WO2024135609A1 - 核酸合成用リンカー、及び担体、並びにそれらの製造方法 - Google Patents
核酸合成用リンカー、及び担体、並びにそれらの製造方法 Download PDFInfo
- Publication number
- WO2024135609A1 WO2024135609A1 PCT/JP2023/045266 JP2023045266W WO2024135609A1 WO 2024135609 A1 WO2024135609 A1 WO 2024135609A1 JP 2023045266 W JP2023045266 W JP 2023045266W WO 2024135609 A1 WO2024135609 A1 WO 2024135609A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- nucleic acid
- solvent
- substitutable position
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/11—Compounds covalently bound to a solid support
Definitions
- the present invention relates to a linker and carrier for nucleic acid synthesis, and a method for producing the same.
- a solid support is used to which a nucleoside is bound via a linker that is cleaved under basic conditions such as aqueous ammonia, etc.
- a linker that is cleaved under basic conditions such as aqueous ammonia, etc.
- a succinyl group is widely used, and a support for solid phase nucleic acid synthesis is used in which the 3'-hydroxyl group of the nucleoside is ester-bonded to the solid support.
- nucleic acids there are eight types: DNA (thymine: T, adenine: A, cytosine: C, guanine: G) and RNA (uracil: U, adenine: A, cytosine: C, guanine: G).
- DNA thymine: T
- adenine: A adenine: A
- cytosine: C guanine: G
- RNA uracil: U, adenine: A, cytosine: C, guanine: G
- Patent documents 1 and 2 disclose a linker for solid-phase synthesis of nucleic acid and a support for solid-phase synthesis of nucleic acid.
- Patent document 3 discloses universal building blocks and supports for use in the preparation of oligomeric compounds.
- Non-Patent Document 1 discloses cleavable spacer (CS) derivatives for tandem synthesis of multiple oligonucleotides in a single column.
- the present invention aims to provide a new linker for nucleic acid synthesis and a support for solid-phase nucleic acid synthesis.
- the inventors have developed a new universal support that can synthesize oligonucleotides with high purity and efficiency without generating by-products derived from non-nucleoside linkers.
- the present invention includes the following linkers for nucleic acid synthesis, supports for solid-phase nucleic acid synthesis, and methods for producing these.
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, at a substitutable position, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group, which may be the same or different.
- a method for producing a linker precursor for nucleic acid synthesis represented by the following formula: In a solvent, In the presence of an oxidizing agent and a reoxidizing agent, The following general formula (1):
- a method for producing a linker precursor for nucleic acid synthesis comprising a step of subjecting a compound represented by the following formula (1):
- Item 2 The method according to item 1, wherein the oxidizing agent is at least one oxidizing agent selected from the group consisting of osmium tetroxide (OsO 4 ) and ozone (O 3 ).
- the oxidizing agent is at least one oxidizing agent selected from the group consisting of osmium tetroxide (OsO 4 ) and ozone (O 3 ).
- Item 3. The method according to item 1, wherein the reoxidizing agent is at least one reoxidizing agent selected from the group consisting of N-methylmorpholine-N-oxide (NMO), trimethylamine-N-oxide (Me 3 NO), and tert-butyl hydroperoxide (TBHP).
- NMO N-methylmorpholine-N-oxide
- Me 3 NO trimethylamine-N-oxide
- TBHP tert-butyl hydroperoxide
- Item 4. The method according to item 1, wherein the solvent is a mixed solvent of water and a hydrophilic organic solvent.
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, at a substitutable position, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group, which may be the same or different.
- a linker precursor for nucleic acid synthesis represented by the formula:
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- a method for producing a linker precursor for nucleic acid synthesis represented by the following formula: In a solvent, The following general formula (2):
- a and B are the same as those in formula (3).
- a compound represented by the formula: A method for producing a linker precursor for nucleic acid synthesis, comprising a step of reacting with 4,4'-dimethoxytrityl chloride (DMTr-Cl), 4-methoxytrityl chloride (MMTr-Cl), or trityl chloride (Tr-Cl).
- DMTr-Cl 4,4'-dimethoxytrityl chloride
- MMTr-Cl 4-methoxytrityl chloride
- Tr-Cl trityl chloride
- Item 7. The method according to item 6, wherein the solvent is at least one organic solvent selected from the group consisting of aprotic polar solvents.
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- a linker precursor for nucleic acid synthesis represented by the formula:
- L is present at one substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for a hydroxyl group.
- a method for producing a linker for nucleic acid synthesis represented by the following formula: The following general formula (3):
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B and Prot 1 are the same as those in formula (4).
- a method for producing a linker for nucleic acid synthesis comprising:
- Item 11 The method according to item 9, wherein the solvent used in step (2) is at least one solvent selected from the group consisting of aprotic polar solvents and ether solvents.
- L is present at one substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for a hydroxyl group.
- a linker for nucleic acid synthesis represented by the formula:
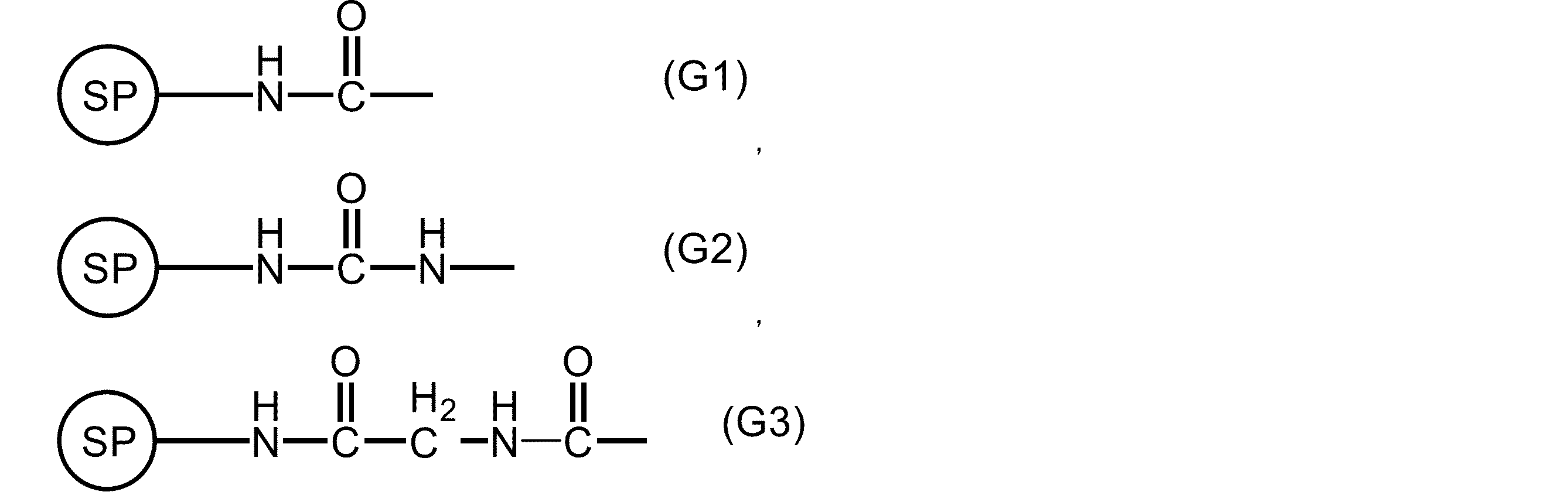
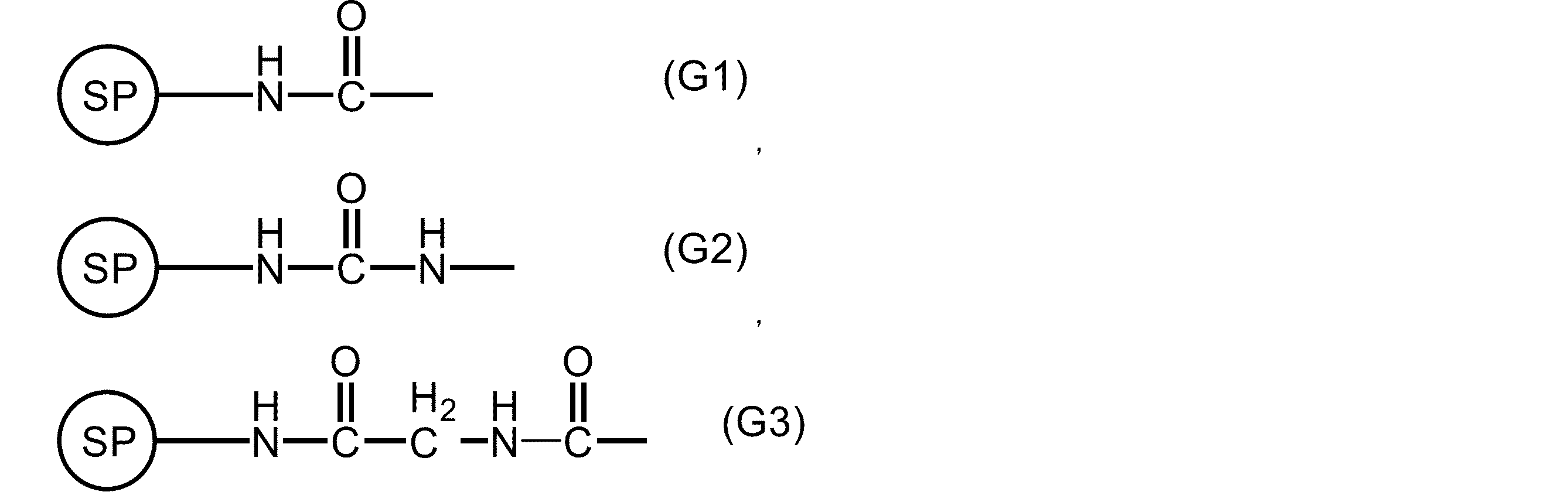
- G is present at one substitutable position and represents the following formula (G1), (G2), or (G3).
- SP represents a solid phase carrier.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for the hydroxyl group.
- a method for producing a support for solid-phase synthesis of nucleic acid comprising the steps of: In a solvent, In the presence of a coupling agent, The following general formula (4):
- L is present at one substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- SP solid phase carrier
- the coupling agent is 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide hexafluorophosphate (HATU), 1-[bis(dimethylamino)methylene]-1H-benzotriazolium-3-oxide hexafluorophosphate (HBTU), O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxide Tetrafluoroborate (TATU), 1-[bis(dimethylamino)methylene]-1H-benzotriazolium-3-oxide tetrafluoroborate (TBTU), N,N'-dicyclohexylcarbodiimide (DCC), 1-e
- the coupling agent is at least one selected from the group consisting of benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate (BOP), 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), and 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP).
- BOP benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate
- PyBOP 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- PyBOP 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- Item 15 The method according to item 13, wherein the solvent is at least one organic solvent selected from the group consisting of aprotic polar solvents and halogenated solvents.
- G is present at one substitutable position and represents the following formula (G1), (G2), or (G3).
- SP represents a solid phase carrier.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for a hydroxyl group.
- the nucleic acid synthesis linker of the present invention can be used as a support for solid-phase nucleic acid synthesis.
- the linker for nucleic acid synthesis of the present invention has no cleavable bond (ester bond) and has a non-cleavable bond (amide bond) between the linker structures (between the solid phase support and the linker structure) under conditions (such as ammonia water) for cleaving the universal linker (compound represented by general formula (4)) from the solid phase support (SP).
