WO2023145341A1 - 熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物および繊維強化複合材料 - Google Patents
熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物および繊維強化複合材料 Download PDFInfo
- Publication number
- WO2023145341A1 WO2023145341A1 PCT/JP2022/047536 JP2022047536W WO2023145341A1 WO 2023145341 A1 WO2023145341 A1 WO 2023145341A1 JP 2022047536 W JP2022047536 W JP 2022047536W WO 2023145341 A1 WO2023145341 A1 WO 2023145341A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- curing agent
- epoxy resin
- composition
- fiber
- resin composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/5033—Amines aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3227—Compounds containing acyclic nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/38—Epoxy compounds containing three or more epoxy groups together with di-epoxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/56—Amines together with other curing agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
Definitions
- the present invention relates to a thermosetting resin curing agent composition, an epoxy resin composition, and a fiber-reinforced composite material.
- Fiber reinforced composite materials are lightweight, high-strength, and high-rigidity. For this reason, FRP is used in a wide range of applications, such as sports and leisure applications such as fishing rods and golf shafts, and industrial applications such as automobiles and aircraft.
- thermosetting resin as a matrix resin
- resin transfer molding ( Hereinafter, it may be referred to as "RTM”.) method is known.
- RTM resin transfer molding
- the RTM method has attracted attention because it has few processes for manufacturing FRP, does not require expensive equipment such as an autoclave, is low cost, and has excellent productivity.
- an epoxy resin composition containing an epoxy resin main agent and a curing agent is used as the liquid resin composition used in this RTM method.
- the epoxy resin composition used in the RTM method is used in a state in which the curing agent is dissolved in the main epoxy resin composition in order to prevent the curing agent contained in the epoxy resin composition from being filtered out when the fiber-reinforced base material is impregnated. often stored.
- Such an epoxy resin composition in which the curing agent is dissolved in the epoxy resin base is called a one-liquid type epoxy resin composition.
- the one-liquid type epoxy resin composition since the curing agent exists in a dissolved state in the epoxy resin main agent, the reaction between the epoxy resin main agent and the curing agent is relatively easy to occur, and the shelf life of the epoxy resin composition ( There is a problem that the shelf life) is shortened. Therefore, the one-liquid type epoxy resin composition needs to be stored frozen.
- This two-liquid type epoxy resin composition is composed of a main liquid containing an epoxy resin as a main component and a curing agent liquid containing a curing agent as a main component, and is obtained by mixing these two liquids just before use. It is an epoxy resin composition.
- the epoxy resin main component liquid and the curing agent liquid are easy to mix.
- the curing agent those used in one-component epoxy resin compositions can also be used.
- the aromatic polyamine curing agent used in one-component epoxy resin compositions It is usually solid and tends to cause poor mixing with the epoxy resin base liquid. Therefore, in the two-liquid type epoxy resin composition, it is desirable that the curing agent is also liquid.
- Patent Documents 2 and 3 describe epoxy resin compositions using a liquid aromatic polyamine as a curing agent. However, resin cured products obtained from these epoxy resin compositions do not have sufficient mechanical properties such as elastic modulus and fracture toughness.
- Patent Document 4 proposes a fast-curing two-component epoxy resin composition using a compound having two or more aromatic rings having phenolic hydroxyl groups.
- a cured resin product that is a two-component epoxy resin composition has sufficiently low viscosity, has a long pot life, and has the heat resistance and mechanical properties required for industrial applications such as automobiles and aircraft.
- An object of the present invention is to provide a thermosetting resin curing agent composition that can produce an epoxy resin composition having a low viscosity and a long usable life.
- the object of the present invention is to provide a curing agent composition for thermosetting resins, which becomes a uniform liquid at a temperature of 200° C. or less by heating, and can then maintain the uniform liquid state at room temperature for one week or more.
- Another object of the present invention is to provide a cured epoxy resin composition and a fiber-reinforced composite material with high mechanical properties.
- the present invention is a curing agent composition for thermosetting resins comprising a curing agent A, a curing agent B and a curing agent C, wherein the curing agent A has substituents at two ortho-positions to an amino group.
- the substituent is selected from an alkyl group, an aromatic group and a halogen group
- the curing agent B is an aromatic polyamine that is liquid at 25 ° C.
- the curing agent C is an aromatic substituent
- a curing agent composition for a thermosetting resin characterized in that it is a monoamine having
- the present invention also provides an epoxy resin composition containing the above curing agent composition for thermosetting resin, epoxy resin D and epoxy resin E, wherein epoxy resin D is a monomer containing 4 or more glycidyl groups.
- Epoxy resin E is an epoxy resin composition, which is an epoxy resin composed of a monomer containing two or three glycidyl groups.
- the present invention is also an epoxy resin cured product obtained by curing any one of the above epoxy resin compositions.
- the present invention is also a fiber-reinforced composite material containing the above epoxy resin cured product and a fiber-reinforced base material.
- the present invention also provides a curing agent composition for a thermosetting resin containing a curing agent C, wherein the curing agent C is a monoamine having an aromatic substituent. It is a curing agent composition for
- the monoamine having an aromatic substituent of the curing agent C has two or more substituents.
- the monoamine having an aromatic substituent of the curing agent C is a benzylamine derivative.
- thermosetting resin curing agent composition that can produce an epoxy resin composition having a low viscosity and a long usable life.
- thermosetting resins which becomes a uniform liquid at a temperature of 200° C. or less by heating, and can then maintain the uniform liquid state at room temperature for one week or more.
- the present invention can further provide a cured epoxy resin composition and a fiber-reinforced composite material with high mechanical properties.
- a fiber reinforced composite material may be abbreviated as "FRP”, and a carbon fiber reinforced composite material as "CFRP”.
- the curing agent composition for thermosetting resins of the present invention is a curing agent composition for thermosetting resins containing curing agent A, curing agent B and curing agent C described later. This curing agent composition for thermosetting resin becomes a uniform liquid by heating to a temperature of 80 to 200°C.
- the curing agent composition for thermosetting resins of the present invention becomes a uniform liquid at a temperature of 80 to 200° C. After raising the liquid temperature to 200° C., lowering the temperature to 25° C., and allowing it to stand at 25° C. for 1 week. Preferably, it is a uniform liquid even after standing still for 2 weeks (3 weeks in total), and particularly preferably after standing still for 1 month.
- the curing agent for thermosetting resin is substantially liquid. It is not preferable because it becomes difficult to handle as a composition, and poor mixing with the main liquid is likely to occur.
- Curing agent A is an aromatic polyamine having two substituents each ortho to the amino group, the substituents being selected from alkyl groups, aromatic groups and halogen groups. Also, curing agent A is solid at 25°C. By containing this curing agent A, when cured as a composition with an epoxy resin, it is possible to obtain an epoxy resin cured product having excellent mechanical properties such as heat resistance, elastic modulus, and fracture toughness.
- a compound represented by the following chemical formula (1) can be used as an aromatic polyamine having substituents at two ortho-positions to an amino group, which is used as the curing agent A.
- R 1 to R 4 are each independently an aliphatic substituent, an aromatic substituent, an alkoxy group or a halogen atom, and at least one substituent has a carbon number Any of 1 to 6 aliphatic substituents, aromatic substituents and halogen atoms.
- aliphatic substituents having 1 to 6 carbon atoms include methyl group, ethyl group, propyl group, isopropyl group, n-butyl group, sec-butyl group, tert-butyl group and n-pentyl group. , neopentyl group, n-hexyl group, and cyclohexyl group.
- a phenyl group and a naphthyl group are illustrated as an aromatic substituent.
- the aromatic polyamine of the curing agent A is preferably an aromatic diamine, with 4,4'-diaminodiphenylmethane derivatives being particularly preferred.
- this aromatic polyamine include compounds represented by the following chemical formulas (2) to (5). These may be used alone or in combination.
- Hardener B is an aromatic polyamine that is liquid at 25°C. By containing this aromatic polyamine, it is possible to obtain a curing agent composition for thermosetting resins that can maintain a liquid state at room temperature.
- a phenylenediamine derivative or a 4,4'-diaminodiphenylmethane derivative is preferably used as the aromatic polyamine for the curing agent B.
- this aromatic polyamine include compounds represented by the following chemical formula (6) or (7).
- R 5 to R 8 are each independently a hydrogen atom, an aliphatic substituent, an alkoxy group or a thioalkoxy group, and at least one substituent has 1 to 6 carbon atoms. is either an aliphatic substituent of or a thioalkoxy group.
- R 9 to R 10 are each independently an aliphatic substituent, a methoxy group, an alkoxy group or a thioalkoxy group.
- aromatic polyamine used as the curing agent B include compounds represented by the following chemical formulas (8) to (12). These may be used alone or in combination.
- Curing agent C is a monoamine having an aromatic substituent.
- the aromatic substituent may be directly attached to the nitrogen atom or may be attached to the nitrogen atom via an alkyl group.
- the monoamine having an aromatic substituent of the curing agent C has two or more substituents on the aromatic ring of the aromatic substituent.
- the aromatic substituent is attached directly to the nitrogen atom of the monoamine.
- the monoamine having an aromatic substituent of curing agent C is a benzylamine derivative.
- This "derivative" means a compound having one or more substituents on the benzene ring of the benzyl group.
- the aromatic-substituted monoamine of the curing agent C is an aromatic compound having one amino group directly bonded to an aromatic ring in one molecule.
- the amino group may be any of primary amine, secondary amine and tertiary amine, preferably primary amine from the viewpoint of heat resistance of the cured product.
- the aromatic ring of the aromatic substituent is, for example, a benzene ring or a naphthalene ring, preferably a benzene ring.
- substituents on the aromatic ring of the aromatic substituents include aliphatic groups, alkoxy groups, hydroxy groups, carbonyl groups, sulfonyl groups, thioalkoxy groups, and halogen atoms. This substituent is preferably an aliphatic group and/or an alkoxy group from the viewpoint of obtaining good reactivity.
- two of the substituents on the aromatic ring are aliphatic groups and/or alkoxy groups.
- the aliphatic group is preferably an aliphatic group having 1 to 4 carbon atoms, ie methyl group, ethyl group, propyl group or butyl group, particularly preferably methyl group.
- the monoamine having an aromatic substituent for the curing agent C include 2,5-dimethylaniline, 3,4-dimethylaniline, 2,4-dimethylaniline, 2,3-dimethylaniline, 3,5-dimethylaniline, -tert-butylaniline, 4-methoxy-2-methylaniline, 5-methoxy-2-methylaniline, 2-methoxy-5-methylaniline, 2,3-dimethoxyaniline, 2,5-dimethoxyaniline, 3,5 -dimethoxyaniline, 3,4-dimethoxyaniline, 3,4-diethoxyaniline, 2,5-diethoxyaniline, 3,4,6-trimethoxyaniline, preferably 2,5-dimethyl Aniline, 3,4-dimethylaniline, 2,4-dimethylaniline, 2,3-dimethylaniline, more preferably 2,5-dimethylaniline.
- the melting point of the monoamine having an aromatic substituent in the curing agent C is preferably 200°C or lower, more preferably 150°C or lower, and particularly preferably 120°C or lower. If the melting point exceeds 200° C., it becomes difficult to obtain a liquid composition when the curing agent C is mixed with the curing agents A and B, and the obtained curing agent composition for thermosetting resins is cooled at room temperature. It is not preferable because it tends to be difficult to maintain the liquid state.
- the curing reaction of the obtained epoxy resin composition is accelerated, and rapid curing can be imparted to the epoxy resin composition.
- the monoamine having an aromatic substituent is a benzylamine derivative.
- the benzylamine derivative is benzylamine (cas number: 100-46-9), a benzylamine having a substituent, and a monoamine having an amino group at the benzylic position.
- the amino group of the benzylamine derivative may be any of primary amine, secondary amine and tertiary amine, and from the viewpoint of heat resistance of the cured product, primary amine is preferred.
- the benzylamine derivative of curing agent C may have a substituent.
- this substituent include an aliphatic group, an alkoxy group, a hydroxy group, a carbonyl group, a sulfonyl group, a thioalkoxy group, and a halogen atom. From the viewpoint of obtaining good reactivity, this substituent is preferably an aliphatic group or an alkoxy group.
- the substituent is an aliphatic group
- the aliphatic group is preferably an aliphatic group having 1 to 4 carbon atoms, such as methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group. , sec-butyl group and tert-butyl group, and particularly preferably methyl group.
- the substituent is an alkoxy group
- the alkoxy group is a methoxy group, an ethoxy group or a phenoxy group, and particularly preferably a methoxy group.
- the substituent may be attached to the aromatic ring of benzylamine or may be attached to the benzylic position.
- Curing agent C may also have a cyclic substituent such as piperonylamine.
- benzylamine derivatives of curing agent C include benzylamine, 2-methylbenzylamine, 3-methylbenzylamine, 4-methylbenzylamine, 2,3-dimethylbenzylamine, 2,4-dimethylbenzylamine, 2,5-dimethylbenzylamine, 2,6-dimethylbenzylamine, 3,4-dimethylbenzylamine, 3,5-dimethylbenzylamine, 2-ethylbenzylamine, 3-ethylbenzylamine, 4-ethylbenzylamine, 4-isopropylbenzylamine, 4-tert-butylbenzylamine, 2-methoxybenzylamine, 3-methoxybenzylamine, 4-methoxybenzylamine, 2,3-dimethoxybenzylamine, 2,4-dimethoxybenzylamine, 2, 5-dimethoxybenzylamine, 2,6-dimethoxybenzylamine, 3,4-dimethoxybenzylamine, 3,5-dimethoxybenzylamine
- -butylbenzylamine cumylamine, 1-(p-tolyl)ethylamine, 2-methoxybenzylamine, 3-methoxybenzylamine, 4-methoxybenzylamine, 2,3-dimethoxybenzylamine, 2,4-dimethoxybenzylamine, 2,5-dimethoxybenzylamine, 2,6-dimethoxybenzylamine, 3,4-dimethoxybenzylamine, 3,5-dimethoxybenzylamine, more preferably 4-methylbenzylamine, 3-methoxybenzylamine, 4-methoxy benzylamine, 3,4-dimethoxybenzylamine. 4-Methoxybenzylamine is particularly preferred.
- the melting point of the benzylamine derivative of the curing agent C is preferably 200°C or lower, more preferably 150°C or lower, and particularly preferably 120°C or lower. If the melting point exceeds 200° C., it becomes difficult to obtain a liquid composition when the curing agent C is mixed with the curing agents A and B, and the obtained curing agent composition for thermosetting resins is cooled at room temperature. It is not preferable because it tends to be difficult to maintain the liquid state.
- the boiling point of the benzylamine derivative of the curing agent C at normal pressure is preferably 180°C or higher, more preferably 200°C or higher, still more preferably 230°C or higher, and particularly preferably 250°C or higher. If the melting point is lower than 180° C., the curing agent C volatilizes in the step of mixing with the epoxy resin and heat-curing to obtain a resin-cured product, which may deteriorate workability, which is not preferable.
- the curing reaction of the obtained epoxy resin composition is accelerated, and rapid curing can be imparted to the epoxy resin composition.
- the mass ratio of curing agent A to curing agent B is preferably 1:99 to 99:1, more preferably 20:80 to 80:20, particularly preferably 40:60 to 70:30. If the proportion of the curing agent A is less than this, mechanical properties such as heat resistance, elastic modulus and fracture toughness of the resin cured product obtained tend to be insufficient, which is not preferable. On the other hand, if the ratio of the curing agent A is higher than this, it becomes difficult for the obtained curing agent composition for thermosetting resins to maintain a liquid state at room temperature, which is undesirable.
- the curing agent C is preferably 1 to 43 parts by mass, more preferably 3 to 30 parts by mass with respect to a total of 100 parts by mass of the curing agent A and the curing agent B. parts, particularly preferably 5 to 20 parts by weight. If the content of the curing agent C is less than 1 part by mass, it becomes difficult to impart rapid curability to the resulting epoxy resin composition, which is not preferred. On the other hand, when it exceeds 43 parts by mass, the reactivity of the obtained epoxy resin composition becomes excessively high, and the pot life in RTM molding becomes extremely short, which is not preferable.
