BR102020009417A2 - Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo - Google Patents
Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo Download PDFInfo
- Publication number
- BR102020009417A2 BR102020009417A2 BR102020009417-3A BR102020009417A BR102020009417A2 BR 102020009417 A2 BR102020009417 A2 BR 102020009417A2 BR 102020009417 A BR102020009417 A BR 102020009417A BR 102020009417 A2 BR102020009417 A2 BR 102020009417A2
- Authority
- BR
- Brazil
- Prior art keywords
- seq
- glyma
- target
- glycine
- plants
- Prior art date
Links
Images





Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H1/00—Processes for modifying genotypes ; Plants characterised by associated natural traits
- A01H1/04—Processes of selection involving genotypic or phenotypic markers; Methods of using phenotypic markers for selection
- A01H1/045—Processes of selection involving genotypic or phenotypic markers; Methods of using phenotypic markers for selection using molecular markers
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H1/00—Processes for modifying genotypes ; Plants characterised by associated natural traits
- A01H1/04—Processes of selection involving genotypic or phenotypic markers; Methods of using phenotypic markers for selection
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H1/00—Processes for modifying genotypes ; Plants characterised by associated natural traits
- A01H1/12—Processes for modifying agronomic input traits, e.g. crop yield
- A01H1/122—Processes for modifying agronomic input traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance
- A01H1/1245—Processes for modifying agronomic input traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance for biotic stress resistance, e.g. pathogen, pest or disease resistance
- A01H1/1255—Processes for modifying agronomic input traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance for biotic stress resistance, e.g. pathogen, pest or disease resistance for fungal resistance
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H6/00—Angiosperms, i.e. flowering plants, characterised by their botanic taxonomy
- A01H6/54—Leguminosae or Fabaceae, e.g. soybean, alfalfa or peanut
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H6/00—Angiosperms, i.e. flowering plants, characterised by their botanic taxonomy
- A01H6/54—Leguminosae or Fabaceae, e.g. soybean, alfalfa or peanut
- A01H6/542—Glycine max [soybean]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/415—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from plants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/6895—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for plants, fungi or algae
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6858—Allele-specific amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/13—Plant traits
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Botany (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Developmental Biology & Embryology (AREA)
- Environmental Sciences (AREA)
- Analytical Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Physics & Mathematics (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Physiology (AREA)
- Natural Medicines & Medicinal Plants (AREA)
- Mycology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes à mancha-alvo, molécula de ácido nucleico e seu uso e kit de detecção. a presente invenção se refere um método de identificação e seleção de plantas resistentes à uma doença fúngica compreendendo as etapas de (a) extração de ácido nucleico de uma planta, (b) análise do ácido nucleico extraído para presença de marcadores associados com aumento de resistência ao fungo dentro de um intervalo de um cromossomo; e (c) seleção das plantas que possuam os referidos marcadores. ademais, a invenção se refere ainda a um método de introgressão em plantas de alelos de resistência à doença fúngica compreendendo as etapas de (a) cruzar parentais de plantas identificadas pelo método da primeira concretização com outros parentais que não possuem a referida resistência; (b) selecionar progênies possuindo marcadores associados com aumento de resistência à doença fúngica através do método como definido na primeira concretização; e (c) retrocruzar em um ou mais ciclos as progênies selecionadas com o genitor recorrente para desenvolver novas progênies.
Description
[001] A presente invenção refere-se ao campo da biologia vegetal e da biotecnologia. Especificamente, a presente invenção refere-se a um método de melhoramento de plantas de modo a identificar plantas, através de marcadores moleculares, com maior resistência a doenças, mais especificamente plantas do gênero Glycine e doenças fúngicas.
[002] A soja pertence ao gênero botânico Glycine, mais precisamente à família Fabaceae (leguminosas). São reconhecidos cerca de 727 gêneros e 19.325 espécies (LEWIS, G. P.; SCHRIRE, B. D.; MACKINDER, B. A.; LOCK, J. M. Legumes of the World. Royal Botanic Gardens, Kew. p. 577, 2005), representando uma das maiores famílias de Angiospermas e também uma das principais, do ponto de vista econômico.
[003] Esta família apresenta distribuição cosmopolita e possui como característica principal, apesar de haver exceções, o fruto do tipo legume (vagem). Ademais, engloba desde espécies arbóreas até espécies herbáceas anuais, muitas de grande importância econômica e, principalmente, alimentar (soja, feijão, entre outras).
[004] Além disso, os representantes desta família ainda possuem grande importância ecológica, pois estão bem adaptadas à primeira colonização e exploração de diversos ambientes, principalmente devido às suas associações com bactérias fixadoras de nitrogênio ou com ectomicorrizas. Bactérias do gênero Rhizobium, localizadas em nódulos radiculares encontrados em muitas espécies, convertem o nitrogênio atmosférico em amônia, forma solúvel que pode ser utilizada por outros vegetais, resultando em espécies extremamente valiosas como fornecedores de adubos naturais (LEWIS, G. P. Legumes of Bahia. Royal Botanic Gardens, Kew. p. 369, 1987).
[005] A soja (Glycine max) é um dos representantes mais importante da família Fabaceae. Na década de 70, a soja se consolidou como a principal cultura do agronegócio brasileiro. O produtor tem usado de todos os meios para incrementar o uso de tecnologia, a fim de reduzir seus custos, aumentar sua produtividade e, dessa forma, melhorar sua rentabilidade. Assim, a produtividade da soja saltou de 2.823 kg/ha na safra 2006/07, para 3.394 kg/ha na safra 2017/18, um incremento de 20% (Acomp. safra bras. grãos, v. 6 - Safra 2018/19 - Décimo levantamento, Brasília). Os dados mais recentes mostram que a soja gera faturamento de R$ 148,6 bilhões em 2018 e a maior receita na exportação, tendo atingido US$ 40 bilhões no mesmo ano (Cleonice de Carvalho, et al. Anuário brasileiro da soja 2019. Santa Cruz do Sul: Editora Gazeta Santa Cruz, P. 14, 2019).
[006] A demanda mundial de proteína animal de qualidade, principalmente aves, vem continuamente aumentando ao redor do mundo (HENCHION, M.; McCARTHY, M.; RESCONI, V.C.; TROY, D. Meat consumption: trends and quality matter. Meat Science, v.98, p.561-568, 2014.). Desta forma, tal crescente demanda também gera um aumento na demanda de farelos proteicos utilizados na fabricação de ração animal, normalmente oriundos de grãos de soja (Embrapa (2011) Tecnologias de Produção de Soja, Região Central do Brasil 2012 e 2013. Londrina PR. Embrapa Soja).
[007] Projeta-se que o consumo mundial de grãos de soja no ano agrícola 2019/20 passe para 352 milhões de toneladas, ante 345 milhões de toneladas consumidos em 2018/19 (Cleonice de Carvalho, et al. Anuário brasileiro da soja 2019. Santa Cruz do Sul: Editora Gazeta Santa Cruz, P. 14, 2019).
[008] Ademais a área cultivada de soja cresceu quando comparado o período 2017/18 com 2018/19 de 124,52 milhões de hectares para 125,64 milhões de hectares (USDA, Global Market Analysis, Fevereiro de 2020).
[009] Devido à importância econômica da soja no cenário agrícola brasileiro, os programas de melhoramento da soja têm como objetivo desenvolver cultivares mais produtivas e resistentes a doenças e pragas presentes nas diferentes regiões do Brasil. Uma parte fundamental do sucesso dos programas de melhoramento para seleção de genótipos resistentes reside no uso de fontes de inóculo (isolados dos fungos) representativas da diversidade local com espectro de virulência e agressividade conhecidos (Bermejo, Gabriela Rastelli. Diversidade genética de isolados brasileiros de Phakopsora pachyrhizi (Sydow & Sydow)/ Gabriela Rastelli Bermejo; orientação Mayra Costa da Cruz Gallo de Carvalho. – Bandeirantes: Universidade Estadual do Norte do Paraná, 2016).
[0010] Neste cenário, o melhoramento da soja para resistência ou tolerância a diversos patógenos é crucial para diminuir fatores restritivos e maximizar a produtividade. Dentre os patógenos, destaca-se o fungo Corynespora cassiicola (Berk. & M.A. Curtis) C.T. Wei, o agente etiológico da doença conhecida como mancha-alvo. É considerada uma das doenças mais importantes economicamente para a produção de soja no Brasil, principalmente na região do Cerrado (Almeida AMR, Ferreira LP, Yorinori JT, Silva JFV, Henning AA, Godoy CV, Costamilan LM, Meyer MC (2005) Doenças da soja. In: Kimati H, Amorim L, Rezende JAM, Bergamin Filho A, Camargo LEA (Eds.). Manual de Fitopatologia - Vol. 2. Doenças de Plantas Cultivadas. 4. ed. São Paulo SP. Editora Agronômica Ceres. pp. 570-588.).
[0011] O referido fungo é encontrado em praticamente todas as regiões de cultivo de soja do Brasil. Acredita-se ser nativo e com habilidade de infectar um grande número de espécies de plantas, a exemplo do algodão, aumentando a sua adaptabilidade em áreas onde a sucessão de culturas sojaalgodão é realizada (GALBIERI, R.; ARAÚJO, D.C.E.B.; KOBAYASTI, L.; GIROTTO, L.; MATOS, J.N.; MARANGONI, M.S.; ALMEIDA, W.P.; MEHTA, Y.R. Corynespora leaf blight of cotton in Brazil and its management. American Journal of Plant Sciences 5: 3805-3811. 2014).
[0012] O referido microrganismo pode sobreviver em restos de cultura e sementes infectadas, essa consistindo em uma forma de disseminação. Estima-se que a doença pode ocasionar uma redução de rendimento de 24%, com variações entre 8 – 42% em lavouras de soja com alta pressão da doença (GALBIERI, R.; ARAÚJO, D.C.E.B.; KOBAYASTI, L.; GIROTTO, L.; MATOS, J.N.; MARANGONI, M.S.; ALMEIDA, W.P.; MEHTA, Y.R. Corynespora leaf blight of cotton in Brazil and its management. American Journal of Plant Sciences 5: 3805-3811. 2014)
[0013] Surtos severos, mas esporádicos, têm sido observados nas regiões mais frias do Sul e nas regiões altas dos Cerrados. Cultivares suscetíveis podem sofrer completa desfolha prematura, apodrecimento das vagens e manchas nas hastes. Através da infecção na vagem, o fungo pode atingir a semente e, desse modo, ser disseminado para outras áreas. A infecção, na região da sutura das vagens em desenvolvimento, pode resultar em necrose, abertura das vagens e germinação ou apodrecimento dos grãos ainda verdes (Embrapa (2011) Tecnologias de Produção de Soja, Região Central do Brasil 2012 e 2013. Londrina PR. Embrapa Soja.).
[0014] As condições de alta umidade relativa e temperaturas amenas são favoráveis à infecção foliar. Os sintomas mais comuns são manchas nas folhas, com halo amarelado e pontuação escura no centro, que causam severa desfolha. Ocorrem também manchas na haste e na vagem. O fungo pode infectar raízes, causando podridão radicular e intensa esporulação (Henning et al., 2005, supra).
[0015] Neste sentido, de maneira geral, a infecção por esse patógeno pode ser observada em todas as partes das plantas acima do solo (GALBIERI, R.; ARAÚJO, D.C.E.B.; KOBAYASTI, L.; GIROTTO, L.; MATOS, J.N.; MARANGONI, M.S.; ALMEIDA, W.P.; MEHTA, Y.R. Corynespora leaf blight of cotton in Brazil and its management. American Journal of Plant Sciences, v.5, p. 3805-3811, 2014; 2. HARTMAN, G.L.; RUPE, J.C.; SIKORA, E.J.; DOMIER, L.L.; DAVIS, J.A.; STEFFEY, K.L. Compendium of soybean diseases and pests. In: HARTMAN et al. (Ed.). 5th. ed. The American Phytopathological Society, St. Paul, MN. 201p., 2015).
[0016] O progresso da mancha alvo em campo é mais lento se comparado à ferrugem asiática, porém, após a doença instalada, é de difícil controle. As estratégias de manejo recomendadas para essa doença são: rotação com culturas não hospedeiras, tratamento de sementes, controle químico em doses e intervalos corretos e utilização de cultivares resistentes. Entretanto, a falta de informação da reação de cultivares de soja a esta doença dificulta o seu manejo, sendo o controle químico utilizado com uma das alternativas mais viáveis (MEYER, M.; GODOY, C.; VENANCIO, W.; TERAMOTO, A. Manejo equilibrado. Revista Cultivar, v.165, p.03-07, 2013). No caso de controle químico, deve-se preconizar sempre a associação de fungicidas multissítios e sempre iniciar o manejo de forma preventiva. A utilização de fungicida isolado e de forma curativa pode eliminar populações mais sensíveis do fungo, aumentando a frequência dos menos sensíveis (Teramoto, A.; Meyer, M.C.; Suassuna, N.D.; Cunha, M.G. In vitro sensitivity of Corynespora cassiicola isolated from soybean to fungicides and field chemical control of target spot. Summa Phytopathologica, v.43, n.4, p.281-289, 2017).
[0017] A arquitetura genética para resistência a doenças tem sido estabelecida por diversos estudos de mapeamento associativo, que apontam para um caráter monogênico ou poligênico, dependendo do tipo de interação entre patógeno e hospedeiro. Os mesmos estudos permitiram a identificação de polimorfismos de DNA nos locci de maior efeito associados às respostas de resistência. Neste contexto, estudos de mapeamento associativo são de grande utilidade para os programas de melhoramento de plantas por possibilitar o mapeamento de loci e obter conhecimento sobre a posição de um gene e a sua região adjacente. Ainda, tais estudos permitem a interpretação dos possíveis mecanismos de resistência e a predição da herança da característica em cruzamentos controlados, além de contribuir para análises de sintenia ou mapeamento comparativo e clonagem de genes (Xuehui Huang and Bin Han, Natural Variations and Genome-Wide Association Studies in Crop Plants, Annual Review of Plant Biology, 65: 531-551, 2014)
[0018] Modelos lineares mistos foram desenvolvidos e aplicados em mapeamento associativo para reduzir o número de associações falso-positivas causadas pela estrutura e relacionamento da população (YU, J.M.; PRESSOIR, G.; BRIGGS, W.H.; VROH BI, I.; YAMASAKI, M.; DOEBLEY, J.F.; MCMULLEN, M.D.; GAUT, B.S.; NIELSEN, D.M.; HOLLAND, J.B.; KRESOVICH, S.; BUCKLER, E.S. A unified mixed‑model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics, v.38, p.203‑208, 2006; ZHANG, Z.; ERSOZ, E.; LAI, C.‑Q.; TODHUNTER, R.J.; TIWARI, H.K.; GORE, M.A.; BRADBURY, P.J.; YU, J.; ARNETT, D.K.; ORDOVAS, J.M.; BUCKLER, E.S. Mixed linear model approach adapted for genome‑wide association studies. Nature Genetics, v.42, p.355‑360, 2010.).
[0019] Marcadores moleculares têm sido utilizados na identificação de polimorfismos associados a resistência a doenças. Em programas de melhoramento genético, a abordagem de seleção assistida por marcadores (SAM) tem sido amplamente utilizada por permitir a identificação de resistência a doenças ou outras características já nas primeiras fases e estádios iniciais do desenvolvimento das plantas.
[0020] Com o uso do SAM, alelos desfavoráveis podem ser eliminados ou bastante reduzidos nas primeiras gerações, o que permite a avaliação e seleção de um número otimizado de plantas no campo. Em outra aplicação, a SAM pode facilitar a introgressão de alelos favoráveis de fontes de resistência em linhagens elite (Shi, Z., Liu, S., Noe, J. et al. SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance. BMC Genomics 16, 314 (2015). https://doi.org/10.1186/s12864-015-1531-3).
[0021] As cultivares resistentes normalmente são desenvolvidas pela transferência de alelos de resistência de germoplasma, muitas vezes não adaptado, para cultivares elite. Devido a ampla variabilidade genética de espécies fúngicas e suas constantes adaptações, é comum o surgimento de novos isolados que desafiam a resistência genética já introduzida em cultivares elite. Desta forma, é fundamental que seja explorada uma ampla base genética no germoplasma para garantir a longevidade da resistência (ALZATE-MARIN, A L.; CERVIGNI, G. D. L.; MOREIRA, M. A; (2005) Marker assisted selection in the development of disease resistant plants, with emphasis on common bean and soybean. Fitopatologia brasileira. v.30, no.4,p.333-342).
[0022] Neste contexto, estudos de associação genômica ampla são de grande utilidade para os programas de melhoramento de plantas por possibilitar o mapeamento de loci que controlam características qualitativas ou quantitativas (QTLs – Quantitative Trait Loci), e por proporcionam o conhecimento sobre a posição de um gene e a sua região adjacente. Ainda, tais estudos permitem a interpretação dos mecanismos evolutivos e a predição de descendências de cruzamentos controlados, além de contribuir para estudos análises de sintenia ou mapeamento genético e clonagem de genes.
[0023] O mapa genético é uma representação gráfica de um genoma (ou uma parte de um genoma, como um único cromossomo), em que as distâncias entre pontos de referência no cromossomo são medidas pelas frequências de recombinação entre os referidos pontos. Um ponto de referência genético pode ser qualquer um de uma variedade de marcadores polimórficos conhecidos, por exemplo, mas não limitado a marcadores moleculares, tais como marcadores do tipo SSR (Simple Sequence Repeats), marcadores do tipo RFLP (Restriction Fragment Length Polymorphism) ou marcadores do tipo SNP (Single nucleotide polymorphism). Além disso, os marcadores do tipo SSR podem ser derivados de ácidos nucleicos genômicos ou expressos (por exemplo, ESTs (Expressed sequence tags)).
[0024] Marcadores associados aos genes ou QTLs, depois de mapeados e avaliados quanto a influência na variação fenotípica, podem ser utilizados para SAM, o que torna o processo de escolha de um determinado genótipo rápido e eficiente, tornando a ferramenta de grande contribuição para o melhoramento genético de plantas (Collins, PJ, et al, Marker assisted breeding for disease resistance in Crop Plants. Biotechnologies of Crop Improvement, v3, 41-47, 2018).
[0025] Recentemente, a seleção assistida por marcadores aumentou a eficiência dos programas tradicionais de melhoramento de soja. Ademais, a disponibilidade de mapas de ligação integrados do genoma da soja contendo densidades crescentes de marcadores públicos de soja facilitou o mapeamento genético da soja e aplicações de SAM (Cregan et al. (1999) "An Integrated Genetic Linkage Map of the Soybean Genome" Crop Sci. 39:1464-1490).
[0026] SNPs (Single nucleotide polymorphism) são marcadores que consistem em uma sequência compartilhada diferenciada com base em um único nucleotídeo.
[0027] Os SNPs entre fragmentos de DNA homólogos e pequenas inserções e deleções (indels), conhecidas coletivamente como polimorfismos de nucleotídeo único (SNPs), demonstraram ser a fonte mais abundante de polimorfismos de DNA em humanos (Kwok P.-Y., Deng Q., Zakeri H., Nickerson D. A., 1996 Increasing the information content of STS-based genome maps: identifying polymorphisms in mapped STSs. Genomics 31: 123–126; Y. L. Zhu, Q. J. Song, D. L. Hyten, C. P. Van Tassell, L. K. Matukumalli, D. R. Grimm, S. M. Hyatt, E. W. Fickus, N. D. Young and P. B. Cregan Genetics March 1, 2003 vol. 163 no. 3 1123-1134).
[0028] Os SNPs são adequados para o desenvolvimento de métodos de genotipagem de alto rendimento e fáceis de automatizar, porque a maioria dos SNPs são bialélicos, simplificando assim abordagens e análises de genotipagem (Lin CH, Yeakley JM, McDaniel TK, Shen R (2009) Mediumto high-throughput SNP genotyping using VeraCode microbeads. Methods Mol Biol 496: 129–142; Yoon MS, Song QJ, Choi IY, Specht JE, Hyten DL, et al. (2007) BARCSoySNP23: a panel of 23 selected SNPs for soybean cultivar identification. Theor Appl Genet 114: 885–899). Com base na análise de SNPs e ferramentas de bioinformática, é possível quantificar desequilíbrio de ligação e a análise de haplótipos. Além disso, outro ponto a ser considerado é que o uso de marcadores moleculares para melhoramento assistido, incluindo SNPs, detecta a informação genética sem interferência do meio ambiente, em regiões transcritas e não transcritas, trazendo a vantagem da possibilidade de eliminar ou diminuir a necessidade de análises fitopatológicas demoradas e trabalhosas. O melhorista pode identificar indivíduos portadores de marcadores ligados ao alelo de interesse, como resistência a doenças, resultando em economia de tempo e recurso (ALZATEMARIN, A L.; CERVIGNI, G. D. L.; MOREIRA, M. A; (2005) Marker assisted selection in the development of disease resistant plants, with emphasis on common bean and soybean. Fitopatologia brasileira. v.30, no.4,p.333-342).
[0029] Atualmente, a principal forma de controle da mancha alvo é através do uso de fungicidas. Contudo, os fungicidas do grupo químico das carboxamidas vêm reduzindo a sua eficiência de controle provavelmente pela presença de isolados resistentes de Corynespora cassiicola aos fungicidas metil-benzimidazol-carbamato (MBC) (GODOY, C.V.; UTIAMADA, C.M.; MEYER, M.C.; CAMPOS, H.D.; PIMENTA, C.B.; JACCOUD-FILHO, D.S. Eficiência de fungicidas para o controle da mancha-alvo, Corynespora cassiicola, na safra 2013/14: resultados sumarizados dos ensaios cooperativos. Londrina: Embrapa Soja, 2014. 6p. (Embrapa Soja. Circular Técnica 104).
[0030] Desta forma, há necessidade de utilização de métodos complementares para manejo efetivo da doença, como a resistência genética em cultivares. Apesar da importância econômica da soja e da ameaça da mancha alvo, até o presente momento, não existem publicações científicas descrevendo fontes (genótipos) para resistência a doença, muito menos estudos de herança genética, descrição de genes/locus de resistência e nem estudos sobre a localização de possíveis genes de resistência à Corynespora cassiicola.
[0031] A presente invenção identifica SNPs do genoma da soja associados à resistência da soja ao fungo Corynespora cassiicola e revela um método para identificação e seleção de plantas resistentes a este patógeno. Além disso, ainda revela método de introgressão em plantas de alelos de resistência ao fungo Corynespora cassiicola em soja.
[0032] As vantagens da invenção serão evidentes na descrição da invenção fornecida neste documento.
[0033] Em um aspecto, a invenção refere-se a um método de identificação, distinção e seleção de plantas do gênero Glycine, resistentes ou suscetíveis, à mancha alvo causada pelo fungo Corynespora cassiicola que compreende:
- (a) Extração de ácido nucleico de uma planta do gênero Glycine;
- (b) Análise do ácido nucleico extraído para presença de um ou mais alelos dos marcadores moleculares associados com aumento de resistência ou suscetibilidade à Corynespora cassiicola dentro de um intervalo de 37,69 – 37,85 Mpb do cromossomo 17;
- (c) Seleção das plantas que possuam os referidos alelos dos marcadores.
[0034] Em uma forma de concretização do método, um ou mais marcadores se encontram na região genômica dos genes ou nos intervalos dos genes Glyma_17g224300 (SEQ ID NO: 1), Glyma_17g223800 (SEQ ID NO: 2), Glyma_17g223900 (SEQ ID NO: 3), Glyma_17g224000 (SEQ ID NO: 4), Glyma_17g224100 (SEQ ID NO: 5), Glyma_17g224200 (SEQ ID NO: 6), Glyma_17g224500 (SEQ ID NO: 8), Glyma_17g224600 (SEQ ID NO: 9), Glyma_17g224700 (SEQ ID NO: 10), Glyma_17g224800 (SEQ ID NO: 11), Glyma_17g224900 (SEQ ID NO: 12), Glyma_17g225000 (SEQ ID NO: 13), Glyma_17g225100 (SEQ ID NO: 14), Glyma_17g225200 (SEQ ID NO: 15), Glyma_17g225300 (SEQ ID NO: 16), Glyma_17g225400 (SEQ ID NO: 17), Glyma_17g225500 (SEQ ID NO: 18). Em uma concretização preferencial, os marcadores se encontram na região genômica dos genes ou nos intervalos dos genes selecionados do grupo consistindo em Glyma_17G224300 (SEQ ID NO: 1), Glyma_17G224400 (SEQ ID NO: 7) e Glyma_17G224500 (SEQ ID NO: 8), e ainda mais preferencialmente, o referido marcador é um SNP selecionado do grupo consistindo em ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações, ou qualquer outro marcador molecular em um intervalo de até 5 cM ou 1Mbp do referido grupo, ainda mais preferencialmente, o referido marcador é um SNP selecionado do grupo consistindo em ss715627288, ss715627273 e ss715627282, ou suas combinações ou qualquer outro marcador molecular em um intervalo de até 5 cM ou 1Mbp do referido grupo.
[0035] Em uma forma de concretização, o método compreende a identificação dos marcadores através de quaisquer metodologias de amplificação, ou por uso de sondas ou por qualquer tipo de sequenciamento (exemplo tGBS ou sequenciamento dirigido).
[0036] Em outra forma de concretização, a planta do gênero Glycine é Glycine max.
[0037] Em outro aspecto, a invenção refere-se a um método de introgressão em plantas do gênero Glycine de alelos de resistência à mancha alvo causada pelo fungo Corynespora cassiicola, que compreende:
[0038] (a) Cruzar parentais de plantas do gênero Glycine identificados pelo método como definido acima com outros parentais que não possuem a referida resistência;
[0039] (b) Selecionar progênies possuindo marcadores associados com aumento de resistência ou redução de suscetibilidade à Corynespora cassiicola através do método como definido acima; e
[0040] (c) Retrocruzar em um ou mais ciclos as progênies selecionadas com o genitor recorrente para desenvolver novas progênies.
[0041] Em outro aspecto adicional, a invenção refere-se a uma molécula de ácido nucleico capaz de hibridizar com qualquer uma das SEQ ID NO: 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 ou 33, ou subsequências das mesmas tendo pelo menos 15 nucleotídeos consecutivos, ou sequências com pelo menos 90% de identidade de sequência com mesmas.
[0042] Em outro aspecto adicional, a invenção também se refere ao uso de uma molécula de ácido nucleico como acima definido nos métodos da invenção.
[0043] Em um outro aspecto, inclui-se na invenção um kit de detecção que compreende pelo menos duas moléculas de ácido nucleico como definidas acima.
[0044] Em um outro aspecto, a invenção refere-se a um método para genotipar plantas alvo de Glycine resistentes à mancha-alvo, que compreende analisar a presença no DNA da planta alvo para um ou mais marcadores associados à resistência à mancha-alvo, selecionado do grupo consistindo de ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações.
[0045] Em um outro aspecto, a invenção refere-se a uma planta de Glycine resistente à mancha-alvo obtida por um método de introgressão como acima definido.
[0046] A Figura 1 se refere a escala diagramática desenvolvida por Soares e colaboradores (2009) e ajustes para escala de notas de 1-9 para avaliação da severidade de Corynespora cassiicola em tecido foliar de soja e algodão, com as respectivas reações do genótipo.
[0047] A Figura 2 se refere ao mapeamento associativo de SNPs associados a resistência a Corynespora cassiicola.
[0048] A Figura 3 se refere ao gráfico do bloco em alto desequilíbrio de ligação sob a região onde os SNPs mais significativos foram mapeados.
[0049] A Figura 4 se refere aos genes identificados no intervalo correspondente ao bloco em desequilíbrio de ligação em que os SNPs mais significativos se encontram.
[0050] A Figura 5 se refere ao efeito de substituição alélica para SNPs detectados por três marcadores na reação (severidade) a Corynespora cassiicola em teste de progênie oriunda de cruzamento entre parental resistente e suscetível.
[0051] A não ser que sejam definidos de maneira diferente, todos os termos técnicos e científicos aqui utilizados têm o mesmo significado entendido por um técnico no assunto ao qual a invenção pertence. A terminologia utilizada na descrição da invenção tem finalidade de descrever concretizações particulares somente, e não tem a intenção de limitar o escopo dos ensinamentos. A não ser que seja indicado de forma diferente, todos os números expressando quantidades, porcentagens e proporções, e outros valores numéricos usados no relatório descritivo e nas reivindicações, devem ser entendidos como sendo modificados, em todos os casos, pelo termo “cerca de”. Assim, a não ser que seja indicado o contrário, os parâmetros numéricos mostrados no relatório descritivo e nas reivindicações são aproximações que podem variar, dependendo das propriedades a serem obtidas.
[0052] A prática da presente invenção irá empregar, a menos que indicado de forma diferente, métodos convencionais de química, bioquímica e técnicas de DNA recombinante, dentro do conhecimento da técnica. Tais técnicas são explicadas completamente na literatura. Veja, por exemplo, Fundamental Virology, 2ª Edição, vols. I & II (B.N. Fields e D. M. Knipe, eds.); T.E. Creighton, Proteins: Structures e Molecular Properties (W.H. Freeman e Company, 1993); A.L. Lehninger, Biochemistry (Worth Publishers, Inc., edição atual); Sambrook, e cols., Molecular Cloning: A Laboratory Manual (2ª Edição, 1989); Methods In Enzymology (S. Colowick e N. Kaplan eds., Academic Press, Inc.).
[0053] Os termos abaixo são definidos, e podem ser utilizados dentro do escopo de presente invenção de modo a facilitar o entendimento geral.
[0054] Gene: unidade física e funcional básica da hereditariedade, sendo compostos por DNA e passíveis de serem transcritos em RNA. Alguns genes agem como instruções para polipeptídeos;
[0055] QTL: sigla em inglês do termo Quantitative Trait Loci, que se refere a um lócus de característica quantitativa. É um lócus que se correlaciona com a variação de uma característica quantitativa no fenótipo de uma população de organismos;
[0056] Lócus: refere-se a uma posição ou local que um determinado gene ou qualquer outro elemento genético ou fator que contribui para uma característica ocupa em um cromossomo de determinada espécie.
[0057] Alelo: formas variantes de um determinado gene, que ocupam a mesma região em cromossomos homólogos, afetando a mesma característica, contudo, de modo diferente. Um mesmo gene pode ter diversos alelos;
[0058] Cromossomo: é um pacote organizado de DNA encontrado no núcleo da célula que pode conter diversos genes;
[0059] Genótipo: referem-se aos alelos, ou formas variantes de um gene, que são compreendidas por um organismo;
[0060] Mapa genético: É uma representação gráfica de um genoma ou uma parte de um genoma, como um único cromossomo. É uma descrição das relações de ligação genética entre os loci em um ou mais cromossomos de uma dada espécie. Para cada mapa genético, as distâncias entre os loci são medidas pelas frequências de recombinação entre eles. A recombinação entre os loci pode ser detectada usando uma variedade de marcadores;
[0061] Desequilíbrio de ligação: é definido no contexto da invenção como a frequência relativa de tipos de gametas em uma população de muitos indivíduos em uma única geração. Se a frequência de um alelo A é p, a é p’, B é q e b é q’, então a frequência esperada (sem desequilíbrio de ligação) do genótipo AB é pq, Ab é pq’, aB é p’q e ab é p’q’. Qualquer desvio da frequência esperada é chamado de desequílibrio de ligação. Dois loci são ditos “geneticamente ligados” quando eles estão em desequilíbrio de ligação
[0062] Ligação genética: refere-se a uma associação de características em herança devido à localização de genes em proximidade no mesmo cromossomo, medido por porcentagem de recombinação entre loci (centi-Morgan, cM). As distâncias entre loci são usualmente medidas pela frequência de recombinação entre os loci no mesmo cromossomo. Quanto mais afastados dois loci estiverem entre si, maior é a probabilidade de ocorrer uma recombinação entre eles. Contrariamente, se dois loci estiverem próximos, uma recombinação é menos provável de acontecer entre eles. Como regra, 1 centi-Morgan é igual a 1% de recombinação entre os loci. Quando um QTL pode ser indicado por múltiplos marcadores, a distância genética entre os marcadores nas extremidades (flanqueadores) é indicativa do tamanho do QTL. Para fins desta invenção, “geneticamente ligado a um marcador” pode ser considerado que o marcador não está distante em mais do que 10cM, preferencialmente 5cM, mais preferencialmente 2cM e ainda mais preferencialmente 1cM do determinante genético que confere a resistência.
[0063] Marcadores moleculares: são fragmentos de DNA que estão associados a uma região específica do genoma, que podem ser monitorados. Referem-se, em outras palavras, a indicadores que são usados em métodos para visualizar diferenças em sequências de ácidos nucleicos. As moléculas marcadoras podem assumir a forma de sequências curtas de DNA, como uma sequência que envolve um único polimorfismo de nucleotídeos, onde ocorre uma única alteração no par de bases. Eles também podem assumir a forma de sequências de DNA mais longas, como microssatélites, com 10 a 60 pares de bases.
[0064] Germoplasma: refere-se à totalidade de genótipos de uma população. Também pode se referir ao material vegetal, por exemplo, um grupo de plantas que são repositórias de vários alelos.
[0065] Resistência: refere-se à habilidade de uma planta de restringir o crescimento e desenvolvimento de um patógeno específico e/ou o sinal/sintoma decorrente, quando comparado com plantas suscetíveis sob condições ambientais e pressão por patógeno similares. Inclui tanto resistência parcial quanto resistência total à infecção (por exemplo, infecção por um patógeno que causa mancha-alvo). Uma planta resistente irá mostrar ausência de ou poucos sintomas da doença. Uma planta suscetível pode ser tanto uma não-resistente ou ter níveis mais baixos de resistência à infecção em comparação à uma planta resistente.
[0066] Introgressão: refere-se a processos naturais ou artificiais em que regiões genômicas de uma espécie, variedade ou cultivar é transferida para o genoma de outra espécie, variedade ou cultivar, pelo cruzamento. O processo pode opcionalmente ser completado pelo retrocruzamento entre um indivíduo e seu genitor recorrente.
[0067] Cruzamento: refere-se à fusão de gametas via polinização para produzir uma progênie, incluindo tanto a autofecundação (quando o pólen e o óvulo são da mesma planta) ou a fecundação cruzada (quando o pólen e o óvulo são de plantas diferentes).
[0068] Seleção assistida por marcador (SAM): é um processo pelo qual fenótipos são selecionados com base nos genótipos moleculares. Seleção assistida por marcador inclui o uso de marcadores moleculares para identificar plantas ou populações que possuam o genótipo de interesse em programas de melhoramento.
[0069] PCR (reação da polimerase em cadeia): refere-se a um método de produzir quantidades relativamente grandes de regiões específicas de DNA, permitindo várias análises baseadas nestas regiões.
[0070] Iniciadores de PCR (“primers”): fragmentos relativamente pequenos de DNA de fita simples usados na amplificação por PCR de regiões específicas de DNA.
[0071] Sonda: refere-se a moléculas ou átomos capazes de reconhecer e ligar a uma molécula alvo específica, permitindo a detecção da molécula alvo. Em particular, para fins desta invenção, “sonda” refere-se a uma sequência de DNA ou de RNA marcados que podem ser usados para detectar e/ou quantificar uma sequência complementar por hibridização molecular.
[0072] A descrição detalhada a seguir refere-se a marcadores genéticos e métodos relacionados para identificação de tais marcadores, genotipagem de plantas do gênero Glycine, e métodos de melhoramento assistido por marcadores destas plantas.
[0073] Os loci pertencentes à presente invenção compreendem sequências genômicas delimitadas que compreendem um ou mais marcadores moleculares, incluindo um polimorfismo identificado na Tabela 5, Tabela 7 ou Tabela 8, conforme mostrados nas SEQ ID NOS: 19 a 33, ou é adjacente a um ou mais destes polimorfismos.
[0074] Em um aspecto da invenção, são providas sequências de ácido nucleico isoladas (oligonucleotídeos) que são capazes de hibridizar aos loci polimórficos da presente invenção. Em certas concretizações, por exemplo, que provêm iniciadores, tais moléculas compreendem pelo menos 15 bases de nucleotídeo. Moléculas úteis como iniciadores podem hibridizar sob condições de alta estringência com uma ou mais fitas de um segmento de DNA em um lócus polimórfico da invenção. Os iniciadores para amplificação de DNA são providos aos pares, i.e., iniciadores “forward (ou F)” ou “reverse (ou R)”. Um iniciador será complementar a uma fita de DNA no lócus e o outro iniciador será complementar à outra fita de DNA no lócus, i.e., de forma preferencial, inclusas estão sequências que sejam pelo menos 90%, mais preferencialmente 95%, ou 100% idênticas a uma sequência como descrita nas SEQ ID Nos: 19 a 48, ou às sub-sequências de pelo menos 15 nucleotídeos. Além disso, é entendido que tais iniciadores podem hibridizar a uma sequência no lócus que está distante do polimorfismo, por exemplo, a pelo menos 5, 10, 20, 50, 100, 200, 500 ou até cerca de 1.000.000 de nucleotídeos distantes do polimorfismo. O desenho de um iniciador da invenção irá depender de fatores bem conhecidos na técnica, por exemplo, evitando uma sequência repetitiva.
[0075] Além disto, deve ser aqui lembrado que, apesar das funções preferidas poderem ser mencionadas em relação a alguns oligonucleotídeos, é óbvio que um dado oligonucleotídeo pode assumir diversas funções, e pode ser utilizado em diferentes formas de acordo com a presente invenção. Como é de conhecimento do técnico no assunto, em algumas situações, um iniciador pode ser usado como sonda e vice-versa, além de ser aplicável em procedimentos de hibridização, detecção etc. Assim, observa-se que os produtos de acordo com a presente invenção, especialmente, inter alia, os oligonucleotídeos, não estão limitados aos usos aqui mostrados, mas, ao contrário, os usos devem ser interpretados de forma ampla, independente do uso aqui indicado. Além disto, quando um oligonucleotídeo é descrito como sendo útil como sonda capaz de se ligar a um amplicon, o técnico no assunto também entende que a sequência complementar deste oligonucleotídeo é igualmente útil como uma sonda para se ligar ao mesmo amplicon. O mesmo ocorre com as sequências descritas como úteis como iniciadores. Adicionalmente, é também óbvio que qualquer iniciador adequado para um protocolo multiplex pode ser também, dentro do significado e escopo da presente invenção, ser utilizado em um protocolo singleplex. O mesmo se aplica a um iniciador adequado para um protocolo de PCR em tempo real, que pode ser usado em um protocolo de PCR convencional, dentro do significado da presente invenção.
[0076] O técnico no assunto, a este respeito, entende que os oligonucleotídeos da presente invenção, isto é, os iniciadores e sondas, não precisam ser completamente complementares a uma parte da sequência do alvo. O iniciador pode apresentar complementaridade suficiente para hibridizar com a sequência alvo e desempenhar as funções intrínsecas de um iniciador. O mesmo se aplica a uma sonda, ou seja, uma sonda pode apresentar complementaridade suficiente para hibridizar com a sequência alvo e desempenhar as funções intrínsecas de uma sonda. Portanto, um iniciador ou uma sonda, em uma concretização, não necessita ser completamente complementar à sequência alvo. Em uma concretização, o iniciador ou a sonda pode se hibridizar ou anelar com uma parte do alvo para formar uma fita dupla. As condições de hibridização de um ácido nucleico são descritas por Joseph Sambrook et al., Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2001) e Haymes et al., Nucleic Acid Hybridization, A Practical Approach, IRL Press, Washington, D.C. (1985).
[0077] Em outro aspecto da invenção, está o kit compreendendo pelo menos dois iniciadores como descritos acima.
[0078] Outro aspecto das moléculas de ácido nucleico da invenção são as sondas de hibridização. Em uma concretização, tais sondas são oligonucleotídeos compreendendo pelo menos 15 bases de nucleotídeo e um marcador detectável. A finalidade de tais moléculas é de hibridizar, por exemplo, sob condições de alta estringência, a uma fita de DNA em um segmento de bases de nucleotídeos que inclui ou é adjacente a um polimorfismo de interesse. Tais oligonucleotídeos são preferencialmente pelo menos 90%, mais preferencialmente 95% idênticas à sequência de um segmento do DNA de Glycine em um lócus polimórfico, ou a um fragmento deste compreendendo pelo menos 15 bases de nucleotídeo. Mas especificamente, o lócus polimórfico é selecionado do grupo que consiste em SEQ ID NO: 19-33.
[0079] O marcador detectável pode ser um elemento radioativo ou um corante. Em aspectos preferenciais, a sonda de hibridização ainda compreende um marcador fluorescente e um quencher, por exemplo, para uso em ensaios de hibridização como os ensaios Taqman®, disponível de AB Biosystems. Neste caso, o marcador detectável e o quencher ficam localizados em extremidades opostas. Para os ensaios de detecção de SNP, é útil prover tais marcadores e quencher em pares, por exemplo, em que cada molécula para detecção de um polimorfismo tem um marcador fluorescente e um quencher distintos, diferentes para cada polimorfismo.
[0080] Mais especificamente, com relação à sonda TaqMan™, um oligonucleotídeo, cuja região 5’ terminal é modificada com um fluoróforo e a região 3’ terminal é modificada com um quencher, é adicionada à reação de PCR. Também se entende que é possível ligar o fluoróforo na região 3’ terminal e o quencher na região 5’ terminal. Os produtos de reação são detectados pela fluorescência gerada após a atividade exonuclease 5’ -> 3’ da DNA polimerase. Os fluoróforos, que se referem a compostos fluorescentes que emitem luz com a excitação por luz tendo um comprimento de onda mais curto que a luz que é emitida, podem ser, mas não estão limitados a, FAM, TAMRA, VIC, JOE, TET, HEX, ROX, RED610, RED670, NED, Cy3, Cy5, e Texas Red. Os quenchers podem ser, mas não estão limitados ao, 6- TAMRA, BHQ-1,2,3 e MGB-NFQ. A escolha do par fluoróforo-quencher pode ser feita de forma que o espectro de excitação do quencher tenha uma sobreposição com o espectro de emissão do fluoróforo. Um exemplo é o par FAM-TAMRA, FAM-MGB, VIC-MGB e assim por diante. Um técnico no assunto saberá reconhecer outros pares apropriados.
[0081] Não é necessário que haja completa complementariedade entre as sequências, desde que as diferenças não prejudiquem completamente a capacidade das moléculas de formarem uma estrutura dupla-fita. Portanto, para que uma molécula de ácido nucleico seja capaz de servir como iniciador ou sonda, ele deve ser suficientemente complementar em sequência para permitir a formação de uma estrutura dupla-fita sob as condições de hibridização utilizadas.
[0082] Em uma concretização preferencial, uma molécula de ácido nucleico irá hibridizar a um segmento do DNA de Glycine mostrada na SEQ ID NO: 1 a 33.
[0083] SNPs são resultados de uma variação na sequência e novos polimorfismos podem ser detectados pelo sequenciamento de DNA genômico ou moléculas de cDNA.
[0084] Em um aspecto, os polimorfismos em um genoma podem ser determinados comparando a sequência de cDNA de diferentes linhagens. Embora a detecção de polimorfismos através da comparação da sequência de cDNA seja relativamente conveniente, a avaliação da sequência de cDNA não permite informações sobre a posição dos íntrons no DNA genômico correspondente. Além disso, polimorfismos na sequência não codificante não podem ser identificados a partir do cDNA. Isso pode ser uma desvantagem, por exemplo ao usar polimorfismos derivados de cDNA como marcadores para genotipagem de DNA genômico. Ensaios de genotipagem mais eficientes podem ser projetados se o escopo dos polimorfismos incluir aqueles presentes na sequência única não codificante.
[0085] A sequência de DNA genômico é mais útil que o cDNA para identificar e detectar polimorfismos. Os polimorfismos em um genoma podem ser determinados comparando a sequência de DNA genômico de diferentes linhagens. No entanto, o DNA genômico de eucariotos superiores normalmente contém uma grande fração de sequência repetitiva e transposons. O DNA genômico pode ser sequenciado de forma mais eficiente se a fração codificadora / única for enriquecida subtraindo ou eliminando as sequências repetitivas.
[0086] Existem várias estratégias bem conhecidas na técnica que podem ser empregadas para enriquecer a amostra em sequências codificadoras / sequências únicas. Exemplos destes incluem o uso de enzimas que são sensíveis à metilação da citosina, o uso da endonuclease McrBC para clivar a sequência repetitiva e a impressão de microarranjos de bibliotecas genômicas que são então hibridizados com sondas de sequência repetitiva.
[0087] Um método para reduzir o DNA repetitivo compreende a construção de bibliotecas de representação reduzida, separando a sequência repetitiva de fragmentos de DNA genômico de pelo menos duas variedades de uma espécie, fracionando os fragmentos de DNA genômico separados com base no tamanho da sequência de nucleotídeos e comparando a sequência de fragmentos em uma fração para determinar polimorfismos. Mais particularmente, esses métodos de identificação de polimorfismos no DNA genômico compreendem a digestão do DNA genômico total de pelo menos duas variantes de uma espécie eucariótica com uma endonuclease sensível à metilação para fornecer um pool de fragmentos de DNA digerido. O comprimento médio dos nucleotídeos dos fragmentos é menor para as regiões de DNA caracterizadas por uma porcentagem menor de citosina 5-metilada. Tais fragmentos são separáveis, p.ex. por eletroforese em gel, com base no comprimento dos nucleotídeos. Uma fração de DNA com comprimento de nucleotídeo menor que a média é separada do pool de DNA digerido. Sequências do DNA em uma fração são comparadas para identificar polimorfismos. Em comparação com a sequência de codificação, é mais provável que a sequência repetitiva compreenda citosina 5-metilada, p.ex. nos segmentos de sequência -CG- e -CNG-. Em uma modalidade do método, o DNA genômico de pelo menos duas variedades consanguíneas diferentes de uma Glycine é digerido com uma endonuclease sensível à metilação selecionada do grupo que consiste em enzimas como Aci I, Apa I, Age I, Bsr FI, BssH II, Eag I, Eae I, Hha I, HinP1 I, Hpa II, Msp I, MspM II, Nar I, Not I, Pst I, Pvu I, Sac II, Sma I, Stu I e Xho I para fornecer um pool de DNA digerido fisicamente separado, por exemplo, por eletroforese em gel. Frações de tamanho comparável de DNA são obtidas do DNA digerido de cada uma das referidas enzimas. As moléculas de DNA das frações comparáveis são inseridas em vetores ou isoladas para construir bibliotecas de representação reduzida de clones genômicos de DNA que são sequenciadas e comparadas para identificar polimorfismos.
[0088] Um outro método para enriquecimento de sequências codificantes / sequência única consiste na construção de bibliotecas de representação reduzida (usando enzimas sensíveis à metilação ou não), imprimindo microarranjos da biblioteca em membrana de nylon, seguidos de hibridação com sondas feitas a partir de elementos repetitivos conhecidamente presentes na biblioteca. Os elementos de sequência repetitiva são identificados e a biblioteca é reorganizada escolhendo apenas os clones negativos. Tais métodos fornecem segmentos de DNA genômico de representação reduzida de uma planta que possui DNA genômico compreendendo regiões de DNA com níveis relativamente mais altos de citosina metilada e regiões de DNA com níveis relativamente mais baixos de citosina metilada.
[0089] Ainda, podem ser usados microarranjos (chip de DNA) de soja disponíveis na técnica, como o SoySNP50K (Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, et al. (2013) Development and Evaluation of SoySNP50K, a High-Density Genotyping Array for Soybean. PLoS ONE 8(1): e54985). Este painel tem sido amplamente explorado para estudos genéticos da soja, permitindo a identificação de associações entre SNPs e resistência a doenças, entre outras características.
[0090] Os polimorfismos nas sequências de DNA podem ser detectados por uma variedade de métodos bem conhecidos na arte. As amostras de DNA incluem, mas não estão limitadas, aos genótipos mostrados na Tabela 1.
[0091] Por exemplo, métodos para detectar SNPs e Indels incluem métodos de extensão de base única (SBE). Exemplos de métodos SBE incluem, mas não estão limitados, aos divulgados nas Pat. US 6.004.744; 6.013.431; 5.595.890; 5.762.876; e 5.945.283. Os métodos SBE são baseados na extensão de um iniciador de nucleotídeo que é imediatamente adjacente a um polimorfismo para incorporar um resíduo de nucleotídeo detectável após a extensão do iniciador. Em certas modalidades, o método SBE usa três oligonucleotídeos sintéticos. Dois dos oligonucleotídeos servem como iniciadores de PCR e são complementares à sequência do local do DNA genômico da soja que flanqueia uma região contendo o polimorfismo a ser testado. Após a amplificação da região do genoma da soja contendo o polimorfismo, o produto de PCR é misturado com o terceiro oligonucleotídeo (chamado de iniciador de extensão), que é projetado para hibridizar com o DNA amplificado imediatamente adjacente ao polimorfismo na presença de DNA polimerase e dois didesoxinucleossidetrifosfatos marcados diferencialmente. Se o polimorfismo estiver presente no molde, um dos didesoxinucleosidetrifosfatos marcados pode ser adicionado ao iniciador em uma única extensão da cadeia de base. O alelo presente é então inferido através da determinação de qual dos dois marcadores diferenciais foi adicionado ao iniciador de extensão. Amostras homozigotas resultarão na incorporação de apenas uma das duas bases marcadas e, portanto, apenas um dos dois marcadores será detectado. Amostras heterozigotas têm ambos os alelos presentes e, portanto, direcionam a incorporação de ambos os marcadores (em diferentes moléculas do iniciador de extensão) e, portanto, ambos os marcadores serão detectados.
[0092] Em um método preferido para detectar polimorfismos, SNPs e Indels podem ser detectados por métodos divulgados na Patente US 5.210.015; 5.876.930; e 6.030.787 na qual se utiliza uma sonda oligonucleotídica com um corante fluorescente a 5' e um quencher a 3' da sonda. Quando a sonda está intacta, a proximidade do corante fluorescente ao quencher resulta na supressão da fluorescência do corante fluorescente, p.ex, por transferência de energia do tipo Forster. Durante a PCR, os iniciadores forward e reverse hibridizam com uma sequência específica do DNA alvo que flanqueia um polimorfismo enquanto a sonda de hibridação hibridiza com a sequência contendo polimorfismo no produto de PCR amplificado. No ciclo de PCR subsequente, a DNA polimerase com atividade de exonuclease 5 '→ 3' quebra a sonda e separa o corante fluoresecente do quencher, resultando em maior fluorescência do corante fluorescente.
[0093] Um teste útil está disponível pela AB Biosystems como o teste Taqman®, que emprega quatro oligonucleotídeos sintéticos em uma única reação que simultaneamente amplifica o DNA genômico da soja, discrimina os alelos presentes e fornece diretamente um sinal para discriminação e detecção. Dois dos quatro oligonucleotídeos servem como iniciadores de PCR e geram um produto de PCR que engloba o polimorfismo a ser detectado. Duas outras são sondas de transferência de energia de ressonância fluorescente específica de alelo (FRET). No ensaio, são utilizadas duas sondas FRET com diferentes corantes repórteres fluorescentes, onde um corante único é incorporado a um oligonucleotídeo que pode anelar com alta especificidade com apenas um dos dois alelos. Os corantes repórteres úteis incluem, entre outros, 6-carboxi-4,7,2′,7′-tetraclorofluoreceína (TET), 2′- cloro-7′-fenil-1,4-dicloro-6-carboxifluoresceína (VIC) e fosforamidito de 6- carboxifluoresceína (FAM). Um inibidor útil é 6-carboxi-N, N, N ', N'- tetrametil-rodamina (TAMRA). Além disso, a extremidade 3' de cada sonda FRET é quimicamente bloqueada para que não possa atuar como um iniciador de PCR. Também está presente um terceiro fluoróforo usado como referência passiva, por exemplo, rodamina X (ROX) para ajudar na normalização posterior dos valores relevantes de fluorescência (corrigindo erros volumétricos na montagem da reação). A amplificação do DNA genômico é iniciada. Durante cada ciclo da PCR, as sondas FRET anelam de maneira alelo-específica aos moldes de moléculas de DNA. As sondas FRET aneladas (mas não as não- aneladas) são degradadas pela TAQ DNA polimerase à medida que a enzima encontra a extremidade 5' da sonda anelada, liberando assim o fluoróforo da proximidade do seu quencher. Após a PCR, a fluorescência de cada um dos dois fluorescentes, bem como a referência passiva, é determinada fluorometricamente. A intensidade normalizada da fluorescência para cada um dos dois corantes será proporcional às quantidades de cada alelo inicialmente presente na amostra e, portanto, o genótipo da amostra pode ser inferido.
[0094] Os iniciadores de PCR são desenhados (a) para ter um tamanho de cerca de 15 a 25 bases e sequências que hibridizem no lócus polimórfico, (b) tenha uma temperatura de melting na faixa de 57°C a 60°C, correspondendo a uma temperatura de anelamento de 52°C a 55°C, (c) produza um produto que inclua o sítio polimórfico e tipicamente tenha um tamanho que varia de 75 a 250 pares de bases. No entanto, existem técnicas de PCR que permitem a amplificação de fragmentos maiores, de 1000 ou mais pares de bases. Os iniciadores são preferencialmente localizados no lócus de forma que o sítio polimórfico esteja pelo menos 1 base distante da extremidade 3’ de cada iniciador. No entanto, entende-se que os iniciadores de PCR podem estar até 1000 pares de bases ou mais de distância do polimorfismo e ainda prover amplificação de um fragmento de DNA correspondente que contém o polimorfismo e que possa ser usado nos ensaios de genotipagem da soja.
[0095] Técnicas de sequenciamento dirigido podem ser aplicadas para detecção de polimorfismo. O desenvolvimento de tecnologias cada vez mais econômicas e rápidas de sequenciamento tem levado à facilitação da detecção em larga escala de polimorfismos em várias espécies de plantas modelo ou não (Kumar S, Banks TW, Cloutier S. SNP Discovery through NextGeneration Sequencing and Its Applications. International journal of plant genomics vol. 2012 (2012): 831460). O desenvolvimento e aprimoramento de softwares de bioinformática de código aberto e disponíveis gratuitamente aceleraram a descoberta de SNPs. Cabe destacar que a facilitação de sequenciamento completo de genomas levou à descoberta de vários milhões de SNPs em diferentes organismos.
[0096] Os polimorfismos nos loci desta invenção podem ser utilizados na identificação de associações de marcadores e resistência à mancha-alvo que são inferidas a partir da análise estatística de dados genotípicos e fenotípicos dos membros de uma população.
[0097] Vários tipos de análises estatísticas podem ser usadas para inferir a associação de marcadores e resistência à mancha-alvo a partir dos dados de fenótipo / genótipo, mas uma ideia básica é detectar marcadores moleculares, isto é, polimorfismos, para os quais genótipos alternativos têm fenótipos médios significativamente diferentes. Por exemplo, se um determinado lócus “A” de marcador tiver três genótipos alternativos (AA, Aa e aa), e se essas três classes de indivíduos tiverem fenótipos significativamente diferentes, então inferiremos que o lócus “A” está associado à característica desejada. O significado das diferenças no fenótipo pode ser testado por vários tipos de testes estatísticos padrão, como regressão linear de genótipos de marcadores moleculares no fenótipo ou análise de variância (ANOVA). Os pacotes de software estatístico disponíveis no mercado, comumente usados para fazer esse tipo de análise, incluem os modelos lineares mistos (MLM) desenvolvidos pelos pacotes MVP (YIN et al., 2018), GAPIT (TANG et al., 2016) e FarmCPU (LIU et al., 2016), com os algoritmos de matrizes Emma (MVP) e VanRaden (GAPIT e FarmCPU). Quando muitos marcadores moleculares são testados simultaneamente, um ajuste, como a correção de Bonferroni, é feito no nível de significância necessário para declarar uma associação.
[0098] Frequentemente, o objetivo de um estudo de associação não é simplesmente detectar associações de marcadores e características desejadas, mas estimar a localização de genes que afetam a característica diretamente em relação às localizações dos marcadores. Em uma abordagem simples para esse objetivo, é feita uma comparação entre os locais dos marcadores da magnitude da diferença entre genótipos alternativos ou o nível de significância dessa diferença. Infere-se que os genes de característica estejam localizados mais próximos do (s) marcador (s) que possuem a maior diferença genotípica associada. A ligação genética de moléculas marcadoras adicionais pode ser estabelecida por um modelo de mapeamento genético, como, sem limitação, o modelo de marcador de flanqueamento relatado por Lander et al. (Lander et al. 1989 Genetics, 121: 185-199) e o mapeamento por intervalos, com base nos métodos de máxima verossimilhança, e implementados no pacote de software MAPMAKER / QTL (Lincoln e Lander, mapeando genes controlando características quantitativas usando MAPMAKER / QTL, Mapping Genes Controlling Quantitative Traits Using MAPMAKER/QTL, Whitehead Institute for Biomedical Research, Massachusetts, (1990).) O software adicional inclui Qgene, Versão 2.23 (1996), Departamento de Melhoramento de Plantas e Biometria, 266 Emerson Hall, Universidade Cornell, Ithaca, NY).
[0099] É calculada uma estimativa de máxima verossimilhança (MV) para a presença de um marcador, juntamente com um MV que não assume efeito QTL, para evitar falsos positivos. Um log10 de uma razão de chances (“odds ratio” ou LOD) é então calculado como: LOD = log10 (MV para a presença de um QTL / MV sem QTL vinculado). O escore LOD indica essencialmente quanto mais provável os dados são de surgirem assumindo a presença de um QTL versus na sua ausência. O valor limite de LOD para evitar um falso positivo com uma determinada confiança, por exemplo, 95%, depende do número de marcadores e do comprimento do genoma.
[00100] Para o desenvolvimento da presente invenção, foi utilizado um conjunto de genótipos (conforme tabela 1), que foram inoculados com isolados de Corynespora cassiicola que apresentaram virulência considerada alta e intermediária (tabela 2). Estes genótipos foram avaliados quanto à resistência à mancha-alvo, tendo sido selecionados os genótipos resistentes conforme descrito na Tabela 4. Dentro do escopo e para fins da presente invenção, estão inclusos todos os genótipos considerados resistentes e altamente resistentes, que podem ser utilizados em programas de melhoramento como fontes de resistência à mancha-alvo. De forma mais preferencial são os genótipos considerados altamente resistentes, selecionado do grupo que consiste de PI 71506, PI 153230, PI 567310B, PI 587802, PI 587860, PI 407999-1 e PI 548984.
[00101] Em outro aspecto da invenção, o polimorfismo nos locais da invenção é mapeado no genoma da soja, como um mapa físico do genoma da soja compreendendo posições no mapa de dois ou mais polimorfismos, como indicado nas Tabelas 6, 7 e 8.
[00102] De forma mais específica, na presente invenção, é descrita a identificação de marcadores genéticos (SNPs ou combinações de dois ou mais SNPs) que podem ser usados para identificar alelos associados à resistência ou tolerância à mancha alvo em plantas. Mais especificamente, os marcadores estão presentes em um intervalo de 110kpb no cromossomo 17 de G. max, associados à resistência à mancha-alvo.
[00103] Quando um lócus foi localizado nas proximidades de marcadores moleculares, esses marcadores podem ser usados para selecionar aspectos melhorados da característica sem a necessidade de análise fenotípica em cada ciclo de seleção. No melhoramento assistido por marcadores e na seleção assistida por marcadores, as associações entre lócus e marcadores são estabelecidas inicialmente por meio de análise de mapeamento. No mesmo processo, determina-se quais alelos dos marcadores moleculares estão ligados a alelos favoráveis do lócus/loci que estão sendo estudados. Posteriormente, alelos dos marcadores associados a alelos favoráveis dos lócus/loci são selecionados na população. Este procedimento melhorará o “valor” da característica a ser selecionada, no presente caso, resistência à mancha-alvo, desde que haja uma ligação suficientemente próxima entre marcadores e o lócus envolvido em resistência. O grau de ligação requerido depende do número de gerações de seleção porque, a cada geração, há uma oportunidade de quebra da associação por recombinação.
[00104] Existem algumas maneiras de quantificar o nível de eficiência dos marcadores moleculares para seleção de genótipos de interesse. Uma das principais formas encontra-se na utilização de cálculos de acurácia e taxas de erro tipo I e II. A acurácia é uma medida que demonstra o quão eficaz é um marcador para detectar indivíduos resistentes e suscetíveis. Este cálculo é utilizado como forma de indicar com precisão a proximidade de um resultado genotípico com os dados fenotípicos da característica estudada. Altos valores de acurácia indicam elevada eficiência na seleção de indivíduos através do uso de marcadores moleculares. Já as taxas de erro tipo I e II são medidas que quantificam as possíveis falhas na correlação dos dados fenotípicos e genotípicos. Erros do tipo I, também denominados de falsos-positivo, tratam de resultados os quais os dados genotípicos indicam a presença de um alelo de resistência, enquanto os dados fenotípicos sugerem que as amostras analisadas são suscetíveis a característica. Em contrapartida, erros do tipo II, ou falsosnegativo, demonstram a presença genotípica de alelos suscetíveis em amostras com fenótipos de resistência a doença. Baixos valores de erro tipo I e II diminuem a probabilidade de eliminação de materiais resistentes e suscetíveis, respectivamente, pela utilização de marcadores moleculares (Maldonado dos Santos, J.V., Ferreira, E.G.C., Passianotto, A.L.d.L. et al (2019). Association mapping of a locus that confers southern stem canker resistance in soybean and SNP marker development. BMC Genomics 20, 798; Bruna Bley Brumer. Caracterização morfológica, molecular e patogênica de isolados de Diaporthe aspalathi e validação de marcadores SNPs associados à resistência ao cancro da haste na soja. Dissertação de Mestrado. Universidade Estadual de Londrina – UEL – PR - 2016; Adriano Consoni Camolese. Podridão radicular de fitóftora em soja: Identificação de um gene recessivo de resistência e validação de SNPs para emprego em seleção assistida por marcadores moleculares. Dissertação de Mestrado. Universidade Estadual de Londrina - UEL - PR - 2015).
[00105] As associações entre alelos marcadores específicos e alelos favoráveis também podem ser usadas para prever quais tipos de progênie podem segregar a partir de um determinado cruzamento. Essa previsão pode permitir a seleção de parentais apropriados para populações de geração a partir das quais novas combinações de alelos favoráveis são reunidas para produzir uma nova linhagem pura. Por exemplo, se a linhagem A possui alelos marcadores previamente associados a alelos favoráveis nos locais 1, 20 e 31, enquanto a linhagem B possui alelos marcadores associados a efeitos favoráveis nos locais 15, 27 e 29, uma nova linhagem pode ser desenvolvida cruzando A × B e selecionando progênies que possuem alelos favoráveis em todos os 6 loci.
[00106] Marcadores moleculares são usados para acelerar a introgressão de genes ou segmentos cromossômicos em novos backgrounds genéticos (isto é, em uma gama diversificada de germoplasma). Introgressão simples envolve cruzar uma linhagem doadora de uma nova característica para uma linhagem elite e, em seguida, selecionar e retrocruzar plantas F1 repetidamente para o parental elite (recorrente), enquanto seleciona a manutenção do gene de interesse / segmento cromossômico. Ao longo de várias gerações de retrocruzamento, o histórico genético da linhagem original é substituído gradualmente pelo histórico genético da elite por recombinação e segregação. Esse processo pode ser acelerado pela seleção dos alelos do parental recorrente através de marcadores moleculares. Esta abordagem é conhecida como retrocruzamento assistido por marcadores.
[00107] Finalmente, é possível estabelecer uma “impressão digital” ou fingerprint de uma linhagem, como a combinação de alelos em um conjunto de dois ou mais loci de marcadores. Fingerprints de alta densidade podem ser usadas para estabelecer e rastrear a identidade do germoplasma, que tem utilidade no estabelecimento de um banco de dados de associações característica-marcador para beneficiar um programa de melhoramento de soja, bem como proteção à propriedade intelectual do germoplasma.
[00108] Assim, de acordo com um primeiro aspecto da invenção, a presente invenção fornece métodos de identificação e seleção de plantas resistentes à uma doença fúngica compreendendo as etapas de:
- (a) Extração de ácido nucleico de uma planta;
- (b) Análise do ácido nucleico extraído para a presença de um ou mais marcadores associados com aumento de resistência ao fungo dentro de um intervalo de um cromossomo;
- (c) Seleção das plantas que possuam os referidos marcadores.
[00109] Preferencialmente, o método é direcionado para identificação de plantas do gênero Glycine, mais especificamente plantas da espécie Glycine max.
[00110] De forma preferencial, a resistência ao fungo é resistência ao Corynespora cassiicola, agente etiológico da mancha-alvo.
[00111] A obtenção de uma amostra de ácido nucleico de uma planta pode ser realizada por métodos de isolamento de DNA padrões bem conhecidos na arte, como descrito supra.
[00112] A análise para a presença de marcadores pode ser feita por PCR, sondas ou sequenciamento. Em uma forma de concretização, as moléculas de ácido nucleico (iniciadores de PCR e sondas) compreendem sequências das SEQ ID Nos: 19-48, ou sub-sequências destas com pelo menos 15 nucleotídeos de comprimento. Ainda, estão inclusas no escopo da invenção, sequências que sejam pelo menos 90% idênticas às SEQ ID Nos: 19-48 ou às suas sub-sequências.
[00113] Em relação à doença fúngica, o método da presente invenção, preferencialmente, se refere ao fungo Corynespora cassiicola, causador da doença chamada Mancha Alvo, sendo que a resistência ou tolerância à referida doença é conferida por um lócus ou QTL.
[00114] Preferencialmente, o marcador é um marcador do tipo SNP (Single nucleotide polymorphism).
[00115] Um marcador corresponde a um produto de amplificação gerado pela amplificação de um ácido nucleico de Glycine sp., por exemplo, por reação da polimerase em cadeia de polimerase (PCR) utilizando dois iniciadores. Neste contexto, “marcador molecular” refere-se a um indicador que é usado nos métodos para visualizar diferenças em características de sequências de ácido nucleico (polimorfismos). Um marcador molecular “ligado a” ou “associado a” um gene capaz de fornecer resistência à manchaalvo pode, portanto, se referir a SNPs.
[00116] Ademais, os marcadores podem ainda ser detectados através do uso de sondas ou sequenciamento dirigido (tGBS)
[00117] A detecção de um marcador molecular pode, em algumas concretizações, compreender o uso de um ou mais conjuntos de iniciadores que podem ser usados para produzir um ou mais produtos de amplificação. Em uma primeira concretização, tais conjuntos de iniciadores podem hibridizar com uma parte das sequências de nucleotídeos como mostrados nas SEQ ID Nos: 19 a 33 (Tabela 10), ou sub-sequências destas com pelo menos 15 nucleotídeos de comprimento. Ainda, estão inclusas no escopo da invenção, sequências que sejam pelo menos 90% idênticas a SEQ ID Nos: 19- 48 ou às suas subsequências.
[00118] Em uma outra concretização da presente invenção, os marcadores se encontram nos genes ou nos intervalos dos genes Glyma_17g224300 (SEQ ID NO: 1), Glyma_17g223800 (SEQ ID NO: 2), Glyma_17g223900 (SEQ ID NO: 3), Glyma_17g224000 (SEQ ID NO: 4), Glyma_17g224100 (SEQ ID NO: 5), Glyma_17g224200 (SEQ ID NO: 6), Glyma_17g224400 (SEQ ID NO: 7), Glyma_17g224500 (SEQ ID NO: 8), Glyma_17g224600 (SEQ ID NO: 9), Glyma_17g224700 (SEQ ID NO: 10), Glyma_17g224800 (SEQ ID NO: 11), Glyma_17g224900 (SEQ ID NO: 12), Glyma_17g225000 (SEQ ID NO: 13), Glyma_17g225100 (SEQ ID NO: 14), Glyma_17g225200 (SEQ ID NO: 15), Glyma_17g225300 (SEQ ID NO: 16), Glyma_17g225400 (SEQ ID NO: 17), Glyma_17g225500 (SEQ ID NO: 18) presentes no cromossomo 17 de Glycine max.
[00119] Em uma terceira concretização da presente invenção, marcadores se encontram, preferencialmente, nas regiões adjacentes dos genes selecionados do grupo consistindo em Glyma_17g224300 (SEQ ID NO: 1), Glyma_17g224400 (SEQ ID NO: 7) e Glyma_17g224500 (SEQ ID NO: 8) presentes no cromossomo 17 de Glycine max.
[00120] Em uma quarta concretização da presente invenção, os marcadores são SNPs selecionados do grupo consistindo em ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações.
[00121] Em uma quinta concretização da presente invenção, os SNPs são preferencialmente ss715627288, ss715627273 e ss715627282.
[00122] Em uma sexta concretização da presente invenção, a planta é preferencialmente da espécie Glycine max.
[00123] Em um outro aspecto, a presente invenção se refere a um método de introgressão em plantas do gênero Glycine de alelos de resistência à mancha alvo causada pelo fungo Corynespora cassiicola, compreendendo as etapas de:
- (a) Cruzar parentais de plantas do gênero Glycine identificados pelo método como definido nas concretizações anteriores com outros parentais que não possuem a referida resistência;
- (b) Selecionar progênies possuindo marcadores associados com aumento de resistência à Corynespora cassiicola através do método como definido nas concretizações anteriores; e
- (c) Retrocruzar em um ou mais ciclos as progênies selecionadas com o genitor recorrente para desenvolver novas progênies.
[00124] Em um outro aspecto, a presente invenção se refere a um método para genotipar plantas alvo de Glycine resistentes à mancha-alvo, compreendendo analisar a presença no DNA da planta alvo para um ou mais marcadores associados à resistência à mancha-alvo, selecionado do grupo consistindo de ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações. Em um outro aspecto, a invenção compreende kits comerciais ou customizados compreendendo tais moléculas de ácido nucleico.
[00125] Em um aspecto adicional, a invenção compreende um método para genotipar plantas alvo de Glycine resistentes à mancha-alvo, compreendendo analisar a presença no DNA da planta alvo para um ou mais marcadores associados à resistência à mancha-alvo, selecionado do grupo consistindo de ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações.
[00126] De uma forma preferencial, a presente invenção se refere a métodos para produzir uma variedade comercial resistente à Corynespora cassiicola a partir de variedades suscetíveis, compreendendo realizar o método de introgressão acima usando técnicas de melhoramento convencionais. A presente invenção é ainda descrita pelos exemplos abaixo, que têm o intuito somente de exemplificar uma das inúmeras maneiras de se realizar a invenção, contudo, sem limitar o escopo da mesma.
[00127] Um total de 520 genótipos de soja foram avaliados neste estudo. Tratam-se de acessos de Glycine max oriundos de diversos centros de origem, com a maior parte originária da Ásia (62,5%) e América (23,4%). A relação das amostras utilizadas neste estudo pode ser observada na Tabela 1
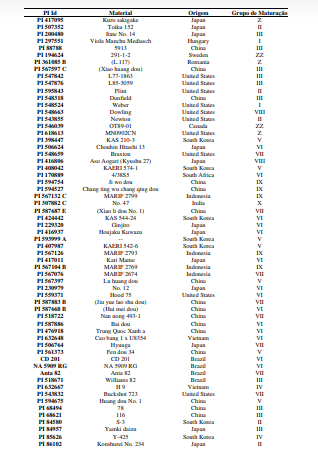



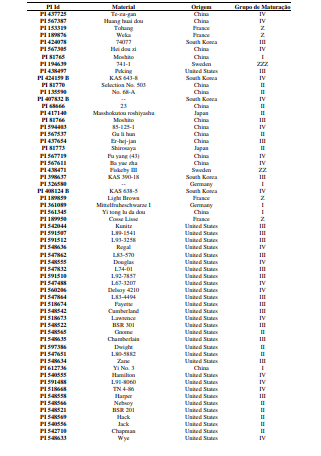


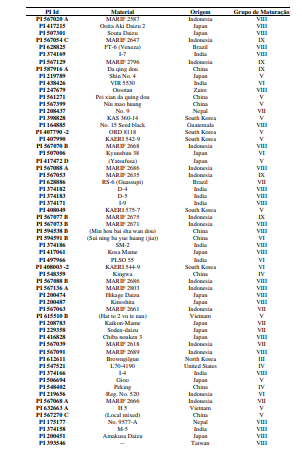
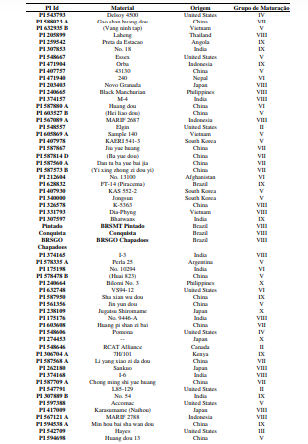
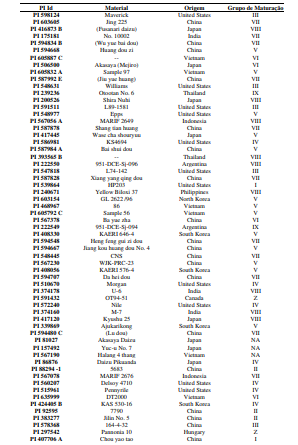

[00128] Foram selecionados 17 isolados de Corynespora cassiicola da micoteca da Titular que apresentaram virulência consideradas alta e intermediária, obtidos em estudos conduzidos nas dependências da titular. Os isolados estão descritos na Tabela 2. 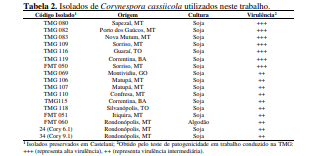
[00129] Culturas puras dos fungos foram obtidas em meio BDA (batata-dextrose-agar) por 7 dias. Uma repetição de cada isolado foi retirada da placa e misturada em um recipiente, adicionando-se 100 mL de água, e procedendo-se com a trituração em liquidificador por 30s, aproximadamente. A solução obtida foi filtrada, através de peneira de 20 mesh. O resíduo que ficou retido na peneira foi descartado e do mix da suspensão de conídios foi retirada uma alíquota para contagem dos esporos. A contagem final de esporos da suspensão foi de 1750 conídios/mL.
[00130] Os materiais selecionados para este estudo foram plantados na casa de vegetação a fim de se avaliar a resistência a doença, com um total de quatro amostras por genótipo. Dois meses após o plantio, os genótipos foram inoculados com o bulk dos 17 isolados de Corynespora cassiicola. Inicialmente, vinte litros de suspensão de esporos foram preparados e pulverizados com o auxílio de bomba costal sob a área foliar das plantas. Duas inoculações foram realizadas, com intervalo de 5 dias. As inoculações foram realizadas no final da tarde, com molhamento foliar nos cinco dias posteriores às inoculações.
[00131] Como forma de avaliar a resposta à doença, duas avaliações foram realizadas. Primeiro avaliou-se a nota média de severidade. Para tal, utilizou-se a escala diagramática desenvolvida por Soares e colaboradores (2009) (SOARES, R.M.; GODOY, C.V.; OLIVEIRA, M.C.N. Escala diagramática para avaliação da severidade da mancha alvo da soja. Tropical Plant Pathology, v.34, p. 333-338, 2009), com algumas modificações (Figura 1). Além disto, o tamanho de lesão também foi observado e notas de 1-5 foram atribuídas, visualmente, ao diâmetro das lesões.
[00132] Após as duas avaliações, selecionou-se os genótipos com reação Altamente Resistente/Imune (AR) ou de Resistência (R) a mancha alvo e com tamanho de lesão variando de 0 a 2mm para um novo plantio. O objetivo desta nova avaliação era confirmar a resistência ou se houve algum escape durante o ensaio. Para isso, dez sementes de cada genótipo foram plantadas em vasos de 8L contendo solo:areia, na proporção de 3:1. Como padrão suscetível utilizou-se a cultivar NA 5909 e alguns genótipos com reação Suscetível (S) ou Altamente Suscetível (AS) do primeiro ensaio. Novamente foi preparada uma suspensão de esporos com contagem dos esporos/mL e procedeu-se com a pulverização/ primeira inoculação, em casa de vegetação no estádio V2.
[00133] A segunda inoculação ocorreu 4 dias depois da primeira inoculação. As inoculações foram realizadas no final da tarde, mantendo-se o molhamento foliar nos cinco dias posteriores às inoculações. A avaliação foi realizada 20 dias após a última inoculação, determinando-se a nota média de severidade e o tamanho de lesão (Tabela 3). 
[00134] Um total de 83 genótipos apresentaram resistência à ação do patógeno. Destes, sete materiais foram altamente resistentes a mancha-alvo: PI 71506, PI 153230, PI 567310B, PI 587802, PI 587860, PI 407999-1 e PI 548984. Estes materiais podem ser trabalhados em programas de melhoramento como fontes de resistência à mancha-alvo. Em contrapartida, 616 materiais apresentaram suscetibilidade à doença, dos quais 67 foram altamente suscetíveis. A classificação dos materiais quanto à resistência a mancha alvo pode ser observado na Tabela 4. 











[00135] O painel escolhido para esta análise foi o SoySNP50K (Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, et al. (2013) Development and Evaluation of SoySNP50K, a High-Density Genotyping Array for Soybean. PLoS ONE 8(1): e54985. https://doi.org/10.1371/journal.pone.0054985). Tal painel possui dados de genotipagem para todos os materiais avaliados neste trabalho. Além disto, possui uma ampla cobertura do genoma da soja, com 42.080 SNPs distribuídos pelos 20 cromossomos da soja.
[00136] Com os dados fenotípicos e genotípicos das amostras utilizadas neste experimento, desenvolveu-se uma análise associativa em busca de SNPs ligados a resistência a mancha-alvo. Para isto, utilizou-se modelos lineares mistos (MLM) desenvolvidos pelos pacotes MVP (YIN et al., 2018), GAPIT (TANG et al., 2016) e FarmCPU (LIU et al., 2016), com os algoritmos de matrizes Emma (MVP)e VanRaden (GAPIT e FarmCPU). Além disto, uma análise de principais componentes foi realizada com um valor de 3. Uma linha de corte de 0,05 para o valor de P foi escolhida a fim de determinar os SNPs mais significativos desta análise.
[00137] Através da análise associativa, foi possível identificar uma região no cromossomo 17 ligada a resistência à mancha-alvo (Figura 2). O intervalo corresponde a 37,69 – 37,85 Mpb e um total de 15 SNPs significativos ao teste de bonferroni elaborado pelo GAPIT foram identificados (Tabela 5).
[00138] Foi observado um grande bloco em desequilíbrio de ligação com 110 kpb, no qual encontram-se 14 dos 15 SNPs (Figura 3). Em uma análise mais aprofundada nesta região, detectou-se a presença de 13 genes neste bloco em desequilíbrio de ligação (Figura 4). A maior parte destes genes são funcionalmente descritos na literatura como auxiliares em mecanismos de resistência, porém nenhum deles até o momento foi associado com resistência à mancha alvo. 
[00139] Os três SNPs mais significativos encontram-se em um intervalo de 27 kpb dentro do bloco identificado. Neste intervalo, há a presença de três genes: Glyma_17G224300, Glyma_17G224400 e Glyma_17G224500. O SNP com maior valor de p-value está localizado na posição 37.772.369 e trata-se de uma mutação não sinônima sob um éxon do gene Glyma_17G224500, uma proteína quinase do tipo LRR. O segundo SNP foi identificado a 1.868 pb a jusante do gene Glyma_17G224400, um polipeptídeo gag de tipo LTR. Por fim, o terceiro SNP foi identificado na posição 37.744.962 e está sob um íntron do gene Glyma_17g224300, uma proteína quinase do tipo LRR (Tabela 6). Ao utilizar o haplótipo dos três SNPs, observou-se uma filtragem com seleção das amostras com maior resistência (Tabela 8). 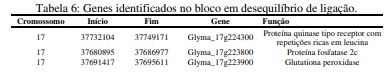
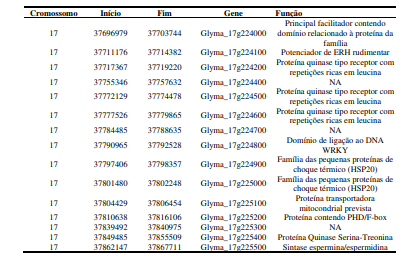
[00140] Com os resultados obtidos, detectou-se que o SNP ss715627273 apresentou uma eficiência de seleção de genótipos de 84,33%. Quando este SNP foi comparado em conjunto com outras variações alélicas, observou-se que não ocorreu um aumento tão relevante na eficiência de seleção, nem na diminuição das porcentagens de erro (Tabela 8). Este resultado demonstra que a marca pode ser utilizada isoladamente para selecionar indivíduos resistentes. 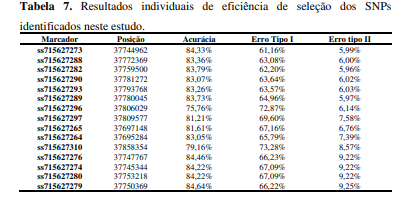

[00141] Uma população segregante foi desenvolvida através do cruzamento entre BRSMG 68 (Vencedora) (resistente à C. cassiicola) e NA 5909 RG (suscetível à C. cassiicola). Esta população foi avançada até a geração F3, a qual foi realizada um teste de progênie em cada indivíduo inferindo sua F2:3. Foram avaliados preliminarmente 96 indivíduos fenotipicamente, através de um experimento desenvolvido em casa-devegetação, com quatro blocos casualizados contendo 5 replicatas por família. Utilizou-se a mesma metodologia de inoculação e avaliação (escala de notas) descrita anteriormente (Soares et al., 2009).
[00142] Os resultados gerados foram analisados através de uma análise de variância (ANOVA) e mostraram que havia diferença significativa entre as famílias fenotipadas (Tabela 9). Com os resultados obtidos, uma análise da herança da característica foi realizada e constatou-se as hipóteses de segregação para a característica de 3:1 (um gene recessivo), 13:3 (um gene dominante e um recessivo) e 55:9 (dois genes dominantes e um recessivo). Para confirmação dos resultados, um número maior de famílias será avaliado em futuras análises. 
[00143] Por fim, os três marcadores com maiores valores de p-value foram sintetizados via tecnologia Taqman e amplificados nas 96 famílias da população segregante. Os resultados demonstraram que os três marcadores possuíam um alto efeito sobre a resistência a doença (Figura 5). A presença do alelo suscetível nos três marcadores esteve associada à altos valores de severidade da doença na população segregante, o que demonstra sua alta eficiência de eliminação de materiais suscetíveis a doença. Desta forma, demonstra-se uma alta aplicabilidade da ferramenta para descarte de genótipos que não possuam o gene de resistência a doença. 

Claims (12)
- Método de identificação, distinção e seleção de plantas do gênero Glycine, resistentes ou suscetíveis, à mancha alvo causada pelo fungo Corynespora cassiicola, caracterizado pelo fato de que compreende:
- (a) Extração de ácido nucleico de uma planta do gênero Glycine;
- (b) Análise do ácido nucleico extraído para presença de um ou mais alelos dos marcadores moleculares associados com aumento de resistência ou suscetibilidade à Corynespora cassiicola dentro de um intervalo de 37,69 – 37,85 Mpb do cromossomo 17;
- (c) Seleção das plantas que possuam os referidos alelos dos marcadores.
- Método de acordo com a reivindicação 1, caracterizado pelo fato de que um ou mais marcadores se encontram na região genômica dos genes ou nos intervalos dos genes Glyma_17g224300 (SEQ ID NO: 1), Glyma_17g223800 (SEQ ID NO: 2), Glyma_17g223900 (SEQ ID NO: 3), Glyma_17g224000 (SEQ ID NO: 4), Glyma_17g224100 (SEQ ID NO: 5), Glyma_17g224200 (SEQ ID NO: 6), Glyma_17g224500 (SEQ ID NO: 8), Glyma_17g224600 (SEQ ID NO: 9), Glyma_17g224700 (SEQ ID NO: 10), Glyma_17g224800 (SEQ ID NO: 11), Glyma_17g224900 (SEQ ID NO: 12), Glyma_17g225000 (SEQ ID NO: 13), Glyma_17g225100 (SEQ ID NO: 14), Glyma_17g225200 (SEQ ID NO: 15), Glyma_17g225300 (SEQ ID NO: 16), Glyma_17g225400 (SEQ ID NO: 17), Glyma_17g225500 (SEQ ID NO: 18).
- Método de acordo com a reivindicação 2, caracterizado pelo fato de que os marcadores se encontram na região genômica dos genes ou nos intervalos dos genes selecionados do grupo consistindo em Glyma_17G224300 (SEQ ID NO: 1), Glyma_17G224400 (SEQ ID NO: 7) e Glyma_17G224500 (SEQ ID NO: 8).
- Método de acordo com qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de que o referido marcador é um SNP selecionado do grupo consistindo em ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações, ou qualquer outro marcador molecular em um intervalo de até 5 cM ou 1Mbp do referido grupo.
- Método de acordo com a reivindicação 4, caracterizado pelo fato de que o referido marcador é um SNP selecionado do grupo consistindo em ss715627288, ss715627273 e ss715627282, ou suas combinações ou qualquer outro marcador molecular em um intervalo de até 5 cM ou 1Mbp do referido grupo.
- Método de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato da identificação dos marcadores ser através de quaisquer metodologias de amplificação, ou por uso de sondas ou por qualquer tipo de sequenciamento (exemplo tGBS ou sequenciamento dirigido).
- Método de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que a planta do gênero Glycine é Glycine max.
- Método de introgressão em plantas do gênero Glycine de alelos de resistência à mancha alvo causada pelo fungo Corynespora cassiicola, caracterizado pelo fato de que compreende:
- (a) Cruzar parentais de plantas do gênero Glycine identificados pelo método como definido em qualquer uma das reivindicações 1 a 6 com outros parentais que não possuem a referida resistência;
- (b) Selecionar progênies possuindo marcadores associados com aumento de resistência ou redução de suscetibilidade à Corynespora cassiicola através do método como definido na reivindicação 1; e
- (c) Retrocruzar em um ou mais ciclos as progênies selecionadas com o genitor recorrente para desenvolver novas progênies.
- Molécula de ácido nucleico, caracterizada pelo fato de ser capaz de hibridizar com qualquer uma das SEQ ID NO: 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 ou 33, ou subsequências das mesmas tendo pelo menos 15 nucleotídeos consecutivos, ou sequências com pelo menos 90% de identidade de sequência com mesmas.
- Uso de uma molécula de ácido nucleico como definido na reivindicação 9, caracterizado pelo fato de ser no método como definido em qualquer uma das reivindicações 1 a 6.
- Kit de detecção, caracterizado pelo fato de que compreende pelo menos duas moléculas de ácido nucleico como definidas na reivindicação 9.
- Método para genotipar plantas alvo de Glycine resistentes à mancha-alvo, caracterizado pelo fato de compreender analisar a presença no DNA da planta alvo para um ou mais marcadores associados à resistência à mancha-alvo, selecionado do grupo consistindo de ss715627273, ss715627288, ss715627282, ss715627290, ss715627293, ss715627289, ss715627296, ss715627297, ss715627265, ss715627264, ss715627310, ss715627276, ss715627274, ss715627280 e ss715627279, ou suas combinações.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR102020009417-3A BR102020009417A2 (pt) | 2020-05-12 | 2020-05-12 | Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo |
| PCT/BR2020/050353 WO2021226686A1 (pt) | 2020-05-12 | 2020-09-02 | Método de identificação, distinção e seleção de plantas do gênero glycine, resistentes ou suscetíveis, à mancha alvo causada pelo fungo corynespora cassiicola, método de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo causada pelo fungo corynespora cassiicola, molécula de ácido nucleico e seu uso, kit de detecção, método para genotipar plantas alvo de glycine resistentes à mancha-alvo e planta de glycine resistente à manchaalvo |
| US17/924,935 US20230189731A1 (en) | 2020-05-12 | 2020-09-02 | Method for identification, distinction and selection of plants of the glycine genus, resistant or susceptible to target spot caused by the fungus corynespora cassiicola , method for introgression into plants of the glycine genus of alleles of resistance to target spot caused by the fungus corynespora cassiicola, nucleic acid molecule and its use, detection kit, method for genotyping target spot-resistant glycine target plants and target spot-resistant glycine plants |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR102020009417-3A BR102020009417A2 (pt) | 2020-05-12 | 2020-05-12 | Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| BR102020009417A2 true BR102020009417A2 (pt) | 2021-11-23 |
Family
ID=78525868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BR102020009417-3A BR102020009417A2 (pt) | 2020-05-12 | 2020-05-12 | Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20230189731A1 (pt) |
| BR (1) | BR102020009417A2 (pt) |
| WO (1) | WO2021226686A1 (pt) |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107012246B (zh) * | 2006-05-25 | 2021-10-26 | 孟山都技术有限公司 | 大豆抗病性数量性状基因座及其组成的鉴定方法 |
| CA2923296A1 (en) * | 2013-10-25 | 2015-04-30 | Pioneer Hi-Bred International, Inc. | Stem canker tolerant soybeans and methods of use |
| US20170298452A1 (en) * | 2014-10-10 | 2017-10-19 | Pioneer Hi-Bred International, Inc. | Carlavirus tolerant soybeans and methods of use |
| CN105603119A (zh) * | 2016-03-31 | 2016-05-25 | 天津科润农业科技股份有限公司 | 用于检测黄瓜棒孢叶斑病抗性位点的snp标记方法 |
-
2020
- 2020-05-12 BR BR102020009417-3A patent/BR102020009417A2/pt unknown
- 2020-09-02 WO PCT/BR2020/050353 patent/WO2021226686A1/pt not_active Ceased
- 2020-09-02 US US17/924,935 patent/US20230189731A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US20230189731A1 (en) | 2023-06-22 |
| WO2021226686A1 (pt) | 2021-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7977533B2 (en) | Genetic loci associated with iron deficiency tolerance in soybean | |
| US9745637B2 (en) | Genetic loci associated with fusarium solani tolerance in soybean | |
| US7790949B2 (en) | Genetic loci associated with Sclerotinia tolerance in soybean | |
| US10337072B2 (en) | Copy number detection and methods | |
| CN101687901A (zh) | 玉米多态性与基因分型方法 | |
| CN104735970A (zh) | 用于小麦中各种性状的分子标记及其使用方法 | |
| US9062352B2 (en) | Loci associated charcoal rot drought complex tolerance in soybean | |
| US20130337442A1 (en) | Genetic loci associated with soybean cyst nematode resistance and methods of use | |
| US9994920B2 (en) | Genetic loci associated with soybean cyst nematode resistance and methods of use | |
| US20100325750A1 (en) | Major QTLS Conferring Resistance Of Corn To Fijivirus | |
| BR112021012597A2 (pt) | Métodos e composições para selecionar e/ou prever plantas de algodão resistentes à resistência ao fusarium race-4 em algodão | |
| BR112015014632B1 (pt) | Método de identificação | |
| BR112015023134B1 (pt) | Método para identificar uma planta de soja ou um germoplasma de soja que exibe resistência aprimorada à mancha foliar olho-de-rã | |
| BRPI0614050A2 (pt) | métodos para peneiramento de polimorfismos de hibridização especìficos de gene (gshps) e seus usos no mapeamento genético e desenvolvimento de marcador | |
| US10982293B2 (en) | Methods for improving plant resistance to soybean cyst nematode and compositions thereof | |
| BR102020009417A2 (pt) | Métodos de identificação, distinção e seleção de plantas do gênero glycine resistentes ou suscetíveis à mancha alvo, de introgressão em plantas do gênero glycine de alelos de resistência à mancha alvo, para genotipar plantas alvo de glycine resistentes àmancha-alvo, molécula de ácido nucleico e seu uso, kit de detecção, e, planta de glycine resistente à manchaalvo | |
| CN112218524B (zh) | 高粱细胞质雄性不育标记和基因座 | |
| CN113736866A (zh) | 用于检测番茄黄化曲叶病毒病抗性的snp位点组合及其应用 | |
| US20140162250A1 (en) | Marker-assisted selection of tolerance to chloride salt stress | |
| CN118441086A (zh) | 一种与丝瓜果实耐褐变连锁的分子标记及应用 | |
| BR112018016164B1 (pt) | Método de seleção de uma primeira planta ou germoplasma de soja | |
| BR122023007756B1 (pt) | Método para identificar uma planta de soja ou um germoplasma de soja que exibe resistência aprimorada à mancha foliar olho-de-rã ou resistência aprimorada à podridão parda da haste (bsr) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B03A | Publication of a patent application or of a certificate of addition of invention [chapter 3.1 patent gazette] |