WO2023054223A1 - ポリプロピレン系樹脂押出発泡粒子、ポリプロピレン系樹脂発泡成形体および積層発泡体 - Google Patents
ポリプロピレン系樹脂押出発泡粒子、ポリプロピレン系樹脂発泡成形体および積層発泡体 Download PDFInfo
- Publication number
- WO2023054223A1 WO2023054223A1 PCT/JP2022/035573 JP2022035573W WO2023054223A1 WO 2023054223 A1 WO2023054223 A1 WO 2023054223A1 JP 2022035573 W JP2022035573 W JP 2022035573W WO 2023054223 A1 WO2023054223 A1 WO 2023054223A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- polypropylene
- extruded
- foam
- polypropylene resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/16—Making expandable particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/18—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by features of a layer of foamed material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/22—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed
- B32B5/32—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed at least two layers being foamed and next to each other
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F255/00—Macromolecular compounds obtained by polymerising monomers on to polymers of hydrocarbons as defined in group C08F10/00
- C08F255/02—Macromolecular compounds obtained by polymerising monomers on to polymers of hydrocarbons as defined in group C08F10/00 on to polymers of olefins having two or three carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0061—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof characterized by the use of several polymeric components
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/122—Hydrogen, oxygen, CO2, nitrogen or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/16—Making expandable particles
- C08J9/18—Making expandable particles by impregnating polymer particles with the blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/22—After-treatment of expandable particles; Forming foamed products
- C08J9/228—Forming foamed products
- C08J9/232—Forming foamed products by sintering expandable particles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/10—Homopolymers or copolymers of propene
- C08L23/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/06—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2266/00—Composition of foam
- B32B2266/02—Organic
- B32B2266/0214—Materials belonging to B32B27/00
- B32B2266/025—Polyolefin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2266/00—Composition of foam
- B32B2266/02—Organic
- B32B2266/0214—Materials belonging to B32B27/00
- B32B2266/0278—Polyurethane
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2201/00—Foams characterised by the foaming process
- C08J2201/02—Foams characterised by the foaming process characterised by mechanical pre- or post-treatments
- C08J2201/03—Extrusion of the foamable blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/06—CO2, N2 or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2205/00—Foams characterised by their properties
- C08J2205/04—Foams characterised by their properties characterised by the foam pores
- C08J2205/052—Closed cells, i.e. more than 50% of the pores are closed
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2351/00—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers
- C08J2351/06—Characterised by the use of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2423/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2423/26—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/14—Applications used for foams
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/20—Applications use in electrical or conductive gadgets
- C08L2203/206—Applications use in electrical or conductive gadgets use in coating or encapsulating of electronic parts
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
Definitions
- the present invention relates to extruded polypropylene resin foam particles, polypropylene resin foam molded articles, and laminated foams.
- a polypropylene resin foam molded article obtained using expanded polypropylene resin particles is characterized by excellent arbitrariness in shape, cushioning properties, lightness, heat insulation, and the like. Therefore, polypropylene-based resin foam molded articles are mainly used for various applications such as automobile interior parts, core materials for automobile bumpers, heat insulating materials, and shock-absorbing packaging materials. For example, in automobile seats, from the viewpoint of cushioning properties, a laminated foam obtained by laminating a polypropylene-based resin foam molded article and a polyurethane foam is used.
- Patent Document 1 there is disclosed a method in which a polyethylene foam is placed in a mold and an unfoamed raw material of polyurethane is injected into the mold to embed the polyethylene foam in a predetermined portion of the polyurethane foam.
- a different hardness pad insert molding method for molding a hardness pad is disclosed.
- the molding method of Patent Literature 1 is characterized by subjecting the surface of the polyethylene foam to an easy-adhesion treatment prior to placing it in a mold.
- Patent Document 2 discloses a method for manufacturing a laminate in which a different material layer made of a material different from that of the molded body is formed on the surface of the molded body obtained by integrally molding a plurality of expandable resin particles. It is In the method for manufacturing the laminate of Patent Document 2, when forming the different material layer, the contact area with the different material layer on the surface of the molded body is heated to a temperature of 80% or more of the softening temperature of the molded body. Characterized by
- One embodiment of the present invention has been made in view of the above problems, and an object of the present invention is to provide a laminated foam containing a polypropylene resin foam molded product and a polyurethane foam with high productivity, and a polypropylene resin extrusion. It is to provide expanded particles.
- the base resin of the extruded polypropylene resin particles is the sum of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- the base resin of the extruded polypropylene resin particles is the sum of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- extruded polypropylene resin expanded particles that can provide a laminated foam containing a polypropylene resin foam molded article and a polyurethane foam with good productivity.
- Patent Literature 1 the surface of the polyethylene foam is treated to facilitate adhesion (for example, roughening treatment, flame treatment, corona discharge treatment, etc.) in order to bond the polyethylene foam and the polyurethane foam together.
- adhesion for example, roughening treatment, flame treatment, corona discharge treatment, etc.
- Patent Document 2 it is necessary to heat the contact area with the different material layer on the surface of the molded body to a temperature of 80% or more of the softening temperature of the molded body in order to bond the molded body and the different material layer. be. That is, in the techniques of Patent Document 1 and Patent Document 2, it is necessary to further physically treat the surface of the obtained polyethylene foam or molded article, and there is room for further improvement from the viewpoint of productivity. was there.
- the present inventors set it as the subject to provide the lamination
- extruded polypropylene resin particles containing a specific amount of can provide a polypropylene resin foam molded article having excellent adhesive strength with polyurethane foam without physically treating the surface;
- extruded resin-based foamed particles can provide a laminated foamed product containing a polypropylene-based resin foam-molded product and a polyurethane foam with good productivity.
- Japanese Patent Application Laid-Open No. 2011-099101 discloses a method of producing expanded beads by a depressurized foaming method using an acid-modified polyolefin resin.
- the present inventors have newly found that the technology described in JP-A-2011-099101 has the following problems: (1) JP-A-2011-099101.
- When producing foamed beads using an acid-modified polyolefin resin, as in the art mutual adhesion between resin particles occurs, so it is necessary to use a large amount of dispersant; A large amount of dispersant remains on the surface of the expanded beads obtained by using a large amount, and the foamed molded article obtained by in-mold foam molding using the expanded beads has (a) a low fusion rate.
- the foamed molding itself becomes fragile, and as a result, (b) the adhesive strength with the polyurethane foam tends to be inferior (the foamed molding is not the bonded part between the foamed molding and the polyurethane foam. (for breakage at interfaces between foamed particles).
- the extruded polypropylene resin particles according to one embodiment of the present invention do not require a dispersant because they are obtained by an extrusion foaming method. That is, the extruded polypropylene-based resin expanded beads according to one embodiment of the present invention have the advantage that the dispersant is not adhered to the surface of the expanded beads. As a result, the polypropylene-based resin extruded expanded particles according to one embodiment of the present invention also have the advantage of being able to provide a polypropylene-based resin foam-molded article having excellent fusion bondability and excellent adhesive strength to polyurethane foam.
- the base resin of the extruded polypropylene resin particles is the sum of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- the base resin of the extruded polypropylene resin particles is the sum of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- the polypropylene-based resin extruded expanded particles according to one embodiment of the present invention have the above-described configuration, and therefore have the advantage of being able to provide a laminated foam containing a polypropylene-based resin foam molded article and a polyurethane foam with good productivity. Specifically, since the polypropylene resin extruded foamed particles according to one embodiment of the present invention have the above-described structure, the polypropylene resin foam molded article has excellent adhesion strength to the polyurethane foam without physically treating the surface.
- the extruded polypropylene-based resin expanded particles according to one embodiment of the present invention may have a structure in which the surface does not contain a dispersant (eg, tribasic calcium phosphate, kaolin, etc.). However, the dispersant may adhere unintentionally to the extruded polypropylene-based resin particles according to one embodiment of the present invention. Therefore, the extruded polypropylene-based resin expanded particles according to one embodiment of the present invention may have a structure in which the surface does not substantially contain a dispersant.
- the expression "substantially free of dispersant on the surface” means that the amount of dispersant adhering to the surface is 100 ppm or less.
- the polypropylene-based resin extruded expanded particles according to one embodiment of the present invention do not contain a dispersant on the surface, or contain a dispersant of more than 0 ppm and not more than 100 ppm on the surface.
- Dispersants include, for example, tricalcium phosphate, calcium carbonate, trimagnesium phosphate, barium sulfate, kaolin and talc.
- the extruded polypropylene resin foam particles according to one embodiment of the present invention can be made into a polypropylene resin foam molded article by subjecting the extruded polypropylene resin foam particles to in-mold foam molding. Also, a laminated foam can be obtained by laminating the polypropylene-based resin foam molded article and a polyurethane foam.
- the "polypropylene resin having a branched structure” may be referred to as "branched polypropylene resin”
- the "extruded expanded polypropylene resin particles” may be referred to as "extruded expanded particles”.
- extruded polypropylene resin expanded particles may be referred to as the "extruded expanded particles", and the "polypropylene resin expanded molded article” may be referred to as the "expanded molded article”.
- a base resin according to one embodiment of the present invention includes a polypropylene resin (A) having a branched structure and an acid-modified polyolefin resin (B) having a weight average molecular weight of 10,000 to 100,000.
- the base resin may further optionally contain additives such as cell nucleating agents.
- the base resin can also be said to be a resin component that substantially constitutes the extruded expanded beads.
- polypropylene resin (A) having a branched structure means (a) a polypropylene resin obtained by partially cross-linking the molecules of a polypropylene resin into which no branched structure has been introduced, and (b) A polypropylene resin obtained by introducing a conjugated diene compound other than (poly)propylene as a branched chain to a polypropylene resin having no branched structure is intended.
- the portion of the branched polypropylene-based resin (A) other than the crosslinked portion may be referred to as the "main chain”.
- the "polypropylene-based resin into which no branched structure has been introduced" that is, the raw material of the branched polypropylene-based resin (A) may be referred to as "linear polypropylene-based resin".
- the polypropylene-based resin means a resin containing 50 mol% or more of structural units derived from a propylene monomer in 100 mol% of all structural units contained in the resin. Therefore, the main chain of the branched polypropylene-based resin (A) contains 50 mol% or more of the structural units derived from the propylene monomer in 100 mol% of the total structural units contained in the main chain.
- structural unit derived from propylene monomer may be referred to as "propylene unit”.
- the backbone may be (a) a homopolymer of propylene, (b) a block or random copolymer of propylene and a monomer other than propylene, or (c) A mixture of two or more of these may be used.
- the main chain may have one or more structural units derived from a monomer other than the propylene monomer, or may have one or more types.
- a "monomer other than a propylene monomer” used in the production of the main chain may be referred to as a "comonomer”, and a "structural unit derived from a monomer other than a propylene monomer” contained in the main chain. is sometimes referred to as a "comonomer unit".
- Comonomers include monomers such as: (a) ethylene, 1-butene, isobutene, 1-pentene, 3-methyl-1-butene, 1-hexene, 4-methyl-1-pentene, ⁇ -olefins having 2 or 4 to 12 carbon atoms such as 3,4-dimethyl-1-butene, 1-heptene, 3-methyl-1-hexene, 1-octene, 1-decene, (b) cyclopentene, norbornene, Cyclic olefins such as tetracyclo[6,2,11,8,13,6]-4-dodecene, (c) 5-methylene-2-norbornene, 5-ethylidene-2-norbornene, 1,4-hexadiene, methyl- dienes such as 1,4-hexadiene, 7-methyl-1,6-octadiene, and (d) vinyl chloride, vinylidene chloride, acrylonitrile, meth
- Acrylic esters include methyl acrylate, ethyl acrylate, butyl acrylate, hexyl acrylate, 2-ethylhexyl acrylate, lauryl acrylate, stearyl acrylate, 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate and and glycidyl acrylate.
- Methacrylates include methyl methacrylate, ethyl methacrylate, butyl methacrylate, hexyl methacrylate, 2-ethylhexyl methacrylate, lauryl methacrylate, stearyl methacrylate, 2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate and and glycidyl methacrylate.
- Styrenic monomers include styrene, methylstyrene, dimethylstyrene, alphamethylstyrene, paramethylstyrene, ethylstyrene, diethylstyrene, isopropylstyrene, t-butylstyrene, bromostyrene, dibromostyrene, tribromostyrene, chlorostyrene. , dichlorostyrene and trichlorostyrene.
- the main chain preferably has a structural unit derived from an ⁇ -olefin having 2 or 4 to 12 carbon atoms as a comonomer unit, such as ethylene, 1-butene, isobutene, 1-pentene, 3-methyl-1-butene, Structural units derived from 1-hexene, 4-methyl-1-pentene, 3,4-dimethyl-1-butene, 1-heptene, 3-methyl-1-hexene, 1-octene and/or 1-decene, etc. It is more preferable to have structural units derived from ethylene, 1-butene, isobutene, 1-pentene, 3-methyl-1-butene, 1-hexene and/or 4-methyl-1-pentene.
- an ⁇ -olefin having 2 or 4 to 12 carbon atoms such as ethylene, 1-butene, isobutene, 1-pentene, 3-methyl-1-butene, Structural units derived from 1-hexene, 4-
- ethylene, 1-butene, isobutene and/or 1-pentene are more preferred, and structural units derived from ethylene and/or 1-butene are even more preferred.
- the main chain is preferably one or more selected from the group consisting of a propylene homopolymer, a polypropylene block copolymer and a polypropylene random copolymer.
- a coalescence is more preferred. According to this configuration, (a) the advantage that a branched polypropylene resin having a high melt tension and a low gel fraction can be obtained, and (b) the obtained branched polypropylene resin has excellent moldability. It has the advantage of being able to provide particles.
- the main chain preferably contains 90 mol% or more, more preferably 93 mol% or more, even more preferably 95 mol% or more of 100 mol% of all structural units contained in the main chain, It is particularly preferable to contain 97 mol % or more.
- This configuration has the advantage of obtaining a branched polypropylene resin having a high melt tension and a low gel fraction.
- the melting point of the branched polypropylene resin (A) is not particularly limited.
- the melting point of the branched polypropylene resin (A) is, for example, preferably 125°C to 170°C, more preferably 130°C to 170°C, more preferably 135°C to 160°C, and 140°C. C. to 155.degree. C. is more preferred, and 145.degree. C. to 150.degree. C. is particularly preferred.
- the melting point of the branched polypropylene-based resin (A) is within the range described above, there is an advantage that the extruded expanded particles have a low open cell ratio.
- the melting point of the branched polypropylene resin (A) is (a) 125° C.
- extruded foam particles can be molded at a relatively low steam pressure. , has the advantage that extruded foamed particles can be molded using general-purpose molding machines for polypropylene-based resin foamed particles.
- the melting point of the branched polypropylene-based resin (A) is a value obtained by measuring with a differential scanning calorimeter method (hereinafter referred to as "DSC method").
- DSC method differential scanning calorimeter method
- the specific operating procedure is as follows: (1) The temperature of 5 mg to 6 mg of the branched polypropylene resin (A) is increased from 40° C. to 220° C. at a rate of 10° C./min. (2) Then, the temperature of the melted branched polypropylene resin (A) is lowered from 220° C. to 40° C. at a rate of 10° C./min. (3) Then, the temperature of the crystallized branched polypropylene resin (A) is increased from 40° C. to 220° C.
- the temperature of the peak (melting peak) of the DSC curve of the branched polypropylene resin (A) obtained during the second heating (that is, at the time of (3)) is taken as the melting point of the branched polypropylene resin (A).
- the peak (melting peak) with the maximum amount of heat of fusion is the melting point of the branched polypropylene resin (A).
- the differential scanning calorimeter for example, DSC6200 type manufactured by Seiko Instruments Inc. can be used.
- the melt flow rate (MFR) of the branched polypropylene resin (A) is not particularly limited.
- the MFR of the branched polypropylene resin (A) is, for example, preferably 0.5 g/10 min to 20.0 g/10 min, and preferably 0.5 g/10 min to 15.0 g/10 min. More preferably, 1.0 g/10 min to 10.0 g/10 min, even more preferably 1.5 g/10 min to 5.0 g/10 min, 1.5 g/10 min ⁇ 3.0 g/10 min is particularly preferred.
- the MFR of the branched polypropylene-based resin (A) is within the range described above, (a) the advantage that the extruded expanded particles have a low open cell rate, (b) the advantage that the extruded expanded particles have excellent moldability, and (c) The extruded foamed particles have the advantage of being able to provide foamed molded articles with excellent breakage resistance.
- the MFR of the branched polypropylene resin (A) is (a) 0.5 g/10 minutes or more, the extruded expanded particles obtained from the branched polypropylene resin (A) have little deformation and good surface properties.
- the MFR of the branched polypropylene resin (A) is a value obtained by measuring under conditions of a temperature of 230°C and a load of 2.16 kg according to ISO 1133.
- the melt tension of the branched polypropylene resin (A) is not particularly limited.
- the melt tension of the branched polypropylene resin (A) is, for example, preferably 1.0 cN to 50.0 cN, more preferably 3.0 cN to 40.0 cN, and 5.0 cN to 30.0 cN. is more preferably 7.0 cN to 20.0 cN, and particularly preferably 9.0 cN to 15.0 cN.
- the melt tension of the branched polypropylene resin (A) is 1.0 cN or more, when the composition containing the branched polypropylene resin (A) and the foaming agent is completely melted and foamed, the tension of the composition is sufficient.
- the extruded expanded beads obtained have the advantage of being excellent in moldability
- the extruded expanded beads have the advantage of being able to provide a foam molded article having excellent resistance to breakage.
- the melt tension of the branched polypropylene resin (A) is measured using Capilograph 1D (manufactured by Toyo Seiki Seisakusho Co., Ltd., Japan). Specifically, it is as follows (1) to (5): (1) A sample resin (branched polypropylene resin) for measurement is placed in a barrel with a diameter of 9.55 mm heated to the test temperature (200 ° C.).
- a polypropylene resin having a branched structure (branched polypropylene resin) (A) can be obtained by introducing a branched structure into a linear polypropylene resin.
- the method for introducing a branched structure into the linear polypropylene resin is not particularly limited, but for example, (a1) a method of irradiating the linear polypropylene resin, and (a2) a linear polypropylene resin and a conjugated diene compound and a method of melt kneading a mixture containing a radical polymerization initiator.
- the method (a2) will be further explained.
- the following (i) to (iv) are performed in order to obtain a branched polypropylene-based resin (A): (i) a linear polypropylene-based resin, a conjugated diene compound, and radical polymerization; (ii) extruding the resulting melt-kneaded mixture through the die; (iii) extruding the extruded melt-kneaded mixture (also referred to as strands). cooling; (iv) chopping the strands simultaneously with or after cooling the strands.
- Specific examples of the method (a2) include the method described in WO2020/004429.
- branched structure can be stably introduced into a linear polypropylene-based resin, and the reproducibility of the introduction of the branched structure is high; and/or (ii) no complicated equipment is required and high productivity Since a branched polypropylene resin can be obtained, in one embodiment of the present invention, the branched polypropylene resin (A) is a branched polypropylene resin obtained by the above method (a2). is preferred.
- Conjugated diene compounds include, for example, butadiene, isoprene, 1,3-heptadiene, 2,3-dimethylbutadiene, and 2,5-dimethyl-2,4-hexadiene. These conjugated diene compounds may be used singly or in combination of two or more. Among these conjugated diene compounds, butadiene and isoprene are particularly preferred from the viewpoints of (a) being inexpensive and easy to handle, and (b) the reaction to proceed uniformly.
- the radical polymerization initiator is an organic peroxide capable of abstracting hydrogen from the polypropylene resin and/or the conjugated diene compound.
- Radical polymerization initiators suitably used in one embodiment of the present invention include organic peroxides such as ketone peroxides, peroxyketals, hydroperoxides, dialkyl peroxides, diacyl peroxides, peroxydicarbonates, and peroxyesters. oxides.
- organic peroxide one with particularly high hydrogen abstraction ability is preferable.
- organic peroxides with high hydrogen abstraction ability include 1,1-bis(t-butylperoxy)3,3,5-trimethylcyclohexane, 1,1-bis(t-butylperoxy)cyclohexane, n- Peroxyketals such as butyl 4,4-bis(t-butylperoxy)valerate and 2,2-bis(t-butylperoxy)butane; dicumyl peroxide, 2,5-dimethyl-2,5-di (t-butylperoxy)hexane, ⁇ , ⁇ '-bis(t-butylperoxy-m-isopropyl)benzene, t-butylcumyl peroxide, di-t-butylperoxide, 2,5-dimethyl-2 , 5-di(t-butylperoxy)-3-hexyne and other dialkyl peroxides; benzoyl peroxide and other
- the amount of the radical polymerization initiator used is not particularly limited, it is preferably 0.01 to 5.00 parts by weight, and 0.10 to 3.00 parts by weight, relative to 100 parts by weight of the linear polypropylene resin. is preferred, 0.10 to 2.50 parts by weight is more preferred, and 0.10 to 2.00 parts by weight is particularly preferred.
- the acid-modified polyolefin-based resin (B) can also be said to be a polyolefin-based resin into which an unsaturated carboxylic acid and/or a derivative thereof is introduced.
- the introduction of the unsaturated carboxylic acid and/or its derivative into the polyolefin resin is carried out by (b1) reacting the polyolefin resin with a radical polymerization initiator, and then reacting the obtained reaction product with the unsaturated carboxylic acid and/or and (b2) a method of thermally decomposing a polyolefin resin and then introducing an unsaturated carboxylic acid and/or a derivative thereof into the resulting reaction product. be done.
- the above method (b2) is preferred because it facilitates the introduction of unsaturated carboxylic acid and/or its derivative to the end of the main chain.
- the acid-modified polyolefin resin (B) is a resin obtained by thermally decomposing a polyolefin resin and then introducing an unsaturated carboxylic acid and/or a derivative thereof into the resulting reaction product.
- the acid-modified polyolefin resin (B) preferably does not have polyolefin branches.
- the weight average molecular weight of the acid-modified polyolefin resin (B) according to one embodiment of the present invention is not particularly limited, but is preferably 15,000 to 65,000, more preferably 16,000 to 60,000, and more preferably 17,000 to 50,000. more preferably 18,000 to 40,000.
- This configuration has the advantage of being able to provide a polypropylene-based resin foam molded article having excellent adhesion strength to polyurethane foam.
- the weight average molecular weight of the acid-modified polyolefin resin (B) is a value calculated by the GPC method.
- the acid means an unsaturated carboxylic acid and/or a derivative thereof.
- Unsaturated carboxylic acids and their derivatives include fumaric acid, fumaric anhydride, maleic acid, maleic anhydride, itaconic acid, itaconic anhydride, citraconic acid, citraconic anhydride, aconitic acid, aconitic anhydride, methyl fumarate, fumaric Ethyl acid, propyl fumarate, butyl fumarate, dimethyl fumarate, diethyl fumarate, dipropyl fumarate, dibutyl fumarate, methyl maleate, ethyl maleate, propyl maleate, butyl maleate, dimethyl maleate, diethyl maleate , dipropyl maleate, dibutyl maleate, and the like.
- the acid-modified polyolefin resin (B) is preferably a resin obtained by introducing itaconic anhydride and/or maleic anhydride into a polyolefin resin, and is a resin obtained by introducing maleic anhydride. is more preferred.
- the acid-modified polyolefin resin (B) preferably contains structural units derived from itaconic anhydride and/or structural units derived from maleic anhydride, and more preferably contains structural units derived from maleic anhydride. I can say.
- a resin obtained by introducing "Z” as an unsaturated carboxylic acid and its derivative into a polyolefin resin may be referred to as "Z-modified polyolefin-based resin”.
- the acid value is measured according to JIS K 0070, and intends the amount (mg) of potassium hydroxide required to neutralize the free acid present in 1 g of the acid-modified polyolefin resin.
- the acid value is one of the numerical values that serve as an indicator of acid denaturation.
- the acid value of the acid-modified polyolefin resin (B) is preferably 1.0 mg/KOH to 200.0 mg/KOH, more preferably 3.0 mg/KOH to 150.0 mg/KOH. More preferably 0 mg/KOH to 100.0 mg/KOH, particularly preferably 10.0 mg/KOH to 80.0 mg/KOH. This configuration has the advantage of being able to provide a foamed molded article having excellent adhesiveness to polyurethane foam.
- the base resin is (i) 70.0% by weight of the branched polypropylene resin (A). 97.5% by weight, and 2.5% by weight to 30.0% by weight of acid-modified polyolefin resin (B), and (ii) 70.0% by weight to 97% by weight of branched polypropylene resin (A). 0% by weight, and 3.0% by weight to 30.0% by weight of the acid-modified polyolefin resin (B), and (iii) 75.0% by weight to 75.0% by weight of the branched polypropylene resin (A).
- the base resin is a resin other than the branched polypropylene-based resin (A) and the acid-modified polyolefin-based resin (B) (sometimes referred to as "other resin") within a range that does not impair the effects of one embodiment of the present invention. Yes.) Or it may further contain rubber.