- the total amount of curing agent A, curing agent B and curing agent C is preferably 70 to 100, based on the total mass of the curing agent composition for thermosetting resins. % by weight, more preferably 80 to 100% by weight. If it is less than 70% by mass, the heat resistance of the cured product may be insufficient, which is not preferred.
- the curing agent composition for thermosetting resins of the present invention may further contain other curing agents and other components as long as the above conditions are satisfied.
- it may contain conductive particles, flame retardants, inorganic fillers, and internal release agents.
- Conductive particles include conductive polymer particles such as polyacetylene particles, polyaniline particles, polypyrrole particles, polythiophene particles, polyisothianaphthene particles and polyethylenedioxythiophene particles; carbon particles; carbon fiber particles; metal particles; Particles in which a core material composed of is coated with a conductive substance can be exemplified.
- a phosphorus-based flame retardant can be exemplified as a flame retardant.
- the phosphorus-based flame retardant may contain a phosphorus atom in the molecule, and examples thereof include organic phosphorus compounds such as phosphoric acid esters, condensed phosphoric acid esters, phosphazene compounds and polyphosphates, and red phosphorus.
- inorganic fillers examples include aluminum borate, calcium carbonate, silicon carbonate, silicon nitride, potassium titanate, basic magnesium sulfate, zinc oxide, graphite, calcium sulfate, magnesium borate, magnesium oxide, and silicate minerals. can do. In particular, it is preferable to use silicate minerals. As a commercially available silicate mineral, THIXOTROPIC AGENT DT 5039 (manufactured by Huntsman Japan Co., Ltd.) can be mentioned.
- internal mold release agents include metal soaps, vegetable waxes such as polyethylene wax and carnauba wax, fatty acid ester mold release agents, silicone oils, animal waxes, and fluorine-based nonionic surfactants.
- the curing agent composition for thermosetting resins of the present invention can be produced by mixing curing agent A, curing agent B, curing agent C, and, if necessary, other components. The order of mixing does not matter.
- the temperature of the composition during mixing is preferably 50-200°C, more preferably 50-150°C, and particularly preferably 80-120°C. If the temperature exceeds 200°C, the added components may be thermally decomposed, which is not preferable. On the other hand, when the temperature is less than 50°C, the solid curing agents A and C do not melt and are difficult to dissolve in the curing agent B, making it difficult to obtain a liquid curing agent composition for thermosetting resins. It is not preferable.
- Examples of devices used for mixing the curing agent include roll mills, planetary mixers, kneaders, extruders, Banbury mixers, mixing vessels equipped with stirring blades, and horizontal mixing tanks. Mixing may be performed in air or under an inert gas atmosphere.
- the epoxy resin composition of the present invention comprises the thermosetting resin curing agent composition of the present invention and an epoxy resin main agent.
- the epoxy resin base contains epoxy resin D and epoxy resin E below.
- the epoxy resin main agent may further contain other optional components.
- the content of the epoxy resin in the epoxy resin main agent is preferably 30 to 100% by weight, more preferably 50 to 100% by weight, based on the total weight of the epoxy resin main agent.
- Epoxy resin D is an epoxy resin composed of a monomer containing four or more glycidyl groups. Epoxy resin D may be a homopolymer composed of one type of monomer, a copolymer composed of two or more types of monomers, or a mixture of homopolymers and/or copolymers. .
- the constituent monomer of the epoxy resin D composed of monomers containing 4 or more glycidyl groups is preferably represented by the following chemical formula (13).
- R 1 to R 4 each independently represent one selected from the group consisting of a hydrogen atom, an aliphatic hydrocarbon group, an alicyclic hydrocarbon group and a halogen atom
- R 1 to R 4 are aliphatic hydrocarbon groups or alicyclic hydrocarbon groups, they preferably have 1 to 4 carbon atoms.
- tetraglycidyl-4,4'-diaminodiphenyl ether As constituent monomers of epoxy resin D, tetraglycidyl-4,4'-diaminodiphenyl ether, tetraglycidyl-4,4'-diaminodiphenylmethane, tetraglycidyl-3,4'-diaminodiphenyl ether and tetraglycidyl-3,3'-diamino
- One or a combination of two or more selected from the group consisting of diphenylmethane is particularly preferred.
- Epoxy resin D is preferably a homopolymer, copolymer or mixture thereof composed of these monomers. It is preferable that R 1 to R 4 are hydrogen atoms, because formation of a special steric structure in the cured resin is less likely to be inhibited. In addition, X is preferably -O- because it facilitates the synthesis of the compound.
- the constituent monomers of epoxy resin D may be synthesized by any method.
- an aromatic diamine and an epihalohydrin such as epichlorohydrin, which are raw materials, are reacted preferably in the presence of an acid catalyst to obtain a tetrahalohydrin body, and then subjected to a cyclization reaction using an alkaline compound.
- an acid catalyst preferably in the presence of an acid catalyst to obtain a tetrahalohydrin body
- an alkaline compound e.g., it can be synthesized by the method described in Examples below.
- aromatic diamines 4,4'-diaminodiphenyl ether, 3,3'-diaminodiphenyl ether, 3,4'-diaminodiphenyl ether, 4,4'-diaminodiphenyl sulfone, 3,3'-diaminodiphenyl sulfone, 3,4 '-diaminodiphenyl sulfone, 4,4'-diaminodiphenyl methane can be exemplified.
- aromatic diamines in which two aromatic rings having amino groups are linked by an ether bond are preferable from the viewpoint of heat resistance. More preferably, it is an ortho-located aromatic diamine.
- aromatic diamines include 3,4'-diaminodiphenyl ether and 3,4'-diaminodiphenyl sulfone.
- epihalohydrin examples include epichlorohydrin, epibromohydrin, and epifluorohydrin.
- epichlorohydrin and epibromohydrin are particularly preferred from the viewpoint of reactivity and handleability.
- the mass ratio of the raw materials, aromatic diamine and epihalohydrin is preferably 1:1 to 1:20, more preferably 1:3 to 1:10.
- Solvents used in the reaction include alcohol solvents such as ethanol and n-butanol, ketone solvents such as methyl isobutyl ketone and methyl ethyl ketone, aprotic polar solvents such as acetonitrile and N,N-dimethylformamide, and aromatic solvents such as toluene and xylene.
- group hydrocarbon solvents can be exemplified.
- Alcohol solvents such as ethanol and n-butanol, and aromatic hydrocarbon solvents such as toluene and xylene are particularly preferred.
- the amount of the solvent used is preferably 1 to 10 times the amount by mass of the aromatic diamine.
- Bronsted acids and Lewis acids can be suitably used as acid catalysts.
- Preferred Bronsted acids are ethanol, water and acetic acid, and preferred Lewis acids are titanium tetrachloride, lanthanum nitrate hexahydrate and boron trifluoride diethyl ether complex.
- the reaction time is preferably 0.1 to 180 hours, more preferably 0.5 to 24 hours.
- the reaction temperature is preferably 20-100°C, more preferably 40-80°C.
- alkaline compounds used during the cyclization reaction include sodium hydroxide and potassium hydroxide.
- the alkaline compound may be added as a solid or as an aqueous solution.
- phase transfer catalyst may be used during the cyclization reaction.
- Phase transfer catalysts such as quaternary ammonium salts such as tetramethylammonium chloride, tetraethylammonium bromide, benzyltriethylammonium chloride, and tetrabutylammonium hydrogen sulfate; phosphonium compounds such as tributylhexadecylphosphonium bromide and tributyldodecylphosphonium bromide; Crown ethers such as 18-crown-6-ether can be exemplified.
- Epoxy resin E is an epoxy resin composed of a monomer containing two or three glycidyl groups. By including this epoxy resin E, the viscosity of the epoxy resin composition can be reduced to improve the resin impregnation property of the reinforcing fiber base material, the pot life can be extended, and it can be used in the RTM molding method. The degree of freedom in mold design can be increased.
- a constituent monomer of epoxy resin E a monomer having two or three glycidyl groups is used.
- it is an aromatic compound.
- Monomers having two glycidyl groups include diglycidylaniline and its derivatives diglycidyl-o-toluidine, diglycidyl-m-toluidine, diglycidyl-p-toluidine, diglycidyl-xylidine, diglycidyl-mesidine, diglycidyl-anisidine, diglycidyl-phenoxy Aniline, diglycidyl-naphthylamine, bisphenol A diglycidyl ether and derivatives thereof are preferably used.
- diglycidyl-aniline, diglycidyl-o-toluidine, diglycidyl-m-toluidine, diglycidyl-p-toluidine, diglycidyl-phenoxyaniline, bisphenol A diglycidyl ether is more preferably used, and diglycidyl-aniline or diglycidyl-o-toluidine is more preferably used. It is more preferable to use
- an epoxy resin having a polycyclic aromatic hydrocarbon skeleton is preferred.
- polycyclic aromatic hydrogen cyclic skeletons examples include naphthalene skeletons and anthracene skeletons, and naphthalene skeletons are preferred from the viewpoint of the physical properties of cured resins.
- the polycyclic aromatic hydrocarbon group may have a substituent in addition to the glycidyl group.
- Monomers having a naphthalene skeleton include 1,6-bis(glycidyloxy)naphthalene, 1,5-bis(glycidyloxy)naphthalene, 2,6-bis(glycidyloxy)naphthalene, and 2,7-bis(glycidyloxy)naphthalene. , 2,2′-bis(glycidyloxy)-1,1′-binaphthalene, and 2,7-bis(glycidyloxy)-1-[2-(glycidyloxy)-1-naphthylmethyl]naphthalene. can.
- the crosslink density of the cured product does not excessively increase, so that the resin can be cured. It is preferable because it can prevent deterioration of the toughness of the object.
- epoxy resins composed of monomers containing two or three glycidyl groups
- aromatic compounds having three glycidyl groups are preferred as constituent monomers of epoxy resin E.
- this epoxy resin a triglycidylaminophenol derivative epoxy resin is preferable.
- triglycidylaminophenol derivative epoxy resins examples include triglycidyl-m-aminophenol and triglycidyl-p-aminophenol.
- this epoxy resin E By containing this epoxy resin E, the viscosity of the epoxy resin composition can be lowered, and the heat resistance of the resin cured product can be improved.
- epoxy resins composed of monomers containing two or three glycidyl groups
- heteroaromatic compounds having three glycidyl groups are also preferable as constituent monomers of epoxy resin E. That is, the epoxy resin E preferably contains a triglycidyl isocyanurate derivative epoxy resin.
- triglycidyl isocyanurate derivative epoxy resins include 1,3,5-triglycidyl isocyanurate, 1,3,5-tri(ethylglycidyl) isocyanurate, and 1,3,5-tri(pentylglycidyl) isocyanurate. be able to.
- the heat resistance and elastic modulus of the cured epoxy resin can be improved. Therefore, by using it in combination with the epoxy resin D, it is possible to obtain a resin cured product and a fiber-reinforced composite material that maintain heat resistance and a high elastic modulus.
- epoxy resin E diglycidylaniline, diglycidyl-o-toluidine, triglycidyl-p-aminophenol, triglycidyl-m-aminophenol, 1,6-bis(2,3-epoxypropan-1-yloxy )
- One or a combination of two or more selected from naphthalene, 1,3,5-triglycidyl isocyanurate and bisphenol A diglycidyl ether is particularly preferred.
- Epoxy resin E is particularly preferably a homopolymer, copolymer, or mixture thereof composed of these monomers.
- epoxy resin E examples include bisphenol F type epoxy resin, bisphenol S type epoxy resin, phenol novolac type epoxy resin, and cresol novolak type epoxy resin.
- the epoxy resin composition of the present invention preferably contains resin particles F.
- the resin particles F are dispersed in the epoxy resin composition without being dissolved, and are dispersed in the cured resin after the epoxy resin composition is cured.
- the resin particles F are present in the cured resin as island components.
- resin particles F By containing resin particles F, high fracture toughness and impact resistance can be obtained in cured resins and fiber-reinforced composite materials.
- thermoplastic resin particles thermosetting resin particles
- rubber particles are preferably used.
- rubber particles include silicone rubber, butadiene rubber, styrene-butadiene rubber, and methyl methacrylate-butadiene-styrene rubber.
- Rubber particles used as resin particles F include MX-153 (bisphenol A type epoxy resin with 33% by mass of butadiene rubber monodispersed, manufactured by Kaneka Corporation), MX-257 (bisphenol A type Epoxy resin with 37% by mass of butadiene rubber monodispersed, manufactured by Kaneka Corporation), MX-154 (bisphenol A type epoxy resin with 40% by mass of butadiene rubber monodispersed, stock Kaneka Company), MX-960 (Bisphenol A epoxy resin with 25% by mass of silicone rubber dispersed in a single dispersion, Kaneka Corporation), MX-136 (Bisphenol F type epoxy resin, 25% by mass) monodispersed butadiene rubber, manufactured by Kaneka Co., Ltd.), MX-965 (bisphenol F type epoxy resin, monodispersed with 25% by mass of silicone rubber, manufactured by Kaneka Co., Ltd.), MX- 217 (25% by mass of butadiene rubber dispersed in phenol novolak type epoxy resin, manufactured by Kaneka Corporation), MX-
- the average particle size of the resin particles F is preferably 1.0 ⁇ m or less, more preferably 0.5 ⁇ m or less, and particularly preferably 0.3 ⁇ m or less.
- the average particle size is preferably 0.03 ⁇ m or more, more preferably 0.05 ⁇ m or more, and particularly preferably 0.08 ⁇ m or more.
- the average particle size is a numerical value calculated by measuring by the following method. Observe the cross section of the resin cured product at 25,000 times with a scanning electron microscope or a transmission electron microscope, measure the diameter of at least 50 particles, and use it as the particle diameter of the resin particles. Determine the particle size. In the above observation, when the particles are not perfectly circular, that is, when the particles are elliptical, the maximum diameter of the particles is taken as the particle diameter of the particles.
- the resin particles F are not filtered out on the surface of the reinforcing fiber base material, and impregnation into the reinforcing fiber bundle can be achieved. become easier. As a result, impregnation failure of the resin can be prevented, and a fiber-reinforced composite material having excellent physical properties can be obtained.
- the resin particles F can also be used as a masterbatch dispersed in an epoxy resin at a high concentration. In this case, it becomes easy to highly disperse the resin particles F in the epoxy resin composition.
- the ratio of the epoxy resin D to the total amount of the epoxy resin (epoxy resin main component) is preferably 50 to 90% by mass, particularly preferably 60 to 80% by mass.
- the proportion of the epoxy resin D is 50% by mass or more, the heat resistance and elastic modulus of the obtained cured resin can be further improved. As a result, various mechanical properties of the resulting fiber-reinforced composite material are also improved.
- the content of the epoxy resin E in the epoxy resin composition of the present invention is preferably 10 to 50% by mass, more preferably 20 to 40% by mass, based on the total mass of the epoxy resin (epoxy resin main ingredient).
- the content of the epoxy resin E relative to the total mass of the epoxy resin within this range, it is possible to produce an epoxy resin composition having a viscosity and pot life suitable for the RTM molding method and having high heat resistance. .
- composition ratio of epoxy resin composition The total amount of the thermosetting resin curing agent composition contained in the epoxy resin composition is an amount suitable for curing all the epoxy resins blended in the epoxy resin composition. It is appropriately adjusted according to the type of curing agent for thermosetting resin.
- the ratio of the total number of epoxy groups of the epoxy resin in the epoxy resin composition to the number of active hydrogens contained in the thermosetting resin curing agent composition is preferably 0.7 to 1.3, More preferably 0.8 to 1.2, particularly preferably 0.9 to 1.1. If the ratio of the number of active hydrogens is less than 0.7 or exceeds 1.3, the molar balance between the epoxy groups and the active hydrogens will be disturbed, and the resulting cured resin will have insufficient crosslink density, resulting in poor heat resistance and elasticity. It is not preferable because the mechanical properties such as modulus and fracture toughness are lowered.
