WO2023190663A1 - 高純度化合物の製造方法および精製方法 - Google Patents
高純度化合物の製造方法および精製方法 Download PDFInfo
- Publication number
- WO2023190663A1 WO2023190663A1 PCT/JP2023/012761 JP2023012761W WO2023190663A1 WO 2023190663 A1 WO2023190663 A1 WO 2023190663A1 JP 2023012761 W JP2023012761 W JP 2023012761W WO 2023190663 A1 WO2023190663 A1 WO 2023190663A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dihydro
- pyrido
- salt
- benzodiazepin
- diethylamino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/10—Ophthalmic agents for accommodation disorders, e.g. myopia
Definitions
- the present invention provides highly purified (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][ 1,4] benzodiazepin-6-one (hereinafter sometimes referred to as "AFDX0250”) or a salt thereof.
- the present invention includes highly purified AFDX0250 or a salt thereof, and 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b ] [1,4] Benzodiazepine-6-one (hereinafter also referred to as "Compound 1”) and the like, relates to a pharmaceutical composition that is substantially free of impurities.
- AFDX0250 is (+)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4] It is a benzodiazepine-6-one, has the structure shown below, and is known to be useful for treating eye diseases such as glaucoma, ocular hypertension, and myopia (Patent Document 1 and Patent Document 2).
- Non-Patent Document 1 describes that 11-(chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1, 4] It is disclosed that AFDX0250 is produced by reacting benzodiazepin-6-one with (-)-2-[(diethylamino)methyl]piperidine.
- Patent Document 3 11-(chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4 ]
- a method for producing AFDX0250 by reacting benzodiazepin-6-one with (-)-2-[(diethylamino)methyl]piperidine is disclosed, but the yield is shown to be as low as 59%. .
- AFDX0250 In order to manufacture and sell AFDX0250 as a pharmaceutical product, it is necessary to produce AFDX0250 in high yield and purify it effectively, and also to remove impurities from the crude product to prevent undesirable effects from occurring. It is important to manufacture with high purity.
- Non-Patent Document 1 and Patent Document 3 disclose methods for producing AFDX0250, but show that the yield of the produced AFDX0250 is low. Therefore, there was room for improvement in the conventional manufacturing method of AFDX0250 in order to use it as a pharmaceutical product.
- the present invention provides a high-yield method that does not contain impurities, especially 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one.
- Another object of the present invention is to provide an effective method for producing and/or purifying highly pure AFDX0250 or a salt thereof.
- Another object of the present invention is to provide a high yield and high purity AFDX0250 or a salt thereof that is substantially free of impurities, and a highly safe pharmaceutical composition containing the high purity AFDX0250.
- the present invention provides the following.
- the organic solvent comprising: A manufacturing method comprising 15 to 50 mL of acetonitrile per gram of 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt. .
- the organic solvent is 15 to 15% per gram of 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt. 50 mL of acetonitrile, the manufacturing method according to item 1.
- the production method according to Item 1 or 2 which includes the step of adjusting the pH by adding water and an acid to the reaction mixture, and then extracting with an organic solvent.
- the production method according to Item 3 wherein the extraction with an organic solvent is performed two or more times.
- the organic solvent is ethyl acetate.
- [Item 13] The pharmaceutical composition according to Item 12, wherein the content of impurities is less than 1%.
- (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4 ] A pharmaceutical composition comprising benzodiazepine-6-one or a salt thereof, comprising 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepine- A pharmaceutical composition substantially free of 6-one or a salt thereof.
- [Item 16] The medicament according to Item 14 or 15, further comprising substantially no 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof.
- Composition [Item 17] The pharmaceutical composition according to Item 16, wherein the content of 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof is less than 1000 ppm. thing.
- the present invention provides the following.
- [Item 18] The pharmaceutical composition according to any one of Items 12 to 17, which is an eye drop.
- [Item 19] The pharmaceutical composition according to any one of Items 12 to 18 for treating glaucoma, ocular hypertension, or myopia.
- -5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof can be produced.
- the content of impurities such as 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one can be suppressed.
- the present invention provides highly purified (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b ][1,4]Benzodiazepin-6-one or its salt, and high purity (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-
- a pharmaceutical composition comprising dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof can be provided.
- the present invention provides (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4 ]
- Provides a method for producing benzodiazepine-6-one or a salt thereof, 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepine -6-one or a salt thereof and (R)-2-(diethylamino)methylpiperidine or a salt thereof are reacted in an organic solvent containing acetonitrile, preferably acetonitrile, in the presence of an alkali metal iodide under basic conditions.
- 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof, (R)-2-( Diethylamino)methylpiperidine or a salt thereof, an organic solvent containing acetonitrile, preferably acetonitrile, an alkali metal iodide, and a base can be mixed, heated and stirred, and then concentrated to obtain a reaction mixture.
- the "organic solvent” in this step includes acetonitrile, and preferably only acetonitrile.
- the amount of acetonitrile added is 15v/w to 50v/w, preferably 15v/w to 40v/w, more preferably 15v/w to 30v/w, even more preferably 18v/w to 25v/w. w, most preferably 20v/w.
- v/w of the amount of acetonitrile added is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the volume (mL) of acetonitrile per mass (1 g). The same applies hereinafter unless otherwise specified.
- alkali metal iodide examples include sodium iodide, potassium iodide, calcium iodide, lithium iodide, and the like, with sodium iodide being preferred. Further, the alkali metal iodides may be used alone or two or more thereof may be used in any combination and ratio.
- the amount of alkali metal iodide added is, for example, 0.01 to 2 equivalents, preferably 0.05 to 1.5 equivalents, more preferably 0.1 to 1 equivalent, and even more preferably 0. .15 to 0.5 equivalent, particularly preferably 0.2 to 0.4 equivalent, most preferably 0.25 equivalent.
- the "equivalent" of the amount of alkali metal iodide added is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepine-6- It means the amount (mol) of alkali metal iodide relative to the amount (1 mol) of iodide. The same applies hereinafter unless otherwise specified.
- the “basic conditions” in this step include, for example, lithium carbonate, sodium carbonate, cesium carbonate, potassium carbonate, lithium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, lithium hydride, sodium hydride, potassium hydride, water Lithium oxide, sodium hydroxide, potassium hydroxide, lithium tert-butoxide, sodium tert-butoxide, potassium tert-butoxide, alkyl lithium (e.g.
- the basic conditions include basic conditions using sodium bicarbonate, triethylamine or diisopropylethylamine.
- the bases may be used alone, or two or more bases may be used in any combination and ratio.
- the amount of the base added is, for example, 1 to 20 equivalents, preferably 1.5 to 15 equivalents, more preferably 2 to 10 equivalents, still more preferably 2.5 to 8 equivalents, and particularly preferably 3 to 5 equivalents, most preferably 3.15 equivalents.
- the "equivalent" of the amount of base added refers to the amount of 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the amount (mol) of the base relative to (1 mol). The same applies hereinafter unless otherwise specified.
- the reaction temperature in this step is, for example, 40 to 100°C, preferably 50 to 90°C, more preferably 60 to 80°C, still more preferably 65 to 78°C, and most preferably 70 to 75°C. It is °C.
- the reaction time in this step is not particularly limited as it varies depending on conditions such as reaction temperature, but is appropriately selected, for example, from 1 to 48 hours, preferably from 3 to 36 hours, more preferably from 6 to 30 hours.
- the time is more preferably 12 to 27 hours, particularly preferably 18 to 24 hours, and most preferably 20 hours.
- the reaction mixture obtained in the above step is, for example, 3 to 12 v/w, preferably 4 to 10 v/w, more preferably 5 to 8 v/w, most preferably 6 to 7 v/w.
- Concentrate, add water and acid to give (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3- b] [1,4]Benzodiazepin-6-one or a salt thereof is adjusted to a pH that allows it to be dissolved in water, and then extracted with an organic solvent.
- water is added to the reaction mixture, and then an acid is added to adjust the pH to acidic, for example, 3.0 or less, more preferably 2.0 or less, most preferably 1.0 or less, one or more times.
- the extraction is carried out with an organic solvent, preferably two or more times, more preferably three or more times, and most preferably four or more times.
- "v/w" of the content of the reaction mixture is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the volume (mL) of the reaction mixture relative to the mass (1 g).
- the amount of water added in this step is, for example, 3 to 50 v/w, preferably 5 to 30 v/w, more preferably 7 to 20 v/w, even more preferably 8 to 15 v/w, Most preferably it is 10v/w.
- v/w of the amount of water added is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the volume of water (mL) relative to mass (1 g). The same applies hereinafter unless otherwise specified.
- the acid in this step includes (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][ 1,4] benzodiazepine-6-one or its salt can be dissolved in water without any particular limitation, examples include hydrochloric acid, sulfuric acid, nitric acid, etc., and hydrochloric acid is preferred.
- the amount of acid added is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1 , 4] is not particularly limited as long as the amount can be adjusted to a pH that allows benzodiazepine-6-one or its salt to be dissolved in water.
- Examples of the organic solvent in this step include chloroform, dichloromethane, diethyl ether, tetrahydrofuran (THF), ethyl acetate, hexane, and toluene, with ethyl acetate being preferred.
- the amount of the organic solvent added is, for example, 5v/w to 50v/w, preferably 10v/w to 40v/w, and most preferably 10v/w.
- "v/w" of the amount of organic solvent added is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the volume (mL) of organic solvent per mass (1 g) of .
- the temperature when adding water and acid in this step is, for example, -10 to 30°C, preferably -5 to 20°C, more preferably 0 to 15°C, and most preferably 0 to 10°C.
- the manufacturing method of the present invention includes the step of adding a solvent to the organic layer obtained in the above step and crystallizing it.
- a solvent is added to the organic layer, and then a base is added to adjust the pH to alkaline, preferably 8 to 12, more preferably 9 to 11, and stirring is performed to (R)-11-[2 -[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt is crystallized can be done.
- Examples of the solvent in this step include water, a mixture of methanol and water, a mixture of ethanol and water, a mixture of acetone and water, a mixture of methyl ethyl ketone and water, a mixture of acetonitrile and water, N,N- Examples include a mixture of dimethylformamide and water, and preferably a mixture of acetonitrile and water.
- the volume ratio of acetonitrile to water is, for example, 1:1 to 1:10, preferably 1:2 to 1:6, most preferably 1:3 to It's 1:5.
- the amount of solvent added is, for example, 5 to 20 v/w, preferably 8 to 16 v/w, and more preferably 10 to 14 v/w.
- "v/w" of the amount of solvent added is 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one.
- the base in this step is not particularly limited as long as it is a base that can adjust the pH to alkaline, preferably 8 to 12, more preferably 9 to 11, but examples include lithium carbonate, sodium carbonate, potassium carbonate, sodium hydroxide, Examples include potassium hydroxide, lithium hydroxide, etc., and potassium hydroxide is preferred.
