WO2023090311A1 - ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 - Google Patents
ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 Download PDFInfo
- Publication number
- WO2023090311A1 WO2023090311A1 PCT/JP2022/042353 JP2022042353W WO2023090311A1 WO 2023090311 A1 WO2023090311 A1 WO 2023090311A1 JP 2022042353 W JP2022042353 W JP 2022042353W WO 2023090311 A1 WO2023090311 A1 WO 2023090311A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polypropylene
- expanded
- particles
- weight
- foamed
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/16—Making expandable particles
- C08J9/18—Making expandable particles by impregnating polymer particles with the blowing agent
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0014—Use of organic additives
- C08J9/0028—Use of organic additives containing nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0014—Use of organic additives
- C08J9/0038—Use of organic additives containing phosphorus
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/22—After-treatment of expandable particles; Forming foamed products
- C08J9/228—Forming foamed products
- C08J9/232—Forming foamed products by sintering expandable particles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/06—CO2, N2 or noble gases
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/14—Copolymers of propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/16—Ethene-propene or ethene-propene-diene copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/122—Hydrogen, oxygen, CO2, nitrogen or noble gases
Definitions
- the present invention relates to polypropylene-based foamed particles, polypropylene-based foamed moldings, and methods for producing them.
- Molded articles made by molding polypropylene-based expanded particles have been widely used as interior materials and cushioning materials for automobiles. Recently, with the shift to electric vehicles, there is an increasing demand for applying polypropylene-based foamed moldings to peripheral members for electrical components, which are increasing in number.
- Patent Document 1 when polypropylene-based expanded beads are molded in a mold, the polypropylene-based expanded beads are impregnated with air to increase the internal pressure of the polypropylene-based expanded beads before molding.
- a technique for obtaining a system foam molded article is disclosed.
- an object of one embodiment of the present invention is to provide expanded polypropylene particles that can provide a polypropylene-based expanded molded article with excellent surface beauty and productivity.
- the present inventors have completed the present invention as a result of diligent studies to solve the above problems.
- one embodiment of the present invention contains polypropylene-based foamed particles having a water content of 1.0% to 60.0%.
- the structural units include a structural unit derived from the X1 monomer, a structural unit derived from the X2 monomer, ... and an Xn monomer (where n is An integer of 2 or more) is also referred to as "X 1 /X 2 /.../X n copolymer".
- the X 1 /X 2 /.../X n copolymer is not particularly limited in its polymerization mode unless otherwise specified, and may be a random copolymer or a block copolymer. It may be an alternating copolymer or a graft copolymer.
- X unit a structural unit derived from an X monomer contained in a polymer or copolymer
- polypropylene-based expanded molded articles if the following polypropylene-based expanded beads could be provided: It is possible to provide a polypropylene-based foamed molded article with excellent surface beauty even without inorganic gas impregnation time (required internal pressure of expanded particles is low) or without impregnation with inorganic gas (without applying internal pressure). Polypropylene foam particles.
- Expanded particles are imparted by molding polypropylene-based expanded particles having a specific moisture content. Even if the internal pressure is low, or even if the internal pressure of the expanded particles is not applied, it is possible to provide a polypropylene-based expanded molded article with excellent surface beauty; It is possible to provide a polypropylene-based expansion molded article with good productivity and excellent surface beauty even when the internal pressure of the expanded beads is low or even when the internal pressure of the expanded beads is not applied.
- the expanded polypropylene particles to be used for molding do not contain water, there is a certain relationship between the bulk density of the expanded polypropylene particles to be used for molding and the density of the molded polypropylene-based foamed article after drying in a dryer. Become. Therefore, when the expanded polypropylene particles to be molded do not contain water or have a very low water content, it is possible to stably produce a polypropylene-based expanded molded article with no variation in the density of the molded article.
- the polypropylene-based foamed particles are molded with the moisture content as low as possible. was taking place.
- the expanded polypropylene particles to be used for molding are dried in a dryer. That is, the moisture content (at the time of molding) of the polypropylene-based expanded particles after drying in a dryer (polypropylene-based expanded particles at the time of molding) in the production of conventional polypropylene-based foamed molded articles is very low (for example, less than 1%).
- the expanded polypropylene particles according to one embodiment of the present invention have a moisture content of 1.0% to 60.0% after drying in a dryer and/or during molding.
- the present foamed particles have the above structure, and thus have the advantage of being able to provide a polypropylene-based foamed molded article with excellent surface beauty with good productivity.
- a polypropylene-based expanded molded product can be provided by molding the expanded polypropylene-based particles according to one embodiment of the present invention by a known method.
- polypropylene-based expanded beads may be referred to as “expanded beads”
- polypropylene-based expanded beads according to one embodiment of the present invention may be referred to as “present expanded beads”
- polypropylene A “base resin foam molded article” may be referred to as a “foam molded article”.
- the "moisture content after drying in a dryer and/or at the time of molding” may be simply referred to as “moisture content”.
- the expanded particles contain a polypropylene-based resin (A).
- the polypropylene resin (A) is a resin containing at least 50 mol%, preferably 75% or more, of structural units derived from a propylene monomer out of 100 mol% of all structural units contained in the resin. Intend.
- structural unit derived from propylene monomer may be referred to as "propylene unit”.
- the polypropylene-based resin (A) may be (a) a homopolymer of propylene, or (b) a block copolymer, alternating copolymer, random copolymer, or It may be a graft copolymer or (c) a mixture of two or more thereof.
- the polypropylene resin (A) may have one or more structural units derived from a monomer other than the propylene monomer, or may have one or more types.
- a "monomer other than a propylene monomer” used in the production of the polypropylene-based resin (A) may be referred to as a "comonomer”.
- a "structural unit derived from a monomer other than a propylene monomer” contained in a polypropylene-based resin may be referred to as a "comonomer unit".
- Comonomers include ethylene, 1-butene, isobutene, 1-pentene, 3-methyl-1-butene, 1-hexene, 4-methyl-1-pentene, 3,4-dimethyl-1-butene, 1-heptene, Examples include ⁇ -olefins having 2 or 4 to 12 carbon atoms such as 3-methyl-1-hexene, 1-octene and 1-decene.
- polypropylene-based resin (A) examples include polypropylene homopolymer, ethylene/propylene random copolymer, 1-butene/propylene random copolymer, 1-butene/ethylene/propylene random copolymer, and ethylene/propylene. block copolymers, 1-butene/propylene block copolymers, propylene/chlorinated vinyl copolymers, propylene/maleic anhydride copolymers and styrene-modified polypropylene resins. As the polypropylene-based resin (A), one of these may be used alone, or two or more thereof may be used in combination.
- ethylene/propylene random copolymers and 1-butene/ethylene/propylene random copolymers are preferred in that the resulting expanded beads have good foaming properties, and that the obtained expanded beads are molded. It is suitable in that the molded article has good moldability.
- the 1-butene is synonymous with butene-1.
- the ethylene content in the ethylene/propylene random copolymer is preferably 0.2 wt% to 15.0 wt%, and preferably 0.5 wt% to 10.0 wt%, based on 100 wt% of each copolymer. % by weight is more preferred, and 0.5% to 4.0% by weight is even more preferred.
- the ethylene content in the copolymer is intended to mean the content (% by weight) of structural units (ethylene units) derived from ethylene contained in 100% by weight of the total amount of all structural units constituting the copolymer. do.
- the content of ethylene units in the ethylene/propylene random copolymer is (i) 0.2% by weight or more, the foamability of the resin beads in the production of the present expanded beads and/or the molding of the obtained expanded beads is affected. (ii) When the content is 15.0% by weight or less, there is no possibility that the mechanical properties of the foamed molded product obtained by molding the present expanded beads are deteriorated.
- the ethylene content in the 1-butene/ethylene/propylene random copolymer is preferably 0.1% by weight to 10.0% by weight, and 0.2% by weight to 5.0% by weight, based on 100% by weight of each copolymer. %, more preferably 0.5 wt % to 1.0 wt %.
- the content of ethylene units in the 1-butene/ethylene/propylene random copolymer is (i) 0.1% by weight or more, the expandability of the expanded beads in the production of the present expanded beads and/or the obtained The moldability of the expanded beads tends to be good, and (ii) when the content is 10.0% by weight or less, the mechanical properties of the expanded molded product obtained from the expanded beads are not likely to deteriorate.
- the content of 1-butene in the 1-butene/ethylene/propylene random copolymer is preferably 0.2% by weight to 15.0% by weight in 100% by weight of the copolymer. 0 wt % to 10.0 wt % is more preferred, and 2.0 wt % to 7.0 wt % is even more preferred.
- the 1-butene content in the copolymer is the content of structural units (1-butene units) derived from 1-butene contained in the total amount of 100% by weight of all structural units constituting the copolymer. (% by weight) is intended.
- the content of 1-butene units in the 1-butene/ethylene/propylene random copolymer is (i) 0.2% by weight or more, the expandability of the expanded beads in the production of the expanded beads and/or The moldability of the resulting expanded beads tends to be good, and (ii) when the amount is 15.0% by weight or less, there is no possibility that the mechanical properties of the expanded molded article formed by molding the present expanded beads are lowered.
- the total content of ethylene units and 1-butene units in the 1-butene/ethylene/propylene random copolymer is 0 in 100% by weight of the 1-butene/ethylene/propylene random copolymer.
- 0.5 wt % to 15.0 wt % is preferred, 0.5 wt % to 10.0 wt % is more preferred, and 2.0 wt % to 6.0 wt % is even more preferred.
- the expandability of the expanded beads in the production of the present expanded beads and/or the moldability of the resulting expanded beads tends to be good, and (ii) when the content is 10.0% by weight or less, the mechanical properties of the expanded molded article obtained from the present expanded beads may deteriorate. do not have.
- the melting point of the polypropylene resin (A) is preferably 125.0° C. to 160.0° C., more preferably 130.0° C. to 158.0° C., more preferably 135.0° C. to 152.0° C., and 138 0°C to 149.0°C is more preferred, 139.0°C to 146.0°C is even more preferred, and 141.0°C to 145.0°C is particularly preferred.
- the melting point of the polypropylene resin (A) is (i) 125.0° C. or higher, a foamed molded article having excellent dimensional stability at high temperatures can be obtained, and (ii) when it is 160.0° C. or lower. In-mold foam molding can be performed at a low steam pressure.
- the melting point of a polypropylene-based resin is a value obtained by measuring with a differential scanning calorimeter method (hereinafter referred to as "DSC method").
- DSC method differential scanning calorimeter method
- the specific operating procedure is as follows: (1) By raising the temperature of 5 mg to 6 mg of polypropylene resin from 40.0° C. to 220.0° C. at a rate of 10.0° C./min. (2) Then, the temperature of the melted polypropylene resin is lowered to 10°C. The polypropylene-based resin is crystallized by lowering the temperature from 220.0°C to 40.0°C at a temperature-lowering rate of 0/min; The temperature is raised from 40.0° C. to 220.0° C. at a heating rate of 1 min.
- the temperature of the peak (melting peak) of the DSC curve of the polypropylene-based resin obtained during the second heating can be obtained as the melting point of the polypropylene-based resin.
- the temperature of the peak (melting peak) with the maximum amount of heat of fusion is It is the melting point of the system resin.
- the differential scanning calorimeter for example, DSC6200 type manufactured by Seiko Instruments Inc. can be used.
- the melt index (MI) of the polypropylene resin (A) is not particularly limited, but is preferably 3.00 g/10 minutes to 30.00 g/10 minutes, and 4.00 g/10 minutes to 20.00 g/10 minutes. It is more preferably 5.00 g/10 minutes to 15.00 g/10 minutes, and particularly preferably 6.00 g/10 minutes to 13.00 g/10 minutes. Note that MI may also be referred to as "melt flow rate (MFR)".
- the MI of the polypropylene-based resin When the MI of the polypropylene-based resin is 3.00 g/10 minutes or more, the polypropylene-based resin has good fluidity during foaming, and foaming is easy. Further, when the MI of the polypropylene-based resin is 30.00 g/10 minutes or less, the polypropylene-based resin has appropriate fluidity, so that expanded beads having a high expansion ratio can be obtained. In particular, when the MI of the polypropylene-based resin (A) is 3.00 g/10 min to 8.00 g/10 min, it is possible to efficiently produce expanded beads capable of suitably providing a relatively high-density foamed article. be able to.
- the value of MI of a polypropylene resin is a value obtained by measuring under the following conditions using an MI measuring instrument described in JIS K7210: 1999: the diameter of the orifice is 2.0959 ⁇ 0.005 mm ⁇ , orifice length of 8.000 ⁇ 0.025 mm, load of 2.16 kgf, and temperature of 230° C. (230 ⁇ 0.2° C.).
- the MI of the polypropylene-based resin (A) means the melting point of the mixture.
- the expanded beads preferably contain 70.0% by weight or more, more preferably 75.0% by weight or more, and 77.0% by weight of the polypropylene-based resin (A) with respect to the total amount of 100% by weight of the expanded beads. % or more, more preferably 79.0% or more by weight.
- the foamed particles contain (i) 70.0% by weight or more of the polypropylene-based resin (A)
- the upper limit of the content of the polypropylene-based resin (A) in the present expanded beads is not particularly limited, but is, for example, 99.0% by weight or less, preferably 90.0% by weight, based on 100% by weight of the total amount of the expanded beads. and more preferably 80.0% by weight or less.
- the polypropylene resin (A) can be obtained by a known method.
- the polymerization catalyst for synthesizing the polypropylene-based resin (A) is not particularly limited, and Ziegler-based catalysts, metallocene catalysts, and the like can be used.
- the expanded beads preferably contain a hydrophilic substance (B).
- a hydrophilic substance B
- the water content of the expanded beads can be increased, and expanded beads having a desired water content (for example, 1.0% to 60.0%) can be stably obtained. can. That is, it has the advantage of being able to stably provide expanded particles that can provide foamed molded articles with excellent surface beauty and productivity.
- the expanded beads preferably contain more than 0.1% by weight and 20.0% by weight or less of the hydrophilic substance (B) with respect to 100% by weight of the total polypropylene-based expanded beads, and 1.0% to 19% by weight. more preferably .0 wt%, more preferably 5.0 wt% to 19.0 wt%, even more preferably 10.0 wt% to 18.0 wt%, more than 10.0 wt% , more preferably 18.0% by weight or less, and particularly preferably 13.0% to 17.0% by weight.
- the expanded beads contain (i) more than 0.1% by weight of the hydrophilic substance (B), it is possible to stably obtain expanded beads having a desired water content, and to obtain a foamed molded article having excellent flame retardancy. (ii) when it is contained in an amount of 20.0% by weight or less, it is possible to obtain a foam-molded article having no sink marks and having excellent strength (for example, compressive strength).
- hydrophilic substance is a compound having polarity in the molecule, and is intended to be a compound that improves the moisture content of the foamed beads obtained in the foaming process described later.
- Hydrophilic substances (B) include phosphonates such as cyclic phosphonates and cyclic bisphosphonates; intumescent flame retardants such as ammonium polyphosphate and melamine polyphosphate; glycerin, diglycerin, and polyethylene.
- C12-C18 aliphatic alcohols such as glycol, pentaerythritol, cetyl alcohol and stearyl alcohol; melamine; isocyanuric acid; melamine-isocyanuric acid condensates; zinc borate;
- phosphonates are preferred, and cyclic bisphosphonates are more preferred, since they can provide foamed molded articles with excellent flame retardancy.
- hydrophilic substances (B) may be used singly or in combination of two or more.
- (phosphonate) Phosphonates are M-PO(OH) 2 groups and/or M-PO(OM 1 ) 2 groups, where M is a monovalent hydrocarbon group such as methyl, benzyl, aryl, alkynyl groups, allenyl groups, vinyl groups or substituted vinyl groups) and M 1 is a hydrocarbyl group (such as an alkyl group or an aryl group), esters and partial esters, and salts thereof intended to
- Phosphonates include alkylphosphonic acids and arylphosphonic acids, and their esters; mono-phosphonates; acyclic bisphosphonates; cyclic phosphonates; Among the phosphonates, cyclic phosphonates are preferred, and cyclic bisphosphonates are particularly preferred.
- cyclic phosphonates include compounds represented by the following structural formula (i):
- R 1 and R 2 are independently a C 1-4 alkyl group, R 3 is H or a C 1-4 alkyl group, and R 4 is a C 9- 22 alkyl group, C 9-22 cycloalkyl group, C 9-22 aryl group or C 9-22 aralkyl group, and n is 0 or 1;
- Cyclic phosphonates include, for example, compounds represented by the following structural formula (ii):
- a 1 and A 2 are independently a C 1-10 alkyl group, a C 2-10 alkenyl group, a benzyl group, a phenylethyl group, a phenyl group or a naphthyl group.
- Cyclic bisphosphonates are disclosed in more detail by US Pat. No. 4,174,343 and British Patent Application Publication No. 1,515,223.
- pentaerythrityl diphosphonate represented by the following structural formula (iii) (for example, Thor GmbH, AFLAMMIT ( (registered trademark) PCO 900 (24% phosphorus content)) is particularly preferred.
- AFLAMMIT (registered trademark) PCO 900 (24% phosphorus content)
- 3,9-dimethyl-2,4,8,10-tetraoxa-3,9-diphosphaspiro[5,5]undecane-3,9-dioxide is particularly preferred as the pentaerythrityl diphosphonate.
- hydrophilic substance (B) a hydrophilic substance whose surface has been subjected to a coating treatment (for example, a surface-coated phosphonate, or a surface-coated ammonium polyphosphate, etc.) can be used.
- a coating treatment for example, a surface-coated phosphonate, or a surface-coated ammonium polyphosphate, etc.
- the expanded beads preferably further contain a hindered amine (C).
- a hindered amine C
- the present foamed beads contain the hindered amine (C)
- flame retardancy standards tend to be stricter, and foam molding with excellent flame retardancy body is required.
- Standards for such flame retardancy include oxygen index (Oxygen Index, OI), vertical combustion test (UL94), and the like.
- OI oxygen index
- UL94 vertical combustion test
- Inclusion of the hindered amine (C) in the foamed particles is preferable because it is possible to provide a foamed molded article that satisfies these criteria and has excellent flame retardancy.
- the expanded beads preferably contain 1.0% to 10.0% by weight, and 2.0% to 8.0% by weight, of the hindered amine (C) with respect to 100% by weight of the total polypropylene-based expanded beads. More preferably, it contains 2.5 wt % to 7.0 wt %, even more preferably 3.0 wt % to 6.0 wt %.
