WO2023234376A1 - ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 - Google Patents
ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 Download PDFInfo
- Publication number
- WO2023234376A1 WO2023234376A1 PCT/JP2023/020372 JP2023020372W WO2023234376A1 WO 2023234376 A1 WO2023234376 A1 WO 2023234376A1 JP 2023020372 W JP2023020372 W JP 2023020372W WO 2023234376 A1 WO2023234376 A1 WO 2023234376A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hyaluronic acid
- acid derivative
- group
- pharmaceutical composition
- chromatogram
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/726—Glycosaminoglycans, i.e. mucopolysaccharides
- A61K31/728—Hyaluronic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/145—Orthomyxoviridae, e.g. influenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0072—Hyaluronic acid, i.e. HA or hyaluronan; Derivatives thereof, e.g. crosslinked hyaluronic acid (hylan) or hyaluronates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55561—CpG containing adjuvants; Oligonucleotide containing adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55583—Polysaccharides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/572—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 cytotoxic response
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/575—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 humoral response
Definitions
- the present invention relates to hyaluronic acid derivatives, pharmaceutical compositions, and methods for producing pharmaceutical compositions.
- This application claims priority based on Japanese Patent Application No. 2022-088995 filed in Japan on May 31, 2022, the contents of which are incorporated herein.
- biopharmaceuticals that contain proteins, peptides, and nucleic acids as active ingredients have been put into practical use, and their number continues to increase year by year.
- Biopharmaceuticals can fill unmet medical needs that could not be met with conventional small molecule drugs. However, they are difficult to absorb from the gastrointestinal tract or mucous membranes, are unstable in the body, and have a short half-life in the blood. Therefore, biopharmaceuticals require frequent administration by injection, which places a heavy burden on both patients and medical personnel. Therefore, there is a need for drug base materials (sustained release drug delivery system base materials) that can encapsulate biopharmaceuticals and gradually release the active ingredients in vivo without impairing pharmacological activity.
- Patent Document 1 proposes a sustained release drug delivery system base material made of a highly safe hyaluronic acid derivative.
- This hyaluronic acid derivative spontaneously associates in an aqueous solution, can efficiently encapsulate drugs, especially biopharmaceuticals, while maintaining their biological activity, and aggregates under physiological saline concentrations (or aggregates under physiological saline concentrations). (disperses even at low temperatures) and has good retention in the blood.
- This hyaluronic acid derivative can be used as a carrier that can efficiently encapsulate many drugs while maintaining pharmacological activity, especially when biopharmaceuticals are used as active ingredients, and as a sustained release carrier and targeting carrier with excellent blood retention. It is said that it can be used as a local (for example, subcutaneous, etc.) sustained release carrier that can continuously release drugs.
- the hyaluronic acid derivative used in Patent Document 2 etc. has a broad and non-uniform particle size distribution, and the particle size distribution of the hyaluronic acid derivative and its delivery to immune cells in lymph nodes are difficult to understand.
- the relationship between gender and activation ability of the immune cells has not been specifically investigated, and there is room for improvement.
- the present invention has been made in view of the above circumstances, and provides a hyaluronic acid derivative which, when formulated with a medicinal ingredient, has excellent delivery properties to immune cells in lymph nodes and the ability to activate the immune cells, and A pharmaceutical composition using a hyaluronic acid derivative and a method for producing the same are provided.
- the present invention includes the following aspects. (1) A hyaluronic acid derivative into which a steryl group has been introduced, A hyaluronic acid derivative having a ratio A1/A2 of areas A1 and A2 of 0.90 or more, calculated by the method shown below from a chromatogram determined by gel permeation chromatography.
- the ratio Da/Db of the distance Da and Db calculated by the method shown below from the chromatogram determined by gel permeation chromatography measurement is more than 0.00 and 1.20 or less, according to (1).
- Hyaluronic acid derivative (i) Draw a perpendicular line from the maximum refractive index intensity point Ka on the chromatogram of 150 kDa polyacrylic acid, which is a standard substance, to the baseline B, and draw a perpendicular line from the point of intersection with the baseline to Ba, and between the maximum refractive index intensity point Ka and the above Ba.
- a hyaluronic acid derivative into which a steryl group has been introduced The weight of the hyaluronic acid derivative calculated from the chromatogram obtained by gel permeation chromatography measurement based on a calibration curve prepared using polyacrylic acid with molecular weights of 2 kDa, 4 kDa, 8 kDa, 18 kDa, 40 kDa, and 150 kDa as standard substances.
- a hyaluronic acid derivative having an average molecular weight of 110,000 or more and less than 500,000.
- R 1 , R 2 , R 3 , and R 4 are each independently selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl; Z represents a direct bond or a peptide linker consisting of 2 to 30 arbitrary amino acid residues;
- X1 is the following formula: -NR b -R, -NR b -COO-R, -NR b -CO-R, -NR b -CO-NR c -R, -COO-R, -O-COO-R, -S-R, -CO-Y a -SR, -O-CO-Y b -SR, -NR b -CO-Y b -SR, and -SSR,
- R a , R b and R c are each independently selected from the group consisting of a hydrogen atom, C 1-20 alkyl,
- a selected group may be inserted;
- R is a steryl group;
- Y is C 2-30 alkylene, or -(CH 2 CH 2 O) m -CH 2 CH 2 -, where the alkylene is a group consisting of -O-, -NR g - and -SS- A group selected from the group consisting of;
- R g is selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl and hydroxy C 2-20 alkyl, and the alkyl portion of the group is selected from the group consisting of -O- and -NH-
- Y a is C 1-5 alkylene;
- Y b is C 2-8 alkylene or C 2-8 alkenylene;
- m is an integer of 1 or more and 100 or less.
- the medicinal ingredient contains at least one selected from the group consisting of cancer antigens, infectious disease-derived antigens, and autoantigens in immune diseases, and further contains an adjuvant (11) or (12) The pharmaceutical composition described in .
- a method for producing a pharmaceutical composition comprising: (16) The method for producing a pharmaceutical composition according to (15), wherein the pH of the aqueous phase containing the hyaluronic acid derivative is 6.00 or more and 11.00 or less.
- the hyaluronic acid derivative of the above embodiment it is possible to provide a hyaluronic acid derivative that, when formulated with a medicinal ingredient, has excellent delivery properties to immune cells in lymph nodes and an ability to activate the immune cells.
- the pharmaceutical composition of the above embodiment contains the hyaluronic acid derivative and has excellent delivery properties to immune cells in lymph nodes and ability to activate the immune cells.
- the method for producing a pharmaceutical composition according to the above aspect uses the hyaluronic acid derivative, and a pharmaceutical composition having excellent delivery properties to immune cells in lymph nodes and ability to activate the immune cells can be obtained.
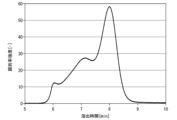
- 1 is a gel permeation chromatogram of the hyaluronic acid derivative in Synthesis Example 1-1.
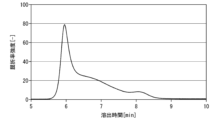
- 1 is a gel permeation chromatogram of a hyaluronic acid derivative in Synthesis Example 1-2.
- 1 is a gel permeation chromatogram of a hyaluronic acid derivative in Synthesis Example 1-3.
- 1 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-1.
- 1 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-2.
- 1 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-3.
- 1 is a gel permeation chromatogram of a hyaluronic acid derivative in Comparative Example 1-1.
- 1 is a gel permeation chromatogram of a hyaluronic acid derivative in Comparative Example 1-2.
- 1 is a gel permeation chromatogram of a hyaluronic acid derivative in Comparative Example 1-3.
- 1 is a gel permeation chromatogram of cholesteryl-modified pullulan (CHP) in Comparative Example 1-4.
- 2 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-1 and the hyaluronic acid derivative in the pharmaceutical composition in Example 2-1.
- CHP cholesteryl-modified pullulan
- Example 2 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-2 and the hyaluronic acid derivative in the pharmaceutical composition in Example 2-2.
- 2 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 1-3 and the hyaluronic acid derivative in the pharmaceutical composition in Example 2-3.
- 12 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide-incorporated DC expressing CD80 to peptide-incorporated dendritic cells (DC) in Test Example 1-1.
- 12 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide-incorporated DCs expressing CD86 to peptide-incorporated DCs in Test Example 1-1.
- FIG. 2 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide-incorporated cDC1 expressing CD80 to peptide-incorporated standard type 1 dendritic cells (cDC1) in Test Example 1-1.
- 12 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide-incorporated cDC1 expressing CD86 to peptide-incorporated cDC1 in Test Example 1-1.
- 12 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide-incorporated Mph expressing CD80 to peptide-incorporated macrophage cells (Mph) in Test Example 1-1.
- FIG. 12 is a graph showing an example of FACS analysis when calculating the ratio (%) of peptide uptake Mph expressing CD86 to peptide uptake Mph in Test Example 1-1.
- 2 is a gel permeation chromatogram of hyaluronic acid derivatives in Test Examples 2-1 to 2-8.
- 3 is a gel permeation chromatogram of hyaluronic acid derivatives in Test Examples 3-1 to 3-6. It is a gel permeation chromatogram of hyaluronic acid in Comparative Example 5-1.
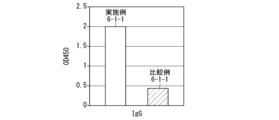
- Photographs of mixtures when attempting to prepare antigen peptide-hyaluronic acid complexes in Comparative Examples 5-1 and 5-2 and when preparing antigen peptide-hyaluronic acid derivative complexes in Example 7-1-1 This is a photograph of the solution. It is a gel permeation chromatogram of hyaluronic acid in Comparative Example 5-2. It is a gel permeation chromatogram of the hyaluronic acid derivative in Comparative Example 6-1. 1 is a gel permeation chromatogram of the hyaluronic acid derivative in Example 6-1. It is a graph showing IgG antibody titer (OD value) in Example 6-1-1 and Comparative Example 6-1-1.
- Example 6-1-1 It is a graph showing the IgG2a antibody titer (OD value) at a dilution factor of 10 (titer 10 ⁇ 1 ) in Example 6-1-1 and Comparative Example 6-1-1. It is a gel permeation chromatogram of the hyaluronic acid derivative in Example 7-1. 1 is a gel permeation chromatogram of a pharmaceutical composition containing an antigen peptide-hyaluronic acid derivative complex in Example 7-1-1. 1 is a gel permeation chromatogram of the hyaluronic acid derivative obtained in Example 1-1. 1 is a gel permeation chromatogram of the hyaluronic acid derivative obtained in Example 1-1. 3 is a graph showing tumor area values (average values) over time in Test Example 6.
- the present embodiment an embodiment of the present invention (hereinafter referred to as “the present embodiment") will be described in detail, but the present invention is not limited thereto, and various modifications can be made without departing from the gist thereof. It is.
- C 1-20 alkyl used herein means a straight or branched alkyl group having 1 to 20 carbon atoms, such as methyl, ethyl, n-propyl, iso - Contains "C 1-4 alkyl” such as propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, and further includes n-pentyl, 3-methylbutyl, 2-methylbutyl, 1-methylbutyl, 1-methylbutyl, -Ethylpropyl, n-hexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3-ethylbutyl, 2-ethylbutyl, and the like.
- C 1-20 alkyl also includes C 1-12 alkyl groups having 1 to 12 carbon atoms and C 1-6 alkyl groups having 1 to 6 carbon atoms.
- C 1-6 alkylcarbonyl as used herein means an alkylcarbonyl group in which the alkyl moiety is C 1-6 alkyl as already mentioned, for example acetyl, propionyl, n-propylcarbonyl, "C 1-4 alkylcarbonyl” such as iso-propylcarbonyl, n-butylcarbonyl, sec-butylcarbonyl, iso-butylcarbonyl, and tert-butylcarbonyl is included.
- amino C 2-20 alkyl used herein means a straight or branched alkyl having 2 to 20 carbon atoms and having an amino group as a substituent, for example, an amino group may be located on the terminal carbon atom of the alkyl group.
- Amino C 2-20 alkyl also includes amino C 2-12 alkyl having 2 or more and 12 or less carbon atoms.
- hydroxyC 2-20 alkyl used herein means a linear or branched alkyl group having 2 to 20 carbon atoms and having a hydroxy group as a substituent, for example, hydroxy The group may be located on the terminal carbon atom of the alkyl group. Hydroxy C 2-20 alkyl also includes hydroxy C 2-12 alkyl having 2 or more and 12 or less carbon atoms.
- C2-30 alkylene used herein means a linear or branched divalent saturated hydrocarbon group having 2 to 30 carbon atoms, such as ethylene, propylene, etc. C2-20 alkylene with 2 to 20 carbon atoms, C2-8 alkylene with 2 to 8 carbon atoms, group "-(CH 2 ) n -" (where n is 2 to 30) and is preferably 2 or more and 20 or less, more preferably 2 or more and 15 or less.).
- C 1-5 alkylene used herein means a linear or branched divalent saturated hydrocarbon group having 1 to 5 carbon atoms, such as methylene, ethylene, Contains propylene etc.
- C 2-8 alkenylene refers to a divalent saturated hydrocarbon group containing one or more double bonds, linear or branched, having 2 to 8 carbon atoms.
- -CH CH-
- -C(CH 3 ) CH-
- 2-butene-1,4-diyl hepta-2,4-diene-1,6-diyl
- octa-2,4 , 6-triene-1,8-diyl etc.
- each isomer and mixtures thereof are also included.
- the hyaluronic acid derivative of the first embodiment of the present invention is a hyaluronic acid derivative into which a steryl group is introduced, From the chromatogram shown in FIG. 1 obtained by gel permeation chromatography measurement, the ratio A1/A2 of areas A1 and A2 calculated by the method shown below is 0.90 or more.
- the hyaluronic acid derivative of the present embodiment has a ratio Da/Db of distance Da and Db of more than 0.00, which is calculated by the method shown below from the chromatogram shown in FIG. 2 determined by gel permeation chromatography measurement. It is preferable that it is .20 or less.
- (i) Draw a perpendicular line from the maximum refractive index intensity point Ka on the chromatogram of 150 kDa polyacrylic acid, which is a standard substance, to the baseline B, and draw a perpendicular line from the point of intersection with the baseline to Ba, and between the maximum refractive index intensity point Ka and the above Ba.
- the hyaluronic acid derivative of this embodiment differs from conventional hyaluronic acid derivatives in that it contains a relatively large amount of hyaluronic acid derivative with a specific particle size in its particle size distribution, and the particle size distribution is sharply controlled.
- this hyaluronic acid derivative with a controlled particle size distribution with a medicinal ingredient By formulating this hyaluronic acid derivative with a controlled particle size distribution with a medicinal ingredient, the inventors have discovered that by formulating this hyaluronic acid derivative with a controlled particle size distribution and a medicinal ingredient, it is possible to improve the effectiveness of hyaluronic acid derivatives with an uncontrolled particle size distribution and the conventional We have completed the present invention by discovering that the delivery to immune cells in lymph nodes and the ability to activate immune cells are significantly superior to that of cholesterol-modified pullulan (CHP), which is used as a carrier. It's arrived.
- CHP cholesterol-modified pullulan
- the immune cells mentioned here are preferably myeloid cells, more preferably macrophages or dendritic cells (DC), still more preferably DCs, and particularly preferably standard type 1 dendritic cells (cDC1 ).
- DC dendritic cells
- cDC1 standard type 1 dendritic cells
- Dendritic cells are the most potent antigen-presenting cells and are responsible for regulating the proliferation and function of T cells and natural killer cells in lymphoid and non-lymphoid tissues.
- cDC1 which specifically expresses the chemokine receptor XCR1 and selectively expresses the C-type lectin endocytosis receptor CLEC9A, has a high cross-presentation ability.
- T cells cytotoxic T cells (CTLs)
- CTLs cytotoxic T cells
- cDC1 cytotoxic T cells
- Macrophages and DCs can also load antigens onto MHC class II molecules. This activates helper T cells and promotes antibody production by B cells. That is, increasing antigen delivery to macrophages or DCs (preferably cDC1) improves the effectiveness of preventing or treating infectious diseases and treating immune diseases.
- the ability to activate immune cells refers to the property of improving, promoting, or enhancing the activity of cells as described above, and in particular, improving, promoting, or enhancing the expression of costimulatory molecules in DCs (preferably cDC1). It is preferable that the property is maintained.
- costimulatory molecules include CD80 and CD86.
- T cells cytotoxic T cells (CTL)
- CTL cytotoxic T cells
- the hyaluronic acid derivative of this embodiment is preferably one that can improve, promote, or maintain the expression of both CD80 and CD86 in DC (preferably cDC1) when formulated with a medicinal ingredient.
- the hyaluronic acid derivative of the present embodiment can form a stable association state with the medicinal ingredient when formulated with the medicinal ingredient, and stably transfer the medicinal ingredient to immune cells (preferably DC, more preferably cDC1). Moreover, it can be efficiently delivered, and furthermore, the uptake of the medicinal ingredient into immune cells can be improved, promoted, or enhanced. It is presumed that this improves, promotes, or enhances the activity of immune cells. Note that even if the desired effect is obtained by a mechanism different from the above-mentioned mechanism, it is still within the technical scope.
- immune cells preferably DC, more preferably cDC1
- the hyaluronic acid derivative of this embodiment can also be referred to as a delivery enhancer, delivery promoter, or delivery enhancer of a medicinal ingredient to immune cells. Alternatively, it can also be referred to as an agent for improving, promoting, or enhancing the uptake of medicinal ingredients into immune cells.
- the below-mentioned pharmaceutical composition containing the hyaluronic acid derivative of the present embodiment and a medicinal ingredient includes a composition for activating immune cells, a composition for improving the expression of costimulatory molecules in DCs (preferably cDC1), and a composition for improving the expression of costimulatory molecules in DCs (preferably cDC1). It can also be referred to as a composition for promoting or a composition for maintaining expression.
- the particle size distribution of the hyaluronic acid derivative of this embodiment can be measured by gel permeation chromatography, and the particle size distribution controlled as described above is calculated from the gel permeation chromatogram by the method shown below. It can be expressed as the ratio A1/A2 of the areas A1 and A2.
- the polyacrylic acid used as a standard substance in the gel permeation chromatography measurement is preferably sodium polyacrylate, and specifically, POLYMER Standards Service-USA, ORDER No. More preferably, it is PSS-Paa series (sodium polyacrylate).
- Measurement of the particle size distribution of a hyaluronic acid derivative by gel permeation chromatography can be performed by the following method.
- a 1 mg/mL hyaluronic acid derivative aqueous solution and a 2 mg/mL polyacrylic acid standard aqueous solution are prepared and measured by gel permeation chromatography under the conditions shown below.
- FIG. 1 is an example of a gel permeation chromatogram of the hyaluronic acid derivative of this embodiment. The method for calculating areas A1 and A2 will be described in detail below with reference to FIG.
- FIG. 1 shows a gel permeation chromatogram of the hyaluronic acid derivative of this embodiment and a gel permeation chromatogram of 50 kDa polyacrylic acid, which is a standard substance.
- Ub the intersection point of the chromatogram of the hyaluronic acid derivative and the perpendicular line drawn from the maximum refractive index intensity point Kb on the chromatogram of 50 kDa polyacrylic acid, which is a standard substance.
- Ub the maximum refractive index intensity point.
- Bb be the intersection of the perpendicular line drawn from point Kb to baseline B and baseline B.
- the refractive index intensity at the start of the measurement is set to zero, and a line drawn horizontally from this is set as the baseline.
- the refractive index intensity is adjusted so that the increase/decrease in the refractive index intensity is within a range of ⁇ 0.5 mV before the start of measurement, and the refractive index intensity is adjusted so that the increase/decrease in the refractive index intensity is within a range of 0.5 mV or less in 5 minutes.
- the first point at which the refractive index intensity increase exceeds an amount equivalent to five times the noise value three times is set as the "starting point" of the chromatogram, and the elution time is set as 0 minutes.
- Tlim' is set as the 'end point'.
- Tlim is the elution time at which maximum refractive index intensity is obtained when 2 kDa polyacrylic acid is measured. In this device, the refractive index intensity is calculated every 0.00167 minutes.
- the ratio A1/A2 of areas A1 and A2 shown in FIG. 1 is 0.90 or more, more preferably 1.00 or more, more preferably 1.10 or more, and 1.20 or more. is more preferable, more preferably 1.30 or more, more preferably 1.40 or more, more preferably 1.50 or more, even more preferably 1.60 or more, and more preferably 1.70 or more. Particularly preferred.
- the ratio A1/A2 of area A1 and A2 to the above lower limit value, it is possible to contain a relatively large amount of hyaluronic acid derivative with a large particle size, and when it is formulated with a medicinal ingredient, it can be stably associated with the medicinal ingredient. state can be formed.
- the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- the upper limit value of A1/A2 it is preferable that the ratio of area A1 is higher than area A2.
- the particle size is larger on the left side of the baseline (shorter elution time), and smaller on the right side (longer elution time), so the ratio of large particle size is It is preferable that the value is higher than that of the smaller value. Therefore, the upper limit value of A1/A2 is not particularly limited, and may be, for example, 7.0 or less, 6.0 or less, or 5.0 or less.
- the shape of the gel permeation chromatogram of the hyaluronic acid derivative of this embodiment is not particularly limited, but there may be two or more maximum points (peak tops) of the refractive index intensity of the hyaluronic acid derivative.
- the refractive index intensity of the hyaluronic acid derivative is indicated on each side (left and right) of a perpendicular line drawn from the maximum refractive index intensity point Kb to the baseline B on the chromatogram of 50 kDa polyacrylic acid, which is a standard substance. It may have one or more local maximum points (peak tops).
- the refractive index intensity at the maximum point (peak top) of the refractive index intensity of the hyaluronic acid derivative on the left side of the perpendicular line drawn from the maximum refractive index intensity point Kb to the baseline B It may be greater than the refractive index intensity at the maximum point (peak top) of the refractive index intensity of the hyaluronic acid derivative on the right side of the perpendicular line drawn to .
- the refractive index intensity at the maximum point (peak top) on the left side with respect to the maximum point (peak top) on the right side is not particularly limited, but is 1.0 times or more, 2.0 times or more, 3.0 times or more.
- It may be more than 1000 times, 5.0 times or more, or 10.0 times or more, and the upper limit may be large, but for example, 1000 times or less, 100 times or less, 50.0 times or less, 20.0 times or less, It may be 10.0 times or less, 2.0 times or less, or 1.5 times or less.
- the fact that the hyaluronic acid derivative of this embodiment has a particle size distribution controlled as described above can also be defined by the ratio Da/Db of the distances Da and Db shown below.
- FIG. 2 is an example of a gel permeation chromatogram of the hyaluronic acid derivative of this embodiment, and the gel permeation chromatogram of the hyaluronic acid derivative shown in FIG. Same as 1.
- FIG. 2 differs from FIG. 1 in that a gel permeation chromatogram of 150 kDa polyacrylic acid, which is a standard substance, is added, and various intersection points are defined. The method for calculating the distances Da and Db will be described in detail below with reference to FIG. 2.
- the ratio Da/Db between the distances Da and Db shown in FIG. 2 is preferably more than 0.00 and less than 1.20, more preferably more than 0.10 and less than 1.00, more than 0.20, and less than 1.00. It is more preferably 8 or less, further preferably 0.30 or more and 0.95 or less, even more preferably 0.40 or more and 0.94 or less, particularly preferably 0.50 or more and 0.93 or less.
- the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- immune cells preferably DC, more preferably cDC1
- the horizontal axis shows the elution time and the vertical axis shows the refractive index intensity obtained using a differential refractometer. good.
- the gel permeation chromatogram of the hyaluronic acid derivative of this embodiment preferably has a particle size distribution with 1 to 5 maximum points, more preferably a particle size distribution with 1 to 3 maximum points. Further, the minimum point may or may not be present. Furthermore, in the gel permeation chromatogram of the hyaluronic acid derivative of this embodiment, whether the chromatogram expressed by the refractive index intensity and elution time obtained using a differential refractometer is asymmetrical or symmetrical, good.
- the steryl groups in the hyaluronic acid derivative self-associate in water, and a single molecule or multiple molecules associate to form a nano-sized hydrogel.
- the steryl group may be bonded directly to hyaluronic acid or may be bonded via a linker.
- the "linker” used herein may be any peptide linker that can be introduced by genetic engineering or a synthetic compound linker, but peptide linkers are preferred for hyaluronic acid derivatives.
- the length of the peptide linker is not particularly limited and can be appropriately selected by those skilled in the art depending on the purpose, but the preferred length is 2 amino acids or more (the upper limit is not particularly limited, but usually 30 amino acids or less, preferably is 20 amino acids or less), particularly preferably 15 amino acids.
- the peptide linkers contained in the hyaluronic acid derivative may all have the same length, or may have different lengths.
- steryl group used herein is not particularly limited as long as it is a group having a steroid skeleton.
- the steroids specifically include cholesterol, cholestanol, campestanol, ergostanol, stigmastanol, coprostanol, stigmasterol, sitosterol, lanosterol, ergosterol, simiarenol, bile acids, testosterone, estradiol, and pro-steroids. Examples include gestrone, cortisol, cortisone, aldosterone, corticosterone, deoxycortisterone, and the like.
- steryl group examples include a cholesteryl group, a stigmasteryl group, a lanosteryl group, an ergosteryl group, and among them, a cholesteryl group (particularly a cholest-5-en-3 ⁇ -yl group) is preferred.
- the weight average molecular weight (absolute molecular weight) of the hyaluronic acid derivative is not particularly limited, but from the viewpoint of increasing the number of steryl groups introduced per molecule of the hyaluronic acid derivative to form a complex with the medicinal ingredient, and increasing molecular entanglement. From the viewpoint of increasing retention in blood, hyaluronic acid derivatives with relatively large molecular weights are preferred.
- the weight average molecular weight (absolute molecular weight) of such a hyaluronic acid derivative is preferably 4000 (4k) or more and 1,000,000 (1,000k) or less, more preferably 5k or more and 500k or less, and further preferably 6k or more and 500k or less.
- the weight average molecular weight (absolute molecular weight) of the hyaluronic acid derivative is more preferably 5k or more and 25k or less.
- the weight average molecular weight (absolute molecular weight) of the hyaluronic acid derivative is equal to or greater than the above lower limit, molecular entanglement can be further enhanced and retention in blood can be further enhanced.
- the weight average molecular weight (absolute molecular weight) of the hyaluronic acid derivative is equal to or less than the above upper limit, an increase in viscosity can be suppressed, and a higher concentration of the hyaluronic acid derivative can be dissolved in the pharmaceutical composition.
- the weight average molecular weight (absolute molecular weight) of a hyaluronic acid derivative can generally be adjusted by using raw materials having a corresponding molecular weight.
- the "molecular weight (absolute molecular weight) of the hyaluronic acid derivative” as used herein is the weight average molecular weight (absolute molecular weight) determined by size exclusion chromatography multi-angle light scattering detector (SEC-MALS).
- the refractive index intensity of the hyaluronic acid derivative with respect to the retention time Pr (elution time Bb) of the maximum refractive index intensity point (peak top) of the standard substance 50 kDa polyacrylic acid The ratio Pt/Pr of the retention time Pt at the maximum point (peak top) is not particularly limited, but Pt/Pr is preferably 0.5 or more and less than 1.0, and 0.7 or more and less than 0.95. It is more preferable that it is 0.75 or more and less than 0.92.
- the ratio Pt/Pr is within the above range, it is possible to contain a relatively large amount of hyaluronic acid derivative with a large particle size, and when formulated with a medicinal ingredient, it is possible to form a stable association state with the medicinal ingredient. can.
- the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- the refractive index intensity maximum points (peak tops) where the refractive index intensity is maximum ) is employed as the retention time Pt of the maximum refractive index intensity point (peak top) of the hyaluronic acid derivative.
- the polyacrylic acid used as a standard substance in the gel permeation chromatography measurement is preferably sodium polyacrylate, and specifically, POLYMER Standards Service-USA, ORDER No. More preferably, it is PSS-Paa series (sodium polyacrylate).
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is not particularly limited, but it is preferable that the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is 110,000 or more, and 110,000 or more and less than 500,000. It is more preferably 130,000 or more and less than 300,000, even more preferably 140,000 or more and less than 250,000.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is within the above range, it is possible to contain a relatively large amount of the hyaluronic acid derivative with a large particle size, and when formulated with a medicinal ingredient, the medicinal ingredient A stable association state can be formed with Thereby, the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- immune cells preferably DC, more preferably cDC1
- the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative exceeds the above upper limit, the viscosity increases and it may be difficult to use it as a preparation.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is determined from the chromatogram determined by the gel permeation chromatography measurement.
- PSS-Paa2k (2kDa), 4k (4kDa), 8k (8kDa), 18k (18kDa), 40k (40kDa), 150k (150kDa) PSS Polymer Standard service GmbH, sodium polyacrylate
- At3+Bt2+Ct+D based on the calibration curve obtained.
- hyaluronic acid derivatives include, for example, hyaluronic acid derivatives having one or more repeating units represented by the following general formula (I) (hereinafter sometimes referred to as “repeat units (I)"). It will be done.
- R 1 , R 2 , R 3 , and R 4 are each independently selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl; Z represents a direct bond or a peptide linker consisting of 2 to 30 arbitrary amino acid residues;
- X1 is the following formula: -NR b -R, -NR b -COO-R, -NR b -CO-R, -NR b -CO-NR c -R, -COO-R, -O-COO-R, -S-R, -CO-Y a -SR, -O-CO-Y b -SR, -NR b -CO-Y b -SR, and -SSR,
- R a , R b and R c are each independently selected from the group consisting of a hydrogen atom, C 1-20 alkyl,
- a selected group may be inserted;
- R is a steryl group;
- Y is C 2-30 alkylene, or -(CH 2 CH 2 O) m -CH 2 CH 2 -, where the alkylene is a group consisting of -O-, -NR g - and -SS- A group selected from the group consisting of;
- R g is selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl and hydroxy C 2-20 alkyl, and the alkyl portion of the group is selected from the group consisting of -O- and -NH-
- Y a is C 1-5 alkylene;
- Y b is C 2-8 alkylene or C 2-8 alkenylene;
- m is an integer of 1 or more and 100 or less.
- the hyaluronic acid derivative preferably contains one or more repeating units represented by the following general formula (Ia) (hereinafter sometimes referred to as “repeat units (Ia)”) as repeating units (I).
- R 1 , R 2 , R 3 , and R 4 are each independently selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl;
- X is a hydrophobic group represented by -NR a -Y-NR b -COO-R;
- R a and R b are each independently selected from the group consisting of a hydrogen atom and C1-6 alkyl;
- R is a steryl group;
- Y is C 2-30 alkylene or -(CH 2 CH 2 O)m-CH 2 CH 2 -, m is an integer of 1 or more and 100 or less.
- the repeating units may be the same or different.
- the hyaluronic acid derivative may be modified at a position other than repeating unit (I) or repeating unit (Ia), for example, the hydroxy group is -O (C 1-6 alkyl), -O (formyl), -
- the carboxy group may be converted to O(C 1-6 alkylcarbonyl), etc., and the carboxy group may be converted to an amide or ester, or may form a salt.
- Z is preferably a direct bond.
- X 1 is -NR b -COO-R.
- Z may be a peptide linker represented by -NH-[CH(-Z a )-CONH] n-1 -CH(-Z a )-CO-, where , n is an integer of 2 or more and 30 or less, and Z a each independently represents a substituent in the ⁇ -amino acid represented as H 2 N-CH(-Z a )-COOH.
- the peptide linker is bonded to the carboxy group of the glucuronic acid moiety at the N-terminus and to the group -N(-R a )-YX 1 at the C-terminus.
- amino acids that can be used as amino acid residues in the peptide linker include ⁇ -amino acids such as alanine, arginine, asparagine (Asn), aspartic acid, cysteine, glutamine, glutamic acid, glycine (Gly), histidine, isoleucine, leucine ( Natural (L-type) amino acids such as Leu), lysine, methionine, phenylalanine (Phe), proline, serine, threonine, tryptophan, tyrosine, and valine, and their D forms, and all synthetic amino acids, including ⁇ -amino acids can be used.
- examples of Z a include -CH 3 , H 2 NC(NH)NH(CH 2 ) 3 -, H 2 NCOCH 2 -, and the like.
- n Z's may be the same or different.
- n is an integer of 2 or more and 30 or less, preferably 2 or more and 10 or less, and more preferably 2 or more and 4 or less.
- Preferred examples of peptide linkers include -Gly-Phe-Leu-Gly-, -Asn-Phe-Phe-, -Phe-Phe-, Phe-Gly-, and the like.
- Y is -(CH 2 ) n1 - and -(CH 2 CH 2 O) m1 -CH 2 CH 2 - (where n1 is an integer of 2 or more and 20 or less, and 2 or more An integer of 15 or less is preferable, an integer of 2 or more and 12 or less is more preferable, and an integer of 2 or more and 6 or less is even more preferable.
- m1 is an integer of 1 or more and 4 or less. Specifically, -(CH 2 ) 2 -, -(CH 2 ) 6 -, -(CH 2 ) 8 -, -(CH 2 ) 12 -, or -(CH 2 CH 2 O) 2 -CH 2 CH 2 - is preferred.
- Y is -(CH 2 ) 2 -, -(CH 2 ) 6 --, --(CH 2 ) 8 -- and --(CH 2 ) 12 -- is preferred, and --(CH 2 ) 6 -- is more preferred.
- Y is, for example, -CH 2 CH 2 O-CH 2 CH 2 -S-S-CH 2 CH 2 O-CH 2 CH 2 -, -(CH 2 CH 2 O) 2 -CH 2 CH 2 -S- S-CH 2 CH 2 O-CH 2 CH 2 -, -CH 2 CH 2 O-CH 2 CH 2 -S-S-(CH 2 CH 2 O) 2 -CH 2 CH 2 -, -(CH 2 CH 2 O) 2 -CH 2 CH 2 -S-S-(CH 2 CH 2 O) 2 -CH 2 CH 2 - and the like.
- Y a is preferably -CH 2 - or -CH 2 -CH 2 -.
- Yb is -CH 2 -CH 2 -, -CH(CH 3 )CH 2 -, 2-butene-1,4-diyl, hepta-2,4-diene-1,6-diyl or octa-2 ,4,6-triene-1,8-diyl is preferred, and --CH 2 --CH 2 -- or --CH(CH 3 )CH 2 -- is more preferred.
- Specific examples of the group "-Z-N(R a )Y-X 1 " include -NH-(CH 2 ) 2 -NH-CO-cholesteryl, -NH-(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH-(CH 2 ) 3 -NH-(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH -(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH-(CH 2 ) 4 -N(-(CH 2 ) 3 -NH 2 )-COO-cholesteryl, -NH- (CH 2 ) 3 -NH-(CH 2 ) 4 -N(-(CH 2 ) 3 -NH 2 )-COO-cholesteryl, -NH- (CH 2 ) 3 -NH-(CH 2
- R a , R b and R c are hydrogen atoms
- Y is linear C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -
- Y a is linear C 1-5 alkylene
- Y b is linear C 2-8 alkylene or linear is a C 2-8 alkenylene.
- Repeat unit (Ia) In the general formula ( Ia ) , -COO-cholesteryl or -NH-(CH 2 CH 2 O) 2 -CH 2 CH 2 -NH-COO-cholesteryl is preferred, -NH-(CH 2 ) 2 -NH-COO-cholesteryl, -NH-(CH 2 ) 6 -NH-COO-cholesteryl or -NH-(CH 2 CH 2 O) 2 -CH 2 CH 2 -NH-COO-cholesteryl is more preferred, and -NH-(CH 2 ) 6 -NH-COO-cholesteryl is even more preferred.
- the hyaluronic acid derivative can further include a repeating unit represented by general formula (II) (hereinafter sometimes referred to as "repeat unit (II)").
- R 1a , R 2a , R 3a , and R 4a are each independently selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl;
- X a is selected from the group consisting of hydroxy and -O-Q + ; where Q + is a countercation.
- the hyaluronic acid derivative when the hyaluronic acid derivative contains two or more repeating units (II), the repeating units may be the same or different.
- the hyaluronic acid derivative may be a hyaluronic acid derivative consisting essentially of repeating unit (I), repeating unit (Ia), and repeating unit (II).
- Q + is not particularly limited as long as it is a counter cation that forms a salt with a carboxy group in water, and if it has a valence of two or more, it forms a salt with a plurality of carboxy groups depending on the valence.
- countercations include metal ions such as lithium ions, sodium ions, rubidium ions, cesium ions, magnesium ions, calcium ions; formula: N + R j R k R l R m (where R j , R k , R l and R m are each independently selected from the group consisting of a hydrogen atom and C 1-6 alkyl).
- Q + is preferably a sodium ion, potassium ion, or tetraalkylammonium ion (eg, tetra n-butylammonium ion, etc.).
- R j , R k , R l and R m are preferably the same group selected from the group consisting of C 1-6 alkyl, preferably n-butyl group.
- R 1 , R 2 , R 3 , and R 4 as well as R 1a , R 2a , R 3a , and R 4a are all hydrogen atoms. Moreover, it is preferable that both R a and R b are hydrogen atoms.
- the hyaluronic acid derivative is preferably a hyaluronic acid derivative consisting essentially of repeating unit (I) and repeating unit (II).
- the hyaluronic acid derivative for example, 80% or more, preferably 90% or more of the disaccharide repeating unit consisting of D-glucuronic acid and N-acetyl-D-glucosamine contained in the derivative is more preferably 95% or more are repeating units (I) and repeating units (II).
- the hyaluronic acid derivative may be composed only of repeating unit (I) and repeating unit (II), or may be composed only of repeating unit (I).
- the introduction rate of steryl groups (hereinafter sometimes simply referred to as "steryl group introduction rate") with respect to the disaccharide repeating unit constituting the hyaluronic acid derivative is 15% or more and 60%.
- the following is preferable, preferably 20% or more and 60% or less, 30% or more and 60% or less, more preferably 30% or more and 55% or less, even more preferably 35% or more and 50% or less, particularly 35% or more and 45% or less. preferable.
- the steryl group introduction rate is equal to or higher than the above lower limit, a stable hydrogel can be maintained without precipitating in vivo.
- the average particle diameter of the hydrogel can be within the above range.
- the steryl group introduction rate can be measured by 1 H-NMR measurement. That is, the integral value of the peak derived from the steryl group of the hyaluronic acid derivative in the 1 H-NMR spectrum of the hyaluronic acid derivative component, and the peak derived from the acetyl group of N-acetyl-D-glucosamine contained in the hyaluronic acid derivative (COCH 3 , 1.6 ppm or more and 2.0 ppm or less, 3H), it can be calculated based on the following formula. Note that in the formula, n H represents the number of hydrogen atoms corresponding to the peak.
- the 1 H-NMR is performed, for example, at a measurement temperature of 85° C.
- the peak derived from the cholesteryl group (5H) overlaps the peak around 1.6 to 2.0 ppm, which includes the peak derived from the acetyl group of glucosamine, so the cholesteryl A value calculated by subtracting 5/3 of the integral value of the peak derived from the group methyl (0.7 ppm) (i.e., integral value (1.6 to 2.0 ppm) - integral value (0.7 ppm) x 5/ 3) can be used to calculate the introduction rate as an integral value of acetyl groups derived from hyaluronic acid.
- Stepryl group introduction rate] (%) [(Peak integral value derived from steryl group ⁇ 3/n H )/(Peak integral value derived from acetyl group of N-acetyl-D-glucosamine)] ⁇ 100
- Hyaluronic acid derivatives can be obtained, for example, by converting the carboxyl group of glucuronic acid into an amide and introducing a steryl group. Furthermore, the rate of steryl group introduction can be controlled by adjusting the amount of the compound having a steryl group to be reacted with the raw material hyaluronic acid or its derivative.
- a method of converting the carboxyl group of glucuronic acid into an amide and introducing a steryl group for example, hyaluronic acid as a raw material or a derivative thereof, preferably hyaluronic acid composed only of repeating unit (II).
- a tetraalkylammonium salt e.g., tetrabutylammonium (TBA) salt
- TSA tetrabutylammonium
- the hyaluronate is combined with the hyaluronate of the formula: "HNR a -Y-NR b -R, NHR a -Y-NR b -COO-R, HNR a -Y-NR b -COO-R, HNR a -Y-NR b -CO-R, HNR a -Y-NR b -CO- NR c -R, HNR a -Y-COO-R, HNR a -Y-O-COO-R, HNR a -Y-S-R, HNR a -Y-CO-Y a -S-R, HNR a -Y-O-CO-Y b -S
- the condensing agent that can be used in the above reaction is not particularly limited, and examples include 4-(4,6-dimethoxy-1,3,5-triazine)-4-methylmorpholium (DMT-MM), N , N'-carbonyldiimidazole (CDI), N,N'-dicyclohexylcarbodiimide (DCC), N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ), 2-benzotriazole-1,1, 3,3-tetramethyluronium tetrafluoroborate (TBTU), 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HODhbt), benzotriazole-1-oxy -Tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP), benzotriazol-1-yl-oxy-tris(dimethylamino)phosphonium hexafluorophosphat
- DMT-MM is preferable in that the reaction proceeds with high efficiency even in a mixed solvent of water and an organic solvent. Furthermore, by using DMT-MM as a condensing agent, it is possible to highly selectively form an amide bond between an amino group and a carboxy group while suppressing the formation of an ester bond in a system where a large number of hydroxy groups coexist.
- the alcohol as a solvent may react with the carboxy group of the hyaluronic acid moiety, or the carboxyl group and hydroxyl group that are simultaneously present in the hyaluronic acid moiety may bond within or intermolecularly. , it is possible to prevent the formation of undesired crosslinks.
- Solvents used in the steryl group introduction reaction include water, DMSO, methanol, ethanol, propanol, butanol, isopropanol, polyhydric alcohol, acetonitrile, DMF, THF, dichloromethane, chloroform, hexane, diethyl ether, ethyl acetate, and mixtures thereof. Examples include solvents.
- the polyhydric alcohol may be a dihydric alcohol or a trihydric alcohol. Examples of the dihydric alcohol include ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, neopentyl glycol, 1,4-butanediol, and 1,6-hexanediol. Examples of the trihydric alcohol include glycerin and trimethylolpropane.
- hyaluronic acid or a derivative thereof as a raw material is ion-exchanged with a tetraalkylammonium salt (e.g., tetrabutylammonium (TBA) salt), and the hyaluronate and spacer moiety are reacted in a solvent in the presence of an appropriate condensing agent. (At this time, protection and deprotection reactions may be performed as necessary) to convert the carboxy group (-COOH) of the raw material hyaluronic acid or its derivative, and then react with an appropriate reagent. . Examples of combinations of groups derived from carboxy groups and reaction reagents are shown below.
- reaction mode examples include dehydrohalogenation reaction, condensation reaction, dehydration reaction, nucleophilic addition reaction such as Michael addition, oxidative disulfide formation reaction, etc. These are well-known reactions and can be selected as appropriate by those skilled in the art. However, the reaction can be carried out by finding preferable reaction conditions.
- the converter or reactant When the converter or reactant has a carboxyl group, it may be converted into an N-hydroxysuccinimide (hereinafter also referred to as "NHS”) ester and reacted.
- NHS N-hydroxysuccinimide
- a hyaluronic acid derivative having a spacer having a mercapto group modified with a leaving group at the end is prepared by reacting 2-aminoethyl 2-pyridyl disulfide with the carboxy group of the raw material hyaluronic acid or its derivative. Then, there is a method in which thiocholesterol is subjected to a nucleophilic substitution reaction to form a disulfide bond.