- the amount of base added is not particularly limited as long as it can adjust the pH to alkaline, preferably 8 to 12, more preferably 9 to 11.
- the temperature during stirring in this step is, for example, -10 to 20°C, preferably -5 to 15°C, and most preferably 0 to 10°C.
- the stirring time in this step is not particularly limited as it varies depending on reaction conditions such as temperature, but is appropriately selected, for example, from 1 to 24 hours, preferably from 1 to 12 hours, more preferably from 2 to 8 hours, most preferably. is 3 to 5 hours.
- the drying temperature in this step is not particularly limited as long as the adhering moisture or solution can be removed, but is, for example, 30 to 100°C, preferably about 50°C.
- the present invention also provides 11-(2-iodoacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof and (R)-2- (R)-11-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl] comprising a step of reacting with (diethylamino)methylpiperidine or a salt thereof in an organic solvent under basic conditions.
- the present invention provides a method for producing -5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof.
- Examples of the "organic solvent” in this production method include acetonitrile, dimethylformamide (DMF), dimethylacetamide (DMA), dimethyl sulfoxide (DMSO), and ethyl acetate.
- the amount of the organic solvent added is, for example, 3v/w to 50v/w, preferably 5v/w to 30v/w, more preferably 7v/w to 20v/w, even more preferably 8v/w to 15v/w, most preferably 10v/w.
- v/w of the amount of organic solvent added is 11-(2-iodoacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the volume (mL) of organic solvent per mass (1 g) of .
- the "basic conditions" in this production method include, for example, sodium carbonate, cesium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, sodium hydride, potassium hydride, sodium hydroxide, potassium hydroxide, sodium tert- butoxide, potassium tert-butoxide, alkyllithium (e.g. n-butyllithium, sec-butyllithium, tert-butyllithium, n-hexyllithium), lithium amide (e.g.
- the bases may be used alone, or two or more bases may be used in any combination and ratio.
- the amount of base added is 1 to 20 equivalents, preferably 1.5 to 15 equivalents, more preferably 2 to 10 equivalents, still more preferably 2.5 to 8 equivalents, particularly preferably 3 equivalents. ⁇ 5 equivalents, most preferably 3.15 equivalents.
- the “equivalent” of the amount of base added refers to the amount of 11-(2-iodoacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one. It means the amount (mol) of the base relative to (1 mol).
- the reaction temperature in this production method is, for example, 40 to 100°C, preferably 50 to 90°C, more preferably 60 to 80°C, still more preferably 65 to 78°C, and most preferably 70 to 78°C.
- the temperature is 75°C.
- the reaction time in this production method is not particularly limited as it varies depending on conditions such as reaction temperature, but is appropriately selected, for example, from 1 to 48 hours, preferably from 3 to 36 hours, more preferably from 6 to 30 hours.
- the time period is more preferably 12 to 27 hours, particularly preferably 18 to 24 hours, and most preferably 20 hours.
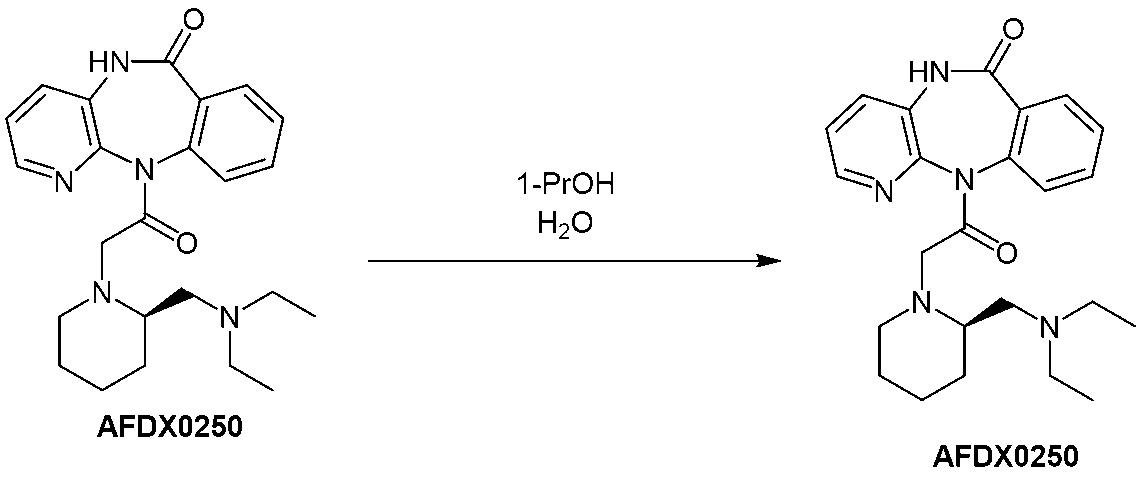
- the present invention relates to the crude product (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H obtained by the production method of the present invention. - crystallizing pyrido[2,3-b][1,4]benzodiazepine-6-one or a salt thereof in a mixture of 1-propanol and water. Purification of 2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt The present invention provides a method.
- the amount of 1-propanol added is, for example, 1 to 10 v/w, preferably 2 to 8 v/w, more preferably 3 to 15 v/w, and most preferably 4 v/w. be.
- the amount of water added is, for example, 1 to 15 v/w, preferably 2 to 10 v/w, more preferably 4 to 8 v/w, and most preferably 6 v/w.
- v/w of the amount of 1-propanol and water added is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro It means the volume (mL) of 1-propanol and water relative to the mass (1 g) of -6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt.
- the temperature of each step in this purification method can be adjusted as appropriate. For example, when stirring in 1-propanol, the mixture is heated to, for example, 80 to 100°C, preferably 85 to 95°C. Thereafter, it is cooled to 40-60°C, preferably 45-55°C, and water is added and stirred. Thereafter, it may be further cooled to -5 to 10°C, preferably 0 to 5°C, for ripening.
- the drying temperature may be any range as long as it can remove the attached moisture or solution, but is, for example, 80°C or lower, preferably about 50°C.
- the time for stirring in 1-propanol in this purification method is not particularly limited, but is appropriately selected, for example, from 1 to 5 hours, preferably from 2 to 4 hours, and most preferably from 3 hours.
- the stirring time after adding water is appropriately selected, for example, from 10 minutes to 5 hours, preferably from 20 minutes to 4 hours, and most preferably from 30 minutes to 3 hours.
- the present invention also provides (R)-11-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro- High purity (R)-11-[2-[2- Provides a method for purifying [(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof It is.
- (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1, 4] A step of treating benzodiazepin-6-one or its salt with activated carbon in an alcoholic solvent and/or a step of recrystallizing it in a mixture of an alcoholic solvent and a ketone solvent.
- the activated carbon treatment step includes a step of mixing AFDX0250 and activated carbon with an alcoholic solvent serving as a good solvent, heating and stirring, filtering through charcoal, and concentrating.
- a ketone solvent as a poor solvent is added to the concentrated mixture, heated and stirred, and cooled to crystallize AFDX0250.
- the precipitated crystals are aged, separated by filtration, washed, and dried.
- high purity AFDX0250 can be obtained.
- the same operation except for the addition of activated carbon and charcoal filtration can be repeated multiple times.
- only the recrystallization step may be performed without performing the activated carbon treatment step.
- Examples of the "alcoholic solvent” in this purification method include methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, and the like, with ethanol and 1-propanol being preferred.
- the amount of alcohol solvent added is, for example, 5 to 50 v/w, preferably 10 to 40 v/w, more preferably 15 to 30 v/w, and most preferably 20 v/w.
- v/w of the amount of alcoholic solvent added is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H- It means the volume (mL) of alcoholic solvent relative to the mass (1 g) of pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt.
- Examples of the "activated carbon" in this purification method include Shirasagi A, Shirasagi P, and the like, with Shirasagi A being preferred.
- the amount of activated carbon added is, for example, 0.01 to 0.3 w/w, preferably 0.03 to 0.2 w/w, and most preferably 0.05 to 0.15 w/w.
- w/w of the amount of activated carbon added is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[ It means the mass (g) of activated carbon relative to the mass (1 g) of 2,3-b][1,4]benzodiazepin-6-one or its salt.
- the mixed solution is concentrated until the alcohol solvent becomes, for example, 0.5 to 3 v/w, preferably 1 to 3 v/w, more preferably 2 to 2.5 v/w.
- a ketone solvent can be added in an amount of, for example, 3 to 15 v/w, preferably 5 to 10 v/w, more preferably 7 to 8 v/w, thereby reducing the concentration of alcohol and ketone solvents. It becomes a mixed liquid.
- the recrystallization solvent is a mixture of an alcohol solvent and a ketone solvent, and the amount of alcohol solvent added is, for example, 0.5 -3v/w, preferably 1-3v/w, more preferably 2-2.5v/w, and the amount of ketone solvent added is, for example, 3-15v/w, preferably 5-10v/w, more Preferably it is 7 to 8 v/w.
- the content of the alcoholic solvent in the mixed solution and the amount of alcoholic solvent and ketone solvent added (v/w) are (R)-11-[2-[2-[(diethylamino)methyl]-1 -piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt by mass (1 g), alcoholic solvent, and ketone Means the volume of solvent (mL).
- ketone solvent examples include acetone, methyl ethyl ketone (MEK), methyl isobutyl ketone (MIBK), diethyl ketone, etc., with acetone, MEK, and MIBK being preferred.
- the temperature of each step in this purification method can be adjusted as appropriate.
- the activated carbon treatment step can be carried out by heating and stirring at a temperature of 50 to 90°C, preferably 60 to 80°C, more preferably 65 to 75°C.
- the recrystallization step after crystallization is performed at a temperature of, for example, 50 to 80°C, preferably 60 to 70°C, more preferably 63 to 67°C, stirring is performed as necessary, and the temperature is, for example, -10 to 15°C, preferably It can be carried out by cooling from -5°C to 10°C, more preferably from 0 to 5°C.
- the aging in the recrystallization step may be carried out at, for example, -10 to 15°C, preferably -5 to 10°C, more preferably 0 to 5°C.
- the heating and stirring time in the activated carbon treatment step in this purification method is not particularly limited as it varies depending on the conditions, but is appropriately selected, for example, between 30 minutes and 24 hours, preferably between 40 minutes and 12 hours, and more preferably.
- the time period is 50 minutes to 6 hours, more preferably 1 hour to 3 hours, and most preferably 1 hour.
- Aging in the recrystallization step is, for example, 6 to 48 hours, preferably 12 to 36 hours, and most preferably 24 hours.
- the starting material, intermediate, and/or production compound may be in the form of a salt.
- the production method of the present invention also includes production methods that include their salt forms.
- the salts of these compounds are not particularly limited as long as they are pharmaceutically acceptable salts. Examples of their salts include salts with inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, sulfuric acid, and phosphoric acid; acetic acid, oxalic acid, fumaric acid, maleic acid, succinic acid, and malic acid.
