WO2023049724A1 - Prc2 inhibitors for use in treating blood disorders - Google Patents
Prc2 inhibitors for use in treating blood disorders Download PDFInfo
- Publication number
- WO2023049724A1 WO2023049724A1 PCT/US2022/076750 US2022076750W WO2023049724A1 WO 2023049724 A1 WO2023049724 A1 WO 2023049724A1 US 2022076750 W US2022076750 W US 2022076750W WO 2023049724 A1 WO2023049724 A1 WO 2023049724A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- equiv
- μmol
- methyl
- mixture
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the present disclosure relates to compounds that inhibit the Polycomb Repressive Complex 2 (PRC2).
- PRC2 Polycomb Repressive Complex 2
- the present disclosure relates to compounds, pharmaceutical compositions comprising the compounds and methods for use therefor in treating blood disorders, including sickle cell disease and thalassemia.
- the Polycomb Repressive Complex 2 is a multiprotein complex that contributes to the epigenetic silencing of target genes to regulate development and homeostasis.
- the PRC2 complex is comprised of three core subunits: enhancer of zeste homolog 2 (EZH2), embryonic ectoderm development protein (EED), and suppressor of zeste 12 (SUZ12).
- EED is a critical regulator of PRC2 in the silencing of expression of genes and gene clusters involved in development including but not limited to fetal orthologues (i.e. gamma globin), Hox genes, X chromosome inactivation, etc.
- EED provides a pharmacologic target for the treatment of diseases or disorders to impact transcription of specific target genes in blood and other tissues.
- a method of treating a blood disorder in a subject comprising administering to the subject a therapeutically effective amount of a compound of Formula (I): or a pharmaceutically acceptable salt thereof: wherein: represents a single or a double bond; Z is O or S; X is O, CR 5 , CR 5 OH, C(R 5 )2, wherein: when X is O, is a single bond; when X is C(R 5 )2, is a single bond; when X is CR 5 OH, is a single bond; or when X is CR 5 , is a double bond; R 1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R 4 ; R 2 is cyano, -COOR 5 or -
- a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is selected from Acute lymphoblastic leukemia (ALL), Acute myeloid leukemia (AML) (e.g., acute promyelocytic leukemia, APL), Amyloidosis, Anemia, Aplastic anemia, Bone marrow failure syndromes, Chronic lymphocytic leukemia (CLL), Chronic myeloid leukemia (CML), Deep vein thrombosis (DVT), Diamond-Blackfan anemia, Dyskeratosis congenita (DKC), Eosinophilic disorder, Essential thrombocythemia, Fanconi anemia, Gaucher disease, Hemochromatosis, Hemolytic anemia, Hemophilia, Hereditary spherocytosis, Hodgkin's lymphoma, Idiopathic thrombocytopen
- ALL Acute lymphoblast
- the thalassemia is alpha thalassemia.
- the thalassemia is beta thalassemia.
- FIG.1A depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32, or hydroxyurea (HU) from Donor A.
- FIG.1B depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B.
- FIG.2A depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A.
- FIG.2B depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A.
- FIG.2C depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B.
- FIG.2D depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B.
- DETAILED DESCRIPTION OF THE DISCLOSURE [00016] Unless defined otherwise, all terms and ranges used herein have the same meaning as is commonly understood by one of skill in the art to which this disclosure belongs, unless expressly defined otherwise. All patents, patent applications, and publications referred to herein are incorporated by reference to the extent they are consistent with the present disclosure.
- a bivalent linking moiety can be “alkyl,” in which case those skilled in the art will understand the alkyl to be a divalent radical (e.g., -CH2-CH2-), which is equivalent to the term “alkylene.”
- alkyl a divalent radical
- aryl a divalent moiety that is required and is stated as being “aryl”
- All atoms are understood to have their normal number of valences for bond formation (i.e., 4 for carbon, 3 for N, 2 for O, and 2, 4, or 6 for S, depending on the oxidation state of the S).
- Polycomb Repressive Complex 2 or “PRC2 complex” refers to a mammalian multiprotein complex comprising three core subunits: enhancer of zeste homolog 2 (EZH2), embryonic ectoderm development protein (EED), and suppressor of zeste 12 (SUZ12) and two additional non- essential subunits, AEBP2, and RbAp48.
- EED refers to the embryonic ectoderm development protein subunit of the PRC2 complex.
- EZH2 or “EZH2 enzyme” refers to a mammalian histone methyltransferase, which is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2), and functions to silence target genes by tri-methylating lysine 27 of histone H3 (H3K27me3).
- PRC2 inhibitor refers to compounds of the present disclosure that are represented by formula (I) as described herein. These compounds are capable of negatively modulating or inhibiting all or a portion of the enzymatic activity of the PRC2 complex.
- inhibitors of the present disclosure may inhibit PRC2 enzymatic activity by binding to EED to prevent assembly of the PRC2 complex on histone H3 tails thereby inhibiting its activity.
- amino refers to –NH2.
- acetyl refers to “-C(O)CH3.
- acyl refers to an alkylcarbonyl or arylcarbonyl substituent wherein the alkyl and aryl portions are as defined herein.
- alkyl as employed herein refers to straight and branched chain aliphatic groups having from 1 to 12 carbon atoms.
- alkyl encompasses C 1 , C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 and C 12 groups.
- alkyl groups include, without limitation, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, and hexyl.
- alkenyl as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon double bonds, having from 2 to 12 carbon atoms.
- alkenyl encompasses C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 and C 12 groups.
- alkenyl groups include, without limitation, ethenyl, propenyl, butenyl, pentenyl, and hexenyl.
- alkynyl as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon triple bonds, having from 2 to 12 carbon atoms.
- alkynyl encompasses C 2 , C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 and C 12 groups.
- alkynyl groups include, without limitation, ethynyl, propynyl, butynyl, pentynyl, and hexynyl.
- An “alkylene,” “alkenylene,” or “alkynylene” group is an alkyl, alkenyl, or alkynyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
- alkylene groups include, without limitation, methylene, ethylene, propylene, and butylene.
- alkenylene groups include, without limitation, ethenylene, propenylene, and butenylene.
- alkynylene groups include, without limitation, ethynylene, propynylene, and butynylene.
- alkoxy refers to –OC 1 –C 6 alkyl.
- cycloalkyl as employed herein is a saturated and partially unsaturated cyclic hydrocarbon group having 3 to 12 carbons.
- cycloalkyl includes C 3 , C 4 , C 5 , C 6 , C 7 , C 8 , C 9 , C 10 , C 11 and C 12 cyclic hydrocarbon groups.
- cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl.
- heteroalkyl refers to an alkyl group, as defined hereinabove, wherein one or more carbon atoms in the chain are independently replaced O, S, or NR x , wherein R x is hydrogen or C 1 –C 3 alkyl.
- heteroalkyl groups include methoxymethyl, methoxyethyl and methoxypropyl.
- An “aryl” group is a C 6 -C 14 aromatic moiety comprising one to three aromatic rings. As such, “aryl” includes C 6 , C 10 , C 13 , and C 14 cyclic hydrocarbon groups.
- An exemplary aryl group is a C 6 -C 10 aryl group.
- aryl groups include, without limitation, phenyl, naphthyl, anthracenyl, and fluorenyl.
- An “aralkyl” or “arylalkyl” group comprises an aryl group covalently linked to an alkylene group wherein the moiety is linked to another group via the alkyl moiety.
- An exemplary aralkyl group is –(C 1 -C 6 )alkyl(C 6 -C 10 )aryl, including, without limitation, benzyl, phenethyl, and naphthylmethyl.
- a “heterocyclyl” or “heterocyclic” group is a mono- or bicyclic (fused or spiro) ring structure having from 3 to 12 atoms, (3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 atoms), for example 4 to 8 atoms, wherein one or more ring atoms are independently –C(O)-, N, NR 5 , O, or S, and the remainder of the ring atoms are quaternary or carbonyl carbons.
- heterocyclic groups include, without limitation, epoxy, oxiranyl, oxetanyl, azetidinyl, aziridinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydrothiophenyl, pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, thiazolidinyl, thiatanyl, dithianyl, trithianyl, azathianyl, oxathianyl, dioxolanyl, oxazolidinyl, oxazolidinonyl, decahydroquinolinyl, piperidonyl, 4- piperidonyl, thiomorpholinyl, dimethyl-morpholinyl, and morpholinyl.
- L-heterocyclyl refers to a heterocyclyl group covalently linked to another group via an alkylene linker L, where L is C 1 –C 4 alkylene.
- heteroaryl refers to a group having 5 to 14 ring atoms, preferably 5, 6, 10, 13 or 14 ring atoms; having 6, 10, or 14 ⁇ electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to three heteroatoms that are each independently N, O, or S.
- Heteroaryl also includes fused multicyclic (e.g., bicyclic) ring systems in which one or more of the fused rings is non- aromatic, provided that at least one ring is aromatic and at least one ring contains an N, O, or S ring atom.
- fused multicyclic e.g., bicyclic
- heteroaryl groups include acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzo[d]oxazol-2(3H)-one, 2H-benzo[b][1,4]oxazin-3(4H)-one, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, furanyl, furazanyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H
- a “L-heteroaryl,” “heteroaralkyl” or “heteroarylalkyl” group comprises a heteroaryl group covalently linked to another group via an alkylene linker.
- heteroalkyl groups comprise a C 1 - C 6 alkyl group and a heteroaryl group having 5, 6, 9, or 10 ring atoms.
- heteroaralkyl groups include pyridylmethyl, pyridylethyl, pyrrolylmethyl, pyrrolylethyl, imidazolylmethyl, imidazolylethyl, thiazolylmethyl, thiazolylethyl, benzimidazolylmethyl, benzimidazolylethyl quinazolinylmethyl, quinolinylmethyl, quinolinylethyl, benzofuranylmethyl, indolinylethyl isoquinolinylmethyl, isoinodylmethyl, cinnolinylmethyl, and benzothiophenylethyl.
- arylene is an bivalent aryl, heteroaryl, or heterocyclyl group, respectively, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
- arylene is an bivalent aryl, heteroaryl, or heterocyclyl group, respectively, as defined hereinabove, that is positioned between and serves to connect two other chemical groups.
- substituents it is meant that the group optionally has from one to four, preferably from one to three, more preferably one or two, non- hydrogen substituents.
- halogen refers to chlorine, bromine, fluorine, or iodine.
- haloalkyl refers to an alkyl chain in which one or more hydrogens have been replaced by a halogen. Exemplary haloalkyls are trifluoromethyl, difluoromethyl, flurochloromethyl, chloromethyl, and fluoromethyl.
- hydroxyalkyl refers to an alkyl chain, as defined herein, wherein at least on hydrogen of the alkyl chain has been replaced by hydroxyl.
- an effective amount” of a compound is an amount that is sufficient to negatively modulate or inhibit the activity of PRC2 complex.
- a “therapeutically effective amount” of a compound is an amount that is sufficient to ameliorate or in some manner reduce a symptom or stop or reverse progression of a condition, or negatively modulate or inhibit the activity of PRC2 complex. Such amount may be administered as a single dosage or may be administered according to a regimen, whereby it is effective.
- treatment means any manner in which the symptoms or pathology of a condition, disorder or disease in a subject are ameliorated or otherwise beneficially altered.
- amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition refers to any lessening, whether permanent or temporary, lasting, or transient, that can be attributed to or associated with administration of the composition.
- a method of treating a blood disorder in a subject comprising administering to the subject a therapeutically effective amount of a compound of Formula (I): or a pharmaceutically acceptable salt thereof: wherein: represents a single or a double bond; Z is O or S; X is O, CR 5 , CR 5 OH, C(R 5 )2, wherein: when X is O, is a single bond; when X is C(R 5 )2, is a single bond; when X is CR 5 OH, is a single bond; or when X is CR 5 , is a double bond; R 1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R 4 ; R 2 is cyano, -COOR
- Z is O. In one embodiment, Z is S.
- X is C(R 5 ) 2 and is a single bond. [00051] In one embodiment, Z is O or S.
- X is O, CR 5 , CR 5 OH or C(R 5 )2, wherein when X is O, is a single bond; when X is C(R 5 )2, is a single bond; when X is CR 5 OH, is a single bond; or when X is CR 5 , is a double bond.
- n is one. In one embodiment, n is two.
- Z is O, X is O, n is one and is a single bond. In another embodiment, Z is O, X is CR 5 and is a double bond. In one embodiment, Z is O, X is C(R 5 )2, n is one, and is a single bond. In one embodiment, Z is O, X is CR 5 OH, n is one, and is a single bond. In another embodiment, Z is O, X is C(R 5 )2, n is two, and is a single bond. In yet another embodiment, Z is S, X is C(R 5 )2, n is one, and is a single bond.
- R 1 is aryl, which may be optionally substituted with one or more R 4 .
- the aryl is phenyl, which may be optionally substituted with one or more R 4 .
- the aryl is substituted with a single R 4 group. In one embodiment, the aryl is substituted with two R 4 groups. In one embodiment, the aryl is substituted with three R 4 groups.
- Exemplary aryl R 4 groups include halogen, hydroxyl, haloalkyl, -Y 1 -C 1 –C 6 alkyl, Y 2 -C 1 –C 6 alkyl, -L- N(R 5 )2, -Y 1 -N(R 5 )2, -Y 2 -N(R 5 )2, Y 2 -haloalkyl, L-heterocyclyl, or Y 1 -heterocyclyl, wherein the heterocyclyl portion of the L-heterocyclyl, or Y 1 -heterocyclyl may be optionally substituted with one or more R 7 .
- the one or more R 4 are each independently halogen, hydroxyl, haloalkyl, - COOR 5 , -Y 1 -C 1 –C 6 alkyl, Y 2 -C 1 –C 6 alkyl, -L-N(R 5 )2, -O-L-N(R 5 )2, -C(CF3)N(R 5 )2, -Y 1 -N(R 5 )2, -Y 2 - N(R 5 )2, Y 2 -haloalkyl, -L-heterocyclyl, or -Y 1 -heterocyclyl, wherein the heterocyclyl portion of the -L- heterocyclyl or -Y 1 -heterocyclyl may be optionally substituted with one or more R 7 .
- R 1 is phenyl substituted with -Y 2 -C 1 –C 6 alkyl.
- Y is a bond and the C 1 –C 6 alkyl is methyl, ethyl or isopropyl.
- R 1 is phenyl substituted with the Y 2 -C 1 –C 6 alkyl, wherein Y 2 is -SO 2 - and the C1– C6 alkyl is methyl.
- R 1 is phenyl, which is disubstituted with methyl and Y 2 -C 1 –C 6 alkyl, wherein Y 2 is -SO 2 - and the C1– C 6 alkyl is methyl.
- R 1 is phenyl substituted one R 4 , wherein R 4 is a cyano group.
- R 1 is phenyl substituted one R 4 , wherein R 4 is L-heteroaryl. In certain embodiments, the L-heteroaryl is tetrazolyl.
- R 1 is phenyl substituted one R 4 , wherein R 4 is PO 3 (C1-C3 alkyl) 2 .
- R 1 is phenyl substituted one R 4 , wherein R 4 is - COOR 5 .
- R 1 is phenyl substituted one R 4 , wherein R 4 is -O-L-N(R 5 ) 2 . In one embodiment, R 1 is phenyl substituted one R 4 , wherein R 4 is aralkyl. [00059] In one embodiment, R 1 is phenyl substituted with at least one R 4 , wherein R 4 is -L-N(R 5 ) 2 . In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R 5 is independently hydrogen. In one embodiment, each R 5 is independently C 1 –C 3 alkyl. In one embodiment, each C 1 –C 3 alkyl is methyl.
- one R 5 is C 1 –C 3 alkyl and the other is hydrogen. In one embodiment, the one C 1 –C 3 alkyl is methyl. In one embodiment, R 1 is phenyl substituted with -L-N(R 5 ) 2 and further substituted with one or more halogen and/or C 1 –C 6 alkyl. [00060] In one embodiment, R 1 is phenyl substituted with one R 4 , wherein R 4 is -Y 1 -N(R 5 ) 2 . In certain embodiments, Y 1 is -C(O)- and each R 5 is independently C 1 –C 3 alkyl. In one embodiment, each C 1 –C 3 alkyl is methyl.
- Y 1 is -C(O)- and each R 5 is hydrogen. In one embodiment, Y 1 is - C(O)- and one R 5 is C 1 –C 3 alkyl and the other is hydrogen. In one embodiment, the one C 1 –C 3 alkyl is methyl. In one embodiment, R 1 is phenyl substituted with -Y 1 -N(R 5 )2 and further substituted with one or more halogen and/or C 1 –C 6 alkyl. [00061] In one embodiment, R 1 is phenyl substituted with the Y 2 -haloalkyl, wherein Y 2 is -S- or -SO 2 - and the haloalkyl is trifluoromethyl.
- R 1 is phenyl substituted with at least one -L-heterocyclyl or -Y 1 - heterocyclyl, each heterocyclyl optionally substituted with one or more R 7 .
- R 1 is phenyl substituted with one R 4 , wherein R 4 is -Y 1 -heterocyclyl optionally substituted with one or more R 7 .
- Y 1 is -C(O)- and the heterocyclyl is piperazinyl optionally substituted with C 1 – C 3 alkyl.
- the R 4 group is L-heterocyclyl optionally substituted with one or more R 7 .
- L is methylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4- methyl-piperazinyl.
- L is methylene and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R 7 .
- R 7 groups include oxo, halogen, hydroxyalkyl and C 1 –C 3 alkyl.
- R 1 is phenyl substituted with Y 1 -heterocyclyl optionally substituted with one or more R 7 .
- Y 1 is -C(O)- and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl, each optionally further substituted with one or more halogen.
- R 1 is phenyl substituted with L-heteroaryl optionally substituted with one or more R 7 .
- the L-heteroaryl is tetrazolyl.
- R 1 is phenyl substituted with PO 3 (C 1 -C 3 alkyl) 2 . In another embodiment, R 1 is phenyl substituted with -COOR 5 . In one embodiment, R 1 is phenyl substituted with hydroxyalkyl, - O-L-N(R 5 ) 2 or aralkyl. [00067] In one embodiment, R 1 is heteroaryl, which may be optionally substituted with one or more R 4 .
- the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, isoxazolyl, isoindolinyl, naphthyridinyl, 1,2,3,4-tetrahydroisoquinolinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R 4 .
- the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R 4 .
- the heteroaryl is substituted with a single R 4 group. In one embodiment, the heteroaryl is substituted with two R 4 groups. In one embodiment, the heteroaryl is substituted with three R 4 groups.
- heteroaryl R 4 groups include amino, cyano, halogen, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, Y 2 -haloalkyl Y 1 -C 1 –C 6 alkyl, Y 2 -C 1 –C 6 alkyl, L-cycloalkyl, L-heteroaryl, L- heterocyclyl, Y 1 -heterocyclyl, -L-N(R 5 )2, or -Y 1 -N(R 5 )2, wherein the ring of the L-cycloalkyl, L- heteroaryl, L-heterocyclyl or Y 1 -heterocyclyl may be optionally substituted with one or more R 7 .
- each R 4 is independently cyano, halogen, -Y 1 -C 1 –C 6 alkyl, -Y 2 -C 1 –C 6 alkyl, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, -L-cycloalkyl, -L-N(R 5 )2, -Y 1 -N(R 5 )2, -L-heteroaryl, - L-heterocyclyl, or -Y 1 -heterocyclyl, wherein the heteroaryl of the -L-heteroaryl or the heterocyclyl portion of the L-heterocyclyl, or Y 1 -heterocyclyl may be optionally substituted with one or more R 7 .
- R 7 is amino, hydroxyl, cyano, alkoxy, or halogen. In one embodiment, R 7 is C 1 –C 3 alkyl. In one embodiment, R 7 is halogen, wherein the halogen is fluorine or chlorine. In one embodiment, R 7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R 7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. [00071] In another embodiment, R 1 is heteroaryl and each R 4 is independently hydroxyalkyl, heteroalkyl or haloalkyl.
- the hydroxyalkyl is hydroxymethyl, hydroxyethyl or 2- methyl, 2-hydroxypropyl.
- the heteroalkyl is methoxymethyl or methoxyethyl.
- the haloalkyl is fluoromethyl, difluoromethyl, trifluoromethyl, fluoroethyl, difluoroethyl, or trifluoroethyl.
- R 1 is heteroaryl and R 4 is -Y 1 -C 1 –C 6 alkyl, wherein Y 1 is a bond and the C 1 –C 6 alkyl is methyl, ethyl or isopropyl.
- R 4 is -Y 1 -C 1 –C 6 alkyl, wherein Y 1 is a -C(O)- and the C 1 –C 6 alkyl is methyl, ethyl or isopropyl. In other embodiments, Y 1 is -NHC(O)- and the C 1 –C 6 alkyl portion is methyl. [00073] In one embodiment, R 1 is heteroaryl and R 4 is -Y 2 -C 1 –C 6 alkyl, wherein Y 2 is -SO 2 - and the C 1 –C 6 alkyl is methyl.
- R 4 is -Y 2 -C 1 –C 6 alkyl, wherein Y 2 is -S- and the C 1 –C 6 alkyl is methyl.
- R 1 is heteroaryl and R 4 is -Y 1 -heterocyclyl, which may be optionally substituted with one or more R 7 .
- Y 1 is a bond.
- Y 1 is -C(O)-.
- Y 1 is a bond and the heterocyclyl is azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl.
- R 7 is C 1 –C 3 alkyl. In one embodiment, R 7 is halogen.
- the heteroaryl is substituted with at least one R 4 that is -L-heterocyclyl, which may be optionally substituted with one or more R 7 .
- L is ethylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl. In one embodiment, L is methylene and the heterocyclyl is azetinidyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R 7 . [00076] In one embodiment, the R 7 is independently -L-N(R 5 ) 2 , hydroxyl, cyano, alkoxy, or halogen.
- R 7 is C 1 –C 3 alkyl. In one embodiment, R 7 is halogen, wherein the halogen is fluorine or chlorine. In one embodiment, R 7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R 7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. In one embodiment, R 7 is -L- N(R 5 )2. In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R 5 is hydrogen. In one embodiment, each R 5 is independently C 1 –C 3 alkyl. In one embodiment, each C 1 –C 3 alkyl is methyl.
- one R 5 is C 1 –C 3 alkyl and the other is hydrogen. In one embodiment, the one C 1 –C 3 alkyl is methyl. [00077] In one embodiment, R 1 is heteroaryl and R 4 is -L-N(R 5 )2. In one embodiment, L is a bond. In one embodiment, L is methylene, ethylene, or propylene. In one embodiment, each R 5 is independently C 1 –C 3 alkyl. In one embodiment, each C 1 –C 3 alkyl is methyl. In one embodiment, one R 5 is C 1 –C 3 alkyl and the other is hydrogen. In one embodiment, the one C 1 –C 3 alkyl is methyl. In one embodiment, each R 5 is hydrogen.
- R 1 is heteroaryl and R 4 is L-heteroaryl, which may be optionally substituted with one or more R 7 .
- L is a bond.
- L is C 1 –C 3 alkylene.
- the C 1 –C 3 alkylene is methylene.
- the heteroaryl of the L-heteroaryl is pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, triazinyl, thiazolyl or pyridazinyl.
- the heteroaryl of the L-heteroaryl is pyridyl.
- R 1 is heteroaryl which is substituted with two R 4 groups independently selected from two -Y 1 -C 1 –C 6 alkyl groups; -Y 1 -C 1 –C 6 alkyl and alkoxy; -Y 1 -C 1 –C 6 alkyl and cycloalkyl; -Y 1 -C 1 –C 6 alkyl and haloalkyl; -Y 1 -C 1 –C 6 alkyl and amino; two alkoxy groups; alkoxy and halogen; alkoxy and cyano, and amino and haloalkyl.