- hyaluronic acid or its derivatives with a part of the spacer introduced into the carboxy group and one with a part of the spacer introduced into the steryl group, and reacting them.
- a hyaluronic acid derivative in which a spacer having a mercapto group at the end is introduced into the carboxy group of hyaluronic acid;
- Another method is to prepare steryl groups into which a spacer having a mercapto group at the end is introduced, and to react them oxidatively to form a disulfide bond. At this time, one mercapto group can be reacted with 2-mercaptopyridine to form a disulfide, and then the other mercapto group can be substituted.
- hyaluronic acid derivative consisting essentially of repeating units (I) and repeating units (II), -CO-X z ,
- a dialysis membrane with a MWCO (molecular weight cutoff) of 300 kDa for 1 to 9 times, preferably 3 to 9 times, more preferably 6 to 9 times.
- MWCO molecular weight cutoff
- a hyaluronic acid derivative having a particle size distribution satisfying an area ratio A1/A2 of 0.9 or more can be obtained.
- Those skilled in the art can appropriately select the number of times of dialysis as long as the area ratio A1/A2 satisfies 0.9 or more. In some cases, fractionation may be performed by dialysis nine or more times.
- the method for obtaining a hyaluronic acid derivative having a particle size distribution satisfying the area ratio A1/A2 of 0.9 or more is not limited to dialysis alone.
- centrifugal filtration using ultrafiltration membrane or microfiltration membrane preparative purification HPLC, preparative purification GPC, separation by ultracentrifugation, separation by ion exchange resin, separation by membrane distillation, membrane separation by organic or inorganic membrane, activated carbon. Separation by adsorption/desorption using zeolite, MOF (Metal Organic Frameworks), separation by TFF (Tangential Flow Filtration), liquid feeding using a filter, pressure or vacuum filtration, precipitation separation using salting out, etc.
- MOF Metal Organic Frameworks
- TFF Tangential Flow Filtration
- separation using hyaluronic acid receptors similarly removes hyaluronic acid derivatives with a particle size distribution below that of the standard substance polyacrylic acid (50 kDa, manufactured by Polymer Standards Service-USA, ORDER No. PSS-Paa50k). By doing so, a hyaluronic acid derivative having a particle size distribution satisfying the area ratio A1/A2 of 0.9 or more can be obtained.
- the obtained hyaluronic acid derivative may be dried.
- the drying method include ventilation drying, drying in a thermostatic oven, reduced pressure drying, hot air circulation drying, and freeze drying. Among these, freeze-drying is preferred.
- the hyaluronic acid derivative preferably further contains a cryoprotectant from the viewpoint of more effectively suppressing an increase in the particle size of the fine particles formed by the hyaluronic acid derivative.
- cryoprotectant is not particularly limited as long as it is known as a "cryoprotectant” or “lyoprotectant” and includes, for example, disaccharides, sorbitol, dextran, polyethylene glycol, propylene glycol, glycerin, glycerol, Examples include polyvinylpyrrolidone and dimethyl sulfoxide.
- Disaccharides are not particularly limited and include, for example, sucrose, lactulose, lactose, maltose, trehalose, cellobiose, cordibiose, nigerose, isomaltose, isotrehalose, neotrehalose, sophorose, laminaribiose, gentiobiose, turanose, maltulose, palatinose,
- Examples include gentiobiulose, mannobiose, melibiose, melibiulose, neolactose, galactosucrose, silabiose, neohesperidose, rutinose, rutinulose, vicyanose, xylobiose, primeverose, and the like.
- sucrose, trehalose, maltose, or lactose are preferred because they are widely used as cryoprotectants. Further, sucrose is more preferred from the viewpoint of its use as a pharmaceutical additive and from the viewpoint of more effectively suppressing the increase in particle size of fine particles formed by hyaluronic acid derivatives during freeze-drying.
- the cryoprotectant may be added in a solid state or in a state dissolved in a solvent such as water.
- the amount of the cryoprotectant added is not particularly limited, but is preferably 20 parts by mass or more based on 100 parts by mass of the hyaluronic acid derivative.
- the amount of the cryoprotectant added is at least the above lower limit, a more sufficient effect of suppressing particle size increase can be obtained.
- the upper limit of the amount of cryoprotectant added is not particularly limited, but may be, for example, 100,000 parts by mass.
- the apparatus used in freeze-drying is not particularly limited, and for example, a commercially available freeze-dryer can be used.
- a freeze dryer that can monitor the degree of vacuum inside the apparatus during freeze-drying is preferable, and from the viewpoint of controlling the product temperature, a shelf-type freeze dryer is preferable.
- the hyaluronic acid derivative of the second embodiment of the present invention is a hyaluronic acid derivative into which a steryl group has been introduced, and in a chromatogram obtained by gel permeation chromatography measurement, the refraction of 50 kDa polyacrylic acid, which is a standard substance, is
- the ratio Pt/Pr of the retention time Pt at the maximum refractive index intensity point (peak top) of the hyaluronic acid derivative to the retention time Pr at the maximum index intensity point (peak top) is 0.5 or more and less than 1.0, and 0 More preferably .7 or more and less than 0.95, most preferably 0.75 or more and less than 0.92.
- the ratio Pt/Pr within the above range, it is possible to contain a relatively large amount of hyaluronic acid derivative with a large particle size, and when formulated with a medicinal ingredient, it is possible to form a stable association state with the medicinal ingredient. .
- the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- the polyacrylic acid used as a standard substance in the gel permeation chromatography measurement is preferably sodium polyacrylate, and specifically, POLYMER Standards Service-USA, ORDER No. More preferably, it is PSS-Paa50k (sodium polyacrylate).
- the measurement by gel permeation chromatography can be performed, for example, by the following method. A 1 mg/mL hyaluronic acid derivative aqueous solution and a 2 mg/mL polyacrylic acid standard aqueous solution are prepared and measured by gel permeation chromatography under the conditions shown below.
- the hyaluronic acid derivative of the third embodiment of the present invention is a hyaluronic acid derivative into which a steryl group has been introduced, and the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is 110,000 or more and less than 500,000. , preferably 130,000 or more and less than 300,000, more preferably 140,000 or more and less than 250,000.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative satisfies the above range, it is possible to contain a relatively large amount of the hyaluronic acid derivative with a large particle size, and when formulated with a medicinal ingredient, it is stable. A state of association can be formed.
- the medicinal ingredient can be stably and efficiently delivered to immune cells (preferably DC, more preferably cDC1), and furthermore, the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- immune cells preferably DC, more preferably cDC1
- the uptake of the medicinal ingredient into the immune cells can be improved, promoted, or enhanced. I can do it.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative exceeds the above upper limit, the viscosity may increase and it may be difficult to use it as a preparation.
- the weight average molecular weight (in terms of polyacrylic acid) of the hyaluronic acid derivative is determined from the chromatogram determined by gel permeation chromatography, using polyacrylic acid having a standard molecular weight of 2 kDa, 4 kDa, 8 kDa, 18 kDa, 40 kDa, and 150 kDa. It is calculated according to the following formula: At3+Bt2+Ct+D based on a calibration curve created as a substance.
- the polyacrylic acid used as the standard substance is preferably sodium polyacrylate, and specifically, POLYMER STANDARDS SERVICE-USA, ORDER No. More preferably, it is PSS-Paa series (sodium polyacrylate).
- the measurement by gel permeation chromatography can be performed by the method described in the first or second embodiment.
- descriptions of the same configurations as the hyaluronic acid derivatives of the first embodiment and/or the second embodiment will be omitted.
- the hyaluronic acid derivatives of the first to third embodiments can be formulated with medicinal ingredients and used as a pharmaceutical composition. That is, the pharmaceutical composition of this embodiment contains the above-described hyaluronic acid derivative and a medicinal ingredient.
- the hyaluronic acid derivative forms a complex with a medicinal ingredient (hereinafter sometimes referred to as "medicinal ingredient-hyaluronic acid derivative complex").
- a medicinal ingredient hereinafter sometimes referred to as "medicinal ingredient-hyaluronic acid derivative complex”
- the steryl group in the hyaluronic acid derivative and the hydrophobic site of the medicinal ingredient form a complex through hydrophobic interaction, and the medicinal ingredient and the hydrophobic site, such as the steryl group, are located in the center.
- the average particle diameter of the spherical structure containing the medicinal ingredient-hyaluronic acid derivative complex can be 20 nm or more and 100 nm or less, 20 nm or more and 95 nm or less, and 20 nm or more
- the thickness can be 90 nm or less.
- the average particle diameter here is a value expressed as a z average.
- the average particle diameter can be measured by, for example, DLS (Dynamic Light Scattering), a nanotracking particle measuring device, size exclusion chromatography, high performance liquid chromatography, electron microscopy, or the like.
- the hyaluronic acid derivative is diluted with a 10 mM phosphate buffer containing 10 w/v% sucrose and measured using a DLS device so that the concentration of the hyaluronic acid derivative becomes 1 mg/mL.
- the weight average molecular weight Mw (in terms of polyacrylic acid) of the hyaluronic acid derivative or the medicinal ingredient-hyaluronic acid derivative complex is preferably 110,000 or more.
- the weight average molecular weight Mw (in terms of polyacrylic acid) can be calculated by creating a calibration curve using a plurality of polyacrylic acid samples having different molecular weights.
- the polyacrylic acid preparation used is preferably sodium polyacrylate, specifically, ORDER No. manufactured by Polymer Standards Service-USA. More preferably, it is PSS-Paa series (sodium polyacrylate).
- the medicinal ingredients such as peptides can be absorbed into immune cells (especially It can be stably and efficiently delivered to cDC1, macrophages), and furthermore, it can improve, promote, or enhance the uptake of the medicinal ingredient into immune cells. It is believed that the expression of the molecules CD80 and CD86) can be improved, promoted, maintained or enhanced.
- Mw can be measured, for example, by DLS (Dynamic Light Scattering), size exclusion chromatography, electron microscopy, or the like. More specifically, for example, the hyaluronic acid derivative is diluted to a concentration of 1 mg/mL using a size exclusion chromatography device, and then measured.
- the peak top time in the particle size distribution of the hyaluronic acid derivative or the medicinal ingredient-hyaluronic acid derivative complex in size exclusion chromatography is greater than the peak top time of the standard substance polyacrylic acid (50 kDa). It is preferable that it is also small.
- the peak top time is within the above range, there are more aggregate sizes that are excellent in activating costimulatory molecules, and by using the pharmaceutical composition of this embodiment, the medicinal ingredients such as peptides can be absorbed into immune cells (especially , cDC1, and macrophages), and further improves, promotes, or enhances the uptake of the medicinal ingredient into immune cells.
- Peak top time can be measured, for example, by size exclusion chromatography. More specifically, for example, the hyaluronic acid derivative is diluted to a concentration of 1 mg/mL using a size exclusion chromatography device, and then measured.
- the content of the hyaluronic acid derivative is not particularly limited, but may be, for example, 0.01 parts by mass or more and 50.00 parts by mass or less based on 100 parts by mass of the pharmaceutical composition. It is preferably 0.10 parts by mass or more and 25.00 parts by mass or less, and even more preferably 0.20 parts by mass or more and 10.00 parts by mass or less.
- medicinal ingredients include, but are not limited to, antigens (cancer antigens, infectious disease-derived antigens, autoantigens in immune diseases, etc.), pharmaceutically active peptides or proteins, nucleic acids, low-molecular compounds, middle-molecular compounds, and the like.
- antigens are preferred, at least one selected from the group consisting of cancer antigens, infectious disease-derived antigens, and self-antigens in immune diseases is more preferred, and cancer antigens or infectious disease-derived antigens are more preferred.
- the pharmaceutical composition of the present embodiment is preferably a pharmaceutical composition for the prevention or treatment of one or more diseases selected from the group consisting of cancer, infectious diseases, and immune diseases; Even more preferably, it is a pharmaceutical composition for the prevention or treatment of.
- the pharmaceutical composition of this embodiment can also be called a vaccine composition when the applicable disease is cancer, infectious disease, or immune disease.
- Cancer antigens are antigens that are often expressed by cancer cells, and in some cases only by cancer cells. Cancer antigens can be expressed within cancer cells or on the surface of cancer cells.
- Antigenic proteins that can be used in the pharmaceutical composition of this embodiment include, but are not limited to, ERK1, ERK2, WT1, MART-1/Melan-A, gp100, adenosine deaminase binding protein (ADAbp), FAP, cyclophilin b, colorectal Related antigen (CRC) - C017-1A/GA733, carcinoembryonic antigen (CEA), CAP-1, CAP-2, etv6, AML1, prostate specific antigen (PSA), PSA-1, PSA-2, PSA -3, prostate specific membrane antigen (PSMA), T cell receptor/CD3-zeta chain, CD20, MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE- A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp2 (MAGE-B2), MAGE-X
- the entire sequence thereof may be used, or a partially deleted sequence may be used.
- the antigenic peptide that can be used in the pharmaceutical composition of the present embodiment is one or more epitopes selected from the group consisting of a CD8-positive cytotoxic T cell recognition epitope and a CD4-positive helper T cell recognition epitope among the antigen protein sequences. It is an antigenic peptide containing.
- the antigenic peptide is preferably an antigenic peptide containing two or more epitopes from the viewpoint of being loaded onto MHC class I molecules or MHC class II molecules after being degraded in antigen-presenting cells.
- the antigenic peptide mentioned above includes an antigenic peptide containing an epitope of an antigenic protein of a tumor cell.
- the antigenic peptide has, for example, 8 to 120 amino acids, preferably 8 to 80 amino acids, more preferably 15 to 80 amino acids, even more preferably 16 to 80 amino acids, even more preferably 23 to 80 amino acids, and even more It preferably has 23 to 60 amino acids, particularly preferably 23 to 50 amino acids.
- the antigen peptide has one epitope recognized by CD8-positive cytotoxic T cells and one epitope recognized by CD4-positive helper T cells. It is an antigenic peptide containing three or more antigenic peptides.
- an amino acid linker may be placed between the epitopes.
- the linker has, for example, 2 to 10 amino acids, preferably 4 to 10 amino acids, more preferably 4 to 8 amino acids.
- Amino acids used in the linker include glycine (G), tyrosine (Y), leucine (L), tryptophan (W), and the like. Preferably they are tyrosine (Y), leucine (L), and tryptophan (W).
- amino acid linkers include a linker (4Y) consisting of four consecutive tyrosines (Y), a linker (4L) consisting of four consecutive leucines (L), and a linker (4L) consisting of four consecutive tryptophans (W).
- a linker of 6 consecutive glycines (G) (6G), a linker of 6 consecutive tyrosines (Y) (6Y), a linker of 6 consecutive leucines (L) (6L), a linker of 6 consecutive tryptophans (W) linker (6W), 8 consecutive tyrosine (Y) linker (8Y), 6 consecutive leucine (L) linker (8L), 8 consecutive tryptophan (W) linker (8W) are mentioned, preferably 6Y, 6L or 6W.
- Cancer antigens may be tumor-associated antigens, cancer testis antigens, viral antigens, or tumor-specific antigens (including neoantigens). Cancer antigens may be used alone or in combination of two or more.
- infectious disease-derived antigen is not particularly limited as long as it is an infectious pathogen or an antigen derived from an infectious pathogen. Infectious pathogens include viruses, bacteria, fungi, nematodes, and the like.
- infectious disease pathogen-derived antigen may be an antigenic protein or an antigenic peptide.
- Diseases contracted from the above infectious pathogens are not particularly limited, and include, for example, adenoviruses, herpesviruses (such as HSV-I, HSV-II, CMV, and VZV), poxviruses (such as variola or vaccinia, and molluscum contagiosum).
- adenoviruses such as HSV-I, HSV-II, CMV, and VZV
- poxviruses such as variola or vaccinia, and molluscum contagiosum
- orthopoxviruses picornaviruses (e.g., rhinoviruses, enteroviruses), orthomyxoviruses (e.g., influenza viruses), paramyxoviruses (e.g., parainfluenza viruses, mumps viruses, measles viruses, respiratory viruses) syncytial virus (RSV)), coronavirus (e.g., SARS coronavirus (SARS-CoV), MERS coronavirus (MERS-CoV), SARS-CoV-2), papovavirus (e.g., genital warts, cystocele vulgaris, papillomaviruses (such as those that cause plantar warts), hepadnaviruses (e.g., hepatitis B virus), flaviviruses (e.g., hepatitis C virus, dengue virus), retroviruses (e.g., lentiviruses such as HIV), etc.
- RSV syncy
- Viral diseases such as those caused by viral infections; Escherichia, Enterobacter, Salmonella, Staphylococcus, Shigella, Listeria, Aerobacter, Helicobacter, Klebsiella, Proteus, Pseudomonas, Streptococcus, Chlamydia, Mycoplasma, Pneumococcus, Neisseria, Clostridium , Bacillus, Corynebacterium, Mycobacterium, Campylobacter, Vibrio, Serratia, Providencia, Chromobacterium, Brucella, Yersinia, Haemophilus, Bordetella, etc.; chlamydia, candidiasis, aspergillosis, Fungal diseases such as histoplasmosis and cryptococcal meningitis; malaria, Pneumocystis carinii pneumonia, leishmaniasis, cryptosporidiosis, toxoplasmosis, trypanosome infection, and the
- the structure of the antigen that can be used in the pharmaceutical composition of the present embodiment is not particularly limited as long as it is at least a part of various components constituting a pathogen, and examples include live vaccines, inactivated whole particles, and the like. Examples include a part of a protein, a protein subunit, a protein, a peptide, and the like. Among these, protein subunits, proteins, or peptides are preferred from the viewpoint of conjugation with hyaluronic acid derivatives.
- the above-mentioned influenza virus is an RNA enveloped virus that belongs to the Orthomyxoviridae family and has a particle size of about 100 nm in diameter, and is divided into types A, B, and C based on the antigenicity of the internal protein.
- the influenza virus consists of a core of ribonucleic acid (RNA) associated with an internal nucleocapsid or nucleoprotein surrounded by a viral envelope with a lipid bilayer structure, and an external glycoprotein.
- the inner layer of the viral envelope is mainly composed of matrix proteins, and the outer layer is mostly composed of host-derived lipid substances.
- the RNA of the influenza virus has a segmented structure. Influenza that is prevalent all over the world is caused by influenza A virus.
- Influenza A virus has two types of envelope glycoproteins, hemagglutinin and neuraminidase, and there are 16 types of hemagglutinin due to antigenic differences. , neuraminidase is classified into nine subtypes.
- antigens derived from influenza A and B viruses are preferably used.
- the subtypes of the above-mentioned influenza A and B viruses are not particularly limited, and may be subtypes that have been isolated so far or subtypes that will be isolated in the future.
- the influenza virus-derived antigen is not particularly limited as long as it is at least a part of the various components constituting the above-mentioned influenza virus.
- purified virus particles are inactivated with an organic solvent/surfactant or other reagent.
- examples include whole virus particles produced by the virus, or virus subunits produced by removing impurities from the whole virus particles and purifying hemagglutinin and/or neuraminidase. From the viewpoint of immunogenicity, hemagglutinin subunits or whole virus particles are preferred.
- the whole virus particles are more preferably inactivated with formalin or the like. Furthermore, it is particularly effective for hemagglutinin subunit (split), which has few impurities and requires an adjuvant such as an immunostimulant.
- the method for preparing the influenza virus antigen described above is not particularly limited, and any known method can be used without limitation.
- a method may be used in which chicken eggs or the like are infected with a virus strain isolated from an influenza-infected animal or an influenza patient, cultured by a conventional method, and an antigen is prepared from the purified virus stock solution.
- a virus-derived antigen prepared in cultured cells by genetic engineering may be used.
- Antigens for immune diseases are not particularly limited as long as they contain epitopes of target proteins of immune diseases.
- Immune diseases are not particularly limited, and include, for example, plaque psoriasis, ankylosing spondylitis, rheumatoid arthritis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, bronchial asthma, chronic urticaria, and hay fever. , atopic dermatitis, etc.
- Target proteins are not particularly limited, and include IL-17A, DPP4, S100A9, PCSK9, IL-23, IgE, TNF ⁇ , IL-12/23p40, IL-6, ⁇ 4 ⁇ 7 integrin, IL-4/13, IL-5, Examples include BLyS and IL-13.
- Reference document 2 International Publication No. 2017/164409) describes a peptide derived from IL-17A.
- the peptide may have not only an epitope of the target protein but also an epitope of a protein other than the target protein. It may be a B cell epitope or a T cell epitope.
- a peptide is a sequence that has an epitope and may have a cyclic structure. The early peptide may have multiple cyclic structures within the molecule.
- the peptide may be conjugated to a protein. A therapeutic effect can be expected by the production of antibodies against the administered protein or peptide within the body.
- pharmaceutically active peptide or protein By pharmaceutically active peptide or protein is meant one that has a positive or beneficial effect on a condition or disease state of a subject when administered to the subject in a therapeutically effective amount.
- Preferred pharmaceutically active peptides or proteins have curative or palliative properties and ameliorate, ameliorate, alleviate, reverse, or delay the onset of symptoms of a disease or disorder, or It can be administered to reduce the severity of symptoms.
- Pharmaceutically active peptides or proteins may also have prophylactic properties and can be used to delay the onset of disease or reduce the severity of such disease or condition.
- pharmaceutically active peptide or protein includes a full-length protein or polypeptide and may also refer to pharmaceutically active fragments thereof. The term also encompasses pharmaceutically active analogs of peptides or proteins.
- cytokines examples include, but are not limited to, cytokines and immune system proteins such as immunoactive compounds (e.g., interleukins, colony stimulating factor (CSF), granulocyte colony stimulating factor (G-CSF), granulocyte Macrophage colony stimulating factor (GM-CSF), erythropoietin, tumor necrosis factor (TNF), interferon, integrin, addressin, seletin, homing receptor, T cell receptor, immunoglobulin, antibody, hormone (insulin, thyroid hormone) , catecholamines, gonadotropins, stimulating hormones, prolactin, oxytocin, dopamine, bovine somatotropin, leptin, etc.), growth hormones (e.g., human growth hormone), growth factors (e.g., epidermal growth factor, nerve growth factor, insulin-like growth factor, etc.) , growth factor receptors, enzymes (tissue plasminogen activator, streptokinase, cholesterol biosy
- proteins that inhibit angiogenesis include structural proteins (collagen, fibroin, fibrinogen, elastin, tubulin, actin, myosin, etc.), blood proteins (thrombin, Serum albumin, factor VII, factor VIII, insulin, factor IX, factor X, tissue plasminogen activator, protein C, von Willebrand factor, antithrombin III, glucocerebrosidase, erythropoietin, modified factor VIII , anticoagulant factors), etc.
- structural proteins collagen, fibroin, fibrinogen, elastin, tubulin, actin, myosin, etc.
- blood proteins thrombin, Serum albumin, factor VII, factor VIII, insulin, factor IX, factor X, tissue plasminogen activator, protein C, von Willebrand factor, antithrombin III, glucocerebrosidase, erythropoietin, modified factor VIII , anticoagulant factors), etc.
- the pharmaceutically active protein is a cytokine involved in the regulation of lymphocyte homeostasis, preferably involved in one or more selected from the group consisting of T cell development, priming, expansion, differentiation and survival. and cytokines that induce or enhance them.
- the cytokine is an interleukin.
- the pharmaceutically active protein is one or more interleukins selected from the group consisting of IL-2, IL-7, IL-12, IL-15 and IL-21.
- the pharmaceutically active peptide may have a cyclic structure. The peptide may have multiple cyclic structures within the molecule.
- nucleic acids examples include DNA, RNA, antisense nucleic acids, decoy nucleic acids, ribozymes, small interfering RNAs, and nucleic acid aptamers. Furthermore, when the antigen is a peptide or protein, nucleic acids (DNA, mRNA, etc.) encoding the antigenic peptide or protein are also preferably used.
- low-molecular compounds examples include anticancer agents (e.g., alkylating agents, antimetabolites, alkaloids, etc.), immunosuppressants, anti-inflammatory agents (steroidal agents, nonsteroidal anti-inflammatory agents, etc.), antirheumatic agents, and antibacterial agents. agents ( ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics, new quinolone antibiotics, sulfa drugs, etc.).
- anticancer agents e.g., alkylating agents, antimetabolites, alkaloids, etc.
- immunosuppressants e.g., anti-inflammatory agents, etc.
- anti-inflammatory agents steroidal agents, nonsteroidal anti-inflammatory agents, etc.
- antirheumatic agents e.g., antibacterial agents.
- agents ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics, new quinolone antibiotics,
- highly hydrophobic, ie, poorly water-soluble ones can also be preferably used because they can sufficiently interact with the steryl group of the above-mentioned hyaluronic acid derivative.
- “poorly water-soluble” refers to the amount of water required to dissolve 1 g of solute in the 17th edition of the Japanese Pharmacopoeia to be 30 mL or more.
- poorly water-soluble solid medicinal ingredients include acetaminophen, ibuprofen, benzoic acid, ethenzamide, caffeine, camphor, quinine, calcium gluconate, dimethyl caprol, sulfamine, theophylline, theopramine, riboflavin, mephenesin, Antipyretic analgesics such as phenobarbital, aminophylline, thioacetazone, quercetin, rutin, salicylic acid, theophylline sodium salt, pirapital, quinine hydrochloride, irgapirin, dioquitoxin, griseofulvin, phenacetin, nervous system drugs, sedative-hypnotics, muscle relaxants, blood pressure stiffening antibiotics such as acetylspiramycin, ampicillin, erythromycin, xatamycin, chloramphenicol, triacetyloleandomycin, nystatin, colistin sulf
- the medicinal ingredient may be in the form of a poorly water-soluble oil or liquid.
- sparingly water-soluble oily or liquid medicinal ingredients include teprenone, indomethacin farnesil, menatetrenone, phytonadione, vitamin A oil, fenipentol, vitamins such as vitamin D and vitamin E, DHA (docosahexaenoic acid), and EPA. (eicosapentaenoic acid), higher unsaturated fatty acids such as cod liver oil, coenzyme Q, oil-soluble flavorings such as orange oil, lemon oil, peppermint oil, etc., according to the Japanese Pharmacopoeia, ⁇ external groups'', and ⁇ USP''.
- vitamin E There are various homologs and derivatives of vitamin E, but there are no particular limitations as long as they are liquid at room temperature. Examples include dl- ⁇ -tocopherol, dl- ⁇ -tocopherol acetate, d- ⁇ -tocopherol, and d- ⁇ -acetate. Examples include tocopherol. One type selected from these medicinal ingredients may be used, or two or more types may be used in combination.
- the medicinal ingredient may be a semi-solid substance that is poorly water soluble.
- Semi-solid medicinal ingredients that are poorly water-soluble include, for example, Jiryu, Liquorice, Keihi, Peony, Botanpi, Valerian, Sansho, Gingerbread, Chinpi, Ephedra, Nantenjitsu, Japanese apricot, Onji, Bellflower, Shazenshi, Chazenso, and Stone.
- One type selected from these medicinal ingredients may be used, or two or more types may be used in combination.
- the content of the medicinal ingredient may be 0.001% by mass or more and 10,000% by mass or less based on the mass of the hyaluronic acid derivative, although it depends on the structure of the medicinal component. It is preferably 0.1% by mass or more and 1000% by mass or less, more preferably 1.0% by mass or more and 100.0% by mass or less, and preferably 1.5% by mass or more and 50.0% by mass or less. It is more preferably 3.0% by mass or more and 30.0% by mass or less, most preferably 5.0% by mass or more and 20.0% by mass or less.
- the content of the medicinal ingredient is more preferably 0.1% by mass or more and 100.0% by mass or less, based on the mass of the hyaluronic acid derivative, and 0.1% by mass. % or more and 50.0 mass% or less, further preferably 0.1 mass% or more and 30.0 mass% or less, and 0.5 mass% or more and 30.0 mass% or less. More preferably, the content is 1.0% by mass or more and 30.0% by mass or less, even more preferably 1.0% by mass or more and 25.0% by mass, and 1.0% by mass or more and 20.0% by mass. It is more preferably not more than 1.0% by mass and not more than 15.0% by mass, even more preferably not less than 1.0% by mass and not more than 10.0% by mass.
- the content of the medicinal ingredient is preferably 0.0001 parts by mass or more and 1.00 parts by mass or less, and 0.001 parts by mass, based on 100 parts by mass of the pharmaceutical composition.
- the amount is more preferably 0.100 parts by mass or more, and even more preferably 0.002 parts by mass or more and 0.500 parts by mass or less.
- the content of the medicinal ingredient is preferably 0.001 parts by mass or more and 1.00 parts by mass or less, and 0.001 parts by mass or less, based on 100 parts by mass of the pharmaceutical composition.
- Parts by weight or more and 0.500 parts by weight or less more preferably 0.001 parts by weight or more and 0.200 parts by weight or less, and 0.005 parts by weight or more and 0.100 parts by weight or less. More preferred.
- the content of the medicinal ingredient is at least the lower limit above, it is possible to activate immune cells more effectively, and on the other hand, when the content is below the upper limit above, the medicinal ingredient is encapsulated in the hyaluronic acid derivative component.
- the structure can be made more stable.
- the pharmaceutical composition of this embodiment when the pharmaceutical composition of this embodiment is a vaccine composition, it can further contain an adjuvant in addition to the above-mentioned medicinal ingredients and the above-mentioned hyaluronic acid derivative. Thereby, immunity can be induced more effectively.
- the immunity induced may be either humoral immunity or cell-mediated immunity. That is, the pharmaceutical composition of this embodiment contains the hyaluronic acid derivative, an antigen (preferably a cancer antigen or an infectious disease-derived antigen) as the medicinal ingredient, and further contains an adjuvant. It is preferable that there be.
- humoral immunity refers to an immune mechanism mainly consisting of B cells and antibodies.
- Cytokines produced by helper T cells (Th2 cells) stimulate B cells, which differentiate into plasma cells and produce large amounts of antibodies, which circulate in body fluids and spread throughout the body. .
- some of the stimulated B cells become memory B cells that remember information about the antigen, and upon reinfection, the response is faster and has a higher affinity for the antigen than the initial response.
- Antibodies can be produced in large quantities.
- cell-mediated immunity refers to an immune mechanism in which cells are the main effectors in eliminating foreign substances such as pathogens themselves, virus-infected cells, and cancer cells. This is an elimination mechanism by immunocompetent cells themselves such as macrophages, cytotoxic T cells (CTL, killer T cells), and natural killer cells (NK cells).
- the adjuvant is not particularly limited as long as it is commonly used in vaccines, but examples include aluminum salts, squalene, and ligands for innate immune receptors.
- ligand here refers to a substance that specifically binds to a receptor, and in particular, substances that specifically bind to a receptor and exhibit various physiological effects can be used. Such substances are also called "agonists.”
- TLRs toll-like receptors
- RLRs RIG-I-like receptors
- NLRs NOD-like receptors
- CLR C-type lectin receptor
- TLR ligand for example, at least one TLR selected from the group consisting of TLR-2, TLR-3, TLR-4, TLR-5, TLR-6, TLR-7, TLR-8, and TLR-9. What is necessary is to select the one that interacts with the appropriate one.
- TLR-2 ligands examples include Pam3CSK4 and the like.
- TLR-3 ligands examples include polyICLC, polyinosine:polycytidylic acid (polyI:C), and the like.
- TLR-4 ligands examples include R-type lipopolysaccharide, S-type lipopolysaccharide, Paclitaxel, lipid A, monophosphoryl lipid A, and the like.
- TLR-5 ligands examples include flagellin and the like.
- TLR-2 and TLR-6 ligands examples include MALP-2.
- TLR-7 and TLR-8 ligands examples include resiquimod (R848), imiquimod (imiquimod, R837), gardiquimod, loxoribine, and the like.
- TLR-9 ligands examples include CpG oligodeoxynucleotides and the like.
- anionic compounds are preferred, and CpG oligodeoxynucleotides are more preferred, since they can further improve the antigen presentation function.
- CpG oligodeoxynucleotides examples include CpG-ODN 1826, CpG-K3, and the like.
- the pharmaceutical composition of this embodiment can be administered alone or together with a pharmacologically acceptable carrier according to conventional methods.
- a pharmacologically acceptable carrier for example, the above-mentioned hyaluronic acid derivative and the above-mentioned medicinal ingredient, and if necessary an adjuvant, water or other physiologically acceptable liquid (for example, physiological saline).
- physiologically acceptable liquid for example, physiological saline.
- Physiologically acceptable buffers, excipients, vehicles, preservatives, stabilizers, binders, etc. which may be mixed with water, aqueous ethanol, phosphate buffered saline (PBS), etc. Freeze-drying aids and the like may also be included.
- buffer examples include Tris, sodium phosphate, potassium phosphate, histidine, or citric acid.
- preservative examples include benzalkonium chloride, methyl paraoxybenzoate, propyl paraoxybenzoate, chlorobutanol, sorbic acid, alkylpolyaminoethylglycine, and the like.
- stabilizer examples include sodium edetate hydrate, polyvinylpyrrolidone (povidone), and polysorbate 80.
- the pharmaceutical composition of this embodiment may be formulated.
- the formulation can be in solid, semi-solid or liquid form.
- examples include powder, granules, pills, pellets, tablets, capsules, and the like.
- the solid is preferably a freeze-dried powder.
- examples include forms such as gels.
- examples include a suspension in which the powder is diluted or suspended in water or a buffer such as phosphate buffered saline (PBS).
- PBS phosphate buffered saline
- the pharmaceutical composition of this embodiment can be manufactured by appropriately mixing the hyaluronic acid derivative and the medicinal ingredient, but it is preferably manufactured by the method shown below.
- the method for producing a pharmaceutical composition of the present embodiment is a method for producing a pharmaceutical composition containing a hyaluronic acid derivative and a medicinal ingredient, and includes the following steps. a preparation step of dissolving the medicinal ingredient in an organic solvent or organic solvent-containing water to prepare an oil phase containing the medicinal ingredient; and A mixing step of mixing the oil phase and the aqueous phase containing the hyaluronic acid derivative at a volume ratio of 20:100 to 0.01:100.
- hyaluronic acid derivatives have been found to have problems such as the interaction between steryl groups being inhibited by the oil phase, causing the particles to disintegrate or some to aggregate. It was difficult to maintain the particle size of the acid derivative. Therefore, pharmaceutical compositions have contained only relatively small-sized hyaluronic acid derivatives in which particles are broken down, or complexes of hyaluronic acid derivatives and medicinal ingredients containing partially aggregated particles. Furthermore, since the pharmaceutical composition was manufactured using a dialysis method, it was difficult to control the weight ratio of the hyaluronic acid derivative to the medicinal ingredient. Furthermore, a concentration step was necessary to achieve the target concentration of medicinal ingredients.
- the method for producing a pharmaceutical composition of the present embodiment does not use the dialysis method, but involves mixing the above composition so that the mixing ratio of the oil phase and the aqueous phase is within the above numerical range.
- This makes it possible to form a complex with the medicinal ingredient while maintaining the particle size distribution of the hyaluronic acid derivative before manufacturing the pharmaceutical composition, and it is also possible to control the weight ratio of the hyaluronic acid derivative to the medicinal ingredient to a constant level. .
- a medicinal ingredient-hyaluronic acid derivative complex can be obtained with a stable structure.
- a pharmaceutical composition can be obtained that has excellent delivery properties to immune cells in lymph nodes and an ability to activate these immune cells.
- the medicinal ingredient is dissolved in an organic solvent or organic solvent-containing water to prepare an oil phase containing the medicinal ingredient.
- a poorly water-soluble medicinal ingredient can be preferably used.
- the medicinal ingredient those exemplified in the above “medicinal ingredient” can be used.
- organic solvents for dissolving medicinal ingredients include known ones that are generally used in the production of pharmaceutical compositions, such as dimethyl sulfoxide (DMSO), methanol, ethanol, tetrahydrofuran, acetone, and acetonitrile. , ethyl acetate, 1-propanol, 2-propanol, 1-butanol, 2-butanol, 1-pentanol, methyl ethyl ketone, heptane, isobutyl acetate, isopropyl acetate, methyl acetate, methyl isobutyl ketone, dimethylformamide and the like.
- DMSO dimethyl sulfoxide
- methanol ethanol
- tetrahydrofuran acetone
- acetonitrile examples include known ones that are generally used in the production of pharmaceutical compositions, such as dimethyl sulfoxide (DMSO), methanol, ethanol, tetrahydrofuran, ace
- the hyaluronic acid derivative is used after being dissolved or dispersed in water or the like in advance to a desired concentration.
- the pH of the aqueous phase containing the hyaluronic acid derivative is preferably 6.00 or more, and preferably 6.00 or more and 11.00 or less.
- the pH is equal to or higher than the above lower limit, it is possible to more effectively suppress the aggregation of the hyaluronic acid derivative and form a complex with the medicinal ingredient, thereby forming a medicinal ingredient-hyaluronic acid derivative complex. A more stable structure is obtained.
- the pH is below the above upper limit, the drug can be complexed while maintaining the dispersion stability of the hyaluronic acid derivative.
- a complex can be formed not only through hydrophobic interactions but also through interactions such as electrostatic interactions and hydrogen bonds.
- the mixing ratio of the oil phase and the aqueous phase is 20:100 to 0.01:100 by volume, preferably 10:100 to 0.05:100, more preferably 5:100 to The ratio is 0.1:100, more preferably 2.5:100 to 0.5:100.
- a sufficient amount of the medicinal ingredient can be encapsulated in the hyaluronic acid derivative to form a medicinal ingredient-hyaluronic acid derivative complex, and hyaluronic acid It is possible to effectively suppress disassociation of acid derivative particles due to inhibition of hydrophobic interaction between steryl groups by the oil phase. However, this is not limited to only hydrophobic interactions, but also the collapse of interactions that can contribute to structural stability, such as electrostatic interactions and hydrogen bonds, can also be effectively suppressed.
- the concentration of the medicinal ingredient dissolved in the oil phase is not particularly limited, but it is preferably 0.1 parts by mass or more and 50 parts by mass or less, and 0.2 parts by mass or more, based on 100 parts by mass of the oil phase. It is more preferably 25 parts by mass or less, and even more preferably 0.5 parts by mass or more and 10 parts by mass or less.
- the concentration of the hyaluronic acid derivative dissolved in the aqueous phase is not particularly limited, but the concentration of the hyaluronic acid derivative is preferably 0.01 parts by mass or more and 10 parts by mass or less with respect to 100 parts by mass of the aqueous phase. , more preferably 0.1 parts by mass or more and 10 parts by mass or less, and still more preferably 0.5 parts by mass or more and 5 parts by mass or less.
- the temperature is preferably 20°C or more and 65°C or less, and preferably 23°C or more and 55°C or less, from the viewpoint of slightly exposing the hydrophobic part of the medicinal ingredient and making it easier to interact with the hyaluronic acid derivative. More preferably, the temperature is 25°C or higher and even more preferably 40°C or lower. Alternatively, the optimum temperature can be appropriately set depending on the structure and properties of the medicinal ingredient.
- the method is not particularly limited, but for example, an oil phase containing a medicinal ingredient is added to the aqueous phase containing the hyaluronic acid derivative in any ratio, or an oil phase containing a medicinal ingredient is added to the aqueous phase containing the hyaluronic acid derivative.
- the aqueous phase containing the above-mentioned hyaluronic acid derivative is added into the mixture at an arbitrary ratio, so that the final mixing ratio of the oil phase and the aqueous phase is 20:100 to 0.01:100 by volume.
- the particle size of the hyaluronic acid derivative can be maintained through the mixing process.
- the mixing step is not limited to a batch method, and the oil phase and the aqueous phase may be mixed by a flow method (continuous method).
- the particle size of the hyaluronic acid derivative can be maintained by setting the mixing ratio of the oil phase and the water phase to a volume ratio of 20:100 to 0.01:100.
- the material and shape of the reactor in the flow method are not particularly limited as long as the material and shape are selected as applicable to the production method of the present invention, but for example, the inner diameter of the piping through which the oil phase and the aqueous phase each pass, They may have different diameters.
- the material and shape of the tube in the flow method may be selected from materials and shapes that are applicable to the method for producing the pharmaceutical composition of this embodiment, and are not particularly limited.
- Teflon (registered trademark) tube stainless steel, etc.
- Examples include tubes, glass tubes, plastic tubes, etc.
- the time is not particularly limited, but can be, for example, 30 minutes or more and 90 hours or less, or 1 hour or more and 80 hours or less.
- the organic solvent in which the medicinal ingredient is dissolved is subjected to tangential flow filtration (TFF), centrifugal filtration, ultrafiltration/diafiltration (UFDF).
- TFF tangential flow filtration
- UFDF ultrafiltration/diafiltration
- a sterilization step may be performed in which the solution containing the obtained medicinal ingredient-hyaluronic acid derivative complex is sterilized to obtain a sterile preparation.
- methods for sterilizing the preparation include filtration sterilization using a sterilization filter, gas sterilization, ⁇ -ray sterilization, and electron beam sterilization. Filtration sterilization using a sterilizing filter is most preferred.
- a drying step may be performed in which the obtained solution containing the medicinal ingredient-hyaluronic acid derivative complex is dried to obtain a dried product.
- Examples of the drying method include the same methods as those exemplified in the above "method for producing hyaluronic acid derivatives.”
- Subjects to which the pharmaceutical composition of the present embodiment is administered include animals classified as mammals including humans (monkeys, marmosets, mice, rats, cows, horses, cats, dogs, pigs, sheep, goats, rabbits, etc.). It will be done.
- Administration routes include, for example, intrathecal injection, intraarterial injection, intravenous injection, subcutaneous injection, as well as intranasal, transbronchial, transpulmonary, intramuscular, percutaneous, or oral administration. This can be done by methods known to those skilled in the art. Among these, when the pharmaceutical composition of this embodiment is a vaccine composition, subcutaneous injection or intramuscular injection is preferred.
- the dosage can be appropriately selected taking into consideration the type of recipient (including age, gender, etc.); For example, for humans (assuming a body weight of 60 kg), the amount of the medicinal ingredient (preferably antigen) per dose can be 0.01 ⁇ g or more and 5 mg or less, and 0.1 ⁇ g or more and 500 ⁇ g or less, The amount can be 1 ⁇ g or more and 100 ⁇ g or less.