- the content of the resin particles F in the epoxy resin composition of the present invention is preferably 0.1 to 50% by mass, more preferably 0.5 to 20% by mass, and particularly preferably 1% by mass of the total amount of the epoxy resin composition. ⁇ 15% by mass. By setting the content to 0.1% by mass or more, the fracture toughness and impact resistance of the cured resin and fiber composite material can be sufficiently improved.
- the epoxy resin composition of the present invention may contain a curing agent other than curing agent A, curing agent B and curing agent C, epoxy resins other than epoxy resin D and epoxy resin E, and epoxy resins other than epoxy resins.
- a thermosetting resin may be contained, and a thermoplastic resin other than the resin particles F and other additives may be contained.
- curing agents other than curing agent A, curing agent B, and curing agent C include aliphatic polyamines, various isomers of aromatic amine-based curing agents, aminobenzoic acid esters, and acid anhydrides.
- aliphatic polyamines examples include 4,4'-diaminodicyclohexylmethane, isophoronediamine, and m-xylylenediamine.
- aminobenzoic acid esters examples include trimethylene glycol di-p-aminobenzoate and neopentyl glycol di-p-aminobenzoate. Cured products and fiber-reinforced composite materials cured using these curing agents have high tensile elongation.
- acid anhydrides examples include 1,2,3,6-tetrahydrophthalic anhydride, hexahydrophthalic anhydride, and 4-methylhexahydrophthalic anhydride.
- epoxy resin other than epoxy resin D and epoxy resin E for example, a monofunctional epoxy resin can be used.
- an epoxy resin containing an aromatic group is preferred, and an epoxy resin containing either a glycidylamine structure or a glycidyl ether structure is more preferred.
- Alicyclic epoxy resins can also be suitably used.
- These epoxy resins may have non-reactive substituents on the aromatic ring structure, etc., if necessary.
- non-reactive substituents include alkyl groups such as methyl group, ethyl group, and isopropyl group, aromatic groups such as phenyl group, alkoxyl groups, aralkyl groups, and halogen groups such as chlorine and bromine.
- thermosetting resins other than epoxy resins examples include vinyl ester resins, benzoxazine resins, bismaleimide resins, and bismaleimide-triazine resins.
- the epoxy resin composition of the present invention may contain a thermoplastic resin as a component to be dissolved in the epoxy resin composition.
- the thermoplastic resin improves the fracture toughness and impact resistance of the resulting fiber-reinforced composite material.
- Such thermoplastic resins may be dissolved in the epoxy resin composition during the curing process of the epoxy resin composition.
- thermoplastic resins examples include polyethersulfone, polysulfone, polyetherimide, and polycarbonate. These may be used alone or in combination of two or more.
- This thermoplastic resin is particularly preferably polyethersulfone or polysulfone having a weight average molecular weight (Mw) in the range of 8000 to 100000 as measured by gel permeation chromatography.
- Mw weight average molecular weight
- the resulting FRP has sufficient impact resistance, and when it is 100,000 or less, the epoxy resin composition exhibits good handleability without significantly increasing viscosity. can be obtained.
- the molecular weight distribution of this thermoplastic resin is preferably uniform, and the polydispersity (Mw/Mn), which is the ratio of the weight average molecular weight (Mw) to the number average molecular weight (Mn), is preferably 1 to 10, more preferably 1.1-5.
- the thermoplastic resin preferably has a reactive group that is reactive with the epoxy resin or a functional group that forms a hydrogen bond.
- Such thermoplastic resins can improve the dissolution stability during the curing process of epoxy resins.
- fracture toughness, chemical resistance, heat resistance, and resistance to moist heat can be imparted to the fiber-reinforced composite material obtained after curing.
- a hydroxyl group, a carboxylic acid group, an imino group, an amino group, etc. are preferable as the reactive group having reactivity with the epoxy resin.
- the use of hydroxyl-terminated polyethersulfone is more preferable because the resulting fiber-reinforced composite material has particularly excellent impact resistance, fracture toughness and solvent resistance.
- the content of the thermoplastic resin contained in the epoxy resin composition is appropriately adjusted according to the viscosity.
- a thermoplastic resin is contained, it is preferably 0.1 to 10 parts by mass, more preferably 0.1 to 10 parts by mass, based on 100 parts by mass of the epoxy resin contained in the epoxy resin composition, from the viewpoint of impregnation into the fiber-reinforced base material. is 0.5 to 5 parts by mass.
- the resulting fiber-reinforced composite material exhibits sufficient fracture toughness and impact resistance.
- the content is 10 parts by mass or less, the viscosity of the epoxy resin composition does not significantly increase, the impregnation of the fiber-reinforced base material is facilitated, and the properties of the resulting fiber-reinforced composite material are improved.
- the thermoplastic resin preferably contains a reactive aromatic oligomer having an amine end group (hereinafter also simply referred to as "aromatic oligomer").
- the epoxy resin composition has a high molecular weight due to the curing reaction between the epoxy resin and the curing agent during heat curing.
- the expansion of the two-phase region due to the increase in the molecular weight causes the aromatic oligomer dissolved in the epoxy resin composition to undergo reaction-induced phase separation. Due to this phase separation, a two-phase resin structure in which the cured epoxy resin and the aromatic oligomer are co-continuous is formed in the matrix resin.
- aromatic oligomers since aromatic oligomers have amine end groups, they also react with epoxy resins. Since each phase in this co-continuous two-phase structure is strongly bonded to each other, solvent resistance is also improved.
- the aromatic oligomer known polysulfones having amine end groups and polyether sulfones having amine end groups can be used.
- the amine end groups are primary amine (--NH 2 ) end groups.
- the aromatic oligomer When blending an aromatic oligomer into the epoxy resin composition, the aromatic oligomer preferably has a weight average molecular weight of 8,000 to 40,000 as measured by gel permeation chromatography. When the weight average molecular weight is 8000 or more, the effect of improving the toughness of the matrix resin is high. Moreover, when the weight average molecular weight is 40,000 or less, the viscosity of the resin composition does not become excessively high, and processing advantages such as facilitating impregnation of the reinforcing fiber base material with the resin composition can be obtained.
- aromatic oligomer As the aromatic oligomer, commercially available products such as "Virantage DAMS VW-30500 RP (registered trademark)" (manufactured by Solvay Specialty Polymers) can be preferably used.
- the form of the thermoplastic resin before being blended into the epoxy resin composition is preferably particulate.
- the particulate thermoplastic resin can be uniformly blended and dissolved in the resin composition.
- the epoxy resin composition of the present invention may contain other additives such as conductive particles, flame retardants, inorganic fillers, and internal release agents.
- Conductive particles include conductive polymer particles such as polyacetylene particles, polyaniline particles, polypyrrole particles, polythiophene particles, polyisothianaphthene particles and polyethylenedioxythiophene particles; carbon particles; carbon fiber particles; metal particles; Particles in which a core material composed of is coated with a conductive substance can be exemplified.
- a phosphorus-based flame retardant can be exemplified as a flame retardant.
- the phosphorus-based flame retardant may be one containing a phosphorus atom in the molecule, and examples thereof include organic phosphorus compounds such as phosphoric acid esters, condensed phosphoric acid esters, phosphazene compounds and polyphosphates, and red phosphorus.
- inorganic fillers examples include aluminum borate, calcium carbonate, silicon carbonate, silicon nitride, potassium titanate, basic magnesium sulfate, zinc oxide, graphite, calcium sulfate, magnesium borate, magnesium oxide, and silicate minerals. be able to. In particular, it is preferable to use silicate minerals. Commercially available silicate minerals include THIXOTROPIC AGENT DT 5039 (manufactured by Huntsman Japan Co., Ltd.).
- Examples of internal release agents include metal soaps, vegetable waxes such as polyethylene wax and carnauba wax, fatty acid ester release agents, silicone oils, animal waxes, and fluorine-based nonionic surfactants.
- the blending amount is preferably 0.1 to 5 parts by mass, more preferably 0.2 to 2 parts by mass, per 100 parts by mass of the epoxy resin. Within this range, the release effect from the mold is favorably exhibited.
- the epoxy resin composition of the present invention can have the following properties.
- the epoxy resin composition has an initial viscosity at 100° C. or 120° C. of preferably 300 mPa ⁇ s or less, more preferably 0.1 to 100 mPa ⁇ s, still more preferably 0.5 to 50 mPa ⁇ s, and even more preferably 0.5 to 30 mPa ⁇ s, particularly preferably 0.5 to 20 mPa ⁇ s.
- the initial viscosity at 100° C. or 120° C. is 300 mPa ⁇ s or less, impregnation of the reinforcing fiber base material with the epoxy resin composition is easy, and formation of voids that cause deterioration of physical properties in the obtained fiber-reinforced composite material is prevented. can be prevented.
- the relationship between viscosity and impregnation is also affected by the composition of the reinforcing fiber base material. If the initial viscosity at 100° C. or 120° C. exceeds 300 mPa ⁇ s, impregnation of the reinforcing fiber base material with the epoxy resin composition becomes difficult, which is undesirable. In this case, voids and the like are likely to be formed in the resulting fiber-reinforced composite material, causing deterioration in physical properties.
- the relationship between the viscosity and the impregnating property depends on the structure of the reinforcing fiber base material, and even if the viscosity is outside the above range, the impregnation of the reinforcing fiber base material may be good.
- the pot life of the epoxy resin composition varies depending on the molding conditions of the composite material.
- the pot life is preferably 20 minutes or more, more preferably 40 minutes or more, more preferably 60 minutes or more, until the viscosity exceeds 50 mPa s when held at 100 ° C. More preferably 90 minutes or longer, more preferably 100 minutes or longer, still more preferably 120 minutes or longer, still more preferably 180 minutes or longer, particularly preferably 300 minutes or longer.
- the pot life is preferably 20 minutes or more, more preferably 40 minutes or more, and still more preferably 60 minutes or more, until the viscosity exceeds 50 mPa ⁇ s when held at 120°C. When the pot life is 20 minutes or more, it can be stably used in the impregnation step.
- the epoxy resin composition of the present invention can be produced by mixing an epoxy resin base liquid and a curing agent liquid.
- the curing agent liquid contains curing agent A, curing agent B and curing agent C described above.
- the epoxy resin base liquid contains the epoxy resin D and the epoxy resin E described above.
- the resin particles F can be contained in the curing agent liquid and/or the epoxy resin base liquid.
- the epoxy resin composition may be in a one-liquid state in which each component is uniformly mixed, and some components are solid, other components are liquid, and the solid component is a liquid component. It may also be in a state of slurry dispersed in the liquid.
- the epoxy resin composition of the present invention can be produced using a conventionally known method.
- the temperature at which each component is mixed that is, the mixing temperature is, for example, 40 to 180°C, preferably 50 to 160°C, more preferably 50 to 120°C. If the temperature exceeds 180° C., the curing reaction proceeds immediately, and the impregnation of the reinforcing fiber substrate may deteriorate, or the physical properties of the cured product may deteriorate. If the temperature is less than 40°C, the viscosity of the epoxy resin base agent is high, and mixing may be substantially difficult.
- a mechanical device used for mixing each component conventionally known devices can be used, and examples include a roll mill, a planetary mixer, a kneader, an extruder, a Banbury mixer, a mixing vessel equipped with a stirring blade, and a horizontal mixing tank. be able to.
- Each component can be mixed in the air or under an inert gas atmosphere.
- an atmosphere with controlled temperature and humidity is preferred.
- a temperature controlled at a constant temperature of 30° C. or less and a relative humidity of 50% RH or less are preferable.
- Epoxy resin base liquid contains an epoxy resin, preferably the epoxy resin is epoxy resin D and epoxy resin E.
- the epoxy resin main agent preferably further contains resin particles F.
- the epoxy resin main agent may contain other optional components in addition to these.
- the content of the epoxy resin in the epoxy resin main agent liquid is preferably 30 to 100% by mass, more preferably 50 to 100% by mass, based on the mass of the entire epoxy resin main agent.
- the epoxy resin base liquid can be preferably produced by mixing epoxy resin D and epoxy resin E, and if necessary, resin particles F and other optional components. The order of these mixtures does not matter.
- the epoxy resin main component liquid may be in the state of one liquid in which each component is uniformly mixed, some components are solid, other components are liquid, and the solid component is a liquid component. It may also be in a state of slurry dispersed in the liquid.
- the mixing temperature is, for example, 40 to 200°C, preferably 50 to 100°C, more preferably 50 to 90°C. If the temperature exceeds 200°C, the self-polymerization reaction of the epoxy resin partially progresses, resulting in a decrease in the impregnation property of the reinforcing fiber base material, and the physical properties of the cured product produced using the resulting epoxy resin base liquid are decreased. may do so. If the temperature is less than 40°C, the viscosity of the epoxy resin base agent is high, and mixing may be substantially difficult.
- a conventionally known one can be used as a mixing machine. Specific examples include roll mills, planetary mixers, kneaders, extruders, Banbury mixers, mixing vessels equipped with stirring blades, and horizontal mixing tanks.
- Each component can be mixed in the air or under an inert gas atmosphere.
- an atmosphere with controlled temperature and humidity is preferred.
- Curing agent A, curing agent B and curing agent C are mixed to form a curing agent liquid. At this time, resin particles F and other optional components may be added as necessary.
- the curing agent liquid is a liquid composition containing curing agent A, curing agent B and curing agent C.
- This curing agent liquid may be in a one-liquid state in which each component is uniformly mixed, and some components are solid, other components are liquid, and the solid component is a liquid component. It may also be in a state of slurry dispersed in the liquid.
- the curing agent liquid can be produced by mixing curing agent A, curing agent B and curing agent C. At this time, a conventionally known method can be used.
- the temperature at which each component is mixed is, for example, 50 to 200°C, preferably 50 to 150°C, more preferably 80 to 120°C. If the temperature exceeds 200°C, the added components may be thermally decomposed. On the other hand, when the temperature is less than 50° C., the solid curing agents A and C do not melt and are difficult to melt into the curing agent B, making it difficult to obtain a liquid curing agent liquid.
- the production of the curing agent liquid can be carried out under the same conditions using the same equipment as the production equipment for the epoxy resin main component liquid described above.
- a resin cured product can be obtained by curing the epoxy resin composition of the present invention.
- the obtained resin cured product can have the following preferable properties.
- the degree of cure of the cured resin is preferably 70% or more, more preferably 80% or more, and particularly preferably 90% or more. Within this range, a fiber-reinforced composite material can be produced with high productivity.
- This degree of curing is the degree of curing of the resin cured product after heating the epoxy resin composition at 180° C. for 40 minutes, which is evaluated by dielectric curing degree measurement.
- the glass transition temperature (dry-Tg) of the cured resin in a dry state is preferably 140° C. or higher, more preferably 170° C. or higher, and particularly preferably 180° C. or higher, from the viewpoint of heat resistance of the resulting fiber-reinforced composite material. is.
- the glass transition temperature (wet-Tg) of the cured resin at saturated water absorption is preferably 120°C or higher, more preferably 150 to 200°C, from the viewpoint of heat resistance of the resulting fiber-reinforced composite material.
- the room temperature dry flexural modulus (RTD-FM) of the cured resin measured by the JIS K7171 method is preferably 3.0 GPa or more, more preferably 3.3 to 10.0 GPa, and still more preferably 3.5 to 9.0 GPa. is.
- the elastic modulus is 3.0 GPa or more
- the fiber-reinforced composite material obtained using the epoxy resin composition of the present invention has excellent mechanical properties.
- the elastic modulus is less than 3.0 GPa, the properties of the fiber-reinforced composite material obtained using the epoxy resin composition tend to deteriorate, which is undesirable.
- the deformation mode I critical stress intensity factor KIc of the cured resin measured by ASTM D5045 is preferably 0.7 MPa ⁇ m 1/2 or more, more preferably 0.8 MPa ⁇ m 1/2 or more, further preferably 0.8 MPa ⁇ m 1/2 or more. 85 MPa ⁇ m 1/2 or more, particularly preferably 0.9 MPa ⁇ m 1/2 or more. If KIc is less than 0.7 MPa ⁇ m 1/2 , the properties of the fiber-reinforced composite material obtained using the epoxy resin composition tend to deteriorate, which is undesirable. Incidentally, the higher the KIc, the better, and the upper limit is, for example, 3.0 MPa ⁇ m 1/2 .
