WO2023085337A1 - バイオジェット燃料製造方法及び該方法に用いるバイオジェット燃料製造用触媒 - Google Patents
バイオジェット燃料製造方法及び該方法に用いるバイオジェット燃料製造用触媒 Download PDFInfo
- Publication number
- WO2023085337A1 WO2023085337A1 PCT/JP2022/041796 JP2022041796W WO2023085337A1 WO 2023085337 A1 WO2023085337 A1 WO 2023085337A1 JP 2022041796 W JP2022041796 W JP 2022041796W WO 2023085337 A1 WO2023085337 A1 WO 2023085337A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- bio
- jet fuel
- oil
- biomass
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/40—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the pentasil type, e.g. types ZSM-5, ZSM-8 or ZSM-11, as exemplified by patent documents US3702886, GB1334243 and US3709979, respectively
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/40—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the pentasil type, e.g. types ZSM-5, ZSM-8 or ZSM-11, as exemplified by patent documents US3702886, GB1334243 and US3709979, respectively
- B01J29/42—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the pentasil type, e.g. types ZSM-5, ZSM-8 or ZSM-11, as exemplified by patent documents US3702886, GB1334243 and US3709979, respectively containing iron group metals, noble metals or copper
- B01J29/44—Noble metals
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G1/00—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal
- C10G1/08—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal with moving catalysts
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
- C10G3/45—Catalytic treatment characterised by the catalyst used containing iron group metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
- C10G3/47—Catalytic treatment characterised by the catalyst used containing platinum group metals or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
- C10G3/44—Catalytic treatment characterised by the catalyst used
- C10G3/48—Catalytic treatment characterised by the catalyst used further characterised by the catalyst support
- C10G3/49—Catalytic treatment characterised by the catalyst used further characterised by the catalyst support containing crystalline aluminosilicates, e.g. molecular sieves
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/50—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids in the presence of hydrogen, hydrogen donors or hydrogen generating compounds
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L1/00—Liquid carbonaceous fuels
- C10L1/02—Liquid carbonaceous fuels essentially based on components consisting of carbon, hydrogen, and oxygen only
-
- C—CHEMISTRY; METALLURGY
- C11—ANIMAL OR VEGETABLE OILS, FATS, FATTY SUBSTANCES OR WAXES; FATTY ACIDS THEREFROM; DETERGENTS; CANDLES
- C11C—FATTY ACIDS FROM FATS, OILS OR WAXES; CANDLES; FATS, OILS OR FATTY ACIDS BY CHEMICAL MODIFICATION OF FATS, OILS, OR FATTY ACIDS OBTAINED THEREFROM
- C11C3/00—Fats, oils, or fatty acids by chemical modification of fats, oils, or fatty acids obtained therefrom
-
- C—CHEMISTRY; METALLURGY
- C11—ANIMAL OR VEGETABLE OILS, FATS, FATTY SUBSTANCES OR WAXES; FATTY ACIDS THEREFROM; DETERGENTS; CANDLES
- C11C—FATTY ACIDS FROM FATS, OILS OR WAXES; CANDLES; FATS, OILS OR FATTY ACIDS BY CHEMICAL MODIFICATION OF FATS, OILS, OR FATTY ACIDS OBTAINED THEREFROM
- C11C3/00—Fats, oils, or fatty acids by chemical modification of fats, oils, or fatty acids obtained therefrom
- C11C3/12—Fats, oils, or fatty acids by chemical modification of fats, oils, or fatty acids obtained therefrom by hydrogenation
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1011—Biomass
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1011—Biomass
- C10G2300/1014—Biomass of vegetal origin
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/20—Technologies relating to oil refining and petrochemical industry using bio-feedstock
Definitions
- the present invention relates to a bio-jet fuel production method and a bio-jet fuel production catalyst for reforming biomass-derived bio-oil to obtain bio-jet fuel.
- biofuels are attracting attention as a carbon-neutral alternative to fossil fuels. Since the carbon dioxide generated by burning biofuels is the carbon dioxide absorbed from the atmosphere during the growth process of biomass, the use of biofuels does not increase the amount of carbon dioxide in the atmosphere. Carbon neutrality can be achieved, the concept that the amount of carbon dioxide emitted is the same as the amount of carbon dioxide absorbed when done.
- Biofuels that achieve such carbon neutrality include bioethanol fuels and biodiesel fuels, which have been widely used in recent years, as well as biojet fuels.
- the spread of bio-jet fuel has lagged compared to bio-ethanol fuel and bio-diesel fuel due to strict product standards, etc.
- the global aviation industry consumes about 1.5 billion to 1.5 billion dollars a year of regular jet fuel. Since 1.7 billion barrels (equivalent to 238-270 Mm 3 ) are being consumed, urgent development of high-quality bio-jet fuel is required.
- biomass-derived raw material oil is decarbonated or deoxygenated, hydrotreated to produce hydrocarbons, and the hydrocarbons are isomerized and cracked. This is done by producing C 7-14 isomerized hydrocarbons, which are then bio-jet fuels.
- Patent Document 1 describes: a) hydrogenation of a renewable feedstock in a first reaction zone using a catalyst under reaction conditions in which hydrogen is present; Treated by deoxygenation to obtain a first reaction zone product stream comprising hydrogen, water, carbon dioxide, and a hydrocarbon fraction comprising diesel fuel boiling range paraffins and aviation fuel boiling range paraffins.
- Patent Document 2 in a first step, biological oil and hydrogen gas are subjected to conditions sufficient to hydrodeoxygenate in the presence of a hydrodeoxygenation catalyst to produce n-paraffins, In a second stage, the n-paraffins and hydrogen gas are subjected to conditions sufficient to effect isomerization in the presence of an isomerization catalyst to produce isoparaffins and a separate fraction, obtained from the second stage through re-isomerization
- a process for producing hydrocarbons comprising recycling fractions boiling above 200° C. under atmospheric pressure and isomerizing in the presence of an isomerization catalyst.
- US Pat. No. 5,300,003 discloses a method for converting triacylglyceride-containing oils into crude precursors and/or distillate hydrocarbon fuels comprising heating a mixture of triacylglyceride-containing oils, water, and diatomic hydrogen to about 250° C. to about reacting at a temperature in the range of 560° C. and a pressure greater than about 75 bar to convert at least a portion of the triacylglycerides, water and one or more of isoolefins, isoparaffins, cycloolefins, cycloparaffins, aromatics; and hydrotreating the reaction effluent to form a hydrotreated effluent.
