WO2024185535A1 - チオールの製造方法 - Google Patents
チオールの製造方法 Download PDFInfo
- Publication number
- WO2024185535A1 WO2024185535A1 PCT/JP2024/006567 JP2024006567W WO2024185535A1 WO 2024185535 A1 WO2024185535 A1 WO 2024185535A1 JP 2024006567 W JP2024006567 W JP 2024006567W WO 2024185535 A1 WO2024185535 A1 WO 2024185535A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- thiol
- producing
- hydrogen
- alkene
- metal
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/02—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B45/00—Formation or introduction of functional groups containing sulfur
- C07B45/06—Formation or introduction of functional groups containing sulfur of mercapto or sulfide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/081—Compounds with Si-C or Si-Si linkages comprising at least one atom selected from the elements N, O, halogen, S, Se or Te
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/0825—Preparations of compounds not comprising Si-Si or Si-cyano linkages
- C07F7/083—Syntheses without formation of a Si-C bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
- C07F7/0825—Preparations of compounds not comprising Si-Si or Si-cyano linkages
- C07F7/0832—Other preparations
Definitions
- the present invention relates to a method for producing a thiol.
- This application claims priority based on Japanese Patent Application No. 2023-034877 filed on March 7, 2023, the contents of which are incorporated herein by reference.
- Thiols are used in a variety of applications, including as gas odorants, fragrances, and ligands.
- gas odorants include thiophenol
- fragrances include 3-mercaptohexanol
- ligands include tert-butylthiol.
- Non-Patent Document 1 a synthesis method using a substitution reaction (SN2 reaction) in which reactants interact in one step
- Non-Patent Document 2 a synthesis method for thiols using hydrogen sulfide from an alcohol as a raw material
- Non-Patent Document 3 a synthesis method for thiols using hydrogen sulfide from an alkene as a raw material
- Non-Patent Document 4 (4) a synthesis method for thiols using ultraviolet light or a platinum electrode from an alkene as a raw material
- Non-Patent Document 5 a synthesis method for thiols using sulfur and a carbonyl compound
- the above synthesis method (1) has low atomic efficiency and produces a large amount of waste as a by-product.
- the above synthesis method (2) requires strict reaction conditions such as high reaction temperature, making production difficult.
- the above synthesis method (3) requires the use of hydrogen sulfide, which is highly toxic and therefore difficult to handle in terms of equipment management, etc.
- the above synthesis method (4) requires an ultraviolet irradiation device and expensive platinum electrodes, which increases production costs.
- the above synthesis method (5) requires the use of CO gas and an expensive organic base, which, like the above synthesis methods (3) and (4), results in complicated handling of raw materials and increased production costs.
- the object of the present invention is to provide a method for producing thiols that can be easily produced with high atomic efficiency, that allows easy handling of the raw materials, and that can reduce production costs.
- the inventors conducted extensive research and discovered that various thiols can be easily obtained by reacting sulfur and hydrogen with the carbon atom at the 2-position of an alkene having a double bond at its terminal or part in the presence of a catalyst containing a metal element.
- hydrogen gas
- the inventors discovered that various thiols can be produced without using hydrogen sulfide gas, which is difficult to handle, or expensive organic bases, and without using ultraviolet irradiation equipment, platinum electrodes, etc.
- a method for producing a thiol comprising reacting an alkene (1) or a derivative thereof with sulfur in the presence of hydrogen and a metal element to obtain a thiol (2).
- the present invention provides a method for producing thiols that can be easily produced with high atomic efficiency, that allows easy handling of raw materials, and that can reduce production costs.
- FIG. 1 is a schematic diagram showing a reaction in a thiol production method according to an embodiment of the present invention.
- FIG. 2 is a schematic diagram showing the reaction in the method for producing thiol (2a) in Example 1.
- FIG. 3 is a graph showing the change over time in the yield of thiol (2a) and the total yield of dialkyl monosulfides (3a) to (5a) in Example 16.
- FIG. 4 shows spectra obtained by performing XRD measurements on CoS and CoS 2 before use in Examples 2 and 3, the cobalt-silicon composite oxide (CoO x -SiO y ) before use in Example 6, CoO x -SiO y after use in Example 6, and ⁇ -Co 2 SiO 4 before use in Example 9.
- FIG. 1 is a schematic diagram showing a reaction in a thiol production method according to an embodiment of the present invention.
- FIG. 2 is a schematic diagram showing the reaction in the method for producing thiol (2a) in Example 1.
- FIG. 5 is a diagram showing an element mapping image obtained by carrying out EDS measurement on CoO x —SiO y before use in Example 6.
- FIG. 6 is a diagram showing an element mapping image obtained by EDS measurement for CoO x —SiO y after use in Example 6.
- FIG. 7 shows the structural formulae of compounds (1b) to (1s) used in Examples 18 to 35.
- FIG. 8 shows the structural formulas of thiols (2b) to (2s) synthesized in Examples 18 to 35.
- the method for producing thiol according to an embodiment of the present invention involves reacting an alkene (1) or a derivative thereof with sulfur in the presence of hydrogen and a metal element to obtain a thiol (2).
- Figure 1 shows the reaction in the method for producing thiol according to this embodiment. Note that the reaction conditions shown in Figure 1, such as the catalyst type, reaction temperature, and heating time, are merely examples and may be changed as appropriate depending on the raw material type, etc.
- the (1) used in the above step is not particularly limited as long as it has a double bond at the end or part thereof, and is, for example, an acyclic olefin (1A), a cyclic olefin (1B), or a derivative thereof.
- the acyclic olefin (1A) used in this embodiment is represented, for example, by R 1 R 2 C ⁇ CH 2 ...(A) (wherein R 1 is an alkyl group, R 2 is a hydrogen atom or an alkyl group, and the total number of carbon atoms in R 1 and R 2 is 2 to 20).
- alkenes (1) From the viewpoint of industrial raw materials making use of the characteristics of sulfur, among alkenes (1), alkenes in which the total number of carbon atoms in R 1 and R 2 is 2 to 18 are preferred, and alkenes in which the total number of carbon atoms in R 1 and R 2 is 2 to 16 are more preferred.
- Examples of the acyclic olefin (1A) used in this embodiment include compounds (1a), (1b), and (1h) described below (see FIG. 9).
- the acyclic olefin (1A) may also have other functional groups such as an aryl group, an alkoxy group, a hydroxy group, a cycloalkyl group, or a silylalkyl group.
- examples of the compounds include the compounds (1f), (1i), (1l) to (1r) described below (see FIG. 9).
