WO2024019127A1 - ヒアルロン酸誘導体医薬組成物及び医薬組成物の製造方法 - Google Patents
ヒアルロン酸誘導体医薬組成物及び医薬組成物の製造方法 Download PDFInfo
- Publication number
- WO2024019127A1 WO2024019127A1 PCT/JP2023/026679 JP2023026679W WO2024019127A1 WO 2024019127 A1 WO2024019127 A1 WO 2024019127A1 JP 2023026679 W JP2023026679 W JP 2023026679W WO 2024019127 A1 WO2024019127 A1 WO 2024019127A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hyaluronic acid
- acid derivative
- pharmaceutical composition
- group
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/726—Glycosaminoglycans, i.e. mucopolysaccharides
- A61K31/728—Hyaluronic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/02—Peptides of undefined number of amino acids; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
- A61K38/13—Cyclosporins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/40—Cyclodextrins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0072—Hyaluronic acid, i.e. HA or hyaluronan; Derivatives thereof, e.g. crosslinked hyaluronic acid (hylan) or hyaluronates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L5/00—Compositions of polysaccharides or of their derivatives not provided for in groups C08L1/00 or C08L3/00
- C08L5/08—Chitin; Chondroitin sulfate; Hyaluronic acid; Derivatives thereof
Definitions
- the present invention relates to a hyaluronic acid derivative pharmaceutical composition and a method for producing the pharmaceutical composition.
- This application is filed in Japanese Patent Application No. 2022-115839 filed in Japan on July 20, 2022, Japanese Patent Application No. 2022-151823 filed in Japan on September 22, 2022, and Patent Application No. 2022-151823 filed in Japan on July 20, 2022. , claims priority based on Japanese Patent Application No. 2022-115841, the contents of which are incorporated herein.
- biopharmaceuticals that contain proteins, peptides, and nucleic acids as active ingredients have been put into practical use, and their number continues to increase year by year. Biopharmaceuticals can fill unmet medical needs that could not be met with conventional small molecule drugs.
- biopharmaceuticals have the problem of being difficult to absorb from the gastrointestinal tract and mucous membranes, being unstable in the body, and having a short half-life in the blood. Therefore, biopharmaceuticals require frequent administration by injection, which places a heavy burden on both patients and medical personnel. Therefore, there is a need for drug base materials (sustained release drug delivery system base materials) that can encapsulate biopharmaceuticals and gradually release the active ingredients in vivo without impairing pharmacological activity.
- Patent Document 1 proposes a sustained release drug delivery system base material made of a highly safe hyaluronic acid derivative.
- the hyaluronic acid derivative disclosed in Patent Document 1 spontaneously associates in an aqueous solution and can efficiently encapsulate drugs, particularly biopharmaceuticals, while maintaining their biological activity. As a result, it aggregates under physiological saline concentration (or disperses even under physiological saline concentration) and has good retention in blood.
- This hyaluronic acid derivative can be used as a carrier that can efficiently encapsulate many drugs while maintaining pharmacological activity, especially when biopharmaceuticals are used as active ingredients, and as a sustained release carrier and targeting carrier with excellent blood retention. It is said that it can be used as a local (for example, subcutaneous) sustained release carrier that can continuously release drugs.
- Method of adding a solubilizing agent A method of adding a surfactant to form micelles, emulsify, and solubilize. Method using serum albumin or plasma proteins.
- the method (a) above involves changing a part of the structure of the active ingredient, which is the active ingredient, and cannot increase the solubility of the active ingredient itself.
- the use of derivatives may cause various problems such as a decrease in the activity as a drug and precipitation of the drug due to changes in pH, which is not a desirable method.
- a poorly water-soluble cyclosporine composition is prepared using an organic solvent, a solubilizing agent, water and an organic solvent, water and a solubilizing agent, or a mixed solvent of water, an organic solvent, and a solubilizing agent.
- a manufacturing method is described that involves a step of dissolving in.
- the obtained solution was a turbid solution, and partial precipitation was confirmed, indicating that solubilization was not sufficient.
- Patent Document 3 when encapsulating a poorly water-soluble active ingredient in a hyaluronic acid derivative, it is necessary to dissolve the poorly water-soluble active ingredient in an organic solvent such as methanol. Therefore, the use of organic solvents is unavoidable. In addition, in Patent Document 3, the amount of solubilized poorly water-soluble active ingredients is insufficient. Furthermore, no specific studies have been made on formulation methods for efficiently solubilizing poorly water-soluble active ingredients from a powdered state, and there is still room for improvement.
- Patent Document 2 even if a solubilizing agent such as polyethylene glycol 300 is used for a poorly water-soluble active ingredient, only a relatively stable suspension containing the poorly water-soluble active ingredient is provided. It is difficult to completely solubilize poorly water-soluble active ingredients at high concentrations.
- a solubilizing agent such as polyethylene glycol 300
- Sustained-release drug bases are required to be able to maintain the concentration of the active ingredient in vivo over a longer period of time and gradually release the active ingredient. In addition, it is required to be able to solubilize active ingredients at high concentrations.
- the present invention has been made in view of the above circumstances, and provides a hyaluronic acid derivative that can maintain the concentration of an active ingredient in the body for a longer period of time, gradually release the active ingredient, and solubilize the active ingredient at a high concentration.
- An object of the present invention is to provide a pharmaceutical composition and a method for producing the same.
- the present invention has been made in view of the above circumstances, and is capable of solubilizing poorly water-soluble active ingredients at high concentrations without using organic solvents and reducing the amount of highly toxic surfactants used.
- An object of the present invention is to provide a hyaluronic acid derivative pharmaceutical composition and a method for producing the same.
- a hyaluronic acid derivative pharmaceutical composition comprising (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient.
- a hyaluronic acid derivative pharmaceutical composition comprising (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient.
- B contains at least 4 or more ether structures (ROR) and has 4 or more carbon atoms.
- the association promoter (B) is polysorbate 80, polysorbate 20, poloxamer, oxyethylene castor oil, polyethylene glycol 300, polyethylene glycol 400, polyethylene glycol 4000, fatty acid sorbitan ester, tocopheryl polyethylene glycol succinate, and polyvinyl
- the content of the active ingredient (C) relative to 100 parts by mass of the (A) hyaluronic acid derivative into which a hydrophobic group has been introduced is 10 parts by mass or more and 100 parts by mass or less, [1] to [11]
- the hyaluronic acid derivative pharmaceutical composition according to any one of the above.
- the hyaluronic acid derivative (A) into which a hydrophobic group has been introduced has one or more repeating units represented by the following general formula (I), according to any one of [1] to [12].
- Hyaluronic acid derivative pharmaceutical composition is one or more repeating units represented by the following general formula (I), according to any one of [1] to [12].
- R 1 , R 2 , R 3 , and R 4 are each independently a group selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl.
- Z represents a direct bond or a peptide linker consisting of 2 to 30 arbitrary amino acid residues.
- X 1 is -NR b -R, -NR b -COO-R, -NR b -CO-R, -NR b -CO-NR c -R, -COO-R, -O-COO-R, - Represented by SR, -CO-Y a -SR, -O-CO-Y b -SR, -NR b -CO-Y b -SR, and -SSR, It is a group selected from the group consisting of groups.
- R a , R b and R c are each independently a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl and hydroxy C 2-20 alkyl.
- the alkyl moieties of R a , R b and R c may have a group selected from the group consisting of -O- and -NR f - inserted therein.
- R f is a group selected from the group consisting of a hydrogen atom, C 1-12 alkyl, amino C 2-12 alkyl, and hydroxy C 2-12 alkyl.
- the alkyl portion of R f may have a group selected from the group consisting of -O- and -NH- inserted therein.
- R is a steryl group.
- Y is C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -.
- the alkylene of Y may have a group selected from the group consisting of -O-, -NR g - and -SS- inserted therein.
- R g is a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl, and hydroxy C 2-20 alkyl.
- the alkyl portion of R g may have a group selected from the group consisting of -O- and -NH- inserted therein.
- Y a is C 1-5 alkylene.
- Y b is C 2-8 alkylene or C 2-8 alkenylene.
- m is an integer of 1 or more and 100 or less.
- hyaluronic acid derivative pharmaceutical composition according to any one of [1] to [16], which can be sterilized by filtration.
- a method for producing a hyaluronic acid derivative pharmaceutical composition comprising (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient, the method comprising: A) A step of mixing the hyaluronic acid derivative into which a hydrophobic group has been introduced and the association promoter (B) to obtain an aqueous hyaluronic acid derivative solution containing the association promoter, and (C) containing the active ingredient and the association promoter.
- a method for producing a hyaluronic acid derivative pharmaceutical composition comprising a mixing step of mixing with a hyaluronic acid derivative aqueous solution.
- a method for producing a hyaluronic acid derivative pharmaceutical composition comprising (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient, the method comprising: C) A step of dispersing the active ingredient in the association promoter (B) to obtain a dispersion (I), and a step of preparing an aqueous hyaluronic acid derivative solution or an aqueous hyaluronic acid solution containing an association promoter to obtain an aqueous solution (II).
- the present invention includes the following aspects.
- [21] Contains (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient, the solubilization aid (B1) having an ether structure (R The content of the solubilizing aid (B1) with respect to the hyaluronic acid derivative containing at least 4 or more -OR) and having 4 or more carbon atoms and into which 100 parts by mass of the hydrophobic group (A1) has been introduced. 0.0001 parts by mass or more and 15,000 parts by mass or less, a hyaluronic acid derivative pharmaceutical composition.
- the solubilization aid (B1) is one or more selected from the group consisting of a nonionic surfactant, a polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/mol or less, and a cyclodextrin derivative.
- a nonionic surfactant e.g., a polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/mol or less
- a cyclodextrin derivative e.g., a nonionic surfactant, a polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/mol or less.
- the solubilization aid (B1) is polysorbate 80, polysorbate 65, polysorbate 60, polysorbate 40, polysorbate 20, poloxamer, polyoxyethylene hydrogenated castor oil, cyclodextrin derivative, polyethylene glycol 300, polyethylene glycol 400, polyethylene
- the solubilization aid (B1) is a nonionic surfactant, and the content of the nonionic surfactant is 0 with respect to 100 parts by mass of the (A) hyaluronic acid derivative into which a hydrophobic group has been introduced.
- the hyaluronic acid derivative pharmaceutical composition according to any one of [21] to [23], which contains .0001 parts by mass or more and 150 parts by mass or less.
- the solubilization aid (B1) is polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/moL or less, and the polyethylene glycol is The hyaluronic acid derivative pharmaceutical composition according to any one of [21] to [24], wherein the content is 25 parts by mass or more and 15,000 parts by mass or less.
- the amount of the poorly water-soluble drug added to 100 parts by mass of the (A1) hyaluronic acid derivative into which a hydrophobic group has been introduced is 21 parts by mass or more and less than 100 parts by mass, [26] to [31]
- hyaluronic acid derivative pharmaceutical composition according to any one of [31] to [32], wherein the hyaluronic acid derivative has one or more repeating units represented by the following general formula (I).
- R 1 , R 2 , R 3 , and R 4 are each independently a group selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl.
- Z represents a direct bond or a peptide linker consisting of 2 to 30 arbitrary amino acid residues.
- X 1 is -NR b -R, -NR b -COO-R, -NR b -CO-R, -NR b -CO-NR c -R, -COO-R, -O-COO-R, - Represented by SR, -CO-Y a -SR, -O-CO-Y b -SR, -NR b -CO-Y b -SR, and -SSR, A group selected from the group consisting of groups.
- R a , R b and R c are each independently a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl and hydroxy C 2-20 alkyl.
- the alkyl moieties of R a , R b and R c may have a group selected from the group consisting of -O- and -NR f - inserted therein.
- R f is a group selected from the group consisting of a hydrogen atom, C 1-12 alkyl, amino C 2-12 alkyl, and hydroxy C 2-12 alkyl.
- the alkyl portion of R f may have a group selected from the group consisting of -O- and -NH- inserted therein.
- R is a steryl group.
- Y is C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -.
- the alkylene of Y may have a group selected from the group consisting of -O-, -NR g - and -SS- inserted therein.
- R g is a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl, and hydroxy C 2-20 alkyl.
- the alkyl portion of R g may have a group selected from the group consisting of -O- and -NH- inserted therein.
- Y a is C 1-5 alkylene.
- Y b is C 2-8 alkylene or C 2-8 alkenylene.
- m is an integer of 1 or more and 100 or less.
- a method for producing a pharmaceutical composition comprising (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient, the method comprising (A1) A step of mixing a hyaluronic acid derivative into which a hydrophobic group has been introduced and (B1) a solubilization aid to obtain a hyaluronic acid derivative aqueous solution containing the solubilization aid; A method for producing a pharmaceutical composition, comprising a mixing step of mixing with an aqueous hyaluronic acid derivative solution.
- a method for producing a pharmaceutical composition comprising (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient, the method comprising: ) A step of dispersing the active ingredient in the solubilization aid (B1) to obtain a dispersion (I), and a step of preparing an aqueous hyaluronic acid derivative solution or an aqueous hyaluronic acid solution containing a solubilizer to obtain an aqueous solution (II). and mixing the dispersion liquid (I) and the aqueous solution (II).
- the hyaluronic acid derivative pharmaceutical composition can maintain the concentration of the active ingredient in vivo for a longer period of time, gradually release the active ingredient, and solubilize the active ingredient at a high concentration. It is possible to provide a product and a method for manufacturing the same.
- the hyaluronic acid derivative pharmaceutical can solubilize a poorly water-soluble active ingredient at a high concentration without using an organic solvent and while reducing the amount of highly toxic surfactant used.
- Compositions and methods for producing the same can be provided.
- the present embodiment an embodiment of the present invention (hereinafter referred to as “the present embodiment") will be described in detail, but the present invention is not limited thereto, and various modifications can be made without departing from the gist thereof. It is.
- C 1-20 alkyl used herein means a straight or branched alkyl group having 1 to 20 carbon atoms, such as methyl, ethyl, n-propyl, iso - Contains "C 1-4 alkyl” such as propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, and further includes n-pentyl, 3-methylbutyl, 2-methylbutyl, 1-methylbutyl, 1-methylbutyl, -Ethylpropyl, n-hexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3-ethylbutyl, 2-ethylbutyl, and the like.
- C 1-20 alkyl also includes C 1-12 alkyl groups having 1 to 12 carbon atoms and C 1-6 alkyl groups having 1 to 6 carbon atoms.
- C 1-6 alkylcarbonyl as used herein means an alkylcarbonyl group in which the alkyl moiety is C 1-6 alkyl as already mentioned, for example acetyl, propionyl, n-propylcarbonyl, Includes “C 1-4 alkylcarbonyl” such as iso-propylcarbonyl, n-butylcarbonyl, sec-butylcarbonyl, iso-butylcarbonyl, and tert-butylcarbonyl.
- amino C 2-20 alkyl used herein means a straight or branched alkyl having 2 to 20 carbon atoms and having an amino group as a substituent, for example, an amino group may be located on the terminal carbon atom of the alkyl group.
- Amino C 2-20 alkyl also includes amino C 2-12 alkyl having 2 or more and 12 or less carbon atoms.
- hydroxyC 2-20 alkyl used herein means a linear or branched alkyl group having 2 to 20 carbon atoms and having a hydroxy group as a substituent, for example, hydroxy The group may be located on the terminal carbon atom of the alkyl group. Hydroxy C 2-20 alkyl also includes hydroxy C 2-12 alkyl having 2 or more and 12 or less carbon atoms.
- C2-30 alkylene used herein means a linear or branched divalent saturated hydrocarbon group having 2 to 30 carbon atoms, such as ethylene, propylene, etc. C 2-20 alkylene with 2 to 20 carbon atoms, C 2-8 alkylene with 2 to 8 carbon atoms, group "-(CH 2 ) n -" (where n is 2 to 30 and preferably 2 or more and 20 or less, more preferably 2 or more and 15 or less).
- C 1-5 alkylene used herein means a linear or branched divalent saturated hydrocarbon group having 1 to 5 carbon atoms, such as methylene, ethylene, Contains propylene etc.
- C 2-8 alkenylene refers to a divalent saturated hydrocarbon group containing one or more double bonds, linear or branched, having 2 to 8 carbon atoms.
- -CH CH-
- -C(CH 3 ) CH-
- 2-butene-1,4-diyl hepta-2,4-diene-1,6-diyl
- octa-2,4 , 6-triene-1,8-diyl etc.
- each isomer and mixtures thereof are also included.
- This embodiment is a hyaluronic acid derivative pharmaceutical composition containing (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient.
- a hyaluronic acid derivative pharmaceutical composition comprising (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient
- “Pharmaceutical Composition 1” It may be abbreviated as.
- Hyaluronic acid derivatives can be formulated with active ingredients and used as pharmaceutical compositions.
- the hyaluronic acid derivative forms a complex with the active ingredient (hereinafter sometimes referred to as "active ingredient-hyaluronic acid derivative complex").
- active ingredient-hyaluronic acid derivative complex the active ingredient-hyaluronic acid derivative complex
- the steryl group in the hyaluronic acid derivative and the hydrophobic site of the active ingredient form a complex through hydrophobic interaction, and the active ingredient and the hydrophobic site such as the steryl group are present in the center.
- the average particle diameter of the structure containing the active ingredient-hyaluronic acid derivative complex can be 20 nm or more and 220 nm or less, 20 nm or more and 150 nm or less, and 30 nm or more.
- the thickness can be set to 100 nm or less.
- the average particle diameter can be measured by, for example, DLS (Dynamic Light Scattering), a nanotracking particle measuring device, size exclusion chromatography, high performance liquid chromatography, electron microscopy, or the like.
- the measurement is performed by diluting the hyaluronic acid derivative with a 10 mM phosphate buffer containing 10 w/v% sucrose at a rate such that the hyaluronic acid derivative concentration is 1 mg/mL using a DLS device, for example.
- association promoter (B) included in the pharmaceutical composition 1 of the present embodiment contains at least 4 or more ether structures (ROR) and has 4 or more carbon atoms.
- composition 1 of this embodiment has (B) an association promoter.
- the association promoter (B) interacts with a hyaluronic acid derivative into which a hydrophobic group has been introduced, and promotes association between hyaluronic acid derivative molecules.
- the association promoter of this embodiment forms a complex with a hyaluronic acid derivative, increases drug conjugation ability, and improves precipitation performance under physiological salt concentration.
- the drug in the gel is sustainedly released by an exchange reaction in which it replaces hydrophobic components such as albumin and other hydrophobic proteins in the body, or by decomposition of the gel. It is thought that the sustained release period can be controlled by controlling the influx of degrading enzymes into the gel, shielding it, and imparting stealth properties.
- composition 1 of the present embodiment includes (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced.
- (A) hyaluronic acid derivative into which a hydrophobic group has been introduced may be referred to as “(A) hyaluronic acid derivative.”
- the hyaluronic acid derivative includes a steryl group as a hydrophobic group.
- the steryl group may be directly bonded to hyaluronic acid or may be bonded via a linker.
- the "linker” here can be any peptide linker that can be introduced by genetic engineering or a synthetic compound linker, but in the hyaluronic acid derivative (A) used in this embodiment, a peptide linker is preferable.
- the length of the peptide linker is not particularly limited and can be appropriately selected by those skilled in the art depending on the purpose, but the preferred length is 2 amino acids or more, particularly preferably 15 amino acids.
- the upper limit of the length of the peptide linker is not particularly limited, but is usually 30 amino acids or less, preferably 20 amino acids or less.
- the peptide linkers contained in the hyaluronic acid derivative may all have the same length, or may have different lengths.
- steryl group used herein is not particularly limited as long as it is a group having a steroid skeleton.
- the steroids specifically include cholesterol, cholestanol, campestanol, ergostanol, stigmastanol, coprostanol, stigmasterol, sitosterol, lanosterol, ergosterol, simiarenol, bile acids, testosterone, estradiol, and pro-steroids. Examples include gestrone, cortisol, cortisone, aldosterone, corticosterone, deoxycortisterone, and the like.
- steryl group examples include a cholesteryl group, a stigmasteryl group, a lanosteryl group, an ergosteryl group, and among them, a cholesteryl group (particularly a cholest-5-en-3 ⁇ -yl group) is preferred.
- Stepryl group introduction rate The introduction rate of steryl groups into the hyaluronic acid derivative (hereinafter sometimes simply referred to as "steryl group introduction rate") is preferably 0.1% or more and less than 50%, more preferably 5% or more and less than 45%, It is more preferably 10% or more and 40% or less, particularly preferably 15% or more and 35% or less.
- the hyaluronic acid derivative (A) can strongly interact with the hydrophobic part of the poorly water-soluble drug or protein and the hydrophobic part of the association promoter. Furthermore, since the steryl group introduction rate is within the above range, the hyaluronic acid derivative-drug complex in which the (A) hyaluronic acid derivative in Pharmaceutical Composition 1 is complexed with a drug has improved formulation stability. It also aggregates and precipitates under physiological salt concentration, allowing sustained release of the drug.
- the steryl group introduction rate can be measured by 1 H-NMR measurement. That is, the integral value of the peak derived from the steryl group of (A) the hyaluronic acid derivative in the 1 H-NMR spectrum of Pharmaceutical Composition 1, and the acetyl group of N-acetyl-D-glucosamine contained in (A) the hyaluronic acid derivative. It can be calculated based on the following formula using the integral value of the peak derived from (COCH 3 , 1.6 ppm or more and 2.0 ppm or less, 3H). In addition, during the ceremony. nH represents the number of hydrogen atoms corresponding to the peak. Specifically, it can be measured, for example, according to the method described in Examples below.
- Stepryl group introduction rate] (%) [(Peak integral value derived from steryl group ⁇ 3/n H )/(Peak integral value derived from acetyl group of N-acetyl-D-glucosamine)] ⁇ 100
- the molecular weight of the (A) hyaluronic acid derivative is not particularly limited, but from the viewpoint of improving the sustained release function derived from diffusion delay in local administration, the (A) hyaluronic acid derivative with a relatively large molecular weight is preferred. On the other hand, when the final dosage form is a solution preparation, (A) hyaluronic acid derivatives having a relatively small molecular weight are preferred from the viewpoint of syringability. (A) The molecular weight of the hyaluronic acid derivative may be adjusted as appropriate depending on the use and dosage form.
- An example of the molecular weight of the hyaluronic acid derivative is preferably 1,000 (1k) or more and 1,000,000 (1,000k) or less, more preferably 5k or more and 300k or less, and even more preferably 5k or more and 120k or less, Particularly preferred is 7k or more and 100k or less.
- the molecular weight of the hyaluronic acid derivative can generally be adjusted by using raw materials having a corresponding molecular weight.
- the weight average molecular weight of the hyaluronic acid derivative is at least the above lower limit, an increase in viscosity can be suppressed, and a higher concentration of the hyaluronic acid derivative can be dissolved in the pharmaceutical composition 1.
- the weight average molecular weight of hyaluronic acid derivatives can generally be adjusted by using raw materials with corresponding molecular weights. More specifically, from the viewpoint of viscosity, the weight average molecular weight of the hyaluronic acid derivative is preferably 100k or less, more preferably 50k or less, and particularly preferably 40k or less. From the viewpoint of producing a hyaluronic acid derivative, 4k to 100k is preferable, and 6k to 50k is particularly preferable.
- the hyaluronic acid derivative strongly exhibits the properties as hyaluronic acid, for example, in order to be strongly recognized by the CD44 receptor, it is preferably 100 kDa or more, particularly preferably 200 kDa or more, and most preferably 300 kDa or more.
- the "molecular weight of the hyaluronic acid derivative” as used herein is the weight average molecular weight determined by size exclusion chromatography multi-angle light scattering detector (SEC-MALS).
- hyaluronic acid derivatives include, for example, hyaluronic acid derivatives having one or more repeating units represented by the following general formula (I) (hereinafter sometimes referred to as “repeat units (I)"). It will be done.
- R 1 , R 2 , R 3 , and R 4 are each independently a group selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl.
- Z represents a direct bond or a peptide linker consisting of 2 to 30 arbitrary amino acid residues.
- X 1 is -NR b -R, -NR b -COO-R, -NR b -CO-R, -NR b -CO-NR c -R, -COO-R, -O-COO-R, - Represented by SR, -CO-Y a -SR, -O-CO-Y b -SR, -NR b -CO-Y b -SR, and -SSR, A group selected from the group consisting of groups.
- R a , R b and R c are each independently a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl and hydroxy C 2-20 alkyl.
- R f is a group selected from the group consisting of a hydrogen atom, C 1-12 alkyl, amino C 2-12 alkyl, and hydroxy C 2-12 alkyl.
- the alkyl portion of R f may have a group selected from the group consisting of -O- and -NH- inserted therein.
- R is a steryl group.
- Y is C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -.
- the alkylene of Y may have a group selected from the group consisting of -O-, -NR g - and -SS- inserted therein.
- R g is a group selected from the group consisting of a hydrogen atom, C 1-20 alkyl, amino C 2-20 alkyl, and hydroxy C 2-20 alkyl.
- the alkyl portion of R g may have a group selected from the group consisting of -O- and -NH- inserted therein.
- Y a is C 1-5 alkylene.
- Y b is C 2-8 alkylene or C 2-8 alkenylene.
- m is an integer of 1 or more and 100 or less.
- the hyaluronic acid derivative preferably includes a hyaluronic acid derivative having one or more repeating units represented by the following general formula (Ia) (hereinafter sometimes referred to as "repeat units (Ia)").
- R 1 , R 2 , R 3 , and R 4 are each independently a group selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl.
- X is a hydrophobic group represented by -NR a -Y-NR b -COO-R.
- R a and R b are each independently selected from the group consisting of a hydrogen atom and C 1-6 alkyl.
- R is a steryl group.
- Y is C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -, and m is an integer from 1 to 100.
- the repeating units may be the same or different.
- the hyaluronic acid derivative may be modified at a position other than repeating unit (I) or repeating unit (Ia), for example, the hydroxy group is -O (C 1-6 alkyl), -O (formyl), -
- the carboxy group may be converted to O(C 1-6 alkylcarbonyl), etc., and the carboxy group may be converted to an amide or ester, or may form a salt.
- Such groups include -NH-(CH 2 ) mz -NH-COO-R; -NH-(CH 2 CH 2 O) m -CH 2 CH 2 -NH-COO-R; and -NH-(CH 2 ) mz -SSR, where mz, R and m are as previously defined herein.
- Z is preferably a direct bond.
- X 1 is -NR b -COO-R.
- Z may be a peptide linker represented by -NH-[CH(-Z a )-CONH] n-1 -CH(-Z a )-CO-, where , n is an integer of 2 or more and 30 or less, and Z a each independently represents a substituent in the ⁇ -amino acid represented as H 2 N-CH(-Z a )-COOH.
- the peptide linker is bonded to the carboxy group of the glucuronic acid moiety at the N-terminus and to the group -N(-R a )-YX 1 at the C-terminus.
- amino acids that can be used as amino acid residues in the peptide linker include ⁇ -amino acids, such as alanine, arginine, asparagine (Asn), aspartic acid, cysteine, glutamine, glutamic acid, glycine (Gly), histidine, isoleucine, and leucine (Leu).
- - Amino acids can be used. That is, examples of Z a include -CH 3 , H 2 NC(NH)NH(CH 2 ) 3 -, H 2 NCOCH 2 -, and the like. Furthermore, n Z's may be the same or different. n is an integer of 2 or more and 30 or less, preferably 2 or more and 10 or less, and more preferably 2 or more and 4 or less.
- Preferred examples of peptide linkers include -Gly-Phe-Leu-Gly-, -Asn-Phe-Phe-, -Phe-Phe-, Phe-Gly-, and the like.
- Y is -(CH 2 ) n1 - and -(CH 2 CH 2 O) m1 -CH 2 CH 2 - (where n1 is an integer of 2 or more and 20 or less, and 2 or more An integer of 15 or less is preferable, an integer of 2 or more and 12 or less is more preferable, and an integer of 2 or more and 6 or less is even more preferable.
- m1 is an integer of 1 or more and 4 or less.
- Y is -(CH 2 ) 2 -, -(CH 2 ) 6 --, --(CH 2 ) 8 -- and --(CH 2 ) 12 -- is preferred, and --(CH 2 ) 6 -- is more preferred.
- Y is, for example, -CH 2 CH 2 O-CH 2 CH 2 -S-S-CH 2 CH 2 O-CH 2 CH 2 -, -(CH 2 CH 2 O) 2 -CH 2 CH 2 -S- S-CH 2 CH 2 O-CH 2 CH 2 -, -CH 2 CH 2 O-CH 2 CH 2 -S-S-(CH 2 CH 2 O) 2 -CH 2 CH 2 -, -(CH 2 CH 2 O) 2 -CH 2 CH 2 -S-S-(CH 2 CH 2 O) 2 -CH 2 CH 2 - and the like.
- Y a is preferably -CH 2 - or -CH 2 -CH 2 -.
- Yb is -CH 2 -CH 2 -, -CH(CH 3 )CH 2 -, 2-butene-1,4-diyl, hepta-2,4-diene-1,6-diyl or octa-2 ,4,6-triene-1,8-diyl is preferred, and --CH 2 --CH 2 -- or --CH(CH 3 )CH 2 -- is more preferred.
- Specific examples of the group "-Z-N(R a )Y-X 1 " include -NH-(CH 2 ) 2 -NH-CO-cholesteryl, -NH-(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH-(CH 2 ) 3 -NH-(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH -(CH 2 ) 4 -NH-(CH 2 ) 3 -NH-COO-cholesteryl, -NH-(CH 2 ) 4 -N(-(CH 2 ) 3 -NH 2 )-COO-cholesteryl, -NH- (CH 2 ) 3 -NH-(CH 2 ) 4 -N(-(CH 2 ) 3 -NH 2 )-COO-cholesteryl, -NH- (CH 2 ) 3 -NH-(CH 2
- R a , R b and R c are hydrogen atoms
- Y is linear C 2-30 alkylene or -(CH 2 CH 2 O) m -CH 2 CH 2 -
- Y a is linear C 1-5 alkylene
- Y b is linear C 2-8 alkylene or linear is a C 2-8 alkenylene.
- Repeat unit (Ia) In the general formula ( Ia ) , -COO-cholesteryl or -NH-(CH 2 CH 2 O) 2 -CH 2 CH 2 -NH-COO-cholesteryl is preferred, -NH-(CH 2 ) 2 -NH-COO-cholesteryl, -NH-(CH 2 ) 6 -NH-COO-cholesteryl or -NH-(CH 2 CH 2 O) 2 -CH 2 CH 2 -NH-COO-cholesteryl is more preferred.
- the hyaluronic acid derivative can further include a repeating unit represented by general formula (II) (hereinafter sometimes referred to as "repeating unit (II)"). .
- R 1a , R 2a , R 3a , and R 4a are each independently a group selected from the group consisting of a hydrogen atom, C 1-6 alkyl, formyl, and C 1-6 alkylcarbonyl.
- X a is a group selected from the group consisting of hydroxy and -O-Q + .
- Q + is a counter cation.
- the hyaluronic acid derivative (A) contains two or more repeating units (II), the repeating units may be the same or different.
- the hyaluronic acid derivative (A) may be a hyaluronic acid derivative consisting essentially of repeating unit (I), repeating unit (Ia), and repeating unit (II).
- Q + is not particularly limited as long as it is a counter cation that forms a salt with a carboxy group in water, and if it has a valence of two or more, it forms a salt with a plurality of carboxy groups depending on the valence.
- countercations include metal ions such as lithium ions, sodium ions, rubidium ions, cesium ions, magnesium ions, calcium ions; formula: N + R j R k R l R m (where R j , R k , R l and R m are each independently selected from the group consisting of a hydrogen atom and C 1-6 alkyl).
- Q + is preferably a sodium ion, potassium ion, or tetraalkylammonium ion (eg, tetra n-butylammonium ion, etc.).
- R j , R k , R l and R m are preferably the same group selected from the group consisting of C 1-6 alkyl, preferably n-butyl group.
- R 1 , R 2 , R 3 , and R 4 , and R 1a , R 2a , R 3a , and R 4a are all hydrogen atoms. Moreover, it is preferable that both R a and R b are hydrogen atoms.
- the hyaluronic acid derivative (A) is preferably a hyaluronic acid derivative consisting essentially of repeating units (I) and repeating units (II).
- A) In the hyaluronic acid derivative for example, 80% or more, preferably 90% or more of the disaccharide repeating units consisting of D-glucuronic acid and N-acetyl-D-glucosamine contained in the derivative are More preferably, 95% or more are repeating units (I) and repeating units (II).
- the hyaluronic acid derivative may be composed only of repeating units (I) and repeating units (II).
- the content of the hyaluronic acid derivative (A) relative to the total amount of the pharmaceutical composition is preferably 1 mg/mL or more and less than 50 mg/mL, more preferably 3 mg/mL or more and 45 mg/mL or less, and further preferably 5 mg/mL or more and 40 mg/mL or less. preferable.
- association promoter can be more specifically measured by gel permeation chromatography, and the association promoter defined in this embodiment is defined as the ratio A2 of the areas A1 and A2 shown below. /A1 to confirm.
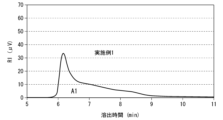
- Figure 1 is a chromatogram of a hyaluronic acid derivative.
- An example of the area A1 shown in FIG. 1 is 1814.
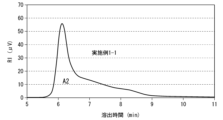
- FIG. 2 is a chromatogram of hyaluronic acid derivative pharmaceutical composition 1.
- An example of the area A2 shown in FIG. 2 is 2813.
- FIG. 2 shows a gel permeation chromatogram of the hyaluronic acid derivative of this embodiment.
- the refractive index intensity at the start of the measurement is set to zero, and a line drawn horizontally from this is set as the baseline.
- the refractive index intensity is adjusted so that the increase or decrease is within the range of ⁇ 0.5 mV, and the refractive index intensity is adjusted so that the increase or decrease is within 0.5 mV or less in 5 minutes.
- the first point where the refractive index intensity increase exceeds an amount equivalent to five times the noise value three times is set as the "starting point" of the chromatogram, and the elution time is set as 0 minutes.
- Tlim' is set as the 'end point'.
- Tlim is the elution time at which maximum refractive index intensity is obtained when 2 kDa polyacrylic acid is measured.
- the refractive index intensity is calculated every 0.00167 minutes.
- each area value is calculated using an analysis application of the GPC workstation EcoSEC Elite-WS.
- a ratio A2/A1 of area A2 to area A1 of 1.2 or more means that association between hyaluronic acid derivative molecules or between the hyaluronic acid derivative and the association promoter is promoted. Note that when the association between hyaluronic acid derivative molecules or between the hyaluronic acid derivative and the association promoter is not promoted, the ratio A2/A1 is in the range of 0.95 to 1.19.
- the ratio A2/A1 is preferably 1.20 or more, more preferably 1.30 or more, even more preferably 1.40 or more, and particularly preferably 1.50 or more.
- a relatively large amount of the hyaluronic acid derivative whose association has been promoted can be contained, and when formulated with (C) the active ingredient, (C) ) A large amount of the active ingredient can be retained, and (C) the active ingredient can be solubilized at a high concentration.
- the association promoter controls the release of the active ingredient (C), making it possible to maintain the sustained release period of the drug in vivo over a long period of time.
- the upper limit of A2/A1 is preferably as high as the ratio of area A2 is than area A1. Therefore, the upper limit of A2/A1 is not particularly limited, but may be 4.0 or less, 3.5 or less, 3.0 or less, 2.5 or less, 2.1 or less, 2.0 or less, 1.9 or less, 1 It may be .5 or less.
- the association promoter is not particularly limited as long as it is a surfactant that can be commonly used in pharmaceutical applications.
- the association promoter is preferably a component containing at least 4 or more ether structures (ROR) and having 4 or more carbon atoms.
- association promoter (B) examples include polysorbate (having 5 or more ether structures and 10 or more carbon atoms), polyoxyethylene fatty acid ester, sorbitan fatty acid ester, polyexyethylene castor oil, and the like.
- Association promoters include, for example, polysorbate 80 (having 20 or more ether structures and having 64 or more carbon atoms), polysorbate 65 (having 20 or more ether structures and having 100 or more carbon atoms), polysorbate 60 (having an ether structure of 20 or more and having a carbon number of 64 or more), Polysorbate 40 (having a 20 or more ether structure and having a carbon number of 62 or more), Polysorbate 20 (having an ether structure of 20 or more and having a carbon number of 62 or more) 57 or more), polyethylene glycol monolaurate (having 7 or more ether structures and having 28 or more carbon atoms), polyoxyl stearate 40 (having 39 or more ether structures and having 98 or more
- the association promoter is not limited to the above-mentioned surfactants, but includes, for example, poloxamer 188 (having an ether structure of 98 or more and a carbon number of 225 or more), poloxamer 124 (having an ether structure of 26 or more), Poloxamer 237 (having an ether structure of 93 or more and having 225 or more carbon atoms), Poloxamer 338 (having an ether structure of 177 or more and having 400 or more carbon atoms), Poloxamer 338 (having an ether structure of 177 or more and having 400 or more carbon atoms) 407 (having 147 or more ether structures and 352 or more carbon atoms), polyethylene glycol 300 (having 5 or more ether structures and having 12 or more carbon atoms), polyethylene glycol 400 (having 7 or more ether structures and having 7 or more carbon atoms) Polyethylene glycol 4000 (having an ether structure of 59 or more and having a carbon number of
- the amount of the association promoter (B) added in Pharmaceutical Composition 1 is preferably 0.001 parts by mass or more and 15,000 parts by mass or less, and 0.05 parts by mass or more and 5,000 parts by mass, based on 100 parts by mass of the hyaluronic acid derivative. The following is more preferred, and 1 part by mass or more and 4000 parts by mass or less is even more preferred.
- the amount of the association promoter (B) added in pharmaceutical composition 1 is 100 parts by mass of the hyaluronic acid derivative. , preferably 0.01 parts by mass or more and 150 parts by mass or less, more preferably 0.05 parts by mass or more and 100 parts by mass or less.
- the amount of the association promoter (B) added in the pharmaceutical composition 1 is 100 parts by mass of the hyaluronic acid derivative. 10 parts by mass or more and 15000 parts by mass or less, more preferably 100 parts by mass or more and 10000 parts by mass or less, even more preferably 1000 parts by mass or more and 5000 parts by mass or less, and particularly preferably 1500 parts by mass or more and 4000 parts by mass or less. Most preferably 2000 parts by mass or more and 3500 parts by mass or less.
- composition 1 of the present embodiment can retain a large amount of the active ingredient by containing (B) the association promoter, and furthermore, the pharmaceutical composition 1 can retain a large amount of the active ingredient.
- the release rate of the active ingredient can be controlled and the active ingredient can be sustainedly released over a long period of time. Note that the desired effect may be obtained by a mechanism different from the above mechanism.
- active ingredients include, but are not limited to, pharmaceutically active peptides or proteins, nucleic acids, low-molecular compounds, middle-molecular compounds, antigens (cancer antigens, infectious disease-derived antigens, autoantigens in immune diseases, etc.), and the like. Among these, low-molecular compounds, middle-molecular compounds, and peptides are more preferred.
- the diseases to which the pharmaceutical composition 1 of the present embodiment can be applied are not particularly limited, and it can be widely used for the prevention and treatment of currently known diseases or diseases that will be discovered in the future. It may be a chronic disease or an acute disease.
- currently known diseases include cancer, infectious diseases, immune diseases, inflammatory relaxation, allergic diseases, skin diseases, hypertension, diabetes, neurological diseases, genetic diseases, cardiovascular diseases, cerebrovascular diseases, and respiratory diseases.
- pharmaceutically active peptide or protein By pharmaceutically active peptide or protein is meant one that has a positive or beneficial effect on the condition or disease state of a subject when administered to the subject in a therapeutically effective amount.
- Preferred pharmaceutically active peptides or proteins have curative or palliative properties and ameliorate, ameliorate, alleviate, reverse, or delay the onset of symptoms of a disease or disorder, or It can be administered to reduce the severity of symptoms.
- Pharmaceutically active peptides or proteins may also have prophylactic properties and can be used to delay the onset of disease or reduce the severity of such disease or condition.
- pharmaceutically active peptide or protein includes a full-length protein or polypeptide and may also refer to pharmaceutically active fragments thereof. The term also encompasses pharmaceutically active analogs of peptides or proteins.