- Patent Documents 1 and 2 are performed in two steps of a hydrodeoxygenation process and an isomerization cracking process, and consume a large amount of energy, and are highly suitable for producing bio jet fuel. There is a problem that cost is required.
- the inventors of the present application have developed bio-oil obtained by pyrolyzing biomass resources using superheated steam and a bifunctional catalyst in which a solid base catalyst is supported on a solid acid catalyst. Focusing attention, the present inventors have invented the method for producing bio-jet fuel and the catalyst for bio-jet fuel production of the present application.
- An object of the present invention is to provide a method for producing bio-jet fuel that is highly energy-saving and capable of easily producing high-quality bio-jet fuel, and a catalyst for producing bio-jet fuel used in this method.
- the invention of claim 1 uses a catalyst obtained by supporting a solid base catalyst on a solid acid catalyst with a biomass-derived oil containing free fatty acids, hydrocarbons and triacylglycerols, It is characterized by having a reforming treatment step in which decarbonation, hydrogenation, isomerization and decomposition are performed under conditions of a reaction temperature of 200°C to 450°C.
- the invention of claim 2 is characterized in that the biomass-derived oil of claim 1 is bio-oil obtained by heating a biomass resource and cooling the generated pyrolysis gas.
- the invention of claim 3 is characterized in that the bio-oil of claim 2 is obtained by cooling the pyrolysis gas generated by heating the biomass resource at 200-450°C.
- the bio-oil of claim 2 or 3 is obtained by cooling pyrolysis gas generated by heating using a heating means including injection of superheated steam to the biomass resource. It is characterized by being
- the bio-oil according to any one of claims 2 to 4 is obtained by cooling pyrolysis gas generated by heating coconut palm fruits as the biomass resource. It is characterized by
- the invention of claim 6 is characterized in that the amount of the catalyst used in any one of claims 1 to 5 is 5 to 15 wt% of the biomass-derived oil.
- the invention of claim 7 is characterized in that the reforming treatment step of any one of claims 1 to 6 is performed in a reactor filled with hydrogen gas at a pressure of 0 to 2 MPa.
- the invention of claim 9 is characterized in that the solid acid catalyst of the catalyst of any one of claims 1 to 8 is zeolite.
- the invention of claim 10 is characterized in that the zeolite, which is the solid acid catalyst, of the catalyst of claim 9 is ZSM-5 type zeolite.
- the invention of claim 11 is characterized in that the solid base catalyst of the catalyst of any one of claims 1 to 10 contains a group 2 metal oxide.
- the oxide of the Group 2 metal which is the solid base catalyst of the catalyst of claim 11, is one or more selected from magnesium oxide, calcium oxide and barium oxide. characterized by being
- the invention of claim 13 is the solid acid catalyst of the catalyst of any one of claims 1 to 12, wherein one or more metals selected from groups 8, 9, and 10 are supported. It is characterized by being
- the invention of claim 14 is characterized in that one or more metals selected from groups 8, 9, and 10 supported on the solid acid catalyst of the catalyst of claim 13 are platinum. Characterized by
- the invention of claim 16 is characterized in that the solid acid catalyst of claim 15 is zeolite.
- the invention of claim 17 is characterized in that the zeolite of claim 16 is ZSM-5 type zeolite.
- the invention of claim 19 is characterized in that the Group 2 metal oxide of claim 18 is one or more selected from magnesium oxide, calcium oxide and barium oxide.
- the solid acid catalyst of any one of claims 15 to 19 supports one or more metals selected from groups 8, 9 and 10. It is characterized by
- the invention of claim 21 is characterized in that the one or more metals selected from groups 8, 9, and 10 supported on the solid acid catalyst of claim 20 are platinum.
- a biomass-derived oil containing free fatty acids, hydrocarbons and triacylglycerol is reacted at a reaction temperature of 200 ° C. to 450 ° C. using a catalyst in which a solid base catalyst is supported on a solid acid catalyst.
- C 7-14 isomerized hydrocarbons suitable for use as bio-jet fuel can be obtained because the reforming process includes decarbonation gas, hydrogenation, isomerization and cracking.
- the bio-oil obtained by heating the biomass resource and cooling the generated pyrolysis gas used in the present invention has little high-molecular fraction such as tar, so the amount of coke deposition on the catalyst surface is small. Reforming to biofuel can be efficiently performed with a small amount.
- bio-oil since bio-oil is in an emulsion state with water generated by thermal decomposition of biomass resources, it can be removed without supplying water or hydrogen in the reforming process, or with a small amount of water or hydrogen. Carbon dioxide gas, hydrogenation, isomerization, and decomposition can be performed, and low-cost production of bio-jet fuel can be realized.
- the bio-oil is preferably produced by cooling pyrolysis gas obtained by heating in the range of 200-450°C. Since such bio-oils contain large amounts of C 6-19 hydrocarbons and free fatty acids, bio-jet fuels composed of C 7-14 isomerized hydrocarbons can be efficiently produced.
- a catalyst consisting of a solid acid catalyst supported by a solid base catalyst is used. Since reforming is possible under pressure, it is possible to save energy and to produce bio-jet fuel at low cost.
- the solid base catalyst of the catalyst preferably one or more selected from group 2 metal oxides, more preferably magnesium oxide, calcium oxide and barium oxide, of free fatty acids and triacylglycerols can be suitably decarboxylated and hydrogenated.
- group 2 metal oxides more preferably magnesium oxide, calcium oxide and barium oxide, of free fatty acids and triacylglycerols can be suitably decarboxylated and hydrogenated.
- bio-oil by reforming bio-oil under conditions of a reaction temperature of 200°C to 450°C, bio-oil can be reliably reformed.
- bio-jet fuel production catalyst used in this method.
- FIG. 4 is an explanatory diagram showing a fixed-bed flow reactor used in Test 5.
- the present invention is a method for producing bio-jet fuel by reforming biomass-derived oil containing free fatty acids, hydrocarbons and triacylglycerols.
- biomass-derived oil containing free fatty acids, hydrocarbons and triacylglycerol is not particularly limited, but bio-oil obtained by heating biomass resources and cooling the generated pyrolysis gas is suitable.
- Biomass resources are not particularly limited as long as they are derived from animals and plants, with the exception of fossil fuels.
- Oil-rich plant seeds are preferably used for stable supply and easy production of bio-oil.
- bintaro, bengowan, poron, jatropha, coconut palm fruit, palm fruit, date palm fruit and the like are suitable.
- coconut palm fruit is more preferably used because its endosperm contains a large amount of oil.