- the foamed particles contain (i) more than 0.1% by weight of the hindered amine (C), it is possible to obtain a foamed molded article having excellent non-ignitability and self-extinguishing properties, and (ii) 10.0% by weight or less. In this case, coalescence (blocking) between resin particles can be suppressed in the later-described foaming step.
- the hindered amine (C) is a hindered amine having an OR group (wherein R is a saturated or unsaturated monovalent hydrocarbon group) substituted directly on the N atom. (hereinafter sometimes referred to as N-substituted hindered amine).
- a hindered amine which does not have an OR group substituted directly on the N atom is not considered a hindered amine (C) herein.
- the hindered amine (C) is particularly limited as long as it is a hindered amine having an OR group (wherein R is a saturated or unsaturated monovalent hydrocarbon group) directly substituted on the N atom.
- the numerous hindered amine subclasses known to those skilled in the art can be used.
- the hindered amine (C) one hindered amine may be used alone, or two or more hindered amines may be used in combination.
- the hindered amine (C) is preferably an N-substituted hindered amine containing a triazine component (hereinafter sometimes referred to as triazine skeleton-containing hindered amine).
- examples of the hindered amine containing a triazine skeleton include (i) the compound of CAS No. 191680-81-6 ((i-1) peroxide-treated N-butyl-2,2,6,6,-tetramethyl-4-piperidinamine and 2,4,6-trichloro-1,3,5-triazine reacted with cyclohexane, and N,N'-bis(3-aminopropyl)ethylenediamine.
- Hindered amines containing a triazine skeleton are disclosed in more detail on page 2, line 32 to page 4, line 6 of EP 0889085.
- triazine skeleton-containing hindered amines Commercially available products can also be suitably used as triazine skeleton-containing hindered amines.
- Commercially available triazine skeleton-containing hindered amines include FLAMSTAB (registered trademark) NOR116 (CAS number 191680-81-6 compound) manufactured by BASF, HOSTAVIN (registered trademark) NOW XP manufactured by CLARIANT, and ADEKA STAB LA-81 manufactured by Adeka. (bis(1-undecaneoxy-2,2,6,6-tetramethylpiperidin-4-yl)carbonate) and the like.
- the hindered amine (C) may be a compound represented by the following structural formula (iv):
- G 1 and G 2 are independently C 1-8 alkyl groups, or pentamethylene; Z 1 and Z 2 are each methyl groups, or Z 1 and Z 2 together form a linking moiety, which may be additionally substituted by an ester, ether, amide, amino, carboxy or urethane group; and E is , C 1-8 alkoxy group, C 5-12 cycloalkoxy group C 7-15 aralkoxy group, —O—C(O)—C 1-18 alkyl group, or —OT—(OH) b group where T is (i) a C 1-18 alkylene chain, (ii) a C 5-18 cycloalkylene chain, (iii) a C 5-18 cycloalkenylene chain, or (iv) a phenyl group or a C 1-4 a C 1-4 alkylene chain substituted by an alkyl-substituted phenyl group; b is 1 to 3 and not more than the number of
- the hindered amine (C) may be a compound represented by the following structural formula (v):
- R is hydrogen or a methyl group
- R 1 is a C 1-18 alkyl group, a C 2-18 alkenyl group, a C 2-18 alkynyl group, a C 5-12 cycloalkyl a C 5-8 cycloalkenyl group, a C 6-10 aryl group or a C 7-9 aralkyl group.
- the hindered amine (C) may be a compound represented by the following structural formula (vi):
- E, k, Y, W, R 1 -R 7 and G 1 -G 4 are as defined in US Pat. No. 8,598,369.
- the expanded beads may contain a phosphonate as a hydrophilic substance (B) and a hindered amine (C). preferable.
- ratio (B)/(C)) is preferably 1.0 to 20.0, more preferably 1.0 to 15.0, and 1.0 to More preferably 10.0, more preferably 2.0 to 10.0, more preferably 2.0 to 7.0, more preferably 2.0 to 5.0 It is preferably 2.0 to 4.0, more preferably 2.0 to 4.0.
- the resulting foamed molded product has (a) excellent flame retardancy and (b) particles that burn during combustion. In addition to suppressing the ignition of surrounding combustibles due to the occurrence of (i.e., excellent self-extinguishing properties), (c) various physical properties (mechanical properties, cushioning properties, etc.) of the foam molding are impaired. No fear.
- the ratio of the total amount (total weight) of the hydrophilic substance (B) and the hindered amine (C) to the weight of the polypropylene resin (A) contained in the expanded beads is preferably 1 to 10, more preferably 1 to 7, more preferably 1 to 5, more preferably 2 to 5, 3 ⁇ 5 is more preferred.
- the expanded beads may optionally contain other additives in addition to the hydrophilic substance (B) and the hindered amine as long as they do not impair the effects of the present invention.
- Other additives include antioxidants, UV light absorbers, peroxide scavengers, nucleating agents, antistatic agents, antioxidants, light stabilizers, conductive agents, lubricants, fillers, carbon black, powder Activated carbon etc. are mentioned.
- such other additives may be (a) added directly to the blend or the polypropylene resin composition described later when producing the resin particles, and (b) adding other additives in advance.
- the additive may be contained in a resin at a high concentration to form a masterbatch, and the obtained masterbatch resin may be added to the blend or the polypropylene resin composition.
- a polypropylene-based resin is preferable as the resin used when producing the masterbatch resin.
- Antioxidants include, for example, alkylated monophenols, alkylthiomethylphenols, hydroquinones and alkylated hydroquinones, tocopherols, thiodiphenyl ether hydroxides, alkylidene bisphenols, O-containing benzyl compounds, N-containing benzyl compounds, S-containing benzyl compounds, hydroxybenzyl hydroxymalonates, aromatic hydroxybenzyl compounds, acylaminophenols, amides and esters of hydroxyphenylpropionic acid, and the like. These antioxidants may be used alone or in combination of two or more.
- the content of the antioxidant in the foamed particles can suppress the deterioration of the resulting foamed molded product at high temperatures (for example, about 110 ° C.), and as a result, the flame retardancy can be maintained for a longer period of time.
- 0.03% to 1.00% by weight, more preferably 0.05% to 0.70% by weight, and 0.10% to 0.50% by weight, relative to the total amount of 100% by weight of the system expanded particles % by weight is more preferred.
- UV light absorbers include, for example, benzotriazoles, benzophenones, benzoic acid esters, nickel complexes, hindered amines without OR groups directly linked to N atoms (included in the hindered amines (C) of the present invention). no), and oxamide and the like. These UV light absorbers may be used alone or in combination of two or more.
- the content of the UV light absorber in the present foamed particles can suppress deterioration of the resulting foamed molded product due to ultraviolet rays, and as a result, can maintain flame retardancy for a longer period of time. 0.01 wt % to 1.00 wt % is preferable, 0.05 wt % to 0.50 wt % is more preferable, and 0.10 wt % to 0.30 wt % is even more preferable.
- Nucleating agents include, for example, talc, titanium oxide, silica (silicon dioxide), silicate, alumina, diatomaceous earth, calcium carbonate, magnesium oxide, magnesium carbonate, magnesium sulfate, calcium phosphate, feldspar apatite, barium sulfate, and the like. .
- silicates include inorganic nucleating agents such as talc, magnesium silicate, kaolin, halloysite, deckite, aluminum silicate, and zeolite, as well as bis(p-methylbenzylidene) sorbitol, dibenzylidene sorbitol, bis (p-ethylbenzylidene) sorbitol, hydroxy(t-butylbenzoate)aluminum, bis(4-tbutyl-phenyl)sodium phosphate, methylenebis(2,4-ditbutyl-phenyl)phosphate sodium salt, potassium rosinate , magnesium rosinate, N,N′-dicyclohexyl-2,6-naphthalene dicarboxamide, N,N′,N′′-tris(2-methylcyclohexan-1-yl)propane-1,2,3-triylcarboxamide
- these nucleating agents commercially available ones may be used, and examples of commercially available products include
- nucleating agents may be used alone or in combination of two or more.
- an inorganic nucleating agent and an organic nucleating agent may be used in combination.
- a nucleating agent may also be referred to as a nucleating agent or a crystal nucleating agent.
- the content of the nucleating agent in the expanded beads is preferably 0.005% to 3.000% by weight with respect to 100% by weight of the total polypropylene-based expanded beads. 0.010% to 2.000% by weight is more preferred, and 0.030% to 1.000% by weight is most preferred.
- Lubricants include, for example, fatty acid esters, polyethylene wax (optionally partially saponified), zinc stearate, glycerol esters, and alkaline earth metal soaps. These lubricants may be used alone or in combination of two or more.
- the content of the lubricant in the foamed beads is preferably 0.01% to 1.00% by weight with respect to 100% by weight of the total polypropylene-based foamed beads, because it can improve the dispersibility of other additives. 0.05% to 0.50% by weight is more preferred, and 0.10% to 0.30% by weight is even more preferred.
- fillers examples include silicate, glass fiber, kaolin, wood flour, graphite, graphene, and cellulose nanofiber. These fillers may be used alone or in combination of two or more.
- the content of the filler in the present expanded beads is, for example, preferably 0.01 wt% to 10.00 wt%, more preferably 0.05 wt% to 7.00 wt%, relative to 100 wt% of the total amount of the present expanded beads. More preferably, 0.10% by weight to 5.00% by weight is even more preferable.
- the foamed particles may further contain a pigment such as carbon black or graphite in order to color the resulting foamed molded product gray or black.
- the content of carbon black in the foamed particles is 0.01% to 6.00% by weight with respect to 100% by weight of the total amount of the foamed particles, from the viewpoint of not impairing the flame retardancy and uniform coloring. preferably 0.30 wt % to 4.00 wt %, even more preferably 0.50 wt % to 3.00 wt %.
- the foamed particles may further contain powdery activated carbon in order to further improve the flame retardancy of the obtained foamed molded product.
- the content of the powdery activated carbon in the foamed particles is preferably 0.8% by weight to 2.8% by weight, more preferably 1.0% by weight to 2.5% by weight.
- the water content of the expanded beads is preferably 1.0% to 60.0%, more preferably 3.0% to 50.0%, and 7.0% to 40.0%. More preferably, 11.0% to 30.0%, even more preferably 15.0% to 30.0%, even more preferably 20.0% to 30.0% is particularly preferred.
- the water content of the present expanded beads is (i) 1.0% or more, no internal pressure is applied to the expanded beads, or even if the internal pressure applied to the expanded beads is small, a foamed molded article having excellent surface beauty is obtained.
- it is 60.0% or less, there is an advantage that shrinkage of expanded beads immediately after expansion can be suppressed.
- the water content of the expanded beads means the water content of the expanded beads after drying with a dryer (sometimes simply referred to as "after drying") and/or the water content during molding. That is, the foamed particles may have a moisture content of 1.0% to 60.0% after drying or at the time of molding.
- the moisture content of the expanded beads immediately after foaming is more than 60.0%
- the expanded beads are dried before molding.
- the water content of the foamed beads at the time of molding may be adjusted to 1.0% to 60.0% by a method such as machine drying.
- the time required for drying the expanded beads can be shortened or eliminated, and the productivity is excellent. It is preferable that both the ratio and the ratio are 1.0% to 60.0%.
- the moisture content of the expanded beads after drying and/or during molding is preferably 1.0% to 60.0%, more preferably 3.0% to 50.0%. 0% to 40.0%, more preferably 11.0% to 30.0%, even more preferably 15.0% to 30.0%; 0% to 30.0% is particularly preferred.
- the water content of the expanded beads immediately after expansion may be 1.0% to 80.0%, preferably 1.0% to 60.0%, and more preferably 3.0% to 50.0%. More preferably 0%, even more preferably 11.0% to 30.0%, even more preferably 15.0% to 30.0%, even more preferably 20.0% to 30.0% % is particularly preferred.
- the term “expanded beads immediately after expansion” refers to expanded beads immediately after expansion of resin beads, and more specifically, expanded beads immediately after (within 120 minutes) the releasing step described later. is intended for expanded particles prior to drying or molding.
- the “expanded particles at the time of molding” refers to the expanded particles immediately before (within 120 minutes) impregnation with air when (i) air is impregnated (internal pressure is applied) when molding the present expanded particles. and (ii) when not impregnated with air, expanded particles immediately before (within 120 minutes) filling into a mold or the like are intended.
- the expanded beads in molding the expanded beads, the expanded beads (expanded beads immediately after expansion) may be dried in a dryer to adjust the moisture content of the expanded beads immediately after the foaming until the time of molding. good.
- the moisture content of the expanded beads after drying may be outside the range of one embodiment of the present invention. This is because, for example, the moisture content of the expanded beads after drying is within the range of one embodiment of the present invention (1.0% to 60.0%), without adjusting the drying time. This is the case of drying. Although it depends on the drying temperature, for example, when drying in a dryer at 100° C.
- the moisture content of the expanded beads before drying is within the scope of one embodiment of the present invention. Even within (1.0% to 60.0%), the moisture content of the expanded beads after drying falls outside the range (less than 1%) of one embodiment of the present invention.
- the moisture content of the expanded beads after drying is within the range of one embodiment of the present invention
- the moisture content of the expanded beads after molding is out of the scope of one embodiment of the present invention
- the dried expanded beads are not regarded as the present expanded beads.
- the "moisture content of expanded beads after drying in a dryer" is a value measured immediately after the drying treatment (within 120 minutes after the drying treatment is finished) and before molding. .
- moisture content of foamed beads means the amount of water contained inside the foamed beads, and such water adhering to the surface of the foamed beads is not regarded as water contained in the foamed beads. . In other words, it is not used to calculate the moisture content of the expanded beads.
- the method for measuring the water content of the expanded beads is as follows (1) to (4).
- the weight (W1) is measured;
- W2) is measured;
- the method for adjusting the water content of the present expanded beads is not particularly limited, but for example, (i) a method of expanding resin beads containing a hydrophilic substance (B) to obtain expanded beads, (ii) the method of (i) above. , furthermore, a method of adjusting the amount of the hydrophilic substance (B) added, (iii) the amount of dispersant used during foaming of the resin beads (during the production of the foamed beads), and the temperature rising-pressurizing step described later. Examples thereof include a method of adjusting the foaming temperature, the holding time in the holding step described later, and/or the grain weight of the expanded beads, and (iv) a method of adjusting drying conditions for the expanded beads after foaming.
- the method (i) of obtaining expanded beads by expanding the resin beads containing the hydrophilic substance (B) is preferred because expanded beads having a high expansion ratio can be obtained.
- the density (bulk density) of the expanded beads is not particularly limited, but is preferably 15.0 g/L to 400.0 g/L, more preferably 60.0 g/L to 200.0 g/L. , 70.0 g/L to 160.0 g/L, and even more preferably 80.0 g/L to 155.0 g/L.
- the bulk density of the expanded beads is (i) 15.0 g/L or more, there is an advantage that the internal pressure required for molding the expanded beads is low, and (ii) when it is 400.0 g/L or less In some cases, there is an advantage that a foam molded article having excellent lightness can be obtained.
- the density of the expanded beads is intended to be the bulk density of the expanded beads before impregnation with air (before applying internal pressure). do.
- the average cell diameter of the expanded beads is not particularly limited, but is preferably 100 ⁇ m to 600 ⁇ m, more preferably 150 ⁇ m to 450 ⁇ m or less, and even more preferably 200 ⁇ m to 400 ⁇ m.
- the average cell diameter of the foamed particles is (i) 100 ⁇ m or more, a foamed molded article having excellent surface beauty can be obtained.
- the particle size is 600 ⁇ m or less, the secondary foamability of the expanded particles during in-mold foam molding is improved, and a foam molded article having excellent surface beauty can be obtained. It can also be said that the average cell diameter of the expanded beads is the cell diameter of the expanded beads.
- the heat of fusion on the high temperature side of the expanded beads is preferably 5.0 J/g to 25.0 J/g, more preferably 8.0 J/g to 22.0 J/g, and 10.0 J/g. More preferably ⁇ 20.0 J/g.
- the heat of fusion of the foamed particles on the high-temperature side is (i) 5.0 J/g or more, the resulting foamed molded product suffers from the occurrence of sink marks on the surface of the foamed molded product during in-mold foam molding, and dimensional changes.
- the shrinkage can be suppressed and (ii) it is 25.0 J/g or less, it is possible to obtain a foamed molded article with excellent internal fusion bondability and a smooth surface. It can also be said that the high temperature side melting heat quantity is the high temperature side melting peak heat quantity.
- the heat of fusion of the expanded beads on the high temperature side was measured by the following procedures (1) to (5): (1) weigh about 5 mg of the expanded beads; The temperature is raised from 10° C. to 190° C. at a rate of temperature increase to melt the expanded beads; (3) In the DSC curve of the expanded beads obtained in the process of (2) above, the point representing the temperature before the start of melting. and the point representing the temperature after the end of melting with a straight line to create a baseline; (5) The amount of heat (J/g) calculated from the area on the high temperature side surrounded by the DSC curve and the straight line passing through the baseline and maximum points calorie.
- One aspect of the present invention is expanded polypropylene beads having a moisture content of 1.0% to 60.0% after drying in a dryer.
- the drying temperature (for example, the temperature in the dryer) is not particularly limited as long as the moisture content of the expanded beads after drying is (becomes) 1.0% to 60.0%, but for example, 40°C. ⁇ 100°C is preferred, 50°C to 95°C is more preferred, and 60°C to 90°C is even more preferred.
- the drying time is not particularly limited as long as the moisture content of the expanded beads after drying is (is) 1.0% to 60.0%, but for example, 10 minutes to 20 hours. Preferably, 20 minutes to 3 hours, more preferably 20 minutes to 2 hours.
- the drying temperature is normal temperature below 40°C (for example, 15°C to 30°C)
- the foamed beads are left in a state of being ventilated (ventilated) for a long period of time, the moisture content of the foamed beads can decrease.
- drying method is not particularly limited, for example, hot air drying is preferable.
- the dryer is preferably a hot air dryer.
- Specific aspects of hot air drying include, for example, a method of blowing hot air into a dryer, and a drying method of blowing hot air into a tank while fluidizing the foamed particles in a fluidized bed.
- the expanded polypropylene particles according to one embodiment of the present invention do not necessarily require drying with a dryer.
- the water content of the expanded beads before drying is within the range of one embodiment of the present invention (1.0% to 60.0%)
- the expanded beads having that water content can be molded without drying. can be used for
- the method for producing polypropylene-based expanded beads according to one embodiment of the present invention is not particularly limited. and a discharging step of discharging the resulting dispersion to a region of lower pressure than the pressure within the vessel.