- cytokines examples include, but are not limited to, cytokines and immune system proteins such as immunoactive compounds (e.g., interleukins, colony stimulating factor (CSF), granulocyte colony stimulating factor (G-CSF), granulocyte Macrophage colony stimulating factor (GM-CSF), erythropoietin, tumor necrosis factor (TNF), interferon, integrin, addressin, seletin, homing receptor, T cell receptor, immunoglobulin, antibody, hormone (insulin, thyroid hormone) , catecholamines, gonadotropins, stimulating hormones, prolactin, oxytocin, dopamine, bovine somatotropin, leptin, etc.), growth hormones (e.g., human growth hormone), growth factors (e.g., epidermal growth factor, nerve growth factor, insulin-like growth factor, etc.) , growth factor receptors, enzymes (tissue plasminogen activator, streptokinase, cholesterol biosy
- proteins that inhibit angiogenesis include structural proteins (collagen, fibroin, fibrinogen, elastin, tubulin, actin, myosin, etc.), blood proteins (thrombin, Serum albumin, factor VII, factor VIII, insulin, factor IX, factor X, tissue plasminogen activator, protein C, von Willebrand factor, antithrombin III, glucocerebrosidase, erythropoietin, modified factor VIII , anticoagulant factors), etc.
- structural proteins collagen, fibroin, fibrinogen, elastin, tubulin, actin, myosin, etc.
- blood proteins thrombin, Serum albumin, factor VII, factor VIII, insulin, factor IX, factor X, tissue plasminogen activator, protein C, von Willebrand factor, antithrombin III, glucocerebrosidase, erythropoietin, modified factor VIII , anticoagulant factors), etc.
- Nucleic acids include DNA and RNA, such as short interfering RNA (siRNA), double-stranded RNA (dsRNA), microRNA (miRNA), short hairpin RNA (shRNA), and nucleic acid aptamers.
- siRNA short interfering RNA
- dsRNA double-stranded RNA
- miRNA microRNA
- shRNA short hairpin RNA
- low-molecular compounds include compounds with a molecular weight of less than about 500, such as anticancer agents (e.g., alkylating agents, antimetabolites, alkaloids, etc.), immunosuppressants, anti-inflammatory agents (steroidal agents, non-steroidal agents, etc.). Anti-inflammatory agents, etc.), anti-rheumatic agents, antibacterial agents ( ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics, new quinolone antibiotics, sulfa drugs, etc.).
- anticancer agents e.g., alkylating agents, antimetabolites, alkaloids, etc.
- immunosuppressants e.g., anti-inflammatory agents, steroidal agents, non-steroidal agents, etc.
- Anti-inflammatory agents, etc. anti-rheumatic agents
- antibacterial agents ⁇ -lactam antibiotics, aminoglycoside antibiotics, macrolide antibiotics, tetracycline antibiotics
- Active ingredients include Rho kinase inhibitors, endothelin A receptor inhibitors, transmembrane conductance regulator (CFTR) regulators, TRPV1 inhibitors, NK1 receptor inhibitors, purinergic receptor inhibitors, and angiotensin. It may be a receptor inhibitor, a peroxisome proliferator-responsive receptor, a P2Y receptor inhibitor, a VEGF inhibitor, etc., or it may be an active ingredient that has two of these inhibitory effects at the same time.
- CFTR transmembrane conductance regulator
- highly hydrophobic that is, sparingly water-soluble substances can also be preferably used, since they can sufficiently interact with the steryl group of the above-mentioned hyaluronic acid derivative.
- “poorly water-soluble” refers to the amount of water required to dissolve 1 g of solute in the 17th edition of the Japanese Pharmacopoeia to be 30 mL or more.
- poorly water-soluble solid active ingredients include acetaminophen, ibuprofen, benzoic acid, ethenzamide, caffeine, camphor, quinine, calcium gluconate, dimethylcaprol, sulfamine, theophylline, theopramine, riboflavin, mephenesin, Antipyretic analgesics such as phenobarbital, aminophylline, thioacetazone, quercetin, rutin, salicylic acid, theophylline sodium salt, pirapital, quinine hydrochloride, irgapirin, dioquitoxin, griseofulvin, phenacetin, nervous system drugs, sedative-hypnotics, muscle relaxants, blood pressure stiffening antibiotics such as acetylspiramycin, ampicillin, erythromycin, xatamycin, chloramphenicol, triacetyloleandomycin, nystatin, colistin sulf
- the active ingredient may be in the form of a poorly water-soluble oil or liquid.
- poorly water-soluble oily or liquid active ingredients include teprenone, indomethacin farnesil, menatetrenone, phytonadione, vitamin A oil, phenipentol, vitamins such as vitamin D and vitamin E, DHA (docosahexaenoic acid), and EPA. (eicosapentaenoic acid), higher unsaturated fatty acids such as cod liver oil, coenzyme Q, oil-soluble flavorings such as orange oil, lemon oil, peppermint oil, etc., according to the Japanese Pharmacopoeia, ⁇ external groups'', and ⁇ USP''. ”, “NF”, and “EP”.
- vitamin E there are various homologs and derivatives of vitamin E, but there are no particular limitations as long as they are liquid at room temperature, such as dl- ⁇ -tocopherol, dl- ⁇ -tocopherol acetate, d- ⁇ -tocopherol, and d- ⁇ -acetate. Examples include tocopherol.
- One type selected from these active ingredients may be used, or two or more types may be used in combination.
- the active ingredient may be a semi-solid active ingredient that is poorly water soluble.
- Semi-solid active ingredients that are poorly water-soluble include, for example, Jiryu, Liquorice, Keihi, Peony, Botanpi, Valerian, Sansho, Gingerbread, Chinpi, Ephedra, Nantenjitsu, Ahi, Onji, Bellflower, Shazenshi, Chazenso, and Stone.
- One type selected from these active ingredients may be used, or two or more types may be used in combination.
- middle molecules refer to peptides and macrolide compounds with a molecular weight of about 500 to 5,000, which are neither low molecules (organic compounds with a molecular weight of up to about 500) nor high molecules (such as proteins with a molecular weight of 10,000 or more).
- the peptide has a molecular weight of about 500 to 2000, ie, a linear or cyclic peptide having about 5 to 20 amino acid residues.
- the peptide is preferably a cyclic peptide, the details of which will be described later.
- a macrolide compound is a macrocyclic lactone, and is a general term for compounds having 12 or more ring members. Examples of middle-molecular compounds include FK506 and rapamycin.
- Cancer antigens are antigens that are often expressed by cancer cells, and in some cases only by cancer cells. Cancer antigens can be expressed within cancer cells or on the surface of cancer cells.
- Antigenic proteins that can be used in the pharmaceutical composition 1 of the present embodiment include, but are not limited to, ERK1, ERK2, WT1, MART-1/Melan-A, gp100, adenosine deaminase binding protein (ADAbp), FAP, cyclophilin b, colon Rectal-related antigen (CRC)-C017-1A/GA733, carcinoembryonic antigen (CEA), CAP-1, CAP-2, etv6, AML1, prostate-specific antigen (PSA), PSA-1, PSA-2, PSA-3, prostate-specific membrane antigen (PSMA), T cell receptor/CD3-zeta chain, CD20, MAGE-A1, MAGE-A2, MAGE-A3, MAGE-A4, MAGE-A5, MAGE-A6, MAGE -A7, MAGE-A8, MAGE-A9, MAGE-A10, MAGE-A11, MAGE-A12, MAGE-Xp2 (MAGE-B2), M
- the entire sequence thereof may be used, or a partially deleted sequence may be used.
- the antigenic peptide that can be used in the pharmaceutical composition 1 of the present embodiment is one or more types selected from the group consisting of a CD8-positive cytotoxic T cell recognition epitope and a CD4-positive helper T cell recognition epitope among the antigen protein sequences. It is an antigenic peptide containing an epitope.
- the antigenic peptide is preferably an antigenic peptide containing two or more epitopes from the viewpoint of being loaded onto MHC class I molecules or MHC class II molecules after being degraded in antigen-presenting cells.
- the antigenic peptide mentioned above includes an antigenic peptide containing an epitope of an antigenic protein of a tumor cell.
- the antigenic peptide has, for example, 8 to 120 amino acids, preferably 8 to 80 amino acids, more preferably 15 to 80 amino acids, even more preferably 16 to 80 amino acids, even more preferably 23 to 80 amino acids, and even more It preferably has 23 to 60 amino acids, particularly preferably 23 to 50 amino acids.
- the antigen peptide has one epitope recognized by CD8-positive cytotoxic T cells and one epitope recognized by CD4-positive helper T cells. It is an antigenic peptide containing three or more antigenic peptides.
- an amino acid linker may be placed between the epitopes.
- the linker has, for example, 2 to 10 amino acids, preferably 4 to 10 amino acids, more preferably 4 to 8 amino acids.
- Amino acids used in the linker include glycine (G), tyrosine (Y), leucine (L), tryptophan (W), and the like. Preferably they are tyrosine (Y), leucine (L), and tryptophan (W).
- amino acid linkers include a linker (4Y) consisting of four consecutive tyrosines (Y), a linker (4L) consisting of four consecutive leucines (L), and a linker (4L) consisting of four consecutive tryptophans (W).
- a linker of 6 consecutive glycines (G) (6G), a linker of 6 consecutive tyrosines (Y) (6Y), a linker of 6 consecutive leucines (L) (6L), a linker of 6 consecutive tryptophans (W) linker (6W), 8 consecutive tyrosine (Y) linker (8Y), 6 consecutive leucine (L) linker (8L), 8 consecutive tryptophan (W) linker (8W) are mentioned, preferably 6Y, 6L or 6W.
- infectious disease-derived antigen is not particularly limited as long as it is an infectious pathogen or an antigen derived from an infectious pathogen. Infectious pathogens include viruses, bacteria, fungi, nematodes, and the like.
- infectious disease pathogen-derived antigen may be an antigenic protein or an antigenic peptide.
- Diseases contracted from the above infectious pathogens are not particularly limited, and include, for example, adenoviruses, herpesviruses (such as HSV-I, HSV-II, CMV, and VZV), poxviruses (such as variola or vaccinia, and molluscum contagiosum).
- adenoviruses such as HSV-I, HSV-II, CMV, and VZV
- poxviruses such as variola or vaccinia, and molluscum contagiosum
- orthopoxviruses picornaviruses (e.g., rhinoviruses, enteroviruses), orthomyxoviruses (e.g., influenza viruses), paramyxoviruses (e.g., parainfluenza viruses, mumps viruses, measles viruses, respiratory viruses) syncytial virus (RSV)), coronavirus (e.g., SARS coronavirus (SARS-CoV), MERS coronavirus (MERS-CoV), SARS-CoV-2), papovavirus (e.g., genital warts, cystocele vulgaris, papillomaviruses (such as those that cause plantar warts), hepadnaviruses (e.g., hepatitis B virus), flaviviruses (e.g., hepatitis C virus, dengue virus), retroviruses (e.g., lentiviruses such as HIV), etc.
- RSV syncy
- Viral diseases such as those caused by viral infections; Escherichia, Enterobacter, Salmonella, Staphylococcus, Shigella, Listeria, Aerobacter, Helicobacter, Klebsiella, Proteus, Pseudomonas, Streptococcus, Chlamydia, Mycoplasma, Pneumococcus, Neisseria, Clostridium , Bacillus, Corynebacterium, Mycobacterium, Campylobacter, Vibrio, Serratia, Providencia, Chromobacterium, Brucella, Yersinia, Haemophilus, Bordetella, etc.; chlamydia, candidiasis, aspergillosis, Fungal diseases including, but not limited to, histoplasmosis, cryptococcal meningitis; malaria, Pneumocystis carinii pneumonia, leishmaniasis, cryptosporidiosis, toxoplasmosis, trypano
- the structure of the antigen that can be used in the pharmaceutical composition 1 of the present embodiment is not particularly limited as long as it is at least a part of various components constituting a pathogen, and includes, for example, a live vaccine, an inactivated whole particle, Some of them include protein subunits, proteins, peptides, and the like. Among these, protein subunits, proteins, or peptides are preferred from the viewpoint of conjugation with hyaluronic acid derivatives.
- influenza virus is an RNA enveloped virus belonging to the Orthomyxoviridae family and having a particle size of about 100 nm in diameter, and is divided into types A, B, and C based on the antigenicity of internal proteins.
- the influenza virus consists of a core of ribonucleic acid (RNA) associated with an internal nucleocapsid or nucleoprotein surrounded by a viral envelope with a lipid bilayer structure, and an external glycoprotein.
- the inner layer of the viral envelope is mainly composed of matrix proteins, and the outer layer is mostly composed of host-derived lipid substances.
- the RNA of the influenza virus has a segmented structure.
- Influenza that is prevalent all over the world is caused by influenza A virus, and this influenza A virus has two types of envelope glycoproteins, hemagglutinin (HA) and neuraminidase (NA), and has antigenic properties. Depending on the differences, HA is divided into 16 subtypes and NA into 9 subtypes.
- HA hemagglutinin
- NA neuraminidase
- antigens derived from influenza A and B viruses are preferably used. Note that the subtypes of the above-mentioned influenza A and B viruses are not particularly limited, and may be subtypes that have been isolated so far or subtypes that will be isolated in the future.
- influenza virus-derived antigen is not particularly limited as long as it is at least a part of the various components constituting the above-mentioned influenza virus.
- examples include inactivated whole virus particles, or virus subunits produced by removing impurities from the whole virus particles and purifying HA and/or NA. From the viewpoint of immunogenicity, HA subunits or whole virus particles are preferred.
- the whole virus particles are more preferably inactivated with formalin or the like. Furthermore, it is particularly effective for HA subunits (split), which have few impurities and require an adjuvant such as an immunostimulant.
- the method for preparing the influenza virus antigen described above is not particularly limited, and any known method can be used without limitation.
- a method may be used in which chicken eggs or the like are infected with a virus strain isolated from an influenza-infected animal or an influenza patient, cultured by a conventional method, and an antigen is prepared from the purified virus stock solution.
- virus-derived antigens prepared in cultured cells by genetic engineering may be used.
- Antigens for immune diseases are not particularly limited as long as they contain epitopes of target proteins of immune diseases.
- Immune diseases are not particularly limited, and include, for example, plaque psoriasis, ankylosing spondylitis, rheumatoid arthritis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, bronchial asthma, chronic urticaria, and hay fever. , atopic dermatitis, etc.
- Target proteins are not particularly limited, and include IL-17A, DPP4, S100A9, PCSK9, IL-23, IgE, TNF ⁇ , IL-12/23p40, IL-6, ⁇ 4 ⁇ 7 integrin, IL-4/13, IL-5, Examples include BLyS and IL-13.
- Reference document 1 International Publication No. 2017/164409) describes a peptide derived from IL-17A.
- the present invention can be utilized more effectively when the active ingredient (C) in the present invention is a poorly water-soluble drug.
- Poorly water-soluble drugs are terms that indicate solubility in the 17th edition of the Japanese Pharmacopoeia, and are defined as extremely soluble, easily soluble, slightly soluble, slightly soluble, difficult to dissolve, extremely difficult to dissolve, and almost insoluble. Refers to drugs that are classified as slightly soluble, slightly soluble, slightly soluble, extremely soluble, or almost insoluble.
- the active ingredient (C) includes a poorly water-soluble drug having a solubility in water of 1 mg/mL or less.
- Pharmaceutical composition 1 of the present invention can solubilize even such poorly water-soluble drugs at high concentrations without using organic solvents and reducing the amount of highly toxic surfactants used.
- the active ingredient preferably has a molecular weight of 200 or more, more preferably 300 or more, more preferably 400 or more, more preferably 500 or more, even more preferably 600 or more, even more preferably 700 or more, and even more preferably 800 or more. It is more preferably 900 or more, even more preferably 1000 or more, particularly preferably 1100 or more, and most preferably 1200 or more.
- the molecular weight of the active ingredient is a value calculated from the molecular formula of the compound.
- the poorly water-soluble drug that is the active ingredient is preferably a poorly water-soluble peptide.
- the poorly water-soluble peptide may have an acidic amino acid, a basic amino acid, or a neutral amino acid, with basic amino acids or neutral amino acids being preferred.
- the pH of the solution may be appropriately selected in consideration of the isoelectric point.
- the amino acids may also include natural amino acids or unnatural amino acids.
- At least one of the nitrogen atoms constituting the amide bond of the poorly water-soluble peptide has a methyl group. Imparting hydrophobicity through methylation increases the interaction with the hydrophobic part of the hyaluronic acid derivative, and is expected to result in more solubilization. It is also presumed that the interaction is enhanced and the stability of the formulation is also improved.
- the poorly water-soluble peptide preferably contains at least one selected from cyclic peptides, long-chain peptides, hydrophobic group-modified peptides, membrane-damaging peptides, and peptide-drug complexes.
- the poorly water-soluble peptide is preferably a cyclic peptide.
- the cyclic and rigid skeleton maximizes interaction with the hydrophobic part of hyaluronic acid and forms a stable structure with hyaluronic acid derivatives.
- the cyclic size is preferably a cyclic peptide consisting of 4 to 49 amino acids, more preferably 6 to 30 amino acids, and most preferably 8 to 25 amino acids.
- the number of rings in the molecule is not particularly limited, but is preferably 1 or more and 8 or less.
- hydrophobized peptide is an alkylated polypeptide.
- “Insulin Detemir” sold by Novo Nordisk Pharma is an insulin analog designed to have a C14 fatty acid side chain attached to the lysine at position 29 of the human insulin B chain and to exhibit affinity for albumin. This fatty acid side chain promotes self-association between insulin detemir hexamers and binds to albumin at the subcutaneous injection site, resulting in a stabilizing effect and reducing the rate of absorption from the administration site.
- the hydrophobic part of the complex of the hyaluronic acid derivative and the association promoter or the solubilization aid and the hydrophobic part of the peptide can be used in vivo.
- the strong interaction between the two components may make it possible to extend the sustained release period.
- hyaluronic acid derivative pharmaceutical composition 1 or pharmaceutical composition 2, which will be described later, which includes the above-mentioned peptide can be expected to be released for a longer period of time because the hyaluronic acid derivative effectively suppresses decomposition by peptide degrading enzymes in the body.
- the blending amount of the active ingredient (C) relative to 100 parts by mass of the (A) hyaluronic acid derivative is preferably 10 parts by mass or more and 100 parts by mass or less, more preferably 15 parts by mass or more and 50 parts by mass or less. More preferably 20 parts by mass or more and 40 parts by mass or less.
- the active ingredient which is not particularly limited, but is not limited to a poorly water-soluble drug, may be used alone or in combination of two or more.
- composition 1 of the present embodiment can be solubilized without using an organic solvent when formulating the active ingredient (C), which is a poorly water-soluble drug, using conventional highly toxic solubilizers. can be reduced. Furthermore, the solubilization aid (B) can promote hydrophobic interaction between the steryl groups of the hyaluronic acid derivative (A). It is presumed that this makes it possible to solubilize the powdered active ingredient (C) at a high concentration. Note that the desired effect may be obtained by a mechanism different from the above mechanism.
- the pharmaceutical composition 1 of the present embodiment can also be called an organic solvent-free composition or a powdered drug solubilizing composition.
- composition 1 of the present embodiment can be administered alone or together with a pharmacologically acceptable carrier according to conventional methods.
- a pharmacologically acceptable carrier for example, the above-mentioned hyaluronic acid derivative and the above-mentioned active ingredient, and optionally an adjuvant, water or another physiologically acceptable liquid (for example, physiological saline) may be used in combination with a pharmacologically acceptable carrier.
- physiologically acceptable liquid for example, physiological saline
- Physiologically acceptable buffers, excipients, vehicles, preservatives, stabilizers, binders, lyophilization aids which may be mixed with water, phosphate buffered saline (PBS), etc. It can also contain agents.
- PBS phosphate buffered saline
- buffer examples include Tris, sodium phosphate, potassium phosphate, histidine, or citric acid.
- the hyaluronic acid derivative is used after being dissolved in a pharmaceutically acceptable medium at any concentration.
- buffer solutions such as water for injection and phosphate buffer are preferred.
- the buffering agent is not particularly limited as long as it is a compound having a buffering capacity within the range of pH 4 to 10 of the composition.
- buffering agents include acetates such as sodium acetate, phosphates such as sodium dihydrogen phosphate, disodium hydrogen phosphate, potassium dihydrogen phosphate, dipotassium hydrogen phosphate, and ⁇ -aminocaproic acid. , amino acid salts such as sodium glutamate, boric acid and its salts, and mixtures thereof.
- particularly preferred are water for injection, phosphate buffer, sucrose-containing water for injection, sucrose-containing phosphate buffer, and glycerol.
- composition 1 of the present embodiment or pharmaceutical composition 2 described later may contain a pH adjuster, and examples of the pH adjuster include hydrochloric acid, citric acid, phosphoric acid, acetic acid, tartaric acid, sodium hydroxide, and sodium hydroxide. Examples include potassium, sodium carbonate, and sodium hydrogen carbonate.
- An acid such as an organic acid or an inorganic acid may be added as necessary. Further, these pH adjusters may be used alone or in any combination of two or more.
- the pH of the pharmaceutical composition 1 of the present embodiment or the pharmaceutical composition 2 described below is not particularly limited as long as it is within a pharmaceutically acceptable range, but is, for example, 4.0 to 9.0, preferably 4.0 to 9.0. It is in the range of 0 to 8.8, more preferably 6.5 to 8.8.
- preservatives for the pharmaceutical composition 1 of the present embodiment or the pharmaceutical composition 2 described later include benzalkonium chloride, methyl paraoxybenzoate, propyl paraoxybenzoate, chlorobutanol, sorbic acid, alkyl polyaminoethylglycine, etc. can be mentioned. If necessary, one or more preservatives may be further added, and there are no particular restrictions as long as they are pharmaceutically acceptable.
- Examples of the preservative in the present invention include benzalkonium chloride, benzethonium chloride, chlorhexidine gluconate, paraoxybenzoic acid esters such as ethyl paraoxybenzoate, benzyl alcohol, m-cresol, phenol, phenethyl alcohol, sorbic acid, and their like. Salt, thimerosal, etc. are used.
- the pharmaceutical composition 1 of this embodiment or the pharmaceutical composition 2 described later may further contain one or more types of thickening agents as needed, and there are no particular restrictions as long as they are pharmaceutically acceptable. It is not something that can be done.
- Examples of the thickening agent for the pharmaceutical composition 1 of the present invention or the pharmaceutical composition 2 described below include cellulose polymers (methylcellulose, ethylcellulose, hydroxyethylcellulose, hydroxypropylmethylcellulose, etc.), vinyl polymers (polyvinylpyrrolidone, polyvinyl alcohol, etc.), saccharides (mucopolysaccharides such as hyaluronic acid and its salts, polysaccharides such as gellan gum, sodium alginate, dextran, and cyclodextrin), oxyalkylene polymers (polyoxyethylene polyoxypropylene block copolymer), etc. Can be mentioned.
- the molecular weight of the thickening agent in the present invention can be selected from, for example, a number average molecular
- stabilizer examples include sodium edetate hydrate, polyvinylpyrrolidone (povidone), and the like.
- composition 1 of the present embodiment or pharmaceutical composition 2 described below may contain a chelating agent, and examples of the chelating agent include disodium edetate, trisodium edetate, tetrasodium edetate, and diethyleneamine. Examples include pentaacetic acid and mixtures thereof, which are used to stabilize drugs or preparations.
- Tonicity agents also include sucrose, glucose, dextrose, lactose, mannitol, calcium or magnesium compounds such as CaCl2 .
- glycerol at a concentration (2 to 2.5% (v/v)) that makes the pressure substantially isotonic. There is. Therefore, it is expected to have not only a function as an isotonic agent but also an antibacterial and preservative effect.
- the content of glycerol is, for example, 0.01 to 10% (w/v), preferably 0.05 to 5% (w/v), more preferably 0.1 to 3.0% (w/v). , more preferably 0.3 to 3.0% (w/v), particularly preferably 0.3 to 2.5% (w/v).
- a base can be added to the pharmaceutical composition 1 of the present embodiment or the pharmaceutical composition 2 described below as necessary.
- the base is not particularly limited as long as it is a pharmaceutically acceptable base, and examples thereof include sodium hydroxide, potassium hydroxide, monoethanolamine, diethanolamine, triethanolamine, trometamol, and meglumine.
- the base is not particularly limited as long as it is a pharmaceutically acceptable base, and examples include zinc chloride, zinc acetate, and the like.
- a formulated product may be used.
- the formulation can be in solid, semi-solid or liquid form.
- examples include powder, granules, pills, pellets, tablets, capsules, and the like.
- the solid is preferably a freeze-dried powder.
- examples include forms such as gel.
- examples include a suspension in which the powder is diluted or suspended in water or a buffer such as phosphate buffer (PB) or phosphate buffered saline (PBS).
- PB phosphate buffer
- PBS phosphate buffered saline
- the pharmaceutical composition of this embodiment is preferably one that produces a precipitate under physiological salt concentration.
- the precipitation rate in vitro is 20%, preferably 50% or more, more preferably 70% or more, even more preferably 80% or more, particularly preferably 90% or more, and most preferably 92% or more. % or more is preferable.
- Precipitation rate (%) ⁇ 1-(Area value of hyaluronic acid derivative of precipitation sample) ⁇ ((Area value of hyaluronic acid derivative of blank sample) x (0.75X)) ⁇ x 100
- the centrifugation conditions and incubation time may be arbitrarily set when preparing the precipitate sample. For example, if the sample satisfies the precipitation rate of 20% or more when left at 37° C. for two weeks and subjected to the above conditions, it is also determined that precipitation occurs under physiological salt concentration.
- the pharmaceutical composition 1 of the present embodiment or the pharmaceutical composition 2 described below has no precipitates visually observed in an environment at 20°C. If precipitates are confirmed, use a light-shielding automatic particle counting device (liquid particle counter "KL -05'') can be detected. Drug crystals on the order of micrometers may precipitate from a supersaturated solution in which an amorphous drug is completely dissolved. If there is no precipitate, no particles are observed by the light-shielding automatic particle measuring device. In addition, the determination can also be made by using a DLS device to verify whether particles of 1 ⁇ m or more are present.
- a light-shielding automatic particle counting device liquid particle counter "KL -05''
- the pharmaceutical composition 1 of this embodiment or the pharmaceutical composition 2 described below can be sterilized by filtration.
- the ability to sterilize by filtration can be determined by evaluating the flow of fluid using the following syringe filter. Specifically, it is evaluated whether liquid can pass through a sterile filtration filter with a pore size of 0.45 ⁇ m or 0.22 ⁇ m. More specifically, when the filtration membrane was made of polyether sulfone (PES) with a 13 mm ⁇ pore size of 0.45 ⁇ m, and a 3 mL sterile syringe filter was used, 2 mL of the hyaluronic acid derivative composition passed through the filtration membrane.
- Hyaluronic acid derivatives can pass 50% or more, preferably 60%, more preferably 70% or more, even more preferably 80% or more, and most preferably 90% or more.
- This embodiment is a hyaluronic acid derivative pharmaceutical composition containing (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient.
- a hyaluronic acid derivative pharmaceutical composition comprising (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilizing agent, and (C1) an active ingredient
- “Pharmaceutical composition 2”. ” may be abbreviated as “.
- the average particle diameter of the spherical structure containing the active ingredient-hyaluronic acid derivative complex can be 20 nm or more and 220 nm or less, 20 nm or more and 150 nm or less, and 30 nm or more.
- the thickness can be set to 100 nm or more.
- the average particle diameter is within the above numerical range, the particles can exist in a stable structure in vivo and can more easily pass through lymph nodes.
- the average particle diameter can be measured by, for example, DLS (Dynamic Light Scattering), a nanotracking particle measuring device, size exclusion chromatography, high performance liquid chromatography, electron microscopy, or the like.
- the hyaluronic acid derivative is diluted with a 10 mM phosphate buffer or a 10 mM phosphate buffer containing 10 w/v% sucrose so that the concentration of the hyaluronic acid derivative becomes 1 mg/mL using a DLS device.
- the solubilization aid (B1) included in the pharmaceutical composition 2 of the present embodiment contains at least 4 or more ether structures (ROR) and has 4 or more carbon atoms.
- Pharmaceutical composition 2 of the present embodiment has a content of (B1) solubilizing agent of 0.0001 parts by mass or more and 15,000 parts by mass or less with respect to 100 parts by mass of (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced. be.
- composition 2 of this embodiment contains a specific amount of a specific solubilization aid.
- the pharmaceutical composition of the present embodiment containing the above-mentioned (B1) solubilization aid can be combined with the above-mentioned (A1) hyaluronic acid derivative into which a hydrophobic group has been introduced, due to the appropriate polarity due to the ether structure and the appropriate hydrophobicity due to the alkyl skeleton.
- the hydrophobic groups in the hyaluronic acid derivative (A1) into which the hydrophobic groups have been introduced can be promoted. This makes it possible to provide a pharmaceutical composition that can solubilize poorly water-soluble active ingredients at high concentrations.
- the active ingredient can be sufficiently solubilized in the pharmaceutical composition.
- the content of the solubilization aid (B1) is below the upper limit, the viscosity of the pharmaceutical composition does not become too high and the active ingredient can be sufficiently incorporated into the pharmaceutical composition without affecting the living body. Can be dissolved.
- Stepryl group introduction rate (A1)
- the introduction rate of steryl groups into the hyaluronic acid derivative is preferably 0.1% or more and less than 50%, more preferably 5% or more and less than 48%, even more preferably 35% or more and less than 47%, and 37% or more and less than 45%. is particularly preferred.
- the hyaluronic acid derivative (A1) can strongly interact with the poorly water-soluble drug and the hydrophobic part of the surfactant. Furthermore, since the steryl group introduction rate is within the above range, the hyaluronic acid derivative-drug complex in which the (A1) hyaluronic acid derivative in Pharmaceutical Composition 2 is complexed with a drug has improved formulation stability. I can do it.
- An example of the molecular weight of the hyaluronic acid derivative is preferably 1,000 (1k) or more and 1,000,000 (1,000k) or less, more preferably 5k or more and 300k or less, and even more preferably 5k or more and 120k or less, Particularly preferred is 7k or more and 100k or less.
- the molecular weight of the hyaluronic acid derivative can generally be adjusted by using raw materials having a corresponding molecular weight.
- the weight average molecular weight of the hyaluronic acid derivative When the weight average molecular weight of the hyaluronic acid derivative is equal to or higher than the above lower limit, molecular entanglement can be further enhanced, and retention in blood can be further enhanced. On the other hand, when the weight average molecular weight of the hyaluronic acid derivative is at most the above upper limit, an increase in viscosity can be suppressed and a higher concentration of the hyaluronic acid derivative can be dissolved in the pharmaceutical composition.
- the weight average molecular weight of hyaluronic acid derivatives can generally be adjusted by using raw materials with corresponding molecular weights.
- the weight average molecular weight of the hyaluronic acid derivative is preferably 100k or less, more preferably 50k or less, particularly preferably 20k or less, and most preferably 15k or less.
- the unit of molecular weight is Da. From the viewpoint of producing hyaluronic acid derivatives, 4k to 20k is preferred, 6k to 16k is particularly preferred, and 8k to 12k is most preferred.
- the hyaluronic acid derivative strongly exhibits the properties as hyaluronic acid, for example, in order to be strongly recognized by the CD44 receptor, it is preferably 100k or more, particularly preferably 200k or more, and most preferably 300k or more.
- the content of the hyaluronic acid derivative (A1) relative to the total amount of pharmaceutical composition 2 is preferably 6 mg/mL or more and less than 65 mg/mL, more preferably 8 mg/mL or more and 50 mg/mL or less, and 10 mg/mL or more and 30 mg/mL or less. More preferred.
- ⁇ (B1) Solubilization aid ⁇ refers to making the active ingredient (C1) described below solubilized in water until it becomes visually transparent.
- the term “solubilization aid” refers to an agent that has the effect of increasing the solubility of (C) the active ingredient when added to water together with (C1) the active ingredient and (A1) the hyaluronic acid derivative.
- the solubilization aid is not particularly limited as long as it has the effect of dissolving (solubilizing) the active ingredient in the aqueous phase (C1), and can be appropriately selected depending on the purpose.
- examples include nonionic surfactants.
- the solubilization aid (B1) used in the present invention is an agent that can be commonly used in pharmaceutical applications, contains at least 4 or more ether structures (R-OR), and has a carbon number of 4 or more. be.
- the solubilization aid is preferably one or more selected from the group consisting of a nonionic surfactant, a polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/mol or less, and a cyclodextrin derivative.
- solubilization aid examples include polysorbate, polyoxyethylene fatty acid ester, sorbitan fatty acid ester, polyexyethylene castor oil, and the like.
- polysorbate 80 having an ether structure of 20 or more and having a carbon number of 64 or more
- polysorbate 65 having a ether structure of 20 or more and having a carbon number of 100 or more
- polysorbate 60 having 20 or more ether structures and having 64 or more carbon atoms
- Polysorbate 40 having 20 or more ether structures and having 62 or more carbon atoms
- Polysorbate 20 having 20 or more ether structures and having 57 carbon atoms) above), poloxamers (having 28 or more ether structures and 74 or more carbon atoms), polyoxyethylene hydrogenated castor oil (having 35 or more ether structures and 57 or more carbon atoms), cyclodextrin derivatives
- Polyethylene glycol 300 having 5 or more ether structures and having 12 or more carbon atoms
- polyethylene glycol 400 having 7 or more ether structures and having 7 or more carbon atoms) 16 or more
- polyethylene glycol 4000 having
- examples include polyethylene glycol monolaurate (having 7 or more ether structures and 28 or more carbon atoms), polyoxyl stearate 40 (having 39 or more ether structures and 98 or more carbon atoms), stearin Acid polyoxyl 45 (having an ether structure of 44 or more and having a carbon number of 108 or more), Stearic acid polyoxyl 55 (having an ether structure of 54 or more and having a carbon number of 128 or more), Cremophor EL (having an ether structure of 35 or more) and carbon number is 127 or more).
- Solubilization aids include, for example, poloxamer 188 (having an ether structure of 98 or more and having a carbon number of 225 or more), poloxamer 124 (having an ether structure of 26 or more and having a carbon number of 74 or more), poloxamer 237 (having an ether structure of 93 or more and a carbon number of 225 or more), poloxamer 338 (having an ether structure of 177 or more and having a carbon number of 400 or more), Poloxamer 407 (having an ether structure of 147 or more and a carbon number of 352 or more), polyethylene glycol 300 (having 5 or more ether structures and having 12 or more carbon atoms), polyethylene glycol 400 (having 7 or more ether structures and having 16 or more carbon atoms), polyethylene glycol 4000 (having 7 or more ether structures and having 16 or more carbon atoms) Polyvinyl alcohol (having an average degree of polymerization of 500, having an ether,
- the content of the solubilizing aid (B1) relative to 100 parts by mass of the (A1) hyaluronic acid derivative is 0.0001 parts by mass or more and 15000 parts by mass or less, preferably 0.01 parts by mass or more and 150 parts by mass or less. , more preferably 0.05 parts by mass or more and 100 parts by mass or less, further preferably 1 part by mass or more and 50 parts by mass or less, particularly preferably 5 parts by mass or more and 30 parts by mass or less, and most preferably 10 parts by mass or more and 20 parts by mass or less. .
- the content of the nonionic surfactant is 0.0001 parts by mass or more and 150 parts by mass or less with respect to 100 parts by mass of (A1) hyaluronic acid derivative. is preferred, more preferably 0.001 parts by mass or more and 100 parts by mass or less, and even more preferably 0.005 parts by mass or more and 50 parts by mass or less.
- the solubilization aid is polyethylene glycol with a molecular weight of 190 g/mol or more and 4000 g/mol or less
- the content of polyethylene glycol is 25 parts by mass or more and 15,000 parts by mass relative to 100 parts by mass of the (A1) hyaluronic acid derivative. parts by weight or less, more preferably 250 parts by weight or more and 10,000 parts by weight or less, and still more preferably 500 parts by weight or more and 5,000 parts by weight or less.
- solubilization aid The smaller the solubilization aid, the better; however, if it is below the above lower limit, the active ingredient cannot be sufficiently solubilized. Moreover, if it exceeds the upper limit, toxicity tends to increase due to too much solubilization aid (B1) and the viscosity of the preparation also improves, so it is preferably within the above range.
- the amount of the poorly water-soluble drug as the active ingredient (C1) relative to 100 parts by mass of the (A1) hyaluronic acid derivative is preferably 21 parts by mass or more and less than 100 parts by mass, and 22 parts by mass or more and 70 parts by mass. It is more preferably 23 parts by mass or more and 50 parts by mass or less.
- the active ingredient although not particularly limited, may be a poorly water-soluble drug, which may be used alone or in combination of two or more.
- composition 2 of the present embodiment can be solubilized without using an organic solvent when formulating the active ingredient (C1), which is a poorly water-soluble drug, and can be solubilized without using a conventional highly toxic solubilizing agent.
- the usage amount can be reduced.
- solubilization aid (B1) hydrophobic interaction between the steryl groups of the hyaluronic acid derivative (A1) can be promoted. This is thought to expand the hydrophobic region of the hyaluronic acid derivative pharmaceutical composition and increase the amount of hydrophobic drug supported.
- the solubilizing agent can act as a hydrophobic part region with high mobility inside the hyaluronic acid derivative pharmaceutical composition.
- C1 It is presumed that the active ingredient can be solubilized at a high concentration. Note that the desired effect may be obtained by a mechanism different from the above mechanism.
- the pharmaceutical composition 2 of this embodiment can also be called an organic solvent-free composition or a powdered drug solubilizing composition.
- the content of organic solvent in the pharmaceutical composition 2 is preferably less than 0.8%.
- the organic solvent contained in the pharmaceutical composition 2 is a solvent that falls under classes 1 to 3 defined by the Pharmaceutical Residual Solvent Guidelines.
- class 1 organic solvents include benzene, carbon tetrachloride, 1,2-dichloroethane, 1,1-dichloroethene, and 1,1,1-trichloroethane.
- Class 2 organic solvents include acetonitrile, chlorobenzene, chloroform, cyclohexane, 1,2-dichloroethene, dichloromethane, 1,2-dimethoxyethane, N,N-dimethylacetamide, N,N-dimethylformamide, 1,4-dioxane. , 2-ethoxyethanol, formamide, hexane, methanol, 2-methoxyethanol, methylbutylketone, methylcyclohexane, N-methylpyrrolidone, nitromethane, pyridine, sulfolane, tetralin, toluene, 1,1,2-trichloroethene, xylene. Can be mentioned.
- Class 3 organic solvents include acetic acid, acetone, anisole, 1-butanol, 2-butanol, n-butyl acetate, t-butyl methyl ether, cumene, dimethyl sulfoxide, ethanol, ethyl acetate, diethyl ether, ethyl formate, formic acid, Heptane, isobutyl acetate, isopropyl acetate, methyl acetate, 3-methyl-1-butanol, methyl ethyl ketone, methyl isobutyl ketone, 2-methyl-1-propanol, pentane, 1-pentanol, 1-propanol, 2-propanol, propyl acetate , tetrahydrofuran.
- organic solvents of classes 1 to 3 are preferably not included in the pharmaceutical composition 2, but are components that inevitably remain in the manufacturing process of hyaluronic acid derivatives. Even if the organic solvent is of Class 1 to Class 3, if the content of the organic solvent in Pharmaceutical Composition 2 is less than 0.8%, it can be used safely.
- composition 2 of the present embodiment can be administered alone or together with a pharmacologically acceptable carrier according to conventional methods.
- a pharmacologically acceptable carrier for example, the above-mentioned hyaluronic acid derivative and the above-mentioned active ingredient, and optionally an adjuvant, water or another physiologically acceptable liquid (for example, physiological saline) may be used in combination with a pharmacologically acceptable carrier.
- physiologically acceptable liquid for example, physiological saline
- Physiologically acceptable buffers, excipients, vehicles, preservatives, stabilizers, binders, lyophilization aids which may be mixed with water, phosphate buffered saline (PBS), etc. It can also contain agents.
- PBS phosphate buffered saline
- Hyaluronic acid derivatives can be obtained, for example, by converting the carboxyl group of glucuronic acid into an amide and introducing a steryl group. Furthermore, the rate of steryl group introduction can be controlled by adjusting the amount of the compound having a steryl group to be reacted with the raw material hyaluronic acid or its derivative.
