WO2023146809A1 - Plasma kallikrein inhibitors - Google Patents
Plasma kallikrein inhibitors Download PDFInfo
- Publication number
- WO2023146809A1 WO2023146809A1 PCT/US2023/011304 US2023011304W WO2023146809A1 WO 2023146809 A1 WO2023146809 A1 WO 2023146809A1 US 2023011304 W US2023011304 W US 2023011304W WO 2023146809 A1 WO2023146809 A1 WO 2023146809A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- halo
- hydrogen
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/537—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- Plasma kallikrein is a zymogen of a trypsin-like serine protease and is present in plasma.
- the gene structure is similar to that of factor XL
- the amino acid sequence of plasma kallikrein has 58% homology to factor XI.
- Proteolytic activation by factor Xlla at an internal I389-R390 bond yields a heavy chain (371 amino acids) and a light chain (248 amino acids).
- the active site of plasma kallikrein is contained in the light chain.
- the light chain of plasma kallikrein reacts wi th protease inhibitors, including alpha 2 macroglobulin and Cl- inhibitor.
- HMWK high molecular weight kininogen
- HAE hereditary angioedema
- the plasma kallikrein-kinin system is abnormally abundant in patients diagnosed with advanced diabetic macular edema (DME).
- DME advanced diabetic macular edema
- Recent publications have shown that plasma kallikrein contributes to observed retinal vascular leakage and dysfunction in diabetic rodent models (A. Clermont, et al., Diabetes, 60: 1590 (2011)), and that treatment with a small molecule plasma kallikrein inhibitor ameliorated the observed retinal vascular permeability and other abnormalities related to retinal blood flow.
- SUBSTITUTE SHEET ( RULE 26 ) utility to treat a wide range of disorders, including hereditary angioedema, diabetic macular edema and diabetic retinopathy.
- the present invention relates to compounds of Formula I: and pharmaceutically acceptable salts thereof.
- the compounds of Formula I are inhibitors of plasma kallikrein, and as such may be useful in the treatment, inhibition or amelioration of one or more disease states that could benefit from inhibition of plasma kallikrein, including hereditary angioedema, uveitis, posterior uveitis, wet age-related macular degeneration, diabetic macular edema, diabetic retinopathy and retinal vein occlusion.
- the compounds of this invention could further be used in combination with other therapeutically effective agents, including but not limited to, other drugs useful for the treatment of hereditary angioedema, uveitis, posterior uveitis, wet age-related macular degeneration, diabetic macular edema, diabetic retinopathy and retinal vein occlusion.
- the invention furthermore relates to processes for preparing compounds of Formula I, and pharmaceutical compositions which comprise compounds of Formula I and pharmaceutically acceptable salts thereof.
- the present invention relates to compounds of Formula I:
- Q is -CH2- or absent
- R 1 is selected from the group consisting of hydrogen, halo, hydroxy and C1-6 alkyl
- R 2 is selected from the group consisting of hydrogen, halo, hydroxy and C1-6 alkyl
- R 3 is selected from the group consisting of hydrogen, halo, hydroxy, C1-6 alkyl and C3-6 cycloalkyl;
- R 4 is selected from the group consisting of hydrogen, halo, hydroxy and C1-6 alkyl
- R 5 is hydrogen, halo or C1-6 alkyl, wherein said alkyl group is optionally substituted with one to three halo;
- R 6 is independently selected from the group consisting of hydrogen, halo, hydroxy, cyclopropyl, C1-6 alkyl and (C1-6 alkyl)cyclopropyl, wherein said alkyl group is optionally substituted with one to three substituents independently selected from the group consisting of halo, phenyl and OR X , and said cyclopropyl groups are optionally substituted with OR X ;
- R 7 is selected from the group consisting of hydrogen, halo, hydroxy and C 1-6 alkyl, wherein said alky l group is optionally substituted with one to three halo or hydroxy; or R 6 and R 7 can be taken together with the carbon atom to which they are attached to form a 3- to 6-membered cycloalkyl group, or a 5- to 6-membered heterocyclyl group;
- R 9 is hydrogen or C1-3 alkyl
- R 10 is hydrogen or C1-3 alkyl
- R x is hydrogen or Ci-s alkyl, which is optionally substituted with one to three substituents selected from the group consisting of halo and hydroxy,
- R y is heteroaryl, heterocyclyl or C3-6 cycloalkyl, wherein said heteroaryl group is optionally substituted with oxo or C 1-6 alkyl, said heterocyclyl group is optionally substituted with one or two oxo and said cycloalkyl group is optionally substituted with C1-6 alkyl; or a pharmaceutically acceptable salt thereof.
- Q is -CH2-. In another embodiment of the invention, Q is absent
- the present invention relates to compounds of the Formula la: the group consisting of hydrogen, halo, hydroxy and Ci-6 alkyl;
- R 2 is selected from the group consisting of hydrogen, halo, hydroxy and Ci-6 alkyl
- R 3 is selected from the group consisting of hydrogen, halo, hydroxy, Ci-6 alkyl and C3-6 cycloalkyl;
- R 4 is selected from the group consisting of hydrogen, halo, hydroxy and C1-6 alkyl
- R 5 is hydrogen, halo or C1-6 alkyl, wherein said alkyl group is optionally substituted with one to three halo;
- R 6 is independently selected from the group consisting of hydrogen, halo, hydroxy, cyclopropyl, C1-6 alkyl and (C1-6 alkyl)cyclopropyl, wherein said alkyl group is optionally substituted with one to three substituents independently selected from the group consisting of halo, phenyl and OR X , and said cyclopropyl groups are optionally substituted with OR X ;
- R 7 is selected from the group consisting of hydrogen, halo, hydroxy and C 1-6 alkyl, wherein said alkyd group is optionally substituted with one to three halo or hydroxy; or R 6 and R 7 can be taken together with the carbon atom to which they are attached to form a 3- to 6-membered cycloalkyl group, or a 5- to 6-membered heterocyclyl group;
- R 8 is selected from the group consisting of phenyl or heteroaryl, which can be monocyclic or bicyclic; wherein said phenyl and heteroaryl groups are optionally substituted
- R 9 is hydrogen or C1-3 alkyl
- R 10 is hydrogen or C1-3 alkyl
- R x is hydrogen or C1-6 alkyl, which is optionally substituted with one to three substituents selected from the group consisting of halo and hydroxy,
- R y is heteroaryl, heterocyclyl or C3-6 cycloalkyl, wherein said heteroaryl group is optionally substituted with oxo or C 1-6 alkyl, said heterocyclyl group is optionally substituted with one or two oxo and said cycloalkyl group is optionally substituted with C1-6 alkyl; or a pharmaceutically acceptable salt thereof.
- A is 0. In another embodiment of the invention, A is -CH 2 -.
- the present invention relates to compounds of the Formula lb:
- R 1 is selected from the group clonsisting of hydrogen, halo, hydroxy and C1-6 alkyl;
- R 2 is selected from the group consisting of hydrogen, halo, hydroxy and C1-6 alkyl
- R 5 is hydrogen, halo or C1-6 alkyl, wherein said alkyl group is optionally substituted with one to three halo;
- R 6 is independently selected from the group consisting of hydrogen, halo, hydroxy, cyclopropyl, C1-6 alkyl and (C1-6 alkyl)cyclopropyl, wherein said alkyl group is optionally substituted with one to three substituents independently selected from the group consisting of halo, phenyl and OR X , and said cyclopropyl groups are optionally substituted with OR X ;
- R 7 is selected from the group consisting of hydrogen, halo, hydroxy and C 1-6 alkyl, wherein
- SUBSTITUTE SHEET ( RULE 26 ) said alkyd group is optionally substituted with one to three halo or hydroxy; or R 6 and R 7 can be taken together with the carbon atom to which they are attached to form a 3- to 6-membered cycloalkyl group, or a 5- to 6-membered heterocyclyl group;
- R 9 is hydrogen or C1-3 alkyl
- R 10 is hydrogen or Ci-s alkyl
- R x is hydrogen or Ci-6 alkyl, which is optionally substituted with one to three substituents selected from the group consisting of halo and hydroxy,
- R y is heteroaryl, heterocyclyl or C3-6 cycloalkyl, wherein said heteroaryl group is optionally substituted with oxo or C 1-6 alkyl, said heterocyclyl group is optionally substituted with one or two oxo and said cycloalkyl group is optionally substituted with C1-6 alkyl; or a pharmaceutically acceptable salt thereof.
- (T) is L N In another embodiment of the invention, . In another embodiment of the invention, is In another embodiment of the invention, In another embodiment of the invention,
- R 1 is halo. In a class of the embodiment, R 1 is chloro.
- R 2 is halo. In a class of the embodiment invention, R 2 is fluoro.
- R? is hydrogen or methyl.
- R 3 is hydrogen.
- R 3 is methyl.
- R 4 is hydrogen or methyl. In a class of the embodiment, R 4 is hydrogen. In a class of the embodiment, R 4 is methyl.
- R 5 is hydrogen or halo. In a class of the embodiment, R 5 is halo. In a subclass of the embodiment, R 5 is fluoro.
- R 6 is hydrogen, hydroxyl, Ci-6-alkyl, and (C i- 6-alkyl)cyclopropyl, wherein said alkyl is optionally substituted with halo or OR X .
- R 6 is hydrogen, hydroxyl, Ci-6-alkyl, and -CHricyclopropyl). wherein said alkyl is optionally substituted with halo or OR X .
- R 6 is Ci-6-alky 1.
- R 6 is methyl.
- R 6 is ethyl.
- R 6 is Ci-6-alkyl, which is substituted with OR X or halo. In a further subclass of the embodiment, R 6 is Ci-6-alkyl, which is substituted with fluoro, hydroxy or methoxy.
- R 7 is hydrogen, hydroxyl or Ci-6-alkyl, which is optionally substituted with hydroxy or halo.
- R 7 is hydrogen.
- R 7 is hydroxyl.
- R 7 is Ci-6- alkyl, which is optionally substituted with hydroxy or halo.
- R 6 and R 7 are taken together with the carbon atom to which they are attached to form a six-membered heterocycle or a Cs/, cycloalkyl group.
- R 8 is phenyl, which is substituted with fluoro or chloro.
- R 8 is a monocyclic or bicyclic heteroaryl ring, which is optionally substituted with halo, Ci-6-alkyl, R x and NR’R 10 .
- Specific embodiments of the present invention include, but are not limited to the compounds identified herein as Examples 1 to 138, or pharmaceutically acceptable salts thereof.
- composition which is compnsed of a compound of Formula I or la as described above and a pharmaceutically acceptable carrier.
- the invention is also contemplated to encompass a
- SUBSTITUTE SHEET ( RULE 26 ) pharmaceutical composition which is comprised of a pharmaceutically acceptable carrier and any of the compounds specifically disclosed in the present application.
- the invention includes compositions for treating diseases or condition in which plasma kallikrein activity is implicated. Accordingly the invention includes compositions for treating impaired visual activity, diabetic retinopathy, diabetic macular edema, retinal vein occlusion, hereditary angioedema, diabetes, pancreatitis, cerebral hemorrhage, nephropathy, cardiomyopathy, neuropathy, inflammatory bowel disease, arthritis, inflammation, septic shock, hypotension, cancer, adult respiratory distress syndrome, disseminated intravascular coagulation, blood coagulation during cardiopulmonary bypass surgery, and bleeding from postoperative surgery in a mammal, comprising a compound of the invention in a pharmaceutically acceptable carrier.
- a class of the invention includes compositions for treating hereditary angioedema, uveitis, posterior uveitis, wet age-related macular degeneration, diabetic macular edema, diabetic retinopathy and retinal vein occlusion. These compositions may optionally include antiinflammatory agents, anti-VEGF agents, immunosuppressive agents, anticoagulants, antiplatelet agents, and thrombolytic agents. The compositions can be added to blood, blood products, or mammalian organs in order to effect the desired inhibitions.
- the invention also includes compositions for preventing or treating retinal vascular permeability associated with diabetic retinopathy and diabetic macular edema in a mammal, comprising a compound of the invention in a pharmaceutically acceptable carrier.
- compositions may optionally include anti-mflammatory agents, anti-VEGF agents, immunosuppressive agents, anticoagulants, antiplatelet agents, and thrombolytic agents
- the invention also includes compositions for treating inflammatory' conditions of the eye, which includes, but is not limited to, uveitis, posterior uveitis, macular edema, acute macular degeneration, wet age-related macular degeneration, retinal detachments, retinal vein occlusion, ocular tumors, fungal infections, viral infections, multifocal choroiditis, diabetic uveitis, diabetic macular edema, diabetic retinopathy, proliferative vitreoretinopathy, sympathetic opthalmia, Vogt Koyanagi -Harada syndrome, histoplasmosis and uveal diffusion.