- the amount of the medicinal ingredient (preferably antigen) per dose can be 0.01 ⁇ g or more and 5 mg or less, and 0.1 ⁇ g or more and 500 ⁇ g or less, The amount can be 1 ⁇ g or more and 100 ⁇ g or less.
- the frequency of administration may be a single administration of the above-mentioned dose, or the above-mentioned dose may be administered once every 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, 2 months, 3 months, or every six months.
- the administration may be multiple times, such as twice or more.
- the drug may be administered to two or more locations in one administration.
- the present invention provides prevention or treatment of one or more diseases selected from the group consisting of cancer, infectious diseases, and immune diseases, which comprises administering an effective amount of the above pharmaceutical composition to a patient or affected animal.
- diseases selected from the group consisting of cancer, infectious diseases, and immune diseases
- infectious diseases include those exemplified under "antigens derived from infectious diseases” under “antigens” above.
- effective amount as used herein includes an amount effective for prevention or treatment, that is, an amount suitable for preventing or treating the onset of the above-mentioned diseases.
- the present invention provides a composition for the prevention or treatment of one or more diseases selected from the group consisting of cancer, infectious diseases, and immune diseases, comprising the above-mentioned medicinal ingredient-hyaluronic acid derivative complex.
- a composition comprising:
- the present invention provides the use of the above medicinal ingredient-hyaluronic acid derivative complex for manufacturing a pharmaceutical composition.
- the pharmaceutical composition is preferably an infectious disease vaccine, a cancer vaccine, or a pharmaceutical composition for immune diseases.
- the present invention further provides a pharmaceutical composition
- a pharmaceutical composition comprising a composition comprising a hyaluronic acid derivative and an antigen, and a lymphocyte expressing an immune receptor for the antigen.
- a pharmaceutical composition containing lymphocytes expressing immunoreceptors for antigens is provided for use in combination administration with a composition containing an early hyaluronic acid derivative and an antigen.
- a pharmaceutical composition characterized in that it consists of a combination of a composition containing a hyaluronic acid derivative and an antigen, and antigen-specific lymphocytes can also be in the form of a kit.
- the pharmaceutical composition can be, for example, a cancer treatment kit comprising a composition containing a hyaluronic acid derivative and an antigen, and antigen-specific lymphocytes.
- the composition containing the hyaluronic acid derivative and the antigen is preferably administered before administering the antigen-specific lymphocytes.
- the unit of administration is one unit or one infusion of antigen-specific lymphocytes after one administration of a composition containing a hyaluronic acid derivative and an antigen.
- the composition containing the hyaluronic acid derivative and the antigen is administered at least once after two units have been administered.
- the interval between the first administration and the second administration of the composition containing a hyaluronic acid derivative and an antigen is, for example, 1 day or more, 2 days or more, 3 days or more, 4 days or more, 5 days or more. , 6 or more days, 7 or more days, 8 or more days, 9 or more days, 10 or more days, 11 or more days, 12 or more days, 13 or more days, or 14 or more days.
- the interval may be, for example, 28 days or less, 24 days or less, 21 days or less, 17 days or less, 14 days or less, 13 days or less, 12 days or less, 11 days or less, 10 days or less, 9 days or less, 8 days or less. , 7 days or less, 6 days or less, 5 days or less, 4 days or less, 3 days or less, or 2 days or less.
- a composition containing a hyaluronic acid derivative and an antigen of the present invention may be administered in combination with one or more antibodies used in cancer treatment.
- the antibody is, for example, one or more antibodies that inhibit immunosuppressive signals by a tumor or activate co-stimulatory signals of immune cells, preferably antibodies that inhibit immunosuppressive signals by a tumor or co-stimulate immune cells. It is an antibody that activates the signal. Specifically, it is one or more antibodies selected from the group consisting of anti-CTLA4 antibody, anti-PD1 antibody, anti-PDL1 antibody, anti-OX40, and anti-4-1BB antibody.
- the dosage of the composition containing the hyaluronic acid derivative and antigen when administered in combination is, for example, 0.01 to 100 mg/time, preferably 0.1 to 50 mg/time, more preferably 0.1 to 20 mg/time.
- the dosage of the antibody is, for example, 0.01 to 200 mg/kg body weight, preferably 0.1 to 100 mg/kg body weight, and more preferably 1 to 40 mg/kg body weight.
- the antibody may be administered at the same timing as the composition containing the hyaluronic acid derivative and the antigen (including when the antibody is included in the formulation) or at a different timing.
- a composition containing a hyaluronic acid derivative and an antigen may be administered in combination with both the adjuvant and antibody described above.
- the dosage of the composition containing the hyaluronic acid derivative and the antigen is, for example, 0.01 to 100 mg/time, preferably 0.1 to 50 mg/time, more preferably 0.1 to 20 mg/time.
- the dosage of the adjuvant is, for example, 0.01 to 100 mg/kg body weight, preferably 0.1 to 50 mg/kg body weight, more preferably 0.1 to 10 mg/kg body weight
- the dosage of the antibody is, for example, 0. .01 to 200 mg/kg body weight, preferably 0.1 to 100 mg/kg body weight, more preferably 1 to 40 mg/kg body weight.
- the adjuvant and antibody may be administered at the same time as the composition containing the hyaluronic acid derivative and antigen (including when the adjuvant is included in the formulation) or at different times. If administered at different times, it is preferred that the vaccine formulation, adjuvant, and antibody are all administered within, eg, 1 minute to 24 hours, preferably 1 minute to 5 hours.
- the present invention provides a composition for improving, promoting, or enhancing the uptake of a medicinal ingredient into immune cells, the composition comprising the above medicinal ingredient-hyaluronic acid derivative complex.
- the present invention provides a composition for improving, promoting, or enhancing the activity of immune cells, the composition comprising the above-mentioned medicinal ingredient-hyaluronic acid derivative complex.
- the present invention provides a composition for improving, promoting, maintaining or enhancing the expression of costimulatory molecules (particularly CD80 and CD86) of DCs (particularly cDC1), the composition comprising: Compositions are provided that include acid derivative complexes.
- the present invention improves, promotes, or enhances the uptake of a medicinal ingredient into immune cells in vivo or in vitro, comprising administering a composition comprising the above medicinal ingredient-hyaluronic acid derivative complex.
- a composition comprising the above medicinal ingredient-hyaluronic acid derivative complex.
- the present invention provides a method for improving, promoting, or enhancing the activity of immune cells in vivo or in vitro, comprising administering a composition comprising the above-described pharmaceutical ingredient-hyaluronic acid derivative complex.
- the present invention provides costimulatory molecules (particularly CD80 and CD86) of DCs (particularly cDC1) in vivo or in vitro, comprising administering a composition comprising the above-mentioned pharmaceutical ingredient-hyaluronic acid derivative complex. ) provides a method for improving, promoting, maintaining or enhancing the expression of
- TBA salt of hyaluronic acid (HA-TBA) was prepared according to step 2-1 shown below, followed by step 2-2.
- TBA salt of hyaluronic acid HA-TBA
- Process 3 An anhydrous DMSO solution (10 mg/mL) of HA-TBA prepared in "2. (2) Step 2-2" was prepared. Thereafter, Chol hydrochloride was added in such a manner that the molar ratio of Chol hydrochloride to the disaccharide repeating unit (HA unit) present in the HA-TBA synthesized in "1. Step 1" was 44/100. Next, the amount of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) added to the HA unit was 48/100 in molar ratio. It was stirred overnight at room temperature (about 25°C).
- DMT-MM 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- the reaction solution was prepared using a Spectrapore 7 dialysis membrane (manufactured by Spectrum Laboratories, molecular weight cutoff (MWCO): 3,500) in the order of 0.3M ammonia acetate/DMSO solution, 0.15M NaCl aqueous solution, and ultrapure water. Dialysis was performed using The obtained dialysate was freeze-dried to obtain the target product (HA-C 6 -Chol) as a white solid.
- MWCO molecular weight cutoff
- the cholesterol introduction rate in the obtained white solid was calculated by the following method.
- the measurement was carried out at 85° C., and a 1 H-NMR spectrum of a white solid was obtained.
- a lyophilized hyaluronic acid derivative (10k HA-C 6 -Chol-44%) was dissolved in water for injection at 1 mg/mL and measured by gel permeation chromatography under the conditions shown below.
- Figure 3A is the resulting chromatogram.
- Example 1-1 The hyaluronic acid derivative obtained in Synthesis Example 1-1 was fractionated as follows. Lyophilized hyaluronic acid derivative (10k HA-C 6 -Chol-44%) was dissolved in water for injection at 5 mg/mL and transferred to a dialysis cassette (Flotalizer G2, MWCO: 300,000, Ieda Boeki Co., Ltd.). , dialysis was performed 9 times against 10 mM phosphate buffer pH 7.4. The hyaluronic acid derivative of the obtained dialysis fluid was measured by gel permeation chromatography. Figure 4A is the resulting chromatogram.
- Example 21 and 22 show the hyaluronic acid derivative obtained in Example 1-1 and the standard substance polyacrylic acid, in order to explain the method of calculating the area ratio A1/A2 and the distance ratio Da/Db, which will be described later. It is a diagram showing overlapping chromatograms of .
- the filtrate was concentrated to a desired concentration using an ultraconcentrator (Vivaspin 20, MWCO: 10,000, Sartorius).
- Example 1-2 The hyaluronic acid derivative obtained in Synthesis Example 1-2 was fractionated using the same procedure as in Example 1-1.
- the hyaluronic acid derivative of the obtained dialysis fluid was measured by gel permeation chromatography.
- Figure 4B is the resulting chromatogram.
- Example 1-3 The hyaluronic acid derivative obtained in Synthesis Example 1-3 was fractionated by the same procedure as in Example 1-1.
- the hyaluronic acid derivative of the obtained dialysis fluid was measured by gel permeation chromatography.
- Figure 4C is the resulting chromatogram.
- hyaluronic acid derivative obtained in Synthesis Example 1-1 was fractionated as follows. Lyophilized hyaluronic acid derivative (10k HA-C 6 -Chol-44%) was dissolved in water for injection at 10 mg/mL and made into 10 mM phosphate buffer (pH 7.4) with 100 mM phosphate buffer. diluted as follows. Thereafter, it was filtered using an ultrafilter (Vivaspin (registered trademark) 20, MWCO: 300,000, Sartorius), and the hyaluronic acid derivative of the obtained filtrate was measured by gel permeation chromatography. Figure 5A is the resulting chromatogram. The filtrate was concentrated to a desired concentration using an ultraconcentrator (Vivaspin 20, MWCO: 10,000, Sartorius).
- the area ratio A1/A2 and the distance ratio Da/Db were calculated from the gel permeation chromatogram of the hyaluronic acid derivative obtained in Example 1-1 and polyacrylic acid as a standard substance.
- the standard substance polyacrylic acid (50 kDa) was manufactured by Polymer Standards Service-USA, ORDER No. PSS-Paa50k (sodium polyacrylate) was used.
- the standard substance polyacrylic acid (150 kDa) was manufactured by Polymer Standards Service-USA, ORDER No. PSS-Paa150k (sodium polyacrylate) was used.
- Standard substance polyacrylic acid powder was dissolved in water for injection to a concentration of 2 mg/mL. All polyacrylic acids that are standard substances described hereinafter were manufactured by Polymer Standards Service-USA (sodium polyacrylate).
- areas A1 and A2 were calculated from the gel permeation chromatogram shown in FIG. 21, and the area ratio A1/A2 was calculated.
- the distances Da and Db were calculated from the gel permeation chromatogram shown in FIG. 22, and the distance ratio Da/Db was calculated.
- the elution time at point R1 was 5.74 minutes
- the elution time at point S1 was 7.68 minutes.
- Bb the intersection of the baseline and a perpendicular line drawn from the maximum refractive index intensity point Kb on the chromatogram of 50 kDa polyacrylic acid, which is a standard substance, to the baseline was defined as Bb.
- the elution time at the maximum refractive index intensity point Kb is as described above.
- the hyaluronic acid derivatives obtained in Synthesis Examples 1-1 to 1-3, Examples 1-2 to 1-3, and Comparative Examples 1-1 to 1-4 were obtained in the same manner as the hyaluronic acid derivatives obtained in Example 1-1.
- the area ratio A1/A2 and the distance ratio Da/Db were calculated from the gel permeation chromatograms of each hyaluronic acid derivative and the standard substance polyacrylic acid.
- the calculated area ratio A1/A2 and distance ratio Da/Db are shown in Table 1 below.
- Table 1 the distance ratio Da/Db of cholesteryl-modified pullulan (CHP) of Comparative Example 3 could not be calculated because the RI value was small.
- Example 2-1 The hyaluronic acid derivative obtained in Example 1-1 was prepared by diluting it to a concentration of 5 mg/mL with 10 mM phosphate buffer pH 7.4. Meanwhile, in another container, a fluorescently labeled peptide was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/mL.
- DMSO dimethyl sulfoxide
- the above hyaluronic acid derivative aqueous solution and the DMSO solution of the fluorescently labeled peptide are mixed at a volume ratio of 100:1 at room temperature (about 25°C), and the fluorescently labeled peptide is mixed at room temperature (about 25°C) for 24 hours.
- a composition containing a hyaluronic acid derivative complex was obtained.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 was added to the composition containing the fluorescently labeled peptide-hyaluronic acid derivative complex at 10% by mass of sucrose based on the total mass of sucrose. and stirred at room temperature (approximately 25°C) for 1 hour or more.
- the peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of approximately 0.3mg/mL, and 0.2 ⁇ m PES (polyether Sulfone) (manufactured by Pall, Acrodisc syringe filter, 25 mm ⁇ ) was used to sterilize and filtrate to obtain a pharmaceutical composition containing a fluorescently labeled peptide-hyaluronic acid derivative complex.
- the content of the fluorescently labeled peptide was 0.0317 parts by mass with respect to 100 parts by mass of the pharmaceutical composition.
- Example 2-2 Using the hyaluronic acid derivative obtained in Example 1-2, a pharmaceutical composition containing a fluorescently labeled peptide-hyaluronic acid derivative complex was obtained by the same procedure as in Example 2-1. The content of the fluorescently labeled peptide was 0.0478 parts by mass with respect to 100 parts by mass of the pharmaceutical composition.
- the content of the fluorescently labeled peptide was 0.0478 parts by mass with respect to 100 parts by mass of the pharmaceutical composition.
- Example 2-3 Using the hyaluronic acid derivative obtained in Example 1-3, a pharmaceutical composition containing a fluorescently labeled peptide-hyaluronic acid derivative complex was obtained by the same procedure as in Example 2-1.
- the content of the fluorescently labeled peptide was 0.0303 parts by mass with respect to 100 parts by mass of the pharmaceutical composition.
- Comparative example 2-1 The hyaluronic acid derivative obtained in Comparative Example 1-1 was prepared by diluting it to a concentration of 5 mg/mL with 10 mM phosphate buffer pH 7.4. On the other hand, in another container, the fluorescently labeled peptide was dissolved in DMSO to a concentration of 50 mg/mL.
- the above hyaluronic acid derivative aqueous solution and the dimethyl sulfoxide solution of the fluorescently labeled peptide are mixed at a volume ratio of 100:1 at room temperature (about 25°C), and the mixture is stirred at room temperature (about 25°C) for 24 hours to obtain the fluorescent label.
- a composition containing a peptide-hyaluronic acid derivative complex was obtained.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 solid purified sucrose
- the mixture was stirred at room temperature (approximately 25° C.) for more than 1 hour.
- the peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of 0.3mg/mL, and 0.2 ⁇ m PES (polyether sulfone) was added.
- the above hyaluronic acid derivative aqueous solution and the dimethyl sulfoxide solution of the fluorescently labeled peptide are mixed at a volume ratio of 100:1 at room temperature (about 25°C), and the mixture is stirred at room temperature (about 25°C) for 24 hours to obtain the fluorescent label.
- a composition containing a peptide-hyaluronic acid derivative complex was obtained.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 solid purified sucrose
- the mixture was stirred at room temperature (approximately 25° C.) for more than 1 hour.
- the peptide was diluted with 25mM phosphate buffer pH 7.4 containing 10% by mass sucrose to a concentration of 0.3mg/mL, and 0.2 ⁇ m PES (polyether sulfone) was added.
- a pharmaceutical composition containing a fluorescently labeled peptide-hyaluronic acid derivative complex (manufactured by Pall, Acrodisc syringe filter, 25 mm ⁇ ) to obtain a pharmaceutical composition containing a fluorescently labeled peptide-hyaluronic acid derivative complex.
- the content of the fluorescently labeled peptide was 0.0316 parts by mass with respect to 100 parts by mass of the pharmaceutical composition.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 solid purified sucrose
- the mixture was stirred at room temperature (approximately 25° C.) for more than 1 hour.
- the peptide was diluted with 30mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of 0.33mg/mL, and 0.2 ⁇ m PES (polyether sulfone) was added.
- Fluorescently labeled peptide-hyaluronic acid derivative complexes of Examples 2-1 to 2-3 and Comparative Examples 2-1 to 2-3, 3, 4-1, and 4-2 were determined by high-performance liquid chromatography measurement under the following conditions. It was confirmed that the fluorescently labeled peptide was complexed and solubilized with the hyaluronic acid derivative in the pharmaceutical compositions containing the following, and the concentration of the fluorescently labeled peptide in each pharmaceutical composition was provided.
- HPLC device JASCO HPLC-EXTREMA
- Flow rate 2 mL/min
- Injection volume 30 ⁇ L
- Gradient program shown in Table 2 below. In Table 2, "%" means "v/v%”.
- Test Example 1-1 Peptide uptake into myeloid cells in lymph nodes and activation analysis
- Materials Adjuvant CpG oligo DNA1668 (purchased from Ajinomoto Biopharma Service Gene Design)
- the fluorescently labeled antibodies used for evaluation are as follows. All were purchased from Biolegend. Brilliant Violet 421 labeled anti-mouse F4/80 antibody (clone BM8); Brilliant Violet 510 labeled anti-mouse CD11c antibody (clone N418); APC-Cy7 labeled anti-mouse XCR1 antibody (clone ZET); APC-labeled anti-mouse CD80 antibody (clone 16-10A1); APC-labeled anti-mouse CD86 antibody (clone GL-1); Purified CD16/32 antibody (clone 93).
- Example 2-1 Comparative Example 2-1 and Comparative Example 4-1 were mixed with 10% by mass sucrose and 10mM phosphorus so that the fluorescent (fluorescein) labeled peptide concentration was about 0.3mg/mL.
- the diluted solution of each pharmaceutical composition was administered to the right dorsal region of BALB/c mice.
- the peptides were subcutaneously administered at 60 ⁇ g each.
- 50 ⁇ g of CpG oligo DNA 1668 (Ajinomoto BioPharma Service Gene Design) dissolved in PBS was administered as an adjuvant.
- the regional lymph node (right inguinal lymph node) at the administration site was collected, and after grinding the lymph node using a slide glass, the cells were suspended in RPMI1640 medium. At this time, cells from two animals per group were pooled. After centrifugation (400 ⁇ g, 5 minutes, 4° C.), the supernatant was removed, washed twice with RPMI1640 medium, and then suspended in RPMI1640 medium containing 10 v/v% FBS. The number of cells was measured using a hemocytometer, and the cell concentration was adjusted to 1.2 x 10 7 cells/mL.
- 50 ⁇ L of the cell suspension was added to a 96-well V-bottom microplate (Nunc, Thermo Fisher Scientific) at 6 ⁇ 10 5 cells per well.
- the cell suspension was centrifuged (2000 rpm, 2 minutes, 4°C), the supernatant was removed, and the cell suspension was washed twice with 200 ⁇ L of staining buffer (PBS containing 0.5 v/v% FBS) and suspended. . After centrifugation and removal of the supernatant, 50 ⁇ L of a solution containing anti-mouse CD16/CD32 antibody diluted 50 times was added, and the mixture was left standing in the dark at 4° C. for 10 minutes.
- the cells were further washed twice with 200 ⁇ L of staining buffer.
- the usage concentration recommended by each antibody manufacturer add 50 ⁇ L of a solution containing the first, second, and third antibodies in the combinations listed in Table 3 above, mix, and store in the dark at 4°C. It was left standing for 15 minutes.
- the cells were further washed twice with 200 ⁇ L of staining buffer.
- the suspension was resuspended in 200 ⁇ L of staining buffer and transferred to a round bottom polystyrene tube (BD Bioscience). Cells were analyzed using a flow cytometer FACS Canto II (BD Biosciences) and the attached analysis software (FACSDiva).
- DC Dendritic cells
- Mph macrophages
- cDC1 standard type 1 DC
- FITC fluorescein
- CD80 and CD86 The expression of costimulatory molecules CD80 and CD86 was analyzed for FITC-positive cells in each cell type.
- the FACS analysis method is shown in FIGS. 7A to 7F. The analysis results are shown in Tables 4-1 to 4-3.
- peptide + CD80 + DC means “peptide-incorporated dendritic cells (DC) that express CD80
- peptide + CD86 + DC means “peptide-incorporated dendritic cells (DC) that express CD86.
- Peptide+DC means "peptide-incorporating dendritic cells (DC).
- Example 1-2 Using the pharmaceutical compositions of Example 2-2, Comparative Example 2-2, and Comparative Example 4-2, 10 mass% sucrose and 10 mM phosphorus were added so that the fluorescent (fluorescein) labeled peptide concentration was about 0.2 mg/mL.
- the test was carried out in the same manner as in Test Example 1-1, except that the peptides were diluted with an acid buffer pH 7.4 and administered subcutaneously at 40 ⁇ g each.
- the analysis results are shown in Tables 5-1 to 5-3.
- Comparative Example 2-2 Similarly to Test Example 1-1, in the mouse group to which the pharmaceutical composition of Example 2-2 was administered, the pharmaceutical compositions of Comparative Example 2-2 and Comparative Example 4-2 (hyaluronic acid derivative before fractionation) were administered. Higher expression of costimulatory molecules (CD80 and CD86) in macrophages and DCs (especially cDC1) was confirmed than in the mouse group. Comparative Example 4-2 had relatively high expression of CD80 and CD86 on cDC1, but low expression of CD80 and CD86 on macrophages, and was inferior to the pharmaceutical composition of Example 2-2.
- Test Example 1-1 was carried out in the same manner as in Test Example 1-1, except that the pharmaceutical composition of Example 2-3 was used and 40 ⁇ g of each peptide was administered subcutaneously. The analysis results are shown in Tables 6-1 to 6-3.
- the pharmaceutical composition of this embodiment it is possible to stably and efficiently deliver medicinal ingredients such as peptides to immune cells (especially cDC1 and macrophages), and furthermore, to deliver the medicinal ingredients such as peptides to immune cells. It is presumed that the uptake of the drug can be improved, promoted, or enhanced, and at the same time, the activity of immune cells (particularly, the expression of costimulatory molecules CD80 and CD86 in cDC1) is improved, promoted, maintained, or enhanced.
- gp100RP2RP1_6Y peptide manufactured by Biologica and having the following amino acid sequence was used.
- the lyophilized hyaluronic acid derivative obtained in Synthesis Example 1-1 was weighed, water for injection was added to the concentration shown in Table 7 below, and the mixture was stirred overnight to ensure sufficient dissolution. I let it happen. Meanwhile, in another container, the peptide was dissolved in DMSO to a concentration of 50 mg/mL. After confirming the dissolution, a 1 mol/L sodium hydroxide aqueous solution (manufactured by Fuji Film Wako Co., Ltd.) was added to the hyaluronic acid derivative aqueous solution according to Table 7 below.
- the hyaluronic acid derivative aqueous solution and the peptide dimethyl sulfoxide solution are mixed in the volume ratio shown in Table 7 below, and the mixture is stirred at room temperature (approximately 25°C) for 24 hours to form the peptide-hyaluron.
- a composition containing an acid derivative complex was obtained.
- the pH of the solution was also measured.
- solid purified sucrose (manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385) was added to the composition containing the peptide-hyaluronic acid derivative complex so that the amount of sucrose was 10% by mass based on the total mass of the sucrose and the composition containing the peptide-hyaluronic acid derivative complex.
- the mixture was added and stirred at room temperature (approximately 25° C.) for 1 hour or more.
- the peptide was diluted with 25mM phosphate buffer pH 7.4 containing 10% by mass sucrose so that the theoretical concentration of the peptide was 0.3mg/mL, and 0.2 ⁇ m PES (polyether sulfone) (manufactured by Pall, Acrodisc) was added.
- the mixture was sterilized and filtered using a syringe filter (25 mm ⁇ ) to obtain a pharmaceutical composition containing a peptide-hyaluronic acid derivative complex.
- the peptide concentration in the pharmaceutical composition containing the peptide-hyaluronic acid derivative complex was determined in the same manner as in Example 2-1 above, and the results are shown in Table 7 above.
- a composition containing a peptide-hyaluronic acid derivative complex produced by the method shown below was measured by dynamic light scattering (DLS) under the following conditions to obtain the z-average particle diameter. The results are shown in Table 7 above.
- DLS device Otsuka Electronics
- ELSZ2000 Cell Micro particle size cell Temperature: 25°C Concentration of peptide-hyaluronic acid derivative complex: 4.95mg/mL
- the particle size of the complex of a medicinal ingredient such as a peptide and a hyaluronic acid derivative can be maintained within a certain range. It has become clear that the stability of the complex in solution can be improved by keeping the temperature within a certain range.
- peptide As the peptide, a peptide manufactured by Biologica and having the following amino acid sequence was used.
- the above hyaluronic acid derivative aqueous solution and peptide DMSO solution are mixed in a volume ratio according to Table 8 below, and stirred at room temperature (approximately 25°C) for 24 hours to form peptide-hyaluronic acid.
- a composition containing a derivative complex was obtained.
- the pH of the solution was also measured.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385 is added to the composition containing the peptide-hyaluronic acid derivative complex and the total mass of sucrose to give a concentration of 10% by mass of sucrose.
- Test Examples 3-1 to 3-6 The mixing ratio for maintaining an appropriate particle size distribution was verified in the same manner as in Test Examples 2-1 to 2-8, except that ethanol was used instead of DMSO for the oil phase. Measurement was performed by gel permeation chromatography, and the area ratio A1/A2 was calculated in the same manner as in Example 1-1, and the results are shown in Table 10 below. The obtained gel permeation chromatogram is shown in FIG.
- hyaluronic acid powder with a weight average molecular weight Mw (absolute molecular weight) of 980 kDa was dissolved in water for injection to a concentration of 10 mg/mL, and then diluted with water for injection to a concentration of 5 mg/mL. I got it. Meanwhile, in another container, the antigen peptide was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/mL to obtain a DMSO solution of the antigen peptide.
- DMSO dimethyl sulfoxide
- the antigen peptide a peptide manufactured by Biologica and having the following amino acid sequence was used.
- the above hyaluronic acid aqueous solution and antigen peptide DMSO solution are mixed at a volume ratio of 100:1, and stirred at room temperature (approximately 25°C) for 24 hours to form antigen peptide-hyaluronic acid.
- the resulting mixed solution was cloudy, as confirmed by the photograph labeled "hyaluronic acid (980 kDa)" in FIG. 11, indicating that the antigenic peptide was not dissolved and a large amount of peptide was not encapsulated. was suggested.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 was added to the sucrose in an amount of 10% by mass based on the total mass of the composition containing the peptide-hyaluronic acid complex and sucrose. and stirred at room temperature for over 1 hour.
- the peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of approximately 0.30mg/mL, and 0.2 ⁇ m PES (polyether Sulfone) (manufactured by Pall, Acrodisc syringe filter, 25 mm ⁇ ) to obtain a pharmaceutical composition containing a peptide-hyaluronic acid complex.
- the peptide content in the pharmaceutical composition was determined by high performance liquid chromatography in the same manner as in Example 2-1. As a result, the peptide content in the pharmaceutical composition was below the detection limit.
- Hyaluronic acid powder having a weight average molecular weight Mw (absolute molecular weight) of 650 kDa was dissolved in water for injection to a concentration of 10 mg/mL, and diluted with water for injection to a concentration of 5 mg/mL to obtain an aqueous hyaluronic acid solution.
- the antigen peptide was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/mL to obtain a DMSO solution of the antigen peptide.
- DMSO dimethyl sulfoxide
- As the antigen peptide a peptide manufactured by Biologica and having the following amino acid sequence was used.
- the above hyaluronic acid aqueous solution and antigen peptide DMSO solution are mixed at a volume ratio of 100:1, and stirred at room temperature (approximately 25°C) for 24 hours to form antigen peptide-hyaluronic acid.
- the resulting mixed solution was cloudy, as confirmed by the photograph labeled "hyaluronic acid (640 kDa)" in Figure 11, suggesting that the antigen peptide was not dissolved and the peptide was not encapsulated. It was done.
- solid purified sucrose manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385
- product number 198-18385 was added to the sucrose in an amount of 10% by mass based on the total mass of the composition containing the peptide-hyaluronic acid complex and sucrose. and stirred at room temperature (approximately 25°C) for 1 hour or more.
- the peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of approximately 0.30mg/mL, and 0.2 ⁇ m PES (polyether Sulfone) (manufactured by Pall, Acrodisc syringe filter, 25 mm ⁇ ) to obtain a pharmaceutical composition containing a peptide-hyaluronic acid complex.
- the content of the peptide in the pharmaceutical composition was determined by high performance liquid chromatography in the same manner as in Example 2-1. As a result, the peptide content in the pharmaceutical composition was below the detection limit.
- Example 6-1 A hyaluronic acid derivative was synthesized in the same manner as in Synthesis Example 1-1. It was used as is without fractionation. The obtained freeze-dried product of the hyaluronic acid derivative was weighed, water for injection was added to give a concentration of 5 mg/mL, and the mixture was stirred overnight at room temperature to fully dissolve. The above hyaluronic acid derivative aqueous solution was diluted five times with water for injection to a concentration of 1 mg/mL, and then measured by gel permeation chromatography. FIG. 13 is the gel permeation chromatogram obtained. In addition, the particle size distribution of the hyaluronic acid derivative was evaluated in the same manner as in Example 1-1, and the results are shown in Table 11 below.
- Example 6-1 A hyaluronic acid derivative synthesized in the same manner as in Synthesis Example 1-1 was fractionated in the same manner as in Example 1-1 to obtain a hyaluronic acid derivative.
- the hyaluronic acid derivative of the obtained dialysis fluid was measured by gel permeation chromatography.
- FIG. 14 is the gel permeation chromatogram obtained.
- the particle size distribution of the hyaluronic acid derivative was evaluated in the same manner as in Example 1-1, and the results are shown in Table 11 below.
- Example 6-1-1 The concentration of the hyaluronic acid derivative in the hyaluronic acid derivative aqueous solution obtained in Example 6-1 was calculated from the area value of the hyaluronic acid derivative of known concentration to be 4.28 mg/mL.
- PBS pH 7.4 (product manufactured by Fujifilm Wako Pure Chemical Industries, Ltd.) so that the concentration of ovalbumin (OVA, low endotoxin product manufactured by Fujifilm Wako Pure Chemical Industries, Ltd.) is 5 mg/mL. No. 166-23555) to obtain an OVA/PBS solution.
- sucrose was added as a solid to make a 10% sucrose isotonic solution, and the mixture was thoroughly stirred at room temperature until a clear solution was obtained.
- the OVA concentration in the formulation was 0.15 mg/mL
- the hyaluronic acid derivative concentration was 3.0 mg/mL
- a 10% sucrose solution containing 10 mM PB (pH 7.4) was used.
- the mixture was incubated at 75° C. for 1 hour to obtain a composition containing an OVA-hyaluronic acid derivative complex.
- Example 6-1-1 The composition containing the OVA-hyaluronic acid derivative complex prepared in Example 6-1-1 and Comparative Example 6-1-1 was administered to BALB/c mice on day 0 so that OVA was 20 ⁇ g. , was administered subcutaneously on the 7th day, and serum was collected on the 10th day.
- OVA-hyaluronic acid derivative complex 50 ⁇ g of CpG1826 (manufacturer: INVIVOGEN, product code: tlrl-1826-1), which is an adjuvant, was administered subcutaneously.
- the antibody titer against OVA was determined using an ELISA kit (Chondrex, Mouse Anti-OVA IgG Antibody Assay Kit, Mouse Anti-OVA IgG1 Antibody Assay Kit, Mouse Anti-OVA IgG2). Measurement using Antibody Assay Kit) according to the attached protocol. did.
- the dilution factors of serum were 10, 50, 250, 1250, 6250, and 31250.
- FIG. 15 shows the IgG antibody titer (OD) calculated using the Mouse Anti-OVA IgG Antibody Assay Kit.
- Figure 16 shows the IgG1 antibody titer (OD) calculated using the Mouse Anti-OVA IgG1 Antibody Assay Kit.
- nti-OVA FIG. 17 shows the IgG2a antibody titer (OD) calculated using the IgG2a Antibody Assay Kit.
- Each antibody titer at a dilution factor of 10 is shown in FIG. 18A (IgG antibody titer), FIG. 18B (IgG1 antibody titer), and FIG. 18C (IgG2a antibody titer).
- the composition containing the OVA-hyaluronic acid derivative complex of Example 6-1-1 is more effective.
- a higher antibody titer was obtained than the composition containing the OVA-hyaluronic acid derivative complex of Comparative Example 6-1-1.
- Example 6-1-1 induces not only humoral immunity but also cell-mediated immunity, and is expected to induce a stronger immune response as an infectious disease vaccine.
- the pharmaceutical composition of this embodiment not only peptides but also the medicinal components of protein antigens can be protected from heating conditions, and they can be applied to immune cells (especially cDC1 and macrophages) while maintaining an appropriate three-dimensional structure. It can be delivered stably and efficiently, and furthermore, it can improve, promote, or enhance the uptake of the medicinal ingredient into immune cells, and at the same time, it can improve the activity of immune cells (in particular, the costimulatory molecules CD80 and CD86 in cDC1). It is presumed that the expression of these substances was improved, promoted, maintained, or enhanced. It is inferred that the antigen directed against the epitope was produced by promoting the activation of helper T cells and further promoting the activation and expansion of B cells.
- hyaluronic acid derivatives of the first to third embodiments it is possible to provide vaccines against foreign antigens (mainly vaccines for infectious diseases) that are superior to those using conventional hyaluronic acid derivatives. At the same time, it is considered possible to provide pharmaceuticals containing excellent therapeutic antibody-inducing antigens.
- a long-chain peptide antigen-loaded hyaluronic acid derivative cancer vaccine was prepared by conjugating a long-chain peptide antigen containing the CD8 epitope of mutant ERK2 antigen (mERK2) with various hyaluronic acid derivatives, and its therapeutic efficacy was investigated.
- mice Female BALB/c mice (CD90.2 positive) aged 6 to 8 weeks were purchased from Japan SLC. Both mice were bred at the Mie University Advanced Science Research Support Center Animal Experiment Facility. The animal experiment protocol was approved by the Ethics Committee of Mie University School of Medicine.
- a long-chain peptide antigen-loaded hyaluronic acid derivative that is a complex of the long-chain peptide antigen and various hyaluronic acid derivatives is sometimes referred to as a "long-chain peptide antigen-loaded HA nanogel cancer vaccine" or "HA nanogel cancer vaccine.” .
- Example 7-1 The hyaluronic acid derivative obtained in Comparative Example 6-1 was fractionated as follows. A lyophilized hyaluronic acid derivative (10k HA-C 6 -Chol-41.3%) was dissolved in water for injection at 5 mg/mL, and a dialysis cassette (Flotalizer G2, MWCO: 300,000, Ieda Boeki Co., Ltd.) was added. The cells were transferred to a 10-mM phosphate buffer solution (pH 7.4), and the hyaluronic acid derivatives in the resulting dialyzed solution were measured by gel permeation chromatography. Figure 19 is the gel permeation chromatogram obtained. The particle size distribution of the hyaluronic acid derivative was evaluated in the same manner as in Example 1-1. The results are shown in Table 12.
- Example 7-1-1 The concentration of the hyaluronic acid derivative in the aqueous hyaluronic acid derivative solution obtained in Example 7-1 was calculated from the area value of the hyaluronic acid derivative at a known concentration to be 3.8 mg/mL. Meanwhile, in another container, the antigen peptide was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/mL to obtain a DMSO solution of the antigen peptide. As the antigen peptide, a peptide manufactured by Biologica and having the following amino acid sequence was used.
- DMSO dimethyl sulfoxide
- the above hyaluronic acid derivative aqueous solution and DMSO solution of antigen peptide were mixed at a ratio of 10 parts by mass of peptide to 100 parts by mass of hyaluronic acid derivative, and the mixture was incubated at room temperature (approximately 25°C) for 2 hours.
- a clear solution was obtained, as confirmed by the photograph labeled "hyaluronic acid derivative" in FIG. 11. This confirmed that the peptide was indeed encapsulated.
- solid purified sucrose (manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only, product number 198-18385) is converted to 10% by mass of sucrose based on the total mass of the composition containing the peptide-hyaluronic acid derivative complex and sucrose. and stirred at room temperature (approximately 25° C.) for more than 1 hour.
- the peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% sucrose to a concentration of approximately 0.30mg/mL, and 0.2 ⁇ m PES (polyether).
- the mixture was sterilized and filtered through a peptide-hyaluronic acid derivative complex (Acrodisc syringe filter, 25 mm ⁇ , manufactured by Pall) to obtain a pharmaceutical composition containing a peptide-hyaluronic acid derivative complex.
- the peptide content in the pharmaceutical composition was determined by high performance liquid chromatography in the same manner as in Example 2-1. As a result, the content of the peptide was 0.383 parts by mass based on 100 parts by mass of the pharmaceutical composition.
- the pharmaceutical composition containing the peptide-hyaluronic acid derivative complex was diluted with a 10 mM phosphate buffer pH 7.4 containing 10% by mass sucrose to give a peptide concentration of 250 ⁇ g/mL to obtain a pharmaceutical composition for administration.
- Ta The pharmaceutical composition containing the above peptide-hyaluronic acid derivative complex was measured by gel permeation chromatography using the method described in Example 1-1 under the measurement conditions shown below.
- FIG. 20 is the gel permeation chromatogram obtained.
- Example 7-1-1 The peptide used in Example 7-1-1 was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 50 mg/mL. The peptide was diluted with 10mM phosphate buffer pH 7.4 containing 10% by mass sucrose to a concentration of 0.25mg/mL.
- DMSO dimethyl sulfoxide
- Tumor Growth Test Mouse fibrosarcoma CMS5a cell line was cultured in RPMI1640 medium containing 10% FBS using T75 culture flasks (Corning). The cultured cells were detached using PBS containing 0.5% trypsin and suspended in RPMI1640 medium containing 10% FBS. The suspension was centrifuged (400 ⁇ g, 5 minutes, 4° C.) and the supernatant was removed. Thereafter, the cells were washed twice with RPMI1640 medium and suspended in RPMI1640 medium at a concentration of 1 ⁇ 10 6 cells/100 ⁇ L. A dose of 100 ⁇ L/individual was subcutaneously implanted in the right anterior dorsal region of BALB/c mice (5 mice per group).
- the long-chain peptide antigen-loaded HA nanogel cancer vaccine When administering the long-chain peptide antigen-loaded HA nanogel cancer vaccine, 50 ⁇ g of the long-chain peptide antigen-loaded HA nanogel cancer vaccine was dissolved in PBS 7, 10, 13, and 16 days after tumor transplantation. It was subcutaneously administered to the right back of the mouse together with 50 ⁇ g of CpG oligo DNA (Ajinomoto BioPharma Service Gene Design). Thereafter, the tumor area was measured over time. Also when administering the long chain peptide antigen of Comparative Example 7-1-1, 50 ⁇ g of the long chain peptide was added to 50 ⁇ g of CpG oligo DNA dissolved in PBS 7 days, 10 days, 13 days, and 16 days after tumor transplantation. (Ajinomoto BioPharma Service Gene Design) was subcutaneously administered to the right hind region of mice. Thereafter, the tumor area was measured over time.
- Figure 23 shows the tumor area values (average values) over time.
- the aggregate size that is superior in the activation of costimulatory molecules is larger.
- the medicinal components of peptides and proteins can be stably and efficiently delivered to immune cells (in particular, cDC1 and macrophages), and furthermore, the medicinal components can be delivered to immune cells. It is presumed that the uptake can be improved, promoted, or enhanced, and at the same time, the activity of immune cells (in particular, the expression of costimulatory molecules CD80 and CD86 in cDC1) is improved, promoted, maintained, or enhanced.
- a calibration curve was created using polyacrylic acid as a standard substance, and Synthesis Examples 1-1 to 1-3, Examples 1-1 to 1-3, 6-1, 7-1, and Comparative Examples 1-1 to The molecular weight of each hyaluronic acid derivative obtained in 1-3 and 6-1 was calculated based on polyacrylic acid.
- the polyacrylic acid standard substances include PSS-Paa2k (2kDa), 4k (4kDa), 8k (8kDa), 18k (18kDa), 40k (40kDa), 150k (150kDa) (manufactured by PSS Polymer Standard service GmbH (polyacrylic Sodium chloride)) was used. The results are shown in Table 14.
- the fractionation method is not limited to the fractionation method using a dialysis membrane.
- the hyaluronic acid derivative obtained in Synthesis Example 1-1 is dissolved in water for injection at 20 mg/mL, and fractionated by gel permeation chromatography under the conditions shown below. For example, aliquots are taken every 0.1 minute.
- the hyaluronic acid derivative of the present embodiment it is possible to provide a hyaluronic acid derivative that is excellent in delivery to immune cells in lymph nodes and activation ability of the immune cells when formulated with a medicinal ingredient.
- the pharmaceutical composition of this embodiment contains the hyaluronic acid derivative and has excellent delivery properties to immune cells in lymph nodes and ability to activate the immune cells.
- the method for producing a pharmaceutical composition of the present embodiment uses the hyaluronic acid derivative, and a pharmaceutical composition having excellent delivery properties to immune cells in lymph nodes and ability to activate the immune cells can be obtained.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Biochemistry (AREA)
- Polymers & Plastics (AREA)
- Materials Engineering (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Dermatology (AREA)
- Inorganic Chemistry (AREA)
- Pulmonology (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
本願は、2022年5月31日に、日本に出願された特願2022-088995号に基づき優先権を主張し、その内容をここに援用する。
(1) ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、以下に示す方法により算出される面積A1とA2の比A1/A2が0.90以上である、ヒアルロン酸誘導体。
(i)標準物質である50kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KbからベースラインBへ引いた垂線と、前記ヒアルロン酸誘導体のクロマトグラムとの交点をUbとする;
(ii)前記ヒアルロン酸誘導体のクロマトグラムで前記Ubから終点までの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA2とし、前記ヒアルロン酸誘導体のクロマトグラムで始点から前記Ubまでの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA1とする。
(2) ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、以下に示す方法により算出される距離DaとDbの比Da/Dbが0.00超1.20以下である、(1)に記載のヒアルロン酸誘導体。
(i)標準物質である150kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KaからベースラインBへ垂線を引き、ベースラインとの交点をBa、前記屈折率強度極大点Kaと前記Baの間の長さをLaとする;
(ii)屈折率強度がLa/20となるクロマトグラム上の2点のうち、溶出時間が早いほうを点R1とし、溶出時間が遅いほうを点S1とする;
(iv)前記点R1と前記点S1を結んだ直線D1と、前記屈折率強度極大点KaからベースラインBへ引いた垂線との交点をTaとし、前記屈折率強度極大点KbからベースラインBへ引いた垂線と前記直線D1との交点をTbとする;
(v)前記点R1と前記Taの距離をDa、前記Taと前記Tbの距離をDbとする。
(3) 前記面積A1とA2の比A1/A2が1.70以上である、(1)又は(2)に記載のヒアルロン酸誘導体。
(4) ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから算出される、標準物質である50kDaのポリアクリル酸の屈折率強度極大点における保持時間Prに対する、ヒアルロン酸誘導体の屈折率強度極大点における保持時間Ptの比Pt/Prが0.5以上1.0未満である、ヒアルロン酸誘導体。
(5) ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、各分子量が2kDa、4kDa、8kDa、18kDa、40kDa、および150kDaであるポリアクリル酸を標準物質として作成した検量線に基づき算出されるヒアルロン酸誘導体の重量平均分子量が11万以上50万未満である、ヒアルロン酸誘導体。
(6) 前記ヒアルロン酸誘導体が、下記一般式(I)で表される繰り返し単位を1以上有する、(1)~(5)のいずれか一つに記載のヒアルロン酸誘導体。