- nucleic acid synthesis linker and nucleic acid solid-phase synthesis support of the present invention When used, they have a bond (amide bond) that does not cleave during the excision step under basic conditions (ammonia water, etc.) in the solid-phase synthesis of nucleic acids, suppressing the production of by-products (linker adducts) that occur when conventional universal linkers are used, making it possible to provide highly pure nucleic acids.
- the linker for nucleic acid synthesis of the present invention has a bicyclic structure that promotes nucleophilic attack by the adjacent hydroxyl group on the phosphate group, allowing the oligonucleic acid to be released from the solid phase support with high efficiency.
- the present invention provides a new linker for nucleic acid synthesis and a support for solid-phase nucleic acid synthesis.
- a method for producing a linker precursor 1 for nucleic acid synthesis according to the present invention comprising the steps of:
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, at a substitutable position, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group, which may be the same or different.
- a method for producing a linker precursor for nucleic acid synthesis represented by the following formula: In a solvent, In the presence of an oxidizing agent and a reoxidizing agent, The following general formula (1):
- the method includes subjecting a compound represented by the following formula (1) to an oxidation reaction.
- the method includes subjecting a compound represented by the following formula (1):
- a linker precursor for nucleic acid synthesis is produced, which is represented by the formula:
- one A is present at a substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- R is preferably a linear, branched or cyclic alkyl group, specifically, for example, a linear or branched alkyl group having 1 to 4 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, 1-ethylpropyl, etc., a benzyl group (C 6 H 5 CH 2 -), etc.
- R is more preferably a linear or branched alkyl group having 1 to 4 carbon atoms (lower alkyl, more preferably a methyl group), a benzyl group, etc.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- the lower alkyl group is preferably a linear, branched or cyclic alkyl group, specifically, for example, a linear or branched alkyl group having 1 to 4 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, 1-ethylpropyl, etc.
- the lower alkoxy group is preferably a straight-chain or branched-chain alkoxy group having 1 to 4 carbon atoms, such as methoxy, ethoxy, n-propoxy, isopropyloxy, n-butoxy, isobutyloxy, sec-butoxy, tert-butoxy, or 1-ethylpropyloxy.
- the lower alkoxy group is preferably methoxy or tert-butoxy.
- the dialkylamino group is preferably a dialkylamino group having two linear or branched alkyl groups each having 1 to 2 carbon atoms, such as a dimethylamino group, a diethylamino group, or an ethylmethylamino group.
- the solvent is preferably a mixed solvent of water and a hydrophilic organic solvent.
- the hydrophilic organic solvent is preferably a hydrophilic organic solvent such as an aprotic polar solvent, an ether solvent, or an ester solvent.
- the aprotic polar solvent is preferably acetone, acetonitrile (CH 3 CN), N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), pyridine, or the like.
- the ether solvent is preferably tetrahydrofuran (THF), dioxane, diethyl ether (Et 2 O), isopropyl ether (IPE), or the like.
- the ester solvent is preferably ethyl acetate (AcOEt) or the like.
- the hydrophilic organic solvent is more preferably an aprotic polar solvent such as acetone, acetonitrile, DMF, or DMSO, or an ether solvent (polar solvent) such as THF, dioxane, or diethyl ether.
- aprotic polar solvent such as acetone, acetonitrile, DMF, or DMSO
- ether solvent such as THF, dioxane, or diethyl ether.
- the hydrophilic organic solvent is more preferably a mixed solvent of water and acetonitrile.
- the amount of hydrophilic solvent used is adjusted so that the concentration (molar concentration M (mol/L)) of the compound represented by general formula (1) (substrate) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
- the oxidizing agent is preferably at least one oxidizing agent selected from the group consisting of osmium tetroxide (OsO 4 ) and ozone (O 3 ).
- an oxidizing agent is used to advance an oxidation reaction, and a hydroxyl group is added to the compound (substrate) represented by general formula (1).
- the amount of oxidizing agent used is adjusted so that the concentration in the solvent (molar concentration M (mol/L)) is preferably 0.01M to 5M, and more preferably 0.1M to 2M.
- the amount of oxidizing agent used is adjusted so that the concentration relative to the compound represented by general formula (1) (substrate) (when the substrate is taken as 100 mol%) is preferably 0.01 mol% to 100 mol%, more preferably 0.1 mol% to 50 mol%, and even more preferably 1 mol% to 10 mol% in molar ratio.
- the reoxidant is preferably at least one reoxidant selected from the group consisting of N-methylmorpholine-N-oxide (NMO), trimethylamine-N-oxide ( Me3NO ), and tert-butyl hydroperoxide (TBHP).
- NMO N-methylmorpholine-N-oxide
- Me3NO trimethylamine-N-oxide
- TBHP tert-butyl hydroperoxide
- a reoxidizing agent is used to advance the oxidation reaction, and a hydroxyl group is added to the compound (substrate) represented by general formula (1).
- the amount of reoxidizing agent used is adjusted so that the concentration in the solvent (molar concentration M (mol/L)) is preferably 0.01M to 5M, and more preferably 0.1M to 2M.
- the amount of reoxidizing agent used is adjusted so that the concentration relative to the compound represented by general formula (1) (substrate) (when the substrate is taken as 100 mol%) is preferably 50 mol% to 10,000 mol%, more preferably 90 mol% to 1000 mol%, and even more preferably 100 mol% to 500 mol% in molar ratio.
- the reaction temperature of the oxidation reaction is preferably 0°C to 100°C, more preferably 10°C to 80°C, and even more preferably 20°C to 60°C.
- the reaction time for the oxidation reaction is preferably 0.1 to 100 hours, more preferably 0.1 to 50 hours, and even more preferably 0.1 to 10 hours.
- a linker precursor for nucleic acid synthesis 1 represented by general formula (2) The present invention relates to The following general formula (2):
- a method for producing a linker precursor 2 for nucleic acid synthesis comprising the steps of:
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- a method for producing a linker precursor for nucleic acid synthesis represented by the following formula: In a solvent, The following general formula (2):
- a compound represented by the formula: The method includes reacting with 4,4'-dimethoxytrityl chloride (DMTr-Cl), 4-methoxytrityl chloride (MMTr-Cl), or trityl chloride (Tr-Cl).
- DMTr-Cl 4,4'-dimethoxytrityl chloride
- MMTr-Cl 4-methoxytrityl chloride
- Tr-Cl trityl chloride
- DMTr-Cl 4,4'-dimethoxytrityl chloride
- MMTr-Cl 4-methoxytrityl chloride
- Tr-Cl trityl chloride
- a linker precursor for nucleic acid synthesis is produced, which is represented by the formula:
- Prot 1 represents a 4,4'-dimethoxytrityl group (DMTr group), a 4-methoxytrityl group (MMTr group), or a trityl group (Tr group).
- DMTr group 4,4'-dimethoxytrityl group
- MMTr group 4-methoxytrityl group
- Tr group trityl group
- the solvent is preferably at least one organic solvent selected from the group consisting of aprotic polar solvents.
- the aprotic polar solvent is more preferably acetone, acetonitrile (CH 3 CN), N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), pyridine, tetrahydrofuran (THF), or the like.
- the organic solvent is more preferably pyridine.
- the amount of solvent used is adjusted so that the concentration (molar concentration M (mol/L)) of the compound represented by general formula (2) (substrate) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
- DMTr-Cl 4,4'-dimethoxytrityl chloride
- MMTr-Cl 4-methoxytrityl chloride
- Tr-Cl trityl chloride