- a steryl group for example, hyaluronic acid or a derivative thereof as a raw material, preferably hyaluronic acid composed only of repeating unit (II) or
- the derivative is ion-exchanged with a tetraalkylammonium salt (e.g., tetrabutylammonium (TBA) salt) and combined with the hyaluronate in a solvent in the presence of a suitable condensing agent to form a compound with the formula: "HNR a -Y-NR b -R, NHR a -Y-NR b -COO-R, HNR a -Y-NR b -COO-R, HNR a -Y-NR b -CO-R, HNR a -Y-NR b -CO-NR c -R, HNR a -Y-NR b -CO-NR c -R, HNR a -Y-NR
- the condensing agent that can be used in the above reaction is not particularly limited, and examples include 4-(4,6-dimethoxy-1,3,5-triazine)-4-methylmorpholium (DMT-MM), N , N'-carbonyldiimidazole (CDI), N,N'-dicyclohexylcarbodiimide (DCC), N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ), 2-benzotriazole-1,1, 3,3-tetramethyluronium tetrafluoroborate (TBTU), 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HODhbt), benzotriazole-1-oxy -Tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP), benzotriazol-1-yl-oxy-tris(dimethylamino)phosphonium hexafluorophosphat
- DMT-MM is preferable in that the reaction proceeds with high efficiency even in a mixed solvent of water and an organic solvent. Furthermore, by using DMT-MM as a condensing agent, it is possible to highly selectively form an amide bond between an amino group and a carboxy group while suppressing the formation of an ester bond in a system where a large number of hydroxy groups coexist.
- the alcohol as a solvent may react with the carboxy group of the hyaluronic acid moiety, or the carboxyl group and hydroxyl group that are simultaneously present in the hyaluronic acid moiety may bond within or intermolecularly. , it is possible to prevent the formation of undesired crosslinks.
- Solvents used in the steryl group introduction reaction include water, DMSO, methanol, ethanol, propanol, butanol, isopropanol, polyhydric alcohol, acetonitrile, DMF, THF, dichloromethane, chloroform, hexane, diethyl ether, ethyl acetate, and mixtures thereof. Examples include solvents.
- the polyhydric alcohol may be a dihydric alcohol or a trihydric alcohol. Examples of the dihydric alcohol include ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, neopentyl glycol, 1,4-butanediol, and 1,6-hexanediol. Examples of the trihydric alcohol include glycerin and trimethylolpropane.
- hyaluronic acid or a derivative thereof as a raw material is ion-exchanged with a tetraalkylammonium salt (e.g., tetrabutylammonium (TBA) salt), and the hyaluronate and spacer moiety are reacted in a solvent in the presence of an appropriate condensing agent. (At this time, protection and deprotection reactions may be performed as necessary) to convert the carboxy group (-COOH) of the raw material hyaluronic acid or its derivative, and then react with an appropriate reagent. . Examples of combinations of groups derived from carboxy groups and reaction reagents are shown below.
- reaction mode examples include dehydrohalogenation reaction, condensation reaction, dehydration reaction, nucleophilic addition reaction such as Michael addition, oxidative disulfide formation reaction, etc. These are well-known reactions and can be selected as appropriate by those skilled in the art. However, the reaction can be carried out by finding preferable reaction conditions.
- the converter or reactant When the converter or reactant has a carboxyl group, it may be converted into an N-hydroxysuccinimide (hereinafter also referred to as "NHS”) ester and reacted.
- NHS N-hydroxysuccinimide
- a hyaluronic acid derivative having a spacer having a mercapto group modified with a leaving group at the end is prepared by reacting 2-aminoethyl 2-pyridyl disulfide with the carboxy group of the raw material hyaluronic acid or its derivative. Then, there is a method in which thiocholesterol is subjected to a nucleophilic substitution reaction to form a disulfide bond.
- hyaluronic acid or its derivatives with a part of the spacer introduced into the carboxy group and one with a part of the spacer introduced into the steryl group, and reacting them.
- a hyaluronic acid derivative in which a spacer having a mercapto group at the end is introduced into the carboxy group of hyaluronic acid;
- Another method is to prepare steryl groups into which a spacer having a mercapto group at the end is introduced, and to react them oxidatively to form a disulfide bond. At this time, one mercapto group can be reacted with 2-mercaptopyridine to form a disulfide, and then the other mercapto group can be substituted.
- the obtained hyaluronic acid derivative may be dried.
- the drying method include ventilation drying, drying in a thermostatic oven, reduced pressure drying, hot air circulation drying, and freeze drying. Among these, freeze-drying is preferred.
- the hyaluronic acid derivative preferably further contains a cryoprotectant from the viewpoint of more effectively suppressing an increase in the particle size of the fine particles formed by the hyaluronic acid derivative.
- cryoprotectant is not particularly limited as long as it is known as a "cryoprotectant” or “lyoprotectant” and includes, for example, disaccharides, sorbitol, dextran, propylene glycol, glycerin, glycerol, polyvinylpyrrolidone, Examples include dimethyl sulfoxide.
- Disaccharides are not particularly limited and include, for example, sucrose, lactulose, lactose, maltose, trehalose, cellobiose, cordibiose, nigerose, isomaltose, isotrehalose, neotrehalose, sophorose, laminaribiose, gentiobiose, turanose, maltulose, palatinose,
- Examples include gentiobiulose, mannobiose, melibiose, melibiulose, neolactose, galactosucrose, silabiose, neohesperidose, rutinose, rutinulose, vicyanose, xylobiose, primeverose, and the like.
- sucrose, trehalose, maltose, or lactose are preferred because they are widely used as cryoprotectants. Further, sucrose is more preferred from the viewpoint of its use as a pharmaceutical additive and from the viewpoint of more effectively suppressing the increase in particle size of fine particles formed by hyaluronic acid derivatives during freeze-drying.
- the cryoprotectant may be added in a solid state or in a state dissolved in a solvent such as water.
- the amount of the cryoprotectant added is not particularly limited, but is preferably 20 parts by mass or more based on 100 parts by mass of the hyaluronic acid derivative.
- the amount of the cryoprotectant added is at least the above lower limit, a more sufficient effect of suppressing particle size increase can be obtained.
- the upper limit of the amount of cryoprotectant added is not particularly limited, but may be, for example, 100,000 parts by mass.
- the apparatus used in freeze-drying is not particularly limited, and for example, a commercially available freeze-dryer can be used.
- a freeze dryer that can monitor the degree of vacuum inside the apparatus during freeze-drying is preferable, and from the viewpoint of controlling the product temperature, a shelf-type freeze dryer is preferable.
- composition 1 of this embodiment can be manufactured by Manufacturing Method 1 or Manufacturing Method 2 of Pharmaceutical Composition 1 below. Each manufacturing method will be explained below.
- Manufacturing method 1 is a method for manufacturing pharmaceutical composition 1 containing (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient.
- Production method 1 includes the steps of: (A) mixing a hyaluronic acid derivative into which a hydrophobic group has been introduced and (B) an association promoter to obtain an aqueous hyaluronic acid derivative solution containing the association promoter; and (C) associating the active ingredient. and a mixing step of mixing with a promoter-containing hyaluronic acid derivative aqueous solution.
- Manufacturing method 2 is a method for manufacturing pharmaceutical composition 1 containing (A) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B) an association promoter, and (C) an active ingredient.
- Production method 2 includes the steps of dispersing (C) an active ingredient in (B) an association promoter to obtain a dispersion (I), preparing an aqueous hyaluronic acid derivative solution or an aqueous hyaluronic acid solution containing an association promoter, and dispersing the aqueous solution (II). ), and a step of mixing the dispersion (I) and the aqueous solution (II).
- the drug may be produced using an appropriate pH and buffering material.
- manufacturing method 1 and manufacturing method 2 do not include the step of removing the organic solvent.
- the pharmaceutical composition 2 of this embodiment can be manufactured by the following manufacturing method 1 or manufacturing method 2 of the pharmaceutical composition 2. Each manufacturing method will be explained below.
- Manufacturing method 1 for pharmaceutical composition 2 includes manufacturing pharmaceutical composition 2 containing (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient. It's a method.
- Manufacturing method 1 of pharmaceutical composition 2 includes a step of mixing (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced and (B1) a solubilizing agent to obtain a hyaluronic acid derivative aqueous solution containing the solubilizing agent; (C1) A mixing step of mixing the active ingredient with a hyaluronic acid derivative aqueous solution containing a solubilization aid.
- Manufacturing method 2 of pharmaceutical composition 2 is manufacturing method 2 of pharmaceutical composition 2, which contains (A1) a hyaluronic acid derivative into which a hydrophobic group has been introduced, (B1) a solubilization aid, and (C1) an active ingredient. It's a method.
- Manufacturing method 2 of pharmaceutical composition 2 includes a step of dispersing (C1) an active ingredient in (B1) a solubilizing agent to obtain a dispersion (I), and a step of dispersing an aqueous hyaluronic acid derivative solution or an aqueous hyaluronic acid solution containing a solubilizing agent. and a step of mixing the dispersion (I) and the aqueous solution (II).
- the manufacturing method 1 and the manufacturing method 2 of the pharmaceutical composition 2 do not include the step of removing the organic solvent.
- the subjects to which the pharmaceutical composition 1 or the pharmaceutical composition 2 of the present embodiment is administered are animals classified as mammals including humans (monkeys, marmosets, mice, rats, cows, horses, cats, dogs, pigs, sheep, goats). , rabbits, etc.).
- administration route of the pharmaceutical composition 1 or pharmaceutical composition 2 of the present embodiment is not particularly limited, and any currently known administration route can be used as appropriate depending on the intended use, the location of the tissue to be treated, etc.
- administration routes include subcutaneous administration, intramuscular administration, intravenous administration, intraarterial administration, intrathecal administration, intracerebral administration, intraarticular administration, intraperitoneal administration, intravaginal administration, intracapsular administration, intrarectal administration, and intravenous administration.
- intracorporeal administration periocular administration, intradermal administration, intraperitoneal administration, intranasal administration, transbronchial administration, transpulmonary administration, transdermal administration, sublingual administration, oral administration, buccal administration, and eye drop administration.
- subcutaneous administration intramuscular administration, intravitreal administration, intrathecal administration, and intraarticular administration are preferred. It may also be administered locally as a powder, cream, ointment, or eye drops.
- Administration by injection includes subcutaneous injection, intramuscular injection, intravenous injection, intraarterial injection, intrathecal injection, intraarticular injection, intraperitoneal injection, intravitreal injection, periocular injection, intradermal injection, intratumoral injection, etc. can be mentioned.
- the use of hyaluronic acid derivatives reduces the initial burst of drugs, suppresses aggregation that may occur between drugs during preparation storage and after administration, reduces variations in drug efficacy, and prevents precipitates under physiological conditions and adheres to and remains in the corneal epithelium.
- the drug By applying the drug, the drug can be released for a longer period of time and the number of eye drops can be reduced.
- promotion of uptake of drugs into corneal epithelial cells promotion of uptake into conjunctival epithelial cells, etc. can also be considered.
- it has high moisturizing properties, viscosity, and biocompatibility derived from hyaluronic acid, and can alleviate inflammation, pain, stress, damage, etc.
- the hyaluronic acid derivative pharmaceutical composition 1 or pharmaceutical composition is excellent in tear film stabilizing effect, corneal epithelial disorder treatment effect, meibomian gland dysfunction treatment effect, and pain suppressing effect by efficiently bringing out drug effects.
- the hyaluronic acid derivative pharmaceutical composition 1 or pharmaceutical composition is excellent in tear film stabilizing effect, corneal epithelial disorder treatment effect, meibomian gland dysfunction treatment effect, and pain suppressing effect by efficiently bringing out drug effects.
- the dosage can be appropriately selected in consideration of the type of recipient (including age, gender, etc.).
- the amount of the active ingredient (preferably protein, peptide, low molecular weight compound) per dose can be 0.01 ⁇ g or more and 20 mg or less, The amount can be 0.1 ⁇ g or more and 15 mg or less, and can be 1 ⁇ g or more and 10 mg or less.
- the frequency of administration may be a single administration of the above-mentioned dose, and the above-mentioned dose may be administered for 1 day, 2 days, 4 days, 1 week, 2 weeks, 3 weeks, 4 weeks, 1 month, or 2 months. Multiple administrations of two or more times, such as once every 3 months, or every 6 months, may be used. Alternatively, the drug may be administered to two or more locations in one administration.
- the present invention comprises administering an effective amount of the above-mentioned pharmaceutical composition 1 or pharmaceutical composition 2 to a patient or an affected animal, from the group consisting of cancer, infectious and immunological diseases, and chronic diseases.
- a method for preventing or treating one or more selected diseases is provided.
- infectious diseases include those exemplified under "antigens derived from infectious diseases” under "antigens” above.
- the term "effective amount" as used herein includes an amount effective for prevention or treatment, that is, an amount suitable for prevention or treatment of the above-mentioned diseases.
- the invention relates to cancer, infectious diseases, immune diseases, inflammatory disorders, allergic diseases, skin diseases, hypertension, diabetes, neurological diseases, genetic diseases, cardiovascular diseases, cerebrovascular diseases, respiratory diseases, etc.
- a composition for the prevention or treatment of one or more diseases selected from the group consisting of diseases, eye diseases, ear diseases, and bone and joint diseases, which comprises the above-mentioned active ingredient-hyaluronic acid derivative complex. provide.
- the present invention provides the use of the above active ingredient-hyaluronic acid derivative complex for producing pharmaceutical composition 1 or pharmaceutical composition 2.
- Hyaluronic acid derivatives were prepared according to the following steps 1-A, 2-A, and 3-A.
- Step 1-A (Synthesis of cholesteryl 6-aminohexylcarbamate hydrochloride) Cholesteryl 6-aminohexyl carbamate hydrochloride (Chol hydrochloride) was synthesized according to the following step 1-1-A, followed by step 1-2-A.
- Step 1-1-A To a solution of cholesteryl chloroformate (3.37 g, 7.5 mmol) in anhydrous dichloromethane (20 mL) was added triethylamine (TEA, 1.05 mL) and stirred under an argon atmosphere. 6-(t-Butoxycarbonyl)amino-1-aminohexane (1.12 mL, 5 mmol) was added dropwise under ice cooling, and after stirring for 30 minutes under ice cooling, the mixture was warmed to room temperature (approximately 25°C). The mixture was stirred overnight.
- TSA triethylamine
- the reaction mixture was washed with ultrapure water and saturated brine, dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 1-2-A The obtained residue was dissolved in ethyl acetate (40 mL), 4N hydrochloric acid/ethyl acetate solution (40 mL) was added, and the mixture was stirred at room temperature (about 25° C.) overnight. The resulting precipitate was collected by centrifugation. The obtained solid was washed four times with ethyl acetate and then dried under reduced pressure to obtain 1.2 g of cholesteryl 6-aminohexylcarbamate hydrochloride (Chol hydrochloride).
- Step 2-A (Preparation of tetrabutylammonium (TBA) salt of hyaluronic acid)
- TBA salt of hyaluronic acid (HA-TBA) was prepared according to the following Step 2-1-A followed by Step 2-2-A.
- Step 2-1-A DOWEX (registered trademark) 50WX-8-400 (manufactured by Aldrich) was suspended in ultrapure water, and the resin was washed with ultrapure water about three times by decantation. A 40% by mass aqueous tetrabutylammonium hydroxide solution (TBA-OH) (manufactured by Aldrich) was added in an amount of about 1.5 times the molar equivalent of the cation exchange capacity of the resin, and the mixture was stirred for 30 minutes. After removing excess TBA-OH solution by decantation, the resin was further washed with excess ultrapure water to obtain a TBA-chlorinated cation exchange resin.
- TBA-OH tetrabutylammonium hydroxide solution
- Step 2-2-A Raw material hyaluronate sodium salt (HA-Na) with a molecular weight of 35,000 (35 kDa) was dissolved in ultrapure water at a concentration of 15 mg/mL.
- the suspension of the cation exchange resin which had been converted into TBA salt was added in an amount equivalent to 5 times the ion exchange capacity of the resin based on the number of moles of the HA unit (unit molecular weight: 401.3). After stirring for 15 minutes, filtration was performed using a 0.45 ⁇ m filter, and the filtrate was freeze-dried to obtain TBA salt of hyaluronic acid (HA-TBA) as a white solid.
- Step 3-A An anhydrous DMSO solution (10 mg/mL) of HA-TBA prepared in the above [Step 2-2-A] was prepared. Thereafter, Chol hydrochloride was added at a molar ratio of 19/100 to the disaccharide repeating unit (HA unit) present in the HA-TBA synthesized in [Step 1-A] above. Next, the amount of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) added to the HA unit was 24/100 in molar ratio. The mixture was added in the following proportions and stirred overnight at room temperature (about 25°C).
- DMT-MM 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- the reaction solution was dialyzed (Spectrapore 7, molecular weight cutoff (MWCO): 3,500) in the order of 0.3M ammonia acetate/DMSO solution, 0.15M NaCl aqueous solution, and ultrapure water.
- the obtained dialysate was freeze-dried to obtain the target product (HA-C 6 -Chol) as a white solid.
- Step 1-B (Synthesis of cholesteryl 6-aminohexylcarbamate hydrochloride) Cholesteryl 6-aminohexyl carbamate hydrochloride (Chol hydrochloride) was synthesized according to the following step 1-1-B, followed by step 1-2-B.
- Step 1-1-B To a solution of cholesteryl chloroformate (3.37 g, 7.5 mmol) in anhydrous dichloromethane (20 mL) was added triethylamine (TEA, 1.05 mL) and stirred under an argon atmosphere. 6-(t-Butoxycarbonyl)amino-1-aminohexane (1.12 mL, 5 mmol) was added dropwise under ice cooling, and after stirring for 30 minutes under ice cooling, the mixture was warmed to room temperature (approximately 25°C). The mixture was stirred overnight.
- TSA triethylamine
- the reaction mixture was washed with ultrapure water and saturated brine, dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 1-2-B The obtained residue was dissolved in ethyl acetate (40 mL), 4N hydrochloric acid/ethyl acetate solution (40 mL) was added, and the mixture was stirred at room temperature (about 25° C.) overnight. The resulting precipitate was collected by centrifugation. The obtained solid was washed four times with ethyl acetate and then dried under reduced pressure to obtain 1.2 g of cholesteryl 6-aminohexylcarbamate hydrochloride (Chol hydrochloride).
- TBA salt of hyaluronic acid was prepared according to the following step 2-1-B followed by step 2-2-B.
- Step 2-1-B DOWEX (registered trademark) 50WX-8-400 (manufactured by Aldrich) was suspended in ultrapure water, and the resin was washed with ultrapure water about three times by decantation. A 40% by mass aqueous tetrabutylammonium hydroxide solution (TBA-OH) (manufactured by Aldrich) was added in an amount of about 1.5 times the molar equivalent of the cation exchange capacity of the resin, and the mixture was stirred for 30 minutes. After removing excess TBA-OH solution by decantation, the resin was further washed with excess ultrapure water to obtain a TBA-chlorinated cation exchange resin.
- TBA-OH tetrabutylammonium hydroxide solution
- Step 2-2-B Raw material hyaluronate sodium salt (HA-Na) with a molecular weight of 35,000 (35 kDa) was dissolved in ultrapure water at a concentration of 15 mg/mL. Add the suspension of the cation exchange resin that was converted to TBA in "(1) Step 2-1-B" in an amount equivalent to 5 times the ion exchange capacity of the resin based on the number of moles of the HA unit (unit molecular weight 401.3). did. After stirring for 15 minutes, filtration was performed using a 0.45 ⁇ m filter, and the filtrate was freeze-dried to obtain TBA salt of hyaluronic acid (HA-TBA) as a white solid.
- HA-TBA TBA salt of hyaluronic acid
- Step 3-B An anhydrous DMSO solution (10 mg/mL) of HA-TBA prepared in [Step 2-2-B] was prepared. Thereafter, Chol hydrochloride was added at a molar ratio of 31/100 to the disaccharide repeating unit (HA unit) present in HA-TBA synthesized in [Step 1-B].
- the amount of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) added to the HA unit was 37/100 in molar ratio.
- the mixture was added in the following proportions and stirred overnight at room temperature (about 25°C).
- the reaction solution was dialyzed (Spectrapore 7, molecular weight cutoff (MWCO): 3,500) in the following order: 0.3M ammonia acetate/DMSO solution, 0.15M NaCl aqueous solution, and ultrapure water.
- the obtained dialysate was freeze-dried to obtain the target product (HA-C 6 -Chol) as a white solid.
- the target product H-C 6 -Chol
- a peak derived from the methyl group in the cholesteryl group (CH 3 , 0.7 ppm, 3H) was confirmed, and the cholesterol introduction rate was 30%.
- Example 1-1 A formulation of CyA using a hyaluronic acid derivative (35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80) was prepared. The following operations were carried out at 20° C. and room temperature. The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 12.0 mg/mL in water for injection by stirring at 20° C. for 24 hours. In a separate vial, polysorbate 80 (Fuji Film Wako, product number: 164-21591) was diluted with water for injection at a ratio of 2 mg/mL.
- Example 1-2 A CyA formulation was prepared using a hyaluronic acid derivative (35k HA-C6-Chol-19%) containing an association promoter (polyethylene glycol 400). The following operations were carried out at 20° C. and room temperature. The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 12.0 mg/mL in water for injection by stirring at 20° C. for 24 hours.
- an association promoter polyethylene glycol 400
- Example 1-3 A CyA formulation was prepared using a hyaluronic acid derivative (35k HA-C6-Chol-19%) containing association promoters (polyethylene glycol 400 and polysorbate 80). The following operations were carried out at 20° C. and room temperature.
- the lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved in water for injection at 12.0 mg/mL by stirring at 20° C. for 24 hours.
- CyA formulation using ⁇ -cyclodextrin was prepared. The following operations were carried out at 20° C. and room temperature. CyA powder was prepared by dissolving it in a 10% by mass sucrose solution containing ⁇ -cyclodextrin (manufactured by Aldrich) at a concentration of 10% by mass at a ratio of 0.5 mg/mL. At the end the solution was visually clear. Further, no precipitate was observed during 2 weeks of refrigerated storage. The final formulation composition is shown in Table 1.
- ⁇ Test Example 2> A pharmacokinetic study of CyA in rats was conducted.
- the solution formulations consisting of the hyaluronic acid derivative pharmaceutical compositions prepared in Examples 1-1, 1-2, 1-3 and Comparative Example 1-2 and the solution formulation of Comparative Example 1-1 were each prepared using a 25G needle.
- the dose (mg/kg) shown in 2 was subcutaneously administered to normal rats (SD, 6 weeks old, male).
- FIG. 3 shows the change in plasma concentration of CyA upon administration of various CyA preparations and the change in plasma concentration of CyA in Comparative Example 1.
- Example 2-1 is a case where a solution preparation consisting of the hyaluronic acid derivative pharmaceutical composition of Example 1-1 was administered.
- Examples 2-2 to 2-3 and Comparative Examples 2-1 to 2-2 are solution formulations and comparisons consisting of the hyaluronic acid derivative pharmaceutical compositions of Examples 1-2 to 1-3 and Comparative Example 1-2, respectively. This is a case where each of the solution formulations of Example 1-1 was administered.
- pharmacokinetic parameters drug half-life (T 1/2 ), mean retention time (MRT), area under the serum drug concentration-(0 to infinity) time curve (AUC inf ) were measured using WinNonlin Ver. 8.3 (manufactured by Pharsight), and the values are shown in Table 2.
- sustained release properties are evaluated by Mean Residual Time (MRT), and the MRT of the hyaluronic acid derivative pharmaceutical composition containing an association promoter is greater than the MRT of the hyaluronic acid derivative pharmaceutical composition. If so, the drug is evaluated as having sustained release properties.
- MRT Mean Residual Time
- Example 2-1, 2-2, and 2-3 which are formulations containing an association promoter of hyaluronic acid derivatives
- the half-life of the blood concentration is shorter than that of Comparative Example 2-2, which does not contain an association promoter. It was long and the MRT was large.
- Example 2-1 was 1.35
- Example 2-2 was 1.42
- Example 2-3 was 1. It was .49. In other words, it was found that the drug remained in the blood for a long period of time. From the above, it was confirmed that the hyaluronic acid derivative composition of the present invention has excellent sustained release properties over a longer period of time.
- Example 3-1 Regarding Example 1-1 prepared in Test Example 1, the degree of association promotion of hyaluronic acid derivatives was evaluated by GPC.
- FIG. 1 is a chromatogram of the hyaluronic acid derivative used in Synthesis Example 1.
- the hyaluronic derivative was dissolved in water for injection at a concentration of 1 mg/mL by stirring for 24 hours, and subjected to GPC measurement. At this time, the area value surrounded by the chromatogram and the baseline was defined as A1.
- FIG. 2 is a chromatogram of the hyaluronic acid derivative composition of Example 1-1.
- the hyaluronic derivative was diluted with water for injection at a rate of 1 mg/mL and measured. At this time, the area value surrounded by the chromatogram and the baseline was defined as A2.
- Example 3-2, 3-3, and Comparative Examples 3-1, 3-2 Examples 1-2, 1-3, and Comparative Example 1-2 were also diluted with water for injection and measured in the same manner as Example 1-1, such that the concentration of the hyaluronic derivative was 1 mg/mL.
- Comparative Example 2-1 was directly subjected to GPC measurement without being diluted. At this time, A2, which is the area value surrounded by each chromatogram and the baseline, was calculated, and the value of the area ratio A2/A1 with A1 is shown in Table 3.
- Example 4 The association promoter was evaluated by GPC.
- Examples 4-1, 4-2, 4-3 and Comparative Examples 4-1, 4-2 In the same manner as in Test Example 1, an aqueous hyaluronic acid derivative solution was prepared according to Table 4 below at the same composition ratios, except that it did not contain CyA as an active ingredient and sucrose as an isotonic agent. Each of the prepared samples was diluted with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and subjected to GPC measurement.
- Examples 5-1 to 5-3, Comparative Examples 5-1 and 5-2 Sample 150 ⁇ L of each formulation obtained in Example 1 into a 1.5 mL microtube. Thereafter, a concentrated buffer solution (40 mM PB (pH 7.4), 600 mM NaCl) was added at a ratio such that the final buffer composition was 10 mM PB (pH 7.4), 150 mM NaCl, and the hyaluronic acid derivative was precipitated. After incubating at 37°C for 20 minutes, centrifuging at 2000G for 5 minutes, sampling 50 ⁇ L of the supernatant, diluting it 2 times with HP- ⁇ -CD aqueous solution (300 mM), and incubating for 1 hour.
- a concentrated buffer solution 40 mM PB (pH 7.4), 600 mM NaCl
- Example 6 was prepared by dissolving the freeze-dried hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 at 12.0 mg/mL in water for injection by stirring at 20°C for 24 hours. -1 to 6-11 and Comparative Examples 6-1 to 6-6.
- Example 6-1 The hyaluronic acid derivative aqueous solution dissolved above was diluted with water for injection at a ratio of 2 mg/mL. In another vial, polyethylene glycol 300 was diluted with water for injection at a concentration of 10 mg/mL. The above hyaluronic acid derivative aqueous solution and polyethylene glycol 300 aqueous solution were mixed at a volume ratio of 1:1 to prepare the compositions shown in Table 6 below. Thereafter, it was incubated at 20°C for 24 hours and then subjected to GPC measurement.
- Examples 6-2 to 6-11, Comparative Examples 6-1 to 6-6 A solution having the composition shown in Table 6 below was prepared using the same method as in Example 6-1 and subjected to GPC measurement.
- Example 7 A hyaluronic acid derivative pharmaceutical composition was produced.
- Example 7 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved in water for injection at 12.0 mg/mL by stirring at 20°C for 24 hours. -1 to 7-10 and Comparative Examples 7-1 to 7-6.
- Example 7-1 A CyA formulation was prepared using a hyaluronic acid derivative (35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80). The following operations were carried out at 20° C. and room temperature. 1.8 mg of powdered CyA (Tokyo Kasei Kogyo, product number: C2408) was weighed into a vial, and then polysorbate 80 was diluted with water for injection to 0.5 mg/mL in another vial. The weighed CyA was suspended in 18 ⁇ L of an aqueous polysorbate 80 solution with a concentration of 0.5 mg/mL.
- Example 7-2 to 7-10 A hyaluronic acid derivative pharmaceutical composition containing an association promoter was prepared in the same manner as in Example 7-1 so that the concentration of the association promoter in the final composition was as shown in Table 7 below. Furthermore, the CyA concentration in the formulation after passing through a 0.45 ⁇ m sterile filtration filter was also determined by HPLC in the same manner as in Example 7-1, and the results are summarized in Table 7.
- a concentrated buffer solution 40 mM PB, 150 mM NaCl aqueous solution
- a preparation consisting of a hyaluronic acid derivative pharmaceutical composition is added. Thereafter, the mixture is mixed with a vortex for 30 seconds, incubated at 37°C for 20 minutes, and then the precipitate is sedimented with a centrifuge (2000G, 5
- Precipitation rate (%) ⁇ 1-(Area value of hyaluronic acid derivative of precipitation sample) ⁇ ((Area value of hyaluronic acid derivative of blank sample) x (0.75X)) ⁇ x 100
- association promoters such as polysorbate 80, polysorbate 20, poloxamer 188, and cremophor EL
- the poorly water-soluble active ingredients can be highly extracted from powder without using organic solvents. It was shown that it could be solubilized at a certain concentration. Furthermore, it was suggested that the association promoter also improves precipitation performance. From the above, it is expected that the association promoter will promote the association of hyaluronic acid derivatives and provide a pharmaceutical composition that is high in active ingredient content and capable of long-term sustained release.
- Example 8 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-30%) obtained in Synthesis Example 2 was dissolved in water for injection at 2.0 mg/mL by stirring at 20°C for 24 hours. -1 to 8-11 and Comparative Examples 8-1 to 8-6.
- Polysorbate 80 was prepared by diluting it with water for injection at a concentration of 10 mg/mL in a glass vial. A 6 mL clean vial was prepared, 400 ⁇ L of the above hyaluronic acid derivative aqueous solution and 10 ⁇ L of polysorbate 80 aqueous solution were added, mixed for 30 seconds with a vortex, and then 390 ⁇ L of water for injection was added to adjust the total to 800 ⁇ L. The ratio was adjusted to give the composition shown in Table 8 below. Thereafter, it was incubated at 20° C. for 24 hours and then subjected to GPC measurement.
- Example 8-2 to 8-4, Comparative Examples 8-1 to 8-6 A solution having the composition shown in Table 8 below was prepared using the same method as in Example 8-1 and subjected to GPC measurement.
- Example 9 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 4.0 mg/mL in water for injection by stirring at 20°C for 24 hours. -1 to 9-2 and used in Comparative Example 9-1.
- Example 9-1 to Example 9-2 An association promoter and water for injection were added to the above hyaluronic acid derivative aqueous solution.
- human growth hormone (hGH: Genotropin (registered trademark) for injection) powder was dissolved in water for injection in a separate vial to a concentration of 2 mg/mL, and then the hyaluronic acid derivative containing the association promoter was prepared. 250 ⁇ L was added to the aqueous solution, and the total volume was adjusted to 1 mL. Thereafter, complexation with hGH was promoted by incubating at 37°C for 24 hours. The composition of the final formulation was adjusted to the concentrations listed in Table 9.
- Example 9-1 to 9-2 and Comparative Example 9-1 the concentration of the hyaluronic acid derivative was adjusted to 1.0 mg/mL, and the concentration of hGH was adjusted to 0.5 mg/mL. After 24 hours, clear solutions were obtained in Examples 9-1 and 9-2. Here, only in Comparative Example 9-1, the solution became cloudy after incubation at 37° C. for 24 hours, making it difficult to obtain a complexed solution of hyaluronic acid derivative and hGH.
- hyaluronic acid derivative composition containing polyethylene glycol 400 which is an association promoter, efficiently complexed hGH, which is a protein drug.
- Long-term sustained release functionality was expected not only for poorly soluble drugs but also for water-soluble drugs.
- peptide drugs such as CyA
- application to protein drugs would enable long-term sustained release in many modalities.
- Examples 10-1 to 10 were prepared by dissolving the hyaluronic acid derivative (10k HA-C6-Chol-40%) obtained in Synthesis Example 3 at 36.0 mg/mL in water for injection by stirring at 20°C for 24 hours. -9 was used.
- polysorbate 80 was diluted with water for injection at a concentration of 10 mg/mL. 600 ⁇ L of the above hyaluronic acid derivative aqueous solution was added to a 6 ml sterile vial, then 10 ⁇ L of 10 mg/mL polysorbate 80 aqueous solution was added and mixed while stirring, and finally 390 ⁇ L of water for injection was added. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-2 First, a 100 mM PB (pH 7.4) phosphate buffer solution was prepared using phosphate buffer powder (manufactured by Fuji Film Wako Co., Ltd.: 167-14491 for Biochemistry).
- Example 10-1 Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 50 ⁇ L of 10 mg/mL polysorbate 80 aqueous solution and mix while stirring, add 100 ⁇ L of 100 mM PB phosphate buffer, and finally add water for injection.
- a hyaluronic acid derivative aqueous solution was prepared by adding 250 ⁇ L to give a final concentration of 10 mM PB. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- Example 10-3 The following solution preparation was performed using the solution prepared in Example 10-1. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 50 ⁇ L of 10 mg/mL polysorbate 80 aqueous solution and mix with stirring, and finally add 350 ⁇ L of water for injection to prepare the hyaluronic acid derivative aqueous solution. Preparation was carried out. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-4 Subsequently, the following solutions were prepared using the solutions prepared in Examples 10-1 and 10-2. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 100 ⁇ L of polyethylene glycol 400 (manufactured by Nacalai Tesque) and mix while stirring, add 100 ⁇ L of 100 mM PB phosphate buffer, and finally inject. A hyaluronic acid derivative aqueous solution was prepared by adding 200 ⁇ L of water to give a final concentration of 10 mM PB. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- polyethylene glycol 400 manufactured by Nacalai Tesque
- Example 10-5 Subsequently, the following solution preparation was performed using the solution prepared in Example 10-1. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add and mix 100 ⁇ L of polyethylene glycol 400 (manufactured by Nacalai Tesque) while stirring, and finally add 300 ⁇ L of water for injection to prepare the hyaluronic acid derivative. An aqueous solution was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-6 Subsequently, the following solution preparation was performed using the solution prepared in Example 10-1. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 300 ⁇ L of polyethylene glycol 400 (manufactured by Nacalai Tesque) and mix while stirring, and finally add 100 ⁇ L of water for injection to prepare the hyaluronic acid derivative. An aqueous solution was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-7 Poloxamer 338 (manufactured by Aldrich) was dissolved in water for injection at a concentration of 150 mg/mL. Subsequently, the following solution preparation was performed using the solution prepared in Example 10-1. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 100 ⁇ L of 150 mg/mL Poloxamer 338 aqueous solution and mix while stirring, and finally add 300 ⁇ L of water for injection to prepare a hyaluronic acid derivative aqueous solution. I did it. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-8 The following solutions were prepared using the solutions prepared in Example 10-1, Example 10-2, and Example 10-7. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 300 ⁇ L of Poloxamer 338 aqueous solution and mix while stirring, add 100 ⁇ L of 100 mM PB phosphate buffer, and add hyaluronic acid at a ratio to give a final concentration of 10 mM PB. An aqueous derivative solution was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-9 The following solutions were prepared using the solutions prepared in Example 10-1 and Example 10-7. Add 600 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 300 ⁇ L of 150 mg/mL Poloxamer 338 aqueous solution and mix while stirring, and finally add 100 ⁇ L of water for injection to prepare a hyaluronic acid derivative aqueous solution. I did it. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-10 Chol-PEG600 (manufactured by Avanti) was dissolved by adding water for injection at a concentration of 50 mg/mL.
- Example 10- 10 to 10-12 Add 750 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 2.8 ⁇ L of 50 mg/mL Chol-PEG600 aqueous solution and mix while stirring, and finally add 747.2 ⁇ L of water for injection.
- a hyaluronic acid derivative aqueous solution was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- 10 mM PB phosphate buffer, pH 7.4
- Example 10-11 The following solution preparation was performed using the solution prepared in Example 10-10. Add 750 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 11.2 ⁇ L of 50 mg/mL Chol-PEG600 aqueous solution and mix while stirring, and finally add 738.8 ⁇ L of water for injection. A hyaluronic acid derivative aqueous solution was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 10-12 The following solution preparation was performed using the solution prepared in Example 10-10. Add 750 ⁇ L of the above hyaluronic acid derivative aqueous solution to a 6 ml sterile vial, then add 28 ⁇ L of 50 mg/mL Chol-PEG600 aqueous solution and mix with stirring, and finally add 722 ⁇ L of water for injection to prepare the hyaluronic acid derivative aqueous solution. was prepared. Thereafter, the mixture was incubated at 20° C. for 24 hours, diluted with 10 mM PB (phosphate buffer, pH 7.4) to give a hyaluronic acid derivative concentration of 1 mg/mL, and then subjected to GPC measurement.
- PB phosphate buffer, pH 7.4
- Example 11-1 A CyA formulation was prepared using a hyaluronic acid derivative (35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80). The following operations were carried out at 20° C. and room temperature. The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved by adding a 10% sucrose aqueous solution at a concentration of 12.0 mg/mL.
- sucrose solution For the 10% sucrose solution, use a solution in which sucrose powder (manufactured by Fujifilm Wako Co., Ltd., for manufacturing purposes only) is sufficiently dissolved in water for injection, and then sterilized and filtered using a 0.22 ⁇ m filter (membrane material: PTFE). did. Weighed 10.0 mg of powdered CyA (Tokyo Kasei Kogyo, product number: C2408) into another vial, then added 0.50 mL of polysorbate 80 aqueous solution dissolved in water for injection at a concentration of 100 mg/mL, and added it using a stirrer tip. The mixture was stirred to disperse CyA.
- Cyclosporin A (manufactured by Tokyo Kasei Kogyo Co., Ltd.) was weighed into a conical tube, 10 mL of 10% sucrose/10% ⁇ -Cyclodextrin aqueous solution was added, and the mixture was stirred and dissolved. Finally, it was filtered through a 0.2 ⁇ m filter to obtain an administration solution.
- the final formulation composition is shown in Table 11.
- ⁇ Test Example 12> A pharmacokinetic study of CyA in rats was conducted.
- the solution formulations consisting of the hyaluronic acid derivative pharmaceutical compositions prepared in Example 11-1 and Comparative Example 11-2 and the solution formulation of Comparative Example 11-1 were administered at the doses shown in Table 12 (mg/kg) using a 25G needle, respectively. ) was administered subcutaneously to normal rats (SD, 6 weeks old, male).
- FIG. 4 shows the changes in the blood concentration of CyA during administration of various CyA preparations and the changes in the blood concentration of CyA in Comparative Example 1.
- Example 12-1 is a case where a solution preparation consisting of the hyaluronic acid derivative pharmaceutical composition of Example 11-1 was administered.
- Comparative Example 12-2 is a case in which a solution formulation consisting of the hyaluronic acid derivative pharmaceutical composition of Comparative Example 11-2 is administered, and Comparative Example 12-1 is a case in which the solution formulation of Comparative Example 11-1 is administered.
- the actual measured value was used for the maximum plasma concentration (C max ).
- the area under the blood concentration-time curve (AUC 0 ⁇ ) and the area under the blood concentration-first moment curve (AUMC 0 ⁇ ) are calculated using the linear trapezoidal method for the area up to the detectable measurement time, and the area under the blood concentration-first moment curve (AUMC 0 ⁇ ) for The area up to infinity was calculated using the disappearance rate constant ( ke ) approximately calculated from three points before the detection limit.
- MRT mean residence time
- Example 12-1 which is a formulation containing an association promoter of a hyaluronic acid derivative
- the value of Cmax/Dose which is an index of initial release abnormality
- Comparative Example 12-2 which does not contain an association promoter. MRT was a large value.
- the sustained release property was evaluated using formula (A)
- the value of Example 12-1 was 2.49 compared to Comparative Example 12-2. In other words, it was found that the drug remained in the blood for a long period of time. From the above, it was confirmed that the hyaluronic acid derivative composition of the present invention has excellent sustained release properties over a longer period of time.
- ⁇ Test Example 13> A hyaluronic acid derivative pharmaceutical composition was produced.
- Example 13 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved in water for injection at 12.0 mg/mL by stirring at 20°C for 24 hours. -1 to 13-2 and used in Comparative Example 13-1. The following operations were carried out at 20° C. and room temperature.