- resins include (a) linear polypropylene resins such as ethylene/propylene random copolymers, ethylene/propylene block copolymers, and propylene homopolymers; Ethylene resins such as density polyethylene, linear low density polyethylene, linear ultra-low density polyethylene, ethylene/vinyl acetate copolymer, ethylene/acrylic acid copolymer, and ethylene/methacrylic acid copolymer, and (c) Styrenic resins such as polystyrene, styrene/maleic anhydride copolymers, and styrene/ethylene copolymers.
- the rubber include olefin rubbers such as ethylene/propylene rubber, ethylene/butene rubber, ethylene/hexene rubber, and ethylene/octene rubber.
- the content of other resins in the base resin is, for example, preferably 60 parts by weight or less per 100 parts by weight in total of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B). , more preferably 40 parts by weight or less, more preferably 30 parts by weight or less, even more preferably 20 parts by weight or less, and particularly preferably 10 parts by weight or less.
- the lower limit of the content of other resins is not particularly limited, and may be, for example, 0 parts by weight with respect to a total of 100 parts by weight of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B). .
- the base resin contains a linear polypropylene resin as another resin, for example, a total of 100% by weight of the branched polypropylene resin (A), the acid-modified polyolefin resin (B), and the linear polypropylene resin
- the content of the branched polypropylene resin (A) is 40.0 wt% to less than 70.0 wt%
- the content of the acid-modified polyolefin resin (B) is 2.5 wt% to 30.0 wt%.
- the content of the linear polypropylene-based resin is preferably more than 0% by weight and less than 30.0% by weight.
- the base resin may contain a cell nucleating agent for the purpose of controlling the number and shape of cells in the extruded foamed particles to be obtained.
- Bubble nucleating agents can include sodium bicarbonate-citric acid mixtures, monosodium citrate, talc, calcium carbonate, and the like. One of these cell nucleating agents may be used alone, or two or more thereof may be used in combination.
- the content of the cell nucleating agent in the base resin is not particularly limited.
- the content of the cell nucleating agent is, for example, 0.01 to 5.00 parts by weight with respect to a total of 100 parts by weight of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B). is preferably 0.01 to 3.50 parts by weight, more preferably 0.01 to 1.00 parts by weight, and 0.01 to 0.50 parts by weight Parts by weight are particularly preferred.
- the base resin may or may not contain a colorant.
- a colorant it is possible to obtain extruded expanded particles having a natural color (for example, a color derived from the base resin).
- the base resin contains a colorant, it is possible to obtain extruded foam particles of a desired color (that is, a color derived from the colorant).
- the coloring agent include perylene-based organic pigments, azo-based organic pigments, quinacridone-based organic pigments, phthalocyanine-based organic pigments, threne-based organic pigments, dioxazine-based organic pigments, isoindoline-based organic pigments, and carbon black. .
- colorants may be used singly or in combination of two or more.
- a foam molded product is used for a member that is visible to people, it may be required that the foam molded product is black from the viewpoint of designability. Therefore, carbon black is preferable as the coloring agent.
- the amount of the coloring agent used in other words, the content of the coloring agent in the base resin, is not particularly limited.
- the base resin may contain other components such as (a) an antioxidant, a metal deactivator, a phosphorus-based processing stabilizer, an ultraviolet absorber, an ultraviolet stabilizer, a fluorescent brightener, a metallic soap, and an antacid. and/or (b) additives such as crosslinkers, chain transfer agents, lubricants, plasticizers, fillers, reinforcements, flame retardants, and antistatic agents. good.
- additives such as crosslinkers, chain transfer agents, lubricants, plasticizers, fillers, reinforcements, flame retardants, and antistatic agents. good.
- These other components may be used individually by 1 type, and may be used in combination of 2 or more type.
- the base resin contained in the extruded expanded beads or the base resin contained in the expanded molded article obtained from the extruded expanded beads that is, the base resin that substantially constitutes the extruded expanded beads or the expanded molded article , branched polypropylene-based resin (A), acid-modified polyolefin-based resin (B), other resins or rubbers, cell nucleating agents, coloring agents and other components, and the types and amounts of the extruded foamed particles or the foamed molded product It does not change substantially even by the operation of melting under reduced pressure and returning to the resin mass.
- the process of melting the extruded foamed particles or the foamed molded article obtained from the extruded foamed particles under reduced pressure to obtain a resin lump may be referred to as "returning the resin”.
- a resin lump may be called “returned resin.”
- "branched polypropylene resin (A), acid-modified polyolefin resin (B), other resins or rubbers, cell nucleating agents, colorants and other components” was changed to "branched polypropylene resin (A) and acid It may be referred to as "modified polyolefin resin (B), etc.”
- the types and amounts of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B) contained in the returned resin are and the type and amount of the acid-modified polyolefin resin (B).
- the types and amounts of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B) contained in the returned resin can be confirmed by
- a specific method for returning the resin is not particularly limited, but includes, for example, a method in which the following (c1) to (c5) are performed in order: (c1) a dryer adjusted to the melting point of the extruded foamed particles or foamed product +10°C; (c2) Then, using a vacuum pump over 5 to 10 minutes, the pressure in the dryer is reduced from -0.05 MPa (cage pressure) to -0.10 MPa. (c3) then leave the extruded foamed particles or foamed molded body in the dryer for 30 minutes to prepare a resin mass (returned resin); (c4) then in the dryer (c5) Then, the resin mass is taken out from the dryer and cooled to room temperature.
- the expanded polypropylene resin particles obtained by the depressurized expansion method have two peaks (melting peaks) in the DSC curve obtained by the DSC method.
- the extruded polypropylene resin particles obtained by the extrusion foaming method have one peak (melting peak) in the DSC curve. Therefore, the present extruded expanded beads can also have one peak (melting peak) in the DSC curve.
- the temperature of the extruded foamed polypropylene resin particles is raised from 40° C. to 220° C. at a rate of 10° C./min to obtain a DSC curve of the extruded foamed particles.
- the DSC curve of the extruded expanded particles is different from the DSC curve obtained during the second temperature increase used when measuring the melting point of the branched polypropylene resin (A), and is the DSC curve obtained during the first temperature increase. be. Therefore, it can be said that the polypropylene-based resin extruded expanded particles can have one peak in the DSC curve obtained during the first temperature increase.
- the extruded expanded particles preferably have a lower open cell ratio.
- the extruded expanded beads preferably have an open cell rate of 10.0% or less, more preferably 8.0% or less, more preferably 6.0% or less, and 5.0% or less. is more preferably 4.0% or less, more preferably 3.0% or less, and particularly preferably 2.0% or less.
- the lower limit of the open cell content of the extruded polypropylene-based resin particles is not particularly limited, and is, for example, 0.0% or more.
- the extruded expanded beads when the extruded expanded beads are molded, the cells hardly break and shrink, so there is an advantage that the extruded expanded beads are excellent in moldability, and (b) the extruded expanded beads are
- the foamed molded article obtained by using it has the advantage that characteristics such as shape arbitrariness, cushioning properties, light weight, compressive strength and heat insulating properties are more exhibited.
- the open cell ratio of the present extruded expanded beads is within the range described above, the extruded expanded beads have the advantage of being able to provide a foamed article having excellent low-pressure moldability and excellent compressive strength.
- the open cell ratio of the extruded polypropylene resin expanded particles is described in ASTM D2856-87 Procedure C (PROCEDURE C) using an air comparison type hydrometer [manufactured by Tokyo Science Co., Ltd., model 1000].
- the expansion ratio of the present extruded expanded beads is not particularly limited, but is preferably 2 times or more, more preferably 3 times or more, and even more preferably 5 times or more.
- the expansion ratio of the extruded expanded beads is preferably 30 times or less, more preferably 28 times or less, and even more preferably 25 times or less. According to the above configuration, there is an advantage that physical properties such as compressive strength of the foamed molded product obtained by molding are excellent, and weight reduction is possible.
- a method for measuring the expansion ratio of the extruded expanded particles will be described in detail in the examples below.
- the extruded foam particles have the advantage of a wide molding width (eg greater than 0). In the present specification, it is intended that the larger the molding width of the extruded expanded beads, the better the moldability of the extruded expanded beads.
- the term "forming width of extruded foam particles” refers to the vapor pressure (gauge pressure ) is intended to have a width of: (x1) the extruded foam particles are sufficiently fused together, (x2) the gaps between the extruded foam particles are sufficiently filled, (x3) the surface is beautiful, and (x4) A foam-molded product whose surface is not melted, (x5) does not shrink, and the shape of the mold (mold) used for the in-mold foam molding is transferred.
- the molding width of the extruded expanded particles is not particularly limited. It is preferable that the molding width of the extruded expanded particles is as wide as possible.
- the molding width of the extruded expanded beads is more preferably 0.02 MPa or more, more preferably 0.03 MPa or more, still more preferably 0.04 MPa or more, and preferably 0.05 MPa or more. More preferably, it is particularly preferably 0.06 MPa or more.
- a method for producing the present extruded foamed particles is not particularly limited, and a known extrusion foaming method can be employed.
- One aspect of the method for producing the present extruded expanded particles includes, for example, the following aspect: (a) branched polypropylene resin (A), (b) acid-modified polyolefin resin (B), (c) optionally (c-1) other resins or rubbers, (c-2) cell nucleating agents, (c-3) colorants, and/or (c-4) other ingredients, and (d) foaming A first step of melt-kneading the agent in a manufacturing apparatus, and a second step of discharging the melt-kneaded product obtained in the first step through a die into a region having a pressure lower than the internal pressure of the manufacturing apparatus.
- a method for producing extruded polypropylene resin foam particles comprising:
- a particularly preferred aspect of the present method for producing extruded expanded beads includes, for example, the following aspects: a first step of melt-kneading a resin composition containing a branched polypropylene-based resin (A) and an acid-modified polyolefin-based resin (B) having a weight average molecular weight of 10,000 to 100,000 and a foaming agent in a manufacturing apparatus; A second step of discharging the melt-kneaded product obtained in the first step through a die to a region having a lower pressure than the internal pressure of the manufacturing apparatus, and the resin composition is included in the resin composition.
- a first step of melt-kneading a resin composition containing a branched polypropylene-based resin (A) and an acid-modified polyolefin-based resin (B) having a weight average molecular weight of 10,000 to 100,000 and a foaming agent in a manufacturing apparatus A second step of discharging the melt-kneaded product obtained in the first step through a die
- the polypropylene resin (A) having the branched structure is 70.0% by weight to A method for producing extruded expanded polypropylene resin particles containing 97.5% by weight and 2.5% to 30.0% by weight of the acid-modified polyolefin resin (B).
- the first step will be specifically described.
- a branched polypropylene resin (A) (b) an acid-modified polyolefin resin (B), and (c) optionally (c-1 ) other resins or rubbers, (c-2) cell nucleating agent, (c-3) colorant, and/or (c-4) other components are melted to form the resin composition
- the melt-kneading step can also be said to be a step of preparing a melt-kneaded product containing the resin composition and the foaming agent.
- the foaming agent used in the production method is not particularly limited, but preferably contains at least carbon dioxide gas. By using carbon dioxide gas, the production method has the advantages of low production cost and low environmental load.
- blowing agents that can be used as blowing agents include (a) physical blowing agents (for example, (a-1) aliphatic hydrocarbons such as propane, normal butane, isobutane, normal pentane, isopentane, and hexane; ( a-2) alicyclic hydrocarbons such as cyclopentane and cyclobutane; (a-3) ethers such as dimethyl ether, diethyl ether and methyl ethyl ether; (a-4) alcohols such as methanol and ethanol; -5) inorganic gases such as air and nitrogen; and (a-6) water, etc.), and (b) a chemical system containing a pyrolytic blowing agent such as sodium bicarbonate, azodicarbonamide, dinitrosopentamethylenetetramine, etc. foaming agent, and the like.
- a pyrolytic blowing agent such as sodium bicarbonate, azodicarbonamide, dinitrosopentamethylenete
- the foaming agent is one or more selected from the group consisting of carbon dioxide gas, butane (for example, normal butane and isobutane) and nitrogen. It is preferable to contain, and it is preferable to consist only of one or more selected from the group.
- the amount of the foaming agent used is not particularly limited, and may be appropriately adjusted according to the type of the foaming agent and/or the target expansion ratio of the extruded polypropylene-based resin expanded particles.
- the amount of the foaming agent used is 0.5 parts by weight to 7.0 parts by weight with respect to 100.0 parts by weight of the total weight of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- stabilizers e.g., antioxidants, metal deactivators, phosphorus-based processing stabilizers, UV absorbers, UV stabilizers, optical brighteners, metal soaps, and antacids
- adsorbents, etc. and additives (e.g. cell nucleating agents, inorganic colorants, organic colorants, crosslinkers, chain transfer agents, lubricants, plasticizers, fillers, reinforcements, pigments, dyes, flame retardants, hydrous materials and antistatic agents) may also be used.
- Additives such as colorants and/or stabilizers may be blended (used) as a masterbatch.
- a masterbatch of additives such as colorants and/or stabilizers is obtained by mixing additives such as colorants and/or stabilizers with an arbitrary resin (for example, polypropylene resin) in an arbitrary ratio. obtain.
- the concentration of additives such as colorants and / or stabilizers in the masterbatch is not particularly limited, for example, 5 to 50% by weight of additives such as colorants and / or stabilizers in 100% by weight of the masterbatch. It may be a masterbatch.
- the resin composition and the foaming agent may be mixed before being supplied to the manufacturing equipment, or may be mixed within the manufacturing equipment.
- the resin composition may be supplied to the manufacturing apparatus, or the resin composition may be prepared (completed) within the manufacturing apparatus.
- the method and order of mixing the resin composition and blowing agent, and optionally other ingredients, or (ii) the resin composition and blowing agent, and optionally The method and order of supplying the other ingredients to the manufacturing equipment are not particularly limited.
- the melt-kneaded product obtained in the first step may be cooled before extruding it into the low-pressure region.
- the second step is a step of extruding the melt-kneaded product obtained in the first step through a die into a region having a lower pressure than the internal pressure of the manufacturing apparatus, and shredding the extruded melt-kneaded product.
- the second step provides extruded foam particles. Therefore, the second step can also be said to be a granulation step of granulating the extruded polypropylene-based resin expanded particles.
- the region in which the melt-kneaded product obtained in the first step is extruded is not particularly limited as long as the pressure is lower than the internal pressure of the manufacturing apparatus.
- the melt-kneaded product obtained in the first step may be extruded into the gas phase or the liquid phase.
- the melt-kneaded material during foaming may be shredded, or the melt-kneaded material that has finished foaming may be shredded. If the melt kneaded material is shredded during foaming, the shredded melt kneaded material may complete foaming in the region beyond which it was extruded.
- the second step can be broadly divided into two methods, a cold cut method and a die face cut method, depending on the region where the melt-kneaded product obtained in the first step is extruded and the method of shredding the extruded melt-kneaded product. can be separated.
- the cold cut method include a method of foaming a melt-kneaded material containing a foaming agent extruded from a die, passing it through a water tank to cool it, taking the strand-shaped foam, and then shredding it (strand cut method). be done.
- the die face cut method is a method in which the molten kneaded material extruded from the die hole is cut by a rotating cutter while being in contact with the surface of the die or ensuring a slight gap.
- the die face cutting method can be further divided into the following three methods according to the difference in cooling method. That is, they are an under water cut (hereinafter also referred to as UWC) method, a water ring cut (hereinafter sometimes referred to as WRC) method, and a hot cut (hereinafter sometimes referred to as HC) method.
- UWC under water cut
- WRC water ring cut
- HC hot cut
- a cooling drum in which cooling water flows along the inner peripheral surface of the cooling drum connected to the die is arranged downstream from the die, and the melted and kneaded material cut by the cutter foams in the air. It is a method of cooling in the cooling water while or after foaming.
- the HC method is a method in which a melt-kneaded material is cut in air with a cutter, and the cut melt-kneaded material is cooled in air while or after foaming.
- the HC method also includes a mist cut method further including a step of spraying mixed mist of water and air.
- polypropylene resin foam molded product The polypropylene-based resin foam molded article according to one embodiment of the present invention is described in [2. Extruded polypropylene resin expanded particles] or the extruded polypropylene resin particles described in [3. Method for producing extruded expanded polypropylene resin particles].
- the "polypropylene-based resin foam-molded article according to one embodiment of the present invention” may be referred to as "the present foam-molded article”.
- the present foamed molded article has the above-described structure, it has the advantage of being excellent in adhesive strength with polyurethane foam.
- the present foamed molded product can provide a laminated foam having excellent adhesive strength between the foamed molded product and the polyurethane foam without requiring a separate member such as an insert material.
- the present foamed molded article can be particularly suitably used as a raw material (foamed molded article) for a laminated foam of a foamed molded article and a polyurethane foam.
- the present foamed molded article can be particularly suitably used as a foamed molded article for integral molding with a polyurethane foam.
- the foamed molded article may be for a laminated foam with polyurethane foam.
- the method for producing the present foamed molded article that is, the method for forming the present extruded expanded particles is not particularly limited, and for example, a known in-mold foaming method using a mold can be employed.
- the extruded expanded particles are pressure-treated with an inorganic gas to impregnate the extruded expanded particles with the inorganic gas to form the extruded expanded particles. After applying a predetermined internal pressure, the extruded foamed particles are filled in a mold, and the extruded foamed particles are heat-fused with steam or the like (for example, Japanese Patent Publication No.
- the density of the present foam molded article is not particularly limited.
- the density of the present foam molded product is, for example, preferably 20 g/L to 300 g/L, more preferably 30 g/L to 200 g/L, and even more preferably 40 g/L to 160 g/L. , 50 g/L to 150 g/L. According to this configuration, there is an advantage that the foam-molded article easily satisfies physical properties (compressive strength, etc.) required as an automobile member.
- the density of a polypropylene-based resin foam molded product is a value obtained by measuring according to the following method. First, the weight W (g) of the foam molded body is measured. Next, the length, width and thickness of the foam-molded body are measured with vernier calipers to calculate the volume V (cm 3 ) of the foam-molded body.
- the present foam molded article has excellent adhesive strength with polyurethane foam.
- the adhesive strength between the present foam molded article and the polyurethane foam is preferably as high as possible. is more preferable, 150 kPa or more is more preferable, and 200 kPa or more is particularly preferable.
- the adhesive strength of a foamed molded product to a polyurethane foam is measured by a peel test performed according to JIS K 6850 (1999). be the value obtained.
- the adhesive strength is a value obtained by converting the unit of the value obtained by dividing the maximum stress (N) obtained in the peel test by the area of the adhesive surface (m 2 ) into kPa.
- the laminated foam according to one embodiment of the present invention has the above-described structure, it has the advantage of being excellent in adhesive strength between the polypropylene-based resin foam molded product and the polyurethane foam.
- the method for producing a laminated foam according to an embodiment of the present invention is not particularly limited.
- (d2) a method of bonding and laminating a polypropylene-based resin foam molded article and a polyurethane foam by pouring a liquid into the gap and foaming the liquid; A method of applying the composition, and adhering and laminating another polypropylene-based resin foam molding or the like on the surface to which the composition has been applied, and the like.
- a laminated foamed body is obtained in which the polypropylene resin foamed product and the polyurethane foam are laminated in the order of polypropylene resin foamed product - polyurethane foam - polypropylene resin foamed product.
- each of the extruded foamed particles, the foamed molded product and the laminated foamed product according to one embodiment of the present invention can be used for various purposes such as automotive interior parts, core materials for automotive bumpers, heat insulating materials and cushioning packaging materials.
- the extruded foamed particles according to one embodiment of the present invention can provide a foamed molded article having excellent adhesion to a polyurethane foam, and can provide a laminated foamed article containing a polypropylene resin foamed molded article and a polyurethane foam with high productivity. Therefore, each of the extruded foamed particles, the foamed molded product and the laminated foamed product according to one embodiment of the present invention can be suitably used for automobile interior parts such as vehicle seats and headrests.
- An embodiment of the present invention may have the following configuration.
- Extruded expanded polypropylene resin particles wherein the base resin of the extruded expanded polypropylene resin particles is a total of 100% by weight of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B). , containing 70% to 97% by weight of a polypropylene resin (A) having a branched structure and 3% to 30% by weight of an acid-modified polyolefin resin (B) having a weight average molecular weight of 10,000 to 100,000. Polypropylene-based resin extruded expanded particles.
- [X4] A polypropylene-based resin foam molded article obtained by molding the extruded polypropylene-based resin expanded particles according to any one of [X1] to [X3].
- a laminated foam obtained by laminating the polypropylene-based resin foam molding described in [X5] and [X4] and a polyurethane foam.
- An embodiment of the present invention may have the following configuration.
- Extruded expanded polypropylene resin particles wherein the base resin of the extruded expanded polypropylene resin particles is a total of 100% by weight of the branched polypropylene resin (A) and the acid-modified polyolefin resin (B).
- the polypropylene resin (A) having a branched structure is 70.0% to 97.5% by weight
- the acid-modified polyolefin resin (B) having a weight average molecular weight of 10000 to 100000 is 2.5% by weight. % to 30.0% by weight of extruded polypropylene resin expanded particles.
- the main chain of the branched polypropylene resin (A) is at least one selected from the group consisting of propylene homopolymers, polypropylene block copolymers and polypropylene random copolymers, [Y1 ] to [Y10].
- the resin composition is the resin composition
- the polypropylene resin (A) having the branched structure is 70.0
- Test method The test methods used for measuring and evaluating various physical properties in Examples and Comparative Examples are as follows.
- the melting point of the branched polypropylene-based resin was obtained by measuring the branched polypropylene-based resin as a sample by a differential scanning calorimeter method. As a differential scanning calorimeter, DSC6200 type manufactured by Seiko Instruments Inc. was used. The specific operating procedure was as follows: (1) The temperature of 5 to 6 mg of a sample (branched polypropylene resin) was raised from 40°C to 220°C at a rate of 10°C/min.
- the temperature of the melted sample was lowered from 220°C to 40°C at a cooling rate of 10°C/min to crystallize the sample; (3) Then Furthermore, the temperature of the crystallized sample was raised from 40°C to 220°C at a heating rate of 10°C/min.
- the temperature of the peak (melting peak) of the DSC curve of the sample (branched polypropylene resin) obtained during the second heating is taken as the melting point of the sample (branched polypropylene resin). bottom.
- MFR of branched polypropylene resin The MFR of the branched polypropylene resin is determined by using the branched polypropylene resin as a sample, conforming to the provisions of B method described in ISO 1133 (1997), using a melt indexer S-01 (manufactured by Toyo Seiki Seisakusho), temperature It was obtained by measuring under conditions of 230° C. and a load of 2.16 kg.
- melt tension of branched polypropylene resin The melt tension of the branched polypropylene resin was measured using Capilograph 1D (manufactured by Toyo Seiki Seisakusho, Ltd., Japan). Specifically, (1) to (5) were as follows: (1) A sample resin (branched polypropylene resin) for measurement was placed in a barrel with a diameter of 9.55 mm heated to the test temperature (200 ° C.).
- the sample resin was then heated for 10 minutes in a barrel heated to the test temperature (200° C.); (3) then from a capillary die (1.0 mm bore, 10 mm length) At a constant piston descent speed (10 mm/min), the sample resin was drawn out in the form of a string, passed through a tension detecting pulley located 350 mm below the capillary die, and then wound up. (4) After the take-up of the string was stabilized, the winding speed of the string was increased from 1.0 m/min to a constant speed of 200 m/min in 4 minutes. (5) The load applied to the pulley with a load cell when the string broke was measured as the melt tension.
- Weight average molecular weight of acid-modified polyolefin resin The weight average molecular weights of the acid-modified polyolefin resin (B) and the acid-modified polyethylene resin and the acid-modified polypropylene resin, which are other resins, were calculated by the GPC method.
- the expansion ratio of the extruded polypropylene resin expanded beads was calculated by the following method: (1) the weight w (g) of the extruded expanded beads was measured; , immersed in ethanol contained in a graduated cylinder, and the volume v (cm 3 ) of the extruded foamed particles was measured based on the rise in the liquid level of the graduated cylinder; (3) weight w (g) was converted to volume v ( cm 3 ) to calculate the density ⁇ 1 (g/cm 3 ) of the extruded expanded beads; (4) the density ⁇ 2 (g/cm 3 ) of the base resin of the extruded expanded beads was divided into It was divided by ⁇ 1 ( ⁇ 2 / ⁇ 1 ), and the resulting value was taken as the foaming ratio (times). As the density ⁇ 2 of the base resin, the density 0.9 g/cm 3 of a general polypropylene-based resin was adopted.
- the open cell ratio of the extruded foamed particles is obtained by measuring using an air comparison type hydrometer [manufactured by Tokyo Science Co., Ltd., model 1000] according to the method described in ASTM D2856-87 Procedure C (PROCEDURE C). rice field.
- a block-shaped mold (having a molding space of 381 mm long, 320 mm wide, and variable thickness) was set to a state in which the thickness of the molding space was 78 mm (cracking rate of 30%).
- the molding space of the mold was filled with extruded polypropylene-based resin foam particles. After that, the mold was moved so that the thickness of the molding space in the mold was 60 mm, and the molding space was compressed. Subsequently, the air in the mold was expelled with water vapor of 0.10 MPa (gauge pressure).
- the mixture was heated for 10 seconds using water vapor having a vapor pressure of 0.20 to 0.36 MPa (gauge pressure), followed by in-mold foam molding to obtain a polypropylene-based resin foam molding.