- the amount of DMTr-Cl, MMTr-Cl, or Tr-Cl used (hydroxyl group protection reaction) is adjusted so that the concentration in the solvent (molar concentration M (mol/L)) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
- the amount of DMTr-Cl, MMTr-Cl, or Tr-Cl used (hydroxyl group protection reaction) is adjusted so that the concentration relative to the compound represented by general formula (2) (substrate) (when the substrate is taken as 100 mol%) is preferably 0.01 mol% to 100 mol%, more preferably 0.1 mol% to 50 mol%, and even more preferably 1 mol% to 10 mol% in molar ratio.
- the reaction temperature for the hydroxyl protection reaction is preferably 0°C to 100°C, more preferably 10°C to 80°C, and even more preferably 20°C to 60°C.
- the reaction time for the hydroxyl group protection reaction is preferably 0.1 to 100 hours, more preferably 0.1 to 50 hours, and even more preferably 0.1 to 20 hours.
- Prot 1 represents a 4,4'-dimethoxytrityl group (DMTr group), a 4-methoxytrityl group (MMTr group), or a trityl group (Tr group).
- DMTr group 4,4'-dimethoxytrityl group
- MMTr group 4-methoxytrityl group
- Tr group trityl group
- the DMTr group in Prot 1 is derived from DMTr-Cl used in the protection reaction of the hydroxyl group.
- the MMTr group in Prot 1 is derived from MMTr-Cl used in the protection reaction of the hydroxyl group.
- Tr group in Prot 1 is derived from Tr-Cl used in the protection reaction of the hydroxyl group.
- a method for producing a linker for nucleic acid synthesis according to the present invention comprising the following general formula (4):
- L is present at one substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for a hydroxyl group.
- the method for producing a linker for nucleic acid synthesis represented by the following formula: The following general formula (3):
- A is present at one substitutable position and represents a -CO-OR group, a -NH-CO-OR group, or a -CO-NH-CH 2 -CO-OR group.
- R represents a protecting group for a carboxylic acid.
- B and Prot 1 are the same as those in formula (4).
- (1) a step of hydrolyzing in a solvent, followed by (2) A step of protecting a hydroxyl group in a solvent is included.
- the present invention produces a linker for nucleic acid synthesis represented by the formula:
- A, B, and Prot 1 are as defined above.
- one L is present at a substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- Prot 2 represents a protecting group for a hydroxyl group.
- Step (1) of hydrolyzing a compound (substrate) represented by general formula (3) in a solvent a hydrolysis reaction is carried out on a compound (substrate) represented by general formula (3) in a solvent (1), and A (-CO-OR, -NH-CO-OR, or -CO-NH- CH2 -CO-OR; R: carboxylic acid protecting group) in formula (3) is hydrolyzed to L (-CO-OH, -NH-CO-OH, or -CO-NH- CH2 -CO-OH) in formula (4).
- the solvent used in step (1) is preferably at least one solvent selected from the group consisting of an alkaline aqueous solution, an ether solvent, an alcohol solvent, and a halogen-based solvent.
- the alkaline aqueous solution is preferably a sodium hydroxide aqueous solution, a sodium carbonate aqueous solution, a calcium hydroxide aqueous solution, a potassium hydroxide aqueous solution, an ammonia aqueous solution, a sodium hydrogen carbonate aqueous solution, etc.
- the ether solvent is preferably tetrahydrofuran (THF), dioxane, diethyl ether (Et 2 O), isopropyl ether (IPE), or the like.
- the alcohol-based solvent is preferably methanol (CH 3 OH), ethanol, propanol, isopropanol, hexafluoroisopropanol (HFIP), trifluoromethanol, pentafluoroethanol, or the like.
- the halogen-based solvent is preferably dichloromethane (DCM, CH 2 Cl 2 ), trichloromethane (CHCl 3 ), or the like.
- the amount of solvent used is adjusted so that the concentration (molar concentration M (mol/L)) of the compound represented by general formula (3) (substrate) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
- the reaction temperature for the hydrolysis reaction is preferably 0°C to 100°C, more preferably 10°C to 80°C, and even more preferably 20°C to 60°C.
- the reaction time for the hydrolysis reaction is preferably 0.1 to 100 hours, more preferably 0.1 to 50 hours, and even more preferably 0.1 to 10 hours.
- step (1) (2) a step of protecting the hydroxyl group in a solvent
- a reaction for protecting the hydroxyl group in a compound (substrate) represented by general formula (3) is carried out in a solvent (2), and the hydroxyl group (-OH) in formula (3) is protected to Prot 2 (a hydroxyl-protecting group, such as an Ac group) in formula (4).
- the solvent used in step (2) is preferably at least one solvent selected from the group consisting of aprotic polar solvents and ether solvents.
- the aprotic polar solvent is preferably acetone, acetonitrile, N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), pyridine, etc.
- the ether solvent is preferably tetrahydrofuran (THF), dioxane, diethyl ether (Et 2 O), isopropyl ether (IPE), or the like.
- the amount of solvent used is adjusted so that the concentration (molar concentration M (mol/L)) of the compound represented by general formula (3) (substrate) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
- Hydroxyl protecting reagents For the reaction of protecting hydroxyl groups, acetic anhydride (Ac 2 O, for Ac group protection), pivaloyl chloride (PvCl, for Pv group protection), p-toluenesulfonyl isocyanate (tosyl isocyanate) (PTSI, for p-toluenesulfonylcarbamate group protection), etc. are used.
- the amount of Ac2O , PvCl, PTSI, etc. used (hydroxyl group protection reaction) is adjusted so that the concentration in the solvent (molar concentration M (mol/L)) is preferably 0.01 M to 5 M, more preferably 0.1 M to 2 M.
- the amount of Ac2O , PvCl, PTSI, etc. used (hydroxyl group protection reaction) is adjusted so that the concentration relative to the compound represented by general formula (3) (substrate) (when the substrate is 100 mol %) is preferably 0.01 mol % to 100 mol %, more preferably 0.1 mol % to 50 mol %, and even more preferably 1 mol % to 10 mol %.
- the reaction temperature for the hydroxyl protection reaction is preferably 0°C to 100°C, more preferably 10°C to 80°C, and even more preferably 20°C to 60°C.
- the reaction time for the hydroxyl group protection reaction is preferably 0.1 to 100 hours, more preferably 0.1 to 50 hours, and even more preferably 0.1 to 30 hours.
- one L is present at a substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- Prot 2 represents a protecting group for a hydroxyl group.
- the hydroxyl protecting group is preferably a protecting group that can be deprotected under alkali (base conditions) and is an acyl protecting group.
- the hydroxyl protecting group is more preferably an acetyl group (Ac group), a pivaloyl group (Pv group), a p-toluenesulfonylcarbamate group, etc.
- Prot 2 when Prot 2 is an Ac group, it is derived from acetic anhydride (Ac 2 O) used in the protection reaction of the hydroxyl group.
- Prot 2 when Prot 2 is a Pv group, it is derived from pivaloyl chloride (PvCl) used in the protection reaction of the hydroxyl group.
- PvCl pivaloyl chloride
- Prot 2 when Prot 2 is a p-toluenesulfonylcarbamate group, it is derived from p-toluenesulfonyl isocyanate (PTSI) used in the protection reaction of the hydroxyl group.
- PTSI p-toluenesulfonyl isocyanate
- a method for producing a support for solid-phase synthesis of nucleic acid according to the present invention comprising the steps of:
- G is present at one substitutable position and represents the following formula (G1), (G2), or (G3).
- SP represents a solid phase carrier.
- B represents, identically or differently, a hydrogen atom, a lower alkyl group, a lower alkoxy group, a cyano group, a nitro group, or a dialkylamino group at a substitutable position.
- Prot 1 represents a 4,4'-dimethoxytrityl group, a 4-methoxytrityl group, or a trityl group.
- Prot 2 represents a protecting group for a hydroxyl group.