- Example 13-1 First, polysorbate 80 (manufactured by Thermo Scientific) was dissolved by adding water for injection at a concentration of 10 mg/mL. Subsequently, glycerin (manufactured by Nacalai Tesque, product code: 17045-94) was dissolved in water for injection to prepare a concentration of 170 mg/mL. Furthermore, 1 mL of 0.5 mol/L-EDTA solution (pH 8.0) (manufactured by Nacalai Tesque, product code: 06894-14) was added to 28.240 mL of water for injection, and 5.0 mg/mL EDTA buffer was added. It was adjusted.
- hyaluronic acid derivative aqueous solution 416.7 ⁇ L of the above hyaluronic acid derivative aqueous solution was added to a 6 ml sterile vial, and then 100 ⁇ L of 10 mg/mL polysorbate 80 aqueous solution was added and mixed while stirring, and then 318.3 ⁇ L of water for injection was added. Finally, 15 ⁇ L of 5.0 mg/mL EDTA buffer was added to prepare a hyaluronic acid derivative aqueous solution. At this time, the EDTA concentration was 0.075 mg/mL, the glycerin concentration was 25.5 mg/mL, the polysorbate 80 concentration was 1 mg/mL, and the hyaluronic acid derivative concentration was 5 mg/mL.
- a hyaluronic acid derivative solution was prepared with the same composition except that it did not contain an association promoter, and the obtained area value was set as A1, and the degree of association promotion by the association promoter A2/A1 was calculated and shown in Table 13.
- CyA formulation using a hyaluronic acid derivative 35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80) was prepared.
- a CyA formulation using a hyaluronic acid derivative 35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80) was prepared.
- 2.5 mg of CyA was weighed into a 6 mL sterile vial. Thereafter, 450 ⁇ L of the hyaluronic acid derivative solution prepared in Example 13-1 was added and stirred at 20° C. for 24 hours. Undissolved CyA powder was observed, and the next operation was carried out in a saturated state.
- the CyA-containing hyaluronic acid derivative pharmaceutical composition prepared above was sterilized and filtered through a 13 mm ⁇ 0.22 ⁇ m PES filter to remove precipitated drugs that could not be encapsulated in the hyaluronic acid derivative.
- Example 13-2 An aqueous solution of a hyaluronic acid derivative containing an association promoter was prepared in the same manner as in Example 13-1, except that the polysorbate concentration was 3 mg/mL. At this time, the EDTA concentration was 0.075 mg/mL, the glycerin concentration was 25.5 mg/mL, the polysorbate 80 concentration was 3 mg/mL, and the hyaluronic acid derivative concentration was 5 mg/mL. Thereafter, the mixture was stirred at 20° C. for 24 hours, diluted with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and subjected to GPC measurement.
- a hyaluronic acid derivative solution was prepared with the same composition except that it did not contain an association promoter, and the degree of association promotion by the association promoter was calculated in the same manner as in Example 13-1, using the obtained area value as A1, and the results are shown in Table 13. Described.
- CyA formulation using a hyaluronic acid derivative 35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80) was prepared.
- a CyA formulation using a hyaluronic acid derivative 35k HA-C6-Chol-19%) containing an association promoter (polysorbate 80) was prepared.
- 2.5 mg of CyA was weighed into a 6 mL sterile vial. Thereafter, 450 ⁇ L of the hyaluronic acid derivative solution prepared in Example 13-2 was added and stirred at 20° C. for 24 hours. Undissolved CyA powder was observed, and the next operation was carried out in a saturated state.
- the CyA-containing hyaluronic acid derivative pharmaceutical composition prepared above was sterilized and filtered through a 13 mm ⁇ 0.22 ⁇ m PES filter to remove precipitated drugs that could not be encapsulated in the hyaluronic acid derivative.
- Example 13-1 A hyaluronic acid derivative aqueous solution was prepared in the same manner as in Example 13-1, except that the polysorbate concentration was 0 mg/mL. At this time, the EDTA concentration was 0.075 mg/mL, the glycerin concentration was 25.5 mg/mL, the polysorbate 80 concentration was 0 mg/mL, and the hyaluronic acid derivative concentration was 5 mg/mL. Thereafter, the mixture was stirred at 20° C. for 24 hours, diluted with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and subjected to GPC measurement.
- CyA formulation using a hyaluronic acid derivative 35k HA-C6-Chol-19%) was prepared.
- 2.5 mg of CyA was weighed into a 6 mL sterile vial. Thereafter, 450 ⁇ L of the hyaluronic acid derivative solution prepared in Comparative Example 13-1 was added and stirred at 20° C. for 24 hours. Undissolved CyA powder was observed, and the next operation was carried out in a saturated state. An attempt was made to sterilize the CyA-containing hyaluronic acid derivative pharmaceutical composition prepared above using a 13 mm ⁇ 0.22 ⁇ m PES filter, but filtration was difficult.
- the GPC peak of the association promoter alone was also verified under conditions not containing the hyaluronic acid derivative, but no peak derived from the association promoter was detected at the peak position of the hyaluronic acid derivative.
- the final composition is shown in Table 13.
- the active ingredient CyA was 29.1 or 37.6 parts by mass per 100 parts by mass of the hyaluronic acid derivative, and it was confirmed that it could be solubilized at a high concentration. Longer-term sustained release is expected.
- Example 14-1 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 12.0 mg/mL in water for injection by stirring at 20° C. for 24 hours. Add 416.7 ⁇ L of the above 12.0 mg/mL hyaluronic acid derivative aqueous solution to a 1.5 mL Eppendorf tube, and then add polyvinyl alcohol (degree of polymerization 500, manufactured by Nacalai Tesque) dissolved in water for injection at a concentration of 100 mg/mL. , product number: 11738-62). Thereafter, water for injection was added at a rate of 1000 ⁇ L in total (Table 14). Thereafter, the mixture was incubated at 20° C. for 2 hours, diluted 5 times with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and then subjected to GPC measurement.
- polyvinyl alcohol degree of polymerization 500, manufactured by Nacal
- Example 14-2 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-40%) obtained in Synthesis Example 3 was dissolved in water for injection at 62.6 mg/mL by stirring at 20° C. for 24 hours. Add 79.9 ⁇ L of the above 62.6 mg/mL hyaluronic acid derivative aqueous solution to a 1.5 mL Eppendorf tube, and then add polyvinyl alcohol (degree of polymerization 500, manufactured by Nacalai Tesque) dissolved in water for injection at a concentration of 100 mg/mL. , product number: 11738-62). Thereafter, water for injection was added at a rate of 1000 ⁇ L in total (Table 14). Thereafter, the mixture was incubated at 20° C. for 2 hours, diluted 5 times with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and then subjected to GPC measurement.
- polyvinyl alcohol degree of polymerization 500, manufactured by Na
- Example 14-3 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-30%) obtained in Synthesis Example 2 was dissolved in water for injection at 40.0 mg/mL by stirring at 20° C. for 24 hours. Add 125.0 ⁇ L of the above 40.0 mg/mL hyaluronic acid derivative aqueous solution to a 1.5 mL Eppendorf tube, and then add polyvinyl alcohol (polymerization degree 500, manufactured by Nacalai Tesque) dissolved in water for injection at a concentration of 100 mg/mL. , product number: 11738-62). Thereafter, water for injection was added at a rate of 1000 ⁇ L in total (Table 14). Thereafter, the mixture was incubated at 20° C. for 2 hours, diluted 5 times with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and then subjected to GPC measurement.
- polyvinyl alcohol polymerization degree 500, manufactured by Nacalai
- the GPC peak of the association promoter alone was also verified under conditions not containing the hyaluronic acid derivative, but no peak derived from PVA, which is the association promoter, was detected at the peak position of the hyaluronic acid derivative.
- Example 14-4 to Example 14-6 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 12.0 mg/mL in water for injection by stirring at 20° C. for 24 hours. Further, using the 10 mg/mL polysorbate 80 aqueous solution prepared in Example 13-1, the 170 mg/mL glycerol aqueous solution, the water for injection, and the 5.0 mg/mL EDTA solution, an association promoter-containing hyaluronic acid derivative aqueous solution was prepared to the concentration shown in Table 14. Adjusted by percentage. Thereafter, it was incubated at 20°C for 24 hours and then subjected to GPC measurement.
- Example 14-7 to Example 14-8 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-40%) obtained in Synthesis Example 3 was dissolved in water for injection at 62.6 mg/mL by stirring at 20° C. for 24 hours. Further, using the 10 mg/mL polysorbate 80 aqueous solution prepared in Example 13-1, the 170 mg/mL glycerol aqueous solution, the water for injection, and the 5.0 mg/mL EDTA solution, an association promoter-containing hyaluronic acid derivative aqueous solution was prepared to the concentration shown in Table 14. Adjusted by percentage. Thereafter, it was incubated at 20°C for 24 hours and then subjected to GPC measurement.
- Example 14-9 to Example 14-11 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-30%) obtained in Synthesis Example 2 was dissolved in water for injection at 40.0 mg/mL by stirring at 20° C. for 24 hours. Further, using the 10 mg/mL polysorbate 80 aqueous solution prepared in Example 13-1, the 170 mg/mL glycerol aqueous solution, the water for injection, and the 5.0 mg/mL EDTA solution, an association promoter-containing hyaluronic acid derivative aqueous solution was prepared to the concentration shown in Table 14. Adjusted by percentage. Thereafter, it was incubated at 20°C for 24 hours and then subjected to GPC measurement.
- Example 14-4 to Example 14-11 Each sample was diluted 5 times with water for injection at a rate such that the concentration of the hyaluronic acid derivative was 1 mg/mL, and then subjected to GPC measurement.
- the GPC peak of the association promoter alone was also verified under conditions not containing the hyaluronic acid derivative, but no peak derived from the association promoter polysorbate 80 was detected at the peak position of the hyaluronic acid derivative.
- the final composition and degree of association promotion are shown in Table 14.
- association promoter has the function of properly interacting and promoting the association of hyaluronic acid derivatives. It is expected that various other buffers, antibacterial preservatives, and stabilizers will have an association-promoting effect, and by expanding the hydrophobic part within nanoparticles, the amount of solubilized poorly soluble drugs can be increased and sustained release after in vivo administration. We can expect the ability to extend the period.
- Example 15-1 The lyophilized hyaluronic acid derivative (35k HA-C6-Chol-19%) obtained in Synthesis Example 1 was dissolved at 12.0 mg/mL in water for injection by stirring at 20° C. for 24 hours. Thereafter, it was dissolved in another vial with water for injection at a glycerol concentration of 170 mg/mL. Furthermore, in another vial, a 0.5 mol/l-EDTA solution (pH 8.0) (manufactured by Nacalai Tesque) was diluted with water for injection at a rate to give an EDTA concentration of 5 mg/mL. Furthermore, in another vial, polysorbate 80 was diluted with water for injection at a concentration of 10 mg/mL. Using the solution prepared above, an aqueous hyaluronic acid derivative solution containing an association promoter was prepared.
- the powder of semaglutide (manufactured by Funakoshi, product code: AG-CP3-0032) was dissolved in a 10 mM phosphate buffer solution in an Eppendorf tube to a concentration of 1 mg/mL, and then the above-mentioned association promoter-containing powder was prepared.
- 150 ⁇ L of a hyaluronic acid derivative aqueous solution and 150 ⁇ L of a 1 mg/mL semaglutide solution were mixed, and then incubated at 20° C. for 1 hour to promote complexation with semaglutide.
- the composition of the final formulation was adjusted to the concentrations listed in Table 15.
- Example 15-2 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-30%) obtained in Synthesis Example 2 was dissolved in water for injection at 40.0 mg/mL by stirring at 20° C. for 24 hours. Thereafter, it was dissolved in another vial with water for injection at a glycerol concentration of 170 mg/mL. Furthermore, in another vial, a 0.5 mol/l-EDTA solution (pH 8.0) (manufactured by Nacalai Tesque) was diluted with water for injection at a rate to give an EDTA concentration of 5 mg/mL. Furthermore, in another vial, polysorbate 80 was diluted with water for injection at a concentration of 10 mg/mL. Using the solution prepared above, an aqueous hyaluronic acid derivative solution containing an association promoter was prepared.
- aqueous solution 125 ⁇ L of a 40.0 mg/mL hyaluronic acid derivative 10k HA-C6-Chol-30%) aqueous solution was added, and then 100 ⁇ L of a 10 mg/mL polysorbate 80 aqueous solution was added while stirring with a stirrer. 150 ⁇ L of mL glycerol solution was added. Thereafter, 610 ⁇ L of water for injection and 15 ⁇ L of 5 mg/mL EDTA solution were added and stirred overnight.
- the powder of semaglutide (manufactured by Funakoshi, product code: AG-CP3-0032) was dissolved in a 10 mM phosphate buffer solution in an Eppendorf tube to a concentration of 1 mg/mL, and then the above-mentioned association promoter-containing powder was prepared.
- 150 ⁇ L of a hyaluronic acid derivative aqueous solution and 150 ⁇ L of a 1 mg/mL semaglutide solution were mixed, and then incubated at 20° C. for 1 hour to promote complexation with semaglutide.
- the composition of the final formulation was adjusted to the concentrations listed in Table 15.
- Example 15-1 to 15-2 the concentration of the hyaluronic acid derivative was adjusted to 2.5 mg/mL, and the concentration of semaglutide was adjusted to 0.5 mg/mL.
- the semaglutide conjugation rate was evaluated by GPC as shown below.
- the hyaluronic acid derivative composition containing polysorbate 80, an association promoter efficiently complexed the peptide drug semaglutide.
- JP6271672B2 mentions that EDTA plays the role of a protease inhibitor and can be included as a stabilizer for API, but even under such a stabilizer, the association promoter may not interact properly. It also suggests that hyaluronic acid derivatives can be encapsulated. From this result, it was inferred that in addition to cyclic peptide drugs such as CyA, application to long-chain peptide drugs would enable long-term sustained release in many modalities.
- Step 1B (Synthesis of cholesteryl 6-aminohexylcarbamate hydrochloride) Cholesteryl 6-aminohexyl carbamate hydrochloride (Chol hydrochloride) was synthesized according to Step 1B-1 shown below, followed by Step 1B-2.
- the reaction mixture was washed with ultrapure water and saturated brine, dried over anhydrous magnesium sulfate, and then the solvent was distilled off under reduced pressure.
- Step 1B-2 The obtained residue was dissolved in ethyl acetate (40 mL), 4N hydrochloric acid/ethyl acetate solution (40 mL) was added, and the mixture was stirred at room temperature (about 25° C.) overnight. The resulting precipitate was collected by centrifugation. The obtained solid was washed four times with ethyl acetate and then dried under reduced pressure to obtain 1.2 g of cholesteryl 6-aminohexylcarbamate hydrochloride (Chol hydrochloride).
- Step 2B Preparation of tetrabutylammonium (TBA) salt of hyaluronic acid
- TBA salt of hyaluronic acid HA-TBA
- Step 2B-1 DOWEX (registered trademark) 50WX-8-400 (manufactured by Aldrich) was suspended in ultrapure water, and the resin was washed with ultrapure water about three times by decantation.
- a 40% by mass aqueous tetrabutylammonium hydroxide solution (TBA-OH) (manufactured by Aldrich) was added in an amount of about 1.5 times the molar equivalent of the cation exchange capacity of the resin, and the mixture was stirred for 30 minutes. After removing the excess TBA-OH solution by decantation, the resin was further washed with excess ultrapure water to obtain a TBA-chlorinated cation exchange resin.
- Step 2B-2 Raw material hyaluronate sodium salt (HA-Na) with a molecular weight of 10,000 (10 kDa) was dissolved in ultrapure water at a concentration of 15 mg/mL. A suspension of the cation exchange resin salted with TBA in "Step 2B-1" was added in an amount equivalent to 5 times the ion exchange capacity of the resin based on the number of moles of the HA unit (unit molecular weight 401.3). After stirring for 15 minutes, filtration was performed using a 0.45 ⁇ m filter, and the filtrate was freeze-dried to obtain TBA salt of hyaluronic acid (HA-TBA) as a white solid.
- HA-TBA TBA salt of hyaluronic acid
- Step 3B An anhydrous DMSO solution (10 mg/mL) of HA-TBA prepared in "Step 2B-2" was prepared. Thereafter, Chol hydrochloride was added in a molar ratio of 44/100 to the disaccharide repeating unit (HA unit) present in HA-TBA synthesized in "Step 1B". Next, the amount of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) added to the HA unit was 48/100 in molar ratio. The mixture was stirred at room temperature (approximately 25°C) overnight.
- DMT-MM 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
- the reaction solution was dialyzed (Spectrapore 7, molecular weight cutoff (MWCO): 3,500) in the order of 0.3M ammonia acetate/DMSO solution, 0.15M NaCl aqueous solution, and ultrapure water.
- the obtained dialysate was freeze-dried to obtain the target product (HA-C 6 -Chol) as a white solid.
- Example 1B-1 Cyclosporine (CyA), a poorly water-soluble peptide, was formulated from powder using a hyaluronic acid derivative (10k HA-C6-Chol-44%).