- the expanded polypropylene particles according to one embodiment of the present invention are formed by expanding polypropylene resin particles. It can also be said that the expanded polypropylene particles according to one embodiment of the present invention contain polypropylene resin particles.
- the production method may include a granulation step of preparing polypropylene-based resin particles before the foaming step.
- polypropylene-based resin particles may be referred to as "resin particles”.
- the granulation step in this production method is not particularly limited as long as resin particles can be obtained, but an example thereof includes a method using an extruder.
- resin particles can be produced by the following methods (1) to (5): (1) a predetermined amount of polypropylene resin (A) and, if necessary, a hydrophilic substance ( B), a hindered amine (C), and other additives are blended to prepare a blend; (2) the blend is introduced into an extruder and melt-kneaded to form a polypropylene (3) extruding the polypropylene-based resin composition through a die provided in an extruder; (4) solidifying the extruded polypropylene-based resin composition by cooling it by, for example, passing it through water; (5) Thereafter, the solidified polypropylene-based resin composition is chopped into desired shapes such as cylindrical, elliptical, spherical, cubic, rectangular parallelepiped, etc.
- melt-kneaded polypropylene resin composition is extruded directly into water from a die provided in an extruder, and the polypropylene resin composition is cut into particles immediately after extrusion, cooled, and solidified. Resin particles may be obtained. By melt-kneading the blend in this manner, more uniform resin particles can be obtained.
- the ratio of the amounts of the polypropylene resin (A), the hydrophilic substance (B), and/or the hindered amine (C) contained in the resin particles is (a) the polypropylene resin in the blend prepared in the step (1) (A), hydrophilic substance (B), and/or hindered amine (C), and (b) polypropylene resin (A), hydrophilic substance (B), and/or It can also be said to be the ratio of the amount of the hindered amine (C).
- the hydrophilic substance (B) and/or the hindered amine (C) are previously melt-kneaded by an extruder. to form a mixture.
- Resin particles can also be produced by melt-kneading the mixture, the polypropylene-based resin (A), and, if necessary, other additives.
- the melting point of the resin particles is not particularly limited, but is preferably 122.0° C. to 159.0° C., more preferably 127.0° C. to 157.0° C., more preferably 132.0° C. to 151.0° C., 135.0°C to 148.0°C is more preferred, 136.0°C to 145.0°C is even more preferred, and 138.0°C to 144.0°C is particularly preferred.
- the melting point of the resin particles is (i) 122.0°C or higher, a foamed molded article having excellent dimensional stability at high temperatures can be obtained, and (ii) when it is 159.0°C or lower, the water vapor pressure is low. Then, the foam-molded product can be foam-molded in the mold.
- the melting point of resin particles is a value obtained by measuring with a differential scanning calorimeter method (hereinafter referred to as "DSC method").
- DSC method differential scanning calorimeter method
- the specific operating procedure is as follows: (1) The temperature of 5 mg to 6 mg of resin particles is raised from 40.0° C. to 220.0° C. at a rate of 10.0° C./min. (2) then lower the temperature of the melted resin particles to 10°C. The resin particles are crystallized by lowering the temperature from 220.0°C to 40.0°C at a temperature lowering rate of 0/min; The temperature is raised from 40.0°C to 220.0°C at a temperature elevation rate.
- the temperature of the peak (melting peak) of the DSC curve of the resin particles obtained during the second heating can be obtained as the melting point of the resin particles.
- the temperature of the peak (melting peak) with the maximum amount of heat of fusion is taken as the temperature of the resin particles.
- the melt index (MI) of the resin particles is not particularly limited, but is preferably 3.0 g/10 minutes to 30.0 g/10 minutes, more preferably 4.0 g/10 minutes to 20.0 g/10 minutes. 0 g/10 min to 15.0 g/10 min is more preferred, and 6.0 g/10 min to 13.0 g/10 min is particularly preferred. Note that MI may also be referred to as "melt flow rate (MFR)".
- the MI of the resin particles When the MI of the resin particles is 3.0 g/10 minutes or more, the flowability of the polypropylene resin during foaming is good, and foaming is easy. Further, when the MI of the resin particles is 30.0 g/10 minutes or less, the resin particles have appropriate fluidity, so that expanded beads having a high expansion ratio can be obtained. In particular, when the MI of the resin particles is 3.0 g/10 min to 8.0 g/10 min, it is possible to suitably provide a relatively high-density (for example, density of more than 92.0 g/L) foamed molded product. , the expanded beads can be produced efficiently.
- a relatively high-density for example, density of more than 92.0 g/L
- the MI value of the resin particles is a value obtained by measuring under the following conditions using an MI measuring instrument described in JIS K7210: 1999: the diameter of the orifice is 2.0959 ⁇ 0. .005 mm ⁇ , orifice length of 8.000 ⁇ 0.025 mm, load of 2.16 kgf, and temperature of 230° C. (230 ⁇ 0.2° C.).
- the weight of one resin particle is not particularly limited, but is preferably 0.3 mg/particle to 10.0 mg/particle, more preferably 0.4 mg/particle to 8.0 mg/particle, and 0 .5 mg/grain to 7.0 mg/grain is more preferable, 0.7 mg/grain to 6.0 mg/grain is more preferable, 1.0 mg/grain to 5.0 mg/grain is more preferable, and 1.2 mg/grain to 5.0 mg/grain is more preferred.
- the particle weight of the resin particles can affect the moisture content of the resulting expanded particles. In other words, it is possible to adjust the water content of the expanded beads by adjusting the particle weight of the resin particles that are the raw material of the expanded beads.
- the particle weight of the resin particles is within the above range, there is an advantage that expanded particles having a water content of 1.0% to 60% can be easily obtained.
- the dispersing step can also be said to be, for example, a step of preparing a dispersion liquid in which resin particles, a foaming agent, and, if necessary, a dispersing agent and/or a dispersing aid are dispersed in an aqueous dispersion medium.
- the container is not particularly limited, it is preferably a container that can withstand the later-described foaming temperature and foaming pressure.
- the container is preferably, for example, a pressure-resistant container, more preferably an autoclave-type pressure-resistant container.
- the container may also be equipped with an agitator within the container.
- the aqueous dispersion medium is not particularly limited as long as it can uniformly disperse the resin particles, foaming agent, and the like.
- aqueous dispersion media include (a) dispersion media obtained by adding methanol, ethanol, ethylene glycol, glycerin, etc. to water, and (b) water such as tap water and industrial water.
- water-based dispersion media include RO water (water purified by reverse osmosis membrane method), distilled water, deionized water (water purified by ion exchange resin), and the like. It is preferable to use pure water, ultrapure water, or the like.
- the amount of the aqueous dispersion medium used is not particularly limited, but is preferably 100 to 500 parts by weight with respect to 100 parts by weight of the resin particles.
- the amount of the aqueous dispersion medium used is (a) 100 parts by weight or more, there is no risk of deterioration in the stability of the dispersion (in other words, the resin particles are well dispersed), and (b) 500 parts by weight or less. In this case, there is no possibility that the productivity is lowered.
- the foaming agent includes (a) (a-1) an inorganic gas such as nitrogen, carbon dioxide, air (a mixture of oxygen, nitrogen, and carbon dioxide), and (a-2) an inorganic foaming agent such as water; (b) (b-1) saturated hydrocarbons having 3 to 5 carbon atoms such as propane, normal butane, isobutane, normal pentane, isopentane and neopentane, (b-2) ethers such as dimethyl ether, diethyl ether and methyl ethyl ether , (b-3) organic foaming agents such as monochloromethane, dichloromethane, dichlorodifluoroethane, chloroethane, and halogenated hydrocarbons such as hydrofluoroolefins; As the foaming agent, at least one or more selected from the group consisting of the above inorganic foaming agents and organic foaming agents can be used.
- the mixing ratio may be appropriately adjusted depending on the purpose.
- the inorganic foaming agent is preferable as the foaming agent among those mentioned above.
- the foaming agent is more preferably an inorganic foaming agent containing carbon dioxide, because it has a moderately high plasticizing effect and tends to improve the expandability of the expanded beads in the production of the present expanded beads. It is even more preferable to have
- the amount of the foaming agent to be used is not particularly limited, and may be used appropriately according to (a) the type of foaming agent and/or (b) the desired expansion ratio of the foamed particles.
- the amount of the foaming agent used is preferably 1 part by weight to 10000 parts by weight, more preferably 1 part by weight to 5000 parts by weight, and even more preferably 1 part by weight to 1000 parts by weight with respect to 100 parts by weight of the resin particles.
- the amount of the foaming agent used is 1 part by weight or more with respect to 100 parts by weight of the resin particles, expanded beads having a suitable density can be obtained.
- the amount of the foaming agent used is 10000 parts by weight or less with respect to 100 parts by weight of the resin particles, an effect corresponding to the amount of the foaming agent used can be obtained, and no economic waste occurs.
- the amount of the foaming agent used may be, for example, 1 to 100 parts by weight or 1 to 10 parts by weight with respect to 100 parts by weight of the resin particles.
- the water in the dispersion liquid in the container can be used as the foaming agent.
- the resin particles contain a water-absorbing substance in advance. This makes it easier for the resin particles to absorb the water in the dispersion liquid in the container, and as a result, it becomes easier to use the water as a blowing agent.
- a dispersant in the present method for producing expanded beads.
- the use of a dispersant has the advantage of reducing coalescence (sometimes referred to as blocking) between resin particles and stably producing expanded beads.
- examples of dispersants include inorganic substances such as tricalcium phosphate, trimagnesium phosphate, basic magnesium carbonate, calcium carbonate, barium sulfate, kaolin, talc, clay, aluminum oxide, titanium oxide, and aluminum hydroxide.
- One type of these dispersants may be used alone, or two or more types may be mixed and used. When two or more dispersants are mixed and used, the mixing ratio may be appropriately adjusted depending on the purpose.
- the amount of the dispersant used in the dispersion used in one embodiment of the present invention is preferably 0.01 to 3.00 parts by weight, preferably 0.05 to 2 parts by weight, relative to 100 parts by weight of the resin particles. 0.00 parts by weight is more preferred, and 0.10 to 1.50 parts by weight is even more preferred.
- the amount of the dispersant used is (a) 0.01 parts by weight or more, there is no risk of causing poor dispersion of the resin particles, and when it is (b) 3.00 parts by weight or less, the obtained expanded beads are used. At the time of in-mold foam molding, there is no possibility of causing poor adhesion between foamed particles.
- a dispersing aid is used to (a) improve the effect of reducing coalescence between resin particles and/or (b) improve the stability of the dispersion in the container.
- Dispersing aids include, for example, anionic surfactants.
- anionic surfactants include sodium alkylbenzenesulfonates such as sodium dodecylbenzenesulfonate, sodium alkanesulfonates, sodium alkylsulfonates, sodium alkyldiphenyletherdisulfonates, and sodium ⁇ -olefinsulfonates.
- anionic surfactants include sodium alkylbenzenesulfonates such as sodium dodecylbenzenesulfonate, sodium alkanesulfonates, sodium alkylsulfonates, sodium alkyldiphenyletherdisulfonates, and sodium ⁇ -olefinsulfonates.
- One type of these dispersing aids may be used alone, or two or more types may be
- the amount of the dispersing aid used in the dispersion used in one embodiment of the present invention is preferably 0.001 to 0.500 parts by weight with respect to 100 parts by weight of the resin particles. It is more preferably from 0.200 parts by weight, and even more preferably from 0.010 parts by weight to 0.200 parts by weight. When the amount of the dispersing aid used is within the above range, there is no risk of poor dispersion of the resin particles.
- This production method includes, after the dispersing step and before the discharging step, (i) a temperature-increasing step of raising the temperature inside the container to a constant temperature and increasing the pressure inside the container to a constant pressure; (ii) a holding step of holding the temperature and pressure in the container at a constant temperature and a constant pressure.
- the holding step is preferably performed after the temperature rising-pressurizing step.
- the (a) constant temperature in the heating-pressurizing step and the holding step may be referred to as the foaming temperature
- the (b) constant pressure may be referred to as the foaming pressure.
- the foaming temperature cannot be defined unconditionally because it varies depending on the type of polypropylene resin (A), hydrophilic substance (B), hindered amine (C), type of foaming agent, etc. contained in the resin particles.
- the foaming temperature is (i) a mixture of (a) polypropylene resin (A), hydrophilic substance (B), and/or hindered amine (C), (b) polypropylene resin composition, or (c) resin particles.
- the melting point is preferably ⁇ 20.0° C. to +20.0° C.
- the foaming pressure is preferably 0.5 MPa (gauge pressure) to 10.0 MPa (gauge pressure), more preferably 0.6 MPa (gauge pressure) to 5.0 MPa (gauge pressure), and 0.6 MPa (gauge pressure) to 2 0.5 MPa (gauge pressure) is more preferred. If the foaming pressure is 0.5 MPa (gauge pressure) or more, expanded beads having a suitable density can be obtained.
- the holding step is a step of holding the dispersion in the container at the foaming temperature and foaming pressure.
- the foaming temperature and foaming pressure held in the holding step do not have to be exact values, and may be within a certain range of values that are allowable from the manufacturing cost and technical point of view.
- the holding step can be said to be a step of holding the dispersion in the container near the foaming temperature and the foaming pressure.
- the production method preferably includes a dispersing step, a retaining step, and a releasing step.
- a preferred embodiment of the present production method may have the following configuration: a dispersing step of dispersing polypropylene resin particles, an aqueous dispersion medium, and a foaming agent in a container; A method for producing polypropylene-based expanded beads, comprising a step of holding the obtained dispersion at an expansion temperature and an expansion pressure, and a step of discharging the dispersion to a region having a pressure lower than the pressure in the container.
- the time (holding time) for holding the dispersion in the container at (near) the foaming temperature and (near) the foaming pressure is not particularly limited.
- the retention time is preferably 5 to 60 minutes, more preferably 10 to 60 minutes, more preferably 12 to 50 minutes, more preferably 15 to 40 minutes, and even more preferably 15 to 30 minutes.
- the holding time is 5 minutes or more, there is a sufficient amount of unmelted crystals (polypropylene-based resin crystals), and as a result, shrinkage of the resulting expanded beads and/or an increase in open cell ratio can be reduced.
- the holding time is 60 minutes or less, there is no excessive amount of unmelted crystals, so there is an advantage that the expanded beads can be molded at a low molding temperature.
- the retention time can affect the moisture content of the resulting foamed particles.
- the shorter the holding time the lower the water content of the obtained expanded beads, and the longer the holding time, the higher the water content of the obtained expanded beads.
- the holding time is within the range described above, there is an advantage that expanded beads having a water content of 1.0% to 60% can be easily obtained.
- the releasing step is (a) after the temperature raising-pressurizing step when the temperature raising-pressurizing step is performed but the holding step is not performed, and (b) when both the temperature raising-pressurizing step and the holding step are performed. It is preferably carried out after the holding step.
- the expulsion step can cause the resin particles to expand, resulting in expanded particles.
- the releasing step can also be said to be a step of releasing one end of the container and releasing the dispersion liquid in the container into a region (space) having a lower pressure than the foaming pressure (that is, the internal pressure of the container).
- area under pressure lower than the foaming pressure intends “area under pressure lower than the foaming pressure” or “space under pressure lower than the foaming pressure”, and “atmosphere at pressure lower than the foaming pressure”. It can also be called “lower”.
- the region of pressure lower than the foaming pressure is not particularly limited as long as the pressure is lower than the foaming pressure, and may be, for example, a region under atmospheric pressure.
- the dispersion In the ejection process, when the dispersion is ejected to a region with a pressure lower than the foaming pressure, the dispersion is passed through an orifice with a diameter of 1 mm to 5 mm for the purpose of adjusting the flow rate of the dispersion and reducing the variation in expansion ratio of the resulting expanded beads. can also be emitted.
- the low-pressure region space may be filled with saturated steam.
- foaming process The process from the dispersion process to the release process may be called a foaming process. Further, the process of producing expanded beads from resin particles in this way is called a “single-stage expansion process", and the obtained expanded beads are called “single-stage expanded beads”.
- method 1 in which a large amount of inorganic foaming agent is used in the first-stage expansion step. Furthermore, as a method other than method 1, after obtaining expanded beads (single-stage expanded beads) with a relatively low expansion ratio (expansion ratio of about 2.0 to 35.0 times) in the first-stage expansion process, A method of increasing the expansion ratio by expanding the stepped expanded particles again (hereinafter referred to as method 2) can also be employed.
- Method 2 includes, for example, a method including the following (a1) to (a3) in order: (a1) using single-stage expanded particles having an expansion ratio of 2.0 to 35.0 times in the single-stage expansion step; (a2) Place the first-stage expanded particles in a pressure vessel and pressurize with nitrogen, air, carbon dioxide, etc. at 0.2 MPa (gauge pressure) to 0.6 MPa (gauge pressure) to produce one stage. (a3) A method in which the pressure inside the expanded bead (hereinafter sometimes referred to as “internal pressure”) is raised above normal pressure; (a3) After that, the first-stage expanded bead with increased internal pressure is heated with steam or the like to further expand.
- the step of increasing the expansion ratio of the first-stage expanded beads as in Method 2 is called the "two-stage expanded process", and the polypropylene-based resin expanded beads obtained by the method of Method 2 are called “two-stage expanded beads”.
- the pressure of steam for heating the first-stage expanded beads is 0.03 MPa (gauge pressure) to 0.20 MPa ( gauge pressure).
- the steam pressure in the two-stage expansion step is 0.03 MPa (gauge pressure) or more, the expansion ratio tends to be improved, and when it is 0.20 MPa (gauge pressure) or less, the obtained two-stage expanded particles are separated from each other. are less likely to coalesce.
- the obtained two-stage expanded beads may not be able to be subjected to subsequent in-mold foam molding.
- the internal pressure of the first-stage expanded particles obtained by impregnating the first-stage expanded particles with nitrogen, air, carbon dioxide, or the like can be appropriately changed in consideration of the expansion ratio of the second-stage expanded particles and the water vapor pressure in the second-stage expansion process. desirable.
- the internal pressure of the first-stage expanded beads is preferably 0.15 MPa (absolute pressure) to 0.60 MPa (absolute pressure), more preferably 0.20 MPa (absolute pressure) to 0.60 MPa (absolute pressure), and 0.30 MPa (absolute pressure). pressure) to 0.60 MPa (absolute pressure) is more preferable.
- the production method may further include a drying step for drying the foamed particles.
- the expanded beads to be dried may be the expanded beads (single-stage expanded beads) obtained in the releasing step (expansion step) or the two-step expanded beads obtained in the two-step expanded step.
