WO2023148783A1 - Process for the preparation of nafamostat, camostat and their derivatives - Google Patents
Process for the preparation of nafamostat, camostat and their derivatives Download PDFInfo
- Publication number
- WO2023148783A1 WO2023148783A1 PCT/IN2023/050123 IN2023050123W WO2023148783A1 WO 2023148783 A1 WO2023148783 A1 WO 2023148783A1 IN 2023050123 W IN2023050123 W IN 2023050123W WO 2023148783 A1 WO2023148783 A1 WO 2023148783A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- fragment
- compound
- mesylate
- nafamostat
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C277/00—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C277/08—Preparation of guanidine or its derivatives, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups of substituted guanidines
Definitions
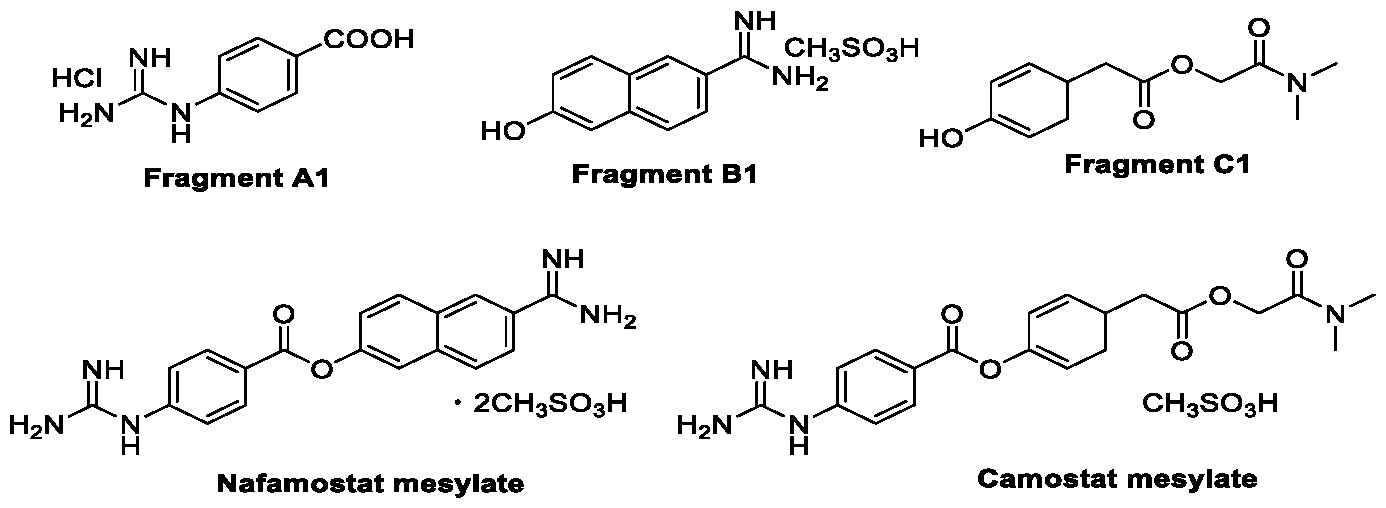
- the present invention provides a process for compound of general Formula (F), or a salt or an isomer thereof. More particularly the present invention provides an effective and economical method for the preparation of highly functionalized ester based drugs namely Nafamostat, Camostat and their derivatives.
- Nafamostat mesylate also known as FUT-175 or 6'-amidino-2-naphthyl-4-guanidinobenzoate dihydrochloride
- FUT-175 6'-amidino-2-naphthyl-4-guanidinobenzoate dihydrochloride
- Fujii et al. Biochim Biophys Acta, 1981, 661, 342
- Japan Tobacco in 1986 for the treatment of inflammatory-related diseases, such as pancreatitis (Surgery, 2001, 130, 175), disseminated intravascular coagulation (DIC), and systemic inflammatory response syndrome.
- DIC disseminated intravascular coagulation
- CN103012214A described the preparation method of a Nafamostat mesylate hydrochloride from amidino-/i-naphthol methane sulfonate and guanidine radicals benzoyl chloride hydrochloride salt at 0-5 °C in pyridine and then converted into Nafamostat mesylate in final step.
- CN103641749B and CN103641749B in 2013 described the preparation method of Nafamostat mesylate from p-guanidinobenzoic acid hydrochloride, 6-amidino-/i-naphthol using 4,5-dicyano imidazole (DCI) as coupling agent in methylene dichloride mix, 0-5 °C is stirred 1 hour, then improve temperature to 18-22 °C, insulation reaction 8-12 h and then finally obtained as mesylate salt.
- DCI 4,5-dicyano imidazole
- KR101595747B 1 claimed the preparation method of Nafamostat mesylate from p-guanidinobenzoic acid hydrochloride and 6-amidino-2-naphthol methanesulfonate in the presence of N,N'- diisopropylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) in pyridine as coupling agent.
- DIC N,N'- diisopropylcarbodiimide
- DMAP 4-dimethylaminopyridine
- EP0465913B1 describes a method for preparing diaminotrifluoromethylpyrimidine derivatives using condensation agents such as dicyclohexylcarbodiimide (DCC), A,A'-carbonyldiimidazole (CDI), 1- ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC).
- DCC dicyclohexylcarbodiimide
- CDI A,A'-carbonyldiimidazole
- EDC 1- ethyl-3-(3-dimethylaminopropyl)carbodiimide
- CN 113999145A describes method for the preparation of Nafamostat from p-chlorobenzoic acid and 6-amidino-2-naphthol hydrochloride utilizing carbodiimide based DCC as coupling reagent.
- EP0048433B1 also describes a method for preparing Nafamostat mesylate wherein DCC is reacted with 4-guanidinobenzoic acid hydrochloride, and then coupled with 6-amidino-2-naphthol methanesulfonate.
- guanidine analogue Camostat also synthesized by using hazardous chemical or coupling reagents such as DIC, EDC, DCC etc.
- the present invention provides an alternate and effective method for the synthesis of Nafamostat and Camostat utilizing the trihalotriazine as inexpensive and effective coupling reagent which also provides the additional advantage towards the ease of operation regarding work up.
- the objective of the invention is to provide a process for compound of general Formula (F).
- Another objective of the invention is to provide a novel, practical and economical route for the preparation of Nafamostat and Camostat drugs and their derivatives.
- Yet another objective of the invention is to explore trihalotriazine more preferably trichlorotriazine (TCT) as the novel and highly economical coupling agent for the preparation of esters and its application in synthesis of Nafamostat and Camostat drugs and their derivatives.
- TCT trichlorotriazine
- the present invention provides a process for the preparation of compound of general
- Ring X and Y is aryl, heteroaryl, extended rings selected from group consisting of naphthalene, phenanthrene, quinoline, isoquinoline;
- Ri is guanidinyl, amidinyl, 2-(dimethylamino)-2-oxoethyl acetyl, halogens, alkyl groups, amide group, cyano group, nitro group, amino group, methoxy group, O-benzyl esters, A-benzyl esters, hydroxyl group, aryl groups, heteroaryl groups;
- R2 is guanidinyl, amidinyl, 2-(dimethylamino)-2-oxoethyl acetyl, halogens, alkyl groups, amide group, cyano group, nitro group, amino group, methoxy group, O-benzyl esters, A-benzyl esters, hydroxyl group, aryl groups, heteroaryl groups, substituted benzenes; wherein the said process comprises the steps; i. reacting 2,4,6-trihalo-l,3,5-triazine and base at 25-40 °C for 5 minutes to obtain activated complex; ii.
- the representative compounds are selected from Nafamostat mesylate and Camostat mesylate.
- the fragment A is prepared by reacting -aminobenzoic acid (1) with thiourea (4) in presence of base in ethanol.
- the base is selected from the group consisting of pyridine (py), A-methylmorpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU), more preferably N-methylmorpholine (NMM).
- the trihalotriazine is selected from the group consisting of trichlorotriazine, tribromotriazine, trifluorotriazine.
- the base is selected from sodium carbonate, cesium carbonate, ammonium carbonate and more preferably potassium carbonate.
- the yield of Nafamostat mesylate is 54 - 70 % and Camostat mesylate is 17 - 28 %
- the representative compounds are selected from
- the base is selected from the group consisting of pyridine (py), A-methylmorpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU), more preferably N-methylmorpholine (NMM).
- the trihalotriazine is selected from the group consisting oftrichlorotriazine, tribromotriazine, trifluorotriazine.
- Fig. 1 depicts the scheme for the process for preparation of Nafamostat, Camostat and their derivatives
- Fig. 2 depicts X H-NMR of Nafamaostat mesylate.
- Fig. 3 depicts 13 C-NMR of Nafamaostat mesylate.
- Fig. 4 depicts DEPT of Nafamaostat mesylate.
- Fig. 5 depicts X H-NMR of Camostat mesylate.
- Fig. 6 depicts 13 C-NMR of Camostat mesylate.
- Fig. 7 depicts DEPT of Camostat mesylate.
- the main aspect of the present invention is to provide a process for compound of general Formula (F) comprising of;
- Ring X and Y is aryl, heteroaryl, extended rings selected from the group consisting of naphthalene, phenanthrene, quinoline, isoquinoline;
- Ri is guanidinyl, amidinyl, 2-(dimethylamino)-2-oxoethyl acetyl, halogens, alkyl groups, amide group, cyano group, nitro group, amino group, methoxy group, O-benzyl esters, /V-benzyl esters, hydroxyl group, aryl groups, heteroaryl groups;
- R2 is guanidinyl, amidinyl, 2-(dimethylamino)-2-oxoethyl acetyl, halogens, alkyl groups, amide group, cyano group, nitro group, amino group, methoxy group, O-benzyl esters, /V-benzyl esters, hydroxyl group, aryl groups, heteroaryl groups, substituted benzenes; i. reacting 2,4,6-trihalo-l,3,5-triazine and base at 25-40 °C for 5 minutes to obtain activated complex; ii. reacting the activated complex as obtained in step-i with fragment A at 40-60 °C for 4 hours to obtain intermediate complex; iii.
- the coupling agent trihalotriazine is selected from trichlorotriazine, tribromotriazine, trifluorotriazine, and more preferably inexpensive trichlorotriazine & base is selected from pyridine (py), /V-methyl morpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU), more preferably N-methylmorpholine (NMM).
- the fragment A is prepared by reacting p- aminobenzoic acid (1) with thiourea (4) in presence of base in ethanol. o i) Trihalotriazine, EtOH, thiourea (4) ii) base, reflux
- base is selected from the list of pyridine (py), N- methylmorpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU), more preferably N- methylmorpholine (NMM).
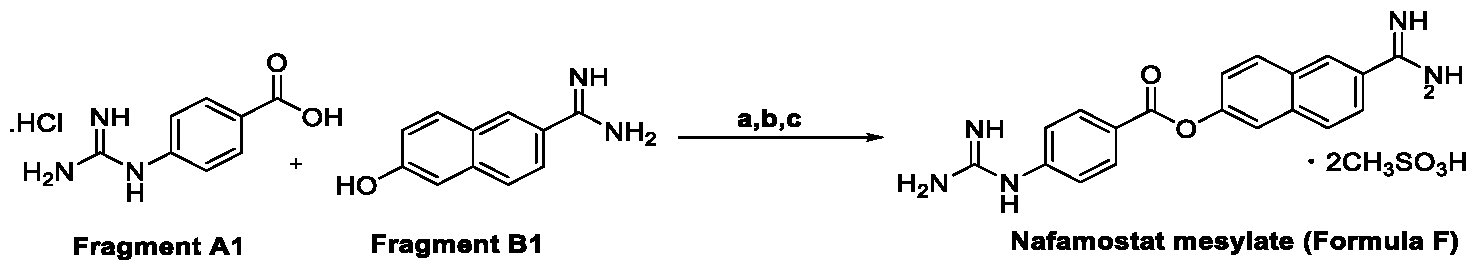
- the present invention provides a process for the preparation of Nafamostat by the coupling of fragment Al namely p-guanidinobenzoic acid and fragment Bl namely 6-hydroxy-2-naphthimidamide using trichlorotriazine, tribromotriazine, trifluorotriazine, and more preferably inexpensive trichlorotriazine (TCT) as coupling reagent in the presence of base such as pyridine (py), A-methylmorpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU) and more preferably A-methyl morpholine (NMM).
- base such as pyridine (py), A-methylmorpholine (NMM) triethylamine (EtsN) Diazabicycloundecene (DBU) and more preferably A-methyl morpholine (NMM).
- base such as pyridine (py), A-methylmorpholine (N
- the present invention provides a process for the preparation of Fragment Al by the reaction of -aminobenzoic acid 1 with thioureatrioxide 2 in presence of base selected from sodium carbonate, cesium carbonate, ammonium carbonate and more preferably potassium carbonate.
- the present invention provides a process for the synthesis of thioureatrioxide 2 from thiourea 4 by oxidation using hydrogen peroxide/peracetic acid system.
- the present invention provides a process for the synthesis of fragment Bl from compound 5 via acid catalysed conversion of cyano to amidine.
- the present invention provides a process for the synthesis of compound 5 from compound 6 via conversion of bromo to cyano group.
- the present invention provides a process for the synthesis of compound 6 from /i-napthol 7 via two step reactions.
- the present invention provides a process for the preparation of Camostat by the coupling of fragment Al namely p-guanidinobenzoic acid and fragment Cl namely 2-(dimethylamino)-2-oxoethyl 2-(4-hydroxyphenyl)acetate using trichlorotriazine, tribromotriazine, trifluorotriazine, and more preferably inexpensive trichlorotriazine as coupling reagent in the presence of base such as pyridine (py), N-methylmorpholine (NMM) triethylamine (EI3N) Diazabicycloundecene (DBU), more preferably N-methylmorpholine (NMM).
- base such as pyridine (py), N-methylmorpholine (NMM) triethylamine (EI3N) Diazabicycloundecene (DBU), more preferably N-methylmorpholine (NMM).
- the Camostat (D2) was precipitated as
- the present invention provides a process for the synthesis of fragment Cl by the coupling of 4-hydroxyphenyl acetic acid 8 with N,N- dimethylbromoacetamide 9.
- the present invention provides a process for the synthesis of compound 9 from the coupling of bromoacetylbromide 10 with VV-dimethylamine hydrochloride.
- the present invention provides a process for the synthesis of fragment A from the coupling of -aminobenzoic acid 1 with thiourea 4 using trichlorotriazine, tribromotriazine, trifluorotriazine, and more preferably inexpensive trichlorotriazine as coupling reagent.
- ES1-MS and HRMS spectra were recorded on LC-MS/MS and HRMS-6540-UHD machines. Optical rotations were measured on a Perkin Elmer polarimeter. Column chromatography was carried out with silica gel (60-120, 230-400 mesh).
- Trichlorotriazine (TCT) as coupling reagent being inexpensive and stable and its use provides cost effective route for the synthesis of Nafamostat, Camostat and their derivatives.
- TCT as a coupling reagent make the preparation of Nafamostat or Camostat simple, high yielding, low impurities and suitable for industrial production.
- Scheme for the synthesis of fragment Al, Bl, Cl and Nafamostat mesylate and Camostat mesylate are provided in examples.
- Table 1 and Table 2 provide the reactants used and the reaction conditions with resulting products and yield thereof.
- thiourea (4) (0.657 mol, 1 eq.) was added into 200 mL of hot water at 40 °C and thoroughly stirred to allow thiourea to dissolve completely.
- the aqueous thiourea solution thus prepared was cooled and then 371.42 mL of hydrogen peroxide (concentration: 30%) (4.6 mol, 7eq.) was added slowly at a rate such that the solution temperature was held below 10 °C. Thereafter, the solution was cooled to 0 °C and stirred for about 30 minutes to allow crystals to be aged. After the crystal ageing, the solid-liquid mixture at 0 °C was immediately filtered and the fractionated crystals were dried at 50 °C.
- /i-naphthol (7) 40 g (1 mole) and 100 ml of glacial acetic acid were taken in a round-bottom flask fitted with a dropping funnel and a reflux condenser. A solution of 88 g (2 mole) of bromine in 30 ml of acetic acid was added through the dropping funnel for a period of 15-30 minutes. The flask was shaken gently during the addition, /i-naphthol dissolves during this period, and heat was evolved; the mixture was cooled somewhat towards the end of the addition to avoid excessive loss of hydrogen bromide, 30 ml of water was then added, and the mixture was heated to boiling.
- the reaction mixture was then cooled to 100 °C, 5 g of mossy tin is added and boiled until the metal is dissolved. A second portion of 5 g of tin was then added and dissolved by boiling, and finally a third portion of 32 g (a total of 42 g) of tin was introduced. The mixture was boiled for 3 h, cooled to 50 °C, and filtered with suction. The crystalline tin salts which were thus removed are washed on the funnel with 100 ml of cold acetic acid, the washings being added to the main portion of the filtrate.
- Example 4 Synthesis of 6-carbamimidoylnaphthalen-2-yl 4-guanidinobenzoate dimesylate (Nafamostat mesylate): 2,4,6-trichloro-l,3,5-triazine (5 g, 26.88 mmol ) and 4-methylmorpholine (70 ml ) were taken in a round-bottom flask and stirred at room temperature for 5 minutes. To the reaction mixture 4- guanidinobenzoic acid, fragment Al (9.62 g, 53.76 mmol) was added and stirred at 40 °C for 4 hours.
- TCT trichlorotriazine
- the present invention also provides ease of operation during work up in contrary to prior art and avoid repeated washing and treatment with solvents.
- TCT trichlorotriazine
- TCT as a coupling reagent make the preparation of Nafamostat or Camostat simple, high yielding, low impurities and suitable for industrial production.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description



































Claims





Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23749463.8A EP4475835A4 (en) | 2022-02-07 | 2023-02-07 | METHOD FOR THE PREPARATION OF NAFAMOSTAT, CAMOSTAT AND THEIR DERIVATIVES |
| US18/836,241 US20250188022A1 (en) | 2022-02-07 | 2023-02-07 | Process for the preparation of nafamostat, camostat and their derivatives |
| JP2024547007A JP2025505208A (en) | 2022-02-07 | 2023-02-07 | Process for the production of nafamostat, camostat and their derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN202211006599 | 2022-02-07 | ||
| IN202211006599 | 2022-02-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023148783A1 true WO2023148783A1 (en) | 2023-08-10 |
Family
ID=87553285
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2023/050123 Ceased WO2023148783A1 (en) | 2022-02-07 | 2023-02-07 | Process for the preparation of nafamostat, camostat and their derivatives |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20250188022A1 (en) |
| EP (1) | EP4475835A4 (en) |
| JP (1) | JP2025505208A (en) |
| WO (1) | WO2023148783A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117986161A (en) * | 2024-02-01 | 2024-05-07 | 华仁医学研究(安徽)有限公司 | A method for synthesizing high-purity anticoagulant drug |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09309873A (en) * | 1996-05-22 | 1997-12-02 | Jiyunsei Kagaku Kk | New production method of camostat mesylate |
| KR102314436B1 (en) * | 2021-01-27 | 2021-10-19 | (주)국전약품 | Process for Preparing Nafamostat Mesilate and Intermediate Thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU527371B2 (en) * | 1980-09-16 | 1983-03-03 | Torii & Co., Ltd. | Amidine |
| CN111574409A (en) * | 2020-05-14 | 2020-08-25 | 河北省医疗器械与药品包装材料检验研究院(河北省医疗器械技术审评中心) | A kind of recrystallization process method of nafamostat mesylate |
-
2023
- 2023-02-07 WO PCT/IN2023/050123 patent/WO2023148783A1/en not_active Ceased
- 2023-02-07 JP JP2024547007A patent/JP2025505208A/en active Pending
- 2023-02-07 US US18/836,241 patent/US20250188022A1/en active Pending
- 2023-02-07 EP EP23749463.8A patent/EP4475835A4/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09309873A (en) * | 1996-05-22 | 1997-12-02 | Jiyunsei Kagaku Kk | New production method of camostat mesylate |
| KR102314436B1 (en) * | 2021-01-27 | 2021-10-19 | (주)국전약품 | Process for Preparing Nafamostat Mesilate and Intermediate Thereof |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP4475835A4 * |
| VENKATARAMAN K.; WAGLE D.R.: "Cyanuric chloride : a useful reagent for converting carboxylic acids into chlorides, esters, amides and peptides", TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM , NL, vol. 20, no. 32, 1 January 1900 (1900-01-01), Amsterdam , NL , pages 3037 - 3040, XP085663861, ISSN: 0040-4039, DOI: 10.1016/S0040-4039(00)71006-9 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117986161A (en) * | 2024-02-01 | 2024-05-07 | 华仁医学研究(安徽)有限公司 | A method for synthesizing high-purity anticoagulant drug |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2025505208A (en) | 2025-02-21 |
| EP4475835A4 (en) | 2026-01-14 |
| US20250188022A1 (en) | 2025-06-12 |
| EP4475835A1 (en) | 2024-12-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6811717B2 (en) | Methods for the preparation of topiroxostat and its intermediates | |
| TWI580675B (en) | Preparation of 4- [5- (pyridin-4-yl) -1H-1,2,4-triazol-3-yl] pyridine-2-carbonitrile and intermediates | |
| US7868207B2 (en) | Process for producing 1-(3,4-dichlorobenzyl)-5-octylbiguanide or a salt thereof | |
| WO2016008461A1 (en) | A new form of sofosbuvir and a method of its preparation | |
| WO2023148783A1 (en) | Process for the preparation of nafamostat, camostat and their derivatives | |
| CN113968815B (en) | A method for synthesizing a thiazaspirocarboxylic acid compound, its intermediate, and the method for synthesizing it. | |
| CN115745874A (en) | Preparation method of o-pyrrolidinyl benzamide compound | |
| CN1251574A (en) | Process for preparing O-(3-amino-2-hydroxy-propyl)-hydroxymic acid halides | |
| RU2193034C2 (en) | Method of synthesis of derivatives of 2-aminothiazole carboxamide | |
| CN115850258B (en) | A synthetic method of masitinib | |
| RU2852723C2 (en) | Method for obtaining jactinibe dihydrochloride monohydrate | |
| RU2825395C1 (en) | Method of producing 2-(1,2,3-thiadiazol-4-yl)benzylamine hydrochloride | |
| US4285878A (en) | N-Phenyl-N'-cyano-O-phenylisoureas | |
| JP5234856B2 (en) | Crystal of compound having NPYY5 receptor antagonistic action | |
| SU1657496A1 (en) | Method for obtaining amide of 4-hydroxychinolone-2-carboxylic-3-acid | |
| JP2025034981A (en) | Method for producing intermediate for the production of cyclaniliprole | |
| SU940471A1 (en) | Process for preparing 4-haloalkyl derivatives of 3-imidazoline-3-oxide | |
| US4321393A (en) | Method for preparing 2-cyanamidobenzimidazoles or 2-cyanamidobenzoxazoles | |
| EP0259140B1 (en) | Cyanoguanidine derivative and process for preparation thereof | |
| CN113979887A (en) | Synthetic method of aromatic amine carboxylic acid derivative | |
| WO2014158641A1 (en) | Preparation of haloalkoxyarylhydrazines and intermediates therefrom | |
| EP1930324A1 (en) | Process for the preparation of atazanavir | |
| WO2000029369A1 (en) | Aminoacrylic acid derivatives and process for producing the same | |
| CN121511233A (en) | A DPP1 inhibitor intermediate, its preparation method, and its pharmaceutical applications | |
| SU977453A1 (en) | Process for producing 1,2-hydroxilaminooximes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23749463 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18836241 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024547007 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023749463 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023749463 Country of ref document: EP Effective date: 20240909 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18836241 Country of ref document: US |