- These compositions may optionally include anti-inflammatory agents, anti-VEGF agents, immunosuppressive agents, anticoagulants, antiplatelet agents, and thrombolytic agents
- the invention also includes compositions treating posterior eye disease, which includes, but is not limited to, uveitis, posterior uveitis, wet age-related macular degeneration, diabetic macular edema, diabetic retinopathy and retinal vein occlusion.
- These compositions may optionally include anti-inflammatory agents, anti-VEGF agents, immunosuppressive agents,
- SUBSTITUTE SHEET ( RULE 26 ) anticoagulants, antiplatelet agents, and thrombolytic agents.
- the invention is directed to the compounds of structural Formula I or la described herein, as well as the pharmaceutically acceptable salts of the compounds of structural Formula I or la and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or their pharmaceutically acceptable salts or in other synthetic manipulations.
- the compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts of basic compounds encompassed within the term “pharmaceutically acceptable salt” refer to non-toxic salts of the compounds of this invention which are generally prepared by reacting the free base with a suitable organic or inorganic acid.
- Representative salts of basic compounds of the present invention include, but are not limited to, the following: acetate, ascorbate, adipate, alginate, aspirate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, camphorate, camphorsulfonate, camsylate, carbonate, chloride, clavulanate, citrate, cyclopentane propionate, diethylacetic, digluconate, dihydrochloride, dodecylsulfanate, edetate, edisylate, estolate, esylate, ethanesulfonate, formic, fumarate, gluceptate, glucoheptanoate, gluconate, glutamate, glycerophosphate, glycollylarsanilate, hemisulfate, heptanoate, hexanoate, hexyl
- suitable pharmaceutically acceptable salts thereof include, but are not limited to, salts derived from inorganic bases including aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, mangamous, potassium, sodium, zinc, and the like.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary', secondary, and tertiary amines, cyclic amines, dicyclohexyl amines and basic ion-exchange resins, such as arginine, betaine, caffeine, choline, N,N-dibenzylethylenediamine, diethylamme, 2-diethylammoethanol, 2-dimethylaminoethanol,
- SUBSTITUTE SHEET (RULE 26 ) ethanolamine, ethylamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamme, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic nitrogencontaining groups which may be quatemized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
- lower alkyl halides such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl
- diamyl sulfates long chain halides
- salts can be obtained by known methods, for example, by mixing a compound of the present invention with an equivalent amount and a solution containing a desired acid, base, or the like, and then collecting the desired salt by filtering the salt or distilling off the solvent.
- the compounds of the present invention and salts thereof may form solvates with a solvent such as water, ethanol, or glycerol.
- the compounds of the present invention may form an acid addition salt and a salt with a base at the same time according to the type of substituent of the side chain.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- the present invention encompasses all stereoisomeric forms of the compounds of Formula I or la. Unless a specific stereochemistry is indicated, the present invention is meant to comprehend all such isomeric forms of these compounds.
- Centers of asymmetry that are present in the compounds of Formula I or la can all independently of one another have (R) configuration or (S) configuration.
- bonds to the chiral carbon are depicted as straight lines in the stmctural Formulas of the invention, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence both each individual enantiomer and mixtures thereof, are embraced within the Formula.
- that entantiomer either (R) or (S), at that center
- enantiomers are a subject of the invention in enantiomerically pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two enantiomers in all ratios.
- the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios.
- the preparation of individual stereoisomers can be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization can be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound of Formula I or la, or it can be done on a final racemic product.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration.
- compounds of this invention are capable of tautomerization, all individual tautomers as well as mixtures thereof are included in the scope of this invention.
- the present invention includes all such isomers, as well as salts, solvates (including hydrates) and solvated salts of such racemates, enantiomers, diastereomers and tautomers and mixtures thereof.
- the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the specifically and generically described compounds.
- different isotopic forms of hydrogen (H) include protium (In) and deuterium (2n).
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the general process schemes and examples herein using appropriate isotopically- ennched reagents and/or intermediates.
- one or more silicon (Si) atoms can be incorporated into the compounds of the instant invention in place of one or more carbon atoms by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art from readily available starting materials.
- Carbon and silicon differ in their covalent radius leading to differences in bond distance and the steric arrangement when comparing analogous C-element and Si-element bonds. These differences lead to subtle changes in the size and shape of silicon-containing compounds when compared to carbon.
- size and shape differences can lead to subtle or dramatic changes in potency, solubility, lack of off-target activity, packaging properties, and so on.
- substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary' skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials. If a substituent is itself substituted with more than one group, it is understood that these multiple groups may be on the same carbon or on different carbons, so long as a stable structure results.
- the phrase “optionally substituted” (with one or more substituents) should be understood as meaning that the group in question is either unsubstituted or may be substituted with one or more substituents.
- compounds of the present invention may exist in amorphous form and/or one or more crystalline forms, and as such all amorphous and crystalline forms and mixtures thereof of the compounds of Formula I or la are intended to be included within the scope of the present invention.
- some of the compounds of the instant invention may form solvates with water (i.e., a hydrate) or common organic solvents.
- solvates and hydrates, particularly the pharmaceutically acceptable solvates and hydrates, of the instant compounds are likewise encompassed within the scope of this invention, along with un-solvated and anhydrous forms.
- esters of carboxylic acid denvatives such as methyl, ethyl, or pivaloyloxymethyl
- acyl derivatives of alcohols such as - 12 -
- SUBSTITUTE SHEET ( RULE 26 ) (9-acetyl, (9-pivaloyl. O-benzoyl. and O-ammoacyl, can be employed. Included are those esters and acyl groups known in the art for modifying the solubility or hydrolysis characteristics for use as sustained-release or prodrug formulations.
- esters can optionally be made by esterification of an available carboxylic acid group or by formation of an ester on an available hydroxy group in a compound.
- labile amides can be made.
- Pharmaceutically acceptable esters or amides of the compounds of this invention may be prepared to act as prodrugs which can be hydrolyzed back to an acid (or -COO- depending on the pH of the fluid or tissue where conversion takes place) or hydroxy form particularly in vivo and as such are encompassed within the scope of this invention.
- Examples of pharmaceutically acceptable prodrug modifications include, but are not limited to, -C i-ealky 1 esters and -Ci-ealkyl substituted with phenyl esters.
- the compounds within the generic structural formulas, embodiments and specific compounds described and claimed herein encompass salts, all possible stereoisomers and tautomers, physical forms (e.g., amorphous and crystalline forms), solvate and hydrate forms thereof and any combination of these forms, as well as the salts thereof, pro-drug forms thereof, and salts of pro-drug forms thereof, where such forms are possible unless specified otherwise.
- alkyl and alkylene are intended to include both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- alkyl groups are used throughout the specification, e.g. methyl, may be represented by conventional abbreviations including “Me” or CH3 or a symbol that is an extended bond as the terminal group, e.g. , ethyl may be represented by “Ef ’ or CH2CH3, propyl may be represented by “Pr” or CH2CH2CH3, buty l may be represented by “Bu” or CH2CH2CH2CH3, etc.
- CM alkyl (or “C1-C4 alkyl”) for example, means linear or branched chain alkyl groups, including all isomers, having the specified number of carbon atoms.
- CM alkyl includes n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. If no number is specified, 1-4 carbon atoms are intended for linear or branched alkyl groups.
- cycloalkyl means a monocyclic or bicyclic saturated aliphatic hydrocarbon group having the specified number of carbon atoms.
- cycloalkyl includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and so on.
- aryl represents a stable monocyclic or bicyclic ring system of up to 10 carbon atoms in each ring, wherein at least one ring is aromatic.
- Bicyclic aryl ring systems include fused nng systems, where two rings share two atoms, and spiro ring systems, where two rings share one atom.
- Aryl groups within the scope of this definition include, but are not limited to: phenyl, indene, isoindene, naphthalene, and tetralin.
- heteroaryl represents a stable monocyclic or bicyclic nng system of up to 10 atoms in each ring, wherein at least one ring is aromatic, and at least one ring contains from 1 to 4 heteroatoms selected from the group consisting of 0, N and S.
- Bicyclic heteroaryl ring systems include fused ring systems, where two rings share two atoms, and spiro ring systems, where two rings share one atom.
- Heteroaryl groups within the scope of this definition include but are not limited to: azaindolyl, benzoimidazolyl, benzisoxazolyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, dihydroindenyl, furanyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthalenyl, naphthpyndinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline, pyranyl, pyrazinyl, pyrazolyl, pyrazolopyrimidinyl,
- heterocycle or “heterocyclyl” as used herein is intended to mean a stable nonaromatic monocyclic or bicyclic ring system of up to 10 atoms in each ring, unless otherwise specified, containing from 1 to 4 heteroatoms selected from the group consisting of 0, N, S, SO, or SO2.
- Bicyclic heterocyclic ring systems include fused ring systems, where two rings - 14 -
- SUBSTITUTE SHEET ( RULE 26 ) share two atoms, and spiro ring systems, where two rings share one atom.
- “Heterocyclyl” therefore includes, but is not limited to the following: azaspirononanyl, azaspirooctanyl, azetidinyl, dioxanyl, isochromanyl, oxadiazaspirodecenyl, oxaspirooctanyl, oxazolidinonyl, piperazinyl, piperidinyl, pyrrolidinyl, morpholinyl, thiomorpholinyl, tetrahydrofumayl, tetrahydropyranyl, dihydropiperidinyl, tetrahydrothiophenyl and the like. If the heterocycle contains a nitrogen, it is understood that the corresponding N-oxides thereof are also encompassed by this definition.
- halogen or “halo” means fluorine, chlorine, bromine or iodine.
- Celite® (Fluka) diatomite is diatomaceous earth, and can be referred to as "celite”.
- variable R shown in the above structure can be attached to any one of 6 bicyclic ring carbon atoms i, ii, iii, iv, v or vi.
- bicyclic nng systems include fused ring systems, where two rings share two atoms, and spiro ring systems, where two rings share one atom.
- the invention also relates to medicaments containing at least one compound of the Formula I or la and/or of a pharmaceutically acceptable salt of the compound of the Formula I or la and/or an optionally stereoisomeric form of the compound of the Formula I or la or a pharmaceutically acceptable salt of the stereoisomeric form of the compound of Formula I or la, together with a pharmaceutically suitable and pharmaceutically acceptable vehicle, additive and/or other active substances and auxiliaries.
- patient used herein is taken to mean mammals such as pnmates, humans, sheep, horses, cattle, pigs, dogs, cats, rats, and mice.
- the medicaments according to the invention can be administered by oral, inhalative, rectal or transdermal administration or by subcutaneous, intraarticular, intraperitoneal
- SUBSTITUTE SHEET ( RULE 26 ) or intravenous injection. Oral administration is preferred. Coating of stents with compounds of the Formulas I and other surfaces which come into contact with blood in the body is possible.
- the invention also relates to a process for the production of a medicament, which comprises bringing at least one compound of the Formula I or la into a suitable administration form using a pharmaceutically suitable and pharmaceutically acceptable carrier and optionally further suitable active substances, additives or auxiliaries.
- Suitable solid or galenical preparation forms are, for example, granules, powders, coated tablets, tablets, (micro)capsules, suppositories, syrups, juices, suspensions, emulsions, drops or injectable solutions and preparations having prolonged release of active substance, in whose preparation customary excipients such as vehicles, dismtegrants, binders, coating agents, swelling agents, glidants or lubricants, flavorings, sweeteners and solubilizers are used.
- auxiliaries which may be mentioned are magnesium carbonate, titanium dioxide, lactose, mannitol and other sugars, talc, lactose, gelatin, starch, cellulose and its derivatives, animal and plant oils such as cod liver oil, sunflower, peanut or sesame oil, polyethylene glycol and solvents such as, for example, sterile water and mono- or polyhydric alcohols such as glycerol.
- the dosage regimen utilizing the plasma kallikrein inhibitors is selected in accordance with a variety of factors including type, species, age, weight, sex and medical condition of the patient; the severity of the condition to be treated; the route of administration; the renal and hepatic function of the patient; and the particular compound or salt thereof employed.
- An ordinarily skilled physician or veterinarian can readily determine and prescribe the effective amount of the drug required to prevent, counter, or arrest the progress of the condition.
- Oral dosages of the plasma kallikrein inhibitors when used for the indicated effects, will range between about 0.01 mg per kg of body weight per day (mg/kg/day) to about 30 mg/kg/day, preferably 0.025-7.5 mg/kg/day, more preferably 0.1-2.5 mg/kg/day, and most preferably 0. 1-0,5 mg/kg/day (unless specificed otherwise, amounts of active ingredients are on free base basis).