- drying temperature drying temperature
- drying time drying time
- drying method in the drying process are the same as those described in the ⁇ Drying> section above, so the description is used and the description is omitted here.
- a polypropylene-based foam-molded article according to one embodiment of the present invention is a foam-molded article obtained by molding the present expanded particles (eg, in-mold foam molding).
- the "polypropylene-based foamed molded article according to one embodiment of the present invention” may be referred to as "this foamed molded article”. It can also be said that the present foamed molded article is a foamed molded article formed by molding polypropylene-based foamed particles having a moisture content of 1.0% to 60.0% or less.
- the present foam molded article has the above structure, it has excellent surface beauty and can be produced with high productivity.
- the "surface beauty" of a foam molded product can be evaluated by surface elongation and edge elongation of the foam molded product.
- the excellent surface elongation of the foam molded product means that there are almost no intergranular spaces (spaces (gaps) between expanded particles constituting the foam molded product) on the surface of the foam molded product. means that there is almost no
- the excellent edge elongation of the foam molded product means that there are almost no intergranular grains at the edge portion (corner portion) of the molded product, that is, the edge portion of the foam molded product has almost no unevenness (no missing grains at the edge portion).
- the foam molded article "excellent in surface beauty" means a foam molded article excellent in surface elongation and edge elongation.
- the surface elongation and edge elongation of the foam molded product can be evaluated by the methods described in Examples.
- the density of the foam molded product is preferably 15.0 g/L to 400.0 g/L, preferably 50.0 g/L to 300.0 g/L, and more preferably 100.0 g/L to 200.0 g/L. 0 g/L is preferred. If the density of the foam-molded article is 15.0 g/L or more, there is an advantage that a foam-molded article having a smooth surface without sink marks and having a more excellent surface beauty can be obtained. /L or less, a sufficiently lightweight foam molded article can be obtained.
- the foam-molded product is preferably a foam-molded product having excellent flame retardancy, since it can be suitably used as a member that requires high flame resistance, such as a peripheral member of an electrical component.
- the flame retardancy of the foam molded product can be evaluated by UL94V "Vertical Combustion Foamed Material Test". The test method of UL94V "Vertical Combustion Foam Material Test" will be described in detail in the examples below.
- the flame retardancy of the foam molded product is evaluated in four grades (V-0, V-1, V-2, NG in order of superior flame retardancy).
- the present foam molded article preferably satisfies at least grade V-2, more preferably grade V-1, and particularly preferably grade V-0.
- the thickness of the foamed molding is one of the factors that affect the evaluation results.
- the thinner the foam molded body the greater the ratio of the surface area to the volume of the foam molded body, so the flame retardant grade tends to deteriorate due to reasons such as an increase in the ratio of the combustion area during combustion. .
- the foam-molded article that satisfies the grade of excellent flame retardancy is the foam-molded article that has superior flame retardancy. I can say.
- the present foamed molded article was grade V- 0 is satisfied and grade V-2 is preferably satisfied when the thickness of the foamed molded article is 5 mm, and grade V-0 is satisfied when the thickness of the foamed molded article is 13 mm and 8 mm. It is more preferable to satisfy Grade V-1 when the thickness of the molded foam is 5 mm, and it is particularly preferable to satisfy Grade V-0 when the thickness of the foam molded product is 13 mm, 8 mm and 5 mm.
- the density of the foam molded body is also one of the factors that affect the test results of the UL94V test.
- the smaller the density of the foamed molded article the smaller the amount of components derived from the raw material expanded particles (present expanded particles) contained in the expanded molded article. Therefore, it becomes difficult to enjoy the effect of improving the flame retardancy due to the component contributing to the flame retardancy (for example, the hydrophilic substance (B) and/or the hindered amine (C)) contained in the foamed beads.
- the test results of the UL94V test for foamed moldings tend to deteriorate. Therefore, it is not possible to simply compare the test results of the UL94V test between foamed molded articles having greatly different densities.
- excellent flame retardancy means that the foamed molded article having an equivalent density (a foamed molded article that does not satisfy one embodiment of the present invention) has superior flame retardancy intended to have sex.
- the method for producing the foamed molded article includes a step of molding the foamed particles. It can also be said that the present method for producing a foamed molded product includes a step of molding polypropylene-based foamed particles having a water content of 1.0% to 60.0% or less. Other aspects of the method for producing the present foam molded article are not particularly limited as long as the above configuration is satisfied.
- the molding method of the present foam molded product is an in-mold foam molding method including a step of filling a mold with polypropylene-based foamed particles and heating the polypropylene-based foamed particles filled in the mold with steam.
- a specific embodiment of the method for producing the present foam molded article by the in-mold foam molding method includes, for example, a production method including the following (b1) to (b6) in order, but is not limited to such a production method: (b1) A mold composed of a fixed mold that cannot be driven and a movable mold that can be driven is mounted on an in-mold foam molding machine.
- the fixed mold and the movable mold can be formed inside the fixed mold and the movable mold by driving the movable mold toward the fixed mold (this operation is sometimes referred to as "mold closing"); (b2) driving the movable mold toward the fixed mold so that a slight gap (also called cracking) is formed so that the fixed mold and the movable mold are not completely closed; (b3) filling the foamed particles into the molding space formed inside the stationary mold and the moving mold, for example through a filling machine; (b4) driving the movable mold so that the fixed mold and the movable mold are completely closed (that is, the mold is completely closed); (b5) performing in-mold foam molding by preheating the mold with steam, heating the mold in one direction and in reverse with steam, and heating both sides of the mold with steam; (b6) The in-mold foam-molded product is removed from the mold and dried (for example, dried at 75° C.) to obtain a foam-molded product.
- mold closing this operation is sometimes referred to as "mold closing”
- a mold having steam holes (core vents) can be suitably used as the mold to be mounted on the in-mold foam molding machine.
- the cracking (%) formed is not particularly limited, but is preferably 1% to 30%, preferably 5% to 25%, and preferably 15% to 25%. preferable. If the cracking is within the above range, there is an advantage that it is easy to obtain a compact having a smooth surface and a beautiful appearance.
- the term "cracking" refers to the distance between the moving mold and the fixed mold in the molding space of the mold during filling of the foamed particles, in other words, the resulting foamed molded product. (thickness of the foam-molded body calculated by dividing the volume of the foam-molded body by the projected area when light is applied to the foam-molded body from the moving direction of the moving mold) was taken as 100%. In this case, the extent of the opening width of the mold is intended.
- Expanded beads are pressurized with an inorganic gas in a container to impregnate the expanded beads with the inorganic gas, and a predetermined internal pressure (expanded beads A method of filling the foamed particles into the molding space after applying internal pressure); (b3-2) A mold whose volume can be changed is used as a mold, and after filling the molding space with foamed particles, the volume inside the mold is compressed by 10% to 75%. how to; (b3-3) A method of compressing foamed particles with gas pressure to fill the molding space; (b3-4) A method of filling a molding space with expanded particles without any particular pretreatment.
- the present expanded particles that is, polypropylene-based expanded particles having a water content of 1.0% to 60.0% or less
- the present method for producing a foamed molded product does not include the step of impregnating the foamed particles with an inorganic gas as in (b3-1) and applying an internal pressure to the foamed particles, or even if the internal pressure is low (for example, 0.25 MPa or less), it is possible to provide a foam molded article having excellent surface beauty. Therefore, the time for air impregnation can be shortened or eliminated, thereby improving the productivity (production efficiency) of the foam molded product.
- the method for producing the foamed molded article does not include a step of impregnating the polypropylene-based foamed particles with an inorganic gas, or impregnates the polypropylene-based foamed particles with an inorganic gas, It is preferable to include a step of applying an internal pressure of 25 MPa (absolute pressure) or less to the expanded polypropylene particles.
- the method for producing the foamed molded article includes a step of impregnating the polypropylene-based foamed particles with an inorganic gas
- the inorganic gas with which the foamed particles are impregnated may be air, nitrogen, oxygen, carbon dioxide, helium, neon, argon, or the like. At least one selected from the group consisting of can be used. Among these inorganic gases, air and/or carbon dioxide are preferred.
- the internal pressure applied to the expanded particles is 0.10 MPa (absolute pressure), preferably 0.25 MPa (absolute pressure) or less, more preferably 0.10 MPa (absolute pressure), 0.20 MPa (absolute pressure) or less, 0.10 MPa (absolute pressure), 0.18 MPa (absolute pressure) or less, and more preferably more than 0.10 MPa (absolute pressure) and 0.15 MPa (absolute pressure) or less.
- the internal pressure of expanded beads that are not impregnated with an inorganic gas (no internal pressure is applied) is usually equivalent to atmospheric pressure (0.10 MPa (absolute pressure)).
- the temperature in the container when impregnating the foamed particles with the inorganic gas is preferably 10°C to 90°C, more preferably 40°C to 90°C.
- the restoring force of the foamed particles compressed by gas pressure is used to fuse the foamed particles.
- water vapor pressure A water vapor pressure during one-way heating and reverse one-way heating
- water vapor pressure B water vapor pressure during double-sided heating
- the pressure of the water vapor pressure B is not particularly limited, but is preferably 0.20 MPa (gauge pressure) to 0.40 MPa (gauge pressure), more preferably 0.22 MPa (gauge pressure) to 0.38 MPa (gauge pressure), 0.24 MPa (gauge pressure) to 0.36 MPa (gauge pressure) is more preferable.
- this configuration there is a tendency to obtain a foam molded article with a high internal fusion bonding rate.
- by setting the steam pressure A to about 1/2 of the steam pressure B at the time of in-mold foam molding excessive pressurization is not required, it is economically advantageous, and the internal fusion rate is improved. It is preferable because it can provide a high foaming molded product.
- An embodiment of the present invention may include the following configuration.
- the hindered amine (C) is 2,4-bis((1-cyclohexyloxy-2,2,6,6-tetramethylpiperidin-4-yl)butylamino)-6-chloro-S-triazine and , N,N'-bis(3-aminopropyl)ethylenediamine.
- the ratio of the total weight of the hydrophilic substance (B) and the hindered amine (C) to the weight of the polypropylene resin (A) contained in the expanded polypropylene particles is 1 to 10 [14] or The expanded polypropylene particles according to [15].
- the polypropylene-based expanded particles are formed by expanding polypropylene-based resin particles, and the particle weight of the polypropylene-based resin particles is 0.3 mg/particle to 10.0 mg/particle.
- the polypropylene-based foamed particles according to any one of .
- a method for producing a polypropylene-based foam-molded product comprising a step of in-mold foam-molding the polypropylene-based foamed particles according to any one of [1] to [21].
- MI of polypropylene resin (A) and resin particles The MI of the polypropylene resin (A) and the resin particles was measured using an MI measuring instrument described in JIS K7210: 1999, with an orifice diameter of 2.0959 ⁇ 0.005 mm ⁇ and an orifice length of 8.000 ⁇ 0.025 mm. , a load of 2160 g and a temperature of 230 ⁇ 0.2°C.
- the melting point of the resin particles was measured using a differential scanning calorimeter (manufactured by Seiko Instruments Inc., model DSC6200). Specific measurement methods were as follows (1) to (3): (1) The temperature of 5 mg to 6 mg of resin particles was increased from 40.0° C. to 220° C. at a rate of 10.0° C./min. The resin particles were melted by raising the temperature to 0°C; The resin particles were crystallized by lowering the temperature from 220.0°C to 40.0°C at a temperature lowering rate of 0/min; The temperature was raised from 40.0° C. to 220.0° C. at a temperature elevation rate of .
- the temperature of the peak (melting peak) of the DSC curve of the resin particles obtained during the second heating was taken as the melting point of the resin particles.
- the temperature of the peak (melting peak) with the maximum amount of heat of fusion is taken as the temperature of the resin particles. was taken as the melting point of
- the method for measuring the average cell diameter of the foamed beads was as follows (1) to (5): (1) Using a razor (high stainless steel double-edged blade manufactured by Feather), so as to pass through the center of the foamed beads.
- the amount of heat of fusion on the high temperature side of the expanded beads was as follows (1) to (5): (1) about 5 mg of the expanded beads was weighed; (2) the temperature of the weighed expanded beads was increased to 10°C/min. (3) In the DSC curve of the expanded beads obtained in the process of (2) above, the temperature before the start of melting is A baseline was created by connecting the point and the point representing the temperature after the end of melting with a straight line; , drawn in the direction perpendicular to the X-axis; side melting heat.
- the method for measuring the moisture content of the expanded beads was as follows (1) to (4): (1) Water adhering to the surface of the expanded beads was dehydrated with an air (compressed air) stream until the temperature of the air stream ceased to change due to the latent heat of vaporization associated with the dehydration of the expanded beads; (2) removing the expanded beads; (3) The foamed beads were dried in an oven at 150°C for 1 hour, and the weight (W2) was measured.
- the method for evaluating the surface elongation of the foam molded body was as follows (1) to (2): (1) In the foam molded body, the ( 2 ) Based on the counted number of intergranules, the surface elongation of the foam molded product was evaluated according to the following criteria: 5.0 (particularly good): less than 5 grains of 1 mm 2 or more 4.5 (better): 5 or more grains of 1 mm 2 or less, less than 7 4.0 (good): 1 mm
- the number of intergranular spaces of 2 or more is 7 or more and less than 10 3.5 (slightly good): The number of intergranular spaces of 1 mm 2 or more is 10 or more and less than 17 3.0 (Pass): 1 mm 2 or more intergranular spaces
- the number is 17 or more and less than 25 2.5 (failed):
- the number of grains of 1 mm 2 or more is 25 or more and less than 37 2.0 (slightly poor):
- the number of grains of 1 mm 2 or more is
- the method for evaluating the edge elongation of the foam molded article was as follows (1) to (2); (2) Based on the counted number of intergranules and the confirmed edge shape, the edge elongation of the foam molded product was evaluated according to the following criteria. : 5 (particularly good): excellent condition with no grain gaps and very beautiful edge shape; 4 (good): almost no intergranular space, good edge shape; 3 (passed): Slight intergranules are observed, but there are no missing grains at the edges. 2 (slightly unsatisfactory): There are grain gaps and missing grains, so that the edges are uneven 1 (poor): There are many grain spaces and missing grains, and the edges are uneven.
- Example 1 (Production of polypropylene resin particles)
- polypropylene resin (A) is 79.6% by weight
- hydrophilic substance (B) is 15.0% by weight
- hindered amine (C) is 5.0% by weight
- other additives Nucleating agent Talcan powder PK-S at 0.10% by weight
- antioxidant Irgafos 168 at 0.133% by weight
- antioxidant Irganox 1010 at 0.067% by weight
- UV inhibitor Tinuvin 622 Each component was dry blended so that the content was 0.10% by weight (the total of other additives was 0.4% by weight).
- the resulting blend was charged into a twin-screw extruder [TEM26SX, manufactured by Shibaura Kikai Co., Ltd.] and melt-kneaded at a resin temperature of 220°C.
- the melt-kneaded polypropylene-based resin composition was extruded into strands through a die having a circular hole attached to the tip of the extruder.
- the extruded polypropylene-based resin composition was cooled with water and then cut with a cutter so that the weight of one resin particle was 1.2 mg to obtain resin particles (granulation step).
- the obtained expanded beads were measured for average cell diameter, heat of fusion on the high temperature side, and water content of the expanded beads immediately after foaming. Table 1 shows the results.
- the obtained expanded beads contained 15.0% by weight of the hydrophilic substance (B) and 5.0% by weight of the hindered amine (C) with respect to 100% by weight of the total amount of the expanded beads.
- the foamed particles impregnated with air are heated and molded with steam at 0.26 MPa (gauge pressure) using a molding machine (polypropylene in-mold expansion molding machine manufactured by Daisen Co., Ltd.) with a cracking rate of 20%.
- a compact was obtained.
- the obtained foamed molded product was left at room temperature for 1 hour, it was cured and dried in a constant temperature room at 75° C. for 12 hours, and then left at room temperature for 4 hours. After that, the density of the resulting foamed molded product was measured by the method described above, and surface elongation and edge elongation were evaluated. Table 1 shows the results.
- Example 2 Resin particles, expanded particles, and an expanded molded article were obtained in the same manner as in Example 1, except that the steam pressure during heat molding of the expanded particles was changed to 0.30 MPa (gauge pressure). was measured and evaluated. Table 1 shows the results.
- Example 3 A resin bead, an expanded bead, and a foamed molded product were obtained in the same manner as in Example 1, except that the steam pressure during heat molding of the expanded beads was changed to 0.34 MPa (gauge pressure), and their physical properties were measured. measured and evaluated. Table 1 shows the results.
- Example 4 A resin bead, an expanded bead, and an expanded molded article were obtained in the same manner as in Example 1, except that no internal pressure was applied to the expanded bead before molding (internal pressure of the expanded bead), and their physical properties were measured and evaluated. Table 1 shows the results.
- Example 5 A resin bead, an expanded bead, and an expanded molded article were obtained in the same manner as in Example 2, except that no internal pressure was applied to the expanded bead before molding (internal pressure of the expanded bead), and their physical properties were measured and evaluated. Table 1 shows the results.
- Example 6 (Production of polypropylene resin particles)
- polypropylene resin (A) is 79.6% by weight
- hydrophilic substance (B) is 15.0% by weight
- hindered amine (C) is 5.0% by weight
- other additives Nucleating agent Talcan powder PK-S at 0.10% by weight
- antioxidant Irgafos 168 at 0.133% by weight
- antioxidant Irganox 1010 at 0.067% by weight
- UV inhibitor Tinuvin 622 Each component was dry blended so that the content was 0.10% by weight (the total of other additives was 0.4% by weight).
- the resulting blend was charged into a twin-screw extruder [TEM26SX, manufactured by Shibaura Kikai Co., Ltd.] and melt-kneaded at a resin temperature of 220°C.
- the melt-kneaded polypropylene-based resin composition was extruded into strands through a die having a circular hole attached to the tip of the extruder.
- the extruded polypropylene-based resin composition was cooled with water and then cut with a cutter so that each resin particle weighed 5.0 mg to obtain resin particles (granulation step).
- the obtained expanded beads were measured for average cell diameter, heat of fusion on the high temperature side, and water content of the expanded beads immediately after foaming. Table 1 shows the results.
- the obtained expanded beads contained 15.0% by weight of the hydrophilic substance (B) and 5.0% by weight of the hindered amine (C) with respect to 100% by weight of the total amount of the expanded beads.