- a fiber-reinforced composite material can be obtained by combining and curing a fiber-reinforced base material and the epoxy resin composition of the present invention.
- fibers for the reinforcing fiber base include carbon fiber, glass fiber, aramid fiber, silicon carbide fiber, polyester fiber, ceramic fiber, alumina fiber, boron fiber, metal fiber, mineral fiber, rock fiber, and slag fiber.
- carbon fiber is more preferable because it has good specific strength and specific modulus, and provides a lightweight and high-strength fiber-reinforced composite material.
- PAN polyacrylonitrile
- PAN-based carbon fibers When PAN-based carbon fibers are used as reinforcing fibers, their tensile modulus is preferably 100 to 600 GPa, more preferably 200 to 500 GPa, and even more preferably 230 to 450 GPa.
- PAN-based carbon fibers When PAN-based carbon fibers are used as reinforcing fibers, their tensile strength is preferably 2,000 to 10,000 MPa, more preferably 3,000 to 8,000 MPa.
- the diameter of the carbon fibers is preferably 4-20 ⁇ m, more preferably 5-10 ⁇ m. By using carbon fibers with this diameter, the mechanical properties of the resulting fiber-reinforced composite material can be improved, which is preferable.
- the reinforcing fibers are preferably treated with a sizing agent.
- the amount of the sizing agent attached is preferably 0.01 to 10% by mass, more preferably 0.05 to 3.0% by mass, more preferably 0% by mass, based on the mass of the reinforcing fibers to which the sizing agent is attached. .1 to 2.0% by mass.
- the adhesion amount of the sizing agent is within this range, since both the adhesiveness between the reinforcing fibers and the matrix resin and the interlayer toughness of the obtained composite material can be achieved.
- the adhesion between the reinforcing fibers and the matrix resin tends to be stronger when the sizing agent is applied in a larger amount. The smaller the adhesion amount, the better the interlaminar toughness of the resulting composite material.
- a reinforcing fiber sheet is preferably used as the fiber-reinforced base material. This is formed by forming reinforcing fibers into a sheet.
- the reinforcing fiber sheet include unidirectional aligned sheets, bidirectional woven fabrics such as plain weaves and twill weaves, multiaxial woven fabrics, nonwoven fabrics, mats, knits, braids, and paper made from reinforced fibers.
- a unidirectional aligned sheet, bidirectional woven fabric, or multiaxial woven fabric is preferably used.
- the unidirectionally aligned sheet is a sheet in which a large number of reinforcing fibers are aligned in one direction.
- the bidirectional woven fabric or multiaxial woven fabric base material may be obtained by laminating and stitching a plurality of unidirectional aligning sheets.
- it may be a woven fabric obtained by arranging a thermoplastic resin fiber nonwoven fabric on one side of the unidirectional aligned sheet and then laminating it.
- thermoplastic resin fibers of the thermoplastic resin fiber nonwoven fabric examples include polyester resin fibers, polyamide resin fibers, polyethersulfone resin fibers, polysulfone resin fibers, polyetherimide resin fibers, and polycarbonate resin fibers. Nonwoven fabrics of these mixtures may also be used.
- the basis weight and the number of layers of the unidirectional alignment sheet can be appropriately set according to the use of the fiber-reinforced composite material.
- the basis weight of the unidirectional aligned sheet is preferably 100 to 300 g/m 2 from the viewpoint of the balance between the shapeability of the fiber-reinforced base material, the molding efficiency of the fiber-reinforced composite material, and the mechanical properties of the obtained fiber-reinforced composite material. More preferably 150 to 250 g/m 2 .
- the thickness of one layer of the unidirectionally aligned sheet of the reinforcing fiber substrate is preferably 0.01 to 3 mm, more preferably 0.05 to 1.5 mm.
- a fiber-reinforced composite material comprising a cured resin obtained by curing the epoxy resin composition of the present invention and a fiber-reinforced base material.
- the fiber-reinforced composite material provided by the present invention has a post-impact compressive strength CAI (impact energy 30.5 J) measured by ASTM D7136 of preferably 240 MPa or more, more preferably 250 to 400 MPa, further preferably 270 to 380 MPa. is.
- CAI impact energy 30.5 J
- the fiber-reinforced composite material provided by the present invention preferably has a room temperature dry open-hole compressive strength (RTD-OHC) measured by SACMA SRM3 of preferably 260 MPa or more, more preferably 280 to 450 MPa, further preferably 315 to 400 MPa. be.
- RTD-OHC room temperature dry open-hole compressive strength
- the fiber-reinforced composite material provided by the present invention preferably has a perforated compressive strength after temperature rise water absorption (HTW-OHC) measured by SACMA SRM3 of preferably 200 MPa or more, more preferably 220 to 400 MPa, further preferably 240 to 240 MPa. 350 MPa.
- HMW-OHC temperature rise water absorption
- the present invention is also a fiber-reinforced composite material comprising the cured epoxy resin and a fiber-reinforced base material.
- This fiber-reinforced composite material can be obtained by compounding and curing a fiber-reinforced base material and the epoxy resin composition of the present invention.
- a carbon fiber reinforced substrate is preferably used as the fiber reinforced substrate. Curing can be done by heating.
- the fiber-reinforced base material and the epoxy resin composition may be compounded in advance before molding, or may be compounded during molding.
- a resin transfer molding method RTM method
- a hand layup method a filament winding method, a pultrusion method, an autoclave molding method, and a press molding method can be used.
- the epoxy resin composition of the present invention is particularly suitable for the RTM method.
- the RTM method is a method for obtaining a fiber-reinforced composite material by impregnating a fiber-reinforced base material placed in a mold with a liquid epoxy resin composition and curing the composition.
- the RTM method is a preferable molding method from the viewpoint of efficiently obtaining a complex-shaped fiber-reinforced composite material.
- the present invention further provides a method for producing a fiber-reinforced composite material, which includes the step of impregnating a fiber-reinforced base material placed in a mold with the epoxy resin composition of the present invention, followed by heat curing.
- a closed mold made of a rigid material may be used, or an open mold made of a rigid material and a flexible film (bag) may be used.
- the fiber reinforced substrate can be placed between an open mold of rigid material and the flexible film.
- rigid materials for example, metals such as steel and aluminum, fiber reinforced plastics (FRP), wood, and gypsum can be used.
- FRP fiber reinforced plastics
- flexible film materials include polyamide, polyimide, polyester, fluororesin, and silicone resin.
- the mold When using a closed mold made of a rigid material in the RTM method, the mold is clamped under pressure, and the epoxy resin composition is injected under pressure. At this time, a suction port may be provided separately from the injection port and connected to a vacuum pump for suction. When suction is performed, the epoxy resin composition can be injected only at atmospheric pressure without using special pressurizing means. This method is preferable because a large member can be manufactured by providing a plurality of suction ports.
- suction may be performed to inject the epoxy resin only at atmospheric pressure without using special pressurization means. It is effective to use a resin diffusion medium to achieve good impregnation by injection at atmospheric pressure only. Additionally, a gel coat may be applied to the surface of the rigid material prior to installation of the fiber reinforced substrate.
- a fiber-reinforced base material is impregnated with an epoxy resin composition, and then heat-cured.
- the mold temperature during heat curing is usually selected to be higher than the mold temperature during injection of the epoxy resin composition.
- the mold temperature during heat curing is, for example, 80 to 200.degree.
- the heat curing time is, for example, 1 minute to 20 hours.
- the mold is demolded and the fiber reinforced composite material is taken out.
- the resulting fiber-reinforced composite material may then be post-cured by heating at a higher temperature.
- the post-curing temperature is, for example, 150 to 200° C.
- the time is, for example, 1 minute to 4 hours.
- the impregnation pressure when the epoxy resin composition is impregnated into the fiber-reinforced base material by the RTM method may be appropriately determined in consideration of the viscosity and resin flow of the resin composition.
- a specific impregnation pressure is preferably 0.001 to 10 MPa, more preferably 0.01 to 1 MPa.
- the epoxy resin composition with which the fiber-reinforced base material is impregnated is produced by mixing the curing agent liquid of the thermosetting resin curing agent composition and the epoxy resin base liquid. This mixing is preferably done immediately before the epoxy resin composition is impregnated into the fiber reinforced substrate.
- thermosetting resin curing agent composition of the present invention and the epoxy resin main agent are mixed immediately before impregnation into the fiber-reinforced base material placed in the mold to form the epoxy resin composition. It is preferable to further include the step of obtaining.
- the curing agent composition for thermosetting resins of the present invention has high reactivity with epoxy resins.
- the viscosity of the epoxy resin composition increases.
- the interior of the reinforcing fiber base material can be impregnated with a sufficient amount of the epoxy resin composition. Therefore, the resulting fiber-reinforced composite material does not contain defects such as voids, and is excellent in compression performance and damage tolerance.
- a method for producing a fiber-reinforced composite material comprising a step of impregnating a fiber-reinforced base material with the epoxy resin composition of the present invention to obtain an impregnated base material, and a step of curing the impregnated base material obtained in the step.
- the step of impregnating a fiber-reinforced base material with the epoxy resin composition of the present invention to form an impregnated base material includes impregnating the fiber-reinforced base material placed in a mold with the epoxy resin composition of the present invention. It is preferable that the step of curing the impregnated base material obtained in the step is a step of heat-curing the impregnated base material.
- a step of preparing the epoxy resin composition of the present invention is included immediately before the step of impregnating the fiber-reinforced base material with the epoxy resin composition of the present invention to form an impregnated base material.
- Resin particles F particle rubber component
- MX-416 manufactured by Kaneka Corporation, MX-416 (product name)
- This is a masterbatch in which a particulate butadiene rubber component is dispersed in a glycidylamine-type tetrafunctional epoxy resin to a concentration of 25% by mass.
- the glycidylamine type tetrafunctional epoxy resin in MX-416 corresponds to epoxy resin D.
- Carbon fiber strand/carbon fiber 1 Tenax (registered trademark) IMS65 E23 830tex (carbon fiber strand, tensile strength 5.8 GPa, tensile modulus 290 GPa, sizing agent adhesion amount 1.2% by mass, manufactured by Teijin Limited )
- Thermoplastic resin nonwoven fabric/nonwoven fabric 1 nonwoven fabric using polyamide 12 resin and having a fiber basis weight of 5 g/m 2 produced by a spunbond method (6)
- Carbon fiber multilayer fabric/carbon fiber multiaxial fabric 1 unidirectional A sheet of 190 g/m 2 per layer of carbon fiber 1 was arranged, and a nonwoven fabric 1 was placed on one side of this sheet-like carbon fiber, (+45/V/90/V/-45/V/0 /V) by laminating and stitching four sheets (760 g/m 2 of carbon fiber total basis weight of
- ⁇ Carbon fiber multiaxial fabric 2 The carbon fibers 1 aligned in one direction are made into a sheet of 190 g / m 2 per layer, and the nonwoven fabric 1 is arranged on one side of the sheet-shaped carbon fibers, (-45 / V / 90/V/+45/V/0/V) and stitched (carbon fiber total basis weight of woven fabric base material: 760 g/m 2 ).
- a resin test piece was prepared with dimensions of 50 mm x 6 mm x 2 mm.
- a pressure cooker (HASTEST PC-422R8, manufactured by Espec Co., Ltd.) was used to subject the prepared resin test piece to water absorption treatment under conditions of 121°C for 24 hours.
- a dynamic viscoelasticity measuring device Rheogel-E400 manufactured by UBM under the conditions of a measurement frequency of 1 Hz, a temperature increase rate of 5 ° C./min, and a strain of 0.0167%, the distance between chucks is 30 mm, and the rubber elastic region is measured from 50 ° C.
- the storage elastic modulus E' of the resin test piece subjected to water absorption treatment was measured.
- Log E' was plotted against temperature, and the temperature obtained from the intersection of the approximate straight line of the flat region of log E' and the approximate straight line of the transition region of E' was recorded as the glass transition temperature (wet-Tg).
- RTD-FM Room temperature dry flexural modulus
- a peel cloth Release Ply C which is a base material with a releasability function
- Resin Flow 90HT manufactured by AIRTECH
- the hose for forming the resin inlet and resin outlet was placed, the whole was covered with nylon bag film, sealed with sealant tape, and the inside was evacuated.
- the aluminum plate was heated to 120°C, and the pressure inside the bag was reduced to 5 torr or less. Injected into the vacuum system.
- CFRP carbon fiber reinforced composite material
- the strength test of the specimen was performed by attaching strain gauges on each side of the specimen at a position of 25.4 mm from the top and 25.4 mm from the side. After attaching the gauge, the crosshead speed of the testing machine (Autograph manufactured by Shimadzu Corporation) was set to 1.27 mm/min, and a load was applied until the specimen fractured.
- the crosshead speed of the testing machine Autograph manufactured by Shimadzu Corporation
- (1-2) Liquid Retention Properties of Curing Agent Liquid The curing agent liquid prepared in (1-1) above was stored at 25° C. for 1 week, and precipitation of solid components was visually observed. A sample with no precipitation was rated as “ ⁇ ”, and a sample with deposition was rated as “x”.
- a resin test piece was prepared with dimensions of 50 mm x 6 mm x 2 mm.
- a pressure cooker (HASTEST PC-422R8, manufactured by Espec Co., Ltd.) was used to subject the prepared resin test piece to water absorption treatment under conditions of 121°C for 24 hours.
- a dynamic viscoelasticity measuring device Rheogel-E400 manufactured by UBM under the conditions of a measurement frequency of 1 Hz, a temperature increase rate of 5 ° C./min, and a strain of 0.0167%, the distance between chucks is 30 mm, and the rubber elastic region is measured from 50 ° C.
- the storage elastic modulus E' of the resin test piece subjected to water absorption treatment was measured.
- Log E' was plotted against temperature, and the temperature obtained from the intersection of the approximate straight line of the flat region of log E' and the approximate straight line of the transition region of E' was recorded as the glass transition temperature (wet-Tg).
- RTD-FM Room temperature dry bark elastic modulus
- a peel cloth Release Ply C which is a base material with a releasability function
- Resin Flow 90HT manufactured by AIRTECH
- the hose for forming the resin inlet and resin outlet was placed, the whole was covered with nylon bag film, sealed with sealant tape, and the inside was evacuated.
- the aluminum plate was heated to 120° C. and the pressure inside the bag was reduced to 5 torr or less, and then the epoxy resin composition prepared in (1-1) above was heated to 100° C. injected into.
- CFRP carbon fiber reinforced composite material
- the strength test of the specimen was performed by attaching strain gauges on each side of the specimen at a position of 25.4 mm from the top and 25.4 mm from the side. After attaching the gauge, the crosshead speed of the testing machine (Autograph manufactured by Shimadzu Corporation) was set to 1.27 mm/min, and a load was applied until the specimen fractured.
- the crosshead speed of the testing machine Autograph manufactured by Shimadzu Corporation
- the test was conducted at an environmental temperature of 25°C in accordance with SACMA SRM3, and the perforated compressive strength was calculated from the maximum point load.
- Example 1 (Preparation of Curing Agent Composition) Curing agents were weighed in proportions shown in Table 1, and mixed using a stirrer at a temperature of 90° C. for 60 minutes to prepare a curing agent composition.
- Table 1 shows the properties of the curing agent composition, the resin composition and the cured resin.
- the hardener composition remained liquid for over a week.
- the resin composition exhibited a low viscosity of 27 mPa ⁇ s at 100° C. and a pot life of 108 minutes.
- the flexural modulus was 3.6 GPa and K1c was 0.95 MPa ⁇ m 1/2 , showing high mechanical properties.
- Examples 2 to 5 The composition was changed as shown in Table 1, and the same procedure as in Example 1 was carried out.
- Table 1 shows the properties of the curing agent composition, the resin composition and the cured resin.
- the hardener composition remained liquid for over a week.
