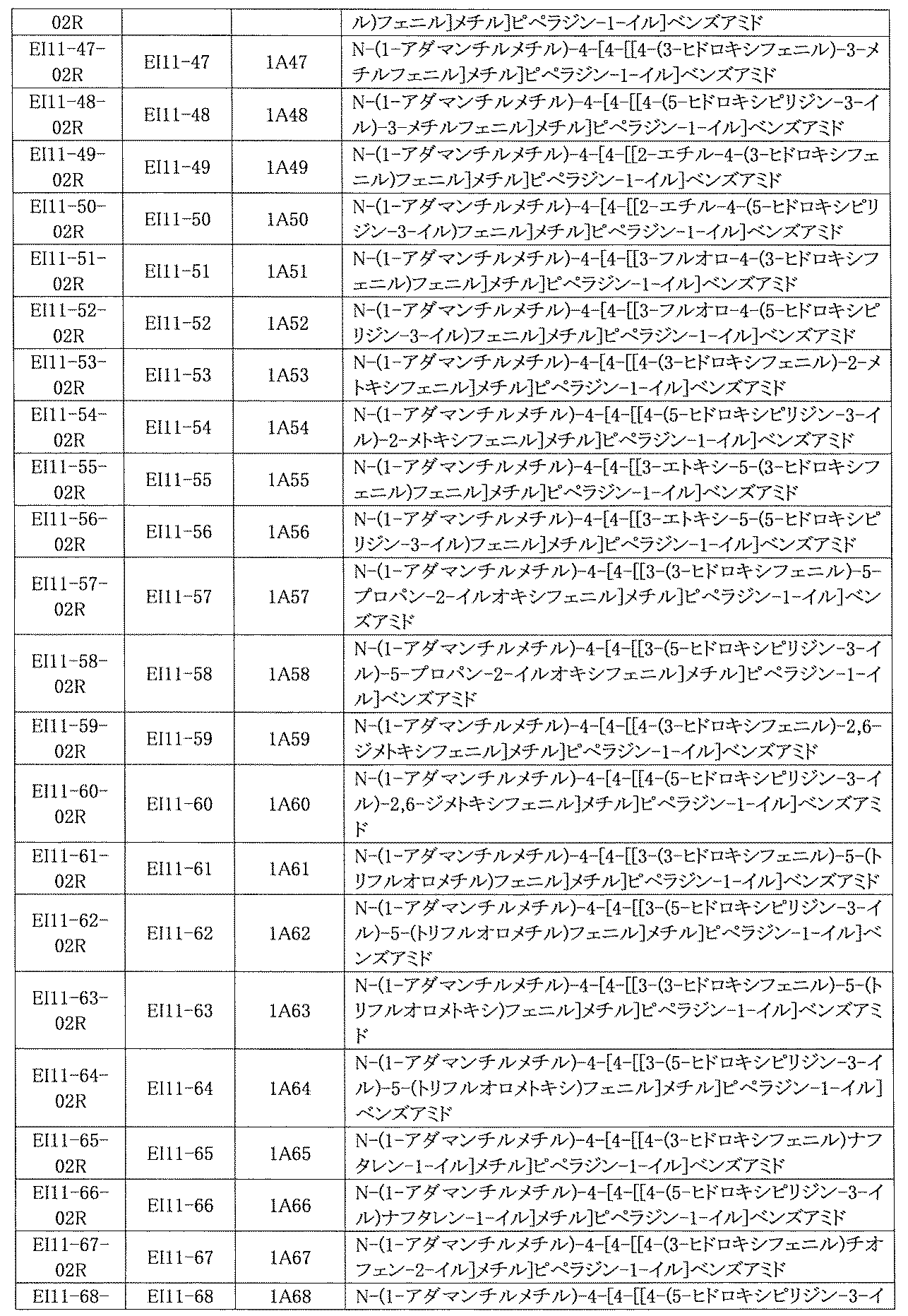

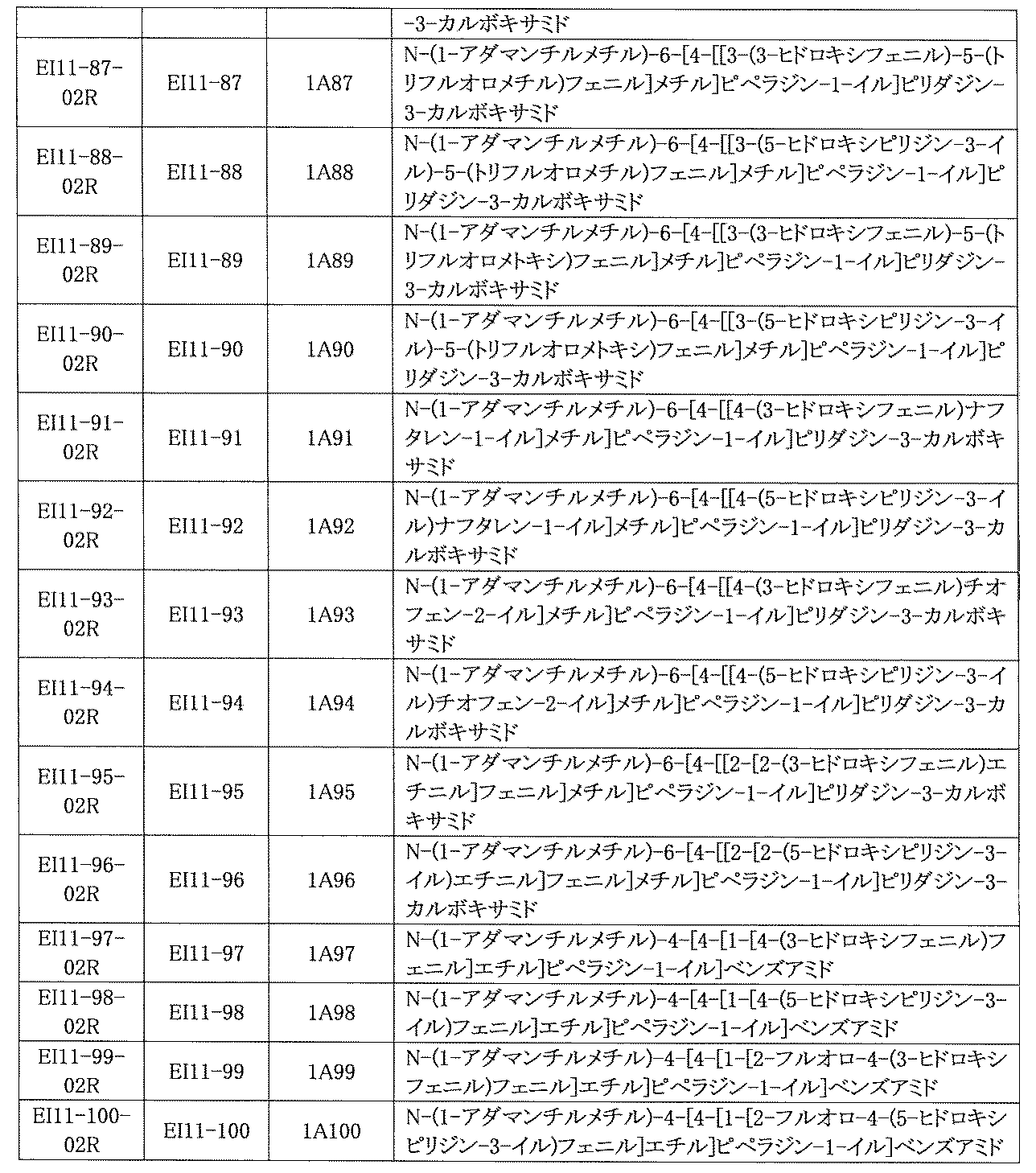
[表EI06]に示した混合物mixEI06−02R(50mg、担持量0.648mmol/g,0.0324mmol)と1−(1−アダマンチル)−N−メチルメタンアミン塩酸塩(BB35)を用いて、実施例4−3−9と同様の方法で合成し[表EI12]に示した標題混合物を得た。得られた混合物に含まれる化合物について、[表EI12]に示す。



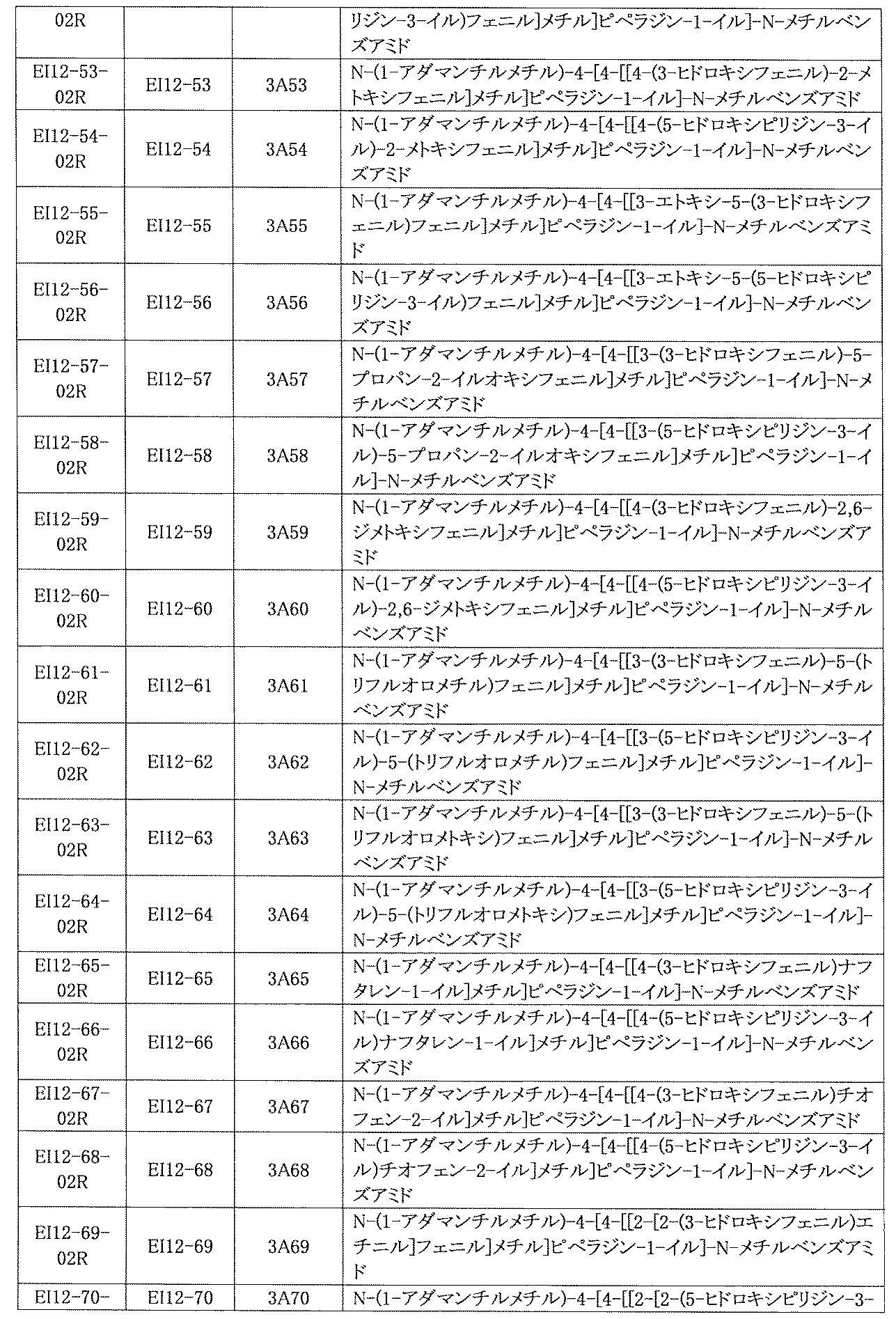



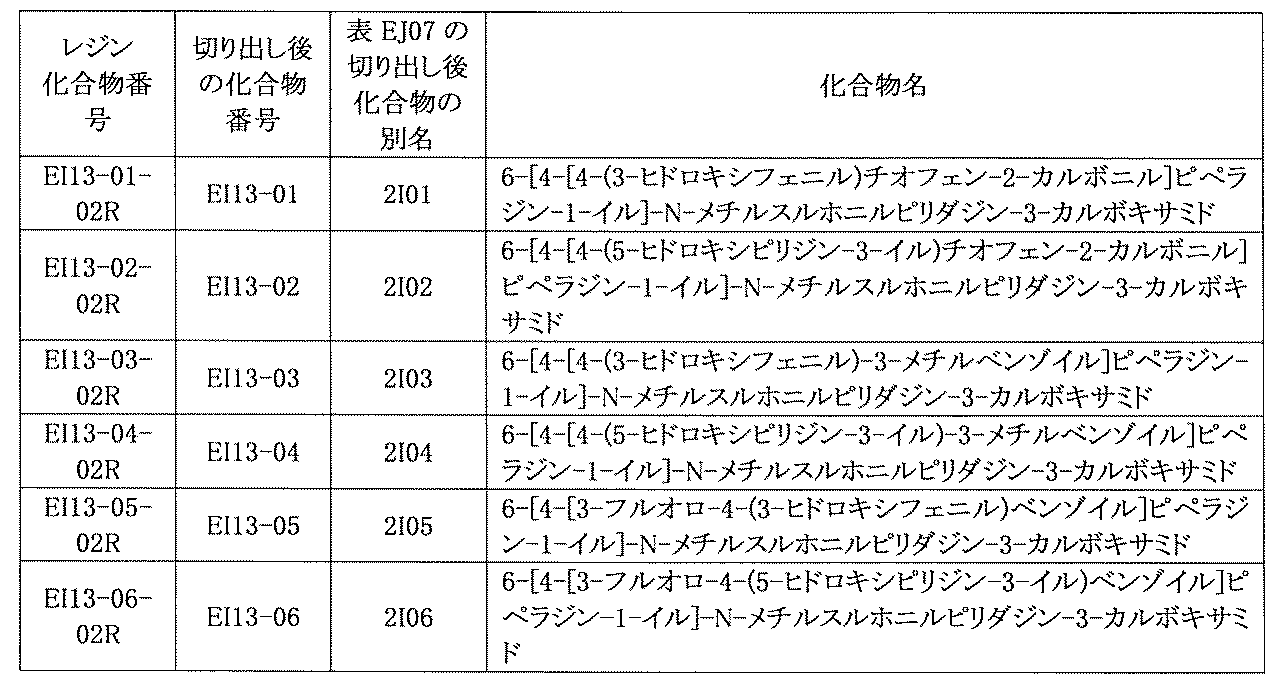
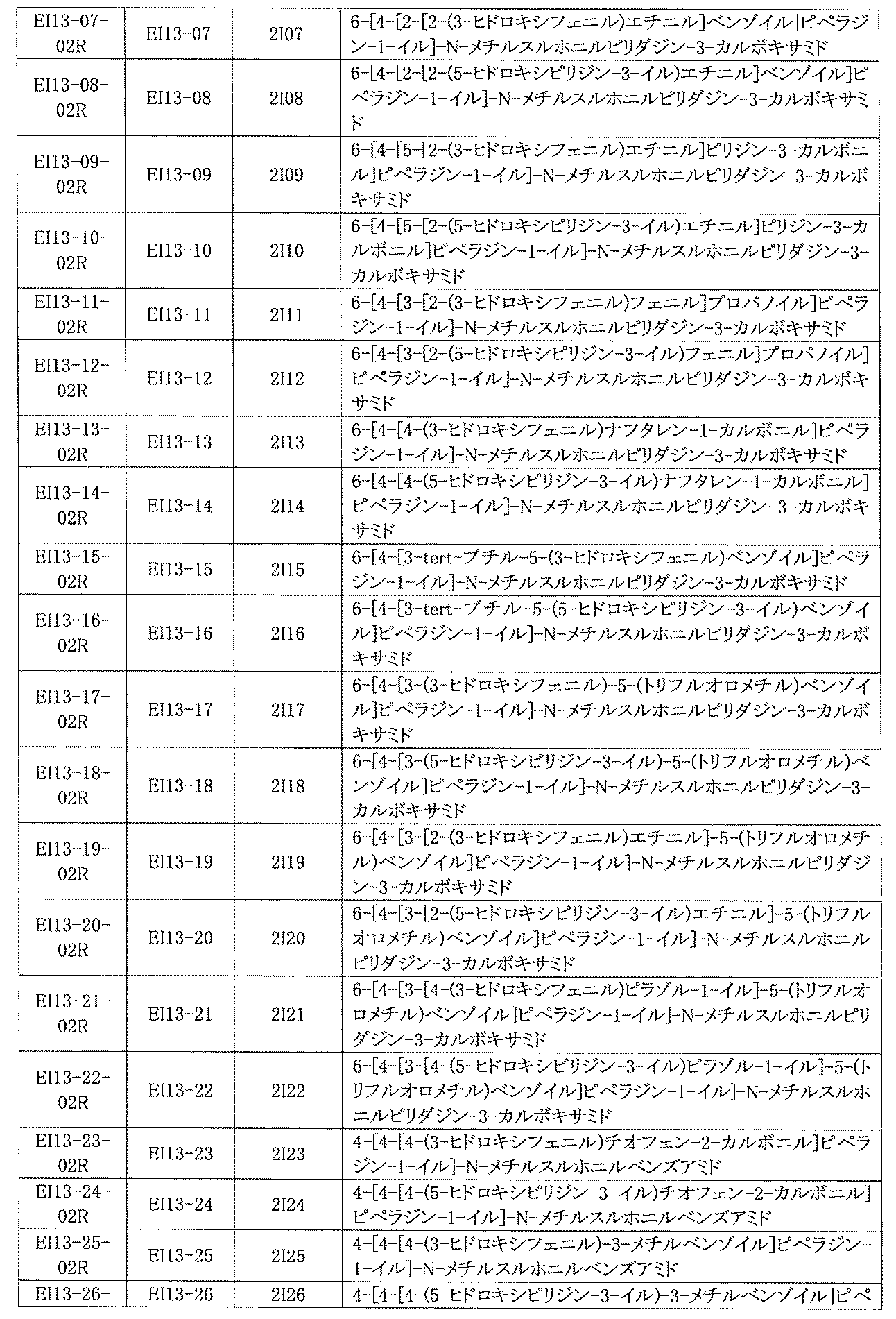




[表EI06]に示した混合物mixEI06−02R(50mg、担持量0.648mmol/g,0.0324mmol)とアミン試薬(BB37~38、40~42)を用いて、実施例4—3−11と同様の方法で合成し、[表EI14]~[表EI18]に示した標題混合物を得た。得られた混合物に含まれる化合物について、[表EI14]~[表EI18]に示す。





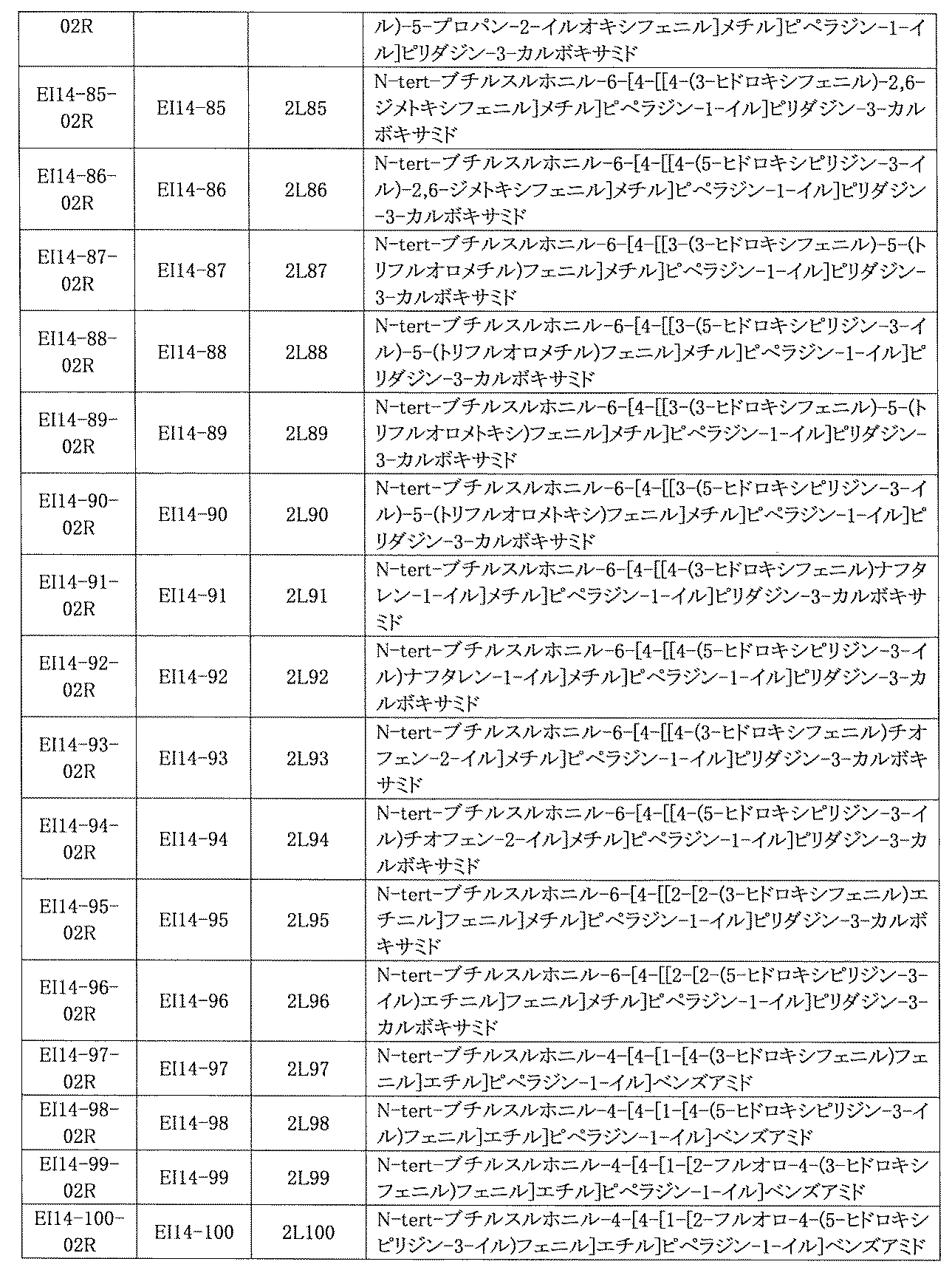


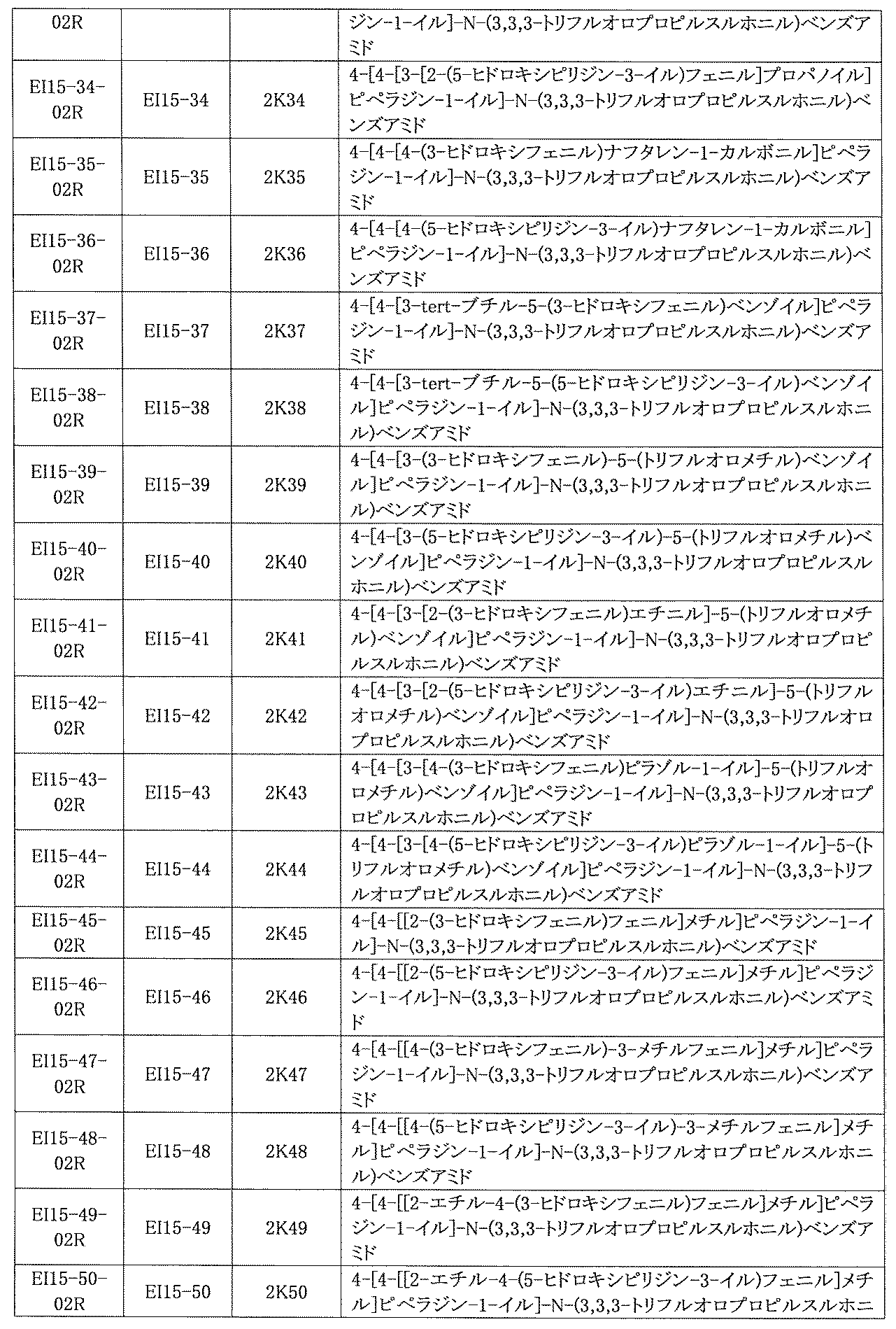
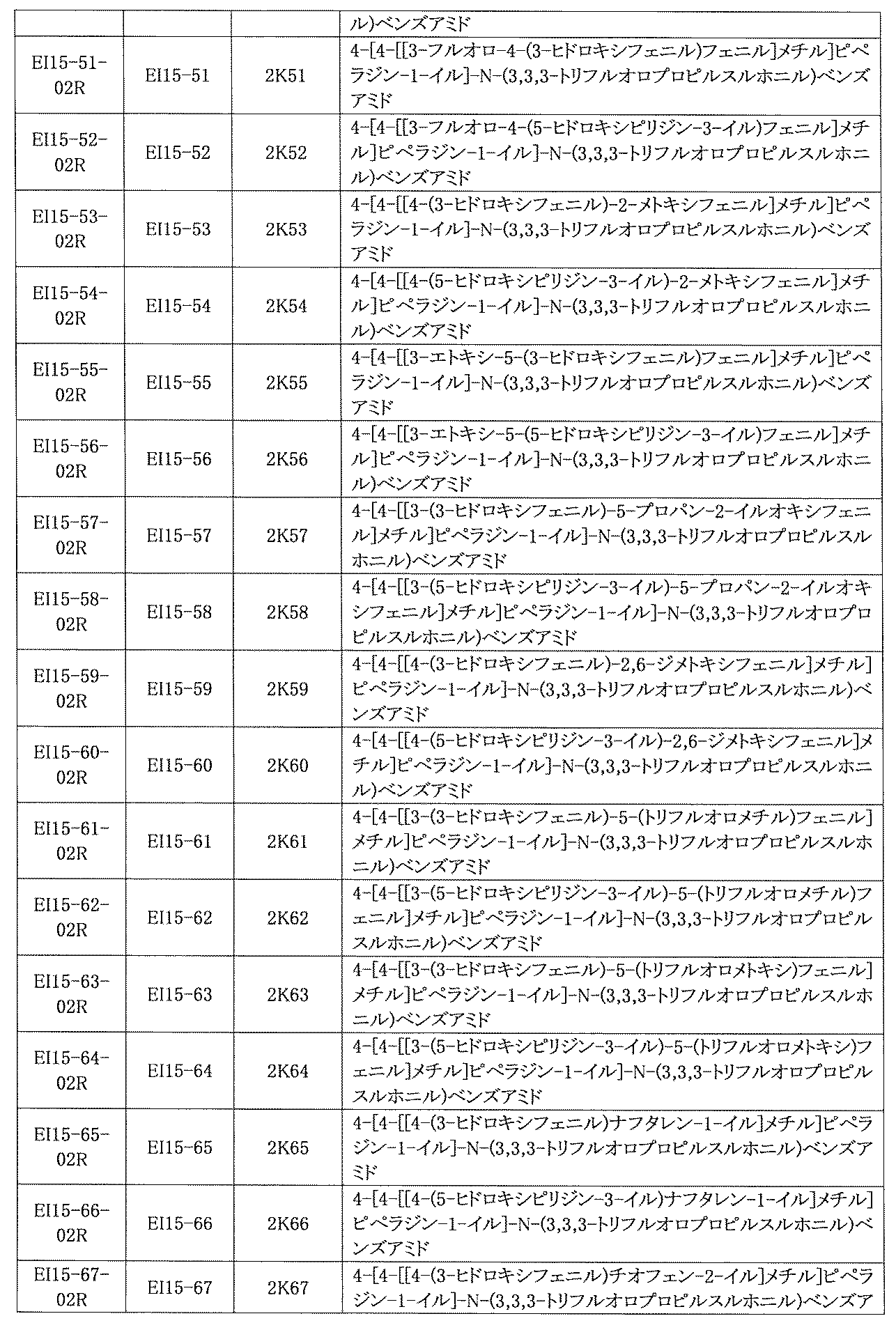









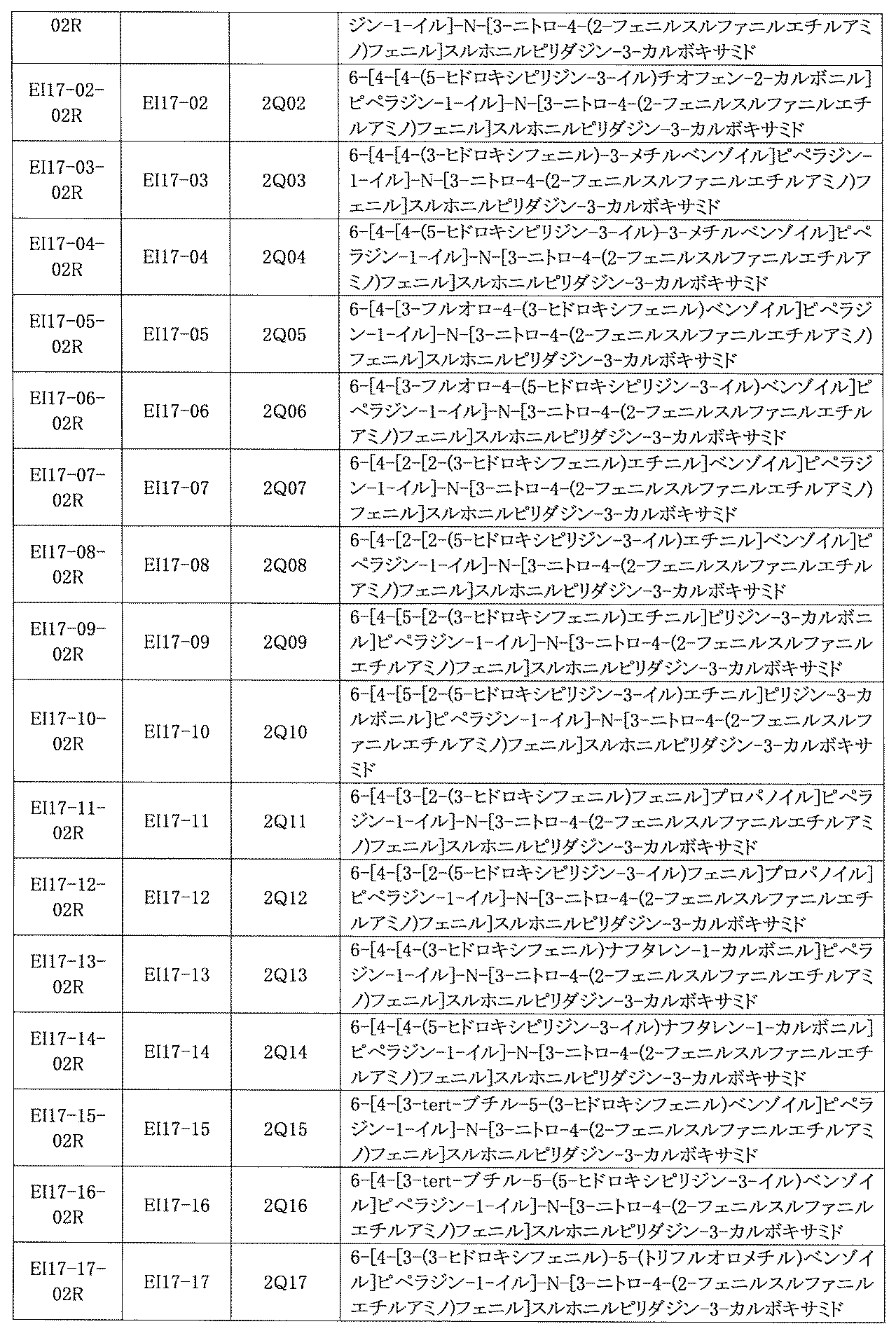




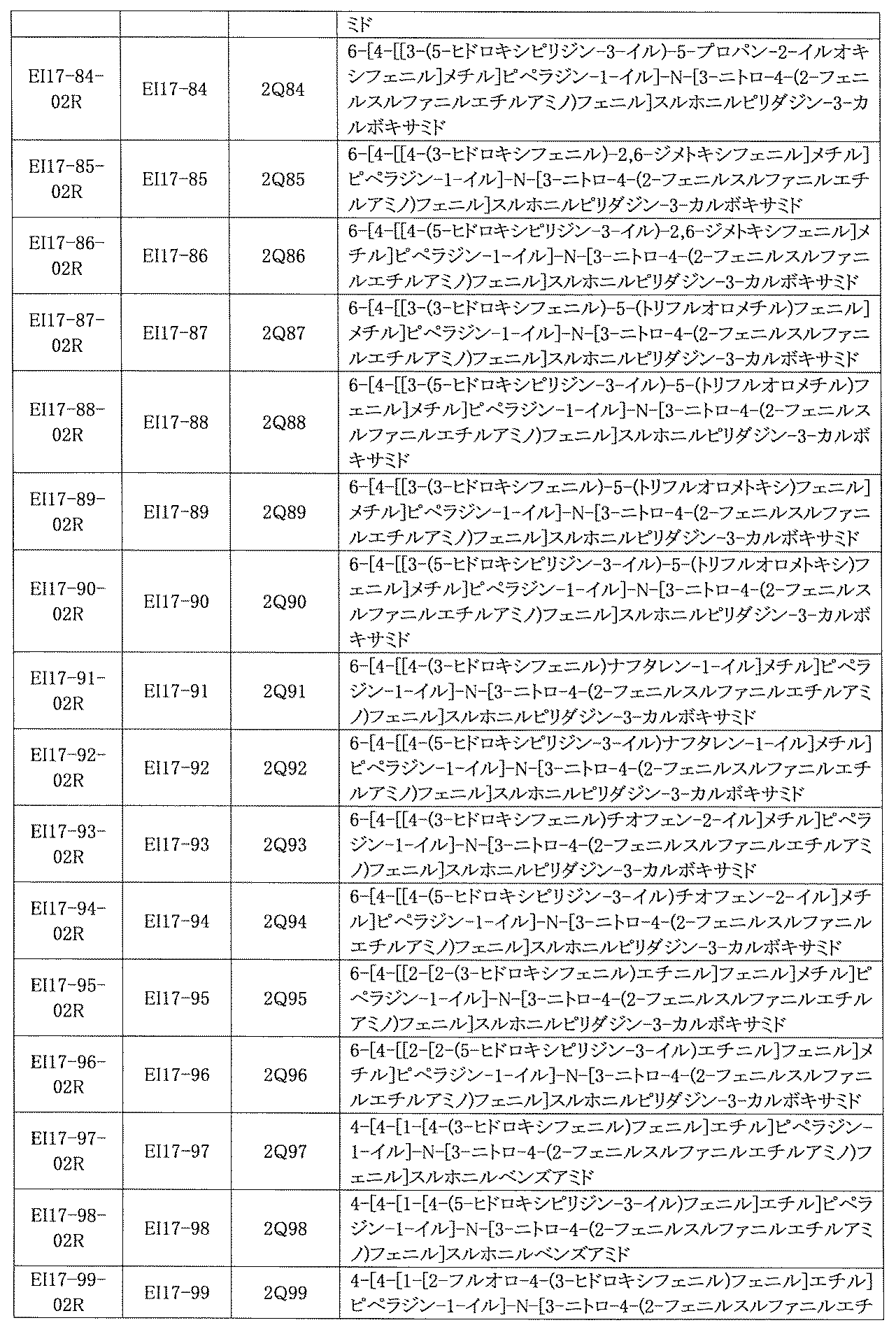


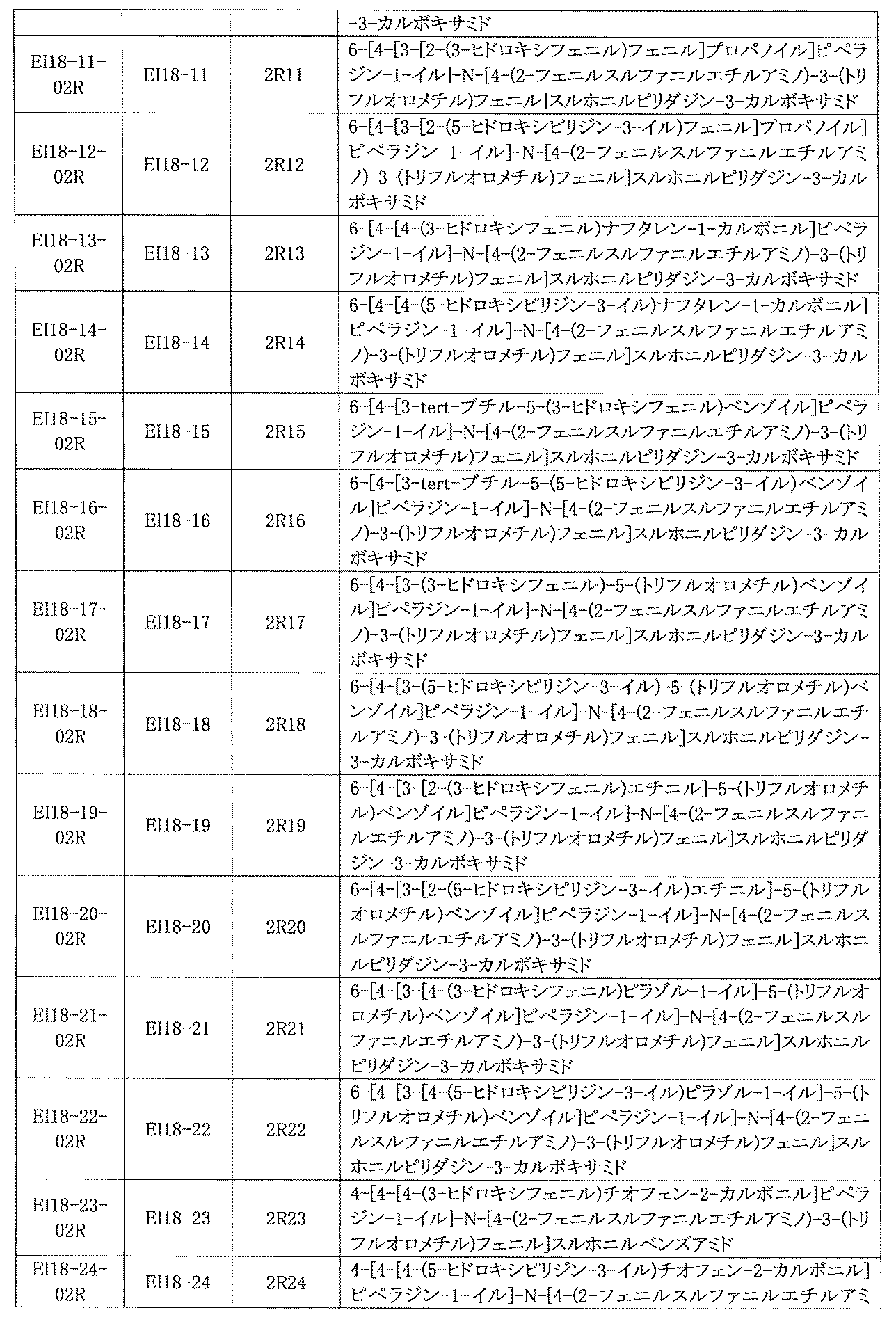

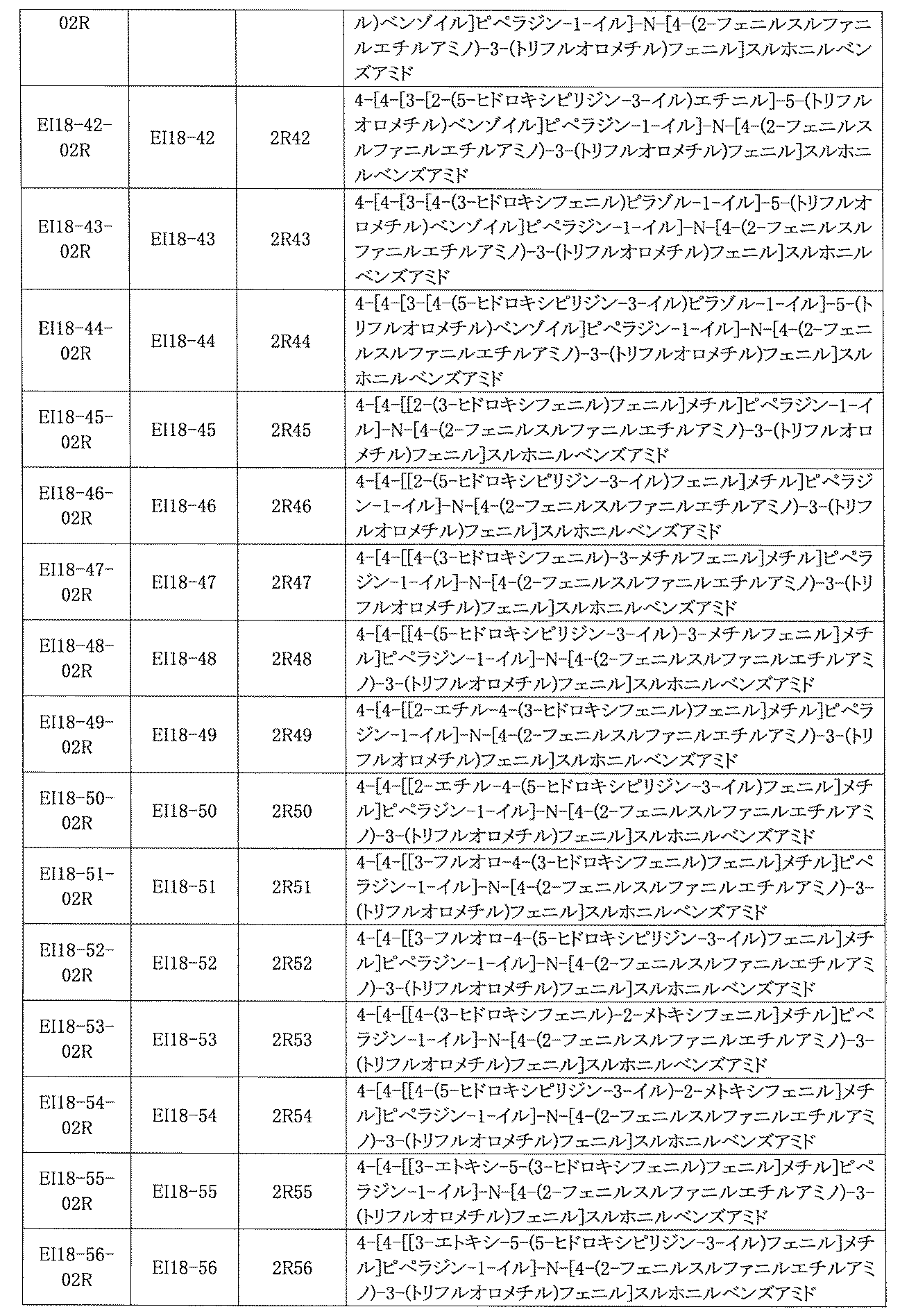

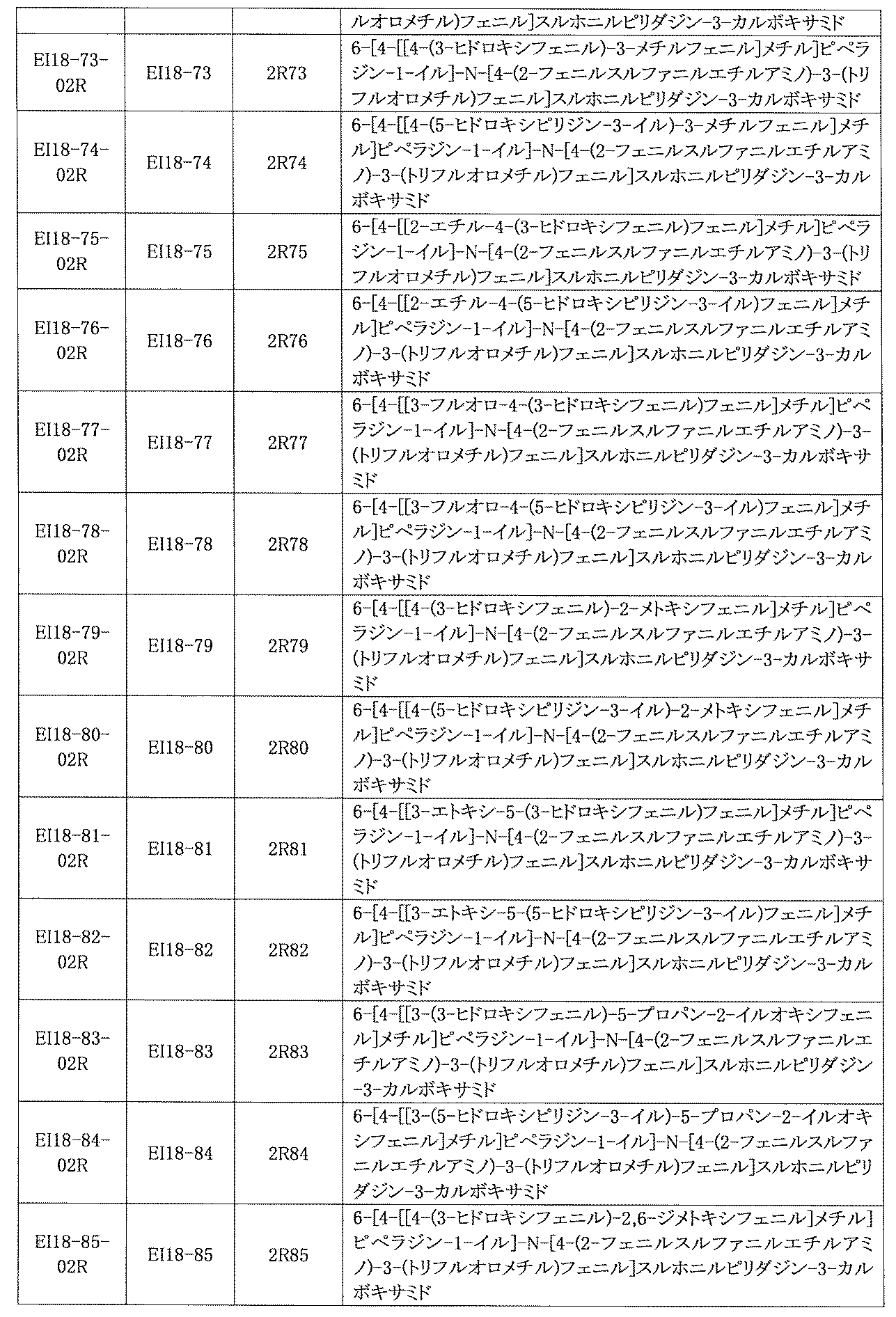









[表EI08]~[表EI18]に示した混合物mixEI08−02R~mixEI18−02R)を用いて、実施例4−4−1と同様の方法で合成し、[表EIJ02]~[表EIJ12]に示す標題混合物mixEJ02~mixEJ12を得た。分析条件SMD−FA05−long−25minにて分析し、UV面積は、UV波長290nmにて確認した。なお混合物の分析結果について、[表EJ02]~[表EJ12]に示す。





























































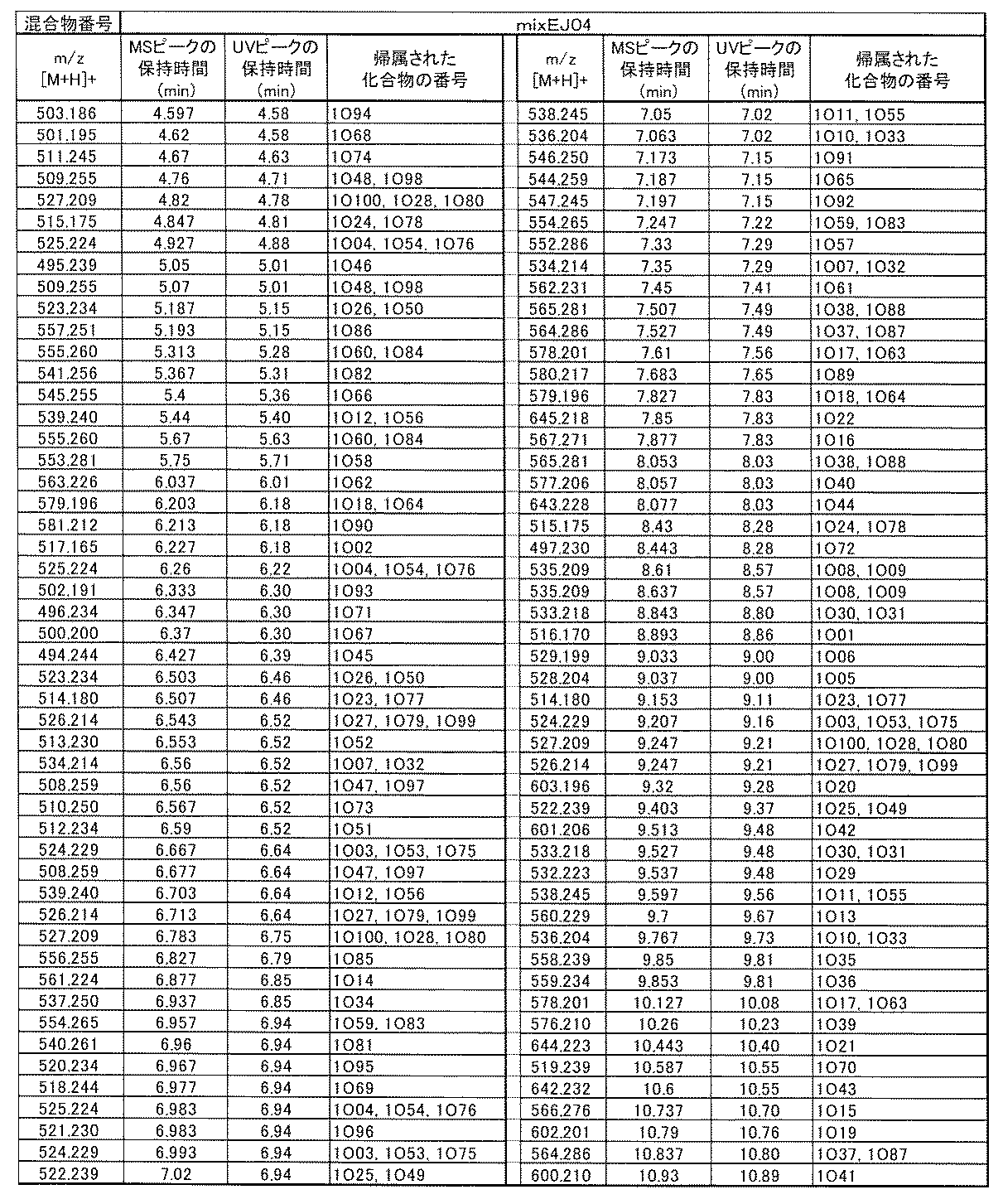
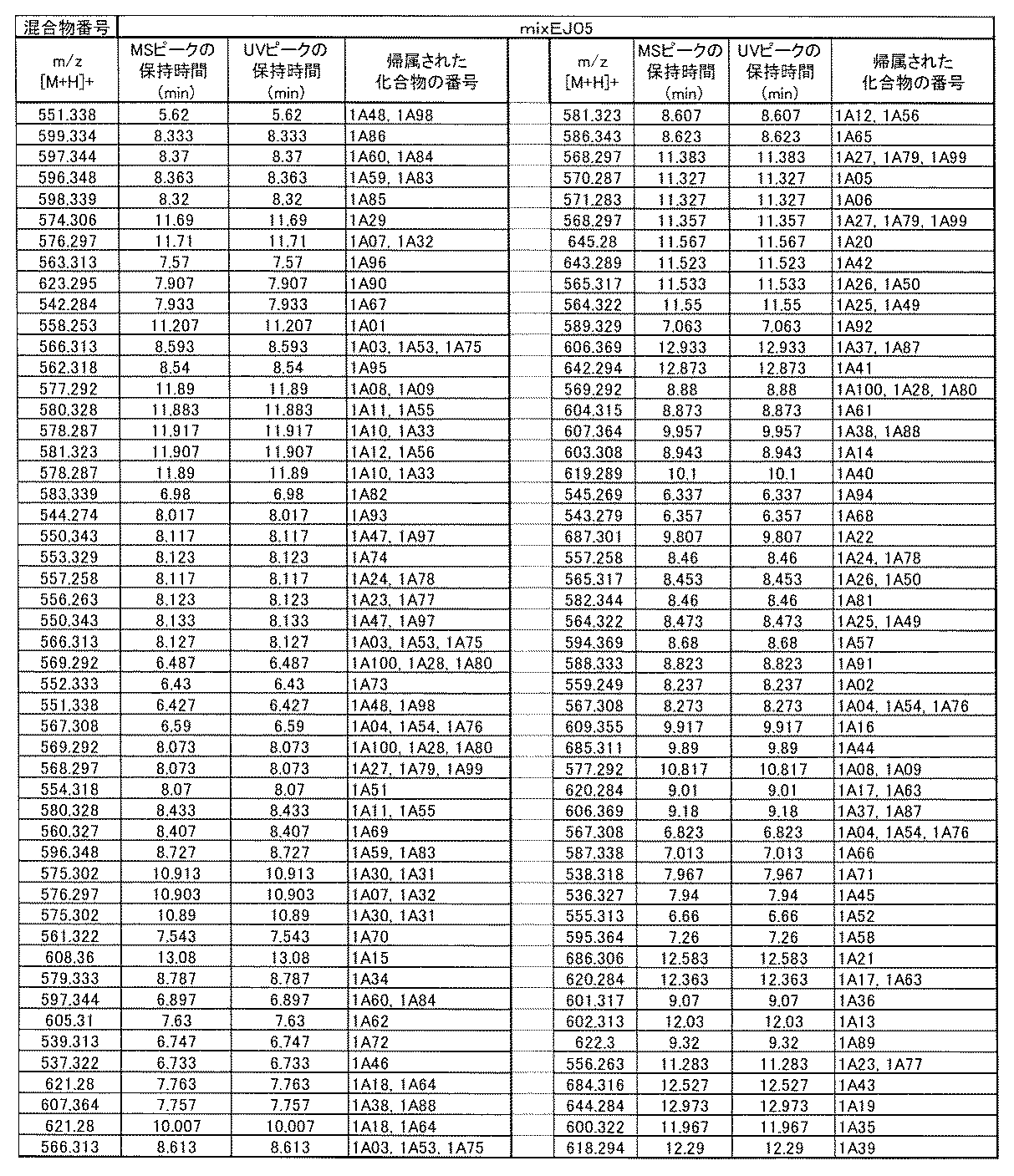



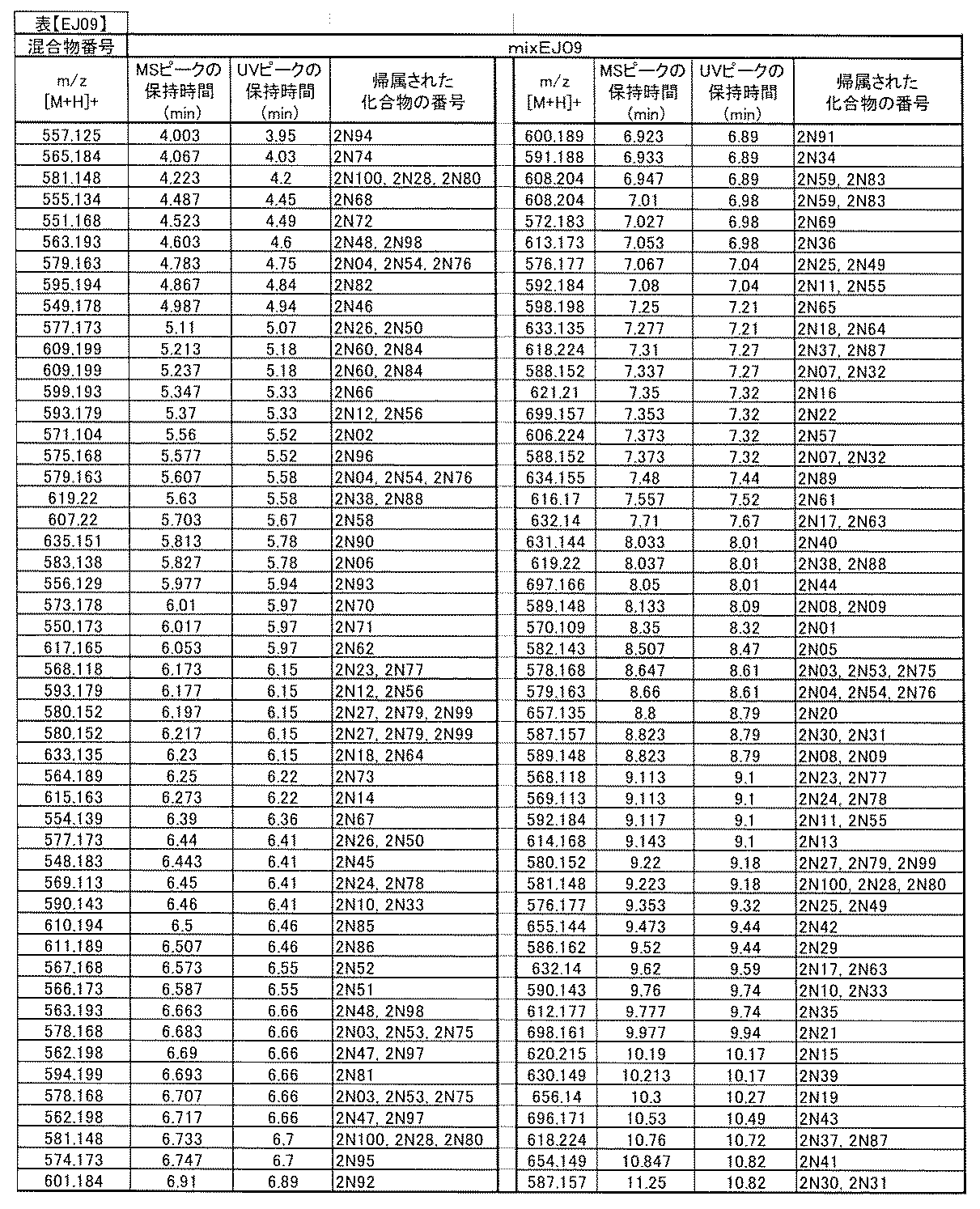
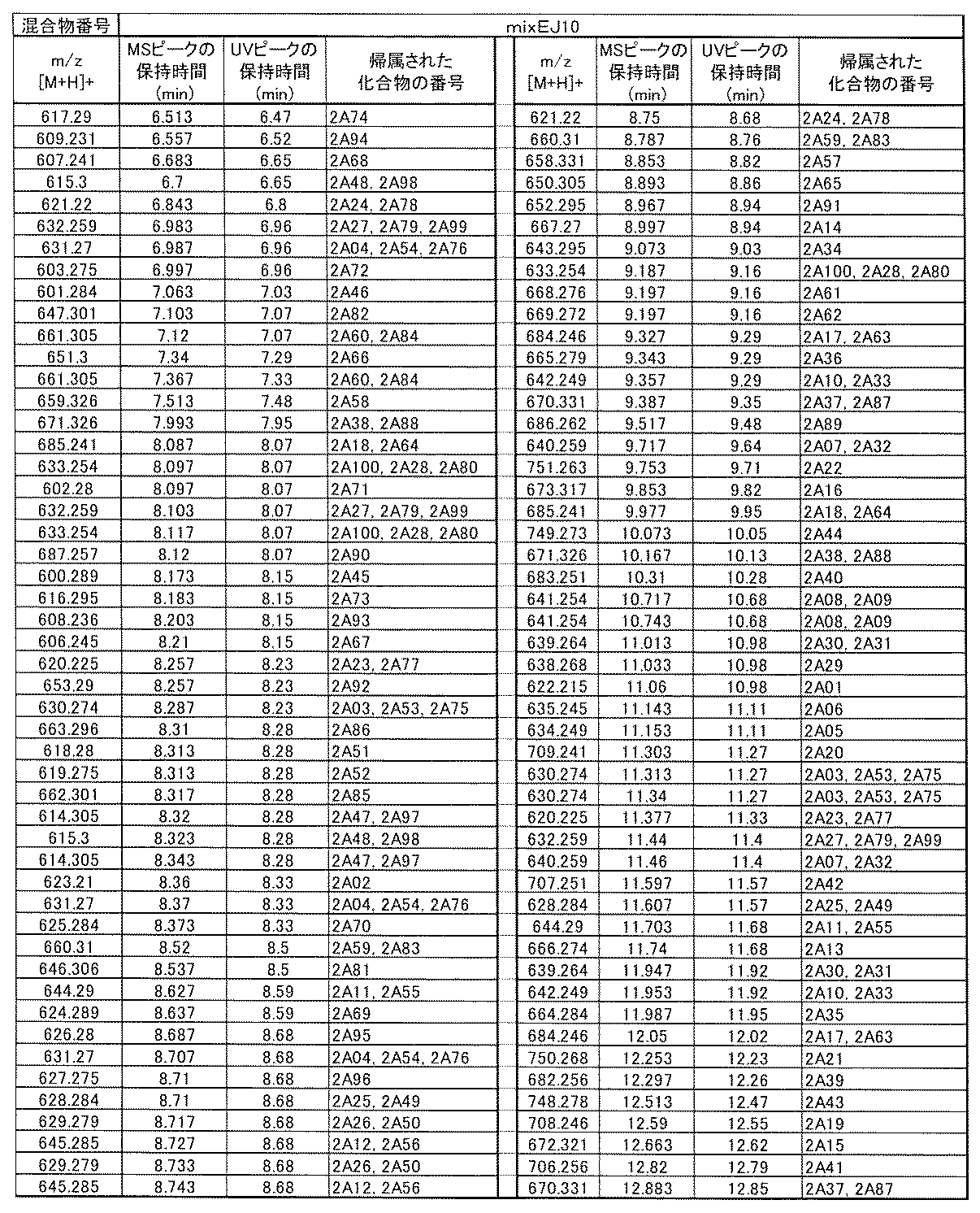



[表EIJ01]~[表EIJ10]および[表EIJ12]に示す混合物mixEJ01~mixEJ10およびmixEJ12についても、実施例4—5−1同様の操作を実施し、[表AI−03−B4J−01]、[表AI−03−B1M−01]、[表AI−03−B1K−01]、[表AI−03−B1O−01]、[表AI−03−B1A−01]、[表AI−03−B3A−01]、[表AI−03−B2I−01]、[表AI−03−B2L−01]、[表AI−03−B2N−01]、[表AI−03−B2A−01]、[表AI−03−B2R−01]に示す混合物を得た。
用いた原料の混合物番号、各々より得られた目的物の混合物番号ならびに目的物の混合物に含まれうる化合物の番号、得られた混合物の重量について[表AI−03−01]に示す。
































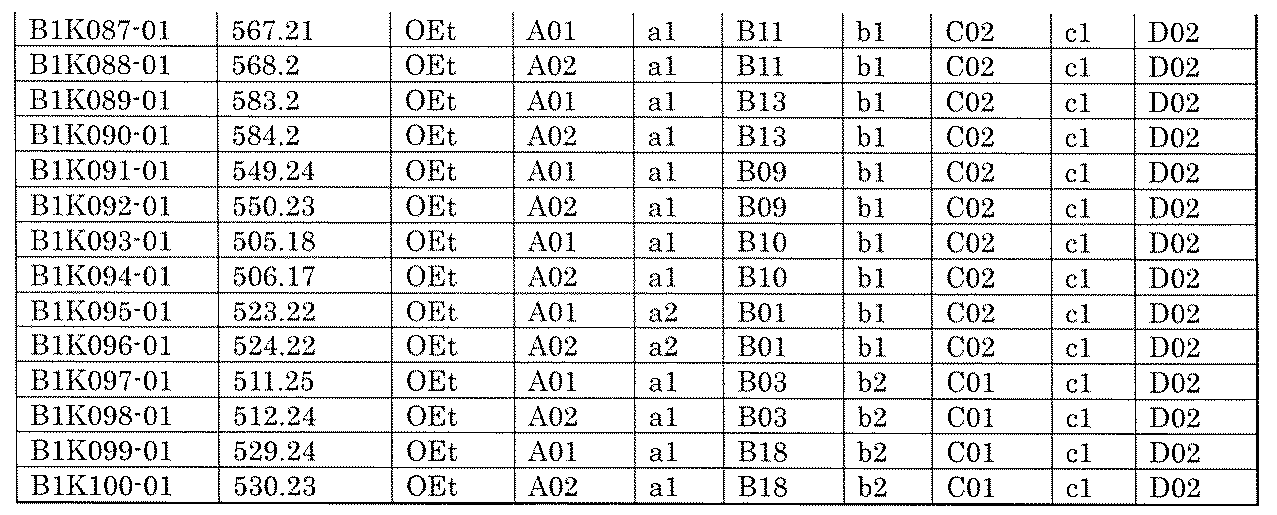
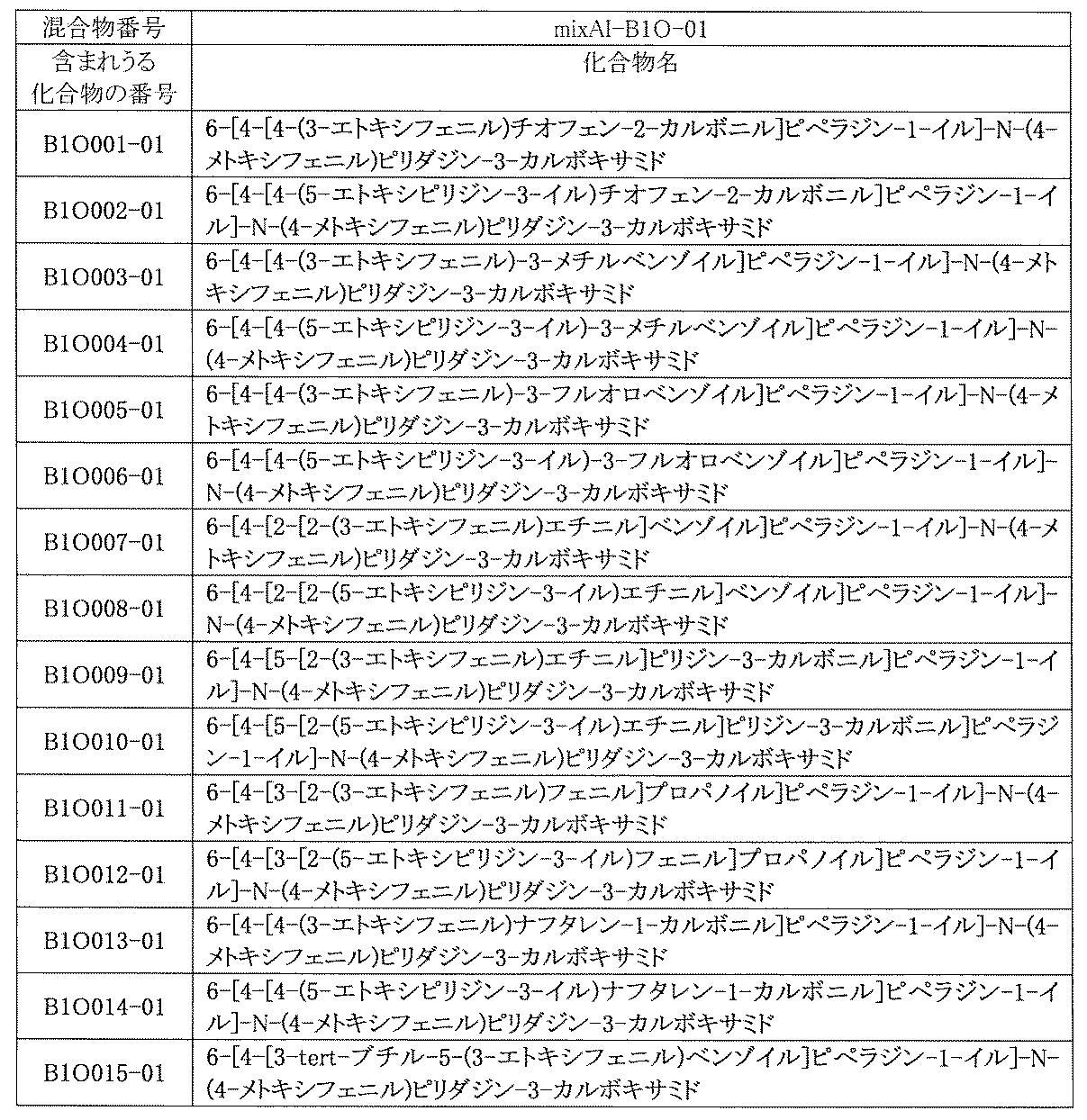




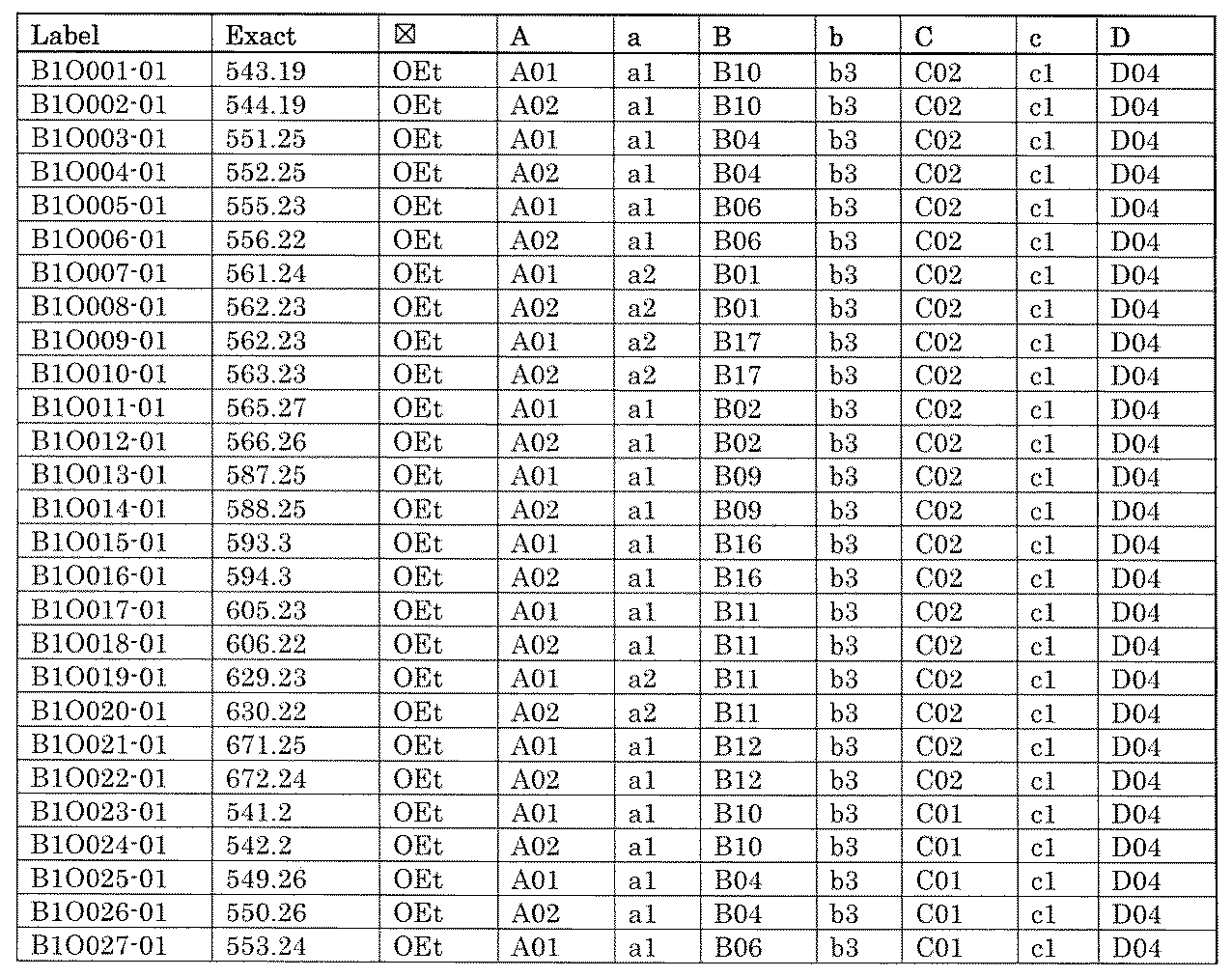


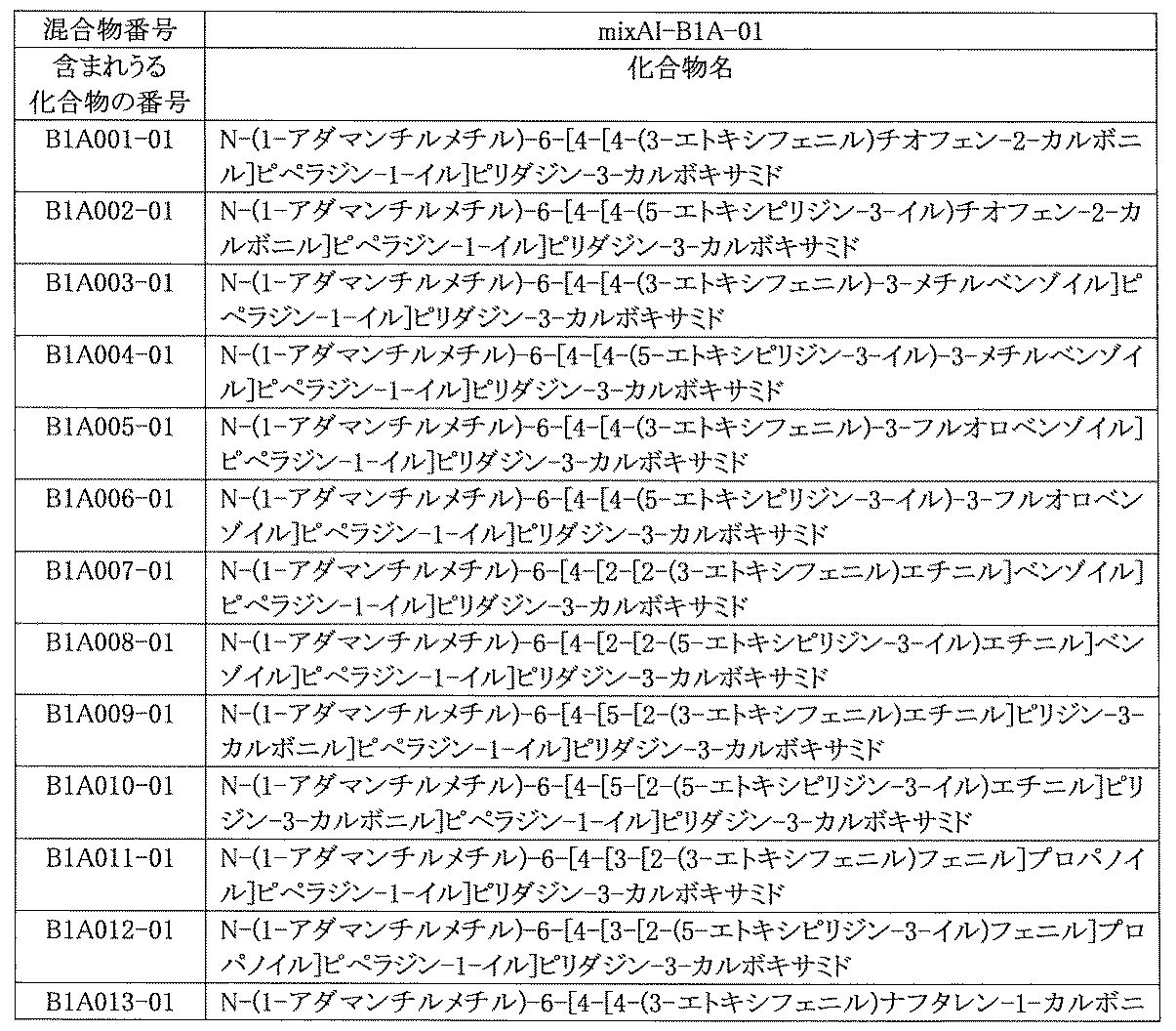



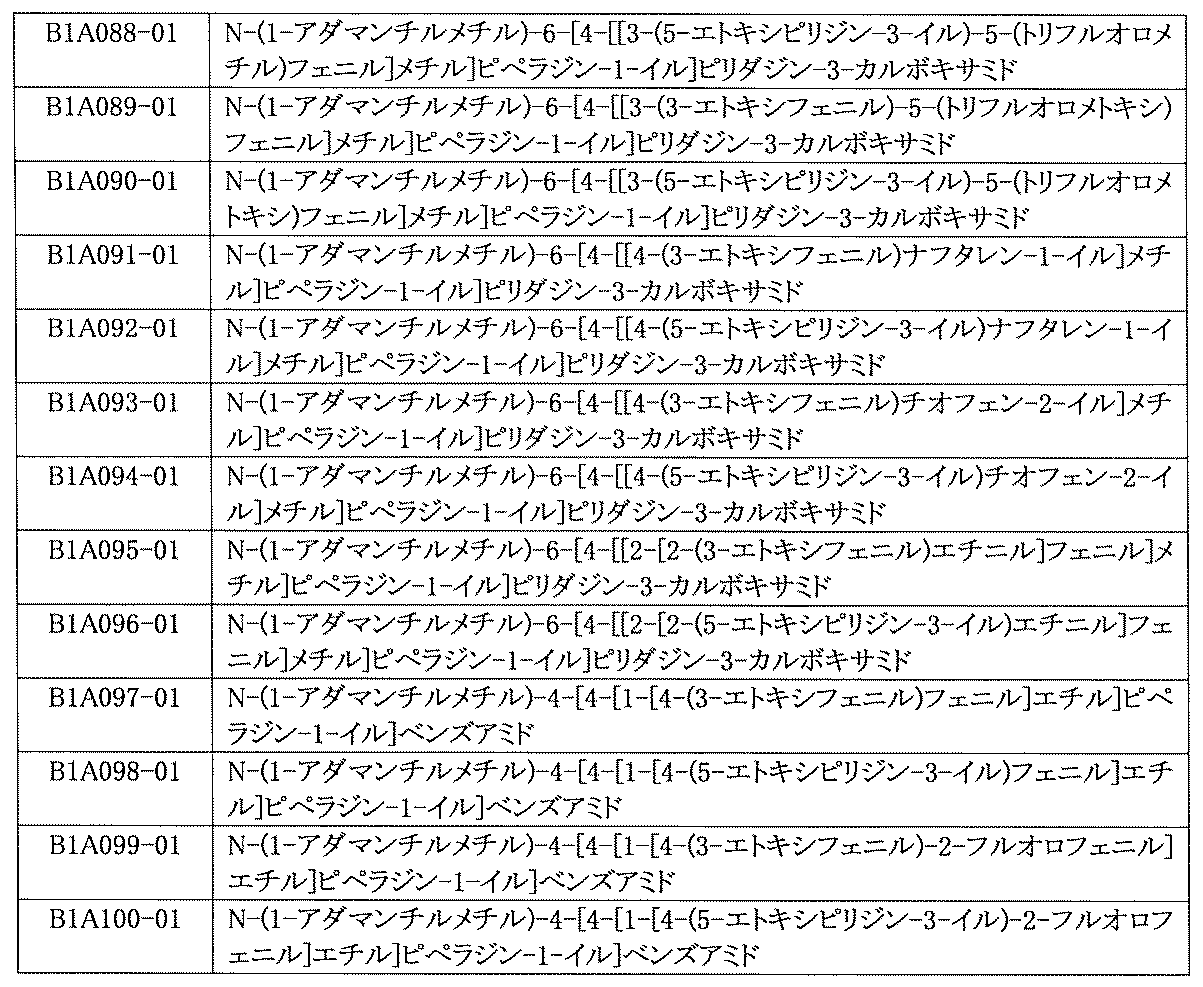

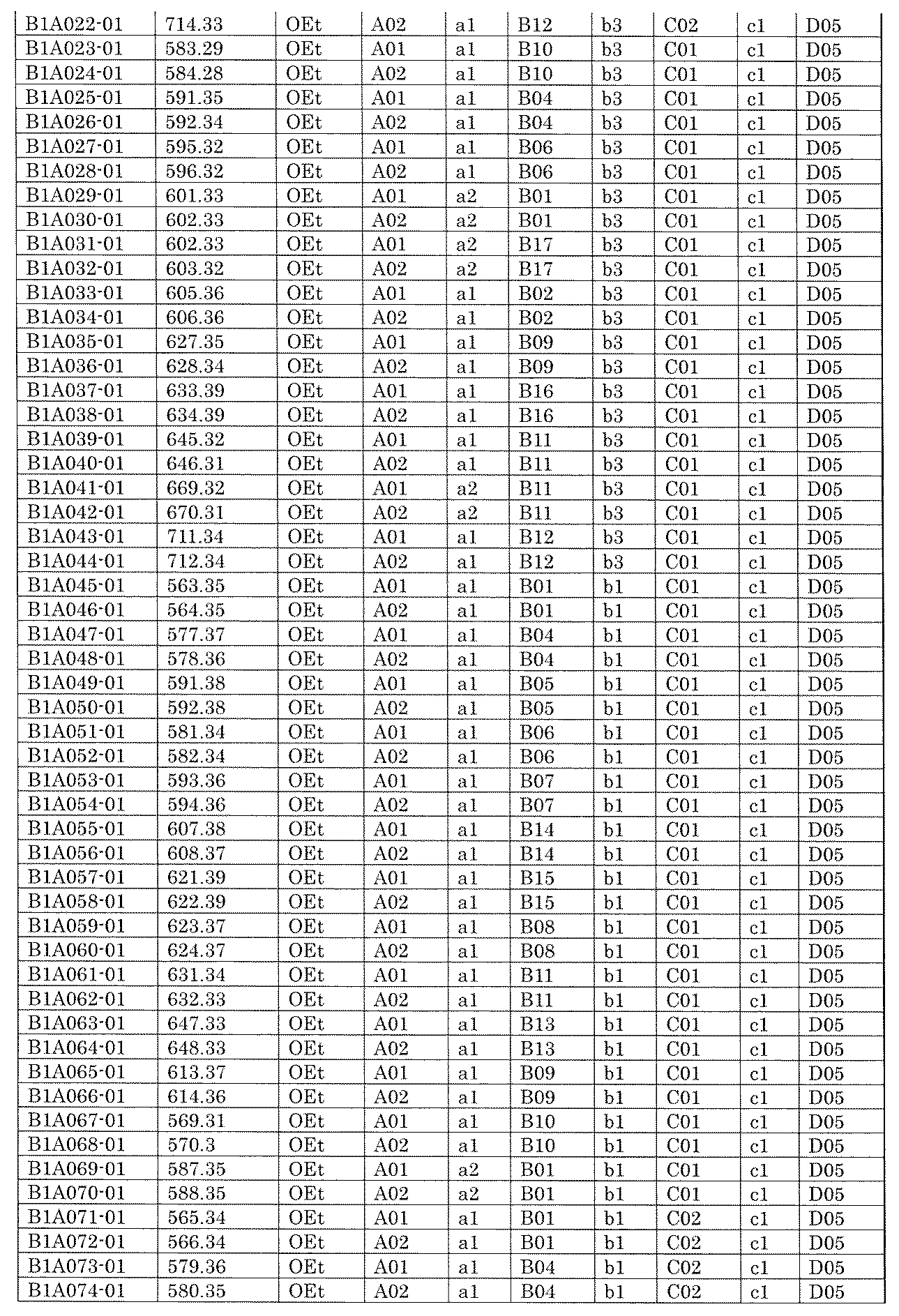











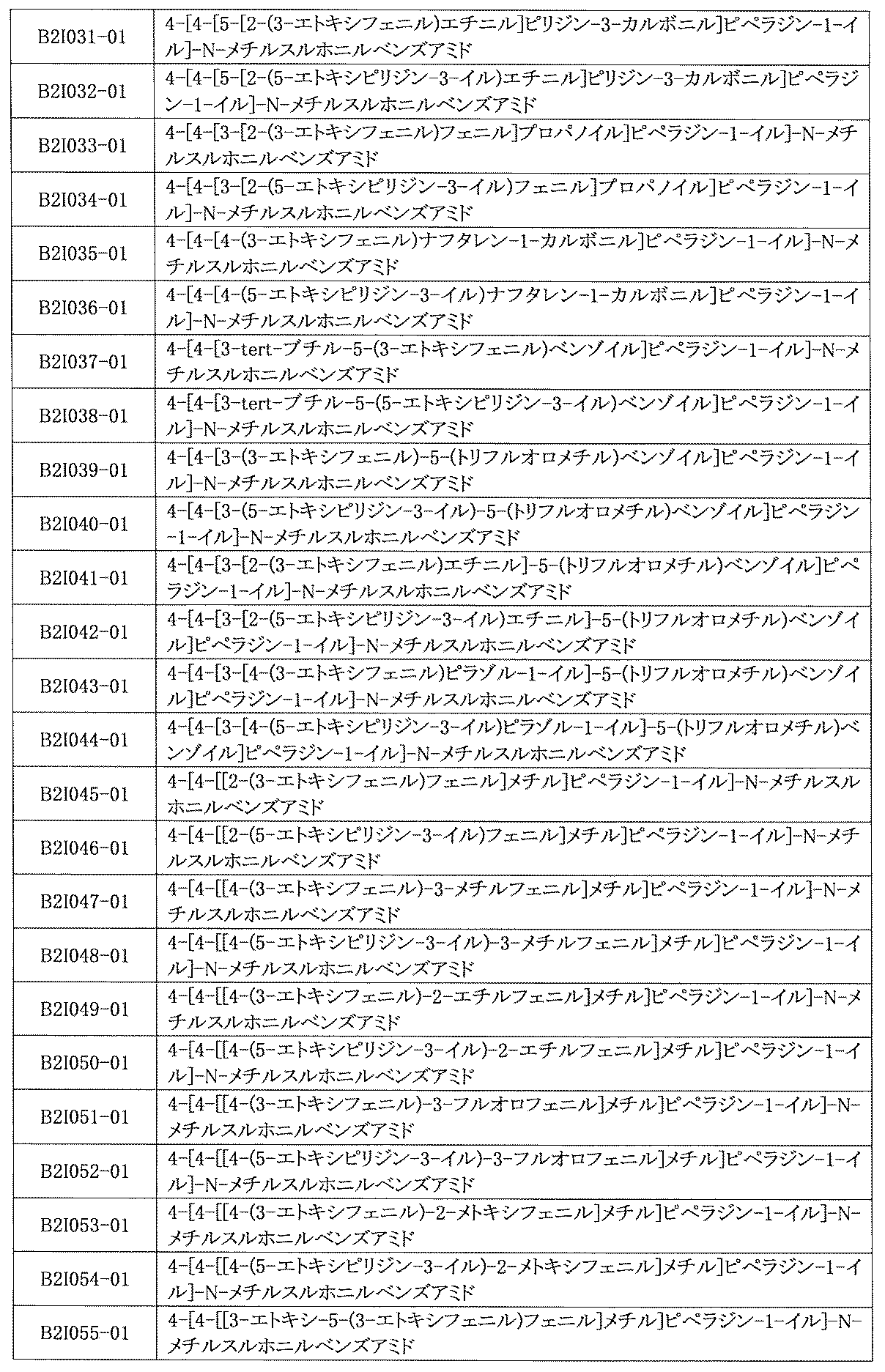
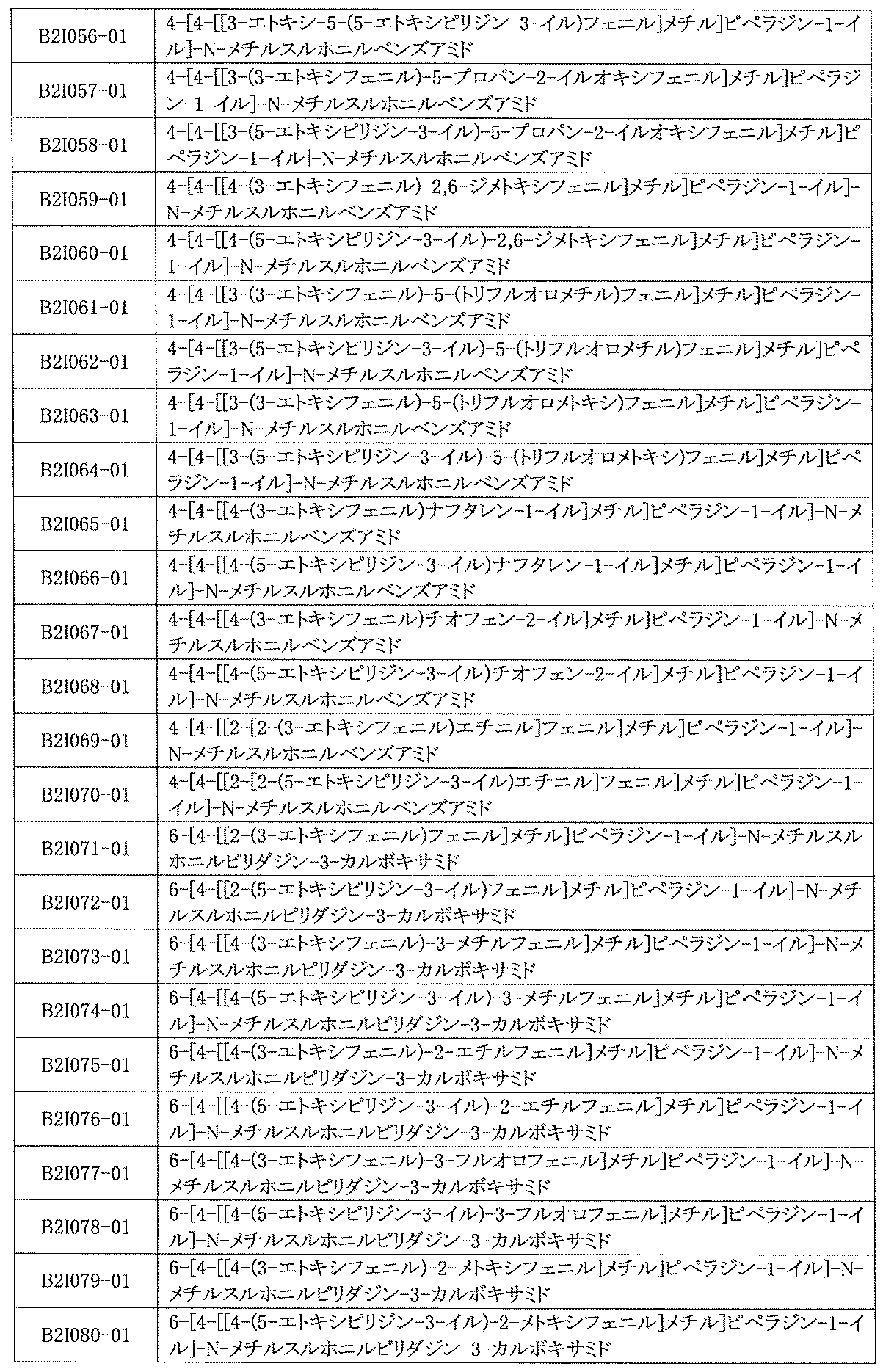
















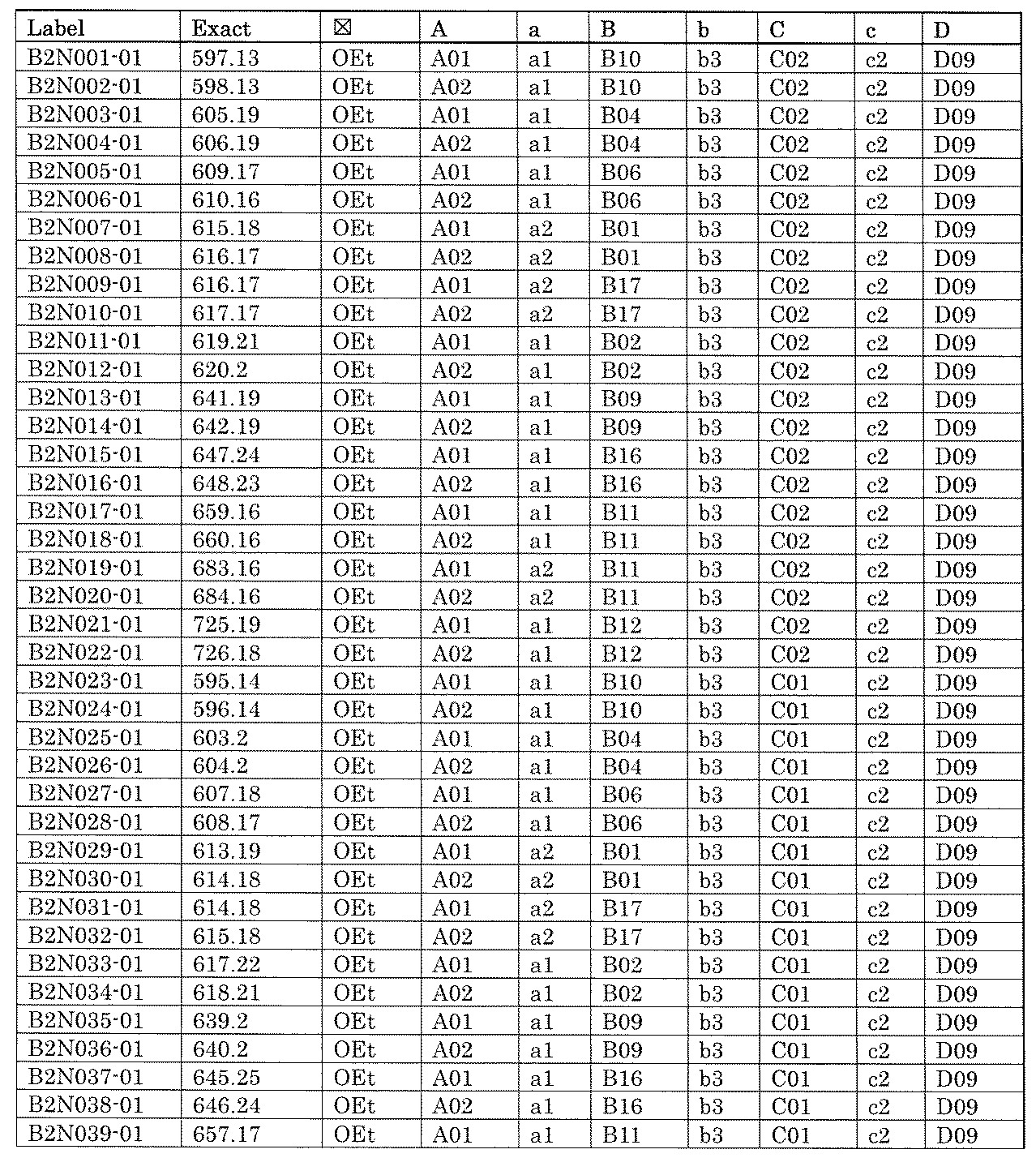

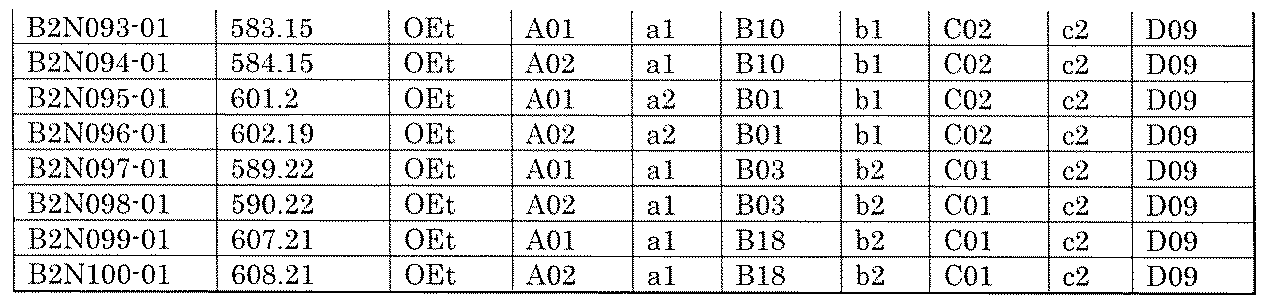





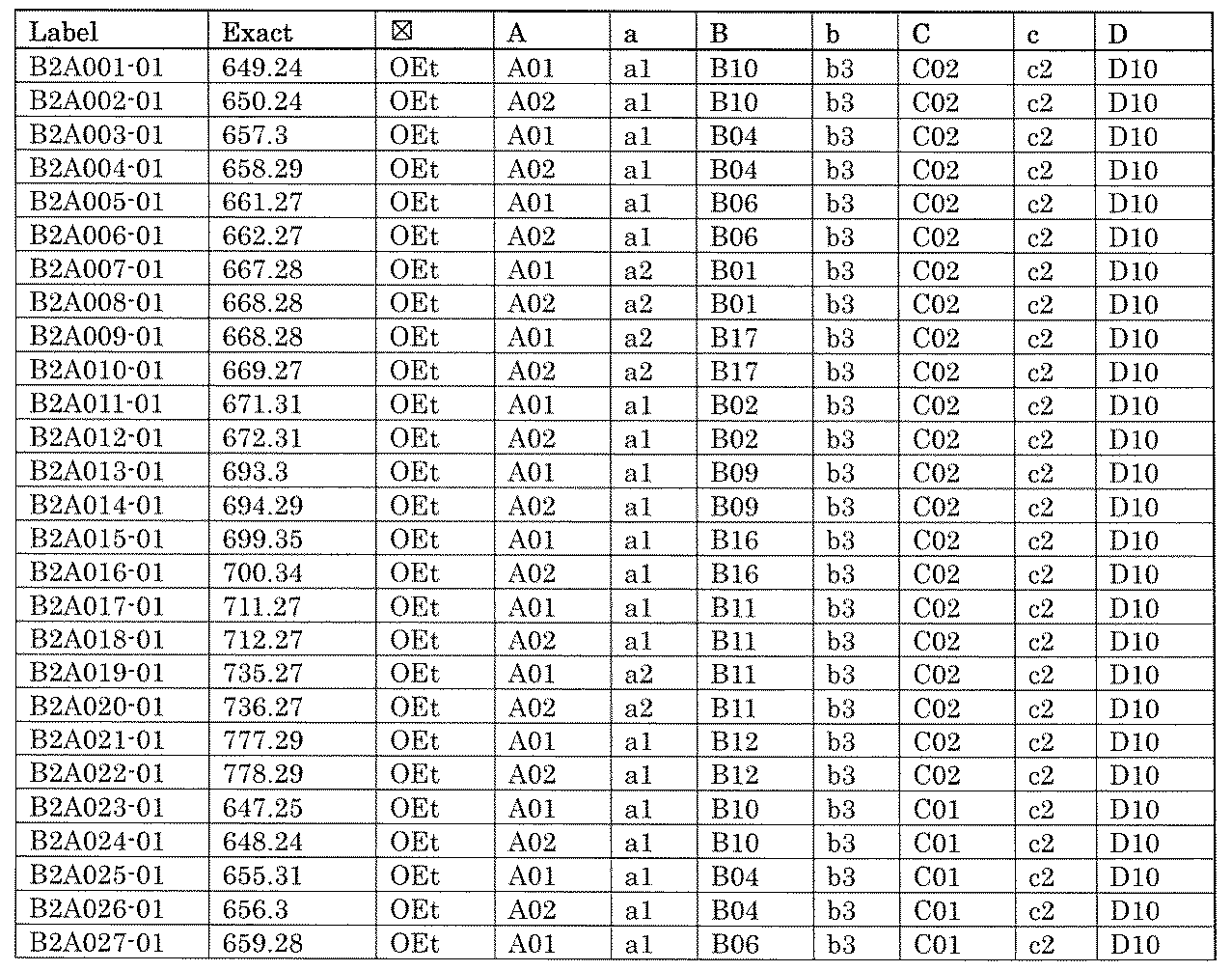
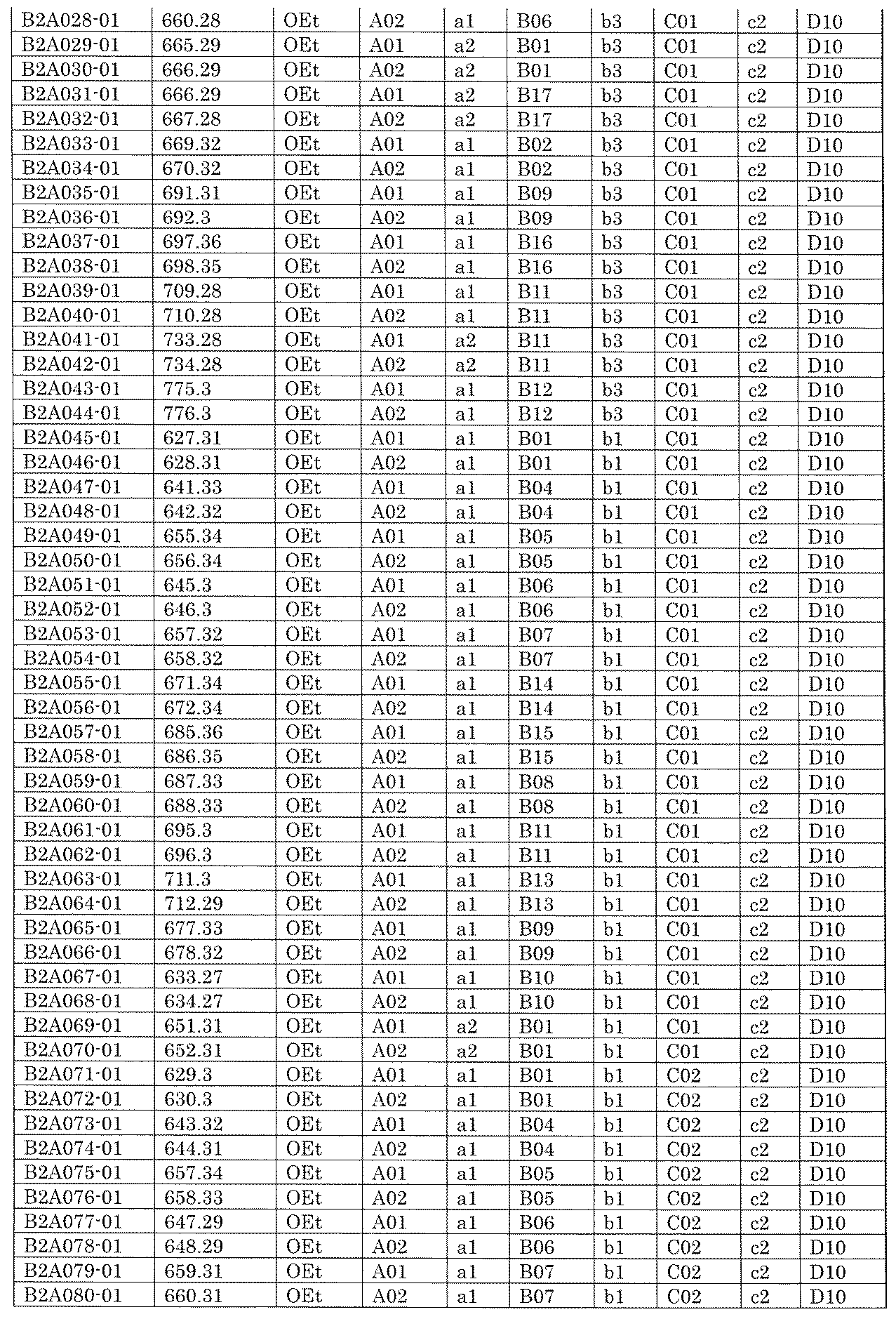


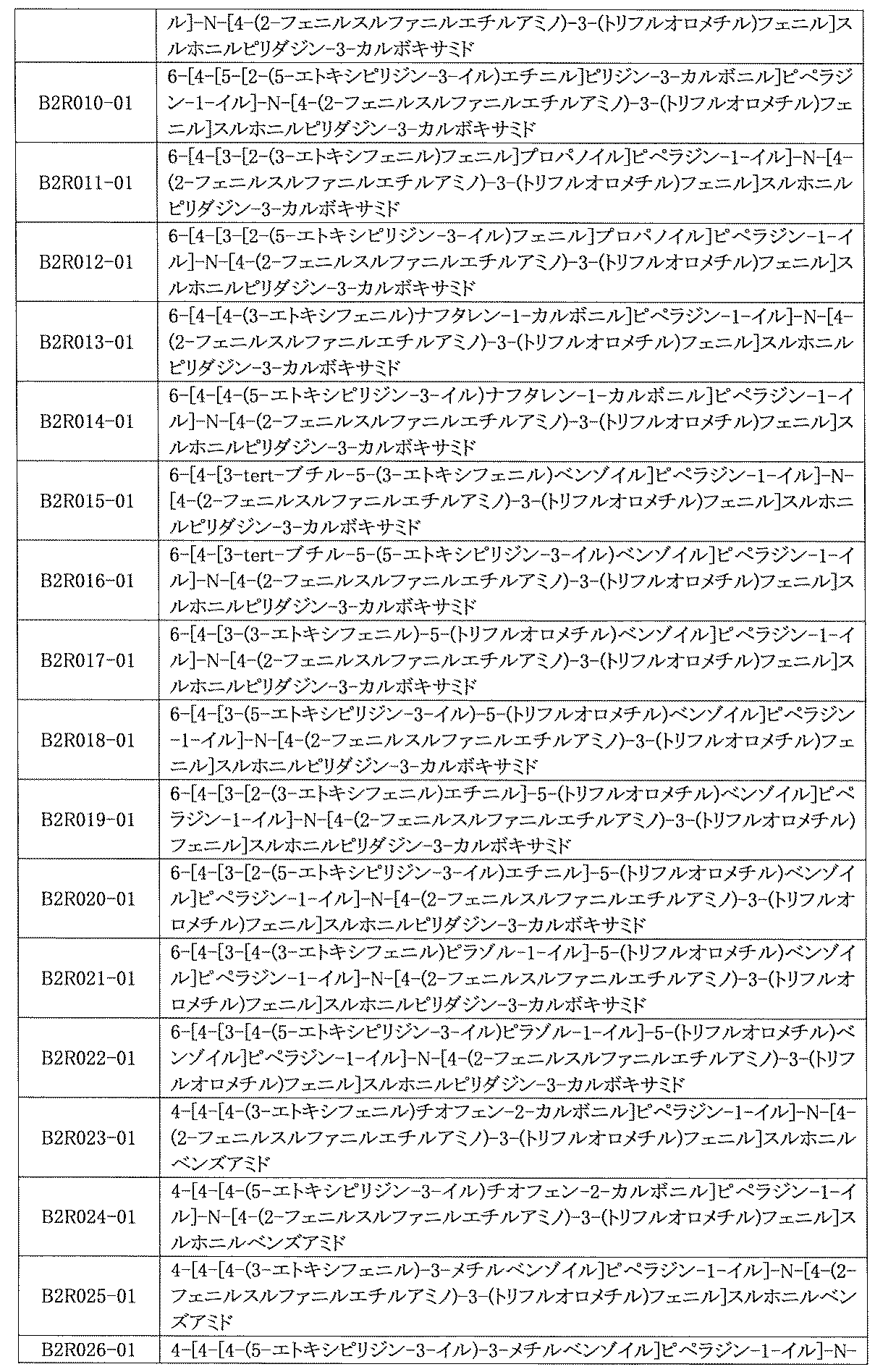


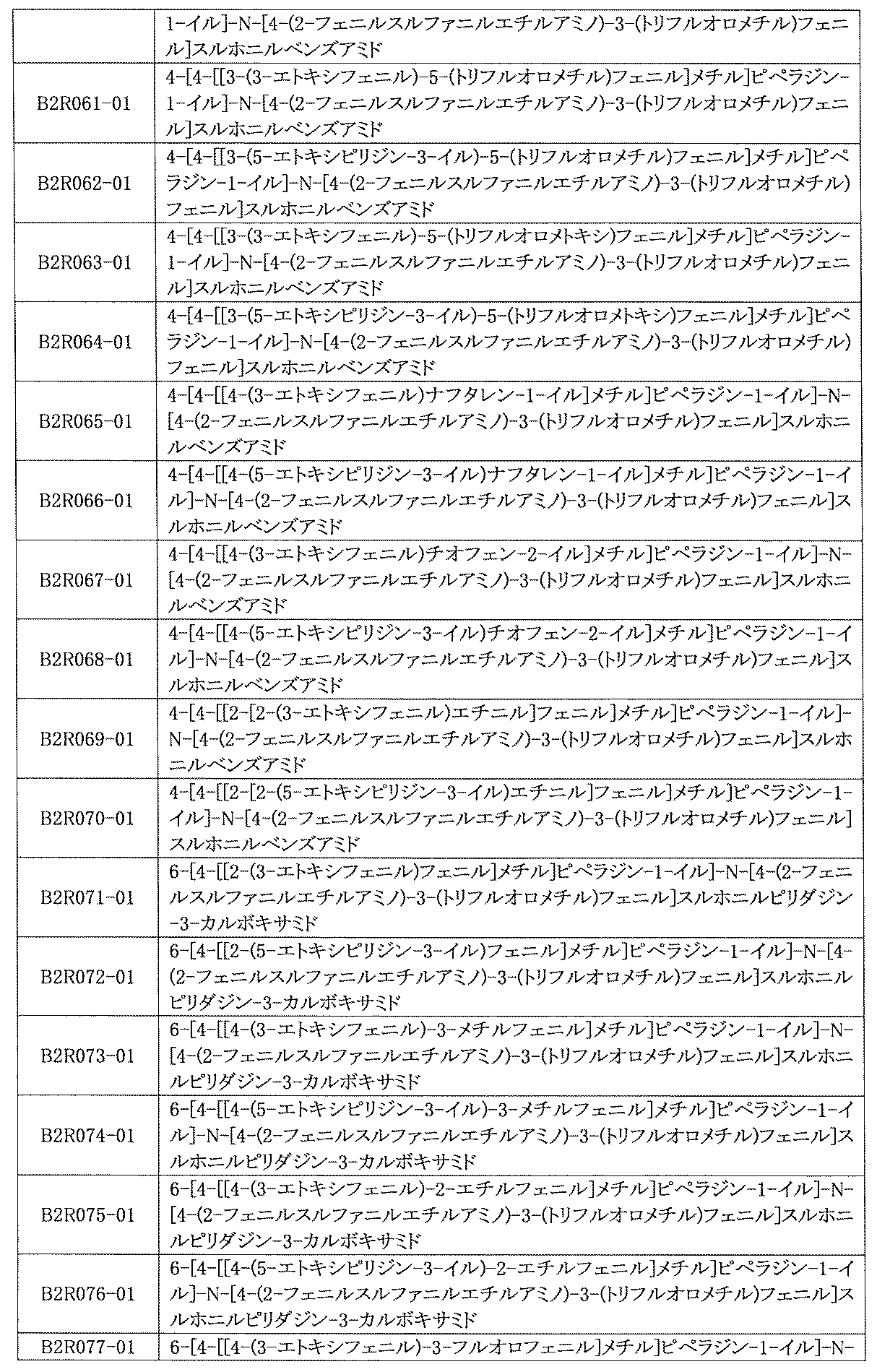




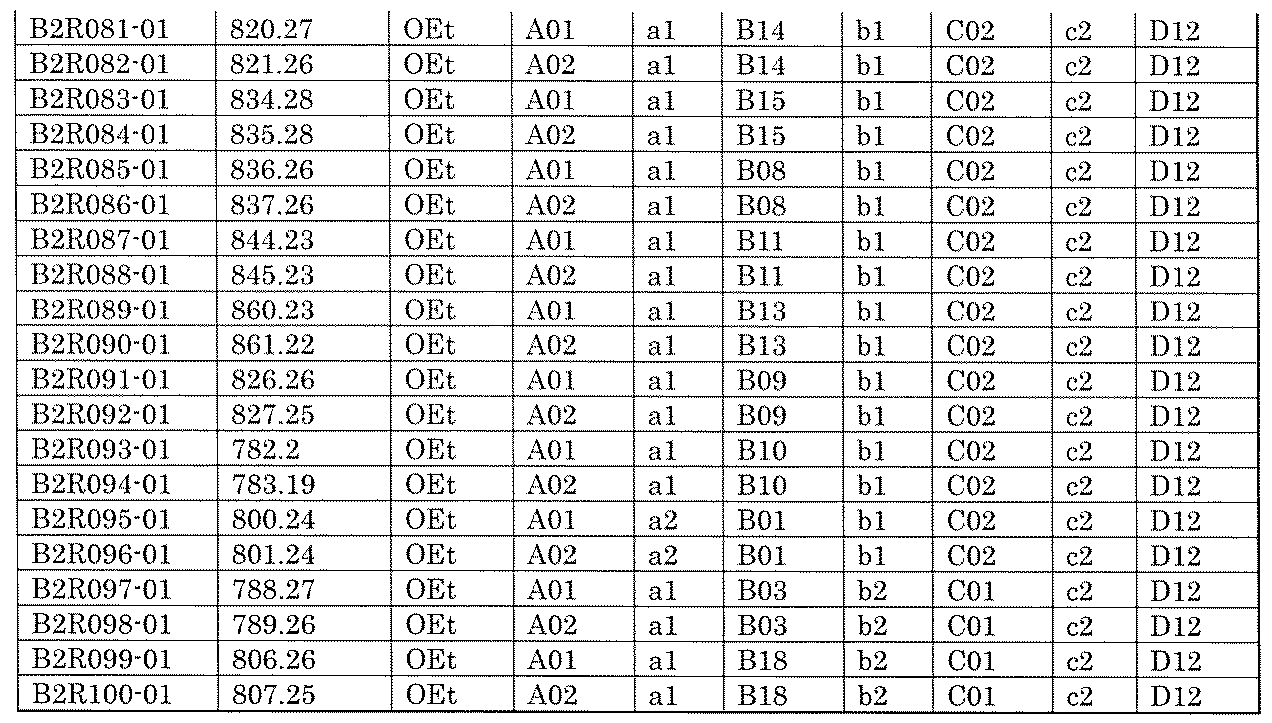
実施例4—5−1および実施例4—5−2にて得られた混合物12種類に対して、各々DMSOを加え、凡そ10mMのDMSO溶液を12種類調製した。そのうち100μLを実施例5で示すAS−MSによるBcl−2に対する結合評価実験に供した。また、12種類の10mMのDMSO溶液の一部をMeCNにて希釈し、LCMSにて分析した。分析条件SMD−FA05−long−25minにて分析し、UV面積は、UV波長280nmにて確認した。分析結果については[表AI−03−02]に示す。[表AI−03−02]には観測されたMSピークのm/zと各保持時間、ならびに各MS−UVピークに帰属することのできる化合物の番号を示す。なお、MSピークのm/zについては小数点以下3桁目までを確認しているが、[表AI−03−02]中の表記においては小数点以下の0を省略して記載していることもある。例えば、「587.000」の場合「587」、「587.100」の場合「587.1」、「587.120」の場合「587.12」と表記していることもある。また、MSピークの保持時間については小数点以下3桁目までを、UVピークの保持時間については小数点以下2桁目までを確認しているが、[表AI−03−02]中の表記においては小数点以下の0を省略して記載していることもある。例えば、「15.000」の場合「15」、「15.100」の場合「15.1」、「15.120」の場合「15.12」と表記していることもある。

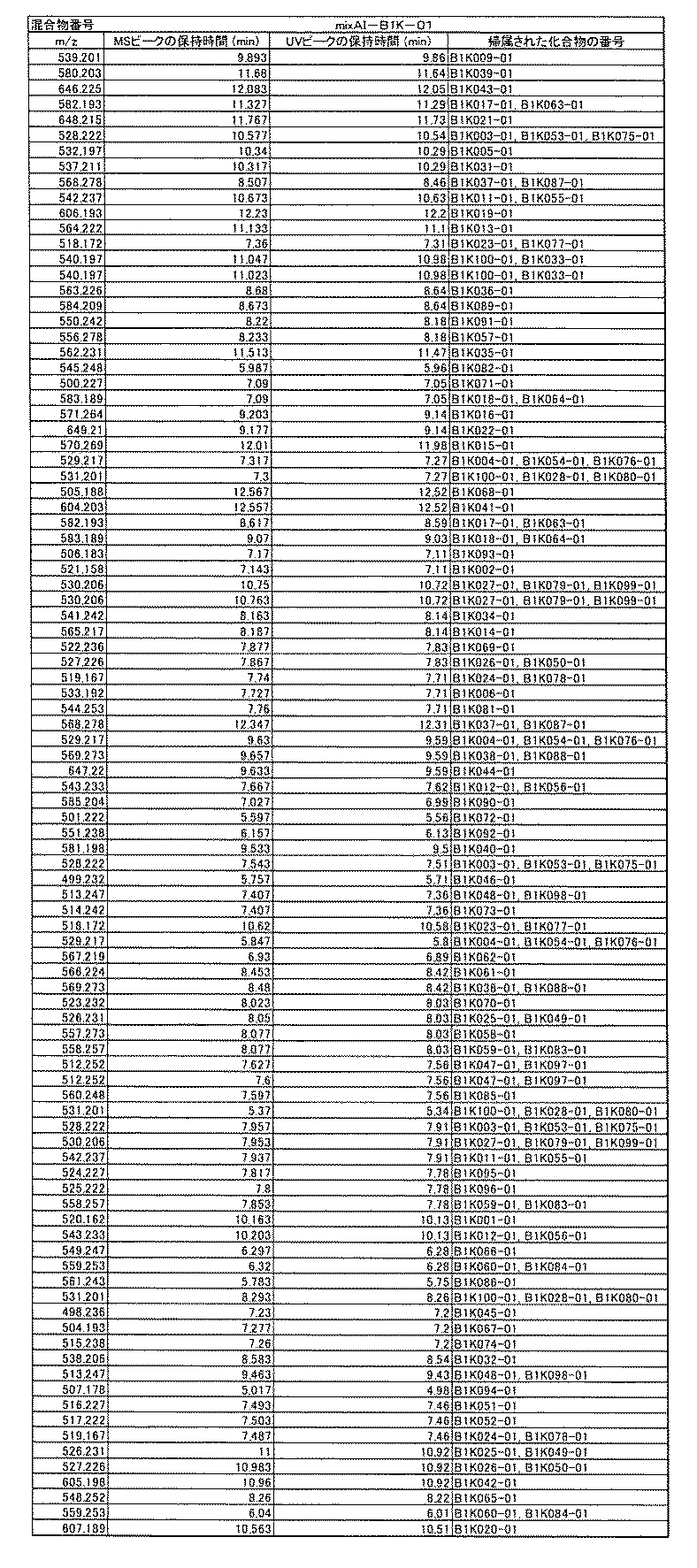






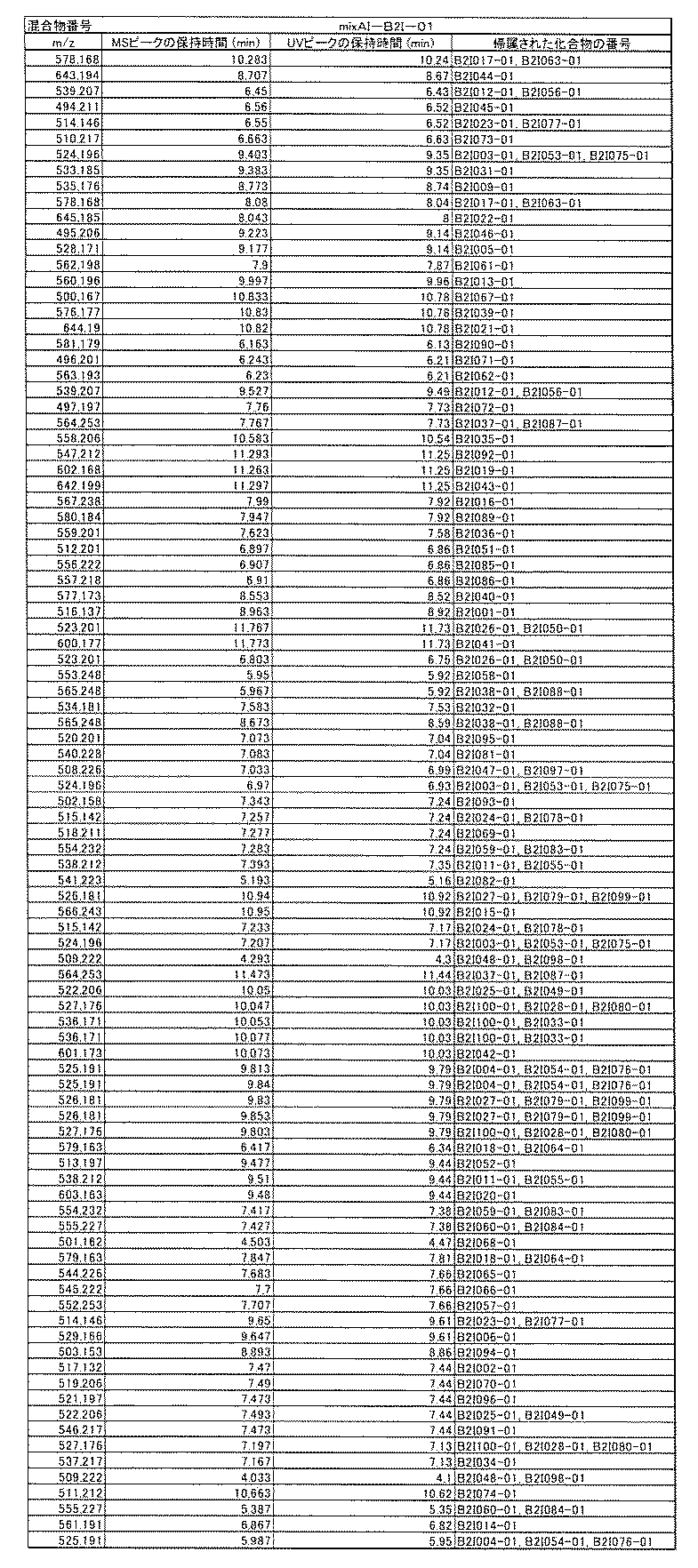



以上の結果より、多様な結合形成に用いる反応条件、洗浄方法、純度担保のためのキャッピング方法について堅牢性の高い方法を明らかにし、適用可能なビルディングブロックの適用範囲を特定する工程を実施することで、高い成功確率でライブラリ合成を実施することが可能になることが明らかになった。
「ビルディングブロックの多様性」に加えて「ビルディングブロック同士を結ぶリンカーの多様性」を持たせ、それらの多様性を掛け合わせたライブラリの合成が実施可能であることを明らかとした。
MICROPLATE,96ウェル,PP,V−BOTTOM(Greiner bio−one,651201)の1ウェルに対して,実施例4中に示す[表AI−03−B4J−01]、[表AI−03−B1M−01]、[表AI−03−B1K−01]、[表AI−03−B1O−01]、[表AI−03−B1A−01]、[表AI−03−B3A−01]、[表AI−03−B2I−01]、[表AI−03−B2L−01]、[表AI−03−B2N−01]、[表AI−03−B2A−01]、[表AI−03−B2Q−01]、AI−03−B2R−01]に記した、各々100化合物が含まれうる12種類の混合物、mixAI—B4J−01、mixAI—B1M−01、mixAI—B1K−01、mixAI—B1O−01、mixAI—B1A−01、mixAI—B3A−01、mixAI—B2I−01、mixAI—B2L−01、mixAI—B2N−01、mixAI—B2A−01、mixAI—B2Q−01、mixAI—B2R−01のDMSO溶液(各混合物の平均分子量からの計算に基づき、凡そ0.58mMに調製)を各々分注し、合計12ウェルに分注した。LABCYTE ECHO(登録商標)555アコースティック分注システム(BECKMAN COULTER)を用いて12ウェルからそれぞれ70nLずつ任意の1ウェル中に集め、1200化合物(実施例4において調製した際には、1152化合物は存在を特定済み)が混合された状態のサンプル溶液を調製した。さらに予めTris−HClバッファー(25mM Tris−HCl pH7.4,150mM NaCl,0.025%Tween−20,1mM DTT)を用いて20.9μMに調製したBcl−2タンパク質(Novus,NBP2−34889,Recombinant Human Bcl−2 Protein,Lot E60145051)(19.2μL)を1200化合物(実施例4において調製した際には、1152化合物は存在を特定済み)が分注されたウェルに加え(最終化合物濃度20nM,最終Bcl−2濃度20μM)、暗所にて23℃で1時間インキュベーションした。つぎに膨潤させておいた96ウェルMacroSpinカラム,P−2 Material(Harvard,74−5655)を用いてアッセイバッファー(25mM Tris−HCl pH7.4,150mM NaCl,0.025%Tween−20,1mM DTT)200μL,2000XG,4℃,2分間遠心にて3回行い96ウェルMacroSpinカラムのコンディショニングを実施した。その次に,インキュベーション後のサンプル20μLをコンディショニング完了後の96ウェルMacroSpinカラム,P−2 Materialに添加し2000XG,2分間遠心にてSEC処理をした。SECからの通液にアセトニトリルとメタノール(LC/MSグレード,富士フイルム和光純薬工業(株))1:1の混合液を32μL加え1800XG,4℃,5分間遠心し上澄みを分析サンプルASMS(A)とし,同様の操作で1200化合物/ウェルで調製し,Bcl−2タンパク質が入っていないTris−HClバッファーのみを加えたサンプルを同一条件でインキュベーションし、96ウェルMacroSpinカラム処理をせず,同一の有機溶媒を加え遠心操作によって得られた上澄み液をSTD(B)とした。次に同様の操作で1200化合物/ウェルで調製し,Bcl−2タンパク質が入っていないTris−HClバッファーのみを加えたサンプルを同一条件でインキュベーションし,同様に96ウェルMacroSpinカラム処理を実施し,同一の有機溶媒を加え遠心操作によって得られた上澄み液をBK(C)とし,逆相LC/MSにて測定した。
計算式 ASMS Ratio(%)=(A−C)/B * 100

[表AJ−01]






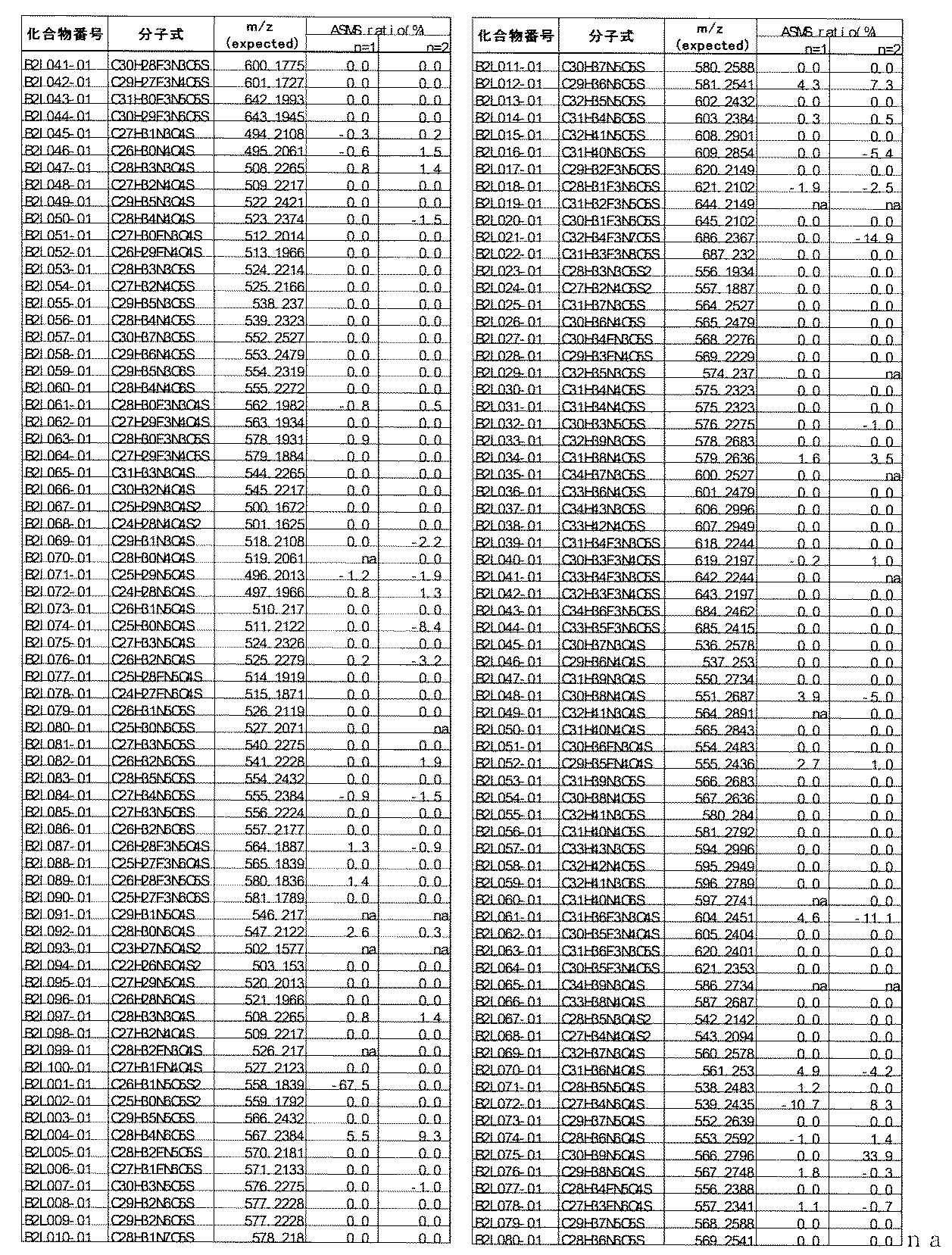


実施例6−1−1:4−[4−[[4−(3−エトキシフェニル)−3−メチルフェニル]メチル]ピペラジン−1−イル]−N−[3−ニトロ−4−(2−フェニルスルファニルエチルアミノ)フェニル]スルホニルベンズアミド[AG016(B2Q047−01)]の合成

LRMS(ESI):m/z 766[M+H]+。

LRMS(ESI):m/z 767[M+H]+。

LRMS(ESI):m/z 258[M+H]+。
保持時間:0.845分(分析条件PH−TFA05−2)。

LRMS(ESI):m/z 783[M+H]+。

LRMS(ESI):m/z 278[M+H]+。
保持時間:0.769分(分析条件PH−FA05−3)。

LRMS(ESI):m/z 803[M+H]+。

LRMS(ESI):m/z 667[M+H]+。
保持時間:1.185分(分析条件PH−FA05−4)。

LRMS(ESI):m/z 567[M+H]+。
保持時間:0.883分(分析条件PH−FA05−4)。
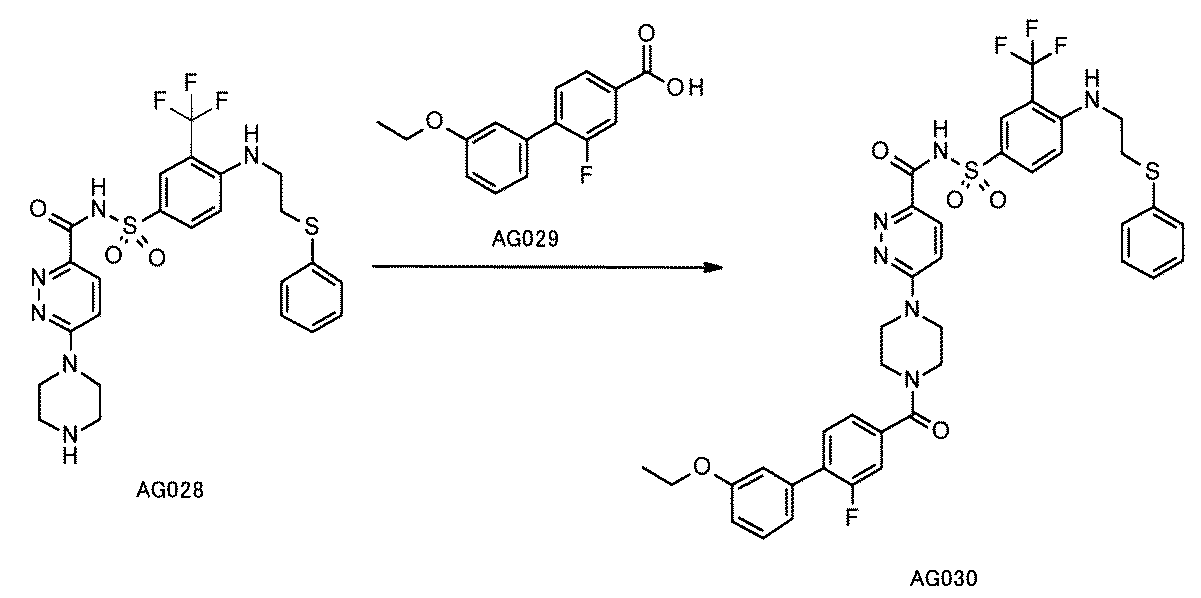
LRMS(ESI):m/z 809[M+H]+。

LRMS(ESI):m/z 859[M+H]+。

LRMS(ESI):m/z 456[M+H]+。
保持時間:1.238分(分析条件PH−TFA05−2)。
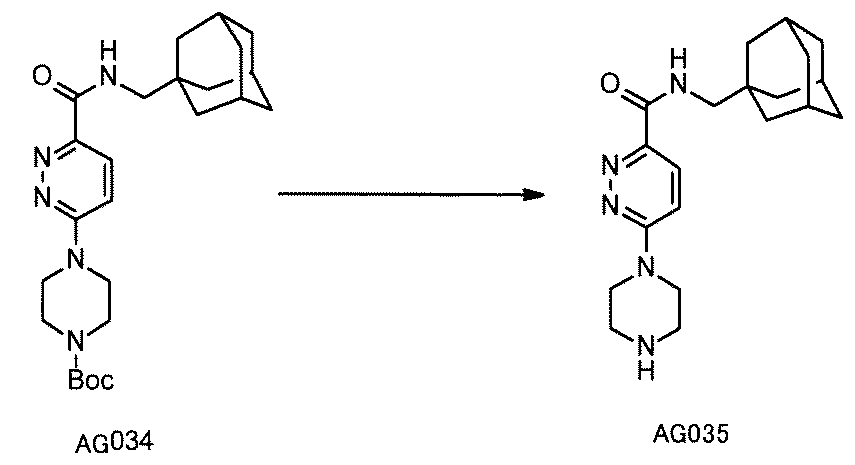
LRMS(ESI):m/z 356[M+H]+。
保持時間:0.917分(分析条件PH−TFA05−2)。

LRMS(ESI):m/z 255[M+H]+。
保持時間:1.184分(分析条件PH−FA05−4)。

LRMS(ESI):m/z 594[M+H]+。

LRMS(ESI):m/z 470[M+H]+。
保持時間:1.155分(分析条件PH−TFA05−2)。

LRMS(ESI):m/z 370[M+H]+。
保持時間:0.882分(分析条件PH−TFA05−2)。

LRMS(ESI):m/z 594[M+H]+。

LRMS(ESI):m/z 257[M+H]+。
保持時間:0.839分(分析条件PH−FA05−3)。

LRMS(ESI):m/z 782[M+H]+。

LRMS(ESI):m/z 286[M+H]+。
保持時間:0.877分(分析条件PH−FA05−1)。

LRMS(ESI):m/z 272[M+H]+。
保持時間:0.765分(分析条件PH−FA05−5)。

LRMS(ESI):m/z 644[M+H]+。
保持時間:1.211分(分析条件PH−TFA05−2)。

LRMS(ESI):m/z 544[M+H]+。
保持時間:0.960分(分析条件PH−FA05−1)。

LRMS(ESI):m/z 797[M+H]+。
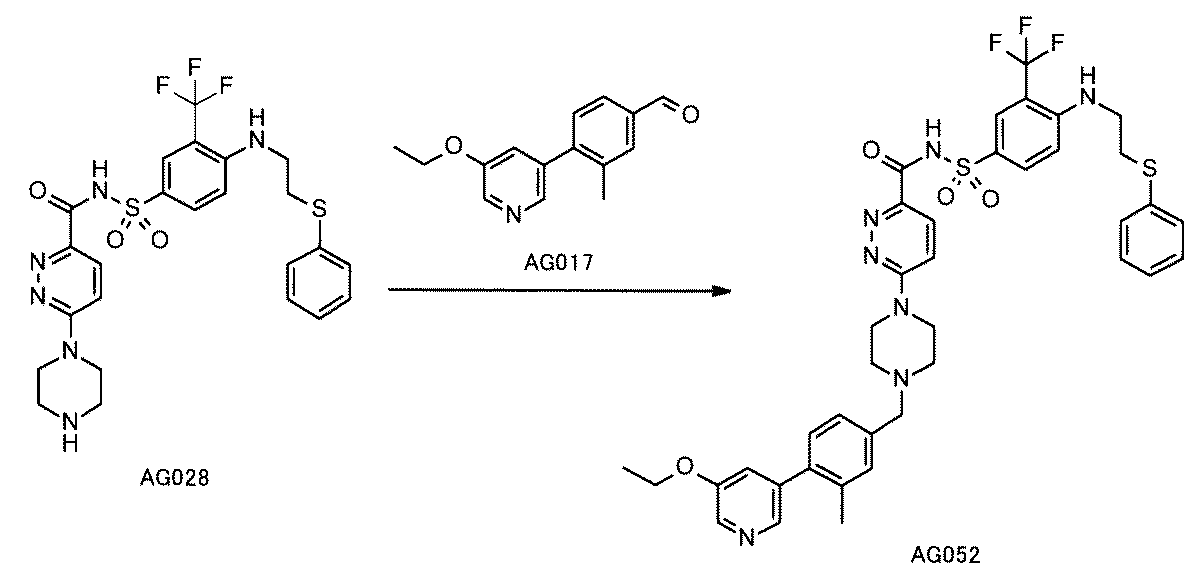
LRMS(ESI):m/z 792[M+H]+。
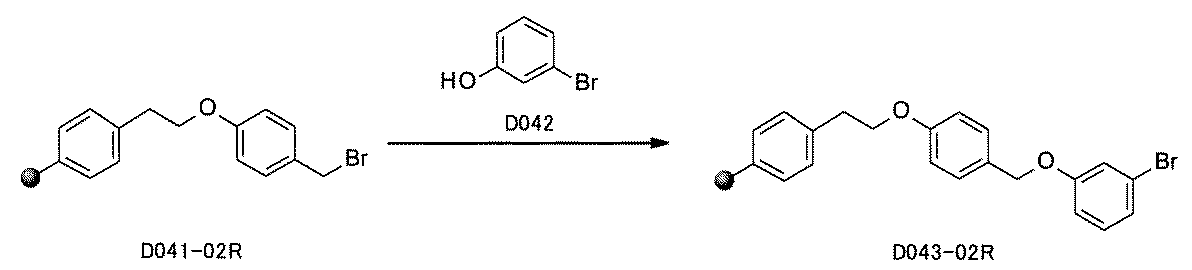
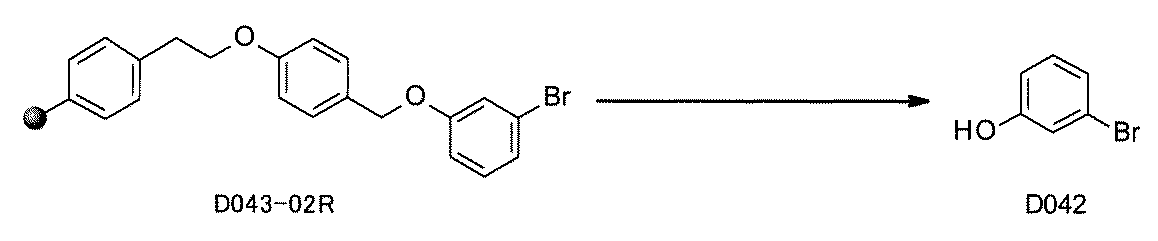
化合物D042
保持時間:0.893分(分析条件 SMD−FA05−1、290nm)。
極大吸収波長: 276 nm。


化合物D044
LCMS:m/z 174[M+H]+。
保持時間:0.573分(分析条件 SMD−FA05−1、290 nm)。


LCMS:m/z 213[M+H]+。
保持時間:0.927分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 214[M+H]+。
保持時間:0.534分(分析条件 SMD−FA05−1、290 nm)。


LCMS:m/z 443[M+H]+。
保持時間:0.797分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 444[M+H]+。
保持時間:0.610分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 403[M+H]+。
保持時間:0.649分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 404[M+H]+。
保持時間:0.457(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 738[M+H]+。
保持時間:0.932分(分析条件 SMD−FA05−1、290nm)。


LCMS:m/z 739[M+H]+。
保持時間:1.866分(分析条件 SMD−TFA05−long、5分、290nm)。




1H−NMR(400MHz,CD2Cl2)δ8.81(d,J=2.0Hz,1H),8.63(t,J=5.2Hz,1H),8.06(dd,J=9.2,2.0Hz,1H),7.64(d,J=8.8Hz,2H),7.56(d,J=8.0Hz,2H),7.41−7.39(m,4H),7.35−7.22(m,4H),7.16(d,J=7.6Hz,1H),7.11(t,J=2.0Hz,1H),6.88−6.81(m,4H),4.08(q,J=7.2Hz,2H),3.59(q,J=6.4Hz,2H),3.50−3.45(m,1H),3.32−3.31(m,2H),3.23(t,J=6.4Hz,2H),3.09−3.04(m,2H),2.68−2.64(m,2H),2.58−2.52(m,2H),1.44−1.40(m,6H)。
LCMS:m/z 766[M+H]+。
保持時間:1.044分(分析条件 SMD−FA05−1、290nm)。
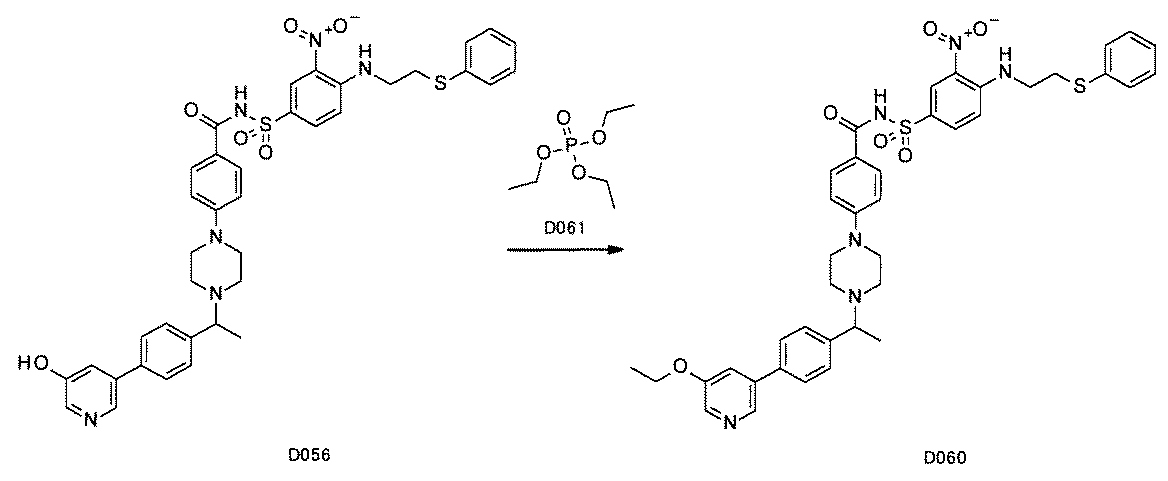

1H−NMR(400MHz,CD2Cl2)δ8.69(d,J=2.0Hz,1H),8.42(d,J=1.6Hz,1H),8.36(t,J=5.6Hz,1H),8.23(d,J=2.8Hz,1H),8.03(dd,J=8.8,2.0Hz,1H),7.89(d,J=8.8Hz,2H),7.56(d,J=8.4Hz,2H),7.47−7.37(m,5H),7.33−7.21(m,3H),6.76(d,J=9.2Hz,2H),6.76(d,J=9.2Hz,1H),4.15(q,J=7.2Hz,2H),3.53(q,J=6.8Hz,2H),3.45(q,J=6.8Hz,1H),3.26−3.18(m,2H),3.13−3.09(m,2H),2.68−2.63(m,2H),2.56−2.51(m,2H),1.81−1.78(m,2H),1.45(t,J=6.8Hz,3H),1.40(d,J=6.8Hz,3H)。
LCMS:m/z 767[M+H]+。
保持時間:2.020分(分析条件 SMD−TFA05−long、290nm)。
表AK−01に示す実施例1ならびに実施例6にて合成した化合物のBcl−2に対する化合物の結合親和性を解析するため、表面プラズモン共鳴法を用いた。装置としてBiacore T200(GE Healthcare)用い、ランニングバッファーには1mM DTT、3mM EDTA、0.01%Tween20及び4%DMSOが添加されたHBS(10mM HEPES−NaOH、150mM NaCl、pH7.4)を用いた。ヒトBcl−2タンパク質(Novus Biologicals)を、EZ−Link Sulfo−NHS−LC−LC−ビオチン(Thermofisher scientific)を用いてビオチン化し、Sensor Chip SA(GE Healthcare)の表面に固定化した。その後、複数の濃度の化合物溶液およびランニングバッファー(ブランク)を、Bcl−2固定化表面に添加し、化合物の結合レスポンスを得た。測定は15℃で行った。
得られたセンサーグラムはBiacore T200 Evalution Softwareにて処理し、各化合物のBcl−2に対する平衡解離定数KDを以下の[表AK−02]、[表AK−03]に示したように決定した。

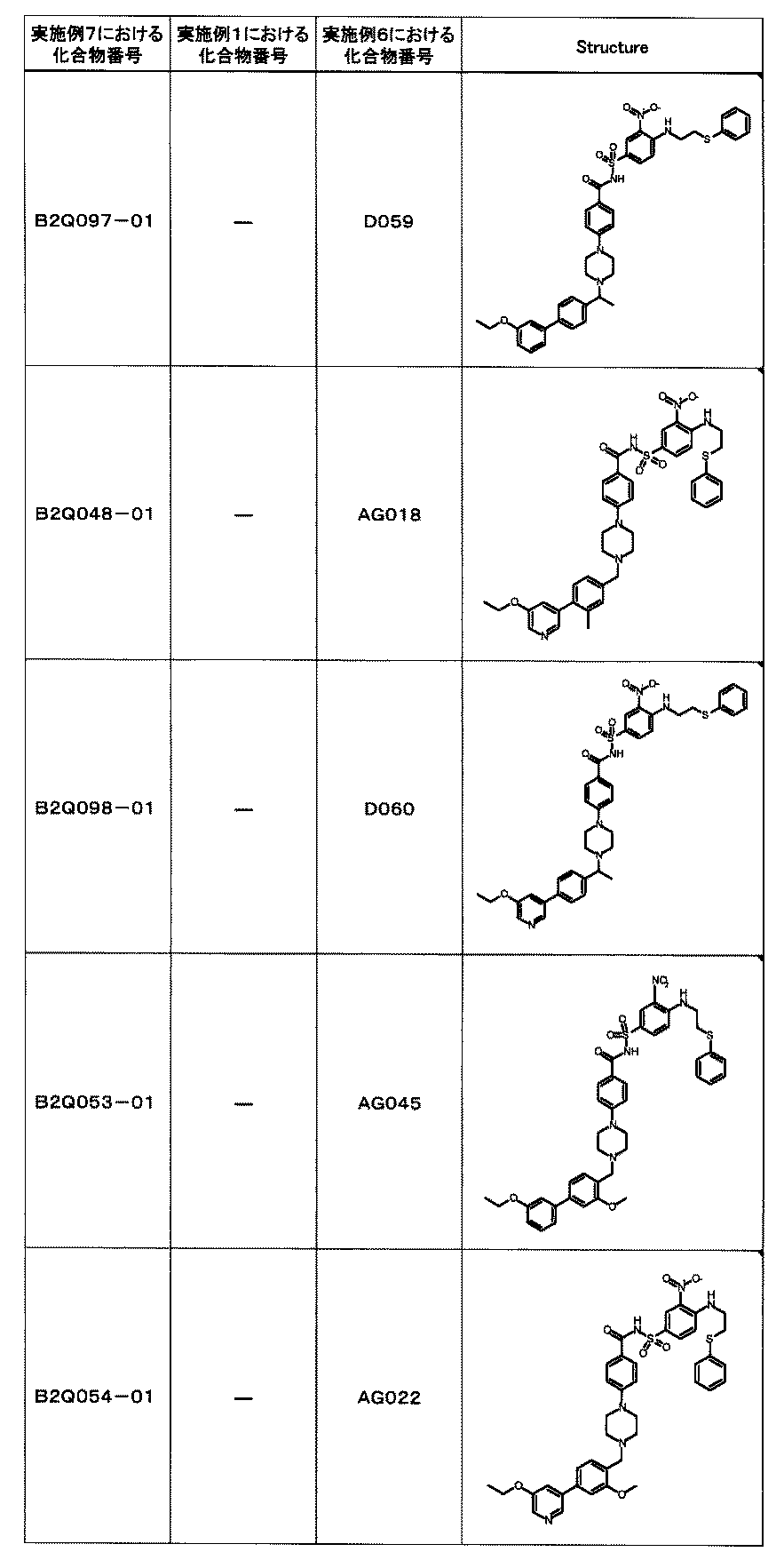


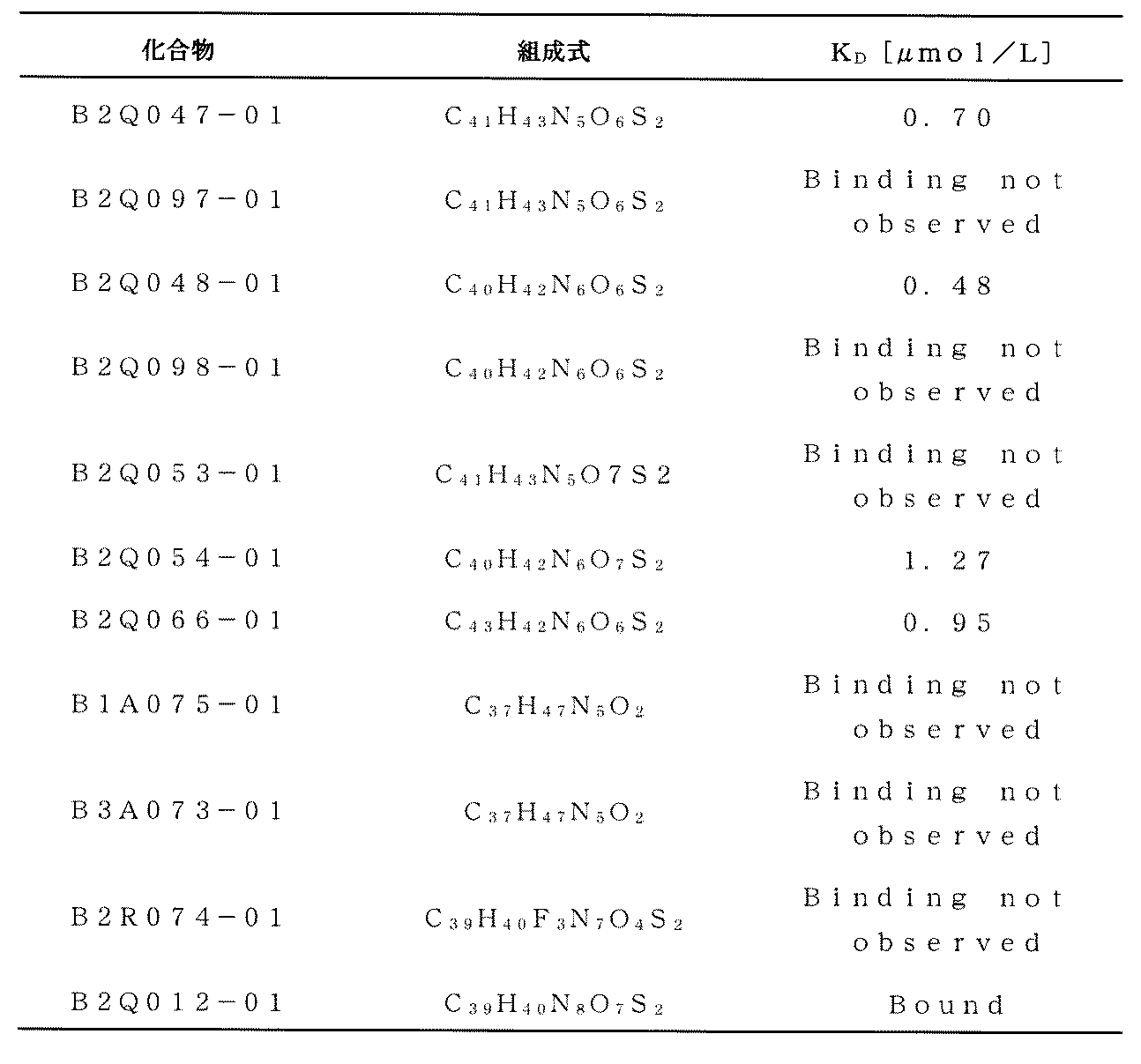
「コアブロックの多様性」と「リンカーの多様性」を掛け合わせ、幅広い範囲を一挙に探索するライブラリを新たに合成し、AS−MSにてBcl−2と結合する化合物を同定した(実施例5)。さらに同定した化合物を個別で合成し、SPRによりBcl−2と特異的に結合することを明らかになった。式AK−01に示すように、SPRにより結合が確認された化合物B2Q047−01、B2Q048−01は共に、ピペラジン環の結合したsp3性炭素原子と分子末端の芳香環の置換様式がp−置換の関係になっている。一方で、既知のBcl−2阻害剤の構造に目を向けると式AK−02に示すように、o−置換の関係の化合物の報告が971化合物に及ぶのに対し、p−置換の化合物の報告は2例に限られる(2020年11月5日時点でのReaxysis(登録商標)による部分構造検索結果)。またp−置換の化合物の活性に関する情報の報告は確認されていない。つまり、特定の理論および推測に限定されないが、p−置換の化合物は誘導体化の過程で活性が減弱する誘導体群として認識された可能性が高く、その後広範囲の誘導体化は実施されていない。
さらに、公知文献(ACS Omega 2019,4,5,8892−8906)にて報告されているo−置換体(ABT−737 analogue)とBcl−2とのX線結晶構造情報(PDB ID;6QGG,図3)に基づいた場合、ピペラジン環の窒素原子が結合したsp3性炭素に対してp−の方向に置換基を導入する発想には至らないばかりか、活性向上を目的にする場合にはp−の方向に置換基を導入することが阻害要因になりうることが示されている。図3は、o−置換の関係の化合物とBclファミリータンパク質との複合体情報の一例であり、この例に限定はされないが、o−置換の関係の化合物の結合様式を表す例である。

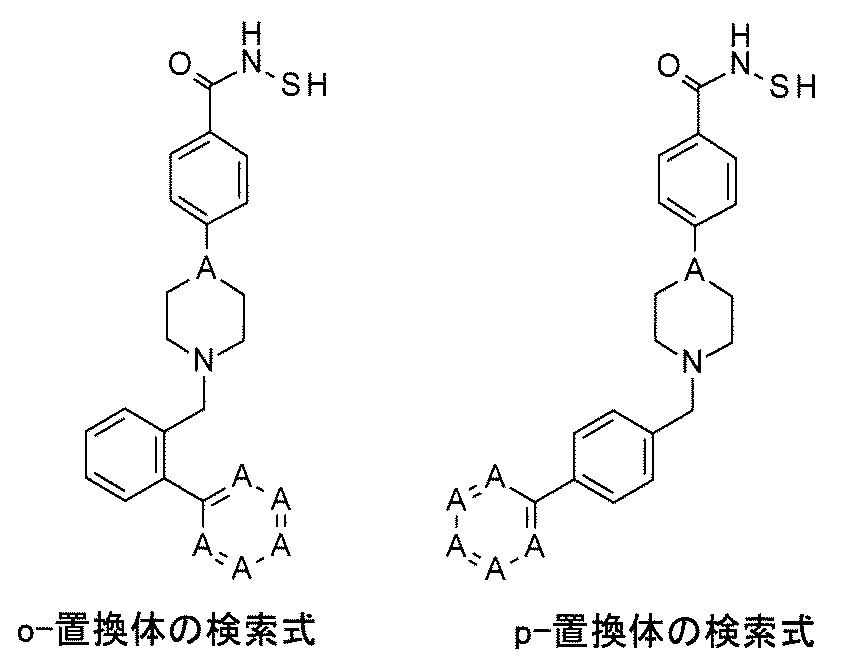

実施例4では実施例3で得た知見を活用した上で、ファーマコフォアを形成する「コアブロックの多様性」と「リンカーの多様性」を掛け合わせ、さらには置換様式の違いを組み合わせた多様な化合物の中から、1,200化合物をライブラリ化合物として設定し、ライブラリの合成を実施した。さらには、AS−MSにてBcl−2と結合する化合物を同定後、同定した化合物を個別で合成し、SPRによりBcl−2と特異的に結合することを明らかにした。従来の手法では探索しきれない範囲に存在し得る化合物の効果を検証することが可能であることを示し、ヒットジェネレーション、スキャホールドホッピングならびにFBDD(Fragment−Based Drug Discovery)の創薬において求められている課題を解決する手法の一態様を示した。
[式08−01]

実施例8にて実施する該ライブラリに含む「コアブロックの多様性」と「リンカーの多様性」について図4に示し、Split & mixによるライブラリ合成ルートに関して図5に示す。実施例8にて合成している混合物を示す場合には、混合物番号を利用することもある。例えば、「2−1−a1B01−0」の「2−1」はライブラリの種類を示し、「a1B01」は混合物の種類を示す。「−0」の「−0」は固相に担持された状態を示し、「−1」の場合には固相から切り出した状態を示す。また、「−2」の場合には固相からの切り出し後に官能基変換を行った状態を示す。混合物に含まれる各化合物を示す際は、IDを利用することもある。例えば、ID「2−1−a1B01−0−0001」の場合、「0001」は混合物「2−1−a1B01−0」中に含まれる任意の1化合物を特定する番号であり、IDにより混合物および含まれる化合物を特定する。また実施例8において、図4中におけるコアブロックAはA01で表される構造を有する。
[表8−0]実施例8で使用したビルディングブロック



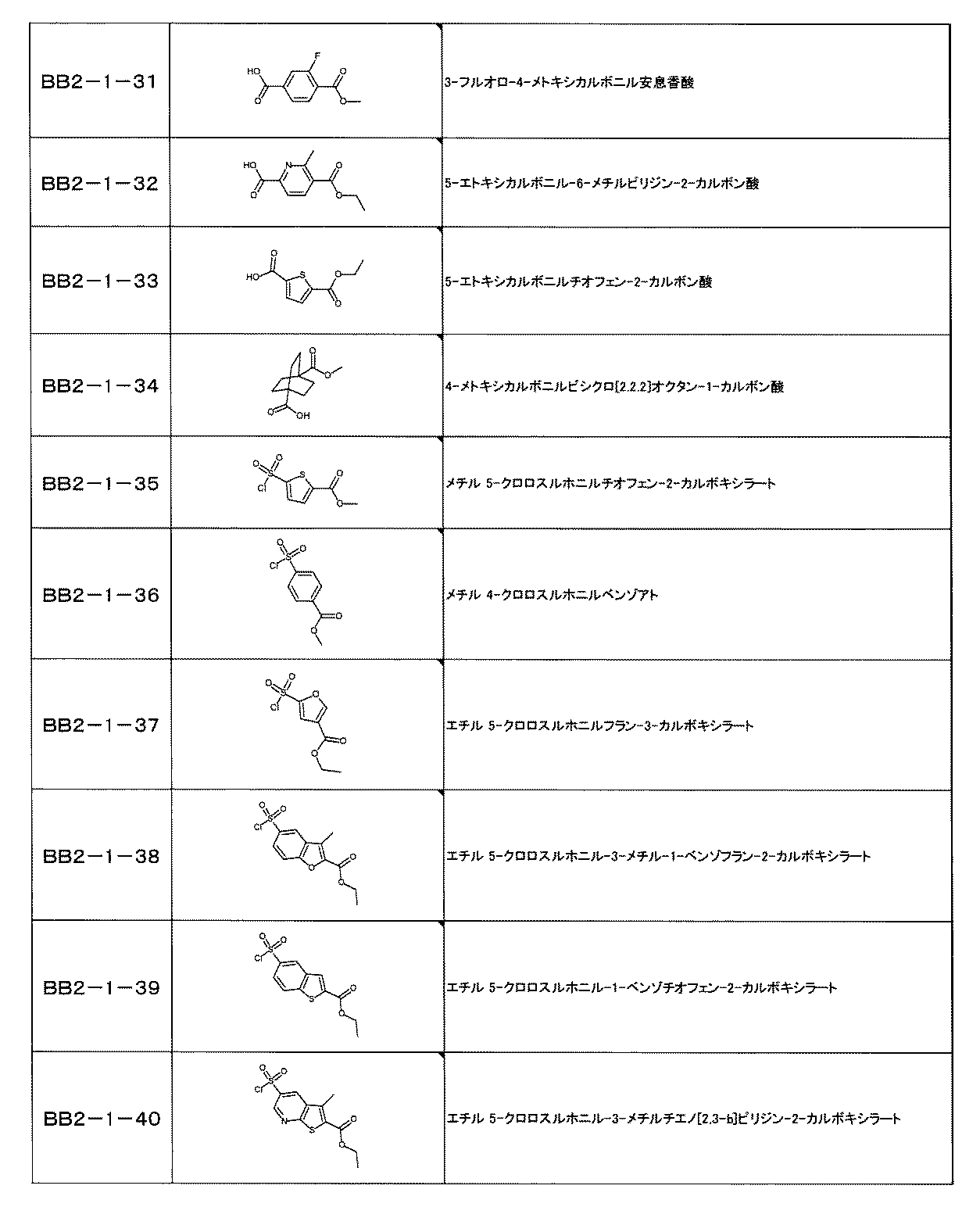

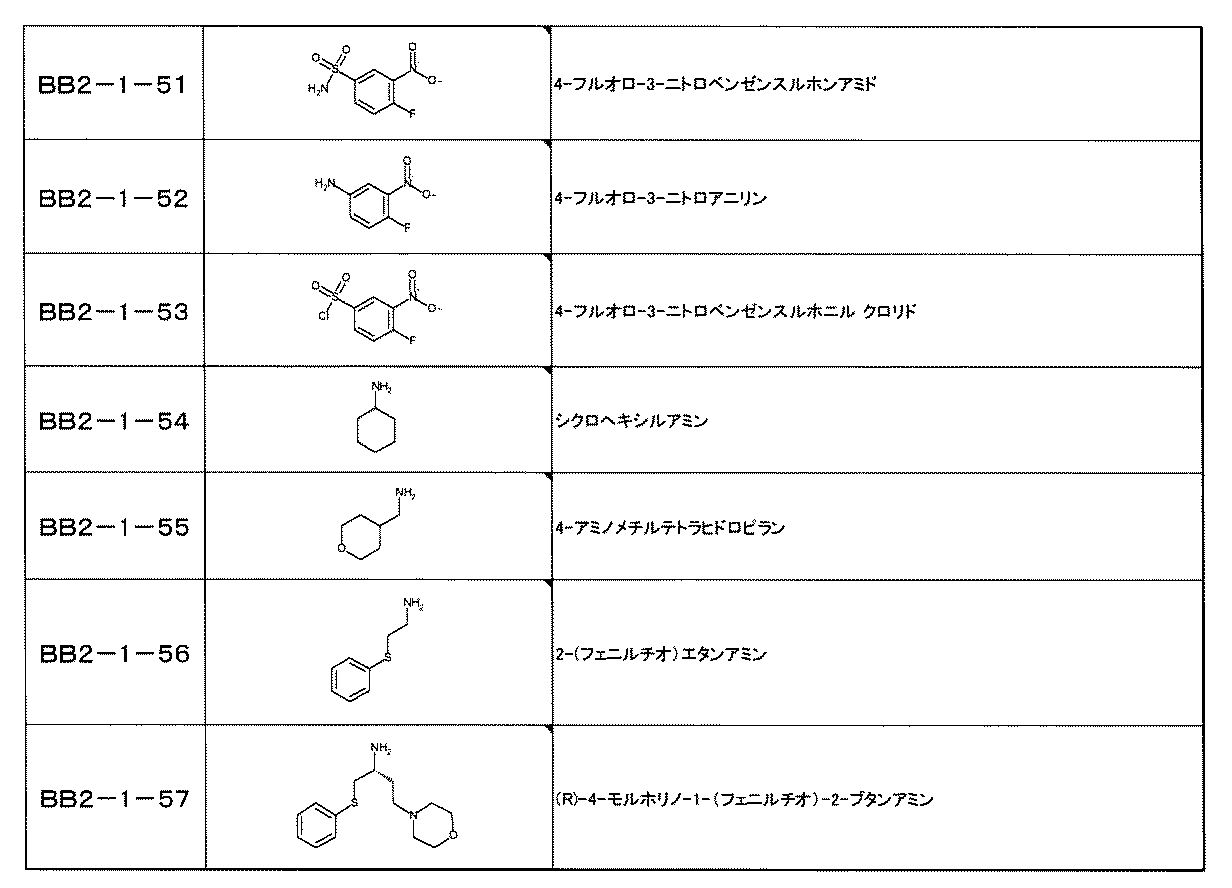
BB2−1−01を用いて工程a1を行った場合の反応式を以下に例示する。
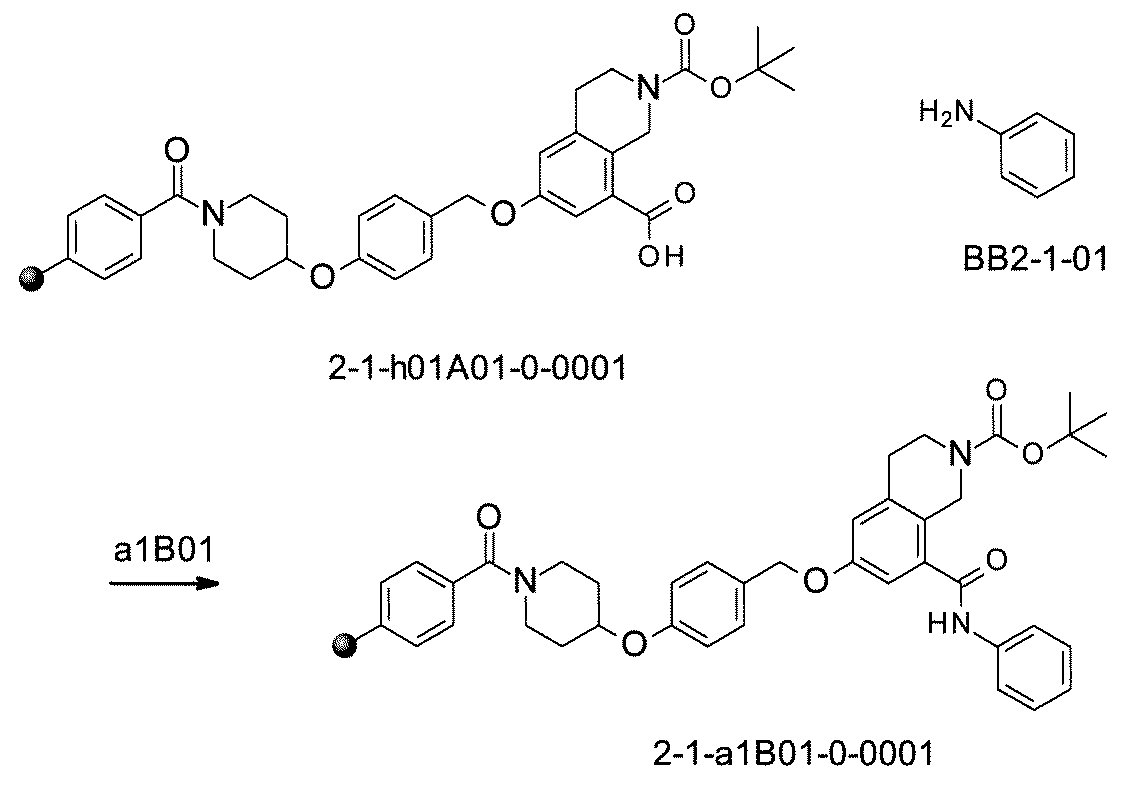
10個の4mLガラスバイアルに化合物2−1−h01A01−0−0001(実施例1においてD166−03Rと記載)(240mg、0.201mmol/g)とDCM(15.4mL)を加えた。[表8−1−1]に記載のアニリン(0.338mmol)を別個にそれぞれのガラスバイアルに加え、反応液が漏れないようにキャップをして室温で1時間振盪した。PipClU(0.145mmol)とNMI(0.145mmol)のMeCN(0.145mL)溶液(PipClUとNMIは共に1M)を加え、室温で3.5時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−1−1]に記載の化合物を得た。
窒素雰囲気下、洗浄後のレジンを少量フィルター付きピペットチップに移し、DMA(100μL)で3回、MeOH(100μL)で3回、DCM(100μL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(50μL)に5分間浸した。切り出し液をろ過し、固相をDMA(50μL)で洗浄した。得られたろ液をMeCN(100μL)で希釈することでLCサンプルを調製し、LCMS測定を行うことで化合物2−1−a1B01−1−0001の生成を確認した。

表中の表記方法について説明する。例えば[表8−1−2]では、工程a1で合成したそれぞれの化合物をIDで示し、化合物2−1−a1B01−1−0001~化合物2−1−a1B10−1−0001の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は[図4]に示す。[表8−1−2]においては、「▲×▼」と「b」は化学式で表す。

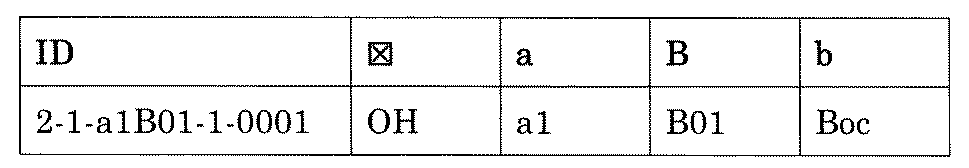

BB2−1−11を用いて工程a2を行った場合の反応式を以下に例示する。

10個の8mLのガラスバイアルに化合物D083−03R(240mg、0.200mmol/g)とNMP(4.8mL)を加え、室温で1時間振盪した。[表8−1−3]に記載のアルキン(0.480mmol)を別個にそれぞれのガラスバイアルに加えた。XPhosPd G4(41.3mg、0.048mmol)とジイソプロピルアミン(101μL、0.720mmol)を加え、60℃で1時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.2M NAC in NMP/水=5/1(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、0.05M NMM in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−1−3]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表8−1−4]では、工程a2で合成したそれぞれの化合物をIDで示し、化合物2−1−a2B11−1−0001~化合物2−1−a2B20−1−0001の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−1−4]においては、「▲×▼」と「b」は化学式で表す。

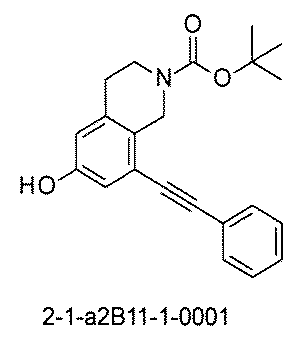


化合物2−1−d01B01−0−0001~化合物2−1−d20B20−0−0001を用いて工程d01~d20を行った後、固相混合によって混合物2−1−C00−0を得る場合の反応式を以下に例示する。

40個の8mLのガラスバイアルに[表8−2−1]に記載の固相原料(180mg、一種類の固相原料につき2バイアルを用いて、合計360mg使用)と2−メチルテトラヒドロフラン(3.6mL)を別個にそれぞれのガラスバイアルに加え、室温で1時間振盪した。DTBP(0.233mL、1.06mmol)とTm(OTf)3(436mg、0.707mmol)を加え、60℃で22時間振盪した。
得られた反応液に1M DMEDA in NMP(1.77mL)を加え、室温で1.5時間振とうした。40個の8mLのガラスバイアルに含まれる反応液と固相の懸濁液を、それぞれNMP(1.8mL)を用いて200mLのフィルター付きカラムに移してろ過し、0.2M DMEDA in NMP(144mL)で3回、NMP(144mL)で3回、0.05M TBAHSO4+DTBP in NMP(144mL)で3回、0.05M BTMG in NMP(144mL)で3回、0.05M DTBP in NMP(144mL)で3回、NMP/水=1/1(144mL)で3回、NMP(144mL)で3回、MeOH(144mL)で3回、DCM(144mL)で3回、ヘプタン(144mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−C00−0(7.49g、混合物の固相担持量:0.200mmol/g)を得た。

表中の表記方法について説明する。例えば[表8−2−2]では、混合物2−1−C00−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−C00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−1−2]においては、「▲×▼」と「b」は化学式で表す。





BB2−1−21を用いて工程b1を行った場合の反応式を以下に例示する。

11個の4mLのガラスバイアルに混合物2−1−c00−0(140mg、混合物の固相担持量:0.200mmol/g)とNMP(2.8mL)を加え、室温で1時間振盪した。[表8−3−1−1]に記載のアリールブロミド(0.280mmol)を別個にそれぞれのバイアルに加えた。RuPhos Pd G4(23.8mg,0.028mmol)と2M P2tBu in THF(0.210mL、0.420mmol)を加え、40℃で3時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(3mL)で3回、NMP(3mL)で3回、0.2M NAC in NMP/水=5/1(3mL)で3回、0.05M TBAHSO4+DTBP in NMP(3mL)で3回、0.05M BTMG in NMP(3mL)で3回、NMP/水=1/1(3mL)で3回、NMP(3mL)で3回、MeOH(3mL)で3回、DCM(3mL)で3回、ヘプタン(3mL)で3回洗浄し、得られた固相を減圧下で乾燥させた。
上述の反応および固相精製を[表8−3−1−1]に記載された回数行った。
上述した固相精製後の混合物が入った6本の6mLフィルターにNMP(2.8mL)を加え室温で1時間振盪した。[表8−3−1−1]に記載のキャッピング試薬を別個にそれぞれのフィルターに加えた。HOAt(38.1mg,0.280mmol)とDIC(87μL、0.560mmol)を加え、室温で4時間振盪することで、未反応のアミンをキャッピングした。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(3mL)で3回、NMP(3mL)で3回、0.05M TBAHSO4+DTBP in NMP(3mL)で3回、0.05M NMM in NMP(3mL)で3回、NMP/水=1/1(3mL)で3回、NMP(3mL)で3回、MeOH(3mL)で3回、DCM(3mL)で3回、ヘプタン(3mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、表に記載の混合物を得た。なお、混合物中にはキャッピングによって生じる、[表8−3−1−1]に記載した成分が含まれうる。
[表8−3−1−1]工程b1で使用した試薬と得られた化合物

表中の表記方法について説明する。[表8−3−1−2]~[表8−3−1−12]では、混合物2−1−b1C08−1~混合物2−1−b1C14−1および2−1−b1C29−1~混合物2−1−b1C32−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−b1C08−1~混合物2−1−b1C14−1および2−1−b1C29−1~混合物2−1−b1C32−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−3−1−2]~[表8−3−1−12]においては、「▲×▼」と「c」は化学式で表す。





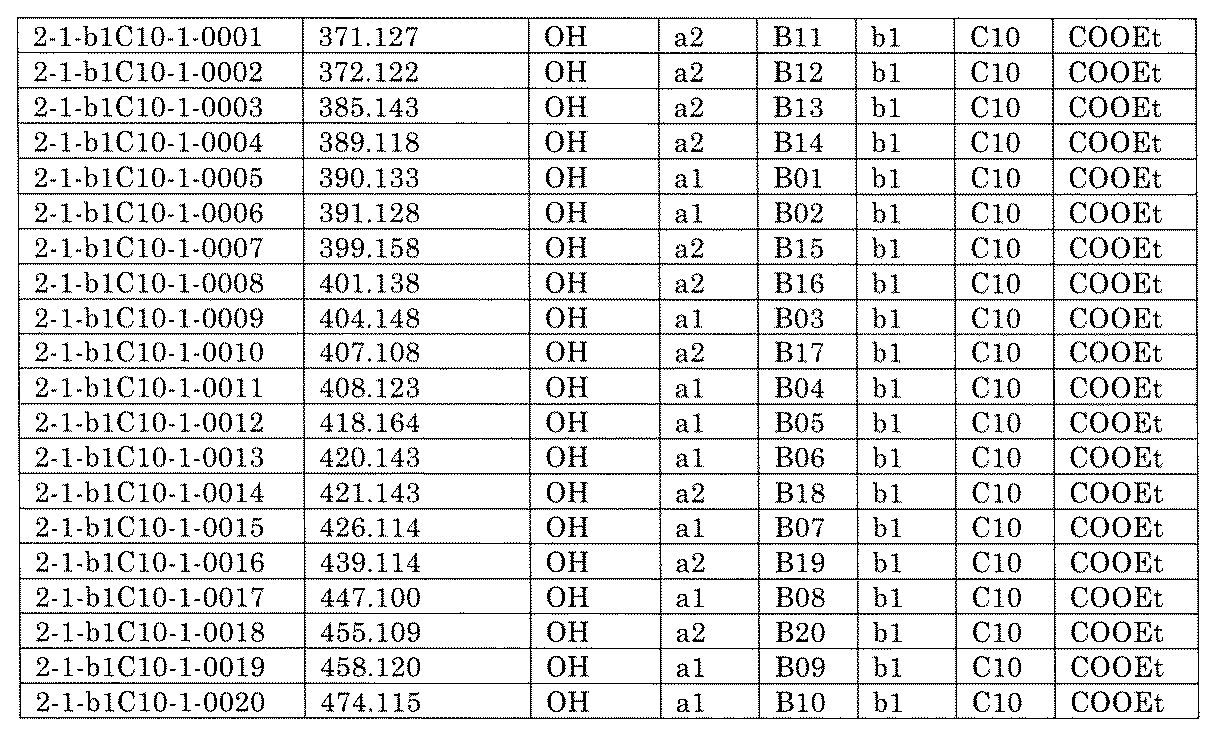

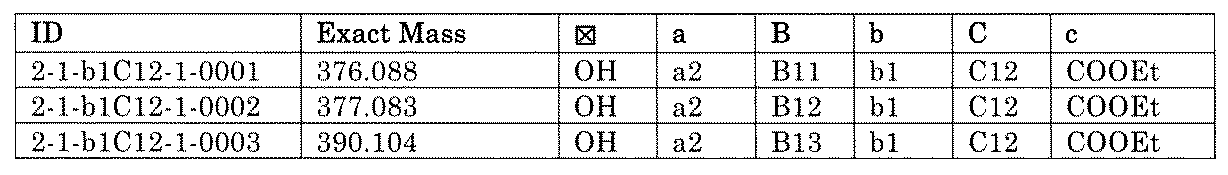




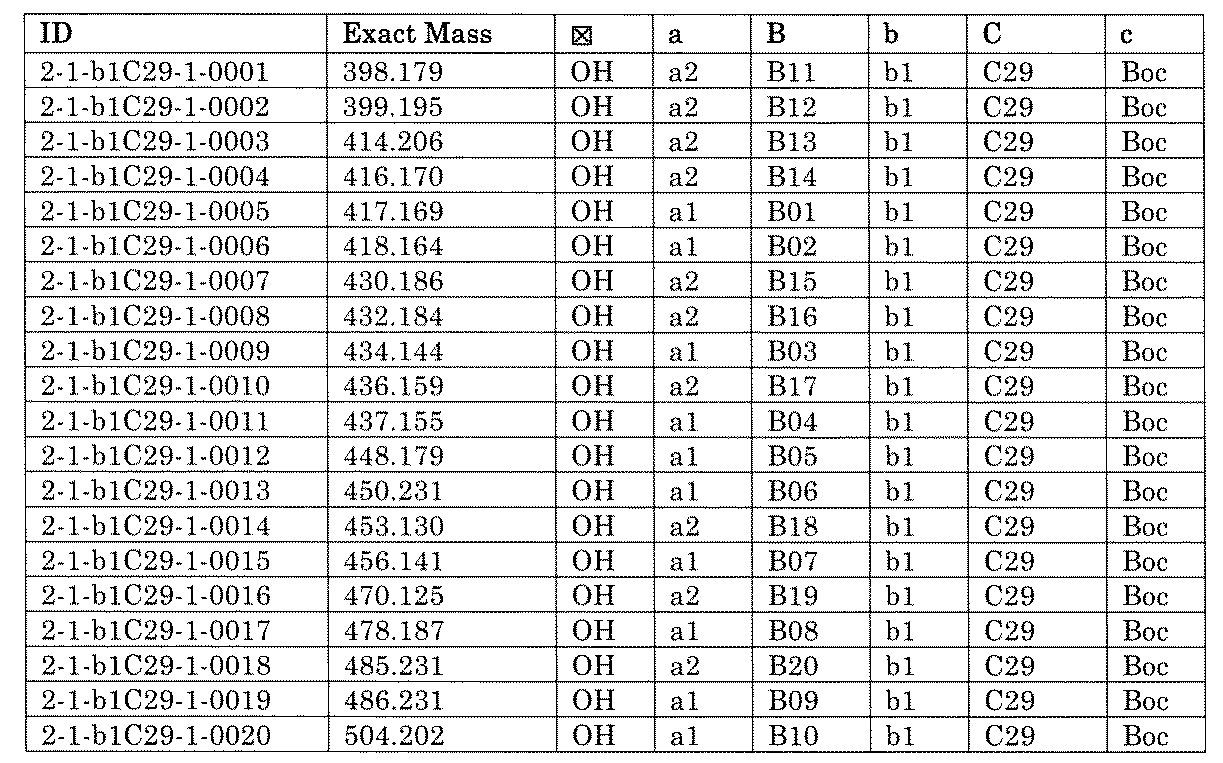
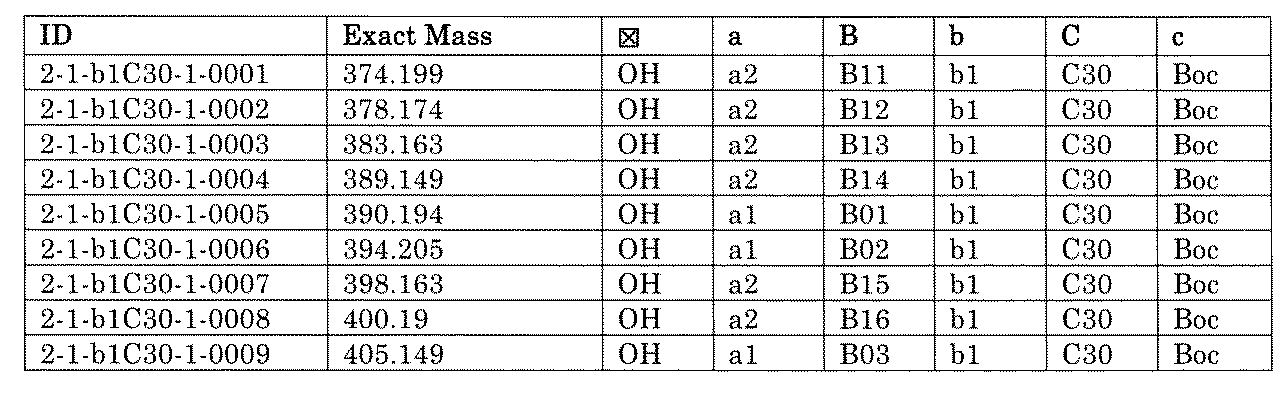




BB2−1−28を用いて工程b2を行った場合の反応式を以下に例示する。

13個の4mLガラスバイアルに混合物2−1−c00−0(135mg、混合物の固相担持量:0.200mmol/g)とNMP(2.7mL)を加え、室温で1時間振盪した。[表8−3−2−1]に記載のカルボン酸(0.270mmol)を別個にそれぞれのバイアルに加えた。HOAt(36.8mg、0.270mmol)とDIC(84μL、0.540mmol)を加え,室温で4時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、0.05M NMM in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−3−2−1]に記載の混合物を得た。
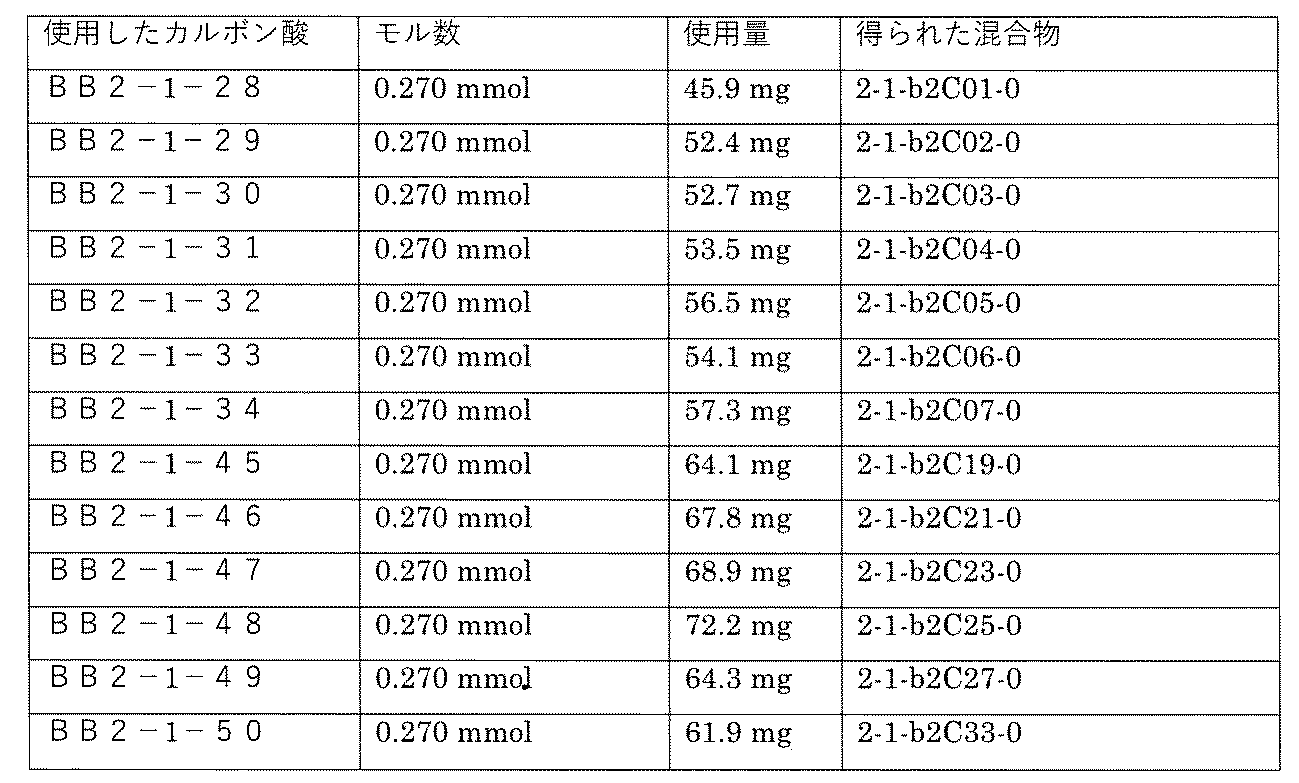

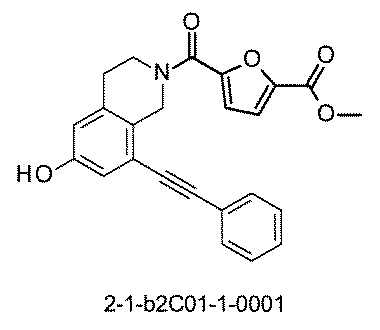


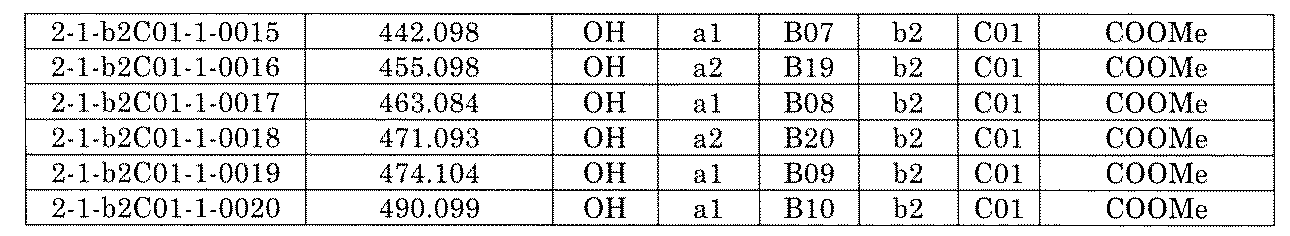















BB2−1−35を用いて工程b3を行った場合の反応式を以下に例示する。

7個の4mLガラスバイアルに2−1−c00−0(135mg、混合物の固相担持量:0.200mmol/g)とテトラヒドロフラン(2.7mL)とDIPEA(70.7μL)を加え、室温で1時間振盪した。[表8−3−3−1]に記載のスルホニルクロリド(0.270mmol)を別個にそれぞれのバイアルに加え、50℃で15時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.7mL)で3回、NMP(2.7mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.7mL)で3回、0.05M NMM in NMP(2.7mL)で3回、NMP/水=1/1(2.7mL)で3回、NMP(2.7mL)で3回、MeOH(2.7mL)で3回、DCM(2.7mL)で3回、ヘプタン(2.7mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、表に記載の化合物を得た。


表中の表記方法について説明する。[表8−3−3−2]~[表8−3−3−7]では、混合物2−1−b3C06−1~混合物2−1−b3C18−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−b3C06−1~混合物2−1−b3C18−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−3−3−2]~[表8−3−3−7]においては、「▲×▼」と「c」は化学式で表す。

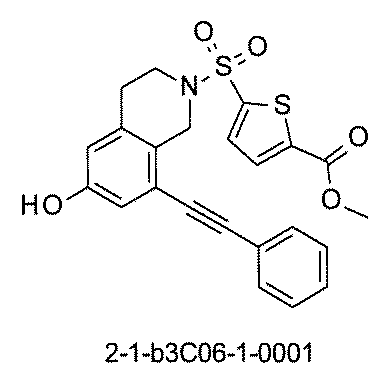

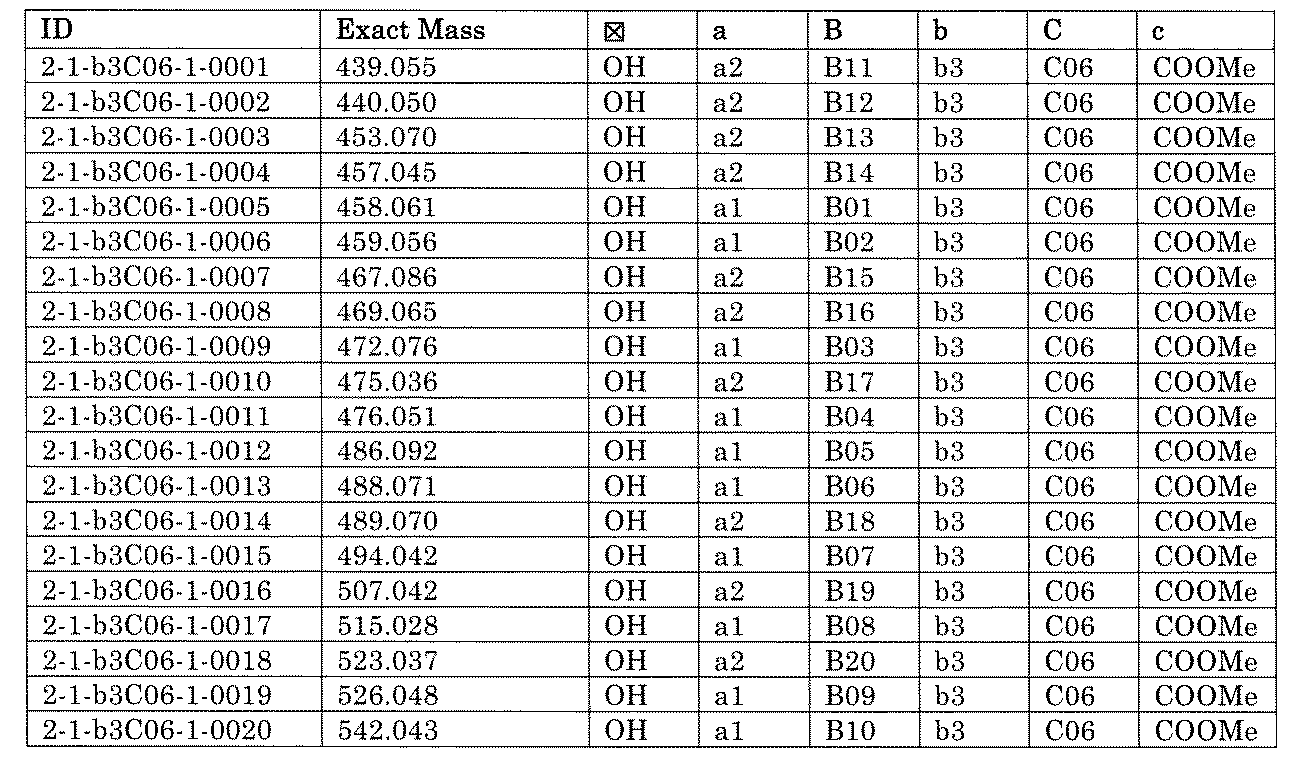





混合物2−1−b1C08−0を用いて工程h02を行った場合の反応式を以下に例示する。
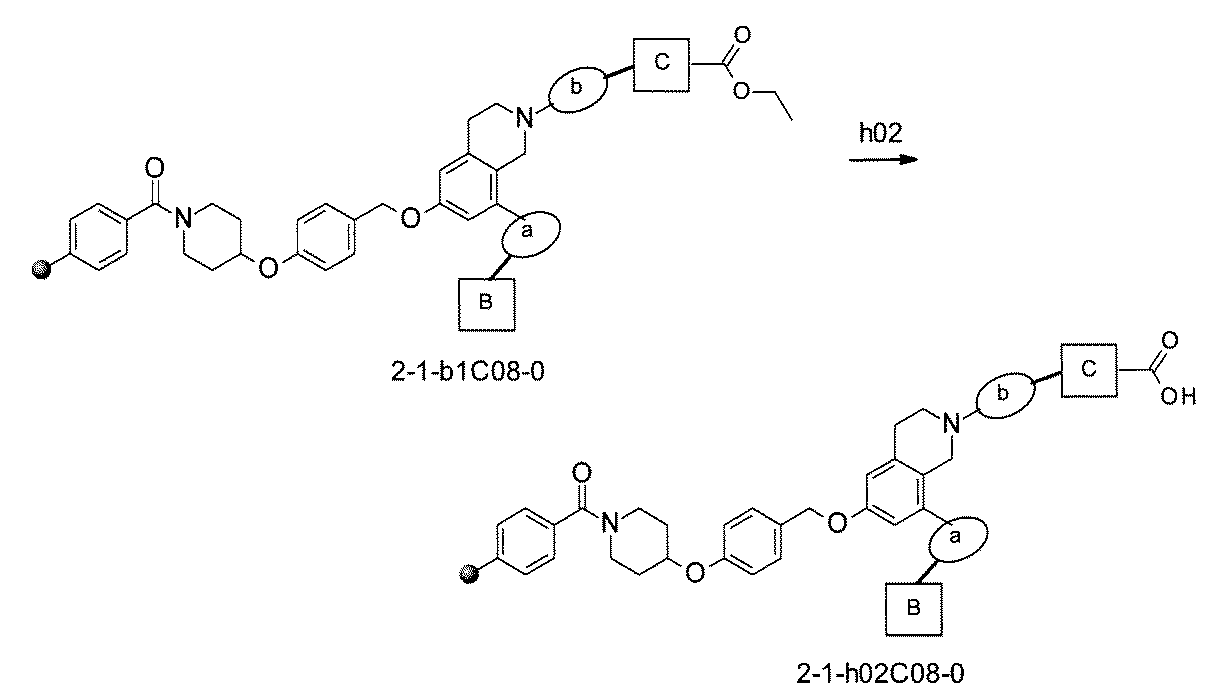
[表8−4−1−1]に記載の混合物が入った12本の6mLフィルター付きカラムに、NMP(1.89mL)と[表8−4−1−1]に示したアルコール(0.540mL)を加え、室温で1時間振盪した。1M TBAOH水溶液を[表8−4−1−1]に記載された量加え、[表8−4−1−1]に記載された温度と時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(2.7mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.7mL)で3回、NMP/水=1/1(2.7mL)で3回、NMP(2.7mL)で3回、MeOH(2.7mL)で3回、DCM(2.7mL)で3回、ヘプタン(2.7mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−4−1−1]に記載の混合物を得た。

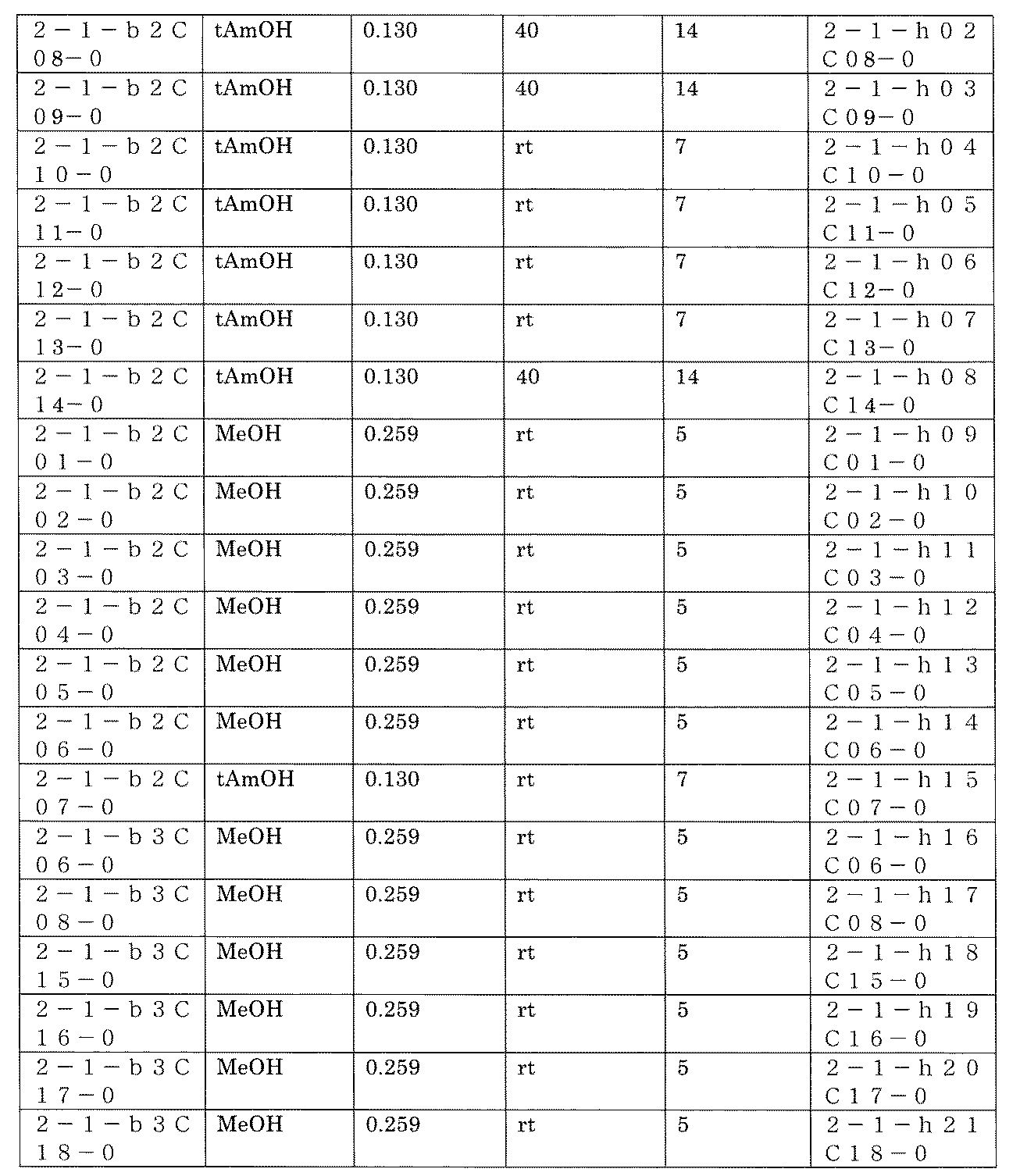
表中の表記方法について説明する。[表8−4−1−2]~[表8−4−1−21]では、混合物2−1−h02C08−1~混合物h21C18−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−h02C08−1~混合物h21C18−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−4−1−2]~[表8−4−1−21]においては、「▲×▼」と「c」は化学式で表す。
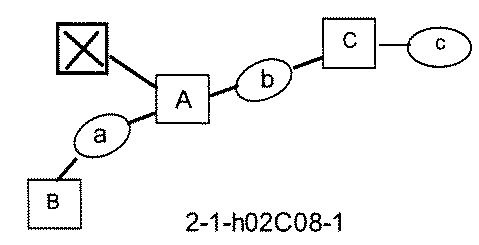
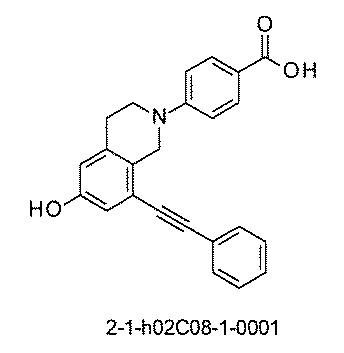



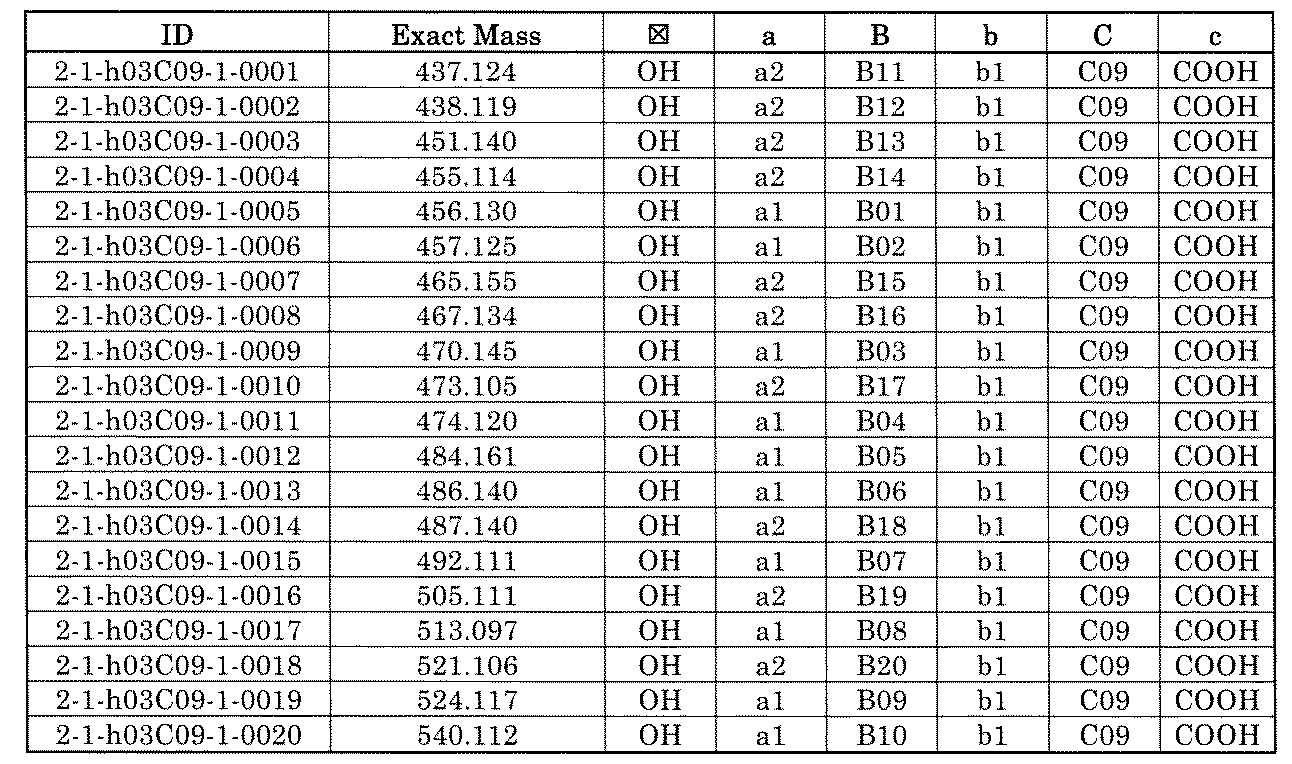



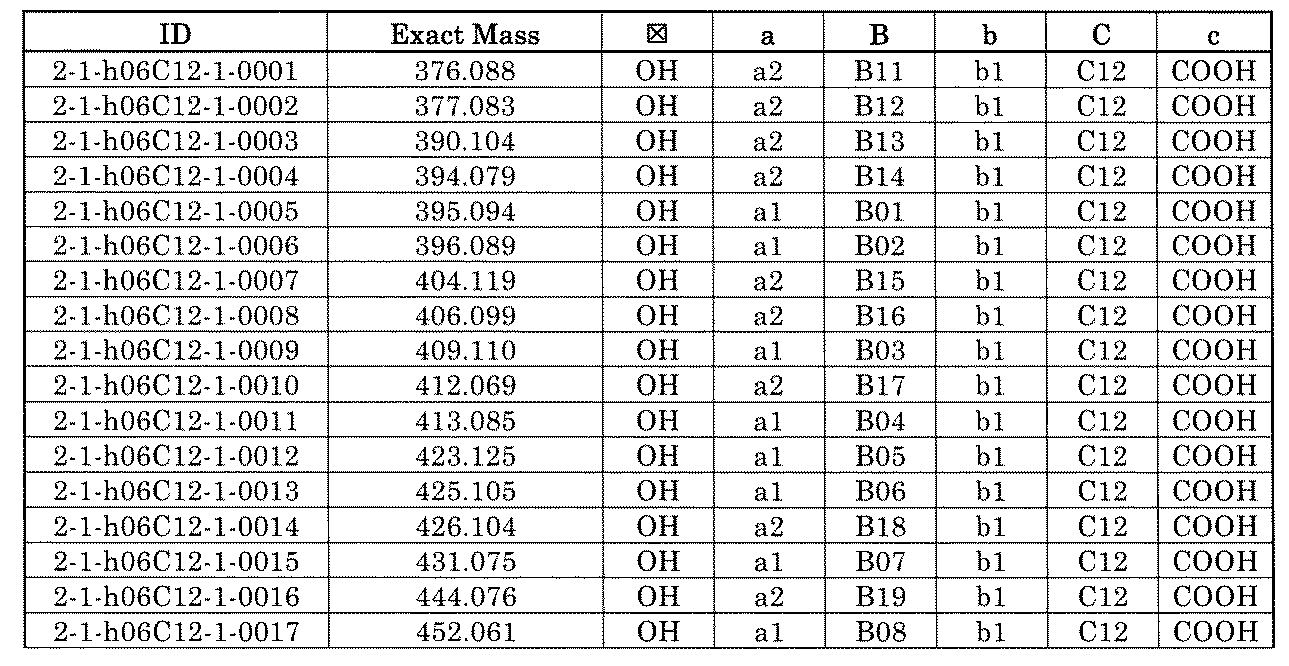




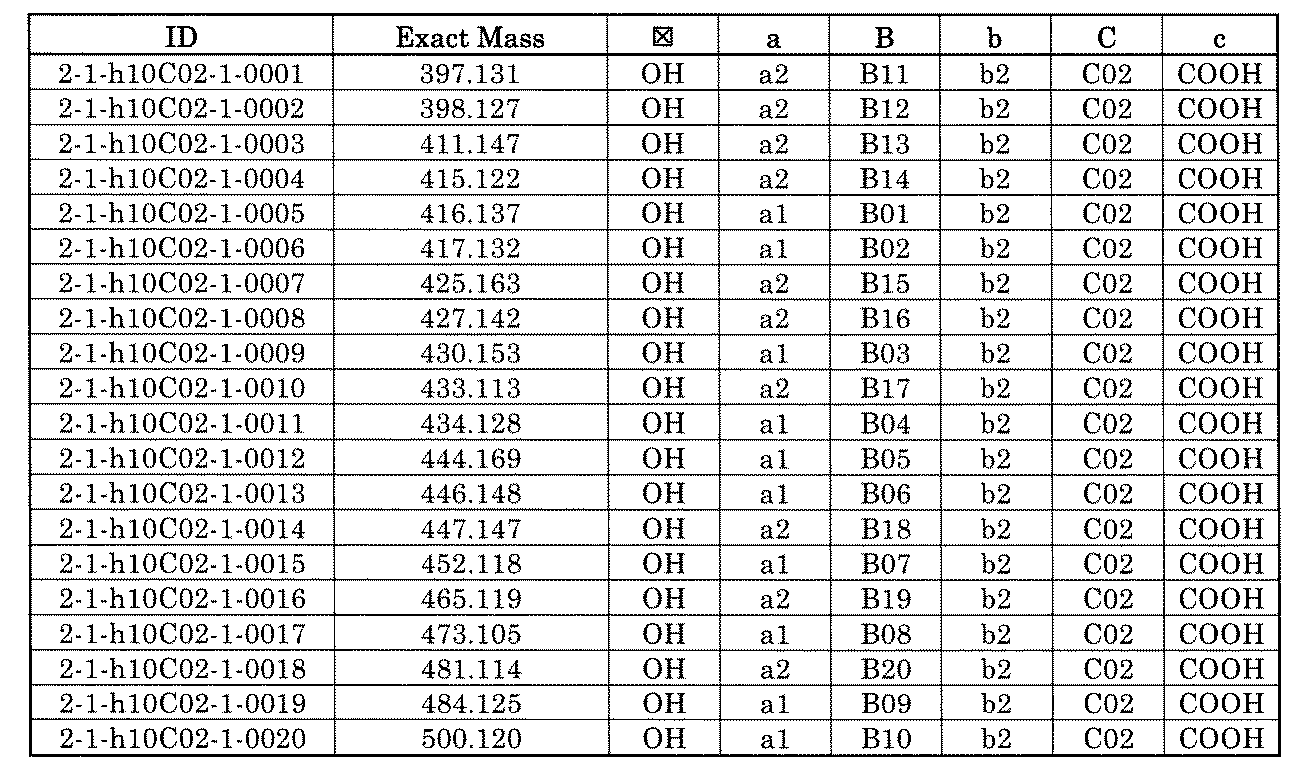
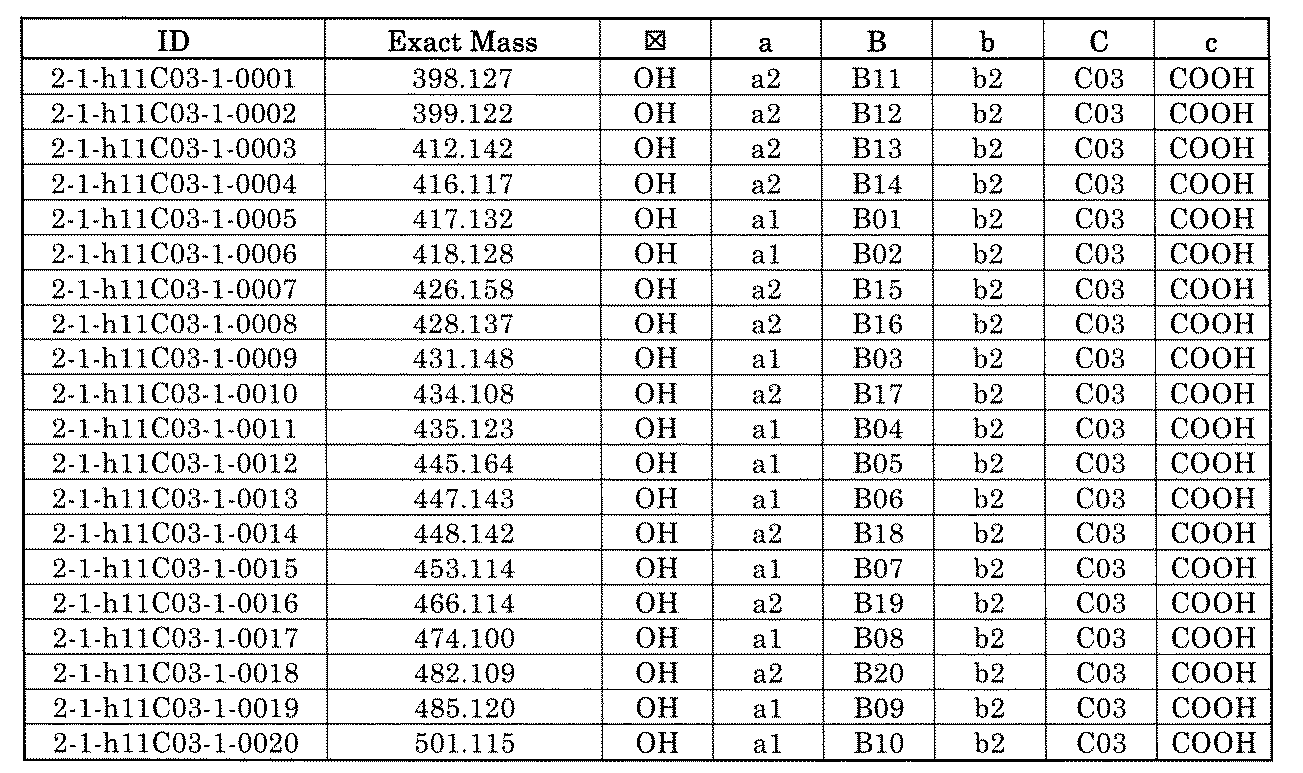
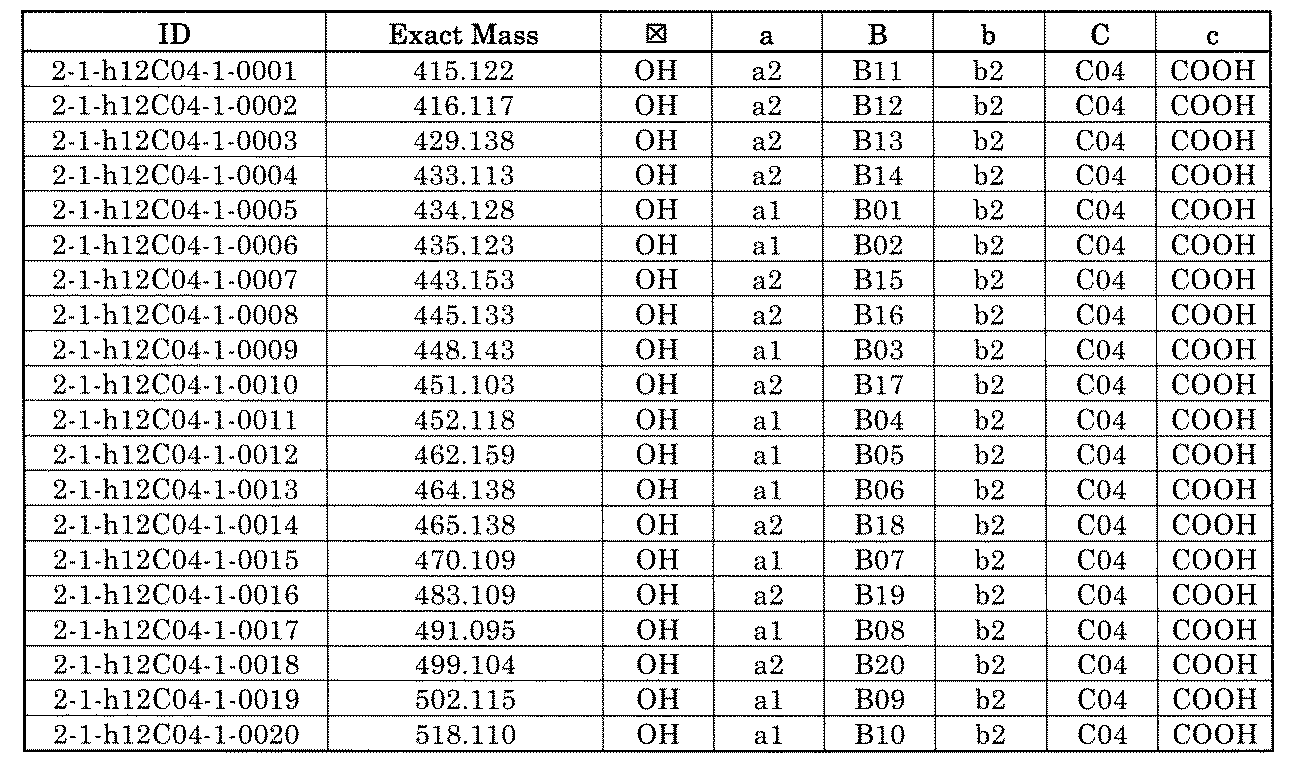

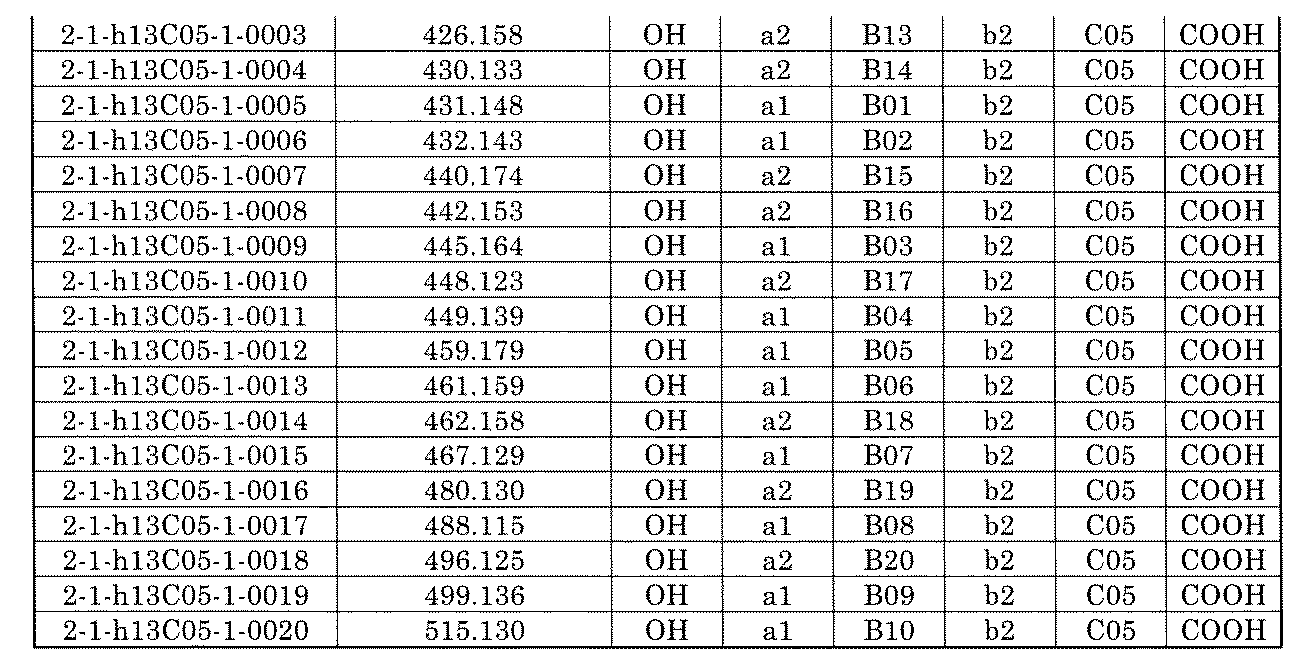


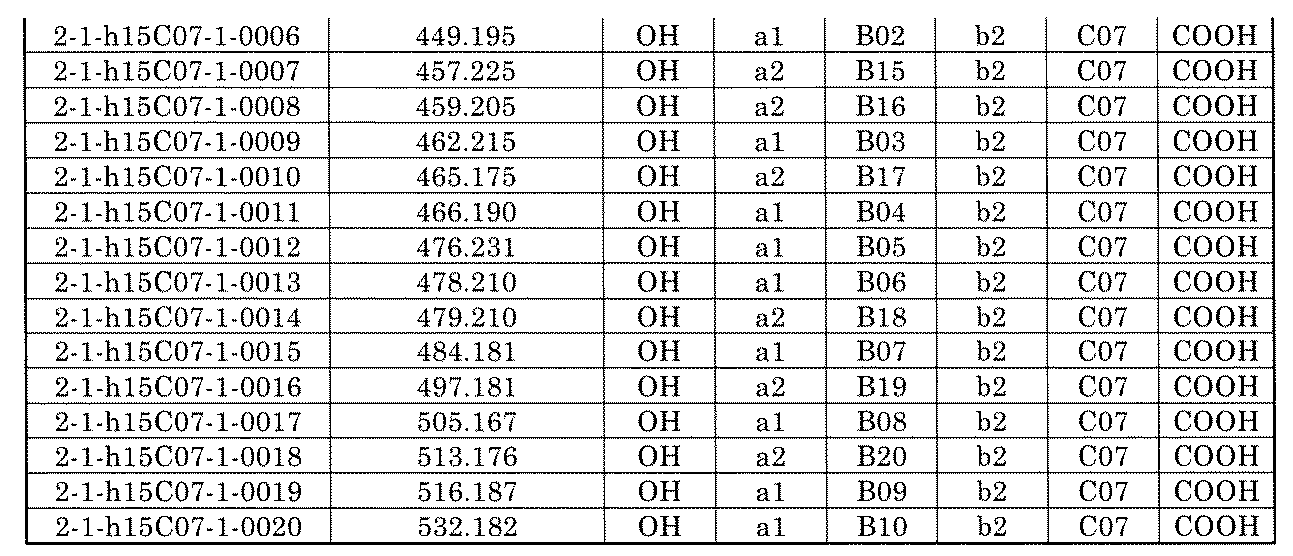


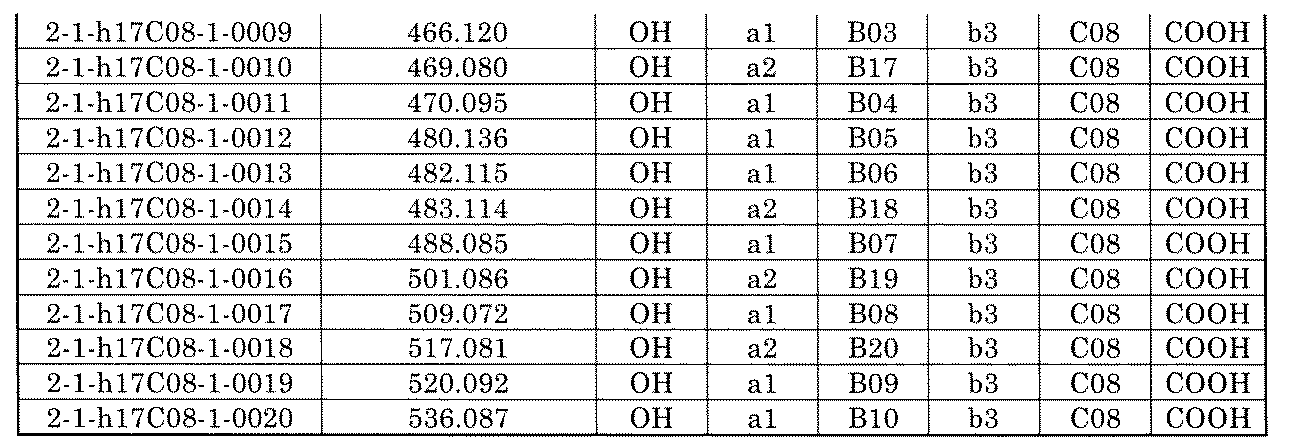
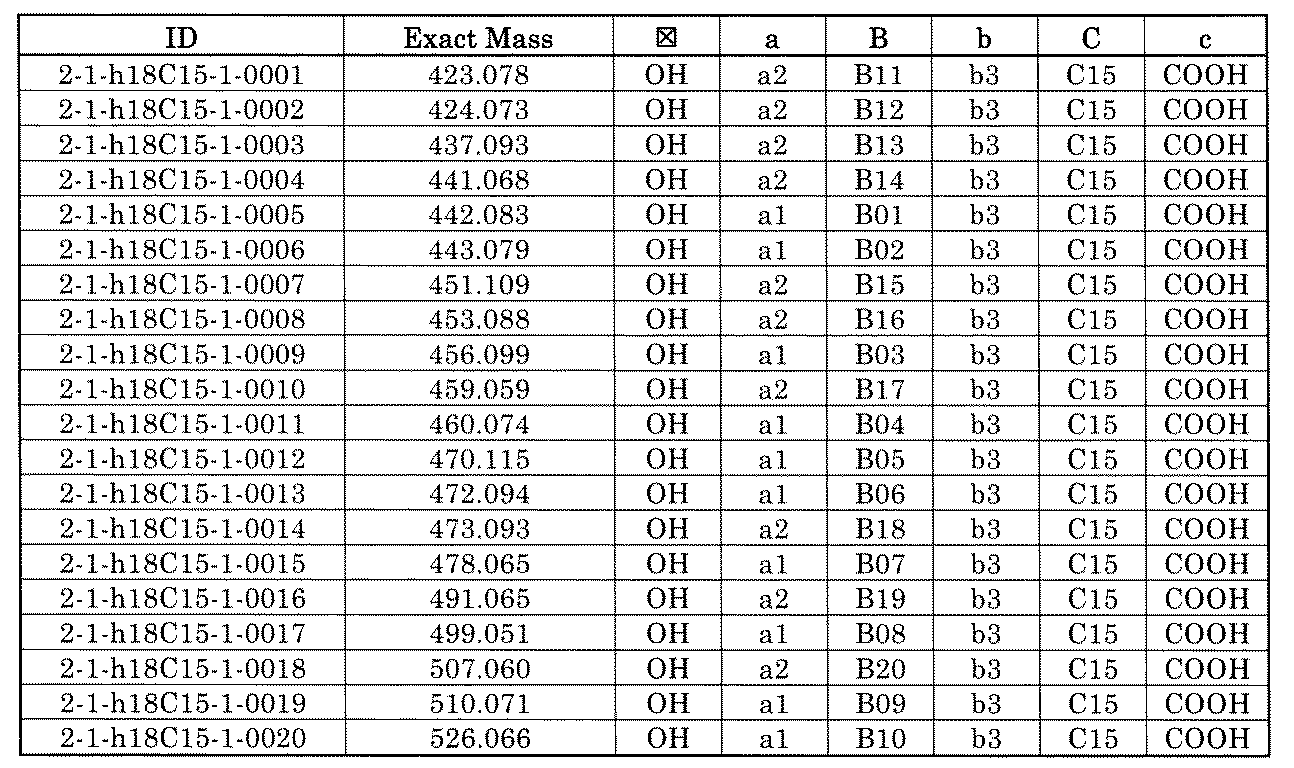



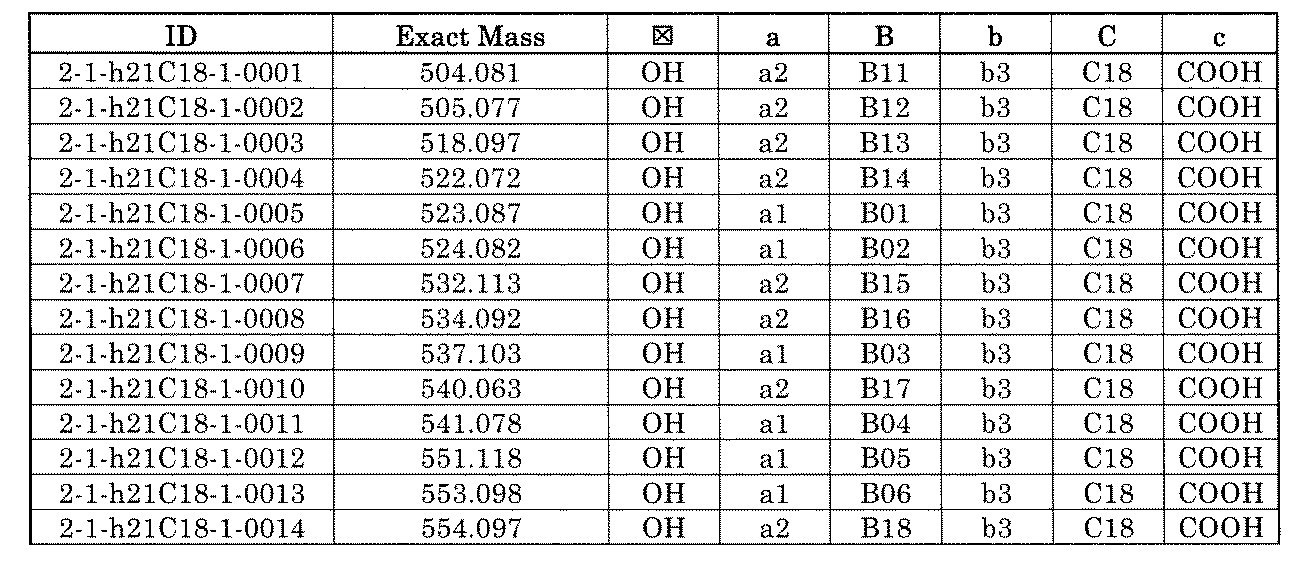

混合物2−1−b1C29−0を用いて工程d21を行った場合の反応式を以下に例示する。
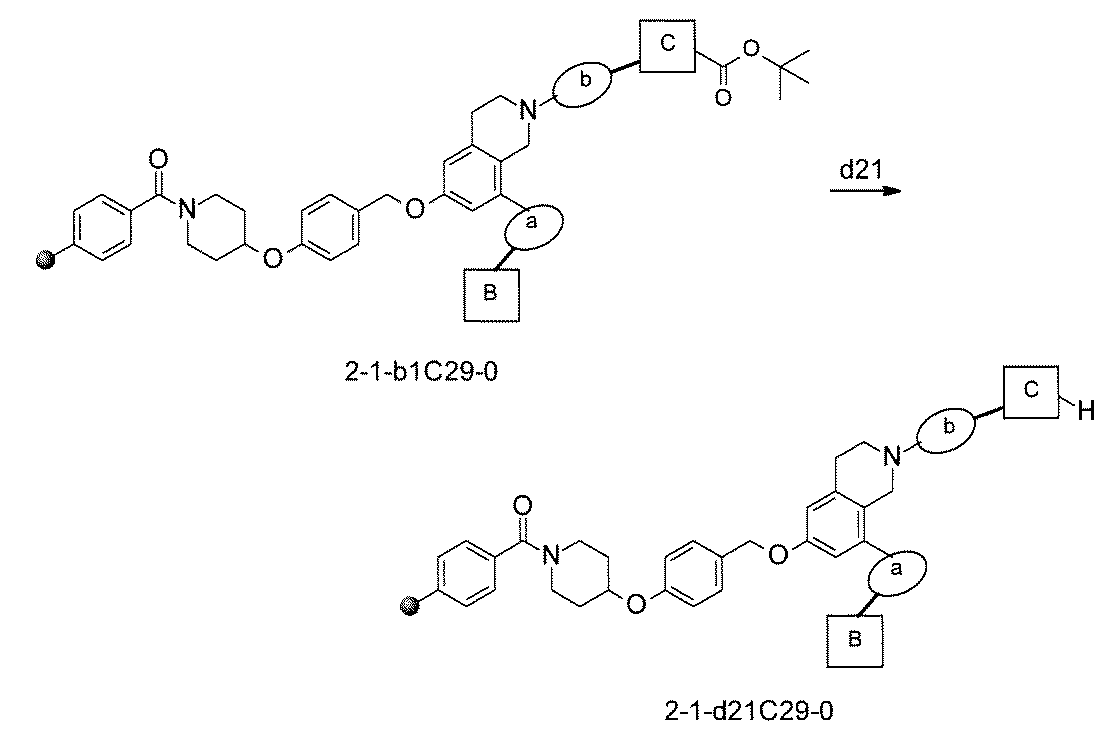
10個の4mLのガラスバイアルに、[表8−4−2−1]に記載の混合物(70mg)と2−メチルテトラヒドロフラン(1.4mL)を別個にそれぞれのガラスバイアルに加え、室温で1時間振盪した。DTBP(88μL、0.40mmol)とTm(OTf)3(165mg、0.267mmol)を加え、[表8−4−2−1]に記載された温度と時間にて振盪した。
得られた反応液に1M DMEDA in NMP(0.669mL)を加え、室温で1.5時間振とうした。NMP(700μL)を用いて反応液と固相の懸濁液を3mLのフィルター付きカラムに移してろ過し、0.2M DMEDA in NMP(1.5mL)で3回、NMP(1.5mL)で3回、0.05M TBAHSO4+DTBP in NMP(1.5mL)で3回、0.05M BTMG in NMP(1.5mL)で3回、0.05M DTBP in NMP(1.5mL)で3回、NMP/水=1/1(1.5mL)で3回、NMP(1.5mL)で3回、MeOH(1.5mL)で3回、DCM(1.5mL)で3回、ヘプタン(1.5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−4−2−1]に記載の混合物を得た。




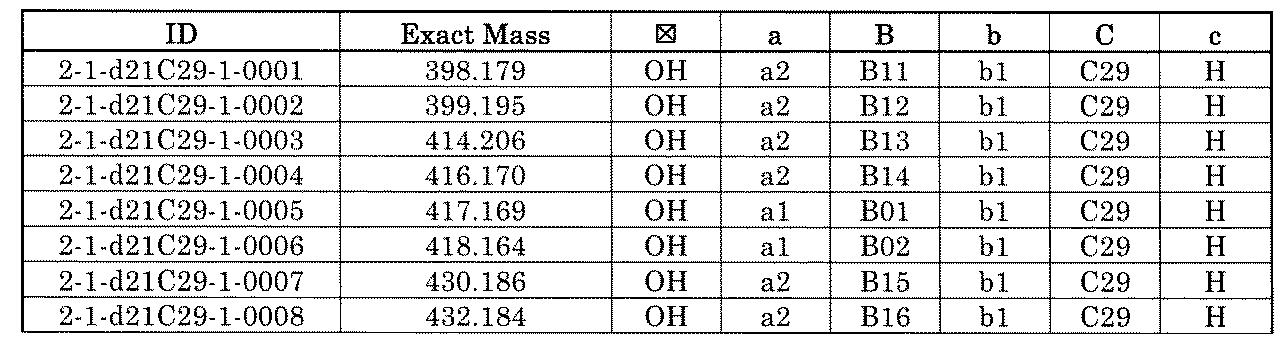



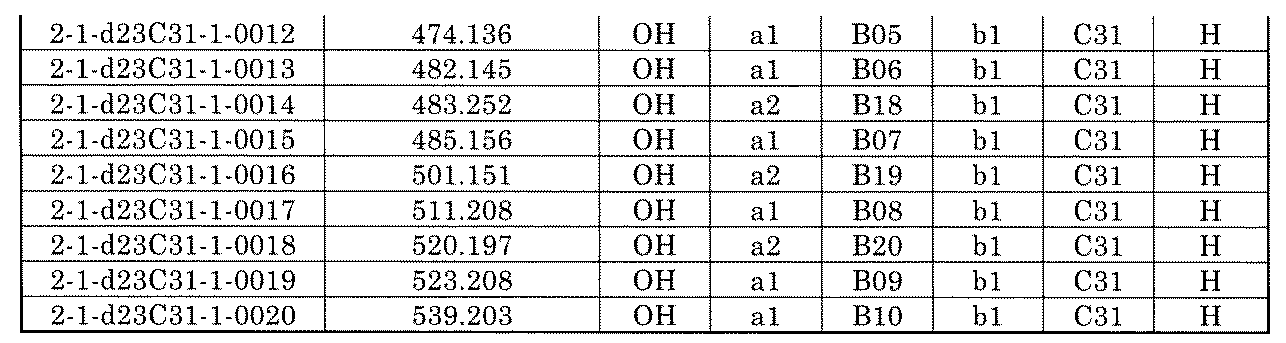


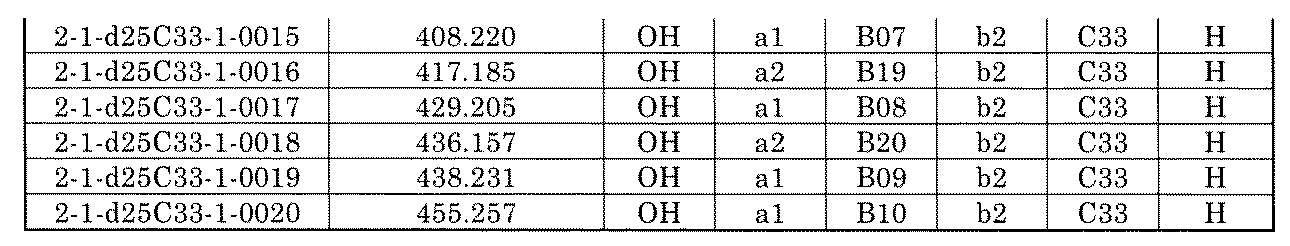






混合物2−1−c1D00−0を用いて工程c1D01を行った場合の反応式を以下に例示する。

20mLのフィルター付きカラムに[表8−5−1−1]に記載の20種類の混合物とDCM(10.5mL)を加え、室温で1時間振盪した。4−フルオロ−3−ニトロアニリン(BB2−1−52、0.211g、1.35mmol)を加えた。PipClU(0.195g,0.540mmol)とNMI(42.7μL、0.540mmol)のMeCN(0.54mL)溶液(PipClUとNMIは共に1M)を加え、室温で20時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(14mL)で3回、NMP(14mL)で3回、0.05M TBAHSO4+DTBP in NMP(14mL)で3回、0.05M NMM in NMP(14mL)で3回、NMP/水=1/1(14mL)で3回、NMP(14mL)で3回、MeOH(14mL)で3回、DCM(14mL)で3回、ヘプタン(14mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−c1D01−0を得た。

[表8−5−1−2]では、混合物2−1−c1D01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−c1D01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−5−1−2]においては、「▲×▼」と「d」は化学式で表す。












混合物2−1−c1D00−0を用いて工程c2D02を行った場合の反応式を以下に例示する。

20mLのフィルター付きカラムに[表8−5−2−1]に記載の20種類の混合物とNMP(10.5mL)を加え、室温で1時間振盪した。2,6−ルチジン(0.094mL、0.811mmol)とHATU(0.308g、0.811mmol)を加え、40℃で18時間振盪した。4−フルオロ−3−ニトロベンゼンスルホンアミド(BB2−1−51、0.208g、0.946mmol)とP1tBu(0.516mL、2.03mmol)を加え、室温で24時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(14mL)で3回、NMP(14mL)で3回、0.05M TBAHSO4+DTBP in NMP(14mL)で3回、NMP/水=1/1(14mL)で3回、NMP(14mL)で3回、MeOH(14mL)で3回、DCM(14mL)で3回、ヘプタン(14mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−c2D02−0を得た。

表中の表記方法について説明する。[表8−5−2−2]では、混合物2−1−c2D02−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−c2D02−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−5−2−2]においては、「▲×▼」と「d」は化学式で表す。
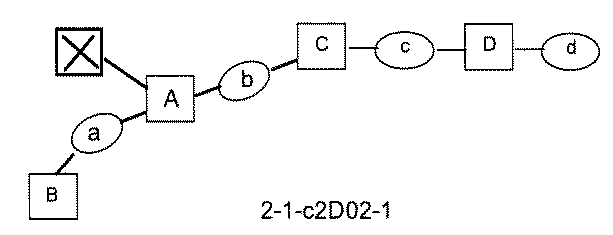











混合物2−1−c3aD00−0を用いて工程c3aD02を行った場合の反応式を以下に例示する。

4mLのガラスバイアルに、[表8−5−3−1]に記載の5種類の混合物とDCE(2.8mL)、DTBP(0.439mL、2.00mmol)を加え、室温で1時間振盪した。DIPEA(17.42μL、100μmol)と4−フルオロ−3−ニトロベンゼンスルホニルクロリド(BB2−1−53、80.0mg、0.333mmol)のDCE(700μL)溶液を加え、60℃で23時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(3.5mL)で3回、NMP(3.5mL)で3回、0.05M TBAHSO4+DTBP in NMP(3.5mL)で3回、0.05M NMM in NMP(3.5mL)で3回、NMP/水=1/1(3.5mL)で3回、NMP(3.5mL)で3回、MeOH(3.5mL)で3回、DCM(3.5mL)で3回、ヘプタン(3.5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−c3aD02−0を得た。







混合物2−1−c3bD00−0を用いて工程c3bD02を行った場合の反応式を以下に例示する。
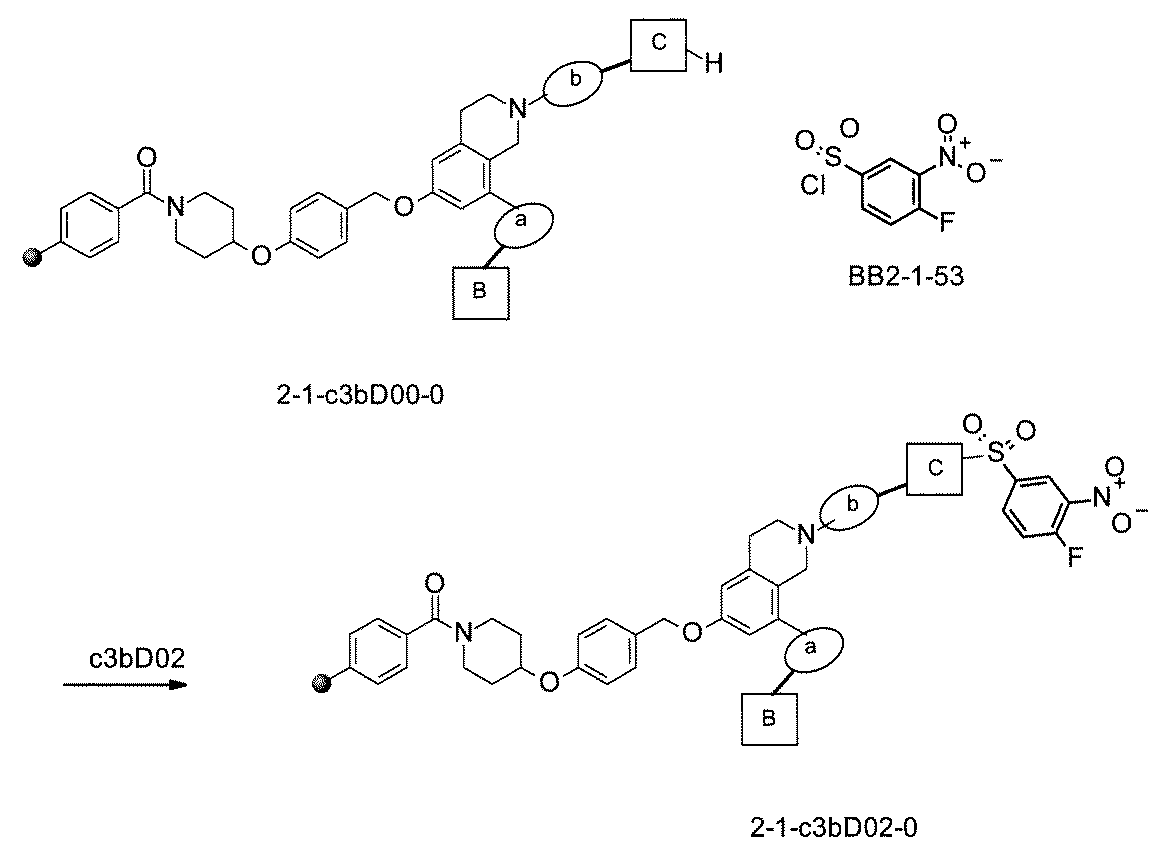
4mLのガラスバイアルに、[表8−5−4−1]に記載の5種類の混合物とDCE(2.8mL)、DTBP(0.441mL、2.01mmol)を加え、室温で1時間振盪した。4−フルオロ−3−ニトロベンゼンスルホニルクロリド(BB2−1−53、80.0mg、0.334mmol)のDCE(700μL)溶液を加え、60℃で23時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(3.5mL)で3回、NMP(3.5mL)で3回、0.05M TBAHSO4+DTBP in NMP(3.5mL)で3回、0.05M NMM in NMP(3.5mL)で3回、NMP/水=1/1(3.5mL)で3回、NMP(3.5mL)で3回、MeOH(3.5mL)で3回、DCM(3.5mL)で3回、ヘプタン(3.5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−c3bD02−0を得た。
[表8−5−4−1]工程c3bD02で使用した原料の混合物

表中の表記方法について説明する。[表8−5−4−2]では、2−1−c3bD02−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−c3bD02−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−5−4−2]においては、「▲×▼」と「d」は化学式で表す。





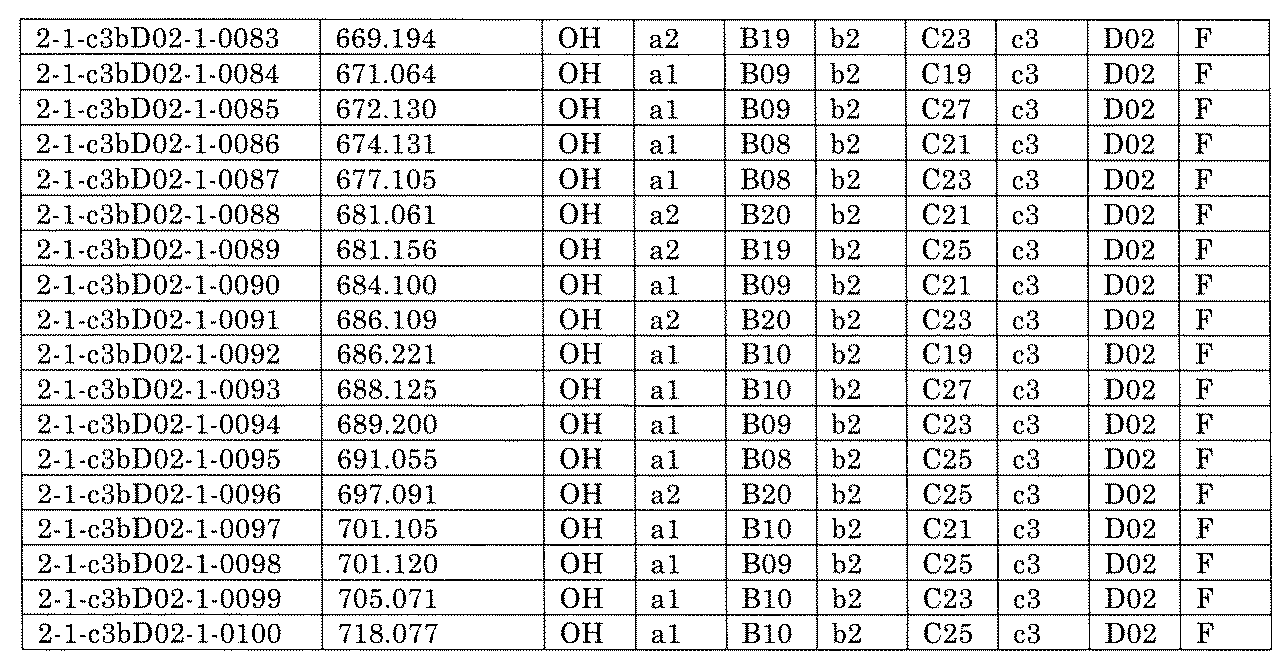
20mLのフィルター付きカラムに[表8−6−1]に記載の4種類の混合物とDCM(15mL)を加え、室温で1時間振盪した。溶媒と固相の懸濁液をカラムのフィルターでろ過し、ジクロロメタン(9mL)で1回、ヘプタン(9mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−1−E00−0(2.12g、混合物の固相担持量:0.186mmol/g)を得た。
[表8−6−1]混合物2−1−E00−0の調製に使用した混合物
表中の表記方法について説明する。[表8−6−2]では、混合物2−1−E00−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−E00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−6−2]においては、「▲×▼」と「d」は化学式で表す。


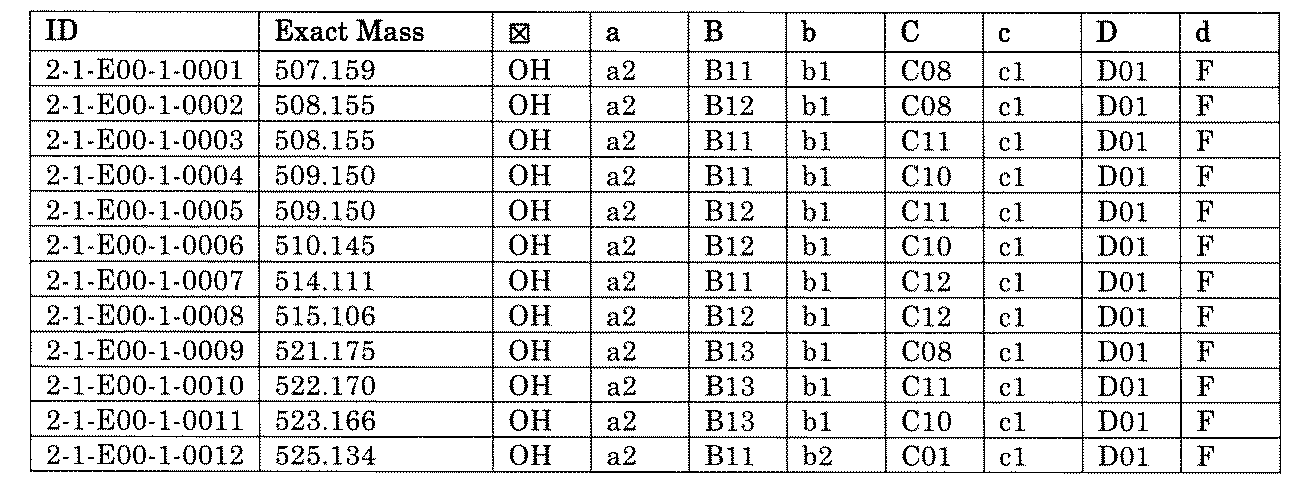
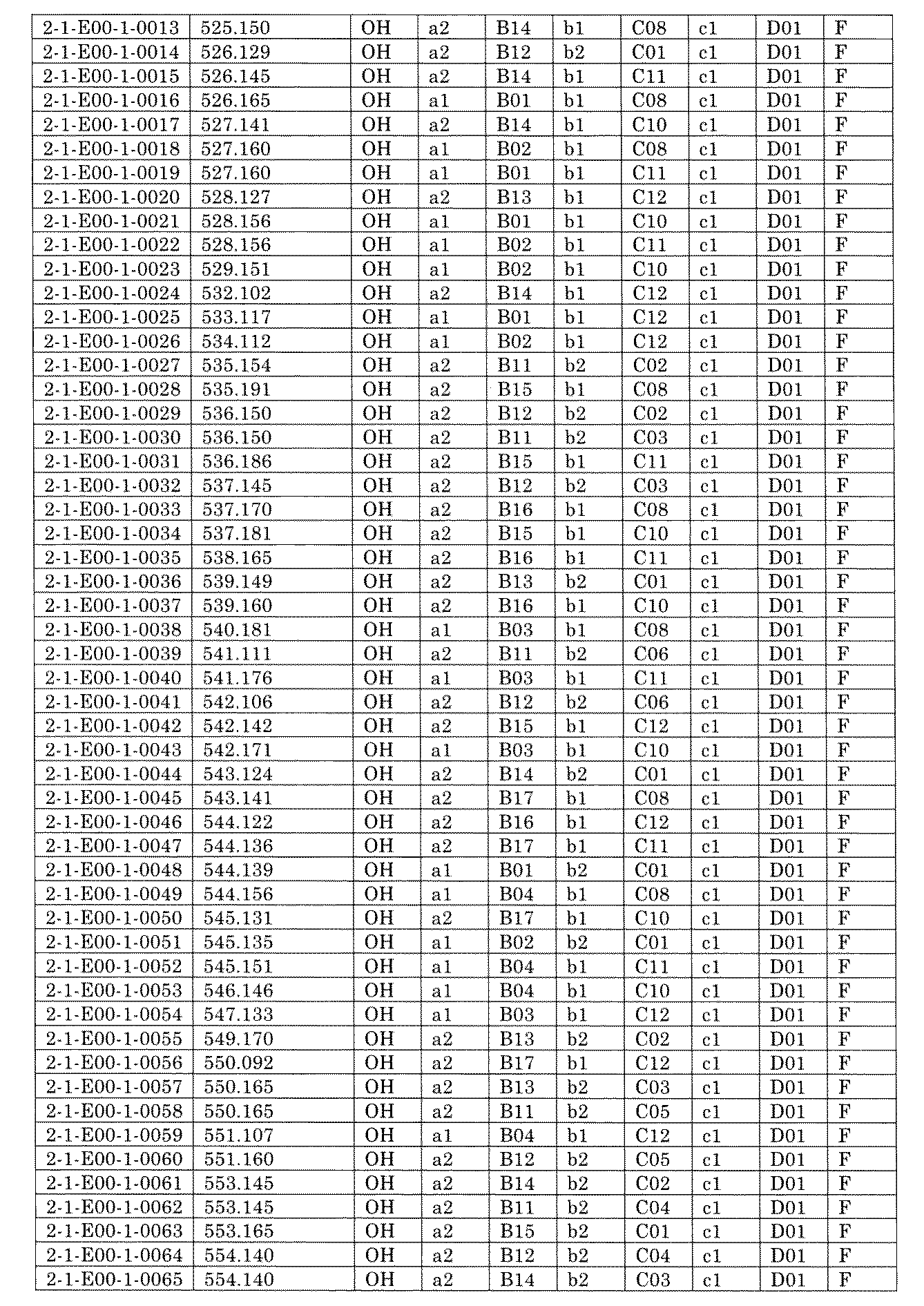

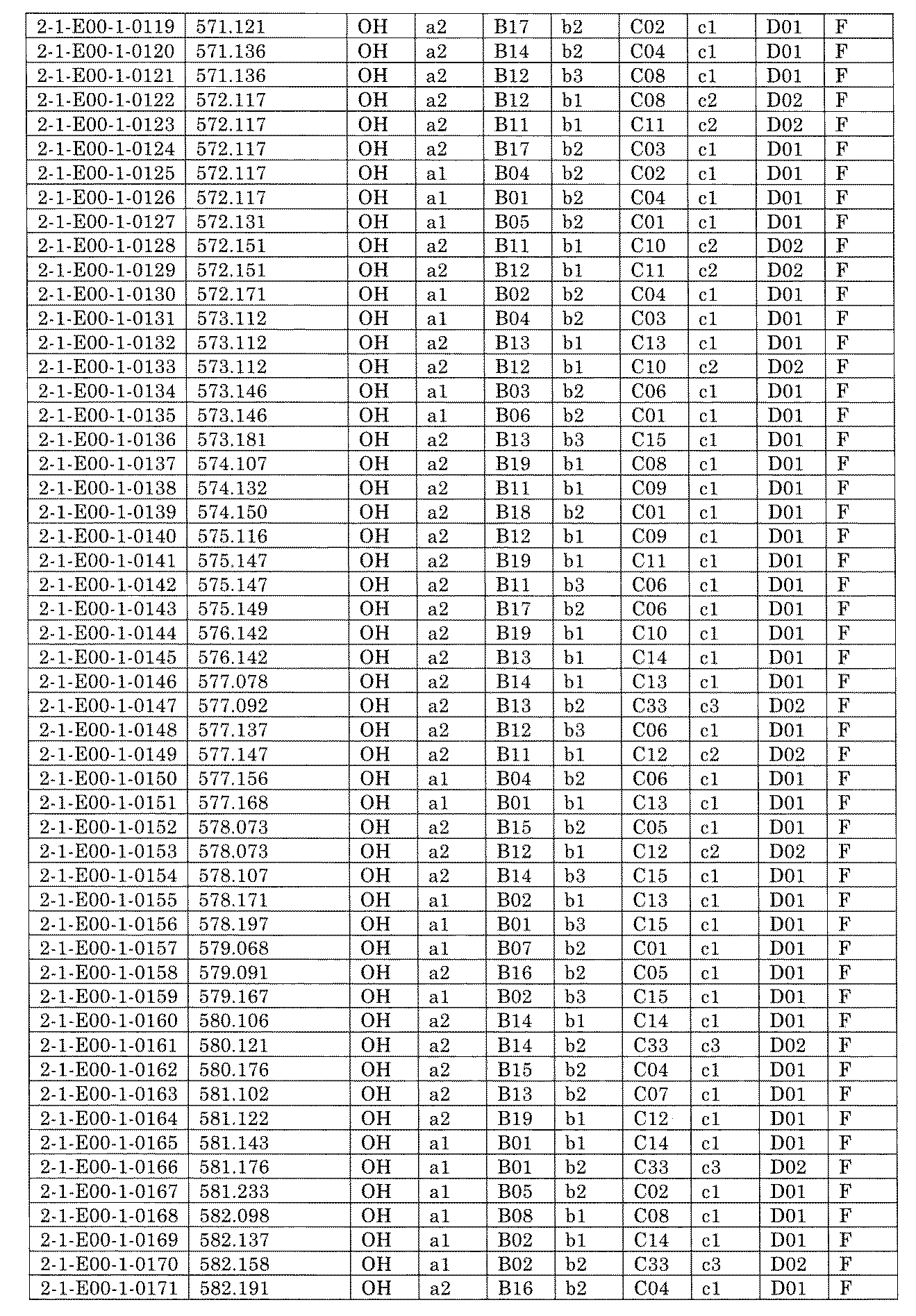

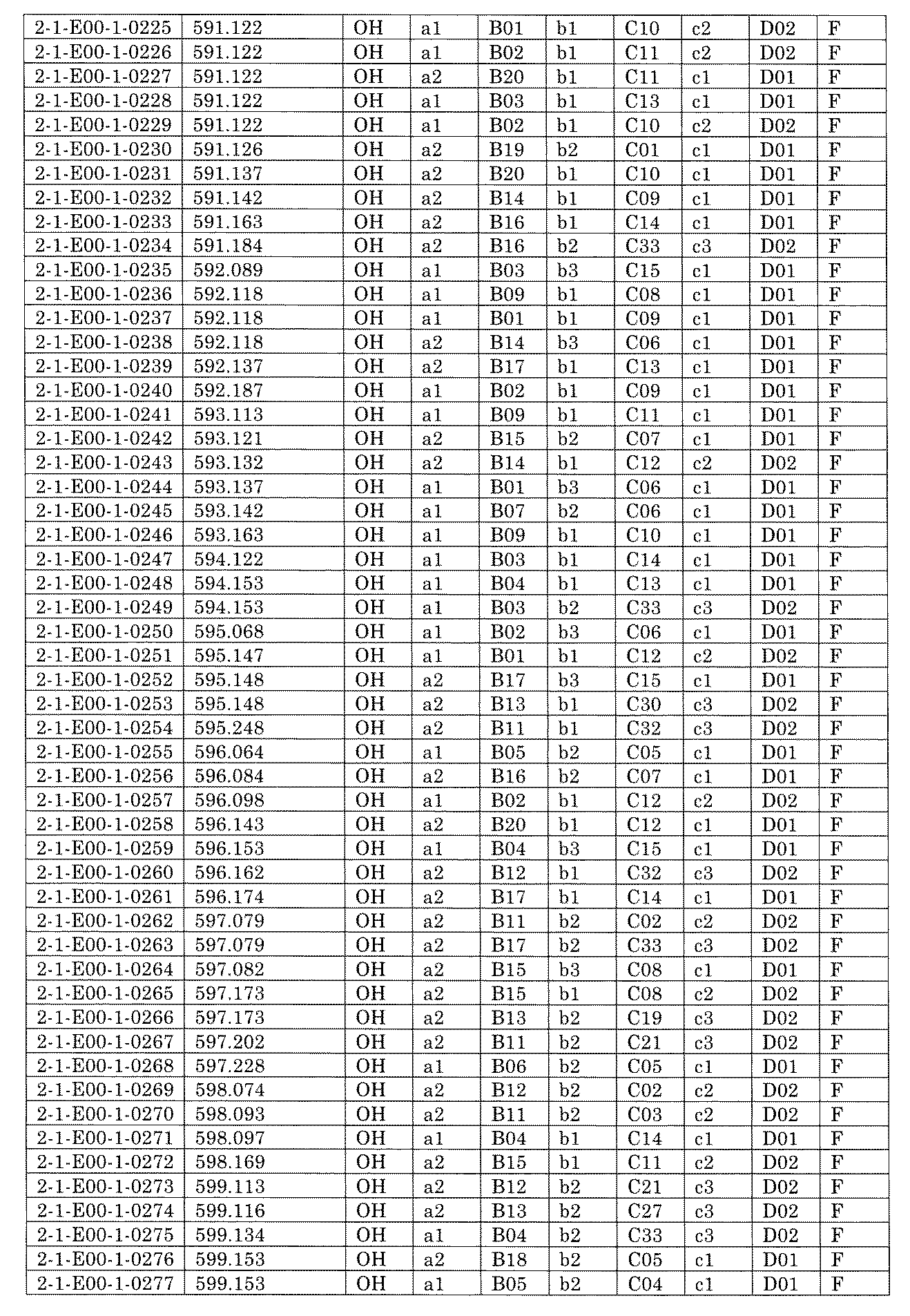
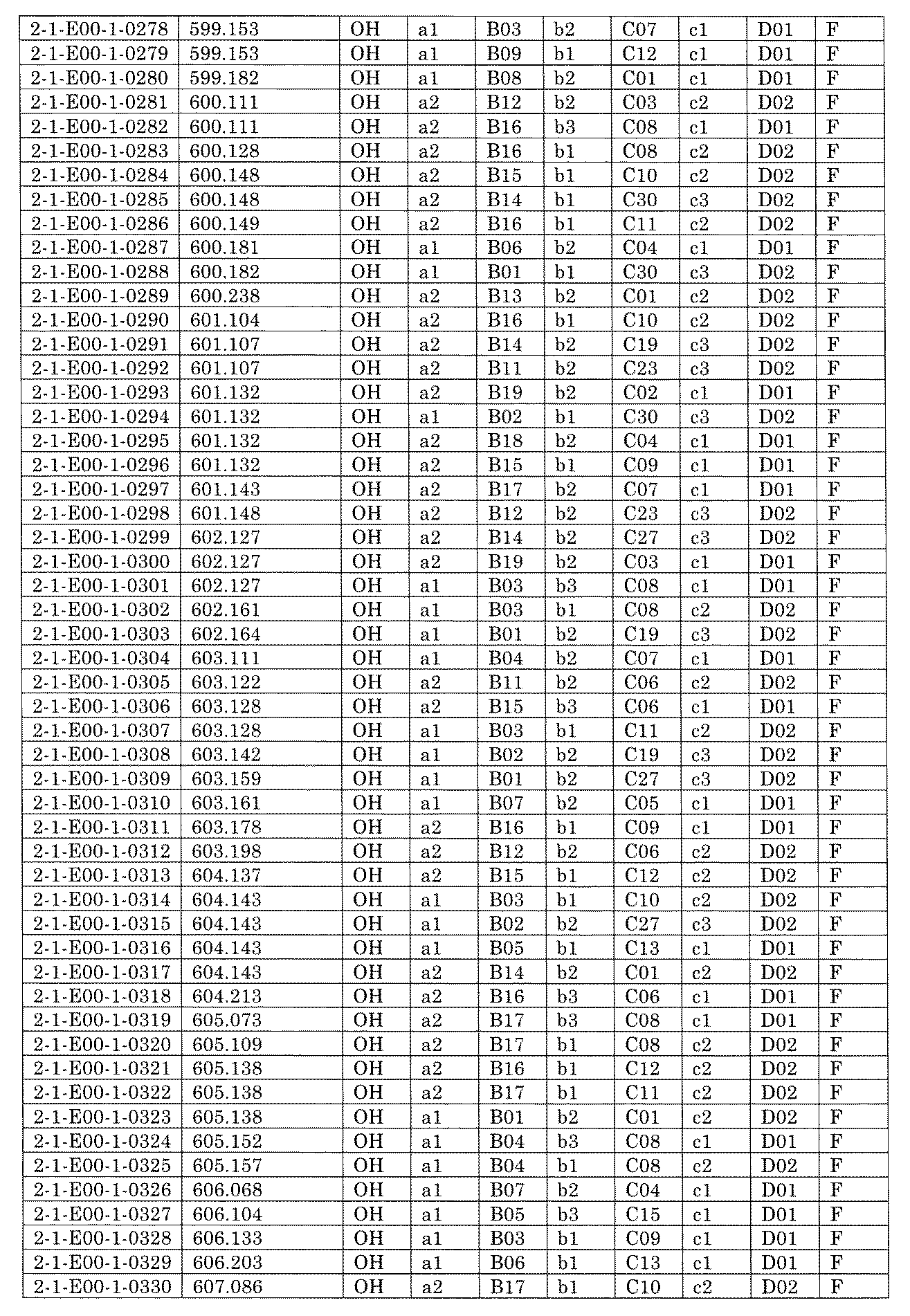

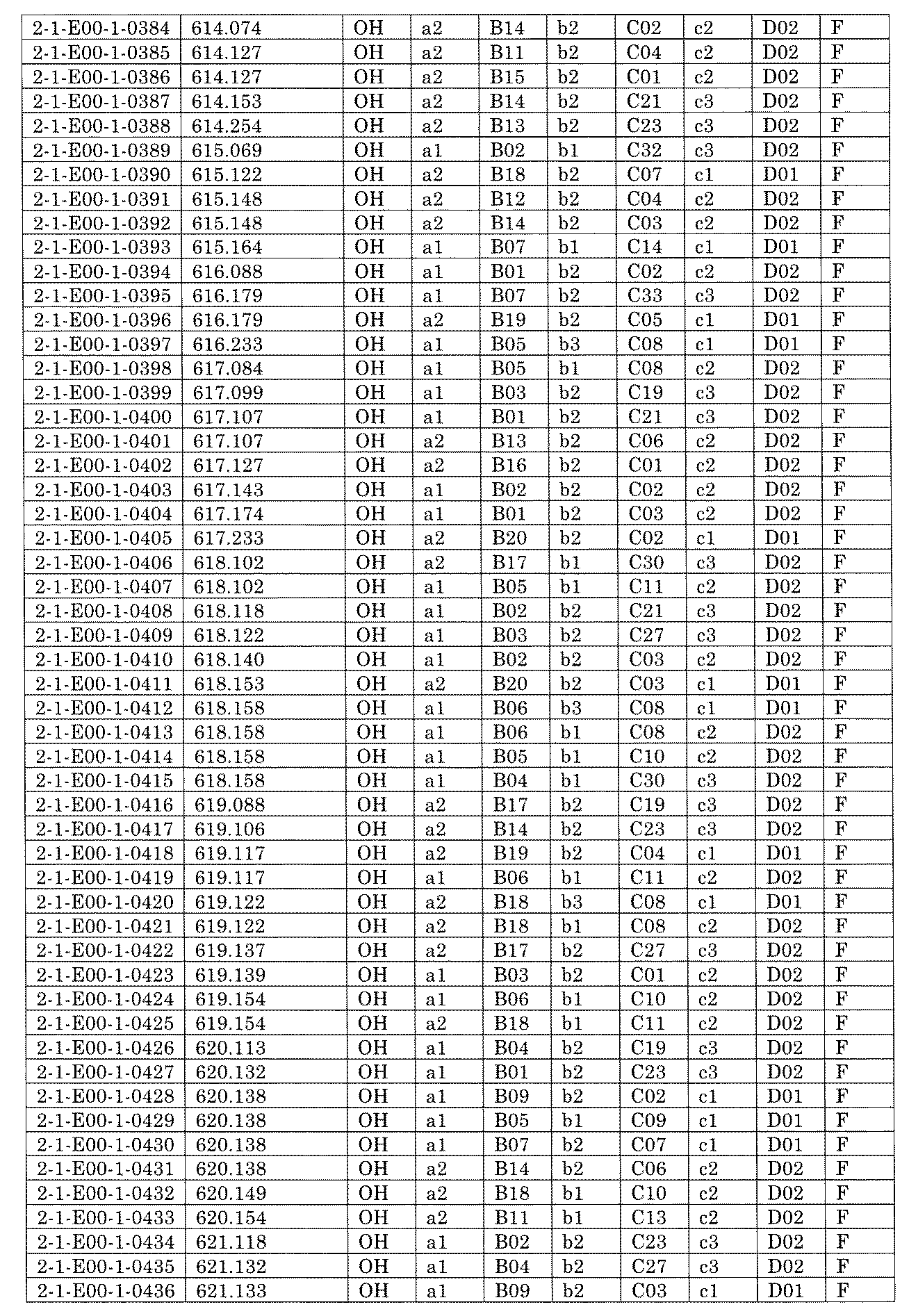


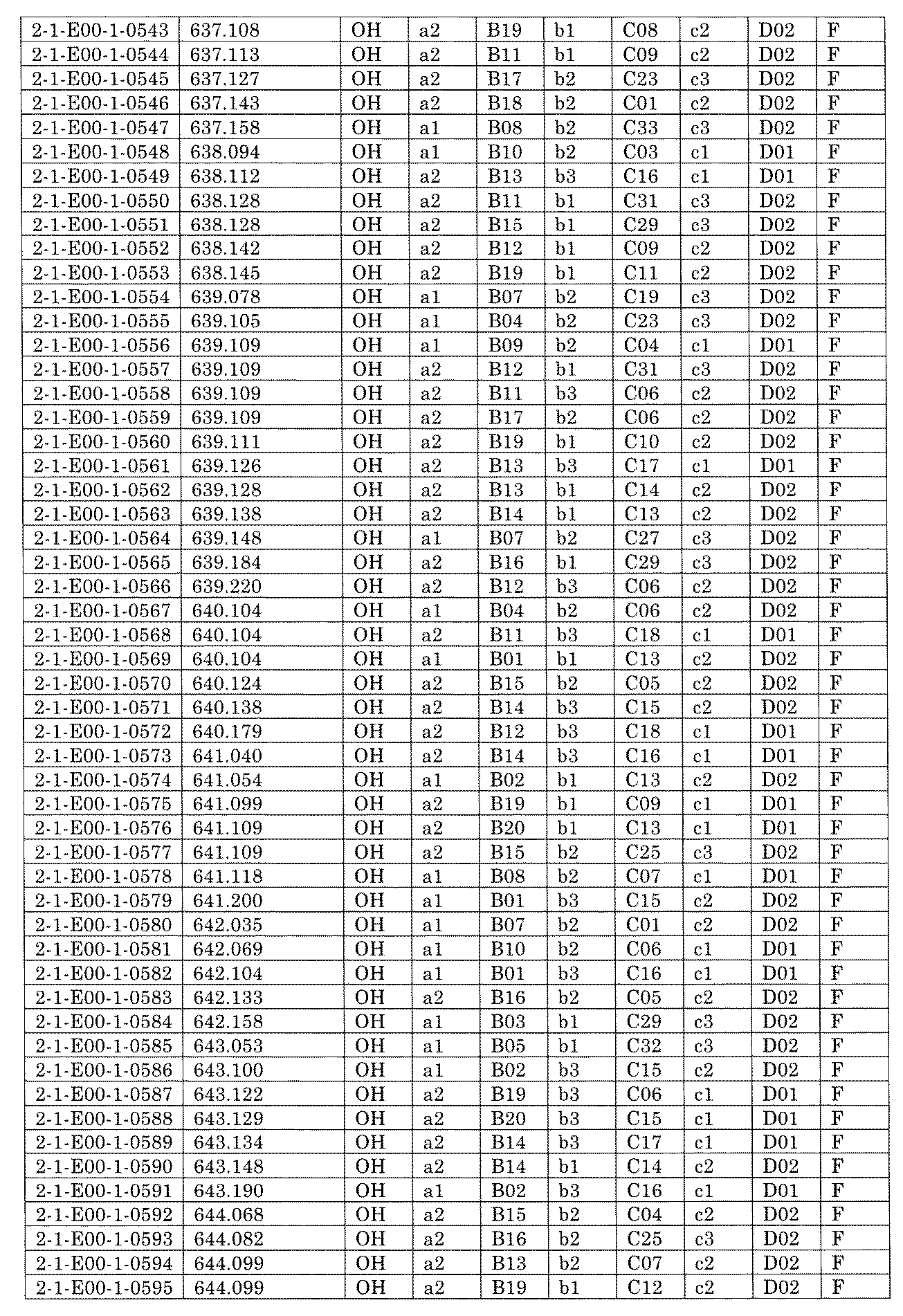



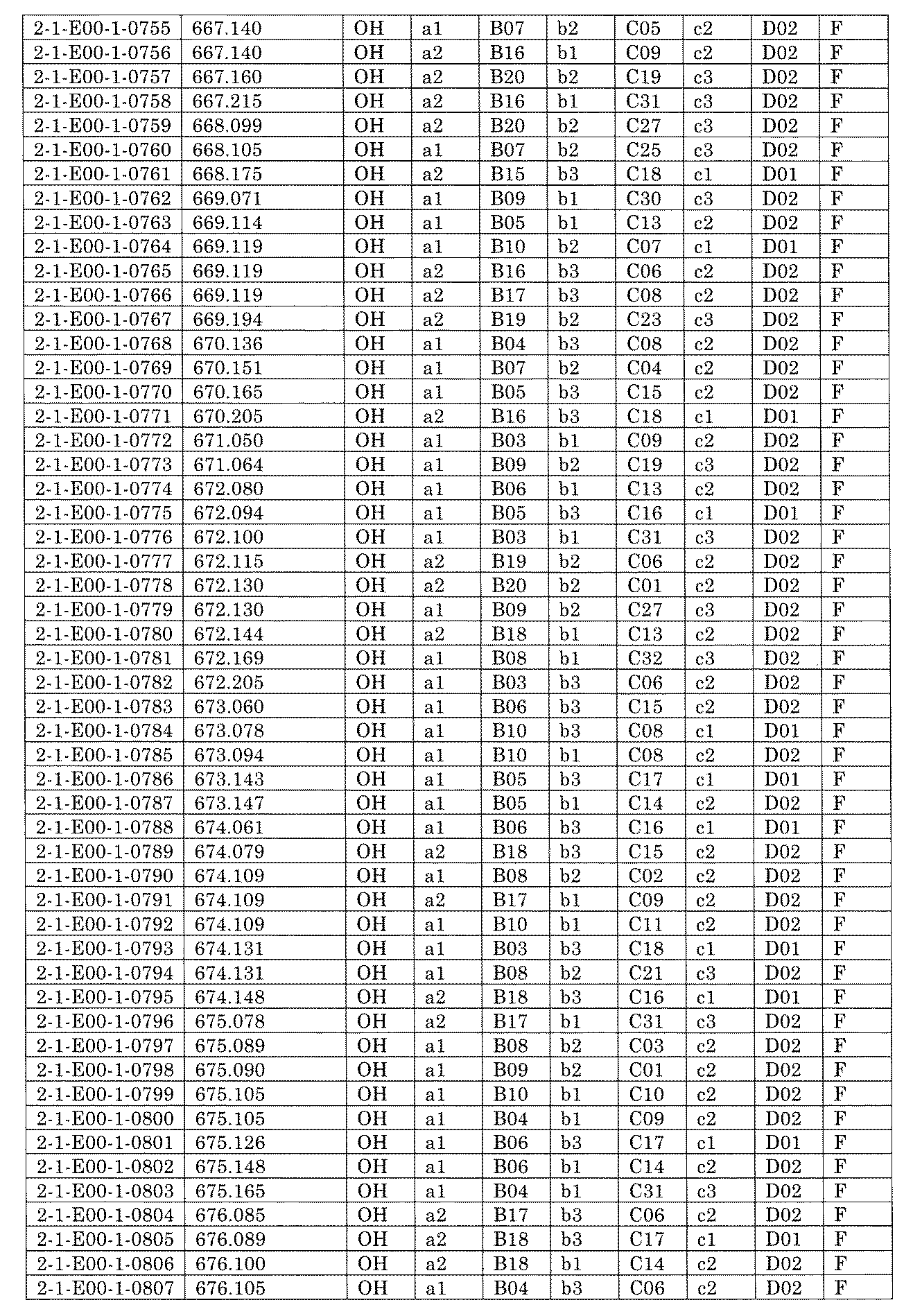
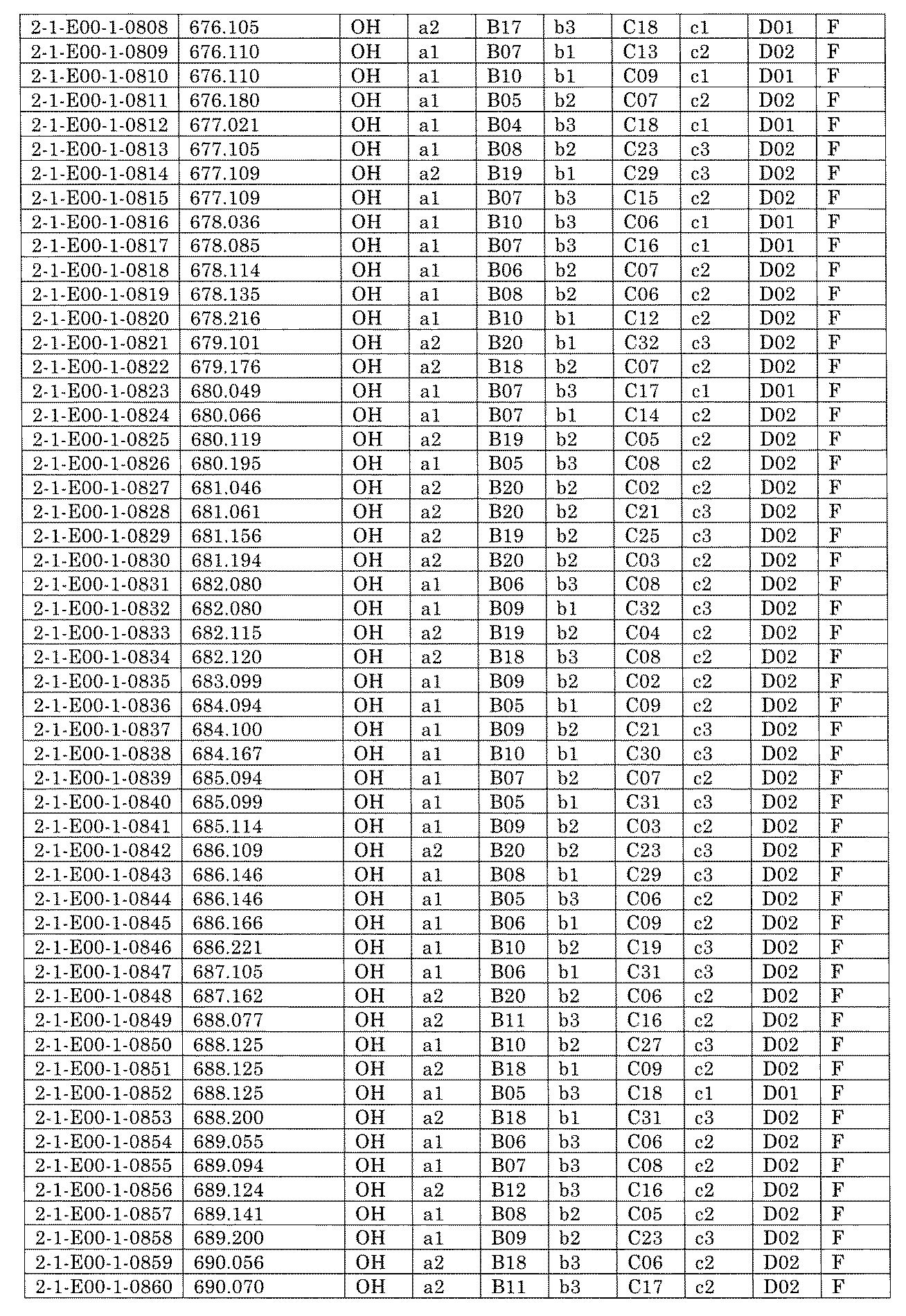



_BB2−1−54を用いて工程d1E01を行った場合の反応式を以下に例示する。

_4個の8mLのガラスバイアルに2−1−E00−0(240mg、混合物の固相担持量:0.186mmol/g)とNMP(2.9mL)を加え、室温で1時間振盪した。[表8−7−1]に記載のアミン(0.446mmol)を別個にそれぞれのガラスバイアルに加えた。1,8−ビス(ジメチルアミノ)ナフタレン(144mg、0.670mmol)を加え、80℃で24時間振盪した。
_反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、0.05M NMM in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、表に記載の混合物を得た。

表中の表記方法について説明する。[表8−7−2]~[表8−7−5]では、混合物2−1−d1E01−1~混合物2−1−d1E04−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−d1E01−1~混合物2−1−d1E04−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。
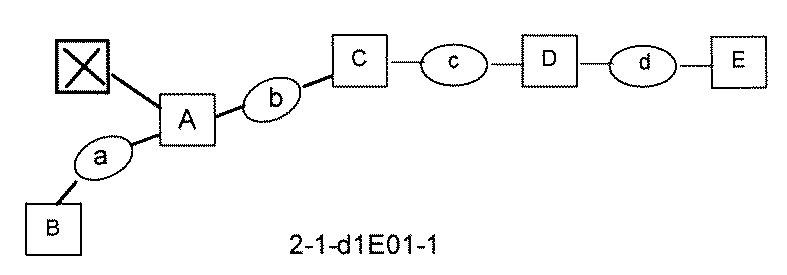


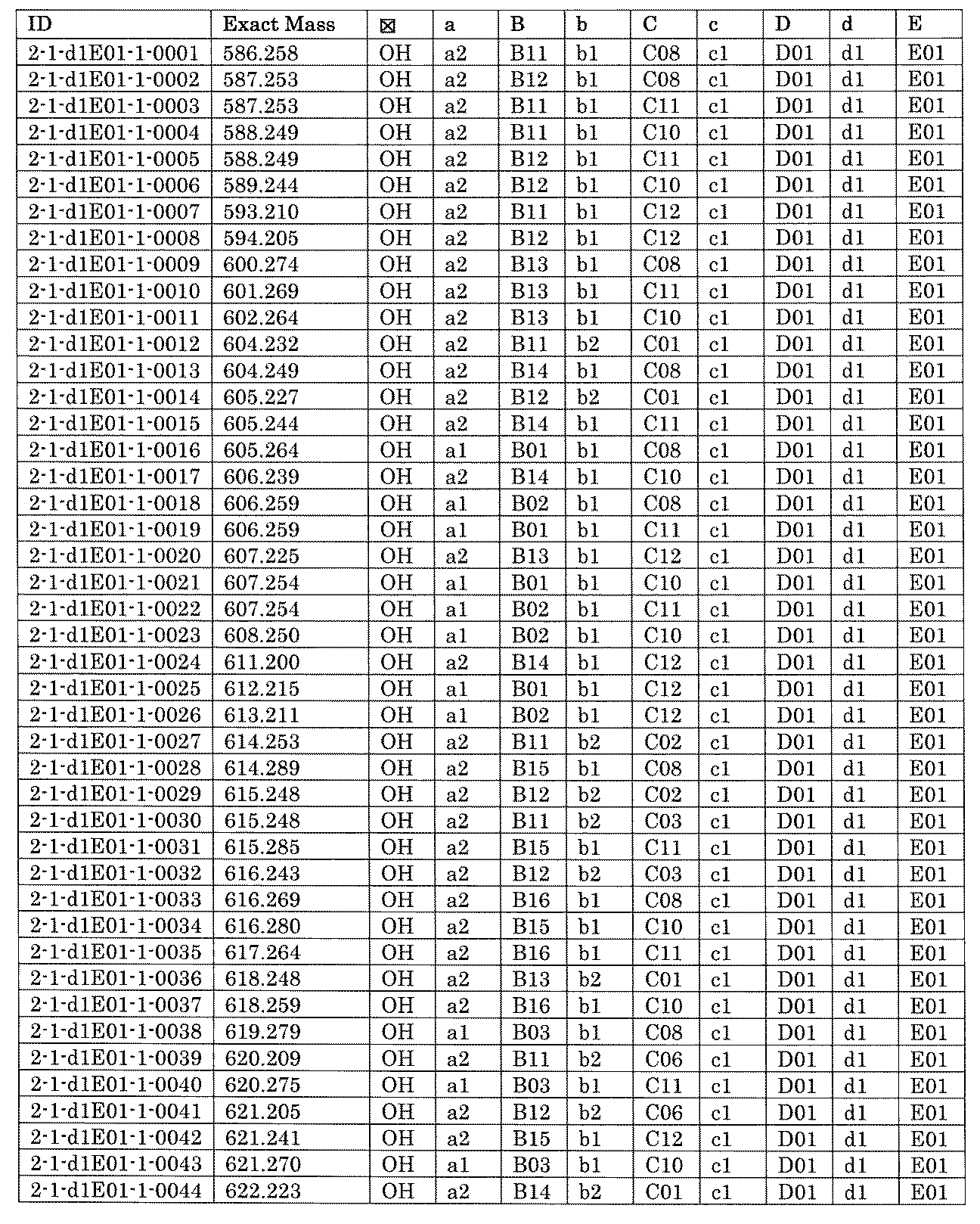




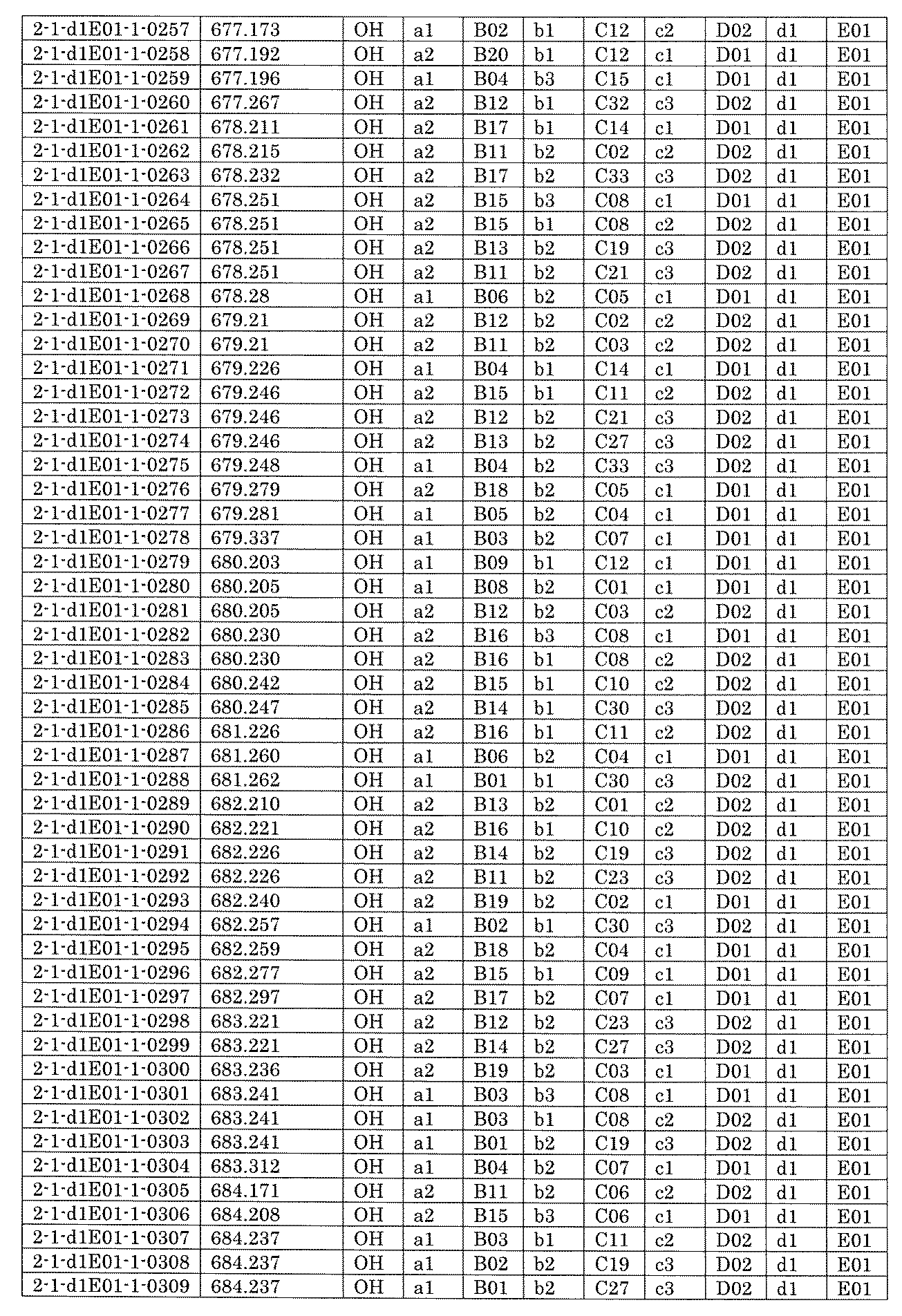

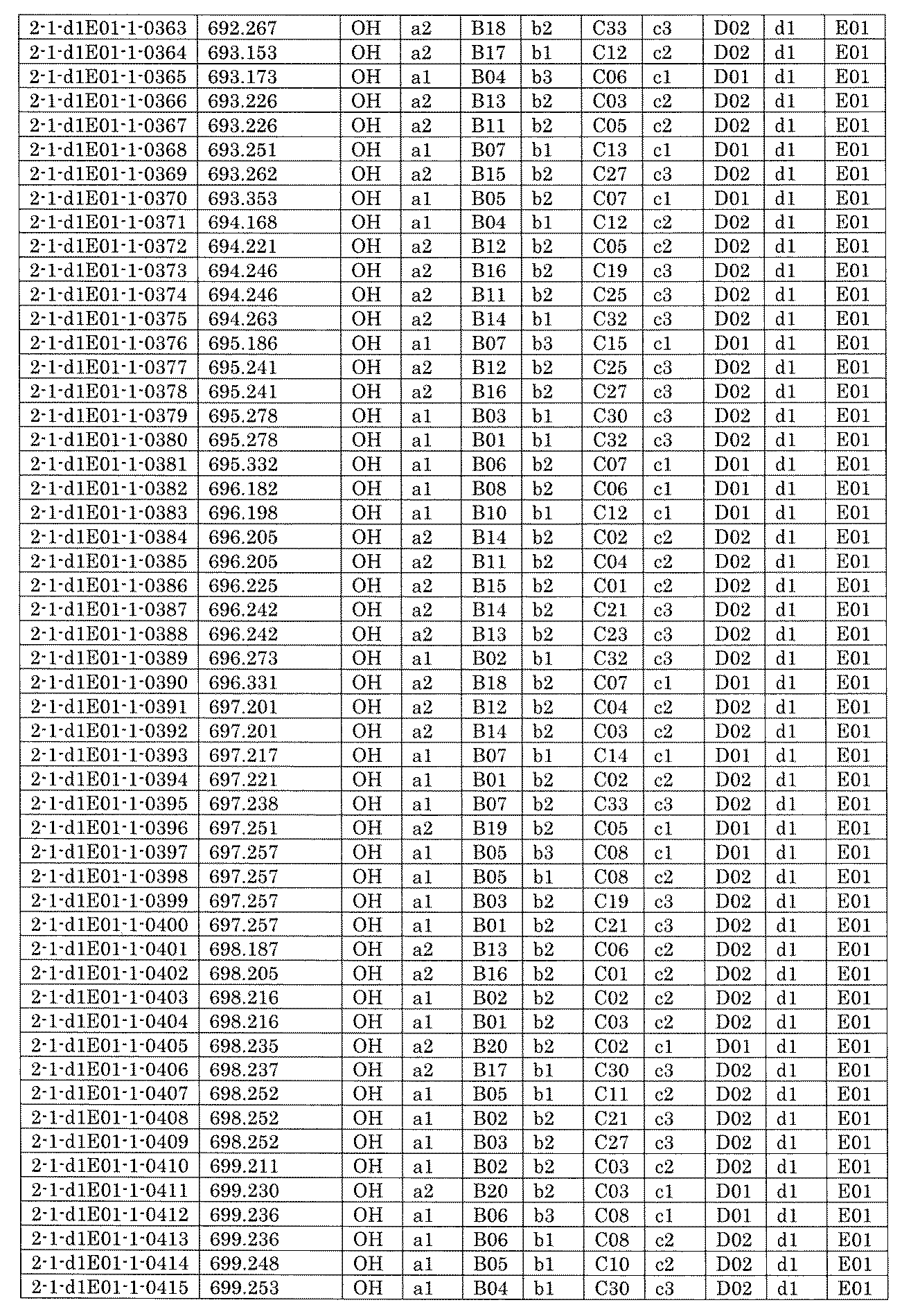






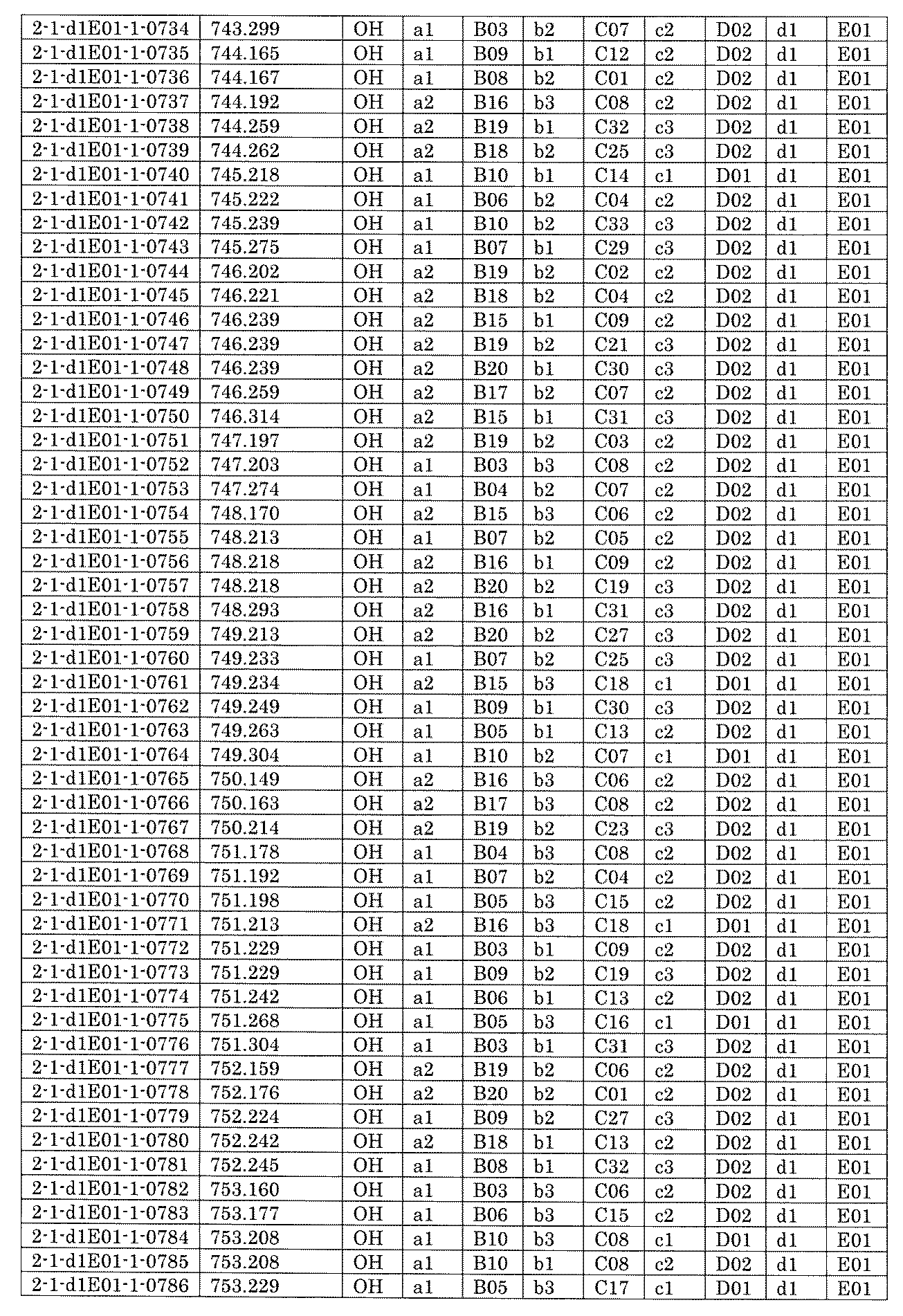
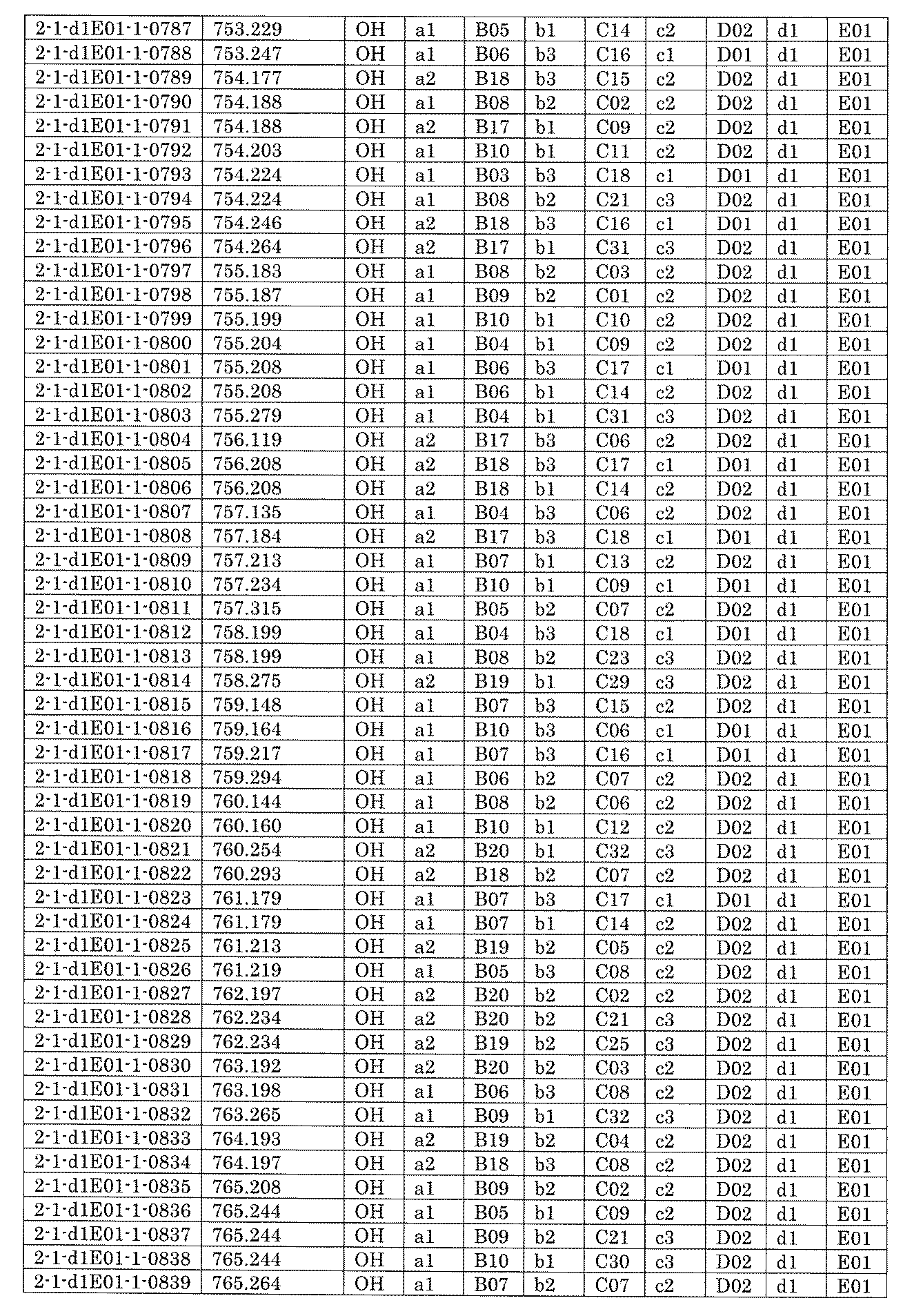










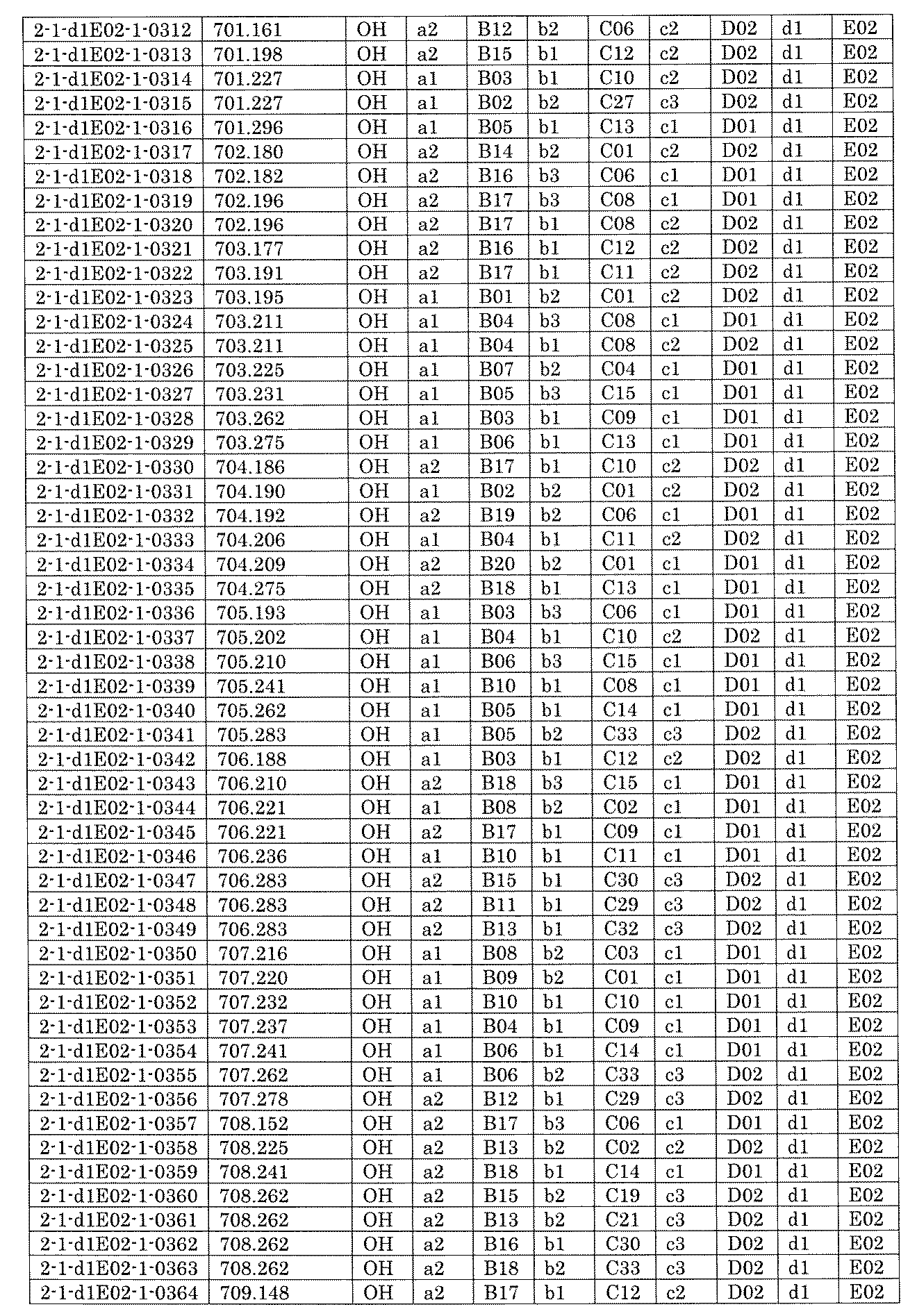

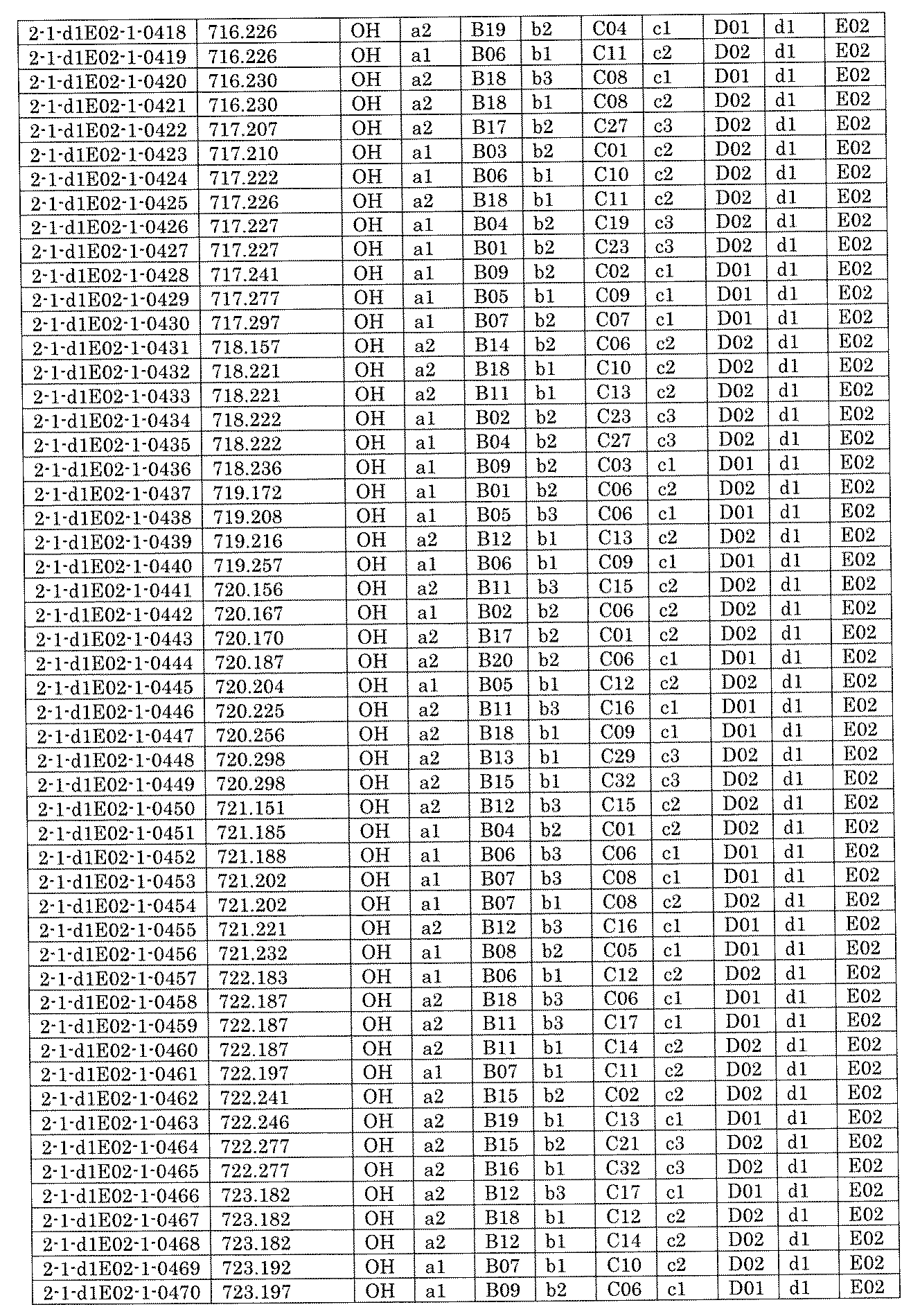


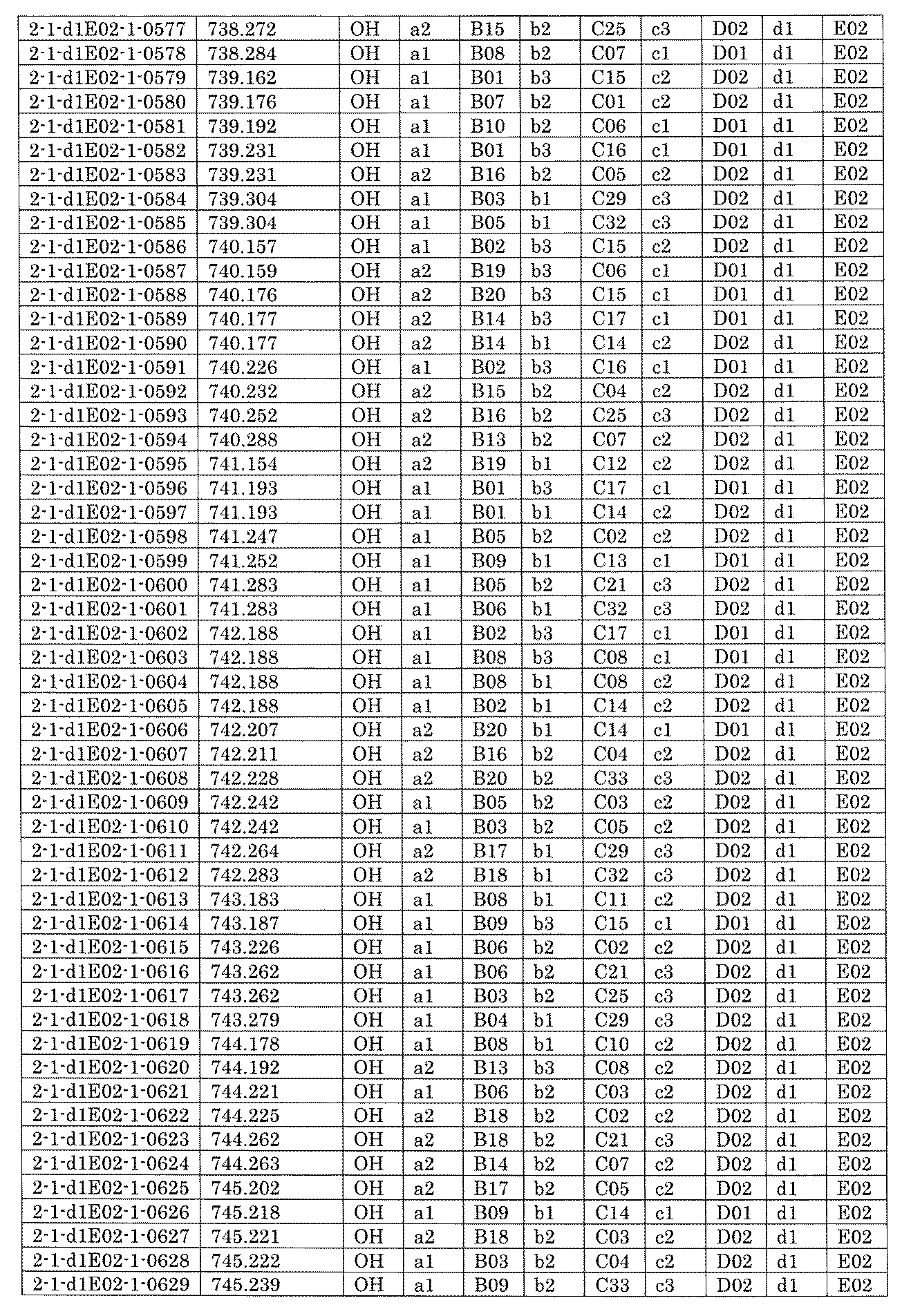
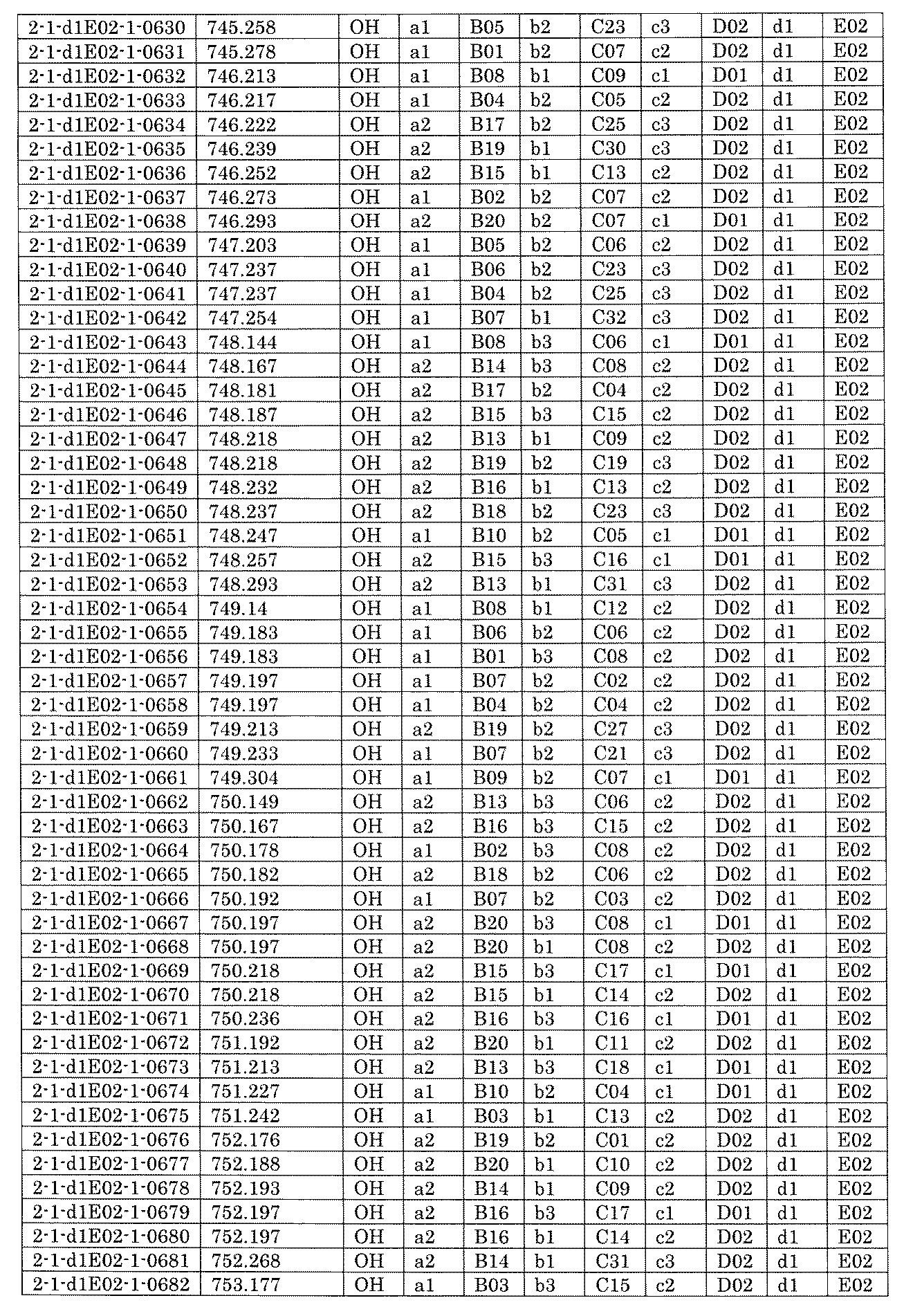
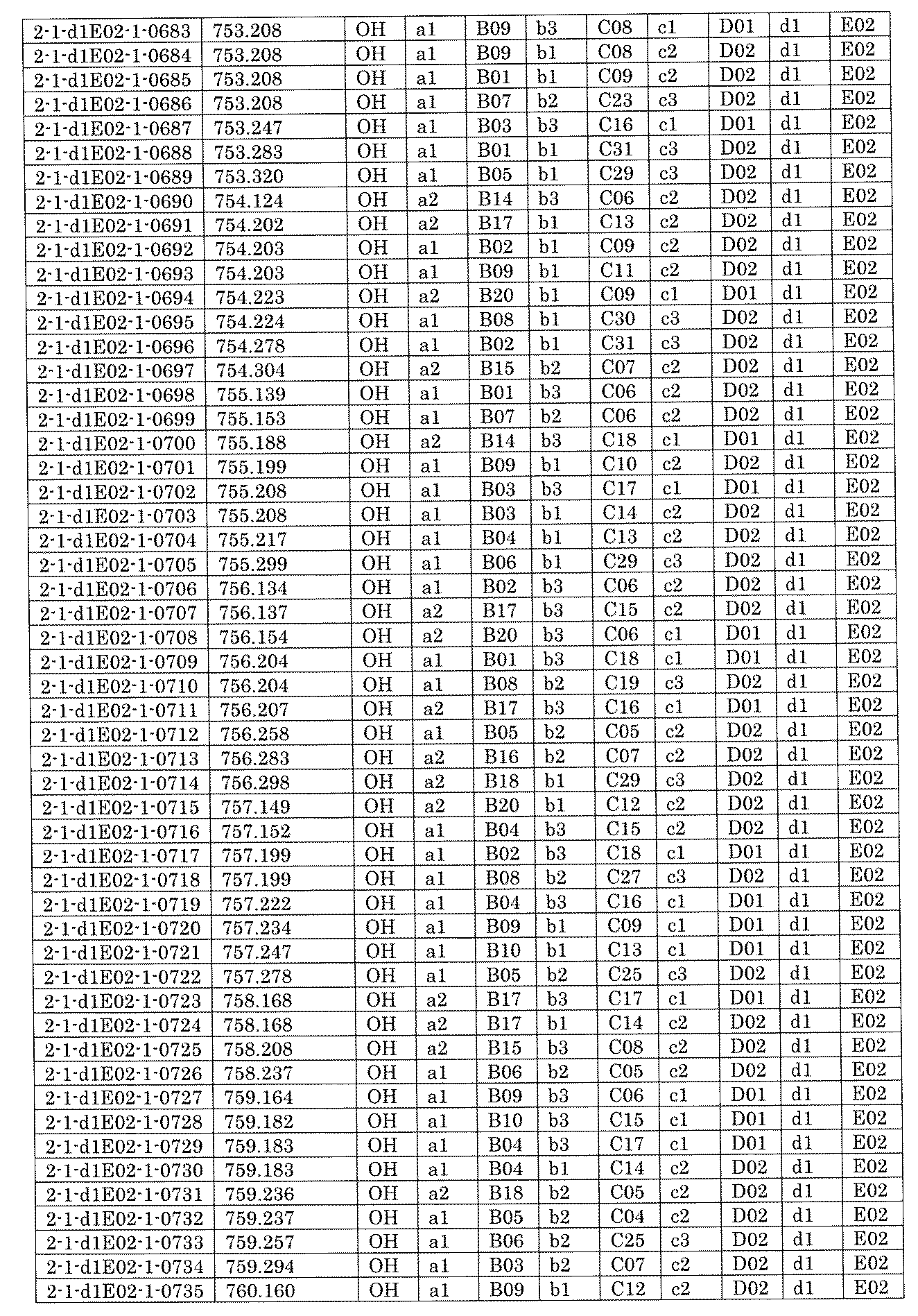










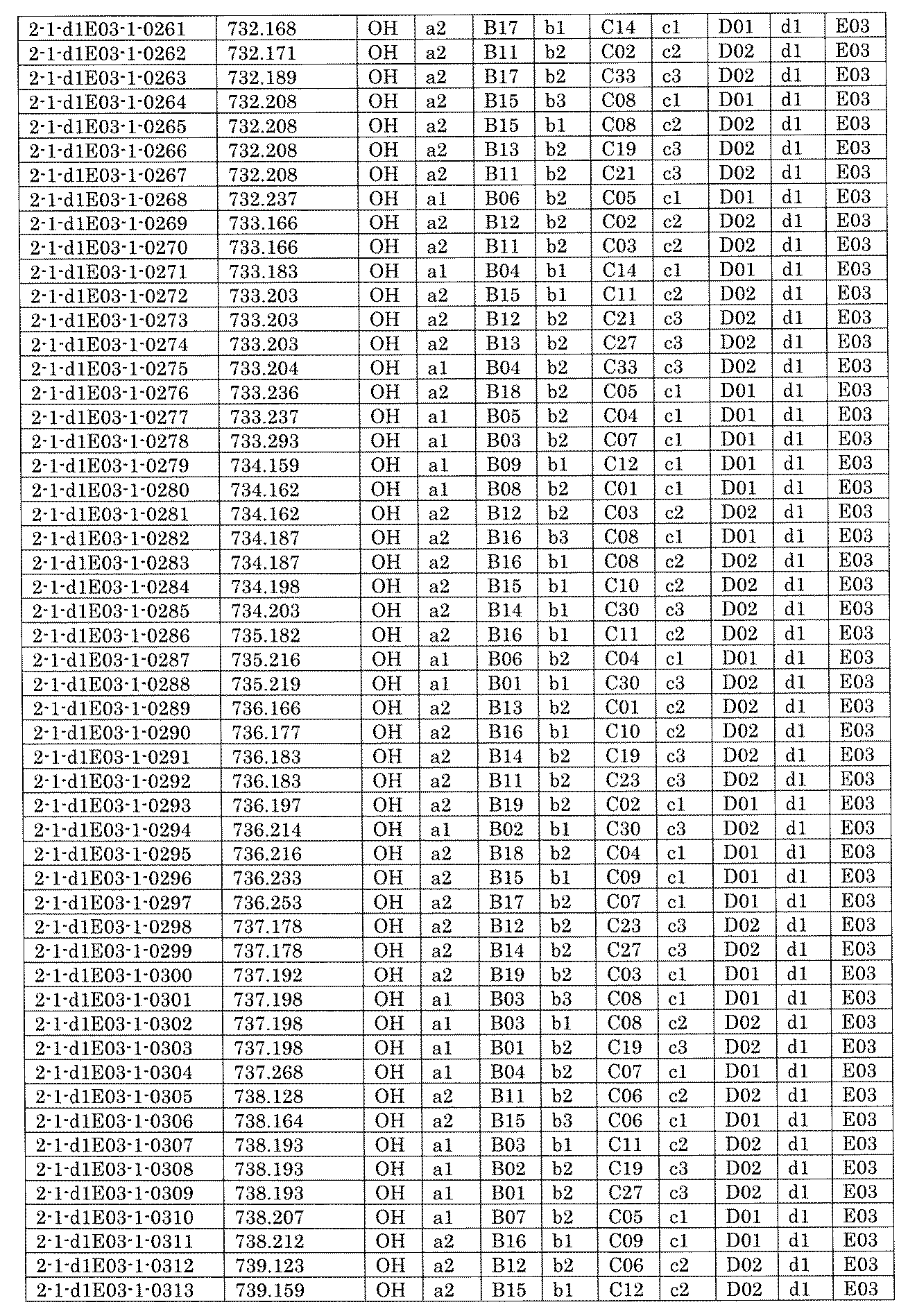

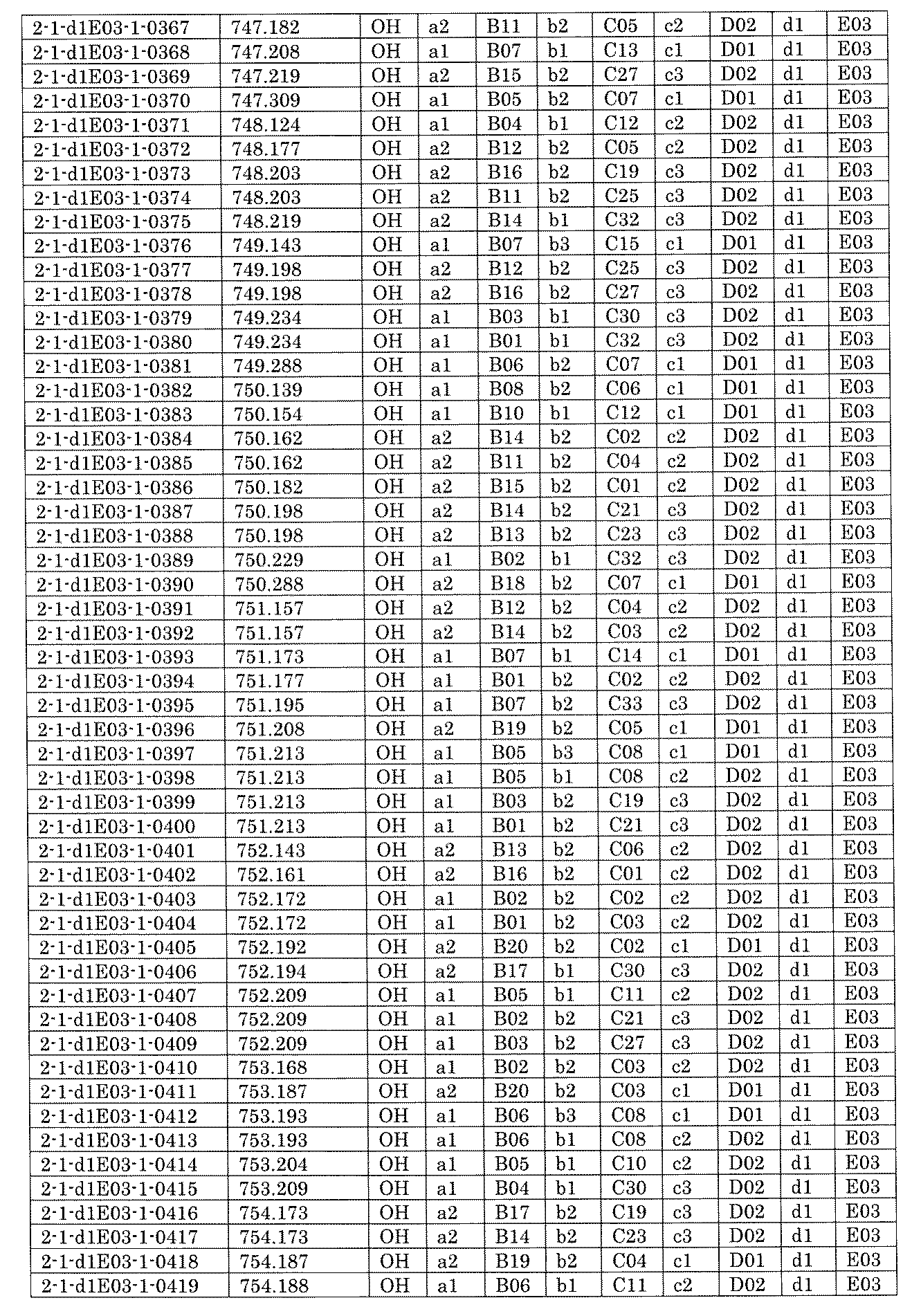


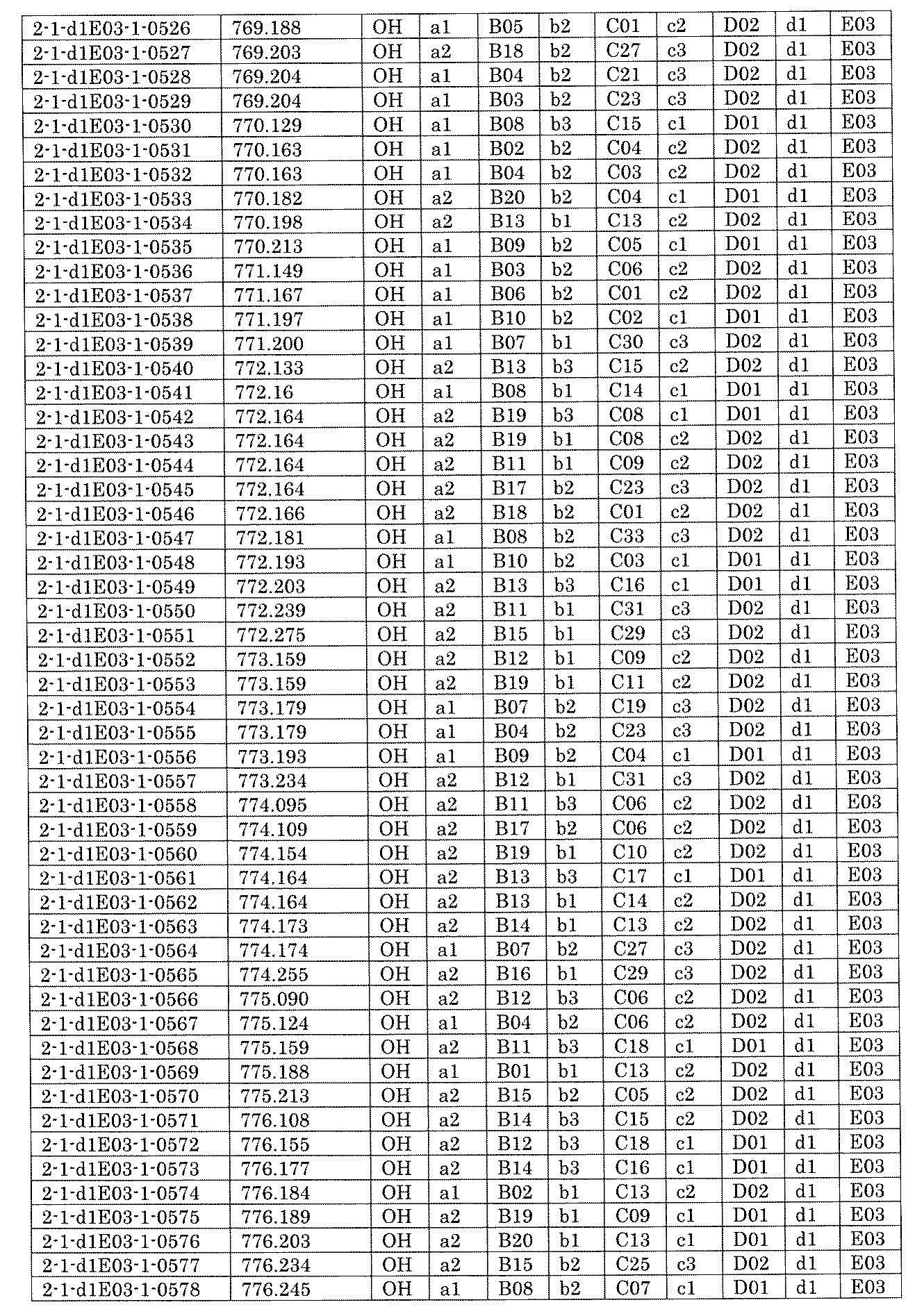




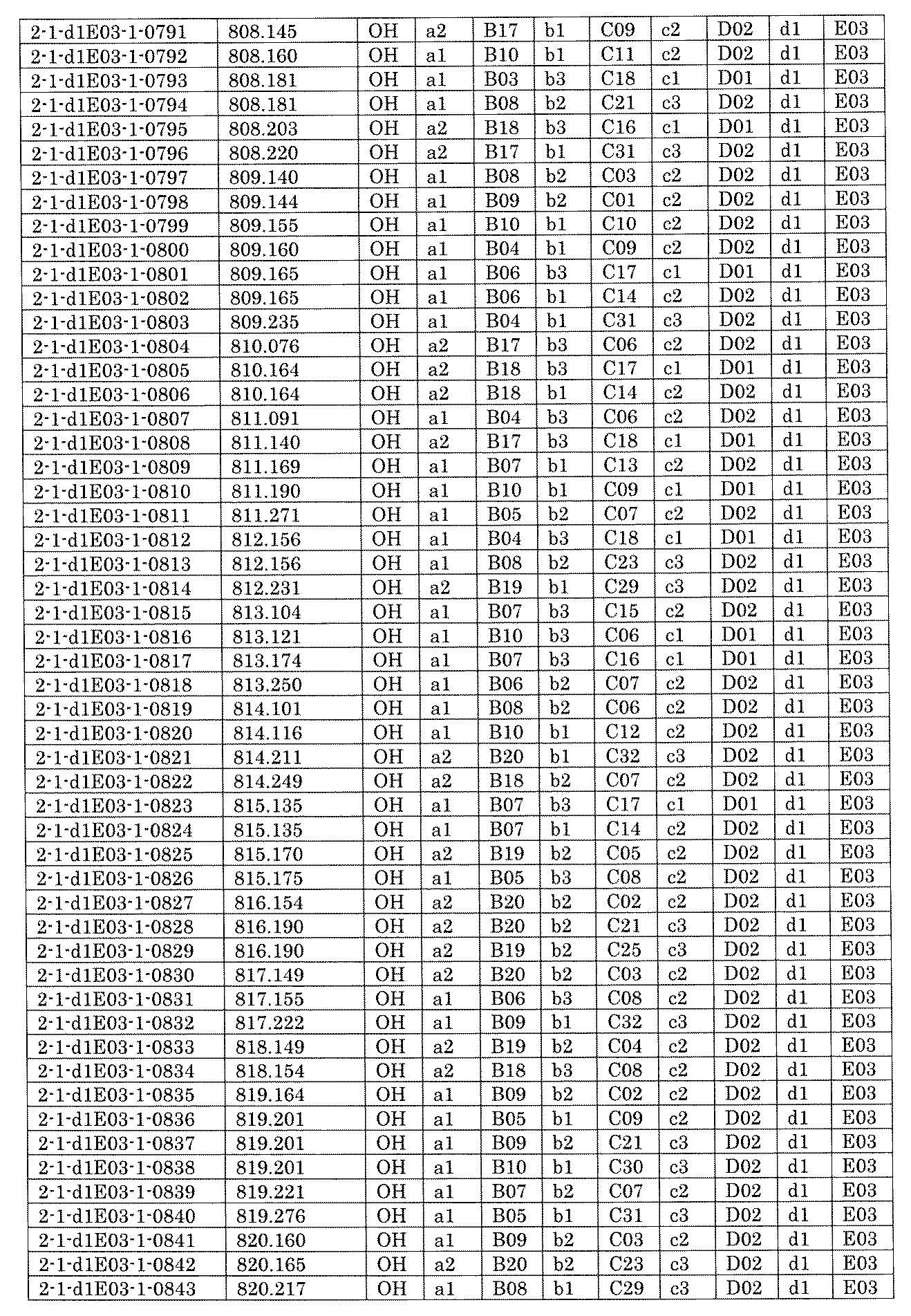




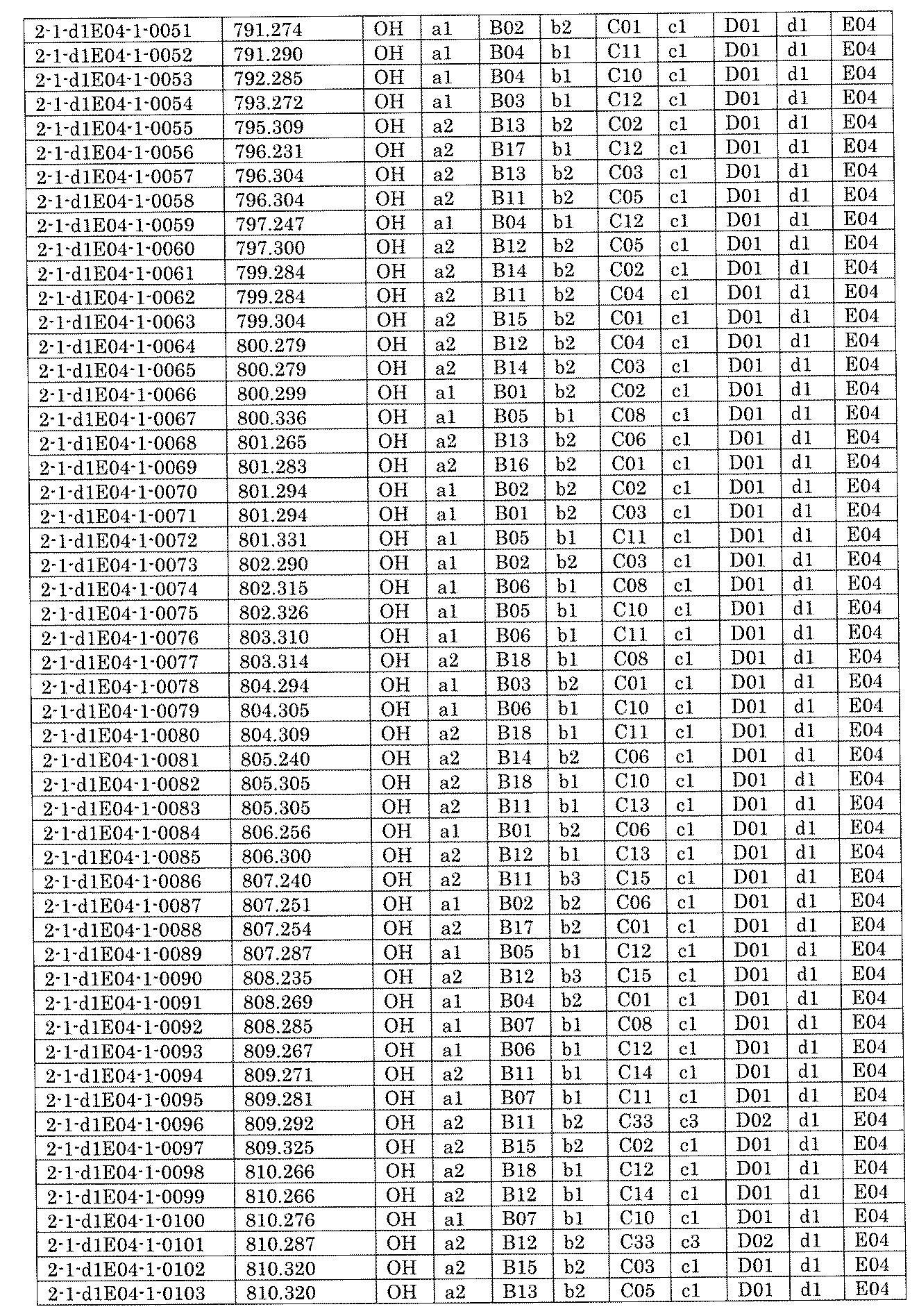










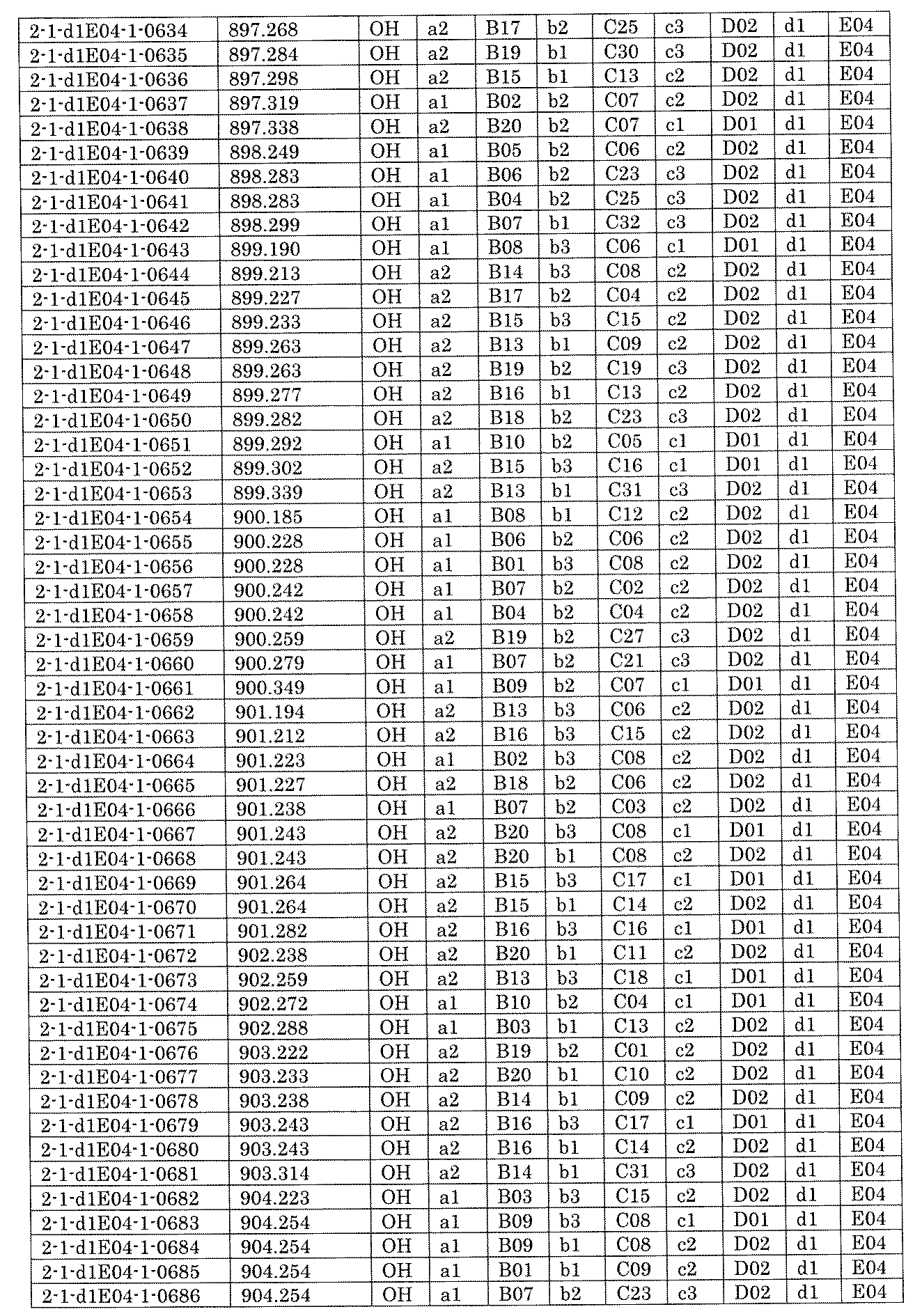
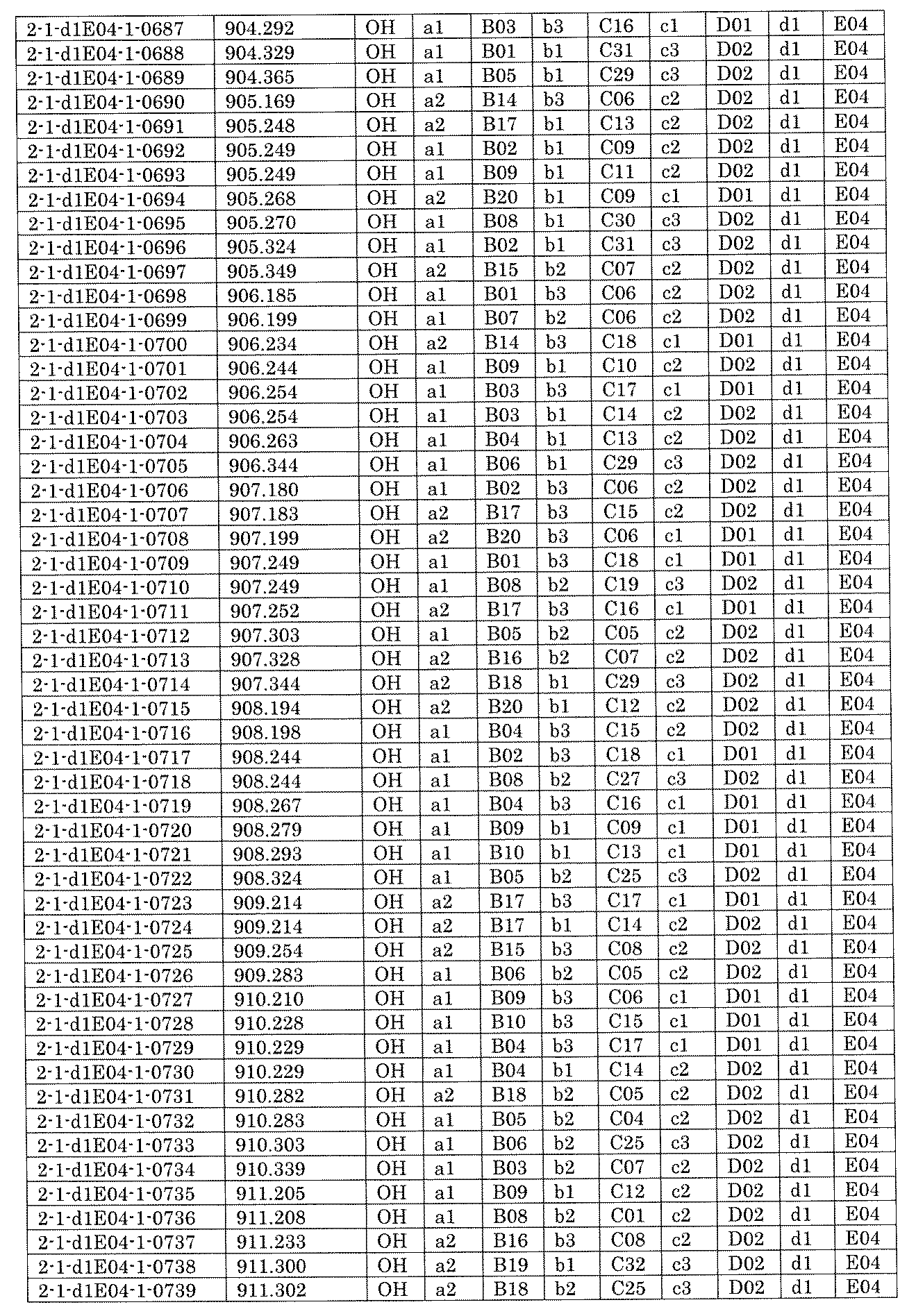





混合物2−1−d1E01−0を用いて工程m1E01を行った場合の反応式を以下に例示する。
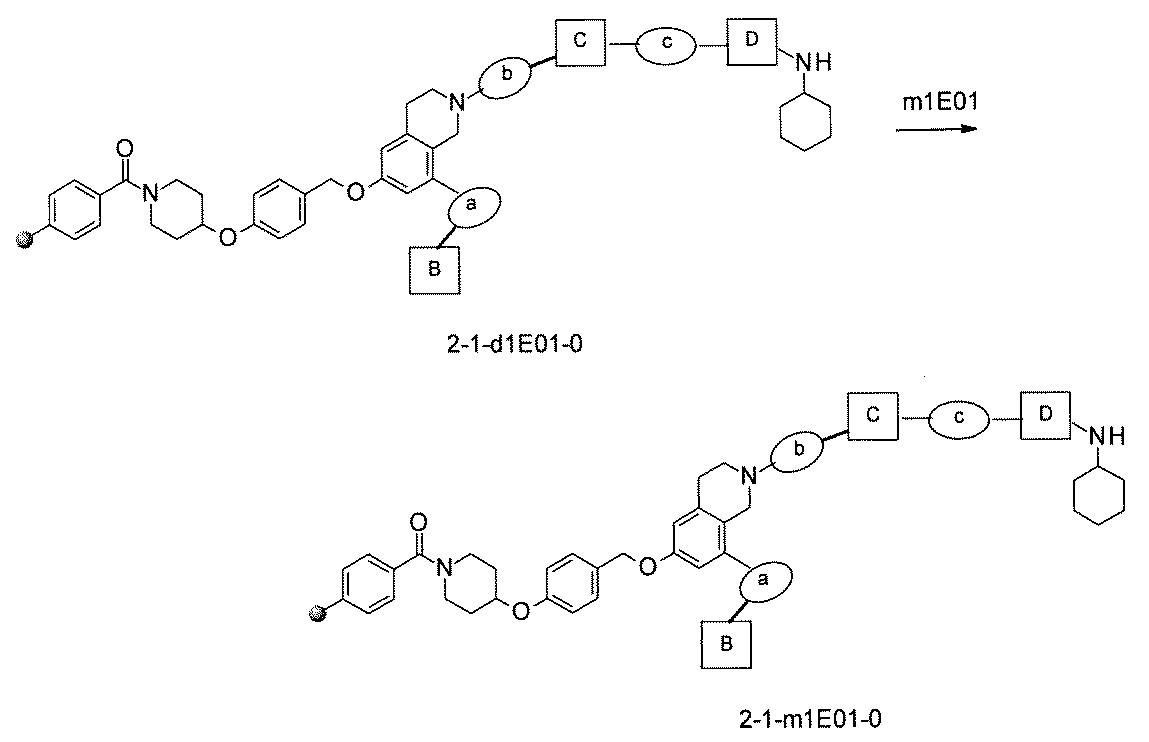
混合物2−1−d1E01−0~混合物2−1−d1E04−0を用いて、混合物2−1−m1E01−0~混合物2−1−m1E04−0は以下のように合成した。
4個の8mLのガラスバイアルに、[表8−8−1]に記載の固相(230mg)と1,4−ジオキサン(2.8mL)を別個にそれぞれのガラスバイアルに加え、室温で1時間振盪した。DIPEA(121μL、690μmol)とリン酸トリメチル(160μL、1.38mmol)を加え、80℃で24時間振盪した。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.05 M TBAHSO4+DTBP in NMP(5mL)で3回、0.05 M NMM in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表8−8−1]に記載の混合物を得た。
[表8−8−2]~[表8−8−5]では、2−1−m1E01−1~混合物2−1−d1E04−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−m1E01−0~混合物2−1−d1E04−0の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図4に示す。[表8−8−2]~[表8−8−5]においては、「▲×▼」は化学式で表す。



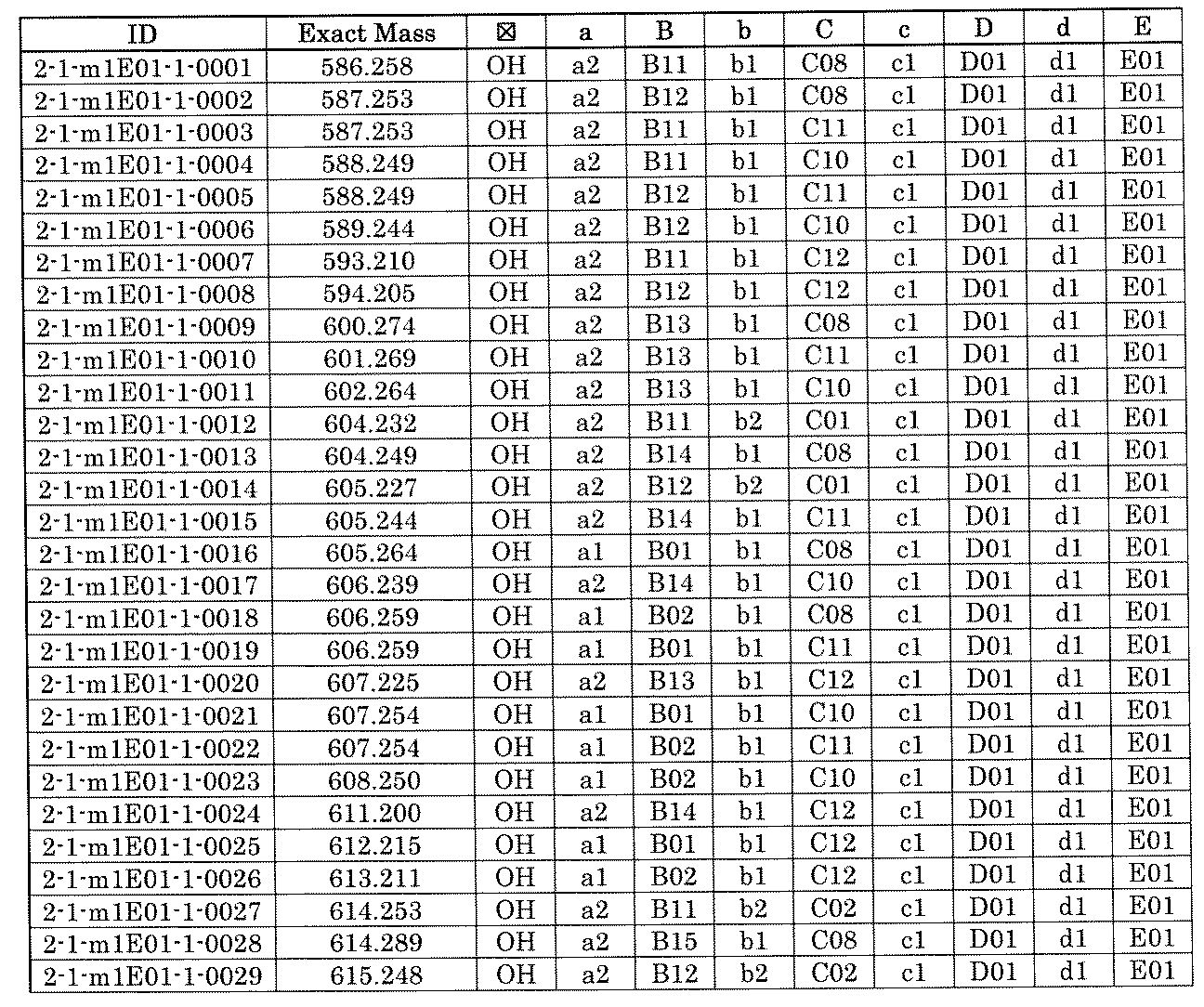







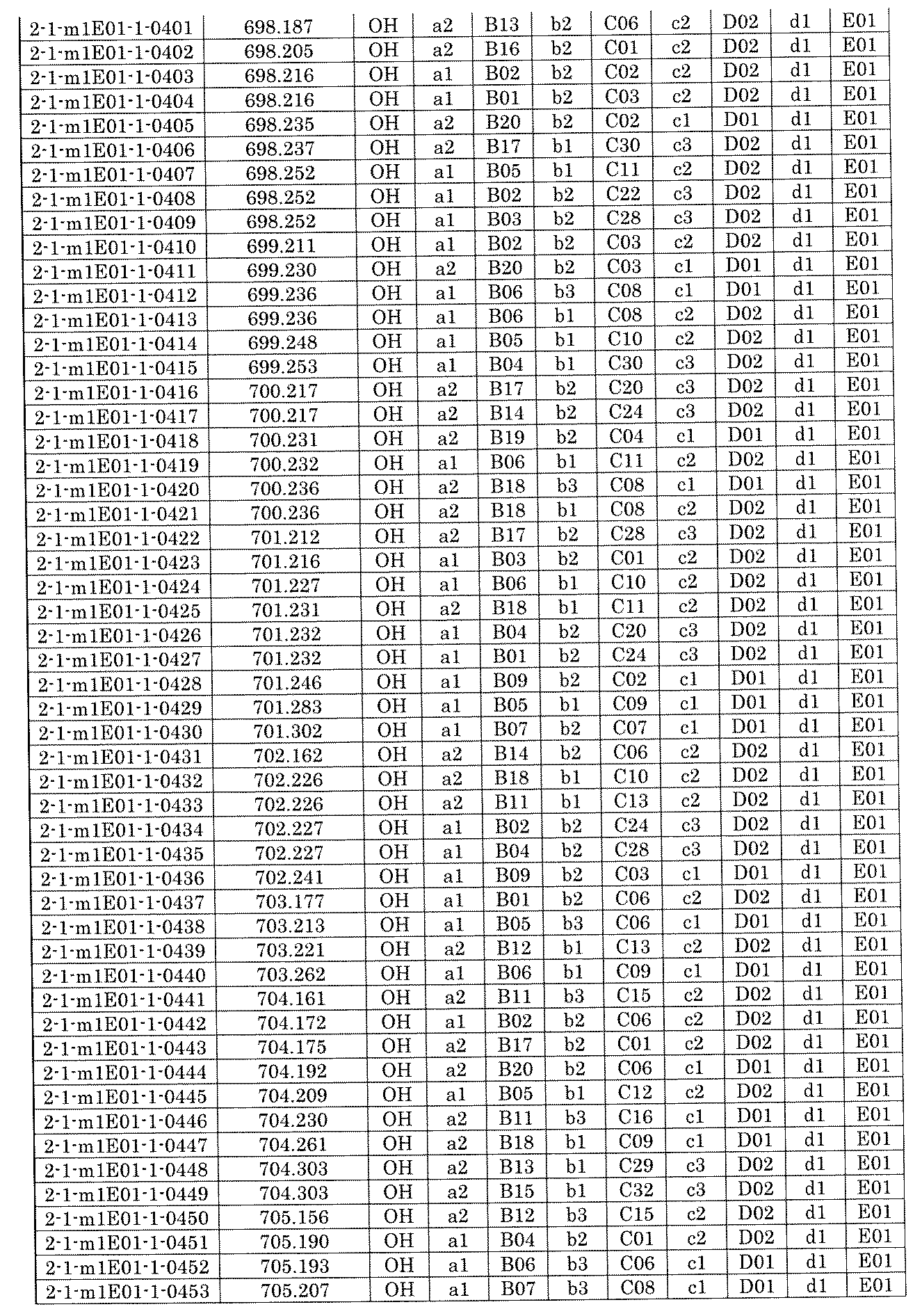


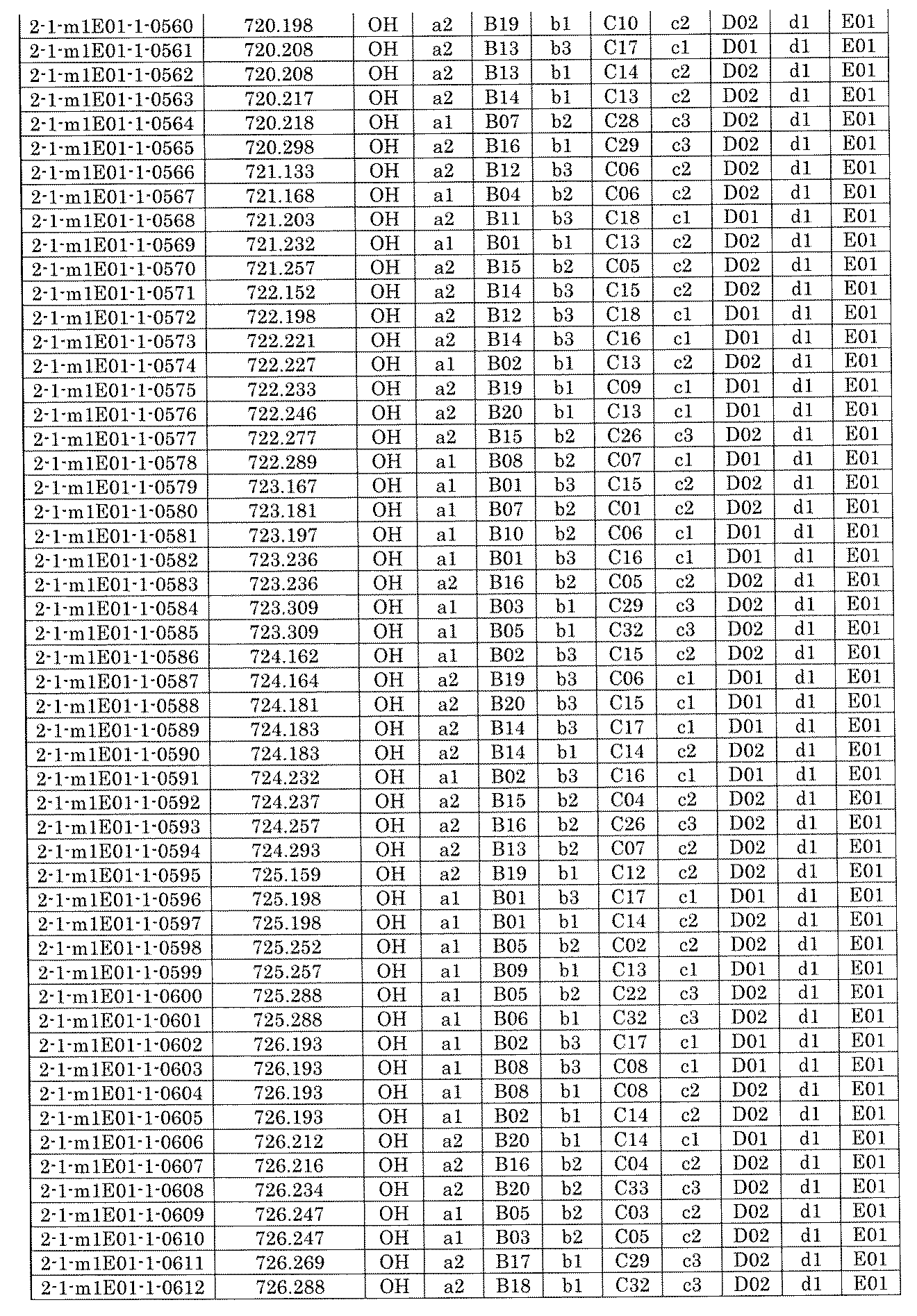







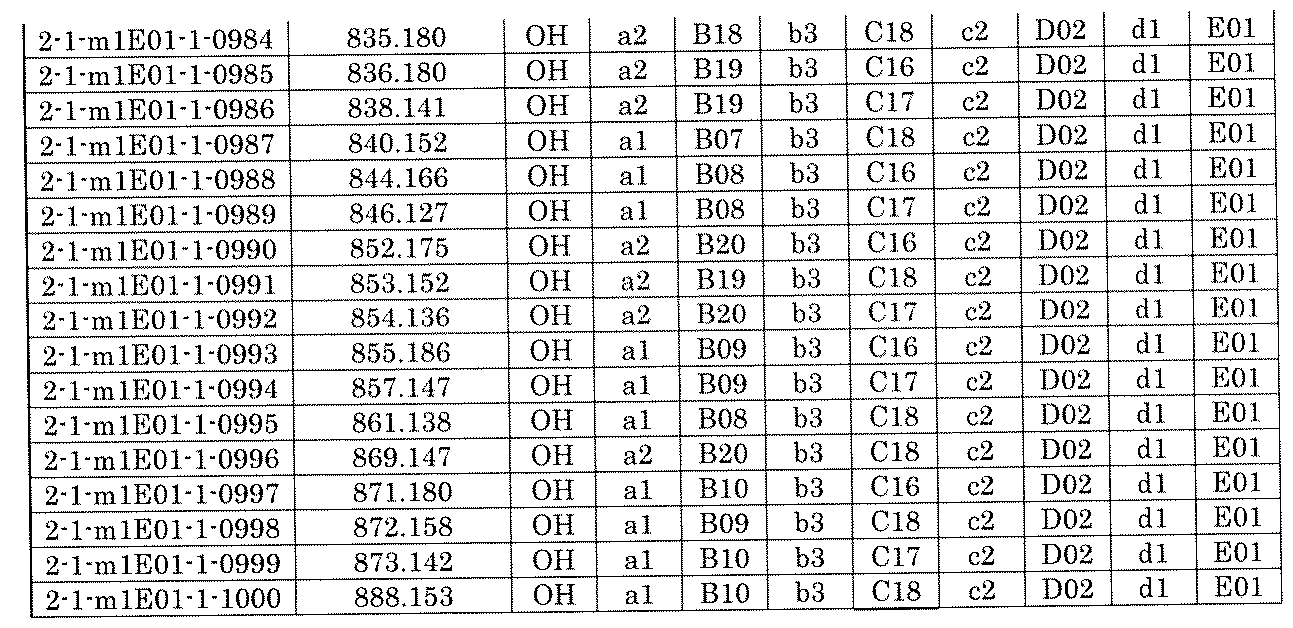












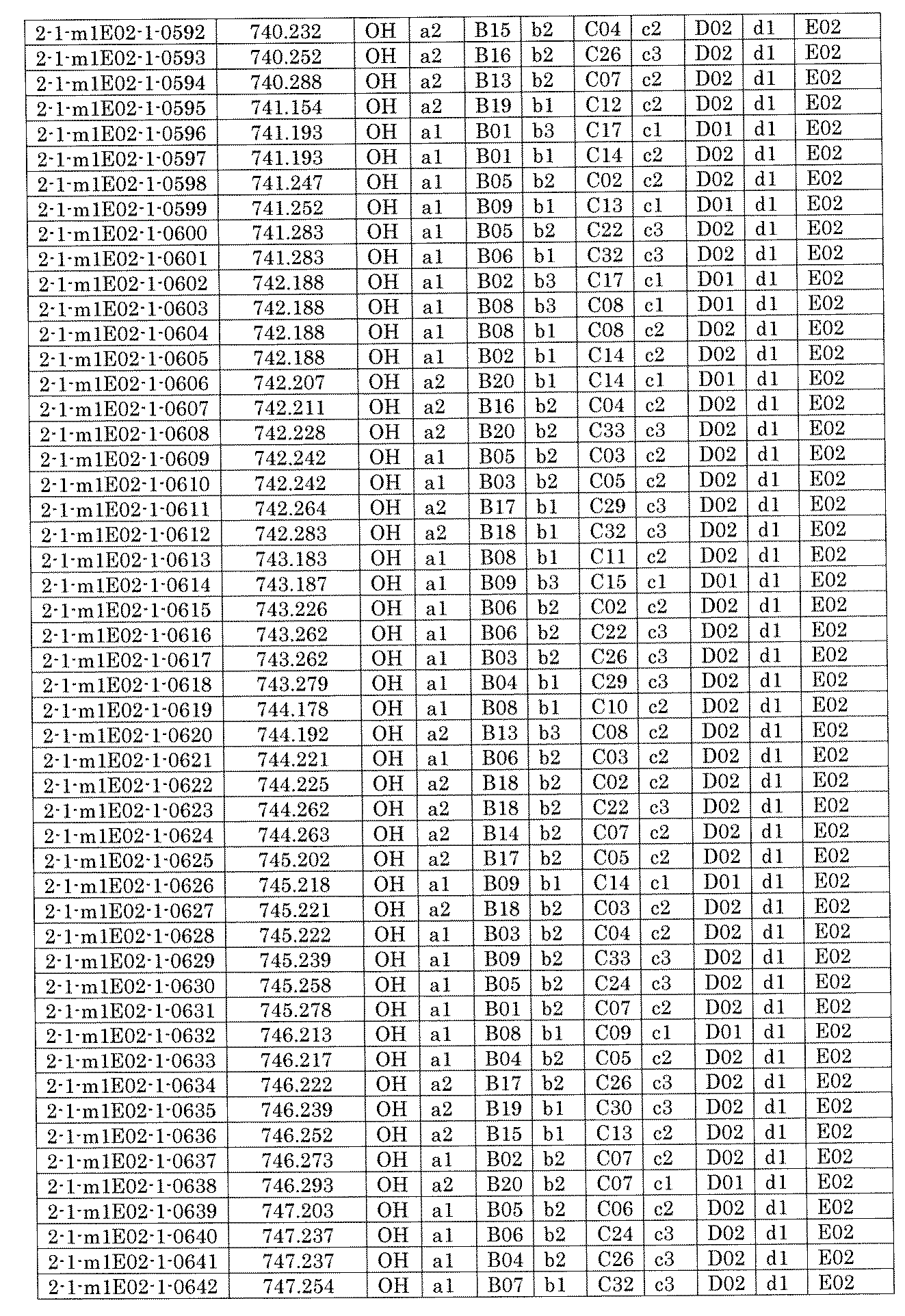











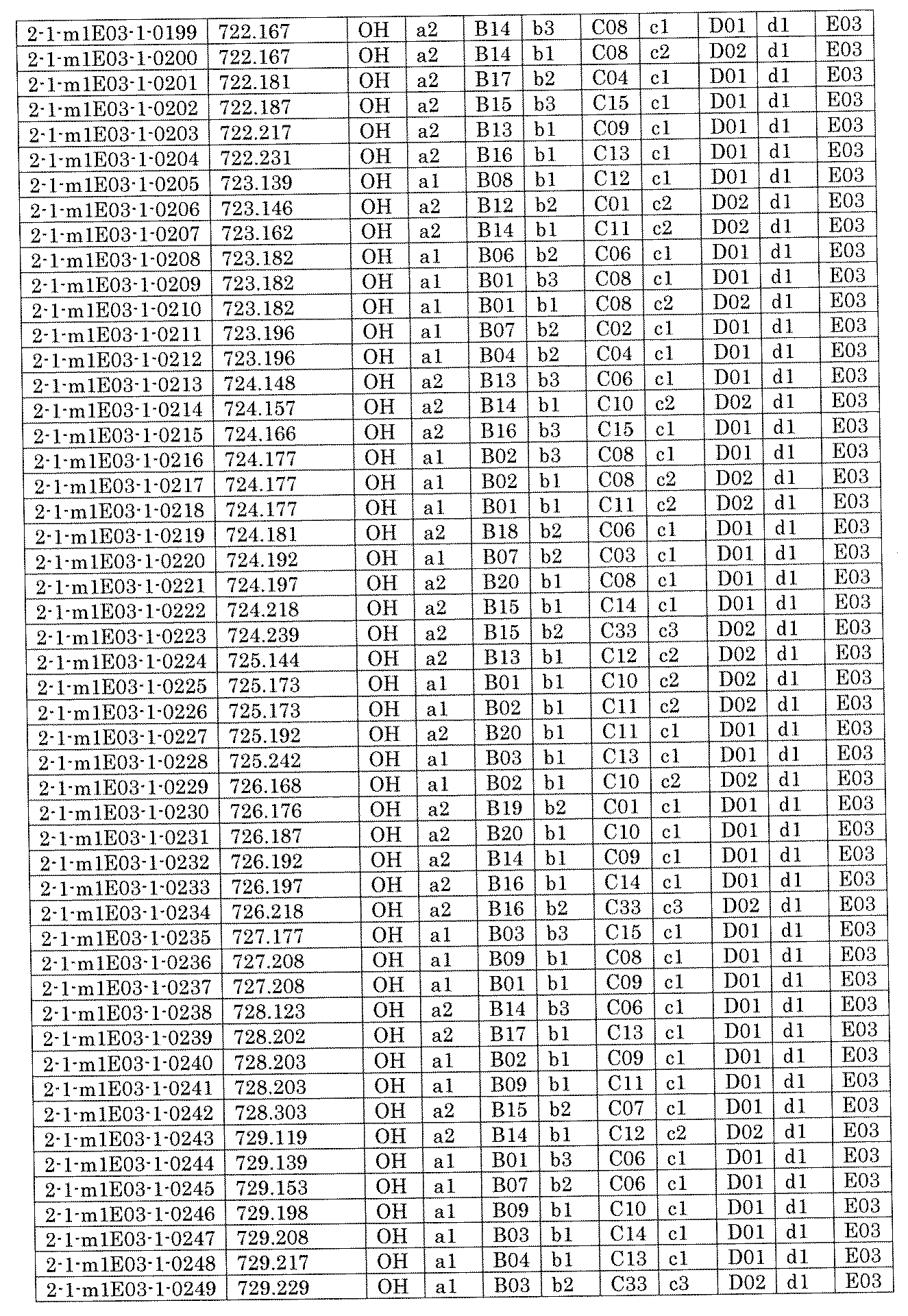


















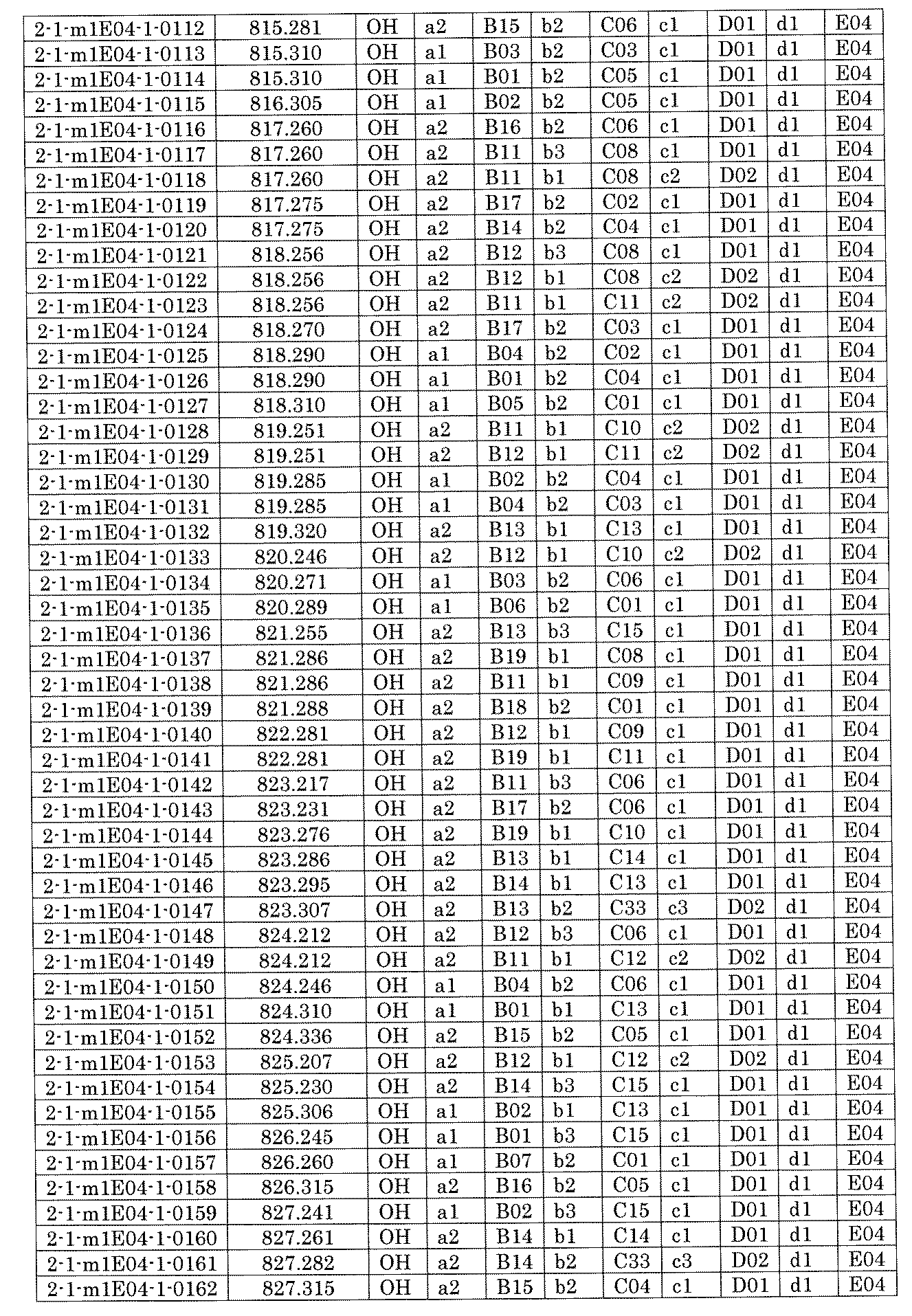
















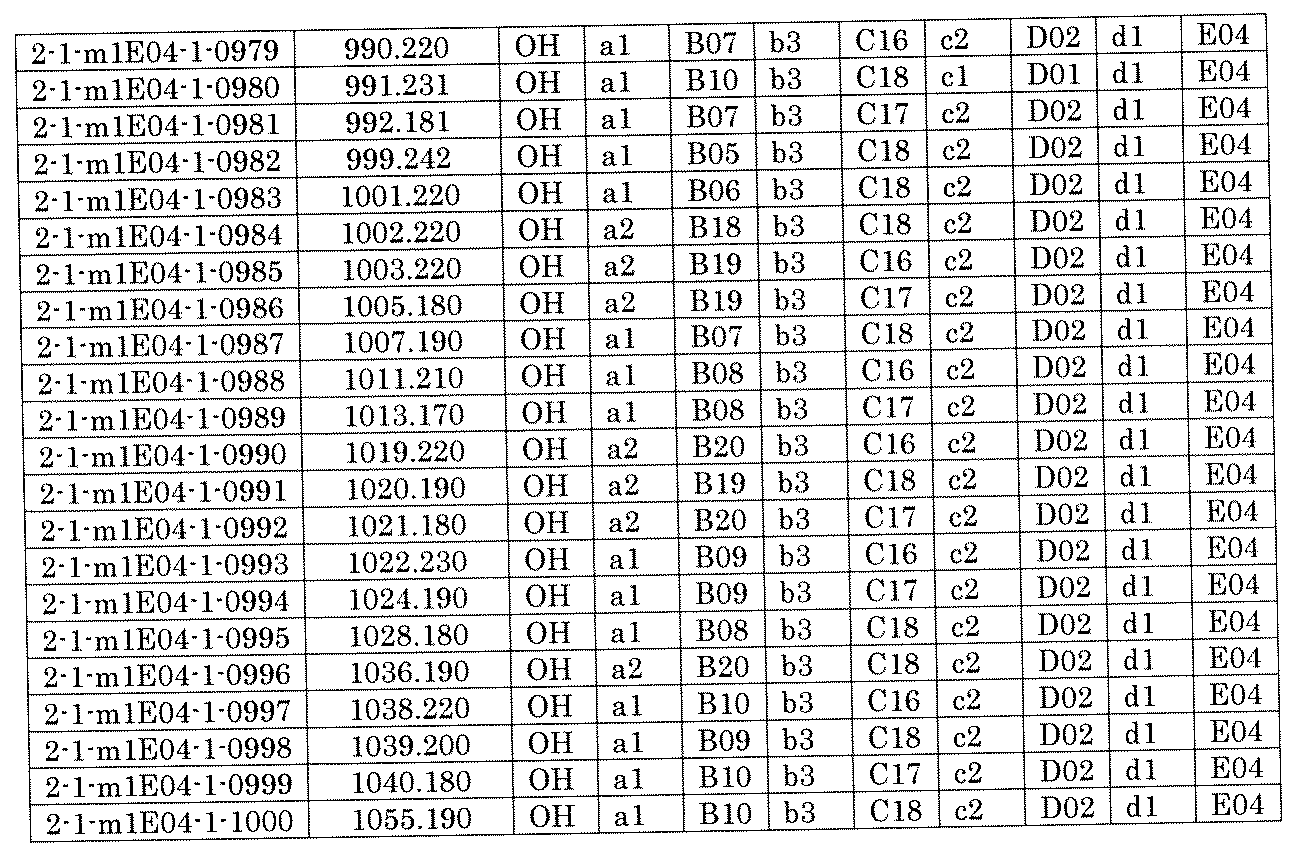
混合物2−1−m1E01−0を用いて工程E01−1を行った場合の反応式を以下に例示する。

4個の4mLのガラスバイアルに[表8−9−1]に記載の混合物(115mg)をそれぞれ別個に加えた。0.08Mペンタメチルベンゼン in DCM(2.3mL)を加え、室温で1時間振盪した。TFA(0.23mL)を加え、室温で1時間振盪した。反応液と固相の懸濁液をDMI(1.1mL)を2回使用して6mLのフィルター付きカラムに移してろ過した。DMI(1.1mL)で3回洗浄し、合わせたろ液を減圧留去した。固相担持モルホリン(Aldrichカタログ番号:493813)(175mg)とDMI(0.92mL)を加えて、室温15分間振盪した。溶液と固相の懸濁液をDMI(1.1mL)を2回使用して6mLのフィルター付きカラムに移してろ過した。DMI(1.1mL)で3回、MeCN(0.5mL)で2回洗浄し、合わせたろ液をGenevacで留去することで、[表8−9−1]に記載の混合物を得た。得られた残渣を[表8−9−1]に記載の量のDMSOに溶解させた。そのうち10μLをAS−MSアッセイ用に分注し、残りの溶液を減圧留去し、次の工程E01−2~E04−2の原料として使用した。混合物2−1−m1E01−1~混合物2−1−m1E04−1の詳細は[表8−8−2]~[表8−8−5]に示す。
[表8−8−2]~[表8−8−5]では、2−1−m1E01−1~混合物2−1−d1E04−1に含まれうるそれぞれの化合物をIDで示し、混合物2−1−m1E01−0~混合物2−1−d1E04−0の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は[図4]に示す。
混合物2−1−m1E01−1を用いて工程E01−2を行った場合の反応式を以下に例示する。

0.5−2mLのマイクロウェーブ容器に入った[表8−10−1]に記載の混合物に、[表8−10−1]に記載の量のDMFとEtOH、BTPP、Et3PO4を加え、70℃で27時間振盪した。反応液をMeCN(0.2mL)を2回使用して、[表8−10−1]に記載した量のISOLUTE(登録商標) CBA(0.7mmol/g)を詰めた6mLのフィルター付きカラムに移した。10分後、MeCN(0.5mL)で5回洗浄し、合わせたろ液を減圧留去し、[表8−10−1]に記載の混合物を得た。混合物の詳細は[表8−10−2]~[表8−10−5]に示す。





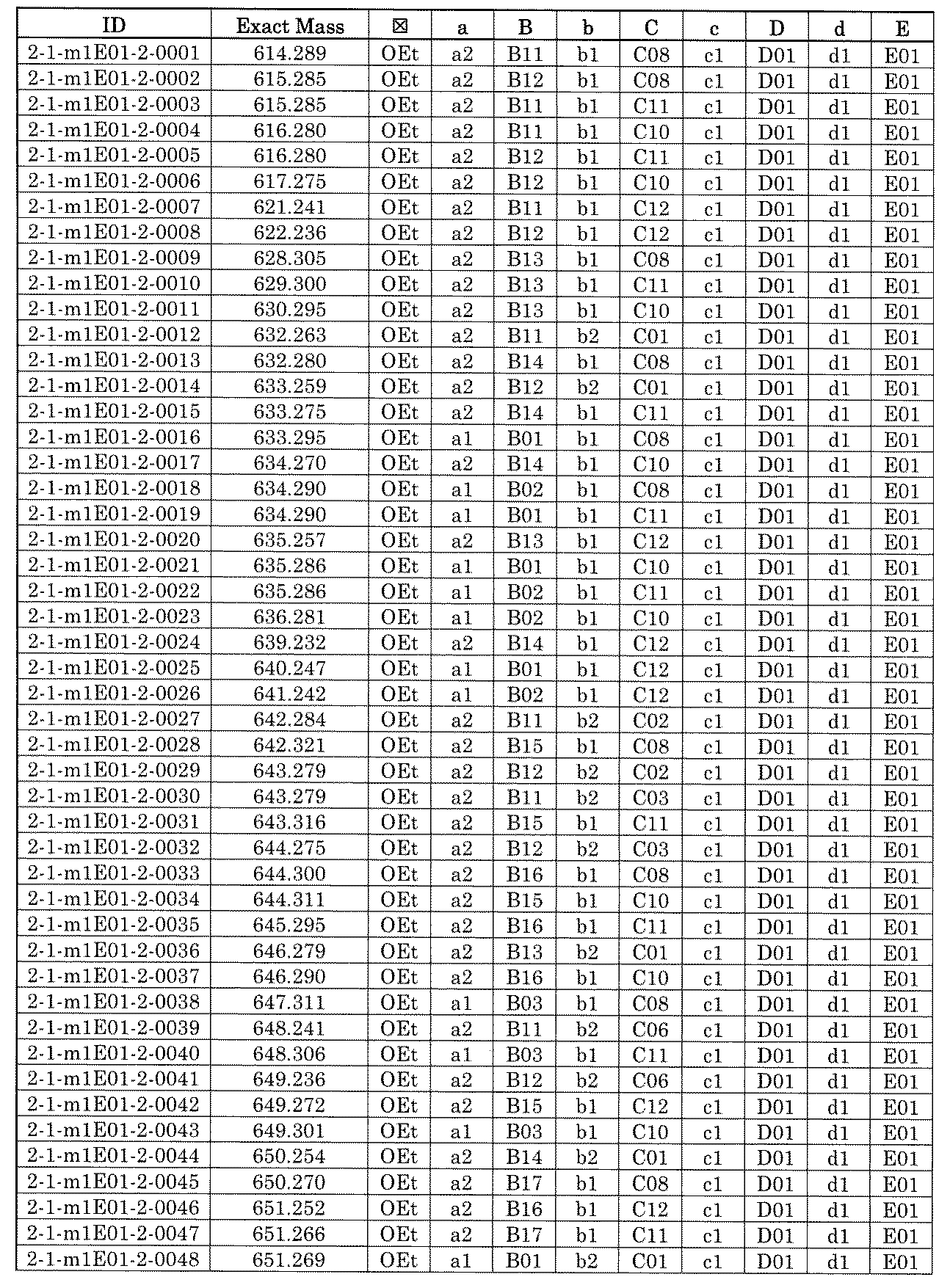


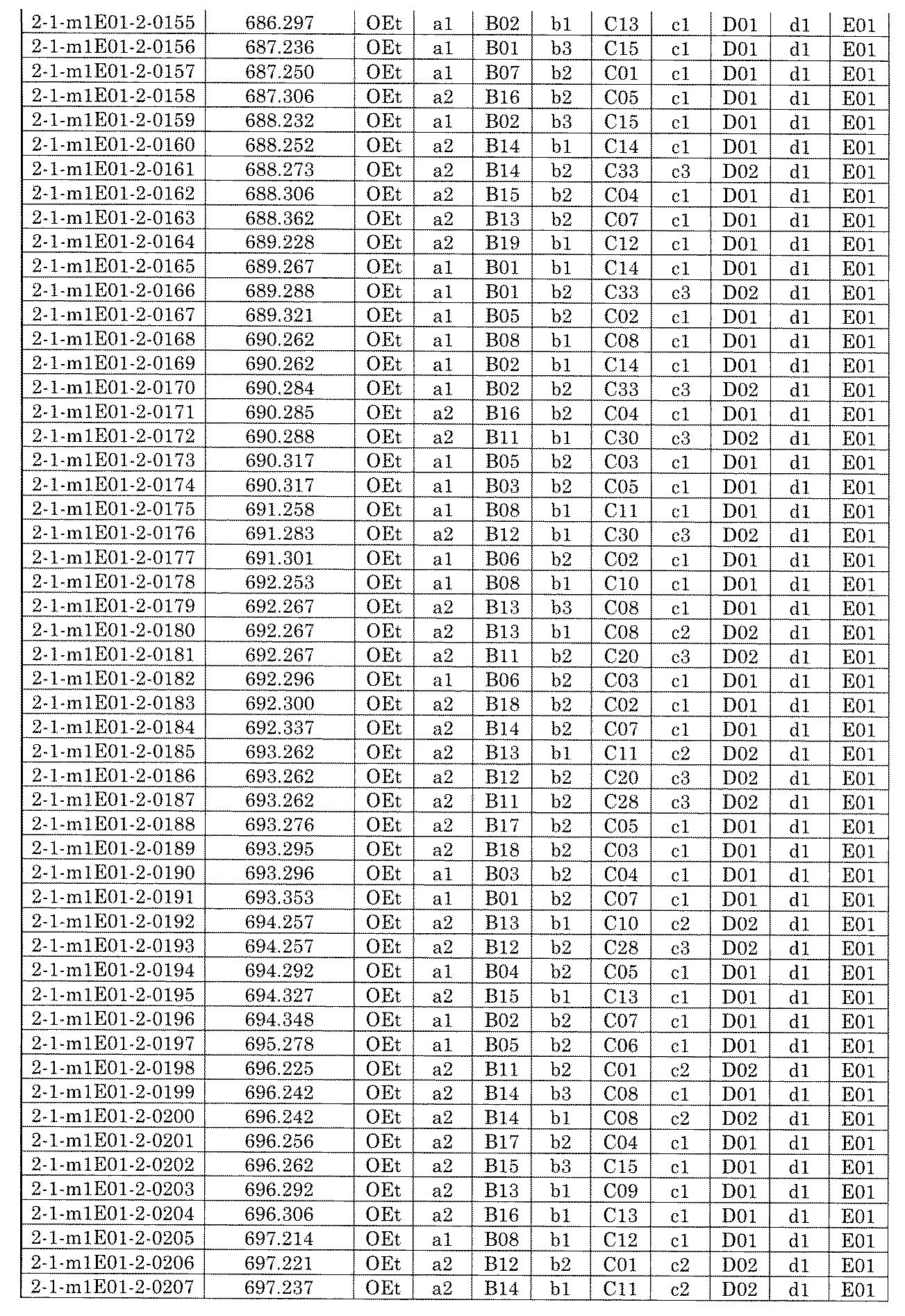



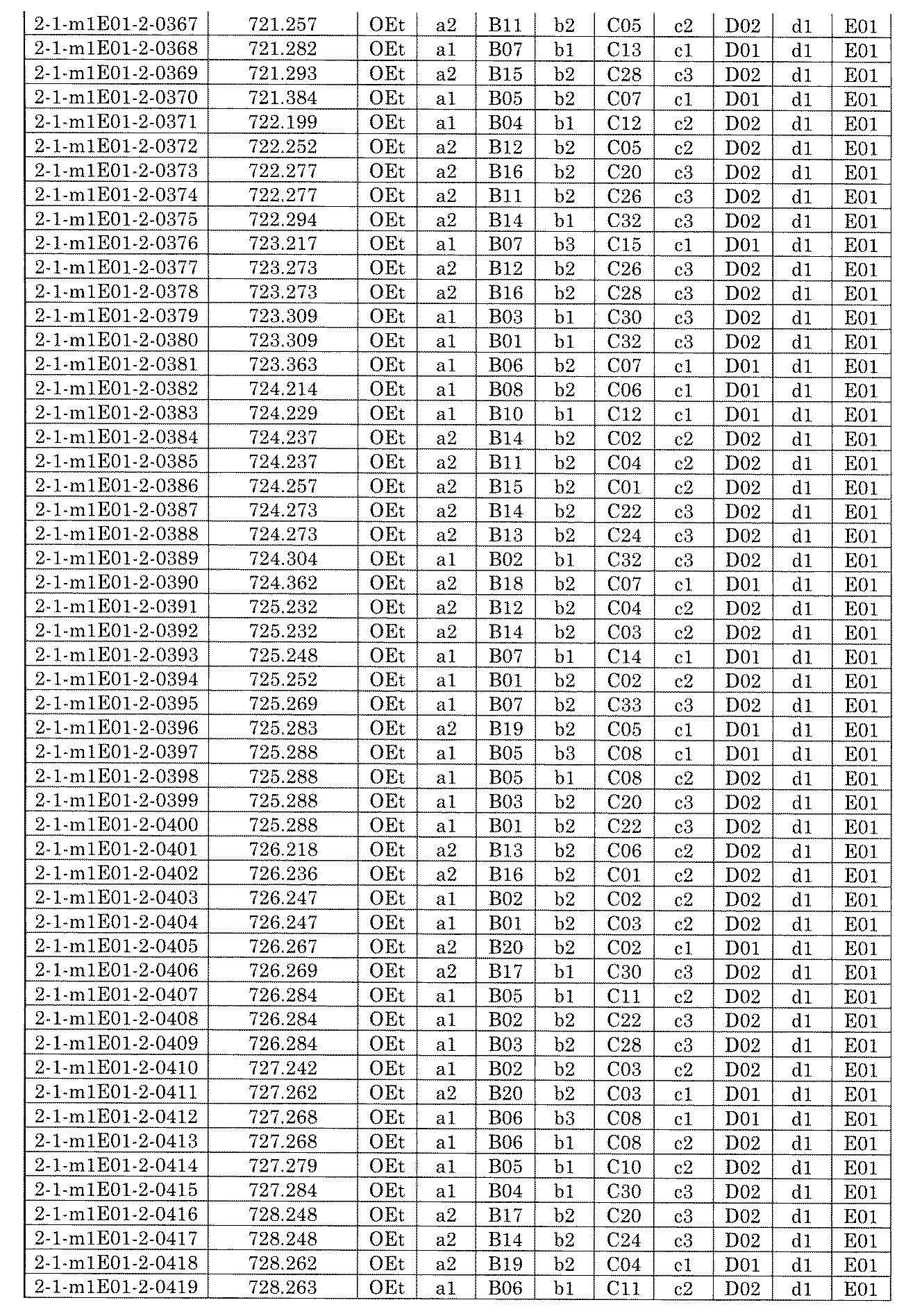





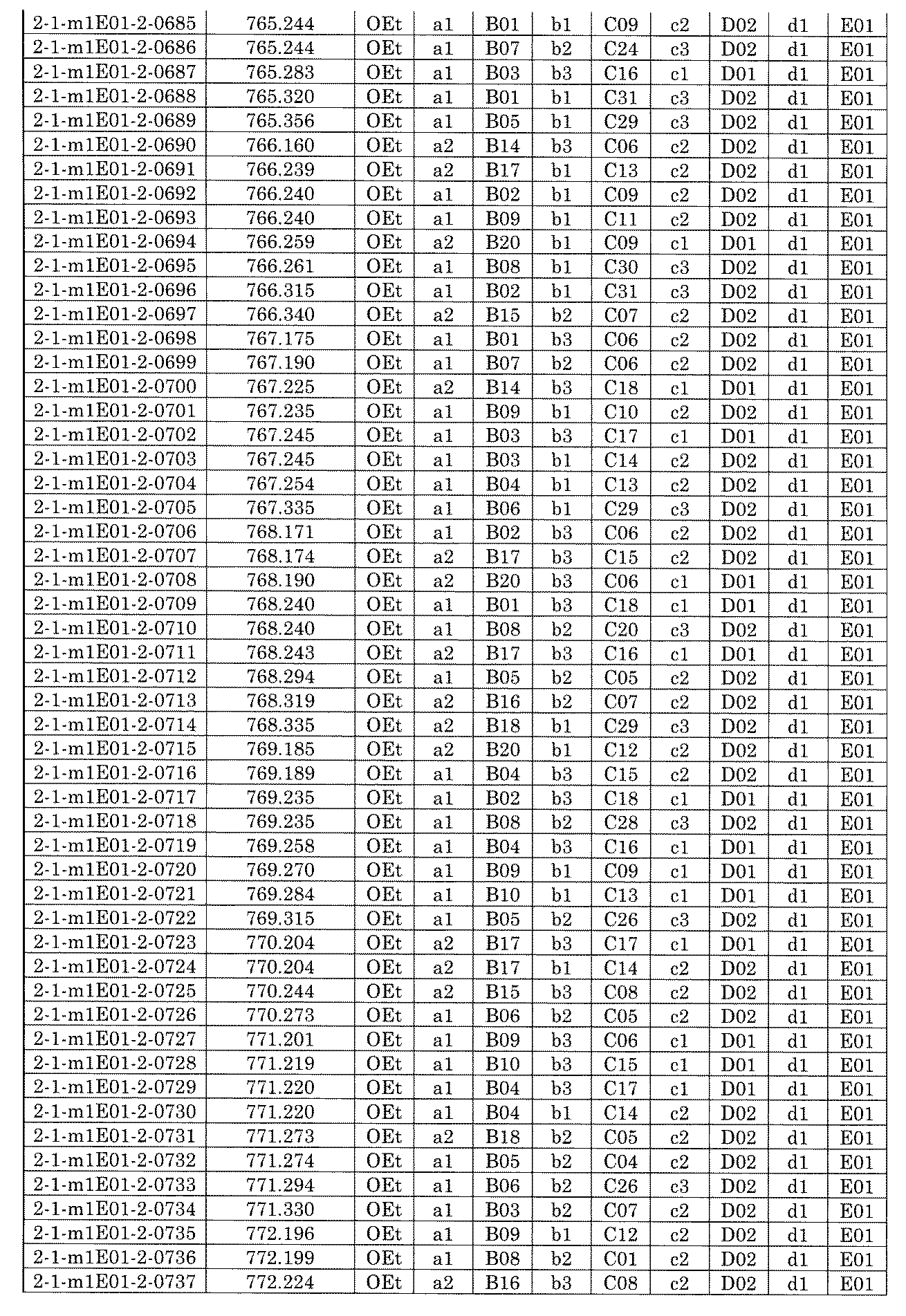





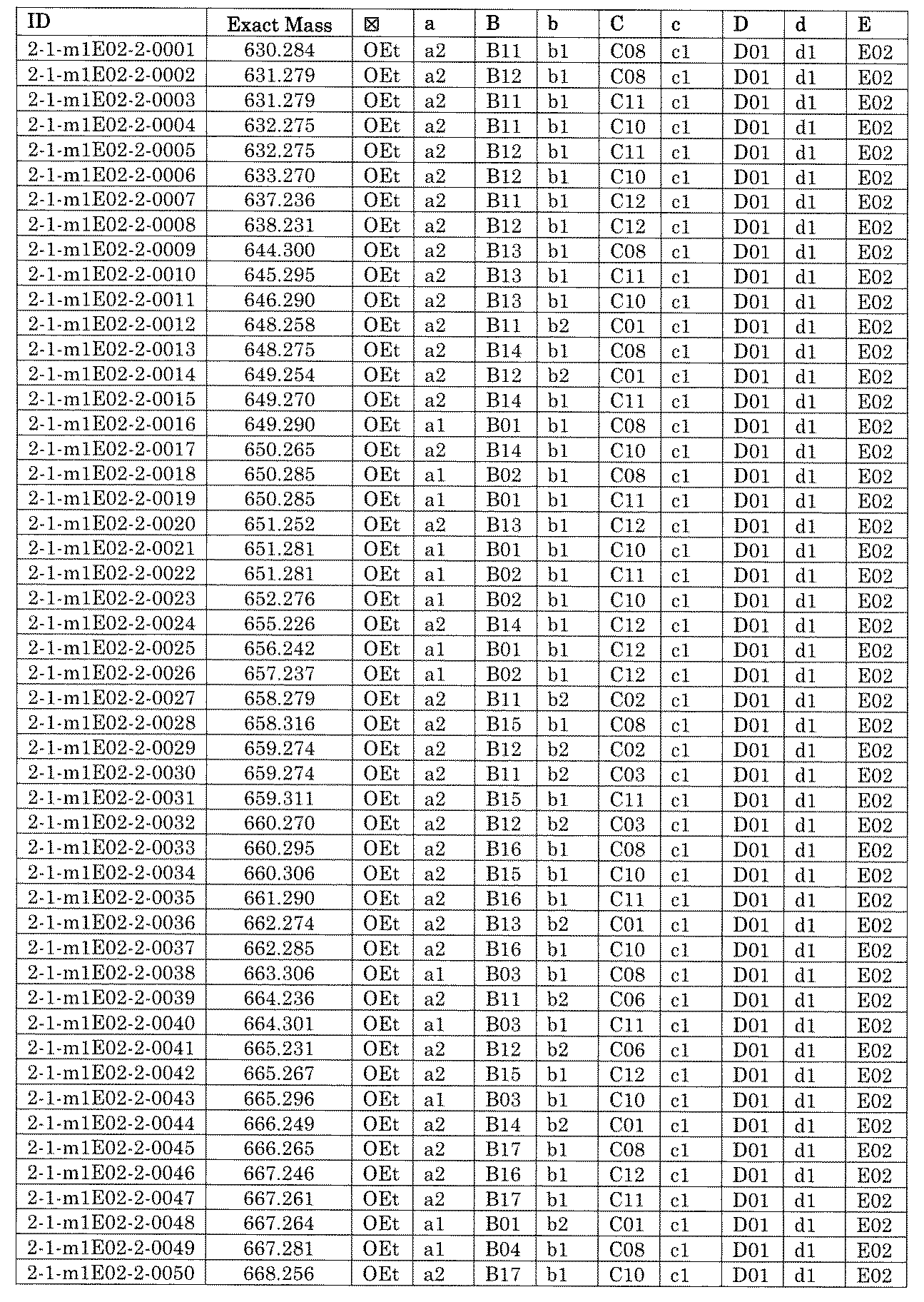






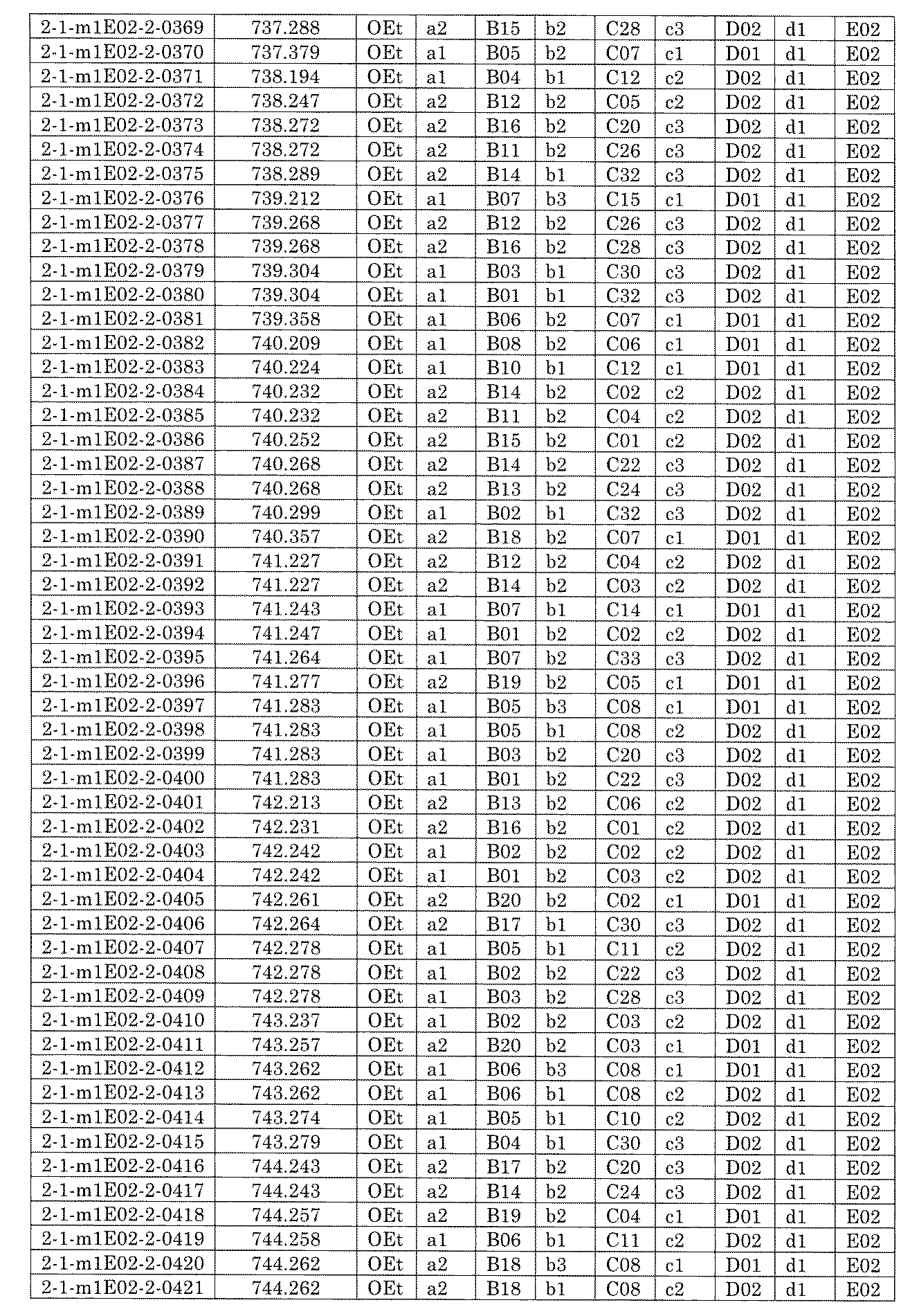

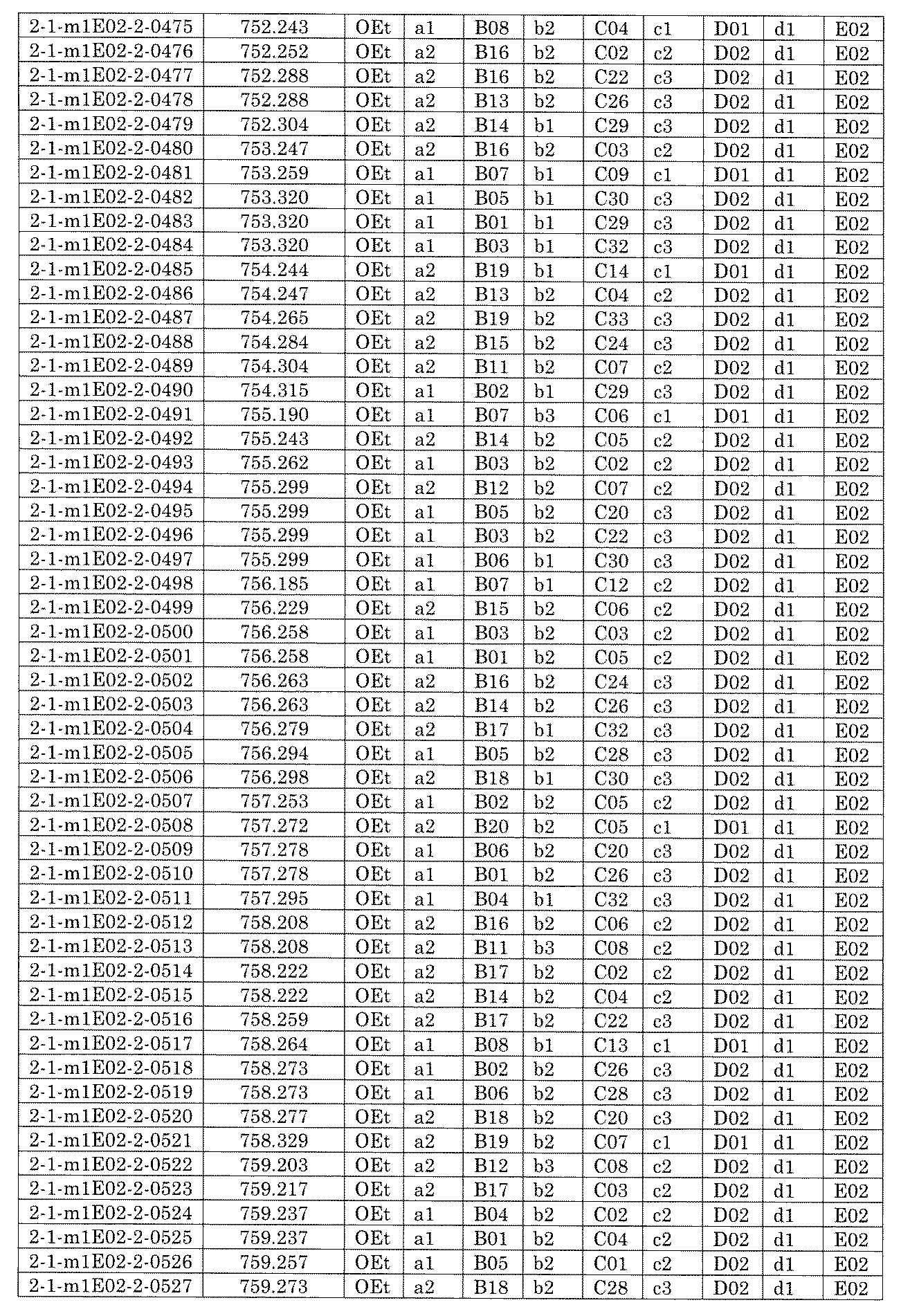




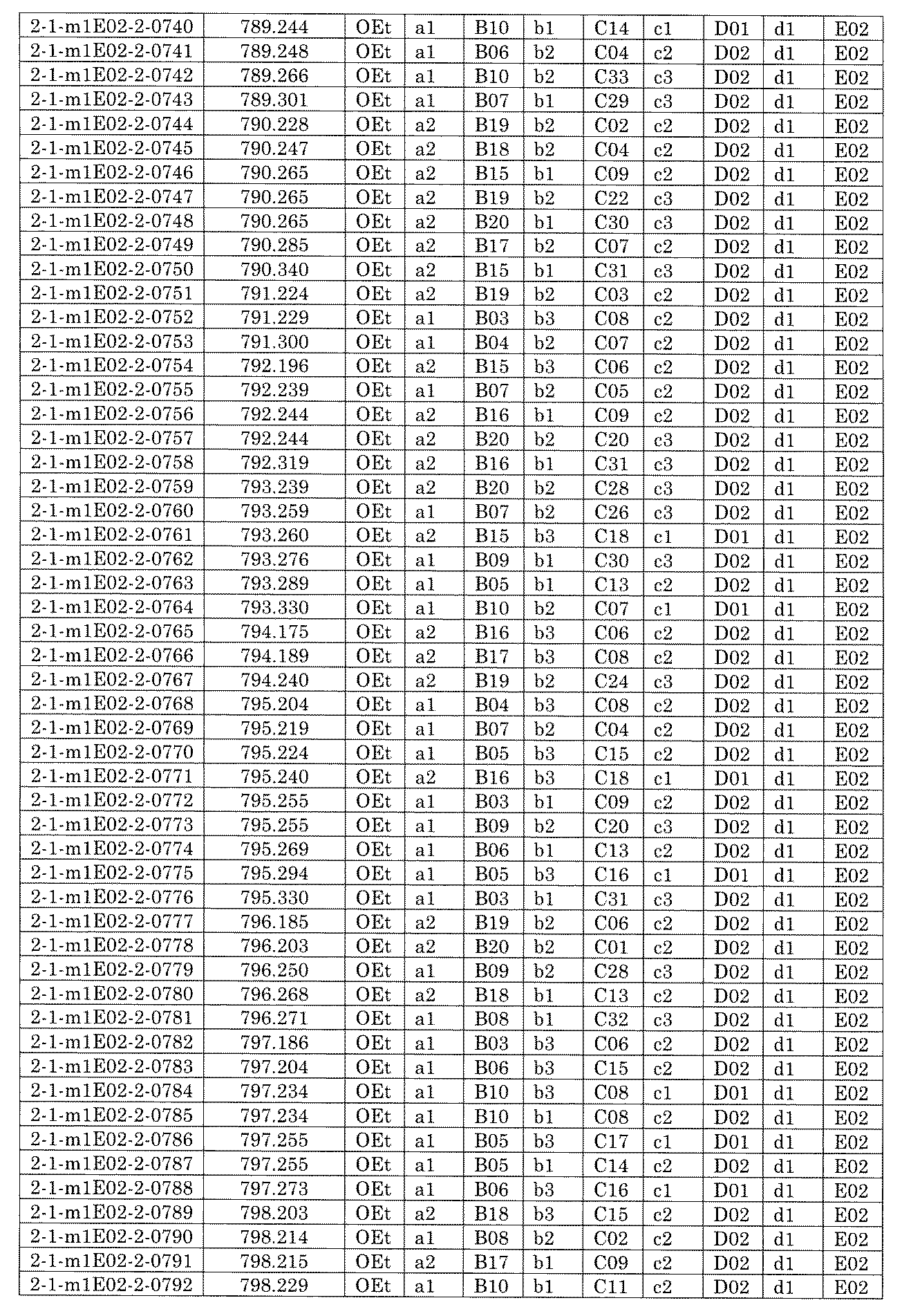



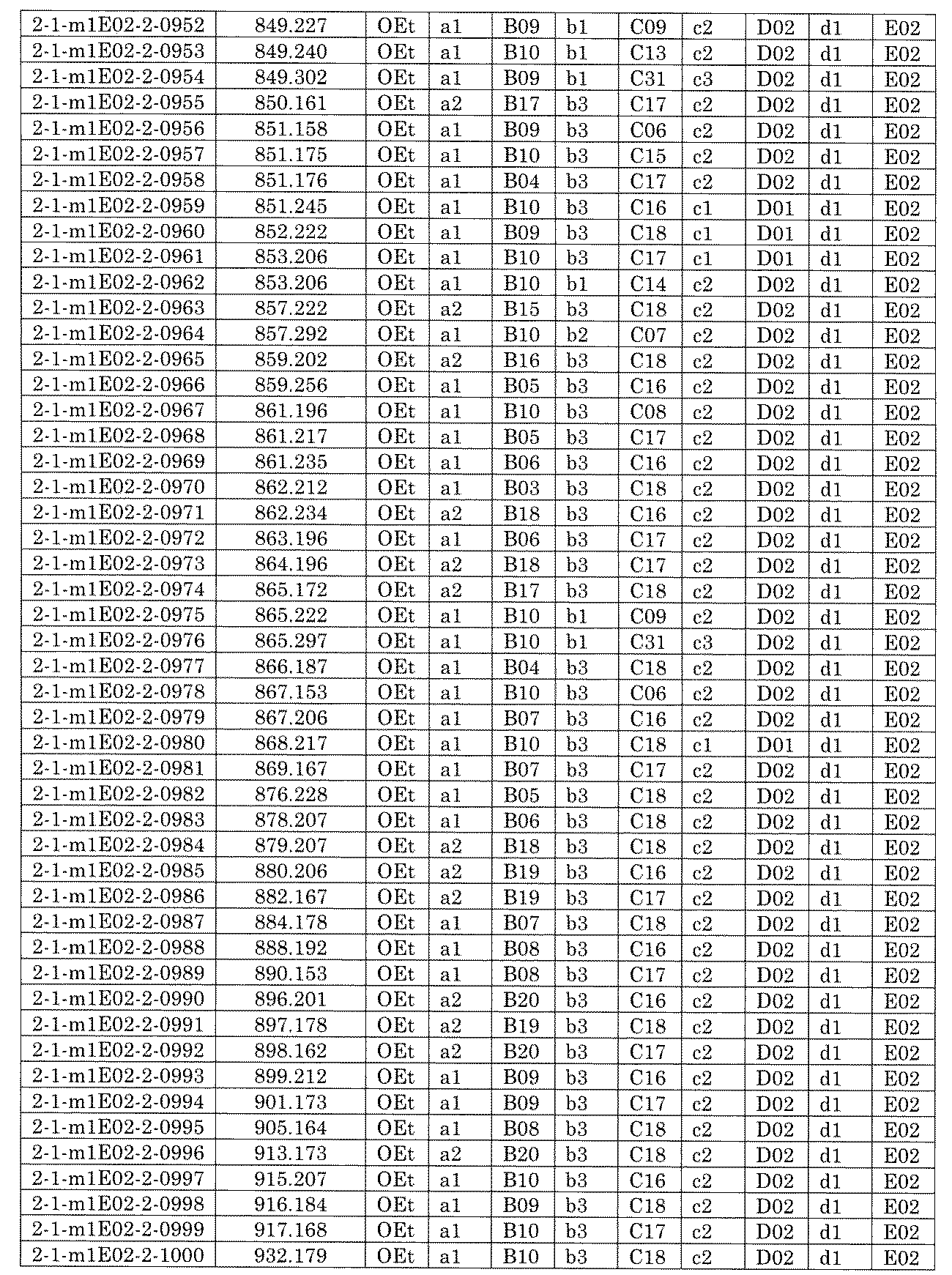


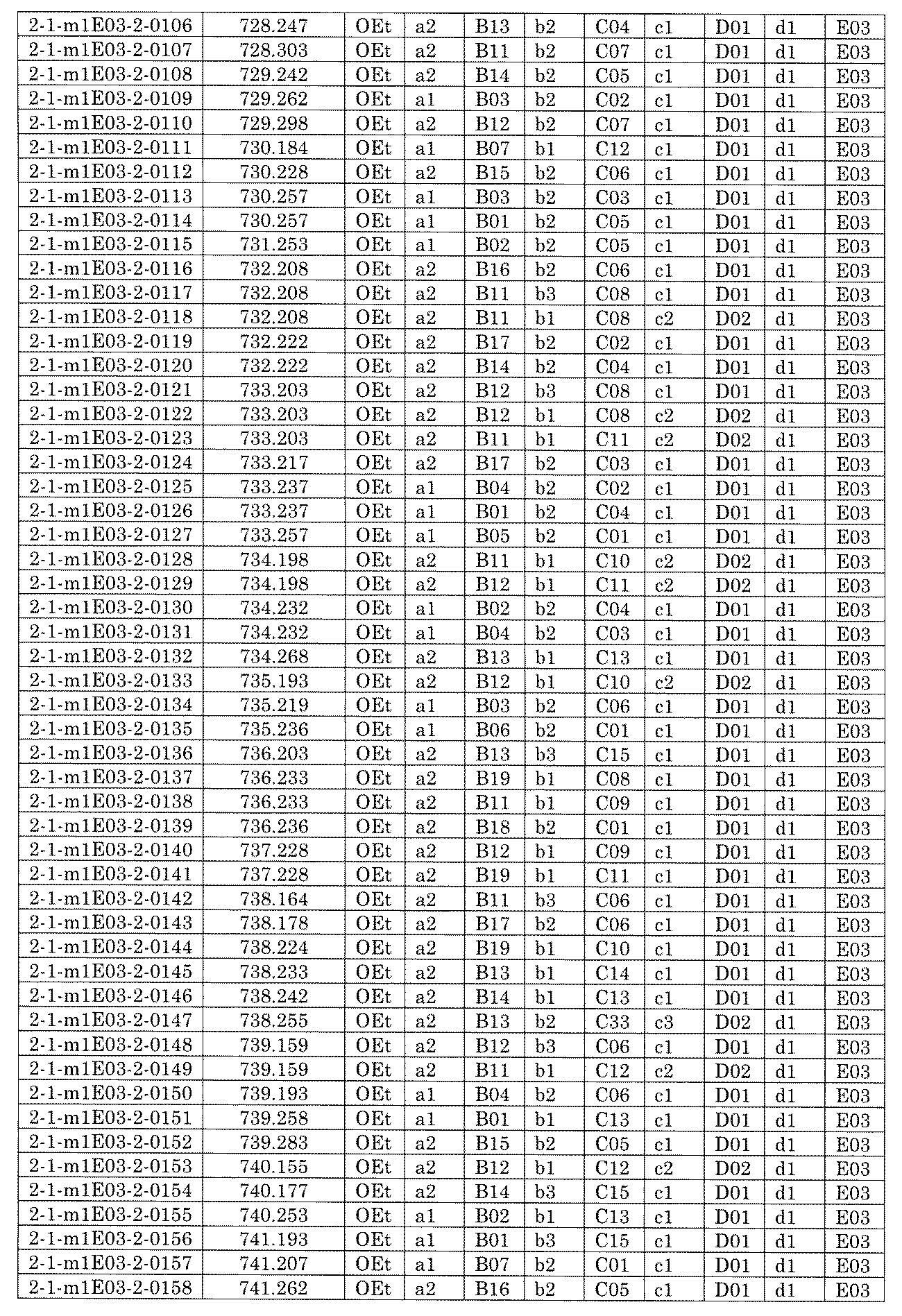




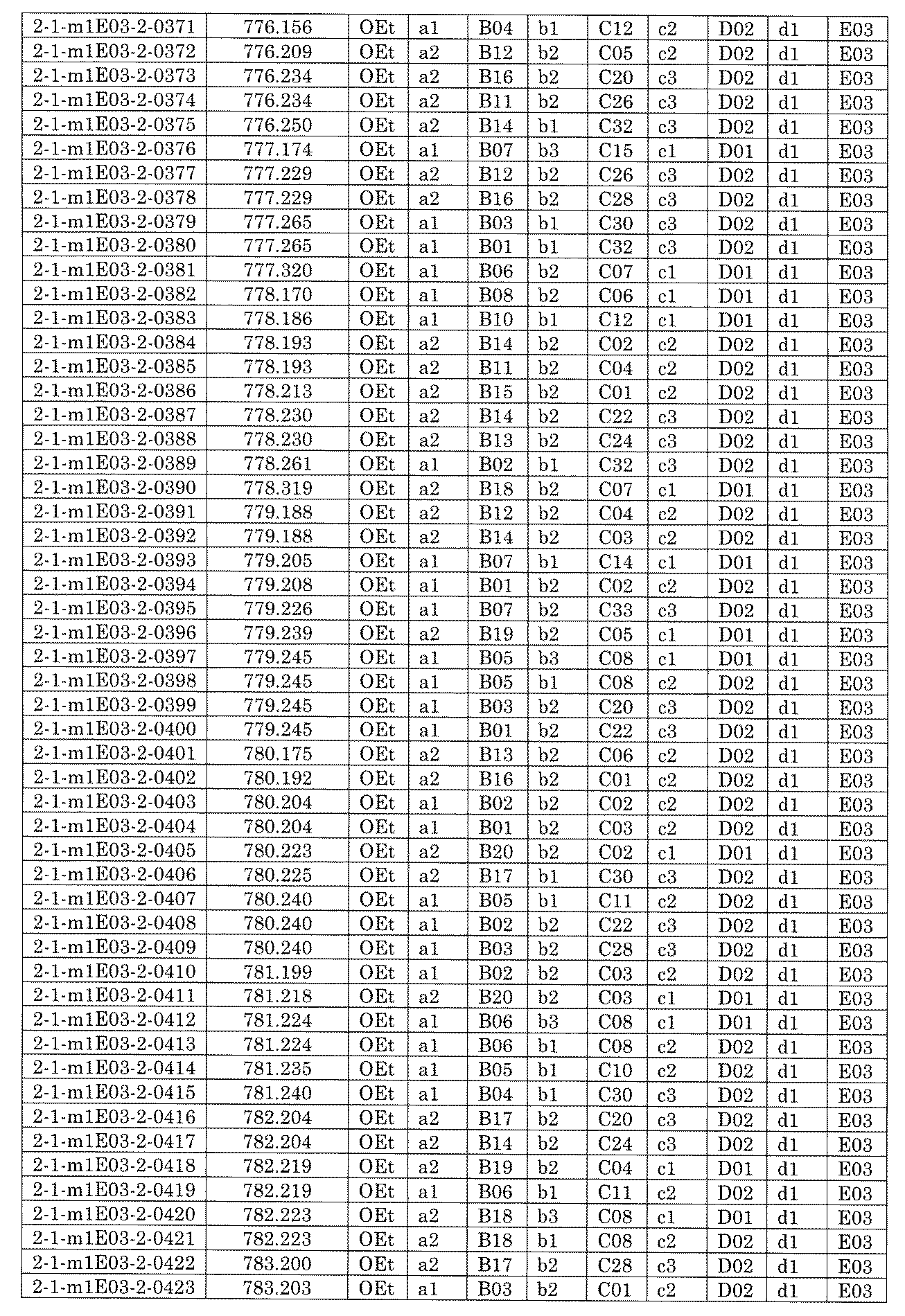































実施例8−9および8−10にて合成した混合物は、以下に示す分析条件により保持時間の測定と質量分析を実施し、Compound Discoverer 3.2(Thermo Fisher Scientific)を用いて解析した。

なお、MSピークのm/zについては小数点以下4桁目までを確認しているが、[表8−11−1]~[表8−11−8]中の表記においては小数点以下の0を省略して記載していることもある。例えば、「587.0000」の場合「587」、「587.100」の場合「587.1」、「587.120」の場合「587.12」、「587.1230」の場合「587.123」と表記していることもある。また、保持時間については小数点以下3桁目までを確認しているが、[表8−11−1]~[表8−11−8]中の表記においては小数点以下の0を省略して記載していることもある。例えば、「15.000」の場合「15」、「15.100」の場合「15.1」、「15.120」の場合「15.12」と表記していることもある。




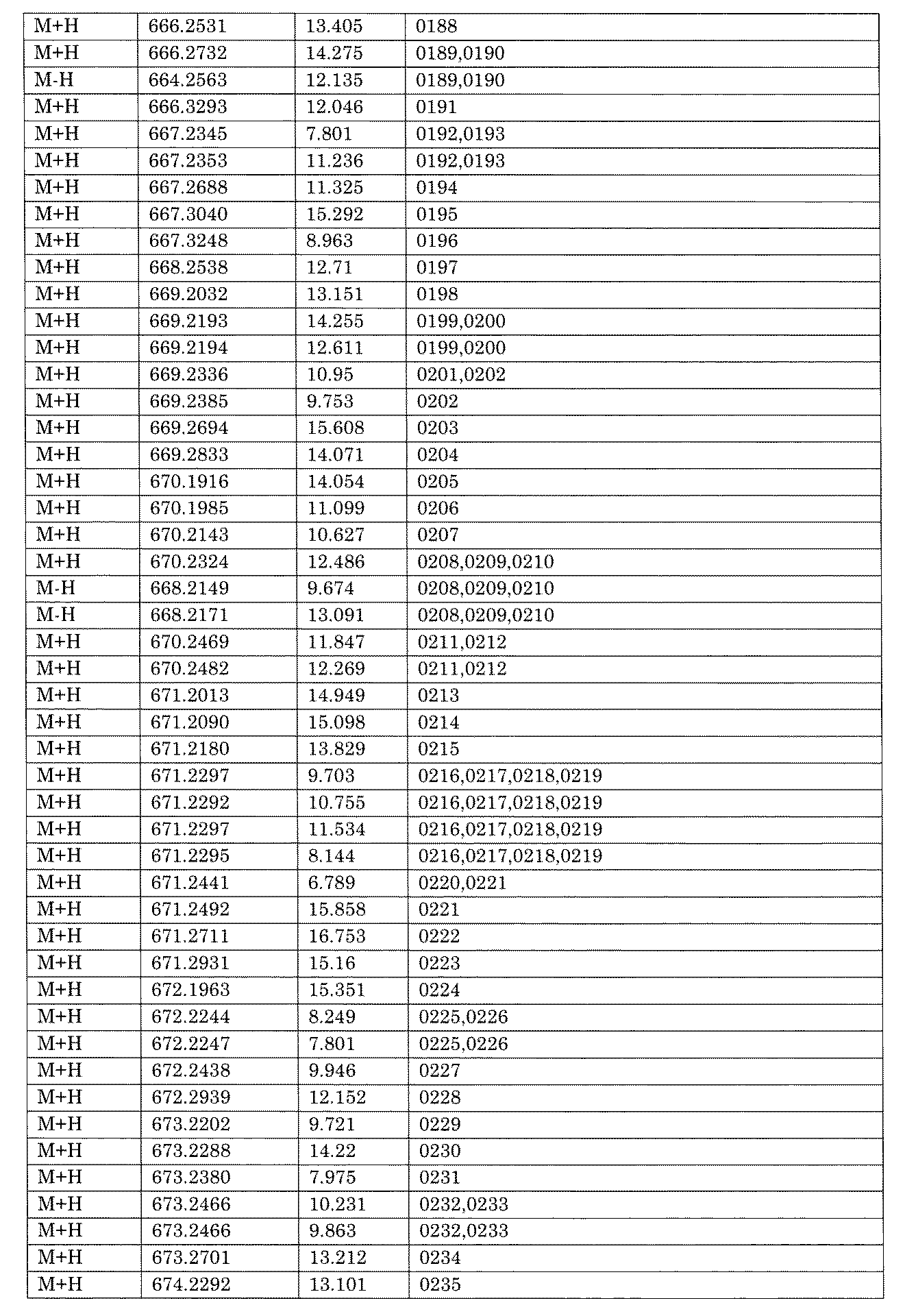



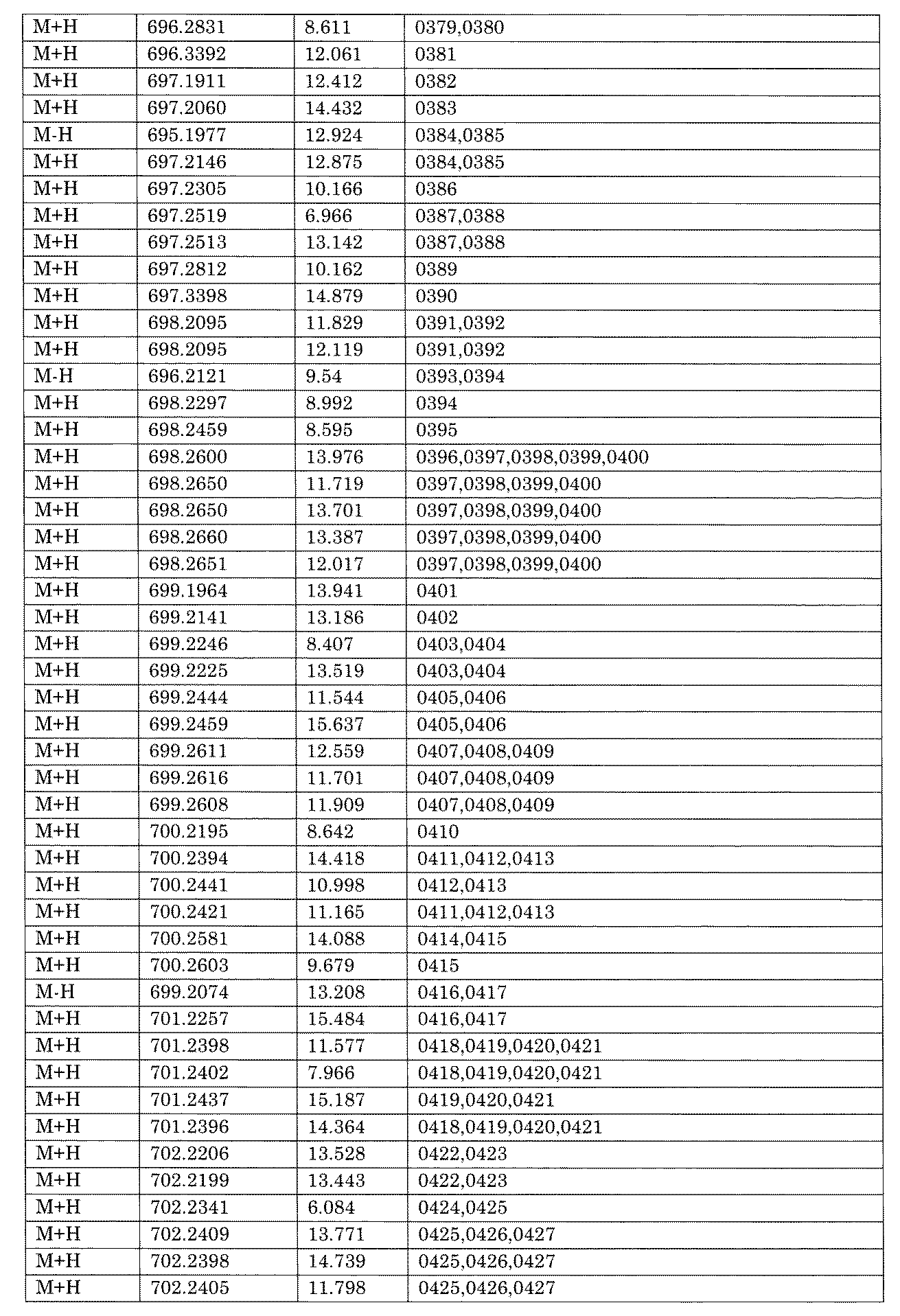
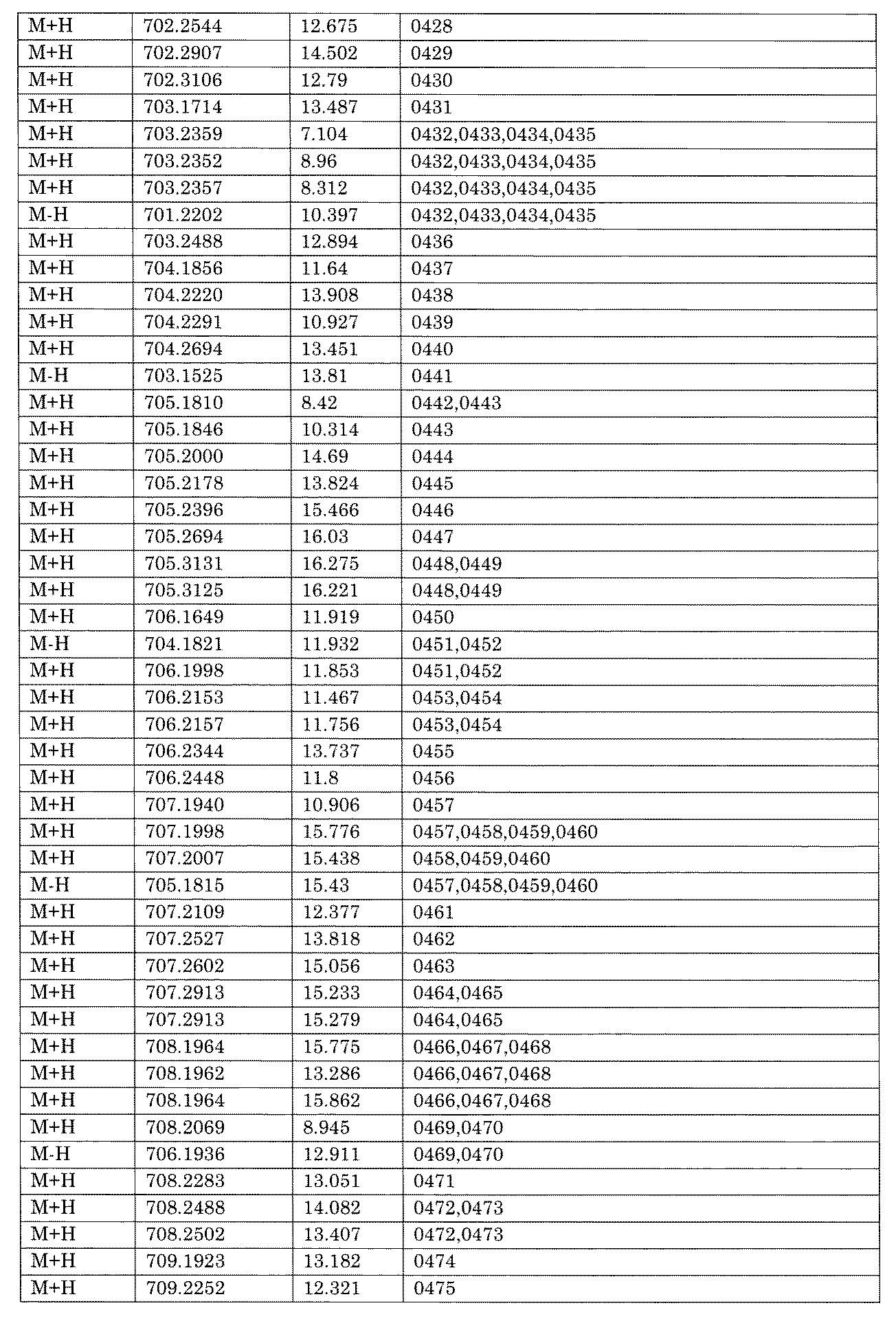

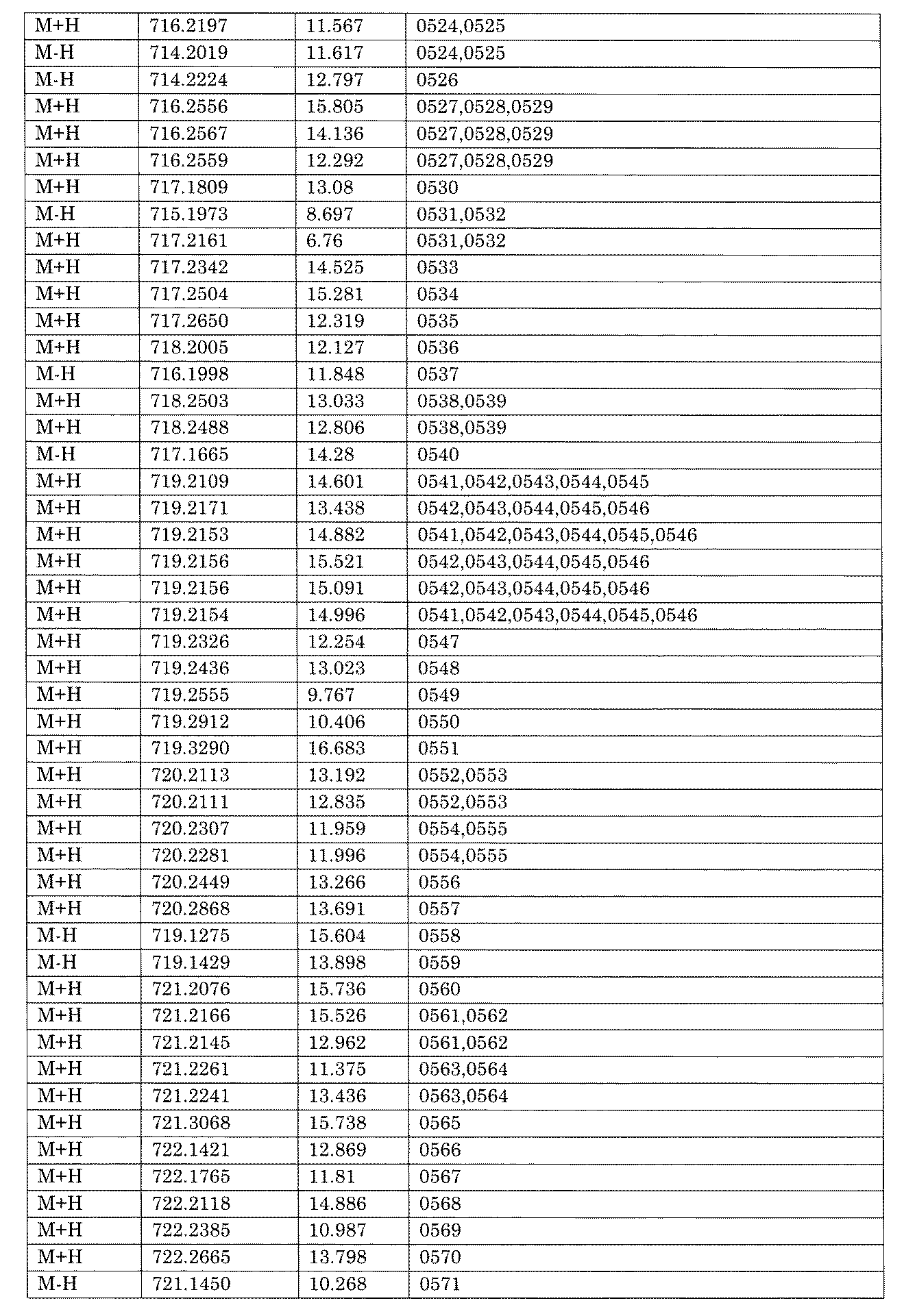

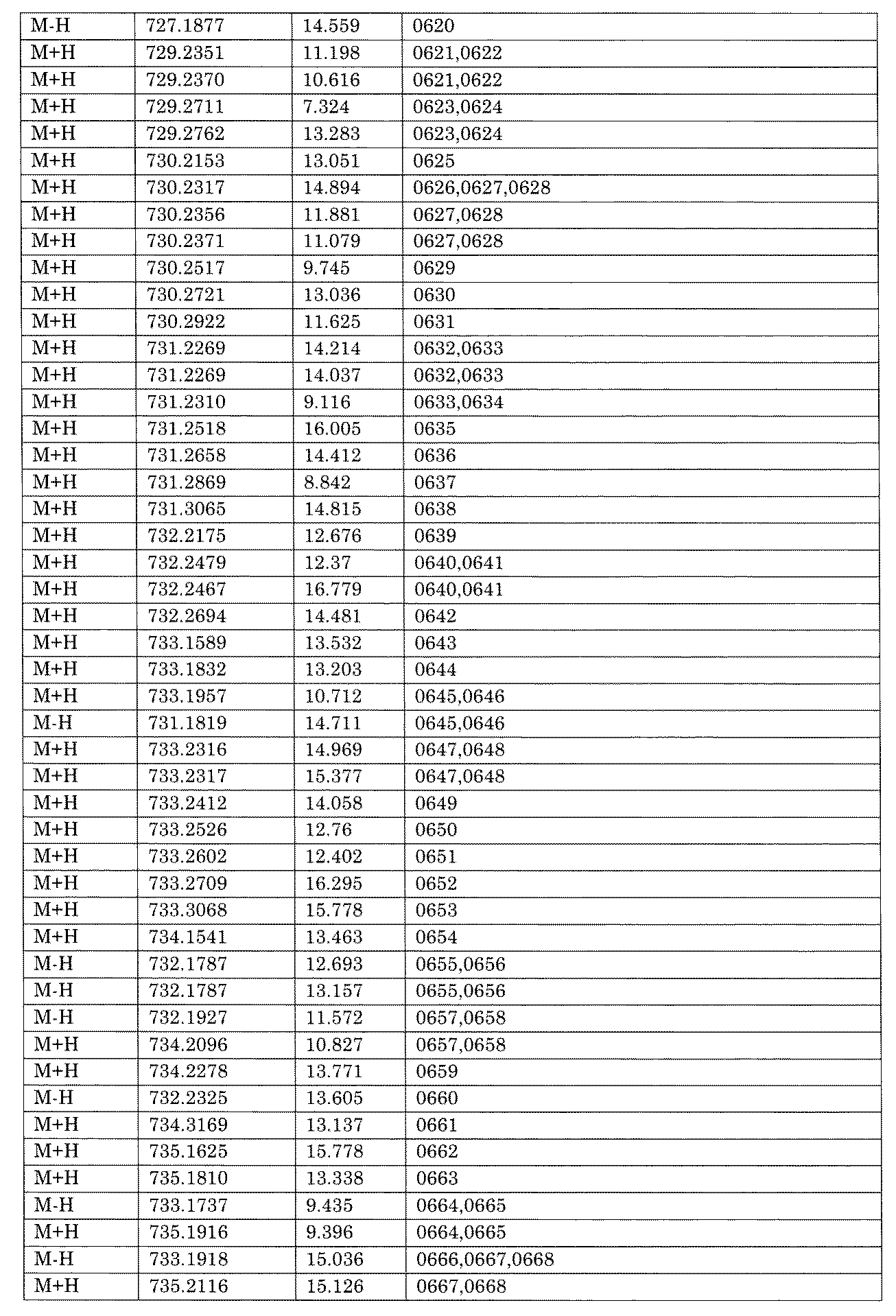
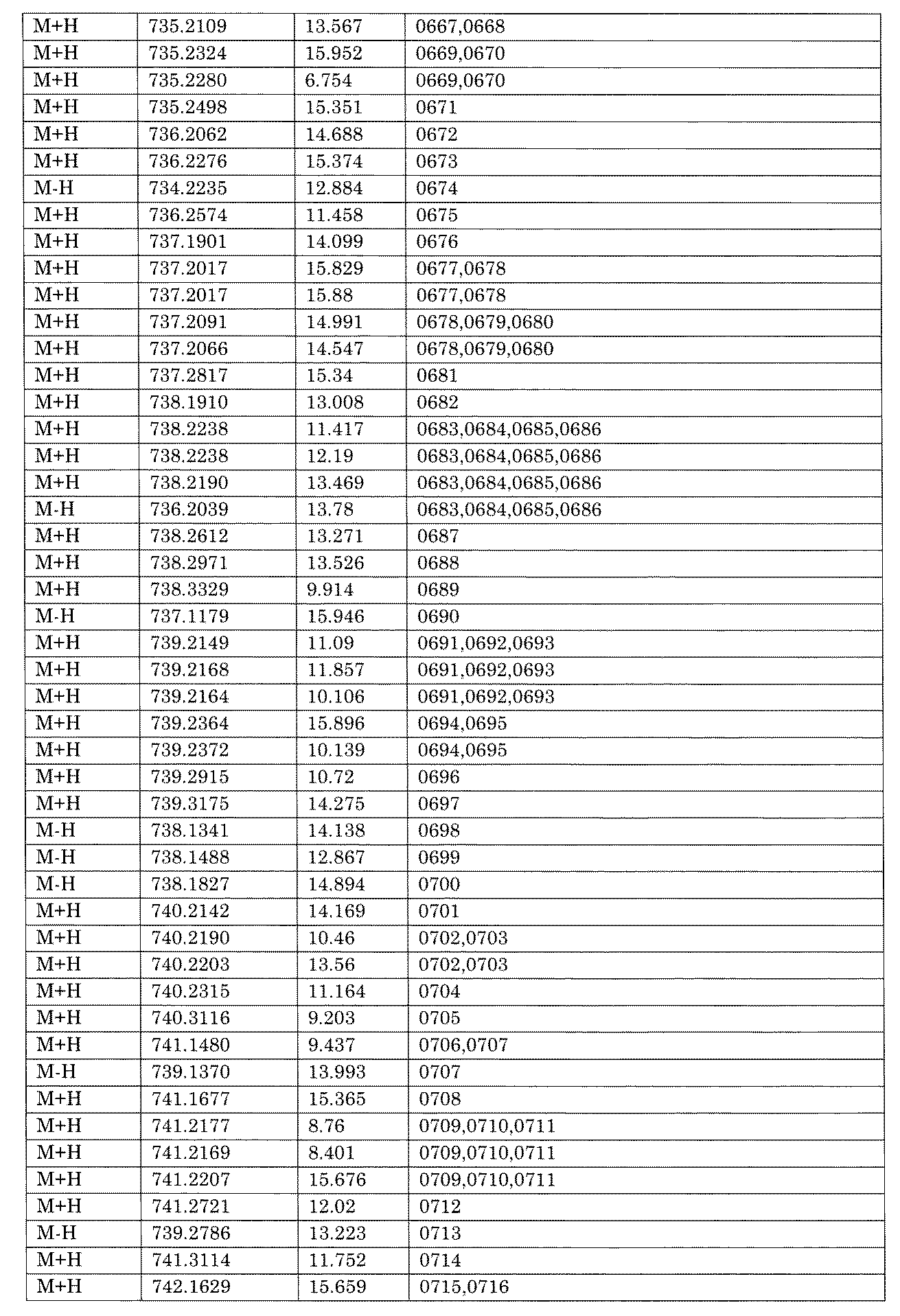





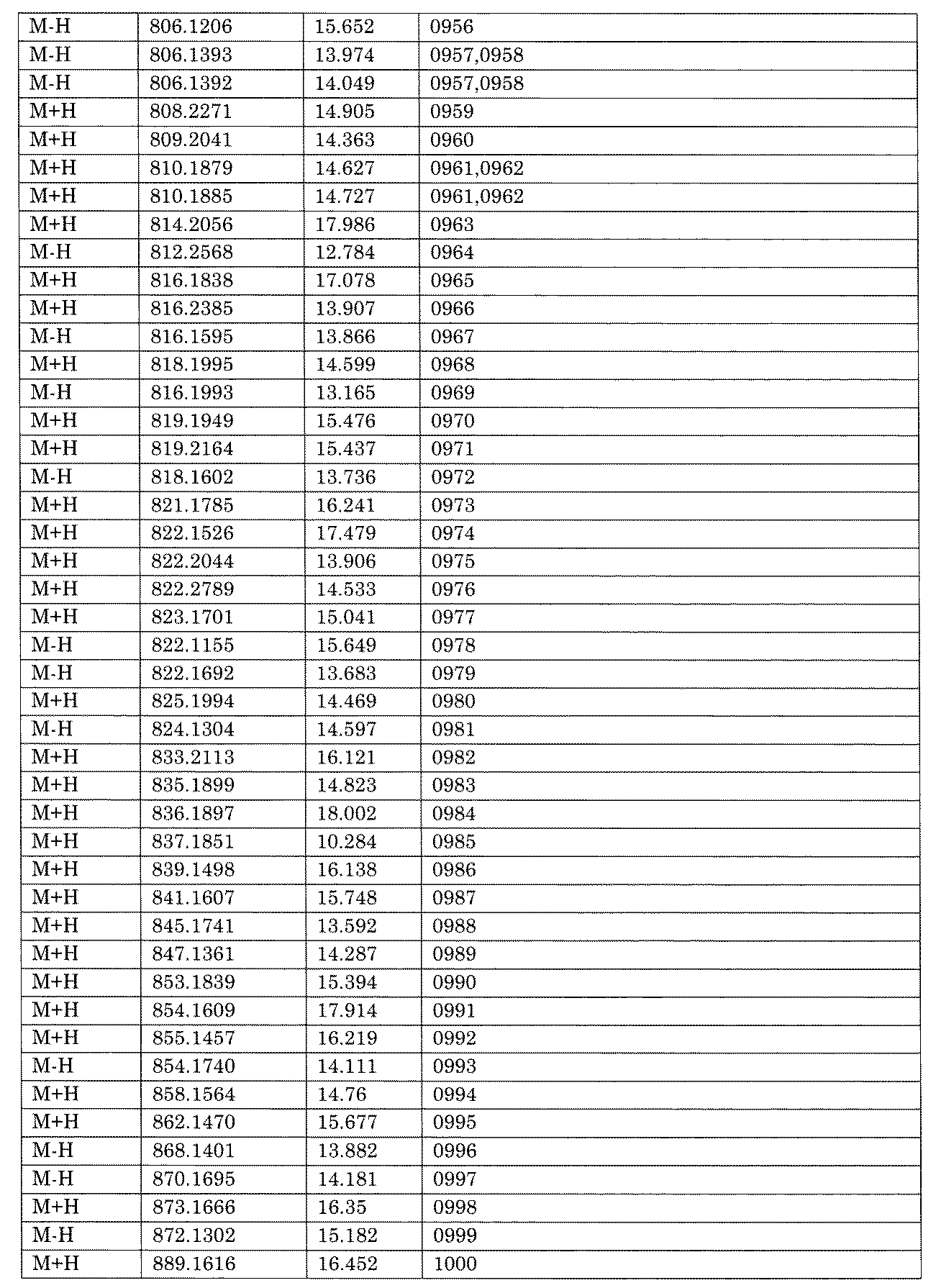









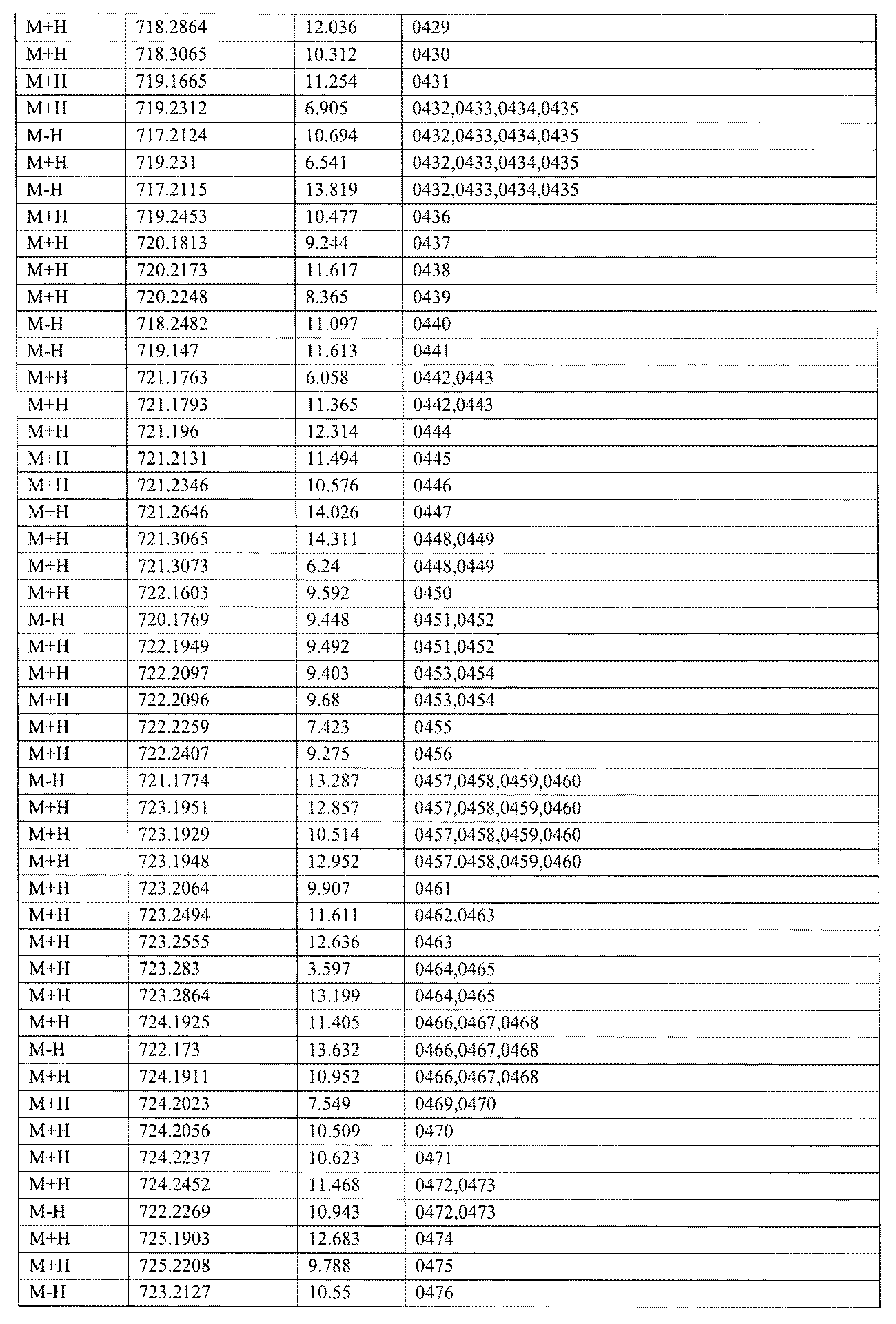

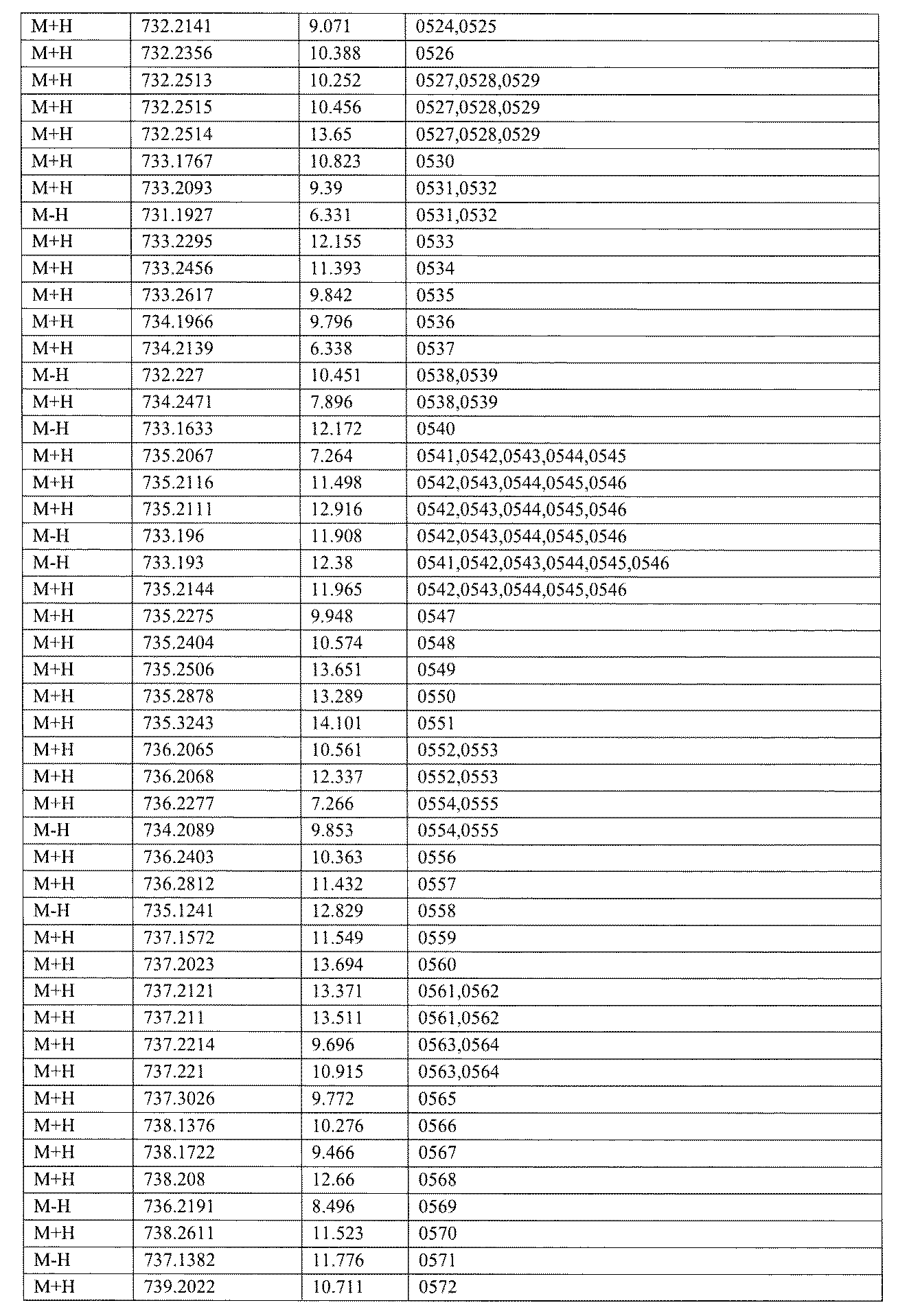





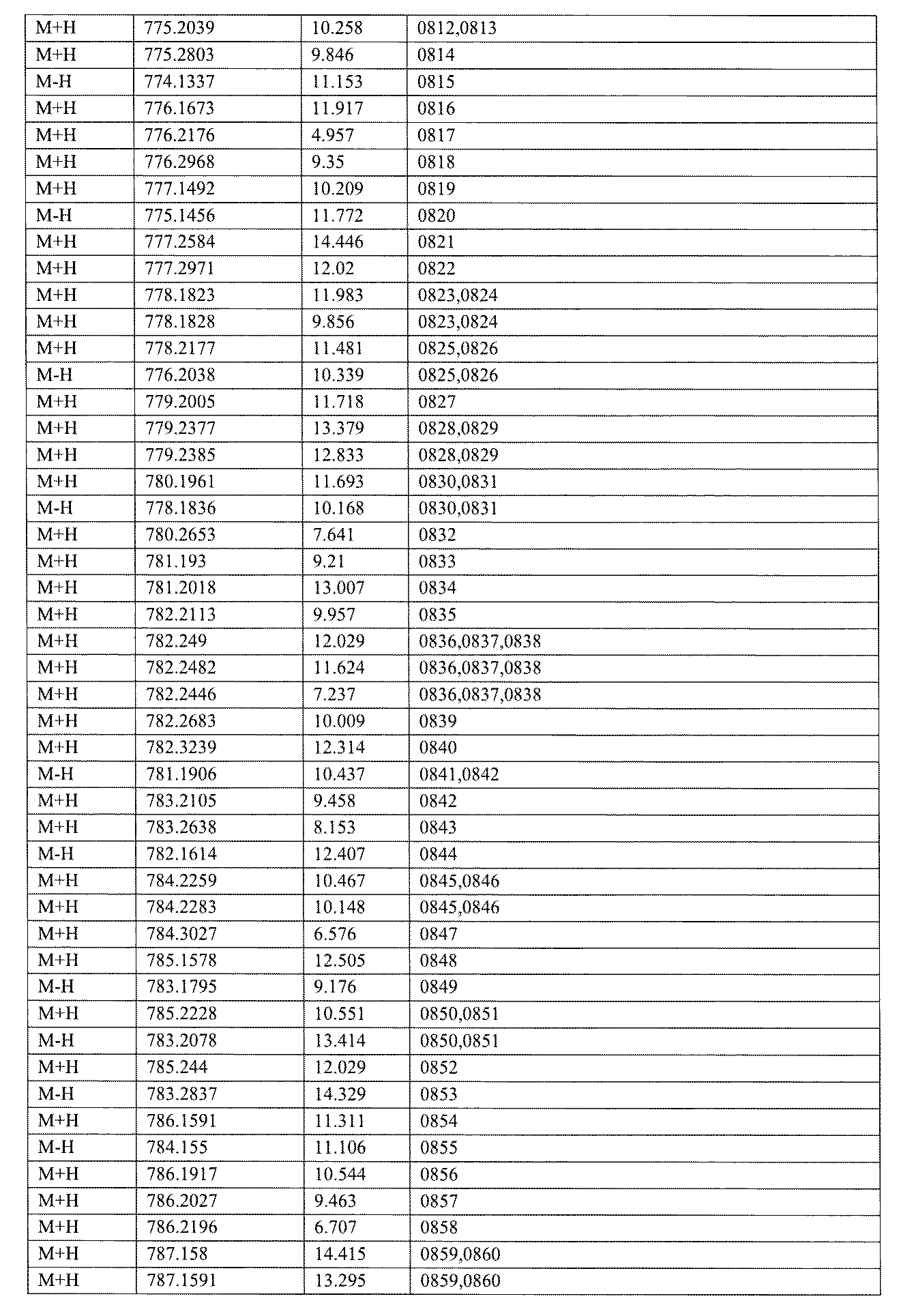

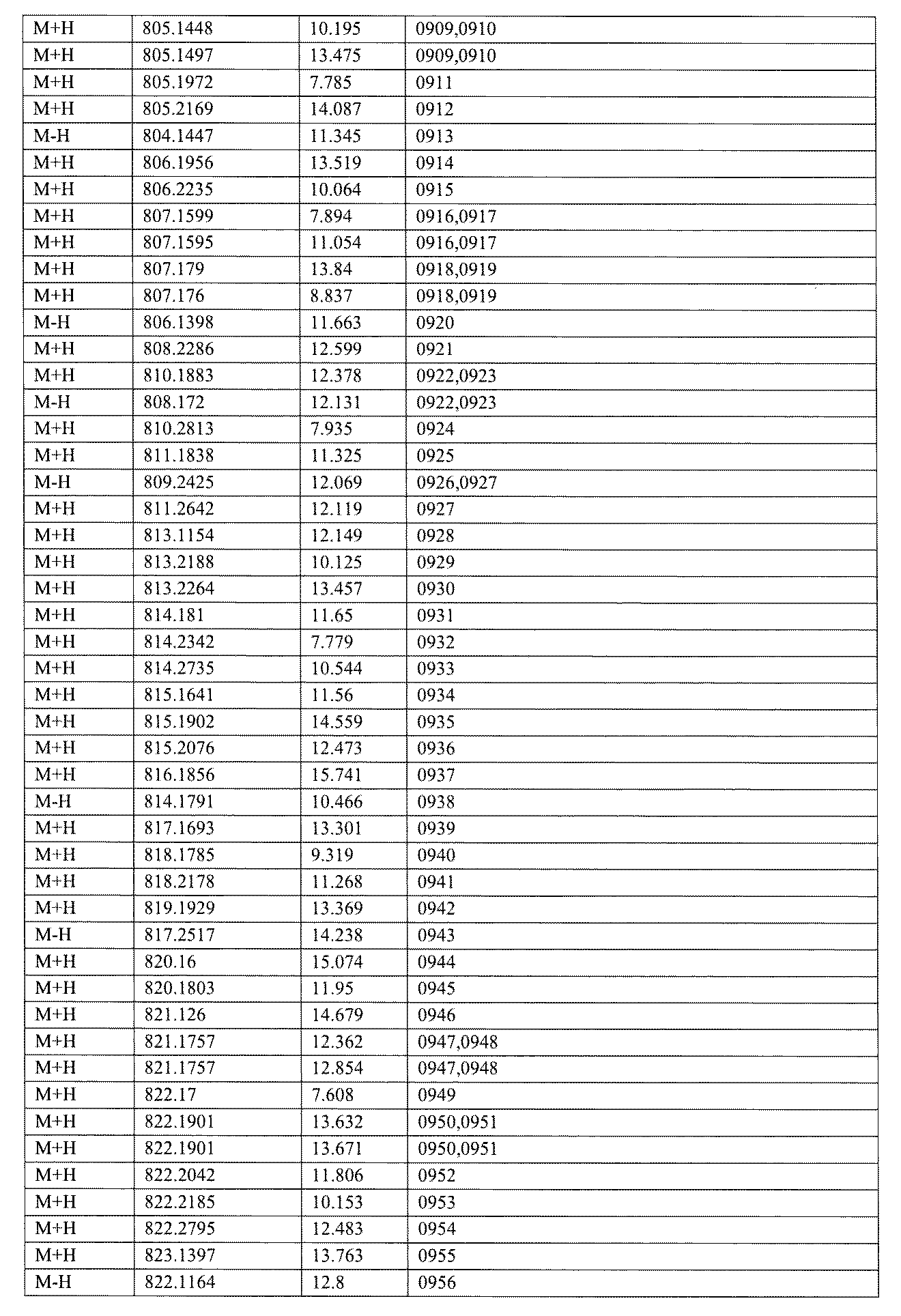


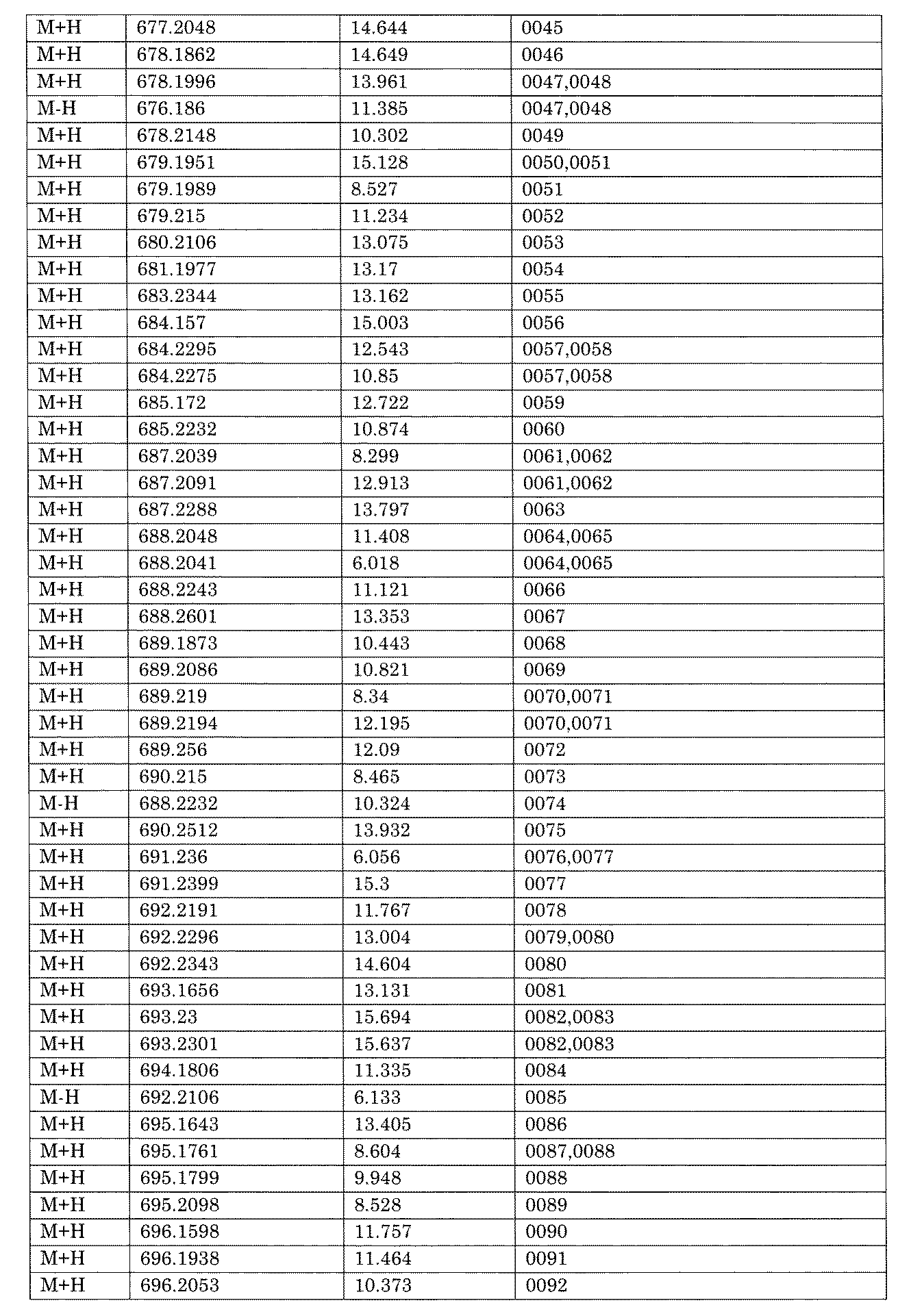








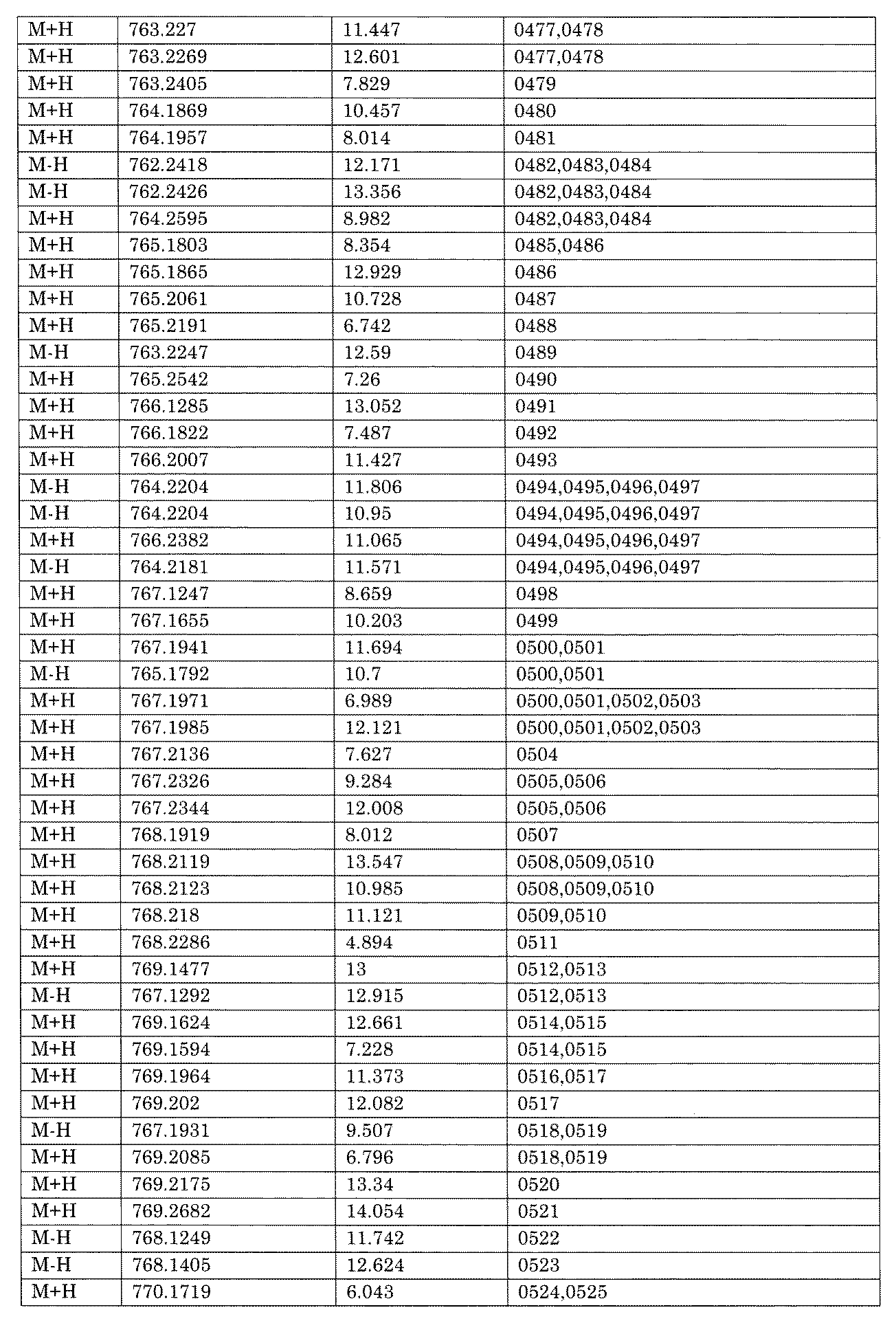



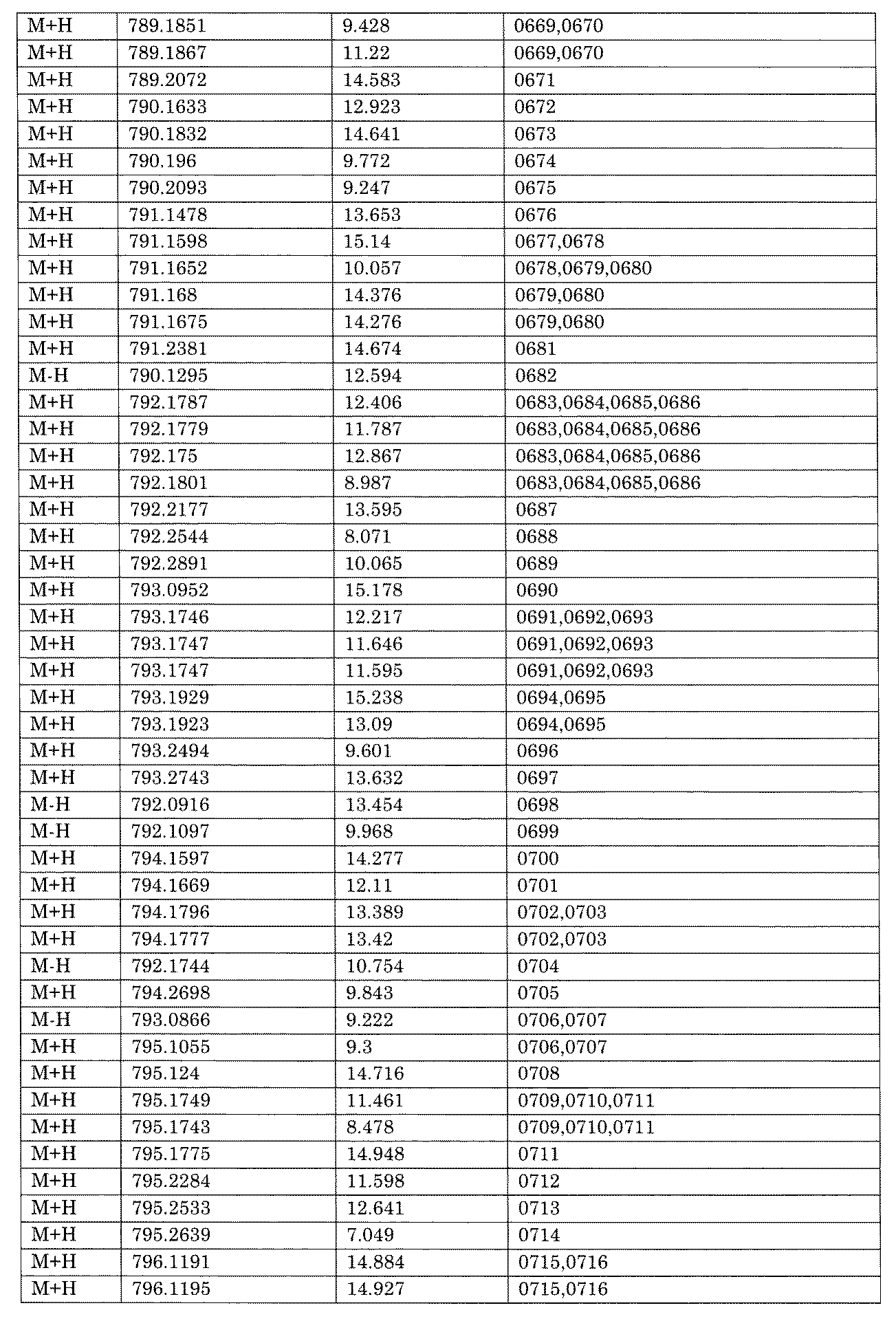
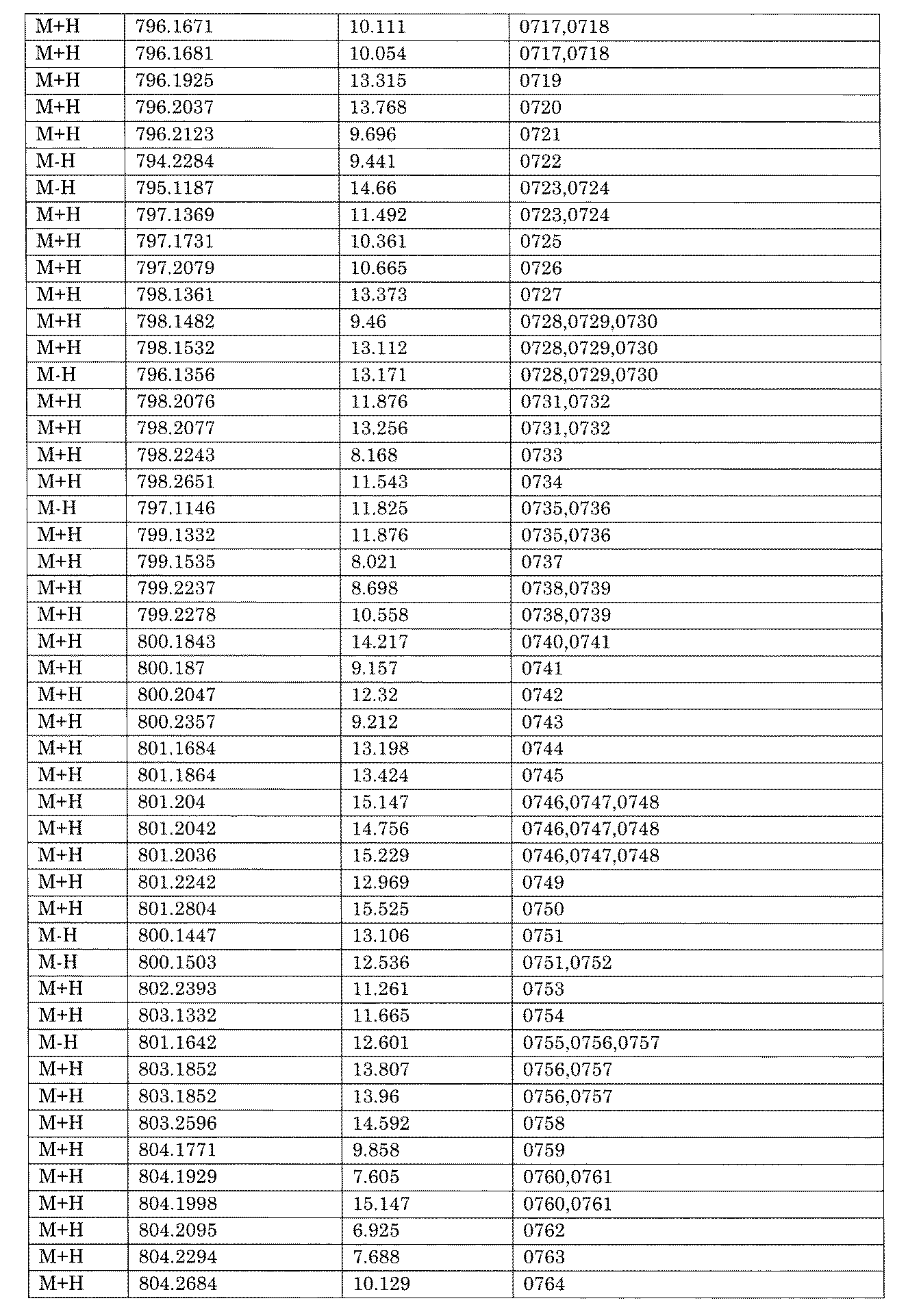
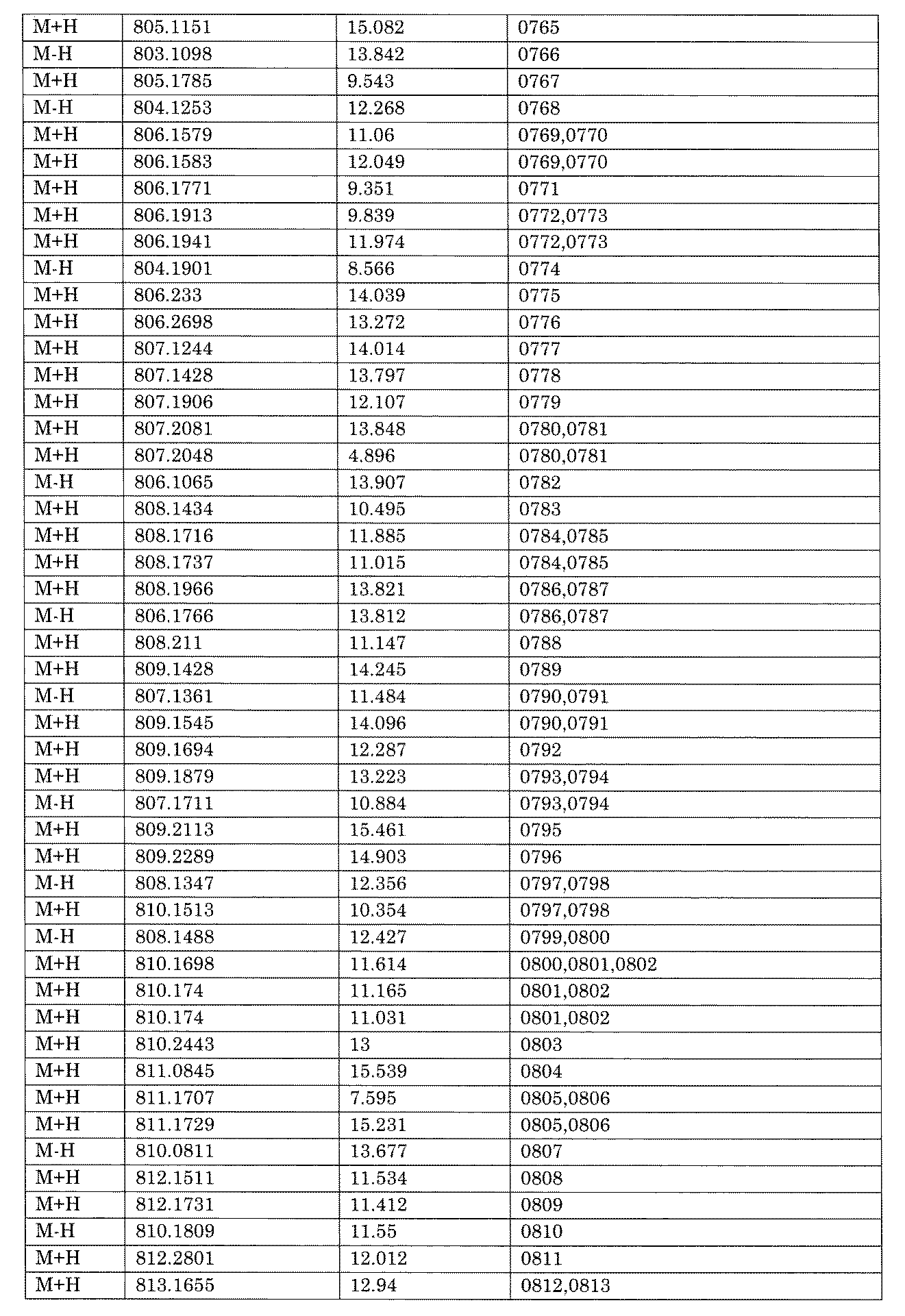

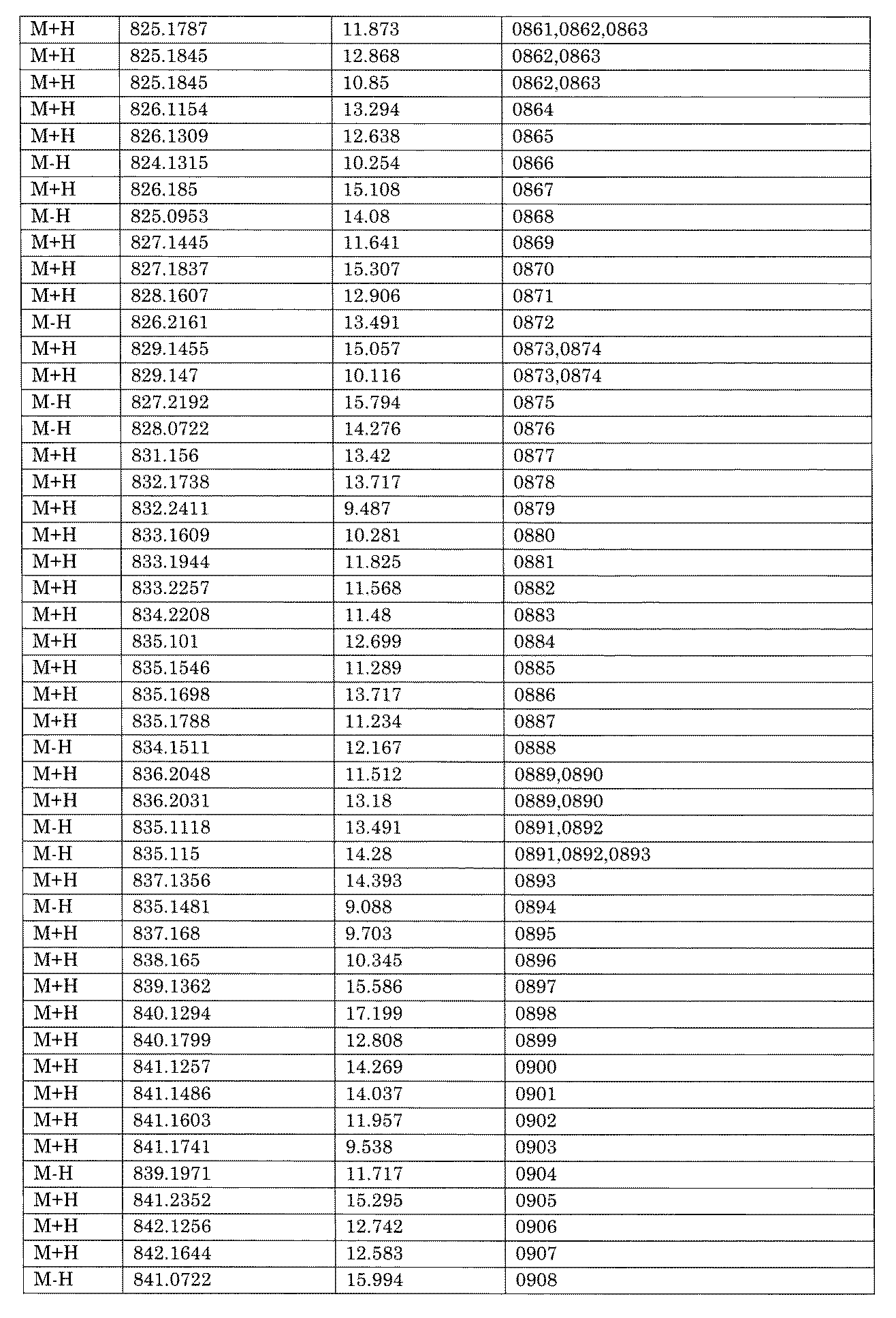
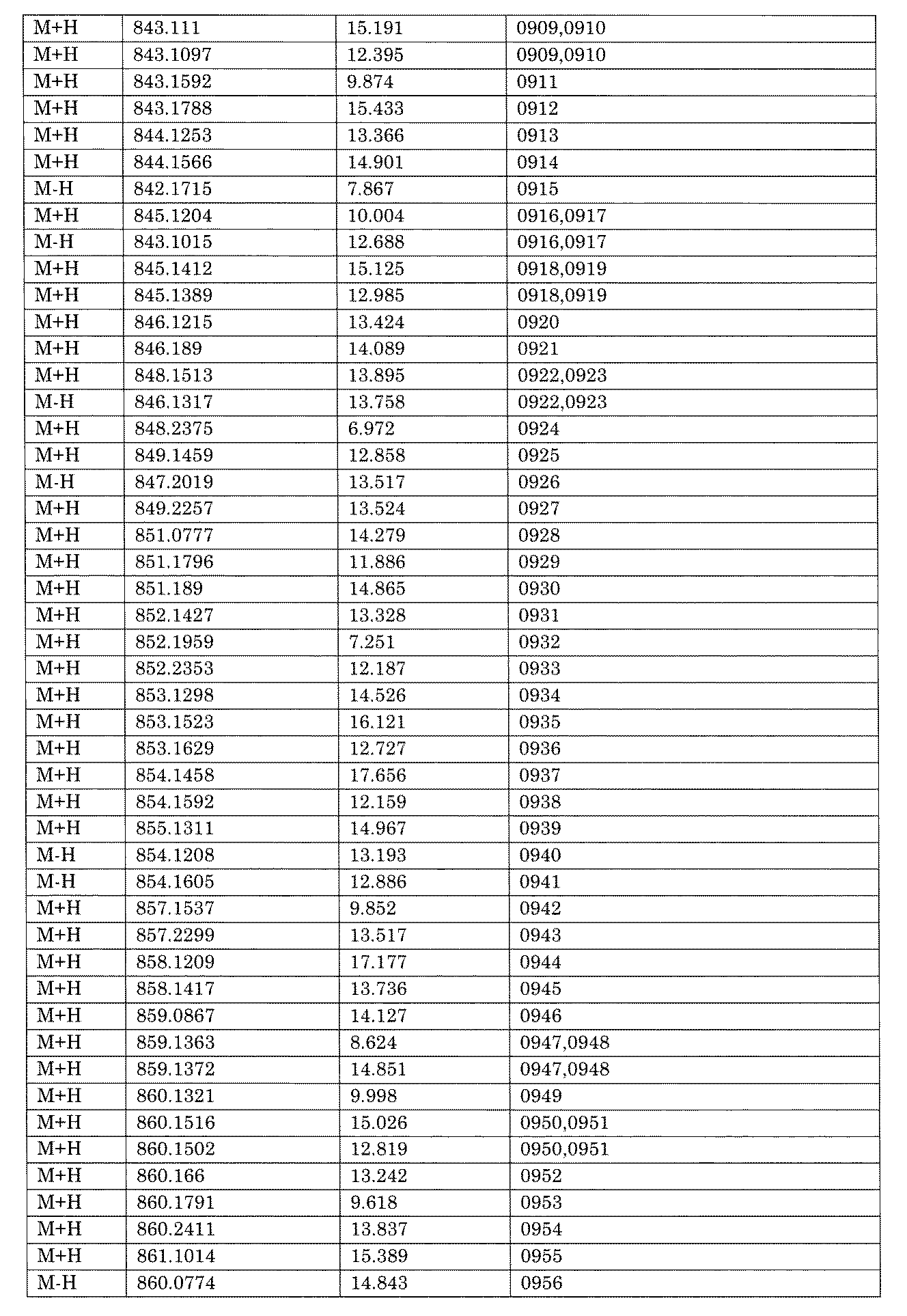
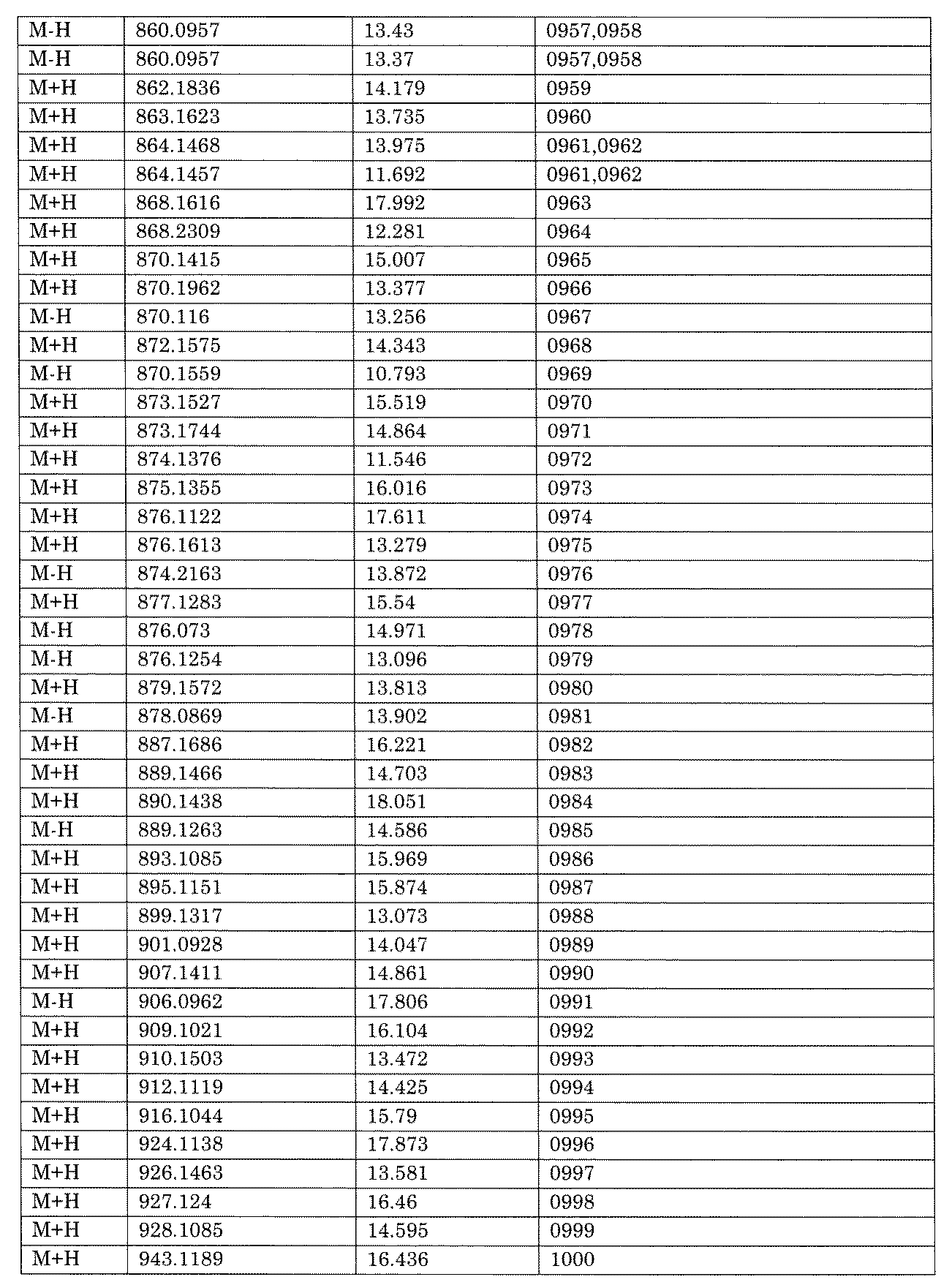

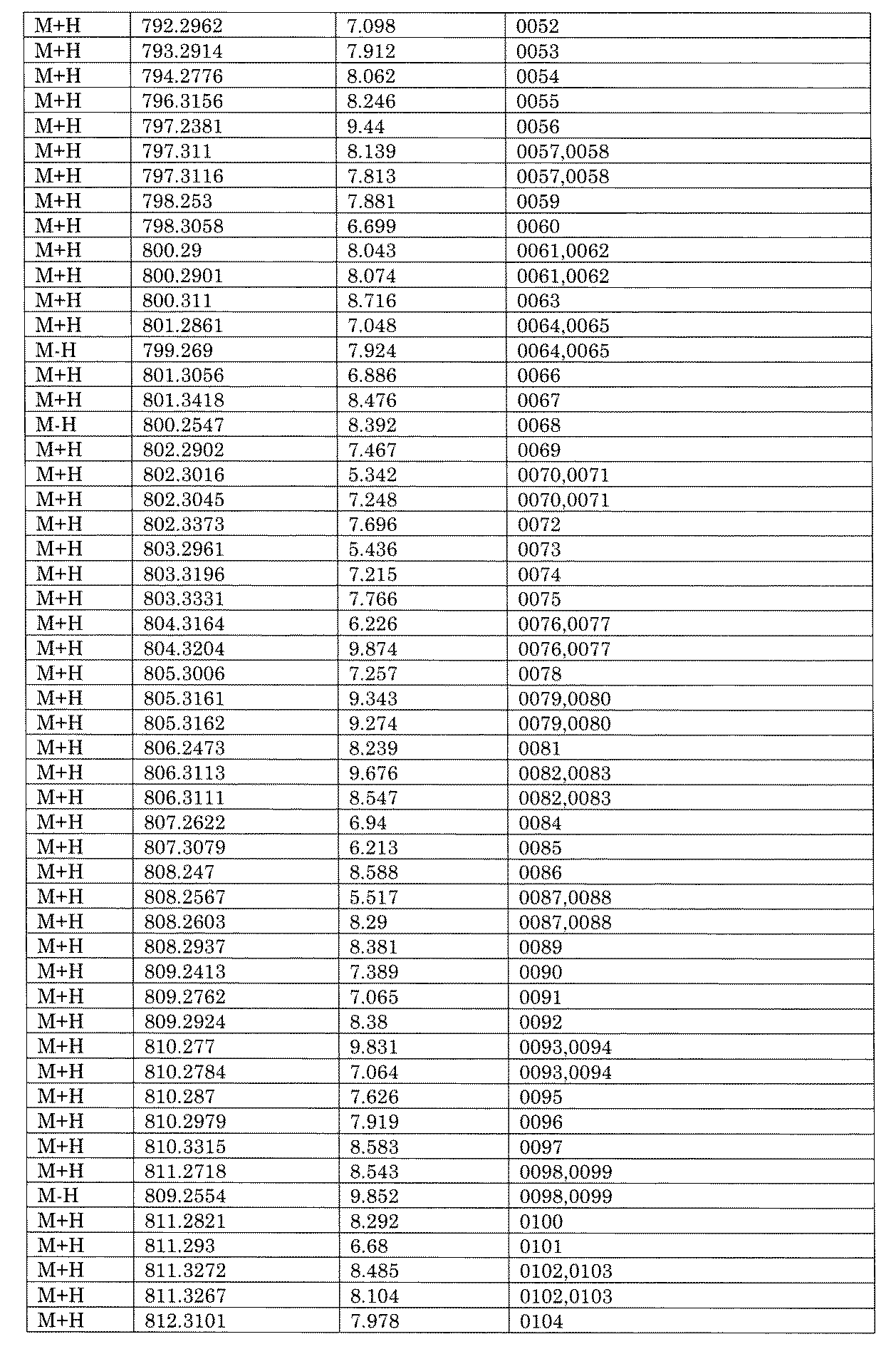














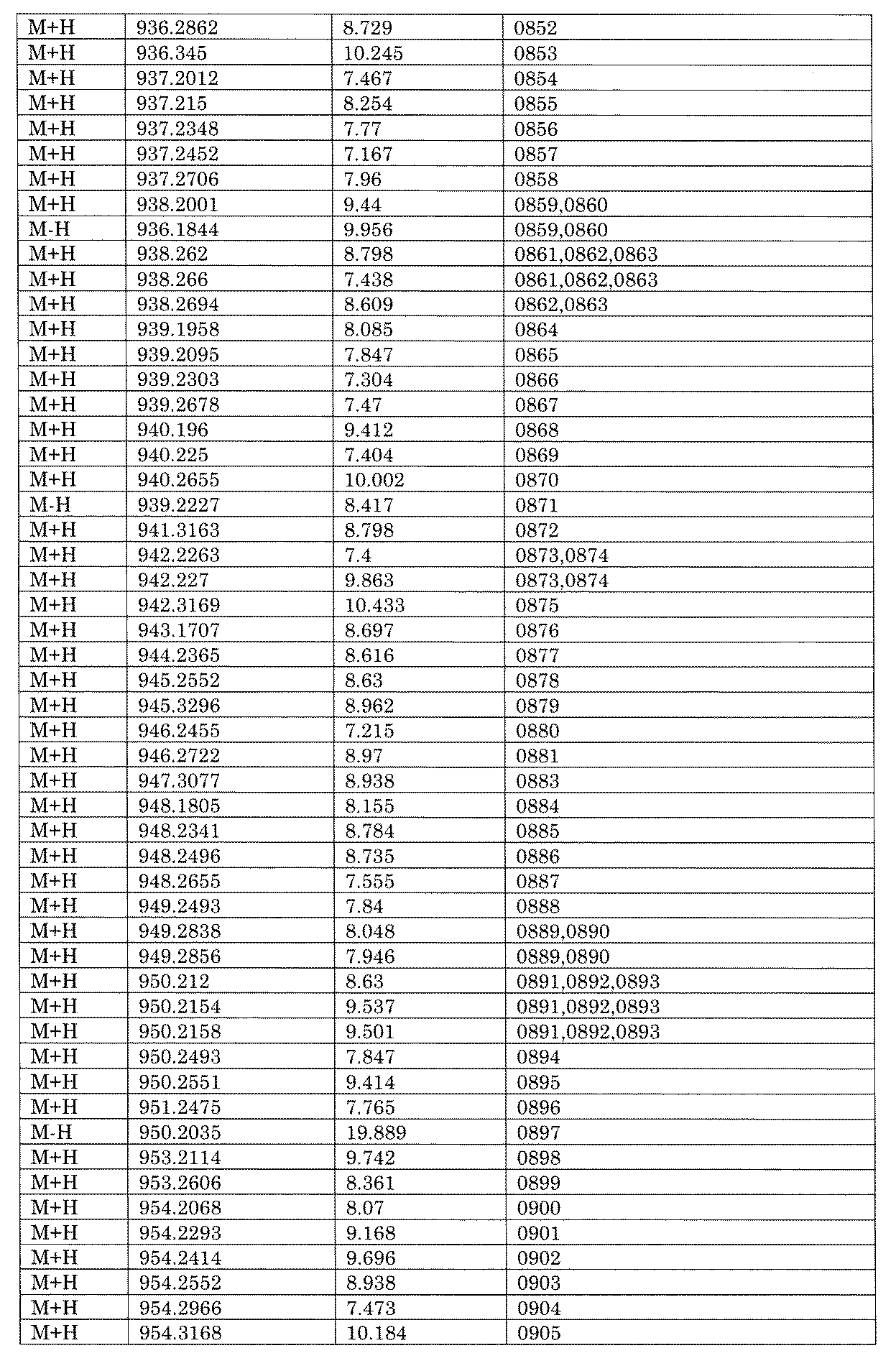
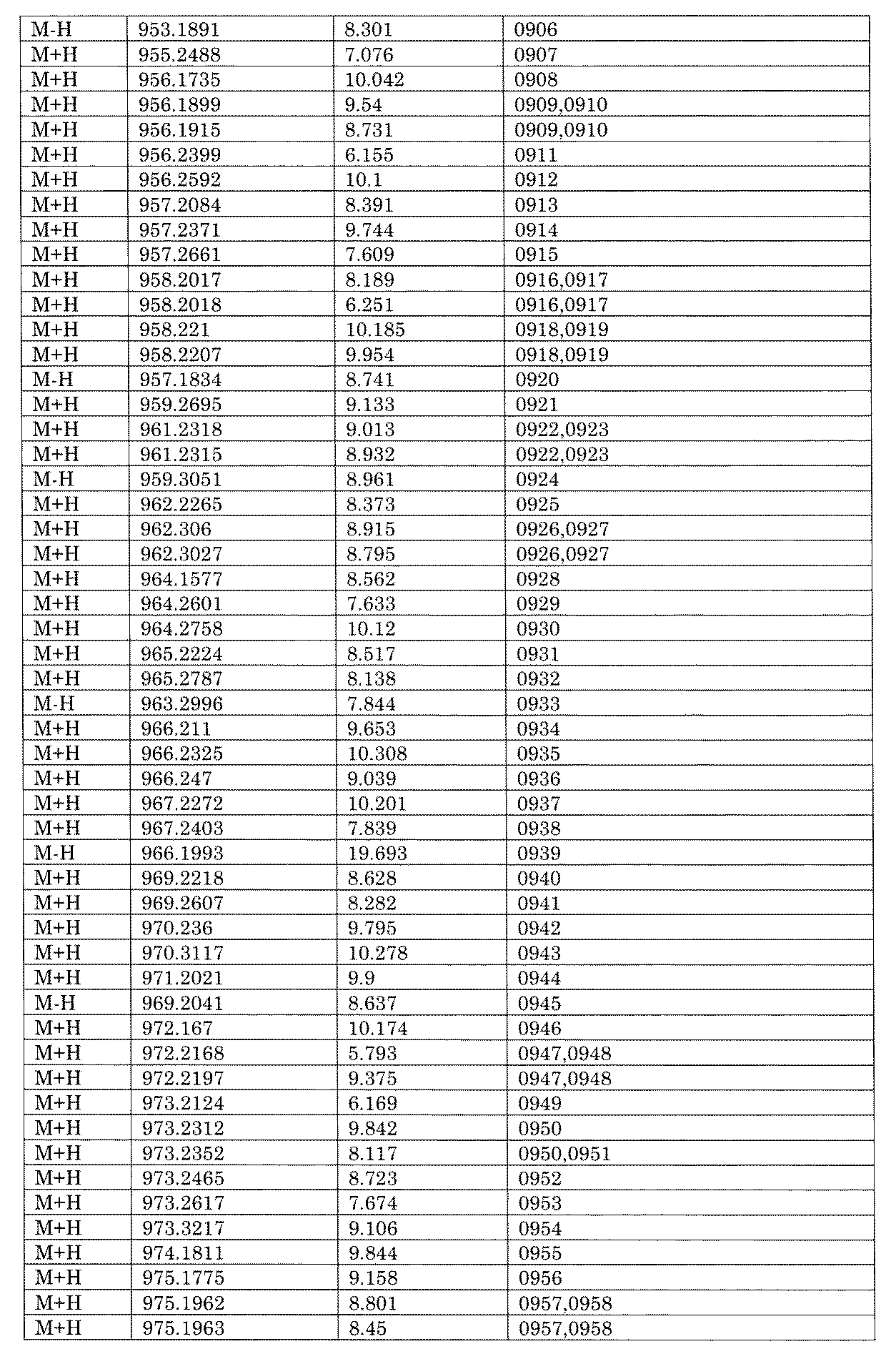


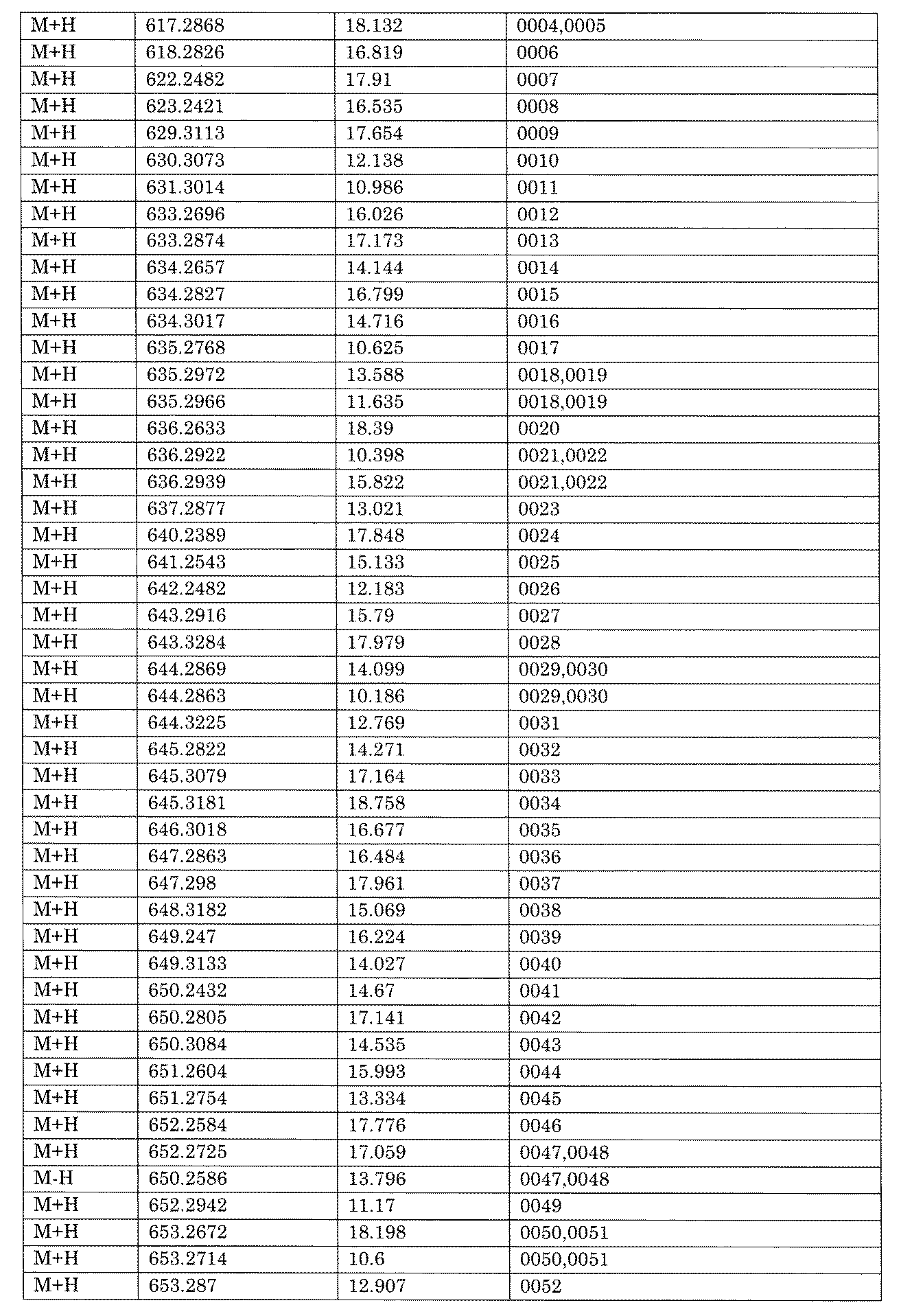




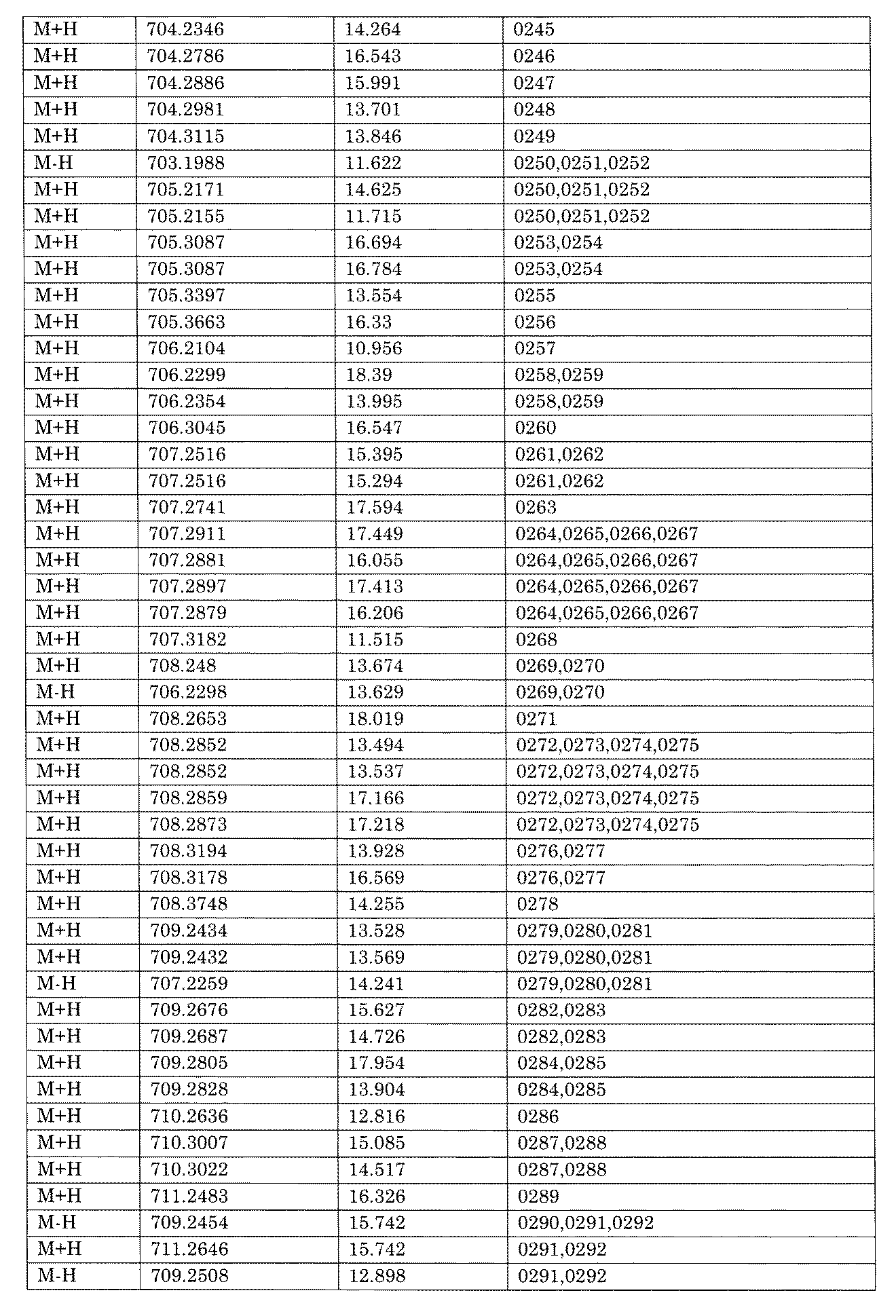





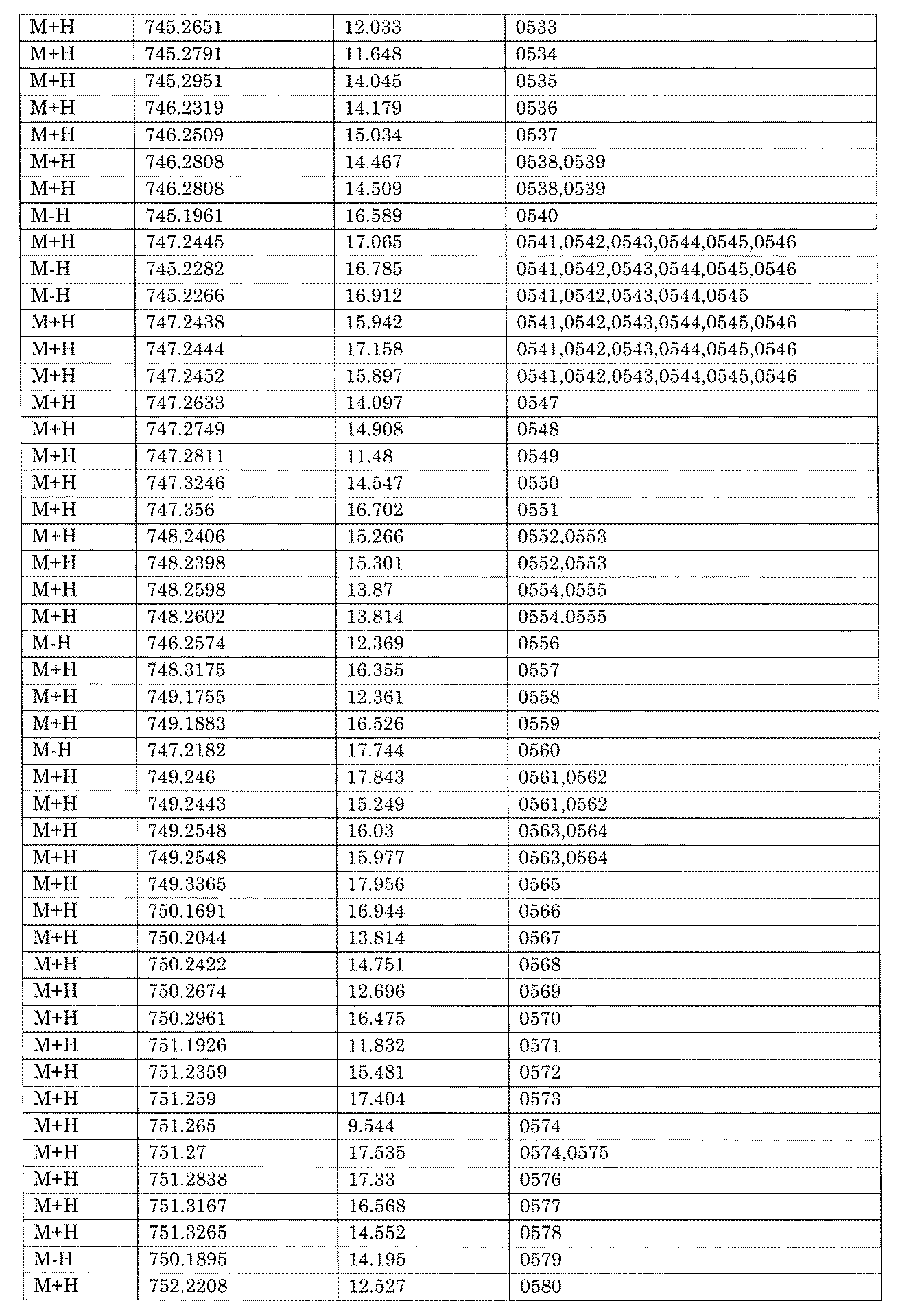



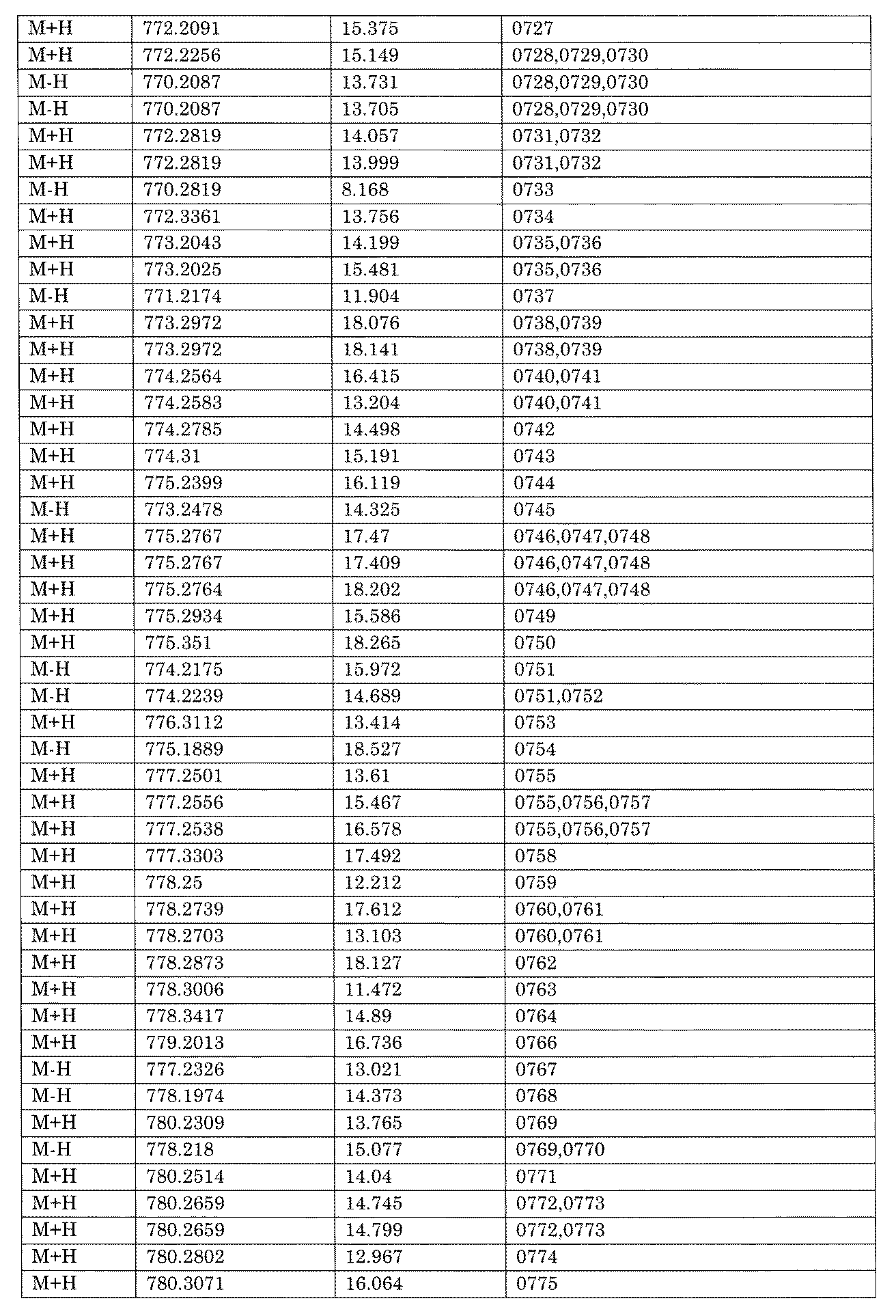



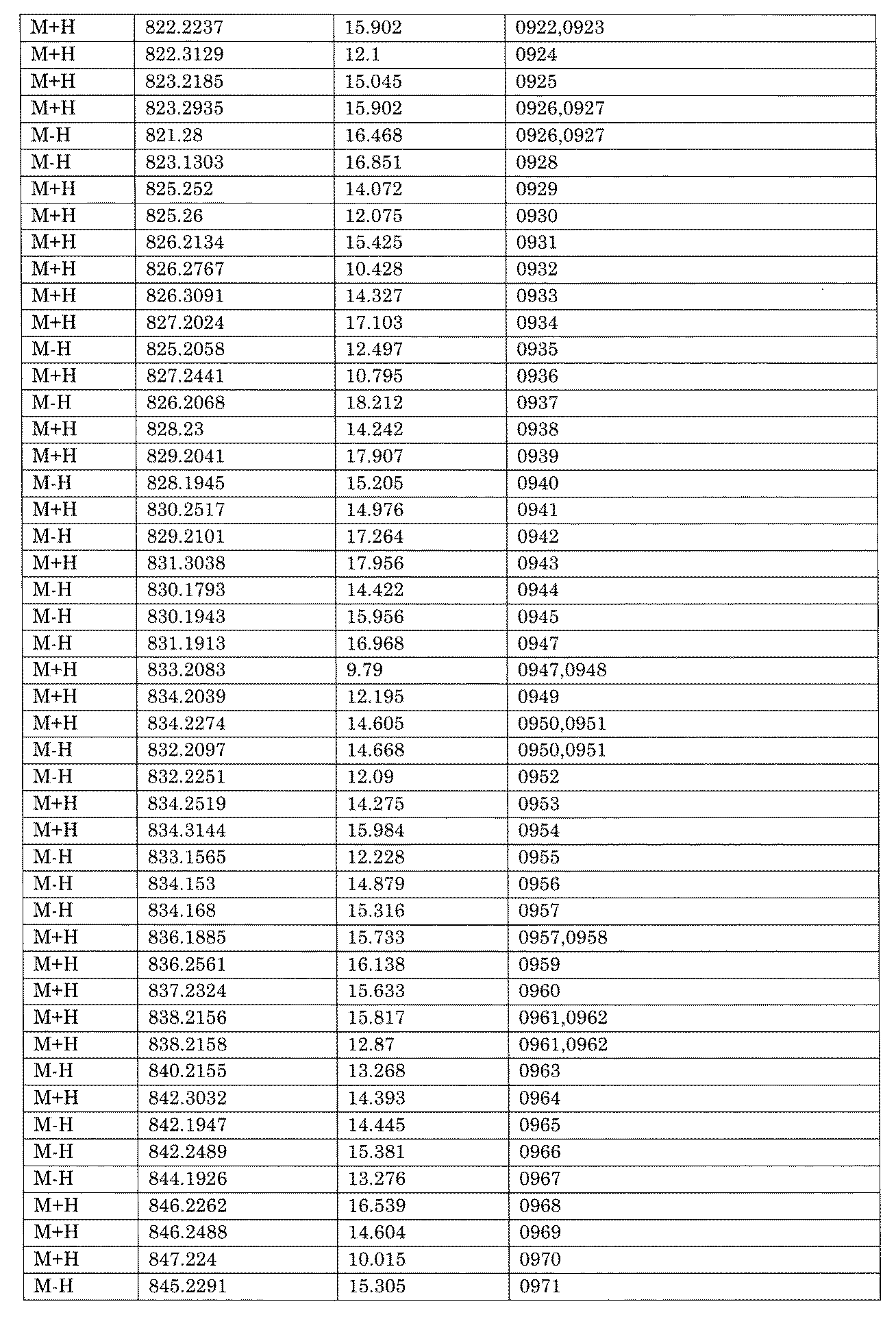

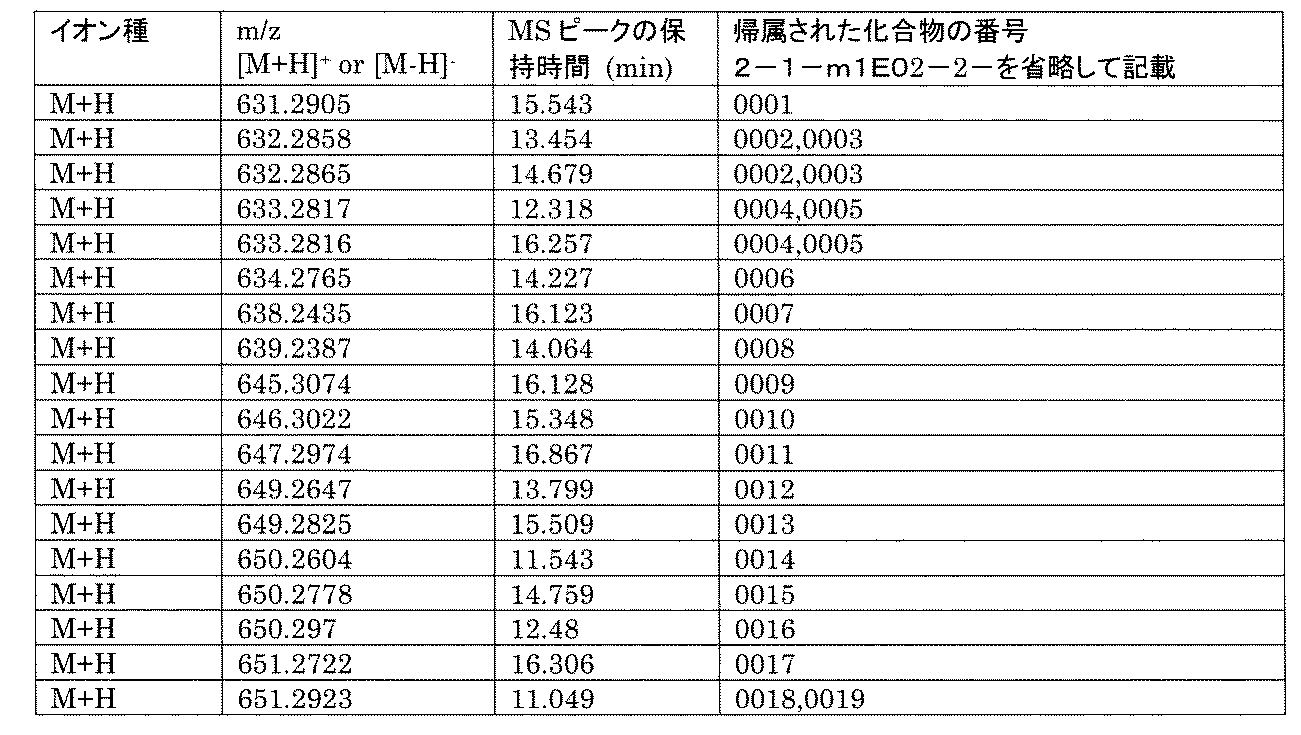








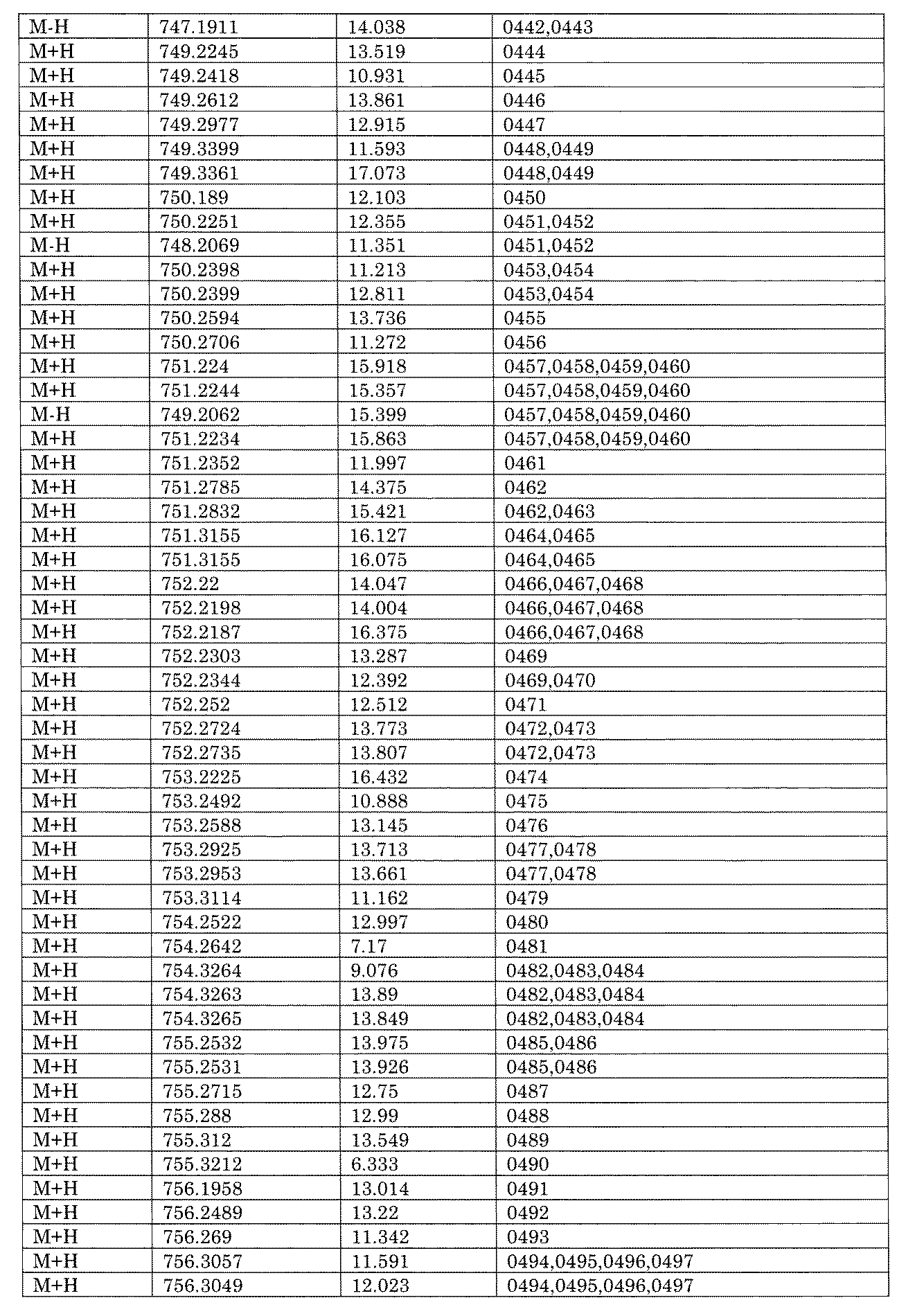



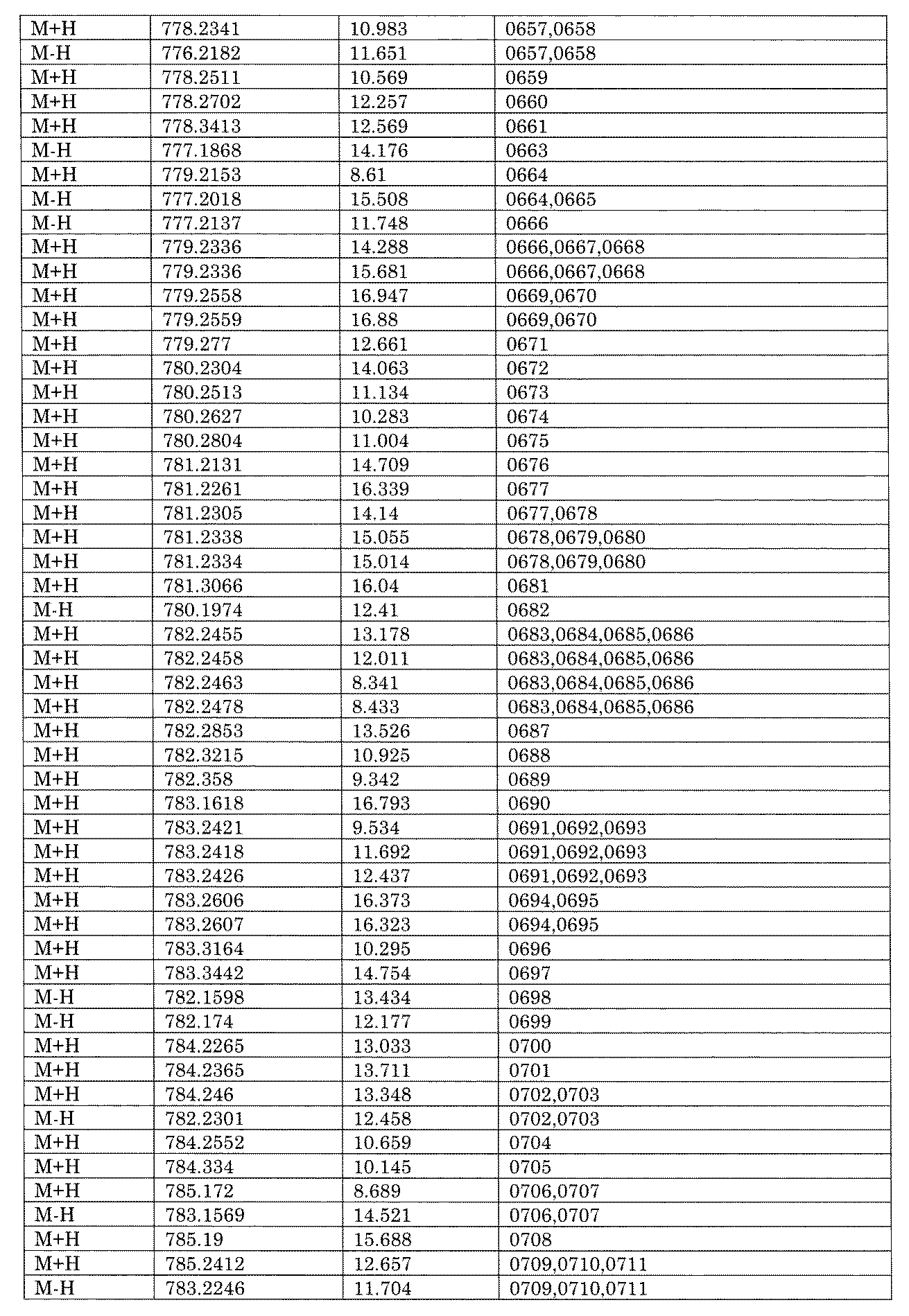


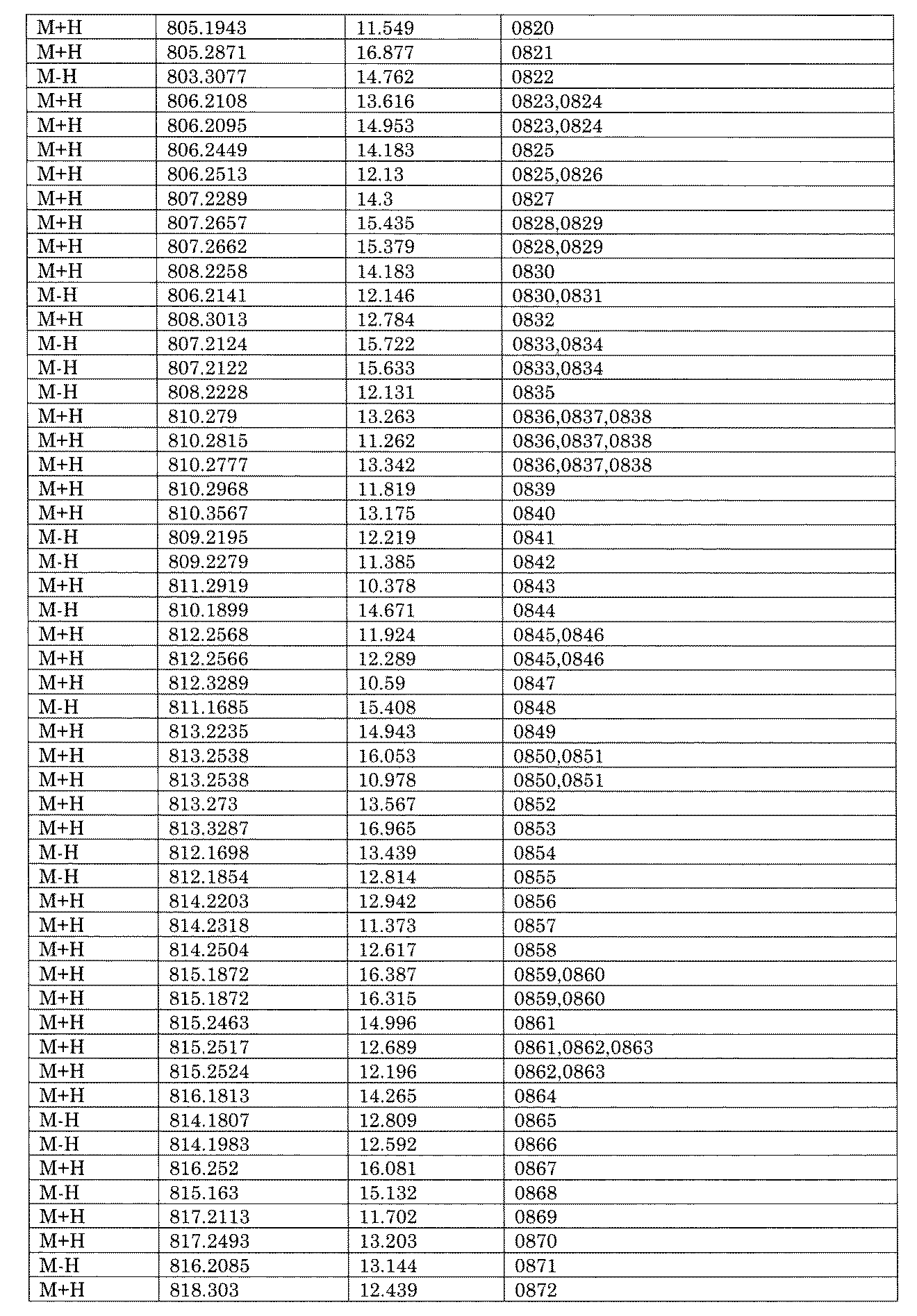
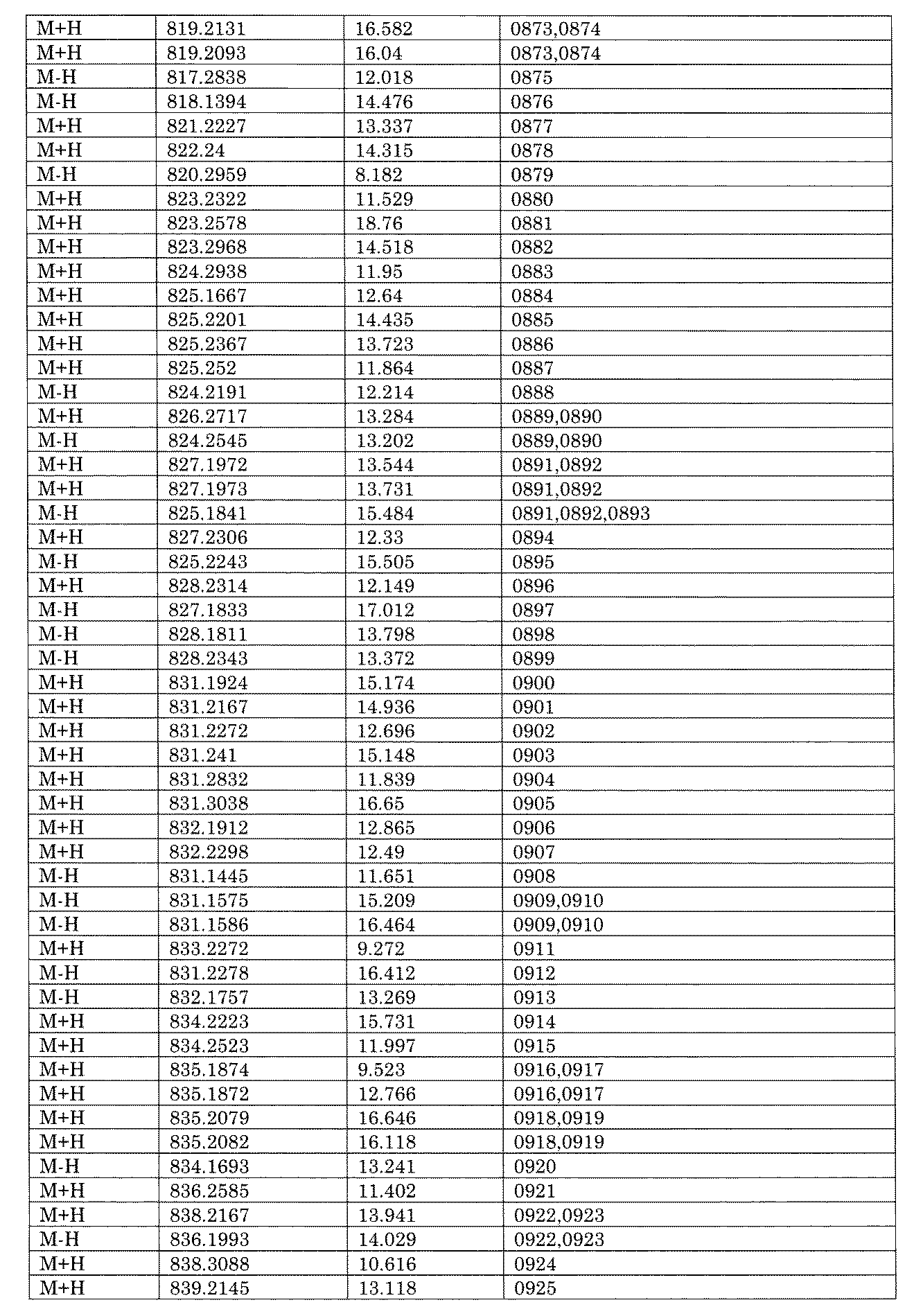
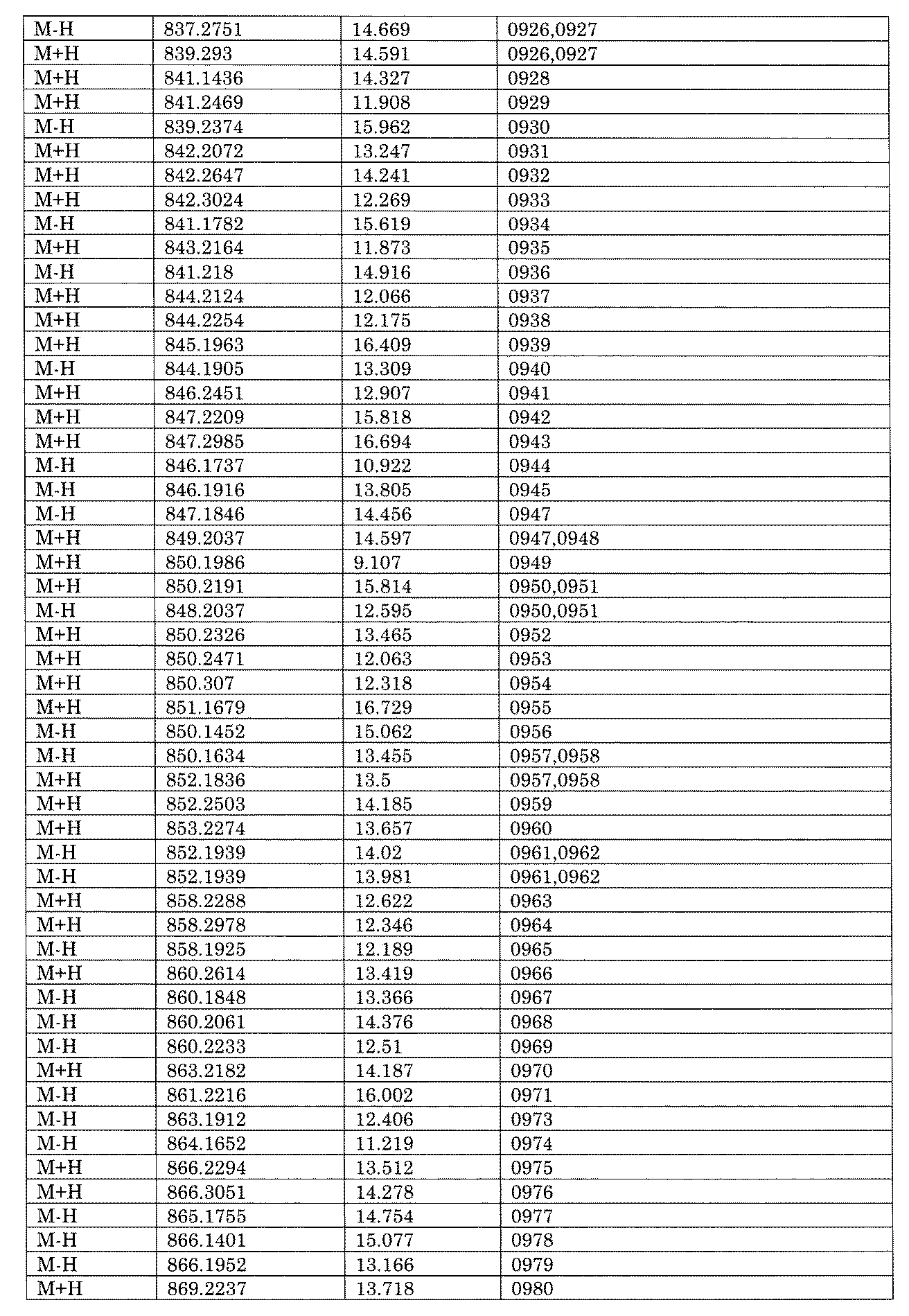


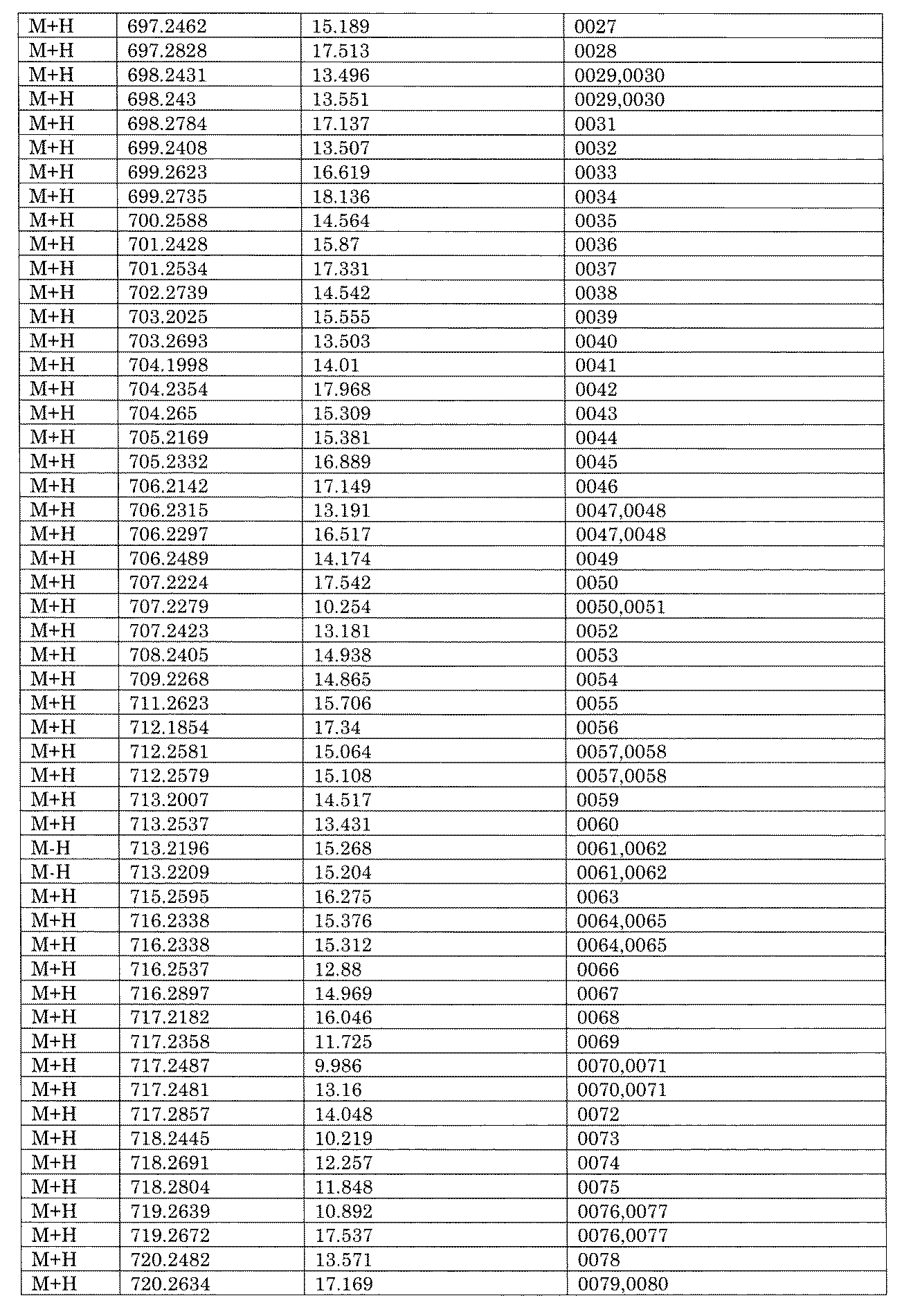

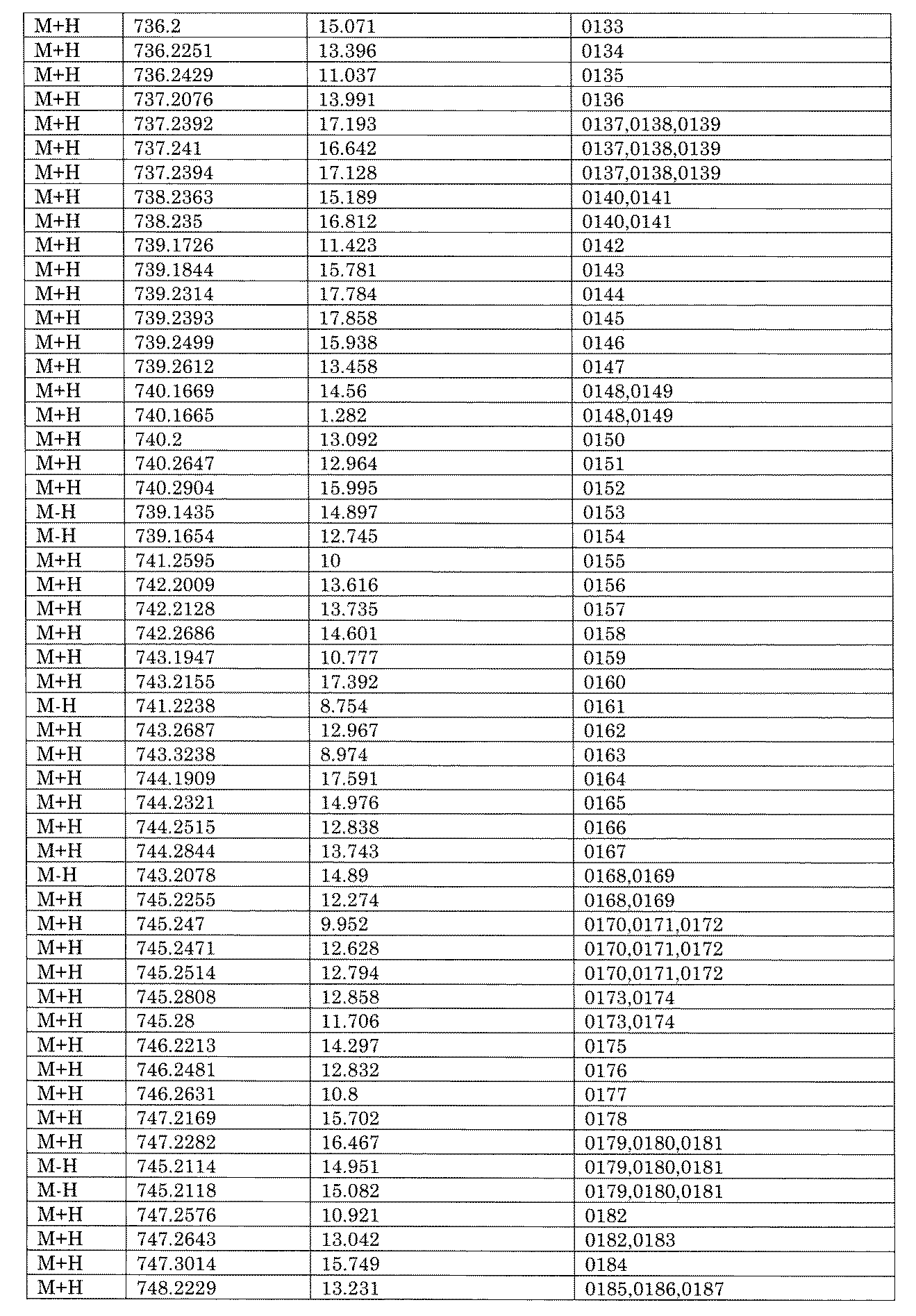
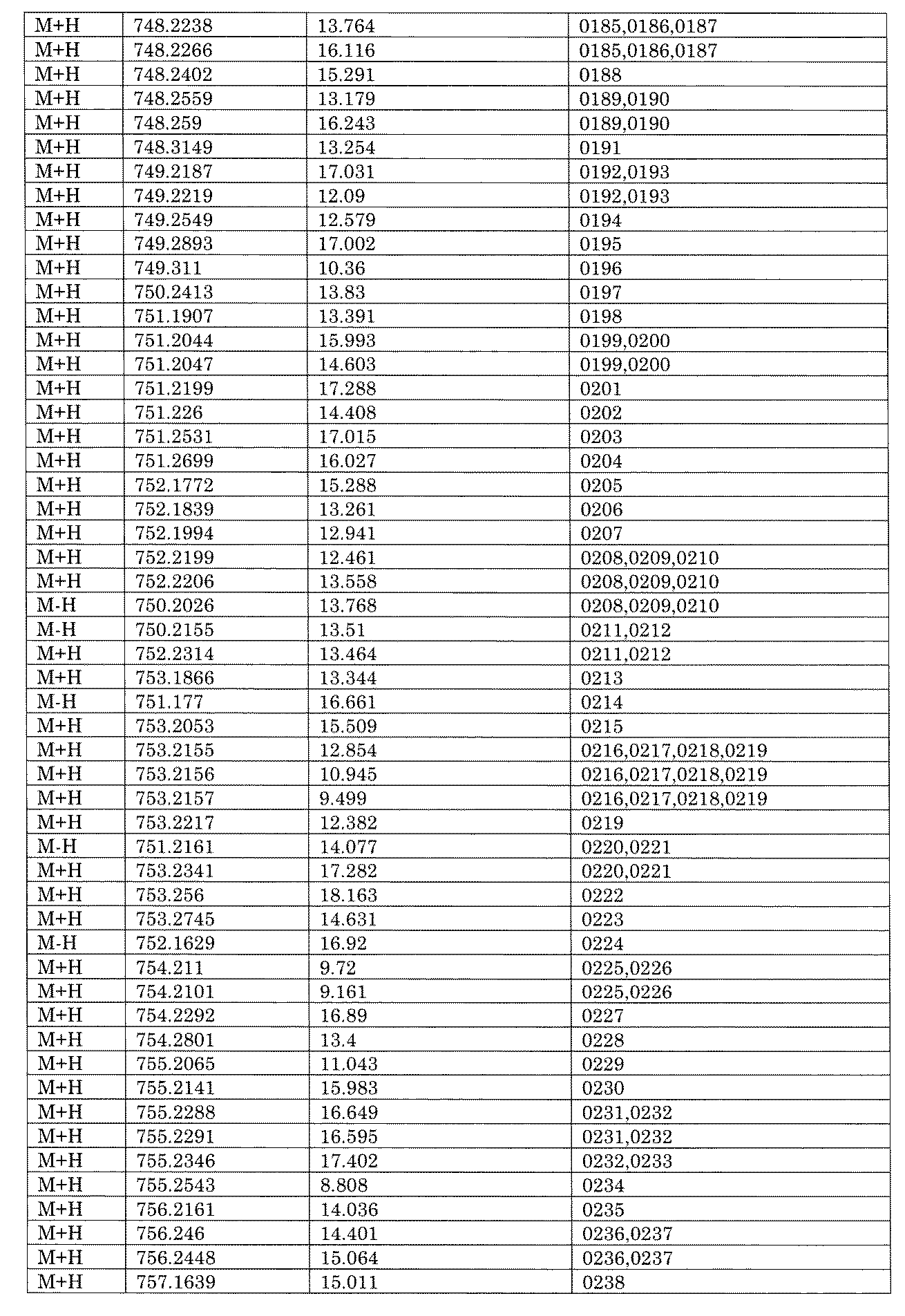

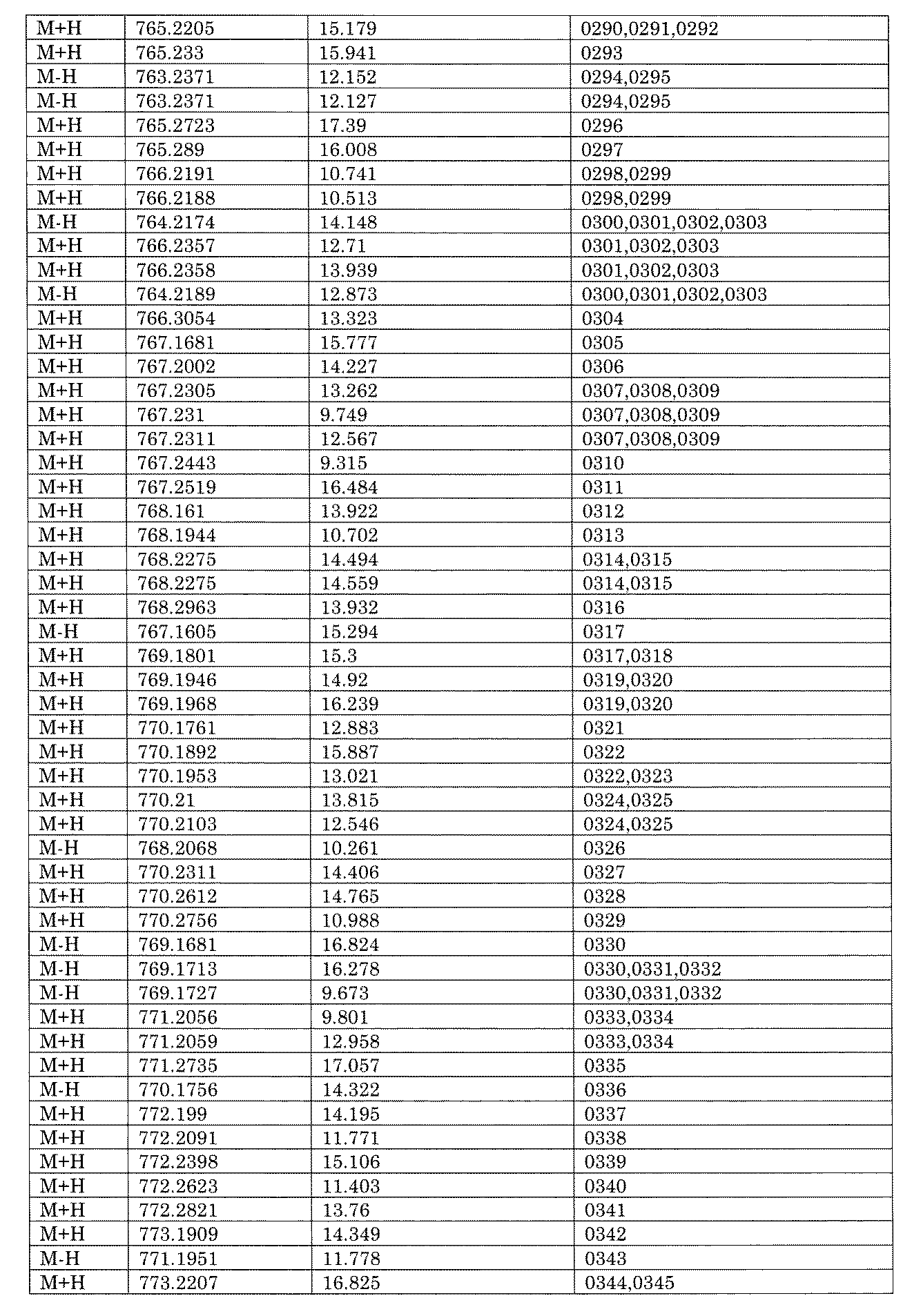


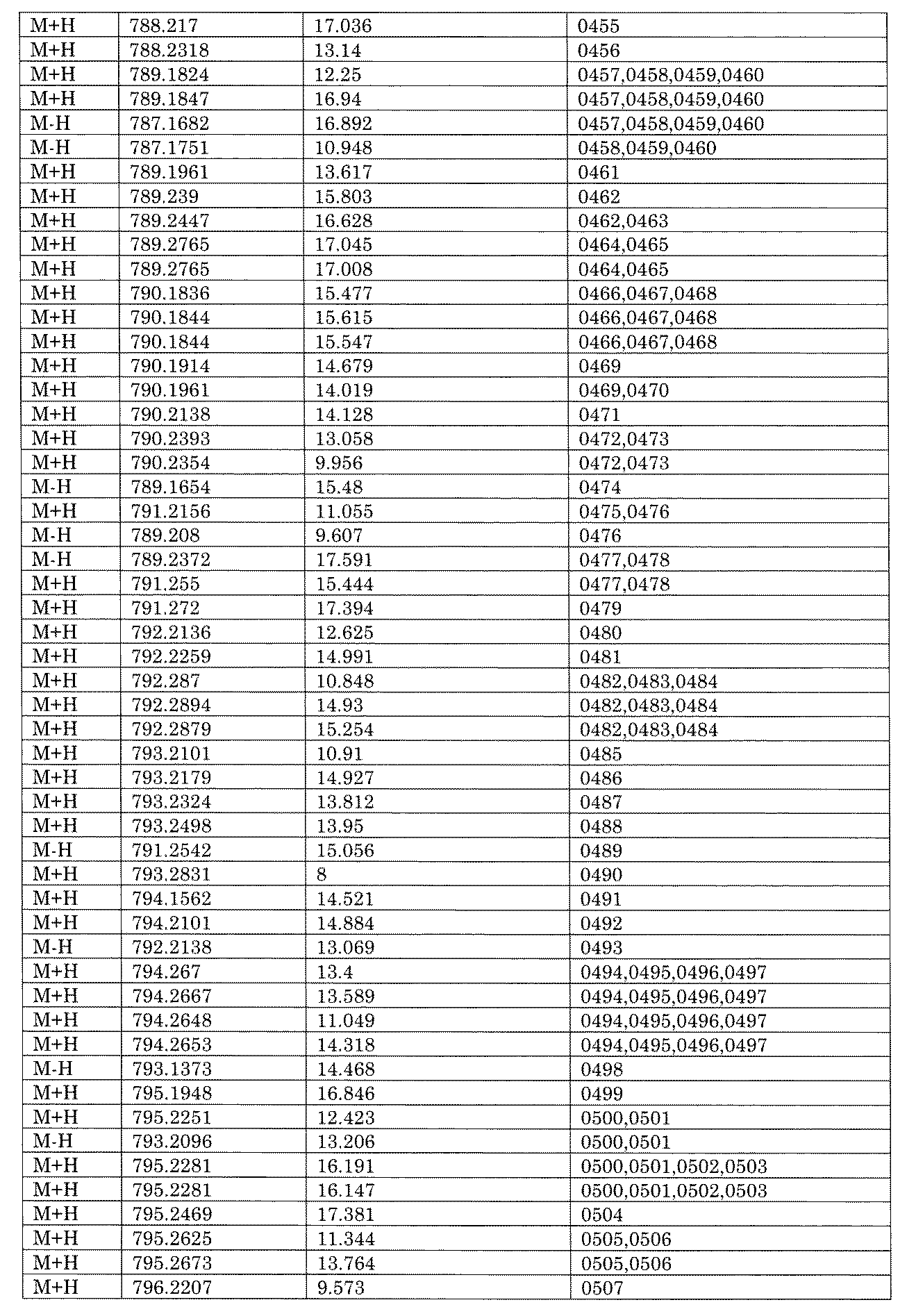

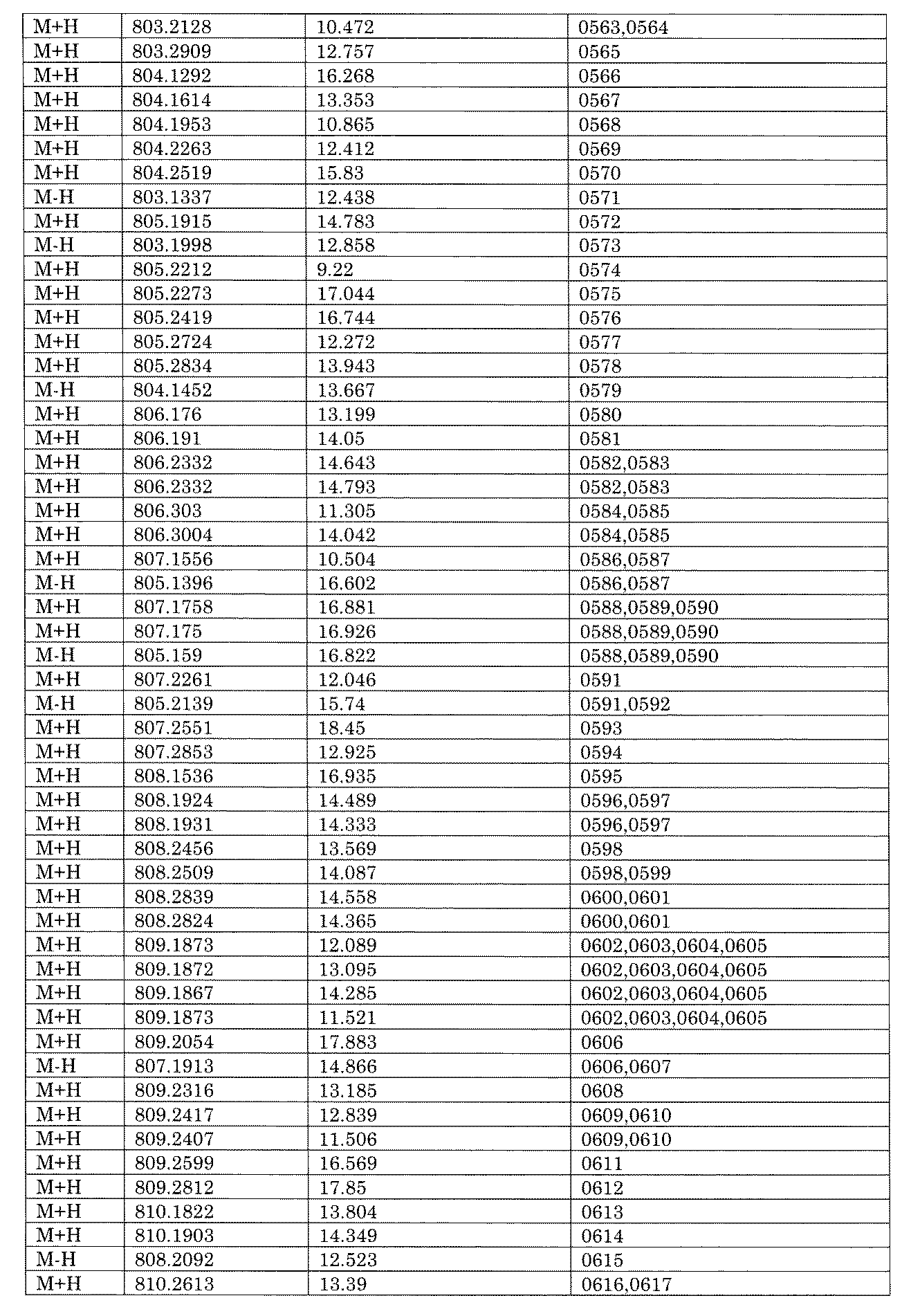
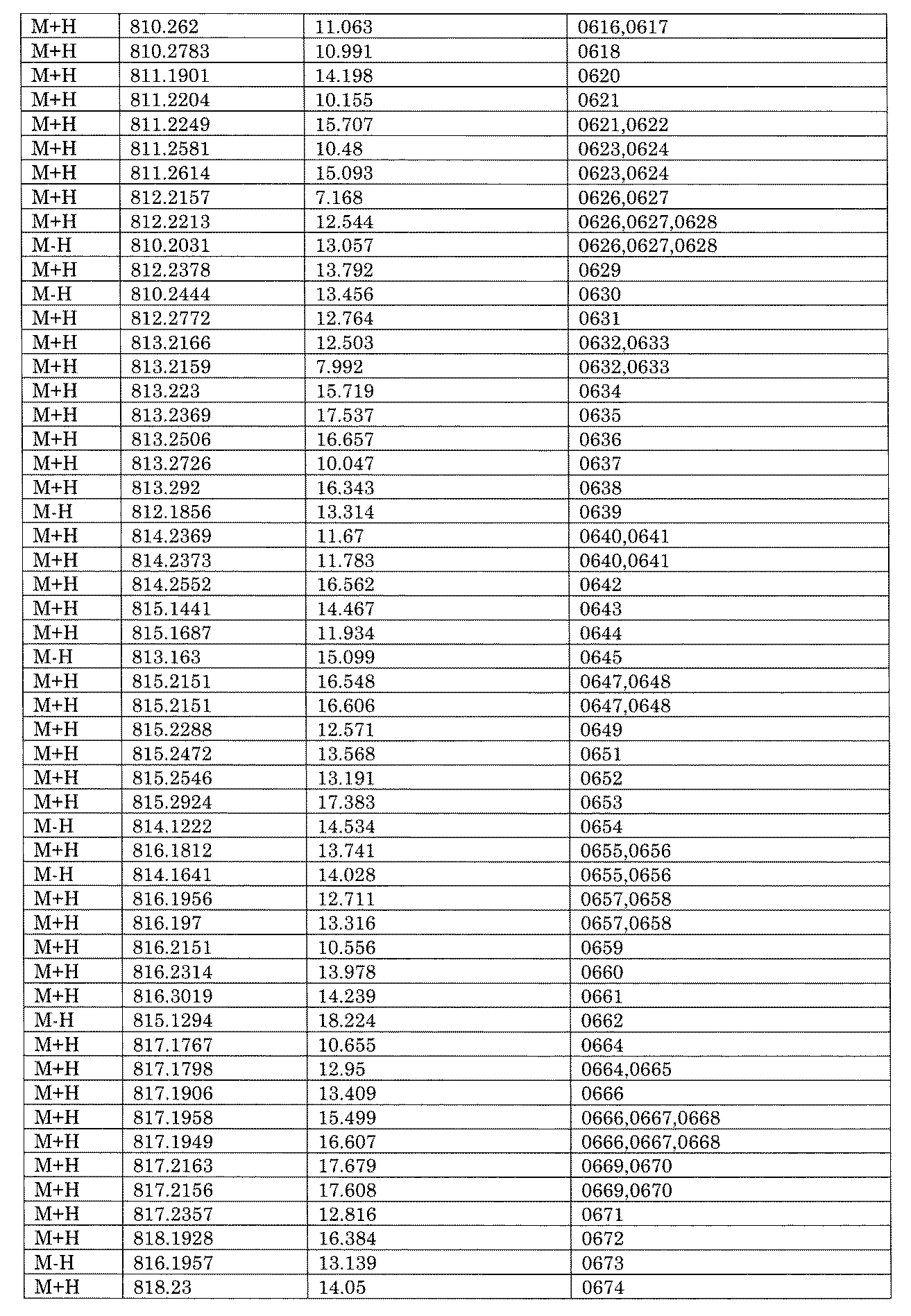








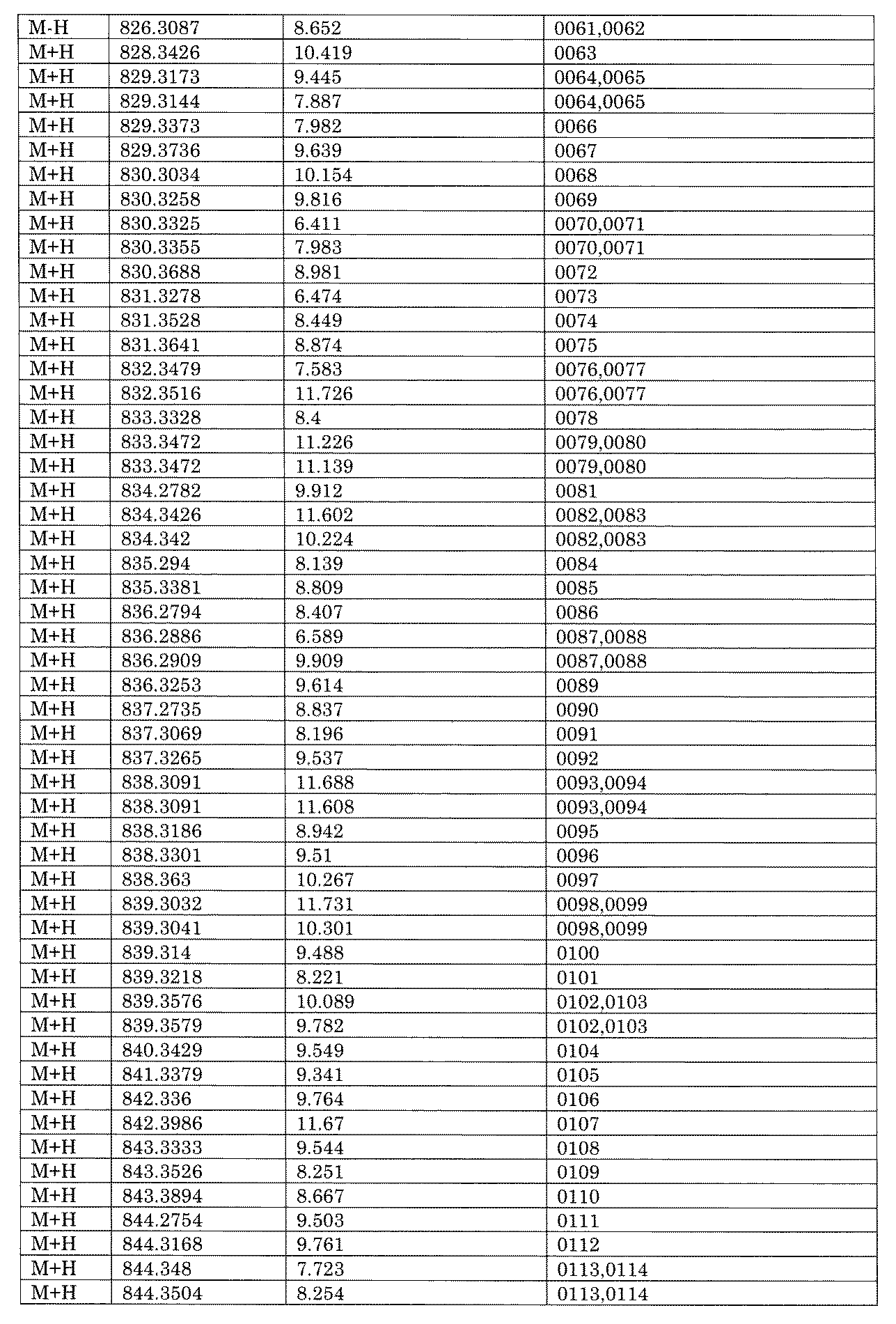

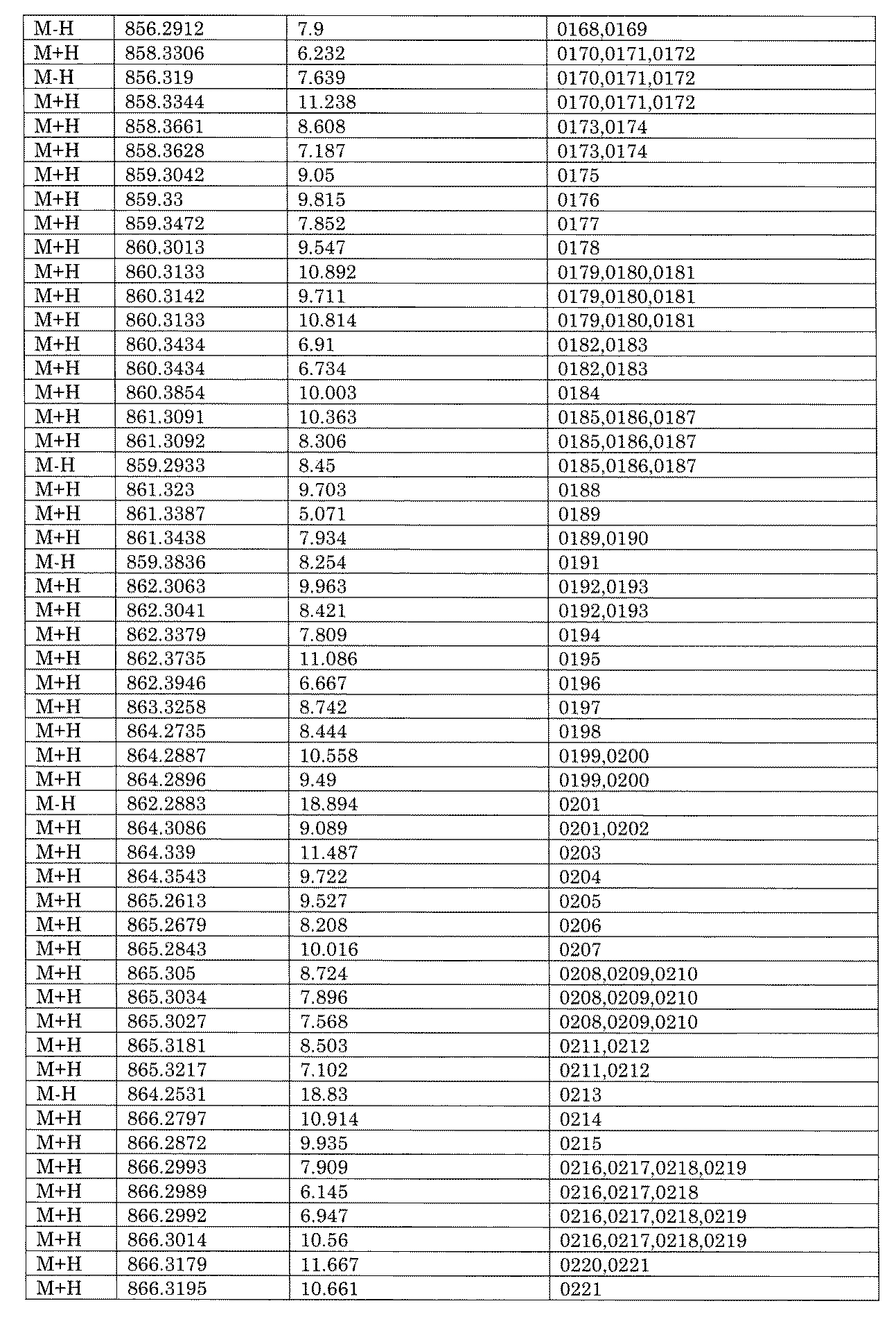

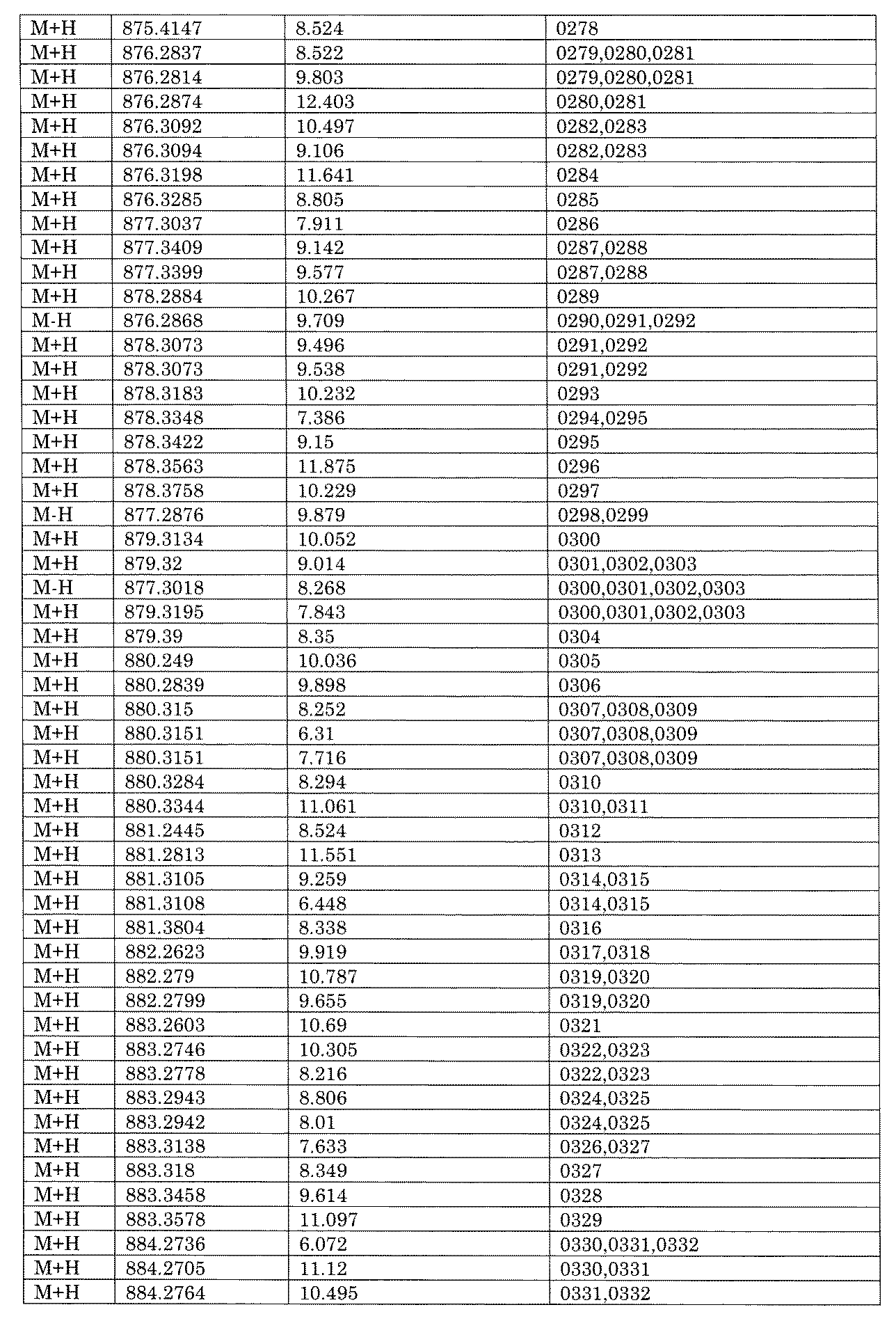
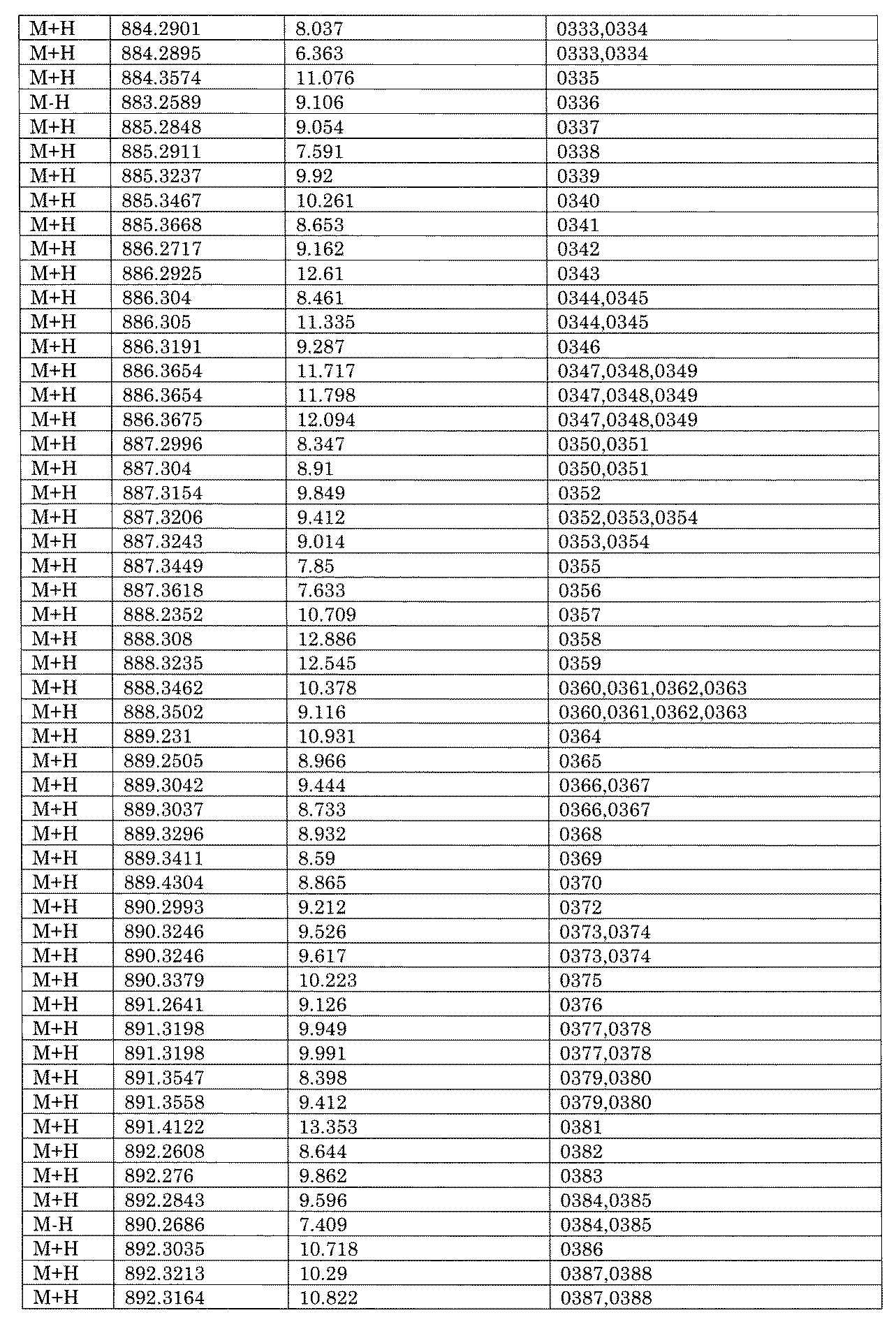




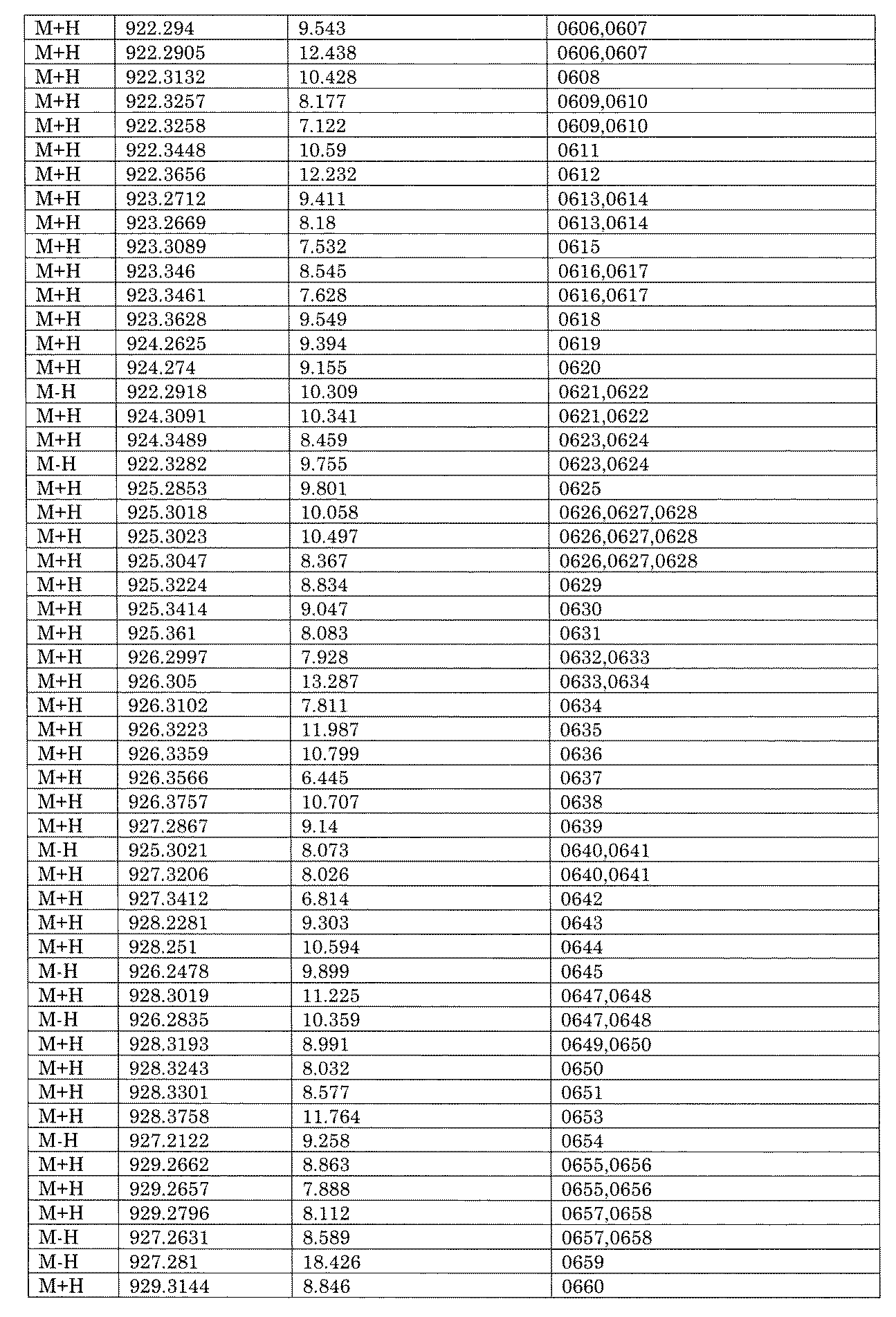


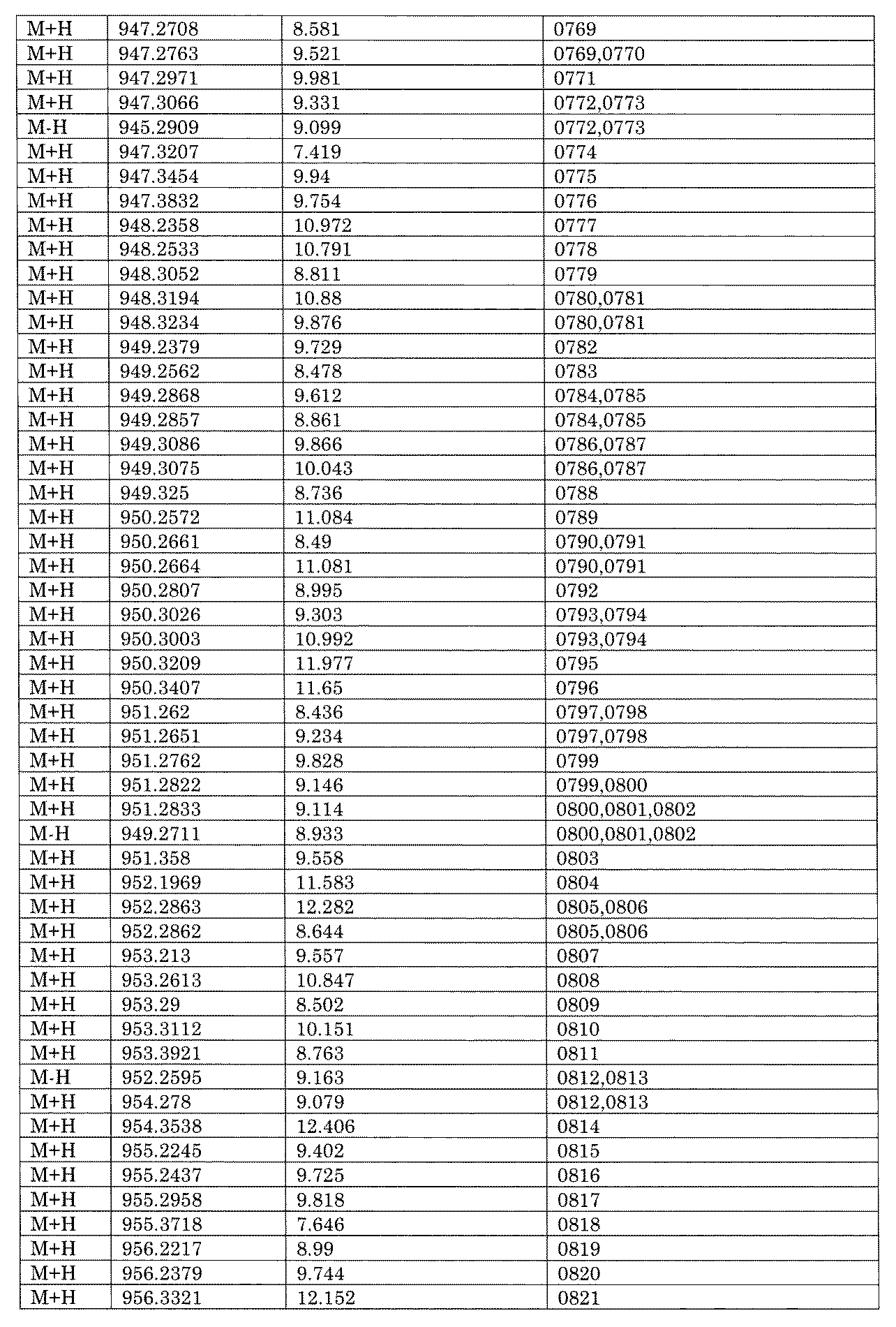






該ライブラリに含む「コアブロックの多様性」と「リンカーの多様性」について図6に示し、Split & mixによるライブラリ合成ルートに関して図7に示す。
実施例9にて合成している混合物を示す場合には、混合物番号を利用することもある。例えば、「2−2−B00−0」の「2−2」はライブラリの種類を示し、「B00」は混合物の種類を示す。「−0」の「−0」は固相に担持された状態を示し、「−1」と記された場合には固相から切り出した状態を示す。また、「−2」の場合には固相からの切り出し後に官能基変換を行った状態を示す。混合物に含まれる各化合物を示す際は、IDを利用することもある。例えば、「2−2−a1B01−0−0001」の「0001」は、混合物「2−2−a1B01−0」に含まれる化合物のうち任意の化合物を示す。
実施例9−1−1:工程a1で利用する2−2−B00−0の調製

反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(128mL)で3回、NMP(128mL)で3回、0.05M TBAHSO4+DTBP in NMP(128mL)で3回、NMP(128mL)で3回、MeOH(128mL)で3回、DCM(128mL)で3回、ヘプタン(128mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−B00−0(6.81g、0.201mmol/g)を得た。
表中の表記方法について説明する。例えば[表9−1−0]では、2−2−B00−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−B00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。[表9−1−0]においては、「▲×▼」と「a」「b」は化学式で表す。





固相精製
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(25mL)で3回、NMP(25mL)で3回、MeOH(25mL)で3回、DCM(25mL)で3回、ヘプタン(25mL)で1回洗浄し、得られた固相を減圧下で乾燥させ、[表9−1−1]に記載の混合物を得た。
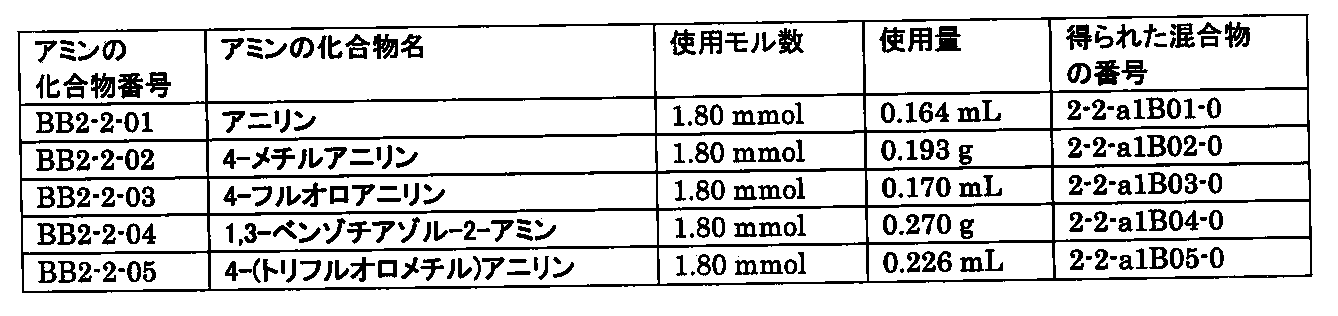
表中の表記方法について説明する。例えば[表9−1−2]では、2−2−a1B01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−a1B01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−1−2]~[表9−1−6]においては、「▲×▼」と「b」は化学式で表す。
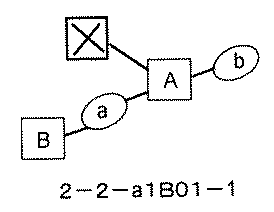

2−2−a1B01−1−0001に対応する表記例




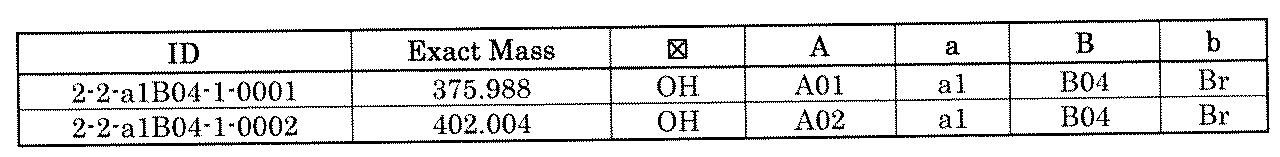

実施例9−2−0:工程bで利用する2−2−C00−0の調製
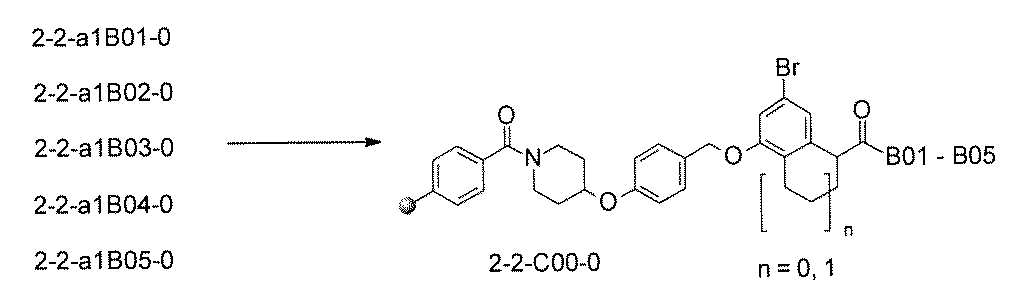


反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−2−1]に記載の混合物を得た。


表中の表記方法について説明する。例えば[表9−2−2]では、2−2−b1C01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b1C01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−2]~[表9−2−6]においては、「▲×▼」と「c」は化学式で表す。






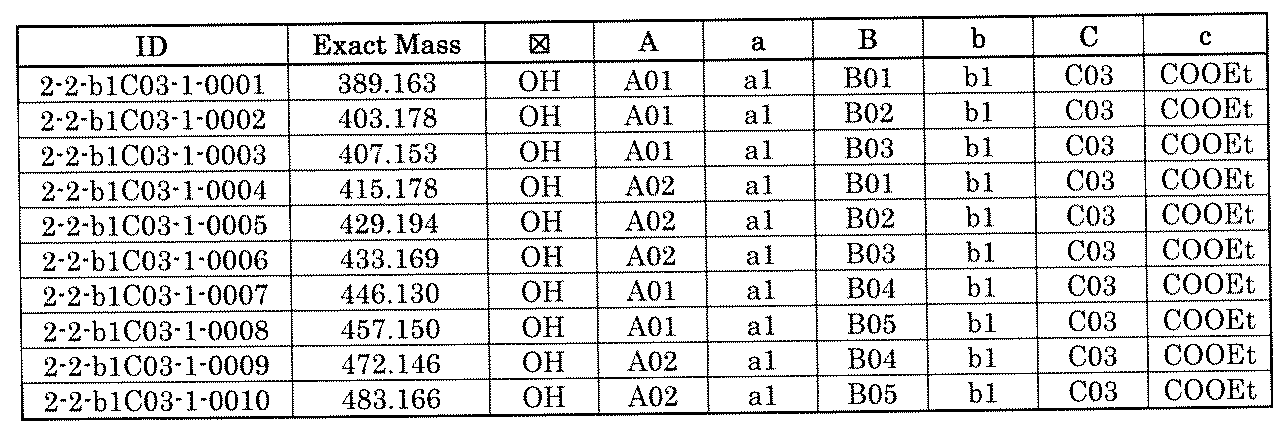
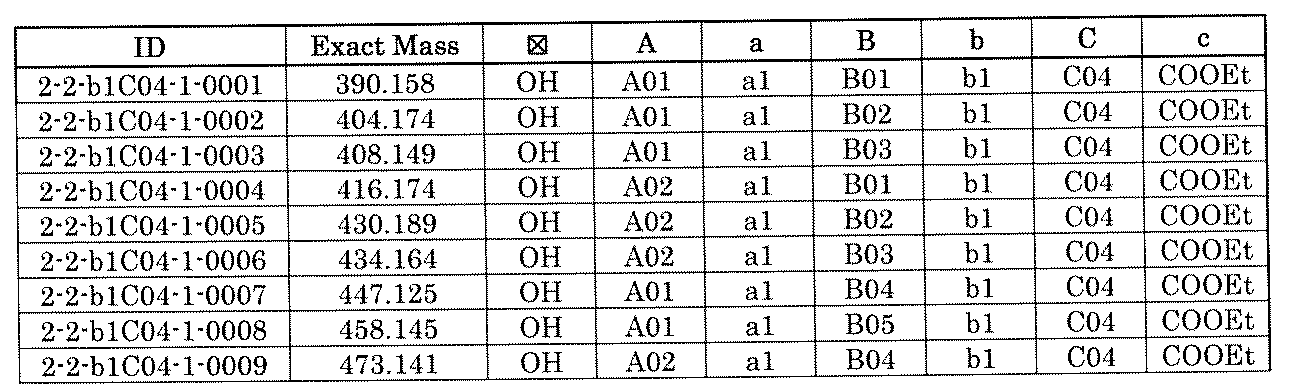

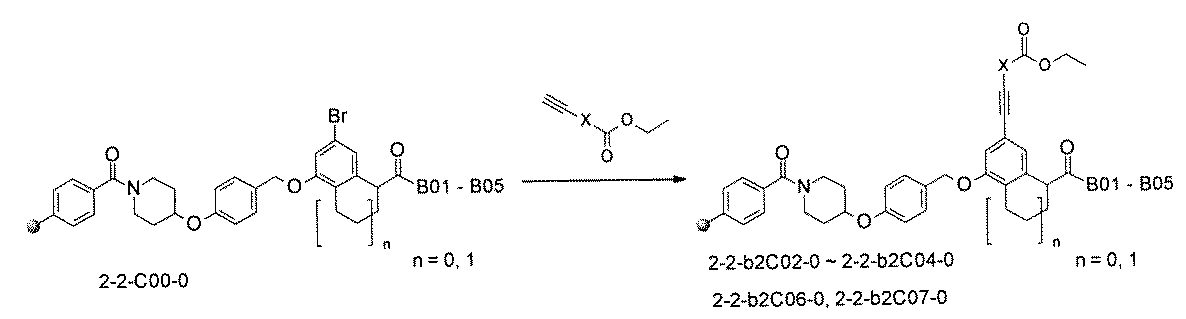
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05 M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−2−7]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−2−8]では、2−2−b2C02−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b2C02−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−8]~[表9−2−12]においては、「▲×▼」と「c」は化学式で表す。
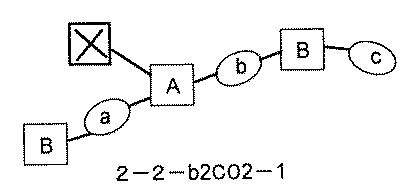

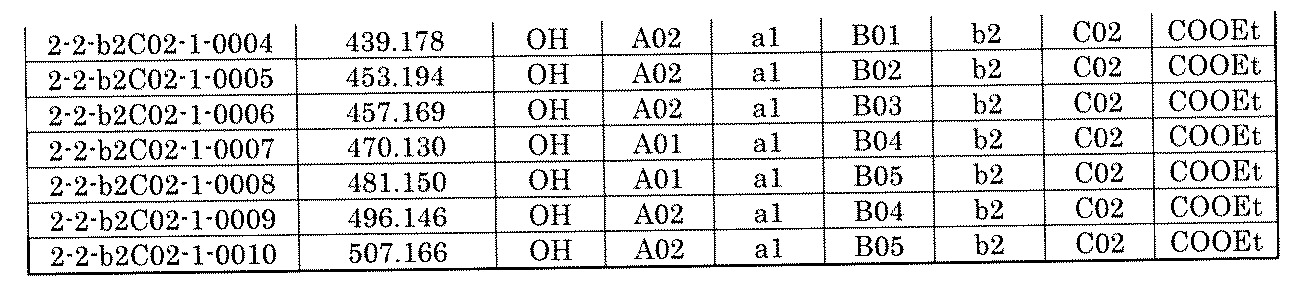





反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−b3C11−0を得た。
表中の表記方法について説明する。例えば[表9−2−13]では、2−2−b3C11−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b3C11−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−13]においては、「▲×▼」と「c」は化学式で表す。



反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP(11mL)で3回、0.2M NAC in NMP/水=5/1(11mL)で3回、0.05M TBAHSO4+DTBP in NMP(11mL)で3回、0.05M NMM in NMP(11mL)で3回、NMP/水=1/1(11mL)で3回、NMP(11mL)で3回、MeOH(11mL)で3回、DCM(11mL)で3回、ヘプタン(11mL)で1回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−b3C00−0を得た。
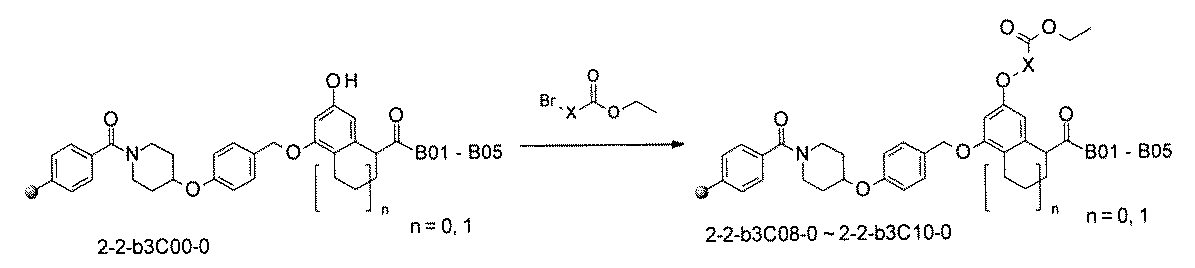
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−2−14]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−2−15]では、2−2−b3C08−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b3C08−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−15]~[表9−2−17]においては、「▲×▼」と「c」は化学式で表す。


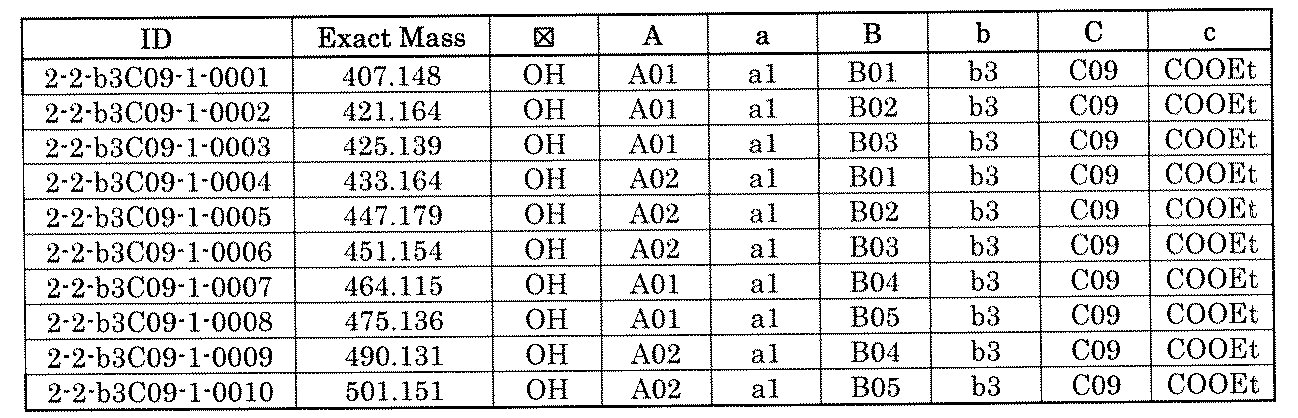


反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05 M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表2−3−18]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−2−19]では、2−2−b4C01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b4C01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−19]~[表9−2−23]においては、「▲×▼」と「c」は化学式で表す。







反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−b1C15−0を得た。
表中の表記方法について説明する。例えば[表9−2−24]では、2−2−b1C15−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b1C15−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−24]においては、「▲×▼」と「c」は化学式で表す。




反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−2−25]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−2−26]では、2−2−b1C14−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b1C14−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−26]~[表9−2−29]においては、「▲×▼」と「c」は化学式で表す。



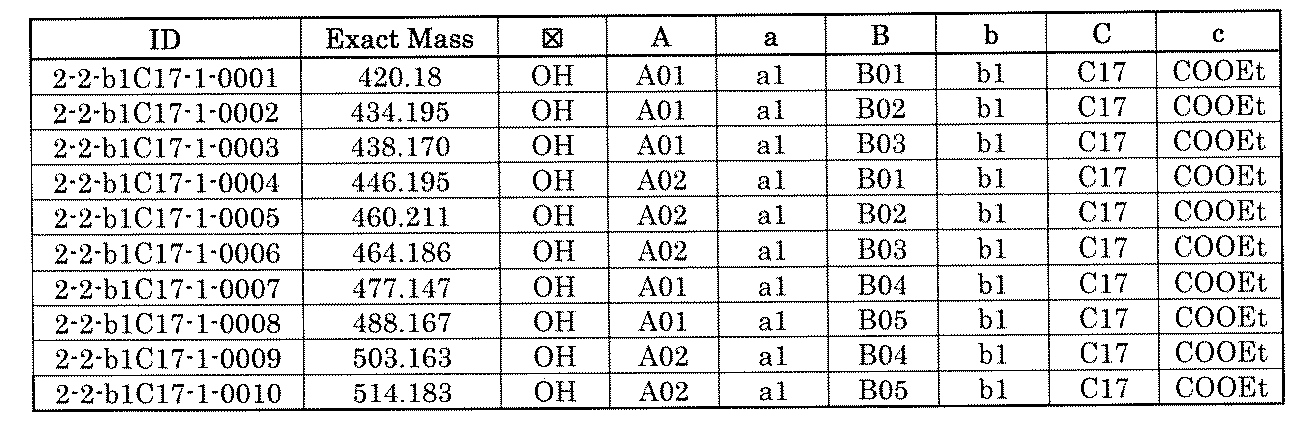


反応液と固相の懸濁液を6mLのフィルターに移してろ過し、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、0.2M NAC in NMP/水=5/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、0.05M NMM in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−b1C19−0を得た。
表中の表記方法について説明する。例えば[表9−2−30]では、2−2−b1C19−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−b1C19−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−2−30]においては、「▲×▼」と「c」は化学式で表す。

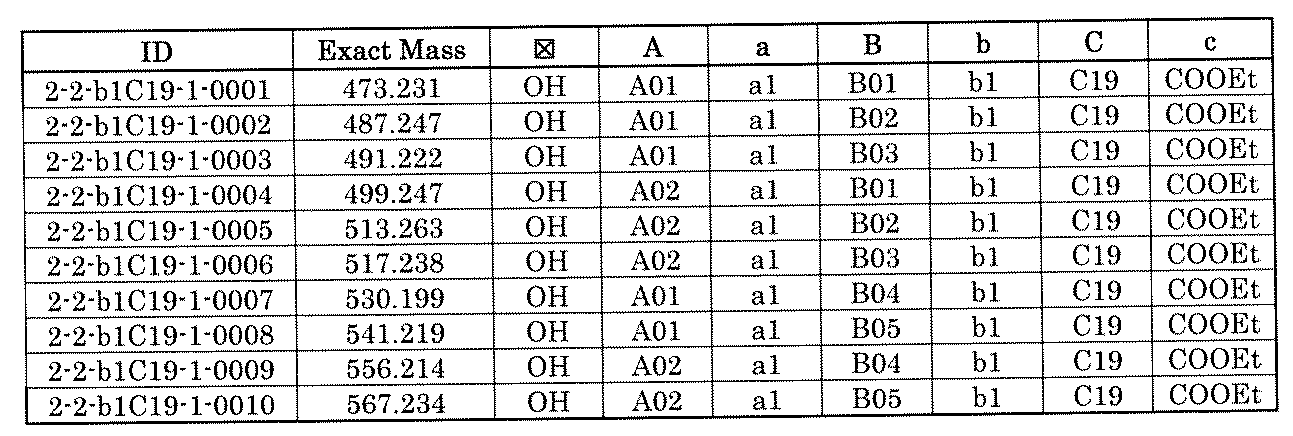
実施例9−3−1:加水分解工程hによる[表9−3−1]記載混合物の合成
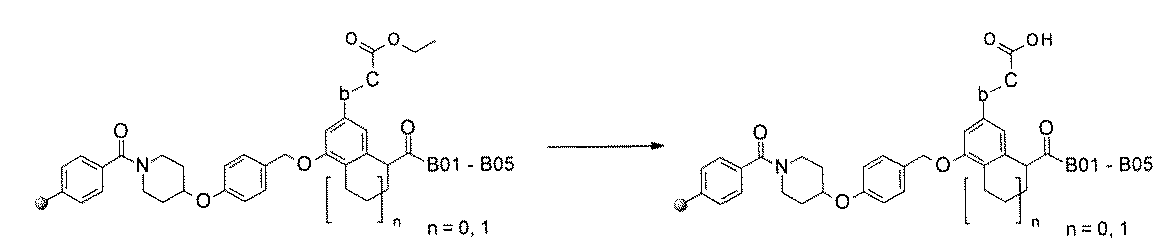
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−3−1]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−3−2]では、2−2−h05C04−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−h05C04−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−3−2]~[表9−3−15]においては、「▲×▼」と「c」は化学式で表す。

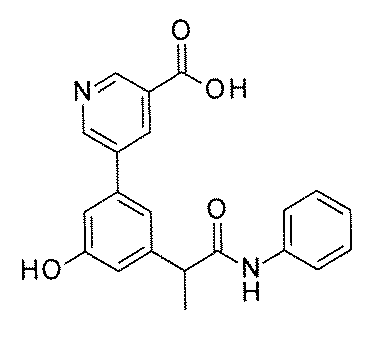

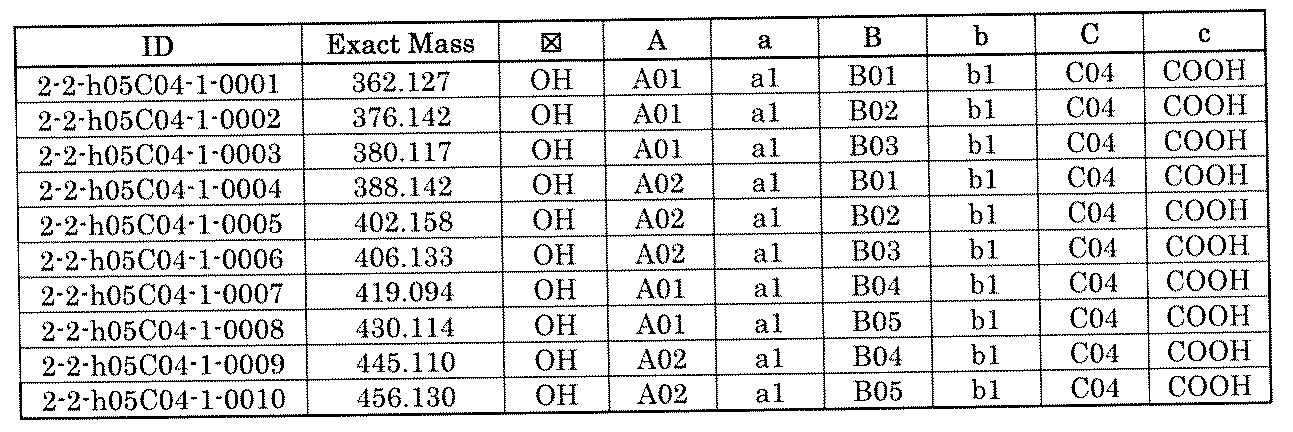



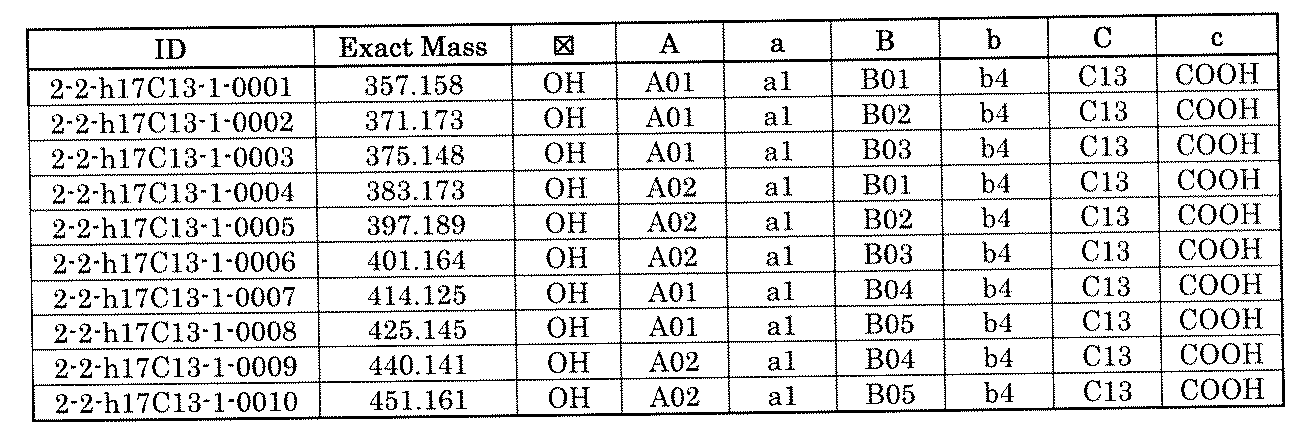
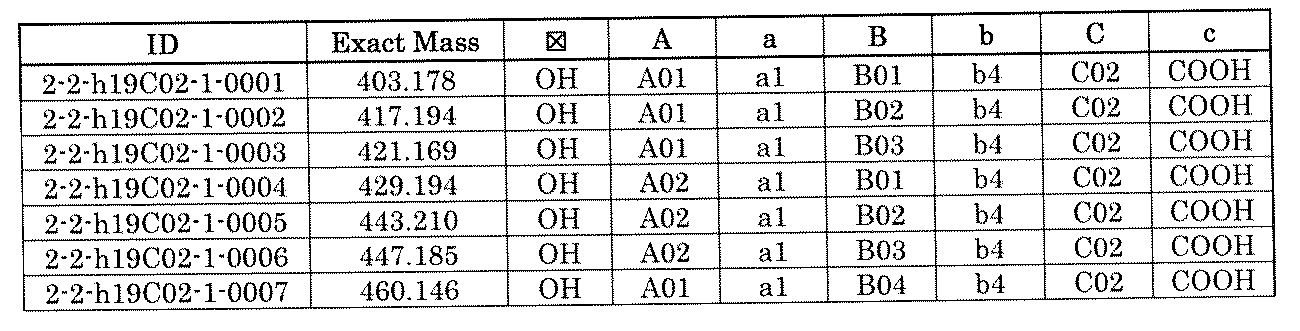




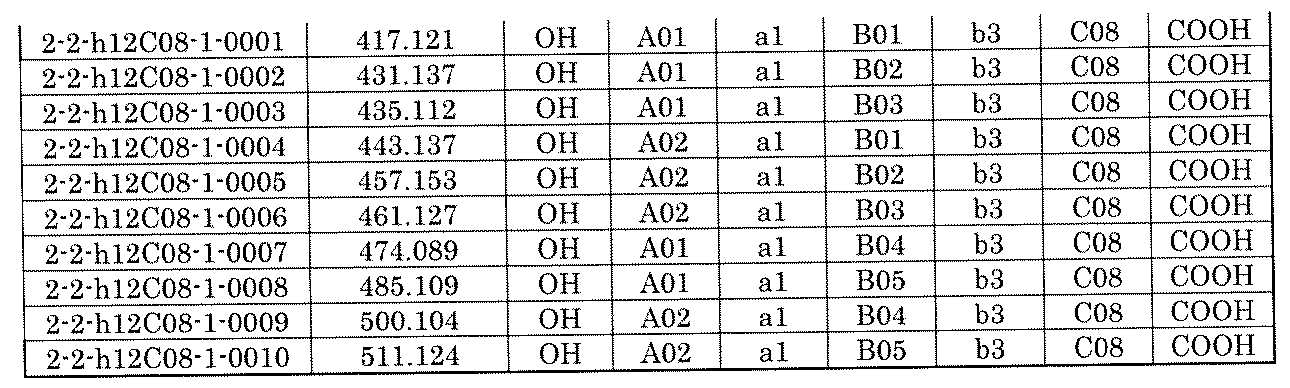

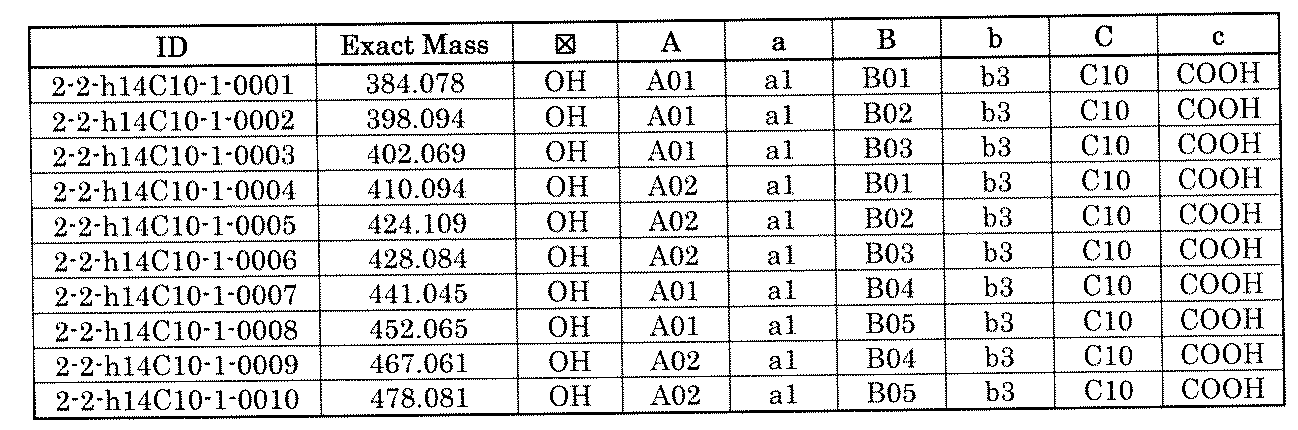



前の行程で得られた[表9−3−16]に記載の混合物の入った9本の6mLのフィルター付きカラムに、NMP(1.79mL)とtAmylOH(0.512mL)、1M TBAOH水溶液(0.074mL、0.074mmol)を加えた。反応液が漏れないようにキャップをして室温で3時間振盪した。
NMP(1.79mL)とtAmylOH(0.512mL)、1M TBAOH水溶液(0.124mL、0.124mmol)を加えた。反応液が漏れないようにキャップをして室温で17時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−3−16]に記載の混合物を得た。
[表9−3−16]

表中の表記方法について説明する。例えば[表9−3−17]では、2−2−h03C02−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−h03C02−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−3−17]~[表9−3−25]においては、「▲×▼」と「c」は化学式で表す。










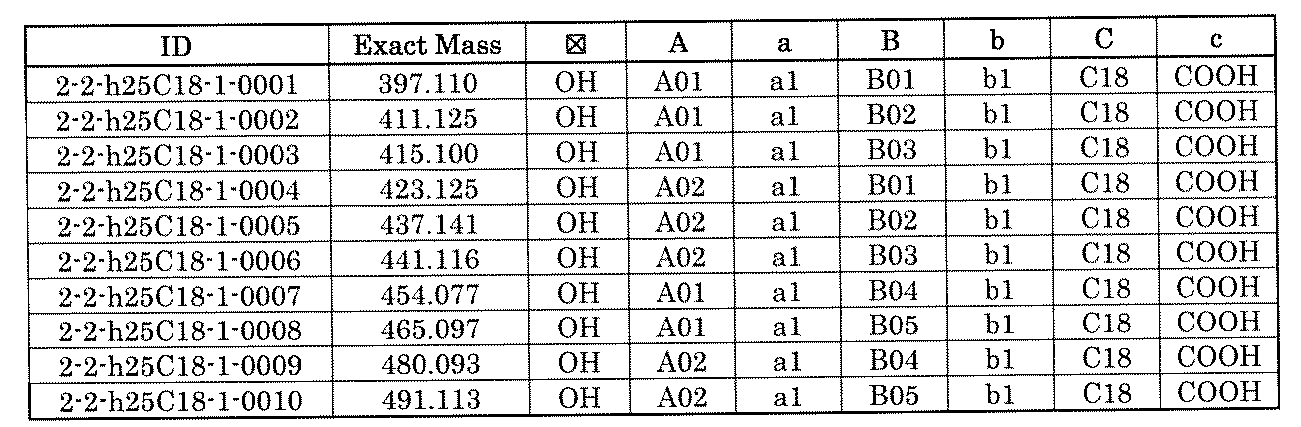
実施例9−2−8で得られた混合物2−2−b1C15−0の入った6mLのフィルター付きカラムに、NMP(1.79mL)とtAmylOH(0.512mL)、1 M TBAOH水溶液(0.074mL、0.074mmol)を加えた。反応液が漏れないようにキャップをして室温で3時間振盪した。
NMP(1.79mL)とtAmylOH(0.512mL)、1M TBAOH水溶液(0.124mL、0.124mmol)を加えた。反応液が漏れないようにキャップをして60℃で17時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(2.6mL)で3回、0.05M TBAHSO4+DTBP in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−h22C15−0を得た。
表中の表記方法について説明する。例えば[表9−3−26]では、2−2−h22C15−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−h22C15−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−3−26]においては、「▲×▼」と「c」は化学式で表す。

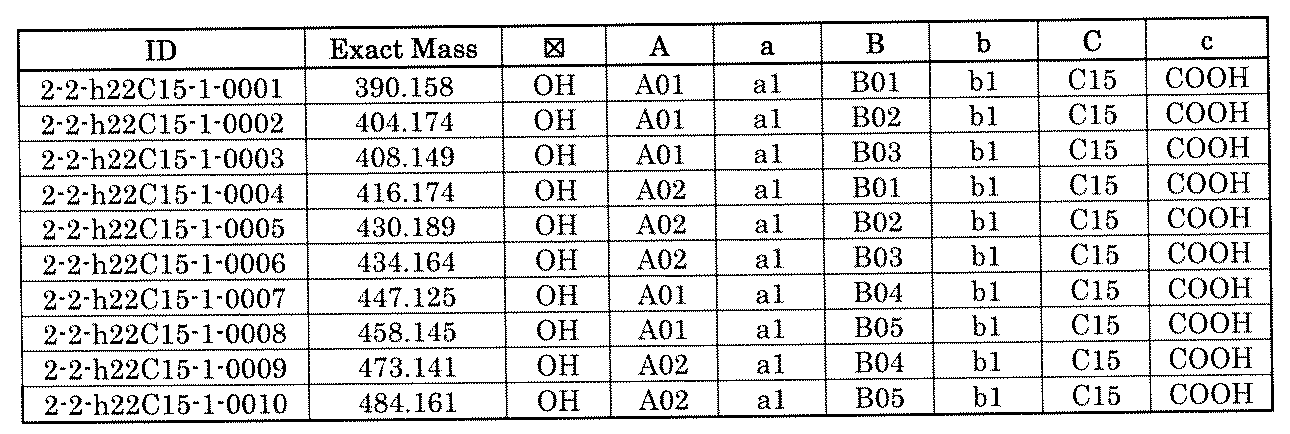
実施例9−2−10で得られた混合物2−2−b1C19−0の入った6mLのフィルター付きカラムに、NMP(1.79mL)とtAmylOH(0.512mL)、1M TBAOH水溶液(0.124mL、0.124 mmol)を加えた。反応液が漏れないようにキャップをして60℃で14時間振盪した。
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(2.6mL)で3回、0.05 M TBAHSO4+DTBP in NMP(2.6mL)で3回、NMP/水=1/1(2.6mL)で3回、NMP(2.6mL)で3回、MeOH(2.8mL)で3回、DCM(2.8mL)で3回、ヘプタン(2.8mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−h26C19−0を得た。
表中の表記方法について説明する。例えば[表9−3−27]では、2−2−h26C19−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−h26C19−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−3−27]においては、「▲×▼」と「c」は化学式で表す。


実施例9−4−1:工程c1D01によるアミドリンカーの形成
アミド化
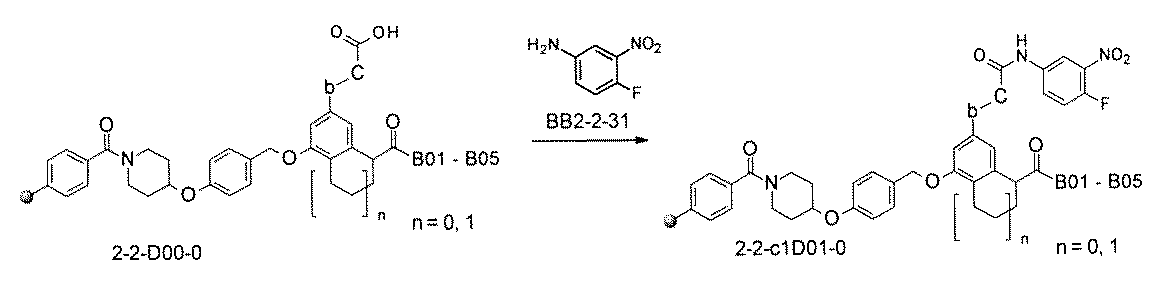
反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP/水=1/1(16mL)で3回、0.05M TBAHSO4+DTBP in NMP(16mL)で3回、0.05M NMM in NMP(16mL)で3回、NMP/水=1/1(16mL)で3回、NMP(16mL)で3回、MeOH(16mL)で3回、DCM(16mL)で3回、ヘプタン(16mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−c1D01−0を得た。
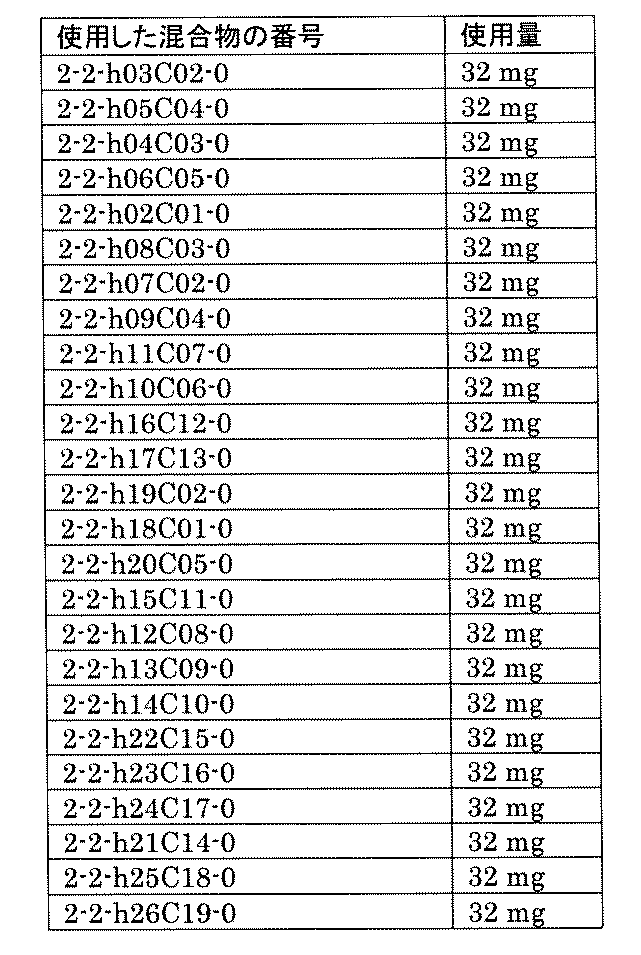
表中の表記方法について説明する。例えば[表9−4−2]では、2−2−c1D01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−c1D01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−4−2]においては、「▲×▼」と「d」は化学式で表す。

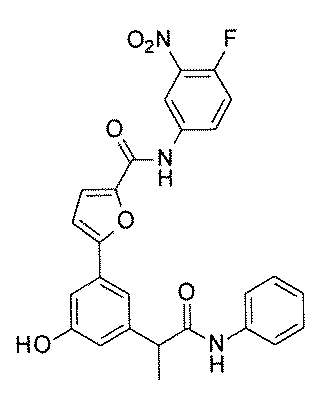








反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP/水=1/1(16mL)で3回、0.05M TBAHSO4+DTBP in NMP(16mL)で3回、0.05M NMM in NMP(16mL)で3回、NMP/水=1/1(16mL)で3回、NMP(16mL)で3回、MeOH(16mL)で3回、DCM(16mL)で3回、ヘプタン(16mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物2−2−c2D02−0を得た。

表中の表記方法について説明する。例えば[表9−4−4]では、2−2−c2D02−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−c2D02−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−4−4]においては、「▲×▼」と「d」は化学式で表す。
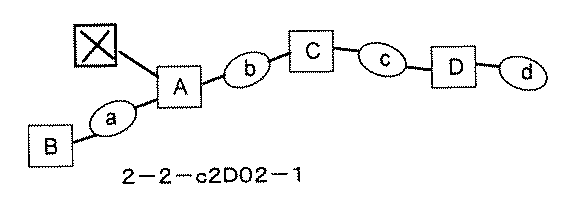
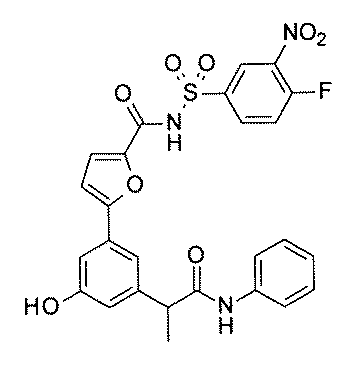

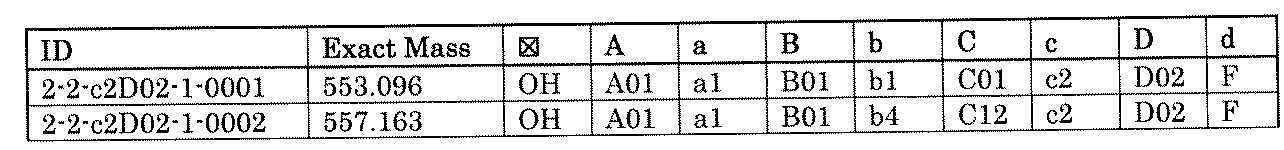
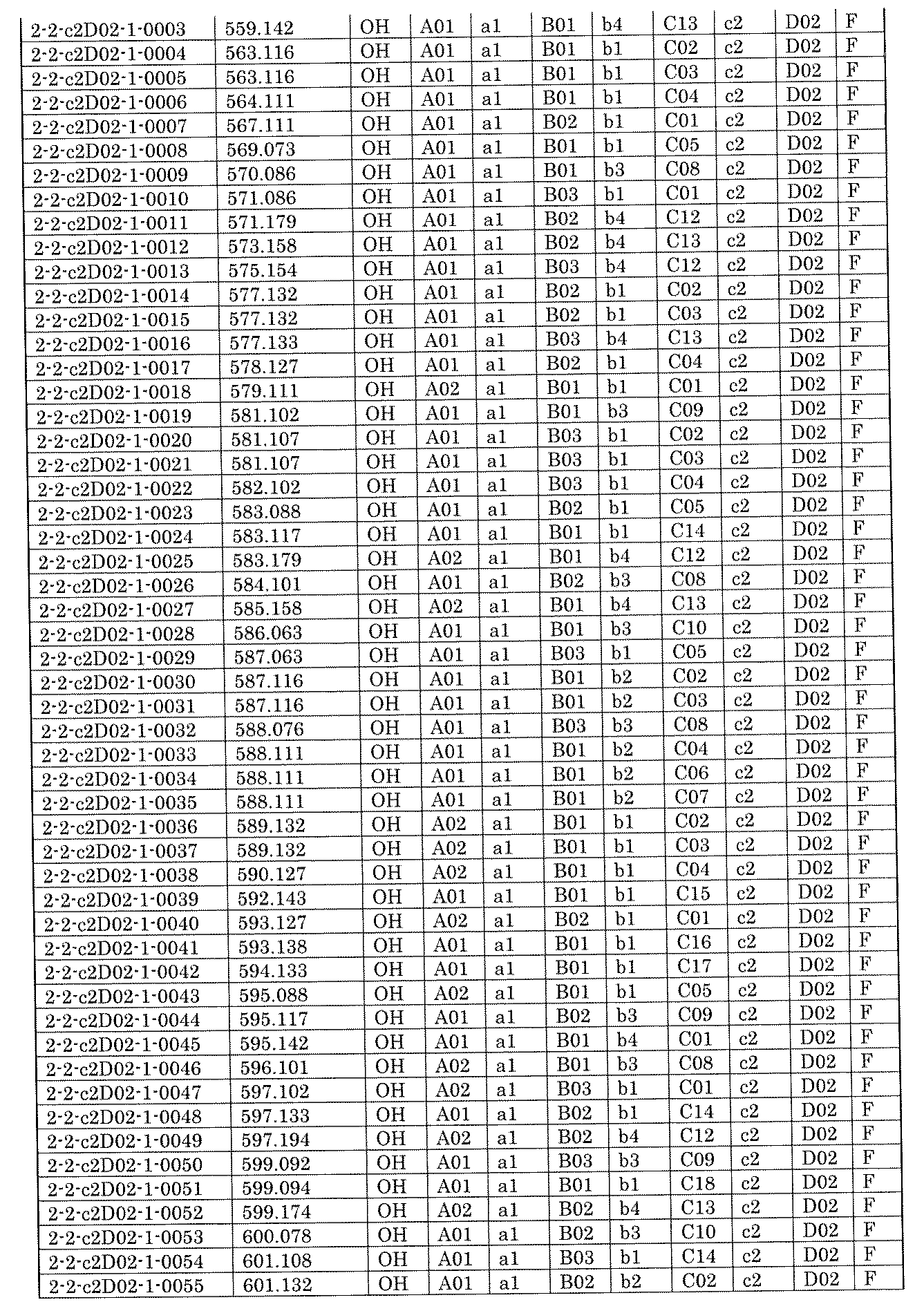




実施例9−5−1:工程d1E01~d1E04による2−2−d1E01−0~2−2−d1E04−0の合成
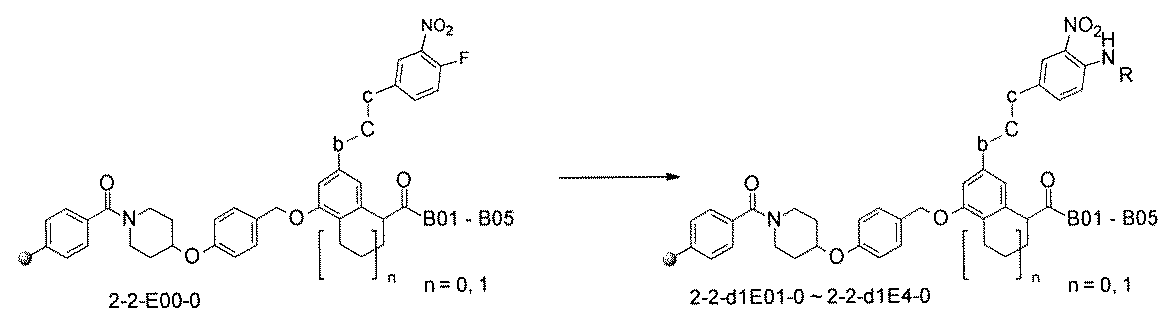
反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP/水=1/1(4mL)で3回、NMP(4mL)で3回、0.05M TBAHSO4+DTBP in NMP(4mL)で3回、0.05M NMM in NMP(4mL)で3回、NMP/水=1/1(4mL)で3回、NMP(4mL)で3回、MeOH(4.5mL)で3回、DCM(4.5mL)で3回、ヘプタン(4.5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、[表9−5−1]に記載の混合物を得た。

表中の表記方法について説明する。例えば[表9−5−2]では、2−2−d1E01−1に含まれうるそれぞれの化合物をIDで示し、混合物2−2−d1E01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図6に示す。
[表9−5−2]~[表9−5−5]においては、「▲×▼」は化学式で表す。




















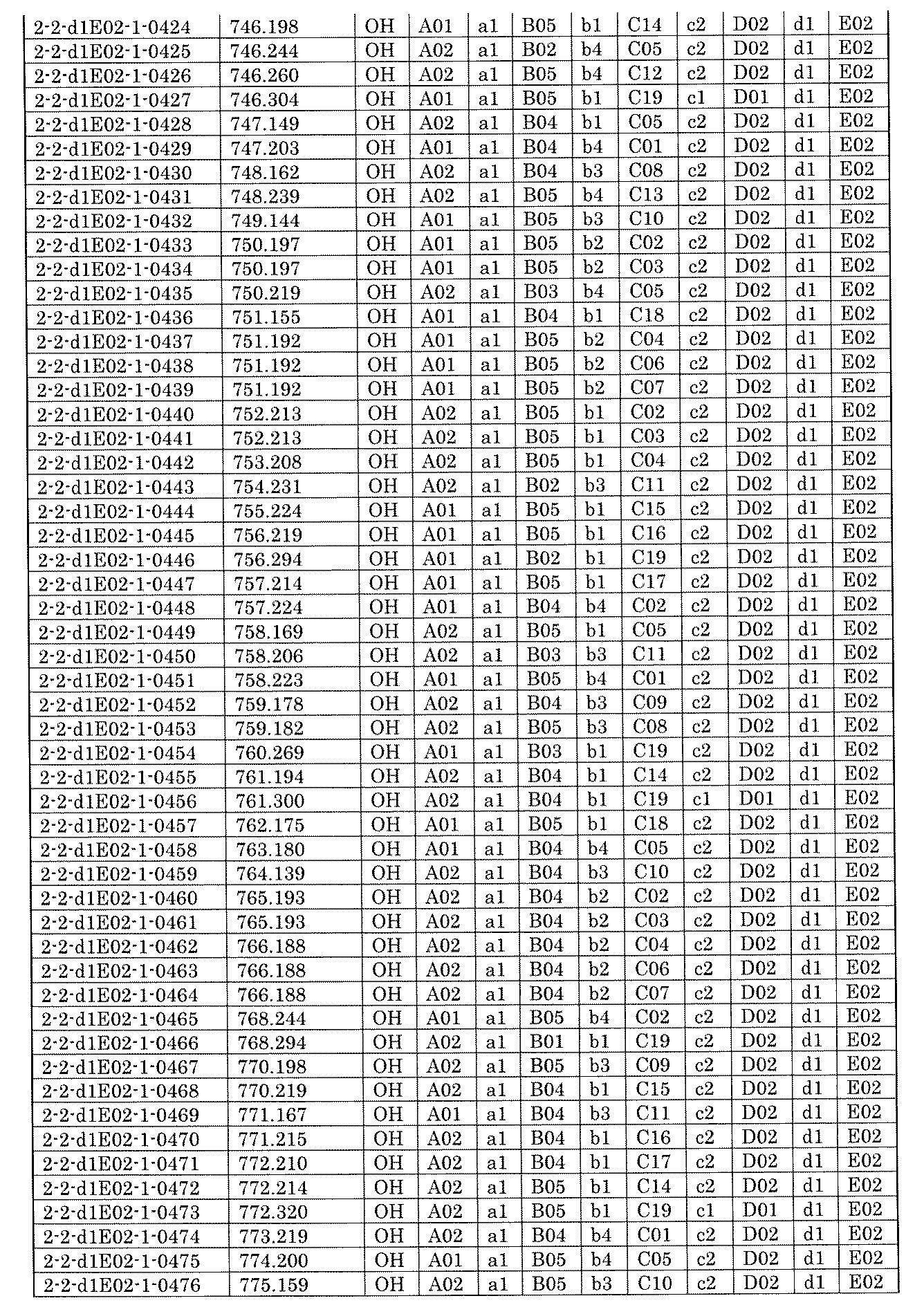



















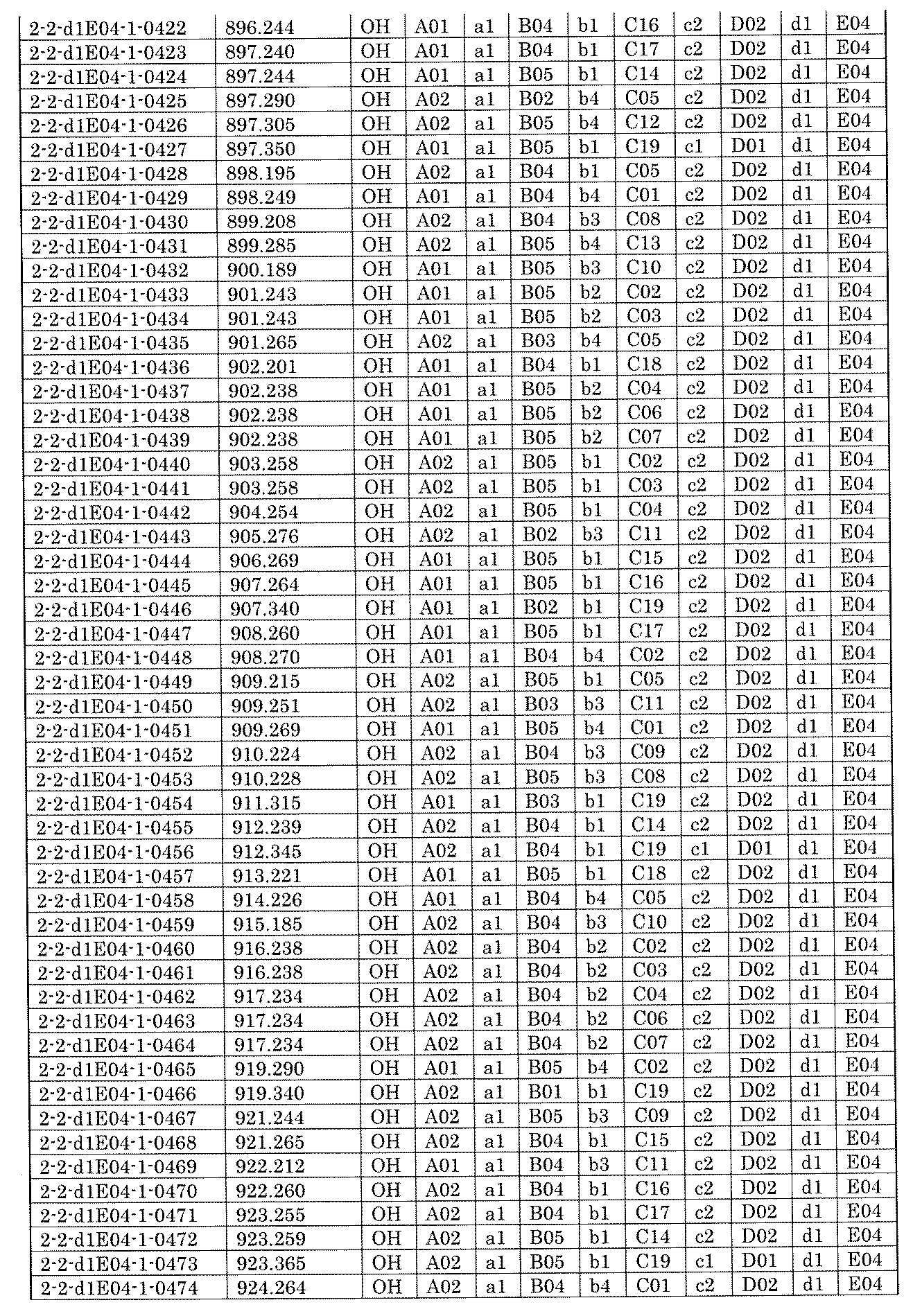
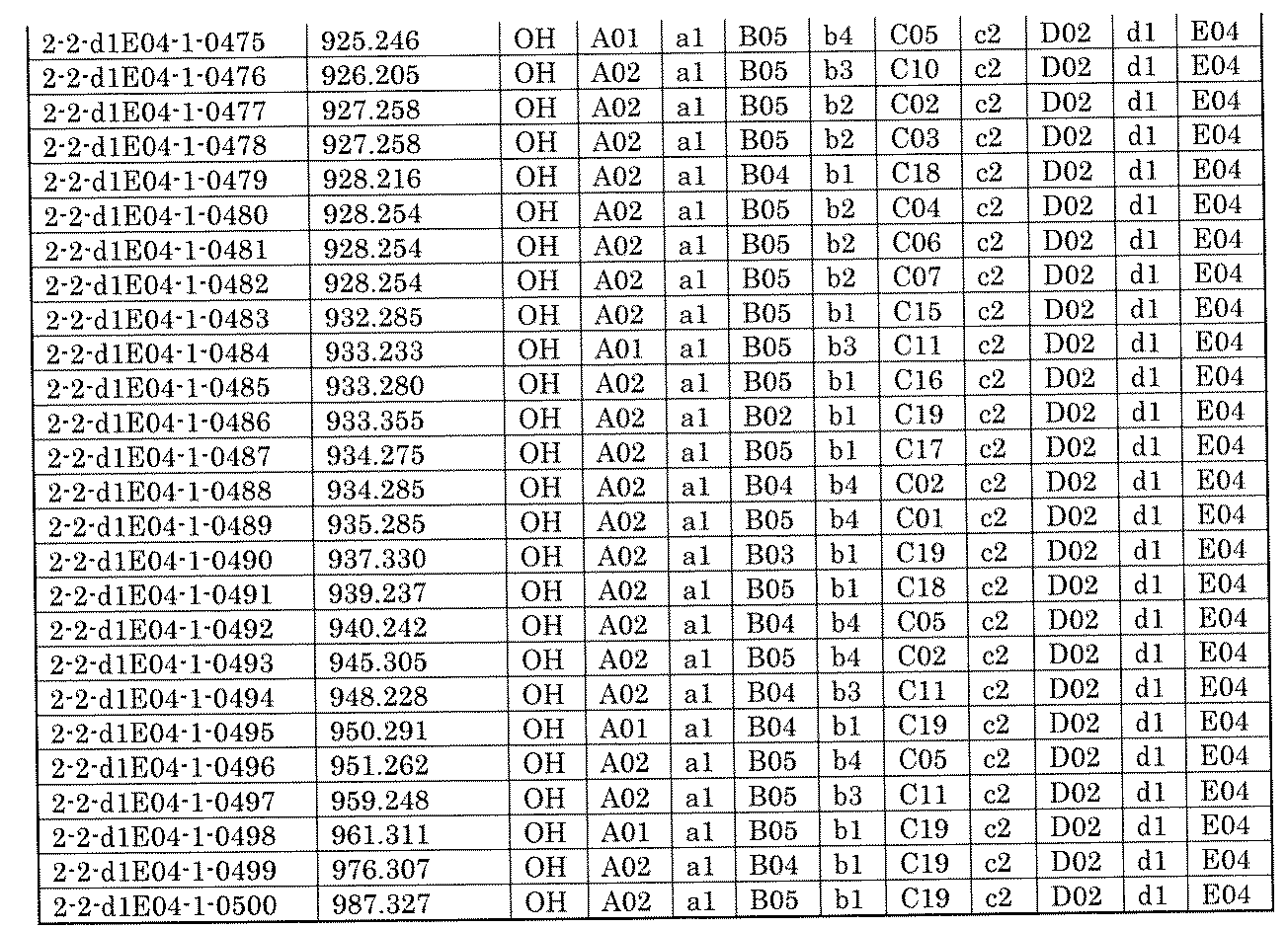

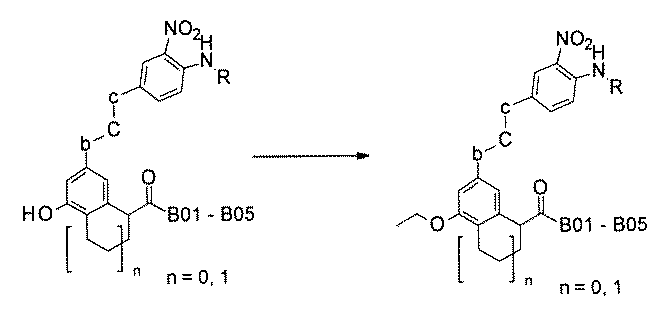

[表9−7−2]~[表9−7−5]においては、「▲×▼」は化学式で表す。
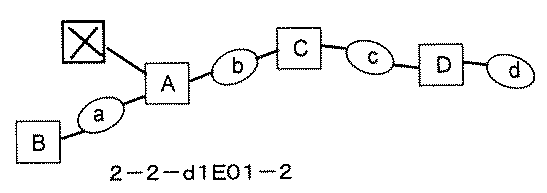
[表9−7−2]中の表記例

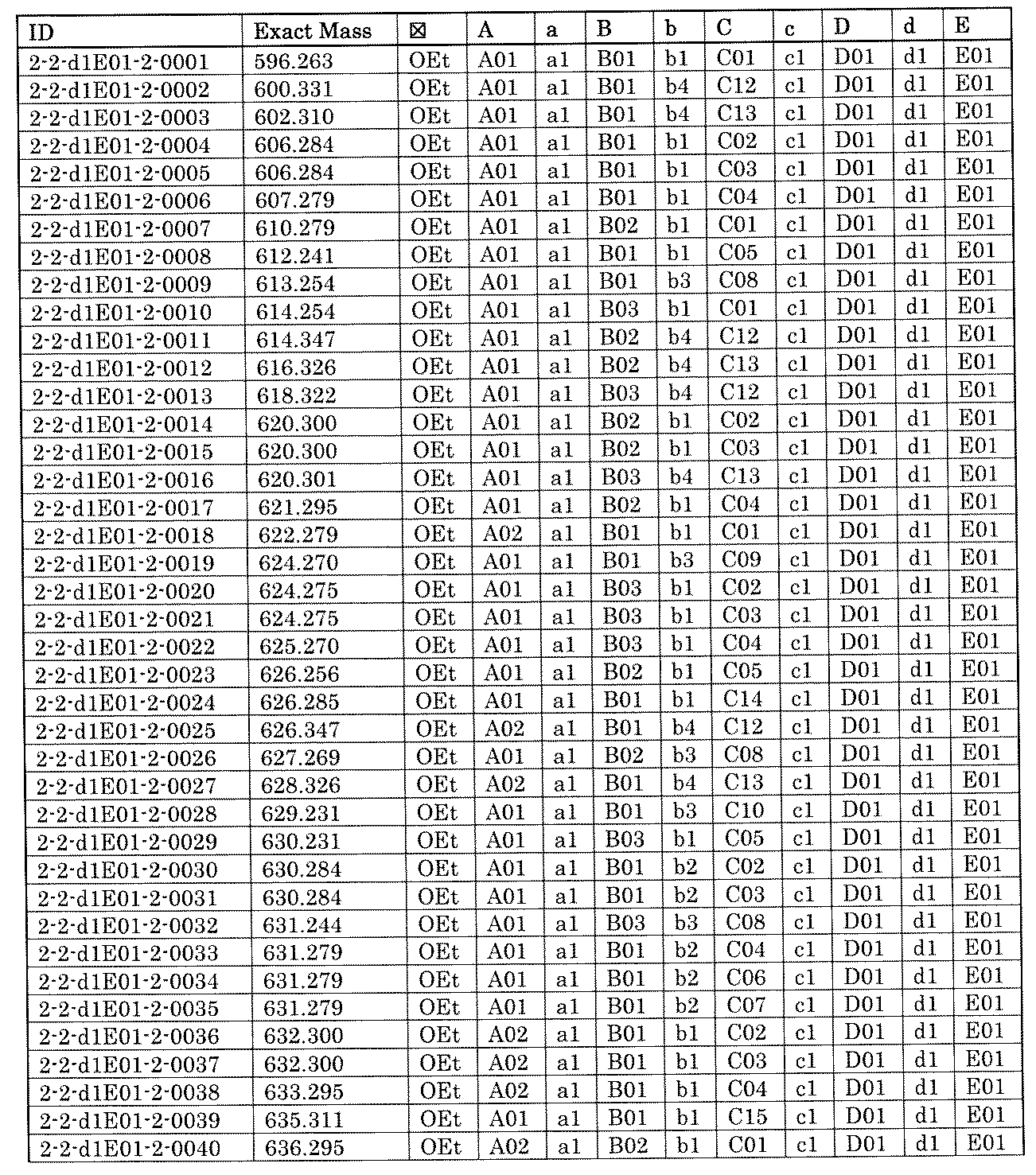









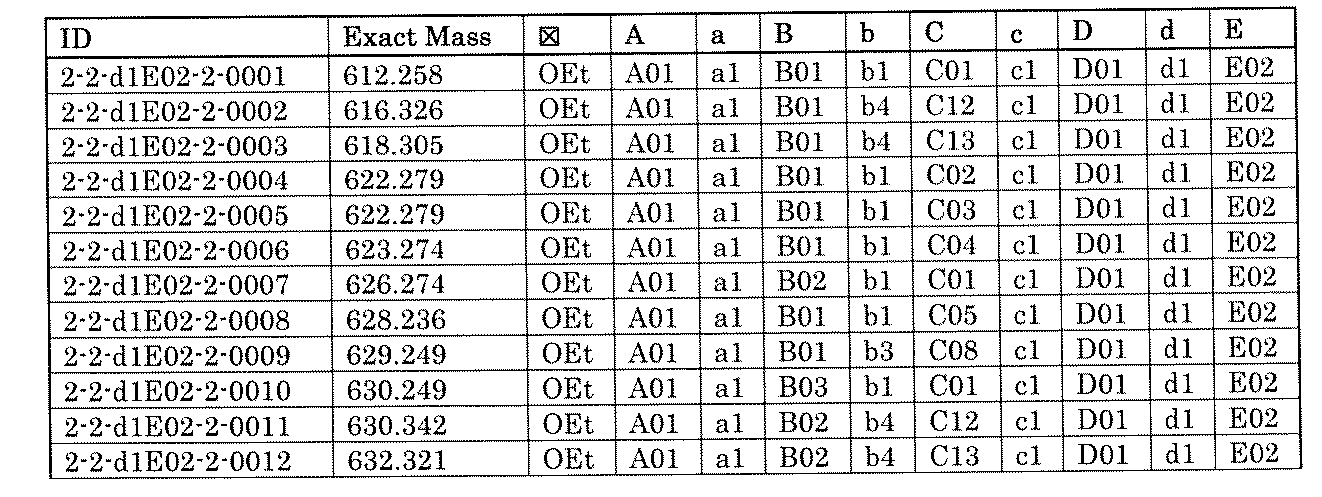












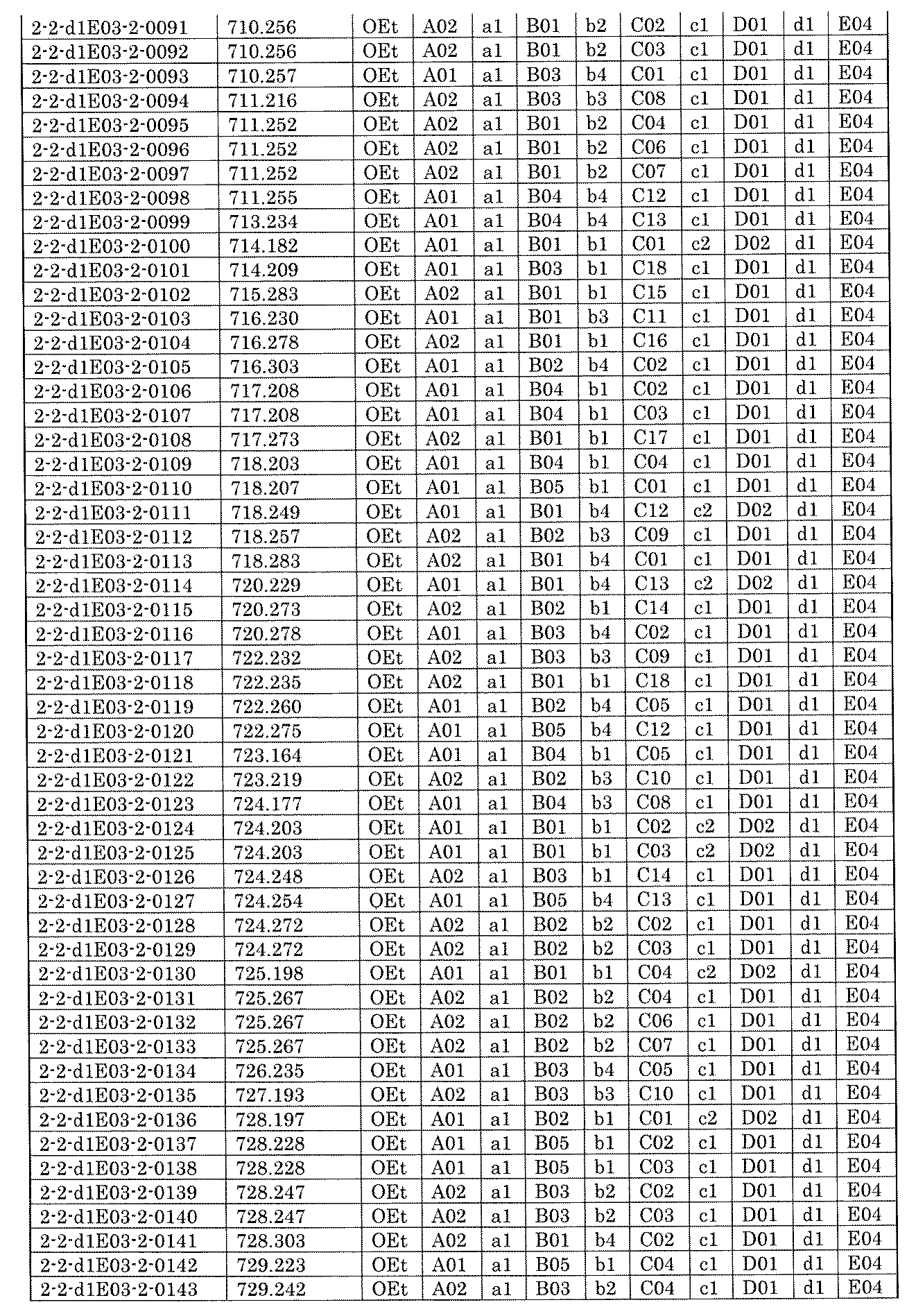




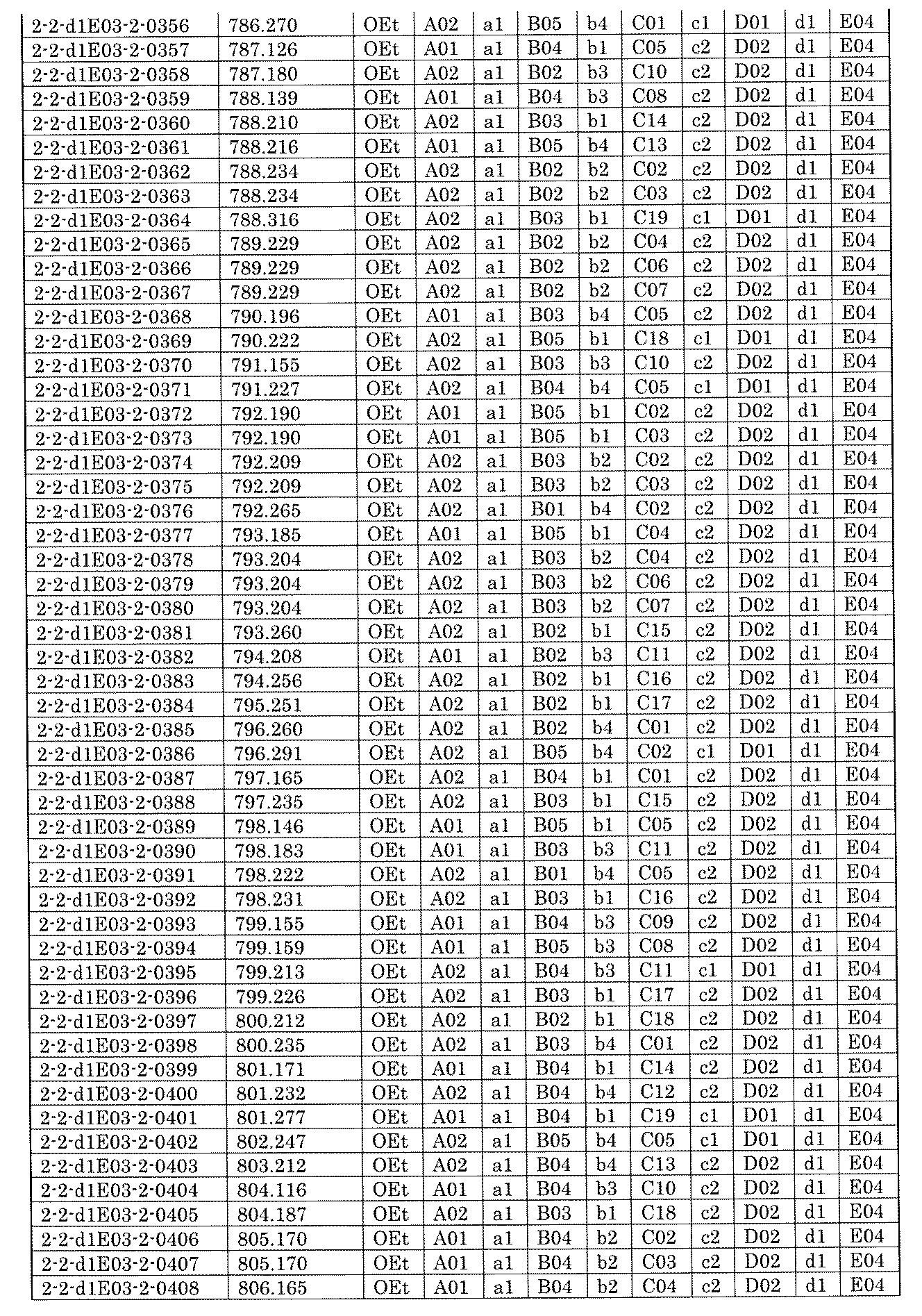




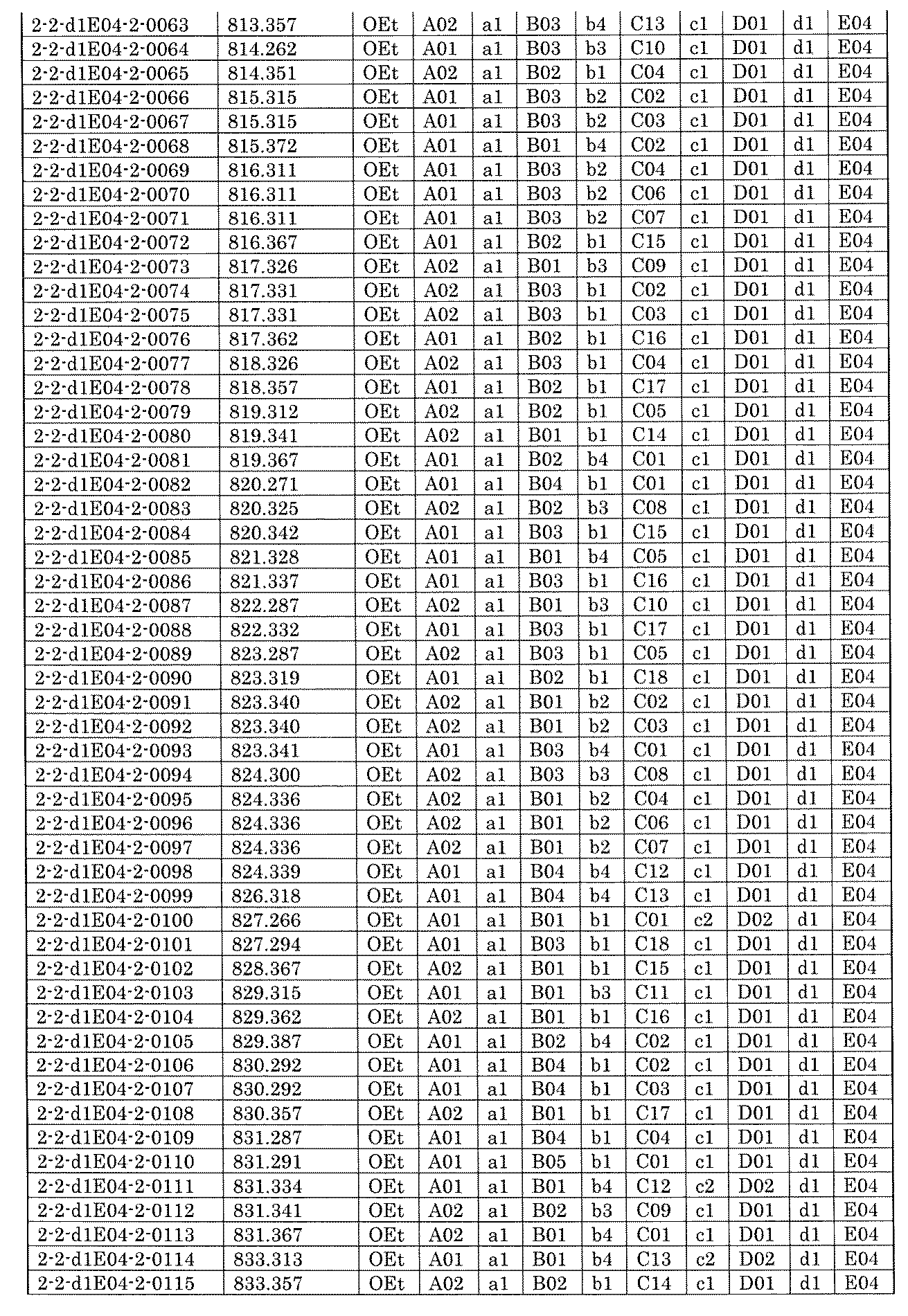





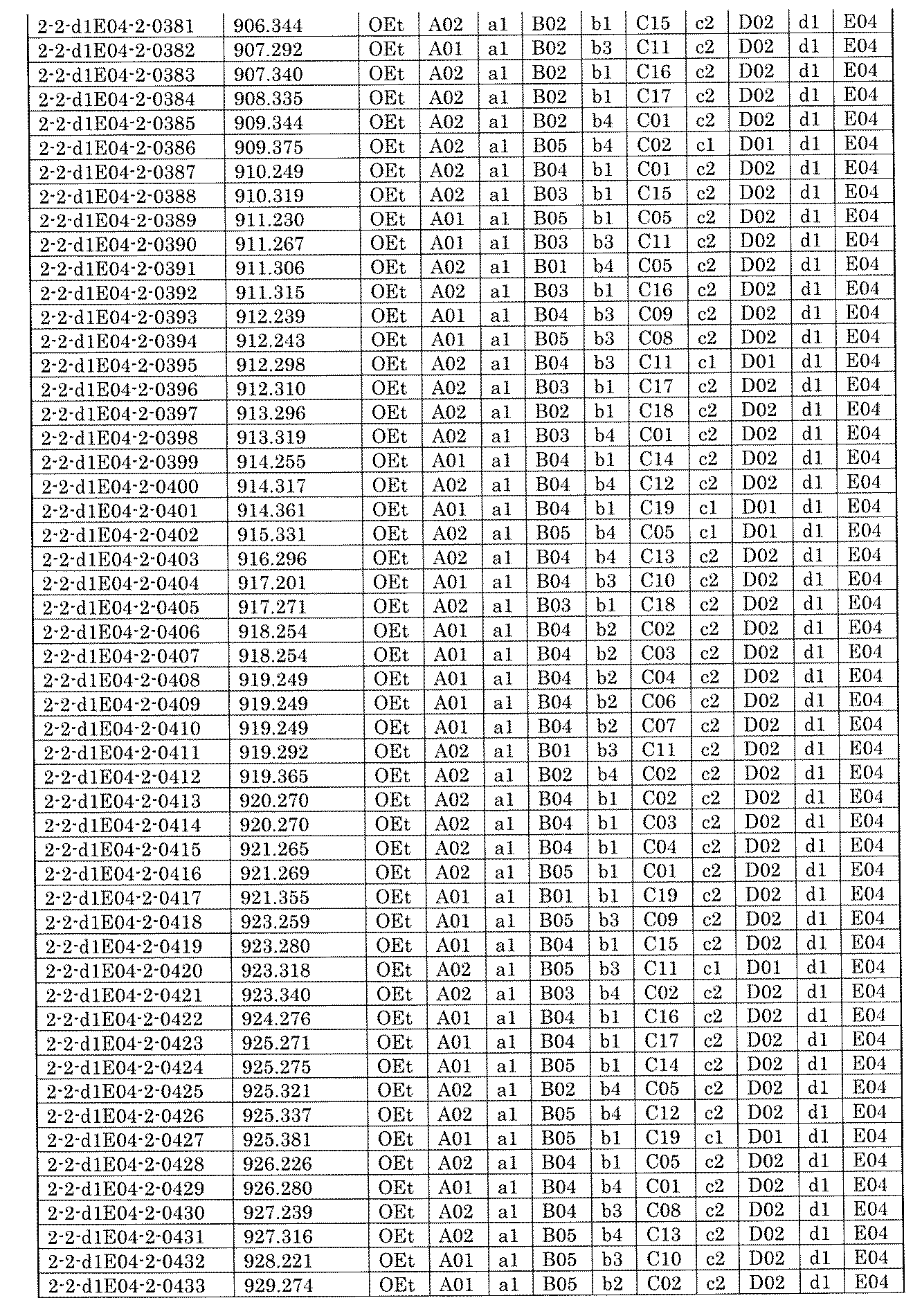

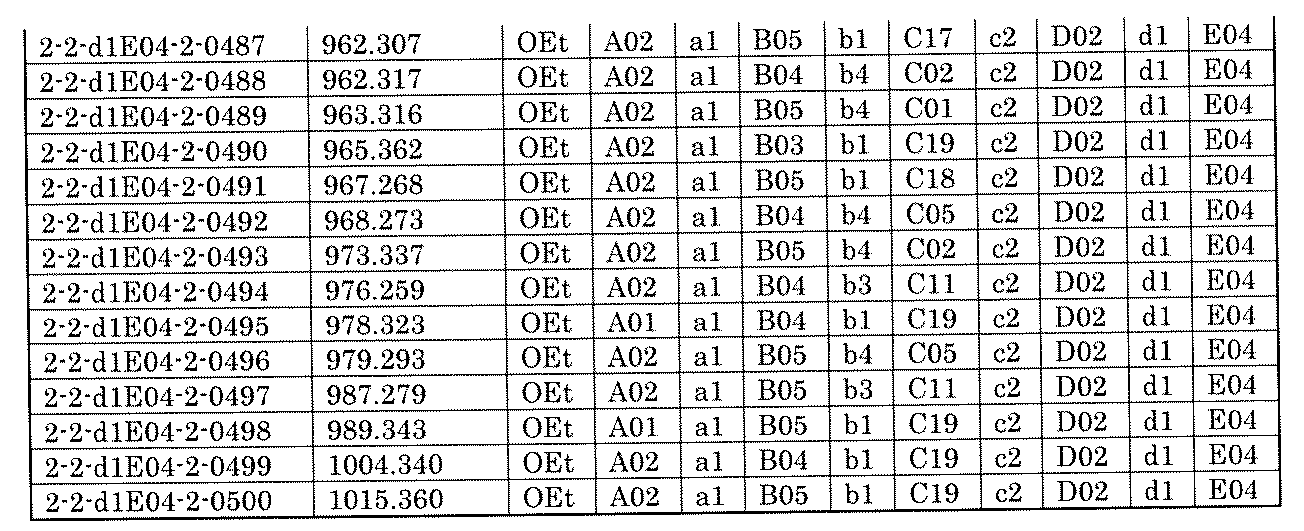
実施例9−6、9−7にて合成した混合物は、以下に示す分析条件により保持時間の測定と質量分析を実施し、Compound Discoverer 3.2(Thermo Fisher Scientific)を用いて解析した。

なお、保持時間については小数点以下3桁目までを確認しているが、[表9−8−1]~[表9−8−8]中の表記においては小数点以下の0を省略して記載していることもある。例えば、「15.000」の場合「15」、「15.100」の場合「15.1」、「15.120」の場合「15.12」と表記していることもある。















































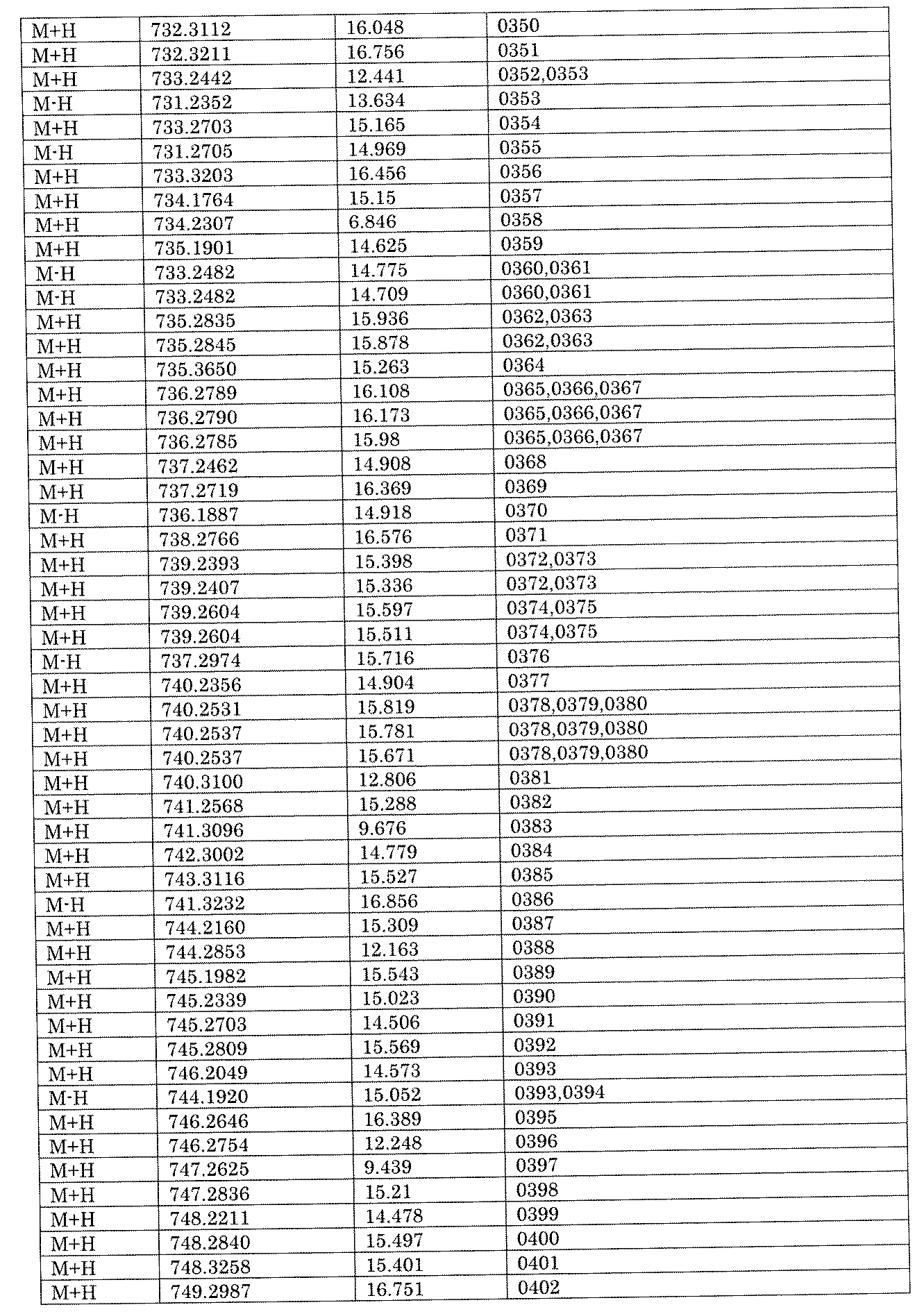

































[表9−9]


実施例4、8、9で合成したライブラリは、ビルディングブロック同士をつなぐ連結部分(リンカー)にも多様性を有し、ファーマコフォアを形成する「コアブロックの多様性」と「リンカーの多様性」を掛け合わせたライブラリの合成に関する例を示した。
Split & mixによるライブラリ合成ルートに関して図10、図11に示す。
実施例10で使用するビルディングブロックを表BB4−1、表BB4−2に示す。
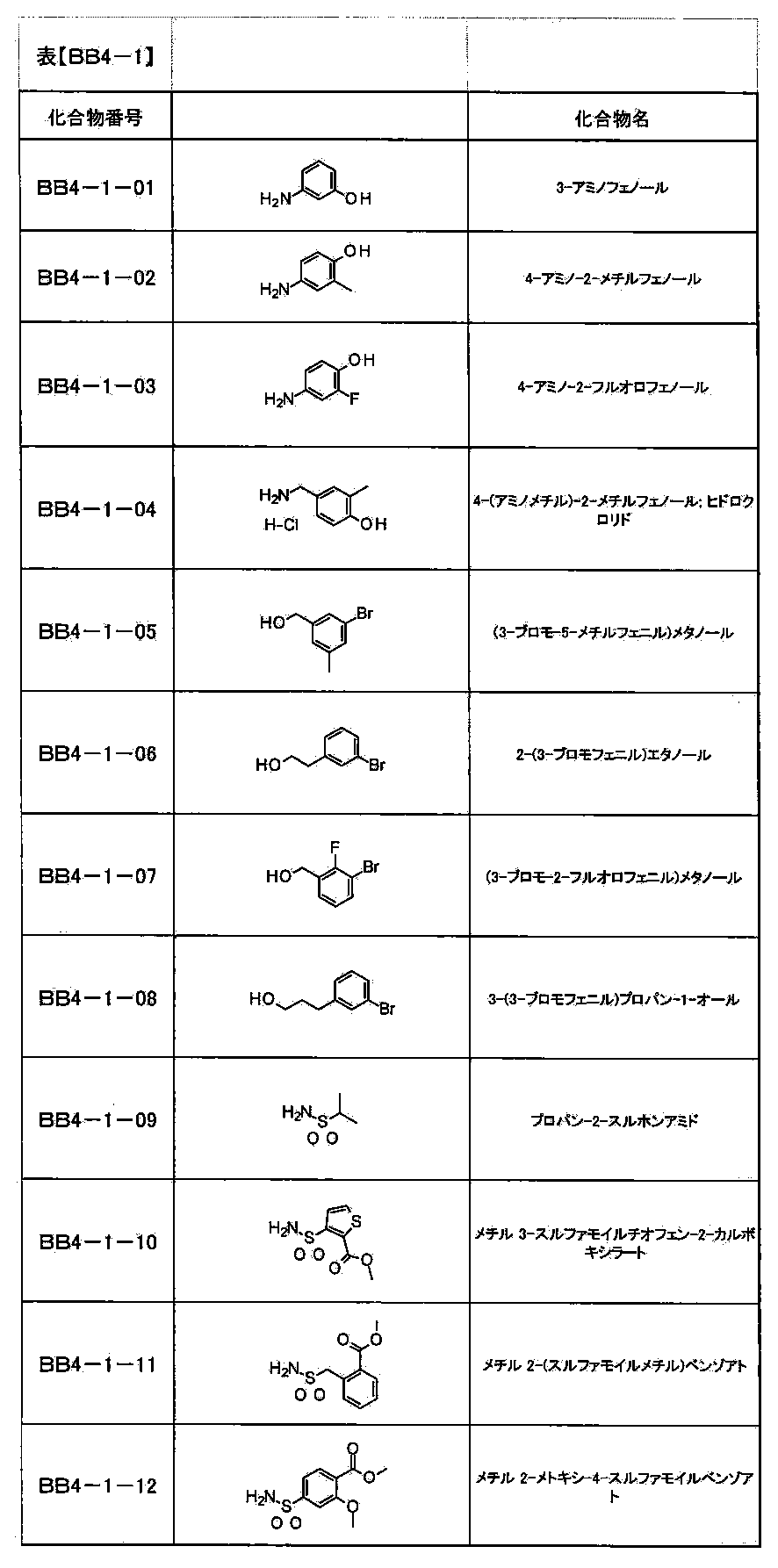

BB4−1−01を用いて工程a1B01を行った場合の反応式を以下に例示する。

窒素雰囲気下8mLガラスバイアルに化合物C005−03R(160mg,0.201mmol/g)と化合物C003−03R(160mg,0.201mmol/g)、NMP(5.0mL)を加え、室温で1時間振とうし、混合物4−1−A00−0を調製した。3−アミノフェノール(BB−4−1−01)(20mg,0.187mmol)とHOAt(25mg,0.187mmol)、DIC(28μL,0.183mmol)を加え、60℃で6時間振とうした。
反応液と固相の懸濁液を全てカラムのフィルター上に移し、NMP(6mL)で3回、NMP−水(1:1,6mL)で3回、NMP(6mL)で3回、メタノール(6mL)で3回、DCM(6mL)で3回、ヘプタン(6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、混合物4−1−a1B01−0(担持量0.197mmol/g,0.322g)を得た。
混合物4−1−a1B01−0を実施例8−1−1と同様の方法で切り出し処理することで混合物4−1−a1B01−1を得ることができる。混合物の詳細は[表10−1−3]に示す。
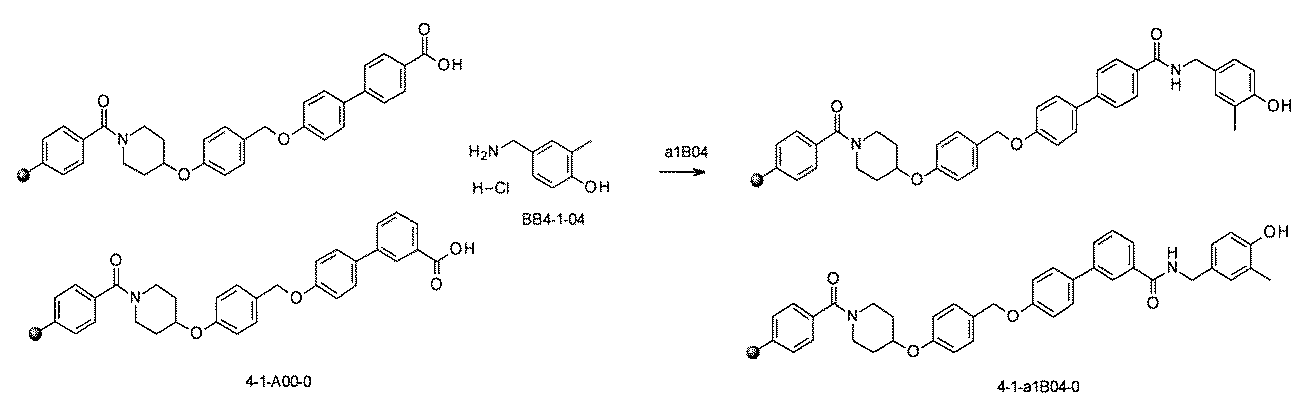
窒素雰囲気下8mLガラスバイアルに化合物C005−03R(160mg,0.201mmol/g)と化合物C003−03R(160mg, 0.201mmol/g)、NMP(4.5mL)を加え、室温で1時間振とうし、混合物4−1−A00−0を調製した。4−(アミノメチル)−2−メチルフェノールヒドロクロリド(BB−4−1−04)(52mg,0.299mmol)、DIPEA(53μL,0.302mmol)とHOAt(41mg,0.302mmol)、DIC(47μL,0.302mmol)を加え、室温で14時間振とうした。
反応液と固相の懸濁液を全てカラムのフィルター上に移し、NMP(6mL)で3回、NMP−水(1:1,6mL)で3回、NMP(6mL)で3回、メタノール(6mL)で3回、DCM(6mL)で3回、ヘプタン(6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、混合物4−1−a1B04−0(担持量0.196mmol/g,0.331g)を得た。
[表10−1−1]に工程a1で使用した試薬と得られた混合物を示す。

表中の表記方法について説明する。例えば[表10−1−23]では、4−1−a1B01−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−a1B01−1中の化合物構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。[表10−1−2]~[表10−1−6]においては、「▲×▼」と「b」は化学式で表す。








[表10−2−1]に工程a1で合成した混合物と使用量を示す。

表中の表記方法について説明する。例えば[表10−2−3]では、4−1−C00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−C00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。
[表10−2−2]、[表10−2−3]においては、「▲×▼」と「b」は化学式で表す。

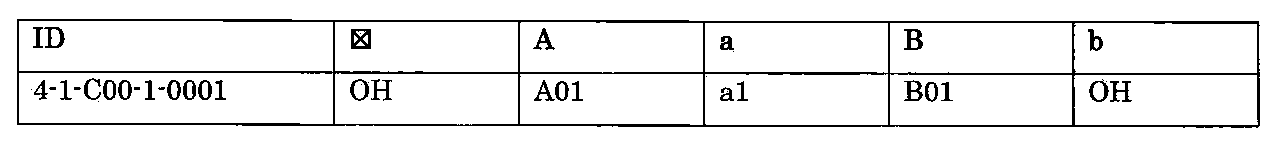
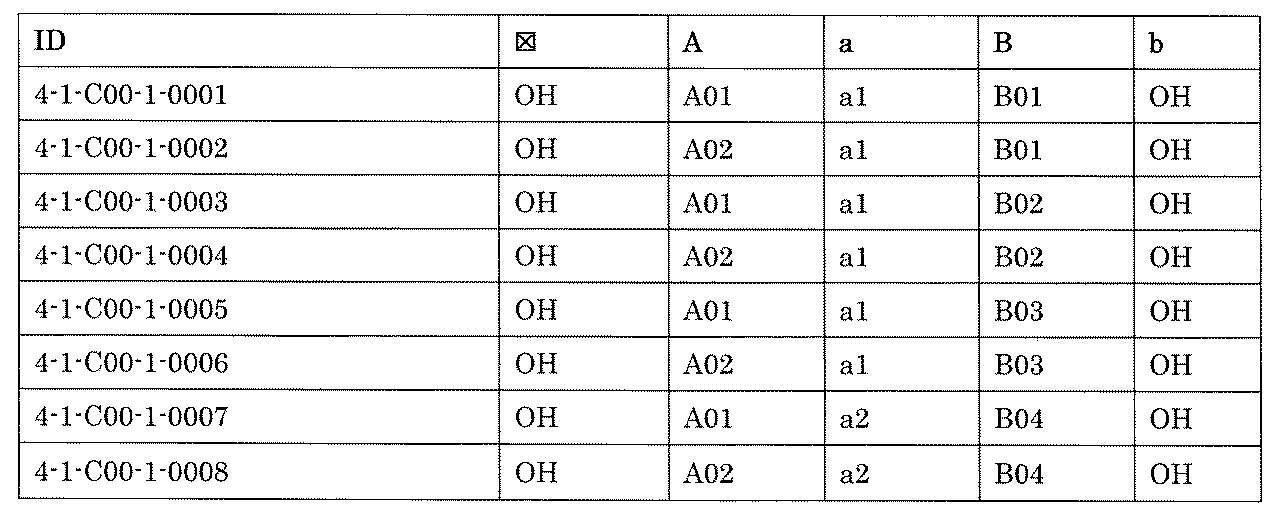
BB4−1−05~08を用いて工程b1を行った場合の反応式を以下に例示する。
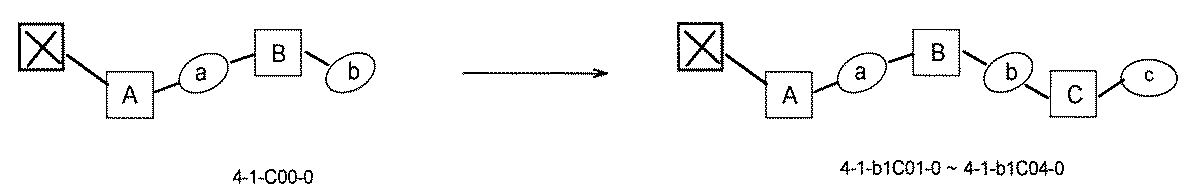
窒素雰囲気下、8mLのガラスバイアルに混合物4−1−C00−0(202mg,0.197mmol/g)とTHF(4.0mL)を加え、室温で1時間振とうした。アルコール試薬(BB4−1−05)(160mg,0.795mmol)をガラスバイアルに加えた。CMMP(シアノメチレントリメチルホスホラン)の0.5M THF溶液(795μL,0.398mmol)を加え、室温で21時間振とうした。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP(4mL)で3回、NMP/水=1/1(4mL)で3回、NMP(4mL)で3回、MeOH(4mL)で3回、DCM(4mL)で3回、ヘプタン(4mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−1−b1C01−0を得た。
混合物4−1−b1C01−0の合成と同様の方法でBB4−1−05の替わりにBB4−1−06~08を用いて工程b1を行い、混合物4−1−b1C02−0~混合物4−1−b1C04−0を得た。
表10−3−1に工程b1で使用したアルコール試薬と使用量を示し、[表10−3−3]~[表10−3−6]に工程b1で得られた混合物の詳細を示す。

表中の表記方法について説明する。例えば[表10−3−3]では、4−1−b1C01−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−b1C01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。[表10−3−2]~[表10−3−6]においては、「▲×▼」と「c」は化学式で表す。
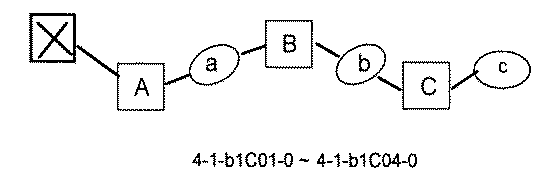


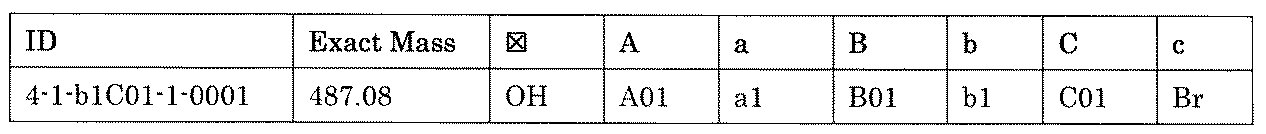
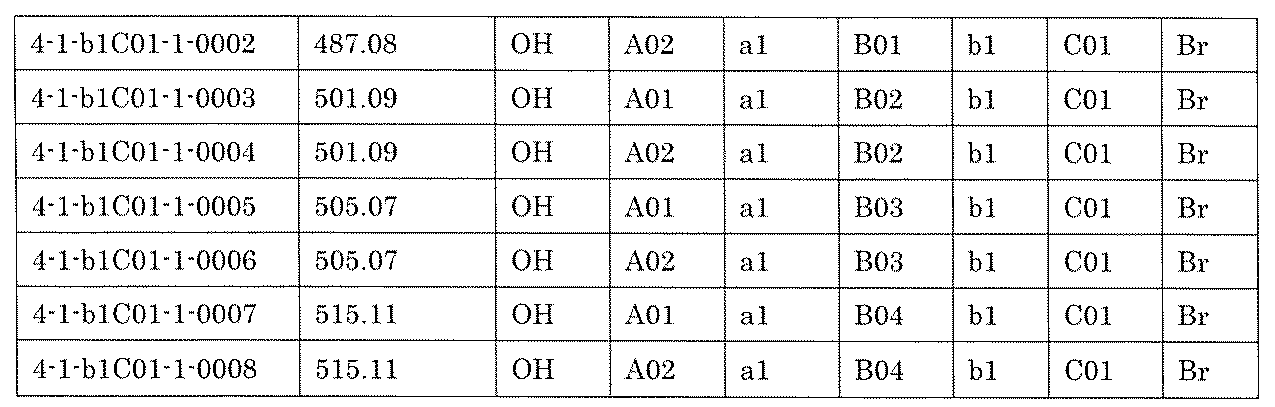


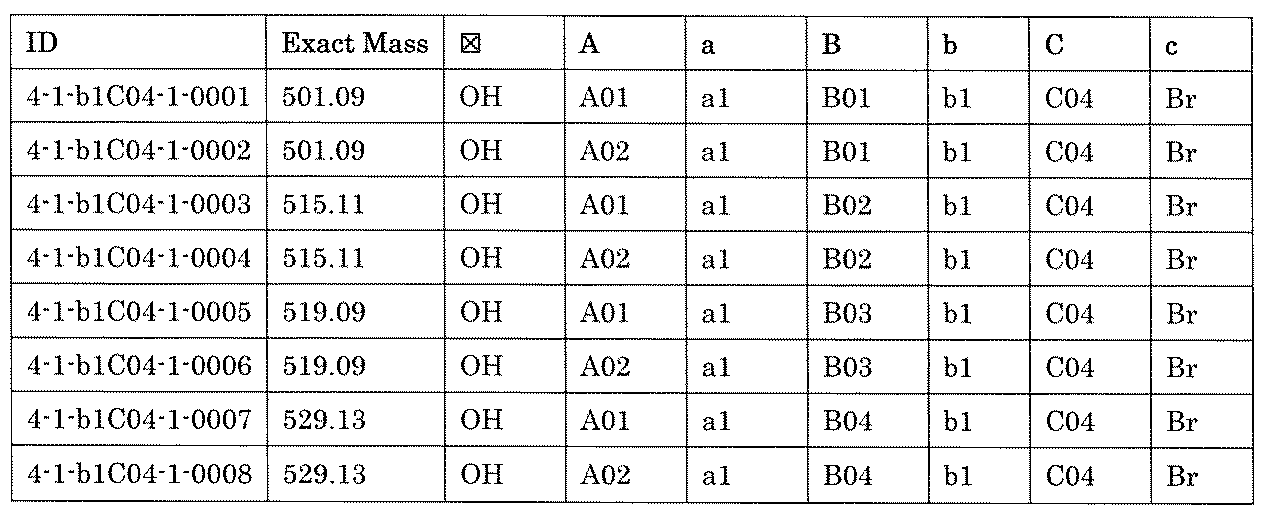

[表10−4−1]に工程b1で合成した混合物と使用量を示す。

表中の表記方法について説明する。例えば[表10−4−3]では、4−1−D00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−D00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。
[表10−4−2]~[表10−4−3]においては、「▲×▼」と「c」は化学式で表す。





BB4−1−09~12を用いて工程c1を行った場合の反応式を以下に例示する。

窒素雰囲気下、4mLガラスバイアルに混合物4−1−D00−0(111mg,0.190mmol/g)とTHF(2.2mL)を加え、室温で1時間振とうした。スルホンアミド試薬(BB4−1−09)(26.0mg,0.211mmol)をガラスバイアルに加えた。tBuBrettphos Pd G4(18.3mg,0.021mmol)とP2Et(107mg,0.316mmol)を加え、60℃で3時間振とうした。
反応液と固相の懸濁液を3mLのフィルター付きカラムに移してろ過し、NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、0.2M NAC in NMP/水=1/1(2mL)で3回、0.05M TBAHSO4+DTBP in NMP(2mL)で3回、0.05M NMM in NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(2mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−1−c1D01−0を得た。
混合物4−1−c1D01−0の合成と同様の方法でBB4−1−09の替わりにBB4−1−10~12を用いて工程c1を行い、混合物4−1−c1D02−0~混合物4−1−c1D04−0を得た。
[表10−5−1]に工程c1で使用したスルホンアミド試薬と使用量を示し、[表10−5−3]~[表10−5−6]に工程c1で得られた混合物を示す。


表中の表記方法について説明する。例えば[表10−5−3]では、4−1−c1D01−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−c1D01−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。[表10−5−2]~[表10−5−6]においては、「▲×▼」は化学式で表す。





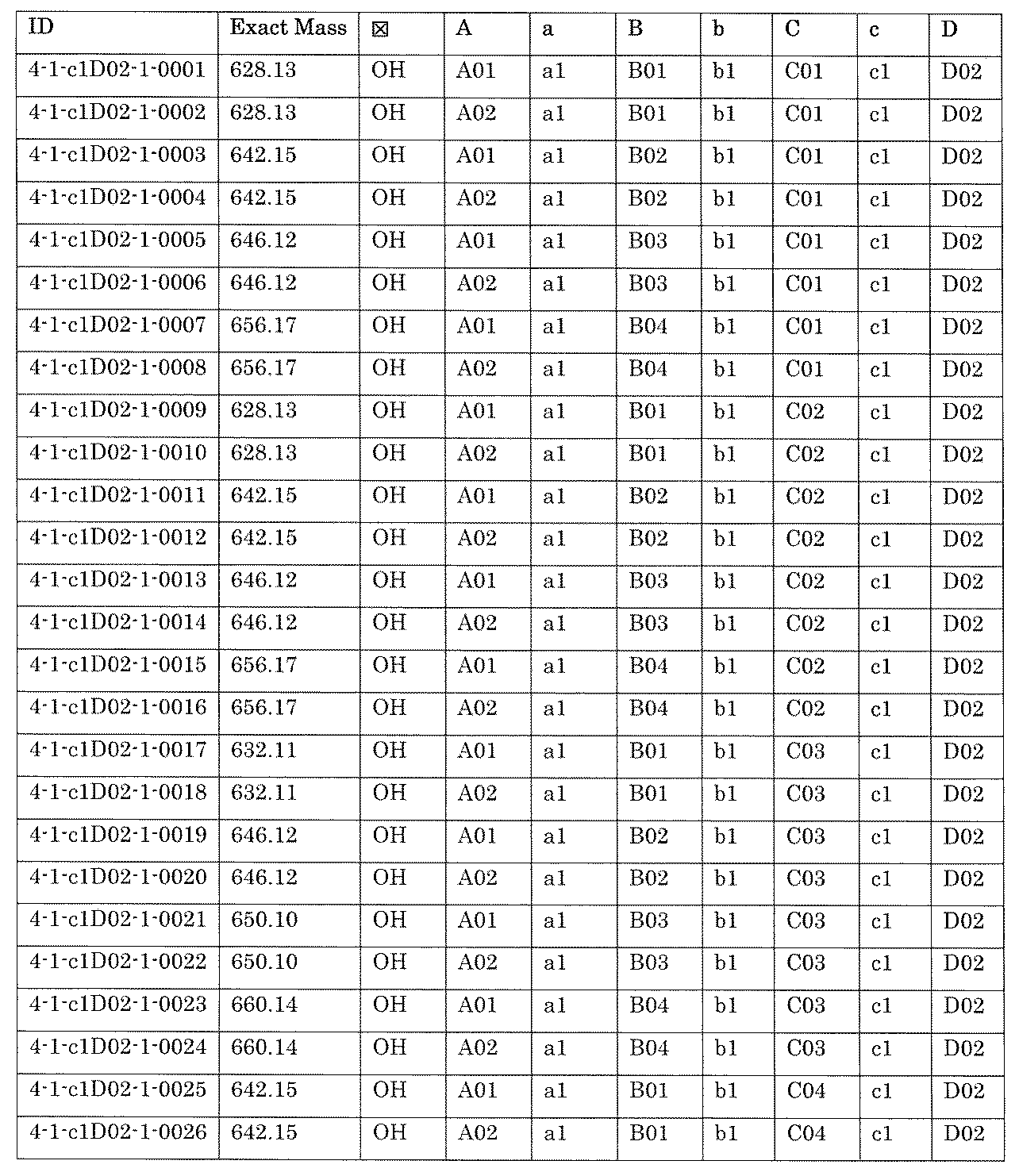

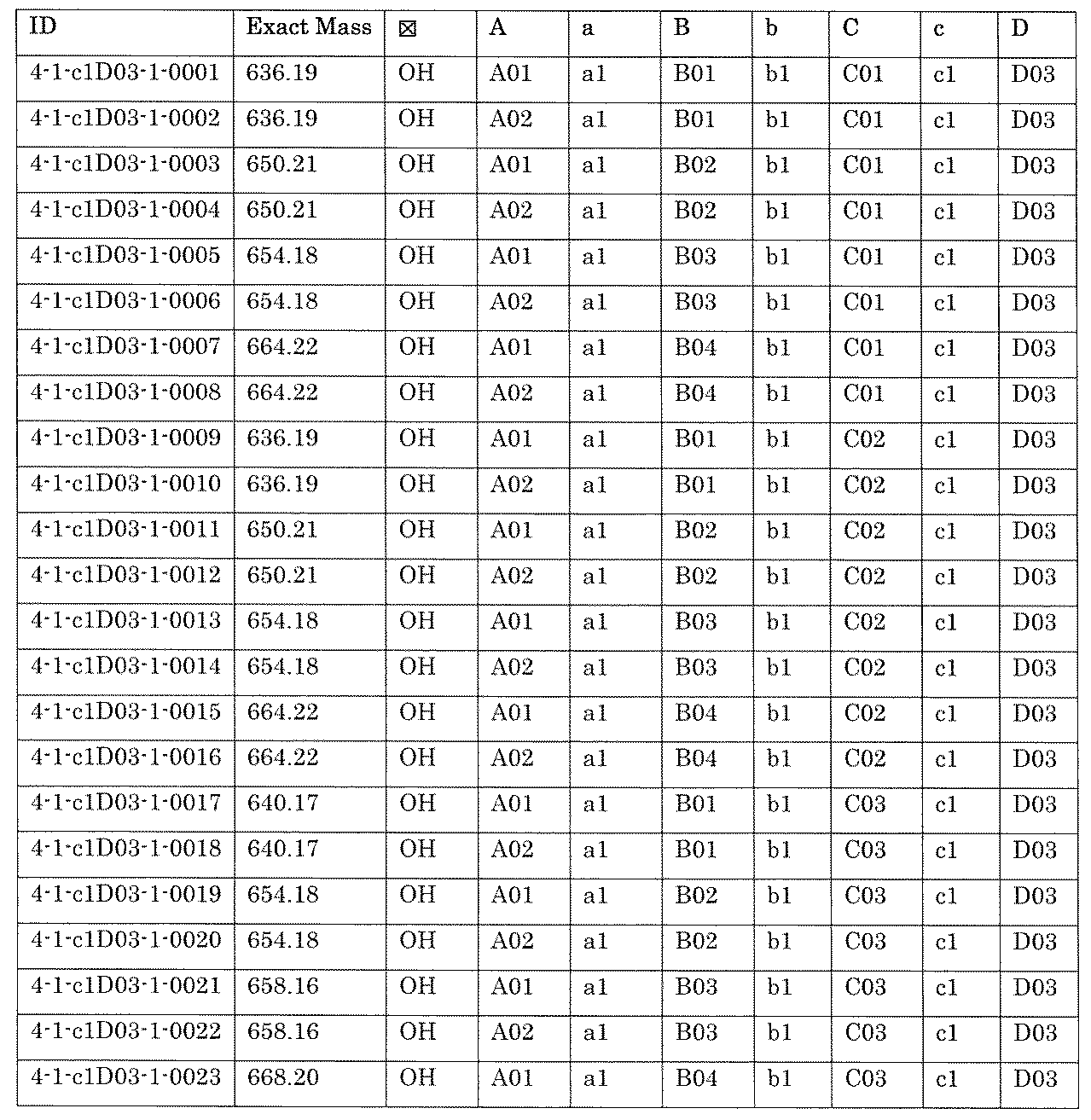



混合物4−1−c1D01−0を用いて工程D01−1を行った場合の反応式を以下に例示する。

窒素雰囲気下、4個の4mLのガラスバイアルに[表10−6−1]に記載の混合物(78mg)をそれぞれ別個に加えた。ペンタメチルベンゼン(17.1mg,0.115mmol)と、DCM(1.4mL)を加え、室温で1時間振盪した。TFA(156μL)を加え、室温で1時間振盪した。反応液と固相の懸濁液をDMF(0.5mL)を2回使用して3mLのフィルター付きカラムに移してろ過した。DMF(0.5mL)で3回洗浄し、合わせたろ液を15mm*75mmの試験管に受け、溶液を減圧留去した(Genevac減圧濃縮装置を用いた)。固相担持モルホリン(Aldrichカタログ番号:493813)(160mg)とDMF(2.0mL)を加えて、室温20分間振盪した。溶液と固相の懸濁液をDMF(0.5mL)を2回使用して3mLのフィルター付きカラムに移してろ過した。DMF(0.5mL)で3回、DCM/DMSO(2/1、0.5mL)で2回洗浄し、合わせたろ液を15mm*75mmの試験管に受けた。溶液を減圧留去(Genevac減圧濃縮装置を用いた)することで、[表10−6−1]に記載の混合物(4−1−c1D01−1~4−1−c1D04−1)を得た。

混合物4−1−c1D02−1を用いて工程h01D09を行った場合の反応式を以下に例示する。

窒素雰囲気下、15mm*75mmの試験管に濃縮された混合物4−1−c1D02−1(9.4mg)に、DMF(658μL)とtAmOH(188μL)を加えた。その溶液に2M NaOH水溶液(29.0μL)を加え、室温で3時間撹拌した。MP−TsOH(Biotageカタログ番号:800462)(78mg)を加え、室温で30分振とうした。反応液と固相の懸濁液をDMF(0.5mL)を2回使用して3mLのフィルター付きカラムに移してろ過した。DMF(0.5mL)で3回、DCM/DMSO(2/1、0.5mL)で2回洗浄し、合わせたろ液を15mm*75mmの試験管に受けた。溶液を減圧留去(Genevac減圧濃縮装置を用いた)することで、混合物4−1−h01D09−2を8.8mg得た。
混合物4−1−h01D09−2の合成と同様の方法でh02D10および工程h03D11を行い、混合物4−1−h02D10−2、および混合物4−1−h03D11−2を得た。
[表10−7−1]に工程h01~h03で使用した原料と使用量、得られた混合物の収量を示し、[表10−7−3]~[表10−7−6]に工程h01~h03で得られた混合物を示す。

表中の表記方法について説明する。例えば[表10−7−3]では、4−1−h01D09−2に含まれうるそれぞれの化合物をIDで示し、混合物4−1−h01D09−2の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。
[表10−7−2]~[表10−7−5]においては、「▲×▼」は化学式で表す。





[表10−7−4]混合物4−1−c1D03−1から工程h02D10で合成した混合物(4−1−h02D10−2)に含まれる化合物
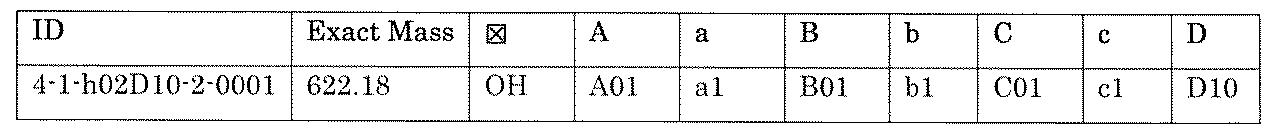



[表10−8−1]に使用した混合物と使用量を示す。

表中の表記方法について説明する。例えば[表10−8−3]では、4−1−E00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−1−E00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図8に示す。
[表10−8−2]~[表10−8−3]においては、「▲×▼」は化学式で表す。

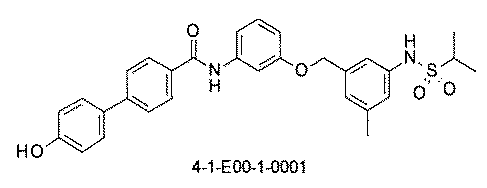

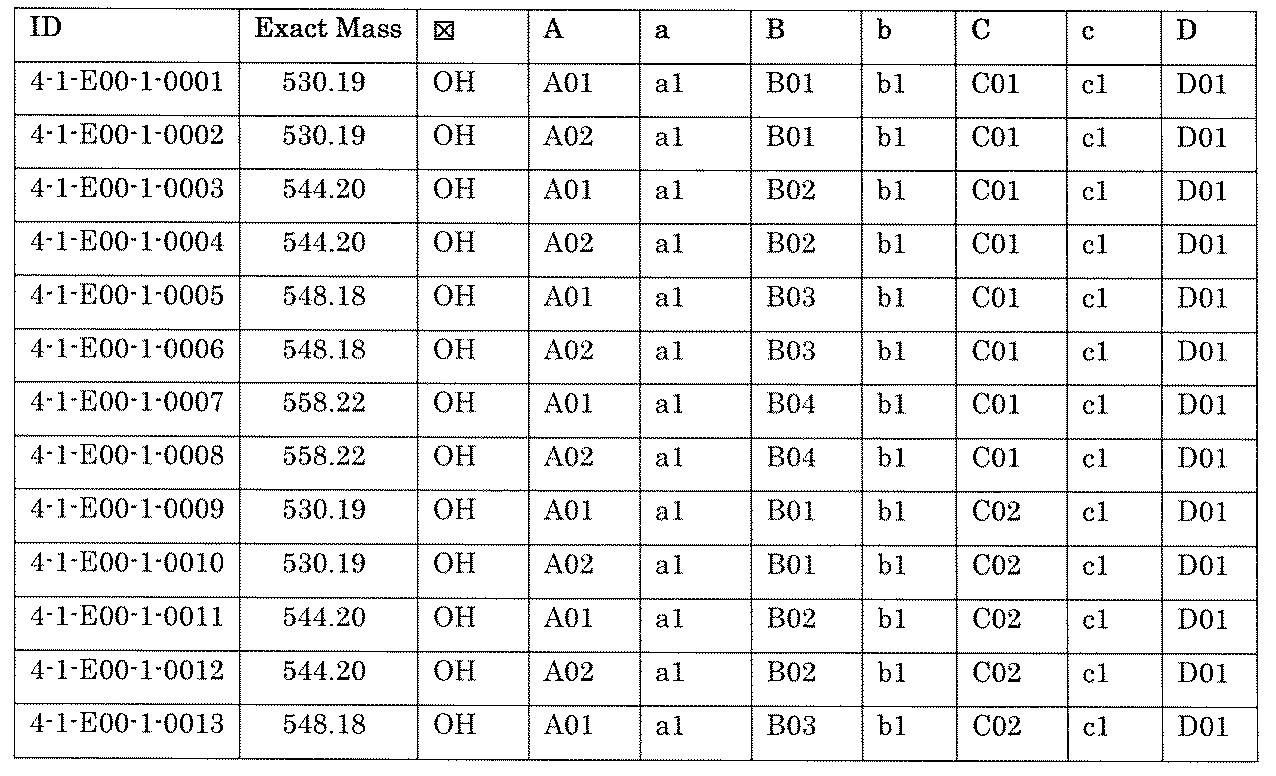




BB4−2−01を用いて工程a1B05を行った場合の反応式を以下に例示する。

窒素雰囲気下8mLガラスバイアルに化合物C005−03R(155mg,0.201mmol/g)と化合物C003−03R(155mg,0.201mmol/g)、DCM(4.5mL)を加え、室温で1時間振とうし、混合物4−2−A00−0を調製した。3−ブロモアニリン(BB−4−2−01)(26.3μL,0.242mmol)と、アセトニトリル(120μL)で1.0Mに調製したPipClU(クロロジピペリジノカルベニウムヘキサフルオロホスフェート)(44mg,0.122mmol)とNMI(1−メチルイミダゾール)(9.6μL,0.120mmol)の混合溶液を加え、25℃で7時間振とうした。
反応液と固相の懸濁液を全てカラムのフィルター上に移し、NMP(6mL)で3回、NMP−水(1:1,6mL)で3回、NMP(6mL)で3回、メタノール(6mL)で3回、DCM(6mL)で3回、ヘプタン(6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、混合物4−2−a1B05−0(担持量0.195mmol/g,0.320g)を得た。

窒素雰囲気、8mLガラスバイアルに化合物C005−03R(160mg,0.201mmol/g)と混合物C003−03R(160mg,0.201mmol/g)、NMP(4.5mL)を加え、室温で1時間振とうした。(3−ブロモフェニル)メタンアミン(BB−4−2−03)(56mg,0.302mmol)とHOAt(41mg,0.302mmol)、DIC(47μL,0.302mmol)を加え、室温で14時間振とうした。
反応液と固相の懸濁液を全てカラムのフィルター上に移し、NMP(6mL)で3回、NMP−水(1:1,6mL)で3回、NMP(6mL)で3回、メタノール(6mL)で3回、DCM(6mL)で3回、ヘプタン(6mL)で3回洗浄し、得られた固相を減圧下で一晩乾燥させ、混合物4−2−a1B07−0(担持量0.194mmol/g,0.330g)を得た。
[表10−9−1]に工程a1で使用した試薬と得られた混合物を示す。

混合物4−2−a1B05−0~4−2−a1B08−0を実施例10−1と同様の方法で切り出し処理することで混合物4−2−a1B05−1~4−2−a1B08−1を得ることができる。
表中の表記方法について説明する。例えば[表10−9−3]では、4−2−a1B05−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−a1B05−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。[表10−9−2]~[表10−9−6]においては、「▲×▼」と「b」は化学式で表す。

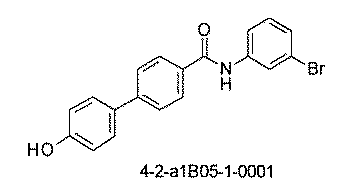


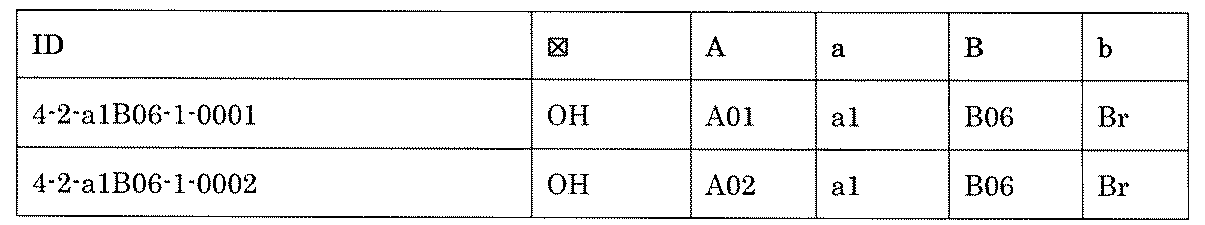

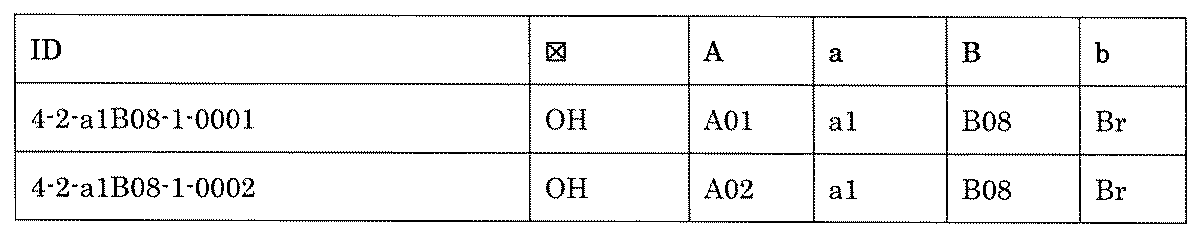

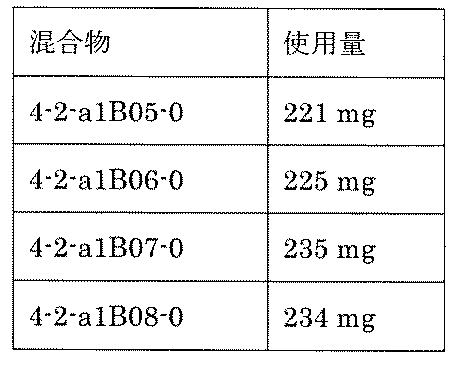
表中の表記方法について説明する。例えば[表10−10−3]では、4−2−C00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−C00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。
[表10−10−2]~[表10−10−3]においては、「▲×▼」と「b」は化学式で表す。





BB4−2−05~08を用いて工程b2を行った場合の反応式を以下に例示する。

窒素雰囲気下、8mLガラスバイアルに混合物4−2−C00−0(213mg,0.194mmol/g)とTHF(4.3mL)を加え、室温で1時間振とうした。アミド試薬(BB4−2−05)(74.1mg,0.414mmol)をガラスバイアルに加えた。tBuBrettphosPd G4(35.9mg,0.041mmol)とMTBD(7−メチル−1,5,7−トリアザビシクロ[4.4.0]デカ−5−エン)(89μL,0.621mmol)を加え、60℃で3時間振とうした。
反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP(4mL)で3回、NMP/水=1/1(4mL)で3回、0.2M NAC in NMP/水=1/1(4mL)で3回、0.05M TBAHSO4+DTBP in NMP(4mL)で3回、0.05M NMM in NMP(4mL)で3回、NMP/水=1/1(4mL)で3回、NMP(4mL)で3回、MeOH(4mL)で3回、DCM(4mL)で3回、ヘプタン(4mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−2−b2C05−0を得た。
混合物4−2−b2C05−0の合成と同様の方法でBB4−2−05の替わりにBB4−2−06~08を用いて工程b2を行い、混合物4−2−b2C06−0~混合物4−2−b2C08−0を得た。
[表10−11−1]に工程b2で使用したアミド試薬と使用量を示し、[表10−11−3]~[表10−11−6]に工程b2で得られた混合物の詳細を示す。
[表10−11−1]に工程b2で使用した試薬と得られた混合物を示す。
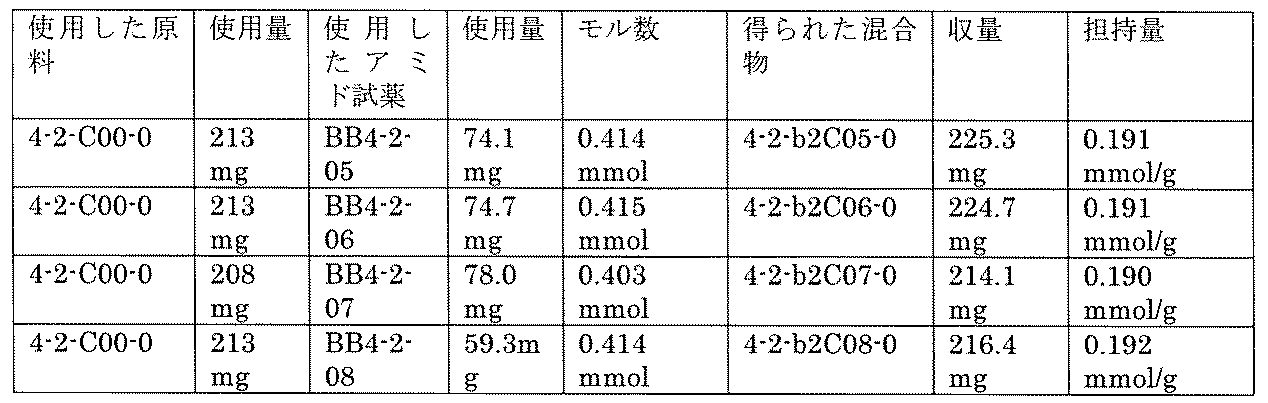
表中の表記方法について説明する。例えば[表10−11−3]では、4−2−b2C05−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−b2C05−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。
[表10−11−2]~[表10−11−6]においては、「▲×▼」と「b」は化学式で表す。
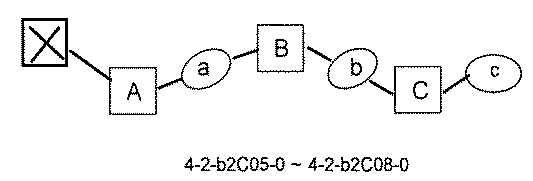



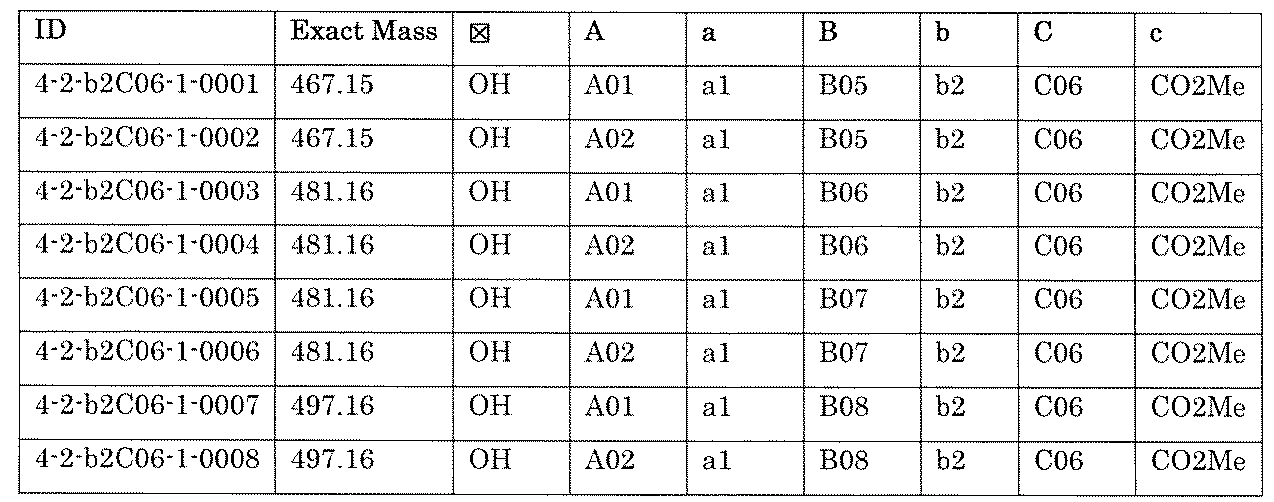
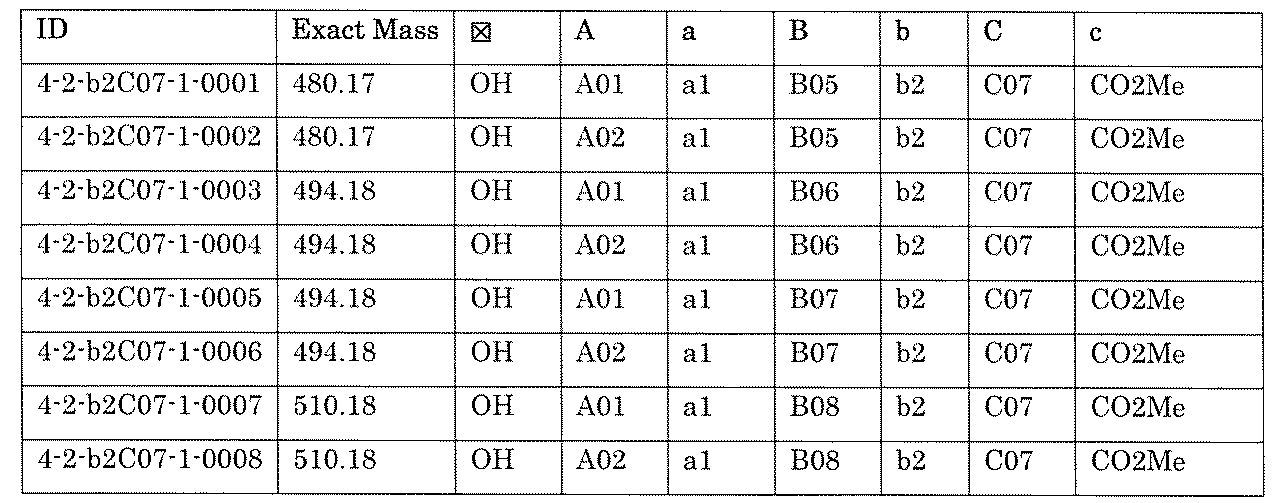
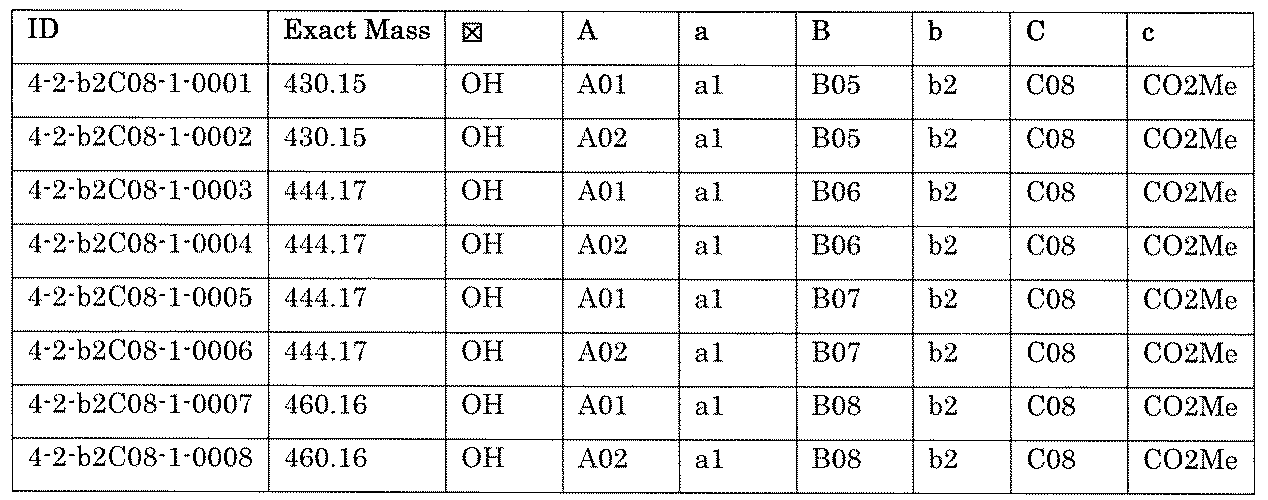
混合物4−2−b2C05−0~4−2−b2C08−0を用いて工程h04~h07を行った場合の反応式を以下に例示する。

窒素雰囲気下、4mLのガラスバイアルに混合物4−2−b2C05−0(144mg,0.191mmol/g)をそれぞれ別個に加えた。NMP(2.0mL)とtAmOH(575μL)を加えて室温で1時間振とうした。1M TBAOH(テトラブチルアンモニウムヒドロキシド)水溶液(137μL)を加え、室温で6時間振とうした。
反応液と固相の懸濁液を3mLのフィルター付きカラムに移してろ過し、NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、0.05M TBAHSO4+DTBP in NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(2mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−2−h04C05−0を得た。
混合物4−2−h04C05−0の合成と同様の方法で工程h05~h07を行い、混合物4−2−h05C06−0~混合物4−2−h07C08−0を得た。
[表10−12−1]に工程h04~h07で使用した混合物、溶媒とTBAOH溶液の使用量を示し、[表10−12−3]~[表10−12−6]に工程h04~h07で得られた混合物を示す。[表10−12−1]に工程h04~h07で使用した試薬と得られた混合物を示す。


表中の表記方法について説明する。例えば[表10−12−3]では、4−2−h04C05−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−h04C05−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。[表10−12−2]~[表10−12−6]においては、「▲×▼」と「c」は化学式で表す。



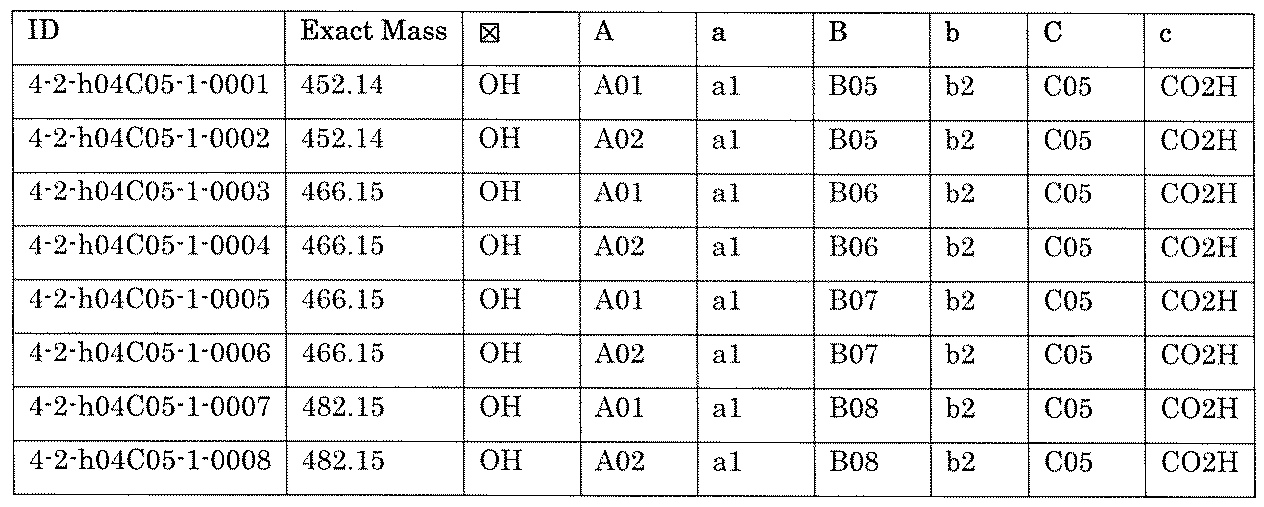
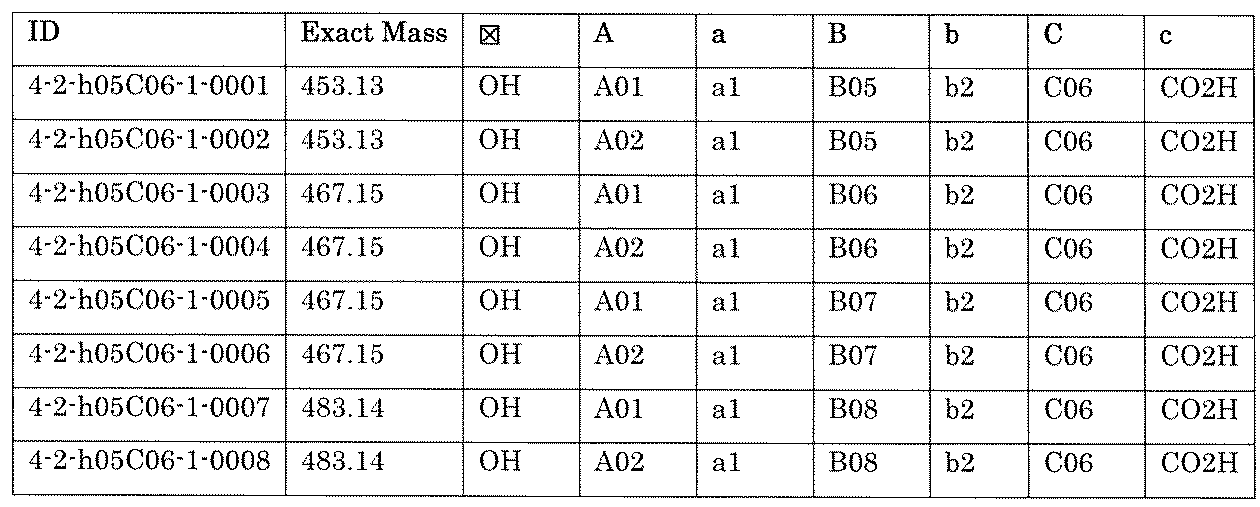

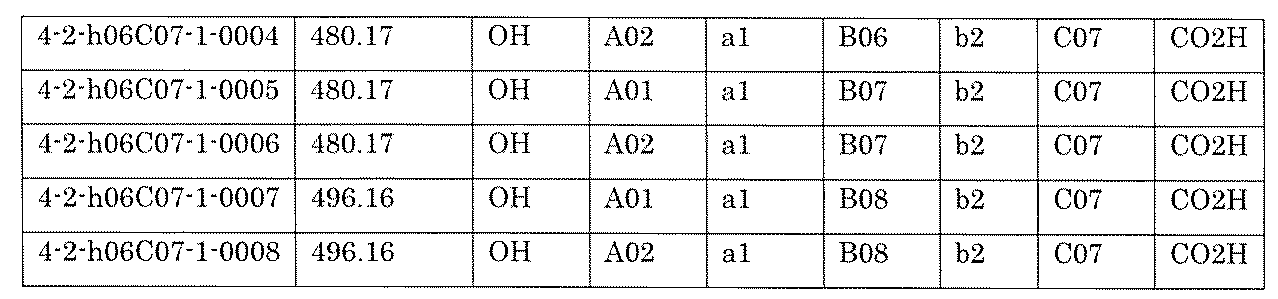
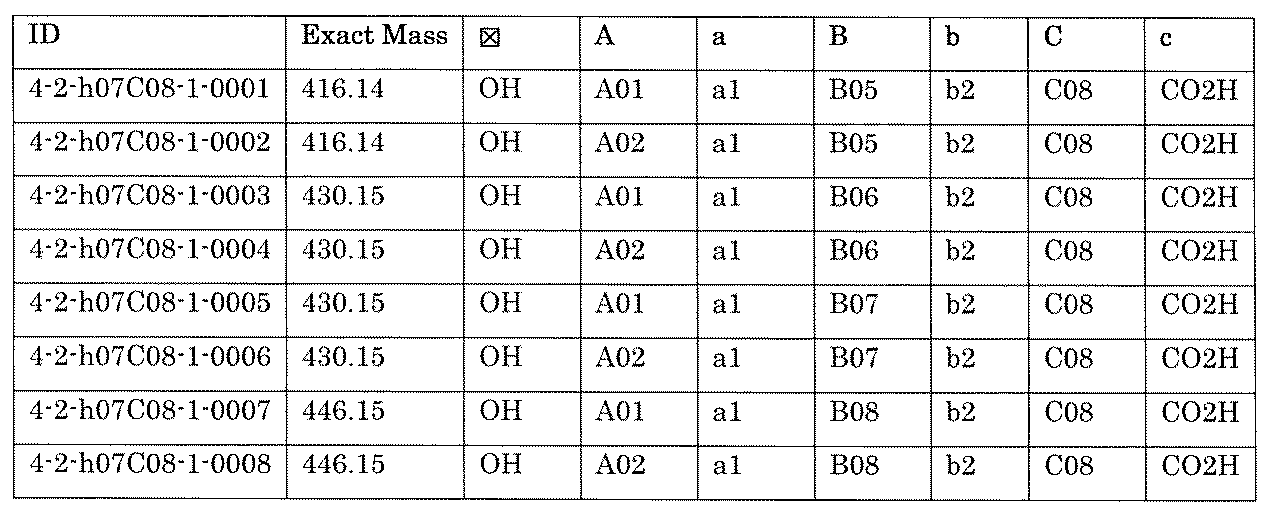

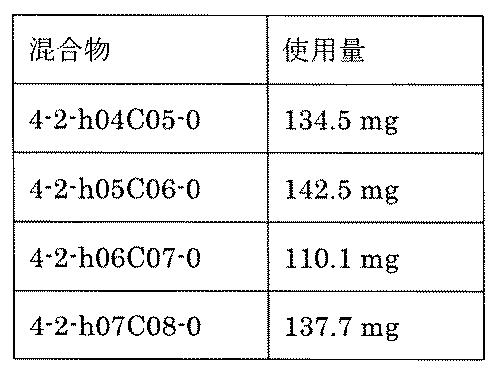
表中の表記方法について説明する。例えば[表10−13−3]では、4−2−D00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−D00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。[表10−13−2]~[表10−13−3]においては、「▲×▼」と「c」は化学式で表す。
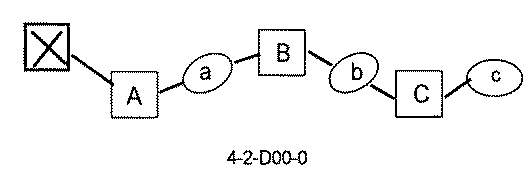
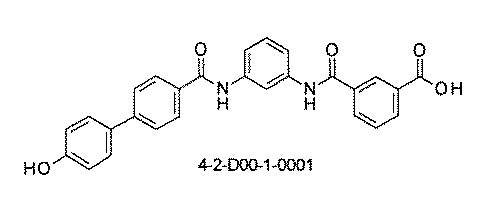


BB4−2−09および10を用いて工程c2を行った場合の反応式を以下に例示する。

窒素雰囲気下、4mLガラスバイアルに混合物4−2−D00−0(100mg,0.192mmol/g)とDCM(2.0mL)を加え、室温で1時間振とうした。アルコール試薬(BB4−2−09)(35.0μL,0.383mmol)をガラスバイアルに加えた。DMAP(4−ジメチルアミノピリジン)(23.4mg,0.192mmol)とWSC−HCl(36.7mg,0.192mmol)を加え、25℃で16.5時間振とうした。
反応液と固相の懸濁液を3mLのフィルター付きカラムに移してろ過し、NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(2mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−2−c2D05−0を得た。
混合物4−2−c2D05−0の合成と同様の方法で工程c2D06を行い、混合物4−2−c2D06−0を得た。
[表10−14−1]に工程c2D05、および工程c2D06で使用した混合物とアルコール試薬の使用量を示し、[表10−14−4]と[表10−14−5]に工程c2D05、およびc2D06で得られた混合物を示す。

窒素雰囲気下、4mLガラスバイアルに混合物4−2−D00−0(100mg,0.192mmol/g)とDCM(2.0mL)を加え、室温で1時間振とうした。フェノール試薬(BB4−2−11)(20.0μL,0.192mmol)をガラスバイアルに加えた。PipClU(クロロジピペリジノカルベニウムヘキサフルオロホスフェート)(69.1mg,0.192mmol)とNMI(1−メチルイミダゾール)(30.5μL,0.383mmol)を加え、25℃で16.5時間振とうした。
反応液と固相の懸濁液を3mLのフィルター付きカラムに移してろ過し、NMP(2mL)で3回、NMP/水=1/1(2mL)で3回、NMP(2mL)で3回、MeOH(2mL)で3回、DCM(2mL)で3回、ヘプタン(2mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、混合物4−2−c2D07−0を得た。
混合物4−2−c2D07−0の合成と同様の方法で工程c2D08を行い、混合物4−2−c2D08−0を得た。
[表10−14−2]に工程c2D07、およびc2D08で使用した混合物とフェノール試薬の使用量を示し、[表10−14−6]~[表10−14−7]に工程c2D07、およびc2D08で得られた混合物を示す。

表中の表記方法について説明する。例えば[表10−14−4]では、4−2−c2D05−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−c2D05−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。[表10−14−3]~[表10−14−7]においては、「▲×▼」は化学式で表す。
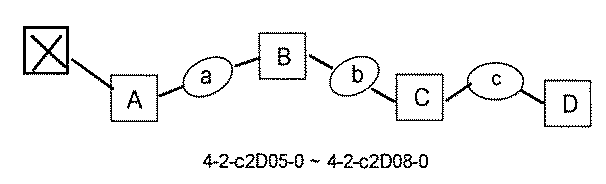
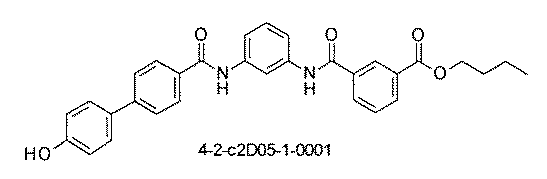



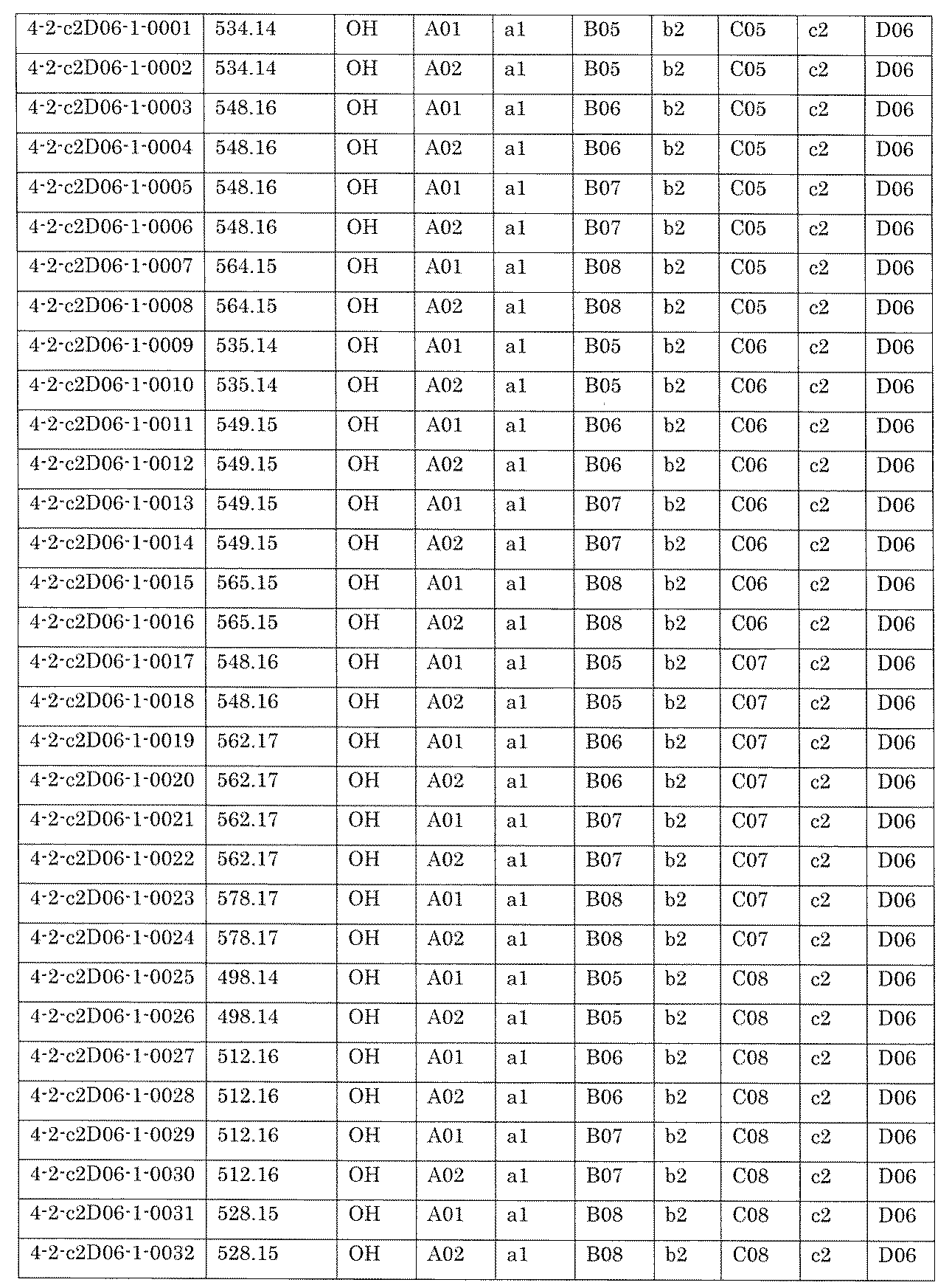
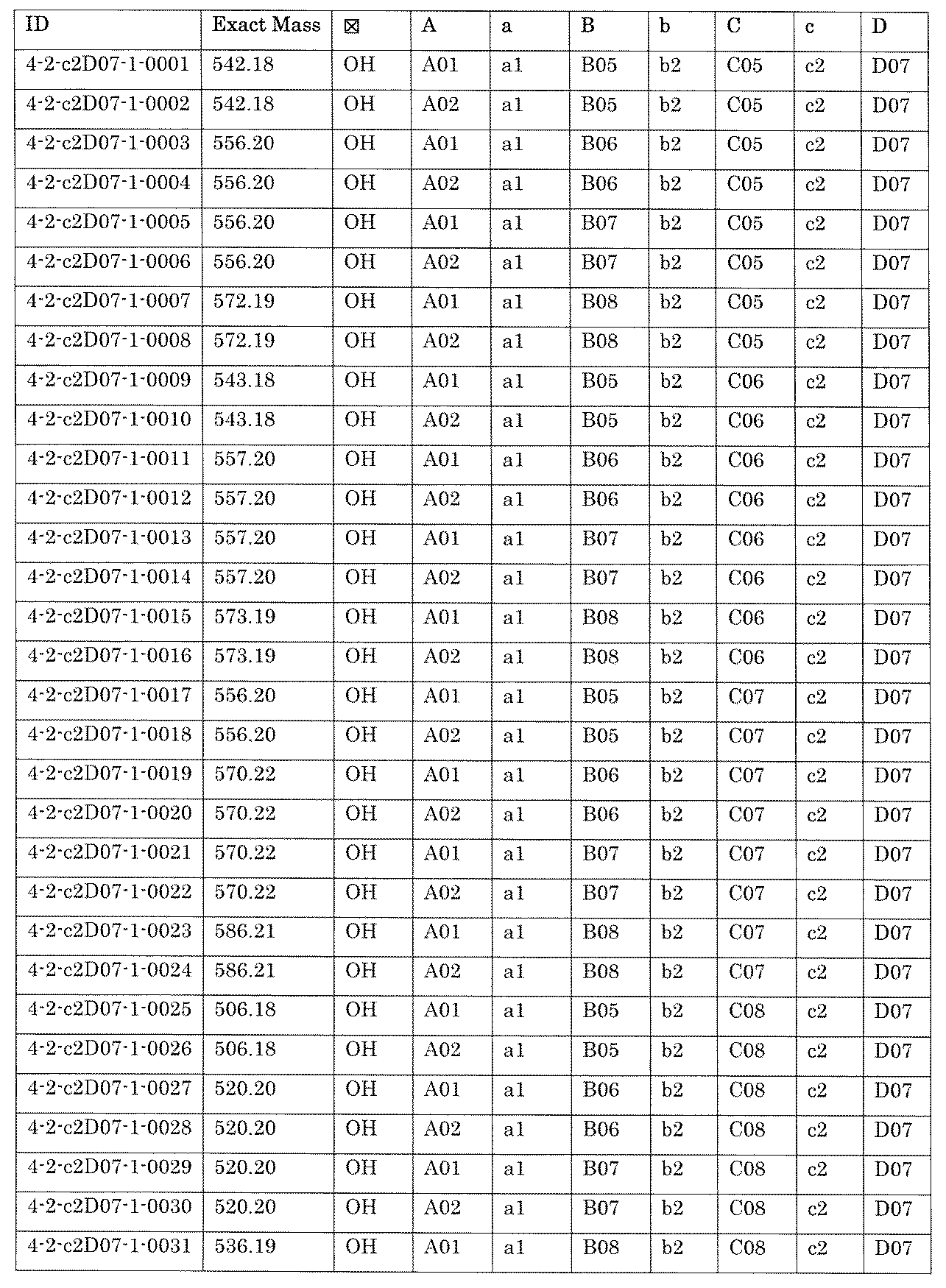

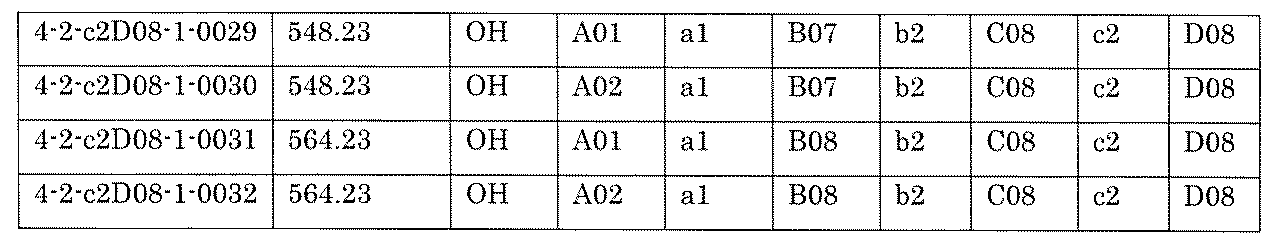
混合物4−2−c2D05−0を用いて工程D05−1を行った場合の反応式を以下に例示する。

窒素雰囲気下、4mLのガラスバイアルに混合物4−2−c2D05−0(80mg,0.190mmol/g)を加えた。ペンタメチルベンゼン(17.1mg,0.115mmol)と、DCM(1.4mL)を加え、室温で1時間振とうした。TFA(160μL)を加え、室温で1時間振とうした。反応液と固相の懸濁液をDMF(0.5mL)を2回使用して3mLのフィルター付きカラムに移してろ過した。DMF(0.5mL)で3回洗浄し、合わせたろ液を15mm*75mmの試験管に受け、溶液をGenevacで留去した。固相担持モルホリン(Aldrichカタログ番号:493813)(160mg)とDMF(2.0mL)を加えて、室温20分間振とうした。溶液と固相の懸濁液をDMF(0.5mL)を2回使用して3mLのフィルター付きカラムに移してろ過した。DMF(0.5mL)で3回、DCM/DMSO(2/1、0.5mL)で2回洗浄し、合わせたろ液を15mm*75mmの試験管に受けた。溶液を減圧留去(Genevac減圧濃縮装置を用いた)することで、混合物4−2−c2D05−1を得た。
[表10−15−1]に工程D05−1~D08−1で使用した試薬と得られた混合物を示す。


[表10−16−1]に使用した混合物と使用量を示す。


表中の表記方法について説明する。[表10−16−3]では、4−2−F00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−2−F00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。[表10−16−2]~[表10−16−3]においては、「▲×▼」は化学式で表す。




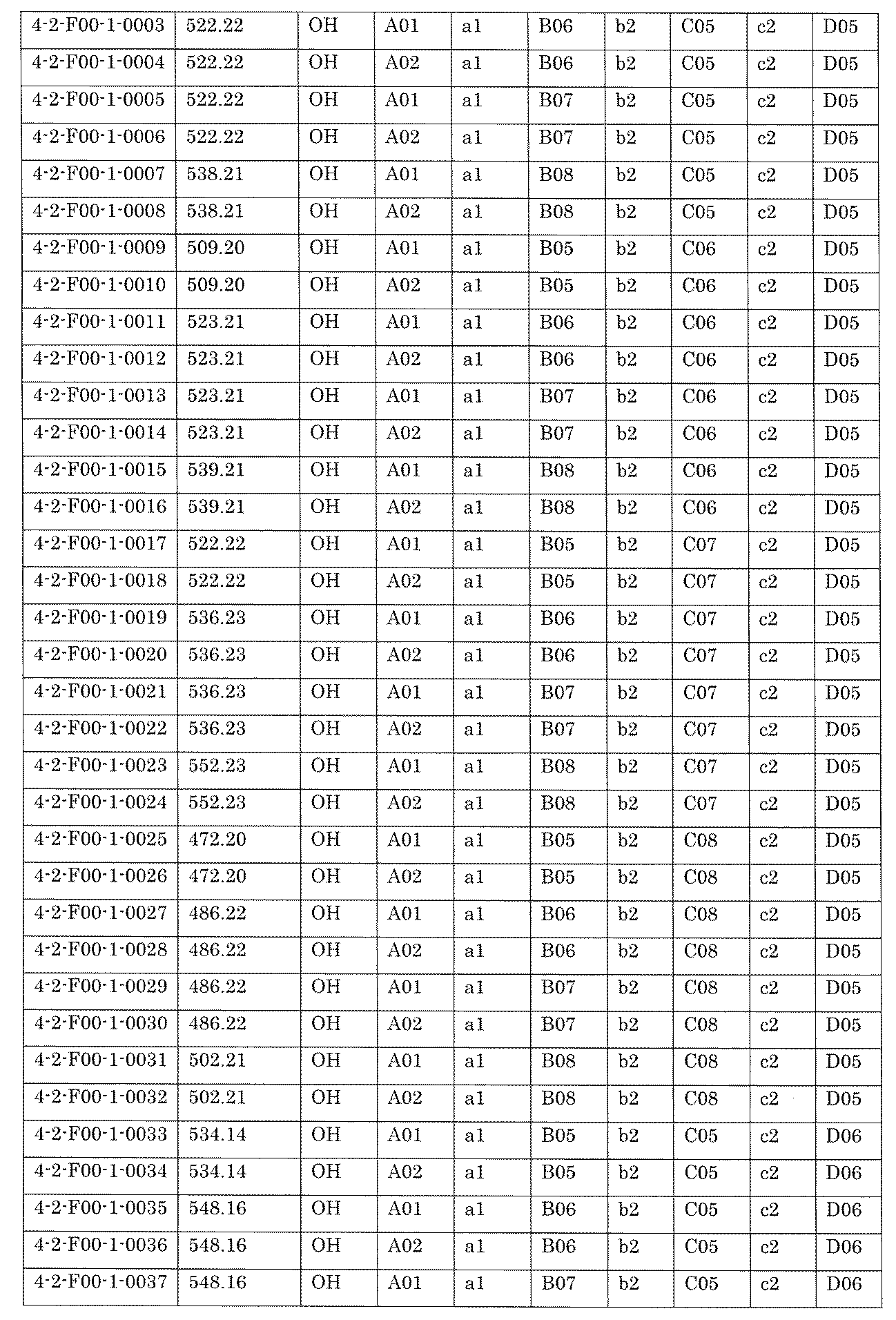



それぞれ独立して合成した混合物4−1−E00−1と混合物4−2−F00−1を混合することで、結合に多様性を持った1つの混合物ライブラリを準備した実例を示す。

[表10−17−1]に使用した混合物と使用量を示す。

表中の表記方法について説明する。例えば[表10−17−3]では、4−M00−1に含まれうるそれぞれの化合物をIDで示し、混合物4−M00−1の構造におけるコアブロックとリンカーの組み合わせを記号もしくは化学式で示している。記号と具体的構造の対応は図9に示す。
[表10−17−2]~[表10−17−3]においては、「▲×▼」は化学式で表す。
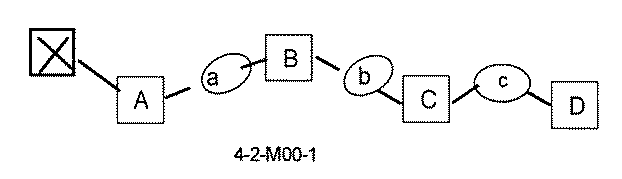




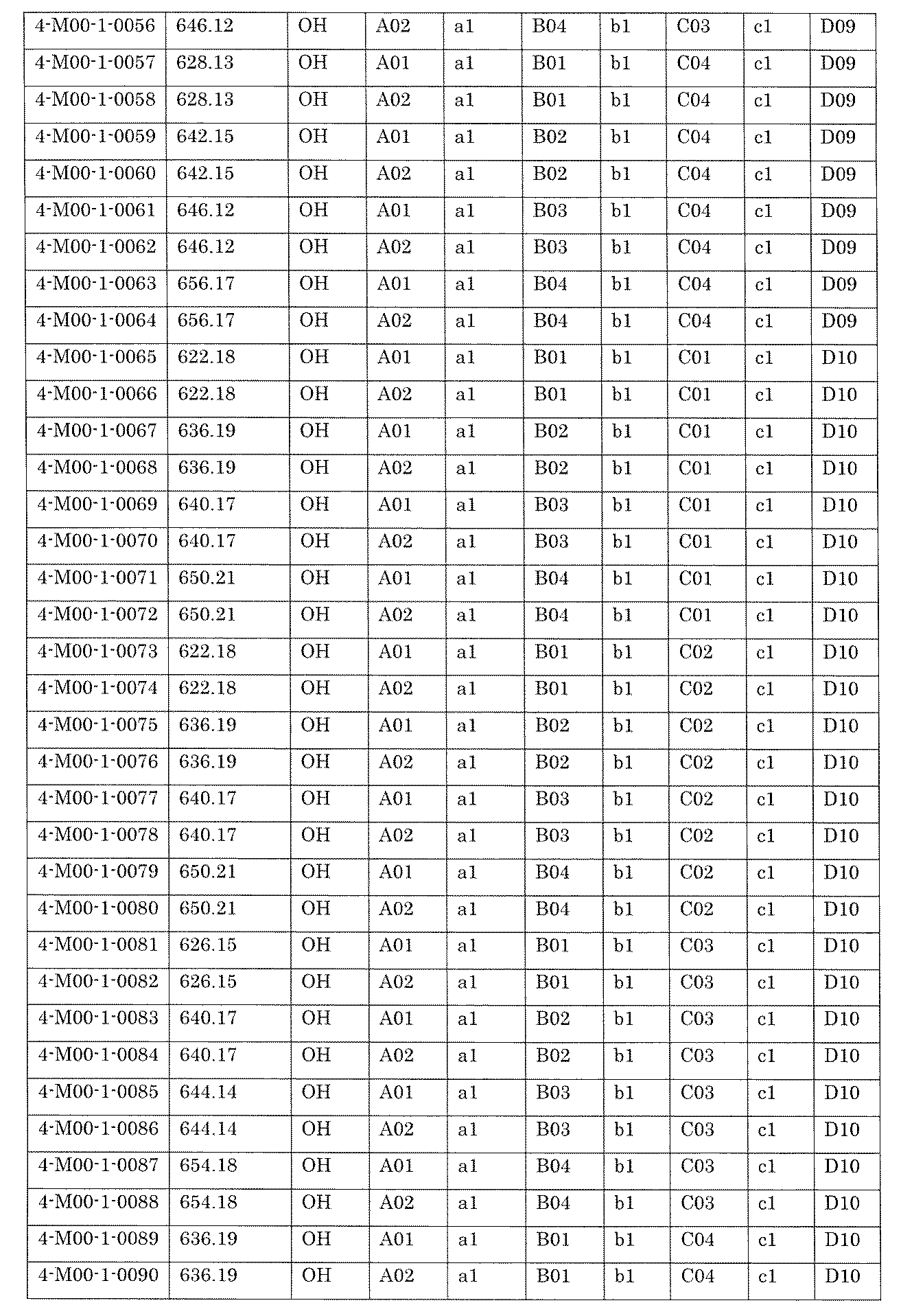




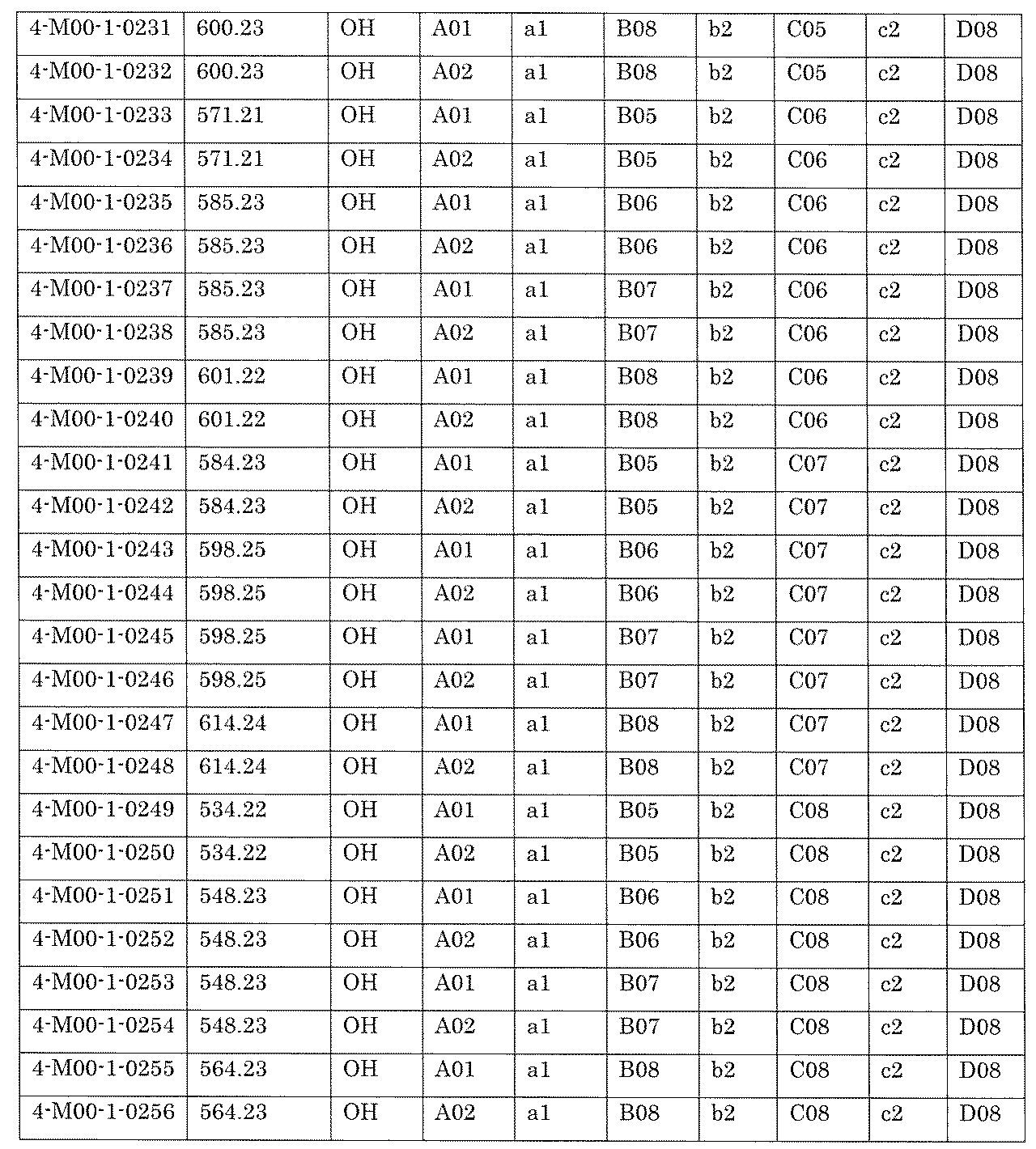









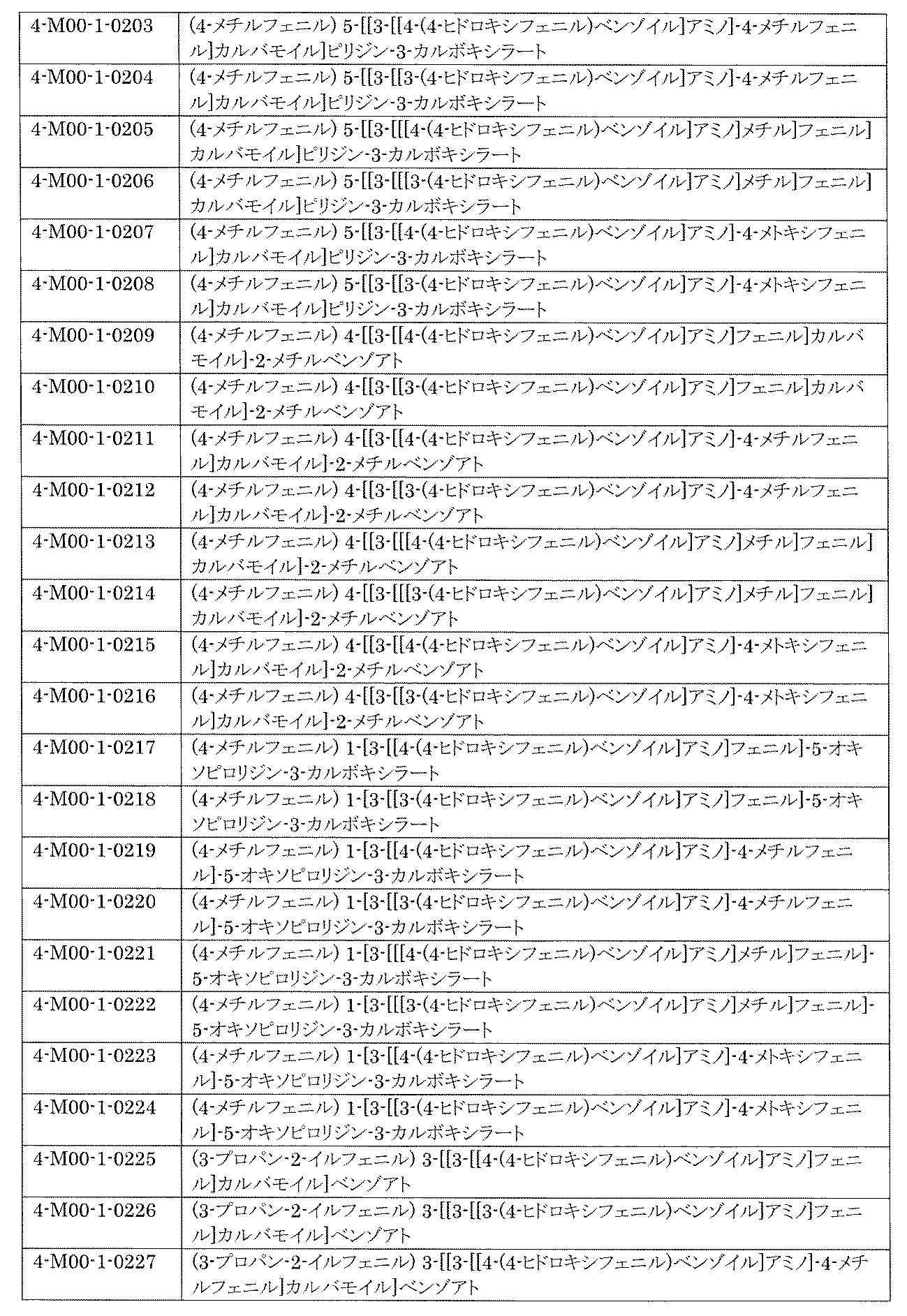


実施例10−17にて得られた混合物(4−M00−1)のDMF(5mL)の溶液から100μLをMeCN(1.0mL)にて希釈し、LCMSにて分析した。分析条件SMD−FA05−long−25minにて分析し、UV面積は、UV波長299nmにて確認した。分析結果については[表10−18−1]に示す。
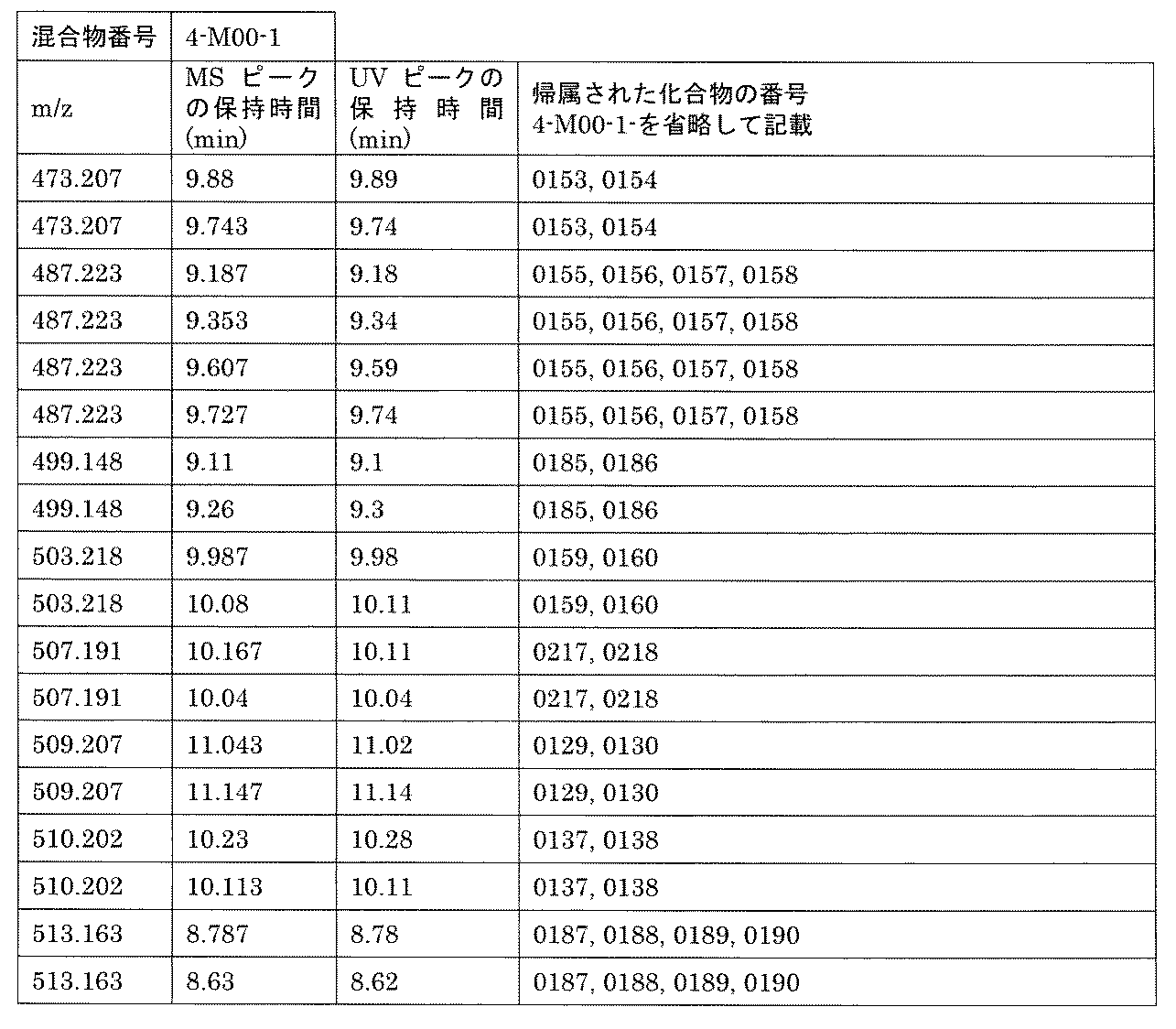





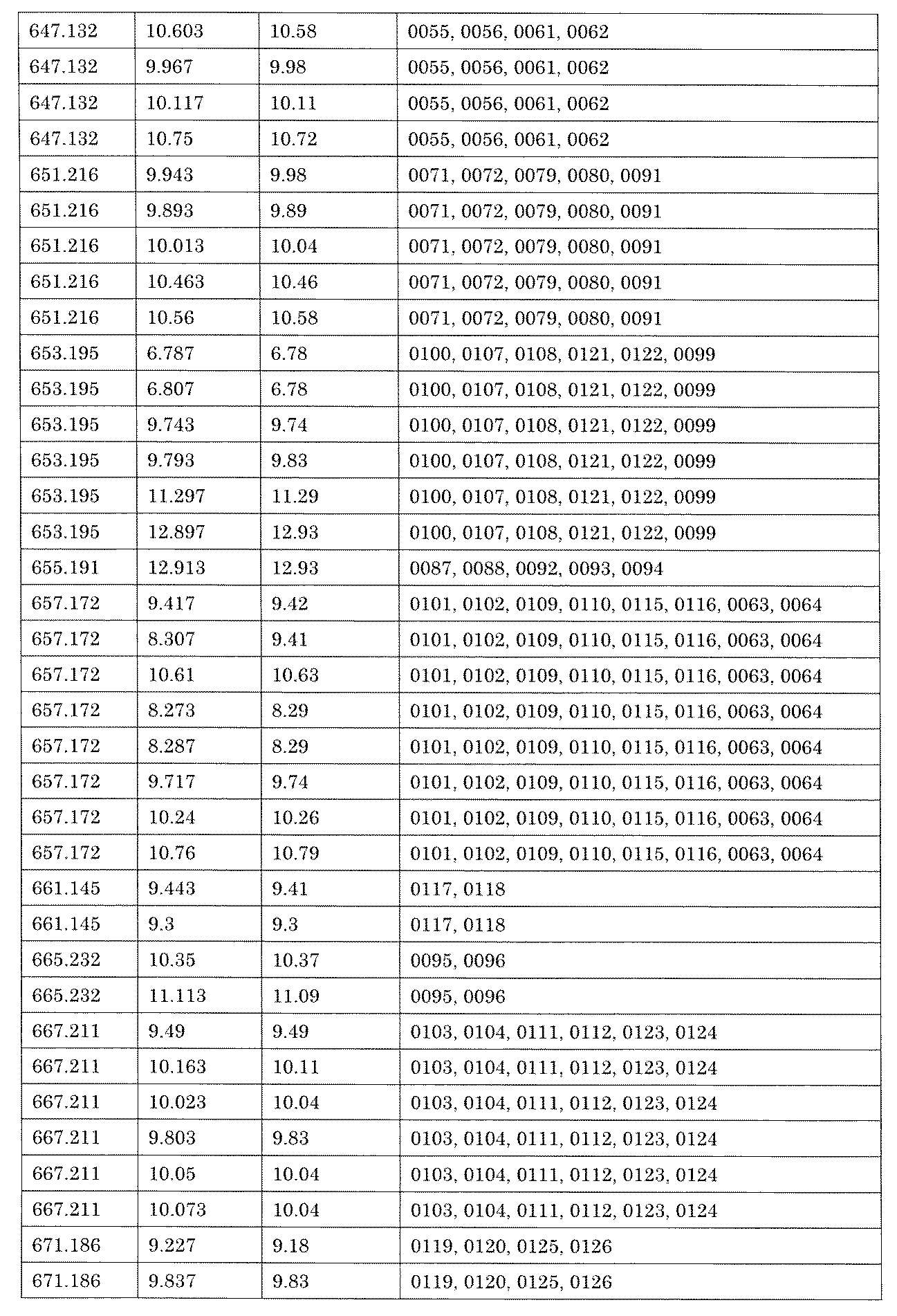

なお、本手法による結合多様性を有するライブラリ調製において結合の種類およびその構築法として実施例10にて使用したものに限定されない。
MICROPLATE,96WELL,PP,V−BOTTOM(Greiner bio−one,651201)の1wellに対して,表11−1−1に示す各混合物のDMSO溶液(各混合物の平均分子量からの計算に基づき、凡そ5mMに調製)を各々8wellに分注した。LABCYTE ECHO(登録商標) 555 アコースティック 分注システム(BECKMAN COULTER)を用いて、実施例8から得た混合物に対しては8wellからそれぞれ400nLずつ指定の3well中に分注し、実施例9から得た混合物に対しては8wellからそれぞれ200nLずつ指定の3well中に分注した。実施例8から得た混合物に対しては2種類の10μM ポジティブコントロール(B2Q045−01、B2Q048−01)を各々200nL分注した。実施例9から得た混合物に対しては2種類の20μMポジティブコントロールを同様に各々100nL、バックフィルでDMSO溶液を400nL分注した。さらに予めTris−HCl Buffer(25mM Tris−HCl pH7.4,150mM NaCl,0.025% Tween−20,1mM DTT)を用いて、実施例8から得た混合物に対しては最終タンパク濃度として5μM、実施例9から得た混合物に対しては10μMになるように調製したBcl−2タンパク(Novus,NBP2−34889,Recombinant Human Bcl−2 Protein,Lot E60145051)(19.2μL)を1000化合物、若しくは500化合物が分注されたwellに加え(最終化合物濃度 100nM)、暗所にて23℃で1時間インキュベーションした。つぎに膨潤させておいた96−well MacroSpinColumn,P−2 Material(Harvard,74−5655)を用いてAssay buffer (25mM Tris−HCl pH7.4,150mM NaCl,0.025% Tween−20,1mM DTT )200μL,2000XG,4℃,2分間遠心にて3回行い96−well MacroSpinColumnのコンディショニングを実施した。その次に,インキュベーション後のサンプル20 μLをコンディショニング完了後の96−well MacroSpinColumn,P−2 Materialに添加し2000XG,2分間遠心にてSEC処理をした。SECからの通液にアセトニトリルとメタノール(LC/MS grade,富士フイルム和光純薬工業(株))1:1の混合液を32μL加え1800XG,4℃,5分間遠心し上澄みを分析サンプルASMS(A)とし,同様の操作で500化合物/well、若しくは1000化合物/wellで調製し,Bcl−2タンパクが入っていないTris−HCl Bufferのみを加えたサンプルを同一条件でインキュベーションし,96−well MacroSpinColumn処理をせず,同一の有機溶媒を加え遠心操作によって得られた上澄み液をSTD(B)とした。次に同様の操作で1000化合物/well、若しくは500化合物/wellで調製し,Bcl−2タンパクが入っていないTris−HCl Bufferのみを加えたサンプルを同一条件でインキュベーションし,同様に96−well MacroSpinColumn処理を実施し,同一の有機溶媒を加え遠心操作によって得られた上澄み液をBK(C)とし,逆相LC/MSにて測定した。
計算式 ASMS Ratio(%)=(A−C)/B * 100
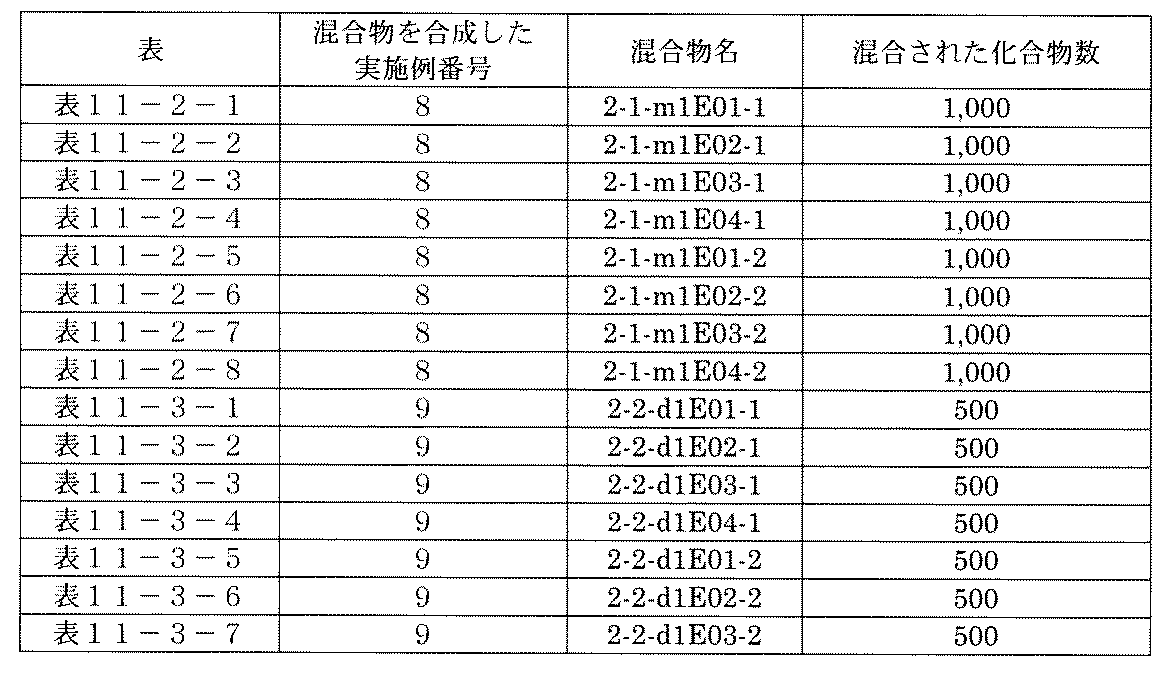

















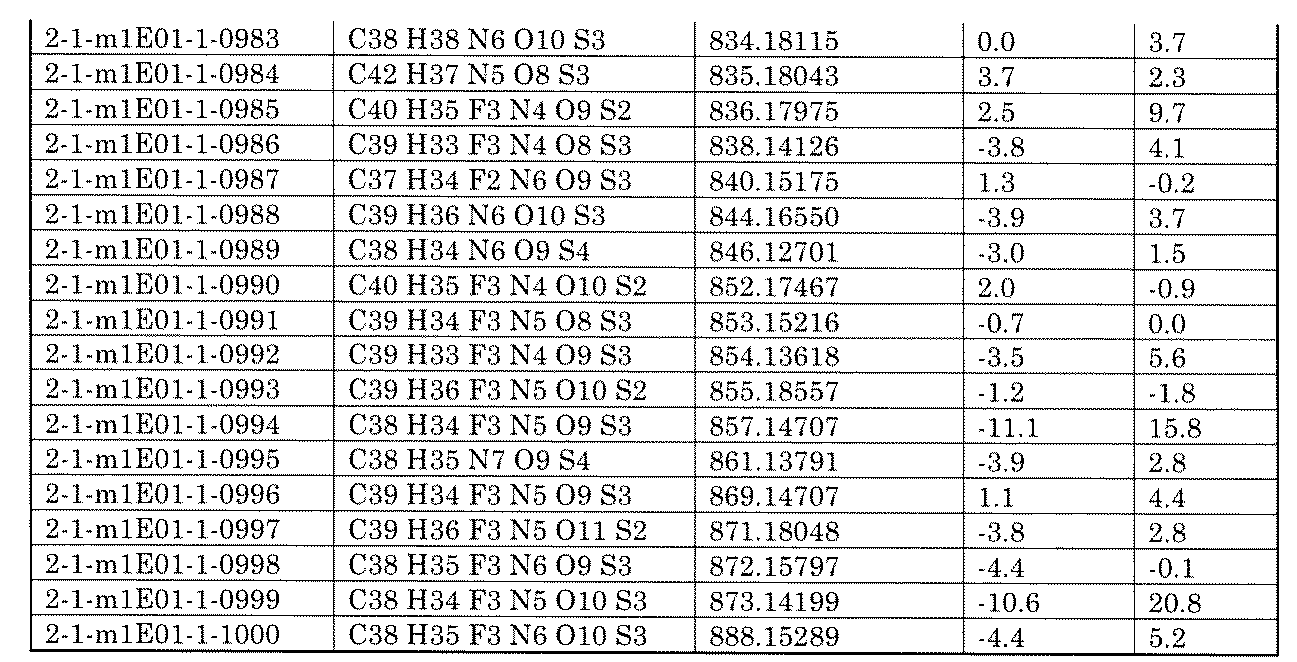


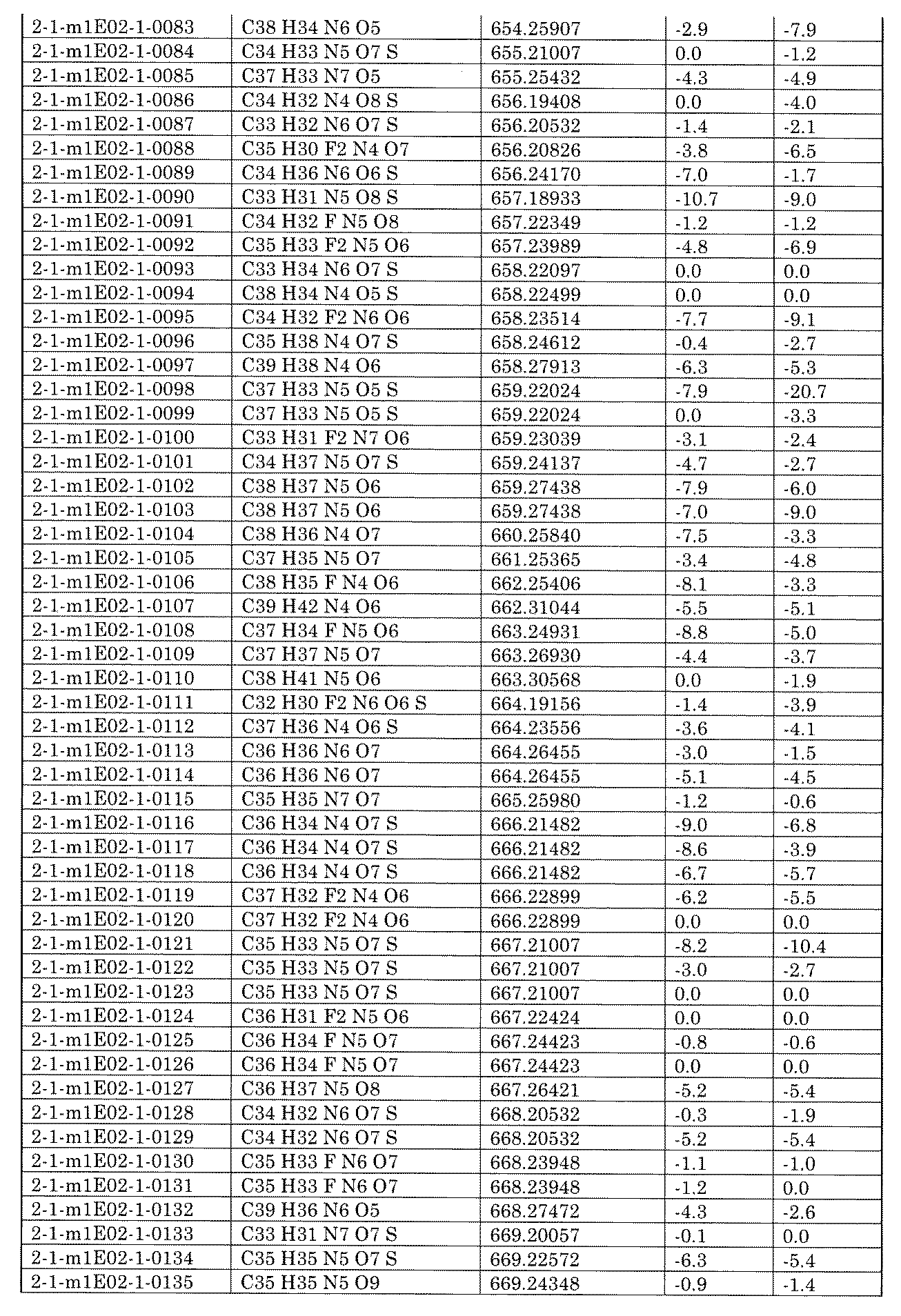
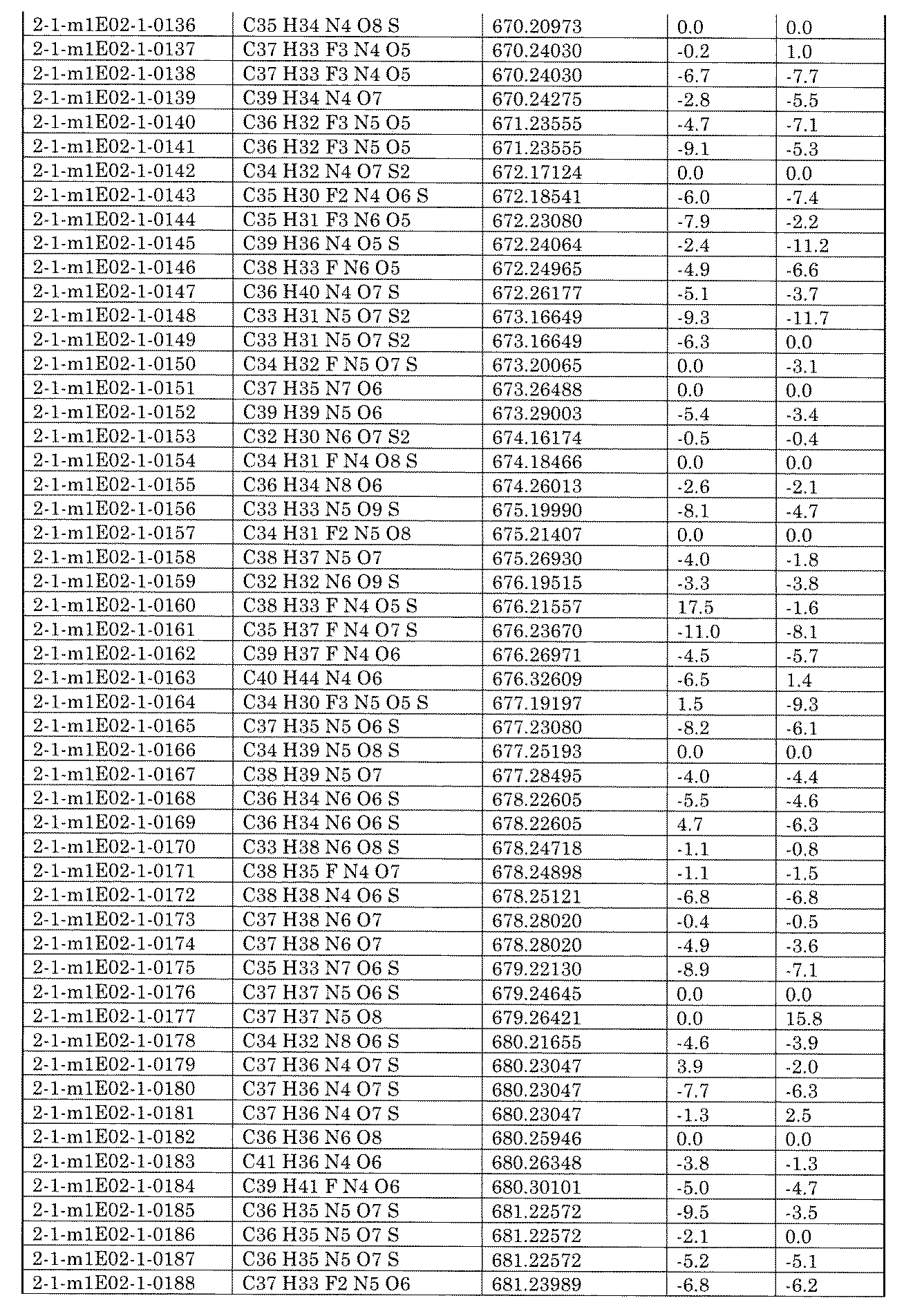


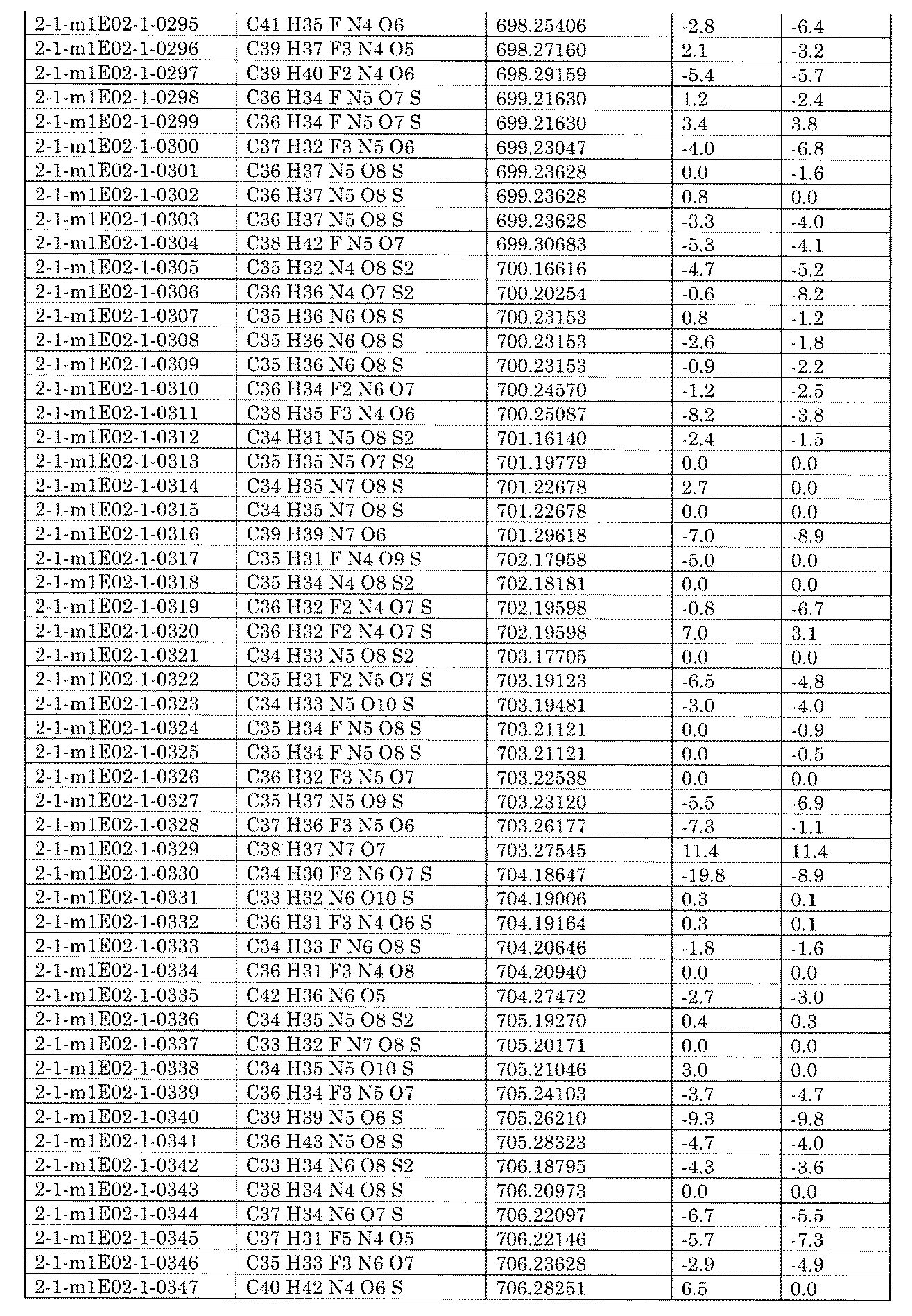

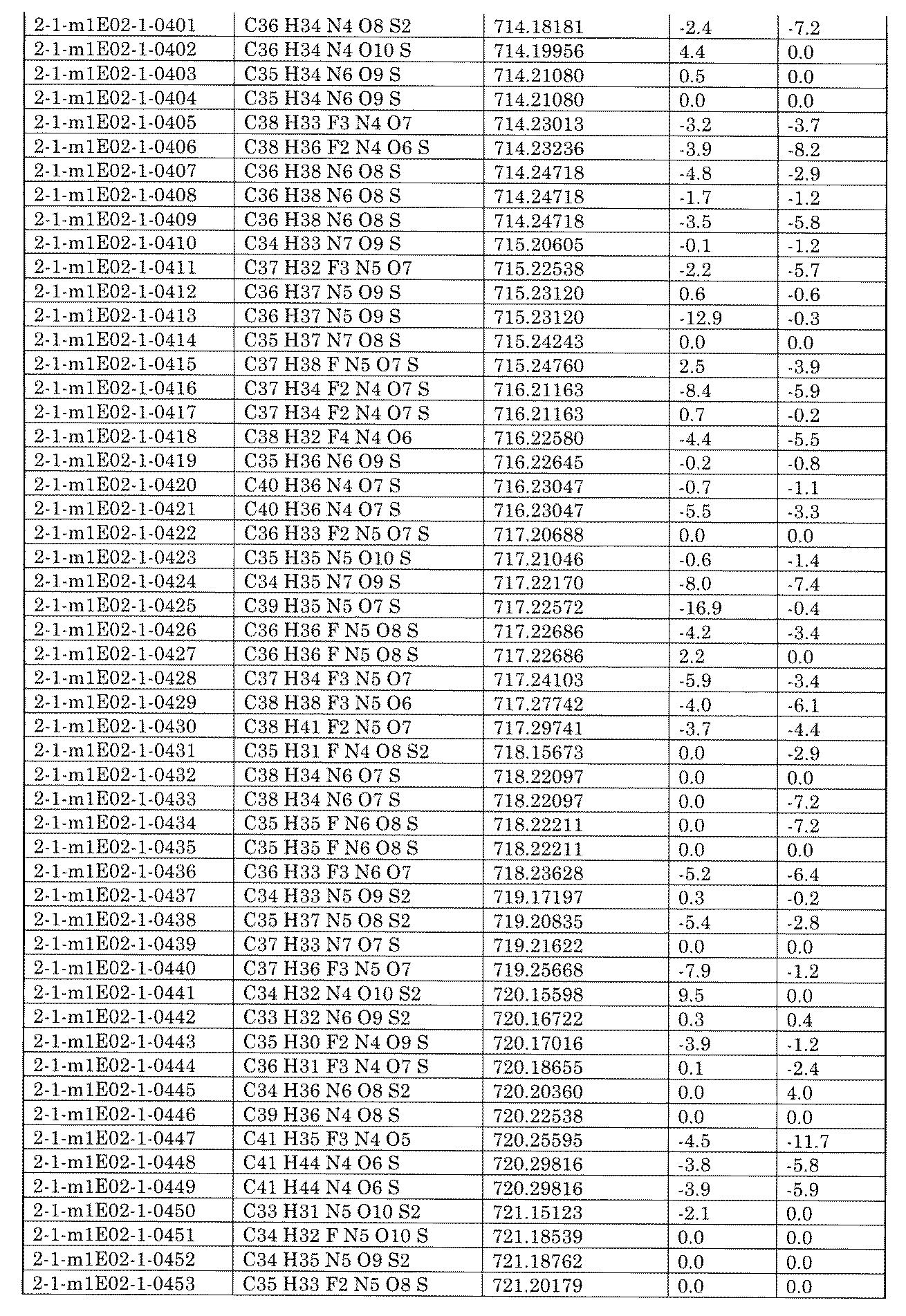



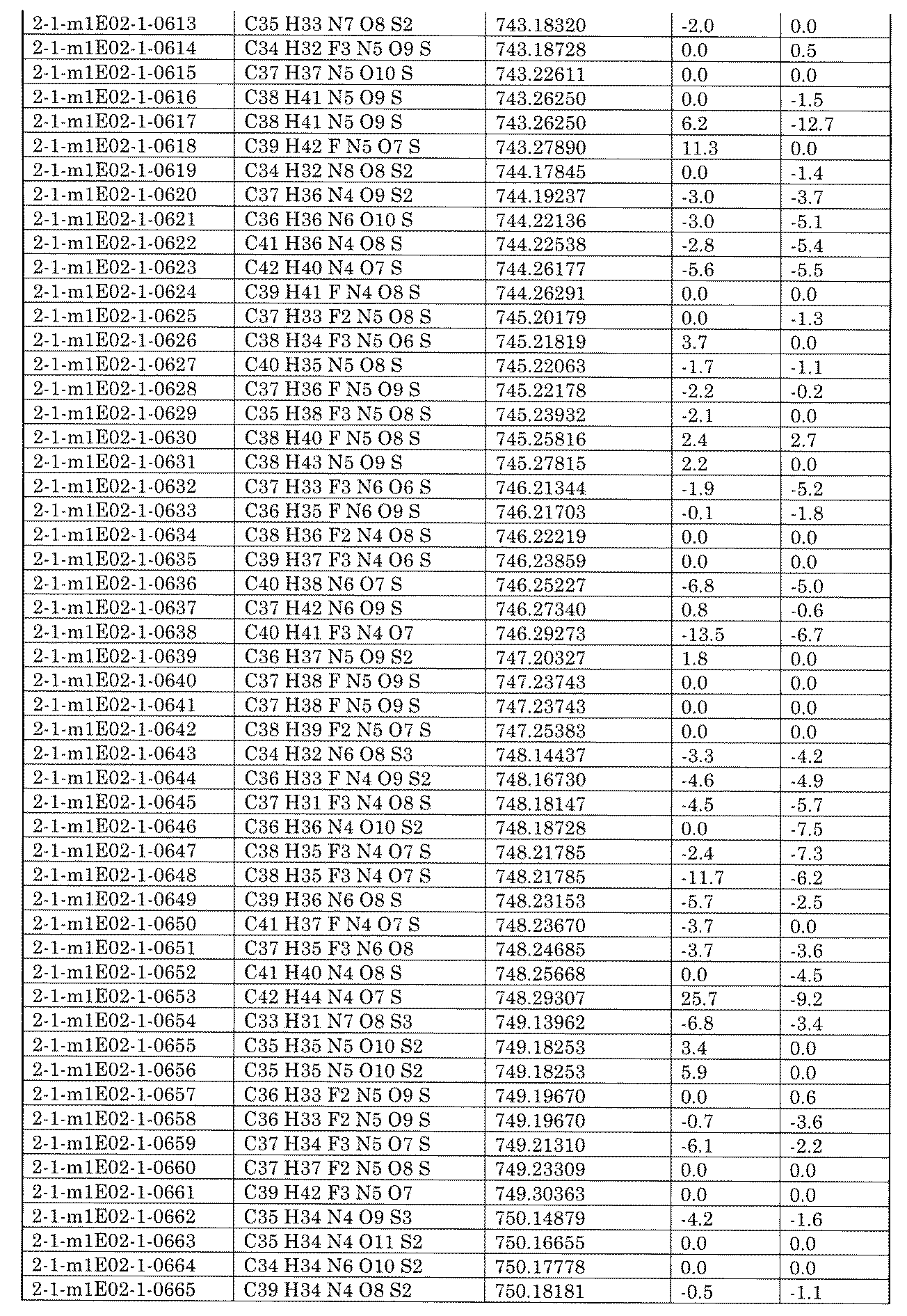






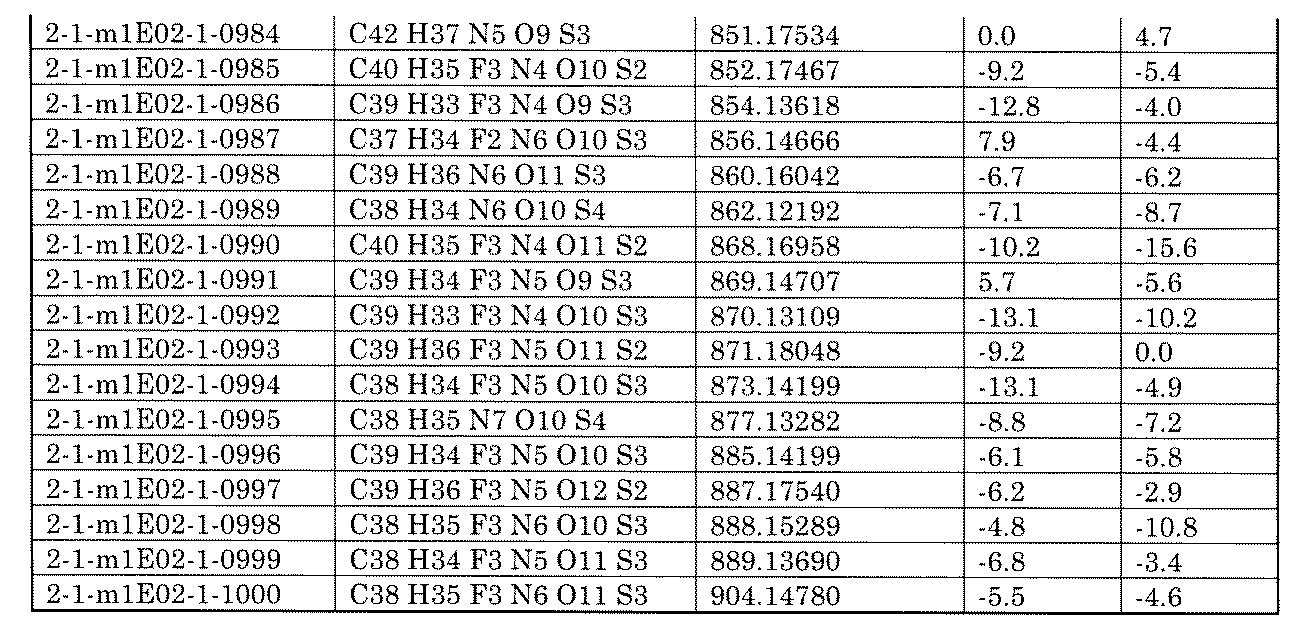





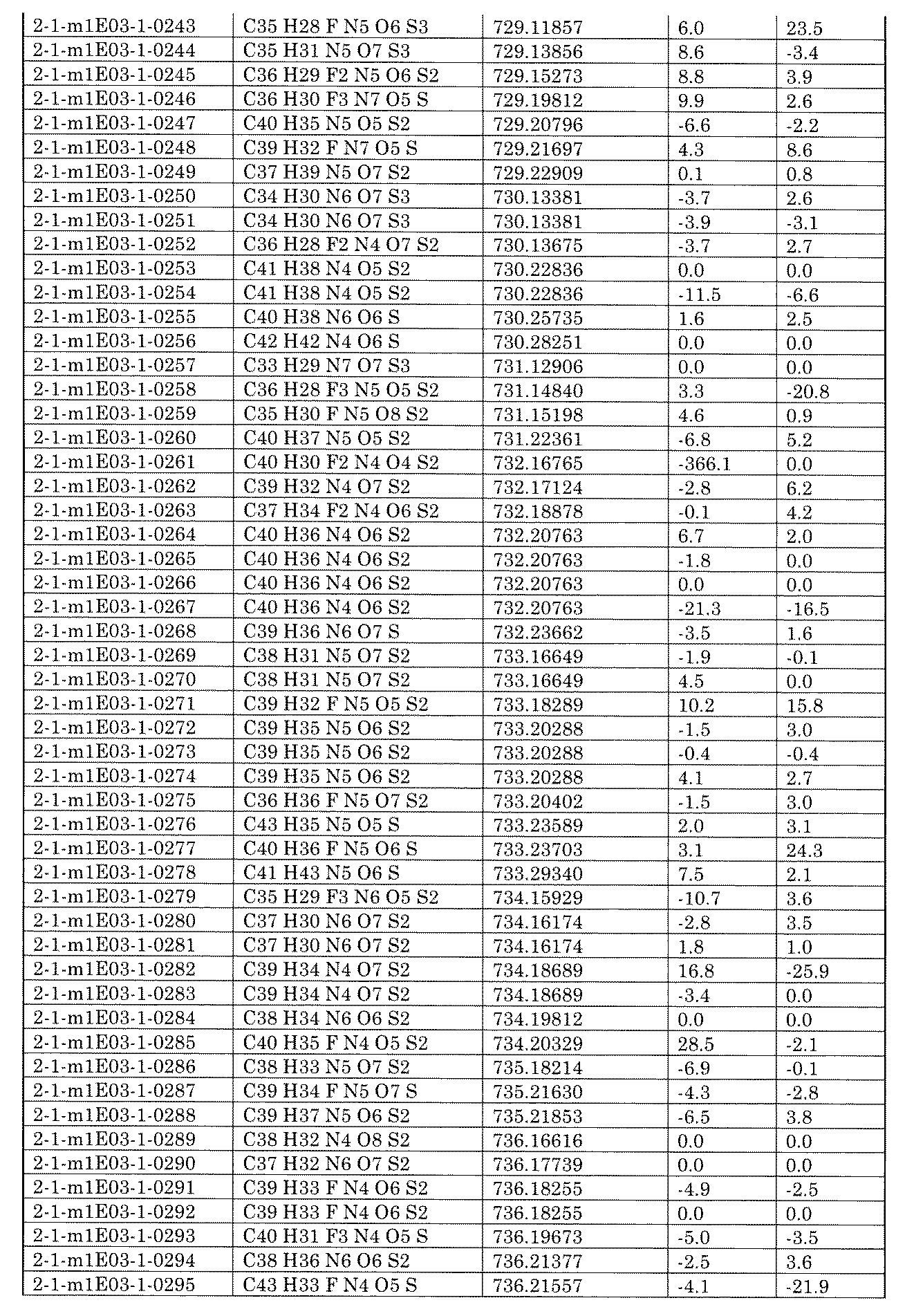



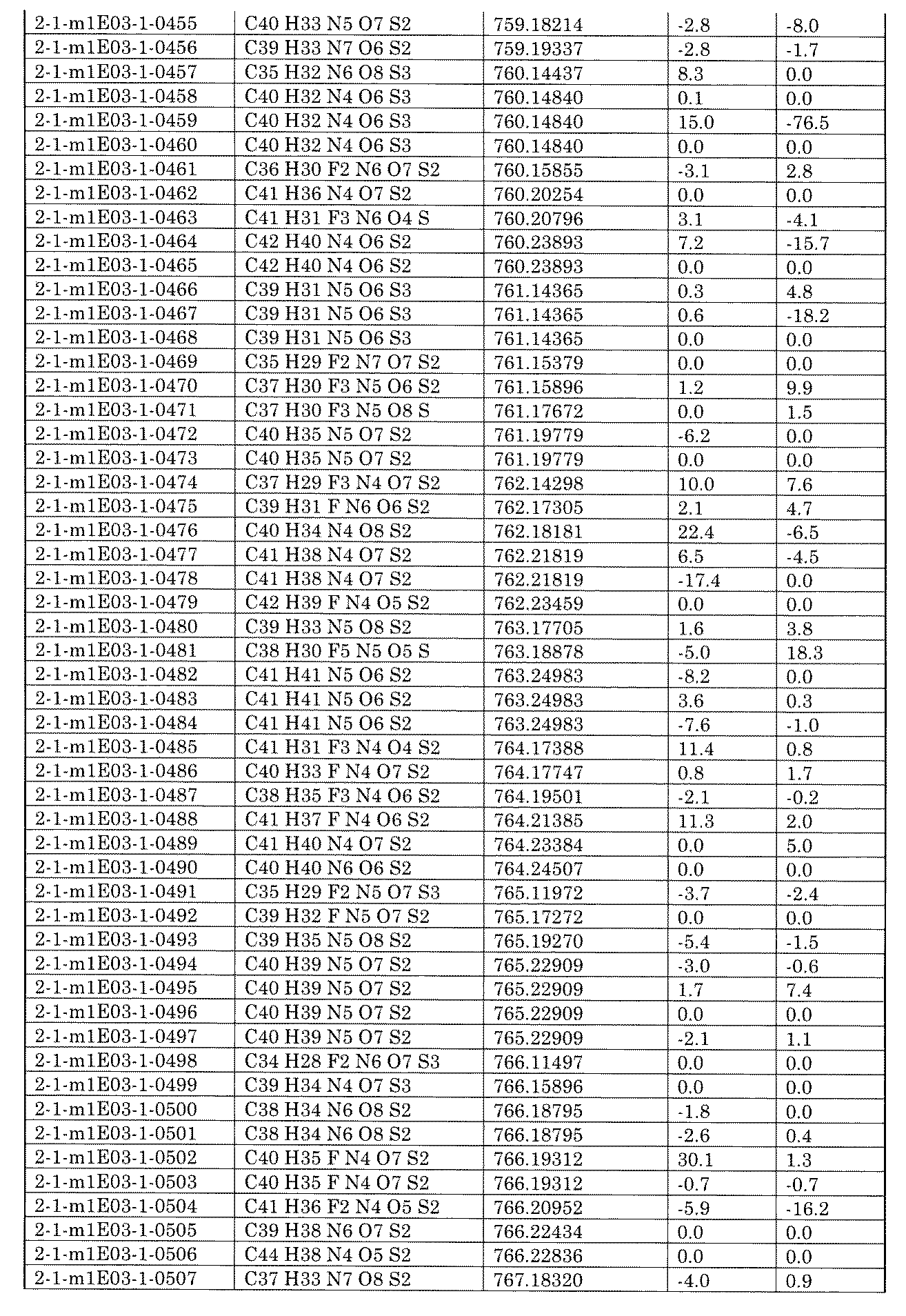




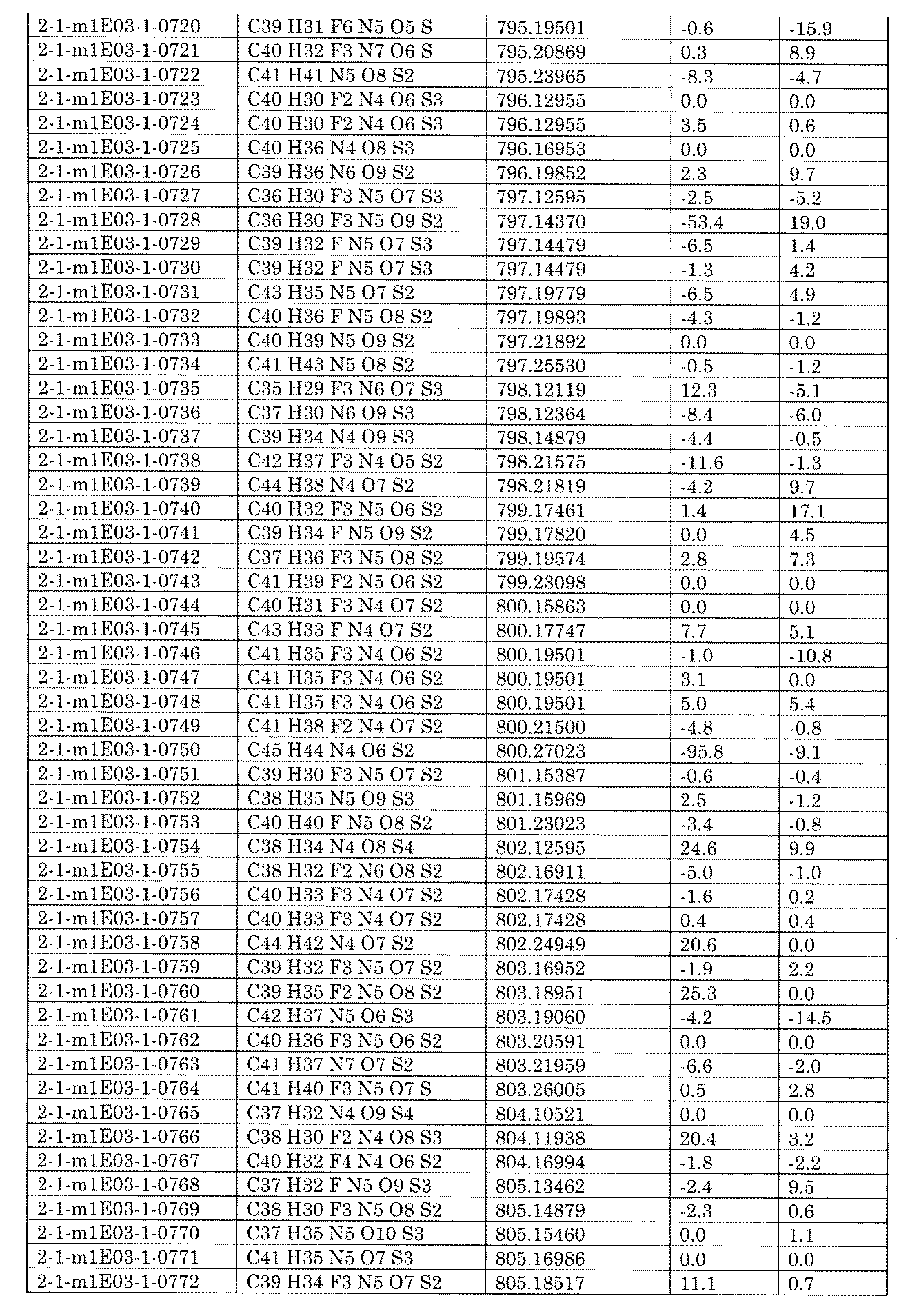








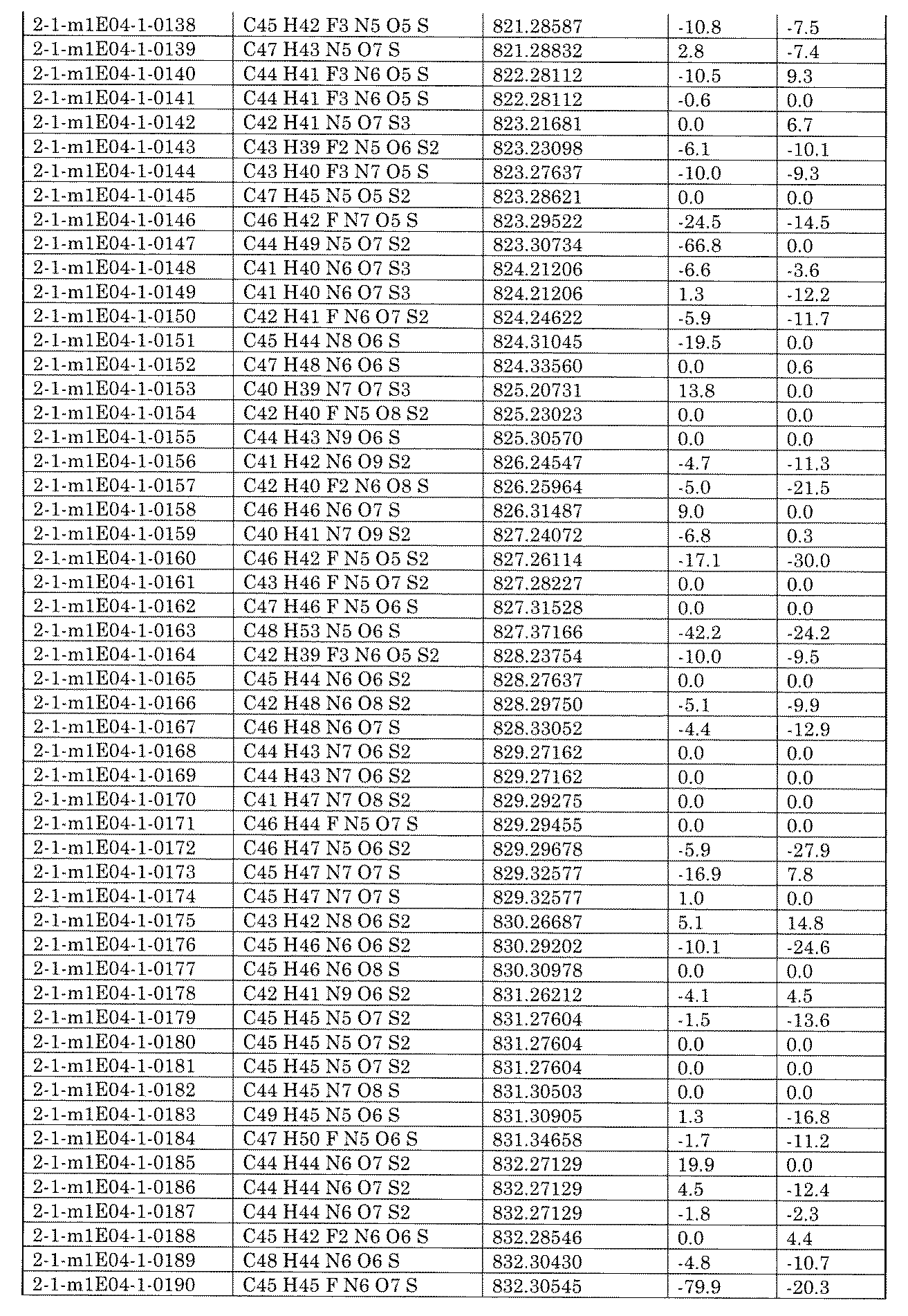
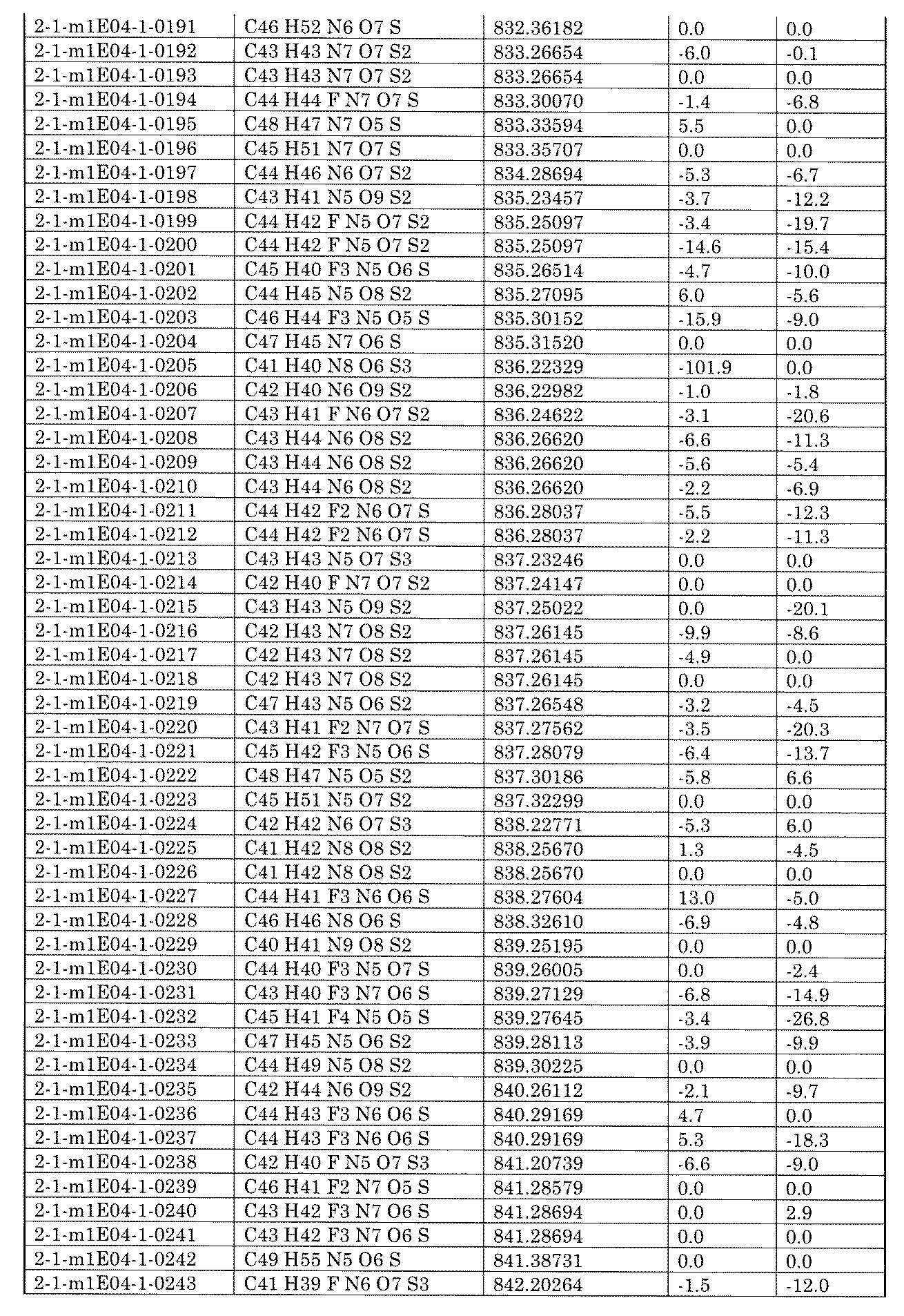







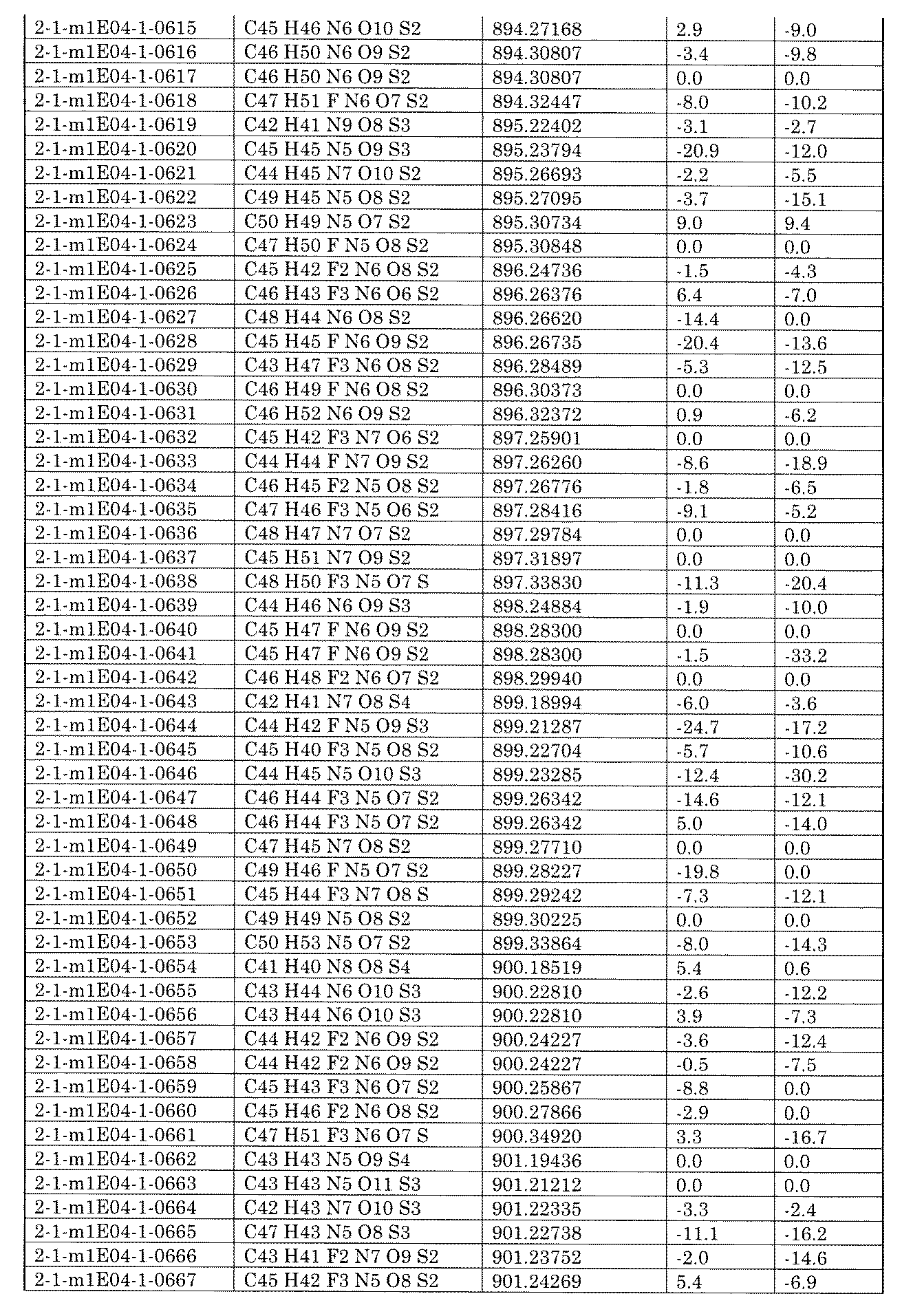

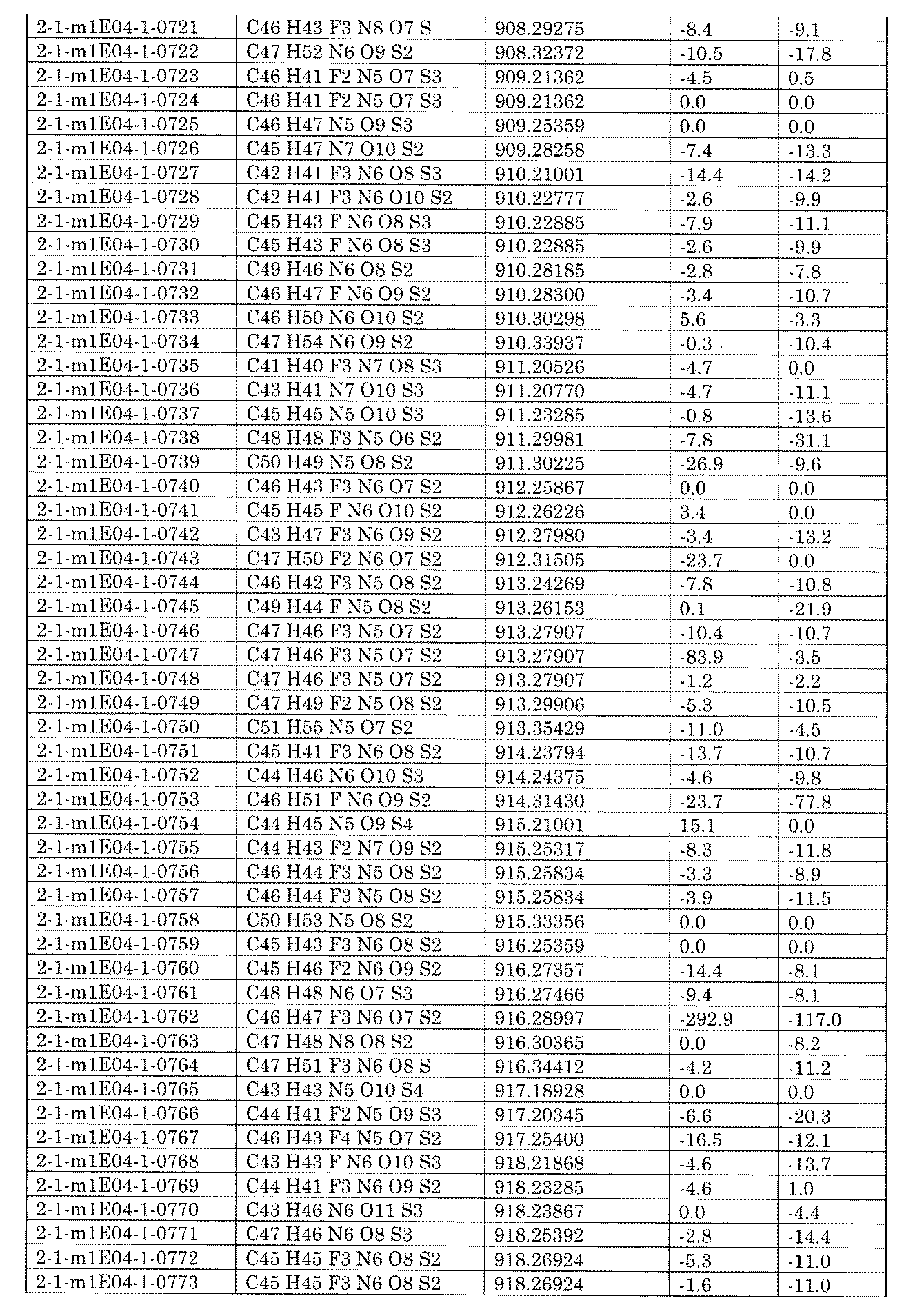



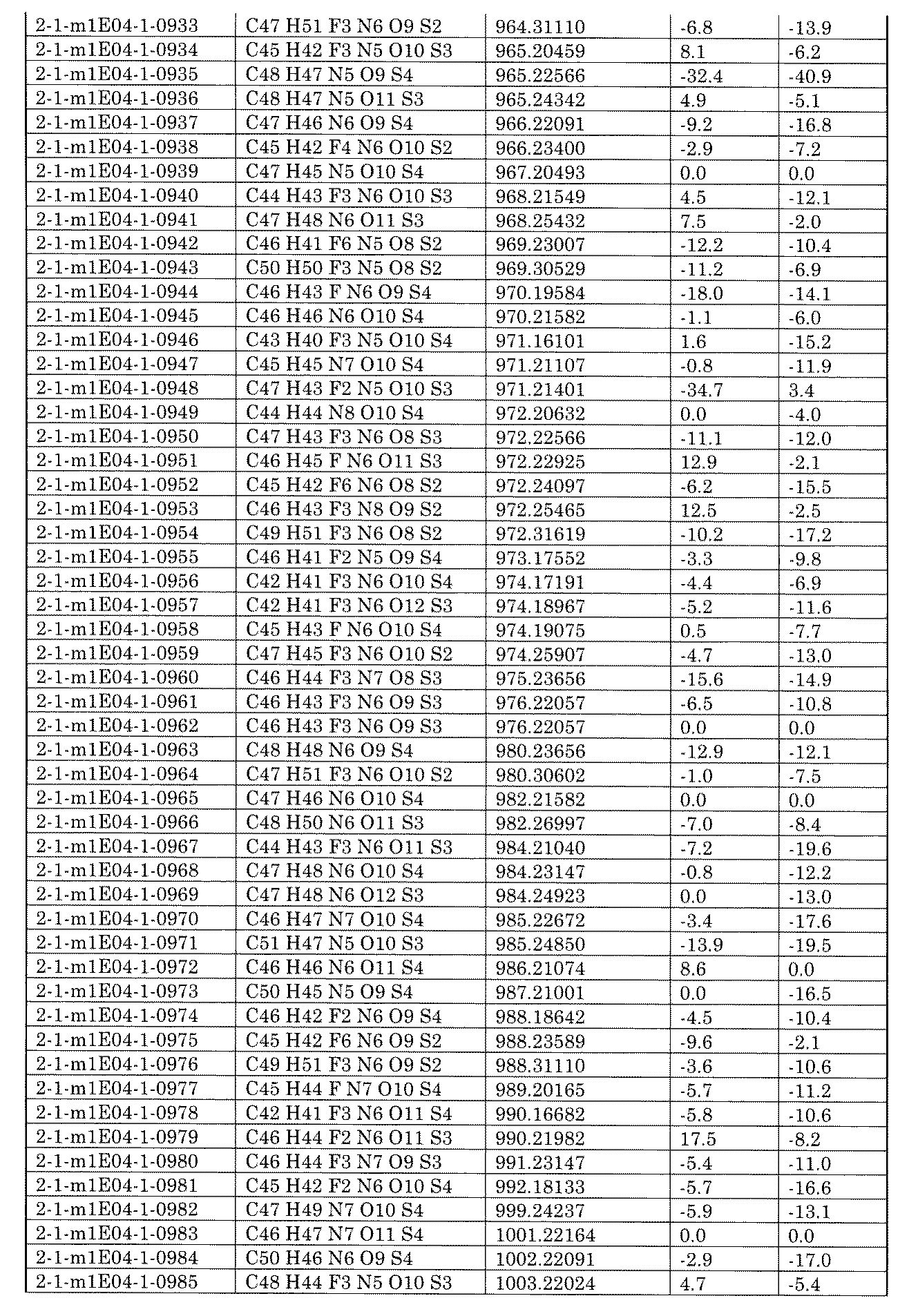


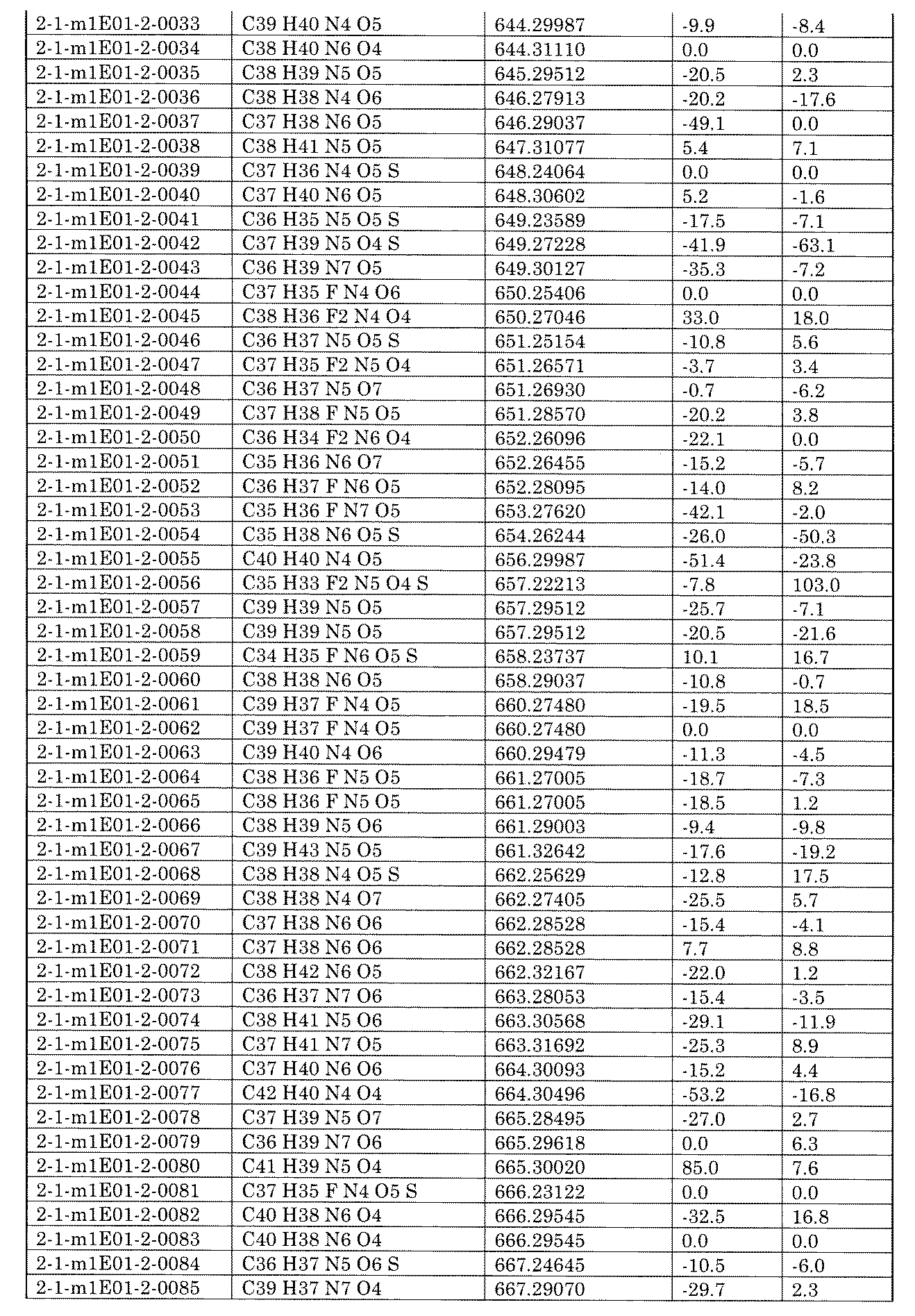


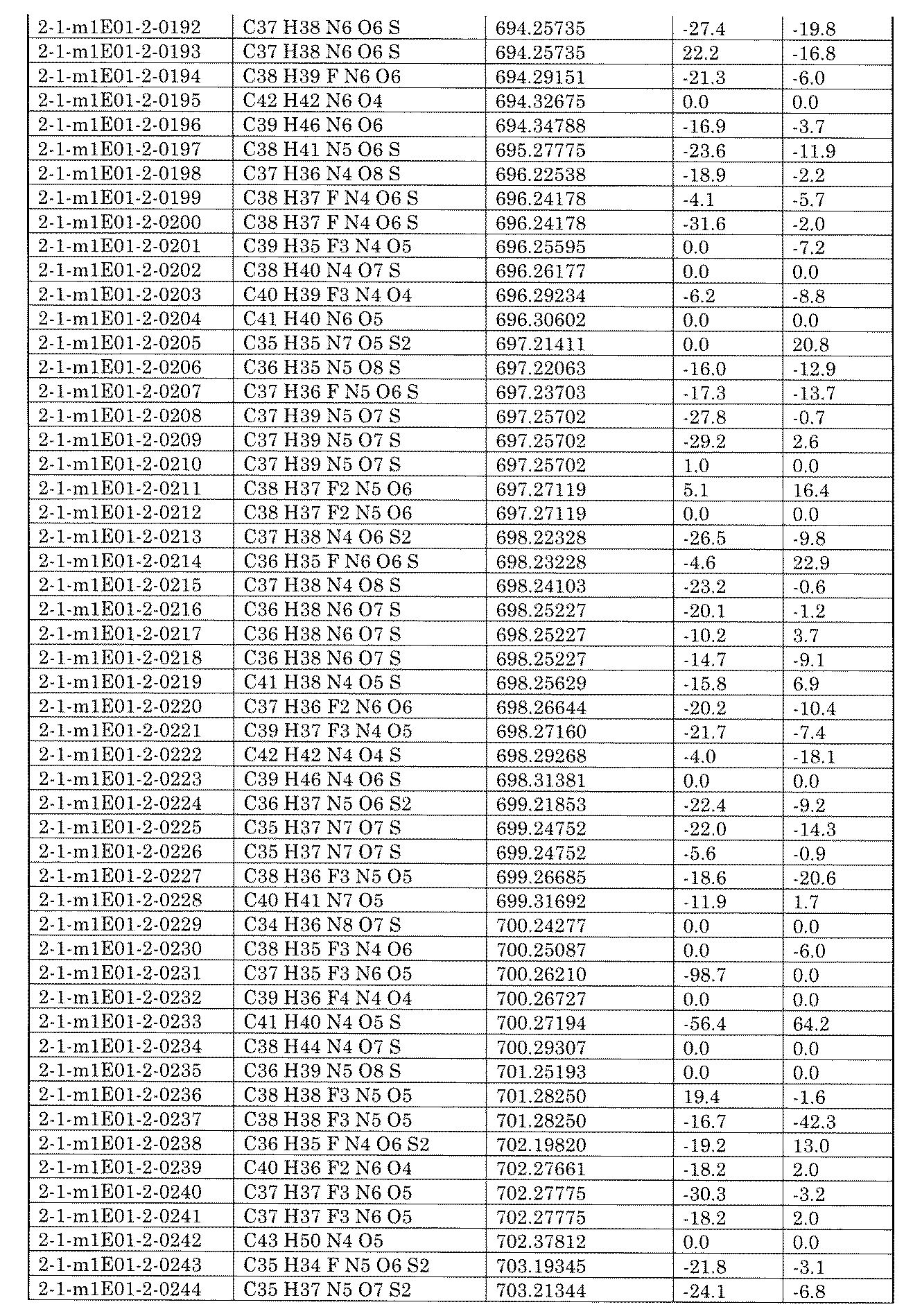


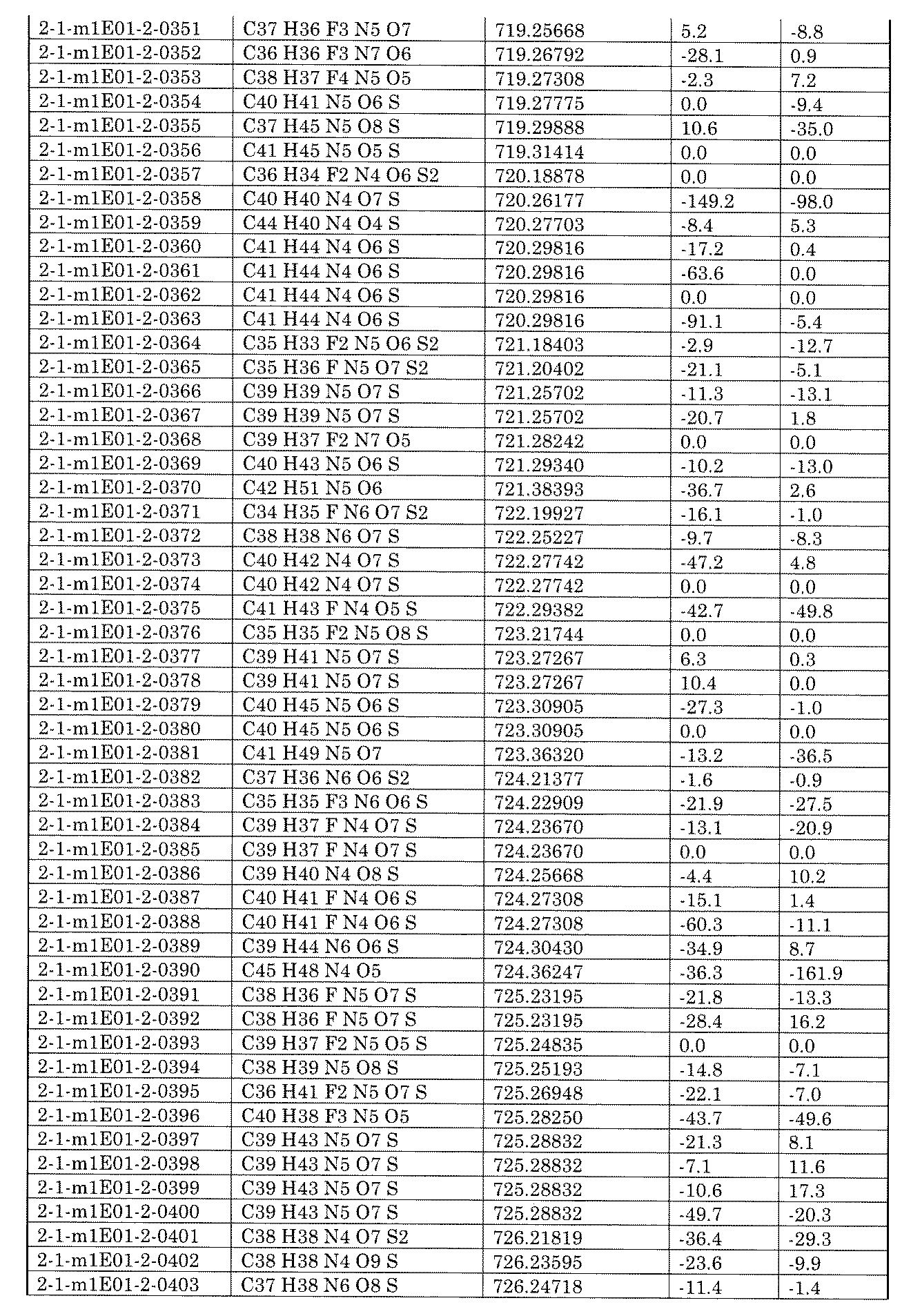

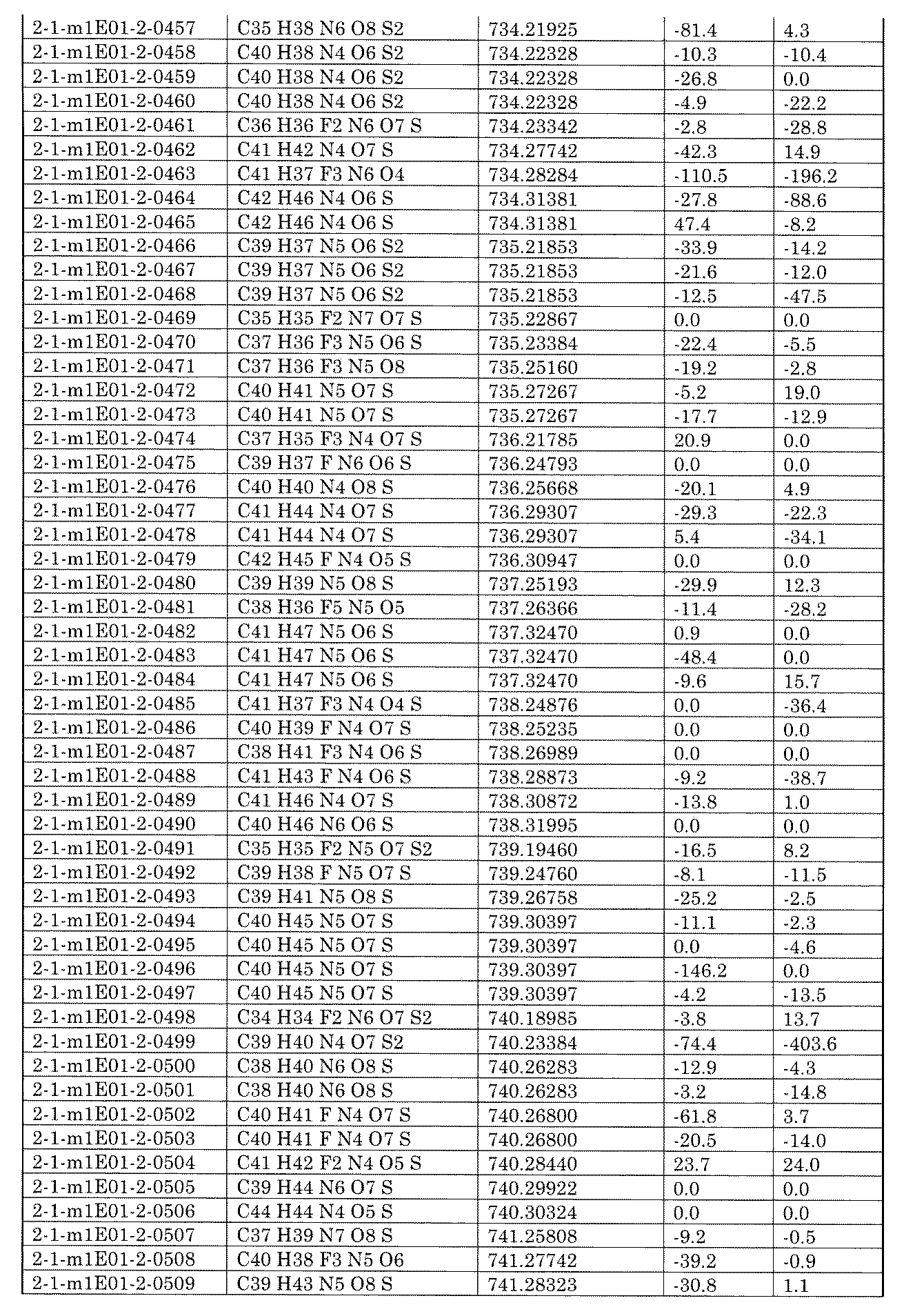
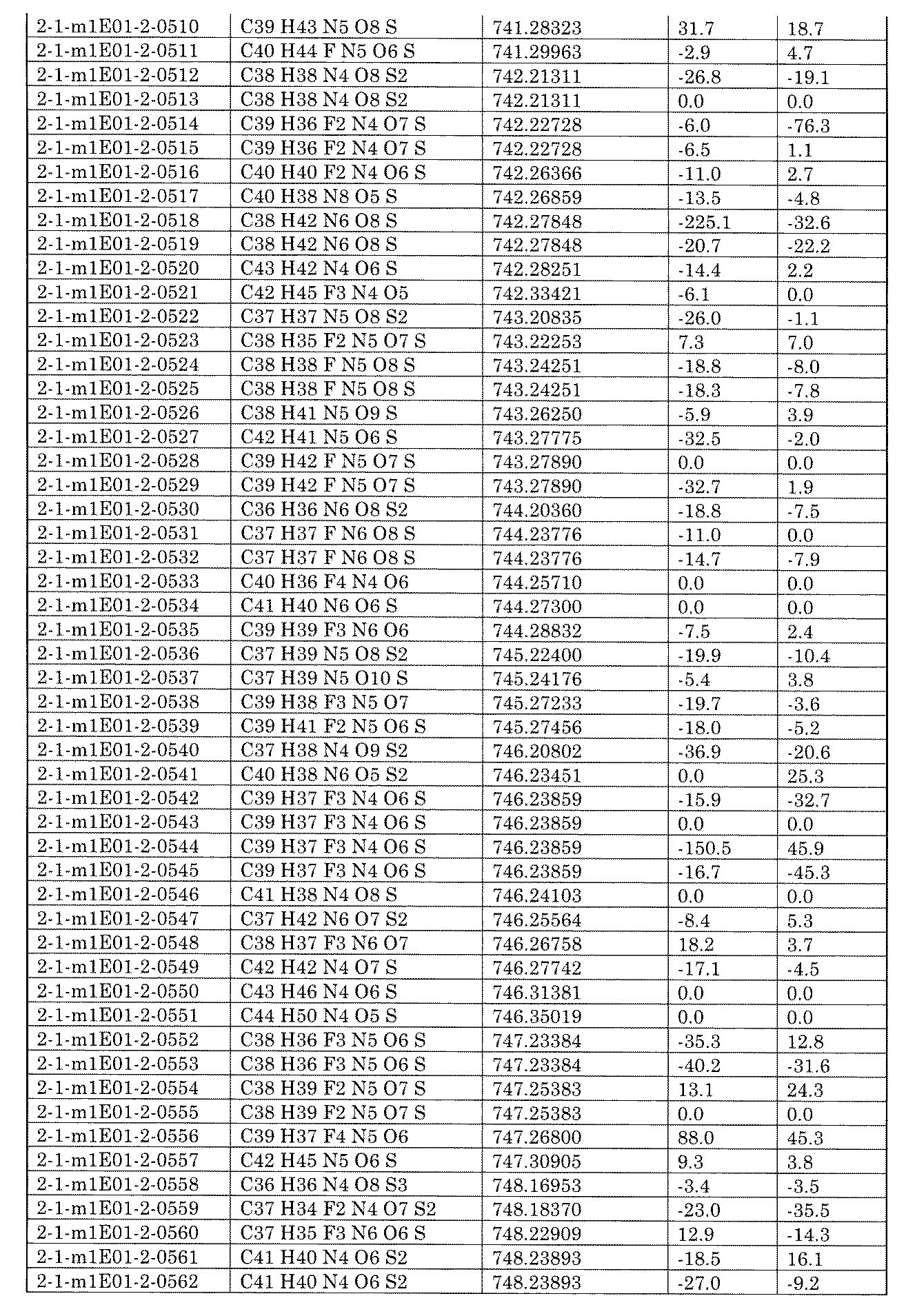


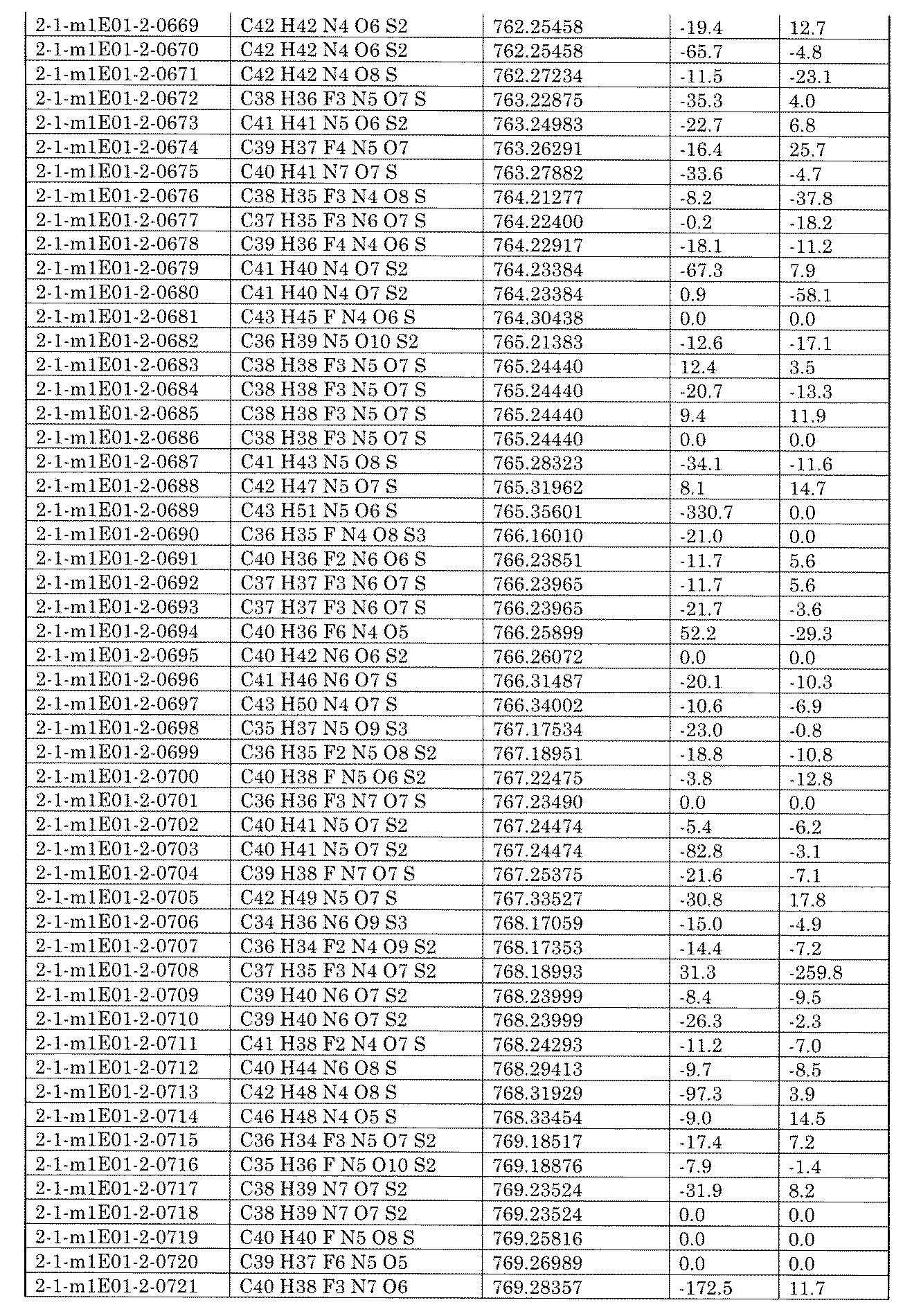
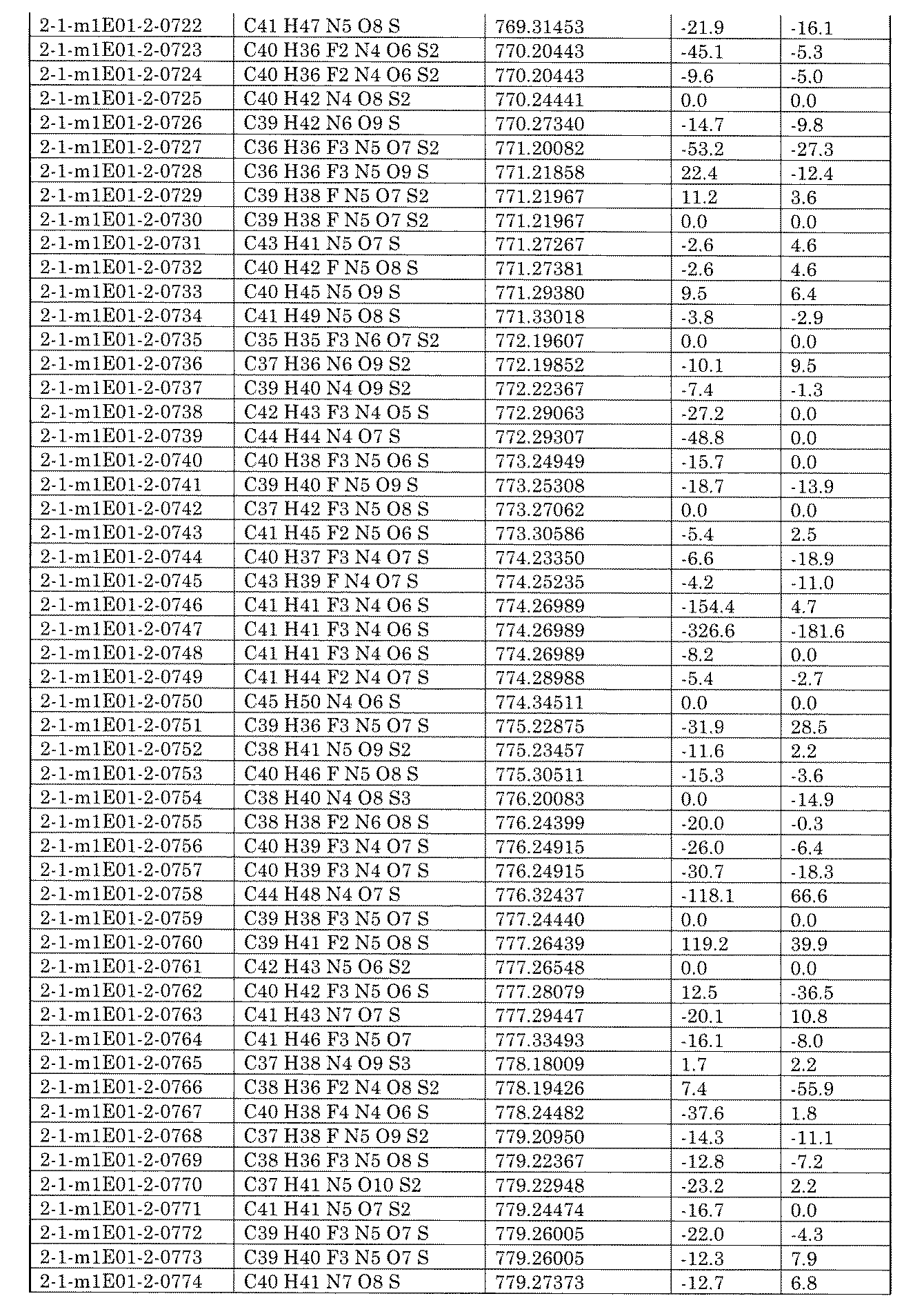


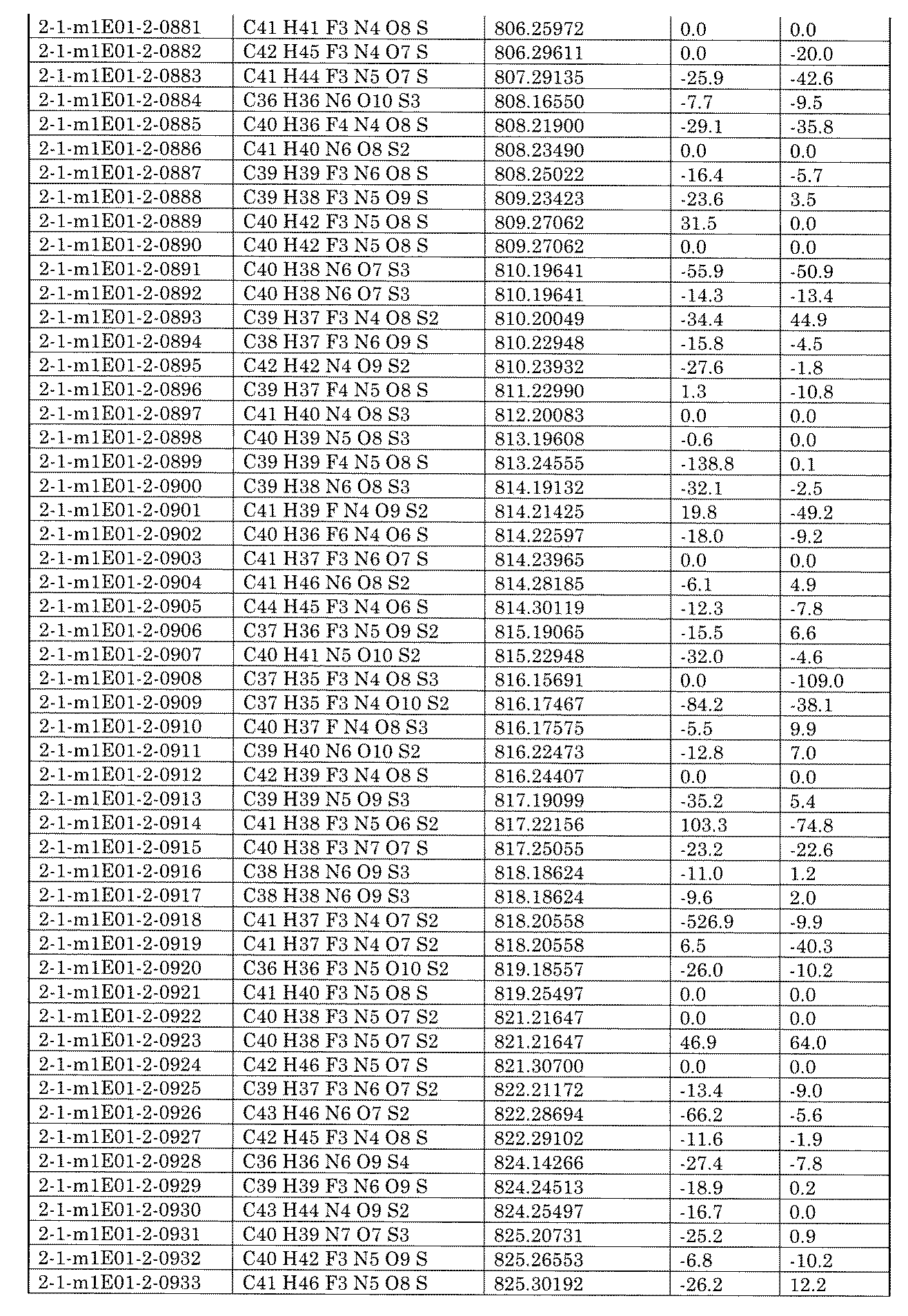
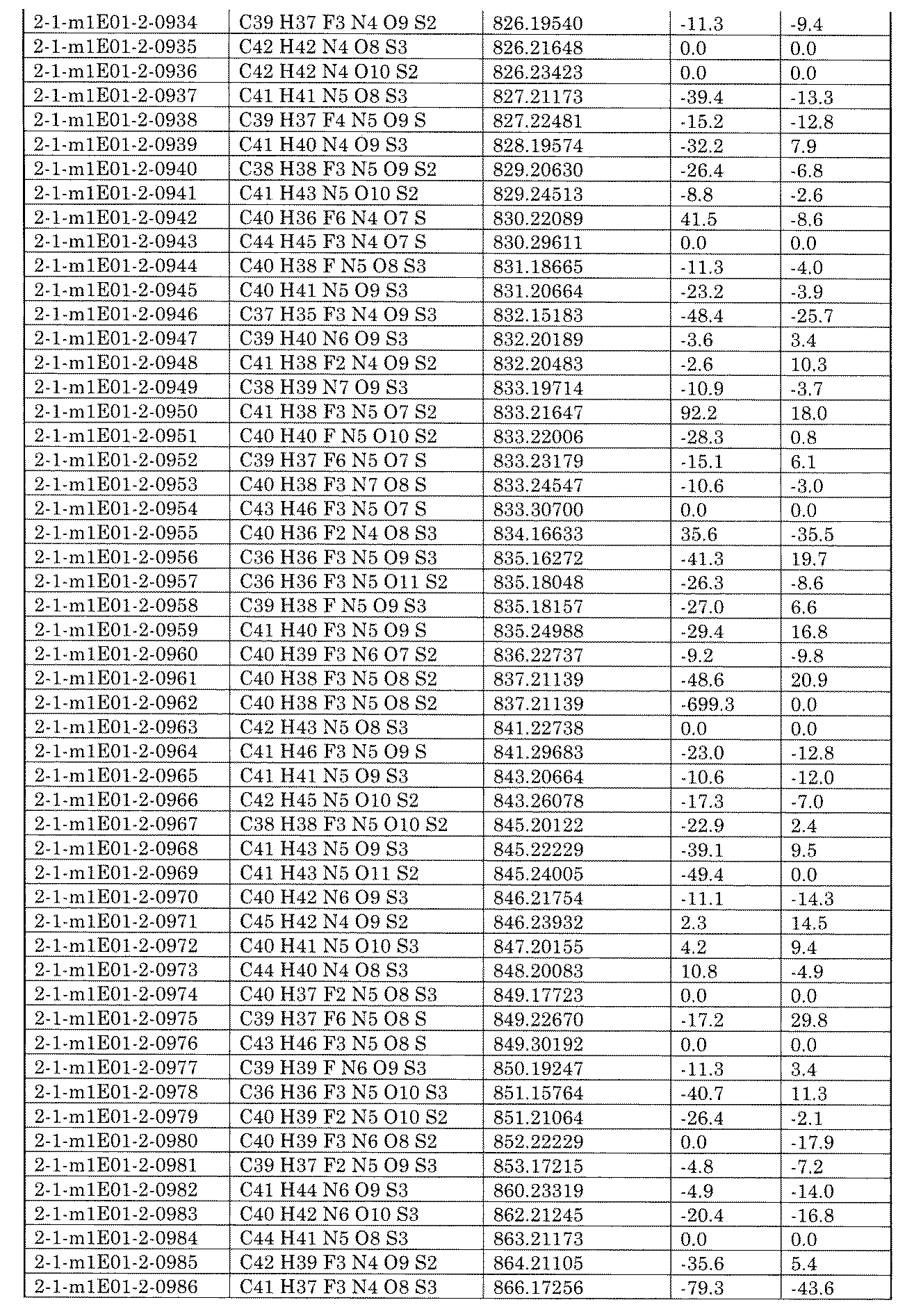


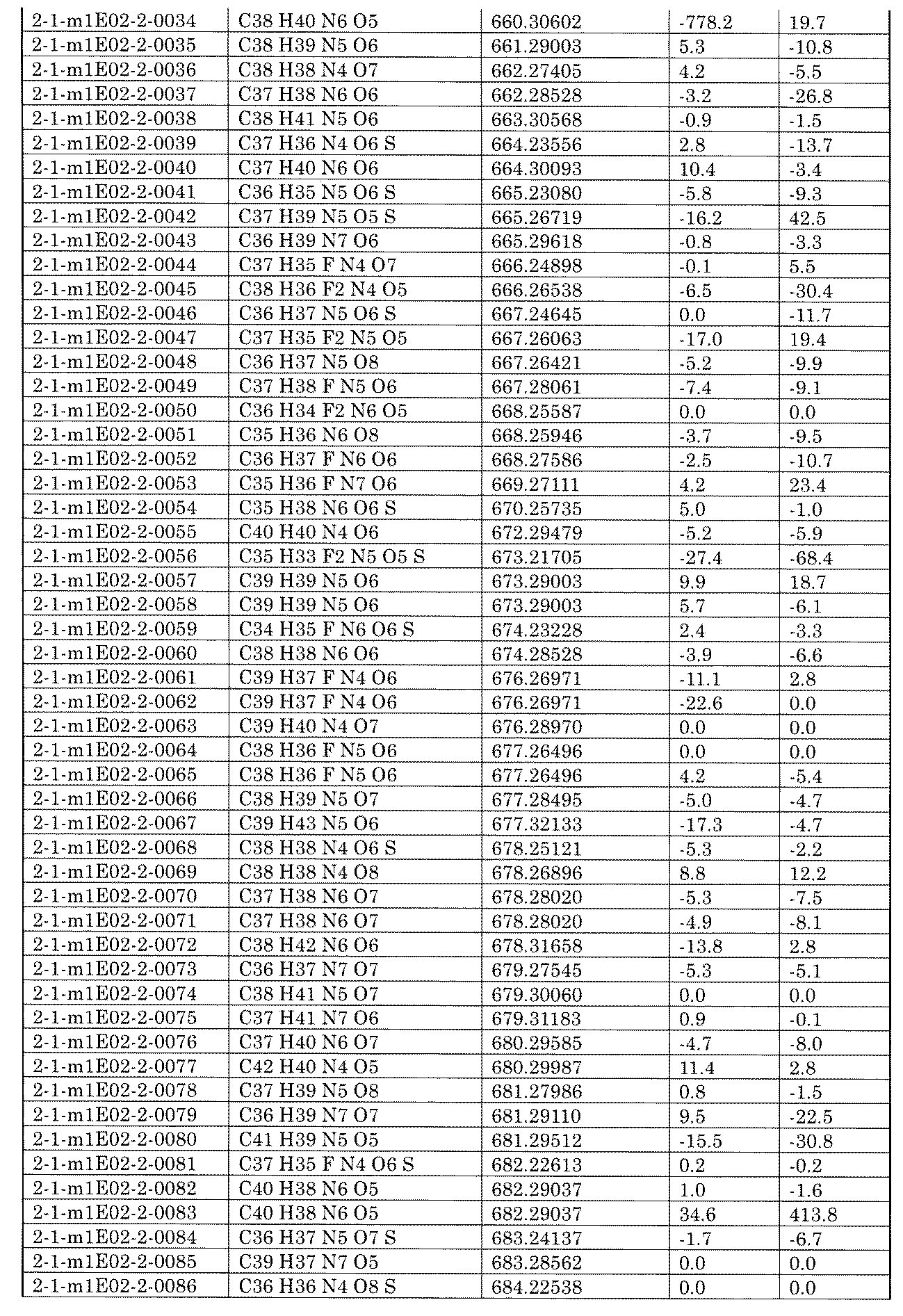

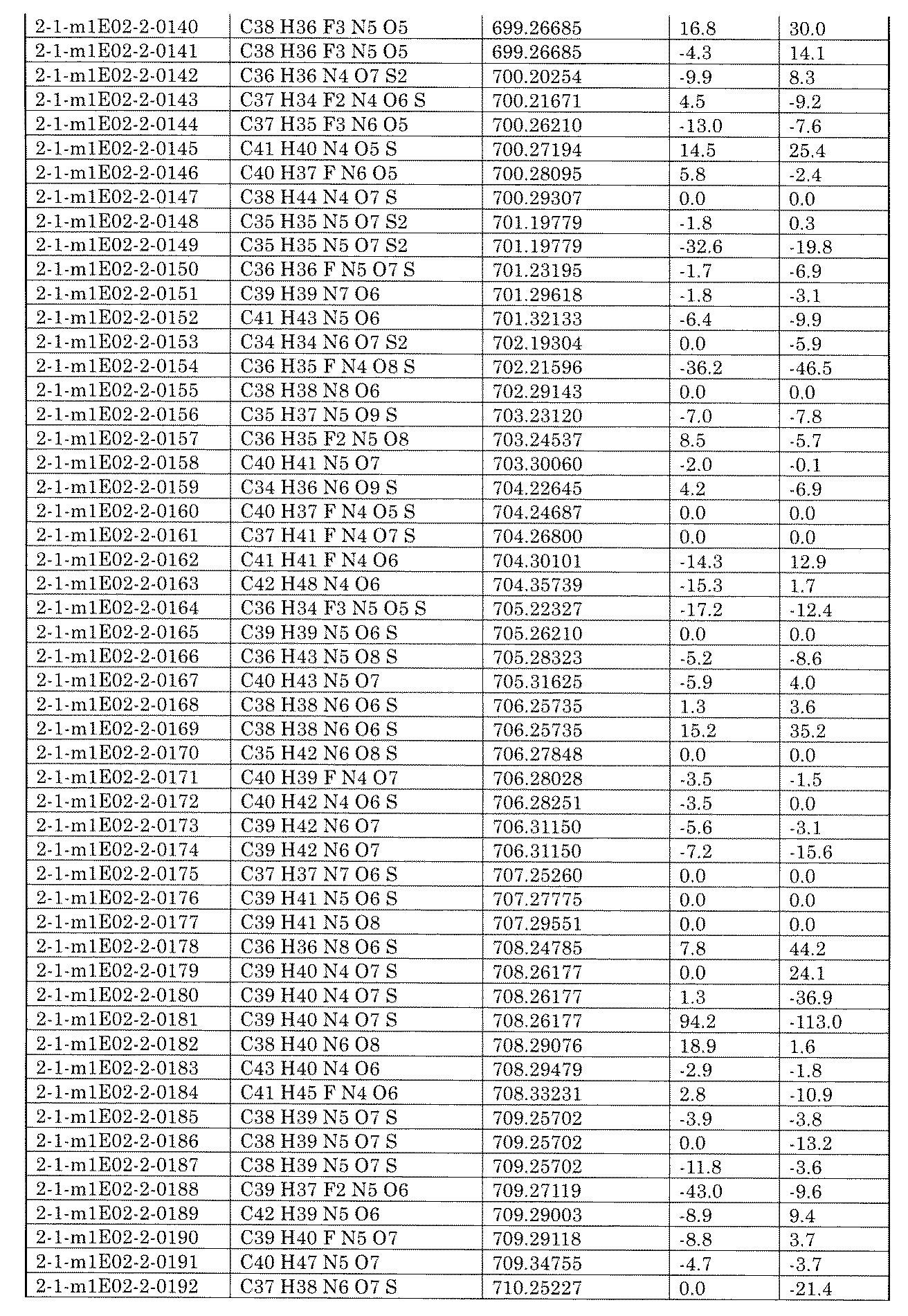
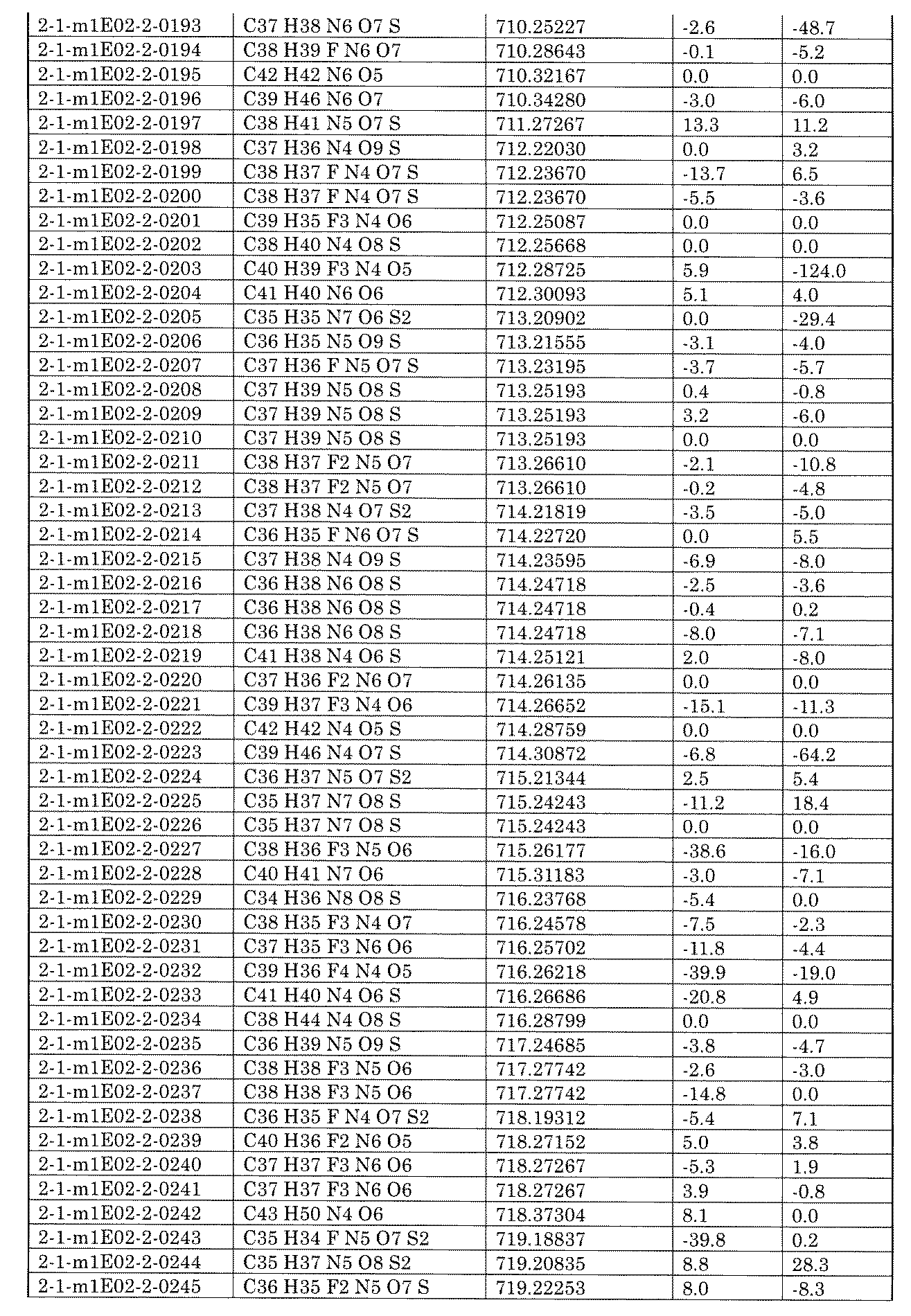
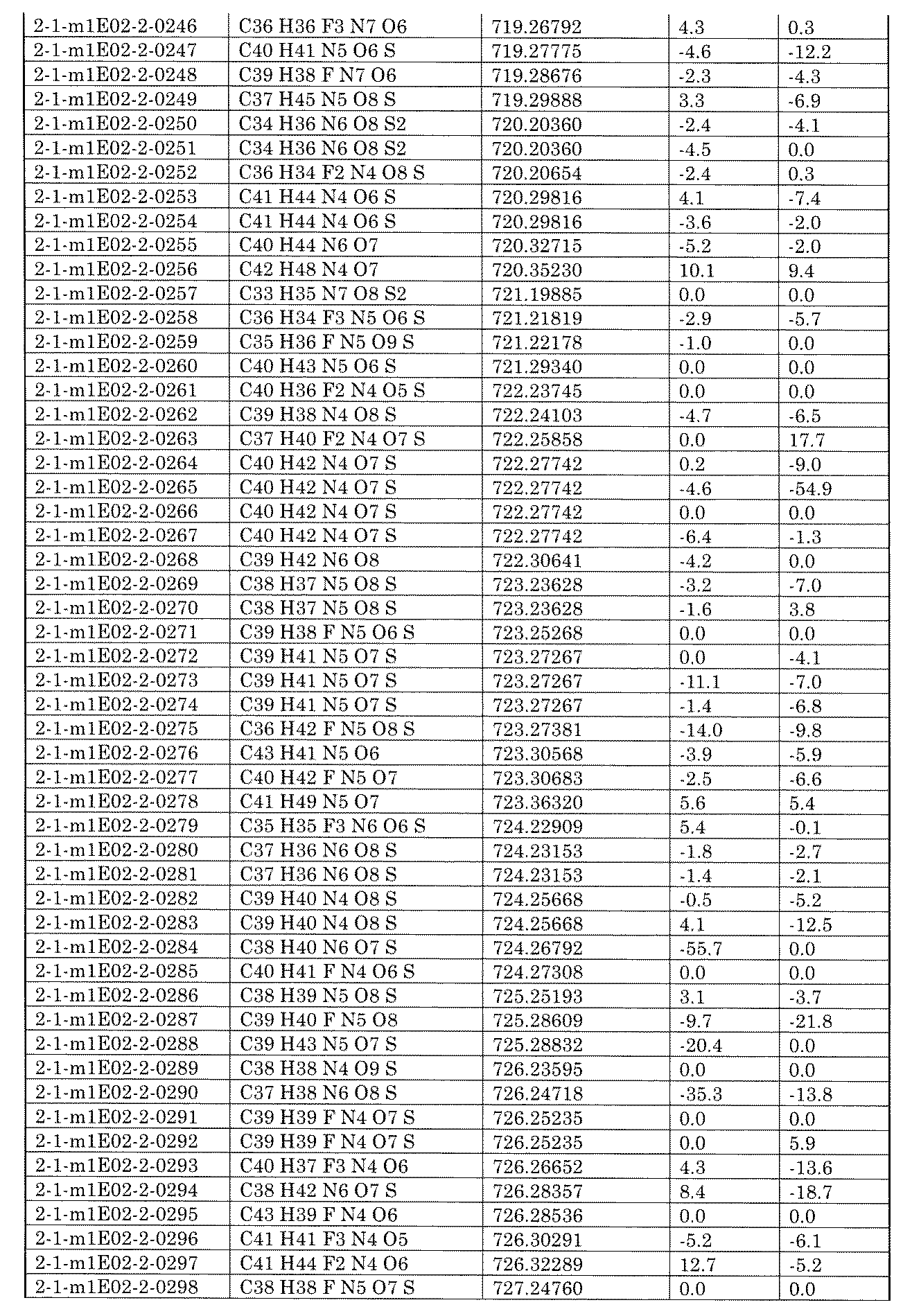



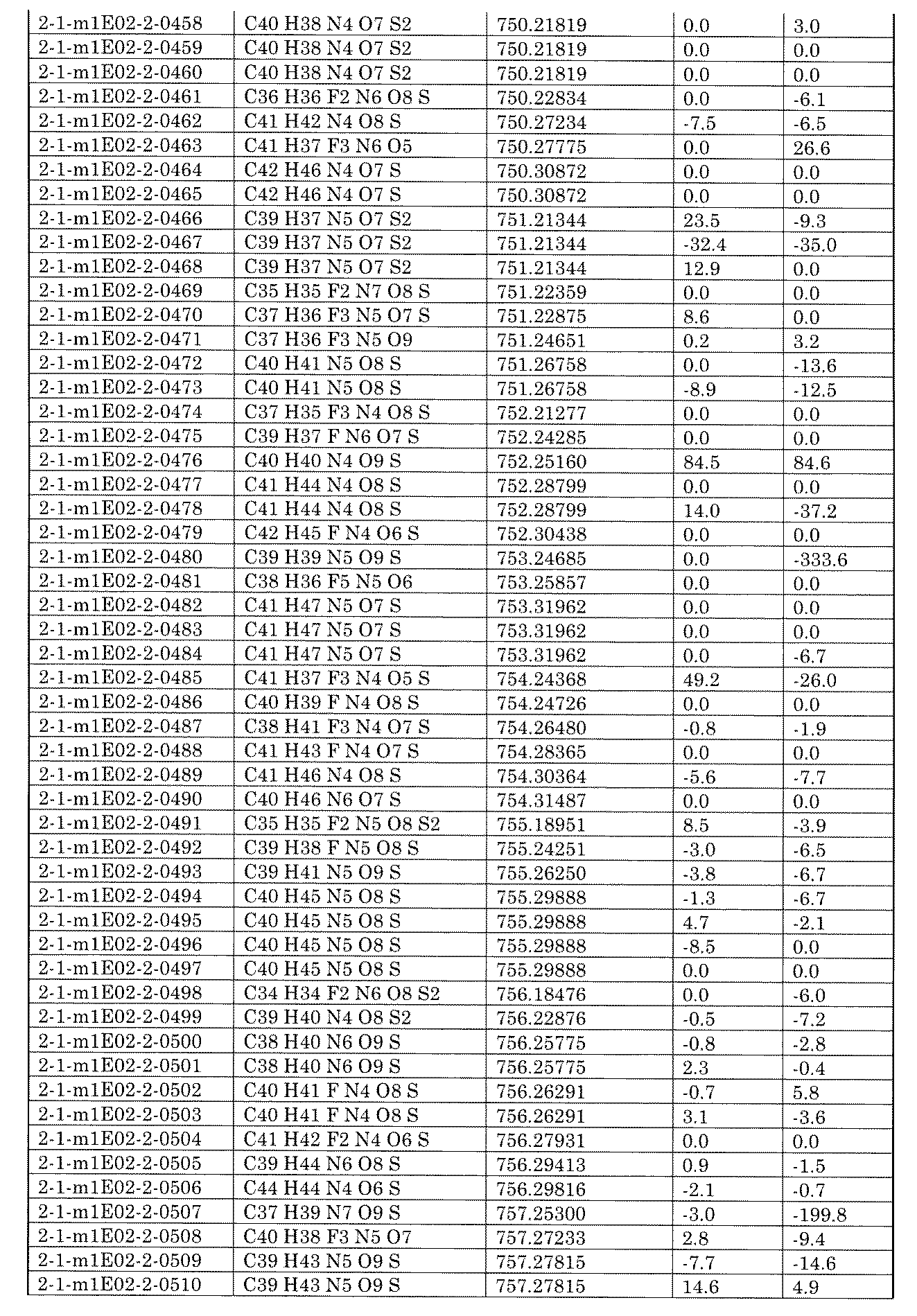

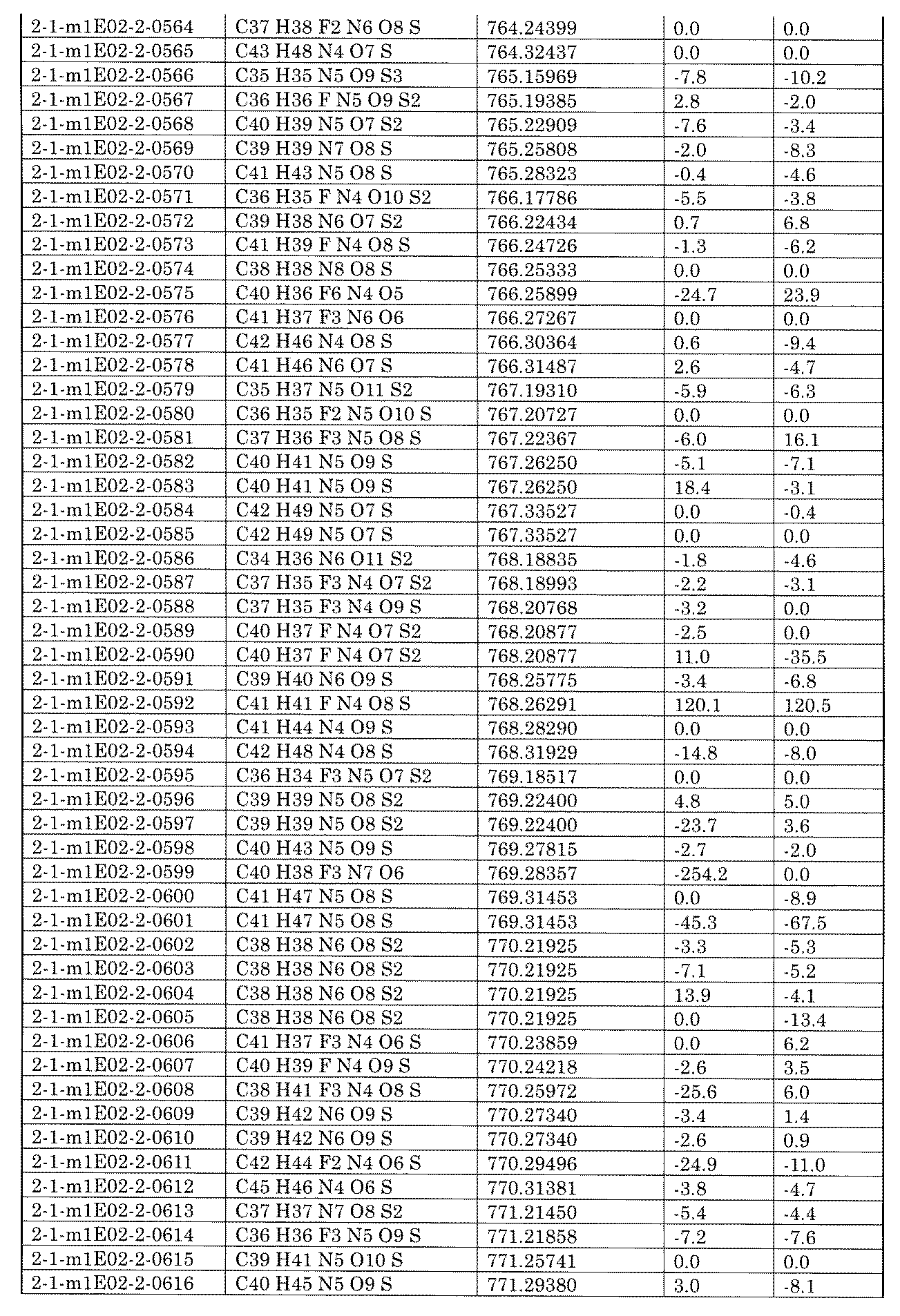




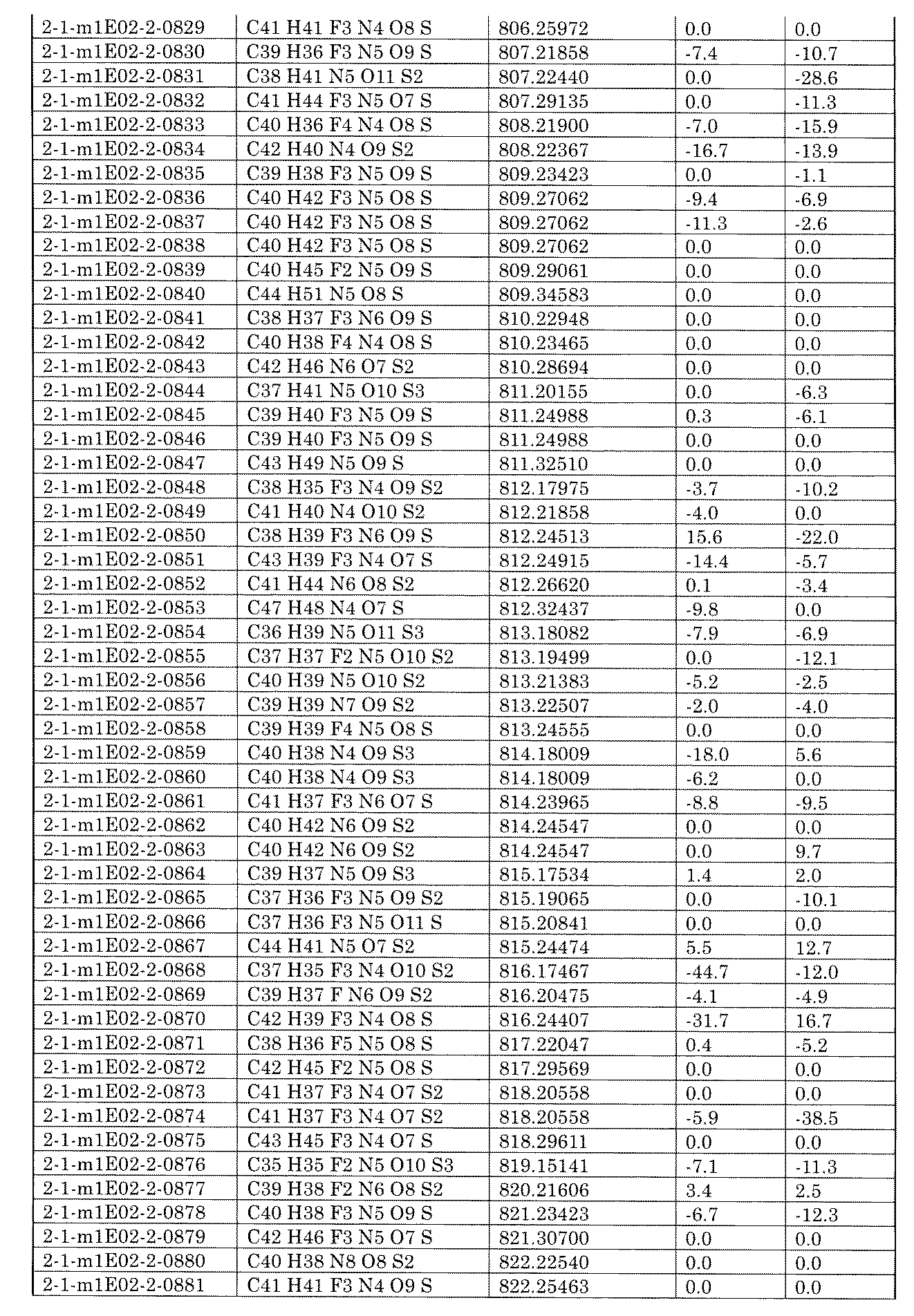
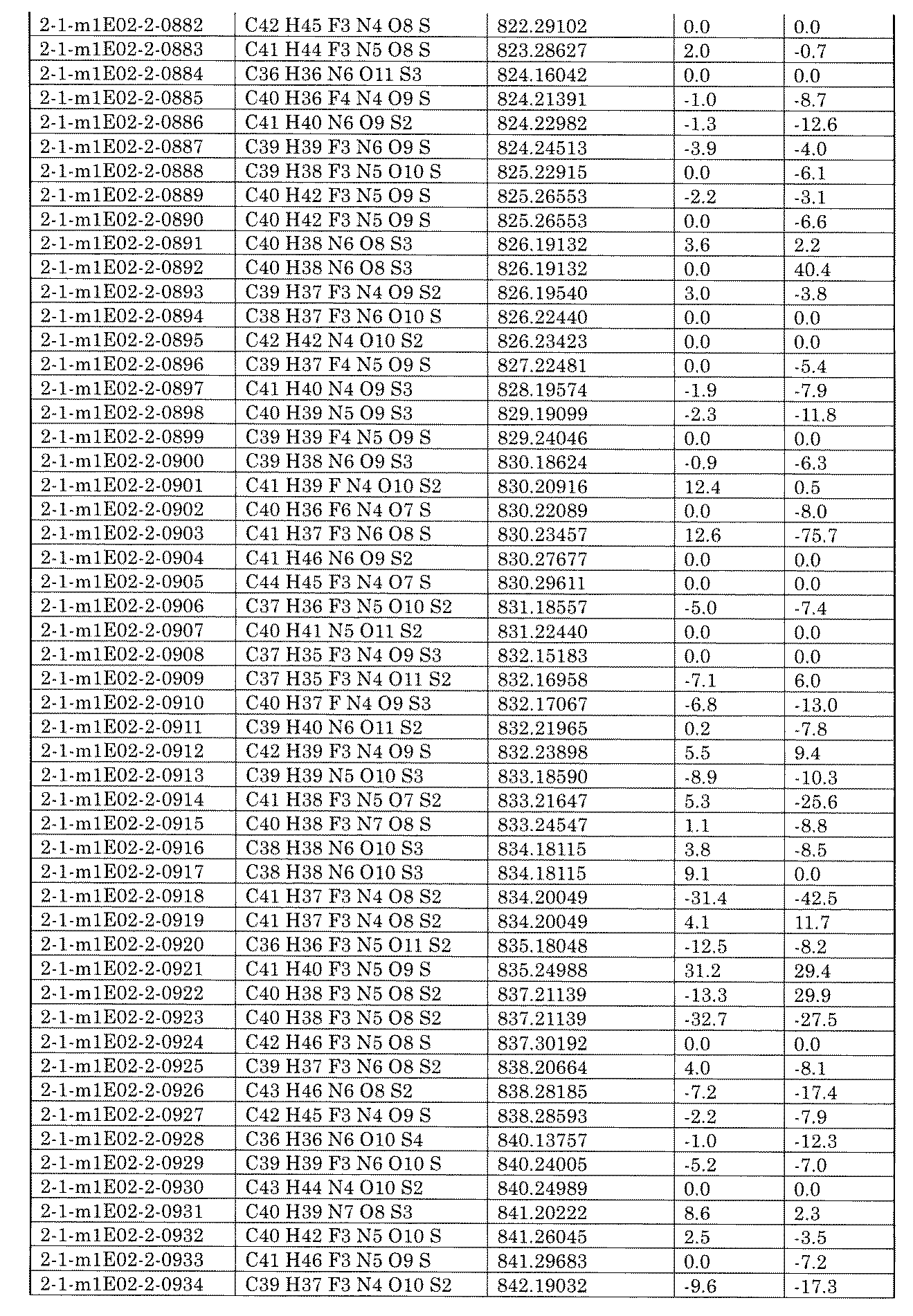






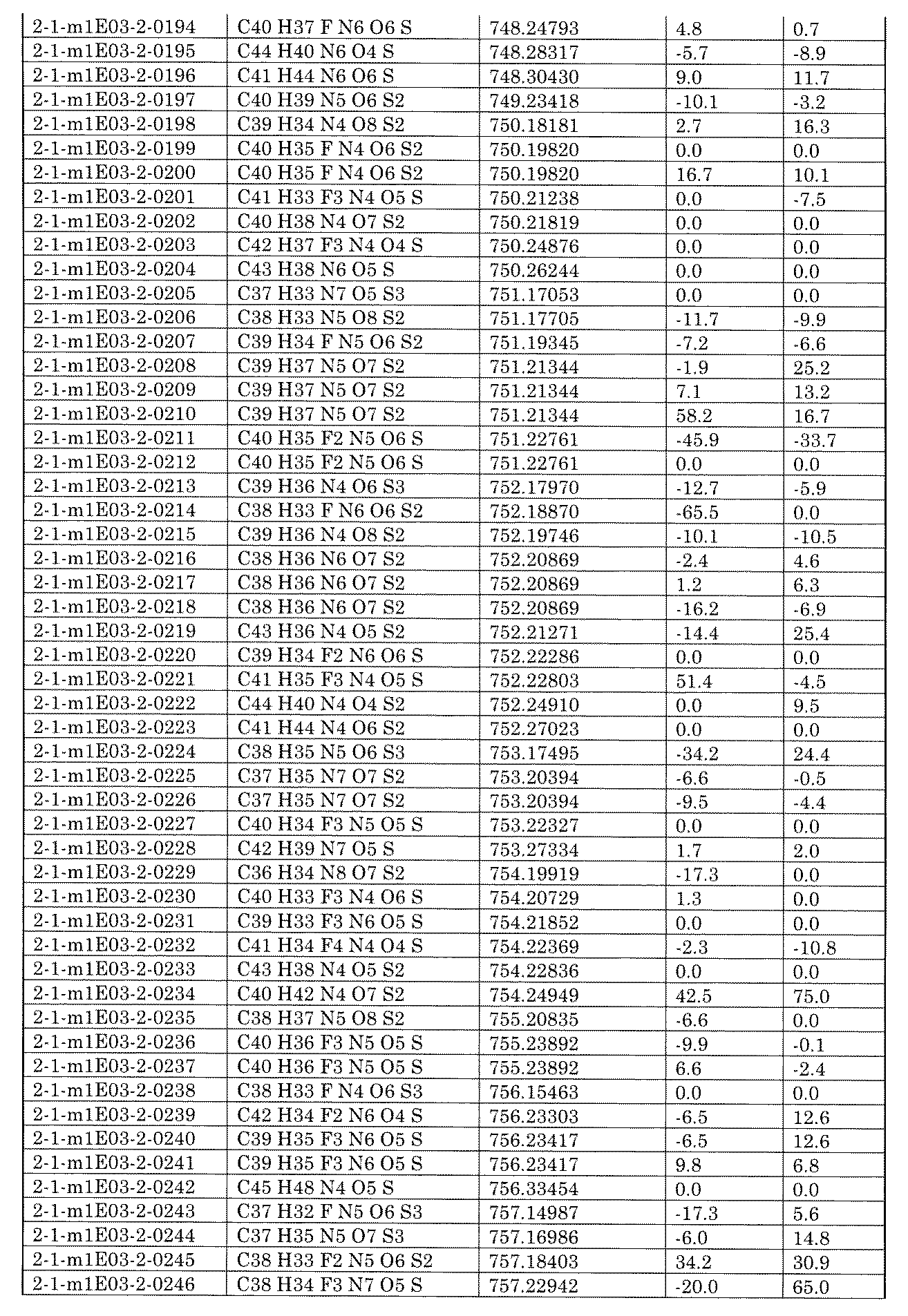
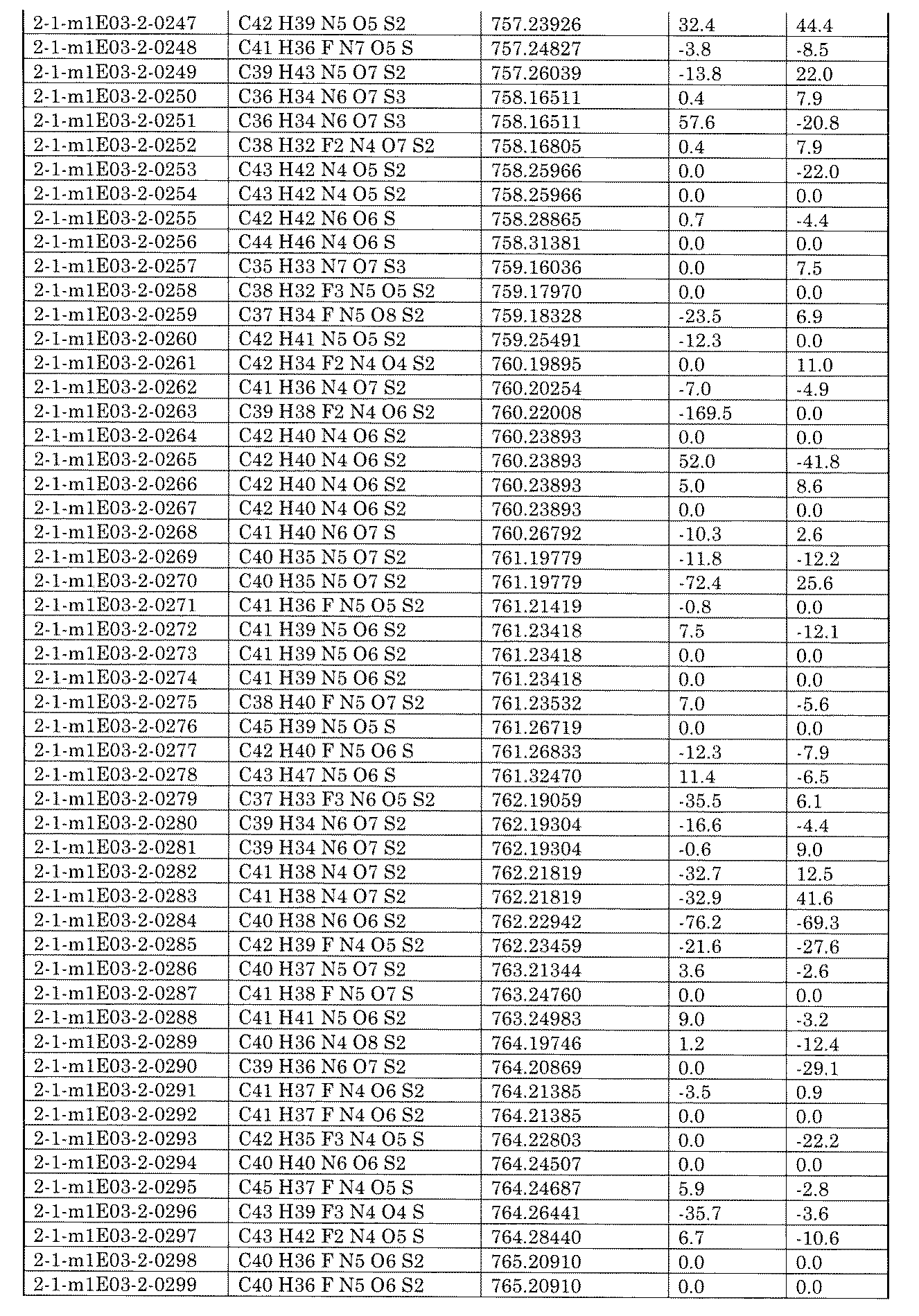









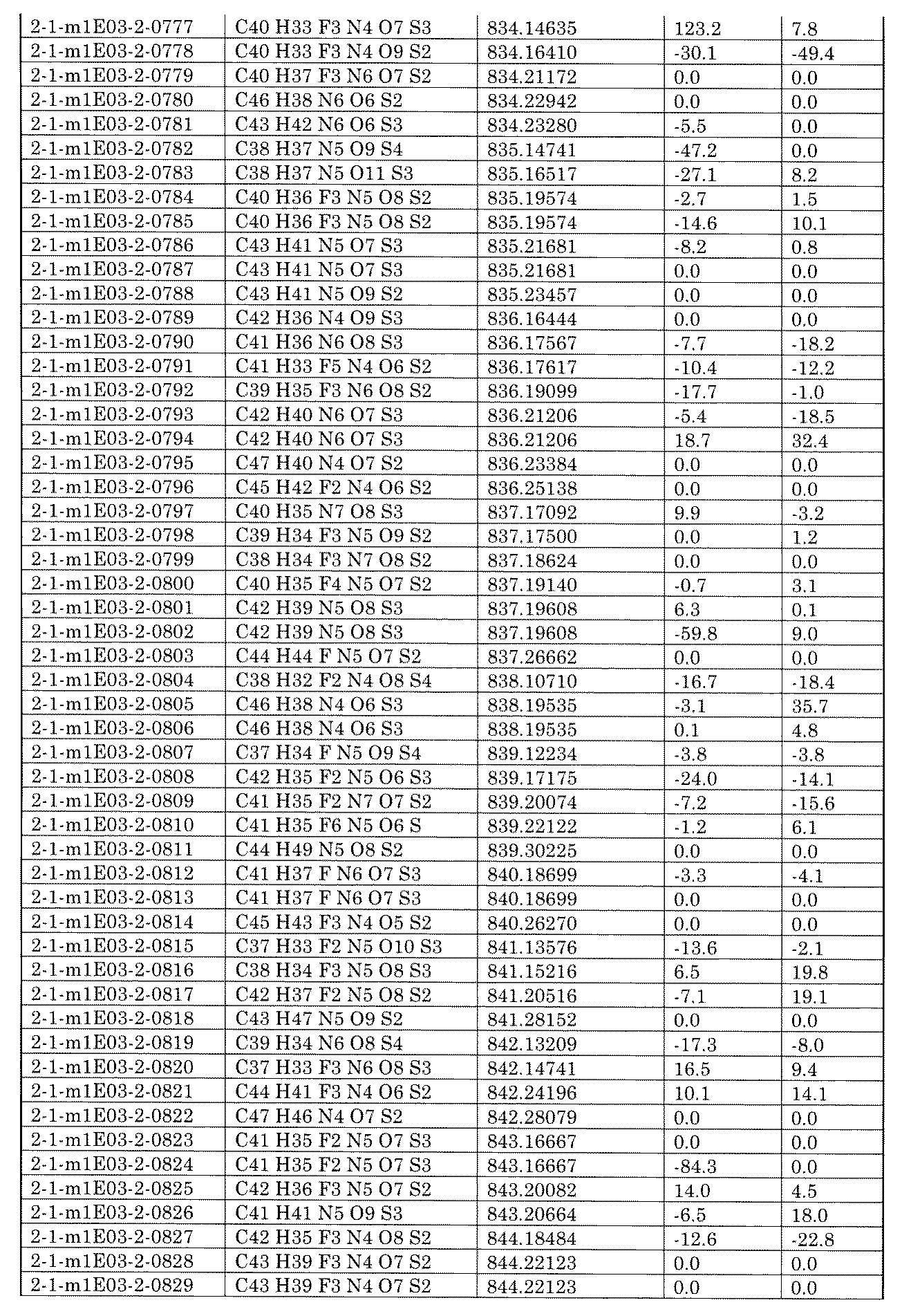












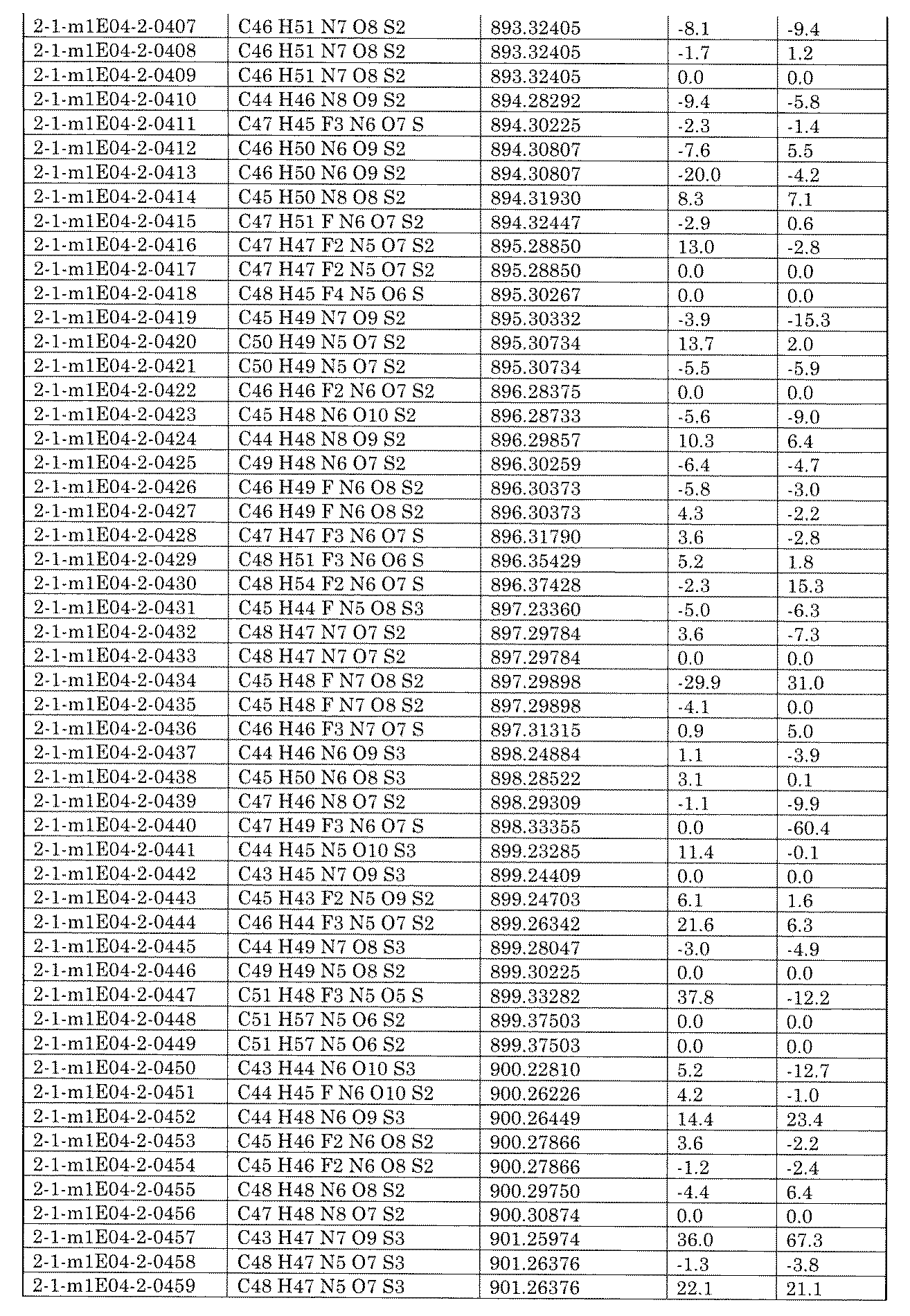

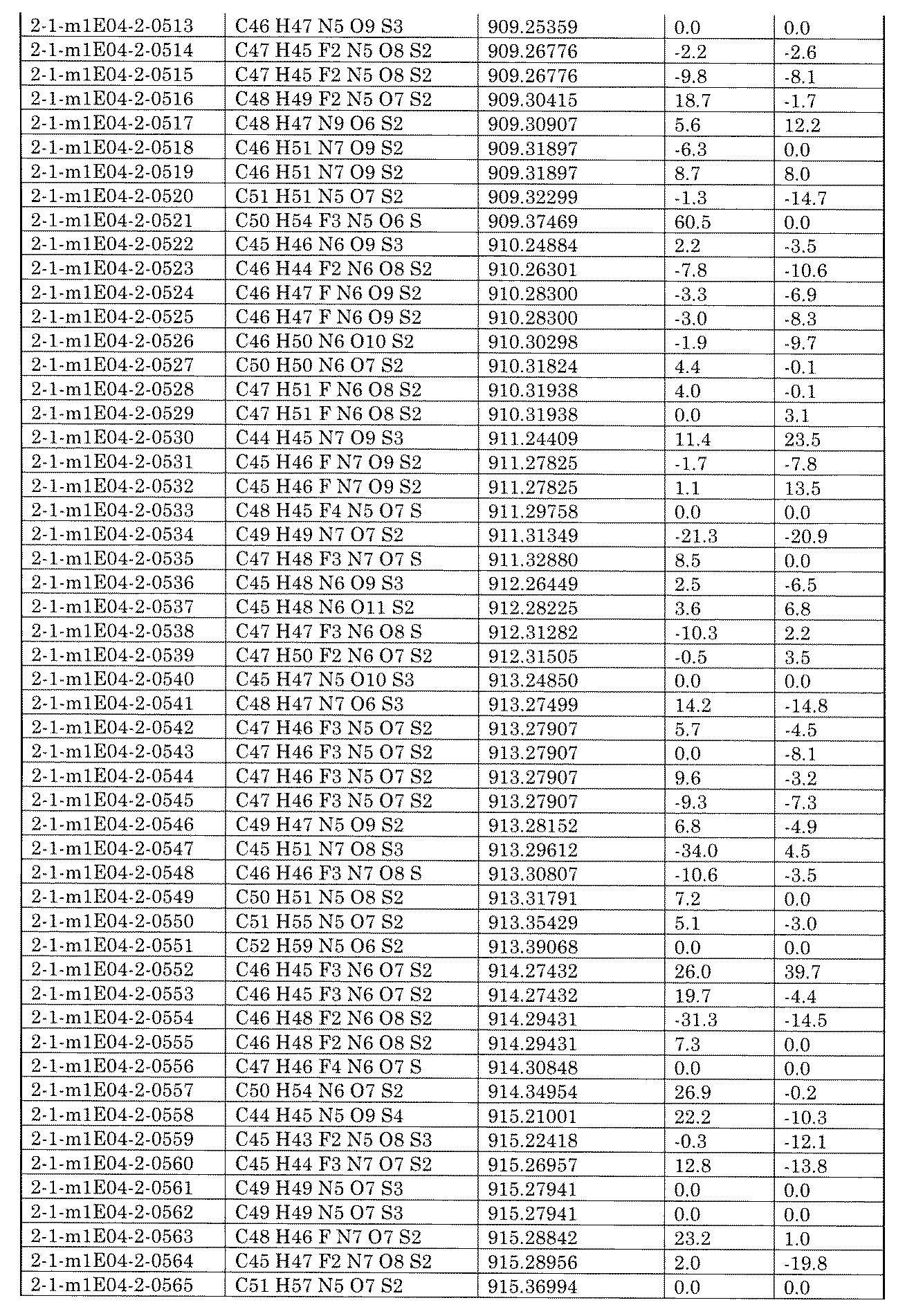



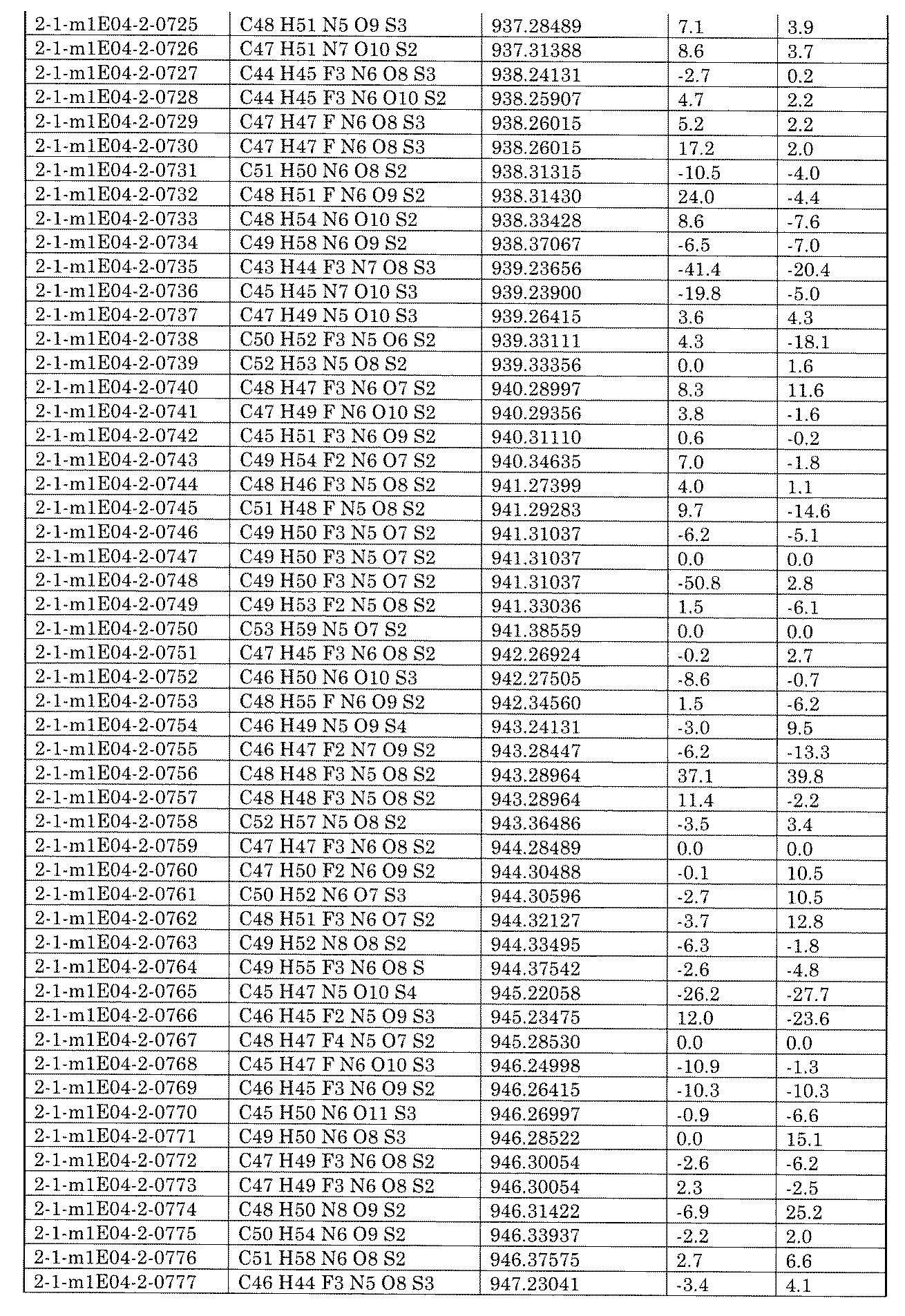







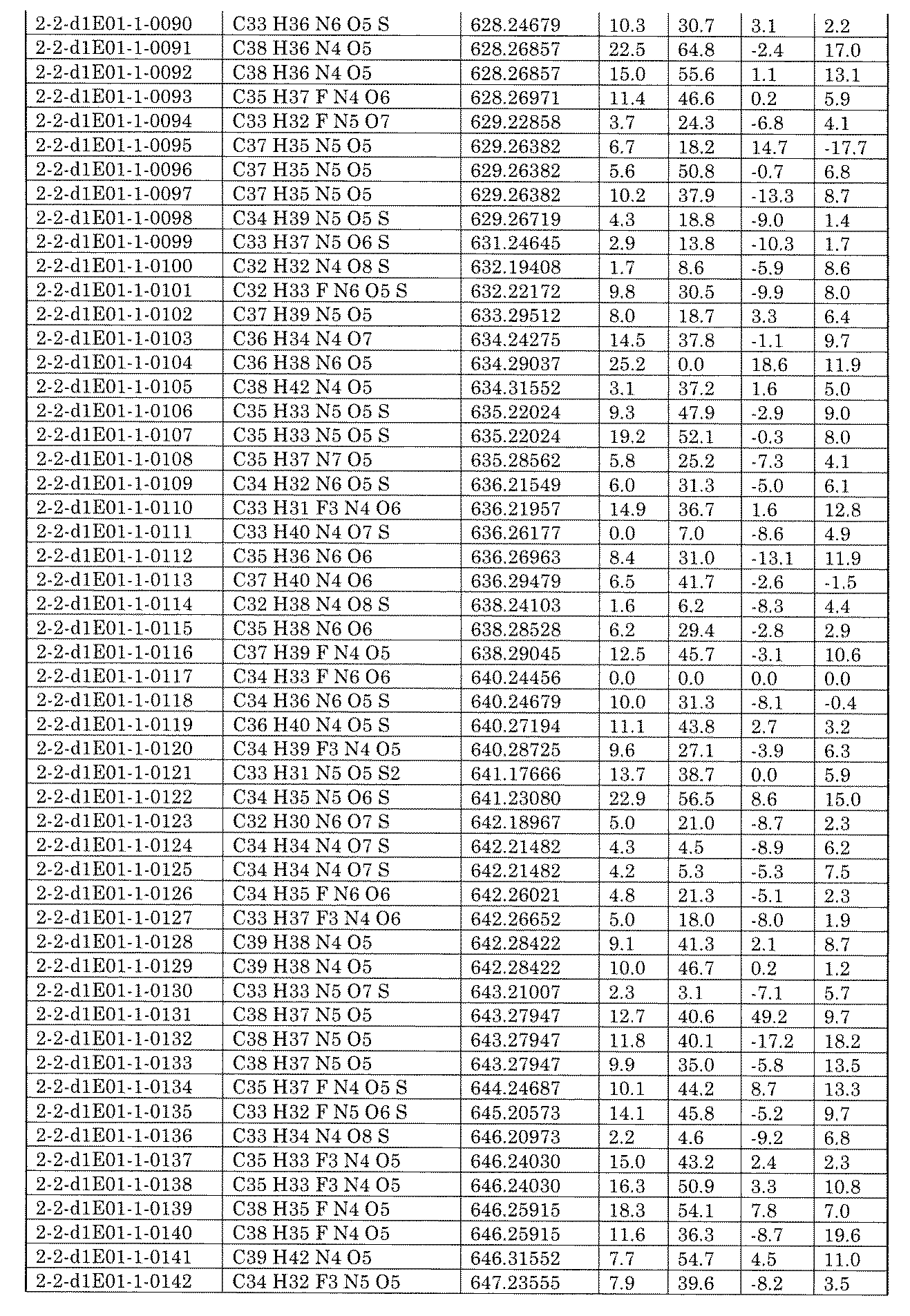














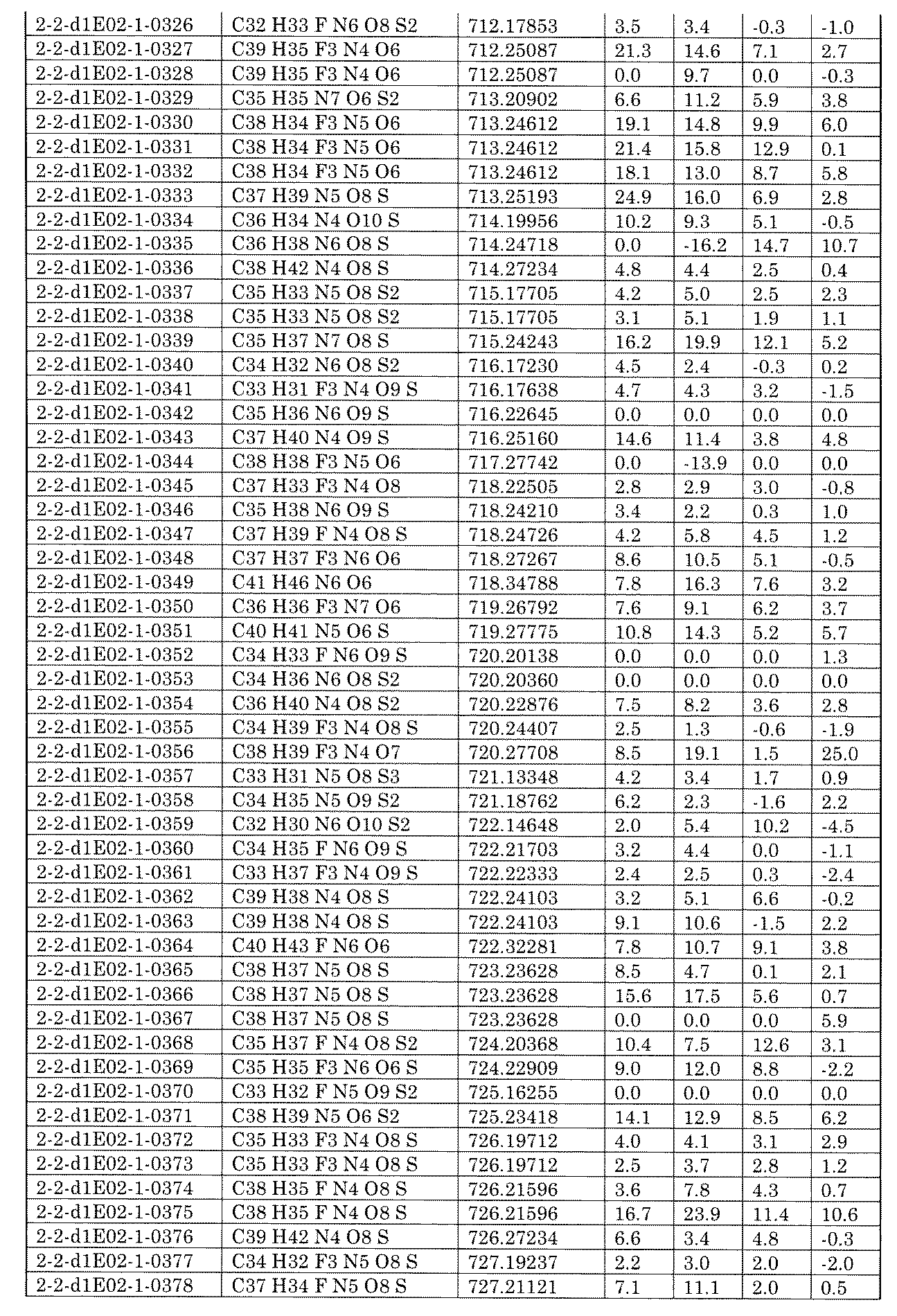

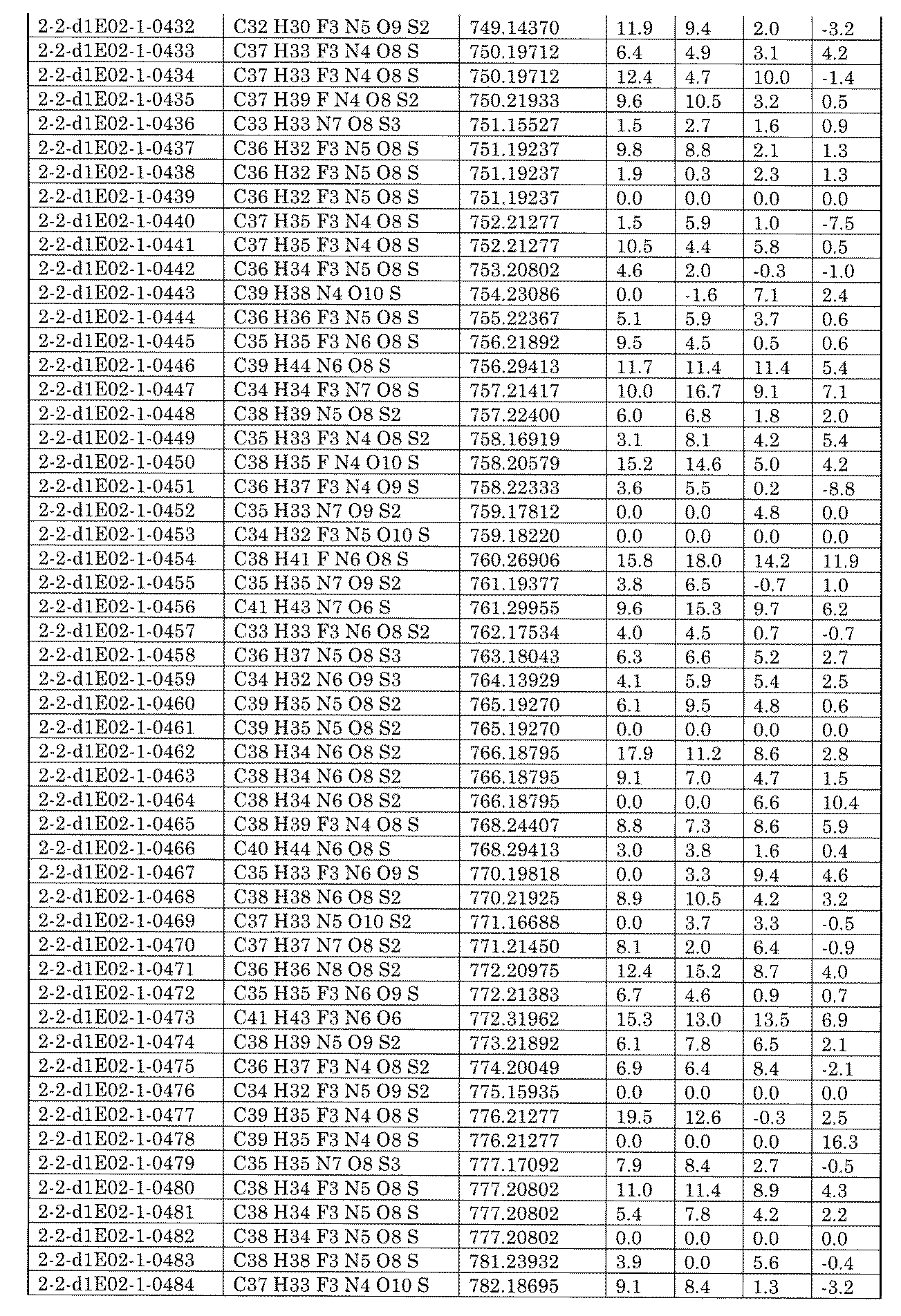





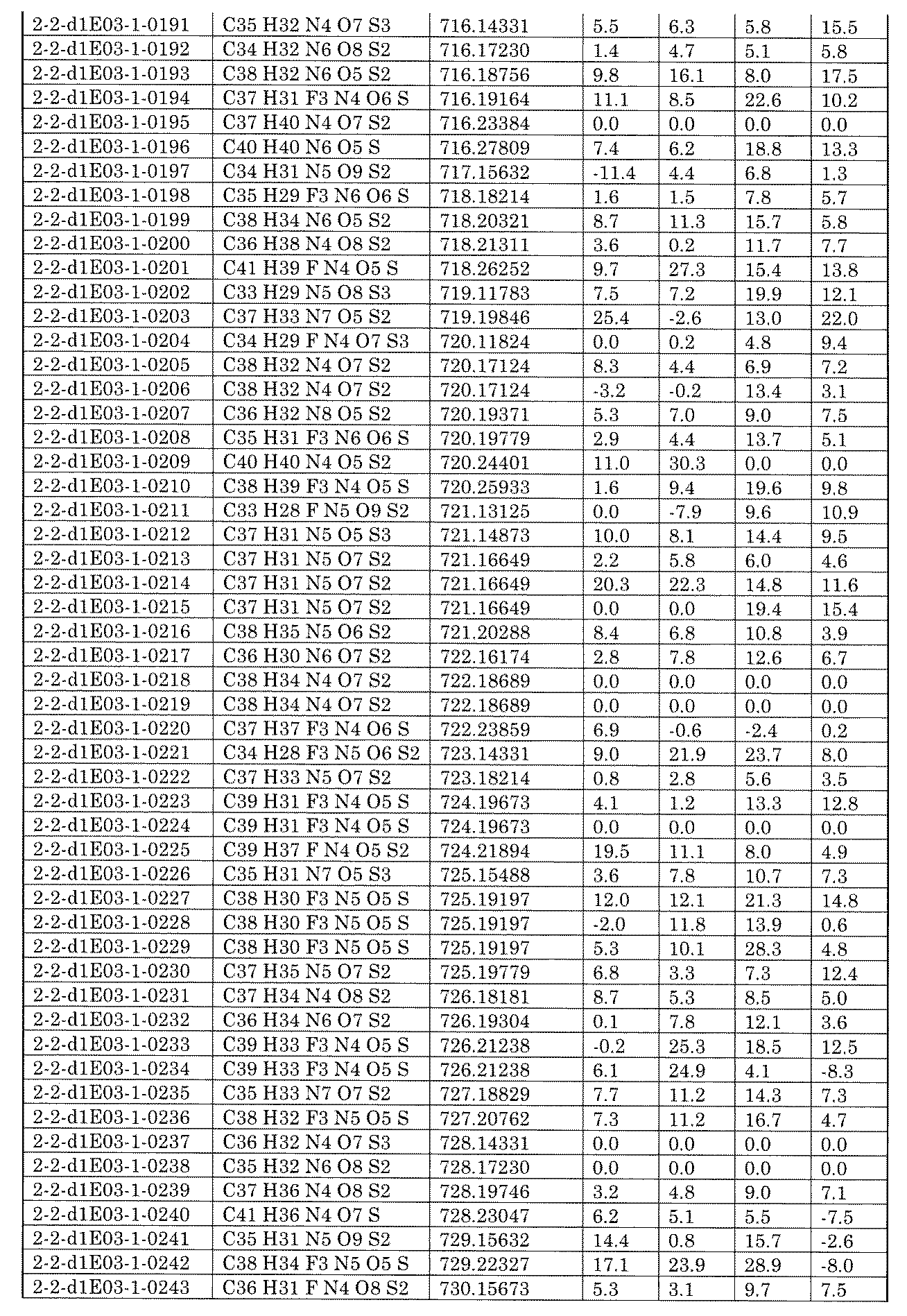
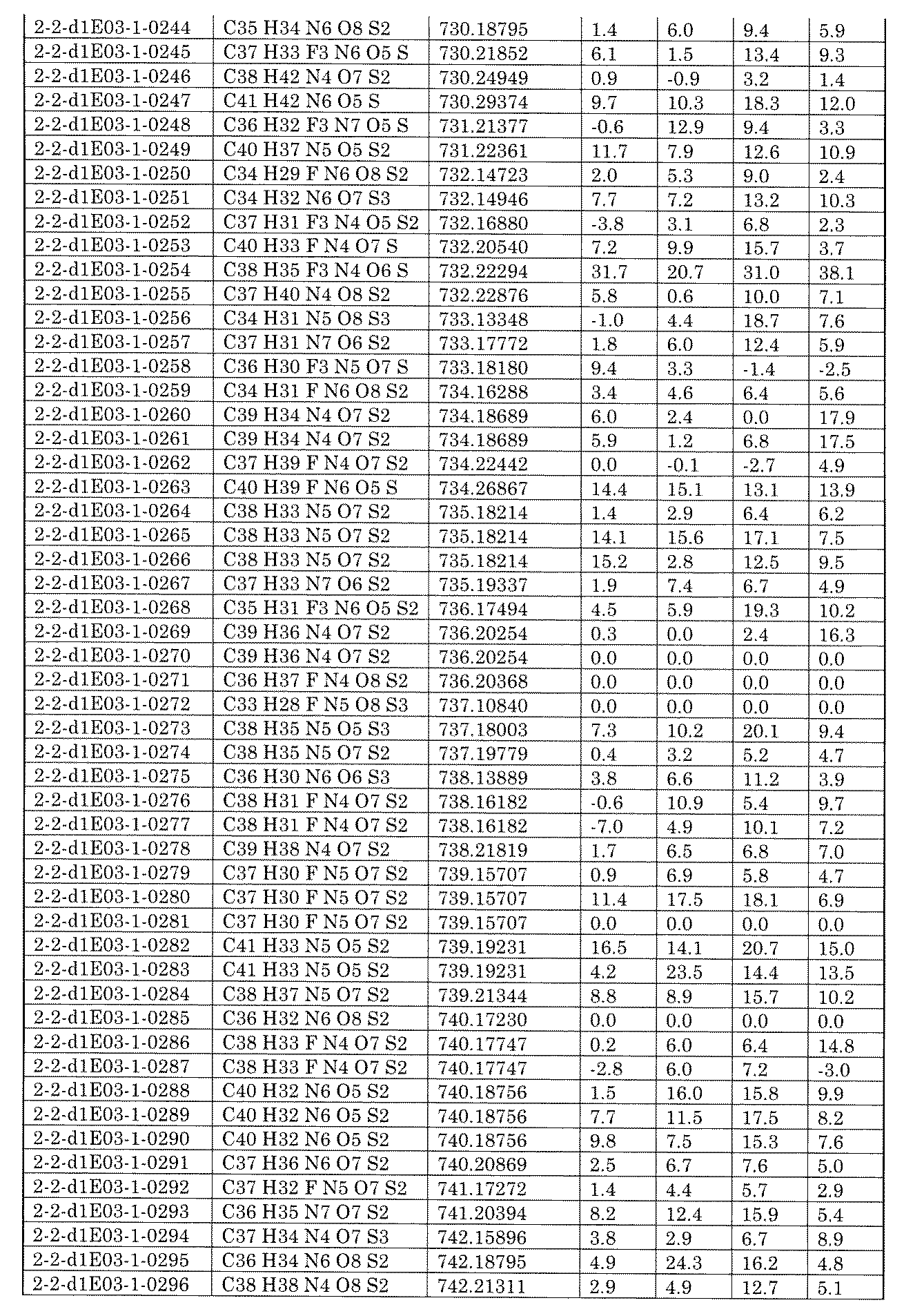
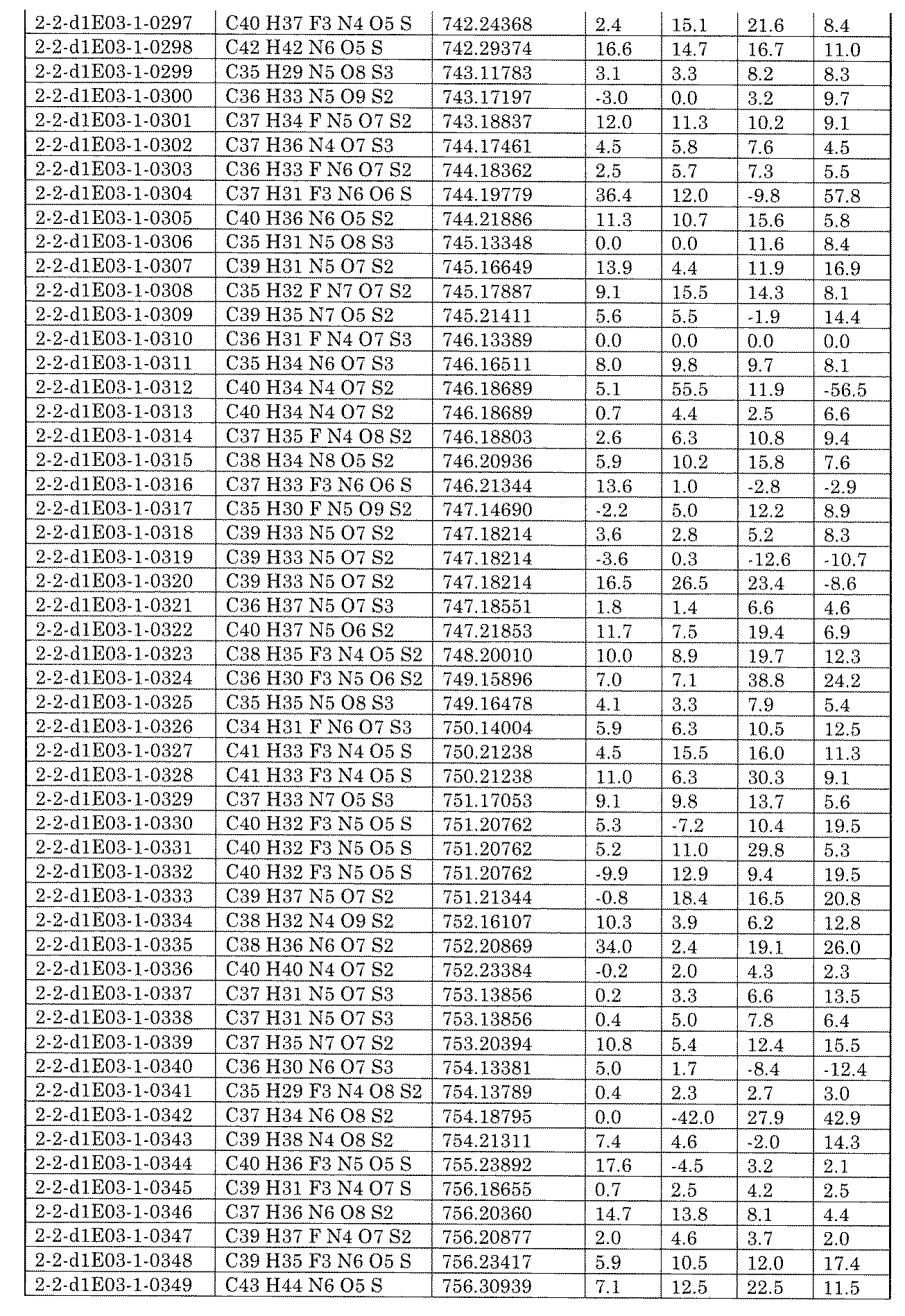




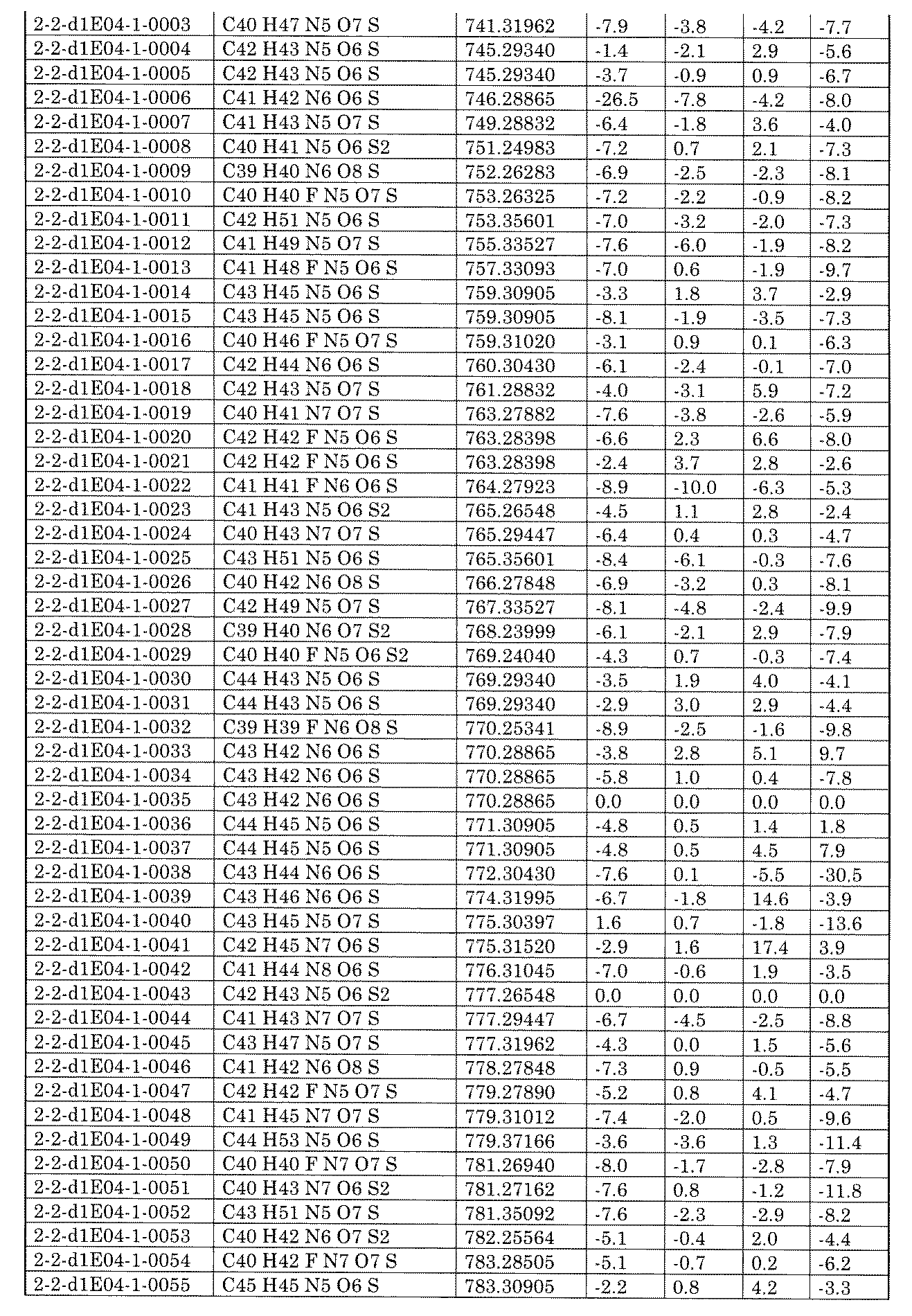



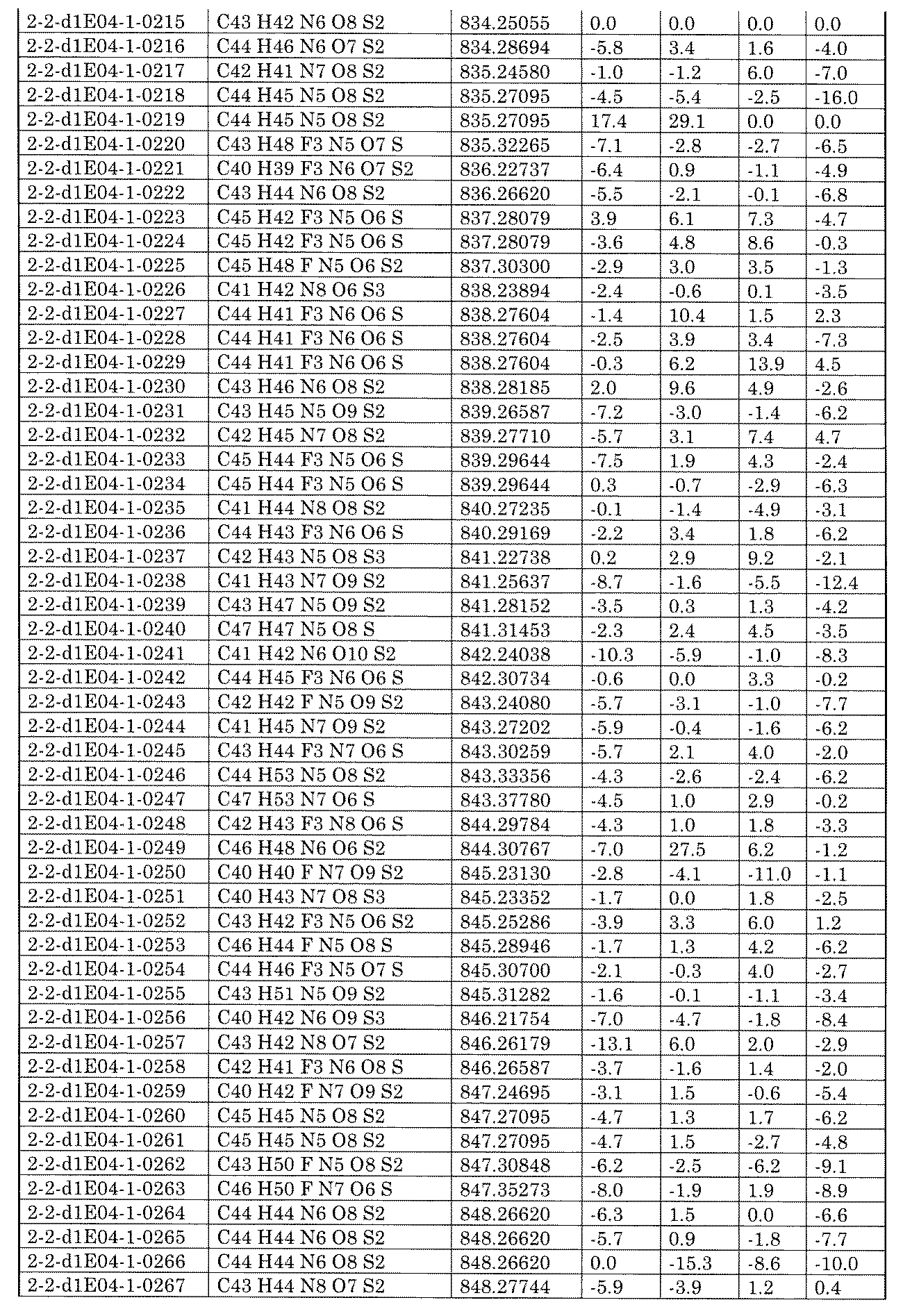







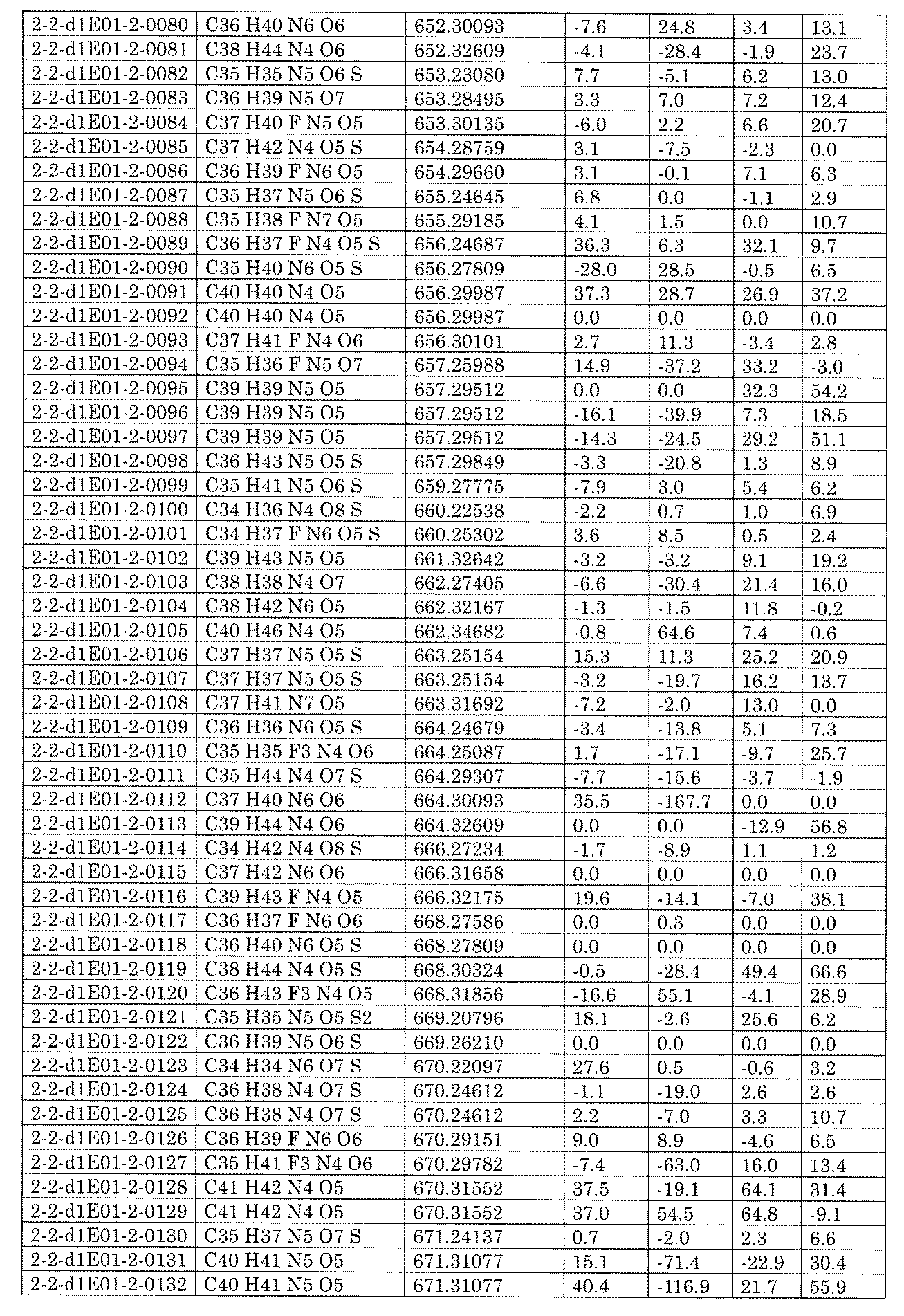



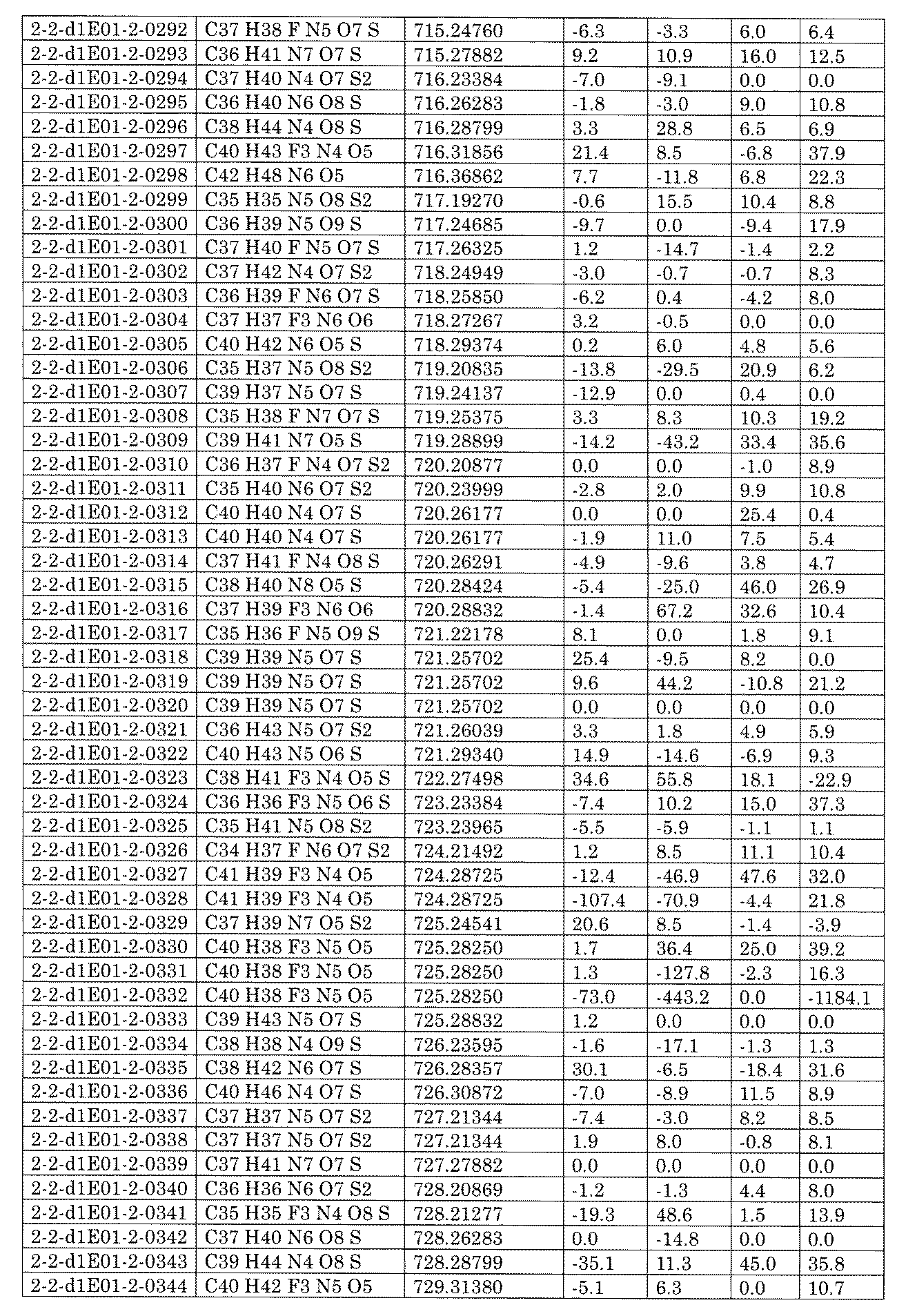


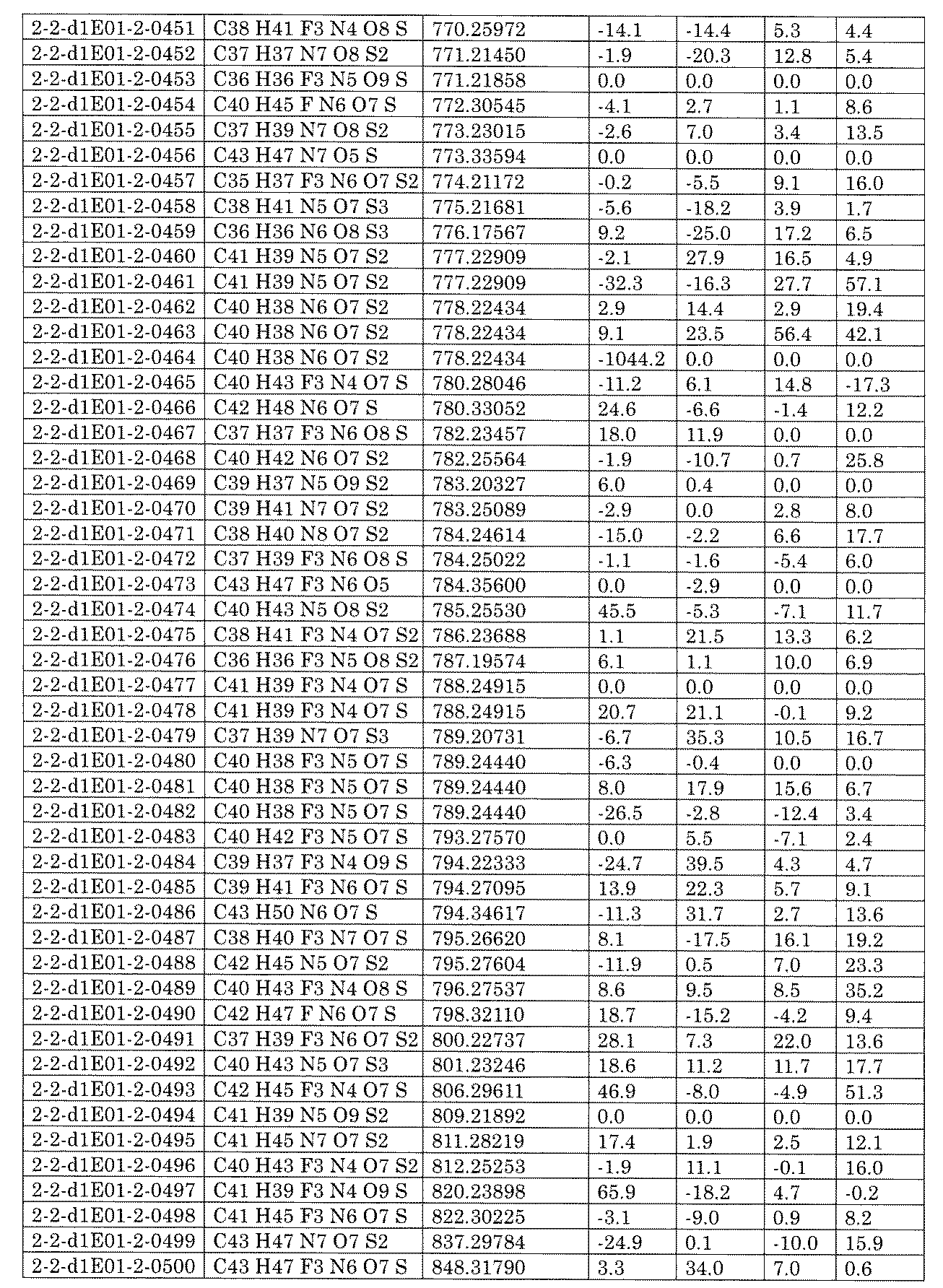



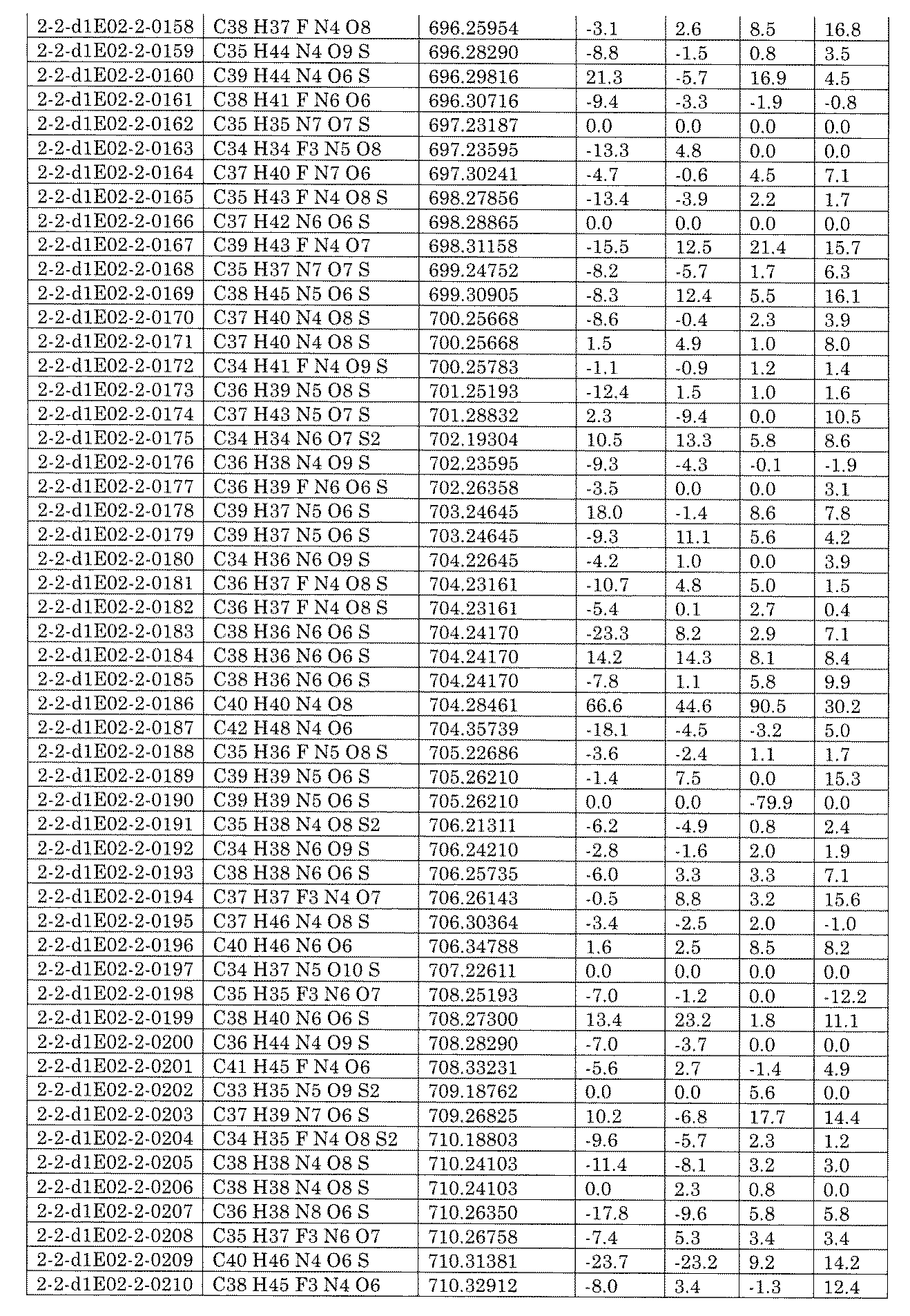

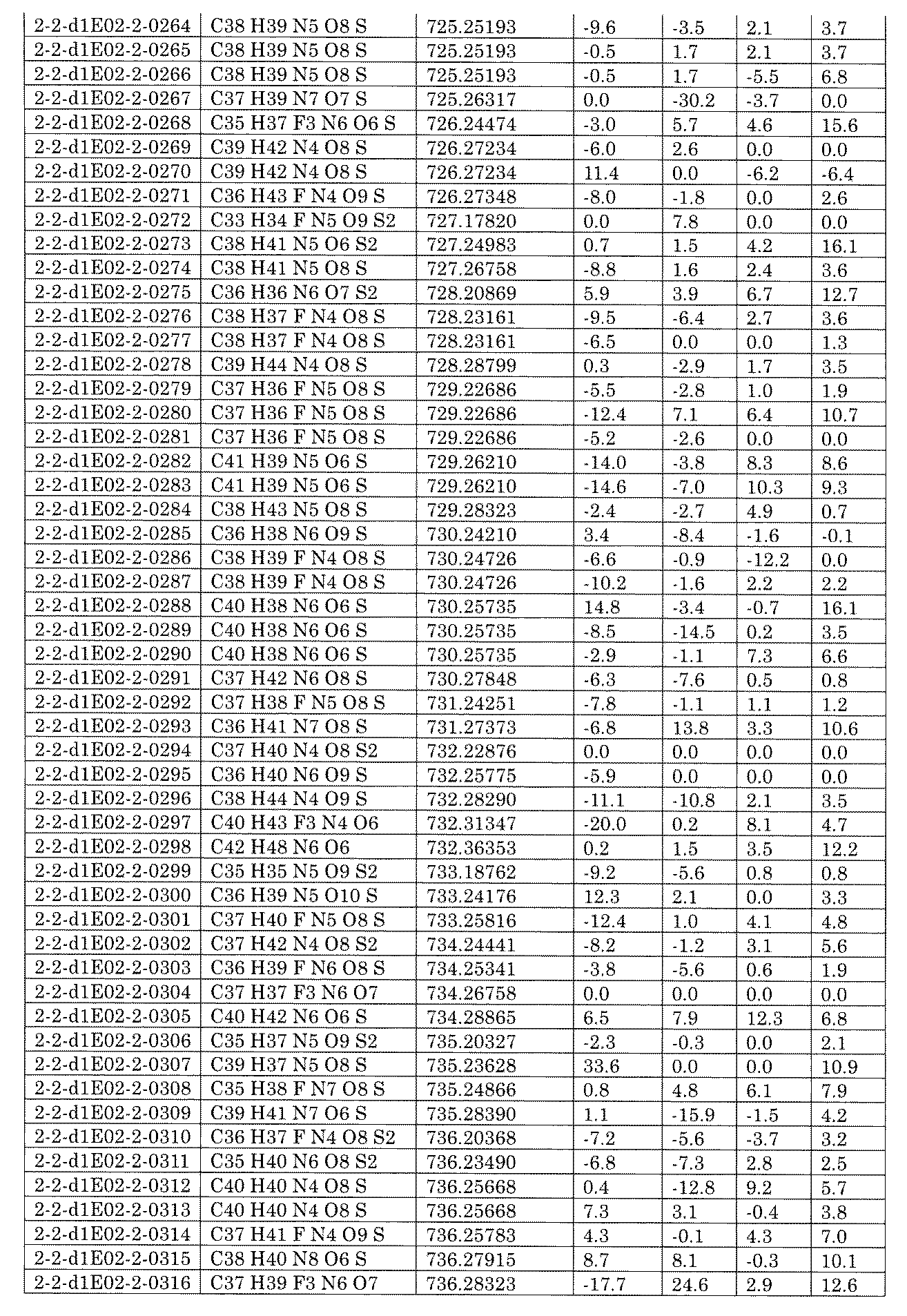

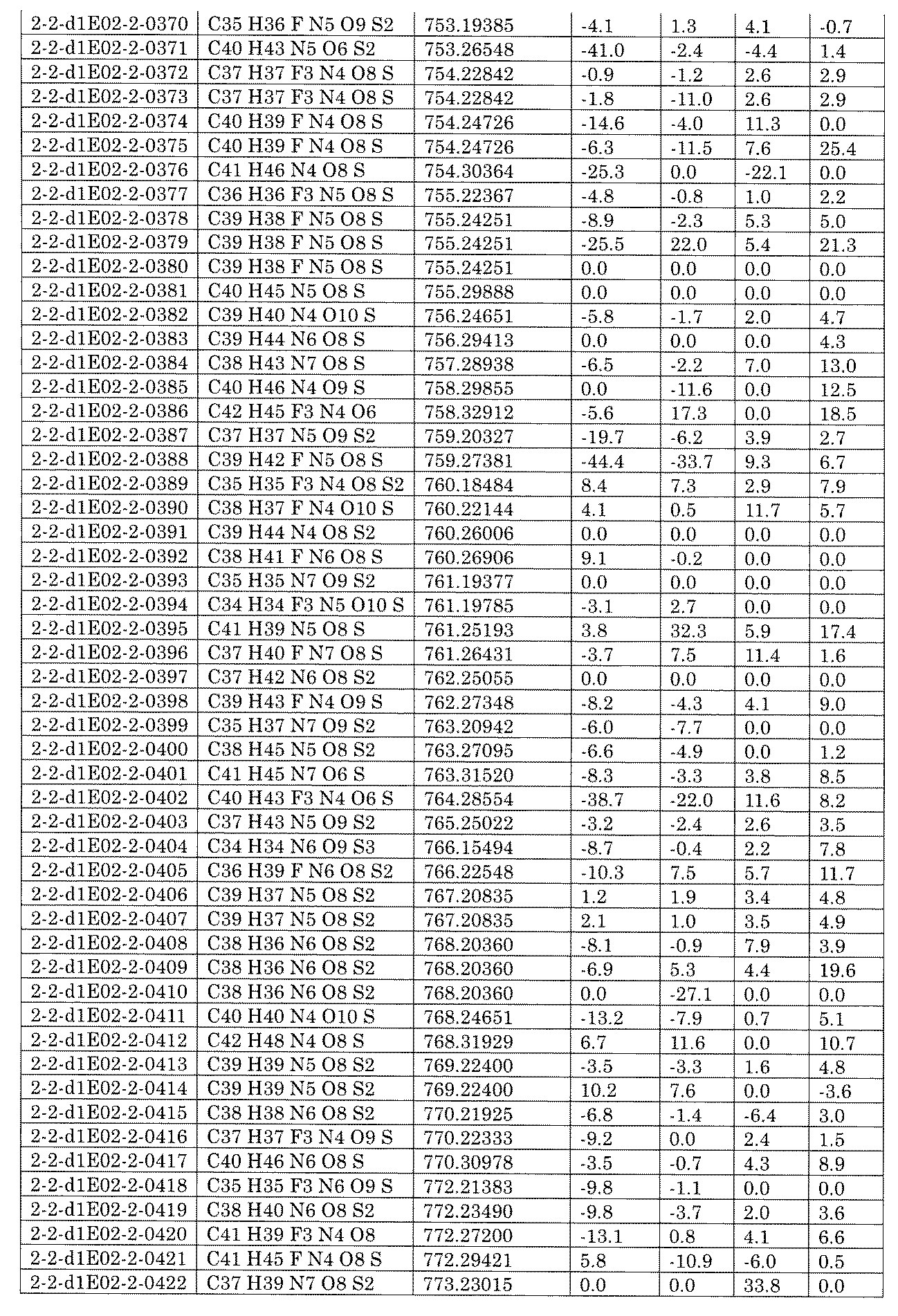













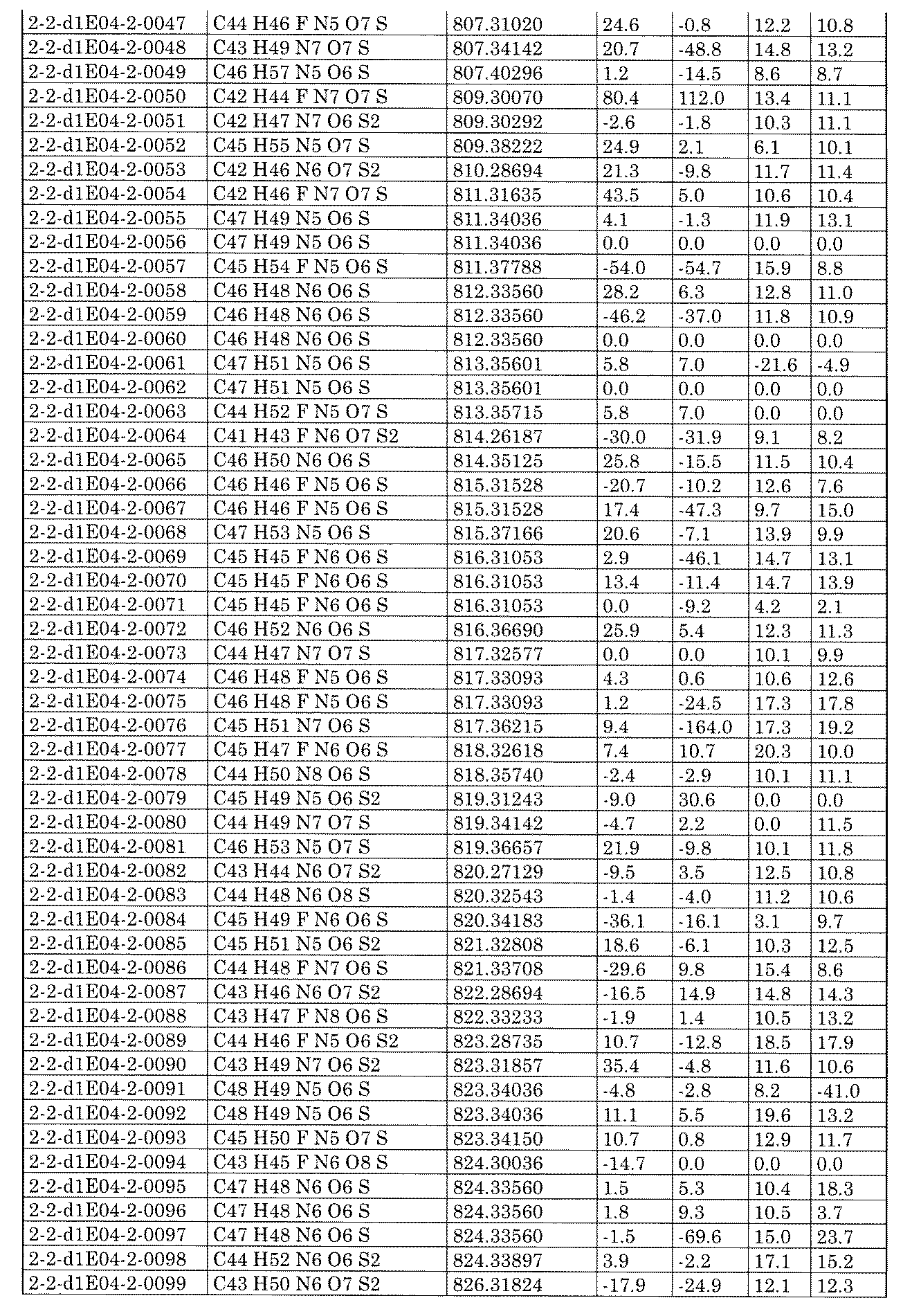
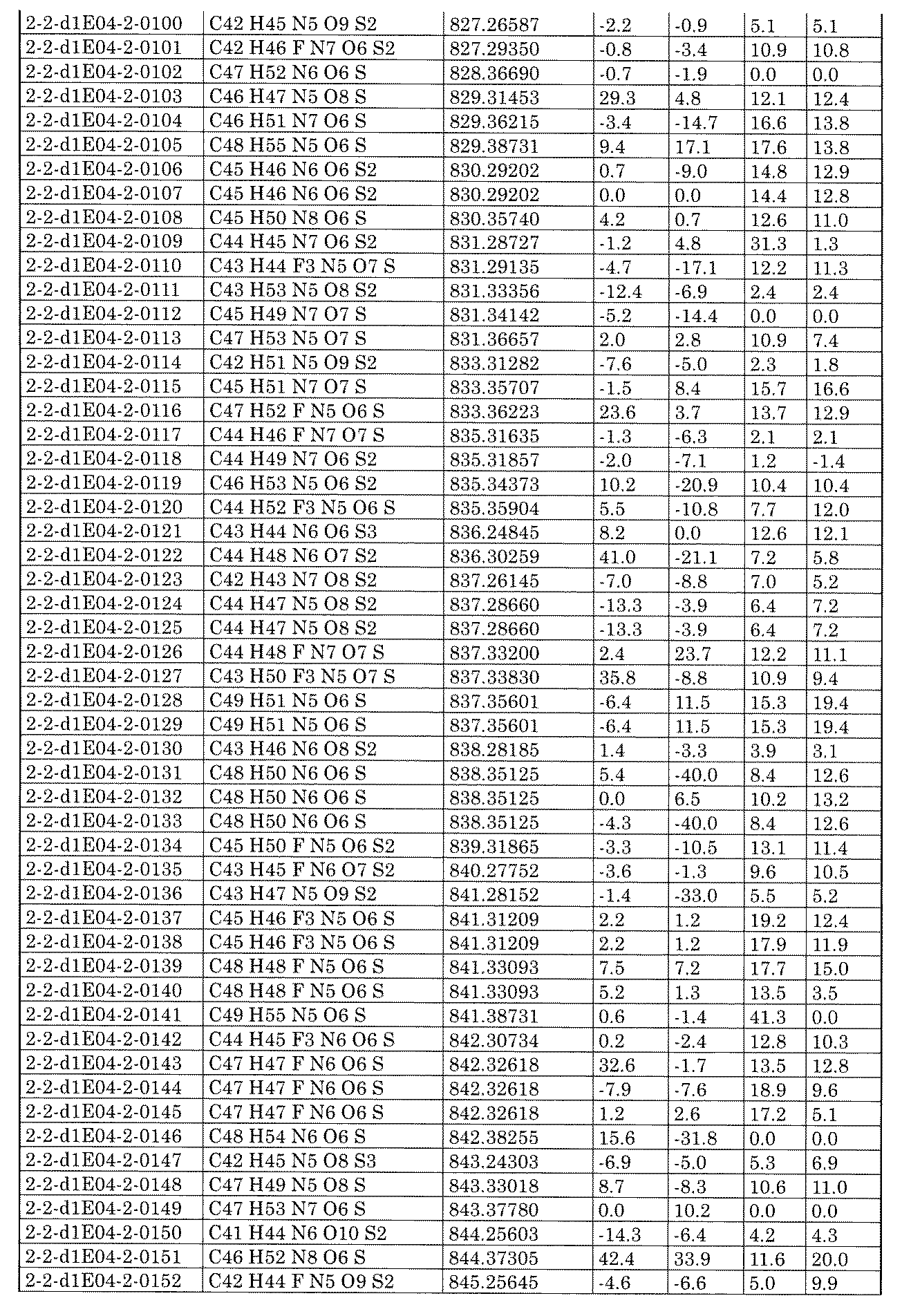



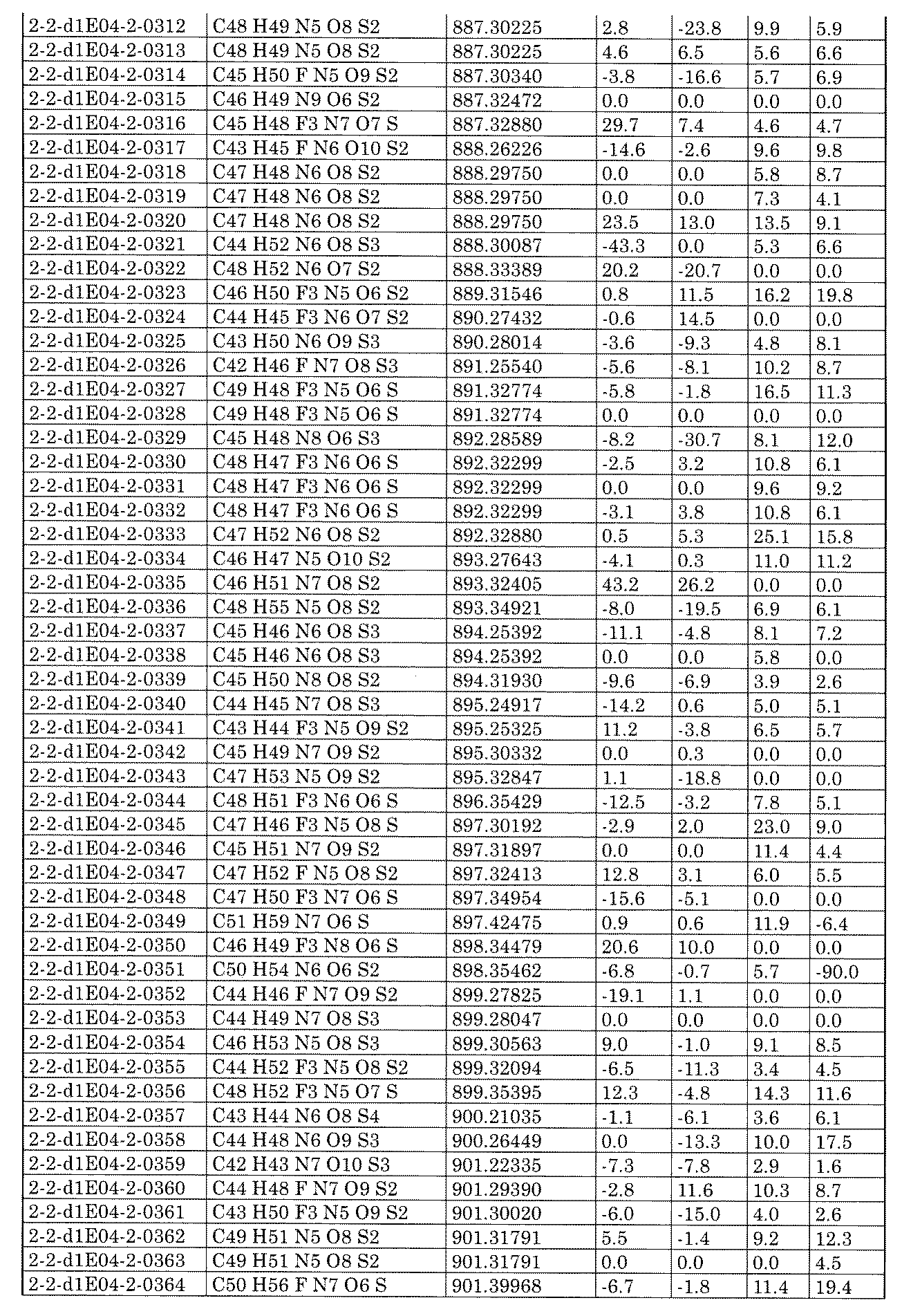

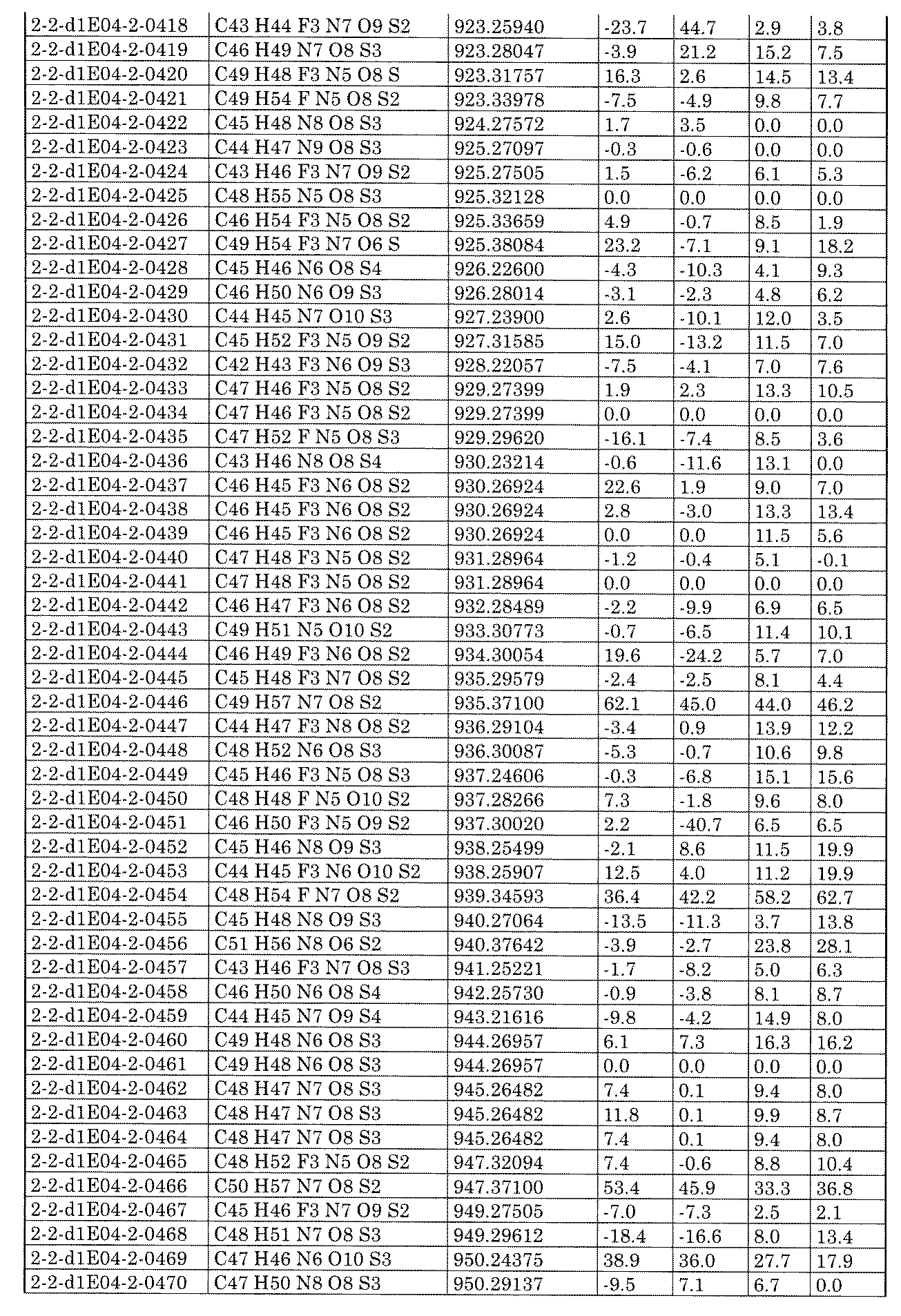

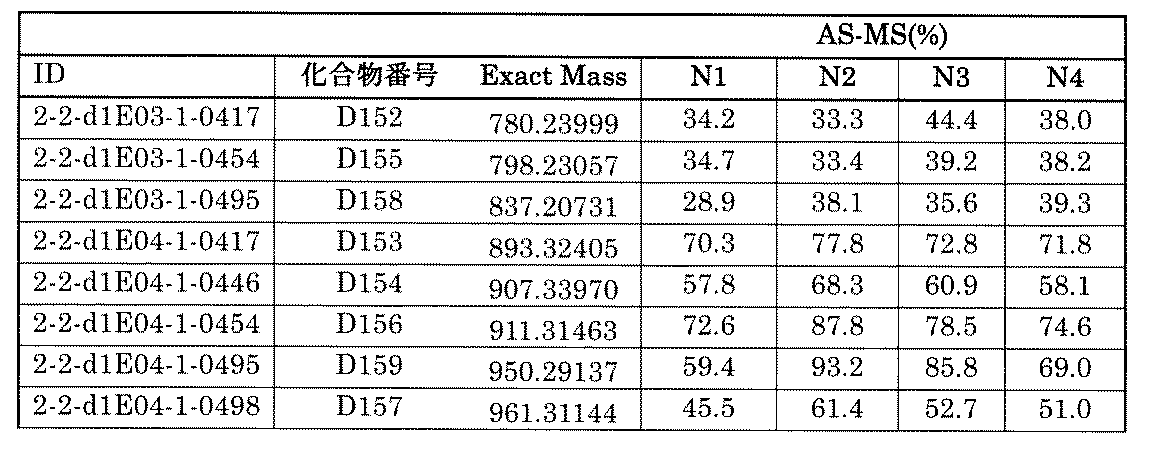

実施例12−1−1:D129−03Rの合成
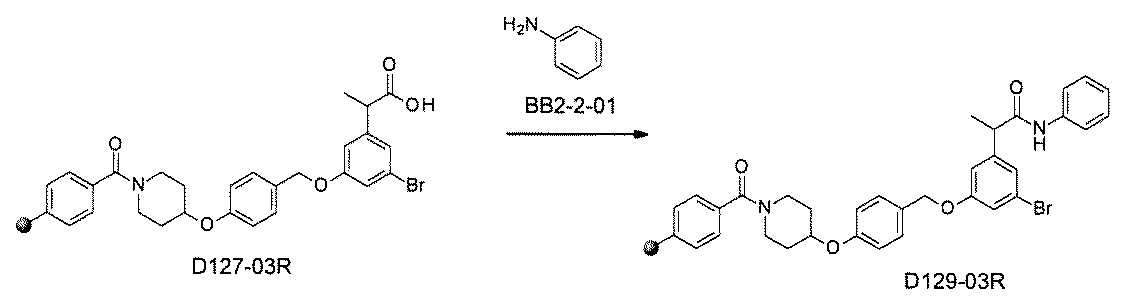
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(36mL)で3回、NMP(36mL)で3回、MeOH(36mL)で3回、DCM(36mL)で3回、ヘプタン(36mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、標題化合物D129−03R(2.07g、0.198mmol/g)を得た。
洗浄後の固相を少量フィルター上に移し、DMA(0.1mL)で3回、メタノール(0.1mL)で3回、DCM(0.1mL)で3回洗浄した後、0.05Mペンタメチルベンゼンを含む10%TFA/DCM溶液(0.05mL)に5分間浸した。ろ過後、フィルター上のレジンをDMA(0.05mL)で洗浄した。得られたろ液にアセトニトリル(0.10mL)を加え、LCサンプルを調製し、LCMS測定を行うことで反応進行度を測定した。その結果、目的物D129を98.5%の純度で得た。

LCMS:m/z 320、322[M+H]+
保持時間:1.027分(分析条件 SMD−FA05−1、280nm)。
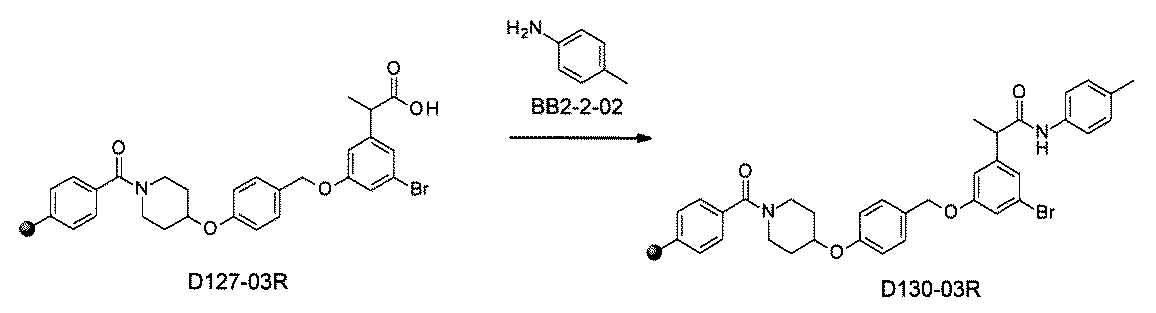

LCMS:m/z 334、336[M+H]+
保持時間:1.085分(分析条件 SMD−FA05−1、280nm)
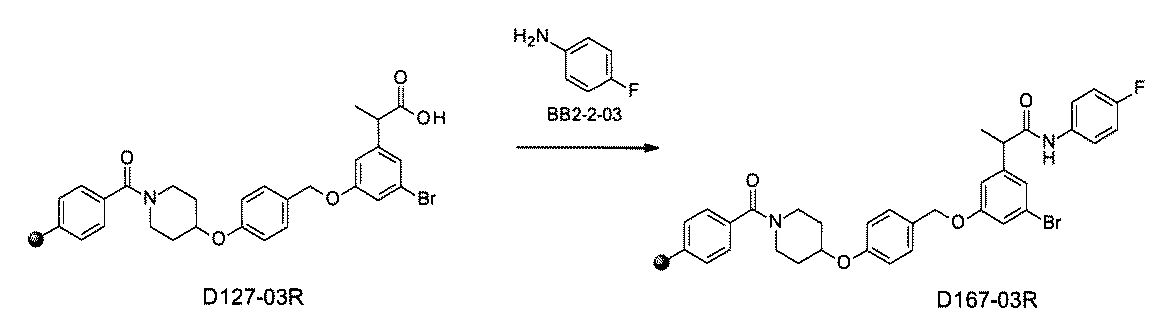

LCMS:m/z 338、340[M+H]+
保持時間:1.048分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 388,390[M+H]+
保持時間:1.187分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 377,379[M+H]+
保持時間:1.00分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 346,348[M+H]+
保持時間:1.052分(分析条件 SMD−FA05−1、290nm)

反応液と固相の懸濁液を6mLのフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.2M NAC in NMP/水=5/1(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、0.05M BTMG in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、標題化合物D134−03R(268.3mg、0.1922mmol/g)を得た。実施例12−1−1と同様の切り出し操作を行い、D134を88.5%の純度で得た。

LCMS:m/z 474[M+H]+
保持時間:1.191分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 488[M+H]+
保持時間:1.211分(分析条件 SMD−FA05−1、280nm)
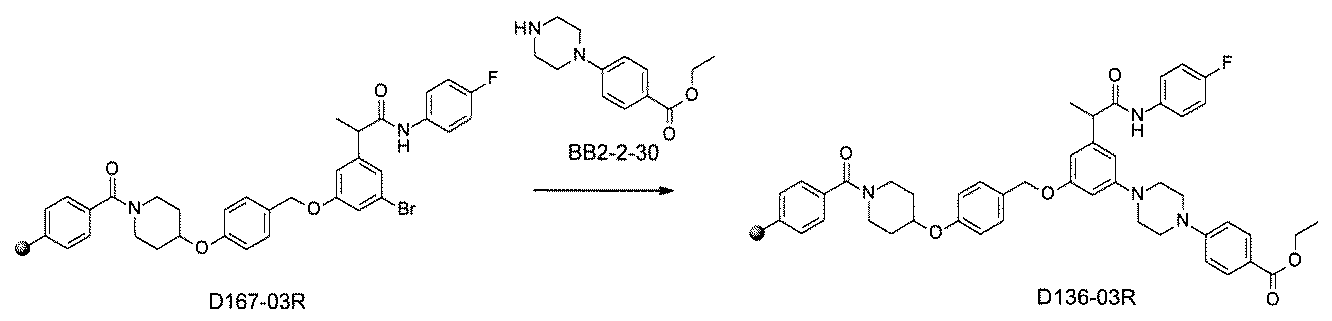

LCMS:m/z 492[M+H]+
保持時間:1.177分(分析条件 SMD−FA05−1、280nm)

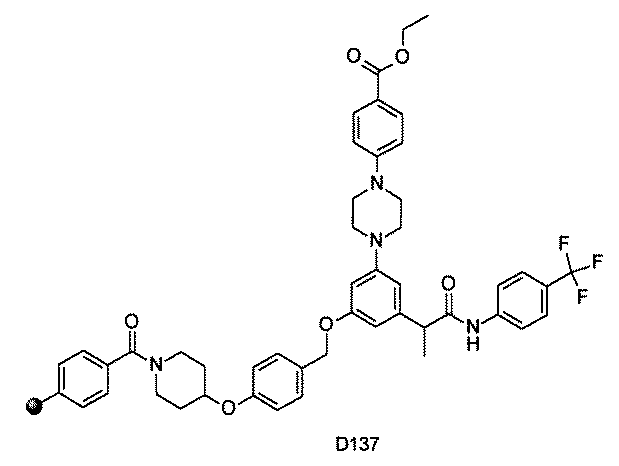
LCMS:m/z 542[M+H]+
保持時間:1.295分(分析条件 SMD−FA05−1、280nm)

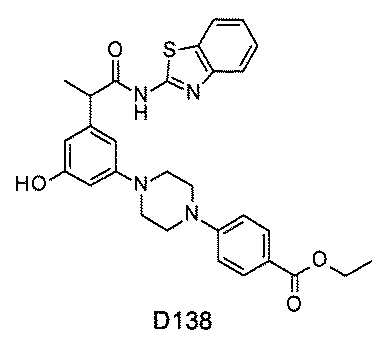
LCMS:m/z 531[M+H]+
保持時間:1.232分(分析条件 SMD−FA05−1、290nm)


LCMS:m/z 500[M+H]+
保持時間:1.163分(分析条件 SMD−FA05−1、280nm)
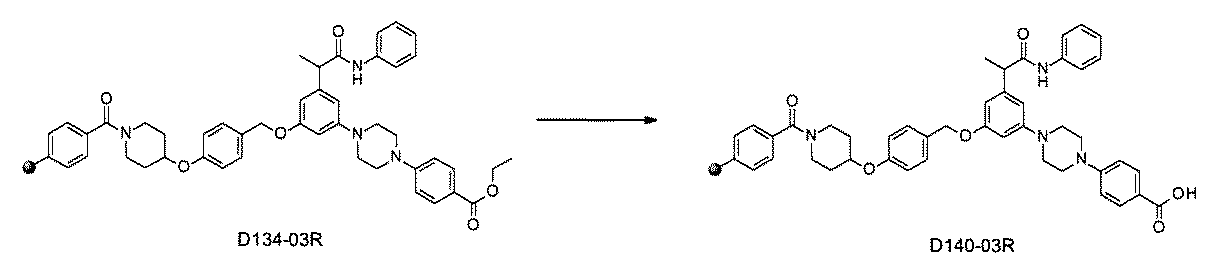
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄し、得られた固相を減圧下で乾燥させ、標題化合物D140−03R(246.8mg、0.1932mmol/g)を得た。実施例12−1−1と同様の切り出し操作を行い、D140を87.4%の純度で得た。

LCMS:m/z 446[M+H]+
保持時間:0.909分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 460[M+H]+
保持時間:0.964分(分析条件 SMD−FA05−1、280nm)

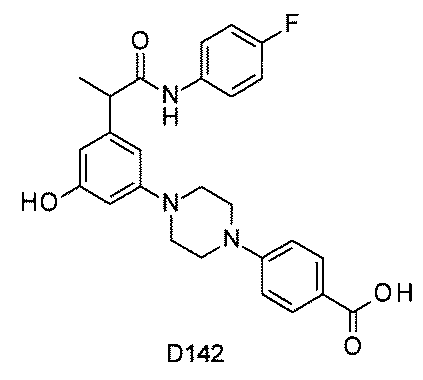
LCMS:m/z 464[M+H]+
保持時間:0.933分(分析条件 SMD−FA05−1、280nm)

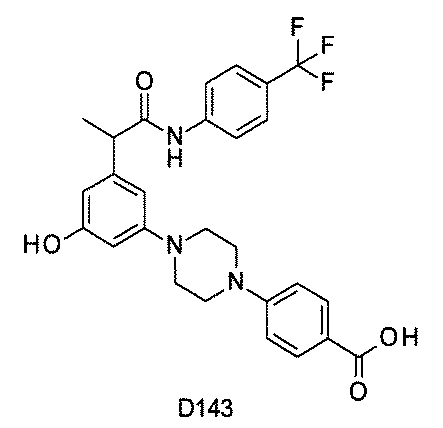
LCMS:m/z 514[M+H]+
保持時間:1.059分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 503[M+H]+
保持時間:0.991分(分析条件 SMD−FA05−1、280nm)
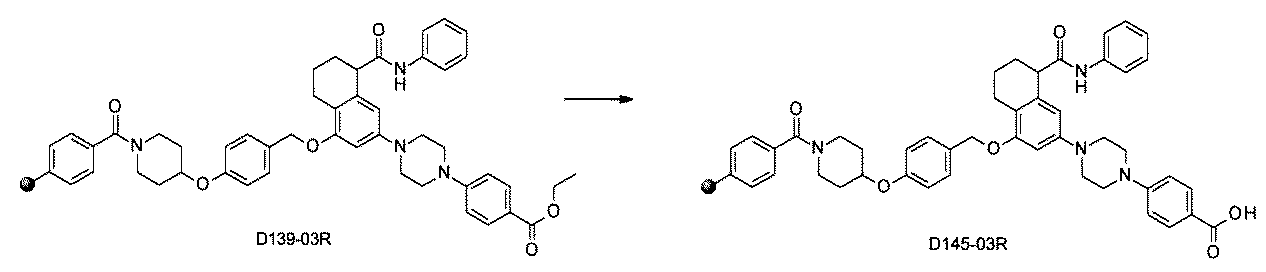

LCMS:m/z 472[M+H]+
保持時間:0.920分(分析条件 SMD−FA05−1、280nm)
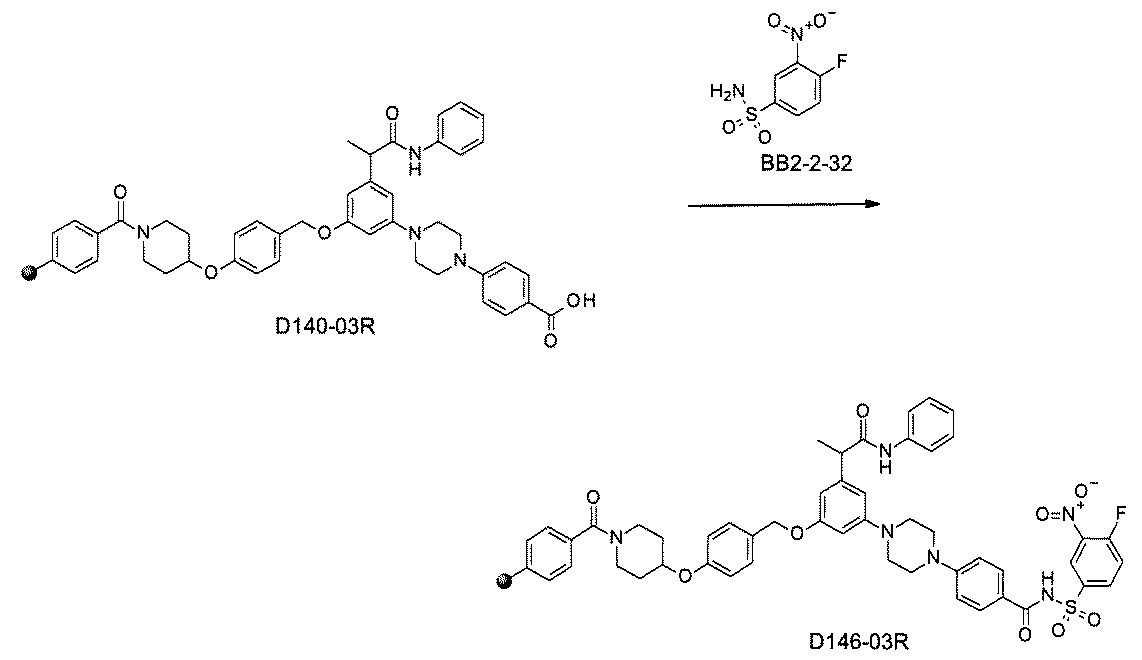
反応液と固相の懸濁液をカラムのフィルターでろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(14mL)で3回、ヘプタン(5mL)で3回洗浄した。得られた固相を減圧下で乾燥させ、標題化合物D146−03R(272.3mg、0.1859mmol/g)を得た。実施例12−1−1と同様の切り出し操作を行い、D146を69.4%の純度で得た。

LCMS:m/z 648[M+H]+
保持時間:1.123分(分析条件 SMD−FA05−1、280nm)

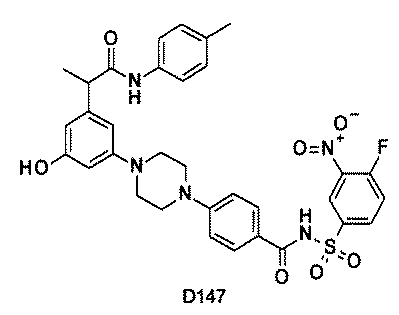
LCMS:m/z 662[M+H]+
保持時間:1.163分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 666[M+H]+
保持時間:1.136分(分析条件 SMD−FA05−1、280nm)
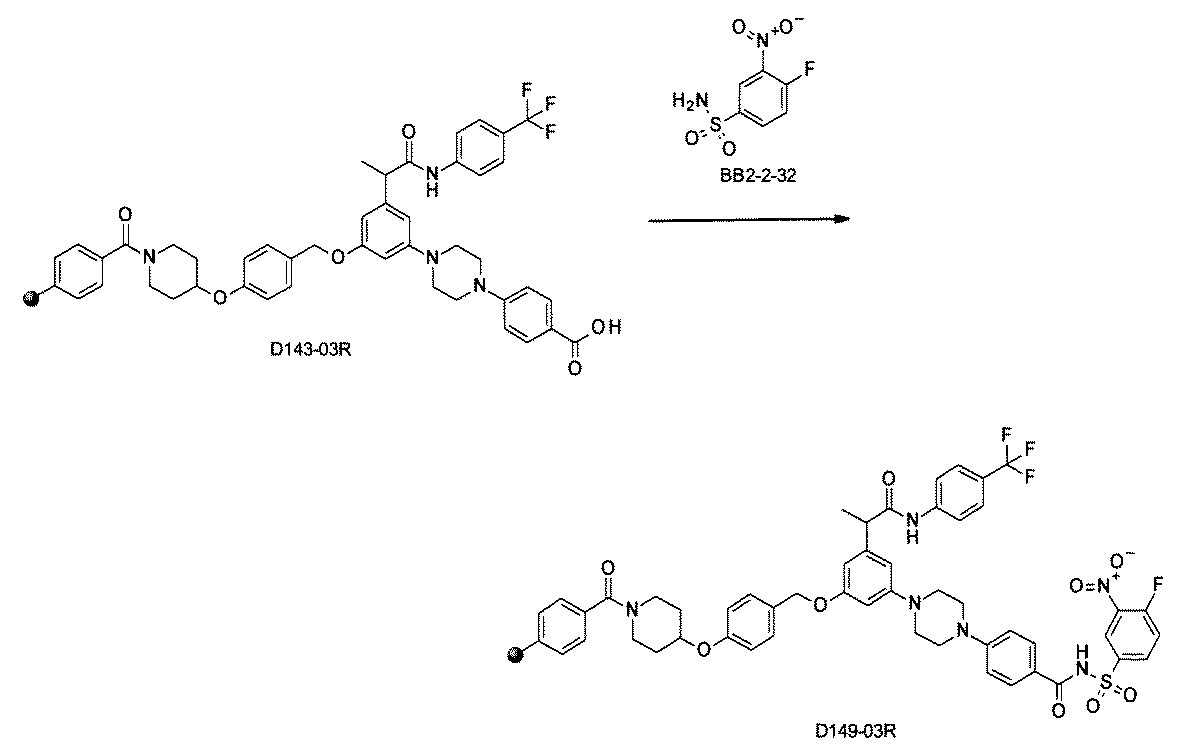
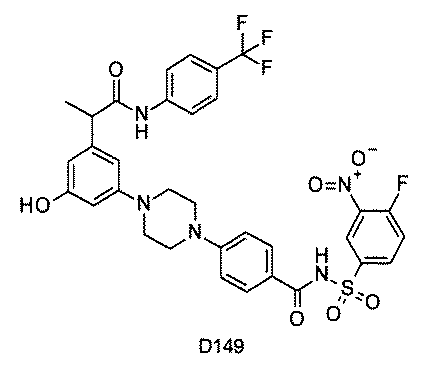
LCMS:m/z 716[M+H]+
保持時間:1.235分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 705[M+H]+
保持時間:1.179分(分析条件 SMD−FA05−1、290nm)
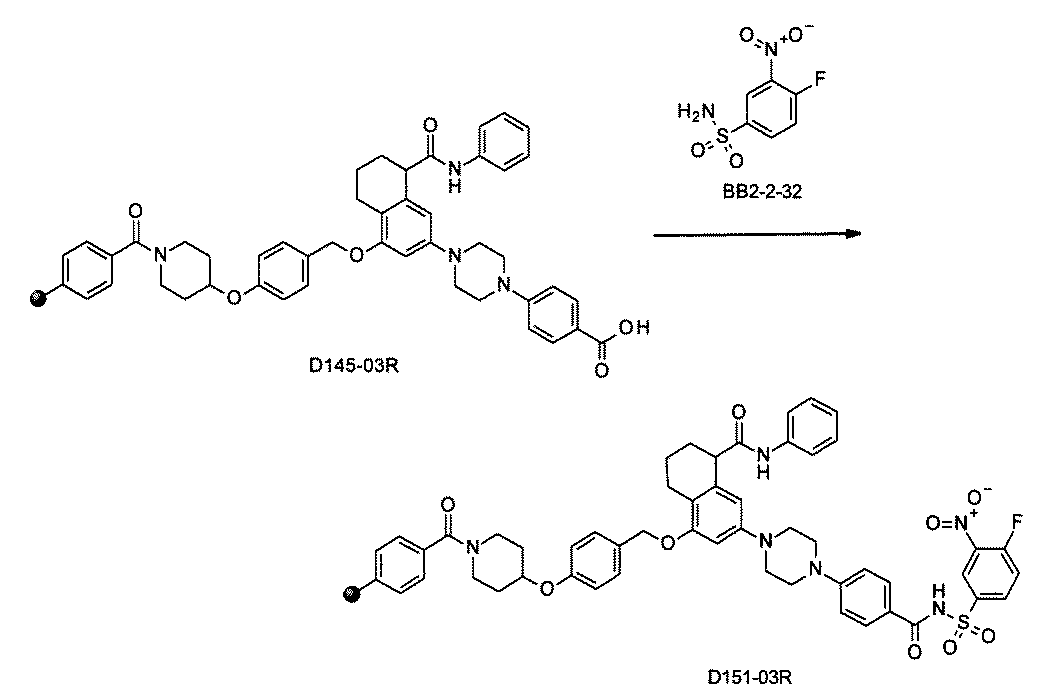

LCMS:m/z 674[M+H]+
保持時間:1.132分(分析条件 SMD−FA05−1、280nm)
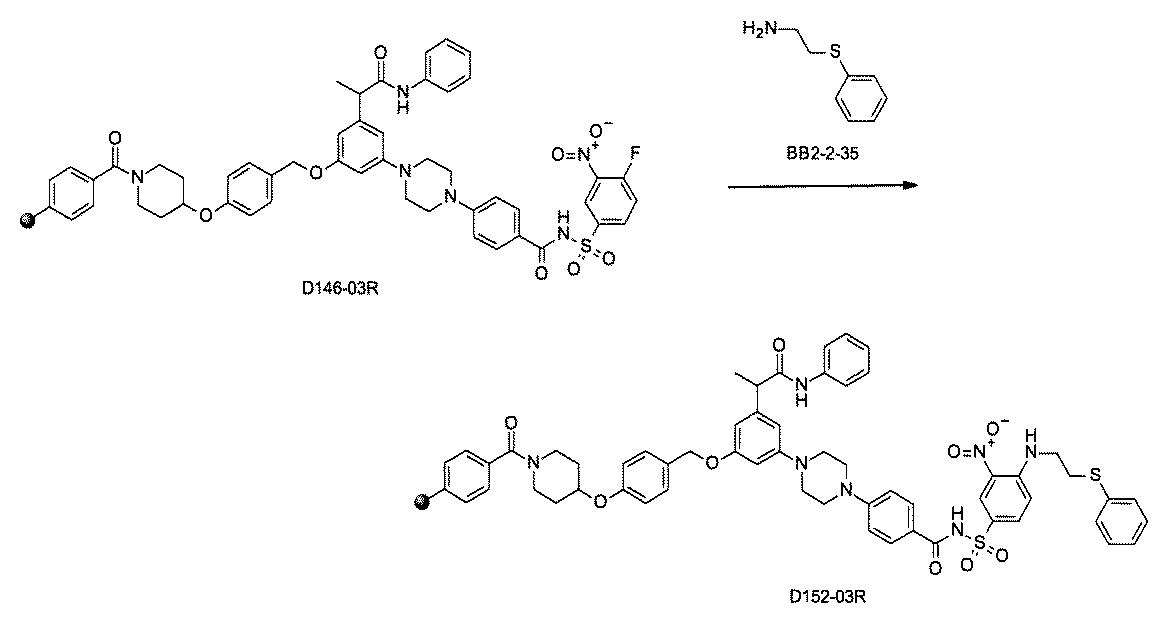
反応液と固相の懸濁液をフィルター付きカラムに移してろ過し、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、0.05M TBAHSO4+DTBP in NMP(5mL)で3回、NMP/水=1/1(5mL)で3回、NMP(5mL)で3回、MeOH(5mL)で3回、DCM(5mL)で3回、ヘプタン(5mL)で3回洗浄した。得られた固相を減圧下で乾燥させ、標題化合物D152−03R(251.3mg、0.181mmol/g)を得た。実施例12−1−1と同様の切り出し操作を行い、D152を81.1%の純度で得た。

LCMS:m/z 781[M+H]+
保持時間:1.265分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 894[M+H]+
保持時間:0.921分(分析条件 SMD−FA05−1、280nm)
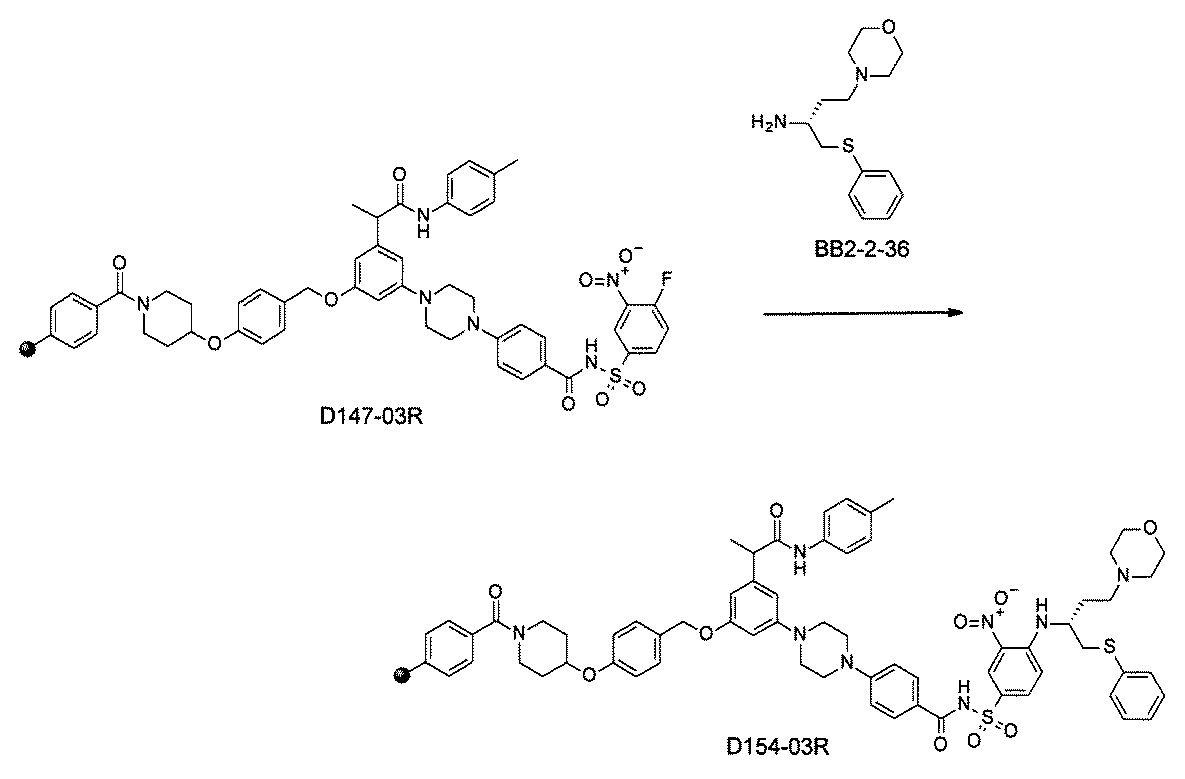
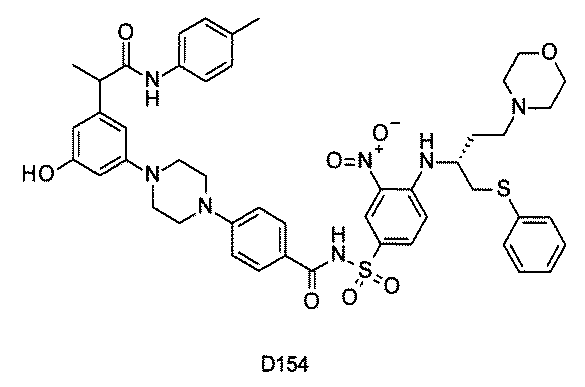
LCMS:m/z 908[M+H]+
保持時間:0.939分(分析条件 SMD−FA05−1、290nm)
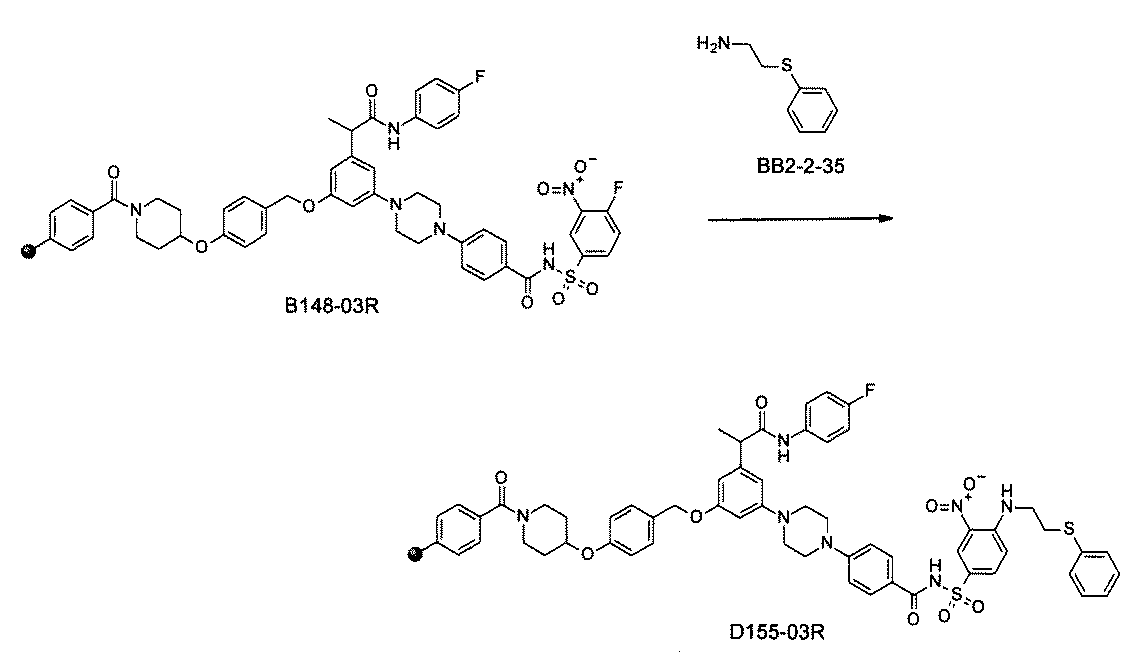

LCMS:m/z 799[M+H]+
保持時間:1.279分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 912[M+H]+
保持時間:0.927分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 962[M+H]+
保持時間:0.999分(分析条件 SMD−FA05−1、280nm)
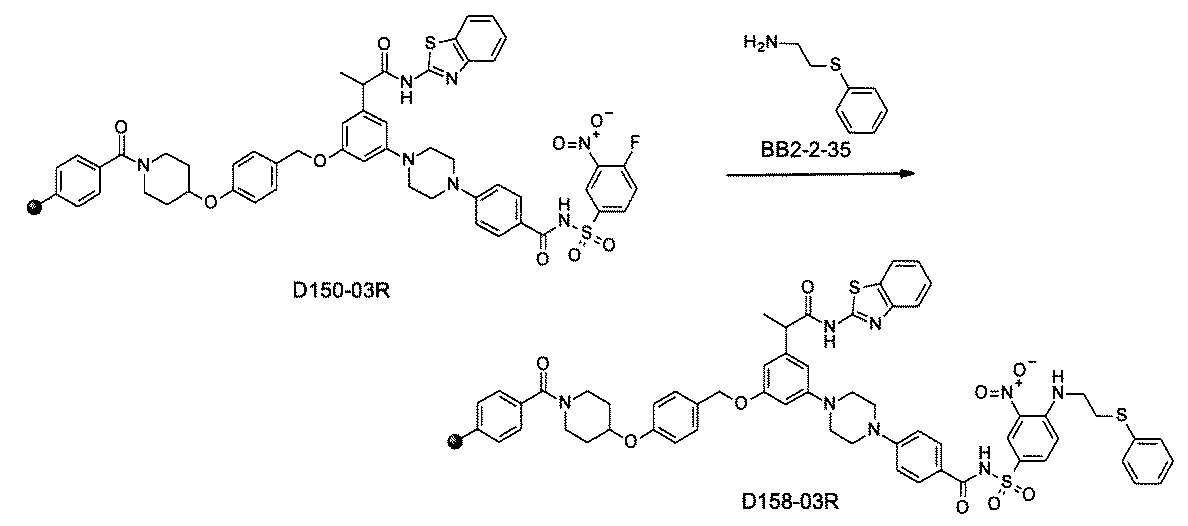
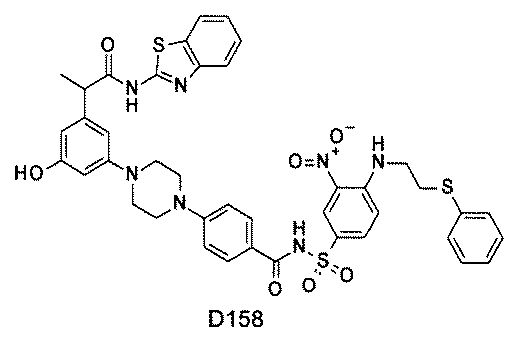
LCMS:m/z 838[M+H]+
保持時間:1.315分(分析条件 SMD−FA05−1、280nm)
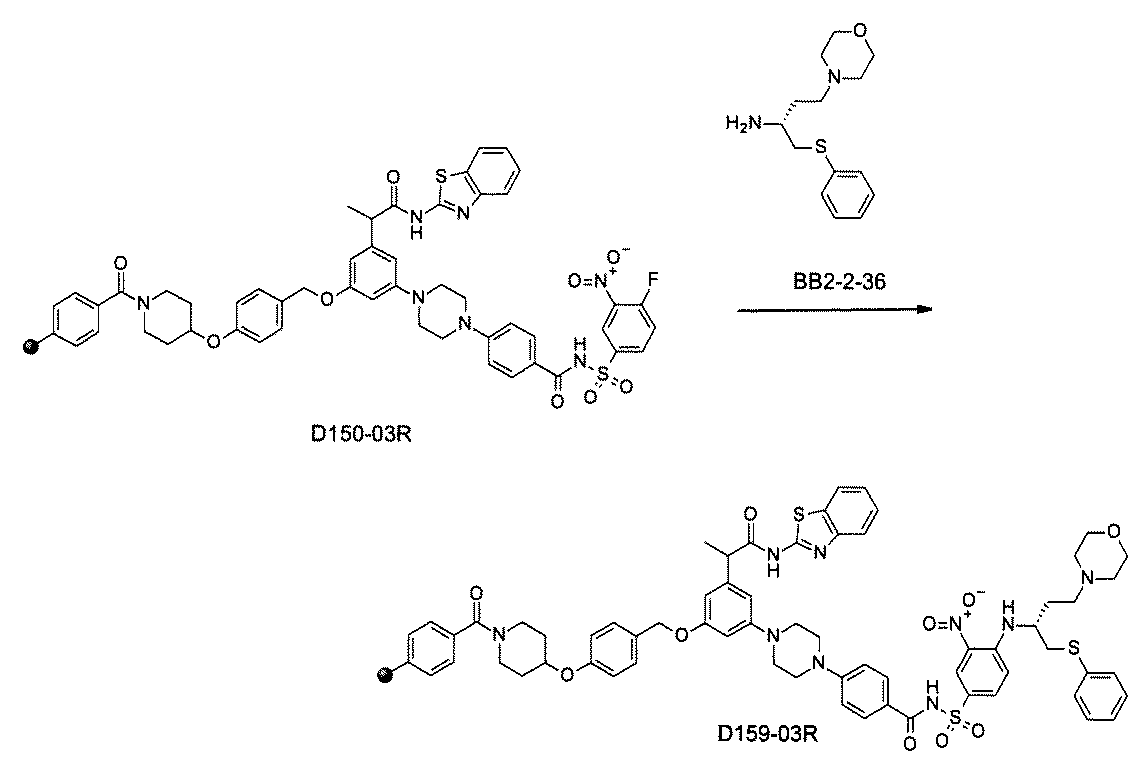

LCMS:m/z 951[M+H]+
保持時間:0.973分(分析条件 SMD−FA05−1、280nm)
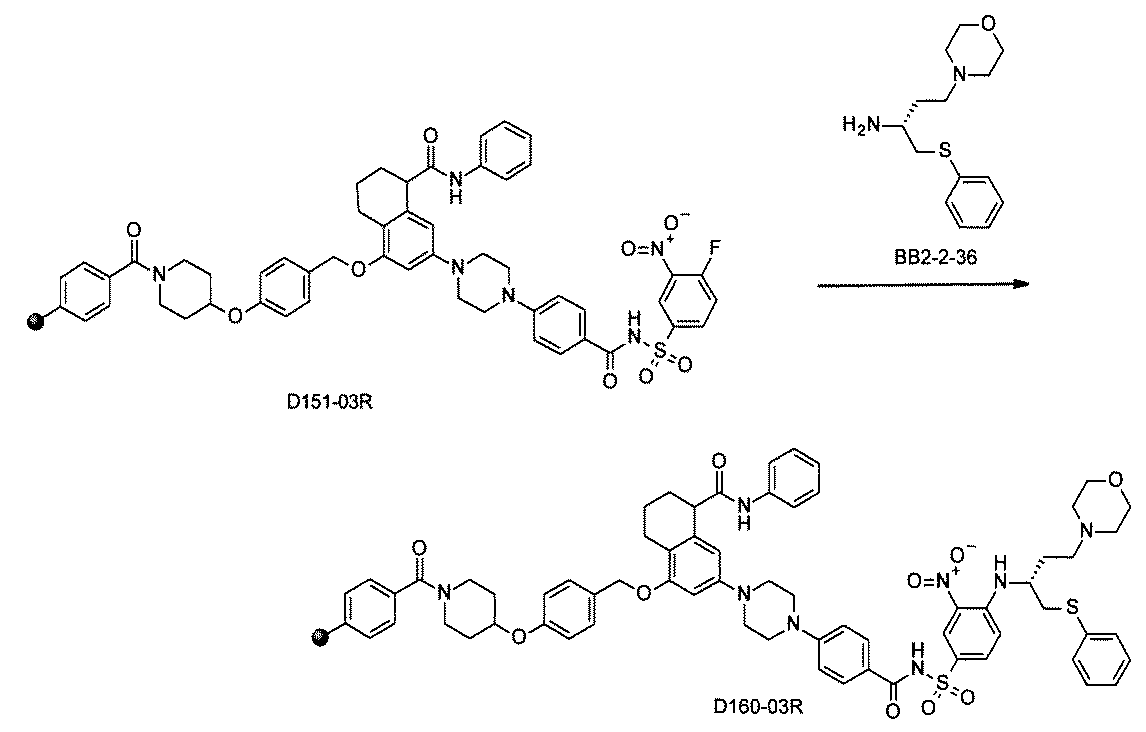

LCMS:m/z 920[M+H]+
保持時間:0.937分(分析条件 SMD−FA05−1、280nm)


LCMS:m/z 781[M+H]+。
保持時間:1.267分(分析条件 SMD−FA05−1、299nm)。


LCMS:m/z 894[M+H]+。
保持時間:0.923分(分析条件 SMD−FA05−1、280nm)。


LCMS:m/z 908[M+H]+。
保持時間:0.928分(分析条件 SMD−FA05−1、299nm)。


LCMS:m/z 799[M+H]+
保持時間:1.277分(分析条件 SMD−FA05−1、299nm)


LCMS:m/z 912[M+H]+
保持時間:0.933分(分析条件 SMD−FA05−1、299nm)
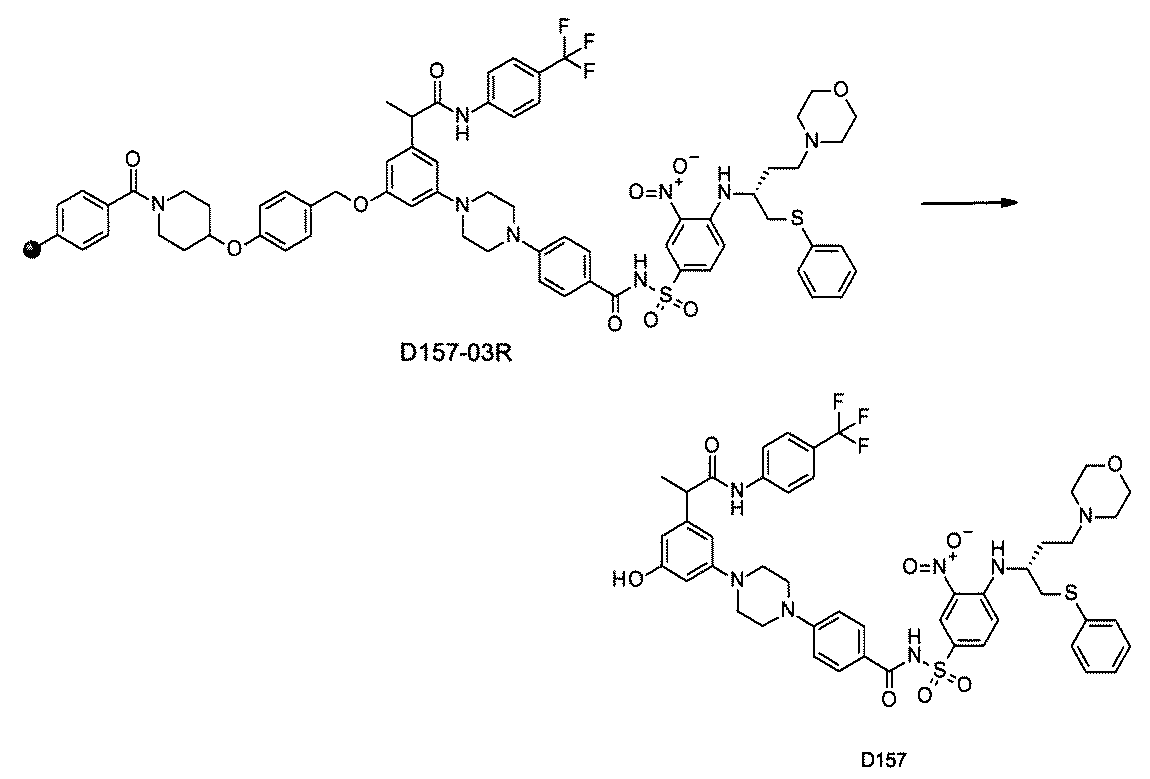
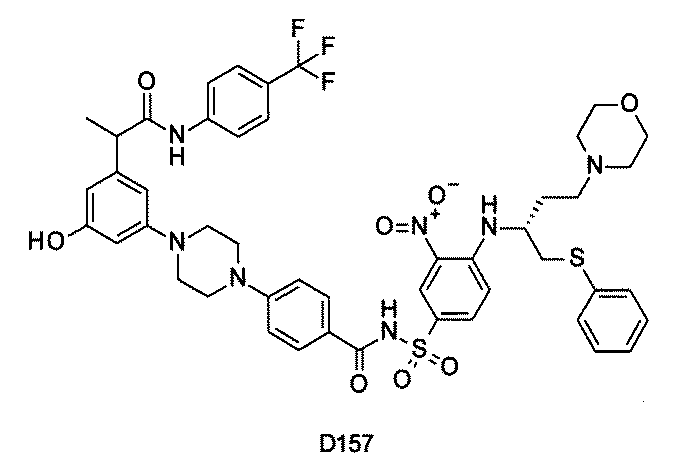
1H NMR(400MHz、DMSO−d6)δ:10.29(1H,s),9.17(1H,s),8.49(1H,s),8.35(1H,s),7.80−7.77(5H,m),7.64(2H,d,J=8.7Hz),7.31−7.14(5H,m),7.00−6.91(3H,m),6.49(2H,d,J=12.4Hz),6.25(2H,d,J=20.9Hz),4.13(1H,s),3.68(1H,q,J=7.1Hz),3.53−3.50(13H,m),3.21−3.19(7H,m),2.01−1.98(1H,m),1.85−1.83(1H,m),1.36(3H,d,J=7.0Hz)。
LCMS:m/z 962[M+H]+
保持時間:1.000分(分析条件 SMD−FA05−1、299nm)
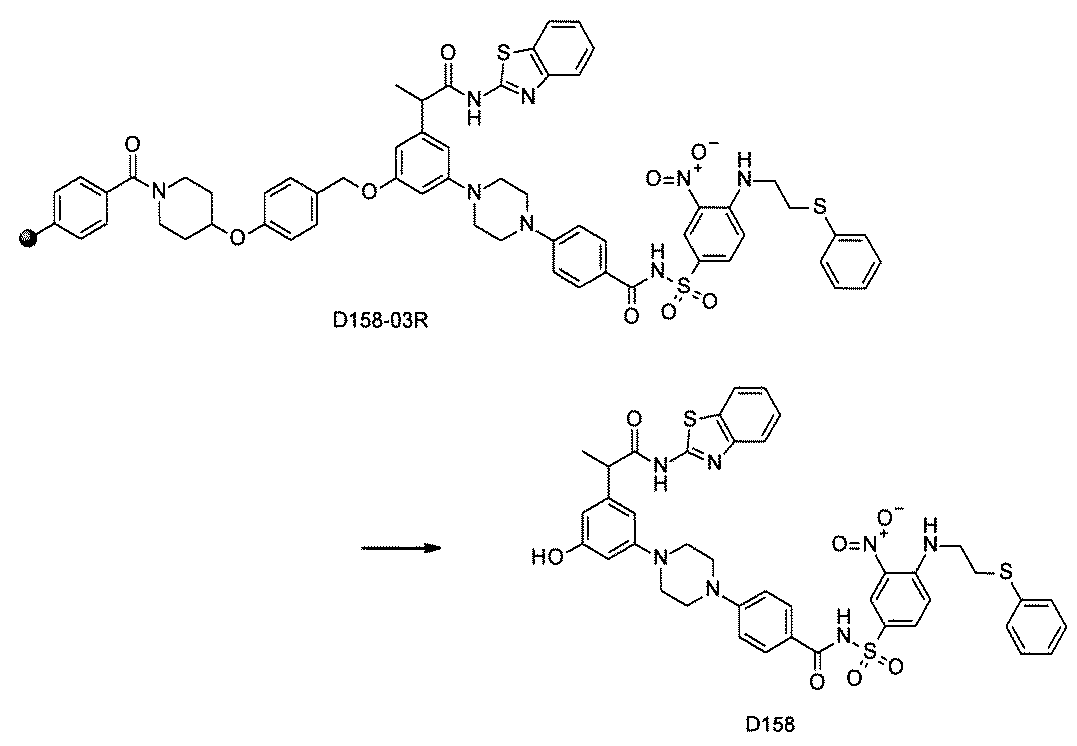
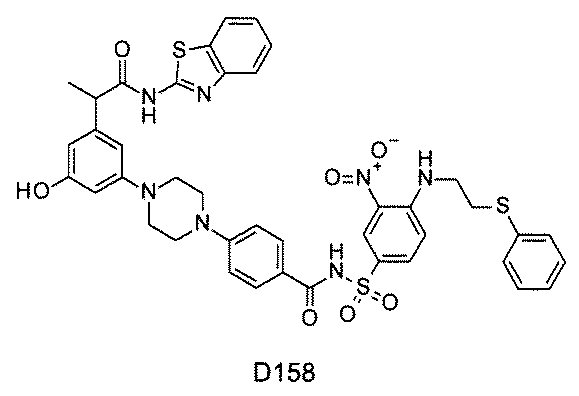
LCMS:m/z 838[M+H]+
保持時間:1.316分(分析条件 SMD−FA05−1、299nm)


LCMS:m/z 951[M+H]+。
保持時間:0.960分(分析条件 SMD−FA05−1、299nm)。


LCMS:m/z 948[M+H]+
保持時間:1.064分(分析条件 SMD−FA05−1、299nm)

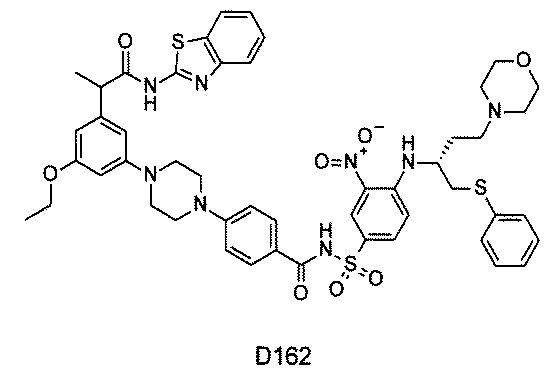
LCMS:m/z 979[M+H]+
保持時間:1.093分(分析条件 SMD−FA05−1、299nm)


LCMS:m/z 922[M+H]+。
保持時間:1.039分(分析条件 SMD−FA05−1、299nm)。
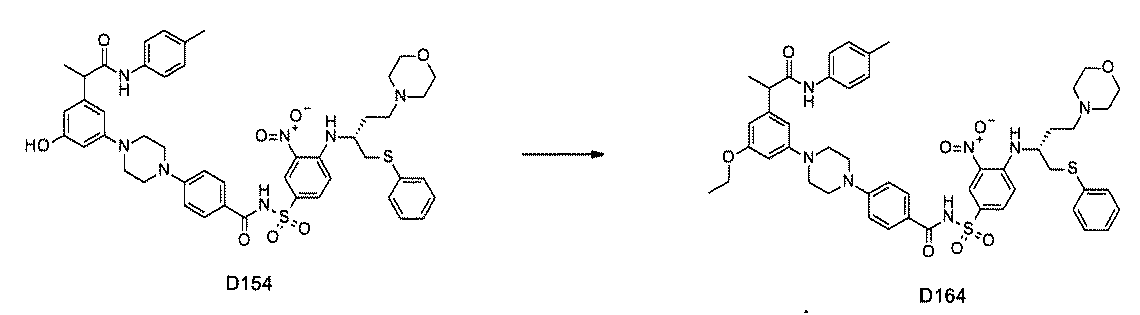
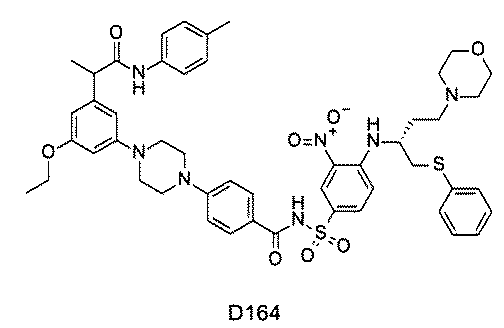
LCMS:m/z 936[M+H]+
保持時間:1.076分(分析条件 SMD−FA05−1、299nm)


LCMS:m/z 940[M+H]+。
保持時間:1.061分(分析条件 SMD−FA05−1、299nm)。
表13−1に示す実施例12にて合成した化合物のBcl−2に対する化合物の結合親和性を解析するため、表面プラズモン共鳴法を用いた。装置としてBiacore T200(GE Healthcare)を用い、ランニングバッファーには1mM DTT、3mM EDTA、0.01%Tween20及び4%DMSOが添加されたHBS(10mM HEPES−NaOH、150mM NaCl、pH7.4)を用いた。ヒトBcl−2タンパク質(Novus Biologicals)を、EZ−Link NHS−PEG4−Biotin(Thermo scientific)を用いてビオチン化し、Sensor Chip SA(Cytiva)の表面に固定化した。その後、複数の濃度の化合物溶液およびランニングバッファー(ブランク)を、Bcl−2固定化表面に添加し、化合物の結合レスポンスを得た。測定は15℃で行った。



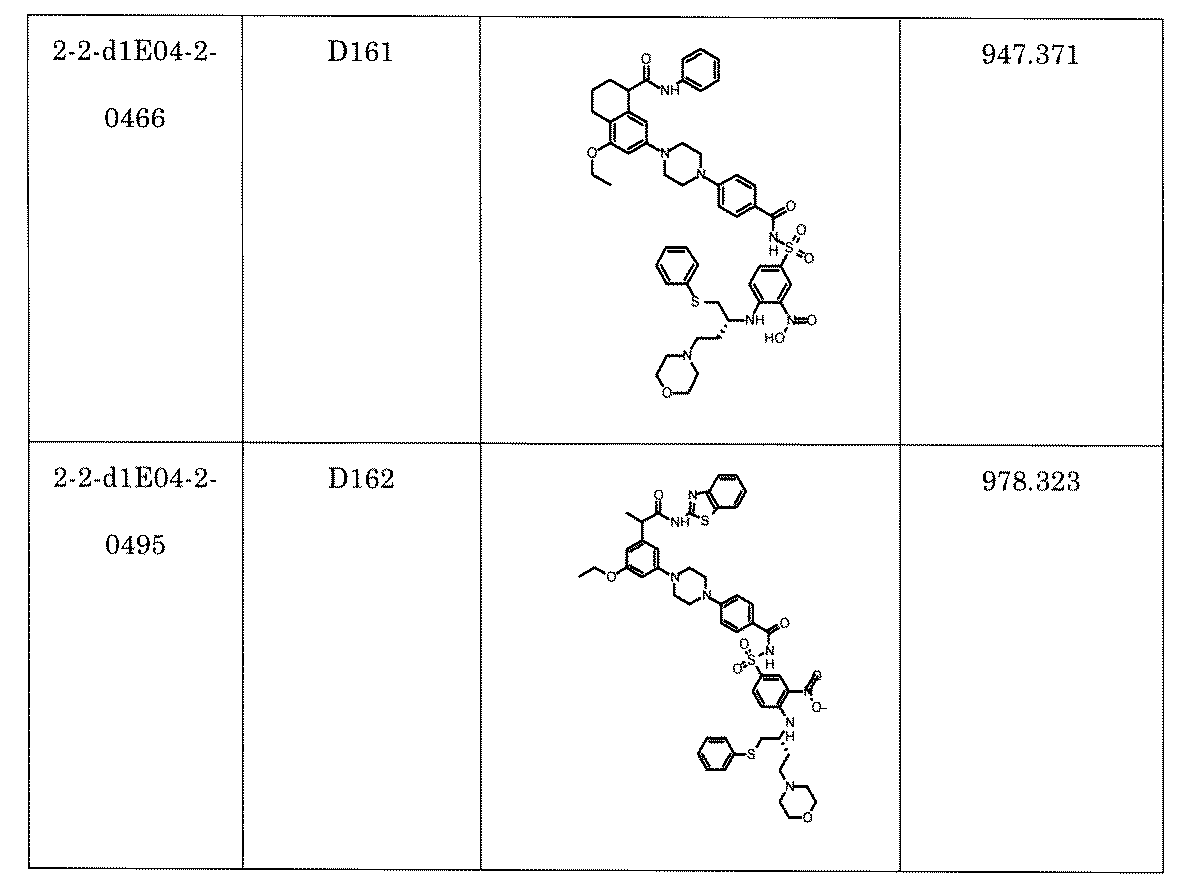


「コアブロックの多様性」と「リンカーの多様性」を掛け合わせ、実施例4で合成した混合物(計1,200化合物)よりも規模を大きく(計12,000化合物)、より幅広い範囲を一挙に探索するライブラリを新たに合成し(実施例8、9)、AS−MSにてBcl−2と結合する化合物を同定した(実施例11)。さらに同定した化合物を個別で合成し、SPRによりBcl−2と特異的に結合することが明らかになった。
Claims (15)
- 1×102~1×108の化合物を含むライブラリであって、
前記化合物は、第1コアブロック(第1CB)、第1リンカー(第1L)、および第2コアブロック(第2CB)が共有結合で連結した以下の構造:
(第1CB)−(第1L)−(第2CB)
を含み、
前記ライブラリは、2種類以上の第1CB、2種類以上の第1L、および2種類以上の第2CBを含み、
前記ライブラリは、1×102~1×105の前記化合物を含む1または2以上の混合物により構成される、
前記ライブラリ。 - 前記化合物が、さらに第2リンカー(第2L)、および第3コアブロック(第3CB)が共有結合で連結した以下の構造:
(第1CB)−(第1L)−(第2CB)−(第2L)−(第3CB)
を含み、
前記ライブラリが、2種類以上の第2L、および2種類以上の第3CBを含む、請求項1に記載のライブラリ。 - 前記化合物が、オリゴヌクレオチドタグを含まない、請求項1または2に記載のライブラリ。
- 第1L、第2L、および第3Lが、アミド、スルホンアミド、アシルスルホンアミド、アミノ、エーテル、チオエーテル、エステル、ケトン、スルホン、単結合、エチン−1,2−ジイル(−C≡C−)、および含窒素5員ヘテロアリーレンからなる群から独立して選択される構造を含む、請求項1~3のいずれか1項に記載のライブラリ。
- 標的分子に結合する化合物をスクリーニングする方法であって
(1)請求項1~4のいずれか1項に記載のライブラリを提供すること;
(2)前記ライブラリを構成する前記1または2以上の混合物をそれぞれ標的分子と接触させること;
(3)標的分子に結合する化合物と同定すること
を含む、前記方法。 - ライブラリの製造方法であって、
(a)第1ビルディングブロック(第1BB)が共有結合で連結する担持担体(第1BB担持担体)を調製すること、ここで第1BB担持担体は、na種類の第1ビルディングブロック(第1BB1~第1BBna)を含む固相担体(第1BB1−na担持担体)であり、第1BB1−na担持担体の第1BB1−na部分はそれぞれ、第2ビルディングブロック(第2BB)との結合を形成するための反応性官能基を有し、第1BB1−na担持担体に含まれる前記反応性官能基は1種類または2種類以上である、
(b)第1BB1−na担持担体のうち、前記反応性官能基が共通する担持担体の混合物を調製し、各混合物を1種類の第2ビルディングブロック導入試薬(第2BB導入試薬)または2種類以上の第2BB導入試薬の混合物と反応させて、第2BBが共有結合で第1BBに連結する担持担体(第2BB−第1BB担持担体)を調製すること、ここで得られる担持担体はnb種類の第2ビルディングブロック(第2BB1~第2BBnb)を含む担持担体(第2BB1−nb−第1BB1−na担持担体)であり、第2BB1導入試薬~第2BBnb導入試薬のいずれかが第1BB1−na担持担体のそれぞれの反応性官能基と反応して第1リンカー(第1L)を形成する、
(e)固相担体から化合物を切り出すこと
を含み、naは2以上の整数であり、nbは2以上の整数であり、得られる化合物ライブラリが100種類以上の化合物を混合物として含む、前記方法。 - (b)において、第1BB1−na担持担体のうち、反応性官能基が共通する担持担体の混合物を調製し、各混合物をnb種類の第2BB導入試薬の1つと反応させる、請求項6に記載の方法。
- 第2BB1−nb−第1BB1−na担持担体がそれぞれ、第3ビルディングブロック(第3BB)とのリンカー形成が可能な反応性官能基を有し、第1BB1−na部分または第2BB1−nb部分のそれぞれに1つ含まれる前記反応性官能基は1種類または2種類以上であり、さらに
(c)第2BB1−nb−第1BB1−na担持担体のうち、反応性官能基が共通する担持担体の混合物を調製し、各混合物を1種類の第3ビルディングブロック導入試薬(第3BB導入試薬)または2種類以上の第3BB導入試薬の混合物と反応させて、第3BBが共有結合で第1BBまたは第2BBに連結する担持担体(第3BB−第2BB−第1BB担持担体、または第2BB−(第3BB−)第1BB担持担体)を調製すること、ここで得られる担持担体はnc種類の第3ビルディングブロック(第3BB1~第3BBnc)を含む担持担体(第3BB1−nc−第2BB1−nb−第1BB1−na担持担体、または第2BB1−nb−(第3BB1−nc−)第1BB1−na担持担体)であり、第3BB1導入試薬~第3BBnc導入試薬のいずれかが第2BB1−nb−第1BB1−na担持担体のそれぞれの反応性官能基と反応して第2リンカー(第2L)を形成する、
を含み、ncは2以上の整数であり、ライブラリに含まれる化合物の第1リンカーまたは第2リンカーが2種類以上である、請求項6または7に記載の方法。 - naが2~1000の整数であり、nbが2~1000の整数であり、かつ、naまたはnbのいずれかが3以上の整数;または
naが2~1000の整数であり、nbが2~1000の整数であり、ncが2~1000の整数であり、かつ、na、nbまたはncのいずれかが3以上の整数である、請求項6~8のいずれか1項に記載の方法。 - 前記反応性官能基の誘導化工程をさらに含む、請求項6~9のいずれかに記載の方法。
- (e)において、固相担体から切り出された化合物に含まれる官能基を修飾することをさらに含む、請求項6~10のいずれか1項に記載の方法。
- 前記ライブラリに含まれる1以上の化合物がUVタグを含む、請求項6~11のいずれか1項に記載の方法。
- 前記ライブラリに含まれる前記化合物が、オリゴヌクレオチドタグを含まない、請求項6~12のいずれか1項に記載の方法。
- 第1リンカー、第2リンカー、および第3リンカーが、アミド化、スルホンアミド化、アシルスルホンアミド化、アミノ化、エーテル化、チオエーテル化、エステル化、炭素—窒素結合形成反応、炭素—炭素結合形成反応、および含窒素5員ヘテロアリーレン形成反応からなる群から独立して選択される反応により形成される、請求項6~13のいずれか1項に記載の方法。
- 得られるライブラリが100以上の化合物を含む、請求項6~14のいずれか1項に記載の方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22736785.1A EP4276092A4 (en) | 2021-01-06 | 2022-01-06 | Compound library |
| US18/270,790 US20250154686A1 (en) | 2021-01-06 | 2022-01-06 | Compound library |
| KR1020237026626A KR102876696B1 (ko) | 2021-01-06 | 2022-01-06 | 화합물 라이브러리 |
| CN202280009278.8A CN116783160A (zh) | 2021-01-06 | 2022-01-06 | 化合物库 |
| KR1020257035255A KR20250159084A (ko) | 2021-01-06 | 2022-01-06 | 화합물 라이브러리 |
| JP2022548055A JP7256338B2 (ja) | 2021-01-06 | 2022-01-06 | 化合物ライブラリ |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021001132 | 2021-01-06 | ||
| JP2021-001132 | 2021-01-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022149617A1 true WO2022149617A1 (ja) | 2022-07-14 |
Family
ID=82357928
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/001007 Ceased WO2022149617A1 (ja) | 2021-01-06 | 2022-01-06 | 化合物ライブラリ |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20250154686A1 (ja) |
| EP (1) | EP4276092A4 (ja) |
| JP (2) | JP7256338B2 (ja) |
| KR (2) | KR102876696B1 (ja) |
| CN (1) | CN116783160A (ja) |
| TW (1) | TWI905355B (ja) |
| WO (1) | WO2022149617A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115626918A (zh) * | 2022-11-08 | 2023-01-20 | 浙江工业大学 | 一种苯基呋喃-四氢异喹啉类化合物及其制备与应用 |
| WO2024032776A1 (en) * | 2022-08-12 | 2024-02-15 | Fochon Biosciences, Ltd. | Compounds as bcl-2 inhibitors |
| US12291524B2 (en) | 2022-05-19 | 2025-05-06 | Astrazeneca Ab | Amido heteroaromatic compounds |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025173784A1 (ja) * | 2024-02-16 | 2025-08-21 | 中外製薬株式会社 | 4-アミノピリジン誘導体を用いるスルホンアミド化合物の製造方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019527672A (ja) | 2016-05-16 | 2019-10-03 | サイクルニウム ファーマ インコーポレイテッド | 多様な大環状化合物のライブラリならびにその製造方法および使用方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU7564296A (en) * | 1995-11-17 | 1997-06-11 | Ciba-Geigy Ag | Solid phase synthesis of heterocyclic compounds and combinatorial compound library |
| US7671052B2 (en) * | 2004-10-05 | 2010-03-02 | Adolor Corporation | Phenyl derivatives and methods of use |
| CN101307029B (zh) * | 2008-07-08 | 2011-04-13 | 浙江大学 | 2,4,6-三取代-1,3,5-三嗪类衍生物库及制备方法 |
| US20150274755A1 (en) * | 2012-09-25 | 2015-10-01 | Shane W. Krska | Compound diversification using late stage functionalization |
| CA3066499A1 (en) | 2017-06-22 | 2018-12-27 | Cyclenium Pharma Inc. | Libraries of pyridine-containing macrocyclic compounds and methods of making and using the same |
-
2022
- 2022-01-06 WO PCT/JP2022/001007 patent/WO2022149617A1/ja not_active Ceased
- 2022-01-06 EP EP22736785.1A patent/EP4276092A4/en active Pending
- 2022-01-06 US US18/270,790 patent/US20250154686A1/en active Pending
- 2022-01-06 TW TW111100495A patent/TWI905355B/zh active
- 2022-01-06 CN CN202280009278.8A patent/CN116783160A/zh active Pending
- 2022-01-06 JP JP2022548055A patent/JP7256338B2/ja active Active
- 2022-01-06 KR KR1020237026626A patent/KR102876696B1/ko active Active
- 2022-01-06 KR KR1020257035255A patent/KR20250159084A/ko active Pending
-
2023
- 2023-03-30 JP JP2023055560A patent/JP2023100613A/ja active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019527672A (ja) | 2016-05-16 | 2019-10-03 | サイクルニウム ファーマ インコーポレイテッド | 多様な大環状化合物のライブラリならびにその製造方法および使用方法 |
Non-Patent Citations (37)
| Title |
|---|
| "Structurally homogeneous and heterogeneous synthetic combinatorial libraries.", MOLECULAR DIVERSITY, vol. 1, 1996, pages 149 - 164 |
| "The ''One-Bead-One-Compound'' Combinatorial Library Method", CHEM. REV., vol. 97, no. 2, 1997, pages 411 - 448 |
| A. BANCET ET AL.: "Fragment Linking Strategies for Structure-Based Drug Design.", J. MED. CHEM., vol. 63, no. 20, 2020, pages 11420 - 11435 |
| A. SATZ: "Analysis of Current DNA Encoded Library Screening Data Indicates Higher False Negative Rates for Numerically Larger Libraries", ACS COMB. SCI., vol. 19, no. 4, 2017, pages 234 - 238 |
| B. C. DOAKJ. KIHLBERG: "Drug discovery beyond the rule of 5 - Opportunities and challenges", EXPERT OPINION ON DRUG DISCOVERY, vol. 12, no. 2, 2017, pages 115 - 119 |
| B. R. LAHUE ET AL.: "Substituted benzimidazoles: A novel chemotype for small molecule hKSP inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 19, 2009, pages 3405, XP026155073, DOI: 10.1016/j.bmcl.2009.05.040 |
| C. S. KOLLMANN ET AL.: "Application of encoded library technology (ELT) to a protein-protein interaction target: Discovery of a potent class of integrin lymphocyte function-associated antigen 1 (LFA-1) antagonists", BIOORG. MED. CHEM., vol. 22, 2014, pages 235, XP028835527, DOI: 10.1016/j.bmc.2014.01.050 |
| CHEN YING-CHU, FAVER JOHN C., KU ANGELA F., MIKLOSSY GABRIELLA, RIEHLE KEVIN, BOHREN KURT M., UCISIK MELEK N., MATZUK MARTIN M., Y: "C–N Coupling of DNA-Conjugated (Hetero)aryl Bromides and Chlorides for DNA-Encoded Chemical Library Synthesis", BIOCONJUGATE CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 31, no. 3, 18 March 2020 (2020-03-18), US , pages 770 - 780, XP055949348, ISSN: 1043-1802, DOI: 10.1021/acs.bioconjchem.9b00863 * |
| CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 11, 2007, pages 518 - 526 |
| D. A. ANNIS ET AL.: "Affinity selection-mass spectrometry screening techniques for small molecule drug discovery", CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 11, 2007, pages 518 - 526, XP022323963, DOI: 10.1016/j.cbpa.2007.07.011 |
| D. A. ANNIS ET AL.: "An affinity selection-mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries: Discovery of a novel antagonist of E. coli dihydrofolate reductase", INTERNATIONAL JOURNAL OF MASS SPECTROMETRY, vol. 238, 2004, pages 77 - 83, XP004639190, DOI: 10.1016/j.ijms.2003.11.022 |
| F. IMRIE ET AL.: "Deep Generative Models for 3D Linker Design.", J. CHEM. INF. MODEL., vol. 60, no. 4, 2020, pages 1983 - 1995, XP055916311, DOI: 10.1021/acs.jcim.9b01120 |
| F. LOVERING ET AL.: "Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success", J. MED. CHEM., vol. 52, no. 21, 2009, pages 6752 - 6756 |
| G. CHESSARI ET AL.: "From fragment to clinical candidate- a historical perspective", DRUG DISCOVERY TODAY, vol. 14, July 2009 (2009-07-01), pages 668 - 675, XP026238300, DOI: 10.1016/j.drudis.2009.04.007 |
| G. SCHNEIDER ET AL.: "Scaffold-Hopping'' by Topological Pharmacophore Search: A Contribution to Virtual Screening", ANGEW. CHEM. INT. ED., vol. 38, 1999, pages 2894 - 2896 |
| H. LU ET AL.: "Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials", SIG TRANSDUCT TARGET THER, vol. 5, 2020, pages 213, Retrieved from the Internet <URL:https://doi.org/10.1038/s41392-020-00315-3> |
| H. SUN ET AL.: "Classification of scaffold-hopping approaches", DRUG DISCOVERY TODAY, vol. 17, April 2012 (2012-04-01), pages 310 - 32 |
| HOUGHTEN RICHARD A., PINILLA CLEMENCIA, APPEL JON R., BLONDELLE SYLVIE E., DOOLEY COLETTE T., EICHLER JUTTA, NEFZI ADEL, OSTRESH J: "Mixture-Based Synthetic Combinatorial Libraries", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 42, no. 19, 1 September 1999 (1999-09-01), US , pages 3743 - 3778, XP055949349, ISSN: 0022-2623, DOI: 10.1021/jm990174v * |
| HOUGHTEN RICHARD A.: "Parallel Array and Mixture-Based Synthetic Combinatorial Chemistry: Tools for the Next Millennium", ANNUAL REVIEW OF PHARMACOLOGY AND TOXICOLOGY., ANNUAL REVIEW INC., PALO ALTO, CA., US, vol. 40, no. 1, 1 April 2000 (2000-04-01), US , pages 273 - 282, XP055949351, ISSN: 0362-1642, DOI: 10.1146/annurev.pharmtox.40.1.273 * |
| J. ORG. CHEM., vol. 76, no. 2, 2011, pages 435 - 440 |
| J. QIAN ET AL.: "Discovery of novel inhibitors of Bcl-xL using multiplehigh-throughput screening platforms", ANALYTICAL BIOCHEMISTRY, vol. 328, 2004, pages 131 - 138, XP004504040, DOI: 10.1016/j.ab.2003.12.034 |
| JOERG HOLENZPATRICK STOY: "Advances in Lead Generation", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 29, 2019, pages 517 - 524 |
| K. M. COMESS ET AL.: "Kinase Drug Discovery by Affinity Selection/Mass Spectrometry (ASMS): Application to DNA Damage Checkpoint Kinase Chkl", JOURNAL OF BIOMOLECULAR SCREENING, vol. 11, no. 7, 2006, pages 755 - 764 |
| KIN-CHUN LUKALEXANDER LEE SATZ: "A Handbook for DNA-Encoded Chemistry", article "DNA-Compatible Chemistry", pages: 67 - 98 |
| M. J. WIGGLESWORTH ET AL.: "Increasing the delivery of next generation therapeutics from high throughput screening libraries", CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 26, 2015, pages 104 - 110 |
| MARIE L. MALONEBRIAN M. PAEGEL: "What is a ''DNA-Compatible'' Reaction?", ACS COMBINATORIAL SCIENCE, vol. 18, no. 4, 2016, pages 182 - 187, XP093047128, DOI: 10.1021/acscombsci.5b00198 |
| P. J. HAJDUK ET AL.: "A decade of fragment-based drug design: strategic advances and lessons learned", NATURE REVIEWS DRUG DISCOVERY, vol. 6, 2007, pages 211 - 219, XP002599578, DOI: 10.1038/NRD2220 |
| R. LIU ET AL.: "Combinatorial peptide library methods for immunobiology research.", EXPERIMENTAL HEMATOLOGY, vol. 31, 2003, pages 11 - 30, XP055639147, DOI: 10.1016/S0301-472X(02)01008-1 |
| R. MACARRON ET AL.: "Impact of high-throughput screening in biomedical research", NATURE REVIEWS DRUG DISCOVERY, vol. 10, 2011, pages 188 - 195 |
| R. SEVERINSEN ET AL.: "Library of Biphenyl Privileged Substructures using a Safety-Catch Linker Approach.", J. CHEM. INF. MODEL., vol. 60, no. 4, 2020, pages 1983 - 1995 |
| RICHARD A. HOUGHTEN ET AL.: "Mixture-Based Synthetic Combinatorial Libraries.", J. MED. CHEM., vol. 42, no. 19, 1999, pages 3743 - 3778, XP055949349, DOI: 10.1021/jm990174v |
| ROBERT A. GOODNOW ET AL.: "DNA-encoded chemistry: enabling the deeper sampling of chemical space", NATURE REVIEWS DRUG DISCOVERY, vol. 16, 2017, pages 131 - 147, XP055672163, DOI: 10.1038/nrd.2016.213 |
| RUIWU LIU ET AL.: "Combinatorial chemistry in drug discovery", CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 38, 2017, pages 117 - 126, XP085070190, DOI: 10.1016/j.cbpa.2017.03.017 |
| SCIENCE, vol. 221, no. 4612, 19 August 1983 (1983-08-19), pages 709 - 13 |
| See also references of EP4276092A4 |
| THE PRACTICE OF MEDICINAL CHEMISTRY, pages 483 |
| Y. C. CHEN ET AL.: "C-N Coupling of DNA-Conjugated (Hetero)aryl Bromides and Chlorides for DNA-Encoded Chemical Library Synthesis", BIOCONJUGATE CHEM, vol. 31, no. 3, 2020, pages 770 - 780, XP055949348, DOI: 10.1021/acs.bioconjchem.9b00863 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12291524B2 (en) | 2022-05-19 | 2025-05-06 | Astrazeneca Ab | Amido heteroaromatic compounds |
| WO2024032776A1 (en) * | 2022-08-12 | 2024-02-15 | Fochon Biosciences, Ltd. | Compounds as bcl-2 inhibitors |
| CN115626918A (zh) * | 2022-11-08 | 2023-01-20 | 浙江工业大学 | 一种苯基呋喃-四氢异喹啉类化合物及其制备与应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20250154686A1 (en) | 2025-05-15 |
| JP2023100613A (ja) | 2023-07-19 |
| CN116783160A (zh) | 2023-09-19 |
| TWI905355B (zh) | 2025-11-21 |
| KR102876696B1 (ko) | 2025-11-03 |
| JPWO2022149617A1 (ja) | 2022-07-14 |
| TW202246224A (zh) | 2022-12-01 |
| EP4276092A4 (en) | 2025-01-01 |
| KR20250159084A (ko) | 2025-11-13 |
| EP4276092A1 (en) | 2023-11-15 |
| JP7256338B2 (ja) | 2023-04-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7256338B2 (ja) | 化合物ライブラリ | |
| Homer et al. | Sulfur fluoride exchange | |
| Zhu et al. | Visible-light-induced direct perfluoroalkylation/heteroarylation of [1.1. 1] propellane to diverse bicyclo [1.1. 1] pentanes (BCPs) under metal and photocatalyst-free conditions | |
| Erdeljac et al. | Exploring physicochemical space via a bioisostere of the trifluoromethyl and ethyl groups (BITE): attenuating lipophilicity in fluorinated analogues of Gilenya® for multiple sclerosis | |
| Gerosa et al. | Joint experimental, in silico, and NMR studies toward the rational design of iminium-based organocatalyst derived from renewable sources | |
| EP3741758A1 (en) | Bromodomain inhibitor compound and use thereof | |
| Yao et al. | Copper-catalyzed reaction of ketenimine and in situ generated immonium ion: access to α, β-unsaturated amidines | |
| Parisi et al. | A greener and efficient access to substituted four-and six-membered sulfur-bearing heterocycles | |
| Shaabani et al. | Scaffold hopping via ANCHOR. QUERY: β-lactams as potent p53-MDM2 antagonists | |
| Mudi et al. | In silico anti-SARS-CoV-2 activities of five-membered heterocycle-substituted benzimidazoles | |
| Han et al. | Diastereoselective [3+ 2] cycloaddition of 3-ylideneoxindoles with in situ generated CF 2 HCHN 2: syntheses of CF 2 H-containing spirooxindoles | |
| Bao et al. | The development of an effective synthetic route of Belinostat | |
| JP7205529B2 (ja) | オキサゾリジノン化合物の製造方法 | |
| Kang et al. | Protecting group-directed annulations of tetra-substituted oxindole olefins and sulfur ylides: regio-and chemoselective synthesis of cyclopropane-and dihydrofuran-fused spirooxindoles | |
| Luo et al. | Trideuteromethylthiolation through Reaction of Arynes, S-Methyl-d 3 Sulfonothioate with Sulfonamides or Amides: Access to Trideuteromethylated Sulfilimines | |
| KR20170129191A (ko) | (4s)-4-[4-사이아노-2-(메틸설폰일)페닐]-3,6-다이메틸-2-옥소-1-[3-(트라이플루오로메틸)페닐]-1,2,3,4-테트라하이드로 피리미딘-5-카보나이트릴의 생성 방법 | |
| JP7236598B2 (ja) | 酸性官能基のアルキル化方法 | |
| CN104945305A (zh) | 一种实现吲哚类化合物选择性芳巯基化的方法 | |
| TW202608855A (zh) | 分子庫 | |
| HK40098303A (zh) | 化合物库 | |
| CN106117104B (zh) | 一种维格列汀的制备方法 | |
| Xiao et al. | Practical Synthesis of Dotinurad and Related Compounds | |
| RU2804686C2 (ru) | СИНТЕЗ 3-МЕТИЛ-1,2,4-ТИАДИАЗОЛ-5-КАРБОГИДРАЗИДА ИЛИ ЕГО МЕТИЛ-d3 ДЕЙТЕРИРОВАННОЙ ФОРМЫ | |
| KR102660894B1 (ko) | 피리미딘-2-아민 화합물의 제조방법 | |
| JP5267096B2 (ja) | N,S−ジ−tert−ブトキシカルボニルグルタチオン蛍光誘導体及びそれを中間体として用いるグルタチオン蛍光誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2022548055 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22736785 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280009278.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020237026626 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022736785 Country of ref document: EP Effective date: 20230807 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18270790 Country of ref document: US |
|
| WWD | Wipo information: divisional of initial pct application |
Ref document number: 1020257035255 Country of ref document: KR |