Zは、直接結合、又は2個以上30個以下の任意のアミノ酸残基からなるペプチドリンカーを表し;
X1は、以下の式:
-NRb-R、
-NRb-COO-R、
-NRb-CO-R、
-NRb-CO-NRc-R、
-COO-R、
-O-COO-R、
-S-R、
-CO-Ya-S-R、
-O-CO-Yb-S-R、
-NRb-CO-Yb-S-R、及び
-S-S-R、
で表される基からなる群より選択される基であり;
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、ここで当該基のアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよく;
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Rは、ステリル基であり;
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-であり、ここで、当該アルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよく;
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Yaは、C1-5アルキレンであり;
Ybは、C2-8アルキレン又はC2-8アルケニレンであり;
mは、1以上100以下の整数である。)
(8) 前記ヒアルロン酸誘導体を構成する二糖の繰り返し単位に対する、ステリル基の導入率が、30%以上60%以下である、(1)~(7)のいずれか一つに記載のヒアルロン酸誘導体。
(9) 前記ヒアルロン酸誘導体の重量平均分子量(絶対分子量)が4,000以上1,000,000以下である、(1)~(8)のいずれか一つに記載のヒアルロン酸誘導体。
(10) 前記ヒアルロン酸誘導体の重量平均分子量(絶対分子量)が5000以上25,000以下である、(9)に記載のヒアルロン酸誘導体。
(11) (1)~(10)のいずれか一つに記載のヒアルロン酸誘導体と、薬効成分と、を含有する、医薬組成物。
(12) がん、感染症、及び免疫疾患からなる群より選ばれる1種以上の疾患の予防又は治療用である、(11)に記載の医薬組成物。
(13) 前記薬効成分として、がん抗原、感染症由来抗原、及び免疫疾患における自己抗原からなる群から選択される少なくとも一つを含み、且つ、アジュバントを更に含む、(11)または(12)に記載の医薬組成物。
(14) 前記薬効成分として、がん抗原又は感染症由来抗原を含み、且つ、アジュバントを更に含む、(11)~(13)のいずれか一つに記載の医薬組成物。
(15) (1)~(10)のいずれか一つに記載のヒアルロン酸誘導体と、薬効成分と、を含有する、医薬組成物の製造方法であって、
前記薬効成分を有機溶媒又は有機溶媒含有水に溶解して、前記薬効成分を含有する油相を調製する調製工程と、
前記油相と前記ヒアルロン酸誘導体を含有する水相との混合比が容量比で20:100~0.01:100となるように混合する混合工程と、
を含む、医薬組成物の製造方法。
(16) 前記ヒアルロン酸誘導体を含有する水相のpHが6.00以上11.00以下である、(15)に記載の医薬組成物の製造方法。
本発明の第1の実施形態のヒアルロン酸誘導体は、ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められる図1に示すクロマトグラムから、以下に示す方法により算出される面積A1とA2の比A1/A2が0.90以上である。
(i)標準物質である50kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KbからベースラインBへ引いた垂線と、前記ヒアルロン酸誘導体のクロマトグラムとの交点をUb、前記屈折率強度極大点KbからベースラインBへ引いた垂線とベースラインとの交点をBbとする;
(ii)前記ヒアルロン酸誘導体のクロマトグラムで前記Ubから終点まで曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA2とし、前記ヒアルロン酸誘導体のクロマトグラムで始点から前記Ubまでの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA1とする。
(i)標準物質である150kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KaからベースラインBへ垂線を引き、ベースラインとの交点をBa、前記屈折率強度極大点Kaと前記Baの間の長さをLaとする;
(ii)屈折率強度がLa/20となるクロマトグラム上の2点のうち、溶出時間が早いほうを点R1とし、溶出時間が遅いほうを点S1とする;
(iii)標準物質である50kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KbからベースラインBへ引いた垂線とベースラインとの交点をBbとする;
(iv)前記点R1と前記点S1を結んだ直線D1と、前記屈折率強度極大点KaからベースラインBへ引いた垂線との交点をTaとし、前記屈折率強度極大点KbからベースラインBへ引いた垂線と前記直線D1との交点をTbとする;
(v)前記点R1と前記Taの距離をDa、前記Taと前記Tbの距離をDbとする。
(ii)において、屈折率強度がLa/20となるクロマトグラム上の点が3点以上ある場合、始点に最も近い交点をR1、終点に最も近い交点をS1とする。
すなわち、マクロファージ又はDC(好ましくはcDC1)への抗原送達性を高めることは、がんの予防又は治療、感染症の予防又は治療の効果を向上させることになる。
すなわち、マクロファージ又はDC(好ましくはcDC1)への抗原送達性を高めることは、感染症の予防又は治療、免疫疾患の治療の効果を向上させることになる。
また、本実施形態のヒアルロン酸誘導体と薬効成分とを含む、後述する医薬組成物は、免疫細胞の活性化用組成物、DC(好ましくはcDC1)における共刺激分子の発現向上用組成物、発現促進用組成物又は発現維持用組成物ということもできる。
前記ゲル浸透クロマトグラフィー測定において標準物質として使用されるポリアクリル酸は、ポリアクリル酸ナトリウムであることが好ましく、具体的には、Pоlymer Standards Service-USA社製、ORDER No.PSS-Paaシリーズ(ポリアクリル酸ナトリウム)であることがより好ましい。
1mg/mLのヒアルロン酸誘導体水溶液、2mg/mLのポリアクリル酸標準物質水溶液を調製し、以下に示す条件にてゲル浸透クロマトグラフィーによる測定を行う。
(測定条件)
装置:高速GPC(ゲル浸透クロマトグラフィー)装置
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率検出器)
温度:30℃
本装置においては、0.00167分おきに屈折率強度を算出する。
ここで、各面積値はGPCワークステーションEcoSEC Elite-WSの解析アプリケーションを用いて算出される。
各面積値を算出する際に、ゲル浸透クロマトグラフィーに使用した展開溶媒などに起因するピークや、使用したカラムや装置に起因するベースラインの揺らぎによる擬似ピークは除く。
さらに、本実施形態のヒアルロン酸誘導体のゲル浸透クロマトグラムにおいて、示差屈折率計を用いて得られた屈折率強度と溶出時間で表されるクロマトグラムが左右非対称であっても対称であってもよい。
本明細書において使用される「ステリル基」という用語は、ステロイド骨格を有する基であれば特に制限されない。ここでステロイドとしては、具体的には、コレステロール、コレスタノール、カンペスタノール、エルゴスタノール、スチグマスタノール、コプロスタノール、スチグマステロール、シトステロール、ラノステロール、エルゴステロール、シミアレノール、胆汁酸、テストステロン、エストラジオール、プロゲストロン、コルチゾール、コルチゾン、アルドステロン、コルチコステロン、デオキシコルチステロン等が挙げられる。ステリル基としては、コレステリル基、スチグマステリル基、ラノステリル基、エルゴステリル基等が挙げられ、中でも、コレステリル基(特に、コレスタ-5-エン-3β-イル基)が好ましい。
なお、前記ゲル浸透クロマトグラフィー測定により求められるクロマトグラムにおいて、ヒアルロン酸誘導体の屈折率強度極大点(ピークトップ)が複数ある場合には、屈折率強度が最大となる屈折率強度極大点(ピークトップ)における溶出時間を、前記ヒアルロン酸誘導体の屈折率強度極大点(ピークトップ)の保持時間Ptとして採用する。
また、前記ゲル浸透クロマトグラフィー測定において標準物質として使用されるポリアクリル酸は、ポリアクリル酸ナトリウムであることが好ましく、具体的には、Pоlymer Standards Service-USA社製、ORDER No.PSS-Paaシリーズ(ポリアクリル酸ナトリウム)であることがより好ましい。
一方、ヒアルロン酸誘導体の重量平均分子量(ポリアクリル酸換算)が前記上限値を超える場合、粘性が増大し、製剤として使用しにくい場合がある。