- citric acid tartaric acid, adipic acid, gluconic acid, glucoheptic acid, glucuronic acid, terephthalic acid, methanesulfonic acid, alanine, lactic acid, hippuric acid, 1,2-ethanedisulfonic acid, isethionic acid, lactobionic acid, oleic acid, gallic acid.
- salts with organic acids such as pamoic acid, polygalacturonic acid, stearic acid, tannic acid, trifluoromethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, lauryl sulfate, methyl sulfate, naphthalenesulfonic acid, sulfosalicylic acid;
- organic acids such as pamoic acid, polygalacturonic acid, stearic acid, tannic acid, trifluoromethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, lauryl sulfate, methyl sulfate, naphthalenesulfonic acid, sulfosalicylic acid
- Examples include salts with metals such as sodium, potassium, calcium, and magnesium; salts with inorganic compounds such as ammonia; and salts with organic amines such
- the starting material, intermediate, and/or manufactured compound (AFDX0250) or a salt thereof may be in the form of a hydrate or a solvate.
- impurity means (R)-11-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2 ,3-b][1,4]It is an impurity derived from benzodiazepin-6-one or its salt, and (R)-11-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl] 11-(2-chloroacetyl)-5, which is mixed during the production or purification process of -5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt.
- the purity and impurity content of benzodiazepine-6-one or its salt can be determined by quantifying the peak area ratio in liquid chromatography (HPLC) using the area percentage method, which is a well-known method in the field of analytical chemistry, and by liquid chromatography. It can be measured by quantifying by mass spectrometry (LC/MS).
- ⁇ Mobile phase B Acetonitrile
- ⁇ Dissolution solution A Mobile phase A: Mobile phase B (1:1) (v/v)
- ⁇ Preparation of AFDX0250 standard solution> Weigh accurately about 10 mg of AFDX0250, and add solution A to make exactly 100 mL.
- ⁇ Preparation of sample solution> Prepare the sample solution using the following method.
- Sample solution A Take 100 to 200 mg of the reaction suspension containing AFDX0250 (reaction solution obtained in step 1 of Example 1 below) and dissolve in 10 mL of solution A.
- ⁇ Sample solution B Take about 200 ⁇ L of the reaction suspension containing AFDX0250 (reaction solution obtained in Step 1 of Example 1 below), and place it in a Kiriyama funnel (S-40), Kiriyama filter paper (No. 3), and a suction bottle. Filter under reduced pressure using a filter, and dissolve about 10 mg of the filtered material in 10 mL of solution A.
- - Sample solution C Accurately weigh approximately 10 mg of the obtained solid (AFDX0250 obtained in Step 3 of Example 1 below), and dissolve by adding solution A to make exactly 100 mL.
- Sample solution D Accurately weigh about 100 ⁇ L of the aqueous layer (obtained in step 2 of Example 1 below), and add solution A to make exactly 10 mL.
- -Mobile phase C 10mM ammonium acetate solution Dissolve 0.77g of ammonium acetate in 1000mL of water.
- ⁇ Mobile phase B Acetonitrile
- ⁇ Dissolution solution B Mobile phase C: Mobile phase B (7:3) (v/v)
- ⁇ Compound 1 standard stock solution Accurately weigh about 20 mg of Compound 1, add solution B to dissolve, and make exactly 100 mL. If it is difficult to dissolve, use ultrasound for about 1 minute to dissolve it. Measure 2 mL of this solution accurately and add solution B to make exactly 20 mL. Accurately measure 1 mL of this solution and add solution B to make exactly 20 mL.
- ⁇ Compound 1 standard solution 1 Accurately measure 4 mL of the standard stock solution, and add solution B to make exactly 20 mL.
- - Compound 1 standard solution 2 Accurately measure 2 mL of standard solution 1, and add solution B to make exactly 10 mL.
- - Compound 1 standard solution 3 Accurately measure 2 mL of standard solution 2, and add solution B to make exactly 20 mL.
- ⁇ Sample solution E (when the measurement target is an aqueous layer): Calculated from the AFDX0250 content (%) of the aqueous layer obtained according to the measurement method in (1) above, and the aqueous layer was adjusted to contain approximately 100 mg of AFDX0250. Measure accurately and add solution B to make exactly 25 mL.
- ⁇ Sample solution F (when the measurement target is a crystal): Accurately weigh about 100 mg of AFDX0250, add 5 mL of solution B and 0.2 mL of diluted hydrochloric acid to dissolve, and add solution B to make exactly 25 mL (AFDX0250 concentration: approximately 4 mg/mL). If it is difficult to dissolve, use ultrasound to dissolve it.
- the present invention also provides (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1, 4]
- the pharmaceutical composition of the present invention has (R)-11-[2-[2-[ (diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof.
- the pharmaceutical composition of the present invention substantially contains 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof.
- it does not contain 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof, and it is even more preferable that it does not substantially contain 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof.
- substantially free means 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one, etc.
- impurities cannot be detected by methods commonly used in pharmaceutical analysis, such as liquid column chromatography using a chiral column (detector: ultraviolet absorption photometer), or its effects and side effects.
- the content of impurities in the pharmaceutical composition is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2 ,3-b][1,4]benzodiazepin-6-one or its salt, less than 5%, preferably less than 3%, more preferably less than 1%, most preferably 0.5 less than %.
- the content of 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof in the pharmaceutical composition is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepine -6-one or a salt thereof, for example less than 1000 ppm, preferably less than 700 ppm, more preferably less than 500 ppm, even more preferably less than 300 ppm, even more preferably less than 100 ppm, especially Preferably it is less than 10 ppm, most preferably less than 2 ppm.
- the content of 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof in the pharmaceutical composition is determined as follows: (R)-11-[ For 2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salts For example, it is less than 1000 ppm, preferably less than 800 ppm, more preferably less than 600 ppm, and most preferably less than 500 ppm.
- the total amount of -6H-pyrido[2,3-b][1,4]benzodiazepin-6-one is (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl ]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or its salt, for example, less than 2000 ppm, preferably less than 1000 ppm, more preferably is less than 700 ppm, most preferably less than 500 ppm.
- composition of the present invention unless otherwise specified, (R)-11-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[ Although it may contain a pharmaceutically acceptable active ingredient other than 2,3-b][1,4]benzodiazepine-6-one or a salt thereof, in one embodiment, (R)-11-[2-[ 2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof. Contains no acceptable active ingredients.
- Additives can be used in the pharmaceutical composition of the present invention as necessary, and additives include surfactants, isotonic agents, stabilizers, preservatives, antioxidants, and high molecular weight polymers. , a pH adjuster, a base, etc. can be added.
- the pharmaceutical composition of the present invention is preferably an aqueous pharmaceutical composition, but may be a non-aqueous pharmaceutical composition.
- the pharmaceutical composition of the present invention can be administered orally or parenterally.
- Administration routes include oral administration, intravenous administration, transdermal administration, topical ocular administration (e.g., eye drops, eye ointment, intraconjunctival sac administration, intravitreal administration, subconjunctival administration, sub-Tenon's administration, intraconjunctival sac insertion, eyelid application), etc.
- the dosage form of the pharmaceutical composition of the present invention is not particularly limited as long as it can be used as a pharmaceutical, and examples thereof include eye drops, eye gels, injections, and the like.
- the pharmaceutical composition of the present invention is particularly preferably used as eye drops.
- the pharmaceutical composition of the present invention when used as a non-aqueous pharmaceutical composition, it may be formulated as a non-aqueous preparation. These can be manufactured according to conventional methods in the art.
- the desired eye drops can be prepared by adding the active ingredient to a medium such as purified water or a buffer solution, stirring, and then adjusting the pH with a pH adjuster.
- a medium such as purified water or a buffer solution
- additives commonly used in eye drops may be used as needed. Examples of additives include tonicity agents, buffering agents, surfactants, stabilizers, preservatives, solubilizing agents, and the like.
- fillers, lubricants, binders, disintegrants, coating agents, film agents, etc. can be added as necessary.
- Extending agents include lactose, crystalline cellulose, starch, vegetable oil, etc.
- lubricants include magnesium stearate, talc, etc.
- binders include hydroxypropylcellulose, polyvinylpyrrolidone, etc.
- disintegrants include carboxymethyl cellulose calcium, low-substituted hydroxypropyl methyl cellulose, etc.
- Coating agents include hydroxypropyl methyl cellulose, macrogol (polyethylene glycol), silicone resins, etc.
- Film agents include gelatin film, etc. can be mentioned.
- the dosage of the pharmaceutical composition of the present invention can be changed as appropriate depending on the dosage form, patient's symptoms, age, body weight, age at onset of disease, physician's judgment, etc.
- the pharmaceutical composition of the present invention is administered topically to the eye as an eye drop, there is no particular restriction on the dosage as long as it is sufficient to exert the desired medicinal effect, but one drop to several drops should be administered once a day.
- the eye drops can be applied several times to several times (eg, 1 to 4 times, 1 to 6 times, 1 to 8 times). It can also be used when wearing contact lenses.
- the pharmaceutical composition of the present invention can be used to treat eye diseases such as glaucoma, ocular hypertension, and myopia.
- HPLC (UHPLC) analysis and LC/MS analysis were performed under the following conditions.
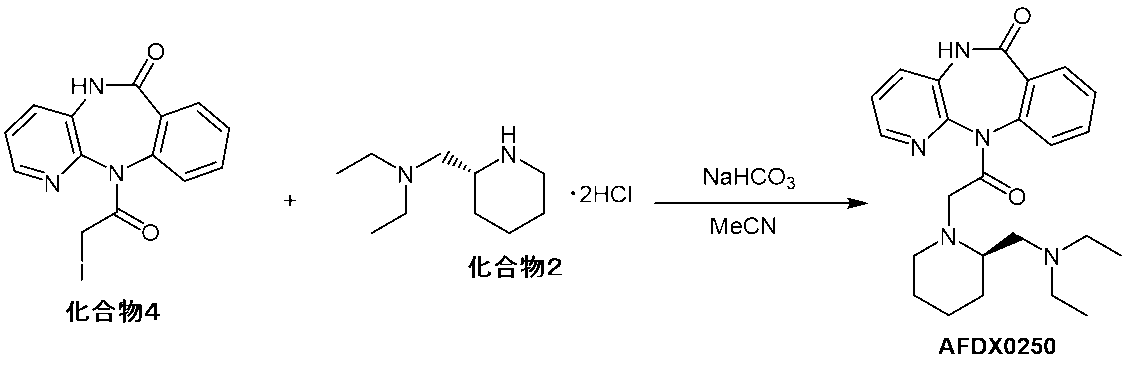
- Example 1 Production of AFDX0250 (1) According to the following steps, (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro-6H-pyrido[2,3-b][1 , 4] benzodiazepin-6-one (AFDX0250) was produced.
- Process 1 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (compound 1) (200 g, 0 .695 mol) in acetonitrile solution (3200 g), (R)-2-(diethylamino)methylpiperidine dihydrochloride (hereinafter sometimes referred to as "Compound 2"; 178 g, 0.732 mol), sodium iodide (26 .0 g, 0.174 mol) and sodium hydrogen carbonate (184 g, 2.19 mol) were added and heated at an internal temperature of 73.0 to 74.5°C.
- Process 2 After allowing the reaction to proceed for 5 hours in step 1, the reaction solution was cooled and concentrated to obtain crude product AFDX0250 (1231.56 g). Water (1800 g) was added, concentrated hydrochloric acid (155 mL) was added dropwise at an internal temperature of 4.0 to 5.5°C until the pH became 1 or less (pH 0.8), and the liquid was separated three times with ethyl acetate (1800 g). . The aqueous layer of each liquid separation was measured by liquid chromatography (UHPLC), and AFDX0250 and Compound 3 were quantified by area percentage method. In addition, the content of Compound 1 was measured by liquid chromatography mass spectrometry (LC/MS).
- LC/MS liquid chromatography mass spectrometry
- Process 3 Water (1400 g) and acetonitrile (780 g) were added to the aqueous layer obtained in step 2, and the mixture was stirred at an internal temperature of 2.0 to 5.0°C, and then a 3.25N aqueous potassium hydroxide solution was added until the pH was 11. It was added dropwise until the amount was below, and stirred for 5 hours.
- the precipitated solid was filtered under reduced pressure, and the filtered product was washed with water (2000 g) and then dried under reduced pressure at 50° C. to obtain crude AFDX0250 (274.35 g, yield 93.3%) as a pale brown solid.
- the obtained pale brown solid was measured by liquid chromatography (UHPLC), and AFDX0250 and Compound 3 were quantified by area percentage method.
- the content of Compound 1 was measured by liquid chromatography mass spectrometry (LC/MS).
- the content of AFDX0250 and compound 3 in the reaction solution obtained in step 1, the aqueous layer obtained in step 2, and the solid obtained in step 3, and the content of compound 1 in the reaction solution obtained in step 1 The content was measured according to the method for measuring the content of (1) AFDX0250 or its salt in this specification. Further, the content of Compound 1 contained in AFDX0250 in the aqueous layer obtained in Step 2 and the solid obtained in Step 3 was measured according to (2) Method for measuring Compound 1 content in this specification.
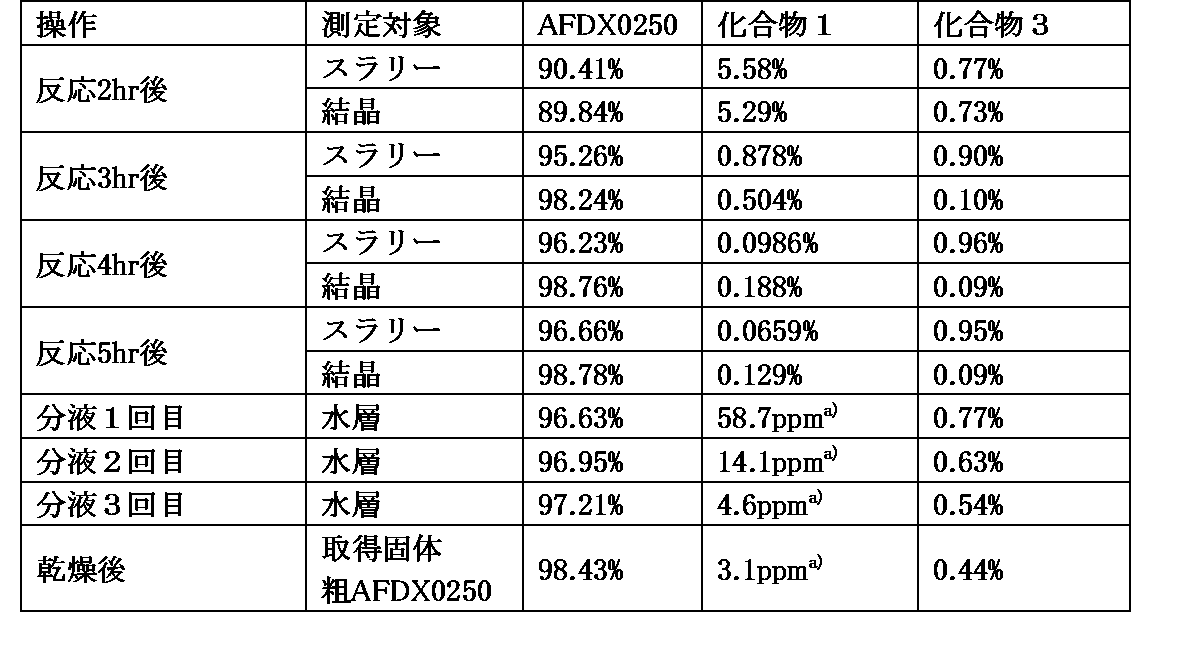
- Table 7 shows the results of UHPLC analysis and LC/MS analysis of the reaction solution and sample solution in each step.
- Table 7 shows the contents of AFDX0250, Compound 1 and Compound 3. Note that a) in the table indicates the content of Compound 1 measured by liquid chromatography mass spectrometry.
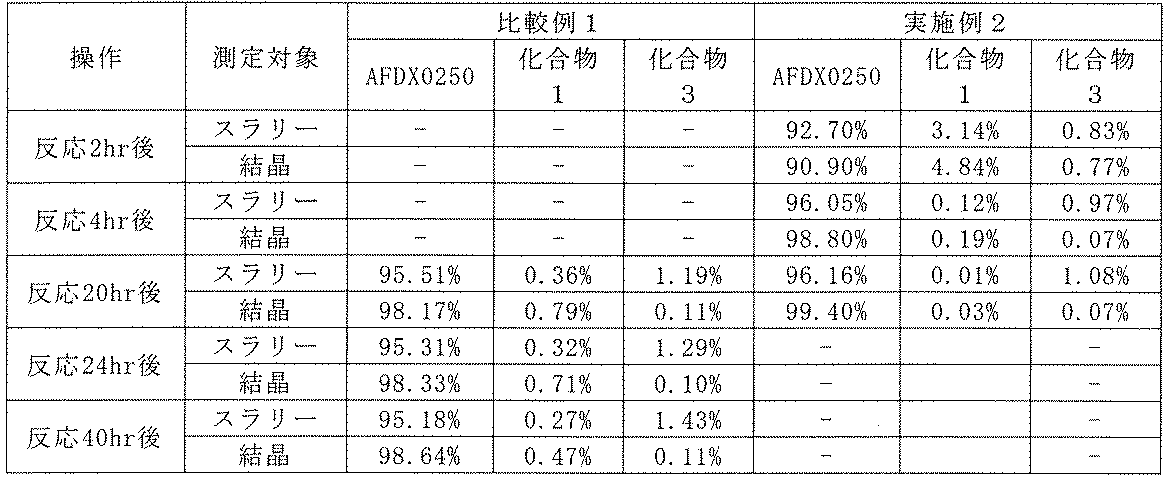
- Example 2 Quantitative analysis of AFDX0250 produced by the production method of the present invention and known production methods. Quantitative analysis was performed on AFDX0250 produced using 10 mL of acetonitrile per 1 g of compound 1, and the area percentages of AFDX0250 and impurities were compared. Specifically, AFDX0250 was manufactured by changing the batch amount of Compound 1 from 200 g to 95 g, and changing the amount of Compound 2 and the reagent (equivalent amount per 1 substance amount of Compound 1) in the same manner as in Example 1. (Example 2).
- AFDX0250 was produced in the same manner as in Example 2, except that 10 mL of acetonitrile was used instead of 20 mL of acetonitrile per 1 g of Compound 1 (Comparative Example 1).
- a test solution was prepared from the obtained reaction solution in the same manner as in Example 1. UHPLC analysis was performed on each test solution.
- Table 8 shows the results of UHPLC analysis of AFDX0250 obtained by the production method of the present invention (Example 2) and the known production method (Comparative Example 1).
- Example 3 Production of AFDX0250 (2) 11-(2-iodoacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one (hereinafter referred to as "compound 4”) at room temperature under a nitrogen atmosphere.
- Compound 2 (135 g, 5.53 mol) and sodium hydrogen carbonate (140 g, 16.6 mol) were added to an acetonitrile solution (40.0 mL) of (2.00 g, 5.27 mol), and the internal temperature was 73. Heated at 0°C for 23 hours. The reaction solution was filtered under reduced pressure, and the filtered material was washed with acetonitrile (2.0 mL) five times.
- Example 4 Purification of AFDX0250 (1) A solution of the crude AFDX0250 (250 g, 0.593 mol) obtained in Example 1 in 1-propanol (800 g) was heated and stirred at an internal temperature of 89.7 to 90.1°C for 3 hours at room temperature under a nitrogen atmosphere. , cooled to an internal temperature of 50.1°C at a cooling rate of 0.5°C/min, added water (1500 g) over 30 minutes at an internal temperature of 48.1 to 50.9°C, and then .Stirred at 9°C for 30 minutes.
- Example 5 Purification of AFDX0250 (2)
- color which is one of the items that indicate the properties of drug substances, needs to be controlled during the manufacturing process of drug substances. Therefore, crude AFDX0250 was heated and dissolved in an organic solvent, activated carbon was added and stirred, and the activated carbon was removed and decolorized by filtration to remove colored components and AFDX0250 was purified.
- Shirasagi A (0.05w/w, 0.10w/w and 0.15w/w) and Shirasagi P (0.10w/w and 0.15w/w) were used as activated carbon, and organic
- Each sample was prepared using ethanol (EtOH), 1-propanol (1-PrOH) and N-methylpyrrolidone (NMP) as solvents. Note that, regarding samples containing NMP as a solvent, those using Shirasagi A (0.15 w/w) and Shirasagi P (0.10 w/w and 0.15 w/w) were not prepared.
- the amount of ethanol and 1-propanol added was 20 mL per 1 g of crude AFDX0250, and the stirring temperature was 70°C.
- the amount of NMP added was 2.5 mL per 1 g of crude AFDX0250, and the stirring temperature was 70°C.
- the properties of samples containing ethanol, 1-propanol, and NMP were checked before and after the activated carbon treatment. Furthermore, HPLC analysis was performed on each sample at each time point (before addition of activated carbon, 1 hour after addition of activated carbon, and 3 hours after addition of activated carbon).
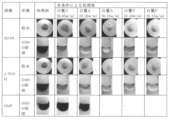
- Figure 1 shows the properties of each sample (powder and DMSO solution) before and after activated carbon treatment.
- each sample containing each organic solvent was filtered under reduced pressure, the filtrate was concentrated under reduced pressure, and the obtained wet solid was dried under reduced pressure at 50 ° C. to confirm its properties.
- DMSO solution the obtained solid was It was dissolved in 10% dimethyl sulfoxide (DMSO) to form a solution and its properties were confirmed.
- EtOH ethanol
- samples containing 1-propanol (1-PrOH) samples containing NMP are shown in Tables 10 to 12.
- Example 6 Purification of AFDX0250 (3) Activated carbon Shirasagi A (30.0 g) was added to a solution of crude AFDX0250 (200 g, 0.474 mol) purified in Example 4 in ethanol (3200 g) at room temperature under a nitrogen atmosphere, and the internal temperature was 69.0. After heating and stirring at ⁇ 72.4°C for 1 hour, the mixture was filtered under reduced pressure using a filtration device filled with a filter aid, and washed with ethanol (320 g) previously heated to 70°C. The filtrate was concentrated under reduced pressure to obtain a concentrated residue (989.04 g).
- the recrystallization process was performed using a combination of the alcohol used in the activated carbon treatment and a poor solvent.
- MEK methyl ethyl ketone
- MIBK methyl isobutyl ketone
- n-heptane n-heptane
- each dried product and DMSO solution obtained in the recrystallization step using ethanol and 1-propanol as alcoholic solvents and the properties of the dried product and DMSO solution before purification are shown in FIGS. 2 and 3, respectively. Further, the results and yields of HPLC analysis of each dried product obtained in the recrystallization step using ethanol and 1-propanol as alcoholic solvents and the dried product before purification are shown in Tables 13 and 14, respectively.
- Example 7 Purification of AFDX0250 (4) Methyl isobutyl ketone (MIBK) (640 g) was added to a solution of AFDX0250 treated with activated carbon in Example 6 in ethanol and concentrated. The mass volume percent of ethanol and MIBK with respect to AFDX0250 was calculated by 1 H-NMR measurement, and the amount of ethanol was adjusted so that the total amount of ethanol was 2.5 v/w. The reaction mixture was then heated to 63-67°C and stirred for 30 minutes. MIBK was added dropwise over 48 minutes to a total of 7.5 v/w, and the mixture was stirred at 63 to 67°C for 1 hour.
- MIBK Methyl isobutyl ketone
- a test solution of the obtained AFDX0250 was prepared according to (1) Method for measuring the content of AFDX0250 or its salt in this specification, and HPLC analysis was performed on the test solution.
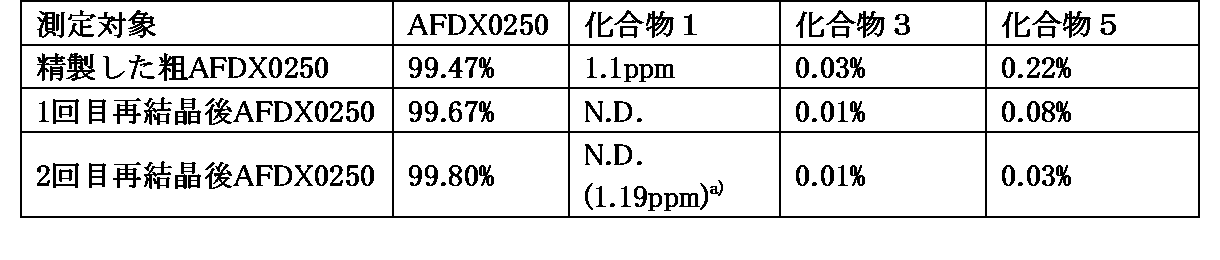
- the obtained AFDX0250 was dissolved in ethanol, and the solution was recrystallized a second time in the same manner as the first recrystallization.
- the obtained wet solid AFDX0250 was dried under reduced pressure at 90° C. for 14 hours and 35 minutes to obtain AFDX0250 (152.27 g, yield 76.1%).
- the obtained AFDX0250 is obtained by preparing a test solution according to (1) method for measuring the content of AFDX0250 or its salt in this specification, and HPLC analysis was performed.
- Table 15 shows the results of HPLC analysis of AFDX0250 obtained in the first and second recrystallizations.
- N.D. in the table means “not detected”, and a) indicates the content of Compound 1 measured by liquid chromatography mass spectrometry.
- (R)-11-[2-[2-[(diethylamino)methyl]-1-piperidinyl]acetyl]-5,11-dihydro -6H-pyrido[2,3-b][1,4]benzodiazepin-6-one or a salt thereof can be produced. Furthermore, the content of impurities such as 11-(2-chloroacetyl)-5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one can be suppressed.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description

[項1] 11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩と(R)-2-(ジエチルアミノ)メチルピペリジンまたはその塩とを有機溶媒中、アルカリ金属のヨウ化物の存在下、塩基性条件下で反応させる工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の製造方法であって、該有機溶媒が、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩1gあたり15~50mLのアセトニトリルを含む、製造方法。
[項2] 有機溶媒が、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩1gあたり15~50mLのアセトニトリルである、項1に記載の製造方法。
[項3] 反応混合物に水および酸を加えてpHを調整し、次いで、有機溶媒で抽出する工程を含む、項1または項2に記載の製造方法。
[項4] 有機溶媒による抽出が、2回以上実施される、項3に記載の製造方法。
[項5] 有機溶媒が、酢酸エチルである、項3または項4に記載の製造方法。
[項6] 11-(2-ヨードアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩と(R)-2-(ジエチルアミノ)メチルピペリジンまたはその塩とを有機溶媒中、塩基性条件下で反応させる工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の製造方法。
[項7] (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を1-プロパノールおよび水の混合液中で結晶化する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
[項8] (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩をアルコール系溶媒中で活性炭処理する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
[項9] (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩をアルコール系溶媒およびケトン系溶媒の混合液中で再結晶化する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
[項10] 純度が95%以上である、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩。
[項11] 不純物の含有量が5%未満である、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩。
[項12] (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物であって、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に由来する不純物の含有量が(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に対して5%未満である、医薬組成物。
[項13] 不純物の含有量が1%未満である、項12に記載の医薬組成物。
[項14] (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物であって、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を実質的に含まない、医薬組成物。
[項15] 11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の含有量が、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に対して1000ppm未満である、項14に記載の医薬組成物。
[項16] さらに5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を実質的に含まない、項14または項15に記載の医薬組成物。
[項17] 5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の含有量が、1000ppm未満である、項16に記載の医薬組成物。
[項18] 点眼剤である、項12~項17のいずれか一項に記載の医薬組成物。
[項19] 緑内障、高眼圧症または近視を治療するための項12~項18のいずれか一項に記載の医薬組成物。
さらに、本発明は、高純度の(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩、および、高純度の(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物を提供することができる。
なお、アセトニトリルの添加量の「v/w」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対するアセトニトリルの体積(mL)を意味する。以下、特に断りがない限り同様とする。
なお、アルカリ金属のヨウ化物の添加量の「当量」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの物質量(1モル)に対するアルカリ金属のヨウ化物の物質量(モル)を意味する。以下、特に断りがない限り同様とする。
なお、塩基の添加量の「当量」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの物質量(1モル)に対する塩基の物質量(モル)を意味する。以下、特に断りがない限り同様とする。
なお、反応混合物の含量の「v/w」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対する反応混合物の体積(mL)を意味する。
なお、水の添加量の「v/w」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対する水の体積(mL)を意味する。以下、特に断りがない限り同様とする。
なお、有機溶媒の添加量の「v/w」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対する有機溶媒の体積(mL)を意味する。
なお、溶媒の添加量の「v/w」は、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対する溶媒の体積(mL)を意味する。
なお、有機溶媒の添加量の「v/w」は、11-(2-ヨードアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの質量(1g)に対する有機溶媒の体積(mL)を意味する。
なお、塩基の添加量の「当量」は、11-(2-ヨードアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの物質量(1モル)に対する塩基の物質量(モル)を意味する。
本精製方法における、水の添加量は、例えば1~15v/wであり、好ましくは2~10v/wであり、より好ましくは4~8v/wであり、最も好ましくは6v/wである。
なお、1-プロパノールおよび水の添加量の「v/w」は、それぞれ(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の質量(1g)に対する1-プロパノールおよび水の体積(mL)を意味する。
なお、アルコール系溶媒の添加量の「v/w」は、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の質量(1g)に対するアルコール系溶媒の体積(mL)を意味する。
なお、活性炭の添加量の「w/w」は、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の質量(1g)に対する活性炭の質量(g)を意味する。
なお、混合液中のアルコール系溶媒の含量、アルコール系溶媒およびケトン系溶媒の添加量の「v/w」は、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の質量(1g)に対する混合液、アルコール系溶媒およびケトン系溶媒の体積(mL)を意味する。
AFDX0250またはその塩の含量は、例えば、以下の方法によって測定することができる。
<移動相および溶解液>
以下の移動相および溶解液を用いる。
・移動相A:0.1mol/L過塩素酸ナトリウム溶液(pH3.0)
過塩素酸ナトリウム一水和物14.0gを水1000mLに溶かし、1mol/L塩酸試液を加えてpH3.0に調整する。
・移動相B:アセトニトリル
・溶解液A:移動相A:移動相B(1:1)(v/v)
<AFDX0250標準溶液の調製>
AFDX0250約10mg精密に量り、溶解液Aを加えて正確に100mLとする。
<試料溶液の調製>
以下の方法で試料溶液を調製する。
・試料溶液A:AFDX0250を含む反応懸濁液(下記実施例1の工程1において得られた反応液)100~200mgを取り、溶解液A10mLに溶かす。
・試料溶液B:AFDX0250を含む反応懸濁液(下記実施例1の工程1において得られた反応液)約200μLを取り、桐山ロート(S-40)、桐山ろ紙(No.3)および吸引瓶を用いて減圧ろ過し、ろ取物約10mgを溶解液A10mLに溶かす。
・試料溶液C:取得固体(下記実施例1の工程3において得られたAFDX0250)約10mgを精密に量り、溶解液Aを加えて溶かし、正確に100mLとする。
・試料溶液D:水層(下記実施例1の工程2において得られた水層)約100μLを精密に量り、溶解液Aを加えて、正確に10mLとする。
<測定方法>
以下の方法、条件の液体クロマトグラフィーにて各試料溶液を測定する。さらに、以下の計算式によりAFDX0250の含量(%)を算出する。
(液体クロマトグラフィー(UHPLC)の試験条件)
・検出器:紫外吸光光度計(測定波長210nm)
・カラム:AQUITY UPLC HSS T3(内径2.1mm、長さ100mm、粒子径1.8μm;Waters社)
・ゴーストトラップ:ゴーストトラップDS-HP(P/N:228-59931-91)(島津製作所)または同等の性能品質を有するもの(移動相の不純物除去のため、ゴーストトラップを移動相ミキサーの後に接続する。)
・カラム温度:40℃付近
・サンプルクーラー温度:10℃付近
・移動相A:0.1mol/L過塩素酸ナトリウム溶液(pH3.0)
・移動相B:アセトニトリル
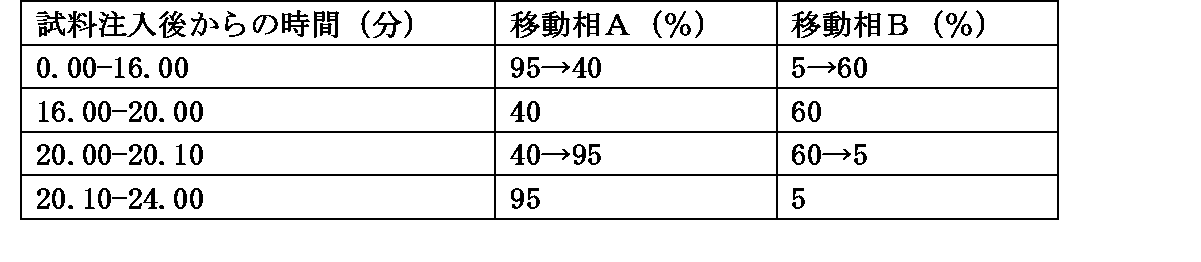
・移動相の送液:移動相Aおよび移動相Bの混合比を表1のように濃度勾配制御する。
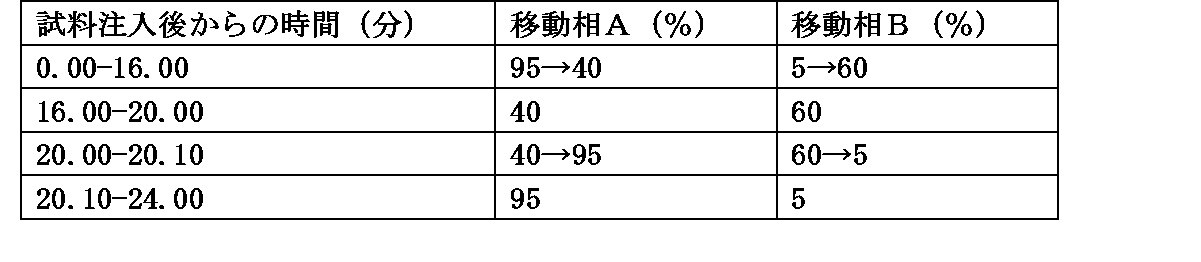
・流量:0.5mL/min
・注入量:2μL
・面積測定範囲:溶媒のピーク後から注入後20分間
(計算式)
試料溶液A~C:
AFDX0250含量(%)=100xAAp/ATp
AAp:試料溶液から得られる、AFDX0250ピーク面積
ATp:試料溶液から得られる、全ピーク面積
試料溶液D:
AFDX0250含量(%)=100xMSxATx10/(MTxASx100)
MS:AFDX0250の採取量(mg)
MT:試料溶液の採取量(mg)
AS:標準溶液から得られる、AFDX0250ピーク面積
AT:試料溶液から得られる、AFDX0250ピーク面積
(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に含まれる化合物1含量は、例えば、以下の方法によって測定することができる。
<移動相および溶解液>
以下の移動相および溶解液を用いる。
・移動相C:10mM酢酸アンモニウム溶液
酢酸アンモニウム0.77gを水1000mLに溶かす。
・移動相B:アセトニトリル
・溶解液B:移動相C:移動相B(7:3)(v/v)
<化合物1標準溶液の調製>
・化合物1標準原液:化合物1約20mgを精密に量り、溶解液Bを加えて溶かし、正確に100mLとする。溶けにくい場合は超音波を約1分間使用して溶解させる。この液2mLを正確に量り、溶解液Bを加えて、正確に20mLとする。この液1mLを正確に量り、溶解液Bを加えて、正確に20mLとする。
・化合物1標準溶液1:標準原液4mLを正確に量り、溶解液Bを加えて正確に20mLとする。
・化合物1標準溶液2:標準溶液1 2mLを正確に量り、溶解液Bを加えて正確に10mLとする。
・化合物1標準溶液3:標準溶液2 2mLを正確に量り、溶解液Bを加えて正確に20mLとする。
<試料溶液の調製>
以下の方法で試料溶液を調製する。
・試料溶液E(測定対象が水層の場合):上記(1)の測定法にしたがって得られた水層のAFDX0250含量(%)から計算し、含有するAFDX0250が約100mgなるように水層を精密に量り、溶解液Bを加えて、正確に25mLとする。
・試料溶液F(測定対象が結晶の場合):AFDX0250約100mgを精密に量り、溶解液B5mLおよび希塩酸0.2mLを加えて溶かし、溶解液Bを加えて、正確に25mLとする(AFDX0250濃度:約4mg/mL)。溶けにくい場合は超音波を使用して溶解させる。
<測定方法>
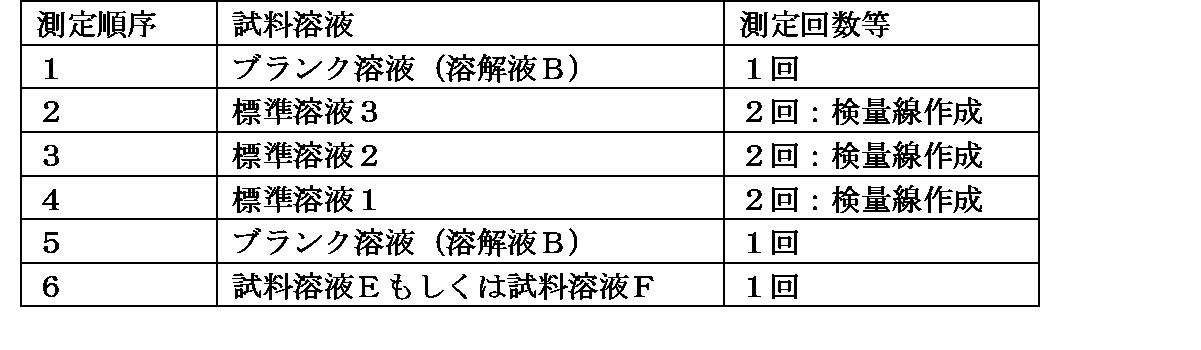
以下の条件の液体クロマトグラフィー質量分析法にて、下表3に示す順序および回数で各試料溶液を測定する。さらに、標準溶液の濃度(μg/mL)とピーク面積から検量線を作成し(標準溶液1~3の合計6個の結果を使用)、以下の計算式により試料溶液中の化合物1濃度(μg/mL)およびAFDX0250中の化合物1含量(ppm)を算出する。
(液体クロマトグラフィー質量分析法(LC/MS)の試験条件)
・検出器:質量分析計
・モニターイオン:m/z 288(ESI、Positive)
・カラム:X Bridge C18(内径2.1mm、長さ150mm、粒子径3.5μm;Waters社)
・カラム温度:40℃付近
・サンプルクーラー温度:成り行き温度
・移動相C:10mM酢酸アンモニウム溶液
・移動相B:アセトニトリル
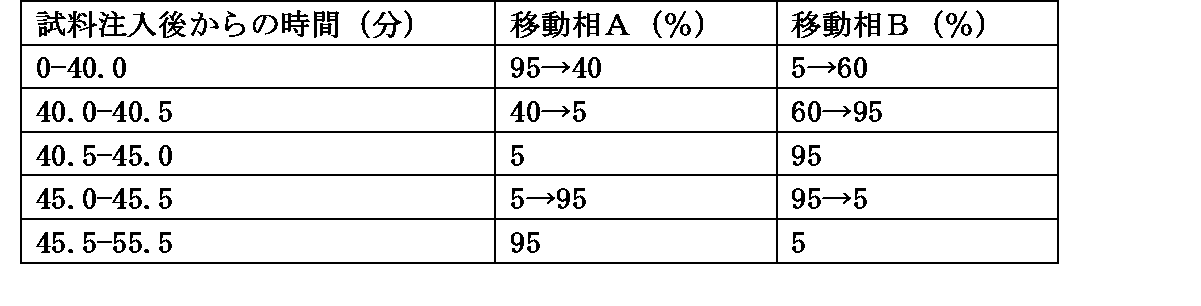
・移動相の送液:移動相Cおよび移動相Bの混合比を表2のように濃度勾配制御する。
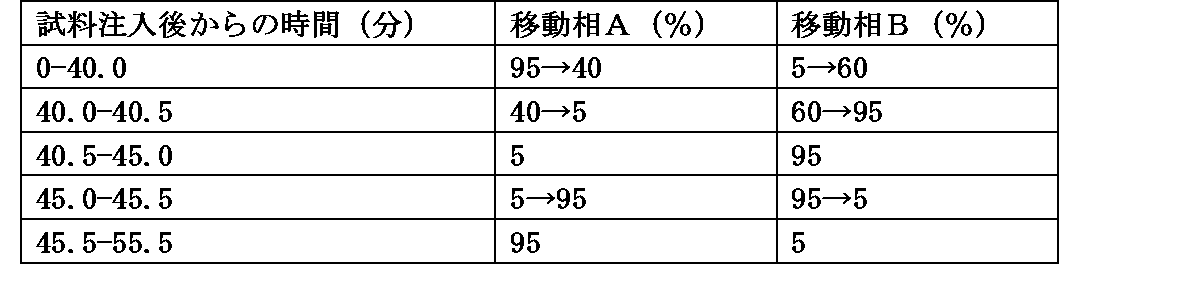
・流量:0.3mL/min
・注入量:5μL
・試料溶液の測定順序および回数:
(計算式)
試料溶液E(測定対象が水層の場合):
試料溶液E中の化合物1濃度(μg/mL)=(AT-b)/a
AFDX0250中の化合物1含量(ppm)=試料溶液E中の化合物1濃度(μg/mL)/MT×P/100×25000
AT:試料溶液Eから得られた化合物1のピーク面積
b:検量線の切片
a:検量線の傾き
MT:試料秤取量(mg)
P:上記(1)の測定法にしたがって得られた水層のAFDX0250含量(%)
試料溶液F(測定対象が結晶の場合):
試料溶液F中の化合物1濃度(μg/mL)=(AT-b)/a
AFDX0250中の化合物1含量(ppm)=試料溶液F中の化合物1濃度(μg/mL)/MT×25000
AT:試料溶液Fから得られた化合物1のピーク面積
b:検量線の切片
a:検量線の傾き
MT:試料秤取量(mg)
これらは当該技術分野における通常の方法に従って製造することができる。
また、必要に応じて点眼剤に汎用されている添加剤を用いてもよい。添加剤の例としては、等張化剤、緩衝化剤、界面活性剤、安定化剤、防腐剤、可溶化剤等が挙げられる。
・検出器:紫外吸光光度計(測定波長210nm)
・カラム:AQUITY UPLC HSS T3(内径2.1mm、長さ100mm、粒子径1.8μm;Waters社)
・ゴーストトラップ:ゴーストトラップDS-HP(P/N:228-59931-91)(島津製作所)または同等の性能品質を有するもの(移動相の不純物除去のため、ゴーストトラップを移動相ミキサーの後に接続する。)
・カラム温度:40℃付近
・サンプルクーラー温度:10℃付近
・移動相A:0.1mol/L過塩素酸ナトリウム溶液(pH3.0)
・移動相B:アセトニトリル
・移動相の送液:移動相Aおよび移動相Bの混合比を表4のように濃度勾配制御する。
・流量:0.5mL/min
・注入量:2μL
・面積測定範囲:溶媒のピーク後から注入後20分間
・検出器:質量分析計
・モニターイオン:m/z 288(ESI、Positive)
・カラム:X Bridge C18(内径2.1mm、長さ150mm、粒子径3.5μm;Waters社)
・カラム温度:40℃付近
・サンプルクーラー温度:成り行き温度
・移動相C:10mM酢酸アンモニウム溶液
・移動相B:アセトニトリル
・移動相の送液:移動相Cおよび移動相Bの混合比を表5のように濃度勾配制御する。
・流量:0.3mL/min
・注入量:5μL
・試料溶液の測定順序および回数:


窒素雰囲気下、室温にて11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オン(化合物1)(200g、0.695mol)のアセトニトリル溶液(3200g)に、(R)-2-(ジエチルアミノ)メチルピペリジン2塩酸塩(以下、「化合物2」とすることもある;178g、0.732mol)、ヨウ化ナトリウム(26.0g、0.174mol)および炭酸水素ナトリウム(184g、2.19mol)を加え、内温73.0~74.5℃で加熱した。各時点(反応2時間後、3時間後、4時間後および5時間後)における反応液を液体クロマトグラフィー(UHPLC)に付して、AFDX0250、化合物1および5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オン(以下、「化合物3」とすることもある)を面積百分率法で定量した。
工程1で5時間反応を進行させた後、反応液を冷却し濃縮して粗生成物AFDX0250(1231.56g)を得た。水(1800g)添加し、内温4.0~5.5℃でpHが1以下になるまで濃塩酸(155mL)を滴下した後(pH0.8)、酢酸エチル(1800g)で3回分液した。各回の分液の水層を液体クロマトグラフィー(UHPLC)で測定し、AFDX0250および化合物3を面積百分率法で定量した。また、液体クロマトグラフィー質量分析法(LC/MS)によって化合物1含量を測定した。
工程2で得られた水層に、水(1400g)およびアセトニトリル(780g)を添加して内温2.0~5.0℃で撹拌し、次いで、3.25N水酸化カリウム水溶液をpHが11以下になるまで滴下し、5時間撹拌した。析出した固体を減圧ろ過し、ろ取物を水(2000g)で洗浄した後、50℃で減圧乾燥し、淡褐色固体の粗AFDX0250(274.35g、収率93.3%)を得た。得られた淡褐色固体を液体クロマトグラフィー(UHPLC)で測定し、AFDX0250および化合物3を面積百分率法で定量した。また、液体クロマトグラフィー質量分析法(LC/MS)によって化合物1含量を測定した。
また、工程2において得られた水層および工程3において得られた固体中のAFDX0250に含まれる化合物1の含量は、本明細書における(2)化合物1含量の測定法に準じて測定した。
さらに、上記の工程1の炭酸水素ナトリウムに代えて、トリエチルアミンを用いた場合であっても、AFDX0250を製造することができることが示された。
本発明の製造方法(反応溶媒として化合物1 1gあたりアセトニトリルを20mL使用)および既知の製造方法(反応溶媒として化合物1 1gあたりアセトニトリルを10mL使用)にて製造されたAFDX0250について定量分析を行い、AFDX0250および不純物の面積%を比較した。
具体的には、化合物1のバッチ量を200gから95gに変更し,化合物2および試薬の仕込量(化合物1 1物質量あたりの当量)は、実施例1と同様の方法にしたがって、AFDX0250を製造した(実施例2)。また、化合物1 1gあたりアセトニトリルを20mL使用する代わりにアセトニトリル10mLを用いたこと以外は実施例2と同様の方法にしたがって、AFDX0250を製造した(比較例1)。得られた反応液に実施例1と同様の方法で試験溶液を調製した。各試験溶液についてUHPLC分析を行った。