- examples of the compounds include the compounds (1s) described below.
- examples of the compounds include the compounds (1j) and (1k) described below.
- the cyclic olefin (1B) used in the present embodiment is represented by, for example, C n H 2n-2 ... (B) (wherein n is 3 or more).
- Examples of the cyclic olefin (1B) include the compound (1g) described later.
- an alkene other than alkene (1) can be used in combination with alkene (1) to the extent that the effect of the present invention is not impaired.
- the raw material used in the production method of this embodiment may contain alkene (1) and an alkene other than alkene (1).
- the sulfur used in the present embodiment is not particularly limited, and may be in a solid state such as a small lump, flake, or powder, or in a molten state (liquid). Among them, molten sulfur is preferred from the viewpoint of ease of charging operation in large-scale production.
- the ratio of alkene (1) and sulfur used is, from the viewpoint of solubility, preferably 1.0 to 5.0 mol of S element per 1 mol of olefin (a) (1.0 to 5.0 molar equivalents as S equivalent, 0.125 to 0.625 molar equivalents as S8 ), more preferably 1.0 to 4.0 mol (1.0 to 4.0 molar equivalents as S equivalent, 0.125 to 0.5 molar equivalents as S8 ), and even more preferably 1.0 to 3.5 mol (1.0 to 3.5 molar equivalents as S equivalent, 0.125 to 0.438 molar equivalents as S8 ).
- the hydrogen used in this embodiment is typically hydrogen gas. Hydrogen gas is widely distributed and is easier to obtain than hydrogen sulfide. Although hydrogen gas is flammable, it is non-toxic and odorless, and therefore easier to handle than hydrogen sulfide.
- the hydrogen gas is not particularly limited, and may be, for example, 99.99% or more, 99.999% or more, or 99.9999% or more.
- hydrogen gas available on the market can be obtained, or hydrogen gas can be produced using a reforming method in which hydrogen is obtained by reacting a hydrocarbon such as methane with water vapor, or an electrolysis method in which hydrogen is obtained by electrolyzing water.
- the pressure of the hydrogen supplied is preferably 0.1 MPa or more and 10.0 MPa or less, more preferably 1.0 MPa or more and 9.0 MPa or less, and even more preferably 2.0 MPa or more and 8.0 MPa or less.
- the heating temperature when reacting alkene (1) with sulfur in the presence of hydrogen is preferably 100°C or higher and 200°C or lower, more preferably 100°C or higher and 160°C or lower, more preferably 100°C or higher and 140°C or lower, and particularly preferably 120°C or higher and 140°C or lower.
- alkene (1) and sulfur are reacted in the presence of hydrogen to obtain thiol (2), but this is not limiting, and alkene (1) and sulfur may also be reacted in the presence of hydrogen and a metal element.
- a metal element as a catalyst and causing a contact reaction between alkene (1) and sulfur in the presence of hydrogen, the yield of the obtained thiol (2) can be improved.
- the metal element used in this embodiment is preferably one or more metal elements selected from Groups 6 to 11.
- metal elements include the following: Group 6: chromium (Cr), molybdenum (Mo), tungsten (W) Group 7: manganese (Mn), technetium (Tc), rhenium (Re) Group 8: iron (Fe), ruthenium (Ru), osmium (Os) Group 9: cobalt (Co), rhodium (Rh), iridium (Ir) Group 10: Nickel (Ni), Palladium (Pd), Platinum (Pt) Group 11: Copper (Cu), silver (Ag), gold (Au)
- the metal element is preferably a metal element that constitutes, for example, a metal oxide, a metal sulfide, or a metal carbonate.
- the metal element is, for example, a metal element that constitutes a metal oxide
- alkene (1) is reacted with sulfur in the presence of hydrogen and the metal oxide.
- the metal element is a metal element that constitutes a metal sulfide
- alkene (1) is reacted with sulfur in the presence of hydrogen and the metal sulfide.
- the valence of the metal element that constitutes the metal oxide or metal sulfide is not particularly limited and can take various values such as +1 or +2.
- metal oxides include, but are not limited to, the following.
- examples of the metal sulfide include, but are not limited to, the following.
- metal sulfides Ni3S2 , PdS , PtS Group 11 metal sulfides: CuS, Ag 2 S, Au 2 S
- cobalt oxide can be used as the metal oxide
- cobalt sulfide can be used as the metal sulfide.
- cobalt oxide is used as the metal oxide
- the alkene (1) is reacted with sulfur in the presence of hydrogen and cobalt oxide.
- the cobalt oxide used is not particularly limited, and includes CoO, Co 2 O 3 , and Co 3 O 4.
- cobalt sulfide is used as the metal sulfide
- the alkene (1) is reacted with sulfur in the presence of hydrogen and cobalt sulfide.
- the cobalt sulfide used is not particularly limited, and includes CoS, Co 9 S 8 , CoS 2 , and Co 3 S 4. Of these, from the viewpoint of improving the yield of dialkyl polysulfide (A), cobalt oxide, particularly Co 3 O 4 , is preferred.
- the metal oxide may have a plurality of metal elements selected from Groups 6 to 11.
- examples of the metal oxide used include NiCo 2 O 4.
- the metal oxide may have a metal element selected from Groups 6 to 11 and other elements. Examples of other elements include silicon (Si).
- examples of the metal oxide used include CoO x -SiO y such as Co 2 SiO 4 and ⁇ -Co 2 SiO 4 , and NiO x -SiO y such as Ni 2 SiO 4 .
- the metal carbonate used may be, for example, CoCO3 .
- the amount of the metal element added to the alkene (1) is preferably 0.1 mol% or more and 10 mol% or less, more preferably 1.0 mol% or more and 10 mol% or less, and even more preferably 2.0 mol% or more and 7.0 mol% or less.
- the amount of the metal element added to the alkene (1) is 0.1 mol% or more and 10 mol% or less, the reaction between the alkene (1) and sulfur in the presence of hydrogen is promoted, and the yield of the obtained thiol (2) can be improved.
- alkene (1) and sulfur are reacted in the presence of hydrogen and a metal element to obtain thiol (2), but the present invention is not limited thereto, and alkene (1) and sulfur may be reacted in the presence of hydrogen, the metal element, and a zeolite.
- a metal element and a zeolite as catalysts and catalytically reacting alkene (1) and sulfur with the two catalysts in the presence of hydrogen, the yield of the obtained thiol (2) can be further improved.