- R 4 is -Y 1 -C 1 –C 6 alkyl, wherein each Y 1 is a bond and each C 1 –C 6 alkyl is methyl, ethyl or isopropyl.
- the cycloalkyl is cyclopropyl.
- the alkoxy is methoxy.
- the halogen is fluorine or chlorine.
- the haloalkyl is trifluoromethyl or trifluoroethyl.
- R 1 is L-heterocyclyl optionally substituted with one or more R 4 .
- L is a bond and the heterocyclyl is tetrahydrofuranyl, piperidinyl, piperazinyl or morpholinyl.
- L is a methylene and the heterocyclyl is azetidinyl, pyrrolidinyl or 3 ⁇ 2 - azabicyclo[3.1.0]hexanyl.
- the heterocyclyl is substituted with one or more R 4 selected from oxo, halogen, alkoxy, hydroxyl and Y 1 -C 1 –C 6 alkyl, wherein Y is a bond or -C(O)-.
- R 2 is cyano.
- R 2 is -COOR 5 .
- the R 5 group is hydrogen.
- R 2 is -C(O)N(R 5 ) 2 .
- each R 5 is independently C 1 –C 3 alkyl.
- each C 1 –C 3 alkyl is methyl.
- one R 5 is C 1 –C 3 alkyl and the other is hydrogen.
- the one C 1 –C 3 alkyl is methyl.
- each R 5 is hydrogen.
- each R 5 together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R 6 .
- n is zero. In one embodiment, n is one and R 3 is halogen. In certain embodiments, the halogen is fluorine or chlorine. In one embodiment, the halogen is fluorine.
- R 6 is hydrogen, C 1 –C 3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl. In certain embodiments, R 6 is hydrogen. In other embodiments, R 6 is methyl, ethyl, or propyl. [00085] In one embodiment, the cyclyl portion of R 4 group is substituted with one R 7 group.
- R 7 is oxo, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L- N(R 5 )2 or C 1 –C 3 alkyl.
- R 7 is C 1 –C 3 alkyl, wherein the C 1 –C 3 alkyl is methyl, ethyl or isopropyl.
- R 7 is halogen, wherein the halogen is fluorine or chlorine.
- R 7 is oxo. [00086] In one embodiment, the cyclyl portion of R 4 group is substituted with two R 7 groups.
- the two R 7 groups are each halogen, wherein each halogen is fluorine.
- each halogen is fluorine.
- compositions comprising a PRC2 inhibitor as disclosed herein and a pharmaceutically acceptable carrier, excipient, or diluent for use in the treatment of a blood disorder in a subject.
- Compounds of the disclosure may be formulated by any method well known in the art and may be prepared for administration by any route, including, without limitation, parenteral, oral, sublingual, transdermal, topical, intranasal, intratracheal, or intrarectal.
- compounds disclosed herein are administered intravenously in a hospital setting.
- administration may preferably be by the oral route.
- the characteristics of the carrier will depend on the route of administration.
- compositions according to the disclosure may contain, in addition to the inhibitor, diluents, fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art.
- diluents such as a cell, cell culture, tissue, or organism
- solubilizers such as a cell, cell culture, tissue, or organism
- the preparation of pharmaceutically acceptable formulations is described in, e.g., Remington's Pharmaceutical Sciences, 18th Edition, ed. A. Gennaro, Mack Publishing Co., Easton, Pa., 1990.
- salts refers to salts that retain the desired biological activity of the above-identified compounds and exhibit minimal or no undesired toxicological effects.
- examples of such salts include, but are not limited to acid addition salts formed with inorganic acids (for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and the like), and salts formed with organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalenesulfonic acid, naphthalenedisulfonic acid, and polygalacturonic acid.
- inorganic acids for example, hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and the like
- organic acids such as acetic acid, oxalic acid, tartaric acid, succinic
- the compounds can also be administered as pharmaceutically acceptable quaternary salts known by those skilled in the art, which specifically include the quaternary ammonium salt of the formula --NR+Z-, wherein R is hydrogen, alkyl, or benzyl, and Z is a counterion, including chloride, bromide, iodide, --O-alkyl, toluenesulfonate, methylsulfonate, sulfonate, phosphate, or carboxylate (such as benzoate, succinate, acetate, glycolate, maleate, malate, citrate, tartrate, ascorbate, benzoate, cinnamoate, mandeloate, benzyloate, and diphenylacetate).
- R is hydrogen, alkyl, or benzyl
- Z is a counterion, including chloride, bromide, iodide, --O-alkyl, toluenesulfonate, methylsulfonate,
- the active compound is included in the pharmaceutically acceptable carrier or diluent in an amount sufficient to deliver to a subject a therapeutically effective amount without causing serious toxic effects in the subject treated.
- a dose of the active compound for all of the above-mentioned conditions is in the range from about 0.01 to 300 mg/kg, preferably 0.1 to 100 mg/kg per day, more generally 0.5 to about 25 mg per kilogram body weight of the recipient per day.
- a typical topical dosage will range from 0.01-3% wt/wt in a suitable carrier.
- the effective dosage range of the pharmaceutically acceptable derivatives can be calculated based on the weight of the parent compound to be delivered.
- the effective dosage can be estimated as above using the weight of the derivative, or by other means known to those skilled in the art.
- METHODS OF USE [00093] In one embodiment is provided a method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I) as disclosed herein.
- a method of treating a blood disorder in a subject comprising administering to the subject a therapeutically effective amount of a compound of Formula (I) as disclosed herein, wherein the blood disorder is selected from Acute lymphoblastic leukemia (ALL), Acute myeloid leukemia (AML) (e.g., acute promyelocytic leukemia, APL), Amyloidosis, Anemia, Aplastic anemia, Bone marrow failure syndromes, Chronic lymphocytic leukemia (CLL), Chronic myeloid leukemia (CML), Deep vein thrombosis (DVT), Diamond-Blackfan anemia, Dyskeratosis congenita (DKC), Eosinophilic disorder, Essential thrombocythemia, Fanconi anemia, Gaucher disease, Hemochromatosis, Hemolytic anemia, Hemophilia, Hereditary spherocytosis, Hodgkin's lymphoma, Id
- the blood disorder is sickle cell disease.
- the blood disorder is thalassemia.
- the thalassemia is alpha thalassemia.
- the thalassemia is beta thalassemia.
- the method results in induction of fetal hemoglobin expression in erythroid cells.
- the method results in upregulation of mRNA levels of fetal hemoglobin protein.
- the method results in increased levels of fetal hemoglobin protein in the subject.
- the concentration and route of administration to the subject will vary depending on the blood disorder to be treated.
- the compounds, pharmaceutically acceptable salts thereof and pharmaceutical compositions comprising such compounds and salts also may be co-administered with other anti- neoplastic compounds, e.g., chemotherapy, or used in combination with other treatments, such as radiation or surgical intervention, either as an adjuvant prior to surgery or post-operatively.
- Other anti- neoplastic compounds e.g., chemotherapy
- other treatments such as radiation or surgical intervention
- the degree of mono- and dimethylation of histone H3K27 may be monitored in the subject using well known methods, including those described in Example A below, to access the effectiveness of treatment, along with other prognostic or biological factors, and dosages may be adjusted accordingly by the attending medical practitioner.
- GENERAL REACTION SCHEME INTERMEDIATES AND EXAMPLES GENERAL REACTION SCHEMES [000101]
- the compounds disclosed herein may be prepared using methods described in United States Patent No.11,091,495, the contents of which are incorporated by reference herein for that purpose.
- the compounds disclosed herein may also be prepared using methods known to those having ordinary skill in the art and commercially available reagents and intermediates in the synthetic methods and reaction schemes described herein, or may be prepared using other reagents and conventional methods well known to those skilled in the art.
- intermediates for compounds and compounds of formula (I) of the present disclosure may be prepared according to General Reaction Schemes I or II:
- R 2 -ester substituted imidazo[1,2-c]pyrimidine A is coupled to R 3 optionally substituted intermediate amine B by nucleophilic substitution to yield Intermediate C.
- a boronic acid derivative (Y)-R 1 D is coupled via a Suzuki reaction with halogen substituted Intermediate C in the presence of a suitable base, e.g., sodium carbonate, and the R 2 ester is converted to the acid by saponification with NaOH to generate intermediate acid E.
- the acid is converted to the corresponding amide, which is dehydrated to form title compound nitrile G.
- An exemplary Intermediate B, Intermediate B-1 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 )2, and is a single bond.
- reaction mixture was diluted with petroleum ether (100 mL) and washed with brine (50.0 mL ⁇ 4), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a residue.
- the crude material was purified by column chromatography (petroleum ether) to afford 1-bromo-3-(2,2- diethoxyethoxy)benzene (17.0 g, 48.2 mmol, 83.4% yield, 82.0% purity) as a light yellow oil.
- a second exemplary Intermediate B, Intermediate B-2 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is CR 5 , is a double bond and one R 3 is fluorine.
- a third exemplary Intermediate B, Intermediate B-3 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 ) 2 , 3 is a single bond and one R is chlorine.
- the vessel was evacuated and purged with hydrogen several times. The mixture was stirred at 25 °C for 12 h under hydrogen (50.0 psi).
- a fourth exemplary Intermediate B, Intermediate B-4 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 ) 2 , is a single bond and one R 3 is fluorine.
- Z is O
- n is one
- X is C(R 5 ) 2
- R 3 is fluorine.
- a fifth exemplary Intermediate B, Intermediate B-5 may be used to synthesize compounds of formula I wherein Z is S, n is one, X is C(R 5 )2, is a single bond and one R 3 is fluorine.
- a sixth exemplary Intermediate B, Intermediate B-6 may be used to synthesize compounds of formula I wherein Z is O, n is two, X is C(R 5 ) 2 , is a single bond and one R 3 is fluorine.
- An exemplary Intermediate C, Intermediate C-1 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 )2, is a single bond and one R 3 is fluorine.
- a second exemplary Intermediate C, Intermediate C-2 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is CR 5 , is a double bond and one R 3 is fluorine.
- a fourth exemplary Intermediate C, Intermediate C-4 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 )2, is a single bond and one R 3 is chlorine.
- a fifth exemplary Intermediate C, Intermediate C-5 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 ) 2 , is a single bond and one R 3 is fluorine.
- Intermediate C-5 may be prepared as follows: [000207] To a solution of 8-bromo-5-chloro-imidazo[1,2-c]pyrimidine-2- carbonitrile (3.00 g, 11.7 mmol, 1.00 equiv) and (5-fluoro-2,3-dihydrobenzofuran-4-yl) methanamine (2.14 g, 12.8 mmol, 1.10 equiv) in DMF (30.0 mL) was added DIEA (3.01 g, 23.3 mmol, 4.06 mL, 2.00 equiv). The resultant mixture was stirred at 85 °C for 1 h, cooled to rt, and poured into water (100 mL).
- a sixth exemplary Intermediate C, Intermediate C-6 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 )2, is a single bond and one R 3 is fluorine.
- a seventh exemplary Intermediate C, Intermediate C-7 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 ) 2 , is a single 3 1 bond, one R is fluorine and R is heteroaryl which may then be further substituted with one or more R 4 .
- An eighth exemplary Intermediate C, Intermediate C-8 may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R 5 )2, is a single bond, one R 3 is fluorine and R 1 is heteroaryl which is substituted two R 4 groups, one of which serves as an intermediate to generate various R 4 groups, e.g., L-N(R 5 )2.
- reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10.0 mL ⁇ 3). The combined organic layer was washed with brine (20.0 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to provide a residue.
- reaction mixture was diluted with water (10.0 mL) and extracted with ethyl acetate (10.0 mL ⁇ 3). The combined organic layer was washed with brine (20.0 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a residue.
- the mixture was stirred at 25 °C for 1 h and was subsequently diluted with water (20.0 mL).
- the suspension was filtered and the solid was dried under reduced pressure to give the crude product.
- the crude product diluted with water (60.0 mL) and extracted with ethyl acetate (60.0 mL ⁇ 2).
- reaction was quenched with saturated aqueous potassium fluoride (20.0 mL) and extracted with ethyl acetate (20.0 mL ⁇ 2). The combined organic phase was washed with brine (20.0 mL ⁇ 2), dried over anhydrous sodium sulfate, filtered, and concentrated under vacuum to provide a residue.
- the aqueous phase was extracted with ethyl acetate (3.00 mL ⁇ 2).
- the combined organic phase was washed with brine (3.00 mL ⁇ 2), dried with anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to afford 8-bromo-5-(((5-fluorobenzo[d][1,3]dioxol-4-yl)methyl)amino)imidazo[1,2- c]pyrimidine-2-carbonitrile (50.0 mg, 128 ⁇ mol, 82.5% yield) as a yellow solid.
- a third exemplary Intermediate D, Intermediate D-3 may be used to synthesize compounds of formula I or formula II wherein R 1 is heteroaryl substituted with two R 4 substituents.
- a fourth exemplary Intermediate D, Intermediate D-4 may be used to synthesize compounds of formula I, wherein R 1 is heteroaryl substituted with two R 4 substituents.
- a fifth exemplary Intermediate D, Intermediate D-5 may be used to synthesize compounds of formula I, wherein R 1 is heteroaryl substituted with two R 4 substituents.
- the reaction mixture was diluted with DCM (20.0 mL), washed with satd aq potassium carbonate (20.0 mL x 3), brine (20.0 mL x 2), and the organic phase was concentrated under reduced pressure to give a residue.
- the crude material was purified by column chromatography (petroleum ether/ ethyl acetate, 1 / 0 to 3 / 1) to afford 1-bromo-2-methyl-4- methylsulfonyl-benzene (370 mg, 1.41 mmol, 34.0% yield, 95.0% purity) as a white solid.
- a seventh exemplary Intermediate D, Intermediate D-7 may be used to synthesize compounds of formula I, wherein R 1 is aryl substituted with an R 4 substituent.
- R 1 is aryl substituted with an R 4 substituent.
- pyrrolidine (1.79 g, 25.1 mmol, 2.10 mL, 5.00 equiv) in methanol (16.0 mL) was added NaBH 3 CN (347 mg, 5.53 mmol, 1.10 equiv). The mixture was stirred at 20 °C for 24 h.
- n- butyllithium (2.50 M, 1.80 mL, 1.00 equiv) was added dropwise over one min to i-PrMgCl (2.00 M, 1.12 mL, 0.500 equiv) in THF (12 mL) at 0 °C under a nitrogen atmosphere.
- the mixture was stirred at 0 °C for 5 min followed by the addition of 5-bromo-4-chloro-2-methoxy-pyridine (1.00 g, 4.50 mmol, 1.00 equiv) after which the mixture was stirred at 0 °C for 45 min.
- a ninth exemplary Intermediate D, Intermediate D-9 may be used to synthesize compounds of formula I, wherein R 1 is heteroaryl substituted with three R 4 substituents.
- An eleventh exemplary Intermediate D, Intermediate D-11 may be used to synthesize compounds of formula I, wherein R 1 is heteroaryl substituted with three R 4 substituents.
- R 1 is heteroaryl substituted with three R 4 substituents.
- (2S)-2-methyloxirane 392 mg, 6.75 mmol, 473 ⁇ L, 15.0 equiv
- cesium carbonate 29.3 mg, 90.1 ⁇ mol, 0.20 equiv).
- a twelfth exemplary Intermediate D, Intermediate D-12 may be used to synthesize compounds of formula I, wherein R 1 is aryl substituted with two R 4 substituents.
- R 1 is aryl substituted with two R 4 substituents.
- DIEA 490 mg, 3.79 mmol, 660 ⁇ L, 3.00 equiv
- N,N-dimethylamine (2.00 M in THF, 1.27 mL, 2.00 equiv) in DMF (3.00 mL) was added HATU (727 mg, 1.91 mmol, 1.50 equiv).
- EXAMPLE 1 8-(1,3-dimethyl-1H-pyrazol-5-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)imidazo[1,2- c]pyrimidine-2-carboxylic acid
- a mixture of ethyl 8-bromo-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carboxylate (0.100 g, 230 ⁇ mol, 1.00 equiv), 1,3-dimethyl- 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole (81.6 mg, 368 ⁇ mol, 1.60 equiv), sodium bicarbonate (77.2
- the reaction mixture was stirred at 105 °C for 1 h under nitrogen.
- the mixture was cooled to 25 °C and filtered.
- the filtrate was concentrated in vacuo to provide a residue.
- the precipitate was filtered to provide the crude material (80 mg) as a brown solid.
- the crude material was purified by prep- HPLC(column: Phenomenex Synergi C 1 8150 ⁇ 25 ⁇ 10 ⁇ m; mobile phase:[water (0.1 %TFA) - ACN]; B%: 12% - 42%, 10 min) to afford 5-(((5-fluorobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3- yl)imidazo[1,2-c]pyrimidine-2-carboxylic acid (10.0 mg, 99.7 % purity) as a gray solid.
- EXAMPLE 12 5-(((5-fluorobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3-yl)imidazo[1,2-c]pyrimidine-2- [000380] A mixture of 5-(((5-fluorobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3- yl)imidazo[1,2-c]pyrimidine-2-carboxylic acid (80.0 mg, 192 ⁇ mol, 1.00 equiv), DIEA (74.3 mg, 575 ⁇ mol, 100 ⁇ L, 3.00 equiv) and ammonium chloride (30.8 mg, 575 ⁇ mol, 3 equiv) in DMF (3.00 mL) was cooled to 0 °C.
- EXAMPLES 13 -21 were prepared following the procedure set forth in Example 12 and using the general reactions schemes and intermediates described herein. TABLE 2 Characterization of EXAMPLES 13-21 EXAMPLE 22 5-(((5-fluorobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3-yl)imidazo[1,2-c]pyrimidine-2- carbonitrile [000383] To a solution of 5-(((5-fluorobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3- yl)imidazo[1,2-c]pyrimidine-2-carboxamide (45.0 mg, 108 ⁇ mol, 1.00 equiv), TEA (219 mg, 2.16 mmol, 301 ⁇ L, 20.0 equiv) in THF (1.50 mL) was added TFAA (136 mg, 648 ⁇ mol, 90.2 ⁇ L, 6.00 equiv) at 0 °C.
- the mixture was subsequently stirred at 25 °C for 1 h.
- the reaction mixture was diluted with ethyl acetate (10.0 mL) and washed with water (10.0 mL x 3).
- the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated at reduced pressure to give a residue.
- the mixture was subsequently stirred at 25 °C for 1 h.
- the reaction mixture was diluted with ethyl acetate (10.0 mL), the pH was adjusted to ⁇ 7 with TFA, and the organic layer was washed with brine (5.00 mL ⁇ 3). Concentration in vacuo provided the crude material.
- EXAMPLES 88 -136 were prepared following the procedure set forth in Example 87 and using the general reactions schemes and intermediates described herein. TABLE 4 Characterization of EXAMPLES 88-136 EXAMPLE 137 5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3-yl)imidazo[1,2- c]pyrimidine-2-carboxamide [000393] A mixture of 5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(2-methylpyridin-3- yl)imidazo[1,2-c]pyrimidine-2-carbonitrile (38.0 mg, 92.5 ⁇ mol, 1.00 equiv) in conc.
- EXAMPLE 140 [000399] A mixture of ethyl 8-bromo-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carboxylate (0.20 g, 460 ⁇ mol, 1.00 equiv), tert-butyl4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydro-2H-pyridine-1-carboxylate(227 mg, 735 ⁇ mol, 1.60 equiv), NaHCO 3 (116 mg, 1.38 mmol, 3.00 equiv), Pd(dppf)Cl 2 (33.6 mg, 46.0 ⁇ mol, 0.100 equiv) in dioxane (2.10 mL) and water (0.700 mL) was purged with nitrogen.
- the vessel was purged with nitrogen, stirred at 105 °C for 1 h and subsequently concentrated in vacuo to provide a residue.
- the residue was purified by Prep- TLC (SiO2, petroleum ether/ethyl acetate, 1/1) to afford ethyl 8-(3,6-dihydro-2H-pyran-4-yl)-5-(((5- fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carboxylate (81.0 mg, 77.7% yield, 96.6% purity) as a white solid.
- the resultant mixture was stirred at 0 - 30 °C for 1 h and was subsequently filtered and concentrated to provide the crude residue.
- the residue was purified by prep-HPLC (column: Gemini 150 ⁇ 255 u; mobile phase: [water (0.04 %NH 3 H 2 O) - ACN]; B %: 35.0 % - 65.0 %, 10 min) to afford 5- (((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(tetrahydro-2H-pyran-4-yl)imidazo[1,2- c]pyrimidine-2-carbonitrile (26.0 mg, 63.1 ⁇ mol, 92.7% yield, 95.5% purity) as a yellow solid.
- reaction mixture was cooled to rt and quenched with sat aq potassium fluoride (2.00 mL).
- the mixture was extracted with ethyl acetate (2.00 mL ⁇ 3) and the combined organic layer was washed with brine (2.00 mL ⁇ 2), dried over sodium sulfate, filtered, and concentrated under reduced pressure to give a residue.
- EXAMPLE 149 8-(4-((dimethylamino)methyl)-3,5-difluorophenyl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000424] To a solution of 8-bromo-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile (50.0 mg, 129 ⁇ mol, 1.00 eq.), (3,5-difluoro-4- formyl-phenyl)boronic acid (28.7 mg, 155 ⁇ mol, 1.20 eq.) in dioxane (1.00 mL) and water (0.20 mL) was added Pd(dppf)Cl2 (9.42 mg, 12.9 ⁇ mol, 0.10
- EXAMPLE 154 was prepared following the procedure set forth in Example 153 and using the general reactions schemes and intermediates described herein. Table 7 Characterization of EXAMPLE 154 EXAMPLE 155 8-(1,3-dimethyl-1H-pyrazol-5-yl)-5-(((5-fluoro-3-hydroxy-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000431] To a solution of tert-butyl (8-bromo-2-cyanoimidazo[1,2-c]pyrimidin-5-yl)((5-fluoro-2,3- dihydrobenzofuran-4-yl)methyl)carbamate (200 mg, 410 ⁇ mol, 1.00 eq.) in acetonitrile (4.50 mL) was added periodic acid (345 mg, 1.52 mmol, 345 ⁇ L, 3.70 eq.) and chro
- the mixture was stirred at 15 °C for 3 h.
- the reaction mixture was filtered through a plug of Celite.
- the filtrate was diluted with water (3.00 mL) and extracted with ethyl acetate (5.00 mL ⁇ 3).
- the combined organic phase was washed with aqueous sodium sulfite solution (2.00 mL), brine (2.00 mL), dried over anh sod sulfate, filtered, and concentrated to provide the crude material.
- EXAMPLE 156 5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(1-methyl-2-oxo-1,2-dihydropyridin-3- yl)imidazo[1,2-c]pyrimidine-2-carbonitrile [000436] To a solution of tert-butyl (2-cyano-8-(2-oxo-1,2-dihydropyridin-3-yl)imidazo[1,2- c]pyrimidin-5-yl)((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)carbamate (45.0 mg, 89.6 ⁇ mol, 1.00 eq.) in DMF (0.50 mL) was added potassium carbonate (24.8 mg, 179 ⁇ mol, 2.00 eq.) at 0 °C.
- EXAMPLES 157 - 184 were prepared following the procedure set forth in EXAMPLE 87 and using the general reactions schemes and intermediates described herein. Table 8 Characterization of EXAMPLES 157 - 184