- the resin composition exhibited a low viscosity of 28 mPa ⁇ s or less at 100° C. and a pot life of 110 minutes or longer.
- the bending elastic modulus was 3.6 GPa or more, and K1c was 0.88 MPa ⁇ m 1/2 or more, showing high mechanical properties.
- Example 6 to 9 The composition was changed as shown in Table 1, and the same procedure as in Example 1 was carried out.
- Table 1 shows the properties of the cured resin.
- Example 1 The composition was changed as shown in Table 1, and the same procedure as in Example 1 was carried out. Properties of the hardener composition are shown in Table 1. Pot life was shortened to 72 minutes. Also, K1c was 0.84 MPa ⁇ m 1/2 , indicating insufficient toughness.
- Example 2 The composition was changed as shown in Table 1, and the same procedure as in Example 1 was carried out. Properties of the hardener composition are shown in Table 1. A solid precipitated within one week, and it was difficult to maintain the liquid state.
- Table 2 shows the properties of the curing agent composition, the resin composition and the resin cured product.
- the hardener composition remained liquid for over a week.
- the resin composition exhibited a low viscosity of 30 mPa ⁇ s at 100° C. and a pot life of 58 minutes.
- the flexural modulus was 3.5 GPa and K1c was 0.96 MPa ⁇ m 1/2 , showing high mechanical properties.
- Example 12 to 18 The composition was changed as shown in Table 2 and the same procedure as in Example 11 was carried out.
- Table 1 shows the properties of the curing agent composition, the resin composition and the cured resin.
- the hardener composition remained liquid for over a week.
- the resin composition exhibited a low viscosity of 39 mPa ⁇ s or less at 100° C. and a pot life of 22 minutes or more.
- the flexural modulus was 3.4 GPa or more, and K1c was 0.91 MPa ⁇ m 1/2 or more, showing high mechanical properties.
- Example 11-12 The composition was changed as shown in Table 3 and the same procedure as in Example 11 was carried out. Table 3 shows the properties of the cured resin. In both cases, the pot life was as short as less than 18 minutes, and K1c was less than 0.89 MPa ⁇ m 1/2 , indicating insufficient toughness.
- Example 14 The composition was changed as shown in Table 3 and the same procedure as in Example 11 was carried out. Table 3 shows the properties of the cured resin. All of them had K1c of less than 0.88 MPa ⁇ m 1/2 and had insufficient toughness.
- Example 17 The composition was changed as shown in Table 3 and the same procedure as in Example 11 was carried out. Properties of the hardener composition are shown in Table 3. A solid precipitated within one week, and it was difficult to maintain the liquid state.
- Example 21 (Preparation of Curing Agent Composition) Curing agents were weighed in proportions shown in Table 4, and mixed using a stirrer at a temperature of 90° C. for 60 minutes to prepare a curing agent composition.
- Epoxy resin and resin particles F were weighed in proportions shown in Table 4 and mixed at 80° C. for 30 minutes using a stirrer to prepare an epoxy resin base liquid.
- Curing agents were weighed in proportions shown in Table 4 and mixed at 80° C. for 30 minutes using a stirrer to prepare a curing agent liquid.
- the separately prepared epoxy resin base liquid and curing agent liquid were mixed at 80° C. for 30 minutes using a stirrer to prepare an epoxy resin composition.
- Table 4 shows the properties of the resulting epoxy resin composition.
- the glycidyl groups of the epoxy resin and the amino groups of the curing agent are equivalent.
- Table 4 shows the properties of the resulting curing agent composition and epoxy resin composition.
- the hardener composition remained liquid for over a week.
- the epoxy resin composition exhibited a low viscosity of 30 mPa ⁇ s at 120° C. and a pot life of 68 minutes.
- Table 4 shows the properties of the obtained cured resin.
- the cured resin had a wet-Tg of 162° C., a flexural modulus of 3.7 GPa, and a K1c of 0.87 MPa ⁇ m 1/2 , showing high mechanical properties.
- the carbon fiber multiaxial fabric 1 and the carbon fiber multiaxial fabric 2 were cut to 300 x 300 mm, and three sheets of the carbon fiber multiaxial fabric 1 were placed on a release-treated aluminum plate of 500 x 500 mm. Three sheets of the multiaxial fabric 2, totaling six sheets, were stacked to form a laminate.
- a peel cloth Release Ply C which is a base material with a releasability function
- Resin Flow 90HT manufactured by AIRTECH
- the hose for forming the resin inlet and resin outlet was placed, the whole was covered with nylon bag film, sealed with sealant tape, and the inside was evacuated.
- the aluminum plate was heated to 120° C., the pressure inside the bag was reduced to 5 torr or less, and then the epoxy resin composition prepared above was heated to 100° C. and injected into the vacuum system through the resin inlet.
- CFRP carbon fiber reinforced composite material
- Table 4 shows the properties of the obtained CFRP.
- CAI of CFRP was 293 MPa and RTD-OHC was 338 MPa, showing high mechanical properties.
- Example 22 to 29 The composition was changed as shown in Table 4 and the same procedure as in Example 21 was carried out.
- Table 4 shows the properties of the curing agent composition, epoxy resin composition, cured resin and CFRP.
- All hardener compositions remained liquid for more than one week. All of the epoxy resin compositions exhibited a low viscosity of 50 mPa ⁇ s or less at 120°C and a pot life of 20 minutes or longer. All of the cured resins had a flexural modulus of 3.5 GPa or more and a K1c of 0.85 MPa ⁇ m 1/2 or more, showing high mechanical properties. All of the CFRPs had a CAI of 281 MPa or more and an RTD-OHC of 318 MPa or more, showing high mechanical properties.
- the curing agent composition for thermosetting resins and the epoxy resin composition of the present invention can be used for producing fiber-reinforced composite materials.
- the fiber-reinforced composite material obtained by the present invention can be used, for example, as members of airplanes and automobiles.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Epoxy Resins (AREA)
Abstract
Description
本発明の熱硬化性樹脂用硬化剤組成物は、後述する硬化剤A、硬化剤Bおよび硬化剤Cを含有してなる熱硬化性樹脂用硬化剤組成物である。この熱硬化性樹脂用硬化剤組成物は、80~200℃の温度に加熱することで均一な液体となる。
硬化剤Aは、アミノ基に対する2つのオルト位にそれぞれ置換基を有する芳香族ポリアミンであり、該置換基はアルキル基、芳香族基およびハロゲン基から選択される。また、硬化剤Aは25℃で固体である。この硬化剤Aを含有することで、エポキシ樹脂との組成物として硬化させたときに、優れた耐熱性や弾性率、破壊靭性などの力学特性を備えるエポキシ樹脂硬化物を得ることができる。