- in-mold foam molding was performed while changing the steam pressure by 0.02 MPa.
- the range of steam pressure during in-mold foam molding that can obtain a polypropylene resin-based in-mold foam molding that satisfies (x1) to (x5) below is determined, and the obtained steam pressure range is P1 to P2: (x1) the extruded polypropylene resin foam particles are sufficiently fused (fusion rate), (x2) the gaps between the extruded polypropylene resin foam particles are sufficiently filled, and (x3) the surface is beautiful, (x4) the surface is not melted, and (x5) the shape of the mold (mold) used for in-mold foam molding is transferred without shrinkage. Foam molding.
- the (x1) fusion rate, the (x2) evaluation of the interparticle gap, the (x3) evaluation of the surface beauty, the (x4) evaluation of the surface melt state, and the (x5) ) was performed as follows.
- the evaluation of (x1) to (x5) the polypropylene resin foam molded article obtained by the above method was allowed to stand in a drying chamber at 75°C for 16 hours or more to dry, and then dried at 23°C for 24 hours. It was carried out using the polypropylene-based resin foam-molded article after standing as described above.
- the polypropylene-based resin foam-molded article immediately after molding was evaluated without drying and allowing the obtained polypropylene-based resin foam-molded article to stand still. Specifically, in the polypropylene-based resin foam-molded article immediately after molding, (a) when the foam-molded article sticks to the mold and cannot be released from the mold, or (b) the foam-molded article can be released from the mold. However, if a part of the surface of the foam-molded article, such as a steam slit portion on the surface of the foam-molded article, remained on the mold side, it was judged as unacceptable, and otherwise it was judged as acceptable.
- the vertical, horizontal and thickness dimensions of the foamed polypropylene resin product were measured. Using the obtained results, the shrinkage rate (%) was evaluated by the following formula for each of the vertical, horizontal and thickness dimensions of the foamed polypropylene resin product: ⁇ (dimension of molding space in mold) ⁇ (dimension of compact) ⁇ 100/dimension of molding space in mold.
- the dimensions of the molding space in the mold were 381 mm long, 320 mm wide and 60 mm thick.
- the density of the foam molded article was measured according to the following method. First, the weight W (g) of the foam molded article was measured. Next, the length, width and thickness of the foam-molded product were measured with vernier calipers to calculate the volume V (cm 3 ) of the foam-molded product. In addition, in the case of a foam molded article having a complicated shape, the volume V (cm 3 ) may be obtained by the submersion method.
- Adhesion strength between foam molded product and polyurethane foam A peel test was carried out according to JIS K 6850 (1999) to measure the adhesive strength between the foam molded article and the polyurethane foam. The value obtained by dividing the maximum stress (N) obtained in the peel test by the area (m 2 ) of the adhesive surface and converting the unit of the value obtained into kPa was taken as the adhesive strength.
- Branched polypropylene resin (A)> A-1: Branched polypropylene resin produced by Production Example 1 below A-2: Branched polypropylene resin produced by Production Example 2 below ⁇ Linear polypropylene resin (X)> ⁇ F-724NPC (manufactured by Prime Polymer Co., Ltd., polypropylene-based random copolymer, melting point: 150 ° C., MFR: 7 g / 10 minutes) ⁇ Acid-modified polyolefin resin (B)> ⁇ B-1: Umex 1010 (manufactured by Sanyo Chemical Industries, maleic anhydride-modified polypropylene resin, weight average molecular weight 30000, acid value 52 mg / KOH) ⁇ B-2: Hi-wax NP0555A (manufactured by Mitsui Chemicals, maleic anhydride-modified polypropylene resin, weight average molecular weight 18000, acid value 61 mg / KOH) ⁇ Other resin
- the prepared resin mixture was further melt-kneaded in the twin-screw extruder at a cylinder temperature of 200°C and a screw rotation speed of 230 rpm to obtain a branched polypropylene resin.
- the resulting branched polypropylene-based resin was extruded from a die in the form of strands at an extruding rate of 70 kg/h.
- the discharged branched polypropylene resin (strand) was (a) cooled with water, and then (b) chopped into pellets (cylindrical).
- the melting point, MFR and melt tension of the resulting branched polypropylene resin (A-1) were measured by the methods described above.
- the branched polypropylene resin (A-1) had a melting point of 147° C., an MFR of 2 g/10 minutes, and a melt tension of 10.8 cN.
- Example 1 Production of extruded foamed particles
- First step As an apparatus for producing extruded expanded particles, an apparatus in which a twin-screw extruder having a shaft diameter of ⁇ 15 mm, a melt cooler, a diverter valve and a die were connected in series was used. Resins and additives (cell nucleating agents) listed in Table 1 were blended in the amounts listed in Table 1 to prepare resin compositions. Then, the resin composition was supplied to a twin-screw extruder and melt-kneaded at a cylinder temperature of 200°C. Further, the amount of blowing agent (carbon dioxide gas) shown in Table 1 was supplied from the press-fitting section provided in the extruder using a metering pump, and the obtained melt-kneaded product was further melt-kneaded.
- blowing agent carbon dioxide gas
- melt-kneaded product was cooled to 150° C. to 160° C. by passing through a melt cooler connected to the tip of the twin-screw extruder. After that, the melt-kneaded product was extruded from a die attached to the tip of the melt cooler into a region having a pressure lower than the internal pressure of the manufacturing apparatus (under atmospheric pressure and in the air) to foam. The melt-kneaded material immediately after passing through the die was finely chopped by a rotating cutter attached to the tip of the die to obtain extruded expanded particles. As mentioned above, the hot cut method was employed in the second step.
- the obtained extruded foamed particles were measured for open cell ratio and bulk density by the methods described above, and the moldable width was evaluated. Those results are shown in Table 1. Further, when the DSC curve was obtained by the method described above for the obtained extruded foamed particles, the DSC curve had one peak.
- a polypropylene-based resin foam-molded article was produced by the following method. First, a block-shaped mold (having a molding space of 381 mm long, 320 mm wide, and variable thickness) was set to a state in which the thickness of the molding space was 78 mm (cracking rate of 30%). Next, the molding space of the mold was filled with extruded polypropylene-based resin foam particles. After that, the mold was moved so that the thickness of the molding space in the mold was 60 mm, and the molding space was compressed. Subsequently, the air in the mold was expelled with water vapor of 0.10 MPa (gauge pressure).
- the fusion rate was evaluated by the method described in the section (Evaluation of fusion rate) for the foamed molded article obtained using steam with a vapor pressure of 0.26 MPa. Table 1 shows the results.
- test piece A A foamed molded article obtained using steam having a vapor pressure of 0.26 MPa was used for the production of the laminated foam. Two specimens with a thickness of 5 mm (single-sided skin) were cut out from the foam molded article.
- the skin surface of one test piece (test piece A) was coated with a raw material liquid for forming polyurethane foam (manufactured by Henkel Japan, trade name: Loctite Green Foam).
- test piece B the skin surface of the other test piece (referred to as test piece B) is applied onto the skin surface of test piece A so that the resulting polyurethane foam has a thickness of 5 mm and a density of approximately 65 g/L.
- the raw material liquid for forming polyurethane foam was foamed and solidified in an open system at 23° C. for 48 hours. After solidification, excess polyurethane foam was removed to obtain a laminated foam with adhesive surface dimensions of 25 mm ⁇ 12.5 mm. The resulting laminated foam was measured for adhesive strength between the foam molded article and the polyurethane foam by the method described above. Table 1 shows the results.
- Examples 2-8) Extruded expanded polypropylene resin particles were produced in the same manner as in Example 1, except that the resins and additives shown in Table 1 were blended in the amounts shown in Table 1.
- a polypropylene-based resin foam-molded product was produced in the same manner as in Example 1, except that the heating steam pressure was changed to the steam pressure described in the column of "Moldable Width" in Table 1.
- a laminated foam was produced in the same manner as in Example 1 using a foamed molded article obtained using steam having a vapor pressure of 0.26 MPa.
- Various evaluations were made on the obtained extruded foamed particles, foamed molded article and laminated foamed article.
- the fusion rate was evaluated by the method described in the section (Evaluation of fusion rate) for the foamed molded article obtained using steam having a vapor pressure of 0.26 MPa. These results are shown in Table 1. Further, when the DSC curve was obtained by the method described above for the extruded foamed particles obtained in Examples 2 to 8, the DSC curve had one peak.
- Extruded expanded polypropylene resin particles were obtained in the same manner as in Example 1, except that the resins and additives shown in Table 2 were blended in the amounts shown in Table 2. Also, a polypropylene-based resin foam-molded article was produced in the same manner as in Example 1, except that the heating steam pressure was set to the steam pressure described in the column of "Moldable Width" in Table 2. Furthermore, a laminated foam was produced in the same manner as in Example 1 using a foamed molded article obtained using steam having a vapor pressure of 0.26 MPa. Various evaluations were made on the obtained extruded foamed particles, foamed molded article and laminated foamed article.
- Polypropylene-based resin foamed particles were produced by the depressurized foaming method (Reference Examples 1 to 4).
- the resulting melt-kneaded product was extruded into strands through a die plate having a plurality of holes with a diameter of 2.2 mm.
- the strand was cut into cylindrical pellets having a predetermined grain weight by a pelletizer. 200 parts by weight of water per 100 parts by weight of the obtained pellets, tricalcium phosphate (manufactured by Taihei Kagaku Sangyo Co., Ltd.) as a dispersant in the amount shown in Table 3, and sodium alkylsulfonate (Latemul PS, Kao Corporation) as a surfactant.
- the dispersion stability in the manufacturing process (unpressurized foaming process) of expanded polypropylene resin particles was evaluated by the method described below. Further, with respect to the expanded polypropylene resin beads obtained, (a) the expansion ratio and (b) the amount of dispersant adhering to the surface of the expanded polypropylene resin beads (amount of adhered dispersant) were determined by the methods described below. It was measured. The bulk density of the obtained foamed polypropylene-based resin particles was evaluated by the method described in the section [Bulk density].
- the method of measuring the expansion ratio of the expanded beads was as follows (1) to (4): (1) the weight w (g) of the expanded beads was measured; The foamed beads used were submerged in ethanol contained in a graduated cylinder, and the volume v (cm 3 ) of the foamed beads was measured based on the rise in the liquid level of the graduated cylinder; (3) Weight w (g ) was divided by the volume v (cm 3 ) to calculate the density ⁇ 1 (g/cm 3 ) of the expanded beads; (4) Density ⁇ 2 (g/cm 3 ) of the resin particles used to produce the expanded beads was divided by the density ⁇ 1 of the foamed beads ( ⁇ 2 / ⁇ 1 ) and the value obtained was taken as the expansion ratio (times) of the foamed beads.
- the dispersant remaining on the surfaces of the obtained foamed polypropylene resin particles is referred to as an adhering dispersant.
- the amount of the adhering dispersant was calculated by ash measurement.
- the amount of the adhered dispersant was obtained by subtracting the ash content of the pellets (resin particles) used for foaming from the ash content of the foamed particles.
- the ash content of each foamed particle or resin particle was measured by the following method. A sample (expanded particles or resin particles) was placed in the crucible whose weight W0 (g) was measured, and its weight W1 (g) was measured. Then, the crucible containing the sample was burned at 750° C. for 1 hour and then left standing in a desiccator for 1 hour. Next, the weight W2 (g) of the crucible was measured, and the ash content was calculated by the following formula.
- Ash content (ppm) (W2-W0) x 1000000/(W1-W0).