- aqueous solution (II) The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL. As a result, an aqueous hyaluronic acid derivative solution (II) was obtained.
- Step of obtaining dispersion liquid (I) In a separate vial, polysorbate 80 was diluted with water for injection at a rate of 10 mg/mL. Furthermore, 4.0 mg of powdered CyA (manufactured by Tokyo Kasei Kogyo Co., Ltd., product number: C2408) was weighed into another vial, and then 80 ⁇ L of 10 mg/mL polysorbate 80 was added. Thereby, a CyA dispersion in which CyA was dispersed in polysorbate 80 was obtained as a dispersion (I).
- Example 1B-2 to Example 1B-11 CyA was formulated into a formulation using a hyaluronic acid derivative and a solubilizing agent in the same manner as in Example 1B-1 except for changing the type and amount of the solubilizing agent.
- the theoretical concentration in the formulation calculated from the added amount and the actual drug concentration in the formulation determined by HPLC measurement are shown in Tables 17 and 18, respectively.
- Example 1B-1 the product was filtered through a 0.45 ⁇ m sterile filter to remove precipitated CyA, and the drug concentration in the preparation was determined by HPLC measurement in the same manner as in Example 1B-1. It was confirmed that all the filtrates were clear.
- Table 4 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- Example 1B-1 the product was filtered through a 0.45 ⁇ m sterile filter to remove precipitated CyA, and the drug concentration in the preparation was determined by HPLC measurement in the same manner as in Example 1B-1. It was confirmed that all the filtrates were clear.
- Table 19 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- the product was filtered through a 0.45 ⁇ m sterile filter to remove precipitated CyA, and the drug concentration in the preparation was determined by HPLC measurement.
- Table 19 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- Span83 used in Comparative Example 1B-9 was manufactured by Tokyo Chemical Industry Co., Ltd., and sucrose stearate used in Comparative Examples 1B-10 to 1B-12 was a reagent manufactured by Mitsubishi Chemical Company.
- the solubilization aid promotes hydrophobic interaction between the steryl groups of the hyaluronic acid derivative, and as a result, the hydrophobic region of the hyaluronic acid derivative is expanded, resulting in a hydrophobic
- the solubilization aid inside the hyaluronic acid derivative composition acts as a highly mobile hydrophobic region, in addition to the hydrophobic region bound by chemical bonds to the hyaluronic acid polymer.
- powdered drugs can be solubilized at high concentrations.
- the amount of poorly water-soluble drugs solubilized generally increases in proportion to the amount of solubilizer added, but in the present invention, even though the total mass of the solubilizer is reduced, it is effective.
- Highly concentrated formulations of ingredients can be prepared.
- Comparative Examples 1B-9 to 1B-12 all satisfy the carbon number of 4 or more, but are solubilization aids that have only one or three ether structures. . In all of these, the drug and solubilization aid could not be stably dissolved in the hyaluronic acid derivative, making filtration almost difficult.
- a composition consisting of a hyaluronic acid derivative and a solubilizing agent that contains at least 4 or more ether structures (R-OR-R) and has a carbon number of 4 or more can be prepared from powder without using any organic solvent. Even if the concentration of the solubilizing agent is reduced, the ability to solubilize poorly water-soluble drugs can be greatly improved, and it is possible to provide a simple formulation method and a pharmaceutical composition with low toxicity.
- Example 2B-1 to 2B-12 The amount of solubilization aid added was verified in detail.
- the lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection to a concentration of 5 mg/mL.
- 1.0 mg of powdered CyA was weighed, 0.2 mL was added so that the final composition of the solubilizing agent concentration in the formulation was the concentration listed in Table 21, and a stirrer tip was added to stir it. .
- solubilizing agents containing at least 4 or more ether structures (R-O-R) and having a carbon number of 4 or more can be used in formulations as shown in Examples 2B-1 to 2B-13. improved drug concentration.
- Example 3B-1 to 3B-14 Next, usable solubilization aids were examined in detail. Specifically, we verified whether polyethylene glycol 300 and polyethylene glycol 400, which contain at least 5 or more ether structures (R-O-R) and have 12 or more carbon atoms, can be used as solubilization aids. .
- the lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection to a concentration of 8 mg/mL.
- the concentration of the solubilizing agent in the finally obtained hyaluronic acid derivative pharmaceutical composition was adjusted as shown in Table 7 below.
- Example 3B-1 is a solubilizing agent that contains at least 4 or more ether structures (ROR) and has a carbon number of 4 or more.
- ROR ether structures
- the drug concentration in the formulation was increased.
- the active ingredient could be solubilized at a higher concentration than the sum of the solubilized amounts of the hyaluronic acid derivative alone and the solubilization aid alone, demonstrating a synergistic effect.
- Example 4B-1 Next, detailed verification of the method for producing a hyaluronic acid derivative pharmaceutical composition was conducted.
- the lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- polysorbate 80 was diluted with water for injection to 10 mg/mL.
- Table 23 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- Example 4B-2 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- polysorbate 80 was diluted with water for injection to 10 mg/mL.
- 320 ⁇ L of 10 mg/mL polysorbate 80 was added to another vial, and while stirring with a stirrer tip in the vial, 0.60 mL of the above 36.0 mg/mL hyaluronic acid derivative aqueous solution was added, and the liquid volume was Water for injection was added to make 1.30 mL.
- the method includes a step (I) of dispersing the active ingredient in a solubilization aid, and a step (II) of preparing the aqueous hyaluronic acid derivative solution or the aqueous hyaluronic acid solution containing the solubilizer, and mixing (I) and (II).
- the solubility of the poorly water-soluble active ingredient in water from the powder can be improved by both methods: the formulation method in which the active ingredient is prepared in the step of mixing the active ingredient with a hyaluronic acid derivative aqueous solution containing a solubilizing agent. It was found that a hyaluronic acid derivative pharmaceutical composition excellent in increasing the In addition, in this production method, even when the solubilization aid is other than polysorbate 80, high solubilization ability is expected without greatly depending on the mixing method.
- Example 5B-1 Next, we verified the types of poorly water-soluble drugs.
- the lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at a concentration of 36.0 mg/mL.
- Table 24 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- Example 5B-2 A formulation was prepared in the same manner as in Example 5B-1, except that the poorly water-soluble drug was changed to fluticasone propionate (manufactured by Tokyo Kasei Kogyo Co., Ltd.) instead of paclitaxel.
- Example 5B-1 A formulation was prepared in the same manner as in Example 5B-1, except that no solubilization aid was added.
- Example 5B-2 A formulation was prepared in the same manner as in Example 5B-2, except that no solubilization aid was added.
- Example 5B-3 A formulation was prepared in the same manner as in Example 5B-1, except that no hyaluronic acid derivative was added.
- Example 5B-4 A formulation was prepared in the same manner as in Example 5B-2, except that no hyaluronic acid derivative was added.
- Table 24 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement. Shown below.
- solubilizing agents that contain at least 4 or more ether structures (R-O-R) and have 4 or more carbon atoms can be used in formulations as shown in Examples 5B-1 and 5B-2. improved drug concentration.
- the solubilizing aid alone had almost no ability to solubilize poorly water-soluble drugs.
- Example 6B-1 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- cholesterol-PEG600 manufactured by Aldrich: C1145-250MG
- 400 ⁇ L of 10 mg/mL cholesterol-PEG600 was added to another vial, and while stirring with a stirrer tip in the vial, 0.60 mL of the above 36.0 mg/mL hyaluronic acid derivative aqueous solution was added.
- Example 1B-1 the precipitated temsirolimus was removed by filtration with a 0.45 ⁇ m sterile filter, and the drug concentration in the preparation was determined by HPLC measurement in the same manner as in Example 1B-1. It was confirmed that all the filtrates were clear.
- Table 26 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- solubilization aids containing at least 4 or more ether structures (ROR) and having 4 or more carbon atoms are as shown in Example 6B-1. Improved drug concentration in formulations.
- the active ingredient could be solubilized at a higher concentration than the sum of the solubilized amounts of the hyaluronic acid derivative alone and the solubilization aid alone, demonstrating a synergistic effect.
- Example 7B-1 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- cholesterol-PEG600 manufactured by Aldrich: C1145-250MG
- 400 ⁇ L of 10 mg/mL cholesterol-PEG600 was added to another vial, and while stirring with a stirrer tip in the vial, 0.60 mL of the above 36.0 mg/mL hyaluronic acid derivative aqueous solution was added.
- Example 7B-2 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- cholesterol-PEG600 manufactured by Aldrich: C1145-250MG
- 10.0 mg of powdered CyA was weighed out, and then 0.60 mL of the above 36.0 mg/mL hyaluronic acid derivative aqueous solution was added, and while stirring with a stirrer tip in the vial, 10 mg/mL of cholesterol was added.
- Example 7B-3 The lyophilized hyaluronic acid derivative (10k HA-C6-Chol-44%) obtained in Synthesis Example 1B was dissolved in water for injection at 36.0 mg/mL.
- HP- ⁇ -CD manufactured by Tokyo Kasei Kogyo Co., Ltd.: H0979
- 10.0 mg of powdered CyA was weighed out, and then 0.60 mL of the above 36.0 mg/mL hyaluronic acid derivative aqueous solution was added, and while stirring with a stirrer tip in the vial, 10 mg/mL of HP was added.
- Example 1B-1 the product was filtered through a 0.45 ⁇ m sterile filter to remove precipitated CyA, and the drug concentration in the preparation was determined by HPLC measurement in the same manner as in Example 1B-1. It was confirmed that all the filtrates were clear.
- Table 26 shows the theoretical drug concentration in the preparation calculated from the added amount and the actual drug concentration in the preparation determined by HPLC measurement.
- the solubilization aid containing at least 4 or more ether structures (ROR) and having 4 or more carbon atoms was found to be Example 7B.
- the drug concentration in the formulation was increased.
- the active ingredient could be solubilized at a higher concentration than the sum of the solubilized amounts of the hyaluronic acid derivative alone and the solubilization aid alone, demonstrating a synergistic effect.
- hyaluronic acid derivative pharmaceutical composition of the present embodiment a large amount of drug can be solubilized from powder, the amount of solubilized can be maximized, and the active ingredient can be absorbed subcutaneously from within the gelled hyaluronic acid derivative composition.
- a pharmaceutical composition using a hyaluronic acid derivative that is excellent in controlling the release rate and sustaining the release of active ingredients over a long period of time can be obtained.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Inorganic Chemistry (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Dermatology (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Materials Engineering (AREA)
- Transplantation (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Abstract
Description
本願は、2022年7月20日に、日本に出願された特願2022-115839号、2022年9月22日に、日本に出願された特願2022-151823号及び2022年7月20日に、日本に出願された特願2022-115841号に基づき優先権を主張し、その内容をここに援用する。
特許文献1に開示されたヒアルロン酸誘導体は、水溶液中で自発的に会合し、薬物、特にバイオ医薬品を、その生物活性を維持したまま効率よく封入することができる。これにより、生理食塩濃度下で凝集し(或いは、生理食塩濃度下でも分散し)、なおかつ血中滞留性が良好である。このヒアルロン酸誘導体は、特にバイオ医薬品を有効成分として使用する場合に、薬理活性を維持したまま多くの薬物を効率よく封入できる担体、及び血中滞留性に優れた血中徐放キャリア並びにターゲティングキャリアとして用いることができ、薬物を持続的に徐放できる局所(例えば、皮下等)徐放キャリアにもなり得るとされている。
本発明は、上記事情に鑑みてなされたものであって、生体内で有効成分の濃度をさらに長期間にわたり維持し徐々に有効成分を放出でき、有効成分を高濃度で可溶化できるヒアルロン酸誘導体医薬組成物及びその製造方法を提供することを目的とする。
さらに、本発明は、上記事情に鑑みてなされたものであって、有機溶媒を使用せず、毒性の高い界面活性剤の使用量を低減しつつ、難水溶性有効成分を高濃度で可溶化できるヒアルロン酸誘導体医薬組成物及びその製造方法を提供することを目的とする。
[1](A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物。
[2]前記(B)会合促進剤はエーテル構造(R-O-R)を少なくとも4つ以上含み、かつ炭素数4以上を満たす、[1]に記載のヒアルロン酸誘導体医薬組成物。
[3]前記(B)会合促進剤が、ポリソルベート80、ポリソルベート20、ポロキサマー、オキシエチレンヒマシ油、ポリエチレングリコール300、ポリエチレングリコール400、ポリエチレングリコール4000、脂肪酸ソルビタンエステル、トコフェリルポリエチレングリコールスクシネート並びにポリビニルアルコールから成る群から選択される1種以上である、[1]又は[2]に記載のヒアルロン酸誘導体医薬組成物。
[4]生理塩濃度下で沈殿物が生じる、[1]~[3]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[5]前記(C)有効成分は、タンパク質または難水溶性薬物から選択される少なくとも1種である、[1]~[4]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。[6]前記難水溶性薬物は水への溶解度が1mg/mL以下である[5]に記載のヒアルロン酸誘導体医薬組成物。
[7]前記難水溶性薬物は分子量200以上である[5]又は[6]に記載のヒアルロン酸誘導体医薬組成物。
[8]前記難水溶性薬物は難水溶性ペプチドである、[5]~[7]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[9]前記難水溶性ペプチドは、アミド結合を構成する窒素原子の少なくとも1つがメチル基を有する、[8]に記載のヒアルロン酸誘導体医薬組成物。
[10]前記難水溶性ペプチドは環状ペプチド、長鎖ペプチドから選択される少なくとも一つ以上を含む、[8]又は[9]に記載のヒアルロン酸誘導体医薬組成物。
[11]前記難水溶性ペプチドは環状ペプチドである、[8]~[10]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[12]100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する前記(C)有効成分の含有量は、10質量部以上100質量部以下である、[1]~[11]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[13]前記(A)疎水性基を導入したヒアルロン酸誘導体は、下記一般式(I)で表される繰り返し単位を1以上有する、[1]~[12]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
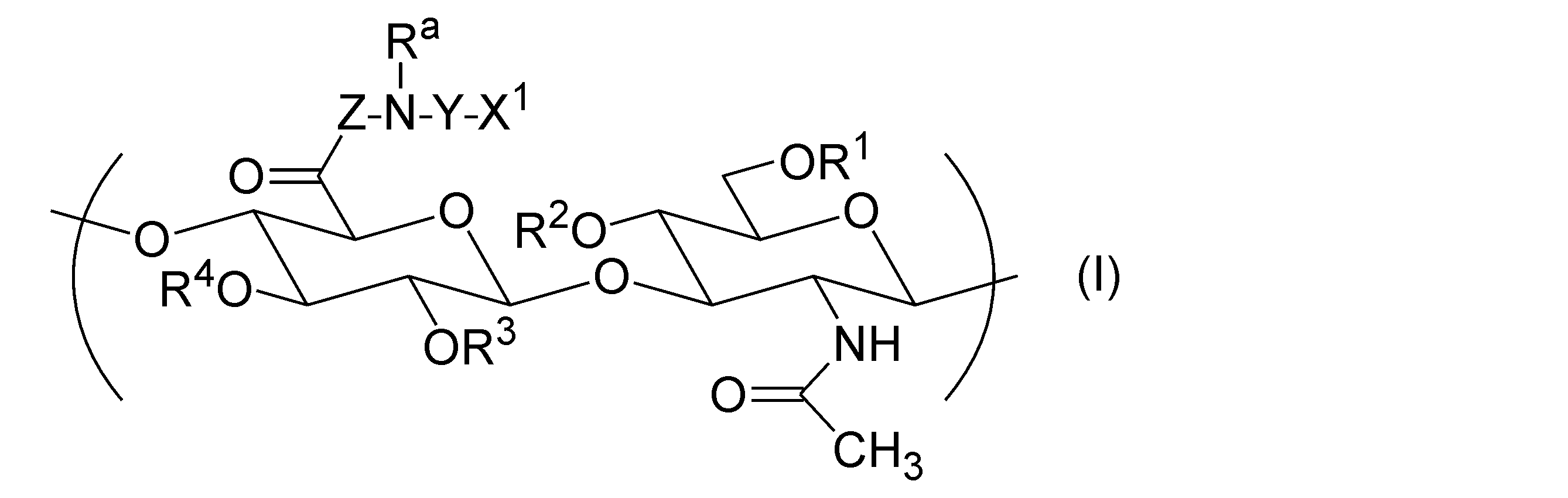
X1は、-NRb-R、-NRb-COO-R、-NRb-CO-R、-NRb-CO-NRc-R、-COO-R、-O-COO-R、-S-R、-CO-Ya-S-R、-O-CO-Yb-S-R、-NRb-CO-Yb-S-R、及び-S-S-R、で表される基からなる群より選択される基である。
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Ra、Rb及びRcのアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよい。
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択される基である。Rfのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Rは、ステリル基である。
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-である。ここで、Yのアルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよい。
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Rgのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Yaは、C1-5アルキレンである。
Ybは、C2-8アルキレン又はC2-8アルケニレンである。
mは、1以上100以下の整数である。)
[14]前記ステリル基はコレステリル基である、[13]に記載のヒアルロン酸誘導体医薬組成物。
[15]前記ヒアルロン酸誘導体に対する前記ステリル基の導入率が7%以上35%未満である、[13]~[14]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。[16]前記ヒアルロン酸誘導体医薬組成物は目視で析出物が観察されない、[1]~[15]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[17]ろ過滅菌可能である、[1]~[16]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[18](A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法であって、前記(A)疎水性基を導入したヒアルロン酸誘導体と、前記(B)会合促進剤とを混合し、会合促進剤含有ヒアルロン酸誘導体水溶液を得る工程と、前記(C)有効成分と前記会合促進剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法。
[19](A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法であって、前記(C)有効成分を前記(B)会合促進剤に分散させ、分散液(I)を得る工程と、ヒアルロン酸誘導体水溶液又は会合促進剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、前記分散液(I)と水溶液(II)とを混合する工程と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法。
[20]有機溶剤を除去する工程を含まない、[18]又は[19]に記載のヒアルロン酸誘導体医薬組成物の製造方法。
[21](A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含み、前記(B1)可溶化助剤は、エーテル構造(R-O-R)を少なくとも4つ以上含み、かつ炭素数4以上を満たし、100質量部の前記(A1)疎水性基を導入したヒアルロン酸誘導体に対する、前記(B1)可溶化助剤の含有量が0.0001質量部以上15000質量部以下である、ヒアルロン酸誘導体医薬組成物。
[22]前記(B1)可溶化助剤が、非イオン界面活性剤、分子量190g/moL以上4000g/moL以下のポリエチレングリコール、シクロデキストリン誘導体、から成る群から選択される1種以上である、[21]に記載のヒアルロン酸誘導体医薬組成物。
[23]前記(B1)可溶化助剤が、ポリソルベート80、ポリソルベート65、ポリソルベート60、ポリソルベート40、ポリソルベート20、ポロキサマー、ポリオキシエチレン硬化ヒマシ油、シクロデキストリン誘導体、ポリエチレングリコール300、ポリエチレングリコール400、ポリエチレングリコール4000、並びにトコフェリルポリエチレングリコールスクシネートから成る群から選択される1種以上である、[21]又は[22]に記載のヒアルロン酸誘導体医薬組成物。
[24]前記(B1)可溶化助剤が非イオン界面活性剤であり、100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記非イオン界面活性剤の含有量が0.0001質量部以上150質量部以下である、[21]~[23]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[25]前記(B1)可溶化助剤が分子量190g/moL以上4000g/moL以下のポリエチレングリコールであり、100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記ポリエチレングリコールの含有量が25質量部以上15000質量部以下である、[21]~[24]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[26]前記(C1)有効成分は、水への溶解度が1mg/mL以下の難水溶性薬物である、[21]~[25]のいずれか1つ記載のヒアルロン酸誘導体医薬組成物。
[27]前記難水溶性薬物は分子量が500以上である、[26]に記載のヒアルロン酸誘導体医薬組成物。
[28]前記難水溶性薬物は難水溶性ペプチドである、[26]又は[27]に記載のヒアルロン酸誘導体医薬組成物。
[29]前記難水溶性ペプチドは、アミド結合を構成する窒素原子の少なくとも1つがメチル基を有する、[28]に記載のヒアルロン酸誘導体医薬組成物。
[30]前記難水溶性ペプチドは、環状ペプチド及び長鎖ペプチドから選択される少なくとも一つ以上を含む、[28]又は[29]に記載のヒアルロン酸誘導体医薬組成物。
[31]前記難水溶性ペプチドは環状ペプチドである、[28]~[30]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[32]100質量部の前記(A1)疎水性基を導入したヒアルロン酸誘導体に対する、前記難水溶性薬物の配合量は、21質量部以上100質量部未満である、[26]~[31]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[33]前記ヒアルロン酸誘導体は、下記一般式(I)で表される繰り返し単位を1以上有する、[31]~[32]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
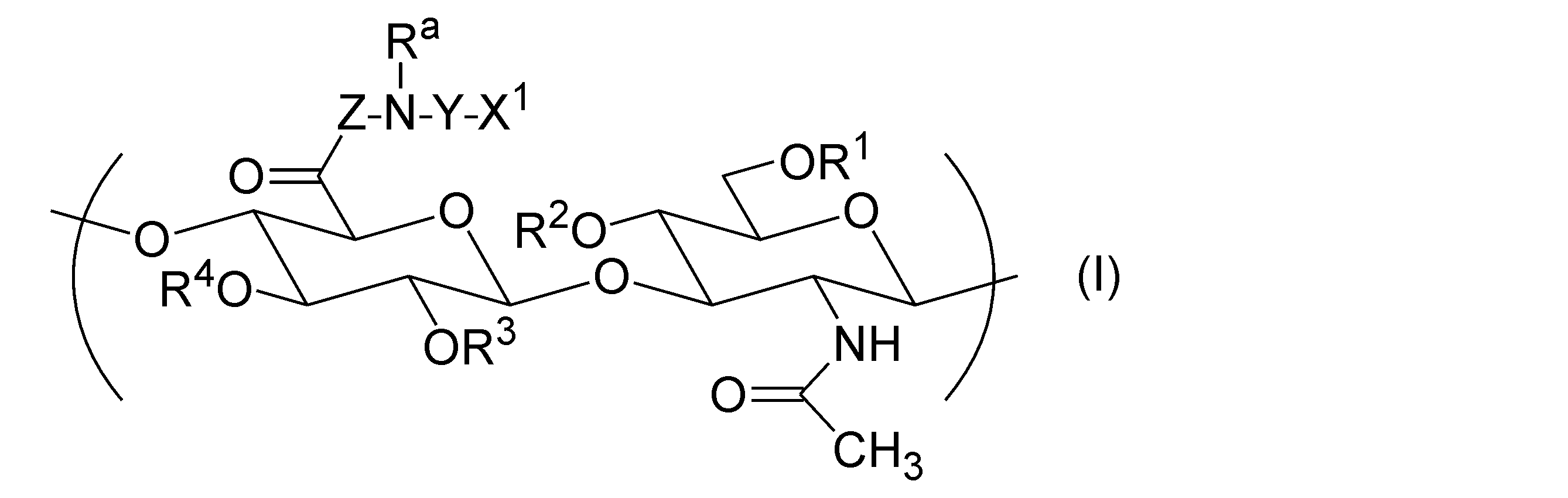
X1は、-NRb-R、-NRb-COO-R、-NRb-CO-R、-NRb-CO-NRc-R、-COO-R、-O-COO-R、-S-R、-CO-Ya-S-R、-O-CO-Yb-S-R、-NRb-CO-Yb-S-R、及び-S-S-R、で表される基からなる群より選択される基である。
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Ra、Rb及びRcのアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよい。
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択される基である。Rfのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Rは、ステリル基である。
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-である。ここで、Yのアルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよい。
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Rgのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Yaは、C1-5アルキレンである。
Ybは、C2-8アルキレン又はC2-8アルケニレンである。
mは、1以上100以下の整数である。)
[34]前記ステリル基がコレステリル基である、[33]に記載のヒアルロン酸誘導体医薬組成物。