Zは、直接結合、又は2個以上30個以下の任意のアミノ酸残基からなるペプチドリンカーを表し;
X1は、以下の式:
-NRb-R、
-NRb-COO-R、
-NRb-CO-R、
-NRb-CO-NRc-R、
-COO-R、
-O-COO-R、
-S-R、
-CO-Ya-S-R、
-O-CO-Yb-S-R、
-NRb-CO-Yb-S-R、及び
-S-S-R、
で表される基からなる群より選択される基であり;
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、ここで当該基のアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよく;
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Rは、ステリル基であり;
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-であり、ここで、当該アルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよく;
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Yaは、C1-5アルキレンであり;
Ybは、C2-8アルキレン又はC2-8アルケニレンであり;
mは、1以上100以下の整数である。)

Xは、-NRa-Y-NRb-COO-Rで表される疎水性基であり;
Ra及びRbは、それぞれ独立に、水素原子及びC1-6アルキルからなる群より選択され;
Rは、ステリル基であり;
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-であり、
mは、1以上100以下の整数である。)
一般式(I)中の基「-Z-N(Ra)Y-X1」は、以下の式:
-NH-(CH2)mz-NH-R;
-NH-(CH2)mz-NH-COO-R;
-NH-(CH2CH2O)m-CH2CH2-NH-COO-R;
-NH-(CH2)mz-COO-R;
-NH-(CH2CH2O)m-CH2CH2-COO-R、
-NH-(CH2)mz-O-COO-R;
-NH-(CH2CH2O)m-CH2CH2-O-COO-R、
-NH-(CH2)mz-S-R;
-NH-(CH2CH2O)m-CH2CH2-S-R;
-NH-(CH2)mz-O-CO-CH(R8)-CH2-S-R;
-NH-(CH2)mz-NHCO-CH(R8)-CH2-S-R;
-NH-(CH2CH2O)m-CH2CH2-NHCO-CH(R8)-CH2-S-R;
-NH-(CH2CH2O)m-CH2CH2-O-CO-CH(R8)-CH2-S-R;
-NH-(CH2)mz-S-S-R;及び
-Z-NRa-Y-NRb-COO-R
(ここで、mzは、2以上30以下の整数であり、R8は、水素原子又はメチル基であり、R及びmは、本明細書で既に定義したとおりである。)
で表される基からなる群より選択されることが好ましく、
-NH-(CH2)mz-NH-COO-R;
-NH-(CH2CH2O)m-CH2CH2-NH-COO-R;及び
-NH-(CH2)mz-S-S-R
(ここで、mz、R、及びmは、本明細書で既に定義したとおりである。)
からなる群より選択されることがより好ましい。
一般式(I)において、Zは直接結合であることが好ましい。
別の態様において、Zがペプチドリンカーである場合に、X1は-NRb-COO-Rであることが好ましい。
さらに、別の態様において、Zは、-NH-[CH(-Za)-CONH]n-1-CH(-Za)-CO-で表されるペプチドリンカーであってもよく、ここで、nは2以上30以下の整数であり、Zaは、それぞれ独立に、H2N-CH(-Za)-COOHとして表されるα-アミノ酸中の置換基を表す。当該ペプチドリンカーは、N末端にてグルクロン酸部分のカルボキシ基に結合し、C末端にて基-N(-Ra)-Y-X1に結合する。当該ペプチドリンカーのアミノ酸残基として利用できるアミノ酸の例としては、α-アミノ酸、例えばアラニン、アルギニン、アスパラギン(Asn)、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン(Gly)、ヒスチジン、イソロイシン、ロイシン(Leu)、リジン、メチオニン、フェニルアラニン(Phe)、プロリン、セリン、スレオニン、トリプトファン、チロシン、バリンといった天然型(L型)のアミノ酸、それらのD体等が挙げられ、合成されたアミノ酸を含む全てのα-アミノ酸を用いることができる。すなわち、Zaとしては、例えば、-CH3、H2NC(NH)NH(CH2)3-、H2NCOCH2-等が挙げられる。また、n個のZは、同一でも異なっていてもよい。nは、2以上30以下の整数であるが、2以上10以下が好ましく、2以上4以下がより好ましい。ペプチドリンカーの好ましい例としては、例えば、-Gly-Phe-Leu-Gly-、-Asn-Phe-Phe-、-Phe-Phe-、Phe-Gly-等が挙げられる。
一般式(I)において、Yは-(CH2)n1-及び-(CH2CH2O)m1-CH2CH2-(ここで、n1は、2以上20以下の整数であり、2以上15以下の整数が好ましく、2以上12以下の整数がより好ましく、2以上6以下の整数がさらに好ましい。m1は、1以上4以下の整数である)からなる群より選択される基が好ましい。具体的には、-(CH2)2-、-(CH2)6-、-(CH2)8-、-(CH2)12-、又は、-(CH2CH2O)2-CH2CH2-が好ましい。また、純水中乃至低塩濃度下では高い溶解性を実現させつつ、生理食塩濃度下では高い沈殿形成能を示させるという観点からは、Yは-(CH2)2-、-(CH2)6-、-(CH2)8-及び-(CH2)12-からなる群より選択される基が好ましく、-(CH2)6-がより好ましい。
Yaとしては、-CH2-又は-CH2-CH2-が好ましい。
Ybとしては、-CH2-CH2-、-CH(CH3)CH2-、2-ブテン-1,4-ジイル、ヘプタ-2,4-ジエン-1,6-ジイル又はオクタ-2,4,6-トリエン-1,8-ジイルが好ましく、-CH2-CH2-又は-CH(CH3)CH2-がより好ましい。
一般式(Ia)において、Xは、-NH-(CH2)2-NH-COO-コレステリル、-NH-(CH2)6-NH-COO-コレステリル、-NH-(CH2)12-NH-COO-コレステリル又は-NH-(CH2CH2O)2-CH2CH2-NH-COO-コレステリルが好ましく、-NH-(CH2)2-NH-COO-コレステリル、-NH-(CH2)6-NH-COO-コレステリル又は-NH-(CH2CH2O)2-CH2CH2-NH-COO-コレステリルがより好ましく、-NH-(CH2)6-NH-COO-コレステリルがより更に好ましい。

Xaは、ヒドロキシ及び-O-Q+からなる群より選択され;ここで、Q+は、カウンターカチオンである。)
別の態様において、ヒアルロン酸誘導体は、繰り返し単位(I)、繰り返し単位(Ia)及び繰り返し単位(II)から実質的になるヒアルロン酸誘導体であってもよい。
一般式(II)において、Q+はカルボキシ基と水中で塩を形成するカウンターカチオンであれば特に限定されず、2価以上の場合は価数に応じて複数のカルボキシ基と塩を形成する。
カウンターカチオンの例としては、リチウムイオン、ナトリウムイオン、ルビジウムイオン、セシウムイオン、マグネシウムイオン、カルシウムイオン等の金属イオン;式:N+RjRkRlRm(式中、Rj、Rk、Rl及びRmは、それぞれ独立に、水素原子及びC1-6アルキルからなる群より選択される)で表されるアンモニウムイオン等が挙げられる。
中でも、Q+は、ナトリウムイオン、カリウムイオン、又はテトラアルキルアンモニウムイオン(例えば、テトラn-ブチルアンモニウムイオン等)が好ましい。
前記式中、Rj、Rk、Rl及びRmは、C1-6アルキルからなる群より選択される同一の基であることが好ましく、n-ブチル基が好ましい。
ステリル基導入率が上記下限値以上であることで、生体内で沈殿せずに安定したハイドロゲルを保つことができる。一方で、上記上限値以下であることで、ハイドロゲルの平均粒子径を上記範囲内とすることができる。
前記1H-NMRは、例えば、測定溶媒として0.02N DCl DMSO-d6/D2O混液(2N DCl D2O:DMSO-d6=1:99)を用い、測定温度85℃にて実施することができる。
なおグルコサミンのアセチル基由来のピークが含まれる1.6~2.0ppm付近のピークにはコレステリル基由来のピーク(5H)が重なるため、1.6~2.0ppm付近のピークの積分値からコレステリル基メチル由来のピーク(0.7ppm)の積分値を5/3したものを差し引いて算出した値(即ち、積分値(1.6~2.0ppm)-積分値(0.7ppm)×5/3)をヒアルロン酸由来のアセチル基の積分値として導入率の計算に使用することができる。
=[(ステリル基に由来するピーク積分値×3/nH)/(N-アセチル-D-グルコサミンのアセチル基に由来するピーク積分値)]×100
ヒアルロン酸誘導体は、例えば、グルクロン酸のカルボキシ基をアミドに変換し、ステリル基を導入することで得られる。また、原料のヒアルロン酸又はその誘導体に対して、反応させるステリル基を有する化合物の配合量を調整することで、ステリル基導入率を制御することができる。
多価アルコールとしては、2価のアルコールであってもよく、3価のアルコールであってもよい。
2価のアルコールとしては、例えば、エチレングリコール、ジエチレングリコール、プロピレングリコール、ジプロピレングリコール、ネオペンチルグリコール、1,4-ブタンジオール、1,6-ヘキサンジオール等が挙げられる。
3価のアルコールとしては、例えば、グリセリン、トリメチロールプロパン等が挙げられる。
-CONRa-Y-NRbH + Hal-R;
-CONRa-Y-NRbH + Hal-COOR;
-CONRa-Y-NRbH + HOCO-R;
-CONRa-Y-NRbH + Hal-CO-R;
-CONRa-Y-NRb-COOH + HNRc-R;
-CONRa-Y-NRb-CO-NRcH + Hal-R;
-CONRa-Y-NRbH + HOCO-NRc-R;
-CONRa-Y-NRbH + Hal-CO-NRc-R;
-CONRa-Y-COOH + HO-R;
-CONRa-Y-OH + Hal-COO-R;
-CONRa-Y-OCOOH + HO-R;
-CONRa-Y-OCOOH + Hal-R;
-CONRa-Y-OCO-Hal + HO-R;
-CONRa-Y-SH + Hal-R;
-CONRa-Y-Hal + HS-R;
-CONRa-Y-CO-Ya-Hal + HS-R;
-CONRa-Y-CO-Ya-SH + Hal-R;
-CONRa-Y-O-CO-CH=CH2 + HS-R;
-CONRa-Y-NRb-CO-CH(CH3)=CH2 + HS-R;
-CONRa-Y-SH + HS-R;
-COZ-OH + HNRa-Y-NRb-COO-R;
-COZ-NRa-Y-NRbH + Hal-COO-R
(式中、Ra、Rb、Rc、Y、Ya、Yb、及びZは本明細書で既に定義したとおりであり、Halは、フッ素原子、塩素原子、臭素原子及びヨウ素からなる群より選択されるハロゲン原子を表す)。
-NH-(CH2)p1-O-CO-C(R17)=CH2;
-NH-(CH2)p1-O-CO-CH(R17)-CH2-S-CH2-CH(OH)-CH(OH)-CH2-SH;
-NH-(CH2)p1-SH;
-NH-(CH2)p1-NH-CO-C(R17)=CH2;
-NH-(CH2)p1-NH-C(=NH)-(CH2)3-SH;
-NH-(CH2)p1-NH-CO-(CH2)r-SH;
-NH-(CH2)p1-NH-CO-CH(R17)-CH2-S-CH2-CH(OH)-CH(OH)-CH2-SH;
-NH-(CH2)p1-NH-CO-CH(NH2)-CH2-SH;
-NH-(CH2)p1-NH-CO-CH(NH2)-(CH2)2-SH;
-NH-NH-CO-(CH2)4-CO-NH-NH-C(=NH)-(CH2)3-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-O-CO-C(R17)=CH2;
-NH-(CH2-CH2-O)q-CH2-CH2-O-CO-CH(R17)-CH2-S-CH2-CH(OH)-CH(OH)-CH2-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-CO-C(R17)=CH2;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-C(=NH)-(CH2)3-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-CO-(CH2)r-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-CO-CH(R17)-CH2-S-CH2-CH(OH)-CH(OH)-CH2-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-CO-CH(NH2)-CH2-SH;
-NH-(CH2-CH2-O)q-CH2-CH2-NH-CO-CH(NH2)-(CH2)2-SH;
-NH-CH(CO2H)-(CH2)-SH;
-NH-CH(CO2H)-(CH2)2-SH;及び
-NH-CH(CO2H)-(CH2)2-CONH-CH(CONH-CH2-CO2H)-CH2-SH
(ここで、R17は、水素原子又はC1-6アルキル基であり、p1は2以上10以下の整数、qは1以上100以下の整数、rは1以上3以下の整数を、それぞれ表す)からなる群より選択される]
に変換することで、分子内或いは他分子を含めた分子間で化学的に架橋させてゲル化することもできる。
前記ゲル浸透クロマトグラフィーによる測定は、例えば、以下の方法で行うことができる。
1mg/mLのヒアルロン酸誘導体水溶液、2mg/mLのポリアクリル酸標準物質水溶液を調製し、以下に示す条件にてゲル浸透クロマトグラフィーによる測定を行う。
(測定条件)
装置:高速GPC(ゲル浸透クロマトグラフィー)装置
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率検出器)
温度:30℃
その他、前記第1の実施形態のヒアルロン酸誘導体と同じ構成等について、その説明を省略する。
一方で、ヒアルロン酸誘導体の重量平均分子量(ポリアクリル酸換算)が前記上限値を超える場合は、粘性が増し、製剤として使用しにくい場合がある。
前記標準物質として使用されるポリアクリル酸は、ポリアクリル酸ナトリウムであることが好ましく、具体的には、Pоlymer Standards Service-USA社製、ORDER No.PSS-Paaシリーズ(ポリアクリル酸ナトリウム)であることがより好ましい。
前記第1~第3の実施形態のヒアルロン酸誘導体は、薬効成分と製剤化して、医薬組成物として使用することができる。
すなわち、本実施形態の医薬組成物は、上述したヒアルロン酸誘導体と、薬効成分と、を含有する。
Mwが上記数値範囲であることで、共刺激分子の活性化に優れる会合体サイズがより多く存在し、本実施形態の医薬組成物を用いることで、ペプチド等の薬効成分を免疫細胞(特に、cDC1、マクロファージ)に安定的且つ効率よく送達させ、さらに、該薬効成分の免疫細胞への取り込みを向上、促進、又は増強させることができ、同時に、免疫細胞の活性(特に、cDC1での共刺激分子CD80及びCD86の発現)を向上、促進、維持又は増強させることができると考えられる。
Mwは、例えば、DLS(Dynamic Light Scattering)、サイズ排除クロマトグラフィー、及び電子顕微鏡法等によって、測定することができる。より具体的には、例えば、サイズ排除クロマトグラフィー装置にてヒアルロン酸誘導体濃度が1mg/mLになるように希釈して測定する。
薬効成分としては、特に限定されないが、抗原(がん抗原、感染症由来抗原、免疫疾患における自己抗原等)、医薬活性ペプチド又はタンパク質、核酸、低分子化合物、中分子化合物等が挙げられる。中でも、抗原が好ましく、がん抗原、感染症由来抗原、及び免疫疾患における自己抗原からなる群から選択される少なくとも一つがより好ましく、がん抗原又は感染症由来抗原がより好ましい。
(がん抗原)
がん抗原とは、がん細胞に多く発現する抗原であり、いくつかの場合には、がん細胞によってのみ発現する。がん抗原は、がん細胞内、又はがん細胞の表面上に発現し得る。
該リンカーに用いるアミノ酸としては、グリシン(G)、チロシン(Y)、ロイシン(L)、トリプトファン(W)等が挙げられる。好ましくは、チロシン(Y)、ロイシン(L)、トリプトファン(W)である。アミノ酸リンカーの具体例としては、連続した4つのチロシン(Y)からなるリンカー(4Y)、連続した4つのロイシン(L)からなるリンカー(4L)、連続した4つのトリプトファン(W)からなるリンカー(4W)、連続した6つのグリシン(G)なるリンカー(6G)、連続した6つのチロシン(Y)なるリンカー(6Y)、連続した6つのロイシン(L)なるリンカー(6L)、連続した6つのトリプトファン(W)なるリンカー(6W)、連続した8つのチロシン(Y)なるリンカー(8Y)、連続した6つのロイシン(L)なるリンカー(8L)、連続した8つのトリプトファン(W)なるリンカー(8W)が挙げられ、好ましくは、6Y、6L又は6Wである。
感染症由来抗原としては、感染性病原体及び感染性病原体由来の抗原であれば特に限定されない。感染性病原体としては、ウイルス、細菌、真菌、線虫等が挙げられる。感染症病原体由来抗原は抗原タンパク質であっても、抗原ペプチドであってもよい。
免疫疾患における抗原としては、免疫疾患の標的タンパク質のエピトープを含むものであれば特に限定されない。免疫疾患としては特に限定されず、例えば、尋常性乾癬、強直性脊椎炎、関節リウマチ、乾癬性関節炎、体軸性脊椎関節炎、クローン病、潰瘍性大腸炎、気管支喘息、慢性蕁麻疹、花粉症、アトピー性皮膚炎等が挙げられる。
標的タンパク質としては特に限定されず、IL-17A、DPP4、S100A9、PCSK9、IL-23、IgE、TNFα、IL-12/23p40、IL-6、α4β7インテグリン、IL-4/13、IL-5、BLyS、IL-13等が挙げられる。参考文献2(国際公開第2017/164409号)にはIL-17A由来のペプチドが記載されている。
ペプチドは、標的タンパク質のエピトープだけではなく、標的タンパク質以外ののエピトープを有しても良い。B細胞エピトープであってもT細胞エピトープであってもよい。
ペプチドはエピトープを有する配列であり、環状構造を有していてもよい。前期ペプチドは分子内に複数の環状構造を有していてもよい。
さらに前記ペプチドはタンパクへコンジュゲートされていてもよい。
投与されたタンパク質やペプチドに対する抗体が体内で産生されることにより治療効果が期待できる。
医薬活性ペプチド又はタンパク質としては、対象に治療有効量を投与した場合に、対象の状態又は病状に対して正の、又は有利な効果を有するものを意味する。好ましい医薬活性ペプチド又はタンパク質としては、根治的又は対症的性質を有し、疾患又は障害の1つ又は複数の症状を改善する、軽快させる、軽減する、元に戻す、症状の発症を遅らせる、又は症状の重症度を減ずるために投与することができるものである。医薬活性ペプチド又はタンパク質は予防的性質を有することもあり、疾患の発症を遅らせるか、又はこのような疾患又は病態の重症度を減ずるために使用することができる。「医薬活性ペプチド又はタンパク質」という用語は、全長タンパク質又はポリペプチドを含意し、又その医薬的に活性な断片を指すこともある。この用語は、ペプチド又はタンパク質の医薬的に活性なアナログも包含する。
一実施形態では、サイトカインはインターロイキンである。
一実施形態では、医薬活性タンパク質は、IL-2、IL-7、IL-12、IL-15及びIL-21からなる群より選択される1種以上のインターロイキンである。
一実施形態では、医薬活性ペプチドは、環状構造を有してもよい。前記ペプチドは分子内に複数の環状構造を有していてもよい。
核酸としては、例えば、DNA、RNA、アンチセンス核酸、デコイ核酸、リボザイム、低分子干渉RNA、核酸アプタマー等が挙げられる。また、上記抗原がペプチド又はタンパク質である場合には、それら抗原ペプチド又はタンパク質をコードする核酸(DNA又はmRNA等)も好ましく用いられる。
低分子化合物としては、例えば、制癌剤(例えば、アルキル化剤、代謝拮抗剤、アルカロイド等)、免疫抑制剤、抗炎症剤(ステロイド剤、非ステロイド剤系抗炎症剤等)、抗リウマチ剤、抗菌剤(β-ラクタム系抗生物質、アミノグリコシド系抗生物質、マクロライド系抗生物質、テトラサイクリン系抗生物質、新キノロン系抗生物質、サルファ剤等)等が挙げられる。
或いは、本実施形態の医薬組成物において、薬効成分の含有量は、ヒアルロン酸誘導体の質量に対して、0.1質量%以上100.0質量%以下であることがより好ましく、0.1質量%以上50.0質量%以下であることがさらに好ましく、0.1質量%以上30.0質量%以下であることがさらに好ましく、0.5質量%以上30.0質量%以下であることがさらに好ましく、1.0質量%以上30.0質量%以下であることがさらに好ましく、1.0質量%以上25.0質量%以下であることがさらに好ましく、1.0質量%以上20.0質量%以下であることがさらに好ましく、1.0質量%以上15.0質量%以下であることがよりさらに好ましく、1.0質量%以上10.0質量%以下であることが特に好ましい。
本実施形態の医薬組成物がワクチン組成物である場合に、上記薬効成分及び上記ヒアルロン酸誘導体に加えて、アジュバントを更に含むことができる。これにより、より効果的に免疫を誘導することができる。ここで、誘導される免疫は液性免疫でも細胞性免疫で合っても良い。すなわち、本実施形態の医薬組成物は、上記ヒアルロン酸誘導体と、上記薬効成分として抗原(好ましくはがん抗原又は感染症由来抗原)と、を含み、且つ、アジュバントを更に含む、医薬組成物であることが好ましい。
一方、細胞性免疫とは、病原体そのものやウイルス感染細胞、癌細胞等の異物の排除において、細胞が主なエフェクターとなる免疫機構のことをいう。マクロファージ、細胞傷害性T細胞(CTL、キラーT細胞)、ナチュラルキラー細胞(NK細胞)等の免疫担当細胞自体による排除機構である。
本実施形態の医薬組成物は、単独で投与することもでき、或いは、薬理学上許容されうる担体とともに常套手段に従って投与することができる。薬理学上許容されうる担体と併用する場合は、例えば、上記ヒアルロン酸誘導体及び上記薬効成分、並びに、必要に応じてアジュバントと、水若しくはそれ以外の生理学的に許容し得る液(例えば、生理食塩水、含水エタノール、リン酸緩衝生理食塩水(PBS))等とを、混合してもよく、生理学的に許容し得る緩衝液、賦形剤、ベヒクル、防腐剤、安定化剤、結合剤、凍結乾燥補助剤等を含むこともできる。
固体の場合、粉末、顆粒、丸剤、ペレット、タブレット、カプセル等の形態が挙げられる。中でも、固体としては、凍結乾燥粉末であることが好ましい。
半固体の場合、ゲル等の形態が挙げられる。
液体の場合、粉末を水又はリン酸緩衝生理食塩水(PBS)等の緩衝液により希釈又は懸濁した懸濁液等の形態が挙げられる。
本実施形態の医薬組成物は、上記ヒアルロン酸誘導体と上記薬効成分を適宜混合することで製造できるが、以下に示す方法で製造することが好ましい。
前記薬効成分を有機溶媒又は有機溶媒含有水に溶解して、前記薬効成分を含有する油相を調製する調製工程;及び、
前記油相と前記ヒアルロン酸誘導体を含有する水相との混合比が容量比で20:100~0.01:100となるように混合する混合工程。
調製工程では、薬効成分を有機溶媒又は有機溶媒含有水に溶解して、薬効成分を含有する油相を調製する。
混合工程では、調製工程で得られた油相と前記ヒアルロン酸誘導体を含有する水相との混合比が容量比で20:100~0.01:100となるように混合する。
混合工程の後に、得られた薬効成分-ヒアルロン酸誘導体複合体を含む溶液を滅菌して、滅菌製剤を得る滅菌工程を行ってもよい。製剤の滅菌方法としては、滅菌用フィルターによるろ過滅菌、ガス滅菌、γ線滅菌または電子線滅菌等が挙げられる。滅菌用フィルターによるろ過滅菌が最も好ましい。
混合工程の後に、得られた薬効成分-ヒアルロン酸誘導体複合体を含む溶液を乾燥して、乾燥体を得る乾燥工程を行ってもよい。乾燥方法としては、上記「ヒアルロン酸誘導体の製造方法」において例示された方法と同様の方法が挙げられる。
本実施形態の医薬組成物を投与する対象は、ヒトを含む哺乳類に分類される動物(サル、マーモセット、マウス、ラット、ウシ、ウマ、ネコ、イヌ、ブタ、ヒツジ、ヤギ、ウサギ等)が挙げられる。
一実施形態において、本発明は、上記医薬組成物の有効量を、患者又は患畜に投与することを含む、がん、感染症及び免疫疾患からなる群より選ばれる1種以上の疾患の予防又は治療方法を提供する。
なお、感染症としては、上記「抗原」の「感染症由来抗原」において例示されたものが挙げられる。
また、ここでいう「有効量」とは、予防又は治療に有効な量、すなわち、上記疾患の発症予防又は治療に適する量が包含される。
前記医薬組成物は、感染症ワクチン、がんワクチン、または免疫疾患用医薬組成物であることが好ましい。
抗体は、例えば腫瘍による免疫抑制シグナルを阻害する抗体、または免疫細胞の共刺激シグナルを活性化する1種類以上の抗体であり、好ましくは腫瘍による免疫抑制シグナルを阻害する抗体または免疫細胞の共刺激シグナルを活性化する抗体である。具体的には、抗CTLA4抗体、抗PD1抗体、抗PDL1抗体、抗OX40、および抗4-1BB抗体からなる群から選択される1種類以上の抗体である。
[合成例1-1]
ヒアルロン酸誘導体を次の工程1~工程3に従って調製した。
(コレステリル 6-アミノヘキシルカーバメート塩酸塩の合成)
コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)を、次に示す工程1-1、続いて工程1-2に従って合成した。
コレステリルクロロホルメート(3.37g、7.5mmol)の無水ジクロロメタン(20mL)の溶液に、アルゴン雰囲気下、トリエチルアミン(TEA、1.05mL)を加えて撹拌した。氷冷下で、6-(t-ブトキシカルボニル)アミノ-1-アミノヘキサン(1.12mL、5mmol)を滴下して加え、そのまま氷冷下で30分間攪拌後、室温(25℃程度)まで昇温し、当該混合物を一晩撹拌した。反応混合物を、超純水及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下で溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液:酢酸エチル:n-ヘキサン=1:4)で精製し、目的物のフラクションを合わせて溶媒を減圧下留去した。
得られた残渣を酢酸エチル(40mL)に溶解し、4N塩酸/酢酸エチル溶液(40mL)を加えて室温(25℃程度)で一晩撹拌した。生じた沈殿物を遠心分離により回収した。得られた固体を酢酸エチルにて4回洗浄後、減圧下で乾燥し、コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)1.2gを得た。
(ヒアルロン酸のテトラブチルアンモニウム(TBA)塩の調製)
ヒアルロン酸(HA)のTBA塩(HA-TBA)を次に示す工程2-1、続いて工程2-2に従って調製した。
DOWEX(登録商標)50WX-8-400(アルドリッチ社製)を超純水に懸濁させ、デカンテーションにより樹脂を超純水で3回程度洗浄した。40wt%テトラブチルアンモニウムヒドロキシド水溶液(TBA-OH)(アルドリッチ社製)を樹脂のカチオン交換能に対し約1.5倍モル等量加え、30分間、室温(25℃程度)で撹拌した。余剰のTBA-OH溶液をデカンテーションにより除去した後、さらに過剰の超純水で洗浄することで、TBA塩化したカチオン交換樹脂を得た。
重量平均分子量(絶対分子量)10,000(10kDa)の原料ヒアルロン酸ナトリウム塩(HA-Na)を15mg/mLの濃度で超純水に溶解した。「(1)工程2-1」でTBA塩化したカチオン交換樹脂の懸濁液をHAユニット(ユニット分子量401.3)のモル数に対し樹脂のイオン交換能換算で5倍モル等量添加した。室温(25℃程度)で15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、ヒアルロン酸のTBA塩(HA-TBA)を白色固体として得た。
「2.(2)工程2-2」で調製したHA-TBAの無水DMSO溶液(10mg/mL)を調製した。その後、「1.工程1」で合成したHA-TBA中に存在する二糖繰り返し単位(HAユニット)に対するChol塩酸塩の添加量がモル比で44/100となるように添加した。次に、HAユニットに対する4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)の添加量がモル比で48/100となるように加え、室温(25℃程度)で一晩撹拌した。反応溶液は、0.3M 酢酸アンモニア/DMSO溶液、0.15M NaCl水溶液、超純水の順で、スペクトラポア7透析膜(Spectrum Laboratories社製製、分画分子量(MWCO):3,500)を用いて透析した。得られた透析液を凍結乾燥して目的物(HA-C6-Chol)を白色固体として得た。
測定溶媒として0.02N DCl DMSO-d6/D2O混液(2N DCl D2O:DMSO-d6=1:99)を用い、NMR装置としてJNM-ECS400(日本電子株式会社製)を用い、85℃で測定を行い、白色個体の1H-NMRスペクトルを得た。得られた1H-NMRスペクトルにおいて、N-アセチル-D-グルコサミンのアセチル基由来のピーク(COCH3、1.6ppm以上2.0ppm以下、3H)、コレステリル基中のメチル基由来のピーク(CH3、0.7ppm、3H)が確認され、コレステロール導入率は44%であった。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
合成例1-1と同様の手順により、ヒアルロン酸誘導体を合成した。
生成物の1H-NMRスペクトルにおいて、N-アセチル-D-グルコサミンのアセチル基由来のピーク(COCH3、1.6ppm以上2.0ppm以下、3H)、コレステリル基中のメチル基由来のピーク(CH3、0.7ppm、3H)が確認され、コレステロール導入率は44%であった。上記合成例1-1に示す条件にてゲル浸透クロマトグラフィーによる測定を行い、得られたクロマトグラムを図3Bに示す。
合成例1-1と同様の手順により、ヒアルロン酸誘導体を合成した。
生成物の1H-NMRスペクトルにおいて、N-アセチル-D-グルコサミンのアセチル基由来のピーク(COCH3、1.6ppm以上2.0ppm以下、3H)、コレステリル基中のメチル基由来のピーク(CH3、0.7ppm、3H)が確認され、コレステロール導入率は44%であった。上記合成例1-1に示す条件にてゲル浸透クロマトグラフィーによる測定を行い、得られたクロマトグラムを図3Cに示す。
合成例1-1で得たヒアルロン酸誘導体の分取を次のとおり行った。凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を5mg/mLで注射用水に溶解し、透析カセット(フロータライザーG2、MWCO:300,000、家田貿易株式会社)に移し、10mMリン酸緩衝液pH7.4に対して9回透析を行った。得られた透析内液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図4Aは、得られたクロマトグラムである。図21及び図22は、後述する面積比A1/A2及び距離の比Da/Dbの算出方法を説明するために、実施例1-1で得られたヒアルロン酸誘導体及び標準物質であるポリアクリル酸のクロマトグラムを重ねて示した図である。
ろ液を所望の濃度まで限外濃縮器(Vivaspin20、MWCO:10,000、ザルトリウス)にて濃縮した。
実施例1-1と同様の手順により、合成例1-2で得たヒアルロン酸誘導体の分取を行った。得られた透析内液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図4Bは、得られたクロマトグラムである。
実施例1-1と同様の手順により、合成例1-3で得たヒアルロン酸誘導体の分取を行った。得られた透析内液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図4Cは、得られたクロマトグラムである。
合成例1-1で得たヒアルロン酸誘導体の分取を次のとおり行った。凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を10mg/mLで注射用水に溶解し、100mMリン酸緩衝液にて、10mMリン酸緩衝液(pH7.4)となるように希釈した。その後、限外ろ過器(Vivaspin(登録商標)20、MWCO:300,000、ザルトリウス)にてろ過し、得られたろ液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図5Aは、得られたクロマトグラムである。ろ液を所望の濃度まで限外濃縮器(Vivaspin20、MWCO:10,000、ザルトリウス)にて濃縮した。
比較例1-1と同様の手順により、合成例1-2で得たヒアルロン酸誘導体の分取を行った。得られたろ液のヒアルロン酸誘導体について、所望の濃度まで限外濃縮器(Vivaspin20、MWCO:10,000、ザルトリウス)にて濃縮した。その後、1mg/mLになるように注射用水で希釈し、ゲル浸透クロマトグラフィーによる測定を行った。図5Bは、得られたクロマトグラムである。
比較例1-2と同様の手順により、合成例1-3で得たヒアルロン酸誘導体の分取を行った。得られたろ液のヒアルロン酸誘導体について、所望の濃度まで限外濃縮器(Vivaspin20、MWCO:10,000、ザルトリウス)にて濃縮した。その後、1mg/mLになるように注射用水で希釈し、ゲル浸透クロマトグラフィーによる測定を行った。図5Cは、得られたクロマトグラムである。
コレステリル修飾プルラン(CHP:日油社製,製品番号CHP-80T)の凍結乾燥体を1mg/mLになるように注射用水に溶解し、ゲル浸透クロマトグラフィーによる測定を行った。図5Dは、得られたクロマトグラムである。
実施例1-1で得られたヒアルロン酸誘導体と標準物質であるポリアクリル酸のゲル浸透クロマトグラムから、面積比A1/A2及び距離の比Da/Dbを算出した。
標準物質であるポリアクリル酸(50kDa)は、Pоlymer Standards Service-USA社製、ORDER No.PSS-Paa50k(ポリアクリル酸ナトリウム)を使用した。
標準物質であるポリアクリル酸(150kDa)は、Pоlymer Standards Service-USA社製、ORDER No.PSS-Paa150k(ポリアクリル酸ナトリウム)を使用した。
標準物質ポリアクリル酸の粉末は2mg/mLの濃度となるように注射用水にて溶解させた。これ以降記載する標準物質であるポリアクリル酸は全て、Pоlymer Standards Service-USA社製(ポリアクリル酸ナトリウム)のものを用いた。
(ii)次に、ヒアルロン酸誘導体のクロマトグラムにおいて、Ubから終点までの曲線と、屈折率強度極大点KbからベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA2とし、前記ヒアルロン酸誘導体のクロマトグラムで始点から前記Ubまでの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA1とした。
(ii)次に、屈折率強度がLa/20となるクロマトグラム上の2点のうち、溶出時間が早いほうを点R1とし、溶出時間が遅いほうを点S1とした。ここで、点R1における溶出時間は5.74分であり、点S1における溶出時間は7.68分であった。
(iii)次に、標準物質である50kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KbからベースラインBへ引いた垂線とベースラインとの交点をBbとした。ここで、屈折率強度極大点Kbにおける溶出時間は前記の通りである。
(iv)次に、前記点R1と前記点S1を結んだ直線D1と、前記屈折率強度極大点KaからベースラインBへ引いた垂線との交点をTaとし、前記屈折率強度極大点KbからベースラインBへ引いた垂線と前記直線D1との交点をTbとした。
(v)最後に、前記点R1と前記Taの距離をDa、前記Taと前記Tbの距離をDbとした。
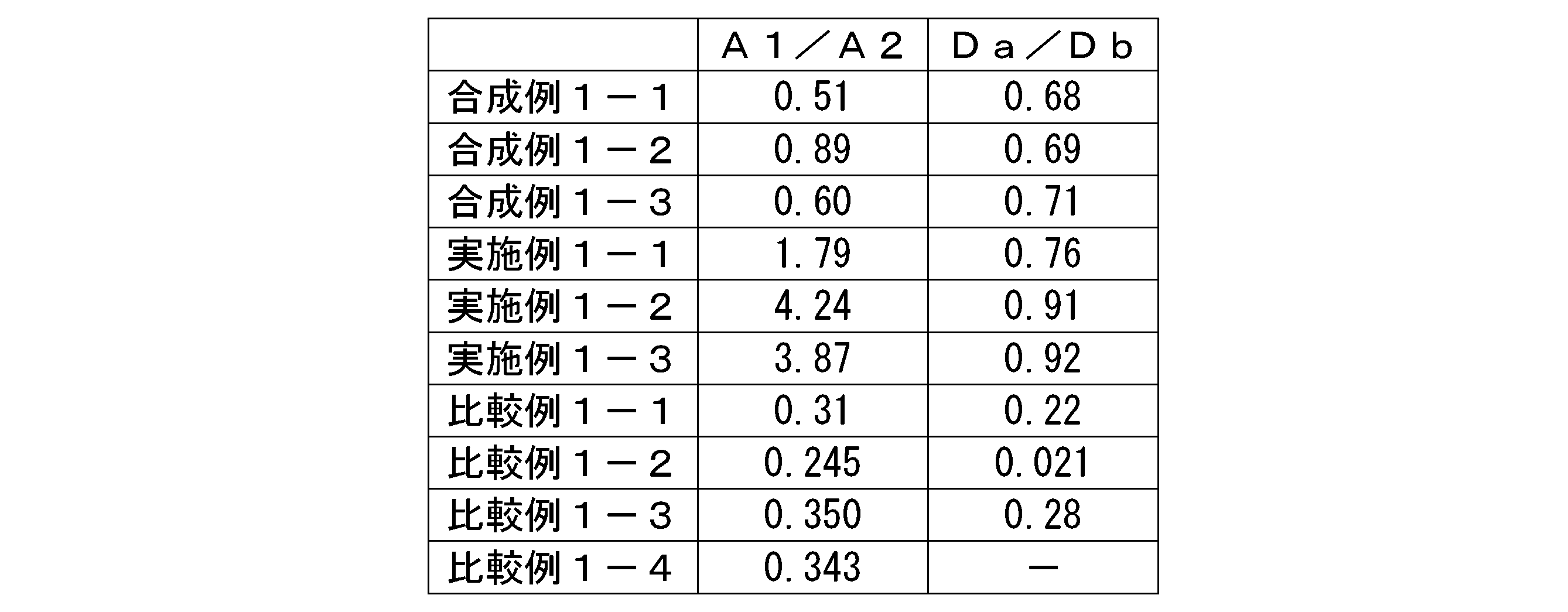
[実施例2-1]
実施例1-1で得られたヒアルロン酸誘導体を5mg/mLの濃度になるように10mMリン酸緩衝液pH7.4にて希釈して調製した。
一方、別の容器にて、蛍光標識ペプチドの濃度が50mg/mLとなるようにジメチルスルホキシド(DMSO)へ溶解させた。蛍光標識ペプチドはバイオロジカ社製のN末にフルオロセインが結合している次のアミノ酸配列を有する蛍光標識ペプチドを用いた。
実施例1-2で得られたヒアルロン酸誘導体を用いて、実施例2-1と同様の手順により、蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物を得た。医薬組成物100質量部に対する、蛍光標識ペプチドの含有量は0.0478質量部であった。
合成例1-1に記載の方法で、ゲル浸透クロマトグラフィーによる測定を行ったところ、ペプチド封入前後でヒアルロン酸誘導体のクロマトグラムに大きな変化は認められなかった(図6B)。
実施例1-3で得られたヒアルロン酸誘導体を用いて、実施例2-1と同様の手順により、蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物を得た。医薬組成物100質量部に対する、蛍光標識ペプチドの含有量は0.0303質量部であった。
合成例1-1に記載の方法で、ゲル浸透クロマトグラフィーによる測定を行ったところ、ペプチド封入前後でヒアルロン酸誘導体のクロマトグラムに大きな変化は認められなかった(図6C)。
比較例1-1で取得したヒアルロン酸誘導体を5mg/mLの濃度になるように10mMリン酸緩衝液pH7.4にて希釈して調製した。
一方、別の容器にて、蛍光標識ペプチドの濃度が50mg/mLとなるようにDMSOへ溶解させた。
比較例1-2で取得したヒアルロン酸誘導体を用いて、比較例2-1と同様の手順により、蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む組成物を得た。医薬組成物100質量部に対する、蛍光標識ペプチドの含有量は0.0332質量部であった。
比較例1-3で取得したヒアルロン酸誘導体を用いて、比較例2-1と同様の手順により、蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む組成物を得た。医薬組成物100質量部に対する、蛍光標識ペプチドの含有量は0.0348質量部であった。
CHP(日油社製,製品番号CHP-80T)の凍結乾燥体を10mg/mLの濃度になるように6M尿素含有リン酸緩衝生理食塩水pH7.4に溶解させた。
一方、別の容器にて、蛍光標識ペプチドの濃度が50mg/mLとなるようにジメチルスルホキシドへ溶解させた。
合成例1-1で取得したヒアルロン酸誘導体の凍結乾燥体を秤量し、5mg/mLの濃度になるように注射用水を添加し、一晩攪拌して十分に溶解させた。
一方、別の容器にて、蛍光標識ペプチドの濃度が50mg/mLとなるようにDMSOへ溶解させた。
合成例1-2で取得したヒアルロン酸誘導体の凍結乾燥体を用いて、比較例4-1と同様の手順により、蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む組成物を得た。
以下の条件の高速液体クロマトグラフィー測定によって、実施例2-1~2-3及び比較例2-1~2-3、3、4-1、4-2の蛍光標識ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物において、蛍光標識ペプチドがヒアルロン酸誘導体に複合化されて可溶化していることを確認し、また、各医薬組成物中の蛍光標識ペプチドの濃度を提供した。
HPLC装置:日本分光HPLC-EXTREMA
カラム:PLRP-S 1000A 8μm 長さ50mm×内径4.6mm(アジレント・テクノロジー株式会社製、製品番号:PL1512-1802)
カラム温度:40℃
移動相:(A)0.1v/v%トリフルオロ酢酸/アセトニトリル
(B)0.1v/v%トリフルオロ酢酸/水
流速:2mL/分
注入量:30μL
検出器:UV(215nm)
グラジエントプログラム:以下の表2に示す。表2中、「%」は「v/v%」を意味する。