- an 80 kg patient would receive between about 0.8 mg/day and 2.4 g/day, preferably 2-600 mg/day, more preferably 8-200 mg/day, and most preferably 8-40 mg/ day.
- a suitably prepared medicament for once a day administration would thus contain between 0.8 mg and 2.4 g, preferably between 2 mg and 600 mg, more preferably between 8 mg and 200 mg, and most preferably 8 mg and 40 mg, e g., 8 mg, 10 mg, 20 mg and 40 mg.
- the plasma kallikrein inhibitors may be administered in divided doses of two, three, or four times daily.
- a suitably prepared medicament would contain between
- SUBSTITUTE SHEET ( RULE 26 ) 0.4 mg and 4 g, preferably between 1 mg and 300 mg, more preferably between 4 mg and 100 mg, and most preferably 4 mg and 20 mg, e.g., 4 mg, 5 mg, 10 mg and 20 mg.
- the patient would receive the active ingredient in quantities sufficient to deliver between 0.025-7.5 mg/kg/day, preferably 0. 1-2.5 mg/kg/day, and more preferably 0.1 -0.5 mg/kg/day.
- Such quantities may be administered in a number of suitable ways, e g. large volumes of low concentrations of active ingredient during one extended period of time or several times a day, low volumes of high concentrations of active ingredient during a short period of time, e.g. once a day.
- a conventional intravenous formulation may be prepared which contains a concentration of active ingredient of between about 0.01-1.0 mg/mL, e.g. 0.
- an 80 kg patient receiving 8 mL twice a day of an intravenous formulation having a concentration of active ingredient of 0.5 mg/mL, receives 8 mg of active ingredient per day.
- Glucuronic acid, L-lactic acid, acetic acid, citric acid or any pharmaceutically acceptable acid/conjugate base with reasonable buffering capacity in the pH range acceptable for intravenous administration may be used as buffers.
- the choice of appropriate buffer and pH of a formulation, depending on solubility of the drug to be administered, is readily made by a person having ordinary skill in the art.
- Compounds of Formula I or la can be administered both as a monotherapy and in combination with other therapeutic agents, including but not limited to anti-inflammatory agents, anti-VEGF agents, immunosuppressive agents, anticoagulants, antiplatelet agents, and thrombolytic agents.
- an “anti-inflammatory agent” is any agent which is directly or indirectly effective in the reduction of inflammation when administered at a therapeutically effective level.
- “Antiinflammatory agent”’ includes, but is not limited to steroidal anti-inflammatory agents and glucocorticoids. Suitable anti-inflammatory agents include, but are not limited to, cortisone, dexamethasone, hydrocortisone, methylprednisolone, prednisolone, prednisone and triamcinolone.
- anti-VEGF agent is any agent which is directly or indirectly effective in inhibiting the activity of VEGF (V ascular Endothelial Growth Factor).
- Suitable anti-VEGF agents include, but are not limited to, bevacizumab, ranibizumab, brolucizumab and aflibercept.
- immunosuppressant agent is any agent which is directly or indirectly effective in suppressing, or reducing, the strength of the body’s immune system.
- Suitable immunosuppressant agents include, but are not limited to, corticosteroids (for example, - 17 -
- SUBSTITUTE SHEET prednisone, budesonide, prednisolone), j anus kinase inhibitors (for example, tofacitinib), calcineurin inhibitors (for example, cyclosporin, tacrolimus), mTOR inhibitors (for example, sirolimus, everolimus), IMDH inhibitors (for example, azathioprine, leflunomide, mycophenolate), biologies (for example, abatacept, adalimumab, anakinra, certolizumab, etanercept, golimumab, infliximab, ixekizumab, natalizumab, rituximab, secukinumab, tocilizumab, ustekinumab, vedolizumab), and monoclonal antibodies (for example, basiliximab, daclizumab).
- calcineurin inhibitors for example,
- Suitable anticoagulants include, but are not limited to, factor Xia inhibitors, thrombin inhibitors, thrombin receptor antagonists, factor Vila inhibitors, factor Xa inhibitors, factor IXa inhibitors, factor Xlla inhibitors, adenosine diphosphate antiplatelet agents (e.g., P2Y12 antagonists), fibrinogen receptor antagonists (e g. to treat or prevent unstable angina or to prevent reocclusion after angioplasty and restenosis), other anticoagulants such as aspirin, and thrombolytic agents such as plasminogen activators or streptokinase to achieve synergistic effects in the treatment of various vascular pathologies.
- factor Xia inhibitors e.g., thrombin inhibitors, thrombin receptor antagonists, factor Vila inhibitors, factor Xa inhibitors, factor IXa inhibitors, factor Xlla inhibitors, adenosine diphosphate antiplatelet agents (e.g., P
- Such anticoagulants include, for example, apixaban, dabigatran, cangrelor, ticagrelor, vorapaxar, clopidogrel, edoxaban, mipomersen, prasugrel, rivaroxaban, and semuloparin.
- apixaban dabigatran
- cangrelor cangrelor
- ticagrelor vorapaxar
- clopidogrel clopidogrel
- edoxaban mipomersen
- prasugrel rivaroxaban
- semuloparin semuloparin
- the anti-inflammatory agents, anti-VEGF agents, immunosuppressant agents, anticoagulants, antiplatelet agents, and thrombolytic agents described herein are employed in their conventional dosage ranges and regimens as reported in the art, including, for example, the dosages described in editions of the Physicians' Desk Reference, such as the 70th edition (2016) and earlier editions.
- the antiinflammatory agents, anti-VEGF agents, immunosuppressant agents, anticoagulants, antiplatelet agents, and thrombolytic agents described herein are employed in lower than their conventional dosage ranges.
- one or more additional pharmacologically active agents may be administered in combination with a compound of the invention.
- the additional active agent (or agents) is intended to mean a pharmaceutically active agent (or agents) that is active in the body, including pro-drugs that convert to pharmaceutically active form after administration, which is different from the compound of the invention, and also includes free- acid, free-base and pharmaceutically acceptable salts of said additional active agents when such forms are sold commercially or are otherwise chemically possible.
- any suitable additional active agent or agents including but not limited to anti-hypertensive agents, additional
- anti-atherosclerotic agents such as a lipid modifying compound, anti-diabetic agents and/or anti-obesity agents may be used in any combination with the compound of the invention in a single dosage formulation (a fixed dose drug combination), or may be administered to the patient in one or more separate dosage formulations which allows for concurrent or sequential administration of the active agents (co-administration of the separate active agents).
- angiotensin converting enzyme inhibitors e.g, alacepril, benazepril, captopril, ceronapril, cilazapril, delapril, enalapril, enalaprilat, fosinopril, imidapril, lisinopril, moveltipril, perindopril, quinapril, ramipril, spirapril, temocapril, or trandolapril); angiotensin II receptor antagonists also known as angiotensin receptor blockers or ARBs, which may be in free-base, free-acid, salt or pro-drug form, such as azilsartan, e.g., azilsartan medoxomil potassium (ED ARBI®), candesartan, e.g., candesartan cilexetil (ATACAND®),
- angiotensin II receptor antagonists also known as an
- calcium channel blockers e.g., amlodipine, nifedipine, verapamil, diltiazem, felodipine, gallopamil, niludipine, nimodipine, nicardipine
- potassium channel activators e.g., nicorandil, pinacidil, cromakalim, minoxidil, aprilkalim, loprazolam
- sympatholitics e.g., beta- adrenergic blocking drugs (e.g., acebutolol, atenolol, betaxolol, bisoprolol, carvedilol, metoprolol, metoprolol tartate, nadolol, propranolol, sotalol, timolol); alpha adrenergic blocking drugs (e.g., doxazosin, prazosin or alpha methyldopa); central alpha a
- lipid lowering agents e.g., HMG-CoA reductase inhibitors such as simvastatin and lovastatin which are marketed as ZOCOR® and MEVACOR® in lactone pro-drug form and function as inhibitors after administration, and pharmaceutically acceptable salts of dihydroxy open ring acid HMG-CoA reductase inhibitors such as atorvastatin (particularly the calcium salt sold in LIPITOR®), rosuvastatin (particularly the calcium salt sold
- SUBSTITUTE SHEET ( RULE 26 ) in CRESTOR®), pravastatin (particularly the sodium salt sold in PRAVACHOL®), and fluvastatin (particularly the sodium salt sold in LESCOL®); a cholesterol absorption inhibitor such as ezetimibe (ZETIA®), and ezetimibe in combination with any other lipid lowering agents such as the HMG-CoA reductase inhibitors noted above and particularly with simvastatin (VYTORIN®) or with atorvastatin calcium; niacin in immediate-release or controlled release forms, and particularly niacin in combination with a DP antagonist such as laropiprant and/or with an HMG-CoA reductase inhibitor; niacin receptor agonists such as acipimox and acifran, as well as niacin receptor partial agonists; metabolic altering agents including insulin sensitizing agents and related compounds for the treatment of diabetes such as biguanides (e.g., metformin),
- Typical doses of the plasma kallikrein inhibitors of the invention in combination with other suitable agents may be the same as those doses of plasma kallikrein inhibitors administered without coadministration of additional agents, or may be substantially less that those doses of plasma kallikrein inhibitors administered without coadministration of additional agents, depending on a patient’s therapeutic needs.
- the compounds are administered to a mammal in a therapeutically effective amount.
- therapeutically effective amount it is meant an amount of a compound of the - 20 -
- SUBSTITUTE SHEET ( RULE 26 ) present invention that, when administered alone or in combination with an additional therapeutic agent to a mammal, is effective to treat (i.e., prevent, inhibit or ameliorate) the disease condition or treat the progression of the disease in a host.
- the compounds of the invention are preferably administered alone to a mammal in a therapeutically effective amount.
- the compounds of the invention can also be administered in combination with an additional therapeutic agent, as defined below, to a mammal in a therapeutically effective amount.
- the combination of compounds is preferably, but not necessarily, a synergistic combination.
- Synergy as described for example by Chou and Talalay, Adv. Enzyme Regul. 1984, 22, 27-55, occurs when the effect (in this case, inhibition of the desired target) of the compounds when administered in combination is greater than the additive effect of each of the compounds when administered individually as a single agent.
- a synergistic effect is most clearly demonstrated at suboptimal concentrations of the compounds.
- Synergy can be in terms of lower cytotoxicity, increased anticoagulant effect, or some other beneficial effect of the combination compared with the individual components.
- administered in combination or “combination therapy” it is meant that the compound of the present invention and one or more additional therapeutic agents are administered concurrently to the mammal being treated.
- each component may be administered at the same time or sequentially in any order at different points in time.
- each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
- the administration of each component does not need to be via the same route of administration; for example, one component can be administered orally, and another can be delivered into the vitreous of the eye.
- Coupling constants (J) are expressed in hertz (Hz), and spin multiplicities are given as s (singlet), d (doublet), dd (double doublet), t (triplet), m (multiplet), and br (broad).
- Chiral resolutions can be performed on either Waters Thar 80 SFC or Berger MG II preparative SFC systems.
- LC-MS data can be recorded on SHIMADAZU LC-MS-2020, SHIMADAZU LC-MS-2010, or Agilent 1100 series LC-MS, Agilent Prime-1260, or Waters Acquity LC-MS instruments using C18 columns employing a MeCN gradient in water containing 0.02 to 0.1% TFA. UV detections were at 220 and/or 254 nm and ESI ionization was used for MS detection.
- UV ultraviolet
- W watts
- wt. % percentage by weight
- x g times gravity
- an is the specific rotation of polarized light at 589 nm
- % w/v percentage in weight of the former agent relative to the volume of the latter agent
- % v/v percentage in volume of the former agent relative to the volume of the latter agent
- cpm counts per minute
- 8H is chemical shift
- a mass spectrum obtained by ES-MS may be denoted herein by “LC-MS”
- m/z mass to charge ratio
- n normal
- nm is nanometer
- nM is nanomolar.
- Scheme A illustrates the synthetic sequence for preparation of substituted spirocarbamates such as A6 from Boc-protected aniline Al and ketones such as A2. Directed lithiation of aniline Al and addition into the heterocyclic ketone A2 occurs in the presence of Lewis acid (eg. LaCk). The tertiary alcohol undergoes in situ cyclization onto the carbamate to give spirocarbamate derivatives such as A3, which can be subjected to chiral separation, preferably using supercritical flow chromatography (SFC) to afford enantiomers A4 and A5. Deprotection of either enantiomer (A4, e.g.) gives the secondary amine A6 that can be carried on to compounds of this invention.
- Scheme B illustrates the synthetic sequence for preparation of substituted spirocarbamates such as A6 from Boc-protected aniline Al and ketones such as A2. Directed lithiation of aniline Al and addition into the heterocyclic ketone A2 occurs in the presence of Lewis acid (e
- Scheme B illustrates a synthetic sequence for the preparation of spirolactams such as B8.
- Difluorophenylacetonitrile Bl is reacted with a bromoacetate input under basic conditions to afford the 3-cyano-3-arylpropionate B2 that is further reacted with methyl acrylate to afford mixed ester B3.
- Cobalt-catalyzed reduction of the nitrile, and in situ cyclization affords a lactam (B4) that is reduced to the piperidine and benzy lated to afford ester B5.
- Ester hydrolysis, followed by formation of the primary amide (B6) produces a synthon that can be further reacted to effect SNAT displacement of an ortho-fluorine to yield the spirolactam skeleton (B7).
- This core structure can be further elaborated to afford the desired aryl ring functionality 7 (B8), and the two enantiomers can be separated by chiral chromatography that can be carried on to compounds of this invention.
- R is any 2
- X suitable alkyl X is a suitable leaving
- C3 group such as Br, I or group, such as
- Scheme C illustrates a synthetic sequence for the preparation of A-alkylpyrazoles such as C5.
- Pyrazole Cl can either be subjected to Mitsunobu reaction with alcohol C2, or reacted with alkyl halides C3 in the presence of a suitable weak base, such as CS2CO3, to afford N-alkylated esters of type C4 that can be deprotected under suitable reaction conditions to afford carboxylic acids (C5) that can be carried on to compounds of this invention.
- a suitable weak base such as CS2CO3
- R is a suitable group as defined in Formula I
- Scheme D illustrates a synthetic sequence for the preparation of epoxides such as D5 from carboxylic acids of type DI.
- Reaction of DI with methoxymethylamine in the presence of a suitable coupling agent, such as CDI can give a Weinreb-type amide D2.
- This intermediate can be reacted with aryllithium reagents (D3), generated by in situ lithium-halogen exchange by treating the corresponding aryl bromide and «-butyl lithium at -78 °C, to give aryl ketones of type D4.
- X is any suitable alkyl group, such as Me R is any suitable group, as defined in Formula I
- R is a suitable group as defined in Formula I
- Scheme F illustrates a synthetic sequence for the preparation of imidazole esters F4 and
- R is a suitable group as defined in Formula I
- Scheme G illustrates a synthetic sequence for the preparation of imidazophenones G4 from imidazolocarboxylates Gl.
- Imidazole G1 can be protected with SEM chloride, which facilitates regiospecific acylation with suitable benzoyl chloride G3 inputs to afford the target imidazolophenones G4, which can be saponified to afford a carboxylic acid intermediate (G5) that can be carried on to compounds of this invention.
- X is an ester, carboxylic acid or amide moiety as defined in Formula I
- Scheme H illustrates a synthetic sequence for the preparation of imidazocarboxaldehyde H4.
- Bromoimidazole Hl can be protected by treatment with a suitable reagent, such as SEM-C1, and coupled with vinyl potassium tetrafluoroborate salt, in the presence of a palladium catalyst, to yield vinylimidazole H3.
- a suitable reagent such as SEM-C1
- vinyl potassium tetrafluoroborate salt in the presence of a palladium catalyst
- R is a suitable group as defined in Formula I
- X is a suitable leaving group, such as Br, I or OMs, for example
- Scheme I illustrates a synthetic sequence for the preparation of substituted phenylacetates 14 from a phenylacetic acid (11) starting material.
- Treatment of II with a suitable base, such as LDA, followed by addition of a suitable alkylating reagent (12) can afford the desired mono- alkylated acid 13.
- Esterification by a number of methods known to those skilled in the art reaction with TMS-CHN2, e.g.
- TMS-CHN2 e.g.
- R is a suitable group as defined in Formula I
- X is a suitable leaving group, such as Br, I or OMs, for example
- Scheme J illustrates a method for generating a-spirophenylacetates J3, where in this instance, an unsubstituted phenylacetate (JI) is reacted with a bifunctional alkylating agent (J2) in the presence of a suitable base, such as NaH, to afford the desired a, a -disubstituted phenylacetate (J3) that can be carried on to compounds of this invention.
- a bifunctional alkylating agent such as NaH
- R is a suitable group as defined in Formula I
- Scheme K illustrates a synthetic sequence for the preparation of 1,2,4-triazoles K3 from esters, such as 14 or J3.
- Reaction of 14 or J3 with hydrazine affords a hydrazide intermediate KI that is subsequently condensed with an imidate (K2) to give an aminocarbazone K3.
- Thermal cyclization of K3 in the presence of a suitable drying agent, such as molecular sieves, can afford the desired 1,2,4-triazole ester K4 that can be carried on to compounds of this invention.
- a suitable drying agent such as molecular sieves
- Scheme L illustrates a synthetic sequence to convert benzyl-substituted heterocycles (LI) into phenone intermediates L2 that can be functionalized further at the benzylic carbon.
- Benzylic oxidation of heterocycles of type LI with an appropriate oxidant, commonly potassium permanganate can directly give the aforementioned phenone intermediate L2.
- Reaction of L2 in the presence of a fluorinating agent, such as DAST can yield a,a-ge»i-di fluorobenzylic heterocycles L3.
- a fluorinating agent such as DAST
- SUBSTITUTE SHEET (RULE 26 ) a 2-step process involving conversion of the hydroxyl to a reactive halide L4 (conversion to a chloride with thionyl chloride, e.g.) followed by reduction with suitable agent, such lithium aluminum hydride, can afford an a-trifluormethyl-substituted benzylic heterocycle L5 that can be carried on to compounds of this invention.
- a reactive halide L4 conversion to a chloride with thionyl chloride, e.g.
- suitable agent such lithium aluminum hydride
- any intermediate examples of L2-L5, wherein X is defined to result in an ester functionality can be treated as described above to yield the corresponding carboxylic acids (not shown) that can likewise be carried on to compounds of this invention.
- Scheme M illustrates a preferred method for generating desired intermediates such as triazolocarboxaldehyde (M5) from aminothi oxoacetate Ml.
- M5 triazolocarboxaldehyde
- Treatment with an appropriate nucleophile such as a Grignard reagent (M6a) or alkyllithium species (M6b), can give a 2°-alcohol M7 that can itself be carried on or reacted further in the presence of carbon tetrachloride and triphenylphosphine to afford a chloromethyl triazolecarboxylate M8 that can be carried on directly or saponified to the carboxylic acid M9, which can be carried on to compounds of this invention.
- an appropriate nucleophile such as a Grignard reagent (M6a) or alkyllithium species (M6b)
- Scheme N illustrates a method for converting secondary alcohol M6 to the corresponding alkylfluoride (Nl) by reacting M6 in the presence of DAST that can be carried on to compounds of this invention.
- Scheme O illustrates a synthetic sequence for the conversion of heteroaryl ketones or aldehydes (01) to benzyl-substituted heterocycles 04 and 05.
- Ketones or aldehydes, such as 01 can be condensed with arylsulfonylhydrzides to afford hydrazones 02, which can be subjected to Barluenga-type coupling with arylboronic acids (03), in the presence of an appropriate base, such as CS2CO3, to afford the desired benzyl-substituted heterocycles 04 that can be carried on directly or saponified to carboxylic acid 05 which can be carried on to compounds of this invention.
- Scheme P illustrates a synthetic sequence for the conversion of heteroaryl ketones or aldehydes (01) to benzyl-substituted heterocycles 04 and 05.
- R is a suitable group as defined in Formula I
- Scheme P illustrates a synthetic sequence for the preparation of sulfones such as P4 in a 2 step sequence from an alcohol starting material (Pl).
- Alcohol Pl can be reacted with thiobenzothiazole under Mitsunobu conditions to give a thioether intermediate P2, which can subsequently be oxidized to afford substituted sulfones P3 that can be carried on to compounds of this invention.
- SUBSTITUTE SHEET ( RULE 26 ) group such as Br, I or Ms. for example
- Scheme Q illustrates a synthetic sequence for the preparation of A-alkylpy rrol idines, and piperidines such as Q3.
- Heterocycle QI can be reacted with an appropriate alky l halide Q2 in the presence of a suitable, non-nucleophilic base, such as NaH or CS2CO3, to afford A-alkylated esters of type Q3 that can be carried on directly or saponified to the carboxylic acid Q4 which can be carried on to compounds of this invention.
- a suitable, non-nucleophilic base such as NaH or CS2CO3
- A is CH, N or a protected nitrogen Scheme R illustrates a synthetic sequence for the preparation of spirocarbamate amides R3 employing the corresponding ester R1 and spirocarbamate A6 synthons.
- Ester R1 can be saponified to afford a carboxylic acid R2, which is subsequently reacted with spirocarbamate A6 in the presence of a suitable coupling agent, such as HATU or T3P, to afford R3, which represents compounds of this invention
- Scheme S illustrates a synthetic sequence for the preparation of a trifluoroborylalkyl-substituted
- SUBSTITUTE SHEET ( RULE 26 ) pyrazole intermediate S2 from deprotected spirocarbamate synthon A6, which can undergo cross-coupling to produce S4.
- Amide formation by reacting spirocarbamate A6 and 4- pyrazolecarboxylic acid under conditions described previously can afford a pyrazolyl amide precursor SI that can be further reacted in the presence of a suitable strong base, such as KHMDS, and potassium bromomethyltrifluoroborate to yield the alkyltrifluoroborate synthon S2 that can be cross-coupled with a suitable aryl halide (S3) in the presence of a palladium catalyst, like CbPd(dppf). to afford S4, which can be carried on to compounds of this invention.
- a suitable strong base such as KHMDS
- KHMDS potassium bromomethyltrifluoroborate
- S3 aryl halide
- Scheme T illustrates a synthetic sequence to convert heteroaryl aldehydes T1 into branched alkyl heteroaryl congeners T6, T8 and T9.
- Reaction of a heteroaryl carboxaldehyde T1 with a suitable Grignard reagent (T2) yields a secondary alcohol T3 that can be oxidized to the corresponding ketone T4, under a variety of known methods, most preferably using manganese dioxide as an oxidant.
- Ketone T4 can be reacted with a second Grignard reagent (T5) to a tertiary alcohol (T6), which can serve as a compound of this invention.
- both tertiary alcohol T6 and ketone T4 can independently be converted to the corresponding vinyl heteroaryl amide T8.
- elimination in the presence of a suitable acid or Lewis acid source can yield vinyl amide T8.
- the olefin moiety can also be synthesized in a single step from ketone T4, by treatment with a sulfone reagent (04) under Julia-Kociensky conditions or using a Wittig olefmation reaction involving reaction with a phosphorus ylide reagent Reduction of the olefin moiety in T8 can be achieved using a variety of conditions, most notably reaction with catalytic Raney Nickel to afford amide T9 which
- SUBSTITUTE SHEET ( RULE 26 ) represents a general compound of this invention.
- Scheme U illustrates an alternate method for synthesizing compounds of this invention (U4).
- Cross-coupling of a SEM-protected triazoloalkylchloride U1 with a suitable aryl bromide (U2) can be achieved using a Nickel-catalyzed reductive coupling procedure to afford a protected benzyltriazole U3.
- SEM deprotection in the presence of a strong acid, such as TFA can give benzyl-substituted triazolyl amides such as U4.
- Scheme V illustrates an alternate method for synthesizing compounds of this invention (V4).
- Scheme W illustrates a method for synthesizing benzylic alcohol compounds of this invention (Wl).
- reducing agents preferably, sodium borohydri de
- intermediate ketones of type T4 can successfully reduce intermediate ketones of type T4.
- chiral reagents and biocatalytic ketoreductases can be employed to effect asymmetric reduction of the ketone moiety. All of which can be carried on further, as necessary, to compounds of this invention.
- Scheme X illustrates an alternative method for synthesizing compounds of this invention (X5).
- the reaction mixture was allowed to stir at -78 °C for an additional 45 min, at which time, a solution of LaCh*2LiCl (22.5 mL of a 0.6 M THF solution, 13.5 mmol) and tert-butyl 3, 3 -dimethyl -5- oxopiperi dine- 1 -carboxylate (3.1 g, 13.5 mmol) was added at -78 °C over a period of 40 min. The reaction mixture was warmed to rt and stirred for 16 h. KO'Bu (5.3 mL of a 1.7 M THF solution, 9.0 mmol) was added to the reaction mixture, and the resulting mixture was heated to 60 °C for 3 h.
- KO'Bu 5.3 mL of a 1.7 M THF solution, 9.0 mmol
- Step 2 6-Chloro-5-fluoro-5'.5'-dimethylspiro[benzo[d
- Step 1 tert-Butyl 3-cvano-3-(2.6-difluorophenyl)propanoate: A solution of 2-(2,6- difluorophenyl) acetonitrile (10.0 g, 65.3 mmol) in THF (15 mL) was added dropwise to a solution of KHMDS (65.3 mL of a 1 M THF solution, 65.3 mmol) at -78 °C. The resulting mixture was allowed to stir at -78 °C for 30 min, at which time, the reaction was warmed to 0 °C and allowed to stir for 30 min. A separate flask charged with tert-butyl 2-bromoacetate (12.7 g,
- Step 2 1 -(tert-Butyl) 6-methyl 3-cyano-3-(2.6-difluorophenyl)hexanedioate: To a mixture of
- Step 4 terf-Butyl 2-(l-benzyl-3-(2,6-difluorophenYl)piperidin-3-yl)acetate: To the solution of tert-butyl 2-(3-(2,6-difluorophenyl)-6-oxopiperidin-3-yl)acetate (11.0 g, 33.8 mmol) in THF (40 mL) at 0 °C was added borane-tetrahydrofuran complex (80 mL of a 1 M THF solution, 80 mmol), and the resulting mixture was warmed to rt and allowed to stir for 2 h. The reaction was cooled to 0 °C , and quenched with AcOH.
- Step 6 /m-Butyl 5'-fluoro-2'-oxo-2'.3'-dihvdro-l'Z7-SDiro[piperidine-3.4'-quinoline1-l- carboxylate:
- the crude 2-(l-Benzyl-3-(2,6-difluorophenyl)piperidin-3-yl)acetamide (14.0 mmol) was dissolved in DMF (12 mL), and NaH (2.79 g, 69.9 mmol) was added portion-wise.
- the resulting mixture was heated to 130 °C for 30 min, at which time, the reaction was cooled to rt and neutralized with HC1.
- the reaction was concentrated, suspended in MeOH and filtered.
- Step 7 terf-Butyl 6'-chloro-5'-fluoro-2'-oxo-2'.3'-dihydro-r7 : 7-spiro[piperidine-3.4'-quinolinel-l- carboxylate: A-Chlorosuccimmide (359 mg, 2.69 mmol) was added to a solution of tert-butyl 5'- fluoro-2'-oxo-2',3'-dihydro-177-spiro[piperidine-3,4'-quinoline]-l-carboxylate (900 mg, 2.69 mmol) in DMF (6 mL), and the resulting mixture was heated to 80 °C.
- Step 1 2-Cvclopropyl-l-phenylethanone: To a solution of phenylmagnesium bromide (2.70 mL of a 3.0 M THF solution, 8.01 mmol) in THF (5 mL) was added 2-cyclopropylacetomtrile (500 mg, 6.16 mmol) in THF (2 mL) at 0 °C. The resulting mixture was allowed to stir at 0 °C for 2 h, at which time, the reaction was quenched with 1 M HC1 and extracted with EtOAc. The combined organic fractions were washed with brine, dried (NarSOi). filtered and the solvent was evaporated under reduced pressure to give a crude residue that was purified by silica gel chromatography, (EtOAc/petroleum ether) to afford the title compound. 'H NMR (500 MHz,
- Step 2 2-Cyclopropyl-l-phenylethanol: To a solution of 2-cyclopropyl-l-phenylethanone (100 mg, 0.624 mmol) in MeOH (5 mL) was added NaBH4 (35 mg, 0 94 mmol) at 0 °C. The reaction was allowed to stir at 0 °C for 1 h, at which time, the mixture was concentrated to give a residue that was suspended in water and extracted with EtOAc. The combined organic fractions were dried (Na2SO4), filtered, and the solvent was evaporated under reduced pressure to afford the crude title compound that was carried on without purification.
- NaBH4 35 mg, 0 94 mmol
- Step 3 Ethyl l-(2-cyclopropyl- l-phenylethyl)-177-pyrazole-4-carboxylate: Di-tert-butyl azodi carboxylate (170 mg, 0.740 mmol) was added to a stirred mixture of tnphenylphosphme (155 mg, 0.592 mmol), 2-cyclopropyl-l -phenyl ethanol (80 mg, 0.493 mmol), ethyl 177-pyrazole- 4-carboxylate (69 mg, 0,49 mmol) in toluene (2 mL), and the resulting mixture was heated 80 °C for 2 h.
- Step 1 2-Cvclopropyl-A-methoxy-A-methylacetamide: To a mixture of 2-cyclopropylacetic acid (5.0 g, 49.9 mmol) in DCM (20.0 mL) was added GDI (9.00 g, 55.5 mmol) at rt under Ni. The mixture was stirred at rt for 1 h. Then N, O-di methylhydroxylamine hydrochloride (5.50 g, 56.4 mmol) was added. The mixture was stirred at rt for another 15 h. The reaction was quenched with 1 N HC1, and the aq. layer was extracted with DCM. The combined organic layer was washed with 50% satd. aq.
- Step 2 2-Cvclopropyl-l-(4-fluorophenyl)ethenone: To a solution of l-bromo-4-fluorobenzene (4.89 g, 27.9 mmol) in anhydrous THF (20 mL) at -78 °C under N2 was added dropwise a solution of n-BuLi (11.2 mL, 27.9 mmol, 2.5 M in hexane). After stirring for 1 h at -78 °C, a solution of 2-cyclopropyl-A r -methoxy-A-methylacetamide (4.00 g, 27.9 mmol) in anhydrous THF (5 mL) was added dropwise.
- Step 3 2-(Cyclopropylmethyl)-2-(4-fluorophenyl)oxirane: Trimethylsulfonium iodide (1.15 g, 5.61 mmol) was suspended added in THF (15 mL). The mixture was cooled to 0 °C, and potassium tert-butoxide (630 mg, 5.61 mmol) was added. The mixture was warmed to rt and stirred for 15 min. 2-Cyclopropyl-l-(4-fluorophenyl)ethanone (500 mg, 2.81 mmol) was added, and the resulting mixture continued stirring at rt for 30 h. The mixture was quenched with satd. aq.
- Step 1 2-(4-Fluorophenyl)-A-hvdroxyacetimidamide: To a mixture of 2-(4- fluorophenyl)acetonitrile (1.0 g, 7.4 mmol) in MeOH (10 mL) was added hydroxylamine
- Step 3 2-(4-Fluorobenzyl)-lff-imidazole-5-carboxylic acid.
- the title compound was prepared following procedures similar to those described in Intermediate C5-b, Step 4.
- Step 1 Methyl l-((2-(trimethylsilyl)ethoxy)methyl)-17f-imidazole-5-carboxylate: To a stirred solution of methyl IH-imidazole-5-carboxvlate (5.00 g, 39.6 mmol) and K2CO3 (11.0 g, 79.0 mmol) in ACN (50 mL) was added SEM-C1 (8.4 mL, 48 mmol), and the resulting mixture was allowed to stir at for 12 h. The reaction mixture was diluted with water and extracted with EtOAc.
- Step 2 Methyl 2-(4-fluorobenzoyl)-l-((2-(trimethylsilyl)ethoxy)methvD-lH-imidazole-5- carboxylate.
- 4-Fluorobenzoyl chloride (1.94 mL, 16.4 mmol) was added to a solution of methyl l-((2-(trimethylsilyl)ethoxy)methyl)-12F-imidazole-5-carboxylate (3.50 g, 13.7 mmol) and TEA (2.3 mL, 16 mmol) in ACN (30 mL) at 0 °C.
- the resulting mixture was warmed to rt and allowed to stir for 12 h.
- the reach on was concentrated to afford a crude residue that was
- Step 3 2-(4-Fluorobenzoyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lK-imidazole-5-carboxylic acid.
- the title compound was prepared following procedures similar to those described in
- Step 1 Ethyl 4-bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-17f-iinidazole-2-carboxylate.
- ethyl 4-bromo- 17/-imidazole-2-carboxylate 3.00 g, 13.7 mmol
- sodium hydride 657 mg, 16.4 mmol, 60% w/w dispersion in mineral spirits
- (2-(Chloromethoxy)ethyl)trimethylsilane (3.43 g, 20.5 mmol) was added, and the resulting mixture was warmed to 30 °C for 10 h.
- Step 2 Lithium 4-bromo-l -((2-(trimethylsilyl)ethoxy)methyl)-l ff-imidazole-2-carboxylate.
- the title compound was prepared following procedures similar to those described in Intermediate C5-b, Step 4.
- LCMS (acid) [M + H] + 321.0 (calcd. 321.0).
- Step 3 (7?)- 1 '-(4-Bromo- 1 -((2-(trimethylsilyl)ethoxy)methy 1)- 177-imidazole-2-carbonyl)-6- chloro-5-fluorospiro[benzo[ ⁇ /
- Step 4 (7?)-6-chloro-5-fluoro-lMl-((2-(trimethylsilyl)ethoxy)methyl)-4-vinyl-lff- imidazole-2- carbonyl)spiro[benzokf
- Step 5 (7?)-2-(6-chloro-5-fluoro-2-oxo-1.2-dihvdrospiro(benzo oxazine-4.3'-piperidin1-r- ylcarbonyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-l#-imidazole-4-carbaldehyde.
- Step 1 Ethyl 4-bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-lEf-iinidazole-2-carboxylate: To a solution of ethyl 4-bromo-l//-imidazole-2-carboxylate (1.00 g, 4.57 mmol) in DMF (8 mL) was
- Step 2 Ethyl l-((2-(trimethylsilyl)ethoxy)methyl)-4-vinyl-l#-imidazole-2-carboxylate: To a solution of ethyl 4-bromo-l-((2-(tnmethylsilyl)ethoxy)methyl)-177-imidazole-2-carboxylate (1.10 g, 3.15 mmol) in EtOH (15 mL) was added potassium vinyltrifluoroborate (548 mg, 4.09 mmol), Pd(dppf)Ch (691 mg, 0,945 mmol) and TEA (1.3 mL, 9.5 mmol). The resulting mixture was heated to 90 °C for 12 h.
- Step 3 Ethyl 4-formyl-l-((2-(trimethylsilyl)ethoxy)methyl)-lJ7-imidazole-2-carboxylate: To a solution of ethyl l-((2-(trimethylsilyl)ethoxy)methyl)-4-vinyl-17f-imidazole-2-carboxylate (600 mg, 2.02 mmol) in acetone (5 mL) and water (5 mL) was added potassium osmate(VI) dihydrate (32 mg, 0.086 mmol) at 0 °C.
- Step 1 2-(4-Fluorophenyl)butanoic acid: To a solution of 2-(4-fluorophenyl) acetic acid (1.00 g, 6.49 mmol) in THF (10 mL) was added LDA (7. 10 mL, 14.3 mmol, 2 M THF solution) at -78 °C. The resulting mixture was warmed to rt over 30 min, at which time, iodoethane (1.21 g, 7.79 mmol) was added in a single portion, and the reaction was stirred at rt for 4 h. The reaction was
- Step 2 Methyl 2-(4-fluorophenyll butanoate.
- (Diazomethyl)trimethylsilane (4.5 mL, 9.1 mmol, 2.0 M toluene solution) was added to a stirred solution of 2-(4-fluorophenyl)butanoic acid (1.1 g, 6.0 mmol) in DCM (7.5 mL) and MeOH (1.5 mL) at 0 °C , and the resulting mixture was stirred at 0 °C for 1 h.
- the reaction was quenched by addition of 5% aq. AcOH, followed by neutralization by addition to satd. aq. NaHCOs.
- Step 1 2-(4-Fluorophenyl)butanehydrazide: Hydrazine hydrate (781 mg, 15.3 mmol) was added
- Step 2 (Z)-Ethyl 2-amino-2-(2-(2-(4-fluorophenyl)butanoyl)hydrazono)acetate.
- Ethyl 2-ethoxy- 2-iminoacetate 148 mg, 1.02 mmol was added to a stirred solution of 2-(4- fluorophenyl)butanehydrazide (100 mg, 0.510 mmol) in EtOH (1 mL), and the resulting mixture was heated to 80 °C for 12 h. The reaction was cooled to rt and concentrated to afford the title compound that was carried on without further purification.
- LCMS [M+H] + 296.2 (calcd. 296.1).
- Step 3 Ethyl 5-(l-(4-fluorophenyl)propyl)-4Ef-L2.4-triazole-3-carboxylate.
- 4A Molecular sieves were added to a stirred solution of (Z)-ethyl 2-amino-2-(2-(2-(4- fluorophenyl)butanoyl)hydrazono)acetate (110 mg, 0.372 mmol) in xylene (1.5 mL), and the resulting mixture was heated to 150 °C for 24 h. The reaction was cooled to rt and concentrated to afford a crude residue that was purified by preparative TLC (EtOAc/ petroleum ether) to give the title compound.
- Step 6 5-(l-(4-Fluorophenyl)propyl)-477-1.2.4-triazole-3-carboxylic acid.
- the title compound was prepared following procedures similar to those described above for Intermediate C5-b, Step 4.
- LCMS [M+H] + 250.1 (calcd. 250.1).
- Step 1 Ethyl 5-(4-fluorobenzoyl )-4//-1.2.4-triazole-3-carboxylate: Potassium permanganate (1.08 g, 6.82 mmol) was added to a stirred solution of ethyl 5-(4-fhiorobenzyl)-477-l,2,4- triazole-3-carboxylate (850 mg, 3.41 mmol) in DCM (15 mL), and the resulting mixture was allowed to stir at rt. After 12 h, the reaction was diluted with 1 M HC1 and extracted with EtOAc. The layers were separated, and the organic layer was concentrated to afford a crude
- Step 2 5-(4-Fluorobenzoyl)-4H-1.2.4-triazole-3-carboxylic acid.
- the title compound was prepared following procedures similar to those described above for Intermediate C5-b, Step 4.
- LCMS [M + H] + 236.0 (calcd. 236.0).
- Step 1 fert-Butyl 2-(2-ethoxy-l-imino-2-oxoethyl)hydrazine-l -carboxylate.
- Boc-hydrazide (19.8 g, 150 mmol) was combined with ethyl 2-amino-2-thioxoacetate (20.0 g, 150 mmol) in EtOH (80 mL), and the resulting mixture was allowed to stir at rt. After 23 h, the reaction was filtered, and the filter cake was washed with EtOH and dried under vacuum to afford the title compound.
- LCMS [M + H] + 232.15 (calcd. 232.3).
- Step 2 Ethyl 5-((benzyloxy)methyl)-4Zf-1.2.4-triazole-3-carboxylate.
- Step 3 Ethyl 5-(hvdroxymethyl)-4-((2-(trimethylsilyl)elhoxy)riiethyl)-4H- 1.2.4-triazole-3- carboxylate.
- Sodium hydride (2.04 g, 50.9 mmol, 60% w/w dispersion in mineral spints) was added in several portions to a stirred solution that was maintained under positive N2 pressure of ethyl 5-((benzyloxy)methyl)-47f-l,2,4-triazole-3-carboxylate (12.1 g, 46.3 mmol) in THF (93 ml) at 0 °C .
- Step 4 Ethyl 5-( l-hvdroxyDropyl)-4-((2-(trimethylsilyl)ethoxy)methyl )-47/-l ,2.4-triazole-3- carboxylate.
- DMP (3.93 g, 9.26 mmol) was added to a stirred solution of ethyl 5- (hydroxymethyl)-4-((2-(trimethylsilyl)ethoxy)methyl)-477- 1 ,2,4-tnazole-3-carboxylate (1.86 g, 6. 17 mmol) in DCM (19 ml), and the resulting mixture was allowed to stir at rt. After 1.5 h, the reaction was quenched with satd. aq.
- Step 6 3-(l-Chloropropyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-lK-1.2.4-triazole-5-carboxylic acid.
- Step 5 Ethyl 4-benzyl-l-((2-(tnmethylsilyl)ethoxy)methyl)-l#-imidazole-2-carboxylate: To a stirred mixture of (Z)-ethyl 4-((2-((4-methoxyphenyl)sulfonyl)hydrazono)methyl)-l-((2- (trimethylsilyl)ethoxy)methyl)-lK-imidazole-2-carboxylate (250 mg, 0.518 mmol) and
- Step 1 2-((( 1 -Methoxy cvclooroDyl tmethyl )thio)benzo
- thiazole: DEAD (930 pL. 5.87 mmol) was added to a solution of (1 -methoxy cy cl opropyl)methanol (300 mg, 2 94 mmol), benzo[c/]thiazole-2-thiol (590 mg, 3.52 mmol) and tnphenylphosphine (925 mg, 3 52 mmol) in THF (5 mL) at 0 °C. The mixture was stirred at 0 °C for 2 h, at which time, the reaction was diluted with water and extracted with EtOAc. The combined organic layers were concentrated, and purification by preparative TLC (EtOAc/petroleum ether) to give the title compound. LCMS [M + H] + 252.0 (calcd. 252.0).
- Step 2 2-(((l -Methoxy cvcloDropyl)methyl)sulfonyl)benzolJ
- thiazole: To a solution of 2-(((l- methoxycyclopropyl)methyl)thio)benzo[ ⁇ f]thiazole (350 mg, 1 39 mmol) in EtOH (5 mL) was added ammonium molybdate tetrahydrate (172 mg, 0.139 mmol) and H2O2 (5.7 mL, 56 mmol, 30% v/v aq. solution). The resulting mixture was allowed to stir at rt. The reaction was concentrated, and the resulting crude residue was purified by preparative TLC (EtOAc/petroleum ether) to give the title compound. LCMS [M + H] + 284.0 (calcd. 284.0).
- Step 1 Methyl pyrrolidine-3-carboxylate hydrochloride: To a mixture of pyridin-2( 1 J7)-one (721 mg, 7.58 mmol) and TBAI (280 mg, 0.758 mmol) in THF (30 mL) was added NaH (303 mg, 7.58 mmol, 60% w/w dispersion in mineral spirits) at 0 °C. The resulting mixture was stirred at rt for 30 min, at which time, l,4-bis(bromomethyl)benzene (2.00 g, 7.58 mmol) was added. After 2 h, the reaction mixture was quenched with satd. aq. NHiCl and extracted with EtOAc.
- Step 2 Methyl l-(4-((2-oxopyridin-l(2H)-yl)methyl)benzyl)pyrrolidine-3-carboxylate: To a mixture of methyl pyrrolidine-3-carboxylate hydrochloride (119 mg, 0.719 mmol) in DMF (2 mL) was added NaH (60 mg, 1.51 mmol, 60% wt dispersion in mineral spirits) at 0 °C.
- Step 3 l-(4-((2-oxopyridin-l -yl)methyl)benzy4)pyrrolidme-3-carboxylic acid: To a mixture of methyl l-(4-((2-oxopyridin-l(2//)-yl)methyl)benzyl)pyrrolidme-3-carboxylate (300 mg, 0.643 mmol) in DMF (1 mL) was added lithium hydroxide hydrate (135 mg, 3.22 mmol) in water (0.2 mL), and the resulting mixture was stirred at rt for 2 h.
- Step 1 -6-Chloro-5-fluoro-F-(lE7-pyrazole-4-carbonyl)spiro[benzolW1.31oxazine-4.3'- piperidinl -2( I H)-one.
- HATU 743 mg, 1.95 mmol
- l/f-pyrazole-4-carboxylic acid 192 mg, 1.71 mmol
- DIEA 0.85 ml, 4.9 mmol
- Step 2 Potassium (7?)-((4-(6-chloro-5-fluoro-2-oxo-1.2-dihydrospiro[benzo[t7][1.31oxazine-4.3'- piperidinl - 1 -ylcarbonyl)- 177-pyrazol - 1 -y l)methyl)trifluoroborate.
- Step 1 (7?)-r-(4-Benzyl-l-((2-(trimethylsilyl)ethoxy)methyl)-177-imidazole-2-carbonyl)-6- chloro-5-fluorospiro[benzo [1.31oxazine-4.3'-piperidin1-2(lF/)-one.
- Intermediate A6-d (105 mg, 0.284 mmol) was added to a solution of Intermediate 05-a (74 mg, 0.22 mmol) and EDC (84 mg, 0.44 mmol) in pyridine (4 mL) at 0 °C. The resulting mixture was warmed to 30 °C and allowed to stir for 16 h. The reaction was diluted with water and extracted with EtOAc.
- Step 2 l'-(4-Benzyl-17f-imidazole-2-carbonyr)-6-chloro-5- lluorospiro
- Trifluoroacetic acid (0.5 mL) was added to a solution of (7?)-l'-(4-benzyl-l-((2-(trimethylsilyl)ethoxy)methyl)-177-imidazole-2- carbonyl)-6-chloro-5-fluorospiro[benzo[ ⁇ 7][l,3]oxazine-4,3'-piperidin]-2(L77)-one (90 mg, 0.154 mmol) in DCM (3 mL). The reaction was allowed to stir at it for 11 h, at which time, the mixture was concentrated to afford a crude residue that was purified by reverse phase HPLC (ACN/water with 0.05% TFA modifier) to the title compound.
- reverse phase HPLC ACN/water with 0.05% TFA modifier
- Step 2 (7?)-r-(4-Benzoyl-l-((2-(trimethylsilyl)ethoxy)methyl)-177-imidazole-2-carbonyl)-6-
- Step 3 (J?.Z)-6-Chl oro-5 -fluoro- 1'-(4-(2-( 1 -methoxy cyclopropyl)- 1 -phenylvinyl)- 1-((2- (trimethylsilyl)ethoxy)methyl)-l H-iniidazole-2-carbori ⁇ l)spiro
- Step 4 -6-Chloro-5-fluoro-r-(4-(2-(l-methoxycyclopropyl)-l-phenylethyl)-l-((2- (tnmethylsilyl) ethoxylmethyl )- 1 /7-imidazole-2-carbonyl )soi rol benzo
- Step 5 (3'7?)-5-Fluoro-T-(4-(2-(l-methoxycvclopropyl)-l-phenylethyl)-l-((2- (tnmethylsilyl)ethoxy) - 1 f/-imidazole-2-carbonyl )spi ro
- Trifluoroacetic acid (0.3 mL) was added to a solution of (47?)-6-chloro-5- fluoro- T-(4-(2-(l -methoxy cyclopropyl)- 1 -phenylethyl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- 177- imidazole-2-carbonyl)spiro[benzo[i/][l,3]oxazine-4,3'-piperidin]-2(lFr)-one (10 mg, 0,015 mmol) in DCM (3 mL) at 0 °C. The resulting mixture was warmed to rt and allowed to stir for - 79 -
- Step 2 -6-Chloro-5-fluoro-142-((4-fluorophenyl)(hydroxy)methyl)-l-((2- (tnmethylsilyl)ethoxy) ineth ⁇ l)- l7/-imidazole-5- l .3
- Step 3 (37?)-6-Chloro-5-fluoro-T-(2-((4-fluorophenyl)(hvdroxy)methyl)-l#-imidazole-5- carbonyDspiro [benzofc/
- the title compound was prepared as a stereochemical mixture following procedures similar to those described above in Examples 75, Step 5.
- Step 1 -6-Chloro-5-fluoro-r-(2-(4-fluorobenzoyl)-l-((2-(trimethylsilyl)ethoxy)methyl)- 17f-imidazole-5-carbonyl)spiro[benzo[J][L3]oxazine-4,3'-piperidin1-2(177)-one.
- the title compound was prepared following procedures similar to those described in Example 1. [M + H] + , 617.2, (calcd. 617.2).
- Step 2 (37?)-6-Chloro-5-fluoro-lM2-(l-(4-fluorophenyl)-l-hvdroxypropyl)-l-((2- (trimethylsilyl)ethoxy) methyl )- 17/-imidazole-5-carbonyl )spi ro
- Step 3 (A7r)-6-Chloro-5-fluoro- 1 '-(2-(l -(4-fluorophenyl)prop- 1 -en-l-yl)-l#-imidazole-5- carbonyl)spiro[benzo -one. BF 3. OEt? (0. 14 mL, 1.
- Step 4 (3'A)-6-Chloro-5-fluoro-r-(2-(l-(4-fluorophenyl)propyl)-177-imidazole-5-carbonyl)spiro
- Step 1 (3'R)-6-Chtoro-5-fluoro-r-(2-(l-(4-fluoroohenvf)-l-hvdroxypentyl)-l-((2- (tnmethylsilyl)ethoxy) methyl )- 1 //-imidazole-5-carbonyl )spi rol benzok/l [ 1.3]oxazine-4.3'- piperidinl -2( 177)-one.
- Step 2 (37?)-6-Chloro-5-fluoro- l '-(2-( l-(4-nLiorophenyl )- l-hvdroxypentyl)- 177-imidazole-5- carbonyl) spiro
- Step 1 (37?)-6-Chloro-5-fluoro- 1 '-(2-(2.2.2-trifluoro- 1 -(4-fluorophenyl)- 1 -hydroxy ethyl)- 1-((2- (trimethylsilyl)ethoxy)methyl)-lK-imidazole-5-carbonyl)spiro[benzo[d
- Step 2 (3'A)-6-Chloro-r-(2-(l-chloro-2.2.2-trifluoro-l-(4-fluorophenyl)ethyl)-12f-imidazole-5- carbom l )-5-fluorospiio
- Step 3 (37?)-6-Chloro-5-fluoro-lM2-(2.2.2-trifluoro-l-(4-fluorophenyl)ethyl)-n/-imidazole-5- carbonyl)spiro[benzo[ ⁇ f
- Step 1 (3'7?)-6-Chloro-5-fluoro-l'-(5-(l-(4-fluorophenyl)propyl)-47/-1.2.4-triazole-3-
- Step 1 (J?)-6-Chloro-l'-(3-((S)-l-chloropropyl)-l-((2-(trimethylsilyl)ethoxy)methyl)-l/f-L2.4- triazole-5-carbonyl)-5-fluorospiro[benzo[d
- 1- Propanephosphonic anhydride (680 pl, 2.30 mmol) was added to a stirred solution of Intermediate M9-a (468 mg, 1.437 mmol), Intermediate A6-d (419 mg, 1.37 mmol) and TEA (600 pL 4.31 mmol) in DCM (9 mL) at rt.
- Step 2 (47?)-6-Chloro-5-fluoro-T-(3-(l-(4-methyl-l/7-benzo[i/
- Step 3 (4ffi-6-Chloro-5-fluoro-r-(3-(l-(4-methyl-17/-benzofcflimidazol-6-yl)propyl)-lff-1.2.4- triazole-5-carbonyl)spi ro
- Step 1 (7?)-6-Chloro-5-fluoro-r-(5-(hYdroxymethyl)-4-((2-(trimethylsilYl)ethoxy)methyl)-4/f-
- Step 2 -lV-((5-(6-chloro-5-fluoro-2-oxo-1.2-dihYdrospiro[benzo[J
- Step 2 (7?)-5-(6-chloro-5-fluoro-2-oxo-1.2-dihvdrospiro[benzo [1.31oxazine-4.3'-pipendin1-r- ylcarbonyl)-l-((2-(trimethylsilYl)ethoxy)methyl)-l#-1.2.4-triazole-3-carbaldehvde.
- the title compound was prepared in two steps following procedures similar to those described for
- Step 3 (3'7?)-6-Chloro-5-fluoro-T-(3-((4-fluorophenyl)(hvdroxy)methyl)-l-((2- (trimethylsilyl)ethoxy) methyl)-lK-L2.4-triazole-5-carbonyl)spiro[benzo[d
- Step 1 (3'7?)-6-Chloro-5-fluoro-T-(3-(l-fluoro-l-(4-fluorophenyl)propyl)-127-L2.4-triazole-5- carbonyl) spiro, benzok/l 1 1 .3
- Step 1 (7?.Z)-6-Chloro-5-fluoro-l'-(3-(2-methoxy-l-phenylvinyl)-177-L2.4-triazole-5- carbonyDspiro I benzol c/
- LHMDS (0.57 mL, 0.75 mmol, 1.4 M THF solution) was added to a stirred solution of (methoxymethyl)triphenylphosphonium chloride (219 mg, 0.638 mmol) in THF (5 mL) at -78 °C.
- Step 2 6-Chloro-5-fluoro-r-(3-(2-methoxy-l-phenylethyl)-lH-1.2.4-triazole-5- carbonyDspirolbenzokfl H.31oxazine-4.3'-piperidin1-2( HD-one. Raney Ni (106 mg, 0.
- Step 1 GS 1 )-6-Cliloro-5-nuoro- l'-( l -(4-((2-oxopyndm- l (2//
- the effectiveness of a compound of the present invention as an inhibitor of Coagulation factor Xia can be determined using a relevant purified serine protease, and an appropriate synthetic substrate.
- the rate of hydrolysis of the chromogenic or Anorogenic substrate by the relevant serine protease was measured both in the absence and presence of compounds of the present invention.
- Assays were conducted at rt or at 37 °C. Hydrolysis of the substrate resulted in release of amino trifluoromethylcoumarin (AFC), which was monitored spectrofluorometrically by measuring the increase in emission at 510 nm with excitation at 405 nm. A decrease in the rate of fluorescence change in the presence of inhibitor is indicative of enzyme inhibition.
- AFC amino trifluoromethylcoumarin
- AFC amino trifluoromethylcoumarin
- AFC amino trifluoromethylcoumarin
- AFC amino trifluoromethylcoumarin
- a decrease in the rate of fluorescence change in the presence of inhibitor is indicative of enzyme inhibition.
- SUBSTITUTE SHEET ( RULE 26 ) parameters equation to determine the half-maximal inhibitory concentrations (IC50).
- IC50 half-maximal inhibitory concentrations
- Ki equilibrium inhibitory constants
- the activities shown by this assay indicate that the compounds of the invention may be therapeutically useful for treating or preventing various cardiovascular and/or cerebrovascular thromboembolic conditions in patients suffering from unstable angina, acute coronary syndrome, refractory angina, myocardial infarction, transient ischemic attacks, atrial fibrillation, stroke such as thrombotic stroke or embolic stroke, venous thrombosis, coronary and cerebral arterial thrombosis, cerebral and pulmonary embolism, atherosclerosis, deep vein thrombosis, disseminated intravascular coagulation, and reocclusion or restenosis of recanalized vessels.
- stroke such as thrombotic stroke or embolic stroke, venous thrombosis, coronary and cerebral arterial thrombosis, cerebral and pulmonary embolism, atherosclerosis, deep vein thrombosis, disseminated intravascular coagulation, and reocclusion or restenosis of recanalized vessels.
- the effectiveness of a compound of the present invention as an inhibitor of plasma kallikrein can be determined using a relevant purified serine protease, and an appropriate synthetic substrate.
- the rate of hydrolysis of the chromogenic or fluorogenic substrate by the relevant serine protease was measured both in the absence and presence of compounds of the present invention.
- Assays were conducted at rt or at 37 °C. Hydrolysis of the substrate resulted in release of amino trifluoromethylcoumarin (AFC), which was monitored spectrofluorometrically by measuring the increase in emission at 510 nm with excitation at 405 nm. A decrease in the rate of fluorescence change in the presence of inhibitor is indicative of enzyme inhibition.
- AFC amino trifluoromethylcoumarin
- AFC amino trifluoromethylcoumarin
- AFC amino trifluoromethylcoumarin
- a decrease in the rate of fluorescence change in the presence of inhibitor is indicative of enzyme inhibition.
- the results of this assay are expressed as the half- maximal
- Plasma kallikrein determinations were made in 50 mM HEPES buffer at pH 7.4 containing 150 mMNaCl, 5 mM CaCh, and 0.1% PEG 8000 (polyethylene glycol; Fisher Scientific). Determinations were made using purified Human plasma kallikrein at a final concentration of 0.5 nM (Enzyme Research Laboratories) and the synthetic substrate, Acetyl-K- P-R-AFC (Sigma # C6608) at a concentration of 100 mM.
- Activity assays were performed by diluting a stock solution of substrate at least tenfold to a final concentration ⁇ 0.2 Km into a solution containing enzyme or enzyme equilibrated with inhibitor. Times required to achieve equilibration between enzyme and inhibitor were determined in control experiments. The reactions were performed under linear progress curve conditions and fluorescence increase measured at 405 Ex/510 Em nm. Values were converted to percent inhibition of the control reaction (after subtracting 100% Inhibition value). IC50 was determined by inflection point from a four parameter logistic curve fit. Ki was
- the activities shown by this assay indicate that the compounds of the invention may be therapeutically useful for treating or preventing various ophthalmic, cardiovascular and/or cerebrovascular thromboembolic conditions in patients suffering from unstable angina, acute coronary' syndrome, refractory angina, myocardial infarction, transient ischemic attacks, atrial fibrillation, stroke such as thrombotic stroke or embolic stroke, venous thrombosis, coronary and cerebral arterial thrombosis, cerebral and pulmonary embolism, atherosclerosis, deep vein thrombosis, disseminated intravascular coagulation, reocclusion or restenosis of recanalized vessels, hereditary angioedema, uveitis, postenor uveitis, wet age-related macular degeneration, diabetic macular edema, diabetic retinopathy and retinal vein occlusion.
- Plasma Kallikrein (PKal) ICso (nM) and Factor Xia ICso (nM) for selected compounds are as follows:
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Ophthalmology & Optometry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
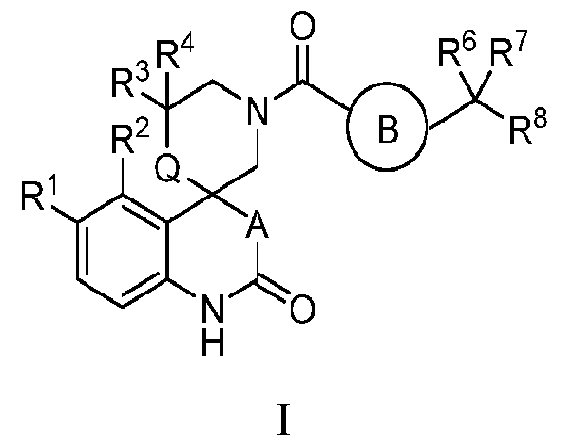



























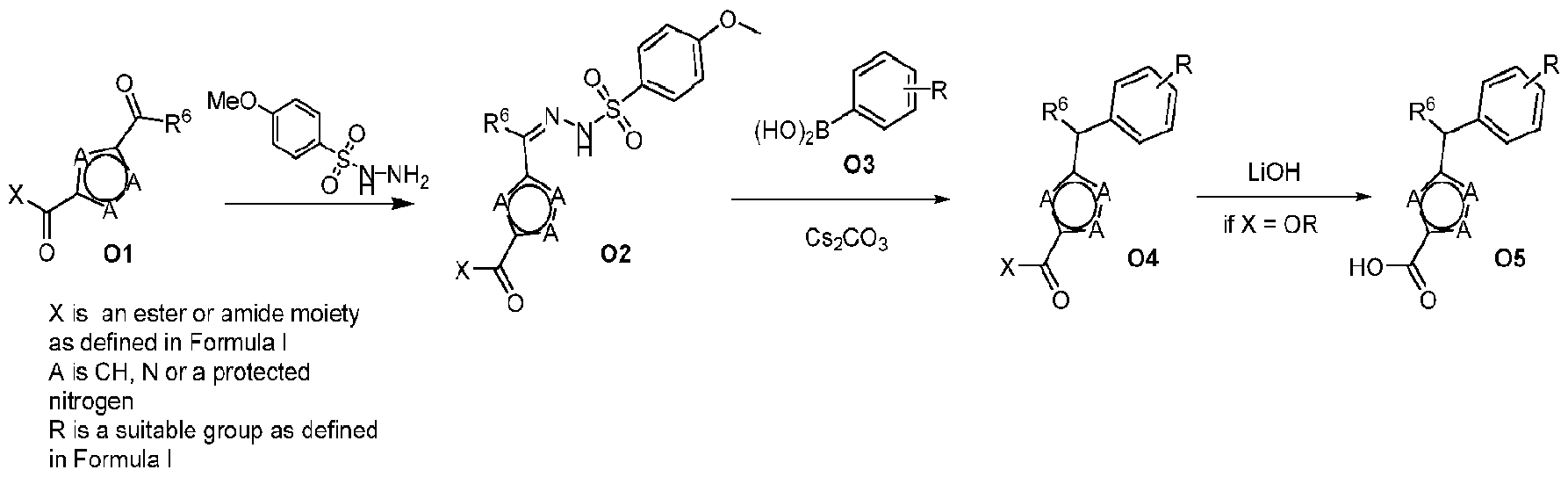

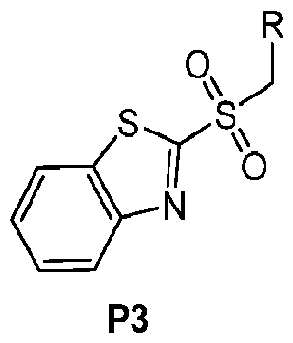





















































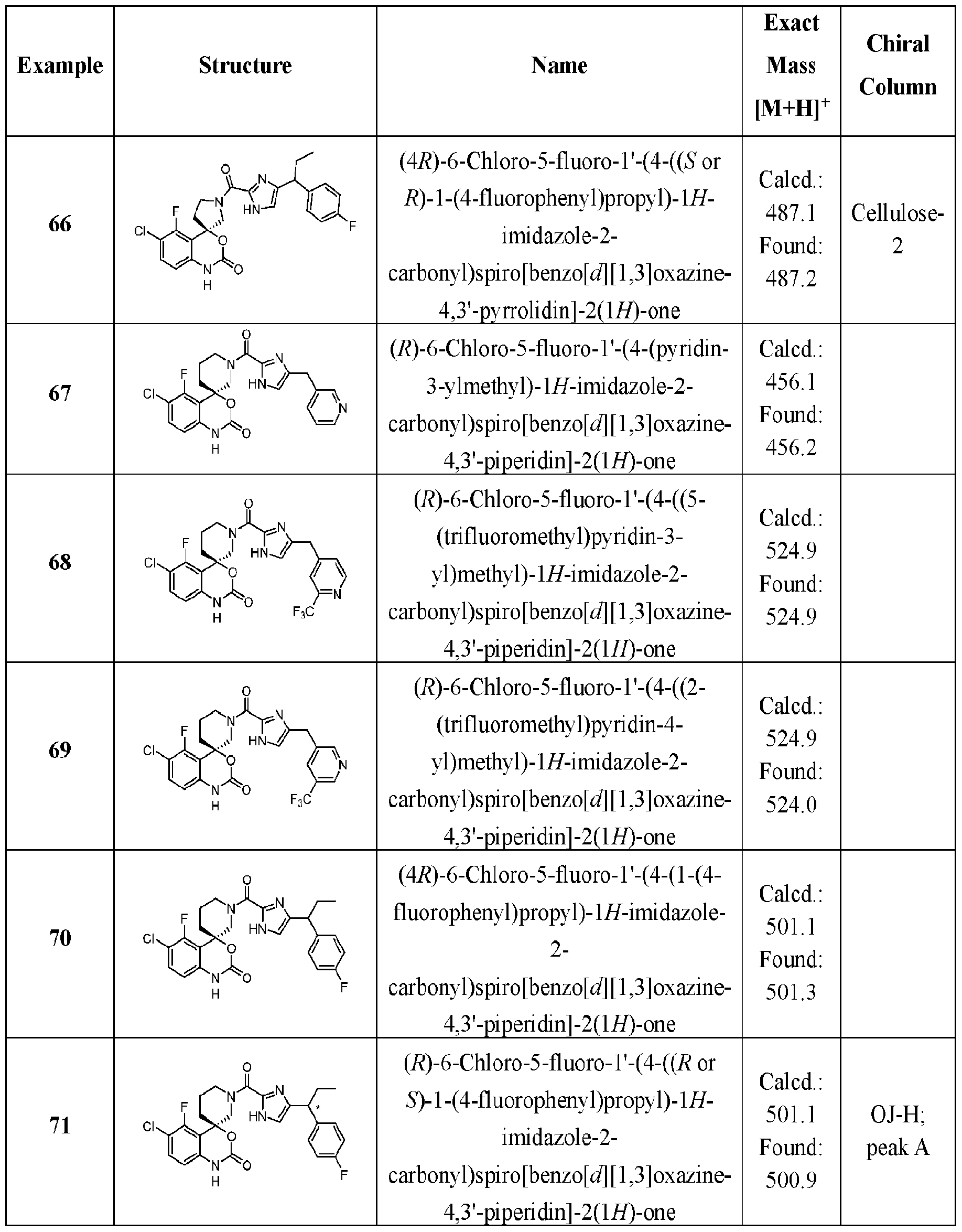

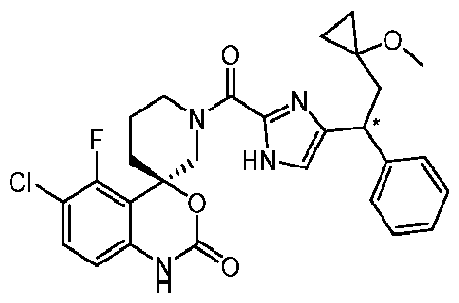






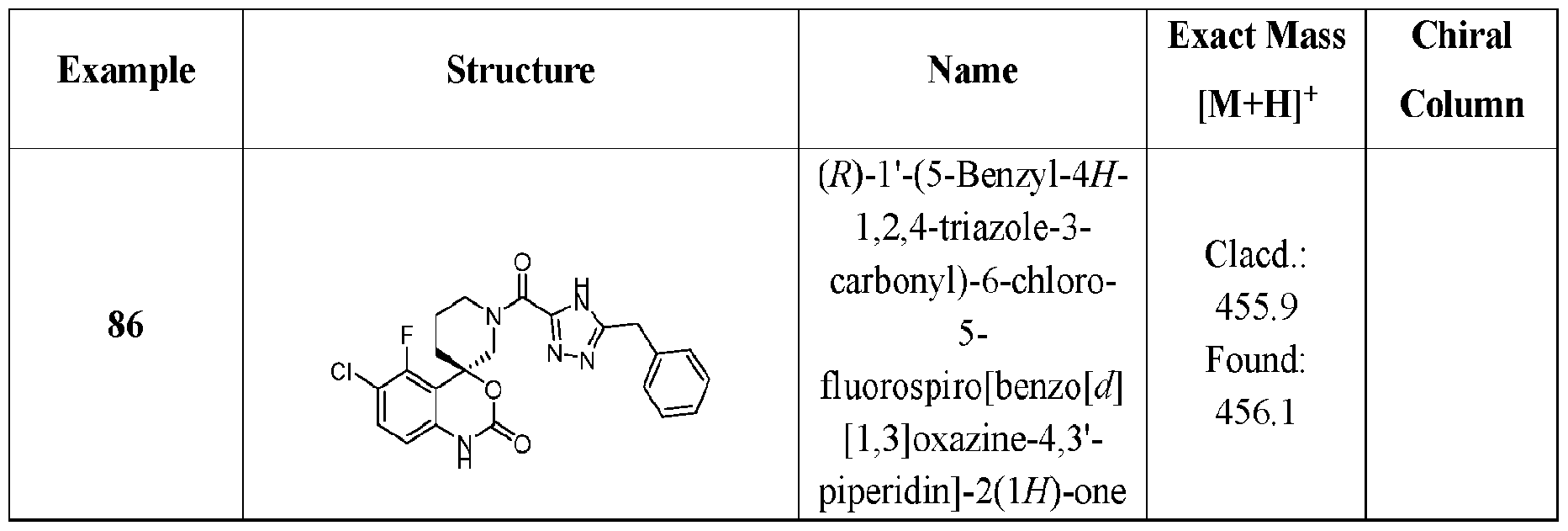






















Claims




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024543515A JP7794991B2 (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
| KR1020247028015A KR20240144234A (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitor |
| MX2024009122A MX2024009122A (en) | 2022-01-25 | 2023-01-23 | PLASMA KALLIKERIN INHIBITORS. |
| US18/728,990 US20250099478A1 (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
| CA3247957A CA3247957A1 (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
| AU2023211555A AU2023211555A1 (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
| CN202380018441.1A CN118613483A (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
| EP23747494.5A EP4469458A4 (en) | 2022-01-25 | 2023-01-23 | PLASMA CALIKINERY INTAKER |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202263302715P | 2022-01-25 | 2022-01-25 | |
| US63/302,715 | 2022-01-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2023146809A1 true WO2023146809A1 (en) | 2023-08-03 |
| WO2023146809A9 WO2023146809A9 (en) | 2024-03-14 |
Family
ID=87472333
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2023/011304 Ceased WO2023146809A1 (en) | 2022-01-25 | 2023-01-23 | Plasma kallikrein inhibitors |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20250099478A1 (en) |
| EP (1) | EP4469458A4 (en) |
| JP (1) | JP7794991B2 (en) |
| KR (1) | KR20240144234A (en) |
| CN (1) | CN118613483A (en) |
| AU (1) | AU2023211555A1 (en) |
| CA (1) | CA3247957A1 (en) |
| MX (1) | MX2024009122A (en) |
| WO (1) | WO2023146809A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4326274B1 (en) * | 2021-04-22 | 2026-01-14 | Merck Sharp & Dohme LLC | Plasma kallikrein inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170044183A1 (en) * | 2014-04-22 | 2017-02-16 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| US20180311250A1 (en) * | 2015-10-29 | 2018-11-01 | Merck Sharp & Dohme Corp. | Macrocyclic spirocarbamate derivatives as factor xia inhibitors, pharmaceutically acceptable compositions and their use |
| WO2022109161A1 (en) * | 2020-11-19 | 2022-05-27 | Merck Sharp & Dohme Corp. | Plasma kallikrein inhibitors |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0944388A4 (en) | 1996-04-03 | 2001-08-16 | Merck & Co Inc | FARNESYL PROTEIN TRANSFERASE INHIBITORS |
| EP3049435A4 (en) | 2013-09-27 | 2017-03-29 | Merck Sharp & Dohme Corp. | Factor xia inhibitors |
-
2023
- 2023-01-23 KR KR1020247028015A patent/KR20240144234A/en active Pending
- 2023-01-23 EP EP23747494.5A patent/EP4469458A4/en active Pending
- 2023-01-23 WO PCT/US2023/011304 patent/WO2023146809A1/en not_active Ceased
- 2023-01-23 AU AU2023211555A patent/AU2023211555A1/en active Pending
- 2023-01-23 US US18/728,990 patent/US20250099478A1/en active Pending
- 2023-01-23 JP JP2024543515A patent/JP7794991B2/en active Active
- 2023-01-23 CN CN202380018441.1A patent/CN118613483A/en active Pending
- 2023-01-23 MX MX2024009122A patent/MX2024009122A/en unknown
- 2023-01-23 CA CA3247957A patent/CA3247957A1/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170044183A1 (en) * | 2014-04-22 | 2017-02-16 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| US20180311250A1 (en) * | 2015-10-29 | 2018-11-01 | Merck Sharp & Dohme Corp. | Macrocyclic spirocarbamate derivatives as factor xia inhibitors, pharmaceutically acceptable compositions and their use |
| WO2022109161A1 (en) * | 2020-11-19 | 2022-05-27 | Merck Sharp & Dohme Corp. | Plasma kallikrein inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE PUBCHEM SUBSTANCE ANONYMOUS : "SID 399795428", XP093083478, retrieved from PUBCHEM * |
| See also references of EP4469458A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3247957A1 (en) | 2023-08-03 |
| JP7794991B2 (en) | 2026-01-06 |
| EP4469458A1 (en) | 2024-12-04 |
| AU2023211555A1 (en) | 2024-07-11 |
| WO2023146809A9 (en) | 2024-03-14 |
| US20250099478A1 (en) | 2025-03-27 |
| MX2024009122A (en) | 2024-08-06 |
| KR20240144234A (en) | 2024-10-02 |
| JP2025503117A (en) | 2025-01-30 |
| EP4469458A4 (en) | 2025-12-31 |
| CN118613483A (en) | 2024-09-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3180327B1 (en) | FACTOR XIa INHIBITORS | |
| EP3180317B1 (en) | FACTOR XIa INHIBITORS | |
| WO2015047973A1 (en) | FACTOR XIa INHIBITORS | |
| US10344039B2 (en) | Macrocyclic spirocarbamate derivatives as factor XIa inhibitors, pharmaceutically acceptable compositions and their use | |
| JP7787890B2 (en) | Plasma kallikrein inhibitors | |
| EP3229801B1 (en) | Factor ixa inhibitors | |
| EP4210694A1 (en) | Plasma kallikrein inhibitors | |
| WO2023146809A1 (en) | Plasma kallikrein inhibitors | |
| EP4308125B1 (en) | Plasma kallikrein inhibitors | |
| EP4178620B1 (en) | Plasma kallikrein inhibitors | |
| RU2854493C2 (en) | Plasma kallikrein inhibitors | |
| EP4326274B1 (en) | Plasma kallikrein inhibitors | |
| RU2852935C1 (en) | Plasma kallikrein inhibitors | |
| WO2024173196A1 (en) | Plasma kallikrein inhibitors | |
| WO2026096331A1 (en) | Plasma kallikrein inhibitors | |
| CA3206548A1 (en) | Factor xia inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23747494 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023211555 Country of ref document: AU Ref document number: AU2023211555 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18728990 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202417055809 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024543515 Country of ref document: JP Ref document number: 202380018441.1 Country of ref document: CN Ref document number: MX/A/2024/009122 Country of ref document: MX |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112024014900 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20247028015 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024118067 Country of ref document: RU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023747494 Country of ref document: EP Effective date: 20240826 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024118067 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 112024014900 Country of ref document: BR Kind code of ref document: A2 Effective date: 20240719 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18728990 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2024118067 Country of ref document: RU |