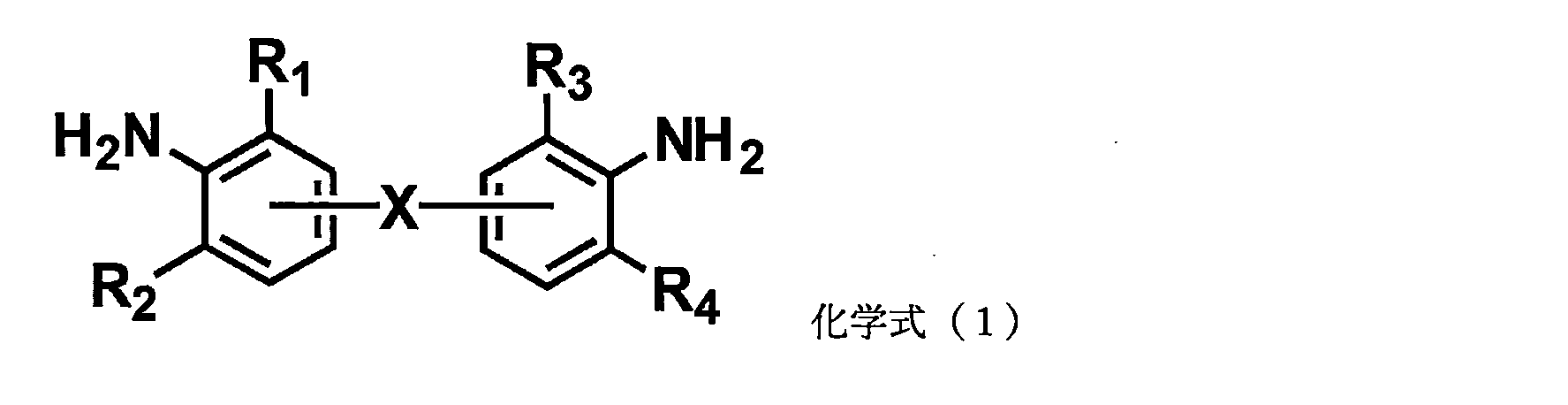
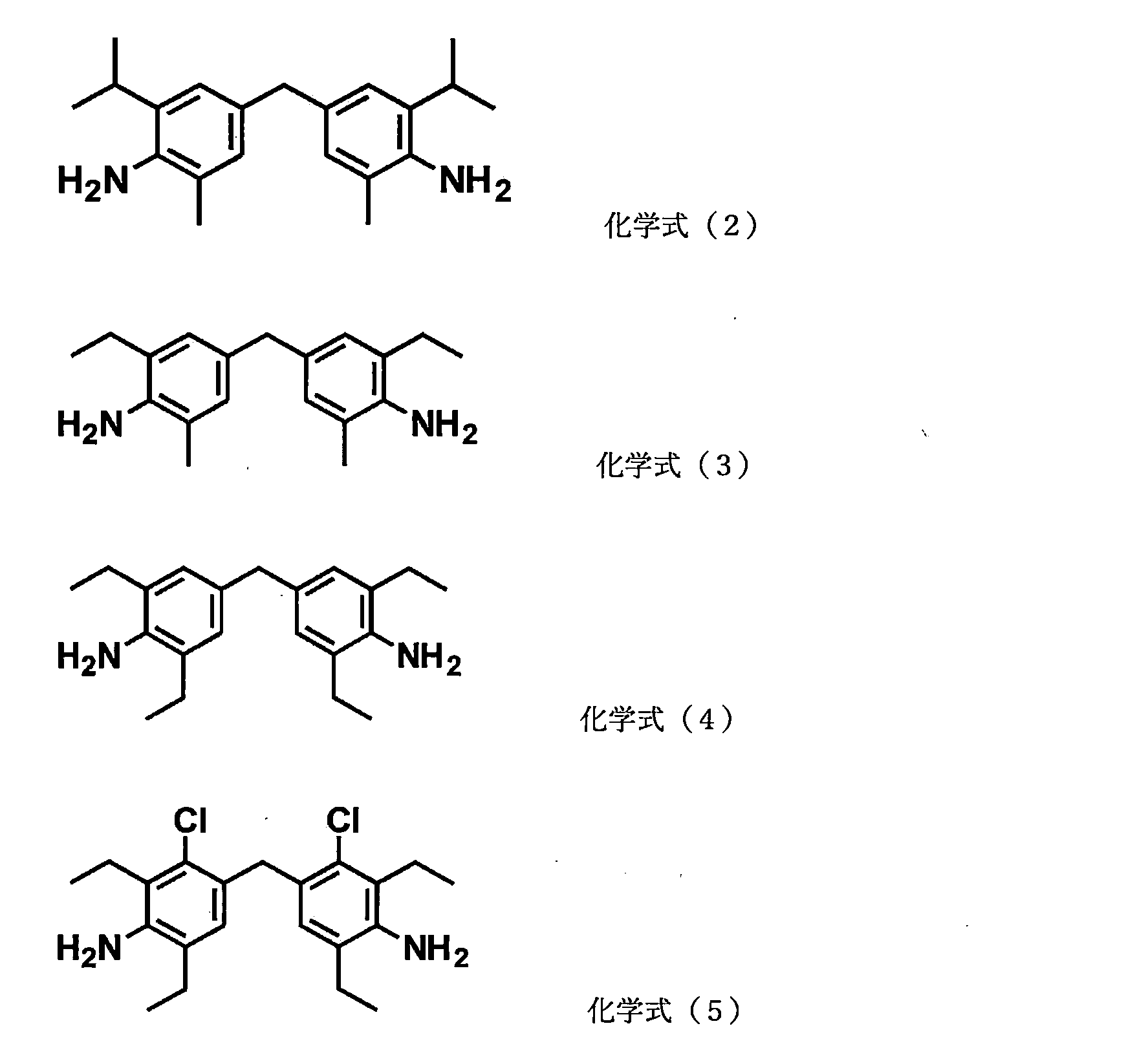
硬化剤Bは、25℃で液体である芳香族ポリアミンである。この芳香族ポリアミンを含有することで、室温で液体の状態を保持することができる熱硬化性樹脂用硬化剤組成物を得ることができる。



硬化剤Cは、芳香族置換基を有するモノアミンである。このモノアミンにおいて、芳香族置換基は、窒素原子に直に結合していてもよく、アルキル基を介して窒素原子に結合していてもよい。
本発明の好ましい第一の態様において、硬化剤Cの芳香族置換基を有するモノアミンは、一分子中に芳香環に直接結合するアミノ基を1つ有する、芳香族化合物である。
本発明の好ましい第二の態様において、芳香族置換基を有するモノアミンは、ベンジルアミン誘導体である。
本発明の熱硬化性樹脂用硬化剤組成物において、硬化剤Aと硬化剤Bの質量比率は、好ましくは1:99~99:1、さらに好ましくは20:80~80:20、特に好ましくは40:60~70:30である。硬化剤Aの割合がこれより少ないと、得られる樹脂硬化物の耐熱性や弾性率、破壊靭性などの力学特性が不十分となりやすく好ましくない。他方、硬化剤Aの割合がこれより多いと、得られる熱硬化性樹脂用硬化剤組成物が室温で液状を保持することが困難となり好ましくない。
本発明の熱硬化性樹脂用硬化剤組成物において、熱硬化性樹脂用硬化剤組成物の全質量を基準として、硬化剤A、硬化剤Bおよび硬化剤Cの合計が、好ましくは70~100質量%、さらに好ましくは80~100質量%を占める。70質量%未満であると硬化物の耐熱性が不十分となる可能性があり好ましくない。
本発明の熱硬化性樹脂用硬化剤組成物は、上記の条件を満足する範囲であれば、さらに他の硬化剤やその他の成分を含有してもよい。例えば、導電性粒子、難燃剤、無機系充填剤、内部離型剤を含有してもよい。
本発明の熱硬化性樹脂用硬化剤組成物は、硬化剤Aと硬化剤Bと硬化剤Cと、必要に応じてその他の成分と、を混合することにより、製造することができる。混合の順序は問わない。
本発明のエポキシ樹脂組成物は、上述の本発明の熱硬化性樹脂用硬化剤組成物とエポキシ樹脂主剤とを含んで成る。
エポキシ樹脂主剤は、さらにその他の任意成分を含んでいてもよい。エポキシ樹脂主剤におけるエポキシ樹脂の含有量は、全エポキシ樹脂主剤の質量を基準として、好ましくは30~100質量%、さらに好ましくは50~100質量%である。
エポキシ樹脂Dは、グリシジル基を4つ以上含むモノマーから構成されるエポキシ樹脂である。エポキシ樹脂Dは、1種類のモノマーから構成されるホモポリマーであってもよく、2種類以上のモノマーから構成されるコポリマーであってもよく、ホモポリマーおよび/またはコポリマーの混合物であってもよい。

R1~R4が、脂肪族炭化水素基または脂環式炭化水素基である場合、その炭素数は1~4であることが好ましい。
エポキシ樹脂Eは、グリシジル基を2つまたは3つ含むモノマーから構成されるエポキシ樹脂である。このエポキシ樹脂Eを含むことで、エポキシ樹脂組成物の粘度を低下させて強化繊維基材への樹脂含浸性を向上させることができ、可使時間を長くすることができ、RTM成形法に使用する金型の設計自由度を高めることができる。
本発明のエポキシ樹脂組成物は、好ましくは樹脂粒子Fを含有する。この場合、樹脂粒子Fは、エポキシ樹脂組成物に溶解せずに分散して存在し、かつエポキシ樹脂組成物が硬化した後の樹脂硬化物においても分散した状態で樹脂硬化物中に存在する。樹脂硬化物を海成分としたときに、樹脂粒子Fは島成分として樹脂硬化物中に存在する。
本発明のエポキシ樹脂組成物における、エポキシ樹脂の総量(エポキシ樹脂主剤)に対する、エポキシ樹脂Dが占める割合は、好ましくは50~90質量%、特に好ましくは60~80質量%である。エポキシ樹脂Dの割合が50質量%以上であることで、得られる樹脂硬化物の耐熱性および弾性率をより向上させることができる。その結果、得られる繊維強化複合材料の各種力学特性も向上する。
エポキシ樹脂組成物に含まれる熱硬化性樹脂用硬化剤組成物の総量は、エポキシ樹脂組成物中に配合されている全てのエポキシ樹脂を硬化させるのに適した量であり、用いられるエポキシ樹脂や熱硬化性樹脂用硬化剤の種類に応じて適宜調節される。
本発明のエポキシ樹脂組成物には、硬化剤A、硬化剤Bおよび硬化剤C以外の硬化剤が含まれていてもよく、エポキシ樹脂Dおよびエポキシ樹脂E以外のエポキシ樹脂や、エポキシ樹脂以外の熱硬化性樹脂が含まれていてもよく、樹脂粒子F以外の熱可塑性樹脂やその他の添加剤が含まれていてもよい。
本発明のエポキシ樹脂組成物、以下の性質を持つことができる。
本発明のエポキシ樹脂組成物は、エポキシ樹脂主剤液と硬化剤液とを混合することにより製造することができる。ここで、硬化剤液は、上述の硬化剤A、硬化剤Bおよび硬化剤Cを含有する。そして、エポキシ樹脂主剤液は、上述のエポキシ樹脂Dおよびエポキシ樹脂Eを含有する。
エポキシ樹脂主剤液は、エポキシ樹脂を含有し、好ましくは、このエポキシ樹脂はエポキシ樹脂Dおよびエポキシ樹脂Eである。エポキシ樹脂主剤は、好ましくは、さらに樹脂粒子Fを含有する。エポキシ樹脂主剤は、これらの他に、その他の任意成分を含んでいてもよい。
エポキシ樹脂主剤液は、好ましくは、エポキシ樹脂Dおよびエポキシ樹脂Eと、必要に応じて樹脂粒子Fとその他の任意成分とを混合することにより製造することができる。これらの混合の順序は問わない。
硬化剤A、硬化剤Bおよび硬化剤Cは、混合して、硬化剤液とする。このとき、必要に応じて樹脂粒子Fとその他の任意成分とを添加してもよい。
硬化剤液は、硬化剤A、硬化剤Bおよび硬化剤Cを混合することで製造することができる。このとき、従来公知の方法を用いることができる。各成分を混合するときの温度は、例えば50~200℃、好ましくは50~150℃、さらに好ましくは80~120℃である。200℃を超えると、添加する成分が熱分解してしまう場合がある。他方、50℃未満であると、固体である硬化剤Aおよび硬化剤Cが融解せず、硬化剤Bへ融解しにくくなるため、液状の硬化剤液を得ることが困難となる。
本発明のエポキシ樹脂組成物を硬化させることにより、樹脂硬化物を得ることができる。得られる樹脂硬化物は、以下の好ましい性質を備えることができる。
繊維強化基材と、本発明のエポキシ樹脂組成物とを複合化して硬化させることにより、繊維強化複合材料を得ることができる。
本発明はまた、上記のエポキシ樹脂硬化物および繊維強化基材を含む繊維強化複合材料である。この繊維強化複合材料は、繊維強化基材と、本発明のエポキシ樹脂組成物とを複合化して硬化させることにより得ることができる。繊維強化基材として、好ましくは炭素繊維強化基材を用いる。硬化は加熱により行うことができる。
1.エポキシ樹脂組成物の原料
(1)硬化剤
(1-1) 硬化剤A
・4,4’-ジアミノ-3,3’-ジイソプロピル-5,5’-ジメチルジフェニルメタン(ロンザ社製 Lonzacure M-MIPA(製品名)、以下「M-MIPA」と略記する、融点70℃、25℃で固体)
(1-2) 硬化剤B
・ジエチルトルエンジアミン(クミアイ化学社製 ハートキュア10(製品名)、以下「DETDA」と略記する、25℃で液体)
(1-3) 硬化剤C
・2,5-ジメチルアニリン(東京化成工業社製、以下「25DMA」と略記する、融点11℃、25℃で液体)
・4-メトキシベンジルアミン(東京化成工業社製、以下「4MxBA」と略記する、25℃で液体)
・3,4-ジメトキシベンジルアミン(東京化成工業社製、以下「34DMxBA」と略記する、25℃で液体)
・3-メトキシベンジルアミン(東京化成工業社製、以下「3MxBA」と略記する、25℃で液体)
・4-メチルベンジルアミン(東京化成工業社製、以下「4MBA」と略記する、融点13℃、25℃で液体)
(1-4) その他の硬化剤
・m-フェニレンジアミン(富士フイルム和光純薬社製、以下「MPD」と略記する、融点65℃、25℃で固体)
・m-トルイジン(東京化成工業社製、以下「mTOL」と略記する、25℃で液体)
・4-フェノキシアニリン(東京化成工業社製、以下「PxAN」と略記する、融点85℃、25℃で固体)
・3-イソプロピルアニリン(東京化成工業社製、以下「iPrAN」と略記する、25℃で液体)
・3-エチルアニリン(東京化成工業社製、以下「EtAN」と略記する、25℃で液体)
・m-キシリレンジアミン(cis-,trans-混合物)(東京化成工業社製、以下「mXDA」と略記する、25℃で液体)
(2) エポキシ樹脂
(2-1) エポキシ樹脂D
・テトラグリシジル-4,4’-ジアミノジフェニルメタン(ハンツマン社製 Araldite MY721(製品名)、以下「4,4’-TGDDM」と略記する)
・テトラグリシジル-3,4’-ジアミノジフェニルエーテル
これは後述の合成例1の方法で合成した。以下「3,4’-TGDDE」と略記する。
・N,N-ジグリシジル-o-トルイジン(日本化薬社製 GOT(製品名)、以下「GOT」と略記する)
・N,N-ジグリシジルアニリン(日本化薬社製 GAN(製品名)、以下「GAN」と略記する)
・トリグリシジル-p-アミノフェノール(ハンツマン社製 Araldite MY0510(製品名)、以下「TG-pAP」と略記する)
・1,6-ビス(グリシジルオキシ)ナフタレン(DIC社製 HP-4032SS(製品名)、以下「1,6-DON」と略記する)
・1,3,5-トリグリシジルイソシアヌレート(日産化学社製 TEPIC-S(製品名)、以下「TEPIC」と略記する)
・ビスフェノールA-ジグリシジルエーテル(三菱化学社製 jER825(製品名)、以下「DGEBA」と略記する)
・テトラグリシジル-3,4’-ジアミノジフェニルエーテル
これは下記の合成例1の方法で合成した。以下「3,4’-TGDDE」と略記する。
以下の方法で合成した。
温度計、滴下漏斗、冷却管および攪拌機を取り付けた四つ口フラスコに、エピクロロヒドリン1110.2g(12.0mol)を仕込み、窒素パージを行いながら温度を70℃まで上げて、これにエタノール1000gに溶解させた3,4’-ジアミノジフェニルエーテル200.2g(1.0mol)を4時間かけて滴下した。
(3)樹脂粒子F(粒子状ゴム成分)
・MX-416(株式会社カネカ製 MX-416(製品名) これは、グリシジルアミン型4官能エポキシ樹脂へ粒子状ブタジエンゴム成分を25質量%の濃度となる様に分散させたマスターバッチである。MX-416中のグリシジルアミン型4官能エポキシ樹脂はエポキシ樹脂Dに相当する。)
(4)炭素繊維ストランド
・炭素繊維1:テナックス(登録商標) IMS65 E23 830tex(炭素繊維ストランド、引張強度 5.8GPa、引張弾性率 290GPa、サイジング剤付着量 1.2質量%、帝人(株)製)
(5)熱可塑性樹脂不織布
・不織布1:ポリアミド12樹脂を使用し、スパンボンド法で作製した繊維目付が5g/m2の不織布
(6)炭素繊維多層織物
・炭素繊維多軸織物1:一方向に引き揃えた炭素繊維1を1層あたり190g/m2のシート状にして、このシート状炭素繊維の片面に不織布1を配置し、(+45/V/90/V/-45/V/0/V)の角度で4枚積層しステッチしたもの(織物基材の炭素繊維総目付760g/m2)。
・炭素繊維多軸織物2:一方向に引き揃えた炭素繊維1を1層あたり190g/m2のシート状にして、シート状炭素繊維の片面に不織布1を配置し、(-45/V/90/V/+45/V/0/V)の角度で4枚積層しステッチしたもの(織物基材の炭素繊維総目付760g/m2)。
2.評価方法(実施例1~9および11~18ならびに比較例1~2および11~17の評価方法)
(1) 硬化剤組成物の特性
(1-1) 硬化剤組成物の調製
表1に記載する割合で硬化剤を計量し、撹拌機を用いて90~110℃の適切な温度で60分間混合し、硬化剤組成物を調製した。
上記の(1-1)で調製した硬化剤組成物を、室温(25℃)で1週間保管し、目視で固体成分の析出を確認した。析出がないものを「○」、析出の認められたものを「×」とした。
(2) 樹脂組成物の特性
(2-1) エポキシ樹脂組成物の調製
上記(1-1)で調製した硬化剤組成物と、エポキシ樹脂、粒子状ゴム成分を表1、表2または表3に記載する割合で計量し、撹拌機を用いて80℃で60分間混合し、エポキシ樹脂組成物を調製した。なお、表1、表2または表3に記載の組成においては、エポキシ樹脂のエポキシ基と硬化剤の活性水素は当量となる。
粘度測定は、東機産業株式会社製B型粘度計TVB-15Mを用い、100℃の条件にて行った。測定開始直後の最小測定値を初期粘度とし、粘度が50mPa・sに到達した時間を可使時間とした。
(3) 樹脂硬化物の特性
(3-1) 樹脂硬化物の作成
上記(2-1)で調製したエポキシ樹脂組成物を真空中で脱泡した後、4mm厚のシリコン樹脂製スペーサーにより厚み4mmになるように設定したステンレス製モールド中に注入した。180℃の温度で30分間硬化させ、厚さ4mmの樹脂硬化物を得た。
SACMA 18R-94法に準じて、ガラス転移温度を測定した。
JIS K7171法に準じて、試験を実施した。その際の、樹脂試験片の寸法は80mm×10mm×4mm(厚みh)で準備した。支点間距離Lは、16×h(厚み)、試験速度2m/minで曲げ試験を行い、室温乾燥曲げ弾性率(RTD-FM)を測定した。
ASTM D5045法に準じて、試験を実施した。その際の、樹脂試験片の寸法は50mm×8mm(幅W)×4mmで準備した。クラック長aは、0.45≦a/W≦0.55となるように調整した。なお、クラック長aは破壊試験後の破断面を光学顕微鏡を用いて観察し、クラックの先端までの長さ、および試験片両表面におけるクラック長さの平均値を採用した。
(4)CFRPの特性
(4-1)CFRPの作成
炭素繊維多軸織物1および炭素繊維多軸織物2を300×300mmにカットし、500×500mmの離型処理したアルミ板の上に、炭素繊維多軸織物1を3枚、炭素繊維多軸織物2を3枚、合計6枚重ねて積層体とした。
上記の(4-1)で得られたCFRPを、幅101.6mm×長さ152.4mmの寸法に切断し、衝撃後圧縮強度(CAI)試験の試験片を得た。試験はASTM D7136に従い実施した。供試体(サンプル)は各試験片の寸法測定後、衝撃試験は落錘型衝撃試験機(インストロン社製 Dynatup)を用いて、30.5Jの衝撃エネルギーを与えた。衝撃後、供試体の損傷面積は、超音波探傷試験機(クラウトクレーマー社製 SDS3600、HIS3/HF)にて測定した。衝撃後、供試体の強度試験は、供試体の上から25.4mmでサイドから25.4mmの位置に、歪みゲージを左右各1本ずつ貼付し、同様に表裏に合計4本/体の歪みゲージを貼付けた後、試験機(島津製作所製オートグラフ)のクロスヘッド速度を1.27mm/minとし、供試体の破断まで荷重を負荷した。
