WO2023204256A1 - アセタール型リリーサブルポリオキシエチレン誘導体、その製造方法及びアセタール型リリーサブルポリオキシエチレン結合体 - Google Patents
アセタール型リリーサブルポリオキシエチレン誘導体、その製造方法及びアセタール型リリーサブルポリオキシエチレン結合体 Download PDFInfo
- Publication number
- WO2023204256A1 WO2023204256A1 PCT/JP2023/015663 JP2023015663W WO2023204256A1 WO 2023204256 A1 WO2023204256 A1 WO 2023204256A1 JP 2023015663 W JP2023015663 W JP 2023015663W WO 2023204256 A1 WO2023204256 A1 WO 2023204256A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- group
- acetal
- added
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/04—1,3-Dioxanes; Hydrogenated 1,3-dioxanes
- C07D319/08—1,3-Dioxanes; Hydrogenated 1,3-dioxanes condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/14—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems
- C07D319/16—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D319/20—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring with substituents attached to the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/331—Polymers modified by chemical after-treatment with organic compounds containing oxygen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/331—Polymers modified by chemical after-treatment with organic compounds containing oxygen
- C08G65/3311—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group
- C08G65/3314—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group cyclic
- C08G65/3315—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group cyclic aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/331—Polymers modified by chemical after-treatment with organic compounds containing oxygen
- C08G65/3311—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group
- C08G65/3314—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group cyclic
- C08G65/3315—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group cyclic aromatic
- C08G65/3317—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing a hydroxy group cyclic aromatic phenolic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/331—Polymers modified by chemical after-treatment with organic compounds containing oxygen
- C08G65/332—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing carboxyl groups, or halides, or esters thereof
- C08G65/3322—Polymers modified by chemical after-treatment with organic compounds containing oxygen containing carboxyl groups, or halides, or esters thereof acyclic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/333—Polymers modified by chemical after-treatment with organic compounds containing nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/333—Polymers modified by chemical after-treatment with organic compounds containing nitrogen
- C08G65/33303—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing amino group
- C08G65/33306—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing amino group acyclic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/333—Polymers modified by chemical after-treatment with organic compounds containing nitrogen
- C08G65/33331—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing imide group
- C08G65/33337—Polymers modified by chemical after-treatment with organic compounds containing nitrogen containing imide group cyclic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/333—Polymers modified by chemical after-treatment with organic compounds containing nitrogen
- C08G65/33396—Polymers modified by chemical after-treatment with organic compounds containing nitrogen having oxygen in addition to nitrogen
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/32—Polymers modified by chemical after-treatment
- C08G65/329—Polymers modified by chemical after-treatment with organic compounds
- C08G65/337—Polymers modified by chemical after-treatment with organic compounds containing other elements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/02—Applications for biomedical use
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to an acetal-type releasable polyoxyethylene derivative, a method for producing the same, and an acetal-type releasable polyoxyethylene conjugate that is a conjugate with a biofunctional molecule.
- biofunctional molecules chemically modified with water-soluble polymers such as PEG can be used to target endogenous molecules and receptors due to the formation of a hydration layer and the steric shielding effect of the active site. It is known that this may lead to a decrease in the interaction with biofunctional molecules, resulting in unfavorable effects such as a decrease in the pharmacological action of biofunctional molecules and changes in internal and intracellular dynamics.
- Patent Documents 1 and 2 disclose that human growth hormone (hGH) and interleukin 2 (IL-2), which are biofunctional molecules, are administered in a non-enzyme-dependent manner under physiological conditions, that is, under neutral conditions.
- hGH human growth hormone
- IL-2 interleukin 2
- Patent Document 3 reports a compound in which a biologically functional molecule chemically modified with PEG temporarily cleaves a bond in a non-enzyme dependent manner to release a biologically functional molecule that is not chemically modified. Specifically, hydrolysis of a linker having an acetal structure that is hydrolyzed under acidic conditions is used as a trigger to cleave a carbamate bond, which is a temporary bond, by elimination of 1,4- or 1,6-benzyl, resulting in chemical modification. It has been reported that biofunctional molecules are released.
- Patent Document 3 exemplify specific structures that are capable of releasing biofunctional molecules in a non-enzyme-dependent manner and that are capable of releasing biofunctional molecules under acidic conditions.
- no prodrug technology has been described that releases biofunctional molecules at an appropriate rate in a non-enzyme-dependent manner under physiological conditions, i.e., neutral conditions around pH 7.4, to express pharmacological effects. .
- the object of the present invention is to obtain a polyoxyethylene derivative having an acetal structure, which is characterized by converting a biofunctional molecule into a prodrug and gradually releasing the biofunctional molecule under physiological conditions, and a stable method for producing the same.
- An object of the present invention is to provide an acetal type releasable polyoxyethylene conjugate.
- B 1 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)E 1
- E 1 is a leaving group
- P 1 is a polyoxyethylene derivative having a dehydroxyl group
- w is an integer from 1 to 8
- R 1 , R 2 , R 3 , R 4 , R 5 and R 11 are each independently a hydrocarbon group having 1 to 10 carbon atoms or a hydrogen atom
- R 6 , R 7 , R 8 , R 9 and R 10 are each independently an electron-withdrawing substituent, an electron-donating substituent or a hydrogen atom
- m is 0 or 1.
- R 1 and R 2 are hydrogen atoms
- R 3 , R 4 , R 5 and R 11 are each independently a hydrogen atom or a methyl group
- a method for producing an acetal-type releasable polyoxyethylene derivative characterized by:
- B 2 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)NHD 1
- D1 is a residue obtained by removing the amino group constituting the carbamate bond from the amino group contained in the biofunctional molecule
- P2 is a polyoxyethylene derivative having a dehydroxylated group, or a combination of a polyoxyethylene derivative having a dehydroxylated group and a biofunctional molecule
- w is an integer from 1 to 8
- R 1 , R 2 , R 3 , R 4 , R 5 and R 11 are each independently a hydrocarbon group having 1 to 10 carbon atoms or a hydrogen atom
- R 6 , R 7 , R 8 , R 9 and R 10 are each independently an electron-withdrawing substituent, an electron-donating substituent or a hydrogen atom
- m is 0 or 1.
- the acetal-type releasable polyoxyethylene derivative of the present invention converts a biofunctional molecule into a prodrug and gradually hydrolyzes the acetal under physiological conditions to induce benzyl elimination, thereby gradually converting the biofunctional molecule into a prodrug. release and improve the pharmacological action of physiologically functional molecules modified with polyoxyethylene derivatives.
- Patent Document 3 a specific structure that can release biofunctional molecules in a non-enzyme-dependent manner and can release biofunctional molecules under acidic conditions is exemplified, but under physiological conditions No specific structure has been described in which a biofunctional molecule can be released by benzyl elimination triggered by acetal hydrolysis under neutral conditions, that is, around pH 7.4.
- the acetal structure in the compound of Patent Document 3 is generally used as a protecting group for a diol or carbonyl group, and it is known that its deprotection is carried out under acidic conditions. It is stable under neutral and basic conditions. Therefore, among the compounds of Patent Document 3, there is no suggestion of creating a compound that is hydrolyzed at an appropriate rate under physiological conditions, that is, neutral conditions, and releases physiologically functional molecules.
- the production method of Patent Document 3 includes a step of coupling the phenolic hydroxyl group of a low molecular weight compound having an acetal structure and the hydroxyl group of PEG under basic conditions, but the acetal type releasable polyester according to the present invention
- the acetal structure in the low molecular weight compound becomes unstable, making it difficult to obtain the acetal type releasable polyoxyethylene derivatives related to the present invention. It turned out to be difficult.
- Figure 3 is the result of HPLC analysis of the purified PEGylated RNase described in Example 13. These are the results of a decomposition test using compounds of formulas (75), (76), (77) and (78) at 40°C in a heavy water buffer with pD 7.4. This is a base sequence of an aptamer having TNF ⁇ inhibitory activity.
- A is the HPLC analysis result of purified PEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ) described in Comparative Example 3.
- B is the HPLC analysis result of purified 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ) described in Example 25.
- PEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ), 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ), and the 5′ end of the aptamer of SEQ ID NO: 1 were modified with a C6 amino linker. This is an evaluation result of TNF ⁇ inhibitory activity using NH 2 -C6-(Apt-TNF ⁇ ).
- A is the HPLC analysis result of purified PEG(20k)-OCO-NH-insulin described in Comparative Example 4.
- B is the HPLC analysis result of purified 2MPEG(20k)-OCO-NH-insulin described in Example 26.
- A is the result of stability evaluation of PEG(20k)-OCO-NH-insulin in buffer solutions with various pHs.
- B is the result of stability evaluation of 2MPEG(20k)-OCO-NH-insulin in buffer solutions with various pHs.
- the present invention is characterized in that acetal is hydrolyzed under physiological conditions, and biofunctional molecules are gradually released through benzyl elimination.
- physiological conditions as used herein means pH 6.0 to 8.0.
- the release rate of biofunctional molecules under physiological conditions can be evaluated as the half-life of acetal in a pH 7.4 buffer.
- the half-life of the acetal in a pH 7.4 buffer is preferably between 0.5 and 35 days.
- half-life refers to the time required for acetal to be hydrolyzed and 1/2 equivalent to become aldehyde.
- the present invention is an acetal type releasable polyoxyethylene derivative represented by the following formula (1), formula (2), formula (3) or formula (4), and characterized in that it is cleaved under physiological conditions. be.
- B 1 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)E 1
- E 1 is a leaving group
- P 1 is a polyoxyethylene derivative having a dehydroxyl group
- w is an integer from 1 to 8
- R 1 , R 2 , R 3 , R 4 , R 5 and R 11 are each independently a hydrocarbon group having 1 to 10 carbon atoms or a hydrogen atom
- R 6 , R 7 , R 8 , R 9 and R 10 are each independently an electron-withdrawing substituent, an electron-donating substituent or a hydrogen atom
- m is 0 or 1.
- B 1 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)E 1 , Preferably it is a hydrogen atom.
- E 1 is a leaving group
- the leaving group include a succinimidyloxy group, a phthalimidyloxy group, a 4-nitrophenoxy group, and a 1-imidazolyl group.
- P 1 is a polyoxyethylene derivative having a dehydroxyl group, specifically, an ether bond (- This is a residue excluding the hydroxy group (OH) that constitutes O-).
- Polyoxyethylene includes both polyoxyethylene with a molecular weight distribution obtained by polymerization of ethylene oxide, and monodisperse polyoxyethylene in which oligooxyethylenes of a single molecular weight are bonded by a coupling reaction.
- Preferred examples of P 1 are preferably the following residues depending on the number of Ws.
- X 1 is a hydrocarbon group having 1 to 24 carbon atoms, an amino group protected with a protective group, or a group capable of reacting with a biofunctional molecule
- Z 1 is a divalent spacer or a single bond
- n and l are each independently from 3 to 2,000
- s is 0 or 1
- t is 2 or 3
- v is 0 or 2.
- * represents the bonding point with the oxygen atom.
- n and l represent the number of added moles of oxyethylene group represented by -(OCH 2 CH 2 )-, and each independently is 3 to 2,000, preferably 20 to 1,500, more preferably 40 to 1,000, and even more preferably 60 to 500, based on the number average molecular weight of the polyoxyethylene derivative determined by size exclusion chromatography, mass spectrometry, etc. It can be calculated by subtracting the molecular weight derived from molecules other than the polyoxyethylene chain represented by -(OCH 2 CH 2 ) n - and dividing it by the molecular weight 44 derived from the oxyethylene group.
- Z1 is a divalent spacer or single bond that connects the polyoxyethylene group and X1 , and as a divalent spacer it is more stable than an acetal structure. There is no particular restriction if it is, but it is preferably an ether bond, ester bond, carbonate bond, urethane bond, amide bond, secondary amino group, or an alkylene group containing these, and the alkylene group has 1 to 24 carbon atoms. Examples include spacers (z1) to (z8) described in group (I).
- q1 and q2 are each independently an integer of 1 to 12; for example, when it is desired to bond the terminal active carbonate group in a hydrophobic environment such as inside a protein, it is preferable that q1 and q2 are larger. , when bonding is desired in a hydrophilic environment, it is preferable that q1 and q2 are smaller.
- Z 1 is an ether bond, ester bond, carbonate bond, urethane bond, amide bond, secondary amino group, or alkylene group containing these, and multiple identical structural units are bonded, the above structural unit The number is 2 or less.
- X1 is a hydrocarbon group having 1 to 24 carbon atoms, an amino group protected with a protective group, or a group capable of reacting with a biofunctional molecule.
- hydrocarbon groups include methyl group, ethyl group, propyl group, isopropyl group, butyl group, t-butyl group, pentyl group, isopentyl group, hexyl group, heptyl group, 2-ethylhexyl group, octyl group.
- ⁇ examples include phenyl group, benzyl group, cresyl group, butylphenyl group, dodecylphenyl group, trityl group, etc., preferably a hydrocarbon group having 1 to 10 carbon atoms, more preferably a methyl group or an ethyl group, still more preferably It is a methyl group.
- the protecting group is a component that prevents or inhibits the reaction of a specific chemically reactive functional group in a molecule under certain reaction conditions.
- Protecting groups will vary depending on the type of chemically reactable functional group being protected, the conditions used, and the presence of other functional groups or protecting groups in the molecule. Specific examples of protecting groups can be found in many popular texts, such as "Wuts, P. G. M.; Greene, T. W. Protective Groups in Organic Synthesis, 4th ed.; Wiley - Interscience: New York, 2007.
- the amino group protected with a protecting group includes, for example, an amino group protected with an acyl-based protecting group or a carbamate-based protecting group, or an azido group, and specific examples of the acyl-based protecting group or carbamate-based protecting group Examples include trifluoroacetyl group, 9-fluorenylmethyloxycarbonyl group, and 2-(trimethylsilyl)ethyloxycarbonyl group.
- Groups that can react with biofunctional molecules include formyl group, epoxy group, maleimidyl group, vinyl sulfone group, acrylic group, sulfonyloxy group, carboxy group, dithiopyridyl group, ⁇ -haloacetyl group, alkynyl group, and allyl group. group, vinyl group, or azido group.
- functional groups that can react with amino groups of biofunctional molecules to form covalent bonds include formyl groups, epoxy groups, maleimidyl groups, vinyl sulfone groups, acrylic groups, sulfonyloxy groups, and It is a carboxy group.
- Functional groups that can react with thiol groups of biofunctional molecules to form covalent bonds include formyl groups, epoxy groups, maleimidyl groups, vinyl sulfone groups, acrylic groups, sulfonyloxy groups, carboxy groups, and dithiopyridyl groups. , ⁇ -haloacetyl group, alkynyl group, allyl group and vinyl group.
- a functional group capable of reacting with an alkynyl group of a biofunctional molecule to form a covalent bond is an azide group.
- Functional groups capable of forming a covalent bond by reacting with the azide group of the biofunctional molecule are alkynyl groups and functional groups containing triple bonds.
- the group capable of reacting with the biofunctional molecule is a group represented by group (II), group (III), group (IV) or group (V). Note that "**" represents the bonding point with Z1 .
- Group (II) Functional group capable of forming a covalent bond by reacting with an amino group of a biofunctional molecule
- Group (III) Functional groups capable of forming covalent bonds by reacting with thiol groups of biofunctional molecules
- Group (IV) Functional group capable of forming a covalent bond by reacting with an alkynyl group of a biofunctional molecule (c), (d) and (g) below
- Y 1 and Y 3 are each independently a hydrogen atom or a hydrocarbon group having 1 to 5 carbon atoms, and specific hydrocarbon groups include methyl group, ethyl group, propyl group, isopropyl group, and butyl group. , t-butyl group and pentyl group.
- Y 2 is a halogen atom selected from chlorine, bromine, and iodine.
- s is 0 or 1
- t is 2 or 3
- v is 0 or 2
- preferred embodiments include the following formulas (p14) to (p19).
- R 1 , R 2 , R 3 , R 4 , R 5 and R 11 are each independently a hydrocarbon group having 1 to 10 carbon atoms or a hydrogen atom, and specifically Preferred hydrocarbon groups include methyl, ethyl, propyl, isopropyl, t-butyl, phenyl, and benzyl, with hydrogen or methyl being more preferred.
- R 6 , R 7 , R 8 , R 9 and R 10 are each independently an electron-withdrawing substituent, an electron-donating substituent or a hydrogen atom.
- electron-withdrawing substituents include acyl groups having 2 to 5 carbon atoms, alkoxycarbonyl groups having 2 to 5 carbon atoms, carbamoyl groups having 2 to 5 carbon atoms, acyloxy groups having 2 to 5 carbon atoms, and acyl groups having 2 to 5 carbon atoms.
- acylamino group C2-5 alkoxycarbonylamino group, fluorine atom, chlorine atom, bromine atom, iodine atom, C1-4 alkylsulfanyl group, C1-4 alkylsulfonyl group, carbon Six to ten arylsulfonyl groups, nitro groups, trifluoromethyl groups, and cyano groups, and preferred examples include acetyl groups, methoxycarbonyl groups, methylcarbamoyl groups, acetoxy groups, acetamido groups, methoxycarbonylamino groups, and fluorine atoms. , chlorine atom, bromine atom, iodine atom, methylsulfanyl group, phenylsulfonyl group, nitro group, trifluoromethyl group and cyano group.
- the electron-donating substituent is an alkyl group having 1 to 4 carbon atoms, and preferable examples thereof include a methyl group, an ethyl group, a propyl group, an isopropyl group, and a t-butyl group.
- Substituents having an electron-withdrawing property at the meta-position, ie, R 7 or R 8 , and electron-donating at the para-position and ortho-position, ie, R 6 , R 9 or R 10 of the phenyl group include those having 1 to 1 carbon atoms; 4 alkoxy group, aryl group having 6 to 10 carbon atoms, and aryloxy group having 6 to 10 carbon atoms, and preferred examples include methoxy group, ethoxy group, propoxy group, isopropoxy group, t-butoxy group, and phenyl group. and phenoxy group.
- m is 0, R 1 and R 2 are hydrogen atoms, and R 3 , R 4 , R 5 and R 11 are hydrogen atoms or More preferably, it is a methyl group having 1 carbon number, R 3 , R 4 and R 5 are hydrogen atoms, and R 11 is a methyl group.
- a method for producing an acetal type releasable polyoxyethylene derivative according to the present invention will be explained.
- a coupling product represented by the following formula (5) or formula (6) is produced by coupling a polyoxyethylene derivative and a hydroxybenzaldehyde derivative.
- the coupling step of a polyoxyethylene derivative and a hydroxybenzaldehyde derivative is a step of obtaining a coupling product of formula (5) or (6) through a reaction step represented by reaction 1-1 or reaction 1-2. There may be a purification step after the reaction step.
- the polyoxyethylene derivative used in the coupling reaction is a compound represented by formula (11), in which the detailed explanation of P 1 and w is the same as above, and E 2 is It is a leaving group or a hydrogen atom.
- the leaving group is not particularly limited as long as it is reactive in the coupling reaction, but examples include chloro group, bromo group, iodo group, mesylate group, tosylate group, chloromethanesulfonate group, etc. or tosylate group is preferred, and mesylate group is more preferred.
- the hydroxybenzaldehyde derivative is a compound represented by formula (12) or formula (13), in which the detailed explanations of R 6 to R 10 are as defined above.
- an organic base such as potassium carbonate, potassium hydroxide, or sodium hydride.
- the proportion of the hydroxybenzaldehyde derivative, organic base, and inorganic base to be used is not particularly limited, but is preferably at least equimolar to the chemically reactable functional group of the compound of formula (11).
- an organic base may be used as a solvent.
- Reaction 1-1 and Reaction 1-2 After the reaction steps of Reaction 1-1 and Reaction 1-2, it is preferable to remove impurities by-produced in the reaction, compounds remaining without being consumed in the reaction, and base catalyst in a purification step, and there are no particular restrictions on the purification method. However, it can be purified by extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, etc.
- the acetalization step of reacting with a phenol having C(R 1 )(R 2 )-OH (B 1 , m, R 1 , and R 2 are as described above) is the reaction 2-1. It is a process of obtaining an acetal structure represented by formula (15), (16), (18) or formula (19) through the reaction process represented by any one of ⁇ 2-4, and a purification process is performed after the reaction process. There may be.
- Reactions 2-1 to 2-4 involve combining the coupling product represented by formula (5) or formula (6) with the phenol represented by formula (14) or formula (17) in toluene, benzene, xylene,
- the reaction is performed in an aprotic solvent such as acetonitrile, ethyl acetate, diethyl ether, t-butyl methyl ether, tetrahydrofuran, chloroform, dichloromethane, dimethyl sulfoxide, dimethylformamide or dimethylacetamide, or in the absence of a solvent in the presence of an acid catalyst to form the formula (
- This is a step of obtaining an acetal structure of formula (15), (16), (18) or formula (19).
- the acid catalyst may be an organic acid or an inorganic acid, and is not particularly limited, but specific examples include p-toluenesulfonic acid, pyridinium p-toluenesulfonate, methanesulfonic acid, 10-camphorsulfonic acid, and chloride. These include hydrogen, iodine, ammonium chloride, oxalic acid, and boron trifluoride diethyl ether complex.
- a dehydrating agent may be added to the reaction system in order to eliminate water molecules generated in the reaction, and there is no particular restriction on the type of dehydrating agent as long as it does not interfere with the reaction, but for example, sodium sulfate , magnesium sulfate, alumina, silica gel or molecular sieve, preferably molecular sieve.
- the purification method is not particularly limited. However, it can be purified by extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, etc.
- the method may include a step of introducing a group capable of reacting with a biofunctional molecule into the deprotected amino group.
- the conditions for the step of deprotecting the protecting group and the step of introducing a group capable of reacting with a biofunctional molecule into the deprotected amino group can be found in many general texts. For example, "Wuts, P. G. M.; Greene, T. W.
- the step of introducing a group capable of reacting with a biofunctional molecule can be performed, for example, according to "Greg T. Hermanso, Bioconjugate Techniques, 3rd ed.”
- the leaving group structure introduction step of introducing a leaving group structure in place of the terminal hydroxy group after the acetalization step or the step of introducing a group capable of reacting with a biofunctional molecule into the deprotected amino group?
- formulas (15), (16), (18) and formula (19) the hydroxy groups in the polyoxyethylene derivatives are converted into succinimidyloxycarbonyloxy groups, phthalimidyloxycarbonyloxy groups, 4-
- the conversion of the hydroxyl group is carried out by using a polyoxyethylene derivative represented by formula (15), (16), (18) or formula (19) and converting it into the respective leaving group listed in Table 1, for example.
- the reagents can be dissolved in an aprotic solvent such as toluene, benzene, xylene, acetonitrile, ethyl acetate, diethyl ether, t-butyl methyl ether, tetrahydrofuran, chloroform, dichloromethane, dimethyl sulfoxide, dimethylformamide or dimethylacetamide, or in the absence of a solvent, triethylamine, Condensation in the presence of an organic base such as N-methylmorpholine, pyridine or 4-dimethylaminopyridine, or an inorganic base such as sodium carbonate, sodium bicarbonate, sodium acetate or potassium carbonate.
- an organic base such as N-methylmorpholine, pyridine or 4-dimethyl
- the proportions of the reagents and base catalysts listed in Table 1 are not particularly limited, but are at least equimolar to the hydroxyl group of the polyoxyethylene derivative of formula (15), (16), (18) or formula (19). is preferred. Furthermore, the reagents listed in Table 1 may be commercially available products, or may be produced using known reactions.
- the purification method is not particularly limited, but It can be purified by extraction, recrystallization, adsorption treatment, reprecipitation, column chromatography, supercritical extraction, etc.
- the acetal-type releasable polyoxyethylene conjugate in the present invention is obtained by reacting one -OC(O)E group of the acetal-type releasable polyethylene glycol derivative with an amino group contained in a biofunctional molecule, and is It is represented by formula (7), (8), (9) or (10).
- B 2 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)NHD 1
- D1 is a residue obtained by removing the amino group constituting the carbamate bond from the amino group contained in the biofunctional molecule
- P2 is a polyoxyethylene derivative having a dehydroxylated group, or a combination of a polyoxyethylene derivative having a dehydroxylated group and a biofunctional molecule
- w is an integer from 1 to 8
- R 1 , R 2 , R 3 , R 4 , R 5 and R 11 are each independently a hydrocarbon group having 1 to 10 carbon atoms or a hydrogen atom
- R 6 , R 7 , R 8 , R 9 and R 10 are each independently an electron-withdrawing or electron-donating substituent or a hydrogen atom
- m is 0 or 1.
- R 1 to R 11 and m are as defined above.
- B 2 is a hydrogen atom or -C(R 1 )(R 2 )OC(O)NHD 1 , preferably a hydrogen atom.
- D 1 is a residue of amino groups contained in the biofunctional molecule excluding the amino group constituting the carbamate bond.
- the biofunctional molecule is not particularly limited, but it is a substance that is involved in the diagnosis, cure, mitigation, treatment, or prevention of diseases in humans or other animals. Specifically, it includes proteins, peptides, nucleic acids, cells, viruses, etc., and suitable proteins or peptides include hormones, cytokines, antibodies, aptamers, enzymes, etc.
- examples of cytokines include interferon type I, type II, and type III that regulate immunity, interleukins, tumor necrosis factors, and receptor antagonists thereof.
- growth factors include erythropoietin, which is a hematopoietic factor, and granulocyte colony stimulating factor (GCSF), which is a stimulating factor.
- blood coagulation factors include factor V, factor VII, factor VIII, factor IX, Examples include factor X and factor XII.
- Hormones include calcitonin, insulin, its analogs, exenatide, GLP-1, somatostatin, and human growth hormone.
- antibodies include full-length antibodies and antibody fragments such as Fab and svFV; examples of aptamers include DNA aptamers and RNA aptamers; and examples of enzymes include superoxide dismutase and uricase.
- Suitable proteins include interferons, interleukins, erythropoietin, GCSF, factor VIII, factor IX, human growth hormone, antibody fragments, and more preferably human growth hormone, interferons, GCSF, erythropoietin, or antibodies. Fragments (especially Fab) may be mentioned.
- Suitable peptides include insulin, bivalirudin, teriparatide, exenatide, enfuvirtide, degarelix, mifamurtide, nesiritide, goserelin, glatiramer, octreotide, lanreotide, icatibant, dicotinide, pramlintide, romiplostim, calcitonin, oxytocin, leuprorelin, glucagon. More preferred examples include insulin, exenatide, and calcitonin (especially salmon calcitonin).
- P 2 is a polyoxyethylene derivative having a dehydroxylated group, or a combination of a polyoxyethylene derivative having a dehydroxylated group and a biofunctional molecule.
- polyoxyethylene derivative having a dehydroxyl group is the same as P 1 of the acetal type releasable polyoxyethylene derivative.
- the conjugate of a polyoxyethylene derivative having a dehydroxylated group and a biofunctional molecule is a group in which X 1 of P 1 of the acetal-type releasable polyoxyethylene derivative is D 2 , and D 2 is a biofunctional molecule.
- the details of the group capable of reacting with a biofunctional molecule are the same as the explanation for X1 of the acetal-type releasable polyoxyethylene derivative, and the biofunctional molecule is not particularly limited, but may be a group capable of reacting with a human or other animal.
- Substances related to the diagnosis, cure, mitigation, treatment, or prevention of diseases includes proteins, peptides, nucleic acids, cells, viruses, etc., and suitable proteins or peptides include hormones, cytokines, antibodies, aptamers, enzymes, etc.
- biofunctional molecules in D2 are target-oriented biofunctional molecules, such as antibodies and aptamers, and examples of antibodies include antibody fragments such as Fab, Fab', and F(ab').
- examples of aptamers include peptide aptamers, RNA aptamers, and DNA aptamers.
- the method for producing the acetal type releasable polyoxyethylene conjugate of the present invention will be explained.
- the acetal-type polyoxyethylene derivative and the biofunctional molecule are mixed in a water-soluble organic solvent such as acetonitrile, dimethyl sulfoxide, or N,N-dimethylformamide.
- a purification step is performed to remove by-products.
- Neutral or basic means pH 6.5 to pH 11.0, preferably pH 7.0 to pH 10.5, more preferably pH 7.0 to pH 10.0, particularly preferably pH 7.0 to pH 10.5.
- the pH is 9.0.
- Neutral or basic buffers are buffers that maintain a nearly constant pH (hydrogen ion index) even if a small amount of acid or base is added from the outside, or even if the concentration is changed by dilution. It is an aqueous solution that has a buffering effect to keep it neutral or basic.
- a water-soluble organic solvent can also be added to the buffer.
- the water-soluble organic solvent includes, for example, methanol, ethanol, propanol, isopropanol, tetrahydrofuran, acetone, acetonitrile, dimethyl sulfoxide, N,N-dimethylformamide, triethylamine, pyridine, or hexamethylphosphoric triamide.
- acetonitrile is methanol, ethanol, propanol, isopropanol, tetrahydrofuran, acetone, acetonitrile, dimethylsulfoxide or N,N-dimethylformamide, more preferably acetonitrile, dimethylsulfoxide or N,N-dimethylformamide.
- Specific methods for the purification step performed after the coupling step include ion exchange chromatography, gel filtration chromatography, hydrophobic interaction chromatography, reversed phase chromatography, or affinity chromatography.
- neutral or basic conditions are pH 6.5 to pH 11.0, preferably pH 7.0 to 10.5, more preferably pH 7.0 to 10.0, particularly preferably pH 7. .0 to 9.0.
- Example 1-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT MEH-20T (40 g, 2.0 mmol), toluene (120 g), and 2 ,6-di-tert-butyl-p-cresol (8 mg) was charged, and water was azeotropically removed with toluene. The mixture was cooled to 45°C, charged with chloroform (200g), cooled to 25°C, and charged with 4-hydroxybenzaldehyde (977mg, 8.0mmol) and triphenylphosphine (2.10g, 8.0mmol).
- diisopropyl azodicarboxylate (1.51 g, 7.0 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 224 mg, 7.0 mmol
- the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 1-2 In a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube, compound of formula (20) (1.0 g, 0.05 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were added. (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg), and after dissolving, add molecular sieve 5A (1.0 g) and p-toluenesulfonic acid monomer. Hydrate (11.4 mg, 0.06 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (12.1 mg, 0.12 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (90 g) to which 2,6-di-tert-butyl-p-cresol (18 mg) was added, and hexane (66 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 1-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (21) (250 mg, 0.0125 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (23.7 mg, 0.30 mmol) and p-nitrophenyl chloroformate (40.3 mg, 0.20 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% brine (2.5 g) at pH 12 to remove some impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 2-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT MEH-20T (40 g, 2.0 mmol), toluene (120 g), and 2, 6-di-tert-butyl-p-cresol (8 mg) was charged, and water was azeotropically removed with toluene. After cooling to 45°C and charging chloroform (200g), cooling to 25°C, and adding 3-fluoro-4-hydroxybenzaldehyde (1.12g, 8.0mmol) and triphenylphosphine (2.62g, 10.0mmol). I prepared it.
- diisopropyl azodicarboxylate (1.84 g, 8.6 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol (276 mg, 8.6 mmol) was added, and after stirring for 30 minutes, the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 2-2 The compound of formula (23) (1.0 g, 0.05 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were placed in a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube. (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg), and after dissolving, add molecular sieve 5A (1.0 g) and p-toluenesulfonic acid monomer. Hydrate (11.4 mg, 0.06 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (12.1 mg, 0.12 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (90 g) to which 2,6-di-tert-butyl-p-cresol (18 mg) was added, and hexane (66 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 2-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (24) (180 mg, 0.009 mmol) and dichloromethane (2.4 g), and after dissolving, pyridine (17.1 mg, 0.216 mmol) and p-nitrophenylchloroformate (29.0 mg, 0.144 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% saline (1.8 g) at pH 12 to remove some impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 3-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT MEH-20T (40 g, 2.0 mmol), toluene (120 g), and 2, 6-di-tert-butyl-p-cresol (8 mg) was charged, and water was azeotropically removed with toluene. After cooling to 45°C and charging chloroform (200g), cooling to 25°C, and adding 4-hydroxy-2-methoxybenzaldehyde (1.22g, 8.0mmol) and triphenylphosphine (2.62g, 10.0mmol). I prepared it.
- diisopropyl azodicarboxylate (1.84 g, 8.6 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol (276 mg, 8.6 mmol) was added, and after stirring for 30 minutes, the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 3-2 The compound of formula (26) (1.0 g, 0.05 mmol), 2,4-bis(hydroxymethyl)-p- was placed in a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube. Cresol (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg) were charged and dissolved, followed by molecular sieve 5A (1.0 g) and p-toluenesulfonic acid. Monohydrate (11.4 mg, 0.06 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (12.1 mg, 0.12 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) of pH 12 to remove some low-molecular impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (90 g) to which 2,6-di-tert-butyl-p-cresol (18 mg) was added, and hexane (66 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 3-3 A 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, and stirrer was charged with the compound of formula (27) (300 mg, 0.015 mmol) and dichloromethane (4.0 g), and after dissolving, pyridine (28.5 mg, 0.360 mmol) was charged. mmol) and p-nitrophenylchloroformate (48.4 mg, 0.240 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% saline (3.0 g) at pH 12 to remove some low-molecular impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 4-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT GL2-400HO (10 g, 0.25 mmol), toluene (30 g), and 2, 6-di-tert-butyl-p-cresol (2 mg) was charged, and water was azeotropically removed with toluene. After cooling to 45°C and charging chloroform (50g), cooling to 25°C, and charging 4-hydroxybenzaldehyde (244mg, 2.0mmol) and triphenylphosphine (524mg, 2.0mmol).
- diisopropyl azodicarboxylate (76 mg, 1.75 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 56 mg, 1.75 mmol was added, and after stirring for 30 minutes, the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (80 g) and hexane (60 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 4-2 In a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube, compound of formula (29) (1.0 g, 0.025 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were added. (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg), and after dissolving, add molecular sieve 5A (1.0 g) and p-toluenesulfonic acid monomer. Hydrate (17.1 mg, 0.09 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (12.1 mg, 0.12 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (20 g). After repeated washing with water using 20% saline (20 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (90 g) to which 2,6-di-tert-butyl-p-cresol (18 mg) was added, and hexane (66 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 4-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (30) (250 mg, 0.00625 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (11.9 mg, 0.15 mmol) was charged. mmol) and p-nitrophenylchloroformate (20.3 mg, 0.10 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% brine (2.5 g) at pH 12 to remove some impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 5-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT PTE-200HO (10 g, 0.5 mmol), toluene (30 g), and 2, 6-di-tert-butyl-p-cresol (2 mg) was charged, and water was azeotropically removed with toluene. After cooling to 45°C and charging chloroform (50g), cooling to 25°C, and charging 4-hydroxybenzaldehyde (977mg, 8.0mmol) and triphenylphosphine (2.10g, 8.0mmol).
- diisopropyl azodicarboxylate (1.51 g, 7.0 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 224 mg, 7.0 mmol
- the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 5-2 The compound of formula (32) (1.0 g, 0.05 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were placed in a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube. (1.34g, 8mmol), tetrahydrofuran (5.0g) and 2,6-di-tert-butyl-p-cresol (1.1mg), and after dissolving, add molecular sieve 5A (1.0g) and p-toluenesulfonic acid monomer. Hydrate (45.6 mg, 0.24 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (48.4 mg, 0.48 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (360 g) to which 2,6-di-tert-butyl-p-cresol (18 mg) was added, and hexane (264 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 5-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (33) (250 mg, 0.0125 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (94.8 mg, 1.20 mmol) was charged. mmol) and p-nitrophenylchloroformate (161 mg, 0.80 mmol), and the reaction was carried out at 25°C for 3 hours.
- Example 6-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT HGEO-400HO (10 g, 0.25 mmol), toluene (30 g), and 2, 6-di-tert-butyl-p-cresol (2 mg) was charged, and water was azeotropically removed with toluene. After cooling to 45°C and charging chloroform (50g), cooling to 25°C, and charging 4-hydroxybenzaldehyde (977mg, 8.0mmol) and triphenylphosphine (2.10g, 8.0mmol).
- diisopropyl azodicarboxylate (1.51 g, 7.0 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 224 mg, 7.0 mmol
- the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 6-2 In a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube, the compound of formula (35) (1.0 g, 0.025 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were added. (1.34g, 8.0mmol), tetrahydrofuran (5.0g) and 2,6-di-tert-butyl-p-cresol (1.1mg), and after dissolving, add molecular sieve 5A (1.0g) and p-toluenesulfonic acid. Monohydrate (45.6 mg, 0.24 mmol) was added, and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (48.4 mg, 0.48 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) of pH 12 to remove some low-molecular impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (360 g) to which 2,6-di-tert-butyl-p-cresol (72 mg) was added, and hexane (264 g) was added to perform crystallization, followed by suction filtration to obtain crystals. I got it.
- Example 6-3 A 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, and stirrer was charged with the compound of formula (36) (250 mg, 0.00625 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (94.8 mg, 1.20 mmol) was charged. mmol) and p-nitrophenylchloroformate (161.2 mg, 0.80 mmol), and the reaction was carried out at 25°C for 3 hours. After washing with water repeatedly using 20% saline (2.5 g) at pH 12 to remove some low-molecular impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 7-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT DKH-20T (20 g, 1.0 mmol), toluene (60 g), and 2, 6-di-tert-butyl-p-cresol (4 mg) was charged, and water was azeotropically removed with toluene. The mixture was cooled to 45°C, charged with chloroform (100g), cooled to 25°C, and charged with 4-hydroxybenzaldehyde (977mg, 8.0mmol) and triphenylphosphine (2.10g, 8.0mmol).
- diisopropyl azodicarboxylate (1.51 g, 7.0 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 224 mg, 7.0 mmol
- the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 7-2 In a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and cooling tube, compound of formula (38) (1.0 g, 0.05 mmol) and 2,4-bis(hydroxymethyl)-p-cresol were added. (672 mg, 4.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg), and after dissolving, add molecular sieve 5A (1.0 g) and p-toluenesulfonic acid monomer. A hydrate (22.8 mg, 0.12 mmol) was added and the reaction was carried out at 40°C for 4 hours.
- compound of formula (38) 1.0 g, 0.05 mmol
- 2,4-bis(hydroxymethyl)-p-cresol were added. (672 mg, 4.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p
- N-methylmorpholine (24.2 mg, 0.24 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with chloroform (10 g). After repeated washing with water using 20% saline (10 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (180 g) to which 2,6-di-tert-butyl-p-cresol (36 mg) was added, and hexane (132 g) was added to perform crystallization, followed by suction filtration. Obtained crystals.
- Example 7-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (39) (250 mg, 0.0125 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (47.4 mg, 0.60 mmol) was charged. mmol) and p-nitrophenylchloroformate (80.6 mg, 0.40 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated washing with water using 20% saline (2.5 g) at pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 8-1 The compound of formula (20) (1.0 g, 0.05 mmol) was added to a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and condenser, as described in the literature (Freeman, J. H.; JAm. Chem. Soc. 1952). , 74, 6257-6260), 2,4-di(hydroxymethyl)phenol (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg).
- Example 8-2 A 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, and stirrer was charged with the compound of formula (45) (250 mg, 0.0125 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (23.7 mg, 0.30 mmol) was charged. mmol) and p-nitrophenylchloroformate (40.3 mg, 0.20 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% brine (2.5 g) at pH 12 to remove some impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 9-1 In a 300 mL four-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, Dean-stark tube, and condenser tube, add NOF SUNBRIGHT MEH-40T (40 g, 2.0 mmol), toluene (120 g), and 2, 6-di-tert-butyl-p-cresol (8 mg) was charged, and water was azeotropically removed with toluene. The mixture was cooled to 45°C, charged with chloroform (200g), cooled to 25°C, and charged with 2-hydroxybenzaldehyde (977mg, 8.0mmol) and triphenylphosphine (2.10g, 8.0mmol).
- diisopropyl azodicarboxylate (1.51 g, 7.0 mmol) was added in three portions so that the internal temperature did not exceed 35°C.
- methanol 224 mg, 7.0 mmol
- the solvent was distilled off under reduced pressure. The residue was dissolved in ethyl acetate (320 g), and hexane (240 g) was added to perform crystallization, followed by suction filtration to obtain crystals.
- Example 9-2 The compound of formula (47) (1.0 g, 0.05 mmol) was added to a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, stirrer, and condenser, as described in the literature (Freeman, J. H.; JAm. Chem. Soc. 1952). , 74, 6257-6260), 2,4-di(hydroxymethyl)phenol (336 mg, 2.0 mmol), tetrahydrofuran (5.0 g) and 2,6-di-tert-butyl-p-cresol (1.1 mg).
- Example 9-3 A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (48) (250 mg, 0.0125 mmol) and dichloromethane (3.6 g), and after dissolving, pyridine (23.7 mg, 0.30 mmol) was charged. mmol) and p-nitrophenylchloroformate (40.3 mg, 0.20 mmol), and the reaction was carried out at 25°C for 3 hours. After repeated water washing with 20% brine (2.5 g) at pH 12 to remove some impurities, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure.
- Example 10 A 20 mg/mL buffer solution of ⁇ -alanine was prepared using 0.1 M sodium phosphate buffer solution (pH 8.5). Charge the compound of formula (22) (70 mg, 0.0035 mmol) and a 20 mg/mL buffer solution of ⁇ -alanine (1.5 g) into a 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, and stirrer, and dissolve. Afterwards, the reaction was carried out at 25°C for 6 hours. After dissolving common salt (500 mg), extraction was performed using chloroform (3.0 g), and the organic layer was dried over anhydrous sodium sulfate and filtered. After diluting the filtrate with ethyl acetate (45 g), hexane (33 g) was added to perform crystallization, followed by filtration and drying under reduced pressure to obtain the compound of formula (50).
- 0.1 M sodium phosphate buffer solution pH 8.5
- Example 11 A 20 mg/mL buffer solution of ⁇ -alanine was prepared using 0.1 M sodium phosphate buffer solution (pH 8.5). A 50 mL three-necked flask equipped with a thermometer, nitrogen blowing tube, and stirrer was charged with the compound of formula (25) (70 mg, 0.0035 mmol) and a 20 mg/mL buffer solution of ⁇ -alanine (1.5 g), and dissolved. Afterwards, the reaction was carried out at 25°C for 6 hours. After dissolving common salt (500 mg), extraction was performed using chloroform (3.0 g), and the organic layer was dried over anhydrous sodium sulfate and filtered. After diluting the filtrate with ethyl acetate (45 g), hexane (33 g) was added to perform crystallization, followed by filtration and drying under reduced pressure to obtain the compound of formula (51).
- Example 12 A 20 mg/mL buffer solution of ⁇ -alanine was prepared using 0.1 M sodium phosphate buffer solution (pH 8.5). A 50 mL three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer was charged with the compound of formula (28) (14 mg, 0.0072 mmol) and a 20 mg/mL buffer solution of ⁇ -alanine (3.0 g). After dissolution, reaction was carried out at 25°C for 6 hours. After dissolving common salt (750 mg), extraction was performed using chloroform (4.5 g), and the organic layer was dried over anhydrous sodium sulfate and filtered. After diluting the filtrate with ethyl acetate (45 g), hexane (33 g) was added to perform crystallization, and after filtration, it was dried under reduced pressure to obtain the compound of formula (52).
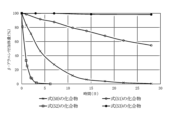
- FIG. 1 shows the amount of ⁇ -alanine adducts at arbitrary timing when the amount of ⁇ -alanine adducts after 0 hours of standing is taken as 100%.
- the half-life (t1/2) calculated from is 3.6 days for the compound of formula (50), 31.5 days for the compound of formula (51), 0.55 days for the compound of formula (52), and 231 days for the compound of formula (53).
- the compounds of the present invention represented by formulas (50), (51), and (52) are under physiological conditions compared with the prior invention, the compound represented by formula (53) and exemplified in Patent Document 3. It was shown to have the desired half-life below.
- NOF ME-200MS ( ⁇ -methyl- ⁇ -[(methylsulfonyl)oxy]poly(oxyethylene), 10 g, 0.5 mmol) was placed in a four-neck flask equipped with a thermometer, nitrogen blowing tube, and stirrer. , 3-methyl-4-hydroxybenzaldehyde (272 mg, 2 mmol), potassium carbonate (0.69 g, 5 mmol), and acetonitrile (50 g) were charged, and the mixture was reacted at 80° C. for 6 hours. After the reaction, it was filtered, concentrated, and redissolved in dichloromethane (80 g).
- Example 14-2 The compound of formula (54) (3.0 g, 0.15 mmol) and 2,4-bis(hydroxymethyl)-p-cresol (1.0 g, 6.0 mmol), tetrahydrofuran (15.0 g) and 2,6-di-tert-butyl-p-cresol (3.3 mg), and after dissolving, add molecular sieve 5A (3.0 g) and p-toluenesulfonic acid monohydrate. hydrate (34.2 mg, 0.18 mmol) was added thereto, and the reaction was carried out at 40°C for 4 hours.
- N-methylmorpholine (36.4 mg, 0.36 mmol) was added, stirred for a while, filtered, and the filtrate was diluted with toluene (33.0 g). After repeated washing with water using 20% brine (30 g) of pH 12, the organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was distilled off under reduced pressure. The residue was dissolved in toluene containing 2,6-di-tert-butyl-p-cresol, and hexane was added to perform crystallization, followed by suction filtration and drying under reduced pressure to obtain the compound of formula (55). Obtained.
- Example 14-3 In a three-necked flask equipped with a thermometer, a nitrogen blowing tube, and a stirrer, the compound of formula (55) (500 mg, 0.025 mmol), 2,6-di-tert-butyl-p-cresol (0.5 mg), and toluene were added. (6.0 g) was charged, and after dissolving, triethylamine (38 mg, 0.375 mmol) and p-nitrophenylchloroformate (50.4 mg, 0.25 mmol) were charged, and the reaction was carried out at 60°C for 3 hours.
- Example 15-1 In the same manner as in Example 14-1 except that 4-hydroxy-3,5-dimethylbenzaldehyde (300 mg, 2.0 mmol) was used instead of 3-methyl-4-hydroxybenzaldehyde, a product represented by formula (57) was prepared. The compound was obtained.
- Example 15-2 A compound represented by formula (58) was obtained in the same manner as in Example 14-2, except that the compound represented by formula (57) (3.0 g, 0.15 mmol) was used instead of the compound represented by formula (54).
- Example 15-3 A compound represented by formula (59) was obtained in the same manner as in Example 14-3, except that the compound represented by formula (58) (500 mg, 0.025 mmol) was used instead of the compound represented by formula (55).
- Example 16-1 A compound represented by formula (60) was obtained in the same manner as in Example 14-1 except that 2-hydroxybenzaldehyde (244 mg, 2.0 mmol) was used instead of 3-methyl-4-hydroxybenzaldehyde.
- Example 16-2 A compound represented by formula (61) was obtained in the same manner as in Example 14-2 except that the compound represented by formula (60) (3.0 g, 0.15 mmol) was used instead of the compound represented by formula (54).
- Example 16-3 A compound represented by formula (62) was obtained in the same manner as in Example 14-3 except that the compound represented by formula (61) (500 mg, 0.025 mg) was used instead of the compound represented by formula (55).
- Example 17-1 A compound represented by formula (63) was obtained in the same manner except that 2-hydroxy-6methoxybenzaldehyde (304 mg, 2.0 mmol) was used instead of 3-methyl-4-hydroxybenzaldehyde in Example 14-1. Ta.
- Example 17-2 A compound represented by formula (64) was obtained in the same manner as in Example 14-2 except that the compound represented by formula (63) (3.0 g, 0.15 mmol) was used instead of the compound represented by formula (54).
- Example 17-3 A compound represented by formula (65) was obtained in the same manner as in Example 14-3 except that the compound represented by formula (64) (500 mg, 0.025 mmol) was used instead of the compound represented by formula (55).
- Example 18-1 A compound represented by formula (66) was obtained in the same manner as Example 17-1 except that NOF ME-050MS (2.5 g, 0.5 mmol) was used instead of NOF ME-200MS. Ta.
- Example 18-2 A compound represented by formula (67) was obtained in the same manner except that compound (66) (0.75 g, 0.15 mmol) was used in place of compound (63) in Example 17-2.
- Example 18-3 A compound represented by formula (68) was obtained in the same manner as in Example 17-3, except that the compound represented by formula (67) (125 mg, 0.025 mmol) was used instead of compound (64).
- Example 19-1 A compound represented by formula (69) was obtained in the same manner as Example 17-1 except that NOF ME-020MS (1 g, 0.5 mmol) was used instead of NOF ME-200MS. .
- Example 19-2 A compound represented by formula (70) was obtained in the same manner except that compound (69) (300 mg, 0.15 mmol) was used in place of compound (63) in Example 17-2.
- Example 19-3 A compound represented by formula (71) was obtained in the same manner as in Example 17-3, except that the compound represented by formula (70) (50 mg, 0.025 mmol) was used instead of compound (64).
- Example 20-1 A compound represented by formula (72) was prepared in the same manner as in Example 14-1 except that 4-hydroxy-2,6 dimethoxybenzaldehyde (364 mg, 2.0 mmol) was used instead of 3-methyl-4-hydroxybenzaldehyde. I got it.
- Example 20-2 A compound represented by formula (73) was obtained in the same manner as in Example 14-2, except that the compound represented by formula (72) (3.0 g, 0.15 mmol) was used instead of the compound represented by formula (54).
- Example 20-3 A compound represented by formula (74) was obtained in the same manner as in Example 14-3 except that the compound represented by formula (73) (500 mg, 0.025 mmol) was used instead of the compound represented by formula (55).
- Example 21 A 20 mg/mL buffer solution of ⁇ -alanine was prepared using 0.1 M sodium phosphate buffer solution (pH 8.5).
- the compound of formula (56) (74 mg, 0.0037 mmol) was dissolved in a 20 mg/mL ⁇ -alanine buffer solution (1.5 mL), and the reaction was carried out at 25° C. for 6 hours. After the reaction, the mixture was diluted with 0.1M sodium phosphate buffer (pH 8.5) containing 20% by weight of sodium chloride, and extracted using chloroform (3 g). The organic layer was dried over anhydrous sodium sulfate and then filtered. After diluting the filtrate with ethyl acetate (45 g), hexane (33 g) was added to perform crystallization, and after filtration, it was dried under reduced pressure to obtain the compound of formula (75).
- Example 22 A compound of formula (76) was obtained in the same manner as in Example 21 except that the compound of formula (59) (74 mg, 0.0037 mmol) was used instead of the compound of formula (56).
- Example 23 A compound of formula (77) was obtained in the same manner as in Example 21, except that the compound of formula (62) (74 mg, 0.0037 mmol) was used instead of the compound of formula (56).
- Example 24 A compound of formula (78) was obtained in the same manner as in Example 21 except that the compound of formula (65) (74 mg, 0.0037 mmol) was used instead of the compound of formula (56).
- FIG. 3 shows the amount of ⁇ -alanine adducts at arbitrary timing when the amount of ⁇ -alanine adducts after 0 hours of standing is taken as 100%.
- the half-life (t1/2) calculated from the formula [Equation 1] described in the degradability test based on the approximate formula in the graph is 2.3 days for the compound of formula (75) and 30.1 days for the compound of formula (76). 23.1 days for the compound of formula (77) and 1.4 days for the compound of formula (78). In comparison, it was shown to have a targeted half-life under physiological conditions.
- FIG. 5(A) shows the HPLC measurement results of fractionated PEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ).
- Comparative example 3 was prepared in the same manner except that the compound of formula (28) (22.5 mg, 1.13 mmol) was used instead of SUNBRIGHT MENP-20T manufactured by NOF Corporation. 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ) was obtained in which the compound of 28) was combined with Apt-TNF ⁇ at a molar ratio of 1:1.
- FIG. 5(B) shows the HPLC measurement results of the fractionated 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ).
- Results are expressed as mean ⁇ standard error, P values were calculated by Student's t test, and a two-tailed P value of less than 0.05 was defined as a statistically significant difference.
- TNF ⁇ inhibitory activity was confirmed for NH 2 -C6-(Apt-TNF ⁇ ) and 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ), while PEG(20k)-OCO-NH TNF ⁇ inhibitory activity could not be confirmed with -C6-(Apt-TNF ⁇ ).
- 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ) of the present invention has a physiological It has been shown that functional substances can be released in a sustained manner and pharmacological activity can be improved.
- Figure 6 uses PEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ), 2MPEG(20k)-OCO-NH-C6-(Apt-TNF ⁇ ), and NH 2 -C6-(Apt-TNF ⁇ ). This is an evaluation result of TNF ⁇ inhibitory activity.
- ⁇ HPLC device Nexera (Shimadzu Corporation) ⁇ Column: Asahipack ES-502N 7C (7.5x100mm, 9um, Showa Denko) ⁇ Flow rate: 1mL/min ⁇ Analysis time: 30 minutes ⁇ Column temperature: 25°C ⁇ Injection volume: 50 ⁇ L ⁇ Detector: PDA (measurement wavelength: 280nm) ⁇ Mobile phase A: 10mM ammonium formate buffer (pH8.5) ⁇ Mobile phase B: 10mM ammonium formate buffer containing 0.25M ammonium sulfate (pH 8.5) ⁇ Gradient program (A/B): 100/0 (0 minutes) ⁇ 67/33 (20 minutes) ⁇ 0/100 (30 minutes)
- Example 26 2MPEG(20k)-OCO-NH-insulin was obtained in the same manner as Comparative Example 4 except that the compound of formula (28) was used instead of SUNBRIGHT MENP-20T manufactured by NOF Corporation.
- FIG. 7(B) shows the HPLC analysis results of purified 2MPEG(20k)-OCO-NH-insulin.
- FIG. 8(A) shows the amount of PEG(20k)-OCO-NH-insulin at an arbitrary timing when the amount of PEG(20k)-OCO-NH-insulin after standing still for 0 hours is taken as 100%.
- B) represents the amount of 2MPEG(20k)-OCO-NH-insulin at an arbitrary timing when the amount of 2MPEG(20k)-OCO-NH-insulin is taken as 100%.
- the acetal-type releasable polyoxyethylene derivative of the present invention is capable of hydrolyzing the acetal structure under physiological conditions and gradually releasing biofunctional molecules. It can improve the pharmacological action of the molecule.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polyethers (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
上記のような課題に対するアプローチとして、生体機能性分子に一時的結合を介して水溶性ポリマーを化学修飾し、生体内でこの一時的結合を開裂させて化学修飾されていない生体機能性分子を放出させる方法、すなわちプロドラッグ化の方法が用いられる。
具体的には、酸性条件下で加水分解されるアセタール構造を有するリンカーの加水分解をトリガーとして1,4-または1,6-ベンジル脱離により一時結合であるカーバメート結合を開裂し、化学修飾されていない生体機能性分子を放出することが報告されている。
[1] 下記の式(1)、式(2)、式(3)または式(4)で表され、生理的条件下で開裂することを特徴とする、アセタール型リリーサブルポリオキシエチレン誘導体。

