BRPI0610520B1 - Vetor, constructo de dna recombinante, e processos para transformar uma célula hospedeira, produzir uma planta, conferir ou aperfeiçoar a resistência a colletotrichum, determinar a presença ou ausência de um polinucleotídeo, alterar o nível de expressão de uma proteína, e de identificação de uma planta de milho - Google Patents
Vetor, constructo de dna recombinante, e processos para transformar uma célula hospedeira, produzir uma planta, conferir ou aperfeiçoar a resistência a colletotrichum, determinar a presença ou ausência de um polinucleotídeo, alterar o nível de expressão de uma proteína, e de identificação de uma planta de milho Download PDFInfo
- Publication number
- BRPI0610520B1 BRPI0610520B1 BRPI0610520-3A BRPI0610520A BRPI0610520B1 BR PI0610520 B1 BRPI0610520 B1 BR PI0610520B1 BR PI0610520 A BRPI0610520 A BR PI0610520A BR PI0610520 B1 BRPI0610520 B1 BR PI0610520B1
- Authority
- BR
- Brazil
- Prior art keywords
- markers
- plant
- gene
- rcg1
- polynucleotide
- Prior art date
Links
- 108090000623 proteins and genes Proteins 0.000 title claims abstract description 467
- 241000196324 Embryophyta Species 0.000 title claims abstract description 350
- 238000000034 method Methods 0.000 title claims abstract description 179
- 102000040430 polynucleotide Human genes 0.000 title claims abstract description 137
- 108091033319 polynucleotide Proteins 0.000 title claims abstract description 137
- 239000002157 polynucleotide Substances 0.000 title claims abstract description 137
- 230000008569 process Effects 0.000 title claims abstract description 75
- 241000222199 Colletotrichum Species 0.000 title claims abstract description 36
- 102000004169 proteins and genes Human genes 0.000 title claims description 103
- 230000014509 gene expression Effects 0.000 title claims description 65
- 241001057636 Dracaena deremensis Species 0.000 title claims description 57
- 108020004511 Recombinant DNA Proteins 0.000 title claims description 25
- 239000013598 vector Substances 0.000 title claims description 23
- 230000001131 transforming effect Effects 0.000 title claims description 7
- 240000008042 Zea mays Species 0.000 claims abstract description 147
- 235000002017 Zea mays subsp mays Nutrition 0.000 claims abstract description 128
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 claims abstract description 61
- 235000005822 corn Nutrition 0.000 claims abstract description 61
- 239000003550 marker Substances 0.000 claims description 120
- 239000002773 nucleotide Substances 0.000 claims description 96
- 210000004027 cell Anatomy 0.000 claims description 95
- 125000003729 nucleotide group Chemical group 0.000 claims description 95
- 230000002759 chromosomal effect Effects 0.000 claims description 93
- 108700028369 Alleles Proteins 0.000 claims description 56
- 210000000349 chromosome Anatomy 0.000 claims description 54
- 230000006798 recombination Effects 0.000 claims description 48
- 238000005215 recombination Methods 0.000 claims description 48
- 108091026890 Coding region Proteins 0.000 claims description 45
- 208000015181 infectious disease Diseases 0.000 claims description 35
- 230000009466 transformation Effects 0.000 claims description 30
- 230000001976 improved effect Effects 0.000 claims description 25
- 230000000295 complement effect Effects 0.000 claims description 22
- 108020004999 messenger RNA Proteins 0.000 claims description 12
- 108091093088 Amplicon Proteins 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 238000001514 detection method Methods 0.000 claims description 5
- 230000008929 regeneration Effects 0.000 claims description 4
- 238000011069 regeneration method Methods 0.000 claims description 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical group C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 3
- 230000033228 biological regulation Effects 0.000 claims description 2
- 230000001172 regenerating effect Effects 0.000 claims description 2
- 235000016383 Zea mays subsp huehuetenangensis Nutrition 0.000 abstract description 67
- 235000009973 maize Nutrition 0.000 abstract description 67
- 201000010099 disease Diseases 0.000 abstract description 34
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 34
- 235000013339 cereals Nutrition 0.000 abstract description 11
- 239000013615 primer Substances 0.000 description 82
- 230000002068 genetic effect Effects 0.000 description 64
- 150000007523 nucleic acids Chemical class 0.000 description 62
- 108020004414 DNA Proteins 0.000 description 53
- 238000003752 polymerase chain reaction Methods 0.000 description 51
- 239000012634 fragment Substances 0.000 description 50
- 102000039446 nucleic acids Human genes 0.000 description 49
- 108020004707 nucleic acids Proteins 0.000 description 49
- 108090000765 processed proteins & peptides Proteins 0.000 description 46
- 102000054766 genetic haplotypes Human genes 0.000 description 45
- 229920001184 polypeptide Polymers 0.000 description 44
- 102000004196 processed proteins & peptides Human genes 0.000 description 44
- 238000009396 hybridization Methods 0.000 description 42
- 238000006243 chemical reaction Methods 0.000 description 38
- 239000000523 sample Substances 0.000 description 38
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 30
- 230000000306 recurrent effect Effects 0.000 description 30
- 238000013507 mapping Methods 0.000 description 25
- 230000009261 transgenic effect Effects 0.000 description 25
- 238000004458 analytical method Methods 0.000 description 24
- 238000003780 insertion Methods 0.000 description 24
- 230000037431 insertion Effects 0.000 description 21
- 230000001105 regulatory effect Effects 0.000 description 21
- 210000001519 tissue Anatomy 0.000 description 21
- 108091028043 Nucleic acid sequence Proteins 0.000 description 20
- 238000009395 breeding Methods 0.000 description 20
- 239000002299 complementary DNA Substances 0.000 description 20
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 19
- 244000053095 fungal pathogen Species 0.000 description 19
- 102000054765 polymorphisms of proteins Human genes 0.000 description 19
- 125000003275 alpha amino acid group Chemical group 0.000 description 18
- 150000001413 amino acids Chemical class 0.000 description 18
- 230000001488 breeding effect Effects 0.000 description 18
- 238000011081 inoculation Methods 0.000 description 18
- 230000003321 amplification Effects 0.000 description 17
- 238000003199 nucleic acid amplification method Methods 0.000 description 17
- 244000052769 pathogen Species 0.000 description 17
- 240000007594 Oryza sativa Species 0.000 description 16
- 235000007164 Oryza sativa Nutrition 0.000 description 16
- 238000003556 assay Methods 0.000 description 16
- 241000894007 species Species 0.000 description 16
- 238000006467 substitution reaction Methods 0.000 description 16
- 238000012217 deletion Methods 0.000 description 15
- 230000037430 deletion Effects 0.000 description 15
- 230000000694 effects Effects 0.000 description 14
- 238000012360 testing method Methods 0.000 description 14
- 241000233866 Fungi Species 0.000 description 13
- 108010006444 Leucine-Rich Repeat Proteins Proteins 0.000 description 13
- 238000005516 engineering process Methods 0.000 description 13
- 108091035707 Consensus sequence Proteins 0.000 description 12
- 208000035240 Disease Resistance Diseases 0.000 description 12
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 12
- 108090000848 Ubiquitin Proteins 0.000 description 12
- 230000001965 increasing effect Effects 0.000 description 12
- 210000004901 leucine-rich repeat Anatomy 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- 238000011160 research Methods 0.000 description 12
- 238000011144 upstream manufacturing Methods 0.000 description 12
- 102000044159 Ubiquitin Human genes 0.000 description 11
- 238000004422 calculation algorithm Methods 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- 238000010367 cloning Methods 0.000 description 10
- 210000002257 embryonic structure Anatomy 0.000 description 10
- 230000001717 pathogenic effect Effects 0.000 description 10
- 235000009566 rice Nutrition 0.000 description 10
- 241000589158 Agrobacterium Species 0.000 description 9
- 108091060211 Expressed sequence tag Proteins 0.000 description 9
- 235000010469 Glycine max Nutrition 0.000 description 9
- 108091034117 Oligonucleotide Proteins 0.000 description 9
- 108700019146 Transgenes Proteins 0.000 description 9
- 238000013461 design Methods 0.000 description 9
- 239000003147 molecular marker Substances 0.000 description 9
- 238000013519 translation Methods 0.000 description 9
- 244000068988 Glycine max Species 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 108091081024 Start codon Proteins 0.000 description 8
- 238000007792 addition Methods 0.000 description 8
- 230000009418 agronomic effect Effects 0.000 description 8
- 125000000539 amino acid group Chemical group 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 230000003902 lesion Effects 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 230000003252 repetitive effect Effects 0.000 description 8
- 238000007894 restriction fragment length polymorphism technique Methods 0.000 description 8
- 238000012163 sequencing technique Methods 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 7
- 239000012014 frustrated Lewis pair Substances 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 239000003471 mutagenic agent Substances 0.000 description 7
- 238000003757 reverse transcription PCR Methods 0.000 description 7
- 238000012216 screening Methods 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 238000012546 transfer Methods 0.000 description 7
- 108700026215 vpr Genes Proteins 0.000 description 7
- 240000005979 Hordeum vulgare Species 0.000 description 6
- 108091092195 Intron Proteins 0.000 description 6
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 6
- 208000027418 Wounds and injury Diseases 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 230000002441 reversible effect Effects 0.000 description 6
- 241001429695 Colletotrichum graminicola Species 0.000 description 5
- 206010061818 Disease progression Diseases 0.000 description 5
- 241000918584 Pythium ultimum Species 0.000 description 5
- 241000813090 Rhizoctonia solani Species 0.000 description 5
- 238000002105 Southern blotting Methods 0.000 description 5
- 206010052428 Wound Diseases 0.000 description 5
- 229920002494 Zein Polymers 0.000 description 5
- 230000005750 disease progression Effects 0.000 description 5
- 101150079179 flp-27 gene Proteins 0.000 description 5
- 238000003205 genotyping method Methods 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 230000021121 meiosis Effects 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 230000008659 phytopathology Effects 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000002864 sequence alignment Methods 0.000 description 5
- 238000013518 transcription Methods 0.000 description 5
- 230000035897 transcription Effects 0.000 description 5
- 239000005019 zein Substances 0.000 description 5
- 229940093612 zein Drugs 0.000 description 5
- 241000223602 Alternaria alternata Species 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 235000007340 Hordeum vulgare Nutrition 0.000 description 4
- 206010020649 Hyperkeratosis Diseases 0.000 description 4
- 108010025815 Kanamycin Kinase Proteins 0.000 description 4
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 4
- 241000209510 Liliopsida Species 0.000 description 4
- 241001495426 Macrophomina phaseolina Species 0.000 description 4
- 241000760038 Monosis Species 0.000 description 4
- 206010034133 Pathogen resistance Diseases 0.000 description 4
- 241000918585 Pythium aphanidermatum Species 0.000 description 4
- 108091028664 Ribonucleotide Proteins 0.000 description 4
- 235000011684 Sorghum saccharatum Nutrition 0.000 description 4
- 238000000137 annealing Methods 0.000 description 4
- 230000004071 biological effect Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 230000007613 environmental effect Effects 0.000 description 4
- 241001233957 eudicotyledons Species 0.000 description 4
- 239000004744 fabric Substances 0.000 description 4
- 230000002349 favourable effect Effects 0.000 description 4
- 230000002363 herbicidal effect Effects 0.000 description 4
- 239000004009 herbicide Substances 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 235000021374 legumes Nutrition 0.000 description 4
- 238000007834 ligase chain reaction Methods 0.000 description 4
- 244000005700 microbiome Species 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 239000002336 ribonucleotide Substances 0.000 description 4
- 125000002652 ribonucleotide group Chemical group 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000007619 statistical method Methods 0.000 description 4
- 230000017260 vegetative to reproductive phase transition of meristem Effects 0.000 description 4
- 230000000007 visual effect Effects 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- 108010000700 Acetolactate synthase Proteins 0.000 description 3
- 241000589155 Agrobacterium tumefaciens Species 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 241000223195 Fusarium graminearum Species 0.000 description 3
- 241000233732 Fusarium verticillioides Species 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 108700026244 Open Reading Frames Proteins 0.000 description 3
- 241000599030 Pythium debaryanum Species 0.000 description 3
- 241000228417 Sarocladium strictum Species 0.000 description 3
- 241000221696 Sclerotinia sclerotiorum Species 0.000 description 3
- 238000012300 Sequence Analysis Methods 0.000 description 3
- 241001250070 Sporisorium reilianum Species 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 230000006978 adaptation Effects 0.000 description 3
- 230000001775 anti-pathogenic effect Effects 0.000 description 3
- 229940121375 antifungal agent Drugs 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 239000005547 deoxyribonucleotide Substances 0.000 description 3
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 3
- 238000002845 discoloration Methods 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 210000005069 ears Anatomy 0.000 description 3
- 238000013213 extrapolation Methods 0.000 description 3
- 108020002326 glutamine synthetase Proteins 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 238000003306 harvesting Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 230000008488 polyadenylation Effects 0.000 description 3
- 239000002987 primer (paints) Substances 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 230000005026 transcription initiation Effects 0.000 description 3
- 230000002103 transcriptional effect Effects 0.000 description 3
- 238000011426 transformation method Methods 0.000 description 3
- IAJOBQBIJHVGMQ-UHFFFAOYSA-N 2-amino-4-[hydroxy(methyl)phosphoryl]butanoic acid Chemical compound CP(O)(=O)CCC(N)C(O)=O IAJOBQBIJHVGMQ-UHFFFAOYSA-N 0.000 description 2
- UHPMCKVQTMMPCG-UHFFFAOYSA-N 5,8-dihydroxy-2-methoxy-6-methyl-7-(2-oxopropyl)naphthalene-1,4-dione Chemical compound CC1=C(CC(C)=O)C(O)=C2C(=O)C(OC)=CC(=O)C2=C1O UHPMCKVQTMMPCG-UHFFFAOYSA-N 0.000 description 2
- 108010085238 Actins Proteins 0.000 description 2
- 101710083587 Antifungal protein Proteins 0.000 description 2
- 241000228438 Bipolaris maydis Species 0.000 description 2
- 241000371633 Bipolaris sorghicola Species 0.000 description 2
- 241000228439 Bipolaris zeicola Species 0.000 description 2
- 241001149956 Cladosporium herbarum Species 0.000 description 2
- 241000384516 Claviceps sorghi Species 0.000 description 2
- 241000223211 Curvularia lunata Species 0.000 description 2
- 108010066133 D-octopine dehydrogenase Proteins 0.000 description 2
- 108020003215 DNA Probes Proteins 0.000 description 2
- 239000003298 DNA probe Substances 0.000 description 2
- 108700003861 Dominant Genes Proteins 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 241000223218 Fusarium Species 0.000 description 2
- 241000223221 Fusarium oxysporum Species 0.000 description 2
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 2
- 241000896246 Golovinomyces cichoracearum Species 0.000 description 2
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 2
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 2
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 2
- 206010061217 Infestation Diseases 0.000 description 2
- 108010044467 Isoenzymes Proteins 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- 241000721701 Lynx Species 0.000 description 2
- 241000710118 Maize chlorotic mottle virus Species 0.000 description 2
- 241000723994 Maize dwarf mosaic virus Species 0.000 description 2
- 244000061176 Nicotiana tabacum Species 0.000 description 2
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 2
- 238000000636 Northern blotting Methods 0.000 description 2
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- 241000760727 Peronosclerospora philippinensis Species 0.000 description 2
- 241000596141 Peronosclerospora sorghi Species 0.000 description 2
- 244000046052 Phaseolus vulgaris Species 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- 241000233624 Phytophthora megasperma Species 0.000 description 2
- 241000233639 Pythium Species 0.000 description 2
- 241001622911 Pythium graminicola Species 0.000 description 2
- 241001505297 Pythium irregulare Species 0.000 description 2
- 241001635622 Pythium splendens Species 0.000 description 2
- 101150090155 R gene Proteins 0.000 description 2
- 241000334150 Ramulispora sorghi f. maydis Species 0.000 description 2
- 244000309516 Ramulispora sorghicola Species 0.000 description 2
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 2
- 240000005384 Rhizopus oryzae Species 0.000 description 2
- 235000013752 Rhizopus oryzae Nutrition 0.000 description 2
- 108010003581 Ribulose-bisphosphate carboxylase Proteins 0.000 description 2
- 241000332749 Setosphaeria turcica Species 0.000 description 2
- 101000611441 Solanum lycopersicum Pathogenesis-related leaf protein 6 Proteins 0.000 description 2
- 244000061456 Solanum tuberosum Species 0.000 description 2
- 235000002595 Solanum tuberosum Nutrition 0.000 description 2
- 241000209072 Sorghum Species 0.000 description 2
- 240000006394 Sorghum bicolor Species 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000266365 Stemphylium vesicarium Species 0.000 description 2
- 241000692746 Stenocarpella maydis Species 0.000 description 2
- 241000723792 Tobacco etch virus Species 0.000 description 2
- 241000723873 Tobacco mosaic virus Species 0.000 description 2
- 241000233791 Ustilago tritici Species 0.000 description 2
- 235000007244 Zea mays Nutrition 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000000843 anti-fungal effect Effects 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000012707 chemical precursor Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000004590 computer program Methods 0.000 description 2
- 108091036078 conserved sequence Proteins 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 231100000676 disease causative agent Toxicity 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 239000000417 fungicide Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000001502 gel electrophoresis Methods 0.000 description 2
- 238000012215 gene cloning Methods 0.000 description 2
- 102000005396 glutamine synthetase Human genes 0.000 description 2
- 239000005090 green fluorescent protein Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 108010002685 hygromycin-B kinase Proteins 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000007689 inspection Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 230000000877 morphologic effect Effects 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 239000002853 nucleic acid probe Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 238000003976 plant breeding Methods 0.000 description 2
- 244000000003 plant pathogen Species 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920000903 polyhydroxyalkanoate Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 2
- 230000008261 resistance mechanism Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 229910001415 sodium ion Inorganic materials 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 230000009897 systematic effect Effects 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 230000017105 transposition Effects 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- 108020004463 18S ribosomal RNA Proteins 0.000 description 1
- OVSKIKFHRZPJSS-UHFFFAOYSA-N 2,4-D Chemical compound OC(=O)COC1=CC=C(Cl)C=C1Cl OVSKIKFHRZPJSS-UHFFFAOYSA-N 0.000 description 1
- 239000005631 2,4-Dichlorophenoxyacetic acid Substances 0.000 description 1
- 229940087195 2,4-dichlorophenoxyacetate Drugs 0.000 description 1
- UPMXNNIRAGDFEH-UHFFFAOYSA-N 3,5-dibromo-4-hydroxybenzonitrile Chemical compound OC1=C(Br)C=C(C#N)C=C1Br UPMXNNIRAGDFEH-UHFFFAOYSA-N 0.000 description 1
- CAAMSDWKXXPUJR-UHFFFAOYSA-N 3,5-dihydro-4H-imidazol-4-one Chemical class O=C1CNC=N1 CAAMSDWKXXPUJR-UHFFFAOYSA-N 0.000 description 1
- 102100026105 3-ketoacyl-CoA thiolase, mitochondrial Human genes 0.000 description 1
- 108010020183 3-phosphoshikimate 1-carboxyvinyltransferase Proteins 0.000 description 1
- 101150101112 7 gene Proteins 0.000 description 1
- WFPZSXYXPSUOPY-ROYWQJLOSA-N ADP alpha-D-glucoside Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=CN=C(C=2N=C1)N)OP(O)(=O)OP(O)(=O)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O WFPZSXYXPSUOPY-ROYWQJLOSA-N 0.000 description 1
- 108010003902 Acetyl-CoA C-acyltransferase Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 241000589156 Agrobacterium rhizogenes Species 0.000 description 1
- 241000919507 Albugo candida Species 0.000 description 1
- 241000724328 Alfalfa mosaic virus Species 0.000 description 1
- 244000291564 Allium cepa Species 0.000 description 1
- 235000002732 Allium cepa var. cepa Nutrition 0.000 description 1
- 241001149961 Alternaria brassicae Species 0.000 description 1
- 241000323764 Alternaria zinnae Species 0.000 description 1
- 241000429811 Alternariaster helianthi Species 0.000 description 1
- 241001444080 Aphanomyces euteiches Species 0.000 description 1
- 241000219195 Arabidopsis thaliana Species 0.000 description 1
- 240000005410 Ascochyta medicaginicola var. medicaginicola Species 0.000 description 1
- 241001414024 Ascochyta sorghi Species 0.000 description 1
- 244000309473 Ascochyta tritici Species 0.000 description 1
- 241000228197 Aspergillus flavus Species 0.000 description 1
- 241001530056 Athelia rolfsii Species 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 241000193388 Bacillus thuringiensis Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- 241000190150 Bipolaris sorokiniana Species 0.000 description 1
- 241000895502 Blumeria graminis f. sp. tritici Species 0.000 description 1
- 241000123650 Botrytis cinerea Species 0.000 description 1
- 235000014698 Brassica juncea var multisecta Nutrition 0.000 description 1
- 235000006008 Brassica napus var napus Nutrition 0.000 description 1
- 240000000385 Brassica napus var. napus Species 0.000 description 1
- 235000006618 Brassica rapa subsp oleifera Nutrition 0.000 description 1
- 235000004977 Brassica sinapistrum Nutrition 0.000 description 1
- 239000005489 Bromoxynil Substances 0.000 description 1
- 229910000906 Bronze Inorganic materials 0.000 description 1
- 238000006037 Brook Silaketone rearrangement reaction Methods 0.000 description 1
- 101100394003 Butyrivibrio fibrisolvens end1 gene Proteins 0.000 description 1
- 241000498608 Cadophora gregata Species 0.000 description 1
- 101100342815 Caenorhabditis elegans lec-1 gene Proteins 0.000 description 1
- 101100150553 Caenorhabditis elegans ssu-1 gene Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920008347 Cellulose acetate propionate Polymers 0.000 description 1
- 240000004385 Centaurea cyanus Species 0.000 description 1
- 235000005940 Centaurea cyanus Nutrition 0.000 description 1
- 241001435629 Cephalosporium gramineum Species 0.000 description 1
- 241001290235 Ceratobasidium cereale Species 0.000 description 1
- 241001658057 Cercospora kikuchii Species 0.000 description 1
- 244000309550 Cercospora medicaginis Species 0.000 description 1
- 241000113401 Cercospora sojina Species 0.000 description 1
- 241000437818 Cercospora vignicola Species 0.000 description 1
- 108010022172 Chitinases Proteins 0.000 description 1
- 102000012286 Chitinases Human genes 0.000 description 1
- 102000040852 Class 2 family Human genes 0.000 description 1
- 108091071896 Class 2 family Proteins 0.000 description 1
- 241000221751 Claviceps purpurea Species 0.000 description 1
- 241001330709 Cochliobolus pallescens Species 0.000 description 1
- 108700010070 Codon Usage Proteins 0.000 description 1
- 241001480648 Colletotrichum dematium Species 0.000 description 1
- 241000222237 Colletotrichum trifolii Species 0.000 description 1
- 241000222239 Colletotrichum truncatum Species 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 1
- 101710190853 Cruciferin Proteins 0.000 description 1
- 241001537312 Curvularia inaequalis Species 0.000 description 1
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 101710096830 DNA-3-methyladenine glycosylase Proteins 0.000 description 1
- 102100039128 DNA-3-methyladenine glycosylase Human genes 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 241000866066 Diaporthe caulivora Species 0.000 description 1
- 241000042001 Diaporthe helianthi Species 0.000 description 1
- 241001508801 Diaporthe phaseolorum Species 0.000 description 1
- 241000588700 Dickeya chrysanthemi Species 0.000 description 1
- 241001422851 Didymella maydis Species 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 241000710188 Encephalomyocarditis virus Species 0.000 description 1
- 241001337814 Erysiphe glycines Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 241000122692 Fusarium avenaceum Species 0.000 description 1
- 241000223194 Fusarium culmorum Species 0.000 description 1
- 241001208371 Fusarium incarnatum Species 0.000 description 1
- 241000221779 Fusarium sambucinum Species 0.000 description 1
- 241000427940 Fusarium solani Species 0.000 description 1
- 241000145502 Fusarium subglutinans Species 0.000 description 1
- 241001508365 Gaeumannomyces tritici Species 0.000 description 1
- 206010071602 Genetic polymorphism Diseases 0.000 description 1
- 101710186901 Globulin 1 Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 108700037728 Glycine max beta-conglycinin Proteins 0.000 description 1
- 239000005562 Glyphosate Substances 0.000 description 1
- 241000308375 Graminicola Species 0.000 description 1
- 244000020551 Helianthus annuus Species 0.000 description 1
- 235000003222 Helianthus annuus Nutrition 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 101000742054 Homo sapiens Protein phosphatase 1D Proteins 0.000 description 1
- 230000010740 Hormone Receptor Interactions Effects 0.000 description 1
- 241000549404 Hyaloperonospora parasitica Species 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010021929 Infertility male Diseases 0.000 description 1
- 241000222058 Kabatiella Species 0.000 description 1
- 241000228457 Leptosphaeria maculans Species 0.000 description 1
- 241001198950 Leptosphaerulina trifolii Species 0.000 description 1
- 244000309551 Leptotrochila medicaginis Species 0.000 description 1
- 241000234280 Liliaceae Species 0.000 description 1
- 241000215452 Lotus corniculatus Species 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 101150050813 MPI gene Proteins 0.000 description 1
- 241000584607 Macrospora Species 0.000 description 1
- 241001598086 Magnaporthiopsis maydis Species 0.000 description 1
- 208000007466 Male Infertility Diseases 0.000 description 1
- 101000763602 Manilkara zapota Thaumatin-like protein 1 Proteins 0.000 description 1
- 101000763586 Manilkara zapota Thaumatin-like protein 1a Proteins 0.000 description 1
- 240000004658 Medicago sativa Species 0.000 description 1
- 235000017587 Medicago sativa ssp. sativa Nutrition 0.000 description 1
- 241001022799 Microdochium sorghi Species 0.000 description 1
- 108091092878 Microsatellite Proteins 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 101000966653 Musa acuminata Glucan endo-1,3-beta-glucosidase Proteins 0.000 description 1
- 208000031888 Mycoses Diseases 0.000 description 1
- 241000131448 Mycosphaerella Species 0.000 description 1
- 102000018463 Myo-Inositol-1-Phosphate Synthase Human genes 0.000 description 1
- 108091000020 Myo-Inositol-1-Phosphate Synthase Proteins 0.000 description 1
- 101710202365 Napin Proteins 0.000 description 1
- 241000244206 Nematoda Species 0.000 description 1
- 241000368696 Nigrospora oryzae Species 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 241001668536 Oculimacula yallundae Species 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 241000233654 Oomycetes Species 0.000 description 1
- 241001147398 Ostrinia nubilalis Species 0.000 description 1
- 238000009004 PCR Kit Methods 0.000 description 1
- 238000002944 PCR assay Methods 0.000 description 1
- 241000932831 Pantoea stewartii Species 0.000 description 1
- 241000218222 Parasponia andersonii Species 0.000 description 1
- 241000787361 Parastagonospora avenae Species 0.000 description 1
- 241000736122 Parastagonospora nodorum Species 0.000 description 1
- 101710096342 Pathogenesis-related protein Proteins 0.000 description 1
- 241000588701 Pectobacterium carotovorum Species 0.000 description 1
- 241000985513 Penicillium oxalicum Species 0.000 description 1
- 241000364057 Peoria Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108091093037 Peptide nucleic acid Proteins 0.000 description 1
- 241000063951 Perconia Species 0.000 description 1
- 241000760719 Peronosclerospora maydis Species 0.000 description 1
- 241001183114 Peronosclerospora sacchari Species 0.000 description 1
- 241001670203 Peronospora manshurica Species 0.000 description 1
- 241000342283 Peronospora trifoliorum Species 0.000 description 1
- 235000010627 Phaseolus vulgaris Nutrition 0.000 description 1
- 101000870887 Phaseolus vulgaris Glycine-rich cell wall structural protein 1.8 Proteins 0.000 description 1
- 244000309499 Phoma insidiosa Species 0.000 description 1
- 241001480007 Phomopsis Species 0.000 description 1
- 241001478707 Phyllosticta sojicola Species 0.000 description 1
- 241000471406 Physoderma maydis Species 0.000 description 1
- 241001246239 Physopella Species 0.000 description 1
- 241000233620 Phytophthora cryptogea Species 0.000 description 1
- 241000709664 Picornaviridae Species 0.000 description 1
- 108700001094 Plant Genes Proteins 0.000 description 1
- 241000886313 Plenodomus lindquistii Species 0.000 description 1
- 229920000331 Polyhydroxybutyrate Polymers 0.000 description 1
- 241000710078 Potyvirus Species 0.000 description 1
- 208000037534 Progressive hemifacial atrophy Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 102100038675 Protein phosphatase 1D Human genes 0.000 description 1
- 240000008296 Prunus serotina Species 0.000 description 1
- 235000014441 Prunus serotina Nutrition 0.000 description 1
- 241001480435 Pseudopeziza medicaginis Species 0.000 description 1
- 241000221300 Puccinia Species 0.000 description 1
- 241000540505 Puccinia dispersa f. sp. tritici Species 0.000 description 1
- 241000567197 Puccinia graminis f. sp. tritici Species 0.000 description 1
- 241001304534 Puccinia polysora Species 0.000 description 1
- 241001304535 Puccinia purpurea Species 0.000 description 1
- 241001123567 Puccinia sorghi Species 0.000 description 1
- 241001123583 Puccinia striiformis Species 0.000 description 1
- 241001192932 Pustula tragopogonis Species 0.000 description 1
- 241000190117 Pyrenophora tritici-repentis Species 0.000 description 1
- 241001622914 Pythium arrhenomanes Species 0.000 description 1
- 239000013614 RNA sample Substances 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 101150075111 ROLB gene Proteins 0.000 description 1
- 101150013395 ROLC gene Proteins 0.000 description 1
- 238000010240 RT-PCR analysis Methods 0.000 description 1
- 108010091086 Recombinases Proteins 0.000 description 1
- 102000018120 Recombinases Human genes 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 241000235546 Rhizopus stolonifer Species 0.000 description 1
- 241001183191 Sclerophthora macrospora Species 0.000 description 1
- 241000342322 Sclerospora graminicola Species 0.000 description 1
- 241001597349 Septoria glycines Species 0.000 description 1
- 241000093892 Septoria helianthi Species 0.000 description 1
- 241000266353 Setosphaeria pedicellata Species 0.000 description 1
- 241000893482 Sporisorium sorghi Species 0.000 description 1
- 108010039811 Starch synthase Proteins 0.000 description 1
- 241000116011 Stenocarpella macrospora Species 0.000 description 1
- 102000002933 Thioredoxin Human genes 0.000 description 1
- 108091036066 Three prime untranslated region Proteins 0.000 description 1
- 241000722093 Tilletia caries Species 0.000 description 1
- 241000167577 Tilletia indica Species 0.000 description 1
- 241000031845 Tilletia laevis Species 0.000 description 1
- 241000218234 Trema tomentosa Species 0.000 description 1
- 241000223261 Trichoderma viride Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 244000098338 Triticum aestivum Species 0.000 description 1
- 241000083901 Urocystis agropyri Species 0.000 description 1
- 241000965658 Uromyces striatus Species 0.000 description 1
- 241000237690 Ustilago cruenta Species 0.000 description 1
- 244000301083 Ustilago maydis Species 0.000 description 1
- 235000015919 Ustilago maydis Nutrition 0.000 description 1
- 241001123669 Verticillium albo-atrum Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 241000209149 Zea Species 0.000 description 1
- 101001036768 Zea mays Glucose-1-phosphate adenylyltransferase large subunit 1, chloroplastic/amyloplastic Proteins 0.000 description 1
- 101001040871 Zea mays Glutelin-2 Proteins 0.000 description 1
- 101000662549 Zea mays Sucrose synthase 1 Proteins 0.000 description 1
- 241001360088 Zymoseptoria tritici Species 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 108091000039 acetoacetyl-CoA reductase Proteins 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000000246 agarose gel electrophoresis Methods 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000012098 association analyses Methods 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 230000000680 avirulence Effects 0.000 description 1
- 229940097012 bacillus thuringiensis Drugs 0.000 description 1
- 210000003578 bacterial chromosome Anatomy 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 101150103518 bar gene Proteins 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 102000005936 beta-Galactosidase Human genes 0.000 description 1
- 102000023732 binding proteins Human genes 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 238000010364 biochemical engineering Methods 0.000 description 1
- 229920000704 biodegradable plastic Polymers 0.000 description 1
- 238000003766 bioinformatics method Methods 0.000 description 1
- 238000005422 blasting Methods 0.000 description 1
- 239000010974 bronze Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 238000009470 controlled atmosphere packaging Methods 0.000 description 1
- KUNSUQLRTQLHQQ-UHFFFAOYSA-N copper tin Chemical compound [Cu].[Sn] KUNSUQLRTQLHQQ-UHFFFAOYSA-N 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000004062 cytokinin Substances 0.000 description 1
- UQHKFADEQIVWID-UHFFFAOYSA-N cytokinin Natural products C1=NC=2C(NCC=C(CO)C)=NC=NC=2N1C1CC(O)C(CO)O1 UQHKFADEQIVWID-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- -1 defined above Substances 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 238000001784 detoxification Methods 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000012631 diagnostic technique Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 235000019621 digestibility Nutrition 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 235000005489 dwarf bean Nutrition 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000010318 early mammalian development Effects 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000003028 elevating effect Effects 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 230000034964 establishment of cell polarity Effects 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000009313 farming Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 244000037666 field crops Species 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- 108091006047 fluorescent proteins Proteins 0.000 description 1
- 102000034287 fluorescent proteins Human genes 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000003008 fumonisin Substances 0.000 description 1
- 244000000004 fungal plant pathogen Species 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 238000011331 genomic analysis Methods 0.000 description 1
- IAJOBQBIJHVGMQ-BYPYZUCNSA-N glufosinate-P Chemical compound CP(O)(=O)CC[C@H](N)C(O)=O IAJOBQBIJHVGMQ-BYPYZUCNSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- XDDAORKBJWWYJS-UHFFFAOYSA-N glyphosate Chemical compound OC(=O)CNCP(O)(O)=O XDDAORKBJWWYJS-UHFFFAOYSA-N 0.000 description 1
- 229940097068 glyphosate Drugs 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- 239000003815 herbicide antidote Substances 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 230000000749 insecticidal effect Effects 0.000 description 1
- 239000002917 insecticide Substances 0.000 description 1
- 230000016507 interphase Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000011005 laboratory method Methods 0.000 description 1
- 101150066555 lacZ gene Proteins 0.000 description 1
- 230000000974 larvacidal effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 108010083942 mannopine synthase Proteins 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 238000007857 nested PCR Methods 0.000 description 1
- 230000003988 neural development Effects 0.000 description 1
- 108091027963 non-coding RNA Proteins 0.000 description 1
- 102000042567 non-coding RNA Human genes 0.000 description 1
- 108010058731 nopaline synthase Proteins 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000012017 passive hemagglutination assay Methods 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 230000029553 photosynthesis Effects 0.000 description 1
- 238000010672 photosynthesis Methods 0.000 description 1
- 230000008121 plant development Effects 0.000 description 1
- 230000008635 plant growth Effects 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 239000005014 poly(hydroxyalkanoate) Substances 0.000 description 1
- 239000005015 poly(hydroxybutyrate) Substances 0.000 description 1
- 229920001690 polydopamine Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 238000012809 post-inoculation Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 230000009979 protective mechanism Effects 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 108010050873 prunasin hydrolase Proteins 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000014493 regulation of gene expression Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000013077 scoring method Methods 0.000 description 1
- 230000008117 seed development Effects 0.000 description 1
- 230000007226 seed germination Effects 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000009958 sewing Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000010902 straw Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 108010050014 systemin Proteins 0.000 description 1
- HOWHQWFXSLOJEF-MGZLOUMQSA-N systemin Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)OC(=O)[C@@H]1CCCN1C(=O)[C@H]1N(C(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]2N(CCC2)C(=O)[C@H]2N(CCC2)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)N)C(C)C)CCC1 HOWHQWFXSLOJEF-MGZLOUMQSA-N 0.000 description 1
- 108060008226 thioredoxin Proteins 0.000 description 1
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical class CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 230000010474 transient expression Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 1
- 229940038773 trisodium citrate Drugs 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 108091005957 yellow fluorescent proteins Proteins 0.000 description 1
Images































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/6895—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for plants, fungi or algae
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H1/00—Processes for modifying genotypes ; Plants characterised by associated natural traits
- A01H1/04—Processes of selection involving genotypic or phenotypic markers; Methods of using phenotypic markers for selection
- A01H1/045—Processes of selection involving genotypic or phenotypic markers; Methods of using phenotypic markers for selection using molecular markers
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H5/00—Angiosperms, i.e. flowering plants, characterised by their plant parts; Angiosperms characterised otherwise than by their botanic taxonomy
- A01H5/10—Seeds
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01H—NEW PLANTS OR NON-TRANSGENIC PROCESSES FOR OBTAINING THEM; PLANT REPRODUCTION BY TISSUE CULTURE TECHNIQUES
- A01H6/00—Angiosperms, i.e. flowering plants, characterised by their botanic taxonomy
- A01H6/46—Gramineae or Poaceae, e.g. ryegrass, rice, wheat or maize
- A01H6/4684—Zea mays [maize]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/415—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from plants
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/82—Vectors or expression systems specially adapted for eukaryotic hosts for plant cells, e.g. plant artificial chromosomes (PACs)
- C12N15/8241—Phenotypically and genetically modified plants via recombinant DNA technology
- C12N15/8261—Phenotypically and genetically modified plants via recombinant DNA technology with agronomic (input) traits, e.g. crop yield
- C12N15/8271—Phenotypically and genetically modified plants via recombinant DNA technology with agronomic (input) traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance
- C12N15/8279—Phenotypically and genetically modified plants via recombinant DNA technology with agronomic (input) traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance for biotic stress resistance, pathogen resistance, disease resistance
- C12N15/8282—Phenotypically and genetically modified plants via recombinant DNA technology with agronomic (input) traits, e.g. crop yield for stress resistance, e.g. heavy metal resistance for biotic stress resistance, pathogen resistance, disease resistance for fungal resistance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/13—Plant traits
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/172—Haplotypes
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Botany (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Developmental Biology & Embryology (AREA)
- Environmental Sciences (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Physiology (AREA)
- Cell Biology (AREA)
- Plant Pathology (AREA)
- Mycology (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicinal Chemistry (AREA)
- Natural Medicines & Medicinal Plants (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Breeding Of Plants And Reproduction By Means Of Culturing (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Pretreatment Of Seeds And Plants (AREA)
Abstract
A presente invenção refere-se a seqfiências de polinucleotídeos que codificam um gene, o qual confere resistência ao patógeno de planta Colletotrichum, o qual causa podridão de caule por antracnose, ressecamento de folha e doença nas pontas em milho e outros cereais. A invenção refere-se ainda a plantas e sementes contendo genes quiméricos que compreendem as referidas seqüências de polinucleotídeos, as quais aumentam ou conferem resistência ao patógeno de planta Colletotrichum, e a processos de produção das referidas plantas e sementes. A invenção refere-se ainda a seqüências que podem ser usadas como marcadores moleculares, os quais são úteis na identificação da região de interesse em linhagens de milho, resultantes de novas cruzas e que podem rápida e eficazmente introgredir o gene de linhagens de milho contendo o referido gene em outras linhagens de milho que não contêm o referido gene, de modo a torná-las resistentes ao Colletotrichum e resistentes à podridão de caule.
Description
[0001] Esta invenção se refere às composições e métodos utilizáveis na criação ou aperfeiçoamento de resistência a patógeno em plantas. Adicionalmente, a invenção refere-se a plantas que tenham sido geneticamente engenheiradas com as composições da invenção.
[0002] Colletotrichum graminicola (Ces) (Cg), mais comumente conhecido como antracnose, é o agente causador da queima das folhas por antracnose, podridão de colmo por antracnose (ASR) e morte descendente que afeta Zea mays (L.), também conhecida como milho (maize ou corn). É a única podridão de colmo comum conhecida que também causa uma queima das folhas (Bergstrom, et al., (1999), Plant Disease, 83:596-608, White, D.G. (1998), Compendium of Corn Diseases, pp. 1-78). Sabe-se que ela ocorre nos Estados Unidos desde 1855 e ela tem sido relatada nas Américas, Europa, África, Ásia e Austrália (McGee, D.C. (1988), Maize Diseases: A Reference Source for Seed Technologists, APS Press, St. Paul, MN; White, (1998) acima mencionado; White, et al., (1979) Proc. Annu. Corn Sorghum Res Conf (34th), 1-15). Somente nos Estados Unidos, mais de 37,5 milhões de acres são infestados anualmente com perdas médias de produção de 6,6% em todo o País (ver Figura 1). As perdas de produção são devidas tanto ao baixo peso de grão em plantas infectadas como ao acamamento, ou seja, a murcha das plantas devida à fraqueza nos colmos causada pela infecção (Dodd, J., (1980), Plant Disease, 64:533-537). As plantas acamadas são mais difíceis de colher e são susceptíveis a outras doenças. Tipicamente após a infecção, a parte superior do colmo morre primeiramente enquanto o colmo mais baixo ainda está verde. Externamente, a infecção pode ser reconhecida por placas manchadas de preto no anel mais externo do colmo, enquanto internamente o tecido da medula da planta fica com aparência descolorida ou preta. A inoculação ocorre por inúmeras maneiras. As raízes podem crescer através dos fragmentos do colmo e se tornarem infectadas. Isso se torna um problema crescente na medida em que os métodos de agricultura de plantio direto são mais amplamente adotados devido aos seus benefícios ambientais. O fungo pode também infectar os colmos através de dano por inseto e outras feridas (ver White (1998) acima). A infecção do colmo pode ser precedida pela infecção da folha que causa a queima da folha e provê o inóculo para a infecção do colmo. Há controvérsia na literatura técnica com referência ao número de diferentes variedades ou raças de Cg presentes na natureza. O patógeno é transmitido pelo vento ou por lotes de semente contaminada. Os esporos permanecem viáveis por até 2 anos (ver McGee (1988) acima mencionado; Nicholson, et al., (1980), Phytopathology, 70:255-261; Warren, H.L. (1977), Phytopathology, 67:160-162; Warren, et al., (1975), Phytopathology, 65:620-623).
[0003] Os agricultores podem combater a infecção por doenças fúngicas de milho, tal como a antracnose, através do uso de fungicidas, mas estes apresentam efeitos ambientais adversos e requerem o monitoramento de campos e técnicas de diagnóstico para determinar qual o fungo que está cusando a infecção de modo que o fungicida correto possa ser usado. Particularmente, para lavouras em campos amplos, tal como a do milho, isto é difícil. O uso de linhagens que portam fontes genéticas ou transgênicas de resistência é mais prático se os genes responsáveis pela resistência puderem ser incorporados em um germoplasma elite de alta produtividade sem reduzir a produção. As fontes genéticas de resistência a Cg têm sido relatadas. Foram identificadas diversas linhagens de milho que portam algum nível de resistência a Cg (ver White et al. (1979) acima). Estas incluem A556, MP305, H21, SP288, CI88A e FR16. Uma análise do tipo testcross de translocação recíproca usando a A556 indicou que os genes que controlam a resistência com relação à posição ASR estão nos longos ramos dos cromossomos 1, 4 e 8, assim como em ambos os ramos do cromossomo 6 (Carson, M.L. (1981), Sources of inheritance of resistance to anthracnose stalk rot of corn. Ph.D. Thesis, University of Illinois, Urbana-Champaign). A introgressão de resistência derivada de tais linhagens é complexa. Foi relatado que uma outra endogâmica, a LB31, porta um único gene dominante que controla a resistência relativa a ASR, mas parece ser instável, especialmente na presença de infestação de broca de milho Europeu (Badu-Apraku et al., (1987) Phytopathology 77 : 957-959). Foi verificado que a linhagen MP305 porta dois genes dominantes para resistência, um com um efeito maior e um com efeito menor (Carson (1981) acima). A MP305 foi tornada disponível pel Universidade do Mississipe através do Sistema Nacional de Germoplasma de Planta (GRIN ID: NSL 250298) operado pelo Departamento de Agricultura dos Estados Unidos. Ver Compilação de Germoplasma de Melhoramento de Milho Norte Americano, J.T. Gerdes et al., Crop Science Society of America, 1993. A semente de MP305 pode ser obtida através de Paul Williams, Geneticista Supervisor de Pesquisa USDA-ARS, “Corn Host Plant Resistance Research Unit”, Box 9555, 340 Dorman Hall, Mississippi State, MS 39762.