- the bio-oil used in the present invention is obtained by heating biomass resources and cooling the generated pyrolysis gas. In this way, since bio-oil contains a small amount of high-molecular fractions such as tar, the amount of coke deposited on the surface of the catalyst is small, and reforming into bio-fuel can be efficiently performed.
- superheated steam it is suitable to use superheated steam to heat biomass resources. It is not necessary to heat the biomass resource with superheated steam alone, and the biomass resource may be heated in combination with other heating means.
- the superheated steam used to heat the biomass oil is mixed with the bio-oil pyrolysis gas. Also, by using superheated steam, the proportion of hydrocarbons in the bio-oil can be increased. It is considered that the pyrolysis gas generated from the biomass resource is thermally decomposed and steam reformed by contacting the superheated steam at a high temperature.
- the temperature for heating the biomass resource is preferably 200 to 450°C.
- Bio-oil obtained by heating biomass resources in this temperature range contains a large amount of hydrocarbons in the range of C6-19 and free fatty acids. can be manufactured to
- the time for heating the biomass resources is appropriately selected depending on the amount of biomass resources, the heating temperature, etc., and is not particularly limited.
- a catalyst made by supporting a solid base catalyst on a solid acid catalyst is used.
- Silica alumina, activated alumina, activated clay, and zeolite can be used as solid acid catalysts. Of these, zeolite is preferred because it is readily available and exhibits high acidity. As the zeolite, mordenite, ⁇ zeolite, ZSM zeolite, A-type zeolite, X-type zeolite, Y-type zeolite, etc. can be used, among which ZSM-5-type zeolite is preferably selected.
- the solid acid catalyst can support one or more metals selected from Groups 8, 9 and 10. Of the group 9 and/or group 10 metals, platinum is most preferably used.
- the catalyst is produced by dissolving the solid base catalyst precursor in distilled water, impregnating the solid acid catalyst with this, drying it, forming it into pellets, and then calcining it to obtain the catalyst.
- the method for producing the catalyst is not limited to this method, and other methods may be used.
- a batch-type reforming process will be described first.
- the bio-oil and the catalyst are placed in the reactor of the above-described reforming apparatus and heated at a reaction temperature of 200° C. to 450° C. to reform the bio-oil.
- a gas injection means may be used to fill the reactor with hydrogen at a pressure of 1-5 MPa.
- the amount of catalyst contained is 1-20 wt% of the bio-oil, preferably 5-15 wt%.
- the continuous reforming process will be described.
- the continuous reforming process is preferably carried out using the above-mentioned flow reactor at a reaction temperature of 200° C. to 450° C.
- the hydrogen pressure should be about 0.5 to 3 MPa, and the volume ratio of hydrogen to biomass-derived oil should be about 100:1 to 1200:1. is preferred.
- the time for which the modification treatment is performed is not particularly limited, but it is preferable to perform it for 1 to 6 hours.
- a catalyst was produced by using ZSM-5 type zeolite as a solid acid catalyst and supporting 5% by mass of magnesium oxide as a solid base catalyst.
- Example 2 Using the above-described method for producing a catalyst, ZSM-5 type zeolite was used as a solid acid catalyst, and a catalyst supporting 5% by mass of calcium oxide was produced as a solid base catalyst.
- a catalyst was produced by using ZSM-5 type zeolite as a solid acid catalyst and supporting 5% by mass of barium oxide as a solid base catalyst.
- the amount of base was measured by the CO 2 -TPD method using ChemBET Pulser (manufactured by Quantachrome). The analysis results are shown in FIG. 2 and Table 1.
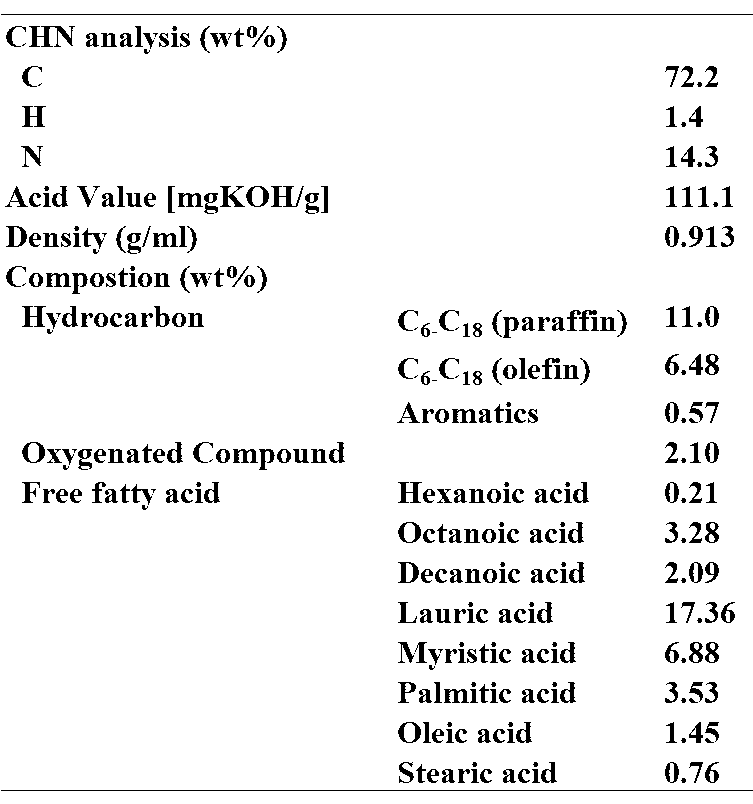
- ⁇ Test 2 Reforming test using the catalyst of Example 1> Next, liquid products obtained by reforming bio-oil using the catalyst of Example 1 of Test 1 were analyzed. In this test, bio-oil obtained by heating copra, which is the endosperm of coconut, using superheated steam and cooling the generated pyrolysis gas was used as bio-oil as a raw material. Table 2 shows the results of CHN elemental analysis of the components of the bio-oil used.
- the catalyst of Example 1 was crushed to 80-120 mesh and dried at 110°C for 1 hour before use.
- the amount of bio-oil to be reformed was 10.0 g, the catalyst was 0.5 g, and the reactor was filled with hydrogen gas at a pressure of 1.0 MPa. This was reacted for 3 hours at a reaction temperature of 300° C. and a stirring speed of 400 rpm.
- the reaction pressure in this specification means the pressure at which hydrogen gas is enclosed in the reactor.