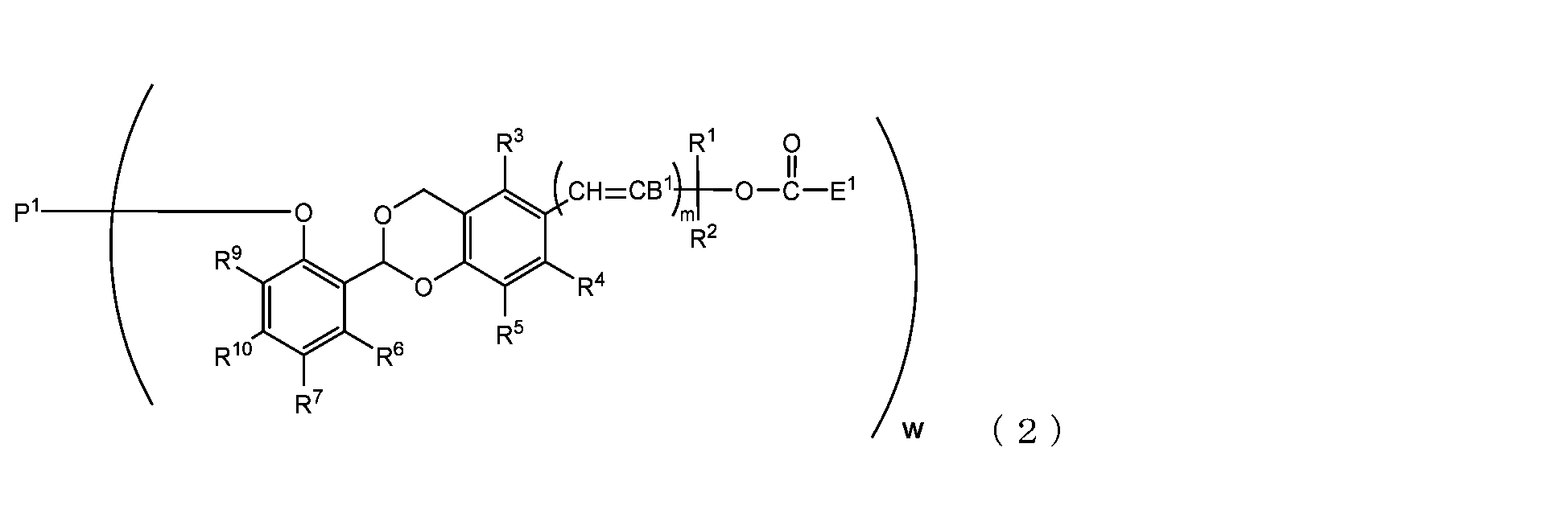
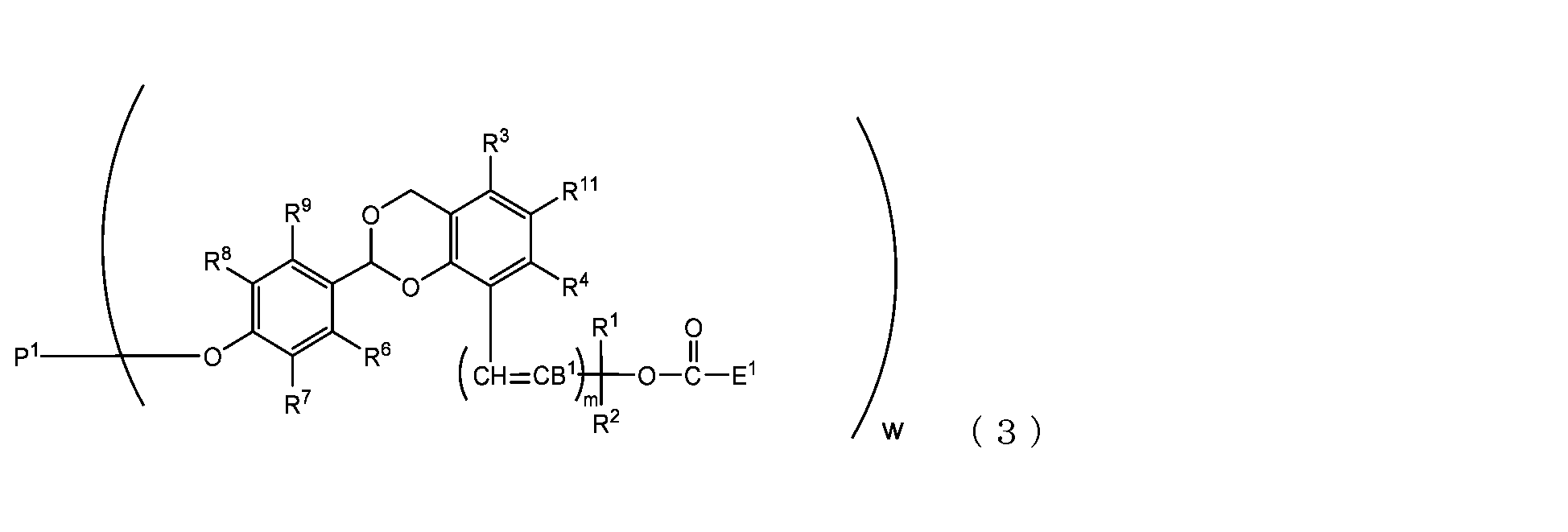
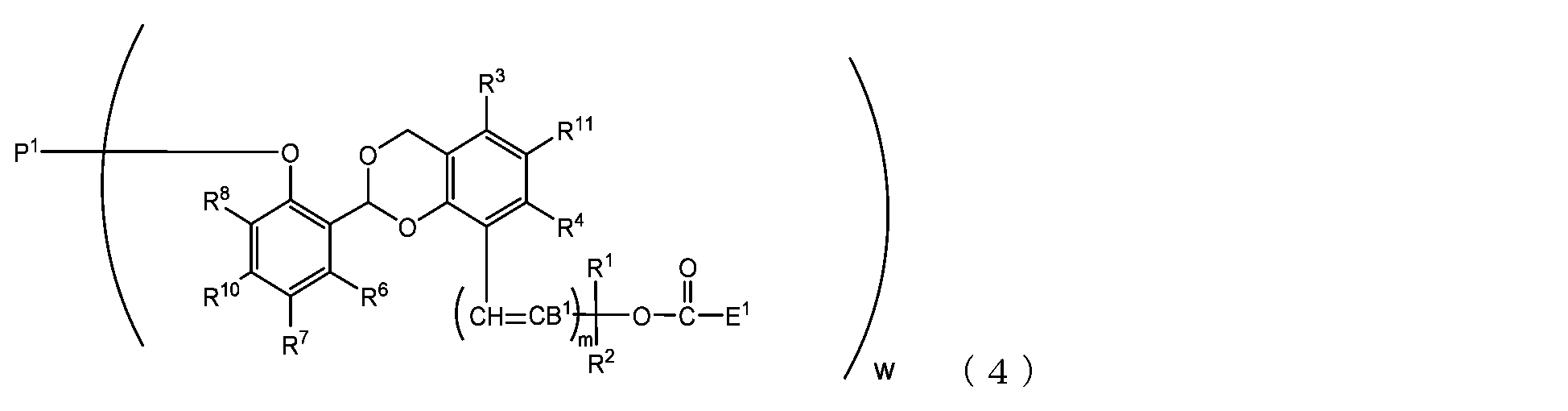
B1は水素原子または-C(R1)(R2)OC(O)E1であり、
E1は脱離基であり、
P1は、脱ヒドロキシ基を有するポリオキシエチレン誘導体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5及びR11は,それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9及びR10は、それぞれ独立して、電子求引性置換基、電子供与性置換基または水素原子であり、
mは0または1である。)
ポリオキシエチレン誘導体とヒドロキシベンズアルデヒド誘導体とをカップリングさせることによって、下記式(5)または式(6)で表されるカップリング生成物を得るカップリング工程、
前記カップリング工程後に、酸性条件下で、前記式(5)または前記式(6)で表される前記カップリング生成物を、2位にヒドロキシメチル基を有し、かつ4位または6位に置換基(-CH=CB1)mC(R1)(R2)-OH(B1、m、R1、R2は前述のとおりである)を有するフェノールと反応させることで、アセタール構造体を得るアセタール化工程、および前記アセタール化工程後に、4位または6位の前記置換基の末端に脱離基構造(-OC(O)E1)を導入する脱離基構造導入工程を備えることを特徴とする、アセタール型リリーサブルポリオキシエチレン誘導体の製造方法。