[35]前記ヒアルロン酸誘導体に対する前記ステリル基の導入率が35%以上50%未満である、[33]~[34]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。[36]前記ヒアルロン酸誘導体医薬組成物に対する前記ヒアルロン酸誘導体の含有量が6mg/mL以上45mg/mL未満である、[21]~[35]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[37]前記ヒアルロン酸誘導体医薬組成物に対する有機溶剤の含有量が0.8%未満である、[21]~[36]のいずれか1つに記載のヒアルロン酸誘導体医薬組成物。
[38](A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含有する、医薬組成物の製造方法であって、(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤とを混合し、可溶化助剤含有ヒアルロン酸誘導体水溶液を得る工程と、前記(C1)有効成分を前記可溶化助剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む、医薬組成物の製造方法。
[39](A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含有する、医薬組成物の製造方法であって、前記(C1)有効成分を前記(B1)可溶化助剤に分散させ、分散液(I)を得る工程と、ヒアルロン酸誘導体水溶液又は可溶化剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、前記分散液(I)と前記水溶液(II)とを混合する工程と、を含む、医薬組成物の製造方法。
[40](A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含有する、医薬組成物の製造方法であって、有機溶剤を除去する工程を含まないことを特徴とする、[38]又は[39]に記載の医薬組成物の製造方法。
本実施形態は、(A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物である。以降において、「(A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物」を「医薬組成物1」と略記する場合がある。
本実施形態の医薬組成物1において、ヒアルロン酸誘導体は、有効成分と複合体(以下、「有効成分-ヒアルロン酸誘導体複合体」と称する場合がある)を形成している。具体的には、ヒアルロン酸誘導体中のステリル基と、有効成分の疎水性部位とが疎水性相互作用により複合体を形成しており、有効成分とステリル基等の疎水性部位が中心部に存在し、一方、ヒアルロン酸誘導体中のヒアルロン酸に由来する部位等の親水性部位が外縁部に存在する、シリンダー構造、又はコア-シェル型様のシリンダー構造を呈しているものと推定される。すなわち、有効成分がヒアルロン酸誘導体に封入又は内包された構造を呈しているものと推定される。
本明細書における(B)会合促進剤とは、疎水性基を導入したヒアルロン酸誘導体と相互作用して、ヒアルロン酸誘導体分子同士の会合を促進するものである。本実施形態の会合促進剤はヒアルロン酸誘導体とコンプレックスを形成し、薬物複合化能を高め、生理塩濃度下での沈殿性能を向上させる。これについて、皮下に沈殿した後に、ゲル内の薬物は生体内のアルブミンや他の疎水タンパク等の疎水的な成分と置き換わる交換反応あるいはゲルの分解により徐放されるが、交換反応による薬物徐放を制御し、ゲルの分解酵素の流入を抑制、遮蔽、ステルス性を付与することで徐放期間をコントロールできるものと考えられる。
本実施形態の医薬組成物1は、(A)疎水性基を導入したヒアルロン酸誘導体を含む。以降において、「(A)疎水性基を導入したヒアルロン酸誘導体」を、「(A)ヒアルロン酸誘導体」と記載する場合がある。
ここでいう「リンカー」とは、遺伝子工学により導入し得る任意のペプチドリンカー、又は合成化合物リンカーを用いることができるが、本実施形態に用いる(A)ヒアルロン酸誘導体においては、ペプチドリンカーが好ましい。
本明細書において使用される「ステリル基」という用語は、ステロイド骨格を有する基であれば特に制限されない。ここでステロイドとしては、具体的には、コレステロール、コレスタノール、カンペスタノール、エルゴスタノール、スチグマスタノール、コプロスタノール、スチグマステロール、シトステロール、ラノステロール、エルゴステロール、シミアレノール、胆汁酸、テストステロン、エストラジオール、プロゲストロン、コルチゾール、コルチゾン、アルドステロン、コルチコステロン、デオキシコルチステロン等が挙げられる。ステリル基としては、コレステリル基、スチグマステリル基、ラノステリル基、エルゴステリル基等が挙げられ、中でも、コレステリル基(特に、コレスタ-5-エン-3β-イル基)が好ましい。
(A)ヒアルロン酸誘導体に対するステリル基の導入率(以下、単に「ステリル基導入率」と称する場合がある)は0.1%以上50%未満が好ましく、5%以上45%未満がより好ましく、10%以上40%以下がさらに好ましく、15%以上35%以下が特に好ましい。
=[(ステリル基に由来するピーク積分値×3/nH)/(N-アセチル-D-グルコサミンのアセチル基に由来するピーク積分値)]×100
より詳細には、ヒアルロン酸誘導体の重量平均分子量が粘度の観点で、100k以下が好ましく、50k以下がさらに好ましく、40k以下が特に好ましい。
ヒアルロン酸誘導体の製造という観点では、4kから100kが好ましく、6kから50kが特に好ましい。
ヒアルロン酸誘導体がヒアルロン酸としての性質を強く発揮するという観点、例えばCD44受容体に強く認識されるためには100kDa以上が好ましく、200kDa以上が特に好ましく、300kDa以上が最も好ましい。
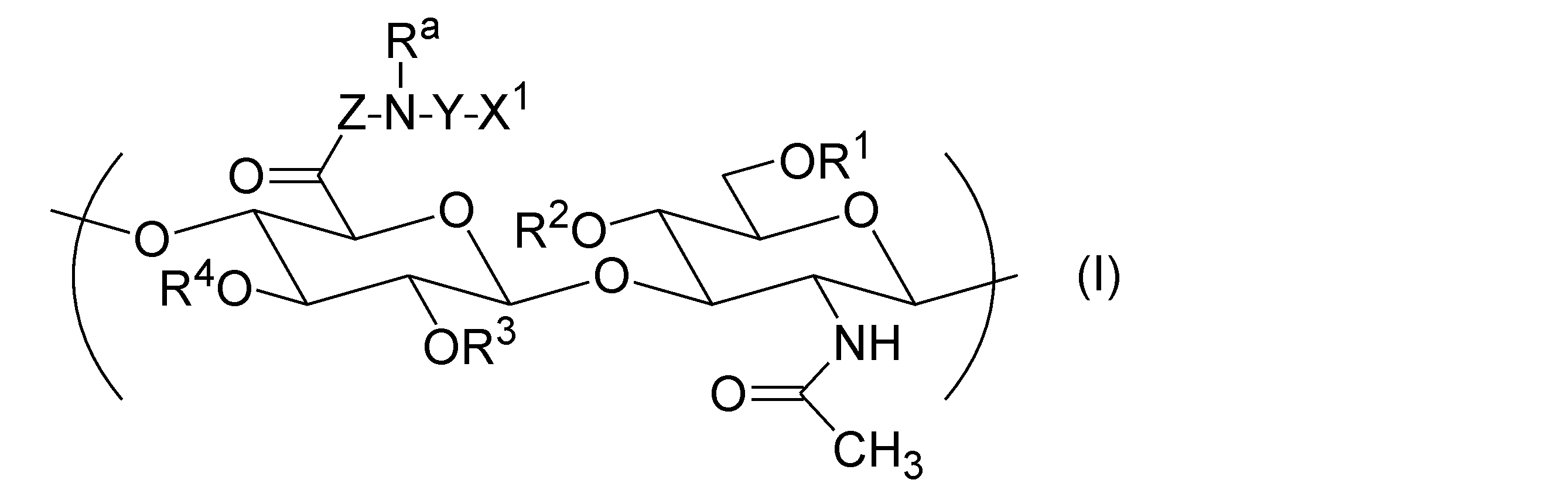
Zは、直接結合、又は2個以上30個以下の任意のアミノ酸残基からなるペプチドリンカーを表す。
X1は、-NRb-R、-NRb-COO-R、-NRb-CO-R、-NRb-CO-NRc-R、-COO-R、-O-COO-R、-S-R、-CO-Ya-S-R、-O-CO-Yb-S-R、-NRb-CO-Yb-S-R、及び-S-S-R、で表される基からなる群より選択される基である。
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。ここでRa、Rb及びRcのアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよい。
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択される基である。Rfのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Rは、ステリル基である。
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-である。ここで、Yのアルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよい。
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Rgのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Yaは、C1-5アルキレンである。
Ybは、C2-8アルキレン又はC2-8アルケニレンである。
mは、1以上100以下の整数である。)

一般式(I)中の基「-Z-N(Ra)Y-X1」は、以下の式:-NH-(CH2)mz-NH-R;-NH-(CH2)mz-NH-COO-R;-NH-(CH2CH2O)m-CH2CH2-NH-COO-R;-NH-(CH2)mz-COO-R;-NH-(CH2CH2O)m-CH2CH2-COO-R、-NH-(CH2)mz-O-COO-R;-NH-(CH2CH2O)m-CH2CH2-O-COO-R、-NH-(CH2)mz-S-R;-NH-(CH2CH2O)m-CH2CH2-S-R;-NH-(CH2)mz-O-CO-CH(R8)-CH2-S-R;-NH-(CH2)mz-NHCO-CH(R8)-CH2-S-R;-NH-(CH2CH2O)m-CH2CH2-NHCO-CH(R8)-CH2-S-R;-NH-(CH2CH2O)m-CH2CH2-O-CO-CH(R8)-CH2-S-R;-NH-(CH2)mz-S-S-R;及び
-Z-NRa-Y-NRb-COO-R(ここで、mzは、2以上30以下の整数であり、R8は、水素原子又はメチル基であり、R及びmは、本明細書で既に定義したとおりである。)
で表される基からなる群より選択される基を含む。
一般式(I)において、Zは直接結合であることが好ましい。また、別の態様において、Zがペプチドリンカーである場合に、X1は-NRb-COO-Rであることが好ましい。さらに、別の態様において、Zは、-NH-[CH(-Za)-CONH]n-1-CH(-Za)-CO-で表されるペプチドリンカーであってもよく、ここで、nは2以上30以下の整数であり、Zaは、それぞれ独立に、H2N-CH(-Za)-COOHとして表されるα-アミノ酸中の置換基を表す。当該ペプチドリンカーは、N末端にてグルクロン酸部分のカルボキシ基に結合し、C末端にて基-N(-Ra)-Y-X1に結合する。当該ペプチドリンカーのアミノ酸残基として利用できるアミノ酸の例としてはα-アミノ酸、例えばアラニン、アルギニン、アスパラギン(Asn)、アスパラギン酸、システイン、グルタミン、グルタミン酸、グリシン(Gly)、ヒスチジン、イソロイシン、ロイシン(Leu)、リジン、メチオニン、フェニルアラニン(Phe)、プロリン、セリン、スレオニン、トリプトファン、チロシン、バリンといった天然型(L型)のアミノ酸、それらのD体等が挙げられ、合成されたアミノ酸を含む全てのα-アミノ酸を用いることができる。すなわち、Zaとしては、例えば、-CH3、H2NC(NH)NH(CH2)3-、H2NCOCH2-等が挙げられる。また、n個のZは、同一でも異なっていてもよい。nは、2以上30以下の整数であるが、2以上10以下が好ましく、2以上4以下がより好ましい。ペプチドリンカーの好ましい例としては、例えば、-Gly-Phe-Leu-Gly-、-Asn-Phe-Phe-、-Phe-Phe-、Phe-Gly-等が挙げられる。
一般式(I)において、Yは-(CH2)n1-及び-(CH2CH2O)m1-CH2CH2-(ここで、n1は、2以上20以下の整数であり、2以上15以下の整数が好ましく、2以上12以下の整数がより好ましく、2以上6以下の整数がさらに好ましい。m1は、1以上4以下の整数である)からなる群より選択される基が好ましい。具体的には、-(CH2)2-、-(CH2)6-、-(CH2)8-、-(CH2)12-、又は、-(CH2CH2O)2-CH2CH2-が好ましい。また、純水中乃至低塩濃度下では高い溶解性を実現させつつ、生理食塩濃度下では高い沈殿形成能を示させるという観点からは、Yは-(CH2)2-、-(CH2)6-、-(CH2)8-及び-(CH2)12-からなる群より選択される基が好ましく、-(CH2)6-がより好ましい。
Yaとしては、-CH2-又は-CH2-CH2-が好ましい。
Ybとしては、-CH2-CH2-、-CH(CH3)CH2-、2-ブテン-1,4-ジイル、ヘプタ-2,4-ジエン-1,6-ジイル又はオクタ-2,4,6-トリエン-1,8-ジイルが好ましく、-CH2-CH2-又は-CH(CH3)CH2-がより好ましい。
一般式(Ia)において、Xは、-NH-(CH2)2-NH-COO-コレステリル、-NH-(CH2)6-NH-COO-コレステリル、-NH-(CH2)12-NH-COO-コレステリル又は-NH-(CH2CH2O)2-CH2CH2-NH-COO-コレステリルが好ましく、-NH-(CH2)2-NH-COO-コレステリル、-NH-(CH2)6-NH-COO-コレステリル又は-NH-(CH2CH2O)2-CH2CH2-NH-COO-コレステリルがより好ましい。
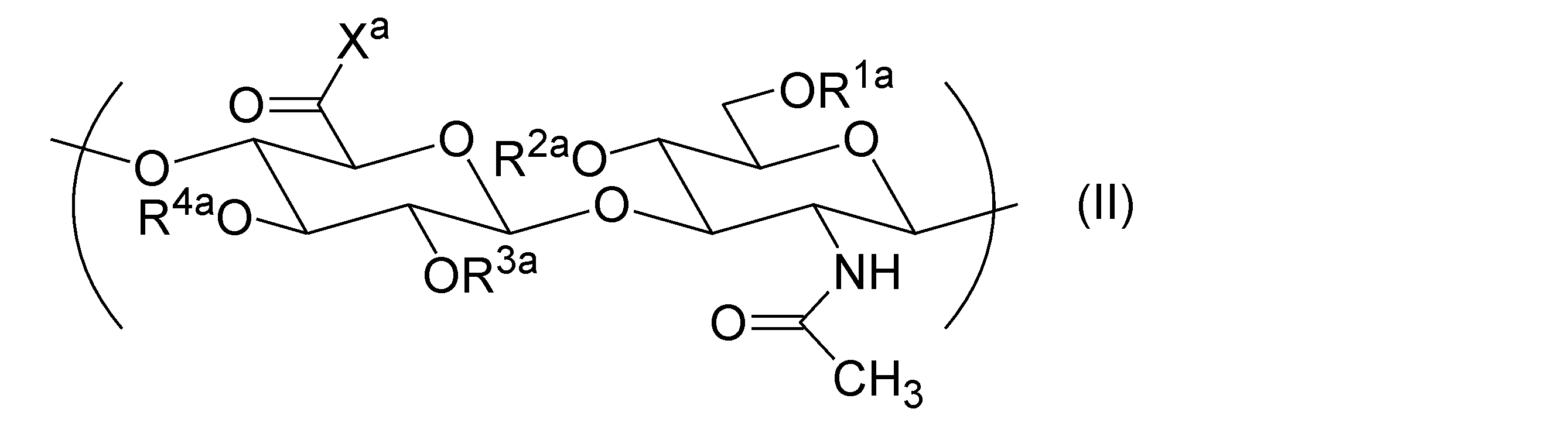
別の態様において、(A)ヒアルロン酸誘導体は、繰り返し単位(I)、繰り返し単位(Ia)及び繰り返し単位(II)から実質的になるヒアルロン酸誘導体であってもよい。
一般式(II)において、Q+はカルボキシ基と水中で塩を形成するカウンターカチオンであれば特に限定されず、2価以上の場合は価数に応じて複数のカルボキシ基と塩を形成する。カウンターカチオンの例としては、リチウムイオン、ナトリウムイオン、ルビジウムイオン、セシウムイオン、マグネシウムイオン、カルシウムイオン等の金属イオン;式:N+RjRkRlRm(式中、Rj、Rk、Rl及びRmは、それぞれ独立に、水素原子及びC1-6アルキルからなる群より選択される)で表されるアンモニウムイオン等が挙げられる。中でも、Q+は、ナトリウムイオン、カリウムイオン、又はテトラアルキルアンモニウムイオン(例えば、テトラn-ブチルアンモニウムイオン等)が好ましい。Rj、Rk、Rl及びRmは、C1-6アルキルからなる群より選択される同一の基であることが好ましく、n-ブチル基が好ましい。
本発明において、会合促進剤とは、より具体的に、ゲル浸透クロマトグラフィーにより測定することができ、本実施形態において定義する会合促進剤であることは、以下に示す面積A1とA2の比A2/A1で確認することができる。
(B)会合促進剤は、エーテル構造(R-O-R)を少なくとも4つ以上含み、かつ炭素数4以上を満たす成分が好ましい。
(B)会合促進剤としては、例えば、ポリソルベート80(エーテル構造を20以上備え、炭素数は64以上である)、ポリソルベート65(エーテル構造を20以上備え、炭素数は100以上である)、ポリソルベート60(エーテル構造を20以上備え、炭素数は64以上である)、ポリソルベート40(エーテル構造を20以上備え、炭素数は62以上である)、ポリソルベート20(エーテル構造を20以上備え、炭素数は57以上である)、ポリエチレングリコールモノラウレート(エーテル構造を7以上備え、炭素数は28以上である)、ステアリン酸ポリオキシル40(エーテル構造を39以上備え、炭素数は98以上である)、ステアリン酸ポリオキシル45(エーテル構造を44以上備え、炭素数は108以上である)、ステアリン酸ポリオキシル55(エーテル構造を54以上備え、炭素数は128以上である)、クレモフォールEL(エーテル構造を35以上備え、炭素数は127以上である)、などが挙げられる。この中でも特に、ポリソルベート20、ポリソルベート80とポリエキシエチレンヒマシ油が好ましい。
有効成分としては、特に限定されないが、医薬活性ペプチド又はタンパク質、核酸、低分子化合物、中分子化合物、抗原(がん抗原、感染症由来抗原、免疫疾患における自己抗原等)等が挙げられる。中でも、低分子化合物、中分子化合物、ペプチドがより好ましい。
医薬活性ペプチド又はタンパク質としては、対象に治療有効量を投与した場合に、対象の状態又は病状に対して正の、又は有利な効果を有するものを意味する。好ましい医薬活性ペプチド又はタンパク質としては、根治的又は対症的性質を有し、疾患又は障害の1つ又は複数の症状を改善する、軽快させる、軽減する、元に戻す、症状の発症を遅らせる、又は症状の重症度を減ずるために投与することができるものである。医薬活性ペプチド又はタンパク質は予防的性質を有することもあり、疾患の発症を遅らせるか、又はこのような疾患又は病態の重症度を減ずるために使用することができる。「医薬活性ペプチド又はタンパク質」という用語は、全長タンパク質又はポリペプチドを含意し、又その医薬的に活性な断片を指すこともある。この用語は、ペプチド又はタンパク質の医薬的に活性なアナログも包含する。
核酸としては、DNA、RNAが含まれ、例えば、短鎖干渉RNA(siRNA)、二本鎖RNA(dsRNA)、マイクロRNA(miRNA)、短鎖ヘアピンRNA(shRNA)及び核酸アプタマー等を含む。
低分子化合物としては、分子量が約500未満の化合物であって、例えば、制癌剤(例えば、アルキル化剤、代謝拮抗剤、アルカロイド等)、免疫抑制剤、抗炎症剤(ステロイド剤、非ステロイド剤系抗炎症剤等)、抗リウマチ剤、抗菌剤(β-ラクタム系抗生物質、アミノグリコシド系抗生物質、マクロライド系抗生物質、テトラサイクリン系抗生物質、新キノロン系抗生物質、サルファ剤等)等が挙げられる。
本明細書において、「中分子」とは、低分子(分子量約500までの有機化合物)でも高分子(分子量1万以上のタンパク質など)でもない、分子量約500~5000程度のペプチド、マクロライド化合物、核酸及び天然物又はそれらの誘導体をいう。好ましくは、分子量500~2000程度のペプチド、すなわち、アミノ酸残基5~20程度の直鎖状又は環状ペプチドである。ペプチドとしては、環状ペプチドが好ましく、その詳細については後述する。マクロライド化合物とは、大環状ラクトンであり、環の員数が12又はそれ以上の化合物の総称をいう。
中分子化合物としては、例えば、FK506やラパマイシンなどが挙げられる。
(がん抗原)
がん抗原とは、がん細胞に多く発現する抗原であり、いくつかの場合には、がん細胞によってのみ発現する。がん抗原は、がん細胞内、又はがん細胞の表面上に発現し得る。
感染症由来抗原としては、感染性病原体及び感染性病原体由来の抗原であれば特に限定されない。感染性病原体としては、ウイルス、細菌、真菌、線虫等が挙げられる。感染症病原体由来抗原は抗原タンパク質であっても、抗原ペプチドであってもよい。
上記感染症由来抗原としては、A型及びB型インフルエンザウイルス由来抗原が好適に用いられる。なお、上述したA型及びB型インフルエンザウイルスの亜型としては特に限定されず、これまで単離された亜型であっても将来単離される亜型であってもよい。
免疫疾患における抗原としては、免疫疾患の標的タンパク質のエピトープを含むものであれば特に限定されない。免疫疾患としては特に限定されず、例えば、尋常性乾癬、強直性脊椎炎、関節リウマチ、乾癬性関節炎、体軸性脊椎関節炎、クローン病、潰瘍性大腸炎、気管支喘息、慢性蕁麻疹、花粉症、アトピー性皮膚炎等が挙げられる。標的タンパク質としては特に限定されず、IL-17A、DPP4、S100A9、PCSK9、IL-23、IgE、TNFα、IL-12/23p40、IL-6、α4β7インテグリン、IL-4/13、IL-5、BLyS、IL-13等が挙げられる。参考文献1(国際公開第2017/164409号)にはIL-17A由来のペプチドが記載されている。
難水溶性薬物とは、第17改正日本薬局方における溶解性を示す用語において、極めて溶けやすい、溶けやすい、やや溶けやすい、やや溶けにくい、溶けにくい、極めて溶けにくい、ほとんど溶けないとされているもののうち、やや溶けやすい、やや溶けにくい、溶けにくい、極めて溶けにくい、またはほとんど溶けないに分類される薬物を意味する。
本発明の医薬組成物1は、このような難水溶性薬物であっても、有機溶媒を使用せず、毒性の高い界面活性剤の使用量を低減しつつ、高濃度で可溶化できる。
分子内に有する環の数は特に限定されるものではないが、1つ以上8つ以下であることが好ましい。
本実施形態の医薬組成物1は、単独で投与することもでき、或いは、薬理学上許容されうる担体とともに常套手段に従って投与することができる。薬理学上許容されうる担体と併用する場合は、例えば、上記ヒアルロン酸誘導体及び上記有効成分、並びに、必要に応じてアジュバントと、水若しくはそれ以外の生理学的に許容し得る液(例えば、生理食塩水、リン酸緩衝生理食塩水(PBS))等とを、混合してもよく、生理学的に許容し得る緩衝液、賦形剤、ベヒクル、防腐剤、安定化剤、結合剤、凍結乾燥補助剤等を含むこともできる。
また、これらのpH調整剤は、1種単独で使用してもよく、また2種以上を任意に組み合わせて使用してもよい。
必要に応じて1または2種以上の防腐剤をさらに配合することができ、医薬として許容されるものであれば特に制限されるものでない。
本発明における防腐剤としては、例えば、塩化ベンザルコニウム、塩化ベンゼトニウム、グルコン酸クロルヘキシジン、パラオキシ安息香酸エチルなどのパラオキシ安息香酸エステル類、ベンジルアルコール、m‐クレゾール、フェノール、フェネチルアルコール、ソルビン酸またはその塩、チメロサールなどが用いられる。
本発明における医薬組成物1又は後述する医薬品組成物2の粘稠化剤としては、例えば、セルロース系高分子(メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルメチルセルロースなど)、ビニル系高分子(ポリビニルピロリドン、ポリビニルアルコールなど)、糖類(ヒアルロン酸、その塩類などのムコ多糖類、ジェランガム、アルギン酸ナトリウム、デキストラン、シクロデキストリンなどの多糖類)、オキシアルキレン系高分子(ポリオキシエチレンポリオキシプロピレンブロックコポリマー)などが挙げられる。本発明における粘稠化剤の分子量は、例えば、数平均分子量0.5×104~100×104程度の範囲から選択できる。
特許4758893号公報によれば、実質的に等浸透圧にする濃度(2~2.5%(v/v))のグリセロールを配合することにより、抗菌防腐の効力を向上させることが記載されている。そのため等張化剤としての機能だけでなく、抗菌防腐の効果も期待される。グリセロールの含有量は、例えば、0.01~10%(w/v)、好ましくは0.05~5%(w/v)、より好ましくは0.1~3.0%(w/v)、さらに好ましくは0.3~3.0%(w/v)、特に好ましくは0.3~2.5%(w/v)である。
固体の場合、粉末、顆粒、丸剤、ペレット、タブレット、カプセル等の形態が挙げられる。中でも、固体としては、凍結乾燥粉末であることが好ましい。
半固体の場合、ゲル等の形態が挙げられる。
液体の場合、粉末を水又はリン酸緩衝液(PB)あるいはリン酸緩衝生理食塩水(PBS)等の緩衝液により希釈又は懸濁した懸濁液等の形態が挙げられる。
本実施形態の医薬品組成物は、生理塩濃度下で沈殿物が生じるものが好ましい。
具体的には、下記の試験条件において、vitroにおける沈殿率が20%、好ましくは50%以上、より好ましくは70%以上、さらに好ましくは80%以上、特に好ましくは90%以上、最も好ましくは92%以上である医薬品組成物であることが好ましい。
・沈殿サンプルの調製
マイクロチューブ(1.5mL)に濃縮緩衝液(40mM PB、600mM NaCl水溶液)を200μL入れ、ヒアルロン酸誘導体医薬組成物からなる製剤を600μL投入する。その後、voltexで30秒混合し、37℃、20minインキュベートした後に、遠心分離機(2000G、5min)で沈殿物を沈降させる。続いて、上澄み中のヒアルロン酸誘導体をGPC測定に供する。
マイクロチューブ(1.5mL)に注射用水を200μL入れ、ヒアルロン酸誘導体医薬組成物からなる製剤(ヒアルロン酸誘導体の理論濃度Xmg/mLとする。X>1.33)を600μL投入する。その後、voltexで30秒混合し、37℃、20minインキュベートした後に、遠心分離機(2000G、5min)に供する。続いて、上澄み中のヒアルロン酸誘導体が1mg/mLとなるよう注射用水で希釈してGPC測定に供する。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
本明細書においては、下式で表される沈殿率が20%以上を満たすヒアルロン酸誘導体医薬組成物を生理塩濃度下で沈殿が生じると評価する。
沈殿率(%)={1-(沈殿サンプルのヒアルロン酸誘導体の面積値)÷((ブランクサンプルのヒアルロン酸誘導体の面積値)×(0.75X))}×100
析出物が確認される場合、日本薬局方で定められる注射剤の不溶性微粒子試験法、第1法「光遮断粒子計数法」で用いられる、光遮蔽型自動微粒子測定装置(液中パーティクルカウンタ「KL-05」)で検出できる。非晶質薬物が完全に溶解した過飽和溶液からはマイクロメートルオーダーサイズの薬物結晶が析出することがある。析出物がない場合、前記光遮蔽型自動微粒子測定装置では粒子は観測されない。
その他、DLS装置を用いて、1μm以上の粒子が存在するか検証することで判断することもできる。
ろ過滅菌可能であることは、以下のシリンジフィルターを用いた通液評価で測定することができ、
具体的には、孔径が0.45μmあるいは0.22μmの滅菌ろ過フィルターを通液可能か評価する。より具体的には、ろ過膜に13mmΦの孔径が0.45μm、ポリエーテルスルフォン(PES)製、および3mLの滅菌シリンジフィルターを用いたときに、2mLのヒアルロン酸誘導体組成物のろ過膜を通過したヒアルロン酸誘導体が50%以上、好ましくは60%、より好ましくは70%以上、さらに好ましくは80%以上、最も好ましくは90%以上通液可能なものである。
本実施形態は、(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物である。以降において、「(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物」を「医薬組成物2」と略記する場合がある。
本実施形態の医薬組成物2は、100質量部の(A1)疎水性基を導入したヒアルロン酸誘導体に対する、(B1)可溶化助剤の含有量が0.0001質量部以上15000質量部以下である。
上記の(B1)可溶化助剤を含む本実施形態の医薬組成物は、エーテル構造による適度な極性とアルキル骨格による適度な疎水性によって、上記(A1)疎水性基を導入したヒアルロン酸誘導体と相互作用することができ、上記(A1)疎水性基を導入したヒアルロン酸誘導体中の疎水性基同士の相互作用を促進できる。これにより、難水溶性有効成分を高濃度で可溶化できる医薬組成物を提供できる。
上記(B1)可溶化助剤の含有量が上記上限値以下であると、医薬組成物の粘度が高くなりすぎず、かつ生体に影響がない範囲で医薬組成物中に有効成分を十分に可溶化できる。
医薬組成物2が含む(A1)疎水性基を導入したヒアルロン酸誘導体に関する説明は、上記医薬組成物1が含む(A)疎水性基を導入したヒアルロン酸誘導体に関する説明と概ね同様である。
(A1)ヒアルロン酸誘導体に対するステリル基の導入率は0.1%以上50%未満が好ましく、5%以上48%未満がより好ましく、35%以上47%以下がさらに好ましく、37%以上45%以下が特に好ましい。
ヒアルロン酸誘導体の製造という観点では、4kから20kが好ましく、6kから16kが特に好ましく、8kから12kが最も好ましい。
本発明において、「可溶化」とは、後述する(C1)有効成分を、目視で透明になるまで水に溶解可能にすることをいう。
「可溶化助剤」とは、(C1)有効成分と(A1)ヒアルロン酸誘導体と共に水に加えた場合に(C)有効成分の溶解度を上昇させる効果を有する剤を言う。
医薬組成物2が含む(C1)有効成分に関する説明は、上記医薬組成物1が含む(C)有効成分に関する説明と概ね同様である。
さらに、(B1)可溶化助剤を含有することにより、(A1)ヒアルロン酸誘導体のステリル基同士の疎水性相互作用を促進させることができる。これにより、ヒアルロン酸誘導体医薬組成物が有する疎水部領域が拡大し、疎水的な薬物の担持量が増加すると考えられる。また、ヒアルロン酸ポリマーに化学結合で束縛されている疎水部に加え、ヒアルロン酸誘導体医薬組成物の内部で可溶化助剤が移動度の高い疎水部領域として働くことができるため、粉末状の(C1)有効成分を高濃度で可溶化することができると推察される。なお、上記メカニズムと異なるメカニズムで所望の効果が得られる場合であってもよい。
ここで、医薬組成物2が含む有機溶剤は、医薬品残溶剤ガイドラインで規定されるクラス1~3に該当する溶剤である。
本実施形態の医薬組成物2は、単独で投与することもでき、或いは、薬理学上許容されうる担体とともに常套手段に従って投与することができる。薬理学上許容されうる担体と併用する場合は、例えば、上記ヒアルロン酸誘導体及び上記有効成分、並びに、必要に応じてアジュバントと、水若しくはそれ以外の生理学的に許容し得る液(例えば、生理食塩水、リン酸緩衝生理食塩水(PBS))等とを、混合してもよく、生理学的に許容し得る緩衝液、賦形剤、ベヒクル、防腐剤、安定化剤、結合剤、凍結乾燥補助剤等を含むこともできる。添加剤に関する説明は、上記医薬品組成物1における≪その他の添加剤≫に関する説明と同様である。
ヒアルロン酸誘導体は、例えば、グルクロン酸のカルボキシ基をアミドに変換し、ステリル基を導入することで得られる。また、原料のヒアルロン酸又はその誘導体に対して、反応させるステリル基を有する化合物の配合量を調整することで、ステリル基導入率を制御することができる。
-CONRa-Y-NRbH + Hal-R;
-CONRa-Y-NRbH + Hal-COOR;
-CONRa-Y-NRbH + HOCO-R;
-CONRa-Y-NRbH + Hal-CO-R;
-CONRa-Y-NRb-COOH + HNRc-R;
-CONRa-Y-NRb-CO-NRcH + Hal-R;
-CONRa-Y-NRbH + HOCO-NRc-R;
-CONRa-Y-NRbH + Hal-CO-NRc-R;
-CONRa-Y-COOH + HO-R;
-CONRa-Y-OH + Hal-COO-R;
-CONRa-Y-OCOOH + HO-R;
-CONRa-Y-OCOOH + Hal-R;
-CONRa-Y-OCO-Hal + HO-R;
-CONRa-Y-SH + Hal-R;
-CONRa-Y-Hal + HS-R;
-CONRa-Y-CO-Ya-Hal + HS-R;
-CONRa-Y-CO-Ya-SH + Hal-R;
-CONRa-Y-O-CO-CH=CH2 + HS-R;
-CONRa-Y-NRb-CO-CH(CH3)=CH2 + HS-R;
-CONRa-Y-SH + HS-R;
-COZ-OH + HNRa-Y-NRb-COO-R;
-COZ-NRa-Y-NRbH + Hal-COO-R
(式中、Ra、Rb、Rc、Y、Ya、Yb、及びZは本明細書で既に定義したとおりであり、Halは、フッ素原子、塩素原子、臭素原子及びヨウ素からなる群より選択されるハロゲン原子を表す)。
本実施形態の医薬組成物1は、以下の医薬組成物1の製造方法1又は製造方法2により製造できる。
以下、各製造方法について説明する。
製造方法1は、(A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含有する、医薬組成物1の製造方法である。
製造方法1は、(A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤とを混合し、会合促進剤含有ヒアルロン酸誘導体水溶液を得る工程と、(C)有効成分を会合促進剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む。
製造方法2は、(A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含有する、医薬組成物1の製造方法である。
製造方法2は、(C)有効成分を(B)会合促進剤に分散させ、分散液(I)を得る工程と、ヒアルロン酸誘導体水溶液又は会合促進剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、前記分散液(I)と前記水溶液(II)とを混合する工程と、を含む。
製造方法1及び2において、薬物の構造によっては、適切なpHや緩衝材を適宜用いて製造しても構わない。
本実施形態の医薬組成物2は、以下の医薬組成物2の製造方法1又は製造方法2により製造できる。
以下、各製造方法について説明する。
医薬品組成物2の製造方法1は、(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含有する、医薬組成物2の製造方法である。
医薬品組成物2の製造方法1は、(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤とを混合し、可溶化助剤含有ヒアルロン酸誘導体水溶液を得る工程と、(C1)有効成分を可溶化助剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む。
医薬品組成物2の製造方法2は、(A1)疎水性基を導入したヒアルロン酸誘導体と、(B1)可溶化助剤と、(C1)有効成分と、を含有する、医薬組成物2の製造方法である。
医薬品組成物2の製造方法2は、(C1)有効成分を(B1)可溶化助剤に分散させ、分散液(I)を得る工程と、ヒアルロン酸誘導体水溶液又は可溶化剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、前記分散液(I)と前記水溶液(II)とを混合する工程と、を含む。
本実施形態の医薬組成物1又は医薬品組成物2を投与する対象は、ヒトを含む哺乳類に分類される動物(サル、マーモセット、マウス、ラット、ウシ、ウマ、ネコ、イヌ、ブタ、ヒツジ、ヤギ、ウサギ等)が挙げられる。
例えば投与経路は、皮下投与、筋肉内投与、静脈内投与、動脈内投与、髄腔内投与、脳内投与、関節腔内投与、腹腔内投与、膣内投与、嚢内投与、直腸内投与、硝子体内投与、眼周囲投与、皮内投与、腹腔内投与、鼻腔内投与、経気管支投与、経肺投与、経皮投与、舌下投与、経口投与、口腔投与、点眼投与が挙げられる。なかでも皮下投与、筋肉内投与、硝子体内投与、髄腔内投与、関節腔内投与が好ましい。
また、局所投与として散剤、クリーム、軟膏、または点眼薬としてもよい。
注射による投与としては、皮下注射、筋肉内注射、静脈内注射、動脈内注射、髄腔内注射、関節腔内注射、腹腔内注射、硝子体内注射、眼周囲注射、皮内注射腫瘍内注射等が挙げられる。
一実施形態において、本発明は、上記医薬組成物1又は医薬品組成物2の有効量を、患者又は患畜に投与することを含む、がん、感染症及び免疫疾患、慢性疾患、からなる群より選ばれる1種以上の疾患の予防又は治療方法を提供する。
なお、感染症としては、上記「抗原」の「感染症由来抗原」において例示されたものが挙げられる。
また、ここでいう「有効量」とは、予防又は治療に有効な量、すなわち、上記疾患の発症予防又は治療に適する量が包含される。
[合成例1]
ヒアルロン酸誘導体を次の工程1-A、工程2-A、工程3-Aに従って調製した。
(コレステリル 6-アミノヘキシルカーバメート塩酸塩の合成)
コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)を次に示す工程1-1-A、続いて工程1-2-Aに従って合成した。
コレステリルクロロホルメート(3.37g、7.5mmol)の無水ジクロロメタン(20mL)の溶液に、アルゴン雰囲気下、トリエチルアミン(TEA、1.05mL)を加えて撹拌した。氷冷下で、6-(t-ブトキシカルボニル)アミノ-1-アミノヘキサン(1.12mL、5mmol)を滴下して加え、そのまま氷冷下で30分間攪拌後、室温(25℃程度)まで昇温し、当該混合物を一晩撹拌した。反応混合物を、超純水及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下で溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液:酢酸エチル:n-ヘキサン=1:4)で精製し、目的物のフラクションを合わせて溶媒を減圧下留去した。
得られた残渣を酢酸エチル(40mL)に溶解し、4N塩酸/酢酸エチル溶液(40mL)を加えて室温(25℃程度)で一晩撹拌した。生じた沈殿物を遠心分離により回収した。得られた固体を酢酸エチルにて4回洗浄後、減圧下で乾燥し、コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)1.2gを得た。
(ヒアルロン酸のテトラブチルアンモニウム(TBA)塩の調製)
ヒアルロン酸のTBA塩(HA-TBA)を次に示す工程2-1-A、続いて工程2-2-Aに従って調製した。
DOWEX(登録商標)50WX-8-400(アルドリッチ社製)を超純水に懸濁させ、デカンテーションにより樹脂を超純水で3回程度洗浄した。40質量%テトラブチルアンモニウムヒドロキシド水溶液(TBA-OH)(アルドリッチ社製)を樹脂のカチオン交換能に対し約1.5倍モル等量加え、30分間撹拌した。余剰のTBA-OH溶液をデカンテーションにより除去した後、さらに過剰の超純水で洗浄することで、TBA塩化したカチオン交換樹脂を得た。
分子量35,000(35kDa)の原料ヒアルロン酸ナトリウム塩(HA-Na)を15mg/mLの濃度で超純水に溶解した。[工程2-1-A]でTBA塩化したカチオン交換樹脂の懸濁液をHAユニット(ユニット分子量401.3)のモル数に対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、ヒアルロン酸のTBA塩(HA-TBA)を白色固体として得た。
上記[工程2-2-A]で調製したHA-TBAの無水DMSO溶液(10mg/mL)を調製した。その後、上記[工程1-A]で合成したHA-TBA中に存在する二糖繰り返し単位(HAユニット)に対するChol塩酸塩の添加量がモル比で19/100となる割合で添加した。次に、HAユニットに対する4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)の添加量がモル比で24/100となる割合で加え、室温(25℃程度)で一晩撹拌した。反応溶液は、0.3M 酢酸アンモニア/DMSO溶液、0.15M NaCl水溶液、超純水の順で透析(スペクトラポア7、分画分子量(MWCO):3,500)した。得られた透析液を凍結乾燥して目的物(HA-C6-Chol)を白色固体として得た。
ヒアルロン酸誘導体を次の工程1-B、工程2-B、工程3-Bに従って調製した。
(コレステリル 6-アミノヘキシルカーバメート塩酸塩の合成)
コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)を次に示す工程1-1-B、続いて工程1-2-Bに従って合成した。
コレステリルクロロホルメート(3.37g、7.5mmol)の無水ジクロロメタン(20mL)の溶液に、アルゴン雰囲気下、トリエチルアミン(TEA、1.05mL)を加えて撹拌した。氷冷下で、6-(t-ブトキシカルボニル)アミノ-1-アミノヘキサン(1.12mL、5mmol)を滴下して加え、そのまま氷冷下で30分間攪拌後、室温(25℃程度)まで昇温し、当該混合物を一晩撹拌した。反応混合物を、超純水及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下で溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液:酢酸エチル:n-ヘキサン=1:4)で精製し、目的物のフラクションを合わせて溶媒を減圧下留去した。
得られた残渣を酢酸エチル(40mL)に溶解し、4N塩酸/酢酸エチル溶液(40mL)を加えて室温(25℃程度)で一晩撹拌した。生じた沈殿物を遠心分離により回収した。得られた固体を酢酸エチルにて4回洗浄後、減圧下で乾燥し、コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)1.2gを得た。
(ヒアルロン酸のテトラブチルアンモニウム(TBA)塩の調製)
ヒアルロン酸のTBA塩(HA-TBA)を次に示す工程2-1-B、続いて工程2-2-Bに従って調製した。
DOWEX(登録商標)50WX-8-400(アルドリッチ社製)を超純水に懸濁させ、デカンテーションにより樹脂を超純水で3回程度洗浄した。40質量%テトラブチルアンモニウムヒドロキシド水溶液(TBA-OH)(アルドリッチ社製)を樹脂のカチオン交換能に対し約1.5倍モル等量加え、30分間撹拌した。余剰のTBA-OH溶液をデカンテーションにより除去した後、さらに過剰の超純水で洗浄することで、TBA塩化したカチオン交換樹脂を得た。
分子量35,000(35kDa)の原料ヒアルロン酸ナトリウム塩(HA-Na)を15mg/mLの濃度で超純水に溶解した。「(1)工程2-1-B」でTBA塩化したカチオン交換樹脂の懸濁液をHAユニット(ユニット分子量401.3)のモル数に対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、ヒアルロン酸のTBA塩(HA-TBA)を白色固体として得た。
[工程2-2-B]で調製したHA-TBAの無水DMSO溶液(10mg/mL)を調製した。その後、[工程1-B]で合成したHA-TBA中に存在する二糖繰り返し単位(HAユニット)に対するChol塩酸塩の添加量がモル比で31/100となる割合で添加した。
試験例1として、難水溶性ペプチドであるシクロスポリン(CyA)の製剤化を行った。
会合促進剤(ポリソルベート80)含有ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyAの製剤を調製した。 以下の操作は20℃、室温下の環境下で実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。別のバイアルに、ポリソルベート80(富士フイルム和光、製品番号:164-21591)を2mg/mLとなる割合で注射用水で希釈した。