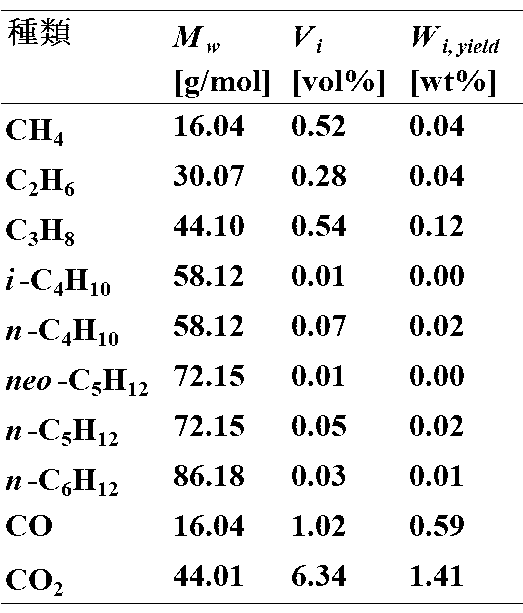
- Table 3 shows the analysis results of the components of the gaseous product obtained by this operation
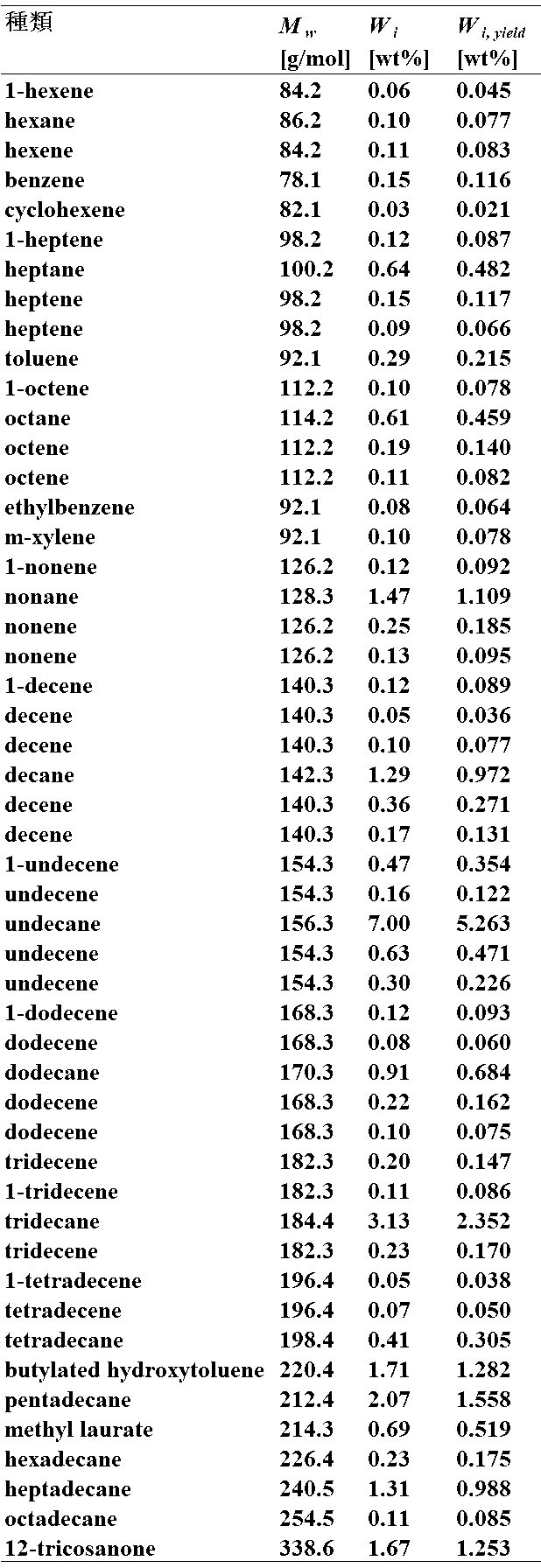
- Table 4 shows the analysis results of the components of the liquid product.
- M w indicates the molecular weight of each component
- V i indicates the volume ratio of each component
- W i yield indicates the weight ratio of each component.
- the gaseous product contains a large amount of carbon dioxide, and the liquid product contains C 7-14 isohydrocarbons with side chains that can be used as bio-jet fuel. It was found that by using the catalyst of Example 1, the bio-oil was hydrogenated, decarboxylated, cracked and isomerized.
- Experiment 1 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 0.5 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction time was 3 hours.
- Experiment 2 a catalyst employing calcium oxide as a solid base catalyst was used, the amount of catalyst was 0.5 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction was carried out for 3 hours.
- Experiment 3 a catalyst employing barium oxide as a solid base catalyst was used, the amount of catalyst was 0.5 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction was carried out for 3 hours.
- Experiment 4 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 1.0 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction was performed for 3 hours.
- Experiment 5 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 1.0 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction was performed for 3 hours.
- Experiment 6 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 1.0 g, 1 MPa of nitrogen was charged without charging hydrogen, and the reaction time was 3 hours.
- Experiment 7 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 1.0 g, hydrogen gas was charged at a reaction pressure of 2.0 MPa, and the reaction time was 3 hours.
- Experiment 8 a catalyst employing magnesium oxide as a solid base catalyst was used, the amount of catalyst was 1.0 g, hydrogen gas was charged at a reaction pressure of 1.0 MPa, and the reaction was performed for 6 hours.
- Experiment 10 the same catalyst as used in Experiment 9 was subjected to hydrogen reduction treatment at a reaction temperature of 400° C. for a reaction time of 3 hours, and the amount of catalyst was 1.0 g, and hydrogen gas was used as the reaction pressure. The pressure was sealed at 1.0 MPa, and the reaction time was 6 hours.
- fatty acid conversion rate x FFA
- S DCO fatty acid decarboxylation rate
- YJF carbon mole fraction of C 7-14 hydrocarbons
- the carbon molar fraction of C 7-14 hydrocarbons is the total molar amount of carbon of C 7-14 hydrocarbons, which is the main component of jet fuel in the liquid product, in the raw material (bio-oil). This is to confirm the ratio in the molar amount of carbon in the compound. This evaluation allows identification of suitable catalysts and processes for producing bio-jet fuel.
- Fatty acid conversion rate (x FFA ) is calculated by Equation 1
- fatty acid decarboxylation rate (S DCO ) is calculated by Equation 2
- carbon mole fraction (Y JF ) of C 7-14 hydrocarbons is calculated by Equation 3.
- the amount of coke on the spent catalyst was measured and each experiment was evaluated. If the amount of coke on the spent catalyst is low, the catalyst has not lost activity and can withstand repeated use. When the amount of coke is large, the catalyst is deactivated early, and not only can it not be used repeatedly, but also there is a possibility that sufficient reforming treatment cannot be performed.
- the amount of coke was measured by washing the remaining used catalyst with tetrahydrofuran and drying it, followed by TG analysis. In the TG analysis, the amount of coke produced during the reaction can be measured by analyzing the weight loss within the analysis temperature range. Using the numerical values obtained in this manner, the coke production rate (W coke ) was calculated by calculating the amount of coke on the used catalyst by calculating with the following formula 2.
- Table 5 shows the results of experiments 1 to 10 evaluated using formulas 1 to 4.
- Experiments 8, 9 and 10 were contrasted to investigate the effect on the reaction with or without supporting one or more metals selected from Groups 8, 9 and 10 in the catalyst.
- the amount of coke attached to the catalyst was found to be smaller than in Experiment 8 in which platinum was not supported.
- Experiment 10 in which hydrogen-reduced platinum was supported on the catalyst the amount of coke deposited was significantly reduced compared to Experiment 8. From the above, it is preferable to support one or more metals selected from Groups 8, 9, and 10 in order to enable long-term use of the catalyst. It was confirmed that one or more metals selected from Groups 8, 9 and 10 are more preferably subjected to hydrogen reduction treatment.
- ⁇ Test 4 Reaction evaluation test by reaction temperature> Next, using the catalyst used in Experiment 10, 10.0 g of bio oil, 1.0 g of catalyst, reaction pressure of 1.0 MPa, stirring speed of 400 rpm, reaction time of 6 hours, reaction temperature of 320 ° C.
- Experiment 11 A reforming experiment was carried out at a reaction temperature of 340° C., and a suitable reaction temperature was confirmed by evaluating the reaction in comparison with Experiment 10. The same evaluation as in test 3 was used. Table 6 shows the results.
- the hydrocarbon isomerization rate was calculated by dividing the carbon molar amount of the C7-14 isomerized hydrocarbons in the liquid product by the carbon molar amount of all the hydrocarbons in the liquid product and multiplying by 100.
- Table 7 shows the results of Test 5 above.
Landscapes
- Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Crystallography & Structural Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Materials Engineering (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
Abstract
Description
次の実施例1~3の触媒について、塩基量および酸量の測定を行った。
前述の触媒の製造方法を用いて、固体酸触媒としてZSM-5型ゼオライトを用い、固体塩基触媒として酸化マグネシウムを5質量%担持させた触媒を製造した。
前述の触媒の製造方法を用いて、固体酸触媒としてZSM-5型ゼオライトを用い、固体塩基触媒として酸化カルシウムを5質量%担持させた触媒を製造した。
前述の触媒の製造方法を用いて、固体酸触媒としてZSM-5型ゼオライトを用い、固体塩基触媒として酸化バリウムを5質量%担持させた触媒を製造した。
その分析結果を図2及び表1に示す。

次に、試験1の実施例1の触媒を用いてバイオオイルを改質して得た液体生成物を解析した。本試験では、原料であるバイオオイルとしてココナッツの胚乳であるコプラを過熱蒸気を使用して加熱して、発生した熱分解ガスを冷却して得たバイオオイルを使用した。使用したバイオオイルの成分をCHN元素分析により分析した結果を表2に示す。
なお、本明細書における反応圧力は反応器内に水素ガスを封入する圧力を意味する。
なお、表中のMw は各成分の分子量、Viは各成分の体積割合、Wi yieldは各成分の重量割合を示している。
次に、触媒、触媒量、反応圧力及び反応時間を異なるものとしたバイオオイルの改質実験1~10を行い、その反応の評価をすることにより好適な触媒、触媒量、反応圧力及び反応時間を確認した。なお、何れの実験もバイオオイルが10.0g、反応温度300℃、撹拌速度400rpmで改質実験を行った。
実験1では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が0.5g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が3時間で行った。
実験2では、固体塩基触媒として酸化カルシウムを採用した触媒を用い、触媒量が0.5g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が3時間で行った。
実験3では、固体塩基触媒として酸化バリウムを採用した触媒を用い、触媒量が0.5g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が3時間で行った。
実験4では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が1.0g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が3時間で行った。
実験5では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が1.0g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が3時間で行った。
実験6では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が1.0g、水素を封入することなく窒素を1MPa封入し、反応時間が3時間で行った。
実験7では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が1.0g、反応圧力として水素ガスを2.0MPaの圧力で封入し、反応時間が3時間で行った。
実験8では、固体塩基触媒として酸化マグネシウムを採用した触媒を用い、触媒量が1.0g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が6時間で行った。
実験9では、固体塩基触媒として酸化マグネシウムを採用し、さらに白金を担持させた触媒を用い、触媒量が1.0g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が6時間で行った。
実験10では、実験9で使用した触媒と同様の触媒に、反応温度400℃、反応時間3時間で水素還元処理を行ったものを使用し、触媒量が1.0g、反応圧力として水素ガスを1.0MPaの圧力で封入し、反応時間が6時間で行った。
活性評価では、脂肪酸転化率(xFFA)、脂肪酸脱炭酸率(SDCO)及びC7-14の炭化水素の炭素モル量率(YJF)の3つの観点で評価を行った。脂肪酸転化率(xFFA)はバイオオイル中の遊離脂肪酸がどの程度炭化水素に転化したかを確認するものである。脂肪酸脱炭酸率(SDCO)は、バイオオイル中の脂肪酸がどの程度脱炭酸反応を生じたかを確認するものである。C7-14の炭化水素の炭素モル量率(YJF)は液体生成物中のジェット燃料の主たる成分となるC7-14の炭化水素の炭素モル量の、原料(バイオオイル)中の全ての化合物中の炭素モル量における割合を確認するものである。この評価により、バイオジェット燃料を製造するのに好適な触媒及び方法を確認することができる。
脂肪酸転化率(xFFA)は式1、脂肪酸脱炭酸率(SDCO)は式2、C7-14の炭化水素の炭素モル量率(YJF)は式3により算出される。



使用済み触媒上のコーク量の測定を行い、各実験の評価を行った。使用済み触媒上のコーク量が少なければ、触媒は活性を失っておらず、繰り返しの使用に耐えることができる。コーク量が多い場合は、触媒が早期に失活し、繰り返しの使用ができないだけではなく、十分な改質処理が行えないおそれがある。コーク量の測定は、残留した使用済み触媒をテトラヒドロフランを使用して洗浄し、乾燥させた後にTG分析を行った。TG分析では分析温度範囲内での重量減少を分析することで、反応中に生成されたコークの生成量を測定することができる。
このようにして得た数値を使用して、次の式2にて算出し、使用済み触媒上のコークの量の測定を行い、コーク生成率(Wcoke)を算出した。

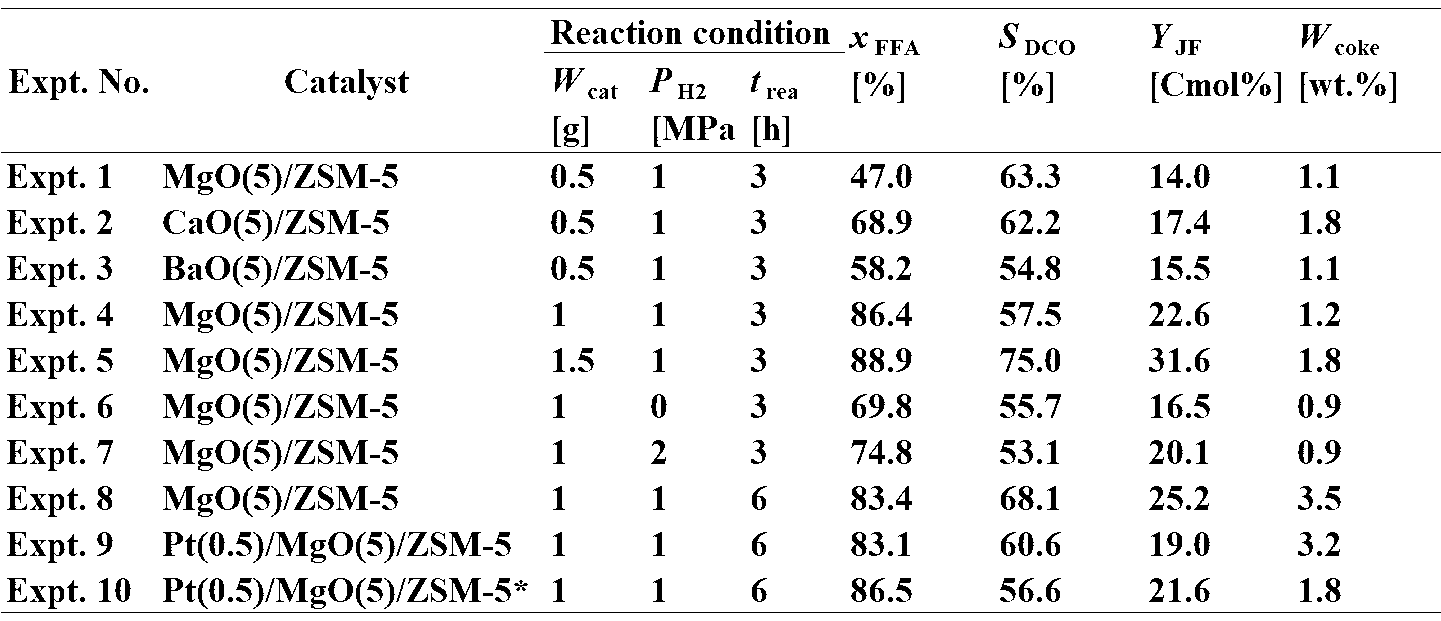
触媒量を異なるものとしたときの評価を得るため、触媒量以外が同一の条件となっている実験1、実験4及び実験5を対比した。実験1では脂肪酸転化率が47.0%であるのに対し、実験4及び実験5では脂肪酸転化率が85%を超えるものとなっている。また、脂肪酸脱炭酸率と炭化水素量については実験5が最も高くなっている。このことからバイオマス由来のオイルに対する比率は5%以上が好適であり、10%以上がより好ましく、15%以上であることが最も好適であることが確認された。
反応圧力を異なるものとしたときの評価を得るため、反応圧力以外が同一の条件となっている実験1、実験6及び実験7を対比した。実験1,6,7何れもC7-14の炭化水素が認められたことから、本発明に係る触媒を使用すれば、水素の封入がなくともC7-14の炭化水素を得られることが認められた。また、実験7では脂肪酸転化率、脂肪酸脱炭酸率および炭化水素量のいずれもが実験1,6よりも優れていたことから、反応圧力を2.0MPaとすることが好適であることも確認された。
反応時間異なるものとしたときの評価を得るため、反応時間以外が同一の条件となっている実験4と実験8を対比した。その結果、実験4及び実験8の脂肪酸転化率、脂肪酸脱炭酸率および炭化水素量に目立った変化は認められなかった。しかし、実験8では触媒に多量のコークスの付着が認められた。以上から、反応時間は6時間以下が好適であり、3時間とすることがより好適であることが確認された。
触媒中の第8族、第9族、第10族から選択された1又は複数の金属の担持の有無による反応への影響を調べるために、実験8、実験9及び実験10を対比した。その結果、白金を担持させた実験9及び実験10では、白金を担持させない実験8よりも触媒のコークスの付着量が減少することが認められた。また、水素還元処理を施した白金を触媒に担持させた実験10では、実験8に比べて顕著にコークスの付着量が減少した。以上から、触媒の長期間の使用を可能とするために第8族、第9族、第10族から選択された1又は複数の金属の担持させることが好適であり、また、担持される第8族、第9族、第10族から選択された1又は複数の金属が水素還元処理を施されていることがより好適であることが確認された。
次に、実験10で使用した触媒を用い、バイオオイルが10.0g、触媒量1.0g、反応圧力1.0MPa、撹拌速度400rpm、反応時間6時間で、反応温度を320℃とした実験11及び反応温度を340℃とした改質実験を行い、実験10と対比してその反応の評価をすることにより好適な反応温度を確認した。評価は試験3における評価と同一のものを使用した。その結果を表6に示す。

次に、試験5では、実験10で用いた、固体酸触媒としてZSM-5型ゼオライト、固体塩基触媒として酸化マグネシウムを用い、さらに白金を担持させ、反応温度400℃、反応時間3時間で水素還元処理を行った触媒を使用し、図5に示すバイオオイルを連続的に反応炉に供給し、反応炉内で改質された液体生成物及び気体生成物を連続的に排出する固定床流通式反応装置を用いて、オイルの異性化率の確認を行った。本試験の試験条件は、重量空間速度が2h-1、反応温度を300℃~400℃の間で段階適に変更し、水素圧を1MPa、水素供給量を原料の体積の500倍として行った。炭化水素異性化率は液体生成物中のC7-14の異性化炭化水素の炭素モル量を、液体生成物中の全炭化水素の炭素モル量で除し、100を乗じて算出した。以上の試験5の結果を表7に示す。

Claims (21)
- 遊離脂肪酸、炭化水素及びトリアシルグリセロールを含有するバイオマス由来のオイルを、固体酸触媒に固体塩基触媒を担持させてなる触媒を用いて、反応温度200℃~450℃の条件の下、脱炭酸ガス、水素化、異性化及び分解を行う改質処理工程を有することを特徴とするバイオジェット燃料製造方法。
- 前記バイオマス由来のオイルは、バイオマス資源を加熱し、発生した熱分解ガスを冷却することにより得られたバイオオイルであることを特徴とする請求項1に記載のバイオジェット燃料製造方法。
- 前記バイオオイルは、前記バイオマス資源を200~450℃で加熱して発生した熱分解ガスを冷却することにより得られたものであることを特徴とする請求項2に記載のバイオジェット燃料製造方法。
- 前記バイオオイルは、前記バイオマス資源への過熱蒸気の噴射を含む加熱手段を使用して加熱して発生した熱分解ガスを冷却することにより得られたものであることを特徴とする請求項2または3に記載のバイオジェット燃料製造方法。
- 前記バイオオイルは、前記バイオマス資源としてココヤシの果実を加熱して発生した熱分解ガスを冷却することにより得られたものであることを特徴とする請求項2から4のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記触媒の使用量は前記バイオマス由来のオイルの5~15wt%であることを特徴とする請求項1から5のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記改質処理工程は、0~2MPaの圧力で水素ガスを封入した反応器内で行うことを特徴とする請求項1から6のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記改質処理工程は、3~6時間の範囲で行うことを特徴とする請求項1から7のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記触媒の前記固体酸触媒は、ゼオライトであることを特徴とする請求項1から8のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記触媒の、前記固体酸触媒である前記ゼオライトは、ZSM-5型ゼオライトであることを特徴とする請求項9に記載のバイオジェット燃料製造方法。
- 前記触媒の前記固体塩基触媒は、第2族の金属の酸化物を含むことを特徴とする請求項1から10のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記触媒の前記固体塩基触媒である前記第2族の金属の酸化物は、酸化マグネシウム、酸化カルシウム又は酸化バリウムから選択される1種または2種以上であることを特徴とする請求項11に記載のバイオジェット燃料製造方法。
- 前記触媒の前記固体酸触媒には、第8族、第9族、第10族から選択された1又は複数の金属が担持されていることを特徴とする請求項1から12のいずれか1項に記載のバイオジェット燃料製造方法。
- 前記触媒の前記固体酸触媒に担持される前記第8族、第9族、第10族から選択された1又は複数の金属は、白金であることを特徴とする請求項13に記載のバイオジェット燃料製造方法。
- 固体酸触媒に固体塩基触媒を担持させてなることを特徴とするバイオジェット燃料製造用触媒。
- 前記固体酸触媒は、ゼオライトであることを特徴とする請求項15に記載のバイオジェット燃料製造用触媒。
- 前記ゼオライトはZSM-5型ゼオライトであることを特徴とする請求項16に記載のバイオジェット燃料製造用触媒。
- 前記固体塩基触媒は、第2族の金属の酸化物を含むことを特徴とする請求項15から17のいずれか1項に記載のバイオジェット燃料製造用触媒。
- 前記第2族の金属の酸化物は、酸化マグネシウム,酸化カルシウム又は酸化バリウムから選択される1種または2種以上であることを特徴とする請求項18に記載のバイオジェット燃料製造用触媒。
- 前記固体酸触媒には第8族、第9族、第10族から選択された1又は複数の金属が担持されていることを特徴とする請求項15から19のいずれか1項に記載のバイオジェット燃料製造用触媒。
- 前記固体酸触媒に担持される前記第8族、第9族、第10族から選択された1又は複数の金属は白金であることを特徴とする請求項20に記載のバイオジェット燃料製造用触媒。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22892835.4A EP4431584A4 (en) | 2021-11-09 | 2022-11-09 | METHOD FOR PRODUCING A BIOJET FUEL AND CATALYST FOR PRODUCING A BIOJET FUEL USED IN SAID METHOD |
| JP2023559877A JPWO2023085337A1 (ja) | 2021-11-09 | 2022-11-09 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-182575 | 2021-11-09 | ||
| JP2021182575 | 2021-11-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023085337A1 true WO2023085337A1 (ja) | 2023-05-19 |
Family
ID=86335752
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/041796 Ceased WO2023085337A1 (ja) | 2021-11-09 | 2022-11-09 | バイオジェット燃料製造方法及び該方法に用いるバイオジェット燃料製造用触媒 |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP4431584A4 (ja) |
| JP (1) | JPWO2023085337A1 (ja) |
| WO (1) | WO2023085337A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024071264A1 (ja) * | 2022-09-27 | 2024-04-04 | 国立大学法人東京農工大学 | バイオジェット燃料製造用触媒及びバイオジェット燃料製造用触媒を用いたバイオジェット燃料製造方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010121072A (ja) * | 2008-11-20 | 2010-06-03 | Nippon Oil Corp | 航空燃料油基材の製造方法 |
| JP2011219708A (ja) * | 2010-04-13 | 2011-11-04 | Ggi Japan Kk | ジェットバイオ燃料の製造システム及び製造方法 |
| CN102513132A (zh) * | 2011-12-06 | 2012-06-27 | 福州东冶能源科技有限公司 | 一步法生物柴油生产专用的dyd催化剂及其生产方法 |
| CN105331387A (zh) * | 2015-11-13 | 2016-02-17 | 北京化工大学 | 生物质热催化转化及精制制备航空燃料的工艺方法 |
| WO2019221287A1 (ja) * | 2018-05-18 | 2019-11-21 | 一般社団法人 HiBD研究所 | バイオジェット燃料の製造方法 |
| WO2021083270A1 (zh) * | 2019-10-31 | 2021-05-06 | 中国石油化工股份有限公司 | 负载型催化剂及其制备方法和应用 |
| JP2021070022A (ja) * | 2019-10-24 | 2021-05-06 | 国立大学法人東京工業大学 | 長鎖直鎖パラフィンの水素化異性化触媒、長鎖直鎖パラフィンの水素化異性化触媒の製造方法、及び長鎖直鎖パラフィンの水素化異性化反応による分岐パラフィンの製造方法 |
| WO2022004267A1 (ja) * | 2020-06-29 | 2022-01-06 | 株式会社レボインターナショナル | 油脂の接触水素化分解による炭化水素製造システム |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8329968B2 (en) * | 2008-04-06 | 2012-12-11 | Uop Llc | Production of blended gasoline aviation and diesel fuels from renewable feedstocks |
| US8377288B2 (en) * | 2009-09-22 | 2013-02-19 | Bp Corporation North America Inc. | Methods and units for mitigation of carbon oxides during hydrotreating |
| CN103314078B (zh) * | 2010-09-14 | 2015-08-19 | Ifp新能源公司 | 将生物油提质为运输级烃燃料的方法 |
| US9534181B2 (en) * | 2012-06-19 | 2017-01-03 | Inaeris Technologies, Llc | Method of using renewable fuel composition |
| EP4257662B1 (en) * | 2014-05-29 | 2025-11-05 | ENI S.p.A. | Process for producing a diesel hydrocarbon fraction starting from a renewable feedstock |
| US10392566B2 (en) * | 2015-04-27 | 2019-08-27 | Gas Technology Institute | Co-processing for control of hydropyrolysis processes and products thereof |
-
2022
- 2022-11-09 JP JP2023559877A patent/JPWO2023085337A1/ja active Pending
- 2022-11-09 WO PCT/JP2022/041796 patent/WO2023085337A1/ja not_active Ceased
- 2022-11-09 EP EP22892835.4A patent/EP4431584A4/en active Pending
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010121072A (ja) * | 2008-11-20 | 2010-06-03 | Nippon Oil Corp | 航空燃料油基材の製造方法 |
| JP2011219708A (ja) * | 2010-04-13 | 2011-11-04 | Ggi Japan Kk | ジェットバイオ燃料の製造システム及び製造方法 |