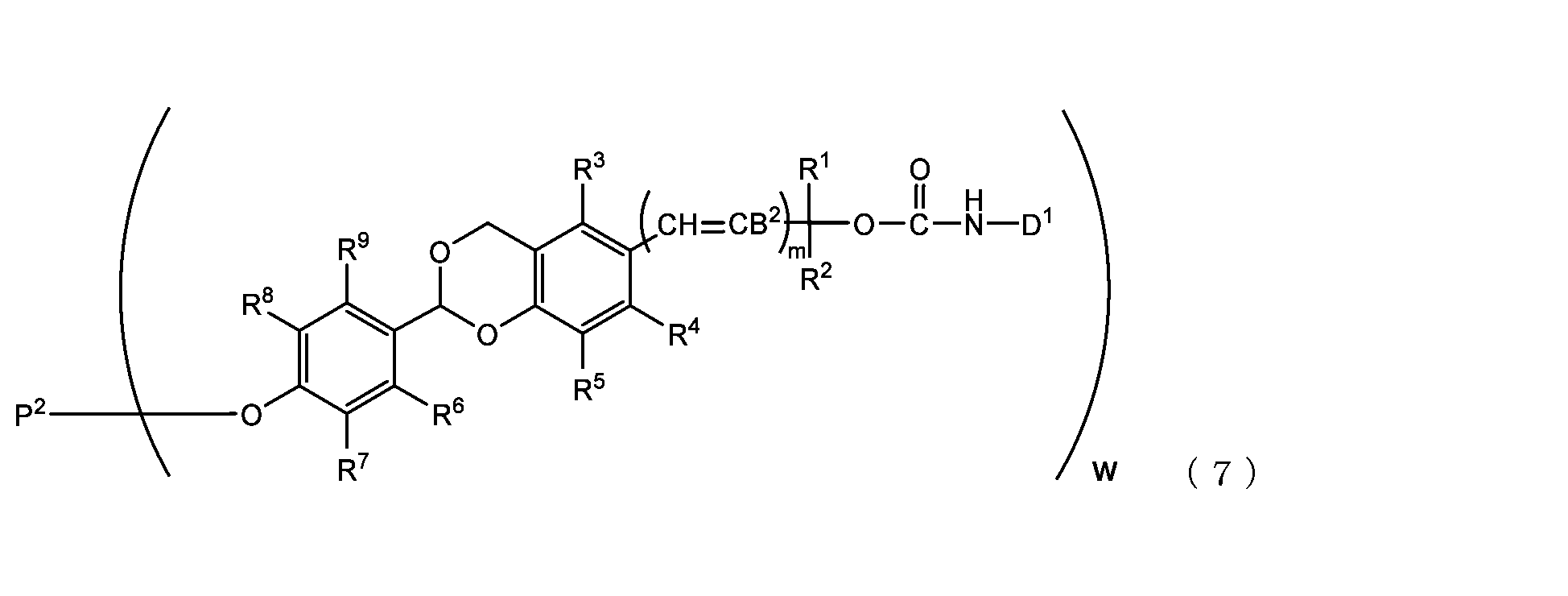

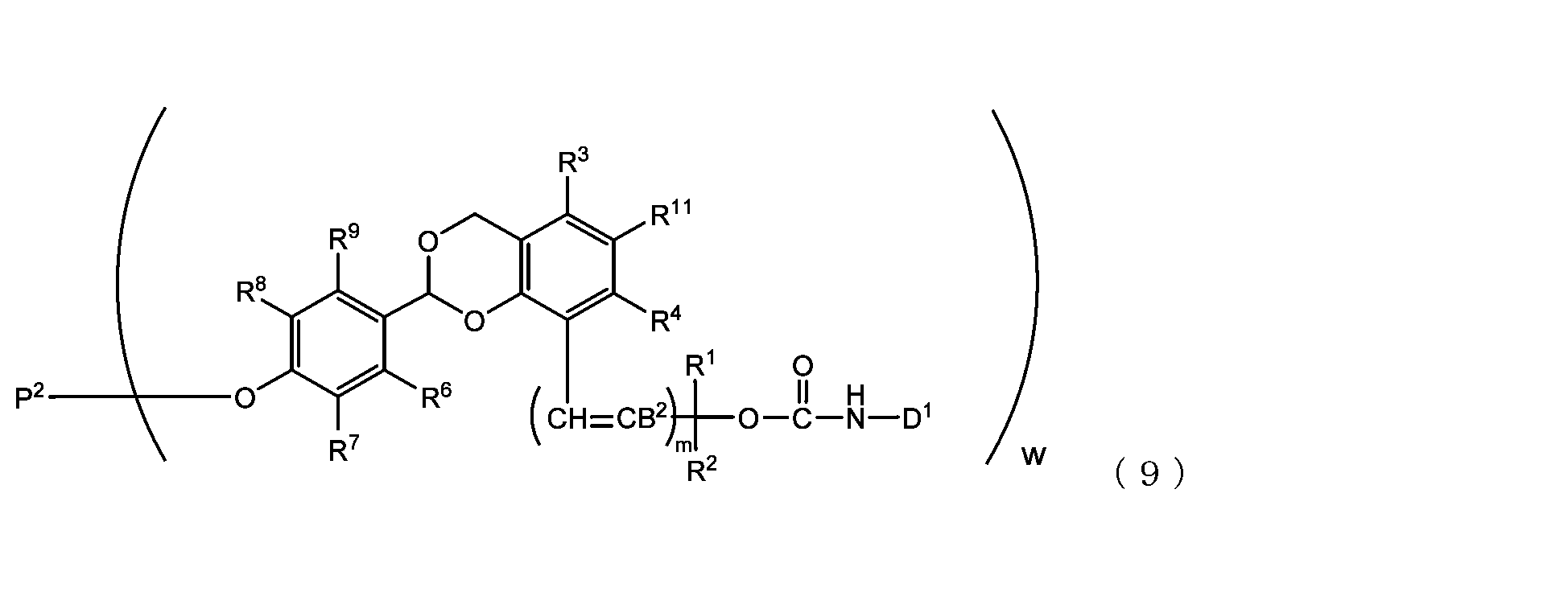

B2は水素原子または-C(R1)(R2)OC(O)NHD1であり、
D1は、生体機能性分子に含まれるアミノ基から、カーバメート結合を構成するアミノ基を除いた残基であり、
P2は、脱ヒドロキシ基を有するポリオキシエチレン誘導体、または脱ヒドロキシ基を有するポリオキシエチレン誘導体および生体機能性分子の結合体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5およびR11は、それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9およびR10は、それぞれ独立して、電子求引性置換基、電子供与性置換基または水素原子であり、
mは0または1である。)
[1]または[2]のアセタール型リリーサブルポリオキシエチレン誘導体と生体機能性分子を、水溶性の有機溶媒が含まれていてもよい中性または塩基性の緩衝液中で反応させるカップリング工程、および
前記カップリング工程後の中性または塩基性条件下での精製工程
を備えることを特徴とする、アセタール型リリーサブルポリオキシエチレン結合体の製造方法。
ここで、特許文献3の化合物中のアセタール構造は、一般的にはジオールやカルボニル基の保護基として用いられるものであり、その脱保護は酸性条件下で行われることが知られており、中性や塩基性条件下では安定である。ゆえに、特許文献3の化合物中、生理的条件下、すなわち中性条件下で適切な速度で加水分解し、生理機能性分子を放出するような化合物の創出は示唆されていない。
本発明は、生理的条件下でアセタールが加水分解し、ベンジル脱離を経て、生体機能性分子が徐々に放出されることを特徴とする。本明細書中における「生理的条件下」とは、pH6.0~8.0である。
また、生理的条件下における生体機能性分子の放出速度は、pH7.4の緩衝液中でのアセタールの半減期として評価することができる。pH7.4の緩衝液中でのアセタールの半減期は、0.5日~35日であることが好ましい。ここで、「半減期」とはアセタールが加水分解され、1/2当量がアルデヒドとなるまでに要する時間を表す。



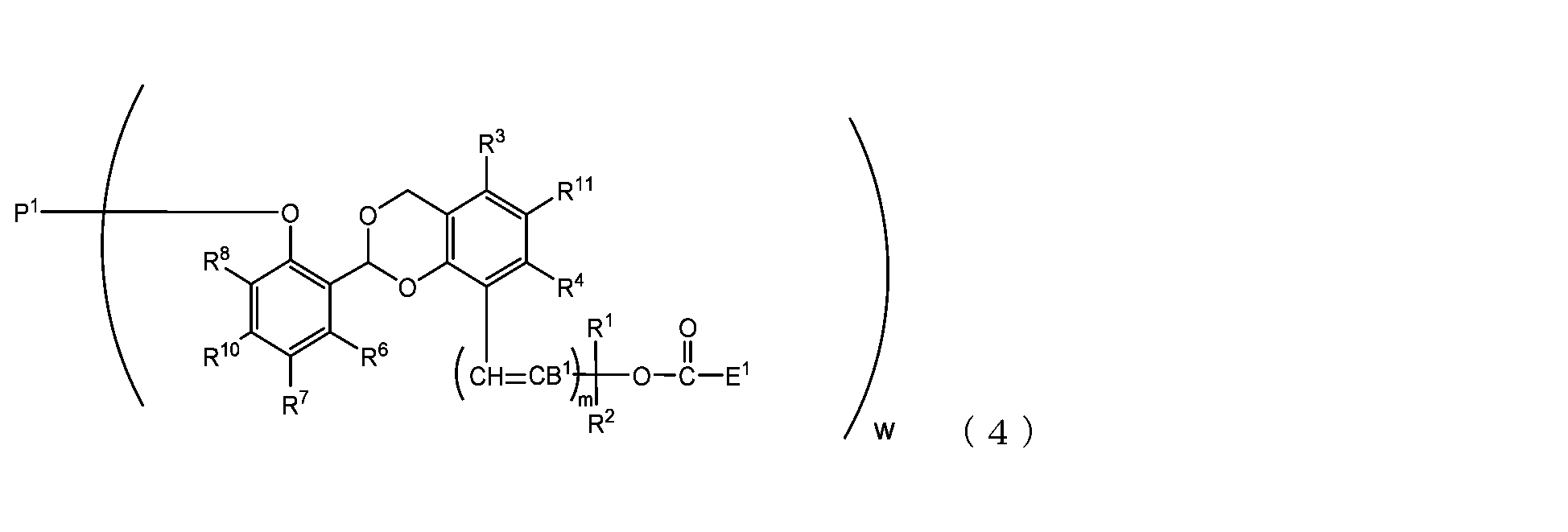
B1は水素原子または-C(R1)(R2)OC(O)E1であり、
E1は脱離基であり、
P1は、脱ヒドロキシ基を有するポリオキシエチレン誘導体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5及びR11は,それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9及びR10は、それぞれ独立して、電子求引性置換基、電子供与性の置換基または水素原子であり、
mは0または1である。
P1の好適例は、Wの数に応じて、以下の残基であることが好ましい。

Z1は2価のスペーサーまたは単結合であり、
n及びlはそれぞれ独立して3~2,000であり、
sは0または1であり、tは2または3であり、vは0または2である。)




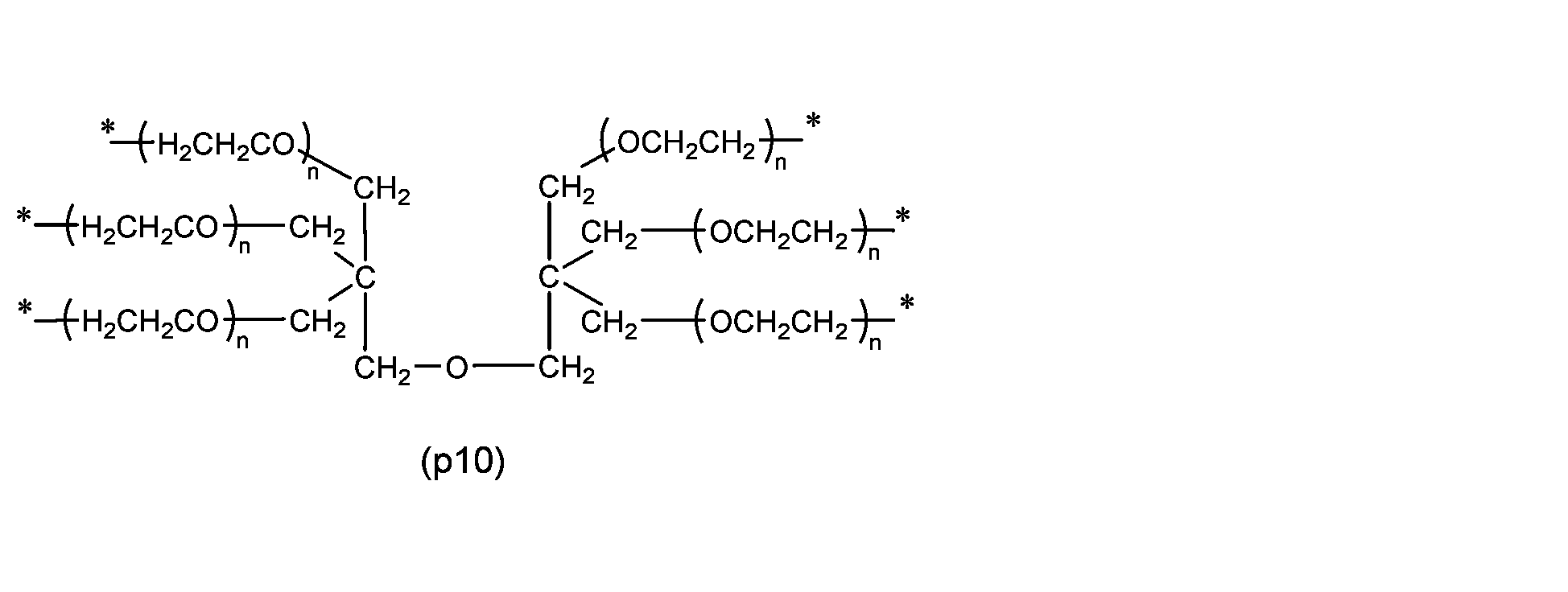
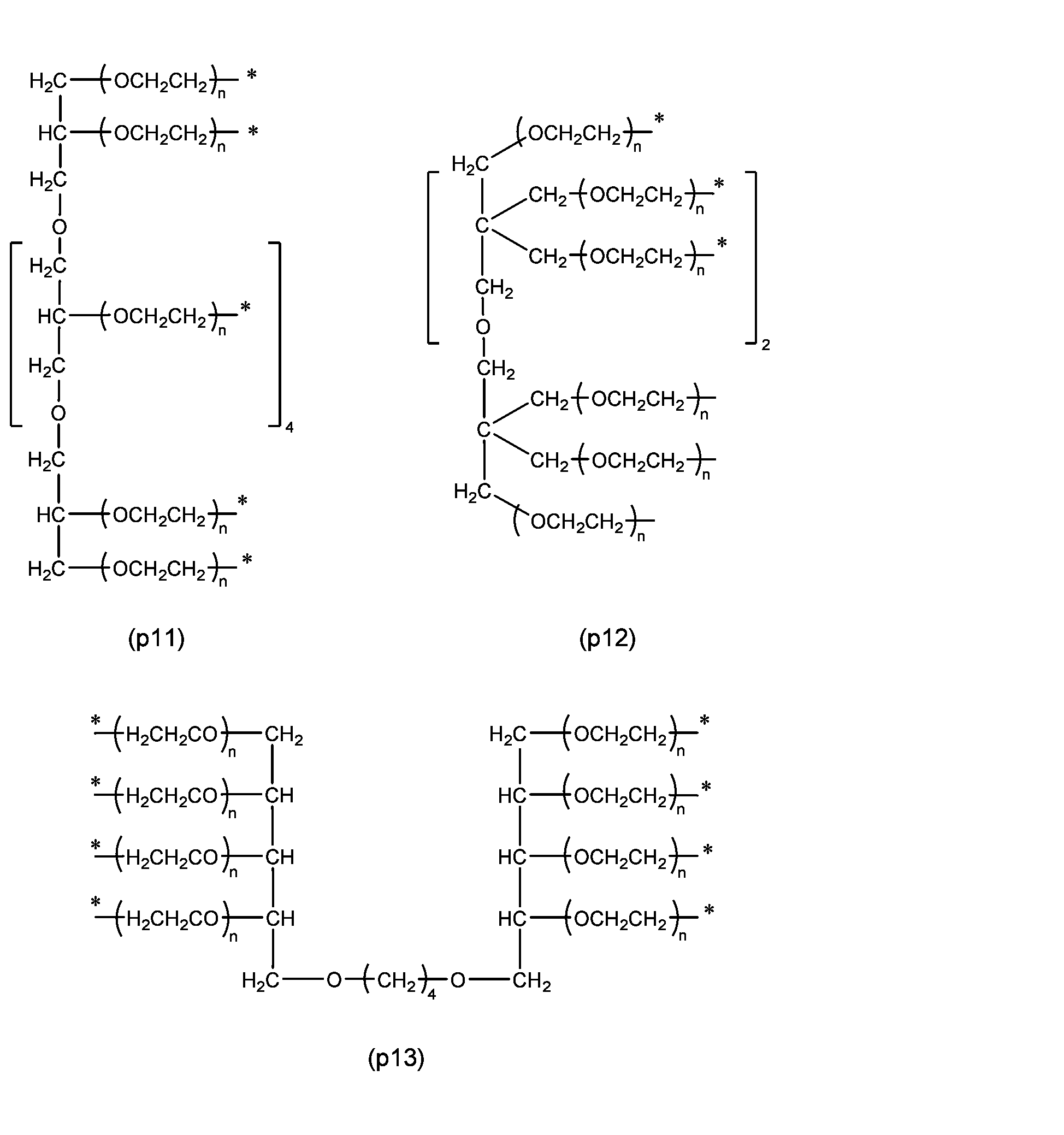
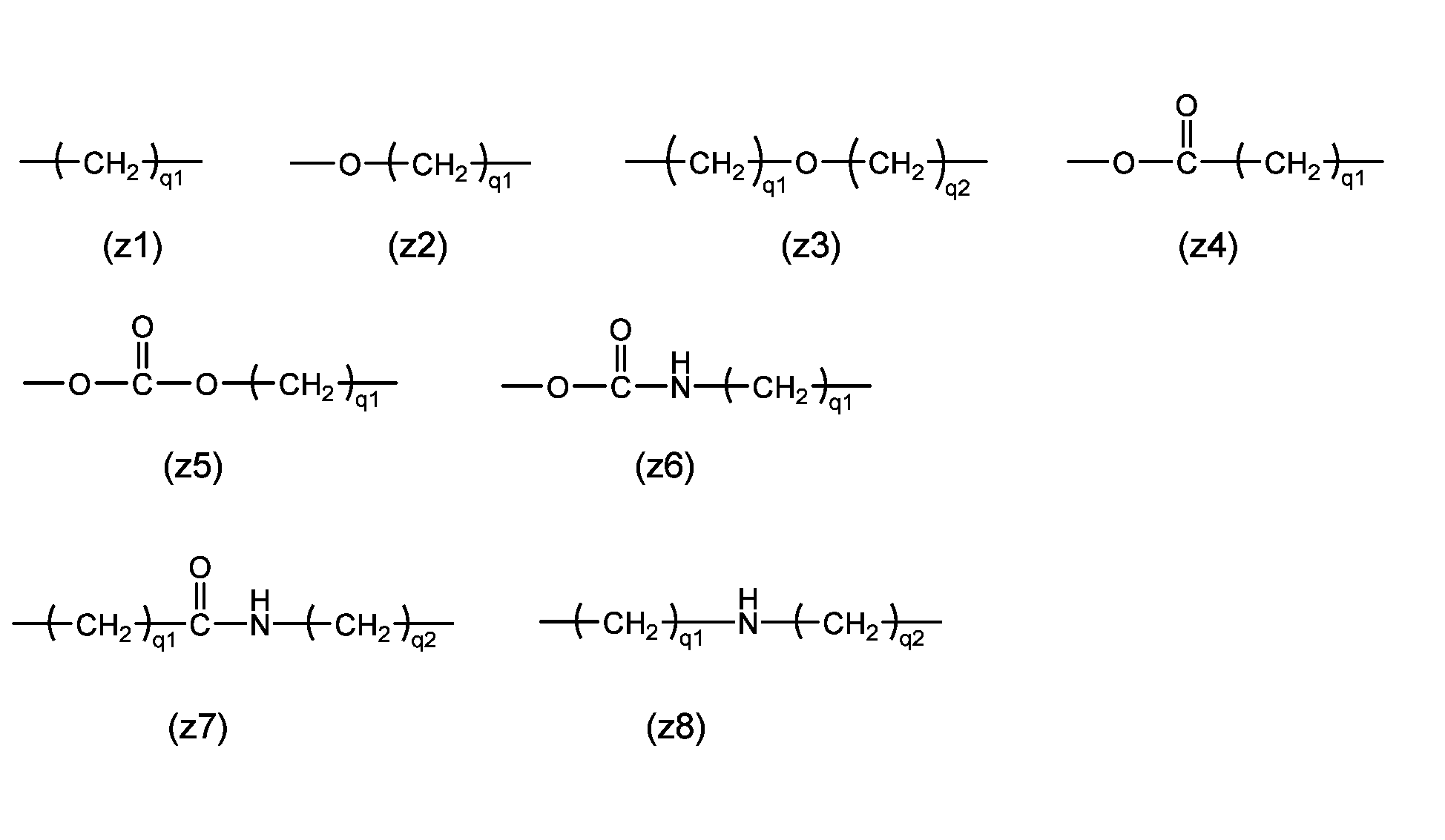
炭化水素基の具体的な例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、t-ブチル基、ペンチル基、イソペンチル基、ヘキシル基、へプチル基、2-エチルヘキシル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基、ノナデシル基、エイコシル基、ヘンエイコシル基、ドコシル基、トイコシル基、テトラコシル基、フェニル基、ベンジル基、クレジル基、ブチルフェニル基、ドデシルフェニル基及びトリチル基などが挙げられ、好ましくは炭素数1~10の炭化水素基、より好ましくはメチル基またはエチル基であり、更に好ましくはメチル基である。
この態様の好適な実施形態において、生体機能性分子と反応可能な基は群(II)、群(III)、群(IV)または群(V)で示される基である。なお、「**」はZ1との結合点を表す。
下記の(a)、(b)、(e)及び(f)
群(III):生体機能性分子のチオール基と反応して共有結合を形成することが可能な官能基
下記の(a)、(b)、(c)、(d)、(e)、(f)及び(h)
群(IV):生体機能性分子のアルキニル基と反応して共有結合を形成することが可能な官能基
下記の(c)、(d)及び(g)
群(V):生体機能性分子のアジド基と反応して共有結合を形成することが可能な官能基
下記の(i)及び(j)
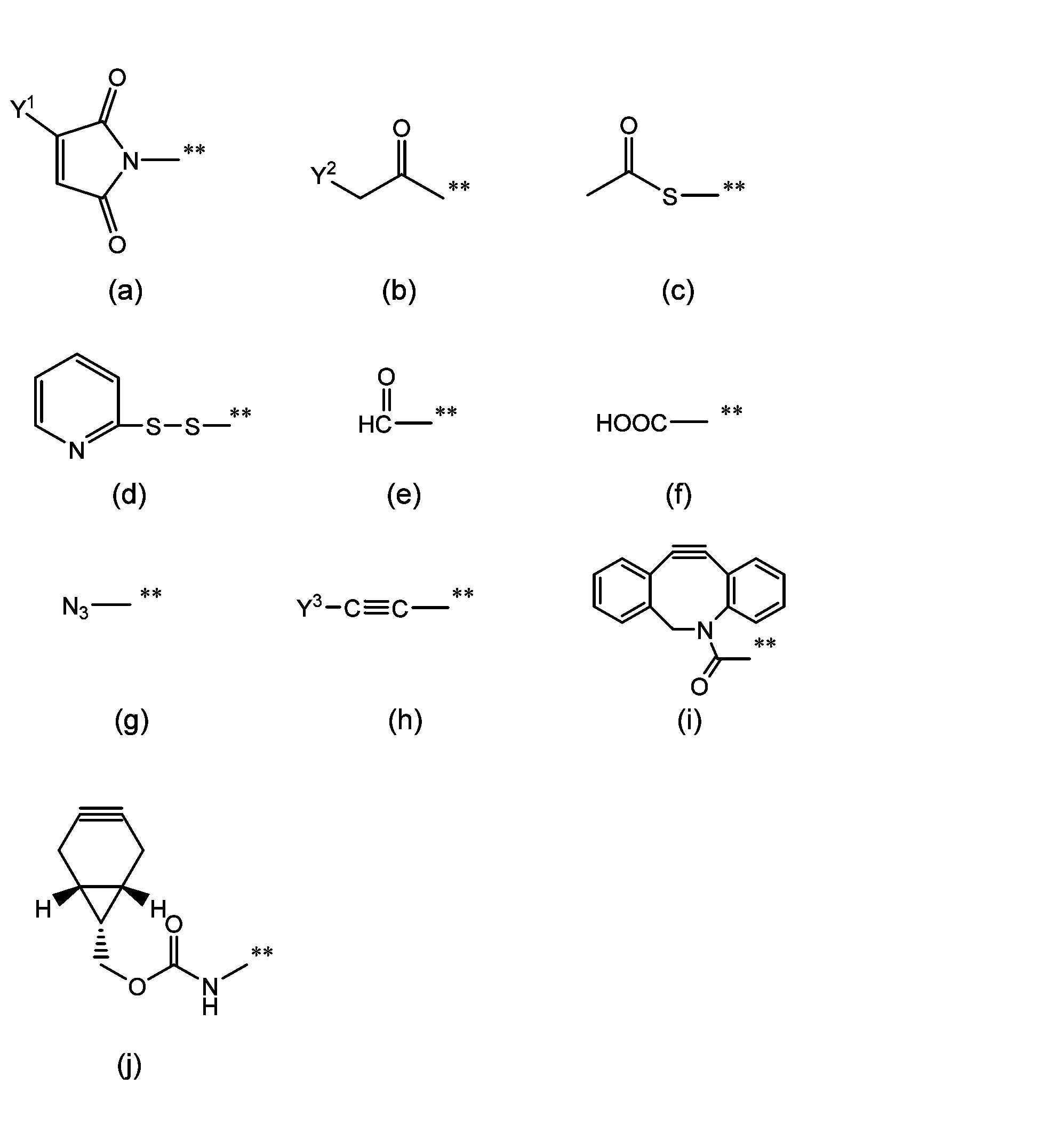
式(p2)、(p4)及び式(p8)において、sは0または1であり、tは2または3であり、vは0または2であり、好ましい実施形態は、下式(p14)~(p19)で表される残基である。
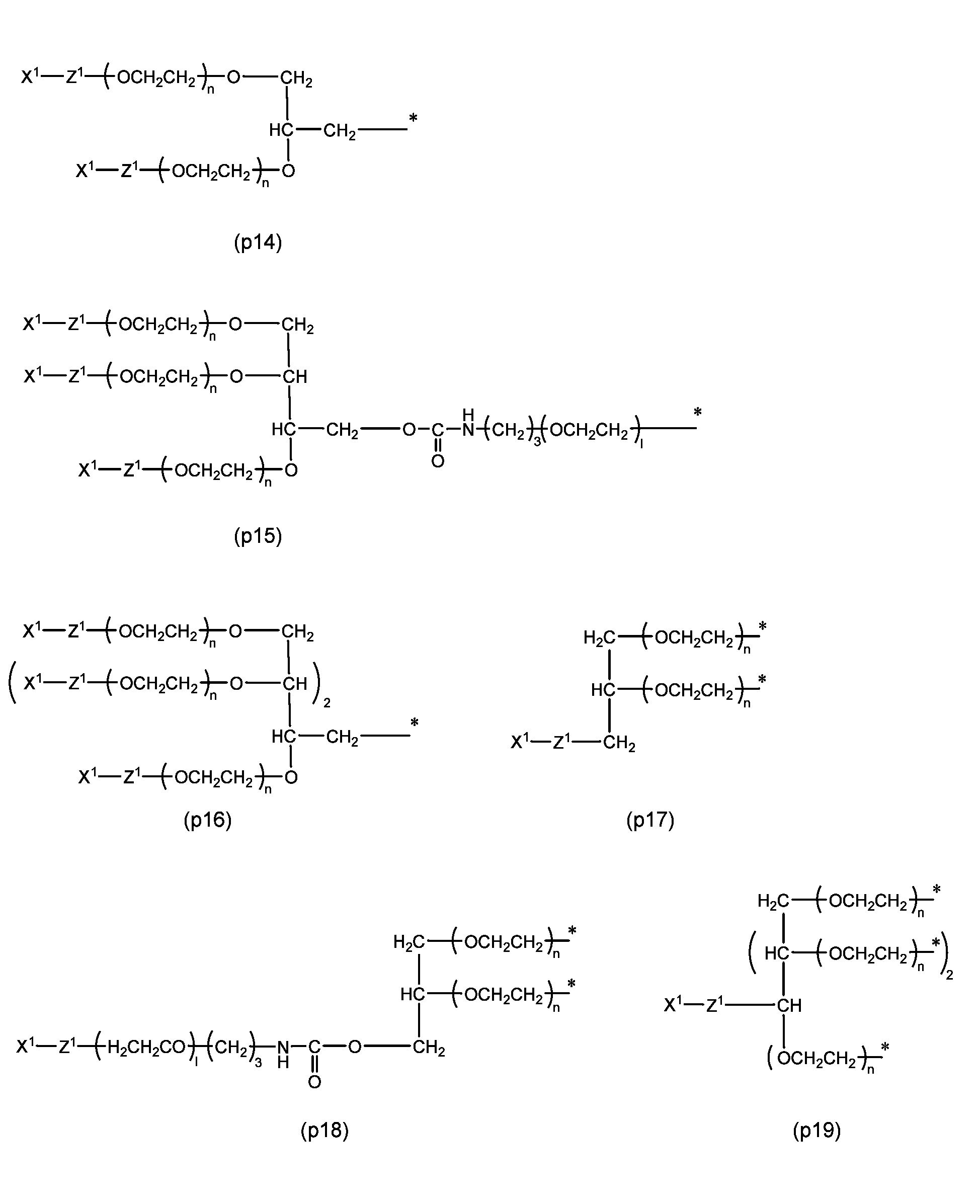


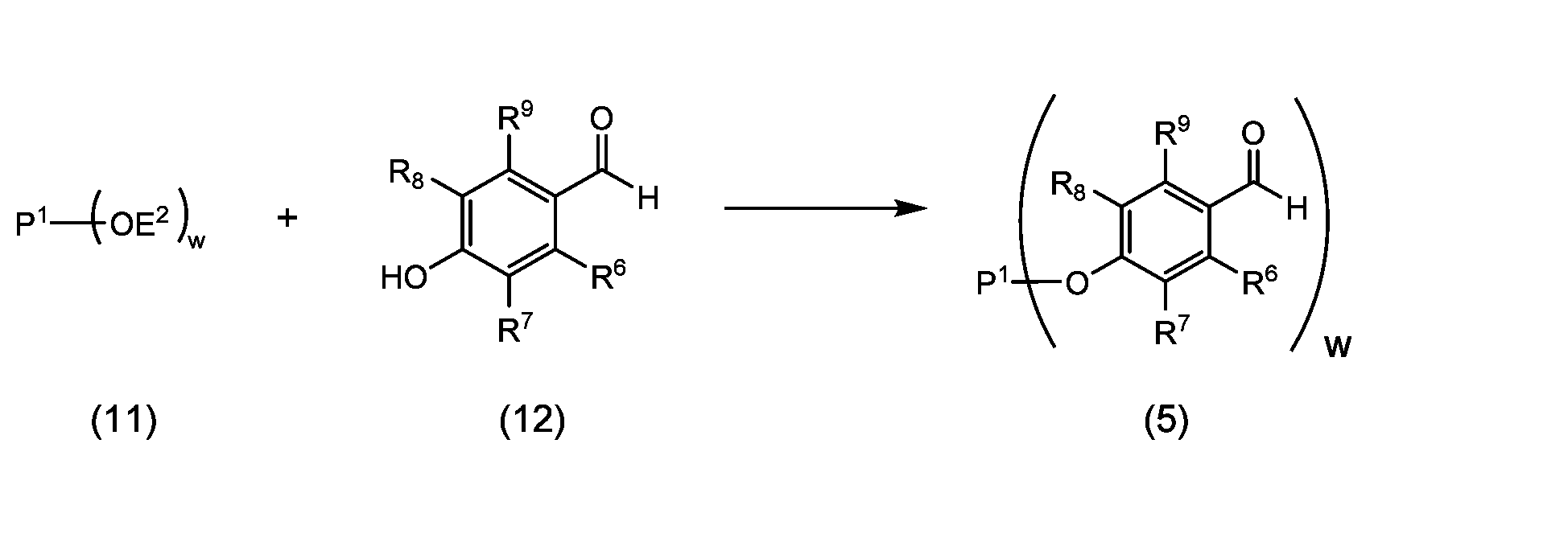
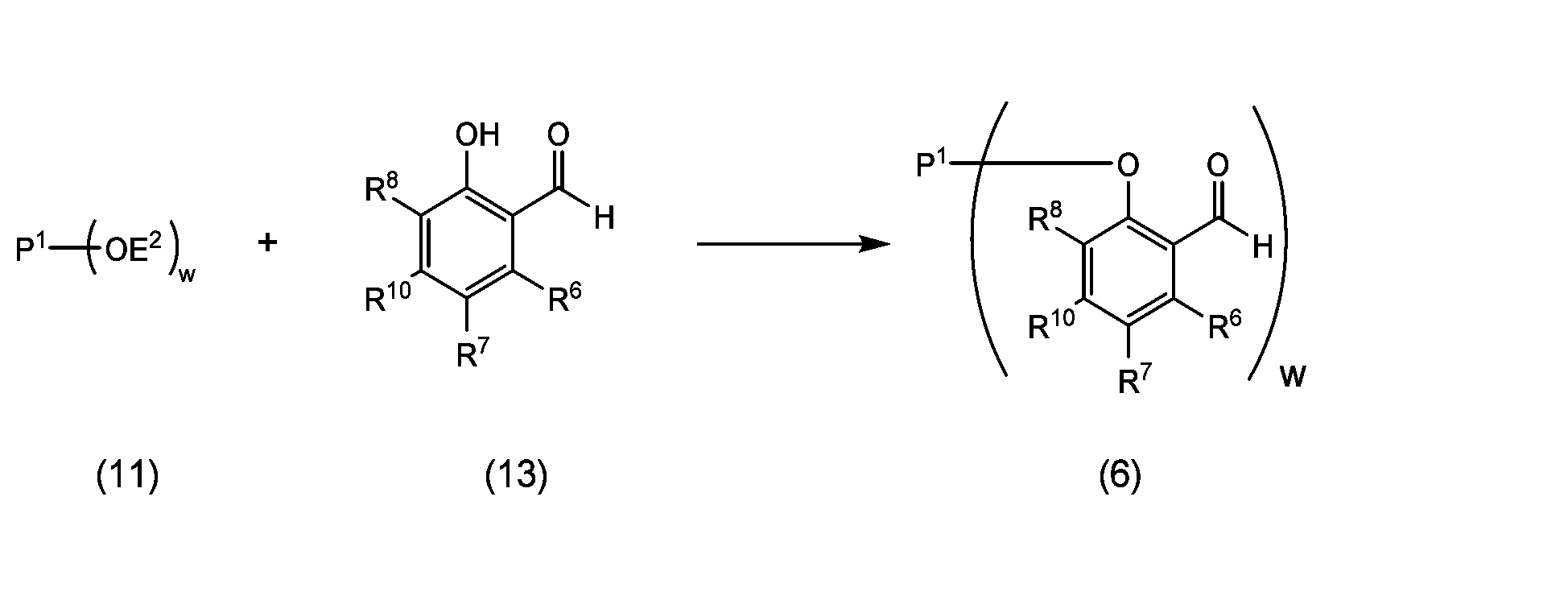

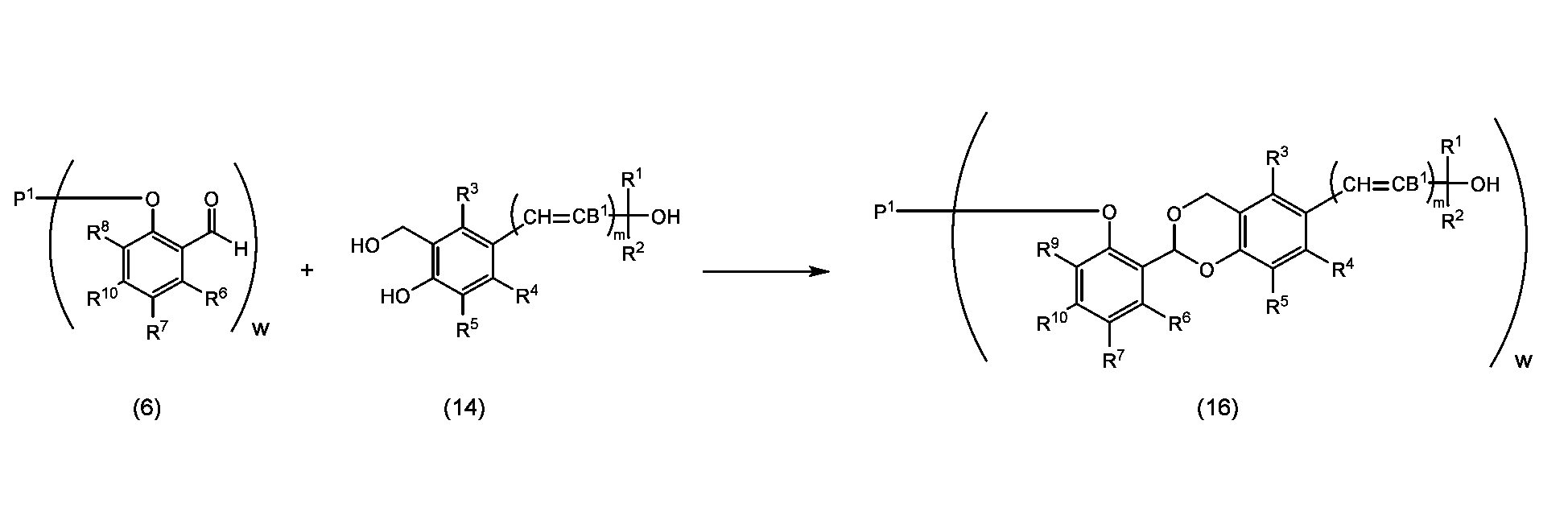
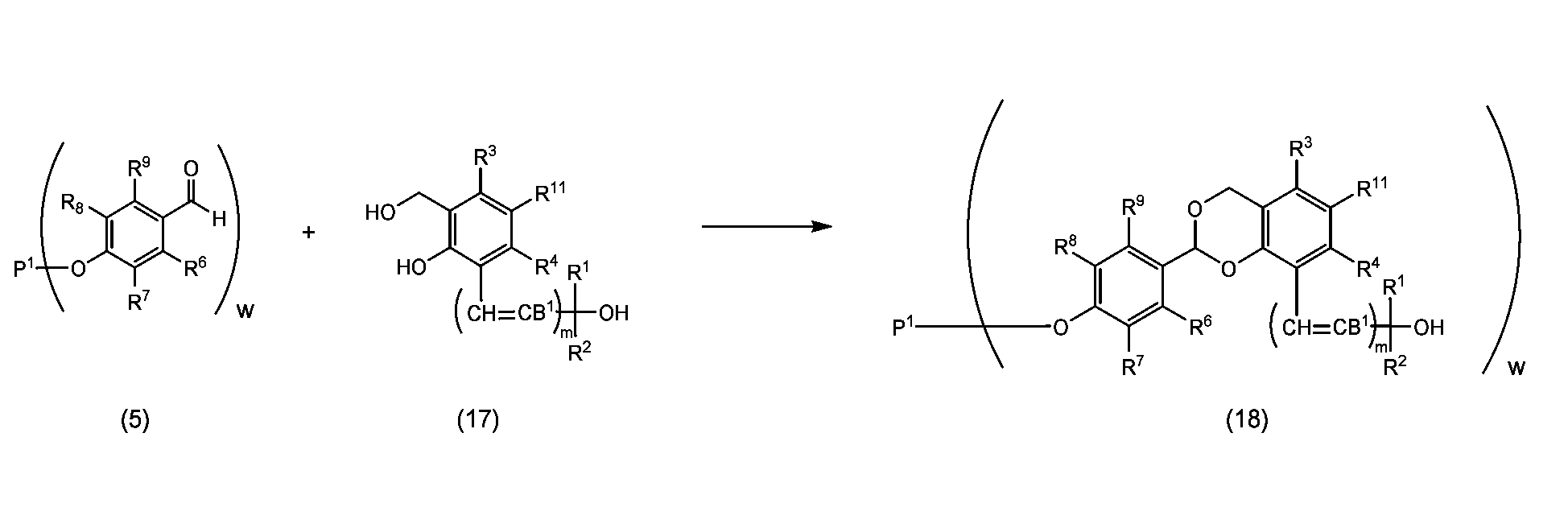



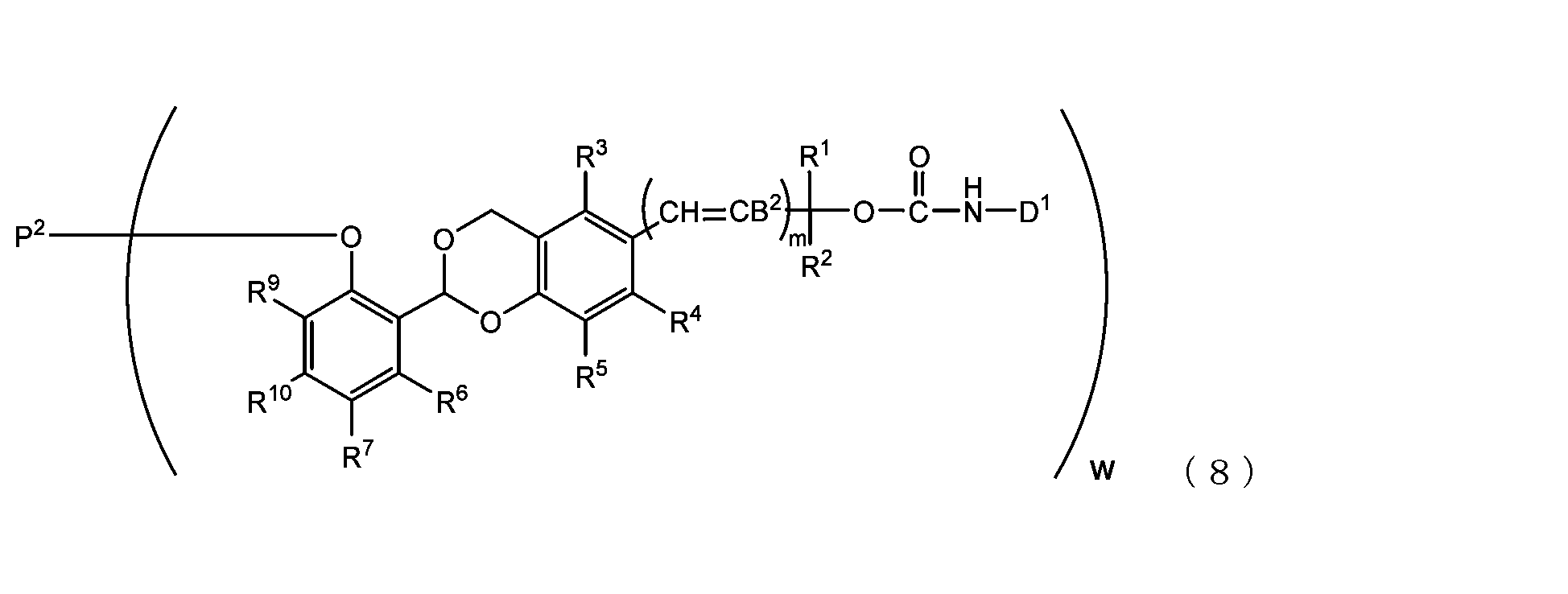
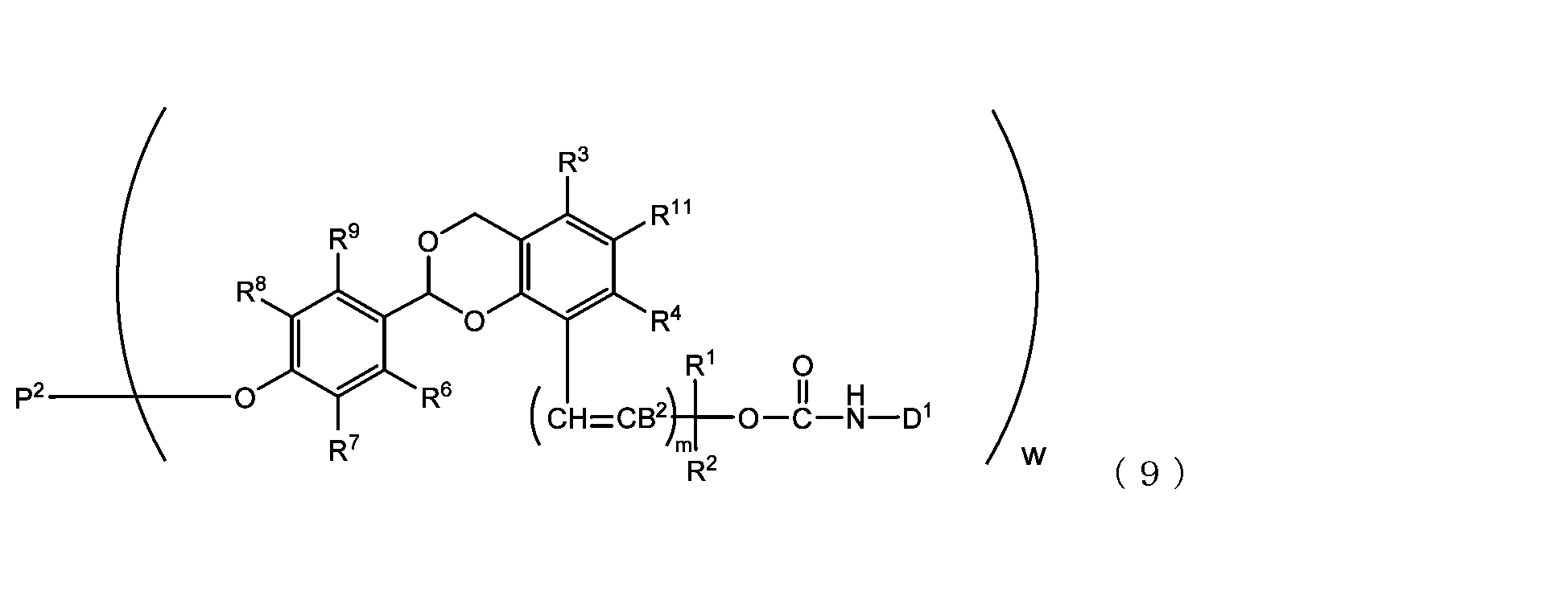
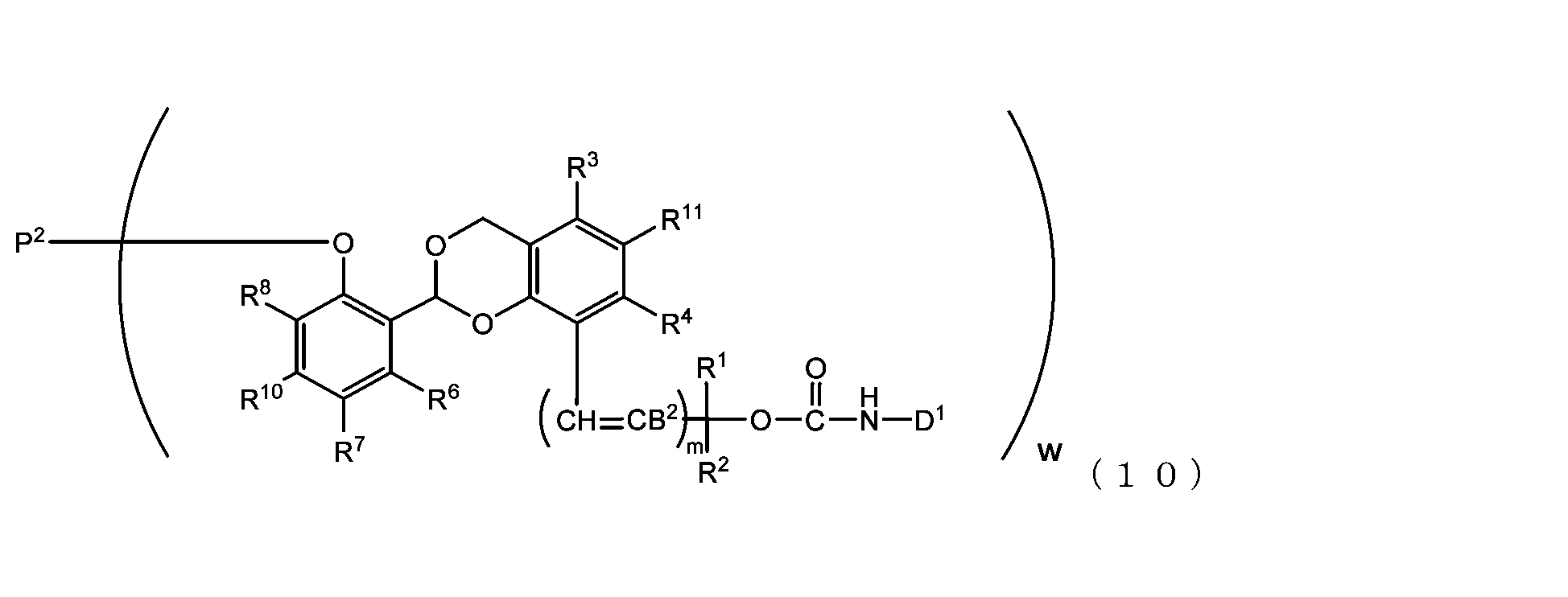
B2は水素原子または-C(R1)(R2)OC(O)NHD1であり、
D1は、生体機能性分子に含まれるアミノ基から、カーバメート結合を構成するアミノ基を除いた残基であり、
P2は、脱ヒドロキシ基を有するポリオキシエチレン誘導体、または脱ヒドロキシ基を有するポリオキシエチレン誘導体および生体機能性分子の結合体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5およびR11は、それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9およびR10は、それぞれ独立して、電子求引性または電子供与性の置換基または水素原子であり、
mは0または1である。
式(7)~(10)中、B2は水素原子または-C(R1)(R2)OC(O)NHD1であり、好ましくは水素原子である。
生体機能性分子としては、特に制限は無いが、ヒト又は他の動物の疾患の診断、治癒、緩和、治療または予防に関わる物質である。具体的にはタンパク質、ペプチド、核酸、細胞、ウィルスなどを含み、好適なタンパク質またはペプチドとしては、ホルモン、サイトカイン、抗体、アプタマー、酵素などが挙げられる。
緩衝液には、水溶性の有機溶媒を添加することもできる。この場合、水溶性の有機溶媒は、例えば、メタノール、エタノール、プロパノール、イソプロパノール、テトラヒドロフラン、アセトン、アセトニトリル、ジメチルスルホキシド、N,N-ジメチルホルムアミド、トリエチルアミン、ピリジンまたはヘキサメチルリン酸トリアミドがあげられ、好ましくはメタノール、エタノール、プロパノール、イソプロパノール、テトラヒドロフラン、アセトン、アセトニトリル、ジメチルスルホキシドまたはN,N-ジメチルホルムアミドであり、より好ましくはアセトニトリル、ジメチルスルホキシドまたはN,N-ジメチルホルムアミドである。
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに、日油社製SUNBRIGHT MEH-20T(40g, 2.0mmol)、トルエン(120g)および2,6-ジ-tert-ブチル-p-クレゾール(8mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(200g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシベンズアルデヒド(977mg, 8.0mmol)、および、トリフェニルホスフィン(2.10g, 8.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.51g, 7.0mmol)を3分割して加えた。25℃にて1時間反応を行った後、メタノール(224mg, 7.0mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(16g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(20)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 7.03(2H, d, arom.H), 7.83(2H, d, arom.H), 9.89(1H, s, -COH)
数平均分子量(Mn): 20,451

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(20)の化合物(1.0g, 0.05mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(11.4mg, 0.06mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(21)の化合物を得た。
2.27(1H, t,-OH), 2.36(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.68 (2H, dd, -CH 2 OH), 4.94(1H, d, -CH 2 O-), 5.15(1H, d, -CH 2 O-), 5.96(1H, s, -CH<), 6.77(1H, s, arom.H), 6.97(2H, d, arom.H), 7.04(1H, s, arom.H), 7.50(2H, d, arom. H)
数平均分子量(Mn): 20,435
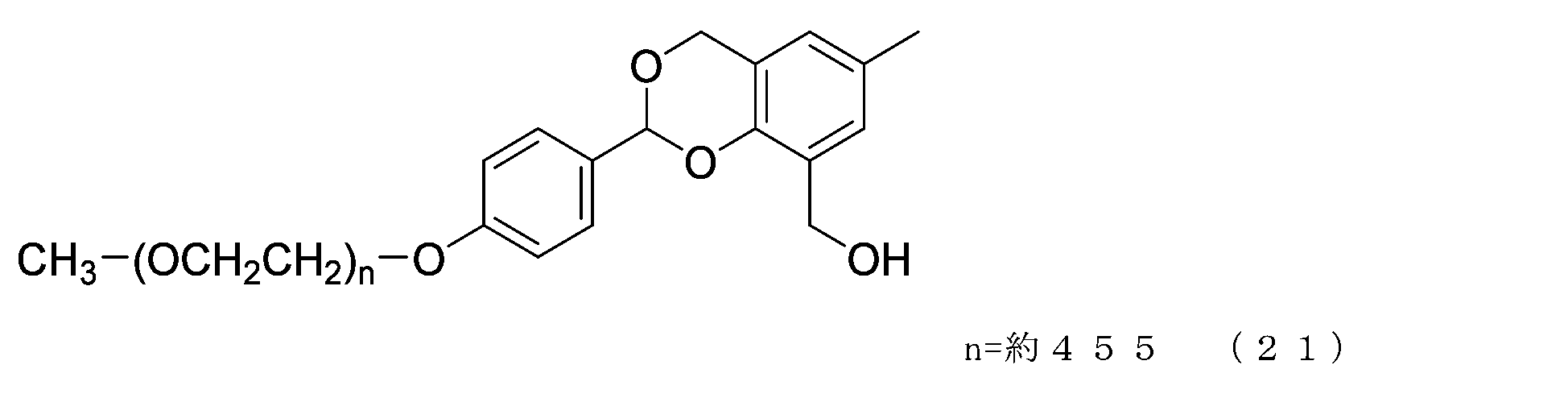
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(21)の化合物(250mg, 0.0125mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(23.7mg, 0.30mmol)およびp-ニトロフェニルクロロホルメート(40.3mg, 0.20mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し、一部の不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析を行い、ろ過後、減圧乾燥して式(22)の化合物を得た。
2.31(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.96(1H, d, -CH 2 O-), 5.16(1H, d, -CH 2 O-), 5.35(2H, dd, -CH 2 OCO-), 5.98(1H, s, -CH<), 6.87(1H, s, arom.H) 6.94(2H, d, arom.H), 7.11(1H, s, arom.H), 7.26-7.29(2H, m, arom.H), 7.52(2H, d, arom.H), 8.24(2H, d, arom.H)
数平均分子量(Mn): 20,204
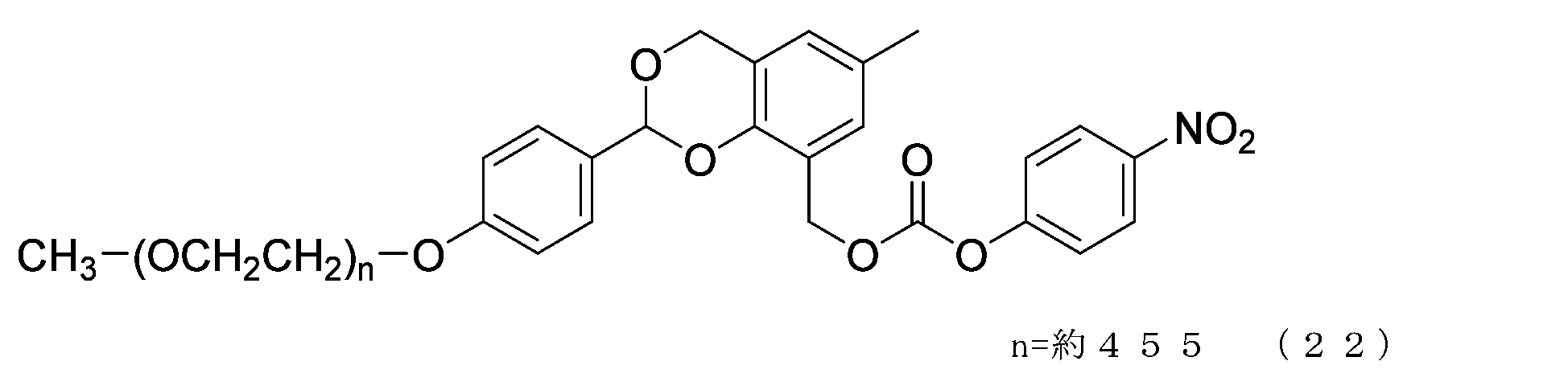
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT MEH-20T(40g, 2.0mmol)、トルエン(120g)および2,6-ジ-tert-ブチル-p-クレゾール(8mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(200g)を仕込んだ後、25℃まで冷却し、3-フルオロ-4-ヒドロキシベンズアルデヒド(1.12g, 8.0mmol)、および、トリフェニルホスフィン(2.62g, 10.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.84g, 8.6mmol)を3分割して加えた。25℃にて2時間反応を行った後、メタノール(276mg, 8.6mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(23)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.30(m, -(OCH 2 CH 2 )n-), 7.13(1H, t, -CH<), 7.60-7.65(2H, m, arom.H), 9.86(1H, s, -COH)
数平均分子量(Mn): 20,201

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(23)の化合物(1.0g, 0.05mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(11.4mg, 0.06mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(24)の化合物を得た。
2.20(1H, t,-OH), 2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.24(m, -(OCH 2 CH 2 )n-), , 4.68(2H, dd, -CH 2 OH), 4.94(1H, d, -CH 2 O-), 5.14(1H, d, -CH 2 O-), 5.93(1H, s, -CH<), 6.77(1H, s, arom.H), 7.04(1H, t, arom.H), 7.06(1H, s, arom.H), 7.34-7.26(2H, m, arom.H)
数平均分子量(Mn): 20,445

温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(24)の化合物(180mg, 0.009mmol)およびジクロロメタン(2.4g)を仕込み、溶解後、ピリジン(17.1mg, 0.216mmol)およびp-ニトロフェニルクロロホルメート(29.0mg, 0.144mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(1.8g)を用いて水洗を繰り返し、一部の不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析を行い、ろ過後、減圧乾燥して式(25)の化合物を得た。
2.32(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.24(m, -(OCH 2 CH 2 )n-), 4.96(1H, d, -CH 2 O-), 5.15(1H, d, -CH 2 O-), 5.34(2H, dd, -CH 2 OCO-), 5.96(1H, s, -CH<), 6.87(1H, s, arom.H), 7.01(1H, t, arom.H), 7.12(1H, s, arom.H), 7.26-7.36(4H, m, arom.H) , 8.26(2H, d, arom.H)
数平均分子量(Mn): 20,298
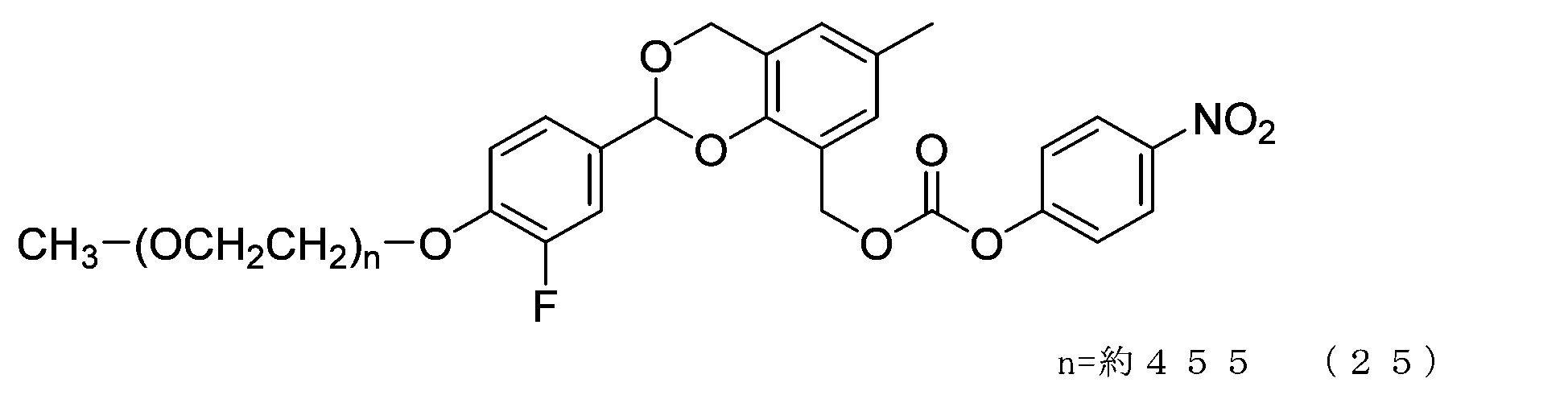
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT MEH-20T(40g, 2.0mmol)、トルエン(120g)および2,6-ジ-tert-ブチル-p-クレゾール(8mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(200g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシ-2-メトキシベンズアルデヒド(1.22g, 8.0mmol)、および、トリフェニルホスフィン(2.62g, 10.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.84g, 8.6mmol)を3分割して加えた。25℃にて2時間反応を行った後、メタノール(276mg, 8.6mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(26)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 6.50(1H, d, arom.H), 6.55(1H, dd, arom.H), 7.79(2H, d, arom.H), 10.3(1H, s, -COH)
数平均分子量(Mn): 20,216

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(26)の化合物(1.0g, 0.05mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(11.4mg, 0.06mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行い、一部の低分子不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(27)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.84(1H, dd,-OH), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.47(1H, dd, -CH 2 OH), 4.75(1H, dd, -CH 2 OH), 4.98(1H, d, -CH 2 O-), 5.17(1H, d, -CH 2 O-), 6.20(1H, s, -CH<), 6.57-6.55(2H, m, arom.H), 6.78(1H, s, arom.H), 6.97(1H, s, arom.H), 7.58(2H, d, arom.H)
数平均分子量(Mn): 20,215
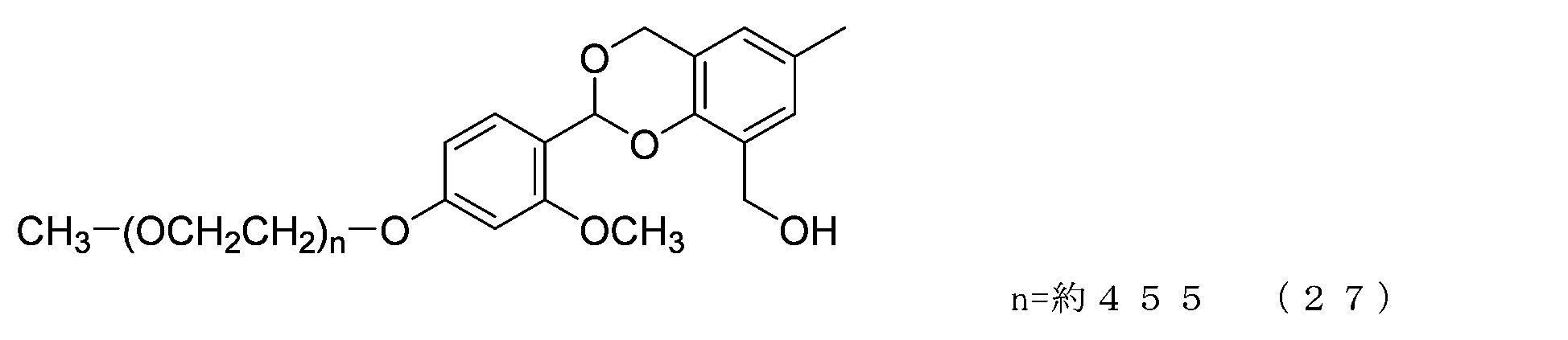
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(27)の化合物(300mg, 0.015mmol)およびジクロロメタン(4.0g)を仕込み、溶解後、ピリジン(28.5mg, 0.360mmol)およびp-ニトロフェニルクロロホルメート(48.4mg, 0.240mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(3.0g)を用いて水洗を繰り返し、一部の低分子不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析を行い、ろ過後、減圧乾燥して式(28)の化合物を得た。
2.31(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.93(1H, d, -CH 2 O-), 5.18(1H, d, -CH 2 O-), 5.33(2H, dd, -CH 2 OCO-), 6.26(1H, s, -CH<), 6.52-6.54(2H, m, arom.H), 6.87(1H, s, arom.H), 7.10(1H, s, arom.H), 7.26-7.27(2H, m, arom.H), 7.59(1H, d, arom.H), 8.22(2H, d, arom.H)
数平均分子量(Mn): 20,315
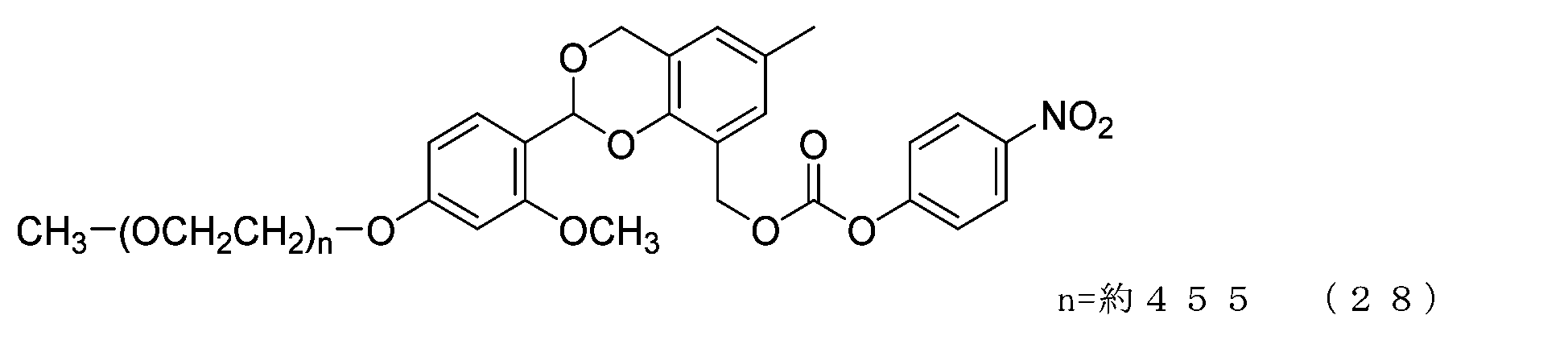
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT GL2-400HO(10g, 0.25mmol)、トルエン(30g)および2,6-ジ-tert-ブチル-p-クレゾール(2mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(50g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシベンズアルデヒド(244mg, 2.0mmol)、および、トリフェニルホスフィン(524mg, 2.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(376mg, 1.75mmol)を3分割して加えた。25℃にて2時間反応を行った後、メタノール(56mg, 1.75mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(80g)に溶解し、ヘキサン(60g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(16mg)を添加した酢酸エチル(80g)に得られた結晶を溶解後、ヘキサン(40g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(40g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(29)の化合物を得た。
3.38(6H, s, -(OCH2CH2)n OCH 3 ), 3.52-3.92(m, -(OCH 2 CH 2 )n-), 4.13(1H, dd, -CH 2 O-), 4.21(1H, dd, -CH 2 O-), 7.03(2H, d, arom.H), 7.83(2H, d, arom.H), 9.89(1H, s, -COH)
数平均分子量(Mn): 38,947
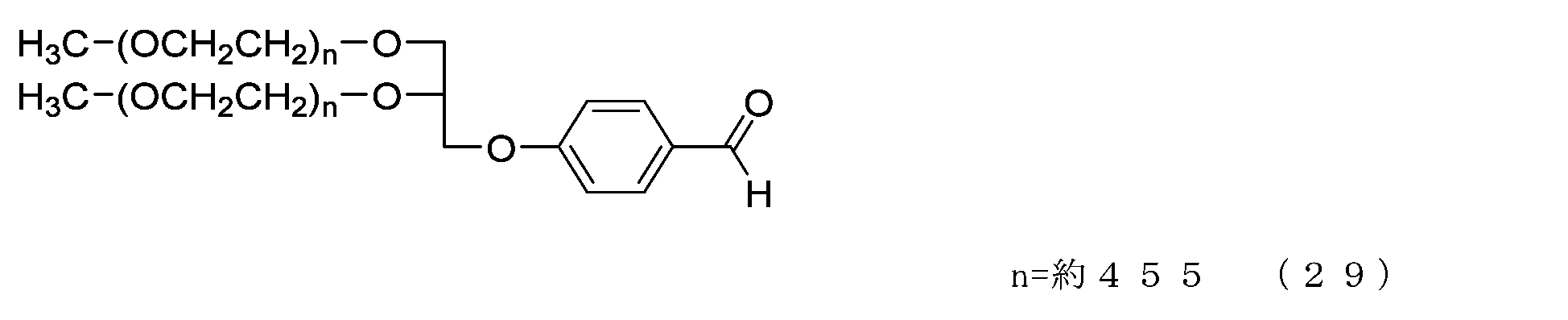
温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(29)の化合物(1.0g, 0.025mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1 mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(17.1mg, 0.09mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(20g)を用いてろ液を希釈した。pH12の20%食塩水(20g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(30)の化合物を得た。
2.29(3H, s, -CH 3 ), 2.35(1H, t,-OH), 3.38(6H, s, -(OCH2CH2)nOCH 3 ), 3.52-3.91(m, -(OCH 2 CH 2 )n-), 4.07(1H, dd, -CH 2 O-), 4.14(1H, dd, -CH 2 O-), 4.68 (2H, dd, -CH 2 OH), 4.94(1H, d, -CH 2 O-), 5.15(1H, d, -CH 2 O-), 5.96(1H, s, -CH<), 6.77(1H, s, arom.H), 6.97(2H, d, arom.H), 7.04(1H, s, arom.H), 7.50(2H, d, arom.H)
数平均分子量(Mn): 39,110
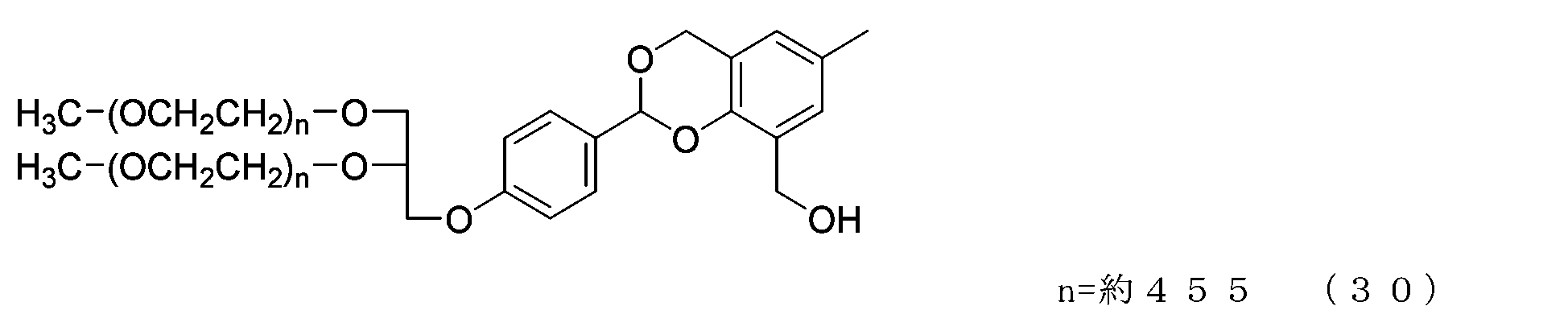
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(30)の化合物(250mg, 0.00625mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(11.9mg, 0.15mmol)およびp-ニトロフェニルクロロホルメート(20.3mg, 0.10mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し、一部の不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(9mg)を添加した酢酸エチル(45g)に残渣を溶解し、ヘキサン(33g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(9mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(31)の化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(6H, s, -(OCH2CH2)n OCH 3 ), 3.52-3.91(m, -(OCH 2 CH 2 )n-), 4.07(1H, dd, -CH 2 O-), 4.14(1H, dd, -CH 2 O-), 4.94(1H, d, -CH 2 O-), 5.16(1H, d, -CH 2 O-), 5.35(1H, d, -CH 2 OCO-), 5.98(1H, s, -CH<), 6.87(1H, s, arom.H) 6.94(2H, d, arom.H), 7.11(1H, s, arom.H), 7.26-7.29(2H, m, arom.H), 7.52(2H, d, arom.H), 8.24(2H, d, arom.H)
数平均分子量(Mn): 39,456
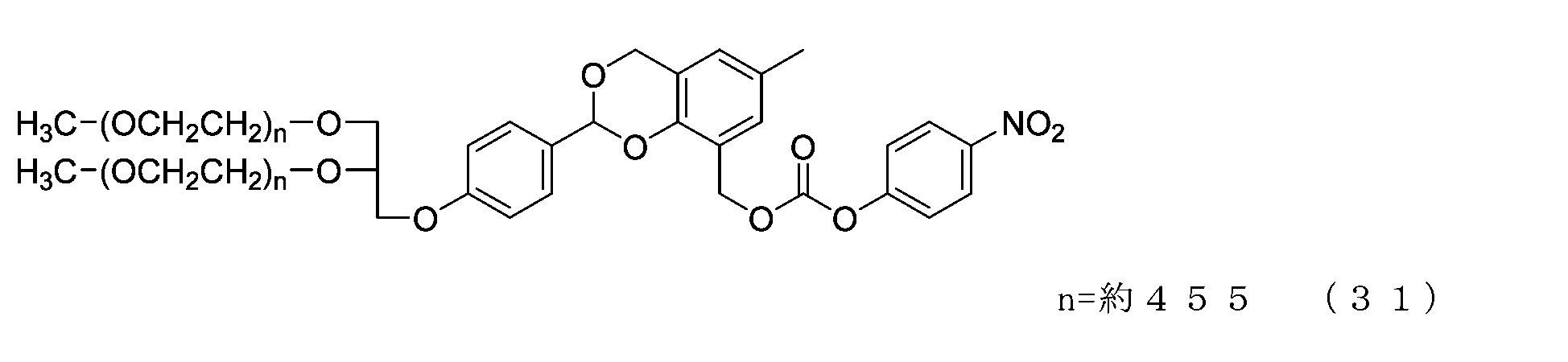
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT PTE-200HO(10g, 0.5mmol)、トルエン(30g)および2,6-ジ-tert-ブチル-p-クレゾール(2mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(50g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシベンズアルデヒド(977mg, 8.0mmol)、および、トリフェニルホスフィン(2.10g, 8.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.51g, 7.0mmol)を3分割して加えた。25℃にて1時間反応を行った後、メタノール(224mg, 7.0mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(80g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ろ過後に得られた結晶をヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(32)の化合物を得た。
3.41-4.23(m, -(OCH 2 CH 2 )n-), 7.03(8H, d, arom.H), 7.83(8H, d, arom.H), 9.89(4H, s, -COH)
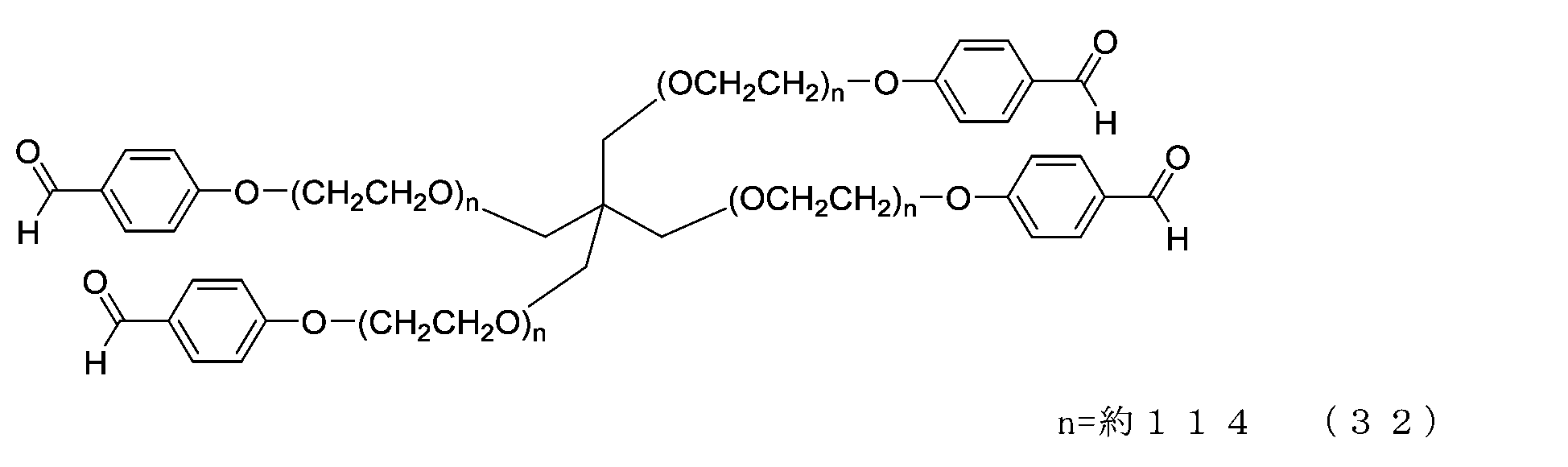
温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(32)の化合物(1.0g, 0.05mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(1.34g, 8mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(45.6mg, 0.24mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(48.4mg, 0.48mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(360g)に残渣を溶解し、ヘキサン(264g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に得られた結晶を溶解後、ヘキサン(264g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(33)の化合物を得た。
2.27(4H, t,-OH), 2.36(12H, s, -CH 3 ), 3.41-4.23(m, -(OCH 2 CH 2 )n-), 4.68(8H, dd, -CH 2 OH), 4.94(4H, d, -CH 2 O-), 5.15(4H, d, -CH 2 O-), 5.96(4H, s, -CH<), 6.77(4H, s, arom.H), 6.97(8H, d, arom.H), 7.04(4H, s, arom.H), 7.50(8H, d, arom.H)

温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(33)の化合物(250mg, 0.0125mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(94.8mg, 1.20mmol)およびp-ニトロフェニルクロロホルメート(161mg, 0.80mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し行い、一部の低分子不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に残渣を溶解し、ヘキサン(264g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に得られた結晶を溶解後、ヘキサン(264g)を添加して晶析を行い、ろ過後、減圧乾燥して式(34)の化合物を得た。
2.31(12H, s, -CH 3 ), 3.41-4.23(m, -(OCH 2 CH 2 )n-), 4.96(4H, d, -CH 2 O-), 5.16(4H, d, -CH 2 O-), 5.35(8H, dd, -CH 2 OCO-), 5.98(4H, s, -CH<), 6.87(4H, s, arom.H), 6.94(8H, d, arom.H), 7.11(4H, s, arom.H), 7.26-7.29(8H, m, arom.H), 7.52(8H, d, arom.H), 8.24(8H, d, arom.H)
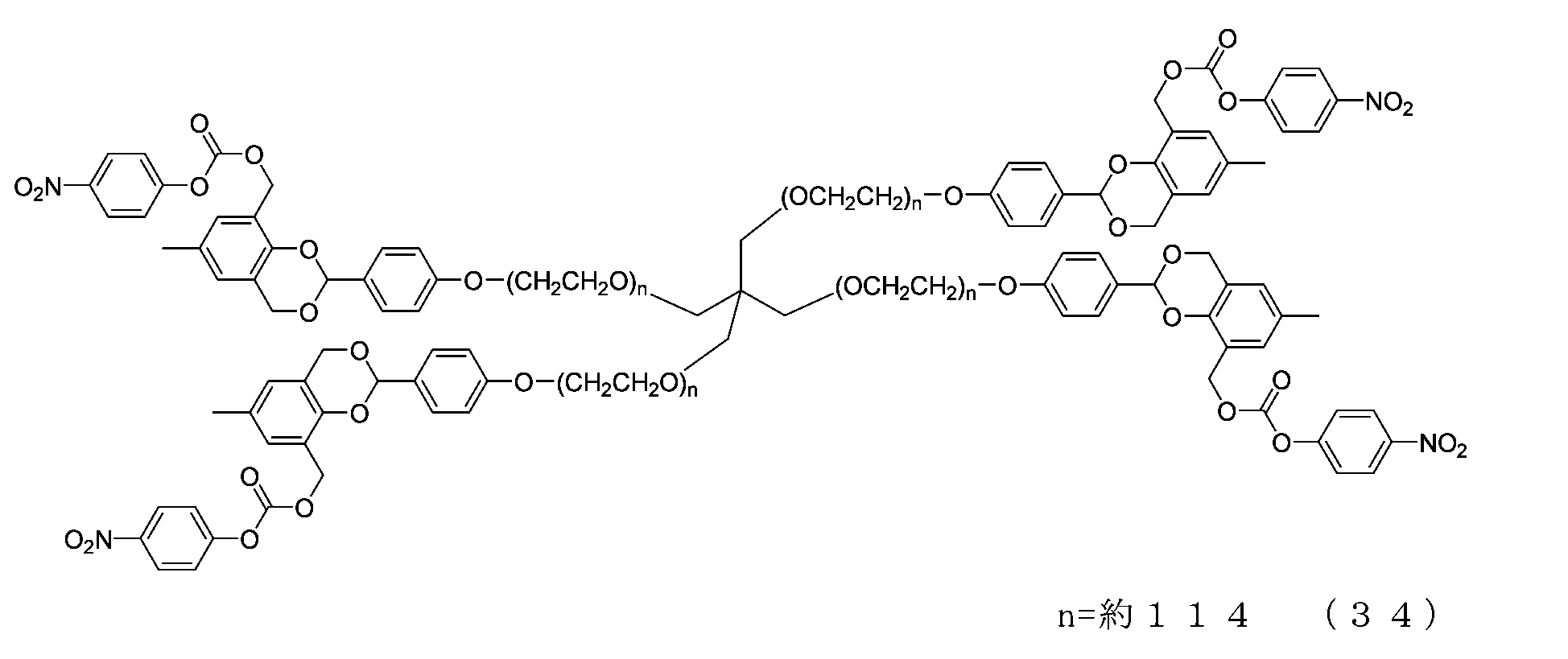
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT HGEO-400HO(10g, 0.25mmol)、トルエン(30g)および2,6-ジ-tert-ブチル-p-クレゾール(2mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(50g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシベンズアルデヒド(977mg, 8.0mmol)、および、トリフェニルホスフィン(2.10g, 8.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.51g, 7.0mmol)を3分割して加えた。25℃にて1時間反応を行った後、メタノール(224mg, 7.0mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(35)の化合物を得た。
2.98-4.23(m, -(OCH 2 CH 2 )n-), 7.03(16H, d, arom.H), 7.83(16H, d, arom.H), 9.89(8H, s, -COH)

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(35)の化合物(1.0g, 0.025mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(1.34g, 8.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(45.6mg, 0.24mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(48.4mg, 0.48mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行い、一部の低分子不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に残渣を溶解し、ヘキサン(264g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に得られた結晶を溶解後、ヘキサン(264g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(36)の化合物を得た。
2.27(8H, t,-OH), 2.36(24H, s, -CH 3 ), 2.98-4.23(m, -(OCH 2 CH 2 )n-), 4.68 (16H, dd, -CH 2 OH), 4.94(8H, d, -CH 2 O-), 5.15(8H, d, -CH 2 O-), 5.96(8H, s, -CH<), 6.77(8H, s, arom.H), 6.97(16H, d, arom.H), 7.04(8H, s, arom.H), 7.50(16H, d, arom.H)
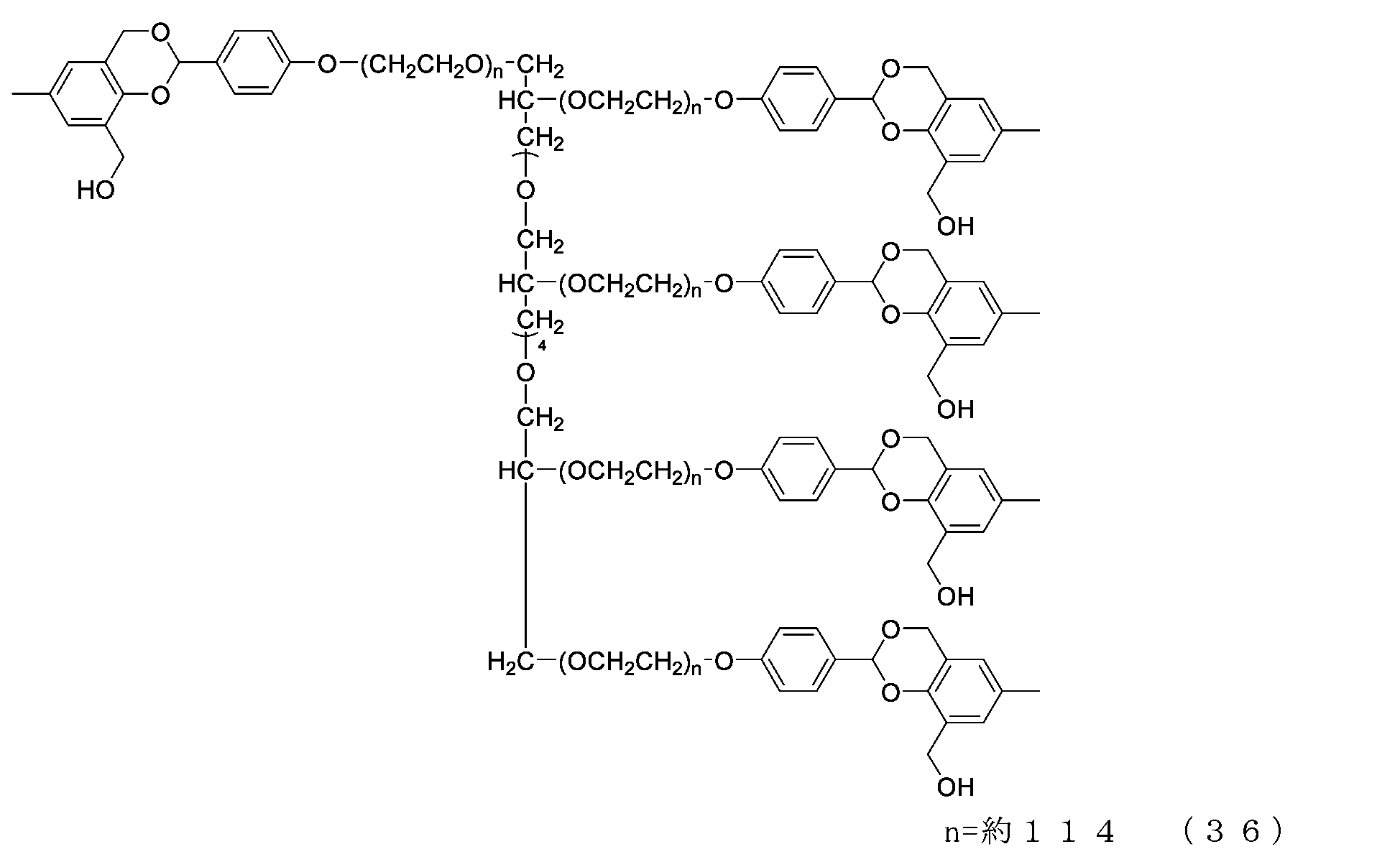
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(36)の化合物(250mg, 0.00625mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(94.8mg, 1.20mmol)およびp-ニトロフェニルクロロホルメート(161.2mg, 0.80mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し行い、一部の低分子不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(72 mg)を添加した酢酸エチル(360g)に残渣を溶解し、ヘキサン(264g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(72mg)を添加した酢酸エチル(360g)に得られた結晶を溶解後、ヘキサン(263g)を添加して晶析を行い、ろ過後、減圧乾燥して式(37)の化合物を得た。
2.31(24H, s, -CH 3 ), 2.98-4.23(m, -(OCH 2 CH 2 )n-), 4.96(8H, d, -CH 2 O-), 5.16(8H, d, -CH 2 O-), 5.35(16H, dd, -CH 2 OCO-), 5.98(8H, s, -CH<), 6.87(8H, s, arom.H), 6.94(16H, d, arom.H), 7.11(8H, s, arom.H), 7.26-7.29(16H, m, arom.H), 7.52(16H, d, arom.H), 8.24(16H, d, arom.H)
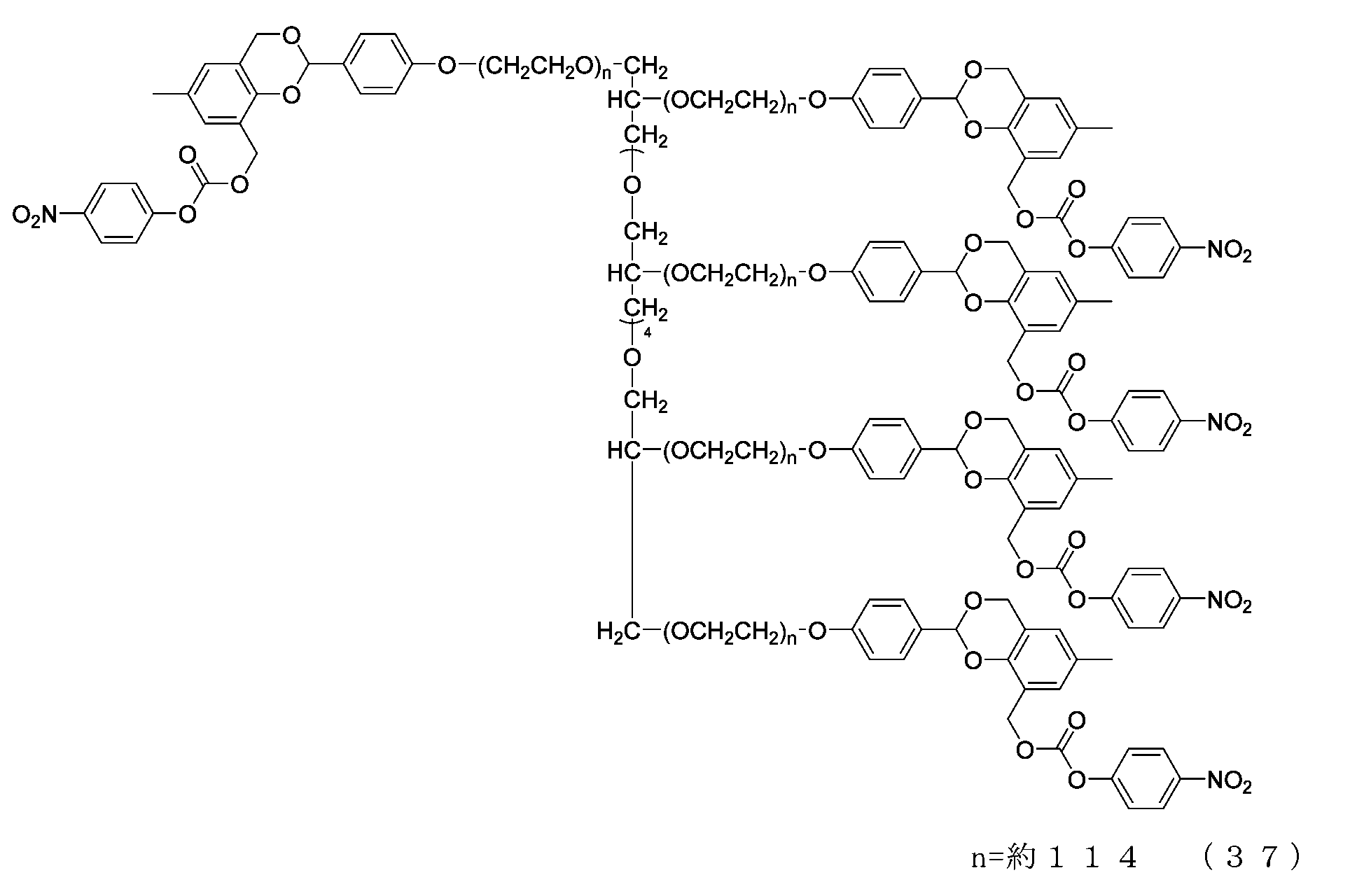
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT DKH-20T(20g, 1.0mmol)、トルエン(60g)および2,6-ジ-tert-ブチル-p-クレゾール(4mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(100g)を仕込んだ後、25℃まで冷却し、4-ヒドロキシベンズアルデヒド(977mg, 8.0mmol)、および、トリフェニルホスフィン(2.10g, 8.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.51g, 7.0mmol)を3分割して加えた。25℃にて1時間反応を行った後、メタノール(224mg, 7.0mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(38)の化合物を得た。
3.52-4.23(m, -(OCH 2 CH 2 )n-), 7.03(4H, d, arom.H), 7.83(4H, d, arom.H), 9.89(2H, s, -COH)

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(38)の化合物(1.0g, 0.05mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(672mg, 4.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(22.8mg, 0.12mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(24.2mg, 0.24mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(36mg)を添加した酢酸エチル(180 g)に残渣を溶解し、ヘキサン(132g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(36mg)を添加した酢酸エチル(180g)に得られた結晶を溶解後、ヘキサン(132g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(39)の化合物を得た。
2.27(2H, t,-OH), 2.36(6H, s, -CH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.68(4H, dd, -CH 2 OH), 4.94(2H, d, -CH 2 O-), 5.15(2H, d, -CH 2 O-), 5.96(2H, s, -CH<), 6.77(2H, s, arom.H), 6.97(4H, d, arom.H), 7.04(2H, s, arom.H), 7.50(4H, d, arom.H)
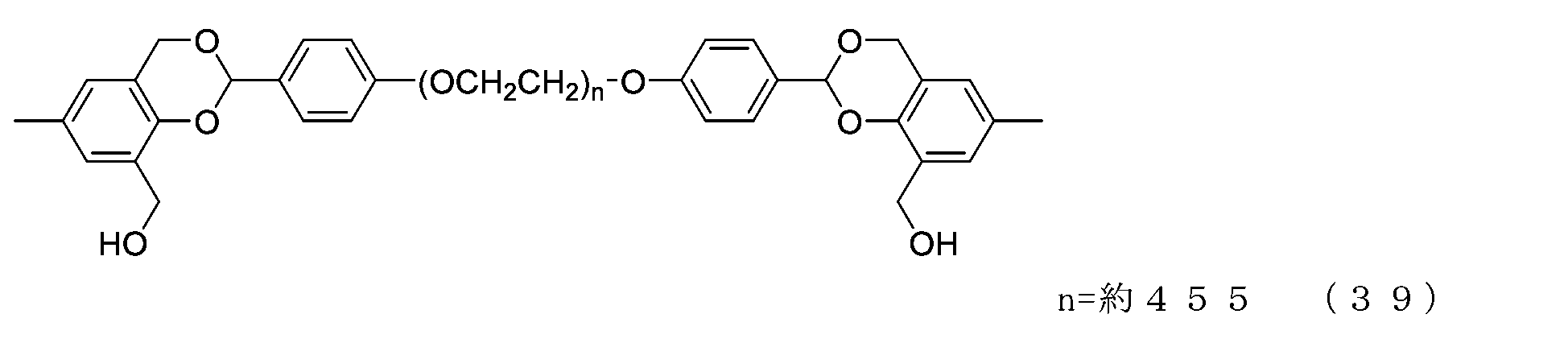
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(39)の化合物(250mg, 0.0125mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(47.4mg, 0.60mmol)およびp-ニトロフェニルクロロホルメート(80.6mg, 0.40mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(36mg)を添加した酢酸エチル(180g)に残渣を溶解し、ヘキサン(132g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(36mg)を溶解添加した酢酸エチル(180g)に得られた結晶を溶解後、ヘキサン(132g)を添加して晶析を行い、ろ過後、減圧乾燥して式(40)の化合物を得た。
2.31(6H, s, -CH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.96(2H, d, -CH 2 O-), 5.16(2H, d, -CH 2 O-), 5.35(4H, dd, -CH 2 OCO-), 5.98(2H, s, -CH<), 6.87(2H, s, arom.H), 6.94(4H, d, arom.H), 7.11(2H, s, arom.H), 7.26-7.29(4H, m, arom.H), 7.52(4H, d, arom.H), 8.24(4H, d, arom.H)
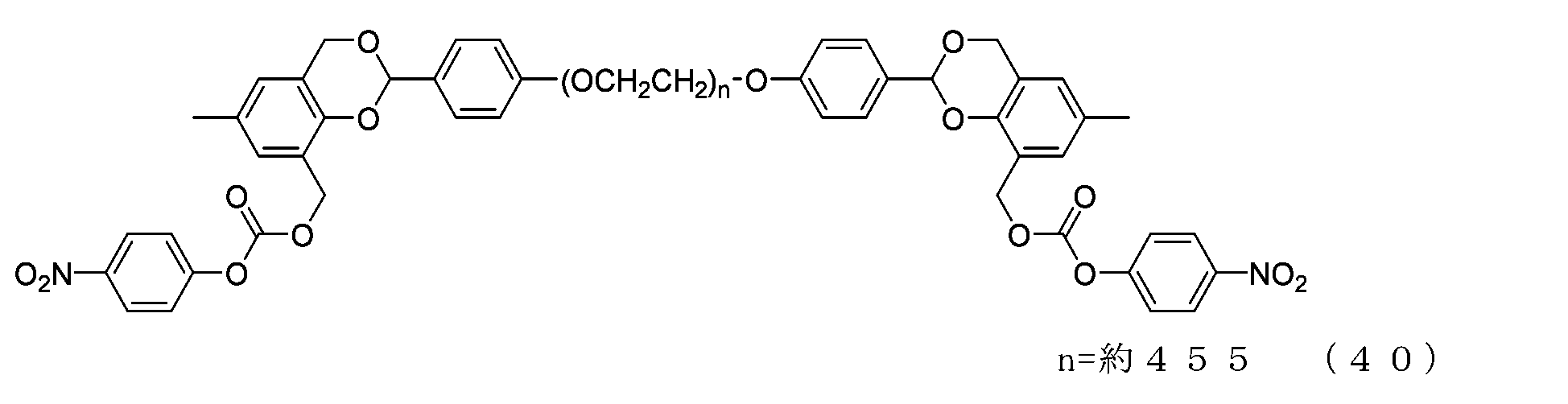
温度計、窒素吹き込み管、攪拌機および冷却管を装備した50mLの三つ口フラスコに3-ヒドロキシベンズアルデヒド(2.00g, 16.4mmol)、オルトギ酸トリメチル(3.48g, 32.8mmol)、メタノール(17g)を仕込み、pトルエンスルホン酸一水和物(0.312mg, 1.64mmol)を加えて25℃にて2時間反応を行った。水酸化ナトリウムを加えてしばらく撹拌した後、溶媒を減圧留去した。残渣をジクロロメタンに溶解し、5wt%炭酸水素ナトリウム水溶液、25wt%食塩水の順で洗浄した後、有機層を無水硫酸ナトリウムで乾燥した。濾過後、溶媒を減圧留去して式(41)の化合物を得た。
3.33(6H, s, -OCH 3 ), 5.35(1H, s, -CH<), 6.81(1H, d, arom.H), 6.95(1H, d, arom.H), 7.23-7.26(1H, m, arom.H)

温度計、窒素吹き込み管、攪拌機および冷却管を装備した50mLの三つ口フラスコに文献(Freeman, J. H.; JAm. Chem. Soc. 1952, 74, 6257-6260)に従い合成した2,4-ジ(ヒドロキシメチル)フェノール(50.0mg, 0.324mmol)、式(41)の化合物(217mg, 1.29mmol)、2,6-ジ-tert-ブチル-p-クレゾール(7.14mg, 0.0324mmol)、無水硫酸ナトリウム(1g)およびシクロペンチルメチルエーテル(10g)を仕込み、p-トルエンスルホン酸一水和物(4.10mg, 0.0212mmol)を加えて40℃にて2時間反応を行った。N-メチルモルホリンを加えてしばらく撹拌した後、濾過を行った。10wt%食塩水で洗浄した後、有機層を無水硫酸ナトリウムで乾燥した。濾過後、溶媒を減圧留去して式(42)の化合物を得た。
4.42(2H, d, -CH 2 OH), 4.93(1H, d, -CH 2 O-), 5.10(1H, t, -CH2OH), 5.15(1H, d, -CH 2 O-), 6.01(1H, s, -CH<), 6.80-7.21(7H, m, arom.H), 9.53(1H, bs, >C-OH)
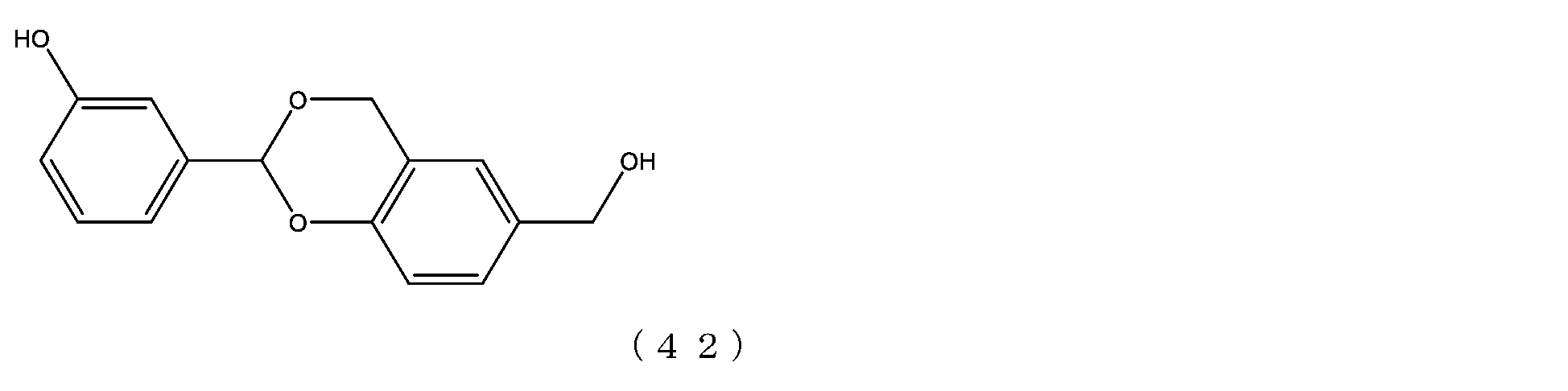
温度計、窒素吹き込み管、攪拌機および冷却管を装備した50mLの三つ口フラスコに式(42)の化合物(37.0mg, 0.141mmol)、日油社製ME-200MS(α-メチル-ω-[(メチルスルホニル)オキシ]ポリ(オキシエチレン),705mg, 0.0353mmol)、炭酸カリウム(97.0mg, 0.705mmol)およびアセトニトリル(3.5g)を仕込み、80℃にて4時間反応を行った。濾過後、溶媒を減圧留去し、残渣をジクロロメタンに溶解した。10wt%食塩水で洗浄した後、有機層を無水硫酸ナトリウムで乾燥した。濾過後、溶媒を減圧留去し、残渣をトルエン(50g)に溶解した。ヘキサン(50g)を添加して晶析を行い、濾過後、減圧乾燥して式(43)の化合物を得た。
3.38(3H, s, -OCH 3 ), 3.52-4.18(m, -(OCH 2 CH 2 )n-), 4.62(2H, s, -CH 2 OH), 4.98(1H, d, -CH 2 O-), 5.18(1H, d, -CH 2 O-), 5.95(1H, s, -CH<), 6.87-7.34(7H, m, arom.H)
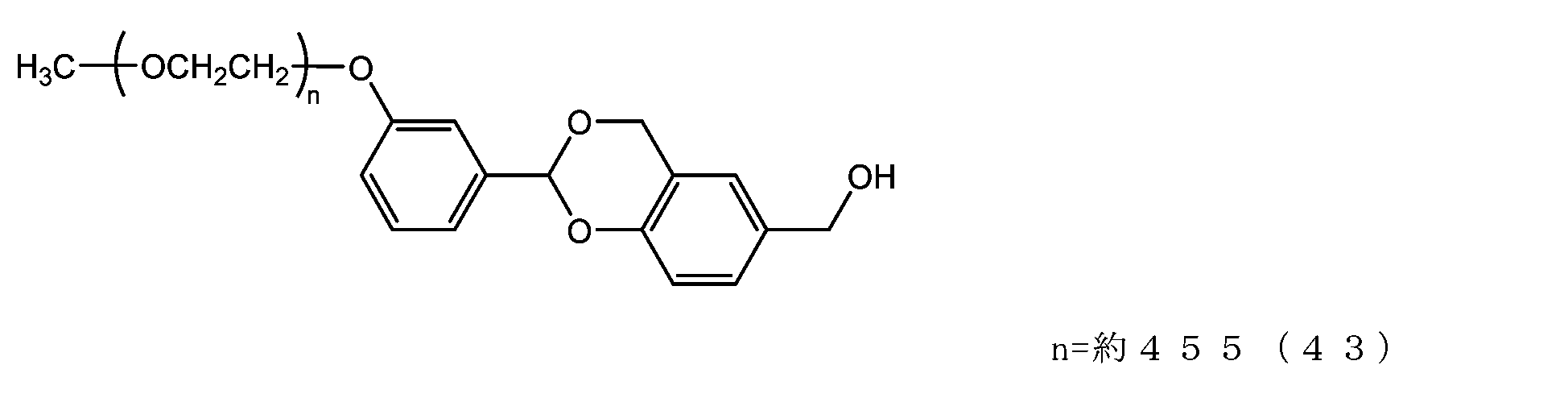
温度計、窒素吹き込み管、攪拌機および冷却管を装備した50mLの三つ口フラスコに式(43)の化合物(300mg, 0.0150mmol)、炭酸ジ(N-スクシンイミジル)(46.0mg, 0.180 mmol)、トリエチルアミン(21.0mg, 0.208mmol)およびジクロロメタン(5g)を仕込み、25℃にて12時間反応を行った。濾過後、5wt%食塩水で洗浄し、有機層の溶媒を減圧留去した。残渣を酢酸エチル(6g)に溶解し、無水硫酸ナトリウムで乾燥した後、濾過を行った。酢酸エチル(44g)を加えた後、ヘキサン(50g)を添加して晶析を行い、濾過後、減圧乾燥して式(44)の化合物を得た。
2.85(4H, s, -COCH 2 CH 2 CO-), 3.38(3H, s, -OCH 3 ), 3.52-4.18(m, -(OCH 2 CH 2 )n-), 5.00(1H, d, -CH 2 OCH<), 5.18(1H, d, -CH 2 O-), 5.25(2H, s, -CH 2 OCO-), 5.97(1H, s, -CH<), 6.96-7.35(7H, m, arom.H)

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(20)の化合物(1.0g, 0.05mmol)、文献(Freeman, J. H.; JAm. Chem. Soc. 1952, 74, 6257-6260)に従い合成した2,4-ジ(ヒドロキシメチル)フェノール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(11.4mg, 0.06mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(45)の化合物を得た。
3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.62(2H, s, -CH 2 OH), 5.00(1H, d, -CH 2 OCH<), 5.18(1H, d, -CH 2 O-), 5.25(2H, s, -CH 2 OCO-), 5.97(1H, s, -CH<), 6.96-7.35(7H, m, arom.H)

温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(45)の化合物(250mg, 0.0125mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(23.7mg, 0.30mmol)およびp-ニトロフェニルクロロホルメート(40.3mg, 0.20mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し、一部の不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析を行い、ろ過後、減圧乾燥して式(46)の化合物を得た。
3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.62(2H, s, -CH 2 OH), 5.00(1H, d, -CH 2 OCH<), 5.18(1H, d, -CH 2 O-), 5.25(2H, s, -CH 2 OCO-), 5.97(1H, s, -CH<), 6.96-7.35(11H, m, arom.H)
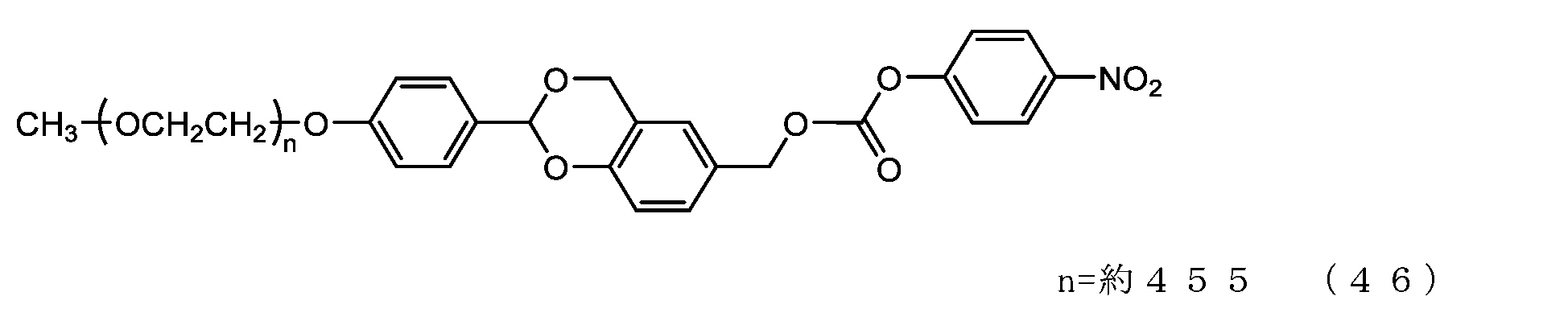
温度計、窒素吹込み管、撹拌機およびDean-stark管および冷却管を装備した300mLの4つ口フラスコに日油社製SUNBRIGHT MEH-40T(40g, 2.0mmol)、トルエン(120g)および2,6-ジ-tert-ブチル-p-クレゾール(8mg)を仕込み、水をトルエンで共沸除去した。45℃へ冷却し、クロロホルム(200g)を仕込んだ後、25℃まで冷却し、2-ヒドロキシベンズアルデヒド(977mg, 8.0mmol)、および、トリフェニルホスフィン(2.10g, 8.0mmol)を仕込んだ。しばらく攪拌させた後、内温が35℃度を越えないように、アゾジカルボン酸ジイソプロピル(1.51g, 7.0mmol)を3分割して加えた。25℃にて1時間反応を行った後、メタノール(224mg, 7.0mmol)を添加し、30分攪拌後、溶媒を減圧留去した。残渣を酢酸エチル(320g)に溶解し、ヘキサン(240g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(64mg)を添加した酢酸エチル(320g)に得られた結晶を溶解後、ヘキサン(160g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した。ヘキサン(160g)を用いて結晶洗浄し、ろ過後、減圧乾燥して式(47)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 9.89(1H, s, -COH)

温度計、窒素吹込み管、撹拌機および冷却管を装備した50mLの3つ口フラスコに式(47)の化合物(1.0g, 0.05mmol)、文献(Freeman, J. H.; JAm. Chem. Soc. 1952, 74, 6257-6260)に従い合成した2,4-ジ(ヒドロキシメチル)フェノール(336mg, 2.0mmol)、テトラヒドロフラン(5.0g)および2,6-ジ-tert-ブチル-p-クレゾール(1.1mg)を仕込み、溶解後、モレキュラーシーブ5A(1.0g)およびp-トルエンスルホン酸一水和物(11.4mg, 0.06mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(12.1mg, 0.12mmol)を加えてしばらく攪拌し、ろ過後、クロロホルム(10g)を用いてろ液を希釈した。pH12の20%食塩水(10g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析する操作を繰り返し行い、低分子不純物を除去した後、減圧乾燥して式(48)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.96(1H, d, -CH 2 O-), 5.16(1H, d, -CH 2 O-), 5.25(2H, dd, -CH 2 OH), 5.97(1H, s, -CH<), 6.96-7.35(7H, m, arom.H)
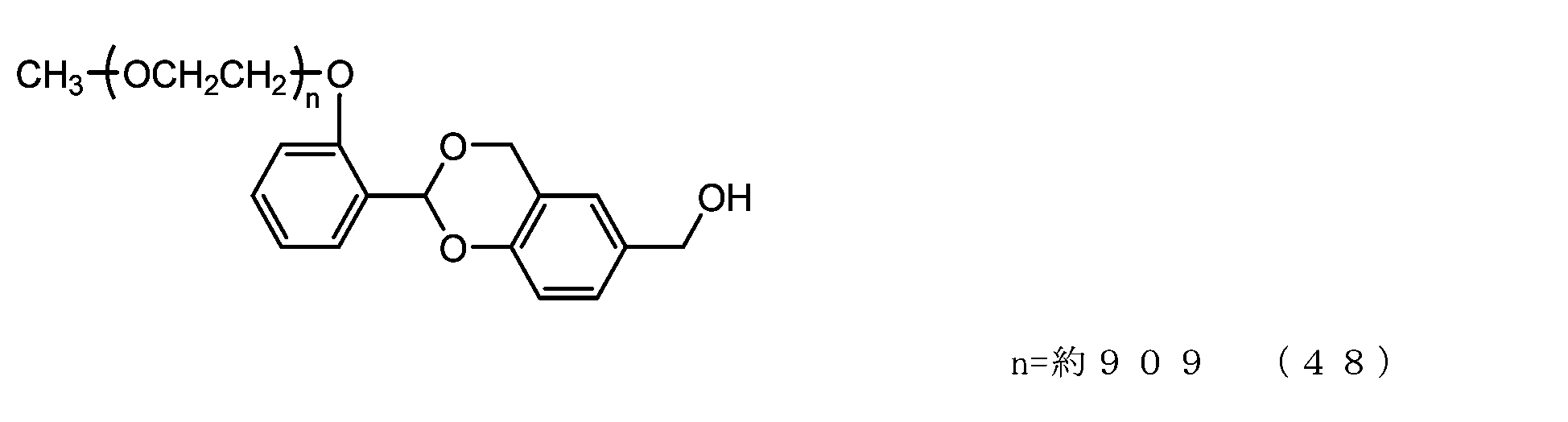
温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(48)の化合物(250mg, 0.0125mmol)およびジクロロメタン(3.6g)を仕込み、溶解後、ピリジン(23.7mg, 0.30mmol)およびp-ニトロフェニルクロロホルメート(40.3mg, 0.20mmol)を仕込んで、25℃にて3時間反応を行った。pH12の20%食塩水(2.5g)を用いて水洗を繰り返し、一部の不純物を除去した後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に残渣を溶解し、ヘキサン(66g)を添加して晶析を行った後、吸引ろ過して結晶を得た。さらに、2,6-ジ-tert-ブチル-p-クレゾール(18mg)を添加した酢酸エチル(90g)に得られた結晶を溶解後、ヘキサン(66g)を添加して晶析を行い、ろ過後、減圧乾燥して式(49)の化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.96(1H, d, -CH 2 O-), 5.16(1H, d, -CH 2 O-), 5.35(2H, dd, -CH 2 OCO-), 5.97(1H, s, -CH<), 6.96-7.35(11H, m, arom.H)
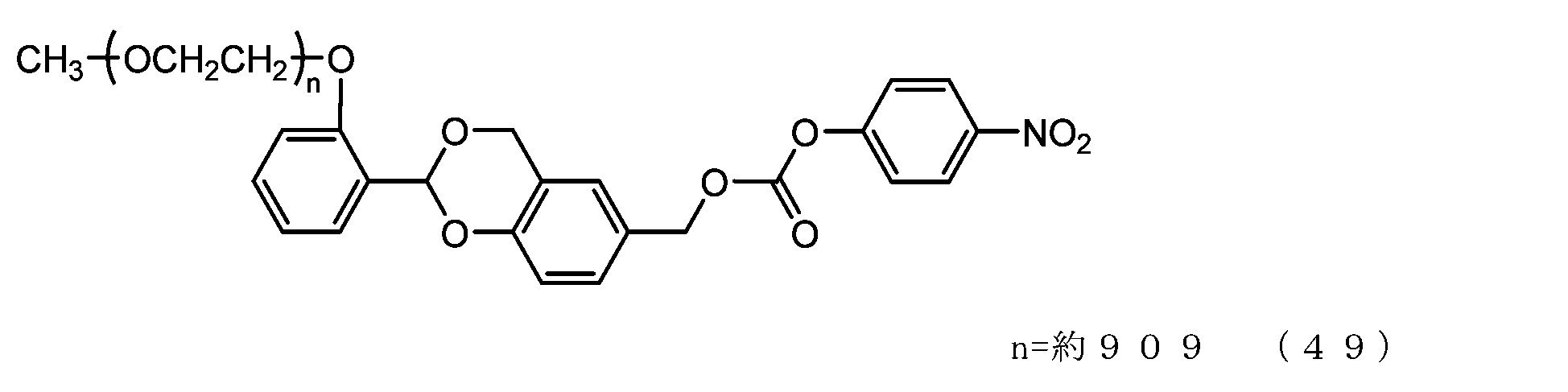
0.1Mリン酸ナトリウム緩衝溶液(pH8.5)を用い、β-アラニンの20mg/mL緩衝溶液を調製した。温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(22)の化合物(70mg, 0.0035mmol)およびβ-アラニンの20mg/mL緩衝溶液(1.5g)を仕込み、溶解後、25℃にて6時間反応を行った。食塩(500mg)を溶解させた後、クロロホルム(3.0g)を用いて抽出を行い、有機層を無水硫酸ナトリウムで乾燥後、ろ過した。酢酸エチル(45g)を用いてろ液を希釈後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(50)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.48(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.39-3.45(2H, m, -CH 2 NH-), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.92(1H, d, -CH 2 O-), 5.08(1H, d, -CH 2 O-), 5.15(2H, dd, -CH 2 OCONH-), 5.96(1H, s, -CH<), 6.79(1H, s, arom.H), 6.96(2H, d, arom.H), 7.04(1H, s, arom.H), 7.50(2H, d, arom.H),
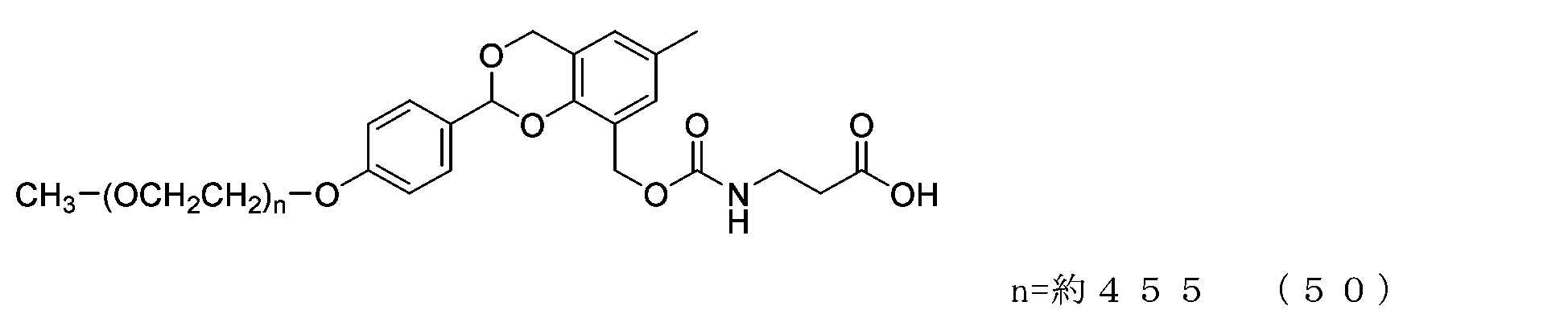
0.1Mリン酸ナトリウム緩衝溶液(pH8.5)を用い、β-アラニンの20mg/mL緩衝溶液を調製した。温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(25)の化合物(70mg, 0.0035mmol)およびβ-アラニンの20mg/mL緩衝溶液(1.5g)を仕込み、溶解後、25℃にて6時間反応を行った。食塩(500mg)を溶解させた後、クロロホルム(3.0g)を用いて抽出を行い、有機層を無水硫酸ナトリウムで乾燥後、ろ過した。酢酸エチル(45g)を用いてろ液を希釈後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(51)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.50(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.25(m, -(OCH 2 CH 2 )n-), 3.41-3.45(2H, m, -CH 2 NH-), 4.91(1H, d, -CH 2 O-), 5.08(1H, d, -CH 2 O-), 5.14(2H, dd, -CH 2 OCONH-), 5.94(1H, s, -CH<), 6.79(1H, s, arom.H), 7.03-7.06(2H, m, arom.H), 7.30-7.34(2H, m, arom.H)

0.1Mリン酸ナトリウム緩衝溶液(pH8.5)を用い、β-アラニンの20mg/mL緩衝溶液を調製した。温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(28)の化合物(14 mg, 0.0072mmol)およびβ-アラニンの20mg/mL緩衝溶液(3.0g)を仕込み、溶解後、25℃にて6時間反応を行った。食塩(750mg)を溶解させた後、クロロホルム(4.5g)を用いて抽出を行い、有機層を無水硫酸ナトリウムで乾燥後、ろ過した。酢酸エチル(45g)を用いてろ液を希釈後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(52)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.47(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.37-3.43(2H, m, -CH 2 NH-), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 4.89(1H, d, -CH 2 O-), 5.05(1H, d, -CH 2 O-), 5.14(2H, dd, -CH 2 OCONH-), 6.22(1H, s, -CH<), 6.52-7.57(5H, arom.H)
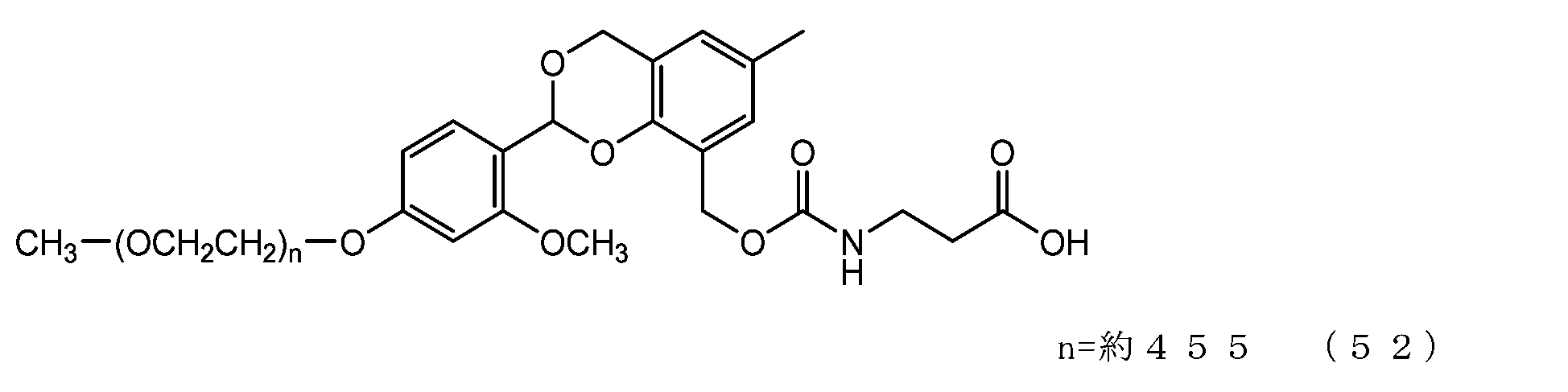
0.1Mリン酸ナトリウム緩衝溶液(pH8.5)を用い、β-アラニンの20mg/mL緩衝溶液を調製した。温度計、窒素吹込み管および撹拌機を装備した50mLの3つ口フラスコに式(44)の化合物(143mg, 0.0072mmol)およびβ-アラニンの20mg/mL緩衝溶液(3.0g)を仕込み、溶解後、25℃にて6時間反応を行った。食塩(750mg)を溶解させた後、クロロホルム(4.5g)を用いて抽出を行い、有機層を無水硫酸ナトリウムで乾燥後、ろ過した。酢酸エチル(45g)を用いてろ液を希釈後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(53)の化合物を得た。
2.47(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.37-3.43(2H, m, -CH 2 NH-), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 4.89(1H, d, -CH 2 O-), 5.05(1H, d, -CH 2 O-), 5.14(2H, dd, -CH 2 OCONH-), 6.22(1H, s, -CH<), 6.52-7.57(7H, arom.H)
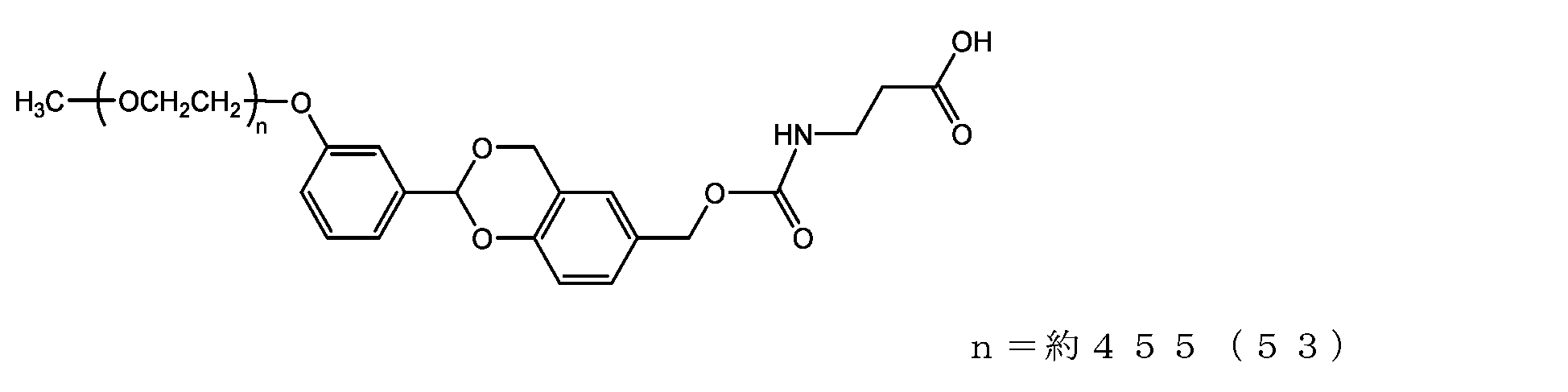
式(50)、(51)、(52)および(53)の化合物(20mg)をそれぞれpD 7.4のリン酸ナトリウム重水緩衝液(0.7mL)に溶解後、40℃の恒温槽で静置し、任意のタイミングでNMR測定を行った。図1は、静置0時間のβアラニン付加体量を100%とした時の任意のタイミングにおけるβアラニン付加体量を表す。グラフの近似式をもとに計算式
から算出した半減期(t1/2)は、式(50)の化合物は3.6日、式(51)の化合物は31.5日、式(52)の化合物は0.55日、式(53)の化合物は231日であり、式(50)、(51)および(52)で表される本発明の化合物は先行発明である式(53)で表される特許文献3に例示の化合物と比較して生理条件下で目的とした半減期を有することが示された。
式(22)の化合物を用い、RNase A(Roche製、 RNase A from bovine pancreas、Mw=13,700)の修飾を行った。0.1Mホウ酸緩衝液(pH=8.5)を用い、RNase Aの10mg/mL緩衝溶液を調製し、RNase Aの10mg/mL緩衝溶液(636.3 μL)および0.1Mホウ酸緩衝液(863.7 μL)を混合後、式(22)の化合物(6.0mg)を加え、20℃で20時間反応を行った。反応溶液をフィルターろ過後、下記条件にて陽イオン交換カラムを用いたHPLC測定を実施した。UVにて溶出液をモニターしながら分取し、式(22)の化合物が1か所修飾された純度96.1%のmono PEG RNase Aを得た。図2は分取したmono PEG RNase AのHPLC測定結果である。
・HPLC装置:Thermo Ultimate3000
・カラム:TSKgel SP-5PW(7.5x75mm, 10um; 東ソー株式会社)
・流速:1mL/分
・分析時間:25分(平衡化含む)
・カラム温度:25℃
・注入量:20 μL
・検出器:PDA(測定波長: 280nm)
・移動相A:5mM リン酸ナトリウム緩衝液 (pH8.5)
・移動相B:5mM リン酸ナトリウム緩衝液+0.5mol/L NaCl (pH8.5)
・グラジエントプログラム(A/B):100/0(-15分)→100/0(0分)→33/67(10分)
温度計、窒素吹込み管および撹拌機を装備した4つ口フラスコに日油社製 ME-200MS(α-メチル-ω-[(メチルスルホニル)オキシ]ポリ(オキシエチレン), 10g, 0.5mmol)、3-メチル-4-ヒドロキシベンズアルデヒド(272mg, 2mmol)、炭酸カリウム(0.69g, 5mmol)およびアセトニトリル(50g)を仕込み80℃にて6時間反応を行った。反応後、ろ過、濃縮し、ジクロロメタン(80g)に再溶解した。その後、1重量%の炭酸カリウムおよび10重量%の食塩を含む水溶液を加え、窒素雰囲気下、25℃で10分間撹拌した。撹拌後、10分間静置し、有機層を回収した。回収した有機層に硫酸マグネシウムを加え窒素雰囲気下、25℃で10分間撹拌した後、吸引ろ過した。得られたろ液を濃縮した後、酢酸エチルに溶解し、ヘキサンを加えて窒素雰囲気下、25℃で結晶を析出させた。析出した結晶を吸引ろ過で回収し、減圧乾燥して式(54)の化合物を得た。
2.28(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 6.93(1H, d, arom.H), 7.69(2H, d, arom.H), 9.85(1H, s, -CHO)

温度計、窒素吹込み管、撹拌機および冷却管を装備した3つ口フラスコに式(54)の化合物(3.0g, 0.15mmol)、2,4-ビス(ヒドロキシメチル)-p-クレゾール(1.0g, 6.0mmol)、テトラヒドロフラン(15.0g)および2,6-ジ-tert-ブチル-p-クレゾール(3.3mg)を仕込み、溶解後、モレキュラーシーブ5A(3.0g)およびp-トルエンスルホン酸一水和物(34.2mg, 0.18mmol)を加えて、40℃にて4時間反応を行った。N-メチルモルホリン(36.4mg, 0.36 mmol)を加えてしばらく攪拌し、ろ過後、トルエン(33.0g)を用いてろ液を希釈した。pH12の20%食塩水(30g)を用いて水洗を繰り返し行った後、有機層を無水硫酸ナトリウムで乾燥し、ろ過後、溶媒を減圧留去した。2,6-ジ-tert-ブチル-p-クレゾールを添加したトルエンに残渣を溶解し、ヘキサンを添加して晶析を行った後、吸引ろ過し、減圧乾燥して式(55)の化合物を得た。
2.23(3H, s, -CH 3 ), 2.28(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.62(2H, s, -CH 2 OH), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 5.92(1H, s, -CH<), 6.8-7.3(5H, arom.H)
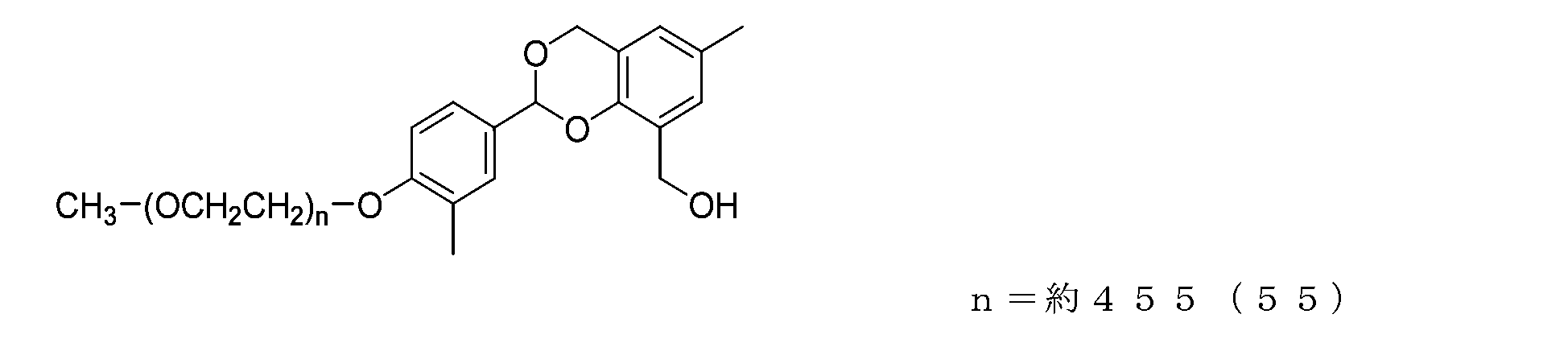
温度計、窒素吹込み管および撹拌機を装備した3つ口フラスコに式(55)の化合物(500mg, 0.025mmol)、2,6-ジ-tert-ブチル-p-クレゾール(0.5mg)およびトルエン(6.0g)を仕込み、溶解後、トリエチルアミン(38mg, 0.375mmol)およびp-ニトロフェニルクロロホルメート(50.4mg, 0.25mmol)を仕込んで、60℃にて3時間反応を行った。反応後、2,6-ジ-tert-ブチル-p-クレゾール(20mg)を添加した酢酸エチル(100g)とアセトニトリル(1.25g)の混合溶媒で希釈し、ヘキサン(50g)を添加して結晶を析出させた後、吸引ろ過して結晶を得た。得られた結晶を減圧乾燥して式(56)の化合物を得た。
2.23(3H, s, -CH 3 ), 2.31(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.24(m, -(OCH 2 CH 2 )n-), 4.96(1H, d, -CH 2 O-), 5.15(1H, d, -CH 2 O-), 5.34(2H, dd, -CH 2 OCO-), 5.96(1H, s, -CH<), 6.8-7.4(5H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):20,223
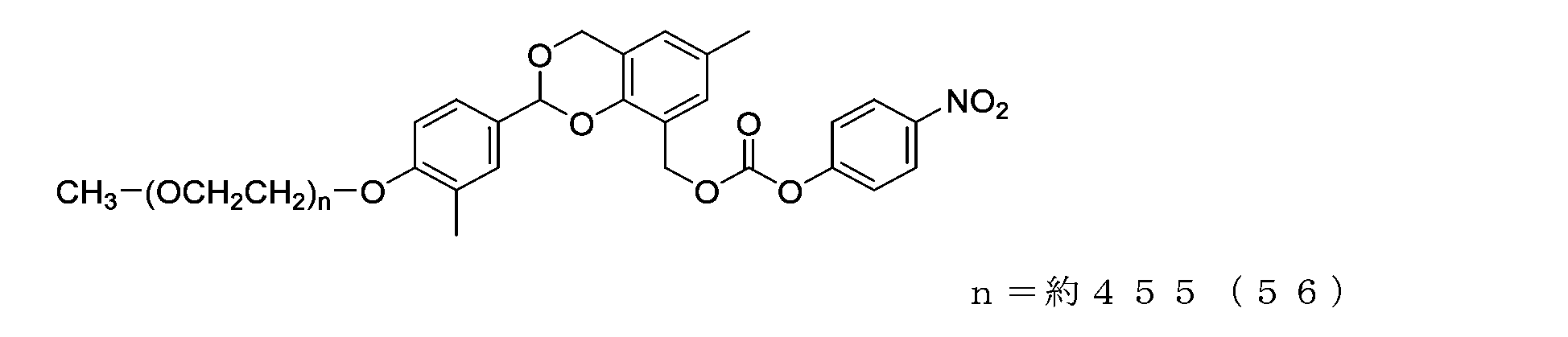
実施例14-1の3-メチル-4-ヒドロキシベンズアルデヒドの代わりに4-ヒドロキシ-3,5-ジメチルベンズアルデヒド(300mg, 2.0mmol)を用いた以外は同様にして、式(57)で表される化合物を得た。
2.36(6H, s), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 7.54(2H, s, arom.H), 9.88(1H, s, -CHO)

実施例14-2の式(54)の化合物の代わりに式(57)の化合物(3.0g, 0.15mmol)を用いた以外は同様にして、式(58)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 2.36(6H, s), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.68(2H, s, -CH 2 OH), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 5.89(1H, s, -CH<), 6.8-7.3(4H, arom.H)
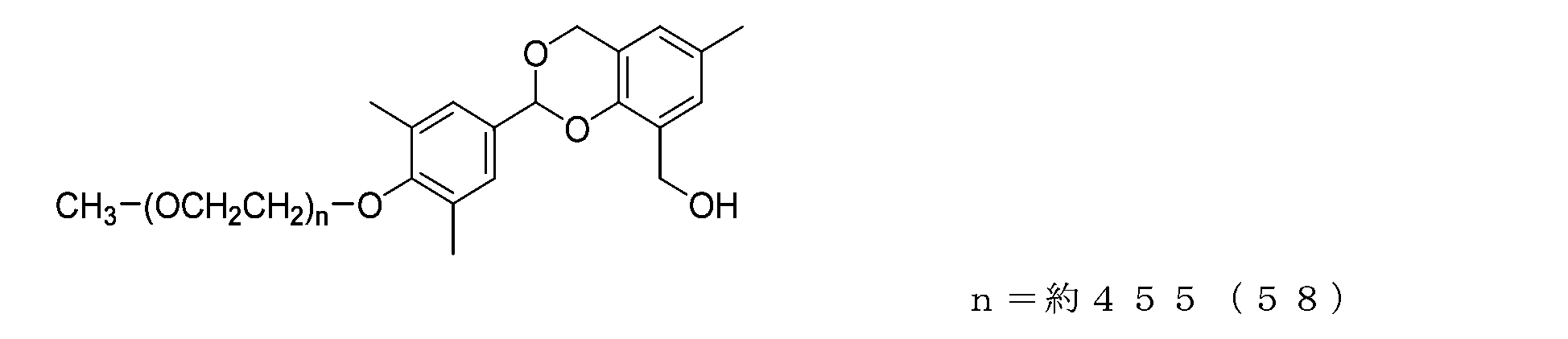
実施例14-3の式(55)の化合物の代わりに式(58)の化合物(500mg, 0.025mmol)を用いた以外は同様にして、式(59)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 2.36(6H, s), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.68(2H, s, -CH 2 O-), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 5.89(1H, s, -CH<), 6.8-7.3(4H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):20,072

実施例14-1の3-メチル-4-ヒドロキシベンズアルデヒドの代わりに2-ヒドロキシベンズアルデヒド(244mg, 2.0mmol)を用いた以外は同様にして、式(60)で表される化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 6.9-7.8(4H, arom.H), 10.52(1H, s, -COH)

実施例14-2の式(54)の化合物の代わりに式(60)の化合物(3.0g, 0.15mmol)を用いた以外は同様にして、式(61)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.49(1H, d, -CH 2 OH), 4.78(1H, d, -CH 2 OH), 4.99(1H, d, -CH 2 OCH<), 5.19(1H, d, -CH 2 O-), 6.32(1H, s, -CH<), 6.8-7.7(6H, arom.H)
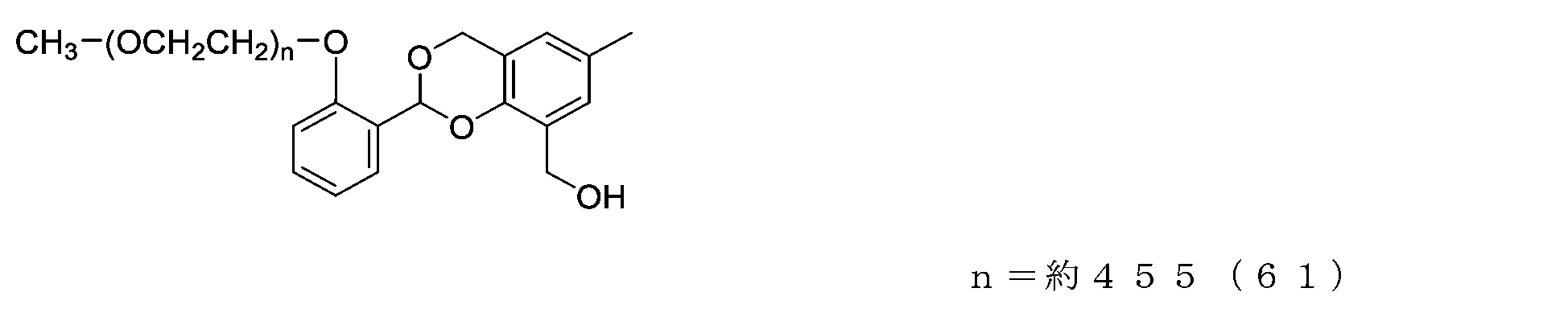
実施例14-3の式(55)の化合物の代わりに式(61)の化合物(500mg, 0.025mg)を用いた以外は同様にして、式(62)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.22(2H, m, -CH 2 O-), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 6.37(1H, s, -CH<), 6.8-7.3(8H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):19,942
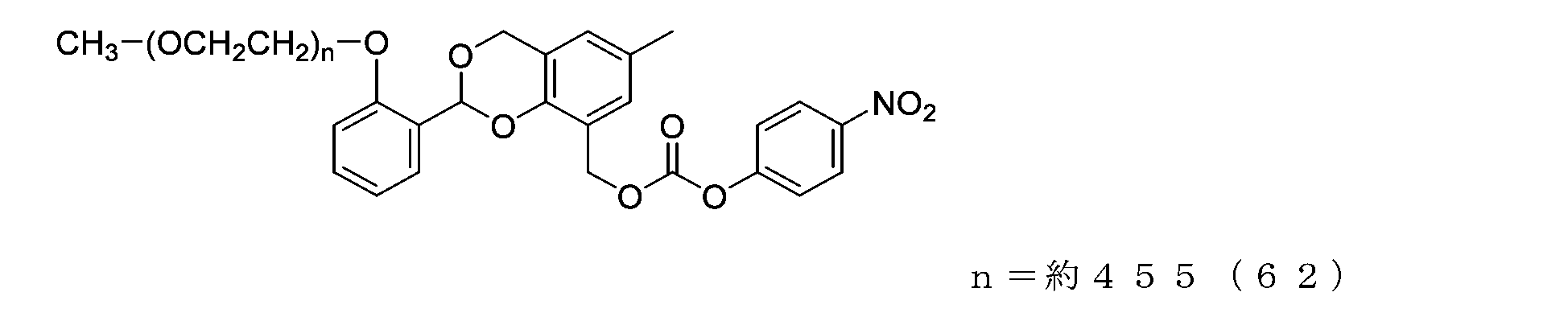
実施例14-1の3-メチル-4-ヒドロキシベンズアルデヒドの代わりに2-ヒドロキシ-6メトキシベンズアルデヒド(304mg, 2.0mmol)を用いた以外は同様にして、式(63)で表される化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 6.6-7.8(3H, arom.H), 10.53(1H, s, -COH)

実施例14-2の式(54)の化合物の代わりに式(63)の化合物(3.0g, 0.15mmol)を用いた以外は同様にして、式(64)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.49(1H, d, -CH 2 OH), 4.78(1H, d, -CH 2 OH), 4.90(1H, d, -CH 2 OCH<), 5.15(1H, d, -CH 2 O-), 6.8-7.7(5H, arom.H)

実施例14-3の式(55)の化合物の代わりに式(64)の化合物(500mg, 0.025mmol)を用いた以外は同様にして、式(65)で表される化合物を得た。
2.30(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.22(2H, m, -CH 2 O-), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 6.6-7.1(5H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):20,915
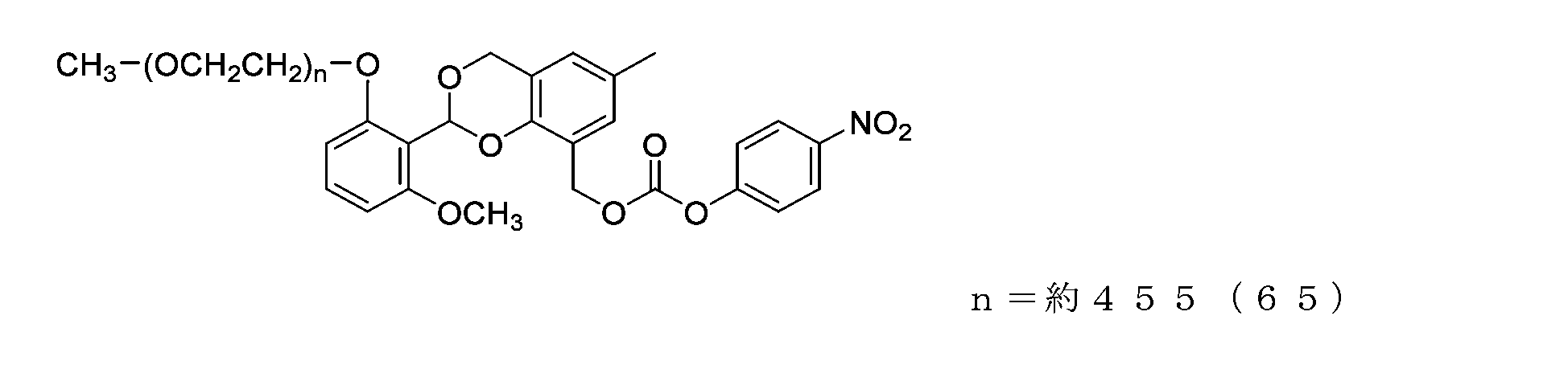
実施例17-1の日油社製ME-200MSの代わりに日油社製ME-050MS(2.5g, 0.5mmol)を用いた以外は同様にして、式(66)で表される化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 6.6-7.8(3H, arom.H), 10.53(1H, s, -COH)

実施例17-2の化合物(63)の代わりに式(66)の化合物(0.75g, 0.15mmol)を用いた以外は同様にして、式(67)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.49(1H, d, -CH 2 OH), 4.78(1H, d, -CH 2 OH), 4.90(1H, d, -CH 2 OCH<), 5.15(1H, d, -CH 2 O-), 6.8-7.7(5H, arom.H)
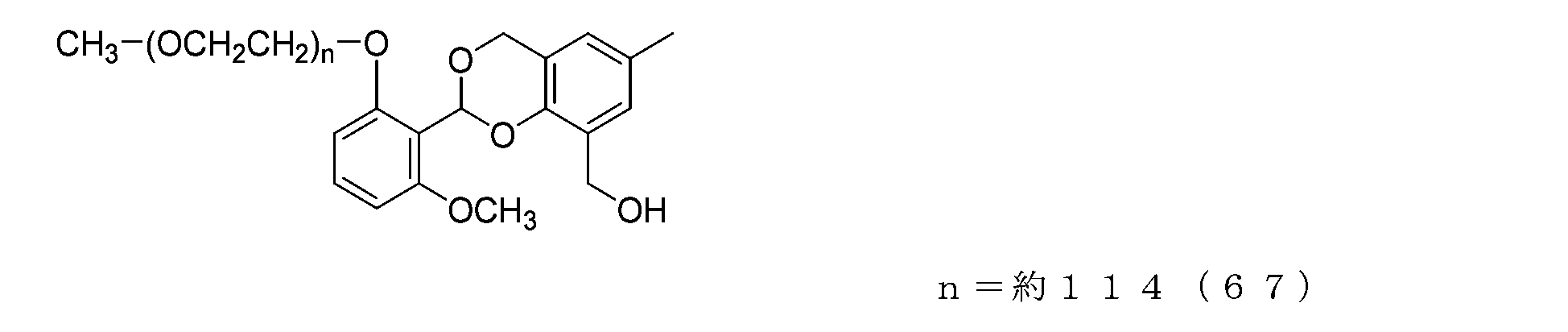
実施例17-3の化合物(64)の代わりに式(67)で表される化合物(125mg, 0.025mmol)を用いた以外は同様にして式(68)で表される化合物を得た。
2.30(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.22(2H, m, -CH 2 O-), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 6.6-7.1(5H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):5,400
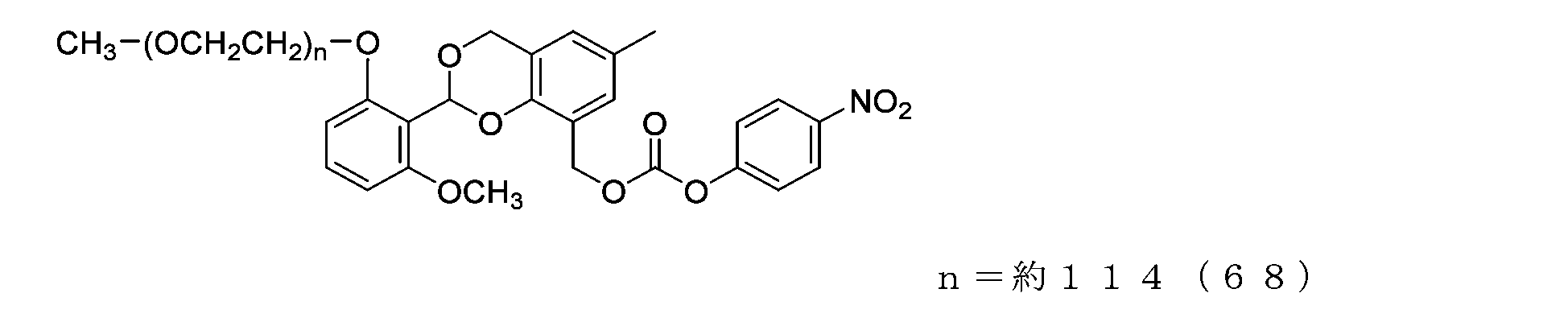
実施例17-1の日油社製ME-200MSの代わりに日油社製ME-020MS(1g, 0.5mmol)を用いた以外は同様にして、式(69)で表される化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 6.6-7.8(3H, arom.H), 10.53(1H, s, -COH)

実施例17-2の化合物(63)の代わりに式(69)の化合物(300mg, 0.15mmol)を用いた以外は同様にして、式(70)で表される化合物を得た。
2.29(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.49(1H, d, -CH 2 OH), 4.78(1H, d, -CH 2 OH), 4.90(1H, d, -CH 2 OCH<), 5.15(1H, d, -CH 2 O-), 6.8-7.7(5H, arom.H)

実施例17-3の化合物(64)の代わりに式(70)で表される化合物(50mg, 0.025mmol)を用いた以外は同様にして式(71)で表される化合物を得た。
2.30(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH2CH2)n-), 4.22(2H, m, -CH 2 O-), 4.95(1H, d, -CH 2 OCH<), 5.16(1H, d, -CH 2 O-), 6.6-7.1(5H, arom.H), 8.23(2H, d, arom.H)
数平均分子量(Mn):2,615
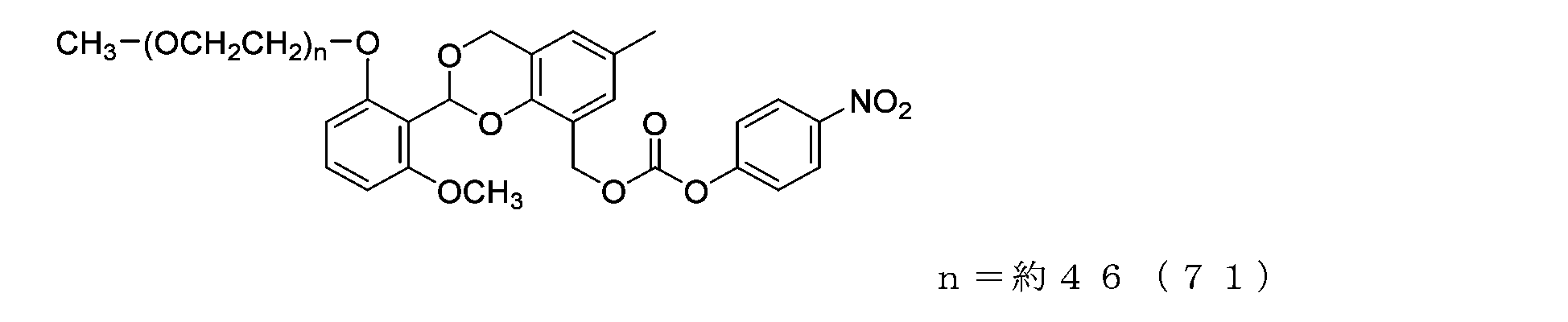
実施例14-1の3-メチル-4-ヒドロキシベンズアルデヒドの代わりに4-ヒドロキシ-2,6ジメトキシベンズアルデヒド(364mg, 2.0mmol)を用いた以外は同様にして、式(72)で表される化合物を得た。
3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.23(m, -(OCH 2 CH 2 )n-), 6.12(2H, arom.H), 10.35(1H, s, -COH)
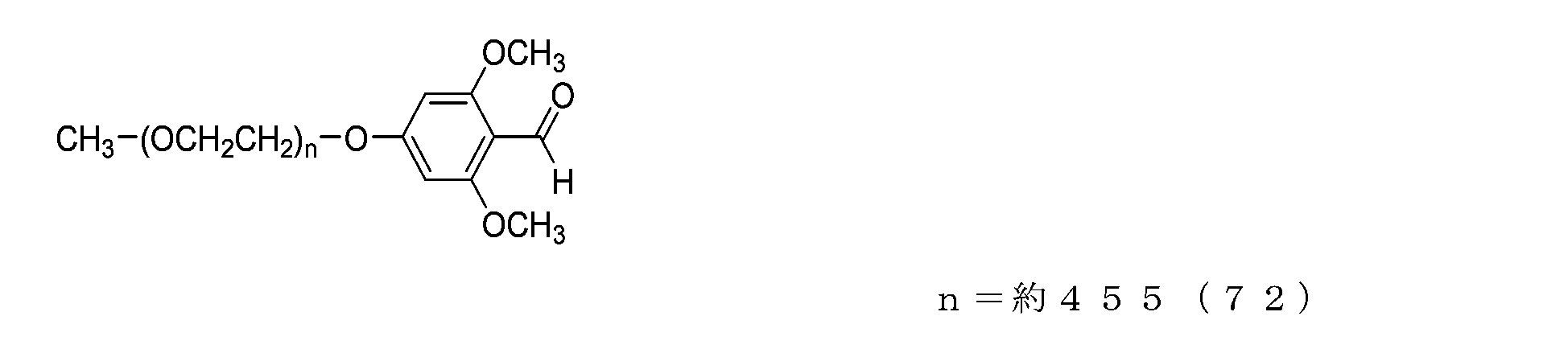
実施例14-2の式(54)の化合物の代わりに式(72)の化合物(3.0g, 0.15mmol)を用いた以外は同様にして、式(73)で表される化合物を得た。
2.28(3H, s, -CH 3 ), 2.84(1H, dd,-OH), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.47(1H, dd, -CH 2 OH), 4.75(1H, dd, -CH 2 OH), 4.98(1H, d, -CH 2 O-), 5.17(1H, d, -CH 2 O-), 6.20(1H, s, -CH<), 6.57-6.55(2H, m, arom.H), 6.78(1H, s, arom.H), 6.97(1H, s, arom.H), 7.58(2H, d, arom.H)

実施例14-3の式(55)の化合物の代わりに式(73)の化合物(500mg, 0.025mmol)を用いた以外は同様にして、式(74)で表される化合物を得た。
2.31(3H, s, -CH 3 ), 3.38(3H, s, -(OCH2CH2)n OCH 3 ), 3.52-4.17(m, -(OCH 2 CH 2 )n-), 4.93(1H, d, -CH 2 O-), 5.18(1H, d, -CH 2 O-), 5.33(2H, dd, -CH 2 OCO-), 6.26(1H, s, -CH<), 6.52-6.54(2H, m, arom.H), 6.87(1H, s, arom.H), 7.10(1H, s, arom.H), 7.26-7.27(2H, m, arom.H), 7.59(1H, d, arom.H), 8.22(2H, d, arom.H)
数平均分子量(Mn):20,560

0.1Mリン酸ナトリウム緩衝溶液(pH8.5)を用い、β-アラニンの20mg/mL緩衝溶液を調製した。式(56)の化合物(74mg, 0.0037mmol)をβ-アラニンの20mg/mL緩衝溶液(1.5mL)に溶解し、25℃にて6時間反応を行った。反応後、20重量%食塩を含む0.1Mリン酸ナトリウム緩衝液(pH8.5)で希釈し、クロロホルム(3g)を用いて抽出を行った。有機層を無水硫酸ナトリウムで乾燥後、ろ過した。酢酸エチル(45g)を用いてろ液を希釈後、ヘキサン(33g)を添加して晶析を行い、ろ過後、減圧乾燥して式(75)の化合物を得た。
2.23(3H, s, -CH 3 ), 2.28(3H, s, -CH 3 ), 2.47(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.37-3.43(2H, m, -CH 2 NH-), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 4.89(1H, d, -CH 2 O-), 5.05(1H, d, -CH 2 O-), 5.14(2H, dd, -CH 2 OCONH-), 5.93(1H, s, -CH<), 6.8-7.7(5H, arom.H)
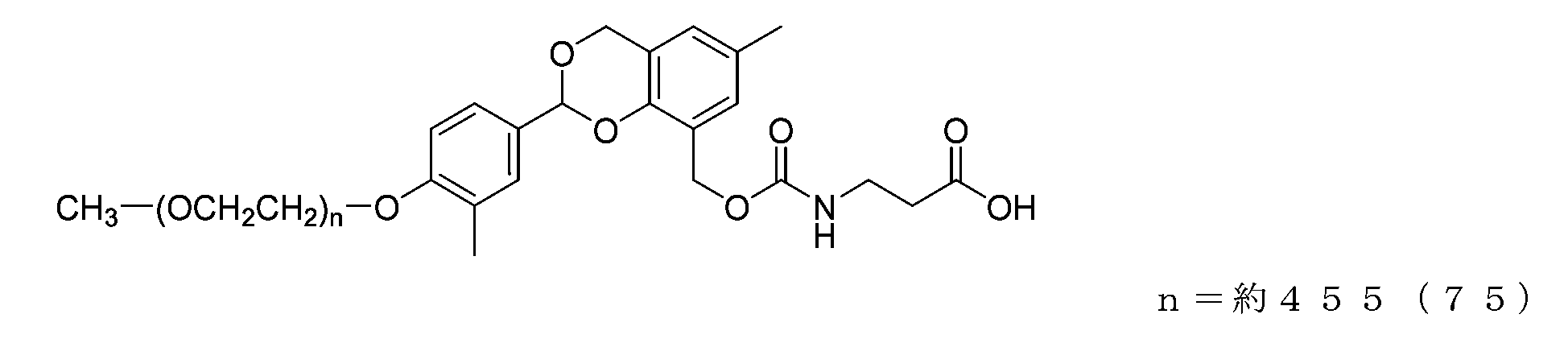
実施例21の式(56)の化合物の代わりに式(59)の化合物(74mg, 0.0037mmol)を用いた以外は同様にして、式(76)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.31(6H, s), 2.52(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 3.93-3.97(2H, m, -CH 2 NH-), 4.92(1H, d, -CH 2 O-), 5.09(1H, d, -CH 2 O-), 5.13(2H, dd, -CH 2 OCONH-), 5.89(1H, s, -CH<), 6.8-7.2(4H, arom.H)
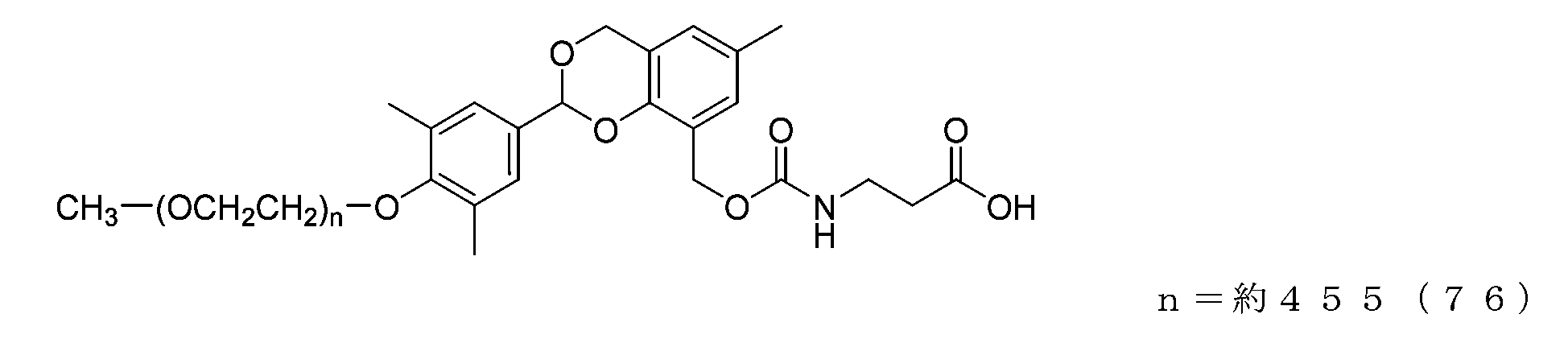
実施例21の式(56)の化合物の代わりに式(62)の化合物(74mg, 0.0037mmol)を用いた以外は同様にして、式(77)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.43(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.37-3.43(2H, m, -CH 2 NH-), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 4.92(1H, d, -CH 2 O-), 4.97(1H, d, -CH 2 O-), 5.19(2H, dd, -CH 2 OCONH-), 6.32(1H, s, -CH<), 6.8-7.7(6H, arom.H)
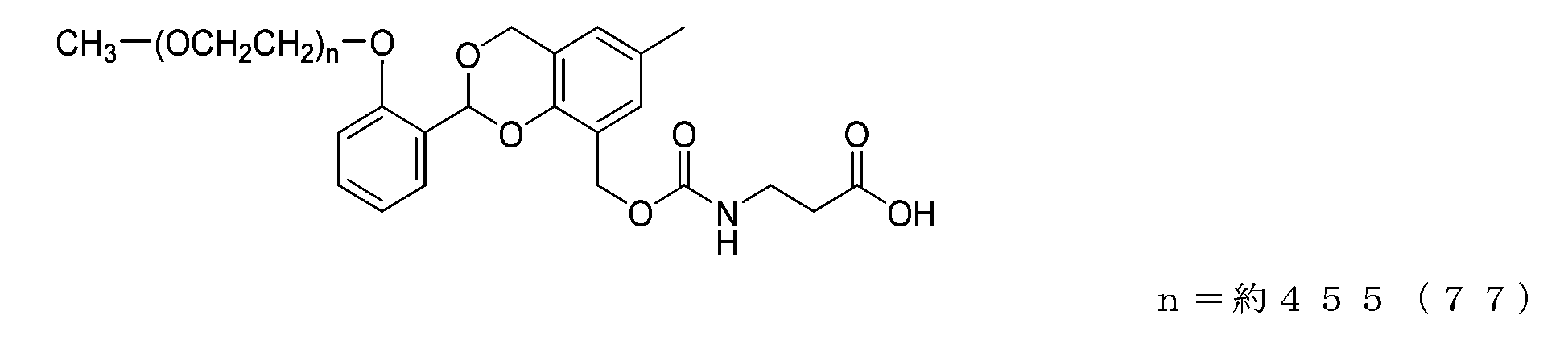
実施例21の式(56)の化合物の代わりに式(65)の化合物(74mg, 0.0037mmol)を用いた以外は同様にして、式(78)の化合物を得た。
2.28(3H, s, -CH 3 ), 2.47(2H, t, -CH 2 COOH), 3.38(3H, s, -(OCH2CH2)nOCH 3 ), 3.52-4.21(m, -(OCH 2 CH 2 )n-), 4.89(1H, d, -CH 2 O-), 5.03(2H, dd, -CH 2 OCONH-), 5.14(1H, d, -CH 2 O-), 6.53(1H, s, -CH<), 6.8-7.7(5H, arom.H)
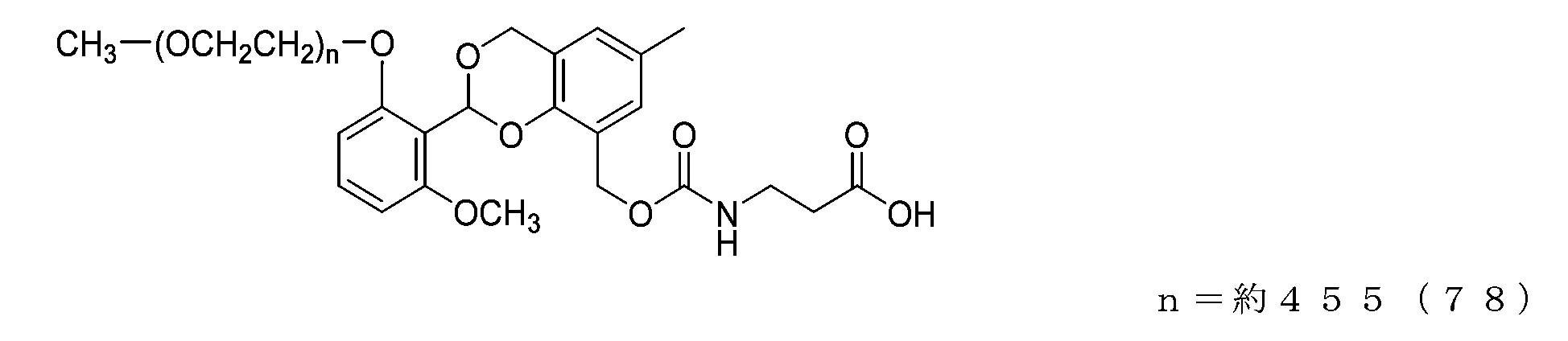
式(75)、(76)、(77)および(78)の化合物(20mg)をそれぞれpD7.4のリン酸ナトリウム重水緩衝液(0.7mL)に溶解後、40℃の恒温槽で静置し、任意のタイミングでNMR測定を行った。図3は、静置0時間のβアラニン付加体量を100%とした時の任意のタイミングにおけるβアラニン付加体量を表す。
グラフの近似式をもとに分解性試験に記載した計算式[数1]から算出した半減期(t1/2)は、式(75)の化合物は2.3日、式(76)の化合物は30.1日、式(77)の化合物は23.1日、式(78)の化合物は1.4日であり、本発明の化合物は、先行発明である式(53)で表される特許文献3に例示の化合物と比較して、生理条件下で目的とした半減期を有することが示された。
日油社製SUNBRIHGT MENP-20T(22.5mg, 1.13mmol)および国際公開2019/203904号でTNFα阻害活性を有し、かつPEG化によりTNFα阻害活性が低下することが報告されている配列番号1で表されるアプタマーの5’末端にC6アミノリンカーを持つ化合物NH2-C6-(Apt-TNFα)(1.8mg, 141.8nmol)を100mM重炭酸ナトリウム緩衝液(pH8.3)125μLに溶解し、窒素雰囲気下、40℃で24時間反応させた。反応後、反応溶液をフィルターろ過後、下記条件にて逆相カラムを用いたHPLC測定を実施した。260nmにて溶出液をモニターしながら分取した溶液を限外ろ過にてPBSに置換し、純度98.9%、GPCのピークトップ分子量33,101の式(28)の化合物がApt-TNFαと1:1のモル比で結合したPEG(20k)-OCO-NH-C6-(Apt-TNFα)を得た。図5(A)は分取したPEG(20k)-OCO-NH-C6-(Apt-TNFα)のHPLC測定結果である。
・HPLC装置:Thermo Ultimate3000
・カラム:Xbridge Oligonucleotide BEH C18 (4.6x50mm, 2.5um, Waters社)
・流速:1mL/分
・分析時間:25分(平衡化含む)
・カラム温度:35℃
・注入量:30μL
・検出器:UV(測定波長: 260nm)
・移動相A:25mMヘキサフルオロイソプロパノール-15mMジブチルアミン/水
・移動相B:25mMヘキサフルオロイソプロパノール-15mMジブチルアミン/メタノール
・グラジエントプログラム(A/B):70/30(0分)→10/90(20分)→10/90(30分)
比較例3の日油社製SUNBRIGHT MENP-20Tの代わりに式(28)の化合物(22.5mg, 1.13mmol)を用いた以外は同様にして、純度84.7%、GPCのピークトップ分子量30,632の式(28)の化合物がApt-TNFαと1:1のモル比で結合した2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)を得た。図5(B)は分取した2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)のHPLC測定結果である。
生細胞濃度が4×105cells/mLのNF-kB Reporter, Luciferase, HEK293細胞(BPS Bioscience Inc.)を96ウェルプレート(Thermo Fisher Scientific)の各ウェルに80μL添加し、22時間培養した。その後、それぞれのウェルに5μM NH2-C6-(Apt-TNFα)溶液、500nM PEG(20k)-OCO-NH-C6-(Apt-TNFα)溶液または500nM 2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)溶液を独立したウェルそれぞれに10μL添加した。添加後、50ng/mL TNFα溶液を10μLずつ添加し、0.5時間培養後、ルシフェラーゼ活性をBio-Glo Luciferase Assay System(Promega)にて測定した。
活性は、TNFα処理なしの系を0%、TNFα処理ありの系を100%として相対値として算出した。
結果は、平均値±標準誤差として表され、P値はスチューデントのt検定によって計算し、両側P値が0.05未満であることを統計的に有意差があると定義した。
評価の結果、NH2-C6-(Apt-TNFα)および2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)ではTNFα阻害活性を確認できた一方で、PEG(20k)-OCO-NH-C6-(Apt-TNFα)ではTNFα阻害活性を確認できなかった。このことから、本発明の2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)は既存技術であるPEG(20k)-OCO-NH-C6-(Apt-TNFα)と比較して、生理機能性物質を徐放でき、薬理活性を改善し得ることが示された。
図6はPEG(20k)-OCO-NH-C6-(Apt-TNFα)、2MPEG(20k)-OCO-NH-C6-(Apt-TNFα)およびNH2-C6-(Apt-TNFα)を用いたTNFα阻害活性の評価結果である。
0.2Mホウ酸緩衝液(pH10.5)に溶解した29mg/mLインスリン溶液0.25mLをアセトニトリルに溶解した100mg/mLの日油社製SUNBRIGHT MENP-20T溶液0.25mLに添加し、窒素雰囲気下、25℃で3時間反応させた。反応後、限外ろ過により溶液を10mMギ酸アンモニウム緩衝液(pH9.0)に交換した後、下記条件にて陰イオン交換クロマトグラフィーによりPEG(20k)-OCO-NH-インスリンを得た。図7(A)は精製したPEG(20k)-OCO-NH-インスリンのHPLC分析結果である。
・HPLC装置:Nexera(株式会社島津製作所)
・カラム:Asahipack ES-502N 7C (7.5x100mm, 9um, 昭和電工)
・流速:1mL/分
・分析時間:30分
・カラム温度:25℃
・注入量:50μL
・検出器:PDA(測定波長: 280nm)
・移動相A:10mMギ酸アンモニウム緩衝液(pH8.5)
・移動相B:0.25M硫酸アンモニウム含有10mMギ酸アンモニウム緩衝液(pH8.5)
・グラジエントプログラム(A/B):100/0(0分)→67/33(20分)→0/100(30分)
比較例4の日油社製SUNBRIGHT MENP-20Tの代わりに式(28)の化合物を用いた以外は同様にして2MPEG(20k)-OCO-NH-インスリンを得た。図7(B)は精製した2MPEG(20k)-OCO-NH-インスリンのHPLC分析結果である。
比較例4で調製した0.5mg/mLのPEG(20k)-OCO-NH-インスリン100μLまたは実施例26で調製した0.5mg/mLの2MPEG(20k)-OCO-NH-インスリン100μLを100mMリン酸緩衝液(pH6.0)100μL、100mMリン酸緩衝液(pH7.0)100μL、100mMギ酸アンモニウム緩衝液(pH8.0)100μLまたは100mMギ酸アンモニウム緩衝液(pH9.0)100μLと混合した後、25℃で静置し、任意のタイミングでHPLC分析を行った。
PEG(20k)-OCO-NH-インスリンは、いずれの測定pHにおいても時間経過に伴うPEG(20k)-OCO-NH-インスリンの減少は確認できなかった。一方で、2MPEG(20k)-OCO-NH-インスリンは、すべての測定pHにおいて時間経過にともない2MPEG(20k)-OCO-NH-インスリンが減少し、特にpH6.0では減少速度が大きく、インスリンの顕著な増加が確認された。以上から、本発明の2MPEG(20k)-OCO-NH-インスリンは時間経過によりアセタールが加水分解して、インスリンを放出することが示された。さらに、アセタール構造の加水分解は中性~塩基性条件で抑制できることが示された。
図8(A)は静置0時間のPEG(20k)-OCO-NH-インスリン量を100%とした時の任意のタイミングにおけるPEG(20k)-OCO-NH-インスリン量を表し、図8(B)は2MPEG(20k)-OCO-NH-インスリン量を100%とした時の任意のタイミングにおける2MPEG(20k)-OCO-NH-インスリン量を表す。
本出願は、2022年4月22日出願の日本特許出願(特願2022-070669)に基づくものであり、その内容はここに参照として取り込まれる。
Claims (6)
- 下記の式(1)、式(2)、式(3)または式(4)で表され、生理的条件下で開裂することを特徴とする、アセタール型リリーサブルポリオキシエチレン誘導体。
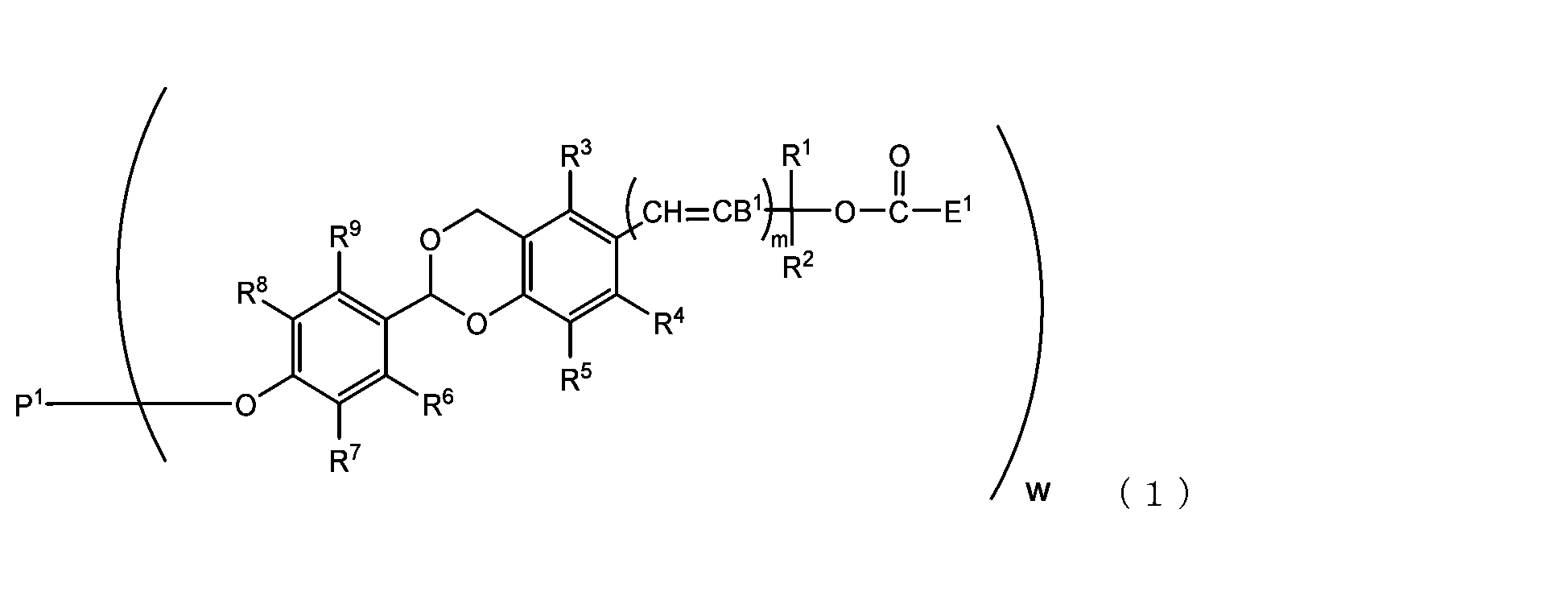
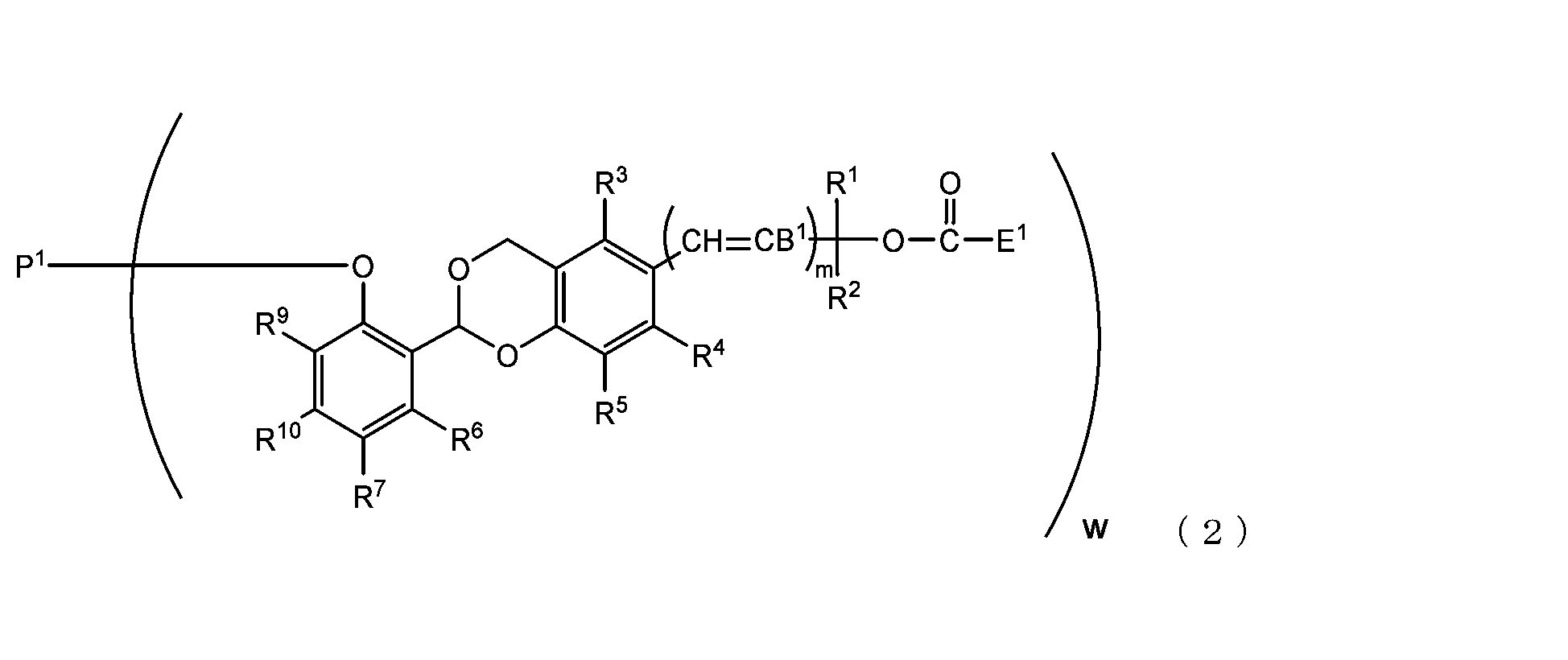
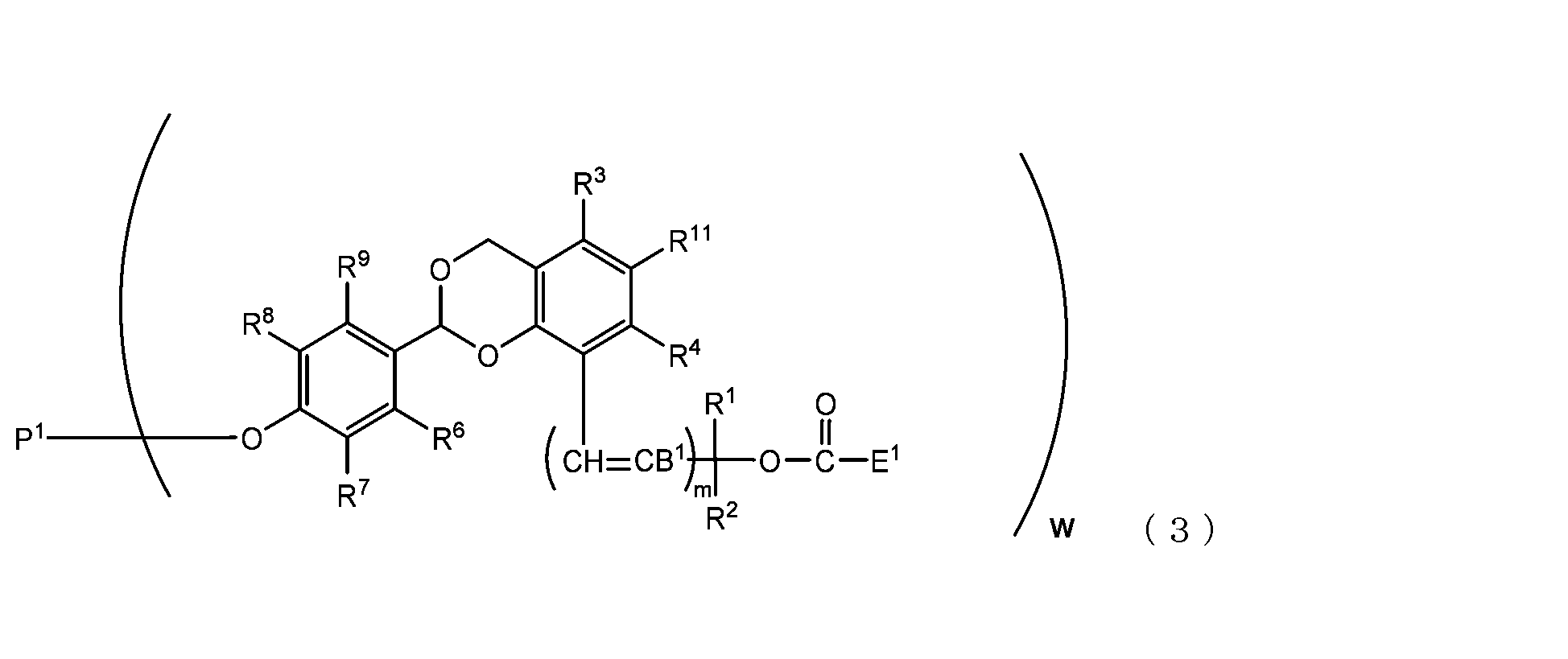
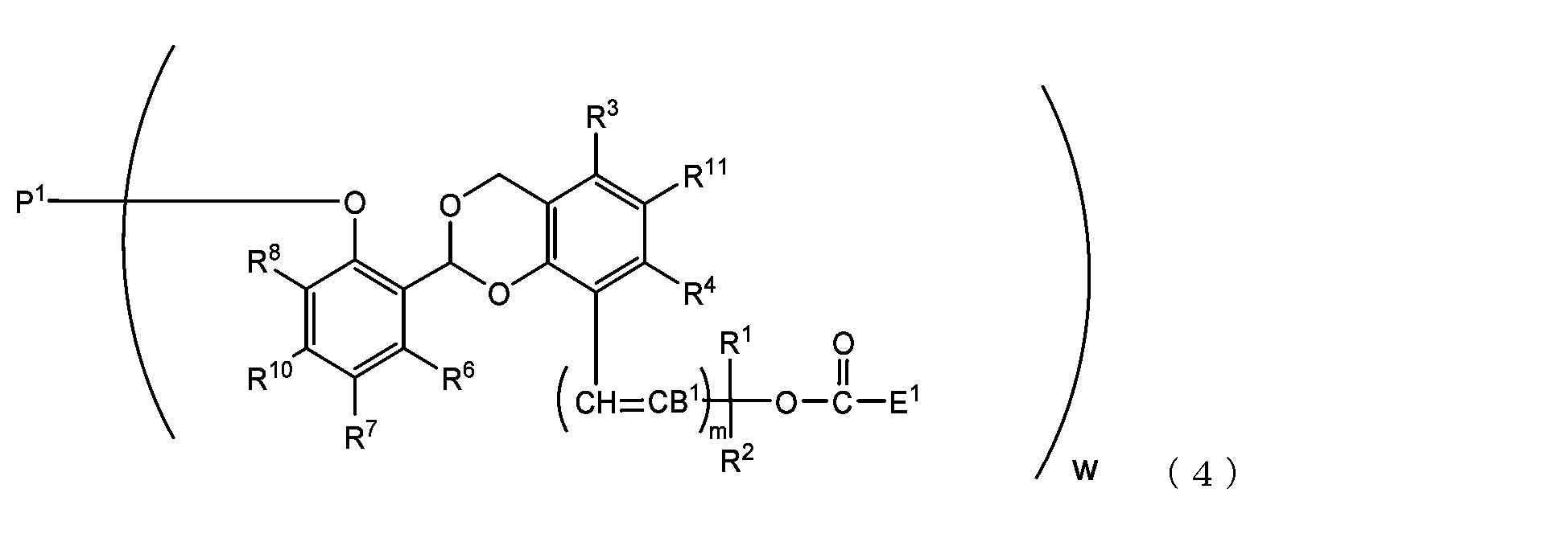
(式(1)、式(2)、式(3)及び式(4)中
B1は水素原子または-C(R1)(R2)OC(O)E1であり、
E1は脱離基であり、
P1は、脱ヒドロキシ基を有するポリオキシエチレン誘導体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5及びR11は,それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9及びR10は、それぞれ独立して、電子求引性置換基、電子供与性置換基または水素原子であり、
mは0または1である。) - mが0であり、R1およびR2が水素原子であり、R3、R4、R5及びR11がそれぞれ独立して水素原子またはメチル基であることを特徴とする、請求項1に記載のアセタール型リリーサブルポリオキシエチレン誘導体。
- 請求項1または2に記載のアセタール型リリーサブルポリオキシエチレン誘導体を製造する方法であって、
ポリオキシエチレン誘導体とヒドロキシベンズアルデヒド誘導体とをカップリングさせることによって、下記式(5)または式(6)で表されるカップリング生成物を得るカップリング工程、
前記カップリング工程後に、酸性条件下で、前記式(5)または前記式(6)で表される前記カップリング生成物を、2位にヒドロキシメチル基を有し、かつ4位または6位に置換基(-CH=CB1)mC(R1)(R2)-OH(B1、m、R1、R2は前述のとおりである)を有するフェノールと反応させることで、アセタール構造体を得るアセタール化工程、および
前記アセタール化工程後に、4位または6位の前記置換基の末端に脱離基構造(-OC(O)E1)を導入する脱離基構造導入工程
を備えることを特徴とする、アセタール型リリーサブルポリオキシエチレン誘導体の製造方法。


(式(5)または(6)中、P1、w、R6、R7、R8、R9およびR10は前述したとおりである) - 前記アセタール化工程と、前記脱離基構造導入工程との間に、前記式(5)及び(6)の前記カップリング生成物中の保護基で保護されたアミノ基の脱保護工程、及び前記脱保護工程後に脱保護されたアミノ基に生体機能性分子と反応可能な基を導入する工程を備えていることを特徴とする、請求項3に記載のアセタール型リリーサブルポリオキシエチレン誘導体の製造方法。
- 下記の式(7)、式(8)、式(9)または式(10)で表され、生理的条件下で開裂することを特徴とする、アセタール型リリーサブルポリオキシエチレン結合体。

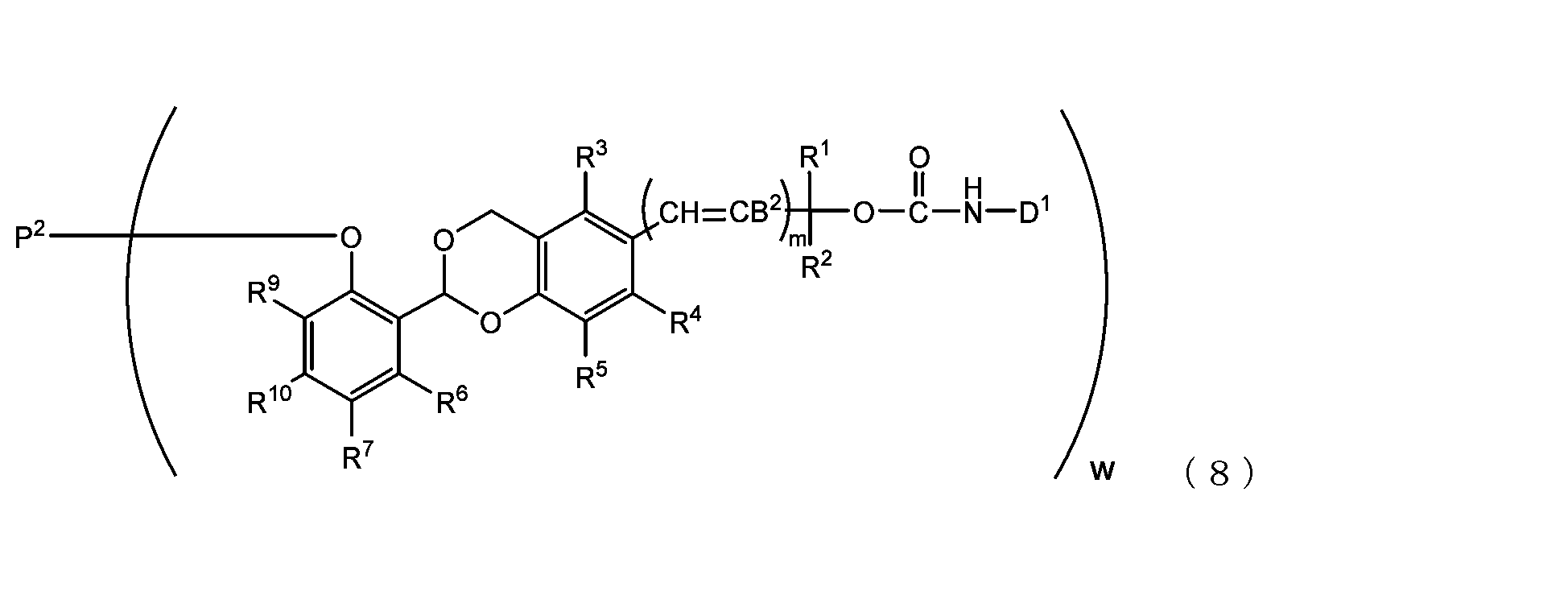
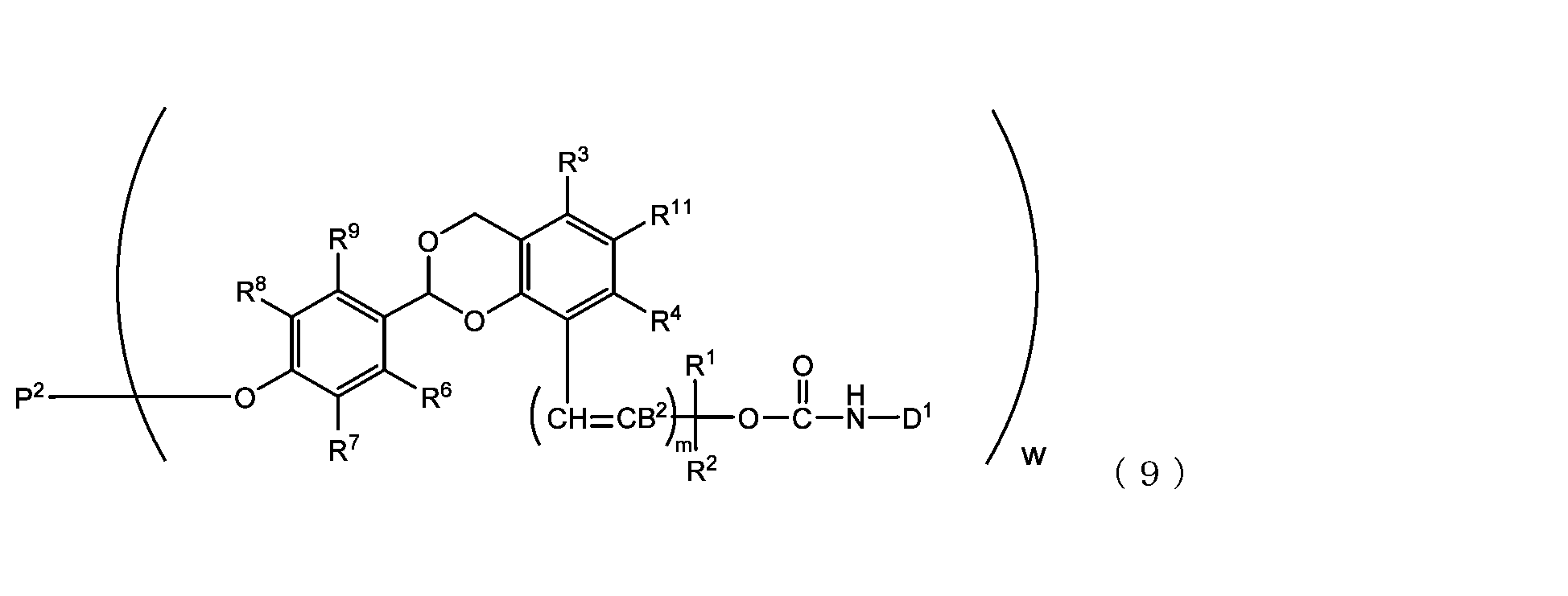

(式(7)、式(8)、式(9)及び式(10)中、
B2は水素原子または-C(R1)(R2)OC(O)NHD1であり、
D1は、生体機能性分子に含まれるアミノ基から、カーバメート結合を構成するアミノ基を除いた残基であり、
P2は、脱ヒドロキシ基を有するポリオキシエチレン誘導体、または脱ヒドロキシ基を有するポリオキシエチレン誘導体および生体機能性分子の結合体であり、
wは1~8の整数であり、
R1、R2、R3、R4、R5およびR11は、それぞれ独立して、炭素数1~10の炭化水素基または水素原子であり、
R6、R7、R8、R9およびR10は、それぞれ独立して、電子求引性置換基、電子供与性置換基または水素原子であり、
mは0または1である。) - 請求項5に記載のアセタール型リリーサブルポリオキシエチレン結合体を製造する方法であって、
請求項1または2記載のアセタール型リリーサブルポリオキシエチレン誘導体と生体機能性分子を、水溶性の有機溶媒が含まれていてもよい中性または塩基性の緩衝液中で反応させるカップリング工程、および
前記カップリング工程後の中性または塩基性条件下での精製工程
を備えることを特徴とする、アセタール型リリーサブルポリオキシエチレン結合体の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202380035693.5A CN119072516A (zh) | 2022-04-22 | 2023-04-19 | 缩醛型可释放聚氧乙烯衍生物及其制备方法和缩醛型可释放聚氧乙烯结合体 |
| EP23791904.8A EP4512845A4 (en) | 2022-04-22 | 2023-04-19 | Acetal-type releaseable polyoxyethylene derivative, its production process and acetal-type releaseable polyoxyethylene conjugate |
| US18/859,066 US20250288684A1 (en) | 2022-04-22 | 2023-04-19 | Acetal-type releasable polyoxyethylene derivative, production method thereof and acetal-type releasable polyoxyethylene conjugate |
| KR1020247035150A KR20250003614A (ko) | 2022-04-22 | 2023-04-19 | 아세탈형 방출 가능 폴리옥시에틸렌 유도체, 그 제조 방법 및 아세탈형 방출 가능 폴리옥시에틸렌 결합체 |
| CA3252159A CA3252159A1 (en) | 2022-04-22 | 2023-04-19 | Acetal-type releaseable polyoxyethylene derivative, its production process and acetal-type releaseable polyoxyethylene conjugate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022-070669 | 2022-04-22 | ||
| JP2022070669 | 2022-04-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023204256A1 true WO2023204256A1 (ja) | 2023-10-26 |
Family
ID=88419929
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/015663 Ceased WO2023204256A1 (ja) | 2022-04-22 | 2023-04-19 | アセタール型リリーサブルポリオキシエチレン誘導体、その製造方法及びアセタール型リリーサブルポリオキシエチレン結合体 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20250288684A1 (ja) |
| EP (1) | EP4512845A4 (ja) |
| JP (1) | JP2023160780A (ja) |
| KR (1) | KR20250003614A (ja) |
| CN (1) | CN119072516A (ja) |
| CA (1) | CA3252159A1 (ja) |
| WO (1) | WO2023204256A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025084230A1 (ja) * | 2023-10-16 | 2025-04-24 | 日油株式会社 | アミノ基を含むアセタール型リリーサブルポリオキシエチレン誘導体、その製造方法およびアミノ基を含むアセタール型リリーサブルポリオキシエチレン結合体 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018150311A (ja) | 2008-04-29 | 2018-09-27 | アセンディス ファーマ エンドクライノロジー ディヴィジョン エー/エス | Peg化された組換えヒト成長ホルモン化合物 |
| WO2018180914A1 (ja) * | 2017-03-30 | 2018-10-04 | 日油株式会社 | 自壊性アセタールリンカーを有する親水性ポリマー誘導体及びそれを用いた複合体 |
| WO2019203904A1 (en) | 2018-04-20 | 2019-10-24 | Academia Sinica | Tnf-targeting aptamers and uses thereof for treatment or diagnosing tnf-related inflammatory diseases |
| JP2022000043A (ja) | 2010-11-12 | 2022-01-04 | ネクター セラピューティクス | Il−2部分とポリマーとのコンジュゲート |
| JP2022070669A (ja) | 2020-10-27 | 2022-05-13 | 株式会社日立製作所 | データベースシステム、及びクエリ実行方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6784932B2 (ja) * | 2015-03-31 | 2020-11-18 | 日油株式会社 | 生体機能性分子または薬物キャリアの化学修飾用生分解性ポリエチレングリコール誘導体 |
-
2023
- 2023-04-19 EP EP23791904.8A patent/EP4512845A4/en active Pending
- 2023-04-19 CA CA3252159A patent/CA3252159A1/en active Pending
- 2023-04-19 JP JP2023068234A patent/JP2023160780A/ja active Pending
- 2023-04-19 KR KR1020247035150A patent/KR20250003614A/ko active Pending
- 2023-04-19 US US18/859,066 patent/US20250288684A1/en active Pending
- 2023-04-19 CN CN202380035693.5A patent/CN119072516A/zh active Pending
- 2023-04-19 WO PCT/JP2023/015663 patent/WO2023204256A1/ja not_active Ceased
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018150311A (ja) | 2008-04-29 | 2018-09-27 | アセンディス ファーマ エンドクライノロジー ディヴィジョン エー/エス | Peg化された組換えヒト成長ホルモン化合物 |
| JP2022000043A (ja) | 2010-11-12 | 2022-01-04 | ネクター セラピューティクス | Il−2部分とポリマーとのコンジュゲート |
| WO2018180914A1 (ja) * | 2017-03-30 | 2018-10-04 | 日油株式会社 | 自壊性アセタールリンカーを有する親水性ポリマー誘導体及びそれを用いた複合体 |
| JP2018172648A (ja) | 2017-03-30 | 2018-11-08 | 日油株式会社 | 自壊性アセタールリンカーを有する親水性ポリマー誘導体及びそれを用いた複合体 |
| WO2019203904A1 (en) | 2018-04-20 | 2019-10-24 | Academia Sinica | Tnf-targeting aptamers and uses thereof for treatment or diagnosing tnf-related inflammatory diseases |
| JP2022070669A (ja) | 2020-10-27 | 2022-05-13 | 株式会社日立製作所 | データベースシステム、及びクエリ実行方法 |
Non-Patent Citations (3)
| Title |
|---|
| FREEMAN, J.H., JAM. CHEM. SOC., vol. 74, 1952, pages 6257 - 6260 |
| See also references of EP4512845A4 |
| WUTS, P. G. M.;GREENE, T. W.: "Protective Groups in Organic Synthesis", 2007, WILEY-INTERSCIENCE |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025084230A1 (ja) * | 2023-10-16 | 2025-04-24 | 日油株式会社 | アミノ基を含むアセタール型リリーサブルポリオキシエチレン誘導体、その製造方法およびアミノ基を含むアセタール型リリーサブルポリオキシエチレン結合体 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA3252159A1 (en) | 2025-07-07 |
| JP2023160780A (ja) | 2023-11-02 |
| EP4512845A4 (en) | 2026-03-18 |
| EP4512845A1 (en) | 2025-02-26 |
| KR20250003614A (ko) | 2025-01-07 |
| CN119072516A (zh) | 2024-12-03 |
| US20250288684A1 (en) | 2025-09-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5951496B2 (ja) | ポリマー−薬物コンジュゲートの酸性塩形態及びアルコキシル化方法 | |
| CA2650035C (en) | Poly (ethylene glycol) containing chemically disparate endgroups | |
| US11529423B2 (en) | Biodegradable polyethylene glycol derivative having cyclic benzylidene acetal linker | |
| CN107573498A (zh) | 末端具有多个羟基的聚氧乙烯衍生物 | |
| WO2015152182A1 (ja) | 環状ベンジリデンアセタールリンカーを有する親水性ポリマー誘導体 | |
| WO2023204256A1 (ja) | アセタール型リリーサブルポリオキシエチレン誘導体、その製造方法及びアセタール型リリーサブルポリオキシエチレン結合体 | |
| WO2014157150A1 (ja) | ベンジリデンアセタールリンカーを有する親水性ポリマー誘導体 | |
| CA2515889A1 (en) | The selective and specific preparation of discrete peg compounds | |
| JP2025068607A (ja) | ケタール型リリーサブルポリオキシエチレン誘導体、その製造方法およびケタール型リリーサブルポリオキシエチレン結合体 | |
| WO2025084230A1 (ja) | アミノ基を含むアセタール型リリーサブルポリオキシエチレン誘導体、その製造方法およびアミノ基を含むアセタール型リリーサブルポリオキシエチレン結合体 | |
| WO2013106097A1 (en) | Poly(ethylene glycol) derivatives for click chemistry | |
| HK1170934B (en) | Acid salt forms of polymer-drug conjugates | |
| HK1170934A (en) | Acid salt forms of polymer-drug conjugates | |
| HK1170931B (en) | Salt form of a multi-arm polymer-drug conjugate | |
| HK1170931A (en) | Salt form of a multi-arm polymer-drug conjugate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23791904 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380035693.5 Country of ref document: CN Ref document number: 202447080124 Country of ref document: IN Ref document number: 18859066 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023791904 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023791904 Country of ref document: EP Effective date: 20241122 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18859066 Country of ref document: US |