- a polypropylene resin foam molded article was produced by the following method. First, a block-shaped mold (having a molding space of 381 mm long, 320 mm wide, and variable thickness) was set to a state in which the thickness of the molding space was 78 mm (cracking rate of 30%). Next, the molding space of the mold was filled with expanded polypropylene resin particles. After that, the mold was moved so that the thickness of the molding space in the mold was 60 mm, and the molding space was compressed. Subsequently, the air in the mold was expelled with water vapor of 0.10 MPa (gauge pressure). After that, by heating and molding with steam having a vapor pressure of 0.26 MPa for 10 seconds, a polypropylene-based resin foam-molded article was obtained.
- a block-shaped mold having a molding space of 381 mm long, 320 mm wide, and variable thickness
- the fusion rate of the obtained polypropylene-based resin foam molded article was evaluated by the method described in the section (Evaluation of fusion rate).
- the laminated foam containing a polypropylene-type resin foaming molding and a polyurethane foam can be provided with sufficient productivity. Therefore, one embodiment of the present invention can be particularly suitably used in the field of automobile interior parts such as vehicle seats and headrests.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
Abstract
ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを積層してなる積層発泡体を生産性よく提供し得る、ポリプロピレン系樹脂押出発泡粒子を提供することを課題とする。ポリプロピレン系樹脂押出発泡粒子の基材樹脂が、分岐状ポリプロピレン系樹脂(A)と、特定の重量平均分子量を有する酸変性ポリオレフィン系樹脂(B)とを、それぞれ特定量含む、ポリプロピレン系樹脂押出発泡粒子とする。
Description
本発明はポリプロピレン系樹脂押出発泡粒子、ポリプロピレン系樹脂発泡成形体および積層発泡体に関する。
ポリプロピレン系樹脂発泡粒子を用いて得られるポリプロピレン系樹脂発泡成形体は、形状の任意性、緩衝性、軽量性および断熱性等に優れるという特徴を有する。そのため、ポリプロピレン系樹脂発泡成形体は、主に自動車内装部材および自動車バンパー用芯材の他、断熱材および緩衝包装材等の様々な用途に用いられている。例えば、自動車のシートでは、クッション性の観点から、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを積層してなる積層発泡体が用いられている。
例えば、特許文献1には、ポリエチレン発泡体を成形型内に収容するとともにこの成形型内にポリウレタンの未発泡原料を注入してポリウレタン発泡体中の所定の部位にポリエチレン発泡体が埋設された異硬度パッドを成形する異硬度パッドのインサート成形法が開示されている。特許文献1の当該成形法は、前記ポリエチレン発泡体の表面を成形型内に収容するに先立って易接着化処理することを特徴とする。
また、特許文献2には、複数の発泡性樹脂粒子が一体的に成形されてなる成形体の表面に、該成形体とは異なる素材からなる異素材層を形成した積層体の製造方法が開示されている。特許文献2の当該積層体の製造方法は、異素材層を形成するに際し、成形体の表面における異素材層との接触領域を、成形体の軟化温度の80%以上の温度で加熱することを特徴とする。
しかしながら、上述のような従来技術は、生産性の観点において、さらなる改善の余地があった。
本発明の一実施形態は、前記問題点に鑑みなされたものであり、その目的は、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供し得る、ポリプロピレン系樹脂押出発泡粒子を提供することである。
本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、前記ポリプロピレン系樹脂押出発泡粒子の基材樹脂が、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する。
本発明の一実施形態によれば、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供し得る、ポリプロピレン系樹脂押出発泡粒子を提供することができるという効果を奏する。
本発明の一実施形態について以下に説明するが、本発明はこれに限定されるものではない。本発明は、以下に説明する各構成に限定されるものではなく、請求の範囲に示した範囲で種々の変更が可能である。また、異なる実施形態または実施例にそれぞれ開示された技術的手段を組み合わせて得られる実施形態または実施例についても、本発明の技術的範囲に含まれる。さらに、各実施形態にそれぞれ開示された技術的手段を組み合わせることにより、新しい技術的特徴を形成することができる。なお、本明細書中に記載された学術文献および特許文献の全てが、本明細書中において参考文献として援用される。また、本明細書において特記しない限り、数値範囲を表す「A~B」は、「A以上(Aを含みかつAより大きい)B以下(Bを含みかつBより小さい)」を意図する。
〔1.本発明の一実施形態に係る技術的思想〕
ポリプロピレン系樹脂発泡成形体を用いた製品には、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームと接着させ、積層発泡体とした製品が多数ある。ただし、ポリプロピレン系樹脂は非極性樹脂であることから、当該積層発泡体を製造する際に、ポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを接着させることには困難性があった。そこで、ポリウレタンフォームに対するポリプロピレン系樹脂発泡成形体の接着性の向上が求められている。
ポリプロピレン系樹脂発泡成形体を用いた製品には、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームと接着させ、積層発泡体とした製品が多数ある。ただし、ポリプロピレン系樹脂は非極性樹脂であることから、当該積層発泡体を製造する際に、ポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを接着させることには困難性があった。そこで、ポリウレタンフォームに対するポリプロピレン系樹脂発泡成形体の接着性の向上が求められている。
上述した通り、特許文献1では、ポリエチレン発泡体とポリウレタン発泡体との接着のために、ポリエチレン発泡体の表面を易接着化処理(例えば、粗面化処理、火炎処理、コロナ放電処理等)する必要がある。また、特許文献2では、成形体と異素材層との接着のために、成形体の表面における異素材層との接触領域を、成形体の軟化温度の80%以上の温度で加熱する必要がある。すなわち、特許文献1および特許文献2の技術では、得られたポリエチレン発泡体または成形体に対して、それらの表面をさらに物理的に処理する必要があり、生産性の観点において、さらなる改善の余地があった。そのため、本発明者らは、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性良く提供することを課題とした。本発明者は、具体的に、ポリプロピレン系樹脂発泡成形体の表面を物理的に処理することなく、ポリウレタンフォームとの接着性に優れるポリプロピレン系樹脂発泡成形体を提供し得る、ポリプロピレン系樹脂押出発泡粒子を開発すべく、鋭意検討を行った。
鋭意検討の結果、本発明者らは、以下の知見を独自に見出し、本発明を完成させるに至った:(1)分岐状ポリプロピレン系樹脂と特定の重量平均分子量を有する酸変性ポリオレフィン系樹脂とを特定量含むポリプロピレン系樹脂押出発泡粒子は、驚くべきことに、表面を物理的に処理することなくポリウレタンフォームとの接着強度に優れるポリプロピレン系樹脂発泡成形体を提供できること;(2)すなわち、ポリプロピレン系樹脂押出発泡粒子は、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供できること。
なお、付言すると、日本国特開2011-099101号には、酸変性ポリオレフィン系樹脂を用い、除圧発泡法により発泡粒子を製造する方法が開示されている。しかしながら、本発明者らは、日本国特開2011-099101号に記載の技術に関して、以下の問題点が存在することを新たに見出した:(1)日本国特開2011-099101号に記載の技術のように、酸変性ポリオレフィン系樹脂を用いて発泡粒子を製造する際には、樹脂粒子同士の互着が生じるため、分散剤を多量に使用する必要があること;(2)分散剤を多量に使用して得られた発泡粒子の表面には多量の分散剤が残存しており、当該発泡粒子を用いた型内発泡成形により得られる発泡成形体は、(a)融着率が低くなるため、発泡成形体自体が壊れやすくなり、その結果(b)ポリウレタンフォームとの接着強度に劣る傾向がある(発泡成形体とポリウレタンフォームとの接着部分ではなく、発泡成形体を構成している発泡粒子同士の界面で破断するため)こと。
これに対し、本発明の一実施形態にかかるポリプロピレン系樹脂押出発泡粒子は、押出発泡法で得られるため、分散剤を必要としない。すなわち、本発明の一実施形態にかかるポリプロピレン系樹脂押出発泡粒子は、発泡粒子の表面に分散剤が付着していないという利点を有する。その結果、本発明の一実施形態にかかるポリプロピレン系樹脂押出発泡粒子は、融着性に優れ、かつポリウレタンフォームとの接着強度に優れるポリプロピレン系樹脂発泡成形体を提供できるとの利点も有する。
〔2.ポリプロピレン系樹脂押出発泡粒子〕
本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、当該ポリプロピレン系樹脂押出発泡粒子の基材樹脂が、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する。
本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、当該ポリプロピレン系樹脂押出発泡粒子の基材樹脂が、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する。
本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、前述した構成を有するため、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供できるという利点を有する。具体的に、本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、前述した構成を有するため、表面を物理的に処理することなくポリウレタンフォームとの接着強度に優れるポリプロピレン系樹脂発泡成形体を提供できる。また、本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、表面に分散剤(例えば、第三リン酸カルシウムおよびカオリンなど)を含まない構成でも有り得る。ただし、本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子に対して、意図せず分散剤が付着する場合も有り得る。それ故、本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、表面に分散剤を実質的に含まない構成でも有り得る。「表面に分散剤を実質的に含まない」とは、表面に付着した分散剤量が、100ppm以下であることを意図する。換言すれば、本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、表面に分散剤を含まないか、表面に0ppm超100ppm以下の分散剤を含む。分散剤は、例えば、第三リン酸カルシウム、炭酸カルシウム、第三リン酸マグネシウム、硫酸バリウム、カオリンおよびタルク等が挙げられる。
本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子は、当該ポリプロピレン系樹脂押出発泡粒子を型内発泡成形することにより、ポリプロピレン系樹脂発泡成形体とすることができる。また、当該ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを積層することにより、積層発泡体とすることができる。本明細書において、「分岐構造を有するポリプロピレン系樹脂」を「分岐状ポリプロピレン系樹脂」と称する場合があり、「ポリプロピレン系樹脂押出発泡粒子」を「押出発泡粒子」と称する場合があり、「本発明の一実施形態に係るポリプロピレン系樹脂押出発泡粒子」を、「本押出発泡粒子」と称する場合があり、「ポリプロピレン系樹脂発泡成形体」を「発泡成形体」と称する場合がある。
(2-1.基材樹脂)
本発明の一実施形態に係る基材樹脂は、分岐構造を有するポリプロピレン系樹脂(A)および、重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む。基材樹脂は、さらに任意で気泡核形成剤等の添加剤を含み得る。基材樹脂とは、押出発泡粒子を実質的に構成している樹脂成分であるともいえる。
本発明の一実施形態に係る基材樹脂は、分岐構造を有するポリプロピレン系樹脂(A)および、重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む。基材樹脂は、さらに任意で気泡核形成剤等の添加剤を含み得る。基材樹脂とは、押出発泡粒子を実質的に構成している樹脂成分であるともいえる。
本明細書において、「分岐構造を有するポリプロピレン系樹脂(A)」とは、(a)分岐構造が導入されていないポリプロピレン系樹脂の分子同士を分子間で一部架橋させたポリプロピレン系樹脂、および(b)分岐構造が導入されていないポリプロピレン系樹脂に対して、(ポリ)プロピレン以外の共役ジエン化合物等を分岐鎖として導入したポリプロピレン系樹脂を意図する。ここで、分岐状ポリプロピレン系樹脂(A)の架橋部分以外の箇所を、「主鎖」と称する場合がある。また、「分岐構造が導入されていないポリプロピレン系樹脂」、すなわち、分岐状ポリプロピレン系樹脂(A)の原料を「線状ポリプロピレン系樹脂」と称する場合がある。
(主鎖)
本明細書において、ポリプロピレン系樹脂とは、樹脂に含まれる全構造単位100モル%中、プロピレン単量体に由来する構造単位を50モル%以上含む樹脂を意図する。それ故、分岐状ポリプロピレン系樹脂(A)の主鎖は、主鎖に含まれる全構造単位100モル%中、プロピレン単量体に由来する構造単位を50モル%以上含む。本明細書において、「プロピレン単量体に由来する構造単位」を「プロピレン単位」と称する場合がある。
本明細書において、ポリプロピレン系樹脂とは、樹脂に含まれる全構造単位100モル%中、プロピレン単量体に由来する構造単位を50モル%以上含む樹脂を意図する。それ故、分岐状ポリプロピレン系樹脂(A)の主鎖は、主鎖に含まれる全構造単位100モル%中、プロピレン単量体に由来する構造単位を50モル%以上含む。本明細書において、「プロピレン単量体に由来する構造単位」を「プロピレン単位」と称する場合がある。
主鎖は、(a)プロピレンの単独重合体であってもよく、(b)プロピレンとプロピレン以外の単量体とのブロック共重合体もしくはランダム共重合体であってもよく、または(c)これらの2種以上の混合物であってもよい。
主鎖は、プロピレン単位に加えて、プロピレン単量体以外の単量体に由来する構造単位を1単位以上有していてもよく、1種以上有していてもよい。主鎖の製造で使用される「プロピレン単量体以外の単量体」を「コモノマー」と称する場合もあり、主鎖に含まれる「プロピレン単量体以外の単量体に由来する構造単位」を「コモノマー単位」と称する場合がある。
コモノマーとしては、以下のような単量体が挙げられる:(a)エチレン、1-ブテン、イソブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセン、4-メチル-1-ペンテン、3,4-ジメチル-1-ブテン、1-ヘプテン、3-メチル-1-ヘキセン、1-オクテン、1-デセンなどの炭素数2または4~12のα-オレフィン、(b)シクロペンテン、ノルボルネン、テトラシクロ[6,2,11,8,13,6]-4-ドデセンなどの環状オレフィン、(c)5-メチレン-2-ノルボルネン、5-エチリデン-2-ノルボルネン、1,4-ヘキサジエン、メチル-1,4-ヘキサジエン、7-メチル-1,6-オクタジエンなどのジエン、並びに(d)塩化ビニル、塩化ビニリデン、アクリロニトリル、メタクリロニトリル、酢酸ビニル、アクリル酸、アクリル酸エステル、メタクリル酸、メタクリル酸エステル、マレイン酸、無水マレイン酸、スチレン系単量体、ビニルトルエン、ジビニルベンゼンなどのビニル系単量体、など。
アクリル酸エステルとしては、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、アクリル酸ヘキシル、アクリル酸2-エチルヘキシル、アクリル酸ラウリル、アクリル酸ステアリル、アクリル酸2-ヒドロキシエチル、アクリル酸2-ヒドロキシプロピルおよびアクリル酸グリシジルなどが挙げられる。
メタクリル酸エステルとしては、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸ヘキシル、メタクリル酸2-エチルヘキシル、メタクリル酸ラウリル、メタクリル酸ステアリル、メタクリル酸2-ヒドロキシエチル、メタクリル酸2-ヒドロキシプロピルおよびメタクリル酸グリシジルなどが挙げられる。
スチレン系単量体としては、スチレン、メチルスチレン、ジメチルスチレン、アルファメチルスチレン、パラメチルスチレン、エチルスチレン、ジエチルスチレン、イソプロピルスチレン、t-ブチルスチレン、ブロモスチレン、ジブロモスチレン、トリブロモスチレン、クロロスチレン、ジクロロスチレンおよびトリクロロスチレンなどが挙げられる。
主鎖は、コモノマー単位として、炭素数2または4~12のα-オレフィンに由来する構造単位を有することが好ましく、エチレン、1-ブテン、イソブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセン、4-メチル-1-ペンテン、3,4-ジメチル-1-ブテン、1-ヘプテン、3-メチル-1-ヘキセン、1-オクテンおよび/または1-デセンなどに由来する構造単位を有することがより好ましく、エチレン、1-ブテン、イソブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセンおよび/または4-メチル-1-ペンテンに由来する構造単位を有することがより好ましく、エチレン、1-ブテン、イソブテンおよび/または1-ペンテンに由来する構造単位を有することがよりさらに好ましく、エチレンおよび/または1-ブテンに由来する構造単位を有することがより特に好ましい。当該構成によると、(a)高い溶融張力および低いゲル分率を有する分岐状ポリプロピレン系樹脂が得られるという利点、並びに(b)得られる分岐状ポリプロピレン系樹脂が成形性に優れるポリプロピレン系樹脂押出発泡粒子を提供できるという利点を有する。
主鎖は、プロピレン単独重合体、ポリプロピレン系ブロック共重合体およびポリプロピレン系ランダム共重合体からなる群から選択される1種以上であることが好ましく、プロピレン単独重合体および/またはポリプロピレン系ランダム共重合体であることがより好ましい。当該構成によると、(a)高い溶融張力および低いゲル分率を有する分岐状ポリプロピレン系樹脂が得られるという利点、並びに(b)得られる分岐状ポリプロピレン系樹脂が成形性に優れるポリプロピレン系樹脂押出発泡粒子を提供できるという利点を有する。
主鎖は、当該主鎖に含まれる全構造単位100モル%中、プロピレン単位を90モル%以上含むことが好ましく、93モル%以上含むことがより好ましく、95モル%以上含むことがさらに好ましく、97モル%以上含むことが特に好ましい。当該構成によると、高い溶融張力および低いゲル分率を有する分岐状ポリプロピレン系樹脂が得られるという利点を有する。
分岐状ポリプロピレン系樹脂(A)の融点は、特に限定されない。分岐状ポリプロピレン系樹脂(A)の融点は、例えば、125℃~170℃であることが好ましく、130℃~170℃であることがより好ましく、135℃~160℃であることがより好ましく、140℃~155℃であることがさらに好ましく、145℃~150℃であることが特に好ましい。分岐状ポリプロピレン系樹脂(A)の融点が上述した範囲内である場合、押出発泡粒子が低い連続気泡率を有するという利点を有する。また、分岐状ポリプロピレン系樹脂(A)の融点が、(a)125℃以上である場合、発泡成形体の寸法安定性が低下する虞がなく、発泡成形体の耐熱性が不十分となる虞がなく、かつ発泡成形体の圧縮強度が高くなる傾向があるという利点を有し、(b)170℃以下である場合、押出発泡粒子を比較的低い蒸気圧で成形することが可能となるため、ポリプロピレン系樹脂発泡粒子用の汎用成形機を使用して押出発泡粒子を成形できるという利点を有する。
本明細書において、分岐状ポリプロピレン系樹脂(A)の融点は、示差走査熱量計法(以降、「DSC法」と称する)により測定して求められる値である。具体的な操作手順は以下の通りである:(1)分岐状ポリプロピレン系樹脂(A)5mg~6mgの温度を10℃/分の昇温速度で40℃から220℃まで昇温することにより当該分岐状ポリプロピレン系樹脂(A)を融解させる;(2)その後、融解された分岐状ポリプロピレン系樹脂(A)の温度を10℃/分の降温速度で220℃から40℃まで降温することにより当該分岐状ポリプロピレン系樹脂(A)を結晶化させる;(3)その後、さらに、結晶化された分岐状ポリプロピレン系樹脂(A)の温度を10℃/分の昇温速度で40℃から220℃まで昇温する。2回目の昇温時(すなわち(3)のとき)に得られる当該分岐状ポリプロピレン系樹脂(A)のDSC曲線のピーク(融解ピーク)の温度を当該分岐状ポリプロピレン系樹脂(A)の融点として求めることができる。なお、上述の方法により、2回目の昇温時に得られる、分岐状ポリプロピレン系樹脂(A)のDSC曲線において、ピーク(融解ピーク)が複数存在する場合、融解熱量が最大のピーク(融解ピーク)の温度を、分岐状ポリプロピレン系樹脂(A)の融点とする。示差走査熱量計としては、例えば、セイコーインスツルメンツ(株)製、DSC6200型を用いることができる。
分岐状ポリプロピレン系樹脂(A)のメルトフローレート(MFR)は、特に限定されない。分岐状ポリプロピレン系樹脂(A)のMFRは、例えば、0.5g/10分~20.0g/10分であることが好ましく、0.5g/10分~15.0g/10分であることがより好ましく、1.0g/10分~10.0g/10分であることがさらに好ましく、1.5g/10分~5.0g/10分であることがよりさらに好ましく、1.5g/10分~3.0g/10分であることが特に好ましい。分岐状ポリプロピレン系樹脂(A)のMFRが上述した範囲内である場合、(a)押出発泡粒子が低い連続気泡率を有するという利点、(b)押出発泡粒子が成形性に優れるという利点、および(c)当該押出発泡粒子は耐破断性に優れる発泡成形体を提供できるという利点、を有する。分岐状ポリプロピレン系樹脂(A)のMFRが、(a)0.5g/10分以上である場合、当該分岐状ポリプロピレン系樹脂(A)から得られる押出発泡粒子は、変形が少なく、表面性が良好(美麗)である発泡成形体を提供できるという利点を有し、(b)20.0g/10分以下である場合、当該分岐状ポリプロピレン系樹脂(A)から得られる組成物は、押出発泡時、発泡性が良好になるという利点を有する。
本明細書において、分岐状ポリプロピレン系樹脂(A)のMFRは、ISO 1133に従い、温度230℃および荷重2.16kgの条件で測定して求められる値である。
分岐状ポリプロピレン系樹脂(A)の溶融張力(メルトテンション)は、特に限定されない。分岐状ポリプロピレン系樹脂(A)のメルトテンションは、例えば、1.0cN~50.0cNであることが好ましく、3.0cN~40.0cNであることがより好ましく、5.0cN~30.0cNであることがより好ましく、7.0cN~20.0cNであることがさらに好ましく、9.0cN~15.0cNであることが特に好ましい。分岐状ポリプロピレン系樹脂(A)の溶融張力が1.0cN以上である場合、分岐状ポリプロピレン系樹脂(A)および発泡剤を含む組成物を完全溶融させて発泡するとき、組成物の張力が十分に高くなり、得られる押出発泡粒子におけるセルの破泡を防ぐことができる。その結果、(a)得られる押出発泡粒子が成形性に優れるという利点、および(b)当該押出発泡粒子は耐破断性に優れる発泡成形体を提供できるという利点、を有する。分岐状ポリプロピレン系樹脂(A)の溶融張力が50.0cN以下である場合、押出発泡工程において、樹脂圧力(溶融混練物が、製造装置に設置された圧力計を押す力)が高くなりすぎず、吐出量を比較的高くすることができる。その結果、生産性が良く押出発泡粒子を得ることができるという利点を有する。
本明細書において、分岐状ポリプロピレン系樹脂(A)の溶融張力は、キャピログラフ1D(日本 株式会社東洋精機製作所製)を用いて測定する。具体的には、以下(1)~(5)の通りである:(1)試験温度(200℃)に加熱された径9.55mmのバレルに測定用の試料樹脂(分岐状ポリプロピレン系樹脂)を充填する;(2)次いで、試料樹脂を10分間、試験温度(200℃)に加熱されたバレル内で加熱する;(3)次いで、キャピラリーダイ(口径1.0mm、長さ10mm)から、一定に保持したピストン降下速度(10mm/分)にて、試料樹脂を紐状に出しながら、この紐状物を前記キャピラリーダイの下方350mmに位置する張力検出のプーリーに通過させた後、巻取りロールを用いる巻取りを開始する;(4)紐状物の引き取りが安定した後、紐状物の巻取り速度を初速1.0m/分から、4分間で200m/分の速度に達するまで一定の割合で増加させる;(5)紐状物が破断したときのロードセル付きプーリーにかかる荷重を溶融張力として測定する。
(分岐状ポリプロピレン系樹脂(A)の製造方法)
分岐構造を有するポリプロピレン系樹脂(分岐状ポリプロピレン系樹脂)(A)は、線状ポリプロピレン系樹脂に分岐構造を導入することによって得ることができる。線状ポリプロピレン系樹脂に分岐構造を導入する方法としては、特に限定されないが、例えば、(a1)線状ポリプロピレン系樹脂に放射線を照射する方法、および(a2)線状ポリプロピレン系樹脂と共役ジエン化合物とラジカル重合開始剤とを含む混合物を溶融混練する方法などが挙げられる。
分岐構造を有するポリプロピレン系樹脂(分岐状ポリプロピレン系樹脂)(A)は、線状ポリプロピレン系樹脂に分岐構造を導入することによって得ることができる。線状ポリプロピレン系樹脂に分岐構造を導入する方法としては、特に限定されないが、例えば、(a1)線状ポリプロピレン系樹脂に放射線を照射する方法、および(a2)線状ポリプロピレン系樹脂と共役ジエン化合物とラジカル重合開始剤とを含む混合物を溶融混練する方法などが挙げられる。
前記(a1)の方法の具体的な方法としては、例えば日本国特表2002-542360に記載の方法が挙げられる。
前記(a2)の方法についてさらに説明する。前記(a2)の方法では、例えば、以下(i)~(iv)を順に行い分岐状ポリプロピレン系樹脂(A)を得ることができる:(i)線状ポリプロピレン系樹脂と共役ジエン化合物とラジカル重合開始剤とを含む混合物を、ダイを備える装置で溶融混練する;(ii)得られた溶融混練物をダイから押出す;(iii)押出された溶融混練物(ストランドとも称される。)を冷却する;(iv)ストランドの冷却と同時にまたは、冷却後に、ストランドを細断する。前記(a2)の方法の具体的な方法としては、例えばWO2020/004429に記載の方法が挙げられる。
(i)線状ポリプロピレン系樹脂に分岐構造を安定して導入でき、かつ分岐構造の導入の再現性が高いことから、および/または(ii)複雑な設備を必要とせず、かつ高い生産性で分岐状ポリプロピレン系樹脂を得ることができるとことから、本発明の一実施形態において、分岐状ポリプロピレン系樹脂(A)は、上述の(a2)の方法によって得られる分岐状ポリプロピレン系樹脂であることが好ましい。
共役ジエン化合物としては、例えば、ブタジエン、イソプレン、1,3-ヘプタジエン、2,3-ジメチルブタジエン、および2,5-ジメチル-2,4-ヘキサジエン、などがあげられる。これら共役ジエン化合物は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。これら共役ジエン化合物の中では、(a)安価で取り扱い点、および(b)反応が均一に進みやすい点から、ブタジエン、およびイソプレンが特に好ましい。
ラジカル重合開始剤は、ポリプロピレン系樹脂および/または共役ジエン化合物からの水素引き抜き能を有する有機過酸化物である。本発明の一実施形態において好適に用いられるラジカル重合開始剤としては、ケトンパーオキサイド、パーオキシケタール、ハイドロパーオキサイド、ジアルキルパーオキサイド、ジアシルパーオキサイド、パーオキシジカーボネート、パーオキシエステルなどの有機過酸化物が挙げられる。
有機過酸化物としては、特に水素引き抜き能が高いものが好ましい。水素引き抜き能が高い有機過酸化物としては、例えば1,1-ビス(t-ブチルパーオキシ)3,3,5-トリメチルシクロヘキサン、1,1-ビス(t-ブチルパーオキシ)シクロヘキサン、n-ブチル4,4-ビス(t-ブチルパーオキシ)バレレート、2,2-ビス(t-ブチルパーオキシ)ブタン等のパーオキシケタール;ジクミルパーオキサイド、2,5-ジメチル-2,5-ジ(t-ブチルパーオキシ)ヘキサン、α,α’-ビス(t-ブチルパーオキシ-m-イソプロピル)ベンゼン、t-ブチルクミルパーオキサイド、ジ-t-ブチルパーオキサイド、2,5-ジメチル-2,5-ジ(t-ブチルパーオキシ)-3-ヘキシン等のジアルキルパーオキサイド;ベンゾイルパーオキサイド等のジアシルパーオキサイド;t-ブチルパーオキシオクテート、t-ブチルパーオキシイソブチレート、t-ブチルパーオキシラウレート、t-ブチルパーオキシ3,5,5-トリメチルヘキサノエート、t-ブチルパーオキシイソプロピルカーボネート、2,5-ジメチル-2,5-ジ(ベンゾイルパーオキシ)ヘキサン、t-ブチルパーオキシアセテート、t-ブチルパーオキシベンゾエート、ジ-t-ブチルパーオキシイソフタレート等のパーオキシエステル;等が好適に挙げられる。これらの中ではt-ブチルパーオキシイソプロピルカーボネートおよび/またはt-ブチルパーオキシベンゾエートが好ましい。これら有機過酸化物は1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
ラジカル重合開始剤の使用量は特に限定されないが、線状ポリプロピレン系樹脂100重量部に対して、0.01重量部~5.00重量部が好ましく、0.10重量部~3.00重量部が好ましく、0.10重量部~2.50重量部がさらに好ましく、0.10重量部~2.00重量部が特に好ましい。
(酸変性ポリオレフィン系樹脂(B))
酸変性ポリオレフィン系樹脂(B)とは、不飽和カルボン酸および/またはその誘導体が導入されたポリオレフィン系樹脂ともいえる。ポリオレフィン系樹脂への不飽和カルボン酸および/またはその誘導体の導入は、(b1)ポリオレフィン系樹脂とラジカル重合開始剤とを反応させた後、得られた反応生成物と不飽和カルボン酸および/またはその誘導体とを反応させて得る方法、および(b2)ポリオレフィン系樹脂を熱分解させた後、得られた反応生成物に不飽和カルボン酸および/またはその誘導体を導入する方法、などが好適に挙げられる。主鎖の末端に不飽和カルボン酸および/またはその誘導体を導入しやすいため、前記(b2)の方法が好ましい。換言すれば、酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂を熱分解させた後、得られた反応生成物に不飽和カルボン酸および/またはその誘導体を導入する方法により得られた樹脂であることが好ましい。また、酸変性ポリオレフィン系樹脂(B)は、ポリオレフィンの分岐を有さないことが好ましい。
酸変性ポリオレフィン系樹脂(B)とは、不飽和カルボン酸および/またはその誘導体が導入されたポリオレフィン系樹脂ともいえる。ポリオレフィン系樹脂への不飽和カルボン酸および/またはその誘導体の導入は、(b1)ポリオレフィン系樹脂とラジカル重合開始剤とを反応させた後、得られた反応生成物と不飽和カルボン酸および/またはその誘導体とを反応させて得る方法、および(b2)ポリオレフィン系樹脂を熱分解させた後、得られた反応生成物に不飽和カルボン酸および/またはその誘導体を導入する方法、などが好適に挙げられる。主鎖の末端に不飽和カルボン酸および/またはその誘導体を導入しやすいため、前記(b2)の方法が好ましい。換言すれば、酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂を熱分解させた後、得られた反応生成物に不飽和カルボン酸および/またはその誘導体を導入する方法により得られた樹脂であることが好ましい。また、酸変性ポリオレフィン系樹脂(B)は、ポリオレフィンの分岐を有さないことが好ましい。
本発明の一実施形態に係る酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、特に限定されないが、15000~65000であることが好ましく、16000~60000であることがより好ましく、17000~50000であることがさらに好ましく、18000~40000であることが特に好ましい。当該構成によれば、ポリウレタンフォームとの接着強度に優れるポリプロピレン系樹脂発泡成形体を提供できる、との利点を有する。
本明細書において、酸変性ポリオレフィン系樹脂(B)の重量平均分子量とは、GPC法によって算出された値である。
また、本明細書において、酸とは、不飽和カルボン酸および/またはその誘導体のことを意図する。不飽和カルボン酸およびその誘導体としては、フマル酸、無水フマル酸、マレイン酸、無水マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、フマル酸メチル、フマル酸エチル、フマル酸プロピル、フマル酸ブチル、フマル酸ジメチル、フマル酸ジエチル、フマル酸ジプロピル、フマル酸ジブチル、マレイン酸メチル、マレイン酸エチル、マレイン酸プロピル、マレイン酸ブチル、マレイン酸ジメチル、マレイン酸ジエチル、マレイン酸ジプロピル、マレイン酸ジブチル等が挙げられる。
本発明の一実施形態において、不飽和カルボン酸およびその誘導体としては、無水イタコン酸および無水マレイン酸がより好ましく、無水マレイン酸がより好ましい。すなわち、酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に、無水イタコン酸および/または無水マレイン酸が導入されてなる樹脂であることが好ましく、無水マレイン酸が導入されてなる樹脂であることがより好ましい。酸変性ポリオレフィン系樹脂(B)は、無水イタコン酸に由来する構成単位および/または無水マレイン酸に由来する構成単位を含むことが好ましく、無水マレイン酸に由来する構成単位を含むことがより好ましいともいえる。これらの構成によると、低コストでポリウレタンフォームとの接着強度に優れるポリプロピレン系樹脂発泡成形体を提供できるという利点を有する。
本明細書において、ポリオレフィン系樹脂に、不飽和カルボン酸およびその誘導体として「Z」が導入されてなる樹脂(換言すれば、不飽和カルボン酸およびその誘導体として「Z」に由来する構成単位を含む酸変性ポリオレフィン系樹脂)を、「Z変性ポリオレフィン系樹脂」と称する場合がある。
本明細書において、酸価とは、JIS K 0070にて測定され、酸変性ポリオレフィン系樹脂1g中に存在する遊離酸を中和するのに必要な水酸化カリウムの量(mg)を意図する。酸価は、酸の変性の指標となる数値の一つである。酸変性ポリオレフィン系樹脂(B)の酸価は、1.0mg/KOH~200.0mg/KOHであることが好ましく、3.0mg/KOH~150.0mg/KOHであることがより好ましく、5.0mg/KOH~100.0mg/KOHであることがさらに好ましく、10.0mg/KOH~80.0mg/KOHであることが特に好ましい。当該構成によれば、ポリウレタンフォームとの接着性に優れる発泡成形体を提供できるという利点を有する。
基材樹脂は、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、(i)分岐状ポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有し、(ii)分岐状ポリプロピレン系樹脂(A)を70.0重量%~97.0重量%、および酸変性ポリオレフィン系樹脂(B)を3.0重量%~30.0重量%含有することが好ましく、(iii)分岐状ポリプロピレン系樹脂(A)を75.0重量%~97.0重量%、および酸変性ポリオレフィン系樹脂(B)を3.0重量%~25.0重量%含有することがより好ましく、(iv)分岐状ポリプロピレン系樹脂(A)を80.0重量%~97.0重量%、および酸変性ポリオレフィン系樹脂(B)を3.0重量%~20.0重量%含有することがより好ましく、(v)分岐状ポリプロピレン系樹脂(A)を85.0重量%~95.0重量%、および酸変性ポリオレフィン系樹脂(B)を5.0重量%~15.0重量%含有することがさらに好ましい。当該構成によると、積層発泡体をより生産性よく提供できるという利点を有する。
(その他の樹脂またはゴム)
基材樹脂は、本発明の一実施形態に係る効果を損なわない範囲で、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)以外の樹脂(「その他の樹脂」と称する場合がある。)またはゴムをさらに含んでいてもよい。その他の樹脂としては、(a)エチレン/プロピレンランダム共重合体、エチレン/プロピレンブロック共重合体、プロピレン単独重合体などの線状のポリプロピレン系樹脂、(b)高密度ポリエチレン、中密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、直鎖状超低密度ポリエチレン、エチレン/酢酸ビニル共重合体、エチレン/アクリル酸共重合体、およびエチレン/メタアクリル酸共重合体などのエチレン系樹脂、並びに(c)ポリスチレン、スチレン/無水マレイン酸共重合体、およびスチレン/エチレン共重合体などのスチレン系樹脂、などが挙げられる。前記ゴムとしては、エチレン/プロピレンゴム、エチレン/ブテンゴム、エチレン/ヘキセンゴム、エチレン/オクテンゴムなどのオレフィン系ゴムが挙げられる。
基材樹脂は、本発明の一実施形態に係る効果を損なわない範囲で、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)以外の樹脂(「その他の樹脂」と称する場合がある。)またはゴムをさらに含んでいてもよい。その他の樹脂としては、(a)エチレン/プロピレンランダム共重合体、エチレン/プロピレンブロック共重合体、プロピレン単独重合体などの線状のポリプロピレン系樹脂、(b)高密度ポリエチレン、中密度ポリエチレン、低密度ポリエチレン、直鎖状低密度ポリエチレン、直鎖状超低密度ポリエチレン、エチレン/酢酸ビニル共重合体、エチレン/アクリル酸共重合体、およびエチレン/メタアクリル酸共重合体などのエチレン系樹脂、並びに(c)ポリスチレン、スチレン/無水マレイン酸共重合体、およびスチレン/エチレン共重合体などのスチレン系樹脂、などが挙げられる。前記ゴムとしては、エチレン/プロピレンゴム、エチレン/ブテンゴム、エチレン/ヘキセンゴム、エチレン/オクテンゴムなどのオレフィン系ゴムが挙げられる。
基材樹脂中のその他の樹脂の含有量は、例えば、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計100重量部に対して60重量部以下であることが好ましく、40重量部以下であることがより好ましく、30重量部以下であることがより好ましく、20重量部以下であることがさらに好ましく、10重量部以下であることが特に好ましい。その他の樹脂の含有量の下限値は特に限定されず、例えば、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計100重量部に対して0重量部であってよい。
基材樹脂がその他の樹脂として線状のポリプロピレン系樹脂を含む場合、例えば、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)と線状のポリプロピレン系樹脂との合計100重量%に対して、分岐状ポリプロピレン系樹脂(A)の含有量が、40.0重量%~70.0重量%未満、酸変性ポリオレフィン系樹脂(B)の含有量が2.5重量%~30.0重量%、および線状のポリプロピレン系樹脂の含有量が0重量%超30.0重量%未満であることが好ましい。
(気泡核形成剤)
基材樹脂は、得られる押出発泡粒子の気泡数および気泡の形状をコントロールする目的で、気泡核形成剤を含んでいてもよい。気泡核形成剤としては、重炭酸ソーダ-クエン酸混合物、クエン酸モノナトリウム塩、タルク、および炭酸カルシウムなどを挙げることができる。これら気泡核形成剤は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
基材樹脂は、得られる押出発泡粒子の気泡数および気泡の形状をコントロールする目的で、気泡核形成剤を含んでいてもよい。気泡核形成剤としては、重炭酸ソーダ-クエン酸混合物、クエン酸モノナトリウム塩、タルク、および炭酸カルシウムなどを挙げることができる。これら気泡核形成剤は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
基材樹脂中の気泡核形成剤の含有量は特に限定されない。気泡核形成剤の含有量は、例えば、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計100重量部に対して、0.01重量部~5.00重量部であることが好ましく、0.01重量部~3.50重量部であることがより好ましく、0.01重量部~1.00重量部であることがさらに好ましく、0.01重量部~0.50重量部であることが特に好ましい。
(着色剤)
基材樹脂は、着色剤を含んでいてもよく、含んでいなくてもよい。基材樹脂が着色剤を含まない場合、ナチュラル色(例えば、基材樹脂に由来する色)の押出発泡粒子を得ることができる。基材樹脂が着色剤を含む場合、所望の色(すなわち、着色剤に由来する色)の押出発泡粒子を得ることができる。着色剤としては、例えば、ペリレン系有機顔料、アゾ系有機顔料、キナクリドン系有機顔料、フタロシアニン系有機顔料、スレン系有機顔料、ジオキサジン系有機顔料、イソインドリン系有機顔料、およびカーボンブラックなどが挙げられる。これら着色剤は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。人の目に触れる部材に発泡成形体を使用する場合、意匠性などの観点からは、当該発泡成形体が黒色であることが求められる場合がある。そのため、着色剤としては、カーボンブラックが好ましい。着色剤の使用量、換言すれば基材樹脂中の着色剤の含有量、は特に限定されない。
基材樹脂は、着色剤を含んでいてもよく、含んでいなくてもよい。基材樹脂が着色剤を含まない場合、ナチュラル色(例えば、基材樹脂に由来する色)の押出発泡粒子を得ることができる。基材樹脂が着色剤を含む場合、所望の色(すなわち、着色剤に由来する色)の押出発泡粒子を得ることができる。着色剤としては、例えば、ペリレン系有機顔料、アゾ系有機顔料、キナクリドン系有機顔料、フタロシアニン系有機顔料、スレン系有機顔料、ジオキサジン系有機顔料、イソインドリン系有機顔料、およびカーボンブラックなどが挙げられる。これら着色剤は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。人の目に触れる部材に発泡成形体を使用する場合、意匠性などの観点からは、当該発泡成形体が黒色であることが求められる場合がある。そのため、着色剤としては、カーボンブラックが好ましい。着色剤の使用量、換言すれば基材樹脂中の着色剤の含有量、は特に限定されない。
(その他成分)
基材樹脂は、必要に応じてその他成分として、(a)酸化防止剤、金属不活性剤、燐系加工安定剤、紫外線吸収剤、紫外線安定剤、蛍光増白剤、金属石鹸、および制酸吸着剤などの安定剤、並びに/または、(b)架橋剤、連鎖移動剤、滑剤、可塑剤、充填材、強化材、難燃剤、および帯電防止剤などの添加剤、をさらに含んでいてもよい。これらその他成分は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
基材樹脂は、必要に応じてその他成分として、(a)酸化防止剤、金属不活性剤、燐系加工安定剤、紫外線吸収剤、紫外線安定剤、蛍光増白剤、金属石鹸、および制酸吸着剤などの安定剤、並びに/または、(b)架橋剤、連鎖移動剤、滑剤、可塑剤、充填材、強化材、難燃剤、および帯電防止剤などの添加剤、をさらに含んでいてもよい。これらその他成分は、1種を単独で用いてもよく、2種以上を組み合わせて用いてもよい。
押出発泡粒子に含まれる基材樹脂または当該押出発泡粒子から得られる発泡成形体に含まれる基材樹脂、すなわち前記押出発泡粒子または前記発泡成形体を実質的に構成する基材樹脂、に含まれる、分岐状ポリプロピレン系樹脂(A)、酸変性ポリオレフィン系樹脂(B)、その他の樹脂またはゴム、気泡核形成剤、着色剤およびその他成分の種類および量は、前記押出発泡粒子または前記発泡成形体を減圧下で融解して樹脂塊に戻す操作によっても、実質的に変化しない。本明細書において、押出発泡粒子、または当該押出発泡粒子から得られる発泡成形体を減圧下で融解して樹脂塊を得ることを「樹脂戻しする」と称する場合があり、樹脂戻しして得られる樹脂塊を「戻し樹脂」と称する場合がある。また、「分岐状ポリプロピレン系樹脂(A)、酸変性ポリオレフィン系樹脂(B)、その他の樹脂またはゴム、気泡核形成剤、着色剤およびその他成分」を「分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)等」と称する場合がある。本明細書において、戻し樹脂に含まれる、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)等の種類および量を、基材樹脂に含まれる、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)等の種類および量と見做すことができる。戻し樹脂に含まれる、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)等の種類および量は、任意の公知の方法により戻し樹脂を解析することで、確認することができる。
樹脂戻しの具体的な方法としては特に限定されないが、例えば以下(c1)~(c5)を順に行う方法が挙げられる:(c1)押出発泡粒子または発泡成形体の融点+10℃に調整した乾燥機中に、押出発泡粒子または発泡成形体を入れる;(c2)次いで、5~10分かけて真空ポンプを使用して、前記乾燥機内の圧力を-0.05MPa(ケージ圧)~-0.10MPa(ケージ圧)になるまで減圧する;(c3)その後、前記乾燥機内で30分間、押出発泡粒子または発泡成形体を放置し、樹脂塊(戻し樹脂)を調製する;(c4)次いで、乾燥機内の圧力を常圧まで戻す;(c5)その後、乾燥機から前記樹脂塊を取り出し、室温まで冷却する。
(2-2.ポリプロピレン系樹脂押出発泡粒子)
(押出発泡粒子の物性)
以下、本押出発泡粒子の物性について説明する。なお、ポリプロピレン系樹脂押出発泡粒子の製造方法については、後で詳述する。
(押出発泡粒子の物性)
以下、本押出発泡粒子の物性について説明する。なお、ポリプロピレン系樹脂押出発泡粒子の製造方法については、後で詳述する。
(DSC曲線)
除圧発泡法により得られるポリプロピレン系樹脂発泡粒子では、DSC法において得られるDSC曲線のピーク(融解ピーク)は、2つである。一方で、押出発泡法により得られるポリプロピレン系樹脂押出発泡粒子では、DSC曲線のピーク(融解ピーク)は、1つである。したがって、本押出発泡粒子においても、DSC曲線のピーク(融解ピーク)は1つであり得る。示差走査熱量計法を用いて、ポリプロピレン系樹脂押出発泡粒子の温度を40℃から220℃まで10℃/分の速度で昇温することにより、押出発泡粒子のDSC曲線を得ることができる。なお、押出発泡粒子の前記DSC曲線は、分岐状ポリプロピレン系樹脂(A)の融点の測定時に用いた2回目の昇温時に得られるDSC曲線と異なり、1回目の昇温時に得られるDSC曲線である。それゆえ、ポリプロピレン系樹脂押出発泡粒子は、1回目の昇温時に得られるDSC曲線のピークは1つであり得る、ともいえる。
除圧発泡法により得られるポリプロピレン系樹脂発泡粒子では、DSC法において得られるDSC曲線のピーク(融解ピーク)は、2つである。一方で、押出発泡法により得られるポリプロピレン系樹脂押出発泡粒子では、DSC曲線のピーク(融解ピーク)は、1つである。したがって、本押出発泡粒子においても、DSC曲線のピーク(融解ピーク)は1つであり得る。示差走査熱量計法を用いて、ポリプロピレン系樹脂押出発泡粒子の温度を40℃から220℃まで10℃/分の速度で昇温することにより、押出発泡粒子のDSC曲線を得ることができる。なお、押出発泡粒子の前記DSC曲線は、分岐状ポリプロピレン系樹脂(A)の融点の測定時に用いた2回目の昇温時に得られるDSC曲線と異なり、1回目の昇温時に得られるDSC曲線である。それゆえ、ポリプロピレン系樹脂押出発泡粒子は、1回目の昇温時に得られるDSC曲線のピークは1つであり得る、ともいえる。
(連続気泡率)
本押出発泡粒子は、連続気泡率が低いほど好ましい。本押出発泡粒子は、連続気泡率が10.0%以下であることが好ましく、8.0%以下であることがより好ましく、6.0%以下であることがより好ましく、5.0%以下であることがより好ましく、4.0%以下であることがより好ましく、3.0%以下であることがより好ましく、2.0%以下であることが特に好ましい。本ポリプロピレン系樹脂押出発泡粒子の連続気泡率の下限値は特に限定されず、例えば0.0%以上である。前記構成によると、(a)押出発泡粒子の成形時に、セルが破泡して収縮することがほとんどないため、当該押出発泡粒子が成形性に優れるという利点、および(b)当該押出発泡粒子を用いて得られた発泡成形体において、形状の任意性、緩衝性、軽量性、圧縮強度および断熱性などの特徴がより発揮されるという利点を有する。更に、本押出発泡粒子の連続気泡率が上述した範囲内である場合、当該押出発泡粒子は、低圧成形性に優れ、かつ圧縮強度に優れる発泡成形体を提供することができるという利点を有する。
本押出発泡粒子は、連続気泡率が低いほど好ましい。本押出発泡粒子は、連続気泡率が10.0%以下であることが好ましく、8.0%以下であることがより好ましく、6.0%以下であることがより好ましく、5.0%以下であることがより好ましく、4.0%以下であることがより好ましく、3.0%以下であることがより好ましく、2.0%以下であることが特に好ましい。本ポリプロピレン系樹脂押出発泡粒子の連続気泡率の下限値は特に限定されず、例えば0.0%以上である。前記構成によると、(a)押出発泡粒子の成形時に、セルが破泡して収縮することがほとんどないため、当該押出発泡粒子が成形性に優れるという利点、および(b)当該押出発泡粒子を用いて得られた発泡成形体において、形状の任意性、緩衝性、軽量性、圧縮強度および断熱性などの特徴がより発揮されるという利点を有する。更に、本押出発泡粒子の連続気泡率が上述した範囲内である場合、当該押出発泡粒子は、低圧成形性に優れ、かつ圧縮強度に優れる発泡成形体を提供することができるという利点を有する。
本明細書において、ポリプロピレン系樹脂押出発泡粒子の連続気泡率は、空気比較式比重計[東京サイエンス(株)製、モデル1000]を用いて、ASTM D2856-87の手順C(PROCEDURE C)に記載の方法に従って、測定して求められる値である。押出発泡粒子の連続気泡率は、具体的には、以下(1)~(3)を順に実施して算出される:(1)空気比較式比重計を用いて押出発泡粒子の体積Vc(cm3)を測定する;(2)次いで、Vcを測定後の押出発泡粒子の全量を、メスシリンダーに入っているエタノール中に沈める;(3)その後、メスシリンダーにおけるエタノールの位置の上昇量から、押出発泡粒子の見かけ上の体積Va(cm3)を求める;(4)以下の式により、押出発泡粒子の連続気泡率を算出する:連続気泡率(%)=((Va-Vc)×100)/Va。なお、体積Vaの測定の方法は水没法とも称される。
(発泡倍率)
本押出発泡粒子の発泡倍率は、特に限定されないが、2倍以上であることが好ましく、3倍以上であることがより好ましく、5倍以上であることがさらに好ましい。本押出発泡粒子の発泡倍率は、30倍以下であることが好ましく、28倍以下であることがより好ましく、25倍以下であることがさらに好ましい。前記構成によると、成形して得られる発泡成形体の圧縮強度等の物性が優れ、軽量化が可能となるという利点を有する。押出発泡粒子の発泡倍率の測定方法は、下記実施例にて詳述する。
本押出発泡粒子の発泡倍率は、特に限定されないが、2倍以上であることが好ましく、3倍以上であることがより好ましく、5倍以上であることがさらに好ましい。本押出発泡粒子の発泡倍率は、30倍以下であることが好ましく、28倍以下であることがより好ましく、25倍以下であることがさらに好ましい。前記構成によると、成形して得られる発泡成形体の圧縮強度等の物性が優れ、軽量化が可能となるという利点を有する。押出発泡粒子の発泡倍率の測定方法は、下記実施例にて詳述する。
(成形幅)
本押出発泡粒子は、成形幅が広い(例えば0を超える)という利点を有する。本明細書において、押出発泡粒子の成形幅が大きいほど、当該押出発泡粒子は成形性に優れることを意図する。本明細書において、「押出発泡粒子の成形幅」とは、押出発泡粒子を型内発泡成形したとき、以下を満たす発泡成形体を得ることができる、型内発泡成形時の蒸気圧(ゲージ圧)の幅を意図する:(x1)押出発泡粒子同士の融着が十分であり、(x2)押出発泡粒子間の隙間が十分に埋まっており、(x3)表面が美麗であり、(x4)表面がメルトしておらず、かつ(x5)収縮することなく、型内発泡成形に使用した型(金型)の形状が転写されている、発泡成形体。本明細書において、上述した(x1)~(x5)を満たす発泡成形体を得ることができる、型内発泡成形時の蒸気圧(ゲージ圧)の幅がP1~P2である場合、「P1~P2」を「押出発泡粒子の成形可能幅」とし、P2-P1で得られる「値」を、「押出発泡粒子の成形幅」とする。なお、成形幅の測定における(x1)~(x5)の各項目の基準については、下記実施例にて詳述する。
本押出発泡粒子は、成形幅が広い(例えば0を超える)という利点を有する。本明細書において、押出発泡粒子の成形幅が大きいほど、当該押出発泡粒子は成形性に優れることを意図する。本明細書において、「押出発泡粒子の成形幅」とは、押出発泡粒子を型内発泡成形したとき、以下を満たす発泡成形体を得ることができる、型内発泡成形時の蒸気圧(ゲージ圧)の幅を意図する:(x1)押出発泡粒子同士の融着が十分であり、(x2)押出発泡粒子間の隙間が十分に埋まっており、(x3)表面が美麗であり、(x4)表面がメルトしておらず、かつ(x5)収縮することなく、型内発泡成形に使用した型(金型)の形状が転写されている、発泡成形体。本明細書において、上述した(x1)~(x5)を満たす発泡成形体を得ることができる、型内発泡成形時の蒸気圧(ゲージ圧)の幅がP1~P2である場合、「P1~P2」を「押出発泡粒子の成形可能幅」とし、P2-P1で得られる「値」を、「押出発泡粒子の成形幅」とする。なお、成形幅の測定における(x1)~(x5)の各項目の基準については、下記実施例にて詳述する。
本押出発泡粒子の成形幅は特に限定されない。本押出発泡粒子の成形幅は広いほど好ましい。本押出発泡粒子の成形幅は、0.02MPa以上であることがより好ましく、0.03MPa以上であることがより好ましく、0.04MPa以上であることがさらに好ましく、0.05MPa以上であることがよりさらに好ましく、0.06MPa以上であることが特に好ましい。
〔3.ポリプロピレン系樹脂押出発泡粒子の製造方法〕
本押出発泡粒子の製造方法としては、特に限定されず、公知の押出発泡方法を採用できる。本押出発泡粒子の製造方法の一態様としては、例えば、以下のような態様が挙げられる:(a)分岐状ポリプロピレン系樹脂(A)と、(b)酸変性ポリオレフィン系樹脂(B)と、(c)任意で(c-1)その他の樹脂またはゴム、(c-2)気泡核形成剤、(c-3)着色剤、および/または(c-4)その他成分と、(d)発泡剤と、を製造装置内で溶融混練する第一の工程、および第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含む、ポリプロピレン系樹脂押出発泡粒子の製造方法。
本押出発泡粒子の製造方法としては、特に限定されず、公知の押出発泡方法を採用できる。本押出発泡粒子の製造方法の一態様としては、例えば、以下のような態様が挙げられる:(a)分岐状ポリプロピレン系樹脂(A)と、(b)酸変性ポリオレフィン系樹脂(B)と、(c)任意で(c-1)その他の樹脂またはゴム、(c-2)気泡核形成剤、(c-3)着色剤、および/または(c-4)その他成分と、(d)発泡剤と、を製造装置内で溶融混練する第一の工程、および第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含む、ポリプロピレン系樹脂押出発泡粒子の製造方法。
本押出発泡粒子の製造方法の特に好ましい一態様(本発明の位置実施形態に係るポリプロピレン系樹脂押出発泡粒子の製造方法、ともいえる)としては、例えば、以下のような態様が挙げられる:
分岐状ポリプロピレン系樹脂(A)および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む樹脂組成物と発泡剤とを製造装置内で溶融混練する第一の工程、および前記第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含み、前記樹脂組成物は、当該樹脂組成物に含まれている前記分岐状ポリプロピレン系樹脂(A)と前記酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、前記分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および前記酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子の製造方法。
分岐状ポリプロピレン系樹脂(A)および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む樹脂組成物と発泡剤とを製造装置内で溶融混練する第一の工程、および前記第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含み、前記樹脂組成物は、当該樹脂組成物に含まれている前記分岐状ポリプロピレン系樹脂(A)と前記酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、前記分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および前記酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子の製造方法。
以下、本発明の位置実施形態に係るポリプロピレン系樹脂押出発泡粒子の製造方法に関する各態様(各工程)について説明するが、以下に詳説した事項以外は、適宜、〔2.ポリプロピレン系樹脂押出発泡粒子〕の記載を援用する。
(第一の工程)
第一の工程について、具体的に説明する。第一の工程の具体例としては、製造装置にて、(a)分岐状ポリプロピレン系樹脂(A)と、(b)酸変性ポリオレフィン系樹脂(B)と、(c)任意で(c-1)その他の樹脂またはゴム、(c-2)気泡核形成剤、(c-3)着色剤、および/または(c-4)その他成分とを含む樹脂組成物を溶融させて、当該樹脂組成物に発泡剤を溶解させる工程が挙げられる。溶融混練工程は、前記樹脂組成物と発泡剤とを含む溶融混練物を調製する工程ともいえる。
第一の工程について、具体的に説明する。第一の工程の具体例としては、製造装置にて、(a)分岐状ポリプロピレン系樹脂(A)と、(b)酸変性ポリオレフィン系樹脂(B)と、(c)任意で(c-1)その他の樹脂またはゴム、(c-2)気泡核形成剤、(c-3)着色剤、および/または(c-4)その他成分とを含む樹脂組成物を溶融させて、当該樹脂組成物に発泡剤を溶解させる工程が挙げられる。溶融混練工程は、前記樹脂組成物と発泡剤とを含む溶融混練物を調製する工程ともいえる。
(発泡剤)
製造方法において、使用する発泡剤は、特に限定されないが、少なくとも炭酸ガスを含むことが好ましい。炭酸ガスを使用することにより、前記製造方法は、生産コストが小さく、かつ環境負荷が小さいという利点を有する。
製造方法において、使用する発泡剤は、特に限定されないが、少なくとも炭酸ガスを含むことが好ましい。炭酸ガスを使用することにより、前記製造方法は、生産コストが小さく、かつ環境負荷が小さいという利点を有する。
本製造方法では、発泡剤として、炭酸ガス以外を用いても良い。発泡剤として使用し得る発泡剤としては、例えば、(a)物理系発泡剤(例えば、(a-1)プロパン、ノルマルブタン、イソブタン、ノルマルペンタン、イソペンタン、ヘキサン等の脂肪族炭化水素類;(a-2)シクロペンタン、シクロブタン等の脂環式炭化水素類;(a-3)ジメチルエーテル、ジエチルエーテル、メチルエチルエーテル等のエーテル類;(a-4)メタノール、エタノール等のアルコール類;(a-5)空気、窒素等の無機ガス;並びに(a-6)水など)、および、(b)重炭酸ナトリウム、アゾジカルボンアミド、ジニトロソペンタメチレンテトラミンなどの熱分解型発泡剤を含む化学系発泡剤、などが挙げられる。
押出発泡時の発泡性、および発泡剤取り扱いの簡便性の観点(利点)から、発泡剤は、炭酸ガス、ブタン(例えば、ノルマルブタンおよびイソブタン)および窒素からなる群から選択される1種以上を含むことが好ましく、当該群から選択される1種以上のみから構成されることが好ましい。
発泡剤の使用量は、特に限定されず、発泡剤の種類に応じて、および/または、目標とするポリプロピレン系樹脂押出発泡粒子の発泡倍率に応じて適宜調整すればよい。一例として、発泡剤の使用量は、分岐状ポリプロピレン系樹脂(A)および酸変性ポリオレフィン系樹脂(B)の合計の重量100.0重量部に対して、0.5重量部~7.0重量部であってもよく、0.5重量部~6.0重量部であってもよく、0.5重量部~5.0重量部であってもよく、0.5重量部~4.0重量部であってもよく、0.5重量部~3.0重量部であってもよい。
第一の工程において、必要に応じて、安定剤(例えば、酸化防止剤、金属不活性剤、燐系加工安定剤、紫外線吸収剤、紫外線安定剤、蛍光増白剤、金属石鹸、および制酸吸着剤など)および添加剤(例えば、気泡核形成剤、無機着色剤、有機着色剤、架橋剤、連鎖移動剤、滑剤、可塑剤、充填材、強化材、顔料、染料、難燃剤、含水材および帯電防止剤など)をさらに使用してもよい。
着色剤および/または安定剤等の添加剤は、マスターバッチとして配合(使用)されてもよい。着色剤および/または安定剤等の添加剤のマスターバッチは、着色剤および/または安定剤等の添加剤と任意の樹脂(例えばポリプロピレン系樹脂)とを、任意の比率で混合することで得られ得る。マスターバッチにおける着色剤および/または安定剤等の添加剤の濃度は特に限定されず、例えば、マスターバッチ100重量%中、5~50重量%の着色剤および/または安定剤等の添加剤を含むマスターバッチであってもよい。
第一の工程において、樹脂組成物および発泡剤は、製造装置に供給される前に混合されていてもよく、製造装置内で混合されてもよい。換言すれば、前記第一の工程において、樹脂組成物が製造装置に供給されてもよく、製造装置内で樹脂組成物が調製(完成)されてもよい。前記第一の工程において、(i)樹脂組成物および発泡剤、並びに、任意で使用され得るその他の成分を混合する方法および順序、または(ii)樹脂組成物および発泡剤、並びに、任意で使用され得るその他の成分を製造装置へ供給する方法および順序、は特に限定されない。
第一の工程で得られた溶融混練物を低圧領域に押出す前に、溶融混練物を冷却してもよい。
(第二の工程)
第二の工程は、第一の工程で得られた溶融混練物を、ダイを通して製造装置の内圧よりも低圧である領域に押出し、押し出された溶融混練物を細断する工程である。第二の工程により、押出発泡粒子が得られる。そのため、第二の工程は、ポリプロピレン系樹脂押出発泡粒子を造粒する造粒工程ともいえる。
第二の工程は、第一の工程で得られた溶融混練物を、ダイを通して製造装置の内圧よりも低圧である領域に押出し、押し出された溶融混練物を細断する工程である。第二の工程により、押出発泡粒子が得られる。そのため、第二の工程は、ポリプロピレン系樹脂押出発泡粒子を造粒する造粒工程ともいえる。
第二工程において、第一工程で得られた溶融混練物を押出す領域は、製造装置の内圧よりも低圧である限り特に限定されない。例えば、第二工程において、第一工程で得られた溶融混練物は、気相中に押出されてもよく、液相中に押出されてもよい。
第二工程において製造装置の内圧よりも低圧である領域に押出された溶融混練物は、直ちに発泡し始める。第二工程では、発泡中の溶融混練物を細断してもよく、発泡し終えた溶融混練物を細断してもよい。発泡中の溶融混練物を細断する場合、細断された溶融混練物は、押出された先の領域中で発泡を完了し得る。
第一工程で得られた溶融混練物を押出す領域および押出された溶融混練物の細断方法によって、第二工程(造粒工程)は、コールドカット法およびダイフェースカット法の2つに大別され得る。コールドカット法としては、ダイから押出された発泡剤を含有する溶融混練物を発泡させ、水槽の中を通して冷却しながらストランド状の発泡体を引き取った後に細断する方法(ストランドカット法)が挙げられる。ダイフェースカット法はダイの孔から押出された溶融混練物をダイの表面に接触しながら又は僅かに隙間を確保しながら回転するカッターで切断する方法である。
ダイフェースカット法は、さらに冷却方法の違いから次の3方式に分けられる。すなわち、アンダーウォータカット(以下、UWCと称する場合もある)法、ウォータリングカット(以下、WRCと称する場合もある)法、およびホットカット(以下、HCと称する場合もある)法である。UWC法は、ダイ先端に取り付けたチャンバー内に所定圧力に調整された冷却水をダイの樹脂吐出面に接するように充満し、ダイの孔から押出された溶融混練物を水中で切断する方法である。また、WRC法は、ダイに連結された冷却ドラムの内周面に沿って冷却水が流れる冷却ドラムをダイから下流側に配置し、空気中にて前記カッターで切断された溶融混練物が発泡しながら、もしくは発泡後に前記冷却水中で冷却される方法である。HC法は、空気中にて溶融混練物をカッターで切断し、切断された溶融混練物が発泡しながら、もしくは発泡後に、空気中にて冷却される方法である。前記HC法としては、水及び空気の混合ミストを噴霧する工程をさらに含むミストカット法も挙げられる。
〔4.ポリプロピレン系樹脂発泡成形体〕
本発明の一実施形態に係るポリプロピレン系樹脂発泡成形体は、〔2.ポリプロピレン系樹脂押出発泡粒子〕の項に記載のポリプロピレン系樹脂押出発泡粒子、または〔3.ポリプロピレン系樹脂押出発泡粒子の製造方法〕の項に記載の製造方法によって得られるポリプロピレン系樹脂押出発泡粒子を成形してなるものである。なお、本明細書において、「本発明の一実施形態に係るポリプロピレン系樹脂発泡成形体」を「本発泡成形体」と称する場合がある。
本発明の一実施形態に係るポリプロピレン系樹脂発泡成形体は、〔2.ポリプロピレン系樹脂押出発泡粒子〕の項に記載のポリプロピレン系樹脂押出発泡粒子、または〔3.ポリプロピレン系樹脂押出発泡粒子の製造方法〕の項に記載の製造方法によって得られるポリプロピレン系樹脂押出発泡粒子を成形してなるものである。なお、本明細書において、「本発明の一実施形態に係るポリプロピレン系樹脂発泡成形体」を「本発泡成形体」と称する場合がある。
本発泡成形体は、前述した構成を有するため、ポリウレタンフォームとの接着強度に優れるという利点を有する。その結果、本発泡成形体は、インサート材等の別部材を必要とすることなく、発泡成形体とポリウレタンフォームとの接着強度に優れる積層発泡体を提供できる。
なお、本発泡成形体は、発泡成形体とポリウレタンフォームとの積層発泡体の原料(発泡成形体)として特に好適に利用できる。本発泡成形体は、ポリウレタンフォームと一体成型するための発泡成形体として、特に好適に利用できる。換言すれば、本発泡成形体は、ポリウレタンフォームとの積層発泡体用であり得る。
本発泡成形体の製造方法、すなわち本押出発泡粒子の成形方法としては、特に限定されず、例えば金型を使用する公知の型内発泡成形方法を採用できる。
本押出発泡粒子からポリプロピレン系樹脂発泡成形体を成形する方法としては、例えば、(A)押出発泡粒子を無機ガスで加圧処理して押出発泡粒子内に無機ガスを含浸させて押出発泡粒子に所定の内圧を付与した後、当該押出発泡粒子を金型に充填し、蒸気等で押出発泡粒子同士を加熱融着させる方法(例えば、日本国特公昭51-22951号)、(B)押出発泡粒子をガス圧力で圧縮して金型に充填し、押出発泡粒子の回復力を利用して、蒸気等で押出発泡粒子同士を加熱融着させる方法(例えば、日本国特公昭53-33996号)、(C)間隙(クラッキング)を広げた金型に押出発泡粒子を充填した後、所定の間隙(クラッキング)まで金型を閉じて充填した押出発泡粒子を圧縮し、蒸気等で押出発泡粒子同士を加熱融着させる方法、等を利用し得る。以上のような方法によって、圧縮強度が高い発泡成形体を低い成形圧力で得ることができる。勿論、本発明は、これらの方法に限定されない。
(ポリプロピレン系樹脂発泡成形体の物性)
以下、発泡成形体の物性について説明する。
以下、発泡成形体の物性について説明する。
(密度)
本発泡成形体の密度は、特に限定されない。本発泡成形体の密度は、例えば、20g/L~300g/Lであることが好ましく、30g/L~200g/Lであることがより好ましく、40g/L~160g/Lであることがさらに好ましく、50g/L~150g/Lであることが特に好ましい。当該構成によれば、発泡成形体が自動車部材として要求される物性(圧縮強度等)を満足しやすいという利点を有する。
本発泡成形体の密度は、特に限定されない。本発泡成形体の密度は、例えば、20g/L~300g/Lであることが好ましく、30g/L~200g/Lであることがより好ましく、40g/L~160g/Lであることがさらに好ましく、50g/L~150g/Lであることが特に好ましい。当該構成によれば、発泡成形体が自動車部材として要求される物性(圧縮強度等)を満足しやすいという利点を有する。
本明細書において、ポリプロピレン系樹脂発泡成形体の密度は、以下の方法に従って測定して得られる値である。まず、発泡成形体の重量W(g)を測定する。次に、当該発泡成形体の縦、横および厚さをノギスで測定し、発泡成形体の体積V(cm3)を算出する。なお、複雑な形状の発泡成形体の場合は、水没法(水を満たした容器に発泡成形体を沈め、容器からあふれた水の重量を、測定時の気温における水の密度で換算し(除し)、発泡成形体発泡体の体積とする方法)にて体積V(cm3)を求めても良い。下記式に従って発泡成形体の密度(g/L)を求める:
発泡成形体の密度(g/L)=W/V×1000。
発泡成形体の密度(g/L)=W/V×1000。
(接着強度)
前述したように、本発泡成形体は、ポリウレタンフォームとの接着強度に優れる。本発泡成形体とポリウレタンフォームとの接着強度は、高いほど好ましく、例えば、60kPa以上が好ましく、65kPa以上がより好ましく、70kPa以上がより好ましく、75kPa以上がより好ましく、80kPa以上がより好ましく、100kPa以上がより好ましく、150kPa以上がさらに好ましく、200kPa以上が特に好ましい。
前述したように、本発泡成形体は、ポリウレタンフォームとの接着強度に優れる。本発泡成形体とポリウレタンフォームとの接着強度は、高いほど好ましく、例えば、60kPa以上が好ましく、65kPa以上がより好ましく、70kPa以上がより好ましく、75kPa以上がより好ましく、80kPa以上がより好ましく、100kPa以上がより好ましく、150kPa以上がさらに好ましく、200kPa以上が特に好ましい。
本明細書において、発泡成形体のポリウレタンフォームに対する接着強度(換言すれば、発泡成形体とポリウレタンフォームとの接着強度)は、JIS K 6850(1999)に準じて行われる剥離試験により、測定して得られる値とする。具体的に、前記接着強度は、剥離試験で得られる最大の応力(N)を接着面の面積(m2)で除して得られる値の単位をkPaに換算して得られる値である。
〔5.積層発泡体〕
本発明の一実施形態に係る積層発泡体は、〔3.ポリプロピレン系樹脂発泡成形体〕の項に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを積層してなる。本発明の一実施形態に係る積層発泡体は、〔3.ポリプロピレン系樹脂発泡成形体〕の項に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを含む、ともいえる。
本発明の一実施形態に係る積層発泡体は、〔3.ポリプロピレン系樹脂発泡成形体〕の項に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを積層してなる。本発明の一実施形態に係る積層発泡体は、〔3.ポリプロピレン系樹脂発泡成形体〕の項に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを含む、ともいえる。
本発明の一実施形態に係る積層発泡体は、前述の構成を有するため、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとの接着強度に優れるという利点を有する。
本発明の一実施形態に係る積層発泡体の製造方法としては特に限定されず、例えば、(d1)ポリプロピレン系樹脂発泡成形体を、隙間のある所定の金型内に入れ、ポリウレタンフォームの原料となる液体を隙間に流し込み、当該液体を発泡させることで、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを接着および積層させる方法、(d2)ポリプロピレン系樹脂発泡成形体にポリウレタンフォームの原料となる液体を塗布し、塗布した面に別のポリプロピレン系樹脂発泡成形体等を接着および積層させる方法、等が挙げられる。前記(d2)の方法では、ポリプロピレン系樹脂発泡成形体およびポリウレタンフォームが、ポリプロピレン系樹脂発泡成形体-ポリウレタンフォーム-ポリプロピレン系樹脂発泡成形体の順で積層してなる積層発泡体が得られる。
〔6.用途〕
本発明の一実施形態に係る押出発泡粒子、発泡成形体および積層発泡体の各々は、自動車内装部材、自動車バンパー用芯材、断熱材および緩衝包装材等、様々な用途に用いることができる。本発明の一実施形態に係る押出発泡粒子は、ポリウレタンフォームとの接着性に優れる発泡成形体を提供でき、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供できる。そのため、本発明の一実施形態に係る押出発泡粒子、発泡成形体および積層発泡体の各々は、特に、車両用シート、および、ヘッドレスト等の自動車内装部材に好適に利用できる。
本発明の一実施形態に係る押出発泡粒子、発泡成形体および積層発泡体の各々は、自動車内装部材、自動車バンパー用芯材、断熱材および緩衝包装材等、様々な用途に用いることができる。本発明の一実施形態に係る押出発泡粒子は、ポリウレタンフォームとの接着性に優れる発泡成形体を提供でき、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供できる。そのため、本発明の一実施形態に係る押出発泡粒子、発泡成形体および積層発泡体の各々は、特に、車両用シート、および、ヘッドレスト等の自動車内装部材に好適に利用できる。
本発明の一実施形態は、以下の様な構成であってもよい。
〔X1〕ポリプロピレン系樹脂押出発泡粒子であって、前記ポリプロピレン系樹脂押出発泡粒子の基材樹脂は、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、分岐構造を有するポリプロピレン系樹脂(A)を70重量%~97重量%、および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を3重量%~30重量%含有する、ポリプロピレン系樹脂押出発泡粒子。
〔X2〕前記酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、15000~65000である、〔X1〕に記載のポリプロピレン系樹脂押出発泡粒子。
〔X3〕前記酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に無水マレイン酸が導入されてなる樹脂である、〔X1〕または〔X2〕に記載のポリプロピレン系樹脂押出発泡粒子。
〔X4〕〔X1〕~〔X3〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子を成形してなる、ポリプロピレン系樹脂発泡成形体。
〔X5〕〔X4〕に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを積層してなる、積層発泡体。
本発明の一実施形態は、以下の様な構成であってもよい。
〔Y1〕ポリプロピレン系樹脂押出発泡粒子であって、前記ポリプロピレン系樹脂押出発泡粒子の基材樹脂は、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子。
〔Y2〕前記酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、15000~65000である、〔Y1〕に記載のポリプロピレン系樹脂押出発泡粒子。
〔Y3〕前記酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に無水マレイン酸が導入されてなる樹脂である、〔Y1〕または〔Y2〕に記載のポリプロピレン系樹脂押出発泡粒子。
〔Y4〕前記分岐状ポリプロピレン系樹脂(A)の融点は、125℃~170℃である、〔Y1〕~〔Y3〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y5〕前記分岐状ポリプロピレン系樹脂(A)のMFRは、0.5g/10分~20.0g/10分である、〔Y1〕~〔Y4〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y6〕前記分岐状ポリプロピレン系樹脂(A)のメルトテンションは、1.0cN~50.0cNである、〔Y1〕~〔Y5〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y7〕前記酸変性ポリオレフィン系樹脂(B)の酸価は、1.0mg/KOH~200.0mg/KOHである、〔Y1〕~〔Y6〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y8〕DSC曲線のピークが1つである、〔Y1〕~〔Y7〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y9〕前記ポリプロピレン系樹脂押出発泡粒子の連続気泡率は、10.0%以下である、〔Y1〕~〔Y8〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y10〕前記ポリプロピレン系樹脂押出発泡粒子は、表面に分散剤を含まないか、表面に0ppm超100ppm以下の分散剤を含む、〔Y1〕~〔Y9〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y11〕前記分岐状ポリプロピレン系樹脂(A)の主鎖は、プロピレン単独重合体、ポリプロピレン系ブロック共重合体およびポリプロピレン系ランダム共重合体からなる群から選択される1種以上である、〔Y1〕~〔Y10〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y12〕前記ポリプロピレン系樹脂押出発泡粒子の発泡倍率は、2倍~30倍である、〔Y1〕~〔Y11〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子。
〔Y13〕〔Y1〕~〔Y12〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子を成形してなる、ポリプロピレン系樹脂発泡成形体。
〔Y14〕前記ポリプロピレン系樹脂発泡成形体は、ポリウレタンフォームとの積層発泡体用である、〔Y13〕に記載のポリプロピレン系樹脂発泡成形体。
〔Y15〕前記ポリプロピレン系樹脂発泡成形体は、ポリウレタンフォームとの接着強度が60kPa以上である、〔Y13〕または〔Y14〕に記載のポリプロピレン系樹脂発泡成形体。
〔Y16〕〔Y13〕~〔Y15〕のいずれかに1つ記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを積層してなる、積層発泡体。
〔Y17〕分岐状ポリプロピレン系樹脂(A)および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む樹脂組成物と発泡剤とを製造装置内で溶融混練する第一の工程、および前記第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含み、前記樹脂組成物は、当該樹脂組成物に含まれている前記分岐状ポリプロピレン系樹脂(A)と前記酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、前記分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および前記酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y18〕前記酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、15000~65000である、〔Y17〕に記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y19〕前記酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に無水マレイン酸が導入されてなる樹脂である、〔Y17〕または〔Y18〕に記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y20〕前記分岐状ポリプロピレン系樹脂(A)の融点は、125℃~170℃である、〔Y17〕~〔Y19〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y21〕前記分岐状ポリプロピレン系樹脂(A)のMFRは、0.5g/10分~20.0g/10分である、〔Y17〕~〔Y20〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y22〕前記分岐状ポリプロピレン系樹脂(A)のメルトテンションは、1.0cN~50.0cNである、〔Y17〕~〔Y21〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y23〕前記酸変性ポリオレフィン系樹脂(B)の酸価は、1.0mg/KOH~200.0mg/KOHである、〔Y17〕~〔Y22〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y24〕DSC曲線のピークが1つである、〔Y17〕~〔Y23〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y25〕前記ポリプロピレン系樹脂押出発泡粒子の連続気泡率は、10.0%以下である、〔Y17〕~〔Y24〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
〔Y26〕前記発泡剤は、炭酸ガス、ブタンおよび窒素からなる群から選択される1種以上を含む、〔Y17〕~〔Y25〕のいずれか1つに記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
以下、実施例により本発明の一実施形態を更に詳細に説明するが、本発明は以下の実施例のみに限定されるものではない。
(試験方法)
実施例および比較例において、各種物性の測定および評価に用いられた試験方法は以下の通りである。
実施例および比較例において、各種物性の測定および評価に用いられた試験方法は以下の通りである。
[分岐状ポリプロピレン系樹脂の融点]
分岐状ポリプロピレン系樹脂の融点は、分岐状ポリプロピレン系樹脂を試料として、示差走査熱量計法により測定して求めた。示差走査熱量計としては、セイコーインスツルメンツ(株)製、DSC6200型を用いた。具体的な操作手順は、以下の通りであった:(1)試料(分岐状ポリプロピレン系樹脂)5~6mgの温度を10℃/分の昇温速度で40℃から220℃まで昇温することにより当該試料を融解させた;(2)その後、融解された試料の温度を10℃/分の降温速度で220℃から40℃まで降温することにより当該試料を結晶化させた;(3)その後、さらに、結晶化された試料の温度を10℃/分の昇温速度で40℃から220℃まで昇温した。2回目の昇温時(すなわち(3)のとき)に得られる当該試料(分岐状ポリプロピレン系樹脂)のDSC曲線のピーク(融解ピーク)の温度を当該試料(分岐状ポリプロピレン系樹脂)の融点とした。なお、以下の実施例および比較例で使用した分岐状ポリプロピレン系樹脂では、2回目の昇温時に得られるDSC曲線のピーク(融解ピーク)は1つであった。
分岐状ポリプロピレン系樹脂の融点は、分岐状ポリプロピレン系樹脂を試料として、示差走査熱量計法により測定して求めた。示差走査熱量計としては、セイコーインスツルメンツ(株)製、DSC6200型を用いた。具体的な操作手順は、以下の通りであった:(1)試料(分岐状ポリプロピレン系樹脂)5~6mgの温度を10℃/分の昇温速度で40℃から220℃まで昇温することにより当該試料を融解させた;(2)その後、融解された試料の温度を10℃/分の降温速度で220℃から40℃まで降温することにより当該試料を結晶化させた;(3)その後、さらに、結晶化された試料の温度を10℃/分の昇温速度で40℃から220℃まで昇温した。2回目の昇温時(すなわち(3)のとき)に得られる当該試料(分岐状ポリプロピレン系樹脂)のDSC曲線のピーク(融解ピーク)の温度を当該試料(分岐状ポリプロピレン系樹脂)の融点とした。なお、以下の実施例および比較例で使用した分岐状ポリプロピレン系樹脂では、2回目の昇温時に得られるDSC曲線のピーク(融解ピーク)は1つであった。
[分岐状ポリプロピレン系樹脂のMFR]
分岐状ポリプロピレン系樹脂のMFRは、当該分岐状ポリプロピレン系樹脂を試料として、ISO 1133(1997)記載のB法の規定に準拠し、メルトインデクサーS-01(東洋精機製作所製)を用い、温度230℃および荷重2.16kgの条件下で、測定して求めた。
分岐状ポリプロピレン系樹脂のMFRは、当該分岐状ポリプロピレン系樹脂を試料として、ISO 1133(1997)記載のB法の規定に準拠し、メルトインデクサーS-01(東洋精機製作所製)を用い、温度230℃および荷重2.16kgの条件下で、測定して求めた。
[分岐状ポリプロピレン系樹脂の溶融張力(メルトテンション)]
分岐状ポリプロピレン系樹脂の溶融張力は、キャピログラフ1D(日本 株式会社東洋精機製作所製)を用いて測定した。具体的には、(1)~(5)の通りであった:(1)試験温度(200℃)に加熱された径9.55mmのバレルに測定用の試料樹脂(分岐状ポリプロピレン系樹脂)を充填した;(2)次いで、試料樹脂を10分間、試験温度(200℃)に加熱されたバレル内で加熱した;(3)次いで、キャピラリーダイ(口径1.0mm、長さ10mm)から、一定に保持したピストン降下速度(10mm/分)にて、試料樹脂を紐状に出しながら、この紐状物を前記キャピラリーダイの下方350mmに位置する張力検出のプーリーに通過させた後、巻取りロールを用いる巻取りを開始した;(4)紐状物の引き取りが安定した後、紐状物の巻取り速度を初速1.0m/分から、4分間で200m/分の速度に達するまで一定の割合で増加させた;(5)紐状物が破断したときのロードセル付きプーリーにかかる荷重を溶融張力として測定した。
分岐状ポリプロピレン系樹脂の溶融張力は、キャピログラフ1D(日本 株式会社東洋精機製作所製)を用いて測定した。具体的には、(1)~(5)の通りであった:(1)試験温度(200℃)に加熱された径9.55mmのバレルに測定用の試料樹脂(分岐状ポリプロピレン系樹脂)を充填した;(2)次いで、試料樹脂を10分間、試験温度(200℃)に加熱されたバレル内で加熱した;(3)次いで、キャピラリーダイ(口径1.0mm、長さ10mm)から、一定に保持したピストン降下速度(10mm/分)にて、試料樹脂を紐状に出しながら、この紐状物を前記キャピラリーダイの下方350mmに位置する張力検出のプーリーに通過させた後、巻取りロールを用いる巻取りを開始した;(4)紐状物の引き取りが安定した後、紐状物の巻取り速度を初速1.0m/分から、4分間で200m/分の速度に達するまで一定の割合で増加させた;(5)紐状物が破断したときのロードセル付きプーリーにかかる荷重を溶融張力として測定した。
[酸変性ポリオレフィン系樹脂の重量平均分子量]
酸変性ポリオレフィン系樹脂(B)、並びにその他の樹脂である酸変性ポリエチレン系樹脂および酸変性ポリプロピレン系樹脂の重量平均分子量は、GPC法によって算出した。
酸変性ポリオレフィン系樹脂(B)、並びにその他の樹脂である酸変性ポリエチレン系樹脂および酸変性ポリプロピレン系樹脂の重量平均分子量は、GPC法によって算出した。
[押出発泡粒子の発泡倍率]
以下の方法によって、ポリプロピレン系樹脂押出発泡粒子の発泡倍率を算出した:(1)押出発泡粒子の重量w(g)を測定した;(2)次に、重量の測定に用いた押出発泡粒子を、メスシリンダー中に入っているエタノール中に沈め、メスシリンダーの液面位置の上昇分に基づき押出発泡粒子の体積v(cm3)を測定した;(3)重量w(g)を体積v(cm3)で除し、押出発泡粒子の密度ρ1(g/cm3)を算出した;(4)押出発泡粒子の基材樹脂の密度ρ2(g/cm3)を押出発泡粒子の密度ρ1で除し(ρ2/ρ1)、その値を発泡倍率(倍)とした。基材樹脂の密度ρ2としては、一般的なポリプロピレン系樹脂の密度0.9g/cm3を採用した。
以下の方法によって、ポリプロピレン系樹脂押出発泡粒子の発泡倍率を算出した:(1)押出発泡粒子の重量w(g)を測定した;(2)次に、重量の測定に用いた押出発泡粒子を、メスシリンダー中に入っているエタノール中に沈め、メスシリンダーの液面位置の上昇分に基づき押出発泡粒子の体積v(cm3)を測定した;(3)重量w(g)を体積v(cm3)で除し、押出発泡粒子の密度ρ1(g/cm3)を算出した;(4)押出発泡粒子の基材樹脂の密度ρ2(g/cm3)を押出発泡粒子の密度ρ1で除し(ρ2/ρ1)、その値を発泡倍率(倍)とした。基材樹脂の密度ρ2としては、一般的なポリプロピレン系樹脂の密度0.9g/cm3を採用した。
[押出発泡粒子のDSC曲線]
示差走査熱量計法を用いて、ポリプロピレン系樹脂押出発泡粒子の温度を40℃から220℃まで10℃/分の速度で昇温し、DSC曲線を得た。
示差走査熱量計法を用いて、ポリプロピレン系樹脂押出発泡粒子の温度を40℃から220℃まで10℃/分の速度で昇温し、DSC曲線を得た。
[嵩密度]
ポリプロピレン系樹脂押出発泡粒子またはポリプロピレン系樹脂発泡粒子の嵩密度は、以下の方法で測定した。まず、乾燥したポリプロピレン系樹脂発泡粒子を容積約10Lのバケツにあふれるまで入れ、その後バケツ表面でポリプロピレン系樹脂発泡粒子を擦切った。次いで、バケツ内のポリプロピレン系樹脂発泡粒子の重量を測定した。次の式にしたがって嵩密度を求めた:
嵩密度(g/L)=ポリプロピレン系樹脂発泡粒子の重量(g)/バケツの容量(L)。
ポリプロピレン系樹脂押出発泡粒子またはポリプロピレン系樹脂発泡粒子の嵩密度は、以下の方法で測定した。まず、乾燥したポリプロピレン系樹脂発泡粒子を容積約10Lのバケツにあふれるまで入れ、その後バケツ表面でポリプロピレン系樹脂発泡粒子を擦切った。次いで、バケツ内のポリプロピレン系樹脂発泡粒子の重量を測定した。次の式にしたがって嵩密度を求めた:
嵩密度(g/L)=ポリプロピレン系樹脂発泡粒子の重量(g)/バケツの容量(L)。
[連続気泡率]
押出発泡粒子の連続気泡率は、空気比較式比重計[東京サイエンス(株)製、モデル1000]を用いて、ASTM D2856-87の手順C(PROCEDURE C)に記載の方法に従って、測定して求めた。より具体的には、押出発泡粒子の連続気泡率は、以下(1)~(3)を順に実施して算出した:(1)空気比較式比重計を用いて押出発泡粒子の体積Vc(cm3)を測定した;(2)次いで、Vcを測定後の押出発泡粒子の全量を、メスシリンダーに入っているエタノール中に沈めた;(3)その後、メスシリンダーにおけるエタノールの位置の上昇量から、押出発泡粒子の見かけ上の体積Va(cm3)を求めた;(4)以下の式により、押出発泡粒子の連続気泡率を算出した:
連続気泡率(%)=((Va-Vc)×100)/Va。
押出発泡粒子の連続気泡率は、空気比較式比重計[東京サイエンス(株)製、モデル1000]を用いて、ASTM D2856-87の手順C(PROCEDURE C)に記載の方法に従って、測定して求めた。より具体的には、押出発泡粒子の連続気泡率は、以下(1)~(3)を順に実施して算出した:(1)空気比較式比重計を用いて押出発泡粒子の体積Vc(cm3)を測定した;(2)次いで、Vcを測定後の押出発泡粒子の全量を、メスシリンダーに入っているエタノール中に沈めた;(3)その後、メスシリンダーにおけるエタノールの位置の上昇量から、押出発泡粒子の見かけ上の体積Va(cm3)を求めた;(4)以下の式により、押出発泡粒子の連続気泡率を算出した:
連続気泡率(%)=((Va-Vc)×100)/Va。
[成形可能幅および成形幅]
まず、ブロック形状の金型(成形空間が、縦381mm×横320mm×厚さ可変)を、成形空間の厚さが78mmとなる状態(クラッキング率30%)とした。次いで、当該金型の成形空間内に、ポリプロピレン系樹脂押出発泡粒子を充填した。その後、金型内の成形空間の厚さが60mmとなるように金型を移動させ、成形空間を圧縮した。
続いて、0.10MPa(ゲージ圧)の水蒸気で金型内の空気を追い出した。その後、0.20~0.36MPa(ゲージ圧)の蒸気圧を示す水蒸気を用いて10秒間加熱し、型内発泡成形することにより、ポリプロピレン系樹脂発泡成形体を得た。
ここで、前記蒸気圧を0.02MPaずつ変化させながら、型内発泡成形を行った。このとき、以下(x1)~(x5)を満たすポリプロピレン系樹脂型内発泡成形体を得ることができる、型内発泡成形時の蒸気圧の幅を求め、得られた蒸気圧の幅をP1~P2とした:(x1)ポリプロピレン系樹脂押出発泡粒子同士の融着(融着率)が十分であり、(x2)ポリプロピレン系樹脂押出発泡粒子間の隙間が十分に埋まっており、(x3)表面が美麗であり、(x4)表面がメルトしておらず、かつ(x5)収縮することなく、型内発泡成形に使用した型(金型)の形状が転写されている、ポリプロピレン系樹脂型内発泡成形体。
まず、ブロック形状の金型(成形空間が、縦381mm×横320mm×厚さ可変)を、成形空間の厚さが78mmとなる状態(クラッキング率30%)とした。次いで、当該金型の成形空間内に、ポリプロピレン系樹脂押出発泡粒子を充填した。その後、金型内の成形空間の厚さが60mmとなるように金型を移動させ、成形空間を圧縮した。
続いて、0.10MPa(ゲージ圧)の水蒸気で金型内の空気を追い出した。その後、0.20~0.36MPa(ゲージ圧)の蒸気圧を示す水蒸気を用いて10秒間加熱し、型内発泡成形することにより、ポリプロピレン系樹脂発泡成形体を得た。
ここで、前記蒸気圧を0.02MPaずつ変化させながら、型内発泡成形を行った。このとき、以下(x1)~(x5)を満たすポリプロピレン系樹脂型内発泡成形体を得ることができる、型内発泡成形時の蒸気圧の幅を求め、得られた蒸気圧の幅をP1~P2とした:(x1)ポリプロピレン系樹脂押出発泡粒子同士の融着(融着率)が十分であり、(x2)ポリプロピレン系樹脂押出発泡粒子間の隙間が十分に埋まっており、(x3)表面が美麗であり、(x4)表面がメルトしておらず、かつ(x5)収縮することなく、型内発泡成形に使用した型(金型)の形状が転写されている、ポリプロピレン系樹脂型内発泡成形体。
ここで、前記(x1)の融着率、前記(x2)の粒子間の隙間の評価、前記(x3)の表面美麗性の評価、前記(x4)の表面メルト状態の評価、および前記(x5)の収縮率の評価については、以下の通り実施した。なお、(x1)~(x5)の評価は、上述の方法で得られたポリプロピレン系樹脂発泡成形体を75℃の乾燥室に16時間以上放置して乾燥させ、さらに、23℃にて24時間以上静置した後のポリプロピレン系樹脂発泡成形体を用いて実施した。
(融着率の評価)
ポリプロピレン系樹脂発泡成形体の一つの角から約100mmの場所から、反対側の約100mmの点までナイフ等で約5mmの切り込みを入れ、切り込みを入れた部分と一つの角で囲まれる部分をハンマー等で叩き、切り込み部を破断させた。破断面を目視し、破断面に存在する全押出発泡粒子数と、破断面において押出発泡粒子内で破断している押出発泡粒子の数とを、それぞれ数えた。破断面に存在する全押出発泡粒子数に対する、破断面において押出発泡粒子内で破断している押出発泡粒子の数の比率(%)を算出し、当該比率が60%以上のものを合格とし、60%未満のものを不合格とした。
ポリプロピレン系樹脂発泡成形体の一つの角から約100mmの場所から、反対側の約100mmの点までナイフ等で約5mmの切り込みを入れ、切り込みを入れた部分と一つの角で囲まれる部分をハンマー等で叩き、切り込み部を破断させた。破断面を目視し、破断面に存在する全押出発泡粒子数と、破断面において押出発泡粒子内で破断している押出発泡粒子の数とを、それぞれ数えた。破断面に存在する全押出発泡粒子数に対する、破断面において押出発泡粒子内で破断している押出発泡粒子の数の比率(%)を算出し、当該比率が60%以上のものを合格とし、60%未満のものを不合格とした。
(粒間の評価)
ポリプロピレン系樹脂発泡成形体の表面のうち、縦×横からなる両面(厚み方向に垂直な面)の中心付近、100mm×100mmにおいて、押出発泡粒子間を目視で観察し、押出発泡粒子間の隙間(表面に対してくぼんでいる部分)の数を数えた。押出発泡粒子間の隙間の数が20個以下であるものを合格とし、21個以上出るものを不合格とした。
ポリプロピレン系樹脂発泡成形体の表面のうち、縦×横からなる両面(厚み方向に垂直な面)の中心付近、100mm×100mmにおいて、押出発泡粒子間を目視で観察し、押出発泡粒子間の隙間(表面に対してくぼんでいる部分)の数を数えた。押出発泡粒子間の隙間の数が20個以下であるものを合格とし、21個以上出るものを不合格とした。
(表面美麗性の評価)
ポリプロピレン系樹脂発泡成形体について、端部のうち最も長い一片において、押出発泡粒子間を目視で観察し、押出発泡粒子間の隙間(表面に対してくぼんでいる部分)の数を数えた。押出発泡粒子間の隙間の数が3個以下のものを合格とし、4個以上のものを不合格とした。
ポリプロピレン系樹脂発泡成形体について、端部のうち最も長い一片において、押出発泡粒子間を目視で観察し、押出発泡粒子間の隙間(表面に対してくぼんでいる部分)の数を数えた。押出発泡粒子間の隙間の数が3個以下のものを合格とし、4個以上のものを不合格とした。
(表面メルト状態の評価)
ポリプロピレン系樹脂発泡成形体の表面メルト状態については、得られたポリプロピレン系樹脂発泡成形体を乾燥および静置することなく、成形直後のポリプロピレン系樹脂発泡成形体について評価した。具体的に、成形直後のポリプロピレン系樹脂発泡成形体において、(a)発泡成形体が金型に張り付く等で離型できない場合、または、(b)発泡成形体の金型からの離型は可能であっても、例えば発泡成形体表面の蒸気スリット部分等の、発泡成形体表面の一部が、金型側に残った場合、は不合格とし、それ以外は合格とした。
ポリプロピレン系樹脂発泡成形体の表面メルト状態については、得られたポリプロピレン系樹脂発泡成形体を乾燥および静置することなく、成形直後のポリプロピレン系樹脂発泡成形体について評価した。具体的に、成形直後のポリプロピレン系樹脂発泡成形体において、(a)発泡成形体が金型に張り付く等で離型できない場合、または、(b)発泡成形体の金型からの離型は可能であっても、例えば発泡成形体表面の蒸気スリット部分等の、発泡成形体表面の一部が、金型側に残った場合、は不合格とし、それ以外は合格とした。
(収縮率の評価)
ポリプロピレン系樹脂発泡成形体の縦、横および厚さの寸法を測定した。得られた結果を用いて、ポリプロピレン系樹脂発泡成形体の縦、横および厚さの寸法のそれぞれについて、以下の式にて収縮率(%)を評価した:
{(金型内の成形空間の寸法)-(成形体寸法)}×100/金型内の成形空間の寸法。縦、横および厚さそれぞれの収縮率が、全て5%以下である場合を合格とし、いずれか1つでも5%より大きい場合は不合格とした。なお、金型内の成形空間の各寸法は、縦381mm、横320mmおよび厚さ60mmであった。
ポリプロピレン系樹脂発泡成形体の縦、横および厚さの寸法を測定した。得られた結果を用いて、ポリプロピレン系樹脂発泡成形体の縦、横および厚さの寸法のそれぞれについて、以下の式にて収縮率(%)を評価した:
{(金型内の成形空間の寸法)-(成形体寸法)}×100/金型内の成形空間の寸法。縦、横および厚さそれぞれの収縮率が、全て5%以下である場合を合格とし、いずれか1つでも5%より大きい場合は不合格とした。なお、金型内の成形空間の各寸法は、縦381mm、横320mmおよび厚さ60mmであった。
蒸気圧の幅「P1~P2」を「押出発泡粒子の成形可能幅」とし、P2-P1で得られる「値」を、「押出発泡粒子の成形幅」とした。「押出発泡粒子の成形可能幅」を、表1および2の「成形可能幅」の欄に記載した。
[発泡成形体の密度]
発泡成形体の密度は、以下の方法に従って測定した。まず、発泡成形体の重量W(g)を測定した。次に、当該発泡成形体の縦、横および厚さをノギスで測定し、発泡成形体の体積V(cm3)を算出した。なお、複雑な形状の発泡成形体の場合は、水没法にて体積V(cm3)を求めても良い。下記式に従って発泡成形体の密度(g/L)を求めた:
発泡成形体の密度(g/L)=W/V×1000。
発泡成形体の密度は、以下の方法に従って測定した。まず、発泡成形体の重量W(g)を測定した。次に、当該発泡成形体の縦、横および厚さをノギスで測定し、発泡成形体の体積V(cm3)を算出した。なお、複雑な形状の発泡成形体の場合は、水没法にて体積V(cm3)を求めても良い。下記式に従って発泡成形体の密度(g/L)を求めた:
発泡成形体の密度(g/L)=W/V×1000。
[発泡成形体とポリウレタンフォームとの接着強度]
JIS K 6850(1999)に準じて剥離試験を行い、発泡成形体とポリウレタンフォームとの接着強度を測定した。剥離試験で得られる最大の応力(N)を接着面の面積(m2)で除して得られた値の単位をkPaに換算して得られる値を接着強度とした。
JIS K 6850(1999)に準じて剥離試験を行い、発泡成形体とポリウレタンフォームとの接着強度を測定した。剥離試験で得られる最大の応力(N)を接着面の面積(m2)で除して得られた値の単位をkPaに換算して得られる値を接着強度とした。
実施例および比較例で使用した原料は以下のとおりである。
<分岐状ポリプロピレン系樹脂(A)>
・A-1:下記製造例1により製造した分岐状ポリプロピレン系樹脂
・A-2:下記製造例2により製造した分岐状ポリプロピレン系樹脂
<線状ポリプロピレン系樹脂(X)>
・F-724NPC(プライムポリマー社製、ポリプロピレン系ランダム共重合体、融点:150℃、MFR:7g/10分)
<酸変性ポリオレフィン系樹脂(B)>
・B-1:ユーメックス1010(三洋化成工業社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量30000、酸価52mg/KOH)
・B-2:ハイワックスNP0555A(三井化学社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量18000、酸価61mg/KOH)
<その他の樹脂(Y)>
・Y-1:ハイワックス2203A(三井化学社製、無水マレイン酸変性ポリエチレン系樹脂、重量平均分子量2700、酸価30.0mg/KOH)
・Y-2:ハイワックス1105A(三井化学社製、無水マレイン酸変性ポリエチレン系樹脂、重量平均分子量1500、酸価60.0mg/KOH)
・Y-3:ユーメックス100TS(三洋化成工業社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量9000、酸価3.5mg/KOH)
<添加剤>
・タルク:林化成社製、タルカンパウダー(登録商標)PK-S。
・A-1:下記製造例1により製造した分岐状ポリプロピレン系樹脂
・A-2:下記製造例2により製造した分岐状ポリプロピレン系樹脂
<線状ポリプロピレン系樹脂(X)>
・F-724NPC(プライムポリマー社製、ポリプロピレン系ランダム共重合体、融点:150℃、MFR:7g/10分)
<酸変性ポリオレフィン系樹脂(B)>
・B-1:ユーメックス1010(三洋化成工業社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量30000、酸価52mg/KOH)
・B-2:ハイワックスNP0555A(三井化学社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量18000、酸価61mg/KOH)
<その他の樹脂(Y)>
・Y-1:ハイワックス2203A(三井化学社製、無水マレイン酸変性ポリエチレン系樹脂、重量平均分子量2700、酸価30.0mg/KOH)
・Y-2:ハイワックス1105A(三井化学社製、無水マレイン酸変性ポリエチレン系樹脂、重量平均分子量1500、酸価60.0mg/KOH)
・Y-3:ユーメックス100TS(三洋化成工業社製、無水マレイン酸変性ポリプロピレン系樹脂、重量平均分子量9000、酸価3.5mg/KOH)
<添加剤>
・タルク:林化成社製、タルカンパウダー(登録商標)PK-S。
(製造例1)分岐状ポリプロピレン系樹脂(A-1)の合成
線状ポリプロピレン系樹脂であるF-724NPC(プライムポリマー社製、ポリプロピレン系ランダム共重合体、融点:150℃、MFR:7g/10分)を二軸押出機に供給した。次に、当該線状ポリプロピレン系樹脂100重量部に対して、ラジカル重合開始剤としてt-ブチルパーオキシイソプロピルカーボネート(日油社製、パーブチル(登録商標)I)1.0重量部を前記二軸押出機に供給した。線状ポリプロピレン系樹脂およびラジカル重合開始剤を溶融混錬した後、線状ポリプロピレン系樹脂100重量部に対して、共役ジエン化合物としてイソプレンモノマー(クラレ社製)0.45重量部を前記二軸押出機に供給し、二軸押出機内で樹脂混合物を調製した。二軸押出機にイソプレンモノマーを供給した時点において、二軸押出機内で調製される樹脂混合物の単位時間当たりの量は、70kg/hであった。
線状ポリプロピレン系樹脂であるF-724NPC(プライムポリマー社製、ポリプロピレン系ランダム共重合体、融点:150℃、MFR:7g/10分)を二軸押出機に供給した。次に、当該線状ポリプロピレン系樹脂100重量部に対して、ラジカル重合開始剤としてt-ブチルパーオキシイソプロピルカーボネート(日油社製、パーブチル(登録商標)I)1.0重量部を前記二軸押出機に供給した。線状ポリプロピレン系樹脂およびラジカル重合開始剤を溶融混錬した後、線状ポリプロピレン系樹脂100重量部に対して、共役ジエン化合物としてイソプレンモノマー(クラレ社製)0.45重量部を前記二軸押出機に供給し、二軸押出機内で樹脂混合物を調製した。二軸押出機にイソプレンモノマーを供給した時点において、二軸押出機内で調製される樹脂混合物の単位時間当たりの量は、70kg/hであった。
次に、シリンダ温度200℃、およびスクリュー回転数230rpmで、調製された樹脂混合物を前記二軸押出機内でさらに溶融混練し、分岐状ポリプロピレン系樹脂を得た。得られた分岐状ポリプロピレン系樹脂を、吐出量70kg/hでダイからストランド状に吐出した。吐出された分岐状ポリプロピレン系樹脂(ストランド)を、(a)水冷し、その後(b)ペレット状(円柱状)に細断した。得られた分岐状ポリプロピレン系樹脂(A-1)の融点、MFRおよびメルトテンションを上述の方法により測定した。その結果、分岐状ポリプロピレン系樹脂(A-1)の融点は147℃であり、MFRは2g/10分であり、メルトテンションは10.8cNであった。
(製造例2)分岐状ポリプロピレン系樹脂(A-2)の合成
ラジカル重合開始剤(t-ブチルパーオキシイソプロピルカーボネート)の使用量を、線状ポリプロピレン系樹脂(F-724NPC)100重量部に対して1.4重量部とし、共役ジエン化合物(イソプレンモノマー)の使用量を線状ポリプロピレン系樹脂(F-724NPC)100重量部に対して0.60重量部としたこと以外は、(製造例1)と同じ方法によって、分岐状ポリプロピレン系樹脂(A-2)を得た。得られた分岐状ポリプロピレン系樹脂(A-2)の融点、MFRおよびメルトテンションを上述の方法により測定した。その結果、分岐状ポリプロピレン系樹脂(A-2)の融点は146℃であり、MFRは2g/10分であり、メルトテンションは11.3cNであった。
ラジカル重合開始剤(t-ブチルパーオキシイソプロピルカーボネート)の使用量を、線状ポリプロピレン系樹脂(F-724NPC)100重量部に対して1.4重量部とし、共役ジエン化合物(イソプレンモノマー)の使用量を線状ポリプロピレン系樹脂(F-724NPC)100重量部に対して0.60重量部としたこと以外は、(製造例1)と同じ方法によって、分岐状ポリプロピレン系樹脂(A-2)を得た。得られた分岐状ポリプロピレン系樹脂(A-2)の融点、MFRおよびメルトテンションを上述の方法により測定した。その結果、分岐状ポリプロピレン系樹脂(A-2)の融点は146℃であり、MFRは2g/10分であり、メルトテンションは11.3cNであった。
(実施例1)
(押出発泡粒子の製造)
(第一の工程)
押出発泡粒子の製造装置としては、軸径φ15mmの二軸押出機とメルトクーラーとダイバーターバルブとダイとが直列に連結された装置を使用した。表1に記載の樹脂および添加剤(気泡核形成剤)を、表1に記載の量にてブレンドし、樹脂組成物を調製した。次いで、当該樹脂組成物を二軸押出機に供給して、シリンダ温度200℃にて当該樹脂組成物を溶融混練した。さらに、押出機に設けた圧入部より、表1に示す量の発泡剤(炭酸ガス)を、定量ポンプを用いて供給し、得られた溶融混練物をさらに溶融混練した。
(押出発泡粒子の製造)
(第一の工程)
押出発泡粒子の製造装置としては、軸径φ15mmの二軸押出機とメルトクーラーとダイバーターバルブとダイとが直列に連結された装置を使用した。表1に記載の樹脂および添加剤(気泡核形成剤)を、表1に記載の量にてブレンドし、樹脂組成物を調製した。次いで、当該樹脂組成物を二軸押出機に供給して、シリンダ温度200℃にて当該樹脂組成物を溶融混練した。さらに、押出機に設けた圧入部より、表1に示す量の発泡剤(炭酸ガス)を、定量ポンプを用いて供給し、得られた溶融混練物をさらに溶融混練した。
(第二の工程)
得られた溶融混練物を、二軸押出機の先端に接続したメルトクーラーを通過させることにより、150℃~160℃に冷却した。その後、メルトクーラーの先端に装着されたダイより、製造装置の内圧よりも低圧である領域(大気圧下であり空気中)に溶融混練物を押出して、発泡させた。ダイ先端に取り付けた回転カッターにより、ダイを通過直後の溶融混練物を細断し、押出発泡粒子を得た。上述のように、第二の工程では、ホットカット法を採用した。得られた押出発泡粒子について、上述の方法で連続気泡率および嵩密度を測定し、成形可能幅を評価した。それらの結果を表1に示した。また、得られた押出発泡粒子について、上述した方法でDSC曲線を得たところ、DSC曲線のピークは1つであった。
得られた溶融混練物を、二軸押出機の先端に接続したメルトクーラーを通過させることにより、150℃~160℃に冷却した。その後、メルトクーラーの先端に装着されたダイより、製造装置の内圧よりも低圧である領域(大気圧下であり空気中)に溶融混練物を押出して、発泡させた。ダイ先端に取り付けた回転カッターにより、ダイを通過直後の溶融混練物を細断し、押出発泡粒子を得た。上述のように、第二の工程では、ホットカット法を採用した。得られた押出発泡粒子について、上述の方法で連続気泡率および嵩密度を測定し、成形可能幅を評価した。それらの結果を表1に示した。また、得られた押出発泡粒子について、上述した方法でDSC曲線を得たところ、DSC曲線のピークは1つであった。
(発泡成形体の製造)
得られたポリプロピレン系樹脂押出発泡粒子を使用して、以下の方法により、ポリプロピレン系樹脂発泡成形体を製造した。まず、ブロック形状の金型(成形空間が、縦381mm×横320mm×厚さ可変)を、成形空間の厚さが78mmとなる状態(クラッキング率30%)とした。次いで、当該金型の成形空間内に、ポリプロピレン系樹脂押出発泡粒子を充填した。その後、金型内の成形空間の厚さが60mmとなるように金型を移動させ、成形空間を圧縮した。続いて、0.10MPa(ゲージ圧)の水蒸気で金型内の空気を追い出した。その後、表1の「成形可能幅」の欄に記載の蒸気圧の水蒸気を用いて10秒間加熱および成形することにより、ポリプロピレン系樹脂発泡成形体を得た。得られたポリプロピレン系樹脂発泡成形体は、上述した(x1)~(x5)の評価が全て合格であった。
得られたポリプロピレン系樹脂押出発泡粒子を使用して、以下の方法により、ポリプロピレン系樹脂発泡成形体を製造した。まず、ブロック形状の金型(成形空間が、縦381mm×横320mm×厚さ可変)を、成形空間の厚さが78mmとなる状態(クラッキング率30%)とした。次いで、当該金型の成形空間内に、ポリプロピレン系樹脂押出発泡粒子を充填した。その後、金型内の成形空間の厚さが60mmとなるように金型を移動させ、成形空間を圧縮した。続いて、0.10MPa(ゲージ圧)の水蒸気で金型内の空気を追い出した。その後、表1の「成形可能幅」の欄に記載の蒸気圧の水蒸気を用いて10秒間加熱および成形することにより、ポリプロピレン系樹脂発泡成形体を得た。得られたポリプロピレン系樹脂発泡成形体は、上述した(x1)~(x5)の評価が全て合格であった。
0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体について、前記(融着率の評価)の項に記載の方法で融着率を評価した。結果を表1に示す。
(積層発泡体の製造)
積層発泡体の製造には、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を使用した。発泡成形体から、厚み5mm(片面スキン)の試験片を2枚切り出した。一方の試験片(試験片Aとする)のスキン面に、ポリウレタンフォーム形成用原料液(ヘンケルジャパン製、商品名:ロックタイト グリーンフォーム)を塗布した。次いで、得られるポリウレタンフォームの厚みが5mm、密度がおおよそ65g/Lになるように、もう一方の試験片(試験片Bとする)のスキン面を、試験片Aのスキン面上に、塗布されたポリウレタンフォーム形成用原料液に触れるように設置し、固定した。次に、ポリウレタンフォーム形成用原料液を23℃にて48時間、開放系にて発泡、固化させた。固化させた後、余分なポリウレタンフォームを除去し、接着面の寸法が25mm×12.5mmの積層発泡体を得た。得られた積層発泡体について、上述した方法により、発泡成形体とポリウレタンフォームとの接着強度を測定した。結果を表1に示す。
積層発泡体の製造には、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を使用した。発泡成形体から、厚み5mm(片面スキン)の試験片を2枚切り出した。一方の試験片(試験片Aとする)のスキン面に、ポリウレタンフォーム形成用原料液(ヘンケルジャパン製、商品名:ロックタイト グリーンフォーム)を塗布した。次いで、得られるポリウレタンフォームの厚みが5mm、密度がおおよそ65g/Lになるように、もう一方の試験片(試験片Bとする)のスキン面を、試験片Aのスキン面上に、塗布されたポリウレタンフォーム形成用原料液に触れるように設置し、固定した。次に、ポリウレタンフォーム形成用原料液を23℃にて48時間、開放系にて発泡、固化させた。固化させた後、余分なポリウレタンフォームを除去し、接着面の寸法が25mm×12.5mmの積層発泡体を得た。得られた積層発泡体について、上述した方法により、発泡成形体とポリウレタンフォームとの接着強度を測定した。結果を表1に示す。
(実施例2~8)
表1に記載の樹脂および添加剤を、表1に記載の量にてブレンドした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂押出発泡粒子を作製した。また、加熱蒸気圧を、表1の「成形可能幅」の欄に記載の蒸気圧とした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂発泡成形体を作製した。さらに、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を用いて、実施例1と同じ方法により、積層発泡体を作製した。得られた押出発泡粒子、発泡成形体および積層発泡体について各種評価した。また、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体について、前記(融着率の評価)の項に記載の方法で融着率を評価した。これらの結果を表1に示した。また、実施例2~8で得られた押出発泡粒子について、上述した方法でDSC曲線を得たところ、DSC曲線のピークは1つであった。
表1に記載の樹脂および添加剤を、表1に記載の量にてブレンドした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂押出発泡粒子を作製した。また、加熱蒸気圧を、表1の「成形可能幅」の欄に記載の蒸気圧とした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂発泡成形体を作製した。さらに、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を用いて、実施例1と同じ方法により、積層発泡体を作製した。得られた押出発泡粒子、発泡成形体および積層発泡体について各種評価した。また、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体について、前記(融着率の評価)の項に記載の方法で融着率を評価した。これらの結果を表1に示した。また、実施例2~8で得られた押出発泡粒子について、上述した方法でDSC曲線を得たところ、DSC曲線のピークは1つであった。
(比較例1~9)
表2に記載の樹脂および添加剤を、表2に記載の量にてブレンドした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂押出発泡粒子を得た。また、加熱蒸気圧を、表2の「成形可能幅」の欄に記載の蒸気圧とした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂発泡成形体を作製した。さらに、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を用いて、実施例1と同じ方法により、積層発泡体を作製した。得られた押出発泡粒子、発泡成形体および積層発泡体について各種評価した。また、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体について、前記(融着率の評価)の項に記載の方法で融着率を評価した。これらの結果を表2に示した。なお、比較例9の押出発泡粒子を用いて発泡成形体の成形を試みたが、上述した(x1)~(x5)の基準を満たす発泡成形体を提供可能な成形可能幅が存在しなかった(表2では「NG」と表記)。すなわち、比較例9については、発泡成形体および積層発泡体を製造できなかった。

表2に記載の樹脂および添加剤を、表2に記載の量にてブレンドした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂押出発泡粒子を得た。また、加熱蒸気圧を、表2の「成形可能幅」の欄に記載の蒸気圧とした以外は、実施例1と同じ方法によって、ポリプロピレン系樹脂発泡成形体を作製した。さらに、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体を用いて、実施例1と同じ方法により、積層発泡体を作製した。得られた押出発泡粒子、発泡成形体および積層発泡体について各種評価した。また、0.26MPaの蒸気圧の水蒸気を用いて得られた発泡成形体について、前記(融着率の評価)の項に記載の方法で融着率を評価した。これらの結果を表2に示した。なお、比較例9の押出発泡粒子を用いて発泡成形体の成形を試みたが、上述した(x1)~(x5)の基準を満たす発泡成形体を提供可能な成形可能幅が存在しなかった(表2では「NG」と表記)。すなわち、比較例9については、発泡成形体および積層発泡体を製造できなかった。
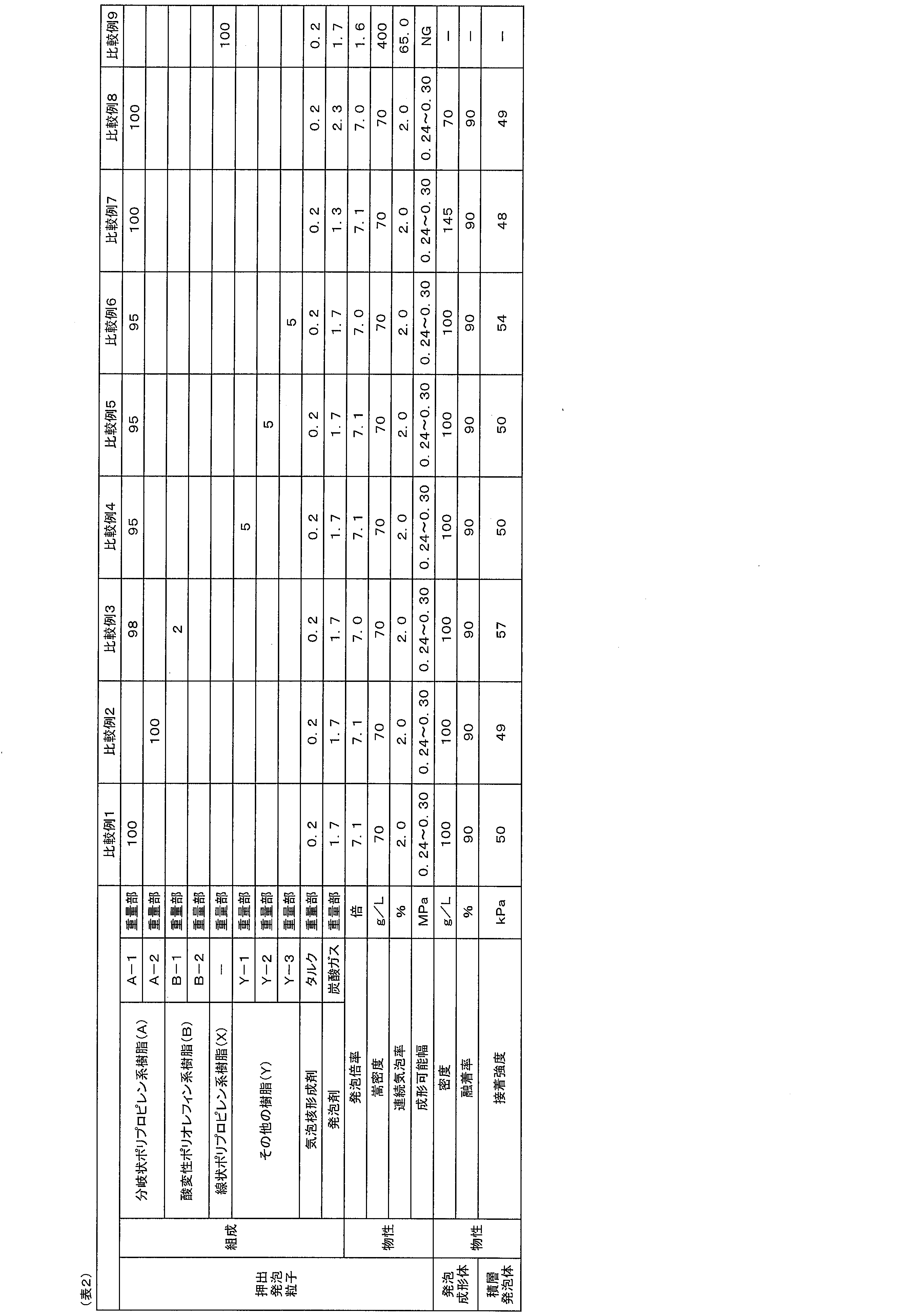
(参考例1~4)
[除圧発泡法によるポリプロピレン系樹脂発泡粒子の作製]
ポリオレフィン系樹脂(E228、プライムポリマー社製)97重量部、変性ポリオレフィン系樹脂(ユーメックス1010、三洋化成工業社製)3重量部、グリセリン0.05重量部、およびタルク0.02重量部をスーパーミキサーでブレンドした。得られたブレンド物を58mm二軸押出機に投入して溶融(200℃)および混錬した。次いで、得られた溶融混練物を、直径2.2mmの穴を複数個有するダイプレートを通してストランド状に押し出した。押し出された溶融混練物(ストランド)を水槽に通して冷却後、当該ストランドをペレタイザーにより所定の粒重量の円柱状ペレットにカットした。得られたペレット100重量部に対して水200重量部、分散剤として第三リン酸カルシウム(太平化学産業社製)を表3に記載の量、界面活性剤としてアルキルスルホン酸ソーダ(ラテムルPS、花王社製)を表3に記載の量、および炭酸ガス5重量部をオートクレーブ型耐圧容器に仕込んだ。続いて、耐圧容器内の原料を撹拌しながら、耐圧容器内の温度を150.5℃(発泡温度)まで昇温し、当該発泡温度で所定の時間保持した。その後、炭酸ガスを耐圧容器内に圧入して耐圧容器内の圧力を2.5MPa(発泡圧力)まで昇圧した。耐圧容器内部を前記発泡温度および前記発泡圧力で30分間保持した。その後、炭酸ガスを耐圧容器内に圧入することで耐圧容器内の圧力を前記発泡圧力に保持しながら、直径4mmの穴を有するオリフィス板を通して、耐圧容器内の混合物(分散液)を大気圧下に放出し、エチレン-プロピレンランダム共重合体発泡粒子を得た。得られたエチレン-プロピレンランダム共重合体発泡粒子は水洗後、60℃のオーブンで乾燥し、後述の評価および成形に供した。
[除圧発泡法によるポリプロピレン系樹脂発泡粒子の作製]
ポリオレフィン系樹脂(E228、プライムポリマー社製)97重量部、変性ポリオレフィン系樹脂(ユーメックス1010、三洋化成工業社製)3重量部、グリセリン0.05重量部、およびタルク0.02重量部をスーパーミキサーでブレンドした。得られたブレンド物を58mm二軸押出機に投入して溶融(200℃)および混錬した。次いで、得られた溶融混練物を、直径2.2mmの穴を複数個有するダイプレートを通してストランド状に押し出した。押し出された溶融混練物(ストランド)を水槽に通して冷却後、当該ストランドをペレタイザーにより所定の粒重量の円柱状ペレットにカットした。得られたペレット100重量部に対して水200重量部、分散剤として第三リン酸カルシウム(太平化学産業社製)を表3に記載の量、界面活性剤としてアルキルスルホン酸ソーダ(ラテムルPS、花王社製)を表3に記載の量、および炭酸ガス5重量部をオートクレーブ型耐圧容器に仕込んだ。続いて、耐圧容器内の原料を撹拌しながら、耐圧容器内の温度を150.5℃(発泡温度)まで昇温し、当該発泡温度で所定の時間保持した。その後、炭酸ガスを耐圧容器内に圧入して耐圧容器内の圧力を2.5MPa(発泡圧力)まで昇圧した。耐圧容器内部を前記発泡温度および前記発泡圧力で30分間保持した。その後、炭酸ガスを耐圧容器内に圧入することで耐圧容器内の圧力を前記発泡圧力に保持しながら、直径4mmの穴を有するオリフィス板を通して、耐圧容器内の混合物(分散液)を大気圧下に放出し、エチレン-プロピレンランダム共重合体発泡粒子を得た。得られたエチレン-プロピレンランダム共重合体発泡粒子は水洗後、60℃のオーブンで乾燥し、後述の評価および成形に供した。
ポリプロピレン系樹脂発泡粒子の製造過程(除圧発泡過程)における分散安定性を後述の方法で評価した。また、得られたポリプロピレン系樹脂発泡粒子について、(a)発泡倍率、および(b)ポリプロピレン系樹脂発泡粒子の表面に付着している分散剤の量(付着分散剤量)をそれぞれ後述の方法で測定した。得られたポリプロピレン系樹脂発泡粒子の嵩密度を、前記[嵩密度]の項に記載の方法で評価した。
(分散安定性)
〇(良好):製造過程(除圧発泡過程)において、耐圧容器内で攪拌している混合物(分散液)中で樹脂粒子の互着および塊化が確認されず、発泡可能である。また、得られた発泡粒子が融着していない。
△(可):製造過程(除圧発泡過程)において、耐圧容器内で攪拌している混合物(分散液)中で樹脂粒子の互着および塊化が確認されず、発泡可能である。しかし、得られた発泡粒子中に、融着しているものおよび発泡粒子表面にキズが認められるものが存在する。
×(不可):製造過程(除圧発泡過程)において、耐圧容器内の樹脂粒子が互着、塊化し、発泡不能である。
〇(良好):製造過程(除圧発泡過程)において、耐圧容器内で攪拌している混合物(分散液)中で樹脂粒子の互着および塊化が確認されず、発泡可能である。また、得られた発泡粒子が融着していない。
△(可):製造過程(除圧発泡過程)において、耐圧容器内で攪拌している混合物(分散液)中で樹脂粒子の互着および塊化が確認されず、発泡可能である。しかし、得られた発泡粒子中に、融着しているものおよび発泡粒子表面にキズが認められるものが存在する。
×(不可):製造過程(除圧発泡過程)において、耐圧容器内の樹脂粒子が互着、塊化し、発泡不能である。
(除圧発泡法で得られた発泡粒子の発泡倍率)
発泡粒子の発泡倍率の測定方法は、以下の(1)~(4)の通りであった:(1)発泡粒子の重量w(g)を測定した;(2)次に、重量の測定に用いた発泡粒子を、メスシリンダー中に入っているエタノール中に沈め、メスシリンダーの液面位置の上昇分に基づき当該発泡粒子の体積v(cm3)を測定した;(3)重量w(g)を体積v(cm3)で除し、発泡粒子の密度ρ1(g/cm3)を算出した;(4)発泡粒子の製造に用いた樹脂粒子の密度ρ2(g/cm3)を発泡粒子の密度ρ1で除し(ρ2/ρ1)て得られた値を発泡粒子の発泡倍率(倍)とした。
発泡粒子の発泡倍率の測定方法は、以下の(1)~(4)の通りであった:(1)発泡粒子の重量w(g)を測定した;(2)次に、重量の測定に用いた発泡粒子を、メスシリンダー中に入っているエタノール中に沈め、メスシリンダーの液面位置の上昇分に基づき当該発泡粒子の体積v(cm3)を測定した;(3)重量w(g)を体積v(cm3)で除し、発泡粒子の密度ρ1(g/cm3)を算出した;(4)発泡粒子の製造に用いた樹脂粒子の密度ρ2(g/cm3)を発泡粒子の密度ρ1で除し(ρ2/ρ1)て得られた値を発泡粒子の発泡倍率(倍)とした。
(付着分散剤量)
得られたポリプロピレン系樹脂発泡粒子の表面に残存している分散剤を、付着分散剤と称する。当該付着分散剤の量は、灰分測定により算出した。発泡粒子の灰分から発泡に使用したペレット(樹脂粒子)の灰分を引いたものを、付着分散剤量とした。発泡粒子または樹脂粒子それぞれの灰分は、以下の方法で測定した。重量W0(g)を測定したるつぼにサンプル(発泡粒子または樹脂粒子)を入れ、重量W1(g)を測定した。ついで、サンプルを含むるつぼを750℃にて1時間燃焼させたのち、デシケーター内で1時間静置した。ついで、るつぼの重量W2(g)を測定し、以下の式で灰分を算出した。
得られたポリプロピレン系樹脂発泡粒子の表面に残存している分散剤を、付着分散剤と称する。当該付着分散剤の量は、灰分測定により算出した。発泡粒子の灰分から発泡に使用したペレット(樹脂粒子)の灰分を引いたものを、付着分散剤量とした。発泡粒子または樹脂粒子それぞれの灰分は、以下の方法で測定した。重量W0(g)を測定したるつぼにサンプル(発泡粒子または樹脂粒子)を入れ、重量W1(g)を測定した。ついで、サンプルを含むるつぼを750℃にて1時間燃焼させたのち、デシケーター内で1時間静置した。ついで、るつぼの重量W2(g)を測定し、以下の式で灰分を算出した。
灰分(ppm)=(W2-W0)×1000000/(W1-W0)。
除圧発泡法により得られたポリプロピレン系樹脂発泡粒子を用いて、以下の方法によりポリプロピレン系樹脂発泡成形体を製造した。まず、ブロック形状の金型(成形空間が、縦381mm×横320mm×厚さ可変)を、成形空間の厚さが78mmとなる状態(クラッキング率30%)とした。次いで、当該金型の成形空間内に、ポリプロピレン系樹脂発泡粒子を充填した。その後、金型内の成形空間の厚さが60mmとなるように金型を移動させ、成形空間を圧縮した。続いて、0.10MPa(ゲージ圧)の水蒸気で金型内の空気を追い出した。その後、0.26MPaの蒸気圧の水蒸気を用いて10秒間加熱および成形することにより、ポリプロピレン系樹脂発泡成形体を得た。
得られたポリプロピレン系樹脂発泡成形体の融着率を、前記(融着率の評価)の項に記載の方法で評価した。

本発明の一実施形態によると、ポリプロピレン系樹脂発泡成形体とポリウレタンフォームとを含む積層発泡体を生産性よく提供できる。そのため、本発明の一実施形態は、車両用シート、および、ヘッドレスト等の自動車内装部材の分野において、特に好適に利用することができる。
Claims (15)
- ポリプロピレン系樹脂押出発泡粒子であって、
前記ポリプロピレン系樹脂押出発泡粒子の基材樹脂は、分岐状ポリプロピレン系樹脂(A)と酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、
分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および
重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子。 - 前記酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、15000~65000である、請求項1に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に無水マレイン酸が導入されてなる樹脂である、請求項1または2に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記分岐状ポリプロピレン系樹脂(A)の融点は、125℃~170℃である、請求項1~3のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記分岐状ポリプロピレン系樹脂(A)のMFRは、0.5g/10分~20.0g/10分である、請求項1~4のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記分岐状ポリプロピレン系樹脂(A)のメルトテンションは、1.0cN~50.0cNである、請求項1~5のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記酸変性ポリオレフィン系樹脂(B)の酸価は、1.0mg/KOH~200.0mg/KOHである、請求項1~6のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 1回目の昇温時のDSC曲線のピークが1つである、請求項1~7のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 前記ポリプロピレン系樹脂押出発泡粒子の連続気泡率は、10.0%以下である、請求項1~8のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子。
- 請求項1~9のいずれか1項に記載のポリプロピレン系樹脂押出発泡粒子を成形してなる、ポリプロピレン系樹脂発泡成形体。
- 前記ポリプロピレン系樹脂発泡成形体は、ポリウレタンフォームとの積層発泡体用である、請求項10に記載のポリプロピレン系樹脂発泡成形体。
- 請求項10または11に記載のポリプロピレン系樹脂発泡成形体と、ポリウレタンフォームとを積層してなる、積層発泡体。
- 分岐状ポリプロピレン系樹脂(A)および重量平均分子量が10000~100000である酸変性ポリオレフィン系樹脂(B)を含む樹脂組成物と発泡剤とを製造装置内で溶融混練する第一の工程、および
前記第一の工程で得られた溶融混練物を、ダイを通して前記製造装置の内圧よりも低圧である領域に吐出する第二の工程、を含み、
前記樹脂組成物は、当該樹脂組成物に含まれている前記分岐状ポリプロピレン系樹脂(A)と前記酸変性ポリオレフィン系樹脂(B)との合計を100重量%とした場合、前記分岐構造を有するポリプロピレン系樹脂(A)を70.0重量%~97.5重量%、および前記酸変性ポリオレフィン系樹脂(B)を2.5重量%~30.0重量%含有する、ポリプロピレン系樹脂押出発泡粒子の製造方法。 - 前記酸変性ポリオレフィン系樹脂(B)の重量平均分子量は、15000~65000である、請求項13に記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
- 前記酸変性ポリオレフィン系樹脂(B)は、ポリオレフィン系樹脂に無水マレイン酸が導入されてなる樹脂である、請求項13または14に記載のポリプロピレン系樹脂押出発泡粒子の製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023551451A JPWO2023054223A1 (ja) | 2021-09-29 | 2022-09-26 | |
| EP22876090.6A EP4410877A4 (en) | 2021-09-29 | 2022-09-26 | EXTRUDED FOAM PARTICLES OF POLYPROPYLENE-BASED RESIN, MOLDED BODY OF POLYPROPYLENE-BASED RESIN FOAM, AND LAMINATED FOAM BODY |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021159952 | 2021-09-29 | ||
| JP2021-159952 | 2021-09-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023054223A1 true WO2023054223A1 (ja) | 2023-04-06 |
Family
ID=85782596
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/035573 Ceased WO2023054223A1 (ja) | 2021-09-29 | 2022-09-26 | ポリプロピレン系樹脂押出発泡粒子、ポリプロピレン系樹脂発泡成形体および積層発泡体 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP4410877A4 (ja) |
| JP (1) | JPWO2023054223A1 (ja) |
| WO (1) | WO2023054223A1 (ja) |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5122951A (ja) | 1974-08-16 | 1976-02-24 | Yoshio Ihara | Eaaenjin |
| JPS5333996A (en) | 1976-09-10 | 1978-03-30 | Central Glass Co Ltd | Production of ultrafine grains of silica |
| JPH01249312A (ja) | 1988-03-30 | 1989-10-04 | Araco Corp | 異硬度パッドのインサート成形法 |
| JPH09249763A (ja) * | 1996-03-15 | 1997-09-22 | Sekisui Plastics Co Ltd | ポリプロピレン系樹脂発泡粒子 |
| JP2002542360A (ja) | 1999-04-19 | 2002-12-10 | バセル テクノロジー カンパニー ベスローテン フェンノートシャップ | 高い溶融強度を持つ軟質プロピレンポリマーブレンド |
| JP2008512502A (ja) * | 2004-09-10 | 2008-04-24 | 株式会社ジェイエスピー | 誘電体成形用ポリプロピレン系樹脂発泡粒子及びそのポリプロピレン系樹脂発泡粒子により成形された誘電体レンズ部材 |
| JP2011099101A (ja) | 2009-11-06 | 2011-05-19 | Kaneka Corp | 空隙を有する発泡成形体の製造方法 |
| JP2012171104A (ja) | 2011-02-17 | 2012-09-10 | Sekisui Plastics Co Ltd | 積層体の製造方法及び積層体 |
| JP2015113403A (ja) * | 2013-12-11 | 2015-06-22 | 株式会社ジェイエスピー | ポリオレフィン系樹脂発泡粒子 |
| WO2020004429A1 (ja) | 2018-06-28 | 2020-01-02 | 株式会社カネカ | 改質ポリプロピレン樹脂およびその製造方法、並びに、当該改質ポリプロピレン樹脂を用いた押出発泡粒子およびその製造方法 |
| WO2021100645A1 (ja) * | 2019-11-21 | 2021-05-27 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 |
| WO2021172016A1 (ja) * | 2020-02-25 | 2021-09-02 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55146230U (ja) * | 1979-04-10 | 1980-10-21 | ||
| JP2002166511A (ja) * | 2000-11-30 | 2002-06-11 | Sumitomo Chem Co Ltd | ポリオレフィン系樹脂発泡シート |
| CN1908053B (zh) * | 2006-08-14 | 2012-07-18 | 华东理工大学 | 超临界二氧化碳技术制备含硅聚丙烯纳米发泡材料的方法 |
| KR20130041091A (ko) * | 2010-06-11 | 2013-04-24 | 다이니폰 인사츠 가부시키가이샤 | 바닥용 화장재 |
-
2022
- 2022-09-26 WO PCT/JP2022/035573 patent/WO2023054223A1/ja not_active Ceased
- 2022-09-26 JP JP2023551451A patent/JPWO2023054223A1/ja active Pending
- 2022-09-26 EP EP22876090.6A patent/EP4410877A4/en active Pending
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5122951A (ja) | 1974-08-16 | 1976-02-24 | Yoshio Ihara | Eaaenjin |
| JPS5333996A (en) | 1976-09-10 | 1978-03-30 | Central Glass Co Ltd | Production of ultrafine grains of silica |
| JPH01249312A (ja) | 1988-03-30 | 1989-10-04 | Araco Corp | 異硬度パッドのインサート成形法 |
| JPH09249763A (ja) * | 1996-03-15 | 1997-09-22 | Sekisui Plastics Co Ltd | ポリプロピレン系樹脂発泡粒子 |
| JP2002542360A (ja) | 1999-04-19 | 2002-12-10 | バセル テクノロジー カンパニー ベスローテン フェンノートシャップ | 高い溶融強度を持つ軟質プロピレンポリマーブレンド |
| JP2008512502A (ja) * | 2004-09-10 | 2008-04-24 | 株式会社ジェイエスピー | 誘電体成形用ポリプロピレン系樹脂発泡粒子及びそのポリプロピレン系樹脂発泡粒子により成形された誘電体レンズ部材 |
| JP2011099101A (ja) | 2009-11-06 | 2011-05-19 | Kaneka Corp | 空隙を有する発泡成形体の製造方法 |
| JP2012171104A (ja) | 2011-02-17 | 2012-09-10 | Sekisui Plastics Co Ltd | 積層体の製造方法及び積層体 |
| JP2015113403A (ja) * | 2013-12-11 | 2015-06-22 | 株式会社ジェイエスピー | ポリオレフィン系樹脂発泡粒子 |
| WO2020004429A1 (ja) | 2018-06-28 | 2020-01-02 | 株式会社カネカ | 改質ポリプロピレン樹脂およびその製造方法、並びに、当該改質ポリプロピレン樹脂を用いた押出発泡粒子およびその製造方法 |
| WO2021100645A1 (ja) * | 2019-11-21 | 2021-05-27 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 |
| WO2021172016A1 (ja) * | 2020-02-25 | 2021-09-02 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4410877A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4410877A1 (en) | 2024-08-07 |
| EP4410877A4 (en) | 2025-08-13 |
| JPWO2023054223A1 (ja) | 2023-04-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2072207B1 (en) | Expanded polypropylene resin beads and foamed molded article thereof | |
| JP7660518B2 (ja) | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 | |
| TWI391435B (zh) | 含碳之改質聚苯乙烯系樹脂粒子、發泡性之含碳之改質聚苯乙烯系樹脂粒子、含碳之改質聚苯乙烯系樹脂發泡粒子、含碳之改質聚苯乙烯系樹脂發泡成形體,以及此等之製造方法 | |
| WO2018016399A1 (ja) | ポリプロピレン系樹脂予備発泡粒子および該予備発泡粒子の製造方法 | |
| WO2016060162A1 (ja) | ポリプロピレン系樹脂発泡粒子、ポリプロピレン系樹脂型内発泡成形体およびその製造方法 | |
| WO2022203036A1 (ja) | ポリプロピレン系樹脂押出発泡粒子およびその製造方法、並びに発泡成形体 | |
| JP2022152955A (ja) | ポリプロピレン系樹脂押出発泡粒子の製造方法 | |
| JP7717047B2 (ja) | ポリプロピレン系樹脂発泡粒子、その製造方法及びポリプロピレン系樹脂発泡成形体 | |
| JP7833419B2 (ja) | 分岐構造を有するポリプロピレン系樹脂の製造方法、押出発泡粒子の製造方法、および、発泡成形体の製造方法 | |
| WO2017169568A1 (ja) | ポリエチレン系樹脂発泡粒子の製造方法、及びポリエチレン系樹脂型内発泡成形体の製造方法 | |
| JP5503123B2 (ja) | スチレン改質ポリオレフィン系樹脂粒子、発泡性樹脂粒子、予備発泡粒子及び発泡成形体 | |
| WO2022154070A1 (ja) | ポリプロピレン系樹脂押出発泡粒子およびその製造方法、並びに発泡成形体 | |
| JP7832171B2 (ja) | 押出発泡用ポリプロピレン系樹脂組成物、押出発泡粒子および発泡成形体 | |
| JP7809692B2 (ja) | ポリプロピレン系樹脂押出発泡粒子 | |
| WO2023054223A1 (ja) | ポリプロピレン系樹脂押出発泡粒子、ポリプロピレン系樹脂発泡成形体および積層発泡体 | |
| WO2022210647A1 (ja) | ポリプロピレン系樹脂押出発泡粒子の製造方法 | |
| JP2025101258A (ja) | ポリプロピレン系樹脂押出発泡粒子、押出発泡粒子成形体、及びポリプロピレン系樹脂押出発泡粒子の製造方法 | |
| WO2025187549A1 (ja) | ポリプロピレン系樹脂押出発泡粒子、その製造方法、および発泡成形体 | |
| WO2022210648A1 (ja) | ポリプロピレン系樹脂押出発泡粒子の製造方法 | |
| JP2025107918A (ja) | ポリプロピレン系樹脂押出発泡粒子、押出発泡粒子成形体、及びポリプロピレン系樹脂押出発泡粒子の製造方法 | |
| WO2022210645A1 (ja) | ポリプロピレン系樹脂押出発泡粒子およびその製造方法、並びに発泡成形体 | |
| WO2024176975A1 (ja) | ポリプロピレン系樹脂二段発泡粒子の製造方法、発泡成形体の製造方法、及びポリプロピレン系樹脂二段発泡粒子 | |
| WO2024101188A1 (ja) | ポリプロピレン系樹脂押出発泡粒子、その製造方法、及び発泡成形体 | |
| WO2025204566A1 (ja) | ポリプロピレン系樹脂溶融混練物、その製造方法、発泡体、及び発泡成形体 | |
| WO2023176911A1 (ja) | ポリプロピレン系樹脂押出発泡粒子の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22876090 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023551451 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022876090 Country of ref document: EP Effective date: 20240429 |