(リンパ節内ミエロイド細胞へのペプチドの取り込み及び活性化解析)
1.材料
アジュバント:CpGオリゴDNA1668(味の素バイオファーマサービスジーンデザインから購入)
投与した組成物:実施例2-1、比較例2-1,比較例3、及び比較例4-1の医薬組成物。
Brilliant Violet 421標識抗マウスF4/80抗体(クローンBM8);
Brilliant Violet 510標識抗マウスCD11c抗体(クローンN418);
APC-Cy7標識抗マウスXCR1抗体(クローンZET);
APC標識抗マウスCD80抗体(クローン16-10A1);
APC標識抗マウスCD86抗体(クローンGL-1);
Purified CD16/32抗体(クローン93)。

蛍光(フルオロセイン)標識ペプチド濃度が0.3mg/mL程度となるように、実施例2-1、比較例2-1及び比較例4-1の医薬組成物を10質量%スクロース10mMリン酸緩衝液pH7.4で希釈し、また比較例3の医薬組成物をリン酸緩衝生理食塩水pH7.4で希釈してから、各医薬組成物の希釈液をBALB/cマウスの右側後背部にペプチドとして各60μgとなるように皮下投与した。同時に、アジュバントとして、PBSに溶解した50μgのCpGオリゴDNA1668(味の素バイオファーマサービスジーンデザイン)を投与した。
細胞種毎に、FITC(フルオロセイン)陽性細胞を、ペプチド取り込み細胞として検出した。
共刺激分子であるCD80及びCD86の発現は、各細胞種におけるFITC陽性細胞に対して解析した。
FACS解析方法を図7A~図7Fに示す。
また、解析結果を表4-1~表4-3に示す。例えば、表4-1において、「ペプチド+CD80+DC」は「CD80を発現するペプチド取り込み樹状細胞(DC)」を意味し、「ペプチド+CD86+DC」は「CD86を発現するペプチド取り込み樹状細胞(DC)」を意味し、「ペプチド+DC」は「ペプチド取り込み樹状細胞(DC)」を意味する。


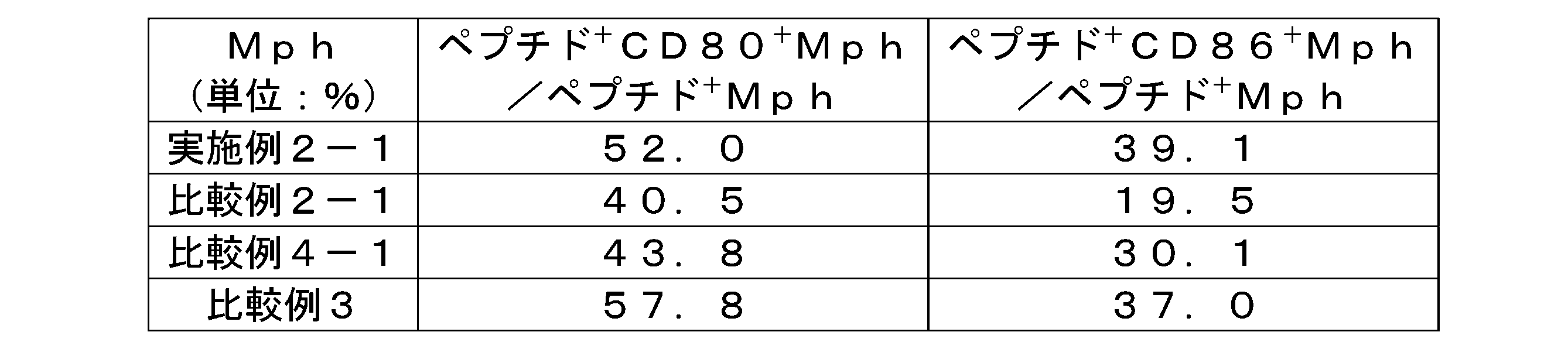
実施例2-2、比較例2-2、比較例4-2の医薬組成物を用いて、蛍光(フルオロセイン)標識ペプチド濃度が0.2mg/mL程度となるように10質量%スクロース10mMリン酸緩衝液pH7.4で希釈し、ペプチドとして各40μgとなるように皮下投与した以外は、試験例1-1と同様の方法で実施した。解析結果を表5-1~表5-3に示す。



実施例2-3の医薬組成物を用い、ペプチドとして各40μgとなるように皮下投与した以外は、試験例1-1と同様の方法で実施した。解析結果を表6-1~表6-3に示す。



[参考例1]
合成例1-1で得たヒアルロン酸誘導体を用いて、ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物の調製を行なった。
一方、別の容器にて、ペプチドの濃度が50mg/mLとなるようにDMSOへ溶解させた。
溶解を確認した後に、上記ヒアルロン酸誘導体水溶液に対して、1moL/Lの水酸化ナトリウム水溶液(富士フイルム和光社製)を以下の表7に従い、添加した。
24時間後のペプチド-ヒアルロン酸誘導体複合体を含む組成物(水溶液)の様子を目視で判断し、白濁しているか検証した。結果は以下の表7に示した。

以下に示す方法で製造したペプチド-ヒアルロン酸誘導体複合体を含む組成物について、以下条件で動的光散乱法(DLS)による測定を行い、z平均粒子径を得た。結果を上記表7に示す。
DLS装置:大塚電子製、ELSZ2000
セル:微量粒径セル
温度:25℃
ペプチド-ヒアルロン酸誘導体複合体の濃度:4.95mg/mL
合成例1-1で得たヒアルロン酸誘導体、及び、参考例1とは異なるペプチドを用いて、ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物の調製を行なった。
一方、別の容器にて、ペプチドの濃度が50mg/mLとなるようにDMSOへ溶解させた。
溶解を確認した後に、上記ヒアルロン酸誘導体水溶液に対して、1moL/Lの水酸化ナトリウム水溶液(富士フイルム和光社製)を以下の表8に従い、添加した。
その後、固体の精製スクロース(富士フイルム和光社製、製造専用、製品番号198-18385)を、ペプチド-ヒアルロン酸誘導体複合体を含む組成物とスクロースの合計質量に対して、10質量%スクロースとなるように添加して、室温(25℃程度)で1時間以上攪拌した。続いて、0.2μmPES(ポリエーテルスルフォン)(Pall社製、アクロディスクシリンジフィルター、25mmΦ)で滅菌ろ過し、ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物を得た。この時のペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物中のペプチド濃度を上述の実施例2-1と同様な方法で定量し、ペプチド-ヒアルロン酸誘導体複合体を含む医薬組成物の平均粒子径を上述の参考例1と同様な方法で測定した。その結果を以下の表8に示した。
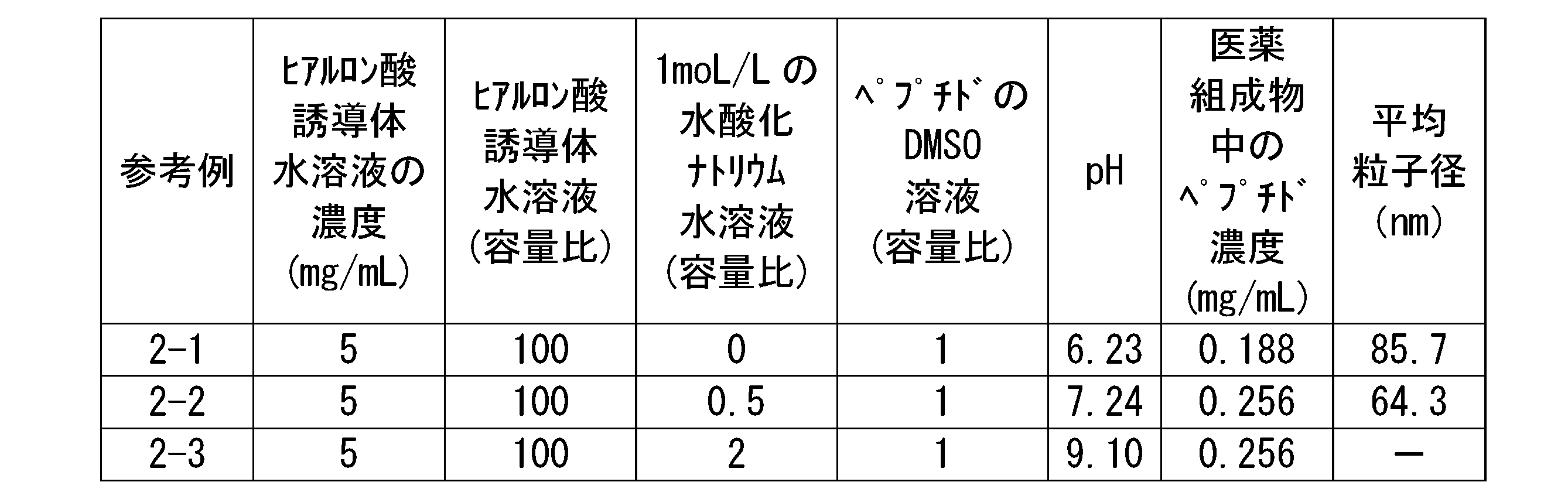
[試験例2-1~2-8]
実施例1-1と同様な手法で分取したヒアルロン酸誘導体について、濃度5mg/mLの水溶液を準備し、ヒアルロン酸誘導体を含有する水相と油相の混合比について詳細な検討を行った。
5mg/mLのヒアルロン酸誘導体400μLに対し、下記表9に従って油相であるDMSOを混合させた。十分に混合した後、最終的なヒアルロン酸誘導体濃度が一定となるように注射用水を下記表9に従い添加する。さらに、24時間室温(25℃)でインキュベートした後に、以下に示す条件にてゲル浸透クロマトグラフィーによる測定を行い、実施例1-1と同様の方法で面積比A1/A2を算出した(表9)。得られたゲル浸透クロマトグラムを図8に示す。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
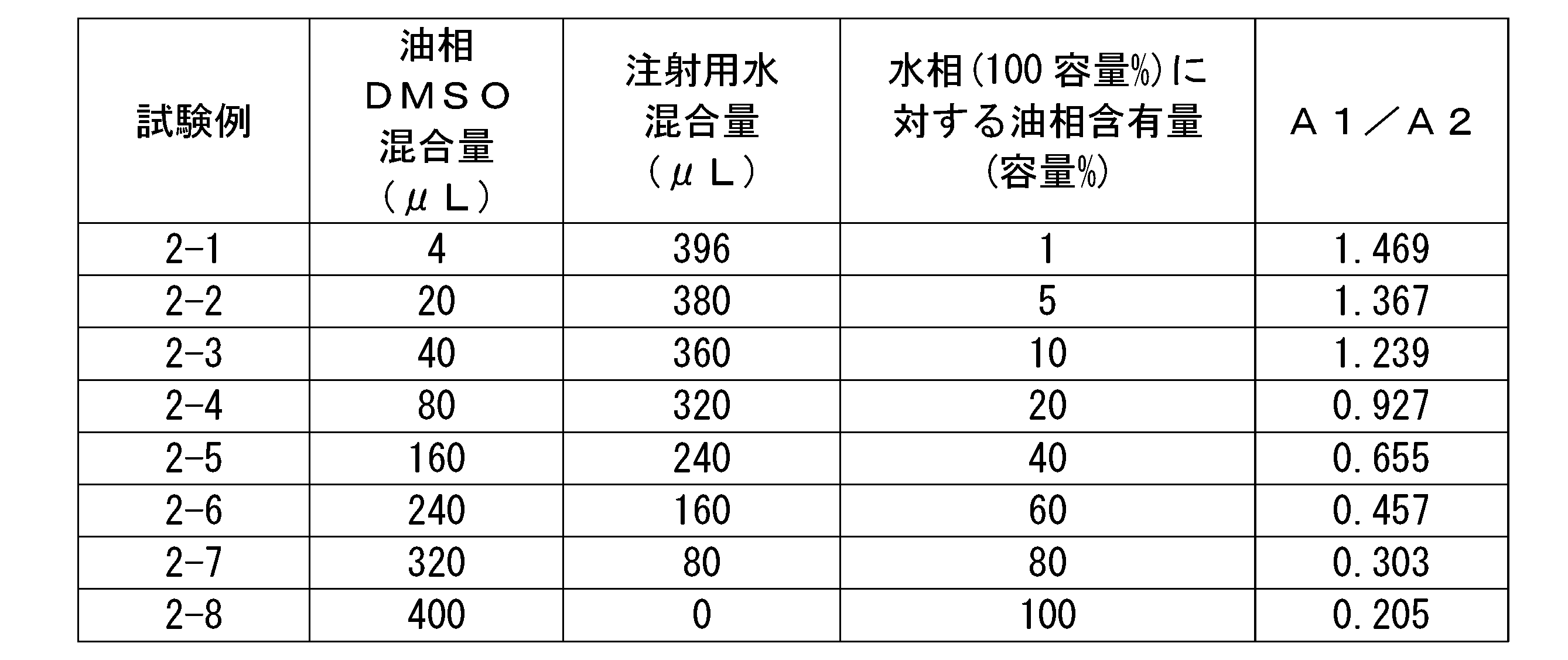
油相をDMSOではなく、エタノールを用いた以外は、前記試験例2-1~2-8と同じ方法で適切な粒子サイズ分布を維持する混合比率の検証を行った。ゲル浸透クロマトグラフィーによる測定を行い、実施例1-1と同様の方法で面積比A1/A2を算出し、下記表10に結果を示した。得られたゲル浸透クロマトグラムを図9に示す。

重量平均分子量Mw(絶対分子量)が980kDaのヒアルロン酸(キッコーマン社製)の粉末を、1mg/mLとなるように注射用水へ溶解し、合成例1-1に示す同様の条件にてゲル浸透クロマトグラフィーによる測定を行い、得られたクロマトグラムを図10に示す。
一方、別の容器にて、抗原ペプチドの濃度が50mg/mLとなるようにジメチルスルホキシド(DMSO)へ溶解させ、抗原ペプチドのDMSO溶液を得た。抗原ペプチドはバイオロジカ社製の次のアミノ酸配列を有するペプチドを用いた。
重量平均分子量Mw(絶対分子量)が650kDaのヒアルロン酸の粉末(キューピー社製)を1mg/mLとなるように注射用水へ溶解し、合成例1-1に示す同様の条件にてゲル浸透クロマトグラフィーによる測定を行い、得られたゲル浸透クロマトグラムを図12に示す。
一方、別の容器にて、抗原ペプチドの濃度が50mg/mLとなるようにジメチルスルホキシド(DMSO)へ溶解させ抗原ペプチドのDMSO溶液を得た。抗原ペプチドはバイオロジカ社製の次のアミノ酸配列を有するペプチドを用いた。
[試験例4]
合成例1-1と同様な方法で、ヒアルロン酸誘導体を合成した。分画は行わず、そのまま用いた。取得したヒアルロン酸誘導体の凍結乾燥体を秤量し、5mg/mLの濃度になるように注射用水を添加し、室温で一晩攪拌して十分に溶解させた。上記ヒアルロン酸誘導体水溶液について、1mg/mLとなるように注射用水で5倍希釈してゲル浸透クロマトグラフィーによる測定を行った。図13は、得られたゲル浸透クロマトグラムである。また、実施例1-1等と同様な方法でヒアルロン酸誘導体の粒子サイズ分布の評価を実施し、結果を下記表11に示した。
合成例1-1と同様な方法で合成したヒアルロン酸誘導体を、実施例1-1と同様な方法で分画し、ヒアルロン酸誘導体を取得した。得られた透析内液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図14は、得られたゲル浸透クロマトグラムである。また、実施例1-1等と同様な方法でヒアルロン酸誘導体の粒子サイズ分布の評価を実施し、結果を下記表11に示した。