1H-NMR(400 MHz,CDCl3)δ0.83-0.97(m,6H),1.07-1.80(m,6H),1.95-2.63(m,7.5H),2.72-2.82(m,0.5H),3.07-3.27(m,0.5H),3.68(s,1H),4.10-4.28(m,0.5H),7.27-7.33(m,1H),7.36-7.48(m,1H),7.55-7.70(m,3H),7.93(d,J=7.8Hz,1H),8.30(d,J=2.5Hz,1H),9.90-10.30(br,1H).


本試験では、活性炭として白鷺A(0.05w/w、0.10w/wおよび0.15w/w)および白鷺P(0.10w/wおよび0.15w/w)を使用し、また、有機溶媒としてエタノール(EtOH)、1-プロパノール(1-PrOH)およびN-メチルピロリドン(NMP)を使用した各試料を調製した。なお、溶媒としてNMPを含む試料については、白鷺A(0.15w/w)および白鷺P(0.10w/wおよび0.15w/w)を使用したものは調製していない。
エタノールおよび1-プロパノールの添加量はそれぞれ、粗AFDX0250 1gあたり20mL、撹拌温度は70℃とした。NMPの添加量は、粗AFDX0250 1gあたり2.5mL、撹拌温度は70℃とした。
着色成分が除去されているかを確認するために、エタノール、1-プロパノールおよびNMPを含む試料について、活性炭処理前後の性状を確認した。さらに、各試料について、各時点(活性炭添加前、活性炭添加1時間後、活性炭添加3時間後)でHPLC分析を行った。
さらに、エタノール(EtOH)を含む試料、1-プロパノール(1-PrOH)を含む試料およびNMPを含む試料のHPLC分析の結果を、表10~12に示す。



窒素雰囲気下、室温にて実施例4で精製された粗AFDX0250(200g、0.474mol)のエタノール(3200g)中溶液に、活性炭である白鷺A(30.0g)を加え、内温69.0~72.4℃で1時間加熱撹拌した後、ろ過助剤が充填されたろ過装置で減圧ろ過し、予め70℃に加熱したエタノール(320g)で洗浄した。ろ液を減圧濃縮し、濃縮残渣(989.04g)を得た。
アルコール系溶媒としてエタノールまたは1-プロパノールを、貧溶媒としてケトン系溶媒であるアセトン、メチルエチルケトン(MEK)、メチルイソブチルケトン(MIBK)またはn-へプタンを用いて、それらの組み合わせについて、それぞれの全溶媒量10v/w(比率1:3)および20v/w(比率1:1)で再結晶化工程を以下のとおり実施した。
AFDX0250のアルコール系溶液中を50℃で0.5時間から1時間加熱撹拌し、その溶液に各貧溶媒を添加した後、さらに1時間加熱撹拌した。その後、5℃に冷却し、24時間熟成した。懸濁溶液をろ過し、ろ取物を90℃で減圧乾燥した。得られた乾燥品について、性状の確認およびHPLC分析を行った。また、乾燥品を10%ジメチルスルホキシド(DMSO)溶液にしてその性状を確認した。



得られたAFDX0250は、本明細書における(1)AFDX0250またはその塩の含量の測定法に準じて、試験溶液を調製し、試験溶液についてHPLC分析を行った。
1回目の再結晶化で得られたAFDX0250と同様に、得られたAFDX0250は、本明細書における(1)AFDX0250またはその塩の含量の測定法に準じて、試験溶液を調製し、試験溶液についてHPLC分析を行った。
1H-NMR(400 MHz,CDCl3)δ0.85-0.97(m,6H),1.08-1.75(m,6H),1.96-2.60(m,7.5H),2.72-2.82(m,0.5H),3.10-3.24(m,0.5H),3.68(s,1H),4.12-4.26(m,0.5H),7.27-7.33(m,1H),7.36-7.43(m,1H),7.59-7.67(m,3H),7.93(d,J=7.8Hz,1H),8.30(d,J=3.2Hz,1H),10.00-11.00(br,1H)
さらに、本発明によれば、高純度の(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩、および、高純度の(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物を提供することができる。
Claims (17)
- 11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩と(R)-2-(ジエチルアミノ)メチルピペリジンまたはその塩とを有機溶媒中、アルカリ金属のヨウ化物の存在下、塩基性条件下で反応させる工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の製造方法であって、該有機溶媒が、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩1gあたり15~50mLのアセトニトリルを含む、製造方法。
- 有機溶媒が、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩1gあたり15~50mLのアセトニトリルである、請求項1に記載の製造方法。
- 反応混合物に水および酸を加えてpHを調整し、次いで、有機溶媒で抽出する工程をさらに含む、請求項1または2に記載の製造方法。
- 有機溶媒による抽出が、2回以上実施される、請求項3に記載の製造方法。
- 有機溶媒が、酢酸エチルである、請求項3または4に記載の製造方法。
- 11-(2-ヨードアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩と(R)-2-(ジエチルアミノ)メチルピペリジンまたはその塩とを有機溶媒中、塩基性条件下で反応させる工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の製造方法。
- (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を1-プロパノールおよび水の混合液中で結晶化する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
- (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩をアルコール系溶媒中で活性炭処理する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
- (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩をアルコール系溶媒およびケトン系溶媒の混合液中で再結晶化する工程を含む、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の精製方法。
- 純度が95%以上である、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩。
- 不純物の含有量が5%未満である、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩。
- (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物であって、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に由来する不純物の含有量が(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に対して5%未満である、医薬組成物。
- 不純物の含有量が1%未満である、請求項12に記載の医薬組成物。
- (R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を含む医薬組成物であって、11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンを実質的に含まない、医薬組成物。
- 11-(2-クロロアセチル)-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンの含有量が、(R)-11-[2-[2-[(ジエチルアミノ)メチル]-1-ピペリジニル]アセチル]-5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩に対して1000ppm未満である、請求項14に記載の医薬組成物。
- さらに5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩を実質的に含まない、請求項14または15に記載の医薬組成物。
- 5,11-ジヒドロ-6H-ピリド[2,3-b][1,4]ベンゾジアゼピン-6-オンまたはその塩の含有量が、1000ppm未満である、請求項16に記載の医薬組成物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024512662A JPWO2023190663A1 (ja) | 2022-03-30 | 2023-03-29 | |
| KR1020247035203A KR20240168375A (ko) | 2022-03-30 | 2023-03-29 | 고순도 화합물의 제조 방법 및 정제 방법 |
| US18/851,293 US20250214992A1 (en) | 2022-03-30 | 2023-03-29 | High-purity compound production method and purification method |
| CN202380031604.XA CN118946564A (zh) | 2022-03-30 | 2023-03-29 | 高纯度化合物的制造方法及纯化方法 |
| EP23780672.4A EP4506347A4 (en) | 2022-03-30 | 2023-03-29 | PROCESS FOR THE PRODUCTION AND PURIFICATION OF HIGH-PURITY COMPOUNDS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022-056754 | 2022-03-30 | ||
| JP2022056754 | 2022-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023190663A1 true WO2023190663A1 (ja) | 2023-10-05 |
Family
ID=88202044
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/012761 Ceased WO2023190663A1 (ja) | 2022-03-30 | 2023-03-29 | 高純度化合物の製造方法および精製方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20250214992A1 (ja) |
| EP (1) | EP4506347A4 (ja) |
| JP (1) | JPWO2023190663A1 (ja) |
| KR (1) | KR20240168375A (ja) |
| CN (1) | CN118946564A (ja) |
| TW (1) | TW202402753A (ja) |
| WO (1) | WO2023190663A1 (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60215683A (ja) | 1984-03-14 | 1985-10-29 | ドクトル.カール トーメー ゲゼルシヤフト ミツト ベシユレンクテル ハフツング | 新規縮合ジアゼピノン |
| JPS6335573A (ja) * | 1986-07-31 | 1988-02-16 | ドクトル カルル ト−マエ ゲゼルシヤフト ミツト ベシユレンクテル ハフツング | 新規置換ピリド〔2,3−b〕〔1,4〕ベンゾジアゼピン−6−オン,その製法及びその化合物を含む医薬 |
| WO1993018772A1 (en) | 1992-03-18 | 1993-09-30 | Allergan, Inc. | Method for reducing intraocular pressure in the mammalian eye by administration of muscarinic antagonists |
| WO2016078770A1 (en) * | 2014-11-21 | 2016-05-26 | Laboratorios Del Dr. Esteve, S.A. | Spiro-isoquinoline-1,4'-piperidine compounds having multimodal activity against pain |
| WO2022030489A1 (en) | 2020-08-04 | 2022-02-10 | Santen Pharmaceutical Co., Ltd. | Agent for treating myopia, preventing myopia and/or suppressing myopia progression |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09318772A (ja) | 1996-05-28 | 1997-12-12 | Rhythm Watch Co Ltd | 装飾装置 |
| JP7253813B2 (ja) | 2020-08-07 | 2023-04-07 | 株式会社ユニバーサルエンターテインメント | 遊技機 |
| WO2022080397A1 (ja) * | 2020-10-14 | 2022-04-21 | 参天製薬株式会社 | 安定な医薬組成物 |
| EP4501329A4 (en) * | 2022-03-30 | 2026-03-25 | Santen Pharmaceutical Co Ltd | METHOD FOR STERILIZING AND PACKAGING A PHARMACEUTICAL PREPARATION |
-
2023
- 2023-03-29 CN CN202380031604.XA patent/CN118946564A/zh active Pending
- 2023-03-29 TW TW112111992A patent/TW202402753A/zh unknown
- 2023-03-29 JP JP2024512662A patent/JPWO2023190663A1/ja active Pending
- 2023-03-29 EP EP23780672.4A patent/EP4506347A4/en active Pending
- 2023-03-29 WO PCT/JP2023/012761 patent/WO2023190663A1/ja not_active Ceased
- 2023-03-29 KR KR1020247035203A patent/KR20240168375A/ko active Pending
- 2023-03-29 US US18/851,293 patent/US20250214992A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60215683A (ja) | 1984-03-14 | 1985-10-29 | ドクトル.カール トーメー ゲゼルシヤフト ミツト ベシユレンクテル ハフツング | 新規縮合ジアゼピノン |
| JPS6335573A (ja) * | 1986-07-31 | 1988-02-16 | ドクトル カルル ト−マエ ゲゼルシヤフト ミツト ベシユレンクテル ハフツング | 新規置換ピリド〔2,3−b〕〔1,4〕ベンゾジアゼピン−6−オン,その製法及びその化合物を含む医薬 |
| WO1993018772A1 (en) | 1992-03-18 | 1993-09-30 | Allergan, Inc. | Method for reducing intraocular pressure in the mammalian eye by administration of muscarinic antagonists |
| JPH07504915A (ja) * | 1992-03-18 | 1995-06-01 | アラーガン、インコーポレイテッド | ムスカリン拮抗物質投与による,哺乳動物の眼における眼内圧低下法 |
| WO2016078770A1 (en) * | 2014-11-21 | 2016-05-26 | Laboratorios Del Dr. Esteve, S.A. | Spiro-isoquinoline-1,4'-piperidine compounds having multimodal activity against pain |
| WO2022030489A1 (en) | 2020-08-04 | 2022-02-10 | Santen Pharmaceutical Co., Ltd. | Agent for treating myopia, preventing myopia and/or suppressing myopia progression |
Non-Patent Citations (6)
| Title |
|---|
| ENGEL W W ET AL: "Tricyclic Compounds as Selective Muscarinic Receptor Antagonists. 3. Structure-Selectivity Relationships in a Series of Cardioselective (M2) Antimuscarinics", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 32, no. 8, 1 January 1989 (1989-01-01), US , pages 1718 - 1724, XP002166482, ISSN: 0022-2623, DOI: 10.1021/jm00128a008 * |
| MANDELLI GIACOMINA R., STEFANO MAIORANA, PATRIZIA TERNI, GIUSEPPINA LAMPERTI, MARIA LUISA COLIBRETTI, BRUNO P. IMBIMBO: "Synthesis of New Cardioselective M2 Muscarinic Receptor Antagonists ", CHEMICAL AND PHARMACEUTICAL BULLETIN, JP, vol. 48, no. 11, 1 November 2000 (2000-11-01), JP , pages 1611 - 1622, XP093095104, ISSN: 1347-5223, DOI: 10.1248/cpb.48.1611 * |
| See also references of EP4506347A4 |
| WATANABE TOSHIHIRO, AKIO KAKEFUDA, ISAO KINOYAMA, KENJI TAKIZAWA, SEIKO HIRANO, HIROSHI SHIBATA, ISAO YANAGISAWA: "Synthesis of Novel Succinamide Derivatives Having a 5, 11-Dihydro-6H-pyrido[2, 3-b][1, 4]benzodiazepin-6-one Skeleton as Potent and Selective M2 Muscarinic Receptor Antagonists. II ", CHEMICAL AND PHARMACEUTICAL BULLETIN, PHARMACEUTICAL SOCIETY OF JAPAN, JP, vol. 45, no. 9, 1 January 1997 (1997-01-01), JP , pages 1458 - 1469, XP093095132, ISSN: 0009-2363, DOI: 10.1248/cpb.45.1458 * |
| WATANABE TOSHIHIRO, ISAO KINOYAMA, KENJI TAKIZAWA, SEIKO HIRANO, TADAO SHIBANUMA: "Synthesis and Biological Evaluation of 1, 2, 3, 4-Tetrahydroisoquinoline Derivatives as Potent and Selective M2 Muscarinic Receptor Antagonist", CHEMICAL AND PHARMACEUTICAL BULLETIN, PHARMACEUTICAL SOCIETY OF JAPAN, JP, vol. 47, no. 5, 15 May 1999 (1999-05-15), JP , pages 672 - 677, XP093095127, ISSN: 0009-2363, DOI: 10.1248/cpb.47.672 * |
| WOLFNARD W. ENGEL ET AL., J. MED. CHEM., vol. 32, no. 8, 1989, pages 1718 - 1724 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4506347A4 (en) | 2026-03-25 |
| US20250214992A1 (en) | 2025-07-03 |
| KR20240168375A (ko) | 2024-11-29 |
| CN118946564A (zh) | 2024-11-12 |
| TW202402753A (zh) | 2024-01-16 |
| JPWO2023190663A1 (ja) | 2023-10-05 |
| EP4506347A1 (en) | 2025-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2024540080A (ja) | Irak4分解剤およびその合成 | |
| KR102442536B1 (ko) | 리나글립틴 결정형 및 이의 제조방법 | |
| JP2017061578A (ja) | 有機化合物 | |
| US7897613B2 (en) | Crystalline polymorphs of clopidogrel | |
| EP2468750A1 (en) | Polymorphic forms of asenapine maleate and processes for their preparation | |
| EP0579681A1 (en) | Crystalline tiagabine hydrochloride monohydrate, its preparation and use | |
| JP2014051517A (ja) | デフェラシロックス(icl670a)の多形形態 | |
| US20210238195A1 (en) | Crystalline polymorphs of a muscarinic acetylcholine receptor agonist | |
| US20210171460A1 (en) | Crystalline form of sofpironium bromide and preparation method thereof | |
| WO2023193563A1 (zh) | 一种噻吩并吡啶化合物的晶型a、制备方法及其药物组合物 | |
| JP6275644B2 (ja) | N−[2−({2−[(2S)−2−シアノピロリジン−1−イル]−2−オキソエチル}アミノ)−2−メチルプロピル]−2−メチルピラゾロ[1,5−a]ピリミジン−6−カルボキサミドの結晶 | |
| CN117377658A (zh) | 喹啉衍生物化合物的制备方法 | |
| NO330528B1 (no) | Nye salter av benzazepinforbindelser, fremgangsmater for fremstilling derav, og anvendelse av slike i farmasoytiske sammensetninger | |
| WO2023190663A1 (ja) | 高純度化合物の製造方法および精製方法 | |
| JP2023536911A (ja) | O-糖タンパク質-2-アセトアミド-2-デオキシ-3-d-グルコピラノシダーゼ阻害剤の結晶形態 | |
| CN111004255A (zh) | 一种头孢卡品内酯化合物或其盐酸盐的制备方法 | |
| CN107540589B (zh) | 一种艾尔骨化醇晶型、药物组合物及制备方法和应用 | |
| CN111670191B (zh) | 吡啶酮衍生物的晶型及制备方法和应用 | |
| KR20180129851A (ko) | 나트륨-포도당 연계된 트랜스포터 억제제의 아민 용매화물 및 그의 제조 방법 및 애플리케이션 | |
| US20120059034A1 (en) | Novel crystalline hydrate, amorphous and polymorphic forms of dihydro-benzoxazole-6-yl-acetamide derivative and processes for their preparation | |
| DE60303603T2 (de) | Benzolsulfonatsalz eines morpholinharnstoff-derivats zur verwendung als ccr-3 antagonist bei der behandlung von entzündlichen erkrankungen | |
| WO2026018176A1 (en) | 2-(4-chloro-2 methoxyphenyl)-2-((3-methoxy-5-(methylsulfonyl)phenyl)amino)-1-(5-(trifluoro¬methoxy)-1h-indol-3-yl)ethanone and pharmaceutical compositions comprising the same | |
| TW202527934A (zh) | 醫藥上活性化合物之固體形式 | |
| WO2024125361A1 (zh) | N-取代苯基磺酰胺类化合物的固体形式 | |
| HK40068368B (zh) | 索吡溴铵的晶型及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23780672 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18851293 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2024512662 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380031604.X Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 20247035203 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023780672 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023780672 Country of ref document: EP Effective date: 20241030 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18851293 Country of ref document: US |