- the foamed particles impregnated with air are heated and molded with steam at 0.30 MPa (gauge pressure) using a molding machine (polypropylene in-mold foam molding machine manufactured by Daisen Co., Ltd.) with cracking of 10%. got a body After the obtained foamed molded product was left at room temperature for 1 hour, it was cured and dried in a constant temperature room at 75° C. for 12 hours, and then left at room temperature for 4 hours. After that, the density of the resulting foamed molded product was measured by the method described above, and surface elongation and edge elongation were evaluated. Table 1 shows the results.
- Example 7 A resin bead, an expanded bead, and an expanded molded product were obtained in the same manner as in Example 6, except that cracking during molding of the expanded bead was changed to 3%, and their physical properties were measured and evaluated. Table 1 shows the results.
- Example 8 The foamed particles were not dried (that is, the moisture content at the time of molding was 21.0%), the foamed particles were not impregnated with air (no internal pressure was applied), and cracking at the time of molding was set at 20%. Resin beads, foamed beads, and foamed moldings were obtained in the same manner as in Example 6 except for the above, and their physical properties were measured and evaluated. Table 1 shows the results.
- Example 9 The same procedure as in Example 6 was performed except that the amount of kaolin ASP-170 used was changed to 0.12 parts by weight, and the temperature in the pressure-resistant sealed container was heated to a foaming temperature of 150.7 ° C. in the temperature rise-pressurization step. Resin particles and expanded particles were obtained by the method. The expanded beads were dried in a dryer at 75° C. for 30 minutes so that the water content of the expanded beads (the water content after drying in a dryer and can also be said to be the water content at the time of molding) was 4.9%.
- the foamed particles after the drying treatment were heat molded in the same manner as in Example 6, except that air was not impregnated (no internal pressure was applied) and cracking was 20%, to obtain a foamed molded product. got Each physical property was measured and evaluated for the obtained expanded beads and expanded molded article. Table 1 shows the results.
- Example 10 The same procedure as in Example 6 was performed except that the amount of kaolin ASP-170 used was changed to 0.10 parts by weight, and the temperature in the pressure-resistant sealed container was heated to a foaming temperature of 150.6 ° C. in the temperature rising-pressurizing step. Resin particles and expanded particles were obtained by the method.
- the expanded beads are dried in a dryer at 75°C for 20 minutes so that the water content of the expanded beads (which is the water content after drying in a dryer and can also be said to be the water content at the time of molding) is 8.1%.
- the foamed particles after the drying treatment were heat-molded and foam-molded in the same manner as in Example 6 except that air was not impregnated (no internal pressure was applied) and cracking was 20%. got a body
- Table 1 shows the results.
- the weight of one resin particle in Examples 2 to 5 is 1.2 mg (1.2 mg/particle), and the weight of one resin particle in Examples 6 to 10 is 5.0 mg (5.0 mg/particle). )Met.
- the holding time in Examples 2-5 was 25 minutes, and the holding time in Examples 6-10 was 10 minutes.
- Example 4 The foamed beads were dried in a dryer at 80°C for 15 hours to adjust the water content (moisture content after drying with a dryer and at the time of molding) to 0.7%. The same operation as in Example 1 was carried out, except that the foamed article was obtained by heat molding without applying an internal pressure (internal pressure of the foamed particles) before molding. As a result, resin particles, foamed particles, and foamed moldings were obtained. Each physical property was measured and evaluated. Table 2 shows the results.
- Example 5 The foamed beads were dried in a dryer at 80°C for 15 hours to adjust the water content (moisture content after drying with a dryer and at the time of molding) to 0.7%. The same operation as in Example 2 was carried out, except that the foamed article was obtained by heat molding without applying an internal pressure (internal pressure of the foamed particles) before molding. As a result, resin particles, foamed particles, and foamed moldings were obtained. Each physical property was measured and evaluated. Table 2 shows the results.
- the resulting blend was charged into a twin-screw extruder [TEM26SX, manufactured by Shibaura Kikai Co., Ltd.] and melt-kneaded at a resin temperature of 220°C.
- the melt-kneaded polypropylene-based resin composition was extruded into strands through a die having a circular hole attached to the tip of the extruder.
- the extruded polypropylene-based resin composition was cooled with water and then cut with a cutter so that the weight of one resin particle was 1.2 mg to obtain resin particles (granulation step).
- the resulting blend was charged into a twin-screw extruder [TEM26SX, manufactured by Shibaura Kikai Co., Ltd.] and melt-kneaded at a resin temperature of 220°C.
- the melt-kneaded polypropylene-based resin composition was extruded into strands through a die having a circular hole attached to the tip of the extruder.
- the extruded polypropylene-based resin composition was cooled with water and then cut with a cutter so that each resin particle weighed 5.0 mg to obtain resin particles (granulation step).
- the obtained expanded beads were measured for average cell diameter, heat of fusion on the high temperature side, and water content of the expanded beads after foaming. Table 1 shows the results.
- the obtained expanded beads contained 15.0% by weight of the hydrophilic substance (B) and 5.0% by weight of the hindered amine (C) with respect to 100% by weight of the total amount of the expanded beads.
- Example 11 The same as in Example 9, except that the expanded particles were dried in a dryer at 80°C for 12 hours before impregnation with air (internal pressure was applied) to adjust the moisture content of the expanded particles to 0.9%. Resin particles, foamed particles, and foamed moldings were obtained by the method of No. 2, and their physical properties were measured and evaluated. Table 2 shows the results.
- the weight of one resin particle in Comparative Examples 1 to 6 is 1.2 mg (1.2 mg/particle), and the weight of one resin particle in Comparative Examples 7 to 11 is 5.0 mg (5.0 mg/particle). )Met.
- Comparative Examples 1-6 The holding time in Comparative Examples 1-6 was 25 minutes, and the holding time in Comparative Examples 7-11 was 10 minutes.
- a foamed molded product obtained by molding foamed particles has good surface elongation and edge elongation, and is a foamed molded product with a beautiful surface.
- the foamed molded articles obtained by molding the foamed particles of Comparative Examples 1 to 11 are poor in both surface elongation and edge elongation, and are foamed articles with poor surface beauty. .
- In-mold expansion molding of the polypropylene-based foamed particles according to one embodiment of the present invention can provide a polypropylene-based foam-molded article having excellent surface beauty and productivity.
- Polypropylene-based foamed molded articles can be suitably used in various applications such as cushioning packaging materials, distribution materials, heat insulating materials, civil engineering and construction members, and automobile members.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
Abstract
Description
特に、内装材のような人目に晒される部材としてポリプロピレン系発泡成形体を適用する場合、当該発泡成形体には、優れた外観(表面美麗性)を有することが要求される。ポリプロピレン系発泡粒子に空気等の無機ガスを含浸させることで、得られるポリプロピレン系発泡成形体の表面美麗性を改善する技術においては、成形前のポリプロピレン系発泡粒子にある程度高い内圧(発泡粒子内圧)を付与する必要がある。必要な内圧が高くなるほど、それに応じて無機ガスの含浸時間も長くなる。そして、当該含浸時間が長くなるほど、ポリプロピレン系発泡成形体の生産に要する時間も増加し、結果的にポリプロピレン系発泡成形体の生産性(生産効率)が悪化するという課題があった。
本発明の一実施形態に係るポリプロピレン系発泡粒子は、乾燥機で乾燥後および/または成形時の含水率が1.0%~60.0%である。
本発泡粒子は、ポリプロピレン系樹脂(A)を含むことが好ましい。本明細書において、ポリプロピレン系樹脂(A)とは、樹脂に含まれる全構成単位100モル%中、プロピレン単量体に由来する構成単位を少なくとも50モル%以上、好ましくは75%以上含む樹脂を意図する。本明細書において、「プロピレン単量体に由来する構成単位」を「プロピレン単位」と称する場合もある。
本発泡粒子は、親水性物質(B)を含むことが好ましい。本発泡粒子が親水性物質を含む場合、発泡粒子の含水率を高めることができ、所望の含水率(例えば、1.0%~60.0%)を有する発泡粒子を安定的に得ることができる。すなわち、表面美麗性および生産性に優れる発泡成形体を提供できる発泡粒子を、安定的に提供できるという利点を有する。
ホスホネートとは、M-PO(OH)2基および/またはM-PO(OM1)2基(ここで、Mは、一価の炭化水素基(例えば、メチル基、ベンジル基、アリール基、アルキニル基、アレニル基、ビニル基または置換されたビニル基など)であり、M1はヒドロカルビル基(例えば、アルキル基またはアリール基など)である)を含有する、エステル、および部分エステル、並びにこれらの塩を意図する。

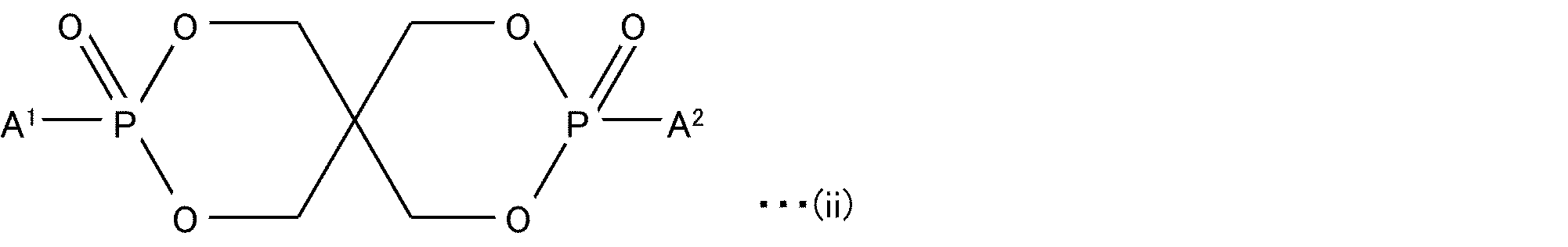

本発泡粒子は、さらに、ヒンダードアミン(C)を含むことが好ましい。本発泡粒子がヒンダードアミン(C)を含む場合、表面美麗性および生産性に加えて、難燃性にも優れる発泡成形体を提供し得る、発泡粒子を提供できる、という利点を有する。


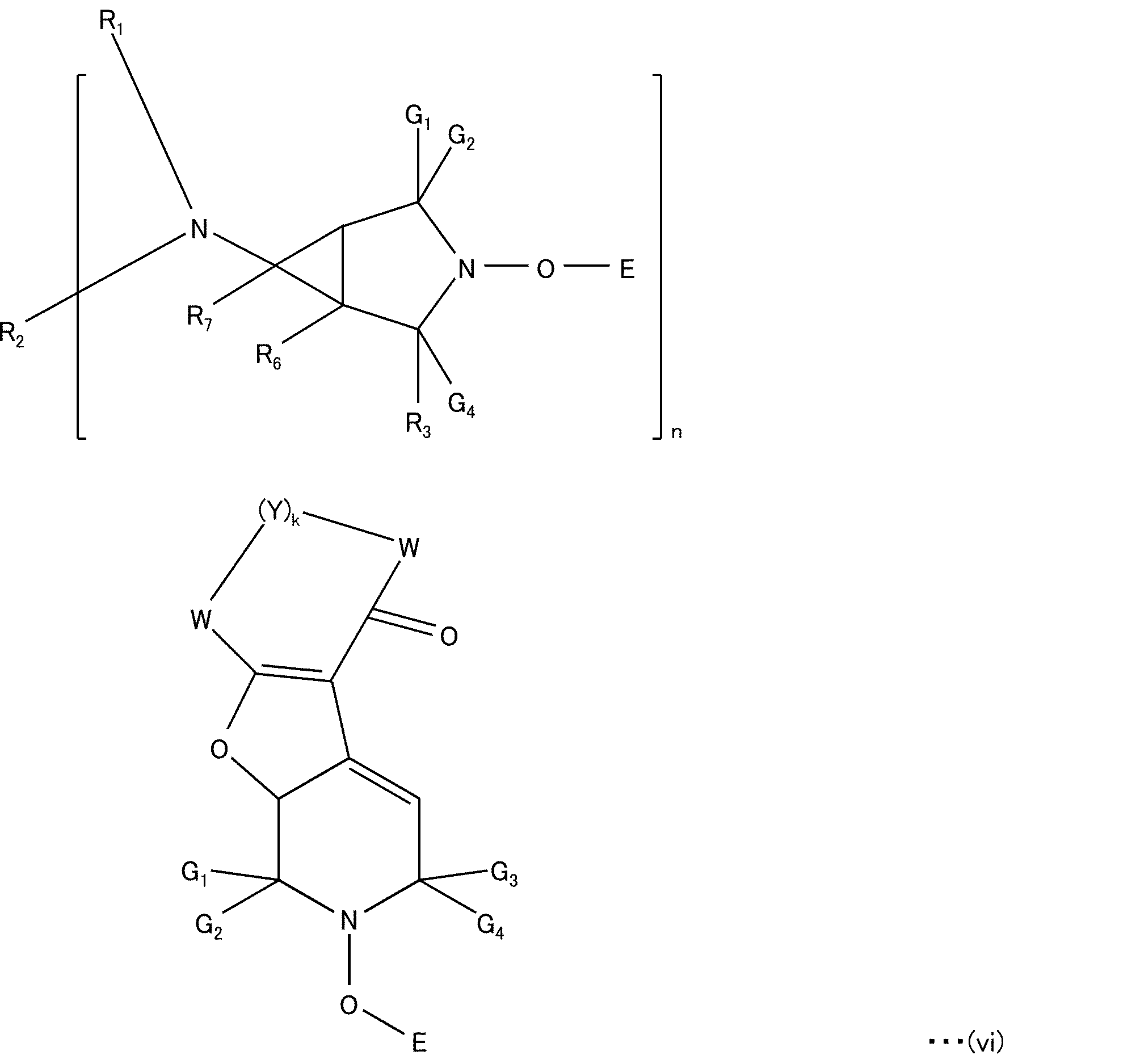
表面美麗性、生産性に加え、より優れる難燃性を有する発泡成形体を提供できることから、本発泡粒子は、親水性物質(B)としてホスホネートを含み、かつ、ヒンダードアミン(C)を含むことが好ましい。
本発泡粒子が親水性物質(B)としてホスホネートを含み、かつ、ヒンダードアミン(C)を含む場合、本発泡粒子の含む親水性物質(B)の重量に対する、本発泡粒子の含むヒンダードアミン(C)の重量の比率(以下、比率(B)/(C)と称する)は、1.0~20.0であることが好ましく、1.0~15.0であることがより好ましく、1.0~10.0であることがより好ましく、2.0~10.0であることがより好ましく、2.0~7.0であることがより好ましく、2.0~5.0であることがより好ましく、2.0~4.0であることがさらに好ましい。比率(B)/(C)が1.0~20.0である場合、得られる発泡成形体は、(a)優れた難燃性を有し、かつ、(b)燃焼時に、燃焼する粒子が発生することによる、周辺の可燃物への着火を抑制することができる(すなわち、自己消火性に優れる)とともに、(c)発泡成形体の諸物性(機械的特性、緩衝性等)を損なう虞がない。
本発泡粒子が親水性物質(B)としてホスホネートを含み、かつ、ヒンダードアミン(C)を含む場合について説明する。この場合、本発泡粒子の含むポリプロピレン系樹脂(A)の重量に対する、親水性物質(B)およびヒンダードアミン(C)の合計量(合計重量)の比率(以下、比率(A)/{(B)+(C)}と称する)は、1~10であることが好ましく、1~7であることがより好ましく、1~5であることがより好ましく、2~5であることがより好ましく、3~5であることがさらに好ましい。発泡粒子における比率(A)/{(B)+(C)}が(i)10以下である場合、得られる発泡成形体の難燃性が良好となる。また、(ii)1以上である場合、得られる発泡成形体の表面美麗性がより良好となる。
本発泡粒子は、親水性物質(B)、ヒンダードアミンの他に、本発明の効果を損なわない限り、任意でその他添加剤を含んでいてもよい。その他添加剤としては、抗酸化剤、UV光吸収剤、過酸化物捕捉剤、造核剤、帯電防止剤、酸化防止剤、光安定剤、導電剤、潤滑剤、フィラー、カーボンブラック、粉状活性炭等が挙げられる。このようなその他添加剤は、本発泡粒子の製造において、(a)樹脂粒子を製造する際に、後述するブレンド物もしくはポリプロピレン系樹脂組成物へ直接添加してもよく、(b)予めその他の樹脂に当該添加剤を高濃度で含有させてマスターバッチ化しておき、得られたマスターバッチ樹脂を、前記ブレンド物もしくは前記ポリプロピレン系樹脂組成物へ添加してもよい。マスターバッチ樹脂を作製する際に用いられる樹脂としては、ポリプロピレン系樹脂が好ましい。
(発泡粒子の含水率)
本発泡粒子の含水率は、1.0%~60.0%であることが好ましく、3.0%~50.0%であることがより好ましく、7.0%~40.0%であることがより好ましく、11.0%~30.0%であることがさらに好ましく、15.0%~30.0%であることがよりさらに好ましく、20.0%~30.0%であることが特に好ましい。本発泡粒子の含水率が、(i)1.0%以上である場合、発泡粒子に内圧を付与しないか、あるいは、発泡粒子に付与する内圧が小さくとも、表面美麗性に優れる発泡成形体を提供できるという利点があり、また、(ii)60.0%以下である場合、発泡直後の発泡粒子の収縮を抑制できるという利点がある。
含水率(%)=(W1-W2)/W2×100。
本発泡粒子の密度(嵩密度)は、特に限定されないが、15.0g/L~400.0g/Lであることが好ましく、60.0g/L~200.0g/Lであることがより好ましく、70.0g/L~160.0g/Lであることがさらに好ましく、80.0g/L~155.0g/Lであることがよりさらに好ましい。本発泡粒子の嵩密度が、(i)15.0g/L以上である場合、当該発泡粒子の成形に必要な内圧が低くなるという利点があり、また、(ii)400.0g/L以下である場合、軽量性に優れる発泡成形体を得ることができるという利点がある。
発泡粒子の嵩密度(g/L)=発泡粒子の重量W(g)/{容器の体積V(cm3)/1000}。
本発泡粒子の平均気泡径は、特に制限されないが、100μm~600μmであることが好ましく、150μm~450μm以下であることがより好ましく、200μm~400μm以下であることがさらに好ましい。本発泡粒子の平均気泡径が、(i)100μm以上である場合、表面美麗性に優れる発泡成形体を得ることができる。また、(ii)600μm以下である場合、発泡粒子の型内発泡成形時の2次発泡性が良好となり、表面美麗性に優れる発泡成形体を得ることができる。発泡粒子の平均気泡径は、発泡粒子のセル径であるとも言える。
本発泡粒子の高温側融解熱量は、5.0J/g~25.0J/gであることが好ましく、8.0J/g~22.0J/gであることがより好ましく、10.0J/g~20.0J/gであることがさらに好ましい。発泡粒子の高温側融解熱量が、(i)5.0J/g以上である場合、得られる発泡成形体の、型内発泡成形時における、発泡成形体表面でのヒケの発生、および、寸法の収縮を抑制でき、(ii)25.0J/g以下である場合、内部融着性に優れ、かつ、表面が平滑な発泡成形体を得ることができる。高温側融解熱量は、高温側融解ピーク熱量であるとも言える。
本発明の一態様は、乾燥機で乾燥後の含水率が1.0%~60.0%であるポリプロピレン系発泡粒子、である。
本発明の一実施形態に係るポリプロピレン系発泡粒子の製造方法は、特に限定されないが、ポリプロピレン系樹脂粒子と、水系分散媒と、発泡剤とを容器中に分散させる分散工程と、前記分散工程にて得られた分散液を、前記容器内の圧力よりも低圧の領域に放出する放出工程と、を含む方法が好ましい。
本製造方法は、発泡工程の前に、ポリプロピレン系樹脂粒子を調製する、造粒工程を含んでいてもよい。本明細書において、「ポリプロピレン系樹脂粒子」を「樹脂粒子」と称する場合がある。
(樹脂粒子の融点)
樹脂粒子の融点としては、特に限定されないが、122.0℃~159.0℃が好ましく、127.0℃~157.0℃がより好ましく、132.0℃~151.0℃がより好ましく、135.0℃~148.0℃がより好ましく、136.0℃~145.0℃がさらに好ましく、138.0℃~144.0℃が特に好ましい。樹脂粒子の融点が、(i)122.0℃以上である場合、高温での寸法安定性に優れる発泡成形体を得ることができ、(ii)159.0℃以下である場合、低い水蒸気圧力で、発泡成形体を型内発泡成形することができる。
分散工程は、例えば、水系分散媒中に樹脂粒子と発泡剤と、必要に応じて分散剤および/または分散助剤とが分散している分散液を調製する工程ともいえる。
本製造方法は、分散工程の後であり、かつ放出工程の前に、(i)容器内温度を一定温度まで昇温し、かつ容器内圧力を一定圧力まで昇圧する昇温-昇圧工程と、(ii)容器内温度および圧力を一定温度かつ一定圧力で保持する保持工程とをさらに含むことが好ましい。保持工程は、昇温-昇圧工程後に実施されることが好ましい。本明細書において、昇温-昇圧工程および保持工程における(a)一定温度を発泡温度と称する場合があり、(b)一定圧力を発泡圧力と称する場合がある。
放出工程は、(a)昇温-昇圧工程を実施するが保持工程を実施しない場合には昇温-昇圧工程後、(b)昇温-昇圧工程および保持工程の両方を実施する場合には保持工程後、に実施されることが好ましい。放出工程により、樹脂粒子を発泡させることができ、結果として発泡粒子が得られる。放出工程は、容器の一端を解放し、容器内の分散液を、発泡圧力(すなわち、容器内圧力)よりも低圧の領域(空間)に放出する工程、ともいえる。
分散工程から放出工程までの工程を発泡工程と称する場合がある。また、このように、樹脂粒子から発泡粒子を製造する工程を「1段発泡工程」と呼び、得られた発泡粒子を「1段発泡粒子」と呼ぶ。
ところで、発泡倍率の高い発泡粒子を得る為には、1段発泡工程において無機系発泡剤の使用量を多量にするという方法(以下、方法1とする)がある。さらに、方法1以外の方法として、1段発泡工程で比較的低倍率(発泡倍率2.0倍~35.0倍程度)の発泡粒子(1段発泡粒子)を得た後、得られた1段発泡粒子を再度発泡させることで発泡倍率を高くする方法(以下、方法2とする)、も採用可能である。
本製造方法は、発泡粒子を乾燥させる乾燥工程を、さらに含んでいてもよい。乾燥工程において、乾燥させる発泡粒子は、放出工程(発泡工程)にて得られた発泡粒子(1段発泡粒子)であってもよく、2段発泡工程にて得られた2段発泡粒子であってもよい。
本発明の一実施形態に係るポリプロピレン系発泡成形体は、本発泡粒子を成形(例えば、型内発泡成形)してなる発泡成形体である。本明細書において、「本発明の一実施形態に係るポリプロピレン系発泡成形体」を、「本発泡成形体」と称する場合がある。本発泡成形体は、含水率が1.0%~60.0%以下であるポリプロピレン系発泡粒子を成形してなる発泡成形体であるとも言える。
本明細書において、発泡成形体の「表面美麗性」は、当該発泡成形体の表面伸び、および、エッジ伸びにより評価することができる。発泡成形体の表面伸びが優れることは、当該発泡成形体の表面に粒間(発泡成形体を構成する発泡粒子間の空間(隙間))がほとんどないこと、すなわち、当該成形体の表面に凹凸がほとんど無いことを意味する。発泡成形体のエッジ伸びが優れることは、成形体のエッジ部分(角部分)において粒間がほとんどないこと、すなわち、当該発泡成形体のエッジ部分にほとんど凹凸が無い(エッジ部の欠粒がない)ことを意味する。本明細書において、「表面美麗性に優れる」発泡成形体とは、表面伸び、および、エッジ伸びに優れる発泡成形体を意図する。なお、発泡成形体の表面伸び、および、エッジ伸びは、実施例に記載の方法により評価することができる。
本発泡成形体の密度は、15.0g/L~400.0g/Lであることが好ましく、50.0g/L~300.0g/Lであることが好ましく、100.0g/L~200.0g/Lであることが好ましい。発泡成形体の密度が15.0g/L以上であれば、発泡成形体の表面にひけがなく、平滑であり、より表面美麗性に優れる発泡成形体を得られるという利点があり、400.0g/L以下であると、十分に軽量化された発泡成形体を得られる。
発泡成形体の密度(g/L)=W/V。
電装部品周辺部材等の高い難燃性が要求される部材として好適に使用可能となることから、本発泡成形体は難燃性に優れる発泡成形体であることが好ましい。本明細書において、発泡成形体の難燃性は、UL94V“垂直燃焼発泡材料試験”によって評価することができる。UL94V“垂直燃焼発泡材料試験”の試験方法については、後述の実施例にて詳説する。
本発泡成形体の製造方法は本発泡粒子を成形する工程を含む。本発泡成形体の製造方法は、含水率が1.0%~60.0%以下であるポリプロピレン系発泡粒子を成形する工程を含む方法であるとも言える。本発泡成形体の製造方法は、上記構成を満たす限り、その他の態様は特に限定されない。本発泡成形体の成型方法は、金型内にポリプロピレン系発泡粒子を充填し、当該金型内に充填したポリプロピレン系発泡粒子を水蒸気で加熱することで成形する工程を含む、型内発泡成形法によることが好ましい。型内発泡成形法による本発泡成形体の製造方法の具体的態様としては、例えば以下(b1)~(b6)を順に含む製造方法が挙げられるが、かかる製造方法に限定されるものではない:
(b1)駆動し得ない固定型と駆動可能な移動型とから構成される金型を型内発泡成形機に搭載する。ここで、固定型および移動型は、固定型に向かって移動型を駆動させる(当該操作を「型閉じ」と称する場合がある)ことにより、固定型および移動型の内部に形成可能である;
(b2)固定型と移動型とが完全に型閉じされないように、わずかな隙間(クラッキングとも称する)が形成されるように、固定型に向かって移動型を駆動させる;
(b3)固定型および移動型の内部に形成された成形空間内に、例えば充填機を通して、発泡粒子を充填する;
(b4)固定型と移動型とが完全に型閉じするように移動型を駆動させる(すなわち、完全に型閉じする);
(b5)金型を水蒸気で予熱した後、金型を水蒸気で一方加熱および逆一方加熱し、さらに金型を水蒸気で両面加熱することにより、型内発泡成形を行う;
(b6)型内発泡成形物を金型から取り出し、乾燥(例えば、75℃で乾燥)することで、発泡成形体を得る。
(b3-1)発泡粒子(上述の2段発泡粒子を含む、以下同じ)を容器内で無機ガスで加圧処理して、当該発泡粒子内に無機ガスを含浸させ、所定の内圧(発泡粒子内圧)を付与した後、当該発泡粒子を成形空間に充填する方法;
(b3-2)金型として、当該金型の体積が変更可能な金型を使用し、発泡粒子を成形空間に充填した後、当該金型内の体積を10%~75%減ずるように圧縮する方法;
(b3-3)発泡粒子をガス圧力で圧縮して成形空間に充填する方法;
(b3-4)特に前処理することなく、発泡粒子を成形空間に充填する方法。
実施例および比較例で使用した物質を以下に示す。
ポリプロピレン(A):エチレンランダムポリプロピレン(エチレン/プロピレンランダム共重合体)、MI=7.00g/10分、融点144℃、エチレン含有量3.2%)
<親水性物質(B)>
Thor GmbH社製 AFLAMMIT(登録商標)PCO900(ペンタエリスリチルジホスホネート)
<ヒンダードアミン(C)>
BASF社製 FLAMESTAB(登録商標)NOR116(2,4-ビス((1-シクロヘキシルオキシ-2,2,6,6-テトラメチルピぺリジン-4-イル)ブチルアミノ)-6-クロロ-S-トリアジンと、N,N’-ビス(3-アミノプロピル)エチレンジアミンとの反応生成物)
<その他の添加剤>
(造核剤)
林化成社製 タルカンパウダーPK-S
(酸化防止剤)
BASF社製 Irgafos168
BASF社製 Irganox1010
(紫外線防止剤)
BASF社製 Tinuvin622
<発泡剤>
二酸化炭素:エア・ウォーター株式会社製
<分散剤>
BASF社製 カオリンASP-170
太平化学社製 第3リン酸カルシウム
<分散助剤>
花王社製DBS G-15
なお、分散剤および分散助剤は、後述する分散工程において、分散液、または耐圧密閉容器中に添加するため、樹脂粒子、および発泡粒子には残存しないか、またはごく微量残存するにすぎない。また、発泡剤として使用した二酸化炭素は、発泡粒子のガス透過性が高いため、得られる発泡粒子から放散される。
実施例および比較例において実施した評価方法に関して、以下に説明する。
ポリプロピレン系樹脂(A)および樹脂粒子のMIは、JIS K7210:1999に記載のMI測定器を用い、オリフィスの直径が2.0959±0.005mmφ、オリフィスの長さが8.000±0.025mm、荷重が2160g、かつ、温度が230±0.2℃の条件下で測定した。
樹脂粒子の融点は示差走査熱量計(セイコーインスツルメンツ(株)製、DSC6200型)を用いて測定した。具体的な測定方法は以下の(1)~(3)の通りであった:(1)樹脂粒子5mg~6mgの温度を10.0℃/分の昇温速度で40.0℃から220.0℃まで昇温することにより当該樹脂粒子を融解させた;(2)その後、融解された樹脂粒子の温度を10℃.0/分の降温速度で220.0℃から40.0℃まで降温することにより当該樹脂粒子を結晶化させた;(3)その後、さらに、結晶化された樹脂粒子の温度を10℃/分の昇温速度で40.0℃から220.0℃まで昇温した。2回目の昇温時(すなわち(3)のとき)に得られる当該樹脂粒子のDSC曲線のピーク(融解ピーク)の温度を当該樹脂粒子の融点とした。なお、上述の方法により、2回目の昇温時に得られる、樹脂粒子のDSC曲線において、ピーク(融解ピーク)が複数存在する場合、融解熱量が最大のピーク(融解ピーク)の温度を、樹脂粒子の融点とした。
発泡粒子の平均気泡径の測定方法は、以下の(1)~(5)の通りであった:(1)カミソリ(フェザー社製ハイステンレス両刃)を用いて、発泡粒子の中心を通るように当該発泡粒子を切断した;(2)得られた発泡粒子の切断面を、光学顕微鏡(キーエンス社製VHX-5000)を用いて、倍率50倍にて観察した;(3)観察によって得られた画像において、当該発泡粒子の切断面の中心または略中心を通る直線を引いた;(4)(4-1)当該直線上に存在する気泡数nを測定し、(4-2)当該直線と当該発泡粒子表面との交点によって当該直線から切り取られた線分の長さを測定し、発泡粒子径Lとした;(5)以下の式により発泡粒子の平均気泡径を算出した:
平均気泡径(μm)=L/n。
発泡粒子の高温側融解熱量は以下の(1)~(5)の通りであった:(1)発泡粒子を約5mg量り取った;(2)量り取った発泡粒子の温度を10℃/分の昇温速度にて10℃から190℃まで昇温して、当該発泡粒子を融解した;(3)前記(2)の過程で得られる発泡粒子のDSC曲線において、融解開始前の温度を表す点と融解終了後の温度を表す点とを直線で結びベースラインを作成した;(4)高温側の融解ピークまたは最も高温の融解ピークと隣の融解ピークとの間の極大点を通る直線を、X軸に対して垂直方向に引いた;(5)ベースラインと極大点を通る直線とDSC曲線とに囲まれる高温側の領域から算出される熱量(J/g)を、発泡粒子の高温側融解熱量とした。
発泡粒子の密度(嵩密度)の測定方法は、以下(1)~(3)の通りであった:(1)発泡粒子を、体積V(cm3)の容器へ、当該容器から前記発泡粒子があふれるまで入れた;(2)容器の粉面(上端)を擦切り、前記容器内の発泡粒子の重量W(g)を測定した;(3)以下の式により発泡粒子の嵩密度を算出した:発泡粒子の嵩密度(g/L)=発泡粒子の重量W(g)/{容器の体積V(cm3)/1000}。
発泡粒子(発泡直後の発泡粒子、乾燥機で乾燥後の発泡粒子および、成形時の発泡粒子)の含水率の測定方法は、以下(1)~(4)の通りであった:(1)発泡粒子の表面に付着した水を、空気(圧縮エアー)気流によって、当該発泡粒子の脱水に伴う蒸発潜熱の影響による前記空気気流の温度変化がなくなるまで脱水させた;(2)前記発泡粒子を、常温常圧の室内に30分放置後、その重量(W1)を測定した;(3)前記発泡粒子を、さらに150℃のオーブン中で1時間乾燥させた時の重量(W2)を測定した;(4)以下の式により発泡粒子の含水率を算出した:
含水率(%)=(W1-W2)/W2×100
(発泡成形体の密度)
発泡成形体の密度(発泡成形体密度)の測定方法は以下の(1)~(3)の通りであった;(1)発泡成形体の長さ方向(mm)、幅方向(mm)、および厚さ方向の長さ(mm)を測定し、発泡成形体の体積V(L)を算出した;(2)当該発泡成形体の重量W(g)を測定した;(3)下記の式に基づき、発泡成形体の密度を算出した:
発泡成形体の密度(g/L)=W/V。
発泡成形体の表面伸びの評価方法は以下の(1)~(2)の通りであった;(1)発泡成形体において、当該発泡成形体の表面の任意の50mm×50mmの範囲内に含まれる1mm2以上の大きさの粒間の数を計数した;(2)計数した粒間の数に基づき、発泡成形体の表面伸びを、以下の基準により評価した:
5.0(特に良好):1mm2以上の粒間数が5個未満
4.5(より良好):1mm2以上の粒間数が5個以上、7個未満
4.0(良好):1mm2以上の粒間数が7個以上、10個未満
3.5(やや良好):1mm2以上の粒間数が10個以上、17個未満
3.0(合格):1mm2以上の粒間数が17個以上、25個未満
2.5(不合格):1mm2以上の粒間数が25個以上、37個未満
2.0(やや不良):1mm2以上の粒間数が37個以上、50個未満
1.0(不良):1mm2以上の粒間数が50個以上。
発泡成形体のエッジ伸びの評価方法は以下の(1)~(2)の通りであった;(1)発泡成形体において、当該発泡成形体のエッジ部分の範囲内に含まれる1mm2以上の大きさの粒間の数を計数し、エッジ形状を目視で確認した;(2)計数した粒間の数および確認したエッジ形状に基づき、発泡成形体のエッジ伸びを、以下の基準により評価した:
5(特に良好):粒間無く、エッジ形状が極めて美麗で優良な状態、
4(良好):粒間はほとんど無く、エッジ形状も良好な状態、
3(合格):粒間は僅かに見られるが、エッジ部の欠粒がない、
2(やや不良):粒間が有り、欠粒も有るためエッジ部に凹凸がある状態
1(不良):粒間、欠粒が多く、エッジが凹凸となっている状態。
発泡成形体の難燃性を、UL94V“垂直燃焼試験”によって、測定、および評価した。
UL94V“垂直燃焼試験”の試験方法は以下の(1)~(5)の通りであった:(1)発泡成形体から、特定の寸法(長さ125±5mm×幅13±0.5mm)で、かつ、特定の厚み(試料の厚み(t)=13mm、8mm、または5mm)を有する試料を切り取った;(2)切り取った試料の上部を固定用クランプによって固定して、試験試料を垂直に保持し、直下に脱脂綿を設置した;(2)保持した試験試料の下端に10秒間ガスバーナーの炎を接炎させ、燃焼させた(1度目の燃焼);(3)試験試料の燃焼時間、および赤熱時間を測定し、30秒以内に燃焼が止まった場合、再度試験試料の下端に10秒間ガスバーナーの炎を接炎させ、燃焼させた(2度目の燃焼);(4)試験試料の燃焼時間、および赤熱時間を再度測定した;(5)試料を変えて(1)~(4)の操作を合計5回繰り返し、以下の評価基準に従って、難燃性を評価した。
[不合格(NG)]:1度目の燃焼、あるいは2度目の燃焼時、試料が30秒以上燃焼を続ける、または、固定用クランプの位置まで燃焼する試料がある;
[V-2]:1度目の接炎の後、30秒以上燃焼を続ける試料がなく、
5個の試料に対する、計10回の接炎後の燃焼時間の合計が250秒以下であり
固定用クランプの位置まで燃焼する試料がなく、
2回目の接炎の後、燃焼時間と赤熱時間の和が250秒以上となる試料がなく、かつ
燃焼の際に落下した試験試料の粒子によって、試験試料の下方に置かれた脱脂綿が発火する試料がある;
[V-1]:1度目および2度目のいずれの接炎の後も、30秒以上燃焼を続ける試料がなく、
5個の試料に対する、計10回の接炎後の燃焼時間の合計が250秒以下であり
固定用クランプの位置まで燃焼する試料がなく、
2回目の接炎の後、燃焼時間と赤熱時間の和が250秒以上となる試料がなく、かつ、
いずれの燃焼の際にも、落下した試験試料の粒子によって、試験試料の下方に置かれた脱脂綿が発火する試料がない。
5個の試料に対する、計10回の接炎後の燃焼時間の合計が50秒以下であり
固定用クランプの位置まで燃焼する試料がなく、
2回目の接炎の後、燃焼時間と赤熱時間の和が30秒以上となる試料がなく、かつ、
いずれの燃焼の際にも、落下した試験試料の粒子によって、試験試料の下方に置かれた脱脂綿が発火する試料がない。
(ポリプロピレン系樹脂粒子の作製)
ブレンド物100重量%中、ポリプロピレン系樹脂(A)が79.6重量%、親水性物質(B)が15.0重量%、ヒンダードアミン(C)が5.0重量%、その他の添加剤として、造核剤であるタルカンパウダーPK-Sが0.10重量%、酸化防止剤であるIrgafos168が0.133重量%、酸化防止剤であるIrganox1010が0.067重量%、および紫外線防止剤であるTinuvin622が0.10重量%(その他の添加剤が合計0.4重量%)となるよう、各成分をドライブレンドした。
得られた樹脂粒子100重量部と、純水442重量部と、分散剤として第3リン酸カルシウムを1.1重量部、ならびにカオリンASP-170を0.22重量部と、分散助剤として、DBSを0.027重量部と、を耐圧密閉容器に投入した。その後、耐圧密閉容器内の原料を攪拌しながら、発泡剤として二酸化炭素1.8重量部を前記耐圧密閉容器内に導入し、分散液を調製した(分散工程)。次いで、耐圧密閉容器内の温度を150.8℃の発泡温度に加熱した。その後、耐圧密閉容器内に二酸化炭素を追加圧入し、耐圧密閉容器内を0.7MPa(ゲージ圧)の発泡圧力まで昇圧した(昇温-昇圧工程)。次いで、耐圧密閉容器内を前記発泡温度、発泡圧力で20分間保持した後(保持工程)、密閉容器下部のバルブを開いて、分散液を、口径3.6mmのオリフィスを通じて、大気圧下の発泡筒に放出して発泡粒子(1段発泡粒子)を得た(放出工程)。この際、分散液の放出中は耐圧密閉容器内の圧力が発泡圧力から低下しないように、二酸化炭素を耐圧密閉容器内に追加圧入して、耐圧密閉容器内の圧力を0.7MPa(ゲージ圧)に保持した。得られた発泡粒子について、平均気泡径、高温側融解熱量、および、発泡直後の発泡粒子含水率を測定した。結果を表1に示す。なお、得られた発泡粒子は、当該発泡粒子の全量100重量%に対して、親水性物質(B)を15.0重量%、ヒンダードアミン(C)を5.0重量%含むものであった。
得られた発泡粒子を80℃の乾燥機[有限会社光輝社製、KF-2000]内で120分間乾燥処理することで、当該発泡粒子の含水率(乾燥機で乾燥後の含水率であり、かつ成形時の含水率ともいえる)を、28.0%に調整した後、当該発泡粒子を耐圧密閉容器内に投入した。耐圧密閉容器内に空気を導入し、耐圧密閉容器内の発泡粒子に加圧空気を含浸させて、0.16MPa(絶対圧)の発泡粒子内圧(絶対圧)を発泡粒子に付与した。空気を含浸させた発泡粒子を、成形機(ダイセン株式会社製ポリプロピレン型内発泡成形機)を用いて、クラッキングを20%とし、0.26MPa(ゲージ圧)の水蒸気で加熱成形することにより、発泡成形体を得た。得られた発泡成形体を室温で1時間放置した後、75℃の恒温室内で12時間養生乾燥を行い、再び室温で4時間放置した。その後、上述の方法により、得られた発泡成形体の密度を測定し、表面伸びおよびエッジ伸びを評価した。結果を表1に示す。
発泡粒子を加熱成形する際の水蒸気圧力を、0.30MPa(ゲージ圧)に変更したこと以外は、実施例1と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
発泡粒子を加熱成形する際の水蒸気圧力を、0.34MPa(ゲージ圧)に変更したこと以外は実施例1と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
成形前に発泡粒子に内圧(発泡粒子内圧)を付与しなかったこと以外は実施例1と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
成形前に発泡粒子に内圧(発泡粒子内圧)を付与しなかったこと以外は実施例2と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
(ポリプロピレン系樹脂粒子の作製)
ブレンド物100重量%中、ポリプロピレン系樹脂(A)が79.6重量%、親水性物質(B)が15.0重量%、ヒンダードアミン(C)が5.0重量%、その他の添加剤として、造核剤であるタルカンパウダーPK-Sが0.10重量%、酸化防止剤であるIrgafos168が0.133重量%、酸化防止剤であるIrganox1010が0.067重量%、および紫外線防止剤であるTinuvin622が0.10重量%(その他の添加剤が合計0.4重量%)となるよう、各成分をドライブレンドした。
得られた樹脂粒子100重量部と、純水442重量部と、分散剤として第3リン酸カルシウムを1.1重量部、ならびにカオリンASP-170を0.15重量部と、分散助剤として、DBSを0.027重量部と、を耐圧密閉容器に投入した。その後、耐圧密閉容器内の原料を攪拌しながら、発泡剤として二酸化炭素2.5重量部を前記耐圧密閉容器内に導入し、分散液を調製した(分散工程)。次いで、耐圧密閉容器内の温度を150.9℃の発泡温度に加熱した。その後、耐圧密閉容器内に二酸化炭素を追加圧入し、耐圧密閉容器内を0.9MPa(ゲージ圧)の発泡圧力まで昇圧した(昇温-昇圧工程)。次いで、耐圧密閉容器内を前記発泡温度、発泡圧力で10分間保持した後(保持工程)、密閉容器下部のバルブを開いて、分散液を、口径4.0mmのオリフィスを通じて、大気圧下の発泡筒に放出して発泡粒子(1段発泡粒子)を得た(放出工程)。この際、分散液の放出中は耐圧密閉容器内の圧力が発泡圧力から低下しないように、二酸化炭素を耐圧密閉容器内に追加圧入して、耐圧密閉容器内の圧力を0.9MPa(ゲージ圧)に保持した。得られた発泡粒子について、平均気泡径、高温側融解熱量、および、発泡直後の発泡粒子含水率を測定した。結果を表1に示す。なお、得られた発泡粒子は、当該発泡粒子の全量100重量%に対して、親水性物質(B)を15.0重量%、ヒンダードアミン(C)を5.0重量%含むものであった。
得られた発泡粒子を80℃の乾燥機で120分間乾燥処理することで、当該発泡粒子の含水率(乾燥機で乾燥後の含水率であり、かつ成形時の含水率ともいえる)を、4.0%に調整した後、当該発泡粒子を耐圧密閉容器内に投入した。耐圧密閉容器内に空気を導入し、耐圧密閉容器内の発泡粒子に加圧空気を含浸させて、0.15MPa(絶対圧)の発泡粒子内圧(絶対圧)を発泡粒子に付与した。空気を含浸させた発泡粒子を、成形機(ダイセン株式会社製ポリプロピレン型内発泡成形機)を用いて、クラッキング10%とし、0.30MPa(ゲージ圧)の水蒸気で加熱成形することにより、発泡成形体を得た。得られた発泡成形体を室温で1時間放置した後、75℃の恒温室内で12時間養生乾燥を行い、再び室温で4時間放置した。その後、上述の方法により、得られた発泡成形体の密度を測定し、表面伸びおよびエッジ伸びを評価した。結果を表1に示す。
発泡粒子を成形する時のクラッキングを3%としたこと以外は実施例6と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
発泡粒子を乾燥処理せず(すなわち、成形時の含水率は21.0%)、発泡粒子に空気を含浸させず(内圧を付与せず)、かつ、成形する時のクラッキングを20%としたこと以外は実施例6と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表1に示す。
カオリンASP-170の使用量を0.12重量部に変更し、昇温-昇圧工程において、耐圧密閉容器内の温度を150.7℃の発泡温度に加熱したこと以外は実施例6と同様の方法により、樹脂粒子および発泡粒子を得た。当該発泡粒子を含水率(乾燥機で乾燥後の含水率であり、かつ成形時の含水率ともいえる)が4.9%となるよう、発泡粒子を75℃の乾燥機で30分間乾燥処理し、空気を含浸させず(内圧を付与せず)、かつ、クラッキングを20%としたこと以外は実施例6と同様の方法により、前記の乾燥処理後の発泡粒子を加熱成形し、発泡成形体を得た。得られた発泡粒子および発泡成形体について、各物性を測定、評価した。結果を表1に示す。
カオリンASP-170の使用量を0.10重量部に変更し、昇温-昇圧工程において、耐圧密閉容器内の温度を150.6℃の発泡温度に加熱したこと以外は実施例6と同様の方法により、樹脂粒子および発泡粒子を得た。当該発泡粒子を含水率(乾燥機で乾燥後のン含水率であり、かつ成形時の含水率ともいえる)が8.1%となるよう、発泡粒子を75℃の乾燥機で20分間乾燥処理し、空気を含浸させず(内圧を付与せず)、かつ、クラッキングを20%としたこと以外は実施例6と同様の方法により、前記の乾燥処理後の発泡粒子を加熱成形し、発泡成形体を得た。得られた発泡粒子および発泡成形体について、各物性を測定、評価した。結果を表1に示す。

空気を含浸(内圧を付与)させる前に、発泡粒子を80℃の乾燥機で15時間乾燥処理して、発泡粒子の含水率(乾燥機で乾燥後の含水率であり、かつ成形時の含水率ともいえる)を、0.7%に調整し、耐圧密閉容器内の発泡粒子に加圧空気を含浸させて、0.21MPa(絶対圧)の発泡粒子内圧(絶対圧)を発泡粒子に付与したこと以外は実施例1と同様の操作を実施した。その結果、樹脂粒子、発泡粒子、および発泡成形体を得た。各物性を測定、評価した。結果を表2に示す。
空気を含浸(内圧を付与)させる前に、発泡粒子を80℃の乾燥機で15時間乾燥処理して、発泡粒子の含水率(乾燥機で乾燥後および成形時の含水率)を、0.7%に調整し、耐圧密閉容器内の発泡粒子に加圧空気を含浸させて、0.21MPa(絶対圧)の発泡粒子内圧(絶対圧)を発泡粒子に付与したこと以外は実施例2と同様の操作を実施した。その結果、樹脂粒子、発泡粒子、および発泡成形体を得た。各物性を測定、評価した。結果を表2に示す。
空気を含浸(内圧を付与)させる前に、発泡粒子を80℃の乾燥機で15時間乾燥処理して、発泡粒子の含水率(乾燥機で乾燥後および成形時の含水率)を、0.7%に調整し、耐圧密閉容器内の発泡粒子に加圧空気を含浸させて、0.21MPa(絶対圧)の発泡粒子内圧(絶対圧)を発泡粒子に付与したこと以外は実施例3と同様の操作を実施した。その結果、樹脂粒子、発泡粒子、および発泡成形体を得た。各物性を測定、評価した。結果を表2に示す。
発泡粒子を80℃の乾燥機で15時間乾燥処理して、含水率(乾燥機で乾燥後および成形時の含水率)を、0.7%に調整した発泡粒子を、空気を含浸させず(成形前に内圧(発泡粒子内圧)を付与せず)に、加熱成形し、発泡成形体を得たこと以外は実施例1と同様の操作を実施した。その結果、樹脂粒子、発泡粒子、および発泡成形体を得た。各物性を測定、評価した。結果を表2に示す。
発泡粒子を80℃の乾燥機で15時間乾燥処理して、含水率(乾燥機で乾燥後および成形時の含水率)を、0.7%に調整した発泡粒子を、空気を含浸させず(成形前に内圧(発泡粒子内圧)を付与せず)に、加熱成形し、発泡成形体を得たこと以外は実施例2と同様の操作を実施した。その結果、樹脂粒子、発泡粒子、および発泡成形体を得た。各物性を測定、評価した。結果を表2に示す。
(ポリプロピレン系樹脂粒子の作製)
ブレンド物100重量%中、ポリプロピレン系樹脂(A)(ポリプロピレン(A)を単独で使用)が94.6重量%、ヒンダードアミン(C)が5.0重量%、その他の添加剤として、造核剤であるタルカンパウダーPK-Sが0.10重量%、酸化防止剤であるIrgafos168が0.133重量%、酸化防止剤であるIrganox1010が0.067重量%、および紫外線防止剤であるTinuvin622が0.10重量%(その他の添加剤が合計0.4重量%)となるよう、各成分をドライブレンドした。
得られた樹脂粒子100重量部と、純水442重量部と、分散剤として第3リン酸カルシウムを1.1重量部、ならびにカオリンASP-170を0.22重量部と、分散助剤として、DBSを0.027重量部と、を耐圧密閉容器に投入した。その後、耐圧密閉容器内の原料を攪拌しながら、発泡剤として二酸化炭素7.1重量部を前記耐圧密閉容器内に導入し、分散液を調製した(分散工程)。次いで、耐圧密閉容器内の温度を151.2℃の発泡温度に加熱した。その後、耐圧密閉容器内に二酸化炭素を追加圧入し、耐圧密閉容器内を2.5MPa(ゲージ圧)の発泡圧力まで昇圧した(昇温-昇圧工程)。次いで、耐圧密閉容器内を前記発泡温度、発泡圧力で20分間保持した後(保持工程)、密閉容器下部のバルブを開いて、分散液を、口径3.6mmのオリフィスを通じて、大気圧下の発泡筒に放出して発泡粒子(1段発泡粒子)を得た(放出工程)。この際、分散液の放出中は耐圧密閉容器内の圧力が発泡圧力から低下しないように、二酸化炭素を耐圧密閉容器内に追加圧入して、耐圧密閉容器内の圧力を2.5MPa(ゲージ圧)に保持した。得られた発泡粒子について、平均気泡径、高温側融解熱量、および、発泡直後の発泡粒子含水率を測定した。結果を表1に示す。なお、得られた発泡粒子は、当該発泡粒子の全量100重量%に対して、ヒンダードアミン(C)を5.0重量%含むものであった。
得られた発泡粒子を80℃で15時間乾燥処理することで、発泡粒子含水率を0.3%に調整した後、空気は含浸せずに(内圧を付与せずに)、当該発泡粒子を、成形機(ダイセン株式会社製ポリプロピレン型内発泡成形機)を用いて、クラッキング20%、0.30MPa(ゲージ圧)の水蒸気で加熱成形することにより、発泡成形体を得た。得られた発泡成形体を室温で1時間放置した後、75℃の恒温室内で12時間養生乾燥を行い、再び室温で4時間放置した。その後、上述の方法により、得られた発泡成形体の密度を測定し、表面伸びおよびエッジ伸びを評価した。結果を表2に示す。
(ポリプロピレン系樹脂粒子の作製)
ブレンド物100重量%中、ポリプロピレン系樹脂(A)(ポリプロピレン(A)を単独で使用)が79.6重量%、親水性物質(B)が15.0重量%、ヒンダードアミン(C)が5.0重量%、その他の添加剤として、造核剤であるタルカンパウダーPK-Sが0.10重量%、酸化防止剤であるIrgafos168が0.133重量%、Irganox1010が0.067重量%、および紫外線防止剤であるTinuvin622を0.10重量%(その他の添加剤が合計0.4重量%)となるよう、各成分をドライブレンドした。
得られた樹脂粒子100重量部と、純水442重量部と、分散剤として第3リン酸カルシウムを1.1重量部、ならびにカオリンASP-170を0.15重量部と、分散助剤として、DBSを0.027重量部と、を耐圧密閉容器に投入した。その後、耐圧密閉容器内の原料を攪拌しながら、発泡剤として二酸化炭素2.5重量部を前記耐圧密閉容器内に導入し、分散液を調製した(分散工程)。次いで、耐圧密閉容器内の温度を150.9℃の発泡温度に加熱した。その後、耐圧密閉容器内に二酸化炭素を追加圧入し、耐圧密閉容器内を0.9MPa(ゲージ圧)の発泡圧力まで昇圧した(昇温-昇圧工程)。次いで、耐圧密閉容器内を前記発泡温度、発泡圧力で10分間保持した後(保持工程)、密閉容器下部のバルブを開いて、分散液を、口径4.0mmのオリフィスを通じて、大気圧下の発泡筒に放出して発泡粒子(1段発泡粒子)を得た(放出工程)。この際、分散液の放出中は耐圧密閉容器内の圧力が発泡圧力から低下しないように、二酸化炭素を耐圧密閉容器内に追加圧入して、耐圧密閉容器内の圧力を0.9MPa(ゲージ圧)に保持した。得られた発泡粒子について、平均気泡径、高温側融解熱量、および、発泡後の発泡粒子含水率を測定した。結果を表1に示す。なお、得られた発泡粒子は、当該発泡粒子の全量100重量%に対して、親水性物質(B)を15.0重量%、ヒンダードアミン(C)を5.0重量%含むものであった。
得られた発泡粒子を80℃で15時間乾燥処理することで、当該発泡粒子の含水率を0.4%に調整した後、空気は含浸せずに(内圧を付与せずに)、当該発泡粒子を、成形機(ダイセン株式会社製ポリプロピレン型内発泡成形機)を用いて、クラッキング10%、0.30MPa(ゲージ圧)の水蒸気で加熱成形することにより、発泡成形体を得た。得られた発泡成形体を室温で1時間放置した後、75℃の恒温室内で12時間養生乾燥を行い、再び室温で4時間放置した。その後、上述の方法により、得られた発泡成形体の密度を測定し、表面伸びおよびエッジ伸びを評価した。結果を表2に示す。
含水率を調整した発泡粒子に、加圧空気を含浸させて、0.13MPa(絶対圧)の発泡粒子内圧(絶対圧)を付与したこと以外は比較例7と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表2に示す。
含水率を調整した発泡粒子に、加圧空気を含浸させて、0.15MPa(絶対圧)の発泡粒子内圧(絶対圧)を付与したこと以外は比較例7と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表2に示す。
発泡粒子を加熱成形する際のクラッキングを3%としたこと以外は比較例7と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表2に示す。
空気を含浸(内圧を付与)させる前に、発泡粒子を80℃の乾燥機で12時間乾燥処理して、発泡粒子の含水率を、0.9%に調整したこと以外は実施例9と同様の方法により、樹脂粒子、発泡粒子、および発泡成形体を得、各物性を測定、評価した。結果を表2に示す。
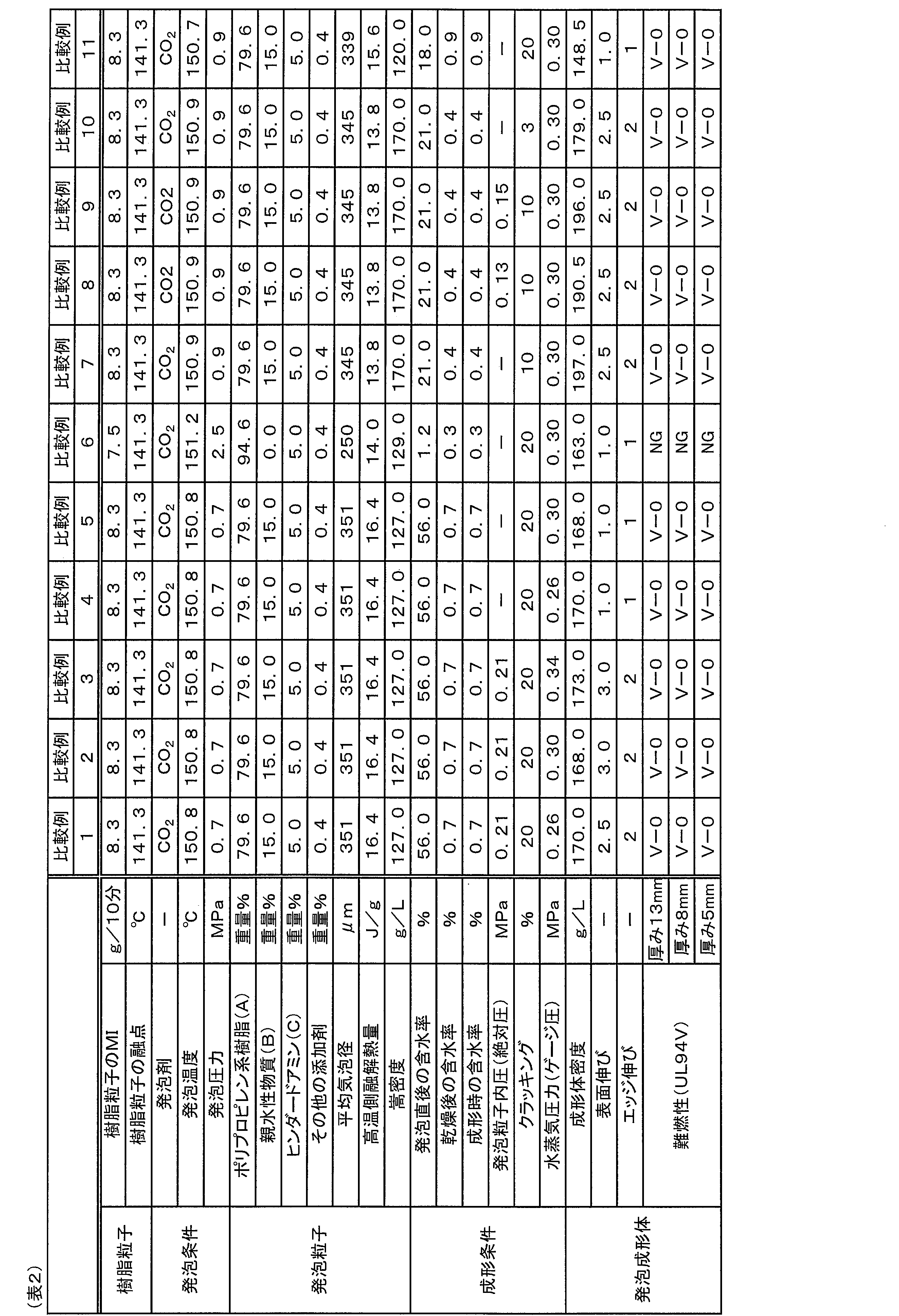
表1より明らかなように、実施例1~3、6および7(低い内圧(0.16MPa(絶対圧)以下)を付与)および実施例4、5および8~10(内圧を付与しない)の発泡粒子を成形してなる発泡成形体は、表面伸び、エッジ伸びともに良好であり、表面美麗な発泡成形体である。一方で、表2より明らかなように、比較例1~11の発泡粒子を成形してなる発泡成形体は、表面伸び、エッジ伸びともに不良であり、表面美麗性が不良な発泡成形体である。以上のように、乾燥機で乾燥後および/または成形時の含水率が本発明の一実施形態の範囲内である発泡粒子を成形することで、発泡粒子に内圧を付与しない、あるいは、発泡粒子に付与する内圧が低い場合であっても、表面美麗な発泡成形体を得られること、すなわち、表面美麗性および生産性に優れる発泡成形体を提供できることが示された。
Claims (15)
- 乾燥機で乾燥後および/または成形時の含水率が1.0%~60.0%である、ポリプロピレン系発泡粒子。
- 前記含水率が11.0%~30.0%である、請求項1に記載のポリプロピレン系発泡粒子。
- 親水性物質(B)を含む、請求項1または2に記載のポリプロピレン系発泡粒子。
- 前記親水性物質(B)は、ホスホネートである、請求項3に記載のポリプロピレン系発泡粒子。
- 前記親水性物質(B)は、ペンタエリスリチルジホスホネートである、請求項3または4に記載のポリプロピレン系発泡粒子。
- 前記ポリプロピレン系発泡粒子の全量100重量%に対して、前記親水性物質(B)を0.1重量%超、20.0重量%以下含む、請求項3~5のいずれか1項に記載のポリプロピレン系発泡粒子。
- ヒンダードアミン(C)を含む、請求項1~6のいずれか1項に記載のポリプロピレン系発泡粒子。
- 前記ヒンダードアミン(C)は、2,4-ビス((1-シクロヘキシルオキシ-2,2,6,6-テトラメチルピぺリジン-4-イル)ブチルアミノ)-6-クロロ-S-トリアジンと、N,N’-ビス(3-アミノプロピル)エチレンジアミンとの反応生成物である、請求項7に記載のポリプロピレン系発泡粒子。
- 前記ポリプロピレン系発泡粒子の全量100重量%に対して、前記ヒンダードアミン(C)を1.0重量%~10.0重量%含む、請求項7または8に記載のポリプロピレン系発泡粒子。
- 前記ポリプロピレン系発泡粒子の全量100重量%に対して、ポリプロピレン系樹脂(A)を70.0重量%以上含む、請求項1~9のいずれか1項に記載のポリプロピレン系発泡粒子。
- 嵩密度が15.0g/L~400.0g/Lである、請求項1~10のいずれか1項に記載のポリプロピレン系発泡粒子。
- 請求項1~11のいずれか1項に記載のポリプロピレン系発泡粒子を型内発泡成形してなる、ポリプロピレン系発泡成形体。
- 前記ポリプロピレン系発泡成形体は、UL94V“垂直燃焼発泡材料試験”においてグレードV-0を満たす、請求項12に記載のポリプロピレン系発泡成形体。
- 請求項1~11のいずれか1項に記載のポリプロピレン系発泡粒子を型内発泡成形する工程を含む、ポリプロピレン系発泡成形体の製造方法。
- 前記ポリプロピレン系発泡粒子に無機ガスを含浸させる工程を含まないか、または、前記ポリプロピレン系発泡粒子に無機ガスを含浸させ、0.10MPa(絶対圧)超、0.25MPa(絶対圧)以下の内圧を前記ポリプロピレン系発泡粒子に付与する工程を含む、請求項14に記載のポリプロピレン系発泡成形体の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280074912.6A CN118234785A (zh) | 2021-11-16 | 2022-11-15 | 聚丙烯系发泡颗粒、和聚丙烯系发泡成型体、以及它们的制造方法 |
| JP2023561594A JPWO2023090311A1 (ja) | 2021-11-16 | 2022-11-15 | |
| EP22895592.8A EP4435041A4 (en) | 2021-11-16 | 2022-11-15 | POLYPROPYLENE FOAM PARTICLES AND MOLDED POLYPROPYLENE FOAM BODY, AND THEIR PRODUCTION PROCESSES |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-186536 | 2021-11-16 | ||
| JP2021186536 | 2021-11-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023090311A1 true WO2023090311A1 (ja) | 2023-05-25 |
Family
ID=86397013
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/042353 Ceased WO2023090311A1 (ja) | 2021-11-16 | 2022-11-15 | ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4435041A4 (ja) |
| JP (1) | JPWO2023090311A1 (ja) |
| CN (1) | CN118234785A (ja) |
| WO (1) | WO2023090311A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7425137B1 (ja) | 2022-08-31 | 2024-01-30 | 株式会社ジェイエスピー | 発泡粒子及び発泡粒子成形体 |
| WO2024247859A1 (ja) * | 2023-05-26 | 2024-12-05 | 株式会社ジェイエスピー | ポリオレフィン系樹脂発泡粒子、及びポリオレフィン系樹脂発泡粒子成形体 |
| WO2025115815A1 (ja) * | 2023-11-30 | 2025-06-05 | ジェイエスピー インターナショナル エスエーアールエル | ポリオレフィン系樹脂発泡粒子、該発泡粒子を型内で成形してなる発泡粒子成形体、該発泡粒子の製造方法、及び該発泡粒子の難燃性の判定方法 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1503429A (fr) | 1965-12-14 | 1967-11-24 | Ciba Geigy | Procédé de préparation d'esters cycliques de l'acide phosphonique |
| GB1515223A (en) | 1975-07-15 | 1978-06-21 | Ciba Geigy Ag | Flame-proofing of polyester fibres |
| US4174343A (en) | 1978-05-05 | 1979-11-13 | American Cyanamid Company | Pentaerythrityl diphosphonate-ammonium polyphosphate combinations as flame retardants for olefin polymers |
| EP0309402A1 (en) | 1987-09-21 | 1989-03-29 | Ciba-Geigy Ag | N-substituted hindered amine stabilizers |
| JPH10152574A (ja) * | 1996-11-25 | 1998-06-09 | Kanegafuchi Chem Ind Co Ltd | ポリオレフィン系樹脂予備発泡粒子の製造方法 |
| EP0889085A2 (en) | 1997-06-30 | 1999-01-07 | Ciba SC Holding AG | N,N',N'''-Tris(2,4-bis(1-hydrocarbyloxy-2,2,6,6,-tetramethylpiperidin-4-yl)alkylamino)-s-triazin-6-yl)-3,3'-ethylenediiminodipropylamines, their isomers and bridged derivatives and polymer compositions stabilized therewith |
| JP2000191860A (ja) * | 1998-12-28 | 2000-07-11 | Kanegafuchi Chem Ind Co Ltd | 含水性ポリプロピレン系樹脂組成物およびそれからなる予備発泡粒子 |
| JP2007137987A (ja) * | 2005-11-17 | 2007-06-07 | Kaneka Corp | ポリプロピレン系樹脂予備発泡粒子 |
| JP2010100313A (ja) * | 2008-10-23 | 2010-05-06 | Kaneka Corp | 集合包装用発泡緩衝材 |
| EP2225318A1 (en) | 2007-12-21 | 2010-09-08 | Basf Se | Flame retardant compositions comprising sterically hindered amines |
| US8598369B2 (en) | 2009-08-11 | 2013-12-03 | Basf Se | Bi—or tricyclic sterically hindered alkoxyamines and process for their preparation |
| WO2016052739A1 (ja) * | 2014-10-03 | 2016-04-07 | カネカ ベルギー ナムローゼ フェンノートシャップ | ポリオレフィン系樹脂予備発泡粒子および型内発泡成形体、並びにそれらの製造方法 |
| WO2017169260A1 (ja) | 2016-03-31 | 2017-10-05 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子およびポリプロピレン系樹脂型内発泡成形体、ならびにその製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4031314B2 (ja) * | 2002-08-02 | 2008-01-09 | 株式会社カネカ | ポリプロピレン系樹脂予備発泡粒子 |
-
2022
- 2022-11-15 EP EP22895592.8A patent/EP4435041A4/en active Pending
- 2022-11-15 JP JP2023561594A patent/JPWO2023090311A1/ja active Pending
- 2022-11-15 WO PCT/JP2022/042353 patent/WO2023090311A1/ja not_active Ceased
- 2022-11-15 CN CN202280074912.6A patent/CN118234785A/zh active Pending
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1503429A (fr) | 1965-12-14 | 1967-11-24 | Ciba Geigy | Procédé de préparation d'esters cycliques de l'acide phosphonique |
| GB1515223A (en) | 1975-07-15 | 1978-06-21 | Ciba Geigy Ag | Flame-proofing of polyester fibres |
| US4174343A (en) | 1978-05-05 | 1979-11-13 | American Cyanamid Company | Pentaerythrityl diphosphonate-ammonium polyphosphate combinations as flame retardants for olefin polymers |
| EP0309402A1 (en) | 1987-09-21 | 1989-03-29 | Ciba-Geigy Ag | N-substituted hindered amine stabilizers |
| JPH10152574A (ja) * | 1996-11-25 | 1998-06-09 | Kanegafuchi Chem Ind Co Ltd | ポリオレフィン系樹脂予備発泡粒子の製造方法 |
| EP0889085A2 (en) | 1997-06-30 | 1999-01-07 | Ciba SC Holding AG | N,N',N'''-Tris(2,4-bis(1-hydrocarbyloxy-2,2,6,6,-tetramethylpiperidin-4-yl)alkylamino)-s-triazin-6-yl)-3,3'-ethylenediiminodipropylamines, their isomers and bridged derivatives and polymer compositions stabilized therewith |
| JP2000191860A (ja) * | 1998-12-28 | 2000-07-11 | Kanegafuchi Chem Ind Co Ltd | 含水性ポリプロピレン系樹脂組成物およびそれからなる予備発泡粒子 |
| JP2007137987A (ja) * | 2005-11-17 | 2007-06-07 | Kaneka Corp | ポリプロピレン系樹脂予備発泡粒子 |
| EP2225318A1 (en) | 2007-12-21 | 2010-09-08 | Basf Se | Flame retardant compositions comprising sterically hindered amines |
| JP2010100313A (ja) * | 2008-10-23 | 2010-05-06 | Kaneka Corp | 集合包装用発泡緩衝材 |
| US8598369B2 (en) | 2009-08-11 | 2013-12-03 | Basf Se | Bi—or tricyclic sterically hindered alkoxyamines and process for their preparation |
| WO2016052739A1 (ja) * | 2014-10-03 | 2016-04-07 | カネカ ベルギー ナムローゼ フェンノートシャップ | ポリオレフィン系樹脂予備発泡粒子および型内発泡成形体、並びにそれらの製造方法 |
| WO2017169260A1 (ja) | 2016-03-31 | 2017-10-05 | 株式会社カネカ | ポリプロピレン系樹脂発泡粒子およびポリプロピレン系樹脂型内発泡成形体、ならびにその製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| no. 191680-81-6 |
| See also references of EP4435041A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7425137B1 (ja) | 2022-08-31 | 2024-01-30 | 株式会社ジェイエスピー | 発泡粒子及び発泡粒子成形体 |
| WO2024048327A1 (ja) * | 2022-08-31 | 2024-03-07 | 株式会社ジェイエスピー | 発泡粒子及び発泡粒子成形体 |
| JP2024033777A (ja) * | 2022-08-31 | 2024-03-13 | 株式会社ジェイエスピー | 発泡粒子及び発泡粒子成形体 |
| WO2024247859A1 (ja) * | 2023-05-26 | 2024-12-05 | 株式会社ジェイエスピー | ポリオレフィン系樹脂発泡粒子、及びポリオレフィン系樹脂発泡粒子成形体 |
| WO2025115815A1 (ja) * | 2023-11-30 | 2025-06-05 | ジェイエスピー インターナショナル エスエーアールエル | ポリオレフィン系樹脂発泡粒子、該発泡粒子を型内で成形してなる発泡粒子成形体、該発泡粒子の製造方法、及び該発泡粒子の難燃性の判定方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN118234785A (zh) | 2024-06-21 |
| EP4435041A1 (en) | 2024-09-25 |
| EP4435041A4 (en) | 2025-11-19 |
| JPWO2023090311A1 (ja) | 2023-05-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107148441B (zh) | 聚烯烃系树脂预发泡粒子和模内发泡成型体以及它们的制造方法 | |
| WO2023090311A1 (ja) | ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 | |
| EP1452559B1 (en) | Flame-retardant expanded polyolefins containing carbon black | |
| JP5927192B2 (ja) | ポリオレフィン系樹脂発泡粒子およびその型内発泡成形体 | |
| CN102388093B (zh) | 聚烯烃系树脂预发泡粒子及由聚烯烃系树脂预发泡粒子形成的聚烯烃系树脂模内发泡成形体 | |
| JP5161663B2 (ja) | 難燃性ポリオレフィン系樹脂予備発泡粒子およびその型内発泡成形体 | |
| JP5914484B2 (ja) | 粉状活性炭含有発泡ポリオレフィン | |
| EP4317288B1 (en) | Polypropylene-based foamed particles and polypropylene-based foamed molded article and manufacturing method thereof | |
| WO2012121163A1 (ja) | ポリエチレン系樹脂発泡粒子、ポリエチレン系樹脂型内発泡成形体、および、ポリエチレン系樹脂発泡粒子の製造方法 | |
| JP2026063159A (ja) | ポリプロピレン系樹脂発泡粒子、ポリプロピレン系樹脂発泡成形体、およびポリプロピレン系樹脂発泡粒子の製造方法 | |
| JP5814687B2 (ja) | ポリオレフィン系樹脂発泡粒子とその型内発泡成形体 | |
| JP7817933B2 (ja) | ポリオレフィン系樹脂発泡粒子及びそのポリオレフィン系樹脂発泡成形体 | |
| WO2023181879A1 (ja) | ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 | |
| JP2018162370A (ja) | ポリプロピレン系樹脂黒色発泡粒子の製造方法 | |
| WO2024189729A1 (ja) | ポリプロピレン系樹脂発泡粒子及び発泡粒子成形体 | |
| WO2024053584A1 (ja) | ポリオレフィン系発泡粒子、および、ポリオレフィン系発泡成形体、並びにそれらの製造方法 | |
| JP7853280B2 (ja) | ポリプロピレン系発泡粒子、および、ポリプロピレン系発泡成形体、並びにそれらの製造方法 | |
| JP7175776B2 (ja) | ポリオレフィン系樹脂発泡粒子の製造方法 | |
| JP2024144291A (ja) | ポリオレフィン系樹脂発泡粒子およびその製造方法 | |
| JP2025099488A (ja) | ポリオレフィン系樹脂発泡粒子 | |
| JP2015168780A (ja) | ポリプロピレン系樹脂予備発泡粒子、及び型内発泡成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22895592 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280074912.6 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2023561594 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022895592 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2022895592 Country of ref document: EP Effective date: 20240617 |