| CN102513132A (zh) * | 2011-12-06 | 2012-06-27 | 福州东冶能源科技有限公司 | 一步法生物柴油生产专用的dyd催化剂及其生产方法 |
| CN105331387A (zh) * | 2015-11-13 | 2016-02-17 | 北京化工大学 | 生物质热催化转化及精制制备航空燃料的工艺方法 |
| WO2019221287A1 (ja) * | 2018-05-18 | 2019-11-21 | 一般社団法人 HiBD研究所 | バイオジェット燃料の製造方法 |
| JP2021070022A (ja) * | 2019-10-24 | 2021-05-06 | 国立大学法人東京工業大学 | 長鎖直鎖パラフィンの水素化異性化触媒、長鎖直鎖パラフィンの水素化異性化触媒の製造方法、及び長鎖直鎖パラフィンの水素化異性化反応による分岐パラフィンの製造方法 |
| WO2021083270A1 (zh) * | 2019-10-31 | 2021-05-06 | 中国石油化工股份有限公司 | 负载型催化剂及其制备方法和应用 |
| WO2022004267A1 (ja) * | 2020-06-29 | 2022-01-06 | 株式会社レボインターナショナル | 油脂の接触水素化分解による炭化水素製造システム |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4431584A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024071264A1 (ja) * | 2022-09-27 | 2024-04-04 | 国立大学法人東京農工大学 | バイオジェット燃料製造用触媒及びバイオジェット燃料製造用触媒を用いたバイオジェット燃料製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4431584A1 (en) | 2024-09-18 |
| EP4431584A4 (en) | 2025-10-01 |
| JPWO2023085337A1 (ja) | 2023-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6752399B2 (ja) | 生物由来油の接触水素添加と接触分解のカップリングによる芳香族炭化水素とオレフィンの製造方法及び装置 | |
| KR101673597B1 (ko) | 니켈 및 몰리브덴에 기초한 촉매를 사용하며, 탈카르복실화 전환이 제한되는, 재생 가능한 공급원으로부터 유래하는 공급물의 수소화탈산소화 방법 | |
| Kostyniuk et al. | Catalytic hydrocracking reactions of tetralin biomass tar model compound to benzene, toluene and xylenes (BTX) over metal-modified ZSM-5 in ambient pressure reactor | |
| ES2433220T3 (es) | Proceso y catalizador de hidroconversión | |
| JP4878824B2 (ja) | 環境低負荷型燃料の製造方法および環境低負荷型燃料 | |
| JP5317644B2 (ja) | 航空燃料油基材の製造方法 | |
| KR101481111B1 (ko) | 수첨탈산소 반응에 사용되는 탄화몰리브덴 촉매 및 이의 제조 방법 | |
| US8282815B2 (en) | Method of converting feedstocks from renewable sources to good-quality diesel fuel bases using a zeolite catalyst without intermediate gas-liquid separation | |
| US20090318740A1 (en) | Method of converting feedstocks from renewable sources to good-quality diesel fuel bases using a zeolite type catalyst | |
| BRPI0811661A2 (pt) | processo de hidrotratamento de uma alimentação líquida e processo para hidrodesoxigenação de um recurso renovável | |
| JP2014224262A (ja) | モリブデンベースの触媒を用いることにより再生可能な起源の流出物を優れた品質の燃料に転化する方法 | |
| CN102482599B (zh) | 航空燃料油组合物 | |
| CN109294623B (zh) | 一种油脂类原料制备柴油馏分的加氢方法 | |
| JP2010121071A (ja) | 航空燃料油基材および航空燃料油組成物 | |
| Galadima et al. | Hydroisomerization of sustainable feedstock in biomass‐to‐fuel conversion: a critical review | |
| WO2023073018A1 (en) | A process for hydrotreatment of aromatic nitrogen compounds | |
| JP2026510763A (ja) | Hefa及び燃料の統合プラントにおける、水素化処理されたエステル及び脂肪酸、低炭素水素並びに二酸化炭素からの燃料の生成 | |
| US20140275670A1 (en) | Process for low-hydrogen-consumption conversion of renewable feedstocks to alkanes | |
| WO2023085337A1 (ja) | バイオジェット燃料製造方法及び該方法に用いるバイオジェット燃料製造用触媒 | |
| Trisunaryanti et al. | Highly selective bio-hydrocarbon production using sidoarjo mud based-catalysts in the hydrocracking of waste palm cooking oil | |
| JP7659247B1 (ja) | バイオジェット燃料の製造方法及びバイオジェット燃料の製造装置 | |
| Vichaphund et al. | Utilization of fly ash-derived HZSM-5: catalytic pyrolysis of Jatropha wastes in a fixed-bed reactor | |
| Mirzayanti et al. | Triglyceride of Kapok seed Oil to biofuel over a synthesised Cu-Mo supported HZSM-5 catalyst | |
| JP2007308566A (ja) | 水素化精製方法及び環境低負荷型ガソリン基材 | |
| KR20250076559A (ko) | 바이오 제트 연료 제조용 촉매 및 바이오 제트 연료 제조용 촉매를 사용한 바이오 제트 연료 제조 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22892835 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18707706 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2023559877 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12024551096 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022892835 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022892835 Country of ref document: EP Effective date: 20240610 |