以上の操作は20℃で行った。
最終的な製剤組成を表1に示した。
会合促進剤(ポリエチレングリコール400)含有ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyA製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。別のバイアルに、粉末のCyA(東京化成工業、品番:C2408)を88.0mg秤量し、その後、ポリエチレングリコール400(富士フイルム和光、製品番号:161-09065)を10.0mL添加して、CyAを均一に溶解させた。
会合促進剤(ポリエチレングリコール400およびポリソルベート80)含有ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyA製剤を調製した。以下の操作は20℃、室温下の環境下で実施した。
αシクロデキストリンを用いたCyA製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
CyAの粉末を0.5mg/mLとなる割合で濃度が10質量%のαシクロデキストリン(アルドリッチ社製)が含有された10質量%スクロース溶液に溶解して調整した。最後に溶液は目視で清澄であった。また、2週間の冷蔵保管中に析出物は認められなかった。最終的な製剤組成を表1に示した。
ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyAの製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。
以上の操作は20℃で行った。
最終的な製剤組成を表1に示した。
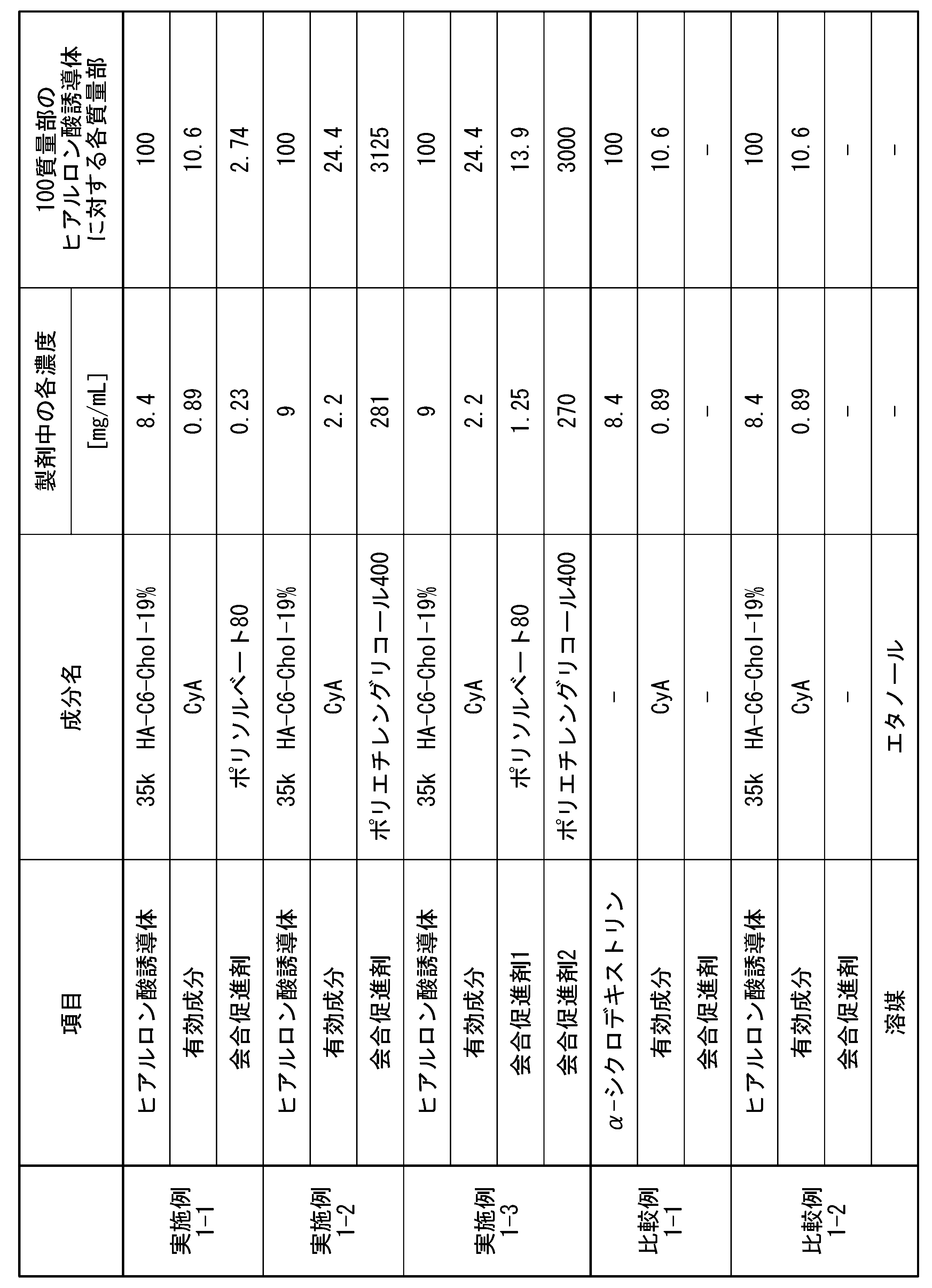
CyAのラットにおける薬物動態試験を実施した。
実施例1-1、1-2、1-3および比較例1-2で調製したヒアルロン酸誘導体医薬組成物からなる溶液製剤および比較例1-1の溶液製剤を、それぞれ25G針を用いて表2に示す用量(mg/kg)で正常ラット(SD、6週齢、オス)の皮下に投与した。

(会合促進剤含有ヒアルロン酸誘導体医薬品組成物のMRT)÷(ヒアルロン酸誘導体医薬品組成物のMRT)≧1.1 ・・・式(A)
GPCによる会合促進度の評価を実施した。
試験例1で調製した実施例1-1について、GPCによるヒアルロン酸誘導体の会合促進度を評価した。
図1は、合成例1で用いたヒアルロン酸誘導体のクロマトグラムである。
ヒアルロン誘導体の濃度が1mg/mLの濃度となる割合で注射用水にて24時間攪拌して溶解させ、GPC測定に供した。この時、クロマトグラムとベースラインで囲まれた面積値をA1とした。
図2は、実施例1-1のヒアルロン酸誘導体組成物のクロマトグラムである。
ヒアルロン誘導体の濃度が1mg/mLの濃度となる割合で注射用水にて希釈して測定した。この時、クロマトグラムとベースラインで囲まれた面積値をA2とした。
装置:HLC8320-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率)
温度:30℃
実施例1-2、1-3、及び比較例1-2も実施例1-1と同様にヒアルロン誘導体の濃度が1mg/mLの濃度となる割合で注射用水にて希釈して測定した。比較例2-1は希釈せずにそのままGPC測定に供した。この時、各クロマトグラムとベースラインで囲まれた面積値であるA2をそれぞれ算出し、A1との面積比A2/A1の値を表3に示した。
添加剤単体の濃度が実施例3-1~3-3、比較例3-2と同様な濃度となる割合で水溶液を調製し、同様にGPC測定に供した。

GPCによる会合促進剤の評価を実施した。
[実施例4-1、4-2、4-3及び比較例4-1、4-2]
試験例1と同様の方法で、有効成分のCyA及び等張剤であるスクロースを含有しないことを除いては同じ組成となる割合で下表4に従ってヒアルロン酸誘導体水溶液を調製した。調整した各サンプルについて、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水にて希釈してGPC測定に供した。
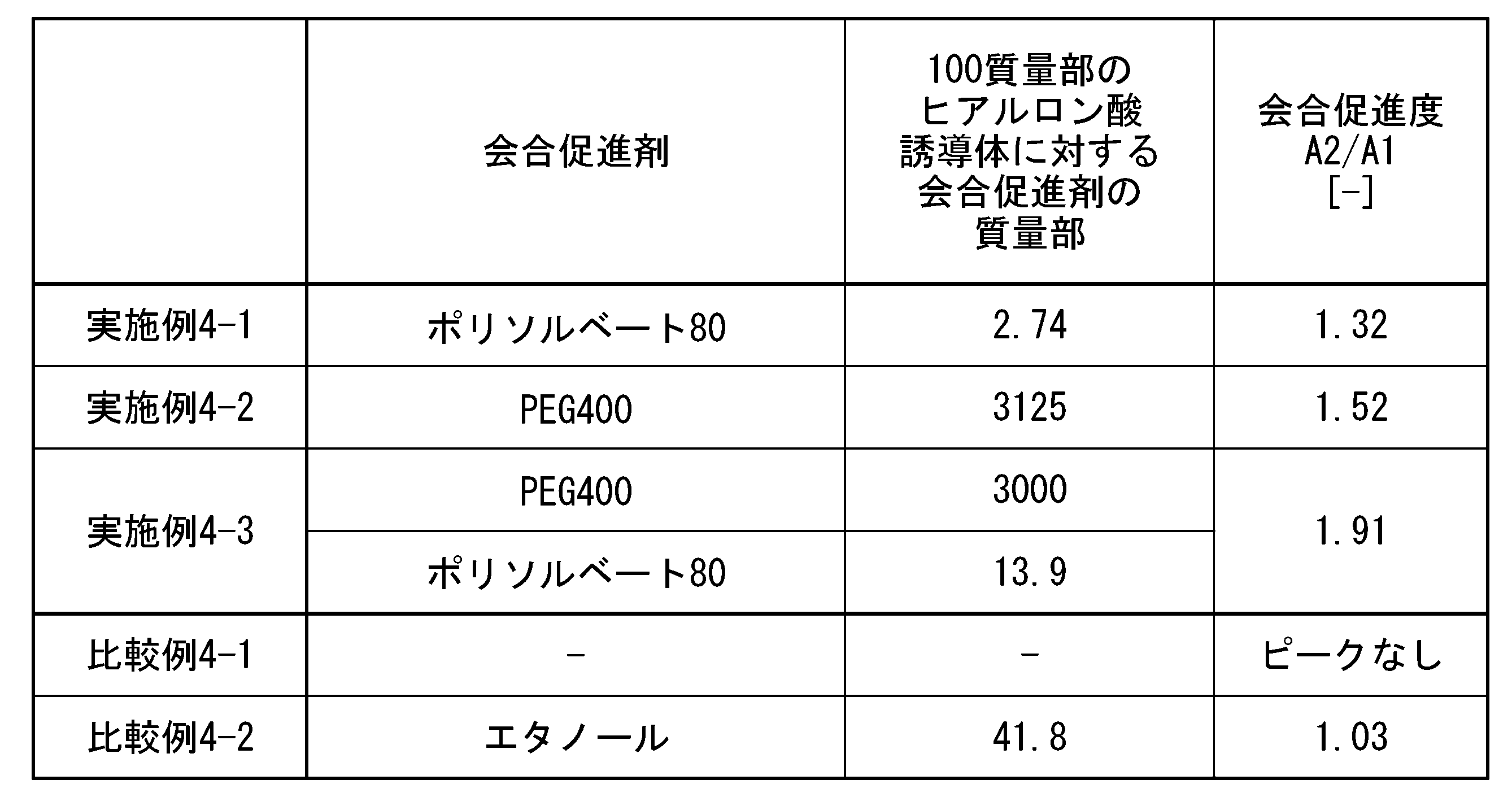
in vitroにおける生理塩濃度下での沈殿挙動を確認した。
1.5mLのマイクロチューブに実施例1で得られた各製剤を150μLサンプリングする。その後、最終緩衝液組成が10mM PB(pH7.4)、150mM NaClとなる割合で濃縮緩衝液(40mM PB(pH7.4)、600mM NaCl)を加え、ヒアルロン酸誘導体を沈殿させた。37℃にて20分間インキュベート後、2000Gにて5分間遠心分離し、上澄みを50μLサンプリングし、HP-β-CD水溶液(300mM)にて2倍希釈し、1時間インキュベート後、HP-β-CD水溶液(10mM)を350μL添加して希釈したのちにGPC測定に供した。検出されたヒアルロン酸誘導体のピーク面積から当初使用量に対するヒアルロン酸誘導体の溶液中の残存率を算出し、沈殿したヒアルロン酸誘導体の割合(沈殿率)を算出した。当該沈殿評価法と、上記[医薬品組成物の物性](試験条件)、沈殿サンプルの調製に記載の沈殿評価法の結果は一致する。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM HP-β-CD/リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率)
温度:30℃
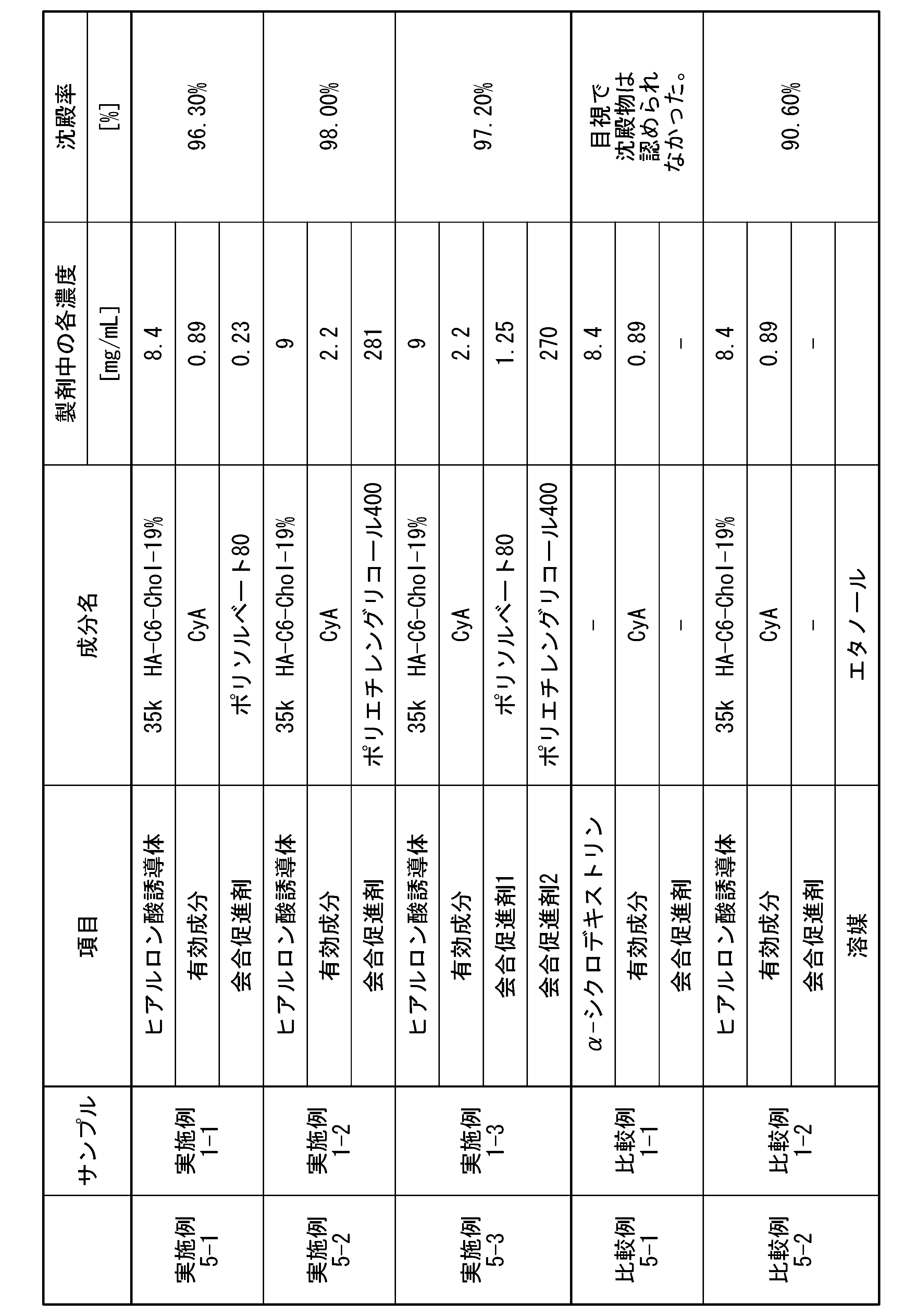
会合促進剤の種類について検討した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例6-1~6-11、比較例6-1~6-6に用いた。
上記で溶解させたヒアルロン酸誘導体水溶液を2mg/mLになる割合で注射用水を入れて希釈した。また別のバイアルに10mg/mLの濃度となる割合でポリエチレングリコール300を注射用水で希釈して調整した。上記ヒアルロン酸誘導体水溶液とポリエチレングリコール300水溶液を体積比1対1で混合して下表6の組成となる割合で調整した。その後、20℃、24時間インキュベートしてからGPC測定に供した。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率)
温度:30℃
実施例6-1と同様な手法で下表6の組成となる割合で溶液を調製してGPC測定に供した。
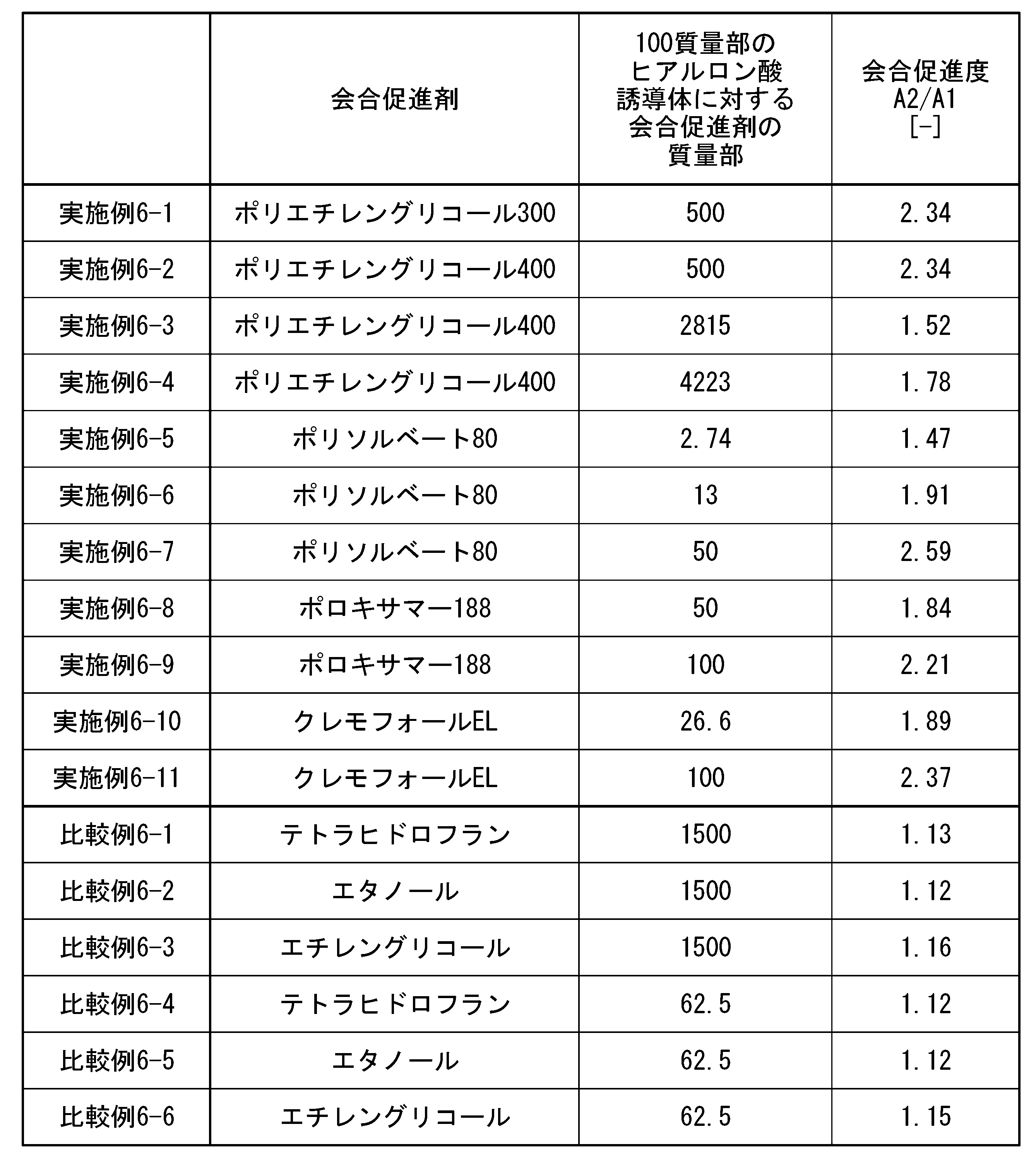
ヒアルロン酸誘導体医薬組成物を製造した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例7-1~7-10、比較例7-1~7-6に用いた。
会合促進剤(ポリソルベート80)含有ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyA製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
バイアルに、粉末のCyA(東京化成工業、品番:C2408)を1.8mg秤量し、その後、別のバイアルにポリソルベート80を0.5mg/mLとなるよう注射用水を添加して希釈した。秤量したCyAを濃度0.5mg/mLのポリソルベート80水溶液18μLで懸濁させた。続けて、12mg/mLのヒアルロン酸誘導体水溶液を攪拌しながら前記CyA含有ポリソルベート80水溶液ポリソルベート80水溶液へ450μL添加して、製剤化時の組成はCyA濃度が2mg/mL、ヒアルロン酸誘導体が6mg/mL、ポリソルベート80が0.01mg/mLとなるよう注射用水を432μL添加して薬物の複合化を行った。20℃、24時間攪拌後に析出物を0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を後述するHPLC測定にて定量した。最終組成は表7に記載した。
実施例7-1と同様な方法で最終組成の会合促進剤濃度が下記表7に記載の濃度となるよう会合促進剤含有ヒアルロン酸誘導体医薬組成物を製造した。また、0.45μm滅菌ろ過フィルター後の製剤中のCyA濃度についても実施例7-1と同様にHPLCにより定量し、結果を表7にまとめた。
カラム :InertSustain C18 粒子径5μm×内径4.6mm×長さ150mm
カラム温度:40℃
移動相 :溶離液A 0.1%TFA/アセトニトリル
溶離液B 0.1%TFA/水
移動相比:溶離液A/溶離液B=9/1
流速 :1mL/min
注入量 :30μL
検出器 :UV(210nm)
試験例7で調製した各種医薬組成物のin vitroにおける生理塩濃度下での沈殿挙動について次に示す方法で評価した。
マイクロチューブ(1.5mL)に濃縮緩衝液(40mM PB、150mM NaCl水溶液)を200μL入れ、ヒアルロン酸誘導体医薬組成物からなる製剤を600μL投入する。その後、voltexで30秒混合し、37℃、20minインキュベートした後に、遠心分離機(2000G、5min)で沈殿物を沈降させる。続いて、上澄み中のヒアルロン酸誘導体をGPC測定に供する。
マイクロチューブ(1.5mL)に注射用水を200μL入れ、ヒアルロン酸誘導体医薬組成物からなる製剤(ヒアルロン酸誘導体の理論濃度Xmg/mLとする。X>1.33)を600μL投入する。その後、voltexで30秒混合し、37℃、20minインキュベートした後に、遠心分離機(2000G、5min)に供する。続いて、上澄み中のヒアルロン酸誘導体が1mg/mLとなるよう注射用水で希釈してGPC測定に供する。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI
温度:30℃
本明細書においては、下式で表される沈殿率が20%以上を満たすヒアルロン酸誘導体医薬組成物を生理塩濃度下で沈殿するものとする。
沈殿率(%)={1-(沈殿サンプルのヒアルロン酸誘導体の面積値)÷((ブランクサンプルのヒアルロン酸誘導体の面積値)×(0.75X))}×100

ヒアルロン酸誘導体の種類について検討した。
合成例2で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-30%)を2.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例8-1~8-11、比較例8-1~8-6に用いた。
ガラスバイアルに10mg/mLの濃度となる割合でポリソルベート80を注射用水で希釈して調整した。6mLのクリーンバイアルを用意し、上記ヒアルロン酸誘導体水溶液を400μLとポリソルベート80水溶液を10μL添加して、voltexで30秒混合後、注射用水を390μL添加して合計800μLとなるよう調整した。下表8の組成となる割合で調整した。その後、20℃、24hインキュベートしてからGPC測定に供した。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率)
温度:30℃
実施例8-1と同様な手法で下表8の組成となる割合で溶液を調製してGPC測定に供した。
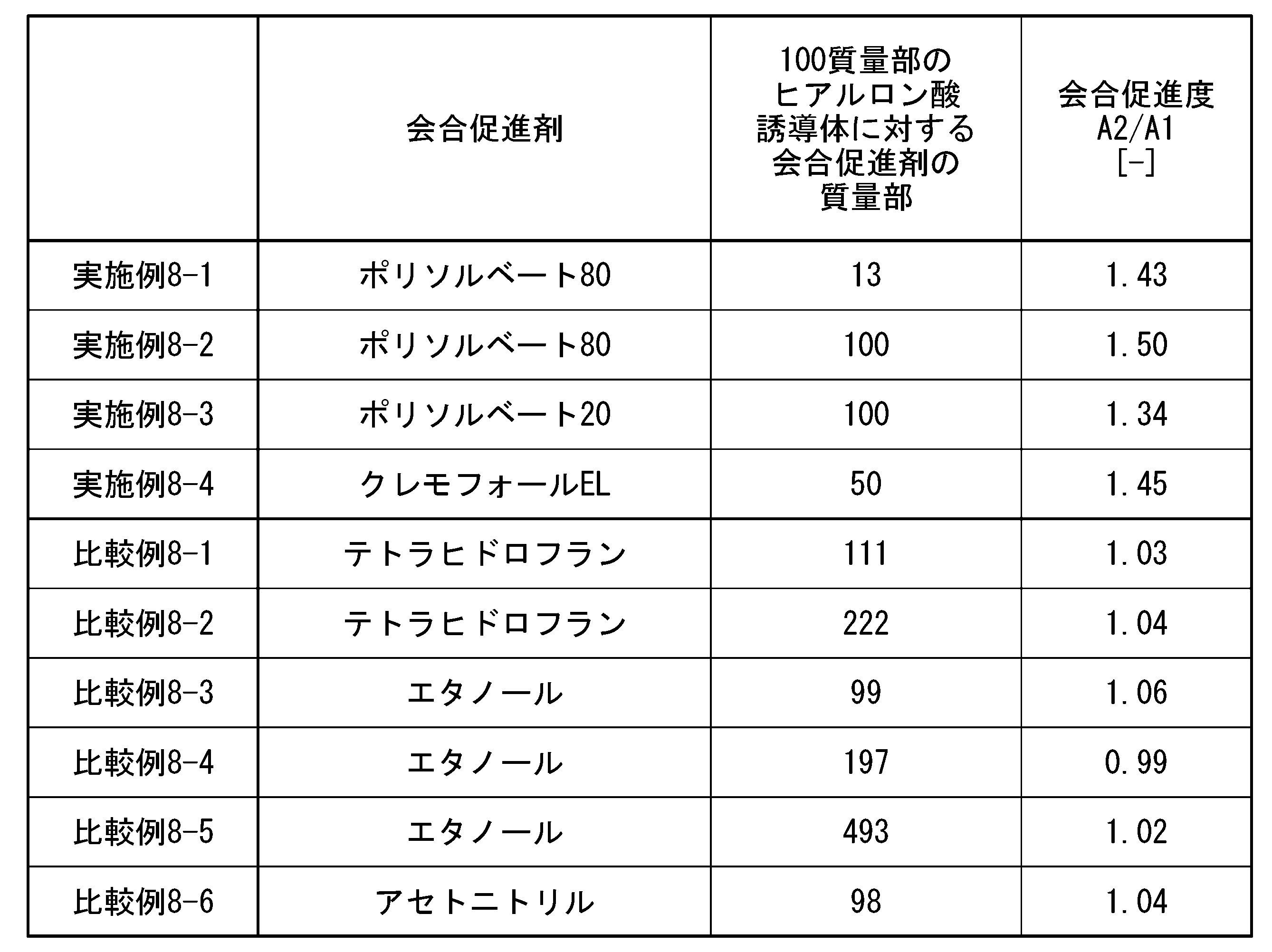
ヒト成長ホルモン(タンパク質)を用いたヒアルロン酸誘導体医薬組成物の調製を実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を4.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例9-1~9-2、比較例9-1に用いた。
会合促進剤及び注射用水を上記ヒアルロン酸誘導体水溶液に添加した。
また、ヒト成長ホルモン(hGH:ジェノトロピン(登録商標)注射用)の粉末を2mg/mLの濃度となるよう別のバイアルで注射用水に溶解し、調製したあとに、前記会合促進剤含有ヒアルロン酸誘導体水溶液に250μL添加し、全量1mLとなるよう調整した。その後、37℃、24時間インキュベートすることでhGHとの複合化を促進させた。最終製剤の組成が表9に記載の濃度となるよう調整した。
テトラヒドロフラン(THF)および注射用水を上記4.0mg/mLの濃度で調製されたヒアルロン酸誘導体水溶液に添加した。加えて、2.0mg/mLのhGH水溶液を前記THF含有ヒアルロン酸誘導体水溶液に250μL添加し、全量1mLとなるよう調整した。その後、37℃、24時間インキュベートすることでhGHとの複合化を促進させた。実施例9-1と同様、最終製剤の組成が表9に記載の濃度となるよう調整した。

装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:UV(280nm)
温度:30℃
以上より、hGHとの複合化も会合促進剤存在下で効率的に行われ、かつ生体内で長期徐放可能なタンパク質-ヒアルロン酸誘導体医薬組成物が取得できるものと期待される。
[合成例3]
試験例6を参考に合成例2と同様の方法で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-40%)について、GPCによるヒアルロン酸誘導体の会合促進度を評価した。
装置:HLC8420-GPC(東ソー社製)
カラム:G4000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:RI(示差屈折率)
温度:30℃
合成例3で得たヒアルロン酸誘導体(10k HA-C6-Chol-40%)を36.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例10-1~10-9に用いた。また別のバイアルに10mg/mLの濃度となる割合でポリソルベート80を注射用水で希釈して調整した。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながら10mg/mLのポリソルベート80水溶液を10μL添加して混合し、最後に注射用水を390μL添加した。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
まず、りん酸緩衝剤粉末(富士フイルム和光社製:167-14491 生化学用 for Biochemistry)を用いて、100mM PB(pH7.4)リン酸緩衝液の調製を行った。
実施例10-1で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながら10mg/mLのポリソルベート80水溶液を50μL添加して混合し、最後に注射用水を350μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
続いて、実施例10-1、実施例10-2で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながらポリエチレングリコール400(ナカライテスク社製)を100μL添加して混合し、100mM PBリン酸緩衝液を100μL加え、最後に注射用水を200μL添加して、終濃度10mM PBとなる割合でヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
続いて、実施例10-1で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながらポリエチレングリコール400(ナカライテスク社製)を100μL添加して混合し、最後に注射用水を300μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24時間インキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
続いて、実施例10-1で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながらポリエチレングリコール400(ナカライテスク社製)を300μL添加して混合し、最後に注射用水を100μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24時間インキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
まず、Poloxamer 338(Aldrich社製)を150mg/mLの濃度となる割合で注射用水を加えて溶解させた。続いて、実施例10-1で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながら150mg/mLのPoloxamer338水溶液を100μL添加して混合し、最後に注射用水を300μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
実施例10-1、実施例10-2、実施例10-7で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながらPoloxamer338水溶液を300μL添加して混合し、100mM PBリン酸緩衝液を100μL加え、終濃度10mM PBとなる割合でヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
実施例10-1、実施例10-7で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を600μL加え、続いて、攪拌しながら150mg/mLのPoloxamer338水溶液を300μL添加して混合し、最後に注射用水を100μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
まず、Chol-PEG600(Avanti社製)を50mg/mLの濃度となる割合で注射用水を加えて溶解させた。続いて、合成例3で得たヒアルロン酸誘導体(10k HA-C6-Chol-40%)を10.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例10-10~10-12に用いた。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を750μL加え、続いて、攪拌しながら50mg/mLのChol-PEG600水溶液を2.8μL添加して混合し、最後に注射用水を747.2μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
実施例10-10で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を750μL加え、続いて、攪拌しながら50mg/mLのChol-PEG600水溶液を11.2μL添加して混合し、最後に注射用水を738.8μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。
実施例10-10で調製した溶液を用いて次の溶液調整を行った。6mlの滅菌バイアルに上記ヒアルロン酸誘導体水溶液を750μL加え、続いて、攪拌しながら50mg/mLのChol-PEG600水溶液を28μL添加して混合し、最後に注射用水を722μL添加して、ヒアルロン酸誘導体水溶液の調製を行った。その後、20℃、24hインキュベートしてから、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で10mM PB(リン酸緩衝液、pH7.4)で希釈してからGPC測定に供した。

試験例11として、難水溶性ペプチドであるシクロスポリン(CyA)の製剤化を行った。
会合促進剤(ポリソルベート80)含有ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyA製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLの濃度となる割合で、10%スクロース水溶液を添加して溶解させた。
αシクロデキストリンを用いたCyA製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。まず、ビーカーにスクロースを2.0g、α-Cyclodextrin(東京化成工業社製)を2.0gを秤量し、15mL程度の注射用水を加え溶解。メスシリンダーに移し、20mLにメスアップした。続いて、0.45μmのフィルターにて濾過し、10%スクロース/10%α-Cyclodextrin水溶液とした。2.0mgのCyclosporin A(東京化成工業社製)をコニカルチューブに秤量し、10mLの10%スクロース/10%α-Cyclodextrin水溶液を加え、攪拌溶解させた。最後に、0.2μmのフィルターにて濾過し、投与溶液とした。最終的な製剤組成を表11に示した。
ヒアルロン酸誘導体(35k HA-C6-Chol-19%)を用いたCyAの製剤を調製した。
以下の操作は20℃、室温下の環境下で実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。
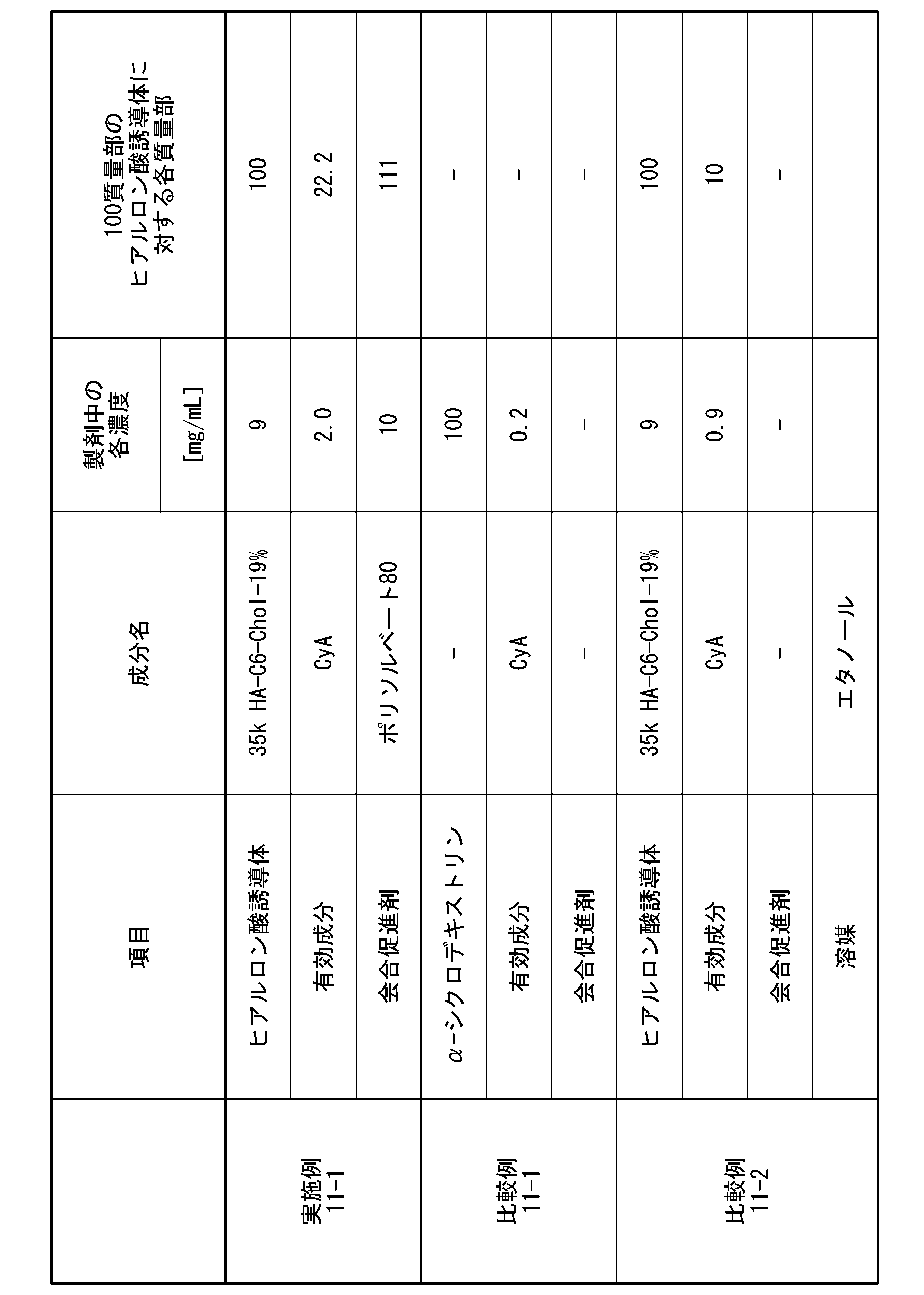
CyAのラットにおける薬物動態試験を実施した。
実施例11-1および比較例11-2で調製したヒアルロン酸誘導体医薬組成物からなる溶液製剤および比較例11-1の溶液製剤を、それぞれ25G針を用いて表12に示す用量(mg/kg)で正常ラット(SD、6週齢、オス)の皮下に投与した。
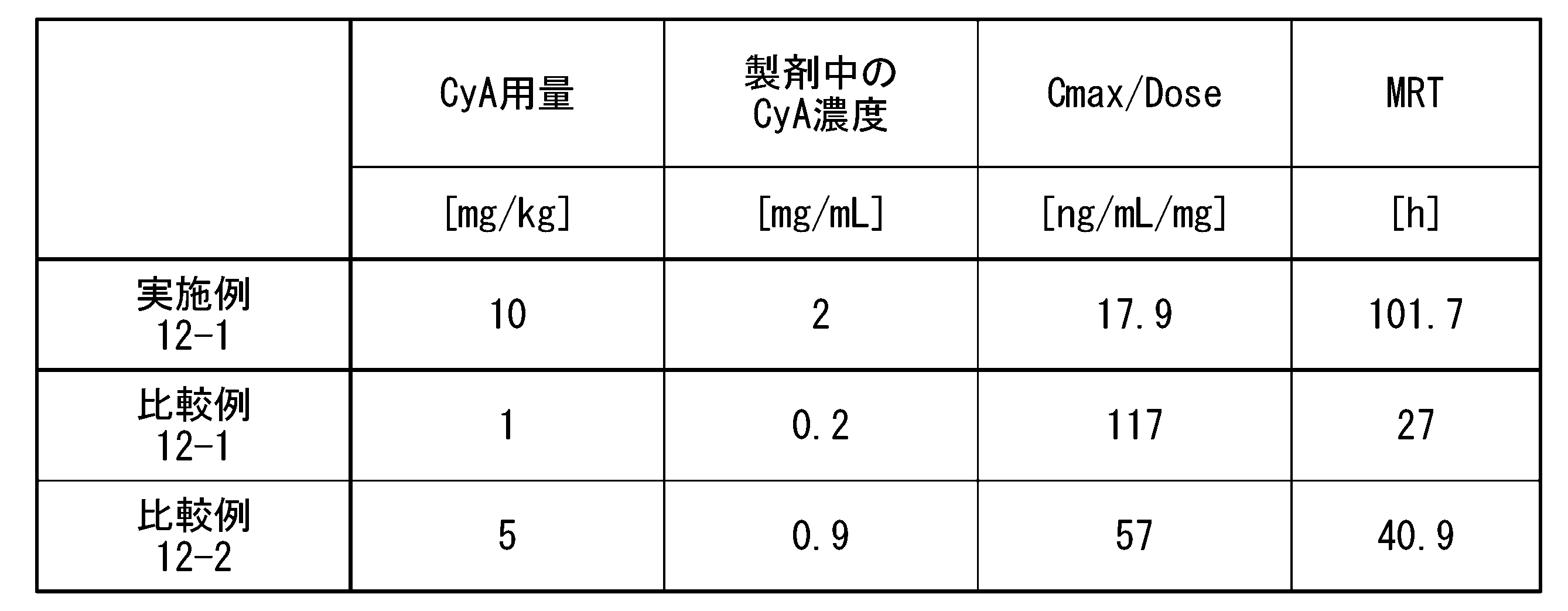
<試験例13>
ヒアルロン酸誘導体医薬組成物を製造した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させたものを実施例13-1~13-2、比較例13-1に用いた。
以下の操作は20℃、室温下の環境下で実施した。
まず、ポリソルベート80(Thermo Scientific社製)を10mg/mLの濃度となる割合で注射用水を加えて溶解させた。続いて、170mg/mLの濃度となる割合でグリセリン(ナカライテスク社製、商品コード:17045-94)を注射用水に溶解して調整した。さらに、注射用水28.240mLに対し、0.5mol/L-EDTA溶液(pH 8.0)(ナカライテスク社製、商品コード:06894-14)を1mL加えて、5.0mg/mLEDTA緩衝液を調整した。
まず、6mLの滅菌バイアルに2.5mgのCyAを秤量した。その後、実施例13-1で調製したヒアルロン酸誘導体溶液を450μL添加して20℃、24時間攪拌した。CyA粉末の溶け残りが認められ、飽和状態で次の操作に移行した。上で調製したCyA含有ヒアルロン酸誘導体医薬組成物に対して、13mmφの0.22μmPESフィルターで滅菌ろ過し、ヒアルロン酸誘導体へ封入できずに析出した薬物を取り除き、得られたろ液の製剤中のCyA濃度を逆相HPLCにて定量した。本実験をn=2で実施し、1.50mg/mLおよび1.41mg/mLであった。製剤中のCyA濃度について、得られた平均値を表13にまとめた。また、試験例7と同様な方法で沈殿するか否か検証した。その結果も表13にまとめた。
ポリソルベート濃度が3mg/mLであること以外すべて同じ濃度で、実施例13-1と同様な方法にて会合促進剤含有ヒアルロン酸誘導体水溶液を調整した。
この時、EDTA濃度は0.075mg/mL、グリセリン濃度は25.5mg/mL、ポリソルベート80濃度は3mg/mL、ヒアルロン酸誘導体の濃度は5mg/mLであった。その後、20℃、24h攪拌し、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で希釈してGPC測定に供した。会合促進剤を含有しないこと以外は同じ組成でヒアルロン酸誘導体溶液を調整し、得られた面積値をA1として会合促進剤による会合促進度を実施例13-1と同様に算出し、表13に記載した。
まず、6mLの滅菌バイアルに2.5mgのCyAを秤量した。その後、実施例13-2で調製したヒアルロン酸誘導体溶液を450μL添加して20℃、24時間攪拌した。CyA粉末の溶け残りが認められ、飽和状態で次の操作に移行した。上で調製したCyA含有ヒアルロン酸誘導体医薬組成物に対して、13mmφの0.22μmPESフィルターで滅菌ろ過し、ヒアルロン酸誘導体へ封入できずに析出した薬物を取り除き、得られたろ液の製剤中のCyA濃度を逆相HPLCにて定量した。本実験をn=2で実施し、1.89mg/mLおよび1.87mg/mLであった。製剤中のCyA濃度について、得られた平均値を表13にまとめた。また、試験例7と同様な方法で沈殿するか否か検証した。その結果も表13にまとめた。
ポリソルベート濃度が0mg/mLであること以外すべて同じ濃度で、実施例13-1と同様な方法にてヒアルロン酸誘導体水溶液を調整した。
この時、EDTA濃度は0.075mg/mL、グリセリン濃度は25.5mg/mL、ポリソルベート80濃度は0mg/mL、ヒアルロン酸誘導体の濃度は5mg/mLであった。その後、20℃、24h攪拌し、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で希釈してGPC測定に供した。
まず、6mLの滅菌バイアルに2.5mgのCyAを秤量した。その後、比較例13-1で調製したヒアルロン酸誘導体溶液を450μL添加して20℃、24時間攪拌した。CyA粉末の溶け残りが認められ、飽和状態で次の操作に移行した。上で調製したCyA含有ヒアルロン酸誘導体医薬組成物に対して、13mmφの0.22μmPESフィルターで滅菌ろ過を試みたが、ろ過が困難であった。
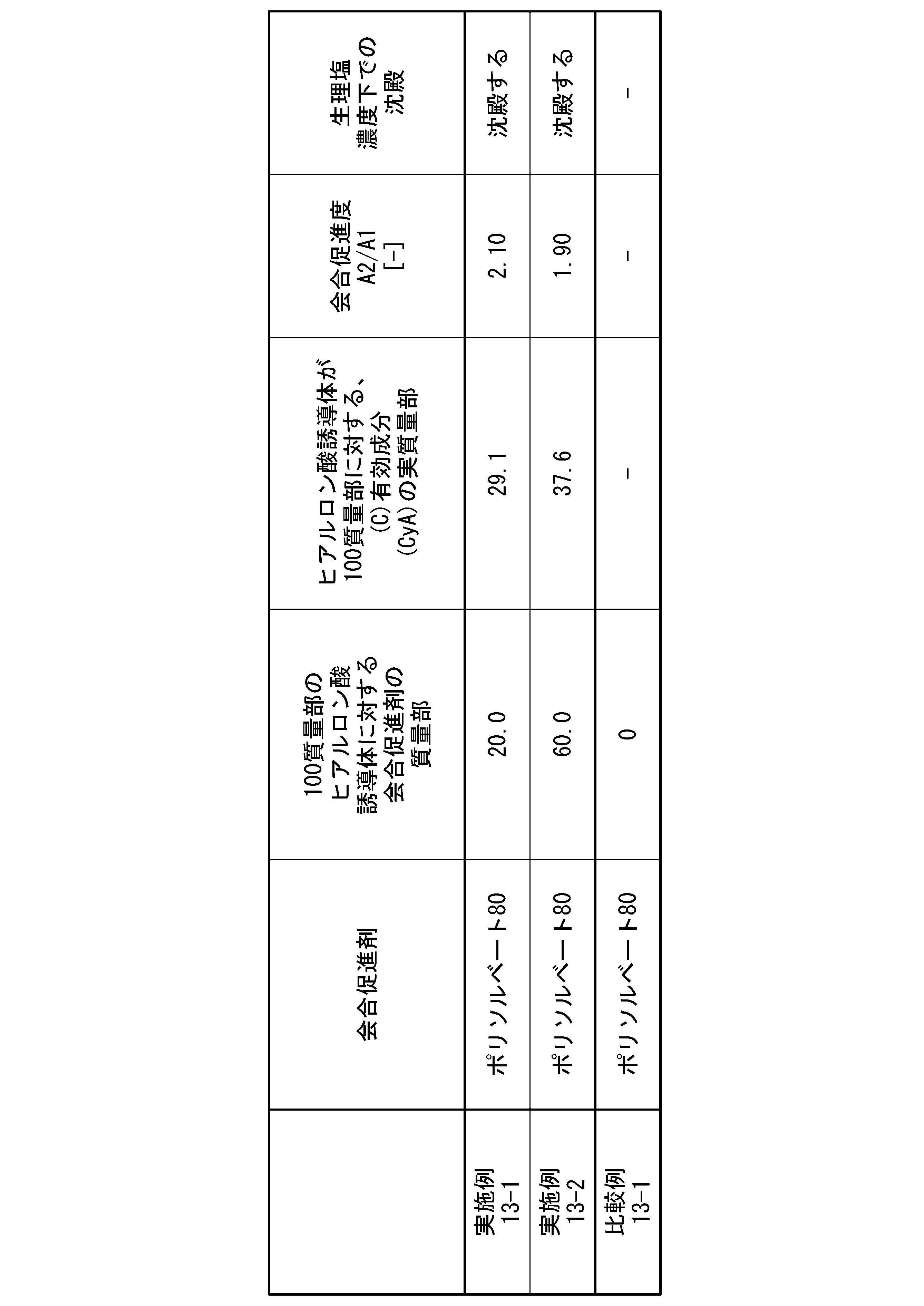
試験例3と同様な方法で、別の会合促進剤について、GPCによる会合促進度の評価を実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。1.5mLのエッペンチューブに12.0mg/mLの前記ヒアルロン酸誘導体水溶液を416.7μL加え、続いて、100mg/mLの濃度で注射用水に溶解させたポリビニルアルコール(重合度500、ナカライテスク社製、製品番号:11738-62)を50μL加える。その後、合計1000μLとなる割合で注射用水を加えて調整した(表14)。その後、20℃、2hインキュベートしてからヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で5倍希釈してからGPC測定に供した。
合成例3で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-40%)を62.6mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。1.5mLのエッペンチューブに62.6mg/mLの前記ヒアルロン酸誘導体水溶液を79.9μL加え、続いて、100mg/mLの濃度で注射用水に溶解させたポリビニルアルコール(重合度500、ナカライテスク社製、製品番号:11738-62)を50μL加える。その後、合計1000μLとなる割合で注射用水を加えて調整した(表14)。その後、20℃、2hインキュベートしてからヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で5倍希釈してからGPC測定に供した。
合成例2で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-30%)を40.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。1.5mLのエッペンチューブに40.0mg/mLの前記ヒアルロン酸誘導体水溶液を125.0μL加え、続いて、100mg/mLの濃度で注射用水に溶解させたポリビニルアルコール(重合度500、ナカライテスク社製、製品番号:11738-62)を50μL加える。その後、合計1000μLとなる割合で注射用水を加えて調整した(表14)。その後、20℃、2hインキュベートしてからヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で5倍希釈してからGPC測定に供した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。さらに実施例13-1で調製した10mg/mLポリソルベート80水溶液、170mg/mLのグリセロール水溶液、注射用水及び5.0mg/mLEDTA溶液を用いて会合促進剤含有ヒアルロン酸誘導体水溶液を表14の濃度となる割合で調整した。
その後、20℃、24時間インキュベートしてからGPC測定に供した。
合成例3で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-40%)を62.6mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。さらに実施例13-1で調製した10mg/mLポリソルベート80水溶液、170mg/mLのグリセロール水溶液、注射用水及び5.0mg/mLEDTA溶液を用いて会合促進剤含有ヒアルロン酸誘導体水溶液を表14の濃度となる割合で調整した。
その後、20℃、24時間インキュベートしてからGPC測定に供した。
合成例2で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-30%)を40.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。さらに実施例13-1で調製した10mg/mLポリソルベート80水溶液、170mg/mLのグリセロール水溶液、注射用水及び5.0mg/mLEDTA溶液を用いて会合促進剤含有ヒアルロン酸誘導体水溶液を表14の濃度となる割合で調整した。
その後、20℃、24時間インキュベートしてからGPC測定に供した。
何れも、ヒアルロン酸誘導体の濃度が1mg/mLとなる割合で注射用水で5倍希釈してからGPC測定に供した。

セマグルチド(ペプチド)を用いたヒアルロン酸誘導体医薬組成物の調製を実施した。
合成例1で得た凍結乾燥品のヒアルロン酸誘導体(35k HA-C6-Chol-19%)を12.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。その後、別のバイアルに170mg/mLのグリセロール濃度となる割合で注射用水で溶解させた。さらに別のバイアルに、0.5mol/l-EDTA溶液(pH 8.0)(ナカライテスク社製)を用いて、5mg/mLのEDTA濃度となる割合で注射用水で希釈した。またさらに別のバイアルにポリソルベート80を10mg/mLの濃度となる割合で注射用水で希釈して調整した。上で調製した溶液を用いて、会合促進剤含有ヒアルロン酸誘導体水溶液の調整を実施した。
合成例2で得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-30%)を40.0mg/mLで注射用水に20℃、24時間攪拌させて溶解させた。その後、別のバイアルに170mg/mLのグリセロール濃度となる割合で注射用水で溶解させた。さらに別のバイアルに、0.5mol/l-EDTA溶液(pH 8.0)(ナカライテスク社製)を用いて、5mg/mLのEDTA濃度となる割合で注射用水で希釈した。またさらに別のバイアルにポリソルベート80を10mg/mLの濃度となる割合で注射用水で希釈して調整した。上で調製した溶液を用いて、会合促進剤含有ヒアルロン酸誘導体水溶液の調整を実施した。
装置:HLC8320-GPC(東ソー社製)
カラム:G3000SWXL(東ソー社製、粒子径8μm、内径7.8mm、長さ30cm、品番:8542)
溶離液:10mM リン酸緩衝液(pH7.4)
流速:1mL/min
注入量:50μL
検出器:UV(220nm)
温度:30℃
以上より、セマグルチドとの複合化も会合促進剤存在下で効率的に行われ、かつ生体内で長期徐放可能な長鎖ペプチド-ヒアルロン酸誘導体医薬組成物が取得できるものと期待される。
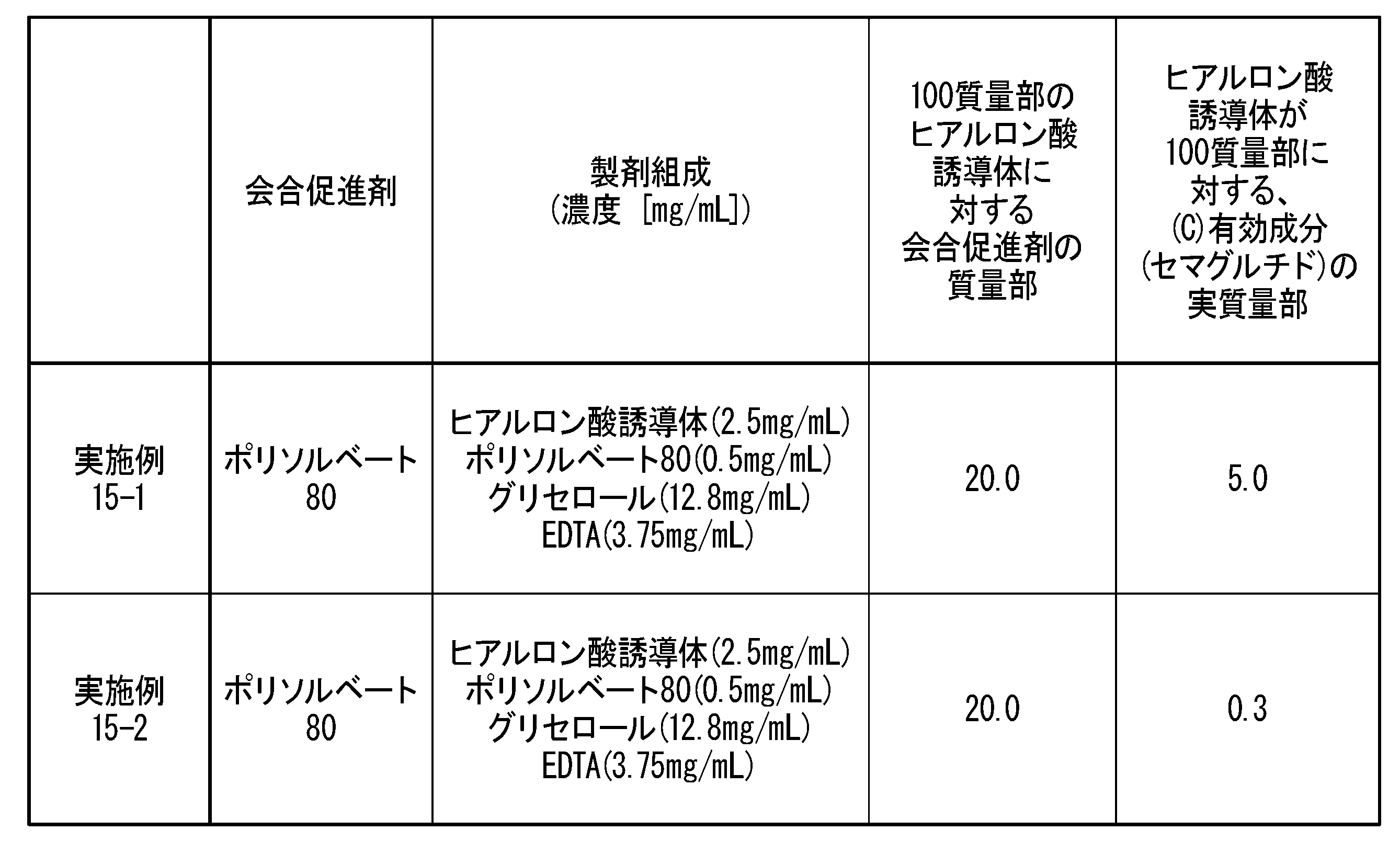
[合成例1B]
ヒアルロン酸誘導体を次の工程1B~工程3Bに従って調製した。
(コレステリル 6-アミノヘキシルカーバメート塩酸塩の合成)
コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)を次に示す工程1B-1、続いて工程1B-2に従って合成した。
コレステリルクロロホルメート(3.37g、7.5mmol)の無水ジクロロメタン(20mL)の溶液に、アルゴン雰囲気下、トリエチルアミン(TEA、1.05mL)を加えて撹拌した。氷冷下で、6-(t-ブトキシカルボニル)アミノ-1-アミノヘキサン(1.12mL、5mmol)を滴下して加え、そのまま氷冷下で30分間攪拌後、室温(25℃程度)まで昇温し、当該混合物を一晩撹拌した。反応混合物を、超純水及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧下で溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶離液:酢酸エチル:n-ヘキサン=1:4)で精製し、目的物のフラクションを合わせて溶媒を減圧下留去した。
得られた残渣を酢酸エチル(40mL)に溶解し、4N塩酸/酢酸エチル溶液(40mL)を加えて室温(25℃程度)で一晩撹拌した。生じた沈殿物を遠心分離により回収した。得られた固体を酢酸エチルにて4回洗浄後、減圧下で乾燥し、コレステリル 6-アミノヘキシルカーバメート塩酸塩(Chol塩酸塩)1.2gを得た。
(ヒアルロン酸のテトラブチルアンモニウム(TBA)塩の調製)
ヒアルロン酸のTBA塩(HA-TBA)を次に示す工程2B-1、続いて工程2B-2に従って調製した。
DOWEX(登録商標)50WX-8-400(アルドリッチ社製)を超純水に懸濁させ、デカンテーションにより樹脂を超純水で3回程度洗浄した。40質量%テトラブチルアンモニウムヒドロキシド水溶液(TBA-OH)(アルドリッチ社製)を樹脂のカチオン交換能に対し約1.5倍モル等量加え、30分間撹拌した。余剰のTBA-OH溶液をデカンテーションにより除去した後、さらに過剰の超純水で洗浄することで、TBA塩化したカチオン交換樹脂を得た。
分子量10,000(10kDa)の原料ヒアルロン酸ナトリウム塩(HA-Na)を15mg/mLの濃度で超純水に溶解した。「工程2B-1」でTBA塩化したカチオン交換樹脂の懸濁液をHAユニット(ユニット分子量401.3)のモル数に対し樹脂のイオン交換能換算で5倍モル等量添加した。15分間撹拌した後、0.45μmのフィルターを用いて濾過を行い、濾液を凍結乾燥し、ヒアルロン酸のTBA塩(HA-TBA)を白色固体として得た。
「工程2B-2」で調製したHA-TBAの無水DMSO溶液(10mg/mL)を調製した。その後、「工程1B」で合成したHA-TBA中に存在する二糖繰り返し単位(HAユニット)に対するChol塩酸塩の添加量がモル比で44/100となるよう添加した。次に、HAユニットに対する4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)の添加量がモル比で48/100となるよう加え、室温(25℃程度)で一晩撹拌した。反応溶液は、0.3M 酢酸アンモニア/DMSO溶液、0.15M NaCl水溶液、超純水の順で透析(スペクトラポア7、分画分子量(MWCO):3,500)した。得られた透析液を凍結乾燥して目的物(HA-C6-Chol)を白色固体として得た。生成物の1H-NMRスペクトルにおいて、N-アセチル-D-グルコサミンのアセチル基由来のピーク(COCH3、1.6ppm以上2.0ppm以下、3H)、コレステリル基中のメチル基由来のピーク(CH3、0.7ppm、3H)が確認され、コレステロール導入率は44%であった。
[実施例1B-1]
ヒアルロン酸誘導体(10k HA-C6-Chol-44%)を用い、難水溶性ペプチドであるシクロスポリン(CyA)を粉末から製剤化した。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。これにより水溶液(II)であるヒアルロン酸誘導体水溶液を得た。
別のバイアルに、ポリソルベート80を10mg/mLとなる割合で注射用水で希釈した。
さらに別のバイアルに、粉末のCyA(東京化成工業社製、製品番号:C2408)を4.0mg秤量し、その後、10mg/mLのポリソルベート80を80μL添加した。これにより、分散液(I)として、CyAがポリソルベート80に分散したCyA分散液を得た。
バイアルにスターラーチップを入れて攪拌しながら、水溶液(II)であるヒアルロン酸誘導体水溶液を0.30mL添加した後に、液量が0.650mLとなる割合で、CyA分散液を添加した。この後、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表16に示した。
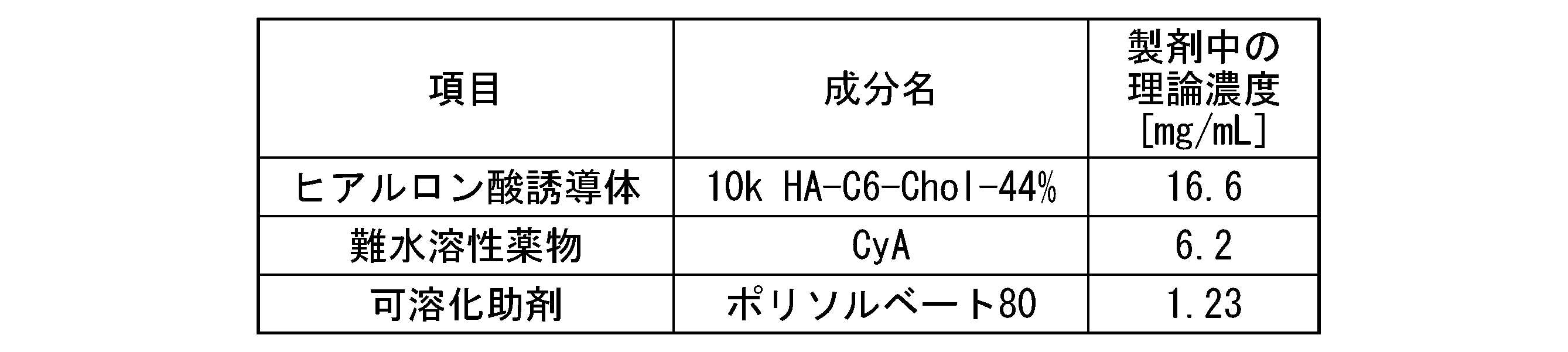
カラム :InertSustain C18 粒子径5μm×内径4.6mm×長さ150mm
カラム温度:40℃
移動相 :溶離液A 0.1%TFA/アセトニトリル
溶離液B 0.1%TFA/水
移動相比:溶離液A:溶離液B=9:1
流速 :1mL/分
注入量 :30μL
検出器 :UV(210nm)
可溶化助剤の種類と添加量を変更する以外は実施例1B-1と同様な手順にて、ヒアルロン酸誘導体と可溶化助剤を用いて、CyAの製剤化を行った。各製剤に対して、添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表17~18にそれぞれ示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。
別のバイアルに、粉末のCyAを4.0mg秤量し、その後、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.30mL添加した。バイアルにスターラーチップを入れて、液量が0.650mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表4に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。
別のバイアルに、粉末のCyAを4.0mg秤量し、その後、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.1783mL添加した。バイアルにスターラーチップを入れて、液量が0.650mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表19に示す。
合成例1Bで使用した分子量10,000(10kDa)の原料ヒアルロン酸ナトリウム塩(HA-Na)を55mg/mLで注射用水に溶解させた。その後、ポリソルベート20、ヒアルロン酸ナトリウムを表4に記載の濃度となるよう適宜注射用水で希釈して水溶液を調製した。
別のバイアルに、粉末のCyAを4.0mg秤量し、その後、上記ポリソルベート20含有のHA-Na水溶液を0.65mL添加した。バイアルにスターラーチップを入れて、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度をHPLC測定にて定量した。添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表19に示す。
ポリソルベート80を100mg/mLとなる割合で注射用水で希釈した。
別のバイアルに、粉末のCyAを4.0mg秤量し、その後、上記100.0mg/mLのポリソルベート80を0.65mL添加した。バイアルにスターラーチップを入れて、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表19に示す。
可溶化助剤の濃度は下表に記載した濃度を用い、それ以外は比較例1B-4又は実施例1B-1と同様な手順で製剤化、薬物濃度の定量を行った。結果を表19~20に示す。
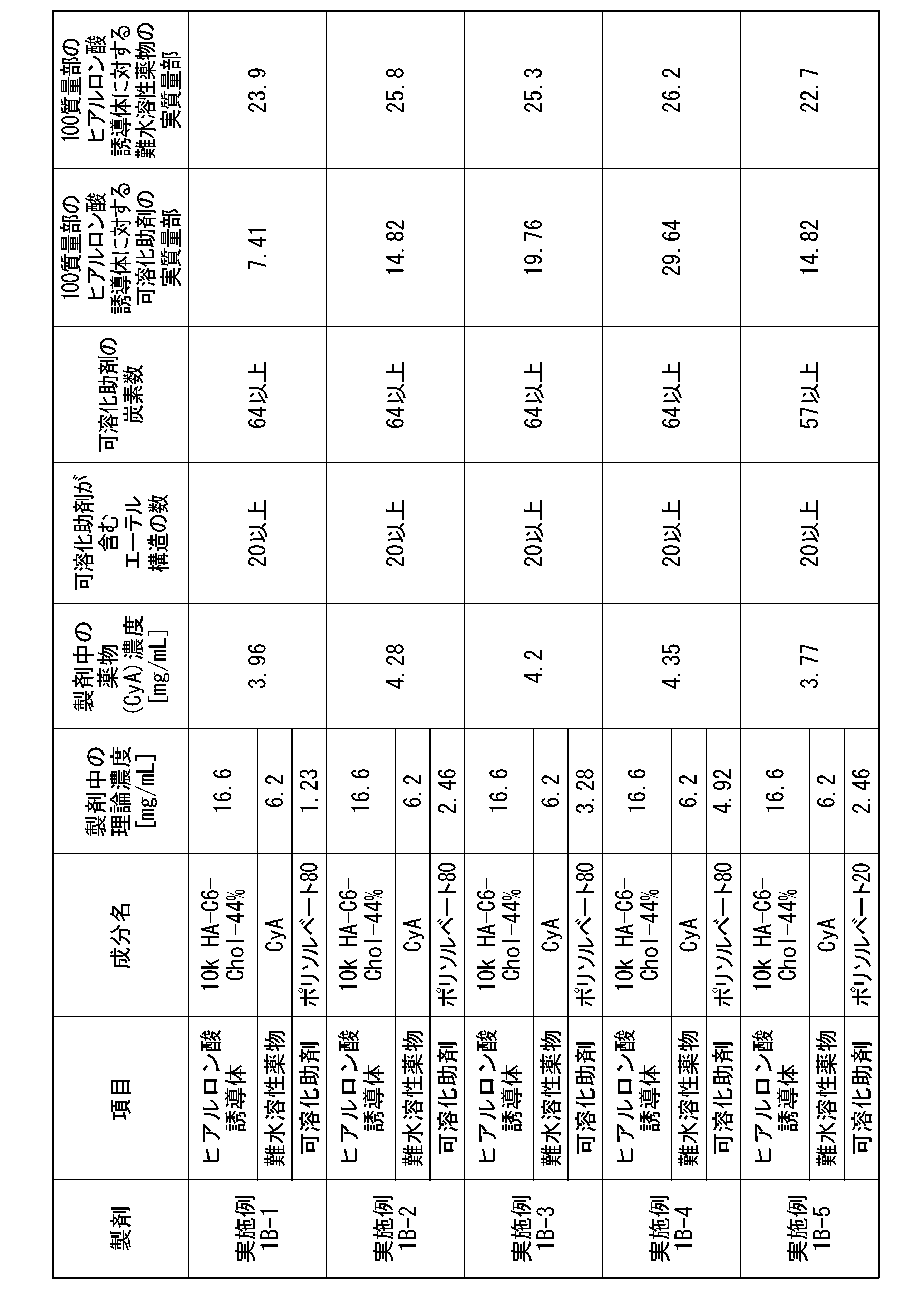

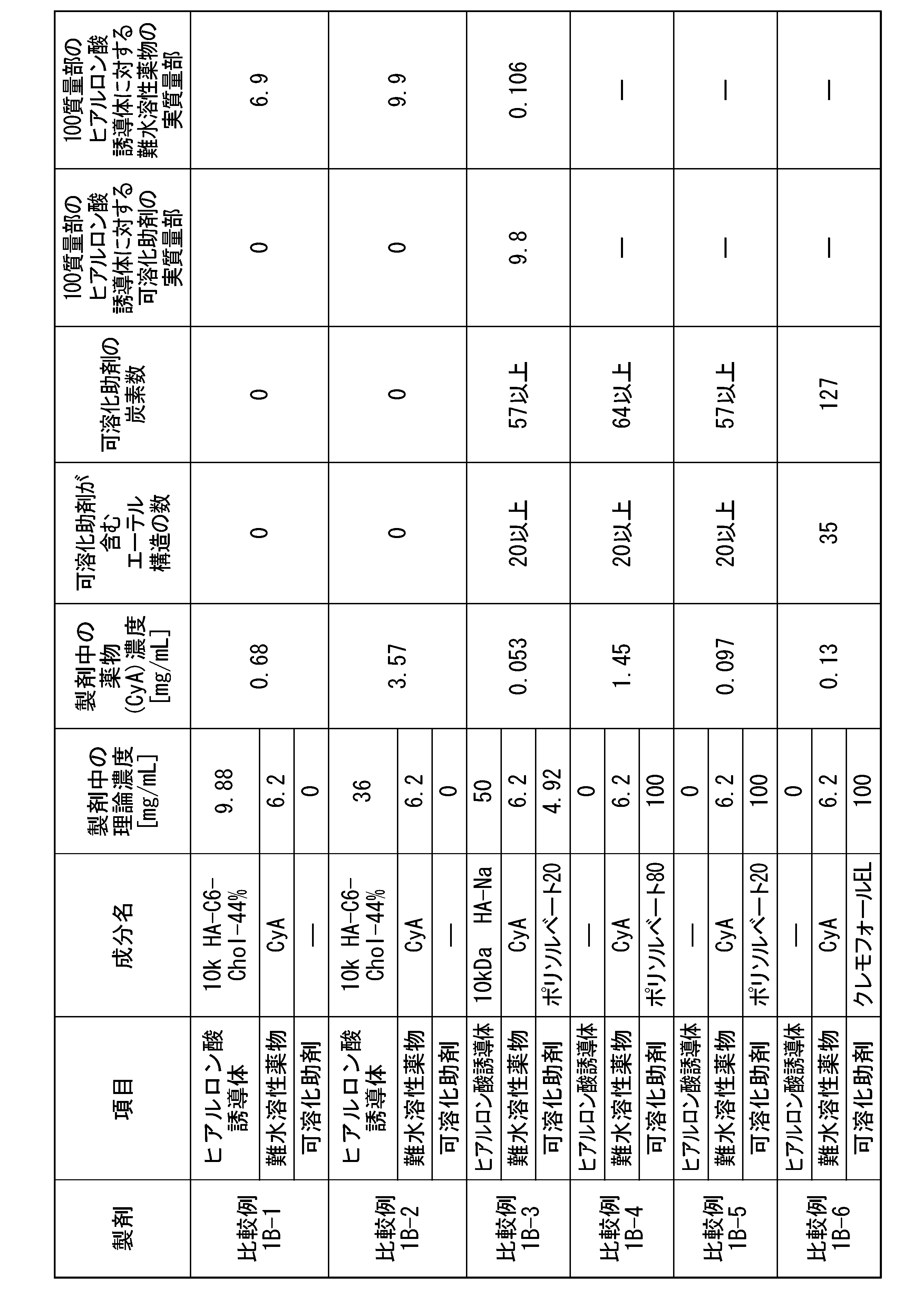

[実施例2B-1~2B-12]
可溶化助剤の添加量を詳細に検証した。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を5mg/mLの濃度となるよう注射用水で溶解させた。別のバイアルに、粉末のCyAを1.0mg秤量し、製剤中の最終組成の可溶化助剤濃度が表21に記載の濃度となるよう0.2mL添加してスターラーチップを入れて攪拌させた。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表21に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を5mg/mLの濃度となるよう注射用水で溶解させた。別のバイアルに、粉末のCyAを1.0mg秤量し、製剤中の最終組成の可溶化助剤濃度が表21に記載の濃度となるよう濃度10mg/mLのステアロイル乳酸ナトリウム水溶液を0.1mL添加してスターラーチップを入れて攪拌させた。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表21に示す。