- Zeolite is a crystalline aluminosilicate, and has a skeletal structure in which silica and alumina are regularly linked, and contains cations as ion exchange sites in the pore structure.
- the pore structure determined by the zeolite skeletal structure is not particularly limited, and examples thereof include LTA (A type), FER (ferrierite), MWW (MCM-22), MFI (ZSM-5), MOR (mordenite), LTL (L type), FAU (Y type, X type), and BEA (beta type).
- LTA A type
- FER ferrierite
- MCM-22 MFI
- ZSM-5 MOR
- MOR mordenite
- LTL L type
- FAU Y type, X type
- BEA beta type
- the zeolite is preferably basic.
- basic zeolites include zeolites that contain cations of alkali metals or alkaline earth metals.
- alkali metals include sodium (Na) and potassium (K)
- alkaline earth metals include magnesium (Mg) and calcium (Ca).
- the above zeolite is preferably Na-X type, which has an X-type skeletal structure and contains sodium ions, or Na-A type, which has an A-type skeletal structure and contains sodium ions, and among these, Na-A type is more preferable.
- the amount of the zeolite added is preferably 2.0 parts by mass or more and 25 parts by mass or less, more preferably 2.0 parts by mass or more and 15 parts by mass or less, even more preferably 3.0 parts by mass or more and 10 parts by mass or less, and particularly preferably 5.0 parts by mass or more and 9.0 parts by mass or less, relative to 100 parts by mass of the alkene (1), and the yield at the first reuse can be maintained at a high value.
- the manufacturing method of this embodiment produces thiol (2).
- thiol (2) and one or more other compounds other than thiol (2) may be produced.
- other compounds include dialkyl monosulfides.
- the content (yield) of thiol (2) and each of the dialkyl monosulfides having a different number of carbon atoms can be determined from the peak area of a chart obtained by gas chromatography (hereinafter also referred to as "GC") measurement.
- GC gas chromatography
- the manufacturing method of this embodiment allows thiol (2) to be obtained efficiently. Furthermore, the obtained thiol (2) can be suitably used, for example, as a gas odorant, fragrance, or ligand.
- Example 1 An autoclave was charged with a stirrer, 6 mmol of compound (1a) (H 2 C ⁇ C-C 12 H 25 ), 0.4125 molar equivalents of sulfur (S 8 ) (3.3 molar equivalents as S element), 6 mol% of Co 3 O 4 (Sigma-Aldrich, nanopowder: 50 nm or less (TEM)), and 100 mg of zeolite (Sigma-Aldrich, molecular sieve 4A, Na-A type), and then hydrogen was charged under pressure up to 7.0 MPa. The autoclave was heated to 130°C while stirring at 800 rpm with a magnetic stirrer, and the reaction was carried out at the same temperature for 16 hours.
- compound (1a) H 2 C ⁇ C-C 12 H 25
- S 8 0.4125 molar equivalents of sulfur
- S 8 3.3 molar equivalents as S element
- 6 mol% of Co 3 O 4 Sigma-Aldrich, nanopowder: 50 nm or less (TEM
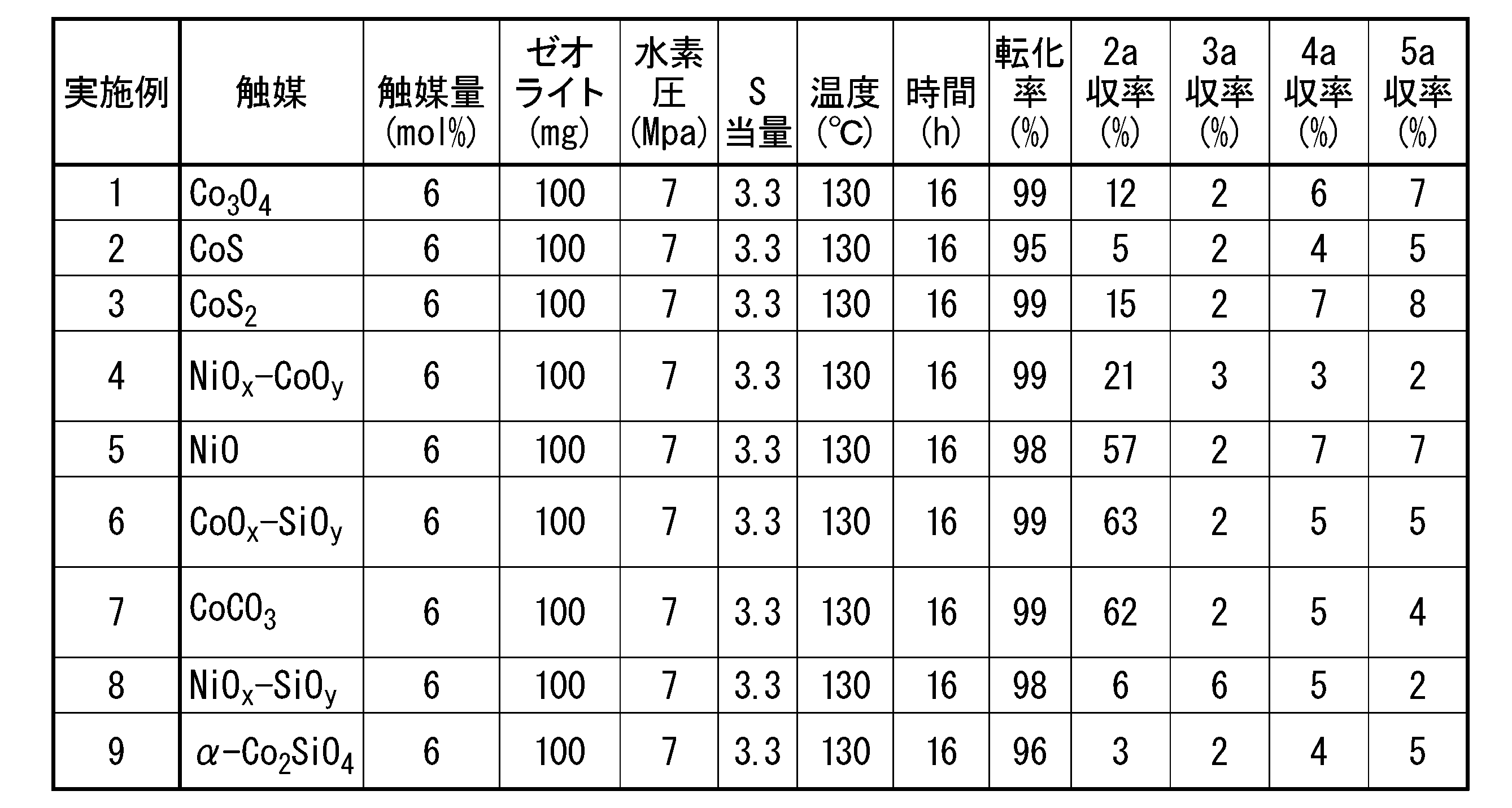
- Example 2 Thiol (2) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to CoS (manufactured by Strem Chemicals).
