BRPI0619894B1 - Composto de amina e composição farmacêutica - Google Patents
Composto de amina e composição farmacêutica Download PDFInfo
- Publication number
- BRPI0619894B1 BRPI0619894B1 BRPI0619894-5A BRPI0619894A BRPI0619894B1 BR PI0619894 B1 BRPI0619894 B1 BR PI0619894B1 BR PI0619894 A BRPI0619894 A BR PI0619894A BR PI0619894 B1 BRPI0619894 B1 BR PI0619894B1
- Authority
- BR
- Brazil
- Prior art keywords
- compound
- reaction
- solvent
- mixture
- added
- Prior art date
Links
- -1 AMINE COMPOUND Chemical class 0.000 title claims abstract description 171
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 13
- 150000001875 compounds Chemical class 0.000 claims abstract description 336
- 239000002253 acid Substances 0.000 claims abstract description 100
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 88
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 59
- 125000005843 halogen group Chemical group 0.000 claims abstract description 59
- 150000003839 salts Chemical class 0.000 claims abstract description 50
- 239000012453 solvate Substances 0.000 claims abstract description 48
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 46
- 125000004430 oxygen atom Chemical group O* 0.000 claims abstract description 25
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 23
- 229910052717 sulfur Chemical group 0.000 claims abstract description 20
- 125000004434 sulfur atom Chemical group 0.000 claims abstract description 20
- 239000003937 drug carrier Chemical class 0.000 claims abstract description 5
- 239000002904 solvent Substances 0.000 claims description 346
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 107
- 229910052739 hydrogen Inorganic materials 0.000 claims description 34
- 239000001257 hydrogen Substances 0.000 claims description 34
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 32
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 27
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 19
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 claims description 19
- 238000011282 treatment Methods 0.000 claims description 14
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 13
- 238000011321 prophylaxis Methods 0.000 claims description 12
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 10
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 9
- JVCPIJKPAKAIIP-UHFFFAOYSA-N 2-amino-2-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol Chemical compound CCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F JVCPIJKPAKAIIP-UHFFFAOYSA-N 0.000 claims description 6
- OIUJCLYDEGSNLF-UHFFFAOYSA-N 2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol Chemical compound CCCCCCCOC1=CC=C(CCC(C)(N)CO)C=C1C(F)(F)F OIUJCLYDEGSNLF-UHFFFAOYSA-N 0.000 claims description 6
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 6
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 6
- 208000024908 graft versus host disease Diseases 0.000 claims description 6
- 238000002054 transplantation Methods 0.000 claims description 6
- 208000023275 Autoimmune disease Diseases 0.000 claims description 5
- 208000026935 allergic disease Diseases 0.000 claims description 5
- 150000002148 esters Chemical class 0.000 claims description 5
- 210000000056 organ Anatomy 0.000 claims description 5
- OIUJCLYDEGSNLF-GOSISDBHSA-N (2r)-2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol Chemical compound CCCCCCCOC1=CC=C(CC[C@@](C)(N)CO)C=C1C(F)(F)F OIUJCLYDEGSNLF-GOSISDBHSA-N 0.000 claims description 4
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 4
- 238000010322 bone marrow transplantation Methods 0.000 claims description 4
- 230000001684 chronic effect Effects 0.000 claims description 4
- ROKBUVCZBDSWOM-MDZDMXLPSA-N 2-amino-2-[(e)-2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethenyl]propane-1,3-diol Chemical compound CCCCCCCOC1=CC=C(\C=C\C(N)(CO)CO)C=C1C(F)(F)F ROKBUVCZBDSWOM-MDZDMXLPSA-N 0.000 claims description 3
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 3
- 208000005777 Lupus Nephritis Diseases 0.000 claims description 3
- 206010029164 Nephrotic syndrome Diseases 0.000 claims description 3
- 206010039085 Rhinitis allergic Diseases 0.000 claims description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 3
- 201000010105 allergic rhinitis Diseases 0.000 claims description 3
- 208000006673 asthma Diseases 0.000 claims description 3
- 201000008937 atopic dermatitis Diseases 0.000 claims description 3
- 201000002491 encephalomyelitis Diseases 0.000 claims description 3
- 201000006417 multiple sclerosis Diseases 0.000 claims description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 3
- 230000001629 suppression Effects 0.000 claims description 3
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 3
- 210000001519 tissue Anatomy 0.000 claims description 3
- IACSWVDQYODOTF-UHFFFAOYSA-N 5-[3-amino-4-hydroxy-3-(hydroxymethyl)butyl]-2-heptoxybenzonitrile Chemical compound CCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C#N IACSWVDQYODOTF-UHFFFAOYSA-N 0.000 claims description 2
- NBFCHCDCCVCHNK-UHFFFAOYSA-N [2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutyl] dihydrogen phosphate Chemical compound CCCCCCCOC1=CC=C(CCC(C)(N)COP(O)(O)=O)C=C1C(F)(F)F NBFCHCDCCVCHNK-UHFFFAOYSA-N 0.000 claims description 2
- 201000009961 allergic asthma Diseases 0.000 claims description 2
- 239000011230 binding agent Substances 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000007884 disintegrant Substances 0.000 claims description 2
- 239000003995 emulsifying agent Substances 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 230000002829 reductive effect Effects 0.000 abstract description 92
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 abstract description 23
- 229910052799 carbon Inorganic materials 0.000 abstract description 23
- 230000000694 effects Effects 0.000 abstract description 8
- 230000036471 bradycardia Effects 0.000 abstract description 4
- 208000006218 bradycardia Diseases 0.000 abstract description 4
- 230000001506 immunosuppresive effect Effects 0.000 abstract description 3
- 150000004677 hydrates Chemical class 0.000 abstract 1
- 238000006243 chemical reaction Methods 0.000 description 276
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 268
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 251
- 239000000203 mixture Substances 0.000 description 207
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 174
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 170
- 239000007795 chemical reaction product Substances 0.000 description 168
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 162
- 238000001816 cooling Methods 0.000 description 139
- 239000000543 intermediate Substances 0.000 description 127
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 120
- 238000010898 silica gel chromatography Methods 0.000 description 120
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 108
- 239000000243 solution Substances 0.000 description 103
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 102
- 239000011541 reaction mixture Substances 0.000 description 97
- 125000006239 protecting group Chemical group 0.000 description 92
- 230000015572 biosynthetic process Effects 0.000 description 87
- 238000003786 synthesis reaction Methods 0.000 description 87
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 85
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 84
- 238000005160 1H NMR spectroscopy Methods 0.000 description 82
- 238000007429 general method Methods 0.000 description 80
- 238000000746 purification Methods 0.000 description 80
- 238000001953 recrystallisation Methods 0.000 description 80
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 79
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 78
- 238000000605 extraction Methods 0.000 description 78
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 78
- 238000001035 drying Methods 0.000 description 77
- 238000005406 washing Methods 0.000 description 77
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 69
- 239000003153 chemical reaction reagent Substances 0.000 description 64
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 63
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 60
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 57
- 239000002585 base Substances 0.000 description 56
- 230000001476 alcoholic effect Effects 0.000 description 53
- 238000001914 filtration Methods 0.000 description 41
- 238000010992 reflux Methods 0.000 description 41
- 239000012044 organic layer Substances 0.000 description 40
- 238000006722 reduction reaction Methods 0.000 description 40
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 38
- 239000012230 colorless oil Substances 0.000 description 38
- 239000003921 oil Substances 0.000 description 37
- 239000000843 powder Substances 0.000 description 37
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 36
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 36
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 33
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 30
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 30
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 30
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 30
- 239000012046 mixed solvent Substances 0.000 description 30
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 30
- 239000007787 solid Substances 0.000 description 30
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 27
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 27
- 150000007529 inorganic bases Chemical class 0.000 description 27
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 27
- 235000017557 sodium bicarbonate Nutrition 0.000 description 27
- 239000012312 sodium hydride Substances 0.000 description 27
- 229910000104 sodium hydride Inorganic materials 0.000 description 27
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 26
- 239000003054 catalyst Substances 0.000 description 26
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 25
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 25
- 150000007530 organic bases Chemical class 0.000 description 25
- 239000003960 organic solvent Substances 0.000 description 25
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- 239000002798 polar solvent Substances 0.000 description 24
- 238000000034 method Methods 0.000 description 23
- 150000007522 mineralic acids Chemical class 0.000 description 23
- 238000004821 distillation Methods 0.000 description 21
- 229910052736 halogen Inorganic materials 0.000 description 21
- 150000002367 halogens Chemical class 0.000 description 21
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 20
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 20
- 239000004215 Carbon black (E152) Substances 0.000 description 19
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 19
- 229930195733 hydrocarbon Natural products 0.000 description 19
- 150000002430 hydrocarbons Chemical class 0.000 description 19
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 19
- 229920006395 saturated elastomer Polymers 0.000 description 19
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 19
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 18
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 18
- 229910000027 potassium carbonate Inorganic materials 0.000 description 18
- 238000005804 alkylation reaction Methods 0.000 description 17
- 230000029936 alkylation Effects 0.000 description 16
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 16
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 15
- 125000002252 acyl group Chemical group 0.000 description 15
- 239000007864 aqueous solution Substances 0.000 description 15
- 238000010511 deprotection reaction Methods 0.000 description 15
- 239000011734 sodium Substances 0.000 description 15
- 229910052708 sodium Inorganic materials 0.000 description 15
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Natural products P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 14
- 150000001241 acetals Chemical class 0.000 description 14
- 125000003277 amino group Chemical group 0.000 description 14
- 239000012280 lithium aluminium hydride Substances 0.000 description 14
- 150000004714 phosphonium salts Chemical class 0.000 description 14
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 14
- 239000010948 rhodium Substances 0.000 description 14
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 14
- 238000007239 Wittig reaction Methods 0.000 description 13
- YGFLCNPXEPDANQ-UHFFFAOYSA-N n-[bis[(2-methylpropan-2-yl)oxy]phosphanyl]-n-propan-2-ylpropan-2-amine Chemical compound CC(C)N(C(C)C)P(OC(C)(C)C)OC(C)(C)C YGFLCNPXEPDANQ-UHFFFAOYSA-N 0.000 description 13
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 12
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 12
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- 239000011259 mixed solution Substances 0.000 description 12
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 12
- QBERHIJABFXGRZ-UHFFFAOYSA-M rhodium;triphenylphosphane;chloride Chemical compound [Cl-].[Rh].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QBERHIJABFXGRZ-UHFFFAOYSA-M 0.000 description 12
- 239000007868 Raney catalyst Substances 0.000 description 11
- 229910000564 Raney nickel Inorganic materials 0.000 description 11
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 11
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 229910052703 rhodium Inorganic materials 0.000 description 11
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 11
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 11
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 10
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 10
- 239000012298 atmosphere Substances 0.000 description 10
- 239000002638 heterogeneous catalyst Substances 0.000 description 10
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 10
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 10
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 10
- 239000007983 Tris buffer Substances 0.000 description 9
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 9
- 229910052801 chlorine Inorganic materials 0.000 description 9
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 238000003682 fluorination reaction Methods 0.000 description 9
- 229910052731 fluorine Inorganic materials 0.000 description 9
- 239000002815 homogeneous catalyst Substances 0.000 description 9
- 229910052751 metal Chemical class 0.000 description 9
- 239000002184 metal Chemical class 0.000 description 9
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 9
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 9
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 8
- HZNVUJQVZSTENZ-UHFFFAOYSA-N 2,3-dichloro-5,6-dicyano-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(C#N)=C(C#N)C1=O HZNVUJQVZSTENZ-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 8
- ABRVLXLNVJHDRQ-UHFFFAOYSA-N [2-pyridin-3-yl-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound FC(C1=CC(=CC(=N1)C=1C=NC=CC=1)CN)(F)F ABRVLXLNVJHDRQ-UHFFFAOYSA-N 0.000 description 8
- 125000003710 aryl alkyl group Chemical group 0.000 description 8
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 8
- 230000003197 catalytic effect Effects 0.000 description 8
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 8
- 125000001153 fluoro group Chemical group F* 0.000 description 8
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 8
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 8
- 229910052987 metal hydride Inorganic materials 0.000 description 8
- 150000004681 metal hydrides Chemical class 0.000 description 8
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 8
- 229910052763 palladium Inorganic materials 0.000 description 8
- 125000003396 thiol group Chemical group [H]S* 0.000 description 8
- 150000003573 thiols Chemical class 0.000 description 8
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 7
- 150000001299 aldehydes Chemical class 0.000 description 7
- PWHMUCVKZZBAJU-UHFFFAOYSA-N benzyl n-[4-[4-heptoxy-3-(trifluoromethyl)phenyl]-1-hydroxy-2-(hydroxymethyl)butan-2-yl]carbamate Chemical compound C1=C(C(F)(F)F)C(OCCCCCCC)=CC=C1CCC(CO)(CO)NC(=O)OCC1=CC=CC=C1 PWHMUCVKZZBAJU-UHFFFAOYSA-N 0.000 description 7
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 7
- 238000009833 condensation Methods 0.000 description 7
- 230000005494 condensation Effects 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 7
- 229910000103 lithium hydride Inorganic materials 0.000 description 7
- AQCVCACIAGRKGW-UHFFFAOYSA-N n-[4-[4-heptoxy-3-(trifluoromethyl)phenyl]-1-hydroxy-2-(hydroxymethyl)butan-2-yl]acetamide Chemical compound CCCCCCCOC1=CC=C(CCC(CO)(CO)NC(C)=O)C=C1C(F)(F)F AQCVCACIAGRKGW-UHFFFAOYSA-N 0.000 description 7
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 7
- 229910052698 phosphorus Inorganic materials 0.000 description 7
- 239000011574 phosphorus Substances 0.000 description 7
- 230000026731 phosphorylation Effects 0.000 description 7
- 238000006366 phosphorylation reaction Methods 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- 125000004665 trialkylsilyl group Chemical group 0.000 description 7
- IOHPVZBSOKLVMN-UHFFFAOYSA-N 2-(2-phenylethyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1CCC1=CC=CC=C1 IOHPVZBSOKLVMN-UHFFFAOYSA-N 0.000 description 6
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 238000006751 Mitsunobu reaction Methods 0.000 description 6
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 6
- 230000003213 activating effect Effects 0.000 description 6
- 229910052783 alkali metal Inorganic materials 0.000 description 6
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 150000004703 alkoxides Chemical class 0.000 description 6
- 239000002168 alkylating agent Substances 0.000 description 6
- 229940100198 alkylating agent Drugs 0.000 description 6
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 6
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 6
- 229910052794 bromium Inorganic materials 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 238000001704 evaporation Methods 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 6
- 239000007800 oxidant agent Substances 0.000 description 6
- 150000002941 palladium compounds Chemical class 0.000 description 6
- 150000008300 phosphoramidites Chemical class 0.000 description 6
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 6
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- OGFAWKRXZLGJSK-UHFFFAOYSA-N 1-(2,4-dihydroxyphenyl)-2-(4-nitrophenyl)ethanone Chemical compound OC1=CC(O)=CC=C1C(=O)CC1=CC=C([N+]([O-])=O)C=C1 OGFAWKRXZLGJSK-UHFFFAOYSA-N 0.000 description 5
- NRCFUYKTFAPUBR-UHFFFAOYSA-N 2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CCC(C)(N)CO)C=C1C(F)(F)F NRCFUYKTFAPUBR-UHFFFAOYSA-N 0.000 description 5
- NVLPDIRQWJSXLZ-UHFFFAOYSA-N 3-hydroxypyridine-4-carbaldehyde Chemical compound OC1=CN=CC=C1C=O NVLPDIRQWJSXLZ-UHFFFAOYSA-N 0.000 description 5
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 5
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 239000005708 Sodium hypochlorite Substances 0.000 description 5
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 5
- 150000001340 alkali metals Chemical class 0.000 description 5
- 150000001342 alkaline earth metals Chemical class 0.000 description 5
- 150000001350 alkyl halides Chemical class 0.000 description 5
- 230000002152 alkylating effect Effects 0.000 description 5
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- NSBNXCZCLRBQTA-UHFFFAOYSA-N dibenzyl bis(phenylmethoxy)phosphoryl phosphate Chemical compound C=1C=CC=CC=1COP(OP(=O)(OCC=1C=CC=CC=1)OCC=1C=CC=CC=1)(=O)OCC1=CC=CC=C1 NSBNXCZCLRBQTA-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 5
- 229940071870 hydroiodic acid Drugs 0.000 description 5
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 5
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 5
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 5
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 5
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 5
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 5
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 5
- YNXLBCWYAAPNQK-UHFFFAOYSA-N (1,3-dihydroxy-2-methylpropan-2-yl)carbamic acid Chemical compound OCC(C)(CO)NC(O)=O YNXLBCWYAAPNQK-UHFFFAOYSA-N 0.000 description 4
- NRCFUYKTFAPUBR-FERBBOLQSA-N (2s)-2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CC[C@](C)(N)CO)C=C1C(F)(F)F NRCFUYKTFAPUBR-FERBBOLQSA-N 0.000 description 4
- ABUSWLOUSQWWLA-UHFFFAOYSA-N 1-[4-methoxy-3-(trifluoromethyl)phenyl]ethanone Chemical compound COC1=CC=C(C(C)=O)C=C1C(F)(F)F ABUSWLOUSQWWLA-UHFFFAOYSA-N 0.000 description 4
- MGDCBOKBTJIJBT-UHFFFAOYSA-N 2,2-difluoro-1,3-dimethylimidazolidine Chemical compound CN1CCN(C)C1(F)F MGDCBOKBTJIJBT-UHFFFAOYSA-N 0.000 description 4
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 4
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 4
- XATIJVWLHOKJLN-UHFFFAOYSA-N 2-amino-2-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]butane-1,4-diol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CCC(N)(CO)CCO)C=C1C(F)(F)F XATIJVWLHOKJLN-UHFFFAOYSA-N 0.000 description 4
- GEDVJGOVRLHFQG-UHFFFAOYSA-N 2-amino-2-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F GEDVJGOVRLHFQG-UHFFFAOYSA-N 0.000 description 4
- QTIKVJYEEWDKHL-UHFFFAOYSA-N 2-amino-2-[2-[4-hexoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol;hydrochloride Chemical compound Cl.CCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F QTIKVJYEEWDKHL-UHFFFAOYSA-N 0.000 description 4
- XUKLNKQQMDAVJF-UHFFFAOYSA-N 2-amino-2-[2-[4-octoxy-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol;hydrochloride Chemical compound Cl.CCCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F XUKLNKQQMDAVJF-UHFFFAOYSA-N 0.000 description 4
- OHDZWQGYWSUUIL-UHFFFAOYSA-N 2-amino-2-ethyl-4-[4-heptoxy-3-(trifluoromethyl)phenyl]butan-1-ol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CCC(N)(CC)CO)C=C1C(F)(F)F OHDZWQGYWSUUIL-UHFFFAOYSA-N 0.000 description 4
- CSDQQAQKBAQLLE-UHFFFAOYSA-N 4-(4-chlorophenyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine Chemical compound C1=CC(Cl)=CC=C1C1C(C=CS2)=C2CCN1 CSDQQAQKBAQLLE-UHFFFAOYSA-N 0.000 description 4
- HYOOWJSLZJKNRO-UHFFFAOYSA-N 5-bromo-2-octoxybenzonitrile Chemical compound CCCCCCCCOC1=CC=C(Br)C=C1C#N HYOOWJSLZJKNRO-UHFFFAOYSA-N 0.000 description 4
- 101100459319 Arabidopsis thaliana VIII-2 gene Proteins 0.000 description 4
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 4
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 4
- 239000005749 Copper compound Substances 0.000 description 4
- 239000002841 Lewis acid Substances 0.000 description 4
- 102100025750 Sphingosine 1-phosphate receptor 1 Human genes 0.000 description 4
- 101710155454 Sphingosine 1-phosphate receptor 1 Proteins 0.000 description 4
- 241001582429 Tetracis Species 0.000 description 4
- RBYGDVHOECIAFC-UHFFFAOYSA-L acetonitrile;palladium(2+);dichloride Chemical compound [Cl-].[Cl-].[Pd+2].CC#N.CC#N RBYGDVHOECIAFC-UHFFFAOYSA-L 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- ODWXUNBKCRECNW-UHFFFAOYSA-M bromocopper(1+) Chemical compound Br[Cu+] ODWXUNBKCRECNW-UHFFFAOYSA-M 0.000 description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- 125000001309 chloro group Chemical group Cl* 0.000 description 4
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 4
- 150000001880 copper compounds Chemical class 0.000 description 4
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 4
- XEIKGKFWHSGXSS-UHFFFAOYSA-N diethyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]-2-[2-(oxan-2-yloxy)ethyl]propanedioate Chemical compound CC(C)(C)OC(=O)NC(C(=O)OCC)(C(=O)OCC)CCOC1CCCCO1 XEIKGKFWHSGXSS-UHFFFAOYSA-N 0.000 description 4
- 229940043279 diisopropylamine Drugs 0.000 description 4
- DQYBDCGIPTYXML-UHFFFAOYSA-N ethoxyethane;hydrate Chemical compound O.CCOCC DQYBDCGIPTYXML-UHFFFAOYSA-N 0.000 description 4
- 229960000556 fingolimod Drugs 0.000 description 4
- SWZTYAVBMYWFGS-UHFFFAOYSA-N fingolimod hydrochloride Chemical compound Cl.CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 SWZTYAVBMYWFGS-UHFFFAOYSA-N 0.000 description 4
- 238000007327 hydrogenolysis reaction Methods 0.000 description 4
- 150000007517 lewis acids Chemical class 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 4
- DUYSYHSSBDVJSM-KRWOKUGFSA-N sphingosine 1-phosphate Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)COP(O)(O)=O DUYSYHSSBDVJSM-KRWOKUGFSA-N 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- GSOIIRXONPJTAK-UHFFFAOYSA-N tert-butyl n-[1-hydroxy-2-(hydroxymethyl)butan-2-yl]carbamate Chemical compound CCC(CO)(CO)NC(=O)OC(C)(C)C GSOIIRXONPJTAK-UHFFFAOYSA-N 0.000 description 4
- VIXYFRQJEPRPAR-UHFFFAOYSA-N tert-butyl n-[5-[2-[4-hexoxy-3-(trifluoromethyl)phenyl]ethyl]-2,2-dimethyl-1,3-dioxan-5-yl]carbamate Chemical compound C1=C(C(F)(F)F)C(OCCCCCC)=CC=C1CCC1(NC(=O)OC(C)(C)C)COC(C)(C)OC1 VIXYFRQJEPRPAR-UHFFFAOYSA-N 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- NRCFUYKTFAPUBR-GMUIIQOCSA-N (2r)-2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CC[C@@](C)(N)CO)C=C1C(F)(F)F NRCFUYKTFAPUBR-GMUIIQOCSA-N 0.000 description 3
- HDPNBNXLBDFELL-UHFFFAOYSA-N 1,1,1-trimethoxyethane Chemical compound COC(C)(OC)OC HDPNBNXLBDFELL-UHFFFAOYSA-N 0.000 description 3
- LSXKDWGTSHCFPP-UHFFFAOYSA-N 1-bromoheptane Chemical compound CCCCCCCBr LSXKDWGTSHCFPP-UHFFFAOYSA-N 0.000 description 3
- MLPGLPVUUORLLP-UHFFFAOYSA-N 2-amino-2-[2-[4-heptylsulfanyl-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol;hydrochloride Chemical compound Cl.CCCCCCCSC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F MLPGLPVUUORLLP-UHFFFAOYSA-N 0.000 description 3
- KJJPLEZQSCZCKE-UHFFFAOYSA-N 2-aminopropane-1,3-diol Chemical class OCC(N)CO KJJPLEZQSCZCKE-UHFFFAOYSA-N 0.000 description 3
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 3
- SZQBLMXBVYWBHM-UHFFFAOYSA-N 4-(2-fluoroethyl)-4-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]-2-methyl-5h-1,3-oxazole Chemical compound C1=C(C(F)(F)F)C(OCCCCCCC)=CC=C1CCC1(CCF)N=C(C)OC1 SZQBLMXBVYWBHM-UHFFFAOYSA-N 0.000 description 3
- IZZYABADQVQHLC-UHFFFAOYSA-N 4-methylbenzenesulfonyl fluoride Chemical compound CC1=CC=C(S(F)(=O)=O)C=C1 IZZYABADQVQHLC-UHFFFAOYSA-N 0.000 description 3
- OZJFNNBFVVQCEV-UHFFFAOYSA-N 5-[3-amino-4-hydroxy-3-(hydroxymethyl)butyl]-2-heptoxybenzonitrile;hydrochloride Chemical compound Cl.CCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C#N OZJFNNBFVVQCEV-UHFFFAOYSA-N 0.000 description 3
- NDNQNUMGGJYDTM-UHFFFAOYSA-N 5-[3-amino-4-hydroxy-3-(hydroxymethyl)butyl]-2-octoxybenzonitrile;hydrochloride Chemical compound Cl.CCCCCCCCOC1=CC=C(CCC(N)(CO)CO)C=C1C#N NDNQNUMGGJYDTM-UHFFFAOYSA-N 0.000 description 3
- GYCNHFWRPJXTSB-UHFFFAOYSA-N 5-bromo-2-fluorobenzonitrile Chemical compound FC1=CC=C(Br)C=C1C#N GYCNHFWRPJXTSB-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 3
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 3
- 239000005977 Ethylene Substances 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- VPIAKHNXCOTPAY-UHFFFAOYSA-N Heptane-1-thiol Chemical compound CCCCCCCS VPIAKHNXCOTPAY-UHFFFAOYSA-N 0.000 description 3
- UXRZLDREKITWRO-UHFFFAOYSA-N P(c1ccccc1)c1ccccc1.CC1(C)c2ccccc2Oc2ccccc12 Chemical compound P(c1ccccc1)c1ccccc1.CC1(C)c2ccccc2Oc2ccccc12 UXRZLDREKITWRO-UHFFFAOYSA-N 0.000 description 3
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 3
- QYTDEUPAUMOIOP-UHFFFAOYSA-N TEMPO Chemical group CC1(C)CCCC(C)(C)N1[O] QYTDEUPAUMOIOP-UHFFFAOYSA-N 0.000 description 3
- NBFCHCDCCVCHNK-GOSISDBHSA-N [(2r)-2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutyl] dihydrogen phosphate Chemical compound CCCCCCCOC1=CC=C(CC[C@@](C)(N)COP(O)(O)=O)C=C1C(F)(F)F NBFCHCDCCVCHNK-GOSISDBHSA-N 0.000 description 3
- LRFKWQGGENFBFO-IBGZPJMESA-N [(2s)-2-amino-2-(hydroxymethyl)-4-(4-octylphenyl)butyl] dihydrogen phosphate Chemical compound CCCCCCCCC1=CC=C(CC[C@](N)(CO)COP(O)(O)=O)C=C1 LRFKWQGGENFBFO-IBGZPJMESA-N 0.000 description 3
- AAZKZGRYBMYXQB-UHFFFAOYSA-N [1-(methoxymethoxy)-2-methyl-3-oxopropan-2-yl]carbamic acid Chemical compound COCOCC(C)(C=O)NC(O)=O AAZKZGRYBMYXQB-UHFFFAOYSA-N 0.000 description 3
- SKPKTJGDLTUMKR-UHFFFAOYSA-N [1-hydroxy-3-(methoxymethoxy)-2-methylpropan-2-yl]carbamic acid Chemical compound COCOCC(C)(CO)NC(O)=O SKPKTJGDLTUMKR-UHFFFAOYSA-N 0.000 description 3
- ZEEBGORNQSEQBE-UHFFFAOYSA-N [2-(3-phenylphenoxy)-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound C1(=CC(=CC=C1)OC1=NC(=CC(=C1)CN)C(F)(F)F)C1=CC=CC=C1 ZEEBGORNQSEQBE-UHFFFAOYSA-N 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- ANRNJQQUHLLCBV-UHFFFAOYSA-N diethyl 2-acetamido-2-[2-[4-methoxy-3-(trifluoromethyl)phenyl]-2-oxoethyl]propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)(NC(C)=O)CC(=O)C1=CC=C(OC)C(C(F)(F)F)=C1 ANRNJQQUHLLCBV-UHFFFAOYSA-N 0.000 description 3
- SWXBJBFZJPQYOG-UHFFFAOYSA-N diethyl 2-acetamido-2-[2-[4-methoxy-3-(trifluoromethyl)phenyl]ethyl]propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)(NC(C)=O)CCC1=CC=C(OC)C(C(F)(F)F)=C1 SWXBJBFZJPQYOG-UHFFFAOYSA-N 0.000 description 3
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000002198 insoluble material Substances 0.000 description 3
- 210000003563 lymphoid tissue Anatomy 0.000 description 3
- 239000002808 molecular sieve Substances 0.000 description 3
- 239000012452 mother liquor Substances 0.000 description 3
- ILRWZHFMKJLWMO-UHFFFAOYSA-N n-[1-hydroxy-2-(hydroxymethyl)-4-[4-methoxy-3-(trifluoromethyl)phenyl]butan-2-yl]acetamide Chemical compound COC1=CC=C(CCC(CO)(CO)NC(C)=O)C=C1C(F)(F)F ILRWZHFMKJLWMO-UHFFFAOYSA-N 0.000 description 3
- KUKSUQKELVOKBH-UHFFFAOYSA-N n-[bis[(2-methylpropan-2-yl)oxy]phosphanyl]-n-ethylethanamine Chemical compound CCN(CC)P(OC(C)(C)C)OC(C)(C)C KUKSUQKELVOKBH-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 229960004624 perflexane Drugs 0.000 description 3
- ZJIJAJXFLBMLCK-UHFFFAOYSA-N perfluorohexane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F ZJIJAJXFLBMLCK-UHFFFAOYSA-N 0.000 description 3
- 239000008177 pharmaceutical agent Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000006268 reductive amination reaction Methods 0.000 description 3
- 229910001923 silver oxide Inorganic materials 0.000 description 3
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 3
- YMMYUCSYHCEJIO-UHFFFAOYSA-N tert-butyl n-[1-[4-heptoxy-3-(trifluoromethyl)phenyl]-3-(hydroxymethyl)pentan-3-yl]carbamate Chemical compound CCCCCCCOC1=CC=C(CCC(CC)(CO)NC(=O)OC(C)(C)C)C=C1C(F)(F)F YMMYUCSYHCEJIO-UHFFFAOYSA-N 0.000 description 3
- WHERGMTWJAMXIK-UHFFFAOYSA-N tert-butyl n-[1-hydroxy-2-(hydroxymethyl)-4-(oxan-2-yloxy)butan-2-yl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CO)(CO)CCOC1CCCCO1 WHERGMTWJAMXIK-UHFFFAOYSA-N 0.000 description 3
- HUMOMCKUXUGIMB-UHFFFAOYSA-N tert-butyl n-[1-hydroxy-2-(hydroxymethyl)-4-[4-hydroxy-3-(trifluoromethyl)phenyl]butan-2-yl]carbamate Chemical compound CC(C)(C)OC(=O)NC(CO)(CO)CCC1=CC=C(O)C(C(F)(F)F)=C1 HUMOMCKUXUGIMB-UHFFFAOYSA-N 0.000 description 3
- BULWJTWDSWWOHF-UHFFFAOYSA-N tert-butyl n-[1-hydroxy-2-(hydroxymethyl)-4-[4-octoxy-3-(trifluoromethyl)phenyl]butan-2-yl]carbamate Chemical compound CCCCCCCCOC1=CC=C(CCC(CO)(CO)NC(=O)OC(C)(C)C)C=C1C(F)(F)F BULWJTWDSWWOHF-UHFFFAOYSA-N 0.000 description 3
- XSCXUSHHTFEZAT-UHFFFAOYSA-N tert-butyl n-[1-hydroxy-2-(methoxymethoxymethyl)-4-(oxan-2-yloxy)butan-2-yl]carbamate Chemical compound COCOCC(CO)(NC(=O)OC(C)(C)C)CCOC1CCCCO1 XSCXUSHHTFEZAT-UHFFFAOYSA-N 0.000 description 3
- WDUIOMIPYHORAT-UHFFFAOYSA-N tert-butyl n-[2-formyl-1-(methoxymethoxy)-4-(oxan-2-yloxy)butan-2-yl]carbamate Chemical compound COCOCC(C=O)(NC(=O)OC(C)(C)C)CCOC1CCCCO1 WDUIOMIPYHORAT-UHFFFAOYSA-N 0.000 description 3
- KSVHRGJHSAYNKY-UHFFFAOYSA-N tert-butyl n-[2-hydroxy-1-(methoxymethoxy)ethyl]-n-propylcarbamate Chemical compound CC(C)(C)OC(=O)N(CCC)C(CO)OCOC KSVHRGJHSAYNKY-UHFFFAOYSA-N 0.000 description 3
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- YGABUCCNCBMODG-UHFFFAOYSA-N 1-(trifluoromethylsulfonyl)imidazole Chemical compound FC(F)(F)S(=O)(=O)N1C=CN=C1 YGABUCCNCBMODG-UHFFFAOYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- MXZROAOUCUVNHX-UHFFFAOYSA-N 2-Aminopropanol Chemical class CCC(N)O MXZROAOUCUVNHX-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- NSLBSFBKBOZRPL-UHFFFAOYSA-N 2-amino-2-[2-[4-octylsulfanyl-3-(trifluoromethyl)phenyl]ethyl]propane-1,3-diol;hydrochloride Chemical compound Cl.CCCCCCCCSC1=CC=C(CCC(N)(CO)CO)C=C1C(F)(F)F NSLBSFBKBOZRPL-UHFFFAOYSA-N 0.000 description 2
- XJTCXIYCDUIGGL-UHFFFAOYSA-N 5-bromo-2-heptoxybenzonitrile Chemical compound CCCCCCCOC1=CC=C(Br)C=C1C#N XJTCXIYCDUIGGL-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- KWOLFJPFCHCOCG-UHFFFAOYSA-N Acetophenone Chemical compound CC(=O)C1=CC=CC=C1 KWOLFJPFCHCOCG-UHFFFAOYSA-N 0.000 description 2
- 238000006218 Arndt-Eistert homologation reaction Methods 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- DTWNZBROXNLDGB-UHFFFAOYSA-N CC#N.CC#N.Cl.Cl Chemical compound CC#N.CC#N.Cl.Cl DTWNZBROXNLDGB-UHFFFAOYSA-N 0.000 description 2
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical group F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 102000011011 Sphingosine 1-phosphate receptors Human genes 0.000 description 2
- 108050001083 Sphingosine 1-phosphate receptors Proteins 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 238000010306 acid treatment Methods 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229930182912 cyclosporin Natural products 0.000 description 2
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 2
- 239000012024 dehydrating agents Substances 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 150000001993 dienes Chemical class 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- ISOLMABRZPQKOV-UHFFFAOYSA-N diethyl 2-acetamidopropanedioate Chemical compound CCOC(=O)C(NC(C)=O)C(=O)OCC ISOLMABRZPQKOV-UHFFFAOYSA-N 0.000 description 2
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- JMUFUWHUKTUAJD-UHFFFAOYSA-N ethane-1,2-diamine methane Chemical compound C.NCCN JMUFUWHUKTUAJD-UHFFFAOYSA-N 0.000 description 2
- LBAQSKZHMLAFHH-UHFFFAOYSA-N ethoxyethane;hydron;chloride Chemical compound Cl.CCOCC LBAQSKZHMLAFHH-UHFFFAOYSA-N 0.000 description 2
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- JQRYUMGHOUYJFW-UHFFFAOYSA-N pyridine;trihydrobromide Chemical compound [Br-].[Br-].[Br-].C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1 JQRYUMGHOUYJFW-UHFFFAOYSA-N 0.000 description 2
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 2
- 229910052709 silver Inorganic materials 0.000 description 2
- 239000004332 silver Substances 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 238000003797 solvolysis reaction Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 2
- 150000003460 sulfonic acids Chemical class 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- 150000003555 thioacetals Chemical class 0.000 description 2
- 210000001541 thymus gland Anatomy 0.000 description 2
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 2
- 125000005147 toluenesulfonyl group Chemical group C=1(C(=CC=CC1)S(=O)(=O)*)C 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 2
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 2
- GUTDVBHLNJEILW-UHFFFAOYSA-M triphenyl-[[4-phenylmethoxy-3-(trifluoromethyl)phenyl]methyl]phosphanium;chloride Chemical compound [Cl-].C=1C=C(OCC=2C=CC=CC=2)C(C(F)(F)F)=CC=1C[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 GUTDVBHLNJEILW-UHFFFAOYSA-M 0.000 description 2
- OIUJCLYDEGSNLF-SFHVURJKSA-N (2s)-2-amino-4-[4-heptoxy-3-(trifluoromethyl)phenyl]-2-methylbutan-1-ol Chemical compound CCCCCCCOC1=CC=C(CC[C@](C)(N)CO)C=C1C(F)(F)F OIUJCLYDEGSNLF-SFHVURJKSA-N 0.000 description 1
- MIFIDEMRXCBMFX-UHFFFAOYSA-N (5-ethynyl-2,2-dimethyl-1,3-dioxan-5-yl)carbamic acid Chemical compound CC1(C)OCC(C#C)(NC(O)=O)CO1 MIFIDEMRXCBMFX-UHFFFAOYSA-N 0.000 description 1
- GVKVSLZKPIHBRK-UHFFFAOYSA-N (5-formyl-2,2-dimethyl-1,3-dioxan-5-yl)carbamic acid Chemical compound CC1(C)OCC(C=O)(NC(O)=O)CO1 GVKVSLZKPIHBRK-UHFFFAOYSA-N 0.000 description 1
- USVVENVKYJZFMW-ONEGZZNKSA-N (e)-carboxyiminocarbamic acid Chemical class OC(=O)\N=N\C(O)=O USVVENVKYJZFMW-ONEGZZNKSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 1
- BLIQUJLAJXRXSG-UHFFFAOYSA-N 1-benzyl-3-(trifluoromethyl)pyrrolidin-1-ium-3-carboxylate Chemical compound C1C(C(=O)O)(C(F)(F)F)CCN1CC1=CC=CC=C1 BLIQUJLAJXRXSG-UHFFFAOYSA-N 0.000 description 1
- MNDIARAMWBIKFW-UHFFFAOYSA-N 1-bromohexane Chemical compound CCCCCCBr MNDIARAMWBIKFW-UHFFFAOYSA-N 0.000 description 1
- VMKOFRJSULQZRM-UHFFFAOYSA-N 1-bromooctane Chemical compound CCCCCCCCBr VMKOFRJSULQZRM-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 1
- GCUOLJOTJRUDIZ-UHFFFAOYSA-N 2-(2-bromoethoxy)oxane Chemical compound BrCCOC1CCCCO1 GCUOLJOTJRUDIZ-UHFFFAOYSA-N 0.000 description 1
- IMSODMZESSGVBE-UHFFFAOYSA-N 2-Oxazoline Chemical compound C1CN=CO1 IMSODMZESSGVBE-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- IOAOAKDONABGPZ-UHFFFAOYSA-N 2-amino-2-ethylpropane-1,3-diol Chemical compound CCC(N)(CO)CO IOAOAKDONABGPZ-UHFFFAOYSA-N 0.000 description 1
- GZTKYOABNIDNQU-UHFFFAOYSA-N 2-amino-2-methylpropane-1,3-diol;hydrochloride Chemical compound Cl.OCC(N)(C)CO GZTKYOABNIDNQU-UHFFFAOYSA-N 0.000 description 1
- ULQQGOGMQRGFFR-UHFFFAOYSA-N 2-chlorobenzenecarboperoxoic acid Chemical compound OOC(=O)C1=CC=CC=C1Cl ULQQGOGMQRGFFR-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 125000003504 2-oxazolinyl group Chemical group O1C(=NCC1)* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- GISCXYLVNFIAEL-UHFFFAOYSA-N 4-(chloromethyl)-1-phenylmethoxy-2-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC(CCl)=CC=C1OCC1=CC=CC=C1 GISCXYLVNFIAEL-UHFFFAOYSA-N 0.000 description 1
- ZDRVLAOYDGQLFI-UHFFFAOYSA-N 4-[[4-(4-chlorophenyl)-1,3-thiazol-2-yl]amino]phenol;hydrochloride Chemical compound Cl.C1=CC(O)=CC=C1NC1=NC(C=2C=CC(Cl)=CC=2)=CS1 ZDRVLAOYDGQLFI-UHFFFAOYSA-N 0.000 description 1
- 101100132433 Arabidopsis thaliana VIII-1 gene Proteins 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- QWOJMRHUQHTCJG-UHFFFAOYSA-N CC([CH2-])=O Chemical compound CC([CH2-])=O QWOJMRHUQHTCJG-UHFFFAOYSA-N 0.000 description 1
- 229940122739 Calcineurin inhibitor Drugs 0.000 description 1
- 102100024123 Calcineurin-binding protein cabin-1 Human genes 0.000 description 1
- 101710192106 Calcineurin-binding protein cabin-1 Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 238000006824 Eschweiler-Clarke methylation reaction Methods 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 241000282414 Homo sapiens Species 0.000 description 1
- 238000006546 Horner-Wadsworth-Emmons reaction Methods 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 102100025749 Sphingosine 1-phosphate receptor 2 Human genes 0.000 description 1
- 101710155462 Sphingosine 1-phosphate receptor 2 Proteins 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- ZENFMHRBKZEKJV-UHFFFAOYSA-N [5-(hydroxymethyl)-2,2-dimethyl-1,3-dioxan-5-yl]carbamic acid Chemical compound CC1(C)OCC(CO)(NC(O)=O)CO1 ZENFMHRBKZEKJV-UHFFFAOYSA-N 0.000 description 1
- RNJCXDJXPXWMDK-UHFFFAOYSA-N [AlH3].COCCO[Na] Chemical compound [AlH3].COCCO[Na] RNJCXDJXPXWMDK-UHFFFAOYSA-N 0.000 description 1
- QQIRAVWVGBTHMJ-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;lithium Chemical compound [Li].C[Si](C)(C)N[Si](C)(C)C QQIRAVWVGBTHMJ-UHFFFAOYSA-N 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- QNFLUQDLGGWVNX-UHFFFAOYSA-N benzyl 4-fluoro-3-(trifluoromethyl)benzoate Chemical compound C1=C(C(F)(F)F)C(F)=CC=C1C(=O)OCC1=CC=CC=C1 QNFLUQDLGGWVNX-UHFFFAOYSA-N 0.000 description 1
- VKFIGHNBHQOVBP-UHFFFAOYSA-N benzyl 4-phenylmethoxy-3-(trifluoromethyl)benzoate Chemical compound FC(F)(F)C1=CC(C(=O)OCC=2C=CC=CC=2)=CC=C1OCC1=CC=CC=C1 VKFIGHNBHQOVBP-UHFFFAOYSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- 229940046731 calcineurin inhibitors Drugs 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 150000001923 cyclic compounds Chemical class 0.000 description 1
- ULRLHEXGRUWQLQ-UHFFFAOYSA-N diethyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)NC(=O)OC(C)(C)C ULRLHEXGRUWQLQ-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000002222 downregulating effect Effects 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 125000003784 fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 231100000304 hepatotoxicity Toxicity 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007056 liver toxicity Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N monoethanolamine hydrochloride Natural products NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 231100000228 neurotoxicity Toxicity 0.000 description 1
- 230000007135 neurotoxicity Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- KZCOBXFFBQJQHH-UHFFFAOYSA-N octane-1-thiol Chemical compound CCCCCCCCS KZCOBXFFBQJQHH-UHFFFAOYSA-N 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- MUMZUERVLWJKNR-UHFFFAOYSA-N oxoplatinum Chemical compound [Pt]=O MUMZUERVLWJKNR-UHFFFAOYSA-N 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- LIGACIXOYTUXAW-UHFFFAOYSA-N phenacyl bromide Chemical group BrCC(=O)C1=CC=CC=C1 LIGACIXOYTUXAW-UHFFFAOYSA-N 0.000 description 1
- 229910003446 platinum oxide Inorganic materials 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001012 protector Effects 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 150000003283 rhodium Chemical class 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 108010035597 sphingosine kinase Proteins 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- UDYFLDICVHJSOY-UHFFFAOYSA-N sulfur trioxide-pyridine complex Substances O=S(=O)=O.C1=CC=NC=C1 UDYFLDICVHJSOY-UHFFFAOYSA-N 0.000 description 1
- 238000004808 supercritical fluid chromatography Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- CJTCZLLNODMGQA-UHFFFAOYSA-N tert-butyl n-[1-[4-hydroxy-3-(trifluoromethyl)phenyl]-3-(methoxymethoxymethyl)-5-(oxan-2-yloxy)pentan-3-yl]carbamate Chemical compound C=1C=C(O)C(C(F)(F)F)=CC=1CCC(NC(=O)OC(C)(C)C)(COCOC)CCOC1CCCCO1 CJTCZLLNODMGQA-UHFFFAOYSA-N 0.000 description 1
- ISPQKURHDKOSDG-UHFFFAOYSA-N tert-butyl n-[3-(methoxymethoxymethyl)-1-(oxan-2-yloxy)-5-[4-phenylmethoxy-3-(trifluoromethyl)phenyl]pentan-3-yl]carbamate Chemical compound C=1C=C(OCC=2C=CC=CC=2)C(C(F)(F)F)=CC=1CCC(NC(=O)OC(C)(C)C)(COCOC)CCOC1CCCCO1 ISPQKURHDKOSDG-UHFFFAOYSA-N 0.000 description 1
- UVJVSOYSJMHQGX-UHFFFAOYSA-N tert-butyl n-[5-[2-(3-cyano-4-heptoxyphenyl)ethyl]-2,2-dimethyl-1,3-dioxan-5-yl]carbamate Chemical compound C1=C(C#N)C(OCCCCCCC)=CC=C1CCC1(NC(=O)OC(C)(C)C)COC(C)(C)OC1 UVJVSOYSJMHQGX-UHFFFAOYSA-N 0.000 description 1
- HRCWIKADZLQHAQ-UHFFFAOYSA-N tert-butyl n-[5-[2-[4-heptoxy-3-(trifluoromethyl)phenyl]ethyl]-2,2-dimethyl-1,3-dioxan-5-yl]carbamate Chemical compound C1=C(C(F)(F)F)C(OCCCCCCC)=CC=C1CCC1(NC(=O)OC(C)(C)C)COC(C)(C)OC1 HRCWIKADZLQHAQ-UHFFFAOYSA-N 0.000 description 1
- KANYXWDVBSATPR-UHFFFAOYSA-N tert-butyl n-[5-[2-[4-hydroxy-3-(trifluoromethyl)phenyl]ethyl]-2,2-dimethyl-1,3-dioxan-5-yl]carbamate Chemical compound C=1C=C(O)C(C(F)(F)F)=CC=1CCC1(NC(=O)OC(C)(C)C)COC(C)(C)OC1 KANYXWDVBSATPR-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 150000007944 thiolates Chemical class 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 125000006000 trichloroethyl group Chemical group 0.000 description 1
- DQWPFSLDHJDLRL-UHFFFAOYSA-N triethyl phosphate Chemical compound CCOP(=O)(OCC)OCC DQWPFSLDHJDLRL-UHFFFAOYSA-N 0.000 description 1
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- PRXNKYBFWAWBNZ-UHFFFAOYSA-N trimethylphenylammonium tribromide Chemical compound Br[Br-]Br.C[N+](C)(C)C1=CC=CC=C1 PRXNKYBFWAWBNZ-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/34—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/02—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/01—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms
- C07C211/26—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing at least one six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/64—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/70—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups and doubly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/72—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups and doubly-bound oxygen atoms bound to the same carbon skeleton with the carbon atoms of the carboxamide groups bound to acyclic carbon atoms
- C07C235/76—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups and doubly-bound oxygen atoms bound to the same carbon skeleton with the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
- C07C235/78—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups and doubly-bound oxygen atoms bound to the same carbon skeleton with the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of an unsaturated carbon skeleton the carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/54—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/16—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/31—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
- C07C323/32—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton having at least one of the nitrogen atoms bound to an acyclic carbon atom of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/39—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton at least one of the nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom
- C07C323/43—Y being a hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/04—1,3-Dioxanes; Hydrogenated 1,3-dioxanes
- C07D319/06—1,3-Dioxanes; Hydrogenated 1,3-dioxanes not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/091—Esters of phosphoric acids with hydroxyalkyl compounds with further substituents on alkyl
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/141—Esters of phosphorous acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4056—Esters of arylalkanephosphonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6527—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and oxygen atoms as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6527—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and oxygen atoms as the only ring hetero atoms
- C07F9/653—Five-membered rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Urology & Nephrology (AREA)
- Otolaryngology (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Transplantation (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
composto de amina e uso do mesmo para fins medicinais. a presente invenção refere-se a um novo composto de amina representado pela seguinte fórmula (i), em que r é um átomo de hidrogênio ou p(=o)(oh) 2, x é um átomo de oxigênio ou um átomo de enxofre, y é ch2 ch2 ou ch=ch, r1 é ciano ou alquila tendo um número de carbono de 1 a 4 e substituída por um ou mais átomos de halogeno, r2 é alquila tendo um número de carbono de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogeno, r3 e r4 podem ser os mesmos ou diferentes e cada um é um átomo de hidrogênio ou alquila tendo um número de carbono de 1 a 4, e n é 5 a 8, o qual é superior em ação imunossupressora, ação supressora de rejeição e similares, e apresenta reduzidos efeitos colaterais tais como bradicardia e similares, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou hidratos do mesmo, ou solvato, bem como uma composição farmacêutica contendo este composto e um veículo farmaceuticamente aceitável.
Description
[0001] A presente invenção se refere a compostos de amina e uso dos mesmos como agentes farmacêuticos.
[0002] Nos últimos anos, inibidores da calcineurina tais como ciclosporina e FK506 são usados para suprimir rejeição de pacientes que são submetidos o transplante de órgãos. No entanto, um certo tipo de inibidor da calcineurina tal como ciclosporina algumas vezes causa efeitos colaterais adversos tais como toxicidade renal, toxicidade hepática, neurotoxicidade e similares. Portanto, o desenvolvimento de um agente farmacêutico mais seguro e altamente eficaz está em andamento para suprimir a rejeição de pacientes de transplante.
[0003] As referências de Patente 1 a 3 descrevem que compostos de 2-aminopropano-1,3-diol são úteis como supressores de rejeição (aguda ou crônica) em transplante de órgãos ou medula óssea, bem como fármacos terapêuticos para várias doenças auto-imunes tais como psoríase, doença de Behcet e similares e doenças de reumatismo.
[0004] Um destes compostos, cloridrato de 2-amino-2-[2-(4-octilfenil)etil]propano-1,3-diol (nas partes que se seguem referido algumas vezes como FTY720) atualmente está sob desenvolvimento clínico como um supressor de rejeição em transplante renal. FTY720 é rapidamente convertido em fosfo-FTY720 (nas partes que se seguem referido algumas vezes como FTY720-P, por exemplo, 2-amino-2- fosforiloximetil-4-(4-octilfenil)butanol) por esfingosina quinase in vivo. FTY720-P age como um agonista de 4 tipos de S1P receptores (diferentes de S1P2) entre 5 tipos de esfingosina-1-fosfato (nas partes que se seguem referido algumas vezes como S1P) receptores nas partes que se seguem referido algumas vezes como S1P1-5, respectivamente) (referência não-patente 1).
[0005] Recentemente, foi sugerido que S1P1 em S1P receptores é essencial para a emigração de linfócitos maduros do timo e tecidos linfóides secundários. FTY720-P age como um S1P1 agonista regulando para baixo S1P1 sobre os linfócitos. Em conseqüência, a emigração de linfócitos maduros do timo e tecidos linfóides secundários é inibida e linfócitos maduros circulantes no sangue são seqüestrados nos tecidos linfóides secundários, por meio do qual a ação imunossupressora é exibida (referência não-patente 2).
[0006] Por outro lado, receia-se que compostos de 2-aminopropano-1,3-diol convencionais apresentem expressão de bradicardia transitória como efeitos colaterais, e para resolver este problema, foram reportadas uma série de novos compostos obtidos modificando as estruturas químicas de compostos de 2-aminopropano-1,3-diol. Entre estes, como um composto tendo um substituinte sobre o anel benzeno de FTY720, a referência de patente 4 descreve um derivado de aminopropanol como um modulador de S1P receptor com um grupo fosfórico, e ambas as referência de patente 5 e 6 descrevem derivados de aminopropanol como moduladores de S1P receptor. No entanto, um grupo trialoalquila, por exemplo, um grupo trifluorometila, não é revelado como um substituinte sobre o anel benzeno no mesmo. Em efeito, o nível de segurança como um produto farmacêutico não atingiu um nível satisfatório conforme a situação permanece.
[0007] referência de patente 1: patente internacional N° WO 94/08943
[0008] referência de patente 2: patente internacional N° WO 96/06068
[0009] referência de patente 3: patente internacional N° WO 98/45429
[00010] referência de patente 4: patente internacional N° WO 02/076995
[00011] referência de patente 5: patente internacional N° WO 2004/096752
[00012] referência de patente 6: patente internacional N° WO 2004/110979
[00013] referência não-patente 1: Science, 2002, vol. 296, pages 346 - 349
[00014] referência não-patente 2: Nature, 2004, vol. 427, pages 355 - 360
[00015] Um objetivo da presente invenção é proporcionar um novo composto de amina superior na ação imunossupressora, ação supressora de rejeição e similares, o qual apresente reduzidos efeitos colaterais tais como bradicardia e similares.
[00016] Os presentes inventores conduziram estudos adicionais em vista da situação mencionada acima e descobriram que um composto de amina tendo a fórmula estrutural em particular mencionada abaixo pode atingir o objetivo, o qual resultou no completamento da presente invenção.
[00017] Por conseguinte, o ponto principal da presente invenção é como se segue.
[00018] 1. Um composto representado pela seguinte fórmula (I) 
[00019] em que R é um átomo de hidrogênio ou P(=O)(OH)2, X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, R3 e R4 podem ser os mesmos ou diferentes e cada um é um átomo de hidrogênio ou alquila tendo um número de carbonos de 1 a 4, e n é 5 a 8, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00020] 2. O composto do 1 supracitado, em que R3 e R4 são cada um um átomo de hidrogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00021] 3. O composto de 1 ou 2, tendo a seguinte fórmula (Ia) ou (Ib)
[00022] em que R é hidrogênio ou P(=O)(OH)2, X é um átomo de oxigênio ou um átomo de enxofre, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00023] 4. O composto de qualquer um de 1 a 3, em que X é um átomo de oxigênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00024] 5. O composto de qualquer um de 1 a 4, em que Y é CH2CH2, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00025] 6. O composto de qualquer um de 1 a 5, em que R1 é metila substituída por um ou mais átomos de halogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00026] 7. O composto de qualquer um de 1 a 6, em que R1 é trifluorometila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00027] 8. O composto de qualquer um de 1 a 7, em que R2 é metila opcionalmente substituída por um ou mais grupos hidroxila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00028] 9. O composto de qualquer um de 1 a 8, em que R2 é hidroximetila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00029] 10. O composto de qualquer um de 1 a 9, em que R é um átomo de hidrogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00030] 11. O composto de qualquer um de 1 a 4, em que o composto da fórmula (I) é qualquer um dos seguintes a - e, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- a. 2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)etil] propano-1,3-diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
- b. (E)-2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)vinil] propano-1,3-diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
- c. 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-metilbutanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
- d. (R)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-metilbutanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
- e. 2-amino-2-[2-(3-ciano-4-heptiloxifenil)etil]propano-1,3- diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
[00031] 12. O composto de qualquer um de 1 a 4, em que o composto da fórmula (I) é qualquer um dos seguintes f - j, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
f. 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
g. (E)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)-3-buten-1-ol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
h. éster de mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)- 2-metilbutílico] do ácido fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
i. éster de mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)- 2-metilbutílico] de ácido (R)-fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
j. 2-amino-4-(3-ciano-4-heptiloxifenil)-2-(fosforiloximetil) butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
f. 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
g. (E)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)-3-buten-1-ol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
h. éster de mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)- 2-metilbutílico] do ácido fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
i. éster de mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)- 2-metilbutílico] de ácido (R)-fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
j. 2-amino-4-(3-ciano-4-heptiloxifenil)-2-(fosforiloximetil) butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo
[00032] 13. 2-Amino-2-[2-(4-heptilóxi-3- trifluorometilfenil)etil]propano-1,3-diol, ou um cloridrato do mesmo.
[00033] 14. Uma composição farmacêutica compreendendo o composto de qualquer um de 1 a 13 e um veículo farmaceuticamente aceitável.
[00034] 15. A composição farmacêutica de 14, a qual é usada para o tratamento ou a profilaxia de doenças auto-imunes; profilaxia ou supressão de resistência ou rejeição aguda ou rejeição crônica de transplante de órgão ou tecido; tratamento ou profilaxia de doença de enxerto-versus-hospedeiro (GvH) devido o transplante de medula óssea; ou tratamento ou profilaxia de doenças alérgicas.
[00035] 16. A composição farmacêutica da 14, em que a doença auto-imune é artrite reumatóide, esclerose múltipla, encefalomielite, lúpus eritematoso sistêmico, nefrite por lúpus, síndrome nefrótica, psoríase ou diabetes melito Tipo I.
[00036] 17. A composição farmacêutica da 14, em que a doença alérgica é dermatite atópica, rinite alérgica ou asma.
[00037] De acordo com a presente invenção, pode ser proporcionado um novo composto tendo uma ação superior de redução de linfócitos do sangue periférico, e redzidos efeitos colaterais tais como bradicardia e similares.
[00038] A presente invenção é explicada em detalhes a seguir.
[00039] O composto da presente invenção é um composto representado pela seguinte fórmula (I) 
[00040] em que R é um átomo de hidrogênio ou P(=O)(OH)2, X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, R3 e R4 podem ser os mesmos ou diferentes e cada um é um átomo de hidrogênio ou alquila tendo um número de carbonos de 1 a 4, e n é 5 a 8, ou um sal de adição de ácido farmaceuticamente aceitável ou um sal de metal do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
[00041] Na presente invenção, o átomo de halogênio é um átomo de flúor, um átomo de cloro, um átomo de bromo ou um átomo de iodo, em que um átomo de flúor é um exemplo preferencial.
[00042] A alquila tendo um número de carbonos de 1 a 4 significa uma alquila de cadeia reta ou de cadeia ramificada tendo um número de carbonos de 1 a 4. Exemplos do mesmo incluem metila, etila, n-propila, isopropila, n-butila, isobutila, butila secundária, butila terciária (nas partes que se seguem "terciária" é algumas vezes indicado como t- ou terc-) e similares. Exemplos preferenciais incluem metila, etila, n-propila e isopropila, e exemplos mais preferenciais incluem metila e etila.
[00043] Exemplos preferenciais de R na fórmula (I) mencionada acima incluem um átomo de hidrogênio.
[00044] Exemplos preferenciais de X incluem um átomo de oxigênio, e exemplos preferenciais de Y incluem CH2CH2.
[00045] Exemplo preferencial de n é 6 ou 7, e exemplo mais preferencial é 6.
[00046] Exemplos preferenciais de R1 incluem fluorometila, difluorometila, trifluorometila, 2,2,2-trifluoroetila e ciano, e exemplos mais preferenciais incluem trifluorometila e ciano, e exemplo ainda mais preferencial é trifluorometila.
[00047] Exemplos preferenciais de R2 incluem metila, etila, hidroximetila, hidroxietila, fluorometila, clorometila, fluoroetila, difluoroetila, trifluoroetila e tricloroetila, exemplos mais preferenciais incluem metila, etila, hidroximetila, 2-hidroxietila e 2-fluoroetila, exemplos ainda mais preferenciais incluem metila e hidroximetila, e hidroximetila é a mais preferida.
[00048] Exemplos preferenciais de R3 e R4 incluem um átomo de hidrogênio, metila e etila, as quais podem ser as mesmas ou diferentes, exemplos mais preferenciais incluem um átomo de hidrogênio e metila, e um átomo de hidrogênio é o mais preferencial.
[00049] Exemplos do sal de adição de ácido farmaceuticamente aceitável do composto da presente invenção incluem sal de ácido inorgânico, sal de ácido orgânico, sal de metal de álcali, sal de metal alcalino-terroso e similares. O composto da presente invenção engloba o composto da fórmula (I) mencionado acima e um sal de adição de ácido farmaceuticamente aceitável do mesmo, e também um isômero geométrico, uma forma oticamente ativa, um hidrato e um solvato do mesmo.
[00050] Exemplos específicos do composto da presente invenção incluem os seguintes.
[00051] 2-amino-2-[[2-(4-heptilóxi-3-trifluorometilfenil)etil]]propano-1,3-diol, e um cloridrato do mesmo,
[00052] (E)-2-amino-2-[2-(4-heptilóxi-3- trifluorometilfenil)vinil]propano-1,3-diol, e um cloridrato do mesmo,
[00053] (R)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutanol, e um cloridrato do mesmo.
[00054] Dos compostos da presente invenção, um composto preferencial é 2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)etil]propano-1,3-diol ou um cloridrato do mesmo.
[00055] O método de síntese do composto da presente invenção é, por exemplo, o seguinte.
[00056] 1) Dos compostos da presente invenção, o composto (I-1) representado pela fórmula (Ia) em que R é um átomo de hidrogênio, X é um átomo de oxigênio e R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio é sintetizado pelo seguinte esquema (II).
[00057] em que Ra, Rb, Rc e Rd são grupos protetores, Xa e Xb são grupos de saída, R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio.
[00058] Ra na fórmula é um átomo de hidrogênio ou um grupo carboxila protetor diverso. Por exemplo, alquila (especificamente metila, etila e similares), aralquil (benzila e similares), o mesmo substituinte como para Rb e similares podem ser mencionados. Rb na fórmula não é particularmente limitado contanto que proteja um grupo hidroxila fenólico. Por exemplo, alquila (especificamente metila, heptila e similares), aralquil (benzila e similares) e similares podem ser mencionados. Quando heptila é usado como Rb , o composto da invenção (I-1) pode ser obtido sem remoção de Rb . Rc na fórmula não é particularmente limitado contanto que proteja um grupo hidroxila. Por exemplo, acila (preferencialmente uma acila tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), trialquilsilila (especificamente trimetilsilila e similares), benzila e um substituinte formador de composto acetal (especificamente metoximetila, tetraidropiranila e similares) podem ser mencionados. Quando R2 tem um grupo hidroxila, seus grupos protetores Re (Re é especificamente similar a Rc) e Rc também podem ser ligados para formar acetal cíclico. O grupo protetor mostrado por Rd na fórmula não é particularmente limitado contanto que proteja um grupo amino. Por exemplo, acila (preferencialmente uma acila tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), um grupo carbamato (especificamente t-butiloxicarbonila, benziloxicarbonila e similares) e similares podem ser mencionados. O grupo de saída para Xa não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por íon alcóxido (Rb-O-). Por exemplo, um átomo de halogênio (especificamente um átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados. O grupo de saída para Xb não é particularmente limitado contanto que seja dissociado durante uma condensação de intermediário (II-4) e trifenilfosfina, e não inibe a reação de Wittig subseqüente. Por exemplo, um átomo de halogênio (especificamente átomo de iodo, átomo de bromo, átomo de cloro e similares), metanossulfonilóxi, toluenossulfonilóxi e similares podem ser mencionados.
[00059] Na primeira etapa, o intermediário (II-2) é obtido condensando derivado do ácido benzóico (II-1) tendo o grupo de saída X a na posição 4 com álcool Rb -OH para introduzir um grupo funcional oxigênio tendo o grupo protetor Rb na posição 4. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio e similares ou uma base orgânica tal como 1,8-diazabiciclo[5.4.0] undec7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 100C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00060] Na segunda etapa, o intermediário (II-3) tendo um grupo hidroxila é obtido reduzindo o grupo carboxila do intermediário (II-2). O reagente a ser usado para a redução não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem metais de álcali tais como sódio e similares, metais alcalino-terrosos, hidretos metálicos tais como hidreto de diisobutilalumínio e similares, compostos de complexo de hidrogênio de metal tais como hidreto de alumínio de lítio, boridreto de sódio e similares, compostos de boro tais como diborano e similares, hidrogenação catalítica usando um catalisador do tipo homogêneo ou do tipo heterogêneo, e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Exemplos específicos dos mesmos incluem redução usando diborano, hidreto de alumínio de lítio ou boridreto de lítio em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por 10 min a 12 horas, redução usando boridreto de sódio ou boridreto de cálcio em um solvente alcoólico tal como etanol e similares ou em um solvente misto de um solvente alcoólico e um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até a temperatura de refluxo por 30 min a 24 horas, e similares. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00061] Na terceira etapa, o grupo hidroxila do intermediário (II-3) é convertido no grupo de saída Xb. O reagente não é particularmente limitado contanto que seja um reagente capaz de converter um grupo hidroxila alcoólico em Xb. Exemplos do reagente usado quando Xb é um átomo de halogênio incluem N-clorossuccinimida, N-bromossuccinimida, tetracloreto de carbono e uma combinação dos mesmos e um auxiliar de reação tal como trifenilfosfina, uma base e similares, ácidos inorgânicos tais como ácido clorídrico, ácido bromídrico, ácido iodídrico, tribrometo de fósforo, pentabrometo de fósforo, tricloreto de fósforo, pentacloreto de fósforo, iodo, bromo, cloro, tionil halogenado e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C até 130°C por 10 min a 6 horas. Quando é usado um ácido inorgânico, a reação pode ser realizada em uma solução aquosa ou um sistema de duas camadas de um solvente orgânico tal como tolueno e similares e água.
[00062] Exemplos do reagente usado quando Xb é sulfonilóxi incluem uma combinação de um cloreto de sulfonila tal como cloreto de metanossulfonila, cloreto de tolueno sulfonila e similares, uma base orgânica tal como trietilamina, piridina e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C a 50°C por cerca de 5 min a 3 horas. Depois da reação, o produtofim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00063] Na quarta etapa, sal de fosfônio (II-5) é obtido reagindo o intermediário (II-4) tendo o grupo de saída Xb com trifenilfosfina. A reação é realizada, por exemplo, em um solvente inativo tal como éter dietílico, benzeno, tolueno e similares a partir da temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por evaporação do solvente, resfriamento e adição de solvente fracamente solúvel tal como éter diisopropílico, hexano e similares, conforme necessário, depois da qual coleta do sólido precipitado por filtração.
[00064] Na quinta etapa, o intermediário fenólico (II-7) é obtido condensando sal de fosfônio (II-5) com o aldeído (II-6) sintetizado separadamente usando reação de Wittig, reduzindo o composto de olefina obtido, e removendo o grupo protetor Rb. As condições da reação de Wittig são aquelas usadas de modo geral para reação de Wittig. Por exemplo, a reação é realizada usando uma base tal como t-butóxido de potássio e similares em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. O reagente a ser usado para a subseqüente redução de uma ligação dupla não é limitado contanto que seja usado para redução de olefina geral. Exemplos do mesmo incluem hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares, um catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina) ródio(I) e similares) e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, sob 1 a 20 atm de pressão de hidrogênio, sob esfriamento com gelo até a temperatura de refluxo a partir de 30 min até 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. As condições da subseqüente remoção do grupo protetor Rb não são particularmente limitadas contanto que sejam usadas para a remoção de Rb . Por exemplo, quando Rb é metila, pode ser usado um método para usar um ácido de Lewis tal como tribrometo de boro e similares em solvente de cloreto de metileno. Quando Rb é acila tal como acetila e similares, pode ser usado um método para usar uma base inorgânica tal como hidróxido de sódio e similares em um solvente misto de um solvente alcoólico e água. Quando Rb é um grupo protetor do tipo éter tal como metoximetila, tetraidropiranila, t-butila e similares, pode ser usado um método para usar um ácido tal como ácido clorídrico, ácido trifluoroacético e similares. Quando Rb é um grupo protetor o qual pode ser removido por hidrogenação catalítica (por exemplo, benzila, benzila substituída, benziloximetila e similares), a desproteção de Rb pode ser realizada simultaneamente com a redução supracitada da ligação dupla. Quando Rb é heptila, a remoção de Rb não é necessária, e a alquilação de fenol na etapa seguinte também pode ser omitida.
[00065] Na sexta etapa, o composto da presente invenção (I-1) é obtido alquilando o grupo hidroxila fenólico do intermediário (II-7), e removendo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. Exemplos do reagente a ser usado para alquilação do grupo hidroxila fenólico o intermediário (II-7) incluem uma combinação de um agente alquilante tal como heptil haleto e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. A reação é realizada, por exemplo, em um solvente polar tal como N,N-dimetilformamida e similares, ou em um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Para alquilação do grupo hidroxila fenólico o intermediário (II-7) tem, reação de Mitsunobu de condensação de álcool heptílico e usando um composto de fosfina tal como trifenilfosfina e similares e um derivado de ácido azodicarboxílico tal como diisopropil azodicarboxilato e similares pode ser usada. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até - 50°C por cerca de 10 min a 6 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. A desproteção subseqüente não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar um acetal cíclico, e Rd é t-butiloxicarbonila, podem ser desprotegidos simultaneamente usando um ácido. Exemplos do ácido para esse fim incluem ácidos inorgânicos tais como ácido clorídrico e similares, ácido trifluoroacético e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00066] 2) Dos compostos da presente invenção, o composto (I-1), isto é, um composto em que, na fórmula (Ia), R é um átomo de hidrogênio, X é um átomo de oxigênio e R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, também é sintetizada usando o intermediário (III-1) o qual é sintetizado pelo seguinte esquema (III) a partir do intermediário (II-2, Ra é H) no esquema (II) ou um composto conhecido representado pela fórmula (III-2).
[00067] em que Rb é um grupo protetor, Rf-OH é um álcool usado para uma reação de solvólise, e R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio.
[00068] Rb é conforme definido no esquema de fórmula (II). Exemplos de Rf incluem metila, etila, benzila e similares. Para a síntese do composto (II-2) no esquema supracitado, podem ser usadas algumas condições de reação gerais para reação de Arndt-Eistert. Além disso, para a redução do éster obtido, podem ser usados o reagente e condições usados para a segunda etapa no esquema (II). Para a síntese do composto (III-2) no esquema supracitado, podem ser usadas as condições para reação geral de Wittig. Para o tratamento ácido subseqüente, um ácido inorgânico tal como ácido clorídrico e similares é usado em água ou um solvente misto de um solvente orgânico tal como tetraidrofurano e similares e água. Para a redução subseqüente, pode ser realizada uma redução usando um composto de complexo de hidrogênio de metal tal como hidreto de alumínio de lítio, boridreto de sódio e similares, uma hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares ou um catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares, ou estas podem ser seqüencialmente realizadas continuamente. O intermediário alcoólico obtido (III-1) no esquema pode ser convertido no composto da presente invenção por um método conhecido (por exemplo, Journal of Medicinal Chemistry vol.43 (2000) 2946-2961).
[00069] 3) O composto (I-2), isto é, um composto em que, a fórmula (Ia), R é um átomo de hidrogênio, X é um átomo de oxigênio e R1 é trifluorometila ou ciano é sintetizado pelo seguinte esquema (IV).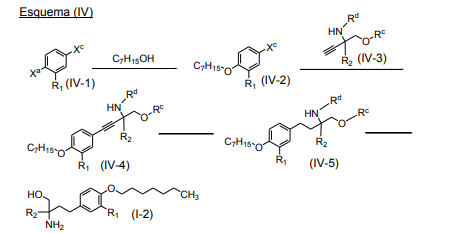
[00070] em que Rc, Rd é um grupo protetor, Xa e Xc são grupos de saída, R1 é trifluorometila ou ciano, e R2 é conforme definido acima
[00071] Rc, Rd e Xa na fórmula são conforme definidos acima. O grupo de saída para Xc não é particularmente limitado contanto que seja ativado por um catalisador e dissociado durante uma reação de Sonogashira. Exemplos do mesmo incluem um átomo de halogênio (preferencialmente um átomo de iodo, um átomo de bromo e similares), trifluorometanossulfonilóxi e similares.
[00072] Na primeira etapa, o intermediário (IV-2) é obtido condensando o composto (IV-1) tendo o grupo de saída Xa com álcool heptílico. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida e dimetilsulfóxido, ou em um solvente etéreo tal como tetraidrofurano e similares, na presença de uma base. Exemplos da base incluem bases inorgânicas tais como hidreto de sódio, hidróxido de potássio e similares e bases orgânicas tais como 1,8- diazabiciclo[5.4.0]undec-7-eno e similares. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 100°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00073] Na segunda etapa, o intermediário (IV-4) tendo uma ligação tripla é obtido condensando o intermediário (IV-2) com o intermediário (IV-3) o qual é sintetizado a partir do intermediário (II-6) por um método conhecido (por exemplo, Tetrahedron vol.57 (2001) 6531-6538, Chemical & Pharmaceutical Bulletin vol.53 (2005) 100-102), sob condição de reação de Sonogashira. Exemplos do catalisador incluem composto de paládio tal como tetracis(trifenilfosfina) paládio(0), tris(dibenzilidenoacetona)dipaládio(0), diclorobis(acetonitrila) paládio(II) e similares. Para promover a reação, podem ser adicionados uma base orgânica tal como trietilamina e similares, uma base inorgânica tal como amônia e similares, um composto de cobre tal como iodeto de cobre, brometo de cobre e similares, um composto de fosfina tal como 2- diciclohexilfosfino-2',4',6'-triisopropilbifenila e similares, e similares. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano, dioxano e similares, um solvente polar tal como acetonitrila, dimetilformamida e similares, ou um solvente de hidrocarboneto tal como benzeno e similares, sob esfriamento com gelo até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00074] Na terceira etapa, o intermediário (IV-5) é obtido reduzindo a ligação tripla do intermediário (IV-4). O reagente a ser usado não é limitado contanto que seja usado para redução geral de ligação de carbono insaturado. Por exemplo, pode ser usada hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney, complexo de paládio carbono-diamina de etileno e similares, ou um catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, sob 1 a 20 atm de pressão de hidrogênio, sob esfriamento com gelo até a temperatura de refluxo por 30 min a 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produtofim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00075] Na quarta etapa, o intermediário (IV-5) é desprotegido para dar o composto da presente invenção (I-2). Remoção de Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos, não é particularmente limitado contanto que seja usado para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar acetal cíclico, e Rd é t-butiloxicarbonila, acetal cíclico é desprotegido por uma quantidade catalítica de um ácido, e em seguida são empregadas condições acidíferas mais fortes, por meio das quais Rd podem ser removidos. As condições empregadas para a desproteção de acetal são, por exemplo, um solvente alcoólico tal como metanol e similares ou uma solução mista de um solvente alcoólico e outro solvente orgânico, uma quantidade catalítica de ácido clorídrico ou ácido toluenossulfônico, sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Por outro lado, as condições da remoção de Rd realizada depois de desproteção do acetal são, por exemplo, não menos da quantidade equivalente de um ácido inorgânico tal como ácido clorídrico e similares, ácido trifluoroacético e similares, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água, ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00076] 4) Um composto (I-3) representado pela fórmula (Ia) em que R é um átomo de hidrogênio, X é um átomo de enxofre, e R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio é sintetizado pelo seguinte esquema (V).
[00077] em que Ra, Rc e Rd são grupos protetores, Xb e Xd são grupos de saída, R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R2 é conforme definido acima.
[00078] Ra, Rc, Rd e Xb na fórmula são conforme definidos acima. O grupo de saída para Xd não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por um íon heptiltio (C7H15S-). Por exemplo, um átomo de halogênio (especificamente um átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados.
[00079] Na primeira etapa, derivado do ácido benzóico (V-1) tendo um grupo de saída Xd na posição 4 é condensado com heptiltiol para introduzir heptiltio na posição 4, por meio do qual o intermediário (V-2) é obtido. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida e sulfóxido de dimetila, um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como carbonato de potássio, hidróxido de sódio e similares ou uma base orgânica tal como trietilamina, 1,8- diazabiciclo[5.4.0]undec-7-eno e similares podem ser usadas. As condições de reação são, por exemplo, cerca de -30 a 80°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00080] Na segunda etapa, o grupo carboxila do intermediário (V-2) é reduzido para dar o intermediário (V-3) tendo um grupo hidroxila. O reagente a ser usado para a redução não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem metais de álcali tais como sódio e similares, metais alcalino-terrosos, hidretos metálicos tais como hidreto de diisobutilalumínio e similares, compostos de complexo de hidrogênio de metal tais como hidreto de alumínio de lítio, boridreto de sódio e similares, compostos de boro tais como diborano e similares, hidrogenação catalítica usando um catalisador do sistema homogêneo ou sistema heterogêneo e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Exemplos específicos dos mesmos incluem redução usando diborano, hidreto de alumínio de lítio ou boridreto de lítio em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por 10 min a 12 horas, redução usando boridreto de sódio ou boridreto de cálcio em um solvente alcoólico tal como etanol e similares ou em um solvente misto de um solvente alcoólico e um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até a temperatura de refluxo por 30 min a 24 horas, e similares. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00081] Na terceira etapa, o grupo hidroxila do intermediário (V-3) é convertido no grupo de saída Xb. O reagente não é particularmente limitado contanto que seja um reagente capaz de converter um grupo hidroxila alcoólico em Xb. Exemplos do reagente usado quando Xb é um átomo de halogênio incluem N-clorossuccinimida, N-bromossuccinimida, tetracloreto de carbono e uma combinação dos mesmos e um auxiliar de reação tal como trifenilfosfina, uma base e similares, ácidos inorgânicos tais como ácido clorídrico, ácido bromídrico, ácido iodídrico, tribrometo de fósforo, pentabrometo de fósforo, tricloreto de fósforo, pentacloreto de fósforo, iodo, bromo, cloro, tionil halogenado e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C até 130°C por 10 min a 6 horas. Quando é usado um ácido inorgânico, a reação pode ser realizada em uma solução aquosa ou um sistema de duas camadas de um solvente orgânico tal como tolueno e similares e água. Exemplos do reagente usado quando Xb é sulfonilóxi incluem uma combinação de cloreto de sulfonila (por exemplo, cloreto de metanossulfonila, cloreto de tolueno sulfonila e similares) e uma base orgânica (por exemplo, trietilamina, piridina e similares). As condições de reação são, por exemplo, um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares), e similares em -30°C até 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[00082] Na quarta etapa, sal de fosfônio (V-5) é obtido reagindo o intermediário (V-4) tendo o grupo de saída Xb com trifenilfosfina. A reação é realizada, por exemplo, em um solvente inativo tal como éter dietílico, benzeno, tolueno e similares a partir da temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 6 horas. Depois da reação, o produto-fim pode ser obtido por evaporação do solvente, resfriamento e adição de solvente fracamente solúvel tal como éter diisopropílico, hexano e similares, conforme necessário, depois da qual coleta do sólido precipitado por filtração.
[00083] Na quinta etapa, o intermediário (V-6) é obtido condensando sal de fosfônio (V-5) com o aldeído (II-6) sintetizado separadamente usando reação de Wittig, e reduzindo o composto de olefina obtido. As condições da reação de Wittig são aquelas usadas de modo geral para reação de Wittig. Por exemplo, a reação é realizada usando uma base tal como t-butóxido de potássio e similares em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. O reagente a ser usado para a subseqüente redução de uma ligação dupla não é limitado contanto que seja usado para redução de olefina geral. Exemplos do mesmo incluem hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares, catalisadores homogêneos tais como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, sob 1 a 20 atm de pressão de hidrogênio, sob esfriamento com gelo até a temperatura de refluxo a partir de 30 min até 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00084] Na sexta etapa, o composto da presente invenção (I-3) é obtido removendo o intermediário (V-6) possuindo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. A desproteção do intermediário (V-6) não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar um acetal cíclico, e Rd é t-butiloxicarbonila, podem ser simultaneamente removidos usando um ácido. Exemplos do ácido para esse fim incluem ácidos inorgânicos tais como ácido clorídrico e similares, ácido trifluoroacético e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00085] 5) Um composto (I-4) representado pela fórmula (Ia) em que R é um átomo de hidrogênio, X é um átomo de enxofre, e R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio é sintetizado pelo seguinte esquema (VI).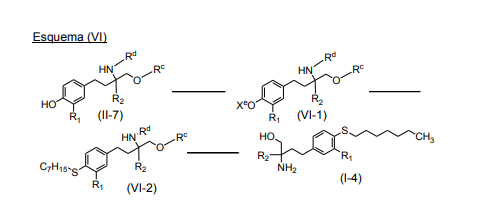
[00086] em que Rc, Rd é um grupo protetor, Xe é um grupo ativador de hidroxila, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R2 é conforme definido acima.
[00087] Rc e Rd na fórmula são conforme definidos acima. Como o grupo ativador de hidroxila para Xe, grupos sulfonila tais como trifluorometanossulfonila, toluenossulfonila e similares podem ser mencionados.
[00088] Na primeira etapa, o intermediário (VI-1) é obtido introduzindo um grupo ativador no grupo hidroxila fenólico do intermediário (II-7) no esquema (II). Esta etapa pode ser realizada em um solvente de halogênio tal como cloreto de metileno e clorofórmio ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como um reagente para esta reação, derivados de ácido sulfônico ativado tais como anidrido de ácido trifluorometanossulfônico, 1-(trifluorometanossulfonil)imidazol, cloreto de tolueno sulfonila são usados. Esta reação também pode ser realizada usando ácido sulfônico e um agente de condensação em combinação. Como a base, é usada uma base orgânica tal como trietilamina, piridina, lutidina e similares. As condições de reação são, por exemplo, -50 a 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00089] Na segunda etapa, o intermediário (VI-1) é condensado com heptiltiol para dar o intermediário (VI-2). Esta etapa pode ser realizada em um solvente etéreo tal como dioxano e similares ou um solvente de hidrocarboneto tal como tolueno e similares, na presença de um catalisador de paládio. Exemplos do catalisador de paládio incluem acetato de paládio(II), tris(dibenzilidenoacetona)dipaládio(0) e similares. Além disso, um composto de fosfina ou uma base pode ser adicionado como um auxiliar de reação a esta reação. Exemplos do composto de fosfina incluem trifenilfosfina, 4, 5-bis(difenilfosfino)-9,9-dimetilxanteno e similares. Por outro lado, como a base, base inorgânica tal como carbonato de césio e similares, e uma base orgânica tal como N,N-diisopropiletilamina e similares podem ser mencionadas. As condições de reação são, por exemplo, temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produtofim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00090] Na terceira etapa, o composto da presente invenção (I-4) é obtido removendo o intermediário (VI-2) possuindo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. Desproteção do intermediário (VI-2) não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc é um grupo protetor que pode ser removido por um ácido tal como metoximetila e similares e Rd é tbutiloxicarbonila, podem ser removidos simultaneamente por um ácido. Exemplos do ácido para esse fim incluem um ácido inorgânico tal como ácido clorídrico e similares, ácido trifluoroacético e similares. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água, ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00091] 6) Dos compostos da presente invenção, um composto (I-5) representado pela fórmula (Ia) em que R é P(=O)(OH)2 é sintetizado pelo seguinte esquema (VII).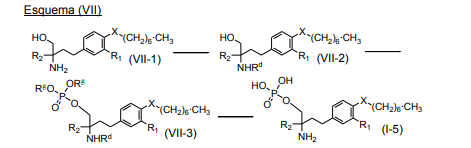
[00092] em que X é um átomo de oxigênio ou um átomo de enxofre, Rd e Rg são grupos protetores, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R2 é conforme definido acima.
[00093] Rd na fórmula são conforme definidos acima. O grupo protetor para Rg na fórmula não é particularmente limitado contanto que proteja um grupo ácido fosfórico. Por exemplo, alquila (preferencialmente tendo um número de carbonos de cerca de 1 - 6, especificamente t-butila e similares), benzila, fenila e similares podem ser mencionados.
[00094] Na primeira etapa, a forma protegida de grupo amino (VII-2) é sintetizada protegendo o grupo amino do composto (VII-1) em que R é um átomo de hidrogênio, dos compostos da presente invenção. Esta etapa pode ser realizada por uma reação de proteção do grupo amino geral. Especificamente, quando acila, alquiloxicarbonila, benziloxicarbonila e similares é usado como um grupo protetor (Rd), esta etapa pode ser realizada em álcool tal como metanol e similares, ou um sistema de duas camadas ou mistura de água e um solvente orgânico tal como acetato de etila, clorofórmio e similares. Exemplos do reagente a ser usado incluem cloreto ácido tal como cloreto de acetila, cloreto de benziloxicarbonila e similares, ou anidrido ácido tal como acético anidrido, dicarbonato de di-t-butila e similares. Uma base orgânica tal como trietilamina e similares ou uma base inorgânica tal como bicarbonato de sódio e similares podem ser adicionadas como um promotor da reação para esta reação. As condições de reação são, por exemplo, sob esfriamento com gelo até 50°C por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[00095] Na segunda etapa, a forma fosforilada (VII-3) é sintetizada reagindo a forma protegida de grupo amino (VII-2) com um reagente de fosforilação (por exemplo, cloreto de fósforo, fosforamidita e oxidante, éster tetrabenzílico de ácido pirofosfórico e similares). Quando éster tetrabenzílico de ácido pirofosfórico é usado como um reagente de fosforilação, esta etapa pode ser realizada sob condições não aquosas preferencialmente em um solvente orgânico tal como tolueno, diclorometano, um solvente misto do mesmo e similares usando um aditivo (por exemplo, óxido de prata, iodeto de tetra-n-hexil amônio e similares). As condições de reação são, por exemplo, sob esfriamento com gelo até 50°C por cerca de 5 a 24 horas. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Para esta reação, um reagente de fosforilação geral (cloreto de fósforo e base, fosforamidita e oxidante e similares) pode ser reagido de acordo com um método conhecido.
[00096] Na terceira etapa, o composto da presente invenção (I-5) é preparado a partir de uma forma fosforilada (VII-3). Esta etapa pode ser realizada por desproteção geral. Especificamente, a etapa pode ser realizada por hidrogenólise, um ácido tal como ácido clorídrico, ácido trifluoroacético e similares, ou um ácido de Lewis tal como brometo de trimetilsilila e similares. Quando hidrogenólise é usada para esta reação, esta etapa é realizada, por exemplo, em um solvente alcoólico tal como metanol e similares usando um catalisador tal como paládio carbono e similares sob uma atmosfera de hidrogênio. As condições de reação são, por exemplo, temperatura ambiente até 60°C por cerca de 1 a 24 horas. O produto-fim pode ser obtido por filtração, concentração e similares da mistura da reação por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. As condições de reação quando um ácido é usado para esta reação são, por exemplo, um solvente alcoólico tal como etanol e similares ou um solvente misto do mesmo com água em temperatura ambiente até 100°C por cerca de 30 min a 12 horas.
[00097] 7) Dos compostos da presente invenção, um composto (I-1a) representado pela fórmula (I) em que R é um átomo de hidrogênio, X é um átomo de oxigênio, Y é CH2CH2, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (VIII).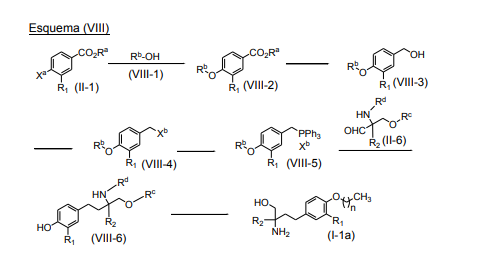
[00098] em que n é 5 a 8, Ra é um átomo de hidrogênio ou um grupo protetor, Rb, Rc e Rd são grupos protetores, Xa e Xb são grupos de saída, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R2 é conforme definido acima.
[00099] Ra na fórmula não é particularmente limitado contanto que proteja um átomo de hidrogênio ou um grupo carboxila. Por exemplo, alquila (especificamente metila, etila e similares), aralquil (benzila e similares), o mesmo substituinte que para Rb e similares podem ser mencionados. Rb na fórmula não é particularmente limitado contanto que proteja um grupo hidroxila fenólico. Por exemplo, alquila (especificamente metila, etila e similares), aralquil (benzila e similares) e similares podem ser mencionados. Quando -(CH2)nCH3 (n é conforme definido acima), o qual é uma estrutura parcial do composto da invenção (I-1a) é usado como Rb, o composto da invenção (I-1a) pode ser obtido sem remoção de Rb. Rc na fórmula não é particularmente limitado contanto que proteja um grupo hidroxila. Por exemplo, acila (preferencialmente uma acila tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), trialquilsilila (especificamente trimetilsilila e similares), benzila e um substituinte formador de composto acetal (especificamente metoximetila, tetraidropiranila e similares) podem ser mencionados. Quando R2 tem um grupo hidroxila, seus grupos protetores Re (Re é especificamente similar a Rc) e Rc também podem ser ligados para formar acetal cíclico. O grupo protetor mostrado por Rd na fórmula não é particularmente limitado contanto que proteja um grupo amino. Por exemplo, acila (preferencialmente uma acila tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), carbamato (especificamente t-butiloxicarbonila, benziloxicarbonila e similares) e similares podem ser mencionados. O grupo de saída para Xa não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por íon alcóxido (Rb-O-). Por exemplo, um átomo de halogênio (especificamente um átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados. O grupo de saída para Xb não é particularmente limitado contanto que seja dissociado durante uma condensação de intermediário (VIII-4) e trifenilfosfina, e não inibe a reação de Wittig subseqüente. Por exemplo, um átomo de halogênio (especificamente um átomo de iodo, um átomo de bromo, um átomo de cloro e similares), metanossulfonilóxi, toluenossulfonilóxi e similares podem ser mencionados.
[000100] Na primeira etapa, o intermediário (VIII-2) é obtido condensando derivado do ácido benzóico (II-1) tendo o grupo de saída X a na posição 4 com álcool (VIII-1) para introduzir um grupo funcional oxigênio tendo o grupo protetor Rb na posição 4. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio e similares ou uma base orgânica tal como 1,8-diazabiciclo[5.4.0] undec7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 100°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000101] Na segunda etapa, o intermediário (VIII-3) tendo um grupo hidroxila é obtido reduzindo o grupo carboxila do intermediário (VIII-2). O reagente a ser usado para a redução não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem metais de álcali tais como sódio e similares ou metais alcalino-terrosos, hidretos metálicos tais como hidreto de diisobutilalumínio e similares, compostos de complexo de hidrogênio de metal tais como hidreto de alumínio de lítio, boridreto de sódio e similares, compostos de boro tais como diborano e similares, hidrogenação catalítica usando um catalisador do tipo homogêneo ou do tipo heterogêneo, e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Exemplos específicos dos mesmos incluem redução usando diborano, hidreto de alumínio de lítio ou boridreto de lítio em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por 10 min a 12 horas, redução usando boridreto de sódio ou boridreto de cálcio em um solvente alcoólico tal como etanol e similares ou em um solvente misto de um solvente alcoólico e um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até a temperatura de refluxo por 30 min a 24 horas, e similares. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000102] Na terceira etapa, o grupo hidroxila do intermediário (VIII-3) é convertido no grupo de saída Xb. O reagente não é particularmente limitado contanto que seja um reagente capaz de converter um grupo hidroxila alcoólico em Xb. Exemplos do reagente usado quando Xb é um átomo de halogênio incluem N-clorossuccinimida, N-bromossuccinimida, tetracloreto de carbono e uma combinação dos mesmos e um auxiliar de reação tal como trifenilfosfina, uma base e similares, ácidos inorgânicos tais como ácido clorídrico, ácido bromídrico, ácido iodídrico, tribrometo de fósforo, pentabrometo de fósforo, tricloreto de fósforo, pentacloreto de fósforo, iodo, bromo, cloro, tionil halogenado e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C até 130°C por 10 min a 6 horas. Quando é usado um ácido inorgânico, a reação pode ser realizada em uma solução aquosa ou um sistema de duas camadas de um solvente orgânico tal como tolueno e similares e água. Exemplos do reagente usado quando Xb é sulfonilóxi incluem uma combinação de cloreto de sulfonila (por exemplo, cloreto de metanossulfonila, cloreto de tolueno sulfonila e similares) e uma base orgânica (por exemplo, trietilamina, piridina e similares). As condições de reação são, por exemplo, um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares), e similares em -30°C até 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000103] Na quarta etapa, sal de fosfônio (VIII-5) é obtido reagindo o intermediário (VIII-4) tendo o grupo de saída Xb com trifenilfosfina. A reação é realizada, por exemplo, em um solvente inativo tal como éter dietílico, benzeno, tolueno e similares a partir da temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por evaporação do solvente, resfriamento e adição de solvente fracamente solúvel tal como éter diisopropílico, hexano e similares, conforme necessário, depois da qual coleta do sólido precipitado por filtração.
[000104] Na quinta etapa, o intermediário fenólico (VIII-6) é obtido condensando sal de fosfônio (VIII-5) com o aldeído (II-6) sintetizado separadamente usando reação de Wittig, reduzindo o composto de olefina obtido, e removendo o grupo protetor Rb . As condições da reação de Wittig são aquelas usadas de modo geral para reação de Wittig. Por exemplo, a reação é realizada usando uma base tal como t-butóxido de potássio e similares em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. O reagente a ser usado para a subseqüente redução de uma ligação dupla não é limitado contanto que seja usado para redução de olefina geral. Exemplos do mesmo incluem hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares, ou alguns catalisadores homogêneos tais como complexo de ródio (clorotris (trifenilfosfina) ródio(I) e similares) e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, sob 1 a 20 atm de pressão de hidrogênio, sob esfriamento com gelo até a temperatura de refluxo a partir de 30 min até 1 semana.
Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. As condições da subseqüente remoção do grupo protetor Rb não são particularmente limitadas contanto que sejam usadas para a remoção de Rb. Por exemplo, quando Rb é metila, um método para usar um ácido de Lewis tal como tribrometo de boro e similares em solvente de cloreto de metileno pode ser usado. Quando Rb é acila tal como acetila e similares, um método para usar uma base inorgânica tal como hidróxido de sódio e similares em um solvente misto de um solvente alcoólico e água pode ser usado. Quando Rb é um grupo protetor do tipo éter tal como metoximetila, tetraidropiranila, t-butila e similares, um método para usar um ácido tal como ácido clorídrico, ácido trifluoroacético e similares pode ser usado. Quando Rb é um grupo protetor o qual pode ser removido por hidrogenação catalítica (por exemplo, benzila, benzila substituída, benziloximetila e similares), a remoção de Rb pode ser realizada simultaneamente com a redução supracitada da ligação dupla. Quando -(CH2)nCH3 (n é conforme definido acima), o qual é uma estrutura parcial do composto da invenção (I-1a), é usado como Rb , a remoção de Rb não é necessária, e a alquilação de fenol na etapa seguinte também pode ser omitida.
Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. As condições da subseqüente remoção do grupo protetor Rb não são particularmente limitadas contanto que sejam usadas para a remoção de Rb. Por exemplo, quando Rb é metila, um método para usar um ácido de Lewis tal como tribrometo de boro e similares em solvente de cloreto de metileno pode ser usado. Quando Rb é acila tal como acetila e similares, um método para usar uma base inorgânica tal como hidróxido de sódio e similares em um solvente misto de um solvente alcoólico e água pode ser usado. Quando Rb é um grupo protetor do tipo éter tal como metoximetila, tetraidropiranila, t-butila e similares, um método para usar um ácido tal como ácido clorídrico, ácido trifluoroacético e similares pode ser usado. Quando Rb é um grupo protetor o qual pode ser removido por hidrogenação catalítica (por exemplo, benzila, benzila substituída, benziloximetila e similares), a remoção de Rb pode ser realizada simultaneamente com a redução supracitada da ligação dupla. Quando -(CH2)nCH3 (n é conforme definido acima), o qual é uma estrutura parcial do composto da invenção (I-1a), é usado como Rb , a remoção de Rb não é necessária, e a alquilação de fenol na etapa seguinte também pode ser omitida.
[000105] Na sexta etapa, o composto da presente invenção (I-1a) é obtido alquilando o grupo hidroxila fenólico do intermediário (VIII-6), e removendo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. Exemplos do reagente a ser usado para alquilação do grupo hidroxila fenólico o intermediário (VIII-6) incluem uma combinação de um agente alquilante tal como heptil haleto e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. A reação é realizada, por exemplo, em um solvente polar tal como N,N-dimetilformamida e similares, ou em um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Para alquilação do grupo hidroxila fenólico o intermediário (VIII-6) tem, reação de Mitsunobu de condensação de álcool heptílico e etc. e usando um composto de fosfina tal como trifenilfosfina e similares e um derivado de ácido azocarboxílico tal como diisopropil azodicarboxilato e similares pode ser usada. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 50°C por cerca de 10 min a 6 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. A desproteção subseqüente não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar um acetal cíclico, e Rd é tbutiloxicarbonila, podem ser simultaneamente removidos usando um ácido. Exemplos do ácido para esse fim incluem ácidos inorgânicos tais como ácido clorídrico e similares, ácido trifluoroacético e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000106] 8) Dos compostos da presente invenção, um composto (I-1a) representado pela fórmula (I) em que R é um átomo de hidrogênio, X é um átomo de oxigênio, Y é CH2CH2, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R3 e R4 são átomos de hidrogênio também pode ser sintetizado usando um intermediário (VIII-2, Ra é H) no esquema (VIII) ou intermediário (IX-1) sintetizado a partir de um composto representado pela fórmula (IX-2) pelo seguinte esquema (IX).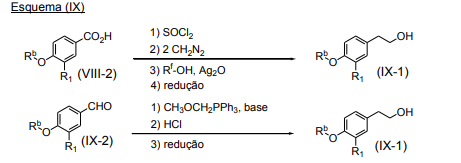
[000107] em que R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, Rb é um grupo protetor, e Rf-OH é álcool usado para uma reação de solvólise.
[000108] Rb na fórmula é o mesmo que no esquema (VIII). Como Rf , metila, etila, benzila e similares podem ser mencionados. No esquema mencionado acima, para síntese a partir do composto (VIII-2), são usadas condições de reação gerais da reação de Arndt-Eistert. Para redução do grupo éster obtido deste modo, o reagente e as condições usadas na segunda etapa do esquema (VIII) podem ser mencionados. No esquema mencionado acima, para síntese a partir do composto (IX-2), são usadas as condições para reação geral de Wittig. Para tratamento ácido depois disso, um ácido inorgânico tal como ácido clorídrico e similares é usado em água ou um solvente misto de um solvente orgânico tal como tetraidrofurano e similares e água. Para a redução subseqüente, um composto de complexo de hidrogênio de metal tal como hidreto de alumínio de lítio, boridreto de sódio e similares, hidrogenação catalítica usando catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares ou catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina) ródio(I) e similares) e similares pode ser realizada, ou estas podem ser seqüencialmente realizadas continuamente. O intermediário alcoólico (IX-1) obtido neste esquema pode ser convertido no composto da presente invenção por um método conhecido (por exemplo, Journal of Medicinal Chemistry vol. 43 (2000) 2946-2961).
[000109] 9) Dos compostos da presente invenção, o composto (I-1c) representado pela fórmula (I) em que R é um átomo de hidrogênio, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é ω-flúor, e R3 e R4 são átomos de hidrogênio também pode ser sintetizado pelo seguinte esquema.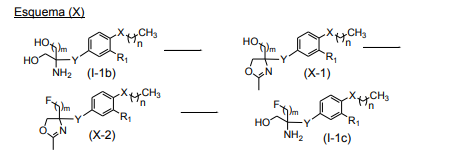
[000110] em que m é um inteiro de 1 a 4, X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e n é conforme definido acima.
[000111] Na primeira etapa, a forma oxazolina (X-1) é sintetizada protegendo o composto (I-1b) da fórmula (I) em que R é um átomo de hidrogênio, e R2 é ω-hidroxialquila. Esta etapa pode ser realizada por reação em um solvente polar tal como acetonitrila, N,N-dimetilformamida e similares, um solvente de halogênio tal como cloreto de metileno e similares, ou um solvente de hidrocarboneto tal como tolueno e similares usando éster de ácido ortoacético como um reagente. Para promover a reação, uma base tal como N,N-diisopropiletilamina e similares, ou um ácido tal como ácido p-toluenossulfônico e similares podem ser adicionados. As condições de reação são, por exemplo, temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000112] Na segunda etapa, a forma fluoreto (X-2) é sintetizada por fluoração do grupo hidroxila do composto (X-1). Exemplos do agente de fluoração incluem trifluoreto de (dietilamino)enxofre (DAST), 2,2-diflúor1,3-dimetilimidazolidina (DFI) e similares. Esta etapa pode ser realizada por reação em um solvente de halogênio tal como cloreto de metileno e similares, ou um solvente de hidrocarboneto tal como hexano e similares. As condições de reação são, por exemplo, -78°C até temperatura ambiente por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Esta etapa também pode ser realizada por um método incluindo converter o grupo hidroxila do composto (X-1) para a forma sulfonato correspondente, e em seguida reagindo a mesma com íon fluoreto. Por exemplo, quando fluoreto de p-toluenossulfonila e fluoreto de tetrabutilamônio (TBAF) são usados, a reação é realizada em um solvente etéreo tal como tetraidrofurano e similares em temperatura ambiente até 80°C por cerca de 1 hora a 24 horas. Nesta reação, um agente desidratante tal como peneiras moleculares e similares pode ser adicionado. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000113] Na terceira etapa, o composto (X-2) é desprotegido para dar o composto da presente invenção (I-1c). Esta etapa pode ser realizada por desproteção geral. Especificamente, pode ser realizada usando um ácido tal como ácido clorídrico, ácido trifluoroacético e similares. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares ou um solvente misto do mesmo com água em temperatura ambiente até 100°C por cerca de 30 min a 12 horas. O produto-fim pode ser obtido por filtração, concentração e similares da mistura da reação por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000114] 10) Um composto (I-2a) representado pela fórmula (I), em que R é um átomo de hidrogênio, X é um átomo de oxigênio, Y é CH2CH2, R1 é trifluorometila ou ciano, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (XI).
[000115] em que R1 é trifluorometila ou ciano, Rc e Rd são grupos protetores, Xa e Xc são grupos de saída, e R2, e n é conforme definido acima.
[000116] Rc, Rd, Xa na fórmula são conforme definidos acima. O grupo de saída para Xc não é particularmente limitado contanto que seja ativado por um catalisador e dissociado durante uma reação de Sonogashira. Por exemplo, um átomo de halogênio (preferencialmente um átomo de iodo, um átomo de bromo e similares), trifluorometanossulfonilóxi e similares podem ser mencionados.
[000117] Na primeira etapa, o intermediário (XI-3) é obtido condensando o composto (XI-1) tendo o grupo de saída Xa com álcool (XI-2). Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio e similares ou uma base orgânica tal como 1,8- diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 100°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000118] Na segunda etapa, o intermediário (XI-4) tendo uma ligação tripla é obtido condensando o intermediário (XI-3) com o intermediário (IV-3) por meio da reação de Sonogashira. Exemplos do catalisador incluem composto de paládio tal como tetracis(trifenilfosfina) paládio(0), tris(dibenzilidenoacetona)dipaládio(0), diclorobis (acetonitrila)paládio(II) e similares. Para promover a reação, uma base orgânica tal como trietilamina e similares, uma base inorgânica tal como amônia e similares, um composto de cobre tal como iodeto de cobre, brometo de cobre e similares, um composto de fosfina tal como 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila e similares, e similares pode ser adicionado. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano, dioxano e similares, um solvente polar tal como acetonitrila, dimetilformamida e similares, ou um solvente de hidrocarboneto tal como benzeno e similares, sob esfriamento com gelo até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000119] Na terceira etapa, o intermediário (XI-5) é obtido reduzindo a ligação tripla do intermediário (XI-4). O reagente a ser usado não é limitado contanto que seja usado para redução geral de ligação de carbono insaturado. Por exemplo, hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney, complexo de paládio carbono-diamina de etileno e similares, catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares podem ser mencionados. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares em 1 a 20 atm de pressão de hidrogênio sob esfriamento com gelo até refluxando por 30 min a 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000120] Na quarta etapa, o intermediário (XI-5) é desprotegido para dar o composto da presente invenção (I-2a). Remoção de Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos, não é particularmente limitado contanto que seja usado para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar acetal cíclico, e Rd é grupo tbutiloxicarbonila, acetal cíclico é desprotegido por uma quantidade catalítica de um ácido, e em seguida são empregadas condições acidíferas mais fortes, por meio das quais Rd podem ser removidos. As condições empregadas para a desproteção de acetal são, por exemplo, um solvente alcoólico tal como metanol e similares ou uma solução mista de um solvente alcoólico e outro solvente orgânico, uma quantidade catalítica de ácido clorídrico ou ácido toluenossulfônico, sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Por outro lado, as condições da remoção de Rd realizada depois de desproteção do acetal são, por exemplo, não menos da quantidade equivalente de um ácido inorgânico tal como ácido clorídrico e similares, ácido trifluoroacético e similares, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água, ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000121] 11) Um composto (I-3a) representado pela fórmula (I) em que R é um átomo de hidrogênio, X é um átomo de enxofre, Y é CH2CH2, R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (XII).
[000122] em que R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, Ra é um átomo de hidrogênio ou um grupo protetor, Rc e Rd são grupos protetores, Xb e Xd são grupos de saída e R2 e n são conforme definidos acima.
[000123] Ra, Rc, Rd, Xb e Xd na fórmula são conforme definidos acima. O grupo de saída para Xd não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por um íon alquiltio (CH3(CH2)nS-). Por exemplo, um átomo de halogênio (especificamente átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados.
[000124] Na primeira etapa, o intermediário (XII-2) é obtido condensando derivado do ácido benzóico (V-1) tendo o grupo de saída X a na posição 4 com tiol (XII-1) para introduzir alquiltio na posição 4. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como carbonato de potássio, hidróxido de sódio e similares ou uma base orgânica tal como trietilamina, 1,8-diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. A reação é realizada, por exemplo, -30 a 80°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000125] Na segunda etapa, o intermediário (XII-3) tendo um grupo hidroxila é obtido reduzindo o grupo carboxila do intermediário (XII-2). O reagente a ser usado para a redução não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem metais de álcali tais como sódio e similares ou metais alcalino-terrosos, hidretos metálicos tais como hidreto de diisobutilalumínio e similares, compostos de complexo de hidrogênio de metal tais como hidreto de alumínio de lítio, boridreto de sódio e similares, compostos de boro tais como diborano e similares, hidrogenação catalítica usando um catalisador do tipo homogêneo ou do tipo heterogêneo, e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Exemplos específicos dos mesmos incluem redução usando diborano, hidreto de alumínio de lítio ou boridreto de lítio em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por 10 min a 12 horas, redução usando boridreto de sódio ou boridreto de cálcio em um solvente alcoólico tal como etanol e similares ou em um solvente misto de um solvente alcoólico e um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até a temperatura de refluxo por 30 min a 24 horas, e similares. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000126] Na terceira etapa, o grupo hidroxila do intermediário (XII-3) é convertido no grupo de saída Xb. O reagente não é particularmente limitado contanto que seja um reagente capaz de converter um grupo hidroxila alcoólico em Xb . Exemplos do reagente usado quando Xb é um átomo de halogênio incluem N-clorossuccinimida, N-bromossuccinimida, tetracloreto de carbono e uma combinação dos mesmos e um auxiliar de reação tal como trifenilfosfina, uma base e similares, ácidos inorgânicos tais como ácido clorídrico, ácido bromídrico, ácido iodídrico, tribrometo de fósforo, pentabrometo de fósforo, tricloreto de fósforo, pentacloreto de fósforo, iodo, bromo, cloro, tionil halogenado e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C a 130°C por 10 min a 6 horas. Quando é usado um ácido inorgânico, a reação pode ser realizada em uma solução aquosa ou um sistema de duas camadas de um solvente orgânico tal como tolueno e similares e água. Exemplos do reagente usado quando Xb é sulfonilóxi incluem uma combinação de cloreto de sulfonila (por exemplo, cloreto de metanossulfonila, cloreto de tolueno sulfonila e similares) e uma base orgânica (por exemplo, trietilamina, piridina e similares). As condições de reação são, por exemplo, um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares), e similares em -30°C a 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000127] Na quarta etapa, sal de fosfônio (XII-5) é obtido reagindo o intermediário (XII-4) tendo o grupo de saída Xb com trifenilfosfina. A reação é realizada, por exemplo, em um solvente inativo tal como éter dietílico, benzeno, tolueno e similares a partir da temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 6 horas. Depois da reação, o produto-fim pode ser obtido por evaporação do solvente, resfriamento e adição de solvente fracamente solúvel tal como éter diisopropílico, hexano e similares, conforme necessário, depois da qual coleta do sólido precipitado por filtração.
[000128] Na quinta etapa, o intermediário (XII-6) é obtido condensando sal de fosfônio (XII-5) com o aldeído (II-6) sintetizado separadamente usando reação de Wittig, e reduzindo o composto de olefina obtido. As condições da reação de Wittig são aquelas usadas de modo geral para reação de Wittig. Por exemplo, a reação é realizada usando uma base tal como t-butóxido de potássio e similares em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. O reagente a ser usado para a subseqüente redução de uma ligação dupla não é limitado contanto que seja usado para redução de olefina geral. Exemplos do mesmo incluem hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney e similares, um catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, sob 1 a 20 atm de pressão de hidrogênio, sob esfriamento com gelo até a temperatura de refluxo a partir de 30 min até 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000129] Na sexta etapa, o composto da presente invenção (I-3a) é obtido removendo o intermediário (XII-6) possuindo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. A desproteção do intermediário (XII-6) não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar um acetal cíclico, e Rd é t-butiloxicarbonila, podem ser simultaneamente removidos usando um ácido. Exemplos do ácido para esse fim incluem ácidos inorgânicos tais como ácido clorídrico e similares, ácido trifluoroacético e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000130] 12) Um composto (I-4a) representado pela fórmula (I) em que R é um átomo de hidrogênio, X é um átomo de enxofre, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (XIII).
[000131] em que R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, Rc e Rd são grupos protetores, Xe é um grupo ativador de hidroxila, Y é CH2CH2 ou CH=CH, e R2 e n são conforme definidos acima.
[000132] Rc, Rd na fórmula são conforme definidos acima. Como o grupo ativador de hidroxila para Xe, sulfonila tal como trifluorometanossulfonila, toluenossulfonila e similares podem ser mencionados.
[000133] Na primeira etapa, o intermediário (XIII-1) é obtido introduzindo um grupo ativador no grupo hidroxila fenólico do intermediário (VIII-6) no esquema (VIII). Esta etapa pode ser realizada em um solvente de halogênio tal como cloreto de metileno e clorofórmio, um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como um reagente para esta reação, derivados de ácido sulfônico ativado tais como anidrido de ácido trifluorometanossulfônico, 1-(trifluorometanossulfonil)imidazol, cloreto de toluenossulfonila são usados. Esta reação também pode ser realizada usando ácido sulfônico e um agente de condensação em combinação. Como a base, uma base orgânica tal como trietilamina, piridina, lutidina e similares é usada. As condições de reação são, por exemplo, -50 até 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000134] Na segunda etapa, o intermediário (XIII-2) é obtido por condensação de intermediário (XIII-1) e tiol (XII-1). Esta etapa pode ser realizada em um solvente etéreo tal como dioxano e similares ou um solvente de hidrocarboneto tal como tolueno e similares na presença de um catalisador de paládio. Exemplos do catalisador de paládio incluem paládio(II) acetato, tris(dibenzilidenoacetona) dipaládio(0) e similares. Além disso, um composto de fosfina ou uma base pode ser adicionado como um auxiliar de reação a esta reação. Exemplos do composto de fosfina incluem trifenilfosfina, 4,5-bis (difenilfosfino)-9,9-dimetilxanteno e similares. Por outro lado, como a base, base inorgânica tal como carbonato de césio e similares, base orgânica tal como N,N-diisopropiletilamina e similares podem ser mencionadas. As condições de reação são, por exemplo, temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produtofim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000135] Na terceira etapa, o composto da presente invenção (I-4a) é obtido removendo o intermediário (XIII-2) possuindo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. A desproteção do intermediário (XIII-2) não é particularmente limitada contanto que seja usada para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc é um grupo protetor que pode ser removido por um ácido tal como metoximetila e similares e Rd é t-butiloxicarbonila, podem ser removidos simultaneamente por um ácido. Exemplos do ácido para esse fim incluem um ácido inorgânico tal como ácido clorídrico e similares e ácido trifluoroacético e similares. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000136] 13) Dos compostos da presente invenção, um composto (I-5a) representado pela fórmula (I) em que R é um átomo de hidrogênio, Y é CH2CH2, R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é hidroximetila, e R3 e R4 são átomos de hidrogênio também pode ser sintetizado pelo seguinte esquema (XIV).
[000137] em que R1 é alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, X é um átomo de oxigênio ou um átomo de enxofre, Xf é um grupo de saída, Rg é um grupo protetor ou -(CH2)nCH3, R2 é hidroximetila, e n é conforme definido acima.
[000138] O grupo de saída para Xf não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por um alcóxido ou ânion tiol. Por exemplo, um átomo de halogênio (especificamente um átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados. Quando Rg na fórmula é um grupo protetor, Rg não é particularmente limitado contanto que proteja um grupo fenol ou um grupo tiol. Exemplos de Rg quando X é um átomo de oxigênio incluem alquil (metila e similares), aralquil (benzila e similares), grupo protetor formando acetal (metoximetila, etoxietila e similares) e similares. Quando X é um átomo de enxofre, alquil (metila e similares), aralquil (4-metoxibenzila e similares), um grupo protetor formando tioacetal (metoximetila, feniltiometila, acetamidometila e similares) e similares podem ser mencionados.
[000139] Na primeira etapa, o intermediário (XIV-3) é obtido por condensação de acetofenona (XIV-1) tendo o grupo de saída Xf na posição 4 e álcool ou tiol (XIV-2). Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio e similares ou uma base orgânica tal como 1,8-diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. As condições de reação são, por exemplo, sob esfriamento com gelo - cerca de 100°C por cerca de 10 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000140] Na segunda etapa, a forma brometo de fenacila (XIV-4) é obtida por brominação do grupo acetila do intermediário (XIV-3). Esta etapa pode ser realizada em um solvente tal como um solvente de halogênio tal como clorofórmio e similares, um solvente etéreo tal como dioxano e similares, um solvente alcoólico tal como etanol e similares ou ácido acético e similares. Como um reagente de brominação, bromo, piridínio tribrometo, feniltrimetilamônio tribrometo e similares podem ser mencionados. As condições de reação são, por exemplo, sob esfriamento com gelo - cerca de 60°C por cerca de 30 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000141] Na terceira etapa, o intermediário (XIV-5) é obtido por condensação de intermediário (XIV-4) e dietil acetamidomalonato. Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, bases inorgânicas tais como hidreto de sódio, hidróxido de potássio, t-butóxido de potássio e similares podem ser usadas. As condições de reação são, por exemplo, sob esfriamento com gelo até cerca de 50°C por cerca de 10 min a 5 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000142] Na quarta etapa, o intermediário (XIV-6) é obtido reduzindo o grupo carbonil do intermediário (XIV-5) para metileno. Como o agente redutor, uma combinação de trialquilsilano e ácido trifluoroacético, uma combinação de trialquilsilano e tetracloreto de titânio e similares pode ser usada em um solvente de halogênio tal como 1,2-dicloroetano e similares, ou sem solvente. As condições de reação são, por exemplo, sob esfriamento com gelo até refluxando por cerca de 1 a 48 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000143] O intermediário obtido pode ser levado para o composto da presente invenção (I-5a) por um método conhecido (por exemplo, Journal of Medicinal Chemistry vol. 43 (2000) 2946-2961).
[000144] 14) Dos compostos da presente invenção, um composto (I-6a) representado pela fórmula (I) em que um de R3 e R4 ou tanto R3 quanto R4 é(são) alquila tendo um número de carbonos de 1 a 4 é sintetizado pelo seguinte esquema (XV).
[000145] em que R é um átomo de hidrogênio ou P(=O)(OH)2, X é um átomo de oxigênio ou um átomo de enxofre, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, um ou ambos R3 e R4 são alquila tendo um número de carbonos de 1 a 4, Y é CH2CH2 ou CH=CH, e n é conforme definido acima.
[000146] Nesta etapa, o composto da presente invenção (1-6a) é sintetizado por alquilação do grupo amino do composto (XV-1) tendo um grupo amino primário, dos compostos da presente invenção. Para esta síntese, reação de aminação redutiva ou reação de alquilação de amina usando haleto de alquila e base podem ser usadas. Quando se usa reação de aminação redutiva, o produto-fim é obtido reagindo o aldeído tendo o mesmo número de carbonos que o de R3 ou R4 com o composto (XV-1) em um solvente alcoólico tal como etanol e similares ou um solvente de halogênio tal como dicloroetano e similares usando um agente redutor tal como boridreto de sódio, cianoboridreto de sódio, triacetoxiboridreto de sódio e similares. A redução também pode ser realizada usando hidrogênio e um catalisador tal como níquel de Raney, óxido de platina e similares. Para esta reação, produção de base de Schiff e reação de redução também podem ser realizadas seqüencialmente. Um ácido tal como ácido acético e similares pode ser adicionado como um promotor da reação para a reação de aminação redutiva. As condições de reação são, por exemplo, sob esfriamento com gelo até cerca de 50°C por cerca de 30 min a 10 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando R3 e R4 são metila, reação de metilação de Eschweiler-Clarke também pode ser realizada usando um agente redutor tal como ácido fórmico e formaldeído, ou formaldeído e cianoboridreto de sódio e similares.
[000147] 15) Dos compostos da presente invenção, um composto (I-7a) representado pela fórmula (I) em que R é um átomo de hidrogênio, Y é CH=CH, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (XVI).
[000148] em que X é um átomo de oxigênio ou um átomo de enxofre, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, Rc e Rd são grupos protetores, Rg é um grupo protetor ou -(CH2)nCH3, Xa e Xb são grupos de saída, Xg é um grupo de saída contendo fósforo, e n é conforme definido acima.
[000149] Rc na fórmula não é particularmente limitado contanto que proteja um grupo hidroxila. Por exemplo, acila (preferencialmente tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), trialquilsilila (especificamente trimetilsilila e similares), benzila e substituinte formando um composto acetal (especificamente metoximetila, tetraidropiranila e similares) podem ser mencionados. Quando R2 tem um grupo hidroxila, seus grupos protetores Re (Re é especificamente similar a Rc) e Rc também podem ser ligados para formar acetal cíclico. O grupo protetor mostrado por Rd na fórmula não é particularmente limitado contanto que proteja um grupo amino. Por exemplo, acila (preferencialmente uma acila tendo um número de carbonos de cerca de 2 a 4, especificamente acetila e similares), carbamato (especificamente t-butiloxicarbonila, benziloxicarbonila e similares) e similares podem ser mencionados. Quando Rg na fórmula é um grupo protetor, Rg não é particularmente limitado contanto que proteja um grupo fenol ou um grupo tiol. Exemplos de Rg quando X é um átomo de oxigênio incluem alquil (metila e similares), aralquil (4- metoxibenzila e similares), grupo protetor formando acetal (metoximetila, etoxietila e similares) e similares. Quando X é um átomo de enxofre, alquil (metila e similares), aralquil (4-metoxibenzila e similares), um grupo protetor formando tioacetal (metoximetila, feniltiometila, acetamidometila e similares) e similares podem ser mencionados. O grupo de saída para Xa não é particularmente limitado contanto que seja dissociado durante uma reação de substituição por um íon alcóxido (Rg-O-) ou tiolato (Rg-S-). Por exemplo, um átomo de halogênio (especificamente um átomo de flúor e similares), toluenossulfonilóxi e similares podem ser mencionados. O grupo de saída para Xb não é particularmente limitado contanto que seja dissociado durante uma reação entre um intermediário (XVI-5) e um composto de fósforo e não inibe a reação subseqüente com o aldeído (II-6). Por exemplo, um átomo de halogênio (especificamente um átomo de iodo, um átomo de bromo, um átomo de cloro e similares), metanossulfonilóxi, toluenossulfonilóxi e similares podem ser mencionados. Exemplos do grupo de saída contendo fósforo para Xg incluem P(C6H5)3 e P(O)(ORh)2 (Rh é alquila tendo um número de carbonos de 1 a 4).
[000150] Na primeira etapa, o intermediário (XVI-3) é obtido condensando ácido benzóico (XVI-1) tendo o grupo de saída Xa na posição 4 com álcool ou tiol (XVI-2). Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio, carbonato de potássio e similares ou uma base orgânica tal como alcóxido (por exemplo, t-butóxido de potássio e similares), 1,8-diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 80°C por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000151] Na segunda etapa, o intermediário (XVI-4) tendo um grupo hidroxila é obtido reduzindo o grupo carboxila do intermediário (XVI-3). O reagente a ser usado para a redução não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem metais de álcali tais como sódio e similares, metais alcalino-terrosos, hidretos metálicos tais como hidreto de diisobutilalumínio e similares, compostos de complexo de hidrogênio de metal tais como hidreto de alumínio de lítio, hidreto bis(2-metoxietóxi)alumínio de sódio e similares, compostos de boro tais como diborano e similares, hidrogenação catalítica usando um catalisador do tipo homogêneo ou do tipo heterogêneo, e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Exemplos específicos dos mesmos incluem redução usando diborano, hidreto de alumínio de lítio em um solvente etéreo tal como tetraidrofurano e similares a partir de -30°C até a temperatura de refluxo por 10 min a 12 horas, redução usando hidreto de bis(2-metoxietóxi)alumínio de sódio em um solvente inativo tal como tolueno e similares, sob esfriamento com gelo até 50°C por cerca de 30 min a 24 horas, e similares. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000152] Na terceira etapa, o grupo hidroxila do intermediário (XVI-4) é convertido no grupo de saída Xb. O reagente não é particularmente limitado contanto que seja um reagente capaz de converter um grupo hidroxila alcoólico em Xb . Exemplos do reagente usado quando Xb é um átomo de halogênio incluem N-clorossuccinimida, N-bromossuccinimida, tetracloreto de carbono e uma combinação dos mesmos e um auxiliar de reação tal como trifenilfosfina, uma base e similares, ácidos inorgânicos tais como ácido clorídrico, ácido bromídrico, ácido iodídrico, tribrometo de fósforo, pentabrometo de fósforo, tricloreto de fósforo, pentacloreto de fósforo, iodo, bromo, cloro, tionil halogenado, αhaloenamina e similares. A reação é realizada, por exemplo, em um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares) e similares a partir de -30°C até 130°C por 10 min a 6 horas. Quando é usado um ácido inorgânico, a reação pode ser realizada em uma solução aquosa ou um sistema de duas camadas de um solvente orgânico tal como tolueno e similares e água. Exemplos do reagente usado quando Xb é sulfonilóxi incluem uma combinação de cloreto de sulfonila (por exemplo, cloreto de metanossulfonila, cloreto de tolueno sulfonila e similares) e uma base orgânica (por exemplo, trietilamina, piridina e similares). As condições de reação são, por exemplo, um solvente orgânico tal como um solvente de halogênio (por exemplo, cloreto de metileno e similares), um solvente etéreo (por exemplo, tetraidrofurano e similares), e similares em -30°C até 50°C por cerca de 5 min a 3 horas. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000153] Na quarta etapa, o intermediário (XVI-6) tendo um grupo de saída Xg contendo fósforo é obtido reagindo o intermediário (XVI-5) tendo um grupo de saída Xb com um fósforo composto. Quando Xg é P(C6H5)3, o intermediário (XVI-6) pode ser obtido reagindo o intermediário (XVI-5) com trifenilfosfina. As condições de reação são, por exemplo, um solvente inativo tal como éter dietílico, benzeno, tolueno e similares em temperatura ambiente até a temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por evaporação do solvente, resfriamento e adição de solvente fracamente solúvel tal como éter diisopropílico, hexano e similares, conforme necessário, depois da qual coleta do sólido precipitado por filtração. Quando Xg é P(O)(ORh)2 (Rh é conforme definido acima), o intermediário (XVI-6) pode ser obtido por reação de Arbuzov reagindo o intermediário (XVI-5) com fosfito de trietila. As condições de reação são, por exemplo, sem solvente ou solvente inativo tal como xileno e similares em 100°C até 170°C por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por evaporação ou destilação do excesso de trietil fosfato.
[000154] Na quinta etapa, o intermediário (XVI-6) contendo fósforo e aldeído sintetizado separadamente (II-6) são condensados para dar a forma olefina (XVI-7). Quando Xg é P(C6H5)3, as condições para reação geral de Wittig são usadas. Por exemplo, reação é realizada em um solvente etéreo tal como tetraidrofurano e similares, usando uma base tal como hidreto de sódio, t-butóxido de potássio e similares em -30°C até temperatura de refluxo por cerca de 30 min a 12 horas. Uma forma Z pode ser obtida preferencialmente por reação em um solvente polar aprótico sob condições livres de sal, ou uma forma E também pode ser obtida preferencialmente por um método de Schlosser aprimorado. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando Xg é P(O)(ORh)2 (Rh é conforme definido acima), são usadas as condições de reação de Horner-Wadsworth-Emmons gerais. Por exemplo, a reação é realizada em um solvente de hidrocarboneto tal como benzeno e similares, ou um solvente etéreo tal como tetraidrofurano e similares usando uma base tal como hidreto de sódio, t-butóxido de potássio, hexametildissilazano de lítio e similares em - 20°C até temperatura de refluxo por cerca de 30 min a 12 horas. Um olefina pode ser obtida preferencialmente como uma forma E. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000155] Na sexta etapa, o composto da presente invenção (I-7a) é obtido removendo o intermediário (XVI-7) possuindo Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos. As condições não são particularmente limitadas contanto que sejam usadas para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc é um grupo protetor que forma um acetal, e Rd é tbutiloxicarbonila, podem ser simultaneamente removidos usando um ácido. Exemplos do ácido para esse fim incluem ácidos inorgânicos tais como ácido clorídrico e similares, ácido trifluoroacético e similares. A reação é realizada, por exemplo, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo, sob esfriamento com gelo até 80°C por cerca de 10 min a a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando Rg é um grupo protetor, antes de remoção dos grupos protetores Rc e Rd , pode ser realizada remoção de Rg e alquilação de fenol ou tiol obtido deste modo. As condições a serem empregadas para remoção de Rg não são particularmente limitadas contanto que sejam usadas para remoção geral de grupos protetores. Por exemplo, quando Rg é 4-metoxibenzila, são empregadas reação de oxidação por 2,3- dicloro-5,6-diciano-1,4-benzoquinona (DDQ) e similares, quando Rg é alila, pode ser empregada uma reação usando um composto de paládio como um catalisador. Exemplos do reagente a ser usado para alquilação do grupo hidroxila fenólico ou grupo tiol que o composto obtido têm incluem uma combinação de um agente alquilante tal como haleto de alquila e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. As condições de reação são, por exemplo, um solvente polar tal como N,N-dimetilformamida e similares, um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Além disso, para alquilação de um grupo hidroxila fenólico, também pode ser usada a reação de Mitsunobu.
[000156] 16) Um composto (I-8a) representado pela fórmula (I) em que R é um átomo de hidrogênio e R1 é difluorometila também pode ser sintetizado pelo seguinte esquema (XVII).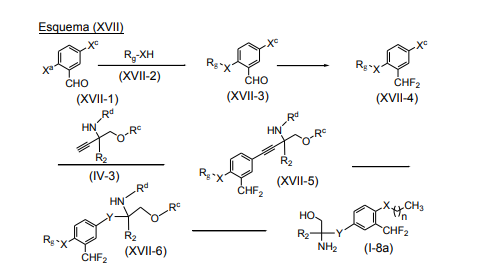
[000157] em que X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, Rc e Rd são grupos protetores, Rg é um grupo protetor ou -(CH2)nCH3, Xa e Xc são grupos de saída, e n é conforme definido acima.
[000158] Rc, Rd, Rg, Xa e Xc são especificamente conforme definidos acima.
[000159] Na primeira etapa, o intermediário (XVII-3) é obtido condensando o material de partida (XVII-1) tendo o grupo de saída Xa com álcool ou tiol (XVII-2). Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio, carbonato de potássio e similares ou uma base orgânica tal como alcóxido tal como t-butóxido de potássio e similares, 1,8-diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 80°C por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Além disso, o composto (XVII-1) em que o grupo de saída Xa é um grupo hidroxila fenólico ou tiol também pode ser usado como o material de partida. Neste caso, a primeira etapa é alquilação de um grupo hidroxila fenólico ou tiol. Exemplos do reagente a ser usado para alquilação incluem uma combinação de um agente alquilante tal como haleto de alquila e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. As condições de reação são, por exemplo, um solvente polar tal como N,Ndimetilformamida e similares ou um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Para alquilação do grupo hidroxila fenólico, além disso, pode ser usada reação de Mitsunobu.
[000160] Na segunda etapa, o intermediário (XVII-4) tendo difluorometila é obtido por fluoração do grupo formila do intermediário (XVII-3). Esta etapa pode ser realizada em um solvente de halogênio tal como cloreto de metileno e similares usando um agente de fluoração tal como trifluoreto de (dietilamino)enxofre (DAST), difluoreto de xenônio e similares. Para esta reação de fluoração, um oxidante tal como N-iodossuccinimida e similares pode ser reagido na presença de íon fluorinante tal como fluoreto de tetrabutilamônio e similares, ao invés de usar um único agente de fluoração. As condições de reação são, por exemplo, sob esfriamento com gelo até cerca de 50°C por cerca de 1 a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000161] Na terceira etapa, o intermediário (XVII-5) tendo uma ligação tripla é obtido condensando o intermediário (XVII-4) com o intermediário (IV-3) por meio da reação de Sonogashira. Exemplos do catalisador incluem composto de paládio tal como tetracis(trifenilfosfina) paládio(0), tris(dibenzilidenoacetona)dipaládio(0), diclorobis (acetonitrila)paládio(II) e similares. Para promover a reação, uma base orgânica tal como trietilamina e similares, uma base inorgânica tal como amônia e similares, um composto de cobre tal como iodeto de cobre, brometo de cobre e similares, um composto de fosfina tal como 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila e similares, e similares podem ser adicionados. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano, dioxano e similares, um solvente polar tal como acetonitrila, dimetilformamida e similares, ou um solvente de hidrocarboneto tal como benzeno e similares, sob esfriamento com gelo até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000162] Na quarta etapa, o intermediário (XVII-6) é obtido reduzindo a ligação tripla do intermediário (XVII-5). O reagente a ser usado quando Y é CH2CH2 não é limitado contanto que seja usado para redução geral de ligação de carbono insaturado. Por exemplo, hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney, complexo de paládio carbono-diamina de etileno e similares, um catalisador homogêneo complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares podem ser mencionados. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares em 1 a 20 atm de pressão de hidrogênio sob esfriamento com gelo até temperatura de refluxo por 30 min a 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Por outro lado, como a reação usada quando Y é CH=CH, hidrogenação catalítica na presença de um catalisador com atividade controlada tal como catalisador de Lindlar, complexo de níquel-grafite-diamina de etileno, composto de dieno complexo diverso e composto de fosfina e ródio, e similares podem ser mencionados. Além disso, uma redução reação usando um hidreto de metal tal como hidreto de diisobutilalumínio e similares também é possível. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000163] Na quinta etapa, o composto da presente invenção (I-8a) é obtido desprotegendo o intermediário (XVII-6). Remoção de Rc e Rd, e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 tem um ou mais grupos hidroxila referidos, não é particularmente limitado contanto que seja usado para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar acetal cíclico e Rd é t-butiloxicarbonila, acetal cíclico é desprotegido por uma quantidade catalítica de um ácido, e em seguida são empregadas condições acidíferas mais fortes, por meio das quais Rd pode ser removido. As condições empregadas para a desprotegido de acetal são, por exemplo, um solvente alcoólico tal como etanol e similares, ou uma solução mista de um solvente alcoólico e outro solvente orgânico, quantidade catalítica de ácido clorídrico ou ácido toluenossulfônico sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Por outro lado, as condições da remoção de Rd a serem realizadas seqüencialmente depois de desproteção do acetal são, por exemplo, não menos da quantidade equivalente de um ácido inorgânico tal como ácido clorídrico e similares, ácido trifluoroacético e similares, em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando Rg é um grupo protetor, alquilação de fenol ou tiol resultante da remoção de Rg pode ser realizada antes de remoção dos grupos protetores Rc e Rd . As condições a serem empregadas para remoção de Rg não são particularmente limitadas contanto que sejam usadas para remoção geral de grupos protetores. Quando, por exemplo, Rg é 4-metoxibenzila, pode ser realizada reação de oxidação por 2,3-dicloro-5,6-diciano-1,4- benzoquinona (DDQ) e similares, e quando Rg é alila, pode ser realizada uma reação usando um composto de paládio como um catalisador. Exemplos do reagente a ser usado para a alquilação do grupo hidroxila fenólico ou grupo tiol do composto obtido incluem uma combinação de um agente alquilante tal como haleto de alquila e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. As condições de reação são, por exemplo, um solvente polar tal como N,N-dimetilformamida e similares ou um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Para alquilação do grupo hidroxila fenólico, também pode ser usada reação de Mitsunobu.
[000164] 17) Um composto (I-9a) representado pela fórmula (I) em que R é um átomo de hidrogênio, e R1 é fluorometila também pode ser sintetizado pelo seguinte esquema (XVIII).
[000165] em que X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, Rc, Rd e Ri são grupos protetores, Xa e Xc são grupos de saída, e n é conforme definido acima.
[000166] Exemplos específicos de Rc, Rd, Xa e Xc na fórmula são conforme os mencionados acima. O grupo protetor para Ri não é particularmente limitado contanto que proteja o grupo hidroxila na fórmula. Por exemplo, trialquilsilila (especificamente t-butildimetilsilila e similares) pode ser mencionado.
[000167] Na primeira etapa, o intermediário (XVIII-3) é obtido condensando o material de partida (XVIII-1) tendo o grupo de saída Xa com álcool ou tiol (XVIII-2). Esta etapa pode ser realizada em um solvente polar tal como N,N-dimetilformamida, sulfóxido de dimetila e similares ou um solvente etéreo tal como tetraidrofurano e similares na presença de uma base. Como a base, uma base inorgânica tal como hidreto de sódio, hidróxido de potássio, carbonato de potássio e similares ou uma base orgânica tal como alcóxido (por exemplo, tbutóxido de potássio e similares), 1,8-diazabiciclo[5.4.0]undec-7-eno e similares pode ser usada. A reação é realizada, por exemplo, sob esfriamento com gelo até cerca de 80°C por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Além disso, o composto (XVIII-1) em que o grupo de saída Xa é um grupo hidroxila fenólico ou tiol também pode ser usado como o material de partida. Neste caso, a primeira etapa é alquilação de um grupo hidroxila fenólico ou tiol. Exemplos do reagente a ser usado para alquilação incluem uma combinação de um agente alquilante tal como haleto de alquila e similares e uma base inorgânica tal como carbonato de potássio, hidreto de sódio e similares. As condições de reação são, por exemplo, um solvente polar tal como N,N-dimetilformamida e similares ou um solvente etéreo tal como tetraidrofurano e similares, sob esfriamento com gelo até 80°C por cerca de 10 min a 12 horas. Para alquilação do grupo hidroxila fenólico, além disso, pode ser usada reação de Mitsunobu.
[000168] Na segunda etapa, o grupo formila do intermediário (XVIII-3) é reduzido para dar hidroximetila, e o grupo protetor Ri é introduzido no mesmo. O reagente a ser usado para a redução do grupo formila não é particularmente limitado contanto que seja usado de modo geral. Exemplos do mesmo incluem hidreto de metal tal como hidreto de diisobutilalumínio e similares, composto de complexo metal hidrogênio tal como hidreto de alumínio de lítio, boridreto de sódio e similares, hidrogenação catalítica usando catalisador de sistema homogêneo ou sistema heterogêneo e similares. Como as condições de reação, são selecionados temperatura e tempo apropriados para o reagente de redução. Especificamente, redução por hidreto de alumínio de lítio ou boridreto de lítio, a qual é realizada em um solvente etéreo tal como tetraidrofurano e similares em -30°C até temperatura ambiente por cerca de 10 min a 3 horas, redução por boridreto de sódio ou boridreto de cálcio, a qual é realizada em um solvente alcoólico tal como etanol e similares ou um solvente misto de um solvente alcoólico e um solvente etéreo tal como tetraidrofurano e similares sob esfriamento com gelo até a temperatura ambiente por cerca de 10 min a 3 horas e similares podem ser mencionados. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares. Para introdução do grupo protetor Ri , é usada reação de introdução geral de grupos protetores. Quando um grupo trialquilsilila é usado para Ri , um agente de sililação tal como t-butildimetilclorossilano e similares é usado como um reagente, e uma base tal como imidazol, trietilamina e similares pode ser adicionada como um promotor de reação. Depois da reação, o produto-fim pode ser obtido por interrupção brusca da reação, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares.
[000169] Na terceira etapa, o intermediário (XVIII-5) tendo uma ligação tripla é obtido condensando o intermediário (XVIII-4) com o intermediário (IV-3) pela reação de Sonogashira. Exemplos do catalisador incluem composto de paládio tal como tetracis(trifenilfosfina) paládio(0), tris(dibenzilidenoacetona)dipaládio(0), diclorobis (acetonitrila)paládio(II) e similares. Para promover a reação, uma base orgânica tal como trietilamina e similares, uma base inorgânica tal como amônia e similares, um composto de cobre tal como iodeto de cobre, brometo de cobre e similares, um composto de fosfina tal como 2- diciclohexilfosfino-2',4',6'-triisopropilbifenila e similares, e similares pode ser adicionado. A reação é realizada, por exemplo, em um solvente etéreo tal como tetraidrofurano, dioxano e similares, um solvente polar tal como acetonitrila, dimetilformamida e similares, ou um solvente de hidrocarboneto tal como benzeno e similares, sob esfriamento com gelo até a temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000170] Na quarta etapa, o intermediário (XVIII-6) é obtido reduzindo a ligação tripla do intermediário (XVIII-5). Quando Y é CH2CH2, o reagente a ser usado não é limitado contanto que seja usado para redução geral de ligação de carbono insaturado. Por exemplo, hidrogenação catalítica usando um catalisador heterogêneo tal como paládio carbono, níquel de Raney, complexo de paládio carbonodiamina de etileno e similares ou um catalisador homogêneo tal como complexo de ródio (clorotris(trifenilfosfina)ródio(I) e similares) e similares podem ser mencionados. As condições de reação são, por exemplo, um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como dioxano e similares, ou um solvente de hidrocarboneto tal como tolueno e similares em 1 a 20 atm de pressão de hidrogênio sob esfriamento com gelo até temperatura de refluxo por 30 min a 1 semana. Um ácido tal como ácido acético e similares ou uma base tal como trietilamina e similares podem ser adicionados à mistura da reação dependendo da velocidade da reação, da estabilidade do composto e similares. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Por outro lado, como uma reação usada quando Y é CH=CH, hidrogenação catalítica na presença de um catalisador tendo atividade controlada tal como catalisador de Lindlar, complexo de níquel-grafite-diamina de etileno, vários complexos de dieno e fosfina e ródio e similares podem ser mencionados. Além disso, reação de redução por hidreto de metal tal como hidreto de diisobutilalumínio e similares também pode ser usada. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000171] Na quinta etapa, a forma fluoreto (XVIII-7) é sintetizada removendo Ri do composto (XVIII-6), e fluoração do grupo hidroxila do composto obtido. O grupo protetor Ri pode ser removido por desproteção geral. Exemplos do reagente a ser usado quando Ri é trialquilsilila incluem compostos de flúor tais como fluoreto de tetrabutilamônio e similares. As condições da reação são, por exemplo, em um solvente etéreo tal como tetraidrofurano e similares, esfriamento com gelo até temperatura de refluxo por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por destilação, cromatografia de coluna de sílica-gel, recristalização e similares. Como o reagente a ser usado para fluoração sucessiva, podem ser mencionados trifluoreto de (dietilamino)enxofre (DAST), 2,2-diflúor-1,3-dimetilimidazolidina (DFI) e similares. Nesta etapa, a reação pode ser realizada em um solvente de halogênio tal como cloreto de metileno e similares ou um solvente de hidrocarboneto tal como hexano e similares. As condições de reação são, por exemplo, -78°C - temperatura ambiente por cerca de 30 min - 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Esta etapa também pode ser realizada por um método incluindo converter o grupo hidroxila para a forma sulfonato correspondente, e reagir o mesmo com íon fluoreto. Por exemplo, quando fluoreto de p-toluenossulfonila e fluoreto de tetrabutilamônio (TBAF) são usados, a reação é realizada em um solvente etéreo tal como tetraidrofurano e similares em temperatura ambiente até 80°C por cerca de 1 hora a 24 horas. Nesta reação, pode ser adicionado um agente desidratante tal como peneiras moleculares e similares. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando Ri é trialquilsilila, pode ser realizada fluoração sem remoção de Ri.
[000172] Na sexta etapa, o composto da presente invenção (I-9a) é obtido desprotegendo o intermediário (XVIII-7). Remoção de Rc e Rd , e o grupo protetor Re (Re é conforme definido acima) o qual protege um ou mais grupos hidroxila quando R2 têm um ou mais grupos hidroxila referidos, não é particularmente limitado contanto que seja usado para remoção geral de grupos protetores, e todos os grupos protetores podem ser removidos de uma vez ou por etapas. Por exemplo, quando Rc e Re são ligados para formar acetal cíclico e Rd é t-butiloxicarbonila, acetal cíclico é desprotegido por uma quantidade catalítica de um ácido, e em seguida são empregadas condições acidíferas mais fortes, por meio das quais Rd podem ser removidos. As condições empregadas para a desproteção de acetal são, por exemplo, um solvente alcoólico tal como metanol e similares ou uma solução mista de um solvente alcoólico e outro solvente orgânico usando um quantidade catalítica de ácido clorídrico ou ácido toluenossulfônico sob esfriamento com gelo até 80°C por cerca de 30 min a 12 horas. Por outro lado, as condições da remoção de Rd para seguinr a desproteção por acetal são, por exemplo, não menos da quantidade equivalente de um ácido inorgânico tal como ácido clorídrico e similares, ácido trifluoroacético e similares em um solvente alcoólico tal como etanol e similares, um solvente etéreo tal como tetraidrofurano e similares, água ou um solvente misto do mesmo sob esfriamento com gelo até a temperatura ambiente por cerca de 10 min a 5 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Além disso, um solvente tendo baixa solubilidade tal como éter diisopropílico e similares pode ser adicionado à solução da reação e o produto-fim precipitado pode ser coletado por filtração.
[000173] 18) Dos compostos da presente invenção, um composto (I-10a) representado pela fórmula (I) em que R é P(=O)(OH)2, e R3 e R4 são átomos de hidrogênio é sintetizado pelo seguinte esquema (XIX).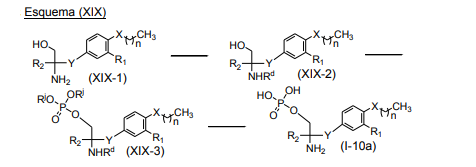
[000174] em que X é um átomo de oxigênio ou um átomo de enxofre, Y é CH2CH2 ou CH=CH, R1 é ciano ou alquila tendo um número de carbonos de 1 a 4 e substituída por um ou mais átomos de halogênio, R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, Rd e Rj são grupos protetores, e n é conforme definido acima.
[000175] Rd na fórmula é conforme definido acima. Quando R2 do composto (XIX-2) contém um grupo hidroxila, o grupo hidroxila pode ser protetido por um grupo protetor Re (Re é conforme definido acima). Quando R2 é hidroximetila ou hidroxietila protegida, seu grupo protetor Re é ligado a Rd ou o átomo de nitrogênio ao qual Rd é ligado para formar o seguinte composto cíclico (XIX-2', XIX-2")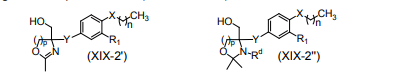
[000176] em que p é 1 ou 2, e outros símbolos são conforme definido para o esquema (XIX), por meio dos quais o grupo amino e o grupo hidroxila são protegidos.
[000177] O grupo protetor para Rj na fórmula não é particularmente limitado contanto que proteja grupo ácido fosfórico. Por exemplo, alquila (preferencialmente tendo um número de carbonos de cerca de 1 a 6, especificamente t-butila e similares), benzila, fenila e similares podem ser mencionados.
[000178] Na primeira etapa, a forma protegida do grupo amino (XIX-2) é sintetizada protegendo o grupo amino do composto (XIX-1) em que R é um átomo de hidrogênio, dos compostos da presente invenção. Esta etapa pode ser realizada por uma reação de proteção de grupo amino geral. Especificamente, quando acila, alquiloxicarbonila, benziloxicarbonila e similares são usados como grupo protetor (Rd ), esta etapa pode ser realizada em álcool tal como metanol e similares, ou um sistema de duas camadas ou mistura de água e um solvente orgânico tal como acetato de etila, clorofórmio e similares. Exemplos do reagente a ser usado incluem cloreto ácido tal como cloreto de acetila, cloreto oxicarbonil de benzila e similares, anidrido ácido tal como anidrido acético, dicarbonato de di-t-butila e similares. Uma base orgânica tal como trietilamina e similares, uma base inorgânica tal como bicarbonato de sódio e similares podem ser adicionadas como um promotor da reação para esta reação. As condições de reação são, por exemplo, sob esfriamento com gelo até 50°C por cerca de 30 min a 24 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Quando o grupo amino e o grupo hidroxila contidos em R2 são protegidos simultaneamente como oxazolina da fórmula (XIX-2'), esta etapa pode ser realizada por reação em um solvente polar tal como acetonitrila, N,N-dimetilformamida e similares, um solvente de halogênio tal como cloreto de metileno e similares, ou um solvente de hidrocarboneto tal como tolueno e similares, usando éster de ácido ortoacético como um reagente. Além disso, para promoção da reação, uma base tal como N,N-diisopropiletilamina e similares, ou um ácido tal como ácido p-toluenossulfônico e similares podem ser adicionados. As condições de reação são, por exemplo, temperatura ambiente até temperatura de refluxo por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000179] Na segunda etapa, a forma fosforilada (XIX-3) é sintetizada reagindo a forma protegida do grupo amino (XIX-2) com um reagente de fosforilação (por exemplo, cloreto de fósforo, fosforamidita e oxidante, éster tetrabenzílico de ácido pirofosfórico e similares). Quando éster tetrabenzílico de ácido pirofosfórico é usado como um reagente de fosforilação, esta etapa pode ser realizada sob condições não aquosas preferencialmente em um solvente orgânico tal como tolueno, diclorometano, um solvente misto do mesmo e similares usando um aditivo (por exemplo, óxido de prata, iodeto de tetra-n-hexil amônio e similares). As condições de reação são, por exemplo, sob esfriamento com gelo até 50°C por cerca de 5 a 24 horas. Depois da reação, o produto-fim pode ser obtido por filtração, extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Para esta reação, um reagente de fosforilação geral (cloreto de fósforo e base, fosforamidita e oxidante e similares) também pode ser reagido por um método conhecido. Por exemplo, quando fosforamidita e um oxidante são usados, a reação é realizada em um solvente de halogênio tal como diclorometano e similares, um solvente etéreo tal como tetraidrofurano e similares, um solvente polar tal como acetonitrila e similares ou um solvente misto do mesmo, usando fosforamidita tal como di-t-butil diisopropilfosforamidita e similares sob esfriamento com gelo até 50°C por cerca de 10 min a 5 horas. 1H-Tetrazol e similares podem ser adicionados como um promotor da reação para esta reação. Para uma reação de oxidação de fósforo realizada sucessivamente depois da fosforilação, um peróxido orgânico tal como ácido m-cloroperbenzóico, hidroperóxido de t-butila e similares ou um peróxido inorgânico tal como peróxido de hidrogênio e similares pode ser usado. A reação é realizada sob esfriamento com gelo até 50°C por cerca de 3 min a 1 hora. Depois da reação, o produtofim pode ser obtido por extração, lavagem, secagem, remoção de solvente e similares por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares.
[000180] Na terceira etapa, o composto da presente invenção (I-10a) é preparado a partir de uma forma fosforilada (XIX-3). Esta etapa pode ser realizada por desproteção geral. Especificamente, a etapa pode ser realizada por hidrogenólise usando um ácido tal como ácido clorídrico, ácido trifluoroacético e similares, um ácido de Lewis tal como brometo de trimetilsilila e similares. Quando se usa hidrogenólise para esta reação, esta etapa é realizada, por exemplo, em um solvente alcoólico tal como metanol e similares usando um catalisador tal como paládio carbono e similares sob uma atmosfera de hidrogênio. As condições de reação são, por exemplo, temperatura ambiente até 60°C por cerca de 1 a 24 horas. O produto-fim pode ser obtido por filtração, concentração e similares da mistura da reação por um método geral e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. As condições de reação quando um ácido é usado para esta reação são, por exemplo, um solvente alcoólico tal como etanol e similares ou um solvente misto do mesmo com água em temperatura ambiente até 100°C por cerca de 30 min a 12 horas. Depois da reação, o produto-fim pode ser obtido adicionando a mistura da reação a água e coletando o produto-fim precipitado por filtração, ou extração, lavagem, secagem, remoção de solvente e similares e, onde necessário, purificação por cromatografia de coluna de sílica-gel, recristalização e similares. Dos compostos da presente invenção, um composto da fórmula (I) em que R é P(=O)(OH)2, e um de R3 e R4 é alquila tendo um número de carbonos de 1 a 4 também é sintetizado por um método similar ao esquema (XIX) mencionado acima. Um composto da fórmula (I) em que R é P(=O)(OH)2, e tanto R3 quanto R4 são alquila tendo um número de carbonos de 1 a 4 é sintetizado por um método similar ao esquema (XIX), sem usar grupo protetor de amino Rd usado no esquema (XIX).
[000181] O composto da presente invenção pode ser convertido em um sal de adição de ácido conforme necessário por meio de tratamento com um ácido em um solvente adequado (água, álcool, éter e similares). Além disso, o composto obtido da presente invenção pode ser convertido em um hidrato ou solvato por meio de tratamento com água, solvente contendo água ou outro solvente (por exemplo, álcool etc.).
[000182] O composto da presente invenção é útil para o tratamento ou a profilaxia de doenças auto-imunes (por exemplo, artrite reumatóide, esclerose múltipla, encefalomielite, lúpus eritematoso sistêmico, nefrite por lúpus, síndrome nefrótica, psoríase, diabetes melito Tipo I e etc.); profilaxia ou supressão de resistência ou rejeição aguda ou rejeição crônica de transplante de órgão ou tecido (por exemplo, inclusive transplante e transplante heterogêneo de coração, rim, fígado, pulmão, medula óssea, córnea, pâncreas, intestino delgado, extremidade, músculo, nervo, medula adiposa, duodeno, pele, células das ilhotas pâncreáticas e similares) em mamíferos tais como ser humano, cão, gato, bovino, cavalo, suíno, macaco, camundongo e similares; doença enxerto-versus-hospedeiro (GvH) devido o transplante de medula óssea; e o tratamento ou a profilaxia de doenças alérgicas (por exemplo, dermatite atópica, rinite alérgica, asma e etc.).
[000183] Na presente invenção, a "profilaxia" significa o ato de administrar o composto da presente invenção ou uma composição farmacêutica contendo o composto a um indivíduo que não tenha desenvolvido uma doença ou sintoma. Além disso, o "tratamento" significa o ato de administrar o composto da presente invenção ou uma composição farmacêutica contendo o composto a um indivíduo que já tenha desenvolvido uma doença ou distúrbio ou sintoma. Por conseguinte, o ato de administração a um indivíduo que já tenha desenvolvido uma doença ou distúrbio ou sintoma para a prevenção de agravação do sintoma e similares, prevenção de ataques ou prevenção de recaída é uma modalidade do "tratamento".
[000184] Quando o composto da presente invenção é usado como um agente farmacêutico, o composto da presente invenção é misturado com um veículo farmaceuticamente aceitável (excipiente, aglutinante, desintegrante, corrigente, aroma, emulsificante, diluente, solubilizante e similares) e a composição ou preparação farmacêutica obtida (preparação oral, injeção e similares) pode ser administrada por via oral ou por via parenteral. Uma composição farmacêutica pode ser preparada de acordo com um método geral.
[000185] No presente relatório descritivo, parenteral inclui injeção subcutânea, injeção intravenosa, injeção intramuscular, injeção intraperitoneal, método de gotejamento ou administração tópica (administração transdérmica, administração transocular, administração transpulmônica-bronquial, administração transnasal, administração transretal e similares) e similares.
[000186] O conteúdo do composto da presente invenção que pode ser combinado com um veículo que pode ser variado dependendo do indivíduo a ser tratado e da forma de dosagem em particular. No entanto, a dose particular de pacientes particulares é determinada dependendo de vários fatores incluindo idade, peso corporal, condições de saúde geral, sexo, dieta, tempo de administração, método de administração, taxa de liberação e gravidade da doença sob tratamento em particular.
[000187] A dose do composto da presente invenção é determinada considerando a idade, peso corporal, condições de saúde geral, sexo, dieta, tempo de administração, método de administração, taxa de clearance e gravidade da doença dos pacientes sob tratamento, bem como outros fatores. O composto da presente invenção não afeta a freqüência cardíaca e pode ser usada com segurança. Sua dose diária varia dependendo da condição e do peso corporal dos pacientes, do tipo de composto, da via de administração e similares. Por exemplo, para administração parenteral, é administrada por via subcutânea, por via intravenosa, por via intramuscular, por via transdérmica, por via transocular, por via transpulmônica ou por via bronquial, por via transnasal ou por via retal a cerca de 0,01 a 50 mg/paciente/dia, e para administração oral, é administrada a cerca de 0,01 a 150 mg/paciente/dia.
[000188] A presente invenção é explicada em mais detalhes a seguir com referência aos Exemplos, os quais não devem ser considerados como limitantes.
[000189] Éster t-butílico de ácido (2,2-dimetil-5-formil-1,3-dioxan-5- il)carbâmico
[000190] Cloridrato de tris(hidroximetil)aminometano (2 g) foi dissolvido em N,N-dimetilformamida (50 ml), 2,2-dimetoxipropano (7,8 ml) e monoidrato de ácido p-toluenossulfônico (229 mg) foram adicionados, e a mistura foi agitada em temperatura ambiente por 15 horas. À solução misturada foram adicionados trietilamina (9,5 ml), metanol (20 ml) e dicarbonato de di-t-butila (4,17 g), e a mistura foi agitada em temperatura ambiente por 12 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (3,11 g) como um sólido incolor.
[000191] 1H-RMN (CDCl3) δ (ppm): 1,44 (3H, s), 1,46(12H, s), 3,73 (2H, d, J = 6,4 Hz), 3,80 (2H, d, J = 11,6 Hz), 3,84 (2H, d, J = 11,6 Hz), 4,20 (1H, brs), 5,32 (1H, brs).
[000192] O composto (2,96 g) do Exemplo de Referência composto 1- 1 foi dissolvido em sulfóxido de dimetila (50 ml), trietilamina (11 ml) e complexo de trióxido de enxofre-piridina (5,4 g) foram adicionados, e a mistura foi agitada em temperatura ambiente por 2 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi lavado com éter dietílico para dar o produto-fim (2,4 g) como um pó incolor.
[000193] 1H-RMN (CDCl3) δ (ppm): 1,46(15H, s), 3,96 (2H, d, J = 11,7 Hz), 4,07 (2H, d, J = 11,7 Hz), 5,54 (1H, brs), 9,64 (1H, s).
[000194] Cloreto de (4-benzilóxi-3-trifluorometilbenzil)trifenilfosfônio
[000195] Ácido 4-flúor-3-trifluorometilbenzóico (100 g) foi dissolvido em N,N-dimetilformamida (400 ml), carbonato de potássio (199 g) e brometo de benzila (84,0 g) foram adicionados sob esfriamento com gelo, e a mistura foi agitada por 20 min sob esfriamento com gelo e em temperatura ambiente por 2 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (144 g) como um óleo amarelo-pálido.
[000196] 1H-RMN (CDCl3) δ (ppm): 5,38 (2H, s), 7,27 (1H, t, J = 9,3 Hz), 7,35 - 7,46 (5H, m), 8,27 (1H, m), 8,35 (1H, dd, J = 6,8, 1,8 Hz).
[000197] Álcool benzílico (52,0 g) foi dissolvido em N,N-dimetilformamida (300 ml), hidreto de sódio (60%, 20,2 g) foi adicionado sob esfriamento com gelo, e a mistura foi agitada por 50 min sob esfriamento com gelo. Uma solução do Exemplo de Referência composto 2-1 (144 g) em N,N-dimetilformamida (400 ml) foi adicionada, e a mistura foi agitada por 2 horas sob esfriamento com gelo. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (198 g, uma mistura com óleo mineral contido em hidreto de sódio) como um sólido amarelo-pálido.
[000198] 1H-RMN (CDCl3) δ (ppm): 5,26 (2H, s), 5,35 (2H, s), 7,06 (1H, d, J = 8,8 Hz), 7,31 - 7,45(10H, m), 8,18 (1H, dd, J = 8,8, 2,0 Hz), 8,32 (1H, d, J = 2,0 Hz).
[000199] O composto (198 g) obtido no Exemplo de Referência composto 2-2 foi dissolvido em tetraidrofurano (1000 ml), boridreto de lítio (15,7 g) foi adicionado, e a mistura foi aquecida sob refluxo por 3 horas. Depois de resfriar uma vez, boridreto de lítio (4,0 g) foi adicionado, e a mistura foi adicionalmente aquecida sob refluxo por 3 horas. A mistura da reação foi resfriada, água (500 ml) foi adicionada para extinguir a reação. A mistura da reação foi adicionada a água, e a mistura foi neutralizada com ácido clorídrico concentrado. A mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. As impurezas (óleo mineral e álcool benzílico) foram removidas da mistura obtida aquecendo sob pressão reduzida a 135°C usando uma bomba a vácuo. O resíduo obtido foi cristalizado a partir de hexano para dar o produto-fim (99,2 g) como um pó branco.
[000200] 1H-RMN (CDCl3) δ (ppm): 1,62 (1H, t, J = 5,7 Hz), 4,66 (2H, d, J = 5,7 Hz), 5,20 (2H, s), 7,02 (1H, d, J = 8,5 Hz), 7,30 - 7,33 (1H, m), 7,38 (2H, t, J = 7,4 Hz), 7,44 (2H, d, J = 7,4 Hz), 7,46 (1H, dd, J = 8,5, 2,0 Hz), 7,61 (1H, d, J = 2,0 Hz).
[000201] O composto (99,2 g) obtido no Exemplo de Referência composto 2-3 foi dissolvido em cloreto de metileno (900 ml), trifenilfosfina (102 g) e N-clorossuccinimida (49,3 g) foram adicionados sob esfriamento com gelo, e a mistura foi agitada sob esfriamento com gelo por 40 min, e adicionalmente em temperatura ambiente por 1 hora. A mistura da reação foi lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. Éter (500 ml) foi adicionado, óxido de trifenilfosfina precipitado primeiro foi removido e o resíduo foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 99:1 a 4:1) para dar o produto-fim (99,5 g) como um óleo incolor.
[000202] 1H-RMN (CDCl3) δ (ppm): 4,56 (2H, s), 5,20 (2H, s), 7,01 (1H, d, J = 8,6 Hz), 7,31 - 7,34 (1H, m), 7,39 (2H, t, J = 7,4 Hz), 7,43 (2H, d, J = 7,4 Hz), 7,48 (1H, dd, J = 8,6, 2,0 Hz), 7,62 (1H, d, J = 2,0 Hz).
[000203] O composto (99,0 g) obtido no Exemplo de Referência composto 2-4 foi dissolvido em tolueno (450 ml), trifenilfosfina (90,7 g) foi adicionado, e a mistura foi refluxada por 8 horas. Depois de resfriamento, os cristais na mistura da reação foram coletados por filtração, e lavados com éter para dar o composto fim (132 g) como um pó branco. O licor-mãe foi concentrado, tolueno (200 ml) foi adicionado, e a operação supracitada foi realizada para dar o composto fim (31,0 g). Além disso, o licor-mãe foi tratado na mesma maneira para dar o composto fim (12,3 g). O rendimento total foi 176 g.
[000204] MS (ESI) m/z: 527 [M+]
[000205] 1H-RMN (DMSO-d6) δ (ppm): 5,17 (2H, d, J = 15,1 Hz), 5,23 (2H, s), 7,02 - 7,04 (1H, m), 7,26 - 7,30 (2H, m), 7,31 - 7,37 (1H, m), 7,38 - 7,42 (4H, m), 7,65 - 7,70 (6H, m), 7,72 - 7,78 (6H, m), 7,90 - 7,94 (3H, m).
[000206] 5-Bromo-2-heptiloxibenzonitrila
[000207] 1-Heptanol (1,55 g) foi dissolvido em N,N-dimetilformamida (24 ml), e hidreto de sódio (0,321 g) foi adicionado em temperatura ambiente. A mistura foi agitada por 1 hora, 5-bromo-2-fluorobenzonitrila (2,43 g) foi adicionado, e a mistura foi adicionalmente agitada por 50 min. A mistura da reação foi vertida em água, extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. Para consumir o material de partida, 5-bromo-2-fluorobenzonitrila, a reação foi realizada de novo sob as mesmas condições, e o resíduo foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 50:1 a 5:1) para dar o produto-fim (3,10 g) como um óleo incolor.
[000208] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,4 Hz), 1,24 - 1,35 (6H, m), 1,48 (2H, quint, J = 7,2 Hz), 1,84 (2H, quint, J = 6,4 Hz), 4,04 (2H, t, J = 6,4 Hz), 6,84 (1H, d, J = 8,8 Hz), 7,59 (1H, dd, J = 8,8, 2,4 Hz), 7,65 (1H, d, J = 2,4 Hz).
[000209] Cloridrato de 2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil) etil]propano-1,3-diol
[000210] O Exemplo de Referência composto 2-5 (70,3 g) foi dissolvido em tetraidrofurano (500 ml), t-butóxido de potássio (13,0 g) foi adicionado, e a mistura foi agitada por 1 hora. À solução misturada foi adicionada gota a gota uma solução do composto (15,0 g) do Exemplo de Referência 1 em tetraidrofurano (100 ml) sob esfriamento com gelo, e a mistura foi agitada por 2 horas sob esfriamento com gelo. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílicagel (hexano: acetato de etila = 3:1) para dar um óleo amarelo-pálido (31,0 g). A proporção de isômeros geométricos do composto obtido foi (E:Z = 1:6).
[000211] O óleo amarelo-pálido foi dissolvido em acetato de etila (200 ml), 10% de paládio carbono (3,00 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 7 horas sob uma atmosfera de hidrogênio. O interior do recipiente da reação foi substituído com nitrogênio, e a solução foi filtrada, e o filtrado foi concentrado. O resíduo foi lavado com éter diisopropílico para dar o produto-fim (22,3 g) como um pó incolor.
[000212] 1H-RMN (CDCl3) δ (ppm): 1,43 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,91 - 1,98 (2H, m), 2,50 - 2,56 (2H, m), 3,69 (2H, d, J = 11,6 Hz), 3,89 (2H, d, J = 11,6 Hz), 5,02 (1H, brs), 5,52 (1H, brs), 6,86 (1H, d, J = 8,2 Hz), 7,22 (1H, dd, J = 8,2, 1,7 Hz), 7,29 (1H, d, J = 1,7 Hz).
[000213] O composto 1-1 (510 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (506 mg) e brometo de n-heptila (0,235 ml) foram adicionados, e a mistura foi agitada em 80°C por 2 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (640 mg) como um óleo incolor.
[000214] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,30 - 1,37 (6H, m), 1,42 - 1,50 (2H, m), 1,42 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,76 - 1,82 (2H, m), 1,91 - 1,98 (2H, m), 2,50 - 2,57 (2H, m), 3,69 (2H, d, J = 11,6 Hz), 3,89 (2H, d, J = 11,6 Hz), 4,00 (2H, t, J = 6,4 Hz), 4,98 (1H, brs), 6,88 (1H, d, J = 8,5 Hz), 7,26 - 7,29 (1H, m), 7,35 (1H, d, J = 1,5 Hz).
[000215] O composto 1-2 (640 mg) foi dissolvido em etanol (15 ml), ácido clorídrico concentrado (3 ml) foi adicionado, e a mistura foi agitada em 80°C por 2 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (492 mg) como pó branco.
[000216] MS (ESI) m/z:378 [M+H]
[000217] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,24 - 1,39 (6H, m), 1,39 - 1,46 (2H, m), 1,68 - 1,78 (4H, m), 2,55 - 2,62 (2H, m), 3,51 (4H, d, J = 5,1 Hz), 4,06 (2H, t, J = 6,2 Hz), 5,38 (2H, t, J = 5,1 Hz), 7,18 (1H, d, J = 8,4 Hz), 7,42 - 7,45 (2H, m), 7,76 (3H, brs).
[000218] 2-Amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)butanol
[000219] Uma mistura de duas camadas do composto 1-3 (290 mg), acetato de etila (5 ml), solução aquosa saturada de bicarbonato de sódio (5 ml) e cloreto de benziloxicarbonila (0,129 ml) foi agitada em temperatura ambiente por 5 horas. A camada de acetato de etila foi separada, e a camada aquosa foi extraída com acetato de etila. A camada de acetato de etila separada e a camada de acetato de etila obtida pela extração foram combinadas, lavadas com salmoura saturada, e secadas sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:3) para dar o produto-fim (230 mg) como um óleo incolor.
[000220] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,26 - 1,39 (6H, m), 1,41 - 1,51 (2H, m), 1,75 - 1,83 (2H, m), 1,84 - 1,91 (2H, m), 2,45 - 2,60 (2H, m), 3,03 (2H, brs), 3,66 - 3,71 (2H, m), 3,88 - 3,93 (2H, m), 3,99 (2H, t, J = 6,3 Hz), 5,09 (2H, s), 5,31 (1H, brs), 6,87 (1H, d, J = 8,5 Hz), 7,22 - 7,26 (2H, m), 7,31 - 7,35 (5H, m).
[000221] O composto 2-1 (230 mg), éster tetrabenzílico de ácido pirofosfórico (485 mg), óxido de prata (208 mg) e iodeto de tetra-n-hexil amônio (433 mg) foram adicionados a um solvente misto de tolueno (4 ml), diclorometano (4 ml) e perfluorohexano (4 ml), e a mistura foi agitada em temperatura ambiente por 15 horas. O material insolúvel foi filtrado e o solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por HPLC do preparativo para dar o produto-fim (210 mg) como um óleo incolor.
[000222] MS (ESI) m/z: 772 [M+H]
[000223] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,4 Hz), 1,29 - 1,44 (6H, m), 1,45 - 1,53 (2H, m), 1,74 - 1,84 (3H, m), 1,85 - 2,02 (1H, m), 2,49 - 2,59 (2H, m), 3,59 (1H, d, J = 11,2 Hz), 3,69 (1H, d, J = 11,2 Hz), 4,02 (2H, t, J = 6,2 Hz), 4,15 - 4,20 (1H, m), 4,26 - 4,31 (1H, m), 4,99 - 5,03 (6H, m), 6,98 (1H, d, J = 8,5 Hz), 7,22 - 7,34(17H, m).
[000224] O composto 2-2 (210 mg) foi dissolvido em metanol (10 ml), 10% de paládio carbono (100 mg) foi adicionado, e o recipiente da reação foi deslocado com hidrogênio. A mistura foi agitada em temperatura ambiente por 4 horas, o recipiente da reação foi deslocado com nitrogênio, e a mistura da reação foi filtrada. O filtrado foi concentrado para dar o produto-fim (33,0 mg) como um pó branco.
[000225] MS (ESI) m/z: 458 [M+H]
[000226] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,4 Hz), 1,29 - 1,44 (6H, m), 1,45 - 1,53 (2H, m), 1,74 - 1,82 (2H, m), 1,90 - 1,99 (2H, m), 2,60 - 2,75 (2H, m), 3,70 (2H, brs), 3,93 - 3,99 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,3 Hz), 7,42 - 7,46 (2H, m).
[000227] (S)-2-Amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)butanol
[000228] A uma mistura do composto 1-1 (3,00 g), clorofórmio (300 ml) e solução aquosa saturada de bicarbonato de sódio (300 ml) foi adicionado anidrido acético (1,03 ml) 8 vezes em intervalos de 10 min com agitação. A mistura foi agitada por 1,5 hora a partir da adição final de anidrido acético. A camada orgânica foi separada, lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (2,96 g) como um sólido branco.
[000229] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,30 - 1,38 (6H, m), 1,46 (2H, quint, J = 7,3 Hz), 1,80 (2H, quint, J = 6,9 Hz), 1,91 - 1,96 (2H, m), 2,02 (3H, s), 2,59 - 2,63 (2H, m), 3,59 (2H, brs), 3,63 (2H, d, J = 11,8 Hz), 3,85 (2H, d, J = 10,4 Hz), 4,00 (2H, t, J = 6,5 Hz), 5,92 (1H, brs), 6,90 (1H, d, J = 8,6 Hz), 7,29 (1H, dd, J = 2,1, 8,6 Hz), 7,36 (1H, d, J = 2,1 Hz).
[000230] A uma solução do composto 3-1 (2,96 g) em diclorometano (70 ml) foram adicionados piridina (0,742 ml) e anidrido acético (0,734 ml) sob esfriamento com gelo, e a mistura foi agitada por 7 horas sob esfriamento com gelo. Piridina (0,371 ml) e anidrido acético (0,367 ml) foram adicionados, e a mistura foi agitada por 1 hora sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 14 horas. A mistura da reação foi diluída com diclorometano (200 ml), lavada sucessivamente com 0,1 M de ácido clorídrico, solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi purificado por cromatografia de sílica-gel para dar o produtofim (1,55 g) como um óleo incolor.
[000231] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,28 - 1,38 (6H, m), 1,46 (2H, quint, J = 7,3 Hz), 1,79 (2H, quint, J = 7,0 Hz), 1,84 - 1,92 (1H, m), 2,01 (3H, s), 2,13 (3H, s), 2,13 - 2,22 (1H, m), 2,53 (1H, dt, J = 5,1, 13,1 Hz), 2,66 (1H, dt, J = 4,9, 13,2 Hz), 3,72 - 3,75 (2H, m), 4,00 (2H, t, J = 6,4 Hz), 4,16 (1H, d, J = 11,5 Hz), 4,38 (1H, d, J = 11,5 Hz), 4,40 (1H, t, J = 6,8 Hz), 5,82 (1H, brs), 6,90 (1H, d, J = 8,6 Hz), 7,28 (1H, dd, J = 1,7, 8,6 Hz), 7,35 (1H, d, J = 1,7 Hz).
[000232] A uma solução do composto 3-2 (1,55 g) e 1H-tetrazol (0,282 g) em diclorometano (50 ml) e acetonitrila (50 ml) foi adicionado di-t-butil diisopropilfosforamidita (1,27 ml) sob esfriamento com gelo, e a mistura foi agitada por 1,5 hora sob esfriamento com gelo. 1H-Tetrazol (0,282 g) e di-t-butil diisopropilfosforamidita (1,27 ml) foram adicionados, e a mistura foi adicionalmente agitada por 2 horas. À mistura da reação foi adicionado ácido m-cloroperbenzóico (contendo 25% de água, 0,994 g) sob esfriamento com gelo, e a mistura foi agitada por 20 min. Ácido mcloroperbenzóico (contendo 25% de água, 0,994 g) foi posteriormente adicionado, e a mistura foi agitada por 10 min. A mistura da reação foi diluída com diclorometano (100 ml), lavada sucessivamente com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi purificado por cromatografia de sílica-gel para dar o produto-fim (1,71 g) como um óleo incolor.
[000233] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,30 - 1,38 (6H, m), 1,42 - 1,50 (2H,m), 1,50 (9H, s), 1,51 (9H, s), 1,79 (2H, quint, J = 7,0 Hz), 1,98 (3H, s), 2,02 - 2,10 (1H, m), 2,07 (3H, s), 2,32 - 2,40 (1H, m), 2,50 - 2,65 (2H, m), 4,00 (2H, t, J = 6,3 Hz), 4,09 (2H, d, J = 8,4 Hz), 4,37 (1H, d, J = 11,1 Hz), 4,47 (1H, t, J = 11,1 Hz), 6,67 (1H, brs), 6,88 (1H, d, J = 8,6 Hz), 7,28 (1H, dd, J = 1,5, 8,6 Hz), 7,35 (1H, d, J = 1,5 Hz).
[000234] O composto 3-3 (1,47 g) foi separado por HPLC usando CHIRALPAK (marca comercial registrada) AD-H (hexano/etanol/ diisopropilamina) para dar ambos os enantiômeros como óleo incolor. O primeiro pico com curto tempo de retenção foi a forma S (0,55 g, composto 3-4-1), e o segundo pico com longo tempo de retenção foi a forma R (0,65 g, composto 3-4-2).
[000235] O composto 3-4-1 (0,55 g) foi dissolvido em etanol (15 ml) e ácido clorídrico (3 ml), e a mistura foi agitada em 50°C por 3 horas. A mistura da reação foi vertida em água (150 ml), e a mistura foi deixada por 7 horas. O sólido precipitado foi coletado por filtração, lavado com água e secado para dar o produto-fim (0,33 g) como um pó branco.
[000236] MS (ESI) m/z: 458 [M+H]
[000237] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,45 - 1,53 (2H, m), 1,74 - 1,81 (2H, m), 1,89 - 1,99 (2H, m), 2,60 - 2,75 (2H, m), 3,70 (2H, brs), 3,94 - 4,02 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,4 Hz), 7,42 - 7,46 (2H, m).
[000238] O composto 3-4-2 (0,65 g) foi dissolvido em etanol (15 ml) e ácido clorídrico (3 ml), e a mistura foi agitada em 50°C por 2,5 horas. A mistura da reação foi vertida em água (150 ml), e a mistura foi deixada por 4 horas. O sólido precipitado foi coletado por filtração, lavado com água e secado para dar o produto-fim (0,35 g) como um pó branco.
[000239] MS (ESI) m/z: 458 [M+H]
[000240] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,45 - 1,53 (2H, m), 1,74 - 1,81 (2H, m), 1,90 - 2,01 (2H, m), 2,61 - 2,74 (2H, m), 3,69 (1H, d, J = 12,0 Hz), 3,70 (1H, d, J = 12,0 Hz), 3,93 - 4,02 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,4 Hz), 7,42 - 7,46 (2H, m).
[000241] Cloridrato de 2-amino-2-[2-(3-ciano-4-heptiloxifenil)etil] propano-1,3-diol
[000242] Exemplo de Referência composto 3-1 (836 mg), éster t-butílico de ácido (2,2-dimetil-5-etinil-1,3-dioxan-5-il)carbâmico (482 mg) sintetizado por um método conhecido (por exemplo, Tetrahedron vol. 57 (2001) 6531-6538), 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila (54 mg), dicloreto de bis(acetonitrila)paládio(II) (10 mg), carbonato de césio (919 mg) foram agitados em um solvente misto de acetonitrila (15 ml) e tetraidrofurano (2 ml) a 70°C por 4 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 4:1) para dar éster t-butílico de ácido [2,2-dimetil-5-(3-ciano-4- heptiloxifenil)etinil-1,3-dioxan-5-il]carbâmico como um óleo marrom (493 mg). O intermediário foi dissolvido em acetato de etila (5 ml), catalisador de Lindlar (80 mg) foi adicionado, e a mistura foi agitada de um dia para o outro sob uma atmosfera de hidrogênio. A mistura da reação foi filtrada e concentrada e o resíduo foi dissolvido em etanol (4 ml), 10% de paládio carbono (envenenamento de diamina de etileno, 40 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 2,5 horas sob uma atmosfera de hidrogênio. A solução foi filtrada, e o filtrado foi concentrado para dar o produto-fim (182 mg).
[000243] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 7,2 Hz), 1,26 - 1,31 (6H, m), 1,35 - 1,37 (2H, m), 1,43 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,78 - 1,86 (2H, m), 1,91 - 1,96 (2H, m), 2,49 - 2,53 (2H, m), 3,68 (2H, d, J = 11,6 Hz), 3,87 (2H, d, J = 11,6 Hz), 4,02 (2H, t, J = 6,4 Hz), 4,99 (1H, brs), 6,85 (1H, d, J = 8,4 Hz), 7,32 (1H, dd, J = 8,4, 1,6 Hz), 7,33 (1H, m).
[000244] O composto 5-1 (255 mg) foi dissolvido em etanol (2 ml), ácido p-toluenossulfônico (19 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 4 horas. À mistura da reação foi adicionado bicarbonato de sódio saturado aquoso, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi purificado por cromatografia de coluna de sílicagel (hexano: acetato de etila = 4:1) para dar um óleo. Ao óleo foi adicionado dioxano contendo cloreto de hidrogênio (4 mols/l), e a mistura foi agitada em temperatura ambiente por 3 horas. O precipitado foi coletado por filtração e secado para dar o produto-fim (45 mg) como um pó branco.
[000245] MS (ESI) m/z: 335 [M+H]
[000246] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,26 - 1,37 (6H, m), 1,38 - 1,44 (2H, m), 1,69 - 1,77 (4H, m), 2,55 - 2,59 (2H, m), 3,50 (4H, d, J = 4,4 Hz), 4,09 (2H, t, J = 6,4 Hz), 5,38 (2H, t, J = 4,4 Hz), 7,18 (1H, d, J = 8,8 Hz), 7,49 (1H, dd, J = 8,8, 2,0 Hz), 7,55 (1H, d, J = 2,0 Hz), 7,82 (3H, brs).
[000247] 2-Amino-4-(3-ciano-4-heptiloxifenil)-2-(fosforiloximetil) butanol
[000248] O composto 5-1 (340 mg) foi dissolvido em etanol (3 ml), monoidrato de ácido p-toluenossulfônico (0,025 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 6 horas. Água foi adicionada à mistura da reação, e a mistura foi neutralizada com bicarbonato de sódio saturado aquoso, extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado para dar o composto desprotegido por acetonida (300 mg) como um óleo incolor. O óleo incolor (205 mg) foi tomado e dissolvido em um solvente misto de diclorometano (2 ml) e tolueno (2 ml), perfluorohexano (2 ml), óxido de prata (219 mg) e éster tetrabenzílico de ácido pirofosfórico (508 mg) foram adicionados, e a mistura foi agitada em temperatura ambiente. 5 min depois, iodeto de tetra-n-hexil amônio (454 mg) foi adicionado, e a mistura foi adicionalmente agitada por 5 horas. O material insolúvel foi filtrado, e o solvente foi evaporado. O resíduo foi purificado por cromatografia de sílica-gel e HPLC do preparativo para dar o produto-fim (81,0 mg) como um óleo incolor.
[000249] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,4 Hz), 1,31 - 1,36 (6H, m), 1,43 (9H, s), 1,64 - 1,71 (2H, m), 1,79 - 1,86 (2H, m), 1,99 - 2,06 (2H, m), 2,39 - 2,49 (2H, m), 3,50 - 3,55 (2H, m), 4,02 (2H, t, J = 6,4 Hz), 4,10 (1H, d, J = 7,2 Hz), 4,13 (1H, d, J = 7,2 Hz), 5,00 (1H, s), 5,03 - 5,09 (4H, m), 6,82 (1H, d, J = 8,4 Hz), 7,26 - 7,27 (2H, m), 7,32 - 7,34 (10H, m).
[000250] O composto 6-1 (81,0 mg) foi dissolvido em acetonitrila (2 ml), iodeto de sódio (140 mg) e clorotrimetilsilano (0,12 ml) foram adicionados, e a mistura foi agitada em temperatura ambiente por 4,5 horas. Água e acetato de etila foram adicionados, e a mistura foi ultrassonicada. O sólido resultante foi coletado por filtração. O sólido foi lavado com água e acetato de etila, e secado para dar o produto-fim (35,0 mg) como um pó amarelo-pálido.
[000251] MS (ESI) m/z: 415 [M+H]
[000252] 1H-RMN (CD3OD) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,32 - 1,39 (6H, m), 1,47 - 1,53 (2H, m), 1,79 - 1,83 (2H, m), 1,89 - 1,95 (2H, m), 2,63 - 2,67 (2H, m), 3,68 (2H, d, J = 2,0 Hz), 3,96 (2H, t, J = 6,4 Hz), 4,10 (2H, t, J = 6,4 Hz), 7,08 (1H, d, J = 9,2 Hz), 7,49 - 7,51 (2H, m).
[000253] Cloridrato de 2-amino-2-[2-(3-ciano-4-octiloxifenil)etil] propano-1,3-diol
[000254] Octanol (0,834 g) foi dissolvido em N,N-dimetilformamida (10 ml), e hidreto de sódio (60%, 0,256 g) foi adicionado. Depois de agitar por 30 min, 5-bromo-2-fluorobenzonitrila (0,640 g) foi adicionado, e a mistura foi adicionalmente agitada em 40 a 50°C por 1 hora. A mistura da reação foi vertida em água, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (1,042 g) como um óleo incolor.
[000255] 1H-RMN (CDCl3) δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,24 - 1,37 (8H, m), 1,44 - 1,51 (2H, m), 1,80 - 1,87 (2H, m), 4,04 (2H, t, J = 6,4 Hz), 6,83 (1H, d, J = 8,8 Hz), 7,59 (1H, dd, J = 8,8, 2,4 Hz), 7,64 (1H, d, J = 2,4 Hz).
[000256] O composto 7-1 (0,636 g), éster t-butílico de ácido (2,2- dimetil-5-etinil-1,3-dioxan-5-il)carbâmico (0,571 g), 2-diciclohexilfosfino2',4',6'-triisopropilbifenila (0,045 g), dicloreto de bis(acetonitrila) paládio(II) (0,008 g) e carbonato de césio (0,668 g) foram agitados em acetonitrila (10 ml) em 70 a 80C por 2 horas. Foi adicionada água à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel para dar éster t-butílico de ácido [2,2-dimetil-5-(3-ciano-4-octiloxifeniletinil)-1,3-dioxan-5- il]carbâmico como um óleo marrom. O óleo foi dissolvido em acetato de etila (6 ml), 10% de paládio carbono (contendo cerca de 50% de água, 0,080 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 3,5 horas sob uma atmosfera de hidrogênio. A solução foi filtrada, e o filtrado foi concentrado para dar o produto-fim (0,610 g) como um pó amarelo-pálido.
[000257] 1H-RMN (CDCl3) δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,27 - 1,34 (8H, m), 1,41 - 1,43 (2H, m), 1,42 (3H, s), 1,43 (3H, s), 1,46 (9H, s), 1,78 - 1,85 (2H, m), 1,92 - 1,96 (2H, m), 2,49 - 2,53 (2H, m), 3,67 (2H, d, J = 11,6 Hz), 3,86 (2H, d, J = 11,6 Hz), 4,02 (2H, t, J = 6,4 Hz), 4,97 (1H, s), 6,84 (1H, d, J = 8,8 Hz), 7,33 (1H, dd, J = 8,8, 2,0 Hz), 7,34 (1H, d, J = 2,0 Hz).
[000258] O composto 7-2 (0,610 g) foi dissolvido em um solvente misto de etanol (5 ml) e tetraidrofurano (2 ml), monoidrato de ácido ptoluenossulfônico (0,043 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 3,5 horas, e adicionalmente em 50 a 60°C por 2,5 horas. À mistura da reação foi adicionado bicarbonato de sódio saturado aquoso, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel para dar o composto desprotegido por acetal do Exemplo (6-2) como um óleo. Ao óleo obtido foi adicionado dioxano contendo cloreto de hidrogênio (4 mols/l, 2 ml), e a mistura foi agitada em temperatura ambiente por 8 horas. O precipitado foi coletado por filtração e secado para dar o produto-fim (145 mg) como um pó branco.
[000259] MS (ESI) m/z: 349 [M+H]
[000260] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,26 - 1,35 (8H, m), 1,40 - 1,46 (2H, m), 1,69 - 1,76 (4H, m), 2,54 - 2,58 (2H, m), 3,49 (4H, d, J = 4,4 Hz), 4,09 (2H, t, J = 6,4 Hz), 5,39 (2H, brs), 7,18 (1H, d, J = 8,8 Hz), 7,49 (1H, d, J = 8,8 Hz), 7,54 (1H, s), 7,63 (3H, brs).
[000261] 2-Amino-4-(3-ciano-4-octiloxifenil)-2-(fosforiloximetil) butanol
[000262] O composto 7-2 (208 mg) foi dissolvido em etanol (2 ml), monoidrato de ácido p-toluenossulfônico (73,0 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 6,5 horas. Água foi adicionada à mistura da reação, e a mistura foi neutralizada com bicarbonato de sódio saturado aquoso, extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado e o resíduo obtido foi dissolvido em um solvente misto de diclorometano (2 ml) e tolueno (2 ml), perfluorohexano (2 ml), óxido de prata (197 mg) e éster tetrabenzílico de ácido pirofosfórico (459 mg) foram adicionados, e a mistura foi agitada em temperatura ambiente. 5 min depois, iodeto de tetra-n-hexil amônio (410 mg) foi adicionado, e a mistura foi adicionalmente agitada por 17 horas. O material insolúvel foi filtrado e o solvente foi evaporado e purificado por cromatografia de sílica-gel e HPLC do preparativo para dar o produto-fim (106 mg) como um óleo incolor.
[000263] 1H-RMN (CDCl3) δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,27 - 1,34 (6H, m), 1,43 (9H, s), 1,46 - 1,51 (2H, m), 1,78 - 1,86 (2H, m), 1,79 - 1,86 (2H, m), 2,00 - 2,06 (2H, m), 2,39 - 2,51 (2H, m), 3,47 - 3,56 (2H, m), 3,90 - 3,92 (1H, m), 3,97 - 4,06 (4H, m), 4,79 (1H, s), 5,03 - 5,07 (4H, m), 6,84 (1H, d, J = 8,0 Hz), 7,26 (1H, d, J = 8,0 Hz), 7,34 - 7,35 (11H, m).
[000264] O composto 8-1 (104 mg) foi dissolvido em acetonitrila (2 ml), iodeto de sódio (110 mg) e clorotrimetilsilano (80,0 mg) foram adicionados, e a mistura foi agitada em temperatura ambiente por 3 horas. Água e acetato de etila foram adicionados, e a mistura foi ultrassonicada. O sólido resultante foi coletado por filtração. O sólido foi lavado com água e acetato de etila, adicionalmente lavado com metanol, e secado para dar o produto-fim (26,0 mg) como um pó branco.
[000265] MS (ESI) m/z: 429 [M+H]
[000266] 1H-RMN (CD3OD) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,31 - 1,35 (8H, m), 1,50 - 1,52 (2H, m), 1,79 - 1,83 (2H, m), 1,89 - 1,92 (2H, m), 2,63 - 2,67 (2H, m), 3,63 - 3,67 (2H, m), 3,95 - 3,97 (2H, m), 4,09 (2H, t, J = 6,4 Hz), 7,07 (1H, d, J = 9,2 Hz), 7,49 - 7,50 (2H, m).
[000267] Cloridrato de 2-amino-2-[2-(4-octilóxi-3-trifluorometilfenil) etil]propano-1,3-diol
[000268] A uma solução de 4'-flúor-3'-trifluorometilacetofenona (25,0 g) em N,N-dimetilformamida (70 ml) foi adicionado metóxido de sódio (7,21 g) sob esfriamento com gelo, e a mistura foi agitada por 2 horas sob esfriamento com gelo e adicionalmente em temperatura ambiente por 1 hora. A mistura da reação foi adicionada a água, e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (24,3 g) como um sólido marrom.
[000269] 1H-RMN (CDCl3)δ (ppm): 2,59 (3H, s), 3,99 (3H, s), 7,06 (1H, d, J = 8,7 Hz), 8,14 (1H, dd, J = 2,1, 8,7 Hz), 8,19 (1H, d, J = 2,1 Hz).
[000270] A uma solução do composto 9-1 (24,3 g) em ácido acético (120 ml) foi adicionado tribrometo de piridínio (90%, 39,6 g) e a mistura foi agitada em 50°C por 1 hora. A mistura da reação foi adicionada a água gelada, extraída com acetato de etila, e a camada orgânica foi lavada sucessivamente com água, 1 M de solução aquosa de hidróxido de sódio, cloreto de amônio saturado e salmoura saturada. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida para dar o produto-fim (34,2 g) como um sólido marrom.
[000271] 1H-RMN (CDCl3) δ (ppm): 4,01 (3H, s), 4,39 (2H, s), 7,09 (1H, d, J = 8,7 Hz), 8,18 (1H, dd, J = 2,2, 8,7 Hz), 8,23 (1H, d, J = 1,9 Hz).
[000272] A uma solução de dietil 2-acetamidomalonato (20,1 g) em N,N-dimetilformamida (100 ml) foi adicionado hidreto de sódio (60%, 4,07 g) em duas porções sob esfriamento com gelo, e a mistura foi agitada por 30 min. À solução foi adicionada uma solução do composto 9-2 (33,0 g) em N,N-dimetilformamida (50 ml) e a mistura foi agitada por 2 horas sob esfriamento com gelo. A mistura da reação foi adicionada a água gelada, extraída com acetato de etila, e a camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produtofim (31,8 g) como um óleo marrom.
[000273] 1H-RMN (CDCl3) δ (ppm): 1,25 (6H, t, J = 7,1 Hz), 1,97 (3H, s), 3,98 (3H, s), 4,22 (2H, s), 4,27 (4H, dq, J = 2,4, 7,1 Hz), 7,05 (1H, d, J = 8,7 Hz), 7,09 (1H, brs), 8,13 (1H, dd, J = 2,2, 8,7 Hz), 8,20 (1H, d, J = 2,0 Hz).
[000274] A uma solução do composto 9-3 (31,5 g) em ácido trifluoroacético (230 ml) foi adicionado trietilsilano (116 ml), e a mistura foi agitada em 70°C por 13 horas. A mistura da reação foi concentrada sob pressão reduzida, água foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa de hidróxido de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar uma mistura do composto do título e o material de partida como um óleo amarelo. A uma solução do óleo em ácido trifluoroacético (230 ml) foi adicionado trietilsilano (116 ml), e a mistura foi agitada em 70°C por 12 horas. A mistura da reação foi concentrada sob pressão reduzida, água foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa de hidróxido de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. Éter dietílico foi adicionado ao resíduo obtido e o sólido precipitado foi coletado por filtração, e secado para dar o produtofim (7,91 g) como um pó branco. O licor-mãe foi concentrado sob pressão reduzida, e o resíduo foi purificado por cromatografia de sílicagel para dar o produto-fim (4,29 g). O rendimento total foi 12,2 g.
[000275] 1H-RMN (CDCl3) δ (ppm): 1,25 (6H, t, J = 7,2 Hz), 2,02 (3H, s), 2,44 - 2,48 (2H, m), 2,62 - 2,68 (2H, m), 3,87 (3H, s), 4,15 - 4,27 (4H, m), 6,78 (1H, brs), 6,90 (1H, d, J = 8,4 Hz), 7,27 (1H, dd, J = 2,0, 8,4 Hz), 7,32 (1H, d, J = 2,0 Hz).
[000276] A uma solução do composto 9-4 (12,2 g) em etanol (200 ml) e água (40 ml) foi adicionado cloreto de cálcio (6,46 g) e a mistura foi dissolvida. Boridreto de sódio (4,40 g) foi adicionado à mistura em duas porções sob esfriamento com gelo, e a mistura foi agitada por 3 horas sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 20 horas. Ácido clorídrico a 1 M (300 ml) foi adicionado à mistura da reação sob esfriamento com gelo, e a mistura foi concentrada sob pressão reduzida, e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (9,88 g) como uma espuma branca.
[000277] 1H-RMN (CDCl3) δ (ppm): 1,92 - 1,96 (2H, m), 2,02 (3H, s), 2,60 - 2,64 (2H, m), 3,57 (2H, brs), 3,64 (2H, brd, J = 11,6 Hz), 3,85 (2H, brd, J = 11,6 Hz), 3,87 (3H, s), 5,94 (1H, brs), 6,92 (1H, d, J = 8,5 Hz), 7,32 (1H, dd, J = 1,9, 8,5 Hz), 7,37 (1H, d, J = 1,9 Hz).
[000278] A uma solução do composto 9-5 (9,70 g) em cloreto de metileno (90 ml) foi adicionada gota a gota em -70°C uma solução a 1 M (116 ml) de tribrometo de boro em cloreto de metileno. A mistura foi aquecida até 0°C durante 1 hora com agitação, e adicionalmente agitada por 2 horas sob esfriamento com gelo. Metanol (200 ml) foi gradualmente adicionado à mistura da reação sob esfriamento com gelo, e a mistura foi concentrada sob pressão reduzida. Ácido clorídrico concentrado (50 ml) foi adicionado a uma solução do resíduo obtido em etanol (50 ml), e a mistura foi agitada em 70°C por 1 hora. A mistura da reação foi concentrada sob pressão reduzida. A uma solução do resíduo obtido e N,N-diisopropiletilamina (12,6 ml) em metanol (80 ml) foi adicionado dicarbonato de di-t-butila (6,94 g) sob esfriamento com gelo, e a mistura foi agitada por 2 horas sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 4 horas. Solução aquosa saturada de bicarbonato de sódio (500 ml) foi adicionada e a mistura foi concentrada sob pressão reduzida, e extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (2,15 g) como um óleo incolor.
[000279] 1H-RMN (CDCl3) δ (ppm): 1,46 (9H, s), 1,84 - 1,89 (2H, m), 2,57 - 2,61 (2H, m), 3,26 (2H, brs), 3,66 (2H, dd, J = 5,9, 11,4 Hz), 3,87 (2H, dd, J = 5,2, 11,4 Hz), 5,04 (1H, brs), 5,58 (1H, brs), 6,87 (1H, d, J = 8,4 Hz), 7,23 (1H, dd, J = 1,8, 8,4 Hz), 7,30 (1H, d, J = 1,8 Hz).
[000280] O composto 9-6 (360 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (263 mg) e 1-bromooctano (0,198 ml) foram adicionados, e a mistura foi agitada em 80°C por 6 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (490 mg) como um óleo incolor.
[000281] 1H-RMN (CDCl3)δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,23 - 1,40 (8H, m), 1,41 - 1,50 (2H, m), 1,47 (9H, s), 1,75 - 1,82 (2H, m), 1,83 - 1,90 (2H, m), 2,57 - 2,62 (2H, m), 3,28 (2H, brs), 3,63 - 3,67 (2H, m), 3,85 - 3,90 (2H, m), 4,00 (2H, t, J = 6,4 Hz), 5,02 (1H, brs), 6,89 (1H, d, J = 8,5 Hz), 7,27 (1H, dd, J = 8,5, 1,9 Hz), 7,36 (1H, d, J = 1,9 Hz).
[000282] O composto 9-7 (490 mg) foi dissolvido em cloreto de metileno (5 ml), dioxano contendo cloreto de hidrogênio (4 mols/l, 5 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 12 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (350 mg) como um pó branco.
[000283] MS (ESI) m/z: 392 [M+H]
[000284] 1H-RMN (CD3OD) δ (ppm): 0,90 (3H, t, J = 6,8 Hz), 1,24 - 1,41 (8H, m), 1,47 - 1,53 (2H, m), 1,75 - 1,81 (2H, m), 1,91 - 1,97 (2H, m), 2,63 - 2,70 (2H, m), 3,69 (4H, s), 4,05 (2H, t, J = 6,2 Hz), 7,03 (1H, d, J = 8,4 Hz), 7,41 (1H, d, J = 8,4 Hz), 7,44 (1H, brs).
[000285] 2-Amino-4-(4-octilóxi-3-trifluorometilfenil)-2- (fosforiloximetil)butanol
[000286] A uma solução do composto 9-8 (270 mg) em N,N-dimetilformamida (7 ml) foram adicionados N,N-diisopropiletilamina (0,340 ml) e ortoacetato de trimetila (0,121 ml), e a mistura foi agitada em 120°C por 5,5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 280 mg de um óleo marrom. A uma solução do óleo marrom (280 mg) em cloreto de metileno (5 ml) e acetonitrila (2 ml) foram adicionados 1H-tetrazol (88 mg) e dietilfosforamidita de di-t-butila (0,377 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, ácido m-cloroperbenzóico (produto contendo 25% de água, 335 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 30 min. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:3 - acetato de etila somente) para dar o produto-fim (190 mg) como um óleo marrom.
[000287] 1H-RMN (CDCl3) δ (ppm): 0,90 (3H, t, J = 6,9 Hz), 1,28 - 1,40 (8H, m), 1,47 - 1,52 (2H, m), 1,48 (9H, s), 1,49 (9H, s), 1,70 - 1,90 (4H, m), 2,01 (3H, s), 2,51 - 2,71 (2H, m), 3,89 - 3,92 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 4,17 (1H, d, J = 9,0 Hz), 4,32 (1H, d, J = 9,0 Hz), 7,05 (1H, d, J = 8,4 Hz), 7,36 - 7,41 (2H, m).
[000288] O composto 10-1 (190 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 3 horas. O solvente foi concentrado sob pressão reduzida, metanol (5 ml), éter dietílico (5 ml) e óxido de propileno (5 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração, lavado com acetato de etila e éter dietílico para dar o produto-fim (137 mg) como um sólido branco.
[000289] MS (ESI) m/z: 472 [M+H]
[000290] 1H-RMN (CD3OD) δ (ppm): 0,90 (3H, t, J = 6,4 Hz), 1,25 - 1,40 (8H, m), 1,45 - 1,53 (2H, m), 1,76 - 1,83 (2H, m), 1,93 - 2,00 (2H, m), 2,63 - 2,74 (2H, m), 3,70 (2H, brs), 3,96 - 4,00 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,3 Hz), 7,42 - 7,46 (2H, m).
[000291] Cloridrato de 2-amino-2-[2-(4-hexilóxi-3-trifluorometilfenil) etil]propano-1,3-diol
[000292] O composto 1-1 (500 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (494 mg) e 1-bromohexano (0,201 ml) foram adicionados, e a mistura foi agitada em 80°C por 2 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (620 mg) como um óleo incolor.
[000293] 1H-RMN (CDCl3) δ (ppm): 0,90 (3H, t, J = 6,9 Hz), 1,30 - 1,36 (4H, m), 1,41 - 1,50 (2H, m), 1,43 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,76 - 1,81 (2H, m), 1,91 - 1,99 (2H, m), 2,51 - 2,56 (2H, m), 3,69 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 4,00 (2H, t, J = 6,4 Hz), 4,98 (1H, brs), 6,88 (1H, d, J = 8,5 Hz), 7,26 - 7,28 (1H, m), 7,35 (1H, d, J = 1,6 Hz).
[000294] O composto 11-1 (620 mg) foi dissolvido em etanol (15 ml), ácido clorídrico concentrado (2,5 ml) foi adicionado, e a mistura foi agitada em 80°C por 3 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (465 mg) como um pó branco.
[000295] MS (ESI) m/z: 364 [M+H]
[000296] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,9 Hz), 1,32 - 1,40 (4H, m), 1,47 - 1,53 (2H, m), 1,73 - 1,81 (2H, m), 1,90 - 1,96 (2H, m), 2,62 - 2,68 (2H, m), 3,68 (4H, d, J = 5,1 Hz), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,4 Hz), 7,41 (1H, dd, J = 8,4, 1,9 Hz), 7,45 (1H, d, J = 1,9 Hz).
[000297] 2-Amino-4-(4-hexilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)butanol
[000298] A uma solução do composto 11-2 (380 mg) em N,N-dimetilformamida (10 ml) foram adicionados N,N-diisopropiletilamina (0,512 ml) e ortoacetato de trimetila (0,180 ml), e a mistura foi agitada em 120°C por 12 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 380 mg de um óleo marrom. 1H-Tetrazol (133 mg) e dietilfosforamidita de di-t-butila (0,569 ml) foram adicionados a uma solução do óleo marrom (380 mg) em cloreto de metileno (5 ml) e acetonitrila (2 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, ácido m-cloroperbenzóico (produto contendo 25% de água, 504 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 1 hora. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:4 - acetato de etila somente) para dar o produto-fim (220 mg) como um óleo amarelo.
[000299] 1H-RMN (CDCl3) δ (ppm): 0,93 (3H, t, J = 6,9 Hz), 1,30 - 1,40 (4H, m), 1,47 - 1,52 (2H, m), 1,48 (9H, s), 1,49 (9H, s), 1,74 - 1,88 (4H, m), 2,01 (3H, s), 2,51 - 2,70 (2H, m), 3,87 - 3,92 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 4,18 (1H, d, J = 8,9 Hz), 4,32 (1H, d, J = 8,9 Hz), 7,05 (1H, d, J = 8,4 Hz), 7,37 - 7,41 (2H, m).
[000300] O composto 12-1 (220 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 3 horas. O solvente foi concentrado sob pressão reduzida, e metanol (5 ml), éter dietílico (5 ml) e óxido de propileno (5 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração, e lavado com acetato de etila e éter dietílico para dar o produto-fim (118 mg) como um sólido branco.
[000301] MS (ESI) m/z: 444 [M+H]
[000302] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,9 Hz), 1,31 - 1,40 (4H, m), 1,45 - 1,54 (2H, m), 1,74 - 1,82 (2H, m), 1,92 - 1,98 (2H, m), 2,60 - 2,75 (2H, m), 3,70 (2H, brs), 3,93 - 3,99 (2H, m), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,2 Hz), 7,42 - 7,46 (2H, m).
[000303] Cloridrato de 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutanol
[000304] Cloridrato de 2-amino-2-metil-1,3-propanodiol (14,0 g) foi dissolvido em metanol (200 ml), N,N-diisopropiletilamina (46.3 ml) e dicarbonato de di-t-butila (43,7 g) foram adicionados sob esfriamento com gelo, e a mistura foi agitada por 40 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 27 horas. Solução aquosa a 1 M de hidróxido de sódio (100 ml) foi adicionada à mistura da reação sob esfriamento com gelo, e a mistura foi agitada por 40 min. Metanol foi evaporado sob pressão reduzida. Água foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (25,3 g) como um pó branco.
[000305] 1H-RMN (CDCl3) δ (ppm): 1,17 (3H, s), 1,44 (9H, s), 3,45 (2H, brs), 3,62 (2H, dd, J = 7,1, 11,3 Hz), 3,78 (2H, dd, J = 5,4, 11,3 Hz), 4,96 (1H, brs).
[000306] A uma solução do composto 13-1 (25,3 g) em cloreto de metileno (300 ml) foram adicionados sob esfriamento com gelo N,N-diisopropiletilamina (26,8 ml) e cloreto de metoximetila (11,6 ml), e a mistura foi agitada por 20 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 22 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (14,2 g) como um óleo incolor.
[000307] 1H-RMN (CDCl3) δ (ppm): 1,26 (3H, s), 1,44 (9H, s), 3,38 (3H, s), 3,57 (1H, d, J = 9,7 Hz), 3,61 (1H, dd, J = 7,8, 11,5 Hz), 3,66 (1H, d, J = 9,7 Hz), 3,71 (1H, dd, J = 5,0, 11,5 Hz), 3,91 (1H, brs), 4,64 (2H, s), 5,10 (1H, brs).
[000308] A uma solução mista do composto 13-2 (14,2 g) e brometo de sódio (5,86 g) em tolueno (100 ml), acetato de etila (100 ml) e água (20 ml) foram adicionados 2,2,6,6-tetrametilpiperidina 1-oxila, radical livre (178 mg) sob esfriamento com gelo. Em seguida, 10% de solução aquosa de hipoclorito de sódio (46,7 g) e uma solução de bicarbonato de sódio (13,8 g) em água (150 ml) foram adicionados gota a gota durante 1,5 hora. A solução foi posteriormente agitada por 1,5 hora sob esfriamento com gelo. A camada orgânica foi separada, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (13,1 g) como um óleo marrom-pálido.
[000309] 1H-RMN (CDCl3) δ (ppm): 1,39 (3H, s), 1,45 (9H, s), 3,34 (3H, s), 3,75 (2H, s), 4,60 (2H, s), 5,39 (1H, brs), 9,51 (1H, s).
[000310] O Exemplo de Referência composto 2-5 (21,8 g) foi suspendido em tetraidrofurano (200 ml), t-butóxido de potássio (4,35 g) foi adicionado sob esfriamento com gelo, e a mistura foi agitada por 1 hora. Uma solução do composto 13-3 (4,80 g) em tetraidrofurano (40 ml) foi adicionada à solução mista, e a mistura foi agitada sob esfriamento com gelo por 1,5 hora e em temperatura ambiente por 1 hora. A mistura da reação foi vertida em água, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar éster t-butílico de ácido [3-(4-benzilóxi-3-trifluorometilfenil)-1-(metoximetóxi)metil-1-metil] alilcarbâmico (8,45 g) como um óleo incolor. A proporção de isômeros geométricos do composto obtido foi E:Z=3:7. 10% de Paládio carbono (contendo cerca de 50% de água, 845 mg) foi adicionado a uma solução deste óleo em 1,4-dioxano (150 ml), e a mistura foi agitada em temperatura ambiente por 24 horas sob uma atmosfera de hidrogênio. A mistura da reação foi filtrada através de celite e concentrada para dar o produto-fim (6,92 g) como um óleo incolor.
[000311] 1H-RMN (CDCl3) δ (ppm): 1,34 (3H, s), 1,45 (9H, s), 1,88 - 1,95 (1H, m), 2,00 - 2,08 (1H, m), 2,52 - 2,60 (2H, m), 3,38 (3H, s), 3,47 (1H, d, J = 9,5 Hz), 3,65 (1H, d, J = 9,5 Hz), 4,65 (2H, s), 4,78 (1H, brs), 5,98 (1H, brs), 6,85 (1H, d, J = 8,4 Hz), 7,18 (1H, dd, J = 1,5, 8,4 Hz), 7,29 (1H, d, J = 1,5 Hz).
[000312] A uma solução do composto 13-4 (1,5 g) em N,N-dimetilformamida (15 ml) foram adicionados carbonato de potássio (1,53 g) e brometo de n-heptila (0,63 ml), e a mistura foi agitada em 50°C por 6 horas. A mistura da reação foi adicionada a água, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (1,69 g) como um óleo incolor.
[000313] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,29 - 1,49 (6H, m), 1,34 (3H, s), 1,42 - 1,50 (2H, m), 1,45 (9H, s), 1,79 (2H, quint, J = 6,9 Hz), 1,84 - 1,95 (1H, m), 2,00 - 2,08 (1H, m), 2,54 - 2,61 (2H, m), 3,38 (3H, s), 3,49 (1H, d, J = 9,5 Hz), 3,64 (1H, d, J = 9,5 Hz), 4,00 (2H, t, J = 6,5 Hz), 4,64 (2H, s), 4,72 (1H, brs), 6,88 (1H, d, J = 8,5 Hz), 7,27 (1H, dd, J = 1,9, 8,5 Hz), 7,35 (1H, d, J = 1,9 Hz).
[000314] A uma solução do composto 13-5 (1,69 g) em etanol (15 ml) foi adicionado ácido clorídrico concentrado (3 ml), e a mistura foi agitada em 50°C por 3 horas. Solução aquosa a 1 M de hidróxido de sódio (50 ml) e salmoura (50 ml) foram adicionados à mistura da reação e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi dissolvido em acetato de etila (50 ml), solução a 1 M de éter de ácido clorídrico (5 ml) foi adicionada, e o solvente foi evaporado. Éter foi adicionado ao resíduo e o sólido precipitado foi filtrado e secado sob pressão reduzida para dar o produto-fim (607 mg) como um pó branco.
[000315] MS (ESI) m/z: 362 [M+H]
[000316] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,4 Hz), 1,20 (3H, s), 1,26 - 1,35 (6H, m), 1,37 - 1,43 (2H, m), 1,67 - 1,83 (4H, m), 2,61 (2H, t, J = 8,7 Hz), 3,39 (1H, dd, J = 4,6, 11,2 Hz), 3,46 (1H, dd, J = 4,6, 11,2 Hz), 4,05 (2H, t, J = 6,1 Hz), 5,52 (1H, t, J = 4,9 Hz), 7,18 (1H, d, J = 8,5 Hz), 7,43 - 7,45 (2H, m), 7,89 (3H, brs).
[000317] Éster de mono[2-amino-4-(4-heptilóxi-3-trifluorometilfe-nil)- 2-metilbutílico] do ácido fosfórico
[000318] A uma solução do composto 13-6 (841 mg) em metanol (25 ml) foram adicionados N,N-diisopropiletilamina (1,10 ml) e dicarbonato de di-t-butila (692 mg), e a mistura foi agitada em temperatura ambiente por 24 horas. A mistura da reação foi concentrada sob pressão reduzida. Bicarbonato de sódio saturado foi adicionado, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (880 mg) como um óleo incolor.
[000319] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,22 (3H, s), 1,28 - 1,39 (6H, m), 1,44 - 1,49(11H, m), 1,79 (2H, quint, J = 6,9 Hz), 1,83 - 1,90 (1H, m), 1,99 - 2,07 (1H, m), 2,50 - 2,58 (1H, m), 2,61 - 2,68 (1H, m), 3,63 - 3,72 (2H, m), 4,00 (2H, t, J = 6,5 Hz), 4,06 (1H, brs), 4,63 (1H, brs), 6,89 (1H, d, J = 8,5 Hz), 7,28 (1H, dd, J = 1,7, 8,5 Hz), 7,36 (1H, d, J = 1,7 Hz).
[000320] A uma solução do composto 14-1 (870 mg) em cloreto de metileno (15 ml) foi adicionada uma solução de 1H-tetrazol (158 mg) em acetonitrila (15 ml). Di-t-butil diisopropilfosforamidita (0,713 ml) foi adicionado à mistura a 0°C, e a mistura foi agitada por 1,5 hora sob esfriamento com gelo. Uma solução de 1H-tetrazol (158 mg) em acetonitrila (15 ml) e di-t-butil diisopropilfosforamidita (0,713 ml) foram adicionados, e a mistura foi adicionalmente agitada por 2 horas sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 600 mg) foi adicionado, e a mistura foi agitada por 40 min sob esfriamento com gelo. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi lavada com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (1,26 g) como um óleo incolor.
[000321] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,28 - 1,35 (6H, m), 1,35 (3H, s), 1,43 (9H, s), 1,49(18H, s), 1,51 - 1,53 (2H, m), 1,79 (2H, quint, J = 6,9 Hz), 1,86 - 1,94 (1H, m), 2,03 - 2,11 (1H, m), 2,52 - 2,62 (2H, m), 3,86 (1H, dd, J = 5,6, 10,2 Hz), 4,00 (2H, t, J = 6,4 Hz), 4,04 (1H, dd, J = 5,5, 10,2 Hz), 4,85 (1H, brs), 6,88 (1H, d, J = 8,5 Hz), 7,27 (1H, dd, J = 1,8, 8,5 Hz), 7,35 (1H, d, J = 1,8 Hz).
[000322] O composto 14-2 (1,24 g) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 1 hora. A mistura da reação foi adicionada a água (30 ml), e o pó precipitado foi coletado por filtração e lavado com água e éter dietílico para dar o produto-fim (648 mg) como um sólido branco.
[000323] MS (ESI) m/z: 442 [M+H]
[000324] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,38 (3H, s), 1,46 - 1,53 (2H, m), 1,75 - 1,81 (2H, m), 1,83 - 1,90 (1H, m), 1,97 - 2,05 (1H, m), 2,60 - 2,68 (1H, m), 2,70 - 2,76 (1H, m), 3,85 (1H, dd, J = 5,4, 11,4 Hz), 3,94 (1H, dd, J = 5,9, 11,4 Hz), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,2 Hz), 7,42 - 7,44 (2H, m).
[000325] Cloridrato de (R)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)- 2-metilbutanol
[000326] Uma solução aquosa saturada de bicarbonato de sódio (100 ml) foi adicionado ao composto 13-6 (1,30 g) e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (1,16 g) como um sólido ceráceo branco.
[000327] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,7 Hz), 1,08 (3H, s), 1,23 - 1,35 (6H, m), 1,38 - 1,45 (2H, m), 1,58 - 1,74 (4H, m), 2,55 - 2,65 (2H, m), 3,28 (1H, d, J = 10,9 Hz), 3,32 (1H, d, J = 10,9 Hz), 4,05 (2H, t, J = 6,2 Hz), 5,09 (1H, brs), 5,52 (2H, brs), 7,16 (1H, d, J = 9,0 Hz), 7,41 - 7,44 (2H, m).
[000328] O composto 15-1 (2,63 g) foi separado por cromatografia de fluido supercrítico (SFC) usando um CHIRALPAK (marca comercial registrada) AD-H (dióxido de carbono/etanol/dietilamina) para dar ambos os enantiômeros como sólidos ceráceos brancos. O primeiro pico com curto tempo de retenção foi forma R (0,91 g, composto 15-2- 1) e o segundo pico com longo tempo de retenção foi forma S (0,95 g, composto 15-2-2).
[000329] O composto 15-2-1 (745 mg) foi suspendido em acetato de etila (5 ml), e uma solução a 4 M de cloreto de hidrogênio em acetato de etila (2 ml) foi adicionada. Hexano (10 ml) foi posteriormente adicionado e a mistura foi deixada por 1 hora. O sólido precipitado foi coletado por filtração para dar o produto-fim (753 mg) como um pó branco.
[000330] MS (ESI) m/z: 362 [M+H]
[000331] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,7 Hz), 1,19 (3H, s), 1,25 - 1,35 (6H, m), 1,37 - 1,45 (2H, m), 1,67 - 1,83 (4H, m), 2,60 (2H, t, J = 8,7 Hz), 3,41 - 3,49 (2H, m), 4,06 (2H, t, J = 6,2 Hz), 5,53 (1H, t, J = 5,1 Hz), 7,18 (1H, d, J = 8,4 Hz), 7,43 - 7,45 (2H, m), 7,78 (3H, brs).
[000332] (R)-mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutil] éster do ácido fosfórico
[000333] A uma solução do composto 15-2-1 (120 mg) em metanol (10 ml) foram adicionados N,N-diisopropiletilamina (0,117 ml) e dicarbonato de di-t-butila (109 mg), e a mistura foi agitada em temperatura ambiente por 24 horas. Bicarbonato de sódio saturado foi adicionada à mistura da reação, e metanol foi evaporado sob pressão reduzida. O resíduo obtido foi diluído com água e extraído com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (159 mg) como um óleo incolor.
[000334] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,22 (3H, s), 1,28 - 1,38 (6H, m), 1,42 - 1,50(11H, m), 1,79 (2H, quint, J = 6,9 Hz), 1,83 - 1,90 (1H, m), 1,99 - 2,07 (1H, m), 2,50 - 2,58 (1H, m), 2,61 - 2,68 (1H, m), 3,63 - 3,72 (2H, m), 4,00 (2H, t, J = 6,4 Hz), 4,03 (1H, brs), 4,62 (1H, brs), 6,89 (1H, d, J = 8,5 Hz), 7,28 (1H, dd, J = 1,6, 8,5 Hz), 7,36 (1H, d, J = 1,6 Hz).
[000335] A uma solução do composto 16-1 (159 mg) em cloreto de metileno (5 ml) foi adicionada uma solução de acetonitrila (5 ml) em 1H-tetrazol (27,9 mg). Di-t-butil diisopropilfosforamidita (0,126 ml) foi adicionado à mistura a 0°C, e a mistura foi agitada por 1 hora sob esfriamento com gelo. 1H-tetrazol (27,9 mg) e di-t-butil diisopropilfosforamidita (0,126 ml) foram adicionados, e a mistura foi adicionalmente agitada por 1 hora sob esfriamento com gelo. 1Htetrazol (55,8 mg) e di-t-butil diisopropilfosforamidita (0,252 ml) foram adicionados, e a mistura foi adicionalmente agitada por 1 hora sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 106 mg) foi adicionado, e a mistura foi agitada por 20 min sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 106 mg) foi adicionado, e a mistura foi adicionalmente agitada por 30 min sob esfriamento com gelo. Uma solução aquosa de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar um óleo incolor (271 mg) contendo essencialmente éster t-butílico de ácido (R)-[1-di(t-butil)fosforiloximetil3-(4-heptilóxi-3-trifluorometilfenil)-1-metilpropil]carbâmico. O óleo foi dissolvido em etanol (10 ml), ácido clorídrico concentrado (3 ml) foi adicionado, e a mistura foi agitada em 50°C por 3 horas. Água (50 ml) foi adicionada à mistura da reação, e o pó precipitado foi coletado por filtração, e lavado com água e éter dietílico para dar o produto-fim (81,9 mg) como um pó branco.
[000336] MS (ESI) m/z: 442 [M+H]
i.1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,37 (3H, s), 1,45 - 1,53 (2H, m), 1,75 - 1,81 (2H, m), 1,82 - 1,90 (1H, m), 1,96 - 2,04 (1H, m), 2,60 - 2,67 (1H, m), 2,69 - 2,77 (1H, m), 3,85 (1H, dd, J = 5,3, 11,4 Hz), 3,94 (1H, dd, J = 5,9, 11,4 Hz), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,1 Hz), 7,42 - 7,44 (2H, m).
i.1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,37 (3H, s), 1,45 - 1,53 (2H, m), 1,75 - 1,81 (2H, m), 1,82 - 1,90 (1H, m), 1,96 - 2,04 (1H, m), 2,60 - 2,67 (1H, m), 2,69 - 2,77 (1H, m), 3,85 (1H, dd, J = 5,3, 11,4 Hz), 3,94 (1H, dd, J = 5,9, 11,4 Hz), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,1 Hz), 7,42 - 7,44 (2H, m).
[000337] (S)-2-Amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-metilbutanol cloridrato
[000338] O composto 15-2-2 (785 mg) foi suspendido em acetato de etila (5 ml), e uma solução a 4 M de cloreto de hidrogênio em acetato de etila (2 ml) foi adicionada. Hexano (10 ml) foi posteriormente adicionado e permaneceu por 1 hora. O sólido precipitado foi coletado por filtração para dar o produto-fim (833 mg) como um pó branco.
[000339] MS (ESI) m/z: 362 [M+H]
[000340] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,7 Hz), 1,20 (3H, s), 1,25 - 1,35 (6H, m), 1,37 - 1,45 (2H, m), 1,67 - 1,83 (4H, m), 2,60 (2H, t, J = 8,7 Hz), 3,41 - 3,49 (2H, m), 4,06 (2H, t, J = 6,2 Hz), 5,53 (1H, t, J = 5,0 Hz), 7,18 (1H, d, J = 8,5 Hz), 7,44 - 7,46 (2H, m), 7,84 (3H, brs).
[000341] (S)-mono[2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutil] éster do ácido fosfórico
[000342] A uma solução do composto 15-2-2 (120 mg) em metanol (10 ml) foram adicionados N,N-diisopropiletilamina (0,117 ml) e dicarbonato de di-t-butila (109 mg), e a mistura foi agitada em temperatura ambiente por 24 horas. Bicarbonato de sódio saturado foi adicionada à mistura da reação, e metanol foi evaporado sob pressão reduzida. O resíduo obtido foi diluído com água e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (139 mg) como um óleo incolor.
[000343] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,22 (3H, s), 1,28 - 1,38 (6H, m), 1,42 - 1,50(11H, m), 1,79 (2H, quint, J = 6,9 Hz), 1,83 - 1,90 (1H, m), 1,99 - 2,07 (1H, m), 2,51 - 2,58 (1H, m), 2,61 - 2,69 (1H, m), 3,63 - 3,72 (2H, m), 4,00 (2H, t, J = 6,3 Hz), 4,02 (1H, brs), 4,63 (1H, brs), 6,89 (1H, d, J = 8,4 Hz), 7,28 (1H, dd, J = 1,7, 8,4 Hz), 7,36 (1H, d, J = 1,7 Hz).
[000344] A uma solução do composto 18-1 (139 mg) em cloreto de metileno (5 ml) foi adicionada uma solução de 1H-tetrazol (27,9 mg) em acetonitrila (5 ml). Di-t-butil diisopropilfosforamidita (0,126 ml) foi adicionado à mistura a 0°C, e a mistura foi agitada por 1 hora sob esfriamento com gelo. 1H-tetrazol (27,9 mg) e di-t-butil diisopropilfosforamidita (0,126 ml) foram adicionados, e a mistura foi adicionalmente agitada por 40 min sob esfriamento com gelo. 1Htetrazol (55,8 mg) e di-t-butil diisopropilfosforamidita (0,252 ml) foram adicionados, e a mistura foi adicionalmente agitada por 50 min sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 106 mg) foi adicionado, e a mistura foi agitada por 20 min sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 106 mg) foi adicionado, e a mistura foi adicionalmente agitada por 50 min sob esfriamento com gelo. Uma solução aquosa de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar um óleo incolor (260 mg) essencialmente contendo éster t-butílico de ácido (S)-[1-di(tbutil)fosforiloximetil-3-(4-heptilóxi-3-trifluorometilfenil)-1- metilpropil]carbâmico. O óleo foi dissolvido em etanol (10 ml), ácido clorídrico concentrado (3 ml) foi adicionado, e a mistura foi agitada em 50°C por 3 horas. Água (50 ml) foi adicionada à mistura da reação, e o pó precipitado foi coletado por filtração e lavado com água e éter dietílico para dar o produto-fim (63,0 mg) como um pó branco.
[000345] MS (ESI) m/z: 442 [M+H]
[000346] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,29 - 1,41 (6H, m), 1,37 (3H, s), 1,45 - 1,53 (2H, m), 1,75 - 1,81 (2H, m), 1,82 - 1,90 (1H, m), 1,96 - 2,04 (1H, m), 2,60 - 2,76 (2H, m), 3,85 (1H, dd, J = 5,3, 11,4 Hz), 3,94 (1H, dd, J = 5,9, 11,4 Hz), 4,04 (2H, t, J = 6,2 Hz), 7,07 (1H, d, J = 8,1 Hz), 7,42 - 7,44 (2H, m).
[000347] Cloridrato de 2-amino-2-etil-4-(4-heptilóxi-3-trifluorometilfenil)butanol
[000348] A uma solução de 2-amino-2-etil-1,3-propanodiol (22,0 g) em metanol (500 ml) e N,N-diisopropiletilamina (64,3 ml) foi adicionado dicarbonato de di-t-butila (60,5 g) sob esfriamento com gelo, e a mistura foi agitada por 40 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 16 horas. solução aquosa a 1 M de hidróxido de sódio (184 ml) foi adicionada à mistura da reação sob esfriamento com gelo e a mistura foi agitada por 40 min. Metanol foi removido sob pressão reduzida. Água foi adicionada e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (41,0 g) como um óleo incolor.
[000349] 1H-RMN (CDCl3) δ (ppm): 0,90 (3H, t, J = 7,5 Hz), 1,45 (9H, s), 1,59 (2H, q, J = 7,5 Hz), 3,45 (2H, brs), 3,60 (2H, dd, J = 6,9, 11,6 Hz), 3,84 (2H, dd, J = 4,8, 11,6 Hz), 4,89 (1H, brs).
[000350] A uma solução do composto 19-1 (41,0 g) em cloreto de metileno (400 ml) foram adicionados N,N-diisopropiletilamina (40,7 ml) e cloreto de metoximetila (17,6 ml) sob esfriamento com gelo, e a mistura foi agitada por 40 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 4 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (21,3 g) como um óleo incolor.
[000351] 1H-RMN (CDCl3)δ (ppm): 0,89 (3H, t, J = 7,5 Hz), 1,44 (9H, s), 1,55 - 1,62 (1H, m), 1,75 - 1,84 (1H, m), 3,38 (3H, s), 3,49 (1H, d, J = 9,8 Hz), 3,68 (2H, d, J = 6,6 Hz), 3,74 (1H, d, J = 9,8 Hz), 4,04 (1H, brs), 4,63 (2H, s), 5,05 (1H, brs).
[000352] A uma solução mista do composto 19-2 (21,3 g) e brometo de sódio (8,32 g) em tolueno (170 ml), acetato de etila (170 ml) e água (30 ml) foram adicionados 2,2,6,6-tetrametilpiperidina 1-oxila, radical livre (253 mg) sob esfriamento com gelo. Em seguida, solução aquosa a 10% de hipoclorito de sódio (66,3 g) e uma solução de bicarbonato de sódio (19,6 g) em água (200 ml) foram adicionadas gota a gota durante 1,5 hora. A mistura foi adicionalmente agitada por 1,5 hora sob esfriamento com gelo, solução aquosa a 10% de hipoclorito de sódio (22,1 g) e uma solução de bicarbonato de sódio (6,53 g) em água (67 ml) foram adicionadas gota a gota durante 30 min, e a mistura foi adicionalmente agitada por 30 min. A camada orgânica foi particionada e diluída com acetato de etila (200 ml). A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (22,0 g) como um óleo marrom-pálido.
[000353] 1H-RMN (CDCl3) δ (ppm): 0,81 (3H, t, J = 7,5 Hz), 1,45 (9H, s), 1,74 - 1,83 (1H, m), 2,04 - 2,11 (1H, m), 3,32 (3H, s), 3,81 (1H, d, J = 10,0 Hz), 4,03 (1H, d, J = 10,0 Hz), 4,59 (2H, s), 5,37 (1H, brs), 9,39 (1H, s).
[000354] O Exemplo de Referência composto 2-5 (26,3 g) foi suspendido em tetraidrofurano (120 ml), t-butóxido de potássio (5,24 g) foi adicionado sob esfriamento com gelo, e a mistura foi agitada por 50 min. Uma solução do composto 19-3 (6,10 g) em tetraidrofurano (80 ml) foi adicionada à solução mista, e a mistura foi agitada por 2 horas sob esfriamento com gelo, e em temperatura ambiente por 4 horas. A mistura da reação foi adicionada a salmoura, e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar éster t-butílico de ácido [1-etil-3-(4- benzilóxi-3-trifluorometilfenil)-1-(metoximetóxi)metil] alilcarbâmico (10,3 g) como um óleo incolor. A proporção de isômeros geométricos do composto obtido foi E:Z = 1:2,8. 10% de Paládio carbono (contendo cerca de 50% de água, 2 g) foi adicionado a uma solução deste óleo em 1,4-dioxano (200 ml), e a mistura foi agitada em temperatura ambiente por 9 horas sob uma atmosfera de hidrogênio. A mistura da reação foi filtrada através de celite e concentrada para dar o produto-fim (8,67 g) como um pó branco.
[000355] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 7,5 Hz), 1,45 (9H, s), 1,65 - 1,74 (1H, m), 1,76 - 1,86 (1H, m), 1,93 - 1,97 (2H, m), 2,52 - 2,56 (2H, m), 3,39 (3H, s), 3,57 (1H, d, J = 9,7 Hz), 3,63 (1H, d, J = 9,7 Hz), 4,64 (3H, m), 5,85 (1H, brs), 6,85 (1H, d, J = 8,3 Hz), 7,20 (1H, brd, J = 8,3 Hz), 7,29 (1H, d, J = 1,4 Hz).
[000356] A uma solução do composto 19-4 (1,5 g) em N,N-dimetilformamida (15 ml) foram adicionados carbonato de potássio (1,48 g) e n-heptilbrometo (0,61 ml), e a mistura foi agitada em 50°C por 6 horas. A mistura da reação foi adicionada a água, e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (1,83 g) como um óleo incolor.
[000357] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,4 Hz), 0,89 (3H, t, J = 7,6 Hz), 1,28 - 1,38 (6H, m), 1,42 - 1,49(11H, m), 1,68 - 1,88 (4H, m), 1,92 - 1,97 (2H, m), 2,52 - 2,57 (2H, m), 3,38 (3H, s), 3,57 (1H, d, J = 9,7 Hz), 3,63 (1H, d, J = 9,7 Hz), 4,00 (2H, t, J = 6,4 Hz), 4,60 (1H, brs), 4,64 (2H, s), 6,88 (1H, d, J = 8,5 Hz), 7,27 (1H, dd, J = 1,6, 8,5 Hz), 7,35 (1H, d, J = 1,6 Hz).
[000358] A uma solução do composto 19-5 (1,83 g) em etanol (15 ml) foi adicionado ácido clorídrico concentrado (3 ml), e a mistura foi agitada em 50°C por 4 horas. solução aquosa a 1 M de hidróxido de sódio (100 ml) foi adicionada à mistura da reação e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi dissolvido em acetato de etila (30 ml), solução a 1 M de éter de ácido clorídrico (10 ml) foi adicionado, e o solvente foi evaporado. Éter dietílico e hexano foram adicionados ao resíduo, e o sólido precipitado foi coletado por filtração e secado para dar o produto-fim (1,22 g) como um pó branco.
[000359] MS (ESI) m/z: 376 [M+H]
[000360] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,4 Hz), 0,90 (3H, t, J = 7,4 Hz), 1,25 - 1,35 (6H, m), 1,37 - 1,45 (2H, m), 1,57 - 1,65 (2H, m), 1,67 - 1,77 (4H, m), 2,56 - 2,61 (2H, m), 3,43 - 3,51 (2H, m), 4,06 (2H, t, J = 6,2 Hz), 5,49 (1H, t, J = 5,0 Hz), 7,18 (1H, d, J = 9,2 Hz), 7,45 - 7,46 (2H, m), 7,90 (3H, brs).
[000361] Éster de mono[2-amino-2-etil-4-(4-heptilóxi-3-trifluorometilfenil)butílico] do ácido fosfórico
[000362] A uma solução do composto 19-6 (1,04 g) em metanol (25 ml) foram adicionados N,N-diisopropiletilamina (1,32 ml) e dicarbonato de di-t-butila (825 mg), e a mistura foi agitada em temperatura ambiente por 13 horas. A mistura da reação foi concentrada sob pressão reduzida, bicarbonato de sódio saturado foi adicionado e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (1,21 g) como um óleo incolor.
[000363] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 0,93 (3H, t, J = 7,5 Hz), 1,28 - 1,39 (6H, m), 1,42 - 1,49(11H, m), 1,64 (2H, q, J = 7,5 Hz), 1,79 (2H, quint, J = 7,0 Hz), 1,82 - 1,94 (2H, m), 2,46 - 2,54 (1H, m), 2,56 - 2,64 (1H, m), 3,72 (2H, d, J = 6,3 Hz), 4,00 (2H, t, J = 6,5 Hz), 4,09 (1H, brs), 4,57 (1H, brs), 6,89 (1H, d, J = 8,5 Hz), 7,28 (1H, dd, J = 1,7, 8,5 Hz), 7,35 (1H, d, J = 1,7 Hz).
[000364] A uma solução do composto 20-1 (1,20 g) em cloreto de metileno (15 ml) foi adicionada uma solução de 1H-tetrazol (212 mg) em acetonitrila (15 ml). Di-t-butil diisopropilfosforamidita (0,956 ml) foi adicionado a esta mistura a 0°C, e a mistura foi agitada por 1 hora sob esfriamento com gelo. Uma solução de 1H-tetrazol (106 mg) em acetonitrila (15 ml) e di-t-butil diisopropilfosforamidita (0,478 ml) foram adicionados, e a mistura foi adicionalmente agitada por 1 hora sob esfriamento com gelo. Ácido m-cloroperbenzóico (produto contendo 25% de água, 804 mg) foi adicionado e a mistura foi agitada por 50 min sob esfriamento com gelo. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi lavada com bicarbonato de sódio saturado aquoso e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (1,56 g) como um óleo incolor.
[000365] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 0,90 (3H, t, J = 7,3 Hz), 1,28 - 1,38 (6H, m), 1,43 (9H, s), 1,47 - 1,51 (2H, m), 1,49(18H, s), 1,70 - 1,82 (4H, m), 1,86 - 2,01 (2H, m), 2,53 - 2,59 (2H, m), 3,95 - 4,06 (4H, m), 4,67 (1H, brs), 6,88 (1H, d, J = 8,5 Hz), 7,28 (1H, dd, J = 1,7, 8,5 Hz), 7,35 (1H, brs).
[000366] O composto 20-2 (1,55 g) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado e a mistura foi agitada em 50°C por 1 hora. Água (50 ml) foi adicionada à mistura da reação, e o pó precipitado foi coletado por filtração e lavado com água e éter dietílico para dar o produto-fim (907 mg) como um pó branco.
[000367] MS (ESI) m/z: 456 [M+H]
[000368] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,7 Hz), 1,03 (3H, t, J = 7,5 Hz), 1,30 - 1,40 (6H, m), 1,49 (2H, quint, J = 7,5 Hz), 1,73 - 2,01 (6H, m), 2,57 - 2,74 (2H, m), 3,88 - 3,96 (2H, m), 4,05 (2H, t, J = 6,2 Hz), 7,08 (1H, d, J = 8,9 Hz), 7,42 - 7,44 (2H, m).
[000369] Cloridrato de dietil 2-amino-2-[2-(4-heptilóxi-3- trifluorometilfenil)etil] butano-1,4-diol
[000370] (t-Butiloxicarbonil)aminomalonato dietila (52,3 g) foi dissolvido em tetraidrofurano (400 ml), t-butóxido de sódio (19,2 g) foi adicionado a isto, uma solução de 2-(2-bromoetóxi)tetraidro-2H-pirano (40,4 g) em tetraidrofurano (100 ml) foi adicionada à mistura da reação a 70°C, e a mistura foi agitada com aquecimento por 10 horas. Depois de esfriar, a mistura da reação foi vertida em salmoura saturada. A mistura foi extraída com acetato de etila, e a camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida, e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produtofim (50,0 g) como um óleo incolor.
[000371] 1H-RMN (CDCl3) δ (ppm): 1,30 (6H, t, J = 7,1 Hz), 1,45 (9H, s), 1,45 - 1,55 (4H, m), 1,58 - 1,78 (2H, m), 2,60 - 2,64 (2H, m), 3,35 - 3,41 (1H, m), 3,46 - 3,50 (1H, m), 3,77 - 3,84 (2H, m), 4,12 - 4,28 (4H, m), 4,49 - 4,51 (1H, m), 6,08 (1H, brs).
[000372] O composto 21-1 (50,0 g) foi dissolvido em uma mistura de etanol (530 ml), tetraidrofurano (130 ml) e água (260 ml). Cloreto de cálcio (27,5 g) foi adicionado a esta solução a 0°C, boridreto de sódio (18,8 g) foi subseqüentemente adicionado em porções, e a mistura foi agitada na mesma temperatura por 2 horas, e adicionalmente em temperatura ambiente por 18 horas. A mistura da reação foi concentrada sob pressão reduzida, adicionada a solução aquosa saturada de cloreto de amônio (3l), e extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produtofim (21,6 g) como um óleo incolor.
[000373] 1H-RMN (CDCl3) δ (ppm): 1,43 (9H, s), 1,53 - 1,62 (4H, m), 1,71 - 1,83 (2H, m), 1,95 (1H, ddd, J = 15,3, 8,0, 2,8 Hz), 2,02 (1H, ddd, J = 15,3, 7,4, 2,8 Hz), 3,46 - 3,59 (4H, m), 3,69 - 3,73 (2H, m), 3,82 - 3,88 (1H, m), 3,91 - 3,96 (1H, m), 4,13 (2H, brs), 4,60 - 4,62 (1H, m), 5,79 (1H, brs).
[000374] A uma solução do composto 21-2 (21,6 g) em cloreto de metileno (250 ml) foram adicionados N,N-diisopropiletilamina (14,7 ml) e cloreto de metoximetila (6,37 ml) sob esfriamento com gelo, e a mistura foi agitada por 1,5 hora sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 17 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (9,61 g) como um óleo incolor.
[000375] 1H-RMN (CDCl3) δ (ppm): 1,42 (9H, s), 1,51 - 1,63 (4H, m), 1,68 - 1,93 (3H, m), 2,03 - 2,10 (1H, m), 3,37 (3H, s), 3,51 - 3,60 (3H, m), 3,69 - 4,00 (5H, m), 4,26, 4,35 (1H, 2×brs), 4,61 - 4,66 (3H, m), 5,61, 5,75 (1H, 2×brs).
[000376] A uma solução mista do composto 21-3 (9,59 g) e brometo de sódio (2,72 g) em tolueno (50 ml), acetato de etila (50 ml) e água (9 ml) foi adicionado 2,2,6,6-tetrametilpiperidina 1-oxila, radical livre (82,5 mg) sob esfriamento com gelo, em seguida uma solução de solução aquosa a 10% de hipoclorito de sódio (21,7 g) e bicarbonato de sódio (3,19 g) em água (75 ml) foram adicionados gota a gota durante 2 horas. A mistura foi adicionalmente agitada por 2 horas sob esfriamento com gelo, uma solução de solução aquosa a 10% de hipoclorito de sódio (10,9 g) e bicarbonato de sódio (3,19 g) em água (35 ml) foi adicionada gota a gota durante 20 min, e a mistura foi adicionalmente agitada por 20 min. A camada orgânica foi particionada e diluída com acetato de etila (200 ml), lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (8,54 g) como um óleo marrom.
[000377] 1H-RMN (CDCl3) δ (ppm): 1,44 (9H, s), 1,45 - 1,78 (6H, m), 2,10 - 2,18 (1H, m), 2,35 - 2,46 (1H, m), 3,31, 3,32 (3H, 2×s), 3,33 - 3,41 (1H, m), 3,47 - 3,51 (1H, m), 3,67 - 3,74 (1H, m), 3,77 - 3,84 (2H, m), 4,05 - 4,13 (1H, m), 4,43 - 4,45, 4,56 - 4,58 (1H, 2×m), 4,58, 4,58 (2H, 2×s), 5,72, 5,74 (1H, 2×brs), 9,40, 9,44 (1H, 2×s).
[000378] O Exemplo de Referência composto 2-5 (10,9 g) foi suspendido em tetraidrofurano (80 ml), t-butóxido de potássio (2,17 g) foi adicionado sob esfriamento com gelo e a mistura foi agitada por 30 min. Uma solução do composto 21-4 (3,50 g) em tetraidrofurano (25 ml) foi adicionada à solução mista e a mistura foi agitada por 20 min sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 5 horas. A mistura da reação foi adicionada a salmoura, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar éster tbutílico de ácido 3-(4-benzilóxi-3-trifluorometilfenil)-1-(metoximetóxi)metil-1-[2-(tetraidro-2H-piran-2-ilóxi)etil]alilcarbâmico (4,95 g) como um óleo amarelo-pálido. A proporção de isômeros geométricos do composto obtido foi E:Z = 1:3. A uma solução deste óleo em tolueno (200 ml) foi adicionado clorotris(trifenilfosfina)ródio(I) (5,0 g), e a mistura foi agitada em 60°C por 19 horas. Clorotris(trifenilfosfina) ródio(I) (2,5 g) foi adicionado e a mistura foi agitada em 60°C por 10 horas sob uma atmosfera de hidrogênio. A mistura da reação foi filtrada através de celite e concentrada. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (4,95 g) como um óleo amarelo.
[000379] 1H-RMN (CDCl3) δ (ppm): 1,43 (9H, s), 1,51 - 1,64 (4H, m), 1,67 - 1,75 (1H, m), 1,79 - 1,88 (1H, m), 1,92 - 2,28 (4H, m), 2,54 - 2,62 (2H, m), 3,36, 3,37 (3H, 2×s), 3,46 - 3,59 (2H, m), 3,71 - 3,78 (2H, m), 3,82 - 4,03 (2H, m), 4,60 - 4,64 (3H, m), 5,15 (2H, s), 5,41, 5,55 (1H, 2×brs), 6,93 (1H, d, J = 8,5 Hz), 7,26 - 7,32 (2H, m), 7,36 - 7,44 (5H, m).
[000380] A uma solução do composto 21-5 (4.94 g) em 1,4-dioxano (150 ml) foi adicionado 10% de paládio carbono (contendo cerca de 50% água, 2 g), e a mistura foi agitada por 22 horas sob uma atmosfera de hidrogênio. A mistura da reação foi filtrada através de celite, e concentrado para dar o produto-fim (4,07 g) como um óleo incolor.
[000381] 1H-RMN (CDCl3) δ (ppm): 1,43 (9H, s), 1,52 - 1,64 (4H, m), 1,68 - 1,75 (1H, m), 1,79 - 1,88 (1H, m), 1,92 - 2,27 (4H, m), 2,55 - 2,61 (2H, m), 3,36, 3,37 (3H, 2×s), 3,46 - 3,60 (2H, m), 3,71 - 4,03 (4H, m), 4,61 - 4,63 (3H, m), 5,45, 5,59 (1H, 2×brs), 5,54 (1H, brs), 6,85 (1H, d, J = 8,4 Hz), 7,21 - 7,23 (1H, m), 7,30 (1H, brs).
[000382] O composto 21-6 (1,24 g) foi dissolvido em N,N-dimetilformamida (20 ml), carbonato de potássio (986 mg) e brometo de nheptila (0,458 ml) foram adicionados, e a mistura foi agitada em 80C por 2,5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (1,49 g) como um óleo amarelo-pálido.
[000383] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,25 - 1,40 (7H, m), 1,40 - 1,49 (2H, m), 1,43 (9H, s), 1,50 - 1,68 (3H, m), 1,69 - 1,90 (4H, m), 1,91 - 2,00 (2H, m), 2,00 - 2,20 (2H, m), 2,56 - 2,61 (2H, m), 3,36, 3,37 (3H, 2×s), 3,45 - 3,60 (2H, m), 3,71 - 3,96 (4H, m), 4,00 (2H, t, J = 6,4 Hz), 4,61 - 4,63 (3H, m), 5,40, 5,57 (1H, 2×brs), 6,88 (1H, d, J = 8,5 Hz), 7,28 (1H, brs), 7,36 (1H, brs).
[000384] O composto 21-7 (1,49 g) foi dissolvido em etanol (20 ml), ácido clorídrico concentrado (3 ml) foi adicionado, e a mistura foi agitada em 80°C por 1,5 hora. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (915 mg) como um pó branco.
[000385] MS (ESI) m/z: 392 [M+H]
[000386] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,25 - 1,34 (6H, m), 1,35 - 1,45 (2H, m), 1,68 - 1,78 (2H, m), 1,79 - 1,83 (4H, m), 2,59 - 2,65 (2H, m), 3,51 (2H, d, J = 4,4 Hz), 3,60 (2H, t, J = 6,4 Hz), 4,05 (2H, t, J = 6,2 Hz), 5,45 (1H, t, J = 4,8 Hz), 7,18 (1H, d, J = 9,2 Hz), 7,43 - 7,46 (2H, m), 7,75 (3H, brs).
[000387] Cloridrato de 2-amino-4-flúor-2-[2-(4-heptilóxi-3-trifluorometilfenil)etil]butanol
[000388] A uma solução do composto 21-8 (830 mg) em N,N-dimetilformamida (20 ml) foram adicionados N,N-diisopropiletilamina (1,04 ml) e trimetil ortoacetato (0,368 ml), e a mistura foi agitada em 120°C por 5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 840 mg de um óleo marrom. Peneiras moleculares 4Å (8,4 g), fluoreto de p-toluenossulfonila (690 mg) e solução a 1 M de fluoreto de tetrabutilamônio/ tetraidrofurano (5,82 ml) foram adicionados a uma solução do óleo marrom em tetraidrofurano (30 ml) e a mistura foi aquecida sob refluxo por um dia. A mistura da reação foi filtrada, e solução aquosa a 1 M de ácido clorídrico foi adicionada ao filtrado. A mistura foi extraída com acetato de etila, lavada com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:2 a 1:3) para dar o produto-fim (400 mg) como um óleo marrom.
[000389] 1H-RMN (DMSO-d6) δ (ppm): 0,86 - 0,94 (3H, m), 1,25 - 1,40 (6H, m), 1,41 - 1,51 (2H, m), 1,76 - 1,95 (4H, m), 1,97 - 2,08 (2H, m), 2,01 (3H, s), 2,50 - 2,66 (2H, m), 4,00 (2H, t, J = 6,2 Hz), 4,06 (1H, d, J = 8,8 Hz), 4,14 (1H, d, J = 8,8 Hz), 4,53 (1H, dd, J = 48, 3,8 Hz), 4,65 (1H, dd, J = 48, 3,8 Hz), 6,89 (1H, d, J = 8,4 Hz), 7,26 (1H, brs), 7,36 (1H, brs).
[000390] O composto 22-1 (400 mg) foi dissolvido em etanol (10 ml), ácido clorídrico concentrado (2 ml) foi adicionado, e a mistura foi agitada em 70°C por 4,5 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter diisopropílico para dar o produto-fim (360 mg) como um pó branco.
[000391] MS (ESI) m/z: 394 [M+H]
[000392] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,7 Hz), 1,24 - 1,38 (6H, m), 1,39 - 1,46 (2H, m), 1,68 - 1,78 (2H, m), 1,79 - 1,86 (2H, m), 2,04 (1H, t, J = 6,0 Hz), 2,10 (1H, t, J = 6,0 Hz), 2,59 - 2,66 (2H, m), 3,53 (2H, d, J = 5,0 Hz), 4,06 (2H, t, J = 6,2 Hz), 4,61 (1H, dt, J = 47, 6,0 Hz), 4,73 (1H, dt, J = 47, 6,0 Hz), 5,56 (1H, t, J = 5,0 Hz), 7,18 (1H, d, J = 8,4 Hz), 7,42 - 7,45 (2H, m), 8,09 (3H, brs).
[000393] Éster de mono[2-amino-2-(2-fluoroetil)-4-(4-heptilóxi-3- trifluorometilfenil)butílico] do ácido fosfórico
[000394] A uma solução do composto 22-2 (290 mg) em metanol (15 ml) foram adicionados trietilamina (0,284 ml) e dicarbonato de di-t-butila (220 mg), e a mistura foi agitada em temperatura ambiente por 18 horas. Além disso, dicarbonato de di-t-butila (220 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 5 horas. A mistura da reação foi concentrada sob pressão reduzida, água foi adicionada ao resíduo e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 400 mg de um óleo amarelo-pálido. A uma solução do óleo amarelo-pálido (400 mg) em cloreto de metileno (5 ml) e acetonitrila (2 ml) foram adicionados 1H-tetrazol (94 mg) e dietilfosforamidita de di-t-butila (0,401 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, uma solução de decano contendo hidroperóxido de t-butila (5 a 6 M, 0,402 ml) foi adicionado e a mistura foi agitada em temperatura ambiente por 30 min. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:1 a 1:2) para dar o produto-fim (530 mg) como um óleo incolor.
[000395] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,29 - 1,40 (6H, m), 1,41 - 1,50 (2H, m), 1,43 (9H, s), 1,49(18H, s), 1,75 - 1,81 (2H, m), 1,90 - 2,01 (2H, m), 2,10 - 2,25 (2H, m), 2,61 (2H, t, J = 8,6 Hz), 4,03 (2H, t, J = 6,2 Hz), 4,04 - 4,18 (2H, m), 4,55 (1H, dt, J = 47, 5,8 Hz), 4,66 (1H, dt, J = 47, 5,8 Hz), 7,37 (1H, d, J = 8,4 Hz), 7,34 - 7,37 (2H, m).
[000396] O composto 23-1 (530 mg) foi dissolvido em cloreto de metileno (5 ml), dioxano contendo cloreto de hidrogênio (4 mols/l, 2 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 3,5 horas. O solvente foi concentrado sob pressão reduzida, e metanol (3 ml), éter dietílico (7 ml) e óxido de propileno (7 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração e lavado com acetato de etila e éter dietílico para dar o produto-fim (182 mg) como um sólido branco.
[000397] MS (ESI) m/z: 474 [M+H]
[000398] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,4 Hz), 1,29 - 1,44 (6H, m), 1,45 - 1,52 (2H, m), 1,73 - 1,82 (2H, m), 1,92 - 2,10 (2H, m), 2,12 - 2,20 (1H, m), 2,21 - 2,26 (1H, m), 2,60 - 2,79 (2H, m), 3,99 (2H, d, J = 5,6 Hz), 4,05 (2H, t, J = 6,2 Hz), 4,68 (1H, t, J = 5,4 Hz), 4,79 - 4,81 (1H, m), 7,07 (1H, d, J = 8,3 Hz), 7,41-7,44 (2H, m).
[000399] Cloridrato de 2-amino-2-[2-(4-heptiltio-3-trifluorometilfenil) etil]propano-1,3-diol
[000400] A uma solução do composto 1-1 (1,00 g) em cloreto de metileno (30 ml) foram adicionados sob esfriamento com gelo trietilamina (0,503 ml), ácido trifluorometanossulfônico anídrico (0,607 ml), a mistura foi agitada por 1 hora sob esfriamento com gelo. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno, lavada com água e salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:2 - 1:4) para dar composto desprotegido por acetonida com o grupo hidroxila fenólico protegido por um triflato (500 mg) como um óleo incolor. Diisopropilamina (0,377 ml), heptanotiol (0,204 ml), 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos) (31 mg) e aducto de tris (dibenzilidenoacetona)paládio(0)-clorofórmio (27 mg) foram adicionados a uma solução do óleo incolor em dioxano (10 ml), e a mistura foi agitada em 120°C por 4 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:2 a 1:3) para dar o produto-fim (390 mg) como um óleo amarelopálido.
[000401] 1H-RMN (CDCl3) δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,23 - 1,38 (6H, m), 1,39 - 1,50 (2H, m), 1,46 (9H, s), 1,60 - 1,68 (2H, m), 1,86 - 1,91 (2H, m), 2,61 - 2,66 (2H, m), 2,92 (2H, t, J = 7,4 Hz), 3,20 (2H, brs), 3,63 - 3,68 (2H, m), 3,85 - 3,90 (2H, m), 5,05 (1H, brs), 7,25 - 7,30 (1H, m), 7,39 (1H, d, J = 8,0 Hz), 7,45 (1H, brs).
[000402] O composto 24-1 (390 mg) foi dissolvido em cloreto de metileno (5 ml), dioxano contendo cloreto de hidrogênio (4 mols/l, 5 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 4 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar um pó branco. O pó branco foi purificado por HPLC do preparativo, éter contendo cloreto de hidrogênio (1 mol/l, 15 ml) foi adicionado ao resíduo obtido para dar um cloridrato. O precipitado foi coletado por filtração e secado para dar o produto-fim (200 mg) como um pó branco.
[000403] MS (ESI) m/z: 394 [M+H]
[000404] 1H-RMN (DMSO-d6) δ (ppm): 0,84 (3H, t, J = 6,8 Hz), 1,19 - 1,31 (6H, m), 1,32 - 1,42 (2H, m), 1,51 - 1,60 (2H, m), 1,75 - 1,80 (2H, m), 2,63 - 2,68 (2H, m), 3,02 (2H, t, J = 7,2 Hz), 3,52 (4H, d, J = 4,0 Hz), 5,36 (2H, brs), 7,47 (1H, d, J = 8,4 Hz), 7,56 - 7,59 (2H, m), 7,74 (3H, brs).
[000405] Cloridrato de 2-amino-2-[2-(4-octiltio-3-trifluorometilfenil) etil]propano-1,3-diol
[000406] A uma solução do composto 1-1 (1,00 g) em cloreto de metileno (30 ml) foi adicionada piridina (0,926 ml), uma solução de ácido trifluorometanossulfônico anídrico (0,480 ml) em cloreto de metileno (5 ml) foi adicionado gota a gota sob esfriamento com gelo e a mistura foi agitada por 2,5 horas sob esfriamento com gelo. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 3:1 a 2:1) para dar um composto (970 mg), em que o grupo hidroxila fenólico é protegido por triflato, como um sólido branco. A uma solução do sólido branco em dioxano (20 ml) foram adicionados diisopropilamina (0,631 ml), octanotiol (0,375 ml), 4,5-bis (difenilfosfino)-9,9-dimetilxanteno (Xantphos) (53 mg) e aducto de tris (dibenzilidenoacetona)paládio(0)- clorofórmio (46 mg), e a mistura foi agitada em 120°C por 2 dias. Além disso, diisopropilamina (0,631 ml), octanotiol (0,375 ml), 4,5- bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos) (53 mg) e aducto de tris(dibenzilidenoacetona)paládio(0)-clorofórmio (46 mg) foram adicionados à solução da reação e a mistura foi agitada em 120°C por 1 dia. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 5:1 a 2:1) para dar o produto-fim (910 mg) como um sólido amarelo-pálido.
[000407] 1H-RMN (CDCl3) δ (ppm): 0,88 (3H, t, J = 6,8 Hz), 1,24 - 1,36 (8H, m), 1,42 - 1,50 (2H, m), 1,43 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,60 - 1,70 (2H, m), 1,96 - 2,00 (2H, m), 2,55 - 2,60 (2H, m), 2,91 (2H, t, J = 7,4 Hz), 3,69 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 4,98 (1H, brs), 7,25 - 7,29 (1H, m), 7,38 (1H, d, J = 8,2 Hz), 7,44 (1H, d, J = 1,0 Hz).
[000408] O composto 25-1 (910 mg) foi dissolvido em etanol (20 ml), ácido clorídrico concentrado (2 ml) foi adicionado, e a mistura foi agitada em 80°C por 2 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (630 mg) como um pó branco.
[000409] MS (ESI) m/z: 408 [M+H]
[000410] 1H-RMN (DMSO-d6) δ (ppm): 0,85 (3H, t, J = 6,8 Hz), 1,19 - 1,32 (8H, m), 1,33 - 1,43 (2H, m), 1,51 - 1,60 (2H, m), 1,76 - 1,81 (2H, m), 2,64 - 2,69 (2H, m), 3,02 (2H, t, J = 7,2 Hz), 3,52 (4H, d, J = 4,8 Hz), 5,38 (2H, t, J = 5,0 Hz), 7,47 (1H, d, J = 8,3 Hz), 7,56 - 7,59 (2H, m), 7,83 (3H, brs).
[000411] 2-Amino-4-(4-octiltio-3-trifluorometilfenil)-2-(fosforiloximetil)butanol
[000412] O composto 25-2 (560 mg) foi dissolvido em metanol (10 ml), trietilamina (0,531 ml) e dicarbonato de di-t-butila (412 mg) foram adicionados, e a mistura foi agitada em temperatura ambiente por 12 horas. Além disso, dicarbonato de di-t-butila (300 mg) foi adicionada à mistura da reação e a mistura foi agitada em temperatura ambiente por 12 horas. Água foi adicionada e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar um óleo amarelo-pálido. A uma solução do óleo em cloreto de metileno (20 ml) foram adicionados N,Ndiisopropiletilamina (0,285 ml) e cloreto de metoximetila (0,121 ml) sob esfriamento com gelo, e a mistura foi agitada por 10 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 14 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (290 mg) e éster tbutílico de ácido [1,1-bis(hidroximetil)-3-(4-octiltio-3- trifluorometilfenil)]propilcarbâmico (280 mg) cada um como um óleo incolor. A uma solução do éster t-butílico de ácido [1,1-bis (hidroximetil)- 3-(4-octiltio-3-trifluorometilfenil)]propilcarbâmico recuperado acima em cloreto de metileno (15 ml) foram adicionados N,N-diisopropiletilamina(0,129 ml) e cloreto de metoximetila (0,063 ml) sob esfriamento com gelo, e a mistura foi agitada por 5 min sob esfriamento com gelo e adicionalmente em temperatura ambiente por 14 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (210 mg) como um óleo incolor. O peso total do produto-fim incluindo o produto-fim obtido pela primeira reação foi 500 mg.
[000413] 1H-RMN (CDCl3) δ (ppm): 0,87 (3H, t, J = 6,8 Hz), 1,25 - 1,38 (8H, m), 1,39 - 1,49 (2H, m), 1,45 (9H, s), 1,59 - 1,70 (2H, m), 1,85 - 1,93 (1H, m), 2,04 - 2,12 (1H, m), 2,54 - 2,64 (1H, m), 2,66 - 2,76 (1H, m), 2,91 (2H, t, 7,4 Hz), 3,39 (3H, s), 3,51 (1H, d, J = 9,6 Hz), 3,70 - 3,79 (3H, m), 3,94 (1H, brs), 4,65 (2H, s), 5,17 (1H, brs), 7,27 - 7,30 (1H, m), 7,39 (1H, d, J = 8,1 Hz), 7,45 (1H, d, J = 1,3 Hz).
[000414] A uma solução do composto 26-1 (500 mg) em cloreto de metileno (3 ml) foram adicionados piridina (2 ml), tetrabrometo de carbono (334 mg) e trimetil fosfito (0,161 ml), e a mistura foi agitada em temperatura ambiente por 4,5 horas. Além disso, trimetil fosfito (0,080 ml) foi adicionada à mistura da reação, e a mistura foi agitada em temperatura ambiente por 2,5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:3 a 1:4) para dar o produto-fim (490 mg) como um óleo marrom.
[000415] 1H-RMN (CD3OD) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,26-1,38 (8H, m), 1,39 - 1,51 (2H, m), 1,45 (9H, s), 1,55 - 1,68 (2H, m), 1,88 - 1,97 (1H, m), 2,05 - 2,16 (1H, m), 2,68 (2H, t, J = 8,5 Hz), 2,96 (2H, t, J = 7,3 Hz), 3,60 (1H, d, J = 9,7 Hz), 3,69 (1H, d, J = 9,7 Hz), 3,77 (3H, s), 3,80 (3H, s), 4,14 - 4,18 (1H, m), 4,30 - 4,34 (1H, m), 4,64 (2H, s), 7,37 - 7,39 (1H, m), 7,49 - 7,52 (2H, m).
[000416] O composto 26-2 (490 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 2 horas. O solvente foi concentrado sob pressão reduzida. A uma solução do resíduo em cloreto de metileno (7 ml) foi adicionado trimetilsilil iodeto (0,527 ml) sob esfriamento com gelo, e a mistura foi agitada sob esfriamento com gelo por 1 hora. O solvente foi concentrado sob pressão reduzida até a metade, e acetonitrila (15 ml) foi adicionado. Pó precipitado foi coletado por filtração, lavado com acetonitrila e éter dietílico para dar o produto-fim (245 mg) como um sólido amarelo-pálido.
[000417] MS (ESI) m/z: 488 [M+H]
[000418] 1H-RMN (CD3OD) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,24-1,38 (8H, m), 1,39-1,50 (2H, m), 1,56-1,68 (2H, m), 1,95-2,01 (2H, m), 2,68- 2,80 (2H, m), 2,97 (2H, t, J = 7,3 Hz), 3,71 (2H, brs), 3,98-4,04 (2H, m), 7,46 (1H, d, J = 8,0 Hz), 7,53 (1H, d, J = 8,2 Hz), 7,57 (1H, brs).
[000419] Cloridrato de 2-amino-2-[2-(4-hexiltio-3-trifluorometilfenil) etil]propano-1,3-diol
[000420] A uma solução do composto 1-1 (1,00 g) em cloreto de metileno (30 ml) foi adicionada piridina (0,926 ml) sob esfriamento com gelo, uma solução de ácido trifluorometanossulfônico anídrico (0,480 ml) em cloreto de metileno (5 ml) foi adicionada gota a gota, e a mistura foi agitada por 2,5 horas sob esfriamento com gelo. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno, lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 3:1 a 2:1) para dar um composto (980 mg), em que o grupo hidroxila fenólico é protegido por triflato, como um sólido branco. A uma solução do sólido branco em dioxano (20 ml) foram adicionados diisopropilamina (0,638 ml), hexanotiol (0,301 ml), 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos) (53 mg) e aducto de tris(dibenzilidenoacetona)paládio(0)- clorofórmio (46 mg), e a mistura foi agitada em 120°C por 2 dias. Além disso, diisopropilamina (0,631 ml), octanotiol (0,375 ml), 4,5- bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos) (53 mg) e aducto de tris(dibenzilidenoacetona)paládio(0)-clorofórmio (46 mg) foram adicionados à solução da reação, e a mistura foi agitada em 120°C por 1 dia. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila, lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida, e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 5:1 a 3:1) para dar éster t-butílico de ácido {2,2-dimetil-5-[2-(4-hexiltio-3-trifluorometilfenil)etil]-1,3-dioxan5-il}carbâmico como um sólido amarelo-pálido. O sólido amarelo-pálido obtido acima foi dissolvido em etanol (15 ml), ácido clorídrico concentrado (1,5 ml) foi adicionado, e a mistura foi agitada em 80°C por 2 horas. A mistura da reação foi concentrada e o resíduo foi lavado com éter dietílico para dar um sólido branco. O sólido branco foi purificado por HPLC do preparativo, éter contendo cloreto de hidrogênio (1 mol/l, 15 ml) foi adicionado ao resíduo obtido para dar um sal de cloridrato. O precipitado foi coletado por filtração, e secado para dar o produto-fim (132 mg) como um pó branco.
[000421] MS (ESI) m/z: 380 [M+H]
[000422] 1H-RMN (DMSO-d6) δ (ppm): 0,85 (3H, t, J = 6,6 Hz), 1,20 - 1,31 (4H, m), 1,32 - 1,43 (2H, m), 1,51 - 1,60 (2H, m), 1,75 - 1,81 (2H, m), 2,63 - 2,69 (2H, m), 3,03 (2H, t, J = 7,2 Hz), 3,52 (4H, d, J = 5,0 Hz), 5,41 (2H, t, J = 5,1 Hz), 7,47 (1H, d, J = 8,7 Hz), 7,57 - 7,59 (2H, m), 7,84 (3H, brs).
[000423] (E)-2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)vinil] propano-1,3-diol
[000424] A uma suspensão de t-butóxido de potássio (20,7 g) em N,Ndimetilformamida (120 ml) foi adicionado n-heptanol (15,6 ml), e a mistura foi agitada em temperatura ambiente por 30 min. Uma solução de ácido 4-flúor-3-trifluorometilbenzóico (16,7 g) em N,N-dimetilformamida (60 ml) foi adicionada gota a gota à mistura da reação em 0°C, e a mistura foi agitada em 70°C por 1 hora. A mistura da reação foi resfriada, água (320 ml) foi adicionado, e ácido clorídrico a 6 M (40 ml) foi adicionado em temperatura ambiente. A mistura foi agitada em temperatura ambiente por 30 min e cristais precipitados foram coletados por filtração. Os cristais foram dissolvidos em etanol (60 ml) a 70°C, água (96 ml) foi adicionada gota a gota na mesma temperatura e a mistura foi agitada por 30 min. A mistura foi deixada para esfriar até a temperatura ambiente, e agitada por 30 min sob esfriamento com gelo. Os cristais precipitados foram coletados por filtração para dar o produtofim (24,1 g) como cristais marrons pálidos.
[000425] 1H-RMN (CDCl3)δ (ppm): 0,90 (3H, t, J = 6,6 Hz), 1,28 - 1,49 (8H, m), 1,80 - 1,90 (2H, m), 4,13 (2H, t, J = 6,3 Hz), 7,04 (1H, d, J = 8,7 Hz), 8,24 (1H, dd, J = 2,1 Hz, 9,0 Hz), 8,33 (1H, d, J = 1,8 Hz).
[000426] A uma solução do composto 28-1 (30,0 g) em N,N-dimetilformamida (240 ml) foi adicionada gota a gota uma solução de hidreto de bis(2-metoxietóxi)alumínio/solução de tolueno (65 % em peso) (20,0 g) em tolueno (80 ml) a 0°C sob uma atmosfera de nitrogênio, e uma solução de hidreto de bis(2-metoxietóxi)alumínio/solução de tolueno (65 % em peso) (80,0 g) em tolueno (80 ml) foi adicionada gota a gota. A mistura foi agitada em temperatura ambiente por 2 horas e resfriada. Solução aquosa a 5N de hidróxido de sódio (200 ml) foi adicionada gota a gota, e a mistura foi agitada em temperatura ambiente por 30 min. A camada orgânica foi particionada e extraída, lavada com solução aquosa a 5N de hidróxido de sódio (100 ml), e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (28,3 g) como cristais brancos.
[000427] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,6 Hz), 1,26 - 1,56 (8H, m), 1,65 (1H, t, J = 5,7 Hz), 1,77 - 1,85 (2H, m), 4,04 (2H, t, J = 6,3 Hz), 4,65 (2H, d, J = 5,7 Hz), 6,97 (1H, d, J = 8,4 Hz), 7,47 (1H, dd, J = 1,5 Hz, 8,4 Hz), 7,57 (1H, d, J = 1,8 Hz).
[000428] A uma solução do composto 28-2 (26,8 g) em cloreto de metileno (107 ml) foram adicionadas várias gotas de N,N-dimetilformamida, e tionil cloreto (8,09 ml) foi adicionado gota a gota a 0°C. A mistura foi agitada na mesma temperatura por 2 horas, e água (50 ml) foi adicionada à mistura da reação. A camada orgânica foi particionada e extraída, lavada com água (50 ml) e bicarbonato de sódio saturado aquoso (70 ml), e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (28,3 g) como cristais brancos.
[000429] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,5 Hz), 1,26 - 1,54 (8H, m), 1,77 - 1,86 (2H, m), 4,04 (2H, t, J = 6,4 Hz), 4,56 (2H, s), 6,96 (1H, d, J = 8,6 Hz), 7,49 (1H, dd, J = 2,0 Hz, 8,5 Hz), 7,58 (1H, d, J = 1,9 Hz).
[000430] Uma solução do composto 28-3 (27,3 g) em fosfito de trietila (29,3 g) foi aquecida sob refluxo por 4 horas sob uma atmosfera de nitrogênio. O fosfito de trietila foi evaporado sob pressão reduzida para dar o produto-fim (36,1 g) como um óleo amarelo-pálido.
[000431] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,6 Hz), 1,23 - 1,54(14H, m), 1,77 - 1,86 (2H, m), 3,10 (2H, d, J = 21,3 Hz), 3,98 - 4,08 (4H, m), 6,93 (1H, d, J = 8,4 Hz), 7,42 (1H, dd, J = 2,4 Hz, 8,4 Hz), 7,45 (1H, d, J = 2,1 Hz).
[000432] A uma solução de t-butóxido de potássio (24,9 g) em tetraidrofurano (177 ml) foram adicionados gota a gota sob esfriamento com gelo uma solução do Exemplo de Referência composto 1-2 (28,8 g) e composto 28-4 (35,1 g) em tetraidrofurano (203 ml), e a mistura foi agitada em 0°C por 5 horas. Heptano (203 ml) e em seguida água (203 ml) foram adicionados à mistura da reação. A camada orgânica foi particionada e extraída, lavada com água, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi suspendido em hexano (50 ml) e coletado por filtração para dar o produto-fim (32,6 g) como cristais brancos.
[000433] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,6 Hz), 1,30 - 1,57 (8H, m), 1,44 (9H, s), 1,47 (3H, s), 1,49 (3H, s), 1,76 - 1,84 (2H, m), 3,90 (2H, d, J = 11,4 Hz), 3,94 (2H, d, J = 13,8 Hz), 4,03 (2H, t, J = 6,3 Hz), 5,21 (1H, brs), 6,10 (1H, d, J = 16,5 Hz), 6,48 (1H, d, J = 16,5 Hz), 6,91 (1H, d, J = 8,4 Hz), 7,43 - 7,46 (1H, m), 7,55 (1H, d, J = 1,8 Hz).
[000434] O composto 28-5 (500 mg) foi dissolvido em etanol (15 ml), ácido clorídrico concentrado (1,5 ml) foi adicionado, e a mistura foi agitada em 80°C por 1,5 hora. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (330 mg) como um pó branco.
[000435] MS (ESI) m/z: 359
[000436] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,25 - 1,39 (6H, m), 1,40 - 1,46 (2H, m), 1,69 - 1,77 (2H, m), 3,59 - 3,67 (4H, m), 4,11 (2H, t, J = 6,2 Hz), 5,48 (2H, t, J = 5,3 Hz), 6,24 (1H, d, J = 16,8 Hz), 6,71 (1H, d, J = 16,8 Hz), 7,27 (1H, d, J = 9,3 Hz), 7,25 - 7,28 (2H, m), 8,12 (3H, brs).
[000437] (E)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2-(fosforiloximetil)-3-buten-1-ol
[000438] A uma solução do composto 28-6 (280 mg) em N,N-dimetilformamida (10 ml) foram adicionados N,N-diisopropiletilamina (0,366 ml) e trimetil ortoacetato (0,129 ml), e a mistura foi agitada em 120°C por 5 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 270 mg de um óleo marrom. A uma solução do óleo marrom (270 mg) em cloreto de metileno (5 ml) e acetonitrila (2 ml) foram adicionados 1H-tetrazol (95 mg) e dietilfosforamidita de di-t-butila (0,407 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, uma solução de decano contendo hidroperóxido de t-butila (5-6M, 0,408 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 30 min. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:3 - acetato de etila somente) para dar o produto-fim (200 mg) como um óleo amarelo.
[000439] 1H-RMN (CD3OD) δ (ppm): 0,90 (3H, t, J = 6,9 Hz), 1,29 - 1,45 (6H, m), 1,47 - 1,53 (2H, m), 1,49 (9H, s), 1,50 (9H, s), 1,75 - 1,83 (2H, m), 2,05 (3H, s), 3,94 - 3,98 (1H, m), 4,00 - 4,05 (1H, m), 4,08 (2H, t, J = 6,2 Hz), 4,23 (1H, d, J = 8,7 Hz), 4,50 (1H, d, J = 8,7 Hz), 6,30 (1H, d, J = 16,2 Hz), 6,64 (1H, d, J = 16,2 Hz), 7,11 (1H, d, J = 8,4 Hz), 7,59 - 7,62 (2H, m).
[000440] O composto 29-1 (200 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 3 horas. O solvente foi concentrado sob pressão reduzida, e metanol (3 ml), éter dietílico (3 ml) e óxido de propileno (5 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração, e lavado com acetato de etila e éter dietílico para dar o produto-fim (45 mg) como um sólido branco.
[000441] MS (ESI) m/z: 456 [M+H]
[000442] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,9 Hz), 1,27 - 1,47 (6H, m), 1,45 - 1,54 (2H, m), 1,76 - 1,83 (2H, m), 3,75 (1H, d, J = 11,6 Hz), 3,83 (1H, d, J = 11,6 Hz), 3,98 - 4,04 (1H, m), 4,07 - 4,15 (1H, m), 4,09 (2H, t, J = 6,3 Hz), 6,22 (1H, d, J = 16,7 Hz), 6,76 (1H, d, J = 16,7 Hz), 7,14 (1H, d, J = 9,3 Hz), 7,65 - 7,67 (2H, m).
[000443] Cloridrato de 2-amino-2-[2-(3-difluorometil-4-heptiloxifenil) etil]propano-1,3-diol
[000444] A uma suspensão de 5-bromossalicilaldeído (25,0 g) e carbonato de potássio (51,4 g) em N,N-dimetilformamida (250 ml) foi adicionado brometo de benzila (15,4 ml) sob esfriamento com gelo, e a mistura foi agitada por 40 min sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 15 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado sucessivamente com 0,1 M de solução aquosa de hidróxido de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi suspendido em hexano (200 ml) e coletado por filtração para dar o produto-fim (32.7 g) como um pó amarelo-pálido.
[000445] 1H-RMN (CDCl3) δ (ppm): 5,19 (2H, s), 6,95 (1H, d, J = 8,8 Hz), 7,34 - 7,43 (5H, m), 7,61 (1H, dd, J = 2,8, 8,8 Hz), 7,95 (1H, d, J = 2,8 Hz), 10,46 (1H, s).
[000446] A uma solução do composto 30-1 (2,70 g) em cloreto de metileno (5 ml) foi adicionada uma solução de trifluoreto de (dietilamino)enxofre (DAST, 1,66 g) em cloreto de metileno (5 ml), e a mistura foi agitada em temperatura ambiente por 21 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com cloreto de metileno. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida e o resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (2,16 g) como um óleo incolor.
[000447] 1H-RMN (CDCl3) δ (ppm): 5,11 (2H, s), 6,86 (1H, d, J = 9,1 Hz), 6,95 (1H, t, J = 55,3 Hz), 7,33 - 7,42 (5H, m), 7,49 (1H, dd, J = 1,6, 9,8 Hz), 7,69 (1H, d, J = 1,9 Hz).
[000448] O composto 30-2 (9,48 g), éster t-butílico de ácido (2,2- dimetil-5-etinil-1,3-dioxan-5-il)carbâmico (7,34 g) sintetizado por um método conhecido (por exemplo, Tetrahedron vol.57 (2001) 6531-6538), 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila (868 mg), dicloreto de bis(acetonitrila)paládio(II) (157 mg) e carbonato de césio (25.6 g) em acetonitrila (200 ml) foram agitados a 80°C por 8 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com salmoura saturada, e secado sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílicagel para dar o produto-fim (11,1 g) como um óleo marrom-pálido.
[000449] 1H-RMN (CDCl3) δ (ppm): 1,45 (3H, s), 1,48 (9H, s), 1,50 (3H, s), 4,03 (2H, d, J = 11,2 Hz), 4,10 (2H, d, J = 11,2 Hz), 5,13 (2H, s), 5,20 (1H, brs), 6,91 (1H, d, J = 8,6 Hz), 6,94 (1H, t, J = 55,4 Hz), 7,33 - 7,40 (5H, m), 7,46 (1H, d, J = 8,6 Hz), 7,65 (1H, s).
[000450] O composto 30-3 (11,1 g) foi dissolvido em 1,4-dioxano (250 ml), 10% de paládio carbono (3,5 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 3,5 horas sob uma atmosfera de hidrogênio. O interior do recipiente da reação foi deslocado com nitrogênio, a solução foi filtrada, e o filtrado foi concentrado. O resíduo foi suspendido em uma solução mista de éter diisopropílico e hexano, e coletado por filtração para dar o produto-fim (8,17 g) como um pó branco.
[000451] 1H-RMN (CDCl3) δ (ppm): 1,43 (3H, s), 1,44 (3H, s), 1,48 (9H, s), 1,92 - 1,96 (2H, m), 2,50 - 2,54 (2H, m), 3,69 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 5,03 (1H, brs), 5,57 (1H, brs), 6,77 (1H, d, J = 8,4 Hz), 6,84 (1H, t, J = 55,5 Hz), 7,12 (1H, d, J = 8,4 Hz), 7,23 (1H, s).
[000452] O composto 30-4 (500 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (516 mg) e brometo de nheptila (0,240 ml) foram adicionados, e a mistura foi agitada em temperatura ambiente por 15 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 620 mg de um óleo incolor. O óleo incolor foi dissolvido em etanol (15 ml), ácido clorídrico concentrado (1,5 ml) foi adicionado, e a mistura foi agitada em 80°C por 1 hora. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar um pó branco. O pó branco foi purificado por HPLC do preparativo, éter contendo cloreto de hidrogênio (1 mol/l, 15 ml) foi adicionado ao resíduo obtido para dar cloridrato. O precipitado foi coletado por filtração, e secado para dar o produto-fim (160 mg) como um pó branco.
[000453] MS (ESI) m/z: 360 [M+H]
[000454] 1H-RMN (DMSO-d6) δ (ppm): 0,87 (3H, t, J = 6,8 Hz), 1,25 - 1,37 (6H, m), 1,38 - 1,45 (2H, m), 1,68 - 1,79 (4H, m), 2,56 - 2,62 (2H, m), 3,52 (4H, d, J = 4,0 Hz), 4,02 (2H, t, J = 6,4 Hz), 5,40 (2H, t, J = 4,5 Hz), 7,05 (1H, t, J = 55,4 Hz), 7,07 (1H, d, J = 8,6 Hz), 7,32 (1H, d, J = 8,6 Hz), 7,36 (1H, s), 7,80 (3H, brs).
[000455] 2-Amino-4-(3-difluorometil-4-heptiloxifenil)-2-(fosforiloxi-metil)butanol
[000456] A uma solução do composto 30-5 (115 mg) em N,N-dimetilformamida (5 ml) foram adicionados N,N-diisopropiletilamina (0,156 ml) e trimetil ortoacetato (0,055 ml), e a mistura foi agitada em 120°C por 12,5 horas. À mistura da reação foram adicionados N,Ndiisopropiletilamina (0,156 ml) e trimetil ortoacetato (0,055 ml) novamente, e a mistura foi agitada em 120°C por 3,5 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 140 mg de um óleo marrom. A uma solução do óleo marrom (140 mg) em cloreto de metileno (3 ml) e acetonitrila (1 ml) foram adicionados 1H-tetrazol (41 mg) e dietilfosforamidita de di-tbutila (0,174 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, uma solução de decano contendo hidroperóxido de t-butila (5-6M, 0,174 ml) foi adicionada, e a mistura foi agitada em temperatura ambiente por 20 min. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:4 - acetato de etila somente) para dar o produto-fim (110 mg) como um óleo amarelo.
[000457] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,7 Hz), 1,30 - 1,42 (6H, m), 1,43 - 1,53 (2H, m), 1,48 (18H, 2×s), 1,70 - 1,90 (4H, m), 2,01 (3H, s), 2,51 - 2,69 (2H, m), 3,87 - 3,92 (2H, m), 4,02 (2H, t, J = 6,4 Hz), 4,17 (1H, d, J = 9,0 Hz), 4,32 (1H, d, J = 9,0 Hz), 6,91 (1H, t, J = 55,8 Hz), 6,97 (1H, d, J = 8,5 Hz), 7,29 (1H, d, J = 8,5 Hz), 7,36 (1H, s).
[000458] O composto 31-1 (110 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 4 horas. O solvente foi concentrado sob pressão reduzida, metanol (1 ml), éter dietílico (1 ml) e óxido de propileno (2 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração, e lavado com acetato de etila e éter dietílico para dar o produto-fim (60 mg) como um sólido amarelo-pálido.
[000459] MS (ESI) m/z: 440 [M+H]
[000460] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,8 Hz), 1,26 - 1,43 (6H, m), 1,44 - 1,52 (2H, m), 1,74 - 1,82 (2H, m), 1,92 - 2,00 (2H, m), 2,59 - 2,71 (2H, m), 3,70 (2H, brs), 3,91 - 4,04 (4H, m), 6,92 (1H, t, J = 55,8 Hz), 6,98 (1H, d, J = 8,5 Hz), 7,33 (1H, d, J = 8,3 Hz), 7,40 (1H, brs).
[000461] Éster t-butílico de ácido 2-amino-2-[2-(3-difluorometil-4- octiloxifenil)etil]propano-1,3-diol cloridrato
[000462] O composto 30-4 (600 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (412 mg) e 1-bromooctano (0,311 ml) foram adicionados, e a mistura foi agitada em temperatura ambiente por 2 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida, e o resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 50:1 a 3:1) para dar o produto-fim (230 mg) como um óleo incolor.
[000463] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,25 - 1,49 (8H, m), 1,40 - 1,50 (2H, m), 1,42 (3H, s), 1,44 (3H, s), 1,47 (9H, s), 1,75 - 1,81 (2H, m), 1,91 - 1,98 (2H, m), 2,50 - 2,57 (2H, m), 3,68 (2H, d, J = 11,8 Hz), 3,89 (2H, d, J = 11,7 Hz), 3,97 (2H, t, J = 6,4 Hz), 4,98 (1H, brs), 6,82 (1H, d, J = 8,4 Hz), 6,93 (1H, t, J = 55,8 Hz), 7,21 (1H, d, J = 8,0 Hz), 7,35 (1H, brs).
[000464] O composto 32-1 (230 mg) foi dissolvido em etanol (10 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 80°C por 2 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar o produto-fim (105 mg) como um pó branco.
[000465] MS (ESI) m/z: 374 [M+H]
[000466] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,24 - 1,38 (8H, m), 1,39 - 1,44 (2H, m), 1,68 - 1,80 (4H, m), 2,55 - 2,62 (2H, m), 3,52 (4H, d, J = 4,6 Hz), 4,01 (2H, t, J = 6,4 Hz), 5,39 (2H, brt, J = 4,8 Hz), 7,05 (1H, t, J = 55,4 Hz), 7,07 (1H, d, J = 8,5 Hz), 7,32 (1H, d, J = 8,5 Hz), 7,36 (1H, s), 7,79 (3H, brs).
[000467] 2-Amino-4-(3-difluorometil-4-octiloxifenil)-2-(fosforiloximetil)butanol
[000468] A uma solução do composto 32-2 (226 mg) em N,N-dimetilformamida (10 ml), N,N-diisopropiletilamina (0,296 ml) e trimetil ortoacetato (0,139 ml) foram adicionados, e a mistura foi agitada em 120°C por 2,5 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 220 mg de um óleo marrom. A uma solução do óleo marrom (220 mg) em cloreto de metileno (5 ml) e acetonitrila (2 ml) foram adicionados 1H-tetrazol (77 mg) e dietilfosforamidita de di-t-butila (0,329 ml), e a mistura foi agitada em temperatura ambiente por 2 horas. A solução da reação foi resfriada, uma solução de decano contendo hidroperóxido de t-butila (5-6M, 0,330 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 20 min. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com clorofórmio. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de coluna de sílica-gel (hexano: acetato de etila = 1:4 - acetato de etila somente) para dar o produto-fim (280 mg) como um óleo amarelo.
[000469] 1H-RMN (CD3OD) δ (ppm): 0,90 (3H, t, J = 7,0 Hz), 1,28 - 1,41 (8H, m), 1,45 - 1,52 (2H, m), 1,48(18H, 2×s), 1,75 - 1,90 (4H, m), 2,01 (3H, s), 2,51 - 2,70 (2H, m), 3,87 - 3,92 (2H, m), 4,02 (2H, t, J = 6,3 Hz), 4,17 (1H, d, J = 9,0 Hz), 4,32 (1H, d, J = 9,0 Hz), 6,92 (1H, t, J = 55,7 Hz), 6,97 (1H, d, J = 8,4 Hz), 7,29 (1H, d, J = 8,9 Hz), 7,36 (1H, brs).
[000470] O composto 33-1 (280 mg) foi dissolvido em etanol (5 ml), ácido clorídrico concentrado (1 ml) foi adicionado, e a mistura foi agitada em 50°C por 3,5 horas. O solvente foi concentrado sob pressão reduzida, metanol (2 ml), éter dietílico (2 ml) e óxido de propileno (5 ml) foram adicionados ao resíduo. O pó precipitado foi coletado por filtração, e lavado com metanol e éter dietílico para dar o produto-fim (175 mg) como um sólido amarelo-pálido.
[000471] MS (ESI) m/z: 454 [M+H]
[000472] 1H-RMN (CD3OD) δ (ppm): 0,90 (3H, t, J = 6,9 Hz), 1,26 - 1,42 (8H, m), 1,44 - 1,52 (2H, m), 1,75 - 1,82 (2H, m), 1,92 - 1,99 (2H, m), 2,62 - 2,72 (2H, m), 3,67 - 3,74 (2H, m), 3,94 - 4,04 (4H, m), 6,92 (1H, t, J = 55,8 Hz), 6,98 (1H, d, J = 8,5 Hz), 7,33 (1H, d, J = 8,5 Hz), 7,40 (1H, brs).
[000473] Cloridrato de 2-amino-2-[2-(3-fluorometil-4-heptiloxifenil) etil]propano-1,3-diol
[000474] A uma suspensão de 5-bromossalicilaldeído (5,00 g) e carbonato de potássio (10,3 g) em N,N-dimetilformamida (50 ml) foi adicionado heptil brometo (4,10 ml), e a mistura foi agitada em temperatura ambiente por 1,5 hora, e adicionalmente a 50°C por 5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com 1 M de solução aquosa de hidróxido de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (7,61 g) como um óleo amarelo.
[000475] 1H-RMN (CDCl3) δ (ppm): 0,90 (3H, t, J = 6,8 Hz), 1,28 - 1,40 (6H, m), 1,44 - 1,51 (2H, m), 1,85 (2H, quint, J = 7,0 Hz), 4,06 (2H, t, J = 6,5 Hz), 6,88 (1H, d, J = 8,9 Hz), 7,60 (1H, dd, J = 2,8, 8,9 Hz), 7,92 (1H, d, J = 2,8 Hz), 10,42 (1H, s).
[000476] A uma solução do composto 34-1 (7,60 g) em etanol (80 ml) foi adicionado boridreto de sódio (0,48 g) sob esfriamento com gelo, e a mistura foi agitada por 1,5 hora sob esfriamento com gelo. 1 M de ácido clorídrico (50 ml) foi adicionada à mistura da reação, e etanol foi evaporado sob pressão reduzida. O resíduo obtido foi diluído com água, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (7,66 g) como um óleo amarelo-pálido.
[000477] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,28 - 1,39 (6H, m), 1,41 - 1,48 (2H, m), 1,80 (2H, quint, J = 7,0 Hz), 2,26 (1H, t, J = 6,3 Hz), 3,98 (2H, t, J = 6,5 Hz), 4,65 (2H, d, J = 5,9 Hz), 6,74 (1H, d, J = 8,6 Hz), 7,34 (1H, dd, J = 2,4, 8,6 Hz), 7,41 (1H, d, J = 2,4 Hz).
[000478] A uma solução do composto 34-2 (7,66 g) e imidazol (4,32 g) em N,N-dimetilformamida (30 ml) foi adicionado t-butildimetilclorossilano (4,59 g), e a mistura foi agitada por 14 horas. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (10,6 g) como um óleo amarelo.
[000479] 1H-RMN (CDCl3) δ (ppm): 0,12 (6H, s), 0,89 (3H, t, J = 6,8 Hz), 0,96 (9H, s), 1,28 - 1,38 (6H, m), 1,40 - 1,47 (2H, m), 1,77 (2H, quint, J = 7,0 Hz), 3,92 (2H, t, J = 6,5 Hz), 4,71 (2H, s), 6,67 (1H, d, J = 8,7 Hz), 7,27 (1H, dd, J = 2,4, 8,7 Hz), 7,55 (1H, d, J = 2,4 Hz).
[000480] O composto 34-3 (10,5 g), éster t-butílico de ácido (2,2- dimetil-5-etinil-1,3-dioxan-5-il)carbâmico (6,46 g) sintetizado por um método conhecido (por exemplo, Tetrahedron vol.57 (2001) 6531-6538), 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila(725 mg), dicloreto de bis(acetonitrila)paládio(II) (131 mg) e carbonato de césio (21,4 g) em acetonitrila (150 ml) foi agitado a 80°C por 12 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com salmoura saturada, e secado sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (11,7 g) como um óleo marrom.
[000481] 1H-RMN (CDCl3) δ (ppm): 0,11 (6H, s), 0,89 (3H, t, J = 6,8 Hz), 0,95 (9H, s), 1,28 - 1,38 (6H, m), 1,40 - 1,47 (2H, m), 1,45 (3H, s), 1,48 (9H, s), 1,50 (3H, s), 1,77 (2H, quint, J = 6,9 Hz), 3,95 (2H, t, J = 6,4 Hz), 4,03 (2H, d, J = 11,5 Hz), 4,09 (2H, d, J = 11,5 Hz), 4,69 (2H, s), 5,19 (1H, brs), 6,71 (1H, d, J = 8,5 Hz), 7,27 (1H, dd, J = 1,9, 8,5 Hz), 7,49 (1H, s).
[000482] O composto 34-4 (11,7 g) foi dissolvido em 1,4-dioxano (150 ml), 10% de paládio carbono (12,0 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 16 horas sob uma atmosfera de hidrogênio. O interior do recipiente da reação foi deslocado com nitrogênio, a solução foi filtrada, e o filtrado foi concentrado. A uma solução do resíduo obtido em tetraidrofurano (100 ml) foi adicionado 1 M de solução de fluoreto de tetrabutilamônio-tetraidrofurano (20 ml) sob esfriamento com gelo, e a mistura foi agitada por 1,5 hora sob esfriamento com gelo. Uma solução de 1 M de solução de fluoreto de tetrabutilamônio- tetraidrofurano (10 ml) foi adicionado, e a mistura foi adicionalmente agitada por 4 horas sob esfriamento com gelo. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, lavada sucessivamente com água salmoura saturada, e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílicagel para dar o produto-fim (5,99 g) como um óleo marrom-pálido.
[000483] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,8 Hz), 1,28 - 1,39 (6H, m), 1,42 - 1,47 (2H, m), 1,42 (3H, s), 1,43 (3H, s), 1,47 (9H, s), 1,79 (2H, quint, J = 7,0 Hz), 1,92 - 1,96 (2H, m), 2,40 (1H, t, J = 6,5 Hz), 2,48 - 2,53 (2H, m), 3,67 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 3,98 (2H, t, J = 6,5 Hz), 4,65 (2H, d, J = 6,8 Hz), 4,97 (1H, brs), 6,77 (1H, d, J = 8,0 Hz), 7,04 - 7,07 (2H, m).
[000484] Uma mistura do composto 34-5 (3,74 g), fluoreto de ptoluenossulfonila (4,08 g), peneiras moleculares de 4A (3,74 g) e solução a 1M de fluoreto de tetrabutilamônio-tetraidrofurano (46,8 ml) foi agitada por 12 horas sob refluxo. Celite foi adicionada à mistura da reação, e a mistura foi filtratada. Acetato de etila (200 ml) e água (200 ml) foram adicionados ao filtrado, e a mistura foi filtratada com celite. A camada orgânica do filtrado foi lavada com salmoura saturada e secada sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílicagel para dar o produto-fim (0,92 g) como um óleo incolor.
[000485] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,26 - 1,38 (6H, m), 1,40 - 1,47 (2H, m), 1,42 (3H, s), 1,43 (3H, s), 1,47 (9H, s), 1,77 (2H, quint, J = 6,9 Hz), 1,93 - 1,97 (2H, m), 2,50 - 2,54 (2H, m), 3,68 (2H, d, J = 11,7 Hz), 3,88 (2H, d, J = 11,7 Hz), 3,95 (2H, t, J = 6,4 Hz), 4,98 (1H, brs), 5,42 (2H, d, J = 47,9 Hz), 6,78 (1H, d, J = 8,5 Hz), 7,11 (1H, d, J = 8,5 Hz), 7,17 (1H, m).
[000486] O composto 34-6 (0,92 g) foi dissolvido em metanol (30 ml), ácido p-toluenossulfônico (15 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 3 horas. Solução aquosa saturada de bicarbonato de sódio (10 ml) e salmoura saturada (100 ml) foram adicionadas à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com salmoura saturada, e secado sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. Acetato de etila (1 ml) e em solução a 4 M de cloreto de hidrogênio- acetato de etila (1 ml) foram adicionados ao resíduo obtido sob esfriamento com gelo, e a mistura foi agitada por 30 min sob esfriamento com gelo. O sólido precipitado foi coletado por filtração, e lavado com éter diisopropílico para dar o produto-fim (44,4 mg) como um pó branco.
[000487] MS (ESI) m/z: 342 [M+H]
[000488] 1H-RMN (DMSO-d6) δ (ppm): 0,87 (3H, t, J = 6,7 Hz), 1,26 - 1,36 (6H, m), 1,37 - 1,44 (2H, m), 1,71 (2H, quint, J = 6,9 Hz), 1,74 - 1,79 (2H, m), 2,53 - 2,57 (2H, m), 3,52 (4H, d, J = 5,2 Hz), 3,97 (2H, t, J = 6,3 Hz), 5,38 (2H, d, J = 48,0 Hz), 5,40 (2H, t, J = 5,1 Hz), 6,97 (1H, d, J = 8,3 Hz), 7,17 - 7,20 (2H, m), 7,85 (3H, brs).
[000489] Cloridrato de 2-amino-2-[2-(3-fluorometil-4-heptiloxifenil) etil]propano-1,3-diol
[000490] A uma suspensão de 5-bromossalicilaldeído (5,00 g) e carbonato de potássio (10,3 g) em N,N-dimetilformamida (50 ml) foi adicionado brometo de octila (4,52 ml), e a mistura foi agitada em temperatura ambiente por 1,5 hora, e adicionalmente a 50°C por 3,5 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada 3 vezes, e secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida para dar o produto-fim (7,72 g) como um sólido branco.
[000491] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,3 Hz), 1,25 - 1,38 (8H, m), 1,44 - 1,51 (2H, m), 1,84 (2H, quint, J = 6,8 Hz), 4,06 (2H, t, J = 6,4 Hz), 6,88 (1H, d, J = 8,9 Hz), 7,61 (1H, d, J = 8,9 Hz), 7,92 (1H, s), 10,42 (1H, s).
[000492] A uma solução do composto 35-1 (7,72 g) em etanol (80 ml) foi adicionado boridreto de sódio (0,47 g) sob esfriamento com gelo, e a mistura foi agitada por 30 min sob esfriamento com gelo. Água (100 ml) e 1 M de ácido clorídrico (30 ml) foram adicionados à mistura da reação, e etanol foi evaporado sob pressão reduzida. O resíduo obtido foi diluído com 0,1 M de ácido clorídrico, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida para dar o produto-fim (7,95 g) como um óleo marrompálido.
[000493] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,25 - 1,38 (8H, m), 1,41 - 1,48 (2H, m), 1,80 (2H, quint, J = 7,0 Hz), 2,25 (1H, t, J = 6,3 Hz), 3,98 (2H, t, J = 6,4 Hz), 4,65 (2H, d, J = 6,0 Hz), 6,74 (1H, d, J = 8,6 Hz), 7,34 (1H, dd, J = 2,4, 8,6 Hz), 7,41 (1H, d, J = 2,4 Hz).
[000494] A uma solução do composto 35-2 (7,95 g) e imidazol (4,19 g) em N,N-dimetilformamida (35 ml) foi adicionado t-butildimetilclorossilano (4,45 g) sob esfriamento com gelo, e a mistura foi agitada por 20 min sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 18 horas. Solução aquosa saturada de bicarbonato de sódio foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (11,1 g) como um óleo marrom-pálido.
[000495] 1H-RMN (CDCl3) δ (ppm): 0,12 (6H, s), 0,89 (3H, t, J = 6,9 Hz), 0,96 (9H, s), 1,24 - 1,38 (8H, m), 1,40 - 1,47 (2H, m), 1,76 (2H, quint, J = 7,0 Hz), 3,92 (2H, t, J = 6,5 Hz), 4,71 (2H, s), 6,67 (1H, d, J = 8,7 Hz), 7,27 (1H, dd, J = 2,5, 8,7 Hz), 7,55 (1H, d, J = 2,5 Hz).
[000496] O composto 35-3 (11,1 g), éster t-butílico de ácido (2,2- dimetil-5-etinil-1,3-dioxan-5-il)carbâmico (6,28 g) sintetizado por um método conhecido (por exemplo, Tetrahedron vol.57 (2001) 6531-6538), 2-diciclohexilfosfino-2',4',6'-triisopropilbifenila(706 mg), dicloreto de bis(acetonitrila)paládio(II) (128 mg) e carbonato de césio (20,8 g) em acetonitrila (150 ml) foram agitados a 80°C por 10 horas. Salmoura foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com salmoura saturada, e secado sobre sulfato de sódio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílicagel para dar o produto-fim (12,4 g) como um óleo marrom.
[000497] 1H-RMN (CDCl3) δ (ppm): 0,11 (6H, s), 0,89 (3H, t, J = 6,8 Hz), 0,95 (9H, s), 1,24 - 1,37 (8H, m), 1,40 - 1,47 (2H, m), 1,45 (3H, s), 1,48 (9H, s), 1,50 (3H, s), 1,77 (2H, quint, J = 6,9 Hz), 3,95 (2H, t, J = 6,5 Hz), 4,03 (2H, d, J = 11,6 Hz), 4,09 (2H, d, J = 11,6 Hz), 4,69 (2H, s), 5,19 (1H, brs), 6,71 (1H, d, J = 8,4 Hz), 7,27 (1H, dd, J = 1,9, 8,4 Hz), 7,49 (1H, s).
[000498] O composto 35-4 (12,4 g) foi dissolvido em 1,4-dioxano (100 ml), 10% de paládio carbono (3,0 g) foi adicionado, e a mistura foi agitada em temperatura ambiente por 5,5 horas sob uma atmosfera de hidrogênio. O interior do recipiente da reação foi deslocado com nitrogênio, a solução foi filtrada, e o filtrado foi concentrado. A uma solução do resíduo obtido em tetraidrofurano (100 ml) foi adicionado solução a 1 M de fluoreto de tetrabutilamônio-tetraidrofurano (30 ml), e a mistura foi agitada por 3 horas. A mistura da reação foi diluída com acetato de etila (200 ml), e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (9,61 g) como um óleo marrom-pálido.
[000499] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,24 - 1,38 (8H, m), 1,41 - 1,47 (2H, m), 1,42 (3H, s), 1,43 (3H, s), 1,47 (9H, s), 1,79 (2H, quint, J = 7,0 Hz), 1,92 - 1,96 (2H, m), 2,41(1Ht, J = 6,6 Hz), 2,48 - 2,53 (2H, m), 3,67 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 3,98 (2H, t, J = 6,5 Hz), 4,65 (2H, d, J = 6,8 Hz), 4,98 (1H, brs), 6,77 (1H, d, J = 8,0 Hz), 7,04 - 7,07 (2H, m).
[000500] Uma mistura do composto 35-5 (9,61 g), fluoreto de ptoluenossulfonila (10,2 g), peneiras moleculares de 4A (9,61 g) e solução a 1 M de fluoreto de tetrabutilamônio-tetraidrofurano (117 ml) foi agitada por 13 horas sob refluxo. A mistura da reação foi adicionada a uma mistura de água e acetato de etila, e a mistura foi agitada por 5 horas. A camada orgânica foi lavada com água e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna de sílica-gel para dar o produto-fim (1,91 g) como um óleo incolor.
[000501] 1H-RMN (CDCl3) δ (ppm): 0,89 (3H, t, J = 6,7 Hz), 1,24 - 1,38 (8H, m), 1,40 - 1,47 (2H, m), 1,42 (3H, s), 1,43 (3H, s), 1,47 (9H, s), 1,77 (2H, quint, J = 7,0 Hz), 1,93 - 1,97 (2H, m), 2,50 - 2,55 (2H, m), 3,68 (2H, d, J = 11,7 Hz), 3,89 (2H, d, J = 11,7 Hz), 3,95 (2H, t, J = 6,5 Hz), 4,98 (1H, brs), 5,42 (2H, d, J = 47,9 Hz), 6,78 (1H, d, J = 8,4 Hz), 7,11 (1H, d, J = 8,4 Hz), 7,17 (1H, s).
[000502] O composto 35-6 (1,91 g) foi dissolvido em metanol (60 ml), monoidrato de ácido p-toluenossulfônico (20 mg) foi adicionado, e a mistura foi agitada em temperatura ambiente por 9 horas. Solução aquosa saturada de bicarbonato de sódio (200 ml) e salmoura saturada (100 ml) foram adicionadas à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de sódio anídrico, e o solvente foi evaporado sob pressão reduzida. Acetato de etila (3 ml) e solução a 4 M de cloreto de hidrogênio-acetato de etila (3 ml) foram adicionados ao resíduo obtido sob esfriamento com gelo, e a mistura foi agitada por 40 min sob esfriamento com gelo. O sólido precipitado foi coletado por filtração, e lavado com acetato de etila para dar o produto-fim (158 mg) como um pó branco.
[000503] MS (ESI) m/z: 356 [M+H]
[000504] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,8 Hz), 1,23 - 1,35 (8H, m), 1,37 - 1,44 (2H, m), 1,67 - 1,78 (4H, m), 2,52 - 2,56 (2H, m), 3,51 (4H, d, J = 4,6 Hz), 3,97 (2H, t, J = 6,4 Hz), 5,38 (2H, d, J = 48,0 Hz), 5,38 (2H, brs), 6,97 (1H, d, J = 8,3 Hz), 7,17 - 7,20 (2H, m), 7,75 (3H, brs).
[000505] Cloridrato de 2-dimetilamino-2-[2-(4-heptilóxi-3-trifluorometilfenil)etil]propano-1,3-diol
[000506] A uma solução do composto 1-3 (1,24 g), 37% de formaldeído (20 ml) e solução aquosa a 30% de ácido acético (3 ml) em acetonitrila (30 ml) foi adicionado cianoboridreto de sódio (0,817 g) sob esfriamento com gelo, e a mistura foi agitada por 1 hora. Solução aquosa saturada de bicarbonato de sódio (50 ml) foi adicionada à mistura da reação, e acetonitrila foi evaporado sob pressão reduzida. Solução aquosa saturada de bicarbonato de sódio foi adicionada à concentração obtida, a mistura foi extraída com acetato de etila, e o extrato foi lavado com salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. Acetato de etila (10 ml) e solução a 4 M de cloreto de hidrogênio-acetato de etila (5 ml) foram adicionados ao resíduo, e o solvente foi evaporado sob pressão reduzida. Éter diisopropílico foi adicionado ao resíduo, e o sólido resultante foi coletado por filtração para dar o produto-fim (0,808 g) como um sólido branco.
[000507] MS (ESI) m/z: 406 [M+H]
[000508] 1H-RMN (DMSO-d6) δ (ppm): 0,86 (3H, t, J = 6,7 Hz), 1,24 - 1,35 (6H, m), 1,38 - 1,45 (2H, m), 1,71 (2H, quint, J = 6,8 Hz), 1,87 - 1,91 (2H, m), 2,60 - 2,67 (2H, m), 2,80 (6H, d, J = 4,8 Hz), 3,70 (2H, dd, J = 4,9, 12,9 Hz), 3,76 (2H, dd, J = 4,8, 12,8 Hz), 4,06 (2H, t, J = 6,2 Hz), 5,71 (2H, t, J = 4,6 Hz), 7,18 (1H, d, J = 8,2 Hz), 7,49 - 7,51 (2H, m).
[000509] Cloridrato de 2-amino-2-[2-(4-heptilóxi-3-metilfenil)etil] propano-1,3-diol
[000510] A uma solução de 4'-hidróxi-3'-metilacetofenona (25,0 g) em N,N-dimetilformamida (120 ml) foram adicionados carbonato de potássio (69,1 g) e metil iodeto (11,4 ml) sob esfriamento com gelo, e a mistura foi agitada por 2 horas sob esfriamento com gelo e adicionalmente em temperatura ambiente por 2 horas. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar o produto-fim (27,5 g) como um óleo amarelo.
[000511] 1H-RMN (CDCl3) δ (ppm): 2,25 (3H, s), 2,55 (3H, s), 3,89 (3H, s), 6,85 (1H, d, J = 8,6 Hz), 7,77 (1H, d, J = 1,7 Hz), 7,82 (1H, dd, J = 2,2, 8,6 Hz).
[000512] A uma solução do composto de comparação 1-1 (27,2 g) em ácido acético (170 ml) foi adicionado tribrometo de piridínio (90%, 59,0 g), e a mistura foi agitada em 50°C por 1 hora. Água foi adicionada à mistura da reação, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com água, solução aquosa a 1 M de hidróxido de sódio, cloreto de amônio saturado e salmoura saturada. A camada orgânica foi secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida para dar o produto-fim (40,3 g) como um sólido marrom.
[000513] 1H-RMN (CDCl3) δ (ppm): 2,26 (3H, s), 3,91 (3H, s), 4,40 (2H, s), 6,87 (1H, d, J = 8,6 Hz), 7,80 (1H, d, J = 1,5 Hz), 7,86 (1H, dd, J = 2,2, 8,6 Hz).
[000514] A uma solução de dietil 2-acetamidomalonate (29,1 g) em N,N-dimetilformamida (140 ml) foi adicionado hidreto de sódio (60%, 5,63 g) em quatro porções sob esfriamento com gelo, e a mistura foi agitada por 1 hora. A esta solução foi adicionada uma solução do composto de comparação 1-2 (39,1 g) em N,N-dimetilformamida (50 ml), e a mistura foi agitada por 3 horas sob esfriamento com gelo. A mistura da reação foi adicionada a água gelada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de sílica-gel para dar o produto-fim (45,0 g) como um óleo marrom.
[000515] 1H-RMN (CDCl3) δ (ppm): 1,24 (6H, t, J = 7,0 Hz), 1,96 (3H, s), 2,23 (3H, s), 3,89 (3H, s), 4,20 (2H, s), 4,26 (4H, dq, J = 1,4, 7,0 Hz), 6,84 (1H, d, J = 8,6 Hz), 7,10 (1H, brs), 7,77 (1H, d, J = 1,8 Hz), 7,83 (1H, dd, J = 2,2, 8,6 Hz).
[000516] A uma solução do composto de comparação 1-3 (45,0 g) em ácido trifluoroacético (260 ml) foi adicionado trietilsilano (133 ml), e a mistura foi agitada em 70°C por 24 horas. A mistura da reação foi concentrada sob pressão reduzida, água foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com água, solução aquosa a 1 M de hidróxido de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. Éter dietílico e hexano foram adicionados ao resíduo obtido, e o sólido precipitado foi coletado por filtração, e secado para dar o produto-fim (31.3 g) como um pó branco.
[000517] 1H-RMN (CDCl3) δ (ppm): 1,25 (6H, t, J = 7,0 Hz), 2,00 (3H, s), 2,18 (3H, s), 2,37 - 2,41 (2H, m), 2,62 - 2,67 (2H, m), 3,79 (3H, s), 4,15 - 4,27 (4H, m), 6,70 - 6,73 (1H, m), 6,75 (1H, brs), 6,90 - 6,93 (2H, m).
[000518] A uma solução do composto de comparação 1-4 (31,3 g) em etanol (300 ml) e água (60 ml) foi adicionado cloreto de cálcio (19,0 g), e o sólido foi dissolvido. Boridreto de sódio (13,0 g) foi adicionado a esta mistura em cinco porções sob esfriamento com gelo, e a mistura foi agitada por 3 horas sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 19 horas. Ácido clorídrico a 1 M (300 ml) foi adicionada à mistura da reação sob esfriamento com gelo, e a mistura foi concentrada sob pressão reduzida. Ácido clorídrico a 0,5 M (700 ml) foi adicionado, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada, e secada sobre sulfato de magnésio anídrico, e o solvente foi evaporado sob pressão reduzida para dar um sólido branco. O produto-fim (22,8 g) foi obtido como um óleo incolor por operação na mesma maneira mencionada acima para este sólido branco.
[000519] 1H-RMN (CDCl3) δ (ppm): 1,91 - 1,95 (2H, m), 1,95 (3H, s), 2,19 (3H, s), 2,55 - 2,59 (2H, m), 3,62 (2H, d, J = 11,6 Hz), 3,80 (3H, s), 3,87 (2H, d, J = 11,6 Hz), 5,84 (1H, brs), 6,74 (1H, d, J = 8,8 Hz), 6,97 - 6,98 (2H, m).
[000520] A uma solução do composto de comparação 1-5 (22,5 g) em cloreto de metileno (200 ml) foi adicionado gota a gota solução a 1 M de tribrometo de boro-cloreto de metileno (320 ml) a -70°C. A mistura foi agitada durante 1 hora até a temperatura aumentar para 0°C, e adicionalmente agitada por 1,5 hora sob esfriamento com gelo. Metanol (300 ml) foi gradualmente adicionada à mistura da reação sob esfriamento com gelo, e a mistura foi concentrada sob pressão reduzida. A uma solução do resíduo obtido em etanol (100 ml) foi adicionado ácido clorídrico concentrado (100 ml), e a mistura foi agitada em 80°C por 4 horas. A mistura da reação foi concentrada sob pressão reduzida. A uma solução do resíduo obtido e N,N-diisopropiletilamina (34,8 ml) em metanol (150 ml) foi adicionado dicarbonato de di-t-butila (19,2 g) sob esfriamento com gelo, e a mistura foi agitada por 1 hora sob esfriamento com gelo, e adicionalmente em temperatura ambiente por 4 horas. A mistura da reação foi concentrada sob pressão reduzida, solução aquosa saturada de bicarbonato de sódio (500 ml) foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada sucessivamente com solução aquosa saturada de bicarbonato de sódio e salmoura saturada, e secada sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida. O resíduo obtido foi lavado com éter dietílico para dar o produto-fim (15,1 g) como um pó branco.
[000521] 1H-RMN (CDCl3) δ (ppm): 1,45 (9H, s), 1,82 - 1,86 (2H, m), 2,21 (3H, s), 2,50 - 2,54 (2H, m), 3,39 (2H, brs), 3,64 (2H, dd, J = 6,8, 11,5 Hz), 3,88 (2H, dd, J = 5,5, 11,5 Hz), 4,83 (1H, brs), 4,99 (1H, brs), 6,68 (1H, d, J = 8,1 Hz), 6,88 (1H, dd, J = 1,9, 8,1 Hz), 6,94 (1H, d, J = 1,9 Hz).
[000522] O composto de comparação 1-6 (500 mg) foi dissolvido em N,N-dimetilformamida (10 ml), carbonato de potássio (425 mg) e brometo de n-heptila (0,296 ml) foram adicionados, e a mistura foi agitada em 80°C por 6 horas. Água foi adicionada à mistura da reação, a mistura foi extraída com acetato de etila, e o extrato foi lavado com água e salmoura saturada, e secado sobre sulfato de magnésio anídrico. O solvente foi evaporado sob pressão reduzida para dar 640 mg de um óleo incolor. O óleo incolor (640 mg) foi dissolvido em cloreto de metileno (5 ml), dioxano contendo cloreto de hidrogênio (4 mols/l, 5 ml) foi adicionado, e a mistura foi agitada em temperatura ambiente por 15 horas. A mistura da reação foi concentrada, e o resíduo foi lavado com éter dietílico para dar um pó branco. O pó branco foi purificado por HPLC do preparativo, éter contendo cloreto de hidrogênio (1 mol/l, 15 ml) foi adicionado ao resíduo obtido para dar cloridrato. O precipitado foi coletado por filtração, e secado para dar o produto-fim (320 mg) como um pó branco.
[000523] MS (ESI) m/z: 324 [M+H]
[000524] 1H-RMN (CD3OD) δ (ppm): 0,91 (3H, t, J = 6,6 Hz), 1,30 - 1,42 (6H, m), 1,43 - 1,52 (2H, m), 1,74 - 1,81 (2H, m), 1,88 - 1,94 (2H, m), 2,16 (3H, s), 2,53 - 2,58 (2H, m), 3,64 - 3,71 (4H, m), 3,94 (2H, t, J = 6,4 Hz), 6,77 (1H, d, J = 8,0 Hz), 6,96 - 6,98 (2H, m).
[000525] As estruturas dos compostos sintetizados são mostradas abaixo.

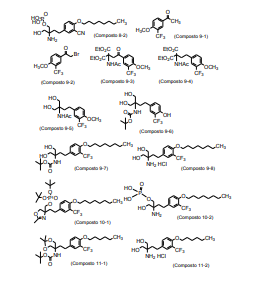


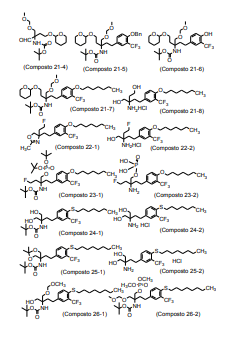


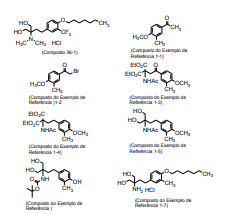
[000526] O composto da presente invenção foi dissolvido ou suspendido em 20% de ciclodextrina (fabricada pela NIHON SHOKUHIN KAKO CO., LTD), e administrado intraperitonealmente a alguns camundongos machos de 7 a 10 semanas de idade BALB/cAnNCrj (CHARLES RIVER LABORATORIES, JAPAN INC.) em uma dose de 0,001 a 10 mg/kg de peso corporal. Depois de 24 horas da administração do composto da presente invenção, foi colhido sangue periférico (cerca de 0,3 ml) da postcava do camundongo com uma seringa de tuberculina (fabricada pela TERUMO CORPORATION) tratada com heparina sódio (fabricada pela NovoNordisk) sob anestesia com éter. O sangue (0,1 ml) foi hemolisado com um equipamento de tratamento de hemólise automático (TQ-Prep, fabricado pela BECKMAN・COULTER), e o número de linfócitos foi contado com um Flow Cytometer (CYTOMICS FC 500, fabricado pela BECKMAN・COULTER) por um método de obstrução usando dispersão na frente e no lado do feixe de laser como índices e usando Fluorospheres Flow-Count® (fabricadas pela BECKMAN-COULTER), cujo padrão de contagem de partículas é conhecido, como padrão interno. Uma dose necessária para 50% de redução da contagem de linfócitos do grupo de veículo como 100% foi calculada e usada como valor de ED50 (mg/kg de peso corporal). O efeito de redução da contagem de linfócitos do sangue periférico de camundongo do composto de comparação 1-7 foi de 0,64 mg/kg de peso corporal no valor de ED50, o efeito de redução da contagem de linfócitos do sangue periférico de camundongo do composto 1-3, do composto 13-6, do composto 15-3 e do composto 28-6 foram de 0,04, 0,02, 0,02 e 0,03 mg/kg de peso corporal, respectivamente, em valor de ED50.
[000527] Ratos machos Sprague-Dawley (IGS) foram anestesiados por administração intraperitoneal de Nembutal (fabricado pela DAINIPPON PHARMACEUTICAL CO., LTD.), um sensor de pressão conectado a um transmissor de telemetria (TL11M2-C50-PTX, fabricado pela Data Sciences International) foi inserido na artéria abdominal, e o transmissor foi introduzido por via subcutânea dentro do abdomen. Os dados de pressão sangüínea e freqüência cardíaca foram registrados por um programa de análise (Dataquest A.R.T., Data Science) através de um receptor (RPC-1, fabricado pela Data Sciences International).
Quando se passaram 10 dias a 2 semanas da cirurgia, foi confirmada a recuperação do ritmo circadiano da freqüência cardíaca, e os ratos foram submetidos ao experimento. O composto da presente invenção foi suspendido em 0,5% de hidroxipropilmetilcelulose (fabricada pela Shin-Etsu Chemical Co., Ltd.) e administrado por via oral. A freqüência cardíaca foi medida a partir de 24 horas antes da administração até 72 horas depois da administração. O Composto 1-3 não afetou a freqüência cardíaca do rato até a dose de 30 mg/kg de peso corporal.
Quando se passaram 10 dias a 2 semanas da cirurgia, foi confirmada a recuperação do ritmo circadiano da freqüência cardíaca, e os ratos foram submetidos ao experimento. O composto da presente invenção foi suspendido em 0,5% de hidroxipropilmetilcelulose (fabricada pela Shin-Etsu Chemical Co., Ltd.) e administrado por via oral. A freqüência cardíaca foi medida a partir de 24 horas antes da administração até 72 horas depois da administração. O Composto 1-3 não afetou a freqüência cardíaca do rato até a dose de 30 mg/kg de peso corporal.
[000528] Ratos machos Sprague-Dawley (IGS) são anestesiados por administração intraperitoneal de Nembutal (fabricado pela DAINIPPON PHARMACEUTICAL CO., LTD.), e fixados na posição dorsal. São montados eletrodos sobre os quatro membros, é registrado o eletrocardiograma por um fio de membro-padrão II usando um amplificador de eletrocardiograma (AC-601G, fabricado pela NIHON KOHDEN CORPORATION). A freqüência cardíaca é contada usando uma unidade medidora da freqüência cardíaca corrente (AT-601G, fabricada pela NIHON KOHDEN CORPORATION) e uma onda eletrocardiográfica como um gatilho. O composto de teste é dissolvido em 20% de ciclodextrina (fabricado pela NIHON SHOKUHIN KAKO CO., LTD) e administrado por via intravenosa durante 30 segundos em uma dose de 0,001 a 10 mg/kg de peso corporal. A freqüência cardíaca é medida antes da administração, e 1, 2, 3, 4, 5, 10 e 15 min depois da administração.
[000529] A partir dos resultados do Exemplo Experimental 1 mencionado acima, como o composto da presente invenção tem uma ação superior de redução de linfócitos do sangue periférico, pode-se esperar que apresente uma ação imunossupressiva superior, ação supressiva de rejeição e ação supressiva de alergia, e é considerado eficaz para o tratamento ou a profilaxia de doenças auto-imunes; profilaxia ou supressão de resistência ou rejeição aguda ou rejeição crônica de transplante de órgão ou tecido; tratamento ou profilaxia de doença enxerto-versus-hospedeiro (GvH) devido o transplante de medula óssea; ou tratamento ou profilaxia de doenças alérgicas. Além disso, a partir dos resultados do Exemplo Experimental 2 mencionado acima, o composto da presente invenção é considerado um composto apresentando reduzidos efeitos colaterais tais como bradicardia e similares.
[000530] Este requerimento se baseia em um requerimento de patente No. 2005-361363 arquivado no Japão, cujo conteúdo é incorporado na totalidade aqui, a este requerimento de patente, por esta referência.
Claims (17)
- Composto, caracterizado pelo fato de que é representado pela seguinte fórmula (I)na qual:
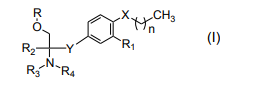
R é um átomo de hidrogênio ou P(=O)(OH)2,
X é um átomo de oxigênio ou um átomo de enxofre,
Y é CH2CH2 ou CH=CH,
R1 é trifluorometila, difluorometila ou ciano,
R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio,
R3 e R4 podem ser os mesmos ou diferentes e cada um é um átomo de hidrogênio ou alquila tendo um número de carbonos de 1 a 4, e
n é 5 a 8,
ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo. - Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que R3 e R4 são cada um um átomo de hidrogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com a reivindicação 1 ou 2, caracterizado pelo fato de que apresenta a seguinte fórmula (Ia) ou (Ib)na qual:

R é hidrogênio ou P(=O)(OH)2,
X é um átomo de oxigênio ou um átomo de enxofre,
R1 é trifluorometila, difluorometila ou ciano,
R2 é alquila tendo um número de carbonos de 1 a 4 e opcionalmente substituído por um ou mais grupos hidroxila ou um ou mais átomos de halogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo. - Composto, de acordo com qualquer uma das reivindicações 1 a 3, caracterizado pelo fato de que X é um átomo de oxigênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 4, caracterizado pelo fato de que Y é CH2CH2, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 5, caracterizado pelo fato de que R1 é trifluorometila ou difluorometila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 6, caracterizado pelo fato de que R1 é trifluorometila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 7, caracterizado pelo fato de que R2 é metila opcionalmente substituída por um ou mais grupos hidroxila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 8, caracterizado pelo fato de que R2 é hidroximetila, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 9, caracterizado pelo fato de que R é um átomo de hidrogênio, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 4, caracterizado pelo fato de que o composto da fórmula (I) é qualquer um dos seguintes (a)-(e), ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo:
- (a) 2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)etil] propano-1,3-diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
- (b) (E)-2-amino-2-[2-(4-heptilóxi-3-trifluorometilfenil)vinil] propano-1,3-diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
- (c) 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
- (d) (R)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- metilbutanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
- (e) 2-amino-2-[2-(3-ciano-4-heptiloxifenil)etil]propano-1,3- diol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo.
- Composto, de acordo com qualquer uma das reivindicações 1 a 4, caracterizado pelo fato de que o composto da fórmula (I) é qualquer um dos seguintes (f)-(j), ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo:
(f) 2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- (fosforiloximetil)butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
(g) (E)-2-amino-4-(4-heptilóxi-3-trifluorometilfenil)-2- (fosforiloximetil)-3-buten-1-ol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
(h) éster de mono[2-amino-4-(4-heptilóxi-3- trifluorometilfenil)-2-metilbutílico] do ácido fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
(i) éster de (R)-mono[2-amino-4-(4-heptilóxi-3- trifluorometilfenil)-2-metilbutil] do ácido fosfórico, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo;
(j) 2-amino-4-(3-ciano-4-heptiloxifenil)-2-(fosforiloximetil) butanol, ou um sal de adição de ácido farmaceuticamente aceitável do mesmo, ou um hidrato do mesmo, ou um solvato do mesmo. - Composto, de acordo com a reivindicação 1, caracterizado pelo fato de que é 2-amino-2-[2-(4-heptilóxi-3- trifluorometilfenil)etil] propano-1,3-diol, ou um cloridrato do mesmo.
- Composição farmacêutica, caracterizada pelo fato de que compreende um composto, como definido em qualquer uma das reivindicações 1 a 13, e um veículo farmaceuticamente aceitável,
em que o veículo farmaceuticamente aceitável é selecionado dentre excipiente, aglutinante, desintegrante, corrigente, aroma, emulsificante, diluente e solubilizante. - Composição farmacêutica, de acordo com a reivindicação 14, caracterizada pelo fato de que é para uso no tratamento ou profilaxia de doenças auto-imunes; profilaxia ou supressão de resistência ou rejeição aguda ou rejeição crônica de transplante de órgão ou tecido; tratamento ou profilaxia de doença enxerto-versus-hospedeiro (GvH) devido o transplante da medula óssea; ou tratamento ou profilaxia de doenças alérgicas.
- Composição farmacêutica, de acordo com a reivindicação 15, caracterizada pelo fato de que a doença auto-imune é artrite reumatóide, esclerose múltipla, encefalomielite, lúpus eritematoso sistêmico, nefrite do lúpus, síndrome nefrótica, psoríase ou diabete melito Tipo I.
- Composição farmacêutica, de acordo com a reivindicação 15, caracterizada pelo fato de que a doença alérgica é dermatite atópica, rinite alérgica ou asma.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005-361363 | 2005-12-15 | ||
| JP2005361363 | 2005-12-15 | ||
| PCT/JP2006/325016 WO2007069712A1 (ja) | 2005-12-15 | 2006-12-15 | アミン化合物及びその医薬用途 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| BRPI0619894A2 BRPI0619894A2 (pt) | 2011-10-25 |
| BRPI0619894B1 true BRPI0619894B1 (pt) | 2021-04-06 |
| BRPI0619894B8 BRPI0619894B8 (pt) | 2021-05-25 |
Family
ID=38163014
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0619894A BRPI0619894B8 (pt) | 2005-12-15 | 2006-12-15 | composto de amina e composição farmacêutica |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8809304B2 (pt) |
| EP (1) | EP1961734B1 (pt) |
| JP (1) | JP4833998B2 (pt) |
| KR (1) | KR101346527B1 (pt) |
| CN (1) | CN101346346B (pt) |
| AT (1) | ATE524432T1 (pt) |
| AU (1) | AU2006325931B8 (pt) |
| BR (1) | BRPI0619894B8 (pt) |
| CA (1) | CA2633374C (pt) |
| DK (1) | DK1961734T3 (pt) |
| ES (1) | ES2369520T3 (pt) |
| PL (1) | PL1961734T3 (pt) |
| PT (1) | PT1961734E (pt) |
| RU (1) | RU2433117C2 (pt) |
| TW (1) | TWI451865B (pt) |
| WO (1) | WO2007069712A1 (pt) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2007217810B2 (en) * | 2006-02-21 | 2012-10-11 | The Ohio State University Research Foundation | Anticancer agents |
| ES2414205T3 (es) * | 2006-06-02 | 2013-07-18 | The Ohio State University Research Foundation | Agentes terapéuticos para el tratamiento de linfoma de células del manto |
| TWI439263B (zh) * | 2007-06-14 | 2014-06-01 | Mitsubishi Tanabe Pharma Corp | 胺化合物及其醫藥用途 |
| WO2009025767A1 (en) | 2007-08-17 | 2009-02-26 | Research Foundation Of The City University Of New York | Boron dipyrromethene difluoro (bodipy) conjugates |
| JP2011136905A (ja) * | 2008-03-27 | 2011-07-14 | Mitsubishi Tanabe Pharma Corp | ベンゼン化合物及びその医薬用途 |
| EP2400960B1 (en) * | 2009-02-24 | 2013-04-17 | Novartis AG | Ceramide-analogous metabolites |
| JP2012523458A (ja) * | 2009-04-13 | 2012-10-04 | ザ オハイオ ステート ユニバーシティー リサーチ ファウンデーション | タンパク質ホスファターゼ2a活性化剤 |
| CA2759011A1 (en) * | 2009-04-17 | 2010-10-21 | The Ohio State University Research Foundation | Antiadhesion agents |
| US8309768B2 (en) | 2010-11-29 | 2012-11-13 | The Ohio State University Research Foundation | FTY720-derived anticancer agents |
| EP2842937B1 (en) | 2012-04-23 | 2018-01-17 | Mitsubishi Tanabe Pharma Corporation | Amine compound and use thereof for medical purposes |
| JP6961595B2 (ja) * | 2016-07-29 | 2021-11-05 | 田辺三菱製薬株式会社 | 4−アルコキシ−3−トリフルオロメチルベンジルアルコールの製造方法 |
| GB201715786D0 (en) * | 2017-09-29 | 2017-11-15 | Univ College Cardiff Consultants Ltd | Compounds |
| AU2020372647B2 (en) | 2019-10-31 | 2026-02-05 | Idorsia Pharmaceuticals Ltd | Combination of a CXCR7 antagonist with an S1P1 receptor modulator |
| US12569455B2 (en) | 2020-02-06 | 2026-03-10 | Mitsubishi Tanabe Pharma Corporation | Therapeutic agent for myalgic encephalomyelitis/chronic fatigue syndrome |
| US20230115921A1 (en) * | 2020-03-31 | 2023-04-13 | Mitsubishi Tanabe Pharma Corporation | Pharmaceutical composition |
| EP4288041A1 (en) | 2021-02-08 | 2023-12-13 | Bausch Health Ireland Limited | Amiselimod for preventing, treating, or ameliorating ulcerative colitis |
| CN118812422B (zh) * | 2023-04-20 | 2025-11-25 | 国科大杭州高等研究院 | 用于单链鞘脂检测的maldi-ft-icr质谱探针及其制备方法和使用方法 |
| WO2025083549A1 (en) | 2023-10-16 | 2025-04-24 | Sun Pharma Advanced Research Company Limited | Methods and combinations of inhibitors of il-23 pathway and modulators of s1p signaling pathway for the treatment of autoimmune disorders |
| WO2025134072A1 (en) | 2023-12-21 | 2025-06-26 | Bausch Health Ireland Limited | Methods for preventing, treating, or ameliorating ulcerative colitis in certain patient populations with amiselimod |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5604229A (en) * | 1992-10-21 | 1997-02-18 | Yoshitomi Pharmaceutical Industries, Ltd. | 2-amino-1,3-propanediol compound and immunosuppressant |
| US5948820A (en) * | 1994-08-22 | 1999-09-07 | Yoshitomi Pharmaceutical Industries, Ltd. | Benzene compound and pharmaceutical use thereof |
| DE69524962D1 (de) * | 1994-08-22 | 2002-02-14 | Welfide Corp | Benzolderivate und deren medizinische verwendung |
| GB9624038D0 (en) | 1996-11-19 | 1997-01-08 | Sandoz Ltd | Organic compounds |
| GB9707307D0 (en) | 1997-04-10 | 1997-05-28 | Ciba Geigy Ag | Polypeptide markers |
| EP1377593B1 (en) * | 2001-03-26 | 2005-12-28 | Novartis AG | 2-amino-propanol derivatives |
| ES2322442T3 (es) | 2001-06-08 | 2009-06-22 | Novartis Ag | Tratamiento o profilaxis del rechazo de injertos de celulas productoras de insulina. |
| US7612238B2 (en) * | 2002-09-13 | 2009-11-03 | Novartis Ag | Amino-propanol derivatives |
| EP1622860B1 (en) | 2003-04-30 | 2012-02-29 | Novartis AG | AMINO-PROPANOL DERIVATIVES AS SPHINGOSINE-1-PHOSPHATE RECEPTOR MODULATORs |
| EP1484057A1 (en) * | 2003-06-06 | 2004-12-08 | Aventis Pharma Deutschland GmbH | Use of 2-amino-1-3-propanediol derivatives for the manufacture of a medicament for the treatment of various types of pain |
| GB0313612D0 (en) * | 2003-06-12 | 2003-07-16 | Novartis Ag | Organic compounds |
| TW200611687A (en) | 2004-07-29 | 2006-04-16 | Sankyo Co | Pharmaceutical compositions used for immunosuppressant |
-
2006
- 2006-12-15 KR KR1020087016615A patent/KR101346527B1/ko not_active Expired - Fee Related
- 2006-12-15 DK DK06834759.0T patent/DK1961734T3/da active
- 2006-12-15 RU RU2008128875/04A patent/RU2433117C2/ru active
- 2006-12-15 WO PCT/JP2006/325016 patent/WO2007069712A1/ja not_active Ceased
- 2006-12-15 PL PL06834759T patent/PL1961734T3/pl unknown
- 2006-12-15 EP EP06834759A patent/EP1961734B1/en active Active
- 2006-12-15 AU AU2006325931A patent/AU2006325931B8/en active Active
- 2006-12-15 TW TW095147069A patent/TWI451865B/zh not_active IP Right Cessation
- 2006-12-15 ES ES06834759T patent/ES2369520T3/es active Active
- 2006-12-15 US US12/086,419 patent/US8809304B2/en active Active
- 2006-12-15 JP JP2007550231A patent/JP4833998B2/ja active Active
- 2006-12-15 CA CA2633374A patent/CA2633374C/en active Active
- 2006-12-15 BR BRPI0619894A patent/BRPI0619894B8/pt active IP Right Grant
- 2006-12-15 CN CN2006800472557A patent/CN101346346B/zh active Active
- 2006-12-15 AT AT06834759T patent/ATE524432T1/de active
- 2006-12-15 PT PT06834759T patent/PT1961734E/pt unknown
-
2014
- 2014-06-17 US US14/306,902 patent/US20140296183A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| CN101346346B (zh) | 2012-08-22 |
| AU2006325931B8 (en) | 2012-09-20 |
| ES2369520T3 (es) | 2011-12-01 |
| AU2006325931A8 (en) | 2012-09-20 |
| US8809304B2 (en) | 2014-08-19 |
| RU2433117C2 (ru) | 2011-11-10 |
| AU2006325931B2 (en) | 2012-09-06 |
| US20140296183A1 (en) | 2014-10-02 |
| KR101346527B1 (ko) | 2013-12-31 |
| DK1961734T3 (da) | 2011-12-05 |
| TW200732284A (en) | 2007-09-01 |
| RU2008128875A (ru) | 2010-01-20 |
| US20090137530A1 (en) | 2009-05-28 |
| CN101346346A (zh) | 2009-01-14 |
| PL1961734T3 (pl) | 2012-02-29 |
| JP4833998B2 (ja) | 2011-12-07 |
| WO2007069712A1 (ja) | 2007-06-21 |
| CA2633374C (en) | 2013-10-01 |
| PT1961734E (pt) | 2011-10-06 |
| TWI451865B (zh) | 2014-09-11 |
| EP1961734A1 (en) | 2008-08-27 |
| BRPI0619894A2 (pt) | 2011-10-25 |
| ATE524432T1 (de) | 2011-09-15 |
| KR20080080167A (ko) | 2008-09-02 |
| AU2006325931A1 (en) | 2007-06-21 |
| JPWO2007069712A1 (ja) | 2009-05-28 |
| CA2633374A1 (en) | 2007-06-21 |
| BRPI0619894B8 (pt) | 2021-05-25 |
| EP1961734A4 (en) | 2009-03-25 |
| EP1961734B1 (en) | 2011-09-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20140296183A1 (en) | Amine compound and use thereof for medical purposes | |
| US8129361B2 (en) | Amine compound and pharmaceutical use thereof | |
| ES2392167T3 (es) | Derivados aminopropanol como moduladores del receptor esfingosina-1-fosfato | |
| US7728020B2 (en) | Amino acid derivatives | |
| ES2491224T3 (es) | Compuesto de 2-aminobutanol y su uso para fines médicos | |
| WO2009119858A1 (ja) | ベンゼン化合物及びその医薬用途 | |
| CN113784754A (zh) | 花生四烯乙醇胺化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06T | Formal requirements before examination [chapter 6.20 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001 |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 10 (DEZ) ANOS CONTADOS A PARTIR DE 06/04/2021, OBSERVADAS AS CONDICOES LEGAIS. |
|
| B16C | Correction of notification of the grant [chapter 16.3 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 15/12/2006 OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF |