WO2022210973A1 - ルチル型の結晶構造を有する粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 - Google Patents
ルチル型の結晶構造を有する粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 Download PDFInfo
- Publication number
- WO2022210973A1 WO2022210973A1 PCT/JP2022/016293 JP2022016293W WO2022210973A1 WO 2022210973 A1 WO2022210973 A1 WO 2022210973A1 JP 2022016293 W JP2022016293 W JP 2022016293W WO 2022210973 A1 WO2022210973 A1 WO 2022210973A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- particles
- dispersion
- film
- titanium
- mass
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D17/00—Pigment pastes, e.g. for mixing in paints
- C09D17/004—Pigment pastes, e.g. for mixing in paints containing an inorganic pigment
- C09D17/007—Metal oxide
- C09D17/008—Titanium dioxide
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/04—Oxides; Hydroxides
- C01G23/047—Titanium dioxide
- C01G23/053—Producing by wet processes, e.g. hydrolysing titanium salts
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/36—Compounds of titanium
- C09C1/3607—Titanium dioxide
- C09C1/3653—Treatment with inorganic compounds
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/04—Oxides; Hydroxides
- C01G23/047—Titanium dioxide
- C01G23/053—Producing by wet processes, e.g. hydrolysing titanium salts
- C01G23/0536—Producing by wet processes, e.g. hydrolysing titanium salts by hydrolysing chloride-containing salts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G23/00—Compounds of titanium
- C01G23/04—Oxides; Hydroxides
- C01G23/047—Titanium dioxide
- C01G23/053—Producing by wet processes, e.g. hydrolysing titanium salts
- C01G23/0536—Producing by wet processes, e.g. hydrolysing titanium salts by hydrolysing chloride-containing salts
- C01G23/0538—Producing by wet processes, e.g. hydrolysing titanium salts by hydrolysing chloride-containing salts in the presence of seeds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09C—TREATMENT OF INORGANIC MATERIALS, OTHER THAN FIBROUS FILLERS, TO ENHANCE THEIR PIGMENTING OR FILLING PROPERTIES ; PREPARATION OF CARBON BLACK ; PREPARATION OF INORGANIC MATERIALS WHICH ARE NO SINGLE CHEMICAL COMPOUNDS AND WHICH ARE MAINLY USED AS PIGMENTS OR FILLERS
- C09C1/00—Treatment of specific inorganic materials other than fibrous fillers; Preparation of carbon black
- C09C1/36—Compounds of titanium
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D1/00—Coating compositions, e.g. paints, varnishes or lacquers, based on inorganic substances
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
- C09D7/61—Additives non-macromolecular inorganic
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/60—Compounds characterised by their crystallite size
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/62—Submicrometer sized, i.e. from 0.1-1 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/64—Nanometer sized, i.e. from 1-100 nanometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/80—Particles consisting of a mixture of two or more inorganic phases
- C01P2004/82—Particles consisting of a mixture of two or more inorganic phases two phases having the same anion, e.g. both oxidic phases
- C01P2004/84—Particles consisting of a mixture of two or more inorganic phases two phases having the same anion, e.g. both oxidic phases one phase coated with the other
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03C—CHEMICAL COMPOSITION OF GLASSES, GLAZES OR VITREOUS ENAMELS; SURFACE TREATMENT OF GLASS; SURFACE TREATMENT OF FIBRES OR FILAMENTS MADE FROM GLASS, MINERALS OR SLAGS; JOINING GLASS TO GLASS OR OTHER MATERIALS
- C03C2217/00—Coatings on glass
- C03C2217/70—Properties of coatings
- C03C2217/73—Anti-reflective coatings with specific characteristics
- C03C2217/732—Anti-reflective coatings with specific characteristics made of a single layer
Definitions
- the present invention relates to particles having a rutile crystal structure, a method for producing the same, and the like.
- a coating liquid containing oxide particles with a high refractive index is used to form a film with a high refractive index on a substrate.
- Such films are used, for example, in eyeglasses, lenses, touch panels of smartphones, and the like.
- the particles have a high refractive index.
- titanium oxide is known to have a high refractive index.
- titanium oxide having a rutile crystal structure has a high refractive index (see, for example, JP-A-2010-42947). Titanium oxide containing tin oxide tends to have a rutile crystal structure.
- titanium oxide particles containing inorganic oxides such as silica are easy to disperse (see, for example, International Publication No. 2018/181241).
- Patent Documents 1 and 2 since rutile-type composite particles containing titanium oxide and silica are used, the titanium oxide content of the particles is low. Therefore, the refractive index of the film becomes low.
- an object of the present invention is to provide particles that are easily dispersed in a solvent and have a high refractive index.
- the present invention relates to particles having a rutile crystal structure and having a crystallite diameter of 7 nm or more.
- Such particles are easily dispersed in solvents.
- the particles contain 90% by weight or more of titanium oxide in terms of TiO 2 and 0.2 to 10% by weight of tin oxide in terms of SnO 2 .
- Such particles have a high refractive index.
- tin is not detected from the particle surface.
- the particle diameter when the particles are dispersed in the solvent is preferably 100 nm or less.
- a method for producing particles having a rutile-type crystal structure includes steps of preparing a dispersion of a titanium-containing compound, preparing a dispersion of core particles having a rutile-type crystal structure, and dispersing the titanium-containing compound.
- the method comprises a step of preparing a liquid mixture by mixing the liquid and a dispersion liquid of core particles, and a step of crystal-growing the core particles by heating the liquid mixture to 80° C. or higher.
- the core particles contain titanium oxide and tin oxide.
- the present invention relates to particles having a rutile-type crystal structure (hereinafter simply referred to as particles) having a crystallite diameter of 7 nm or more.
- particles a rutile-type crystal structure
- the present invention relates to particles having a rutile-type crystal structure (hereinafter simply referred to as particles) having a crystallite diameter of 7 nm or more.
- a film containing such particles has a high refractive index (hereinafter referred to as film refractive index).
- the crystallite size of the particles is preferably 9 nm or more, more preferably 12 nm or more.
- this content is 90% by weight or more in terms of TiO2 .
- This content is preferably 92% by weight or more, more preferably 95% by weight or more in terms of TiO 2 .
- the tin oxide content of the particles should be low. Therefore, the tin oxide content is 10 % by weight or less in terms of SnO2. This tin oxide content is more preferably 5 % by weight or less in terms of SnO2.
- the proportion of titanium oxide on the particle surface side (hereinafter referred to as shell) is high. Therefore, the content of titanium oxide tends to be high in the particles as a whole.
- the center side (hereinafter referred to as core) of the particles contains tin oxide, the particles tend to have a rutile crystal structure. That is, the shell may be free of tin oxide when the core contains a sufficient amount of tin oxide so that the crystal structure of the particle is of the rutile type. The lower the percentage of tin in the core, the higher the titanium oxide content of the particles.
- the ratio of tin in the core is preferably 6.5 atomic % (at %) or less.
- the proportion of tin in the core is the number of tin atoms relative to the total number of atoms of titanium and tin.
- this ratio is preferably 0.8 to 3.0, more preferably 0.8 to 2.0, even more preferably 0.8 to 1.5.
- the aspect ratio the smaller the average value of the aspect ratios of the particles (hereinafter referred to as the aspect ratio), the easier the particles are dispersed in the solvent or binder. Therefore, the haze of the film tends to be low. Also, the smaller the aspect ratio, the smaller the specific surface area of the particles. Therefore, it is preferable that the aspect ratio is 1.0 to 2.3. In particular, when the crystallite diameter is 7 nm or more and the aspect ratio is 2.3 or less, the particles are easily dispersed in the solvent.
- the aspect ratio is preferably 1 to 2.1, more preferably 1 to 1.9, even more preferably 1 to 1.6. 1 to 1.5 is more preferred.
- the aspect ratio is preferably 1 to 1.9, and when the crystallite diameter is 12.5 nm or more, the aspect ratio is preferably 1 to 1.8. is 17 nm or more, the aspect ratio is preferably 1 to 1.5.
- the aspect ratio is the ratio of the average short diameter to the average long diameter of the particles.
- the average particle size of the particles is preferably 100 nm or less.
- the dispersed particle size is 80 nm or less.
- the dispersed particle diameter is 20 nm or more, the particles are easily dispersed in the solvent or the binder.
- the crystallite size is 7 nm or more and the dispersed particle size is 20 nm or more.
- the particle refractive index varies depending on factors such as particle composition, density, crystal structure, and crystallite size.
- the refractive index is preferably 1.95 or higher, more preferably 2.05 or higher, and even more preferably 2.10 or higher. It is difficult to obtain particles with a refractive index of 2.8 or higher.
- a dispersion of particles comprises the particles described above and a solvent.
- the solvent is water, 20% by weight or more of the particles can be stably dispersed in water.
- the particle surfaces must be treated with a surface treatment agent. A portion of this surface treatment agent may be dispersed in the organic solvent without bonding to the particles.
- the solvent is an organic solvent (in the case of an organic solvent dispersion)
- two or more surface treatment agents may be included. Examples of surface treatment agents include silicon compounds, titanium compounds, zirconium compounds, and aluminum compounds. Among these, silicon compounds are industrially easy to handle.
- the organic solvent dispersion contains a surface treatment agent having an alkoxy group
- the alkoxy group is hydrolyzed.
- the hydrolyzed alkoxy group and the OH group on the particle surface undergo a dehydration condensation reaction (hereinafter referred to as a chemical bond). Therefore, the particles are easily dispersed in the organic solvent.
- the hydrolysis reaction of the alkoxy group is promoted by containing the catalyst, water, and the like in the organic solvent dispersion. Along with this, chemical bonding is promoted.
- an alkyl group is bonded to the oxygen atom of the alkoxy group.
- Surface treatment agents include molecules of the general formula (RO) n M(X) 4-n .
- n is an integer of 1-4.
- M represents either Si, Ti or Zr.
- X is a hydrocarbon group.
- R is a hydrocarbon group. When M is Si and R is Me or Et, it is easy to control the reaction rate of this molecule. Therefore, it is industrially easy to handle.
- the surface treatment agent has 3-4 alkoxy groups.
- the surface treatment agent has four alkoxy groups.
- a surface treating agent having four alkoxy groups is hereinafter referred to as a first surface treating agent.
- the dispersion contains 20 to 85 parts by mass of the first surface treatment agent per 100 parts by mass of the particles (3 to 30 parts by mass in terms of oxide ⁇ in terms of SiO 2 if the surface treatment agent is a silicon compound ⁇ ) of the first surface treatment agent, Particles are easily dispersed in alcohol.
- the amount of the surface treatment agent can be reduced as the crystallite size of the particles increases.
- the particles are treated with a surface treatment agent having a hydrocarbon group (hereinafter referred to as a second surface treatment agent), the particles are dispersed in an organic solvent that is more hydrophobic than alcohol (hereinafter referred to as a hydrophobic solvent). easy.
- a hydrophobic solvent an organic solvent that is more hydrophobic than alcohol
- the second surface treatment agent is different from the first surface treatment agent.
- the first surface treatment agent does not have a hydrocarbon group.
- a hydrocarbon group is not an alkyl group attached to the oxygen atom of an alkoxy group.
- hydrophobic solvents examples include organic solvents having at least one of an ester bond, an ether bond, and a ketone group.
- An organic solvent having such bonds and functional groups easily dissolves an ultraviolet curable binder.
- organic solvents polyethylene glycol monomethyl ether acetate (PGMEA) is preferred.
- the particles are easily dispersed in the hydrophobic solvent and UV-curable binder.
- the particles can be bound to the ultraviolet curable binder by ultraviolet irradiation.
- the film becomes dense and the film refractive index increases. Therefore, it is preferable that the hydrocarbon group of the second surface treatment agent has a (meth)acrylate group, or the hydrocarbon group of the second surface treatment agent is a (meth)acrylate group.
- a more dense layer is formed on the particle surface with a surface treatment agent because the particle surface is treated with silica.
- Such particles are easily dispersed in an organic solvent (particularly alcohol) even if the amount of the surface treatment agent is small. That is, with such particles, it is easy to replace the solvent of the dispersion liquid from water with an organic solvent.
- An aqueous dispersion of these particles has an isoelectric point of 3 or less (however, this is the isoelectric point when the solid content concentration is 0.5% by mass).
- the isoelectric point of the aqueous dispersion of the particles is in the range of 4-6.
- a crystal structure other than the rutile type may be observed.
- the surface of the particles is preferably treated with silica in an amount of 2 parts by mass or more in terms of SiO 2 per 100 parts by mass of the particles. That is, it is preferable that the silica content of the particles is 2 parts by mass or more per 100 parts by mass of the particles, and the isoelectric point of the aqueous dispersion is 3 or less. Even with a small amount of surface treatment agent, such particles are readily dispersed in organic solvents (especially alcohols). As for this silica content, 4 mass parts or more are more preferable. On the other hand, if the silica content is too high, the particle refractive index will be low. Therefore, the silica content is preferably 20 parts by mass or less per 100 parts by mass of the particles.
- the particle surface is treated with 2 parts by mass or more of silica with respect to 100 parts by mass of the particles, 10 to 30 parts by mass per 100 parts by mass of the particles (calculated as oxide ⁇ if the surface treatment agent is a silicon compound, SiO It is preferable that the surface of the particles is treated with the first surface treating agent in an amount of 3 to 10 parts by mass in terms of 2 ).
- the coating liquid contains particles, a surface treatment agent, a binder, and an organic solvent.
- a film can be formed by using a coating liquid containing a binder. If part of the surface treatment agent functions as a binder, the binder may not be included.
- binders include monomers before polymerization, oligomers, and polymers after they are polymerized. Of these, monomers or oligomers are preferred. When a film is cured, the film tends to be more dense when a coating liquid containing a monomer or oligomer is used than when a coating liquid containing a polymer is used.
- the use of an adamantane derivative as a monomer or oligomer increases the film refractive index.
- the organic solvent can be appropriately selected depending on the type of binder added when preparing the coating liquid.
- this concentration is preferably 10% by weight or more, more preferably 20% by weight or more. On the other hand, if this concentration is 50% by weight or less, the viscosity of the coating liquid tends to be low. This concentration is preferably 30% by weight or less.
- the coating liquid can be dried slowly, resulting in a dense film.
- This boiling point is more preferably 100° C. or higher.
- the organic solvent is less likely to remain, and the film tends to shrink. Therefore, the hardness of the film increases.
- This boiling point is more preferably 180° C. or lower.
- a dispersion of a titanium-containing compound and a dispersion of core particles having a rutile crystal structure (hereinafter referred to as core particles) are prepared ⁇ preparation step>. Thereafter, the dispersion of the titanium-containing compound and the dispersion of the core particles are mixed to prepare a mixture (mixing step). By heating the mixture to 80° C. or higher, crystals are grown based on the core particles ⁇ crystal growth step>. By crystal growth based on the core particles, the tin oxide content of the particles is reduced. Therefore, the titanium oxide content of the particles is relatively high. Also, the crystallite diameter increases. After the crystal growth step, the core particle becomes the core of the particle and the titanium-containing compound becomes the shell of the particle. Each step will be described in detail below.
- the core particles serving as starting points for crystal growth contain tin oxide and have a rutile crystal structure. Even if the titanium-containing compound does not contain tin oxide, crystal growth of such core particles results in the particles having a rutile crystal structure. Here, since the titanium-containing compound does not contain tin oxide, the particle refractive index is increased.
- the core particles When the core particles contain tin oxide, the core particles tend to become rutile.
- the tin oxide content of the core particles may be any amount as long as the crystal structure of the core particles becomes the rutile type. If the crystal structure of the core particles is other than the rutile type, the particles will have a mixed crystal or a crystal structure other than the rutile type.
- the crystallite size of the core particles may be any size as long as it is dispersed in the solvent. When this solvent is water, the core particles are easily dispersed.
- the titanium-containing compound may contain oxides and hydroxides of titanium. After obtaining a gel by neutralizing the titanium compound, the gel is deflocculated to obtain a dispersion of the titanium-containing compound.
- the titanium compound is not particularly limited as long as it is water-soluble. Specific examples of titanium compounds include titanium tetrachloride, titanium trichloride, titanium sulfate, titanyl sulfate, and titanium hydride.
- the resulting gel contains titanium hydroxide.
- the salt in the gel lowers the film refractive index and particle dispersibility. Therefore, it is preferable to wash the gel with water.
- hydrogen peroxide is used to deflocculate the gel, crystal growth tends to occur while the core particles are in the rutile state. After adding hydrogen peroxide to the gel, if the gel is kept at a temperature of 50° C. to 100° C., the gel is easily deflocculated.
- a liquid mixture is prepared by mixing a dispersion liquid of a titanium-containing compound and a dispersion liquid of core particles.
- the weight ratio of the titanium-containing compound to the core particles is 7 or less, crystals other than rutile crystals are less likely to form.
- the solid content and the weight of the core particles are the weights when the amounts of Ti and Sn in the raw material are converted to TiO 2 and SnO 2 , respectively.
- the titanium-containing compound is the solid content of the dispersion of the titanium-containing compound.
- the mixed solution is heated to 80° C. or higher to cause crystal growth based on the core particles. If the temperature of the mixed liquid is lower than 80° C., the crystallite size of the particles becomes small because the speed of crystal growth is slow. Moreover, in this case, the yellow titanium-containing compound remains because the reaction becomes insufficient.
- the mixed liquid is heated to 80° C. or higher, it is preferable to subject the mixed liquid to hydrothermal synthesis (autoclave treatment).
- the higher the hydrothermal synthesis temperature the larger the crystallite size. Therefore, the temperature is preferably 100° C. or higher, more preferably 130° C. or higher. On the other hand, if this temperature is 300° C. or less, the production efficiency will be high.
- This temperature is more preferably 250°C. Also, the longer the hydrothermal synthesis time, the higher the density of the particles. Therefore, this time is preferably 1 hour or longer, more preferably 5 hours or longer, and even more preferably 10 hours or longer. On the other hand, if this time is 50 hours or less, production efficiency will be high. This time is more preferably 40 hours or less, more preferably 20 hours or less.
- the titanium oxide content of the particles increases.
- the crystallite size and particle size increase.
- the aspect ratio becomes low.
- the number of times of crystal growth is preferably 2 to 5 times.
- the particle film has a low haze.
- the film refractive index is high.
- the crystal growth is performed 4 to 5 times, the refractive index of the particles becomes higher than when the crystal is grown 2 to 3 times.
- the number of times of crystal growth is 4 to 5 times, the haze of the film is higher than in the case of 2 to 3 times.
- the crystal-grown particles are used as core particles to perform the mixing step and the crystal growth step.
- silica it is preferable to treat the particle surface with silica before treating the particle surface with the surface treatment agent. Specifically, after silica is added to an aqueous dispersion of crystal-grown particles, this aqueous dispersion is hydrothermally synthesized. Thereby, a layer of silica is densely formed on the particle surface.
- the specific surface area of silica is 100 to 600 m 2 /g, fine particles and coarse particles are less likely to occur.
- the specific surface area of silica is more preferably 200 to 550 m 2 /g, still more preferably 300 to 550 m 2 /g.
- the lower the solid content concentration of the aqueous dispersion the denser the silica layer is formed on the particle surface.
- the solid content concentration is preferably 5% by weight or less, more preferably 3% by weight or less, and even more preferably 1.5% by weight or less.
- this temperature is preferably 100° C. or higher, more preferably 150° C. or higher.
- this temperature is 200° C. or less, the production efficiency will be high.
- This temperature is more preferably 180° C. or lower.
- the longer the hydrothermal synthesis time the denser the silica particles are treated on the particle surface. Therefore, this time is preferably 1 hour or longer, more preferably 15 hours or longer.
- this time is 40 hours or less, the production efficiency is high. This time is more preferably 20 hours or less.
- a method for producing an organic solvent dispersion of particles will be described below.
- a surface treatment agent By adding a surface treatment agent to an aqueous dispersion of crystal-grown particles, the surface of the particles is treated with the surface treatment agent. After that, the solvent of the dispersion liquid is replaced from water with an organic solvent.
- the surface treatment agent preferably has an alkoxy group. Since the surface treatment agent having an alkoxy group can be chemically bonded to the particle surface, the particles can be easily dispersed in the organic solvent.
- the retention time is preferably 20 hours or less.
- an alcohol dispersion can be prepared by replacing the solvent with alcohol.
- the particle surfaces can be treated with the second surface treating agent.
- the solvent (alcohol) of the alcohol dispersion is replaced with a hydrophobic solvent.
- the amount of the second surface treatment agent added is 10 parts by mass or more with respect to 100 parts by mass of the particles, the particles are easily dispersed in the hydrophobic solvent.
- the amount added is more preferably 20 parts by mass or more, more preferably 30 parts by mass or more.
- the amount added is preferably 50 parts by mass or less, more preferably 40 parts by mass or less per 100 parts by mass of the particles. Further, when the second surface treating agent is applied to the particle surface, adding water or a catalyst accelerates the hydrolysis reaction of the second surface treating agent.
- a coating liquid is obtained by adding a binder to an organic solvent dispersion of particles.
- an organic solvent dispersion may be used as the coating liquid.
- a film is formed on the substrate using the coating liquid described above to produce a film-coated substrate. Specifically, after the coating liquid is applied onto the substrate, the coating liquid is dried to form a film.
- coating methods include spin coating, bar coating, gravure coating, and slit coating. Drying means volatilizing and removing the solvent. If the drying temperature is 60°C or higher, the drying time will be shortened. Also, the solvent is less likely to remain in the film. Therefore, a dense film can be obtained. On the other hand, if this temperature is 120° C. or less, the base material is difficult to deform. This temperature is more preferably 100° C. or lower, more preferably 80° C. or lower. Moreover, in order to increase production efficiency, it is preferable to cure the film after drying the coating liquid.
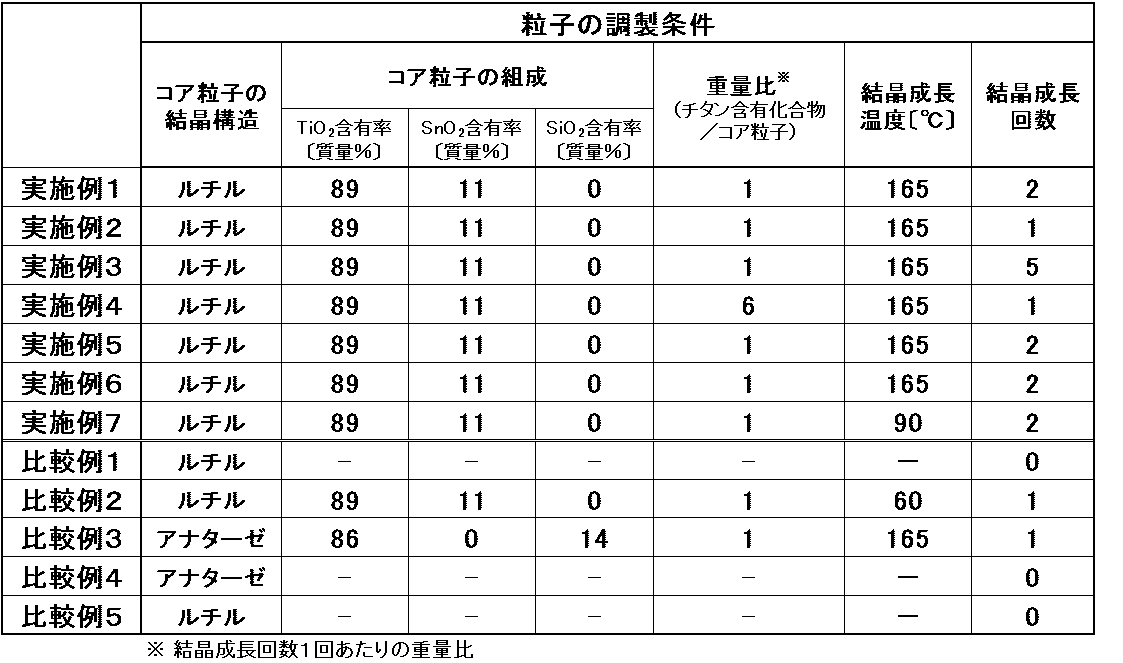
- Example 1 The method for preparing the particles will be specifically described below. Table 1 shows the preparation conditions of the particles.
- a dispersion of a titanium-containing compound and a dispersion of core particles were prepared as follows. 523 g of titanium tetrachloride aqueous solution of 7.66% by mass in terms of TiO 2 and 523 g of ammonia water of 7.66% by mass are mixed. This prepared a white slurry (gel) of pH 9.2. This slurry was filtered. By washing the gel with pure water, 400.5 g of cake with a solid content concentration of 10% by mass was obtained. A slurry was obtained again by diluting the cake with pure water to 1.5% by mass. 457.7 g of 35 mass % hydrogen peroxide water was added to this slurry.
- the dispersion was heated at a temperature of 80° C. for 1 hour. By adding 877 g of pure water to this dispersion, a dispersion of a titanium-containing compound (having a titanium oxide concentration of 1.0% by weight in terms of TiO 2 ) was obtained.
- This dispersion had a pH of 7.8 and a laser particle diameter of 37 nm.
- the laser particle size was measured by diluting this dispersion with water to 0.01% by weight and measuring it by an electrophoretic light scattering method using ELSZ-2000S manufactured by Otsuka Electronics Co., Ltd. In the following examples and comparative examples, all laser particle sizes were measured at this concentration.
- a cation exchange resin (manufactured by Mitsubishi Chemical Corporation) was added to 4005 g of a titanium-containing compound dispersion. 495 g of an aqueous potassium stannate solution diluted with pure water to 1% by weight was added to this dispersion. The ion exchange resin was separated from this dispersion. By hydrothermally synthesizing this dispersion in an autoclave at 165° C. for 18 hours, 4500 g of a dispersion of core particles was obtained.
- a mixed liquid was prepared by mixing 4500 g of the dispersion liquid of the titanium-containing compound and 4500 g of the dispersion liquid of the core particles.
- the laser particle diameter of the mixed liquid was 37 nm.
- Core particles were crystal-grown by hydrothermally synthesizing this mixture in an autoclave.
- the hydrothermal synthesis conditions were 165° C. for 18 hours.
- the laser particle diameter of the dispersion after the first crystal growth was 37 nm.
- the core particles were crystal-grown twice.
- particles were crystal-grown in the same manner as in the first crystal growth, except that the dispersion after the first crystal growth was used as the core particle dispersion.
- the laser particle diameter of the dispersion after the second crystal growth was 43 nm.