- Example 3 Thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to CoS 2 (manufactured by Alfa Aesar).
- Example 4 A thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to nickel-cobalt composite oxide (NiO x —CoO y ).
- the nickel-cobalt composite oxide was prepared by the following method.
- Nickel acetate tetrahydrate (Kishida Chemical Co., Ltd., 2.48 g, 10 mmol) and cobalt nitrate nonahydrate (Fujifilm Wako Pure Chemical Industries, Ltd., 5.82 g, 20 mmol) were dissolved in 150 mL of distilled water, and a solution of ammonium carbonate (Fujifilm Wako Pure Chemical Industries, Ltd., 2.88 g, 30 mmol) dissolved in 50 mL of distilled water was added dropwise over 5 minutes while stirring at room temperature, and the mixture was left at room temperature for 3 hours. After the reaction was completed, the resulting precipitate was filtered and washed with 2 x 50 mL of distilled water, and dried at 70 ° C.
- the black powder was then crushed in a mortar and sieved to 125 ⁇ m or less and collected.
- the mixture was then transferred to a baking dish and baked.
- the baking conditions were to heat the mixture to 300 ° C. in 1 hour (heating program: 5 ° C. / min) and hold the mixture at the same temperature for 6 hours. After calcination, the mixture was cooled to room temperature and a black powder was collected (2.16 g).
- the cobalt and nickel contents were evaluated by a microwave plasma atomic emission spectrometer (MP-AES) and found to be 38% by mass and 11% by mass, respectively.
- MP-AES microwave plasma atomic emission spectrometer
- Example 5 Thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to NiO (Sigma-Aldrich).
- Example 6 A thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to cobalt-silicon composite oxide (CoO x —SiO y ).
- the cobalt-silicon composite oxide was prepared by the following method. Cobalt nitrate hexahydrate (Fujifilm Wako Pure Chemical Industries, Ltd., 2.91 g, 10 mmol) was dissolved in 90 mL of distilled water, and a solution of sodium silicate (Sigma-Aldrich, 1.22 g, 10 mmol) dissolved in 10 mL of distilled water was added dropwise over 3 minutes while stirring at room temperature, and the mixture was left at room temperature for 3 hours.
- the resulting precipitate was filtered and washed twice with 50 mL of distilled water, and dried at 70 ° C. for 14 hours in air to obtain a purple solid.
- the resulting mixture was then crushed in a mortar and sieved to 125 ⁇ m or less to recover a pink powder (1.6 g).
- the cobalt content was evaluated by MP-AES, and the cobalt content was 30% by mass.
- Example 7 Thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to CoCO 3 (manufactured by Fujifilm Wako Pure Chemical Industries, Ltd.).
- Example 8 A thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to nickel-silicon composite oxide (NiO x —SiO y ).
- NiO x —SiO y was prepared by the following method. Nickel acetate tetrahydrate (Kishida Chemical Co., Ltd., 4.98 g, 20 mmol) was dissolved in 90 mL of distilled water, and a solution of sodium silicate (Sigma-Aldrich Co., Ltd., 2.44 g, 20 mmol) dissolved in 10 mL of distilled water was added dropwise over 3 minutes while stirring at room temperature, and the mixture was left at room temperature for 3 hours.
- the resulting precipitate was filtered and washed with 2 x 50 mL of distilled water, and dried at 70 ° C. for 14 hours under air to obtain a purple solid. Then, the mixture was crushed in a mortar and sieved to 125 ⁇ m or less to recover a green powder (3.97 g).
- the nickel content was evaluated by MP-AES, and the nickel content was 29% by mass.
- Example 9 Thiol (2a) was obtained in the same manner as in Example 1, except that the type of metal catalyst was changed from Co 3 O 4 to ⁇ -Co 2 SiO 4.
- ⁇ -Co 2 SiO 4 was prepared by the following method.
- CoO x -SiO y (1.1 g) was transferred to a quartz or alumina board, charged into a vacuum sintering furnace equipped with a hydrogen generator, substituted with hydrogen three times, and heated to 1000° C. in a hydrogen atmosphere for 3 hours and 18 minutes (heating program: 5° C./min). The temperature was then maintained for 3 hours under a hydrogen atmosphere. After sintering, the mixture was cooled to room temperature, and purple powder was collected (1 g). The cobalt content was evaluated by MP-AES, and was found to be 25% by mass.
- Example 10 Thiol (2a) was obtained in the same manner as in Example 6, except that the amount of powdered sulfur (S 8 ) was changed from 3.3 molar equivalents to 1.7 molar equivalents in terms of S element.
- Example 11 Thiol (2a) was obtained in the same manner as in Example 6, except that zeolite was not used.
- Example 12 Thiol (2a) was obtained in the same manner as in Example 6, except that zeolite was not used and the hydrogen pressure during charging was changed from 7.0 MPa to 5.0 MPa.
- Example 13 Thiol (2a) was obtained in the same manner as in Example 6, except that zeolite was not used and the hydrogen pressure during charging was changed from 7.0 MPa to 3.0 MPa.
- Example 15 Thiol (2a) was obtained in the same manner as in Example 6, except that zeolite was not used and the heating temperature of the autoclave, i.e., the reaction temperature, was changed from 130°C to 140°C.
- Example 10 Even when the amount of powdered sulfur (S 8 ) was changed from 3.3 molar equivalents to 1.7 molar equivalents as S element, thiol (2a) could be obtained with a yield of 27%.
- Example 11 it was found that even when zeolite was not used, thiol (2a) could be obtained with a yield of 64%, similar to that of Example 1.
- Examples 12 and 13 it was found that even when zeolite was not used and the hydrogen pressure during charging was changed from 7.0 MPa to 5.0 MPa or 3.0 MPa, thiol (2a) could be obtained with a yield of 54% or 18%.
- Examples 14 and 15 it was found that even when zeolite was not used and the reaction temperature was changed from 130° C. to 120° C. or 140° C., thiol (2a) could be obtained with a yield of 55% or 62%.
- Comparative Example 1 when no catalyst was used, the conversion rate was not particularly low at 92%, but the yield of thiol (2a) was low at 2%, and the ratio of the yield of thiol (2a) to the total yield of thiol (2a) and dialkyl monosulfides (3a) to (5a) was low at 18.1%.
- Example 16 A stirrer, 6 mmol of compound (1a) (H 2 C ⁇ C-C 12 H 25 ), 0.4125 molar equivalents of powdered sulfur (S 8 ) (3.3 molar equivalents as S element), and 6.6 mol% of CoO x -SiO y were charged into the autoclave, and hydrogen was charged under pressure up to 7.0 MPa. The autoclave was heated to 130° C. while stirring at 800 rpm with a magnetic stirrer, and the reaction was carried out at the same temperature for a predetermined time (t).
- Example 6 when CoO x -SiO y is used as a catalyst in the synthesis reaction of thiol (2a), its crystal structure changes to one close to the crystal structure of Co x S y used in Examples 2 and 3, and it was confirmed that the crystal structure is different from the crystal structure of ⁇ -Co 2 SiO 4 used in Example 9.
- Example 6 In addition, several samples of CoO x -SiO y after use in Example 6 were prepared, and the organic matter was oxidized using sulfuric acid and aqua regia, and cobalt (Co) was extracted in an aqueous solution. The cobalt content in the resulting aqueous solution was measured, and the cobalt content was less than 0.09 ppm in all samples. From this result, it was confirmed that the cobalt (Co) contained in CoO x -SiO y was hardly dissolved in the synthesis reaction, and that the production method can maintain the catalytic activity that the catalyst originally has.
- Example 18 to 35 Thiols (2b) to (2s) shown in Figure 8 were synthesized in the same manner as in Example 6, except that compound (1a) (H 2 C ⁇ C—C 12 H 25 ) was replaced with compounds (1b) to (1s) shown in Figure 7. The yields of the obtained thiols (2b) to (2s), respectively, are shown in Figure 8. As shown in FIG. 8, in all of Examples 18 to 35, it was found that by reacting any one of the compounds (1b) to (1s) with powdered sulfur in the presence of hydrogen, CoO x —SiO y , and zeolite, thiols (2b) to (2s) could be obtained in yields of 36 to 94%.
- the above-mentioned method for producing thiol does not require hydrogen sulfide gas, which is difficult to handle, or expensive organic bases, and does not require the installation of ultraviolet light irradiation equipment or platinum electrodes, etc.
- Thiol can be obtained using hydrogen gas and a catalyst, which are relatively easy to obtain and handle, making it extremely useful as thiol can be easily produced in a variety of regions.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
本出願は、2023年3月7日に、日本に出願された特願2023-034877に基づき優先権を主張し、その内容をここに援用する。
[1]アルケン(1)又はその誘導体と硫黄とを、水素及び金属元素の存在下で反応させてチオール(2)を得る、チオールの製造方法。
上記工程で使用される(1)は、末端又はその一部に二重結合を有していれば特に制限されないが、例えば非環式オレフィン(1A)、環式オレフィン(1B)又はこれらの誘導体である。
本実施形態で使用される非環式オレフィン(1A)は、例えばR1R2C=CH2 ・・・(A)(式中、R1はアルキル基であり、R2は水素原子またはアルキル基であり、R1及びR2の炭素原子数の合計は2~20である。)で表される。硫黄の特性を活かした工業用原料の観点からは、アルケン(1)の中でも、R1とR2の炭素原子数の合計が2~18であるアルケンが好ましく、R1とR2の炭素原子数の合計が2~16であるアルケンがより好ましい。本実施形態で使用される非環式オレフィン(1A)としては、例えば、後述の化合物(1a)、(1b)、(1h)等が挙げられる(図9参照)。
本実施形態で使用される硫黄としては、特に限定されるものではなく、例えば、小塊状、フレーク状、粉末状の固形状態であっても、溶融状態(液体)であっても良い。中でも、大スケールでの製造での仕込み作業が容易である観点からは、溶融状態の硫黄が好ましい。
本実施形態で使用される水素は、典型的には水素ガスである。水素ガスは流通量が多く、硫化水素と比べて入手し易い。また、水素ガスは、可燃性ではあるものの毒性がなく無臭であるため、硫化水素と比べて取扱いが容易である。水素ガスは、特に制限されないが、例えば99.99%以上、99.999%以上、或いは99.9999%以上とすることができる。
本実施形態で使用される金属元素は、第6族~第11族から選択された1又は複数の金属元素であるのが好ましい。このような金属元素としては、例えば以下のものが挙げられる。
第6族:クロム(Cr)、モリブデン(Mo)、タングステン(W)
第7族:マンガン(Mn)、テクネチウム(Tc)、レニウム(Re)
第8族:鉄(Fe)、ルテニウム(Ru)、オスミウム(Os)
第9族:コバルト(Co)、ロジウム(Rh)、イリジウム(Ir)
第10族:ニッケル(Ni)、パラジウム(Pd)、白金(Pt)
第11族:銅(Cu)、銀(Ag)、金(Au)
第6族の金属酸化物:CrO3、MoO3、WO3
第7族の金属酸化物:Mn2O3、Tc2O7、ReO3
第8族の金属酸化物:Fe2O3、RuO2、OsO4
第9族の金属酸化物:CoO、Rh2O3、IrO2
第10族の金属酸化物:NiO、PdO、PtO2
第11族の金属酸化物:CuO、Ag2O、Au2O3
第6族の金属硫化物:Cr2S3、MoS2、WS2
第7族の金属硫化物:MnS、ReS2
第8族の金属硫化物:FeS、RuS2、OsS2
第9族の金属硫化物:CoS、Rh2S3、IrS2
第10族の金属硫化物:Ni3S2、PdS、PtS
第11族の金属硫化物:CuS、Ag2S、Au2S
本実施形態の製造方法では、アルケン(1)と硫黄とを水素及び金属元素の存在下で反応させてチオール(2)を得るが、これに限らず、アルケン(1)と硫黄とを、水素、前記金属元素及びゼオライトの存在下で反応させてもよい。金属元素及びゼオライトを触媒とし、水素の存在下で、当該2つの触媒によってアルケン(1)と硫黄とを接触反応させることにより、得られるチオール(2)の収率をより向上することができる。
ゼオライトは、結晶性アルミノケイ酸塩であり、シリカとアルミナが規則的に連結した骨格構造を有すると共に、当該細孔構造中にイオン交換サイトとしての陽イオンを含有している。ゼオライトの骨格構造によって規定される細孔構造としては、特に制限されず、LTA(A型)、FER(フェリエライト)、MWW(MCM-22)、MFI(ZSM-5)、MOR(モルデナイト)、LTL(L型)、FAU(Y型、X型)、BEA(ベータ型)などが挙げられる。これらのうち、入手容易さおよび経済的な観点から、ゼオライトがX型又はA型の細孔構造を有するのが好ましい。
オートクレーブに攪拌子、化合物(1a)(H2C=C-C12H25)6mmol、硫黄(S8)0.4125モル当量(S元素として3.3モル当量)、Co3O4(シグマアルドリッチ社製、ナノパウダー:50nm以下(TEM))6mol%、ゼオライト(シグマアルドリッチ社製、モレキュラーシーブ4A、Na-A型)100mgを仕込んだ後、水素を7.0MPaまで加圧仕込を行った。マグネチックスターラーで800rpmで攪拌しながら、オートクレーブを130℃まで加温し、同温度で16時間反応を行った。その後、室温に冷却して圧力弁を開放した後、内部標準物質としてトリデカンを20mg(化合物(1a)100質量部に対して1.7質量部)を反応溶液に加え、さらに、空気を吹き込んで残留する硫化水素を留去した。未反応硫黄と触媒を遠心分離機にて除いた後、チオール(2a)を得た。実施例1のチオール(2a)の製造方法における反応を図2に示す。
金属触媒の種類をCo3O4からCoS(Strem Chemicals社製)に変更したこと以外は、実施例1と同様にしてチオール(2)を得た。
金属触媒の種類をCo3O4からCoS2(Alfa Aesar社製)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。
金属触媒の種類をCo3O4からニッケル-コバルト複合酸化物(NiOx-CoOy)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。ニッケル-コバルト複合酸化物は、以下の方法で調製した。
酢酸ニッケル四水合物(キシダ化学社製、2.48g、10mmol)と硝酸コバルト九水合物(富士フィルム和光純薬工業社製、5.82g、20mmol)を150mLの蒸留水に溶解し室温下で炭酸アンモニウム(富士フィルム和光純薬工業社製、2.88g、30mmol)を50mL蒸留水に溶かした溶液を攪拌しながら、5分かけて滴下し、そのまま3時間室温下で放置した。反応終了後、得られた沈殿物を濾別し蒸留水2×50mLで洗浄し、70℃、15時間空気下で乾燥して黒の固体を得た。その後、擂鉢で粉砕し、篩で125μm以下に細かくした黒の粉末を回収した。その後、焼成皿に移して、焼成を行った。焼成条件は、1時間で(昇温プログラム:5℃/min)300度まで昇温し、そのまま6時間同温度で保持した。焼成後、室温に冷却し、黒の粉末を回収した(2.16g)。コバルト、ニッケルの含有量をマイクロ波プラズマ原子発光分光分析装置(MP-AES)で評価したところ、コバルトの含有量は38質量%、ニッケルの含有量は11質量%であった。
金属触媒の種類をCo3O4からNiO(シグマアルドリッチ社製)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。
金属触媒の種類をCo3O4からコバルト-ケイ素複合酸化物(CoOx-SiOy)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。コバルト-ケイ素複合酸化物は、以下の方法で調整した。
硝酸コバルト六水合物(富士フィルム和光純薬工業社製、2.91g、10mmol)を90mLの蒸留水に溶解し、室温下でケイ酸ナトリウム(シグマアルドリッチ製、1.22g、10mmol)を10mL蒸留水に溶かした溶液を攪拌しながら、3分かけて滴下し、そのまま3時間室温下で放置した。反応終了後、得られた沈殿物を濾別し蒸留水50mLで2回洗浄し、70℃、14時間空気下で乾燥して紫色の固体を得た。その後、擂鉢で粉砕し、篩で125μm以下に細かくしたピンク色の粉末(1.6g)を回収した。コバルトの含有量をMP-AESで評価したところ、コバルト含有量は30質量%であった。
金属触媒の種類をCo3O4からCoCO3(富士フィルム和光純薬工業社製)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。
金属触媒の種類をCo3O4からニッケル-ケイ素複合酸化物(NiOx-SiOy)に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。NiOx-SiOyは、以下の方法で調製した。
酢酸ニッケル四水合物(キシダ化学社製、4.98g、20mmol)を90mLの蒸留水に溶解し室温下でケイ酸ナトリウム(シグマアルドリッチ社製、2.44g、20mmol)を10mL蒸留水に溶かした溶液を攪拌しながら、3分かけて滴下し、そのまま3時間室温下で放置した。反応終了後、得られた沈殿物を濾別し蒸留水2×50mLで洗浄し、70℃、14時間空気下で乾燥して紫色の固体を得た。その後、擂鉢で粉砕し、篩で125μm以下に細かくした緑色の粉末(3.97g)を回収した。ニッケルの含有量をMP-AESで評価したところ、ニッケル含有量は29質量%であった。
金属触媒の種類をCo3O4からα-Co2SiO4に変更したこと以外は、実施例1と同様にしてチオール(2a)を得た。α-Co2SiO4は、以下の方法で調製した。 CoOx-SiOy(1.1g)を石英又はアルミナボードに移して、水素発生装置付きの真空焼成炉に装入し、三回水素置換して、水素雰囲気下、3時間18分で(昇温プログラム:5℃/min)1000度まで昇温した。その後3時間水素雰囲気下、同温度で保持した。焼成後は室温に冷却し、紫色の粉末を回収した(1g)。コバルトの含有量をMP-AESで評価したところ、コバルト含有量は25質量%であった。
チオール(2a)及びジアルキルモノスルフィド(3a)~(5a)の収率は、ガスクロマトグラフ(アジレントテクノロジー社製、6850シリーズII)及びカラム(アジレントテクノロジー社製、HP-1、30m、直径0.32mm、膜厚0.25μm)を用い、以下の測定条件にてチオール(2)及びジアルキルモノスルフィド(3a)~(5a)のチャートを取得し、それぞれのピーク面積から求めた。転化率は、化合物(1a)の収率から求めた。実施例1~9の測定、評価結果を表1に示す。
(昇温プログラム)
1.40℃、5min保持
2.10℃/minで240℃まで昇温
3.240℃に到達後、15分保持
(GC各種設定)
・使用ガス:窒素(キャリアガス)、水素(検出器用)、空気(検出器用)
・注入口の設定:ヒーター200℃、圧力93kPa、トータルフロー(N2)24.5mL/min、スプリット比5.3:1。
・カラムの設定:圧力93kPa、流量(N2)3.6mL/min、平均線速度52cm/sec
・検出器設定:ヒーター250℃、水素流量30.0mL/min、空気流量250.0mL/min、メークアップ流量(N2)10.0mL/min
(各化合物の同定に使用した標準物質のGC保持時間)
・内部標準(トリデカン):13.5min
・テトラデセン:14.9min
・チオール:18.8min
・ジアルキルモノスルファン類(3a:38.7min、4a:36.0min、5a:34.8min)
粉末硫黄(S8)をS元素として3.3モル当量から1.7モル当量に変更したこと以外は、実施例6と同様にしてチオール(2a)を得た。
ゼオライトを使用しなかったこと以外は、実施例6と同様にしてチオール(2a)を得た。
ゼオライトを使用せず、且つ仕込み時の水素圧力を7.0MPaから5.0MPaに変更したこと以外は、実施例6と同様にしてチオール(2a)を得た。
ゼオライトを使用せず、且つ仕込み時の水素圧力を7.0MPaから3.0MPaに変更したこと以外は、実施例6と同様にしてチオール(2a)を得た。
ゼオライトを使用せず、且つオートクレーブの加熱温度、すなわち反応温度を130℃から120℃に変更したこと以外は、実施例6と同様にしてチオール(2a)を得た。
ゼオライトを使用せず、且つオートクレーブの加熱温度、すなわち反応温度を130℃から140℃に変更したこと以外は、実施例6と同様にしてチオール(2a)を得た。
触媒を使用しなかったこと以外は、実施例6と同様にしてチオール(2a)を得た。 実施例10~15及び比較例1の測定、評価結果を表2に示す。

オートクレーブに攪拌子、化合物(1a)(H2C=C-C12H25)6mmol、粉末硫黄(S8)0.4125モル当量(S元素として3.3モル当量)、CoOx-SiOy6.6mol%を仕込んだ後、水素を7.0MPaまで加圧仕込を行った。マグネチックスターラーで800rpmで攪拌しながら、オートクレーブを130℃まで加温し、同温度で所定時間(t)で反応を行った。その後、室温に冷却して圧力弁を開放した後、内部標準物質としてトリデカンを20mg(化合物(1a)100質量部に対して1.7質量部)を反応溶液に加え、さらに、空気を吹き込んで残留する硫化水素を留去した。未反応硫黄とCo3O4を遠心分離機にて除いた後、チオール(2a)を得た。実施例16におけるチオール(2a)の収率、及びジアルキルモノスルフィド(3a)~(5a)の合計収率の経時変化を図3に示す。
実施例2,3での使用前のCoS、CoS2、実施例6での使用前のCoOx-SiOy、実施例6での使用後のCoOx-SiOy、及び実施例9での使用前のα-Co2SiO4に対し、X線回折装置(Rigaku社製、装置名「MiniFlex600」)を用いてXRD測定を行った。XRD測定は、以下の条件で行った。結果を図4に示す。
Cu Kα線源使用(λ=0.15418nm)
測定速度:1.5°/min
室温、高真空条件下
データ取り込み時間:20mmsec
分解能 128×128
化合物(1a)(H2C=C-C12H25)を図7に示す化合物(1b)~(1s)に代えたこと以外は実施例6と同様にして、図8に示すチオール(2b)~(2s)を合成した。得られたチオール(2b)~(2s)のそれぞれの収率を図8に示す。
図8に示すように、実施例18~35のいずれでも、化合物(1b)~(1s)のいずれかと粉末硫黄とを、水素、CoOx-SiOy及びゼオライトの存在下で反応させることにより、収率36~94%でチオール(2b)~(2s)を得られることが分かった。
Claims (12)
- アルケン(1)又はその誘導体と硫黄とを、水素及び金属元素の存在下で反応させてチオール(2)を得る、チオールの製造方法。
- アルケン(1)又はその誘導体と硫黄とを反応させる際、供給される水素の圧力が0.1MPa以上10MPa以下である、請求項1に記載のチオールの製造方法。
- アルケン(1)又はその誘導体と硫黄とを水素の存在下で反応させる際の加熱温度が、100℃以上200℃以下である、請求項1又は2に記載のチオールの製造方法。
- 前記金属元素が、第6族~第11族から選択された1又は複数の金属元素である、請求項1に記載のチオールの製造方法。
- 前記金属元素が、金属酸化物又は金属硫化物を構成する金属元素である、請求項4に記載のチオールの製造方法。
- 前記アルケン(1)又はその誘導体に対する前記金属元素の添加量が、0.1mol%以上10mol%以下である、請求項1に記載のチオールの製造方法。
- 前記アルケン(1)又はその誘導体と硫黄とを、水素、前記金属元素及びゼオライトの存在下で反応させる、請求項1に記載のチオールの製造方法。
- 前記ゼオライトが塩基性を有する、請求項7に記載のチオールの製造方法。
- 前記ゼオライトが、X型又はA型の細孔構造を有する、請求項8に記載のチオールの製造方法。
- 前記ゼオライトの添加量が、前記アルケン(1)100質量部に対して2.0質量部以上25質量部以下である、請求項7に記載のチオールの製造方法。
- 前記アルケン(1)が、R1R2C=CH2 ・・・(A)(式中、R1はアルキル基であり、R2は水素原子またはアルキル基であり、R1及びR2の炭素原子数の合計は2~20である。)で表される、請求項1に記載のチオールの製造方法。
- 上記一般式(A)中のR1とR2の炭素原子数の合計が2~16である、請求項11に記載のチオールの製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP24766906.2A EP4678631A1 (en) | 2023-03-07 | 2024-02-22 | Thiol production method |
| JP2024571297A JP7697643B2 (ja) | 2023-03-07 | 2024-02-22 | チオールの製造方法 |
| CN202480013627.2A CN120693320A (zh) | 2023-03-07 | 2024-02-22 | 硫醇的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023034877 | 2023-03-07 | ||
| JP2023-034877 | 2023-03-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024185535A1 true WO2024185535A1 (ja) | 2024-09-12 |
Family
ID=92674906
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/006567 Ceased WO2024185535A1 (ja) | 2023-03-07 | 2024-02-22 | チオールの製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4678631A1 (ja) |
| JP (1) | JP7697643B2 (ja) |
| CN (1) | CN120693320A (ja) |
| WO (1) | WO2024185535A1 (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS388314B1 (ja) * | 1959-07-18 | 1963-06-07 | ||
| JPH0255210A (ja) * | 1988-08-17 | 1990-02-23 | Jgc Corp | 硫化水素の製造方法 |
| JP2001354643A (ja) * | 2000-04-28 | 2001-12-25 | Atofina | 硫化オレフィン類の製造方法 |
| US20060247475A1 (en) * | 2005-04-27 | 2006-11-02 | Hasenberg Daniel M | Method of making 2-thiols |
| JP2010524660A (ja) * | 2007-04-17 | 2010-07-22 | エボニック デグサ ゲーエムベーハー | メチルメルカプタン製造用触媒 |
-
2024
- 2024-02-22 JP JP2024571297A patent/JP7697643B2/ja active Active
- 2024-02-22 CN CN202480013627.2A patent/CN120693320A/zh active Pending
- 2024-02-22 EP EP24766906.2A patent/EP4678631A1/en active Pending
- 2024-02-22 WO PCT/JP2024/006567 patent/WO2024185535A1/ja not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS388314B1 (ja) * | 1959-07-18 | 1963-06-07 | ||
| JPH0255210A (ja) * | 1988-08-17 | 1990-02-23 | Jgc Corp | 硫化水素の製造方法 |
| JP2001354643A (ja) * | 2000-04-28 | 2001-12-25 | Atofina | 硫化オレフィン類の製造方法 |
| US20060247475A1 (en) * | 2005-04-27 | 2006-11-02 | Hasenberg Daniel M | Method of making 2-thiols |
| JP2010524660A (ja) * | 2007-04-17 | 2010-07-22 | エボニック デグサ ゲーエムベーハー | メチルメルカプタン製造用触媒 |
Non-Patent Citations (7)
| Title |
|---|
| "33", J. ORG. CHEM, 1968, pages 1275 - 1276 |
| CATALYSIS LETT., vol. 64, 2000, pages 197 - 200 |
| IND. ENG. CHEM., vol. 40, no. 12, 1948, pages 2308 - 2313 |
| J. ORG. CHEM., 6 July 1942 (1942-07-06), pages 472 - 476 |
| NIPPON KAGAKU KAISHI, vol. 7, 1987, pages 1502 - 1504 |
| PAVELKO G. F.: "Mechanochemical reactions of elementary sulfur and iron sulfides with hydrogen, oxygen, and water", RUSSIAN JOURNAL OF INORGANIC CHEMISTRY., CHEMICAL SOCIETY, LONDON., GB, vol. 53, no. 7, 1 July 2008 (2008-07-01), GB , pages 981 - 987, XP093208325, ISSN: 0036-0236, DOI: 10.1134/S0036023608070012 * |
| See also references of EP4678631A1 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7697643B2 (ja) | 2025-06-24 |
| EP4678631A1 (en) | 2026-01-14 |
| JPWO2024185535A1 (ja) | 2024-09-12 |
| CN120693320A (zh) | 2025-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6125023B2 (ja) | エチレンへのエタンの酸化脱水素及びこのようなプロセスのための触媒としての多金属混合酸化物の調製 | |
| JP5991553B2 (ja) | 炭素担持コバルト及びモリブデン触媒 | |
| CN101511472B (zh) | 含Mo催化剂、其制备方法和甲硫醇的制备方法 | |
| JP2007044602A (ja) | 触媒担体、金属固定化触媒、その製造方法およびそれを用いたアルコールの酸化方法 | |
| EP3868763B1 (en) | Compound and method for producing same | |
| US20040152791A1 (en) | Catalyst | |
| Kalong et al. | Continuous flow hydrogenolysis of 5-hydroxymethylfurfural into 2, 5-dimethylfuran over alumina-supported nickel–iron alloy catalysts | |
| JP4211900B2 (ja) | 金属微粒子担持炭化水素改質用触媒及びその製造方法 | |
| US10676419B2 (en) | Production of products from natural resources | |
| CN105618151B (zh) | 锶‑锗钨氧簇催化剂、制备方法及其用途 | |
| JP7697643B2 (ja) | チオールの製造方法 | |
| Li et al. | Ni–Fe nanoparticles supported on UiO-66-X catalyst for hydrogenation of fatty acid esters to alcohols | |
| CN105481666B (zh) | 一种用于合成气催化转化的方法 | |
| CN114409548A (zh) | 一种光催化制备苯甲胺类化合物的方法 | |
| CN107790173B (zh) | 一种制备二甲基硫醚的催化剂以及合成二甲基硫醚的方法 | |
| CN108558792A (zh) | 4-(6-氨基吡啶-3-基)哌嗪-1-羧酸叔丁酯的制备方法 | |
| Yang et al. | Aerobic Thiols Oxidative Coupling to Disulfides over Robust CoO x Nanoclusters Confined within Hierarchical Silicalite-1 Zeolite | |
| KR102103039B1 (ko) | CoCrMo-합금을 포함하는 금속 분말형 촉매 | |
| Sun et al. | High-nuclearity Ag-substituted tetrameric sandwich-type polyoxometalates for excellent catalytic performance in C–C bond formation | |
| Trinh et al. | Alternative pathways to α, β-unsaturated ketones via direct oxidative coupling transformation using Sr-doped LaCoO3 perovskite catalyst | |
| CN119386930B (zh) | 基于Ni-MOF金属节点部分还原制备的贵金属-Ni共存负载型催化剂、制备方法及应用 | |
| Anbari et al. | MOF-derived cobalt catalysts for sustainable tandem hydroformylation–acetalization in green solvents: experimental and DFT calculations | |
| CN100389872C (zh) | 一种含高浓度硫化氢的合成气一步法制甲硫醇的催化剂 | |
| Yang | Organic coupling reactions by heterogeneous photocatalysis | |
| 許柯 | Novel Synthetic Method of Thiols using Heterogeneous Cobalt Catalysts from Hydrogen, Elemental Sulfur and Alkenes and Other Substrates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24766906 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024571297 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202480013627.2 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 202480013627.2 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024766906 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024766906 Country of ref document: EP Effective date: 20251007 |
|
| ENP | Entry into the national phase |
Ref document number: 2024766906 Country of ref document: EP Effective date: 20251007 |
|
| ENP | Entry into the national phase |
Ref document number: 2024766906 Country of ref document: EP Effective date: 20251007 |
|
| ENP | Entry into the national phase |
Ref document number: 2024766906 Country of ref document: EP Effective date: 20251007 |