[0004] Foi relatado que há dois genes ligados no ramo longo do cromossomo 4 que confere resistência a Cg (Toman, et al., (1993), Phytopathology, 83:981-986; Cowen, N et al. (1991) Maize Genetics Conference Abstracts 33). Foi também relatada a existência de um lócus de caractere quantitativo (QTL) de significativa resistência no cromossomo 4 (Jung, et al., (1994), Theoretical and Applied Genetics, 89:413-418). Jung et al. (acima) relatou que o UMC15 pode ser usado para selecionar o QTL no cromossomo 4 na MP305 e sugeriu que o QTL está em uma região 12cM do cromossomo 4 entre o UMC15 e o UMC66. De fato, como discutido em maiores detalhes mais adiante, a região entre o UMC15 e o UMC66, como relatado no mapa genético neighbors 4 IBM2, é de aproximadamente 129 cM, e a seleção com relação ao QTL, na maneira sugerida por Jung et al. (1994, acima), seria, na melhor das hipóteses, selecionar um grande intervalo cromossômico com considerável arraste de ligação e efeito fenotípico negativo, e, na pior das hipóteses, uma recombinação dupla poderia ocorrer entre os dois marcadores resultando em uma seleção falso positiva para o lócus de Rcg1.
[0005] Muito trabalho tem sido feito no mecanismo de resistência à doença em plantas em geral. Alguns mecanismos de resistência são não-patogênicos específicos por natureza, ou a chamada “resistência de não-hospedeiro”. Esses podem estar baseados na estrutura da parede celular ou mecanismos de proteção similares. Entretanto, apesar de em algumas plantas estar ausente um sistema imunológico com anticorpos circulantes e os outros atributos de um sistema imunológico de mamífero, elas possuem outros mecanismos para se proteger especificamente contra patógenos. O mais importante e mais estudado desses mecanismos é o dos genes de resistência da planta a doenças, ou genes “R”. Uma das muitas revisões deste mecanismo de resistência e os genes R pode ser encontrada em Bekhadir et al., (2004), Current Opinion in Plant Biology 7:391-399. Há cinco classes conhecidas de genes R: proteínas intracelulares com um sítio de ligação a nucleotídeo (NBS) e uma repetição rica em leucina (LRR); proteínas transmembrana com um domínio extracelular LRR (TM-LRR); LRR transmembrana e extracelular com um domínio quinase citoplásmica (TM-CK-LRR); proteína sustentada no sinal de membrana com um domínio citoplásmico espiral-espiralada (MSAP-CC); e quinases associadas a membrana com um sítio de miristilação N-terminal (MAK-N) (ver, por exemplo: Cohn, et al., (2001), Immunology, 13:55-62; Dangl, et al. (2001), Nature, 411:826-833).
[0006] O gene de resistência das concretizações da presente invenção codifica um novo gene R relacionado ao Apesar de múltiplos genes NBS-LRR terem sido descritos, eles diferem amplamente em sua resposta a diferentes patógenos e ação exata. No conhecimento do Depositante, o novo gene R aqui descrito é o único que comprovadamente provê resistência a Cg.
[0007] As concretizações desta invenção estão baseadas no mapeamento refinado, clonagem e caracterização do gene responsável pela maior porção do fenótipo de resistência a partir da linhagem MP305, na introgressão de um intervalo cromossômico truncado com o lócus de resistência de MP305 em outras linhagens com pouco ou nenhum arraste de genes, na demonstração do uso daquele gene como um transgene e no uso de marcadores moleculares para mover o gene ou o transgene para dentro das linhagens elite usando técnicas de melhoramento.
[0008] As concretizações incluem um polinucleotídeo isolado compreendendo uma sequência de nucleotídeos codificante de um polipeptídeo capaz de conferir resistência a Colletotrichum, sendo que o polipeptídeo possui uma sequência de aminoácidos de pelo menos 50%, pelo menos 75%, pelo menos 80%, pelo menos 85%, pelo menos 90%, e pelo menos 95% de identidade, quando comparada com a SEQ ID NO:3 ou as sequências depositadas junto à Agricultural Research Service (ARS) Culture Collection em 22 de fevereiro de 2006, como Depósito de Patente No. NRRL B- 30895, com base no algoritmo de alinhamento de Needleman- Wunsch, ou um complemento da sequência de nucleotídeos, sendo que o complemento e a sequência de nucleotídeos são compostos do mesmo número de nucleotídeos e são 100% complementares.
[0009] Concretizações adicionais da presente invenção incluem um vetor compreendendo o polinucleotídeo de uma concretização da presente invenção, tal como a SEQ ID NO:3, ou as sequências do plasmídio depositado como Depósito de Patente No. NRRL-30895, e um constructo de DNA recombinante compreendendo o polinucleotídeo de uma concretização da presente invenção operacionalmente ligado a pelo menos uma sequência de regulação. Uma célula de planta, bem como uma planta, cada uma compreendendo o constructo de DNA recombinante de uma concretização da presente invenção, e uma semente compreendendo o constructo de DNA recombinante que também são concretizados pela presente invenção.
[0010] Os métodos concretizados pela presente invenção incluem: (1) um método para transformar uma célula hospedeira, incluindo uma célula de planta, compreendendo a transformação da célula hospedeira com o polinucleotídeo de uma concretização da presente invenção e regeneração de uma planta a partir da célula de planta transformada, e (3) métodos para conferir ou melhorar a resistência a Colletotrichum e/ou podridão do colmo, compreendendo a transformação de uma planta com o constructo de DNA recombinante de uma concretização da presente invenção, conferindo e/ou melhorando, por esse meio, a resistência a Colletotrichum ou à podridão do colmo.
[0011] Concretizações adicionais incluem métodos de determinação da presença ou ausência dos polinucleotídeos de uma concretização da presente invenção, ou o lócus do Rcg1, em uma planta de milho, compreendendo pelo menos um dentre: (a) isolar moléculas de ácido nucléico a partir da planta de milho e determinar se um gene Rcg1 está presente ou ausente pela amplificação de sequências homólogas ao polinucleotídeo, (b) isolar moléculas de ácido nucléico a partir da planta de milho e realizar uma hibridização Southern, (c) isolar proteínas a partir da planta de milho e realizar um western blot usando anticorpos para a proteína Rcg1, (d) isolar proteínas a partir da planta de milho e realizar um ensaio ELISA usando anticorpos para a proteína Rcg1, ou (e) demonstrar a presença de sequências de mRNA derivadas do transcripto de mRNA de Rcg1 e único para o Rcg1, determinando, por esse meio a presença do polinucleotídeo ou do lócus de Rcg1 na planta de milho.
[0012] Também são concretizados pela presente invenção os métodos para alterar o nível de expressão de uma proteína capaz de conferir resistência a Colletotrichum ou à podridão do colmo em uma planta ou célula de planta compreendendo: (a) transformar uma célula de planta com o constructo de DNA recombinante de uma concretização da presente invenção e (b) deixar crescer a célula de planta transformada sob condições que são apropriadas para expressão do constructo de DNA recombinante, sendo que a expressão do constructo de DNA recombinante resulta na produção de níveis alterados de uma proteína capaz de conferir resistência a Colletotrichum ou à podridão do colmo no hospedeiro transformado.
[0013] Um método adicional concretizado pela presente invenção é um método para conferir ou melhorar a resistência a Colletotrichum e/ou podridão do colmo em uma planta de milho, compreendendo: (a) cruzar uma primeira planta de milho em que está ausente o lócus de Rcg1 com uma segunda planta de milho contendo o lócus de Rcg1 para produzir uma população segregante, (b) realizar o screening da população segregante com relação a um membro contendo o lócus de Rcg1 com um primeiro ácido nucléico, não incluindo o UMC15a ou o UMC66, capaz de hibridizar com um segundo ácido nucléico ligado a ou localizado dentro do lócus de Rcg1, e (c) selecionar um membro para posterior cruzamento e seleção.
[0014] São também concretizações da presente invenção os métodos para melhorar a resistência a Colletotrichum e/ou podridão do colmo, ou para fazer a introgressão de resistência a Colletotrichum e/ou podridão do colmo em uma planta de milho, compreendendo a realização da seleção assistida por marcador da planta de milho com um marcador de ácido nucléico, sendo que o marcador de ácido nucléico especificamente hibridiza com uma molécula de ácido nucléico tendo uma primeira sequência de ácido nucléico que está ligada a uma segunda sequência de ácido nucléico que está localizada no lócus de Rcg1 da MP305 e selecionar a planta de milho com base na seleção assistida por marcador. Os marcadores específicos FLP, MZA e o SNP específico de Rcg1 aqui revelados são aspectos adicionais da invenção.
[0015] As concretizações adicionais são: uma fonte doadora aperfeiçoada de germoplasma para realizar a introgressão de resistência ou melhorar a resistência a Colletotrichum ou à podridão do colmo em uma planta de milho, dito germoplasma compreendendo a DE811ASR (BC5) e progênie derivada da mesma. A dita progênie pode ser ainda caracterizada como contendo as sequências de Rcg1 da DE811ASR (BC5) aqui reveladas, os marcadores moleculares em ou geneticamente ligados ao Rcg1, resistência ou resistência aperfeiçoada a Colletotrichum ou quaisquer combinações das mesmas.
[0016] As concretizações adicionais incluem processos para identificar plantas de milho que apresentam resistência recentemente conferida ou melhorada a Colletotrichum pela detecção dos alelos de pelo menos 2 marcadores na planta de milho, sendo que pelo menos um dos marcadores está em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1, e pelo menos um dos marcadores está em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200.
[0017] As concretizações adicionais incluem processos para identificar plantas de milho que apresentam resistência recentemente conferida ou melhorada a Colletotrichum pela detecção de alelos de pelo menos 2 marcadores na planta de milho, sendo que pelo menos um dos marcadores está em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1, e pelo menos um dos marcadores está em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200. Concretizações semelhantes englobadas por este processo incluem pelo menos um dos marcadores estando em ou dentro do intervalo cromossômico abaixo do UMC1086 e acima do gene Rcg1, em ou dentro do intervalo cromossômico abaixo do UMC2285 e acima do gene Rcg1, e pelo menos um dos marcadores está em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200, em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2187, ou em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC15a. Concretizações adicionais relacionadas ao mesmo processo incluem aquelas em que pelo menos um dos marcadores é capaz de detectar um polimorfismo localizado em uma posição correspondente aos nucleotídeos 7230 e 7535 da SEQ ID NO: 137, nucleotídeos 11293 e 12553 da SEQ ID NO: 173, nucleotídeos 25412 e 29086 da SEQ ID NO: 137, ou nucleotídeos 43017 e 50330 da SEQ ID NO: 137.
[0018] Concretizações adicionais incluem processos para identificar plantas de milho que apresentam resistência recentemente conferida ou melhorada a Colletotrichum pela detecção de alelos de pelo menos 2 marcadores na planta de milho, sendo que pelo menos um dos marcadores em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1 é selecionado dos marcadores listados na Tabela 16, e pelo menos um dos marcadores em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200 é também selecionado dos marcadores listados na Tabela 16. As concretizações incluem processos para identificar plantas de milho que apresentam resistência recentemente conferida ou melhorada a Colletotrichum pela seleção de pelo menos quatro marcadores ou pelo menos seis, sendo que pelo menos dois ou três dos marcadores estão em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1, e pelo menos dois ou três dos marcadores estão em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200. Concretizações adicionais incluem este mesmo processo quando os dois ou três marcadores em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1, bem como os dois ou três marcadores em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200, são selecionados daqueles listados na Tabela 16. Uma outra concretização deste processo inclui a detecção do alelo 7 no MZA1112, detecção do alelo 2 no MZA2591, ou detecção do alelo 8 no MZA3434. As plantas e sementes de milho produzidas pelos processos concretizados são também concretizações da invenção, incluindo aquelas plantas de milho que não compreendem alelos iguais aos da MP305 em ou acima do UMC2041, ou em ou abaixo do UMC2200 nos lócus mostrados na Tabela 16.
[0019] Outras concretizações incluem processos para identificar plantas de milho que apresentam resistência recentemente conferida ou melhorada a Colletotrichum pela detecção de alelos de pelo menos dois marcadores na planta de milho, sendo que pelo menos um dos marcadores está em ou dentro do intervalo cromossômico abaixo do UMC2041 e acima do gene Rcg1, e pelo menos um dos marcadores está em ou dentro do intervalo abaixo do gene Rcg1 e acima do UMC2200, e quando o processo detecta a presença ou ausência de pelo menos um marcador localizado dentro do gene Rcg1. Tal concretização adicional inclui uma modificação deste processo na qual quatro marcadores são selecionados, em que dois dos marcadores estão dentro do intervalo cromossômico abaixo do UMC2285 e acima do gene Rcg1, e pelo menos dois dos marcadores estão dentro do intervalo abaixo do gene Rcg1 e acima do UMC15a. Uma concretização adicional deste processo inclui o gene Rcg1 que tenha sido introgredido a partir de uma planta de milho doadora, incluindo a MP305 ou a DE811ASR(BC5), em uma planta de milho receptora para produzir uma planta de milho introgredida. Este processo também inclui a possibilidade de quando a planta de milho introgredida for selecionada para um evento de recombinação abaixo do gene Rcg1 e acima do UMC15a, de modo que a planta de milho introgredida retenha o primeiro intervalo cromossômico derivado da MP305 abaixo do gene Rcg1 e acima do UMC15a, e não retenha um segundo intervalo cromossômico derivado da MP305 em ou abaixo do UMC15a. As plantas e sementes de milho produzidas por esses processos são também concretizações da invenção. As plantas de milho introgredidas concretizadas pela invenção incluem aquelas que são conversões de lócus de Rcg1 de PH705, PH5W4, PH51K ou PH87P, ou progênie das mesmas.
[0020] Uma concretização adicional da invenção é um processo de identificação de uma planta de milho que apresenta resistência melhorada a infecção por Colletotrichum, pela detecção, na planta de milho, da presença ou ausência de pelo menos um marcador no lócus do Rcg1, e seleção da planta de milho na qual pelo menos um marcador está presente. As concretizações incluem aquelas quando pelo menos um marcador está em ou dentro da SEQ ID NO:137, e também quando o pelo menos um marcador é capaz de detectar um polimorfismo localizado em uma posição na SEQ ID NO:137 correspondente à posição entre os nucleotídeos 1 e 536, entre os nucleotídeos 7230 e 7535, entre os nucleotídeos 11293 e 12553, entre os nucleotídeos 25412 e 29086; e entre os nucleotídeos 43017 e 50330, e também quando pelo menos um marcador está em ou dentro da sequência codificante de Rcg1, ou localizado em ou dentro do polinucleotídeo apresentado na SEQ ID NO: 1. Uma outra concretização inclui aquela quando o processo detecta um único polimorfismo de nucleotídeo em uma posição na SEQ ID NO:1 correspondente a uma ou mais das posições 413, 958, 971, 1099, 1154, 1235, 1250, 1308, 1607, 2001, 2598, e 3342. Os marcadores incluídos pelos processos dessas concretizações incluem os marcadores de SNP C00060-01 e C00060-02, marcadores que detectam uma sequência de mRNA derivada do transcripto de mRNA do Rcg1 e única para Rcg1, e marcadores de FLP em um amplicon gerado por um par de primers apresentado nesta descrição, tais como aqueles das SEQ ID NOs: 35-42, e seus complementos. Uma outra concretização inclui aquela quando o processo detecta a presença ou ausência de pelo menos dois marcadores dentro do lócus de Rcg1, incluindo o C00060-01 e o C00060-02. As plantas e sementes de milho produzidas por esses processos são também concretizações da invenção. Plantas de milho introgredidas concretizadas pela invenção incluem aquelas que são conversões do lócus de Rcg1 da PH705, PH5W4, PH51K ou PH87P, ou progênie das mesmas. Tais concretizações incluem semente de milho compreendendo um primeiro intervalo cromossômico derivado da MP305 definido por BNLG2162 e UMC1051, e não compreendendo um segundo intervalo cromossômico derivado da MP305 acima do UMC2041 ou abaixo do UMC1051, e quando a semente de milho compreende o gene de Rcg1 e, quando crescida, produz uma planta de milho que apresenta resistência à infecção por Colletotrichum. Sementes das concretizações também incluem semente de milho compreendendo um primeiro intervalo cromossômico derivado da MP305 entre, mas não incluindo, o UMC2285 e o UMC15a, e não compreendendo um segundo intervalo cromossômico derivado da MP305 em ou acima do UMC2285 ou em ou abaixo do UMC15a, e, além disso, tal semente de milho que compreende o gene Rcg1 e, quando crescida, produz uma planta de milho que apresenta resistência à infecção por Colletotrichum. As plantas e células de planta de milho oriundas desta semente estão também incluídas nas concretizações da invenção.
[0021] Concretizações adicionais incluem semente de uma variedade de milho designada DE811ASR(BC5), ou a semente de milho depositada como número de acesso ATCC PTA-7434, ou uma semente de progênie derivada a partir daquela variedade, que compreende o gene Rcg1, que quando crescida, produz uma planta que apresenta resistência melhorada ou recentemente conferida a infecção por Colletotrichum. As plantas e células de planta crescidas a partir desta semente são também concretizações, assim como a semente da progênie que retém um primeiro intervalo cromossômico derivado da MP305 ou da DE811ASR(BC5) dentro, mas não incluindo, o UMC2285 e o UMC15a, e a semente da progênie que não compreende um segundo intervalo cromossômico da MP305 em ou acima do UMC2285 ou em ou abaixo do UMC15a. As plantas e células de planta da semente acima estão incluídas como concretizações. A semente da progênie que é uma conversão do lócus de Rcg1 da PH705, PH5W4, PH51K ou PH87P, ou uma progênie das mesmas é também concretizada na invenção, assim como o são as sementes da progênie que compreendem pelo menos dois ou mais dos alelos 7 na MZA11123, alelo 2 na MZA2591, ou alelo 8 na MZA3434. As concretizações adicionais incluem a semente da progênie que compreende um nucleotídeo citosina na MZA2591.32, um nucleotídeo timina na MZA2591.35, e um nucleotídeo citosina na MZA3434.17.
[0022] Concretizações adicionais incluem um sistema de computador para identificar uma planta de milho que apresenta resistência recentemente conferida ou melhorada a infecção por Colletotrichum compreendendo uma base de dados que compreende uma informação de escore de alelo para uma ou mais plantas de milho com relação a quatro ou mais lócus de marcador que estão proximamente ligados a ou dentro do lócus de Rcg1, e instruções que examinam a dita base de dados para determinar a herança do intervalo cromossômico ou porções do mesmo definido para quatro ou mais lócus de marcador e computar se sim ou se não uma ou mais plantas de milho compreende o gene Rcg1. Concretizações adicionais incluem um sistema de computador para identificar uma planta de milho que apresenta resistência recentemente conferida ou melhorada para infecção por Colletotrichum compreendendo uma base de dados que compreende informação de escore de alelos para uma ou mais plantas de milho com relação a um ou mais lócus de marcador dentro do lócus de Rcg1, e instruções que examinam dita base de dados para determinar a herança do lócus de Rcg1. A informação de escore de alelos para uma ou mais plantas de milho para tais sistemas de computador podem adicionalmente compreender dois, três, ou mais lócus de marcador dentro do lócus de Rcg1.
[0023] As concretizações também incluem marcadores em ou dentro das SEQ ID NOs: 140 até 146 para a MZA3434, MZA2591, MZA11123, MZA15842, MZA1851, MZA8761 e MZA11455, respectivamente. Outras concretizações incluem marcadores genéticos localizados em ou no lócus de Rcg1 ou no gene Rcg1, incluindo aquelas localizadas na SEQ ID NO: 137, por exemplo, aquelas localizadas nas regiões correspondentes aos nucleotídeos entre 1 e 536, entre 7230 e 7535, entre 11293 e 12553, entre 25412 e 29086, e a região entre os nucleotídeos 43017 e 50330. Os marcadores concretizados também incluem aqueles localizados na SEQ ID NO: 1, tais como aqueles localizados em ou dentro das posições de nucleotídeos 550-658 da SEQ ID NO: 1, ou aquelas localizadas em ou dentro das posições de nucleotídeos 1562-1767 da SEQ ID NO: 1. Os marcadores das concretizações incluem aqueles marcadores localizados nos amplicons gerados por um par de primers, sendo que o primeiro primer é uma sequência de número ímpar oriunda da SEQ ID NO: 23 até 41, e sendo que o segundo primer é uma sequência de número par oriunda da SEQ ID NO: 24 até 42.
[0024] Concretizações adicionais incluem um método para obtenção de plantas de milho compreendendo: cruzamento da MP305 ou DE811ASR(BC5) [No. de Depósito PTO-7434] como uma primeira planta parental, com uma planta diferente na qual está ausente um lócus de Rcg1, como uma segunda planta parental, para obter, por esse meio, a progênie compreendendo o lócus de Rcg1 da primeira parental; e opcionalmente, ainda, compreendendo uma ou mais etapas adicionais de melhoramento para obter a progênie de uma ou mais gerações posteriores que compreendem o lócus de Rcg1 da primeira parental. Tais plantas de milho concretizadas incluem ambas as plantas endogâmicas e híbridas. As sementes de tais plantas, incluindo aquelas que são homozigotas e heterozigotas com relação ao lócus de Rcg1, e são concretizados pela invenção os métodos de obtenção de produtos de milho resultante do processamento daquelas sementes. É também uma concretização da invenção o uso de tal semente em alimento ou ração ou na produção de um produto de milho, tais como farinha de milho, cereal de milho e óleo de milho.
[0025] A Figura 1 é o mapa dos Estados Unidos mostrando a gravidade de infestação pela podridão do colmo por antracnose por município, em 2002.
[0026] A Figura 2 (a, b, c) é um alinhamento de uma sequência de polipeptídeo das concretizações (SEQ ID NO:3) comparando-a com outros polipeptídeos NBS-LRR conhecidos.
[0027] A Figura 3 é um gráfico produzido pelo software Windows QTL Cartographer mostrando uma análise estatística da chance (eixo dos Y) de que o lócus responsável pelo fenótipo de resistência a Cg esteja localizado em uma posição particular ao longo do cromossomo (eixo dos X) como definido pelos marcadores de FLP.
[0028] A Figura 4 é um blot de gel de eletroforese de alíquotas de reações de RT-PCR que revelam a presença de uma banda de 260 pb presente nas amostras derivada de ambas as plantas resistentes, a infectada e a não-infectada, mas ausente para as amostras susceptíveis. Os fragmentos de RT-PCR foram obtidos a partir de 12,5 ng de RNA total oriundo do tecido de colmo da DE811 e da DE811ASR. O cDNA obtido por transcrição reversa foi amplificado usando primers específicos de Rcg1 e primers de rRNA 18S como um padrão interno.
[0029] A Figura 5 é um diagrama esquemático da estratégia de marcação com Mu (Mu-tagging) usada para validar o gene Rcg1.
[0030] A Figura 6 é a estrutura do Rcg1 mostrando a localização de quatro diferentes sítios de inserção do mutator.
[0031] A Figura 7 (a-b) é uma série de imagens de mapa genético com resolução aumentada do mapa da região próxima ao gene Rcg1. As distâncias no mapa para 7(a) no mapa assinalado “A” estão em cM e em relação ao mapa genético 4 Neighbors IBM2. As distâncias no mapa para 7(b) no mapa assinalado “B” foram desenvolvidas usando 184 indivíduos oriundos da população BC7, e as distâncias no mapa para 7(b) no mapa assinalado “C” foram desenvolvidas usando 1060 indivíduos oriundos da população BC7. O mapeamento genético na população BC7 aumentou a resolução do mapa mais do que 10 vezes, quando comparado com o mapa publicado. A localização dos marcadores mostrada à direita de cada mapa é baseada na extrapolação de sua localização no mapa físico.
[0032] A Figura 8(a-b) é uma imagem do mapa genético mostrando o intervalo cromossômico com o gene Rcg1 em DE811ASR (BC3), o tamanho reduzido do intervalo cromossômico com o gene Rcg1 obtido em DE811ASR (BC5) e o adicional tamanho reduzido do intervalo cromossômico em endogâmicas obtido por meio do uso, inicialmente, da DE811ASR (BC5) como a fonte doadora. Para todos os marcadores, as distâncias do mapa mostradas foram relatadas no mapa neighbors IBM2 disponível publicamente no GDB de milho, separadamente para a MZA15842, FLP27 e FLP56 para as quais as posições no mapa foram extrapoladas usando a análise de regressão relativa aos mapas de alta resolução na Figura 7(b), para os mapas B e C, usando as posições de UMC2285, PHI093 e CSU166a as quais foram comuns em ambos os mapas.
[0033] A Figura 9 (a-b) mostra em: (a) o alinhamento da região não-colinear oriunda de DE811ASR (BC5) com relação a B73 e a Mo17. Os tamanhos do BAC na Figura 9(a) são estimativos; (b) uma porção da região não-colinear como apresentada na SEQ ID NO: 137 na qual reside o Rcg1, incluindo as regiões repetitivas situadas nele, assim como os exons 1 e 2 de Rcg1.
[0034] A Figura 10 (a-b) mostra as distribuições de tamanho médio de lesão de folha em diferentes plantas individuais em 15 dias após a inoculação com Cg nas linhagens DE811ASR(BC5) e DE811, respectivamente.
[0035] A Figura 11 mostra uma comparação de tamanho médio de lesão de folha em plantas de DE811 e DE811ASR(BC5) infectadas com Cg nos dias 7 e 15 após a inoculação.
[0036] A Figura 12 mostra a gravidade média da doença quatro a cinco semanas após a inoculação com Cg em colmos de híbridos derivados do cruzamento de DE811ASR(BC5) e DE811 para a linhagem indicada.
[0037] A Figura 13 mostra o aperfeiçoamento da produtividade na maturidade após a inoculação com Cg em híbridos derivados do cruzamento de DE811ASR(BC5) para a linhagem indicada, em comparação com a produtividade de híbridos derivados do cruzamento de DE811 para a linhagem indicada.
[0038] A Figura 14 mostra a gravidade da doença em 5 diferentes localidades causada por Cg em colmos de linhagens endogâmicas derivadas de DE811ASR(BC5) ou de MP305, quatro a cinco semanas após a inoculação. As diferenças entre as linhagens que foram positivas e negativas com relação ao gene Rcg1 são estatisticamente significativas em um valor de P de menos que 0,05.
[0039] A figura 15 mostra a progressão da doença em colmos representativos oriundos de linhagens PH705 endogâmicas que são positivas e negativas com relação a Rcg1.
[0040] A Figura 16 mostra a progressão da doença em colmos representativos oriundos de linhagens PH87P endogâmicas que são positivas e negativas com relação a Rcg1.
[0041] A Figura 17 mostra a gravidade da doença quatro a cinco semanas após a inoculação em 5 diferentes localidades causada por Cg em colmos de híbridos derivados do cruzamento de DE811ASR(BC5) para a linhagem indicada. As diferenças entre as linhagens que são positivas e negativas com relação ao gene Rcg1 são estatisticamente significativas em um valor P de menos que 0,05, exceto para a localidade 5.
[0042] A Figura 18 mostra a progressão da doença em colmos representativos oriundos de híbridos criados a partir das linhagens PH4CV e PH705 que são positivas e negativas com relação a Rcg1.
[0043] A Figura 19 mostra a progressão da doença em colmos representativos oriundos de híbridos criados a partir das linhagens PH705 e PH87P que são positivas e negativas com relação a Rcg1.
[0044] A Figura 20 mostra o método de contagem de escore para a gravidade da doença em colmos de milho. Aos colmos é dado um escore, designado antgr75, que representa o número de internódulos (até 5, incluindo o internódulo inoculado) que estão mais que 75% descoloridos. Estes resultados em um escore variando de 0 a 5, sendo que 0 indica menos que 75% de descoloração no internódulo inoculado, e 5 indica 75% ou mais de descoloração dos primeiros cinco internódulos, incluindo o internódulo inoculado.
[0045] A Figura 21 mostra um contig no mapa físico de B73 que é homólogo à região no interior da qual é inserida a DE811ASR (BC5) contendo a região não-colinear de Rcg1, o que demonstra que muitas B73 derivadas de cromossomos artificiais bacterianos (BACs) estão disponíveis na região de interesse a partir da qual a informação de sequência pode ser obtida.
[0046] A Figura 22 mostra o alinhamento do mapa genético contendo os marcadores MZA e público com os mapas físicos de Mo17 e B73. As distâncias de mapa genético foram desenvolvidas pela utilização de 1060 indivíduos a partir da população de mapeamento BC7. Uma análise de uma biblioteca BAC de Mo17 também mostrou o lócus de Rcg1 como sendo não-colinear com a correspondente região de Mo17. A localização dos marcadores mostrada por linhas pontilhadas com relação ao mapa de B73 são extrapolações a partir da localização no mapa físico de Mo17. A localização dos marcadores mostrada pelas linhas pontilhadas com relação ao mapa de Mo17 são extrapolações da localização no mapa físico de B73.
[0047] A Figura 23 mostra os oligos para os marcadores de hibridização de Rcg1 desenhado para uso com reações Invader™.
[0048] A Figura 24 mostra os oligos para os marcadores de hibridização de Rcg1 desenhados para uso com reações TaqMan®.
[0049] A Figura 25 mostra os resultados de um northern blot obtido a partir de aproximadamente 1,5 mg de RNA rico em polyA isolado a partir de plantas resistentes e susceptíveis nos dias 0, 3, 6, 9 e 13 após a inoculação (dpi). A membrana foi sondada com um fragmento de primer aleatório de Rcg1 marcado de 420 pb. O tecido resistente é oriundo de DE811ASR(BC5) e o tecido susceptível é oriundo de DE811.
[0050] A Figura 26 mostra que a amplificação por PCR usando pares de primers específicos para Rcg1 somente amplifica na linhagem resistente DE811ASR(BC5) e na parental doadora MP305, mas não na linhagem susceptível DE811, com exceção de FLP110F- R, a qual amplifica a região do sítio de ligação espiral espiralada-nucleotídeo, a qual é altamente conservada, e, portanto, amplifica uma região em qualquer lugar no genoma que não seja Rcg1 na linhagem DE811. Foi usada uma ladder de 100 pb para fazer o fragmento sob medida.
[0051] As concretizações da presente invenção provêem composições e métodos (ou processos) dirigidos à indução de resistência em plantas a patógeno, particularmente resistência a fungos. As composições são novas sequências de nucleotídeos e de aminoácidos que conferem ou melhoram a resistência de plantas a patógenos fúngicos. Especificamente, certas concretizações provêem polipeptídeos tendo a sequência de aminoácidos apresentada em SEQ ID NO:3, e variantes e fragmentos da mesma. São ainda providas moléculas de ácido nucléico isoladas, e variantes e fragmentos das mesmas, que compreendem sequências de nucleotídeos que codificam a sequência de aminoácidos mostrada na SEQ ID NO:3.
[0052] As sequências de nucleotídeos que codificam o polipeptídeo da SEQ ID NO:3 são apresentadas nas SEQ ID NOs: 1 e 4. As plantas, células de planta, sementes e microrganismos compreendendo uma sequência de nucleotídeos que codifica um polipeptídeo das concretizações são também aqui reveladas.
[0053] Foi feito um depósito da molécula de ácido nucléico de Rcg1 em 22 de fevereiro de 2006 junto à Agricultural Research Service (ARS) Culture Collection, guardada na Microbial Genomics and Bioprocessing Research Unit do National Center for Agricultural Utilization Research (NCAUR), sob as disposições do Tratado de Budapeste. Foi dado ao depósito o seguinte número de acesso: NRRL B-30895. O endereço do NCAUR é 1815 N. University Street, Peoria, IL, 61604. Este depósito será mantido sob os termos do tratado de Budapeste para o Reconhecimento Internacional do Depósito de Microrganismos para os Propósitos de Procedimento de Patente. Este depósito foi feito meramente como uma conveniência para aqueles técnicos no assunto e não é uma admissão de que o depósito seja requerido de acordo com o §112 do U.S.C. 35. O depósito ficará disponível ao público irrevogavelmente e sem restrição ou condição após a publicação de uma patente. Entretanto, deve ser compreendido que a disponibilidade de um depósito não constitui uma licença para praticar o objeto da invenção em detrimento dos direitos de patente concedidos por ação governamental.
[0054] Uma amostra de 2500 sementes de DE811ASR (BC5) foram depositadas junto à American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, VA 20110-2209, USA em 13 de março de 2006 e atribuído o No. de Depósito PTO-7434. O acesso a este depósito ficará disponível, durante a pendência do pedido, ao Representante de Patentes e de Marcas Registradas, a pessoas indicadas pelo Representante para serem habilitadas para tanto sob solicitação, e correspondentes funcionários em repartições de patentes estrangeiras nas quais seja depositado o pedido de patente. Este depósito será mantido sob os termos do Tratado de Budapeste sobre o Reconhecimento Internacional de Depósito de Microrganismos para os Propósitos de Procedimento de Patente. O depósito ficará disponível ao público irrevogavelmente e sem restrição ou condição após a publicação de uma patente. No entanto, deve ficar entendido que a disponibilidade do depósito não constitui uma licença para praticar o objeto da invenção ou métodos em detrimento dios direitos de patente.
[0055] O polipeptídeo de comprimento completo das concretizações (SEQ ID NO:3) compartilha variados graus de homologia com polipeptídeos conhecidos da família NBS-LRR. Em particular, o novo polipetídeo das concretizações compartilha homologia com as proteínas NBS-LRR isoladas de Oryza sativa (Nos. De Acesso NP_910480 (SEQ ID NO: 14), NP_910482 (SEQ ID NO: 16), NP_921091 (SEQ ID NO: 17) e NP_910483 (SEQ ID NO: 15)) e de Hordeum vulgare (No. de Acesso AAG37354 (SEQ ID NO: 18); Zhou et al., (2001) Plant Cell 13:337-350). A Figura 1 provê um alinhamento da sequência de aminoácidos apresentada na SEQ ID NO: 3 com as proteínas antifúngicas de O. sativa e de H. vulgare (SEQ ID NOs: 14-18).
[0056] Os alinhamentos de aminoácidos usando o programa GAP indicam que a SEQ ID NO:3 compartilha aproximadamente 42,3% de similaridade de sequência com a proteína antifúngica de O. sativa NP_910480 (SEQ ID NO: 14), 41,7% de similaridade de sequência com a proteína de O. sativa NP_910482 (SEQ ID NO: 16), 56,9% de similaridade com a proteína de O. sativa NP_921091 (SEQ ID NO: 17) e 42,1% de similaridade de sequência com a proteína de O. sativa NP_910483 (SEQ ID NO: 15). Além disso, a SEQ ID NO: 3 compartilha aproximadamente 42,8% de similaridade de sequência com a proteína de H. vulgare AAG37354 (SEQ ID NO: 18).
[0057] O grupo NBS-LRR de genes-R é a maior classe de genes- R descobertos até hoje. Em Arabidopsis thaliana, estima-se que mais de 150 estejam presentes no genoma (Meyers, et al., (2003), Plant Cell, 15:809-834; Monosi, et al., (2004), Theoretical and Applied Genetics, 109:1434-1447), enquanto que em arroz, estima-se em aproximadamente 500 genes NBS-LRR (Monosi, (2004) supra). A classe NBS-LRR de genes R está compreendida por duas subclasses. A classe 1 de genes NBS-LRR contém um domínio semelhante a TIR-Toll/Interleucina-1 na sua extremidade N’, a qual, até hoje, foi encontrada em dicotiledôneas (Meyers, (2003) acima mencionado; Monosi, (2004) acima mencionado). A segunda classe de NBS-LRR contém tanto um domínio espiral-espiralado ou um domínio (nt) na sua extremidade N (Bai, et al. (2002) Genome Research, 12:18711884; Monosi, (2004) acima mencionado; Pan, et al., (2000), Journal of Molecular Evolution, 50:203-213). A classe 2 de NBS-LRR foi encontrada e ambas as espécies, monocotiledôneas e dicotiledôneas (Bai, (2002) acima mencionado; Meyers, (2003) acima mencionado; Monosi, (2004) acima mencionado; Pan, (2000) acima mencionado).
[0058] O domínio NBS do gene parece desempenhar um papel de sinalização nos mecanismos de defesa da planta (van der Biezen, et al., (1998), Current Biology: CB, 8:R226-R227). A região LRR parece ser a região que interage com os produtos AVR do patógeno (Michelmore, et al., (1998), Genome Res., 8:1113-1130; Meyers, (2003) acima citado). Esta região LRR, em comparação com o domínio NBS está sob uma pressão de seleção muito maior para a diversificação (Michelmore, (1998) acima citado; Meyers, (2003) acima citado; Palomino, et al., (2002), Genome Research, 12:1305-1315). Os domínios LRR também são encontrados em outros contextos; esses motifs de resíduos 20-29 estão presentes em arranjos in tandem em inúmeras proteínas com funções diversas, tais como interações hormônio- receptor, inibição de enzima, adesão celular e tráfego celular. Inúmeros estudos recentes revelaram o envolvimento de proteínas LRR no desenvolvimento inicial de mamíferos, desenvolvimento neural, polarização celular, regulação de expressão de gene e sinalização de apoptose.
[0059] O gene das concretizações está nitidamente relacionado ao NBS-LRR da família de classe 2, mas não se ajusta completamente ao molde clássico. A extremidade amino possui homologia com os chamados sítios de ligação a nucleotídeo (NBS). Há uma região rica em leucina também localizada, como esperado, a jusante do NBS. Entretanto, diferentemente das proteínas NBS-LRR previamente estudadas, a região rica em leucina carece da natureza repetitiva sistemática encontrada nos domínios LRR mais clássicos, muito menos consistentemente seguindo o padrão de repetição Lxx típico e, em particular, não tendo exemplos de sequências de consenso descritas por Wang et al. ((1999) Plant J. 19:55-64; ver, especialmente, a Figura 5) ou Bryan et al. ((2000), Plant Cell 12:2033-2045; ver, especialmente, a Figura 3).
[0060] Como a região LRR é a porção receptora de um NBS-LRR, quando uma nova LRR, tal como a desta descrição, for encontrada, a faixa de sua atividade, ou seja, a faixa de patógenos aos quais ela responderá, não é imediatamente óbvia a partir da sequência. O gene das concretizações foi isolado com base no fenótipo de resistência a Cg, e, portanto, a nova LRR responde a Cg. Entretanto, não fica excluído o fato de que ela responde a outros patógenos não testados no trabalho feito até agora.
[0061] Os ácidos nucléicos e polipeptídeos das concretizações encontram uso em métodos para conferir ou melhorar a resistência em plantas a fungos. Consequentemente, as composições e métodos revelados aqui são utilizáveis na proteção de plantas contra patógenos fúngicos. Os termos “resistência a patógeno”, “resistência a fungo” e “resistência a doença” têm a intenção de significar o fato de que a planta evita os sintomas da doença que são o resultado de interações planta-patógeno. Ou seja, é feita a prevenção de patógenos causarem doenças em plantas e os sintomas de doença associados, ou, alternativamente, os sintomas de doença causados pelo patógeno são minimizados ou diminuídos, tais como, por exemplo, a redução de estresse e perda de produtividade associada. Os especialistas na técnica saberão reconhecer que as composições e métodos aqui revelados podem ser usados com outras composições e métodos disponíveis na técnica para proteger plantas do ataque de patógenos.
[0062] Em consequência, os métodos das concretizações podem ser utilizados para proteger plantas de doenças, particularmente aquelas doenças que são causadas por patógenos de fungos de planta. Como aqui usado, o termo “resistência a fungo” se refere a resistência melhorada ou tolerância com relação a um patógeno de fungo em comparação com aquela de uma planta do tipo selvagem. Os efeitos podem variar desde um leve aumento na tolerância aos efeitos do patógeno fúngico (por exemplo, inibição parcial) até resistência total, tal como a planta não ser afetada pela presença do patógeno fúngico. Um nível aumentado de resistência contra um patógeno fúngico particular ou contra um espectro mais amplo de patógenos fúngicos constitui a resistência “melhorada” ou aperfeiçoada contra fungos. As concretizações da invenção também melhorarão ou aperfeiçoarão a resistência da planta a patógeno de fungo, de modo que a resistência da planta a um patógeno ou a patógenos fúngicos aumentará. O termo “melhorar” se refere a aperfeiçoar, aumentar, amplificar, multiplicar, elevar, fazer surgir e semelhantes. Aqui, as plantas da invenção são descritas como sendo resistentes a infecção por Cg ou tendo ‘resistência melhorada’ a infecção por Cg, como resultado do lócus de Rcg1 da invenção. Consequentemente, elas tipicamente apresentam resistência melhorada à doença quando comparadas com plantas equivalentes que são susceptíveis à infecção por Cg porque lhes falta o lócus de Rcg1. Por exemplo, usando o sistema de contagem de escore descrito no Exemplo 11 (ver também a Figura 20), elas tipicamente apresentam um decréscimo de um ponto, de dois pontos ou de três pontos ou de mais pontos no escore de infecção, ou mesmo uma redução do escore para 1 ou 0, quando comparadas com plantas equivalentes que são susceptíveis a infecção por Cg porque lhes falta o lócus de Rcg1.
[0063] Em aspectos particulares, os métodos para conferir ou melhorar resistência de uma planta a fungos compreendem a introdução, em uma planta, de pelo menos um cassete de expressão, sendo que o cassete de expressão compreende uma sequência de nucleotídeos codificante de um polipeptídeo antifúngico das concretizações operacionalmente ligado a um promotor que dirige a expressão na planta. A planta expressa o polipeptídeo, conferindo, por esse meio, a resistência antifúngica sobre a planta, ou aperfeiçoando o nível de resistência inerente da planta. Em concretizações particulares, o gene confere resistência ao patógeno fúngico Cg.
[0064] A expressão de um polipeptídeo antifúngico das concretizações pode ser direcionada para tecidos específicos de planta onde a resistência ao patógeno é particularmente importante, tais como, as folhas, as raízes, os colmos, ou os tecidos vasculares. Tal expressão tecido-preferida pode ser realizada por meio de promotores raiz-preferidos, folha- preferidos, tecido vascular-preferidos, colmo-preferidos ou semente-preferidos.
[0065] Como aqui usado, o termo “ácido nucléico” inclui referência a um polímero de desoxirribonucleotídeos ou de ribonucleotídeos, tanto na forma de simples-fita como na de dupla-fita, e a menos que seja limitado de outra forma, engloba análogos conhecidos (por exemplo, ácidos nucléicos de peptídeo) tendo a natureza essencial de nucleotídeos naturais na medida em que eles hibridizam com ácidos nucléicos de simples-fita de uma maneira semelhante aos nucleotídeos de ocorrência natural.
[0066] Os termos “polipeptídeo”, “peptídeo”, e “proteína” são aqui usados indistintamente para se referir a um polímero de resíduos de aminoácidos. Os termos se aplicam aos polímeros de aminoácidos nos quais um ou mais resíduos de aminoácido é um análogo químico artificial de um aminoácido correspondente de ocorrência natural, assim como aos polímeros de aminoácidos de ocorrência natural. Os polipeptídeos das concretizações podem ser produzidos tanto a partir de um ácido nucléico aqui revelado, como pelo uso de técnicas de biologia molecular padrão. Por exemplo, uma proteína truncada das concretizações pode ser produzida por expressão de um ácido nucléico recombinante das concretizações em uma célula hospedeira apropriada, ou alternativamente, pela combinação de procedimentos ex vivo, tal como digestão com protease e purificação.
[0067] Como aqui usados, os termos “codificar” ou “codificado”, quando usados no contexto de um ácido nucléico específico, significa que o ácido nucléico compreende a informação de requisito para dirigir a tradução da sequência de nucleotídeos para uma proteína especificada. A informação pela qual uma proteína é codificada é especificada pelo uso de códons. Um ácido nucléico codificante de uma proteína pode compreender sequências não-traduzidas (por exemplo, introns) dentro de regiões traduzidas do ácido nucléico ou tais sequências não-traduzidas interpostas (por exemplo, tal como no cDNA) podem estar ausentes.
[0068] As concretizações da invenção englobam polinucleotídeos isolados ou substancialmente purificados ou composições de proteína. Um polinucleotídeo ou proteína “isolado(a)” ou “purificado(a)”, ou porção biologicamente ativa do mesmo, está substancialmente ou essencialmente isento(a) de componentes que normalmente acompanham ou interagem com o polinucleotídeo ou proteína como encontrado(a) no seu ambiente de ocorrência natural. Portanto, um polinucleotídeo ou proteína isolado(a) ou purificado(a) está substancialmente isento(a) de outro material celular, ou meio de cultura produzido por técnica recombinante (por exemplo, amplificação por PCR), ou substancialmente isento(a) de precursores químicos, quando sintetizados(as) quimicamente. De modo ótimo, um polinucleotídeo “isolado” está isento de sequências (por exemplo, sequências codificantes de proteína) que naturalmente flanqueiam o polinucleotídeo (ou seja, sequências localizadas nas extremidades 5' e 3' do polinucleotídeo) no DNA genômico do organismo a partir do qual o polinucleotídeo é derivado. Por exemplo, nas várias concretizações, o polinucleotídeo isolado pode conter menos que cerca de 5 kb, cerca de 4 kb, cerca de 3 kb, cerca de 2 kb, cerca de 1 kb, cerca de 0,5 kb, ou cerca de 0,1 kb da sequência de nucleotídeos que flanqueia naturalmente o polinucleotídeo no DNA genômico da célula a partir da qual o polinucleotídeo é derivado. Uma proteína que é substancialmente isenta de material celular inclui preparações de proteína tendo menos do que cerca de 30%, cerca de 20%, cerca de 10%, cerca de 5%, ou cerca de 1% (em peso seco) de proteína contaminante. Quando a proteína das concretizações, ou uma porção biologicamente ativa da mesma, for recombinantemente produzida, de modo ótimo, o meio de cultura representa menos do que cerca de 30%, cerca de 20%, cerca de 10%, cerca de 5%, ou cerca de 1% (em peso seco) de precursores químicos ou substâncias químicas não-protéicas de interesse.
[0069] Os fragmentos e variantes das sequências de nucleotídeos reveladas e proteínas codificadas por esse meio estão também englobadas pelas concretizações. O termo fragmento tem a intenção de significar uma porção da sequência de nucleotídeos ou uma porção da sequência de aminoácidos e, portanto, da proteína codificada por esse meio. Os fragmentos de uma sequência de nucleotídeos podem codificar fragmentos de proteína que retenham a atividade biológica da proteína natural e, assim, a capacidade de conferir, na planta, resistência a fungos. Alternativamente, os fragmentos de uma sequência de nucleotídeos que são utilizáveis como sondas de hibridização não necessariamente codificam proteínas de fragmentos que retenham a atividade biológica. Portanto, os fragmentos de uma sequência de nucleotídeos pode variar de pelo menos cerca de 15 nucleotídeos, cerca de 50 nucleotídeos, cerca de 100 nucleotídeos, e até a sequência de nucleotídeos de comprimento completo codificante dos polipeptídeos das concretizações.
[0070] Um fragmento de uma sequência de nucleotídeos que codifica uma porção biologicamente ativa de um polipeptídeo das concretizações codificará pelo menos cerca de 15, cerca de 25, cerca de 30, cerca de 40, ou cerca de 50 aminoácidos contíguos, ou até o número total de aminoácidos presente no polipeptídeo de comprimento completo das concretizações (por exemplo, 980 aminoácidos para o peptídeo codificado pela SEQ ID NO:1). Os fragmentos de uma sequência de nucleotídeos que são utilizáveis como sondas ou primers de PCR, em geral, não necessitam codificar uma porção biologicamente ativa de uma proteína.
[0071] Como aqui usado, o termo “sequência de comprimento completo”, com referência a um polinucleotídeo especificado, significa que ela possui a sequência inteira de ácido nucléico de uma sequência natural. O termo “sequência natural” tem a intenção de significar uma sequência endógena, ou seja, uma sequência não-engenheirada encontrada no genoma de um organismo.
[0072] Portanto, um fragmento de uma sequência de nucleotídeos das concretizações pode codificar uma porção biologicamente ativa de um polipeptídeo, ou ele pode ser um fragmento que pode ser usado como uma sonda de hibridização ou primer de PCR usando métodos revelados mais adiante. Uma porção biologicamente ativa de um polipeptídeo antipatogênico pode ser preparada pelo isolamento de uma porção de uma das sequências de nucleotídeos das concretizações, expressando a porção codificada da proteína e avaliando a capacidade de a porção codificada da proteína conferir ou melhorar a resistência a fungos em uma planta. As moléculas de ácido nucléico que são fragmentos de uma sequência de nucleotídeos das concretizações compreendem pelo menos cerca de 15, cerca de 20, cerca de 50, cerca de 75, cerca de 100, ou cerca de 150 nucleotídeos, ou até o número de nucleotídeos presente na sequência de nucleotídeos de comprimento completo aqui revelada (por exemplo, 4212 nucleotídeos para a SEQ ID NO: 1).
[0073] O termo “variantes” tem a intenção de significar sequências substancialmente similares. Para polinucleotídeos, uma variante compreende uma deleção e/ou adição de um ou mais nucleotídeos em um ou mais sítios internos dentro do polinucleotídeo natural e/ou uma substituição de um ou mais nucleotídeos em um ou mais sítios no polinucleotídeo natural. Como aqui usado, um polinucleotídeo ou polipeptídeo “natural” compreende uma sequência de nucleotídeos ou sequência de aminoácidos de ocorrência natural, respectivamente. Os especialistas na técnica saberão reconhecer que variantes dos ácidos nucléicos das concretizações serão construídos de modo que o quadro aberto de leitura seja mantido. Para polinucleotídeos, as variantes conservativas incluem aquelas sequências que, por causa da degeneração do código genético, codificam a sequência de aminoácidos de um dos polipeptídeos das concretizações. As variantes alélicas de ocorrência natural, tais como aquelas que podem ser identificadas com o uso de técnicas de biologia molecular amplamente conhecidas, como, por exemplo, com as técnicas de reação em cadeia da polimerase (PCR) e de hibridização como esboçadas mais adiante. Os polinucleotídeos variantes também incluem polinucleotídeos derivados sinteticamente, tais como aqueles gerados, por exemplo, pelo uso de mutagênese sítio-dirigida, mas que ainda codificam uma proteína das concretizações. Em geral, as variantes de um polinucleotídeo particular das concretizações terá pelo menos cerca de 40%, cerca de 45%, cerca de 50%, cerca de 55%, cerca de 60%, cerca de 65%, cerca de 70%, cerca de 75%, cerca de 80%, cerca de 85%, cerca de 90%, cerca de 91%, cerca de 92%, cerca de 93%, cerca de 94%, cerca de 95%, cerca de 96%, cerca de 97%, cerca de 98%, cerca de 99% ou mais de identidade de sequência com relação àquele polinucleotídeo particular, como determinado pelos programas e parâmetros de alinhamento de sequência descritos aqui em algum lugar.
[0074] As variantes de um polinucleotídeo particular das concretizações (ou seja, o polinucleotídeo de referência) podem também ser avaliadas por comparação da percentagem de identidade de sequência entre o polipeptídeo codificado por um polinucleotídeo variante e o polipeptídeo codificado pelo polinucleotídeo de referência. Assim, por exemplo, são revelados os polinucleotídeos isolados que codificam um polipeptídeo com uma dada percentagem de identidade de sequência com relação ao polipeptídeo de SEQ ID NO:3. A identidade de sequência percentual entre quaisquer dois polipeptídeos pode ser calculada usando os programas e parâmetros de alinhamento de sequências descritos aqui em algum lugar. Quando qualquer dado par de polinucleotídeos das concretizações é avaliado por comparação da identidade de sequência percentual compartilhada pelos dois polipeptídeos que ele codifica, a identidade de sequência percentual entre os dois polipeptídeos codificados tem pelo menos cerca de 40%, cerca de 45%, cerca de 50%, cerca de 55%, cerca de 60%, cerca de 65%, cerca de 70%, cerca de 75%, cerca de 80%, cerca de 85%, cerca de 90%, cerca de 91%, cerca de 92%, cerca de 93%, cerca de 94%, cerca de 95%, cerca de 96%, cerca de 97%, cerca de 98%, cerca de 99% ou mais de identidade de sequência.
[0075] O termo proteína “variante” tem a intenção de significar uma proteína derivada da proteína nativa por deleção ou adição de um ou mais aminoácidos em um ou mais sítios internos na proteína natural e/ou substituição de um ou mais aminoácidos em um ou mais sítios na proteína natural. As proteínas variantes englobadas pelas concretizações são biologicamente ativas, ou seja, elas continuam a possuir a atividade biológica desejada da proteína natural, isto é, a capacidade de conferir ou melhorar a resistência da planta a patógeno de fungo como aqui descrito. Tais variantes podem resultar, por exemplo, de polimorfismo genético ou de manipulação pelo homem. As variantes biologicamente ativas de uma proteína natural das concretizações terão pelo menos cerca de 40%, cerca de 45%, cerca de 50%, cerca de 55%, cerca de 60%, cerca de 65%, cerca de 70%, cerca de 75%, cerca de 80%, cerca de 85%, cerca de 90%, cerca de 91%, cerca de 92%, cerca de 93%, cerca de 94%, cerca de 95%, cerca de 96%, cerca de 97%, cerca de 98%, cerca de 99% ou mais de identidade de sequência com relação à sequência de aminoácidos para a proteína natural como determinada pelos programas e parâmetros de alinhamento de sequências aqui descritos em algum lugar. Uma variante biologicamente ativa de uma proteína das concretizações pode diferir daquela proteína por tão pouco quanto cerca de 1-15 resíduos de aminoácido, tão pouco quanto cerca de 1-10, tal como cerca de 6-10, tão pouco quanto cerca de 5, tão pouco quanto 4, 3, 2, ou até mesmo 1 resíduo de aminoácido.
[0076] As proteínas das concretizações podem ser alteradas de diversas maneiras, incluindo substituições, deleções, truncagens e inserções de aminoácidos. Os métodos para realizar tais manipulações são, em geral, conhecidos do estado da técnica. Por exemplo, as variantes e fragmentos da sequência de aminoácidos das proteínas antipatogênicas podem ser preparadas por meio de mutações no DNA. Os métodos para mutagênese e alterações de polinucleotídeos são bem conhecidos no estado da técnica. Ver, por exemplo, Kunkel (1985) Proc. Natl. Acad. Sci. USA 82:488-492; Kunkel et al. (1987) Methods in Enzymol. 154:367-382; patente US 4.873.192; Walker e Gaastra, eds. (1983) Techniques in Molecular Biology (MacMillan Publishing Company, New York) e as referências aqui citadas. A orientação de como realizar apropriadas substituições de aminoácidos que não afetem a atividade biológica da proteína de interesse pode ser encontrada no modelo de Dayhoff et al. (1978) Atlas of Protein Sequence and Structure (Natl. Biomed. Res. Found., Washington, D.C.), aqui incorporado a título de referência. As substituições conservativas, tal como a troca de um aminoácido por um outro tendo propriedades similares podem ser o modo ótimo.
[0077] Portanto, os genes e polinucleotídeos das concretizações incluem ambas, as sequências de ocorrência natural e as formas mutantes. Da mesma maneira, as concretizações englobam ambas, as proteínas de ocorrência natural e as variações e formas modificadas das mesmas. Tais variantes continuarão a possuir a capacidade desejada de conferir ou melhorar a resistência da planta a patógeno de fungo. Obviamente, as mutações que serão feitas no DNA codificante da variante não devem ocorrer fora do quadro de leitura e, de modo ótimo, não criarão regiões complementares que possam produzir estrutura secundária de mRNA (ver EP 0075444).
[0078] Não se espera que as deleções, inserções e substituições das sequências de proteína englobadas aqui produzam mudanças radicais nas características da proteína. Entretanto, quando for difícil predizer o efeito exato da substituição, deleção ou inserção antes de realizá-la, os especialistas na técnica saberão reconhecer que o efeito será avaliado pelo screening de plantas transgênicas que tenham sido transformadas com a proteína variante para averiguar o efeito na capacidade da planta resistir ao ataque patogênico do fungo.
[0079] Os polinucleotídeos e proteínas variantes também englobam as sequências e proteínas derivadas de procedimentos mutagênicos ou recombinogênicos, incluindo, mas não estando limitados a, procedimentos tal como o de combinações aleatórias de DNA. O especialista na técnica pode imaginar modificações que alterem a faixa de patógenos com relação aos quais a proteína responde. Com tal procedimento, uma ou mais diferentes sequências codificantes de proteína podem ser manipuladas para criar uma nova proteína que possua as propriedades desejadas. Desta maneira, são geradas bibliotecas de polinucleotídeos recombinantes a partir de uma população de polinucleotídeos de sequências relacionadas compreendendo regiões de sequência que possuem substancial identidade de sequência e podem ser homologamente recombinadas in vitro ou in vivo. Por exemplo, usando esta abordagem, os motifs de sequência codificantes de um domínio de interesse podem ser combinados aleatoriamente entre o gene de proteína das concretizações e outros genes de proteína conhecidos para obter um novo gene codificante de uma proteína com uma propriedade de interesse aperfeiçoada, tal como capacidade aumentada de conferir ou melhorar a resistência da planta a patógeno de fungo. As estratégias para combinar DNA aleatoriamente são conhecidas no estado da técnica. Ver, por exemplo, Stemmer (1994) Proc. Natl. Acad. Sci. USA 91:1074710751; Stemmer (1994) Nature 370:389-391; Crameri et al. (1997) Nature Biotech. 15:436-438; Moore et al. (1997) J. Mol. Biol. 272:336-347; Zhang et al. (1997) Proc. Natl. Acad. Sci. USA 94:4504-4509; Crameri et al. (1998) Nature 391:288-291; e patentes US 5.605.793 e US 5.837.458.
[0080] Os polinucleotídeos das concretizações podem ser usados para isolar as sequências correspondentes a partir de outros organismos, particularmente outras plantas. Desta maneira, métodos tais como PCR, hibridização e semelhantes podem ser usados para identificar tais sequências com base na sua homologia de sequência com relação às sequências aqui apresentadas. As sequências isoladas com base na sua identidade de sequência com relação às sequências inteiras aqui apresentadas ou com relação a variantes ou fragmentos das mesmas estão englobados pelas concretizações. Tais sequências incluem sequências que são ortólogas das sequências reveladas. O termo “ortólogas” tem a intenção de significar genes derivados de um gene ancestral comum e que são encontrados em diferentes espécies como resultado de uma especialização. Os genes encontrados em diferentes espécies são considerados ortólogos quando suas sequências de nucleotídeos e/ou suas sequências de proteína codificada compartilham pelo menos cerca de 60%, cerca de 70%, cerca de 75%, cerca de 80%, cerca de 85%, cerca de 90%, cerca de 91%, cerca de 92%, cerca de 93%, cerca de 94%, cerca de 95%, cerca de 96%, cerca de 97%, cerca de 98%, cerca de 99%, ou mais de identidade de sequência. As funções dos ortólogos são, com frequência, altamente conservadas entre as espécies. Portanto, polinucleotídeos isolados que codificam uma proteína que confere ou melhora a resistência da planta a patógeno de fungo e que hibridiza sob condições estringentes com as sequências aqui reveladas, ou com variantes ou fragmentos das mesmas, estão englobados pelas concretizações.
[0081] Em uma abordagem de PCR, os primers de oligonucleotídeos podem ser desenhados para uso em reações de PCR para amplificar as correspondentes sequências de DNA a partir do cDNA ou do DNA genômico extraído de qualquer organismo de interesse. Os métodos para desenhar primers de PCR e clonagem de PCR são, em geral, conhecidos no estado da técnica e estão descritos em Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual (2d ed., Cold Spring Harbor Laboratory Press, Plainview, New York). Ver também Innis et al., eds. (1990) PCR Protocols: A Guide to Methods and Applications (Academic Press, New York); Innis e Gelfand, eds. (1995) PCR Strategies (Academic Press, New York); e Innis e Gelfand, eds. (1999) PCR Methods Manual (Academic Press, New York). Os métodos de PCR conhecidos incluem, mas não estão limitados a, métodos usando primers emparelhados, primers nested, primers únicos específicos, primers degenerados, primers gene-específicos, primers vetor-específicos, primers com emparelhamento desigual parcial, e semelhantes.
[0082] Nas técnicas de hibridização, o todo ou parte de um polinucleotídeo conhecido é usado como uma sonda que seletivamente hibridiza com outros polinucleotídeos correspondentes presentes em uma população de fragmentos de DNA genômico clonado ou fragmentos de cDNA (ou seja, bibliotecas genômicas ou de cDNA) a partir de um organismo conhecido. As sondas de hibridização podem ser fragmentos de DNA genômico, fragmentos de cDNA, fragmentos de RNA, ou outros oligonucleotídeos, e podem ser marcados com um grupo detectável, tal como 32P, ou qualquer outro marcador detectável. Assim, por exemplo, as sondas para hibridização podem ser feitas por meio de marcação de oligonucleotídeos sintéticos baseados nos polinucleotídeos das concretizações. Os métodos de preparação das sondas para hibridização e para a construção do cDNA e bibliotecas genômicas são, em geral, conhecidos no estado da técnica e estão descritos em Sambrook et al. (1989) acima citado.
[0083] Por exemplo, o polinucleotídeo inteiro aqui revelado, ou uma ou mais porções do mesmo, pode ser usado como uma sonda capaz de especificamente hibridizar com os correspondentes polinucleotídeos e RNAs mensageiros. Para alcançar a hibridização específica sob uma variedade de condições, tais sondas incluem sequências que são únicas e, de modo ótimo, têm um comprimento de pelo menos cerca de 10 nucleotídeos, um comprimento de pelo menos cerca de 15 nucleotídeos, ou um comprimento de pelo menos cerca de 20 nucleotídeos. Tais sondas podem ser usadas para amplificar os correspondents polinucleotídeos a partir de um organismo conhecido por meio de PCR. Esta técnica pode ser usada para isolar sequências codificantes adicionais a partir de um organismo desejado ou como um ensaio de diagnóstico para determinar a presença de sequências codificantes em um organismo. As técnicas de hibridização incluem screening de hibridização de bibliotecas de DNA plaqueado (tanto placas como colônias); ver, por exemplo, Sambrook et al. (1989) acima citado.
[0084] A hibridização de tais sequências pode ser realizada sob condições estringentes. Os termos “condições estringentes” ou “condições de hibridização estringentes” tem a intenção de significar condições sob as quais uma sonda hibridizará com sua sequência alvo com relação a um grau detectavelmente maior do que outras sequências (por exemplo, pelo menos duas vezes acima do background). As condições estringentes são sequência- dependentes e serão diferentes em diferentes circunstâncias. Pelo controle da estringência da hibridização e/ou das condições de lavagem, as sequências alvo que forem 100% complementares com relação à sonda podem ser identificadas (sondagem homóloga). Alternativamente, as condições de estringência podem ser ajustadas para permitir algum emparelhamento desigual em sequências de modo que graus mais baixos de similaridade sejam detectados (sondagem heteróloga). Em geral, uma sonda tem um comprimento menor do que cerca de 1000 nucleotídeos, de modo ótimo, um comprimento menor que 500 nucleotídeos.
[0085] Tipicamente, condições estringentes serão aquelas nas quais a concentração de sal é menor do que cerca de 1,5 M de íon Na, tipicamente concentração de cerca de 0,01 a 1,0 M de íon Na (ou outros sais) em pH 7,0 a 8,3 e a temperatura é de pelo menos cerca de 30°C para sondas curtas (por exemplo, 10 a 50 nucleotídeos) e pelo menos cerca de 60°C para sondas longas (por exemplo, maiores do que 50 nucleotídeos). Condições estringentes também podem ser alcançadas com a adição de agentes desestabilizantes, tal como formamida. Condições de baixa estringência exemplificativas incluem a hibridização com uma solução tampão de formamida a 30 a 35%, NaCl 1 M, SDS a 1% (dodecil sulfato de sódio) a 37°C, e uma lavagem em 1X a 2X SSC (20X SSC = NaCl 3,0 M /citrato trissódico 0,3 M) a uma temperatura de 50 a 55°C. Condições de moderada estringência exemplificativas incluem hibridização em formamida a 40 a 45%, NaCl 1,0 M, SDS a 1% a 37°C, e uma lavagem em 0,5X a 1X SSC a uma temperatura de 55 a 60°C. Condições de alta estringência exemplificativas incluem hibridização em formamida a 50%, NaCl 1 M, SDS 1% a a 37°C, e uma lavagem final em 0,1X SSC a umatemperatura de 60 a 65°C por pelo menos 30 minutos. De modo ótimo, os tampões de lavagem podem compreender cerca de 0,1% a cerca de 1% de SDS. A duração de hibridização é, em geral, menor do que cerca de 24 horas, usualmente de cerca de 4 a cerca de 12 horas. A duração do tempo de lavagem será pelo menos um período de tempo suficiente para alcançar o equilíbrio.
[0086] Especificidade é tipicamente uma função das lavagens pós-hibridização, sendo os fatores críticos a força iônica e a temperatura da solução de lavagem final. Para os híbridos de DNA-DNA, o ponto de fusão térmico (Tm) pode ser aproximado a partir da equação de Meinkoth e Wahl (1984) Anal. Biochem. 138:267-284: Tm = 81,5°C + 16,6 (log M) + 0,41 (%GC) — 0,61 (% form) - 500/L; onde M é a molaridade de cátions monovalentes, %GC é a percentagem de nucleotídeos guanosina e citosina no DNA, % form é a percentagem de formamida na solução de hibridização, e L é o comprimento do híbrido em pares de base. A Tm é a temperatura (sob força iônica e pH definidos) na qual 50% de uma sequência alvo complementar hibridiza com uma sonda perfeitamente emparelhada. A Tm é reduzida por cerca 1°C para cada 1% de emparelhamento desigual; assim, a Tm, a hibridização, e/ou as condições de lavagem podem ser ajustadas para hibridizar com sequências da identidade desejada. Por exemplo, se forem desejadas sequências com >90% de identidade, a Tm pode ser diminuída 10°C. Em geral, condições estringentes são selecionadas para serem cerca de 5°C mais baixas do que a Tm para a sequência específica e seu complemento em uma força iônica e pH definido. Entretanto, condições severamente estringentes podem utilizar uma hibridização e/ou lavagem a 1, 2, 3, ou 4°C mais baixa do que a Tm; condições moderadamente estringentes podem utilizar uma hibridização e/ou lavagem em 6, 7, 8, 9, ou 10°C mais baixa do que a Tm; condições de baixa estringência podem utilizar uma hibridização e/ou lavagem em 11, 12, 13, 14, 15, ou 20°C mais baixa do que a Tm. Usando a equação, as composições de hibridização e lavagem, e a Tm desejada, aqueles especialistas na técnica saberão reconhecer que as variações na estringência das soluções de hibridização e/ou lavagem estão inerentemente descritas. Se o grau desejado de emparelhamento desigual resultar em uma Tm menor que 45°C (solução aquosa) ou 32°C (solução de formamida), o modo ótimo será aumentar a concentração de SSC de forma que uma temperatura mais alta possa ser usada. Uma orientação detalhada para a hibridização de ácidos nucléicos é encontrada em Tijssen (1993) Laboratory Techniques in Biochemistry and Molecular Biology—Hybridization with Nucleic Acid Probes, Part I, Capítulo 2 (Elsevier, New York); e Ausubel et al., eds. (1995) Current Protocols in Molecular Biology, Capítulo 2 (Greene Publishing and Wiley-Interscience, New York). Ver Sambrook et al. (1989) acima citado.
[0087] Vários procedimentos podem ser usados para checar a presença ou ausência de uma sequência particular de DNA, RNA ou de uma proteína. Eles incluem, por exemplo, Southern blots, northern blots, western blots, e análise ELISA. Técnicas tais como essas são bastante conhecidas no estado da técnica e existem muitas referências que fornecem protocolos detalhados. Tais referências incluem Sambrook et al. (1989) acima citado, e Crowther, J.R. (2001), The ELISA Guidebook, Humana Press, Totowa, NJ, USA.
[0088] Os seguintes termos são usados para descrever as relações de sequência entre dois ou mais polinucleotídeos ou polipeptídeos: (a) “sequência de referência”, (b) “janela de comparação”, (c) “identidade de sequência” e (d) “percentagem de identidade de sequência”. (a) Como aqui usada, a “sequência de referência” é uma sequência definida usada como base para a comparação de sequências. Uma sequência de referência pode ser um subconjunto ou a integralidade de uma sequência especificada; por exemplo, como um segmento de um cDNA de comprimento completo ou sequência de gene, ou o cDNA completo ou sequência de gene. (b) Como aqui usada, a “janela de comparação” faz referência a um segmento contíguo e especificado de uma sequência de polinucleotídeo, sendo que a sequência de polinucleotídeo na janela de comparação pode compreender adições ou deleções (ou seja, gaps) comparada com a sequência de referência (a qual não compreende adições ou deleções) para o alinhamento ótimo de dois polinucleotídeos. Em geral, a janela de comparação tem comprimento de pelo menos cerca de 20 nucleotídeos contíguos, e opcionalmente, pode ter cerca de 30, cerca de 40, cerca de 50, cerca de 100, ou ser mais longa. Aqueles especialistas na técnica entendem que para evitar uma alta similaridade com uma sequência de referência devido à inclusão de gaps na sequência do polinucleotídeo, é tipicamente introduzida uma penalidade de gap e é subtraída do número de pareamentos.
[0089] Os métodos de alinhamento de sequências para comparação são bastante conhecidos no estado da técnica. Portanto, a determinação da identidade de sequência percentual entre quaisquer duas sequências pode ser realizada usando um algoritmo matemático. Exemplos não limitativos de tais algoritmos matemáticos são os algoritmos de Myers e Miller (1988) CABIOS 4:11-17; o algoritmo de alinhamento local de Smith et al. (1981) Adv. Appl. Math. 2:482; o algoritmo de alinhamento global de Needleman e Wunsch (1970) J. Mol. Biol. 48:443-453; o método de alinhamento de busca-por-local de Pearson e Lipman (1988) Proc. Natl. Acad. Sci. 85:2444-2448; o algoritmo de Karlin e Altschul (1990) Proc. Natl. Acad. Sci. USA 872264, modificado como em Karlin e Altschul (1993) Proc. Natl. Acad. Sci. USA 90:5873-5877.
[0090] As implementações em computador desses algoritmos matemáticos podem ser utilizadas para comparação de sequências para determinar a identidade de sequência. Tais implementações incluem, mas não estão limitadas a: CLUSTAL no programa PC/Gene (disponível a partir de Intelligenetics, Mountain View, California); o programa ALIGN (Versão 2.0) e GAP, BESTFIT, BLAST, FASTA, e TFASTA no GCG Wisconsin Genetics Software Package, Versão 10 (disponível a partir de Accelrys Inc., 9685 Scranton Road, San Diego, California, USA). Os alinhamentos usando esses programas podem ser realizados usando os parâmetros de default. O programa CLUSTAL está bem descrito por Higgins et al. (1988) Gene 73:237-244 (1988); Higgins et al. (1989) CABIOS 5:151-153; Corpet et al. (1988) Nucleic Acids Res. 16:10881-90; Huang et al. (1992) CABIOS 8:155-65; e Pearson et al. (1994) Meth. Mol. Biol. 24:307-331. O programa ALIGN é baseado no algoritmo de Myers e Miller (1988) acima citado. Podem ser usadas uma tabela de peso de resíduo PAM120, uma penalidade de comprimento de gap de 12, e uma penalidade de gap de 4 com o programa ALIGN para comparar sequências de aminoácidos. Os programas BLAST de Altschul et al (1990) J. Mol. Biol. 215:403 são baseados no algoritmo de Karlin e Altschul (1990) acima citado. As buscas de nucleotídeos BLAST podem ser realizadas com o programa BLASTN, escore = 100, comprimento de palavra = 12, para obter sequências de nucleotídeos homólogas a uma sequência de nucleotídeos codificante de uma proteína das concretizações. As buscas de proteína BLAST podem ser realizadas com o programa BLASTX, escore = 50, comprimento de palavra = 3, para obter sequências de aminoácidos homólogas a uma proteína ou polipeptídeo das concretizações. Para obter os alinhamentos com gap para finalidade de comparação, pode ser usado o Gapped BLAST (em BLAST 2.0) como descrito em Altschul et al. (1997) Nucleic Acids Res. 25:3389. Alternativamente, pode ser usado o PSI-BLAST (em BLAST 2.0) para realizar uma busca iterada que detecta relações distantes entre moléculas (ver Altschul et al. (1997) acima citado). Quando se utiliza o BLAST, Gapped BLAST, PSI-BLAST, podem ser usados os parâmetros de default dos programas respectivos (por exemplo, BLASTN para sequências de nucleotídeos, BLASTX para proteínas) (ver www.ncbi.nlm.nih.gov). O alinhamento também pode ser realizado manualmente por inspeção.
[0091] A menos que seja estabelecido diferentemente, os valores de identidade/similaridade de sequência aqui providos se referem ao valor obtido usando o GAP Versão 10 utilizando os seguintes parâmetros: % de identidade e % de similaridade para uma sequência de nucleotídeos usando o Peso de Gap de 50 e o Peso de Comprimento de 3, e a matriz de contagem de escore nwsgapdna.cmp; % de identidade e % de similaridade para uma sequência de aminoácidos usando o Peso de Gap de 8 e o Peso de Comprimento de 2, e a matriz de contagem de escore BLOSUM62; ou qualquer programa equivalente ao mesmo. O termo “programa equivalente” tem a intenção de significar qualquer programa de comparação de sequência que, para quaisquer duas sequências em questão, gera um alinhamento tendo nucleotídeo idêntico ou pareamentos de resíduos de aminoácido e uma idêntica identidade de sequência percentual quando comparado com o alinhamento correspondente gerado pelo GAP Versão 10.
[0092] O GAP usa o algoritmo de Needleman e Wunsch (1970) J. Mol. Biol. 48:443-453 para encontrar o alinhamento de duas sequências completas que maximize o número de pareamentos e minimize o número de gaps. O GAP considera todos os alinhamentos e posições de gap possíveis e cria o alinhamento com o maior número de bases emparelhadas e o menor número de gaps. Ele possibilita a provisão de uma penalidade de criação de gap e uma penalidade de extensão de gap em unidades de bases emparelhadas. O GAP precisa ter um ganho de número de pareamentos de penalidade de criação de gap para cada gap que ele insere. Se for escolhida uma penalidade de extensão de gap maior do que zero, o GAP precisa, adicionalmente, ter um ganho para cada gap inserido do comprimento do gap vezes a penalidade de extensão de gap. Os valores de penalidade de criação de gap de default e os valores de penalidade de extensão de gap na Versão 10 do GCG Wisconsin Genetics Software Package para sequências de proteína são 8 e 2, respectivamente. Para sequências de nucleotídeos a penalidade de criação de gap de default é 50 enquanto que a penalidade de extensão de gap de default é 3. As penalidades de criação de gap e de extensão de gap podem ser expressas como um número inteiro selecionado do grupo de números inteiros consistindo de 0 até 200. Portanto, por exemplo, as penalidades de criação de gap e de extensão de gap podem ser 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65 ou maiores.
[0093] O GAP apresenta um membro da família dos melhores alinhamentos. Pode haver muitos membros nesta família, e nenhum outro membro possui a melhor qualidade. O GAP apresenta quatro valores de mérito para alinhamentos: Qualidade, Razão, Identidade e Similaridade. A qualidade é o métrico maximizado de modo a alinhar as sequências. A Razão é a qualidade dividida pelo número de bases no segmento mais curto. A Identidade Percentual é a percentagem dos símbolos que realmente emparelham. A Similaridade Percentual é a percentagem dos símbolos que são similares. Os símbolos que atravessam a partir dos gaps são ignorados. É contado o escore de uma similaridade quando o valor da matriz de contagem de escore para um par de símbolos for maior que ou igual a 0,50, a similaridade limiar. A matriz de contagem de escore usada na Versão 10 do GCG Wisconsin Genetics Software Package é BLOSUM62 (ver Henikoff e Henikoff (1989) Proc. Natl. Acad. Sci. USA 89:10915). (c) Como aqui usada, a “identidade de sequência” ou “identidade” no contexto de duas sequências de polinucleotídeo ou polipeptídeo faz referência aos resíduos nas duas sequências que são as mesmas quando alinhadas para a máxima correspondência ao longo de uma janela de comparação especificada. Quando a percentagem de identidade de sequência é usada com referência a proteínas, é reconhecido que as posições de resíduo que não forem idênticas frequentemente diferem por substituições conservativas de aminoácidos, onde resíduos de aminoácido são substituídos por outros resíduos de aminoácidos com propriedades químicas semelhantes (por exemplo, carga ou hidrofobicidade) e, portanto, não mudam as propriedades funcionais da molécula. Quando as sequências diferem em substituições conservativas, a identidade de sequência percentual pode ser ajustada para cima para corrigir a natureza conservativa da substituição. Sequências que diferem por tais substituições conservativas são ditas possuírem “similaridade de sequência” ou “similaridade”. Os meios para fazer este ajuste são bastante conhecidos daqueles especialistas na técnica. Tipicamente isto envolve a contagem de escore de uma substituição conservativa como um emparelhamento desigual parcial ao invés de um emparelhamento desigual total, aumentando, por esse meio, a percentagem de identidade de sequência. Assim, por exemplo, quando a um aminoácido idêntico for dado um escore de 1 e a uma substituição não-conservativa for dado um escore de zero, a uma substituição conservativa é dado um escore entre zero e 1. A contagem de escore de substituições conservativas é calculada, por exemplo, como implementada no programa PC/GENE (Intelligenetics, Mountain View, California). (d) Como aqui usada, a “percentagem de identidade de sequência” significa o valor determinado por comparação de duas sequências alinhadas de modo ótimo ao longo de uma janela de comparação, sendo que a porção da sequência de polinucleotídeo na janela de comparação pode compreender adições ou deleções (ou seja, gaps) quando comparada com a sequência de referência (a qual não compreende adições ou deleções) para o alinhamento ótimo das duas sequências. A percentagem é calculada para determinar o número de posições nas quais ocorre a idêntica base de ácido nucléico ou o idêntico resíduo de aminoácido em ambas as sequências para produzir o número de posições pareadas, dividindo o número de posições pareadas pelo número total de posições na janela de comparação, e multiplicando o resultado por 100 para produzir a percentagem de identidade de sequência.
[0094] O uso do termo “polinucleotídeo” não tem a intenção de limitar as concretizações a polinucleotídeos compreendendo DNA. Aqueles especialistas na técnica reconhecerão que polinucleotídeos podem compreender ribonucleotídeos e combinações de ribonucleotídeos e desoxirribonucleotídeos. Tais desoxirribonucleotídeos e ribonucleotídeos incluem ambos, as moléculas de ocorrência natural e os análogos sintéticos. Os polinucleotídeos das concretizações também englobam todas as formas de sequências incluindo, mas não estando limitadas a, formas de simples-fita, formas de dupla-fita e semelhantes.
[0095] Os polinucleotídeos isolados das concretizações podem ser incorporados em constructos de DNA recombinante capaz de introdução e replicação em uma célula hospedeira. Um “vetor” pode ser um constructo tal que inclui um sistema de replicação e sequências que são capazes de transcrição e tradução de uma sequência codificante de polipeptídeo em uma dada célula hospedeira. Inúmeros vetores apropriados para a transfecção estável de plantas transgênicas têm sido descritos, por exemplo, Pouwels et al., Cloning Vectors: A Laboratory Manual, 1985, supp. 1987; Weissbach e Weissbach, Methods for Plant Molecular Biology, Academic Press, 1989; e Flevin et al., Plant Molecular Biology Manual, Kluwer Academic Publishers, 1990. Tipicamente, os vetores de expressão em planta incluem, por exemplo, um ou mais genes de planta clonados sob o controle de transcrição de sequências de regulação 5' e 3' e um marcador selecionável dominante. Tais vetores de expressão em planta também podem conter uma região de regulação de promotor (por exemplo, uma expressão de região de regulação de controle induzível ou constitutivo, regulada por condições ambientais ou de desenvolvimento, ou célula- ou tecido- específica), um sítio de partida de iniciação de transcrição, um sítio de ligação a ribossomo, um sinal de processamento de RNA, um sítio de terminação de transcrição, e/ou um sinal de poliadenilação.
[0096] Os termos “constructo recombinante”, “cassete de expressão”, “constructo de expressão”, “constructo quimérico”, “constructo”, “constructo de DNA recombinante” e “fragmento de DNA recombinante” são aqui usados indiferentemente e são fragmentos de ácido nucléico. Um constructo recombinante compreende uma combinação artificial de fragmentos de ácido nucléico, incluindo, mas não estando limitado a, sequências de regulação e codificantes que não são encontradas juntas na natureza. Por exemplo, um constructo de DNA recombinante pode compreender sequências de regulação e sequências codificantes que são derivadas de diferentes fontes e arrumadas em uma maneira diferente daquela encontrada na natureza. Tais constructos podem ser usados por si mesmos ou podem ser usados em conjugação com um vetor. Se for usado um vetor, então a escolha do vetor é dependente do método que será usado para transformar as células hospedeiras, como é bastante conhecido dos especialistas na técnica. Por exemplo, pode ser usado um vetor plasmidial. Os especialistas na técnica estão bem familiarizados com os elementos genéticos que devem estar presentes no vetor de de modo a transformar com sucesso, selecionar e propagar células hospedeiras que compreendem quaisquer dos fragmentos de ácido nucléico isolado das concretizações. O screening para obter as linhagens que apresentam o nível e padrão de expressão desejado dos polinucleotídeos ou do lócus de Rcg1 pode ser realizado por amplificação, análise Southern de DNA, análise northern da expressão de mRNA, análise immunoblotting de expressão de proteína, análise fenotípica, e semelhantes.
[0097] O termo “constructo de DNA recombinante” se refere a um constructo de DNA montado a partir de fragmentos de ácido nucléico obtido de diferentes fontes. Os tipos e origens dos fragmentos de ácido nucléico podem ser muito diversos.
[0098] Em algumas concretizações, são adicionalmente providos cassetes de expressão compreendendo um promotor operacionalmente ligado a uma sequência de nucleotídeos heteróloga das concretizações. Os cassetes de expressão das concretizações encontram uso na geração de plantas transformadas, células de planta e microrganismos e na prática de métodos para induzir resistência da planta a patógeno de fungo aqui revelada. O cassete de expressão incluirá sequências de regulação 5' e 3' operacionalmente ligadas a um polinucleotídeo das concretizações. O termo “operacionalmente ligado” tem a intenção de significar uma ligação funcional entre dois ou mais elementos. As “sequências de regulação” se referem a nucleotídeos localizados a montante (sequências não- codificantes 5'), dentro ou a jusante (sequências não- codificantes 3') de uma sequência codificante, e que podem influenciar a transcrição, o processamento de RNA, a estabilidade, a tradução da sequência codificante associada. As sequências de regulação podem incluir, mas não estão limitadas a, promotores, sequências líder de tradução, introns e sequências de reconhecimento de poliadenilação. Por exemplo, uma ligação operável entre um polinucleotídeo de interesse e uma sequência de regulação (um promotor, por exemplo) é uma ligação funcional que permite a expressão do polinucleotídeo de interesse. Os elementos operacionalmente ligados podem ser contínuos ou não-contínuos. Quando usado para se referir a uma união de duas regiões codificantes de proteína, o termo operacionalmente ligado tem a intenção de significar que as regiões codificantes estão no mesmo quadro de leitura. O cassete pode ainda conter pelo menos um gene adicional para ser co-transformado dentro do organismo. Alternativamente, o(s) gene(s) adicional(ais) pode(m) ser provido(s) em cassetes múltiplos de expressão. Tal cassete de expressão é provido com uma pluralidade de sítios de restrição e/ou sítios de recombinação para inserção do polinucleotídeo que codifica um polipeptídeo antipatogênico para ficar sob a regulação de transcrição das regições de regulação. O cassete de expressão pode adicionalmente conter genes marcadores selecionáveis.
[0099] O cassete de expressão incluirá, na direção de transcrição 5'-3', uma região de iniciação de transcrição (ou seja, um promotor), região de iniciação de tradução, um polinucleotídeo das concretizações, uma região de terminação da tradução e, opcionalmente, uma região de terminação de transcrição funcional no organismo hospedeiro. As regiões de regulação (ou seja, promotores, regiões de regulação de transcrição e regiões de terminação de tradução) e/ou o polinucleotídeo das concretizações podem ser nativos/análogos com relação à célula hospedeira ou de uma em relação à outra. Alternativamente, as regiões de regulação e/ou os polinucleotídeos das concretizações podem ser heteterólogas com relação à célula hospedeira ou de uma em relação à outra. Como aqui usado, o termo “heterólogo”, com referência a uma sequência é uma sequência que se origina de espécies estrangeiras, ou, se a partir da mesma espécie, está substancialmente modificada com relação à sua forma natural em composição e/ou lócus genômico por deliberada intervenção humana. Por exemplo, um promotor operacionalmente ligado a um polinucleotídeo heterólogo é oriundo de uma espécie diferente da espécie a partir da qual o polinucleotídeo foi derivado, ou se da mesma/análoga espécie, um ou ambos estão substancialmente modificados com relação à sua forma original e/ou lócus genômico, ou o promotor não é o promotor natural para o polinucleotídeo operacionalmente ligado.
[0100] A região de terminação opcionalmente incluída pode ser natural com relação à região de iniciação de transcrição, pode ser natural com relação ao polinucleotídeo de interesse operacionalmente ligado, pode ser natural com relação à planta hospedeira, ou pode ser derivada de uma outra fonte (ou seja, estrangeira ou heteróloga) com relação ao promotor, ao polinucleotídeo de interesse, ao hospedeiro, ou a qualquer combinação dos mesmos. Estão disponíveis regiões de terminação convenientes a partir do plasmídio-Ti de A. tumefaciens, tais como as regiões de terminação da octopina sintase e nopalina sintase. Ver também Guerineau et al. (1991) Mol. Gen. Genet. 262:141-144; Proudfoot (1991) Cell 64:671-674; Sanfacon et al. (1991) Genes Dev. 5:141-149; Mogen et al. (1990) Plant Cell 2:1261-1272; Munroe et al. (1990) Gene 91:151-158; Ballas et al. (1989) Nucleic Acids Res. 17:7891-7903; e Joshi et al. (1987) Nucleic Acids Res. 15:9627-9639. Em concretizações particulares, é usado o terminador do gene II do inibidor de protease de batata (PinII). Ver, por exemplo, Keil et al. (1986) Nucl. Acids Res. 14:5641-5650; e An et al. (1989) Plant Cell 1:115-122, aqui incorporados a título de referências em sua integralidade.
[0101] Inúmeros promotores podem ser usados na prática das concretizações, incluindo o promotor natural da sequência de polinucleotídeo de interesse. Os promotores podem ser selecionados com base no resultado desejado. Uma ampla faixa de promotores de planta é discutida na recente revisão de Potenza et al. (2004) In Vitro Cell Dev Biol - Plant 40:1-22, aqui incorporada a título de referência. Por exemplo, os ácidos nucléicos podem ser combinados com promotores constitutivos, tecido-preferidos, patógeno-induzíveis ou outros promotores para expressão em plantas. Tais promotores constitutivos incluem, por exemplo, o promotor de núcleo do promotor Rsyn7 e outros promotores constitutivos descritos no WO 99/43838 e patente US 6.072.050; o promotor de núcleo de 35S de CaMV (Odell et al. (1985) Nature 313:810-812); actina de arroz (McElroy et al. (1990) Plant Cell 2:163-171); ubiquitina (Christensen et al. (1989) Plant Mol. Biol. 12:619632 e Christensen et al. (1992) Plant Mol. Biol. 18:675-689); pEMU (Last et al. (1991) Theor. Appl. Genet. 81:581-588); MAS (Velten et al. (1984) EMBO J. 3:2723-2730); promotor da ALS (US 5.659.026), e semelhantes. Outros promotores constitutivos incluem, por exemplo, os descritos nas patentes US 5.608.149; US 5.608.144; US 5.604.121; US 5.569.597; US 5.466.785; US 5.399.680; US 5.268.463; US 5.608.142; e US 6.177.611.
[0102] Algumas vezes, pode trazer benefício a expressão do gene a partir de um promotor induzível, particularmente a partir de um promotor patógeno-induzível. Tais promotores incluem aqueles oriundos de proteínas patogênese-relacionadas (proteínas PR), as quais são induzidas a seguir à infecção por um patógeno, por exemplo, proteínas PR, proteínas SAR, beta- 1,3-glucanase, chitinase, etc. Ver, por exemplo, Redolfi et al. (1983) Neth. J. Plant Pathol. 89:245-254; Uknes et al. (1992) Plant Cell 4:645-656; e Van Loon (1985) Plant Mol. Virol. 4:111-116. Ver também WO 99/43819, aqui incorporado a título de referência.
[0103] São de interesse os promotores que resultam em expressão de uma proteína localmente em ou perto do sítio de infecção pelo patógeno. Ver, por exemplo, Marineau et al. (1987) Plant Mol. Biol. 9:335-342; Matton et al. (1989) Molecular Plant-Microbe Interactions 2:325-331; Somsisch et al. (1986) Proc. Natl. Acad. Sci. USA 83:2427-2430; Somsisch et al. (1988) Mol. Gen. Genet. 2:93-98; e Yang (1996) Proc. Natl. Acad. Sci. USA 93:14972-14977. Ver também, Chen et al. (1996) Plant J. 10:955-966; Zhang et al. (1994) Proc. Natl. Acad. Sci. USA 91:2507-2511; Warner et al. (1993) Plant J. 3:191-201; Siebertz et al. (1989) Plant Cell 1:961-968; patente US 5.750.386 (nematódeo-induzível); e as referências aqui citadas. De particular interesse é o promotor induzível para o gene PRms de milho, cuja expressão é induzida pelo patógeno Fusarium moniliforme (ver, por exemplo, Cordero et al. (1992) Physiol. Mol. Plant Path. 41:189-200).
[0104] Adicionalmente, como os patógenos entram nas plantas através de feridas ou danos provocados por insetos, pode ser usado um promotor ferida-induzível nas construções das concretizações. Tais promotores ferida-induzíveis incluem gene de inibidor de proteinase de batata (pin II) (Ryan (1990) Ann. Rev. Phytopath. 28:425-449; Duan et al. (1996) Nature Biotechnology 14:494-498); wun1 e wun2, patente US 5.428.148; win1 e win2 (Stanford et al. (1989) Mol. Gen. Genet. 215:200 208); sistemina (McGurl et al. (1992) Science 225:1570-1573); WIP1 (Rohmeier et al. (1993) Plant Mol. Biol. 22:783-792; Eckelkamp et al. (1993) FEBS Letters 323:73-76); gene MPI (Corderok et al. (1994) Plant J. 6(2):141-150); e semelhantes, aqui incorporados a título de referência.
[0105] Os promotores químico-regulados podem ser usados para modular a expressão de um gene em uma planta através da aplicação de um regulador químico exógeno. Dependendo do objetivo, o promotor pode ser um promotor químico-induzível, quando a aplicação da substância química induz a expressão gênica, ou um promotor químico-suprimível, quando a aplicação da substância química suprime a expressão gênica. Os promotores químico-induzíveis são conhecidos no estado da técnica e incluem, mas não estão limitados a, promotor In2-2 de milho, que é ativado por antídotos de herbicida de benzeno- sulfonamida, promotor GST de milho, que é ativado por compostos eletrofílicos hidrofóbicos que são usados como herbicidas pré-emergentes, e promotor PR-1a de tabaco, que é ativado por ácido salicílico. Outros promotores químico- regulados de interesse incluem promotores esteróide- responsivos (ver, por exemplo, o pomotor glucocorticóide- induzível em Schena et al. (1991) Proc. Natl. Acad. Sci. USA 88:10421-10425 e McNellis et al. (1998) Plant J. 14(2):247- 257) e promotores tetraciclina-induzível e tetraciclina- suprimível (ver, por exemplo, Gatz et al. (1991) Mol. Gen. Genet. 227:229-237, e patentes US 5.814.618 e US 5.789.156), aqui incorporados a título de referência.
[0106] Os promotores tecido-preferidos podem ser utilizados para direcionar a expressão melhorada dos polipeptídeos das concretizações dentro de um tecido de planta particular. Por exemplo, um promotor tecido-preferido pode ser usado para expressar um polipepetídeo em um tecido de planta onde a resistência à doença é particularmente importante, tais como, por exemplo, as raízes, colmo ou as folhas. Os promotores tecido-preferidos incluem Yamamoto et al. (1997) Plant J. 12(2):255-265; Kawamata et al. (1997) Plant Cell Physiol. 38(7):792-803; Hansen et al. (1997) Mol. Gen Genet. 254(3):337-343; Russell et al. (1997) Transgenic Res. 6(2):157-168; Rinehart et al. (1996) Plant Physiol. 112(3):1331-1341; Van Camp et al. (1996) Plant Physiol. 112(2):525-535; Canevascini et al. (1996) Plant Physiol. 112(2):513-524; Yamamoto et al. (1994) Plant Cell Physiol. 35(5):773-778; Lam (1994) Results Probl. Cell Differ. 20:181196; Orozco et al. (1993) Plant Mol Biol. 23(6):1129-1138; Matsuoka et al. (1993) Proc Natl. Acad. Sci. USA 90(20):9586- 9590; e Guevara-Garcia et al. (1993) Plant J. 4(3):495-505. Tais promotores podem ser modificados, se necessário, para expressão fraca.
[0107] Os promotores tecido vascular-preferidos são conhecidos no estado da técnica e incluem aqueles promotores que seletivamente dirigem a expressão em, por exemplo, tecido de xilema e tecido de floema. Os promotores tecido vascular- preferidos incluem, mas não estão limitados a, promotor do gene de prunasin hidrolase de Prunus serotina (ver, por exemplo, Pedido de Patente Internacional publicado WO 03/006651), e também aqueles encontrados no Pedido de Patente de No. de série 10/109.488.
[0108] Os promotores colmo-preferidos podem ser usados para dirigir a expressão de um polipeptídeo das concretizações. Promotores colmo-preferidos exemplificativos incluem o promotor do gene MS8-15 de milho (ver, por exemplo, patente US 5.986.174 e Pedido de Patente Internacional publicado WO 98/00533), e aqueles encontrados em Graham et al. (1997) Plant Mol Biol 33(4): 729-735.
[0109] Os promotores folha-preferidos são conhecidos no estado da técnica. Ver, por exemplo, Yamamoto et al. (1997) Plant J. 12(2):255-265; Kwon et al. (1994) Plant Physiol. 105:357-67; Yamamoto et al. (1994) Plant Cell Physiol. 35(5):773-778; Gotor et al. (1993) Plant J. 3:509-18; Orozco et al. (1993) Plant Mol. Biol. 23(6):1129-1138; e Matsuoka et al. (1993) Proc. Natl. Acad. Sci. USA 90(20):9586-9590.
[0110] Os promotores raiz-preferidos são conhecidos no estado da técnica e podem ser selecionados dos muito disponíveis a partir da literatura ou isolados pela primeira vez a partir de várias espécies compatíveis. Ver, por exemplo, Hire et al. (1992) Plant Mol. Biol. 20(2):207-218 (gene de glutamina sintetase raiz-específico de soja); Keller e Baumgartner (1991) Plant Cell 3(10):1051-1061 (elemento de controle raiz-específico no gene GRP 1.8 de feijão francês); Sanger et al. (1990) Plant Mol. Biol. 14(3):433-443 (promotor raiz-específico do gene da manopina sintase (MAS) de Agrobacterium tumefaciens); e Miao et al. (1991) Plant Cell 3(1):11-22 (clone de cDNA de comprimento completo codificante de glutamina sintetase (GS) citosólica, que é expressado em raízes e nódulos de raiz de soja). Ver também Bogusz et al. (1990) Plant Cell 2(7):633-641, onde são descritos dois promotores raiz-específicos isolados de genes de hemoglobina oriundos do não-legume fixador de nitrogênio Parasponia andersonii e o não-legume não-fixador de nitrogênio relacionado Trema tomentosa. Os promotores desses genes foram ligados a um gene repórter de β-glucuronidase e introduzidos em ambos, o não-legume Nicotiana tabacum e o legume Lotus corniculatus, e em ambas as circunstâncias, a atividade de promotor raiz-específico ficou preservada. Leach e Aoyagi (1991) descrevem sua análise dos promotores dos genes raiz- induzíveis rolC e rolD altamente expressados de Agrobacterium rhizogenes (ver Plant Science (Limerick) 79(1):69-76). Eles concluíram que o enhancer e os determinantes do DNA tecido- preferido estão dissociados naqueles promotores. Teeri et al. (1989) usaram a fusão de gene ao lacZ para mostrar que o gene de T-DNA de Agrobacterium codificante da octopina sintase é especialmente ativo na epiderme da ponta da raiz e que o gene TR2' é específico para raiz na planta intacta e é estimulado por meio de ferimento em tecido de folha, uma combinação especialmente desejável de características para uso com um gene inseticida ou larvicida (ver EMBO J. 8(2):343-350). O gene TR1', fundido ao nptII (neomicina fosfotransferase II) mostrou semelhantes características. Promotores raiz- preferidos adicionais incluem o promotor do gene VfENOD-GRP3 (Kuster et al. (1995) Plant Mol. Biol. 29(4):759-772); e o promoter de rolB (Capana et al. (1994) Plant Mol. Biol. 25(4):681-691. Ver também patentes US 5.837.876; US 5.750.386; US 5.633.363; US 5.459.252; US 5.401.836; US 5.110.732; e US 5.023.179.
[0111] Os promotores semente-preferidos incluem ambos, os promotores “semente-específicos” (aqueles promotores ativos durante o desenvolvimento de semente, tais como os promotores de proteínas de reserva de semente), bem como os promotores “semente-germinadores” (aqueles promotores ativos durante a germinação da semente). Ver Thompson et al. (1989) BioEssays 10:108, aqui incorporado a título de referência. Tais promotores semente-preferidos incluem, e não estão limitados a, Cim1 (mensagem citocinina-induzida); cZ19B1 (zeína de 19 kDa de milho); milps (mio-inositol-1-fosfato sintase) (ver WO 00/11177 e patente US 6.225.529; aqui incorporadas a título de referência). O gamma-zeína é um promotor endosperma-específico preferido. O glob-1 é um promotor embrião-específico preferido. Para dicotiledôneas, os promotores semente- específicos incluem, e não estão limitados a, β-faseolin de feijão, napin, β-conglicinin, lectina de soja, cruciferina, e semelhantes. Para as monocotiledôneas, os promotores semente- específicos incluem, e não estão linmitados a, zeína de 15 kDa de milho, zeína de 22 kDa, zeína de 27 kDa, g-zeína, waxy, shrunken 1, shrunken 2, globulin 1, etc. Ver também WO 00/12733, onde são descritos promotores semente-preferidos oriundos dos genes end1 e end2; aqui incorporados a título de referência.
[0112] São conhecidas modificações adicionais de sequência para melhorar a expressão gênica em um hospedeiro celular. Elas incluem a eliminação de sequências codificantes de sinais espúrios de poliadenilação, sinais de sítio de processamento exon-intron, repetições semelhantes a transposon e outras tais sequências bem caracterizadas que podem ser prejudiciais à expressão gênica. O teor de G-C da sequência pode ser ajustado a níveis médios para um dado hospedeiro celular, como calculado com referência aos genes conhecidos expressados na célula hospedeira. Quando possível, a sequência é modificada para evitar estruturas hairpin de mRNA secundário.
[0113] Os cassetes de expressão podem adicionalmente conter sequências líder 5'. Tais sequências líder podem atuar para melhorar a tradução. Os líderes de tradução são conhecidos do estado da técnica e incluem: líderes de picornavirus, por exemplo, líder de EMCV (região não-codificante 5' de Encefalomiocardite) (Elroy-Stein et al. (1989) Proc. Natl. Acad. Sci. USA 8 6:612 6-6130); líderes de potyvirus, por exemplo, líder de TEV (Tobacco Etch Virus) (Gallie et al. (1995) Gene 165(2):233-238), líder MDMV (Maize Dwarf Mosaic Virus), e proteína de ligação de cadeia-longa de imunoglobulina humana (BiP) (Macejak et al. (1991) Nature 353:90-94); líder não-traduzido oriundo do mRNA de proteína de capa do vírus de mosaico de alfafa (AMV RNA 4) (Jobling et al. (1987) Nature 325:622-625); líder de vírus de mosaico de tabaco (TMV) (Gallie et al. (1989) in Molecular Biology of RNA, ed. Cech (Liss, New York), pp. 237-256); e líder de vírus mosqueado clorótico de milho (MCMV) (Lommel et al. (1991) Virology 81:382-385). Ver também, Della-Cioppa et al. (1987) Plant Physiol. 84:965-968. Também podem ser utilizados outros métodos conhecidos de melhorar a tradução, por exemplo, introns, e semelhantes.
[0114] No preparo do cassete de expressão, os vários fragmentos de DNA podem ser manipulados de modo a prover as sequências de DNA na orientação própria e, quando apropriado, no quadro de leitura próprio. Para esta finalidade, podem ser empregados adaptadores e linkers para unir os fragmentos de DNA ou podem estar envolvidas outras manipulações para prover os sítios de restrição convenientes, a remoção do DNA supérfluo, a remoção dos sítios de restrição, ou semelhantes. Para este propósito podem estar envolvidos, mutagênese in vitro, reparo de primer, restrição, anelamento, re- substituições, por exemplo, transições e transversões.
[0115] O cassete de expressão também pode compreender um gene de marcador selecionável para a seleção de células transformadas. Os genes de marcador selecionável são utilizados para a seleção de células ou tecidos transformados. Os genes de marcador incluem genes codificantes de resistência a antibiótico, tais como aqueles codificantes de neomicina fosfotransferase II (NEO) e higromicina fosfotransferase (HPT), assim como os genes que conferem resistência a compostos herbicidas, tais como glufosinato de amônio, bromoxinil, imidazolinonas, e 2,4-diclorofenoxiacetato (2,4-D). Os marcadores selecionáveis adicionais incluem marcadores fenotípicos tais como β-galactosidase e proteínas fluorescentes tais como proteína verde fluorescente (GFP) (Su et al. (2004) Biotechnol Bioeng 85:610-9 e Fetter et al. (2004) Plant Cell 16:215-28), proteína ciano fluorescente (CYP) (Bolte et al. (2004) J. Cell Science 117:943-54 e Kato et al. (2002) Plant Physiol 129:913-42), e proteína amarelo fluorescente (PhiYFPa a partir de Evrogen, ver, Bolte et al. (2004) J. Cell Science 117:943-54). Para marcadores selecionáveis adicionais, ver, em geral, Yarranton (1992) Curr. Opin. Biotech. 3:506-511; Christopherson et al. (1992) Proc. Natl. Acad. Sci. USA 89:6314-6318; Yao et al. (1992) Cell 71:6372; Reznikoff (1992) Mol. Microbiol. 6:2419-2422; Barkley et al. (1980) em The Operon, pp. 177-220; Hu et al. (1987) Cell 48:555566; Brown et al. (1987) Cell 49:603-612; Figge et al. (1988) Cell 52:713-722; Deuschle et al. (1989) Proc. Natl. Acad. Aci. USA 86:5400-5404; Fuerst et al. (1989) Proc. Natl. Acad. Sci. USA 86:2549-2553; Deuschle et al. (1990) Science 248:480-483; Gossen (1993) Tese de Ph.D., Universidade de Heidelberg; Reines et al. (1993) Proc. Natl. Acad. Sci. USA 90:1917-1921; Labow et al. (1990) Mol. Cell. Biol. 10:3343-3356; Zambretti et al. (1992) Proc. Natl. Acad. Sci. USA 89:3952-3956; Baim et al. (1991) Proc. Natl. Acad. Sci. USA 88:5072-5076; Wyborski et al. (1991) Nucleic Acids Res. 19:4647-4653; Hillenand-Wissman (1989) Topics Mol. Struc. Biol. 10:143-162; Degenkolb et al. (1991) Antimicrob. Agents Chemother. 35:1591-1595; Kleinschnidt et al. (1988) Biochemistry 27:1094-1104; Bonin (1993) Tese de Ph.D., Universidade de Heidelberg; Gossen et al. (1992) Proc. Natl. Acad. Sci. USA 89:5547-5551; Oliva et al. (1992) Antimicrob. Agents Chemother. 36:913-919; Hlavka et al. (1985) Handbook of Experimental Pharmacology, Vol. 78 (Springer-Verlag, Berlin); Gill et al. (1988) Nature 334:721-724. Tais descrições são aqui incorporadas a título de referência.
[0116] A lista acima de genes de marcador selecionável não significa ser limitativa. Qualquer gene de marcador selecionável pode ser usado nas concretizações.
[0117] O gene das concretizações pode ser expressado como um transgene de modo a produzir plantas resistentes a Cg. Usando os diferentes promotores descritos em algum lugar nesta descrição, será possibilitada a sua expressão em uma forma modulada em diferentes circunstâncias. Por exemplo, podem ser desejados níveis mais altos de expressão em colmos para melhorar a resistência à podridão do colmo causada por Cg. Em ambientes onde a queima das folhas causada por Cg for mais do que um problema, as linhagens com níveis mais altos de expressão em folhas podem ser usadas. Entretanto, também se pode inserir o gene inteiro, tendo ambos, o promotor e a sequência codificante, naturais, como um transgene. Finalmente, o uso do gene das concretizações como um transgene possibilitará a rápida combinação com outros caracteres, tal como a resistência a inseto ou a herbicida.
[0118] Em certas concretizações, as sequências de ácido nucléico das concretizações pode ser montada com qualquer combinação de sequências de polinucleotídeo de interesse de forma a criar plantas com um desejado fenótipo. Esta montagem pode ser realizada por uma combinação de genes dentro do constructo de DNA, ou por meio de cruzamento de Rcg1 com uma outra linhagem que compreende a combinação. Por exemplo, os polinucleotídeos das concretizações podem ser montados com quaisquer outros polinucleotídeos das concretizações ou com outros genes. As combinações geradas também podem incluir cópias múltiplas de qualquer um dos polinucleotídeos de interesse. Os polinucleotídeos das concretizações também podem ser montados com qualquer outro gene ou combinação de genes para produzir plantas com uma variedade de combinações de caracteres desejados, incluindo, mas não estando limitados a, caracteres desejáveis para alimentação animal, tal como genes de alto teor de óleo (por exemplo, patente US 6.232.529); aminoácidos equilibrados (por exemplo hordotioninas (patentes US 5.990.389; US 5.885.801; US 5.885.802; e US 5.703.409); cevada de alto teor de lisina (Williamson et al. (1987) Eur. J. Biochem. 165:99-106; e WO 98/20122); e proteínas de alto teor de metionina (Pedersen et al. (1986) J. Biol. Chem. 261:6279; Kirihara et al. (1988) Gene 71:359; e Musumura et al. (1989) Plant Mol. Biol. 12: 123)); aumentada digestibilidade (por exemplo, proteínas de reserva modificadas (Pedido de Patente US de No. de Série 10/053.410, depositado em 7 de novembro de 2001); e tioredoxinas (Pedido de Patente US de No. de Série 10/005.429, depositado em 3 de Dezembro de 2001)), cujas descrições são aqui incorporadas a título de referência. Os polinucleotídeos das concretizações também podem ser montados com os caracteres desejáveis para resistência a insetos, a doenças ou a herbicidas (por exemplo, proteínas tóxicas de Bacillus thuringiensis (patentes US 5.366.892; US 5.747.450; US 5.737.514; US 5.723.756; US 5.593.881; Geiser et al (1986) Gene 48:109); lectinas (Van Damme et al. (1994) Plant Mol. Biol. 24:825); genes de desintoxicação de fumonisina (patente US 5.792.931); genes de avirulêcia e de resistência a doenças (Jones et al. (1994) Science 266:789; Martin et al. (1993) Science 262:1432; Mindrinos et al. (1994) Cell 78:1089); mutantes de acetolactato sintase (ALS) que conduzem a resistência a herbicida, tal como as mutações S4 e/ou Hra; inibidores de glutamina sintase, tais como fosfinotricina ou basta (por exemplo, gene bar); e resistência a glifosato (genes EPSPS, genes GAT tais como aqueles descritos no Pedido de Patente publicado US US2004/0082770, também o WO02/36782 e o WO03/092360)); e caracteres desejáveis para processamento ou produtos de processo tal como alto teor em óleo (por exemplo, patente US 6.232.529 ); óleos modificados (por exemplo, genes de ácido graxo desnaturase (patente US 5.952.544; WO 94/11516)); amidos modificados (por exemplo, ADPG pirofosforilases (AGPase), amido sintases (SS), enzimas de ramificação de amido (SBE) e enzimas de des-ramificação de amido (SDBE)); e polímeros ou bioplásticos (por exemplo, patente US 5.602.321; beta-cetotiolase, poliidroxibutirato sintase, e acetoacetil-CoA redutase (Schubert et al. (1988) J. Bacteriol. 170:5837-5847) facilitam a expressão de poliidroxialcanoatos (PHAs)), cujas descrições são aqui incorporadas a título de referência. Também pode-se combinar os polinucleotídeos das concretizações com polinucleotídeos que forneçam caracteres agronômicos, tais como esterilidade masculina (por exemplo, ver patente US 5.583.210), resistência de colmo, tempo de florescência, ou caracteres de tecnologia de transformação tais como regulação do ciclo da célula ou direcionamento do gene (por exemplo, WO 99/61619; WO 00/17364; WO 99/25821), cujas descrições são aqui incorporadas a título de referência.
[0119] Essas combinações montadas podem ser criadas por qualquer método, incluindo, mas não estando limitado a, melhoramento por cruzamento de plantas por qualquer metodologia convencional ou por TopCross®, ou transformação genética. Se os caracteres forem montados por transformação genética de plantas, as sequências de polinucleotídeo de interesse podem ser combinadas em qualquer momento e em qualquer ordem. Por exemplo, uma planta transgênica compreendendo um ou mais caracteres desejados pode ser usada como o alvo para introduzir caracteres adicionais por subsequente transformação. Os caracteres podem ser introduzidos simultaneamente em um protocolo de co- transformação com os polinucleotídeos de interesse providos por qualquer combinação de cassetes de transformação. Por exemplo, se duas sequências forem introduzidas, as duas sequências podem estar contidas em cassetes de transformação separados (trans) ou contidos no mesmo cassete de transformação (cis). A expressão das sequências pode ser dirigida pelo mesmo promotor ou por promotores diferentes. Em certos casos, pode ser desejável introduzir um cassete de transformação que reprimirá a expressão do polinucleotídeo de interesse. Isto pode ser aliado a qualquer combinação de outros cassetes de supressão ou cassetes de super-expressão para gerar a combinação desejada de caracteres na planta.
[0120] Os métodos das concretizações podem envolver, e não ficarem limitados a, introdução de um polipeptídeo ou polinucleotídeo em uma planta. O termo “introdução” tem a intenção de significar a apresentação de um polinucleotídeo a uma planta. Em algumas concretizações, o polinucleotídeo será apresentado de tal maneira que a sequência ganha acesso ao interior da célula da planta, incluindo sua inserção potencial no genoma de uma planta. Os métodos das concretizações não dependem de um método particular para introduzir uma sequência em uma planta, dependem somente do fato de que o polinucleotídeo tenha condições de ganhar acesso ao interior de pelo menos uma célula da planta. Os métodos para introduzir polinucleotídeos no interior de plantas são conhecidos no estado da técnica, incluindo, mas não estado limitados a, métodos de transformação estável, métodos de transformação transiente e métodos mediados por vírus.
[0121] O termo “transformação” se refere à transferência de um fragmento de ácido nucléico para dentro do genoma de um organismo hospedeiro, resultando em herança geneticamente estável. Os organismos hospedeiros contendo os fragmentos de ácido nucléico transformado são referidos como organismos “transgênicos”. O termo “célula hospedeira” se refere à célula no interior da qual a transformação do constructo de DNA recombinante acontece e pode incluir uma célula de levedura, uma célula bacteriana e uma célula de planta. Exemplos de métodos de transformação incluem a tranformação Agrobacterium- mediada (De Blaere et al., 1987, Meth. Enzymol. 143:277) e a transformação por partícula acelerada ou tecnologia de transformação “arma de gene” (Klein et al., 1987, Nature (London) 327:70~73; patente US 4.945.050), entre outras.
[0122] O termo “transformação estável” tem a intenção de significar o fato de que o constructo de nucleotídeos introduzido em uma planta integra o genoma da planta e é capaz de ser herdado pela progênie da mesma. Os termos “transformação transiente” ou “expressão transiente” tem a intenção de significar o fato de que o polinucleotídeo é introduzido na planta e não se integra no genoma da planta ou que um polipeptídeo é introduzido em uma planta.
[0123] Os protocolos de transformação, assim como os protocolos de introdução de polipeptídeos ou de sequências de polinucleotídeos em plantas pode variar dependendo do tipo de planta ou célula de planta, ou seja, monocotiledônea ou dicotiledônea, que é o alvo da transformação. Métodos apropriados para introduzir polipeptídeos e polinucleotídeos em células de planta incluem a microinjeção (Crossway et al. (1986) Biotechniques 4:320-334), a eletroporação (Riggs et al. (1986) Proc. Natl. Acad. Sci. USA 83:5602-5606, a transformação Agrobacterium-mediada (patentes US 5.563.055 e US 5.981.840), transferência direta de gene (Paszkowski et al. (1984) EMBO J. 3:2717-2722), e aceleração balística de partícula (ver, por exemplo, Sanford et al., patentes US 4.945.050; US 5.879.918; US 5.886.244; e US 5.932.782; Tomes et al. (1995) in Plant Cell, Tissue, and Organ Culture: Fundamental Methods, ed. Gamborg and Phillips (Springer-Verlag, Berlin); McCabe et al. (1988) Biotechnology 6:923-926); e transformação de Lec1 (WO 00/28058). Ver também Weissinger et al. (1988) Ann. Rev. Genet. 22:421-477; Sanford et al. (1987) Particulate Science and Technology 5:27-37 (cebola); Christou et al. (1988) Plant Physiol. 87:671-674 (soja); McCabe et al. (1988) Bio/Technology 6:923-926 (soja); Finer e McMullen (1991) In Vitro Cell Dev. Biol. 27P:175-182 (soja); Singh et al. (1998) Theor. Appl. Genet. 96:319-324 (soja); Datta et al. (1990) Biotechnology 8:736-740 (arroz); Klein et al. (1988) Proc. Natl. Acad. Sci. USA 85:4305-4309 (milho); Klein et al. (1988) Biotechnology 6:559-563 (milho); patentes US 5.240.855; US 5.322.783 e US 5.324.646; Klein et al. (1988) Plant Physiol. 91:440-444 (milho); Fromm et al. (1990) Biotechnology 8:833-839 (milho); Hooykaas-Van Slogteren et al. (1984) Nature (London) 311:763764; patente US 5.736.369 (cereais); Bytebier et al. (1987) Proc. Natl. Acad. Sci. USA 84:5345-5349 (Liliaceae); De Wet et al. (1985) in The Experimental Manipulation of Ovule Tissues, ed. Chapman et al. (Longman, New York), pp. 197-209 (pólen); Kaeppler et al. (1990) Plant Cell Reports 9:415-418 e Kaeppler et al. (1992) Theor. Appl. Genet. 84:560-566 (transformação mediada por sonda); D'Halluin et al. (1992) Plant Cell 4:14951505 (eletroporação); Li et al. (1993) Plant Cell Reports 12:250-255 e Christou e Ford (1995) Annals of Botany 75:407413 (arroz); Osjoda et al. (1996) Nature Biotechnology 14:745 750 (milho via Agrobacterium tumefaciens); todas as quais são aqui incorporadas a título de referência.
[0124] Os métodos são bem conhecidos no estado da técnica para a inserção direcionada de um polinucleotídeo em uma localização específica no genoma da planta. Em uma concretização, a inserção do polinucleotídeo em uma desejada localização genômica é alcançada usando um sistema de recombinação sítio-específica. Ver, por exemplo, WO99/25821, WO99/25854, WO99/25840, WO99/25855 e WO99/25853, todos os quais são aqui incorporados a título de referência. Em resumo, o polinucleotídeo das concretizações pode estar contido em cassete de transferência flanqueado por dois sítios de recombinação não-idênticos. O cassete de transferência que é introduzido em uma planta possui estavelmente incorporado em seu genoma um sítio alvo que é flanqueado por dois sítios de recombinação não-idênticos que correspondem aos sítios do cassete de transferência. É provida uma recombinase apropriada e o cassete de transferência é integrado no sítio alvo. O polinucleotídeo de interesse é, por esse meio, integrado em uma posição cromossômica específica no genoma da planta.
[0125] As células que foram transformadas podem ser deixadas a crescer até plantas de acordo com maneiras convencionais. Ver, por exemplo, McCormick et al. (1986) Plant Cell Reports 5:81-84. Essas plantas podem, então, ser deixadas a crescer e serem tanto polinizadas com a mesma estirpe transformada ou com estirpes diferentes, e ser identificada a progênie resultante tendo a expressão constitutiva da característica fenotípica desejada. Duas ou mais gerações podem ser crescidas para assegurar que a expressão da característica fenotípica desejada fique estavelmente mantida e seja herdada e, depois, sementes sejam colhidas para assegurar que a expressão da característica fenotípica desejada tenha sido alcançada. Desta maneira, as concretizações fornecem semente transformada (também referidas como “semente transgênica”) tendo um constructo de nucleotídeos das concretizações, por exemplo, um cassete de expressão das concretizações, estavelmente incorporado no seu genoma.
[0126] Como aqui usado, o termo “planta” pode ser uma planta completa, qualquer parte da mesma, ou uma célula ou cultura de tecido derivado de uma planta. Portanto, o termo “planta” pode se referir a qualquer um entre: plantas inteiras, componentes ou órgãos de planta (incluindo, mas não limitados a embriões, pólen, óvulos, sementes, folhas, flores, ramos, fruta, grãos (kernels), espigas, sabugos, palha, colmos, raízes, pontas de raiz, anteras, e semelhantes), tecidos de planta, células de planta, protoplastos de planta, culturas de tecido de célula de planta a partir das quais a planta de milho pode ser regenerada, calos de planta, aglomerados de planta e sementes de planta. Uma célula de planta é uma célula que é retirada tanto diretamente de uma semente de uma planta quanto da planta, ou derivada através do cultivo de uma célula retirada de uma planta. O termo grão (grain) tem a intenção de significar a semente madura produzida para agricultores comerciais para propósitos diferentes do crescimento ou reprodução da espécie. A progênie, as variantes e as mutantes das plantas regeneradas também estão incluídas dentro do escopo das concretizações, desde que essas partes compreendam os polinucleotídeos introduzidos.
[0127] As concretizações da invenção podem ser usadas para conferir ou melhorar a resistência a patógeno fúngico de planta ou para proteger plantas do ataque de patógemo fúngico, especialmente milho (Zea mays). Serão protegidas diferentes partes da planta do ataque de patógenos, incluindo, mas não estando limitadas a colmos, espigas, folhas, raízes e pendões. Outras espécies de planta também podem ser de interesse na prática das concretizações da invenção, incluindo, mas não estando limitadas a, outras plantas monocotiledôneas de cultivo.
[0128] Quando apropriado, os polinucleotídeos podem ser otimizados para se obter expressão aumentada no organismo transformado. Por exemplo, os polinucleotídeos podem ser sintetizados usando códons planta-preferidos para expressão melhorada. Ver, por exemplo, Campbell e Gowri (1990) Plant Physiol. 92:1-11 para uma discussão da utilização de códon hospedeiro-preferido. Estão disponíveis no estado da técnica os métodos para sintetizar genes planta-preferidos. Ver, por exemplo, patentes US 5.380.831 e US 5.436.391, e Murray et al. (1989) Nucleic Acids Res. 17:477-498, aqui incorporados a título de referência.
[0129] As concretizações da presente invenção podem ser eficazes contra uma diversidade de patógenos de planta, particularmente patógenos fúngicos, tais como, por exemplo, Colletotrichum, incluindo Cg. As concretizações da presente invenção também podem ser eficazes contra a podridão do colmo de milho, incluindo podridão do colmo por antracnose, sendo que o agente causador é o Colletotrichum. Outros fungos patogênicos de planta e oomicetos (muitos, destes últimos, historicamente têm sido considerados fungos, apesar de que agora os taxonomistas modernos estão classificando-os separadamente) incluem, mas não estão limitados aos seguintes: Soja: Phytophthora megasperma fsp. glycinea, Macrophomina phaseolina, Rhizoctonia solani, Sclerotinia sclerotiorum, Fusarium oxysporum, Diaporthe phaseolorum var. sojae (Phomopsis sojae), Diaporthe phaseolorum var. caulivora, Sclerotium rolfsii, Cercospora kikuchii, Cercospora sojina, Peronospora manshurica, Colletotrichum dematium (Colletotichum truncatum), Corynespora cassiicola, Septoria glycines, Phyllosticta sojicola, Alternaria alternata, Microsphaera diffusa, Fusarium semitectum, Phialophora gregata, Glomerella glycines, Phakopsora pachyrhizi, Pythium aphanidermatum, Pythium ultimum, Pythium debaryanum, Fusarium solani; Canola: Albugo candida, Alternaria brassicae, Leptosphaeria maculans, Rhizoctonia solani, Sclerotinia sclerotiorum, Mycosphaerella brassiccola, Pythium ultimum, Peronospora parasitica, Fusarium roseum, Alternaria alternata; Alfalfa: Pythium ultimum, Pythium irregulare, Pythium splendens, Pythium debaryanum, Pythium aphanidermatum, Phytophthora megasperma, Peronospora trifoliorum, Phoma medicaginis var. medicaginis, Cercospora medicaginis, Pseudopeziza medicaginis, Leptotrochila medicaginis, Fusarium oxysporum, Verticillium albo-atrum, Aphanomyces euteiches, Stemphylium herbarum, Stemphylium alfalfae, Colletotrichum trifolii, Leptosphaerulina briosiana, Uromyces striatus, Sclerotinia trifoliorum, Stagnospora meliloti, Stemphylium botryosum, Leptotrochila medicaginis; Trigo: Urocystis agropyri, Alternaria alternata, Cladosporium herbarum, Fusarium graminearum, Fusarium avenaceum, Fusarium culmorum, Ustilago tritici, Ascochyta tritici, Cephalosporium gramineum, Collotetrichum graminicola, Erysiphe graminis f.sp. tritici, Puccinia graminis f.sp. tritici, Puccinia recondita f.sp. tritici, Puccinia striiformis, Pyrenophora tritici- repentis, Septoria nodorum, Septoria tritici, Septoria avenae, Pseudocercosporella herpotrichoides, Rhizoctonia solani, Rhizoctonia cerealis, Gaeumannomyces graminis var. tritici, Pythium aphanidermatum, Pythium arrhenomanes, Pythium ultimum, Bipolaris sorokiniana, Claviceps purpurea, Tilletia tritici, Tilletia laevis, Ustilago tritici, Tilletia indica, Rhizoctonia solani, Pythium arrhenomannes, Pythium gramicola, Pythium aphanidermatum; Girassol: Plasmophora halstedii, Sclerotinia sclerotiorum, Septoria helianthi, Phomopsis helianthi, Alternaria helianthi, Alternaria zinniae, Botrytis cinerea, Phoma macdonaldii, Macrophomina phaseolina, Erysiphe cichoracearum, Rhizopus oryzae, Rhizopus arrhizus, Rhizopus stolonifer, Puccinia helianthi, Verticillium dahliae, Erwinia carotovorum pv. carotovora, Cephalosporium acremonium, Phytophthora cryptogea, Albugo tragopogonis; Milho: Fusarium moniliforme var. subglutinans, Erwinia stewartii, Fusarium moniliforme, Gibberella zeae (Fusarium graminearum), Stenocarpella maydi (Diplodia maydis), Pythium irregulare, Pythium debaryanum, Pythium graminicola, Pythium splendens, Pythium ultimum, Pythium aphanidermatum, Aspergillus flavus, Bipolaris maydis O, T (Cochliobolus heterostrophus), Helminthosporium carbonum I, II & III (Cochliobolus carbonum), Exserohilum turcicum I, II & III, Helminthosporium pedicellatum, Physoderma maydis, Phyllosticta maydis, Kabatiella maydis, Cercospora sorghi, Ustilago maydis, Puccinia sorghi, Puccinia polysora, Macrophomina phaseolina, Penicillium oxalicum, Nigrospora oryzae, Cladosporium herbarum, Curvularia lunata, Curvularia inaequalis, Curvularia pallescens, Trichoderma viride, Claviceps sorghi, Erwinia chrysanthemi pv. zea, Erwinia carotovora, Diplodia macrospora, Sclerophthora macrospora, Peronosclerospora sorghi, Peronosclerospora philippinensis, Peronosclerospora maydis, Peronosclerospora sacchari, Sphacelotheca reiliana, Physopella zeae, Cephalosporium maydis, Cephalosporium acremonium; Sorgo: Exserohilum turcicum, Colletotrichum graminicola (Glomerella graminicola), Cercospora sorghi, Gloeocercospora sorghi, Ascochyta sorghina, Puccinia purpurea, Macrophomina phaseolina, Perconia circinata, Fusarium moniliforme, Alternaria alternata, Bipolaris sorghicola, Helminthosporium sorghicola, Curvularia lunata, Phoma insidiosa, Ramulispora sorghi, Ramulispora sorghicola, Phyllachara sacchari, Sporisorium reilianum (Sphacelotheca reiliana), Sphacelotheca cruenta, Sporisorium sorghi, Claviceps sorghi, Rhizoctonia solani, Acremonium strictum, Sclerophthona macrospora, Peronosclerospora sorghi, Peronosclerospora philippinensis, Sclerospora graminicola, Fusarium graminearum, Fusarium oxysporum, Pythium arrhenomanes, Pythium graminicola, etc.
[0130] O termo “germoplasma” se refere ao material genético de um indivíduo ou oriundo de um indivíduo (por exemplo, uma planta), de um grupo de indivíduos (por exemplo, uma linhagem, variedade ou família de planta), ou de um clone derivado de uma linhagem, variedade, espécie ou cultura. O germoplasma pode ser parte de um organismo ou célula, ou pode ser separado a partir do organismo ou célula. Em geral, o germoplasma provê o material genético com uma composição molecular específica que provê uma fundamentação (base) física para uma ou todas as qualidades hereditárias de um organismo ou cultura de célula. Como aqui usado, o germoplasma inclui células, sementes ou tecidos a partir dos quais novas plantas podem ser crescidas, ou partes de planta, tais como folhas, caules, pólen, ou células que podem ser cultivadas até se tornarem uma planta completa.
[0131] O termo “alelo” se refere a uma ou mais diferentes sequências de nucleotídeos que ocorrem em um lócus específico. Um primeiro alelo é encontrado em um cromossomo, enquanto que um segundo alelo ocorre na mesma posição no homólogo daquele cromossomo, por exemplo, como ocorre para diferentes cromossomos de um indivíduo heterozigoto, ou entre diferentes homozigotos ou indivíduos heterozigotos em uma população. Um “alelo favorável” é o alelo em um lócus particular que confere, ou contribui para, um fenótipo agronomicamente desejável, por exemplo, resistência à infecção por Cg. Um alelo favorável de um marcador é um alelo de marcador que segrega com o fenótipo favorável. Uma forma alélica favorável de um segmento de cromossomo é um segmento de cromossomo que inclui uma sequência de nucleotídeo que contribui para o desempenho agronômico superior em um ou mais lócus genéticos fisicamente localizados no segmento do cromossomo. O termo “frequência de alelo” se refere à frequência (proporção ou percentagem) de um alelo dentro de uma população, ou uma população de linhagens. Pode-se estimar a frequência de alelos dentro de uma população pela média de frequências de alelos de uma amostra de indivíduos a partir daquela população.
[0132] Um alelo se correlaciona “positivamente” com um caractere quando o alelo está ligado ao caractere e quando a presença do alelo é um indicador de que o caractere ou forma de caractere desejada ocorrerá em uma planta que compreende o alelo.
[0133] Um indivíduo é “homozigoto” se ele possuir somente um tipo de alelo em um dado lócus (por exemplo, um indivíduo diplóide possui uma cópia do mesmo alelo em um lócus para cada um dos dois cromossomos homólogos). Um indivíduo é “heterozigoto” se mais do que um tipo de alelo estiver presente em um dado lócus (por exemplo, um indivíduo diplóide com uma cópia de cada um dos dois diferentes alelos). Um caso especial de uma situação heterozigota é quando um cromossomo possui um alelo de um gene e no outro cromossomo falta completamente aquele gene, lócus ou região - em outras palavras, tem uma deleção com relação ao primeiro cromossomo. Esta situação é referida como “hemizigota”. O termo “homogeneidade” indica que membros de um grupo possuem o mesmo genótipo em um ou mais lócus específicos. Em contraste com isso, o termo “heterogeneidade” é usado para indicar que indivíduos dentro do grupo diferem em genótipo em um ou mais lócus específicos.
[0134] As concretizações provêem não somente um gene e suas variantes funcionais para uso em aplicações transgênicas, mas também sequências e processos que permitem que o gene de resistência Rcg1 seja transportado entre linhagens de milho usando melhoramento assistido por marcador. As concretizações também se referem às plantas produzidas por esses processos que retêm um intervalo cromossômico truncado compreendendo o gene de resistência Rcg1.
[0135] Um mapa genético é uma representação gráfica de um genoma (ou uma porção de um genoma tal como um cromossomo simples) onde as distâncias entre os marcos de limite em um cromossomo são medidas pelas frequências de recombinação entre os marcos de limite. As recombinações entre marcos de limite genéticos podem ser detectadas usando uma diversidade de marcadores genéticos moleculares (também chamados de marcadores moleculares) que são aqui descritos em maiores detalhes.
[0136] Para que os marcadores sejam utilizáveis em detectar recombinações, eles precisam detectar diferenças, ou polimorfismos, dentro de uma população que está sendo monitorada. Para marcadores moleculares, isto significa diferenças ao nível de DNA devido às diferenças de sequência de polinucleotídeo (por exemplo, SSRs, RFLPs, FLPs, SNPs). A variabilidade genética pode ser de qualquer origem, por exemplo, inserções, deleções, duplicações, elementos de repetição, mutações pontuais, eventos de recombinação, ou a presença e sequência de elementos de transposição. Os marcadores moleculares podem ser derivados de ácidos nucléicos genômicos ou expressados (por exemplo, ESTs). Os ESTs são em geral bem conservados dentro de uma espécie, enquanto que outras regiões de DNA (tipicamente não-codificante) tendem a acumular polimorfismos e, portanto, podem ser mais variáveis entre indivíduos da mesma espécie. Um grande número de marcadores moleculares de milho é conhecido no estado da técnica e estão publicados ou disponíveis a partir de várias fontes, tais como o recurso de internet Maize GDB e o recurso de internet Arizona Genomics Institute rodado pela Universidade do Arizona.
[0137] Os marcadores moleculares podem ser usados em uma diversidade de aplicações de melhoramento de planta (ver, por exemplo, Staub et al. (1996) Hortscience 31: 729-741; Tanksley (1983) Plant Molecular Biology Reporter. 1: 3-8). Uma das principais areas de interesse é aumentar a eficiência de retrocruzamento e introgressão de genes usando a seleção marcador-assistida (MAS). Um marcador molecular que mostra ligação com um lócus que afeta um desejado caractere fenotípico provê uma ferramenta utilizável para a seleção do caractere em uma população de plantas. Isto é particularmente verdade quando o fenótipo é difícil de ser analisado, por exemplo, muitos caracteres de resistência a doenças, ou ocorre em um estágio mais tardio no desenvolvimento de plantas, por exemplo, características de grão. Na medida em que os ensaios de marcador de DNA são menos trabalhosos e ocupam menos espaço físico do que a fenotipagem de campo, populações muito grandes podem ser analisadas, aumentando as chances de encontrar um recombinante com o segmento alvo oriundo da linhagem doadora transportado para a linhagem receptora. Quanto mais próxima for a ligação, mais utilizável será o marcador, na medida em que a recombinação é menos provável ocorrer entre o marcador e o gene causador do caractere, o que pode resultar em falsos positivos. Com o uso de marcadores flanqueadores diminuem as chances de que a seleção de falso positivo ocorra, tanto quanto seja necessário um evento de dupla recombinação. A situação ideal é ter um marcador no próprio gene, de modo que a recombinação não possa ocorrer entre o marcador e o gene. Tal marcador é chamado um “marcador perfeito”.
[0138] Quando um gene é introgredido por MAS, não é somente o gene que é introduzido mas também as regiões flanqueadoras (Gepts. (2002). Crop Sci; 42: 1780-1790). Isto é referido como “arraste de ligação”. No caso em que a planta doadora for altamente não-relacionada com relação à planta receptora, como no caso do lócus de Rcg1 que está sendo introgredido a partir da MP305, uma fonte exótica, em endogâmicas elite, essas regiões flanqueadoras portam genes adicionais que podem codificar caracteres agronomicamente indesejáveis. Este “arraste de ligação” também pode resultar em produtividade reduzida ou outras características agronômicas negativas mesmo após múltiplos ciclos de retrocruzamento na linhagem de milho elite. Isto também é algumas vezes referido como “arraste de produtividade”. O tamanho da região flanqueadora pode ser diminuído por retrocruzamento adicional, apesar de que isto não é sempre bem sucedido na medida em que os melhoristas não têm controle sobre o tamanho da região ou sobre os breakpoints de recombinação (Young et al. (1998) Genetics 120:579-585). No melhoramento clássico, usualmente, é somente por acaso que recombinações que são selecionadas contribuem para a redução no tamanho do segmento da doadora (Tanksley et al. (1989). Biotechnology 7: 257-264). Mesmo após 20 retrocruzamentos deste tipo, espera-se encontrar uma porção bastante grande do cromossomo da doadora ainda ligada ao gene que está sendo selecionado. Com marcadores, entretanto, é possível selecionar aqueles raros indivíduos que tenham experimentado recombinação perto do gene de interesse. Em 150 plantas retrocruzadas, há 95% de chance de que pelo menos uma planta tenha experimentado um crossover (cruzamento) dentro de 1 cM do gene, com base em uma distância no mapa de meiose simples. Os marcadores possibilitarão identificação inequívoca desses indivíduos. Com um retrocruzamento adicional de 300 plantas, pode haver uma chance de 95% de um crossover dentro de uma distância no mapa de meiose simples de 1 cM, gerando um segmento em torno do gene alvo de menos que 2 cM com base em uma distância no mapa de meiose simples. Isto pode ser realizado em duas gerações com marcadores, enquanto que requereria uma média de 100 gerações sem marcadores (ver Tanksley et al., acima mencionado). Quando a localização exata de um gene é conhecida, uma série de marcadores flanqueadores no entorno do gene pode ser utilizada para selecionar recombinações em diferentes tamanhos de população. Por exemplo, em recombinações de tamanhos menores de população pode ser esperada uma distância adicional do gene, de modo que marcadores flanqueadores mais distais devem ser requeridos para detectar a recombinação.
[0139] A disponibilidade de mapas de ligação integrada do genoma de milho contendo densidades aumentadas de marcadores de milho públicos facilitou o mapeamento genético de milho e do MAS. Ver, por exemplo, o mapa Neighbors 4 IBM2 [online], [recuperado em 21-3-2006]. Recuperação feita a partir do sítio de Internet: <URL: http:// www.maizegdb.org/ cgi-bin/ displaymaprecord.cgi? id=871214>.
[0140] Os componentes chave para a implementação do MAS são: (i) definir a população dentro da qual a associação marcador- caractere será determinada, a qual pode ser uma população segregante, ou uma população aleatória ou estruturada; (ii) monitorar a segregação ou associação de marcadores polimórficos com relação ao caractere e determinar a ligação ou associação usando métodos estatísticos; (iii) definir um conjunto de marcadores desejáveis com base nos resultados da análise estatística e (iv) usar e/ou extrapolar esta informação ao conjunto atual de germoplasma de melhoramento para possibilitar as decisões da seleção baseada em marcador. Os três tipos de marcador descritos neste relatório podem ser usados em protocolos de seleção assistida por marcador; marcadores de repetição simples de sequência (SSR, também conhecida como microssatélite), marcadores de polimorfismo simples de nucleotídeos (SNP) e marcadores polimórficos de comprimento de fragmento (FLP). As SSRs podem ser definidas como corridas relativamente curtas de DNA repetido em tandem com comprimentos de 6 pb ou menos (Tautz (1989) Nucleic Acid Research 17: 6463-6471; Wang et al. (1994) Theoretical and Applied Genetics, 88:1-6). Os polimorfismos surgem devido à variação no número de unidades de repetição, provavelmente causada por deslizamento durante a replicação de DNA (Levinson e Gutman (1987) Mol Biol Evol 4: 203-221). A variação no comprimento da repetição pode ser detectada pelo desenho de primers de PCR para as regiões flanqueadoras não-repetitivas conservadas (Weber e May (1989) Am J Hum Genet 44:388-396). As SSRs são altamente apropriadas para o mapeamento e para o MAS na medida em que elas são multi-alélicas, co-dominantes, reproduzíveis e receptivas à automação de alta produção (Rafalski et al. (1996) Generating and using DNA markers in plants. In: Non-mammalian genomic analysis: a practical guide. Academic press. pp 75-135).
[0141] Por exemplo, no Exemplo 5 mais adiante, é provido um perfil de marcador de SSR de MP305. Este perfil de marcador foi gerado por eletroforese em gel dos produtos de amplificação gerados pelos pares de primers para esses marcadores. A contagem de escore de genótipo de marcador é baseada no tamanho do fragmento amplificado, o qual, neste caso, foi medido por meio de peso de par de bases do fragmento. Apesar do fato de que a variação no primer usado ou nos procedimentos de laboratório podem afetar o peso do par de bases relatado, os valores relativos permanecerão constantes independentemente do primer específico ou do laboratório usado. Portanto, quando se compara linhagens, os perfis de SSR que estão sendo comparados devem ser obtidos a partir do mesmo laboratório, de modo que sejam usados os mesmos primers e equipamento. Por esta razão, quando se compara plantas ou marcadores específicos de linhagens vis à vis, é preferível determinar que tais plantas linhagens possuam os mesmos (ou diferentes) alelos em lócus específicos (por exemplo, pode-se dizer que se uma planta não compreende o intervalo cromossômico derivado da MP305 em ou abaixo do UMC15a, ela não compreenderá alelos iguais à MP305 em todos os lócus em ou abaixo do UMC15a listados na Tabela 6 no Exemplo 5). Um serviço de SSR para milho está disponível ao público em uma base contratual por meio de marcos de DNA em Saint-Jean-sur- Richelieu, Quebec, Canadá.
[0142] Vários tipos de marcadores de FLP podem ser gerados. Mais comumente, primers de amplificação são usados para gerar polimorfismos de comprimento de fragmento. Tais marcadores de FLP são, em muitas maneiras, similares aos marcadores de SSR, exceto pelo fato de que a região amplificada pelos primers não é tipicamente uma região altamente repetitiva. Além disso, a região amplificada, ou amplicon, terá suficiente variabilidade entre o germoplasma, frequentemente devido a inserções ou deleções, de modo que os fragmentos gerados pelos primers de amplificação podem ser distinguidos entre indivíduos polimórficos, e de que é reconhecido que tais indels ocorrem frequentemente em milho (Bhattramakki et al. (2002). Plant Mol Biol 48, 539-547; Rafalski (2002b), acima citado). O termo “indel” se refere a uma inserção ou deleção, sendo que uma linhagem pode ser referida como tendo uma inserção com relação a uma segunda linhagem, ou a segunda linhagem pode ser referida como tendo uma deleção com relação à primeira linhagem. Os marcadores de MZA aqui revelados são exemplos de marcadores de FLP amplificados que haviam sido selecionados porque eles estão em estreita proximidade com relação ao gene Rcg1.
[0143] Os marcadores de SNP detectam substituições de nucleotídeos em par de bases simples. De todos os tipos de marcadores moleculares, os SNPs são os mais abundantes, tendo, portanto, o potencial para prover a resolução mais alta de mapa genético (Bhattramakki et al. 2002 Plant Molecular Biology 48:539-547). Os SNPs podem ser analisados em um nível ainda mais alto de produção do que as SSRs, em um assim chamado modo ‘ultra-alta-produção’ (‘ultra-high-throughput’), na medida em que eles não requerem grandes quantidades de DNA e a automação do ensaio pode ser direta. Os SNPs também têm o compromisso de serem sistemas de custo relativamente baixo. Esses três fatores juntos tornam os SNPs altamente atrativos para uso em MAS. Diversos métodos estão disponíveis para a genotipagem do SNP, incluindo, mas não estando limitados a, hibridização, extensão de primer, ligação de oligonucleotídeo, clivagem por nuclease, minisequenciamento e esferas codificadas. Tais métodos foram revisados em: Gut (2001) Hum Mutat 17 pp. 475-492; Shi (2001) Clin Chem 47, pp. 164-172; Kwok (2000) Pharmacogenomics 1, pp. 95-100; Bhattramakki e Rafalski (2001) Discovery and application of single nucleotide polymorphism markers in plants. In: R.J. Henry, Ed, Plant Genotyping: The DNA Fingerprinting of Plants, CABI Publishing, Wallingford. Uma ampla faixa de tecnologias comercialmente disponíveis utilizam esses e outros métodos para examinar os SNPs, incluindo Masscode™ (Qiagen), Invader® (Third Wave Technologies), SnapShot® (Applied Biosystems), Taqman® (Applied Biosystems) e Beadarrays™ (Illumina).
[0144] Inúmeros SNPs juntos dentro de uma sequência, ou através de sequências ligadas, podem ser usados para descrever um haplótipo para qualquer genótipo particular (Ching et al. (2002), BMC Genet. 3:19 pp Gupta et al. 2001, Rafalski (2002b), acima citado). Os haplótipos podem ser mais informativos do que os SNPs simples e podem ser mais descritivos do que qualquer genótipo particular. Por exemplo, um SNP simples pode ser o alelo ‘T’ para a MP305, mas o alelo ‘T’ também pode ocorrer na população de melhoramento de milho que está sendo utilizada para os parentais recorrentes. Neste caso, um haplótipo, por exemplo, uma série de alelos em marcadores de SNP ligados, pode ser mais informativo. Uma vez que um haplótipo único tenha sido designado para uma região cromossômica da doadora, esse haplótipo pode ser usado naquela população ou em qualquer subconjunto da mesma para se determinar se um indivíduo possui um gene particular. Ver, por exemplo, WO2003054229. O uso de plataformas de detecção de marcador de alta produção automatizadas conhecidas daqueles especialistas na técnica torna este processo altamente eficiente e eficaz.
[0145] Como aqui descrito, muitos dos primers listados nas Tabelas 1 e 2 podem ser usados como marcadores de FLP para selecionar lócus de Rcg1. Esses primers também podem ser convertidos nesses marcadores para SNP ou outros marcadores estruturalmente semelhantes ou funcionalmente equivalentes (SSRs, CAPs, indels, etc), nas mesmas regiões. Uma abordagem muito produtiva para a conversão de SNP está descrita em Rafalski (2002a) Current opinion in plant biology 5 (2): 94100 e também em Rafalski (2002b) Plant Science 162: 329-333. No uso de PCR, os primers são utilizados para amplificar segmentos de DNA a partir de indivíduos (preferencialmente endogâmicos) que representam a diversidade na população de interesse. Os produtos de PCR são sequenciados diretamente em uma ou ambas as direções. As sequências resultantes são alinhadas e os polimorfismos são identificados. Os polimorfismos não estão limitados a Polimorfismos Simples de Nucleotídeos (SNPs), mas também incluem indels, CAPS, SSRs, e VNTRs (número variável de repetições em tandem). Especificamente com relação à informação refinada de mapa aqui descrita, pode-se prontamente usar a informação aqui provida para obter SNPs (e outros marcadores) polimórficos adicionais dentro da região amplificada pelos primers listados nesta descrição. Os marcadores dentro da região descrita do mapa pode ser hibridizada com BACs ou outras bibliotecas genômicas, ou alinhadas, por meios eletrônicos, com sequências genômicas para encontrar novas sequências na mesma localização aproximada com relação aos marcadores descritos.
[0146] Em adição aos SSR’s, FLPs e SNPs acima descritos, outros tipos de marcadores moleculares são também amplamente utilizados, incluindo, mas não estando limitados a, marcadores (tags) de sequências expressadas (ESTs) e marcadores de SSR derivados de sequências EST, e DNA polimórfico aleatoriamente amplificado (RAPD). Como aqui usado, o termo “Marcador Genético” se referirá a qualquer marcador baseado em ácido nucléico, incluindo, mas não estando limitado a, Polimorfismo de Comprimento de Fragmento de Restrição (RFLP), Repetição Simples de Sequência (SSR), DNA Polimórfico Aleatoriamente Amplificado (RAPD), Sequências Polimórficas Clivadas Amplificadas (CAPS) (Rafalski e Tingey, 1993, Trends in Genetics 9:275-280), Polimorfismo de Comprimento de Fragmento Amplificado (AFLP) (Vos et al., 1995, Nucleic Acids Res. 23:4407-4414), Polimorfismo Simples de Nucleotídeo (SNP) (Brookes, 1999, Gene 234:177-186), Região Amplificada de Sequência Caracterizada (SCAR) (Paran e Michelmore, 1993, Theor. Appl. Genet. 85:985-993), Sítio Marcado (Tagged) de Sequência (STS) (Onozaki et al., 2004, Euphytica 138:255-262), Polimorfismo de Conformação de Simples Fita (SSCP) (Orita et al. , 1989, Proc Natl Acad Sci USA 8 6:27 66-2770), Repetição de Sequência Inter-Simples (ISSR) (Blair et al., 1999, Theor. Appl. Genet. 98:780-792), Polimorfismo Amplificado Inter- Retrotransposon (IRAP), Polimorfismo Amplificado de Retrotransposon-Microsatélite (REMAP) (Kalendar et al., 1999, Theor. Appl. Genet. 98:704-711), um produto de clivagem de RNA (tal como um tag Lynx) e semelhantes.
[0147] Mais genericamente, o termo “marcador molecular” pode ser usado para se referir a um marcador genético, definido acima, ou a um produto codificado pelo mesmo (por exemplo, uma proteína) usado como um ponto de referência quando se identifica um lócus ligado. Um marcador pode ser derivado de sequências genômicas de nucleotídeos ou de sequências expressadas de nucleotídeos (por exemplo, oriundo de um RNA processado (spliced), de um cDNA, etc.), ou a partir de um polipeptídeo codificado. O termo também se refere às sequências de ácido nucléico complementares a ou flanqueando as sequências de marcador, tais como os ácidos nucléicos usados como sondas ou pares de primer capazes de amplificar a sequência de marcador. Uma “sonda de marcador molecular” é uma sequência de ácido nucléico ou molécula que pode ser usada para identificar a presença de um lócus de marcador, por exemplo, uma sonda de ácido nucléico que é complementar a uma sequência de lócus de marcador. Alternativamente, em alguns aspectos, uma sonda de marcador se refere a uma sonda de qualquer tipo que é capaz de distinguir (ou seja, genótipo) o alelo particular que está presente em um lócus de marcador. Ácidos nucléicos são “complementares” quando eles hibridizam especificamente em solução, por exemplo, de acordo com as regras de pareamento de bases de Watson-Crick. Alguns dos marcadores aqui descritos também são referidos como marcadores de hibridização quando localizados em uma região de indel, tal como a região não-colinear aqui descrita. Isto é devido ao fato de que a região de inserção é, por definição, um polimorfismo vis à vis uma planta sem a inserção. Portanto, o marcador precisa somente indicar se a região de indel está presente ou ausente. Qualquer apropriada tecnologia de detecção de marcador pode ser usada para identificar tal marcador de hibridização, por exemplo, a tecnologia de SNP é usada nos exemplos aqui fornecidos.
[0148] Um “ácido nucléico genômico” é um ácido nucléico que corresponde, em sequência, a um ácido nucléico hereditário em uma célula. Exemplos comuns incluem DNA genômico de núcleo e amplicons do mesmo. Um ácido nucléico genômico é, em alguns casos, diferente de um RNA processado (spliced), ou um correspondente cDNA, em que o RNA spliced ou o cDNA é processado, por exemplo, por meio da maquinaria de processamento, para remover introns. Os ácidos nucléicos genômicos opcionalmente compreendem sequências não-transcritas (por exemplo, sequências estruturais do cromossomo, regiões de promotor, regiões de enhancer, etc.) e/ou não-traduzidas (por exemplo, introns), enquanto que o RNA spliced/cDNA tipicamente não possuem sequências não transcritas ou introns. Um “ácido nucléico template” é um ácido nucléico que serve como um molde em uma reação de amplificação (por exemplo, uma reação de amplificação baseada em polimerase, tal como PCR, uma reação de amplificação mediada por ligase, tal como LCR, uma reação de transcrição, ou semelhante). Um ácido nucléico template pode ser genômico na origem, ou, alternativamente, pode ser derivado de sequências expressadas, por exemplo, um cDNA ou um EST.
[0149] O termo “amplificar”, no contexto de amplificação de ácido nucléico é qualquer processo pelo qual são produzidas cópias adicionais de um ácido nucléico selecionado (ou de uma forma transcrita do mesmo). Métodos típicos de amplificação incluem vários métodos de replicação baseados em polimerase, incluindo a reação em cadeia de polimerase (PCR), métodos mediados por ligase, tais como a reação em cadeia de ligase (LCR) e métodos de amplificação baseada em polimerase de RNA (por exemplo, por transcrição). Um “amplicon” é um ácido nucléico amplificado, por exemplo, um ácido nucléico que é produzido por amplificação de um ácido nucléico template por qualquer método de amplificação disponível (por exemplo, PCR, LCR, transcrição ou semelhantes).
[0150] Os perfis de isozima e as características morfológicas ligadas também podem, em alguns casos, ser indiretamente usados como marcadores. Mesmo que eles não detectem diretamente diferenças de DNA, eles frequentemente são influenciados por diferenças genéticas específicas. Entretanto, os marcadores que detectam variação de DNA são muito mais numerosos e polimórficos do que os marcadores de isozima ou morfológicos (Tanksley (1983) Plant Molecular Biology Reporter 1:3-8).
[0151] Os alinhamentos de sequência ou contigs também podem ser usados para encontrar sequências a montante ou a jusante dos marcadores específicos listados aqui. Essas novas sequências, próximas dos marcadores descritos aqui, são depois usadas para descobrir e desenvolver marcadores funcionalmente equivalentes.
[0152] Por exemplo, diferentes mapas físicos ou genéticos são alinhados para localizar marcadores equivalentes não descritos neste relatório, mas que estão dentro de regiões similares. Esses mapas podem estar dentro de espécies de milho, ou mesmo ao longo de outras espécies, as quais não tenham sido geneticamente ou fisicamente alinhadas com milho, tais como arroz, trigo, cevada ou sorgo.
[0153] Como observado no Exemplo 2, pelo uso de sequências comuns oriundas da região flanqueadora do lócus de Rcg1 que hibridizou com BACs nas bibliotecas do Mo17 de do BAC de B73, os BACs oriundos de ambas as bibliotecas foram dispostos com o BAC oriundo da região flanqueadora homóloga DE811ASR(BC5) do lócus de Rcg1 em uma rota diferente (tiling path) como mostrado na Figura 9(a). Os BACs de B73 públicos c0113f01 e c0117e18 foram identificados como diretamente norte e sul, respectivamente, do lócus de Rcg1.
[0154] Com esta informação, uma tiling path não-contígua estendida de BACs de B73 entre os marcadores genéticos UMC2285 e UMC15a, UMC2285 e UMC2187, UMC1086 e UMC2200, ou UMC2041 e UMC2200, pode ser criada por alinhamento de marcadores genéticos dentro desta região com o mapa físico do BAC de B73. A informação de alinhamento dos mapas genético e físico de B73 é obtida a partir da base de dados do genoma de milho do Arizona Genomics Institute disponível na world wide web, acessado pela entrada do seguinte endereço na web com o prefixo “www.”>: genome.arizona.edu/fpc/maize/#webagcol. Na vista WebChrom, pode-se selecionar os marcadores genéticos nas vizinhanças do gene Rcg1 e obter um link com o contig físico onde esses marcadores estão localizados. Pelo alinhamento do mapa físico com o mapa genético feito dessa maneira, pode-se encontrar um plethora de BACs de B73 na região entre os intervalos cromossômicos definidos pelos marcadores genéticos UMC2285 e UMC15a, UMC2285 e UMC2187, UMC1086 e UMC2200, ou UMC2041 e UMC2200. Os BACs podem ser usados pelos especialistas na técnica para desenvolver novos marcadores para introgressão do lócus de Rcg1 no germoplasma de milho. Em particular, tais marcadores genéticos são utilizáveis no rastreamento do lócus de Rcg1 em quaisquer linhagens nas quais o lócus de Rcg1 ou o gene Rcg1 tenha sido introgredido, e na seleção do genoma de parental recorrente em um programa de retrocruzamento.
[0155] Por exemplo, a informação de sequência da região nas vizinhanças do lócus de Rcg1 pode ser usada para desenhar marcadores polimórficos que serão utilizáveis em introgressão e seleção do gene ou lócus de Rcg1 em outro germoplasma de milho. Há muitos B73 derivados de cromossomos artificiais bacterianos (BACs) disponíveis na região de interesse a partir da qual a informação de sequência pode ser obtida. Um exemplo de BACs na região de interesse é ilustrada na Figura 21, a qual mostra um contig no mapa físico de B73 que é homólogo à região de Rcg1 em DE811ASR (BC5) [Figura 21 encontrada em 1003-2006]. Buscada a partir do sítio de Internet <URL: http:// www.genome.arizona.edu/ cgi-bin// WebAGCoL/ WebFPC/ WebFPC_Direct_v2.1.cgi? name=maize&contig=187&marker=ssu1>. A informação de sequência é obtida tanto através de informação que já está disponível ao público (por exemplo, sequência- terminal de BAC, sequência de Tags de Sequência Expressada (ESTs) que hibridizam com BACs nesta região, sondas do tipo overgo (sobreposição) que frequentemente se relacionam com esses ESTs, etc.) ou para obter nova sequência pelo sequenciamento direto de clones de BAC nesta região. A partir desta sequência podem ser determinadas quais regiões são, em maioria, únicas usando diversos métodos diferentes conhecidos dos especialistas na técnica. Por exemplo, pelo uso do software de predição de gene ou pela técnica de blasting da sequência contra todas as sequências disponíveis de milho, pode-se selecionar a sequência não-repetitiva. A sequência de baixo número de cópias pode ser usada para desenvolver uma ampla série de marcadores baseados em ácido nucléico. Esses marcadores são usados para realizar o screening do material de planta no qual o lócus de Rcg1 está presente e o material de planta no qual o lócus de Rcg1 está ausente. Se for desejado um marcador fora do lócus de Rcg1, então os marcadores são usados para realizar o screening do material de planta no qual o lócus de Rcg1 está presente e o material de planta no qual o lócus de Rcg1 está ausente para se determinar se o marcador é polimórfico em tal germoplasma. Os marcadores polimórficos são então usados na introgressão assistida por marcador e na seleção da região de Rcg1 e, de modo ótimo, também na seleção do genoma do parental recorrente, em outro germoplasma de milho. Portanto, com a localização do lócus de Rcg1 identificada e estabelecida sua associação com a resistência a Colletotrichum, os especialistas na técnica podem utilizar qualquer número de marcadores existentes, ou prontamente desenvolver novos marcadores, que podem ser usados na introgressão ou identificação da presença ou ausência do lócus de Rcg1 em germoplasma, e selecionar o genoma do parental recorrente em um programa de retrocruzamento.
[0156] Em um mapa genético, a ligação de um marcador molecular a um gene ou a um outro marcador molecular é medida como uma frequência de recombinação. Em geral, os dois lócus mais próximos (por exemplo, os dois marcadores de SSR) estão no mapa genético mais próximos do que eles ficam um do outro no mapa físico. Uma distância genética relativa (determinada pela sobreposição de frequências, medida em centimorgans; cM) pode ser proporcional à distância física (medida em pares de base, por exemplo, em pares de quilobases [kb] ou em megapares de bases [Mbp]) que separa dois lócus ligados entre si em um cromossomo. A ausência de proporcionalidade precisa entre cM e a distância física pode resultar da variação em frequências de recombinação para diferentes regiões cromossômicas, por exemplo, algumas regiões cromossômicas são recombinação do tipo “hot spots”, enquanto outras regiões não mostram qualquer recombinação, ou somente demonstram raros eventos de recombinação. Alguns dos dados de introgressão e de informação de mapeamento sugerem que a região em torno do lócus de Rcg1 é uma daquelas que possuem uma grande quantidade de recombinação.
[0157] Em geral, quanto mais próximo um marcador estiver de um outro marcador, se medido em termos de recombinação ou distância física, mais fortemente eles estão ligados. Quanto mais próximo um marcador estiver de um gene que codifica um polipeptídeo que confere um fenótipo particular (resistência a doença), se medido em termos de recombinação ou de distância física, melhor aquele marcador será para marcar o caractere fenotípico desejado. Se possível, o melhor marcador é aquele que fica dentro do próprio gene, na medida em que ele sempre permanecerá ligado com o gene causador do fenótipo desejado.
[0158] A variabilidade genética de mapeamento também pode ser observada entre diferentes populações da mesma espécie de cultura, incluindo milho. A despeito desta variabilidade no mapa genético que pode ocorrer entre populações, o mapa genético e a informação de marcador derivada de uma população geralmente permanece utilizável ao longo de múltiplas populações na identificação de plantas com caracteres desejados, contra-seleção de plantas com caracteres indesejáveis e na orientação de MAS.
[0159] Na localização de marcadores equivalentes ao longo de mapas genéticos, uma população de mapeamento pode ser usada para confirmar se qualquer marcador equivalente está dentro da região aqui descrita e, portanto, utilizável na seleção de Rcg1. Usando este método, o marcador equivalente, juntamente com os marcadores listados aqui, é mapeado na dita população de mapeamento. Qualquer marcador equivalente que fique dentro da mesma região pode ser usado para selecionar o Rcg1. As populações de mapeamento conhecidas do estado da técnica e que podem ser usadas para esta finalidade incluem, mas não ficam limitadas a, populações IBM e a população T218 X GT119 IF2 descrita em Sharopova, N. et al. (2002) Plant Mol Biol 48(5):463-481 e Lee, M. et al. (1999): Tools for high resolution genetic mapping in maize - status report. Proc. Plant Animal Genome VII, January 17-21, 1999, San Diego, USA, P. 146; a população UMC 98, descrita em Davis, G.L. et al. (1999) Genetics 152(3):1137-72 e em Davis, M.D. et al., (1998) The 1998 UMC Maize Genetic Map: ESTs, Sequenced Core Markers, and Nonmaize Probes as a Foundation for Gene Discovery, Maize Genetics Conference Abstracts 40.
[0160] Como aqui usado, o termo “introgressão” e “introgredir” se referirá a mover um gene ou lócus de uma linhagem para uma outra por: (1) cruzamento de indivíduos de cada linhagem para criar uma população; e (2) seleção de indivíduos que portam o gene ou lócus desejado. Após cada cruzamento, o processo de seleção é repetido. Por exemplo, o gene das concretizações, ou o lócus contendo-o, pode ser introgredido para um parental recorrente que não seja resistente, ou seja, somente parcialmente resistente, significando que é sensível ou susceptível ou parcialmente sensível ou susceptível a Cg. A linhagem parental recorrente com o gene ou lócus introgredido possui, então, resistência melhorada ou recentemente conferida a Cg. Esta linhagem na qual o lócus de Rcg1 foi introgredido é referida aqui como uma conversão de lócus de Rcg1.
[0161] O processo de introgressão é frequentemente referido como “retrocruzamento” quando o processo é repetido duas ou mais vezes. Na introgressão ou retrocruzamento, a parental “doadora” se refere à planta parental com o gene ou lócus desejado a ser introgredida. A parental “receptora” (usada uma ou mais vezes) ou parental “recorrente” (usada duas ou mais vezes) se refere à planta parental na qual o gene ou lócus está sendo introgredido. Por exemplo, ver Ragot, M. et al. (1995) Marker-assisted backcrossing: a practical example, in Techniques et Utilisations des Marqueurs Moleculaires (Les Colloques, Vol. 72, pp. 45-56) e Openshaw et al., (1994) Marker- assisted Selection in Backcross Breeding, Analysis of Molecular Marker Data, pp. 41-43. O cruzamento inicial dá origem à geração F1; o termo “BC1” então se refere ao segundo uso do parental recorrente, o “BC2” se refere ao terceiro uso do parental recorrente e assim por diante.
[0162] No caso do Rcg1, onde a sequência do gene e as regiões muito próximas estão disponíveis, os marcadores de DNA baseados no próprio gene ou em sequências proximamente ligadas podem ser desenvolvidos para seleção direta do gene doador no background do parental recorrente. Apesar de poder ser usada qualquer sequência de DNA polimórfico oriundo da região cromossômica que porta o gene, as sequências providas nas concretizações permitem o uso de marcadores de DNA dentro ou próximas do gene, minimizando a seleção de falso positivo para o gene. Os marcadores de flanqueamento limitam o tamanho dos fragmentos do genoma doador introduzidos no background do receptor, minimizando, assim, o chamado “arraste de ligação” que significa a introdução de sequências indesejáveis da linhagem doadora e que pode impactar no desempenho da planta em germoplasma elite diferente. As concretizações provêem múltiplos exemplos de marcadores de DNA que podem ser assim usados, e os especialistas na técnica serão capazes de usar as sequências genômicas providas para criarem até mesmo mais marcadores. Um exemplo é usar marcadores que hibridizam (no caso de ensaios de RFLP) ou anelam (no caso de ensaios de PCR) especificamente (exclusivamente) com sequências proximamente ligadas, incluindo aquelas dentro do lócus. Em princípio, as sequências que também hibridizam ou anelam em algum lugar no genoma podem ser usadas se diversos marcadores forem usados em combinação. Quando são usadas reações de PCR, na prática, o comprimento dos primers usados na reação de amplificação devem ter pelo menos cerca de 15 nucleotídeos, mas dependendo das sequências e das condições de hibridização, qualquer comprimento que forneça anelamento específico pode ser usado, tal como cerca de 16, cerca de 17, cerca de 18, cerca de 19, cerca de 20, cerca de 21, cerca de 22, cerca de 23, cerca de 24, cerca de 25, cerca de 26, cerca de 27, cerca de 28 ou mais longas. Para reações de PCR, o termo “anelar” é comumente usado, e, como aqui usado, deve ser entendido como tendo o mesmo significado de “hibridizar”.
[0163] Portanto, com o uso de marcadores e processos aqui descritos, pode-se produzir uma planta compreendendo um intervalo cromossômico truncado que compreende o lócus de Rcg1 e/ou o gene Rcg1. O termo “intervalo cromossômico” ou “segmento cromossômico” se refere a um intervalo linear contíguo de DNA genômico que reside na planta em um único cromossomo, usualmente definido com referência a dois marcadores que definem os dois pontos extremos do intervalo cromossômico. O intervalo especificado pode incluir os marcadores nos pontos extremos (por exemplo, um ou mais marcadores em ou dentro do intervalo cromossômico definido por marcador A e marcador B) ou pode excluir os marcadores nos pontos extremos do intervalo (por exemplo, um ou mais marcadores dentro do intervalo cromossômico definido pelo marcador A e marcador B). Um intervalo cromossômico truncado se refere a um intervalo cromossômico que foi reduzido em tamanho pela seleção de um ou mais eventos de recombinação que reduziram o tamanho do intervalo cromossômico. Um “evento cromossômico” se refere à ocorrência de recombinação entre cromossomos homólogos, e se refere a uma localização cromossômica específica onde tal recombinação tenha ocorrido (por exemplo, uma recombinação do intervalo cromossômico interno aos pontos extremos do cromossomo terão um evento de recombinação em cada extremo do intervalo cromossômico). O intervalo cromossômico truncado pode ser definido com referência a um ou ambos os novos marcadores nos pontos extremos do segmento. O comprimento dos dois segmentos cromossômicos pode ser medido tanto em centimorgans como em pares de bases. Os elementos genéticos ou genes localizados em um único intervalo cromossômico são fisicamente ligados. O tamanho de um intervalo cromossômico não é particularmente limitado, mas, no contexto das concretizações da presente invenção, em geral, os elementos genéticos localizados dentro de um intervalo cromossômico também são ligados geneticamente.
[0164] Pelo uso dos processos das concretizações, é possível selecionar uma planta que compreende um intervalo cromossômico truncado compreendendo o gene Rcg1. Especificamente, com respeito à invenção descrita em maiores detalhes nos exemplos mais adiante, o intervalo cromossômico pode ser reduzido a um comprimento de 12cM ou menos, 10 cM ou menos, 8 cM ou menos, 6 cM ou menos, 4 cM ou menos, 3cM ou menos, 2,5 cM ou menos, 2 cM ou menos, 1,5 cM ou menos, 1 cM ou menos, 0,75 cM ou menos, 0,50 cM ou menos, ou 0,25 cM ou menos, em cada caso como medido com respeito às distâncias do mapa como mostrado no mapa genético Neighbors 4 do IBM2 colocado em prática em 21 de março de 2006. Quando medido em pares de bases, o intervalo cromossômico pode ser reduzido a um comprimento de 15 mpb ou menos, 10 mpb ou menos, 5 mpb ou menos, 3 mpb ou menos, 1 mpb ou menos, 500 kpb ou menos, ou 250 kpb ou menos. Os especialistas na técnica podem compreender que é desejável causar uma quebra na região cromossômica bem próxima à sequência codificante de Rcg1 (por exemplo, dentro de 5kpb ou menos, dentro de 4 kpb ou menos, 3 kpb ou menos, 2 kpb ou menos, 1 kpb ou menos, ou 0,5 kpb ou menos), de modo que o promotor e os outros elementos de regulação a montante fiquem não-ligados a partir da sequência codificante.
[0165] O termo “lócus”, em geral, se refere a uma região geneticamente definida de um cromossomo que porta um gene ou, possivelmente, dois ou mais genes tão proximamente ligados que geneticamente eles se comportam como um único lócus responsável por um fenótipo. Quando aqui usado com respeito ao Rcg1, o “lócus de Rcg1” se referirá à região definida do cromossomo que porta o gene Rcg1 incluindo suas sequências de regulação associadas, mais a região vizinha do gene Rcg1 que é não-colinear com B73, ou qualquer porção menor do mesmo que retém o gene Rcg1 e as sequências de regulação associadas. Este lócus também foi referido em algum lugar como o lócus de ASR, e será aqui referido como o lócus de Rcg1.
[0166] Um “gene” deve se referir a uma região genética codificante específica dentro de um lócus, incluindo as suas sequências de regulação associadas. A região codificante do transcripto primário de Rcg1, referida aqui como a “sequência codificante de Rcg1”, será usada para definir a posição do gene Rcg1, e os especialistas na técnica compreendem que as sequências de regulação associada estarão dentro de uma distância de cerca de 4 kb a partir da sequência codificante de Rcg1, com o promotor localizado a montante. Uma concretização da presente invenção é o isolamento do gene Rcg1 e a demonstração de que ele é o gene responsável pelo fenótipo conferido pela presença do lócus.
[0167] Como aqui usados, os termos “ligado” ou “ligação” (como distinguido do termo “operacionalmente ligado”) deve se referir à ligação genética ou física dos lócus ou genes. Os lócus ou genes são considerados geneticamente ligados se a frequência de recombinação entre eles for menor do que cerca de 50%, como determinado em um mapa de meiose simples. Eles são progressivamente mais ligados se a frequência de recombinação for cerca de 40%, cerca de 30%, cerca de 20%, cerca de 10% ou menos, como determinado em um mapa de meiose simples. Dois ou mais genes são fisicamente ligados (ou sintênicos) se ficar demonstrado que eles estão localizados em uma única porção de DNA, tal como um cromossomo. Genes geneticamente ligados, na prática, estarão fisicamente ligados (ou sintênicos), mas a distância física exata (número de nucleotídeos) pode não ter sido ainda calculada. Como aqui usado, o termo “proximamente ligado” se refere a marcadores geneticamente ligados dentro de 15 cM ou menos, incluindo sem limitação 12 cM ou menos, 10 cM ou menos, 8 cM ou menos, 7 cM ou menos, 6 cM ou menos, 5 cM ou menos, 4 cM ou menos, 3 cM ou menos, 2 cM ou menos, 1 cM ou menos e 0,5 cM ou menos, como calculado no mapa genético neighbors 4 de IBM2 disponível ao público no website Maize GDB anteriormente referenciado nesta descrição. Uma sequência de DNA, tal como um oligonucleotídeo curto representando uma sequência dentro de um lócus ou uma complementar da mesma, é também ligada àquele lócus.
[0168] Uma “linhagem” ou “estirpe” é um grupo de indivíduos de parentesco idêntico que são geralmente endogâmicos em algum grau e que são geralmente homozigotos e homogêneos na maioria dos lócus.
[0169] Uma “linhagem ancestral” ou “progenitor” é uma linhagem parental usada como fonte de genes, por exemplo, para o desenvolvimento de linhagens elite. A “progênie” são os descendentes da linhagem ancestral, e podem estar separados dos seus ancestrais por muitas gerações de melhoramento. Por exemplo, muitas linhagens elite são progênies de B73 ou Mo17. Uma “estrutura pedigree” define a relação entre um descendente e cada ancestral que deu origem àquele descendente. Uma estrutura pedigree pode se expandir por uma ou mais gerações, descrevendo relações entre os descendentes e seus pais, avós, bisavós, etc.
[0170] Uma “linhagem elite” ou “variedade elite” é uma linhagem ou variedade agronomicamente superior que resultou de muitos ciclos de melhoramento e seleção para o desempenho agronômico superior. Uma “linhagem elite endogâmica” é uma linhagem elite que é uma endogâmica e que mostrou ser utilizável para produzir variedades híbridas de produtividade suficientemente alta e agronomicamente ajustadas (uma “variedade híbrida elite”). Numerosas linhagens e variedades elite estão disponíveis e são conhecidas daqueles especialistas na técnica de melhoramento de milho. Semelhantemente, “germoplasma elite” é um germoplasma agronomicamente superior, tipicamente derivado de e/ou capaz de dar origem a uma planta com desempenho agronômico superior, tal como uma linhagem elite de milho existente ou recentemente desenvolvida.
[0171] Por outro lado, uma “linhagem de milho exótica” ou “germoplasma de milho exótico” é o germoplasma derivado de milho não pertencente a uma linhagem elite disponível, variedade elite ou germoplasma elite. No contexto de um cruzamento entre duas plantas de milho, uma linhagem exótica ou germoplasma exótico não é proximamente relacionada pelo descendente da linhagem elite, variedade elite ou germoplasma elite com o qual é cruzado. Mais comumente, a linhagem exótica ou germoplasma exótico é selecionada para introduzir novos elementos genéticos (tipicamente novos alelos) em um programa de melhoramento.
[0172] Unidades, prefixos e símbolos podem ser indicados por sua forma aceita do SI. A menos que seja indicado de modo diferente, os ácidos nucléicos são escritos da esquerda para a direita na orientação 5' para 3'; as sequências de aminoácidos são escritas da esquerda para a direita em orientação amino para carboxil, respectivamente. As faixas numéricas são inclusivas dos números que definem a faixa. Os aminoácidos podem ser aqui referidos tanto por seus símbolos de três letras comumente conhecidos ou pelos símbolos de uma letra recomendado pela Comissão de Nomenclatura Bioquímica da IUPAC- IUB. Da mesma forma, os nucleotídeos podem ser referidos por seus códigos de uma letra comumente aceitos. Os termos acima definidos são mais completamente definidos por referência à descrição como um todo.
[0173] Com relação às direções do mapa aqui observadas, ao invés dos termos 5' e 3', são usados os termos “norte” e “acima” (por exemplo, um marcador norte do gene Rcg1 se refere a um marcador acima do gene Rcg1, como determinado com referência aos mapas providos em uma orientação vertical, tal como as Figuras 7 e 8, e à esquerda do gene Rcg1, como determinado com referência aos mapas providos em uma orientação horizontal, tal como a Figura 22). Da mesma forma, são usados os termos “sul” e “abaixo” (por exemplo, um marcador sul do gene Rcg1 se refere a um marcador abaixo do gene Rcg1, como determinado com referência aos mapas orientados verticalmente aqui providos, e para a direita do gene Rcg1, como determinado com referência aos mapas orientados horizontalmente providos aqui). Mais especificamente, acima da sequência codificante de Rcg1 se refere ao cromossomo acima, ou norte do transcripto primário na SEQ ID NO:1 (perto de FLP110F), e abaixo da sequência codificante de Rcg1 se refere ao cromossomo abaixo ou sul do transcripto primário na SEQ ID NO:1 (perto de FLPA1R). Ver Figura 26. Os termos “proximal” e “distal” são termos relativos, significando, respectivamente, mais perto e mais afastado de uma localização especificada (por exemplo, o gene Rcg1) quando usado para comparar dois pontos em um mapa com relação à localização especificada.
[0174] O termo “sistemas de computador” se refere, em geral, a vários sistemas automatizados usados para realizar algumas ou todas as etapas dos métodos aqui descritos. O termo “instruções” se refere ao código computacional que instrui o sistema de computador para realizar algumas ou todas as etapas do método. Adicionalmente, para praticar algumas ou todas as etapas do método, sistemas digitais ou analógicos, por exemplo, compreendendo um computador digital ou analógico, também pode controlar uma variedade de outras funções tal como um mostrador visível ao usuário (por exemplo, para permitir a visualização dos resultados do método pelo usuário) e/ou o controle dos aspectos de saída (por exemplo, para assistir na seleção assistida por marcador ou no controle do equipamento de campo automatizado).
[0175] Certos métodos aqui descritos são opcionalmente (e tipicamente) implementados via um programa ou programas de computador (por exemplo, que armazenam e podem ser usados para analisar dados de marcador molecular). Portanto, as concretizações provêem sistemas digitais, por exemplo, meios legíveis por computador, e/ou sistemas integrados compreendendo instruções (por exemplo, concretizados em software apropriado) para realizar os métodos aqui revelados. O sistema digital incluirá informação (dados) correspondente ao genótipo de planta para um conjunto de marcadores genéticos e, opcionalmente, valores fenotípicos e/ou relações de família. O sistema também pode ajudar um usuário a realizar a seleção assistida por marcador para Rcg1 de acordo com os métodos aqui revelados, ou pode controlar o equipamento de campo com seleção, colheita e/ou esquemas de melhoramento automatizados.
[0176] As aplicações desktop padrão, tal como o software de processamento de palavra (por exemplo, Microsoft Word™ ou Corel WordPerfect™) e/ou software de base de dados (por exemplo, software de planilha eletrônica, tal como Microsoft Excel™, Corel Quattro Pro™, ou programas de base de dados tal como Microsoft Access™ ou Paradox™) podem ser adaptados às concretizações pela entrada de dados os quais são alimentados na memória de um sistema digital, e pela realização de uma operação como observado aqui nos dados. Por exemplo, os sistemas podem incluir o software precedente tendo os dados genotípicos apropriados e, opcionalmente, os dados de pedigree, que são usados em conjugação com uma interface de usuário (por exemplo, um GUI em um sistema de operação padrão tal como o sistema Windows, Macintosh ou LINUX) para realizar qualquer análise observada aqui, ou simplesmente para adquirir os dados (por exemplo, em uma planilha eletrônica) a serem usados nos métodos aqui revelados. O computador pode ser, por exemplo, um PC (máquina Intel x86 ou Pentium chip-compatível DOS™, OS2™, WINDOWS™, WINDOWS NT™, WINDOWS95™, WINDOWS98™, LINUX, compatível com o Apple, compatível com o MACINTOSH™, compatível com o Power PC, ou um compatível com o UNIX (por exemplo, estação de trabalho SUN™)) ou outro computador comercialmente comum que é conhecido dos especialistas na técnica. O software para realizar análise de associação e/ou predição de valor fenotípico pode ser construído por um especialista na técnica usando linguagem de programação padrão tal como Visualbasic, Fortran, Basic, Java, ou semelhantes, de acordo com os métodos aqui revelados.
[0177] Qualquer controlador de sistema ou computador opcionalmente inclui um monitor o qual pode incluir, por exemplo, um mostrador de tubo de raios catódicos (“CRT”), um mostrador de painel plano (por exemplo, um mostrador de cristal líquido de matriz ativa) ou outros. Os circuitos de computador são frequentemente colocados em uma caixa, a qual inclui numerosos chips de circuito integrado, tais como microprocessador, memória, circuitos de interface e outros. A caixa também, opcionalmente, inclui uma unidade de disco rígido, uma unidade de disco flexível, uma unidade removível de alta capacidade, tal como um CD-ROM gravável, e outros elementos periféricos comuns. Os dispositivos de entrada, tais como um teclado ou mouse opcionalmente fornecem a entrada para o usuário e para a seleção pelo usuário de genótipo de marcador genético, valor fenotípico os semelhantes no sistema computacional relevante.
[0178] O computador tipicamente inclui software apropriado para receber instruções do usuário, tanto na forma de entrada de usuário em um conjunto de campos de parâmetro, por exemplo, em um GUI, ou na forma de instruções programadas, por exemplo, programadas para uma variedade de diferentes operações específicas. O software então converte essas instruções em uma linguagem apropriada para instruir o sistema a executar qualquer operação desejada. Por exemplo, um sistema digital pode instruir a seleção de plantas compreendendo certos marcadores, ou controlar a maquinaria de campo para colheita, seleção, cruzamento ou preservação de culturas de acordo com o método relevante aqui revelado.
[0179] A invenção também pode ser concretizada dentro da rede de circuitos de um circuito integrado específico de aplicação (ASIC) ou dispositivo lógico programável (PLD). Em tal caso, a invenção é concretizada em uma linguagem de descritor legível por computador que pode ser usada para criar um ASIC ou PLD. A invenção também pode ser concretizada dentro da rede de circuitos ou processadores lógicos de uma variedade de outros aparelhos digitais, tais como PDAs, sistemas de computador laptop, mostradores, equipamento de edição de imagem, etc.
[0180] As concretizações da invenção são adicionalmente definidas nos exemplos a seguir, nos quais todas as partes e percentagens são em peso e graus Celsius, a menos que seja estabelecido diferentemente. Deve ficar compreendido que esses exemplos, apesar de indicarem concretizações da invenção, são dados a título de ilustração somente. A partir da discussão acima e desses exemplos, os especialistas na técnica podem determinar as características essenciais das concretizações desta invenção e, sem se afastar do espírito e escopo da mesma, podem fazer várias mudanças e modificações para adaptá- la a várias utilizações e condições. Portanto, várias modificações das concretizações da invenção, em adição àquelas mostradas e descritas aqui, ficarão evidentes àqueles especialistas na técnica a partir da descrição precedente. Tais modificações também têm a intenção de ficar dentro do escopo das reivindicações em anexo. A descrição de cada referência apresentada aqui é incorporada a título de referência em sua integralidade. Os Exemplos 1-4 e 7-12 são reais. Os Exemplos 5, 6 e 13 são reais em parte e proféticos em outra parte.
[0181] De modo a mapear e clonar o gene responsável pela resistência da linhagem de milho MP305 a Cg, linhagens que tinham sido previamente criadas e que diferiam geneticamente tão pouco quanto possível uma da outra, com exceção da presença do lócus responsável pelo fenótipo resistente. Tais linhagens são chamadas linhagens isogênicas próximas. Para esta finalidade, a DE811 foi cruzada com a MP305 e a progênie foi retrocruzada com a linhagem sensível DE811, por três vezes, sendo feita a cada retrocruzamento a seleção com relação à resistência a Cg e de diferença com relação às características de DE811 (Weldekidan and Hawk, (1993), Maydica, 38:189-192). A linhagem resultante foi designada de DE811ASR (BC3) (Weldekidan and Hawk, (1993) acima mencionado). Esta linhagem foi usada como ponto de partida para o mapeamento refinado do lócus de Rcg1. Foi primeiro necessário saber aproximadamente onde, no genoma de milho, ele estava localizado. Usando métodos genéticos padrão, Jung et al. ((1994) acima citado) localizou previamente o lócus no ramo longo do cromossomo 4.
[0182] Na medida em que o lócus de Rcg1 havia sido previamente mapeado no ramo longo do cromossomo 4 de milho, usando a informação sobre marcadores próximos ao lócus obtidos por Jung et al. (1994) acima citado, todos os marcadores de repetição de sequência simples (SSR), disponíveis ao público ou privados, localizados na região do cromossomo designado 4.06-4.08, foram analisados para determinar se esses marcadores eram polimórficos entre as duas linhagens isogênicas próximas DE811 e DE811ASR (BC5). A linhagem DE811ASR (BC5) foi derivada da linhagem DE811ASR (BC3) descrita por Weldekidan e Hawk (1993), acima mencionado, através de dois retrocruzamentos com a DE811 sob seleção com relação à resistência a Cg, seguidos por 5 gerações de autofecundação e seleção para obter a linhagem BC5. A linhagem BC5 foi retrocruzada duas vezes mais com a DE811 para criar a população segregante BC7 usada para o mapeamento refinado. De forma a possibilitar a condução da avaliação fenotípica em base familiar, os indivíduos BC7 foram autofecundados para criar famílias BC7S1.
[0183] A partir desta análise, foi descoberto que dois marcadores de SSR, PHI093 e UMC2041, são polimórficos. Usando o mapa neighbors de B73 X Mo17 (IBM) (Lee et al. (2002) Plant Mol Biol 48:453-61; Sharopova et al., (2002) Plant Mol Biol 48:463-81) inter-pareado (inter-mated) publicamente disponível (Coe et al. (2002) Plant Physiol. 128:9-12; Gardiner, et al., (2004), Plant Physiol., 134:1317-1326; Yim et al., (2002) Plant Physiol. 130:1686-1696), as sequências de três marcadores próximos de Polimorfismo de Comprimento de Fragmento de Restrição (RFLP), CDO365, CSU166 e CDO127, foram usadas para criar marcadores polimórficos de Comprimento de Fragmento (aqui designados FLPs). Os FLPs são marcadores que podem ser analisados usando eletroforese em gel ou qualquer método similar de separação de fragmentos de alta resolução seguindo-se uma reação de PCR usando os primers de uma sequência definida. Foi verificado que os três marcadores são polimórficos. Os FLPs usados no mapeamento do lócus de Rcg1 estão resumidos na Tabela 1. Quaisquer primers para os FLPs de MZA mostrados na Tabela 1, a qual também tem os mesmos nomes de marcadores MZA mostrados na Tabela 2, amplificarão a região interna de FLP para a sequência interna mostrada na Tabela 2. A temperatura de anelamento para todos os primers listados na Tabela 1 é de 60°C.
[0184] De modo a determinar se a presença desses três FLPs polimórficos e dois SSRs polimórficos estava associada com o fenótipo de resistência, indicando que a região portadora do lócus de Rcg1 foi localizada em um segmento cromossômico contendo esses três marcadores, foi criada uma tabela na qual foram alimentados os status fenotípicos de 4784 indivíduos determinados por observação de campo e os status genotípicos com relação a cada um dos cinco marcadores, determinado pela análise de tamanho de fragmento. Estes dados foram submetidos sequencialmente aos programas de software Joinmap (Van Ooijen, et al., (2001), Plant Research International, Wageningen, the Netherlands) e Windows QTL Cartographer (Wang, et al., (2004), (online, versão 2.0 recuperada em 14-06-2004 e versão 2.5 recuperada em 22-02-2005)); recuperado a partir da North Carolina State University Statistical Genetics e do website de Bioinformática na Internet em <URL: http:// statgen.ncsu.edu/ qtlcart/ WQTLCart.htm>. O primeiro programa determina a ordem dos marcadores ao longo da região cromossômica. O segundo programa determina se um alelo particular de um marcador (uma forma particular das duas formas polimórficas do marcador) está significativamente associado com a presença do fenótipo. Os marcadores para os quais a presença de um ou dos outros alelos é mais significativamente associada com o fenótipo de resistência mais provavelmente estão mais próximos do gene responsável pelo fenótipo de resistência. A Figura 3 representa um gráfico produzido pelo QTL de Windows Cartographer mostrando uma análise estatística da chance (eixo dos Y) de que o lócus responsável pela resistência a Cg esteja localizado em uma posição particular ao longo do cromossomo (eixo dos X) como definido pelos marcadores FLP.
[0185] A partir do mapa físico e genético integrado, como descrito por Fengler, et al., ((2004) Plant and Animal Genome XII Abstract Book, Página 192 (Poster número P487), 10-14 de janeiro, San Diego, California) e Gardiner, (2004) acima mencionado, é possível identificar dois contigs de cromossomo bacteriano (BAC), derivados de uma biblioteca de BAC de Mo17, cultivando os acima mencionados marcadores genéticos.
[0186] Entretanto, os dois contigs de BAC contendo os marcadores flanqueadores da região de interesse continham um gap de tamanho desconhecido. De modo a identificar adicionais BACs para fazer a ponte deste gap, foi usado um denso mapa genético contendo marcadores (Fengler, (2004) acima citado) com posições conhecidas no mapa físico para encontrar marcadores adicionais geneticamente ligados a marcadores previamente ligados identificados nos dois contigs de BAC. Esses marcadores adicionais na Tabela 2, foram usados para identificar contigs de BAC a partir de uma biblioteca de B73 a qual fechou o gap físico entre os previamente encontrados contigs de BAC Mo17-derivados (Coe et al. (2002) acima citado; Gardiner (2004) acima citado; Yim et al. (2002) acima citado. Quatro marcadores, MZA11455, MZA6064, MZA2591 e MZA15842, foram usados para finalidades de mapeamento. Na Tabela 2, “E” significa “externo” e “I” significa “interno”, os quais, respectivamente, se referem aos primers externo e interno usados durante o PCR nested. O conjunto externo é usado na primeira rodada de PCR, após a qual as sequências internas são usadas para uma segunda rodada de PCR nos produtos da primeira rodada. Isto aumenta a especificidade da reação. As letras em caixa-alta indicam porções do primer baseadas em sequências de vetor, as quais são mais tarde usadas para sequenciar os produtos de PCR. Elas não são sequências de milho. Para os primers de MZA nested internos forward, a porção em caixa-alta da sequência é a SEQ ID NO:126, e para os primers de MZA nested internos reversos, a porção em caixa-alta é a SEQ ID NO:127. As sequências mostradas na Tabela 2 para os primers de MZA nested internos forward são, portanto, uma combinação da SEQ ID NO:126 mais a SEQ ID NO: para cada respectivo primer. De modo semelhante, as sequências mostradas na Tabela 2 para os primers nested de MZA internos reversos são uma combinação da SEQ ID NO: 127 mais a SEQ ID NO: para cada respectivo primer. Essas combinações estão indicadas na coluna de SEQ ID NO: da Tabela 2. A temperatura de anelamento para todos os primers listados na Tabela 2 é de 55°C. Todos os marcadores apresentados na Tabela 2 mostraram polimorfismo dentro de um painel diverso de germoplasma de milho, incluindo a MP305 e as linhagens de milho mostradas na Tabela 18.
[0187] As sequências das extremidades de diversos desses BACs, assim como os ESTs conhecidos a serem localizados nesses BACs, foram usados de modo a identificar novos marcadores com os quais é feito o posterior estreitamento de faixa na qual o lócus está localizado. Os marcadores adicionais usados para esta finalidade são designados FLP8, FLP27, FLP33, FLP41, FLP56 e FLP95 na Tabela 1. De forma similar àquela descrita acima, foram feitas as correlações fenotípicas e genotípicas. Foi determinado que o lócus muito provavelmente estava localizado entre o FLP 8 e o FLP 27 (ver Figura 3). Tabela 1: Marcadores e pares de primers usados nos Exemplos 1, 4 e 5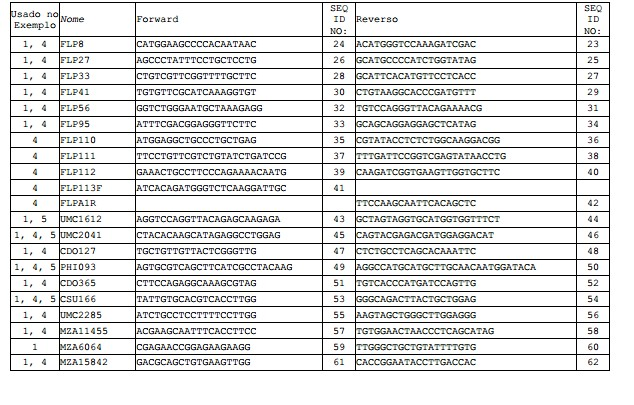
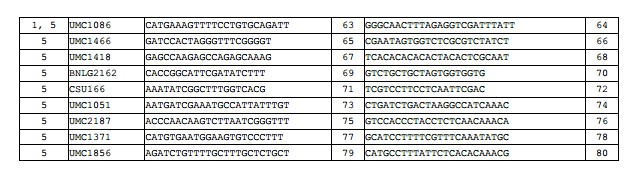 Tabela 2: Pares de Primers de MZA Nested Usados no Examplo 1
Tabela 2: Pares de Primers de MZA Nested Usados no Examplo 1 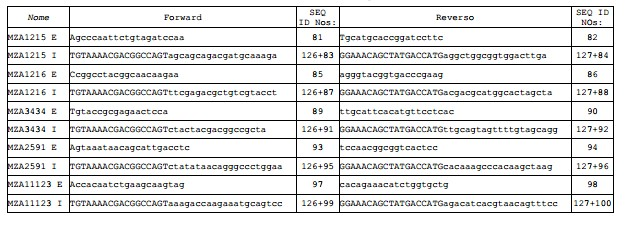

[0188] De maneira a isolar o gene responsável pelo fenótipo conferido pelo lócus de Rcg1, os BACs contendo a região entre os marcadores FLP 8 e FLP 27 foram isolados a partir de uma biblioteca BAC preparada a partir da linhagem resistente DE811ASR (BC5). Esta biblioteca foi preparada usando técnicas padrão para a preparação de DNA genômico (Zhang et al. (1995) Plant Journal 7:175-184) seguida pela digestão parcial com HindIII e ligação dos fragmentos de tamanho selecionado para uma forma modificada do vetor comercialmente disponível pCCIBACa (Epicentre, Madison, USA). Após a transformação em células de E. coli EPI300a seguindo as instruções do fornecedor (Epicentre, Madison, USA), 125.184 clones recombinantes foram dispostos em 326 placas de microtitulação de 384 poços. Esses clones foram então colocados em filtros de nylon (Hybond N+, Amersham Biosciences, Piscataway, USA).
[0189] A biblioteca foi sondada com sondas de oligonucleotídeos de sobreposição (sondas overgo; Ross et al. (1999) Screening large-insert libraries by hybridization, p. 5.6.1-5.6.52, In A. Boyl, ed. Current Protocols in Human Genetics. Wiley, New York) desenhadas na base de sequências encontradas nas sequências BAC mostradas no exemplo anterior para estarem presentes entre o FLP8 e o FLP27. Foi feita análise de busca BLAST para submeter a screening as sequências repetidas e identificar as sequências únicas para o desenho da sonda. A posição e o inter-espaçamento das sondas ao longo do contig foi verificada por PCR. Para cada sonda, foram desenhados dois oligos de 24-meros auto-complementares sobre 8 pb. Seu anelamento resultou em um overgo de 40 pb, no qual os dois overhangs de 16 pb foram preenchidos. As sondas usadas desta maneira são apresentadas na Tabela 4. Observe-se que algumas dessas sondas foram baseadas em marcadores também usados no Exemplo 1 e Tabela 1, mas as sequências exatas são diferentes na medida em que elas foram produzidas para serem usadas como sondas overgo ao invés de somente primers de PCR. As sondas para hibridização foram preparadas como descrito (Ross et al. (1999) acima citado), e os filtros preparados pela colocação em rede da biblioteca BAC foram hibridizados como descrito por (Ross et al. (1999) acima citado). Foi usada a análise de Phosphorimager para a detecção de sinais de hibridização. Depois disso, as membranas foram separadas das sondas pela colocação das mesmas em uma solução recém-fervida de 0,1X SSC e 0,1% SDS e deixadas a resfriar até a temperatura ambiente na solução durante a noite.
[0190] Os BACs que deram um sinal positivo foram isolados a partir das placas. Foram usados o mapeamento de restrição, os experimentos de PCR com primers correspondentes aos marcadores previamente usados e as sequências obtidas a partir das extremidades de cada BAC para determinar a ordem dos BACs que cobrem a região de interesse. Quatro BACs que se espalharam pela região inteira foram selecionados para sequenciamento. Esses BACs foram sequenciados usando técnicas de sequenciamento do tipo shotgun padrão e as sequências foram montadas usando o Phred/Phrap/Consed software package (Ewing et al. (1998) Genome Research, 8:175-185).
[0191] Após a montagem, as sequências consideradas como estando na região mais próxima do lócus com base nos dados de mapeamento foram anotadas, significando que foram deduzidas as possíveis regiões codificantes de gene e as regiões representando elementos repetitivos. As regiões codificantes de genes (regiões gênicas) foram buscadas usando o fGenesH software package (Softberry, Mount Kisco, New York, USA). O fGenesH permitiu a predição de uma porção de uma proteína que, quando submetida ao BLAST (BLASTx/nr), mostrou parcial homologia ao nível de aminoácido com relação a uma porção de uma proteína de arroz que foi verificada como codificante de uma proteína que confere resistência, em arroz, à doença. A porção da sequência de milho que apresentou homologia com relação a esta proteína foi localizada como estando na extremidade de uma extensão contígua da sequência consenso de BAC e pareceu ser truncada. De modo a obter a representação completa do gene no BAC de milho, foi usada a sequência de aminoácidos de arroz em uma análise de tBLASTn contra todas as outras sequências consenso oriundas do mesmo clone de BAC de milho. Isto resultou na identificação de uma sequência consenso representando a extremidade 3' do gene de milho. Entretanto, a porção central do gene não foi representada nas sequências assim obtidas. Os primers de PCR foram desenhados com base nas regiões 5' e 3' do gene putativo e usados em um experimento de PCR com DNA do BAC de milho original como um template. A sequência do produto de PCR resultante continha a sequência que faz a ponte dos fragmentos 5' e 3' previamente isolados.
[0192] A DE811ASR (BC5) foi depositada junto à ATCC, e os métodos aqui descritos podem ser usados para obter o clone de BAC compreendendo o lócus de Rcg1. Como mostrado na Figura 9(a), o intervalo cromossômico de DE811ASR (BC5) com o lócus de Rcg1 é não-colinear com relação à região correspondente de B73 e Mo17 (ver Figuras 9 e 22), como determinado pela análise de bibliotecas de BAC.
[0193] Usando a sequência comum que hibridiza com BACs nas bibliotecas de BAC da Mo17 e da B73, os correspondentes BACs oriundos de ambas as bibliotecas foram dispostas em uma rota tracejada como mostrado na Figura 22. Foram dados nomes mais curtos aos BACs de B73 na Figura 22 para finalidade de ilustração. A Tabela 3, a seguir, mostra a ID de BAC para cada designação indicada na Figura 22. Os BACs de B73 disponíveis ao público, c0113f01 e c0117e18 são diretamente norte e sul, respectivamente, da região indel do lócus de Rcg1, com a deleção ocorrendo na B73. A informação desses dois BACs pode ser vista em diversos sítios da Web, incluindo o website de GDB de milho (maizegdb.org), o website Gramene (gramene.org) e a base de dados do genoma de milho do Arizona Genomics Institute (genome.arizona.edu). O website do Arizona Genomics Institute também provê o Mapa FPC de Agarose de Milho, versão 19 de julho de 2005, o qual identifica os BACs contíguos em relação a c0113f01 e c0117e18. Pela busca nessas bases de dados, foi identificada uma multidão de BACs que formam um contig das regiões flanqueadoras do lócus de Rcg1. Portanto, a localização precisa do lócus de Rcg1 e do gene Rcg1 foi agora identificada em ambos os mapas físico e genético de milho (ver Figuras 7(a,b) e 22). Tabela 3: Designações de BAC na Figura 22, as quais foram parte tanto do contig 187 (B73a até B73p) como do contig 188 (B73q até B73af) do B73 como mostrado no website do Arizona Genomics Institute acima mencionado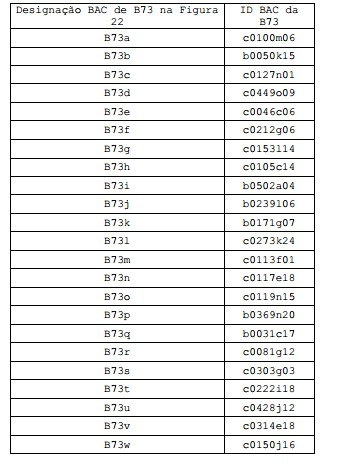
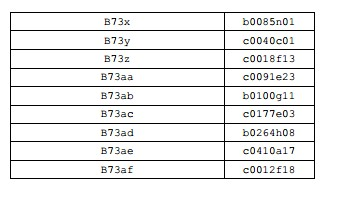
[0194] A sequência completa do gene putativo é apresentada na SEQ ID NO: 1. O gene contém um intron, desde o nucleotídeo 950 até o nucleotídeo 1452 da SEQ ID NO: 1. Foi usada PCR de tanscriptase reversa utilizando o RNA preparado a partir de plantas da DE811ASR (BC5) para determinar os limites do intron. A sequência codificante da proteína do gene é apresentada na SEQ ID NO: 2, e a tradução em aminoácidos é apresentada na SEQ ID NO: 3. A proteína predita possui um peso molecular de 110,76 kD.
[0195] A extremidade amino a partir aproximadamente dos aminoácidos 157 até 404 tem homologia com relação aos chamados sítios de ligação a nucleotídeo (NBS). Há uma região com homologia vaga com relação aos domínios de LRR localizados aproximadamente a partir dos aminoácidos 528 até 846. Entretanto, diferentemente das proteínas de NBS-LRR anteriormente estudadas, a região rica em leucina carece de natureza repetitiva sistemática (Lxx) encontrada em domínios de LRR mais clássicos e, em particular, sem circunstâncias das sequências consenso descritas por Wang et al. ((1999), Plant J. 19:55-64) ou Bryan et al. ((2000), Plant Cell 12:2033-2045). Como mostrado na Figura 2 (a, b e c), o gene possui vaga homologia com relação à família dos genes de arroz e com relação a um gene de cevada. A maior parte da homologia está na extremidade amino terminal da proteína; a extremidade carboxil é bastante distinta. Isto é demonstrado pelo uso do estilo negrito, na Figura 2 (a, b e c), os quais são aminoácidos idênticos ao gene das concretizações, enquanto que aqueles não idênticos não estão mostrados em negrito. Tabela 4. Oligonucleotídeos anelados para sintetizar sondas Overgo
[0196] Tendo encontrado um gene candidato na região geneticamente definida para portar o lócus responsável pelo fenótipo de resistência à antracnose, foram feitos esforços para primeiramente determinar se haveriam outros genes presentes na região e, em segundo lugar para determinar se os padrões de expressão do gene candidato eram consistentes com o seu papel putativo. Fu e Dooner ((2002), Proc Natl Acad Sci 99:9573-9578) e Brunner et al. ((2005), Plant Cell 17:343-360) demonstraram que diferentes linhagens endogâmicas de milho podem ter significativos rearranjos e ausência de colinearidade entre si. A comparação de tais genomas ao longo das regiões maiores pode, portanto, ser complexa. Tal comparação dos genomas de Mo17 (Missouri 17) e DE811ASR (BC5) revelou que, na região onde o gene candidato é encontrado na DE811ASR (BC5), está presente uma grande inserção com relação à Mo17. As regiões dentro e nas vizinhanças da inserção foram sequenciadas e escaneadas com relação a possíveis genes. Um gene codificante de uma subunidade de Ribulose bisfosfato carboxilase (Rubisco, uma proteína envolvida na fixação de carbono após a fotossíntese, cujo gene está presente em múltiplas cópias no genoma de milho) foi encontrado em ambos os genomas, da DE811ASR (BC5) e da Mo17, exatamente a jusante da posição do gene Rcg1. Foi encontrado um pseudo-gene (um gene tornado não-funcional devido a mutações que rompem a sequência codificante) relacionado com uma proteína de reserva vegetativa, presente somente no genoma de DE811ASR (BC5) a alguma distância a montante do gene Rcg1. O único gene estruturalmente intacto provavelmente para codificar uma proteína com uma função possivelmente relacionada com resistência a doença foi o gene Rcg1 isolado no exemplo anterior. Do mesmo modo, foram localizados outros genes improváveis de estarem envolvidos com a resistência à doença a uma maior distância da posição mais provável do lócus, assim como um grande número de sequências repetitivas.
[0197] Para determinar se um gene Rcg1 foi transcrito, e quando, foram usadas duas técnicas. Primeiramente foram examinados os perfis de RNA de materiais de planta resistente e susceptível usando o Sequenciamento de Assinatura Massivamente Paralela (Massively Parallel Signature Sequencing - MPSS; Lynx Therapeutics, Berkeley, USA). Resumidamente, bibliotecas de cDNA foram construídas e imobilizadas em microcontas como descrito em Brenner et al. (Brenner, S. et al. (2000) Nat. Biotechnol. 18(6): 630-634). A construção da biblioteca sobre um suporte sólido permite que a biblioteca seja disposta em uma monocamada e milhares de clones sejam submetidos à análise de sequência de nucleotídeos em paralelo. A análise resulta em uma sequência de “assinatura” de 17-meros cuja frequência de ocorrência é proporcional à abundância daquele transcripto no tecido da planta. O cDNA derivado de RNA preparado a partir da DE811ASR(BC5) e da DE811 (linhagem controle, susceptível a Cg) foi submetida a análise de MPSS. A inspeção por bioinformática das assinaturas resultantes mostrou que a sequência de assinatura, aqui referida como Lynx19, (SEQ ID NO: 19) estava presente em 43 partes por milhão (ppm) nas amostras de RNA oriundas de colmos não- infectados de DE811ASR (BC5) e em 65 ppm em colmos resistentes infectados 9 dias após a inoculação (DPI) com Cg. Esta sequência de assinatura não foi detectada em bibliotecas de cDNA de colmos não-infectados ou infectados por Cg da linhagem de milho susceptível DE811. Uma análise da sequência de Rcg1 indica que o marcador (tag) de 17-meros está presente nos nucleotídeos 3945 a 3961 da SEQ ID NO: 1 na região não- traduzida 3' putativa do gene.
[0198] A prova adicional de que o Rcg1 é exclusivamente expressado em linhagens de milho que são derivadas da MP305 e resistentes à podridão do colmo por antracnose foi obtida pelos experimentos de RT-PCR. O RNA total foi isolado a partir de colmos não-infectado e infectado por Cg de linhagens de milho, resistente (DE811ASR1 (BC5)) e susceptível (DE811), usando RNA STAT-60TM (Iso-Tex Diagnostics, Friendswood, Texas, USA). O RNA total (250 ng) de amostras resistente e susceptível de 0, 3, 6, 9, e 13 DPI foi copiado para o cDNA e amplificado usando um kit GeneAmp® RNA-PCR (Applied Biosystems, Foster City, California, USA). A reação de síntese de cDNA foi montada de acordo com o protocolo do kit usando hexâmeros aleatórios como primers e incubada a 42°C por 45 minutos. Para realizar a PCR, foram usados o KEB131 (SEQ ID NO: 20) e o KEB138 (SEQ ID NO: 21), ambos desenhados a partir da sequência não-traduzida 3' putativa do Rcg1, como os primers a montante e a jusante, respectivamente. O cDNA foi amplificado por 30 ciclos consistindo de 1 minuto a 94°C, 2 minutos a 50°C e 3 minutos a 72°C seguido por uma extensão de 7 minutos a 72°C. Como mostrado na Figura 4, a eletroforese em gel de agarose de uma alíquota dos RT-PCRs revelou a presença de uma banda de 260 pb presente nas amostras derivadas de ambas as plantas resistentes, a infectada e a não-infectada, mas ausente nas amostras susceptíveis. A análise de sequência de DNA confirmou o fato de que este fragmento correspondia ao nt 3625 até 3884 da sequência de Rcg1 consistente com o produto de amplificação predito a partir dos primers KEB131 e KEB138.
[0199] Um método para determinar se um gene é responsável por um fenótipo, é romper geneticamente o gene através da inserção de um elemento de transposição (a chamada marcação por transposon) e depois determinar se o fenótipo relevante da planta está alterado, neste caso, de resistente a Cg para susceptível a Cg. Em milho, isto pode ser feito usando o elemento mutator (Mu) (Walbot, V. (1992) Annu. Rev. Plant Physiol. Plant Mol. Biol. 43:49-82). A estratégia básica, esboçada na Figura 5, foi a de introduzir elementos mutator ativos em linhagens portadoras do gene de resistência, isolando as plantas homozigotas com relação ao gene de resistência através da análise de marcadores de DNA associados, assim como de resistência a Cg pela inoculação com Cg, depois cruzando aquelas plantas homozigotas com uma linhagem susceptível de “teste”. Se o gene de resistência for dominante, em princípio, toda a progênie resultante deve ser resistente, mas heterozigota com relação ao gene. Entretanto, se um elemento Mu inserido no gene resistente em um modo que rompe sua função, aquele indivíduo deve ser susceptível a Cg. O gene rompido pode depois ser isolado e caracterizado.
[0200] A MP305 foi cruzada com quinze diversos estoques mutator (linhagens portadoras de elementos mutator ativos). As F1s resultantes foram inter-casadas (cruzadas uma com a outra) em todas as combinações possíveis. Para rastrear a região cromossômica 4L na qual foi descoberto que o lócus de resistência reside (ver Exemplo 1), uma variedade de marcadores de DNA conhecidos como estando nas vizinhanças do lócus, a partir do trabalho descrito no Exemplo 1, foi selecionada e usada nas materiais marcados com Mu. Cerca de 1500 plantas da progênie oriundas do processo de inter- cruzamento foram examinadas com relação à resistência a Cg e com relação à presença desses marcadores. A análise dos marcadores foi feita usando tanto Southern blots (Botstein et al., (1980) Am. J. Hum. Gen. 32:314-331) para marcadores de RFLP como por PCR para marcadores de FLP como descrito no Exemplo 1. As plantas que foram homozigotas para todos os marcadores testados e resistentes a Cg foram selecionadas e submetidas a teste de cruzamento com linhagens de teste susceptíveis (A63, EH6WA e EF09B). Cerca de 16.000 sementes de teste de cruzamento geradas a partir dessas plantas homozigotas e resistentes foram depois plantadas e foram usadas como parentais femininos (significando que os pendões produtores de pólen foram removidos) e cruzadas com as linhagens de teste susceptíveis usadas como masculinas. Todas as plantas femininas foram submetidas a screening com relação à susceptibilidade a Cg. Mais de dez plantas susceptíveis (mutantes knockout putativas) foram identificadas. A semente polinizada aberta de cada uma dessas plantas susceptíveis foi colhida, juntamente com oito irmãs resistentes como controle.
[0201] Foi extraído o DNA de um conjunto de 24 mudas (crescidas em toalhas de papel) de cada um dos knockouts putativos e das irmãs resistentes controle. Este DNA foi usado como tamplate para a amplificação da sequência flanqueadora oriunda do sítio de inserção-de-Mu usando primers gene- específicos em combinação com um primer consenso desenhado a partir de repetições invertidas terminais (TIR) oriundas da sequência do elemento Mutator (SEQ ID NO: 125). Em outras palavras, os produtos de PCR devem somente ser observados se um elemento Mu tiver sido inserido no gene candidate isolado no Exemplo 2. Os primers FLP110F, FLP110R, FLP111F, FLP111R, FLP112F, FLP112R, FLP113F, e FLPA1R foram usados como primers gene-específicos (ver Tabela 1). Os produtos amplificados por PCR foram submetidos a blotting sobre membranas e hibridizados com uma sonda de DNA a partir do gene candidato isolado no Exemplo 2. Os produtos de PCR que mostraram forte hibridização foram excisados a partir do gel, purificados, clonados e sequenciados. As sequências resultantes foram analisadas por meio de alinhamento com sequências oriundas do gene candidato e Mu-TIR. Os elementos Mutator causam uma duplicação direta de 9 pb e o sítio de inserção. Com base na informação da sequência flanqueadora e em uma duplicação direta de 9 pb, foram identificadas quatro inserções independentes no exon 1 do gene candidato (Figura 5). Uma inserção (m177) foi detectada aproximadamente 97 pb a montante do códon de iniciação, na região não-traduzida 5' do gene. Um evento de inserção comum, 270 pb a jusante do códon de iniciação, foi detectado em três plantas susceptíveis: m164, m159 e m179. Foi descoberto que a planta susceptível m171 contém duas inserções-Mu, 556 pb e 286 pb a jusante do códon de iniciação. Quando foram realizados Southern blots usando a região do exon1 do gene como uma sonda de DNA, o padrão de hibridização modificado observado posteriormente confirmou esses resultados.
[0202] Este e os exemplos precedentes podem ser resumidos como se segue. O trabalho inicial citado no Exemplo 1 mostrou que um lócus previamente observado como conferindo resistência a Cg foi localizado no ramo longo do cromossomo 4 de milho. A natureza deste lócus, sua exata localização ou gene(s) codificado(s) por ele eram completamente desconhecidos. O trabalho feito no Exemplo 1 demonstra que o lócus pode ser mapeado para uma região muito pequena do ramo longo do cromossomo 4. O Exemplo 2 demonstra que há somente um gene a ser encontrado nesta região cromossômica provavelmente sendo dito gene de resistência. Ele codifica uma nova forma de uma proteína NBS-LRR, uma família de proteínas conhecidas por estarem envolvidas na resistência a patógenos, mas que variam amplamente em sua sequência e especificidade de resistência. O Exemplo 3 mostra que este gene está presente somente na linhagem de resistência, e não na linhagem susceptível isogênica, e que os transcriptos correspondentes a este gene são encontrados na linhagem resistente, indicando que o gene é expressado, e que esses transcriptos são encontrados somente na linhagem resistente. O Exemplo 4 demonstra que, nos quatro eventos independentemente isolados de inserção Mu, quando o gene é rompido pela inserção de um elemento Mu, o fenótipo dessas plantas é mudado de resistente para susceptível a Cg. Tomados em conjunto, esses dados fornecem evidência decisiva de que o objeto das concretizações desta invenção é um gene que pode melhorar ou conferir resistência a Cg em plantas de milho.
[0203] Foi feita a introgressão de um lócus de Rcg1 em uma endogâmica para confirmar o fato de que o lócus de Rcg1 pode ser retrocruzado com sucesso em endogâmicas, e que os híbridos produzidos com a linhagem endogâmica com o lócus de Rcg1 deve ter resistência a Cg melhorada ou conferida. A DE811ASR (BC5) também foi desenvolvida e usada como uma fonte doadora aperfeiçoada para a introgressão do lócus de Rcg1. Em seguida, diversas endogâmicas adicionais foram utilizadas como parentais recorrentes de modo a usar métodos, aqui descritos, de melhoramento assistido por marcador para introgredir eficientemente o lócus de Rcg1 em uma diversidade de backgrounds genéticos endogâmicos e híbridos, melhorando ou conferindo, por esse meio, resistência a Cg. Cada um desses exemplos é discutido em maiores detalhes a seguir.
[0204] A MP305 é uma linhagem endogâmica de cor de grão branca com forte resistência a Cg, mas seu florescimento tardio, baixa produtividade e fracas características agronômicas a tornam uma parental doadora pobre na ausência do uso dos métodos aqui descritos de melhoramento assistidos por marcador. Um perfil de marcador molecular da MP305 é provido na Tabela 6. Os primers usados para as SSRs relatadas na tabela podem ser construídos a partir de sequências disponíveis ao público encontradas no GDB de Milho na World Wide Web em maizegdb.org (patrocinado pelo USDA Agricultural Research Service), em Sharopova et al. (Plant Mol. Biol. 48(5- 6):463-481), e/ou em Lee et al. (Plant Mol. Biol. 48(5-6); 453-461). Um UMC15a é um marcador de RFLP, e o escore relatado é baseado no sítio de restrição EcoR1.
[0205] Para demonstrar o valor fenotípico do lócus de Rcg1, o lócus foi primeiramente introgredido na linhagem PH09B (patente US 5.859.354) até o estágio BC3 como se segue. A população F1 derivada do cruzamento entre a MP305 e a linhagem PH09B foi retrocruzada uma vez mais com a linhagem PH09B, resultando em uma população BC1. As mudas foram plantadas e retrocruzadas novamente com a linhagem PH09B para desenvolver uma população BC2. O DNA foi preparado a partir de punções de folha das famílias BC2. Para determinar quais famílias BC2 plantar para posteriores retrocruzamentos, foi realizada genotipagem no DNA das famílias BC2 usando ou primers para marcadores flanqueadores da região de interesse, UMC2041, PHI093 e CSU166 (ver Tabela 1). As sementes oriundas da família BC2 foram plantadas e as plantas individuais foram genotipadas contra a presença da versão MP305 daquela região do cromossomo usando os mesmos três marcadores acima anotados. As plantas positivas foram retrocruzadas com a linhagem PH09B uma vez mais para desenvolver as populações BC3. As sementes dessas populações BC3 foram plantadas e as plantas foram autofecundadas para obter famílias BC3S1 segregantes para a região de interesse, assim como as famílias BC3S1 faltando a região de interesse. Essas famílias foram usadas para a comparação fenotípica (BC3S1 segregantes versus BC3S1 sem a região de interesse).
[0206] De forma a observar o desempenho do gene Rcg1, em uma situação heterozigota, tal como seria encontrada em um híbrido comercial, foram feitos cruzamentos de teste apropriados. Especificamente, famílias BC3S1 segregantes para a região de interesse foram plantadas e plantas individuais BC3S1 foram genotipadas. Foram usadas as plantas homozigotas para o gene Rcg1, assim como as plantas homozigotas para o alelo nulo (faltando o gene em ambos os cromossomos) dentro de cada família, para realizar os cruzamentos de teste com as endogâmicas PH2EJ (patente US 6.333.453), PH2NO (patente US 6.124.533), PH4CV (patente US 6.897.363) e PH8CW (patente US 6.784.349).
[0207] No caso de ambas as linhagens BC3S1 e os híbridos, as diferenças fenotípicas observadas indicaram significativo aperfeiçoamento com relação à resistência de ASR em linhagens e híbridos contendo a região portadora de Rcg1. O efeito do lócus de Rcg1 introgredido nas famílias BC3S1 e os híbridos de cruzamento de teste derivados resultou em um aperfeiçoamento em termos de ambos, no número de internódulos infectados e no número de internódulos infectados em mais do que 75%. Os escores usando um sistema de contagem de escore visual comumente usado pelos melhoristas de planta são mostrados na Tabela 5 mais adiante. Os dados nitidamente demonstram que o uso de técnicas de cruzamento para transportar o gene das concretizações para outras linhagens geneticamente competentes em usar o gene resulta em resistência melhorada a Cg. Tabela 5: Efeito da região Rcg1 introgredida no grau de resistência à podridão do colmo por antracnose em famílias BC3S1 e cruzamentos de teste derivados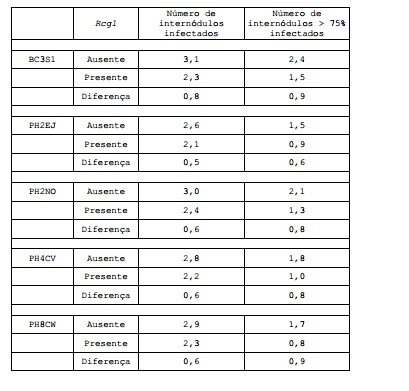 Tabela 6: Perfil de marcador molecular de MP305
Tabela 6: Perfil de marcador molecular de MP305 
[0208] Apesar de a MP305 ter sido utilizada no experimento acima, como está ilustrado na Figura 8(a), a DE811ASR(BC5) retém um intervalo cromossômico da MP305 menor com o lócus de Rcg1 do que a DE811ASR(BC3) (e, é claro, também a MP305), e, portanto, é particularmente utilizável como uma fonte doadora do gene Rcg1. Foi mostrado que o intervalo cromossômico estreitado oriundo a fonte DE811ASR(BC5) está associado com um fenótipo agronômico aperfeiçoado. Vinte e duas plantas oriundas da linhagem derivada DE811ASR(BC3), 20 plantas oriundas da linhagem derivada the DE811ASR(BC5), cinco plantas DE811 e cinco plantas MP305 foram crescidas em uma casa de vegetação de novembro de 2005 até março de 2006 e foram recolhidos dados com relação à altura da planta e altura da espiga; foram observadas as datas quando 50% das plantas soltaram pólen (midshed), quando 50% das plantas tinham um visual de brotos de espiga (midves) e quando 50% das plantas tinham os estilo-estigmas apontando nas brotações de espiga (midslk); e a cor do grão. Na media, a linhagem DE811ASR(BC5) foi mais curta que a DE811ASR(BC3) (293 cm versus 345 cm) e a localização da espiga foi mais baixa na DE811ASR(BC5) do que na DE811ASR(BC3) (146 cm versus 183 cm), os quais são caracteres positivos em termos de desenvolvimento de variedade elite. A DE811ASR(BC5) foi mais precoce com relação a midshed, midves e midslk quando comparada com a DE811ASR(BC3). A característica de midshed foi aproximadamente 1 dia mais cedo, a midves foi aproximadamente 6 dias mais cedo e a midslk foi aproximadamente 3 dias mais cedo para a DE811ASR(BC5) quando comparada à DE811ASR(BC3). Os grãos da DE811ASR(BC5) tiveram uma cor marrom-amarelada (bronze) enquanto que os grãos de DE811ASR(BC3) tiveram uma capa amarelo pálida. As datas para midshed, midves e midslk foram similares para a DE811ASR(BC5) e para a DE811, enquanto que a MP305 foi aproximadamente 11 dias mais tarde para midshed e não produziu 50% de brotos de espiga visíveis, nem 50% de estilo-estigmas durante o período de crescimento. Apesar de que esses dados são baseados somente em umas poucas plantas para a DE811 e para a MP305, e as espigas não foram produzidas naquelas poucas linhagens, esses resultados de casa de vegetação se assemelham com observações dessas linhagens no campo. Esses dados indicam que a DE811ASR(BC5) se assemelha à parental recorrente DE811 muito mais proximamente do que a DE811ASR(BC3). Portanto, a DE811ASR(BC5) é uma excelente fonte doadora inicial do lócus de Rcg1 e do gene Rcg1, em ambos os aspectos, genotipicamente e fenotipicamente. Adicionalmente, a DE811ASR(BC5) é particularmente utilizável quando da introgressão do lócus de Rcg1 no germoplasma com adaptação semelhante à DE811.
[0209] A DE811 foi desenvolvida por J. Hawk (Hawk, J.A. (1985). Crop Science Vol 25: p716) e foi descrita como uma linhagem endogâmica dentada amarela originada de autofecundação e seleção de seis gerações em um programa de pedigree escolhidas dentre aquelas de um cruzamento de B68 com uma endogâmica derivada de [B37 Ht X (duplo cruzamento de C103 X Mp3204) sel.]. A DE811 desenvolveu o estilo-estigma de 1 a 2 dias mais tarde do que a B73 em testes em Delaware, mas 4 dias mais tarde do que a B73 em Missouri. Os ensaios limitados de produtividade indicaram que a DE811 possui satisfatória capacidade de combinação. É uma boa formadora de estilo- estigma (forma bons estilo-estigmas, um componente da flor fêmea de milho importante para a fertilização) e liberadora de pólen e pode ser cruzada com germoplasma de maturidade precoce para adaptação ao nordeste dos Estados Unidos e para germoplasma de maturidade mais tardia para adaptação ao sudeste dos Estados Unidos. Portanto, a DE811ASR (BC5), em combinação com os marcadores e métodos de melhoramento aqui revelados, é utilizável como uma fonte doadora inicial para introgressão do gene Rcg1 em uma ampla diversidade de germoplasma, incluindo o germoplasma adaptado a todas as regiões dos Estados Unidos onde o Cg está presente.
[0210] Seguindo-se aos testes para a introgressão bem- sucedida de lócus de Rcg1 em PH09B descrita acima, foram realizadas conversões de lócus de Rcg1 adicionais em outras linhagens endogâmicas. A primeira série tinha 5 retrocruzamentos, sendo a MP305 e a DE811ASR(BC5) as doadoras. Para a segunda série de retrocruzamentos, foram usados marcadores moleculares para reduzir o intervalo cromossômico nas conversões de BC5 oriundas da primeira série. Essas conversões de BC5 foram selecionadas para permutas (crossovers) abaixo do gene Rcg1. Aquelas plantas selecionadas foram depois retrocruzadas para criar a geração BC6. As plantas com crossovers acima do gene foram selecionadas na geração BC6.
[0211] Na primeira série, foi usada a DE811ASR(BC5) como a primeira fonte doadora, mas introgressões paralelas também foram feitas para as mesmas endogâmicas usando a MP305 como uma fonte doadora. Esses dados, descritos em maiores detalhes mais adiante, mostram que enquanto a DE811ASR(BC5) é a doadora preferida em muitas situações, a MP305 também pode ser eficazmente usada com os métodos de melhoramento assistido com marcador das concretizações ensinadas aqui.
[0212] As linhagens endogâmicas elite primariamente adaptadas às condições de crescimento norte-americanas foram selecionadas para uso como parentais recorrentes. As linhagens endogâmicas inicialmente selecionadas para uso como parentais recorrentes foram as linhagens PH0R8 (US 6.717.036), PH7CH (US 6.730.835), PH705 (US 6.903.25), PH5W4 (US 6.717.040), PH51K (US 6.881.881) e PH87P (US 6.888.051). Cada uma dessas linhagens foi cruzada com a DE811ASR (BC5) assim como com a MP305. A geração F1 derivada de cada um desses cruzamentos foi retrocruzada uma vez com a respectiva linhagem endogâmica, resultando em uma primeira geração de retrocruzamento (a parental recorrente BC1). As mudas foram plantadas e o DNA foi preparado a partir de punções de folha. As reações de PCR foram realizadas usando primers para marcadores que flanqueiam a região de interesse; foram usados UMC1466, UMC1418, BNLG2162, UMC1086, UMC2041, UMC1612, CSU166, UMC1051, UMC2187, UMC1371, e UMC1856 nas primeiras rodadas de BC (ver Tabela 1) enquanto que, nas últimas rodadas de BC, foram usados UMC1418, BNLG2162, UMC1051, UMC2041, UMC2187, UMC1371 e UMC1856. As mudas cujas reações de PCR deram um resultado positivo (significando que o lócus de Rcg1 derivado de MP305 estava presente) foram depois adicionalmente retrocruzadas com as respectivas linhagens endogâmicas para produzir uma BC2. Este procedimento, chamado “genotipagem”, identifica a composição genética de uma planta no sítio de um marcador particular. Essas etapas foram repetidas para o desenvolvimento das parentais recorrentes BC3, BC4 e BC5. A análise mostra que, após cinco retrocruzamentos, essas linhagens retinham um intervalo cromossômico significativamente truncado compreendendo o lócus de Rcg1, e, com base em observações visuais, não foi observada nenhuma indicação de efeitos negativos resultantes da presença do lócus de Rcg1.
[0213] A seleção de parental recorrente também foi realizada pela seleção das plantas fenotipicamente mais parecidas com a parental recorrente. Usando esses métodos genotípicos e fenotípicos, foram selecionadas conversões de alta qualidade com uma alta percentagem de parental recorrente ao longo de todo o genoma.
[0214] Este exemplo também ilustra o fato de que os marcadores flanqueadores não são usados exclusivamente para selecionar tanto dentro como fora do gene Rcg1. As mudas cujas reações de PCR deram um resultado positivo (significando que o lócus de Rcg1 derivado da MP305 estava presente) foram depois adicionalmente retrocruzadas com as respectivas linhagens endogâmicas para produzir o retrocruzamento final (a geração BC5 parental recorrente) nesta primeira série. Quando os marcadores polimórficos flanqueadores mais próximos determinaram que o gene estava presente, foi usado o próximo conjunto de marcadores polimórficos flanqueadores duplos mais distais com relação ao gene para a seleção do parental recorrente. Portanto, o uso de marcadores que flanqueiam o gene Rcg1 ou o lócus do Rcg1 serve para clarear a recombinação que ocorre na região.
[0215] As conversões do lócus de Rcg1 endogâmicas feitas usando as SSRs que flanqueiam o lócus de Rcg1 na primeira série de retrocruzamento foram depois usadas como doadoras em uma rodada sucessiva de retrocruzamentos. Para esta primeira série de retrocruzamentos, foram desenvolvidos marcadores de SNP para o gene Rcg1 que possibilita a seleção assistida com marcador em um modo de alta produção, como descrito no Exemplo 13, para selecionar com relação ao gene Rcg1. Os marcadores de SNP também foram desenhados na região em torno do lócus de Rcg1, permitindo que os marcadores flanqueadores sejam usados para selecionar fora do intervalo cromossômico de MP305 que rodeia o lócus de Rcg1, e para selecionar o genótipo parental recorrente, reduzindo, por esse meio, grandemente o arraste de ligação. É somente através do mapeamento físico e clonagem do gene que é possível tal seleção precisa de parental recorrente assistida com marcador.
[0216] Primeiramente, as plantas BC5 parentais recorrentes resultantes da primeira série de retrocruzamentos foram mais uma vez submetidas a screening com um conjunto de marcador mais preciso, e a recombinação foi selecionada para o sul do gene Rcg1. Os marcadores flanqueadores firmemente ligados ao gene Rcg1 (MZA8761, MZA1851, UMC1051, e UMC2187) foram usados para selecionar o parental recorrente para o sul do gene em pequenos tamanhos de população de aproximadamente 40 progênies (ver Figura 8(a-b)). Essas progênies foram depois analisadas usando os marcadores de FLP aqui revelados, para mais precisamente determinar o ponto de recombinação. Estes dados mostraram que foram selecionadas algumas progênies com genoma parental recorrente de menos que 1 cM (com base nas distâncias de mapa genético Neighbors IBM2) do sul do gene Rcg1, como mostrado na Figura 8(b). Outra progênie tinha genoma parental recorrente de menos que 4 cM do sul do gene Rcg1. Essas conversões de BC5 selecionadas com marcador foram depois usadas como doadores, e cruzadas com contra-partes aproximadamente isogênicas de PH705, PH5W4, PH51K e PH87P como as parentais recorrentes para dar a população BC6. Os marcadores no gene Rcg1 foram novamente usados para selecionar com relação ao Rcg1, com marcadores flanqueadores com relação ao norte do Rcg1, desta vez sendo usado para selecionar o parental recorrente. Nesta rodada de seleções, as recombinações foram detectadas em cada uma das populações entre o Rcg1 e o marcador MZA15842. A posição do MZA15842 no mapa genético Neighbors IBM2 pode ser extrapolada a partir da sua posição no mapa de alta resolução mostrado na Figura 7(b), mapa B, usando regressão com relação aos marcadores flanqueadores UMC2285 e PHI093. Isto colocou o MZA15842 em 520,5 cM no mapa genético Neighbors IBM2. Portanto, como mostrado na Figura 8(b), em duas rodadas de retrocruzamento, o genoma doador foi reduzido a um segmento de menos que 6 cM em cada população, ou menos que 0,8% do cromossomo 4, com base nas distâncias do mapa genético Neighbors IBM2, e em algumas progênies o segmento foi de menos que 2,1 cM, ou menos que 0,25% do cromossomo 4. Para fins de comparação, o intervalo cromossômico de MP305 com o lócus de Rcg1 em DE811ASR (BC3) foi de 131 cM, ou aproximadamente 16% do cromossomo 4, com base nas distâncias do mapa genético Neighbors IBM2. É somente através do mapeamento físico e clonagem do gene que é possível tal seleção precisa e eficiente de parental recorrente assistida com marcador.
[0217] Portanto, como resultado do mapeamento refinado da localização do gene Rcg1, pode-se utilizar quaisquer dois marcadores flanqueadores que são geneticamente ligados com o gene Rcg1 para selecionar uma pequena região cromossômica com crossovers ao norte e ao sul do gene Rcg1. Isto traz como benefício a redução do arraste de ligação, o qual pode ser um fator de confusão quando se tenta introgredir um gene específico oriundo de germoplasma não adaptado, tal como a MP305, para um germoplasma elite, tal como as linhagens endogâmicas observadas acima. As Figuras 7 e 22 e a Tabela 16 mostram muitas combinações de marcadores que flanqueiam o gene e o lócus de Rcg1 que podem ser usadas para este propósito. Alguns marcadores flanqueadores específicos que podem ser usados para selecionar intervalos cromossômicos truncados que incluem o gene ou o lócus de Rcg1 são UMC2285 e UMC15a, UMC2285 e UMC2187, UMC1086 e UMC2200, UMC2041 e UMC2200, UMC2041 e PHI093, MZA11455 e UMC15a, MZA11455 e MZA3434, MZA15842 e MZA3434, e FLP8 e FLP33. Opcionalmente, em ou dentro de cada um desses intervalos cromossômicos, pode-se utilizar pelo menos 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 ou mais marcadores de modo a localizar o evento de recombinação e selecionar o gene Rcg1 ou o lócus de Rcg1 com a máxima quantidade de genótipo parental recorrente. Adicionalmente, pode-se ter pelo menos 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, ou mais marcadores entre a extremidade norte de tal intervalo cromossômico e o topo do gene Rcg1 e/ou entre a extremidade sul de tal intervalo cromossômico e o fundo do gene Rcg1.
[0218] É vantajoso ter marcadores flanqueadores estreitamente ligados para a seleção de um gene, e é altamente vantajoso ter marcadores dentro do próprio gene. Isto é um aperfeiçoamento sobre o uso de um único marcador ou de marcadores flanqueadores distantes, na medida em que, com um único marcador ou com marcadores flanqueadores distantes, a ligação associada com o Rcg1 pode ser quebrada e, por seleção de tais marcadores é mais provável que ocorra a seleção inadvertidamente de plantas sem o gene Rcg1. Na medida em que a seleção assistida com marcador é frequentemente usada, ao invés da seleção fenotípica, uma vez que a associação marcador-caractere tenha sido confirmada, o resultado inauspicioso de tal erro pode ser a seleção de plantas que não são resistentes a Cg e descartar plantas que são resistentes a Cg. Neste aspecto, os marcadores dentro do gene Rcg1 são particularmente utilizáveis, já que eles, por definição, permanecerão ligados com resistência a Cg como melhorado ou conferido pelo gene. Além disso, os marcadores dentro do lócus de Rcg1 são justamente utilizáveis por uma razão similar. Devido à sua proximidade muito estreita com relação ao gene Rcg1, muito provavelmente eles permanecem ligados ao gene Rcg1. Uma vez introgredidas com o gene Rcg1, tais endogâmicas elite podem ser usadas para a produção de semente híbrida e como fonte doadora de posterior introgressão do gene Rcg1 em outras linhagens endogâmicas.
[0219] Portanto, os dados nitidamente mostram que a progênie endogâmica convertida pelo uso da DE811ASR(BC5) como uma fonte doadora retém o intervalo cromossômico truncado da MP305. As endogâmicas compreendendo o intervalo cromossômico truncado da MP305 são muito úteis como as próprias fontes doadoras e não há necessidade de reverter para a DE811ASR(BC5) como uma fonte doadora. Pelo uso de melhoramento assistido com marcador como acima descrito, o intervalo cromossômico truncado de MP305 pode ser ainda reduzido em tamanho quando for necessário sem preocupação de perder a ligação entre os marcadores e o gene Rcg1. Fenotipicamente, um intervalo cromossômico reduzido está associado com o desempenho agronômico aperfeiçoado, como foi demonstrado para a DE811ASR(BC5) versus a DE811ASR(BC3) acima descritas.
[0220] O gene Rcg1 também pode ser expressado como um transgene, permitindo a modulação de sua expressão em diferentes circunstâncias. Os exemplos a seguir mostram como o gene Rcg1 pode ser expressado em diferentes maneiras para combater diferentes doenças ou proteger diferentes porções da planta, ou simplesmente mover o gene Rcg1 para diferentes linhagens de milho como um transgene, como uma alternativa para o método descrito no Exemplo 5.
[0221] Neste exemplo, o gene Rcg1 é expressado usando seu próprio promotor. A região a montante do gene Rcg1 foi sequenciada usando os mesmos BACs que, no Exemplo 2, forneceram as sequências da seção codificante de proteína do gene. A sequência 5' de 1684 pb até o ATG está apresentada na SEQ ID NO: 24. De modo a transformar o gene Rcg1 completo, incluindo o promotor e a região codificante de proteína, um fragmento de 5910 pb que se estende desde a posição 41268 até a posição 47176 na SEQ ID NO: 137 foi amplificado por PCR usando o clone de BAC #24 (pk257m7) como template de DNA. Para possibilitar a clonagem usando a Tecnologia Gateway® (Invitrogen, Carlsbad, USA), sítios attB foram incorporados em primers de PCR, e o produto amplificado foi clonado no vetor pDONR221 por reação de recombinação BP Gatewayâ. O fragmento resultante, flanqueado por sítios attL, foi transportado pela reação de recombinação LR Gatewayâ em um vetor binário. O DNA do constructo foi depois usado para a transformação de milho como descrito no Exemplo 7.
[0222] De modo a expressar o gene Rcg1 através da planta em um baixo nível, a região codificante do gene e seu terminador são colocados atrás dos promotores tanto do gene de actina de arroz (patentes US 5.641.876 e US 5.684.239) como do gene F3.7 (patente US 5.850.018). Para possibilitar a clonagem usando a Tecnologia Gateway® (Invitrogen, Carlsbad, USA), sítios attB são incorporados nos primers de PCR que são usados para amplificar o gene Rcg1 começando 35 pb a montante do seu códon de iniciação. É adicionado um sítio NotI ao primer attB1. O produto de Rcg1 amplificado é clonado no vetor pDONR221 pela reação de recombinação BP Gatewayâ (Invitrogen, Carlsbad, USA). Após a clonagem, o gene Rcg1 resultante é flanqueado por sítios attL e possui um único sítio NotI no 35 pb a montante do códon de iniciação. Depois disso, os fragmentos de promotor são amplificados por PCR usando primers que contêm sítios NotI. Cada promotor é fundido ao sítio NotI do Rcg1. Na etapa final, o gene quimérico é transportado pela reação de recombinação LR Gatewayâ (Invitrogen, Carlsbad, USA) para o vetor binário PHP20622. Isto é usado para a transformação de milho como descrito no Exemplo 7.
[0223] De modo a expressar o gene Rcg1 através da planta em um alto nível, a região codificante do gene e seu terminador foram colocados atrás do promotor da região 5' não-traduzida e de um intron de um gene de ubiquitina de milho (Christensen et al. (1989) Plant Mol. Biol. 12:619-632; Christensen et al. (1992) Plant Mol. Biol. 18:675-689). Para possibilitar a clonagem usando a Tecnologia Gateway® (Invitrogen, Carlsbad, USA), sítios attB foram incorporados nos primers de PCR que foram usados para amplificar o gene Rcg1 começando no pb 142 a montante do códon de iniciação. O produto de amplificação foi clonado no pDONR221 (Invitrogen, Carlsbad, USA) usando uma reação de recombinação BP Gatewayâ (Invitrogen, Carlsbad, USA). Após a clonagem, o gene Rcg1 resultante foi flanqueado por sítios attL. Na etapa final, o clone Rcg1 foi transportado por meio de uma reação de recombinação LR Gatewayâ (Invitrogen, Carlsbad, USA) para um vetor que continha o promotor de ubiquitina de milho, a região 5' não-traduzida e o primeiro intron do gene de ubiquitina como descrito por Christensen et al.(acima mencionado) seguido por sítios ATTR1 e R2 Gatewayâ para a inserção do gene Rcg1, atrás do cassete de expressão de ubiquitina. O vetor também continha um gene marcador apropriado para transformação de milho, de modo que o plasmídeo resultante, portanto o gene quimérico (promotor de ubiquitina de milho - região não-traduzida 5' de ubiquitina - intron 1 de ubiquitina - Rcgl), foi apropriado para a transformação de milho como descrito no Exemplo 7.
[0224] De modo a expressar o gene de Rcg1 sob condições de baixo nível de expressão colmo-preferida, a região codificante do gene e seu terminador são colocados atrás do promotor do gene Br2 (Pedido de Patente US No. de Série 10/931.077). O fragmento descrito no Exemplo 6b contendo a região codificante de Rcg1 flanqueada pelos sítios attL e contendo um sítio único NotI 35 pb a montante do códon de iniciação de Rcg1 é usado para possibilitar a clonagem usando a Tecnologia Gateway® (Invitrogen, Carlsbad, USA). Os fragmentos de promotor tanto do Br2 como do ZM-419 são amplificados por PCR usando primers que contêm sítios NotI. Cada promotor é fundido ao sítio NotI de Rcg1. Na etapa final, o constructo de gene quimérico é transportado por meio de reação de recombinação LR Gatewayâ (Invitrogen, Carlsbad, USA) para o vetor binário PHP20622. Este é usado para a transformação de milho como descrita no Exemplo 7.
[0225] Os constructos de DNA recombinante preparados no Exemplo 6a e 6c foram usados para preparar plantas transgênicas de milho como se segue.
[0226] O milho foi transformado com constructos de polinucleotídeo selecionados descritos no Exemplo 6a e 6c usando o método de Zhao (patente US 5.981.840, e Pedido de Patente PCT publicado WO98/32326). Resumidamente, embriões imaturos foram isolados a partir de milho e os embriões foram contatados com uma suspensão de Agrobacterium, na qual as bactérias foram capazes de transferir o constructo de polinucleotídeo a pelo menos uma célula de pelo menos um dos embriões imaturos (etapa 1: a etapa de infecção). Nesta etapa, os embriões imaturos foram imersos em uma suspensão de Agrobacterium para a iniciação da inoculação. Os embriões foram co-cultivados por um tempo com o Agrobacterium (etapa 2: a etapa de co-cultivo). Os embriões imaturos foram cultivados em meio sólido seguindo-se a etapa de infecção. A seguir deste período de co-cultivo, foi realizada uma etapa opcional de “descanso”. Nesta etapa de descnaso, os embriões foram incubados na presença de pelo menos um antibiótico conhecido por inibir o crescimento de Agrobacterium sem a adição de um agente de seleção para os transformantes de planta (etapa 3: etapa de descanso). Os embriões imaturos foram cultivados em meio sólido com antibiótico, mas sem um agente de seleção, para a eliminação do Agrobacterium e para uma fase de descanso das células infectadas. Em seguida, os embriões inoculados foram cultivados em meio contendo um agente de seleção, e o calo transformado crescido foi recuperado (etapa 4: a etapa de seleção). O calo foi então regenerado para plantas (etapa 5: a etapa de regeneração), e os calos crescidos no meio de seleção foram cultivados em meio sólido para regenerar as plantas.
[0227] As plantas transgênicas foram produzidas como descrito no Exemplo 7 usando constructos descritos nos Exemplos 6a e 6c, respectivamente. Para ambos os constructos, do gene Rcg1 natural e do gene Rcg1 de ubiquitina, foram gerados 30 eventos independentes e 10 eventos de controle de vetor somente.
[0228] Discos de folha de cada evento transgênico de gene natural foram colhidos para isolamento do RNA total. O RT-PCR foi realizado usando os primers específicos de gene FLP111F e FLP111R apresentados nas SEQ ID NOs: 37 e 38. Em 30 dos 30 eventos transgênicos, a banda de RT-PCR esperada de 637 pb estava presente, indicando a expressão do constructo de gene natural. Os ensaios de doença foram realizados na casa de vegetação nos mesmos 30 eventos transgênicos de Rcg1 natural para determinar se as plantas eram resistentes a Cg. Para realizar isto, primeiramente foram feitos os ensaios de queima das folhas em 5 plantas irmãs de cada evento usando os procedimentos descritos no Exemplo 10. Foi verificado que um único evento mostrou uma significativa redução na doença com relação às plantas controle nas quais está ausente o constructo do gene Rcg1 natural. As plantas que foram submetidas ao ensaio de queima de folha foram deixadas a desenvolver por duas semanas após a antese e, depois, adicionalmente testadas pela inoculação de Cg no interior do primeiro internódulo de colmo alongado. Esses ensaios de infecção de colmo mostraram um único evento transgênico que expressa o transgene Rcg1 natural como sendo mais resistente à infecção por Cg quando comparado com as plantas controle. Entretanto, este evento diferiu do evento positivo identificado via os ensaios de infecção de folha.
[0229] As plantas transformadas com o constructo de Rcg1 de ubiquitina descritos no Exemplo 6c foram analisadas de um modo semelhante. A análise de RT-PCR mostrou que 28 dos 30 eventos transgênicos continham a esperada banda de transcripto, indicando a expressão do constructo de Rcg1 de ubiquitina. Quando os ensaios de infecção de folha foram realizados em 5 plantas a partir de cada um dos 30 eventos, foi identificado um único evento que mostrou uma redução estatisticamente significativa na doença comparada com as plantas controle. As plantas transgênicas foram adicionalmente analisadas pelos ensaios de infecção de colmo. Foram encontrados três eventos como apresentando aumenta resistência à podridão do colmo quando comparados com as plantas controle nas quais está ausente o gene Rcg1 de ubiquitina. Esses eventos transgênicos não incluem o primeiro evento positivo identificado no ensaio de queima de folha.
[0230] Os resultados desses experimentos foram considerados encorajadores com relação aos eventos que mostraram alguma resistência, mas inconclusos, no geral, por diversas razões. Eventos positivos mostrando resistência aumentada à doença pelo ensaio de queima da folha falharam em correlacionar tais eventos com aqueles identificados pelo ensaio de infecção do colmo. Isto é o oposto do controle positivo DE811ASR(BC5) que mostra um nítido aumento de resistência com relação à DE811 em ambos os ensaios de queima de folha e de infecção de colmo. Adicionalmente, os ensaios dos tansgênicos primários mostraram um grau mais alto de variabilidade quando comparados com os ensaios de controles de DE811 ou DE811ASR(BC5). Isto foi frequentemente visto nas replicatas e também através dos eventos de controle negativo. Esta última observação pode tornar difícil a discriminação dos eventos positivos dos negativos. As causas possíveis para a natureza inconclusa dos resultados dos ensaios de doença incluem, mas não estão limitadas, às que se seguem. É bem conhecido dos especialistas na técnica que aquelas plantas transgênicas que são derivadas de cultura de tecido apresentam maior variabilidade de planta para planta com relação às plantas controle que são derivadas de semente. Além disso, a expressão em transformantes primários, ou seja, plantas que foram submetidas ao processo de transformação e regeneração como descrito no Exemplo 7, é frequentemente não previsível devido ao estresse dos procedimentos de cultura de tecido. Se, de fato, os eventos forem negativos, os quais não podem ser determinados neste ponto, há diversas razões técnicas para que isto ocorra. Os ensaios realizados também não determinaram se a proteína codificada pelo gene Rcg1 está realmente presente nas linhagens transgênicas - somente a presença de um segmento do mRNA predito foi analisado usando o RT-PCR. Pode ser que artefatos tenham sido introduzidos no cassete gênico durante a transformação - não foram realizados exaustivos Southern blots e sequenciamento para determinar a integridade do constructo completo nas linhagens transgênicas. De modo a se estudar mais apuradamente essas linhagens transgênicas, as plantas das últimas gerações serão crescidas em grandes quantidades sob condições de campo e analisadas com relação à resistência à doença. Pode ser antecipado que essas futuras plantas transgênicas apresentarão mais nitidamente resistência aumentada a Cg.
[0231] Em seguida à identificação, sequenciamento e mapeamento refinado de Rcg1, outras linhagens foram submetidas a screening com relação ao gene Rcg1. Para determinar a presença do gene Rcg1 em outro germoplasma de milho, foram usadas combinações dos primers específicos FLP111F e FLP111R assim como FLP113F e FLPA1R para amplificar o DNA genômico a partir de um painel diverso de linhagens endogâmicas de milho, incluindo aquelas listadas na Tabela 18 e a F2834T, por reação em cadeia da polimerase. Em somente 14 (incluindo a MP305), de um painel de linhagens endogâmicas de milho, foi detectado um produto de amplificação, indicando que o gene Rcg1 é o único presente em uma percentagem muito pequena das linhagens endogâmicas que foram submetidas a screening. Portanto, em adição ao uso da MP305 ou da DE811ASR (BC5) como fonte doadora, outras fontes contendo o gene Rcg1 também podem ser usadas como uma fonte doadora. Por exemplo, podem ser usadas as linhagens endogâmicas disponíveis ao público TX601 (disponível sob a ID ‘Ames 22763' a partir do National Plant Germplasm System (NPGS)) e F2834T (disponível sob a ID ’Ames 27112’ a partir do NPGS) as quais contêm o gene Rcg1 como fontes doadoras em cruzamentos com outras linhagens endogâmicas de milho que não contêm o gene Rcg1, e seleção com relação ao gene Rcg1 pelo uso de marcadores como aqui descrito.
[0232] Variantes do gene Rcg1 também foram identificadas e analisadas com relação a polimorfismos simples de nucleotídeos (SNPs). Os SNPs foram identificados em posições na Sequência ID número 1 correspondentes a uma ou mais das posições 413, 958, 971, 1099, 1154, 1235, 1250, 1308, 1607, 2001, 2598 e 3342 (ver Tabela 7). Nem todas as variantes alélicas do gene Rcg1 indicaram um fenótipo resistente. Consequentemente, esses SNPs podem ser usados como marcadores para identificar e rastrear precisamente a sequência de Rcg1 em um programa de melhoramento de planta e para distinguir entre variantes alélicas resistentes e susceptíveis. Além disso, esses SNPs indicam que há sequências que mostram o fenótipo resistente e que podem ser usadas em métodos e produtos aqui revelados. Também pode ser verificado que quatro outras linhagens contêm um alelo de Rcg1: BYD10, 7F11, CML261 e CML277. O teste realizado em 10 plantas não forneceu dados suficientes para determinar conclusivamente se a linhagem 7F11 é resistente. Não há dados disponíveis com relação à resistência das linhagens BYD10, CML261 e CML277, e o sequenciamento desses alelos não foi completado. Tabela 7: SNPs identificados em variantes alélicas do gene Rcg1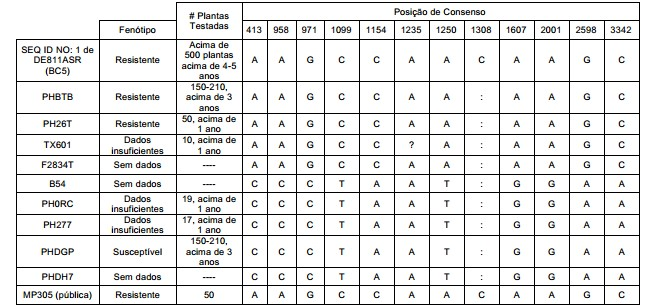 Comprimento da sequência Consenso = 4212 nucleotídeos. SEQ ID NO: 1 é a sequência de Rcg1. Para as linhagens restantes, a sequência disponível se expandiu desde o códon de início “atg” no primeiro exon até o códon de parada “tga” no segundo exon. A posição consenso é baseada na SEQ ID NO:1.
Comprimento da sequência Consenso = 4212 nucleotídeos. SEQ ID NO: 1 é a sequência de Rcg1. Para as linhagens restantes, a sequência disponível se expandiu desde o códon de início “atg” no primeiro exon até o códon de parada “tga” no segundo exon. A posição consenso é baseada na SEQ ID NO:1.
[0233] As linhagens aproximadamente isogênicas DE811 e DE811ASR descritas no Exemplo 1 foram testadas com relação às diferenças de resistência à queima de folha causada por Cg usando o procedimento a seguir. Quatro agulhas comuns de costura caseira foram coladas a um suporte de metal de modo que os orifícios para a linha se estendessem para fora da peça de metal, sendo que todas as quatro agulhas se estenderam por igual distância. Esse conjunto foi mergulhado em uma suspensão de esporos de Cg a 5 X 106 esporos/mL e depois empurrado através da superfície de uma folha jovem de milho de modo que a folha ficasse ferida e as feridas fossem simultaneamente inoculadas com os esporos. Uma mecha de algodão úmido foi colocada na nervura central perto do sítio de inoculação e a área inteira foi coberta com filme plástico e, sobre este, um pano refletor, ambos amarrados com uma faixa para mantê-la úmida e protegida. As plantas foram deixadas nesse estado por 50-54 horas em uma casa de vegetação padrão, após o que a faixa, o pano e o filme plástico foram removidos. Nos dias 7 e 15 após a inoculação, o tamanho da lesão foi medido e registrado em unidades de centímetros quadrados.
[0234] A Figura 10 (a-b) mostra a distribuição de tamanhos de lesão 15 dias após a inoculação ao longo do todo das folhas individuais. Os tamanhos de lesão variam em cada conjunto de dados, mas virtualmente todos os das folhas da DE811 (Figura 10b) tinham tamanhos de lesão significativamente maiores do que as maiores lesões a serem encontradas nas folhas da DE811ASR(BC5) (Figura 10a). Os dados estão resumidos para ambos os conjuntos de dados, os do dia 7 e os do dia 15 após a inoculação, como visto na Figura 11. Em ambos os dias 7 e 15, o tamanho médio de lesão foi menor nas folhas portadoras do gene Rcg1. A diferença se tornou maior ao longo do tempo, à medida que o fungo tivesse tempo para crescer e causar dano adicional, de modo que enquanto a diferença é de aproximadamente duas vezes nos dias 7, nos dias 15 ela é mais do que quatro vezes e, de fato, o fungo fez somente progresso mínimo nas folhas da DE811ASR(BC5). Esses resultados demonstram claramente que a presença do lócus contendo o gene Rcg1 confere resistência à queima de folha por antracnose.
[0235] De forma a demonstrar que as híbridas de milho contendo o gene Rcg1 possuem mais alto potencial de produtividade quando infectados com Cg em comparação com as linhagens híbridas sem Rcg1, a DE811ASR (BC5) e a DE811, as linhagens isogênicas descritas no Exemplo 1, foram, cada uma delas, cruzadas com as linhagens endogâmicas B73Ht e Mo17Ht, as quais são ambas susceptíveis a Cg.
[0236] As linhagens híbridas foram crescidas e avaliadas com relação à resposta a Cg em 2005, em seis localidades em cinco estados diferentes dos Estados Unidos da América. Para cada linhagem híbrida, foram plantadas três replicações de quatro fileiras a aproximadamente 74.000 plantas por hectare. As plantas foram inoculadas com Cg na base do colmo aproximadamente 10 dias após a florescência. A primeira fileira de cada parcela de quatro fileiras foi avaliada para verificar se as inoculações tinham sido bem sucedidas, através da determinação da resposta a Cg quatro a cinco semanas após a inoculação. Os colmos foram rachados e a progressão da doença foi contabilizada em escore por meio de observação da cor preta característica do fungo à medida que ele se espalha ascendentemente no colmo. Os índices de doença foram conduzidos como descrito por Jung et al. (1994) Theoretical and Applied Genetics, 89:413-418). O número total de internódulos descoloridos maior do que 75% (antgr75) foi registrado nos primeiros cinco internódulos (ver Figura 20). Isto forneceu um escore de doença variando de 0 a 5, sendo que zero indica sem internódulos que sejam mais do que 75% descoloridos e 5 indicando a completa descoloração dos primeiros cinco internódulos. As duas parcelas centrais foram colhidas na maturidade fisiológica através da máquina de ceifar e debulhar e foi determinada a produtividade de grão em kg/ha.
[0237] Os resultados resumidos oriundos de todas as localidades são mostrados na Figura 12 com relação à gravidade da doença e na Figura 13 com relação à produtividade. Os dados mostram que os híbridos contendo o Rcg1 (DE811ASR(BC5)/B73Ht e DE811ASR(BC5)/Mo17Ht) possuem muito menos progresso de doença do que os híbridos sem o Rcg1 (DE811/B73Ht e DE811/Mo17Ht). Os altos escores relativos ao progresso da doença nos híbridos susceptíveis (ausência do Rcg1) mostram a infecção bem sucedida do experimento com Cg. Além disso, os dados mostram que, quando infectados com Cg, os híbridos contendo Rcg1 possuem uma produtividade maior do que os híbridos com ausência do Rcg1. As diferenças das comparações em pares individuais são significantes em P<0,05.
[0238] Esses resultados demonstram claramente que, por meio do uso dos métodos das concretizações, podem ser criados híbridos que produzam mais kg de grão por hectare quando infectados com Cg.
[0239] De modo a demonstrar que as linhagens comerciais de milho podem ser tornadas resistentes a podridão do colmo induzida por Cg, as MP305 e DE811ASR (BC5) foram cruzadas com PH87P, PH5W4, e PH705. A progênie resultante foi cruzada novamente com as três linhagens (ou seja, as linhagens foram usadas como parentais recorrentes em um programa de retrocruzamento) mais três vezes, a cada vez sendo feita a seleção com relação à presença do gene Rcg1 usando marcadores moleculares, como descrito no Exemplo 5 acima. Como controles, foram também coletadas linhagens selecionadas de retrocruzamento nas quais estava ausente o gene Rcg1 a partir do mesmo programa de retrocruzamento. Após terem sido completados três retrocruzamentos, diversas versões foram selecionadas e autofecundadas para obter as famílias BC3S1. Plantas individuais de BC3S1 foram genotipadas e as plantas positivas homozigotas e negativas homozigotas com relação ao Rcg1 foram autofecundadas para obter famílias BC3S2, as quais foram depois fenotipadas. As versões de BC3S2 contendo Rcg1 e, como controles, versões selecionadas sem o gene, foram plantadas em parcelas de fileiras simples contendo aproximadamente 25 plantas por fileira. O experimento foi reproduzido em cinco diferentes localidades em cinco diferentes estados dos Estados Unidos, designadas Localidades 1, 2, 3, 4 e 5. A aproximadamente duas semanas após a florescência, as plantas foram inoculadas com Cg na base do colmo. Quatro a cinco semanas mais tarde os colmos foram rachados e foi avaliado o progresso da doença por estimativa visual da quantidade de doença no colmo. Um escore visual foi assinalado para cada colmo com base no grau de infecção de cada internódulo com relação ao internódulo infectado e os quatro internódulos acima do internódulo de inoculação. Portanto, um baixo escore indica resistência à doença. Os resultados compilados para todas as fileiras e localidades estão resumidos na Figura 14. Imagens representativas de duas linhagens são mostradas nas Figuras 15 e 16. Os dados mostram que em todas as localidades, cada uma das linhagens endogâmicas elite foi tornada mais resistente à doença pela presença do gene Rcg1.
[0240] A semente de milho vendida aos agricultores é “híbrida”, indicando que ela é mais comumente o resultado de um cruzamento de duas parentais endogâmicas, referidas como um híbrido de cruzamento simples. Muitos anos de melhoramento e experiência de produção mostraram que o uso de híbridos de cruzamento simples resulta em produtividades mais altas. Portanto, é importante, para aplicações comerciais, que o gene Rcg1 funcione nas plantas híbridas (aquelas do campo de produção do agricultor), mesmo que esteja presente em somente um dos dois parentais usado para produzir a semente híbrida de cruzamento simples. Uma das linhagens endogâmicas com a qual a linhagem de Rcg1 tenha sido cruzada, a PH705, foi assim usada para criar a semente híbrida por cruzamento com a PH4CV, uma endogâmica elite que não porta o gene Rcg1. As sementes híbridas resultantes foram usadas em experimentos idênticos àqueles descritos para as linhagens endogâmicas como discutido acima e contabilizado em escore da mesma maneira em todas as cinco localidades. Os dados estão resumidos para todas as localidades na Figura 17, a qual também mostra o desempenho da endogâmica PH705, e imagens representativas mostradas nas Figuras 18 e 19. Como pode ser visto, uma nítida diferença no progresso da doença foi observado em todas as localidades para o híbrido PH705 x PH5W4 e em quatro das cinco localidades para o PH705 x PH87P. Na quinta localidade, as condições ambientais foram muito estressantes para o crescimento da planta, resultando em plantas que estavam em fraca condição. Sob essas condições, as medidas de resistência da planta à doença frequentemente não são confiáveis.
[0241] Os resultados com ambas as linhagens, a endogâmica e combinações híbridas contendo Rcg1, nitidamente demonstram que, usando os métodos das concretizações, pode-se criar linhagens utilizáveis comercialmente, as quais são resistentes a podridão do colmo induzida por Cg.
[0242] Três níveis de localização de marcador podem ser utilizados como o resultado de um mapeamento refinado e clonagem do gene Rcg1, marcadores desenhados dentro da sequência codificante de Rcg1, marcadores desenhados dentro da região não-colinear que identifica o lócus de Rcg1 (mas fora da sequência codificante de Rcg1) e marcadores desenhados dentro da região colinear flanqueadora.
[0243] Seguindo-se à identificação e ao mapeamento refinado do gene Rcg1, foram desenhados marcadores de hibridização que funcionarão em plataformas de SNP. Na medida em que o gene Rcg1 ocorre em uma região não co-linear do genoma de milho, os marcadores de hibridização estarão presentes em linhagens compreendendo o gene Rcg1 e ausentes em linhagens que não compreendem o gene Rcg1. Esses marcadores identificam sequências de polinucleotídeo específicas para a sequência codificante de Rcg1 listada na SEQ ID NO: 1. Como observado na Tabela 7, há outras linhagens de milho com variantes da sequência codificante de Rcg1 apresentada na SEQ ID NO: 1, e esses marcadores também foram desenhados para também identificar essas variantes de sequência codificante de Rcg1.
[0244] Para realizar isso, foi criado um mapa consenso da sequência codificante de Rcg1 a partir de diferentes fontes, como mostrado na Tabela 7. Este mapa consenso alinhou 4209 bases da sequência codificante de Rcg1 isolada da MP305 com 3451 bases oriundas da PHBTB e 3457 bases oriundas da PH26T. O gene Rcg1 em ambas, PHBTB e PH26T, mostrou resistência à antracnose. Em seguida, os segmentos da sequência codificante de Rcg1 foram submetidos ao BLAST contra diversas bases de dados incluindo a NT (DNA Público a partir do NCBI) e os mais altos sucessos de homologia foram alinhados com a sequência consenso de Rcg1 para determinar os segmentos que compartilhavam alta homologia e tinham segmentos comuns com outros genes de resistência na família NBS-LRR. As regiões únicas para a sequência codificante de Rcg1 e comuns entre as diferentes fontes de Rcg1 foram selecionadas para o desenho do marcador. Especificamente, na medida em que os primers FLP111F e FLP111R produziram um único amplicon que confiavelmente diagnosticou a presença de Rcg1 oriundo de diferentes fontes, as regiões em que FLP111F e FLP111R hibridizaram foram, portanto, tomadas como alvo para o desenvolvimento de um desenho de marcador de SNP.
[0245] Um marcador InvaderTM (Third Wave Technologies, Madison, WI) foi desenhado usando um segmento de 1413pb a partir da sequência consenso que continha ambos os sítios do primer, sendo que as regiões do primer em si foram tornadas alvo com relação à sonda e hibridização do oligo do InvaderTM. Os primers foram desenhados em torno de cada sítio de sonda para dar um tamanho de amplicon abaixo de 150 pb. Este marcador indicou a presença da sequência codificante de Rcg1 com fluorescência devida à hibridização, sendo que a ausência da sequência codificante de Rcg1 resultou em não- fluorescência. Um sinal de fluorescência controle também pode ser gerado pelo desenho de um marcador que hibridiza com um segundo gene de milho altamente conservado, de modo que a presença da sequência codificante de Rcg1 resulta em fluorescência de dois corantes (Rcg1 e o gene conservado) e a ausência de Rcg1 resulta em fluorescência devida ao gene conservado somente. Esta fluorescência ‘controle’ pode ser usada para reduzir o erro laboratorial por meio da distinção entre as situações em que o Rcg1 está de fato ausente e a situação em que um falso negativo tenha ocorrido por causa de uma reação falha. Tais marcadores não estão limitados a uma plataforma de detecção de marcador específico. Os marcadores Taqman® (Applied Biosystems) também foram desenhados para a mesma localização (par de primers FLP111F e FLP111R), que foram usados da mesma meneira que para os marcadores InvaderTM. Os marcadores estão mostrados na Tabela 15 e Figuras 23 e 24.
[0246] Os desenhos de marcador C00060-01-A e C00060-02-A foram testados para uma ampla variedade de fontes e foram altamente bem sucedidos em identificar plantas que continham o lócus de Rcg1 e o gene Rcg1, independentemente da fonte do lócus de Rcg1 ou do gene Rcg1. Esses marcadores também foram usados contra um conjunto controle de perto de 100 linhagens endogâmicas diversas conhecidas como não portadoras do gene, e nenhuma fluorescência foi detectada no conjunto controle. As plantas em que um ou ambos os desenhos de marcador, C00060-01- A e C00060-02-A, confirmadas como tendo o Rcg1 incluem aquelas mostradas na Tabela 7.
[0247] Portanto, este exemplo mostra que, com base no ensinamento aqui provido, podem ser construídos marcadores que identificam a sequência codificante de Rcg1 em uma diversidade de fontes.
[0248] Podem ser desenhados marcadores para o lócus de Rcg1 em adição a ou ao invés de usar marcadores dentro da própria sequência codificante de Rcg1. A distância física próxima entre a sequência codificante de Rcg1 e a região não-colinear torna improvável o fato de que a ligação entre marcadores dentro da região não-colinear, mas fora da sequência codificante de Rcg1, seja perdida através de recombinação. Da mesma forma que com marcadores para a sequência codificante de Rcg1, um marcador mostrando como presente ou ausente seria suficiente para identificar o lócus de Rcg1.
[0249] Para desenhar marcadores para esta região, foi sequenciado um segmento de 64.460 pb da região não-colinear incluindo o gene Rcg1 e a região diretamente ao norte do gene Rcg1. Os BACs nesta região foram rompidos em sub-clones com comprimento de aproximadamente 800 nucleotídeos e sequenciados. Estas sequências foram depois montadas para construir a sequência BAC, e as regiões gênicas e repetitivas foram identificadas. As regiões repetitivas foram identificadas de modo a evitar a colocação de marcadores em regiões repetitivas. Semelhantemente, as sequências com alta homologia com relação a sequências de milho conhecidas foram facilmente evitadas por meio de uma busca simples de BLAST. Foram evitadas as sequências potenciais que contivessem SSRs, corridas de As, Ts ou Gs, ou que resultassem na geração de sondas de baixo teor de GC que podem causar problemas dentro da plataforma Invader™ (ver Figura 9(b) e Tabela 17).
[0250] Os segmentos selecionados foram depois colocados no software Invader Creator™ (Third Wave, Madison, WI), o qual gera oligos para uma reação Invader™. Isto produziu um desenho sense e um antisense para todos os SNPs. Foram selecionados os desenhos sense com os melhores escores e sem penalidades. Apesar de esses marcadores terem sido desenhados, eles ainda não foram testados.
[0251] Foram desenhados primers usando o Primer3 (Steve Rozen e Helen J. Skaletsky (2000) Primer3 disponível na Web para usuários em geral e para programadores em biologia. In: Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press, Totowa, NJ, pp 365-386). Os primers foram selecionados fora dos components Invader™, e foram selecionados os primers preferidos perto de ou abaixo do comprimento de 150 pb. A temperatura e o comprimento do primer foram ajustados para ser o mais utilizável pela plataforma Invader™, apesar de que, se plataformas de detecção, os primers
[0252] Marcadores proximamente ligados flanqueando o lócus de Rcg1 podem ser eficazmente usados para selecionar uma planta de progênie que tenha herdado o lócus de Rcg1 de uma parental que compreende o lócus de Rcg1. Os marcadores aqui descritos, tais como aqueles listados na Tabela 16, assim como outros marcadores geneticamente ou fisicamente mapeados com relação ao mesmo segmento cromossômico, podem ser usados para selecionar um segmento cromossômico truncado compreendendo o lócus de Rcg1. Tipicamente, um conjunto desses marcadores será usado (por exemplo, 2 ou mais, 3 ou mais, 4 ou mais, 5 ou mais) na região flanqueadora acima do gene e conjunto semelhante na região flanqueadora abaixo do gene. Opcionalmente, como descrito acima, um marcador dentro do gene Rcg1 e/ou do lócus de Rcg1 também pode ser usado. Os parentais e sua progênie foram submetidos a screening com relação a esses conjuntos de marcadores, e os marcadores que são polimórficos entre os dois parentais são usados para seleção. Os marcadores polimórficos mais próximos do gene Rcg1 ou do lócus de Rcg1 são usados para selecionar com relação ao gene ou ao lócus. Em um programa de introgressão, isto permite a seleção do genótipo do gene Rcg1 ou do lócus de Rcg1 nos marcadores polimórficos mais próximos, e seleção com relação ao genótipo do parental recorrente nos marcadores polimórficos mais distantes. Como descrito em maiores detalhes no Exemplo 5 acima, este processo permitiu a seleção eficiente de um segmento cromossômico truncado compreendendo o lócus de Rcg1.
[0253] O processo descrito acima requer conhecimento dos genótipos dos parentais usados no cruzamento. Opcionalmente, podem ser usados haplótipos de maneira que o gene Rcg1 ou o lócus de Rcg1 possa ser selecionado sem primeiramente genotipar os parentais específicos usados no cruzamento. Esta é uma maneira altamente eficiente de selecionar o lócus de Rcg1, especialmente na ausência de uso de marcadores dentro do gene Rcg1 ou do lócus de Rcg1.
[0254] Todas as plantas a serem usadas no programa de melhoramento, tal como um programa de introgressão de gene, são submetidas a screening com marcadores. Podem ser usados os marcadores aqui revelados ou marcadores equivalentes no mesmo segmento cromossômico. Os haplótipos de planta (uma série de SNP ou outros marcadores em desequilíbrio de ligação) são observados. O haplótipo da planta resistente em torno do lócus de Rcg1 é comparado com o haplótipo das outras plantas a serem usadas que não compreendem o lócus de Rcg1. Um haplótipo único com relação à planta resistente em torno do lócus de Rcg1 é então usado para seleção, e este haplótipo identificará especificamente o segmento cromossômico oriundo da planta resistente com o lócus de Rgc1.
[0255] Com base em uma análise da MP305 e um conjunto diverso de várias centenas de linhagens de milho, incluindo as 50 linhagens de milho públicas mostradas na Tabela 18, foi identificado um único haplótipo de SNP para o segmento cromossômico da MP305 com o lócus de Rcg1. Este haplótipo de SNP identifica de modo único o segmento cromossômico da MP305 que se estende ao longo de MZA3434, MZA2591 e MZA11123. Ver Figura 22, SEQ ID NO: 140, 141 e 142, e Tabelas 8, 9 e 10.
[0256] Primeiramente, os pares de primer denscritos na Tabela 2 para os três MZA’s foram usados para identificar os haplótipos. Os pares de primers MZA3434 E forward e reverse foram usados para amplificar o DNA genômico do conjunto de linhagens de milho. Os fragmentos de PCR foram posteriormente purificados por amplificação com os pares de primers MZA3434 I forward e reverse. Este processo foi repetido para a MZA2591 e a MZA11123. Os fragmentos de PCR resultants foram sequenciados na direção forward e reverse e as sequências foram alinhadas para dar uma sequência consenso (ver as sequências apresentadas em SEQ ID NOs: 140, 141 e 142). Os SNPs e indels dentro dessas sequências consenso são mostrados nas Tabelas 8, 9 e 10. Essas séries de SNPs e indels foram comparados com o conjunto de genótipos.
[0257] Para a MZA3434, o haplótipo 8 foi um raro alelo de haplótipo e foi o único para a MP305 e para somente uma outra linhagem de milho. Este processo foi repetido para a MZA2591, e foi verificado que a MP305 possui o haplótipo 2 em MZA2591, o qual foi compartilhado por somente duas outras linhagens de milho. A MP305 foi a única linhagem de milho a possuir ambos, o haplótipo 8 em MZA3434 e o haplótipo 2 em MZA2591 e, portanto, a combinação desses dois haplótipos, 8 em MZA3434 e 2 em MZA2591, identifica de modo único a região cromossômica da MP305 compreendendo o lócus de Rcg1. A MP305 também tinha um haplótipo informativo em MZA11123. Foi verificado que a MP305 possui o haplótipo 7, que era compartilhado com 66 outras linhagens de milho, mas nenhuma dessas linhagens de milho tinha o haplótipo 8 em MZA3434, ou haplótipo 2 em MZA2591. Portanto, qualquer combinação de 2 haplótipos em MZA3434, MZA2591 ou MZA11123 pode ser usado para identificar de modo único a MP305 dentre esses genótipos. Os haplótipos podem depois ser interrogados por sequenciamento do fragmento ou pelo desenho de marcadores para cada SNP ou indel dentro de um fragmento.
[0258] Os polimorfismos dentro dos haplótipos podem ser usados para marcar o haplótipo. Os chamados ‘Tag-SNPs’, ou ‘haplótipo-tags’ podem ser muito úteis no melhoramento de plantas, na medida em que pode ser determinada mais informação do que o polimorfismo em si via extrapolação para o haplótipo. Um haplótipo pode ser definido como uma série de polimorfismos ao longo das sequências e eles podem ser denomonados ‘haplótipos de longa-faixa’.
[0259] Foram observados raros polimorfismos dentro de haplótipos que devem ser usados como ‘marcadores (tags) de haplótipo’. Por exemplo, tanto os SNPs MZA2591.32 (alelo c) ou MZA2591.35 (alelo t) podem ser usados para marcar o haplótipo 2 em MZA2591, e da mesma maneira que o haplótipo 2, ambos foram únicos para a MP305 e duas outras linhagens de milho. A combinação dos SNPs MZA2591.32 (alelo c) e MZA2591.35 (alelo t) combinada com o MZA3434.17 (alelo c) deu um haplótipo de ‘longa-faixa’ que pode ser usado para distinguir a MP305 de todos os outros genótipos no estudo.
[0260] Adicionalmente, outros marcadores, MZA15842, MZA11455, MZA8761 e MZA1851 também mostraram polimorfismo com a MP305. Para o MZA15842, somente 18 das outras linhagens de milho compartilharam haplótipo igual ao da MP305; para o MZA11455, somente 43 das outras linhagens de milho compartilharam haplótipo igual ao da MP305; para o MZA8761, somente cerca de metade das outras linhagens de milho compartilhou haplótipo igual ao da MP305; e para o MZA1851, somente cerca de metade das outras linhagens de milho compartilhou haplótipo igual ao da MP305. Foram desenvolvidas sequências consenso para esses marcadores que estão apresentadas nas SEQ ID NOs: 143-146. Os SNPs e indels dentro de três sequências consenso estão mostrados nas Tabelas 11-14. Quatro exemplos de haplótipos únicos usando os marcadores de MZA são: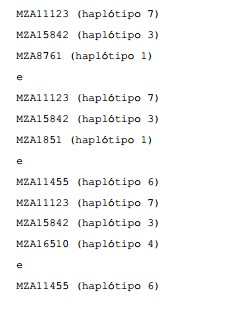

[0261] Múltiplas combinações no seio de todos os marcadores aqui revelados, ou outros marcadores dentro da região também conterão haplótipos únicos que identificam o lócus de Rcg1. Tabela 8: Polimorfismos de MZA3434 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso Tabela 9: Polimorfismos de MZA2591 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 9: Polimorfismos de MZA2591 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso Tabela 9 (cont.): Polimorfismos de MZA2591 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 9 (cont.): Polimorfismos de MZA2591 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso Tabela 10: Polimorfismos de MZA11123 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 10: Polimorfismos de MZA11123 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso 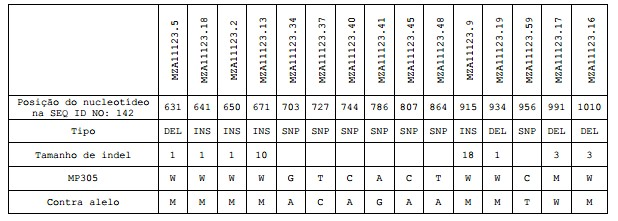 Tabela 11: Polimorfismos de MZA15842 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 11: Polimorfismos de MZA15842 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso 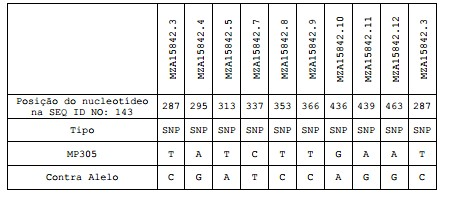 Tabela 12: Polimorfismos de MZA8761 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 12: Polimorfismos de MZA8761 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso  Tabela 13: Polimorfismos de MZA1851 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 13: Polimorfismos de MZA1851 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso  Tabela 14: Polimorfismos de MZA11455 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso
Tabela 14: Polimorfismos de MZA11455 M=‘Mutante’: difere do consenso; W=‘tipo selvagem’: igual ao consenso 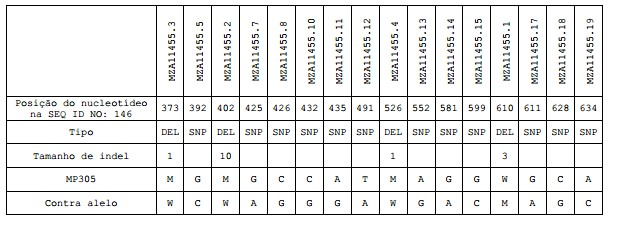 Tabela 15: Marcadores dentro da Sequência Codificante de Rcg1
Tabela 15: Marcadores dentro da Sequência Codificante de Rcg1 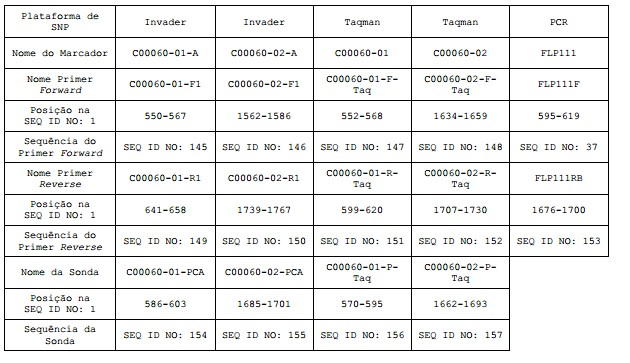 Tabela 16: Marcadores contidos dentro dos intervalos cromossômicos definidos que podem ser usados para selecionar o Rcg1
Tabela 16: Marcadores contidos dentro dos intervalos cromossômicos definidos que podem ser usados para selecionar o Rcg1
[0262] Os marcadores públicos são retirados do mapa 4 neighbors IBM2, enquanto que as localizações relativas dos marcadores Pioneer (prefixo ‘MZA’) foram determinados por meio de mapeamento com relação ao mesmo mapa genético e por meio da localização no mapa físico.
 Tabela 17: Marcadores dentro do Lócus de Rcg1
Tabela 17: Marcadores dentro do Lócus de Rcg1 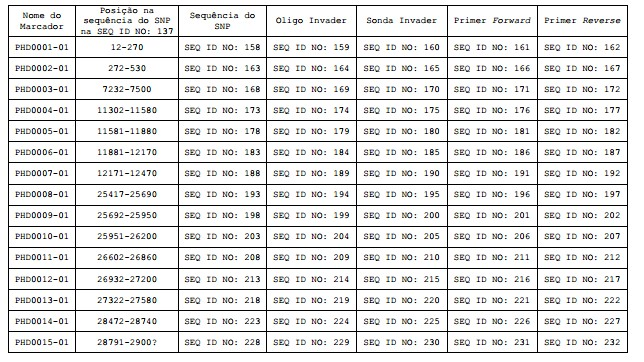 Tabela 18: Lista de Linhagens Públicas usadas em Análise de Haplótipos
Tabela 18: Lista de Linhagens Públicas usadas em Análise de Haplótipos 
Claims (34)
1. Vetor, caracterizado pelo fato de que compreende um polinucleotídeo selecionado do grupo consistindo em: (a) sequência de nucleotídeos que compreende a SEQ ID NO: 1 ou 2; e (b) um complemento da sequência de nucleotídeos de (a), sendo que o complemento e a sequência de nucleotídeos consistem no mesmo número de nucleotídeos e são 100% complementares, em que o referido polinucleotídeo está operacionalmente ligado a pelo menos uma sequência de regulação que é heteróloga em relação ao referido polinucleotídeo.
2. Constructo de DNA recombinante, caracterizado pelo fato de que compreende o vetor conforme definido na reivindicação 1.
3. Processo para transformar uma célula hospedeira, caracterizado pelo fato de que compreende a transformação da célula hospedeira com o vetor conforme definido na reivindicação 1 ou o constructo de DNA recombinante conforme definido na reivindicação 2.
4. Processo de acordo com a reivindicação 3, caracterizado pelo fato de que a célula hospedeira é uma célula de planta.
5. Processo para produzir uma planta, caracterizado pelo fato de que compreende a transformação de uma célula de planta com o constructo de DNA recombinante conforme definido na reivindicação 2 e a regeneração de uma planta a partir da célula da planta transformada.
6. Processo de acordo com a reivindicação 5, caracterizado pelo fato de que a célula de planta é oriunda de milho.
7. Processo para conferir ou aperfeiçoar a resistência a Colletotrichum, caracterizado pelo fato de que compreende a transformação de uma planta com o constructo de DNA recombinante conforme definido na reivindicação 2, conferindo ou aperfeiçoando, por esse meio, a resistência a Colletotrichum.
8. Processo para alterar o nível de expressão de uma proteína capaz de conferir resistência a Colletotrichum ou podridão do colmo em uma célula de planta, caracterizado pelo fato de que compreende: (a) transformar uma célula de planta com o constructo de DNA recombinante conforme definido na reivindicação 2; e (b) colocar para crescer a célula de planta transformada sob condições que são apropriadas para a expressão do constructo de DNA recombinante sendo que a expressão do constructo de DNA recombinante resulta na produção de níveis alterados de uma proteína capaz de conferir resistência a Colletotrichum ou podridão do colmo na célula de planta transformada.
9. Processo para alterar o nível de expressão de uma proteína capaz de conferir resistência a Colletotrichum ou podridão do colmo em uma planta, caracterizado pelo fato de que compreende: (a) transformar uma célula de planta com o constructo de DNA recombinante conforme definido na reivindicação 2; (b) regenerar a planta transformada a partir da célula de planta transformada; e (c) colocar para crescer a planta transformada sob condições que são apropriadas para a expressão do constructo de DNA recombinante sendo que a expressão do constructo de DNA recombinante resulta na produção de níveis alterados de uma proteína que é capaz de conferir resistência a Colletotrichum ou podridão do colmo na planta transformada.
10. Processo de identificação de uma planta de milho que apresenta resistência recentemente conferida ou aperfeiçoada a infecção por Colletotrichum em comparação com uma planta de milho que não possui o polinucleotídeo conforme definido na reivindicação 1, caracterizado pelo fato de que dito processo compreende detectar, nos alelos de planta de milho, pelo menos dois marcadores, sendo que pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido por UMC2041 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200.
11. Processo de acordo com a reivindicação 10, caracterizado pelo fato de que: (a) pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido por UMC1086 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200; (b) pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido por UMC2285 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2187; ou (c) pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido por UMC2285 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos um de ditos marcadores está em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC15a.
12. Processo de acordo com a reivindicação 11, caracterizado pelo fato de que o item (c) compreende ainda a seleção de pelo menos quatro marcadores, sendo que pelo menos dois de ditos marcadores estão dentro de um intervalo cromossômico definido por UMC2285 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos dois de ditos marcadores estão dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC15a.
13. Processo de acordo com a reivindicação 12, caracterizado pelo fato de que pelo menos um de ditos marcadores está em ou dentro da SEQ ID NO: 137, sendo que pelo menos um marcador é capaz de detectar um polimorfismo localizado em uma posição selecionada do grupo consistindo em: (a) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 7230 e 7535; (b) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 11293 e 12553; (c) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 25412 e 29086; e (d) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 43017 e 50330.
14. Processo de acordo com a reivindicação 13, caracterizado pelo fato de que compreende ainda a seleção de pelo menos quatro marcadores, sendo que pelo menos dois de ditos marcadores estão em ou dentro de um intervalo cromossômico definido por UMC2041 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos dois de ditos marcadores estão em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200.
15. Processo de acordo com a reivindicação 10 ou 14, caracterizado pelo fato de que o pelo menos um marcador em ou dentro do intervalo cromossômico definido por UMC2041 e o polinucleotídeo conforme definido na reivindicação 1 é selecionado a partir dos marcadores: UMC2041, AY112127, UMC1086, AY110631, UMC2285, MZA8136, MZA6064, NPI270, NPI300C, PHP20071, CDO127a, RGPl102, UAZ122, BNL17.05, MZA11455, MZA15842, MZA11123, MZA2591; e sendo que pelo menos um marcador em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200 é selecionado a partir dos marcadores: PHI093, MZA1215, MZA1216, MZA3434, CL12681_1, NPI444, UMC15a, MZA8761, CSU166a, CDO365, CSU1038b, CSU1073b, CSU597a, RGPG111, UMN433, PHP20562, C2, NPI910, CSU178a, CSU202, TDA44, MZA1851, UMC1051, MZA11394, PCO136722, UMC2187, NPI410, PSR109B, UMC1371, UMC1842, UMC1856, AY109980, UMC1132, NFD106, AY105971, AY110989, ENSl002A, RZ596B, BNL23A, BNL29, UMC2200.
16. Processo de acordo com a reivindicação 10, caracterizado pelo fato de que compreende ainda a seleção de pelo menos seis marcadores, sendo que pelo menos três de ditos marcadores estão em ou dentro de um intervalo cromossômico definido por UMC2041 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos três de ditos marcadores estão em ou dentro do intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200.
17. Processo de acordo com a reivindicação 16, caracterizado pelo fato de que os pelo menos três marcadores em ou dentro de um intervalo cromossômico definido por UMC2041 e o polinucleotídeo conforme definido na reivindicação 1 são selecionados a partir dos marcadores: UMC2041, AY112127, UMC1086, AY110631, UMC2285, MZA8136, MZA6064, NPI270, NPI300C, PHP20071, CDO127a, RGPl102, UAZ122, BNL17.05, MZA11455, MZA15842, MZA11123, MZA2591, e em que os pelo menos três marcadores que estão em ou dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC2200 são selecionados a partir dos marcadores: PHI093, MZA1215, MZA1216, MZA3434, CL12681_1, NPI444, UMC15a, MZA8761, CSU166a, CDO365, CSU1038b, CSU1073b, CSU597a, RGPG111, UMN433, PHP20562, C2, NPI910, CSU178a, CSU202, TDA44, MZA1851, UMC1051, MZA11394, PCO136722, UMC2187, NPI410, PSR109B, UMC1371, UMC1842, UMC1856, AY109980, UMC1132, NFD106, AY105971, AY110989, ENSl002A, RZ596B, BNL23A, BNL29, UMC2200.
18. Processo de acordo com a reivindicação 10, caracterizado pelo fato de que dito processo compreende ainda detectar pelo menos dois ou mais dentre: (a) alelo 7 em MZA11123 (SEQ ID NO: 140), (b) alelo 2 em MZA2591 (SEQ ID NO: 139) e (c) alelo 8 em MZA3434 (SEQ ID NO: 138).
19. Processo de acordo com a reivindicação 10, caracterizado pelo fato de que compreende detectar ainda, na planta de milho, a presença ou ausência de pelo menos um marcador dentro do polinucleotídeo conforme definido na reivindicação 1.
20. Processo de acordo com a reivindicação 19, caracterizado pelo fato de compreende ainda a seleção de pelo menos quatro marcadores, sendo que pelo menos dois de ditos marcadores estão dentro de um intervalo cromossômico definido por UMC2285 e o polinucleotídeo conforme definido na reivindicação 1, e pelo menos dois de ditos marcadores estão dentro do intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC15a.
21. Processo de acordo com a reivindicação 19, caracterizado pelo fato de que a planta de milho foi obtida por um processo de introgressão do polinucleotídeo conforme definido na reivindicação 1, a partir de uma planta de milho doadora, em uma planta de milho receptora para produzir uma planta de milho introgredida.
22. Processo de acordo com a reivindicação 21, caracterizado pelo fato de que a planta de milho doadora é a MP305 ou a DE811ASR(BC5) (Número de acesso ATCC PTA-7434).
23. Processo de acordo com a reivindicação 21, caracterizado pelo fato de que a planta de milho introgredida é selecionada para um evento de recombinação dentro de um intervalo cromossômico definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC15a, de modo que a planta de milho introgredida retém um primeiro intervalo cromossômico derivado da MP305 definido pelo polinucleotídeo conforme definido na reivindicação 1 e UMC15a, e não retém um segundo intervalo cromossômico derivado da MP305.
24. Processo de identificação de uma planta de milho que apresenta resistência aperfeiçoada a infecção por Colletotrichum comparada a uma planta de milho que não possui o polinucleotídeo conforme definido na reivindicação 1, caracterizado pelo fato de que dito processo compreende detectar na planta de milho a presença ou ausência de pelo menos um marcador, em que o pelo menos um marcador está em ou dentro da sequência codificante do Rcg1, em que dita sequência codificante do Rcg1 é a SEQ ID NO: 2, ou o pelo menos um marcador está em ou dentro do polinucleotídeo definido na SEQ ID NO: 1.
25. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que o pelo menos um marcador está em ou dentro da SEQ ID NO: 137.
26. Processo de acordo com a reivindicação 25, caracterizado pelo fato de que o pelo menos um marcador é capaz de detectar um polimorfismo localizado em uma posição selecionada do grupo consistindo da: (a) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 1 e 536; (b) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 7230 e 7 535; (c) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 11293 e 12553; (d) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 25412 e 29086; e (e) posição na SEQ ID NO: 137 correspondente aos nucleotídeos entre 43017 e 50330.
27. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que pelo menos um marcador detecta um polimorfismo simples de nucleotídeos em uma posição na sequência de nucleotídeos definida como SEQ ID NO: 1 correspondente a uma ou mais posições 413, 958, 971, 1099, 1154, 1235, 1250, 1308, 1607, 2001, 2598, e 3342.
28. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que o pelo menos um marcador é um marcador de SNP selecionado do grupo consistindo em C00060-01 (SEQ ID NOs: 145 a 152) e C00060-02 (SEQ ID NOs: 154 a 157).
29. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que o pelo menos um marcador é um marcador de FLP em um amplicon gerado por um par de primers compreendendo um primeiro e um segundo primer, sendo que o primeiro primer é selecionado do grupo consistindo em: (a) sequência definida na SEQ ID NO: 35 e o complemento da mesma; (b) sequência definida na SEQ ID NO: 37 e o complemento da mesma; (c) sequência definida na SEQ ID NO: 39 e o complemento da mesma; e (d) sequência definida na SEQ ID NO: 41 e o complemento da mesma; e sendo que o segundo primer é selecionado do grupo consistindo em: (e) sequência definida na SEQ ID NO: 36 e o complemento da mesma; (f) sequência definida na SEQ ID NO: 38 e o complemento da mesma; (g) sequência definida na SEQ ID NO: 40 e o complemento da mesma; e (h) sequência definida na SEQ ID NO: 42 e o complemento da mesma.
30. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que o pelo menos um marcador detecta uma sequência de mRNA derivada do transcripto de mRNA da dita sequência codificante do Rcg1, e único para dita sequência codificante do Rcg1, em que dita sequência codificante do Rcg1 é a SEQ ID NO: 2.
31. Processo de acordo com a reivindicação 24, caracterizado pelo fato de que dito processo compreende ainda a detecção na planta de milho da presença ou ausência de pelo menos dois dos ditos marcadores.
32. Processo de acordo com a reivindicação 31, caracterizado pelo fato de que os pelo menos dois marcadores são o C00060-01 (SEQ ID NOs: 145 a 152) e C00060-02 (SEQ ID NOs: 154 a 157).
33. Processo de acordo com a reivindicação 32, caracterizado pelo fato de que a planta de milho foi obtida por um processo de introgressão a partir de uma planta de milho doadora, em uma planta de milho receptora para produzir uma planta de milho introgredida.
34. Processo de acordo com a reivindicação 33, caracterizado pelo fato de que a planta de milho doadora é a MP305 ou a DE811ASR(BC5) (Número de acesso ATCC PTA-7434).
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66824105P | 2005-04-04 | 2005-04-04 | |
| US60/668,241 | 2005-04-04 | ||
| US67566405P | 2005-04-28 | 2005-04-28 | |
| US60/675,664 | 2005-04-28 | ||
| PCT/US2006/012443 WO2006107931A2 (en) | 2005-04-04 | 2006-04-04 | Polynucleotides and methods for making plants resistant to fungal pathogens |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BRPI0610520A2 BRPI0610520A2 (pt) | 2010-06-22 |
| BRPI0610520B1 true BRPI0610520B1 (pt) | 2022-04-05 |
Family
ID=36940190
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0610520-3A BRPI0610520B1 (pt) | 2005-04-04 | 2006-04-04 | Vetor, constructo de dna recombinante, e processos para transformar uma célula hospedeira, produzir uma planta, conferir ou aperfeiçoar a resistência a colletotrichum, determinar a presença ou ausência de um polinucleotídeo, alterar o nível de expressão de uma proteína, e de identificação de uma planta de milho |
Country Status (6)
| Country | Link |
|---|---|
| US (10) | US20060225151A1 (pt) |
| EP (1) | EP1874935B2 (pt) |
| BR (1) | BRPI0610520B1 (pt) |
| CA (1) | CA2604351C (pt) |
| MX (1) | MX2007012302A (pt) |
| WO (1) | WO2006107931A2 (pt) |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011109661A1 (en) | 2010-03-04 | 2011-09-09 | Mendel Biotechnology Inc. | Transcription regulators for improving plant performance |
| US20060225151A1 (en) * | 2005-04-04 | 2006-10-05 | E.I. Du Pont De Nemours And Company | Polynucleotides and methods for making plants resistant to fungal pathogens |
| WO2008157432A1 (en) | 2007-06-15 | 2008-12-24 | E. I. Dupont De Nemours & Company | Polynucleotides and methods for making plants resistant to fungal pathogens |
| US8115055B2 (en) | 2007-12-18 | 2012-02-14 | E.I. Du Pont De Nemours And Company | Down-regulation of gene expression using artificial microRNAs |
| US8900808B2 (en) * | 2008-07-15 | 2014-12-02 | E.I. Du Pont De Nemours And Company | Genetic loci associated with mechanical stalk strength in maize |
| AR075465A1 (es) | 2008-10-22 | 2011-04-06 | Basf Se | Uso de herbicidas de sulfonilurea en plantas cultivadas |
| AR075466A1 (es) | 2008-10-22 | 2011-04-06 | Basf Se | Uso de herbicidas tipo auxina en plantas cultivadas |
| EP2584887B1 (en) | 2010-06-25 | 2018-05-30 | E. I. du Pont de Nemours and Company | Compositions and methods for enhancing resistance to northern leaf blight in maize |
| TWI667347B (zh) | 2010-12-15 | 2019-08-01 | 瑞士商先正達合夥公司 | 大豆品種syht0h2及偵測其之組合物及方法 |
| US9062317B2 (en) | 2011-05-09 | 2015-06-23 | E I Du Pont De Nemours And Company | Methods and compositions for silencing gene families using artificial microRNAs |
| US20150250174A1 (en) | 2012-10-01 | 2015-09-10 | Basf Se | Use of n-thio-anthranilamide compounds on cultivated plants |
| KR101640278B1 (ko) * | 2012-10-05 | 2016-07-15 | 경희대학교 산학협력단 | 흰잎 마름병 및 도열병 저항성 인산화 효소 및 이를 이용한 형질전환 벼 |
| WO2014079820A1 (en) | 2012-11-22 | 2014-05-30 | Basf Se | Use of anthranilamide compounds for reducing insect-vectored viral infections |
| WO2014116989A1 (en) | 2013-01-25 | 2014-07-31 | Pioneer Hi-Bred International, Inc. | Maize event dp-032218-9 and methods for detection thereof |
| US8902663B1 (en) | 2013-03-11 | 2014-12-02 | Monolithic 3D Inc. | Method of maintaining a memory state |
| WO2014159306A1 (en) | 2013-03-13 | 2014-10-02 | Pioneer Hi-Bred International, Inc. | Glyphosate application for weed control in brassica |
| MX369750B (es) | 2013-03-14 | 2019-11-20 | Pioneer Hi Bred Int | Composiciones y metodos para controlar plagas de insectos. |
| US10023877B2 (en) | 2013-03-15 | 2018-07-17 | Pioneer Hi-Bred International, Inc. | PHI-4 polypeptides and methods for their use |
| MX359026B (es) | 2013-08-16 | 2018-09-12 | Pioneer Hi Bred Int | Proteinas insecticidas y metodos de uso. |
| US10667524B2 (en) | 2013-09-13 | 2020-06-02 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2015120276A1 (en) | 2014-02-07 | 2015-08-13 | Pioneer Hi Bred International Inc | Insecticidal proteins and methods for their use |
| CN114763376A (zh) | 2014-02-07 | 2022-07-19 | 先锋国际良种公司 | 杀昆虫蛋白及其使用方法 |
| WO2016044092A1 (en) | 2014-09-17 | 2016-03-24 | Pioneer Hi Bred International Inc | Compositions and methods to control insect pests |
| EP3207143B1 (en) | 2014-10-16 | 2023-11-22 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3028573A1 (en) | 2014-12-05 | 2016-06-08 | Basf Se | Use of a triazole fungicide on transgenic plants |
| WO2016091674A1 (en) | 2014-12-12 | 2016-06-16 | Basf Se | Use of cyclaniliprole on cultivated plants |
| US20170359965A1 (en) | 2014-12-19 | 2017-12-21 | E I Du Pont De Nemours And Company | Polylactic acid compositions with accelerated degradation rate and increased heat stability |
| EP3244727B1 (en) | 2015-01-15 | 2025-10-22 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| WO2016162371A1 (en) | 2015-04-07 | 2016-10-13 | Basf Agrochemical Products B.V. | Use of an insecticidal carboxamide compound against pests on cultivated plants |
| CN108064233B (zh) | 2015-05-19 | 2022-07-15 | 先锋国际良种公司 | 杀昆虫蛋白及其使用方法 |
| US20160355840A1 (en) | 2015-06-03 | 2016-12-08 | E I Du Pont De Nemours And Company | Methods of identifying and selecting maize plants with resistance to anthracnose stalk rot |
| MX2017016119A (es) | 2015-06-16 | 2018-03-07 | Pioneer Hi Bred Int | Composiciones y metodos para controlar plagas de insectos. |
| EP3943602A1 (en) | 2015-08-06 | 2022-01-26 | Pioneer Hi-Bred International, Inc. | Plant derived insecticidal proteins and methods for their use |
| JP2018527931A (ja) | 2015-08-28 | 2018-09-27 | パイオニア ハイ−ブレッド インターナショナル, インコーポレイテッド | 植物のオクロバクテリウム(ochrobactrum)媒介形質転換 |
| EA037614B1 (ru) * | 2015-10-16 | 2021-04-21 | Пайонир Хай-Бред Интернэшнл, Инк. | Получение растений маиса с повышенной устойчивостью к северной пятнистости листьев |
| WO2017192560A1 (en) | 2016-05-04 | 2017-11-09 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins and methods for their use |
| EP3472323A1 (en) | 2016-06-16 | 2019-04-24 | Pioneer Hi-Bred International, Inc. | Compositions and methods to control insect pests |
| US11155829B2 (en) | 2016-07-01 | 2021-10-26 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins from plants and methods for their use |
| US20210292778A1 (en) | 2016-07-12 | 2021-09-23 | Pioneer Hi-Bred International, Inc. | Compositions and methods to control insect pests |
| CN109844121A (zh) | 2016-10-13 | 2019-06-04 | 先锋国际良种公司 | 产生北方叶枯病抗性玉蜀黍 |
| BR112019008800A2 (pt) | 2016-11-01 | 2019-07-16 | Pioneer Hi-Bred International, Inc. | polipeptídeo inseticida, composição inseticida, polinucleotídeo recombinante, construto de dna, célula de planta ou planta transgênica, método para inibir o crescimento ou exterminar uma população de praga de inseto agrícola, método para inibir o crescimento ou exterminar uma praga de inseto, método para controlar infestação de inseto lepidoptera e/ou coleoptera em uma planta transgênica e fornecer gerenciamento de resistência de inseto e uso de pelo menos um polipeptídeo inseticida |
| EP3338552A1 (en) | 2016-12-21 | 2018-06-27 | Basf Se | Use of a tetrazolinone fungicide on transgenic plants |
| CA3092078A1 (en) | 2018-03-14 | 2019-09-19 | Pioneer Hi-Bred International, Inc. | Insecticidal proteins from plants and methods for their use |
| CN116410286A (zh) | 2018-03-14 | 2023-07-11 | 先锋国际良种公司 | 来自植物的杀昆虫蛋白及其使用方法 |
| EP3833182A1 (en) * | 2018-06-06 | 2021-06-16 | Huazhong Agricultural University | Methods of identifying, selecting, and producing southern corn rust resistant crops |
| CN109234431B (zh) * | 2018-09-27 | 2022-11-01 | 北京大北农生物技术有限公司 | 玉米抗茎腐病qtl的分子标记及其应用 |
| CA3109984A1 (en) | 2018-10-16 | 2020-04-23 | Pioneer Hi-Bred International, Inc. | Genome edited fine mapping and causal gene identification |
| US12371706B2 (en) | 2019-08-23 | 2025-07-29 | Pioneer Hi-Bred International, Inc. | Methods of identifying, selecting, and producing anthracnose stalk rot resistant crops |
| BR112022020714A2 (pt) | 2020-04-15 | 2022-12-20 | Pioneer Hi Bred Int | Identificação de gene efetor de patógeno vegetal e de resistência à doença, composições e métodos de uso |
| CN115867564A (zh) | 2020-07-14 | 2023-03-28 | 先锋国际良种公司 | 杀昆虫蛋白及其使用方法 |
| WO2025191167A1 (en) | 2024-03-15 | 2025-09-18 | Limagrain Europe | Chimeric resistance genes for maize resistance against pathogens |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6479731B1 (en) | 1998-08-04 | 2002-11-12 | E. I. Du Pont De Nemours And Company | Pi-ta gene conferring fungal disease resistance to plants |
| US20060225151A1 (en) * | 2005-04-04 | 2006-10-05 | E.I. Du Pont De Nemours And Company | Polynucleotides and methods for making plants resistant to fungal pathogens |
| EP1867723A1 (en) | 2006-06-16 | 2007-12-19 | Genoplante-Valor | Plants with increased tolerance to water deficit |
| WO2008157432A1 (en) * | 2007-06-15 | 2008-12-24 | E. I. Dupont De Nemours & Company | Polynucleotides and methods for making plants resistant to fungal pathogens |
-
2006
- 2006-04-04 US US11/397,153 patent/US20060225151A1/en not_active Abandoned
- 2006-04-04 CA CA2604351A patent/CA2604351C/en not_active Expired - Lifetime
- 2006-04-04 WO PCT/US2006/012443 patent/WO2006107931A2/en not_active Ceased
- 2006-04-04 US US11/397,275 patent/US20060223102A1/en not_active Abandoned
- 2006-04-04 US US11/397,247 patent/US20060225152A1/en not_active Abandoned
- 2006-04-04 EP EP06740467.3A patent/EP1874935B2/en not_active Expired - Lifetime
- 2006-04-04 MX MX2007012302A patent/MX2007012302A/es active IP Right Grant
- 2006-04-04 BR BRPI0610520-3A patent/BRPI0610520B1/pt not_active IP Right Cessation
-
2007
- 2007-01-29 US US11/699,105 patent/US20080072341A1/en not_active Abandoned
- 2007-07-06 US US11/774,121 patent/US8084671B2/en active Active
- 2007-07-20 US US11/780,499 patent/US7619133B2/en not_active Expired - Lifetime
- 2007-10-26 US US11/924,639 patent/US8062847B2/en active Active
-
2011
- 2011-10-11 US US13/270,722 patent/US8884126B2/en active Active
-
2014
- 2014-11-11 US US14/538,238 patent/US9884896B2/en active Active
-
2017
- 2017-12-27 US US15/855,435 patent/US20180112280A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US9884896B2 (en) | 2018-02-06 |
| US8084671B2 (en) | 2011-12-27 |
| US20080072341A1 (en) | 2008-03-20 |
| US7619133B2 (en) | 2009-11-17 |
| EP1874935B1 (en) | 2013-11-06 |
| US20060225151A1 (en) | 2006-10-05 |
| US20080022426A1 (en) | 2008-01-24 |
| EP1874935A2 (en) | 2008-01-09 |
| MX2007012302A (es) | 2008-04-14 |
| US20090035765A1 (en) | 2009-02-05 |
| US20080016595A1 (en) | 2008-01-17 |
| US20180112280A1 (en) | 2018-04-26 |
| US20060223102A1 (en) | 2006-10-05 |
| WO2006107931A3 (en) | 2006-12-07 |
| US8884126B2 (en) | 2014-11-11 |
| EP1874935B2 (en) | 2023-10-18 |
| CA2604351C (en) | 2012-08-28 |
| US8062847B2 (en) | 2011-11-22 |
| US20120054921A1 (en) | 2012-03-01 |
| WO2006107931A2 (en) | 2006-10-12 |
| BRPI0610520A2 (pt) | 2010-06-22 |
| US20160201079A1 (en) | 2016-07-14 |
| US20060225152A1 (en) | 2006-10-05 |
| CA2604351A1 (en) | 2006-10-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10731225B2 (en) | Methods for making plants resistant to fungal pathogens | |
| US9884896B2 (en) | Polynucleotides and methods for making plants resistant to fungal pathogens | |
| US20230416774A1 (en) | Wheat stripe resistance genes and method of use | |
| CN102165072A (zh) | 玉米中与丝黑穗病抗性相关联的基因座 | |
| CN107338254B (zh) | 用于制备抗真菌病原体的植物的多核苷酸和方法 | |
| Butler et al. | 11111111111111111111111111111111111111111111111111111111111111111111111111 II OI IIi | |
| BRPI0813360B1 (pt) | Polinucleotídeo isolado, vetor, processo para transformar uma célula hospedeira, processo para a produção de uma planta, processo para conferir ou melhorar a resistência a colletotrichum, processo para alterar o nível de expressão de proteínas capazes de conferir resistência a colletotrichum e a podridão no caule de uma célula vegetal e processo de identificação de uma planta de milho |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09B | Patent application refused [chapter 9.2 patent gazette] | ||
| B12B | Appeal against refusal [chapter 12.2 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 04/04/2006, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |
|
| B21F | Lapse acc. art. 78, item iv - on non-payment of the annual fees in time |
Free format text: REFERENTE A 18A ANUIDADE. |
|
| B24D | Patent annual fee: restoration after fee payment |