上記の(4-1)で得られたCFRPを、幅38.1mm×長さ304.8mmの寸法に切断し、試験片中心に直径6.35mmの穴あけ加工を施し、室温乾燥有孔圧縮強度(RTD-OHC)試験の試験片を得た。
3.評価方法(実施例21~29の評価方法)
(1)樹脂組成物の特性
(1-1)エポキシ樹脂組成物の調製
表4に記載する割合でエポキシ樹脂および樹脂粒子を計量し、撹拌機を用いて80℃で30分間混合し、エポキシ樹脂主剤液を調製した。表4に記載する割合で硬化剤成分を計量し、撹拌機を用いて80℃で30分間混合し、硬化剤液を調製した。これら別々に調製したエポキシ樹脂主剤液と硬化剤液とを、撹拌機を用いて80℃で30分間混合し、エポキシ樹脂組成物を調製した。なお、表4に記載の組成において、エポキシ樹脂のグリシジル基と硬化剤のアミノ基は当量となる。
上記の(1-1)で調製した硬化剤液を、25℃で1週間静置保管し、目視で固体成分の析出を確認した。析出がないものを「○」、析出の認められたものを「×」とした。
粘度測定は、東機産業株式会社製B型粘度計TVB-15Mを用い、120℃の条件にて行った。測定開始直後の最小測定値を初期粘度とし、粘度が50mPa・sに到達した時間を可使時間とした。
(2)樹脂硬化物の特性
(2-1)樹脂硬化物の作成
上記の(1-1)で調製したエポキシ樹脂組成物を真空中で60分間脱泡した後、4mm厚のテフロン(登録商標)樹脂製スペーサーにより厚み4mmになるように設定したステンレス製モールド中に注入した。180℃の温度で40分間、加熱硬化させ、厚さ4mmの樹脂硬化物を得た。
SACMA 18R-94法に準じて、ガラス転移温度を測定した。
JIS K7171法に準じて、試験を実施した。上記の(2-1)で得た樹脂硬化板を用いて、樹脂試験片を寸法80mm×10mm×4mm(厚みh)で準備した。25℃の環境温度で、支点間距離Lは、16×h(厚み)、試験速度2mm/minで曲げ試験を行い、室温乾燥曲げ弾性率(RTD-FM)を測定した。
ASTM D5045に従い、万能試験機(島津製作所製オートグラフ)を用いて靱性(KIc)を測定した。ASTM D5045法に準じて、試験を実施した。上記の(2-1)で得た樹脂硬化板を用いて、樹脂試験片を寸法50mm×8mm(幅W)×4mmで準備した。クラック長aは、0.45≦a/W≦0.55となるように調整した。なお、クラック長aは破壊試験後の破断面を光学顕微鏡を用いて観察し、クラックの先端までの長さ、および試験片両表面におけるクラック長さの平均値を採用した。
(3)CFRPの特性
(3-1)CFRPの作成
炭素繊維多軸織物1および炭素繊維多軸織物2を300×300mmにカットし、500×500mmの離型処理したアルミ板の上に、炭素繊維多軸織物1を3枚、炭素繊維多軸織物2を3枚、合計6枚重ねて積層体とした。
上記の(3-1)で得られたCFRPを、幅101.6mm×長さ152.4mmの寸法に切断し、衝撃後圧縮強度(CAI)試験の試験片を得た。試験はASTM D7136に従い実施した。供試体(サンプル)は各試験片の寸法測定後、衝撃試験は落錘型衝撃試験機(インストロン社製 Dynatup)を用いて、30.5Jの衝撃エネルギーを与えた。衝撃後、供試体の損傷面積は、超音波探傷試験機(クラウトクレーマー社製 SDS3600、HIS3/HF)にて測定した。衝撃後、供試体の強度試験は、供試体の上から25.4mmでサイドから25.4mmの位置に、歪みゲージを左右各1本ずつ貼付し、同様に表裏に合計4本/体の歪みゲージを貼付けた後、試験機(島津製作所製オートグラフ)のクロスヘッド速度を1.27mm/minとし、供試体の破断まで荷重を負荷した。
上記の(3-1)で得られたCFRPを、幅38.1mm×長さ304.8mmの寸法に切断し、試験片中心に直径6.35mmの穴あけ加工を施し、室温乾燥有孔圧縮強度(RTD-OHC)試験の試験片を得た。
(硬化剤組成物の調製)
表1に記載する割合で硬化剤を計量し、撹拌機を用いて90℃の温度で60分間混合し、硬化剤組成物を調製した。
上記で調製した硬化剤組成物と、エポキシ樹脂、粒子状ゴム成分を表1に記載する割合で計量し、撹拌機を用いて80℃で60分間混合し、エポキシ樹脂組成物を調製した。なお、表1に記載の組成においては、エポキシ樹脂のエポキシ基と硬化剤の活性水素は当量となる。
上記で調製したエポキシ樹脂組成物を真空中で脱泡した後、4mm厚のシリコン樹脂製スペーサーにより厚み4mmになるように設定したステンレス製モールド中に注入した。180℃の温度で30分間硬化させ、厚さ4mmの樹脂硬化物を得た。
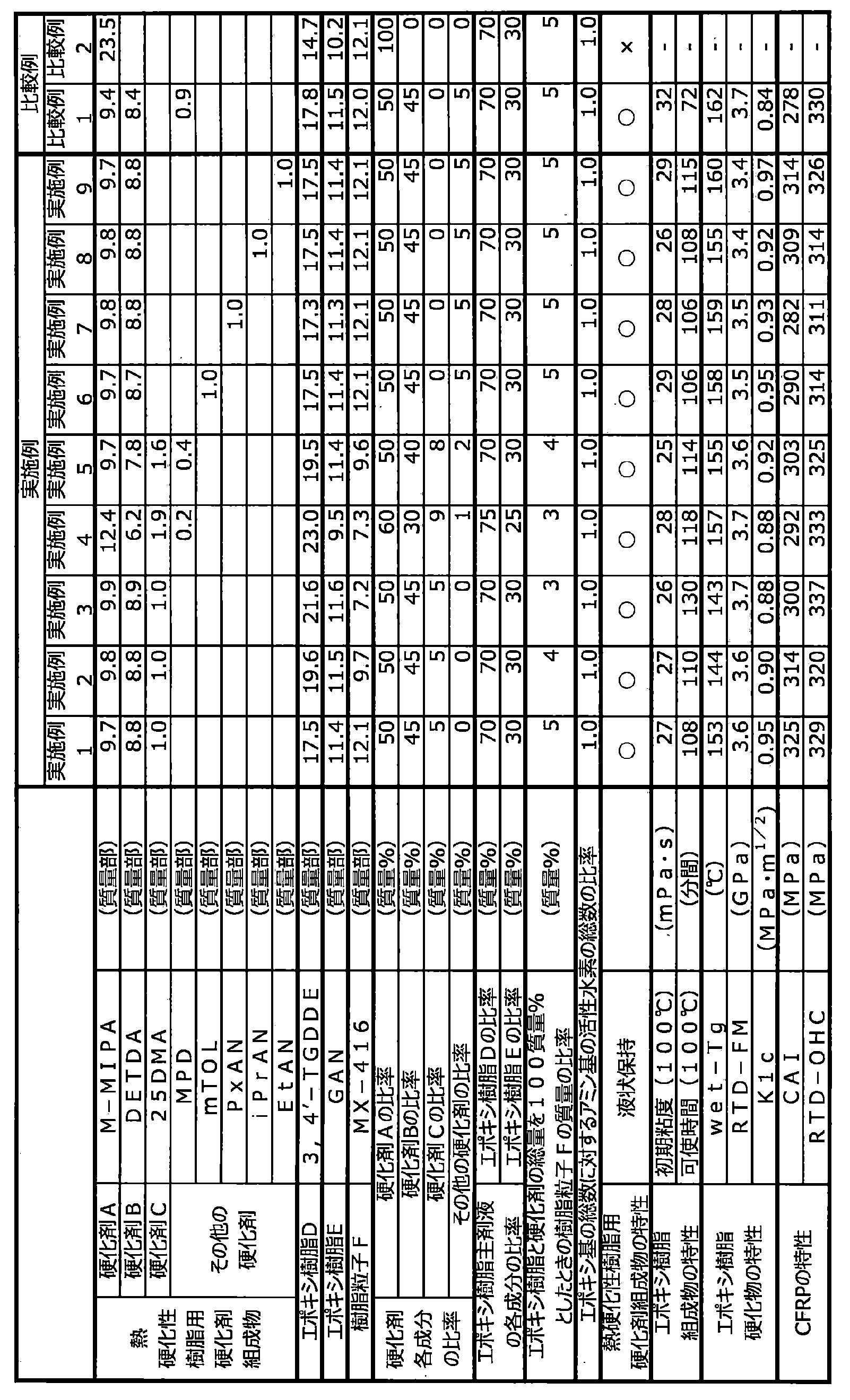
表1に記載のとおり組成を変更して実施例1と同様に実施した。硬化剤組成物、樹脂組成物および樹脂硬化物の特性を表1に示した。硬化剤組成物は、1週間以上液状を保持していた。樹脂組成物は、100℃において28mPa・s以下の低粘度を示し、可使時間は110分間以上となった。曲げ弾性率は3.6GPa以上、K1cは0.88MPa・m1/2以上と高い力学特性を示した。
表1に記載のとおり組成を変更して実施例1と同様に実施した。樹脂硬化物の特性を表1に示した。
表1に記載のとおり組成を変更して実施例1と同様に実施した。硬化剤組成物の特性を表1に示した。可使時間が72分間と短くなった。またK1cが0.84MPa・m1/2となり、靭性が不十分であった。
表1に記載のとおり組成を変更して実施例1と同様に実施した。硬化剤組成物の特性を表1に示した。1週間以内に固体が析出してきたため、液状を保持することが困難であった。
(硬化剤組成物の調製)
表2に記載する割合で硬化剤を計量し、撹拌機を用いて90℃の温度で60分間混合し、硬化剤組成物を調製した。
上記で調製した硬化剤組成物と、エポキシ樹脂、粒子状ゴム成分を表2に記載する割合で計量し、撹拌機を用いて80℃で60分間混合し、エポキシ樹脂組成物を調製した。なお、表2に記載の組成においては、エポキシ樹脂のエポキシ基と硬化剤の活性水素は当量となる。
上記で調製したエポキシ樹脂組成物を真空中で脱泡した後、4mm厚のシリコン樹脂製スペーサーにより厚み4mmになるように設定したステンレス製モールド中に注入した。180℃の温度で30分間硬化させ、厚さ4mmの樹脂硬化物を得た。
表2に記載のとおり組成を変更して実施例11と同様に実施した。硬化剤組成物、樹脂組成物および樹脂硬化物の特性を表1に示した。硬化剤組成物は、1週間以上液状を保持していた。樹脂組成物は、100℃において39mPa・s以下の低粘度を示し、可使時間は22分間以上となった。曲げ弾性率は3.4GPa以上、K1cは0.91MPa・m1/2以上と高い力学特性を示した。

表3に記載のとおり組成を変更して実施例11と同様に実施した。樹脂硬化物の特性を表3に示した。いずれも可使時間が18分間未満と短くなり、またK1cが0.89MPa・m1/2未満となり、靭性が不十分であった。
表3に記載のとおり組成を変更して実施例11と同様に実施した。樹脂硬化物の特性を表3に示した。いずれもK1cが0.88MPa・m1/2未満なり、靭性が不十分であった。
表3に記載のとおり組成を変更して実施例11と同様に実施した。樹脂硬化物の特性を表3に示した。いずれも曲げ弾性率が3.3GPa未満と低く剛性が不十分であり、またK1cが0.89MPa・m1/2未満となり、靭性が不十分であった。
表3に記載のとおり組成を変更して実施例11と同様に実施した。硬化剤組成物の特性を表3に示した。1週間以内に固体が析出してきたため、液状を保持することが困難であった。
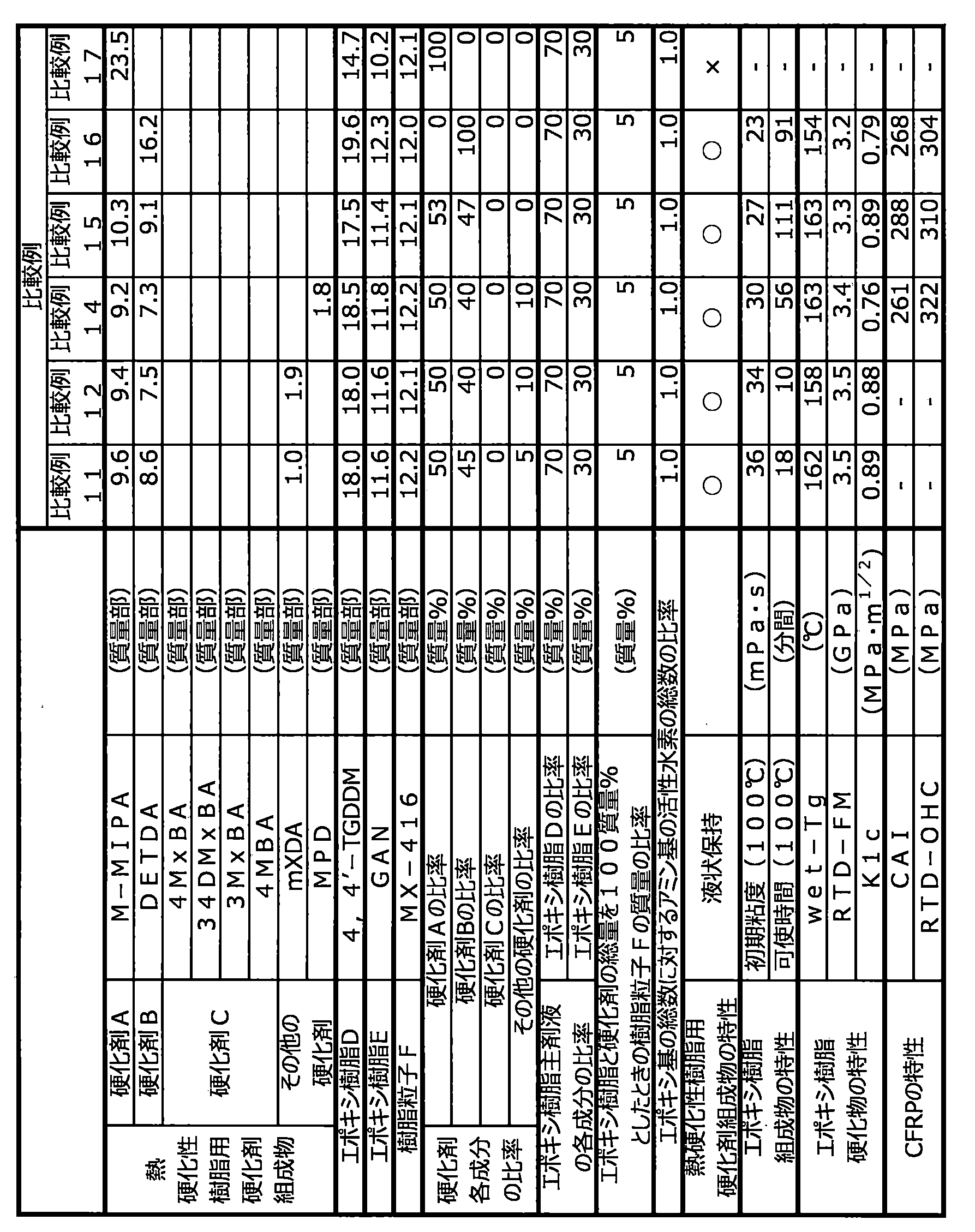
(硬化剤組成物の調製)
表4に記載する割合で硬化剤を計量し、撹拌機を用いて90℃の温度で60分間混合し、硬化剤組成物を調製した。
表4に記載する割合でエポキシ樹脂、樹脂粒子Fを計量し、撹拌機を用いて80℃で30分間混合し、エポキシ樹脂主剤液を調製した。表4に記載する割合で硬化剤を計量し、撹拌機を用いて80℃で30分間混合し、硬化剤液を調製した。これら別々に調製したエポキシ樹脂主剤液と硬化剤液とを、撹拌機を用いて80℃で30分間混合して、エポキシ樹脂組成物を調製した。
上記で得られたエポキシ樹脂組成物を真空中で60分間脱泡した後、4mm厚のテフロン(登録商標)樹脂製スペーサーにより厚み4mmになるように設定したステンレス製モールド中に注入した。180℃の温度で40分間、加熱硬化させ、厚さ4mmの樹脂硬化物を得た。
つぎに、炭素繊維多軸織物1および炭素繊維多軸織物2を300×300mmにカットし、500×500mmの離型処理したアルミ板の上に、炭素繊維多軸織物1を3枚、炭素繊維多軸織物2を3枚、合計6枚重ねて積層体とした。
表4に記載のとおり組成を変更して実施例21と同様に実施した。硬化剤組成物、エポキシ樹脂組成物、樹脂硬化物およびCFRPの特性を表4に示す。
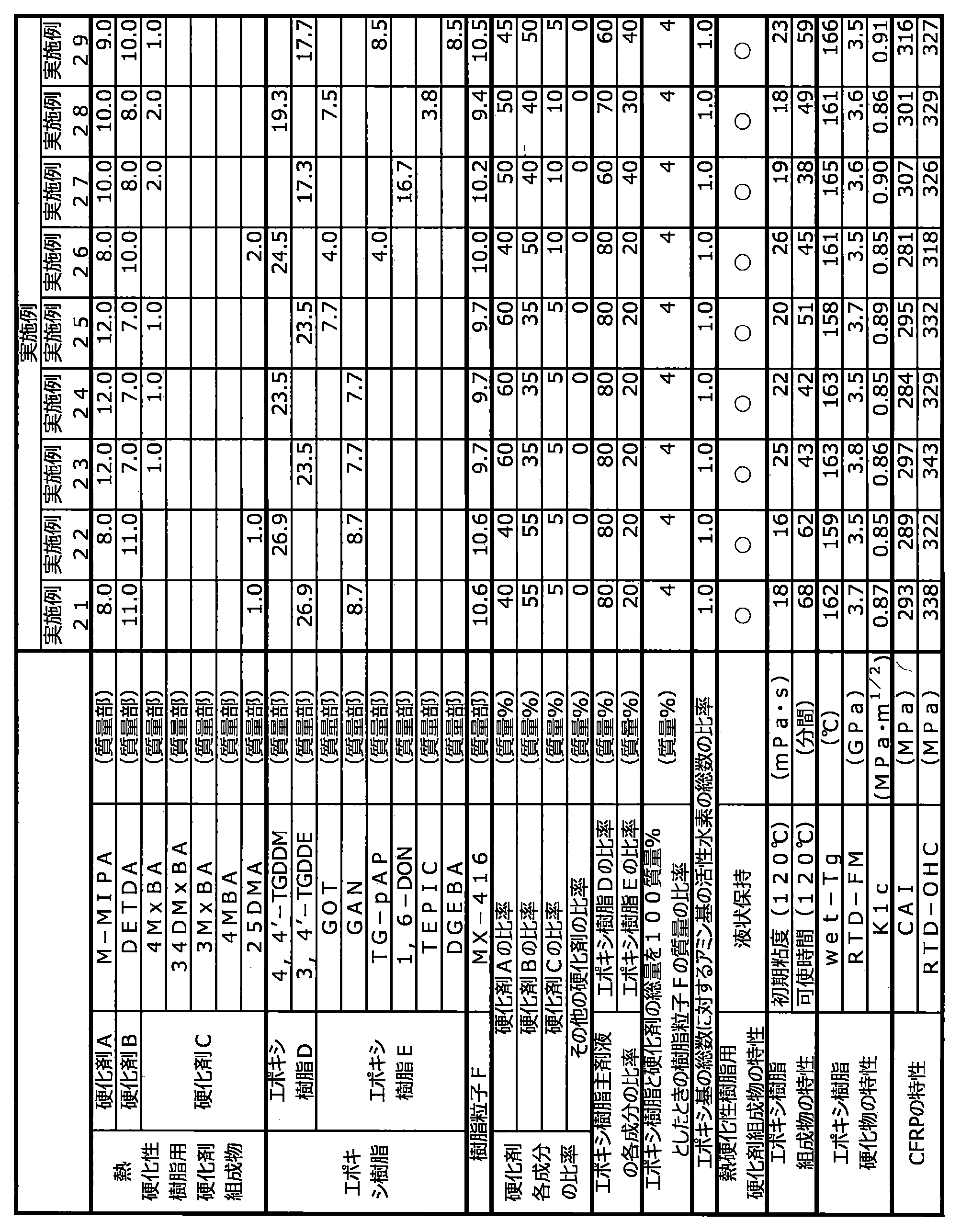
Claims (28)
- 硬化剤A、硬化剤Bおよび硬化剤Cを含有してなる熱硬化性樹脂用硬化剤組成物であって、硬化剤Aは、アミノ基に対する2つのオルト位にそれぞれ置換基を有する芳香族ポリアミンであり、該置換基はアルキル基、芳香族基およびハロゲン基から選択され、硬化剤Bは、25℃で液体である芳香族ポリアミンであり、硬化剤Cは、芳香族置換基を有するモノアミンであることを特徴とする、熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンが、2つ以上の置換基を有する、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンの置換基がいずれも脂肪族基および/またはアルコキシ基である、請求項2に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンの置換基がいずれも炭素数1~4の脂肪族基である、請求項2に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンの置換基がいずれもメチル基である、請求項2に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンが、ベンジルアミン誘導体である、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cのベンジルアミン誘導体が、脂肪族基および/またはアルコキシ基を有するベンジルアミン誘導体である、請求項6に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Aの芳香族ポリアミンが芳香族ジアミンである、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Aの芳香族ポリアミンが4,4’-ジアミノジフェニルメタン誘導体である、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Bの芳香族ポリアミンが、フェニレンジアミン誘導体または4,4’-ジアミノジフェニルメタン誘導体である、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 熱硬化性樹脂用硬化剤組成物の全質量を基準として、硬化剤A、硬化剤Bおよび硬化剤Cの合計が70~100質量%を占め、硬化剤Aと硬化剤Bとの質量比率が1:99~99:1であり、硬化剤Aと硬化剤Bとの合計100質量部に対して硬化剤Cが1~43質量部である、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 80~200℃の温度で均一な液体となり、かつ液温を200℃に昇温後に25℃に降温させて25℃で1週間静置した後において均一な液体である、請求項1に記載の熱硬化性樹脂用硬化剤組成物。
- 請求項1乃至12のいずれかに記載の熱硬化性樹脂用硬化剤組成物、ならびにエポキシ樹脂D、エポキシ樹脂Eを含有するエポキシ樹脂組成物であって、エポキシ樹脂Dは、グリシジル基を4つ以上含むモノマーから構成されるエポキシ樹脂であり、エポキシ樹脂Eは、グリシジル基を2つまたは3つ含むモノマーから構成されるエポキシ樹脂である、エポキシ樹脂組成物。
- エポキシ樹脂Dのグリシジル基を4つ以上含むモノマーが、下記化学式で表される、請求項13に記載のエポキシ樹脂組成物。

- エポキシ樹脂Dのグリシジル基を4つ以上含むモノマーが、テトラグリシジル-4,4’-ジアミノジフェニルエーテル、テトラグリシジル-4,4’-ジアミノジフェニルメタン、テトラグリシジル-3,4’-ジアミノジフェニルエーテルおよびテトラグリシジル-3,3’-ジアミノジフェニルメタンからなる群から選択される1種または2種以上の組み合わせである、請求項13に記載のエポキシ樹脂組成物。
- エポキシ樹脂Eのグリシジル基を2つまたは3つ含むモノマーが、ジグリシジルアニリン、ジグリシジル-o-トルイジン、トリグリシジル-p-アミノフェノール、トリグリシジル-m-アミノフェノール、1,6-ビス(2,3-エポキシプロパン-1-イルオキシ)ナフタレン、1,3,5-トリグリシジルイソシアヌレートおよびビスフェノールAジグリシジルエーテルからなる群から選択される1種または2種以上の組み合わせである、請求項13に記載のエポキシ樹脂組成物。
- エポキシ樹脂組成物中の総エポキシ基の数と熱硬化性樹脂用硬化剤組成物に含まれる活性水素の数の比率が0.7~1.3である、請求項13に記載のエポキシ樹脂組成物。
- エポキシ樹脂組成物に含まれるエポキシ樹脂の全質量を基準として、エポキシ樹脂Dが50~90質量%含有される、請求項13に記載のエポキシ樹脂組成物。
- エポキシ樹脂組成物に含まれる硬化剤の全質量を基準として、硬化剤A、硬化剤Bおよび硬化剤Cの合計が70~100質量%を占め、硬化剤Aと硬化剤Bの質量比率が1:99~99:1であり、硬化剤Aと硬化剤Bとの合計100質量部に対して硬化剤Cが1~43質量部である、請求項13に記載のエポキシ樹脂組成物。
- 請求項13に記載のエポキシ樹脂組成物を硬化させて得られるエポキシ樹脂硬化物。
- 請求項20に記載のエポキシ樹脂硬化物および繊維強化基材を含む繊維強化複合材料。
- 繊維強化基材が炭素繊維強化基材である、請求項21に記載の繊維強化複合材料。
- 繊維強化基材と、請求項13に記載のエポキシ樹脂組成物を複合化して硬化させる繊維強化複合材料の製造方法。
- 請求項13に記載のエポキシ樹脂組成物を、型内に配置した繊維強化基材へ含浸させ、加熱硬化する工程を含む繊維強化複合材料の製造方法。
- 請求項24に記載の繊維強化複合材料の製造方法において、請求項1~12のいずれかに記載の熱硬化性樹脂用硬化剤組成物とエポキシ樹脂主剤とを、型内に配置した繊維強化基材へ含浸させる直前に混合してエポキシ樹脂組成物を得る工程をさらに含む、繊維強化複合材料の製造方法。