バイアルに、粉末のCyAを1.0mg秤量し、製剤中の最終組成の可溶化助剤濃度が表21に記載の濃度となるよう0.2mL添加してスターラーチップを入れて攪拌させた。 続いて、注射用水を攪拌しながら0.8mL添加し、24時間攪拌させ、可溶化助剤のみで薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表21に示す。
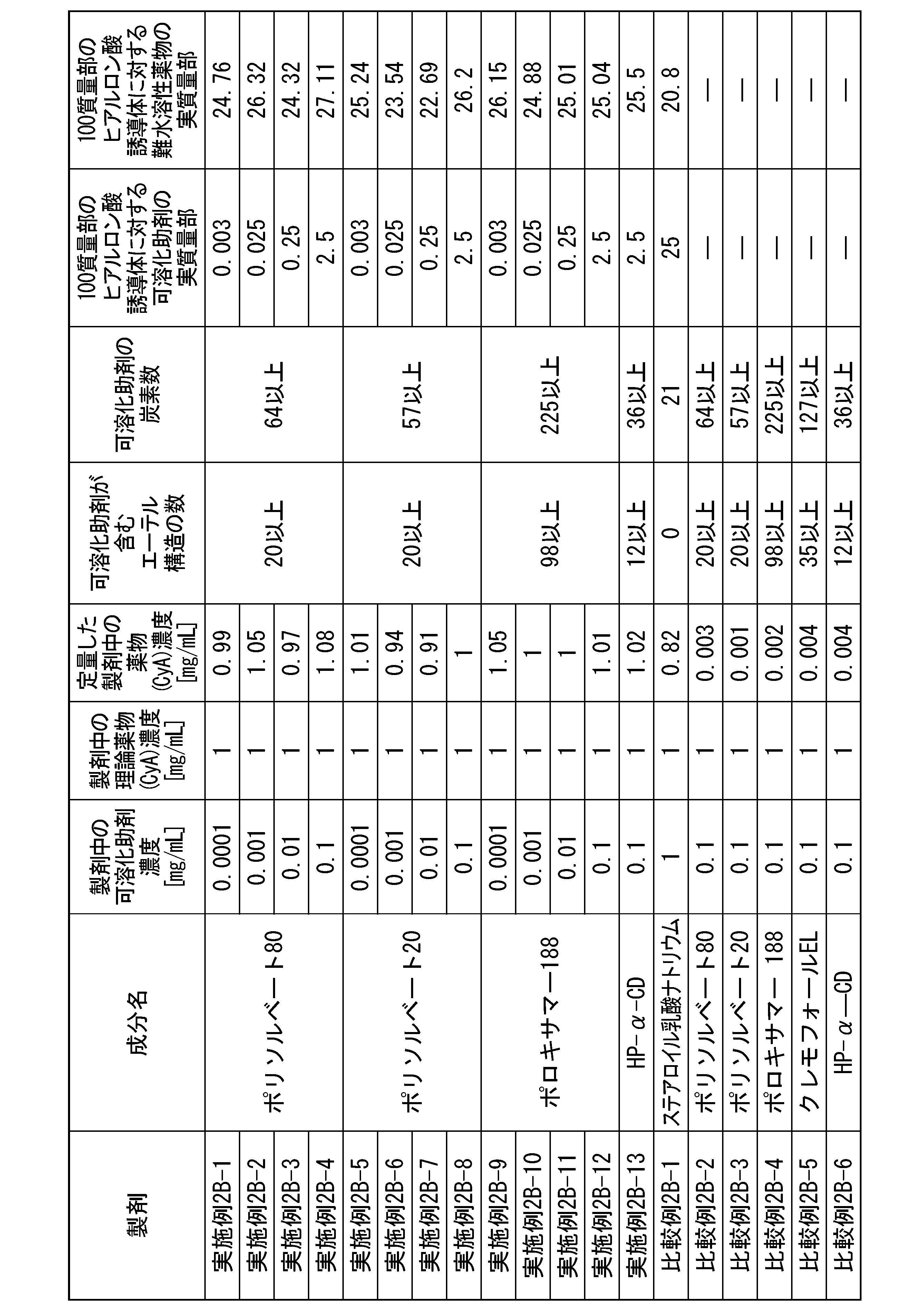
上記表21に示すように、可溶化助剤はヒアルロン酸誘導体が100質量部に対し、0.003質量部以上を添加するだけで粉末から有機溶剤を使用せずに可溶化能を向上させる。
[実施例3B-1~3B-14]
次に、使用可能な可溶化助剤を詳細に検証した。
具体的には、エーテル構造(R-O-R)を少なくとも5以上含み、かつ炭素数12以上である、ポリエチレングリコール300及びポリエチレングリコール400が可溶化助剤として使用可能であるか否か検証した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表22に示す。
バイアルに、粉末のCyAを1.0mg秤量し、可溶化助剤および注射用水を合わせて0.5mLとなるよう添加してスターラーチップを入れて攪拌させた。この時、最終的に得られる可溶化助剤を含む医薬組成物中の可溶化助剤濃度が下表22の通りになるよう調整した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表22に示す。
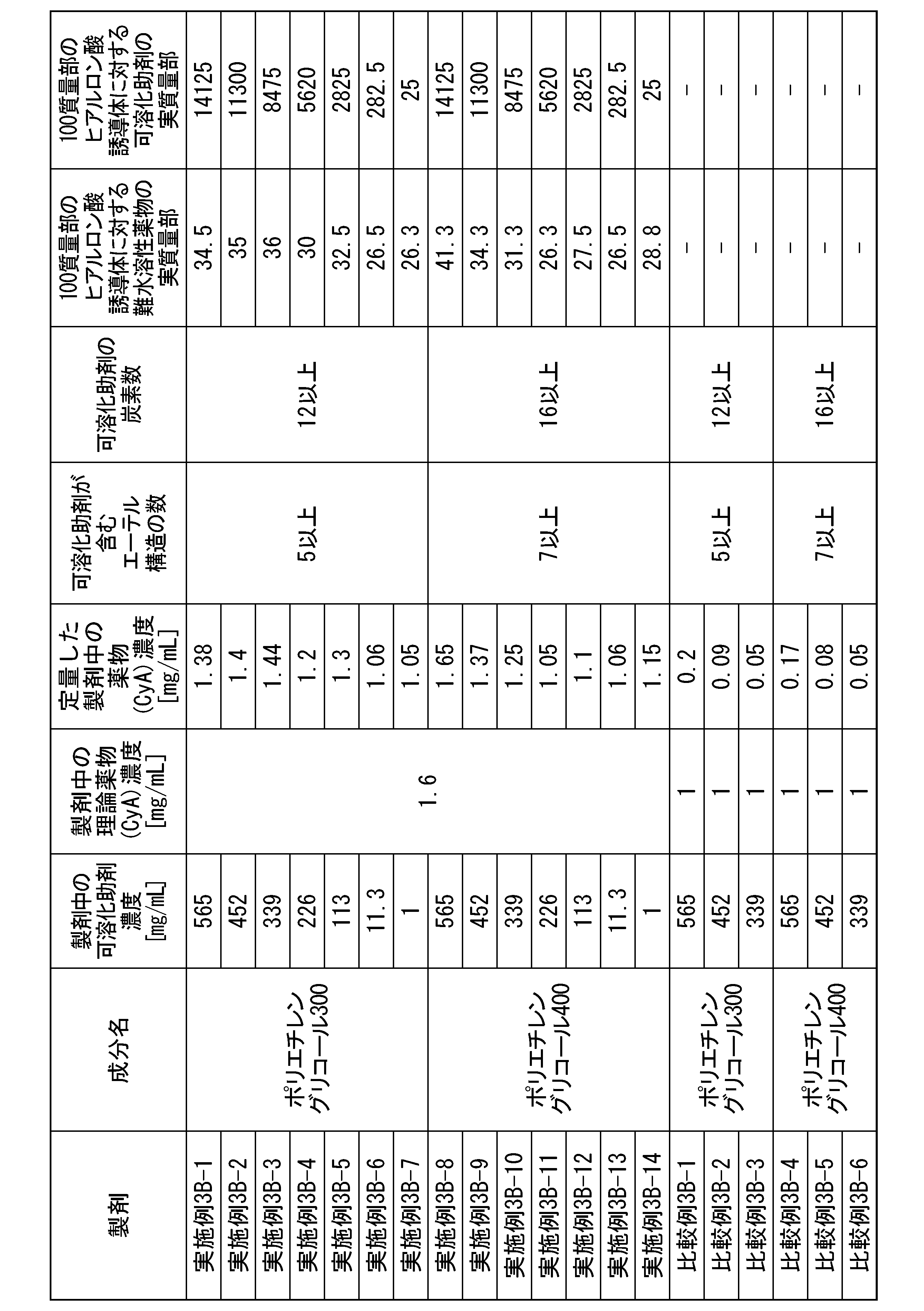
[実施例4B-1]
次に、ヒアルロン酸誘導体医薬組成物の製造方法の詳細な検証を実施した。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、ポリソルベート80を10mg/mLとなるよう注射用水で希釈した。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、ポリソルベート80を10mg/mLとなるよう注射用水で希釈した。
その後、別のバイアルに10mg/mLのポリソルベート80を320μL添加し、バイアルにスターラーチップを入れて攪拌しながら、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加した後に、液量が1.30mLとなるよう注射用水を添加した。またさらに別のバイアルに、粉末のCyAを4.0mg秤量し、上記可溶化助剤含有ヒアルロン酸誘導体を0.65mL添加し、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表20に示した。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表23に示す。
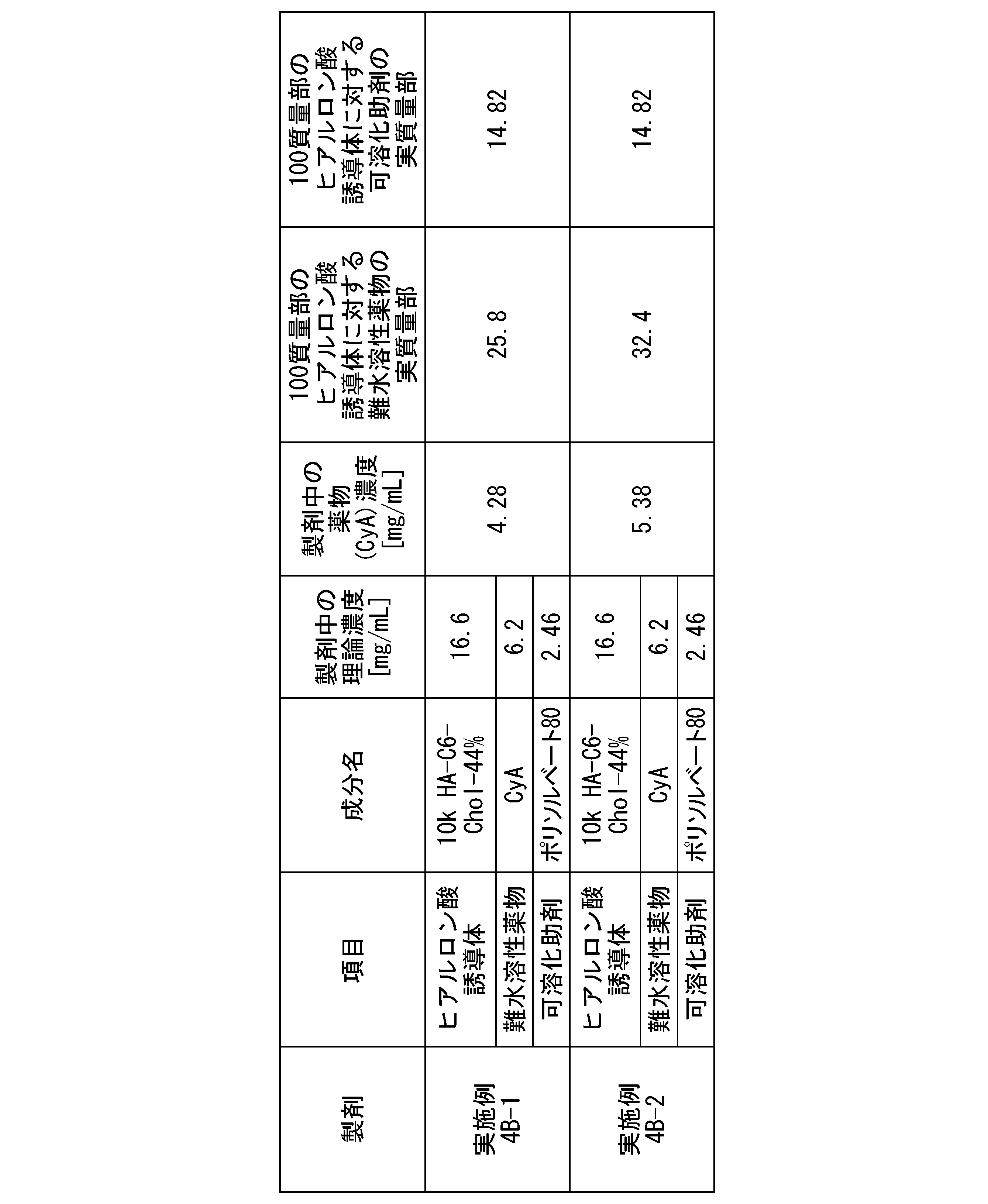
[実施例5B-1]
次に、難水溶性薬物の種類について検証を実施した。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を、濃度が36.0mg/mLとなる割合で、注射用水に溶解させた。
難水溶性薬物をパクリタキセルではなく、フルチカゾンプロピオン酸エステル(東京化成工業社製)に変更した以外は実施例5B-1と同様の手順で製剤調製を行った。
可溶化助剤を添加しない以外は、実施例5B-1と同様の手順で製剤調製を行った。
可溶化助剤を添加しない以外は、実施例5B-2と同様の手順で製剤調製を行った。
ヒアルロン酸誘導体を添加しない以外は、実施例5B-1と同様の手順で製剤調製を行った。
ヒアルロン酸誘導体を添加しない以外は、実施例5B-2と同様の手順で製剤調製を行った。
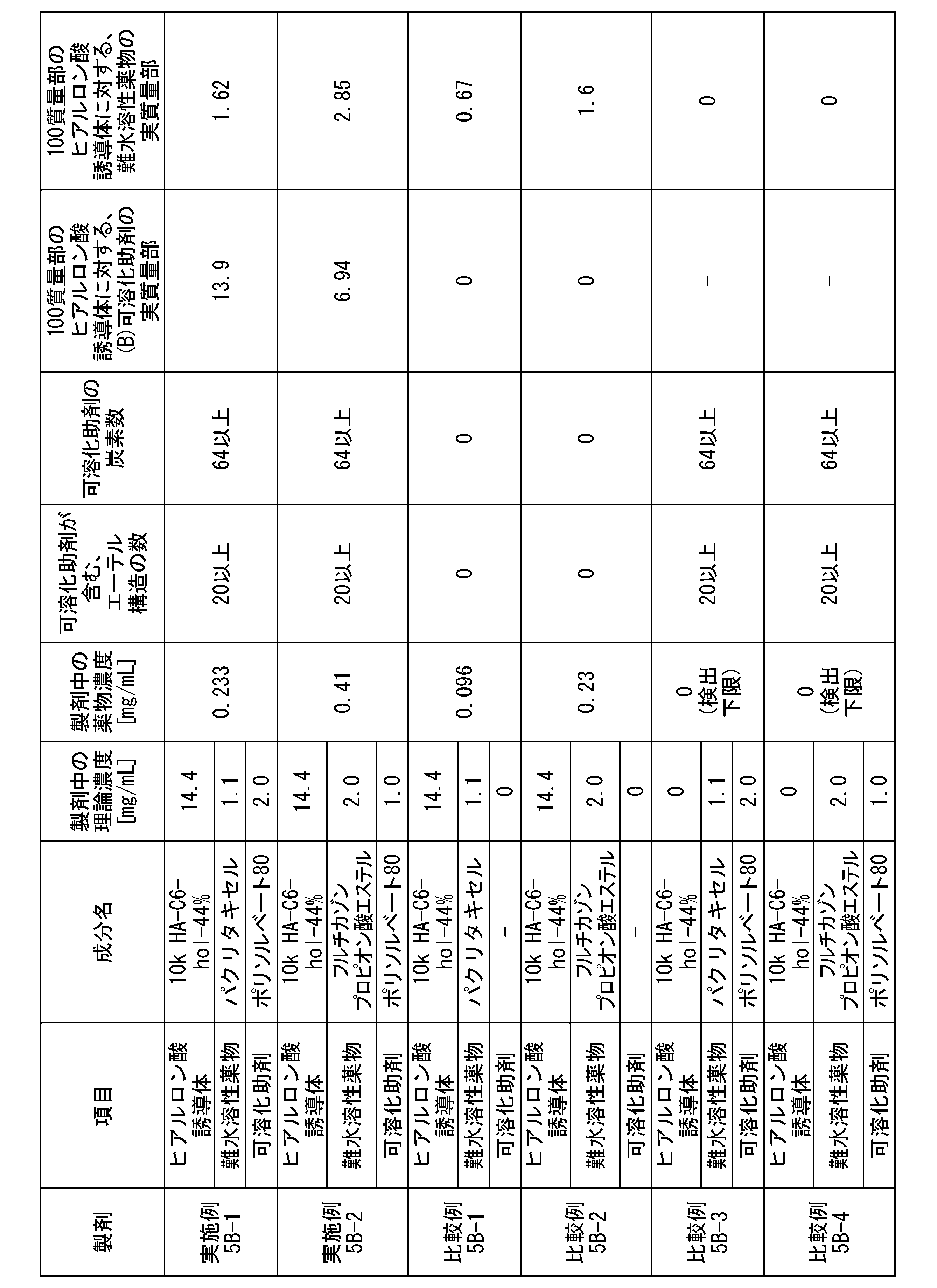
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、コレステロール-PEG600(Aldrich社製:C1145-250MG)を10mg/mLとなるよう注射用水で希釈した。
その後、別のバイアルに10mg/mLのコレステロール-PEG600を400μL添加し、バイアルにスターラーチップを入れて攪拌しながら、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加した。またさらに別のバイアルに、粉末のテムシロリムス(東京化成工業社製、製品番号:T3574)を5.0mg秤量し、上記可溶化助剤含有ヒアルロン酸誘導体を0.5mL添加し、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表X1に示した。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したテムシロリムスを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表25に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。
別のバイアルに、テムシロリムス(東京化成工業社製、 製品番号:T3574)を5.0mg秤量し、その後、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.30mL添加した。バイアルにスターラーチップを入れて、液量が0.50mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したテムシロリムスを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
バイアルに、テムシロリムス(東京化成工業社製、 製品番号:T3574)を5.0mg秤量し、その後、バイアルにスターラーチップを入れて、10mg/mLのコレステロール-PEG600を200μL添加し、液量が0.50mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したテムシロリムスを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表25に示す。

合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、コレステロール-PEG600(Aldrich社製:C1145-250MG)を10mg/mLとなるよう注射用水で希釈した。
その後、別のバイアルに10mg/mLのコレステロール-PEG600を400μL添加し、バイアルにスターラーチップを入れて攪拌しながら、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加した。またさらに別のバイアルに、粉末のCyAを4.0mg秤量し、を5.0mg秤量し、上記可溶化助剤含有ヒアルロン酸誘導体を0.5mL添加し、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表Y1に示した。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、コレステロール-PEG600(Aldrich社製:C1145-250MG)を10mg/mLとなるよう注射用水で希釈した。
その後、粉末のCyAを10.0mg秤量し、続いて上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加したのちに、バイアルにスターラーチップを入れて攪拌しながら、10mg/mLのコレステロール-PEG600水溶液を40μL添加し、注射用水を360μL加えた。その後、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表26に示した。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。別のバイアルに、HP-β-CD(東京化成工業社製:H0979)を10mg/mLとなるよう注射用水で希釈した。
その後、粉末のCyAを10.0mg秤量し、続いて上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加したのちに、バイアルにスターラーチップを入れて攪拌しながら、10mg/mLのHP-β-CD水溶液を272.2μL添加した。その後、注射用水を127.8μL添加し、24時間攪拌させて薬物の可溶化を行った。最終的な製剤組成を表Y1に示した。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
合成例1Bで得た凍結乾燥品のヒアルロン酸誘導体(10k HA-C6-Chol-44%)を36.0mg/mLで注射用水に溶解させた。
別のバイアルに、粉末のCyAを10.0mg秤量し、その後、上記36.0mg/mLのヒアルロン酸誘導体水溶液を0.60mL添加した。バイアルにスターラーチップを入れて、液量が1.00mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
バイアルに、粉末のCyAを10.0mg秤量し、その後、バイアルにスターラーチップを入れて、10mg/mLのコレステロール-PEG600を400μL添加し、液量が1.0mLになるよう注射用水を添加した。この後、24時間攪拌させて薬物の可溶化を行った。続いて、0.45μm滅菌ろ過フィルターでろ過し、析出したCyAを除去し、製剤中の薬物濃度を実施例1B-1と同様にHPLC測定にて定量した。ろ液は全て透明であることを確認した。
添加した量から算出される製剤中の理論濃度およびHPLC測定で定量した実際の製剤中の薬物濃度結果を表26に示す。
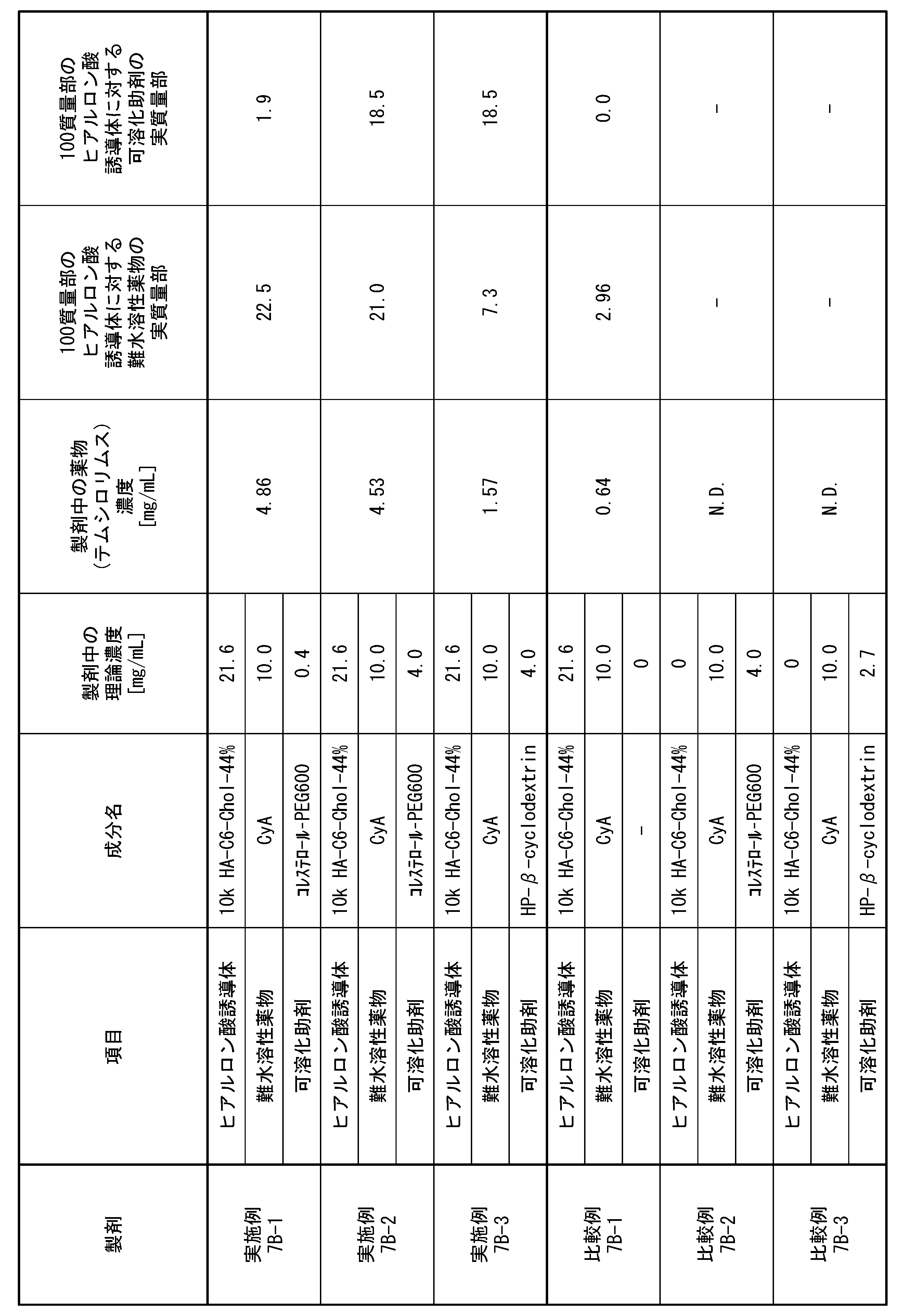
Claims (40)
- (A)疎水性基を導入したヒアルロン酸誘導体と、
(B)会合促進剤と、
(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物。 - 前記(B)会合促進剤はエーテル構造(R-O-R)を少なくとも4つ以上含み、かつ炭素数4以上を満たす、請求項1に記載のヒアルロン酸誘導体医薬組成物。
- 前記(B)会合促進剤が、ポリソルベート80、ポリソルベート20、ポロキサマー、オキシエチレンヒマシ油、ポリエチレングリコール300、ポリエチレングリコール400、ポリエチレングリコール4000、脂肪酸ソルビタンエステル、トコフェリルポリエチレングリコールスクシネート並びにポリビニルアルコールから成る群から選択される1種以上である、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- 生理塩濃度下で沈殿物が生じる、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- 前記(C)有効成分は、タンパク質または難水溶性薬物から選択される少なくとも1種である、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性薬物は水への溶解度が1mg/mL以下である請求項5に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性薬物は分子量200以上である請求項5に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性薬物は難水溶性ペプチドである、請求項5に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは、アミド結合を構成する窒素原子の少なくとも1つがメチル基を有する、請求項8に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは環状ペプチド、長鎖ペプチドから選択される少なくとも一つ以上を含む、請求項8に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは環状ペプチドである請求項8に記載のヒアルロン酸誘導体医薬組成物。
- 100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する前記(C)有効成分の含有量は、10質量部以上100質量部以下である、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- 前記(A)疎水性基を導入したヒアルロン酸誘導体は、下記一般式(I)で表される繰り返し単位を1以上有する、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。

X1は、-NRb-R、-NRb-COO-R、-NRb-CO-R、-NRb-CO-NRc-R、-COO-R、-O-COO-R、-S-R、-CO-Ya-S-R、-O-CO-Yb-S-R、-NRb-CO-Yb-S-R、及び-S-S-R、で表される基からなる群より選択される基である。
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Ra、Rb及びRcのアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよい。
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択される基である。Rfのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Rは、ステリル基である。
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-である。ここで、Yのアルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよい。
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Rgのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Yaは、C1-5アルキレンである。
Ybは、C2-8アルキレン又はC2-8アルケニレンである。
mは、1以上100以下の整数である。) - 前記ステリル基はコレステリル基である、請求項13に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体に対する前記ステリル基の導入率が7%以上35%未満である、請求項13に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体医薬組成物は目視で析出物が観察されない、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- ろ過滅菌可能である、請求項1又は2に記載のヒアルロン酸誘導体医薬組成物。
- (A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法であって、
前記(A)疎水性基を導入したヒアルロン酸誘導体と、前記(B)会合促進剤とを混合し、会合促進剤含有ヒアルロン酸誘導体水溶液を得る工程と、
前記(C)有効成分と前記会合促進剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法。 - (A)疎水性基を導入したヒアルロン酸誘導体と、(B)会合促進剤と、(C)有効成分と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法であって、
前記(C)有効成分を前記(B)会合促進剤に分散させ、分散液(I)を得る工程と、 ヒアルロン酸誘導体水溶液又は会合促進剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、
前記分散液(I)と水溶液(II)とを混合する工程と、を含む、ヒアルロン酸誘導体医薬組成物の製造方法。 - 有機溶剤を除去する工程を含まない、請求項18又は19に記載のヒアルロン酸誘導体医薬組成物の製造方法。
- (A)疎水性基を導入したヒアルロン酸誘導体と、
(B)可溶化助剤と、
(C)有効成分と、を含み、
前記(B)可溶化助剤は、エーテル構造(R-O-R)を少なくとも4つ以上含み、かつ炭素数4以上を満たし、
100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記(B)可溶化助剤の含有量が0.0001質量部以上15000質量部以下である、ヒアルロン酸誘導体医薬組成物。 - 前記(B)可溶化助剤が、非イオン界面活性剤、分子量190g/moL以上4000g/moL以下のポリエチレングリコール、シクロデキストリン誘導体、から成る群から選択される1種以上である、請求項21に記載のヒアルロン酸誘導体医薬組成物。
- 前記(B)可溶化助剤が、ポリソルベート80、ポリソルベート65、ポリソルベート60、ポリソルベート40、ポリソルベート20、ポロキサマー、ポリオキシエチレン硬化ヒマシ油、シクロデキストリン誘導体、ポリエチレングリコール300、ポリエチレングリコール400、ポリエチレングリコール4000、並びにトコフェリルポリエチレングリコールスクシネートから成る群から選択される1種以上である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。
- 前記(B)可溶化助剤が非イオン界面活性剤であり、
100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記非イオン界面活性剤の含有量が0.0001質量部以上150質量部以下である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。 - 前記(B)可溶化助剤が分子量190g/moL以上4000g/moL以下のポリエチレングリコールであり、
100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記ポリエチレングリコールの含有量が25質量部以上15000質量部以下である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。 - 前記(C)有効成分は、水への溶解度が1mg/mL以下の難水溶性薬物である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性薬物は分子量が200以上である、請求項26に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性薬物は難水溶性ペプチドである、請求項26に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは、アミド結合を構成する窒素原子の少なくとも1つがメチル基を有する、請求項28に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは、環状ペプチド及び長鎖ペプチドから選択される少なくとも一つ以上を含む、請求項28に記載のヒアルロン酸誘導体医薬組成物。
- 前記難水溶性ペプチドは環状ペプチドである、請求項28に記載のヒアルロン酸誘導体医薬組成物。
- 100質量部の前記(A)疎水性基を導入したヒアルロン酸誘導体に対する、前記難水溶性薬物の配合量は、21質量部以上100質量部未満である、請求項26に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体は、下記一般式(I)で表される繰り返し単位を1以上有する、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。
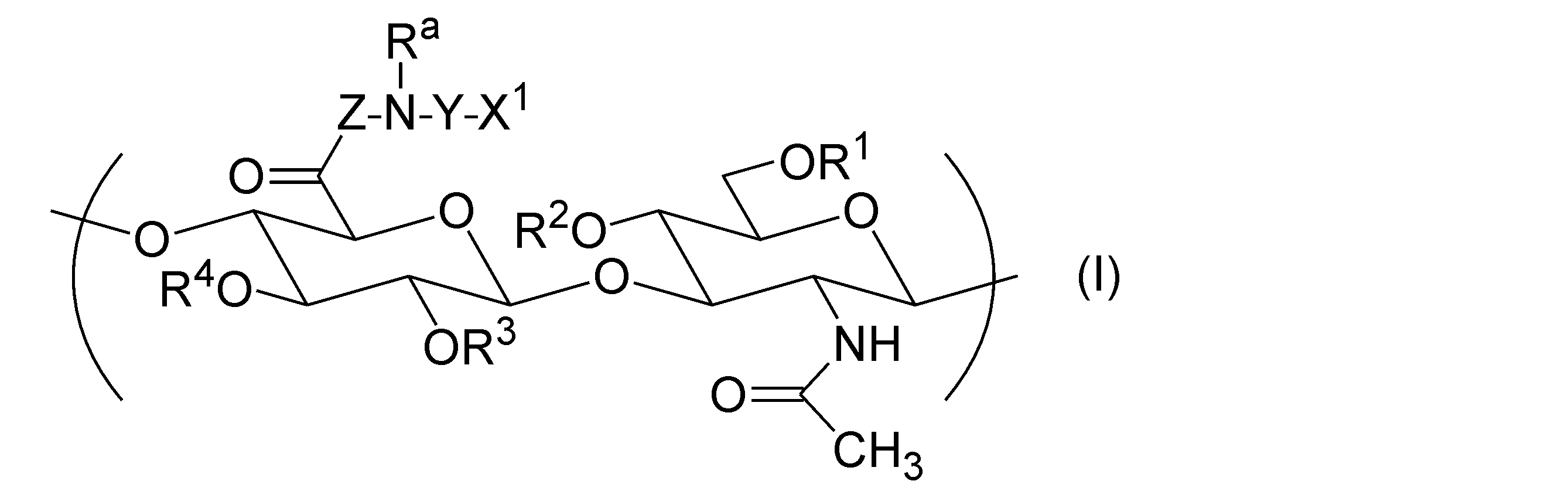
X1は、-NRb-R、-NRb-COO-R、-NRb-CO-R、-NRb-CO-NRc-R、-COO-R、-O-COO-R、-S-R、-CO-Ya-S-R、-O-CO-Yb-S-R、-NRb-CO-Yb-S-R、及び-S-S-R、で表される基からなる群より選択される基である。
Ra、Rb及びRcは、それぞれ独立に、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Ra、Rb及びRcのアルキル部分は、-O-及び-NRf-からなる群より選択される基が挿入されていてもよい。
Rfは、水素原子、C1-12アルキル、アミノC2-12アルキル及びヒドロキシC2-12アルキルからなる群より選択される基である。Rfのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Rは、ステリル基である。
Yは、C2-30アルキレン、又は-(CH2CH2O)m-CH2CH2-である。ここで、Yのアルキレンは、-O-、-NRg-及び-S-S-からなる群より選択される基が挿入されていてもよい。
Rgは、水素原子、C1-20アルキル、アミノC2-20アルキル及びヒドロキシC2-20アルキルからなる群より選択される基である。Rgのアルキル部分は-O-及び-NH-からなる群より選択される基が挿入されていてもよい。
Yaは、C1-5アルキレンである。
Ybは、C2-8アルキレン又はC2-8アルケニレンである。
mは、1以上100以下の整数である。) - 前記ステリル基がコレステリル基である、請求項33に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体に対する前記ステリル基の導入率が35%以上50%未満である、請求項33に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体医薬組成物に対する前記ヒアルロン酸誘導体の含有量が6mg/mL以上45mg/mL未満である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。
- 前記ヒアルロン酸誘導体医薬組成物に対する有機溶剤の含有量が0.8%未満である、請求項21又は22に記載のヒアルロン酸誘導体医薬組成物。
- (A)疎水性基を導入したヒアルロン酸誘導体と、(B)可溶化助剤と、(C)有効成分と、を含有する、医薬組成物の製造方法であって、
(A)疎水性基を導入したヒアルロン酸誘導体と、(B)可溶化助剤とを混合し、可溶化助剤含有ヒアルロン酸誘導体水溶液を得る工程と、
前記(C)有効成分を前記可溶化助剤含有ヒアルロン酸誘導体水溶液と混合する混合工程と、を含む、医薬組成物の製造方法。 - (A)疎水性基を導入したヒアルロン酸誘導体と、(B)可溶化助剤と、(C)有効成分と、を含有する、医薬組成物の製造方法であって、
前記(C)有効成分を前記(B)可溶化助剤に分散させ、分散液(I)を得る工程と、 ヒアルロン酸誘導体水溶液又は可溶化剤含有ヒアルロン酸水溶液を調製し、水溶液(II)を得る工程を含み、
前記分散液(I)と前記水溶液(II)とを混合する工程と、を含む、医薬組成物の製造方法。 - (A)疎水性基を導入したヒアルロン酸誘導体と、(B)可溶化助剤と、(C)有効成分と、を含有する、医薬組成物の製造方法であって、
有機溶剤を除去する工程を含まないことを特徴とする、請求項38又は39に記載の医薬組成物の製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024535139A JPWO2024019127A1 (ja) | 2022-07-20 | 2023-07-20 | |
| KR1020257001420A KR20250023547A (ko) | 2022-07-20 | 2023-07-20 | 히알루론산 유도체 의약 조성물 및 의약 조성물의 제조 방법 |
| EP23843050.8A EP4559484A4 (en) | 2022-07-20 | 2023-07-20 | PHARMACEUTICAL COMPOSITION BASED ON A HYALURONIC ACID DERIVATIVE, AND PROCESS FOR PRODUCING THE PHARMACEUTICAL COMPOSITION |
| CN202380054267.6A CN119584989A (zh) | 2022-07-20 | 2023-07-20 | 透明质酸衍生物药物组合物以及药物组合物的制造方法 |
| CA3262457A CA3262457A1 (en) | 2022-07-20 | 2023-07-20 | Hyaluronic acid derivative pharmaceutical composition and method of producing pharmaceutical composition |
| US18/995,468 US20250360217A1 (en) | 2022-07-20 | 2023-07-20 | Hyaluronic acid derivative pharmaceutical composition and method of producing pharmaceutical composition |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022-115841 | 2022-07-20 | ||
| JP2022-115839 | 2022-07-20 | ||
| JP2022115839 | 2022-07-20 | ||
| JP2022115841 | 2022-07-20 | ||
| JP2022151823 | 2022-09-22 | ||
| JP2022-151823 | 2022-09-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024019127A1 true WO2024019127A1 (ja) | 2024-01-25 |
Family
ID=89617852
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/026679 Ceased WO2024019127A1 (ja) | 2022-07-20 | 2023-07-20 | ヒアルロン酸誘導体医薬組成物及び医薬組成物の製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20250360217A1 (ja) |
| EP (1) | EP4559484A4 (ja) |
| JP (1) | JPWO2024019127A1 (ja) |
| KR (1) | KR20250023547A (ja) |
| CN (1) | CN119584989A (ja) |
| CA (1) | CA3262457A1 (ja) |
| TW (1) | TWI868812B (ja) |
| WO (1) | WO2024019127A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025216306A1 (ja) * | 2024-04-11 | 2025-10-16 | 旭化成株式会社 | 担体組成物 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN120899772A (zh) * | 2025-09-26 | 2025-11-07 | 中国热带农业科学院农产品加工研究所 | 一种抗血栓的微生物制剂及其制备方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001316284A (ja) * | 2000-04-07 | 2001-11-13 | Lab Medidom Sa | 眼科用製剤 |
| JP2009079043A (ja) * | 2007-09-04 | 2009-04-16 | Rohto Pharmaceut Co Ltd | 線維芽細胞増殖促進能を有する組成物 |
| WO2010053140A1 (ja) | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| JP4758893B2 (ja) | 2003-06-26 | 2011-08-31 | 参天製薬株式会社 | キノロンを含む眼科用組成物及びその使用の方法 |
| WO2017164409A1 (ja) | 2016-03-25 | 2017-09-28 | 国立大学法人大阪大学 | 疾患の要因となる生体内タンパク質を標的とするコンジュゲートワクチン |
| JP6271672B2 (ja) | 2011-04-12 | 2018-01-31 | ノヴォ ノルディスク アー/エス | 二重アシル化されたglp−1誘導体 |
| US20200237859A1 (en) | 2019-01-25 | 2020-07-30 | Newport Research, Inc. | Aqueous suspensions of cyclosporin |
| JP2022044579A (ja) * | 2020-09-07 | 2022-03-17 | 旭化成株式会社 | 点眼剤 |
| JP2022115839A (ja) | 2021-01-28 | 2022-08-09 | ケミプロ化成株式会社 | 新規な1,10-フェナントロリン化合物を用いた有機エレクトロルミネッセンス素子、及び有機エレクトロルミネッセンス照明 |
| JP2022115841A (ja) | 2021-01-28 | 2022-08-09 | 三菱マテリアル株式会社 | 炭素材料の製造方法、炭素材料、二酸化炭素の分解方法、還元剤 |
| JP2022151823A (ja) | 2021-03-26 | 2022-10-07 | 関西ペイント株式会社 | 低光沢塗料組成物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140067052A (ko) | 2011-09-09 | 2014-06-03 | 암젠 인코퍼레이티드 | 식도암 및 위암 환자들에서 항-간세포 성장 인자(“hgf”) 항체들의 유효성을 예측하기 위한 c―met 단백질의 용도 |
| CN104888224B (zh) * | 2015-03-25 | 2019-02-01 | 中山大学 | 一种两亲性多糖衍生物/泊洛沙姆温敏型原位水凝胶及其制备方法 |
-
2023
- 2023-07-20 WO PCT/JP2023/026679 patent/WO2024019127A1/ja not_active Ceased
- 2023-07-20 KR KR1020257001420A patent/KR20250023547A/ko active Pending
- 2023-07-20 JP JP2024535139A patent/JPWO2024019127A1/ja active Pending
- 2023-07-20 CN CN202380054267.6A patent/CN119584989A/zh active Pending
- 2023-07-20 EP EP23843050.8A patent/EP4559484A4/en active Pending
- 2023-07-20 TW TW112127200A patent/TWI868812B/zh active
- 2023-07-20 CA CA3262457A patent/CA3262457A1/en active Pending
- 2023-07-20 US US18/995,468 patent/US20250360217A1/en active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001316284A (ja) * | 2000-04-07 | 2001-11-13 | Lab Medidom Sa | 眼科用製剤 |
| JP4758893B2 (ja) | 2003-06-26 | 2011-08-31 | 参天製薬株式会社 | キノロンを含む眼科用組成物及びその使用の方法 |
| JP2009079043A (ja) * | 2007-09-04 | 2009-04-16 | Rohto Pharmaceut Co Ltd | 線維芽細胞増殖促進能を有する組成物 |
| WO2010053140A1 (ja) | 2008-11-05 | 2010-05-14 | 国立大学法人 東京医科歯科大学 | ヒアルロン酸誘導体、およびその医薬組成物 |
| JP6271672B2 (ja) | 2011-04-12 | 2018-01-31 | ノヴォ ノルディスク アー/エス | 二重アシル化されたglp−1誘導体 |
| WO2017164409A1 (ja) | 2016-03-25 | 2017-09-28 | 国立大学法人大阪大学 | 疾患の要因となる生体内タンパク質を標的とするコンジュゲートワクチン |
| US20200237859A1 (en) | 2019-01-25 | 2020-07-30 | Newport Research, Inc. | Aqueous suspensions of cyclosporin |
| JP2022044579A (ja) * | 2020-09-07 | 2022-03-17 | 旭化成株式会社 | 点眼剤 |
| JP2022115839A (ja) | 2021-01-28 | 2022-08-09 | ケミプロ化成株式会社 | 新規な1,10-フェナントロリン化合物を用いた有機エレクトロルミネッセンス素子、及び有機エレクトロルミネッセンス照明 |
| JP2022115841A (ja) | 2021-01-28 | 2022-08-09 | 三菱マテリアル株式会社 | 炭素材料の製造方法、炭素材料、二酸化炭素の分解方法、還元剤 |
| JP2022151823A (ja) | 2021-03-26 | 2022-10-07 | 関西ペイント株式会社 | 低光沢塗料組成物 |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP4559484A4 |
| TOXICOLOGY, vol. 216, 2005, pages 106 - 121 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2025216306A1 (ja) * | 2024-04-11 | 2025-10-16 | 旭化成株式会社 | 担体組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4559484A4 (en) | 2025-11-26 |
| US20250360217A1 (en) | 2025-11-27 |
| JPWO2024019127A1 (ja) | 2024-01-25 |
| EP4559484A1 (en) | 2025-05-28 |
| TW202412822A (zh) | 2024-04-01 |
| CA3262457A1 (en) | 2025-06-12 |
| KR20250023547A (ko) | 2025-02-18 |
| CN119584989A (zh) | 2025-03-07 |
| TWI868812B (zh) | 2025-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Guan et al. | N-trimethyl chitosan nanoparticle-encapsulated lactosyl-norcantharidin for liver cancer therapy with high targeting efficacy | |
| WO2024019127A1 (ja) | ヒアルロン酸誘導体医薬組成物及び医薬組成物の製造方法 | |
| CN102037058B (zh) | 多西紫杉醇高分子衍生物、及其制造方法及其用途 | |
| US20170267727A1 (en) | Conjugates of pH Low Insertion Peptide and Monomethyl Auristatins in the Treatment of Solid Tumors | |
| Kim et al. | Bioimaging and pulmonary applications of self-assembled Flt1 peptide–hyaluronic acid conjugate nanoparticles | |
| KR20220020821A (ko) | 이중 glp1/2 아고니스트의 약학적 비경구 조성물 | |
| JP7818130B2 (ja) | ヒアルロン酸誘導体、医薬組成物、及び医薬組成物の製造方法 | |
| EP4237012A1 (en) | Fap-activated serum extended half-life therapeutic conjugates | |
| CN100544769C (zh) | 包含喜树碱衍生物的液体制剂以及冻干该制剂制得的药物组合物 | |
| WO2024041535A1 (zh) | 纳米组合物及其制备方法和用途 | |
| KR102270619B1 (ko) | 고분자화 약물 함유 약제학적 조성물 | |
| WO2022242527A1 (zh) | 一种单克隆抗体-细胞因子融合蛋白制剂 | |
| ITMI20111866A1 (it) | Polietilenglicoli modificati e loro complessi supramolecolari con macromolecole biologicamente attive | |
| US20260000611A1 (en) | Hyaluronic acid derivative pharmaceutical composition and pharmaceutical composition | |
| Cheng et al. | Synthesis, characterization and in vivo activity of salmon calcitonin coconjugated with lipid and polyethylene glycol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23843050 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024535139 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20257001420 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020257001420 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18995468 Country of ref document: US Ref document number: 202380054267.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202517003913 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257001420 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023843050 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023843050 Country of ref document: EP Effective date: 20250220 |
|
| WWP | Wipo information: published in national office |
Ref document number: 202380054267.6 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023843050 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 18995468 Country of ref document: US |