- EXAMPLES 193 and 194 [000440] EXAMPLES 193 and 194 were prepared following the procedure set forth in EXAMPLE 142 and using the general reactions schemes and intermediates described herein. Table 9 Characterization of EXAMPLES 193 and 194 EXAMPLES 195 -202 [000441] EXAMPLES 195 -202 were prepared following the procedure set forth in EXAMPLE 149 and using the general reactions schemes and intermediates described herein. Table 10 Characterization of EXAMPLES 195 - 202
- EXAMPLE 207 8-(4-((dimethylamino)methyl)-2-methylphenyl)-5-(((5-fluoro-3-hydroxy-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000452]
- EXAMPLE 210 8-(3-((dimethylamino)methyl)-1-methyl-1H-pyrazol-5-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000460] To a solution of 5-(2-cyano-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidin-8-yl)-N,N,1-trimethyl-1H-pyrazole-3-carboxamide (100 mg, 217 ⁇ mol, 1.00 equiv) in THF (1.00 mL) was added di-tert-butyl dicarbonate (56.9 mg, 261 ⁇ mol, 59.9 ⁇ L, 1.20 equiv) and DMAP (2.65 mg, 21.7 ⁇ mol, 0.10 e
- EXAMPLE 212 8-(3-cyano-1-methyl-1H-pyrazol-5-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000468] To a solution of 5-[2-cyano-5-[(5-fluoro-2,3-dihydrobenzofuran -4- yl)methylamino]imidazo[1,2-c]pyrimidin-8-yl]-1-methyl-pyrazole-3-carboxylic acid (100 mg, 185 ⁇ mol, 1.00 equiv) and ammonium chloride (30.0 mg, 561 ⁇ mol, 3.04 equiv) in DMF (2.00 mL) was added HATU (105 mg, 276 ⁇ mol, 1.50 equiv) and DIEA (72.0 mg, 557 ⁇ mol, 97.0 ⁇ L, 3.02 equiv).
- EXAMPLE 216 8-(2-((dimethylamino)methyl)pyrimidin-5-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000483] To a solution of tert-butyl ((5-(2-cyano-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidin-8-yl)pyrimidin-2-yl)methyl)carbamate (90.0 mg, 156 ⁇ mol, 1.00 equiv) in DCM (2.00 mL) was added TFA (890 mg, 7.81 mmol, 578 ⁇ L, 50.0 equiv).
- EXAMPLE 217 8-(3-(2-(dimethylamino)ethyl)phenyl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000486] A mixture of 8-bromo-5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)imidazo[1,2- c]pyrimidine-2-carbonitrile (50.0 mg, 129 ⁇ mol, 1.00 equiv), [3-(2-hydroxyethyl)phenyl]boronic acid (32.1 mg, 193 ⁇ mol, 1.50 equiv), sodium bicarbonate (32.5 mg, 386 ⁇ mol, 3.00 equiv) and Pd(dppf)Cl2 (9.42 mg, 12.9 ⁇ mol, 0.10 equiv) in dioxane (2.00
- EXAMPLE 218 5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(2-oxo-1,2-dihydropyridin-4- yl)imidazo[1,2-c]pyrimidine-2-carbonitrile [000491] A mixture of 5-[(5-fluoro-2,3-dihydrobenzofuran-4-yl)methylamino]-8-(2-methoxy-4- pyridyl)imidazo[1,2-c]pyrimidine-2-carbonitrile (55.0 mg, 125 ⁇ mol, 1.00 equiv) and pyridine hydrochloride (71.5 mg, 619 ⁇ mol, 4.93 equiv) was heated at 130 °C for 0.5 h.
- the reaction was stirred at 25 °C for 1 h and was subsequently quenched by the addition of water (30.0 mL) at 25 °C.
- the aqueous layer was diluted with ethyl acetate (50.0 mL), at which time a white precipitate formed.
- EXAMPLE 230 8-(1-(2-(dimethylamino)ethyl)-1H-pyrazol-5-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4- yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile [000496] To a solution of tert-butyl (E)-(2-cyano-8-(3-(dimethylamino)acryloyl)imidazo[1,2- c]pyrimidin-5-yl)((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)carbamate (13.0 mg, 25.0 ⁇ mol, 1.00 equiv) in ethanol (2.00 mL) was added 2-hydrazino-N,N-dimethyl-ethanamine-HCl (14.0 mg, 99.9 ⁇ mol, 4.00 equiv), the reaction was stirred at 80 °C
- EXAMPLE 231 5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)-8-(1-(2-hydroxyethyl)-1H-pyrazol-5- yl)imidazo[1,2-c]pyrimidine-2-carbonitrile [000499] To a solution of tert-butyl (E)-(2-cyano-8-(3-(dimethylamino)acryloyl)imidazo[1,2- c]pyrimidin-5-yl)((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)carbamate (16.0 mg, 31.6 ⁇ mol, 1.00 equiv) in ethanol (1.00 mL) was added 2-hydrazinoethanol (4.81 mg, 63.2 ⁇ mol, 4.29 ⁇ L, 2.00 equiv).
- EXAMPLE 232 8-(4-bromo-1H-pyrazol-3-yl)-5-(((5-fluoro-2,3-dihydrobenzofuran-4-yl)methyl)amino)imidazo[1,2- c]pyrimidine-2-carbonitrile [000502] To a solution of tert-butyl (2-cyano-8-(1H-pyrazol-3-yl)imidazo[1,2-c]pyrimidin-5-yl)((5- fluoro-2,3-dihydrobenzofuran-4-yl)methyl)carbamate (95.0 mg, 200 ⁇ mol, 1.00 equiv) in dry chloroform (1.00 mL) was added portionwise NBS (53.5 mg, 300 ⁇ mol, 1.50 equiv).
- the compounds of the present disclosure may have one or more chiral center and, if so, are synthesized as stereoisomeric mixtures, isomers of identical constitution that differ in the arrangement of their atoms in space.
- the compounds may be used as mixtures or the individual components/isomers may be separated using commercially available reagents and conventional methods for isolation of stereoisomers and enantiomers well-known to those skilled in the art, e.g., using CHIRALPAK® (Sigma- Aldrich) or CHIRALCEL® (Diacel Corp) chiral chromatographic HPLC columns according to the manufacturer’s instructions, as well as methods described herein, e.g., EXAMPLES 213 and 214.
- compounds of the present disclosure may be synthesized using optically pure, chiral reagents and intermediates to prepare individual isomers or enantiomers. Unless otherwise indicated, all chiral (enantiomeric and diastereomeric) and racemic forms are within the scope of the disclosure. [000511] Also contemplated within the scope of the disclosure are variants of compounds of the present disclosure in which one or more hydrogen atoms have been replaced with deuterium. As exemplified herein, Intermediates C-11 and D-31 have one or more hydrogen atom replaced with deuterium and were used to generated EXAMPLE 165 and 173, respectively. Intermediates B-7, B-8, and C18 also contain deuteriums substituted as specifies location(s).
- EXAMPLE 202 illustrates deuterated compounds of the present disclosure wherein R 7 is deuterated.
- R 7 is deuterated.
- deuterated versions of the compounds of the present disclosure can be readily generated using methods well known in the art.
- EXAMPLE A This Example illustrates that exemplary compounds of the present disclosure inhibit PRC2 enzymatic activity.
- Ten-point dose-response curves for compounds of the present disclosure were determined using a Hot Spot HMT assay (Reaction Biology Corp; see Horiuchi et al., Assay Drug Dev Technol. (2013) 4: 227-236 doi: 10.1089/adt.2012.480).
- the assay uses purified human, His-tagged PRC2 complex, including N-terminal His-tagged EZH2 enzyme, N-terminal Flag-tagged embryonic ectoderm development protein (EED), N-terminal His-tagged suppressor of zeste 12 (SUZ12), N-terminal His- tagged AEBP2, and N-terminal His-tagged RbAp48.
- EED embryonic ectoderm development protein
- SAM radiolabeled S-adenosyl methionine
- Example B [000516] This Example illustrates that treatment of bone marrow derived CD34+ cells from two healthy human donors with compound Example 32 (8-(4-((dimethylamino)methyl)-2-methylphenyl)-5-(((5- fluoro-2,3-dihydrobenzofuran-2-yl)methyl)amino)imidazo[1,2-c]pyrimidine-2-carbonitrile) increases fetal hemoglobin levels in the cells. [000517] Cell Culture and differentiation protocol. Human bone marrow derived CD34+ cells from healthy donors were purchased from StemCell (#70002.2).
- the CD34+ cells were differentiated using the STEMdiff TM Erythroid kit (StemCell #100-0074) according to manufacturer’s instructions.
- the CD34+ cells were differentiated for 14 days, and the differentiation medium consisted of StemSpanTM SFEM II media and 10% of STEMdiffTM Erythroid Supplement E2 (10X).
- the CD34+ cells were thawed and plated in a 12-well plate (Corning #3512) at a density of 40,000 cells/well in 1mL of differentiation media.100 ⁇ L of STEMdiffTM Erythroid Supplement E2 (10X) was added to each well on Day 2, 4, 9 and 11 during differentiation. On day 7, cells were passaged and replated at a density of 167,000 cells/well in a 12-well plate.
- RNA extraction and RT-qPCR analysis were performed with vehicle, 7 concentrations of compound Example 32 in concentrations ranging from 15.625 nM to 1 ⁇ M (1:5-fold serial dilution) and 20 ⁇ M of HU.100 ⁇ L of STEMdiffTM Erythroid Supplement E2 (10X) was added to each well on Day 2 and 4.
- Cells were treated with vehicle, compound Example 32, or HU for 7 days and were then harvested for RT-qPCR and flow cytometry. [000519] RNA extraction and RT-qPCR analysis.
- RT qPCR reactions were set up using 3 ⁇ L of 3 ng of RNA in 7 ⁇ L of pre-mixed SensiFASTTM SYBR No-ROX One-Step Kit reagents (BIOLINE, Cat# BIO-98005) and RT-qPCR was run on a Bio-Rad CFX384 Real-Time PCR System. HBG1 and HBG2 mRNA levels were normalized to housekeeping gene RPL27 and are plotted relative to mRNA levels in vehicle treated cells. Primer sequences for RT-qPCR [000520] Flow Cytometry. The cell pellets harvested for flow cytometry analysis were collected in a V- shaped 96-well polypropylene plate (ThermoFisher #249944).
- the cells were washed with PBS and resuspended in 100 ⁇ L of PBS.
- the cells were first stained with eFluor 450 viability dye (diluted 1:250 in cold PBS; Invitrogen #65-0863-14) for 5 minutes on ice in the dark followed by incubation in Fc receptor blocking solution for 10 minutes (BioLegend #422302).
- the cells were then washed with FACS buffer (1X PBS, 0.5% BSA, 0.02% NaAzide and 2 mM EDTA) and resuspend in 100 ⁇ L of FACS buffer.
- the cells were next stained with fluorochrome conjugated antibodies targeted against cell surface receptors such as anti-human CD235a-FITC (StemCell #60152FI) and/or anti-human CD71-APC (StemCell #60106AZ) for 25 minutes on ice.
- the cells were washed twice using FACS buffer and then fixed using Fixation buffer from the Fixation/Permeabilization kit (BD biosciences #554714) for 10 minutes on ice.
- the cells were further washed twice with 1X Permeabilization/Wash buffer and then incubated in the same buffer for 5 minutes.
- the permeabilized cells were stained using anti-human Fetal Hemoglobin-APC (ThermoFisher #MHFH05) for 15 minutes on ice in 1X Permeabilization/Wash buffer. Cells were washed twice and resuspended in FACS buffer and then analyzed by flow cytometry. Data was acquired using SONY SA3800 cytometer (software version 2.0.4.13263) and analyzed using FlowJo. [000521] Bone marrow derived CD34+ cells from two healthy human donors (donor A and donor B) were differentiated into erythroid progenitor cells (erythroblasts) expressing Glycophorin A and CD71.
- donor A and donor B erythroid progenitor cells
- HBG1 and HBG2 are the genes that encode the gamma-globin subunit that is unique to fetal hemoglobin. Consistent with treatment-induced increases in the percentage of fetal hemoglobin containing cells, HBG1 and HBG2 mRNA levels also increased in a dose-dependent manner when cells were treated with compound Example 32 (FIG.2A-2D). Donor A showed a 4-fold increase in HBG1 (FIG. 2A) and a 9-fold increase in HBG2 mRNA levels (FIG.2B) following treatment with compound Example 32. Donor B showed a 4-fold increase in both HBG1 (FIG.
- HBG2 mRNA levels (FIG. 2D) following treatment with compound Example 32.
- Treatment of the cells with hydroxyurea (HU) increased HBG2 levels by 2.7-fold (FIG.2B) but not HBG1 levels in Donor A (FIG.2A).
- a modest increase (1.7-fold) with HU treatment was observed for HBG1 mRNA in Donor B (FIG. 2C), but not for HBG2 (FIG.2D).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure relates to methods of treating a subject having a blood disorder, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I). Also disclosed herein are such methods wherein the blood disorder is sickle cell disease, or thalassemia, including alpha thalassemia, and beta thalassemia.
Description
PRC2 INHIBITORS FOR USE IN TREATING BLOOD DISORDERS
CROSS-REFERENCE
[0001] This application claims the benefit of U. S. Provisional Application Serial No. 63/248,237 filed September 24, 2021, and U. S. Provisional Application Serial No. 63/298,531 filed January 11, 2022 which are hereby incorporated by reference in their entirety.
FIELD OF THE DISCLOSURE
[0002] The present disclosure relates to compounds that inhibit the Polycomb Repressive Complex 2 (PRC2). In particular, the present disclosure relates to compounds, pharmaceutical compositions comprising the compounds and methods for use therefor in treating blood disorders, including sickle cell disease and thalassemia.
BACKGROUND OF THE DISCLOSURE
[0003] The Polycomb Repressive Complex 2 (PRC2) is a multiprotein complex that contributes to the epigenetic silencing of target genes to regulate development and homeostasis. The PRC2 complex is comprised of three core subunits: enhancer of zeste homolog 2 (EZH2), embryonic ectoderm development protein (EED), and suppressor of zeste 12 (SUZ12). EED is a critical regulator of PRC2 in the silencing of expression of genes and gene clusters involved in development including but not limited to fetal orthologues (i.e. gamma globin), Hox genes, X chromosome inactivation, etc. Thus, EED provides a pharmacologic target for the treatment of diseases or disorders to impact transcription of specific target genes in blood and other tissues.
SUMMARY OF THE DISCLOSURE
[0004] In one embodiment is provided a method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I):
 or a pharmaceutically acceptable salt thereof:
wherein:
or a pharmaceutically acceptable salt thereof:
wherein:
 represents a single or a double bond; Z is O or S; X is O, CR5, CR5OH, C(R5)2, wherein: when X is O,
represents a single or a double bond; Z is O or S; X is O, CR5, CR5OH, C(R5)2, wherein: when X is O,
 is a single bond; when X is C(R5)2,
is a single bond; when X is C(R5)2,
 is a single bond; when X is CR5OH,
is a single bond; when X is CR5OH,
 is a single bond; or when X is CR5,
is a single bond; or when X is CR5,
 is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or -C(O)N(R5)2; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-; Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or Y1-heterocyclyl, wherein the Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. [0005] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is selected from Acute lymphoblastic leukemia (ALL), Acute myeloid leukemia (AML) (e.g., acute promyelocytic leukemia, APL), Amyloidosis, Anemia, Aplastic anemia, Bone marrow failure syndromes, Chronic lymphocytic leukemia (CLL), Chronic myeloid leukemia (CML), Deep vein thrombosis (DVT), Diamond-Blackfan anemia, Dyskeratosis congenita (DKC), Eosinophilic disorder, Essential thrombocythemia, Fanconi anemia, Gaucher disease, Hemochromatosis, Hemolytic anemia, Hemophilia, Hereditary spherocytosis, Hodgkin's lymphoma, Idiopathic thrombocytopenic purpura (ITP), Inherited bone marrow failure syndromes, Iron-deficiency anemia, Langerhans cell
histiocytosis, Large granular lymphocytic (LGL) leukemia, Leukemia, Leukopenia, Mastocytosis, Monoclonal gammopathy, Multiple myeloma, Myelodysplastic syndromes (MDS), Myelofibrosis, Myeloproliferative neoplasms (MPN), Non-Hodgkin's lymphoma, Paroxysmal nocturnal hemoglobinuria (PNH), Pernicious anemia (B12 deficiency), Polycythemia vera, Porphyria, Post-transplant lymphoproliferative disorder (PTLD), Pulmonary embolism (PE), Shwachman-Diamond syndrome (SDS), sickle cell disease (SCD), Thalassemia (e.g., .beta.-thalassemia), Thrombocytopenia, Thrombotic thrombocytopenic purpura (TTP), Venous thromboembolism, Von Willebrand disease, and Waldenstrom's macroglobulinemia (lymphoplasmacytic lymphoma). [0006] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is sickle cell disease. [0007] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is thalassemia. In one embodiment, the thalassemia is alpha thalassemia. In another embodiment, the thalassemia is beta thalassemia. [0008] Also provided herein is a use of a compound of Formula I, or a pharmaceutically acceptable salt or solvate thereof, as defined herein in the manufacture of a medicament for the inhibition of activity of PRC2. [0009] Also provided herein is the use of a compound of Formula I, or a pharmaceutically acceptable salt or solvate thereof, as defined herein, in the manufacture of a medicament for the treatment of a blood disorder. BRIEF DESCRIPTION OF THE FIGURES [00010] FIG.1A depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32, or hydroxyurea (HU) from Donor A. [00011] FIG.1B depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B. [00012] FIG.2A depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A. [00013] FIG.2B depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A. [00014] FIG.2C depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B. [00015] FIG.2D depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B.
DETAILED DESCRIPTION OF THE DISCLOSURE [00016] Unless defined otherwise, all terms and ranges used herein have the same meaning as is commonly understood by one of skill in the art to which this disclosure belongs, unless expressly defined otherwise. All patents, patent applications, and publications referred to herein are incorporated by reference to the extent they are consistent with the present disclosure. [00017] For simplicity, chemical moieties are defined and referred to throughout primarily as univalent chemical moieties (e.g., alkyl, aryl, etc.). Nevertheless, such terms may also be used to convey corresponding multivalent moieties under the appropriate structural circumstances clear to those skilled in the art. For example, while an “alkyl” moiety generally refers to a monovalent radical (e.g. CH3-CH2- ), in certain circumstances a bivalent linking moiety can be “alkyl,” in which case those skilled in the art will understand the alkyl to be a divalent radical (e.g., -CH2-CH2-), which is equivalent to the term “alkylene.” (Similarly, in circumstances in which a divalent moiety is required and is stated as being “aryl,” those skilled in the art will understand that the term “aryl” refers to the corresponding divalent moiety, arylene.) All atoms are understood to have their normal number of valences for bond formation (i.e., 4 for carbon, 3 for N, 2 for O, and 2, 4, or 6 for S, depending on the oxidation state of the S). [00018] As used herein, “Polycomb Repressive Complex 2” or “PRC2 complex” refers to a mammalian multiprotein complex comprising three core subunits: enhancer of zeste homolog 2 (EZH2), embryonic ectoderm development protein (EED), and suppressor of zeste 12 (SUZ12) and two additional non- essential subunits, AEBP2, and RbAp48. [00019] As used herein, “EED” refers to the embryonic ectoderm development protein subunit of the PRC2 complex. [00020] As used herein, “EZH2” or “EZH2 enzyme” refers to a mammalian histone methyltransferase, which is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2), and functions to silence target genes by tri-methylating lysine 27 of histone H3 (H3K27me3). [00021] As used herein, an “PRC2 inhibitor” refers to compounds of the present disclosure that are represented by formula (I) as described herein. These compounds are capable of negatively modulating or inhibiting all or a portion of the enzymatic activity of the PRC2 complex. While not wanting to be bound by any theory, we theorize that the inhibitors of the present disclosure may inhibit PRC2 enzymatic activity by binding to EED to prevent assembly of the PRC2 complex on histone H3 tails thereby inhibiting its activity. [00022] The term “amino” refers to –NH2. [00023] The term “acetyl” refers to “-C(O)CH3. [00024] As herein employed, the term “acyl” refers to an alkylcarbonyl or arylcarbonyl substituent wherein the alkyl and aryl portions are as defined herein. [00025] The term “alkyl” as employed herein refers to straight and branched chain aliphatic groups having from 1 to 12 carbon atoms. As such, “alkyl” encompasses C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkyl groups include, without limitation, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, and hexyl.
[00026] The term “alkenyl” as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon double bonds, having from 2 to 12 carbon atoms. As such, “alkenyl” encompasses C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkenyl groups include, without limitation, ethenyl, propenyl, butenyl, pentenyl, and hexenyl. [00027] The term “alkynyl” as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon triple bonds, having from 2 to 12 carbon atoms. As such, “alkynyl” encompasses C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkynyl groups include, without limitation, ethynyl, propynyl, butynyl, pentynyl, and hexynyl. [00028] An “alkylene,” “alkenylene,” or “alkynylene” group is an alkyl, alkenyl, or alkynyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups. Examples of alkylene groups include, without limitation, methylene, ethylene, propylene, and butylene. Exemplary alkenylene groups include, without limitation, ethenylene, propenylene, and butenylene. Exemplary alkynylene groups include, without limitation, ethynylene, propynylene, and butynylene. [00029] The term “alkoxy” refers to –OC1–C6 alkyl. [00030] The term “cycloalkyl” as employed herein is a saturated and partially unsaturated cyclic hydrocarbon group having 3 to 12 carbons. As such, “cycloalkyl” includes C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 cyclic hydrocarbon groups. Examples of cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl. [00031] The term “heteroalkyl” refers to an alkyl group, as defined hereinabove, wherein one or more carbon atoms in the chain are independently replaced O, S, or NRx, wherein Rx is hydrogen or C1–C3 alkyl. Examples of heteroalkyl groups include methoxymethyl, methoxyethyl and methoxypropyl. [00032] An “aryl” group is a C6-C14 aromatic moiety comprising one to three aromatic rings. As such, “aryl” includes C6, C10, C13, and C14 cyclic hydrocarbon groups. An exemplary aryl group is a C6-C10 aryl group. Particular aryl groups include, without limitation, phenyl, naphthyl, anthracenyl, and fluorenyl. [00033] An “aralkyl” or “arylalkyl” group comprises an aryl group covalently linked to an alkylene group wherein the moiety is linked to another group via the alkyl moiety. An exemplary aralkyl group is –(C1-C6)alkyl(C6-C10)aryl, including, without limitation, benzyl, phenethyl, and naphthylmethyl. [00034] A “heterocyclyl” or “heterocyclic” group is a mono- or bicyclic (fused or spiro) ring structure having from 3 to 12 atoms, (3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 atoms), for example 4 to 8 atoms, wherein one or more ring atoms are independently –C(O)-, N, NR5, O, or S, and the remainder of the ring atoms are quaternary or carbonyl carbons. Examples of heterocyclic groups include, without limitation, epoxy, oxiranyl, oxetanyl, azetidinyl, aziridinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydrothiophenyl, pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, thiazolidinyl, thiatanyl, dithianyl, trithianyl, azathianyl, oxathianyl, dioxolanyl, oxazolidinyl, oxazolidinonyl, decahydroquinolinyl, piperidonyl, 4- piperidonyl, thiomorpholinyl, dimethyl-morpholinyl, and morpholinyl. Specifically excluded from the scope of this term are compounds having adjacent ring O and/or S atoms.
[00035] As used herein, “L-heterocyclyl” refers to a heterocyclyl group covalently linked to another group via an alkylene linker L, where L is C1–C4 alkylene. [00036] As used herein, the term “heteroaryl” refers to a group having 5 to 14 ring atoms, preferably 5, 6, 10, 13 or 14 ring atoms; having 6, 10, or 14 π electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to three heteroatoms that are each independently N, O, or S. “Heteroaryl” also includes fused multicyclic (e.g., bicyclic) ring systems in which one or more of the fused rings is non- aromatic, provided that at least one ring is aromatic and at least one ring contains an N, O, or S ring atom. [00037] Examples of heteroaryl groups include acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzo[d]oxazol-2(3H)-one, 2H-benzo[b][1,4]oxazin-3(4H)-one, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, furanyl, furazanyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolinyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5- thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, and xanthenyl. [00038] A “L-heteroaryl,” “heteroaralkyl” or “heteroarylalkyl” group comprises a heteroaryl group covalently linked to another group via an alkylene linker. Examples of heteroalkyl groups comprise a C1- C6 alkyl group and a heteroaryl group having 5, 6, 9, or 10 ring atoms. Examples of heteroaralkyl groups include pyridylmethyl, pyridylethyl, pyrrolylmethyl, pyrrolylethyl, imidazolylmethyl, imidazolylethyl, thiazolylmethyl, thiazolylethyl, benzimidazolylmethyl, benzimidazolylethyl quinazolinylmethyl, quinolinylmethyl, quinolinylethyl, benzofuranylmethyl, indolinylethyl isoquinolinylmethyl, isoinodylmethyl, cinnolinylmethyl, and benzothiophenylethyl. Specifically excluded from the scope of this term are compounds having adjacent ring O and/or S atoms. [00039] An “arylene,” “heteroarylene,” or “heterocyclylene” group is an bivalent aryl, heteroaryl, or heterocyclyl group, respectively, as defined hereinabove, that is positioned between and serves to connect two other chemical groups. [00040] As employed herein, when a moiety (e.g., cycloalkyl, aryl, heteroaryl, heterocyclyl, urea, etc.) is described as “optionally substituted” without expressly stating the substituents it is meant that the
group optionally has from one to four, preferably from one to three, more preferably one or two, non- hydrogen substituents. [00041] The term “halogen” or “halo” as employed herein refers to chlorine, bromine, fluorine, or iodine. [00042] The term “haloalkyl” refers to an alkyl chain in which one or more hydrogens have been replaced by a halogen. Exemplary haloalkyls are trifluoromethyl, difluoromethyl, flurochloromethyl, chloromethyl, and fluoromethyl. [00043] The term “hydroxyalkyl” refers to an alkyl chain, as defined herein, wherein at least on hydrogen of the alkyl chain has been replaced by hydroxyl. [00044] As used herein, “an effective amount” of a compound is an amount that is sufficient to negatively modulate or inhibit the activity of PRC2 complex. [00045] As used herein, a “therapeutically effective amount” of a compound is an amount that is sufficient to ameliorate or in some manner reduce a symptom or stop or reverse progression of a condition, or negatively modulate or inhibit the activity of PRC2 complex. Such amount may be administered as a single dosage or may be administered according to a regimen, whereby it is effective. [00046] As used herein, “treatment” means any manner in which the symptoms or pathology of a condition, disorder or disease in a subject are ameliorated or otherwise beneficially altered. [00047] As used herein, “amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition” refers to any lessening, whether permanent or temporary, lasting, or transient, that can be attributed to or associated with administration of the composition. COMPOUNDS [00048] In one embodiment is provided a method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I):
is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or -C(O)N(R5)2; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-; Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or Y1-heterocyclyl, wherein the Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. [0005] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is selected from Acute lymphoblastic leukemia (ALL), Acute myeloid leukemia (AML) (e.g., acute promyelocytic leukemia, APL), Amyloidosis, Anemia, Aplastic anemia, Bone marrow failure syndromes, Chronic lymphocytic leukemia (CLL), Chronic myeloid leukemia (CML), Deep vein thrombosis (DVT), Diamond-Blackfan anemia, Dyskeratosis congenita (DKC), Eosinophilic disorder, Essential thrombocythemia, Fanconi anemia, Gaucher disease, Hemochromatosis, Hemolytic anemia, Hemophilia, Hereditary spherocytosis, Hodgkin's lymphoma, Idiopathic thrombocytopenic purpura (ITP), Inherited bone marrow failure syndromes, Iron-deficiency anemia, Langerhans cell
histiocytosis, Large granular lymphocytic (LGL) leukemia, Leukemia, Leukopenia, Mastocytosis, Monoclonal gammopathy, Multiple myeloma, Myelodysplastic syndromes (MDS), Myelofibrosis, Myeloproliferative neoplasms (MPN), Non-Hodgkin's lymphoma, Paroxysmal nocturnal hemoglobinuria (PNH), Pernicious anemia (B12 deficiency), Polycythemia vera, Porphyria, Post-transplant lymphoproliferative disorder (PTLD), Pulmonary embolism (PE), Shwachman-Diamond syndrome (SDS), sickle cell disease (SCD), Thalassemia (e.g., .beta.-thalassemia), Thrombocytopenia, Thrombotic thrombocytopenic purpura (TTP), Venous thromboembolism, Von Willebrand disease, and Waldenstrom's macroglobulinemia (lymphoplasmacytic lymphoma). [0006] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is sickle cell disease. [0007] In another embodiment is provided a method of treating a blood disorder in a subject by administering to the subject a therapeutically effective amount of a compound of Formula (I), wherein the blood disorder is thalassemia. In one embodiment, the thalassemia is alpha thalassemia. In another embodiment, the thalassemia is beta thalassemia. [0008] Also provided herein is a use of a compound of Formula I, or a pharmaceutically acceptable salt or solvate thereof, as defined herein in the manufacture of a medicament for the inhibition of activity of PRC2. [0009] Also provided herein is the use of a compound of Formula I, or a pharmaceutically acceptable salt or solvate thereof, as defined herein, in the manufacture of a medicament for the treatment of a blood disorder. BRIEF DESCRIPTION OF THE FIGURES [00010] FIG.1A depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32, or hydroxyurea (HU) from Donor A. [00011] FIG.1B depicts the percentage of F cells (cells containing fetal hemoglobin) in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B. [00012] FIG.2A depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A. [00013] FIG.2B depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor A. [00014] FIG.2C depicts HBG1 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B. [00015] FIG.2D depicts HBG2 mRNA levels in 14-day differentiated cells post 7-day treatment with vehicle (veh), compound Example 32 or hydroxyurea (HU) in Donor B.
DETAILED DESCRIPTION OF THE DISCLOSURE [00016] Unless defined otherwise, all terms and ranges used herein have the same meaning as is commonly understood by one of skill in the art to which this disclosure belongs, unless expressly defined otherwise. All patents, patent applications, and publications referred to herein are incorporated by reference to the extent they are consistent with the present disclosure. [00017] For simplicity, chemical moieties are defined and referred to throughout primarily as univalent chemical moieties (e.g., alkyl, aryl, etc.). Nevertheless, such terms may also be used to convey corresponding multivalent moieties under the appropriate structural circumstances clear to those skilled in the art. For example, while an “alkyl” moiety generally refers to a monovalent radical (e.g. CH3-CH2- ), in certain circumstances a bivalent linking moiety can be “alkyl,” in which case those skilled in the art will understand the alkyl to be a divalent radical (e.g., -CH2-CH2-), which is equivalent to the term “alkylene.” (Similarly, in circumstances in which a divalent moiety is required and is stated as being “aryl,” those skilled in the art will understand that the term “aryl” refers to the corresponding divalent moiety, arylene.) All atoms are understood to have their normal number of valences for bond formation (i.e., 4 for carbon, 3 for N, 2 for O, and 2, 4, or 6 for S, depending on the oxidation state of the S). [00018] As used herein, “Polycomb Repressive Complex 2” or “PRC2 complex” refers to a mammalian multiprotein complex comprising three core subunits: enhancer of zeste homolog 2 (EZH2), embryonic ectoderm development protein (EED), and suppressor of zeste 12 (SUZ12) and two additional non- essential subunits, AEBP2, and RbAp48. [00019] As used herein, “EED” refers to the embryonic ectoderm development protein subunit of the PRC2 complex. [00020] As used herein, “EZH2” or “EZH2 enzyme” refers to a mammalian histone methyltransferase, which is the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2), and functions to silence target genes by tri-methylating lysine 27 of histone H3 (H3K27me3). [00021] As used herein, an “PRC2 inhibitor” refers to compounds of the present disclosure that are represented by formula (I) as described herein. These compounds are capable of negatively modulating or inhibiting all or a portion of the enzymatic activity of the PRC2 complex. While not wanting to be bound by any theory, we theorize that the inhibitors of the present disclosure may inhibit PRC2 enzymatic activity by binding to EED to prevent assembly of the PRC2 complex on histone H3 tails thereby inhibiting its activity. [00022] The term “amino” refers to –NH2. [00023] The term “acetyl” refers to “-C(O)CH3. [00024] As herein employed, the term “acyl” refers to an alkylcarbonyl or arylcarbonyl substituent wherein the alkyl and aryl portions are as defined herein. [00025] The term “alkyl” as employed herein refers to straight and branched chain aliphatic groups having from 1 to 12 carbon atoms. As such, “alkyl” encompasses C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkyl groups include, without limitation, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, and hexyl.
[00026] The term “alkenyl” as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon double bonds, having from 2 to 12 carbon atoms. As such, “alkenyl” encompasses C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkenyl groups include, without limitation, ethenyl, propenyl, butenyl, pentenyl, and hexenyl. [00027] The term “alkynyl” as used herein means an unsaturated straight or branched chain aliphatic group with one or more carbon-carbon triple bonds, having from 2 to 12 carbon atoms. As such, “alkynyl” encompasses C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 groups. Examples of alkynyl groups include, without limitation, ethynyl, propynyl, butynyl, pentynyl, and hexynyl. [00028] An “alkylene,” “alkenylene,” or “alkynylene” group is an alkyl, alkenyl, or alkynyl group, as defined hereinabove, that is positioned between and serves to connect two other chemical groups. Examples of alkylene groups include, without limitation, methylene, ethylene, propylene, and butylene. Exemplary alkenylene groups include, without limitation, ethenylene, propenylene, and butenylene. Exemplary alkynylene groups include, without limitation, ethynylene, propynylene, and butynylene. [00029] The term “alkoxy” refers to –OC1–C6 alkyl. [00030] The term “cycloalkyl” as employed herein is a saturated and partially unsaturated cyclic hydrocarbon group having 3 to 12 carbons. As such, “cycloalkyl” includes C3, C4, C5, C6, C7, C8, C9, C10, C11 and C12 cyclic hydrocarbon groups. Examples of cycloalkyl groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl. [00031] The term “heteroalkyl” refers to an alkyl group, as defined hereinabove, wherein one or more carbon atoms in the chain are independently replaced O, S, or NRx, wherein Rx is hydrogen or C1–C3 alkyl. Examples of heteroalkyl groups include methoxymethyl, methoxyethyl and methoxypropyl. [00032] An “aryl” group is a C6-C14 aromatic moiety comprising one to three aromatic rings. As such, “aryl” includes C6, C10, C13, and C14 cyclic hydrocarbon groups. An exemplary aryl group is a C6-C10 aryl group. Particular aryl groups include, without limitation, phenyl, naphthyl, anthracenyl, and fluorenyl. [00033] An “aralkyl” or “arylalkyl” group comprises an aryl group covalently linked to an alkylene group wherein the moiety is linked to another group via the alkyl moiety. An exemplary aralkyl group is –(C1-C6)alkyl(C6-C10)aryl, including, without limitation, benzyl, phenethyl, and naphthylmethyl. [00034] A “heterocyclyl” or “heterocyclic” group is a mono- or bicyclic (fused or spiro) ring structure having from 3 to 12 atoms, (3, 4, 5, 6, 7, 8, 9, 10, 11 or 12 atoms), for example 4 to 8 atoms, wherein one or more ring atoms are independently –C(O)-, N, NR5, O, or S, and the remainder of the ring atoms are quaternary or carbonyl carbons. Examples of heterocyclic groups include, without limitation, epoxy, oxiranyl, oxetanyl, azetidinyl, aziridinyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydrothiophenyl, pyrrolidinyl, piperidinyl, piperazinyl, imidazolidinyl, thiazolidinyl, thiatanyl, dithianyl, trithianyl, azathianyl, oxathianyl, dioxolanyl, oxazolidinyl, oxazolidinonyl, decahydroquinolinyl, piperidonyl, 4- piperidonyl, thiomorpholinyl, dimethyl-morpholinyl, and morpholinyl. Specifically excluded from the scope of this term are compounds having adjacent ring O and/or S atoms.
[00035] As used herein, “L-heterocyclyl” refers to a heterocyclyl group covalently linked to another group via an alkylene linker L, where L is C1–C4 alkylene. [00036] As used herein, the term “heteroaryl” refers to a group having 5 to 14 ring atoms, preferably 5, 6, 10, 13 or 14 ring atoms; having 6, 10, or 14 π electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to three heteroatoms that are each independently N, O, or S. “Heteroaryl” also includes fused multicyclic (e.g., bicyclic) ring systems in which one or more of the fused rings is non- aromatic, provided that at least one ring is aromatic and at least one ring contains an N, O, or S ring atom. [00037] Examples of heteroaryl groups include acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzo[d]oxazol-2(3H)-one, 2H-benzo[b][1,4]oxazin-3(4H)-one, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, furanyl, furazanyl, imidazolinyl, imidazolyl, 1H-indazolyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isoxazolyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolidinyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyrimidinyl, pyrrolinyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, tetrazolyl, 6H-1,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5- thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, and xanthenyl. [00038] A “L-heteroaryl,” “heteroaralkyl” or “heteroarylalkyl” group comprises a heteroaryl group covalently linked to another group via an alkylene linker. Examples of heteroalkyl groups comprise a C1- C6 alkyl group and a heteroaryl group having 5, 6, 9, or 10 ring atoms. Examples of heteroaralkyl groups include pyridylmethyl, pyridylethyl, pyrrolylmethyl, pyrrolylethyl, imidazolylmethyl, imidazolylethyl, thiazolylmethyl, thiazolylethyl, benzimidazolylmethyl, benzimidazolylethyl quinazolinylmethyl, quinolinylmethyl, quinolinylethyl, benzofuranylmethyl, indolinylethyl isoquinolinylmethyl, isoinodylmethyl, cinnolinylmethyl, and benzothiophenylethyl. Specifically excluded from the scope of this term are compounds having adjacent ring O and/or S atoms. [00039] An “arylene,” “heteroarylene,” or “heterocyclylene” group is an bivalent aryl, heteroaryl, or heterocyclyl group, respectively, as defined hereinabove, that is positioned between and serves to connect two other chemical groups. [00040] As employed herein, when a moiety (e.g., cycloalkyl, aryl, heteroaryl, heterocyclyl, urea, etc.) is described as “optionally substituted” without expressly stating the substituents it is meant that the
group optionally has from one to four, preferably from one to three, more preferably one or two, non- hydrogen substituents. [00041] The term “halogen” or “halo” as employed herein refers to chlorine, bromine, fluorine, or iodine. [00042] The term “haloalkyl” refers to an alkyl chain in which one or more hydrogens have been replaced by a halogen. Exemplary haloalkyls are trifluoromethyl, difluoromethyl, flurochloromethyl, chloromethyl, and fluoromethyl. [00043] The term “hydroxyalkyl” refers to an alkyl chain, as defined herein, wherein at least on hydrogen of the alkyl chain has been replaced by hydroxyl. [00044] As used herein, “an effective amount” of a compound is an amount that is sufficient to negatively modulate or inhibit the activity of PRC2 complex. [00045] As used herein, a “therapeutically effective amount” of a compound is an amount that is sufficient to ameliorate or in some manner reduce a symptom or stop or reverse progression of a condition, or negatively modulate or inhibit the activity of PRC2 complex. Such amount may be administered as a single dosage or may be administered according to a regimen, whereby it is effective. [00046] As used herein, “treatment” means any manner in which the symptoms or pathology of a condition, disorder or disease in a subject are ameliorated or otherwise beneficially altered. [00047] As used herein, “amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition” refers to any lessening, whether permanent or temporary, lasting, or transient, that can be attributed to or associated with administration of the composition. COMPOUNDS [00048] In one embodiment is provided a method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I):
 or a pharmaceutically acceptable salt thereof: wherein:
or a pharmaceutically acceptable salt thereof: wherein:
 represents a single or a double bond;
Z is O or S; X is O, CR5, CR5OH, C(R5)2, wherein: when X is O, is a single bond; when X is C(R5)2,
represents a single or a double bond;
Z is O or S; X is O, CR5, CR5OH, C(R5)2, wherein: when X is O, is a single bond; when X is C(R5)2,
 is a single bond; when X is CR5OH,
is a single bond; when X is CR5OH,
 is a single bond; or when X is CR5,
is a single bond; or when X is CR5,
 is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or -C(O)N(R5) 2; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-; Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or Y1-heterocyclyl, wherein the Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. [00049] In one embodiment, Z is O. In one embodiment, Z is S. [00050] In one embodiment, X is C(R5)2 and
is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or -C(O)N(R5) 2; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-; Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or Y1-heterocyclyl, wherein the Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. [00049] In one embodiment, Z is O. In one embodiment, Z is S. [00050] In one embodiment, X is C(R5)2 and
 is a single bond. [00051] In one embodiment, Z is O or S. In one embodiment, X is O, CR5, CR5OH or C(R5)2, wherein when X is O,
is a single bond. [00051] In one embodiment, Z is O or S. In one embodiment, X is O, CR5, CR5OH or C(R5)2, wherein when X is O,
 is a single bond; when X is C(R5)2,
is a single bond; when X is C(R5)2,
 is a single bond; when X is CR5OH,
is a single bond; when X is CR5OH,
 is a single bond; or when X is CR5,
is a single bond; or when X is CR5,
 is a double bond. In one embodiment, n is one. In one embodiment, n is two. [00052] In one embodiment, Z is O, X is O, n is one and
is a double bond. In one embodiment, n is one. In one embodiment, n is two. [00052] In one embodiment, Z is O, X is O, n is one and
 is a single bond. In another embodiment, Z is O, X is CR5 and
is a single bond. In another embodiment, Z is O, X is CR5 and
 is a double bond. In one embodiment, Z is O, X is C(R5)2, n is one, and
is a double bond. In one embodiment, Z is O, X is C(R5)2, n is one, and
 is a single bond. In one embodiment, Z is O, X is CR5OH, n is one, and
is a single bond. In one embodiment, Z is O, X is CR5OH, n is one, and
 is a single bond. In another embodiment, Z is O, X is C(R5)2, n is two, and
is a single bond. In another embodiment, Z is O, X is C(R5)2, n is two, and
 is a single bond. In yet another embodiment, Z is S, X is C(R5)2, n is one, and is a single bond.
[00053] In one embodiment, R1 is aryl, which may be optionally substituted with one or more R4. In certain embodiments, the aryl is phenyl, which may be optionally substituted with one or more R4. [00054] In one embodiment, the aryl is substituted with a single R4 group. In one embodiment, the aryl is substituted with two R4 groups. In one embodiment, the aryl is substituted with three R4 groups. Exemplary aryl R4 groups include halogen, hydroxyl, haloalkyl, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L- N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2, Y2-haloalkyl, L-heterocyclyl, or Y1-heterocyclyl, wherein the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. [00055] In one embodiment, the one or more R4 are each independently halogen, hydroxyl, haloalkyl, - COOR5, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L-N(R5)2, -O-L-N(R5)2, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2- N(R5)2, Y2-haloalkyl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heterocyclyl portion of the -L- heterocyclyl or -Y1-heterocyclyl may be optionally substituted with one or more R7. [00056] In one embodiment, R1 is phenyl substituted with -Y2-C1–C6 alkyl. In one embodiment, Y is a bond and the C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, R1 is phenyl substituted with the Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1– C6 alkyl is methyl. In one embodiment, R1 is phenyl, which is disubstituted with methyl and Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1– C6 alkyl is methyl. [00057] In one embodiment, R1 is phenyl substituted one R4, wherein R4 is a cyano group. [00058] In one embodiment, R1 is phenyl substituted one R4, wherein R4 is L-heteroaryl. In certain embodiments, the L-heteroaryl is tetrazolyl. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is PO3(C1-C3 alkyl)2. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is - COOR5. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is -O-L-N(R5)2. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is aralkyl. [00059] In one embodiment, R1 is phenyl substituted with at least one R4, wherein R4 is -L-N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R5 is independently hydrogen. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, R1 is phenyl substituted with -L-N(R5)2 and further substituted with one or more halogen and/or C1–C6 alkyl. [00060] In one embodiment, R1 is phenyl substituted with one R4, wherein R4 is -Y1-N(R5)2. In certain embodiments, Y1 is -C(O)- and each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, Y1 is -C(O)- and each R5 is hydrogen. In one embodiment, Y1 is - C(O)- and one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, R1 is phenyl substituted with -Y1-N(R5)2 and further substituted with one or more halogen and/or C1–C6 alkyl. [00061] In one embodiment, R1 is phenyl substituted with the Y2-haloalkyl, wherein Y2 is -S- or -SO2- and the haloalkyl is trifluoromethyl. [00062] In one embodiment, R1 is phenyl substituted with at least one -L-heterocyclyl or -Y1- heterocyclyl, each heterocyclyl optionally substituted with one or more R7. In one embodiment, R1 is
phenyl substituted with one R4, wherein R4 is -Y1-heterocyclyl optionally substituted with one or more R7. In one embodiment, Y1 is -C(O)- and the heterocyclyl is piperazinyl optionally substituted with C1– C3 alkyl. [00063] In certain embodiments, the R4 group is L-heterocyclyl optionally substituted with one or more R7. In one embodiment, L is methylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4- methyl-piperazinyl. In one embodiment, L is methylene and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R7. Exemplary R7 groups include oxo, halogen, hydroxyalkyl and C1–C3 alkyl. [00064] In one embodiment, R1 is phenyl substituted with Y1-heterocyclyl optionally substituted with one or more R7. In certain embodiments, Y1 is -C(O)- and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl, each optionally further substituted with one or more halogen. [00065] In one embodiment, R1 is phenyl substituted with L-heteroaryl optionally substituted with one or more R7. In certain embodiments, the L-heteroaryl is tetrazolyl. [00066] In one embodiment, R1 is phenyl substituted with PO3(C1-C3 alkyl)2. In another embodiment, R1 is phenyl substituted with -COOR5. In one embodiment, R1 is phenyl substituted with hydroxyalkyl, - O-L-N(R5)2 or aralkyl. [00067] In one embodiment, R1 is heteroaryl, which may be optionally substituted with one or more R4. In certain embodiments, the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, isoxazolyl, isoindolinyl, naphthyridinyl, 1,2,3,4-tetrahydroisoquinolinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R4. In certain embodiments, the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R4. [00068] In one embodiment, the heteroaryl is substituted with a single R4 group. In one embodiment, the heteroaryl is substituted with two R4 groups. In one embodiment, the heteroaryl is substituted with three R4 groups. Exemplary heteroaryl R4 groups include amino, cyano, halogen, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, Y2-haloalkyl Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, L-cycloalkyl, L-heteroaryl, L- heterocyclyl, Y1-heterocyclyl, -L-N(R5)2, or -Y1-N(R5)2, wherein the ring of the L-cycloalkyl, L- heteroaryl, L-heterocyclyl or Y1-heterocyclyl may be optionally substituted with one or more R7. [00069] In one embodiment, each R4 is independently cyano, halogen, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, -L-cycloalkyl, -L-N(R5)2, -Y1-N(R5)2, -L-heteroaryl, - L-heterocyclyl, or -Y1-heterocyclyl, wherein the heteroaryl of the -L-heteroaryl or the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. [00070] In one embodiment, R7 is amino, hydroxyl, cyano, alkoxy, or halogen. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen, wherein the halogen is fluorine or chlorine. In one
embodiment, R7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. [00071] In another embodiment, R1 is heteroaryl and each R4 is independently hydroxyalkyl, heteroalkyl or haloalkyl. In certain embodiments, the hydroxyalkyl is hydroxymethyl, hydroxyethyl or 2- methyl, 2-hydroxypropyl. In certain embodiments, the heteroalkyl is methoxymethyl or methoxyethyl. In certain embodiments, the haloalkyl is fluoromethyl, difluoromethyl, trifluoromethyl, fluoroethyl, difluoroethyl, or trifluoroethyl. [00072] In certain embodiments, R1 is heteroaryl and R4 is -Y1-C1–C6 alkyl, wherein Y1 is a bond and the C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, R4 is -Y1-C1–C6 alkyl, wherein Y1 is a -C(O)- and the C1–C6 alkyl is methyl, ethyl or isopropyl. In other embodiments, Y1is -NHC(O)- and the C1–C6 alkyl portion is methyl. [00073] In one embodiment, R1 is heteroaryl and R4 is -Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1–C6 alkyl is methyl. In another embodiment, R4 is -Y2-C1–C6 alkyl, wherein Y2 is -S- and the C1–C6 alkyl is methyl. [00074] In one embodiment, R1 is heteroaryl and R4 is -Y1-heterocyclyl, which may be optionally substituted with one or more R7. In one embodiment, Y1 is a bond. In another embodiment, Y1 is -C(O)-. In one embodiment, Y1 is a bond and the heterocyclyl is azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen. [00075] In one embodiment, the heteroaryl is substituted with at least one R4 that is -L-heterocyclyl, which may be optionally substituted with one or more R7. In one embodiment, L is ethylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl. In one embodiment, L is methylene and the heterocyclyl is azetinidyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R7. [00076] In one embodiment, the R7 is independently -L-N(R5)2, hydroxyl, cyano, alkoxy, or halogen. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen, wherein the halogen is fluorine or chlorine. In one embodiment, R7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. In one embodiment, R7 is -L- N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R5 is hydrogen. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. [00077] In one embodiment, R1 is heteroaryl and R4 is -L-N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene, ethylene, or propylene. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, each R5 is hydrogen.
[00078] In one embodiment, R1 is heteroaryl and R4 is L-heteroaryl, which may be optionally substituted with one or more R7. In one embodiment, L is a bond. In one embodiment, L is C1–C3 alkylene. In one embodiment, the C1–C3 alkylene is methylene. In certain embodiments, the heteroaryl of the L-heteroaryl is pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, triazinyl, thiazolyl or pyridazinyl. In one embodiment, the heteroaryl of the L-heteroaryl is pyridyl. [00079] In one embodiment, R1 is heteroaryl which is substituted with two R4 groups independently selected from two -Y1-C1–C6 alkyl groups; -Y1-C1–C6 alkyl and alkoxy; -Y1-C1–C6 alkyl and cycloalkyl; -Y1-C1–C6 alkyl and haloalkyl; -Y1-C1–C6 alkyl and amino; two alkoxy groups; alkoxy and halogen; alkoxy and cyano, and amino and haloalkyl. In certain embodiments, R4 is -Y1-C1–C6 alkyl, wherein each Y1 is a bond and each C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, the cycloalkyl is cyclopropyl. In one embodiment, the alkoxy is methoxy. In one embodiment, the halogen is fluorine or chlorine. In one embodiment, the haloalkyl is trifluoromethyl or trifluoroethyl. [00080] In one embodiment, R1 is L-heterocyclyl optionally substituted with one or more R4. In one embodiment, L is a bond and the heterocyclyl is tetrahydrofuranyl, piperidinyl, piperazinyl or morpholinyl. In one embodiment, L is a methylene and the heterocyclyl is azetidinyl, pyrrolidinyl or 3ƛ2- azabicyclo[3.1.0]hexanyl. In certain embodiments, the heterocyclyl is substituted with one or more R4 selected from oxo, halogen, alkoxy, hydroxyl and Y1-C1–C6 alkyl, wherein Y is a bond or -C(O)-. [00081] In one embodiment, R2 is cyano. In one embodiment, R2 is -COOR5. In certain embodiments, the R5 group is hydrogen. [00082] In one embodiment, R2 is -C(O)N(R5)2. In one embodiment, each R5 is independently C1–C3 alkyl. In certain embodiments, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In certain embodiments, the one C1–C3 alkyl is methyl. In one embodiment, each R5 is hydrogen. In one embodiment, each R5 together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6. [00083] In one embodiment, n is zero. In one embodiment, n is one and R3 is halogen. In certain embodiments, the halogen is fluorine or chlorine. In one embodiment, the halogen is fluorine. [00084] In one embodiment, R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl. In certain embodiments, R6 is hydrogen. In other embodiments, R6 is methyl, ethyl, or propyl. [00085] In one embodiment, the cyclyl portion of R4 group is substituted with one R7 group. In certain embodiments, R7 is oxo, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L- N(R5)2 or C1–C3 alkyl. In certain embodiments, R7 is C1–C3 alkyl, wherein the C1–C3 alkyl is methyl, ethyl or isopropyl. In certain embodiments, R7 is halogen, wherein the halogen is fluorine or chlorine. In certain embodiments, R7 is oxo. [00086] In one embodiment, the cyclyl portion of R4 group is substituted with two R7 groups. In certain embodiments, the two R7 groups are each halogen, wherein each halogen is fluorine. [00087] It is understood that in the compounds of Formula (I), when
is a single bond. In yet another embodiment, Z is S, X is C(R5)2, n is one, and is a single bond.
[00053] In one embodiment, R1 is aryl, which may be optionally substituted with one or more R4. In certain embodiments, the aryl is phenyl, which may be optionally substituted with one or more R4. [00054] In one embodiment, the aryl is substituted with a single R4 group. In one embodiment, the aryl is substituted with two R4 groups. In one embodiment, the aryl is substituted with three R4 groups. Exemplary aryl R4 groups include halogen, hydroxyl, haloalkyl, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L- N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2, Y2-haloalkyl, L-heterocyclyl, or Y1-heterocyclyl, wherein the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. [00055] In one embodiment, the one or more R4 are each independently halogen, hydroxyl, haloalkyl, - COOR5, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L-N(R5)2, -O-L-N(R5)2, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2- N(R5)2, Y2-haloalkyl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heterocyclyl portion of the -L- heterocyclyl or -Y1-heterocyclyl may be optionally substituted with one or more R7. [00056] In one embodiment, R1 is phenyl substituted with -Y2-C1–C6 alkyl. In one embodiment, Y is a bond and the C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, R1 is phenyl substituted with the Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1– C6 alkyl is methyl. In one embodiment, R1 is phenyl, which is disubstituted with methyl and Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1– C6 alkyl is methyl. [00057] In one embodiment, R1 is phenyl substituted one R4, wherein R4 is a cyano group. [00058] In one embodiment, R1 is phenyl substituted one R4, wherein R4 is L-heteroaryl. In certain embodiments, the L-heteroaryl is tetrazolyl. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is PO3(C1-C3 alkyl)2. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is - COOR5. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is -O-L-N(R5)2. In one embodiment, R1 is phenyl substituted one R4, wherein R4 is aralkyl. [00059] In one embodiment, R1 is phenyl substituted with at least one R4, wherein R4 is -L-N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R5 is independently hydrogen. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, R1 is phenyl substituted with -L-N(R5)2 and further substituted with one or more halogen and/or C1–C6 alkyl. [00060] In one embodiment, R1 is phenyl substituted with one R4, wherein R4 is -Y1-N(R5)2. In certain embodiments, Y1 is -C(O)- and each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, Y1 is -C(O)- and each R5 is hydrogen. In one embodiment, Y1 is - C(O)- and one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, R1 is phenyl substituted with -Y1-N(R5)2 and further substituted with one or more halogen and/or C1–C6 alkyl. [00061] In one embodiment, R1 is phenyl substituted with the Y2-haloalkyl, wherein Y2 is -S- or -SO2- and the haloalkyl is trifluoromethyl. [00062] In one embodiment, R1 is phenyl substituted with at least one -L-heterocyclyl or -Y1- heterocyclyl, each heterocyclyl optionally substituted with one or more R7. In one embodiment, R1 is
phenyl substituted with one R4, wherein R4 is -Y1-heterocyclyl optionally substituted with one or more R7. In one embodiment, Y1 is -C(O)- and the heterocyclyl is piperazinyl optionally substituted with C1– C3 alkyl. [00063] In certain embodiments, the R4 group is L-heterocyclyl optionally substituted with one or more R7. In one embodiment, L is methylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4- methyl-piperazinyl. In one embodiment, L is methylene and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R7. Exemplary R7 groups include oxo, halogen, hydroxyalkyl and C1–C3 alkyl. [00064] In one embodiment, R1 is phenyl substituted with Y1-heterocyclyl optionally substituted with one or more R7. In certain embodiments, Y1 is -C(O)- and the heterocyclyl is azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl, each optionally further substituted with one or more halogen. [00065] In one embodiment, R1 is phenyl substituted with L-heteroaryl optionally substituted with one or more R7. In certain embodiments, the L-heteroaryl is tetrazolyl. [00066] In one embodiment, R1 is phenyl substituted with PO3(C1-C3 alkyl)2. In another embodiment, R1 is phenyl substituted with -COOR5. In one embodiment, R1 is phenyl substituted with hydroxyalkyl, - O-L-N(R5)2 or aralkyl. [00067] In one embodiment, R1 is heteroaryl, which may be optionally substituted with one or more R4. In certain embodiments, the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, isoxazolyl, isoindolinyl, naphthyridinyl, 1,2,3,4-tetrahydroisoquinolinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R4. In certain embodiments, the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, or 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazolyl, each of which may be optionally substituted with one or more R4. [00068] In one embodiment, the heteroaryl is substituted with a single R4 group. In one embodiment, the heteroaryl is substituted with two R4 groups. In one embodiment, the heteroaryl is substituted with three R4 groups. Exemplary heteroaryl R4 groups include amino, cyano, halogen, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, Y2-haloalkyl Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, L-cycloalkyl, L-heteroaryl, L- heterocyclyl, Y1-heterocyclyl, -L-N(R5)2, or -Y1-N(R5)2, wherein the ring of the L-cycloalkyl, L- heteroaryl, L-heterocyclyl or Y1-heterocyclyl may be optionally substituted with one or more R7. [00069] In one embodiment, each R4 is independently cyano, halogen, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, -L-cycloalkyl, -L-N(R5)2, -Y1-N(R5)2, -L-heteroaryl, - L-heterocyclyl, or -Y1-heterocyclyl, wherein the heteroaryl of the -L-heteroaryl or the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. [00070] In one embodiment, R7 is amino, hydroxyl, cyano, alkoxy, or halogen. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen, wherein the halogen is fluorine or chlorine. In one
embodiment, R7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. [00071] In another embodiment, R1 is heteroaryl and each R4 is independently hydroxyalkyl, heteroalkyl or haloalkyl. In certain embodiments, the hydroxyalkyl is hydroxymethyl, hydroxyethyl or 2- methyl, 2-hydroxypropyl. In certain embodiments, the heteroalkyl is methoxymethyl or methoxyethyl. In certain embodiments, the haloalkyl is fluoromethyl, difluoromethyl, trifluoromethyl, fluoroethyl, difluoroethyl, or trifluoroethyl. [00072] In certain embodiments, R1 is heteroaryl and R4 is -Y1-C1–C6 alkyl, wherein Y1 is a bond and the C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, R4 is -Y1-C1–C6 alkyl, wherein Y1 is a -C(O)- and the C1–C6 alkyl is methyl, ethyl or isopropyl. In other embodiments, Y1is -NHC(O)- and the C1–C6 alkyl portion is methyl. [00073] In one embodiment, R1 is heteroaryl and R4 is -Y2-C1–C6 alkyl, wherein Y2 is -SO2- and the C1–C6 alkyl is methyl. In another embodiment, R4 is -Y2-C1–C6 alkyl, wherein Y2 is -S- and the C1–C6 alkyl is methyl. [00074] In one embodiment, R1 is heteroaryl and R4 is -Y1-heterocyclyl, which may be optionally substituted with one or more R7. In one embodiment, Y1 is a bond. In another embodiment, Y1 is -C(O)-. In one embodiment, Y1 is a bond and the heterocyclyl is azetidinyl, oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen. [00075] In one embodiment, the heteroaryl is substituted with at least one R4 that is -L-heterocyclyl, which may be optionally substituted with one or more R7. In one embodiment, L is ethylene and the heterocyclyl is pyrrolidinyl, piperidinyl, piperazinyl or 4-methyl-piperazinyl. In one embodiment, L is methylene and the heterocyclyl is azetinidyl, pyrrolidinyl, piperidinyl, piperazinyl, piperazinone, tetrahydropyranyl, morpholinyl, thiomorpholinyl or diazapanyl, each optionally substituted with one or more R7. [00076] In one embodiment, the R7 is independently -L-N(R5)2, hydroxyl, cyano, alkoxy, or halogen. In one embodiment, R7 is C1–C3 alkyl. In one embodiment, R7 is halogen, wherein the halogen is fluorine or chlorine. In one embodiment, R7 is alkoxy, wherein the alkoxy is methoxy or ethoxy. In one embodiment, R7 is cycloalkyl, wherein the cycloalkyl is cyclopropyl. In one embodiment, R7 is -L- N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene. In one embodiment, each R5 is hydrogen. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. [00077] In one embodiment, R1 is heteroaryl and R4 is -L-N(R5)2. In one embodiment, L is a bond. In one embodiment, L is methylene, ethylene, or propylene. In one embodiment, each R5 is independently C1–C3 alkyl. In one embodiment, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In one embodiment, the one C1–C3 alkyl is methyl. In one embodiment, each R5 is hydrogen.
[00078] In one embodiment, R1 is heteroaryl and R4 is L-heteroaryl, which may be optionally substituted with one or more R7. In one embodiment, L is a bond. In one embodiment, L is C1–C3 alkylene. In one embodiment, the C1–C3 alkylene is methylene. In certain embodiments, the heteroaryl of the L-heteroaryl is pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, triazinyl, thiazolyl or pyridazinyl. In one embodiment, the heteroaryl of the L-heteroaryl is pyridyl. [00079] In one embodiment, R1 is heteroaryl which is substituted with two R4 groups independently selected from two -Y1-C1–C6 alkyl groups; -Y1-C1–C6 alkyl and alkoxy; -Y1-C1–C6 alkyl and cycloalkyl; -Y1-C1–C6 alkyl and haloalkyl; -Y1-C1–C6 alkyl and amino; two alkoxy groups; alkoxy and halogen; alkoxy and cyano, and amino and haloalkyl. In certain embodiments, R4 is -Y1-C1–C6 alkyl, wherein each Y1 is a bond and each C1–C6 alkyl is methyl, ethyl or isopropyl. In one embodiment, the cycloalkyl is cyclopropyl. In one embodiment, the alkoxy is methoxy. In one embodiment, the halogen is fluorine or chlorine. In one embodiment, the haloalkyl is trifluoromethyl or trifluoroethyl. [00080] In one embodiment, R1 is L-heterocyclyl optionally substituted with one or more R4. In one embodiment, L is a bond and the heterocyclyl is tetrahydrofuranyl, piperidinyl, piperazinyl or morpholinyl. In one embodiment, L is a methylene and the heterocyclyl is azetidinyl, pyrrolidinyl or 3ƛ2- azabicyclo[3.1.0]hexanyl. In certain embodiments, the heterocyclyl is substituted with one or more R4 selected from oxo, halogen, alkoxy, hydroxyl and Y1-C1–C6 alkyl, wherein Y is a bond or -C(O)-. [00081] In one embodiment, R2 is cyano. In one embodiment, R2 is -COOR5. In certain embodiments, the R5 group is hydrogen. [00082] In one embodiment, R2 is -C(O)N(R5)2. In one embodiment, each R5 is independently C1–C3 alkyl. In certain embodiments, each C1–C3 alkyl is methyl. In one embodiment, one R5 is C1–C3 alkyl and the other is hydrogen. In certain embodiments, the one C1–C3 alkyl is methyl. In one embodiment, each R5 is hydrogen. In one embodiment, each R5 together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6. [00083] In one embodiment, n is zero. In one embodiment, n is one and R3 is halogen. In certain embodiments, the halogen is fluorine or chlorine. In one embodiment, the halogen is fluorine. [00084] In one embodiment, R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl. In certain embodiments, R6 is hydrogen. In other embodiments, R6 is methyl, ethyl, or propyl. [00085] In one embodiment, the cyclyl portion of R4 group is substituted with one R7 group. In certain embodiments, R7 is oxo, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L- N(R5)2 or C1–C3 alkyl. In certain embodiments, R7 is C1–C3 alkyl, wherein the C1–C3 alkyl is methyl, ethyl or isopropyl. In certain embodiments, R7 is halogen, wherein the halogen is fluorine or chlorine. In certain embodiments, R7 is oxo. [00086] In one embodiment, the cyclyl portion of R4 group is substituted with two R7 groups. In certain embodiments, the two R7 groups are each halogen, wherein each halogen is fluorine. [00087] It is understood that in the compounds of Formula (I), when
 represents a single bond, there are two R6 present and each is independently selected from the groups set forth herein. It is further
understood that in the compounds of Formula (I), when
represents a single bond, there are two R6 present and each is independently selected from the groups set forth herein. It is further
understood that in the compounds of Formula (I), when
 represents a single bond, there is one R6 present selected from the groups set forth herein. [00088] In one embodiment, the compound is:
represents a single bond, there is one R6 present selected from the groups set forth herein. [00088] In one embodiment, the compound is:





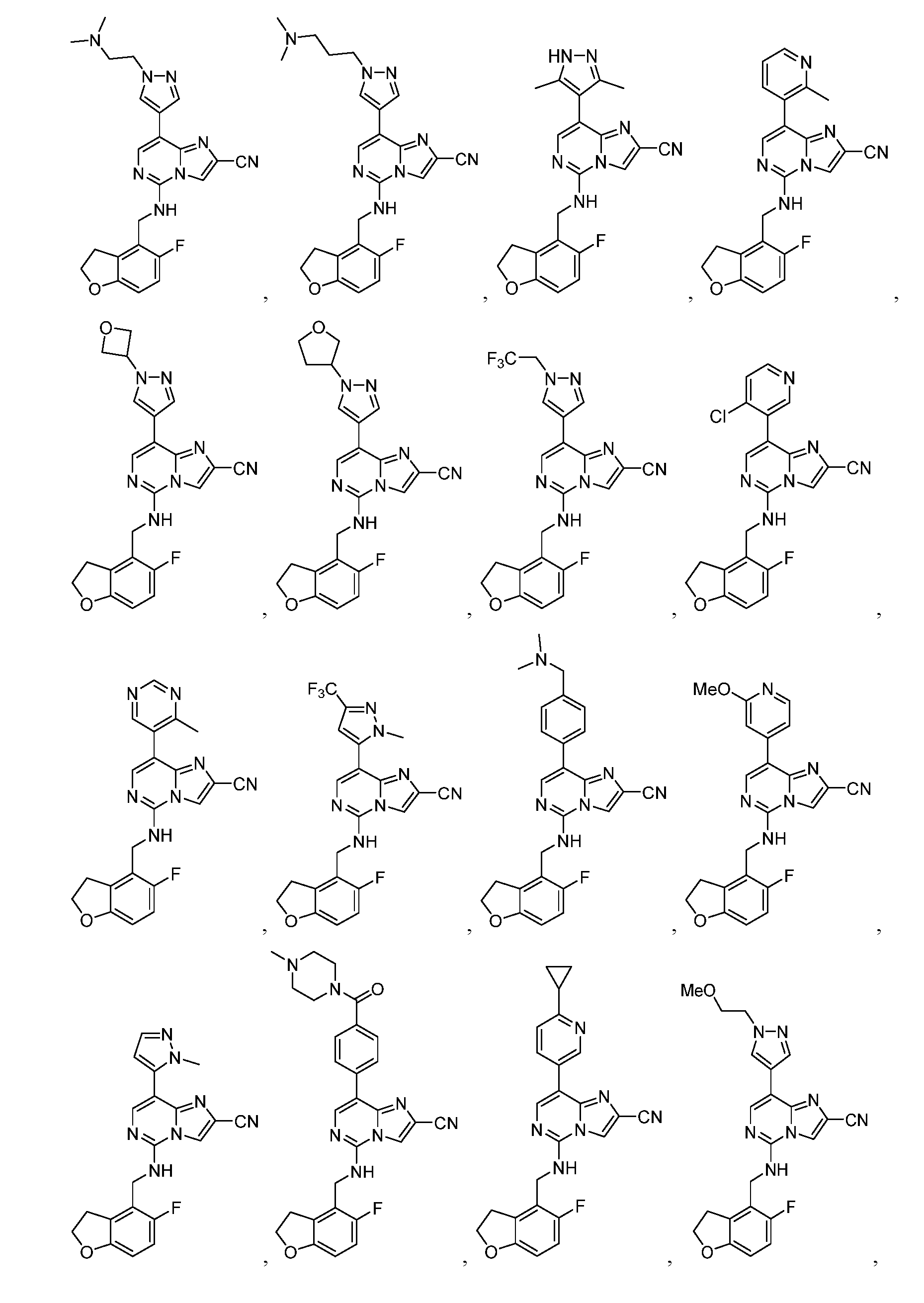






















































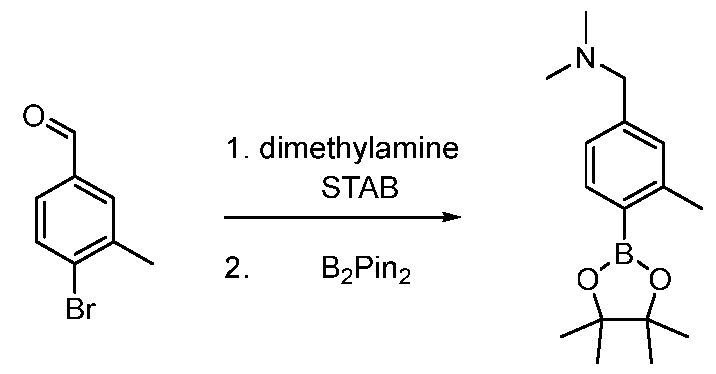






































Table 3 Characterization of EXAMPLES 24-85




















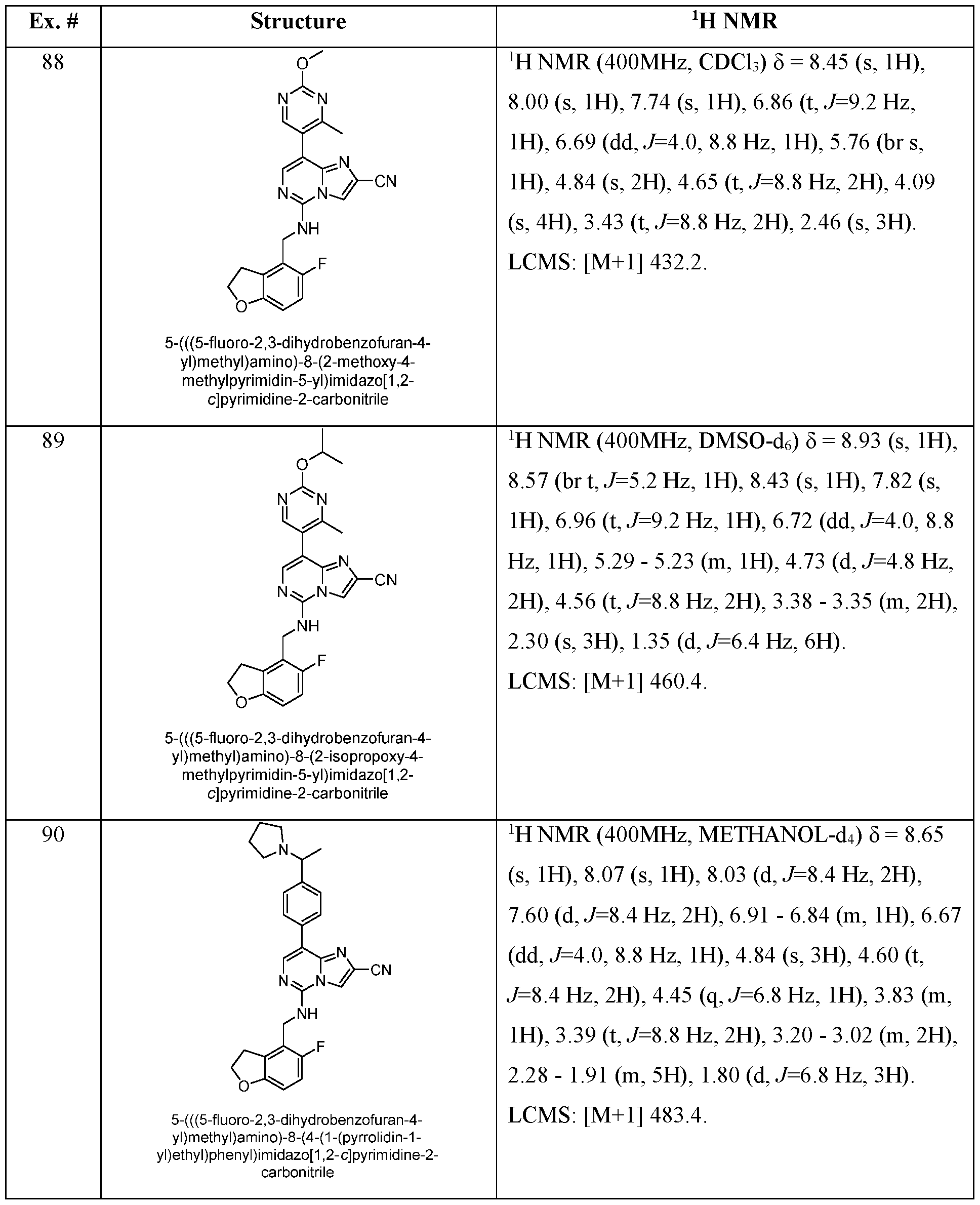




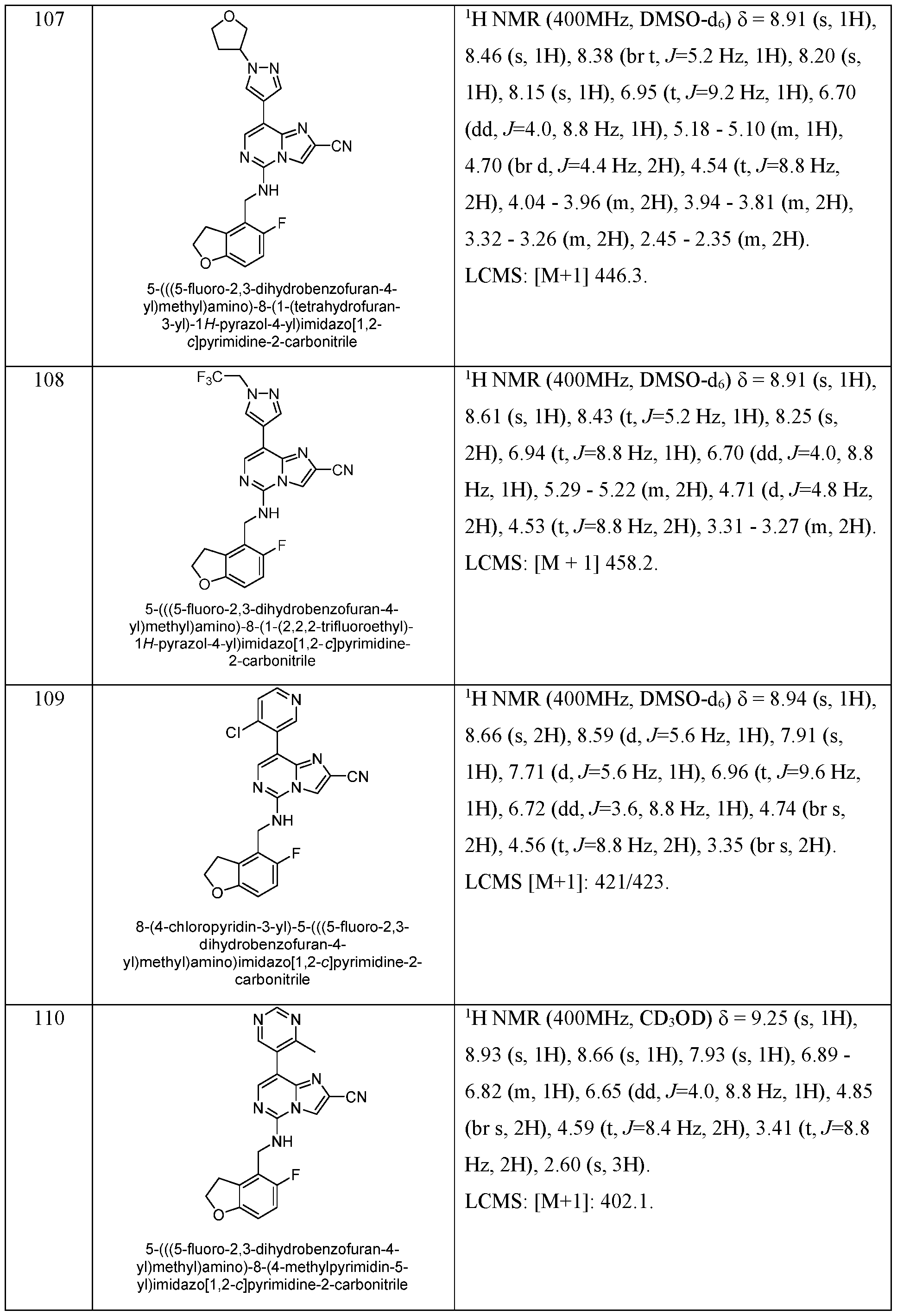


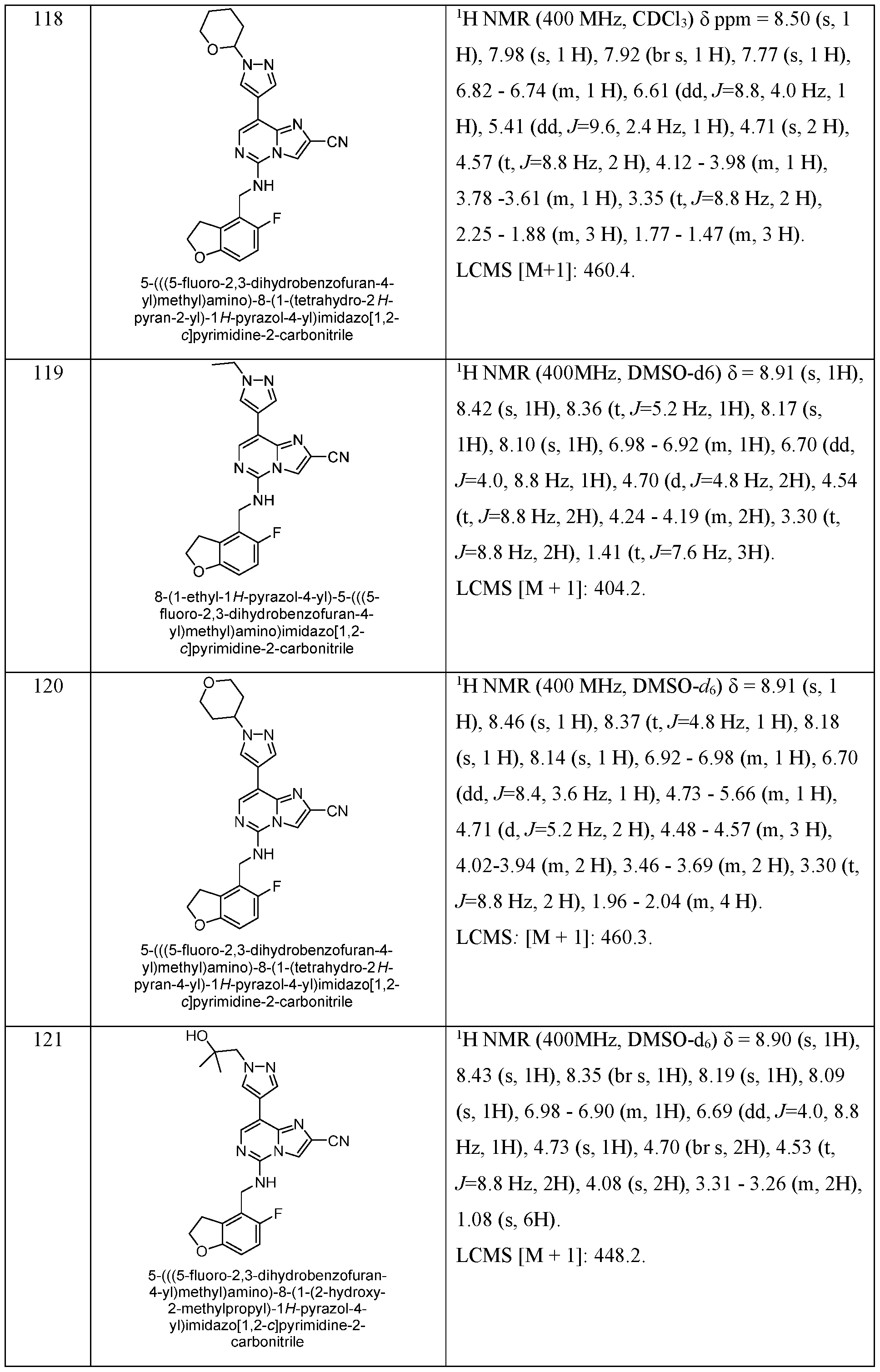







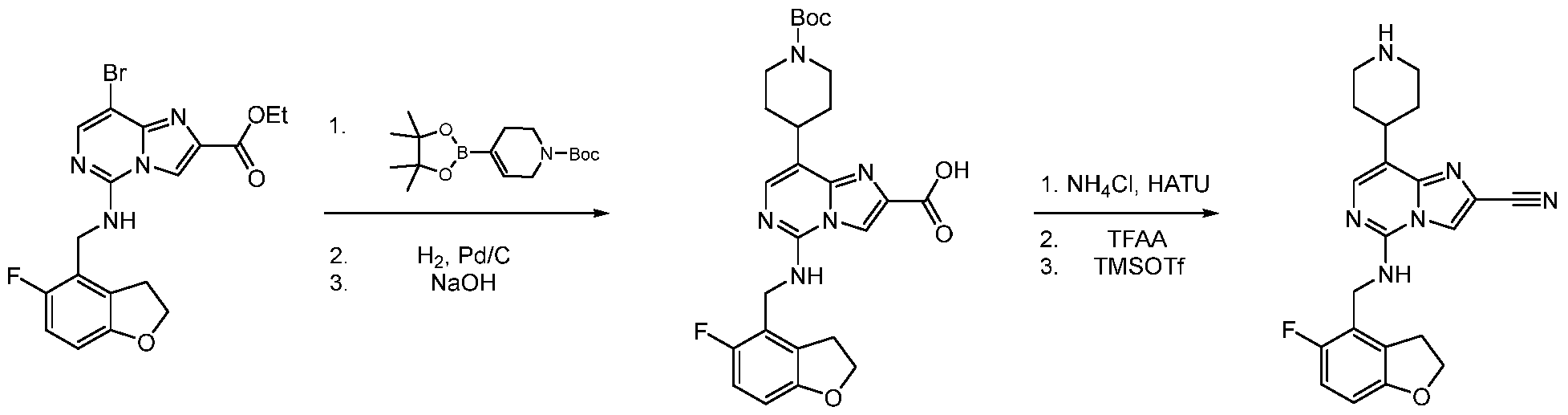




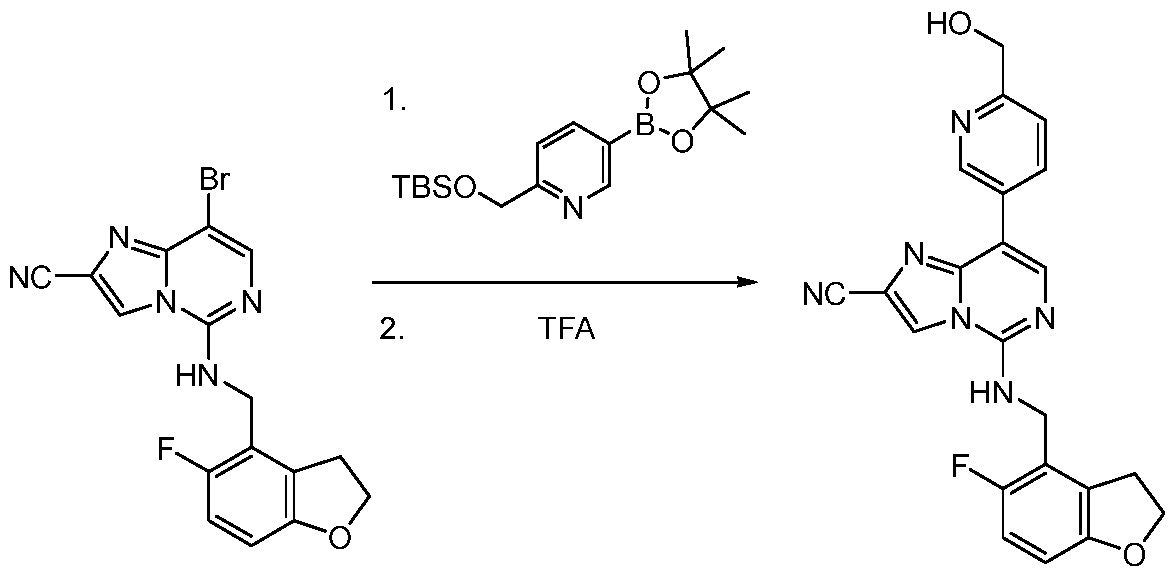


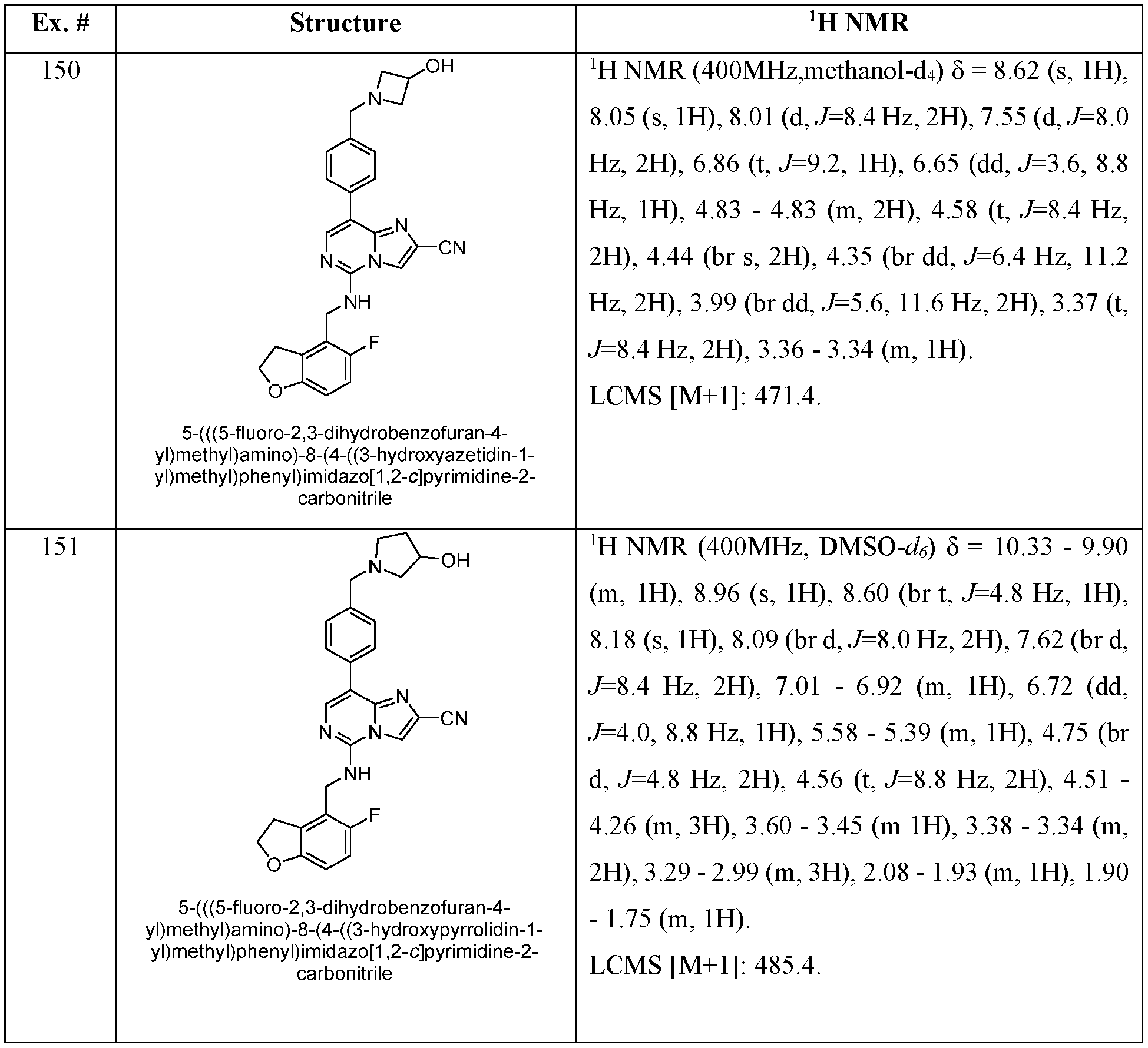








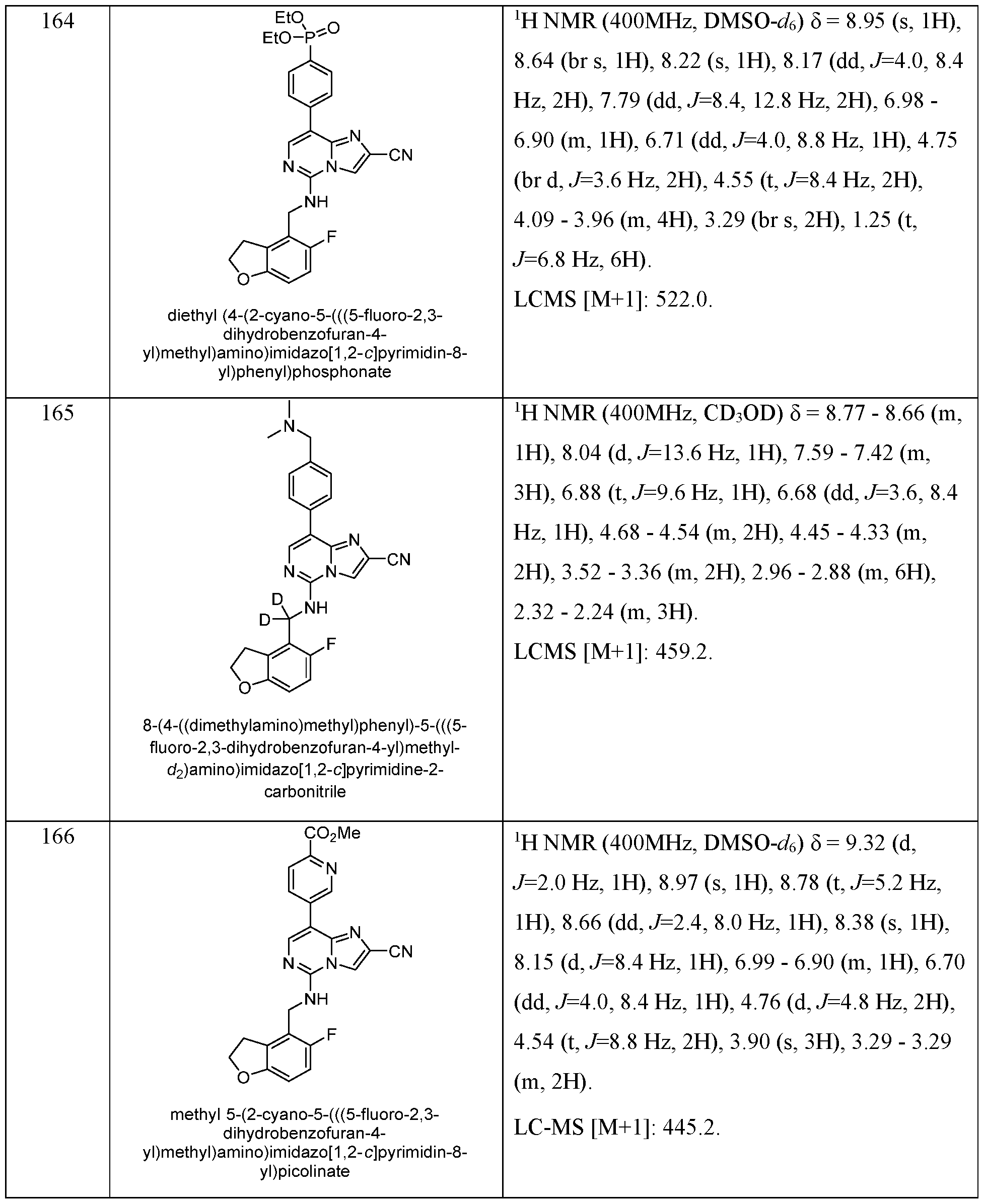
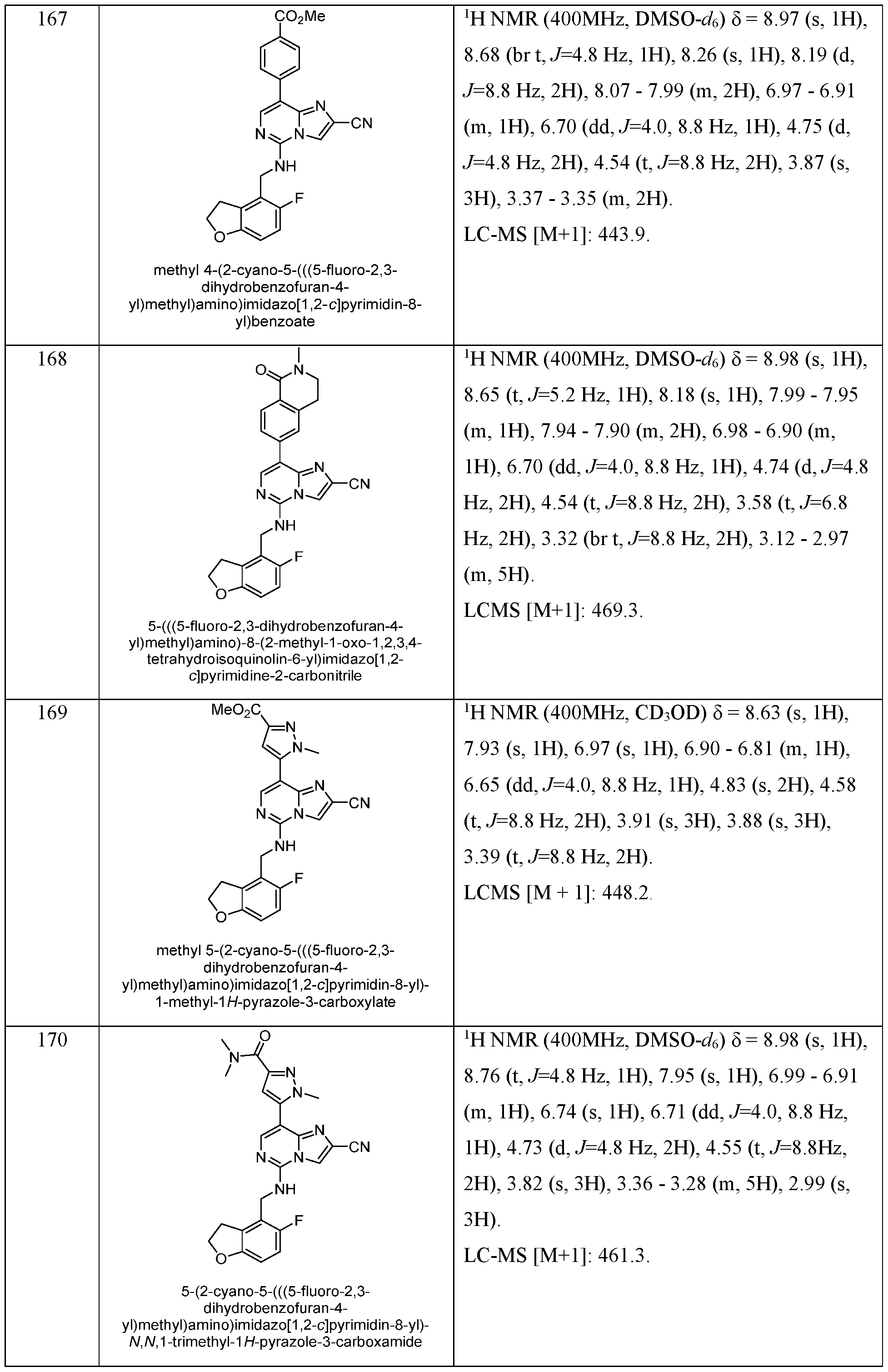















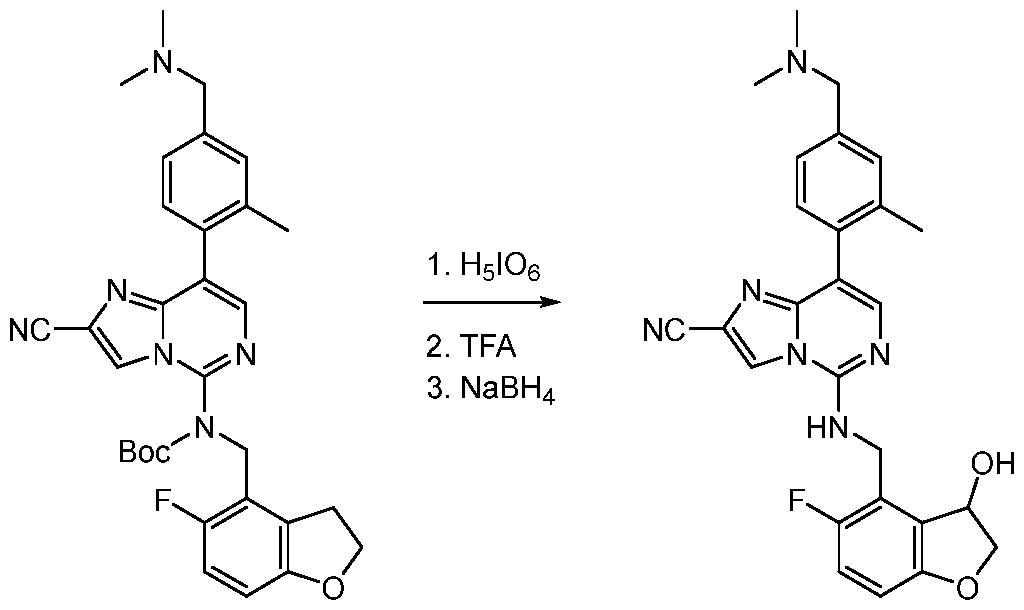









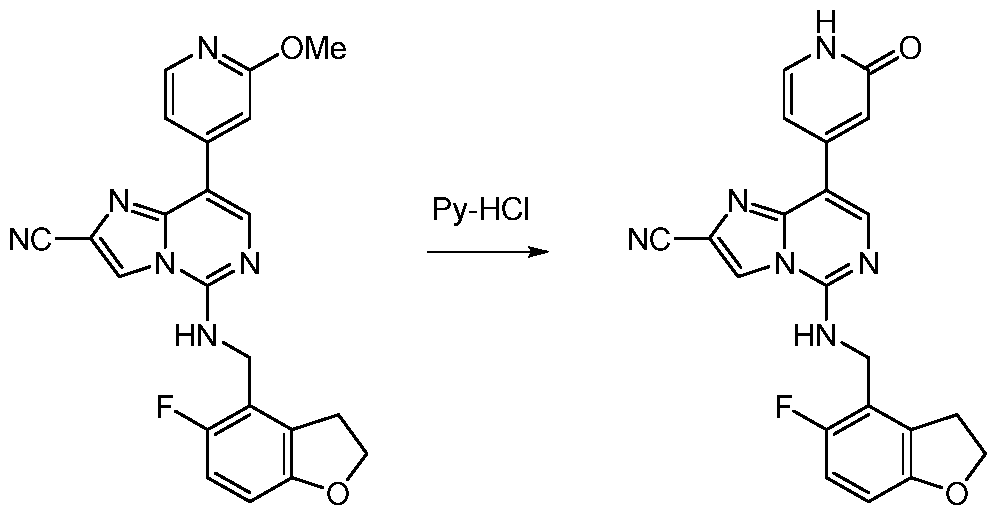
Table 12



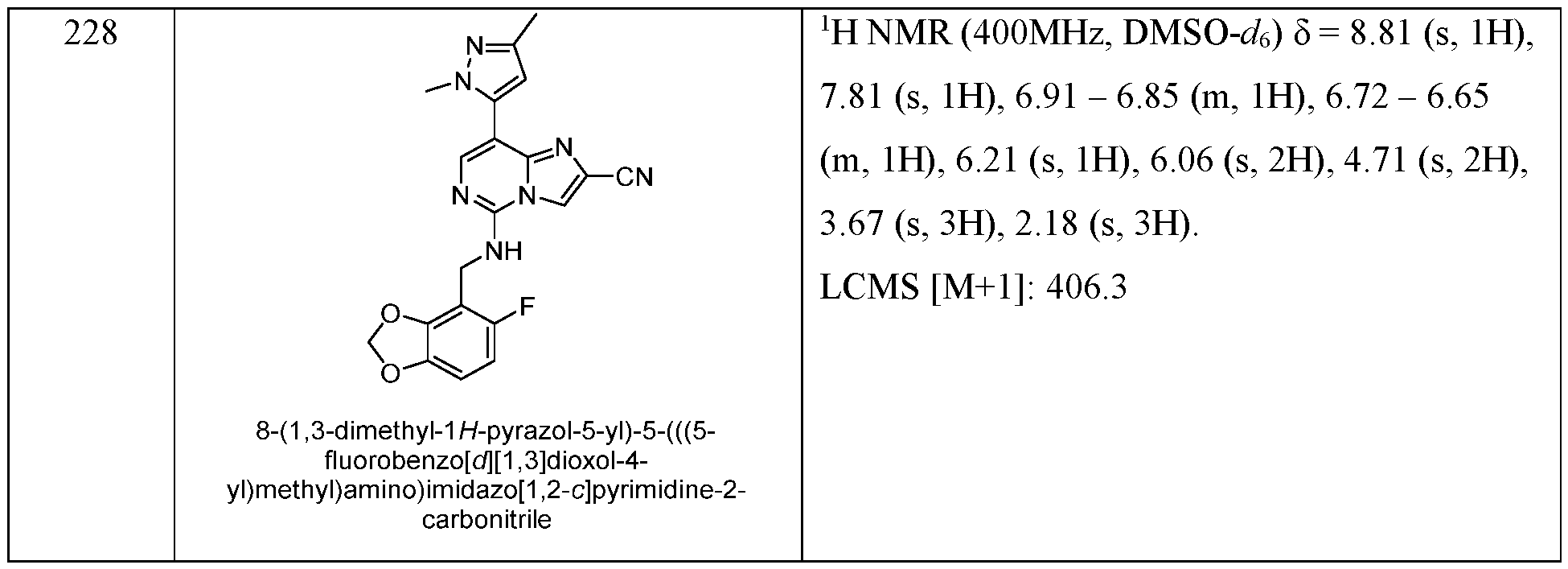










Claims
WE CLAIM: 1. A method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound of Formula (I):
 Formula (I) or a pharmaceutically acceptable salt thereof: wherein:
Formula (I) or a pharmaceutically acceptable salt thereof: wherein:
 represents a single or a double bond; Z is O or S; X is O, CR5, CR5OH, or C(R5)2, wherein: when X is O, is a single bond; when X is C(R5)2,
represents a single or a double bond; Z is O or S; X is O, CR5, CR5OH, or C(R5)2, wherein: when X is O, is a single bond; when X is C(R5)2,
 is a single bond; when X is CR5OH,
is a single bond; when X is CR5OH,
 is a single bond; or when X is CR5, is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, -N(R5)heterocyclyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl, -N(R5)heterocyclyl, and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or-C(O)N(R5)2, ; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-;
Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or -Y1-heterocyclyl, wherein -Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. 2. The method according to claim 1, wherein Z is O. 3. The method according to claim 1, wherein Z is S. 4. The method according to any of claims 1-3, wherein n is 1. 5. The method according to any of claims 1-4, wherein R2 is cyano. 6. The method according to any one of claims 1-5, wherein R3 is fluorine. 7. The method according to any of claims 1-6, wherein X is C(R5)2 and
is a single bond; or when X is CR5, is a double bond; R1 is aryl, heteroaryl, L-cycloalkyl, -N(R5)heterocyclyl, or L-heterocyclyl, wherein the aryl, the heteroaryl and the cyclyl portion of the L-cycloalkyl, -N(R5)heterocyclyl, and L-heterocyclyl may be optionally substituted with one or more R4; R2 is cyano, -COOR5 or-C(O)N(R5)2, ; R3 is C1-C3 alkyl or halogen; each R4 is independently oxo, cyano, halogen, -P(O)(OC1-C3)2, alkoxy, hydroxyl, hydroxyalkyl, heteroalkyl, aralkyl, haloalkyl, -COOR5, -Y2-haloalkyl, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, -L- cycloalkyl, -L-heteroaryl, -L-heterocyclyl, -Y1-heterocyclyl, -Y2-heterocyclyl, -L-N(R5)2, -O-L- N(R5)2, -N(R5)CO(R6), -O-L-OR5, -C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2 wherein the ring portion of the aralkyl, -L-cycloalkyl, -L-heteroaryl, -L-heterocyclyl and -Y1-heterocyclyl may be optionally substituted with one or more R7; L is a bond or C1–C4 alkylene; Y1 is a bond, -C(O)-, or -NHC(O)-;
Y2 is a bond, -S-, -SO-, -SO2-, or -NR5SO2-, each R5 is independently hydrogen or C1–C3 alkyl; or each R5 taken together with the nitrogen atom to which they are attached form a 5 – 8 membered heterocyclic ring optionally substituted with one or more R6; R6 is hydrogen, C1–C3 alkyl, halogen, haloalkyl, hydroxyalkyl, or heteroalkyl; each R7 is independently oxo, cyano, hydroxyl, alkoxy, halogen, haloalkyl, hydroxyalkyl, heteroalkyl, cycloalkyl, -L-N(R5)2, C1–C6 alkyl or -Y1-heterocyclyl, wherein -Y1-heterocyclyl may be optionally substituted with one or more R6; and n is 1 or 2. 2. The method according to claim 1, wherein Z is O. 3. The method according to claim 1, wherein Z is S. 4. The method according to any of claims 1-3, wherein n is 1. 5. The method according to any of claims 1-4, wherein R2 is cyano. 6. The method according to any one of claims 1-5, wherein R3 is fluorine. 7. The method according to any of claims 1-6, wherein X is C(R5)2 and
 is a single bond. 8. The method according to any of claims 1-7, wherein R1 is aryl optionally substituted with one or more R4. 9. The method according to claim 8, wherein R1 is phenyl optionally substituted with one or more R4. 10. The method according to claim 9, wherein the one or more R4 are each independently halogen, hydroxyl, haloalkyl, -COOR5, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L-N(R5)2, -O-L-N(R5)2, - C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2, Y2-haloalkyl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heterocyclyl portion of the -L-heterocyclyl or -Y1-heterocyclyl may be optionally substituted with one or more R7. 11. The method according to any of claims 1-7, wherein R1 is heteroaryl optionally substituted with one or more R4. 12. The method according to claim 11, wherein the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, isoxazolyl, isoindolinyl, naphthyridinyl, 1,2,3,4-tetrahydroisoquinolinyl, or 5,6-dihydro-4H- pyrrolo[1,2-b]pyrazolyl, each optionally substituted with one or more R4. 13. The method according to claim 12, wherein each R4 is independently cyano, halogen, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, -L-cycloalkyl, -L-N(R5)2, - Y1-N(R5)2, -L-heteroaryl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heteroaryl of the -L- heteroaryl or the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. 14. A method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound selected from
is a single bond. 8. The method according to any of claims 1-7, wherein R1 is aryl optionally substituted with one or more R4. 9. The method according to claim 8, wherein R1 is phenyl optionally substituted with one or more R4. 10. The method according to claim 9, wherein the one or more R4 are each independently halogen, hydroxyl, haloalkyl, -COOR5, -Y1-C1–C6 alkyl, Y2-C1–C6 alkyl, -L-N(R5)2, -O-L-N(R5)2, - C(CF3)N(R5)2, -Y1-N(R5)2, -Y2-N(R5)2, Y2-haloalkyl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heterocyclyl portion of the -L-heterocyclyl or -Y1-heterocyclyl may be optionally substituted with one or more R7. 11. The method according to any of claims 1-7, wherein R1 is heteroaryl optionally substituted with one or more R4. 12. The method according to claim 11, wherein the heteroaryl is pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, triazinyl, pyridyl, pyridinyl-2-one, pyrazinyl, pyridazinyl, pyrimidinyl, isoxazolyl, isoindolinyl, naphthyridinyl, 1,2,3,4-tetrahydroisoquinolinyl, or 5,6-dihydro-4H- pyrrolo[1,2-b]pyrazolyl, each optionally substituted with one or more R4. 13. The method according to claim 12, wherein each R4 is independently cyano, halogen, -Y1-C1–C6 alkyl, -Y2-C1–C6 alkyl, alkoxy, hydroxyalkyl, heteroalkyl, haloalkyl, -L-cycloalkyl, -L-N(R5)2, - Y1-N(R5)2, -L-heteroaryl, -L-heterocyclyl, or -Y1-heterocyclyl, wherein the heteroaryl of the -L- heteroaryl or the heterocyclyl portion of the L-heterocyclyl, or Y1-heterocyclyl may be optionally substituted with one or more R7. 14. A method of treating a blood disorder in a subject, comprising administering to the subject a therapeutically effective amount of a compound selected from













Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22873813.4A EP4404932A4 (en) | 2021-09-24 | 2022-09-21 | PRC2 INHIBITORS FOR USE IN THE TREATMENT OF BLOOD DISORDERS |
| US18/694,105 US20240398811A1 (en) | 2021-09-24 | 2022-09-21 | Prc2 inhibitors for use in treating blood disorders |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202163248237P | 2021-09-24 | 2021-09-24 | |
| US63/248,237 | 2021-09-24 | ||
| US202263298531P | 2022-01-11 | 2022-01-11 | |
| US63/298,531 | 2022-01-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023049724A1 true WO2023049724A1 (en) | 2023-03-30 |
Family
ID=85719675
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2022/076750 Ceased WO2023049724A1 (en) | 2021-09-24 | 2022-09-21 | Prc2 inhibitors for use in treating blood disorders |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20240398811A1 (en) |
| EP (1) | EP4404932A4 (en) |
| WO (1) | WO2023049724A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024215699A1 (en) * | 2023-04-11 | 2024-10-17 | Oric Pharmaceuticals, Inc. | Treatment of t-cell lymphoma |
| WO2025043191A1 (en) * | 2023-08-24 | 2025-02-27 | Oric Pharmaceuticals, Inc. | Prc2 inhibitors for use in treating sickle cell disease |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019152419A1 (en) * | 2018-01-31 | 2019-08-08 | Mirati Therapeutics, Inc | Prc2 inhibitors |
| WO2020190754A1 (en) * | 2019-03-15 | 2020-09-24 | Fulcrum Therapeutics, Inc. | Macrocyclic azolopyridine derivatives as eed and prc2 modulators |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3980422A1 (en) * | 2019-06-05 | 2022-04-13 | Mirati Therapeutics, Inc. | Imidazo[1,2-c]pyrimidine derivatives as prc2 inhibitors for treating cancer |
-
2022
- 2022-09-21 EP EP22873813.4A patent/EP4404932A4/en active Pending
- 2022-09-21 US US18/694,105 patent/US20240398811A1/en active Pending
- 2022-09-21 WO PCT/US2022/076750 patent/WO2023049724A1/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019152419A1 (en) * | 2018-01-31 | 2019-08-08 | Mirati Therapeutics, Inc | Prc2 inhibitors |
| WO2020190754A1 (en) * | 2019-03-15 | 2020-09-24 | Fulcrum Therapeutics, Inc. | Macrocyclic azolopyridine derivatives as eed and prc2 modulators |
Non-Patent Citations (3)
| Title |
|---|
| ANONYMOUS: "Fulcrum Therapeutics Presents Published Structure of Investigational Small Molecule FTX6058 at the American Chemical Society (ACS) Spring 2021 Virtual Conference", FULCRUM THERAPEUTICS, 9 April 2021 (2021-04-09), XP093059746, Retrieved from the Internet <URL:https://www.globenewswire.com/news-release/2021/04/09/2207657/0/en/Fulcrum-Therapeutics-Presents-Published-Structure-of-Investigational-Small-Molecule-FTX-6058-at-the-American-Chemical-Society-ACS-Spring-2021-Virtual-Conference.html> [retrieved on 20230630] * |
| EFREMOV IVAN: "Discovery of clinical candidate FTX-6058: a potent, orally bioavailable upregulator of fetal hemoglobin for treatment of sickle cell disease", 5 April 2021 (2021-04-05) - 1 May 2021 (2021-05-01), XP055944156 * |
| See also references of EP4404932A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024215699A1 (en) * | 2023-04-11 | 2024-10-17 | Oric Pharmaceuticals, Inc. | Treatment of t-cell lymphoma |
| WO2025043191A1 (en) * | 2023-08-24 | 2025-02-27 | Oric Pharmaceuticals, Inc. | Prc2 inhibitors for use in treating sickle cell disease |
Also Published As
| Publication number | Publication date |
|---|---|
| US20240398811A1 (en) | 2024-12-05 |
| EP4404932A4 (en) | 2025-07-23 |
| EP4404932A1 (en) | 2024-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7495555B2 (en) | PRC2 inhibitors | |
| JP7541538B2 (en) | Imidazo[1,2-C]pyrimidine derivatives as prc2 inhibitors for treating cancer - Patents.com | |
| JP7624404B2 (en) | Naphthyridine Derivatives as PRC2 Inhibitors | |
| WO2023049724A1 (en) | Prc2 inhibitors for use in treating blood disorders | |
| WO2023049723A1 (en) | Prc2 inhibitors for use in treating blood disorders | |
| WO2025043191A1 (en) | Prc2 inhibitors for use in treating sickle cell disease | |
| HK40077182B (en) | Prc2 inhibitors | |
| HK40077182A (en) | Prc2 inhibitors | |
| HK40039589A (en) | Prc2 inhibitors | |
| HK40039589B (en) | Prc2 inhibitors | |
| EA043605B1 (en) | PRC2 INHIBITORS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22873813 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022873813 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022873813 Country of ref document: EP Effective date: 20240424 |