- the method for producing a support for solid-phase synthesis of nucleic acid represented by the following formula (1) is: In a solvent, In the presence of a coupling agent, The following general formula (4):
- L is present at one substitutable position and represents a -CO-OH group, a -NH-CO-OH group, or a -CO-NH-CH 2 -CO-OH group.
- the method includes a step of reacting the compound represented by the formula:
- the present invention produces a support for solid-phase synthesis of nucleic acids.
- SP stands for solid phase carrier.
- G is present at one substitutable position and represents the following formula (G1), (G2), or (G3).
- SP represents a solid phase carrier.
- the solvent is preferably at least one organic solvent selected from the group consisting of aprotic polar solvents and halogenated solvents.
- the aprotic polar solvent is preferably N,N-diisopropylethylamine (DIPEA), acetonitrile (ACN), acetone, N,N-dimethylformamide (DMF), dimethylsulfoxide (DMSO), pyridine, etc.
- DIPEA N,N-diisopropylethylamine
- ACN acetonitrile
- DMF N,N-dimethylformamide
- DMSO dimethylsulfoxide
- pyridine pyridine
- the halogen-based solvent is preferably dichloromethane (CH 2 Cl 2 ), trichloromethane (CHCl 3 ), or the like.
- the amount of solvent used is adjusted so that the concentration (molar concentration M (mol/L)) of the compound represented by general formula (4) (substrate) is preferably 0.01M to 5M, more preferably 0.1M to 2M.
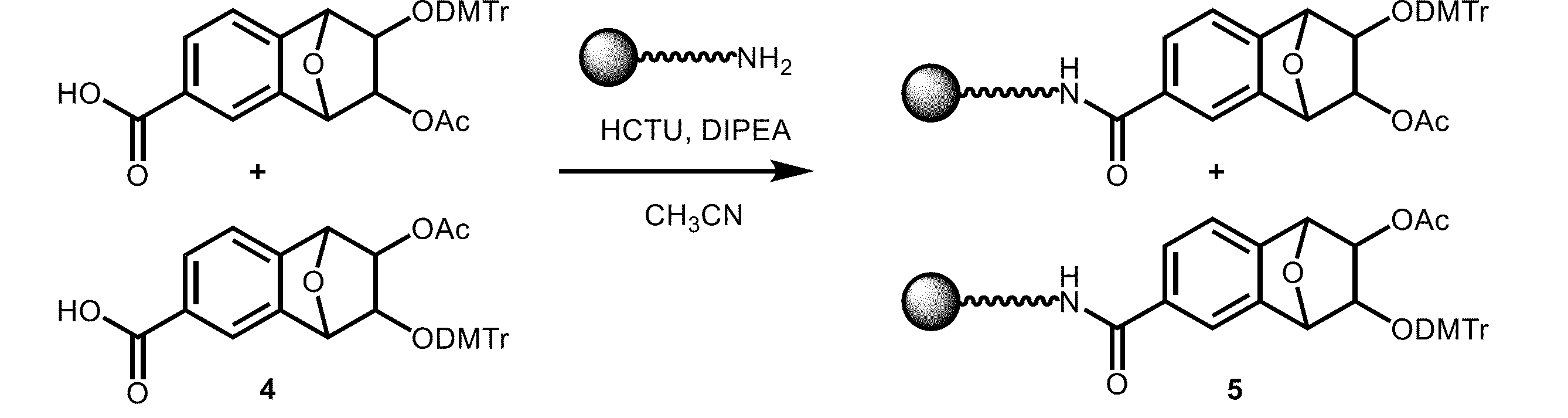
- [4-3] Coupling Agent In the manufacturing method of the present invention, a coupling agent is used to carry out a coupling reaction, and the compound represented by general formula (4) is reacted with a solid phase support (SP- NH2 ) to produce a solid phase synthesis support for nucleic acid represented by general formula (5).
- a coupling agent is used to carry out a coupling reaction, and the compound represented by general formula (4) is reacted with a solid phase support (SP- NH2 ) to produce a solid phase synthesis support for nucleic acid represented by general formula (5).
- the coupling agent is preferably 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), 1-[Bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate (HBTU), O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), or O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU).
- HATU 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HBTU 1-[Bis(d
- HCTU O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium Hexafluorophosphate
- TATU 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide tetrafluoroborate
- TBTU 1-[Bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide tetrafluoroborate
- TBTU oborate
- DCC N,N'-Dicyclohexylcarbodiimide
- the amount of coupling agent used is adjusted so that its concentration in the solvent (molar concentration M (mol/L)) is preferably 0.01M to 5M, and more preferably 0.1M to 2M.
- the amount of coupling agent used is adjusted so that the concentration relative to the compound (substrate) represented by general formula (4) (when the substrate is taken as 100 mol%) is preferably 50 mol% to 10,000 mol%, more preferably 90 mol% to 1000 mol%, and even more preferably 100 mol% to 500 mol%, in molar ratio.
- a compound (substrate) represented by general formula (4) is coupled with a solid phase support in a solvent in the presence of a coupling agent, so that the compound (substrate) represented by general formula (4) is bonded with the solid phase support (SP- NH2 ) via an amide bond (a bond that does not cleave under basic conditions) to produce a nucleic acid solid phase synthesis support represented by general formula (5).
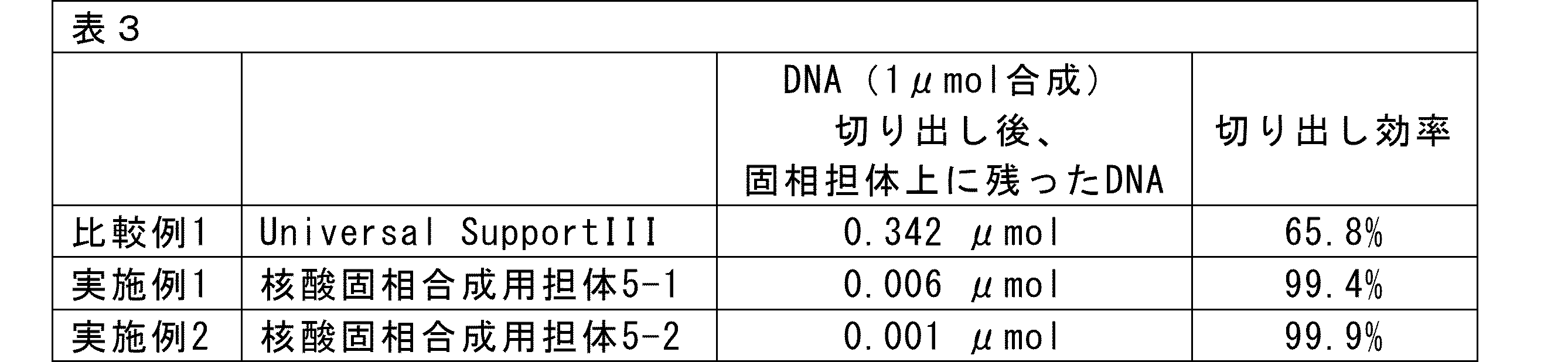
- the nucleic acid solid-phase synthesis support represented by the general formula (5) of the present invention is capable of suppressing the generation of by-products (linker adducts) by binding the solid phase support (SP) and the universal linker (compound represented by the general formula (4)) via a bond (amide bond, etc.) that is not cleaved under excision conditions (ammonia water, etc.).
- the reaction temperature for the coupling reaction is preferably 0°C to 100°C, more preferably 10°C to 80°C, and even more preferably 20°C to 60°C.
- the reaction time for the coupling reaction is preferably 0.1 to 100 hours, more preferably 0.1 to 50 hours, and even more preferably 0.1 to 10 hours.
- G is present at one substitutable position and represents the following formula (G1), (G2), or (G3).
- SP represents a solid phase carrier.
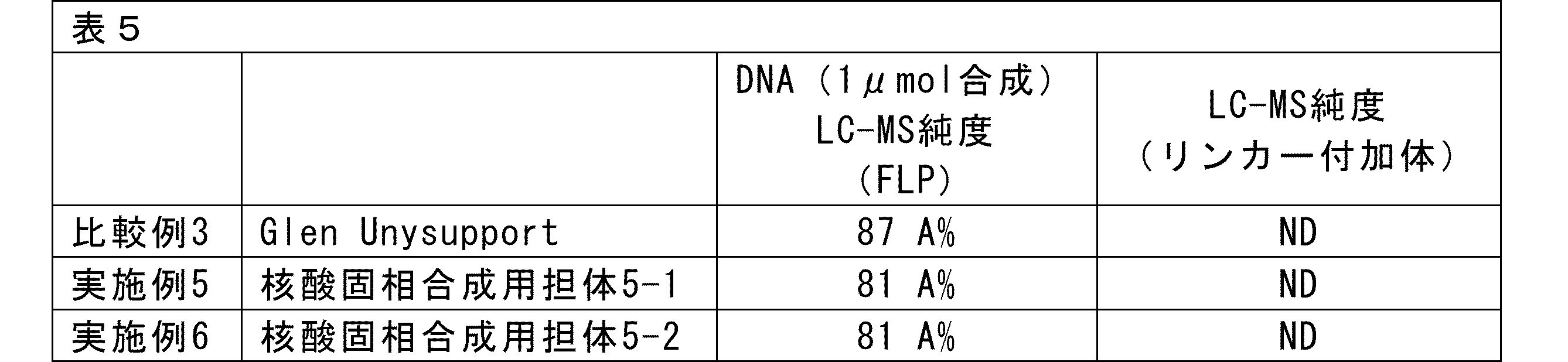
- the present invention is a linker for nucleic acid synthesis and a support for solid-phase nucleic acid synthesis that can suppress the production of by-products (linker adducts) that are generated when using conventional universal supports.
- the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention are characterized in that the solid-phase support and the linker for nucleic acid synthesis are connected via a bond (such as an amide bond) that is not cleaved under cleavage conditions such as in aqueous ammonia.
- the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention do not generate linker adducts, and high-purity oligonucleic acids can be obtained by using the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention.
- the present invention is not limited to these.
- reaction solution was then neutralized by adding 25% aqueous acetic acid (50 ⁇ L).
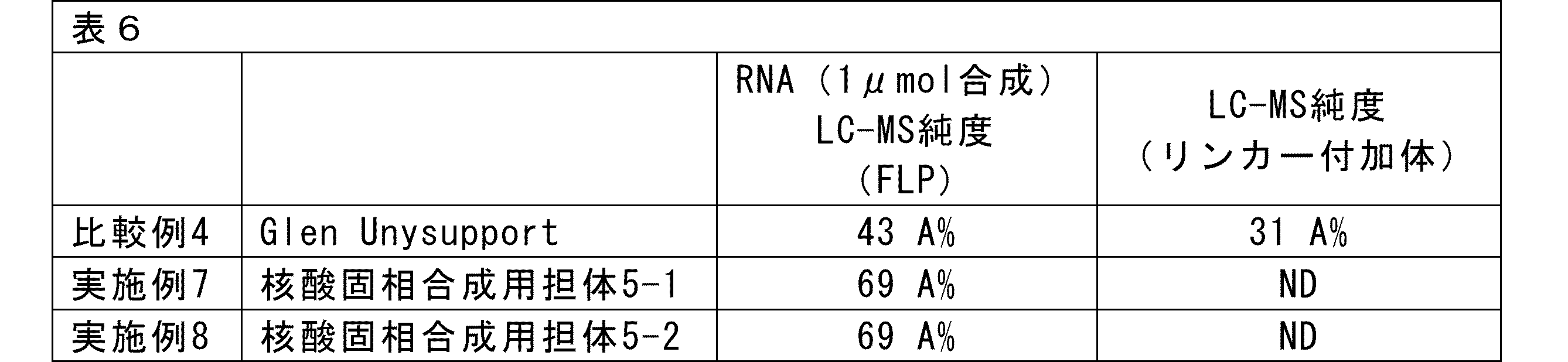
- nucleic acid solid phase synthesis support 5-1 Compound 4 (9.5 mg, 16.8 ⁇ mol), O-(1H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) (7.0 mg, 16.8 ⁇ mol), and N,N-diisopropylethylamine (DIPEA) (5.9 ⁇ L, 33.6 ⁇ mol) were dissolved in acetonitrile (CH 3 CN) (1 mL).
- DIPEA N,N-diisopropylethylamine
- this mixed solution was added to the amino group-containing CPG solid phase support (105 ⁇ mol/g, 200 mg, 21 ⁇ mol of amino groups) and shaken for 3 hours.
- reaction solution was then filtered under suction, and the solid phase carrier was washed with acetonitrile.
- reaction solution was then filtered by suction, and the solid phase carrier was washed with acetonitrile and dried under reduced pressure.
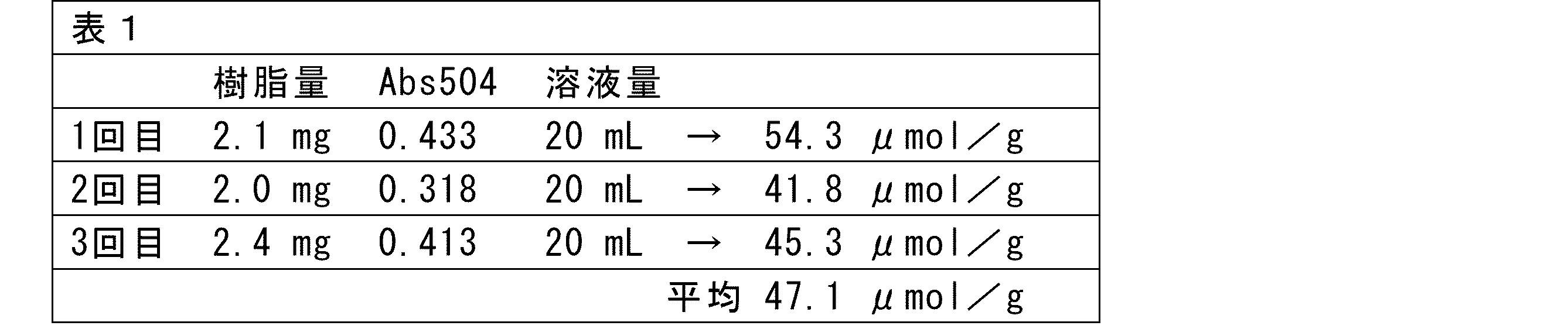
- the measurement results showed that the nucleic acid solid-phase synthesis support 5 obtained was 47.1 ⁇ mol/g.
- reaction solution was then filtered under suction, and the solid phase carrier was washed with acetonitrile.
- reaction solution was then filtered by suction, and the solid phase carrier was washed with acetonitrile and dried under reduced pressure.
- nucleic acid solid phase synthesis support 5-2 carrying compound 4 was prepared in the same manner as nucleic acid solid phase synthesis support 5-1.
- the measurement results showed that the nucleic acid solid-phase synthesis support 5-2 obtained was 37.5 ⁇ mol/g.
- the present invention is a linker for nucleic acid synthesis and a support for solid-phase nucleic acid synthesis that can suppress the production of by-products (linker adducts) that are generated when using conventional universal supports.
- the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention are characterized in that the solid-phase support and the linker for nucleic acid synthesis are connected via a bond (such as an amide bond) that is not cleaved under cleavage conditions such as in aqueous ammonia.
- the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention do not generate linker adducts, and high-purity oligonucleic acids can be obtained by using the linker for nucleic acid synthesis and the support for solid-phase nucleic acid synthesis of the present invention.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Saccharide Compounds (AREA)
Abstract
Description
2:カップリング工程(活性化剤存在下で、核酸アミダイトを縮合し、伸長させる)
3:キャッピング工程(未反応水酸基を無水酢酸等で保護する)
4:酸化、又は硫化工程(ホスファイトを酸化剤、又は硫化剤と反応させる)
これらの工程を行った後、アンモニア水等に依り、固相担体からオリゴ核酸を溶液中へ放出させる。従って、上記固相合成法では、アンモニア水等の塩基性条件下で開裂するリンカーを介してヌクレオシドを結合させた固相担体を使用する。この開裂性リンカーとしては、スクシニル基が広く使用されており、ヌクレオシド3'位水酸基と固相担体とをエステル結合させた核酸固相合成用担体が使用されている。
下記一般式(2):

で表される、核酸合成用リンカー前駆体の製造方法であって、
溶媒中で、
酸化剤、及び再酸化剤の存在下、
下記一般式(1):

で表される化合物を酸化反応させる工程
を含む、核酸合成用リンカー前駆体の製造方法。
前記酸化剤は、四酸化オスミウム(OsO4)、及びオゾン(O3)から成る群から選択される少なくとも一種の酸化剤である、前記項1に記載の製造方法。
前記再酸化剤は、N-メチルモルホリン-N-オキシド(NMO)、トリメチルアミン-N-オキシド(Me3NO)、及びtert-ブチルヒドロペルオキシド(TBHP)から成る群から選択される少なくとも一種の再酸化剤である、前記項1に記載の製造方法。
前記溶媒は、水と親水性有機溶媒との混合溶媒である、前記項1に記載の製造方法。
下記一般式(2):

で表される、核酸合成用リンカー前駆体。
下記一般式(3):

で表される、核酸合成用リンカー前駆体の製造方法であって、
溶媒中で、
下記一般式(2):

で表される化合物と、
4,4'-ジメトキシトリチルクロリド(DMTr-Cl)、4-メトキシトリチルクロリド(MMTr-Cl)、又はトリチルクロリド(Tr-Cl)とを反応させる工程
を含む、核酸合成用リンカー前駆体の製造方法。
前記溶媒は、非プロトン性極性溶媒から成る群から選択される少なくとも一種の有機溶媒である、前記項6に記載の製造方法。
下記一般式(3):

で表される、核酸合成用リンカー前駆体。
下記一般式(4):

で表される、核酸合成用リンカーの製造方法であって、
下記一般式(3):

で表される化合物(2 steps)に対して、
(1)溶媒中で、加水分解する工程、次いで、
(2)溶媒中で、水酸基を保護する工程、
を含む、核酸合成用リンカーの製造方法。
前記工程(1)で使用する溶媒は、アルカリ水溶液、エーテル系溶媒、アルコール系溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の溶媒である、前記項9に記載の製造方法。
前記工程(2)で使用する溶媒は、非プロトン性極性溶媒、及びエーテル系溶媒から成る群から選択される少なくとも一種の溶媒である、前記項9に記載の製造方法。
下記一般式(4):

で表される、核酸合成用リンカー。
下記一般式(5):


で表される、核酸固相合成用担体の製造方法であって、
溶媒中で、
カップリング剤の存在下、
下記一般式(4):

で表される化合物と、
下記:
で表される固相担体とを反応させる工程
を含む、核酸固相合成用担体の製造方法。
前記カップリング剤は、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム-3-オキシドヘキサフルオロホスフェート(HATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキサイドヘキサフルオロホスフェート(HBTU)、O-(1H-6-クロロベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HCTU)、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム-3-オキシド テトラフルオロボレート(TATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドテトラフルオロボレート(TBTU)、N,N'-ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC・HCl)、エチル-2-シアノ-2-((ジメチルイミニオ)(モルホリノ)メチルオキシイミノ)アセテートヘキサフルオロホスフェート(COMU)、ジフェニルホスホリルアジド(DPPA)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)、ベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)-ホスホニウムヘキサフルオロホスフェート(BOP)、及び1H-ベンゾトリアゾール-1-イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)から成る群から選択される少なくとも一種のカップリング剤である、前記項13に記載の製造方法。
前記溶媒は、非プロトン性極性溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の有機溶媒である、前記項13に記載の製造方法。
下記一般式(5):

で表される、核酸固相合成用担体。
本発明の
下記一般式(2):

で表される、核酸合成用リンカー前駆体の製造方法は、
溶媒中で、
酸化剤、及び再酸化剤の存在下、
下記一般式(1):

で表される化合物を酸化反応させる工程を含む。
本発明は、溶媒中で、酸化剤、及び再酸化剤の存在下、
下記一般式(1):

下記一般式(2):

本発明の製造方法では、溶媒は、好ましくは、水と親水性有機溶媒との混合溶媒である。
本発明の製造方法では、酸化剤は、好ましくは、四酸化オスミウム(OsO4)、及びオゾン(O3)から成る群から選択される少なくとも一種の酸化剤である。
本発明の製造方法では、再酸化剤は、好ましくは、N-メチルモルホリン-N-オキシド(NMO)、トリメチルアミン-N-オキシド(Me3NO)、及びtert-ブチルヒドロペルオキシド(TBHP)から成る群から選択される少なくとも一種の再酸化剤である。
酸化反応の反応温度は、好ましくは、0℃~100℃であり、より好ましくは、10℃~80℃であり、更に好ましくは、20℃~60℃である。
本発明は、
下記一般式(2):

本発明の
下記一般式(3):

で表される、核酸合成用リンカー前駆体の製造方法は、
溶媒中で、
下記一般式(2):

で表される化合物と、
4,4'-ジメトキシトリチルクロリド(DMTr-Cl)、4-メトキシトリチルクロリド(MMTr-Cl)、又はトリチルクロリド(Tr-Cl)とを反応させる工程を含む。
本発明は、溶媒中で、
下記一般式(2):

4,4'-ジメトキシトリチルクロリド(DMTr-Cl)、4-メトキシトリチルクロリド(MMTr-Cl)、又はトリチルクロリド(Tr-Cl)とを反応させる工程を含み、
下記一般式(3):

本発明の製造方法では、溶媒は、好ましくは、非プロトン性極性溶媒から成る群から選択される少なくとも一種の有機溶媒である。
本発明の製造方法では、DMTr-Cl、MMTr-Cl、又はTr-Clを用いて、一般式(2)で表される化合物(基質)の水酸基を保護する。
水酸基の保護反応の反応温度は、好ましくは、0℃~100℃であり、より好ましくは、10℃~80℃であり、更に好ましくは、20℃~60℃である。
本発明は、
下記一般式(3):

本発明の
下記一般式(4):

で表される、核酸合成用リンカーの製造方法は、
下記一般式(3):

で表される化合物(2 steps)に対して、
(1)溶媒中で、加水分解する工程、次いで、
(2)溶媒中で、水酸基を保護する工程を含む。
本発明は、
下記一般式(3):

(1)溶媒中で、加水分解する工程、次いで、
(2)溶媒中で、水酸基を保護する工程を含み、
下記一般式(4):

本発明の製造方法では、一般式(3)で表される化合物(基質)に対して、(1)溶媒中で、加水分解反応を進め、式(3)中のA(基-CO-OR、基-NH-CO-OR、又は基-CO-NH-CH2-CO-OR、R:カルボン酸の保護基)を加水分解し、式(4)中のL(基-CO-OH、基-NH-CO-OH、又は基-CO-NH-CH2-CO-OH)に加水分解する。
本発明の製造方法では、工程(1)で使用する溶媒は、好ましくは、アルカリ水溶液、エーテル系溶媒、アルコール系溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の溶媒である。
加水分解反応の反応温度は、好ましくは、0℃~100℃であり、より好ましくは、10℃~80℃であり、更に好ましくは、20℃~60℃である。
本発明の製造方法では、工程(1)の後、一般式(3)で表される化合物(基質)に対して、(2)溶媒中で、水酸基を保護する反応を進め、式(3)中の水酸基(-OH)を保護し、式(4)中のProt2(水酸基の保護基、Ac基等)に保護する。
本発明の製造方法では、工程(2)で使用する溶媒は、好ましくは、非プロトン性極性溶媒、及びエーテル系溶媒から成る群から選択される少なくとも一種の溶媒である。
水酸基を保護する反応には、無水酢酸(Ac2O、Ac基保護)、ピバロイルクロリド(PvCl、Pv基保護)、イソシアン酸p-トルエンスルホニル(イソシアン酸トシル)(PTSI、p-トルエンスルホニルカルバマート基保護)等を使う。
水酸基の保護反応の反応温度は、好ましくは、0℃~100℃であり、より好ましくは、10℃~80℃であり、更に好ましくは、20℃~60℃である。
本発明は、
下記一般式(4):

本発明の
下記一般式(5):


で表される、核酸固相合成用担体の製造方法は、
溶媒中で、
カップリング剤の存在下、
下記一般式(4):

で表される化合物と、
下記:
で表される固相担体とを反応させる工程を含む。
本発明は、溶媒中で、カップリング剤の存在下、
下記一般式(4):

下記:
下記一般式(5):


本発明の製造方法では、溶媒は、好ましくは、非プロトン性極性溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の有機溶媒である。
本発明の製造方法では、カップリング剤を用いて、カップリング反応を進め、一般式(4)で表される化合物と固相担体(SP-NH2)とを反応させて、一般式(5)で表される、核酸固相合成用担体を製造する。
本発明の製造方法では、溶媒中で、カップリング剤の存在下、一般式(4)で表される化合物(基質)と固相担体とをカップリングさせる事に依り、一般式(4)で表される化合物(基質)と固相担体(SP-NH2)とが、アミド結合(塩基性条件下で開裂しない結合)を介して結合させて、一般式(5)で表される核酸固相合成用担体を製造すする。
本発明は、
下記一般式(5):

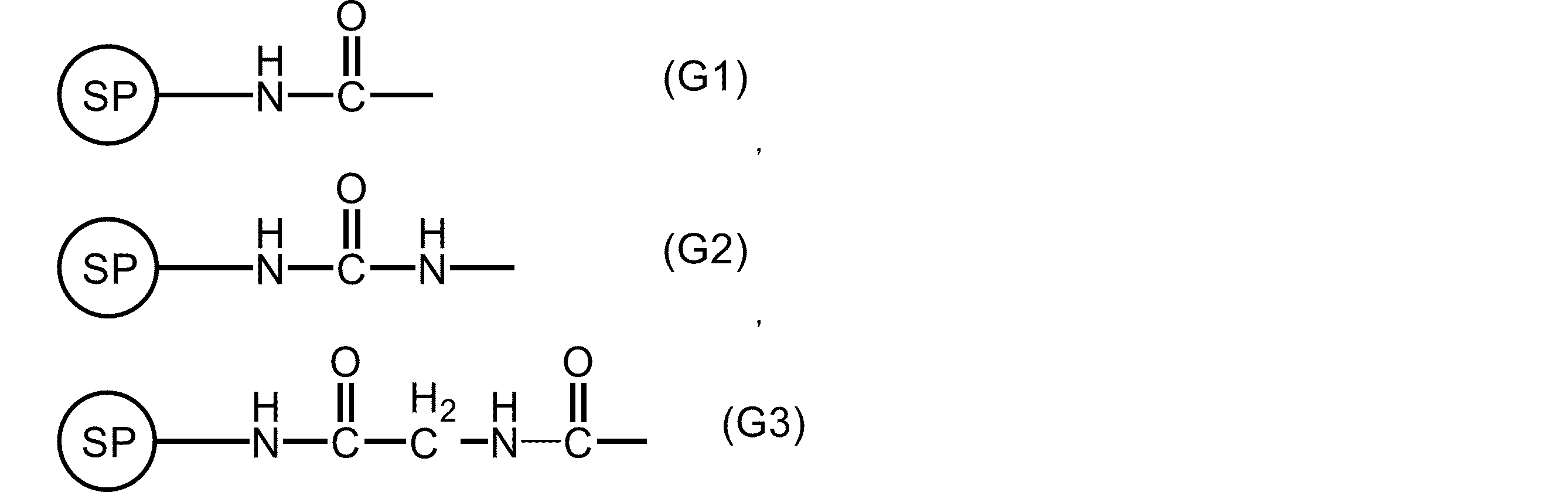
本発明の一般式(2)で表される核酸合成用リンカー前駆体1を製造した。
化合物1(1.03 g, 5.09 mmol)のアセトニトリル(CH3CN)溶液(10 mL)に、0.1M 四酸化オスミウム(Os4O)/t-ブチルアルコール溶液(50.94 μL, 5.094 μmol)、50%N-メチルモルホリンオキシド(NMO)水溶液(2.65 mL, 12.735 mmol)を加えて、40℃で10分間撹拌した。
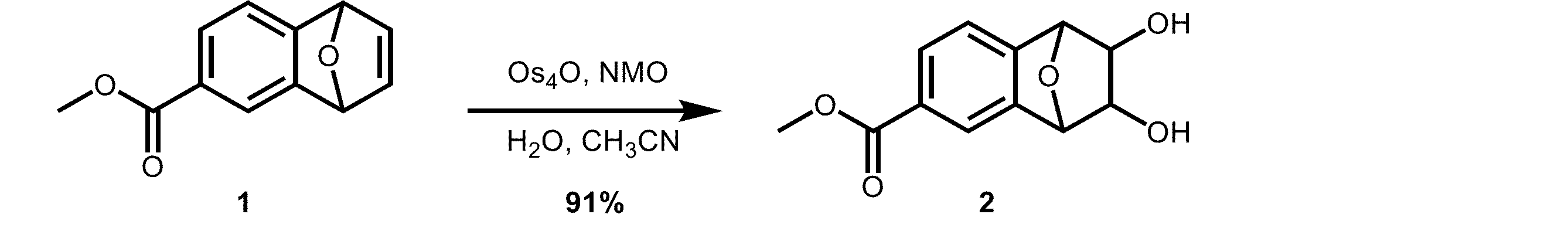
本発明の一般式(3)で表される核酸合成用リンカー前駆体2を製造した。
化合物2(112 mg, 0.475 mmol)のピリジン(pyridine)溶液(4 mL)に、4,4’-ジメトキシトリチルクロリド(DMTrCl)(193 mg, 0.570 mmol)を加えて、室温で20時間撹拌した。
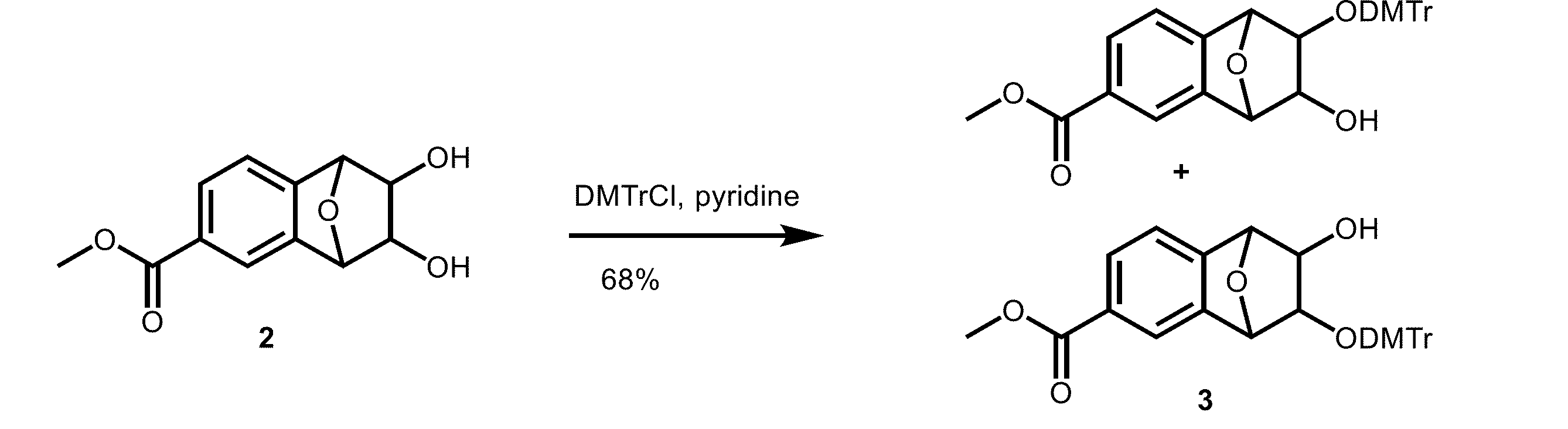
本発明の一般式(4)で表される核酸合成用リンカーを製造した。

(1)溶媒中で、加水分解する工程
化合物3(54 mg, 0.10mmol)のジクロロメタン(DCM)溶液(0.54mL)に、メタノール(CH3OH)(0.54mL)、1M水酸化ナトリウム水溶液(NaOH aq)(0.20mL)を加えて、45℃で2.5時間撹拌した。
次いで、得られた残渣を、ピリジン(pyridine)(1.0mL)に溶かし、4-ジメチルアミノピリジン(11.2mg)、無水酢酸(Ac2O)(86μL)加えて、室温で23時間撹拌した。
[4]核酸固相合成用担体の製造1
本発明の一般式(5)で表される核酸固相合成用担体を製造した。
化合物4(9.5 mg, 16.8 μmol)、O-(1H-6-クロロベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HCTU)(7.0 mg, 16.8 μmol)、N,N-ジイソプロピルエチルアミン(DIPEA)(5.9μL, 33.6 μmol)を、アセトニトリル(CH3CN)(1 mL)溶解させた。

グリシンFmoc保護体(47 mg, 158 μmol)、HCTU(59 mg, 189 μmol)、N,N-ジイソプロピルエチルアミン(55 μL, 420 μmol)を、N,N’-ジメチルホルムアミド(1 mL)溶解させた。



DNA(5’-TTT TTT TTT T-3’)
切り出し条件:アンモニア水、15時間(55℃)
切り出し条件:アンモニア水/メチルアミン(v/v=1/1)、90分(室温)
→10分(65℃)

DNA(5’-TTT TTT TTT T-3’)
Claims (16)
- 下記一般式(2):

式(2)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。)
で表される、核酸合成用リンカー前駆体の製造方法であって、
溶媒中で、
酸化剤、及び再酸化剤の存在下、
下記一般式(1):

で表される化合物を酸化反応させる工程
を含む、核酸合成用リンカー前駆体の製造方法。 - 前記酸化剤は、四酸化オスミウム(OsO4)、及びオゾン(O3)から成る群から選択される少なくとも一種の酸化剤である、請求項1に記載の製造方法。
- 前記再酸化剤は、N-メチルモルホリン-N-オキシド(NMO)、トリメチルアミン-N-オキシド(Me3NO)、及びtert-ブチルヒドロペルオキシド(TBHP)から成る群から選択される少なくとも一種の再酸化剤である、請求項1に記載の製造方法。
- 前記溶媒は、水と親水性有機溶媒との混合溶媒である、請求項1に記載の製造方法。
- 下記一般式(2):

式(2)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。)
で表される、核酸合成用リンカー前駆体。 - 下記一般式(3):

式(3)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。
式(3)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。)
で表される、核酸合成用リンカー前駆体の製造方法であって、
溶媒中で、
下記一般式(2):

で表される化合物と、
4,4'-ジメトキシトリチルクロリド(DMTr-Cl)、4-メトキシトリチルクロリド(MMTr-Cl)、又はトリチルクロリド(Tr-Cl)とを反応させる工程
を含む、核酸合成用リンカー前駆体の製造方法。 - 前記溶媒は、非プロトン性極性溶媒から成る群から選択される少なくとも一種の有機溶媒である、請求項6に記載の製造方法。
- 下記一般式(3):

式(3)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。
式(3)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。)
で表される、核酸合成用リンカー前駆体。 - 下記一般式(4):

式(4)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。
式(4)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。
式(4)中、Prot2は、水酸基の保護基を示す。)
で表される、核酸合成用リンカーの製造方法であって、
下記一般式(3):

式(3)中、B、及びProt1は、前記式(4)と同じ。)
で表される化合物に対して、
(1)溶媒中で、加水分解する工程、次いで、
(2)溶媒中で、水酸基を保護する工程、
を含む、核酸合成用リンカーの製造方法。 - 前記工程(1)で使用する溶媒は、アルカリ水溶液、エーテル系溶媒、アルコール系溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の溶媒である、請求項9に記載の製造方法。
- 前記工程(2)で使用する溶媒は、非プロトン性極性溶媒、及びエーテル系溶媒から成る群から選択される少なくとも一種の溶媒である、請求項9に記載の製造方法。
- 下記一般式(4):

式(4)中、Bは、置換可能な位置に、同一又は異なって、水素原子、低級アルキル基、低級アルコキシ基、シアノ基、ニトロ基、又はジアルキルアミノ基を示す。
式(4)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。
式(4)中、Prot2は、水酸基の保護基を示す。)
で表される、核酸合成用リンカー。 - 下記一般式(5):

式(G1)、(G2)、又は(G3)中、SPは、固相担体を示す。

式(5)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。
式(5)中、Prot2は、水酸基の保護基を示す。)
で表される、核酸固相合成用担体の製造方法であって、
溶媒中で、
カップリング剤の存在下、
下記一般式(4):

式(4)中、B、Prot1、及びProt2は、前記式(5)と同じ。)
で表される化合物と、
下記:

で表される固相担体とを反応させる工程
を含む、核酸固相合成用担体の製造方法。 - 前記カップリング剤は、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム-3-オキシドヘキサフルオロホスフェート(HATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキサイドヘキサフルオロホスフェート(HBTU)、O-(1H-6-クロロベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HCTU)、1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム-3-オキシド テトラフルオロボレート(TATU)、1-[ビス(ジメチルアミノ)メチレン]-1H-ベンゾトリアゾリウム-3-オキシドテトラフルオロボレート(TBTU)、N,N'-ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDC・HCl)、エチル-2-シアノ-2-((ジメチルイミニオ)(モルホリノ)メチルオキシイミノ)アセテートヘキサフルオロホスフェート(COMU)、ジフェニルホスホリルアジド(DPPA)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)、ベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)-ホスホニウムヘキサフルオロホスフェート(BOP)、及び1H-ベンゾトリアゾール-1-イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)から成る群から選択される少なくとも一種のカップリング剤である、請求項13に記載の製造方法。
- 前記溶媒は、非プロトン性極性溶媒、及びハロゲン系溶媒から成る群から選択される少なくとも一種の有機溶媒である、請求項13に記載の製造方法。
- 下記一般式(5):

式(G1)、(G2)、又は(G3)中、SPは、固相担体を示す。
式(5)中、Prot1は、4,4'-ジメトキシトリチル基、4-メトキシトリチル基、又はトリチル基を示す。
式(5)中、Prot2は、水酸基の保護基を示す。)
で表される、核酸固相合成用担体。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP23906969.3A EP4640687A1 (en) | 2022-12-20 | 2023-12-18 | Linker for nucleic acid synthesis, carrier, and methods for producing same |
| JP2024566041A JPWO2024135609A1 (ja) | 2022-12-20 | 2023-12-18 | |
| KR1020257019301A KR20250121547A (ko) | 2022-12-20 | 2023-12-18 | 핵산 합성용 링커, 및 담체, 그리고 그들의 제조방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022203402 | 2022-12-20 | ||
| JP2022-203402 | 2022-12-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024135609A1 true WO2024135609A1 (ja) | 2024-06-27 |
Family
ID=91588688
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/045266 Ceased WO2024135609A1 (ja) | 2022-12-20 | 2023-12-18 | 核酸合成用リンカー、及び担体、並びにそれらの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP4640687A1 (ja) |
| JP (1) | JPWO2024135609A1 (ja) |
| KR (1) | KR20250121547A (ja) |
| TW (1) | TW202430648A (ja) |
| WO (1) | WO2024135609A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040152905A1 (en) * | 2003-01-31 | 2004-08-05 | Guzaev Andrei P. | Universal building blocks and support media for synthesis of oligonucleotides and their analogs |
| JP2013177371A (ja) * | 2012-01-30 | 2013-09-09 | Nitto Denko Corp | 核酸固相合成用リンカー及び担体 |
| WO2022191172A1 (ja) * | 2021-03-09 | 2022-09-15 | リードファーマ株式会社 | 核酸固相合成のためのリンカー及び担体 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016204316A (ja) | 2015-04-24 | 2016-12-08 | 日東電工株式会社 | 核酸固相合成用リンカー及び担体 |
| JP6537338B2 (ja) | 2015-04-24 | 2019-07-03 | 日東電工株式会社 | 核酸固相合成用リンカー及び担体 |
-
2023
- 2023-12-13 TW TW112148461A patent/TW202430648A/zh unknown
- 2023-12-18 WO PCT/JP2023/045266 patent/WO2024135609A1/ja not_active Ceased
- 2023-12-18 JP JP2024566041A patent/JPWO2024135609A1/ja active Pending
- 2023-12-18 EP EP23906969.3A patent/EP4640687A1/en active Pending
- 2023-12-18 KR KR1020257019301A patent/KR20250121547A/ko active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040152905A1 (en) * | 2003-01-31 | 2004-08-05 | Guzaev Andrei P. | Universal building blocks and support media for synthesis of oligonucleotides and their analogs |
| JP2013177371A (ja) * | 2012-01-30 | 2013-09-09 | Nitto Denko Corp | 核酸固相合成用リンカー及び担体 |
| WO2022191172A1 (ja) * | 2021-03-09 | 2022-09-15 | リードファーマ株式会社 | 核酸固相合成のためのリンカー及び担体 |
Non-Patent Citations (5)
| Title |
|---|
| DATABASE Registry 25 August 2021 (2021-08-25), ANONYMOUS: "INDEX NAME NOT YET ASSIGNED", XP093184214, retrieved from STNext Database accession no. 2680694-63-5 * |
| DATABASE Registry 29 April 2021 (2021-04-29), ANONYMOUS: "INDEX NAME NOT YET ASSIGNED", XP093184218, retrieved from STNext Database accession no. 2639457-09-1 * |
| See also references of EP4640687A1 |
| SYNTHESIS, vol. 53, no. 23, 2021, pages 4440 - 4448 |
| YAMAMOTO KAZUKI, FUCHI YASUFUMI, OKABE MASAYA, OSAWA TAKASHI, ITO YUTA, HARI YOSHIYUKI: "New cleavable spacers for tandem synthesis of multiple oligonucleotides", SYNTHESIS, GEORG THIEME VERLAG, STUTTGART, DE., vol. 53, no. 23, 1 December 2021 (2021-12-01), STUTTGART, DE. , pages 4440 - 4448, XP009555690, ISSN: 0039-7881, DOI: 10.1055/a-1538-9883 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20250121547A (ko) | 2025-08-12 |
| EP4640687A1 (en) | 2025-10-29 |
| JPWO2024135609A1 (ja) | 2024-06-27 |
| TW202430648A (zh) | 2024-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6281599B2 (ja) | 擬似固相保護基およびヌクレオチド | |
| AU651289B2 (en) | Process of linking nucleosides with a siloxane bridge | |
| EP1427743A2 (en) | Process for the synthesis of pyrazolopyrimidines | |
| CN115215921A (zh) | 一种连接基药物偶联物的制备方法及其中间体 | |
| JP2025111536A (ja) | オリゴ核酸化合物の製造方法 | |
| CN115385926B (zh) | 一种连接基药物偶联物的制备方法及其中间体 | |
| CA2730622C (en) | Process for the synthesis of carbonucleoside and intermediates for use therein | |
| US11390641B2 (en) | 5′S-LNA nucleotides and oligonucleotides | |
| US12269841B2 (en) | Production method for oligonucleotides | |
| WO2024135609A1 (ja) | 核酸合成用リンカー、及び担体、並びにそれらの製造方法 | |
| JP7475056B2 (ja) | 光応答性ヌクレオチドアナログの製造方法 | |
| CN116554237B (zh) | 一种氟代核糖中间体的制备方法与应用 | |
| JP2000327694A (ja) | ヌクレオシド化合物 | |
| JPH10114788A (ja) | Dna/pnaコ・オリゴマーの構築用ブロツク | |
| JP4885963B2 (ja) | ヌクレオシドの選択的o−アシル化 | |
| US20250236633A1 (en) | Synthesis of isomerically pure polyol-based phosphoramidites | |
| Efimov et al. | N-azidomethylbenzoyl blocking group in the phosphotriester synthesis of oligoribonucleotides | |
| JP7423533B2 (ja) | 配糖体化合物の製造方法 | |
| JP2008531487A5 (ja) | ||
| JP2007137843A (ja) | リボフラノース化合物およびプリンヌクレオシド化合物の製造方法 | |
| CN118852303A (zh) | 胞苷乙烯基磷酸酯化合物的制备方法及其应用 | |
| WO2001079248A1 (fr) | Procede de preparation de derives de cytidine | |
| JP4627625B2 (ja) | N−アセチルシチジン類の製造方法 | |
| JPH072846A (ja) | テトラヒドロフルフリルアルコール誘導体 | |
| JPWO2000039144A1 (ja) | ヌクレオシド又は糖のフッ素化誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23906969 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024566041 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023906969 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2023906969 Country of ref document: EP Effective date: 20250721 |
|
| ENP | Entry into the national phase |
Ref document number: 2023906969 Country of ref document: EP Effective date: 20250721 |
|
| ENP | Entry into the national phase |
Ref document number: 2023906969 Country of ref document: EP Effective date: 20250721 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257019301 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2023906969 Country of ref document: EP |