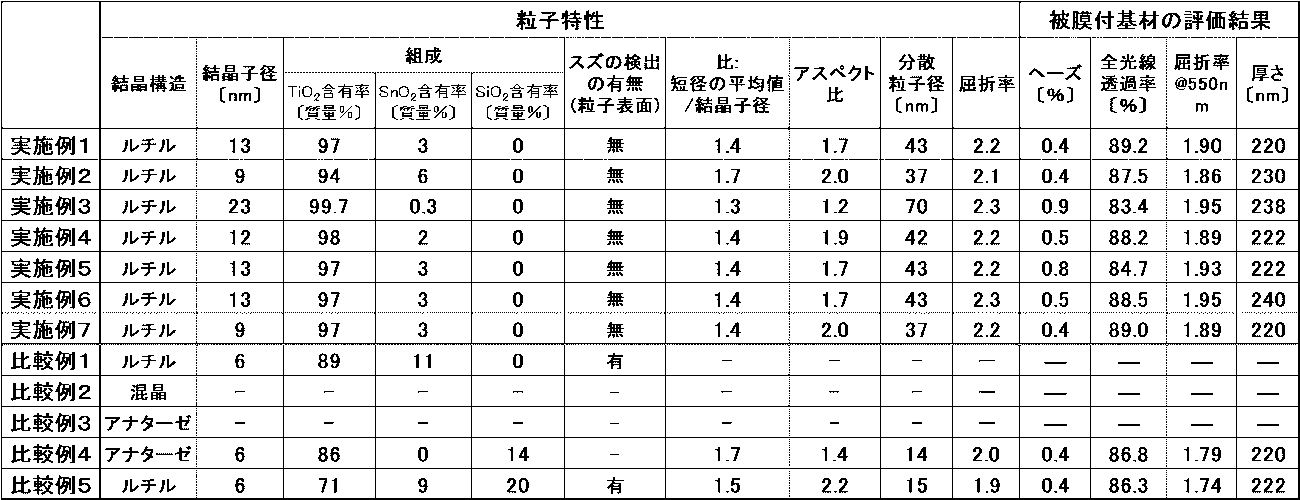
- Table 3 also shows the laser particle diameters (dispersed particle diameters) of the dispersions of the particles of the other examples and comparative examples.
- an alcohol dispersion of particles was prepared as follows. To 2250 g of the aqueous dispersion of particles were added 2250 g of methanol and 56.3 g of normal ethyl silicate (containing 28.8% by mass of silicon in terms of SiO 2 ) as a first surface treatment agent. By heating and stirring this dispersion at 50° C. for 18 hours, a water/methanol dispersion of particles was obtained. After cooling the dispersion to room temperature, the solvent of the dispersion was replaced with methanol using an ultrafiltration membrane. By concentrating this dispersion, 531 g of an alcohol dispersion of particles (with a solid concentration of 20% by mass) was obtained. The amount of water contained in this dispersion was 0.3% by mass.
- aqueous dispersion of particles was diluted to a solid content concentration of 1%. By drying 100 g of this aqueous dispersion at 100° C. for 10 minutes, a powder of particles was obtained. The powder was put through a burner to incinerate the organics. The powder was melted by adding the powder, sodium peroxide and sodium hydroxide. Further, the powder was dissolved by adding sulfuric acid and hydrochloric acid to the powder. Using ICP-OES (SII SPS5520 or Shimadzu ICPS-8100), the Sn, Ti, and Si contents of this solution were measured. These contents were converted into SnO 2 , TiO 2 and SiO 2 contents of the particles, respectively.
- ICP-OES SII SPS5520 or Shimadzu ICPS-8100
- the rutile Miller index (110) was chosen as the diffraction peak.
- D is the crystallite size (nm)
- K is the Scherrer constant
- ⁇ is the X-ray wavelength (nm)
- ⁇ is the diffraction line width (rad)
- ⁇ is the Bragg angle (rad).
- tin oxide is not detected on the particle surface or whether the core contains tin oxide can be determined using X-ray photoelectron spectroscopy (XPS).
- XPS X-ray photoelectron spectroscopy
- a film is prepared as a measurement sample as follows. First, 1.1 g of diphenyl (2,4,6-trimethylbenzoyl)-phenylphosphine oxide (manufactured by IGM Resins B.V.: Omnirad (registered trademark) TPO-H) was added as a photopolymerization initiator to 300 g of an organic solvent dispersion of particles. A dispersion liquid (coating liquid) was prepared.
- This dispersion liquid was applied onto a silicon wafer substrate, and the coating liquid was dried at 80° C. for 2 minutes.
- a sample (film) for XPS measurement was prepared by irradiating the dried coating liquid with ultraviolet rays under the condition of 3000 mJ/cm 2 using a high-pressure mercury lamp (manufactured by GS Yuasa: EYEUVMETER). It was measured by XPS as follows, and it was confirmed that tin oxide was not detected from the particle surface.
- Titanium may not be detected on the outermost surface of the film due to the presence of a surface treatment agent. Therefore, the percentage of tin on the particle surface is measured by etching to a depth where titanium is detected. For example, in this embodiment, etching is performed to a depth of 1.2 nm where titanium is detected. For this depth, Ar etching is performed for 20 seconds. That is, Ar etching is performed at 0.06 nm/sec. Titanium is detected when the proportion of titanium is 1.0 atomic % or more. This ratio is the number of atoms of titanium to the total number of atoms of carbon, oxygen, titanium, and tin.
- tin does not exist on the particle surface when it is lower than this. Therefore, when the proportion of tin at the depth where titanium is detected is less than 0.1 atomic % (that is, no tin is detected), it is assumed that no tin oxide exists on the particle surface. In this example, tin was not detected from the particle surface because this ratio was less than 0.1 at %. If tin is present in the core, continuing the etching will detect tin (the proportion of tin becomes 0.1 at % or more).
- ESCALAB220Xi manufactured by Thermo Fisher Scientific was used as an X-ray photoelectron spectroscopic analyzer.
- the spectrum measurement conditions are X-Ray: 190 W, Pass Energy: Wide 100 eV, Narrow 20 eV, analysis diameter: 250 ⁇ 1000 ⁇ m, Charge Neutralizer: On.
- Ion gun used Ar Gass Cluster Ion to perform measurements at an acceleration voltage of 3 kV and an etching rate of 3.5 nm/min (silicon oxide film).
- the 1s(CH) binding energy calibration was performed at 284.8 eV.
- the energy intensity of the C1s peak is observed at 284-286 eV.
- O1s is observed at 532-534 eV, Ti2p at 454-467 eV, and Sn3d5 at 483-490 eV.
- Particle refractive index Np' The particle refractive index Np' was determined by the following method. In the following examples and comparative examples, the refractive index Np' was similarly determined.
- coating liquids (a) to (c) were prepared, and then the coating liquids were applied onto a silicon wafer (manufactured by Matsuzaki Seisaku Co., Ltd.: 6-inch dummy wafer (P type), thickness: 625 ⁇ m) by spin coating. was applied.
- This coating liquid was dried at 80° C. for 2 minutes.
- a film-coated substrate (silicon wafer) was produced by irradiating the dried coating liquid with ultraviolet light under the condition of 3000 mJ/cm 2 using an EYEUVMETER. Using spectroscopic ellipsometry (manufactured by Japan Semilab Co., Ltd.: SE-2000), the refractive index Nav' of these film-coated substrates (silicon wafers) was actually measured.
- Three levels of coating liquids (a) to (c) were prepared as follows.
- (a) Preparation of coating liquid with weight ratio [particles:ADDA 6:4] 60.0 g of an organic solvent dispersion of particles having a solid content concentration of 20% by mass, 8.0 g of ADDA, and 0.5 g of Omnirad TPO-H Mixed.
- a coating liquid (a) was prepared by adding 60.0 g of PGMEA to this.
- (b) Preparation of coating liquid with weight ratio [particles:ADDA 7:3] Mix 70.0 g of an organic solvent dispersion of particles with a solid content concentration of 20% by mass, 6.0 g of ADDA, and 0.4 g of Omnirad TPO-H. did.
- a coating liquid (b) was prepared by adding 70.0 g of PGMEA to this.
- a coating liquid (c) was prepared by adding 80.0 g of PGMEA to this.
- the film refractive index Nav (calculated value) is calculated using Equation 1 (volume fraction/weight fraction conversion equation) and Equation 2 (Maxwell-Garnett equation).
- Equation 1 f(m) is the volume fraction of particles to total solids.
- m is the weight fraction of the particles to the total solid content
- dm is the specific gravity of the binder (here, 1.1 g/ml, which is the specific gravity of ADDA)
- dp is the specific gravity of the particles.
- the specific gravity dp is the total product of the content of each component contained in the particles and the specific gravity.
- the specific gravities dp of the components TiO 2 , SiO 2 , and SnO 2 contained in these particles are 4.3 (3.8 g/ml only in Comparative Example 4 because it is an anatase type), 2.2, and 7.0 g/ml, respectively.
- ml The content of each component is a value obtained by dividing the content (% by mass) obtained in (1) Particle composition by 100.
- Equation 2 Nav is the film refractive index, Nm is the binder refractive index (here, the refractive index of ADDA is 1.7), and Np is the particle refractive index.
- Equation (1) The weight fraction m of the particles, the specific gravity dm of the binder, and the specific gravity dp of the particles in the coating liquids (a) to (c) were substituted into Equation (1). Since there are three levels of weight fraction m from (a) to (c), three levels of f(m) are obtained.
- a film refractive index Nav was calculated by substituting values in increments of 0.01 in the range of 1.70 to 2.70 for the particle refractive index Np in Equation 2. These film refractive indices Nav were calculated for each of the three levels of f(m).
- a deviation ⁇ (Nav-Nav') between the film refractive index Nav (calculated value) and the film refractive index Nav' (actual measurement) was determined.
- the particle refractive index Np at which the sum of squares of deviations ⁇ 2 is the minimum value was defined as the particle refractive index Np′. That is, the particle refractive index Np' was determined by the method of least squares.
- the particle refractive index Np′ determined by this method is based on the affinity of the particles for the binder in the film, the particle diameter, the shape of the particles, the properties of the particle surfaces (the amount of the surface treatment agent, the composition and structure of the particle surfaces, etc.), etc. contains a factor of .
- a film was formed on each of the glass base material and the silicon wafer using the coating liquid as described below to prepare a base material with the film (glass base material) and a base material with the film (silicon wafer).
- a haze meter manufactured by Nippon Denshoku Industries Co., Ltd.: NDH-5000
- the total light transmittance and haze of the film-coated substrate (glass substrate) were measured.
- the refractive index and film thickness of the film-coated substrate (silicon wafer) were evaluated using spectroscopic ellipsometry (manufactured by Japan Semilab Co., Ltd.: SE-2000).
- Table 3 also shows the measurement and evaluation results of other examples and comparative examples.
- the coating liquid was applied to a glass substrate (made by Hamashin Co., Ltd.: FL glass, thickness: 3 mm, refractive index: 1.51) by a spin coating method. After drying at 80° C. for 2 minutes, the film was irradiated with ultraviolet light under the condition of 3000 mJ/cm 2 using a high-pressure mercury lamp (EYEUVMETER manufactured by GS Yuasa) to prepare a film-coated substrate (glass substrate). did.
- the uncoated glass substrate had a total light transmittance of 99.0% and a haze of 0.1%.
- the coating liquid was applied to a silicon wafer (manufactured by Matsuzaki Manufacturing Co., Ltd.: 6-inch dummy wafer (P type), thickness: 625 ⁇ m) by spin coating. After drying at 80° C. for 2 minutes, the film was irradiated with ultraviolet light under the condition of 3000 mJ/cm 2 using EYEUVMETER to produce a film-coated substrate (silicon wafer).
- Example 2 By concentrating the dispersion after the first crystal growth in Example 1 using an ultrafiltration membrane device, 2250 g of an aqueous dispersion of particles (with a solid concentration of 4% by mass) was obtained. In the preparation of an organic solvent dispersion of particles, an alcohol dispersion of particles (with a solid concentration of 20 mass %) was obtained. 11.2 g of 5 mass % aqueous ammonia was added to this alcohol dispersion. Further, 33.5 g of KBM-503 was added to this alcohol dispersion as a second surface treatment agent. After adding KBM-503, an organic solvent dispersion of particles (solid concentration: 20% by mass) was obtained in the same manner as in Example 1. A coating liquid was prepared in the same manner as in Example 1, except that this organic solvent dispersion was used in the preparation of the coating liquid.
- Example 3 Crystal growth was carried out in the same manner as in Example 1, except that the number of times of crystal growth was changed to 5 in the crystal growth process. In the n+1-th crystal growth, the dispersion after the n-th crystal growth was used as the core particle dispersion. Other than this, crystal growth was carried out in the same manner as the first time (where n is 1 to 4). By concentrating the dispersion after the fifth crystal growth using an ultrafiltration membrane device, 2250 g of an aqueous dispersion of particles (solid concentration: 4% by mass) was obtained. In the preparation of the organic solvent dispersion of the particles, an alcohol dispersion of the particles (solid content concentration: is 20% by mass).
- Example 1 A coating liquid was prepared in the same manner as in Example 1, except that this organic solvent dispersion was used in the preparation of the coating liquid.
- Example 4 In the mixing step, core particles were crystal-grown in the same manner as in Example 1, except that the amount of the titanium-containing compound dispersion was changed to 7716 g and the amount of the core particle dispersion was changed to 1286 g. Crystal growth was performed once. By concentrating this dispersion using an ultrafiltration membrane device, 2250 g of an aqueous dispersion of particles (with a solid concentration of 4% by mass) was obtained. A coating liquid was prepared in the same manner as in Example 1, except that this aqueous dispersion was used in the preparation of the organic solvent dispersion of the particles.
- Example 5 In the preparation of the organic solvent dispersion of the particles, the added amount of orthoethyl silicate was set to 43.8 g, the added amount of ammonia water was set to 10.3 g, and the added amount of KBM-503 was set to 30.8 g. A coating liquid was prepared in the same manner as in Example 1 except for the above.
- Example 6 An aqueous dispersion of the particles of Example 1 was diluted so that the solid content concentration was 1% by mass. To 9000 g of this aqueous dispersion was added 36 g of silica sol (manufactured by Nikki Shokubai Kasei Co., Ltd.: Cataloid (registered trademark) SN-350, having a specific surface area of silica of 375 m 2 /g and containing 15% by weight of silica). This dispersion was hydrothermally synthesized at 165° C. for 18 hours using an autoclave. The dispersion was cooled to room temperature.
- silica sol manufactured by Nikki Shokubai Kasei Co., Ltd.: Cataloid (registered trademark) SN-350, having a specific surface area of silica of 375 m 2 /g and containing 15% by weight of silica.
- This dispersion was hydrothermally synthesized at 165° C. for 18 hours using an autoclave. The
- a coating liquid was prepared in the same manner as in Example 1, except that this organic solvent dispersion was used in the preparation of the coating liquid.
- XPS measurement samples were prepared using particles of silica before surface treatment. That is, the same XPS measurement sample as in Example 1 was used.
- the isoelectric point of the aqueous dispersion of the particles obtained in this example was measured as follows. Using a zeta potential measuring device ("ZETASIZER Nano-ZS" manufactured by Malvern), the zeta potential was measured at each pH while adjusting the pH at intervals of pH 1 or less. The isoelectric point was defined as the pH at which the zeta potential is ⁇ 0 mV on the straight line connecting two points sandwiching the zeta potential ⁇ 0 mV. At this time, the concentration of particles to be measured was set to 0.25% by mass, and pure water was used for dilution. For pH adjustment, an automatic titrator (“MPT-2” manufactured by Malvern) was used. A 0.1 M sodium hydroxide aqueous solution was used to raise the pH, and a 0.1 M hydrochloric acid solution was used to lower the pH. The isoelectric point was 2.3.
- Example 7 In the crystal growth step, core particles were crystal-grown by heating and stirring at 90° C. for 40 hours with reflux instead of hydrothermal synthesis. A coating liquid was prepared in the same manner as in Example 1 except for the above.
- Example 1 The core particle dispersion of Example 1 was concentrated using an ultrafiltration membrane device to obtain 1125 g of an aqueous dispersion of particles (with a solid concentration of 4% by mass). 1125 g of methanol and 28.2 g of orthoethyl silicate (manufactured by Tama Chemical Co., Ltd., containing 28.8% by mass of silicon in terms of SiO 2 ) were added to this aqueous dispersion. When this aqueous dispersion was heated at 50°C, the viscosity of the dispersion increased and the dispersion became cloudy.
- Example 2 The mixed liquid of Example 1 was stirred at 60° C. for 40 hours to obtain an aqueous dispersion of particles. This aqueous dispersion was pale yellow. The particles in the aqueous dispersion were mixed crystals of rutile and anatase.
- the pH of this aqueous solution was 7.8.
- a dealkalization treatment was performed by adding a cation exchange resin to 15,000 g of this dispersion.
- a 1% by weight aqueous solution of potassium stannate was added to 1901 g of the dispersion.
- the ion exchange resin was separated from the dispersion. 634 grams of Cataloid SN-350 and 3591 grams of pure water were added to this dispersion.
- This dispersion was hydrothermally synthesized at 165° C. for 18 hours using an autoclave. This dispersion was concentrated using an ultrafiltration membrane device.
- Dealkalization was carried out by adding a cation exchange resin to 2641 g of this dispersion.
- An aqueous dispersion of the particles was prepared by separating the ion exchange resin from the dispersion.
- the laser particle diameter of this aqueous dispersion was 15 nm.
- an alcohol dispersion of particles (with a solid concentration of 20 mass%) was obtained. 31.2 g of 5% by weight aqueous ammonia was added to this alcohol dispersion. Further, 93.9 g of KBM-503 was added to this alcohol dispersion as a second surface treatment agent. After adding KBM-503, an organic solvent dispersion of particles (solid concentration: 20% by mass) was obtained in the same manner as in Example 1.
- a coating liquid was prepared in the same manner as in Example 1, except that this organic solvent dispersion was used in the preparation of the coating liquid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Inorganic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Geology (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Environmental & Geological Engineering (AREA)
- Paints Or Removers (AREA)
- Inorganic Compounds Of Heavy Metals (AREA)
- Liquid Crystal Substances (AREA)
Abstract
Description
結晶成長の起点となるコア粒子は、酸化スズを含み、且つルチル型の結晶構造を有する。チタン含有化合物に酸化スズが含まれていなくても、このようなコア粒子を結晶成長させることにより、粒子はルチル型の結晶構造になる。ここで、チタン含有化合物が酸化スズを含まないことにより、粒子屈折率が高くなる。
本工程では、チタン含有化合物の分散液とコア粒子の分散液とを混合することにより、混合液を調製する。このチタン含有化合物とコア粒子の重量比(チタン含有化合物の量/コア粒子の量)が7以下であることにより、ルチル型以外の結晶が生成し難くなる。ここで、固形分とコア粒子の重量は、原料中のTi、Snの量をそれぞれTiO2、SnO2換算したときの重量である。チタン含有化合物は、チタン含有化合物の分散液の固形分である。
本工程では、混合液を80℃以上にすることにより、コア粒子を元に結晶成長させる。混合液が80℃未満の場合、結晶成長の速度が遅いため、粒子の結晶子径が小さくなる。また、この場合、反応が不十分となるため、黄色いチタン含有化合物が残存してしまう。混合液を80℃以上にするとき、混合液を水熱合成(オートクレーブ処理)することが好ましい。水熱合成の温度が高いほど、結晶子径が大きくなる。そのため、この温度は100℃以上が好ましく、130℃以上がさらに好ましい。一方、この温度が300℃以下だと、生産効率が高くなる。この温度は、250℃がより好ましい。また、水熱合成の時間が長いほど、密度の高い粒子となる。そのため、この時間は1時間以上が好ましく、5時間以上がより好ましく、10時間以上がさらに好ましい。一方、この時間が50時間以下だと、生産効率が高くなる。この時間は、40時間以下がより好ましく、20時間以下がさらに好ましい。
以下、粒子の調製方法について具体的に説明する。粒子の調製条件を表1に示す。
<準備工程>
まず、以下のようにチタン含有化合物の分散液とコア粒子の分散液とを準備した。TiO2換算で7.66質量%の四塩化チタン水溶液523gと、7.66質量%のアンモニア水523gとを混合する。これにより、pH9.2の白色のスラリー(ゲル)を調製した。このスラリーをろ過した。純水を用いてゲルを洗浄することにより、固形分濃度10質量%のケーキ400.5gを得た。ケーキを純水で1.5質量%に希釈することにより、再度スラリーを得た。このスラリーに35質量%の過酸化水素水457.7gを加えた。80℃の温度で1時間この分散液を加熱した。この分散液に純水877gを添加することにより、チタン含有化合物の分散液(TiO2換算で酸化チタン濃度が1.0重量%)を得た。この分散液のpHは7.8、レーザー粒子径は、37nmであった。レーザー粒子径は、この分散液を水で0.01重量%に希釈して、大塚電子社製のELSZ-2000Sで電気泳動光散乱法により測定した。以下の実施例および比較例では、レーザー粒子径を全てこの濃度で測定した。
次に、チタン含有化合物の分散液4500gとコア粒子の分散液4500gを混合することにより、混合液を調製した。混合液のレーザー粒子径は37nmであった。
この混合液をオートクレーブ中で水熱合成することにより、コア粒子を結晶成長させた。水熱合成の条件は、165℃で18時間とした。1回目の結晶成長後の分散液のレーザー粒子径は、37nmであった。
以下、粒子の有機溶媒分散液の調製について具体的に説明する。分散液の調製条件を表2に示す。
以下、塗布液の調製方法について説明する。他の実施例と比較例の塗布液の調製条件も併せて表2に示す。粒子の有機溶媒分散液100.0gと、バインダとしてダイヤピュレスト(登録商標)ADDA(三菱瓦斯化学社製)6.0gと、光重合開始剤としてOmnirad TPO-H0.4gを添加・混合した。
粒子の水分散液を固形分濃度が1%となるように希釈した。この水分散液100gを100℃で10分間乾燥することにより、粒子の粉末を得た。粉末をバーナーにかけて、有機物を灰化した。この粉末と過酸化ナトリウムと水酸化ナトリウムを添加することにより、粉末を溶融した。さらに、粉末に硫酸と塩酸を添加することにより、粉末を溶解した。ICP-OES(SII社製SPS5520または島津製作所社製ICPS-8100)を用いて、この溶液のSn、Ti、およびSi含有率を測定した。これらの含有率をそれぞれ粒子のSnO2、TiO2、およびSiO2含有率に換算した。
粒子の水分散液を固形分濃度が1%となるように希釈した。この水分散液200gを110℃で20時間(実施例7のみ60℃で48時間)乾燥することにより、測定用のサンプルを調製した。リガク社製のRINT(登録商標)1400を用いて、サンプルのX線構造解析測定を行った。PDXLを用いて回折ピークのパターンを解析し、粒子の結晶構造がルチル型であることを確認した。Scherrerの式(D=K×λ/(β×cosθ))を用いて、回折ピークの半値幅から、粒子の結晶子径を算出した。回折ピークとして、ルチル型のミラー指数(110)を選択した。Dは結晶子サイズ(nm)、Kはシェラー定数、λはX線の波長(nm)、βは回折線幅の広がり(rad)、θはブラッグ角(rad)である。
粒子の水分散液を固形分濃度が0.002%となるように希釈した。希釈した分散液をコロジオン膜上に一滴たらした。これを50℃で10分間乾燥した。日立ハイテクノロジー社製の走査電子顕微鏡S-5500(SEM)を用いてこの粒子の写真を撮影した。SEMを用いて撮影した写真に投影された粒子を無作為に100個選出し、これらの短径と長径を測定した。これら短径の平均値と長径の平均値を算出した。この短径の平均値と長径の平均値の比(短径の平均値/長径の平均値)をアスペクト比とした。
粒子表面から酸化スズが検出されないことや、コアが酸化スズを含むことは、X線光電子分光分析(XPS)を用いて測定できる。しかし、粒子のみでは測定が困難であるため、測定試料として、以下のように膜を作製する。まず、粒子の有機溶媒分散液300gに光重合開始剤としてジフェニル(2,4,6トリメチルベンゾイル)-フェニルフォスフィンオキサイド(IGMResinsB.V.製:Omnirad(登録商標) TPO-H)を1.1g添加した分散液(塗布液)を調製した。この分散液をシリコンウエハ基材上に塗布し、80℃、2分で塗布液を乾燥した。高圧水銀ランプ(GSユアサ社製:EYEUVMETER)を用いて、3000mJ/cm2の条件で紫外線を乾燥した塗布液に照射することにより、XPS測定用試料(膜)を作製した。以下のとおりにXPSで測定し、粒子表面から酸化スズが検出されないことを確認した。
以下の方法で粒子屈折率Np’を決定した。以下の実施例および比較例においても、同様に屈折率Np’を決定した。
(a)重量比〔粒子:ADDA=6:4〕の塗布液の調製
固形分濃度20質量%の粒子の有機溶媒分散液60.0gと、ADDA8.0gと、Omnirad TPO-H 0.5gを混合した。これにPGMEA60.0gを添加することにより、塗布液(a)を調製した。
(b)重量比〔粒子:ADDA=7:3〕の塗布液の調製
固形分濃度20質量%の粒子の有機溶媒分散液70.0gと、ADDA6.0gと、Omnirad TPO-H0.4gを混合した。これにPGMEA70.0gを添加することにより、塗布液(b)を調製した。
(c)重量比〔粒子:ADDA=8:2〕の塗布液の調製
固形分濃度20質量%の粒子の有機溶媒分散液80.0gと、ADDA4.0gと、Omnirad TPO-H0.3gを混合した。これにPGMEA80.0gを添加することにより、塗布液(c)を調製した。


以下のとおりに、ガラス基材と、シリコンウエハそれぞれに、塗布液を用いて膜を形成し、膜付基材(ガラス基材)と膜付基材(シリコンウエハ)を作製した。ヘーズメーター(日本電色工業社製:NDH-5000)を用いて、膜付基材(ガラス基材)の全光線透過率およびヘーズを測定した。また、分光エリプソメトリー(日本セミラボ社製:SE―2000)を用いて、膜付基材(シリコンウエハ)の屈折率と膜厚を評価した。他の実施例と比較例の測定・評価結果も併せて表3に示す。
塗布液をガラス基材(浜新社製:FL硝子、厚さ:3mm、屈折率:1.51)にスピンコート法で塗布した。80℃で2分間乾燥した後、高圧水銀ランプ(GSユアサ社製:EYEUVMETER)を用いて、3000mJ/cm2の条件で紫外光を膜に照射し、膜付基材(ガラス基材)を作製した。なお、未塗布のガラス基材は全光線透過率が99.0%、ヘーズが0.1%であった。
塗布液をシリコンウエハ(松崎製作社製:6インチダミーウエハ(P型)、厚さ:625μm)にスピンコート法で塗布した。80℃で2分間乾燥した後、EYEUVMETERを用いて3000mJ/cm2の条件で紫外光を膜に照射し、膜付基材(シリコンウエハ)を作製した。
限外濾過膜装置を用いて実施例1の1回目の結晶成長後の分散液を濃縮することにより、粒子の水分散液2250g(固形分濃度が4質量%)を得た。粒子の有機溶媒分散液の調製において、この粒子の水分散液2250gに、メタノール2250gと正系酸エチル75gを添加したこと以外は実施例1と同様に粒子のアルコール分散液(固形分濃度が20質量%)を得た。5質量%アンモニア水をこのアルコール分散液に11.2g添加した。さらに、第二表面処理剤としてKBM-503を33.5gこのアルコール分散液に添加した。KBM-503を添加した後は、実施例1と同様に粒子の有機溶媒分散液(固形分濃度20質量%)を得た。塗布液の調製において、この有機溶媒分散液を用いたこと以外は実施例1と同様に塗布液を調製した。
結晶成長工程において、結晶成長回数を5回にした以外は、実施例1と同様に結晶成長させた。n+1回目の結晶成長では、コア粒子の分散液としてn回目の結晶成長後の分散液を用いた。これ以外は、1回目と同様に結晶成長を行った(ただし、nは1~4)。限外濾過膜装置を用いて5回目の結晶成長後の分散液を濃縮することにより、粒子の水分散液(固形分濃度が4質量%)2250gを得た。粒子の有機溶媒分散液の調製において、この粒子の水分散液2250gに、メタノール2250gと正系酸エチル37.5gを添加したこと以外は実施例1と同様に粒子のアルコール分散液(固形分濃度が20質量%)を得た。5質量%アンモニア水をこのアルコール分散液に10.1g添加した。さらに、第二表面処理剤としてKBM-503を30.2gこのアルコール分散液に添加した。KBM-503を添加した後は、実施例1と同様に粒子の有機溶媒分散液(固形分濃度20質量%)を得た。塗布液の調製において、この有機溶媒分散液を用いたこと以外は実施例1と同様に塗布液を調製した。
混合工程において、チタン含有化合物の分散液の量を7716gに、コア粒子の分散液の量を1286gに変更した以外は実施例1と同様にコア粒子を結晶成長させた。結晶成長回数は1回とした。限外濾過膜装置を用いてこの分散液を濃縮することにより、粒子の水分散液(固形分濃度が4質量%)2250gを得た。粒子の有機溶媒分散液の調製において、この水分散液を用いたこと以外は実施例1と同様に塗布液を調製した。
粒子の有機溶媒分散液の調製において、正珪酸エチルの添加量を43.8gに、アンモニア水の添加量を10.3gに、KBM-503の添加量を30.8とした。それ以外は実施例1と同様に塗布液を調製した。
実施例1の粒子の水分散液を固形分濃度が1質量%となるように希釈した。この水分散液9000gにシリカゾル(日揮触媒化成社製:カタロイド(登録商標)SN-350、シリカの比表面積が375m2/g、シリカを15重量%含む)36gを添加した。オートクレーブを用いて165℃で18時間この分散液を水熱合成した。この分散液を室温まで冷却した。限外濾過膜装置を用いてこの分散液を濃縮することにより、粒子の水分散液(固形分濃度が4質量%)2385gを得た。すなわち、シリカで粒子を表面処理した。粒子の有機溶媒分散液の調製において、この粒子の水分散液2250gに、メタノール2250gと正系酸エチル19.9gを添加したこと以外は実施例1と同様に粒子のアルコール分散液(固形分濃度が20質量%)を得た。5質量%アンモニア水をこのアルコール分散液に10.1g添加した。さらに、第二表面処理剤としてKBM-503を30.1gこのアルコール分散液に添加した。KBM-503を添加した後は、実施例1と同様に粒子の有機溶媒分散液(固形分濃度20質量%)を得た。塗布液の調製において、この有機溶媒分散液を用いたこと以外は実施例1と同様に塗布液を調製した。また、XPSの測定試料は、シリカを表面処理する前の粒子を用いて作製した。すなわち、実施例1と同様のXPS測定試料を用いた。
結晶成長工程において、水熱合成する代わりに90℃で40時間リフラックスしながら加熱撹拌することによりコア粒子を結晶成長させた。それ以外は、実施例1と同様に塗布液を調製した。
限外濾過膜装置を用いて実施例1のコア粒子の分散液を濃縮して、粒子の水分散液(固形分濃度が4質量%)1125gを得た。メタノール1125gと、正珪酸エチル(多摩化学社製、SiO2換算でケイ素が28.8質量%含まれる)28.2gをこの水分散液に添加した。この水分散液を50℃で加熱すると、分散液の粘度が高くなり、分散液が白濁した。
実施例1の混合液を60℃で40時間攪拌し、粒子の水分散液を得た。この水分散液は薄い黄色であった。水分散液中の粒子はルチルとアナターゼの混晶であった。
TiO2換算で2重量%の四塩化チタン水溶液2250gと、15重量%のアンモニア水880gとを混合した。これにより、pH8.6の白色のスラリー(ゲル)を調製した。このスラリーを濾過した後、純水を用いてゲルを洗浄した。これにより、固形分濃度が5重量%のケーキ900gを得た。ケーキ900gに、35重量%の過酸化水素水514gと純水2100gを加えた。これを80℃で1時間加熱した。さらに、これに純水を986g添加した。この分散液4500gに、カタロイドSN-350を48.8gと純水683.8gとを添加した。オートクレーブ中にて165℃で18時間この分散液を水熱合成することにより、コア粒子の分散液5232.6gを得た。この分散液4500gに、実施例1のチタン含有化合物の分散液4500gを添加した。オートクレーブを用いて165℃で18時間この分散液を水熱合成した。水熱合成後の分散液のレーザー粒子径は400nmで、白濁していた。
限外濾過膜装置を用いて比較例3で得たコア粒子の分散液を濃縮することにより、粒子の水分散液(固形分濃度が10重量%)を得た。この水分散液に陽イオン交換樹脂を添加することにより、脱アルカリを行った。イオン交換樹脂を水分散液から分離した。粒子の有機溶媒分散液の調製において、この水分散液2250gに、メタノール2250gと正系酸エチル65.5gを添加したこと以外は実施例1と同様に粒子のアルコール分散液(固形分濃度が20質量%)を得た。5質量%アンモニア水を12.4gこのアルコール分散液に添加した。さらに、第二表面処理剤としてKBM-503を77.3gこのアルコール分散液に添加した。KBM-503を添加した後は、実施例1と同様に粒子の有機溶媒分散液(固形分濃度20質量%)を得た。塗布液の調製において、この有機溶媒分散液を用いたこと以外は実施例1と同様に塗布液を調製した。
TiO2換算で7.5重量%の四塩化チタン水溶液2000gと7.5重量%のアンモニア水2000gとを混合した。これにより、pH9.2の白色のスラリー(ゲル)を調製した。このスラリーを濾過した後、純水を用いてゲルを洗浄した。これにより、固形分が10重量%であるケーキ1500gを得た。純水を用いてこのケーキ1500gを1.5重量%に希釈した。35重量%の過酸化水素水1714gをこれに添加した。これを80℃で1時間加熱した。これに純水3286gを添加することより、チタン含有化合物の分散液を得た。この水溶液のpHは7.8であった。この分散液15000gに、陽イオン交換樹脂を添加することにより、脱アルカリ処理を行った。1重量%の錫酸カリウム水溶液を1901g分散液に添加した。イオン交換樹脂を分散液から分離した。カタロイドSN-350を634gと純水3591gとをこの分散液に添加した。オートクレーブを用いて165℃で18時間この分散液を水熱合成した。限外濾過膜装置を用いてこの分散液を濃縮した。この分散液2641gに陽イオン交換樹脂を添加することにより、脱アルカリを行った。イオン交換樹脂を分散液から分離することにより、粒子の水分散液を調製した。この水分散液のレーザー粒子径は15nmであった。粒子の有機溶媒分散液の調製において、この水分散液2250gに、メタノール2250gと正系酸エチル165.1gを添加したこと以外は実施例1と同様に粒子のアルコール分散液(固形分濃度が20質量%)を得た。5質量%アンモニア水を31.2gこのアルコール分散液に添加した。さらに、第二表面処理剤としてKBM-503を93.9gこのアルコール分散液に添加した。KBM-503を添加した後は、実施例1と同様に粒子の有機溶媒分散液(固形分濃度20質量%)を得た。塗布液の調製において、この有機溶媒分散液を用いたこと以外は実施例1と同様に塗布液を調製した。

Claims (9)
- TiO2換算で酸化チタンを90重量%以上、SnO2換算で酸化スズを0.2~10重量%含み、
結晶子径が7nm以上であり、
ルチル型の結晶構造を有する粒子。 - 前記粒子の表面からスズが検出されないことを特徴とする請求項1に記載の粒子。
- 前記粒子を溶媒に分散させたとき、当該粒子の平均粒子径が100nm以下であることを特徴とする請求項1または2に記載の粒子。
- ルチル型の結晶構造を有する粒子と、溶媒と、を含む粒子の分散液であって、
前記粒子がTiO2換算で酸化チタンを90重量%以上、SnO2換算で酸化スズを0.2~10重量%含み、
前記粒子の結晶子径が7nm以上であり、
前記粒子の平均粒子径が100nm以下であることを特徴とする分散液。 - 前記溶媒が水であり、
前記粒子が、前記粒子100質量部に対してSiO2換算で2質量部以上のシリカを含み、
当該分散液の等電点が3以下であることを特徴とする請求項4に記載の分散液。 - チタン含有化合物の分散液と、ルチル型の結晶構造を有するコア粒子の分散液と、を準備する工程と、
前記チタン含有化合物の分散液と、前記コア粒子の分散液と、を混合することにより、混合液を調製する工程と、
前記混合液を80℃以上にすることにより、前記コア粒子を元に結晶成長させる工程と、を順に備え、
前記コア粒子が酸化チタンと酸化スズとを含むことを特徴とする粒子の製造方法。 - 前記コア粒子を元に結晶成長させる工程の後の分散液にシリカを添加する工程と、
前記シリカを添加した後の分散液を水熱合成する工程と、を順に備えることを特徴とする請求項6に記載の粒子の製造方法。 - 請求項1に記載の粒子、または請求項6に記載の製造方法により得た粒子を含むことを特徴とする塗布液。
- 請求項8に記載の塗布液を用いて基材上に膜を形成することを特徴とする膜付基材の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22781183.3A EP4317070A4 (en) | 2021-03-31 | 2022-03-30 | PARTICLES HAVING RUTILE CRYSTAL STRUCTURE, PRODUCTION METHOD THEREOF, AND PRODUCTION METHOD FOR PARTICLE DISPERSION LIQUID, COATING LIQUID, AND SUBSTRATE WITH FILM |
| CN202280017046.7A CN116997528B (zh) | 2021-03-31 | 2022-03-30 | 具有金红石型晶体结构的粒子及其制造方法以及粒子的分散液、涂布液、带膜基材的制造方法 |
| US18/547,770 US20240301211A1 (en) | 2021-03-31 | 2022-03-30 | Particles having rutile crystal structure, production method therefor, and production method for particle dispersion liquid, coating liquid, and substrate with film |
| JP2023511528A JPWO2022210973A1 (ja) | 2021-03-31 | 2022-03-30 | |
| KR1020237031445A KR20230161959A (ko) | 2021-03-31 | 2022-03-30 | 루틸형의 결정 구조를 갖는 입자 및 그 제조 방법, 그리고 입자의 분산액, 도포액, 막 구비 기재의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021061695 | 2021-03-31 | ||
| JP2021-061695 | 2021-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022210973A1 true WO2022210973A1 (ja) | 2022-10-06 |
Family
ID=83459628
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/016293 Ceased WO2022210973A1 (ja) | 2021-03-31 | 2022-03-30 | ルチル型の結晶構造を有する粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20240301211A1 (ja) |
| EP (1) | EP4317070A4 (ja) |
| JP (1) | JPWO2022210973A1 (ja) |
| KR (1) | KR20230161959A (ja) |
| CN (1) | CN116997528B (ja) |
| TW (1) | TW202302463A (ja) |