- 硬化剤Cを含有してなる熱硬化性樹脂用硬化剤組成物であって、硬化剤Cは、芳香族置換基を有するモノアミンであることを特徴とする、熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンが、2つ以上の置換基を有する、請求項26に記載の熱硬化性樹脂用硬化剤組成物。
- 硬化剤Cの芳香族置換基を有するモノアミンが、ベンジルアミン誘導体である、請求項26に記載の熱硬化性樹脂用硬化剤組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023576714A JPWO2023145341A1 (ja) | 2022-01-28 | 2022-12-23 | |
| US18/729,633 US20250171582A1 (en) | 2022-01-28 | 2022-12-23 | Curing agent composition for thermosetting resin, epoxy resin composition, and fiber-reinforced composite material |
| CN202280090170.6A CN118742586A (zh) | 2022-01-28 | 2022-12-23 | 热固化性树脂用固化剂组合物、环氧树脂组合物及纤维增强复合材料 |
| AU2022437899A AU2022437899A1 (en) | 2022-01-28 | 2022-12-23 | Curing agent composition for thermosetting resin, epoxy resin composition, and fiber-reinforced composite material |
| EP22924188.0A EP4471073A4 (en) | 2022-01-28 | 2022-12-23 | Curing agent composition for thermosetting resin, epoxy resin composition and fiber-reinforced composite material |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022011654 | 2022-01-28 | ||
| JP2022-011654 | 2022-01-28 | ||
| JP2022-039840 | 2022-03-15 | ||
| JP2022039840 | 2022-03-15 | ||
| JP2022-140557 | 2022-09-05 | ||
| JP2022140557 | 2022-09-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023145341A1 true WO2023145341A1 (ja) | 2023-08-03 |
Family
ID=87471646
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/047536 Ceased WO2023145341A1 (ja) | 2022-01-28 | 2022-12-23 | 熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物および繊維強化複合材料 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20250171582A1 (ja) |
| EP (1) | EP4471073A4 (ja) |
| JP (1) | JPWO2023145341A1 (ja) |
| AU (1) | AU2022437899A1 (ja) |
| WO (1) | WO2023145341A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025204944A1 (ja) * | 2024-03-29 | 2025-10-02 | 帝人株式会社 | エポキシ樹脂組成物、エポキシ樹脂硬化物、プリプレグ、繊維強化複合材料、エポキシ樹脂硬化物の修復方法、繊維強化複合材料の修復方法、エポキシ樹脂硬化物の再成形方法、繊維強化複合材料の再成形方法、エポキシ樹脂硬化物の分解方法、及び繊維強化複合材料の強化繊維回収方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62292823A (ja) * | 1986-06-09 | 1987-12-19 | シエル・インタ−ナシヨネイル・リサ−チ・マ−チヤツピイ・ベ−・ウイ | 熱硬化加工特性を有する熱可塑性重合体組成物 |
| JP2009227907A (ja) * | 2008-03-25 | 2009-10-08 | Toray Ind Inc | エポキシ樹脂組成物およびそれを用いた繊維強化複合材料 |
| JP2012041486A (ja) * | 2010-08-23 | 2012-03-01 | Toray Ind Inc | エポキシ樹脂組成物および繊維強化複合材料の製造方法 |
| WO2016208618A1 (ja) * | 2015-06-25 | 2016-12-29 | 東レ株式会社 | エポキシ樹脂組成物、繊維強化複合材料、成形品および圧力容器 |
| JP2019157096A (ja) * | 2018-03-16 | 2019-09-19 | 帝人株式会社 | エポキシ樹脂組成物、プリプレグ、及び繊維強化複合材料 |
| JP6617559B2 (ja) | 2014-07-31 | 2019-12-11 | 東レ株式会社 | 繊維強化複合材料用2液型エポキシ樹脂組成物および繊維強化複合材料 |
-
2022
- 2022-12-23 AU AU2022437899A patent/AU2022437899A1/en active Pending
- 2022-12-23 US US18/729,633 patent/US20250171582A1/en active Pending
- 2022-12-23 JP JP2023576714A patent/JPWO2023145341A1/ja active Pending
- 2022-12-23 WO PCT/JP2022/047536 patent/WO2023145341A1/ja not_active Ceased
- 2022-12-23 EP EP22924188.0A patent/EP4471073A4/en not_active Withdrawn
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62292823A (ja) * | 1986-06-09 | 1987-12-19 | シエル・インタ−ナシヨネイル・リサ−チ・マ−チヤツピイ・ベ−・ウイ | 熱硬化加工特性を有する熱可塑性重合体組成物 |
| JP2009227907A (ja) * | 2008-03-25 | 2009-10-08 | Toray Ind Inc | エポキシ樹脂組成物およびそれを用いた繊維強化複合材料 |
| JP2012041486A (ja) * | 2010-08-23 | 2012-03-01 | Toray Ind Inc | エポキシ樹脂組成物および繊維強化複合材料の製造方法 |
| JP6617559B2 (ja) | 2014-07-31 | 2019-12-11 | 東レ株式会社 | 繊維強化複合材料用2液型エポキシ樹脂組成物および繊維強化複合材料 |
| WO2016208618A1 (ja) * | 2015-06-25 | 2016-12-29 | 東レ株式会社 | エポキシ樹脂組成物、繊維強化複合材料、成形品および圧力容器 |
| JP2019157096A (ja) * | 2018-03-16 | 2019-09-19 | 帝人株式会社 | エポキシ樹脂組成物、プリプレグ、及び繊維強化複合材料 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4471073A4 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025204944A1 (ja) * | 2024-03-29 | 2025-10-02 | 帝人株式会社 | エポキシ樹脂組成物、エポキシ樹脂硬化物、プリプレグ、繊維強化複合材料、エポキシ樹脂硬化物の修復方法、繊維強化複合材料の修復方法、エポキシ樹脂硬化物の再成形方法、繊維強化複合材料の再成形方法、エポキシ樹脂硬化物の分解方法、及び繊維強化複合材料の強化繊維回収方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4471073A1 (en) | 2024-12-04 |
| US20250171582A1 (en) | 2025-05-29 |
| AU2022437899A1 (en) | 2024-07-18 |
| JPWO2023145341A1 (ja) | 2023-08-03 |
| EP4471073A4 (en) | 2025-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102348735B (zh) | 纤维增强复合材料用环氧树脂组合物、预浸料坯及纤维增强复合材料 | |
| CN111187395B (zh) | 环氧树脂组合物、纤维增强复合材料、成型品及压力容器 | |
| CN102822227A (zh) | 碳纤维增强复合材料用环氧树脂组合物、预浸料以及碳纤维增强复合材料 | |
| CN107531879A (zh) | 环氧树脂组合物、预浸料坯及纤维增强复合材料 | |
| JP7431841B2 (ja) | エポキシ樹脂組成物、樹脂硬化物、繊維強化複合材料、及びこれらの製造方法 | |
| JP7190258B2 (ja) | エポキシ樹脂組成物、プリプレグ、及び繊維強化複合材料 | |
| WO2023145341A1 (ja) | 熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物および繊維強化複合材料 | |
| TW201945462A (zh) | 環氧樹脂組成物、預浸體及纖維強化複合材料,以及此等之製造方法 | |
| JP2023005406A (ja) | エポキシ樹脂組成物、樹脂硬化物、繊維強化複合材料、及びこれらの製造方法 | |
| JP7693337B2 (ja) | エポキシ樹脂組成物、樹脂硬化物および繊維強化複合材料 | |
| WO2023017729A1 (ja) | 繊維強化複合材料およびその製造方法 | |
| WO2022186101A1 (ja) | 熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物および繊維強化複合材料 | |
| JP7315304B2 (ja) | エポキシ樹脂組成物、プリプレグ、及び繊維強化複合材料 | |
| JP7783035B2 (ja) | エポキシ樹脂組成物、プリプレグ、繊維強化複合材料および製造方法 | |
| EP3835336A1 (en) | Epoxy compound, epoxy resin, epoxy resin composition, cured resin product, prepreg, fiber-reinforced composite material, and production method for these | |
| JP2020023630A (ja) | エポキシ化合物、エポキシ樹脂、エポキシ樹脂組成物、樹脂硬化物、プリプレグ、繊維強化複合材料、及びこれらの製造方法 | |
| JP7072466B2 (ja) | エポキシ化合物、エポキシ樹脂、エポキシ樹脂組成物、樹脂硬化物、プリプレグ、繊維強化複合材料、及びこれらの製造方法 | |
| CN118742586A (zh) | 热固化性树脂用固化剂组合物、环氧树脂组合物及纤维增强复合材料 | |
| JP2021178902A (ja) | プリプレグ | |
| JP2021195473A (ja) | プリプレグ | |
| JP7693334B2 (ja) | 熱硬化性樹脂用硬化剤組成物、エポキシ樹脂組成物、および繊維強化複合材 | |
| WO2025192675A1 (ja) | エポキシ樹脂組成物、樹脂硬化物、及び繊維強化複合材料、並びに繊維強化複合材料の製造方法 | |
| JP7224800B2 (ja) | エポキシ樹脂組成物、プリプレグ、繊維強化複合材料、及びそれらの製造方法 | |
| WO2019177131A1 (ja) | エポキシ樹脂組成物、プリプレグ及び繊維強化複合材料、並びにこれらの製造方法 | |
| TW202112875A (zh) | 環氧樹脂組成物、樹脂硬化物、纖維強化複合材料及彼等之製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22924188 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2023576714 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AU2022437899 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18729633 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2022437899 Country of ref document: AU Date of ref document: 20221223 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280090170.6 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022924188 Country of ref document: EP Effective date: 20240828 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18729633 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2022924188 Country of ref document: EP |