[実施例6-1-1]
実施例6-1で取得したヒアルロン酸誘導体水溶液におけるヒアルロン酸誘導体の濃度を、既知濃度のヒアルロン酸誘導体の面積値から算出すると、4.28mg/mLであった。
別の容器にて、オボアルブミン(OVA、富士フイルム和光純薬株式会社製低エンドトキシン品)の濃度が5mg/mLとなるようにPBS(pH7.4)(富士フイルム和光純薬株式会社製、製品番号:166-23555)へ溶解させ、OVA/PBS溶液を得た。
注射用水、ヒアルロン酸誘導体水溶液、OVA/PBS溶液の順で混合したのちに、10%スクロース等張液となるようにスクロースを固体添加して清澄な溶液になるまで十分に室温で攪拌した。製剤中のOVA濃度は0.15mg/mL、ヒアルロン酸誘導体濃度は3.0mg/mLであり、10mM PB(pH7.4)を含む10%スクロース溶液とした。75℃、1時間の加熱条件下でインキュベートし、OVA-ヒアルロン酸誘導体複合体を含む組成物を得た。
比較例6-1で取得したヒアルロン酸誘導体水溶液を用い、実施例6-1-1と同様の方法でOVAをヒアルロン酸誘導体に封入し、OVA-ヒアルロン酸誘導体複合体を含む組成物を得た。
実施例6-1-1、比較例6-1-1で調製したOVA-ヒアルロン酸誘導体複合体を含む組成物を、BALB/cマウスに対して、OVAが20μgとなるように、0日目、7日目に皮下投与し、10日目に血清を採取した。なお、OVA-ヒアルロン酸誘導体複合体投与時にアジュバントであるCpG1826(製造元:INVIVOGEN、製品コード:tlrl-1826-1)を50μg皮下投与した。OVAに対する抗体価をELISAキット(Chondrex社製、Mouse Anti-OVA IgG Antibody Assay Kit、Mouse Anti-OVA IgG1 Antibody Assay Kit、Mouse Anti-OVA IgG2a Antibody Assay Kit)を用いて、添付のプロトコールに準じて測定した。血清の希釈倍率は10、50、250、1250、6250、31250とした。
ヘルパーT細胞の活性化を促し、さらにB細胞の活性化・増大の促すことにより、エピトープに対する抗原が産生されたと推察される。
[試験例5]
面積比A1/A2の異なるヒアルロン酸誘導体のがんワクチンによる抗腫瘍効果について明らかにするため、免疫チェックポイント阻害剤(ICI)抵抗性繊維肉腫細胞株CMS5a担癌マウスを用いて、CMS5aに発現する変異型ERK2抗原(mERK2)のCD8エピトープを含む長鎖ペプチド抗原と種々のヒアルロン酸誘導体を複合化した長鎖ペプチド抗原搭載ヒアルロン酸誘導体がんワクチンを作製し、治療効果を検討した。
(1)細胞
RPMI1640培地(2-メルカプトエタノール添加)は細胞科学研究所から購入した。
ウシ胎児血清(FBS)はGibcoから購入した。
マウス線維肉腫CMS5a細胞株はメモリアルスローンケタリングがん研究所より分譲され、三重大学で継代したものを用いた。マウス線維肉腫CMS5a細胞株は、変異型ERK2蛋白質を発現している。変異型ERK2蛋白質の変異部分を含むペプチド(QYIHSANVL(配列番号4))は、BALB/cマウスのCD8陽性細胞傷害性T細胞に認識される。このペプチドを認識するT細胞受容体(TCR)が単離され、TCRを遺伝子導入したトランスジェニックマウス(DUC18マウス)が作出されている。
雌性BALB/cマウス(CD90.2陽性)は6週齢~8週齢のものを日本SLCから購入した。
両マウスは三重大学先端科学研究支援センター動物実験施設にて飼育した。動物実験のプロトコールは、三重大学医学部の倫理委員会の承認を得た。
合成長鎖ペプチド抗原はシグマジェノシスから購入した。配列は次の通りであった。
NDHIAYFLYQILRGLQYIHSANVLHRDLKPSNLLLNT(配列番号1)。
当該配列は、前記変異型ERK2のCD8陽性細胞障害性T細胞認識エピトープ配列として16番目のQから24番目のLまでの9個のアミノ酸配列(QYIHSANVL(配列番号4))を、CD4陽性ヘルパーT細胞認識エピトープ配列として13番目のRから29番目のKまでの17個のアミノ酸配列(RGLQYIHSANVLHRDLK(配列番号5))を含む。
当該長鎖ペプチド抗原と種々のヒアルロン酸誘導体を複合化した長鎖ペプチド抗原搭載ヒアルロン酸誘導体を、「長鎖ペプチド抗原搭載HAナノゲルがんワクチン」又は「HAナノゲルがんワクチン」と示すこともある。
比較例6-1で得たヒアルロン酸誘導体を次のとおり分画した。凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-41.3%)を5mg/mLで注射用水に溶解し、透析カセット(フロータライザーG2、MWCO:300,000、家田貿易株式会社)に移し、10mMリン酸緩衝液pH7.4に対して透析を行い、得られた透析内液のヒアルロン酸誘導体について、ゲル浸透クロマトグラフィーによる測定を行った。図19は、得られたゲル浸透クロマトグラムである。実施例1-1等と同様な方法で、ヒアルロン酸誘導体の粒子サイズ分布の評価を実施した。結果を表12に示す。

[実施例7-1-1]
実施例7-1で得られたヒアルロン酸誘導体水溶液中におけるヒアルロン酸誘導体の濃度を、既知濃度のヒアルロン酸誘導体の面積値から算出すると、3.8mg/mLであった。
一方、別の容器にて、抗原ペプチドの濃度が50mg/mLとなるようにジメチルスルホキシド(DMSO)へ溶解させ、抗原ペプチドのDMSO溶液を得た。抗原ペプチドはバイオロジカ社製の次のアミノ酸配列を有するペプチドを用いた。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
実施例7-1-1で用いたペプチドを50mg/mLとなるようにジメチルスルホキシド(DMSO)へ溶解させた。10質量%スクロース含有10mMリン酸緩衝液pH7.4にてペプチドの濃度が0.25mg/mLとなるように希釈した。
腫瘍増殖試験
T75培養フラスコ(コーニング)を用い、マウス線維肉腫CMS5a細胞株を10%FBS含有RPMI1640培地中で培養した。培養した細胞は0.5%トリプシン含有PBSを用いて剥離し、10%FBS含有RPMI1640培地に懸濁した。懸濁液を遠心分離(400×g、5分、4℃)した後に上清を除いた。その後、RPMI1640培地を用いて2回洗浄し、1×106個/100μLの濃度でRPMI1640培地に懸濁した。100μL/個体の用量でBALB/cマウスの右側前背部に皮下移植した(1群あたり5匹)。
比較例7-1-1の長鎖ペプチド抗原を投与する際にも、腫瘍移植7日後、10日後、13日後、及び16日後に、長鎖ペプチド50μgを、PBSに溶解した50μgのCpGオリゴDNA(味の素バイオファーマサービスジーンデザイン)と共にマウスの右側後背部に皮下投与した。その後、経時的に腫瘍面積を測定した。
実施例7-1のヒアルロン酸誘導体(A1/A2=6.62)と前記長鎖ペプチド抗原とを複合化して得た長鎖ペプチド抗原搭載HAナノゲルがんワクチンとCpGオリゴDNAを組み合わせた医薬組成物は、比較例7-1の前記長鎖ペプチド抗原とCpGオリゴDNAを組み合わせた医薬組成物よりも、CMS5a腫瘍の増殖を抑制することが明らかとなった。このことからA1/A2=6.62であるヒアルロン酸誘導体を用いることで、優れたがんワクチンを提供できると考えられる。
[試験例7]
[ヒアルロン酸誘導体の粒子サイズ分布におけるピークトップの保持時間比]
合成例1-1~1-3、実施例1-1~1-3、6-1、7-1、及び、比較例1-1~1-3、6-1、7-1、7-2で得たヒアルロン酸誘導体と、標準物質であるポリアクリル酸(50kDa)について、実施例1-1に記載の方法にてサンプルを調製し、以下に示す条件にてゲル浸透クロマトグラフィーによる測定を行った。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃

[試験例8]
合成例1-1~1-3、実施例1-1~1-3、6-1、7-1、及び、比較例1-1~1-3、6-1で得たヒアルロン酸誘導体の重量平均分子量(Mw)および数平均分子量(Mn)(ポリアクリル酸換算)を以下の条件で測定を行った。ポリアクリル酸を標準物質として、検量線を作成し、合成例1-1~1-3、実施例1-1~1-3、6-1、7-1、及び、比較例1―1~1-3、6-1で得たヒアルロン酸誘導体のポリアクリル酸を基準とした各分子量を算出した。前記ポリアクリル酸標準物質としてはPSS-Paa2k(2kDa)、4k(4kDa)、8k(8kDa)、18k(18kDa)、40k(40kDa)、150k(150kDa)(PSS Polymer Standard service GmbH社製(ポリアクリル酸ナトリウム))を用いた。結果を表14に示す。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
サンプル濃度:合成例、実施例、比較例で得たヒアルロン酸誘導体は1mg/ml、ポリアクリル酸標準物質は2mg/ml
検量線条件:近似式として3次:At3+Bt2+Ct+Dを用いた。

[実施例8]
合成例1-1で取得したヒアルロン酸誘導体を20mg/mLで注射用水に溶解し、以下に示す条件にてゲル浸透クロマトグラフィーによる分取を行う。例えば、0.1分おきに分取する。
装置:HITACHI, CHROMASTER
カラム:G4000SWXL(東ソー社製、粒子径8mm、内径7.8mm、長さ30cm、品番:8542)
ガードカラム:TSKGEL GUARDCOLUMN SWXL (6.0mm I.D.X 4CM)
溶離液:10mm リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:100mL
検出器:RI
[実施例9-1~9-4、比較例9-1~9-4]
実施例1-1と同様な手法で分取したヒアルロン酸誘導体について、濃度5mg/mLの水溶液を準備し、ヒアルロン酸誘導体を含有する水相と油相の混合比について詳細な検討を行った。
6mLのクリーンバイアルに、ペプチド薬物であるシクロスポリン(東京化成工業社製、製品番号:C2408)を秤量し、ペプチドが50mg/mLの濃度となるようにDMSO(富士フイルム和光社製、製品番号:045-24511)へ溶解させた。
続いて、5mg/mLのヒアルロン酸誘導体400μLに対し、薬物を含有した油相である50mg/mLのシクロスポリンを4μL添加した。その後、下記表15に従って油相DMSOを混合させた。十分に混合した後、最終的なヒアルロン酸誘導体濃度が一定となるように注射用水を下記表15に従い添加する。さらに、24時間、室温(25℃)でインキュベートした後に、以下に示す条件にてゲル浸透クロマトグラフィーによる測定を行い、実施例1-1と同様の方法で面積比A1/A2を算出した(表15)。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
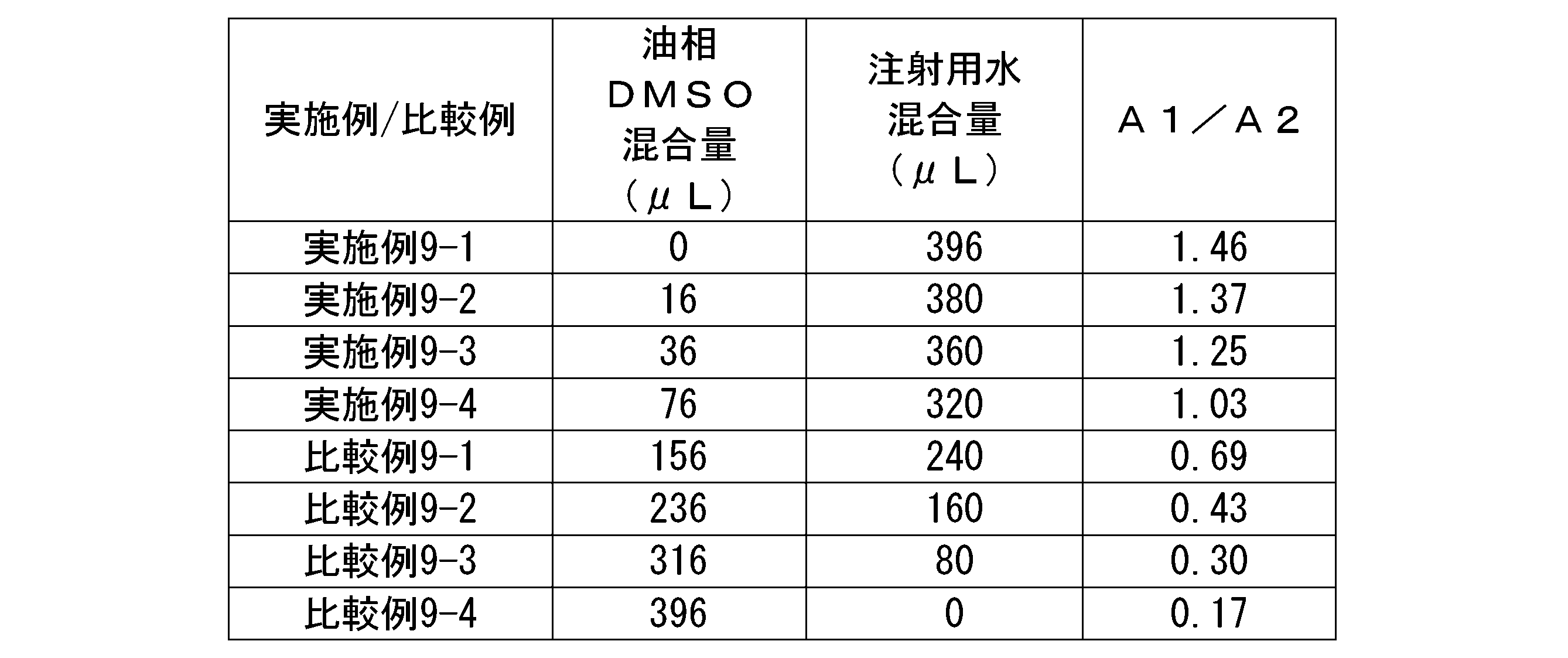
Claims (16)
- ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、以下に示す方法により算出される面積A1とA2の比A1/A2が0.90以上である、ヒアルロン酸誘導体。
(i)標準物質である50kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KbからベースラインBへ引いた垂線と、前記ヒアルロン酸誘導体のクロマトグラムとの交点をUbとする;
(ii)前記ヒアルロン酸誘導体のクロマトグラムで前記Ubから終点までの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA2とし、前記ヒアルロン酸誘導体のクロマトグラムで始点から前記Ubまでの曲線と、前記屈折率強度極大点Kbから前記ベースラインBへ引いた垂線と、ベースラインで囲まれた面積値をA1とする。 - ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、以下に示す方法により算出される距離DaとDbの比Da/Dbが0.00超1.20以下である、請求項1に記載のヒアルロン酸誘導体。
(i)標準物質である150kDaのポリアクリル酸のクロマトグラム上の屈折率強度極大点KaからベースラインBへ垂線を引き、ベースラインとの交点をBa、前記屈折率強度極大点Kaと前記Baの間の長さをLaとする;
(ii)屈折率強度がLa/20となるクロマトグラム上の2点のうち、溶出時間が早いほうを点R1とし、溶出時間が遅いほうを点S1とする;
(iv)前記点R1と前記点S1を結んだ直線D1と、前記屈折率強度極大点KaからベースラインBへ引いた垂線との交点をTaとし、前記屈折率強度極大点KbからベースラインBへ引いた垂線と前記直線D1との交点をTbとする;
(v)前記点R1と前記Taの距離をDa、前記Taと前記Tbの距離をDbとする。 - 前記面積A1とA2の比A1/A2が1.70以上である、請求項1又は2に記載のヒアルロン酸誘導体。
- ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから算出される、標準物質である50kDaのポリアクリル酸の屈折率強度極大点における保持時間Prに対する、ヒアルロン酸誘導体の屈折率強度極大点における保持時間Ptの比Pt/Prが0.5以上1.0未満である、ヒアルロン酸誘導体。 - ステリル基が導入されたヒアルロン酸誘導体であって、
ゲル浸透クロマトグラフィー測定により求められるクロマトグラムから、各分子量が2kDa、4kDa、8kDa、18kDa、40kDa、および150kDaであるポリアクリル酸を標準物質として作成した検量線に基づき算出されるヒアルロン酸誘導体の重量平均分子量が11万以上50万未満である、ヒアルロン酸誘導体。 - 前記ヒアルロン酸誘導体が、下記一般式(I)で表される繰り返し単位を1以上有する、請求項1、4又は5に記載のヒアルロン酸誘導体。

Zは、直接結合、又は2個以上30個以下の任意のアミノ酸残基からなるペプチドリンカーを表し;
X1は、以下の式:
-NRb-R、
-NRb-COO-R、
-NRb-CO-R、
-NRb-CO-NRc-R、
-COO-R、
-O-COO-R、
-S-R、
-CO-Ya-S-R、
-O-CO-Yb-S-R、
-NRb-CO-Yb-S-R、及び
-S-S-R、
で表される基からなる群より選択される基であり;
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、ここで当該基のアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよく;
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Rは、ステリル基であり;
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-であり、ここで、当該アルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよく;
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択され、当該基のアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよく;
Yaは、C1-5アルキレンであり;
Ybは、C2-8アルキレン又はC2-8アルケニレンであり;
mは、1以上100以下の整数である。) - 前記ステリル基がコレステリル基である、請求項1、4又は5に記載のヒアルロン酸誘導体。
- 前記ヒアルロン酸誘導体を構成する二糖の繰り返し単位に対する、ステリル基の導入率が、30%以上60%以下である、請求項1、4又は5に記載のヒアルロン酸誘導体。
- 前記ヒアルロン酸誘導体の重量平均分子量(絶対分子量)が4,000以上1,000,000以下である、請求項1、4又は5に記載のヒアルロン酸誘導体。
- 前記ヒアルロン酸誘導体の重量平均分子量(絶対分子量)が5,000以上25,000以下である、請求項9に記載のヒアルロン酸誘導体。
- 請求項1、4又は5に記載のヒアルロン酸誘導体と、薬効成分と、を含有する、医薬組成物。
- がん、感染症、及び免疫疾患からなる群より選ばれる1種以上の疾患の予防又は治療用である、請求項11に記載の医薬組成物。
- 前記薬効成分として、がん抗原、感染症由来抗原、及び免疫疾患における自己抗原からなる群から選択される少なくとも一つを含み、且つ、アジュバントを更に含む、請求項11に記載の医薬組成物。
- 前記薬効成分として、がん抗原又は感染症由来抗原を含む、請求項13に記載の医薬組成物。
- 請求項1、4又は5に記載のヒアルロン酸誘導体と、薬効成分と、を含有する、医薬組成物の製造方法であって、
前記薬効成分を有機溶媒又は有機溶媒含有水に溶解して、前記薬効成分を含有する油相を調製する調製工程と、
前記油相と前記ヒアルロン酸誘導体を含有する水相との混合比が容量比で20:100~0.01:100となるように混合する混合工程と、
を含む、医薬組成物の製造方法。 - 前記ヒアルロン酸誘導体を含有する水相のpHが6.00以上11.00以下である、請求項15に記載の医薬組成物の製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18/861,810 US20250339460A1 (en) | 2022-05-31 | 2023-05-31 | Hyaluronic acid derivative, pharmaceutical composition, and method for producing pharmaceutical composition |
| KR1020247033916A KR20240164786A (ko) | 2022-05-31 | 2023-05-31 | 히알루론산 유도체, 의약 조성물 및 의약 조성물의 제조 방법 |
| EP23816133.5A EP4534566A4 (en) | 2022-05-31 | 2023-05-31 | HYALURONIC ACID DERIVATIVE, PHARMACEUTICAL COMPOSITION AND METHOD FOR PRODUCING A PHARMACEUTICAL COMPOSITION |
| JP2024524929A JP7684519B2 (ja) | 2022-05-31 | 2023-05-31 | ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 |
| CN202380043355.6A CN119301160A (zh) | 2022-05-31 | 2023-05-31 | 透明质酸衍生物、药物组合物以及药物组合物的制造方法 |
| JP2025082087A JP7818130B2 (ja) | 2022-05-31 | 2025-05-15 | ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022088995 | 2022-05-31 | ||
| JP2022-088995 | 2022-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023234376A1 true WO2023234376A1 (ja) | 2023-12-07 |
Family
ID=89024934
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/020372 Ceased WO2023234376A1 (ja) | 2022-05-31 | 2023-05-31 | ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20250339460A1 (ja) |
| EP (1) | EP4534566A4 (ja) |
| JP (2) | JP7684519B2 (ja) |
| KR (1) | KR20240164786A (ja) |
| CN (1) | CN119301160A (ja) |
| TW (2) | TW202515587A (ja) |
| WO (1) | WO2023234376A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025216306A1 (ja) * | 2024-04-11 | 2025-10-16 | 旭化成株式会社 | 担体組成物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010053140A1 (ja) | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| WO2017164409A1 (ja) | 2016-03-25 | 2017-09-28 | 国立大学法人大阪大学 | 疾患の要因となる生体内タンパク質を標的とするコンジュゲートワクチン |
| WO2020158771A1 (ja) | 2019-01-29 | 2020-08-06 | 国立大学法人三重大学 | がんワクチン製剤 |
| WO2021157665A1 (ja) * | 2020-02-07 | 2021-08-12 | 旭化成株式会社 | ヒアルロン酸誘導体、医薬組成物及びヒアルロン酸誘導体-薬物結合体 |
| JP2022088995A (ja) | 2020-12-03 | 2022-06-15 | 株式会社椿本チエイン | ワーク保持装置 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014038641A1 (ja) * | 2012-09-05 | 2014-03-13 | 中外製薬株式会社 | アミノ酸およびステリル基が導入されたヒアルロン酸誘導体 |
-
2023
- 2023-05-31 KR KR1020247033916A patent/KR20240164786A/ko active Pending
- 2023-05-31 CN CN202380043355.6A patent/CN119301160A/zh active Pending
- 2023-05-31 TW TW113148283A patent/TW202515587A/zh unknown
- 2023-05-31 US US18/861,810 patent/US20250339460A1/en active Pending
- 2023-05-31 EP EP23816133.5A patent/EP4534566A4/en active Pending
- 2023-05-31 TW TW112120351A patent/TWI870882B/zh active
- 2023-05-31 WO PCT/JP2023/020372 patent/WO2023234376A1/ja not_active Ceased
- 2023-05-31 JP JP2024524929A patent/JP7684519B2/ja active Active
-
2025
- 2025-05-15 JP JP2025082087A patent/JP7818130B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010053140A1 (ja) | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| WO2017164409A1 (ja) | 2016-03-25 | 2017-09-28 | 国立大学法人大阪大学 | 疾患の要因となる生体内タンパク質を標的とするコンジュゲートワクチン |
| WO2020158771A1 (ja) | 2019-01-29 | 2020-08-06 | 国立大学法人三重大学 | がんワクチン製剤 |
| WO2021157665A1 (ja) * | 2020-02-07 | 2021-08-12 | 旭化成株式会社 | ヒアルロン酸誘導体、医薬組成物及びヒアルロン酸誘導体-薬物結合体 |
| JP2022088995A (ja) | 2020-12-03 | 2022-06-15 | 株式会社椿本チエイン | ワーク保持装置 |
Non-Patent Citations (7)
| Title |
|---|
| NOUBADE R ET AL.: "Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity", FRONT IMMUNOL., vol. 10, 2019, pages 1 - 13 |
| SANSOM DM ET AL.: "What's the difference between CD80 and CD86?", TRENDS IN IMMUNOLOGY, vol. 24, 2003, pages 313 - 318, XP004430847, DOI: 10.1016/S1471-4906(03)00111-X |
| See also references of EP4534566A4 |
| SHIGEHARU FUJITA ET AL.: "Regulatory dendritic cells protect against allergic airway inflammation in a murine asthmatic model", J ALLERGY CLIN IMMUNOL, vol. 121, 2008, pages 95 - 104 |
| TAAMS LS ET AL.: "Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells", HUM IMMUNOL., vol. 66, 2005, pages 222 - 230, XP004803416, DOI: 10.1016/j.humimm.2004.12.006 |
| YAMANE, SETSUKO; OHNUMA, KIYOSHI; SAWADA, SHIN-ICHI; SASAKI, YOSHIHIRO; AKIYOSHI, KAZUNARI: "2B6-29 Synthesis of hydrophobized hyaluronic acid-calcium phosphate hybrid fine particle", PROCEEDINGS OF THE 96TH ANNUAL SPRING CONFERENCE OF THE CHEMICAL SOCIETY OF JAPAN (CSJ); KYOTANABE, JAPAN; MARCH 24-27, 2016, vol. 96, 30 November 2015 (2015-11-30) - 27 March 2016 (2016-03-27), pages 2B6 - 29, XP009538320 * |
| YUTAKA MASUDA, HIROKI MATSUSHIMA, SETSUKO YAMANE, KIYOSHI ONUMA, SHINICHI SAWADA, YOSHIHIRO SASAKI, KAZUNARI AKIYOSHI: "1PB-030 Synthesis of Silica-Hydrophobized Hyaluronic Acid Hybrid Hydrogel", PROCEEDINGS OF THE 99TH ANNUAL SPRING MEETING OF THE CHEMICAL SOCIETY OF JAPAN (CSJ); KOBE, JAPAN; MARCH 16-19, 2019, vol. 99, 30 November 2018 (2018-11-30) - 19 March 2019 (2019-03-19), pages 1PB - 030, XP009538319 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025216306A1 (ja) * | 2024-04-11 | 2025-10-16 | 旭化成株式会社 | 担体組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI870882B (zh) | 2025-01-21 |
| JP2025113317A (ja) | 2025-08-01 |
| KR20240164786A (ko) | 2024-11-20 |
| JP7684519B2 (ja) | 2025-05-27 |
| TW202515587A (zh) | 2025-04-16 |
| EP4534566A1 (en) | 2025-04-09 |
| EP4534566A4 (en) | 2025-09-03 |
| JP7818130B2 (ja) | 2026-02-19 |
| CN119301160A (zh) | 2025-01-10 |
| JPWO2023234376A1 (ja) | 2023-12-07 |
| US20250339460A1 (en) | 2025-11-06 |
| TW202402308A (zh) | 2024-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12097252B2 (en) | Formulation of a peptide vaccine | |
| JP6041314B2 (ja) | がん治療用ワクチン製剤 | |
| JP6792452B2 (ja) | 免疫原性化合物 | |
| RS52115B (sr) | Nove formulacije tumor-asociranih peptida koji se vezuju za molekule humanog leukocitnog antigena (hla) klase i ili ii za vakcine | |
| JP7818130B2 (ja) | ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 | |
| US20250360217A1 (en) | Hyaluronic acid derivative pharmaceutical composition and method of producing pharmaceutical composition | |
| Ji et al. | Multi-functional nanocomplex codelivery of Trp2 and R837 to activate melanoma-specific immunity | |
| Liu et al. | Preparation and evaluation of antigen/N-trimethylaminoethylmethacrylate chitosan conjugates for nasal immunization | |
| KR20090112590A (ko) | 백신을 위한 인간 조직 적합성 항원(hla) 종류 i 또는 ii 분자에 결합하는 종양 관련 펩티드의 새로운 제형 | |
| Yu et al. | Conjugation with loxoribine and mannan improves the immunogenicity of Mycobacterium tuberculosis CFP10-TB10. 4 fusion protein | |
| JP2023089973A (ja) | ワクチン組成物 | |
| KR20250073419A (ko) | 테르페노이드 및 거대분자를 포함하는 신규한 조성물, 및 그의 용도 | |
| WO2024019118A1 (ja) | ヒアルロン酸誘導体医薬品組成物及び医薬品組成物 | |
| HK1223290A1 (zh) | 结合到人类白细胞抗原(hla)i类或ii类分子上用作疫苗的肿瘤相关肽新制剂 | |
| TW202412847A (zh) | 包含類萜和大分子的新穎組合物及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23816133 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2024524929 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20247033916 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020247033916 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202417090090 Country of ref document: IN |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112024023894 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380043355.6 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023816133 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2023816133 Country of ref document: EP Effective date: 20250102 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024135382 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380043355.6 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024135382 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112024023894 Country of ref document: BR Free format text: APRESENTE NOVAS FOLHAS DO RELATORIO DESCRITIVO E RESUMO ADAPTADAS AOS ARTS. 26 E 40 DA PORTARIA NO 14/2024, UMA VEZ QUE O CONTEUDO ENVIADO NA PETICAO NO 870240097885 ENCONTRA-SE FORA DA NORMA: OS DOCUMENTOS DEVEM SER INICIADOS PELO TITULO CENTRALIZADO SEM O USO DE PALAVRAS ADICIONAIS (RELATORIO DESCRITIVO DE?, PATENTE DE INVENCAO...). A EXIGENCIA DEVE SER RESPONDIDA EM ATE 60 (SESSENTA) DIAS DE SUA PUBLICACAO E DEVE SER REALIZADA POR MEIO DA PETICAO GRU CODIGO DE SERVICO 207. |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023816133 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 112024023894 Country of ref document: BR Kind code of ref document: A2 Effective date: 20241114 |
|
| WWP | Wipo information: published in national office |
Ref document number: 18861810 Country of ref document: US |