| WO (1) | WO2022210973A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20250103794A (ko) | 2023-12-20 | 2025-07-07 | 닛산 가가쿠 가부시키가이샤 | 입도 분포가 고른 코어 셸 구조를 갖는 금속 산화물 입자 및 그 제조 방법 |
| WO2025164644A1 (ja) * | 2024-01-31 | 2025-08-07 | 日産化学株式会社 | 酸化物コロイド粒子、そのゾル及びその製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02255532A (ja) * | 1989-03-30 | 1990-10-16 | Catalysts & Chem Ind Co Ltd | ルチル型酸化チタンゾルの製造法 |
| JP2007197278A (ja) * | 2006-01-27 | 2007-08-09 | Mitsui Chemicals Inc | 無機酸化物超微粒子およびその製造法 |
| JP2010042947A (ja) | 2008-08-11 | 2010-02-25 | Jgc Catalysts & Chemicals Ltd | 酸化チタン系複合粒子の分散液および該分散液の製造方法 |
| JP2012056816A (ja) * | 2010-09-10 | 2012-03-22 | Jgc Catalysts & Chemicals Ltd | コアシェル型無機酸化物微粒子の分散液、その製造方法および該分散液を含む塗料組成物 |
| WO2018181241A1 (ja) | 2017-03-31 | 2018-10-04 | 日揮触媒化成株式会社 | 鉄含有ルチル型酸化チタン微粒子分散液の製造方法、鉄含有ルチル型酸化チタン微粒子およびその用途 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997015526A1 (en) * | 1995-10-27 | 1997-05-01 | E.I. Du Pont De Nemours And Company | Hydrothermal process for making ultrafine metal oxide powders |
| JP5514487B2 (ja) * | 2008-12-27 | 2014-06-04 | 日揮触媒化成株式会社 | 高屈折率金属酸化物微粒子の水分散ゾル、その調製方法および該金属酸化物微粒子の有機溶媒分散ゾル |
| JP5854584B2 (ja) * | 2009-11-30 | 2016-02-09 | 日揮触媒化成株式会社 | 高屈折率金属酸化物微粒子を含む水分散ゾルの調製方法、該方法から得られる水分散ゾルおよび前記微粒子を含む有機溶媒分散ゾル並びに塗料組成物 |
| GB201300810D0 (en) * | 2013-01-16 | 2013-02-27 | Llika Technologies Ltd | Composite Materials |
| JP2014196215A (ja) * | 2013-03-29 | 2014-10-16 | 日揮触媒化成株式会社 | 改質金属酸化物微粒子粉末およびその製造方法 |
-
2022
- 2022-03-30 EP EP22781183.3A patent/EP4317070A4/en active Pending
- 2022-03-30 US US18/547,770 patent/US20240301211A1/en active Pending
- 2022-03-30 WO PCT/JP2022/016293 patent/WO2022210973A1/ja not_active Ceased
- 2022-03-30 CN CN202280017046.7A patent/CN116997528B/zh active Active
- 2022-03-30 JP JP2023511528A patent/JPWO2022210973A1/ja active Pending
- 2022-03-30 KR KR1020237031445A patent/KR20230161959A/ko active Pending
- 2022-03-31 TW TW111112561A patent/TW202302463A/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02255532A (ja) * | 1989-03-30 | 1990-10-16 | Catalysts & Chem Ind Co Ltd | ルチル型酸化チタンゾルの製造法 |
| JP2007197278A (ja) * | 2006-01-27 | 2007-08-09 | Mitsui Chemicals Inc | 無機酸化物超微粒子およびその製造法 |
| JP2010042947A (ja) | 2008-08-11 | 2010-02-25 | Jgc Catalysts & Chemicals Ltd | 酸化チタン系複合粒子の分散液および該分散液の製造方法 |
| JP2012056816A (ja) * | 2010-09-10 | 2012-03-22 | Jgc Catalysts & Chemicals Ltd | コアシェル型無機酸化物微粒子の分散液、その製造方法および該分散液を含む塗料組成物 |
| WO2018181241A1 (ja) | 2017-03-31 | 2018-10-04 | 日揮触媒化成株式会社 | 鉄含有ルチル型酸化チタン微粒子分散液の製造方法、鉄含有ルチル型酸化チタン微粒子およびその用途 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4317070A4 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20250103794A (ko) | 2023-12-20 | 2025-07-07 | 닛산 가가쿠 가부시키가이샤 | 입도 분포가 고른 코어 셸 구조를 갖는 금속 산화물 입자 및 그 제조 방법 |
| WO2025164644A1 (ja) * | 2024-01-31 | 2025-08-07 | 日産化学株式会社 | 酸化物コロイド粒子、そのゾル及びその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN116997528B (zh) | 2026-04-07 |
| US20240301211A1 (en) | 2024-09-12 |
| EP4317070A4 (en) | 2025-07-30 |
| CN116997528A (zh) | 2023-11-03 |
| JPWO2022210973A1 (ja) | 2022-10-06 |
| EP4317070A1 (en) | 2024-02-07 |
| TW202302463A (zh) | 2023-01-16 |
| KR20230161959A (ko) | 2023-11-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4276077B2 (ja) | ルチル様結晶相を有するナノ粒子およびその調製方法 | |
| JP5754943B2 (ja) | 高屈折率金属酸化物微粒子を含む塗料組成物および該塗料組成物を基材上に塗布して得られる硬化性塗膜 | |
| JP4279465B2 (ja) | 透明な金属酸化物のコロイド及びセラマーを製造するためのナノサイズ金属酸化物の粒子 | |
| US7276231B2 (en) | Lower-energy process for preparing passivated inorganic nanoparticles | |
| JP5717252B2 (ja) | 高屈折率粉末、その製造方法及び用途 | |
| CN102625817B (zh) | 球状核壳型氧化铈/高分子杂化纳米粒子的聚集体及其制造方法 | |
| JP5854584B2 (ja) | 高屈折率金属酸化物微粒子を含む水分散ゾルの調製方法、該方法から得られる水分散ゾルおよび前記微粒子を含む有機溶媒分散ゾル並びに塗料組成物 | |
| JP5514487B2 (ja) | 高屈折率金属酸化物微粒子の水分散ゾル、その調製方法および該金属酸化物微粒子の有機溶媒分散ゾル | |
| WO2022210973A1 (ja) | ルチル型の結晶構造を有する粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 | |
| JP5922231B2 (ja) | ジルコニアコロイドを生成するための方法 | |
| JP4681879B2 (ja) | 光触媒の透明な膜における被覆剤として使用される溶液を生産する工程 | |
| JP7399599B2 (ja) | ルチル型酸化チタンオルガノゾルおよびルチル型酸化チタンオルガノゾルの製造方法並びにこのルチル型酸化チタンオルガノゾルを用いた高屈折率被膜形成用組成物および光学素子 | |
| WO2008007708A1 (en) | Method for producing modified zirconium oxide-tin oxide composite sol | |
| JP5511368B2 (ja) | 高屈折率金属酸化物微粒子を含む有機溶媒分散ゾルの調製方法並びにその有機溶媒分散ゾル、および該有機溶媒分散ゾルを用いて得られる塗料組成物 | |
| JP7169493B1 (ja) | 塗布液及びその製造方法、膜付基材の製造方法 | |
| JP2024052629A (ja) | ルチル型の結晶構造を有する粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 | |
| JP7551597B2 (ja) | シリカ-チタニア複合酸化物粉末 | |
| JP2023152781A (ja) | 中空シリカ粒子の分散液、及びその製造方法 | |
| WO2023189785A1 (ja) | 中空シリカ粒子の分散液、及びその製造方法 | |
| JP2024051854A (ja) | 光触媒活性が低い粒子およびその製造方法、並びに粒子の分散液、塗布液、膜付基材の製造方法 | |
| JP2023147930A (ja) | 粒子の分散液と、その製造方法、塗布液および膜付基材の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22781183 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18547770 Country of ref document: US Ref document number: 202280017046.7 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023511528 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022781183 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022781183 Country of ref document: EP Effective date: 20231031 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |